
JHS 502 地盤の平板載荷試験 記録用紙(試験様式502-4 平板載荷試験(logP-logS曲線) 報告用紙 調査件名 調査場所 試験年月日 試験位置 地区土質調査

医薬品の生物学的同等性試験 表1:182×257 背:14×257 表4:182×257
医薬品の生物学的同等性試験
―
ガイドライン対応―緒方 宏泰
医薬品の生物学的同等性試験
緒方 宏泰[編著]
﹇編著﹈
-ガイドライン対応-
9784840744249
1923047100009
ISBN978-4-8407-4424-9 C3047 ¥10000E

医薬品の生物学的同等性試験
緒方 宏泰[編著]
-ガイドライン対応-

目 次
第 1章 生物学的同等性試験概説
Ⅰ 生物学的同等性試験の目的㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀1
1 治療学的同等性と生物学的同等性試験との関係からの考察 3
(1)後発医薬品を評価する 2つのポイント 3
(2)治療学的同等性を評価する方法 5
2 わが国における生物学的同等性試験とその関連事項の歴史的経過 8
Ⅱ バイオアベイラビリティパラメータの算出㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀14
1 全身適用医薬品の場合 14
(1)血中薬物濃度を決定している因子 14
(2)バイオアベイラビリティの算出法 15
2 局所皮膚適用医薬品の場合 24
(1)製剤中薬物量の低下がわずかで,薬物放出速度が一定の場合 24
(2)製剤中薬物量の低下が無視できず,薬物放出速度が変化する場合 25
Ⅲ 薬力学的指標を用いたパラメータの算出㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀26
Ⅳ 生物学的同等性試験法をつくり上げるための基礎検討㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀27
1 わが国の製剤に認められた生物学的同等性の課題 28
2 バイオアベイラビリティに影響を与えるヒトの生理的要因 35
(1)胃液酸性度 35
(2)食事 37
Ⅴ 生物学的同等性試験の概要と課題㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀39
1 ヒト試験,動物試験 39
2 ヒトを対象とした試験の免除 40
3 対照製剤の定義 40
4 標準製剤の選択 40
5 基準処方 41
6 試験製剤の選択 42
7 試験計画(プロトコール) 42

8 被験者の選択 44
(1)健康成人志願者 44
(2)患者あるいは低胃酸の被験者 45
(3)局所皮膚適用製剤における被験者 45
9 投与条件 46
(1)投与量 46
(2)投与法 47
(3)単回投与試験 47
(4)多回投与試験 47
(5)絶食投与,食後投与 48
(6)服用時の水 48
10 測定対象 49
11 分析法 50
12 予試験 50
13 生物学的同等性の評価 50
(1)評価パラメータ 50
(2)生物学的同等性の許容域 51
(3)統計学的解析 53
(4)追加試験 55
(5)体内動態において高い個体内変動性を有する薬物 55
14 in vitro試験の利用 56
(1)溶出試験データの考え方と利用 56
(2)放出試験,透過試験データの考え方と利用 64
(3)医薬品の試験法としての溶出試験規格 65
第 2章 生物学的同等性試験
Ⅰ 医薬品の製造販売承認における生物学的同等性試験㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀71
1 医薬品の製造販売承認に係るこれまでの経緯 71
2 医療用医薬品の製造販売承認申請に際して添付すべき資料について 73
(1)医療用医薬品の分類 73
(2)医療用医薬品の添付資料 76
3 後発医薬品と生物学的同等性 77
(1)後発医薬品の製造販売承認申請 77
vi 目 次

(2)後発医薬品の添付文書と生物学的同等性試験 78
4 生物学的同等性試験を利用した医薬品製造販売承認申請 81
(1)後発医薬品の新規承認申請 81
(2)既承認製剤または開発段階の製剤の処方変更 82
(3)含量違い製剤の新規承認申請 84
Ⅱ 生物学的同等性試験について㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀85
1 生物学的同等性試験の種類 85
(1)経口固形製剤 85
(2)局所皮膚適用製剤 87
(3)その他の製剤 88
2 経口固形製剤における生物学的同等性試験
(バイオアベイラビリティ比較試験)の試験基準 89
(1)試験法 89
(2)評価法 89
3 局所皮膚適用製剤における皮膚薬物動態学的試験の試験基準 90
(1)試験法 90
(2)評価法 91
Ⅲ 生物学的同等性試験ガイドラインとその改正㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀93
1 生物学的同等性試験ガイドラインとその改正 94
(1)用語の改正 97
(2)標準製剤の選定における改正点 97
(3)生物学的同等性試験の評価法 98
(4)溶出試験の試験条件 99
(5)溶出挙動の類似性の判定 103
2 経口固形製剤の含量違い,処方変更および剤形追加に係る
生物学的同等性試験ガイドラインとその変更点 108
(1)「含量が異なる経口固形製剤の生物学的同等性試験ガイドライン」
における変更点 111
(2)経口固形製剤の処方変更の生物学的同等性試験ガイドライン
に特有な改正点 114
(3)剤形が異なる製剤の追加のための生物学的同等性試験ガイドライン 116
Ⅳ 諸外国の生物学的同等性試験ガイドラインの動き㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀118
1 米国食品医薬品局(FDA)の生物学的同等性試験に係る
一連のガイドライン 118
目 次 vii

(1)経口固形製剤のバイオアベイラビリティや生物学的同等性
の試験ガイダンス 118
(2)BCS にもとづいた経口固形製剤のバイオアベイラビリティや
生物学的同等性試験の免除に関するガイドライン 121
(3)SUPAC-IRおよび SUPAC-MR 123
2 欧州医薬品庁(EMA)の生物学的同等性関連ガイドライン 126
(1)導入部分 127
(2)ヒト生物学的同等性試験 127
(3)含量違い製剤 128
(4)治療濃度域の狭い医薬品の取り扱い 129
(5)ばらつきの大きい医薬品,医薬品製剤に関する取り扱い 129
(6)本文中の溶出試験に関する重要な記載 130
(7)変更管理 130
3 即放性製剤における 3極の生物学的同等性に係る
ガイドラインの比較 133
Ⅴ 他極における種々の製剤の後発医薬品のためのガイドライン㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀136
1 米国食品医薬品局(FDA)の各種製剤に対するガイドライン 136
2 欧州医薬品庁(EMA)のガイドライン 137
(1)口腔内崩壊錠(ODT) 137
(2)経口液剤 138
(3)注射剤 138
(4)静脈注射用リポソーム,ミセル,エマルション製剤 138
(5)ミセル形成製剤 139
(6)局所適用製剤 139
3 リポソーム製剤に関するガイドライン 139
(1)FDAドラフトガイダンス 139
(2)ドキシルの後発医薬品のためのガイドライン 141
(3)EMAのリポソーム製剤に関するリフレクションペーパー 143
Ⅵ 局所皮膚適用製剤の生物学的同等性試験ガイドライン㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀145
(1)局所皮膚ガイドラインにおける後発医薬品の定義および剤形の考え方 145
(2)局所皮膚適用製剤におけるバイオアベイラビリティの考え方 146
(3)製剤の物理化学的特性の影響 146
(4)生物学的同等性の許容域 147
(5)局所皮膚適用製剤の生物学的同等性試験 148
viii 目 次

