
· proporcionando de este modo incremento en los niveles plasmáticos de lopinavir. ... La...
68
I .; MINISTERIO DE SALUD DIRECCION DE ATENCIQN AL CLiENTE Telefax: (506) 2222-51-49 Apartado: 10123-1000, San Jose DAC·3889·10·RM·CP 25 de agosto de 2010 Licda. Michelle Ortiz de Medrano Gerente de Asuntos Regulatorlos ABBOTT FAX: 2420·9748 Eslimada licenciada: En atenci6n a su nota recibida con contrasena MS·DR·41731·10, segun el anlfllisis de los evaluadores, se Ie informa que: PRODUCTO: N° DE REGISTRO: LABORATORIO; TRAMITE 1: CONDlCI6N: TRAMITE2: EMPAQUE PRIMA RIO: EMP. SECUNDARIO: INSERTO: Atentamente, DTNmem t!: Archivo Kalelra Soluci6n Oral 1005·AP·7303 Abbott Laboratories USA Actualizacion de Monografia Aprobado Actualizacion de Arte AprObado Aprobado Aprobado "De fa arendon de fa enfermedad hada 10 promoclon de 10 salud" --"""-
Transcript of · proporcionando de este modo incremento en los niveles plasmáticos de lopinavir. ... La...

I .;
MINISTERIO DE SALUD
DIRECCION DE ATENCIQN AL CLiENTE
Telefax: (506) 2222-51-49 Apartado: 10123-1000, San Jose
DAC·3889·10·RM·CP 25 de agosto de 2010
Licda. Michelle Ortiz de Medrano Gerente de Asuntos Regulatorlos ABBOTT FAX: 2420·9748
Eslimada licenciada:
En atenci6n a su nota recibida con contrasena MS·DR·41731·1 0, segun el anlfllisis de los evaluadores, se Ie informa que:
PRODUCTO: N° DE REGISTRO: LABORATORIO;
TRAMITE 1: CONDlCI6N:
TRAMITE2: EMPAQUE PRIMA RIO: EMP. SECUNDARIO: INSERTO:
Atentamente,
DTNmem t!: Archivo
Kalelra Soluci6n Oral 1005·AP·7303 Abbott Laboratories USA
Actualizacion de Monografia Aprobado
Actualizacion de Arte AprObado Aprobado Aprobado
"De fa arendon de fa enfermedad hada 10 promoclon de 10 salud"
--"""-

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
1. NOMBRE COMERCIAL DEL MEDICAMENTO Y DENOMINACION COMUN INTERNACIONAL.
KALETRA LOPINAVIR/RITONAVIR CAPSULAS BLANDAS / SOLUCION ORAL 2. FORMULA QUIMICA ESTRUCTURAL MOLECULAR Y EMPIRICA DEL PRINCIPIO
ACTIVO Lopinavir se describe químicamente como [1S-[1R*,(R*),3R*,4R*]]-N-[4-[[2,6-dimetilfenoxi)acetil]amino]-3-hidroxi-5-fenil-1-(fenilmetil)pentil]tetrahidroalfa-(1-metiletil)-2-oxo-1(2H)-pirimidinacetamida. Su fórmula molecular es C37H48N4O5, y su peso molecular es 628.80. Lopinavir tiene la siguiente fórmula estructural:
Ritonavir se describe químicamente como ácido 10-Hidroxi-2-metil-5-(1-metiletil)-1-[2-(1-metiletil)-4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraazotriadecan-13-oico, 5-tiazolilmetil éster, [5S-(5R*,8R*,10R*,11R*)]. Su fórmula molecular es C37H48N6O5S2, y su peso molecular es 720.95. Ritonavir tiene la siguiente fórmula estructural:
Lopinavir es un polvo blanco o ligeramente oscurecido. Es soluble libremente en metanol y etanol, soluble en isopropanol y prácticamente insoluble en agua. 3. DESCRIPCION DEL PRODUCTO TERMINADO KALETRA® (lopinavir/ritonavir) es una coformulación de lopinavir y ritonavir. Lopinavir es un inhibidor de la proteasa del VIH-1 y VIH-2. Al venir coformulado en KALETRA®, ritonavir

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
2
inhibe el metabolismo de lopinavir mediado por el CYP3A (Citocromo P450 3A), proporcionando de este modo incremento en los niveles plasmáticos de lopinavir. KALETRA® cápsulas está disponible para administración oral a concentraciones de 133.3 mg de lopinavir y 33.3 mg de ritonavir con los siguientes ingredientes inactivos: Amarillo FD&C N° 6, gelatina, glicerina, ácido oleico, aceite de ricino hidrogenado polioxil 35, propilenglicol, sorbitol especial, dióxido de titanio y agua. KALETRA® solución oral está disponible para administración oral como 80 mg de lopinavir y 20 mg de ritonavir por mililitro, con los siguientes ingredientes inactivos: Acesulfame de potasio, alcohol, aroma artificial de dulce de algodón, ácido cítrico, glicerina, jarabe de maíz alto en fructuosa, sabor Magnasweet-110, mentol, sabor a vainilla natural y artificial, aceite de menta, aceite de ricino polioxil 40 hidrogenado, povidona, propilenglicol, sacarina sódica, cloruro de sodio, citrato de sodio y agua. KALETRA® solución oral contiene 42.4% de alcohol (v/v). 4. CLASIFICACION FARMACOLOGICA Y TERAPEUTICA KALETRA es un inhibidor de la proteasa del HIV con actividad contra el virus de la inmunodeficiencia adquirida. 5. ACCIONES TERAPEUTICAS FARMACOLOGÍA CLINICA Microbiología Mecanismo de acción: El Lopinavir es un inhibidor de la proteasa del VIH-1 y VIH-2, impide el corte de la poliproteína gag-pol, lo que da como resultado la producción de partículas virales inmaduras, no infecciosas. Actividad antiviral in vitro: La actividad antiviral in vitro de lopinavir contra las cepas VIH de laboratorio y las aisladas del VIH fue evaluada en líneas de células linfoblásticas y linfocitos sanguíneos periféricos infectadas agudamente. En ausencia de suero humano el promedio de la concentración efectiva 50% (CE50) de lopinavir contra cinco diferentes cepas del VIH-1 de laboratorio estuvo en el rango de 10-27 nM (0.006-0.017 μg/mL, 1 μg/mL = 1.6 μM) y en el rango de 4-11 nM (0.003-0.007 μg/mL) contra varios aislados clínicos del VIH-1 (n = 6). En la presencia de suero humano al 50%, la CE50 promedio de lopinavir contra estas cinco cepas de laboratorio estuvo en el rango de 65-289 nM (0.04-0.18 μg/mL), representando una atenuación de 7-11 veces. Los estudios de actividad farmacológica en combinación con lopinavir y otros inhibidores de proteasa o inhibidores de la transcriptasa inversa no han sido completados. Resistencia: Aislados de VIH-1 con susceptibilidad reducida a lopinavir han sido seleccionados in vitro. Al parecer la presencia de ritonavir no tiene incidencia sobre la selección de virus resistente a lopinavir in vitro. La selección de resistencia a KALETRA® en pacientes no experimentados para tratamiento con antirretroviral todavía no ha sido caracterizada. En un estudio fase III con 653 pacientes

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
3
no experimentados para tratamiento antirretroviral (estudio 863), los virus aislados de los pacientes con ARN viral cuantificable (> 400 copias/mL) fueron analizados en las semanas 24, 32, 40 y 48. No existe evidencia de resistencia genotípica o fenotípica a KALETRA® en 37 pacientes evaluables (0%). La evidencia de resistencia genotípica a nelfinavir, definida como presencia de mutación D30N y/o L90M en la proteasa del VIH se detectó en 25/76 (33%) de los pacientes tratados con nelfinavir evaluables. La selección de resistencia a KALETRA® en pacientes pediátricos no experimentados (estudio 940) es consistente con lo observado en adultos (estudio 863). Se ha observado que la resistencia a KALETRA® aparece en pacientes tratados con otros inhibidores de las proteasas, previos a la terapia con KALETRA®. En un estudio fase II de 227 pacientes no experimentados para tratamiento antirretroviral y experimentados con inhibidor de proteasa, los aislados de 4 de 23 pacientes con ARN viral cuantificable (> 400 copias/mL) después de tratamiento con KALETRA® por 12 a 100 semanas reveló una susceptibilidad significativamente reducida a lopinavir comparados con los aislados virales basales correspondientes. Tres de estos pacientes habían recibido previamente tratamiento con un solo inhibidor de proteasa (nelfinavir, indinavir, o saquinavir) y un paciente había recibido tratamiento con múltiples inhibidores de proteasa (indinavir, saquinavir y ritonavir). Todos los pacientes tenían al menos 4 mutaciones asociadas con resistencia al inhibidor de proteasa inmediatamente antes a la terapia con KALETRA®. Después del rebote viral, todos los aislados de estos pacientes contenían mutaciones adicionales, algunas de las cuales son reconocidas o se asocian con resistencia al inhibidor de proteasa. Sin embargo, existen datos insuficientes en este momento para identificar patrones mutacionales asociados con lopinavir en aislados de pacientes bajo terapia con KALETRA®. La evaluación de estos patrones mutacionales está bajo estudio. Resistencia Cruzada Estudios Preclínicos. Se han observado grados variables de resistencia cruzada entre inhibidores de proteasa. Se ha determinado la actividad in vitro de lopinavir contra los aislados clínicos de pacientes previamente tratados con un solo inhibidor de proteasa. Los aislados que revelaron susceptibilidad reducida > 4 veces a nelfinavir (n = 13) y saquinavir (n = 4), mostraron susceptibilidad < 4 veces reducida a lopinavir. Los aislados con susceptibilidad > 4 veces a indinavir (n = 16) y ritonavir (n = 3) revelaron un promedio de susceptibilidad reducida de 5.7 y 8.3 veces a lopinavir, respectivamente. Los aislados de pacientes previamente tratados con dos o más inhibidores de proteasa mostraron mayores reducciones en la susceptibilidad a lopinavir como se describe en el siguiente párrafo. Resistencia cruzada durante tratamiento con KALETRA® Hay poca información disponible sobre resistencia cruzada de virus seleccionados durante la terapia con KALETRA®. Aislados de cuatro pacientes tratados previamente con uno o más inhibidores de proteasa que desarrollaron resistencia fenotípica a lopinavir durante el tratamiento con KALETRA®, permanecieron con la resistencia cruzada o desarrollaron resistencia cruzada a ritonavir, indinavir y nelfinavir. Todos los virus obtenidos después del rebote permanecieron sensibles completamente o tuvieron solamente una modesta reducción de la susceptibilidad a amprenavir (más de 8.5 veces concurrente con 99 veces de aumento

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
4
de la resistencia a lopinavir). Los virus obtenidos de dos de estos pacientes que no eran previamente resistentes a saquinavir permanecieron totalmente susceptibles a saquinavir. Correlación genotípica de la reducción en la respuesta virológica en pacientes bajo terapia antirretroviral que inician un régimen combinado con KALETRA® Se ha demostrado que la respuesta virológica a KALETRA® se ve afectada por la presencia de tres o más de las siguientes sustituciones de aminoácidos en la línea de base de proteasas: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V. La tabla 1 muestra la respuesta virológica a la semana 48 (VIH ARN<400 copias/mL de acuerdo al número de mutaciones resistentes al inhibidor de proteasas en los estudios 888, 765 y 957 (Tabla 1) Tabla 1: Respuesta virológica (ARN VIH <400 copias/mL) a la semana 48 por susceptibilidad a KALETRA® en línea base y por el número de sustituciones de proteasa asociada con una respuesta reducida a KALETRA®.1 Número de mutaciones de inhibidores de proteasa en línea base1
Estudio 888 (Experimentado2 con un solo Inhibidor de proteasa, Nuevo para ITRNN) n=130
Estudio 765 (Experimentado3 con un solo Inhibidor de proteasa, Nuevo para ITRNN) n=56
Estudio 957 (Experimentado4 con Múltiples inhibidores de proteasa-, Nuevo para ITRNN) n=50
0-2 76/103 (74%) 34/45 (76%) 19/20 (95%) 3-5 13/26 (50%) 8/11 (73%) 18/26 (69%) 6 o más 0/1 (0%) N/A 1/4 (25%) 1 Sustituciones consideradas en el análisis incluido L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, e I84V. 2 43% indinavir, 42% nelfinavir, 10% ritonavir, 15% saquinavir. 3 41% indinavir, 38% nelfinavir, 4% ritonavir, 16% saquinavir. 4 86% indinavir, 54% nelfinavir, 80% ritonavir, 70% saquinavir. La tabla 2 muestra la respuesta virológica a la semana 48 (VIH-1 ARN <50 copias/mL) en el estudio 802 de acuerdo con el número de mutaciones resistentes asociados a lopinavir enumerados en la tabla 1 presentes en el inicio del estudio. No hay datos suficientes para apoyar una administración única diaria de Lopinavir/Ritonavir en pacientes adultos con tres o más mutaciones asociadas a lopinavir. Tabla 2. Respuesta virológica semanas (VIH-1 ARN <50 copias/mL) a la semana 48 por punto de inicio por número de sustituciones asociadas a la proteasa con respuesta reducida a Lopinavir/Ritonavir. Número de sustituciones
de inhibidores de proteasa Estudio 802 (tratamiento-
Estudio 802 (tratamiento-

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
5
en el punto de inicio experimentado2) Lop/Rit una vez al día + ITRN n=268
experimentado3) Lop/Rit BID + ITRN n=264
0-2 167/255 (65%) 154/250 (62%) 3-5 4/13 (31%) 8/14 (57%)
6 o más NA NA 1 Sustituciones consideradas en el análisis incluyen L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, e I84V 2 88% experimentados con ITRNN, 47% experimentados con inhibidores de la proteasa (24% nelfinavir, 19% indinavir, 13% atazanavir) 3 81% experimentados con ITRNN, 45% experimentados con inhibidores de la proteasa (20% nelfinavir, 17% indinavir, 13% atazanavir) 6. DESTINO EN EL ORGANISMO (FARMACOCINETICA) Las propiedades farmacocinéticas de lopinavir coadministrado con ritonavir han sido evaluadas en voluntarios adultos sanos y en pacientes infectados con el VIH; no se observaron diferencias sustanciales entre los dos grupos. Lopinavir es metabolizado de forma esencialmente completa por el CYP3A. Ritonavir inhibe el metabolismo de lopinavir, incrementando por esto los niveles plasmáticos de lopinavir. A través de los estudios, la administración de KALETRA® 400/100 mg, dos veces al día da lugar a concentraciones plasmáticas promedio de lopinavir en estado de equilibrio 15 a 20 veces más altas que las de ritonavir en pacientes infectados con el VIH. Los niveles plasmáticos de ritonavir son menos del 7% de los obtenidos después de la dosis de ritonavir de 600 mg dos veces al día. La CE50 antiviral in vitro de lopinavir es aproximadamente 10 veces más baja que la de ritonavir. Por lo tanto, la actividad antiviral de KALETRA® se debe a lopinavir. La Figura 1 muestra las concentraciones plasmáticas promedio en estado de equilibrio de lopinavir y ritonavir después de tomar KALETRA® 400/100 mg, dos veces al día con alimentos por 3 semanas de un estudio farmacocinético en sujetos adultos infectados con el VIH (n = 19).

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
6
Absorción: En un estudio farmacocinético en sujetos VIH positivo (n = 19) a quienes se les administraron múltiples dosis de KALETRA® 400/100 mg, dos veces al día con los alimentos por tres semanas se produjo una concentración máxima promedio en plasma de lopinavir ± DE (Cmáx) de 9.8 ± 3.7 μg/mL, presentándose aproximadamente 4 horas después de la administración. La concentración promedio en el valle en estado de equilibrio antes de la dosis matutina fue 7.1 ± 2.9 μg/mL y la concentración mínima en el intervalo de las dosis fue de 5.5±2.7 μg/mL. El ABC de lopinavir sobre un intervalo de dosificación de 12 horas tuvo un promedio de 92.6 ± 36.7 μg•h/mL. No se ha establecido la biodisponibilidad absoluta de lopinavir coformulado con ritonavir en humanos. Bajo condiciones de no ayuno (500 Kcal, 25% de grasa), las concentraciones de lopinavir fueron similares después de la administración de KALETRA® coformulada, cápsulas de gelatina blanda y solución oral. Cuando se administró bajo condiciones en ayunas, tanto el ABC como la Cmáx promedio de lopinavir fueron 22% más bajas para KALETRA® solución oral con relación a la forma farmacéutica en cápsulas de gelatina blanda. Efectos de los alimentos sobre la absorción oral: Las Cápsulas de gelatina blanda y la Solución Oral de Kaletra® co-formulado, fueron bioequivalentes bajo condiciones de no ayuno (comida moderada en grasa). La administración de una sola dosis de 400/100 mg de KALETRA® cápsulas de gelatina blanda con una comida moderada en grasa (500-682 Kcal, 23 a 25% calorías de grasa) se asoció con incremento promedio de 48 y 23% en el ABC y la Cmáx de lopinavir, respectivamente, con relación al estado en ayunas. Para KALETRA® solución oral, los incrementos correspondientes en el ABC y la Cmáx de lopinavir fueron 80 y 54%, respectivamente. Con relación al estado en ayunas, la administración de KALETRA® con comida alta en grasa (872 Kcal, 56% de grasa) incrementó el ABC y la Cmáx de lopinavir en 97 y 43%, respectivamente, para las cápsulas de gelatina blanda, y 130 y 56%,

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
7
respectivamente, para la solución oral. Para aumentar la biodisponibilidad y minimizar la variabilidad farmacocinética se debe tomar KALETRA® con los alimentos. Distribución: En estado de equilibrio lopinavir se une aproximadamente 98-99% a las proteínas plasmáticas. Lopinavir se une tanto a la glicoproteína ácida alfa-1 (GAA) y a la albúmina; sin embargo, tiene afinidad más alta por la GAA. En estado de equilibrio, la unión de lopinavir a las proteínas permanece constante sobre el rango de concentraciones observadas después de 400/100 mg de KALETRA® dos veces al día, y es similar entre voluntarios sanos y pacientes VIH positivo. Metabolismo: Experimentos in vitro con microsomas hepáticos humanos indican que lopinavir sufre primariamente metabolismo oxidativo. Lopinavir es metabolizado principalmente por el sistema hepático del citocromo P450, casi exclusivamente por la isoenzima CYP3A. Ritonavir es un potente inhibidor del CYP3A lo que retrasa el metabolismo de lopinavir, y por lo tanto incrementa los niveles plasmáticos de lopinavir. Un estudio con 14C-lopinavir en humanos mostró que 89% de la radioactividad en plasma después de una dosis única de 400/100 mg de KALETRA® se debió al fármaco padre. Se han identificado al menos 13 metabolitos oxidativos de lopinavir en el hombre. Se ha demostrado que ritonavir induce las enzimas metabólicas, lo que resulta en inducción de su propio metabolismo. Las concentraciones pre dosis de lopinavir declinan con el tiempo durante la dosificación múltiple, estabilizándose después de aproximadamente 10 a 16 días. Eliminación: Después de una dosis de 400/100 mg de 14C-lopinavir/ritonavir, aproximadamente 10.4 ± 2.3% y 82.6 ± 2.5% de una dosis administrada de 14C-lopinavir puede ser detectada en la orina y las heces, respectivamente, después de 8 días. Lopinavir sin cambios es aproximadamente 2.2 y 19.8% de la dosis administrada en orina y heces, respectivamente. Después de dosificación múltiple, menos del 3% de la dosis de lopinavir se excreta sin cambios en la orina. La depuración oral aparente (CL/F) de lopinavir es 5.98 ± 5.75 L/h (media ± DE, N=19). Dosificación una vez al día: La farmacocinética de la administración una sola vez al día de KALETRA® ha sido evaluada en pacientes VIH positivo no experimentados para tratamiento anti-retroviral. KALETRA® 800/200 mg fue administrado en combinación con emtricitabina 200 mg y tenofovir DF 300 mg como parte de un tratamiento administrado una sola vez al día. La dosificación múltiple de KALETRA® 800/200 mg, una sola vez al día por cuatro semanas junto con comida (n=24) produjo una concentración máxima promedio de 11.8 ± 3.7 μg/mL seis horas después de su administración. La concentración mínima de lopinavir observada antes de la dosis de la mañana fue de 3.2 ± 2.1 μg/mL, la concentración mínima en el intervalo de las dosis fue 1.7 ± 1.6 μg/mL. El ABC de lopinavir con la dosificación diaria tuvo un promedio de 154.1 ± 61.4 μg•h/mL. Efectos sobre el electrocardiograma Se evaluó el Intervalo QTcF en un estudio, randomizado, cruzado controlado activo y con placebo (moxifloxacina 400 mg una vez al día) en 39 adultos saludables con 10 medidas durante 12 horas en el día 3. El promedio máximo de diferencia (95% del l[imite de confianza superior) en el QTcF con placebo fue de 3.6(6.3) mseg y 13.1(15.8) mseg para la dosis de lopinavir/ritonavir 400/100 mg dos veces al día y supraterapéutica 800/200 mg dos veces al día lopinavir/ritonavir respectivamente. Los dos regímenes resultaron para el día tres en exposiciones que fueron aproximadamente 1.5 a 3 veces más altas que las observadas en las dosis una vez al día y dos veces al día en estado de equilibrio. Ningún sujeto experimentó

