ASEGURAMIENTO SANITARIO REGISTROS …€¦ · 1. OBJETIVO Dar a conocer el ... 01 Fecha de...
41
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES ASOCIADOS GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN LA MODALIDAD DE FABRICAR Y VENDER Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 1 de 41 ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos 1. OBJETIVO Dar a conocer el procedimiento seguido para otorgar el Registro Sanitario de Medicamentos, en la Modalidad de Fabricar y Vender, ya sea por primera vez o en el caso de las renovaciones. 2. ALCANCE Dirigido al grupo de Registros Sanitarios de Medicamentos y Productos Biológicos de la Dirección de Medicamentos y Productos Biológicos 3. RESPONSABILIDAD Es responsabilidad de las personas a quienes está dirigido el presente documento conocerlo y cumplirlo, y de la coordinación del grupo de verificar su cumplimiento, y de los directivos del Instituto suministrar las herramientas bibliográficas, técnicas y logísticas para consulta y aplicación de los conocimientos por parte de los evaluadores. 4. DOCUMENTOS DE REFERENCIA Procedimiento registro sanitario nuevo o renovación con estudio previo. ASS-RSA-PR001 Ver Matriz de Requisitos Legales y Otros Ver Requisitos Normas del Sistema de Gestión Integrado 5. RECURSOS PARA EL DESARROLLO Y EJECUCIÓN DE LA VALORACIÓN TÉCNICA Y LEGAL 5.1. APLICATIVO DE REGISTRO SANITARIO Este aplicativo tiene como objeto el apoyo, generación, gestión y trazabilidad de los trámites que son radicados ante la Dirección de Medicamentos y Productos Biológicos. Para el ingreso al aplicativo, el coordinador del Grupo de medicamentos debe solicitar la creación del usuario a la Oficina de tecnologías de la información. Creado el usuario, el funcionario responsable debe cambiar la contraseña y con esto queda habilitado para el ingreso. Si ya posee usuario del aplicativo de registro sanitario puede ingresar directamente a él de la siguiente manera: 1. Ingresar con el usuario y la contraseña asignadas.
Transcript of ASEGURAMIENTO SANITARIO REGISTROS …€¦ · 1. OBJETIVO Dar a conocer el ... 01 Fecha de...

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 1 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
1. OBJETIVO
Dar a conocer el procedimiento seguido para otorgar el Registro Sanitario de Medicamentos, en la
Modalidad de Fabricar y Vender, ya sea por primera vez o en el caso de las renovaciones.
2. ALCANCE
Dirigido al grupo de Registros Sanitarios de Medicamentos y Productos Biológicos de la Dirección de Medicamentos y Productos Biológicos
3. RESPONSABILIDAD
Es responsabilidad de las personas a quienes está dirigido el presente documento conocerlo y cumplirlo, y de la coordinación del grupo de verificar su cumplimiento, y de los directivos del Instituto suministrar las herramientas bibliográficas, técnicas y logísticas para consulta y aplicación de los conocimientos por parte de los evaluadores.
4. DOCUMENTOS DE REFERENCIA
Procedimiento registro sanitario nuevo o renovación con estudio previo. ASS-RSA-PR001 Ver Matriz de Requisitos Legales y Otros Ver Requisitos Normas del Sistema de Gestión Integrado
5. RECURSOS PARA EL DESARROLLO Y EJECUCIÓN DE LA VALORACIÓN TÉCNICA Y LEGAL
5.1. APLICATIVO DE REGISTRO SANITARIO
Este aplicativo tiene como objeto el apoyo, generación, gestión y trazabilidad de los trámites que son radicados ante la Dirección de Medicamentos y Productos Biológicos. Para el ingreso al aplicativo, el coordinador del Grupo de medicamentos debe solicitar la creación del usuario a la Oficina de tecnologías de la información. Creado el usuario, el funcionario responsable debe cambiar la contraseña y con esto queda habilitado para el ingreso. Si ya posee usuario del aplicativo de registro sanitario puede ingresar directamente a él de la siguiente manera:
1. Ingresar con el usuario y la contraseña asignadas.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 2 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
2. Ingresar a la opción registro sanitario y desplegar la lista de opciones. Elegir área de trabajo
3. En el área de trabajo se presentan los trámites asignados divididos en carpetas por tipos de trámites y en cada carpeta van presentados por orden de radicación, respetando el derecho de turno. Las carpetas son: Autorizaciones, Certificaciones, Modificaciones, R.S. Nuevo, Renovaciones revisiones de Oficio, Correcciones, Otros trámites (desgloses, cancelaciones, etc.) La visualización de la pantalla es la siguiente:
4. Desplegar las carpetas en las cuales va a realizar el estudio, allí aparecerá un listado que relaciona el expediente y el número de radicado, con el historial de los pasos que ha recorrido el trámite, desde el momento de la radicación hasta la asignación en la pantalla con las fechas en que estos pasos se realizaron. Identificar el trámite que tenga la fecha más antigua.
5.2. APLICATIVO DE VISUALIZACIÓN DE EXPEDIENTES ELECTRÓNICOS:
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 3 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
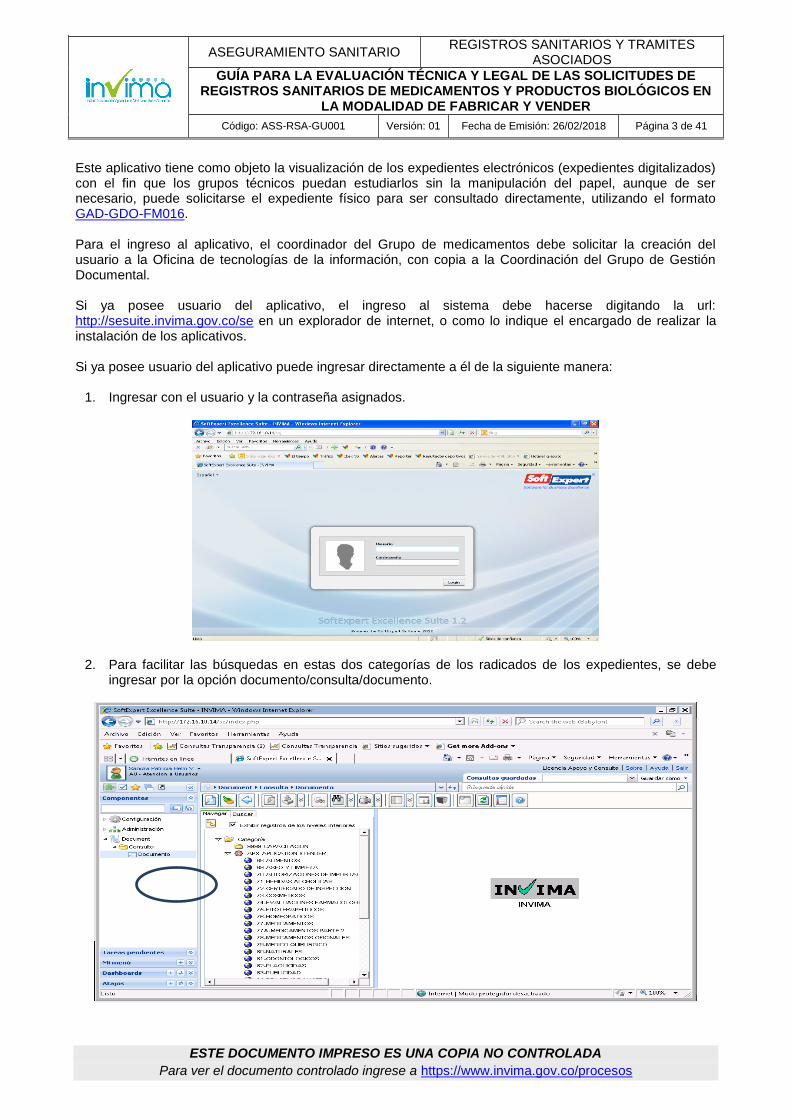
Este aplicativo tiene como objeto la visualización de los expedientes electrónicos (expedientes digitalizados) con el fin que los grupos técnicos puedan estudiarlos sin la manipulación del papel, aunque de ser necesario, puede solicitarse el expediente físico para ser consultado directamente, utilizando el formato GAD-GDO-FM016. Para el ingreso al aplicativo, el coordinador del Grupo de medicamentos debe solicitar la creación del usuario a la Oficina de tecnologías de la información, con copia a la Coordinación del Grupo de Gestión Documental. Si ya posee usuario del aplicativo, el ingreso al sistema debe hacerse digitando la url: http://sesuite.invima.gov.co/se en un explorador de internet, o como lo indique el encargado de realizar la instalación de los aplicativos. Si ya posee usuario del aplicativo puede ingresar directamente a él de la siguiente manera:
1. Ingresar con el usuario y la contraseña asignados.
2. Para facilitar las búsquedas en estas dos categorías de los radicados de los expedientes, se debe ingresar por la opción documento/consulta/documento.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 4 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
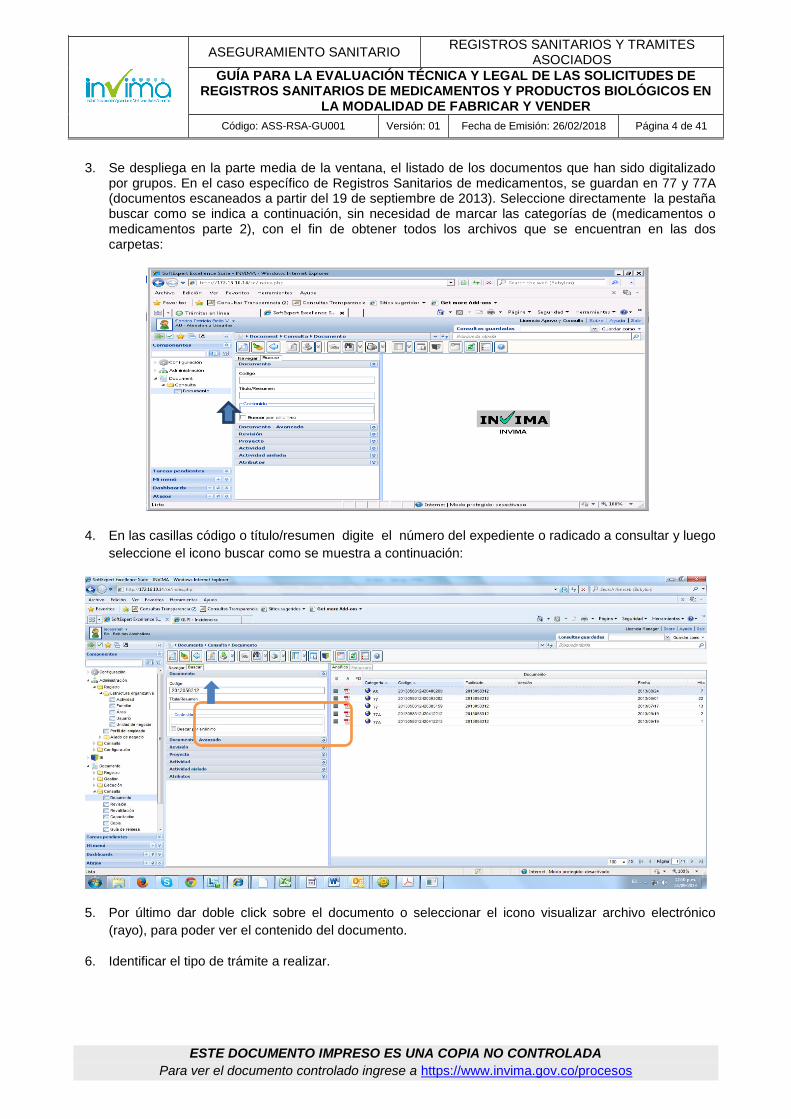
3. Se despliega en la parte media de la ventana, el listado de los documentos que han sido digitalizado por grupos. En el caso específico de Registros Sanitarios de medicamentos, se guardan en 77 y 77A (documentos escaneados a partir del 19 de septiembre de 2013). Seleccione directamente la pestaña buscar como se indica a continuación, sin necesidad de marcar las categorías de (medicamentos o medicamentos parte 2), con el fin de obtener todos los archivos que se encuentran en las dos carpetas:
4. En las casillas código o título/resumen digite el número del expediente o radicado a consultar y luego
seleccione el icono buscar como se muestra a continuación:
5. Por último dar doble click sobre el documento o seleccionar el icono visualizar archivo electrónico
(rayo), para poder ver el contenido del documento.
6. Identificar el tipo de trámite a realizar.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 5 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
5.3. DILIGENCIAMIENTO DE FORMATO PARA INFORME DE EVALUACIÓN (FIE):
El FIE es una herramienta para evaluar la calidad de la información enviada por el interesado, que se basa en algunas listas de chequeo, cuya finalidad tiene llevar al evaluador por una serie de documentos que debe evaluar con ciertos aspectos claves. Al final de cada requisito evaluado, deberá plasmarse el concepto del profesional que evalúa el trámite, conforme a su criterio y experiencia. Es importante señalar que cada evaluador debe dejar expreso 1. Qué evalúa: Esto debe ser coherente con los requisitos de ley y los requisitos publicados en la web del Instituto para los usuarios. 2. Qué conceptúa respecto a lo evaluado y 3. Qué decisión toma ante el trámite evaluado. Adicionalmente cada evaluador debe firmar con su nombre como responsable del informe de evaluación emitido y lo entrega diligenciado y firmado en archivo PDF, al Coordinador o delegado, quien lo cargará en un aplicativo para que se guarde automáticamente en expediente digital que se puede visualizar en el aplicativo establecido para ello. Los objetivos de la implementación de los formatos para informe de evaluación son: a. Facilitar la unificación de criterios. b. Incrementar la formación de criterios y facilitar los procesos de inducción en el nuevo personal. c. Facilitar evaluación por pares. d. Garantizar el derecho del usuario a que se le evalúen todos los requisitos. e. Establecer trazabilidad en las decisiones de los evaluadores. f. Dejar evidencia del estudio del trámite conforme al sistema de Gestión de calidad del Instituto.
En el caso de la evaluación de un Registro Sanitario nuevo o renovación, debe remitirse a los formatos:
Formato para informe de evaluación técnica Registro Sanitario Medicamentos Fabricación Nacional ASS-RSA-FM011
El Informe debe evidenciar claramente la evaluación de todos y cada uno de los requisitos de ley para el
trámite, debe plasmar los conceptos técnico científicos y farmacéuticos emitidos por el evaluador en cada
punto y debe reflejar claramente que decisión toma el evaluador, respecto a la calidad de la información
enviada por el solicitante.
Al final cada informe debe quedar debidamente firmado por el evaluador correspondiente.
Los Formatos para Informe de Evaluación (FIE), recopilan la evaluación, y existen herramientas para cada uno de las partes, técnicas y legales. El informe de evaluación es una herramienta digital que sigue el flujo de relacionado en el diagrama 1.
Diagrama 1. Flujo del Formato de Informe de Evaluación