(6)曝露量試験 152
(7)剤形追加における生物学的同等性試験 153
(8)処方変更のための生物学的同等性試験 153
Ⅶ 医療用配合剤の生物学的同等性試験の考え方㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀162
1 医療用配合剤 162
(1)医療用配合剤の分類 162
(2)医療用配合剤の承認申請に添付すべき資料 163
(3)医療用配合剤の承認状況 164
2 医療用配合剤の生物学的同等性試験に関する Q & Aについて 166
(1)検討の経緯 166
(2)医療用配合剤の後発医薬品の生物学的同等性試験 167
(3)含量が異なる医療用配合剤及び医療用配合剤の処方変更
の生物学的同等性試験 170
第 3章 実 施 方 法
Ⅰ ヒトを対象とする生物学的同等性試験の実際㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀179
1 試験に影響を与えるヒトの生物学的要因
∼プロトコールを組むにあたっての留意点∼ 179
(1)被験者の選抜 179
(2)試験の実際 181
(3)実施上の信頼性の配慮∼GCP 上の留意点∼ 185
2 OD錠の生物学的同等性試験の実施にあたっての留意点 187
(1)試験デザイン 188
(2)投与時の飲水制限 189
(3)投与方法 189
3 局所皮膚適用製剤の生物学的同等性試験の実施にあたっての留意点 189
Ⅱ 生物学的同等性試験の統計解析㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀191
1 生物学的同等の許容域 192
(1)評価パラメータ 192
(2)生物学的同等の許容域 192
2 一般的な解析手順 193
(1)実験計画 193
(2)測定値の変換 194
目 次 ix

(3)対数変換を行う場合の解析 194
(4)対数変換を行わない場合の解析 199
(5)計算例 199
3 例数設計 203
4 欠測値,外れ値 207
5 その他の統計解析および実験計画 207
Ⅲ 生物学的同等性試験と溶出試験㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀209
1 溶出試験 209
(1)溶出試験 209
(2)生物学的同等性試験における溶出試験の意義 210
(3)後発医薬品ガイドラインの溶出試験条件 211
(4)溶出試験実施における留意点 214
(5)含量違い製剤,処方変更製剤のガイドラインの溶出試験条件 216
(6)溶出挙動の評価 216
2 生物学的同等性試験における溶出試験の役割 221
(1)標準製剤の選定 221
(2)即放性製剤において健康成人でヒト試験できる試験製剤の要件 222
(3)徐放性製剤においてヒト試験するための試験製剤の要件 224
(4)生物学的同等性評価のための補強データ(第 2 の判定基準) 225
(5)含量違い,処方変更ガイドラインでのヒト試験免除 227
(6)今後の溶出試験の役割 228
第 4章 生物学的同等性試験をめぐるよくある質問
Q-1 生物学的同等性試験のデータのばらつきについて 235
Q-2 米国FDAの SUPACガイドラインとわが国の処方変更ガイドライン,
含量違いガイドラインの類似点と相違点について 237
Q-3 処方変更ガイドライン,含量違いガイドラインにおける
「基準処方」について 240
付 録
2012年 2月 29 日付改正のポイント 245
後発医薬品の生物学的同等性試験ガイドライン(原文)
本文 247
Q & A 267
x 目 次

含量違い,処方変更の生物学的同等性試験ガイドライン(原文)
「含量が異なる経口固形製剤の生物学的同等性試験ガイドライン」
本文 293
「経口固形製剤の処方変更の生物学的同等性試験ガイドライン」
本文 303
Q & A 313
剤形違いの生物学的同等性試験ガイドライン(原文)
「剤型が異なる製剤の追加のための生物学的同等性試験ガイドライン」
本文 333
Q & A 335
配合剤の生物学的同等性試験ガイドライン(原文)
医療用配合剤の後発医薬品の生物学的同等性試験について Q & A 337
含量が異なる医療用配合剤及び医療用配合剤の処方変更
の生物学的同等性試験について Q & A 339
局所皮膚適用製剤の生物学的同等性試験ガイドライン(原文)
「局所皮膚適用製剤の後発医薬品のための生物学的同等性試験
ガイドライン」本文 343
「局所皮膚適用製剤の後発医薬品のための生物学的同等性試験
ガイドライン」Q & A 353
「局所皮膚適用製剤の剤形追加のための生物学的同等性試験
ガイドライン」本文 365
「局所皮膚適用製剤の剤形追加のための生物学的同等性試験
ガイドライン」Q & A 367
「局所皮膚適用製剤(半固形製剤及び貼付剤)の処方変更のための
生物学的同等性試験ガイドライン」本文 369
「局所皮膚適用製剤(半固形製剤及び貼付剤)の処方変更のための
生物学的同等性試験ガイドライン」Q & A 373
索 引㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀㌀381
目 次 xi

こととされた。
Q-1 は総論であり,医療用配合剤(経口固形製剤)の含量が異なる製剤間,
および,処方変更前後の製剤間の生物学的同等性試験は,含有する有効成分ご
とに既存の「含量違いガイドライン」および「処方変更ガイドライン」にした
がい,以下Q-2∼6 およびAppendix を参照して実施するとの原則を述べてい
る。
Q-2 および 3 では,医療用配合剤の処方変更の程度の計算方法と変更水準
の計算例が製剤設計別に述べられ,具体的な計算方法の例はAppendix に示さ
れている。複数の有効成分を単一層に含有する配合剤(単層錠)では,生物学
Ⅶ 医療用配合剤の生物学的同等性試験の考え方 171
表 2.25 含量違いの医療用配合剤の処方変更の程度の計算例(単層錠)
(「含量が異なる医療用配合剤及び医療用配合剤の処方変更の生物学的同等性試験について Q& A」(平
成 24 年 2 月 29 日付 事務連絡,別紙 5)Appendix(2))
(a)処方の変更
基準処方 試験製剤
有効成分 A 20 mg(4.00%)*1) 10 mg (2.22%)
有効成分 B 10 mg(2.00%) 2.5 mg (0.56%)
崩壊剤 トウモロコシデンプン 40 mg(8.00%) 40 mg (8.89%)
結合剤 ポピドン 5 mg(1.00%) 5 mg(1.111%)
滑沢剤 ステアリン酸Mg 5 mg(1.00%) 5 mg(1.111%)
賦形剤 乳糖水和物 380 mg(76.0%) 347.5 mg(77.22%)
結晶セルロース 40 mg(8.00%) 40 mg (8.89%)
製剤の総質量 500 mg 450 mg
*1) 括弧内は製剤の総質量に対する各成分の質量%。
(b)有効成分 Aを対象とした場合
添加剤の使用目的(成分) 含有率の差 水準
崩壊剤(トウモロコシデンプン) 0.89% (B)
結合剤(ポピドン) 0.111% (B)
滑沢剤(ステアリン酸Mg) 0.111% (B)
賦形剤(有効成分B) −1.44%
賦形剤(乳糖水和物) 1.22%
賦形剤(結晶セルロース) 0.89%
(c)有効成分 Bを対象とした場合
添加剤 含有率の差 水準
同左 0.89% (B)
同左 0.111% (B)
同左 0.111% (B)
同左(有効成分A) −1.78%
同左 1.22%
同左 0.89%
賦形剤で変更した成分の含有率の差の絶対値の和 3.55%(B) 3.89%(B)
変更した成分の含有率の差の絶対値の和 4.66%(B) 5.00%(B)
➡
すべての変更の水準は Bであり,有効成分 Aおよび Bを対象とした処方変更水準は B水準。