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
8
aumento en el intervalo QTcF ≥60 mseg sobre la línea basal o un intervalo QTcF que excediera de manera clínicamente significativa el límite de 500 mseg. Una prolongación modesta del intervalo PR también se evidenció en sujetos recibiendo lopinavir/ritonavir en el mismo estudio para el día 3. El intervalo PR máximo fue de 286 mseg y no se observó bloqueo de segundo o tercer grado (Ver Advertencias y Precauciones). Poblaciones especiales: Género, raza y edad: La farmacocinética de lopinavir no ha sido estudiada en pacientes ancianos. No se han observado diferencias farmacocinéticas relacionadas con el género en pacientes adultos. No se han identificado diferencias farmacocinéticas clínicamente importantes debidas a la raza. Pacientes pediátricos: La farmacocinética de KALETRA® 300/75 mg/m2 dos veces al día y 230/57.5 mg/m2 dos veces al día ha sido estudiada en un total de 53 pacientes pediátricos, con rango de edad de 6 meses a 12 años. El régimen de 230/57.5 mg/m2 dos veces al día sin nevirapina y el régimen de 300/75 mg/m2 dos veces al día con nevirapina proporcionó concentraciones plasmáticas de lopinavir similares a las obtenidas en pacientes adultos que recibían el régimen 400/100 mg BID (sin nevirapina). KALETRA® una vez al día no ha sido evaluado en pacientes pediátricos. Los valores promedio de lopinavir del ABC, Cmáx y Cmín en estado de equilibrio fueron 72.6 ± 31.1 μg•h/mL, 8.2 ± 2.9 y 3.4 ± 2.1 μg/mL, respectivamente. Para KALETRA® 230/57.5 mg/m2 dos veces al día sin nevirapina (n = 12), fueron 85.8 ± 36.9 μg•h/mL, 10.0 ± 3.3 y 3.6 ± 3.5 μg/mL, respectivamente después de KALETRA® 300/75 mg/m2 dos veces al día con nevirapina (n = 12). El régimen con nevirapina fue 7 mg/Kg dos veces al día (6 meses a 8 años) ó 4 mg/Kg dos veces al día (> 8 años). Insuficiencia renal: La farmacocinética de lopinavir no ha sido estudiada en pacientes con insuficiencia renal; sin embargo, ya que la depuración renal de lopinavir es insignificante, no se espera disminución en la depuración corporal total en pacientes con insuficiencia renal. Insuficiencia hepática: Lopinavir es metabolizado y eliminado principalmente por el hígado. La administración de múltiples dosis de lopinavir/ritonavir 400/100 mg dos veces al día a pacientes coinfectados con VIH y VHC con compromiso hepático leve a moderado resultó en un incremento del 30% en el ABC de lopinavir y 20% de incremento de la Cmax comparados con pacientes infectados con VIH con función hepática normal. Adicionalmente, la fijación a las proteínas plasmáticas de lopinavir fue menor en pacientes con compromiso hepático leve a moderado comparado con los controles (99.09 vs. 99.31%, respectivamente). KALETRA® no ha sido estudiado en pacientes con compromiso hepático severo (ver PRECAUCIONES). Interacciones fármaco-fármaco: véase también CONTRAINDICACIONES, ADVERTENCIAS y PRECAUCIONES: Interacciones medicamentosas. KALETRA® es inhibidor del isómero CYP3A del P450 in vitro. La coadministración de KALETRA® y de fármacos metabolizados primariamente por el CYP3A puede resultar en incremento de las concentraciones plasmáticas del otro fármaco, las cuales pueden aumentar o prolongar sus efectos terapéuticos y adversos (véase CONTRAINDICACIONES). KALETRA® no inhibe el CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 o CYP1A2 a las concentraciones clínicamente relevantes.
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
9
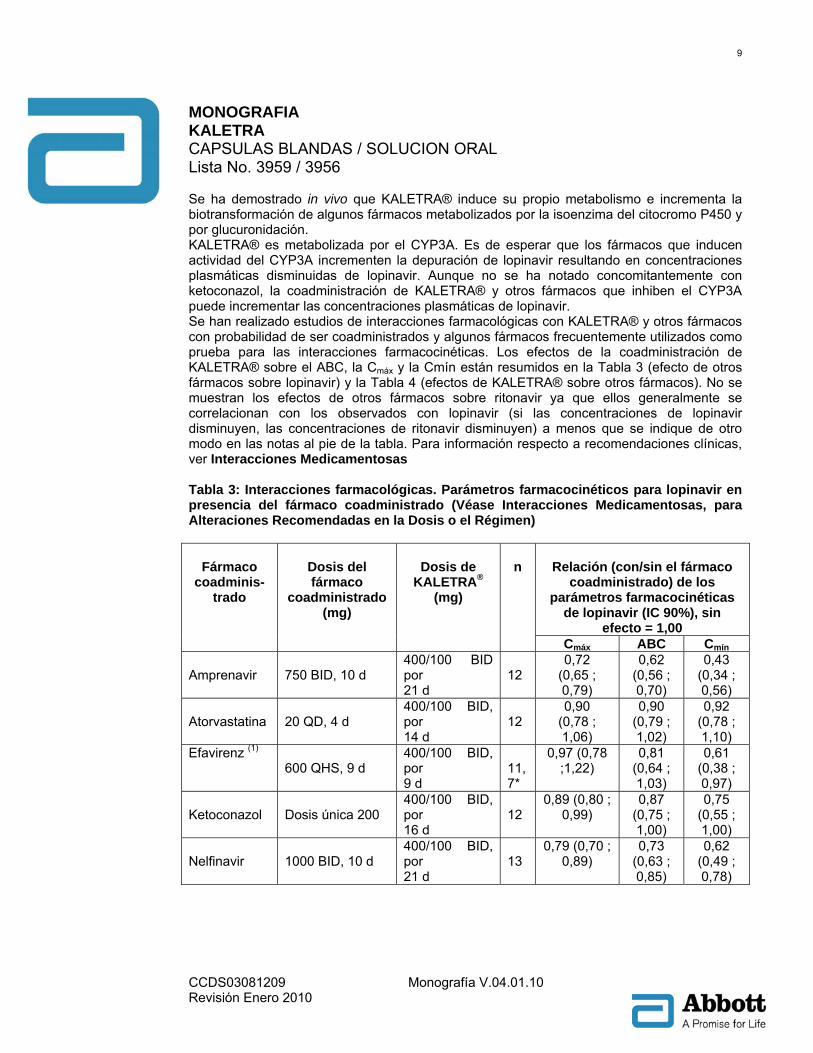
Se ha demostrado in vivo que KALETRA® induce su propio metabolismo e incrementa la biotransformación de algunos fármacos metabolizados por la isoenzima del citocromo P450 y por glucuronidación. KALETRA® es metabolizada por el CYP3A. Es de esperar que los fármacos que inducen actividad del CYP3A incrementen la depuración de lopinavir resultando en concentraciones plasmáticas disminuidas de lopinavir. Aunque no se ha notado concomitantemente con ketoconazol, la coadministración de KALETRA® y otros fármacos que inhiben el CYP3A puede incrementar las concentraciones plasmáticas de lopinavir. Se han realizado estudios de interacciones farmacológicas con KALETRA® y otros fármacos con probabilidad de ser coadministrados y algunos fármacos frecuentemente utilizados como prueba para las interacciones farmacocinéticas. Los efectos de la coadministración de KALETRA® sobre el ABC, la Cmáx y la Cmín están resumidos en la Tabla 3 (efecto de otros fármacos sobre lopinavir) y la Tabla 4 (efectos de KALETRA® sobre otros fármacos). No se muestran los efectos de otros fármacos sobre ritonavir ya que ellos generalmente se correlacionan con los observados con lopinavir (si las concentraciones de lopinavir disminuyen, las concentraciones de ritonavir disminuyen) a menos que se indique de otro modo en las notas al pie de la tabla. Para información respecto a recomendaciones clínicas, ver Interacciones Medicamentosas Tabla 3: Interacciones farmacológicas. Parámetros farmacocinéticos para lopinavir en presencia del fármaco coadministrado (Véase Interacciones Medicamentosas, para Alteraciones Recomendadas en la Dosis o el Régimen)
Relación (con/sin el fármaco
coadministrado) de los parámetros farmacocinéticas
de lopinavir (IC 90%), sin efecto = 1,00
Fármaco
coadminis-trado
Dosis del fármaco
coadministrado (mg)
Dosis de
KALETRA® (mg)
n
Cmáx ABC Cmín Amprenavir
750 BID, 10 d
400/100 BID por 21 d
12
0,72 (0,65 ; 0,79)
0,62 (0,56 ; 0,70)
0,43 (0,34 ; 0,56)
Atorvastatina
20 QD, 4 d
400/100 BID, por 14 d
12
0,90 (0,78 ; 1,06)
0,90 (0,79 ; 1,02)
0,92 (0,78 ; 1,10)
Efavirenz (1) 600 QHS, 9 d
400/100 BID, por 9 d
11, 7*
0,97 (0,78 ;1,22)
0,81 (0,64 ; 1,03)
0,61 (0,38 ; 0,97)
Ketoconazol
Dosis única 200
400/100 BID, por 16 d
12
0,89 (0,80 ; 0,99)
0,87 (0,75 ; 1,00)
0,75 (0,55 ; 1,00)
Nelfinavir
1000 BID, 10 d
400/100 BID, por 21 d
13
0,79 (0,70 ; 0,89)
0,73 (0,63 ; 0,85)
0,62 (0,49 ; 0,78)
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
10
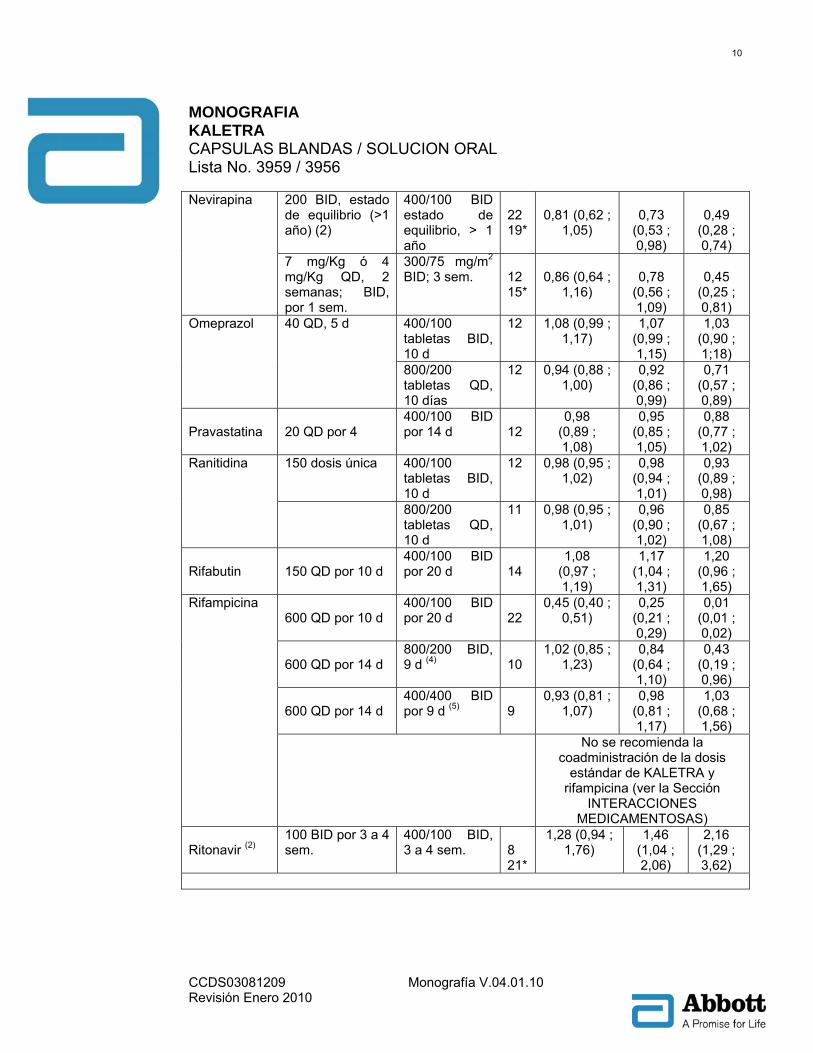
200 BID, estado de equilibrio (>1 año) (2)
400/100 BID estado de equilibrio, > 1 año
22 19*
0,81 (0,62 ;
1,05)
0,73
(0,53 ; 0,98)
0,49
(0,28 ; 0,74)
Nevirapina
7 mg/Kg ó 4 mg/Kg QD, 2 semanas; BID, por 1 sem.
300/75 mg/m2 BID; 3 sem.
12 15*
0,86 (0,64 ;
1,16)
0,78
(0,56 ; 1,09)
0,45
(0,25 ; 0,81)
400/100 tabletas BID, 10 d
12 1,08 (0,99 ; 1,17)
1,07 (0,99 ; 1,15)
1,03 (0,90 ; 1;18)
Omeprazol 40 QD, 5 d
800/200 tabletas QD, 10 días
12 0,94 (0,88 ; 1,00)
0,92 (0,86 ; 0,99)
0,71 (0,57 ; 0,89)
Pravastatina
20 QD por 4
400/100 BID por 14 d
12
0,98 (0,89 ; 1,08)
0,95 (0,85 ; 1,05)
0,88 (0,77 ; 1,02)
150 dosis única 400/100 tabletas BID, 10 d
12 0,98 (0,95 ; 1,02)
0,98 (0,94 ; 1,01)
0,93 (0,89 ; 0,98)
Ranitidina
800/200 tabletas QD, 10 d
11 0,98 (0,95 ; 1,01)
0,96 (0,90 ; 1,02)
0,85 (0,67 ; 1,08)
Rifabutin
150 QD por 10 d
400/100 BID por 20 d
14
1,08 (0,97 ; 1,19)
1,17 (1,04 ; 1,31)
1,20 (0,96 ; 1,65)
600 QD por 10 d
400/100 BID por 20 d
22
0,45 (0,40 ; 0,51)
0,25 (0,21 ; 0,29)
0,01 (0,01 ; 0,02)
600 QD por 14 d
800/200 BID, 9 d (4)
10
1,02 (0,85 ; 1,23)
0,84 (0,64 ; 1,10)
0,43 (0,19 ; 0,96)
600 QD por 14 d
400/400 BID por 9 d (5)
9
0,93 (0,81 ; 1,07)
0,98 (0,81 ; 1,17)
1,03 (0,68 ; 1,56)
Rifampicina
No se recomienda la coadministración de la dosis
estándar de KALETRA y rifampicina (ver la Sección
INTERACCIONES MEDICAMENTOSAS)
Ritonavir (2)
100 BID por 3 a 4 sem.
400/100 BID, 3 a 4 sem.
8 21*
1,28 (0,94 ; 1,76)
1,46 (1,04 ; 2,06)
2,16 (1,29 ; 3,62)

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
11
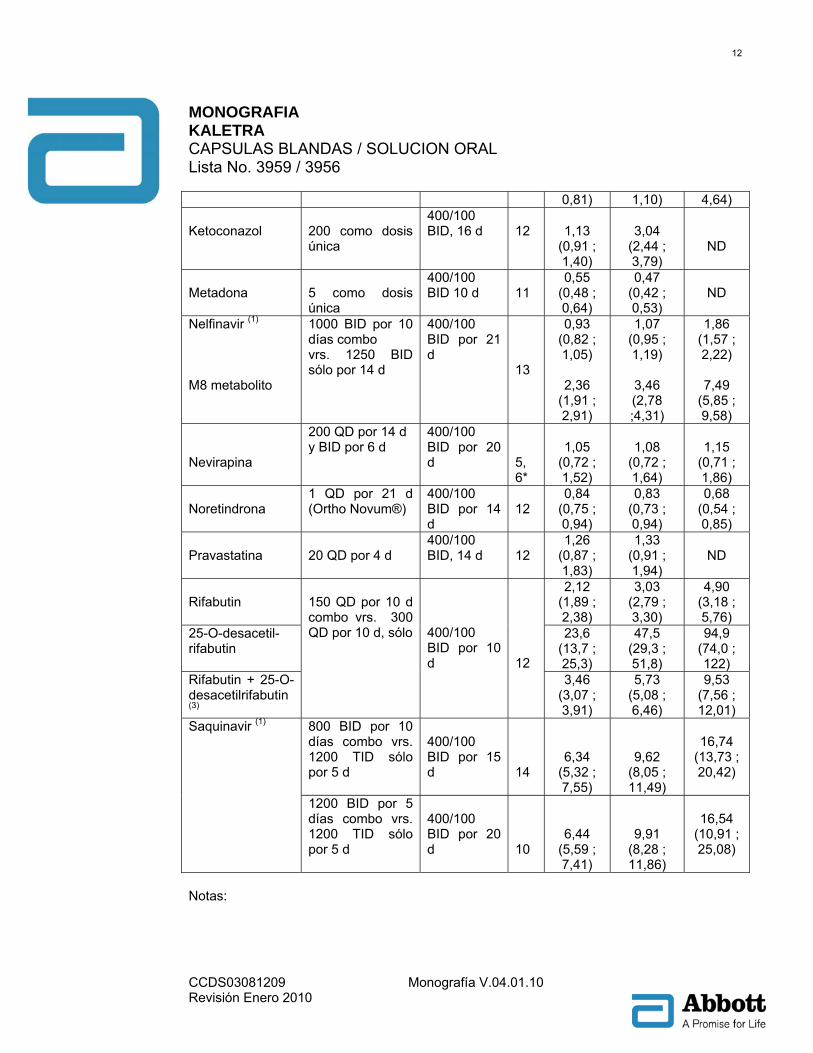
Notas: Todos los estudios de interacción fueron hechos en sujetos sanos, VIH negativo a menos que se indique de otro modo. 1 Los parámetros farmacocinéticos de ritonavir no son afectados por la administración concomitante de efavirenz. 2 Estudio conducido en sujetos ADULTOS VIH positivo. 3 Estudio conducido en sujetos pediátricos VIH positivo con rango en edad de 6 meses a 12 años. 4 La titulación a 800/200 BID como 533/133 BID por 1 día, 667/167 BID por 1 día, después 800/200 BID por 7 días, comparado con 400/100 BID por 10 días solo. 5 Titulado a 400/400 BID como 400/200 BID por 1 día, 400/300 BID por 1 día, después 400/400 BID por 7 días, comparado con 400/100 BID por 10 días solo. * Diseño con grupo paralelo; n para KALETRA® + fármacos coadministrados, n para KALETRA® sola. Tabla 4: Interacciones Farmacológicas: Parámetros farmacocinéticos para fármacos coadministrados en presencia de KALETRA® (Véase Precauciones para Alteraciones Recomendadas en la Dosis o el Régimen)
Relación (con/sin KALETRA) de los parámetros
farmacocinéticos del fármaco coadministrado (IC 90%), sin
efecto = 1,00
Fármaco
coadminis-trado
Dosis fármaco coadministrado
(mg)
Dosis de
KALETRA® (mg)
n
Cmáx ABC Cmín Amprenavir (1)
750 BID , 10 días combo vrs. 1200 BID solo por 14 d
400/100 BID por 21 d
11
1,12 (0,91 ; 1,39)
1,72 (1,41 ; 2,09)
4,57 (3,51 ; 5,95)
Atorvastatina
20 QD, 4 d 400/100 BID por 14 d
12
4,67 (3,35 ; 6,51)
5,88 (4,69 ; 7,37)
2,28 (1,91 ; 2,71)
Desipramina (2)
100 como dosis única
400/100 BID por 10 d
15
0,91 (0,84 ; 0,97)
1,05 (0,96 ; 1,16)
ND
Efavirenz 600 QHS, 9 d 400/100 BID, 9 d
11 12*
0,91 (0,72 ; 1,15)
0,84 (0,62 ; 1,15)
0,84 (0,58 ; 1,20)
Etinil estradiol
35 μg QD, por 21 d (Ortho Novum®)
400/100 BID 14 d
12
0,59 (0,52 ; 0,66)
0,58 (0,54 ; 0,62)
0,42 (0,36 ; 0,49)
Indinavir (1)
Combo sin ayuno 600 BID por 10 d vrs. 800 TID por 5 d, sólo ayuno
400/100 BID 15 d
13
0,71 (0,63 ;
0,91 (0,75 ;
3,47 (2,60 ;
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
12
0,81) 1,10) 4,64) Ketoconazol
200 como dosis única
400/100 BID, 16 d
12
1,13
(0,91 ; 1,40)
3,04
(2,44 ; 3,79)
ND
Metadona
5 como dosis única
400/100 BID 10 d
11
0,55 (0,48 ; 0,64)
0,47 (0,42 ; 0,53)
ND
Nelfinavir (1)
M8 metabolito
1000 BID por 10 días combo vrs. 1250 BID sólo por 14 d
400/100 BID por 21 d
13
0,93 (0,82 ; 1,05)
2,36
(1,91 ; 2,91)
1,07 (0,95 ; 1,19)
3,46 (2,78 ;4,31)
1,86 (1,57 ; 2,22)
7,49
(5,85 ; 9,58)
Nevirapina
200 QD por 14 d y BID por 6 d
400/100 BID por 20 d
5, 6*
1,05
(0,72 ; 1,52)
1,08
(0,72 ; 1,64)
1,15
(0,71 ; 1,86)
Noretindrona
1 QD por 21 d (Ortho Novum®)
400/100 BID por 14 d
12
0,84 (0,75 ; 0,94)
0,83 (0,73 ; 0,94)
0,68 (0,54 ; 0,85)
Pravastatina
20 QD por 4 d
400/100 BID, 14 d
12
1,26 (0,87 ; 1,83)
1,33 (0,91 ; 1,94)
ND
Rifabutin
2,12 (1,89 ; 2,38)
3,03 (2,79 ; 3,30)
4,90 (3,18 ; 5,76)
25-O-desacetil-rifabutin
23,6 (13,7 ; 25,3)
47,5 (29,3 ; 51,8)
94,9 (74,0 ; 122)
Rifabutin + 25-O-desacetilrifabutin (3)
150 QD por 10 d combo vrs. 300 QD por 10 d, sólo
400/100 BID por 10 d
12
3,46 (3,07 ; 3,91)
5,73 (5,08 ; 6,46)
9,53 (7,56 ; 12,01)
800 BID por 10 días combo vrs. 1200 TID sólo por 5 d
400/100 BID por 15 d
14
6,34 (5,32 ; 7,55)
9,62 (8,05 ; 11,49)
16,74
(13,73 ; 20,42)
Saquinavir (1)
1200 BID por 5 días combo vrs. 1200 TID sólo por 5 d
400/100 BID por 20 d
10
6,44 (5,59 ; 7,41)
9,91 (8,28 ; 11,86)
16,54
(10,91 ; 25,08)
Notas:

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
13
Todos los estudios de interacción conducidos en sujetos sanos, VIH negativo a menos que se indique de otro modo. 1 Índice de parámetros para amprenavir, indinavir, nelfinavir y saquinavir no son normalizados por dosis 2 Desipramina es un substrato probado para valorar efectos sobre el metabolismo mediado por CYP2D6. 3 Efecto de la suma normalizada de la dosis de la molécula rifabutín y 25-O-desacetil rifabutín metabolito activo. * Diseño con grupo paralelo; n para KALETRA® + fármaco coadministrado, n para fármaco coadministrado solo. ND = no disponible. 7. INDICACIONES Y USO CLINICO KALETRA® está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por VIH. Esta indicación se basa en análisis de los niveles plasmáticos de ARN de VIH y conteos de células CD4 en un estudio controlado de KALETRA® de 48 semanas de duración y estudios más pequeños de rango de dosis no controlados de KALETRA® por 144-360 semanas. Actualmente, no existen resultados de estudios controlados evaluando el efecto de KALETRA® sobre la progresión clínica del VIH. 8. CONTRAINDICACIONES KALETRA® está contraindicada en pacientes con hipersensibilidad conocida a lopinavir y ritonavir o a cualquiera de los excipientes. La coadministración de KALETRA® está contraindicada con fármacos que son altamente dependientes del CYP3A para su depuración y para los cuales las concentraciones plasmáticas elevadas se asocian con eventos serios o que amenacen la vida. Estos fármacos se muestran en la Tabla 5. Tabla 5: Fármacos que no deben ser administrados concomitantemente con KALETRA®
Tabla 5: Fármacos que no deben coadministrarse con KALETRA Clase Agente dentro de la clase de fármacos
que no deben ser coadministrados Antihistamínicos Astemizol, Terfenadina Antipsicóticos Blonaserin Benzodiazepinas Midazolam, triazolam Derivados ergotamínicos Ergotamina, dihidroergotamina,
ergonovina, metilergonovina Agentes que actúan sobre la motilidad gastrointestinal.
Cisaprida

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
14
Productos herbales St. John’s Wort (Hypericum perforatum) Inhibidores de la HMG-CoA reductasa Lovastatina, simvastatina Neurolépticos Pimozida Antagonistas de larga acción de los receptores beta adrenérgicos
Salmeterol
Inhibidores de la enzima PDE5 Sildenafil* solamente cuando se utiliza para el tratamiento de la hipertensión arterial pulmonar
ver ADVERTENCIAS Y PRECAUCIONES e INTERACCIONES para información sobre la coadministración de sildenafil en pacientes con disfunción
eréctil 9. INTERACCIONES MEDICAMENTOSAS KALETRA® es un inhibidor del CYP3A (citocromo P450 3A) tanto in vitro como in vivo. La coadministración de KALETRA® y fármacos metabolizados primariamente por el CYP3A (p. ej., bloqueadores de los canales de calcio tipo dihidropiridina, inhibidores de la HMG-CoA reductasa, inmunosupresores e inhibidores de PDE5 ) puede resultar en un incremento de las concentraciones plasmáticas de los otros fármacos que pueden aumentar o prolongar sus efectos terapéuticos y adversos (véase PRECAUCIONES Y ADVERTENCIAS). Los agentes que son metabolizados extensamente por el CYP3A y tienen mucho metabolismo de primer paso parecen ser los más susceptibles a grandes incrementos en el ABC (> 3 veces) cuando se coadministran con KALETRA®. Los medicamentos que están contraindicados específicamente debido a las importantes interacciones y potencial de efectos adversos se listan en la tabla 5 en CONTRAINDICACIONES. KALETRA® es metabolizado por el CYP3A. La coadministración de KALETRA® y fármacos que inducen el CYP3A puede disminuir las concentraciones plasmáticas de lopinavir y reducir su efecto terapéutico (véase PRECAUCIONES Y ADVERTENCIAS). Aunque no se han observado con la administración concomitante con ketoconazol, la coadministración de KALETRA® y otros fármacos que inhiben el CYP3A puede incrementar las concentraciones plasmáticas de lopinavir. Agentes anti VIH Inhibidores nucleósidos de la transcriptasa reversa INTR Estavudina y Lamivudina No se ha observado cambio en la farmacocinética de lopinavir cuando se administra KALETRA® con estavudina y lamivudina. Didanosina Se recomienda que la didanosina se administre con el estómago vacío; por esto debe administrarse una hora antes o dos horas después de KALETRA® administrado junto con comida. Zidovudina y Abacavir KALETRA® induce la glucoronidación y por esto tiene el potencial de reducir las concentraciones plasmáticas de abacavir y zidovudina. Se desconoce las consecuencias de esta interacción. Tenofovir

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
15
Se realizó un estudio que demostró que KALETRA® incrementa los niveles plasmáticos de tenofovir. No se conoce su mecanismo causal. Los pacientes que estén recibiendo Kaletra® y tenofovir, deben ser controlados buscando reacciones adversas asociadas a tenofovir. Todos La administración de inhibidores de proteasa simultáneamente con INTR causa incremento en CPK (creatina fosfoquinasa), mialgia, miositis y raramente rabdomiolisis. Inhibidores no nucleósidos de la transcriptasa reversa INNTR. Nevirapina No se han observado cambios en la farmacocinética de lopinavir en adultos sanos a los que también se les ha administrado nevirapina. En un estudio realizado con niños infectados con VIH se produjo disminución en las concentraciones de lopinavir al usarse simultáneamente con nevirapina; estos resultados probablemente también ocurran en adultos VIH positivo y por lo tanto las concentraciones de lopinavir pueden disminuir. El resultado clínico de esta interacción es desconocido. En los pacientes que tienen larga experiencia con IP o evidencia fenotípica o genotípica de pérdida significativa de la susceptibilidad a lopinavir, se recomienda incrementar la dosis a 533/133 mg BID de KALETRA® cuando es coadministrado con Nevirapina. KALETRA® no debe ser administrado una vez al día en combinación con Nevirapina. Efavirenz El uso simultáneo de KALETRA® y efavirenz con dos inhibidores nucleósidos de la transcriptasa reversa en pacientes multi-experimentados, incrementando la dosis en un 33,3% de 400/100 mg (3 cápsulas) dos veces al día a 533/133 mg (4 cápsulas) dos veces al día, produjo concentraciones plasmáticas de lopinavir comparables a las históricamente obtenidas con lopinavir 400/100mg dos veces al día. Los pacientes multi-experimentados a IP o evidencias de fenotipo o genotipo de resistencia también deben incrementar la dosis a 533/133 mg dos veces al día al coadministrar con efavirenz. Nota: efavirenz y nevirapina inducen la actividad de CYP3A y tiene el potencial de disminuir las concentraciones plasmáticas de otros IP cuando se usan en combinación con KALETRA®. KALETRA® no debe ser administrada una vez al día en combinación con efavirenz. Delavirdina La delarvidina tiene el potencial de aumentar las concentraciones de lopinavir en plasma. Inhibidores de proteasa IP. Amprenavir KALETRA® incrementa la concentración de amprenavir (amprenavir 750 mg BID más KALETRA® produce incremento de ABC, similar Cmax, incrementa Cmin, similar a lo obtenido con amprenavir 1200 mg BID). La co-administración de KALETRA® y amprenavir reduce la concentración plasmática de lopinavir. La dosis de KALETRA® puede ser incrementada si se coadministra con amprenavir, particularmente en pacientes con experiencia extensiva a inhibidores de proteasa o reducción de la susceptibilidad viral a lopinavir (ver DOSIFICACION Y ADMINISTRACIÓN y FARMACOLOGÍA CLINICA: Tablas 3 y 4). KALETRA® no debe ser administrada una vez al día en combinación con amprenavir Fosamprenavir

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
16
Un estudio de coadministración de KALETRA® y fosamprenavir demostró la reducción de niveles de amprenavir y lopinavir. No se ha establecido la dosificación adecuada de KALETRA® y fosamprenavir cuando se hace esta coadministración. Indinavir KALETRA® incrementa las concentraciones de indinavir (indinavir 600 mg BID más KALETRA® produce ABC similar, disminuye Cmax, incrementa Cmin si se compara con indinavir 800 mg TID). La dosis de indinavir puede requerir disminuirse durante su coadministración con KALETRA® 400/100 mg BID (ver FARMACOLOGIA CLINICA: Tabla 4). La administración de KALETRA® una vez al día no ha sido estudiada en combinación con indinavir. Nelfinavir KALETRA® incrementa las concentraciones de nelfinavir y su metabolito M8 (nelfinavir 1000 mg BID más KALETRA® produce ABC similar, similar Cmax, incremento de Cmin comparando con nelfinavir 1250 mg BID). La co-administración de KALETRA® y nelfinavir resulta en disminución de las concentraciones de lopinavir. La dosis de KALETRA® puede ser incrementado cuando se coadministre con nelfinavir, particularmente en pacientes con VIH muy expuestos a inhibidores de proteasa o con reducida susceptibilidad al lopinavir (ver DOSIFICACION Y ADMINISTRACIÓN y FARMACOLOGÍA CLINICA: Tablas 3 y 4). KALETRA® no debe ser administrada una vez al día en combinación con nelfinavir. Ritonavir Cuando KALETRA® se coadministra con 100 mg adicionales de ritonavir dos ves al día el ABC de lopinavir se incrementa en 33% y la Cmin se incrementa en 64% si se compara con KALETRA® 400/100 mg (tres (3) cápsulas de gelatina blanda dos veces al día). Saquinavir KALETRA® incrementa las concentraciones de saquinavir (saquinavir 800 mg BID más KALETRA® produce incremento del ABC, incrementa Cmax, incrementa Cmin al comparar con saquinavir 1200 mg TID). La dosis de saquinavir puede necesitar disminuirse cuando se coadministra con KALETRA® 400/100 mg BID (ver FARMACOLOGÍA CLINICA: Tabla 4). KALETRA® no debe ser administrada una vez al día en combinación con saquinavir. Otros medicamentos Analgésicos Fentanilo: Lopinavir/Ritonavir inhibe el CYP3A4 y como resultado se espera que incremente las concentraciones plasmáticas de fentanilo. Se recomienda un monitoreo cuidadoso de los efectos terapéuticos y adversos (incluyendo depresión respiratoria) cuando el fentanilo es administrado concomitantemente con Lopinavir/Ritonavir. Antiarrítmicos: La amiodarona, bepridil, lidocaína sistémica y quinidina pueden aumentar su concentración si son coadministrados con KALETRA®. Se recomienda cuidado y control de las concentraciones terapéuticas. Digoxina: La literatura ha reportado que la coadministración de ritonavir (300 mg cada 12 horas) y digoxina resultó en un incremento significativo de los niveles de digoxina: Se debe tener precaución cuando se coadministre lopinavir/ritonavir y digoxina; se requiere el apropiado monitoreo de los niveles séricos de digoxina. Agentes anticancerígenos (ej. dasatinib, nilotinib, vincristina, vinblastina): Pueden aumentar sus concentraciones séricas cuando se co-administran con lopinavir/ritonavir, aumentando potencialmente los eventos adversos usualmente asociados con los agentes anticancerígenos. Para nilotinib y dasatinib, referirse a la información de prescripción de instrucciones de dosificación.