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 6 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Para diligenciar el FIE remítase al Instructivo para el manejo del formato de informe de evaluación en el estudio de registros sanitarios de medicamentos y trámites asociados, ASS-RSA-IN013 Para el diligenciamiento del formato debe tener en cuenta las siguientes observaciones:
1. R = requerimiento. A = Aprobación. F.F = formas farmacéuticas. P.A = principio activo, PC = presentaciones comerciales FIE = Formato para Informe de Evaluación. NA = no aplica NR = No reporta. 2. Si para cualquier punto se genera una consulta, a otra dependencia colocar el radicado(s) de la(s) consulta(s) realizada por correspondencia _____________________________ y la fecha de la misma (s):________________________ 3. Si para cualquier punto se genera un requerimiento diligenciar la casilla de resumen de requerimiento, las casillas están dispuestas de tal manera que se amplían de acuerdo al texto escrito, si el diligenciamiento es manual cada evaluador debe dar los espacios que considere necesarios antes de la impresión. 4. En caso de aprobación diligenciar la casilla de aprobación 5. Si alguno de los numerales no aplican, favor indicarlo con NA y dejarlo en letra número 6 y sombrearlo en gris claro. 6. Si diligencia el formato en computador escriba sus apreciaciones o respuestas en cursiva para diferenciarlas del formato. 7. Si en el estudio se genera una resolución de aprobación, debe continuar con la generación del
documento en PDF, y seguir con los pasos establecidos en el Diagrama No. 1. 8. Recuerde la finalidad de este formato (ASS-RSA-FM011) es emplearlo como una herramienta para
evaluar la calidad de la información enviada por el interesado, más allá de una simple lista de chequeo, por tanto el concepto técnico/legal siempre debe ser diligenciado.
9. Si de los hallazgos encontrados el evaluador genera un requerimiento, debe continuar con la
generación del documento en PDF, y seguir con los pasos establecidos en el Diagrama No. 1. Una vez llegue la respuesta de auto a la pantalla del evaluador, éste deberá diligenciar el formato para informe de evaluación respuesta a requerimiento o evaluación de recurso de reposición medicamentos ASS-RSA-FM035, siguiendo el procedimiento descrito anteriormente.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 7 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
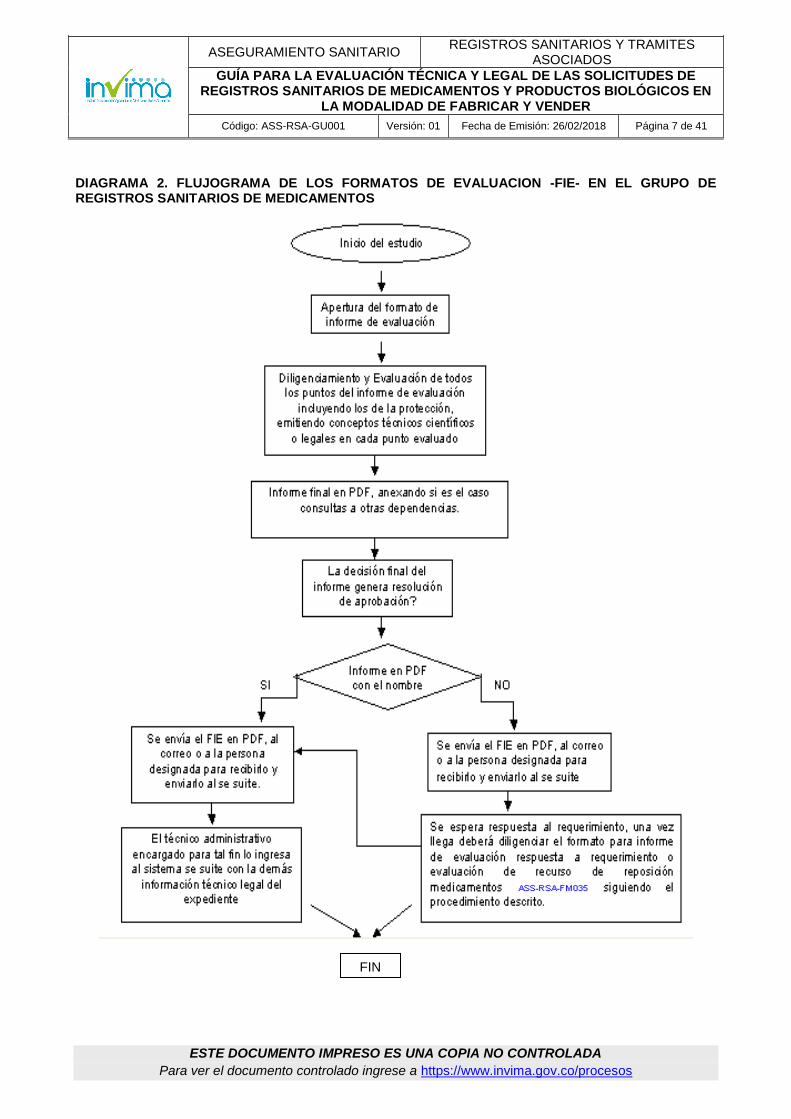
DIAGRAMA 2. FLUJOGRAMA DE LOS FORMATOS DE EVALUACION -FIE- EN EL GRUPO DE REGISTROS SANITARIOS DE MEDICAMENTOS
FIN

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 8 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
5.4. COMPLETAR DATOS
En el aplicativo de Registros Sanitarios, en el área de trabajo, completar datos del producto, llenando la base de datos a partir de la información radicada para el trámite correspondiente. 5.5. REALIZACIÓN DEL ACTO ADMINISTRATIVO
Ver lo establecido en el numeral 6.26 5.6. OFICIO A FONDO NACIONAL DE ESTUPEFACIENTES
Enviar oficio al Fondo Nacional de Estupefacientes cada vez que se otorgue un Registro Sanitario o Renovación a un medicamento de control especial monopolio del estado, en aplicación de la Resolución 1478 de 2006, Artículo 4.
6. DESARROLLO DE LA GUIA
En este paso, se inicia propiamente el análisis de la calidad de la información enviada por el interesado y para ello los evaluadores deben tener en cuenta todos los soportes técnico – científicos que reporta la literatura y las redes de información internacional, por ejemplo: reportes tanto nacionales como internacionales sobre alertas de medicamentos emitidos por las Agencias Nacionales Regulatorias (ANR): FDA, EMEA, ANVISA, ANMAT, OPS, OMS, etc., las normas farmacológicas vigentes, el manual de normas técnicas colombianas vigente, las farmacopeas oficiales en Colombia: la de Estados Unidos de América (USP), Británica (BP), Europea (EP) Alemana (DAB), etc., los criterios de grupo adoptados mediante actas, los conceptos emitidos por la Comisión Revisora, los conceptos emitidos por la oficina jurídica o cualquier dependencia del Invima, o del Ministerio de Salud y Protección Social (o quien haga sus veces).
Los criterios de grupo adoptados mediante actas resultantes de las reuniones del grupo de Registros
Sanitarios de Medicamentos y Productos Biológicos, los conceptos emitidos por la Sala Especializada de
Medicamentos y Productos Biológicos de Comisión Revisora, la Sala Especializada de Dispositivos Médicos
si es el caso, los conceptos emitidos por la Oficina Asesora Jurídica o cualquier dependencia del Invima, o
del Ministerio Salud y la Protección Social, son las herramientas de trabajo para evaluar el trámite en
estudio y es obligación estar actualizado. Así mismo, de ser necesario, el evaluador deberá elevar consultas escritas a otras dependencias tales como la Sala Especializada de Medicamentos y Productos Biológicos de Comisión Revisora, la Oficina de Laboratorios y Control de Calidad del Instituto, Grupo de BPM, Farmacovigilancia, etc., en aras de dar claridad y solventar cualquier duda sobre la calidad, coherencia e integralidad de la información que está evaluando. De igual forma, tiene el deber de solicitar recolección de muestras o solicitar y de ser posible realizar visitas en caso que el trámite evaluado lo amerite. En todo caso el evaluador no puede dejar pasar ninguna inconsistencia sin haber hecho lo correspondiente. Cuando se requiera realizar una consulta a la Sala Especializada de Medicamentos y Productos Biológicos, el profesional evaluador deberá seguir el INSTRUCTIVO PARA TOMA DE DECISIONES Y DIRECCIONAMIENTO DE TRAMITES A COMISIÓN REVISORA ASS-RSA-IN014 Si se trata de consultas a otras dependencias y/o grupos, se preferirá realizar formalmente una correspondencia, tomando el consecutivo de la Dirección de Medicamentos, inclusive, si se trata de consultas entre grupos, la cual puede firmar únicamente el coordinador, siguiendo el Instructivo para el manejo de la Comunicación al interior del grupo de Medicamentos.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 9 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Una vez la otra dependencia responde la consulta realizada, el profesional responsable estudia el concepto y decide, emitiendo el concepto de aprobado o emitiendo un requerimiento, como se indica en el numeral 7.26.2. Si de los grupos mencionados se recibe correspondencia que en su contenido afecta el estudio particular de los Registros Sanitarios, estos se darán a conocer al grupo mediante el Formato para Socialización de Información ASS-RSA-FM025 vinculado al Instructivo para el manejo de la comunicación al interior del grupo de medicamentos y Productos Biológicos ASS-RSA-IN015
También debe tener en cuenta las decisiones tomadas en las reuniones de unificación de criterios y que
quedan registradas en actas, las cuales se pueden consultar en la herramienta de Documentos Internos de
Trabajo del Grupo de Registros Sanitarios de Medicamentos y Productos Biológicos (Y:\Documentos de
Trabajo Grupo de Registros Sanitarios de Medicamentos y Productos Biológicos).
Si se hace necesaria la toma de muestra para análisis en el laboratorio del Invima, el evaluador podrá tomar muestras de medicamento, dando aplicación a lo establecido en el numeral 2º del Artículo 23 del Decreto 677 de 1995 o solicitando apoyo directo a la Dirección de Operaciones Sanitarias. Dando aplicación a lo establecido en el numeral dos del Artículo 23 del Decreto 677 de 1995, el evaluador realizará una visita a las instalaciones del fabricante y/o del encargado de realizar los estudios de estabilidad, para verificar lo que considere conveniente basado en la información recibida. Cuando se estudie un trámite que involucre la fabricación de un medicamento que requiera áreas especiales de manufactura o que se trate de un medicamento que contenga un principio activo de estrecho margen terapéutico, o formas farmacéuticas inyectables, el evaluador de la parte técnica podrá realizar visita e inspeccionar la validación de procesos de acuerdo INSTRUCTIVO DE VISITA A LABORATORIOS FARMACÉUTICOS DESDE EL GRUPO DE REGISTROS SANITARIOS DE MEDICAMENTOS DE LA DIRECCIÓN DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS ASS-RSA-IN009 Cuando se trate de productos nuevos de origen biológico o biotecnológico, como por ejemplo Hormonas peptídicas, Hemoderivados, Vacunas, entre otros, debe seguir la Guía de evaluación técnica y legal para concesión de registro sanitario nuevo para productos biológicos y biotecnológicos ASS-RSA-GU025 y diligenciar el Formato para informe de evaluación técnica - Registro sanitario productos biológicos ASS-RSA-FM014 6.1. INFORMACIÓN GENERAL DEL PRODUCTO
Se compone del nombre del producto, principio activo, forma farmacéutica, roles de las empresas registradas con su domicilio (titular, fabricante, envasador, acondicionador, importador, etc). Esta información se diligencia en el formato de informe de evaluación de los registros sanitarios nuevos o renovaciones y tiene la finalidad de resumir los parámetros a tener en cuenta durante la revisión. 6.2. INFORMACIÓN FARMACOLÓGICA
6.2.1. Verificación que el principio activo o combinación de principios activos, la concentración y la
forma farmacéutica se encuentre en normas farmacológicas y reportes internacionales. En este numeral, se debe revisar la inclusión en normas farmacológicas, en actas de la Sala Especializada de Medicamentos y Productos Biológicos de Comisión Revisora. Se sugiere la búsqueda de la información en los aplicativos disponibles para su uso: Actas de Comisión Revisora, las cuales se encuentra en el controlador de dominios y en dado caso la información publicada en la web por agencias sanitarias de referencia para Colombia, Ministerio de Salud y Protección social (o quien haga sus veces), bases de datos a las que se tenga acceso, OMS, entre otros. Verificar que el principio activo o combinación de principios activos, la concentración y la forma farmacéutica se encuentre en la norma farmacológica asignada en la evaluación farmacológica, o verificar si hubo alguna

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 10 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
actualización o manifestación de la Sala Especializada de Medicamentos y Productos Biológicos de Comisión Revisora respecto al producto o productos similares. En caso de no encontrarse en el mismo listado o incluido mediante acta, se requerirá al interesado mediante auto para que solicite el estudio de la evaluación farmacológica ante la Sala Especializada de Medicamentos y Productos Biológicos y que en la respuesta del requerimiento adjunte la evidencia de solicitud realizada a la Sala, (ésta última facilitará la trazabilidad al trámite de evaluación farmacológica internamente). 6.2.2. Llamado a revisión de oficio Verificar en la base de datos de las actas de comisión revisora si hay algún llamado a revisión de oficio para el principio activo relacionado con indicaciones, contraindicaciones, precauciones y advertencias. De ser así, hay dos opciones:
Que la evaluación farmacológica tenga una fecha de expedición anterior a la fecha del llamado a revisión de oficio, por lo cual, no hay lugar a una corrección y la información se incluirá en el registro sanitario dejando tres posibilidades para dar continuidad al trámite:
Llamado a revisión de oficio por indicaciones: se incluirá directamente en el registro sanitario
y sólo en el caso que no soliciten inserto o información para prescribir, no será necesario realizar requerimiento para que se incluyan las nuevas indicaciones.
Llamado a revisión de oficio por contraindicaciones: se realizará requerimiento para que se incluyan en los artes envase y empaque y de ser necesario en los insertos, información para prescribir, etc.
Llamado a revisión de oficio por seguridad y eficacia: se realizará requerimiento para que se allegue la documentación de seguridad y sea evaluado por la Sala Especializada.
Que la evaluación farmacológica tenga una fecha de expedición posterior a la fecha del llamado a revisión de oficio y la información no se encuentre contenida dentro de la evaluación farmacológica, debe emitirse un requerimiento en donde le solicite al interesado que envíe la corrección a la evaluación farmacológica y se realice la aclaración para continuar con el trámite.
6.2.3. Alertas internacionales Verificar las páginas web de otras agencias tales como EMA, FDA, OMS, entre otras, para saber si existen alertas farmacológicas de seguridad del medicamento. Si se encuentra una alerta emitida por estas agencias, se debe informar al grupo de farmacovigilancia al correo [email protected], el grupo se encargará de realizar la gestión de acuerdo con el instructivo para la gestión de la información de seguridad sobre medicamentos y productos biológicos. Para realizar un seguimiento adecuado y evidenciar la comunicación entre los dos grupos, se creó el formulario ubicado en la URL: goo.gl/IWi1ay. Allí, el grupo de Farmacovigilancia, se encargará de indicar la respuesta y el procedimiento a seguir, en caso dado que la consulta tenga que hacerse directamente a la Sala Especializada de Medicamentos y Productos Biológicos, desde el grupo de Registros Sanitarios. La notificación de esta respuesta de Farmacovigilancia, estará a cargo del coordinador del grupo de Registros Sanitarios de Medicamentos y Productos Biológicos o encargado, para indicar al evaluador qué procedimiento debe seguir. 6.2.4. Indicaciones y contraindicaciones del producto Hace referencia a la información para el principio activo de las indicaciones, contraindicaciones y advertencias, dosis y frecuencia de administración, condición de venta, condición del medicamento (esencial, controlado), vía de administración, ATC, entre otros aspectos farmacológicos que el interesado solicite, en donde se debe corroborar con la información aprobada para el producto en estudio y/o otros productos previamente aprobados.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 11 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Si el código de ATC no está incluido en el aplicativo de Registros, deberá seguir el Instructivo asignación del ATC a los medicamentos incluidos en la base de datos ASS-RSA-IN003, por favor diligencie el Formato para Reportar un ATC no Creado en el Aplicativo de Registros Sanitarios ASS-RSA-FM027 a través del link http://goo.gl/2Bvg2B e informe al responsable de la creación por correo electrónico, para que se pueda actualizar. El encargado, deberá seguir el Instructivo para el Ingreso de ATC que no se encuentre creado en la Base de Datos ASS-RSA-IN026 En este punto se debe tener en cuenta que coincidan, entre otros, los aspectos relacionados a continuación, informados en la solicitud en comparación con los aprobados en la evaluación farmacológica y dejar evidencia en el FIE de la información que se aprueba en cuanto a: Forma farmacéutica, Vía de administración, Dosis y frecuencia de la administración, Indicaciones farmacológicas y uso terapéutico, Contraindicaciones, Advertencias, Condición de venta y Efectos secundarios. 6.3. BIODISPONIBILIDAD Y/O EQUIVALENCIA TERAPÉUTICA
En este numeral se debe revisar si es necesario contar con estudios de biodisponibilidad y/o equivalencia,
de acuerdo con la normatividad vigente, siguiendo los lineamientos del Acta 19 de 2002, numeral 2.3.13,
Acta 05 de 2014, numeral 3.11.1 y demás normatividad y lineamientos relacionados. Se verifica
principalmente si están aprobados dichos estudios por la Sala Especializada de Medicamentos y Productos
Biológicos, en qué actas y el concepto de la Sala.
Si no se encuentra el acta, deberá requerirse y solicitar al interesado que envíe los Estudios
Farmacocinéticos o de Biodisponibilidad y Bioequivalencia a la Sala Especializada de Medicamentos y
Productos Biológicos de la Comisión Revisora para que sean aprobados y que se envíe una copia de la
solicitud para que se dé continuidad al trámite una vez se tenga un concepto.
Si el interesado allega los estudios requeridos y estos no han sido previamente revisados por la Comisión
Revisora, se enviará la consulta ante este organismo para que evalúen y emitan concepto respecto a la
información allegada.
Si el interesado allega los estudios requeridos y estos ya han sido previamente revisados por la Comisión
Revisora, se revisará las Actas de comisión Revisora publicadas verificando que efectivamente ya se ha
emitido un concepto sobre estos.
Si el medicamento objeto de estudio no está contemplado dentro de los grupos obligatorios para presentar
biodisponibilidad pero el interesado los allega, estos se enviarán para evaluación de la Comisión Revisora, a
no ser que el interesado específicamente declare prescindir de esta evaluación.
Todos los productos nuevos de origen biológico o biotecnológico deben presentar evaluación farmacológica
ante la Comisión Revisora. Si no es así, se procederá a rechazar la solicitud de registro sanitario.
6.4. PROTECCIÓN DE DATOS
Este es un numeral de carácter informativo, el cual indica si será necesario diligenciar el Formato para
informe de evaluación-registro sanitario con solicitud de protección decreto 2085 de 2002 ASS-RSA-FM009
conforme a la Guía de evaluación técnica y legal para concesión de registro sanitario con solicitud de
protección. Decreto 2085 de 2002 ASS-RSA-GU004.
6.5. EVALUACIÓN DE LA CARTA AVAL
La carta aval es una declaración del Director Técnico del laboratorio fabricante e implica que el conoce la
información y es idónea de acuerdo con la legislación vigente para la obtención de un registro sanitario, la
cual debe tener una fecha de expedición reciente y debe coincidir el Director Técnico con el reportado en la