的同等性評価の対象とする有効成分以外の有効成分は賦形剤とし,「臨床試験
で有効性および安全性が確認された」,または「ヒトを対象とした生物学的同
等性試験により先発医薬品との同等性が確認された」製剤の処方(基準処方)
172 第 2章 生物学的同等性試験
表 2.26 医療用配合剤の処方変更の程度の計算例(二層錠)
(「含量が異なる医療用配合剤及び医療用配合剤の処方変更の生物学的同等性試験について
Q& A」(平成 24 年 2 月 29 日付 事務連絡,別紙 5)Appendix(3))
(a)処方の変更
基準処方 試験製剤
有効成分 A 20 mg (7.69%)*1) 20 mg(10.53%)
崩壊剤 トウモロコシデンプン 20 mg (7.69%) 15 mg (7.89%)
結合剤 ポピドン 5 mg(1.923%) 4 mg(2.105%)
滑沢剤 ステアリン酸Mg 1 mg(0.384%) 1 mg(0.526%)
賦形剤 乳糖水和物 194 mg(74.62%) 135 mg(71.05%)
結晶セルロース 20 mg (7.69%) 15 mg (7.89%)
A層の総質量 260 mg 190 mg
有効成分 B 10 mg(4.17%) 10 mg(4.17%)
崩壊剤 トウモロコシデンプン 20 mg(8.33%) 20 mg(8.33%)
結合剤 ポピドン 5 mg(2.083%) 5 mg(2.083%)
滑沢剤 ステアリン酸Mg 2 mg(0.833%) 2 mg(0.833%)
賦形剤 乳糖水和物 183 mg(76.25%) 183 mg(76.25%)
結晶セルロース 20 mg(8.33%) 20 mg(8.33%)
B層の総質量 240 mg 240 mg
製剤の総質量 500 mg 430 mg
*1) 括弧内は製剤の総質量に対する各成分の質量%。
(b)変更する A層について,含有率の差の計算
添加剤の使用目的と成分 含有率の差 水準
崩壊剤 トウモロコシデンプン +0.20% (B)
結合剤 ポピドン +0.182% (B)
滑沢剤 ステアリン酸Mg +0.142% (B)
賦形剤 乳糖水和物 −3.57% (B)
結晶セルロース +0.20% (B)
賦形剤で変更した成分の含有率の差の絶対値の和 3.77%(B)
変更した成分の含有率の差の絶対値の和 4.29%(B)
➡
すべての変更の水準は Bであり,有効成分 Aを対象とした場合の処方変更水準は Bである。
有効成分 Bを含有する B層については,変更がなく,処方変更水準は A水準。

からの処方変更の程度を既存のガイドラインにもとづき計算する。一方,複層
錠では,各層を 1つの製剤とみなして,層ごとに基準処方からの処方変更の程
度を同様に計算する(単層錠は表 2.24,複層錠は表 2.26,および図 2.32参
照)。
Q-4∼6 では,医療用配合剤の後発医薬品の含量違い製剤において,生物学
的同等性を評価する際の標準製剤の選択,処方変更の程度の計算方法と変更水
準の例,および溶出試験またはヒト生物学的同等性試験を実施する場合に想定
される問題点が述べられている。有効成分ごとに高含量製剤が異なる場合,た
とえば有効成分Aおよび Bをそれぞれ 10 mg および 1 mg,または,5 mg お
よび 2 mg 含有する配合剤の場合,高含量製剤は,有効成分 A については
10 mg/1 mg であるが,Bについては 5 mg/2 mg となる。このような含量違
い製剤間における標準製剤の選択は,複数の有効成分のうち,臨床上の有用性
や溶出試験の識別性の観点から,より重要度が高いと判断される有効成分につ
Ⅶ 医療用配合剤の生物学的同等性試験の考え方 173
方法 1:同含量の先発品との間で生物学的同等性試験を実施
方法 2:処方変更水準により,標準製剤①′と含量違い製剤間で生物学的同等性評価(ヒト生物学的同等性試験が必要な水準の場合,②′と③′は①′と含量比が異なり,同じ投与量で試験を実施できないため,方法 1により検討する)
①薬物A:8 mg 薬物 B:2 mg
②薬物A:8 mg 薬物 B:1 mg
薬物A: ①′1錠,②′1錠薬物B: ①′1錠,②′1錠 or 2 錠
ヒトBE試験
ヒトBE試験
ヒトBE試験
ヒトBE試験
配合剤(先発品) 配合剤(後発品)
溶出試験の実施方法
薬物A: ①′1錠,③′1錠 or 2 錠薬物 B: ①′1錠,③′1錠
薬物A: ①′1錠,④′1錠 or 2 錠薬物 B: ①′1錠,④′1錠 or 2 錠
方法 1
方法 2
③薬物A:4 mg 薬物 B:2 mg
④薬物A:4 mg 薬物 B:1 mg
②′薬物A:8 mg 薬物 B:1 mg
③′薬物A:4 mg 薬物 B:2 mg
④′薬物A:4 mg 薬物 B:1 mg
①′薬物A:8 mg 薬物 B:2 mg
有効成分A,Bいずれも倍数の関係にあるのは①と④のみ。
図 2.32 含量が異なる医療用配合剤の生物学的同等性試験
(「含量が異なる医療用配合剤及び医療用配合剤の処方変更の生物学的同等性試験についてQ & A」(平
成 24 年 2 月 29 日付 事務連絡,別紙 5)Q-4∼6)

いての高含量製剤が標準製剤となる。また,含量違い製剤間の処方変更は,含
量違い製剤ガイドラインを適用して処方変更の程度を計算する(表 2.25およ
び図 2.32 参照)。
以上の留意点について,図 2.32 により具体例を説明する(第 2 章Ⅰ.4(3)
図 2.7 の単剤の場合も参照のこと〔84 ページ〕)。有効成分Aおよび Bそれぞ
れの高含量を含有し,臨床試験で有効性および安全性が確認された医療用配合
剤の先発医薬品①に対し,ヒトを対象とした生物学的同等性試験により生物学
的同等性が確認された医療用配合剤の後発医薬品①′*7 の含量違い製剤②′∼④′
について考える。
生物学的同等性を検討する場合,次の 2通りの方法が考えられる。
方法 1 同含量の先発医薬品②∼④を標準製剤として「後発医薬品ガイドラ
イン」にしたがい試験を実施する。
方法 2 先発医薬品と生物学的同等性が証明された高含量の製剤①′を標準
製剤として,含量違い製剤②′∼④′を試験製剤として「含量違いガイ
ドライン」にしたがい試験を実施する。基準処方(当該事例において
は,後発医薬品の高含量の製剤処方①′)との処方変更水準を有効成分
ごとに計算し,得られた水準の程度に応じて要求される試験の内容,
および判定基準が決定される。水準の程度が小さい場合には,溶出挙
動の同等性により生物学的に同等とみなされ,ヒトを対象とした生物
学的同等性試験が免除される。有効成分ごとの水準が異なる場合には,
有効成分それぞれについて水準に対応する試験を実施する。たとえば,
有効成分AおよびBともに溶出試験結果にもとづき生物学的同等性を
判定する水準である場合,有効成分AおよびBの特性に対応する試験
条件で溶出試験を実施する(配合剤の後発医薬品Q & A)3)。
有効成分 Aについては溶出試験,一方,B についてはヒトによる試験が必
要な変更水準の場合,有効成分 Aについては溶出挙動が同等であること,一
方,Bについてはヒトを対象とした生物学的同等性試験を実施する(配合剤の
含量違い及び処方変更Q & A)6)。
有効成分Aおよび Bともにヒトを対象とした生物学的同等性試験の実施が
必要な変更水準の場合,有効成分Aおよび Bの含量比が異なる製剤では,同
じ投与量で後発医薬品ガイドラインにしたがった生物学的同等性試験を実施す
ることができないため,同含量の先発医薬品の後発医薬品として,後発医薬品
174 第 2章 生物学的同等性試験
*7 平成 24 年 2 月 29 日付 厚生労働省医薬食品局審査管理課 事務連絡の別紙 2「含量が異なる経口固形製剤の生物学的同等
性試験ガイドラインQ&A,経口固形製剤の処方変更の生物学的同等性試験ガイドラインQ&A」のQ-10 により,原則
として,自社の高含量の製剤を選択することとされている。
なお,有効成分ごとに高含量製剤が異なる場合,複数の有効成分のうち,臨床上の有用性や溶出試験の識別性の観点
から,「より重要度が高いと判断される有効成分」についての高含量製剤を選択する(「含量が異なる医療用配合剤及び
医療用配合剤の処方変更の生物学的同等性試験についてQ & A」のQ-55)。