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
17
Anticoagulantes: La concentración de warfarina se afecta al coadministarse con KALETRA®. Se recomienda control de RIN (Relación Internacional Normalizada). Antidepresivos: Bupropión: La administración concomitante de bupropión con lopinavir/ritonavir disminuye los niveles plasmáticos tanto del bupropión como de su metabolito activo (hidroxibupropión) Trazodona: el uso concomitante de ritonavir y trazodona puede incrementar los niveles de esta última. Eventos adversos de nausea, mareo, hipotensión y síncope se han observado. Si se usa trazodona con un inhibidor de CYP3A4 como lopinavir/ritonavir, la combinación debe ser usada con precaución y se debe considerar una disminución en la dosis de trazodona. Anticonvulsivantes: Fenobarbital, fenitoína y carbamazepina inducen CYP3A4 y disminuyen las concentraciones de lopinavir. KALETRA® no debe ser administrado una vez al día en combinación con fenobarbital, fenitoína o carbamazepina. Además, la co-administración de fenitoína y lopinavir/ritonavir resulta en una disminución de las concentraciones en estado de equilibrio de fenitoína. Los niveles de fenitoína deben ser monitoreados cuando se administra con lopinavir/ritonavir Antifúngicos: Ketoconazol e Itraconazol: pueden incrementar su concentración sérica, no se recomiendan dosis mayores a 200 mg/día. Voriconazol: Un estudio ha demostrado que la coadministración de ritonavir 100 mg cada 12 horas disminuye el ABC en estado de equilibrio de voriconazol en un promedio de 39%. Por lo tanto la co-administración de lopinavir/ritonavir y voriconazol se debe evitar, a menos que una valoración del riesgo/beneficio del paciente justifique el uso de voriconazol. Anti- Infecciosos: Se ha observado un incremento moderado del ABC de claritromicina al coadministrarse con KALETRA®. Para pacientes con insuficiencia renal y hepática se debe considerar reducción de la dosis de claritromicina. Antimicobacterianos: Rifabutina: Al coadministrarla con KALETRA® por diez días se produce incremento de rifabutina (fármaco padre) y de su metabolito principal (25-O-desacetil) de 3.5 y 5.7 veces en Cmáx y ABC. Con estos datos se recomienda disminuir la rifabutina en un 75% (150 mg cada dos días o tres veces por semana). Puede ser necesario reducirla aún más. Rifampicina: Debido a la gran disminución de las concentraciones de lopinavir no se recomienda usar Rifampicina en combinación con la dosis estándar de KALETRA® (ver PRECAUCIONES Y ADVERTENCIAS e INTERACCIONES MEDICAMENTOSAS). El uso de rifampicina con la dosis estándar de KALETRA® puede conllevar a una pérdida de la respuesta virológica y a una posible resistencia a KALETRA®, a los inhibidores de proteasas o a otros agentes antiretrovirales administrados conjuntamente. La coadministración de rifampicina con 800/200mg de lopinavir/ritonavir BID resultó en una disminución de lopinavir hasta del 57% y con lopinavir/ritonavir 400/400mg BID hubo una disminución hasta del 7% cuando se comparó con lopinavir/ritonavir 400/100mg BID administrado en ausencia de rifampicina. Se han encontrado elevaciones de ALT y AST con dosis más altas de lopinavir/ritonavir coadministrados con rifampicina y esto puede ser dependiente de la secuencia en la administración de la dosis. Si se considera la coadministración , se debe iniciar KALETRA® a la dosis estándar por aproximadamente 10 días previos a la adición de rifampicina. La dosis de KALETRA® debe ser titulada de forma ascendente. Se debe monitorizar cuidadosamente la función hepática (Ver FARMACOLOGÍA CLÍNICA: Tabla 3 para la magnitud de la interacción).

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
18
Antiparasitarios: Atovaquona: Se produce disminución en la concentración de Atovaquona al coadministrarse con KALETRA®, puede ser necesario incrementar la dosis de atovaquona. Corticosteroides: Dexametasona: Induce la CYP3A4 y disminuye las concentraciones de lopinavir. Propionato de Fluticasona: El uso concomitante de propionato de fluticasona y KALETRA® puede incrementar las concentraciones del primero. Utilizar con precaución. Se deben considerar alternativas al propionato de fluticasona especialmente para uso a largo plazo. (Ver Precauciones y Advertencias: interacciones medicamentosas). Dihidropiridina: Los bloqueadores de canales de calcio como felodipina, nifedipina, nicardipina pueden incrementar sus concentraciones séricas al administrarse con KALETRA®. Disulfiram/Metronidazol: KALETRA® solución oral contiene alcohol, que puede producir reacciones tipo disulfiram cuando se coadministra con disulfiram u otro fármaco que produce esta reacción, como metronidazol. Agentes para disfunción eréctil (Inhibidores de PDE5): Sildenafil: Para el tratamiento de la disfunción eréctil se debe reducir la dosis a 25 mg cada 48 horas e incrementar el control de los efectos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) El uso concomitante de sildenafil y lopinavir/ritonavir está contraindicado en los pacientes con Hipertensión Arterial Pulmonar (HAP) (Ver CONTRAINDICACIONES) Tadalafil: Usar con precaución en dosis reducidas de no más de 10 mg cada 72 horas e incrementar el control de eventos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) Vardenafil: Usar con precaución en dosis reducidas de no más de 2,5 mg cada 72 horas e incrementar el control de eventos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) Productos herbales: Hierba de San Juan (Hypericum perforatum): Los pacientes en tratamiento con KALETRA® no deben usar productos que la contengan porque reduce la concentración sérica de KALETRA®. El efecto es debido a inducción de CYP3A4 y puede causar la pérdida de su efecto terapéutico y la aparición de resistencia (Ver CONTRAINDICACIONES, ADVERTENCIAS Y PRECAUCIONES: INTERACCIONES MEDICAMENTOSAS). Inhibidores de la HMG-CoA reductasa: Los que son muy dependientes para su metabolismo de CYP3A4 como lovastatina y simvastatina tienen incrementos marcados en su concentración plasmática al coadministrase con KALETRA®. Esto causa miopatía, incluso rabdomiolisis. El uso de estos medicamentos con lopinavir/ritonavir está contraindicado (ver Contraindicaciones). La atorvastatina es menos dependiente de CYP3A para su metabolismo y al administrase con KALETRA® produce incremento de 4.7 y 5.9 veces en la Cmáx de atorvastatina y ABC, respectivamente. Cuando se use con KALETRA® se recomienda usar la menor dosis posible. Los estudios con pravastatina no han demostrado interacciones clínicas significativas. El metabolismo de pravastatina y fluvastatina no depende de CYP3A4 y no se esperan interacciones con KALETRA®.

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
19
Si el tratamiento con inhibidores de la HMG-CoA reductasa está indicado se debe usar pravastatina o fluvastatina. (Ver Precauciones y Advertencias: Interacciones medicamentosas) Inmunomoduladores: Las concentraciones de ciclosporina, tacrolimus y sirolimus (rapamicina) se incrementan al coadministrarse con KALETRA®. Se recomienda monitoreo frecuente de sus niveles hasta que se estabilicen. Metadona: KALETRA® disminuye las concentraciones plasmáticas de metadona, se recomienda control de niveles plasmáticos. Anticonceptivos orales y parches: KALETRA® disminuye las concentraciones de etinil-estradiol, por lo que se debe usar medidas anticonceptivas adicionales. No se esperan otras Interacciones clínicamente significativas: Los estudios de interacción farmacológica no revelan interacción clínicamente significativa entre desipramina (CYP2D6) omeprazol o ranitidina. Con base en los perfiles metabólicos conocidos, no es de esperar interacciones farmacológicas clínicamente significativas entre KALETRA® y fluvastatina, dapsona, trimetoprim / sulfametoxazol, azitromicina, o fluconazol en pacientes con función renal y hepática normal. 10. ADVERTENCIAS Y PRECAUCIONES Interacciones Medicamentosas KALETRA® es inhibidor de la isoenzima P450 CYP3A. La coadministración de KALETRA® y fármacos primariamente metabolizados por el CYP3A puede resultar en incremento de las concentraciones plasmáticas del otro fármaco que pueden incrementar o prolongar sus efectos terapéuticos y adversos (véase Interacciones medicamentosas y Farmacocinética: Interacciones fármaco-fármaco. Contraindicaciones en la tabla 5). Antimicóticos KALETRA® no debe ser coadministrado con rifampicina debido a que la rifampicina disminuye mucho las concentraciones de lopinavir, lo que puede causar disminución de su efecto terapéutico (Ver Interacciones medicamentosas). Corticosteroides El uso concomitante de KALETRA® con propionato de fluticasona puede incrementar significativamente la concentración en plasma de fluticasona lo que causa la reducción de las concentraciones plasmáticas de cortisol. El efecto sistémico de los corticosteroides como el Síndrome de Cushing y supresión adrenal han sido reportados en pacientes que recibían administración concomitante de ritonavir y propionato de fluticasona administrada por inhalación o por vía intranasal. También se ha reportado este efecto en pacientes que reciben corticosteroides inhalados que son metabolizados en forma similar a la fluticasona como budesonida. Se debe tener particular precaución cuando se coadministre lopinavir/ritonavir y alguno de estos glucocorticoides administrados por inhalación o intranasalmente. Ver Interacciones Medicamentosas. Agentes para disfunción eréctil (Inhibidores de PDE5) Se debe tener precaución particular cuando se prescribe sildenafil, tadalafil,vardenafil para el tratamiento de la disfunción eréctil en pacientes que reciben KALETRA®. Es de esperar que la coadministración de KALETRA® incremente sustancialmente las concentraciones de estos agentes de disfunción eréctil, y puede dar como resultado incremento en los eventos adversos asociados incluyendo hipotensión y erección prolongada. El uso concomitante de

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
20
sildenafil con lopinavir/ritonavir está contraindicado en los pacientes con Hipertensión Arterial Pulmonar (HAP) (véase CONTRAINDICACIONES e INTERACCIONES MEDICAMENTOSAS). Productos Herbales No se recomienda el uso concomitante de KALETRA® y Hierba de San Juan (Hypericum perforatum), o productos que contienen la Hierba de San Juan. Es de esperar que la coadministración de inhibidores de proteasa, incluyendo KALETRA®, con Hierba de San Juan disminuya sustancialmente las concentraciones en plasma de inhibidor de proteasa y puede resultar en niveles subóptimos de lopinavir y conducir a pérdida de la respuesta virológica y posible resistencia a lopinavir o a los inhibidores de proteasa (ver CONTRAINDICACIONES E INTERACCIONES MEDICAMENTOSAS). Inhibidores de la HMG-CoA Reductasa La administración simultánea de KALETRA® con lovastatina o simvastatina está contraindicada (Ver contraindicaciones) Se debe tener precaución con la coadministración simultánea de inhibidores de proteasa, incluyendo KALETRA®, Rosuvastatina u otros inhibidores de la HMG-CoA reductasa que son metabolizados por la vía CYP3A4 ( ej, atorvastatina) porque pueden ocurrir reacciones serias potenciales como miopatia que incluyen rabdomiólisis (Ver interacciones medicamentosas) Tipranavir En un estudio clínico con doble potenciación de la terapia combinada del inhibidor de proteasa en adultos con VIH positivo experimentados multitratados, el tipranavir (500 mg dos veces al día) con ritonavir (200 mg dos veces al día), co-administrado con lopinavir/ritonavir (400/100 mg dos veces al día), resultó en una reducción del 47% y 70% en la ABC y la Cmin respectivamente. La administración concomitante de lopinavir/ritonavir y tipranavir con bajas dosis de ritonavir por lo tanto no está recomendada. Pancreatitis Se ha observado pancreatitis en pacientes que reciben terapia con KALETRA®, incluyendo aquellos que desarrollaron elevaciones marcadas de los triglicéridos. En algunos casos, se han presentado muertes. Aunque no se ha establecido una relación causal de KALETRA®, las elevaciones marcadas en los triglicéridos son un factor de riesgo para el desarrollo de pancreatitis (véase PRECAUCIONES Y ADVERTENCIAS - Elevaciones de los lípidos). Los pacientes con enfermedad avanzada por el VIH pueden tener un riesgo incrementado de triglicéridos elevados y pancreatitis, y los pacientes con una historia de pancreatitis pueden tener un mayor riesgo de recurrencia durante la terapia con KALETRA®. Diabetes mellitus/hiperglicemia Se ha informado inicio de diabetes mellitus, exacerbación de diabetes mellitus pre-existente, e hiperglicemia en la vigilancia post-mercadeo en pacientes infectados con VIH que reciben terapia con inhibidor de proteasa. Algunos pacientes requirieron inicio o ajuste de la dosis de insulina o agentes hipoglicemiantes orales para el tratamiento de estos eventos. En algunos casos, ha ocurrido cetoacidosis diabética. En algunos de aquellos pacientes que descontinuaron la terapia con inhibidor de proteasa la hiperglicemia persistió. Debido a que estos eventos han sido informados voluntariamente durante la práctica clínica, no se pueden hacer estimados de la frecuencia y no se ha establecido una relación causal entre terapia con inhibidor de proteasa y estos eventos. Insuficiencia hepática KALETRA® es metabolizado principalmente por el hígado; por lo tanto, se debe tener precaución cuando se administra este fármaco a pacientes con insuficiencia hepática.

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
21
KALETRA® no ha sido estudiado en pacientes con disfunción hepática severa. Los datos farmacocinéticos sugieren que las concentraciones plasmáticas de lopinavir se incrementan aproximadamente en un 30% pero también se reduce la fijación a las proteínas plasmáticas en pacientes coinfectados con VIH y VHC con falla hepática de leve a moderada (ver FARMACOLOGIA CLINICA: FARMACOCINETICA). Los pacientes con hepatitis B o C subyacente o elevaciones marcadas en las transaminasas previo al tratamiento pueden tener mayor riesgo de desarrollar elevaciones adicionales en las transaminasas. Se han hecho reportes en la etapa postmercadeo de disfunción hepática, incluyendo mortalidades. Las complicaciones han ocurrido en pacientes con infecciones avanzadas por VIH que toman múltiples medicamentos concomitantemente en el contexto de hepatitis crónica o cirrosis. La relación causal con KALETRA® no se ha establecido. Se debe considerar vigilar cuidadosamente las transaminasas (AST, ALT) en estos pacientes, especialmente durante los primeros meses del tratamiento con KALETRA®. Resistencia/Resistencia Cruzada Se han observado varios grados de resistencia cruzada entre los inhibidores de proteasa. El efecto de la terapia con KALETRA® sobre la eficacia de los inhibidores de proteasa administrados subsecuentemente está bajo investigación (véase MICROBIOLOGÍA). Hemofilia Ha habido informes de incremento en el sangrado, incluyendo hematomas cutáneos espontáneos y hemartrosis, en pacientes con hemofilia tipo A y B tratados con inhibidores de proteasa. En algunos pacientes se administró factor VIII adicional. En más de la mitad de los casos informados, el tratamiento con inhibidores de proteasa fue continuado o reintroducido. No se ha establecido relación causal entre terapia con inhibidor de proteasa y estos eventos. Prolongación del intervalo PR Se ha demostrado que lopinavir/ritonavir causa una modesta prolongación asintomática del intervalo PR en algunos pacientes. Se han reportado casos de bloqueo auriculoventricular de segundo o tercer grado con patología cardíaca estructural de base y anormalidades preexistentes del sistema de conducción o en pacientes recibiendo medicaciones con efectos conocidos de causar prolongación del intervalo PR (tales como el Verapamilo o el atazanavir) en pacientes recibiendo lopinavir/ritonavir. Lopinavir/ritonavir debe ser utilizado con cautela en este grupo de pacientes (ver FARMACOLOGÍA CLÍNICA) Redistribución de Grasa La redistribución/acumulación de grasa corporal incluyendo obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), atrofia periférica, atrofia facial, aumento de los senos, “apariencia cushingoide” han sido observadas en pacientes recibiendo terapia antirretroviral. El mecanismo y las consecuencias a largo plazo de estos eventos se desconocen actualmente. No se ha establecido una relación causal. Elevaciones en los Lípidos El tratamiento con KALETRA® ha resultado en grandes incrementos en la concentración de colesterol total y triglicéridos (véase REACCIONES ADVERSAS - Tabla 8 y 9). Se debe realizar exámenes de triglicéridos y colesterol antes de iniciar la terapia con KALETRA® y a intervalos periódicos durante la terapia. Las alteraciones de los lípidos se deben manejar en forma clínicamente apropiada. Véase Advertencias y precauciones para información adicional sobre interacciones farmacológicas potenciales con KALETRA® e inhibidores de la HMG-CoA reductasa. Síndrome de Reconstitución Inmunológico

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
22
Se ha reportado la ocurrencia de síndrome de reconstitución inmunológico en pacientes infectados con VIH tratados con terapia combinada que incluye KALETRA®. Durante la fase inicial del tratamiento cuando el sistema inmunológico responde los pacientes pueden desarrollar una respuesta inflamatoria a infecciones oportunistas asintomáticas o residuales (como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocistis jiroveci o tuberculosis) que pueden necesitar evaluación adicional y tratamiento. Uso geriátrico Los estudios clínicos con KALETRA® no incluyeron suficientes sujetos de 65 años edad y mayores para determinar si ellos responden de forma diferente que los sujetos más jóvenes. En general, se debe ejercer precaución apropiada sobre la administración y monitoreo de KALETRA® en pacientes ancianos quienes tienen mayor frecuencia de disminución de la función hepática, renal o cardiaca, y de enfermedad concomitante u otra terapia farmacológica. Uso Pediátrico Los perfiles de seguridad y farmacocinéticos de KALETRA® en pacientes pediátricos menores de 6 meses de edad no han sido establecidos. En pacientes de 6 meses a 12 años de edad infectados con VIH, el perfil de eventos adversos observado durante una prueba clínica fue similar al de los pacientes adultos. La evaluación de la actividad antiviral de KALETRA® en pacientes pediátricos en ensayos clínicos está en curso. La administración una vez al día de KALETRA® no se ha evaluado en la población pediátrica. EMBARAZO Y LACTANCIA Embarazo, fertilidad y reproducción Lopinavir en combinación con ritonavir a una relación de 2:1 no produjo efectos sobre la fertilidad en ratas macho y hembra con las dosis máximas obtenibles produciendo exposición medicamentosa comparable o ligeramente menor que las posibles con las dosis terapéuticas recomendadas 10/5, 30/15 ó 100/50 mg/Kg/día. Basados en mediciones de ABC la exposición en ratas a dosis altas, es aproximadamente 0.7 veces para lopinavir y 1.8 veces para ritonavir de la lograda al exponer a humanos a las dosis recomendadas (400/100 mg BID). No se han observado malformaciones asociadas al tratamiento con lopinavir/ritonavir en ratas o conejas preñadas. Se pueden observar desarrollo de toxicidad embrionaria y fetal en ratas (reabsorción temprana, disminución de la viabilidad fetal, disminución del peso corporal fetal, incremento en la incidencia de variaciones esqueléticas o retraso en la osificación esquelética) en ratas a las que se les administró una dosis maternalmente tóxica de 100/50 mg/Kg/día. Si se compara el ABC la exposición alcanzada de estas drogas es aproximadamente 0,7 veces para lopinavir y 1,8 veces para ritonavir con la lograda al administrar en humanos (400/100 mg BID). Sin embargo, no existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios en animales no son siempre predictivos de la respuesta humana KALETRA® se debe administrar durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Madres en período de lactancia Por el potencial de transmisión de VIH y por los efectos adversos serios posibles, se debe instruir a las madres para no amamantar si reciben KALETRA®. Estudios con ratas han

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
23
demostrado la presencia de lopinavir en la leche. No se sabe si lopinavir es excretado en la leche materna. Carcinogénesis y mutagénesis Los estudios a largo plazo de carcinogenicidad de lopinavir/ritonavir en ratones revelaron la no genotoxicidad, inducción mitogénica o inducción de tumores en hígado, generalmente considerados de tener muy poca relevancia de riesgo humano. Los estudios de carcinogenicidad en ratas no revelaron hallazgos tumorigénicos. Lopinavir no es teratogénico ni clastogénico en una batería de ensayos in vitro incluyendo la prueba reversa de mutación en bacterias de Ames, la prueba de linfoma murino, la prueba de micronúcleo de ratón y las pruebas de aberraciones cromosómicas de linfocitos humanos. Lopinavir/ritonavir no se encontró mutagénico o clastogénico en ensayos in vivo usando ensayos de micronúcleo de ratón. 11. REACCIONES ADVERSAS Adultos Eventos adversos emergentes con el tratamiento KALETRA® ha sido estudiada en 2,154 pacientes con VIH-1 con terapia de combinación en ensayos clínicos Fase I/II y Fase III. El evento adverso más común asociado a la terapia con KALETRA® fue diarrea, la cual generalmente fue de severidad leve a moderada. Las tasa de descontinuación de la terapia aleatoria debida a eventos adversos incluyendo muerte fue 5.8% en los pacientes tratados con KALETRA® y 4.9% en los que recibieron nelfinavir en el Estudio 863. La incidencia de diarrea fue mayor en el grupo que recibió KALETRA® cápsulas una vez al día, comparado con los que lo recibieron dos veces al día en el estudio 418. (Tabla 6) Los eventos adversos clínicos relacionados con el fármaco de intensidad moderada o severa en ≥2% de los pacientes tratados con la terapia de combinación incluyendo KALETRA® hasta por 48 semanas (estudios 863, 418 y 730) y hasta por 360 semanas (estudio 720) se presentan en la Tabla 6 (pacientes sin experiencia a antiretrovirales) y hasta por 48 semanas (estudios 888 y 802), 84 semanas (estudio 957) y 144 semanas (estudio 765) en la Tabla 7 (pacientes con experiencia a antiretrovirales). Para más información de eventos adversos serios observados o potenciales ver advertencias y precauciones. Tabla 6: Porcentaje de pacientes con eventos adversos emergentes1 con el tratamiento de intensidad moderada o severa informados en ≥2% de los pacientes adultos no experimentados a antiretrovirales Estudio 863
(48 Semanas) Estudio 418
(48 Semanas) Estudio 720 (360
Semanas)
Estudio 730 (48 Semanas)

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
24
Lop/Rit 400/100 mg BID + d4T+
3TC (N=326)
Nelfinavir 750 mg
TID + d4T + 3TC
(N=327)
Lop/Rit Cap
800/200 mg QD + TDF+
FTC (N=115)
Lop/Rit Cap
400/100 mg BID + TDF+
FTC (N=75)
Lop/Rit BID(2) + d4T + 3TC
(N=100)
Lop/Rit Tab
800/200 mg QD + TDF+
FTC (N=333)
Lop/Rit Tab
400/100 mg BID + TDF+
FTC (N=331)
Trastornos Gastrointestinales
Distensión abdominal
0.3% 0.6% 0.9% 0.0% 4.0% 0.3% 0.3%
Dolor Abdominal 4.0% 3.1% 2.6% 2.7% 11.0% 0.6% 0.9%
Heces anormales 0.0% 0.3% 0.0% 0.0% 8.0% 0.0% 0.0%
Diarrea 15.6% 17.1% 15.7% 5.3% 28.0% 16.5% 15.1% Dispepsia 2.1% 0.3% 0.0% 1.3% 6.0% 0.0% 0.0% Flatulencia 1.5% 1.2% 1.7% 1.3% 4.0% 0.9% 0.6% Náusea 6.7% 4.6% 8.7% 8.0% 16.0% 7.2% 5.4% Vómito 2.5% 2.4% 3.5% 4.0% 6.0% 3.3% 3.9% Trastornos generales y condiciones del sitio de administración Astenia 4.0% 3.4% 0.0% 0.0% 9.0% 0.3% 0.3% Dolor 0.6% 0.0% 0.0% 0.0% 3.0% 0.0% 0.0% Trastornos del sistema nervioso Cefalea 2.5% 1.8% 2.6% 2.7% 6.0% 1.5% 0.6% Parestesia 0.9% 0.9% 0.0% 0.0% 2.0% 0.0% 0.0% Trastornos psiquiátricos Insomnio 1.5% 1.2% 0.0% 0.0% 3.0% 1.2% 0.0% Libido disminuida 0.3% 0.3% 0.0% 1.3% 2.0% 0.0% 0.3%
Depresión 0.6% 1.5% 1.0% 0.0% 0.0% 0.0% 0.0% Trastornos vasculares Vasodilatación 0.0% 0.0% 0.0% 0.0% 3.0% 0.0% 0.0% Trastornos de la piel y del tejido subcutáneo Lipodistrofia adquirida 0.6% 0.6% 0.0% 0.0% 12.0% 0.0% 0.0%
Rash cutáneo 0.6% 1.5% 0.9% 0.0% 5.0% 0.3% 0.6% Trastornos músculo-esqueléticos y del tejido conectivo Mialgia 0.6% 0.9% 0.0% 0.0% 2.0% 0.0% 0.0% Infecciones e infestaciones Bronquitis 0.0% 0.0% 0.0% 0.0% 2.0% 0.0% 0.3% Trastornos endocrinos Hipogonadismo masculino 0.0% 0.0% 0.0% 0.0% 2.1% 0.0% 0.0%

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
25
Trastornos del metabolismo y de la nutrición Anorexia 0.9% 0.3% 0.9% 1.3% 2.0% 0.3% 0.9% Investigación Pérdida de peso
0.6% 0.3% 0.0% 0.0% 2.0% 0.0% 0.3%
Trastornos del sistema reproductivo y del tejido mamario Amenorrea 0.0% 0.0% 4.5% 0.0% 0.0% 0.0% 0.0%
(1) Incluye los eventos adversos, con relación posible o probable con el fármaco en estudio
(2) Incluye los datos de los eventos adversos de los pacientes del grupo I que recibieron 200/100 mg BID (N=16) y 400/100 mg BID solo (N=16) y los pacientes del grupo II que recibieron 400/100 mg BID (N=35) y 400/200 mg BID (N=33). En ambos grupos, se presento náusea moderada a severa probablemente/posiblemente relacionada con Lopinavir/Ritonavir en una tasa más alta en el brazo con dosis 400/200 mg comparado con el brazo 400/100 mg en el grupo II.
Definiciones: Lop/Rit = Lopinavir/Ritonavir; d4T = estavudina; 3TC = Lamivudina; TDF = Tenofovir; FTC = Emtricitabina.
Tabla 7: Porcentaje de pacientes con eventos adversos emergentes1 con el tratamiento de intensidad moderada o severa informados en ≥2% de los pacientes adultos con experiencia previa a antiretrovirales
Estudio 888 (48 Semanas)
Estudio 957(2) y Estudio
765(3)
(84-144 Semanas)
Estudio 802 (48 Semanas)
Lop/Rit 400/100 mg BID + NVP
+ INTR (N=148)
Inhibidor(es) de la
proteasa, seleccionado
por el investigador + NVP + INTR
(N=140)
Lop/Rit BID + INNTR + INTR
(N=127)
Lop/Rit 800/200 mg Una
vez al día + INTR (N=300)
Lop/Rit 400/100 mg BID + INTR
(N=299)
Trastornos Gastrointestinales Dolor Abdominal
2.0% 2.1% 3.9% 2.0% 0.3%
Dolor abdominal superior
NA NA NA 0.7% 2.0%
Heces anormales
0.0% 0.0% 2.4% 0% 0%
Diarrea 7.4% 9.3% 22.8% 14.0% 11.0%