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 12 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
base de datos de Invima. De ser necesario se debe indicar que partes del proceso se avalan: Fabricación,
envase, acondicionamiento y control de calidad.
6.6. EVALUACIÓN DE LOS CONTRATOS
Los contratos de fabricación, acondicionamiento, control de calidad, estudios de estabilidad, en donde se
deberá revisar las responsabilidades establecidas, los productos que cobija el contrato. Dependiendo de la
modalidad del Registro Sanitario, el FIE describe que tipo de contrato se debe revisar. Esta información es
muy importante, cuando el Invima requiere algún soporte por parte del interesado, cuando es necesario
realizar inspección, vigilancia y control o cuando se requiere tomar medidas sancionatorias.
6.6.1. Entre el titular y el laboratorio fabricante (únicamente para fabricar y vender) La evaluación farmacéutica de este documento debe enfocarse hacia los siguientes aspectos:
Debe indicar quién suministrará las materias primas, incluyendo materiales de envase y empaque, quién estará a cargo del análisis y liberación de las mismas.
Debe establecer quién será el responsable de suministrar los estándares primarios y secundarios o en su defecto, el Working Estándar.
Debe señalar quién se encargará de las materias primas, los productos a granel, intermedios y terminados, en caso de que sean rechazados.
El nombre del laboratorio fabricante referido en este documento debe coincidir con el señalado en la carta aval y en el certificado de BPM.
Debe coincidir la planta específica en caso de áreas especiales.
Debe incluir la responsabilidad sobre todo el proceso productivo o si fuera el caso deben haber contratos para los responsables de los procesos que serán contratados con otro laboratorio.
Debe especificar quién será el responsable de los análisis de producto en proceso y producto terminado y quién efectuará la respectiva toma de muestras.
Debe informar el producto sobre el cual se está contratando la fabricación.
Debe establecer la responsabilidad de la realización de los estudios de estabilidad acelerados y los estudios de estabilidad natural y los estudios de estabilidad de seguimiento “on going”.
Debe establecer la responsabilidad sobre las muestras de retención.
Debe establecer la responsabilidad del control de calidad del producto.
Debe establecer quién será el responsable de liberar el producto terminado al mercado.
La modificación de las responsabilidades o parte de ellas solo mediará con autorización previa del contratante y con el consecuente aviso al Invima como autoridad sanitaria.
6.6.2. Entre el titular y el laboratorio envasador y/o acondicionador La evaluación farmacéutica de este documento debe enfocarse hacia los siguientes aspectos:
El nombre del laboratorio envasador y/o acondicionador referido en este documento debe coincidir con el señalado en el certificado de BPM.
Debe coincidir la planta específica en caso de áreas especiales.
Debe señalar las actividades de envase y/o acondicionamiento específicos.
Debe informar el producto sobre el cual se está contratando el envase y/o acondicionamiento.
Debe establecer la responsabilidad de la realización de los estudios de estabilidad acelerados y los estudios de estabilidad natural.
Debe establecer la responsabilidad sobre las muestras de retención.
Debe establecer la responsabilidad del control de calidad del producto.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 13 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.6.3. Entre el titular y el laboratorio que realizará control de calidad y/o estudios de estabilidad Si los análisis los realiza un tercero, debe mediar un contrato escrito entre el titular y el laboratorio que abarque los análisis de productos y defina la responsabilidad de cada una de las partes, así, entre las responsabilidades definidas estarán:
El nombre del laboratorio envasador y/o acondicionador referido en este documento debe coincidir con el señalado en el certificado de BPM (cuando alleguen el certificado)
Debe informar el producto que sobre el cual se está contratando.
Debe establecer la responsabilidad de ambas partes: Tipo de análisis contratado, esto es, análisis de materias primas, productos en proceso, producto terminado, estudios de estabilidad acelerados, naturales, y/o estudios de estabilidad de seguimiento (“ongoing”).
Periodicidad de la evaluación de idoneidad del contratista por parte del contratante.
Ubicación de los certificados de análisis originales. 6.7. EVALUACIÓN DEL CERTIFICADO DE CUMPLIMIENTO DE LAS BPM
El laboratorio fabricante debe contar con certificado de Buenas Prácticas de Manufactura, así como cada uno
de los integrantes de la cadena de fabricación (envasadores, acondicionadores). El certificado siempre debe
indicar si el producto se realiza en un área común o requiere un área específica, si cuenta con áreas para la
fabricación de productos estériles, las formas farmacéuticas autorizadas, si se requieren áreas con cadena de
frío, entre otros.
Para todo laboratorio nacional, las BPM son certificadas por Invima y deberá cumplir con lo establecido en la
Resolución 3028 de 2008 en donde se definen las áreas técnicas de producción de los establecimientos
farmacéuticos y se establecen otras disposiciones.
Para los productos en la modalidad FABRICAR y VENDER, no es necesario que el interesado allegue el
certificado de BPM, por tanto deberá realizarse la consulta en la base de datos y/o realizar la consulta
directamente a los encargados del grupo técnico, para consultar los datos del certificado, de ninguna manera
será procedente realizar el requerimiento al interesado para que allegue el certificado emitido por Invima.
Verificar en el certificado, como mínimo la siguiente información:
a) Tipo de principio activo: Común, Corticosteroide, Antibiótico betalactámico, Cefalosporínico, etc.
b) Áreas de fabricación y Tipo de producto: Estéril o no estéril.
c) Forma(s) farmacéutica(s) autorizada(s).
d) Razón social y domicilio del fabricante.
e) Fecha de las inspecciones realizadas al fabricante por el Invima.
f) Número de la Resolución o Certificado de BPM y vigencia de las mismas.
g) Certificación de que las instalaciones industriales y las operaciones de fabricación se ajustan a las
Buenas Prácticas de Manufactura aceptadas en el país.
El documento debe encontrarse vigente al momento de la expedición del Registro Sanitario, lo cual se indica en
la parte considerativa de la Resolución con un párrafo de redacción similar a la siguiente:
Que mediante certificado o resolución de BPM No. XXXXXXXXX de dd/mm/aaaa expedida por
(ENTIDAD REGULATORIA QUE AVALA: INVIMA) se concedió certificación de Buenas Prácticas
de Manufactura - BPM al laboratorio fabricante XXXXXXXXXX (RAZÓN SOCIAL DEL
LABORATORIO FABRICANTE) con domicilio en XXXXXXXXXX (DIRECCIÓN), ubicada en
(CIUDAD, PAÍS), para fabricar en área de producto (ESTERIL / NO ESTERIL), los principios
activos (COMUNES/ BETALACTAMICOS / CON ÁREAS ESPECIALES), en formas farmacéuticas
XXXXXXXXX, con una vigencia hasta el dd/mm/aaaa. Por lo tanto, puede fabricar el producto de la

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 14 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
referencia.
Cuando se trate de productos de origen Biológico o Biotecnológico se deberá documentar el fabricante del
principio activo, del diluente (si aplica) y del producto terminado. En el certificado de fabricante del principio
activo no debe incluirse los productos en investigación.
En caso que el certificado de BPM no coincida, o no estén vigentes las Buenas Prácticas de Manufactura, se
deberá realizar un requerimiento de solicitud del certificado que coincida con lo indicado por la normatividad
vigente.
6.8. INFORMACIÓN QUÍMICA Y FÓRMULAS
Esta información hace referencia al principio activo, su estructura y la fórmula cualicuantitativa, y para su evaluación deberá tener en cuenta los siguientes aspectos:
Verificar que la composición cualicuantitativa sea expresada según lo que se establece en el literal c del
artículo 22 del decreto 677/1995. Se debe verificar que cada sustancia incluida en la fórmula del
medicamento esté identificada con nombre genérico y químico (IUPAC), y no con nombres de marca; para
el activo se escribirá su nombre según la Denominación Común Internacional (DCI).
Un resumen de la interpretación de la evidencia de la estructura e isomerismo, información tal como el
potencial de isomerismo, la identificación de estereoquímica o el potencial para formar polimorfos debe
estar incluida.
Cuando un principio activo es quiral, debe indicarse si se trata de un estéreo-isómero específico o una
mezcla de estéreo-isómeros que fueron utilizados en los estudios clínicos y no clínicos. Por lo tanto, si es
necesario, deberá compararse la información farmacológica allegada, principalmente en el caso de los
productos nuevos.
A continuación se señala la letra con la que se identifica la composición y será la completada en la base de
datos en la pestaña otros, en el campo “Concentr”:
A. Por unidad en formas de presentación dosificada en caso de tabletas, grageas, cápsulas,
óvulos, supositorios, inyectables y similares. B. Por cada 100 mL en caso de composiciones líquidas no inyectables. C. Por cada mililitro en líquidos para administración por gotas en inyectables en multidosis. D. Por cada 100 gramos en caso de productos presentados en forma de polvos, cremas,
ungüentos, geles, emulgeles, etc. E. Por gramos de polvo para reconstituir a 100 mL, en caso de polvos para reconstituir a solución o
suspensión. F. En porcentaje de peso o volumen en el caso de aerosoles.
Si la expresión de la fórmula cuali-cuantitativa no coincide, deberá emitirse un requerimiento señalando que debe corregirse y expresarse como lo indica el decreto, dado que será la formulación que figurará en la base de datos. Revisar que se presenten de forma separada los excipientes de los principios activos. El activo debe ser descrito con certeza indicando si corresponde a una sal, éster o solvato. Si es una sal, se deberá expresar su contenido como tal, o por el contrario aclarar si corresponde al equivalente de la forma ácida, básica o anhidra de la sustancia activa. En ningún caso los excipientes pueden tener actividad terapéutica. El incumplimiento de estos parámetros dará lugar a un requerimiento. Verificar que la sal, éster o solvato, sea la aprobada en normas farmacológicas, si no es así, deberá requerirse al interesado indicando que no es posible continuar con el estudio hasta que sea incluido en normas farmacológicas por la Sala Especializada de Medicamentos y Productos Biológicos

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 15 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
En la fórmula cuali-cuantitativa también se describen las cápsulas empleadas y los recubrimientos en caso de ser empleados. Si no se encuentran, deberá emitirse un requerimiento para que las incluyan. Verificar que no se infrinja el Artículo 78 del decreto 677/1995 parágrafo 1 que establece: “No se otorgará registro sanitario a medicamentos de igual composición, pero con diferente nombre, a favor de un mismo titular”, haciendo la respectiva búsqueda en la base de datos de registros sanitarios del Invima. 6.9. PRODUCTO TERMINADO
6.9.1. Formula del lote estandarizado La fórmula maestra o fórmula del lote estandarizado es la fórmula directamente proporcional a la fórmula cualitativa que ha sido estandarizada para los equipos, condiciones y áreas de producción por el laboratorio que fabrica un medicamento y que siendo repetida permite garantizar la reproducibilidad entre lotes y la eficacia y seguridad del medicamento. El interesado deberá indicar cuál es el lote estandarizado para la producción del producto, expresado bien sea en peso o volumen de granel o en unidades (tabletas, cápsulas, etc.). La fórmula del lote estandarizado debe ser coherente con la fórmula cuali-cuantitativa y si es necesario el empleo de excesos, éste debe ser justificado. En caso de presentar diferencias o no justificar los excesos, deberá requerirse al interesado para que corrija la fórmula o presente justificación de las diferencias. La fórmula del lote estandarizado debe coincidir con lo dispensado para cada materia prima en el batch record (registro de fabricación de los lotes).
Verificar que corresponda proporcionalmente a la del lote industrial proyectado y a la formulación por unidad
posológica (por ejemplo: Por tableta). No se aceptarán los excesos en el principio activo para compensar la
degradación en los estudios de estabilidad. Si el fabricante agrega excesos para compensar pérdidas en el
proceso productivo debe establecer claramente la magnitud de estos excesos y justificarlo debidamente. 6.9.2. Descripción del proceso de fabricación
En el caso de los productos de fabricación nacional, deberá revisarse la descripción detallada del proceso
de fabricación del medicamento, el cual debe estar en orden lógico y estar acorde con el principio activo,
excipientes y la forma farmacéutica producida. Además, debe ser coherente con lo contemplado en las
B.P.M. y con el Informe 32 respecto la calidad de la información. El procedimiento debe incluir:
Nombre completo del producto.
Descripción de la fórmula y tamaño del lote.
Lista de los materiales a ser utilizados y la cantidad de cada uno.
Las especificaciones esperadas para el producto final, con los límites de aceptación, y las de los productos intermedios cuando sea relevante.
Especificaciones de humedad y temperatura de las áreas cuando se requiera.
Descripción de los procesos, localización y descripción de los equipos a emplear.
El método o referencia de los métodos utilizados en la preparación crítica de los equipos. Ej. Limpieza (especialmente después del cambio de producto), calibración, esterilización, etc.
Descripción detallada paso a paso, de las instrucciones del proceso (chequeo de materiales, pre-tratamiento, secuencia de adición de materiales, tiempo de mezclas, temperaturas, etc.)
Las instrucciones para los controles en proceso, con sus respectivos límites.
Cuando sea necesario, los requisitos para almacenamiento del producto, incluyendo envase, etiquetado y las condiciones especiales de almacenamiento.
El control del medio ambiente o de los equipos pueden considerarse también parte del control en proceso, se verificará los registros en los respectivos formatos de los controles realizados al producto durante su

