
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y … · MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL....
28
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES ASOCIADOS GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 1 de 28 ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos 1. OBJETIVO: Dar a conocer la guía para realizar la evaluación de los documentos radicados con la solicitud del trámite de Registro Sanitario en la modalidad Importar y Vender para un medicamento. 2. ALCANCE: Dirigido al grupo de Registros Sanitarios de Medicamentos y Productos Biológicos de la Dirección de Medicamentos y Productos Biológicos. 3. RESPONSABILIDADES: Es responsabilidad de las personas a quienes está dirigido el presente documento conocerlo y cumplirlo, y de la coordinación del grupo de verificar su cumplimiento, y de los directivos del Instituto suministrar las herramientas bibliográficas, técnicas y logísticas para consulta y aplicación de los conocimientos por parte de los evaluadores. 4. DOCUMENTOS DE REFERENCIA. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 677 de 1995. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/decretos/decreto_677_1995.pdf Recuperado: 06 de junio de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 162 de 2004. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-162-de-2004.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 549 de 2001. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-549-de-2001.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 426 de 2009. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-549-de-2001.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL. Decreto 2078 del 8 de Octubre de 2012. [En Línea]. Disponible en: http://wsp.presidencia.gov.co/Normativa/Decretos/2012/Documents/OCTUBRE/08/DECRETO%202078%20DEL %2008%20DE%20OCTUBRE%20DE%202012.pdf. Recuperado: 31 de diciembre de 2012. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución 2009036096 del 27 de noviembre de 2009.. [En línea] Disponible en: https://www.invima.gov.co/images/stories/normatividad/Resolucion_2009_27_22.pdf Recuperado: Recuperado: 15 de Septiembre de 2014. REPÚBLICA DE COLOMBIA. MINIST ERIO DE SALUD. Resolución No. 3028 de 2008. Por la cual se definen las áreas técnicas de producción de los establecimientos farmacéuticos y se establecen otras disposiciones. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion-3028-de2008.pdf Recuperado: 15 de Septiembre de 2014.
Transcript of ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y … · MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL....

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 1 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
1. OBJETIVO:
Dar a conocer la guía para realizar la evaluación de los documentos radicados con la solicitud del trámite de Registro Sanitario en la modalidad Importar y Vender para un medicamento.
2. ALCANCE: Dirigido al grupo de Registros Sanitarios de Medicamentos y Productos Biológicos de la Dirección de Medicamentos y Productos Biológicos.
3. RESPONSABILIDADES: Es responsabilidad de las personas a quienes está dirigido el presente documento conocerlo y cumplirlo, y de la coordinación del grupo de verificar su cumplimiento, y de los directivos del Instituto suministrar las herramientas bibliográficas, técnicas y logísticas para consulta y aplicación de los conocimientos por parte de los evaluadores. 4. DOCUMENTOS DE REFERENCIA. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 677 de 1995. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/decretos/decreto_677_1995.pdf Recuperado: 06 de junio de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 162 de 2004. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-162-de-2004.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 549 de 2001. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-549-de-2001.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. PRESIDENCIA DE LA REPUBLICA. Decreto 426 de 2009. [En Línea]. Disponible en: https://www.invima.gov.co/images/pdf/productos-fitoterapeuticos/decretos/decreto-549-de-2001.pdf Recuperado: 15 de Septiembre de 2014. COLOMBIA. MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL. Decreto 2078 del 8 de Octubre de 2012. [En Línea]. Disponible en: http://wsp.presidencia.gov.co/Normativa/Decretos/2012/Documents/OCTUBRE/08/DECRETO%202078%20DEL%2008%20DE%20OCTUBRE%20DE%202012.pdf. Recuperado: 31 de diciembre de 2012. