Acumulaciones y depósitos intracelulares y extracelulares
53
Acumulaciones y Depósitos Intracelulares y extracelulares Juliana Zapata M.V, MSc
-
Upload
julianazapatacardona -
Category
Documents
-
view
3.293 -
download
14
Transcript of Acumulaciones y depósitos intracelulares y extracelulares

Acumulaciones y Depósitos Intracelulares y extracelulares
Juliana ZapataM.V, MSc
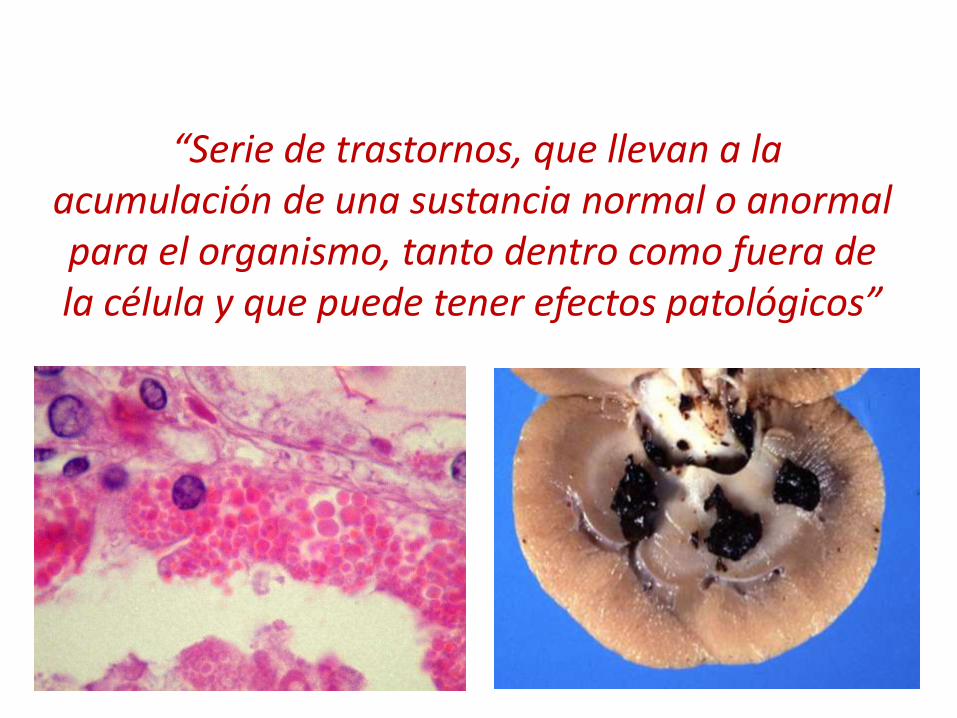
“Serie de trastornos, que llevan a la acumulación de una sustancia normal o anormal para el organismo, tanto dentro como fuera de la célula y que puede tener efectos patológicos”
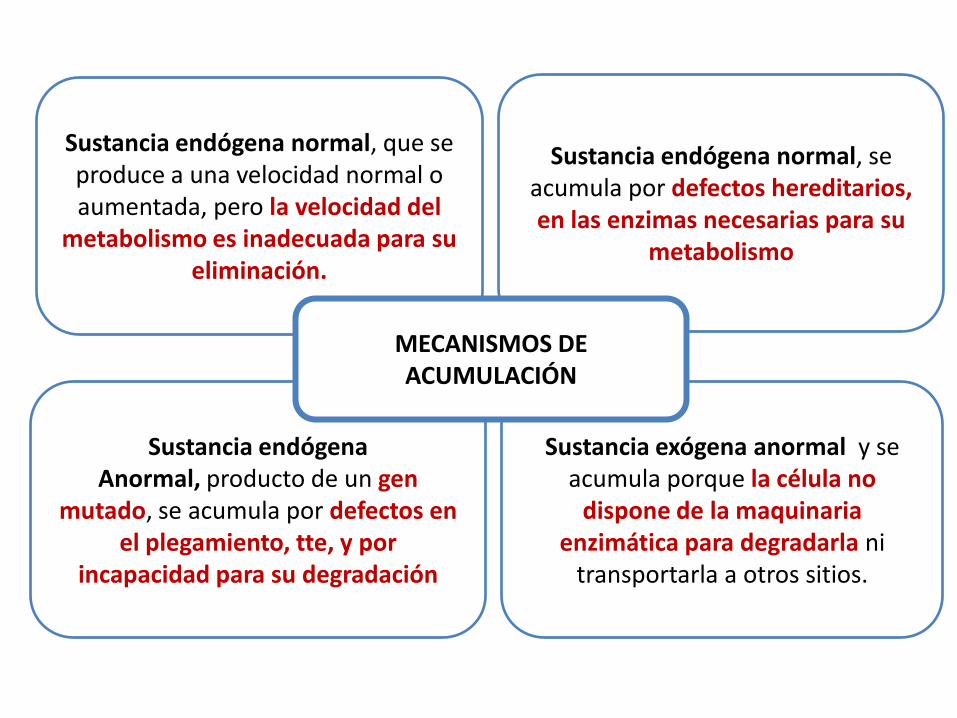
Sustancia endógena normal, que se produce a una velocidad normal o aumentada, pero la velocidad del
metabolismo es inadecuada para su eliminación.
Sustancia endógena Anormal, producto de un gen
mutado, se acumula por defectos en el plegamiento, tte, y por
incapacidad para su degradación
Sustancia exógena anormal y se acumula porque la célula no
dispone de la maquinaria enzimática para degradarla ni
transportarla a otros sitios.
Sustancia endógena normal, se acumula por defectos hereditarios, en las enzimas necesarias para su
metabolismo
MECANISMOS DE ACUMULACIÓN

Acumulación de agua: cambio hidrópico
Cuando la cantidad de líquido que difunde al interior de la célula, es mayor de la que debe salir.
Edema Celular
Trastorno osmótico reversible.