ガイドラインにしたがい試験を実施する(配合剤の含量違い及び処方変更 Q
& A)4)。図 2.6 において,標準製剤①′と,含量違い製剤②′または③′では含
量比が異なり,同一投与量でヒトによる試験を実施できないため,上述の方法
1 と同様に,有効成分 Aおよび Bがともに同含量の先発医薬品②および③を
標準製剤として,後発医薬品ガイドラインにしたがい試験を実施する。
(永井 尚美)
■文 献
Ⅲ 生物学的同等性試験ガイドラインとその改正
1) 緒方宏泰,医薬品研究,29, 818(1998).
2) 村主教行,第 36回薬事エキスパート研修会−医薬品の生物学的同等性確保における溶出試験の有
用性と限界−要旨集,2008,p. 35.
3) Morihara, M., Aoyagi, N., Kaniwa, N., Kojima, S., Biol. Pharm. Bull., 24, 313(2001).
4) Ogata, H., Aoyagi, N., Kaniwa, N., Koibuchi, M., Shibazaki, T., Int. J. Pharm. Ther. Tox., 20, 166
(1982).
5) 青柳伸男,医薬品研究,28, 355(1997).
6) 青柳伸男,医薬ジャーナル,39, 66(2003).
7) 伊井直人,濱浦健司,脇山尚樹,四方田千佳子,溶出試験パドル法と回転バスケット法の攪拌力の
差異と溶出性に及ぼす影響,日本薬剤学会第 26 年会講演要旨集,2011,p. 200.
8) Hollenbeck R. Gary, Bioavailability of phenylpropanolamine HCl from tablet dosage forms containing
croscarmellose sodium, International Journal of Pharmaceutics, 47, 89-93(1988).
9) 四方田千佳子,厚生労働科学研究報告,後発医薬品の同等性ガイドラインにおける試験条件の最適
化に関する研究平成 23 年度総括・分担研究報告書,75-209(2012).
10) http://www.nihs.go.jp/drug/DrugDiv-E.html.
Ⅳ 諸外国の生物学的同等性試験ガイドラインの動き
1) 小林征雄,「日米欧(3極)との比較」,2012改正ガイドラインをふまえた生物学的同等性試験,情
報機構,2012, pp. 371-393.
2) 四方田千佳子,後発医薬品を語るための知識(3), PHARM TECH JAPAN, 27, 443-449(2010).
3) FDA, Guidance for Industry, Bioavailability and Bioequivalence Studies for Orally Administered
Drug Products - General Considerations(2003).
4) FDA, Guidance for Industry, Immediate Release Solid Oral Dosage Forms : Scale-Up and Post-
Approval Changes : Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In
Vivo Bioequivalence Documentation(1995).
5) FDA, Guidance for Industry, Modified Release Solid Oral Dosage Forms : Scale-Up and Post-
Approval Changes : Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In
Vivo Bioequivalence Documentation(1997).
6) FDA, Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for
Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System
(2000).
7) EMA, Guideline on the Investigation of Bioequivalence(2010).
8) EMA, Note for Guidence on Modified Release Oral and Transdermal Dosage Forms : Section II
(Pharmacokinetics and clinical evaluation)(1999).
9) EMA, Concept paper on the need for revision of the note for guidance on modified release oral and
Ⅶ 医療用配合剤の生物学的同等性試験の考え方 175

H :μ
μ≤θ or
μ
μ≥θ
H : θ<μ
μ<θ (3.1)
(θ1, θ2)は生物学的同等の許容域で,対数変換を行って Cmax および AUCt の評
価を行うときには,(0.80, 1.25)である。
式(3.1)における帰無仮説 H0 は「生物学的に非同等である状態」を表してお
り,対立仮説 H1 が「生物学的に同等である状態」を表していることに注目し
てほしい。多くの生物学的検定では,「比較する 2つの処理間に差がない」と
いう帰無仮説を設定し,「処理間に差がある」という対立仮説をおく。このよ
うな生物学的検定では,試験を行って帰無仮説を棄却し対立仮説を採択できる
ときには,積極的に「2 つの処理間に有意に差がある」と主張できるが,帰無
仮説を棄却できないときには,「2つの処理間に有意差がない」とは積極的に
はいえない。なぜなら,試験のばらつきが大きいために実際には意味のある処
理間の差を検出できなかったのか,本当に 2つの処理間に差がなかったために
試験でも差が検出できなかったのか識別ができないからである。「差がない」
という判定が得られたときのこのようなあいまいさを避けるために,生物学的
同等性試験では,帰無仮説には「生物学的に非同等である状態」を設定し,対
立仮説には「生物学的に同等である状態」を設定して,試験を行って帰無仮説
を棄却し対立仮説を採択することにより,積極的に試験製剤は標準製剤と生物
学的に同等であると判定できるようにしたのである。なお,等号が帰無仮説の
側についているのは,「後発医薬品の生物学的同等性試験ガイドライン Q &
A」(平成 24 年 2 月 29 日付 事務連絡,別紙 1)(以下,「後発医薬品 Q & A」
という)のQ-36 等にしたがっている。1, 3)
具体的には,PK パラメータを対数変換した後に得られる試験製剤と標準製
剤の平均値を X, X とするとき,下記式(3.2)がともに成立するときに試験製
剤は標準製剤と生物学的に同等であると結論づけられる。なお,ここでは常用
対数で変換した例を示すことにする。
log θ−(X−X)
s 2n>t (1−α, n−2)
(X−X)−log θ
s 2n>t (1−α, n−2) (3.2)
ここで,α は有意水準,n は生物学的同等性試験の全被験者数,また,s は分
散分析表における残差平均平方の平方根である。α は,PK パラメータで評価
を行うときには 5% に設定するが,薬力学的試験で評価を行うときには 2.5%4)
である。式(3.2)は,有意水準 α で 2回の t 検定を行うことを意味しており,
「2つの片側検定」の言葉の由来となっている。
Ⅱ 生物学的同等性試験の統計解析 195