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
26
Disfagia 2.0% 0.7% 0.0% 0% 0% Flatulencia 0.7% 2.1% 1.6% 1.0% 1.0% Náusea 6.8% 16.4% 4.7% 2.7% 7.4% Vómito 4.1% 12.1% 1.6% 2.0% 2.7% Trastornos generales y condiciones del sitio de administración Astenia 2.7% 6.4% 9.4% 0.3% 0.3% Escalofríos 2.0% 0.0% 0.0% 0% 0% Pirexia 2.0% 1.4% 1.6% 0% 0.3% Dolor 0.0% 0.0% 3.9% 0% 0% Trastornos del sistema nervioso Cefalea 2.0% 2.9% 2.4% 0.3% 0% Parestesia 0.0% 1.4% 2.4% 0% 0% Trastornos vasculares Hipertensión 0.0% 0.0% 2.4% 0% 0% Trastornos del metabolismo y de la nutrición Anorexia 0.7% 2.9% 0.0% 0% 0.7% Investigaciones Pérdida de peso
0.0% 1.4% 3.1% 0.3% 0.3%
Trastornos de la piel y del tejido subcutáneo Lipodistrofia adquirida 0.7% 1.4% 6.3% 0.7% 1.0%
Rash cutáneo 2.0% 1.4% 2.4% 0% 0% Trastornos psiquiátricos Insomnio 0.0% 2.1% 2.4% 0% 0.3% Depresión 0.7% 2.1% 3.1% 0.3% 0%
(1) Incluye los eventos adversos, con relación posible o probable con el fármaco en estudio
(2) Incluye los datos de los eventos adversos de los pacientes que recibieron 400/100 mg BID (n=29) ó 533/133 mg BID (n=28) por 84 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y efavirenz
(3) Incluye los datos de los eventos adversos de los pacientes que recibieron 400/100 mg BID (n=36) ó 400/200 mg BID (n=34) por 144 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y nevirapina
Definiciones: Lop/Rit = Lopinavir/Ritonavir; NVP = Nevirapina; INTR = Inhibidores Nucleósidos de la Transcriptasa Reversa; INNTR = Inhibidores No Nucleósidos de la Transcriptasa Reversa
Los eventos adversos emergentes con el tratamiento que ocurrieron en menos del 2% de los pacientes adultos recibiendo KALETRA® en todos los ensayos clínicos Fase II/III considerados al menos posiblemente relacionados o de relación desconocida al tratamiento

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
27
con KALETRA® y aquellos calificados como de por lo menos intensidad moderada, se mencionan a continuación por sistema corporal. Infecciones e infestaciones: Infección bacteriana, bronconeumonía, celulitis, foliculitis, furunculosis, gastroenteritis, influenza, otitis media, abscesos perianales, faringitis, rinitis, sialoadenitis, sinusitis e infección viral. Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos): Neoplasma benigno de la piel, y neoplasias. Alteraciones del sistema sanguíneo y linfático: Anemia, leucopenia y linfadenopatía, neutropenia y esplenomegalia. Alteraciones del sistema inmunológico: Hipersensibilidad al fármaco, hipersensibilidad, síndrome de reconstitución inmune Alteraciones del Sistema endocrino: Síndrome de Cushing e hipotiroidismo. Alteraciones metabólicas y nutricionales: Disminución del apetito, deshidratación, diabetes mellitus, hiperamilasemia, hiperlipasemia, hipovitaminosis, aumento de apetito, acidosis láctica, lipomatosis, obesidad. Alteraciones Psiquiátricas: Sueños anormales, labilidad afectiva, agitación, ansiedad, apatía, estado de confusión, desorientación, cambios de humor, nerviosismo y pensamiento anormal. Alteraciones del sistema nervioso: pérdida del sentido del gusto, amnesia, desorden del balance, ataxia, infarto cerebral, convulsión, vértigo, alteración del gusto, discinesia, encefalopatía, síndrome extrapiramidal, parálisis facial, hipertonía, migraña, neuropatía, neuritis periférica, somnolencia y tremor. Desórdenes oculares: Visión anormal, alteración ocular. Alteraciones del oído y laberinto: Hiperacusia, tinnitus y vértigo. Desórdenes Cardíacos: Angina de pecho, fibrilación atrial, bloqueo atrioventricular, infarto miocardio y palpitaciones, incompetencia de la válvula tricúspide. Alteraciones vasculares: Trombosis de las venas profundas, hipotensión ortostática, tromboflebitis, venas varicosas y vasculitis. Alteraciones respiratorias, torácicas y del mediastino: asma, tos, disnea, edema pulmonar. Alteraciones gastrointestinales: Malestar abdominal, dolor abdominal inferior, constipación, boca seca, duodenitis, enteritis, enterocolitis, enterocolitis hemorrágica, eructos, esofagitis, incontinencia fecal, gastritis, gastritis ulcerativa, enfermedad de reflujo gastroesofágico,
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
28
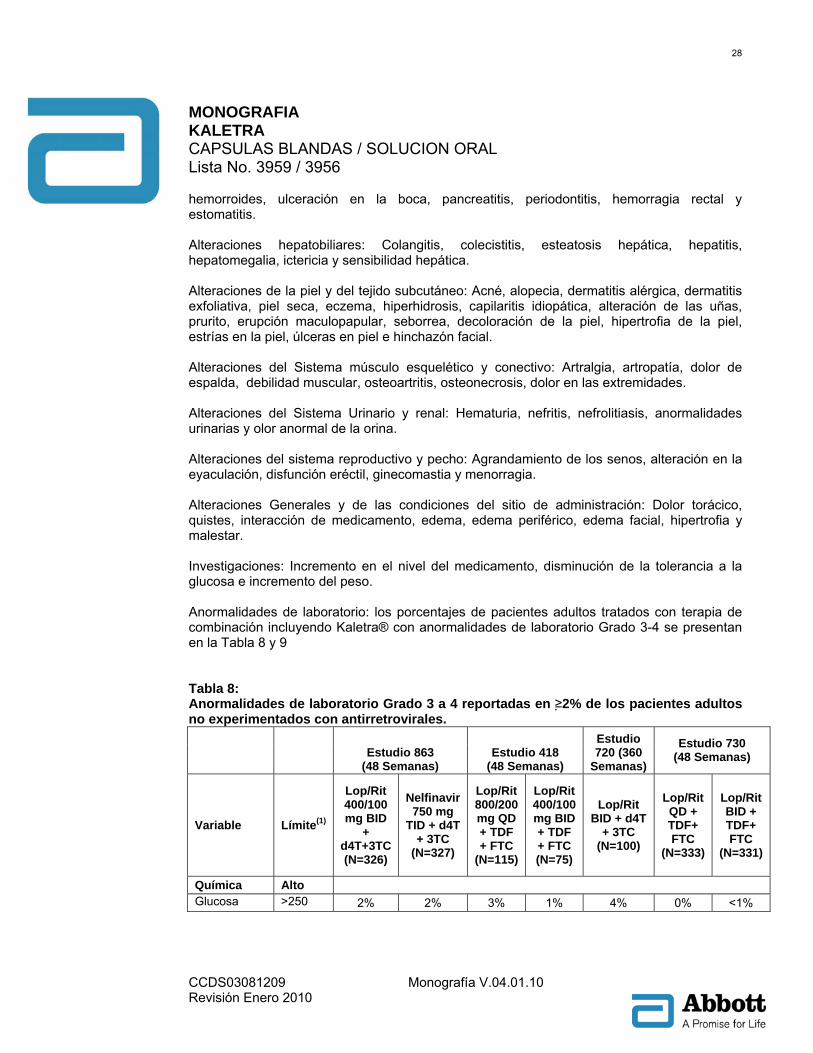
hemorroides, ulceración en la boca, pancreatitis, periodontitis, hemorragia rectal y estomatitis. Alteraciones hepatobiliares: Colangitis, colecistitis, esteatosis hepática, hepatitis, hepatomegalia, ictericia y sensibilidad hepática. Alteraciones de la piel y del tejido subcutáneo: Acné, alopecia, dermatitis alérgica, dermatitis exfoliativa, piel seca, eczema, hiperhidrosis, capilaritis idiopática, alteración de las uñas, prurito, erupción maculopapular, seborrea, decoloración de la piel, hipertrofia de la piel, estrías en la piel, úlceras en piel e hinchazón facial. Alteraciones del Sistema músculo esquelético y conectivo: Artralgia, artropatía, dolor de espalda, debilidad muscular, osteoartritis, osteonecrosis, dolor en las extremidades. Alteraciones del Sistema Urinario y renal: Hematuria, nefritis, nefrolitiasis, anormalidades urinarias y olor anormal de la orina. Alteraciones del sistema reproductivo y pecho: Agrandamiento de los senos, alteración en la eyaculación, disfunción eréctil, ginecomastia y menorragia. Alteraciones Generales y de las condiciones del sitio de administración: Dolor torácico, quistes, interacción de medicamento, edema, edema periférico, edema facial, hipertrofia y malestar. Investigaciones: Incremento en el nivel del medicamento, disminución de la tolerancia a la glucosa e incremento del peso. Anormalidades de laboratorio: los porcentajes de pacientes adultos tratados con terapia de combinación incluyendo Kaletra® con anormalidades de laboratorio Grado 3-4 se presentan en la Tabla 8 y 9 Tabla 8: Anormalidades de laboratorio Grado 3 a 4 reportadas en ≥2% de los pacientes adultos no experimentados con antirretrovirales.
Estudio 863
(48 Semanas) Estudio 418
(48 Semanas)
Estudio 720 (360
Semanas)
Estudio 730 (48 Semanas)
Variable Límite(1)
Lop/Rit 400/100 mg BID
+ d4T+3TC (N=326)
Nelfinavir 750 mg
TID + d4T + 3TC
(N=327)
Lop/Rit 800/200 mg QD + TDF + FTC
(N=115)
Lop/Rit 400/100 mg BID + TDF + FTC (N=75)
Lop/Rit BID + d4T
+ 3TC (N=100)
Lop/Rit QD + TDF+ FTC
(N=333)
Lop/Rit BID + TDF+ FTC
(N=331)
Química Alto Glucosa >250 2% 2% 3% 1% 4% 0% <1%

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
29
mg/dL
Ácido úrico >12 mg/dL
2% 2% 0% 3% 5% <1% 1%
SGOT/AST2 >180 U/L
2% 4% 5% 3% 10% 1% 2%
SGPT/ALT2 >215 U/L
4% 4% 4% 3% 11% 1% 1%
GGT >300 U/L
N/A N/A N/A N/A 10% NA NA
Colesterol total
>300 mg/dL 9% 5% 3% 3% 27% 4% 3%
Triglicéridos >750 mg/dL
9% 1% 5% 4% 29% 3% 6%
Amilasa >2x LSN
3% 2% 7% 5% 4% NA NA
Lipasa >2x LSN
NA NA NA NA NA 3% 5%
Química Bajo
Aclaramiento de creatinina calculado
<50 mL/min
NA NA NA NA NA 2% 2%
Hematología Bajo
Neutrófilos 0.75 x 109 /L
1% 3% 5% 1% 5% 2% 1%
(1) LSN = límite superior del rango normal; N/A = No Aplicable. (2) El criterio para el estudio 730 fue >5xLSN (AST/ALT)
Tabla 9: Anormalidades de laboratorio Grado 3 a 4 reportadas en ≥ 2% de los pacientes adultos con experiencia previa a inihibidores de la proteasa.
Estudio 888
(48 Semanas)
Estudio 957(2) y Estudio 765(3)
(84-144 Semanas)
Estudio 802 (48 Semanas)
Variable Límite (1)
Lop/Rit 400/100
mg BID + NVP + INTR
(N=148)
Inhibidor(es) de la
proteasa, seleccionado
por el investigador + NVP + INTR
(N=140)
Lop/Rit BID + INNTR + INTR
(N=127)
Lop/Rit 800/200 mg Una vez al día + INTR
(N=300)
Lop/Rit 400/100
mg BID + INTR
(N=299)
Química Alto
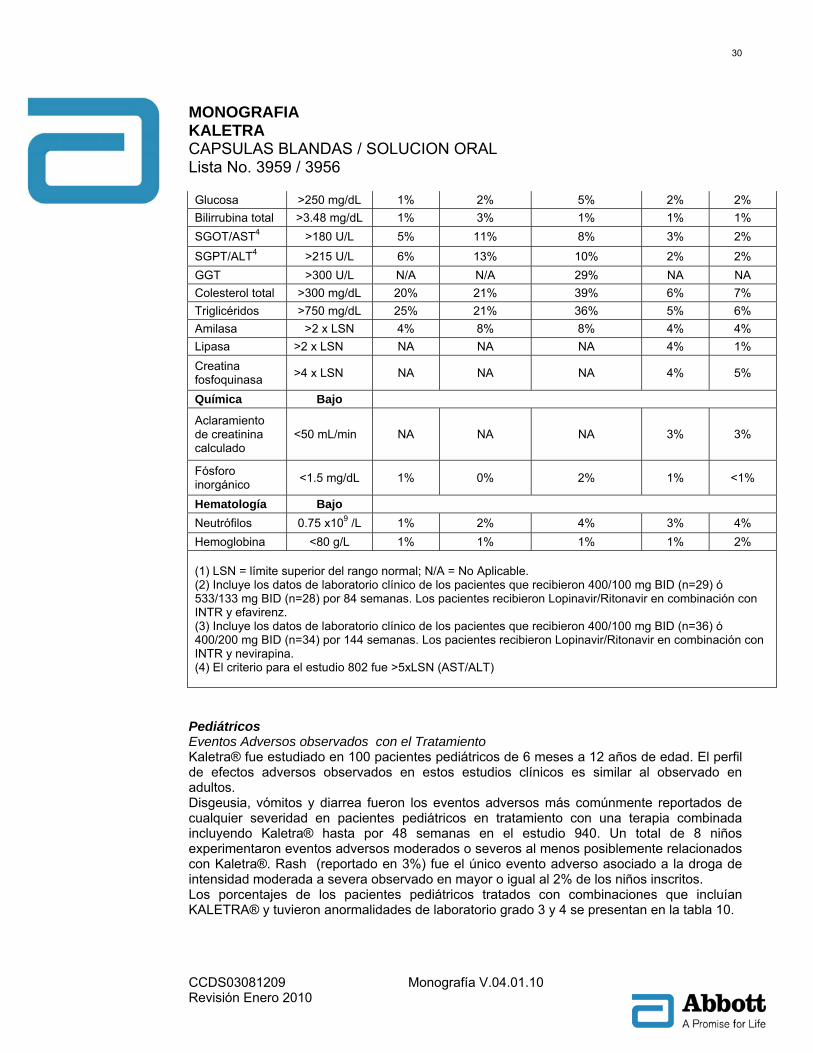
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
30
Glucosa >250 mg/dL 1% 2% 5% 2% 2%
Bilirrubina total >3.48 mg/dL 1% 3% 1% 1% 1%
SGOT/AST4 >180 U/L 5% 11% 8% 3% 2%
SGPT/ALT4 >215 U/L 6% 13% 10% 2% 2%
GGT >300 U/L N/A N/A 29% NA NA
Colesterol total >300 mg/dL 20% 21% 39% 6% 7%
Triglicéridos >750 mg/dL 25% 21% 36% 5% 6%
Amilasa >2 x LSN 4% 8% 8% 4% 4%
Lipasa >2 x LSN NA NA NA 4% 1%
Creatina fosfoquinasa
>4 x LSN NA NA NA 4% 5%
Química Bajo
Aclaramiento de creatinina calculado
<50 mL/min NA NA NA 3% 3%
Fósforo inorgánico
<1.5 mg/dL 1% 0% 2% 1% <1%
Hematología Bajo
Neutrófilos 0.75 x109 /L 1% 2% 4% 3% 4%
Hemoglobina <80 g/L 1% 1% 1% 1% 2%
(1) LSN = límite superior del rango normal; N/A = No Aplicable. (2) Incluye los datos de laboratorio clínico de los pacientes que recibieron 400/100 mg BID (n=29) ó 533/133 mg BID (n=28) por 84 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y efavirenz. (3) Incluye los datos de laboratorio clínico de los pacientes que recibieron 400/100 mg BID (n=36) ó 400/200 mg BID (n=34) por 144 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y nevirapina. (4) El criterio para el estudio 802 fue >5xLSN (AST/ALT)
Pediátricos Eventos Adversos observados con el Tratamiento Kaletra® fue estudiado en 100 pacientes pediátricos de 6 meses a 12 años de edad. El perfil de efectos adversos observados en estos estudios clínicos es similar al observado en adultos. Disgeusia, vómitos y diarrea fueron los eventos adversos más comúnmente reportados de cualquier severidad en pacientes pediátricos en tratamiento con una terapia combinada incluyendo Kaletra® hasta por 48 semanas en el estudio 940. Un total de 8 niños experimentaron eventos adversos moderados o severos al menos posiblemente relacionados con Kaletra®. Rash (reportado en 3%) fue el único evento adverso asociado a la droga de intensidad moderada a severa observado en mayor o igual al 2% de los niños inscritos. Los porcentajes de los pacientes pediátricos tratados con combinaciones que incluían KALETRA® y tuvieron anormalidades de laboratorio grado 3 y 4 se presentan en la tabla 10.

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
31
Tabla 10: Anormalidades de Laboratorio Grado 3-4 informadas en ≥ 2% de los pacientes pediátricos
Variable Límite (1) LOP/RIT BID +ITR (N=100) Química Alto
Sodio > 149 mEq/L 3,0% Bilirrubina total > 2,9 x LSN 3,0% SGOT/AST > 180 U/L 8,0% SGPT/ALT > 215 U/L 7,0% Colesterol total >300 mg/dL ó
> 7,77 mmol/L 3,0%
Amilasa >2,5 x LSN 7,0% (2) Química Bajo
Sodio < 130 mEq/L 3,0% Hematología Bajo
Recuento de plaquetas < 50 x 109/L 4,0% Neutrófilos < 0,40 x 109/L 2,0%
(1) LSN = límite superior del rango normal (2) Individuos con amilasa grado 3 a 4 confirmada con las elevaciones de la amilasa
pancreática Experiencia post-mercadeo Se ha reportado hepatitis en los pacientes que tienen terapia con KALETRA®. Se ha reportado necrólisis epidérmica tóxica, síndrome de Stevens Johnson y eritema multiforme. 12. SINTOMAS Y TRATAMIENTO DE DOSIS EXCESIVA La experiencia en humanos de la sobredosis aguda con KALETRA® es limitada. El tratamiento de la sobredosis con KALETRA® consiste en medidas de apoyo general incluyendo monitoreo de los signos vitales y observación del estado clínico del paciente. No existe antídoto específico para la sobredosis con KALETRA®. Si está indicada, la eliminación del fármaco no absorbido debe ser lograda por emesis o lavado gástrico. La administración de carbón activado también puede ser utilizada para ayudar a retirar el fármaco no absorbido. Ya que KALETRA® se une altamente a las proteínas, es improbable que la diálisis sea benéfica en la remoción significativa del fármaco. KALETRA® solución oral contiene 42.4% de alcohol (v/v). La ingestión accidental del producto por un niño de corta edad podría resultar en toxicidad significativa relacionada con alcohol. 13. VIA DE ADMINISTRACION Y DOSIFICACION (POSOLOGIA) Adultos

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
32
Las cápsulas de gelatina blanda y la solución oral de KALETRA® deben ser tomadas con alimentos. La dosis diaria de KALETRA® es: Pacientes no experimentados: KALETRA® 400/100 mg (3 cápsulas ó 5 mL) dos veces al día con las comidas, KALETRA® 800/200 mg (6 cápsulas ó 10 mL) una vez al día con la comida, en pacientes con menos de tres mutaciones asociados a lopinavir. No hay datos suficientes para respaldar el uso de una administración una vez al día de Kaletra® para pacientes adultos con tres o más mutaciones asociados al lopinavir. Kaletra no debe ser administrado una vez al día en combinación con carbamazepina, fenobarbital o fenitoína (ver Interacciones Medicamentosas). Terapia concomitante: Omeprazol y Ranitidina: Las capsulas de KALETRA® y la solución oral pueden ser usadas en combinación con agentes reductores de ácido (omeprazol y ranitidina) sin ajuste de dosis (ver tabla 3). Efavirenz, Nevirapina, Amprenavir o Nelfinavir Se debe incrementar la dosis de KALETRA® a 533/133 mg (4 cápsulas ó 6.5 mL de solución oral) tomadas dos veces al día con alimentos si se toma en combinación con efavirenz o nevirapina, amprenavir, o nelfinavir en pacientes experimentados porque se sospecha reducción en la susceptibilidad a lopinavir (por historia y laboratorio) (véase FARMACOLOGÍA CLINICA: Interacciones fármaco-fármaco y/o Interacciones medicamentosas). La administración de KALETRA® una vez al día no debe hacerse en combinación con efavirenz, nevirapina, amprenavir o nelfinavir. Pacientes pediátricos Tomar atención especial para realizar el cálculo exacto de la dosis pediátrica. En niños de seis meses a doce años de edad, la dosis recomendada de KALETRA® solución oral es 12/3 mg/Kg para los que pesan entre 7 y menos de 15 Kg y 10/2.5 mg/Kg para los que pesan entre 15 y 40 Kg (aproximadamente 230/57.5 mg/m2); administrada dos veces al día con las comidas, hasta una dosis máxima de 400/100 mg en niños de más de 40 Kg (3 cápsulas ó 5.0 mL) dos veces al día. KALETRA® una vez al día no ha sido evaluado en pacientes pediátricos. Es preferible que se calcule la dosis apropiada en mg para los niños menores o igual a doce años y determinar el correspondiente volumen de solución o cantidad de cápsulas de gelatina blanda. Como alternativa se puede usar la siguiente tabla que contiene las guías de dosificación de acuerdo al peso corporal. Guía de dosificación pediátrica basada en el peso sin efavirenz, nevirapina, nelfinavir o amprenavir concomitante. Peso (Kg) Dosis (mg/Kg)* Volumen de solución oral dos veces
al día (80 mg de lopinavir / 20 mg de ritonavir por mL)

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
33
7 a menos de 15 Kg 12 mg/Kg dos veces al día
7 a 10 Kg 1.25 mL Más de 10 a menos de 15 Kg
1.75 mL
15 a 40 Kg 10 mg/Kg dos veces al día
15 a 20 Kg 2.25 mL Más de 20 a 25 Kg 2.75 mL Más de 25 a 30 Kg 3.50 mL Más de 30 a 35 Kg 4.00 mL Más de 35 a 40 Kg 4.75 mL Más de 40 Kg Dosis de adultos 5.0 mL (o tres cápsulas) * Dosificación basada en el componente lopinavir de la solución KALETRA (80 mg/20 mg por mL). Nota: Emplear la recomendación posológica para adultos, en niños mayores de 12 años. Terapia concomitante: Efavirenz, Nevirapina, amprenavir o nelfinavir Un incremento de la dosis de KALETRA® solución oral a 13/3.25 mg/Kg para niños que pesan entre 7 y menos de 15 Kg y 11/2.75 mg/Kg para los que pesan entre 15 y 45 Kg (aproximadamente 300/75 mg/m2) dos veces al día, tomado con alimentos; hasta un máximo de 533/133 mg dos veces al día en niños de más de 45 Kg se debe considerar cuando es utilizado en combinación con efavirenz, nevirapina, amprenavir o nelfinavir; en niños de 6 meses a 12 años habitualmente tratados en los que clínicamente se sospeche susceptibilidad reducida al lopinavir (por antecedentes terapéuticos o evidencia de laboratorio). La siguiente tabla contiene las guías de dosificación de KALETRA® solución oral basadas en el peso corporal cuando se combina con nevirapina o efavirenz, amprenavir o nelfinavir. (Ver FARMACOLOGIA CLINICA: Interacciones fármaco-fármaco y/o INTERACCIONES MEDICAMENTOSAS) Guía de dosificación pediátrica basada en el peso y administración concomitante con efavirenz, nevirapina, amprenavir o nelfinavir Peso (Kg) Dosis (mg/Kg)* Volumen de solución oral dos veces
al día (80 mg de lopinavir / 20 mg de ritonavir por mL)
7 a menos de 15 Kg 13 mg/Kg dos veces al día
7 a 10 Kg 1.50 mL Más de 10 a menos de 15 Kg
2.00 mL
15 a 45 Kg 11 mg/Kg dos veces al día
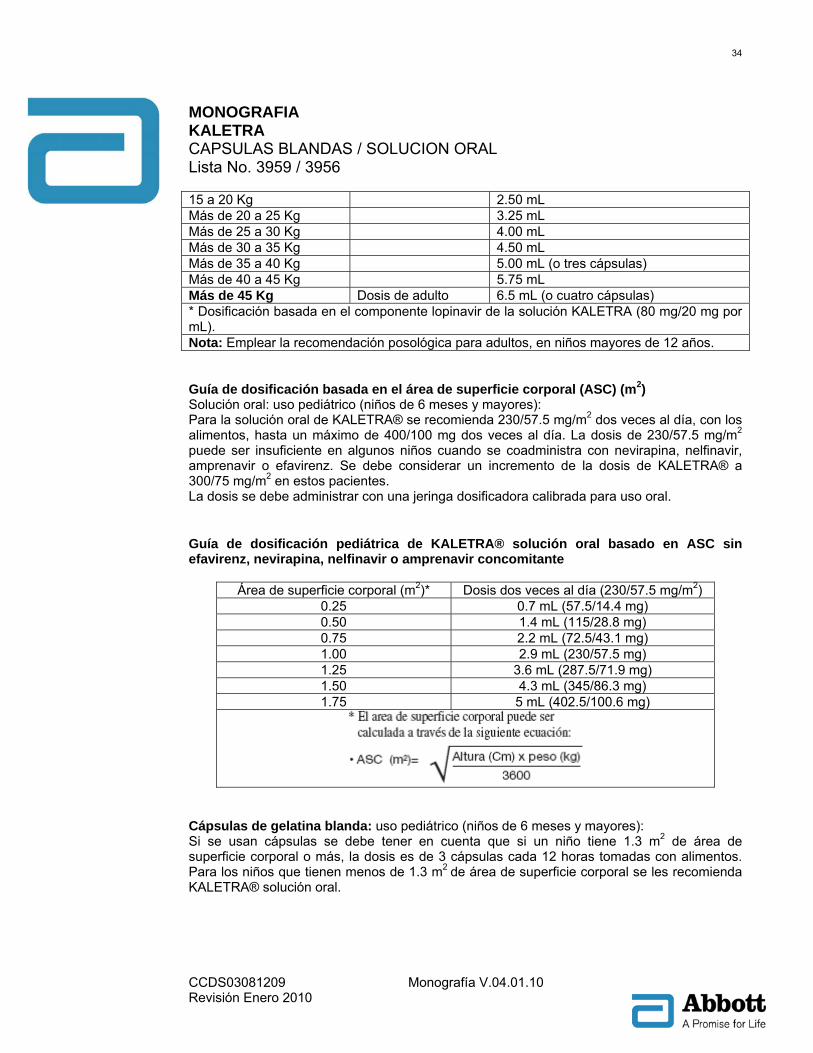
MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
34
15 a 20 Kg 2.50 mL Más de 20 a 25 Kg 3.25 mL Más de 25 a 30 Kg 4.00 mL Más de 30 a 35 Kg 4.50 mL Más de 35 a 40 Kg 5.00 mL (o tres cápsulas) Más de 40 a 45 Kg 5.75 mL Más de 45 Kg Dosis de adulto 6.5 mL (o cuatro cápsulas) * Dosificación basada en el componente lopinavir de la solución KALETRA (80 mg/20 mg por mL). Nota: Emplear la recomendación posológica para adultos, en niños mayores de 12 años. Guía de dosificación basada en el área de superficie corporal (ASC) (m2) Solución oral: uso pediátrico (niños de 6 meses y mayores): Para la solución oral de KALETRA® se recomienda 230/57.5 mg/m2 dos veces al día, con los alimentos, hasta un máximo de 400/100 mg dos veces al día. La dosis de 230/57.5 mg/m2 puede ser insuficiente en algunos niños cuando se coadministra con nevirapina, nelfinavir, amprenavir o efavirenz. Se debe considerar un incremento de la dosis de KALETRA® a 300/75 mg/m2 en estos pacientes. La dosis se debe administrar con una jeringa dosificadora calibrada para uso oral. Guía de dosificación pediátrica de KALETRA® solución oral basado en ASC sin efavirenz, nevirapina, nelfinavir o amprenavir concomitante
Área de superficie corporal (m2)* Dosis dos veces al día (230/57.5 mg/m2) 0.25 0.7 mL (57.5/14.4 mg) 0.50 1.4 mL (115/28.8 mg) 0.75 2.2 mL (72.5/43.1 mg) 1.00 2.9 mL (230/57.5 mg) 1.25 3.6 mL (287.5/71.9 mg) 1.50 4.3 mL (345/86.3 mg) 1.75 5 mL (402.5/100.6 mg)
Cápsulas de gelatina blanda: uso pediátrico (niños de 6 meses y mayores): Si se usan cápsulas se debe tener en cuenta que si un niño tiene 1.3 m2 de área de superficie corporal o más, la dosis es de 3 cápsulas cada 12 horas tomadas con alimentos. Para los niños que tienen menos de 1.3 m2 de área de superficie corporal se les recomienda KALETRA® solución oral.