Causas
Hipoxia Exceso de glucosa
Deja de Funcionar la bomba de Na+
Efecto Osmótico
Aspecto macroscópico: En algunas ocasiones, los órganos afectados pueden apreciarse aumentados de tamaño o turgentes.
Aspecto Microscópico

Cambio hidrópico SNC
Cambio hidrópico hígado

Acumulación de agua: cambio hidrópico
Consecuencias:
Si el daño hipóxico es grave o prolongado, lacélula quedará desprovista de ATP y de energía;se llenará de agua y sus organelos y la membrananuclear sufrirán daños irreversibles → Muertecelular

Acumulación de triglicéridos: Cambio Graso
Acumulación anormal en células parenquimatosas ppal de órganos involucrados en el metabolismo de las grasas,
Hígado, corazón, riñón.
• En etapas iniciales, es una lesión reversible, que puede desaparecer sin dejar lesiones, si no se corrige la causa: Muerte celular
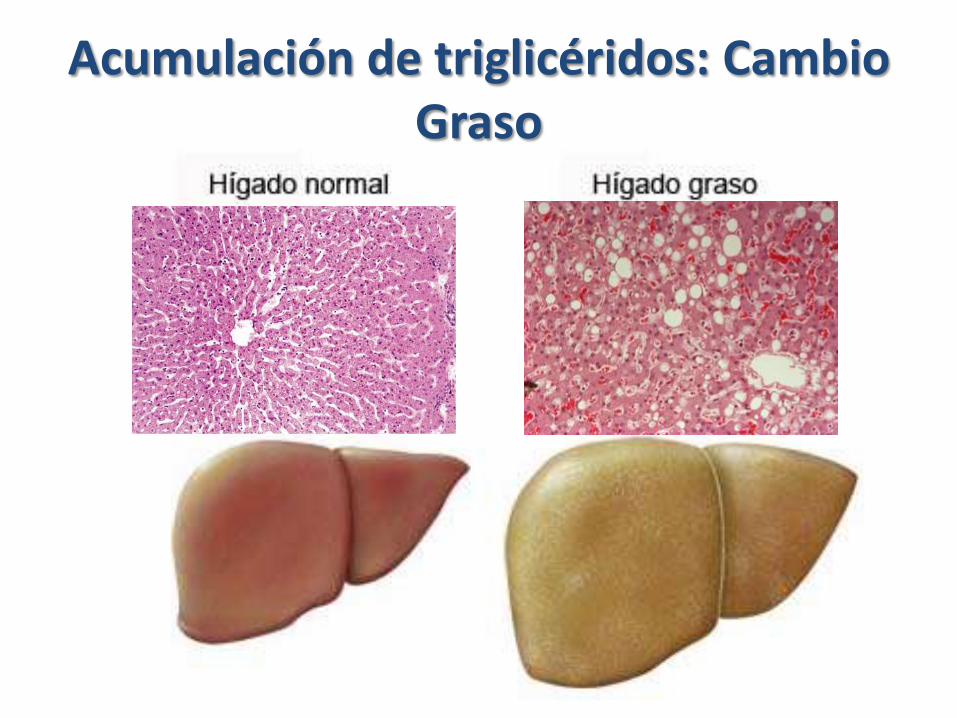
Acumulación de triglicéridos: Cambio Graso