有意水準 5% で 2つの片側検定を実施することは,信頼係数 90% で推定し
た試験製剤と標準製剤の PK パラメータの母平均の差の信頼区間を,生物学的
同等の許容域と比較することと等価である。log μT−log μR の信頼区間の下限
値と上限値は次の式(3.3)で計算できる。
δ=(X−X)−t (1−2α, n−2) s 2n
δ=(X−X)+t (1−2α, n−2) s 2n (3.3)
ここで,δL が log(0.80)より大きく,かつ,δU が log(1.25)未満のときに試験製
剤は標準製剤と生物学的に同等であると結論づけられる。
図 3.6 に,推定された信頼区間と生物学的同等の許容域との関係を示した。
信頼区間の左端が式(3.3)に示す δL,右端が δU である。ケース a の結果が得ら
れたときには,信頼区間は完全に生物学的同等の許容域に含まれるので試験製
剤と標準製剤は同等と判定される。ケース b の場合は,信頼区間が log(1.0)
を含んでいないので通常の検定では製剤間のバイオアベイラビリティの差は有
意であると判定されるが,信頼区間は完全に生物学的同等の許容域に含まれて
いるので,生物学的同等性試験では試験製剤と標準製剤は同等と結論づけられ
る。一方,ケース c は,信頼区間が完全に生物学的同等の許容域の外にあるの
で,試験製剤は標準製剤と生物学的に非同等であると判定される。またケース
d と e はともに例数不足のために,試験製剤は標準製剤と生物学的に同等であ
るとも非同等であるとも結論づけられない状態にある。このようなときにも,
試験製剤と標準製剤は同等であると判定することはできない。ケース d と e
の場合には,1 回に限り追加の試験を行うことが可能で,例数を増やしてケー
ス a または bの結果が得られれば,試験製剤と標準製剤は同等と判定される。
196 第 3章 実施方法
logθ1 log 1 logθ2 μT/μR
ケース a
ケース b
ケース c
ケース d
ケース e
図 3.6 生物学的同等性の許容域と信頼区間
生物学的同等の許容域
生物学的同等の非許容域
生物学的同等性試験から得られた製剤間の
バイオアベイラビリティの差の信頼区間
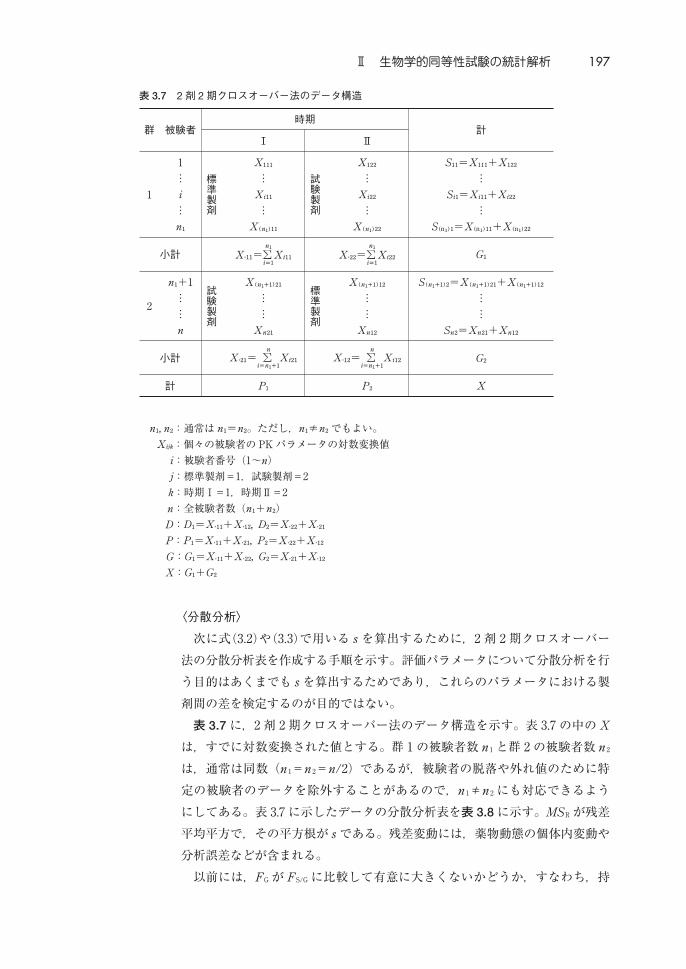
〈分散分析〉
次に式(3.2)や(3.3)で用いる s を算出するために,2 剤 2期クロスオーバー
法の分散分析表を作成する手順を示す。評価パラメータについて分散分析を行
う目的はあくまでも s を算出するためであり,これらのパラメータにおける製
剤間の差を検定するのが目的ではない。
表 3.7 に,2 剤 2期クロスオーバー法のデータ構造を示す。表 3.7の中の X
は,すでに対数変換された値とする。群 1 の被験者数 n1 と群 2 の被験者数 n2
は,通常は同数(n1=n2=n/2)であるが,被験者の脱落や外れ値のために特
定の被験者のデータを除外することがあるので,n1≠n2 にも対応できるよう
にしてある。表 3.7に示したデータの分散分析表を表 3.8 に示す。MSR が残差
平均平方で,その平方根が s である。残差変動には,薬物動態の個体内変動や
分析誤差などが含まれる。
以前には,FGが FS/Gに比較して有意に大きくないかどうか,すなわち,持
Ⅱ 生物学的同等性試験の統計解析 197
表 3.7 2剤 2期クロスオーバー法のデータ構造
群 被験者時期
計Ⅰ Ⅱ
1
1
⋮
i
⋮
n
標準製剤
X
⋮
X
⋮
X
試験製剤
X
⋮
X
⋮
X
S=X+X
⋮
S=X+X
⋮
S=X+X
小計 X∙=∑
X X∙=∑
X G
2
n+1
⋮
⋮
n
試験製剤
X
⋮
⋮
X
標準製剤
X
⋮
⋮
X
S=X+X
⋮
⋮
S=X+X
小計 X∙= ∑
X X∙= ∑
X G
計 P P X
n, n:通常は n=n。ただし,n≠n でもよい。
X:個々の被験者の PKパラメータの対数変換値
i:被験者番号(1∼n)
j:標準製剤=1,試験製剤=2
k:時期Ⅰ=1,時期Ⅱ=2
n:全被験者数(n+n)
D:D=X∙+X∙, D=X∙+X∙
P:P=X∙+X∙, P=X∙+X∙
G:G=X∙+X∙, G=X∙+X∙
X:G+G
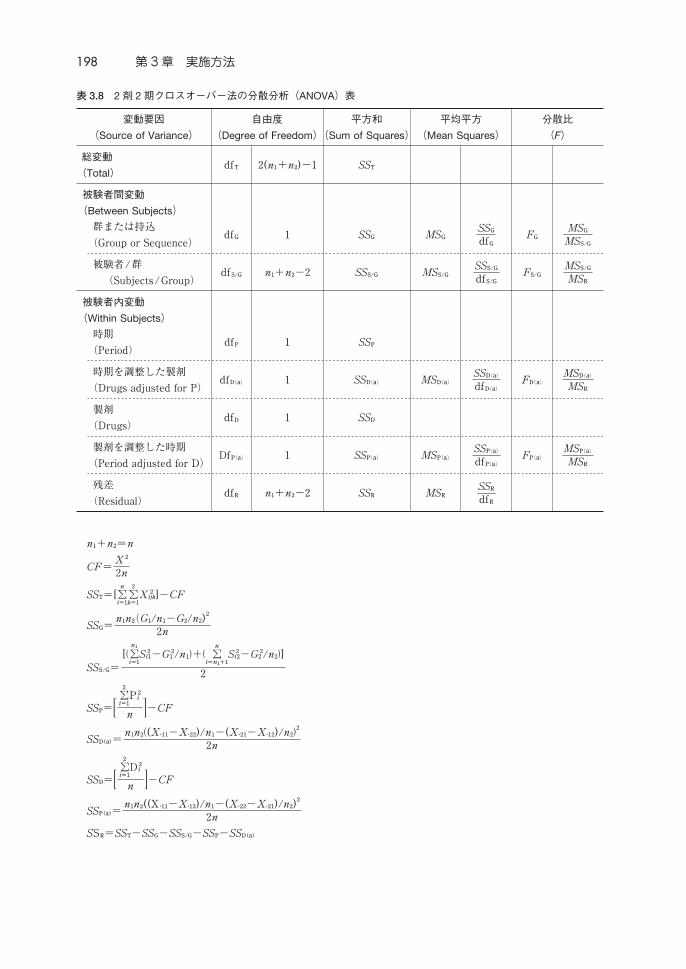
198 第 3章 実施方法
表 3.8 2剤 2期クロスオーバー法の分散分析(ANOVA)表
変動要因
(Source of Variance)
自由度
(Degree of Freedom)
平方和
(Sum of Squares)
平均平方
(Mean Squares)
分散比
(F)
総変動
(Total)df 2(n+n)−1 SS
被験者間変動
(Between Subjects)
群または持込
(Group or Sequence)df 1 SS MS
SS
dfF
MS
MS/
被験者/群
(Subjects/Group)df/ n+n−2 SS/ MS/
SS/
df/F/
MS/
MS
被験者内変動
(Within Subjects)
時期
(Period)df 1 SS
時期を調整した製剤
(Drugs adjusted for P)df() 1 SS() MS()
SS()
df()F()
MS()
MS
製剤
(Drugs)df 1 SS
製剤を調整した時期
(Period adjusted for D)Df() 1 SS() MS()
SS()
df()F()
MS()
MS
残差
(Residual)df n+n−2 SS MS
SS
df
n+n=n
CF=X
2n
SS=[∑
∑
X
]−CF
SS=nn(G/n−G/n)
2n
SS/=
[∑
S
−G /n+ ∑
S
−G /n]
2
SS=∑
P
n −CF
SS=nn(X∙−X∙)/n−(X∙−X∙)/n
2n
SS=∑
D
n −CF
SS=nn((X∙−X∙)/n−(X∙−X∙)/n)
2n
SS=SS−SS−SS/−SS−SS