MONOGRAFIA KALETRA CAPSULAS BLANDAS / SOLUCION ORAL Lista No. 3959 / 3956
CCDS03081209 Monografía V.04.01.10 Revisión Enero 2010
35
14. PRESENTACION Las cápsulas de KALETRA® (lopinavir/ritonavir) son de gelatina blanda, son color naranja. Contienen 133.3 mg de lopinavir/33.3 mg ritonavir. La solución oral de KALETRA® (lopinavir/ritonavir) es un líquido de color ligeramente amarillento a naranja que se suministra en frascos de color ámbar de dosis múltiples; contienen 400 mg de lopinavir/100 mg de ritonavir por cada 5 mL (80 mg de lopinavir/20 mg de ritonavir por mL) empacado con una copa de dosificación marcada. 15. CONDICIONES DE ALMACENAMIENTO Cápsulas de gelatina blanda de KALETRA Almacenar a 2°C a 8°C (36°F a 46°F) hasta cuando se entregue el medicamento. Evite la exposición a calor excesivo. No se requiere que las cápsulas de gelatina blanda de Kaletra se refrigeren si los pacientes las toman antes de 42* días y se mantiene a menos de 25°C (77°F). La Solución Oral de Kaletra se debe mantener refrigerada entre 2° - 8°C (36°F - 46° F ) antes de su dispensación. Evitar la exposición del producto al calor excesivo. No se requiere que el paciente guarde el producto en refrigerador si se va a consumir dentro de los 2 meses y se mantiene a temperaturas inferiores a 25°C. Si el producto se mantiene refrigerado, es estable hasta su fecha de vencimiento. MANTENER FUERA DEL ALCANCE DE LOS NIÑOS BIBLIOGRAFIA I. U.S. FOOD AND DRUG ADMINISTRATION (FDA) CENTER FOR DRUG EVALUATION AND RESEARCH FDA Approved Drug Products. KALETRA, LOPINAVIR; RITONAVIR. II. Abbott Laboratories. 2010.

PROYECTO DE PROSPECTO
KALETRA® (lopinavir/ritonavir) cápsulas de gelatina blanda (lopinavir/ritonavir) solución oral DESCRIPCIÓN KALETRA® (lopinavir/ritonavir) es una coformulación de lopinavir y ritonavir. Lopinavir es un inhibidor de la proteasa del VIH-1 y VIH-2. Al venir coformulado en KALETRA®, ritonavir inhibe el metabolismo de lopinavir mediado por el CYP3A (Citocromo P450 3A), proporcionando de este modo incremento en los niveles plasmáticos de lopinavir. Lopinavir se describe químicamente como [1S-[1R*,(R*),3R*,4R*]]-N-[4-[[2,6-dimetilfenoxi)acetil]amino]-3-hidroxi-5-fenil-1-(fenilmetil)pentil]tetrahidroalfa-(1-metiletil)-2-oxo-1(2H)-pirimidinacetamida. Su fórmula molecular es C37H48N4O5, y su peso molecular es 628.80. Lopinavir tiene la siguiente fórmula estructural:
Ritonavir se describe químicamente como ácido 10-Hidroxi-2-metil-5-(1-metiletil)-1-[2-(1-metiletil)-4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraazotriadecan-13-oico, 5-tiazolilmetil éster, [5S-(5R*,8R*,10R*,11R*)]. Su fórmula molecular es C37H48N6O5S2, y su peso molecular es 720.95. Ritonavir tiene la siguiente fórmula estructural:
Lopinavir es un polvo blanco o ligeramente oscurecido. Es soluble libremente en metanol y etanol, soluble en isopropanol y prácticamente insoluble en agua. KALETRA® cápsulas está disponible para administración oral a concentraciones de 133.3 mg de lopinavir y 33.3 mg de ritonavir con los siguientes ingredientes inactivos: Amarillo FD&C N° 6, gelatina, glicerina, ácido oleico, aceite de ricino hidrogenado polioxil 35, propilenglicol, sorbitol especial, dióxido de titanio y agua. KALETRA® solución oral está disponible para administración oral como 80 mg de lopinavir y 20 mg de ritonavir por mililitro, con los siguientes ingredientes inactivos:

Acesulfame de potasio, alcohol, aroma artificial de dulce de algodón, ácido cítrico, glicerina, jarabe de maíz alto en fructuosa, sabor Magnasweet-110, mentol, sabor a vainilla natural y artificial, aceite de menta, aceite de ricino polioxil 40 hidrogenado, povidona, propilen glicol, sacarina sódica, cloruro de sodio, citrato de sodio y agua. KALETRA® solución oral contiene 42.4% de alcohol (v/v). FARMACOLOGÍA CLÍNICA Microbiología Mecanismo de acción: El Lopinavir es un inhibidor de la proteasa del VIH-1 y VIH-2, impide el corte de la poliproteína gag-pol, lo que da como resultado la producción de partículas virales inmaduras, no infecciosas. Actividad antiviral in vitro: La actividad antiviral in vitro de lopinavir contra las cepas VIH de laboratorio y las aisladas del VIH fue evaluada en líneas de células linfoblásticas y linfocitos sanguíneos periféricos infectadas agudamente. En ausencia de suero humano el promedio de la concentración efectiva 50% (CE50) de lopinavir contra cinco diferentes cepas del VIH-1 de laboratorio estuvo en el rango de 10-27 nM (0.006-0.017 μg/mL, 1 μg/mL = 1.6 μM) y en el rango de 4-11 nM (0.003-0.007 μg/mL) contra varios aislados clínicos del VIH-1 (n = 6). En la presencia de suero humano al 50%, la CE50 promedio de lopinavir contra estas cinco cepas de laboratorio estuvo en el rango de 65-289 nM (0.04-0.18 μg/mL), representando una atenuación de 7-11 veces. Los estudios de actividad farmacológica en combinación con lopinavir y otros inhibidores de proteasa o inhibidores de la transcriptasa inversa no han sido completados. Resistencia: Aislados de VIH-1 con susceptibilidad reducida a lopinavir han sido seleccionados in vitro. Al parecer la presencia de ritonavir no tiene incidencia sobre la selección de virus resistente a lopinavir in vitro. La selección de resistencia a KALETRA® en pacientes no experimentados para tratamiento con antirretroviral todavía no ha sido caracterizada. En un estudio fase III con 653 pacientes no experimentados para tratamiento antirretroviral (estudio 863), los virus aislados de los pacientes con ARN viral cuantificable (> 400 copias/mL) fueron analizados en las semanas 24, 32, 40 y 48. No existe evidencia de resistencia genotípica o fenotípica a KALETRA® en 37 pacientes evaluables (0%). La evidencia de resistencia genotípica a nelfinavir, definida como presencia de mutación D30N y/o L90M en la proteasa del VIH se detectó en 25/76 (33%) de los pacientes tratados con nelfinavir evaluables. La selección de resistencia a KALETRA® en pacientes pediátricos no experimentados (estudio 940) es consistente con lo observado en adultos (estudio 863). Se ha observado que la resistencia a KALETRA® aparece en pacientes tratados con otros inhibidores de las proteasas, previos a la terapia con KALETRA®. En un estudio fase II de 227 pacientes no experimentados para tratamiento antirretroviral y experimentados con inhibidor de proteasa, los aislados de 4 de 23 pacientes con ARN viral cuantificable (> 400 copias/mL) después de tratamiento con KALETRA® por 12 a 100 semanas reveló una susceptibilidad significativamente reducida a lopinavir comparados con los aislados virales basales correspondientes. Tres de estos pacientes habían recibido previamente tratamiento con un solo inhibidor de proteasa (nelfinavir, indinavir, o saquinavir) y un paciente había recibido tratamiento con múltiples inhibidores de proteasa (indinavir, saquinavir y ritonavir). Todos los pacientes tenían al menos 4 mutaciones asociadas con resistencia al inhibidor de proteasa inmediatamente antes a la terapia con KALETRA®. Después del rebote viral, todos los aislados de estos pacientes contenían mutaciones adicionales, algunas de las cuales son reconocidas o se asocian con resistencia al inhibidor de proteasa. Sin embargo,

existen datos insuficientes en este momento para identificar patrones mutacionales asociados con lopinavir en aislados de pacientes bajo terapia con KALETRA®. La evaluación de estos patrones mutacionales está bajo estudio. Resistencia Cruzada Estudios Preclínicos. Se han observado grados variables de resistencia cruzada entre inhibidores de proteasa. Se ha determinado la actividad in vitro de lopinavir contra los aislados clínicos de pacientes previamente tratados con un solo inhibidor de proteasa. Los aislados que revelaron susceptibilidad reducida > 4 veces a nelfinavir (n = 13) y saquinavir (n = 4), mostraron susceptibilidad < 4 veces reducida a lopinavir. Los aislados con susceptibilidad > 4 veces a indinavir (n = 16) y ritonavir (n = 3) revelaron un promedio de susceptibilidad reducida de 5.7 y 8.3 veces a lopinavir, respectivamente. Los aislados de pacientes previamente tratados con dos o más inhibidores de proteasa mostraron mayores reducciones en la susceptibilidad a lopinavir como se describe en el siguiente párrafo. Resistencia cruzada durante tratamiento con KALETRA® Hay poca información disponible sobre resistencia cruzada de virus seleccionados durante la terapia con KALETRA®. Aislados de cuatro pacientes tratados previamente con uno o más inhibidores de proteasa que desarrollaron resistencia fenotípica a lopinavir durante el tratamiento con KALETRA®, permanecieron con la resistencia cruzada o desarrollaron resistencia cruzada a ritonavir, indinavir y nelfinavir. Todos los virus obtenidos después del rebote permanecieron sensibles completamente o tuvieron solamente una modesta reducción de la susceptibilidad a amprenavir (más de 8.5 veces concurrente con 99 veces de aumento de la resistencia a lopinavir). Los virus obtenidos de dos de estos pacientes que no eran previamente resistentes a saquinavir permanecieron totalmente susceptibles a saquinavir. Correlación genotípica de la reducción en la respuesta virológica en pacientes bajo terapia antirretroviral que inician un régimen combinado con KALETRA® Se ha demostrado que la respuesta virológica a KALETRA® se ve afectada por la presencia de tres o más de las siguientes sustituciones de aminoácidos en la línea de base de proteasas: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V. La tabla 1 muestra la respuesta virológica a la semana 48 (VIH ARN<400 copias/mL de acuerdo al número de mutaciones resistentes al inhibidor de proteasas en los estudios 888, 765 y 957 (Tabla 1) Tabla 1: Respuesta virológica (ARN VIH <400 copias/mL) a la semana 48 por susceptibilidad a KALETRA® en línea base y por el número de sustituciones de proteasa asociada con una respuesta reducida a KALETRA®.1 Número de mutaciones de inhibidores de proteasa en línea base1
Estudio 888 (Experimentado2 con un solo Inhibidor de proteasa, Nuevo para ITRNN) n=130
Estudio 765 (Experimentado3
con un solo Inhibidor de proteasa, Nuevo para ITRNN) n=56
Estudio 957 (Experimentado4 con Múltiples inhibidores de proteasa-, Nuevo para ITRNN) n=50
0-2 76/103 (74%) 34/45 (76%) 19/20 (95%)

3-5 13/26 (50%) 8/11 (73%) 18/26 (69%) 6 o más 0/1 (0%) N/A 1/4 (25%) 1 Sustituciones consideradas en el análisis incluido L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, e I84V. 2 43% indinavir, 42% nelfinavir, 10% ritonavir, 15% saquinavir. 3 41% indinavir, 38% nelfinavir, 4% ritonavir, 16% saquinavir. 4 86% indinavir, 54% nelfinavir, 80% ritonavir, 70% saquinavir. La tabla 2 muestra la respuesta virológica a la semana 48 (VIH-1 ARN <50 copias/mL) en el estudio 802 de acuerdo con el número de mutaciones resistentes asociados a lopinavir enumerados en la tabla 1 presentes en el inicio del estudio. No hay datos suficientes para apoyar una administración única diaria de Lopinavir/Ritonavir en pacientes adultos con tres o más mutaciones asociadas a lopinavir. Tabla 2. Respuesta virológica semanas (VIH-1 ARN <50 copias/mL) a la semana 48 por punto de inicio por número de sustituciones asociadas a la proteasa con respuesta reducida a Lopinavir/Ritonavir. Número de sustituciones
de inhibidores de proteasa en el punto de
inicio
Estudio 802 (tratamiento-
experimentado2) Lop/Rit una vez al día + ITRN
n=268
Estudio 802 (tratamiento-
experimentado3) Lop/Rit BID + ITRN n=264
0-2 167/255 (65%) 154/250 (62%) 3-5 4/13 (31%) 8/14 (57%)
6 o más NA NA 1 Sustituciones consideradas en el análisis incluyen L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, e I84V 2 88% experimentados con ITRNN, 47% experimentados con inhibidores de la proteasa (24% nelfinavir, 19% indinavir, 13% atazanavir) 3 81% experimentados con ITRNN, 45% experimentados con inhibidores de la proteasa (20% nelfinavir, 17% indinavir, 13% atazanavir) Farmacocinética Las propiedades farmacocinéticas de lopinavir coadministrado con ritonavir han sido evaluadas en voluntarios adultos sanos y en pacientes infectados con el VIH; no se observaron diferencias sustanciales entre los dos grupos. Lopinavir es metabolizado de forma esencialmente completa por el CYP3A. Ritonavir inhibe el metabolismo de lopinavir, incrementando por esto los niveles plasmáticos de lopinavir. A través de los estudios, la administración de KALETRA® 400/100 mg, dos veces al día da lugar a concentraciones plasmáticas promedio de lopinavir en estado de equilibrio 15 a 20 veces más altas que las de ritonavir en pacientes infectados con el VIH. Los niveles plasmáticos de ritonavir son menos del 7% de los obtenidos después de la dosis de ritonavir de 600 mg dos veces al día. La CE50 antiviral in vitro de lopinavir es aproximadamente 10 veces más baja que la de ritonavir. Por lo tanto, la actividad antiviral de KALETRA® se debe a lopinavir.

La Figura 1 muestra las concentraciones plasmáticas promedio en estado de equilibrio de lopinavir y ritonavir después de tomar KALETRA® 400/100 mg, dos veces al día con alimentos por 3 semanas de un estudio farmacocinético en sujetos adultos infectados con el VIH (n = 19).
Absorción: En un estudio farmacocinético en sujetos VIH positivo (n = 19) a quienes se les administraron múltiples dosis de KALETRA® 400/100 mg, dos veces al día con los alimentos por tres semanas se produjo una concentración máxima promedio en plasma de lopinavir ± DE (Cmáx) de 9.8 ± 3.7 μg/mL, presentándose aproximadamente 4 horas después de la administración. La concentración promedio en el valle en estado de equilibrio antes de la dosis matutina fue 7.1 ± 2.9 μg/mL y la concentración mínima en el intervalo de las dosis fue de 5.5±2.7 μg/mL. El ABC de lopinavir sobre un intervalo de dosificación de 12 horas tuvo un promedio de 92.6 ± 36.7 μg•h/mL. No se ha establecido la biodisponibilidad absoluta de lopinavir coformulado con ritonavir en humanos. Bajo condiciones de no ayuno (500 Kcal, 25% de grasa), las concentraciones de lopinavir fueron similares después de la administración de KALETRA® coformulada, cápsulas de gelatina blanda y solución oral. Cuando se administró bajo condiciones en ayunas, tanto el ABC como la Cmáx promedio de lopinavir fueron 22% más bajas para KALETRA® solución oral con relación a la forma farmacéutica en cápsulas de gelatina blanda. Efectos de los alimentos sobre la absorción oral: Las Cápsulas de gelatina blanda y la Solución Oral de Kaletra® co-formulado, fueron bioequivalentes bajo condiciones de no ayuno (comida moderada en grasa). La administración de una sola dosis de 400/100 mg de KALETRA® cápsulas de gelatina blanda con una comida moderada en grasa (500-682 Kcal, 23 a 25% calorías de grasa) se asoció con incremento promedio de 48 y 23% en el ABC y la Cmáx de lopinavir, respectivamente, con relación al estado en ayunas. Para KALETRA® solución oral, los incrementos correspondientes en el ABC y la Cmáx de lopinavir fueron 80 y 54%, respectivamente. Con relación al estado en ayunas, la administración de KALETRA® con comida alta en grasa (872 Kcal, 56% de grasa) incrementó el ABC y la Cmáx de lopinavir en 97 y 43%, respectivamente, para las cápsulas de gelatina blanda, y 130 y 56%, respectivamente, para la solución oral. Para

aumentar la biodisponibilidad y minimizar la variabilidad farmacocinética se debe tomar KALETRA® con los alimentos. Distribución: En estado de equilibrio lopinavir se une aproximadamente 98-99% a las proteínas plasmáticas. Lopinavir se une tanto a la glicoproteína ácida alfa-1 (GAA) y a la albúmina; sin embargo, tiene afinidad más alta por la GAA. En estado de equilibrio, la unión de lopinavir a las proteínas permanece constante sobre el rango de concentraciones observadas después de 400/100 mg de KALETRA® dos veces al día, y es similar entre voluntarios sanos y pacientes VIH positivo. Metabolismo: Experimentos in vitro con microsomas hepáticos humanos indican que lopinavir sufre primariamente metabolismo oxidativo. Lopinavir es metabolizado principalmente por el sistema hepático del citocromo P450, casi exclusivamente por la isoenzima CYP3A. Ritonavir es un potente inhibidor del CYP3A lo que retrasa el metabolismo de lopinavir, y por lo tanto incrementa los niveles plasmáticos de lopinavir. Un estudio con 14C-lopinavir en humanos mostró que 89% de la radioactividad en plasma después de una dosis única de 400/100 mg de KALETRA® se debió al fármaco padre. Se han identificado al menos 13 metabolitos oxidativos de lopinavir en el hombre. Se ha demostrado que ritonavir induce las enzimas metabólicas, lo que resulta en inducción de su propio metabolismo. Las concentraciones pre dosis de lopinavir declinan con el tiempo durante la dosificación múltiple, estabilizándose después de aproximadamente 10 a 16 días. Eliminación: Después de una dosis de 400/100 mg de 14C-lopinavir/ritonavir, aproximadamente 10.4 ± 2.3% y 82.6 ± 2.5% de una dosis administrada de 14C-lopinavir puede ser detectada en la orina y las heces, respectivamente, después de 8 días. Lopinavir sin cambios es aproximadamente 2.2 y 19.8% de la dosis administrada en orina y heces, respectivamente. Después de dosificación múltiple, menos del 3% de la dosis de lopinavir se excreta sin cambios en la orina. La depuración oral aparente (CL/F) de lopinavir es 5.98 ± 5.75 L/h (media ± DE, N=19). Dosificación una vez al día: La farmacocinética de la administración una sola vez al día de KALETRA® ha sido evaluada en pacientes VIH positivo no experimentados para tratamiento anti-retroviral. KALETRA® 800/200 mg fue administrado en combinación con emtricitabina 200 mg y tenofovir DF 300 mg como parte de un tratamiento administrado una sola vez al día. La dosificación múltiple de KALETRA® 800/200 mg, una sola vez al día por cuatro semanas junto con comida (n=24) produjo una concentración máxima promedio de 11.8 ± 3.7 μg/mL seis horas después de su administración. La concentración mínima de lopinavir observada antes de la dosis de la mañana fue de 3.2 ± 2.1 μg/mL, la concentración mínima en el intervalo de las dosis fue 1.7 ± 1.6 μg/mL. El ABC de lopinavir con la dosificación diaria tuvo un promedio de 154.1 ± 61.4 μg•h/mL. Efectos sobre el electrocardiograma Se evaluó el Intervalo QTcF en un estudio, randomizado, cruzado controlado activo y con placebo (moxifloxacina 400 mg una vez al día) en 39 adultos saludables con 10 medidas durante 12 horas en el día 3. El promedio máximo de diferencia (95% del límite de confianza superior) en el QTcF con placebo fue de 3.6(6.3) mseg y 13.1(15.8) mseg para la dosis de lopinavir/ritonavir 400/100 mg dos veces al día y supraterapéutica 800/200 mg dos veces al día lopinavir/ritonavir respectivamente. Los dos regímenes resultaron para el día tres en exposiciones que fueron aproximadamente 1.5 a 3 veces más altas que las observadas en las dosis una vez al día y dos veces al día en estado de equilibrio. Ningún sujeto experimentó aumento en el intervalo QTcF ≥60 mseg sobre la línea basal o un intervalo QTcF que excediera de manera clínicamente significativa el límite de 500 mseg.

Una prolongación modesta del intervalo PR también se evidenció en sujetos recibiendo lopinavir/ritonavir en el mismo estudio para el día 3. El intervalo PR máximo fue de 286 mseg y no se observó bloqueo de segundo o tercer grado (Ver Advertencias y Precauciones). Poblaciones especiales: Género, raza y edad: La farmacocinética de lopinavir no ha sido estudiada en pacientes ancianos. No se han observado diferencias farmacocinéticas relacionadas con el género en pacientes adultos. No se han identificado diferencias farmacocinéticas clínicamente importantes debidas a la raza. Pacientes pediátricos: La farmacocinética de KALETRA® 300/75 mg/m2 dos veces al día y 230/57.5 mg/m2 dos veces al día ha sido estudiada en un total de 53 pacientes pediátricos, con rango de edad de 6 meses a 12 años. El régimen de 230/57.5 mg/m2
dos veces al día sin nevirapina y el régimen de 300/75 mg/m2 dos veces al día con nevirapina proporcionó concentraciones plasmáticas de lopinavir similares a las obtenidas en pacientes adultos que recibían el régimen 400/100 mg BID (sin nevirapina). KALETRA® una vez al día no ha sido evaluado en pacientes pediátricos. Los valores promedio de lopinavir del ABC, Cmáx y Cmín en estado de equilibrio fueron 72.6 ± 31.1 μg•h/mL, 8.2 ± 2.9 y 3.4 ± 2.1 μg/mL, respectivamente. Para KALETRA® 230/57.5 mg/m2 dos veces al día sin nevirapina (n = 12), fueron 85.8 ± 36.9 μg•h/mL, 10.0 ± 3.3 y 3.6 ± 3.5 μg/mL, respectivamente después de KALETRA® 300/75 mg/m2 dos veces al día con nevirapina (n = 12). El régimen con nevirapina fue 7 mg/Kg dos veces al día (6 meses a 8 años) ó 4 mg/Kg dos veces al día (> 8 años). Insuficiencia renal: La farmacocinética de lopinavir no ha sido estudiada en pacientes con insuficiencia renal; sin embargo, ya que la depuración renal de lopinavir es insignificante, no se espera disminución en la depuración corporal total en pacientes con insuficiencia renal. Insuficiencia hepática: Lopinavir es metabolizado y eliminado principalmente por el hígado. La administración de múltiples dosis de lopinavir/ritonavir 400/100 mg dos veces al día a pacientes coinfectados con VIH y VHC con compromiso hepático leve a moderado resultó en un incremento del 30% en el ABC de lopinavir y 20% de incremento de la Cmax comparados con pacientes infectados con VIH con función hepática normal. Adicionalmente, la fijación a las proteínas plasmáticas de lopinavir fue menor en pacientes con compromiso hepático leve a moderado comparado con los controles (99.09 vs. 99.31%, respectivamente). KALETRA® no ha sido estudiado en pacientes con compromiso hepático severo (ver PRECAUCIONES). Interacciones fármaco-fármaco: véase también CONTRAINDICACIONES, ADVERTENCIAS y PRECAUCIONES: Interacciones medicamentosas. KALETRA® es inhibidor del isómero CYP3A del P450 in vitro. La coadministración de KALETRA® y de fármacos metabolizados primariamente por el CYP3A puede resultar en incremento de las concentraciones plasmáticas del otro fármaco, las cuales pueden aumentar o prolongar sus efectos terapéuticos y adversos (véase CONTRAINDICACIONES). KALETRA® no inhibe el CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 o CYP1A2 a las concentraciones clínicamente relevantes. Se ha demostrado in vivo que KALETRA® induce su propio metabolismo e incrementa la biotransformación de algunos fármacos metabolizados por la isoenzima del citocromo P450 y por glucuronidación.