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 16 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
fabricación y envase. Los formatos especificarán nombre de producto, No. de lote, variable medida con sus respectivos límites, hora de toma de muestras, nombre del equipo empleado en la operación.
Los controles en proceso incluyen para sólidos dureza, peso, friabilidad, dimensiones, humedad, aspecto. Los anteriores se controlaran con una periodicidad basada en lo establecido por el fabricante.
Para líquidos y semisólidos se incluyen pH del producto, aspecto, viscosidad, olor y el completar a volumen final, estos ensayos se determinan solamente hasta cuando se observe el cumplimiento de la especificación, se debe hacer con periodicidad establecida por el interesado, en el envasado del producto.
6.9.3. Revisión de los Registros de Producción Los registros de producción o batch record son todos los documentos inherentes a la manufactura de un medicamento, por tanto, se identifican como el soporte del proceso de fabricación por eso los documentos deberán estar debidamente identificados, fechados, firmados indicando en forma clara la hora de inicio y finalización de cada proceso, los cuales deben tener los siguientes documentos así: Orden de producción, dispensación, orden envase y empaque, rótulos de limpieza de los equipos y áreas de trabajo, durante el proceso y en el momento que se lleva a cabo cada acción deben ser indicadas fechas y horas de inicio de las etapas intermedias y de la conclusión de la fabricación, el nombre de la persona responsable de cada etapa de producción, así como de a persona que verificó la operación, los controles en proceso efectuados (indicar quién los realizó y los resultados obtenidos), cantidad de producto obtenido en las diferentes etapas (rendimiento) y explicación si se han presentado desviaciones significativas de lo esperado, notas de los problemas presentados y firma de la persona que autoriza toda desviación de la fórmula maestra. De igual forma, se debe indicar el número de lote y/o número de análisis de control y las cantidades dispensadas de cada materia prima. Se debe incluir el registro de envasado de lotes, en el cual se debe registrar durante la operación, horas de operación de envasado, indicarse el nombre de la persona responsable, cantidad de producto que se espera obtener, la cantidad real obtenida y la conciliación. Tenga en cuenta que un registro sanitario nuevo puede solicitarse con los documentos correspondientes al lote piloto, el cual es fabricado con cantidad directamente proporcional a la fórmula cualitativa o composición básica que le confiere las características principales al producto, usando equipos y condiciones similares (tiempos mezcla, temperatura, humedad, velocidad equipo agitador/bombo, etc.) que permitan su reproducibilidad a escala industrial, conservando las especificaciones de calidad. La información correspondiente a los registros de producción se debe presentar para la solicitud de:
Registro Sanitario para modalidad fabricar y vender.
Registro Sanitario para modalidad fabricar y exportar.
Toda modificación de un Registro Sanitario en la modalidad fabricar y vender o fabricar y exportar que implique cambio de la fórmula cualicuantitativa.
Los documentos que conforman la historia de un lote de producción son: a) Orden de producción, la cual debe contener:
Nombre o logo de laboratorio fabricante.
Código interno o de identificación del producto asignado por la compañía.
Nombre del producto y forma farmacéutica.
Tamaño lote de fabricación (peso, volumen, número de unidades)
Número de lote, Número de orden producción, fecha de emisión.
Firma y fecha de recibo por producción.
Cantidad obtenida en producción.
Lista de materias primas a usar indicando código, nombre genérico, cantidad con unidades, número de entrada almacén o número de análisis o control.
Debe ser directamente proporcional a la fórmula cualicuantitativa propuesta.
Listado de material de envase e insumos a usar.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 17 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Firma y fecha de dispensación.
Firma y fecha de recibo por producción.
Cantidad obtenida en producción. b) Rótulos de pesadas, las cuales deben contener:
Nombre o logo de laboratorio que fabrica o pesa.
Nombre del producto y forma farmacéutica.
Número de lote del producto y número de orden producción.
Nombre y código de la materia prima.
Nombre fabricante materia prima y su número de lote.
Número de entrada al almacén, potencia, si aplica.
Número de control o análisis y fecha aprobación.
Cantidad que contiene el recipiente.
Firma y fecha de dispensación.
Firma y fecha de chequeo por producción. c) Orden de fabricación o manufactura - no se aceptan tachones ni enmendaduras, las cuales deben
contener:
Nombre o logo de laboratorio fabricante, en cada hoja del procedimiento.
Nombre del producto, concentración y forma farmacéutica, en cada hoja del procedimiento.
Código del producto, número de lote y tamaño lote, en cada hoja del procedimiento.
Fecha y hora iniciación manufactura.
Precauciones hacia el producto y de seguridad para el personal (máscara para polvos o gases, uso cabina, extracción especial, instrucciones en caso de contacto directo, etc.).
Identifica activos potencialmente peligrosos / potentes según normas nacionales de seguridad.
Informa condiciones ambientales de temperatura, humedad relativa y diferenciales de presión.
Describe completamente los equipos usados (tanques, mezclador, granulador, bombo, etc.), tipo, capacidad, balanzas, velocidad, inclinación agitador, entre otros.
Expresa la cantidad de sal equivalente a base.
Calcula la cantidad de activo de acuerdo con la potencia de la materia prima usada.
Describe el proceso de fabricación es realmente detallada y numerada.
Cada etapa del proceso de manufactura está firmado por quien lo realiza y por el responsable
Presenta diagrama de flujo.
Cuando se trata de áreas estériles: Informa clases de aires y relaciones presión en zonas general, fabricación y llenado, reporta esterilización de cada elemento de envase primario, reporta esterilización de cada elemento de envase primario.
Indica el rendimiento en cada fase del proceso.
Fecha y hora finalización manufactura. d) Formatos de controles en proceso, las cuales deben contener:
Especificaciones para productos líquidos. (Densidad, pH, viscosidad, volumen final y de llenado, etc).
Especificaciones para productos sólidos. (Peso, forma, altura, largo, ancho, diámetro, dureza, no. de cápsula, etc.
Especificaciones para productos semisólidos. (Viscosidad, volumen o peso final y de llenado, etc).
Graficas de los ciclos de esterilización de cada parte en productos inyectables. Condiciones.
Clases de aires y relación de presión en área estéril.
Los controles en proceso están documentados.
Firma de control de calidad cuando hace controles.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 18 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
e) Rótulos de demarcación de áreas, en los cuales está documentado el uso, limpieza, sanitización y rotación de desinfectantes de las áreas usadas para cada operación de cada proceso (manufactura, envase, empaque).
f) Rótulos de identificación de equipos, en los cuales se describen adecuadamente los equipos usados.
g) Orden de empaque o acondicionamiento, las cuales deben contener:
Nombre o logo de laboratorio fabricante.
Nombre del producto y presentación comercial.
Número de lote, número de orden de empaque, fecha de emisión.
Tamaño de lote: Indica si es parcial o total.
Fecha y hora iniciación y finalización.
Listado de material de envase y empaque a usar.
Cada presentación tiene su propia orden empaque.
Listado de material de envase y empaque a usar.
Fecha y horas de iniciación y finalización.
Hay procedimiento para envasado indicando empaque primario, cantidad y especificaciones.
Hay conciliación de los materiales recibidos con la orden empaque.
Documenta la destrucción de etiquetas y empaques sobrantes y deteriorados. h) Orden de marcado del material de empaque, las cuales deben contener:
Documenta la aprobación de la codificación de número de lote y fecha expiración en etiquetas, frascos y/o material de empaque.
La muestra codificada tiene la aprobación de producción y control de calidad autorizada.
Hay conciliación de los materiales codificados y no codificados recibidos con la orden empaque.
Documenta la destrucción de etiquetas y empaques codificados sobrantes y deteriorados. 6.10. (3.2.P.6) ESTÁNDARES Y MATERIALES DE REFERENCIA
Los estándares y materiales de referencia son utilizados para el control de calidad del principio activo, producto en proceso, terminado y estudios de estabilidad tanto de corto como de largo plazo. Respecto a los patrones, se deben tener en cuenta los siguientes criterios (basados en las B.P.M.) y considerando los avances técnico-científicos en el campo de los medicamentos. El objetivo de contar con sustancias químicas de referencia es conseguir PRECISIÓN y REPRODUCIBILIDAD en los resultados analíticos exigidos por los ensayos de las farmacopeas oficiales en el país y del control farmacéutico en general.
Patrones de referencia primarios: Farmacopeicos
Los estándares de trabajo o “Working Standard”, son aquellas sustancias cuyas características se aseguran o calibran por comparación con una sustancia química de referencia primaria.
En el evento que el producto no sea farmacopéico, el “Working Standard” (WS) o estándar de trabajo se constituye en el único patrón disponible y no se podrá calibrar otro patrón comparándolo con éste, es decir, éste se utilizará en todos los ensayos, a menos que se posean todos los soportes de análisis para garantizar la idoneidad de un WS, éste se podrá tomar como uno de referencia primario y se podrá establecer con base en éste, otro para trabajar. Las impurezas sólo serán patrones de referencia primario, nunca secundarios. Con el objetivo de conocer la trazabilidad del WS, el interesado deberá anexar el certificado de análisis del fabricante de dicha sustancia, con todos los soportes de lo consignado en el documento. Dentro de los

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 19 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
soportes allegados, estarán los destinados principalmente a identificar la sustancia y los utilizados para determinar su pureza. Su identidad puede ser realizada aplicando varias técnicas analíticas, entre ellas pueden figurar: Estudios cristalográficos, espectrometría de masas, espectroscopía con resonancia magnética nuclear - RMN, análisis de grupos funcionales y espectrofotometría de infrarrojo - IR o ultravioleta - UV, así como otras pruebas suplementarias necesarias para decidir que la sustancia propuesta está plenamente caracterizada. Por tanto, se anexarán soportes de algunos de estos ensayos o de varios de ellos, según lo realizado por el fabricante del WS comparados contra una sustancia que sirve como referencia.
Además, se adicionará la información referente a las condiciones óptimas de almacenamiento de la sustancia, métodos de pruebas utilizados, los valores encontrados y el número de réplicas utilizadas y los espectros o cromatogramas, e identificación de las impurezas detectadas.
NOTA: El porcentaje de pureza de los WS dependerá del uso a que se destine la sustancia. Una sustancia utilizada en una prueba de identificación, no necesita ser de alta pureza, mientras que una sustancia utilizada en ensayos (valoración) debe poseer un alto grado de pureza de al menos 99,5% calculada en su material como forma anhidra o libre de sustancias volátiles. 6.11. CONTROLES (PRINCIPIO ACTIVO, MATERIAS PRIMAS, PRODUCTO A GRANEL, PRODUCTO
TERMINADO)
6.11.1. (2.2.2.1) Especificaciones de calidad del principio activo, materias primas, producto a granel,
producto terminado Las especificaciones describen con detalle un conjunto de características definidas o condiciones mínimas de calidad requerida para una materia prima o producto, las cuales se controlan o verifican mediante una lista de ensayos, referente a los procedimientos analíticos y criterios de aceptación apropiados, tales como límites numéricos, rangos u otros criterios para las pruebas descritas. Estas se establecen a partir de un conjunto de criterios para un principio activo, excipiente o producto, de acuerdo a lo que se considere como aceptable para el uso para el cual se ha diseñado. Se define que el producto está “conforme a las especificaciones” cuando al principio activo o al producto farmacéutico se le han realizado las pruebas de acuerdo a los procedimientos analíticos, los cuales se han encontrado dentro de los criterios de aceptación. Las especificaciones son estándares críticos de calidad que son propuestos y justificados por el fabricante y aprobados por las autoridades regulatorias como condiciones de aprobación.
En el caso de una materia prima que se comercialice como una mezcla de excipientes, se solicitará al interesado la información proveniente del fabricante de la misma, correspondiente a identificación, valoración, descripción de los componentes de la mezcla, viscosidad en el caso de materiales para cubierta, etc., y lo que a criterio del evaluador sea requerido para lograr la apropiada utilización del excipiente.
De estar consignado en la literatura que de la materia prima en evaluación existen varias formas polimórficas, se solicitará al interesado que especifique y demuestre el tipo de cristal utilizado. Una vez demostrada la apropiada validación del proveedor o fabricante de las materias primas, se aceptará a los interesados allegar con la información radicada con la solicitud, el certificado de análisis de la materia prima utilizada en la fabricación de los lotes piloto industrial o industriales soportada, con un número de ensayos reducidos que incluyan su identificación, valoración y determinación de impurezas, complementada con la copia de un certificado de la materia prima a la cual se le ha realizado el 100% de los ensayos.
6.11.1.1. Cuando el principio activo, materias primas, producto terminado es farmacopéico
En el caso del principio activo y del producto terminado que se encuentre dentro de las farmacopeas aceptadas en Colombia, deberá cumplir con lo establecido por las especificaciones que establece la respectiva monografía.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 20 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Teniendo en cuenta lo consignado en la USP, edición 30, la cual menciona en su página 7, con referencia a impurezas y sustancias extrañas: “Si bien uno de los objetivos de la farmacopea es asegurar al usuario la identidad, el contenido, la calidad y la pureza de los artículos oficiales, resulta prácticamente imposible incluir en cada monografía una prueba para determinar la presencia de cada impureza que pudiera estar presente…” Otras impurezas las sustancias oficiales pueden obtenerse por más de un proceso y por ello pueden contener impurezas que no han sido consideradas durante la preparación de las pruebas y valoraciones de la monografía. Por lo anterior, en el caso de impurezas, compuestos relacionados, impurezas orgánicas volátiles o solventes residuales, se solicitarán las consignadas en el certificado del fabricante, en el caso que éste mencione otras diferentes a las consignadas en las farmacopeas.
Se solicitará, si no lo allegan, la evidencia de la confirmación de la descripción de la materia prima, lo cual se podrá soportar con las evidencias enviadas por el fabricante de la materia prima y confirmadas (si es posible, por el interesado) mediante difracción de rayos X para determinar con certeza si se trata del mismo amorfo, hidrato o cristal, y en el caso de resultar ser éste último, confirmar el tipo de cristal, para lo cual se deberá contar con un patrón para realizar la comparación y determinación.
Para las formas farmacéuticas tabletas, suspensiones, polvo para reconstituir a suspensión, semisólidos donde el principio activo está suspendido, se solicitarán los resultados de los ensayos de tamaño y distribución de partícula.
6.11.1.2. Cuando el principio activo, materias primas, producto a granel, producto terminado NO es farmacopéico
Se solicitará toda la información proveniente del fabricante de la materia prima, referente al perfil de impurezas y descripción detallada de la materia prima, todo con los respectivos soportes. En el caso de que el interesado manifieste que los resultados fueron obtenidos aplicando el método del fabricante, se solicitará toda la información proveniente de éste.
6.11.2. (2.2.2.4) Certificados de análisis del principio activo, materias primas, producto a granel,
producto terminado El certificado de análisis indica la conformidad del principio activo o del medicamento, con las especificaciones previamente establecidas. 6.11.3 Productos semielaborados
Para los productos cuya modalidad sea Importar (Ejemplo: Importación de principios activos procesados en forma de microgránulos para productos destinados a compresión directa), Semielaborar y Vender, el interesado deberá anexar la siguiente información:
Composición cualicuantitativa del semielaborado, si no es procedente como mínimo que anexen un rango de los componentes y cuáles son.
Fórmula del lote estandarizado de fabricación
Descripción detallada del proceso de fabricación
Especificaciones de calidad de las materia primas
Especificaciones de calidad del producto en proceso y terminado
Certificado de análisis del lote utilizado.
Certificado de cumplimiento de B.P.M. emitido por la entidad regulatoria del país de origen (No es necesario que sea de país de referencia, pero si debe estar apostillado y/o consularizado y legalizado y estar vigente)
Indicar cuál es la compañía responsable de la importación del semielaborado
En este caso, deberá diligenciarse el Formato para informe de evaluación técnica Registro Sanitario Medicamentos Fabricación Nacional ASS-RSA-FM011 y evaluarlo con las pautas dadas con anterioridad, como si fuera un producto diferente.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 21 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.12. CONTROLES EN PROCESO
El control del medio ambiente o de los equipos puede considerarse también parte del control en proceso, lo cual se verificará en los registros de los respectivos formatos de los controles realizados al producto durante su fabricación y envase. Los formatos especificarán nombre de producto, número de lote, variable medida con sus respectivos límites, hora de toma de muestras y nombre del equipo empleado en la operación.
Los controles en proceso para formas farmacéuticas sólidas incluyen los ensayos de: Dureza, peso, friabilidad, dimensiones, humedad, aspecto. Los anteriores, se controlaran con una periodicidad basada en lo establecido por el fabricante.
Para formas farmacéuticas líquidas y semisólidas se incluyen los ensayos de: pH del producto, aspecto, viscosidad, olor y el completar a volumen final. Estos ensayos se determinan solamente hasta cuando se observe el cumplimiento de la especificación, se debe hacer con periodicidad establecida por el interesado, en el envasado del producto. 6.13. (2.2.4.4) CONTROL DEL PRODUCTO TERMINADO
6.13.1. (2.2.4.4.1) Especificaciones de los controles en proceso
El interesado deberá anexar el certificado de análisis de los lotes cuya información se anexa con la solicitud este debe incluir las especificaciones y resultados de: dureza, friabilidad, disolución, dimensiones, descripción del núcleo y de la tableta recubierta (si lo es), que incluya color y aspecto en general, contenido o valoración de sustancia activa y de compuestos relacionados o impurezas, si lo establece una de las farmacopeas oficialmente aceptadas en Colombia, (en el caso que exista monografía del producto en una de ellas).
Para líquidos descripción específica del color, olor, sabor y aspecto (translúcido u opaco), viscosidad, rango de pH, densidad, valoración, carga microbiana. En el caso de líquidos estériles adicionalmente endotoxinas bacterianas, esterilidad.
Para los polvos que se reconstituyen: Descripción de aspecto, color, olor, humedad cuando está en forma de polvo y descripción de aspecto, color, olor, viscosidad, densidad y rango de pH unas vez reconstituido.
Además, es importante tener en cuenta los análisis referidos en el manual de normas técnicas colombianas vigente, para los productos que sean de fabricación nacional. 6.13.2. (2.2.4.4.2) Metodologías analíticas a. Productos Farmacopéico
El fabricante debe realizar todas las pruebas descritas en la farmacopea oficial escogida. Es importante que las pruebas realizadas sean adecuadas para la forma farmacéutica del medicamento y para el principio activo. En el caso que el producto sea oficial en más de una Farmacopea, todos los análisis deberán corresponder a la Farmacopea que haya sido elegida.
En el caso que el interesado manifieste que utiliza un método de análisis alterno a uno farmacopeico, deberá enviar los documentos que demuestren la equivalencia de los dos métodos. b. Productos No Farmacopéicos Se aceptará la metodología analítica establecida por el fabricante del medicamento, en donde se incluya la descripción detallada de todos los ensayos que se realicen y sus respectivos límites o especificaciones. Es importante que las pruebas realizadas sean adecuadas para la forma farmacéutica del medicamento y para el principio activo. 6.13.3. (2.2.4.4.3) Validación de los procedimientos analíticos Para la VALIDACIÓN DEL MÉTODO DE CUANTIFICACIÓN O VALORACIÓN DEL PRINCIPIO ACTIVO: No será necesario realizar una validación completa del método de cuantificación o valoración del principio