越し効果(群効果と交絡している)の有無に関する検定が求められていたが,
健康な被験者に同一の有効成分を投与する生物学的同等性試験では,一般的に
持越し効果は起こりえないと考えられるので,現行ガイドラインでは持越し効
果の検定は行わない。
(4)対数変換を行わない場合の解析
対数変換を行わない場合の仮説を次の式(3.4)に示す。
H : μ−μ≤θ μ or μ−μ≥θ μ
H : θ μ<μ−μ<θ μ (3.4)
生物学的同等の許容域(θ1, θ2)は,Cmax および AUCt の評価を行うときに
は,(−0.20, +0.20)で表される。試験製剤と標準製剤の PK パラメータの平
均値を Y, Y とするとき,2つの片側検定では次の式(3.5)がともに成立する
ときに,試験製剤と標準製剤とは生物学的に同等と判定される。
θY−(Y−Y)
s 2n>t (1−α, n−2)
(Y−Y)−θY
s 2n>t (1−α, n−2) (3.5)
また,信頼区間法を適用するときには,次の式(3.6)で示す δL が θY より
大きく,δU が θY 未満のときに,試験製剤は標準製剤と生物学的に同等であ
ると結論づけられる。
δ=(Y−Y)−t (1−2α, n−2) s 2n
δ=(Y−Y)+t (1−2α, n−2) s 2n (3.6)
また,分散分析表の作成手順は,対数変換したデータを用いるときと同様
で,Xを Yに置き換えればよい。
(5)計算例
以下に,具体的な数値を用いて解析手順を示す。
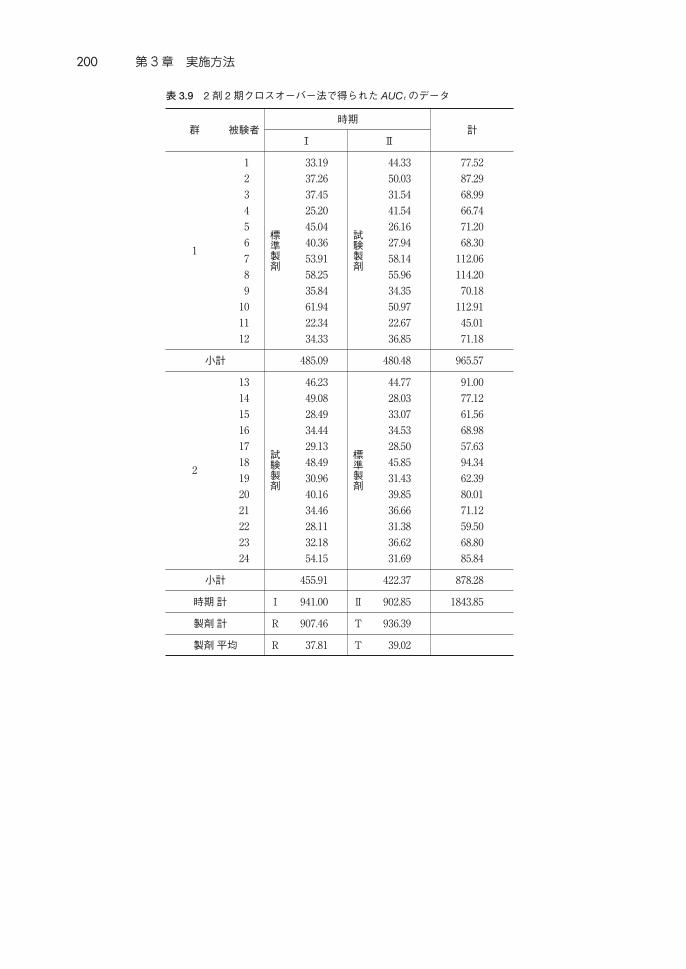
表 3.9 は,1群 12 例の 2 剤 2期クロスオーバー法による生物学的同等性試
験を実施して得られた AUCt の値である。試験製剤の平均値は 39.02,標準製
剤の平均値は 37.81 である。このデータについて,常用対数変換して得られた
データが表 3.10である。X,X はそれぞれ,1.576 および 1.565 である。
表 3.8 にしたがい,次のような手順で各平方和を計算する。
Ⅱ 生物学的同等性試験の統計解析 199
200 第 3章 実施方法
表 3.9 2剤 2期クロスオーバー法で得られた AUCtのデータ
群 被験者時期
計Ⅰ Ⅱ
1
1
2
3
4
5
6
7
8
9
10
11
12
標準製剤
33.19
37.26
37.45
25.20
45.04
40.36
53.91
58.25
35.84
61.94
22.34
34.33
試験製剤
44.33
50.03
31.54
41.54
26.16
27.94
58.14
55.96
34.35
50.97
22.67
36.85
77.52
87.29
68.99
66.74
71.20
68.30
112.06
114.20
70.18
112.91
45.01
71.18
小計 485.09 480.48 965.57
2
13
14
15
16
17
18
19
20
21
22
23
24
試験製剤
46.23
49.08
28.49
34.44
29.13
48.49
30.96
40.16
34.46
28.11
32.18
54.15
標準製剤
44.77
28.03
33.07
34.53
28.50
45.85
31.43
39.85
36.66
31.38
36.62
31.69
91.00
77.12
61.56
68.98
57.63
94.34
62.39
80.01
71.12
59.50
68.80
85.84
小計 455.91 422.37 878.28
時期計 Ⅰ 941.00 Ⅱ 902.85 1843.85
製剤計 R 907.46 T 936.39
製剤平均 R 37.81 T 39.02