KALETRA® es metabolizada por el CYP3A. Es de esperar que los fármacos que inducen actividad del CYP3A incrementen la depuración de lopinavir resultando en concentraciones plasmáticas disminuidas de lopinavir. Aunque no se ha notado concomitantemente con ketoconazol, la coadministración de KALETRA® y otros fármacos que inhiben el CYP3A puede incrementar las concentraciones plasmáticas de lopinavir. Se han realizado estudios de interacciones farmacológicas con KALETRA® y otros fármacos con probabilidad de ser coadministrados y algunos fármacos frecuentemente utilizados como prueba para las interacciones farmacocinéticas. Los efectos de la coadministración de KALETRA® sobre el ABC, la Cmáx y la Cmín están resumidos en la Tabla 3 (efecto de otros fármacos sobre lopinavir) y la Tabla 4 (efectos de KALETRA® sobre otros fármacos). No se muestran los efectos de otros fármacos sobre ritonavir ya que ellos generalmente se correlacionan con los observados con lopinavir (si las concentraciones de lopinavir disminuyen, las concentraciones de ritonavir disminuyen) a menos que se indique de otro modo en las notas al pie de la tabla. Para información respecto a recomendaciones clínicas, ver Interacciones Medicamentosas Tabla 3: Interacciones farmacológicas. Parámetros farmacocinéticos para lopinavir en presencia del fármaco coadministrado (Véase Interacciones Medicamentosas, para Alteraciones Recomendadas en la Dosis o el Régimen)
Relación (con/sin el fármaco
coadministrado) de los parámetros farmacocinéticas
de lopinavir (IC 90%), sin efecto = 1,00
Fármaco
coadminis-trado
Dosis del fármaco
coadministrado (mg)
Dosis de
KALETRA® (mg)
n
Cmáx ABC Cmín Amprenavir
750 BID, 10 d
400/100 BID por 21 d
12
0,72 (0,65 ; 0,79)
0,62 (0,56 ; 0,70)
0,43 (0,34 ; 0,56)
Atorvastatina
20 QD, 4 d
400/100 BID, por 14 d
12
0,90 (0,78 ; 1,06)
0,90 (0,79 ; 1,02)
0,92 (0,78 ; 1,10)
Efavirenz (1) 600 QHS, 9 d
400/100 BID, por 9 d
11, 7*
0,97 (0,78 ;1,22)
0,81 (0,64 ; 1,03)
0,61 (0,38 ; 0,97)
Ketoconazol
Dosis única 200
400/100 BID, por 16 d
12
0,89 (0,80 ; 0,99)
0,87 (0,75 ; 1,00)
0,75 (0,55 ; 1,00)
Nelfinavir
1000 BID, 10 d
400/100 BID, por 21 d
13
0,79 (0,70 ; 0,89)
0,73 (0,63 ; 0,85)
0,62 (0,49 ; 0,78)
Nevirapina 200 BID, estado de equilibrio (>1 año) (2)
400/100 BID estado de equilibrio, > 1 año
22 19*
0,81 (0,62
; 1,05)
0,73
(0,53 ; 0,98)
0,49
(0,28 ; 0,74)
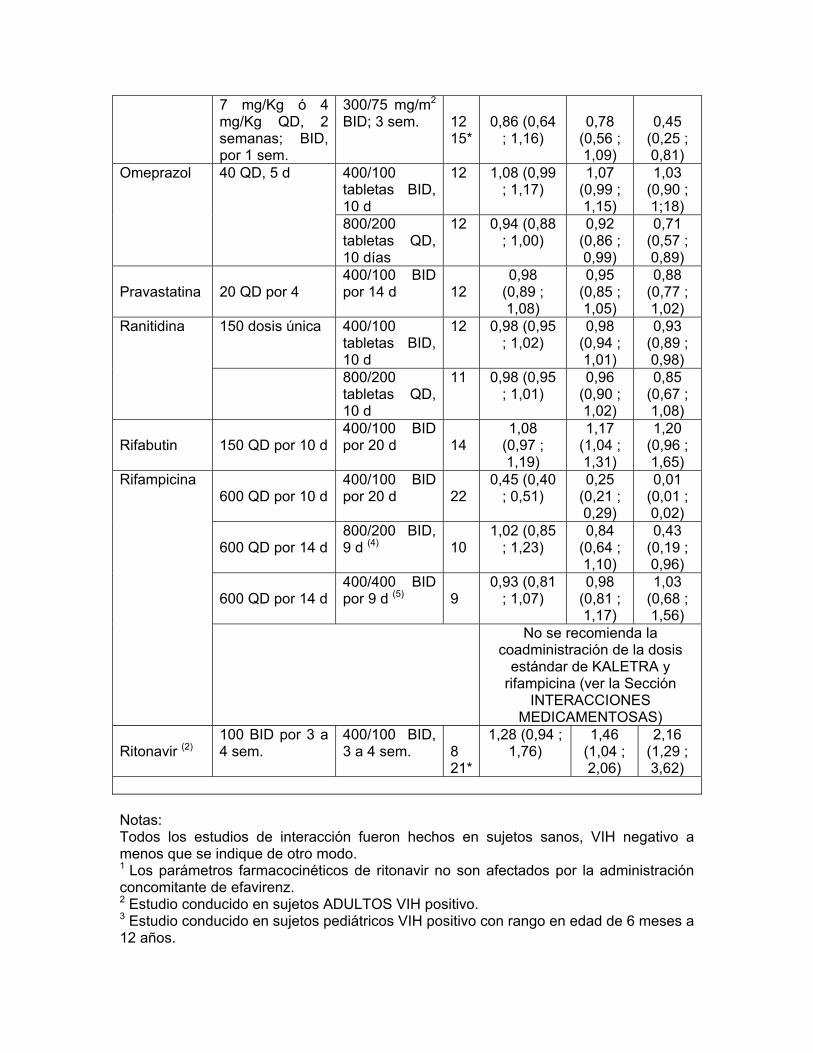
7 mg/Kg ó 4 mg/Kg QD, 2 semanas; BID, por 1 sem.
300/75 mg/m2 BID; 3 sem.
12 15*
0,86 (0,64
; 1,16)
0,78
(0,56 ; 1,09)
0,45
(0,25 ; 0,81)
400/100 tabletas BID, 10 d
12 1,08 (0,99 ; 1,17)
1,07 (0,99 ; 1,15)
1,03 (0,90 ; 1;18)
Omeprazol 40 QD, 5 d
800/200 tabletas QD, 10 días
12 0,94 (0,88 ; 1,00)
0,92 (0,86 ; 0,99)
0,71 (0,57 ; 0,89)
Pravastatina
20 QD por 4
400/100 BID por 14 d
12
0,98 (0,89 ; 1,08)
0,95 (0,85 ; 1,05)
0,88 (0,77 ; 1,02)
150 dosis única 400/100 tabletas BID, 10 d
12 0,98 (0,95 ; 1,02)
0,98 (0,94 ; 1,01)
0,93 (0,89 ; 0,98)
Ranitidina
800/200 tabletas QD, 10 d
11 0,98 (0,95 ; 1,01)
0,96 (0,90 ; 1,02)
0,85 (0,67 ; 1,08)
Rifabutin
150 QD por 10 d
400/100 BID por 20 d
14
1,08 (0,97 ; 1,19)
1,17 (1,04 ; 1,31)
1,20 (0,96 ; 1,65)
600 QD por 10 d
400/100 BID por 20 d
22
0,45 (0,40 ; 0,51)
0,25 (0,21 ; 0,29)
0,01 (0,01 ; 0,02)
600 QD por 14 d
800/200 BID, 9 d (4)
10
1,02 (0,85 ; 1,23)
0,84 (0,64 ; 1,10)
0,43 (0,19 ; 0,96)
600 QD por 14 d
400/400 BID por 9 d (5)
9
0,93 (0,81 ; 1,07)
0,98 (0,81 ; 1,17)
1,03 (0,68 ; 1,56)
Rifampicina
No se recomienda la coadministración de la dosis
estándar de KALETRA y rifampicina (ver la Sección
INTERACCIONES MEDICAMENTOSAS)
Ritonavir (2)
100 BID por 3 a 4 sem.
400/100 BID, 3 a 4 sem.
8 21*
1,28 (0,94 ; 1,76)
1,46 (1,04 ; 2,06)
2,16 (1,29 ; 3,62)
Notas: Todos los estudios de interacción fueron hechos en sujetos sanos, VIH negativo a menos que se indique de otro modo. 1 Los parámetros farmacocinéticos de ritonavir no son afectados por la administración concomitante de efavirenz. 2 Estudio conducido en sujetos ADULTOS VIH positivo. 3 Estudio conducido en sujetos pediátricos VIH positivo con rango en edad de 6 meses a 12 años.
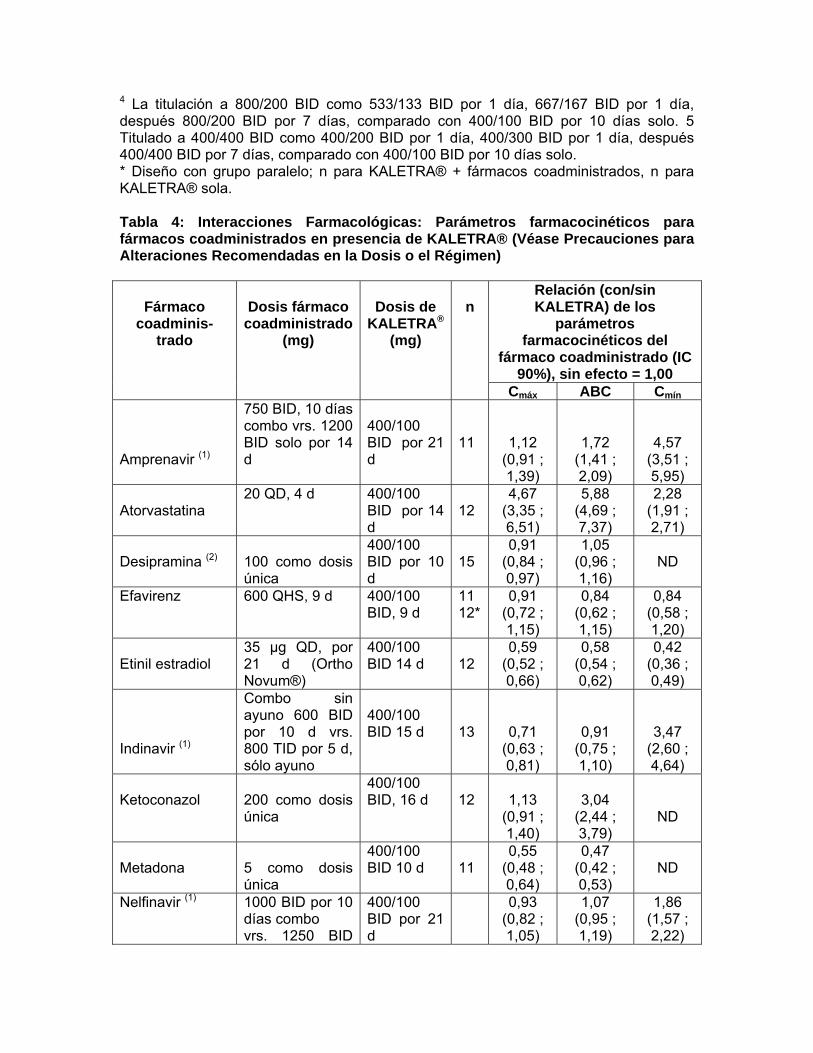
4 La titulación a 800/200 BID como 533/133 BID por 1 día, 667/167 BID por 1 día, después 800/200 BID por 7 días, comparado con 400/100 BID por 10 días solo. 5 Titulado a 400/400 BID como 400/200 BID por 1 día, 400/300 BID por 1 día, después 400/400 BID por 7 días, comparado con 400/100 BID por 10 días solo. * Diseño con grupo paralelo; n para KALETRA® + fármacos coadministrados, n para KALETRA® sola. Tabla 4: Interacciones Farmacológicas: Parámetros farmacocinéticos para fármacos coadministrados en presencia de KALETRA® (Véase Precauciones para Alteraciones Recomendadas en la Dosis o el Régimen)
Relación (con/sin KALETRA) de los
parámetros farmacocinéticos del
fármaco coadministrado (IC 90%), sin efecto = 1,00
Fármaco
coadminis-trado
Dosis fármaco
coadministrado (mg)
Dosis de
KALETRA® (mg)
n
Cmáx ABC Cmín Amprenavir (1)
750 BID, 10 días combo vrs. 1200 BID solo por 14 d
400/100 BID por 21 d
11
1,12 (0,91 ; 1,39)
1,72 (1,41 ; 2,09)
4,57 (3,51 ; 5,95)
Atorvastatina
20 QD, 4 d 400/100 BID por 14 d
12
4,67 (3,35 ; 6,51)
5,88 (4,69 ; 7,37)
2,28 (1,91 ; 2,71)
Desipramina (2)
100 como dosis única
400/100 BID por 10 d
15
0,91 (0,84 ; 0,97)
1,05 (0,96 ; 1,16)
ND
Efavirenz 600 QHS, 9 d 400/100 BID, 9 d
11 12*
0,91 (0,72 ; 1,15)
0,84 (0,62 ; 1,15)
0,84 (0,58 ; 1,20)
Etinil estradiol
35 μg QD, por 21 d (Ortho Novum®)
400/100 BID 14 d
12
0,59 (0,52 ; 0,66)
0,58 (0,54 ; 0,62)
0,42 (0,36 ; 0,49)
Indinavir (1)
Combo sin ayuno 600 BID por 10 d vrs. 800 TID por 5 d, sólo ayuno
400/100 BID 15 d
13
0,71 (0,63 ; 0,81)
0,91 (0,75 ; 1,10)
3,47 (2,60 ; 4,64)
Ketoconazol
200 como dosis única
400/100 BID, 16 d
12
1,13
(0,91 ; 1,40)
3,04
(2,44 ; 3,79)
ND
Metadona
5 como dosis única
400/100 BID 10 d
11
0,55 (0,48 ; 0,64)
0,47 (0,42 ; 0,53)
ND
Nelfinavir (1)
1000 BID por 10 días combo vrs. 1250 BID
400/100 BID por 21 d
0,93 (0,82 ; 1,05)
1,07 (0,95 ; 1,19)
1,86 (1,57 ; 2,22)
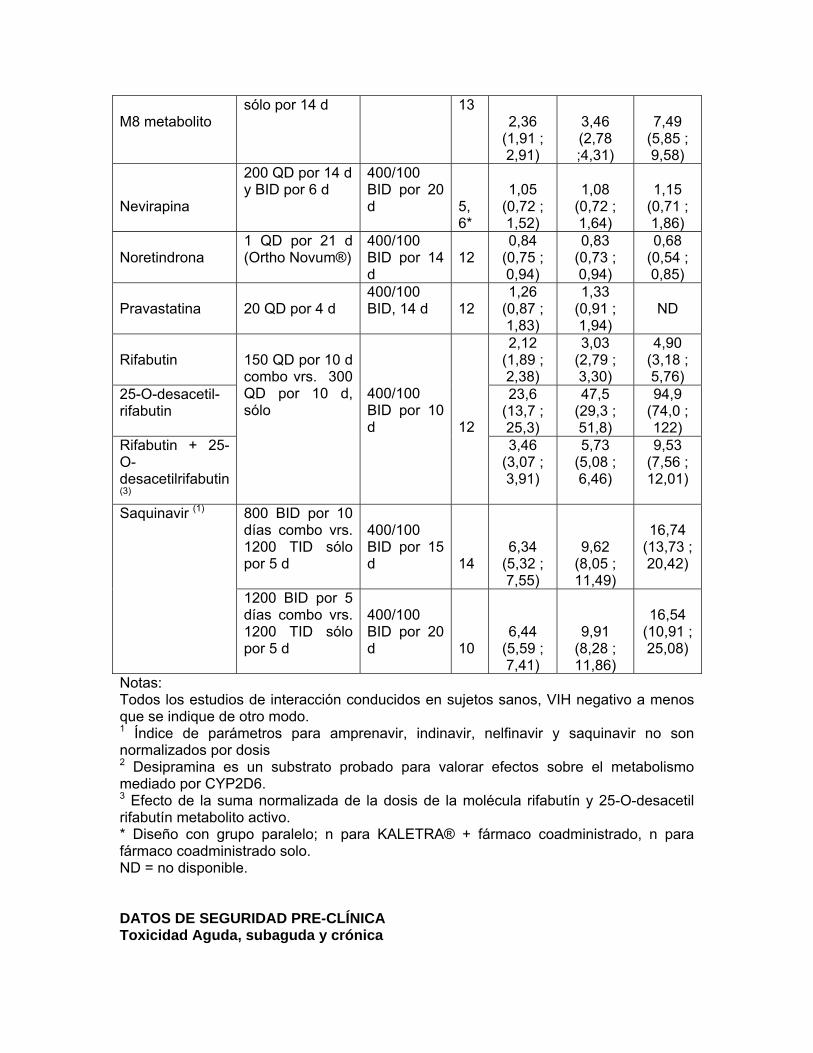
M8 metabolito
sólo por 14 d 13 2,36
(1,91 ; 2,91)
3,46 (2,78 ;4,31)
7,49
(5,85 ; 9,58)
Nevirapina
200 QD por 14 dy BID por 6 d
400/100 BID por 20 d
5, 6*
1,05
(0,72 ; 1,52)
1,08
(0,72 ; 1,64)
1,15
(0,71 ; 1,86)
Noretindrona
1 QD por 21 d (Ortho Novum®)
400/100 BID por 14 d
12
0,84 (0,75 ; 0,94)
0,83 (0,73 ; 0,94)
0,68 (0,54 ; 0,85)
Pravastatina
20 QD por 4 d
400/100 BID, 14 d
12
1,26 (0,87 ; 1,83)
1,33 (0,91 ; 1,94)
ND
Rifabutin
2,12 (1,89 ; 2,38)
3,03 (2,79 ; 3,30)
4,90 (3,18 ; 5,76)
25-O-desacetil-rifabutin
23,6 (13,7 ; 25,3)
47,5 (29,3 ; 51,8)
94,9 (74,0 ; 122)
Rifabutin + 25-O-desacetilrifabutin (3)
150 QD por 10 d combo vrs. 300 QD por 10 d, sólo
400/100 BID por 10 d
12
3,46 (3,07 ; 3,91)
5,73 (5,08 ; 6,46)
9,53 (7,56 ; 12,01)
800 BID por 10 días combo vrs. 1200 TID sólo por 5 d
400/100 BID por 15 d
14
6,34 (5,32 ; 7,55)
9,62 (8,05 ; 11,49)
16,74
(13,73 ; 20,42)
Saquinavir (1)
1200 BID por 5 días combo vrs. 1200 TID sólo por 5 d
400/100 BID por 20 d
10
6,44 (5,59 ; 7,41)
9,91 (8,28 ; 11,86)
16,54
(10,91 ; 25,08)
Notas: Todos los estudios de interacción conducidos en sujetos sanos, VIH negativo a menos que se indique de otro modo. 1 Índice de parámetros para amprenavir, indinavir, nelfinavir y saquinavir no son normalizados por dosis 2 Desipramina es un substrato probado para valorar efectos sobre el metabolismo mediado por CYP2D6. 3 Efecto de la suma normalizada de la dosis de la molécula rifabutín y 25-O-desacetil rifabutín metabolito activo. * Diseño con grupo paralelo; n para KALETRA® + fármaco coadministrado, n para fármaco coadministrado solo. ND = no disponible. DATOS DE SEGURIDAD PRE-CLÍNICA Toxicidad Aguda, subaguda y crónica

Los estudios con dosis repetidas en roedores y perros han identificado como órganos blanco principales al hígado, riñones, tiroides, bazo y eritrocitos circulantes. En el hígado se observa edema celular y degeneración focal. La exposición que causa estos cambios fue comparable a la exposición en humanos, las dosis en animales fueron seis veces superior a las dosis clínicas recomendadas. En ratones se ha observado degeneración tubular renal leve con dosis al menos el doble de las recomendadas a humanos; los riñones no se afectaron en ratas y perros. La reducción de la tiroxina sérica conllevó a un aumento en la liberación de TSH con hipertrofia celular folicular resultante en la glándulas tiroides de las ratas. Estos cambios se revirtieron completamente al suspender el medicamento y no se observaron en ratones y perros. Anisocitosis Coombs negativa y poiquilocitosis se observaron en ratas pero no en ratones ni en perros. El bazo se observó aumentado con histiocitosis en las ratas pero no en las otras especies. El colesterol sérico estaba elevado en los roedores pero no en los perros, mientras los triglicéridos sólo se elevan en los ratones. Carcinogénesis y Mutagénesis Los estudios a largo plazo de carcinogenicidad de lopinavir/ritonavir en ratones revelaron la no genotoxicidad, inducción mitogénica o inducción de tumores en hígado, generalmente considerados de tener muy poca relevancia de riesgo humano. Los estudios de carcinogenicidad en ratas no revelaron hallazgos tumorigénicos. Lopinavir no es teratogénico ni clastogénico en una batería de ensayos in vitro incluyendo la prueba reversa de mutación en bacterias de Ames, la prueba de linfoma murino, la prueba de micronúcleo de ratón y las pruebas de aberraciones cromosómicas de linfocitos humanos. Lopinavir/ritonavir no se encontró mutagénico o clastogénico en ensayos in vivo usando ensayos de micronucleo de ratón. INDICACIONES Y USO KALETRA® está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por VIH. Esta indicación se basa en análisis de los niveles plasmáticos de ARN de VIH y conteos de células CD4 en un estudio controlado de KALETRA® de 48 semanas de duración y estudios más pequeños de rango de dosis no controlados de KALETRA® por 144-360 semanas. Actualmente, no existen resultados de estudios controlados evaluando el efecto de KALETRA® sobre la progresión clínica del VIH. CONTRAINDICACIONES KALETRA® está contraindicada en pacientes con hipersensibilidad conocida a lopinavir y ritonavir o a cualquiera de los excipientes. La coadministración de KALETRA® está contraindicada con fármacos que son altamente dependientes del CYP3A para su depuración y para los cuales las concentraciones plasmáticas elevadas se asocian con eventos serios o que amenacen la vida. Estos fármacos se muestran en la Tabla 5. Tabla 5: Fármacos que no deben ser administrados concomitantemente con KALETRA®

*Ver Advertencias y Precauciones e Interacciones Medicamentosas para la coadministración de sildenafil en pacientes con disfunción eréctil. ADVERTENCIAS Y PRECAUCIONES Interacciones Medicamentosas KALETRA® es inhibidor de la isoenzima P450 CYP3A. La coadministración de KALETRA® y fármacos primariamente metabolizados por el CYP3A puede resultar en incremento de las concentraciones plasmáticas del otro fármaco que pueden incrementar o prolongar sus efectos terapéuticos y adversos (véase Interacciones medicamentosas y Farmacocinética: Interacciones fármaco-fármaco. Contraindicaciones en la tabla 5). Antimicóticos KALETRA® no debe ser coadministrado con rifampicina debido a que la rifampicina disminuye mucho las concentraciones de lopinavir, lo que puede causar disminución de su efecto terapéutico (Ver Interacciones medicamentosas). Corticosteroides El uso concomitante de KALETRA® con propionato de fluticasona puede incrementar significativamente la concentración en plasma de fluticasona lo que causa la reducción de las concentraciones plasmáticas de cortisol. El efecto sistémico de los corticosteroides como el Síndrome de Cushing y supresión adrenal han sido reportados en pacientes que recibían administración concomitante de ritonavir y propionato de fluticasona administrada por inhalación o por vía intranasal. También se ha reportado este efecto en pacientes que reciben corticosteroides inhalados que son metabolizados en forma similar a la fluticasona como budesonida. Se debe tener particular precaución cuando se coadministre lopinavir/ritonavir y alguno de estos glucocorticoides administrados por inhalación o intranasalmente. Ver Interacciones Medicamentosas. Agentes para disfunción eréctil (Inhibidores de PDE5) Se debe tener precaución particular cuando se prescribe sildenafil, tadalafil, vardenafil para el tratamiento de la disfunción eréctil en pacientes que reciben KALETRA®. Es de esperar que la coadministración de KALETRA® incremente sustancialmente las concentraciones de estos agentes de disfunción eréctil, y puede dar como resultado

incremento en los eventos adversos asociados incluyendo hipotensión y erección prolongada. El uso concomitante de sildenafil con lopinavir/ritonavir está contraindicado en los pacientes con Hipertensión Arterial Pulmonar (HAP) (véase CONTRAINDICACIONES e INTERACCIONES MEDICAMENTOSAS). Productos Herbales No se recomienda el uso concomitante de KALETRA® y Hierba de San Juan (Hypericum perforatum), o productos que contienen la Hierba de San Juan. Es de esperar que la coadministración de inhibidores de proteasa, incluyendo KALETRA®, con Hierba de San Juan disminuya sustancialmente las concentraciones en plasma de inhibidor de proteasa y puede resultar en niveles subóptimos de lopinavir y conducir a pérdida de la respuesta virológica y posible resistencia a lopinavir o a los inhibidores de proteasa (ver CONTRAINDICACIONES E INTERACCIONES MEDICAMENTOSAS). Inhibidores de la HMG-CoA Reductasa La administración simultánea de KALETRA® con lovastatina o simvastatina está contraindicada (Ver contraindicaciones) Se debe tener precaución con la coadministración simultánea de inhibidores de proteasa, incluyendo KALETRA®, Rosuvastatina u otros inhibidores de la HMG-CoA reductasa que son metabolizados por la vía CYP3A4 ( ej, atorvastatina) porque pueden ocurrir reacciones serias potenciales como miopatia que incluyen rabdomiólisis (Ver interacciones medicamentosas) Tipranavir En un estudio clínico con doble potenciación de la terapia combinada del inhibidor de proteasa en adultos con VIH positivo experimentados multitratados, el tipranavir (500 mg dos veces al día) con ritonavir (200 mg dos veces al día), co-administrado con lopinavir/ritonavir (400/100 mg dos veces al día), resultó en una reducción del 47% y 70% en la ABC y la Cmin respectivamente. La administración concomitante de lopinavir/ritonavir y tipranavir con bajas dosis de ritonavir por lo tanto no está recomendada. Pancreatitis Se ha observado pancreatitis en pacientes que reciben terapia con KALETRA®, incluyendo aquellos que desarrollaron elevaciones marcadas de los triglicéridos. En algunos casos, se han presentado muertes. Aunque no se ha establecido una relación causal de KALETRA®, las elevaciones marcadas en los triglicéridos son un factor de riesgo para el desarrollo de pancreatitis (véase PRECAUCIONES Y ADVERTENCIAS - Elevaciones de los lípidos). Los pacientes con enfermedad avanzada por el VIH pueden tener un riesgo incrementado de triglicéridos elevados y pancreatitis, y los pacientes con una historia de pancreatitis pueden tener un mayor riesgo de recurrencia durante la terapia con KALETRA®. Diabetes mellitus/hiperglicemia Se ha informado inicio de diabetes mellitus, exacerbación de diabetes mellitus pre-existente, e hiperglicemia en la vigilancia post-mercadeo en pacientes infectados con VIH que reciben terapia con inhibidor de proteasa. Algunos pacientes requirieron inicio o ajuste de la dosis de insulina o agentes hipoglicemiantes orales para el tratamiento de estos eventos. En algunos casos, ha ocurrido cetoacidosis diabética. En algunos de aquellos pacientes que descontinuaron la terapia con inhibidor de proteasa la hiperglicemia persistió. Debido a que estos eventos han sido informados voluntariamente durante la práctica clínica, no se pueden hacer estimados de la frecuencia y no se ha establecido una relación causal entre terapia con inhibidor de proteasa y estos eventos. Insuficiencia hepática

KALETRA® es metabolizado principalmente por el hígado; por lo tanto, se debe tener precaución cuando se administra este fármaco a pacientes con insuficiencia hepática. KALETRA® no ha sido estudiado en pacientes con disfunción hepática severa. Los datos farmacocinéticos sugieren que las concentraciones plasmáticas de lopinavir se incrementan aproximadamente en un 30% pero también se reduce la fijación a las proteínas plasmáticas en pacientes coinfectados con VIH y VHC con falla hepática de leve a moderada (ver FARMACOLOGIA CLINICA: FARMACOCINETICA). Los pacientes con hepatitis B o C subyacente o elevaciones marcadas en las transaminasas previo al tratamiento pueden tener mayor riesgo de desarrollar elevaciones adicionales en las transaminasas. Se han hecho reportes en la etapa postmercadeo de disfunción hepática, incluyendo mortalidades. Las complicaciones han ocurrido en pacientes con infecciones avanzadas por VIH que toman múltiples medicamentos concomitantemente en el contexto de hepatitis crónica o cirrosis. La relación causal con KALETRA® no se ha establecido. Se debe considerar vigilar cuidadosamente las transaminasas (AST, ALT) en estos pacientes, especialmente durante los primeros meses del tratamiento con KALETRA®. Resistencia/Resistencia Cruzada Se han observado varios grados de resistencia cruzada entre los inhibidores de proteasa. El efecto de la terapia con KALETRA® sobre la eficacia de los inhibidores de proteasa administrados subsecuentemente está bajo investigación (véase MICROBIOLOGÍA). Hemofilia Ha habido informes de incremento en el sangrado, incluyendo hematomas cutáneos espontáneos y hemartrosis, en pacientes con hemofilia tipo A y B tratados con inhibidores de proteasa. En algunos pacientes se administró factor VIII adicional. En más de la mitad de los casos informados, el tratamiento con inhibidores de proteasa fue continuado o reintroducido. No se ha establecido relación causal entre terapia con inhibidor de proteasa y estos eventos. Prolongación del intervalo PR Se ha demostrado que lopinavir/ritonavir causa una modesta prolongación asintomática del intervalo PR en algunos pacientes. Se han reportado casos de bloqueo auriculoventricular de segundo o tercer grado con patología cardíaca estructural de base y anormalidades preexistentes del sistema de conducción o en pacientes recibiendo medicaciones con efectos conocidos de causar prolongación del intervalo PR (tales como el Verapamilo o el atazanavir) en pacientes recibiendo lopinavir/ritonavir. Lopinavir/ritonavir debe ser utilizado con cautela en este grupo de pacientes (ver FARMACOLOGÍA CLÍNICA) Redistribución de Grasa La redistribución/acumulación de grasa corporal incluyendo obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), atrofia periférica, atrofia facial, aumento de los senos, “apariencia cushingoide” han sido observadas en pacientes recibiendo terapia antirretroviral. El mecanismo y las consecuencias a largo plazo de estos eventos se desconocen actualmente. No se ha establecido una relación causal. Elevaciones en los Lípidos El tratamiento con KALETRA® ha resultado en grandes incrementos en la concentración de colesterol total y triglicéridos (véase REACCIONES ADVERSAS - Tabla 8 y 9). Se debe realizar exámenes de triglicéridos y colesterol antes de iniciar la terapia con KALETRA® y a intervalos periódicos durante la terapia. Las alteraciones de los lípidos se deben manejar en forma clínicamente apropiada. Véase Advertencias y precauciones para información adicional sobre interacciones farmacológicas potenciales con KALETRA® e inhibidores de la HMG-CoA reductasa.