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 22 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
activo, si éste es farmacopéico. Sin embargo, se exigirán los parámetros de adecuabilidad del sistema y especificidad para efectos de estabilidad frente al blanco y al placebo, allegando soportes debidamente identificados. Los métodos no farmacopeicos de cuantificación del principio activo deben venir totalmente validados de acuerdo a los lineamientos de la Norma Compendial <1225> de la USP vigente. En el caso que la metodología para producto terminado sea la misma utilizada en los estudios de estabilidad, ésta debe estar validada en el parámetro de especificidad con fines de estabilidad, es decir, en donde se garantice que los productos de degradación del principio activo, obtenidos mediante degradación forzada o por enriquecimiento del medicamento con sustancias de degradación y/o impurezas conocidas que no deben interferir con la cuantificación del principio activo, tal y como se establece en la Norma Compendial USP <1225>. En el caso que el interesado manifieste que utiliza dos o más métodos de cuantificación del principio activo, debe establecer cuál utiliza para el estudio de estabilidad y deberá cumplir con lo expuesto anteriormente, enviando soportes debidamente identificados.
Para el ensayo de disolución, el cual es requerido por tabletas con y sin cubierta, tabletas masticables, tabletas dispensables, cápsulas (duras y blandas), suspensiones (a la fecha las que sí lo requieren según la monografía), si la farmacopea escogida por el interesado incluye una metodología analítica diferente a la del producto terminado, deberá validarla. 6.14. (2.2.4.5) SISTEMA DE ENVASE - CIERRE
6.14.1. Presentaciones comerciales y material de envase y empaque Confirmar que la presentación comercial esté claramente descrita, la cual debe indicar con precisión el color, clase y tipo de material de envase y empaque. En el caso de que el producto se presente en forma de blíster, cada presentación indicará el número de unidades por blister. Además, se debe revisar si la forma farmacéutica solicitada (teniendo en cuenta su composición y descripción) corresponde con las consignadas en la literatura farmacéutica como en las farmacopeas oficiales en Colombia u otras formas farmacéuticas nuevas aprobadas en Actas de Comisión Revisora. Si la presentación comercial incluye dispositivos médicos, deberá incluirlos en la presentación comercial y describirlos completamente. La información presentada en este punto, servirá como soporte para evaluar los certificados de calidad del material de envase, empaque y los dispositivos que acompañan el producto, lo cual deberá coincidir en su totalidad. Para medicamentos unidosis inyectables, no se aprobarán presentaciones comerciales que impliquen dosificaciones diferentes aunque la concentración sea la misma. Es decir, volúmenes diferentes a los cuales corresponden cada uno a un Registro Sanitario distinto (ver Decreto 677 de 1995, Artículo 22, numeral c), para lo cual se tendrá en cuenta lo siguiente: Si se otorga el Registro Sanitario bajo la categoría A: Por unidad, en formas de presentación dosificada, en caso de tabletas, grageas, cápsulas, óvulos, supositorios, inyectables y similares es unidosis y no permite dosis diferentes. Si se otorga bajo la categoría C: (Por cada mililitro, en líquidos para administración por gotas e inyectables en multidosis) se permiten varios volúmenes distintos. 6.14.2. Especificaciones para los materiales de envase Se verificarán las especificaciones de calidad del material de envase primario. Las especificaciones de calidad deben ser adecuadas para el tipo de envase y material del mismo. Por ejemplo: Si es plástico, se solicitará que el interesado haya confirmado lo indicado por el fabricante como es el tipo de polímero del material plástico, color, dimensiones, indicar el volumen de rebose, determinación de permeabilidad, sustancias extractables (según Norma Compendial <661> de la USP) o Farmacopeas BP y Europea. Para

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 23 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
los goteros, la determinación del número de gotas por cada 1 mL se debe hacer con producto. Por tanto, si lo han realizado con agua, el evaluador deberá solicitar el resultado del ensayo con la solución de producto. Se verificará el certificado del fabricante del material de envase, contra el del laboratorio encargado del análisis de materias primas o el mismo laboratorio fabricante del medicamento, si es éste quien realiza los análisis. 6.14.3. Especificaciones de los dispositivos médicos Se considera como dispositivo complementario todo implemento de preparación o administración que se comercializará conjuntamente con el medicamento. Ejemplos de dichos dispositivos: Cucharas y jeringas para administración de productos orales, sets o kits de transferencia y preparación de soluciones y polvos estériles, entre otros. El interesado debe allegar la descripción detallada del dispositivo, si es posible allegar diagramas o fotos y las especificaciones de calidad adecuadas para el uso del dispositivo. En caso de alguna inquietud el evaluador deberá consultar de manera escrita (e-mail o carta) al Grupo de Dispositivos Médicos y otras Tecnologías, sobre si el dispositivo requiere o no Registro Sanitario de acuerdo al Decreto 4725 de 2005. Si los mismos cuentan con características especiales definidas en el Decreto 4725 de 2005, deberá complementar la información de los mismos: Nombre genérico o marca del dispositivo médico, clasificación de acuerdo al riesgo, indicaciones y uso, indicar el código internacional (ECRTI, GMDN u otro de igual reconocimiento internacional), advertencias, precauciones y contraindicaciones, descripción del dispositivo médico (listado de principales partes, funcionamiento, información descriptiva, estudios técnicos y comprobaciones analíticas), resumen de los documentos de verificación y validación (informe de pruebas) de diseño o certificado de análisis. Para el sistema de infusión estéril por ser catalogado como dispositivo médico clase IIa, deberá allegar adicionalmente la información científica necesaria que respalde la seguridad del producto y un análisis de riesgos del dispositivo médico según sus indicaciones, descripción de soluciones adoptadas para cumplir con los requisitos esenciales de seguridad y funcionamiento y los respectivos estudios de estabilidad que sustenten el tiempo de vida útil. 6.15. (2.2.4.6) ESTUDIOS DE ESTABILIDAD Y PERÍODO DE VIDA ÚTIL DEL PRODUCTO
Tomando como referencia el Artículo 102 del Decreto 677 de 1995 y el Informe 32 de la OMS, los titulares y fabricantes de Medicamentos que deberán establecer y poner en conocimiento del Invima, las responsabilidades que tendrán en el proceso de fabricación, acondicionamiento primario (envase) y secundario (empaque). 6.15.1. Información del producto farmacéutico terminado – PFT
6.15.1.1. Información Mínima Suministrada
Nombre del Ingrediente Farmacéutico Activo - IFA (en Denominación Común Internacional).
Forma farmacéutica, concentración y presentación comercial, incluyendo la fórmula cualicuantitativa
completa, con la denominación IUPAC, el CI (Color Index) o las referencias particulares en el caso
de excipientes tales como saborizantes, esencias y colorantes.
En caso que se modifiquen los artes de etiquetas en cuanto a las condiciones de almacenamiento o
que no figuren en el expediente, deberá presentarse copia del proyecto de etiqueta del producto y si
aplica, del empaque secundario en donde se puedan verificar las condiciones de almacenamiento
propuestas (aplica para modificaciones).
Composición completa del material empleado en el envase primario y secundario. Debe incluir,
entre otros, los gramajes empleados en los materiales laminados, el tipo, el tamaño del envase
primario, el color y el sistema de cierre (describiendo detalladamente el material que entra en
contacto con el producto y el que no).
Fabricante del producto.
6.15.1.2. Información sobre las especificaciones del producto

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 24 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
El interesado debe presentar las especificaciones físicas, químicas, microbiológicas, terapéuticas y
toxicológicas del Producto Farmacéutico Terminado (PFT) en estudio. Tenga en cuenta que si el
producto es oficial en alguna de las farmacopeas vigentes en Colombia, las especificaciones
deberán corresponder con lo estipulado en dicha farmacopea. En caso contrario, es el fabricante del
medicamento quien fija las especificaciones del producto terminado como consecuencia lógica del
desarrollo del medicamento y por lo tanto deberán justificarse de tal manera que correspondan a la
forma farmacéutica y al Ingrediente Activo Farmacéutico.
El fabricante deberá fijar las especificaciones del producto farmacéutico tanto para la liberación del
mismo (control de calidad), como para el final de la vida útil propuesta (estabilidad), las cuales
podrán diferir, pero siempre deberá existir una justificación técnica de tal diferencia.
6.15.2. Metodología de ensayo
Se debe indicar la metodología de ensayo utilizada para cada una de las especificaciones antes mencionadas y esta información deberá corresponder con la que se encuentra en el expediente. La metodología de análisis debe estar acorde con los parámetros descritos a continuación y con lo establecido en el Decreto 19 de 2012, Artículos 129 y 130.
Deberá presentarse como documento complementario del estudio, la correspondiente validación de la metodología analítica utilizada para la disolución, uniformidad, contenido, potencia y compuestos de degradación, entre otros. Tenga en cuenta los siguientes aspectos para la presentación del informe de validación de la metodología analítica:
a) En el caso de encontrarse la metodología analítica en alguna de las farmacopeas oficiales vigentes en Colombia, el interesado evaluará la adecuabilidad del método para corroborar la aplicabilidad del mismo en el PFT y dentro de los parámetros estudiados como mínimo estarán precisión, exactitud, linealidad y especificidad.
b) La información obtenida de la validación de la metodología analítica, se debe presentar de una manera tabulada y gráfica. Los soportes analíticos presentados deben contener la información suficiente para identificar el medicamento y el IFA, la concentración, la forma farmacéutica, el tipo de ensayo, el equipo empleado, la fecha, la hora y el analista responsable del análisis.
6.15.3. Respecto al diseño del estudio de estabilidad
Debe indicar el tipo de estudio a realizar: Estabilidad acelerada y/o natural.
Definir el objetivo de los estudios de estabilidad: Esto es, si se trata de determinar la vida útil de un medicamento por primera vez, como consecuencia de un cambio en el proceso de fabricación, para la adición de un fabricante alternativo del producto, después de realizar una reformulación (cambio de excipientes), para realizar un seguimiento de la estabilidad (on going) de un producto, entre otros casos.
Definir las condiciones de temperatura y humedad relativa bajo las cuales se realizará el estudio de estabilidad, tomando en consideración que Colombia se encuentra ubicada en la Zona Climática IV.
Presentar el cronograma, indicando la fecha de inicio y la estimada de finalización de los estudios de estabilidad.
Indicar el número de lotes del estudio y la identificación de los mismos la cual debe ser clara y suficiente para hacer la trazabilidad del medicamento en cada etapa de su análisis. Los lotes deberán ser consecutivos en su fabricación y claramente deberá indicarse si se trata de lotes piloto o industriales y la fecha de manufactura de los mismos. Allegar con la solicitud, el sistema de numeración de lote establecido en el laboratorio farmacéutico para la identificación de los productos realizados.
Incluir la fecha de fabricación de los lotes y el inicio del estudio de estabilidad.
Señalar la cantidad total de las muestras de acuerdo a la forma farmacéutica, y aclarar si los ensayos serán realizados sobre unidades individuales o sobre compuestos de unidades individuales (muestra compuesta).