Acumulación de triglicéridos: Cambio Graso
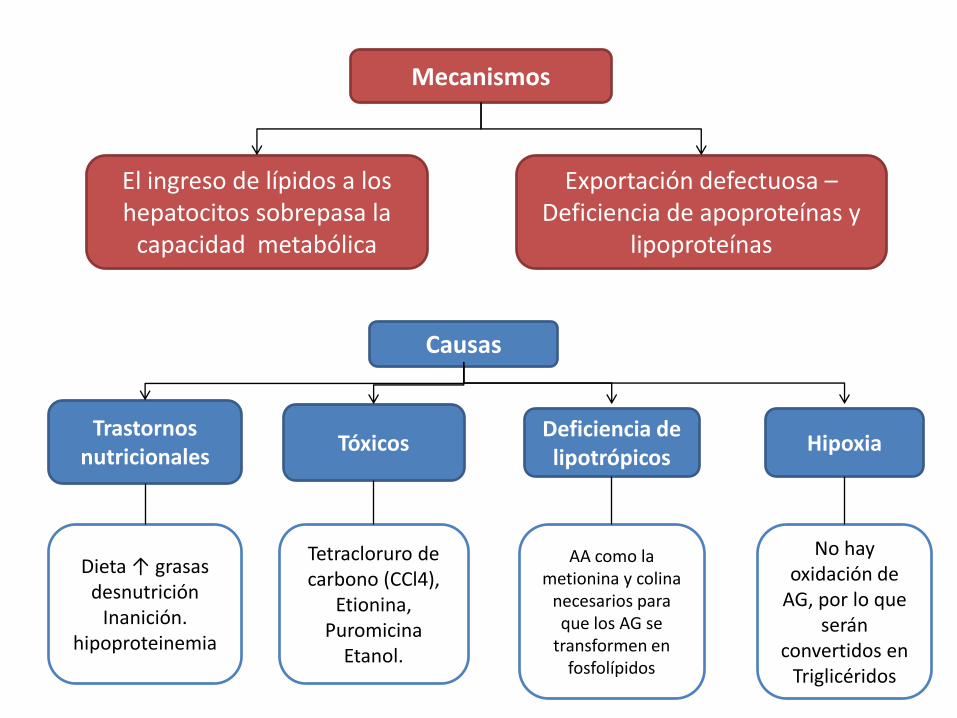
Mecanismos
El ingreso de lípidos a los hepatocitos sobrepasa la
capacidad metabólica
Exportación defectuosa –Deficiencia de apoproteínas y
lipoproteínas
Causas
Trastornos nutricionales
TóxicosDeficiencia de lipotrópicos
Hipoxia
Dieta ↑ grasasdesnutrición
Inanición. hipoproteinemia
Tetracloruro de carbono (CCl4),
Etionina, Puromicina
Etanol.
AA como la metionina y colina
necesarios para que los AG se
transformen en fosfolípidos
No hay oxidación de
AG, por lo que serán
convertidos en Triglicéridos
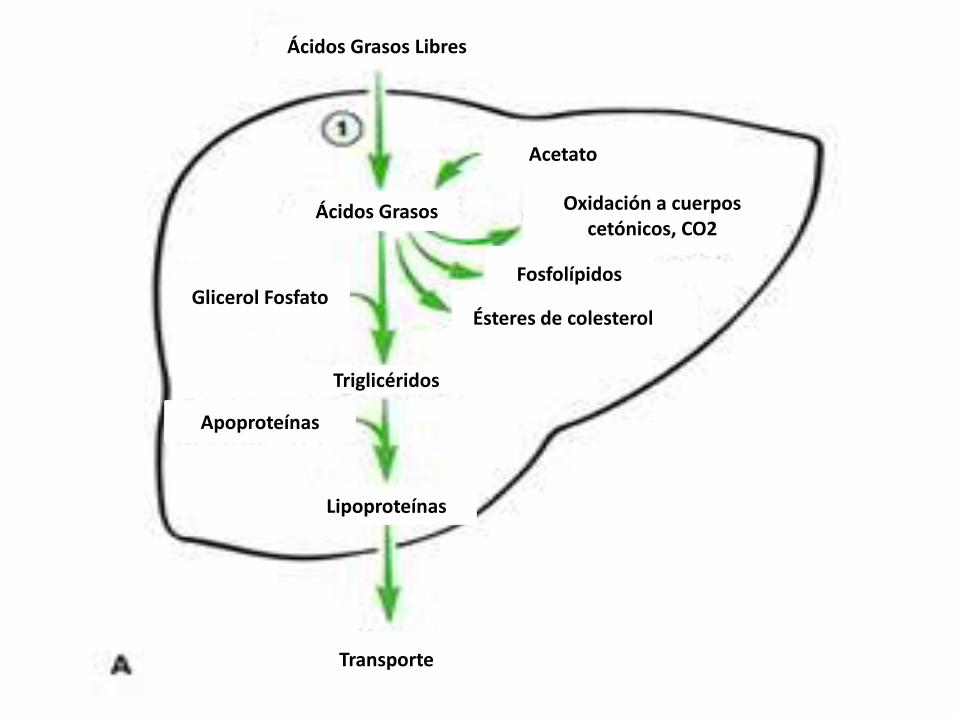
Acetato
Ácidos Grasos
Ácidos Grasos Libres
Oxidación a cuerpos cetónicos, CO2
Fosfolípidos
Ésteres de colesterol
Triglicéridos
Lipoproteínas
Glicerol Fosfato
Apoproteínas
Acetato
Transporte

Acumulación de triglicéridos: Cambio Graso
Consecuencias:• Alteración en la función de los hepatocitos,
principalmente disminuye la síntesis de albúmina y transaminasas.
• Atrofia o muerte de las células del parénquima hepático.
En los animales la principal causa es desnutrición, seguida por el daño hepatotóxico.En los humanos, las principales causas son el alcoholismo y la desnutrición.

Colesterol
Molécula que no puede ser destruida ni desdoblada dentro del organismo, únicamente puede ser eliminada a través del hígado, incorporándose a micelas que contienen bilis.
El colesterol puede ser de origen exógeno, a través de la ingesta de alimentos; o también puede ser endógeno producido en el hígado y circula unido a lipoproteínas.
Cuando el aporte es superior al requerido, o cuando existe una deficiencia en su metabolismo, puede ocurrir hipercolesterolemia, las células deben almacenar el colesterol excedente en forma de vacuolas intracitoplásmicas.