2 生物学的同等性試験における溶出試験の役割
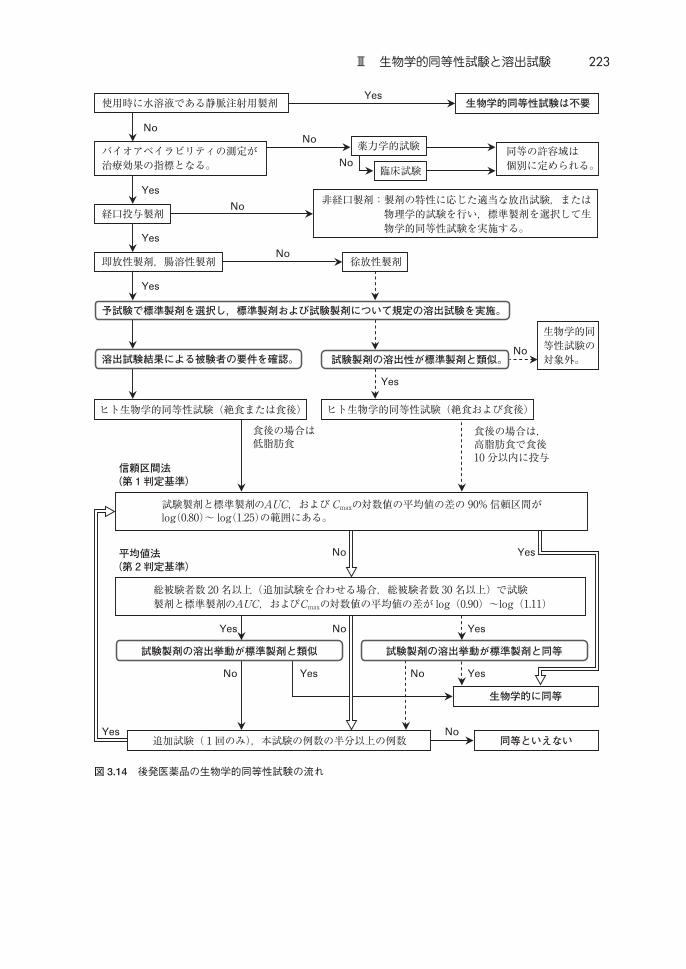
生物学的同等性試験の各ガイドラインの構成と溶出試験の役割を図 3.13に,
後発医薬品の生物学的同等性試験の流れを図 3.14に示す。剤形追加に係る医
薬品の生物学的同等性試験は,ほとんどのケースで後発医薬品ガイドラインに
したがった試験をすることになる(「剤形が異なる製剤の追加のための生物学
的同等性試験ガイドライン」(平成 24 年 2 月 29 日付 薬食審査発 0229 第 10
号,別紙 4)(以下「剤形追加ガイドライン」という))。後発医薬品ガイドラ
インはすべての製剤を対象としているが,後発医薬品ガイドライン中で具体的
に示されている試験手順は,経口投与製剤を対象としたものである(局所皮膚
適用製剤については,本書では,第 2 章Ⅵ(145 ページ)を参照)。図 3.13 の
①∼⑤に示すように日本の生物学的同等性試験に関するガイドラインにおいて
は,溶出試験が大きな役割を担っている。
(1)標準製剤の選定
後発医薬品ガイドラインおよび剤形追加ガイドラインは先発医薬品,含量違
いガイドラインは既承認の製剤,処方変更ガイドラインは処方変更前の製剤,
それぞれ 3 ロットにつき中間の溶出性を示すロットの製剤を標準製剤*4 とす
る。「規格及び試験方法」に溶出試験が設定されている場合には,その溶出試
験液で,パドル法 50 回転の試験を行う。「規格及び試験方法」に溶出試験が設
定されていない場合は,後発医薬品ガイドラインの溶出試験条件の試験液を用
いる。3 ロットとも 15 分以内に平均 85% 以上溶出する場合,いずれのロット
を標準製剤にしてよい。標準製剤選定のための溶出試験は緩和な試験条件でな
されるので対照製剤のロット間で溶出が異なれば対照となるロットの違いに
よって溶出比較結果が異なってくることが考えられ,3ロットにつき中間の溶
出性を示すロットの製剤を対照として試験溶出比較を行い,また,ヒト生物学
的同等性試験が実施される。
標準製剤(対照製剤)が 3ロット入手困難な場合,困難である妥当な理由が
あれば,2以下のロットから標準製剤を選択してよい(後発医薬品ガイドライ
ンQ & A,Q-4)。
Ⅲ 生物学的同等性試験と溶出試験 221
*4 「標準製剤」は日本独自の用語である。海外では「対照製剤(Reference product)」という用語が使われている。「標準製
剤」という用語は,平成 9年 12 月 22 日付 医薬審第 487 号別添「後発医薬品の生物学的同等性試験ガイドライン」から
使われるようになった。対照製剤 3ロットから中間の溶出性を示すロットを標準製剤と呼んで,「対照製剤」と区別する
ためにつくられた用語と思われるが,生物学的同等性試験関連では実際にはとくに区別せずに「標準製剤」という用語
が「対照製剤」の意味で使われている。

標準製剤を選定する溶出試験において,3 ロットとも 15 分以内に 85% 以上
溶出する場合,いずれのロットを標準製剤にしてよい。
含量違い,処方変更ガイドラインにおいて,パドル法 50 回転で,15 分以内
に 85% 以上溶出することが規格試験結果から確認できる製剤については,規
格試験のデータをもとに標準製剤を選択してよい。また,治療濃度域の狭い薬
物を除き,パドル法 50 回転で,30 分以内に 85% 以上溶出することが規格試
験結果から確認できる製剤についても,規格試験のデータをもとに標準製剤を
選択してよい(含量違い,処方変更ガイドラインQ & A,Q-38)。
(2)即放性製剤において健康成人でヒト試験できる試験製剤の要件
後発医薬品ガイドラインで即放性製剤の試験製剤について以下の記載があ
る。
医薬品の適用集団が限られている医薬品では,第 3 章,A.Ⅴ. に従った溶出試験
の一つ以上の条件において,標準製剤と試験製剤の溶出率の間に「著しい差」が
ある場合には,適用集団を対象とした生物学的同等性試験の実施が必要となる。
適用集団が限られていない医薬品の即放性製剤では,第 3 章,A.Ⅴ. に従った溶出
試験により pH 6.8 付近(ただし,塩基性薬物は pH 3.0∼6.8)の試験液で,標準製
剤と試験製剤の溶出率の間に「特異的に著しい差」が認められる場合には,低胃
酸の被験者で試験する。
222 第 3章 実施方法
後発医薬品の生物学的同等性試験ガイドライン
ガイドラインの構成
含量違い 処方変更後発医薬品
・溶出試験
・ヒト生物学的同等性試験
処方変更の程度(A~E水準)
大きい(C~E水準)
小さい(B~D水準) 小さい
(A水準)
規格の溶出試験条件
剤形追加
①標準製剤を3ロットから選定
②即放性製剤で健康成人でヒト試験できる試験製剤の要件
③徐放性製剤:ヒト試験するための試験製剤の要件
⑤溶出挙動の同等性によるヒト生物学的同等性試験免除
④生物学的同等性評価のための補強データ(第 2判定基準)
図 3.13 生物学的同等性試験の各ガイドラインの構成と溶出試験の役割
Ⅲ 生物学的同等性試験と溶出試験 223
経口投与製剤
使用時に水溶液である静脈注射用製剤 生物学的同等性試験は不要
バイオアベイラビリティの測定が治療効果の指標となる。
薬力学的試験
臨床試験
同等の許容域は個別に定められる。
非経口製剤:製剤の特性に応じた適当な放出試験,または物理学的試験を行い,標準製剤を選択して生物学的同等性試験を実施する。
即放性製剤,腸溶性製剤 徐放性製剤
予試験で標準製剤を選択し,標準製剤および試験製剤について規定の溶出試験を実施。
溶出試験結果による被験者の要件を確認。 試験製剤の溶出性が標準製剤と類似。
生物学的同等性試験の対象外。
NoNo
No
No
No
No
Yes
Yes
Yes
Yes
Yes
試験製剤の溶出挙動が標準製剤と類似
ヒト生物学的同等性試験(絶食および食後)
生物学的に同等
同等といえない
Yes
Yes
Yes
No Yes
No
ヒト生物学的同等性試験(絶食または食後)
追加試験(1回のみ),本試験の例数の半分以上の例数
信頼区間法(第 1判定基準)
平均値法(第 2判定基準)
試験製剤と標準製剤のAUC,およびCmaxの対数値の平均値の差の 90%信頼区間がlog(0.80)~ log(1.25)の範囲にある。
総被験者数 20 名以上(追加試験を合わせる場合,総被験者数 30 名以上)で試験製剤と標準製剤のAUC,およびCmaxの対数値の平均値の差が log(0.90)~log(1.11)
YesNo
食後の場合は,高脂肪食で食後10 分以内に投与
食後の場合は低脂肪食
試験製剤の溶出挙動が標準製剤と同等
No
No
Yes
図 3.14 後発医薬品の生物学的同等性試験の流れ