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 25 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Presentar los métodos estadísticos y/o matemáticos a ser utilizados en el análisis de los datos y la obtención de resultados.
Se deberá tener en cuenta por parte de los laboratorios que, para efectos de la planeación del estudio, se deberá incluir una cantidad adicional de muestras equivalente a un período de muestreo, para ser suministrado al Invima, en el evento de ser necesaria la toma de muestras, de conformidad con lo establecido en el numeral 2º, Artículo 23 del Decreto 677 de 1995. 6.15.4. Respecto a las formas farmacéuticas que requieren reconstitución Además de lo indicado anteriormente, se debe presentar el protocolo del estudio indicando entre otros aspectos los siguientes:
Fecha de inicio del estudio y la estimada de finalización respecto a la forma farmacéutica reconstituida.
Cronograma del estudio de estabilidad a realizarse, indicando cada uno de los tiempos de muestreo, incluyéndose el tiempo de vida útil propuesto después de la reconstitución del producto.
Cantidad de muestras incluidas en el estudio y criterios de selección de las mismas.
Vehículo empleado en la reconstitución. En caso de proponerse más de uno, deberá realizarse el estudio de estabilidad para cada vehículo / solvente referido.
En caso de recomendarse más de una condición de almacenamiento (ºT y H.R.), para cada condición, deberá realizarse el estudio de estabilidad correspondiente.
Como mínimo, debe realizarse la valoración del producto y evaluarse la calidad microbiológica del mismo. En el caso de los medicamentos para multidosis, deberá garantizarse la uniformidad de contenido hasta la última dosificación.
6.15.5. Presentación de los resultados de estabilidad
Los datos primarios deben ser presentados en forma tabulada individual indicando el lote, envase,
compuesto, etc. No se aceptarán datos promedio y se tendrá como parámetro fundamental la
concentración remanente del (los) IFA(s) durante el tiempo de estudio.
Los promedios resultantes de los datos deben ser tabulados con los límites de confianza
debidamente identificados y justificados.
Si corresponde al tipo de producto, se deberá presentar los resultados de las pruebas estadísticas
utilizadas para llegar a la estimación de la potencia microbiológica.
Complementar la información de los Certificados de Análisis de Producto terminado, con los
parámetros evaluados, para cada lote y cada tiempo de estudio.
Para presentar los resultados del estudio de estabilidad de seguimiento ("on going"), se aceptarán
los estudios de estabilidad de los dos ó tres lotes consecutivos más recientemente incluidos en el
programa de estabilidad.
Deberá existir una conclusión acerca de la vida útil propuesta para el medicamento, indicando las
condiciones de almacenamiento y su justificación correspondiente respecto al material de envase y
empaque en el que será comercializado, el cual deberá quedar completamente caracterizado como
parte determinante del medicamento. En las formas farmacéuticas que requieren reconstitución, se
deberá incluir el tiempo de vida útil sugerido para cada vehículo de reconstitución y las condiciones
específicas de temperatura y humedad relativa recomendadas para el almacenamiento durante la
vida útil propuesta dentro de las conclusiones del estudio.
La validación de la metodología analítica y el estudio de estabilidad con las respectivas conclusiones, deberá estar firmado por el Director Técnico responsable.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 26 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.15.6. Validación de la metodología analítica empleada
Si el método corresponde al consignado en la monografía del producto, cuando éste sea oficial, el interesado deberá demostrar que el mismo es indicativo de estabilidad (Norma Compendial <1151> de la USP).
Si se emplea un método carente de especificidad (Ejemplo: Espectrofotométrico, titulación, etc.), el interesado anexará la información correspondiente al método que suple dicha carencia de selectividad. La información será evaluada con base en lo indicado en la literatura respecto al método complementario, el cual igualmente deberá estar validado.
Si el método para estabilidad es el mismo empleado para analizar el producto terminado, tan solo se complementará la información de validación ya mencionada en el literal k, con el protocolo de los ensayos de degradación forzada con sus respectivos soportes, es decir, los cromatogramas representativos que permitan verificar que el activo intacto se puede diferenciar de los demás componentes de la matriz (excipientes, impurezas y/o productos de degradación), el límite de detección (categoría II de la Norma Compendial <1225> de la USP).y se deberá realizar el parámetro de robustez cuando el interesado contrata el servicio de una entidad externa y luego va a realizar los ensayos en sus instalaciones.
En el caso que el interesado contrate la validación por un tercero y luego lo aplique en su fábrica, deberá allegar el documento de transferencia de método.
6.15.7. Descripción de los procedimientos usados para garantizar la cadena de frío En caso que los medicamentos requieran garantizar dentro de su almacenamiento la cadena de frío, deberá allegarse una validación de los procedimientos en donde se realiza un estudio de estabilidad en las condiciones en las cuales se transportó el producto y en las condiciones donde se debe almacenar. 6.15.8. Información a tener en cuenta para la evaluación
El material de envase primario debe corresponder con las especificaciones de calidad reportadas en la sección respectiva y/o sección: “Sistema de envase / cierre”.
En general, las condiciones ambientales de almacenamiento de los lotes sometidos a estabilidad, deben corresponder con las de zona climática IVa: (30±2°C, 65±5% HR) o IVb: (30±2°C, 75±5% HR). El número de lotes sometidos al estudio y el intervalo de muestreo en general, debe corresponder con lo establecido por la ICH o el Decreto 677 de 1995. Otros tiempos de muestreo deben estar justificados según criterio del evaluador y del fabricante.
En general, las condiciones ambientales de almacenamiento de los lotes sometidos a estabilidad acelerada deben corresponder a 40±2°C y 75±5 % de HR. El número de lotes sometidos al estudio y el intervalo de muestreo en general, debe corresponder a lo establecido por la ICH o Decreto 677 de 1995. Otros tiempos de muestreo deben estar justificados según criterio del evaluador y del fabricante.
Si el producto en estudio está indicado para almacenamiento en refrigeración o congelamiento, las condiciones ambientales de almacenamiento de los lotes sometidos a estabilidad deben corresponder a 5±3°C (refrigeración) y/o congelamiento (-20°C).
Los lotes de productos envasados en contenedores semipermeables deben ser almacenados bajo condiciones especiales de humedad relativa según las Guía ICH Q1A (R2), o presentar las pruebas de pérdida de agua correspondientes.
Dependiendo del tipo de producto en evaluación, otros estudios de estabilidad complementarios deben allegarse (estabilidad del producto reconstituido, estudios para materiales de envase semipermeables, fotoestabilidad, estabilidad después de abierto, entre otros).
Cuando el medicamento se acompaña en su presentación de un solvente o diluyente, se requiere la presentación de estudios de estabilidad del producto diluido, disuelto o dispersado según proceda en las condiciones de utilización.
Si se utiliza más de un vehículo para su preparación, deben presentar los datos de los estudios de estabilidad realizados para cada uno de ellos.
Si un producto es envasado en dos o más sistemas de envase/cierre, se presentarán los estudios de estabilidad realizados para cada uno de ellos.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 27 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Si un producto es envasado en dos a más volúmenes, se presentarán los estudios de estabilidad por bracketing, conforme a la guía ICH Q1D.
Los métodos de análisis usados en el estudio de estabilidad deben estar debidamente validados.
Las pruebas realizadas en el estudio de estabilidad deben ser indicadoras de estabilidad. En general las especificaciones deben corresponder a las especificaciones del producto terminado. Sin embargo, la exclusión o inclusión de parámetros de análisis podría aceptarse previa justificación del fabricante o criterio del evaluador.
Teniendo en cuenta los datos de estabilidad presentados, el fabricante debe establecer compromiso de continuar los estudios naturales.
6.16. EVALUACIÓN DEL NOMBRE
Se debe evaluar si el nombre: a) Se ajusta a términos de moderación científica y, por lo tanto, no serán admitidas en ningún caso las
denominaciones que estén dentro de las siguientes circunstancias (Artículo 78 del Decreto 677):
Las que induzcan a engaño, sean estrambóticas o exageradas.
Las que se presten a confusión con los nombres de otros productos.
Las que indiquen expresamente la utilización o indicaciones farmacológicas.
Las exclusivamente formadas por iniciales o números.
Las acompañadas o adicionadas con una cifra, con excepción de las que se refieren a la concentración de los principios activos y de aquellos en que la cifra se utilice para diferenciarlas de otros productos de nombre básico o prefijo igual.
Las que utilicen los nombres del santoral de cualquier religión o secta religiosa, se identifiquen con los de las llamadas deidades o pertenezcan al orden mitológico, así como aquéllas vinculadas a creencias o temas religiosos, de superstición o hechicería.
Las que sin conexión alguna con los efectos reales del producto, según lo determine el Invima, usen palabras tales como: Tónico, confortativo, vigor, enérgico, vida, extra, súper, mejor, ideal, hermoso, maravilloso y único, ya sea como nombre o marca o simplemente como explicación.
Los que incluyan la palabra doctor o se refieran a otros títulos o dignidades o sus abreviaturas.
Las que utilicen nombres o apellidos de personas naturales, a menos que se trate de productos que en la literatura científica mundial, figuren con los nombres de sus autores, tales como solución de Ringer, Pasta de Lassar, Bota de Unna, los cuales podrán ser utilizados por cualquier fabricante.
b) No se otorgará Registro Sanitario a medicamentos de igual composición, pero con diferente nombre, a
favor de un mismo titular. c) No se autorizarán cambios de nombre de los productos cuando obedezcan solamente a cambios de
marca de sus componentes, sin que la composición del producto hubiere variado, o induzcan a creer que se trata de un producto nuevo o diferente, sin que se haya presentado variación fundamental que justifique el cambio.
d) Tampoco se permitirá adicionar el nombre de los productos con marcas registradas correspondientes a un excipiente de la fórmula.
e) Cuando el Invima otorgue Registro Sanitario a un medicamento bajo su nombre genérico, no autorizará posteriormente el cambio a nombre de marca.
f) Si revisada la base de datos del Instituto el nombre puede confundirse con el nombre de otro medicamento previamente aprobado, si el nombre va acompañado de siglas tales como NF (nueva fórmula), HR, etc. En todo caso, debe haber explicación del significado y si hace referencia a una propiedad de la forma farmacéutica esto debe ser verificado con los datos del expediente.
Así se apruebe la marca en la Superintendencia de Industria y Comercio – SIC, no deben contrariar ninguno de los conceptos anteriores. 6.17. ARTES DE ETIQUETAS DE MATERIAL DE ENVASE Y EMPAQUE Los bocetos de etiquetas para los materiales de envase y empaque incluirán la siguiente información:

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 28 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Nombre del producto: Si es genérico, se escribirá el nombre según la Denominación Común Internacional (DCI), nombre y domicilio del fabricante del producto, composición del producto por unidad posológica. En el caso de polvos para reconstituir, expresar por unidad posológica una vez reconstituido, fecha de vencimiento, número de lote, gotas contenidas por mililitro, cantidad contenida en el envase, condiciones especiales de almacenamiento cuando el producto lo requiera, el número de Registro Sanitario, la frase venta bajo fórmula medica o libre venta, la vía de administración, una franja verde y la leyenda medicamento esencial cuando así se requiera (basados en los listados incluidos en los Acuerdos 228, 236, 263, 282 y 336 del CNSSS) y la leyenda: “Manténgase fuera del alcance de los niños”.
Para los productos biológicos, además de incluir las características biológicas e inmunológicas del producto, la indicación de su actividad y de sus unidades protectoras y de capacidad, así como la dosis floculante o título del germen, la indicación del estado biológico del microorganismo vivo, modificado o muerto.
Tener en cuenta que los productos de control especial deben tener una franja vertical de color violeta y la leyenda “MEDICAMENTO DE CONTROL ESPECIAL. ÚSESE BAJO ESTICTA VIGILANCIA MEDICA”, y si fuera el caso “MEDICAMENTO SUSCEPTIBLE DE CAUSAR DEPENDENCIA.”
En términos generales, los artes de etiquetas para los materiales de envase y empaque deben incluir lo establecido en el Artículo 72 y adicionalmente, lo establecido en el Artículo 76 para el caso de Muestras Médicas y en el Artículo 73 para los Medicamentos de Control Especial del Decreto 677 de 1995. Al momento de la evaluación, es importante que el evaluador tenga en cuenta los aspectos relacionados en el formato de informe de evaluación, en donde está detallado cada uno de ellos, dada la importancia del etiquetado en la comercialización y en la seguridad del paciente. 6.18. OBSERVACIONES Y CONSIDERACIONES EN CASOS PARTICULARES SOBRE LA
EVALUACIÓN DE LA INFORMACIÓN TÉCNICA
a. Toma de muestra para análisis en el laboratorio del Invima: El evaluador podrá tomar muestras de medicamento, dando aplicación a lo establecido en el numeral 2º del Artículo 23 del Decreto 677 o solicitando apoyo directo a la Dirección de Medicamentos. Adicionalmente, en el caso de medicamentos nuevos o cuando el grupo de Registros Sanitarios de Medicamentos y Productos Biológicos lo decida, podrá enviarse el dossier para que sea estudiado por la Oficina de Control de Calidad, tal como indica el procedimiento ASS-RSA-PR001.
b. Dando aplicación a lo establecido en el numeral 2º del Artículo 23 del Decreto 677, el evaluador realizará, cuando considere pertinente, una visita a las instalaciones del fabricante y/o del encargado de realizar los estudios de estabilidad, para verificar lo que considere conveniente basado en la información recibida. Cuando se estudie un trámite que involucre la fabricación de un medicamento que requiera áreas especiales de manufactura o que se trate de un medicamento que contenga un activo de estrecho margen terapéutico, o formas farmacéuticas inyectables, el evaluador de la parte técnica podrá realizar visita e inspeccionar la validación de procesos, de acuerdo al anexo Validación de procesos de manufactura, del Instructivo de Visita a Laboratorios Farmacéuticos desde el Grupo de Registros Sanitarios de Medicamentos y Productos Biológicos ASS-RSA-IN009.
c. En el caso de que el producto contenga ciertos excipientes como: Alcohol bencílico, metabisulfito, tartrazina (Amarillo FD&C No. 5), aspartame, entre otros, se debe tener precaución ya que estos requieren la inclusión de leyendas especiales en las contraindicaciones y advertencias.
d. En el caso de materias primas (principios activos, excipientes e intermediarios de fabricación) de origen animal, se debe establecer claramente el origen de estas y de ser necesario debe presentar certificado del proveedor o de la autoridad sanitaria del país, en donde se establezca la ausencia agentes infecciosos y/o patógenos como la Encefalitis Espongiforme Bovina.
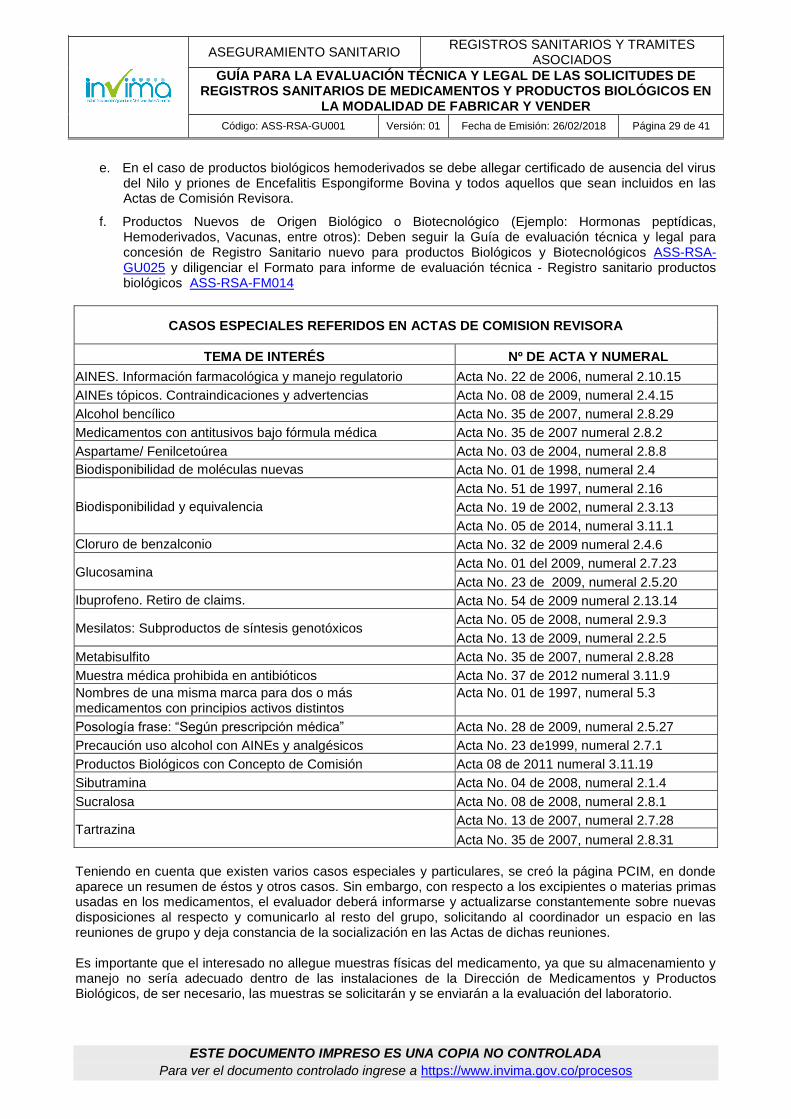
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 29 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
e. En el caso de productos biológicos hemoderivados se debe allegar certificado de ausencia del virus del Nilo y priones de Encefalitis Espongiforme Bovina y todos aquellos que sean incluidos en las Actas de Comisión Revisora.
f. Productos Nuevos de Origen Biológico o Biotecnológico (Ejemplo: Hormonas peptídicas, Hemoderivados, Vacunas, entre otros): Deben seguir la Guía de evaluación técnica y legal para concesión de Registro Sanitario nuevo para productos Biológicos y Biotecnológicos ASS-RSA-GU025 y diligenciar el Formato para informe de evaluación técnica - Registro sanitario productos biológicos ASS-RSA-FM014
CASOS ESPECIALES REFERIDOS EN ACTAS DE COMISION REVISORA
TEMA DE INTERÉS Nº DE ACTA Y NUMERAL
AINES. Información farmacológica y manejo regulatorio Acta No. 22 de 2006, numeral 2.10.15
AINEs tópicos. Contraindicaciones y advertencias Acta No. 08 de 2009, numeral 2.4.15
Alcohol bencílico Acta No. 35 de 2007, numeral 2.8.29
Medicamentos con antitusivos bajo fórmula médica Acta No. 35 de 2007 numeral 2.8.2
Aspartame/ Fenilcetoúrea Acta No. 03 de 2004, numeral 2.8.8
Biodisponibilidad de moléculas nuevas Acta No. 01 de 1998, numeral 2.4
Biodisponibilidad y equivalencia
Acta No. 51 de 1997, numeral 2.16
Acta No. 19 de 2002, numeral 2.3.13
Acta No. 05 de 2014, numeral 3.11.1
Cloruro de benzalconio Acta No. 32 de 2009 numeral 2.4.6
Glucosamina Acta No. 01 del 2009, numeral 2.7.23
Acta No. 23 de 2009, numeral 2.5.20
Ibuprofeno. Retiro de claims. Acta No. 54 de 2009 numeral 2.13.14
Mesilatos: Subproductos de síntesis genotóxicos Acta No. 05 de 2008, numeral 2.9.3
Acta No. 13 de 2009, numeral 2.2.5
Metabisulfito Acta No. 35 de 2007, numeral 2.8.28
Muestra médica prohibida en antibióticos Acta No. 37 de 2012 numeral 3.11.9
Nombres de una misma marca para dos o más medicamentos con principios activos distintos
Acta No. 01 de 1997, numeral 5.3
Posología frase: “Según prescripción médica” Acta No. 28 de 2009, numeral 2.5.27
Precaución uso alcohol con AINEs y analgésicos Acta No. 23 de1999, numeral 2.7.1
Productos Biológicos con Concepto de Comisión Acta 08 de 2011 numeral 3.11.19
Sibutramina Acta No. 04 de 2008, numeral 2.1.4
Sucralosa Acta No. 08 de 2008, numeral 2.8.1
Tartrazina Acta No. 13 de 2007, numeral 2.7.28
Acta No. 35 de 2007, numeral 2.8.31
Teniendo en cuenta que existen varios casos especiales y particulares, se creó la página PCIM, en donde aparece un resumen de éstos y otros casos. Sin embargo, con respecto a los excipientes o materias primas usadas en los medicamentos, el evaluador deberá informarse y actualizarse constantemente sobre nuevas disposiciones al respecto y comunicarlo al resto del grupo, solicitando al coordinador un espacio en las reuniones de grupo y deja constancia de la socialización en las Actas de dichas reuniones. Es importante que el interesado no allegue muestras físicas del medicamento, ya que su almacenamiento y manejo no sería adecuado dentro de las instalaciones de la Dirección de Medicamentos y Productos Biológicos, de ser necesario, las muestras se solicitarán y se enviarán a la evaluación del laboratorio.
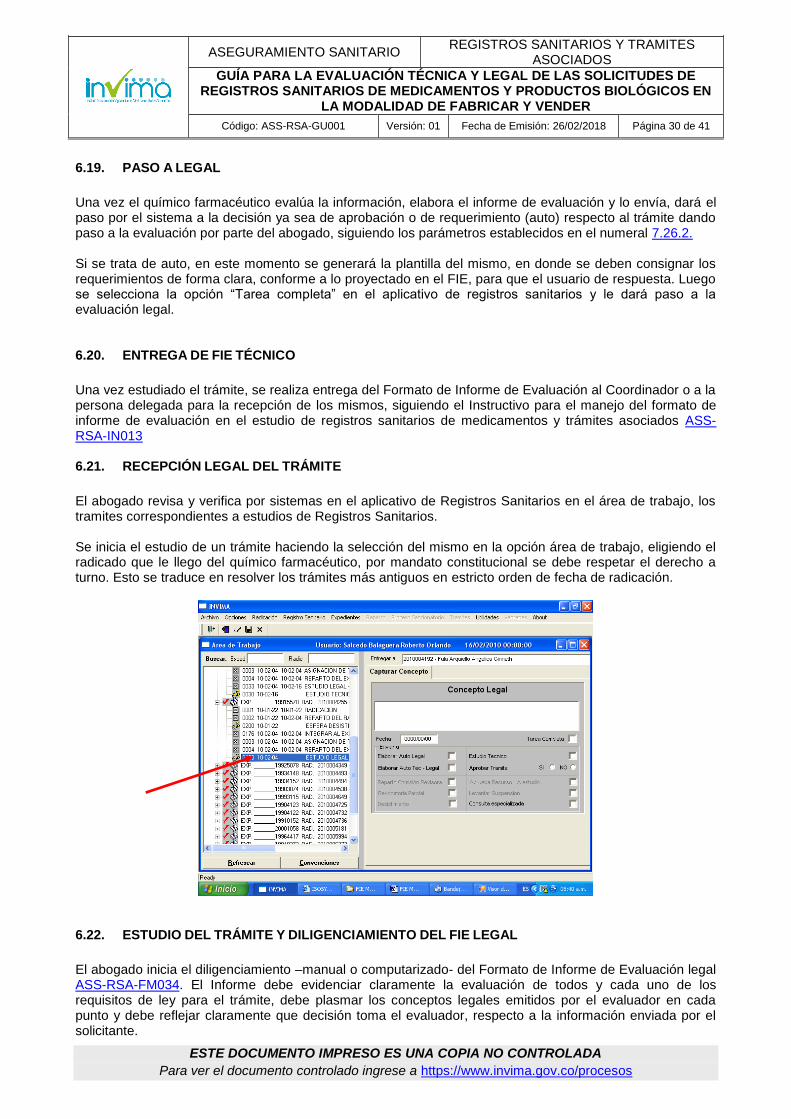
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 30 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.19. PASO A LEGAL
Una vez el químico farmacéutico evalúa la información, elabora el informe de evaluación y lo envía, dará el paso por el sistema a la decisión ya sea de aprobación o de requerimiento (auto) respecto al trámite dando paso a la evaluación por parte del abogado, siguiendo los parámetros establecidos en el numeral 7.26.2. Si se trata de auto, en este momento se generará la plantilla del mismo, en donde se deben consignar los requerimientos de forma clara, conforme a lo proyectado en el FIE, para que el usuario de respuesta. Luego se selecciona la opción “Tarea completa” en el aplicativo de registros sanitarios y le dará paso a la evaluación legal. 6.20. ENTREGA DE FIE TÉCNICO
Una vez estudiado el trámite, se realiza entrega del Formato de Informe de Evaluación al Coordinador o a la persona delegada para la recepción de los mismos, siguiendo el Instructivo para el manejo del formato de informe de evaluación en el estudio de registros sanitarios de medicamentos y trámites asociados ASS-RSA-IN013 6.21. RECEPCIÓN LEGAL DEL TRÁMITE
El abogado revisa y verifica por sistemas en el aplicativo de Registros Sanitarios en el área de trabajo, los tramites correspondientes a estudios de Registros Sanitarios. Se inicia el estudio de un trámite haciendo la selección del mismo en la opción área de trabajo, eligiendo el radicado que le llego del químico farmacéutico, por mandato constitucional se debe respetar el derecho a turno. Esto se traduce en resolver los trámites más antiguos en estricto orden de fecha de radicación.
6.22. ESTUDIO DEL TRÁMITE Y DILIGENCIAMIENTO DEL FIE LEGAL
El abogado inicia el diligenciamiento –manual o computarizado- del Formato de Informe de Evaluación legal ASS-RSA-FM034. El Informe debe evidenciar claramente la evaluación de todos y cada uno de los requisitos de ley para el trámite, debe plasmar los conceptos legales emitidos por el evaluador en cada punto y debe reflejar claramente que decisión toma el evaluador, respecto a la información enviada por el solicitante.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 31 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
El abogado debe verificar los documentos correspondientes a: 6.22.1. Formulario de solicitud (ASS-RSA-FM004) Debe corresponder al trámite solicitado (Registro, modificación) y que esté firmado por el representante legal o apoderado. 6.22.2. Verificación del recibo de pago En donde debe comprobar que la tarifa corresponda al trámite solicitado. Si la tarifa no corresponde con la del trámite solicitado, deberá realizarse un oficio dirigido al coordinador o encargado de la Oficina de atención al ciudadano para devolver el trámite y que se tomen las medidas pertinentes. 6.22.3. La carta aval firmada por el Director Técnico La carta aval firmada por el Director Técnico sólo aplica para los Registros Sanitarios en la modalidad Fabricar y Vender. El Director Técnico es el responsable de la información técnica, soporte de la solicitud del trámite de evaluación farmacéutica (igual que técnico). 6.22.4. Contratos Cuando el fabricante es diferente al titular (en el caso de fabricar y vender), revisar el (los) contrato(s) entre el titular y el fabricante en el(los) cual(es) debe estar incluido el nombre del producto, vigencia, prórrogas, el objeto del contrato, responsabilidades, condiciones, que esté firmado por el representante legal o apoderado de cada una de las partes. Cuando se trate de otros contratos (estabilidad, acondicionamiento, envase, control de calidad), en cualquier modalidad de Registro Sanitario, también deberá verificarse la información señalada anteriormente. 6.22.5. Certificados de cámara y comercio No es un requisito que se allegue dentro de la solicitud el Certificado de Cámara y Comercio, ya que, conforme al Decreto 19 de 2012, se debe hacer la verificación al interior del Instituto. La revisión de la información que se realiza es la misma, tanto si allega el certificado de Cámara y Comercio en físico como si se debe consultar la información en línea. La información que debe revisar es la siguiente: verificar la existencia de la empresa, verificar que el representante legal coincida con lo reportado a lo largo de la documentación. Para este último caso deberá seguir los siguientes pasos:
a) Ingresar a la página del RUES (Registro Único Empresarial y Social de Cámaras de Comercio) http://www.rues.org.co/
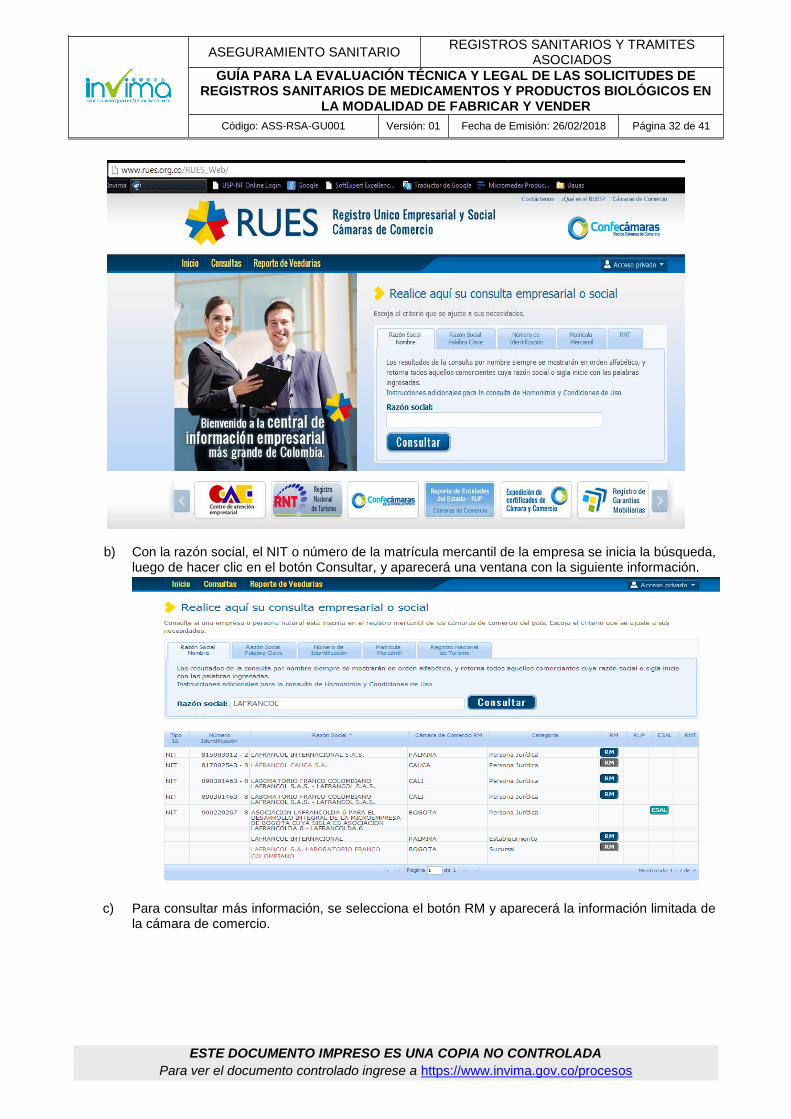
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 32 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
b) Con la razón social, el NIT o número de la matrícula mercantil de la empresa se inicia la búsqueda, luego de hacer clic en el botón Consultar, y aparecerá una ventana con la siguiente información.
c) Para consultar más información, se selecciona el botón RM y aparecerá la información limitada de la cámara de comercio.
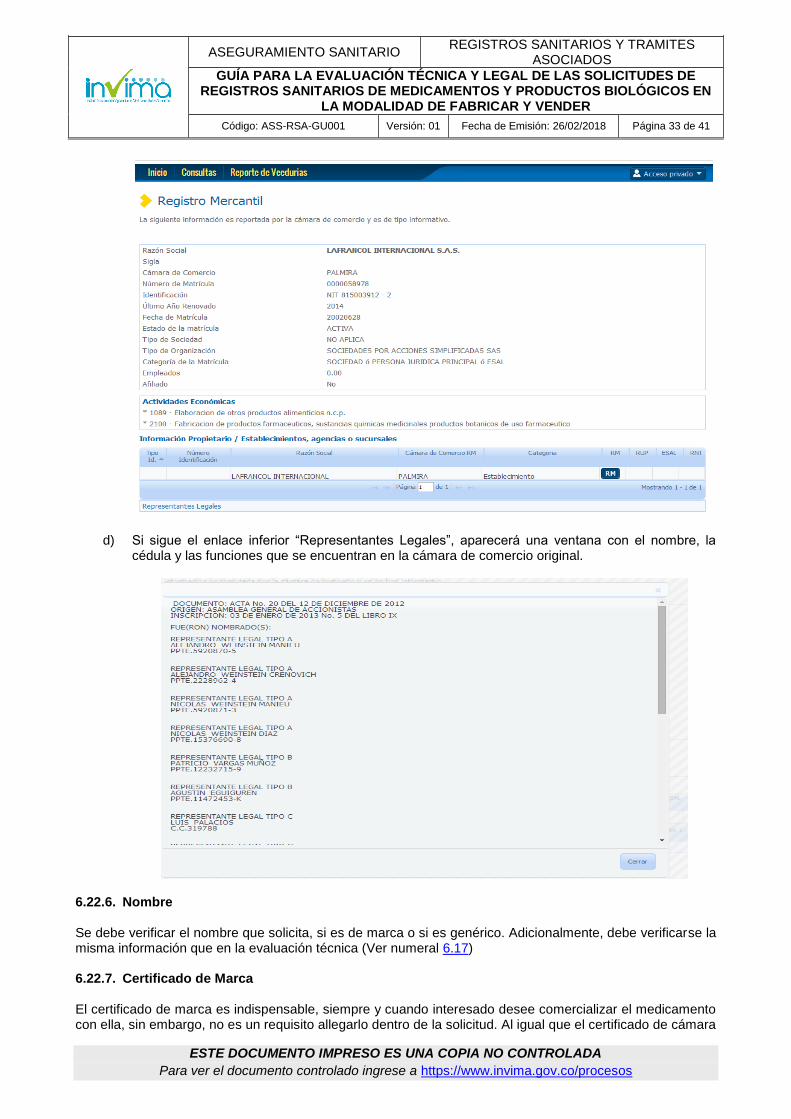
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 33 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
d) Si sigue el enlace inferior “Representantes Legales”, aparecerá una ventana con el nombre, la cédula y las funciones que se encuentran en la cámara de comercio original.
6.22.6. Nombre Se debe verificar el nombre que solicita, si es de marca o si es genérico. Adicionalmente, debe verificarse la misma información que en la evaluación técnica (Ver numeral 6.17) 6.22.7. Certificado de Marca El certificado de marca es indispensable, siempre y cuando interesado desee comercializar el medicamento con ella, sin embargo, no es un requisito allegarlo dentro de la solicitud. Al igual que el certificado de cámara