Proteínas
• Gotículas de reabsorción en los túbulos renales proximales.Nefropatías asociadas a proteinuria, por aumento en la reabsorción. Proceso reversible.
• Acumulación de proteínas del citoesqueleto.Filamentos de queratina (Cells epiteliales)Neurofilamentos (Neuronas)Filamentos de desmina (Músculo)Filamentos de vimentina (Tejido Conjuntivo)Filamentos gliales (astrocitos)
Alzheimer está constituido por neurofilamentos y otras proteínas.

Proteínas
• Agregación de proteínas anormales. Las proteínas anormales o plegadas de forma errónea pueden depositarse en los tejidos e interferir con la función normal. Los depósitos pueden ser intracell, extracell. - Priones
– Amiloidosis: Material proteínaceo hialino que se deposita en las paredes de los vasos y extracelularmente en el glomérulo. Principalmente por discrasias de las células plasmáticas
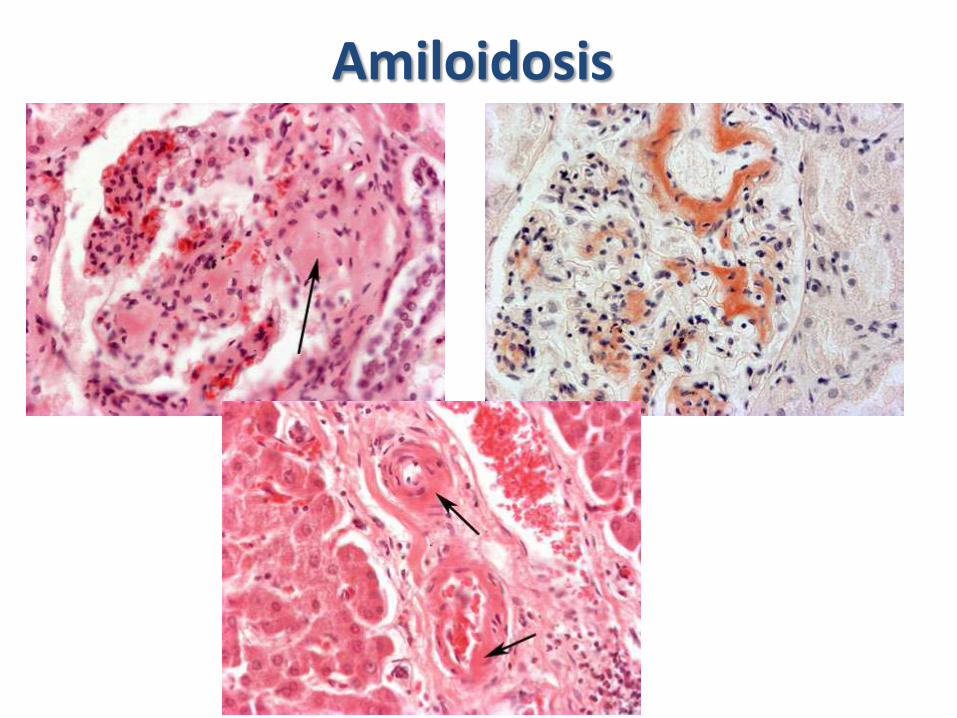
Amiloidosis
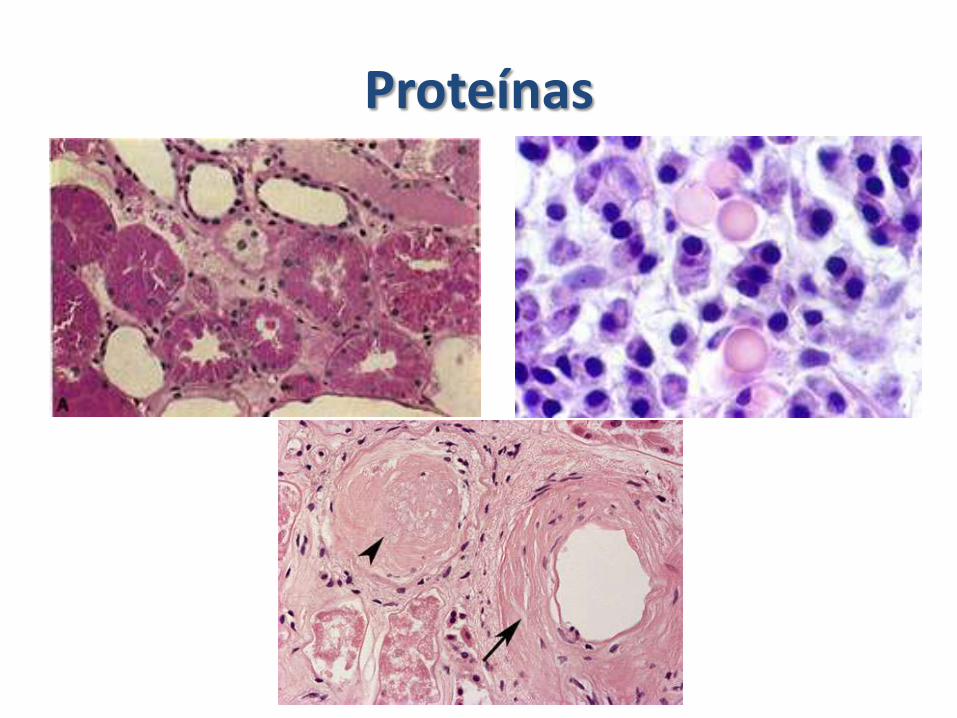
Proteínas

Acumulación de Glucógeno
• Los carbohidratos viajan en la sangre en forma de glucosa (dextrosa) y son almacenados en el hígado y músculo en forma de glucógeno, que constituye una reserva de energía.
• La causa más frecuente es la hiperglucemia que se presenta en la diabetes mellitus.