Síndrome de Reconstitución Inmunológico Se ha reportado la ocurrencia de síndrome de reconstitución inmunológico en pacientes infectados con VIH tratados con terapia combinada que incluye KALETRA®. Durante la fase inicial del tratamiento cuando el sistema inmunológico responde los pacientes pueden desarrollar una respuesta inflamatoria a infecciones oportunistas asintomáticas o residuales (como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocistis jiroveci o tuberculosis) que pueden necesitar evaluación adicional y tratamiento. Uso geriátrico Los estudios clínicos con KALETRA® no incluyeron suficientes sujetos de 65 años edad y mayores para determinar si ellos responden de forma diferente que los sujetos más jóvenes. En general, se debe ejercer precaución apropiada sobre la administración y monitoreo de KALETRA® en pacientes ancianos quienes tienen mayor frecuencia de disminución de la función hepática, renal o cardiaca, y de enfermedad concomitante u otra terapia farmacológica. Uso Pediátrico Los perfiles de seguridad y farmacocinéticos de KALETRA® en pacientes pediátricos menores de 6 meses de edad no han sido establecidos. En pacientes de 6 meses a 12 años de edad infectados con VIH, el perfil de eventos adversos observado durante una prueba clínica fue similar al de los pacientes adultos. La evaluación de la actividad antiviral de KALETRA® en pacientes pediátricos en ensayos clínicos está en curso. La administración una vez al día de KALETRA® no se ha evaluado en la población pediátrica. INTERACCIONES MEDICAMENTOSAS KALETRA® es un inhibidor del CYP3A (citocromo P450 3A) tanto in vitro como in vivo. La coadministración de KALETRA® y fármacos metabolizados primariamente por el CYP3A (p. ej., bloqueadores de los canales de calcio tipo dihidropiridina, inhibidores de la HMG-CoA reductasa, inmunosupresores e inhibidores de PDE5 ) puede resultar en un incremento de las concentraciones plasmáticas de los otros fármacos que pueden aumentar o prolongar sus efectos terapéuticos y adversos (véase PRECAUCIONES Y ADVERTENCIAS). Los agentes que son metabolizados extensamente por el CYP3A y tienen mucho metabolismo de primer paso parecen ser los más susceptibles a grandes incrementos en el ABC (> 3 veces) cuando se coadministran con KALETRA®. Los medicamentos que están contraindicados específicamente debido a las importantes interacciones y potencial de efectos adversos se listan en la tabla 5 en CONTRAINDICACIONES. KALETRA® es metabolizado por el CYP3A. La coadministración de KALETRA® y fármacos que inducen el CYP3A puede disminuir las concentraciones plasmáticas de lopinavir y reducir su efecto terapéutico (véase PRECAUCIONES Y ADVERTENCIAS). Aunque no se han observado con la administración concomitante con ketoconazol, la coadministración de KALETRA® y otros fármacos que inhiben el CYP3A puede incrementar las concentraciones plasmáticas de lopinavir. Agentes anti VIH Inhibidores nucleósidos de la transcriptasa reversa INTR Estavudina y Lamivudina No se ha observado cambio en la farmacocinética de lopinavir cuando se administra KALETRA® con estavudina y lamivudina. Didanosina Se recomienda que la didanosina se administre con el estómago vacío; por esto debe administrarse una hora antes o dos horas después de KALETRA® administrado junto con comida.

Zidovudina y Abacavir KALETRA® induce la glucoronidación y por esto tiene el potencial de reducir las concentraciones plasmáticas de abacavir y zidovudina. Se desconoce las consecuencias de esta interacción. Tenofovir Se realizó un estudio que demostró que KALETRA® incrementa los niveles plasmáticos de tenofovir. No se conoce su mecanismo causal. Los pacientes que estén recibiendo Kaletra® y tenofovir, deben ser controlados buscando reacciones adversas asociadas a tenofovir. Todos La administración de inhibidores de proteasa simultáneamente con INTR causa incremento en CPK (creatina fosfoquinasa), mialgia, miositis y raramente rabdomiólisis. Inhibidores no nucleósidos de la transcriptasa reversa INNTR. Nevirapina No se han observado cambios en la farmacocinética de lopinavir en adultos sanos a los que también se les ha administrado nevirapina. En un estudio realizado con niños infectados con VIH se produjo disminución en las concentraciones de lopinavir al usarse simultáneamente con nevirapina; estos resultados probablemente también ocurran en adultos VIH positivo y por lo tanto las concentraciones de lopinavir pueden disminuir. El resultado clínico de esta interacción es desconocido. En los pacientes que tienen larga experiencia con IP o evidencia fenotípica o genotípica de pérdida significativa de la susceptibilidad a lopinavir, se recomienda incrementar la dosis a 533/133 mg BID de KALETRA® cuando es coadministrado con Nevirapina. KALETRA® no debe ser administrado una vez al día en combinación con Nevirapina. Efavirenz El uso simultáneo de KALETRA® y efavirenz con dos inhibidores nucleósidos de la transcriptasa reversa en pacientes multi-experimentados, incrementando la dosis en un 33,3% de 400/100 mg (3 cápsulas) dos veces al día a 533/133 mg (4 cápsulas) dos veces al día, produjo concentraciones plasmáticas de lopinavir comparables a las históricamente obtenidas con lopinavir 400/100mg dos veces al día. Los pacientes multi-experimentados a IP o evidencias de fenotipo o genotipo de resistencia también deben incrementar la dosis a 533/133 mg dos veces al día al coadministrar con efavirenz. Nota: efavirenz y nevirapina inducen la actividad de CYP3A y tiene el potencial de disminuir las concentraciones plasmáticas de otros IP cuando se usan en combinación con KALETRA®. KALETRA® no debe ser administrada una vez al día en combinación con efavirenz. Delavirdina La delavidina tiene el potencial de aumentar las concentraciones de lopinavir en plasma. Inhibidores de proteasa IP. Amprenavir KALETRA® incrementa la concentración de amprenavir (amprenavir 750 mg BID más KALETRA® produce incremento de ABC, similar Cmax, incrementa Cmin, similar a lo obtenido con amprenavir 1200 mg BID). La co-administración de KALETRA® y amprenavir reduce la concentración plasmática de lopinavir. La dosis de KALETRA® puede ser incrementada si se coadministra con amprenavir, particularmente en pacientes con experiencia extensiva a inhibidores de proteasa o reducción de la susceptibilidad viral a lopinavir (ver DOSIFICACION Y ADMINISTRACION y FARMACOLOGIA CLINICA: Tablas 3 y 4). KALETRA® no debe ser administrada una vez al día en combinación con amprenavir Fosamprenavir

Un estudio de coadministración de KALETRA® y fosamprenavir demostró la reducción de niveles de amprenavir y lopinavir. No se ha establecido la dosificación adecuada de KALETRA® y fosamprenavir cuando se hace esta coadministración. Indinavir KALETRA® incrementa las concentraciones de indinavir (indinavir 600 mg BID más KALETRA® produce ABC similar, disminuye Cmax, incrementa Cmin si se compara con indinavir 800 mg TID). La dosis de indinavir puede requerir disminuirse durante su coadministración con KALETRA® 400/100 mg BID (ver FARMACOLOGIA CLINICA: Tabla 4). La administración de KALETRA® una vez al día no ha sido estudiada en combinación con indinavir. Nelfinavir KALETRA® incrementa las concentraciones de nelfinavir y su metabolito M8 (nelfinavir 1000 mg BID más KALETRA® produce ABC similar , similar Cmax, incremento de Cmin
comparando con nelfinavir 1250 mg BID). La co-administración de KALETRA® y nelfinavir resulta en disminución de las concentraciones de lopinavir. La dosis de KALETRA® puede ser incrementado cuando se coadministre con nelfinavir, particularmente en pacientes con VIH muy expuestos a inhibidores de proteasa o con reducida susceptibilidad al lopinavir (ver DOSIFICACIÓN Y ADMINISTRACIÓN y FARMACOLOGÍA CLINICA: Tablas 3 y 4). KALETRA® no debe ser administrada una vez al día en combinación con nelfinavir. Ritonavir Cuando KALETRA® se coadministra con 100 mg adicionales de ritonavir dos ves al día el ABC de lopinavir se incrementa en 33% y la Cmin se incrementa en 64% si se compara con KALETRA® 400/100 mg (tres (3) cápsulas de gelatina blanda dos veces al día). Saquinavir KALETRA® incrementa las concentraciones de saquinavir (saquinavir 800 mg BID más KALETRA® produce incremento del ABC, incrementa Cmax, incrementa Cmin al comparar con saquinavir 1200 mg TID). La dosis de saquinavir puede necesitar disminuirse cuando se coadministra con KALETRA® 400/100 mg BID (ver FARMACOLOGÍA CLÍNICA: Tabla 4). KALETRA® no debe ser administrada una vez al día en combinación con saquinavir. Otros medicamentos Analgésicos Fentanilo: Lopinavir/Ritonavir inhibe el CYP3A4 y como resultado se espera que incremente las concentraciones plasmáticas de fentanilo. Se recomienda un monitoreo cuidadoso de los efectos terapéuticos y adversos (incluyendo depresión respiratoria) cuando el fentanilo es administrado concomitantemente con Lopinavir/Ritonavir. Antiarrítmicos: La amiodarona, bepridil, lidocaína sistémica y quinidina pueden aumentar su concentración si son coadministrados con KALETRA®. Se recomienda cuidado y control de las concentraciones terapéuticas. Digoxina: La literatura ha reportado que la coadministración de ritonavir (300 mg cada 12 horas) y digoxina resultó en un incremento significativo de los niveles de digoxina: Se debe tener precaución cuando se coadministre lopinavir/ritonavir y digoxina; se requiere el apropiado monitoreo de los niveles séricos de digoxina. Agentes anticancerígenos (ej. Dasatinib, Nilotinib Vincristina, Vinblastina): Pueden aumentar sus concentraciones séricas cuando se co-administran con lopinavir/ritonavir, aumentando potencialmente los eventos adversos usualmente asociados con los agentes anticancerígenos. Para nilotinib y dasatinib, referirse a la información de prescripción de instrucciones de dosificación.

Anticoagulantes: La concentración de warfarina se afecta al coadministrarse con KALETRA®. Se recomienda control de RIN (Relación Internacional Normalizada). Antidepresivos: Bupropión: La administración concomitante de bupropión con lopinavir/ritonavir disminuye los niveles plasmáticos tanto del bupropión como de su metabolito activo (hidroxibupropión) Trazodona: el uso concomitante de ritonavir y trazodona puede incrementar los niveles de esta última. Eventos adversos de nausea, mareo, hipotensión y síncope se han observado. Si se usa trazodona con un inhibidor de CYP3A4 como lopinavir/ritonavir, la combinación debe ser usada con precaución y se debe considerar una disminución en la dosis de trazodona. Anticonvulsivantes: Fenobarbital, fenitoína y carbamazepina inducen CYP3A4 y disminuyen las concentraciones de lopinavir. KALETRA® no debe ser administrado una vez al día en combinación con fenobarbital, fenitoína o carbamazepina. Además, la co-administración de fenitoína y lopinavir/ritonavir resulta en una disminución de las concentraciones en estado de equilibrio de fenitoína. Los niveles de fenitoína deben ser monitoreados cuando se administra con lopinavir/ritonavir Antifúngicos: Ketoconazol e Itraconazol: pueden incrementar su concentración sérica, no se recomiendan dosis mayores a 200 mg/día. Voriconazol: Un estudio ha demostrado que la coadministración de ritonavir 100 mg cada 12 horas disminuye el ABC en estado de equilibrio de voriconazol en un promedio de 39%. Por lo tanto la co-administración de lopinavir/ritonavir y voriconazol se debe evitar, a menos que una valoración del riesgo/beneficio del paciente justifique el uso de voriconazol. Anti- Infecciosos: Se ha observado un incremento moderado del ABC de claritromicina al coadministrarse con KALETRA®. Para pacientes con insuficiencia renal y hepática se debe considerar reducción de la dosis de claritromicina. Antimicobacterianos: Rifabutina: Al coadministrarla con KALETRA® por diez días se produce incremento de rifabutina (fármaco padre) y de su metabolito principal (25-O-desacetil) de 3.5 y 5.7 veces en Cmáx y ABC. Con estos datos se recomienda disminuir la rifabutina en un 75% (150 mg cada dos días o tres veces por semana). Puede ser necesario reducirla aún más. Rifampicina: Debido a la gran disminución de las concentraciones de lopinavir no se recomienda usar Rifampicina en combinación con la dosis estándar de KALETRA® (ver PRECAUCIONES Y ADVERTENCIAS e INTERACCIONES MEDICAMENTOSAS). El uso de rifampicina con la dosis estándar de KALETRA® puede conllevar a una pérdida de la respuesta virológica y a una posible resistencia a KALETRA®, a los inhibidores de proteasas o a otros agentes antiretrovirales administrados conjuntamente. La coadministración de rifampicina con 800/200mg de lopinavir/ritonavir BID resultó en una disminución de lopinavir hasta del 57% y con lopinavir/ritonavir 400/400mg BID hubo una disminución hasta del 7% cuando se comparó con lopinavir/ritonavir 400/100mg BID administrado en ausencia de rifampicina. Se han encontrado elevaciones de ALT y AST con dosis más altas de lopinavir/ritonavir coadministrados con rifampicina y esto puede ser dependiente de la secuencia en la administración de la dosis. Si se considera la coadministración , se debe iniciar KALETRA® a la dosis estándar por aproximadamente 10 días previos a la adición de rifampicina. La dosis de KALETRA® debe ser titulada de forma ascendente. Se debe monitorizar cuidadosamente la función hepática (Ver FARMACOLOGÍA CLÍNICA: Tabla 3 para la magnitud de la interacción). Antiparasitarios:

Atovaquona: Se produce disminución en la concentración de Atovaquona al coadministrarse con KALETRA®, puede ser necesario incrementar la dosis de atovaquona. Corticosteroides: Dexametasona: Induce la CYP3A4 y disminuye las concentraciones de lopinavir. Propionato de Fluticasona: El uso concomitante de propionato de fluticasona y KALETRA® puede incrementar las concentraciones del primero. Utilizar con precaución. Se deben considerar alternativas al propionato de fluticasona especialmente para uso a largo plazo. (Ver Precauciones y Advertencias: interacciones medicamentosas). Dihidropiridina: Los bloqueadores de canales de calcio como felodipina, nifedipina, nicardipina pueden incrementar sus concentraciones séricas al administrarse con KALETRA®. Disulfiram/Metronidazol: KALETRA® solución oral contiene alcohol, que puede producir reacciones tipo disulfiram cuando se coadministra con disulfiram u otro fármaco que produce esta reacción, como metronidazol. Agentes para disfunción eréctil (Inhibidores de PDE5): Sildenafil: Para el tratamiento de la disfunción eréctil se debe reducir la dosis a 25 mg cada 48 horas e incrementar el control de los efectos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) El uso concomitante de sildenafil y lopinavir/ritonavir está contraindicado en los pacientes con Hipertensión Arterial Pulmonar (HAP) (Ver CONTRAINDICACIONES) Tadalafil: Usar con precaución en dosis reducidas de no más de 10 mg cada 72 horas e incrementar el control de eventos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) Vardenafil: Usar con precaución en dosis reducidas de no más de 2,5 mg cada 72 horas e incrementar el control de eventos adversos. (Ver Precauciones y Advertencias: interacciones medicamentosas) Productos herbales: Hierba de San Juan (Hypericum perforatum): Los pacientes en tratamiento con KALETRA® no deben usar productos que la contengan porque reduce la concentración sérica de KALETRA®. El efecto es debido a inducción de CYP3A4 y puede causar la pérdida de su efecto terapéutico y la aparición de resistencia (Ver CONTRAINDICACIONES, ADVERTENCIAS Y PRECAUCIONES: INTERACCIONES MEDICAMENTOSAS). Inhibidores de la HMG-CoA reductasa: Los que son muy dependientes para su metabolismo de CYP3A4 como lovastatina y simvastatina tienen incrementos marcados en su concentración plasmática al coadministrase con KALETRA®. Esto causa miopatía, incluso rabdomiólisis. El uso de estos medicamentos con lopinavir/ritonavir está contraindicado (ver CONTRAINDICACIONES). La atorvastatina es menos dependiente de CYP3A para su metabolismo y al administrase con KALETRA® produce incremento de 4.7 y 5.9 veces en la Cmáx de atorvastatina y ABC, respectivamente. Cuando se use con KALETRA® se recomienda usar la menor dosis posible. Los estudios con pravastatina no han demostrado interacciones clínicas significativas. El metabolismo de pravastatina y fluvastatina no depende de CYP3A4 y no se esperan interacciones con KALETRA®. Si el tratamiento con inhibidores de la HMG-CoA reductasa está indicado se debe usar pravastatina o fluvastatina. (Ver Precauciones y Advertencias: Interacciones medicamentosas)

Inmunomoduladores: Las concentraciones de ciclosporina, tacrolimus y sirolimus (rapamicina) se incrementan al coadministrarse con KALETRA®. Se recomienda monitoreo frecuente de sus niveles hasta que se estabilicen. Metadona: KALETRA® disminuye las concentraciones plasmáticas de metadona, se recomienda control de niveles plasmáticos. Anticonceptivos orales y parches: KALETRA® disminuye las concentraciones de etinil-estradiol, por lo que se debe usar medidas anticonceptivas adicionales. No se esperan otras Interacciones clínicamente significativas: Los estudios de interacción farmacológica no revelan interacción clínicamente significativa entre desipramina (CYP2D6) omeprazol o ranitidina. Con base en los perfiles metabólicos conocidos, no es de esperar interacciones farmacológicas clínicamente significativas entre KALETRA® y fluvastatina, dapsona, trimetoprim / sulfametoxazol, azitromicina, o fluconazol en pacientes con función renal y hepática normal. EMBARAZO Y LACTANCIA Embarazo, fertilidad y reproducción Lopinavir en combinación con ritonavir a una relación de 2:1 no produjo efectos sobre la fertilidad en ratas macho y hembra con las dosis máximas obtenibles produciendo exposición medicamentosa comparable o ligeramente menor que las posibles con las dosis terapéuticas recomendadas 10/5, 30/15 ó 100/50 mg/Kg/día. Basados en mediciones de ABC la exposición en ratas a dosis altas, es aproximadamente 0.7 veces para lopinavir y 1.8 veces para ritonavir de la lograda al exponer a humanos a las dosis recomendadas (400/100 mg BID). No se han observado malformaciones asociadas al tratamiento con lopinavir/ritonavir en ratas o conejas preñadas. Se pueden observar desarrollo de toxicidad embrionaria y fetal en ratas (reabsorción temprana, disminución de la viabilidad fetal, disminución del peso corporal fetal, incremento en la incidencia de variaciones esqueléticas o retraso en la osificación esquelética) en ratas a las que se les administró una dosis maternalmente tóxica de 100/50 mg/kg/día. Si se compara el ABC la exposición alcanzada de estas drogas es aproximadamente 0,7 veces para lopinavir y 1,8 veces para ritonavir con la lograda al administrar en humanos (400/100 mg BID). Sin embargo, no existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios en animales no son siempre predictivos de la respuesta humana KALETRA® se debe administrar durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Madres en período de lactancia Por el potencial de transmisión de VIH y por los efectos adversos serios posibles, se debe instruir a las madres para no amamantar si reciben KALETRA®. Estudios con ratas han demostrado la presencia de lopinavir en la leche. No se sabe si lopinavir es excretado en la leche materna. REACCIONES ADVERSAS Adultos Eventos adversos emergentes con el tratamiento KALETRA® ha sido estudiada en 2,154 pacientes con VIH-1 con terapia de combinación en ensayos clínicos Fase I/II y Fase III. El evento adverso más común asociado a la terapia con KALETRA® fue diarrea, la cual generalmente fue de severidad leve a moderada. Las tasa de descontinuación de la terapia aleatoria debida a eventos adversos incluyendo muerte fue 5.8% en los pacientes tratados con KALETRA® y 4.9% en los que recibieron nelfinavir en el Estudio 863. La incidencia de diarrea fue mayor en el grupo que recibió KALETRA® cápsulas una vez al día, comparado con los que lo recibieron dos veces al día en el estudio 418. (Tabla 6)

Los eventos adversos clínicos relacionados con el fármaco de intensidad moderada o severa en ≥2% de los pacientes tratados con la terapia de combinación incluyendo KALETRA® hasta por 48 semanas (estudios 863, 418 y 730) y hasta por 360 semanas (estudio 720) se presentan en la Tabla 6 (pacientes sin experiencia a antiretrovirales) y hasta por 48 semanas (estudios 888 y 802), 84 semanas (estudio 957) y 144 semanas (estudio 765) en la Tabla 7 (pacientes con experiencia a antiretrovirales). Para más información de eventos adversos serios observados o potenciales ver advertencias y precauciones. Tabla 6: Porcentaje de pacientes con eventos adversos emergentes1 con el tratamiento de intensidad moderada o severa informados en ≥2% de los pacientes adultos no experimentados a antiretrovirales
Estudio 863 (48 Semanas)
Estudio 418 (48 Semanas)
Estudio 720 (360
Semanas)
Estudio 730 (48 Semanas)
Lop/Rit 400/100 mg BID + d4T+
3TC (N=326)
Nelfinavir 750 mg TID + d4T + 3TC
(N=327)
Lop/Rit Cap
800/200 mg QD + TDF+
FTC (N=115)
Lop/Rit Cap
400/100 mg BID + TDF+
FTC (N=75)
Lop/Rit BID(2) + d4T
+ 3TC (N=100)
Lop/Rit Tab
800/200 mg QD + TDF+
FTC (N=333)
Lop/Rit Tab
400/100 mg BID + TDF+
FTC (N=331)
Trastornos Gastrointestinales
Distensión abdominal
0.3% 0.6% 0.9% 0.0% 4.0% 0.3% 0.3%
Dolor Abdominal 4.0% 3.1% 2.6% 2.7% 11.0% 0.6% 0.9%
Heces anormales 0.0% 0.3% 0.0% 0.0% 8.0% 0.0% 0.0%
Diarrea 15.6% 17.1% 15.7% 5.3% 28.0% 16.5% 15.1% Dispepsia 2.1% 0.3% 0.0% 1.3% 6.0% 0.0% 0.0% Flatulencia 1.5% 1.2% 1.7% 1.3% 4.0% 0.9% 0.6% Náusea 6.7% 4.6% 8.7% 8.0% 16.0% 7.2% 5.4% Vómito 2.5% 2.4% 3.5% 4.0% 6.0% 3.3% 3.9% Trastornos generales y condiciones del sitio de administración Astenia 4.0% 3.4% 0.0% 0.0% 9.0% 0.3% 0.3% Dolor 0.6% 0.0% 0.0% 0.0% 3.0% 0.0% 0.0% Trastornos del sistema nervioso Cefalea 2.5% 1.8% 2.6% 2.7% 6.0% 1.5% 0.6% Parestesia 0.9% 0.9% 0.0% 0.0% 2.0% 0.0% 0.0% Trastornos psiquiátricos Insomnio 1.5% 1.2% 0.0% 0.0% 3.0% 1.2% 0.0% Libido disminuida 0.3% 0.3% 0.0% 1.3% 2.0% 0.0% 0.3%
Depresión 0.6% 1.5% 1.0% 0.0% 0.0% 0.0% 0.0% Trastornos vasculares Vasodilatación 0.0% 0.0% 0.0% 0.0% 3.0% 0.0% 0.0% Trastornos de la piel y del tejido subcutáneo Lipodistrofia adquirida 0.6% 0.6% 0.0% 0.0% 12.0% 0.0% 0.0%