したがって,即放性製剤において健康成人でヒト試験するためには,溶出試
験を実施して,試験製剤が上記に該当していないことを確認しておく必要があ
る。
「著しい差」の具体的な内容は後発医薬品ガイドラインの記載を参照してほ
しい。なお,「著しい差」は,溶出挙動の評価を行った条件での 15 分以上の時
点での比較で判定することでよい(後発医薬品ガイドライン Q & A,Q-55,
パブコメ 19)。
(3)徐放性製剤においてヒト試験するための試験製剤の要件
後発医薬品ガイドラインで徐放性製剤の試験製剤について以下の記載があ
る。
試験製剤は,その大きさ,形状,比重,放出機構が先発医薬品のものと著しく
異ならないものとする。試験製剤の溶出挙動は,標準製剤の溶出挙動と類似して
いなければならない。
したがって,徐放性製剤において,試験製剤の溶出挙動が標準製剤の溶出挙
動と類似していないと,ヒト試験が実施できない。放出機構の類似性は,マト
リックスタイプか膜制御タイプか,シングルユニットかマルチプルユニット
か,崩壊型か非崩壊型か,などから説明し(後発医薬品ガイドラインQ & A,
Q-67),放出機構などが類似していることの証明として溶出挙動の類似性が示
されなければ,先発医薬品の後発品とは認められない(後発医薬品ガイドライ
ンQ & A,Q-42)。
また,含量違い,処方変更ガイドラインにも,同じような試験製剤について
以下の記載がある。
標準製剤と大きさ,形状,比重,放出機構などが著しく異ならず,標準製剤と
溶出挙動が類似していなければならない。
すなわち,
・ 形状:相似形であること。
・ 大きさ:錠剤の場合,杵の直径の差が 25% 以内であること。顆粒状の
徐放性粒子を充填したカプセル剤の場合は,カプセルの大きさの違いは
問わない。
224 第 3章 実施方法

・ 比重:溶出試験において製剤の崩壊状況を観察するとき,試験液上に浮
遊する粒子,沈殿堆積する粒子,その中間に浮遊する粒子の割合が同程
度であること。
・ 放出機構:製剤設計の概念が同じであること,および溶出挙動の類似性
で判定する(含量違い,処方変更ガイドラインQ & A,Q-19)。
となる。
(4)生物学的同等性評価のための補強データ(第 2の判定基準)
図 3.15に後発医薬品ガイドラインでの生物学的同等性の判定法を図示し
た*5。信頼区間法,平均値法は 2つの判定方法を区別するために便宜的に付け
た名称で正式な名称ではない。信頼区間法で試験製剤と標準製剤の AUCおよ
び Cmaxの対数値の平均値の差の 90%信頼区間が log(0.80)∼log(1.25)の範囲に
あるとき生物学的同等と判定される。この判定基準は第 1判定基準と呼ばれて
いる。
平均値法*5 では総被験者数 20人以上(追加試験を合わせる場合 30人以上)
で試験製剤と標準製剤の AUCおよび Cmaxの対数値の平均値の差が log(0.90)
∼log(1.11)の範囲にあり,かつ,溶出挙動が即放性製剤/腸溶性製剤で類似,
徐放性製剤で同等のとき,生物学的同等と判定される(図 3.13)。表 3.16は
90%信頼区間で,80% の検出力を達成するのに必要な 1群あたりの例数を示
している。つまり,表中の試験製剤と標準製剤との母平均の比(μ / μ)と,
被験者内変動係数(CV)のときに信頼区間法で 80%の確率で生物学的同等と
なる 1群あたりの例数を示しており,生物学的同等性試験の例数設計する際の
参考にする表である。CV値が大きな医薬品では,信頼区間法で生物学的同等
とするためには,多くの被験者数が必要となる(2群の試験が行われるので,
総被験者数は 1群の例数の 2倍になる)。平均値法は,そのような医薬品の生
物学的同等性試験で被験者数が多くなってしまわないように設けられている。
平均値法は第 2判定基準とも呼ばれているが,ヒト試験で「生物学的に同等で
あることがきわめて強い」と推定できる条件下で,ヒトの追加試験を免除する
条件という考え方6, 8)である。ヒト試験で AUCと Cmaxの幾何平均値の比に大き
な差がなく吸収性の大きな差がなく,さらに溶出挙動が同等であるとのデータ
から,「ヒトの追加試験を行って同等を確認する条件にはない」と判断し,追加
試験を免除するという考え方である(本書第 1 章Ⅴ.13.(5)(55 ページ)および
Ⅲ 生物学的同等性試験と溶出試験 225
*5 この平均値法による生物学的同等の判定基準は,日本独自のもので海外にはない。海外では,CV値が 30% 以上の医薬
品を highly valuable drugsと定義し,対照製剤を同じ被験者に投与し求めた CV値に応じて,90%信頼区間の許容域を
広げる scaled-average-BEを適用することがある。9, 10) FDAと EMAで許容域を広げる計算方法が若干異なり 11),EMA
のガイドラインでは Cmaxのみに対して適用される。日本では scaled-average-BEは採用されていない。

医薬品の生物学的同等性試験 表1:182×257 背:14×257 表4:182×257
医薬品の生物学的同等性試験
―
ガイドライン対応―
緒方
宏泰
医薬品の生物学的同等性試験
緒方 宏泰[編著]
﹇編著﹈
-ガイドライン対応-
9784840744249
1923047100009
ISBN978-4-8407-4424-9 C3047 ¥10000E


![留学生 入学試験に関する要項 - Kurume U · 試験種別 学 部 出願期間[必着] 試験日 合格発表日 入学申込締切日 入学手続締切日[必着] 留学生入試](https://static.fdocuments.es/doc/165x107/6030e7e0e2511f43f40cba52/cc-eeeee-kurume-u-eec-ef-eoeec.jpg)