Acumulación de Glucógeno
Macroscópicamente no se observa ningún cambio en los órganos con esta alteración.
Aspecto microscópico: El glucógeno se observa como vacuolas claras dentro del citoplasma; para identificarlo la tinción de elección es el Carmín de Best, aunque también puede emplearse el ácido peryódico de Schiff (PAS).

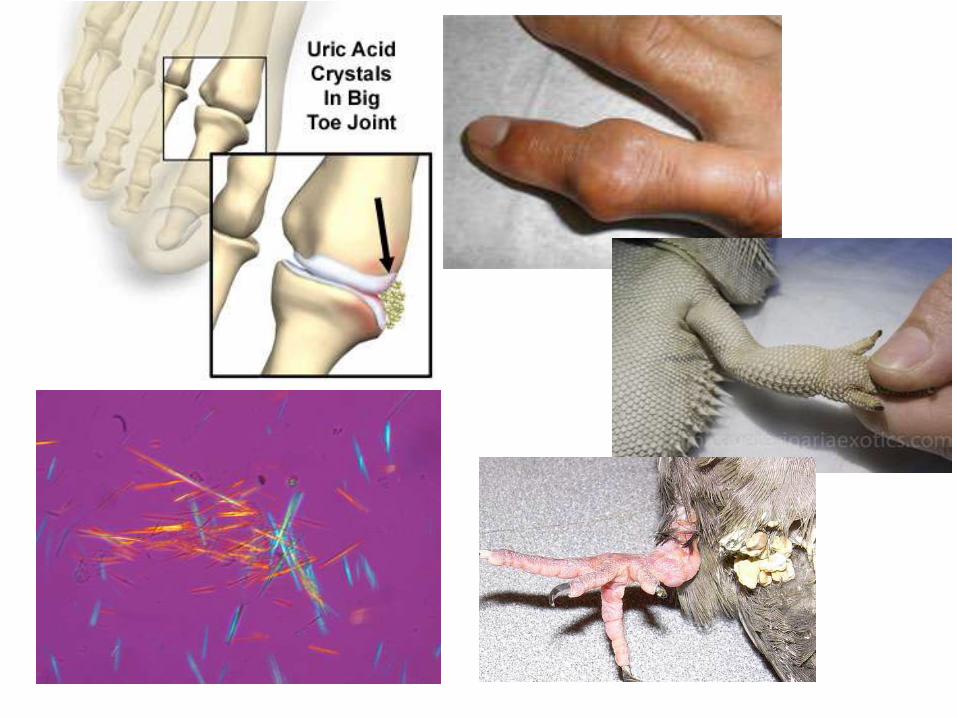
Depósito de uratos
El depósito de uratos y uratosis se da cuando hay un aumento de ácido úrico sanguíneo (hiperuricemia).
Es producto del catabolismo de las bases púricas o purinas (adenina y guanina) en las aves, reptiles, primates y perros dálmata; el resto de los mamíferos poseen la enzima urato-oxidasa, que hidroliza el ácido úrico hasta convertirlo en alantoína y así se elimina por la orina.

Depósito de uratos
Aumento en la ingesta de alimentos ricos en nucleoproteínasDaño en la función renal que impida la excreción de ácido úrico
Favorecen el depósito de cristales de ácido úrico en membranas serosas, túbulos renales y uréteres, en las superficies articulares y tejido periarticular, (gota artrítica); dichos cristales producen irritación, inducen reacción inflamatoria y también pueden producir reacción a cuerpo extraño.

Depósito de uratos
• Aspecto macroscópico: en su forma visceral, se aprecia como una delgada capa granular, gris brillante, en la superficie de serosas. En su presentación articular, se observan nodulaciones con aspecto de yeso, también puede haber úlceras e intenso dolor en las articulaciones interfalángicas, metacarpianas y metatarsianas.
• Aspecto microscópico: Se observan pequeños cristales en forma de agujas birrefringentes, rodeados por neutrófilos, macrófagos y células gigantes de cuerpo extraño.