Rash cutáneo 0.6% 1.5% 0.9% 0.0% 5.0% 0.3% 0.6% Trastornos músculo-esqueléticos y del tejido conectivo Mialgia 0.6% 0.9% 0.0% 0.0% 2.0% 0.0% 0.0% Infecciones e infestaciones Bronquitis 0.0% 0.0% 0.0% 0.0% 2.0% 0.0% 0.3% Trastornos endocrinos Hipogonadismo masculino 0.0% 0.0% 0.0% 0.0% 2.1% 0.0% 0.0%
Trastornos del metabolismo y de la nutrición Anorexia 0.9% 0.3% 0.9% 1.3% 2.0% 0.3% 0.9% Investigación Pérdida de peso 0.6% 0.3% 0.0% 0.0% 2.0%
0.0% 0.3% Trastornos del sistema reproductivo y del tejido mamario Amenorrea 0.0% 0.0% 4.5% 0.0% 0.0% 0.0% 0.0%
(1) Incluye los eventos adversos, con relación posible o probable con el fármaco en estudio (2) Incluye los datos de los eventos adversos de los pacientes del grupo I que recibieron 200/100
mg BID (N=16) y 400/100 mg BID solo (N=16) y los pacientes del grupo II que recibieron 400/100 mg BID (N=35) y 400/200 mg BID (N=33). En ambos grupos, se presento náusea moderada a severa probablemente/posiblemente relacionada con Lopinavir/Ritonavir en una tasa más alta en el brazo con dosis 400/200 mg comparado con el brazo 400/100 mg en el grupo II.
Definiciones: Lop/Rit = Lopinavir/Ritonavir; d4T = estavudina; 3TC = Lamivudina; TDF = Tenofovir; FTC = Emtricitabina.
Tabla 7: Porcentaje de pacientes con eventos adversos emergentes1 con el tratamiento de intensidad moderada o severa informados en ≥2% de los pacientes adultos con experiencia previa a antiretrovirales
Estudio 888 (48 Semanas)
Estudio 957(2) y Estudio 765(3)
(84-144 Semanas)
Estudio 802 (48 Semanas)
Lop/Rit 400/100 mg
BID + NVP + INTR
(N=148)
Inhibidor(es) de la proteasa, seleccionado
por el investigador + NVP + INTR
(N=140)
Lop/Rit BID + INNTR + INTR
(N=127)
Lop/Rit 800/200 mg Una vez al día + INTR
(N=300)
Lop/Rit 400/100 mg
BID + INTR (N=299)
Trastornos Gastrointestinales Dolor Abdominal
2.0% 2.1% 3.9% 2.0% 0.3%
Dolor abdominal superior NA NA NA 0.7% 2.0%
Heces anormales 0.0% 0.0% 2.4% 0% 0% Diarrea 7.4% 9.3% 22.8% 14.0% 11.0% Disfagia 2.0% 0.7% 0.0% 0% 0% Flatulencia 0.7% 2.1% 1.6% 1.0% 1.0%
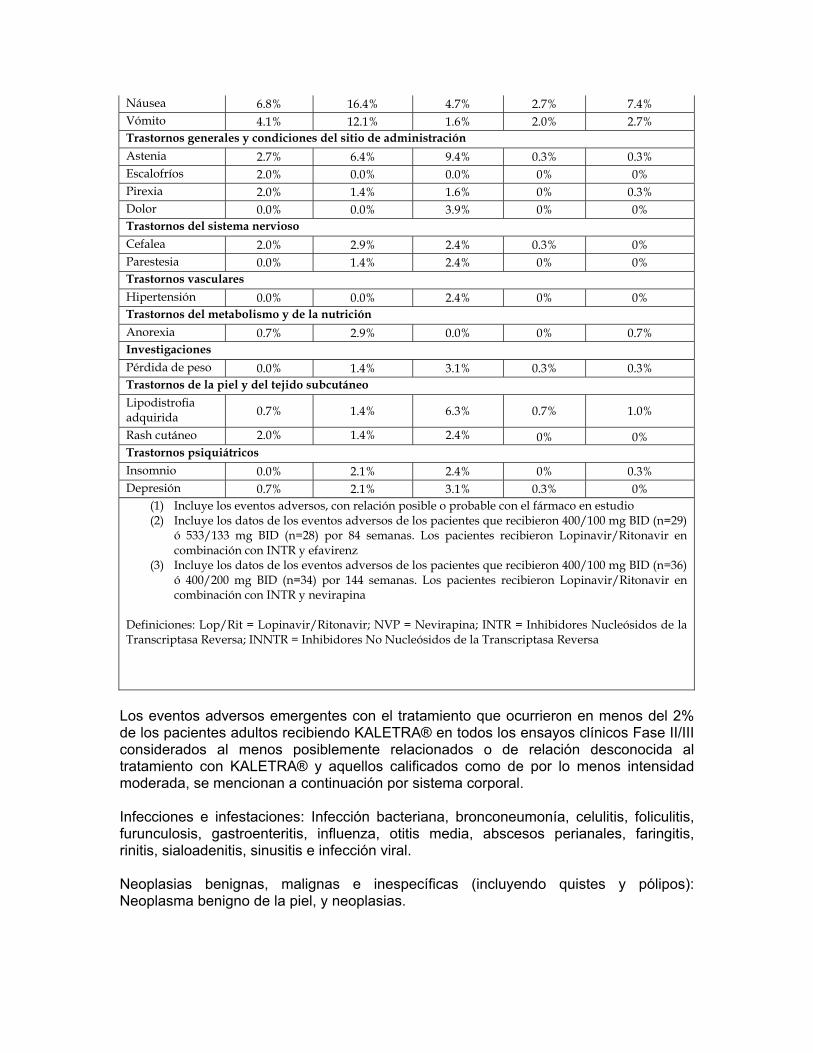
Náusea 6.8% 16.4% 4.7% 2.7% 7.4% Vómito 4.1% 12.1% 1.6% 2.0% 2.7% Trastornos generales y condiciones del sitio de administración Astenia 2.7% 6.4% 9.4% 0.3% 0.3% Escalofríos 2.0% 0.0% 0.0% 0% 0% Pirexia 2.0% 1.4% 1.6% 0% 0.3% Dolor 0.0% 0.0% 3.9% 0% 0% Trastornos del sistema nervioso Cefalea 2.0% 2.9% 2.4% 0.3% 0% Parestesia 0.0% 1.4% 2.4% 0% 0% Trastornos vasculares Hipertensión 0.0% 0.0% 2.4% 0% 0% Trastornos del metabolismo y de la nutrición Anorexia 0.7% 2.9% 0.0% 0% 0.7% Investigaciones Pérdida de peso 0.0% 1.4% 3.1% 0.3% 0.3% Trastornos de la piel y del tejido subcutáneo Lipodistrofia adquirida 0.7% 1.4% 6.3% 0.7% 1.0%
Rash cutáneo 2.0% 1.4% 2.4% 0% 0% Trastornos psiquiátricos Insomnio 0.0% 2.1% 2.4% 0% 0.3% Depresión 0.7% 2.1% 3.1% 0.3% 0%
(1) Incluye los eventos adversos, con relación posible o probable con el fármaco en estudio (2) Incluye los datos de los eventos adversos de los pacientes que recibieron 400/100 mg BID (n=29)
ó 533/133 mg BID (n=28) por 84 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y efavirenz
(3) Incluye los datos de los eventos adversos de los pacientes que recibieron 400/100 mg BID (n=36) ó 400/200 mg BID (n=34) por 144 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y nevirapina
Definiciones: Lop/Rit = Lopinavir/Ritonavir; NVP = Nevirapina; INTR = Inhibidores Nucleósidos de la Transcriptasa Reversa; INNTR = Inhibidores No Nucleósidos de la Transcriptasa Reversa
Los eventos adversos emergentes con el tratamiento que ocurrieron en menos del 2% de los pacientes adultos recibiendo KALETRA® en todos los ensayos clínicos Fase II/III considerados al menos posiblemente relacionados o de relación desconocida al tratamiento con KALETRA® y aquellos calificados como de por lo menos intensidad moderada, se mencionan a continuación por sistema corporal. Infecciones e infestaciones: Infección bacteriana, bronconeumonía, celulitis, foliculitis, furunculosis, gastroenteritis, influenza, otitis media, abscesos perianales, faringitis, rinitis, sialoadenitis, sinusitis e infección viral. Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos): Neoplasma benigno de la piel, y neoplasias.

Alteraciones del sistema sanguíneo y linfático: Anemia, leucopenia y linfadenopatía, neutropenia y esplenomegalia. Alteraciones del sistema inmunológico: Hipersensibilidad al fármaco, hipersensibilidad, síndrome de reconstitución inmune. Alteraciones del Sistema endocrino: Síndrome de Cushing e hipotiroidismo. Alteraciones metabólicas y nutricionales: Disminución del apetito, deshidratación, diabetes mellitus, hiperamilasemia, hiperlipasemia, hipovitaminosis, aumento de apetito, acidosis láctica, lipomatosis, obesidad. Alteraciones Psiquiátricas: Sueños anormales, labilidad afectiva, agitación, ansiedad, apatía, estado de confusión, desorientación, cambios de humor, nerviosismo y pensamiento anormal. Alteraciones del sistema nervioso: pérdida del sentido del gusto, amnesia, desorden del balance, ataxia, infarto cerebral, convulsión, vértigo, alteración del gusto, discinesia, encefalopatía, síndrome extrapiramidal, parálisis facial, hipertonía, migraña, neuropatía, neuritis periférica, somnolencia y tremor. Desórdenes oculares: Visión anormal, alteración ocular. Alteraciones del oído y laberinto: Hiperacusia, tinnitus y vértigo. Desórdenes Cardíacos: Angina de pecho, fibrilación atrial, bloqueo atrioventricular, infarto miocardio y palpitaciones, incompetencia de la válvula tricúspide. Alteraciones vasculares: Trombosis de las venas profundas, hipotensión ortostática, tromboflebitis, venas varicosas y vasculitis. Alteraciones respiratorias, torácicas y del mediastino: asma, tos, disnea, edema pulmonar. Alteraciones gastrointestinales: Malestar abdominal, dolor abdominal inferior, constipación, boca seca, duodenitis, enteritis, enterocolitis, enterocolitis hemorrágica, eructos, esofagitis, incontinencia fecal, gastritis, gastritis ulcerativa, enfermedad de reflujo gastroesofágico, hemorroides, ulceración en la boca, pancreatitis, periodontitis, hemorragia rectal y estomatitis. Alteraciones hepatobiliares: Colangitis, colecistitis, esteatosis hepática, hepatitis, hepatomegalia, ictericia y sensibilidad hepática. Alteraciones de la piel y del tejido subcutáneo: Acné, alopecia, dermatitis alérgica, dermatitis exfoliativa, piel seca, eczema, hiperhidrosis, capilaritis idiopática, alteración de las uñas, prurito, erupción maculopapular, seborrea, decoloración de la piel, hipertrofia de la piel, estrías en la piel, úlceras en piel e hinchazón facial. Alteraciones del Sistema músculo esquelético y conectivo: Artralgia, artropatía, dolor de espalda, debilidad muscular, osteoartritis, osteonecrosis, dolor en las extremidades.

Alteraciones del Sistema Urinario y renal: Hematuria, nefritis, nefrolitiasis, anormalidades urinarias y olor anormal de la orina. Alteraciones del sistema reproductivo y pecho: Agrandamiento de los senos, alteración en la eyaculación, disfunción eréctil, ginecomastia y menorragia. Alteraciones Generales y de las condiciones del sitio de administración: Dolor torácico, quistes, interacción de medicamento, edema, edema periférico, edema facial, hipertrofia y malestar. Investigaciones: Incremento en el nivel del medicamento, disminución de la tolerancia a la glucosa e incremento del peso. Anormalidades de laboratorio: los porcentajes de pacientes adultos tratados con terapia de combinación incluyendo Kaletra® con anormalidades de laboratorio Grado 3-4 se presentan en la Tabla 8 y 9 Tabla 8: Anormalidades de laboratorio Grado 3 a 4 reportadas en ≥2% de los pacientes adultos no experimentados con antirretrovirales.
Estudio 863
(48 Semanas) Estudio 418
(48 Semanas)
Estudio 720 (360
Semanas)
Estudio 730 (48 Semanas)
Variable Límite(1)
Lop/Rit 400/100 mg BID
+ d4T+3TC (N=326)
Nelfinavir 750 mg
TID + d4T + 3TC
(N=327)
Lop/Rit 800/200 mg QD + TDF + FTC
(N=115)
Lop/Rit 400/100 mg BID + TDF + FTC (N=75)
Lop/Rit BID + d4T
+ 3TC (N=100)
Lop/Rit QD + TDF+ FTC
(N=333)
Lop/Rit BID + TDF+ FTC
(N=331)
Química Alto Glucosa >250
mg/dL 2% 2% 3% 1% 4% 0% <1%
Acido úrico >12 mg/dL
2% 2% 0% 3% 5% <1% 1%
SGOT/AST2 >180 U/L
2% 4% 5% 3% 10% 1% 2%
SGPT/ALT2 >215 U/L
4% 4% 4% 3% 11% 1% 1%
GGT >300 U/L
N/A N/A N/A N/A 10% NA NA
Colesterol total
>300 mg/dL 9% 5% 3% 3% 27% 4% 3%
Triglicéridos >750 mg/dL
9% 1% 5% 4% 29% 3% 6%
Amilasa >2x LSN
3% 2% 7% 5% 4% NA NA
Lipasa >2x LSN
NA NA NA NA NA 3% 5%
Química Bajo
Aclaramiento de creatinina calculado
<50 mL/min
NA NA NA NA NA 2% 2%
Hematología Bajo
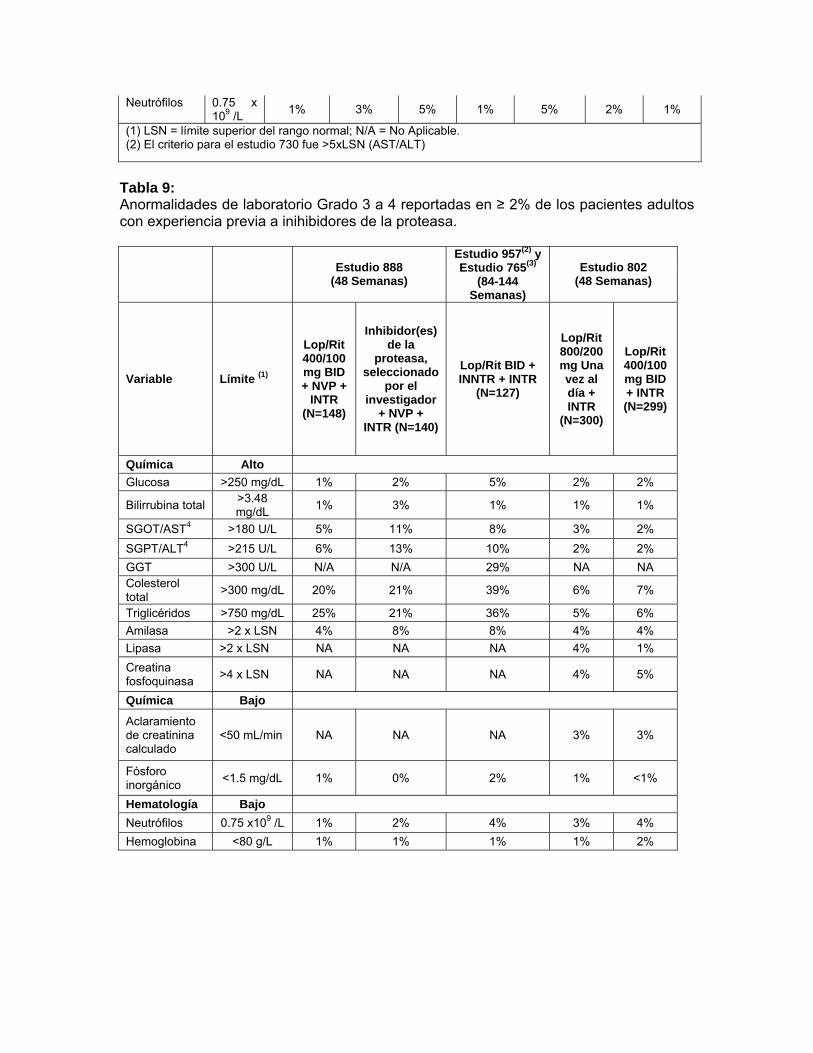
Neutrófilos 0.75 x 109 /L
1% 3% 5% 1% 5% 2% 1%
(1) LSN = límite superior del rango normal; N/A = No Aplicable. (2) El criterio para el estudio 730 fue >5xLSN (AST/ALT)
Tabla 9: Anormalidades de laboratorio Grado 3 a 4 reportadas en ≥ 2% de los pacientes adultos con experiencia previa a inihibidores de la proteasa.
Estudio 888
(48 Semanas)
Estudio 957(2) y Estudio 765(3)
(84-144 Semanas)
Estudio 802 (48 Semanas)
Variable Límite (1)
Lop/Rit 400/100 mg BID + NVP +
INTR (N=148)
Inhibidor(es) de la
proteasa, seleccionado
por el investigador
+ NVP + INTR (N=140)
Lop/Rit BID + INNTR + INTR
(N=127)
Lop/Rit 800/200 mg Una vez al día + INTR
(N=300)
Lop/Rit 400/100 mg BID + INTR (N=299)
Química Alto
Glucosa >250 mg/dL 1% 2% 5% 2% 2%
Bilirrubina total >3.48 mg/dL
1% 3% 1% 1% 1%
SGOT/AST4 >180 U/L 5% 11% 8% 3% 2%
SGPT/ALT4 >215 U/L 6% 13% 10% 2% 2%
GGT >300 U/L N/A N/A 29% NA NA Colesterol total
>300 mg/dL 20% 21% 39% 6% 7%
Triglicéridos >750 mg/dL 25% 21% 36% 5% 6%
Amilasa >2 x LSN 4% 8% 8% 4% 4%
Lipasa >2 x LSN NA NA NA 4% 1%
Creatina fosfoquinasa
>4 x LSN NA NA NA 4% 5%
Química Bajo
Aclaramiento de creatinina calculado
<50 mL/min NA NA NA 3% 3%
Fósforo inorgánico
<1.5 mg/dL 1% 0% 2% 1% <1%
Hematología Bajo
Neutrófilos 0.75 x109 /L 1% 2% 4% 3% 4%
Hemoglobina <80 g/L 1% 1% 1% 1% 2%

(1) LSN = límite superior del rango normal; N/A = No Aplicable. (2) Incluye los datos de laboratorio clínico de los pacientes que recibieron 400/100 mg BID (n=29) ó 533/133 mg BID (n=28) por 84 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y efavirenz. (3) Incluye los datos de laboratorio clínico de los pacientes que recibieron 400/100 mg BID (n=36) ó 400/200 mg BID (n=34) por 144 semanas. Los pacientes recibieron Lopinavir/Ritonavir en combinación con INTR y nevirapina. (4) El criterio para el estudio 802 fue >5xLSN (AST/ALT)
Pediátricos Eventos Adversos Emergentes con el Tratamiento KALETRA® fue estudiado en 100 pacientes pediátricos de 6 meses a 12 años de edad. El perfil de efectos adversos observados en estos estudios clínicos es similar al observado en adultos. Disgeusia, vómitos y diarrea fueron los eventos adversos más comúnmente reportados de cualquier severidad en pacientes pediátricos en tratamiento con una terapia combinada incluyendo Kaletra® hasta por 48 semanas en el estudio 940. Un total de 8 niños experimentaron eventos adversos moderados o severos al menos posiblemente relacionados con Kaletra®. Rash (reportado en 3%) fue el único evento adverso asociado a la droga de intensidad moderada a severa observado en mayor o igual al 2% de los niños inscritos. Anormalidades de Laboratorio Los porcentajes de los pacientes pediátricos tratados con combinaciones que incluían KALETRA® y tuvieron anormalidades de laboratorio grado 3 y 4 se presentan en la tabla 10. Tabla 10: Anormalidades de Laboratorio Grado 3-4 informadas en ≥ 2% de los pacientes pediátricos
Variable Límite (1) LOP/RIT BID +ITR (N=100) Química Alto
Sodio > 149 mEq/L 3,0% Bilirrubina total > 2,9 x LSN 3,0% SGOT/AST > 180 U/L 8,0% SGPT/ALT > 215 U/L 7,0% Colesterol total >300 mg/dL ó
> 7,77 mmol/L 3,0%
Amilasa >2,5 x LSN 7,0% (2) Química Bajo
Sodio < 130 mEq/L 3,0% Hematología Bajo
Recuento de plaquetas < 50 x 109/L 4,0% Neutrófilos < 0,40 x 109/L 2,0%
(1) LSN = límite superior del rango normal (2) Individuos con amilasa grado 3 a 4 confirmada con las elevaciones de la amilasa pancreática
Experiencia post-mercadeo Se ha reportado hepatitis en los pacientes que tienen terapia con KALETRA®. Se ha reportado necrólisis epidérmica tóxica, el síndrome de Stevens Johnson y eritema multiforme.

SOBREDOSIS La experiencia en humanos de la sobredosis aguda con KALETRA® es limitada. El tratamiento de la sobredosis con KALETRA® consiste en medidas de apoyo general incluyendo monitoreo de los signos vitales y observación del estado clínico del paciente. No existe antídoto específico para la sobredosis con KALETRA®. Si está indicada, la eliminación del fármaco no absorbido debe ser lograda por emesis o lavado gástrico. La administración de carbón activado también puede ser utilizada para ayudar a retirar el fármaco no absorbido. Ya que KALETRA® se une altamente a las proteínas, es improbable que la diálisis sea benéfica en la remoción significativa del fármaco. KALETRA® solución oral contiene 42.4% de alcohol (v/v). La ingestión accidental del producto por un niño de corta edad podría resultar en toxicidad significativa relacionada con alcohol. DOSIS Y ADMINISTRACION Adultos Las cápsulas de gelatina blanda y la solución oral de KALETRA® deben ser tomadas con alimentos. La dosis diaria de KALETRA® es: KALETRA® 400/100 mg (3 cápsulas ó 5 mL) dos veces al día con las comidas, KALETRA® 800/200 mg (6 cápsulas ó 10 mL) una vez al día con la comida, en pacientes con menos de tres mutaciones asociados a lopinavir. No hay datos suficientes para respaldar el uso de una administración una vez al día de Kaletra® para pacientes adultos con tres o más mutaciones asociados al lopinavir. Kaletra® no debe ser administrado una vez al día en combinación con carbamazepina, fenobarbital o fenitoína (ver Interacciones Medicamentosas). Terapia concomitante: Omeprazol y Ranitidina: Las cápsulas de KALETRA® y la solución oral pueden ser usadas en combinación con agentes reductores de ácido (omeprazol y ranitidina) sin ajuste de dosis (ver tabla 3). Efavirenz, Nevirapina, Amprenavir o Nelfinavir Se debe incrementar la dosis de KALETRA® a 533/133 mg (4 cápsulas ó 6.5 mL de solución oral) tomadas dos veces al día con alimentos si se toma en combinación con efavirenz o nevirapina, amprenavir, o nelfinavir en pacientes experimentados porque se sospecha reducción en la susceptibilidad a lopinavir (por historia y laboratorio) (véase FARMACOLOGÍA CLINICA: Interacciones fármaco-fármaco y/o Interacciones medicamentosas). La administración de KALETRA® una vez al día no debe hacerse en combinación con efavirenz, nevirapina, amprenavir o nelfinavir. Pacientes pediátricos Tomar atención especial para realizar el cálculo exacto de la dosis pediátrica. En niños de seis meses a doce años de edad, la dosis recomendada de KALETRA® solución oral es 12/3 mg/Kg para los que pesan entre 7 y menos de 15 Kg y 10/2.5 mg/Kg para los que pesan entre 15 y 40 Kg (aproximadamente 230/57.5 mg/m2); administrada dos veces al día con las comidas, hasta una dosis máxima de 400/100 mg en niños de más de 40 Kg (3 cápsulas ó 5.0 mL) dos veces al día. KALETRA® una vez al día no ha sido evaluado en pacientes pediátricos. Es preferible que se calcule la dosis apropiada en mg para los niños menores o igual a doce años y determinar el correspondiente volumen de solución o cantidad de cápsulas de gelatina

blanda Como alternativa se puede usar la siguiente tabla que contiene las guías de dosificación de acuerdo al peso corporal. Guía de dosificación pediátrica basada en el peso sin efavirenz, nevirapina, nelfinavir o amprenavir concomitante. Peso (Kg) Dosis (mg/Kg)* Volumen de solución oral dos
veces al día (80 mg de lopinavir / 20 mg de ritonavir por mL)
7 a menos de 15 Kg 12 mg/Kg dos veces al día
7 a 10 Kg 1.25 mL Más de 10 a menos de 15 Kg
1.75 mL
15 a 40 Kg 10 mg/Kg dos veces al día
15 a 20 Kg 2.25 mL Más de 20 a 25 Kg 2.75 mL Más de 25 a 30 Kg 3.50 mL Más de 30 a 35 Kg 4.00 mL Más de 35 a 40 Kg 4.75 mL
Más de 40 Kg Dosis de adultos 5.0 mL (ó tres cápsulas)
* Dosificación basada en el componente lopinavir de la solución KALETRA (80 mg/20 mg por mL). Nota: Emplear la recomendación posológica para adultos, en niños mayores de 12 años. Terapia concomitante: Efavirenz, Nevirapina, amprenavir o nelfinavir Un incremento de la dosis de KALETRA® solución oral a 13/3.25 mg/Kg para niños que pesan entre 7 y menos de 15 Kg y 11/2.75 mg/Kg para los que pesan entre 15 y 45 Kg (aproximadamente 300/75 mg/m2) dos veces al día, tomado con alimentos; hasta un máximo de 533/133 mg dos veces al día en niños de más de 45 Kg se debe considerar cuando es utilizado en combinación con efavirenz, nevirapina, amprenavir o nelfinavir; en niños de 6 meses a 12 años habitualmente tratados en los que clínicamente se sospeche susceptibilidad reducida al lopinavir (por antecedentes terapéuticos o evidencia de laboratorio). La siguiente tabla contiene las guías de dosificación de KALETRA® solución oral basadas en el peso corporal cuando se combina con nevirapina o efavirenz, amprenavir o nelfinavir. (Ver FARMACOLOGIA CLINICA: Interacciones fármaco-fármaco y/o INTERACCIONES MEDICAMENTOSAS) Guía de dosificación pediátrica basada en el peso y administración concomitante con efavirenz, nevirapina, amprenavir o nelfinavir Peso (Kg) Dosis (mg/Kg)* Volumen de solución oral dos
veces al día (80 mg de lopinavir / 20 mg de ritonavir por mL)
7 a menos de 15 Kg 13 mg/Kg dos

veces al día 7 a 10 Kg 1.50 mL Más de 10 a menos de 15 Kg
2.00 mL
15 a 45 Kg 11 mg/Kg dos veces al día
15 a 20 Kg 2.50 mL Más de 20 a 25 Kg 3.25 mL Más de 25 a 30 Kg 4.00 mL Más de 30 a 35 Kg 4.50 mL Más de 35 a 40 Kg 5.00 mL (o tres cápsulas) Más de 40 a 45 Kg
5.75 mL
Más de 45 Kg Dosis de adulto 6.5 mL (o cuatro cápsulas)
* Dosificación basada en el componente lopinavir de la solución KALETRA (80 mg/20 mg por mL). Nota: Emplear la recomendación posológica para adultos, en niños mayores de 12 años. Guía de dosificación basada en el área de superficie corporal (ASC) (m2) Solución oral: uso pediátrico (niños de 6 meses y mayores): Para la solución oral de KALETRA® se recomienda 230/57.5 mg/m2 dos veces al día, con los alimentos, hasta un máximo de 400/100 mg dos veces al día. La dosis de 230/57.5 mg/m2 puede ser insuficiente en algunos niños cuando se coadministra con nevirapina, nelfinavir, amprenavir o efavirenz. Se debe considerar un incremento de la dosis de KALETRA® a 300/75 mg/m2 en estos pacientes. La dosis se debe administrar con una jeringa dosificadora calibrada para uso oral. Guía de dosificación pediátrica de KALETRA® solución oral basado en ASC sin efavirenz, nevirapina, nelfinavir o amprenavir concomitante
Área de superficie corporal (m2)* Dosis dos veces al día (230/57.5 mg/m2)
0.25 0.7 mL (57.5/14.4 mg) 0.50 1.4 mL (115/28.8 mg) 0.75 2.2 mL (72.5/43.1 mg) 1.00 2.9 mL (230/57.5 mg) 1.25 3.6 mL (287.5/71.9 mg) 1.50 4.3 mL (345/86.3 mg) 1.75 5 mL (402.5/100.6 mg)
Cápsulas de gelatina blanda: uso pediátrico (niños de 6 meses y mayores):

Si se usan cápsulas se debe tener en cuenta que si un niño tiene 1.3 m2 de área de superficie corporal o más, la dosis es de 3 cápsulas cada 12 horas tomadas con alimentos. Para los niños que tienen menos de 1.3 m2 de área de superficie corporal se les recomienda KALETRA® solución oral. ALMACENAMIENTO: Cápsulas de gelatina blanda de KALETRA Almacenar a 2°C a 8°C (36°F a 46°F) hasta cuando se entregue el medicamento. Evite la exposición a calor excesivo. No se requiere que las cápsulas de gelatina blanda de Kaletra se refrigeren si los pacientes las toman antes de 42* días y se mantiene a menos de 25°C (77°F). La Solución Oral de Kaletra se debe mantener refrigerada entre 2° - 8°C (36°F - 46° F ) antes de su dispensación. Evitar la exposición del producto al calor excesivo. No se requiere que el paciente guarde el producto en refrigerador si se va a consumir dentro de los 2 meses y se mantiene a temperaturas inferiores a 25°C. Si el producto se mantiene refrigerado, es estable hasta su fecha de vencimiento. MANTENER FUERA DEL ALCANCE DE LOS NIÑOS PRESENTACIÓN Las cápsulas de KALETRA® (lopinavir/ritonavir) son de gelatina blanda, son color naranja. Contienen 133.3 mg de lopinavir/33.3 mg ritonavir. La solución oral de KALETRA® (lopinavir/ritonavir) es un líquido de color ligeramente amarillento a naranja que se suministra en frascos de color ámbar de dosis múltiples; contienen 400 mg de lopinavir/100 mg de ritonavir por cada 5 mL (80 mg de lopinavir/20 mg de ritonavir por mL) empacado con una copa de dosificación marcada. Cápsulas Blandas: Fabricado por Catalent Pharma Solutions, LLC 27-25 Scherer Drive , St Peterburg, FL 33716-1016 U.S.A. Para Abbott Laboratories, North Chicago, IL 60064, U.S.A. Empacado por Abbott Laboratories, Abbott Park, IL 60064 U.S.A. Solución Oral Hecho en EUA por: Abbott Laboratories, Abbott Park, IL 60064-3500 Abbott Laboratorios, S.A. 5ta. Avenida 5-55, Zona 14 Europlaza, Torre 1, Piso 10 Guatemala, Guatemala C.A. Venta bajo receta médica. Vía de administración oral. Manténgase fuera del alcance de los niños. CCDS No. 03081209


