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 34 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
y comercio, la información puede ser consultada a través del sitio web de la Superintendencia de Industria y Comercio. Ya sea en el certificado o en la consulta en el sitio web, deberá verificar la siguiente información:
La marca debe estar vigente.
El estado de la marca (concedido, en trámite, vencido, entre otros).
El titular de la marca corresponda al titular del Registro Sanitario, en caso que no corresponda, acompañado al certificado de marca se debe allegar autorización para el uso de la marca emitida por el titular de la marca a favor del titular del Registro Sanitario.
Debe corresponder a la clase 5 según la clasificación de NIZA1, la cual sirve para identificar productos farmacéuticos, medicamentos.
La marca puede ser nominativa o mixta. En caso que sea mixta se verifica que el logo de la marca sea el allegado en las etiquetas.
Se concede cuando está la marca en trámite o concedida (pero debe estar en firme). Si la marca se encuentra negada, pero se ha interpuesto recurso de apelación contra dicha negación, podrá otorgarse el registro sanitario haciendo la observación que en caso de que la negación sea confirmada, se le informa al interesado en la parte considerativa de la Resolución que deberá solicitar modificación de cambio de nombre, cambiando la marca y allegando la información respectiva. Para el ingreso de la consulta en el sitio web de la SIC, puede seguir las instrucciones que se desarrollan a continuación:
a) Ingrese a la página web de la SIC. En el enlace Propiedad industrial aparece una lista desplegable, seleccione la opción “Marcas y otros signos distintivos”.
b) Aparecerá la siguiente página web. Siga el enlace Consulta en base de datos, en la opción Marcas y otros Signos Distintivos.
1 La Clasificación de Niza, establecida por el Arreglo de Niza (1957), es una clasificación internacional de productos y servicios que se aplica para el registro de marcas.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 35 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
c) Aparecerá una página con la siguiente información, siga el enlace: Consulta de Marcas, Lemas, Nombres, Enseñas Comerciales y Denominaciones de Origen.
d) Ingrese la información con la denominación y haga clic en el botón Enviar consulta.
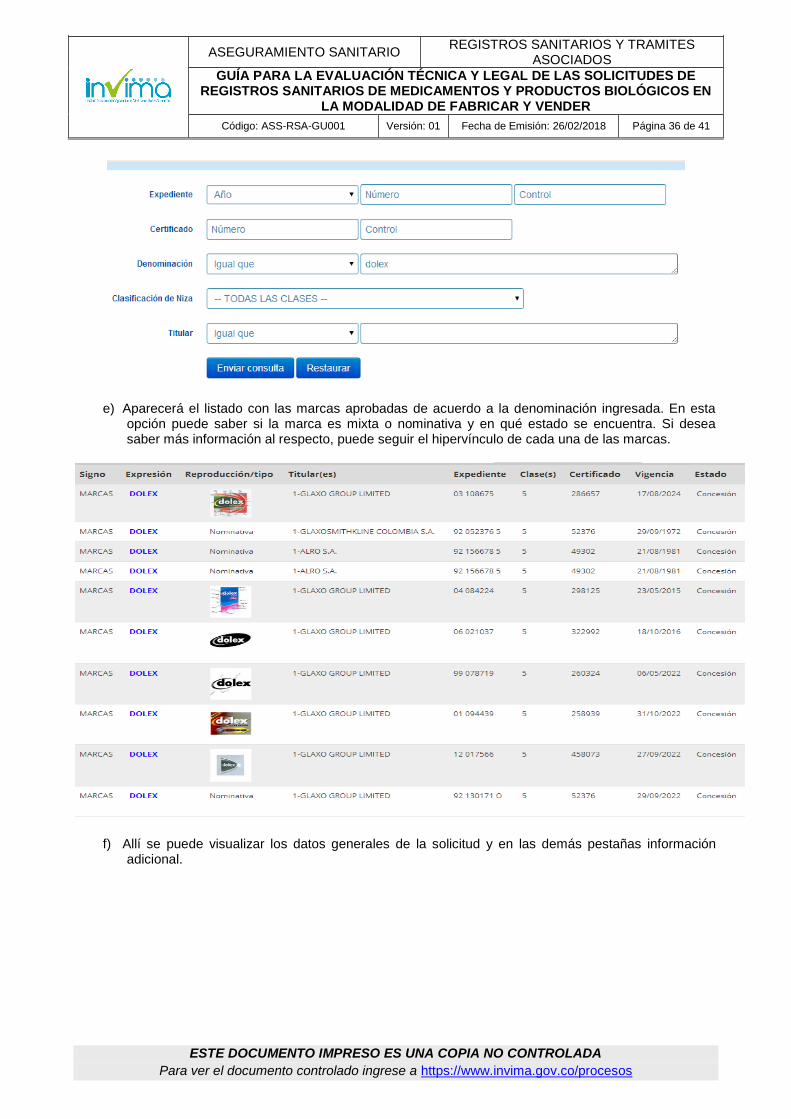
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 36 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
e) Aparecerá el listado con las marcas aprobadas de acuerdo a la denominación ingresada. En esta opción puede saber si la marca es mixta o nominativa y en qué estado se encuentra. Si desea saber más información al respecto, puede seguir el hipervínculo de cada una de las marcas.
f) Allí se puede visualizar los datos generales de la solicitud y en las demás pestañas información
adicional.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 37 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
g) Si desea conocer el historial de la marca, puede hacer ir a la pestaña “Actuaciones y Actos administrativos” y hacer clic en cada actuación para visualizar la resolución y tomar la decisión pertinente.
6.22.8. Autorización del fabricante al importador (Sólo para la modalidad Importar y vender e
Importar, Semielaborar y Vender) Este documento se allega en las solicitudes de los Registros Sanitarios de modalidad Importar y Vender e Importar, Semielaborar y Vender, en donde se verifica que el importador esté autorizado para importar, distribuir, comercializar y/o ser el titular del Registro Sanitario en Colombia. 6.22.9. Poder Cuando el que realiza la solicitud es una persona diferente al titular, debe existir un poder emitido por la sociedad titular y/o importadora a favor de quien lo va a representar, en donde debe incluir como mínimo que lo autorice para realizar la solicitud del Registro Sanitario. En el caso de las modificaciones, debe decir que puede realizar la modificación (si ya lo ha allegado durante el expediente correspondiente al Registro

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 38 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Sanitario no es necesario allegar poder). El poder debe estar firmado por la persona que lo otorga y debe ser otorgado a un abogado. 6.22.10. Certificado de Buenas Prácticas de Manufactura Conforme al Decreto 677 de 1995, la Certificación de Buenas Prácticas de Manufactura, asegura además de la capacidad del fabricante, la calidad de los medicamentos, es ésta la razón por la cual el fabricante del medicamento solicitado y amparado en un Registro Sanitario debe mantener el Certificado de Buenas Prácticas de Manufactura (BPM) vigente, al momento de la radicación de la solicitud, de la expedición del acto administrativo y durante toda la vigencia del registro sanitario.
a) El Certificado de Buenas Prácticas de Manufactura (BPM) debe ser emitido por cualquiera de las autoridades sanitarias de referencia señaladas en el Decreto 162 de 2004, para el producto terminado. En el caso de los productos semielaborados se acepta el certificado del país de origen.
b) Debe incluir el fabricante, acondicionador, envasador y/o semielaborador, según corresponda, con su domicilio y dirección, el cual debe corresponder con el solicitado en el Registro Sanitario.
c) El Certificado de BPM, si se trata de una entidad diferente a Invima, debe traer el sello de apostille o dado el caso, estar consularizado y legalizado y de requerirse, ser traducido oficialmente al español.
d) El Certificado de BPM debe encontrarse vigente al momento de la expedición del Registro Sanitario o Modificación.
6.23. ENTREGA DE FIE LEGAL
Una vez estudiado el trámite, se realiza entrega del Formato de informe de evaluación legal ASS-RSA-FM034 al Coordinador o a la persona delegada para la recepción de los mismos, siguiendo el Instructivo para el manejo del formato de informe de evaluación en el estudio de registros sanitarios de medicamentos y trámites asociados ASS-RSA-IN013 6.24. PASO A LEGAL
Una vez el químico farmacéutico evalúa la información, elabora el informe de evaluación y lo envía, dará el paso por el sistema a la decisión ya sea de aprobación o de requerimiento respecto al trámite dando paso a la evaluación por parte del abogado 6.25. GENERACIÓN DE ACTO ADMINISTRATIVO
6.25.1. Generación de Resolución Si de acuerdo a los hallazgos y conceptos planteados en el informe de evaluación los evaluadores (químico farmacéutico y abogado) emiten una aprobación, generan una resolución, en una plantilla predefinida por el sistema, la cual pasa por sistema al Coordinador o a la persona encargada por éste para revisar el concepto emitido por el profesional. Si el Coordinador o persona responsable no está de acuerdo con el concepto, este lo devuelve por sistema al profesional que lo estudió para que realice la corrección correspondiente, indicándolo en la resolución con una nota resaltada de un color diferente, para que el evaluador ajuste la resolución. En la parte considerativa de la resolución se deben describir los hallazgos efectuados y soportar, conforme a lo plasmado en el Formato de Informe de Evaluación, las razones por las cuales se toma la decisión de aprobar o negar el Registro Sanitario, con el fundamento legal correspondiente.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 39 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.25.2. Generación de Requerimiento o Auto Si por el contrario y de acuerdo a los hallazgos y conceptos planteados en el informe de evaluación, los evaluadores establecen que la información está incompleta o no es clara se emitirán un requerimiento -auto- para que el interesado aclare o complemente la información allegada.
6.26. RECEPCIÓN DE LA RESPUESTA AUTO
Si el usuario da respuesta al requerimiento dentro de los términos establecidos, los profesionales responsables estudian la respuesta tal como si estuviera entrando por primera vez (el que entra primero se evalúa primero), en donde debe diligenciar el Formato para informe de Evaluación respuesta a requerimiento o Evaluación de Recurso de Reposición Medicamentos ASS-RSA-FM035, expresando claramente los resultados encontrados en la respuesta de auto y las razones por las cuales se aprueba o no el Registro Sanitario. Se debe realizar el acto administrativo siguiendo las instrucciones descritas en el numeral 6.26.1.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 40 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Deberá realizarse la entrega de los formatos de informe de evaluación de respuesta a requerimiento (tanto técnico como legal) siguiendo lo descrito en el numeral 7.24. 6.27. RECEPCIÓN DEL DESISTIMIENTO
Si el usuario pasados los términos no ha respondido el requerimiento se da por negada la solicitud de acuerdo a lo establecido en el código contencioso administrativo, Artículo 13, o en caso de que el mismo usuario cierre el tramite sin haber contestado el requerimiento, el abogado será el encargado de generar la resolución de abandono de la solicitud de acuerdo con el Artículo 8 del mismo código tal como se describe en el numeral 6.26.1. 6.28. CORRECCIONES
Las correcciones que el usuario realice, deberán tratarse prioritariamente, una vez aparezcan en el área de trabajo. Deberá diligenciarse el Formato de informe de evaluación correcciones a resoluciones de registro sanitario de medicamentos y trámites asociados ASS-RSA-FM015 y realizar la entrega correspondiente, siguiendo el Instructivo para el manejo del formato de informe de evaluación en el estudio de registros sanitarios de medicamentos y trámites asociados ASS-RSA-IN013.
7. PERIODICIDAD DE REVISIÓN Y ACTUALIZACIÓN DEL PRESENTE DOCUMENTO
Este documento se revisará y actualizará cada vez que surja un avance en el campo de los medicamentos, que modifique y obligue a solicitar nuevo documento no incluido en el presente documento. Cuando alguno de los Decretos o Resoluciones reglamentarias de la legislación sanitaria de medicamentos sea modificada.
8. DOCUMENTOS RELACIONADOS
Guía de evaluación técnica y legal para concesión de registro sanitario con solicitud de protección. Decreto 2085 de 2002 ASS-RSA-GU004 Guía de evaluación técnica y legal para concesión de registro sanitario nuevo para productos biológicos y biotecnológicos ASS-RSA-GU025 Instructivo asignación del ATC a los medicamentos incluidos en la base de datos ASS-RSA-IN003 Instructivo de Visita a Laboratorios Farmacéuticos desde el Grupo de Registros Sanitarios de Medicamentos y Productos Biológicos ASS-RSA-IN009 Instructivo para el manejo del formato de informe de evaluación en el estudio de registros sanitarios de medicamentos y trámites asociados ASS-RSA-IN013 Instructivo para la toma de decisiones y direccionamiento de trámites a Comisión Revisora ASS-RSA-IN014 Instructivo para el manejo de la comunicación al interior del grupo de medicamentos y Productos Biológicos ASS-RSA-IN015 Instructivo para el Ingreso de ATC que no se encuentre creado en la Base de Datos ASS-RSA-IN026 Formato único de medicamentos ASS-RSA-FM004 Formato para informe de evaluación-registro sanitario con solicitud de protección decreto 2085 de 2002 ASS-RSA-FM009 Formato de informe de evaluación – registro sanitario modalidad importar y vender ASS-RSA-FM010 Formato para informe de evaluación técnica Registro Sanitario Medicamentos Fabricación Nacional ASS-RSA-FM011 Formato para informe de evaluación técnica - Registro sanitario productos biológicos ASS-RSA-FM014 Formato de informe de evaluación correcciones a resoluciones de Registro Sanitario de medicamentos y trámites asociados ASS-RSA-FM015

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS EN
LA MODALIDAD DE FABRICAR Y VENDER
Código: ASS-RSA-GU001 Versión: 01 Fecha de Emisión: 26/02/2018 Página 41 de 41
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA
Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Formato de informe de evaluación – modificación a registro sanitário – cambio de nombre del medicamento ASS-RSA-FM022 Formato para Socialización de Información ASS-RSA-FM025 Formato para Reportar un ATC no Creado en el Aplicativo de Registros Sanitarios ASS-RSA-FM027 Formato para control de entrega de FIES del evaluador a auxiliar administrativo ASS-RSA-FM033 Formato de informe de evaluación legal ASS-RSA-FM034 Formato para informe de Evaluación respuesta a requerimiento o Evaluación de Recurso de Reposición Medicamentos ASS-RSA-FM035