Pigmentos Exógenos
Carbón: Mas frecuente - AntracosisCaptado por los macrófagos alveolares y se transporta por canales linfáticos regionales de la región traqueo bronquial.
Carotenoides: Pigmentos de origen vegetal, se encuentran ppalmente en la zanahorias. Suelen emplearse como aditivos en los alimentos para aves. Se depositan, principalmente en las células epiteliales, el tejido adiposo, las glándulas adrenales, el cuerpo lúteo, el epitelio testicular y la yema del huevo.
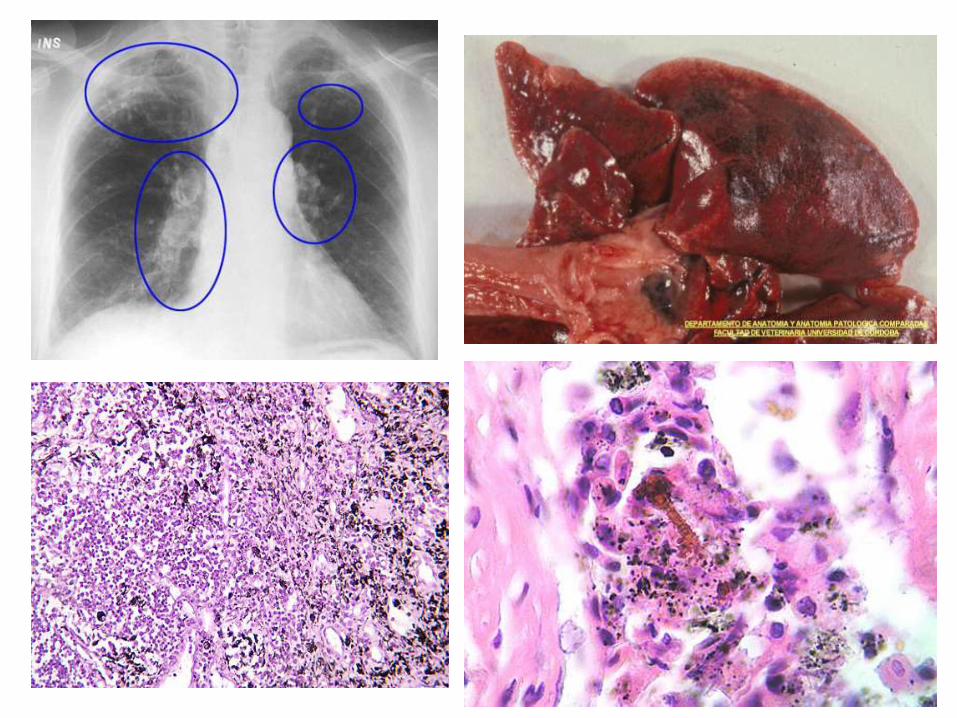

Pigmentos Exógenos
• Sílice (silicosis):
Se produce por inhalación crónica de dióxido de sílice. Quienes están más expuestos son los mineros, los pulidores de mármol y quienes trabajan con granito y cuarzo, por lo que se considera un padecimiento de tipo ocupacional.
• Asbesto (asbestosis)
• Hierro (siderosis)

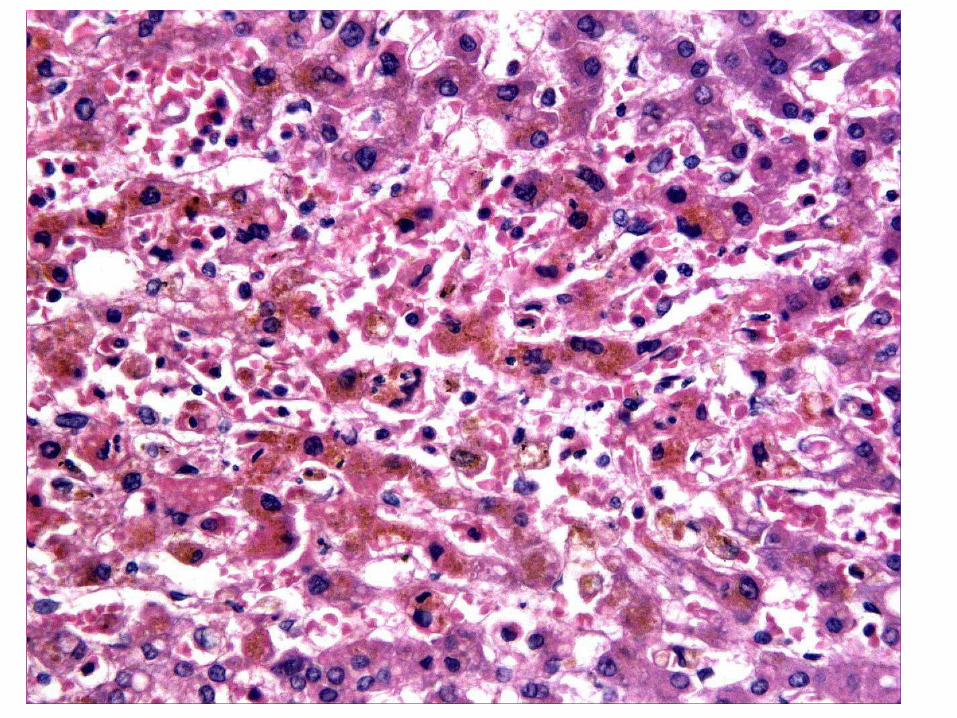
Pigmentos Endógenos
• La lipofuscina: pigmento de envejecimiento. Signo de lesiones por radicales libres o peroxidación lipídica, aparece como un pigmento citoplasmático pardo amarillento finamente granular y con frecuencia perinuclear, residuos insolubles e indigestibles de organelos que la propia célula ha autofagocitado.
En pacientes envejecidos, con malnutrición grave o caquexia tumoral.

Melanina
Pigmento negro parduzco, producido por los melanocitos.


Efélides
Melanosis
Léntigo
Aumento Disminución
Vitiligo
Albinismo
Nevus
Melanina

Efélides
Efélides
Léntigo

Melanosis

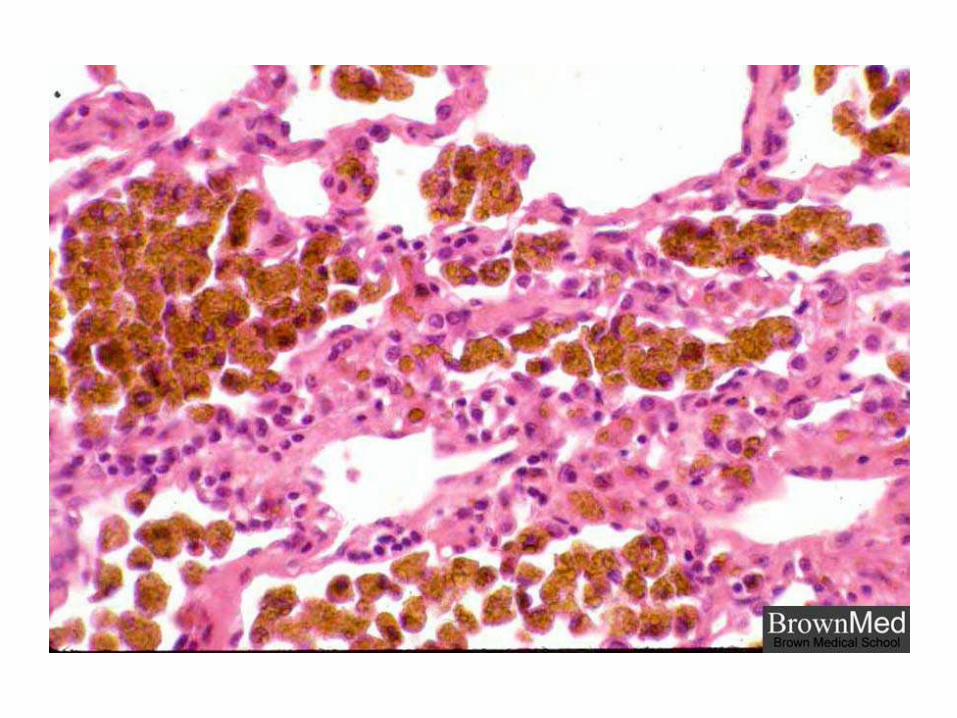
Hemosiderina
Pigmento cristalino o granular, amarillento o pardo, derivado de la hemoglobina, es una de las principales formas de depósito de hierro.
Se observa en las células que se encargan de la degradación de eritrocitos y en Hemorragias

CALCIFICACIONES PATOLÓGICAS
• Depósito de sales de Ca+.
• Se produce lesión irreversible en la célula.
Tipos de sales:
Fosfato cálcico.
Carbonato de calcio.
Oxalato de calcio.
Pirofosfatos de calcio.
• Están acompañadas de hierro, magnesio y otras sales minerales.

TIPOS DE CALCIFICACIONES
-Calcificación distrófica
-Calcificación metastásica
-Calcinosis
-Osteopatías metabólicas
- Litiasis

Calcificación Distrófica
Siempre es local, en tejidos que han sufrido daño o necrosis, los niveles plasmáticos de calcio son normales.
Es uno de los mecanismos con los que el organismo desecha o delimita el tejido muerto, volviéndolo inerte desde un punto de vista funcional, los fosfatos de calcio tienen afinidad con proteínas desnaturalizadas, y fosfolípidos de las membranas celulares.
Los depósitos de calcio son relativamente permanentes y dependiendo de su localización y pueden interferir en la función de los tejidos.


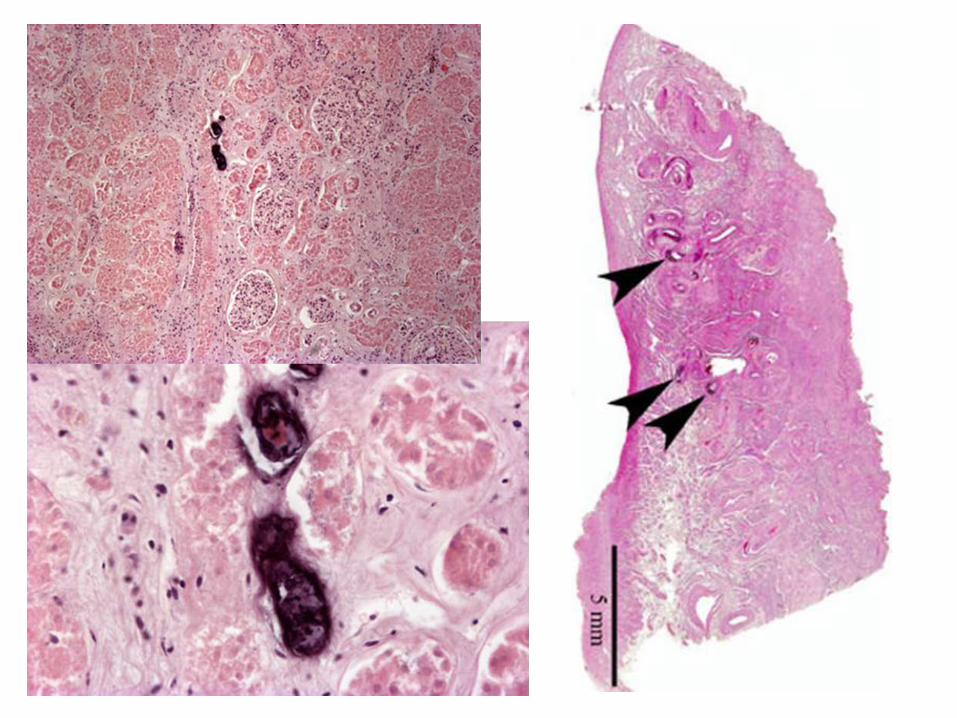
Calcificación Metastásica
Sucede en tejidos que no presentan daño previo, la condición es que el animal presente hipercalcemia prolongada asociada a varios factores:
Aumento en la secreción de hormona paratirodea (PTH) –hiperparatiroidismo
Destrucción del tejido óseo, secundaria a tumores medulares primarios o metástasis esqueléticas difusas, recambio óseo acelerado o inmovilización
Trastornos relacionados con la vitamina D

CALCINOSIS
• Depósitos de sales de Ca (carbonatos)
dermis, tej subcutáneo, músculo, fascias
• CALCINOSIS CIRCUNSCRITA: Calcinosis cutis
• CALCINOSIS UNIVERSAL

CALCINOSIS (“calcinosis cutis”)

Osteopatías metabólicas
- -Raquitismo
-Osteomalacia
-Osteodistrofia fibrosa

LITIASIS
• Formación de masas sólidas de aspecto ycomposición variables en el interior de conductosexcretores.
• Urolitiasis.
• Colelitiasis.
• Sialolitiasis.
• Pancreolitiasis.
• Enterolitiasis.

Inclusiones
• Son estructuras que pueden observarse en el citoplasma o el núcleo de las células, pueden tener diversos orígenes: cúmulos de proteínas, restos de membranas o de otras células, partículas de metales pesados, parásitos intracelulares o bien partículas virales. Estas inclusiones son de gran valor diagnóstico para el patólogo.

Cuerpos de inclusión de origen viral
• Son restos de proteínas de virus, Desafortunadamente no todos los virus dan lugar a cuerpos de inclusión y cuando lo hacen, sólo son visibles durante ciertas fases de la infección.
• Intranucleares o intracitoplasmáticos


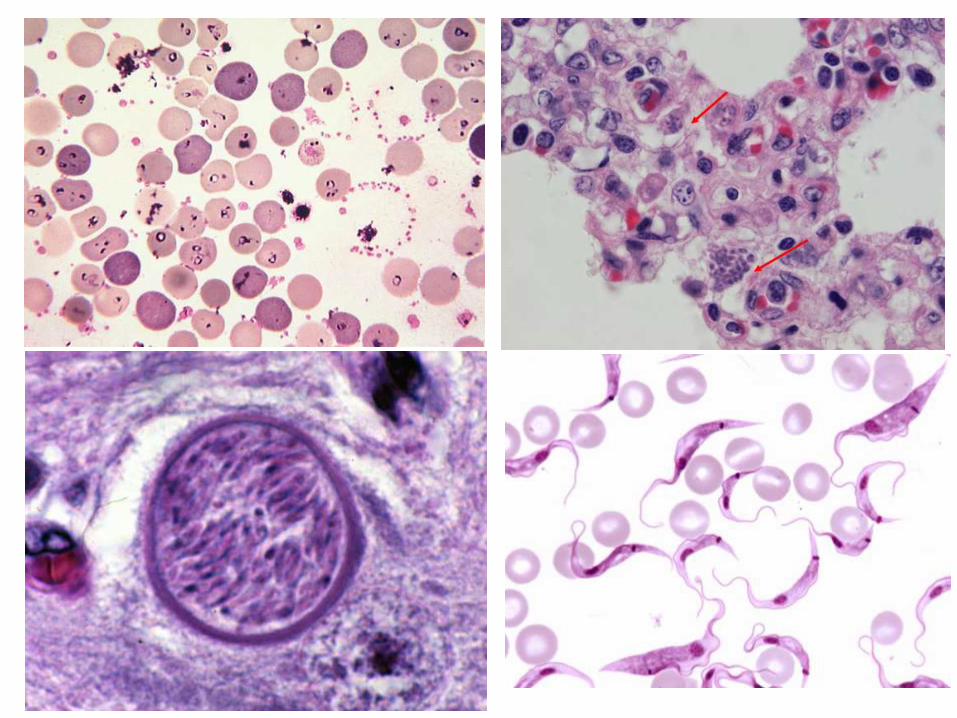
Cuerpos de Inclusión - Infecciones
• Cuerpos elementales intracitoplásmicos, por infección por Chlamydia spp, e.
• Protozoarios intracelulares de los géneros: Toxoplasma, Trypanosoma, Eimeria, Isospora, Neospora, Babesia,
• Anaplasma, Haemobartonella, Leishmania, etc.
• Bacterias intracelulares como las de los géneros Mycobacterium y Brucella.

Gracias….