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución 2009036096 del 27 de noviembre de 2009.. [En línea] Disponible en: https://www.invima.gov.co/images/stories/normatividad/Resolucion_2009_27_22.pdf Recuperado: Recuperado: 15 de Septiembre de 2014. REPÚBLICA DE COLOMBIA. MINIST ERIO DE SALUD. Resolución No. 3028 de 2008. Por la cual se definen las áreas técnicas de producción de los establecimientos farmacéuticos y se establecen otras disposiciones. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion-3028-de2008.pdf Recuperado: 15 de Septiembre de 2014.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 2 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Acuerdo No. 228 de 2002. Por medio del cual se actualiza el Manual de Medicamentos del Plan Obligatorio de Salud y se dictan otras disposiciones. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/acuerdos/ministerio-de-salud.pdf . Recuperado: 15 de septiembre de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Acuerdo No. 282 de 2004. Por el cual se fija el valor de la Unidad de Pago por Capitación del Plan Obligatorio de Salud de los Regímenes Contributivo y Subsidiado para el año 2005 y se dictan otras disposiciones. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/acuerdos/acuerdo_282_2004.pdf. Recuperado: 15 de Septiembre de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Acuerdo No. 228 de 2002. Por medio del cual se actualiza el Manual de Medicamentos del Plan Obligatorio de Salud y se dictan otras disposiciones. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/acuerdos/ministerio-de-salud.pdf. Recuperado: 15 de Septiembre de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución 5521 de 2013. Por la cual se define, aclara y actualiza integralmente el Plan Obligatorio de Salud (POS). [En línea] Disponible en: http://www.minsalud.gov.co/sites/rid/Lists/BibliotecaDigital/RIDE/DE/DIJ/Resoluci%C3%B3n%205521%20de%202013.pdf. Recuperado: 15 de Septiembre de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución Nº 1400 de agosto de 2001. Por la cual se establece la Guía Biodisponibilidad y de Bioequivalencia de Medicamentos que trata el Decreto 677 de 1995. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion-1400.pdf Recuperado: 15 de Septiembre de 2014. SUIZA. ORGANIZACIÓN MUNDIAL DE LA SALUD. Serie de Informes técnicos de la OMS 823. Informe 32, acogido por la Resolución No. 3183 de 23 de agosto 1995. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/informes/informe32delaOMScompleto.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Guía técnica de análisis de medicamentos, acogida por el decreto 677 de 1995, artículo 89. [En línea] Disponible en: http://apps.who.int/medicinedocs/documents/s18381es/s18381es.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Decreto 019 de 2012. Por el cual se dictan normas para suprimir o reformar regulaciones, procedimientos y trámites innecesarios existentes en la administración pública. [En línea] Disponible en: http://wsp.presidencia.gov.co/Normativa/Decretos/2012/Documents/Enero/10/Dec1910012012.pdf Recuperado: 4 de marzo de 2014 REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución No. 1890 de noviembre de 2001. Por la cual se modifica la Resolución No. 1400 de 2001. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion_1890_2001.pdf Recuperado: 4 de marzo de 2014 REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD Y PROTECCIÓN SOCIAL. LISTADO DE MEDICAMENTOS DE CONTROL ESPECIAL USO HUMANO. [En línea] Disponible en: http://www.fne.gov.co/Index.aspx?Id=14097 Recuperado: 4 de marzo de 2014

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 3 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. MEDICAMENTOS POS. [En línea] Disponible en: http://www.minsalud.gov.co/salud/POS/Paginas/pospopuli.aspx Recuperado: 4 de marzo de 2014. INSTITUTO NACIONAL DE VIGILANCIA DE MEDICAMENTOS Y ALIMENTOS – INVIMA - Actas de Comisión Revisora (Sala Especializada de Medicamentos). [En línea] Disponible en: https://www.invima.gov.co/index.php?option=com_content&view=article&id=1076%3Asala-especializada-de-medicamentos-y-productos-biologicos&catid=239%3Asala-especializada-de-medicamentos-y-productos-bio&Itemid=581. Recuperado: 4 de marzo de 2014 REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución No. 0255 de 2007. Por la cual se adopta el Código Único de Medicamentos, CUM. [En línea] Disponible en: http://www.minsalud.gov.co/Normatividad/RESOLUCI%C3%93N%200255%20DE%202007.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución 2004008172 de 2004. Por la cual se adopta una fe de erratas del Manual de Normas Técnicas de Calidad Guía Técnica de Análisis. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion_2004008172_2004.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución 2514 de julio de 1995. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion_2514_1995.pdf Recuperado: 4 de marzo de 2014. Guía Práctica de Requisitos para el Desarrollo de Estudios de Estabilidad de Medicamentos acogida por la Resolución 2514 de 1995. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/Guia_Estabilidad_para_Colombia.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE SALUD. Resolución No. 3183 de 23 de agosto 1995. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion_3183_1995.pdf REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución No. 1672 de 2004. Por la cual se adopta el Manual de Buenas Prácticas de Manufactura de los Gases Medicinales. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion_001672_2004.pdf Recuperado: 4 de marzo de 2014. REPÚBLICA DE COLOMBIA. MINISTERIO DE LA PROTECCIÓN SOCIAL. Resolución No. 1478 de mayo 10 de 2006. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/resoluciones/resolucion%20001478%20de%202006.pdf Recuperado: 4 de marzo de 2014. Circular Externa DG-100-007-07. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/circular-externa/circularexterna_DG-100-007-07.pdf CIRCULAR INTERNA No 003 DE 2003 DE MEDICAMENTOS. [En línea] Disponible en: https://www.invima.gov.co/images/pdf/medicamentos/circular-interna/circular_interna_003_2003.pdf Conceptos Internos y Externos Vigentes emitidos por las Direcciones Misionales del INVIMA, el Ministerio de la Protección Social, la Sala especializada de Medicamentos y Productos Biológicos de la Comisión Revisora Social y otras entidades cuyos conceptos apliquen para la presente guía.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 4 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Guía ICH Q1A(R2), Stability Testing of New Drug Substances and ProductsGuía ICH Q1D Evaluation for Stability DataGuía ICH Q1E Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and ProductsGuía ICH Q1F Stability Data Package for Registration Applications in Climatic Zones III and IV. Farmacopeas Oficiales en Colombia: USP Estados Unidos, BP Británica, DAB Alemana, WHO/Farmacopea Internacional y Codex Francés. 5. DOCUMENTOS REQUERIDOS PARA REALIZAR LA EVALUACIÓN TECNICA: 5.1. Todos los citados en los artículos 22 y 31 del decreto 677 de 1995 y los establecidos en el Decreto 426 de 2009 a saber:
Certificado de Producto Farmacéutico y/o Certificado de Venta Libre.
Certificado de Cumplimiento de Buenas Prácticas de Manufactura.
Descripción de la forma farmacéutica y presentación comercial.
Fórmula estructural y condensada de los principios activos.
Composición cualitativa y cuantitativa del producto.
Formula del lote estandarizado de fabricación.
Descripción detallada del proceso de fabricación.
Especificaciones de calidad de las materias primas.
Especificaciones de calidad y descripción de los controles en proceso realizados al producto durante el proceso de fabricación.
Especificaciones de calidad para el producto terminado.
Metodología de análisis del producto terminado y Validación de este si lo requiere.
Muestra Física y/o Boceto del arte para impresión de etiquetas y empaques (por duplicado y a color),
Resumen de la información farmacológica.
Estudios de estabilidad y periodo de vida útil del producto.
Resultados de los estudios de biodisponibilidad (farmacocinéticos) para los productos que así lo defina Comisión revisora.
5.2 RECURSOS PARA EL DESARROLLO Y EJECUCIÓN DE LA VALORACIÓN TÉCNICA Y LEGAL 5.2.1 Aplicativo de Registro Sanitario
Este aplicativo tiene como objeto el apoyo, generación, gestión y trazabilidad de los trámites que son radicados ante la Dirección de Medicamentos y Productos Biológicos. Para el ingreso al aplicativo, el coordinador del Grupo de medicamentos debe solicitar la creación del usuario a la Oficina de tecnologías de la información. Creado el usuario, el funcionario responsable debe cambiar la contraseña y con esto queda habilitado para el ingreso. Si ya posee usuario del aplicativo de registro sanitario puede ingresar directamente a él de la siguiente manera: 1. Ingresar con el usuario y la contraseña asignados.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 5 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
2. Ingresar a la opción registro sanitario y desplegar la lista de opciones. Elegir área de trabajo
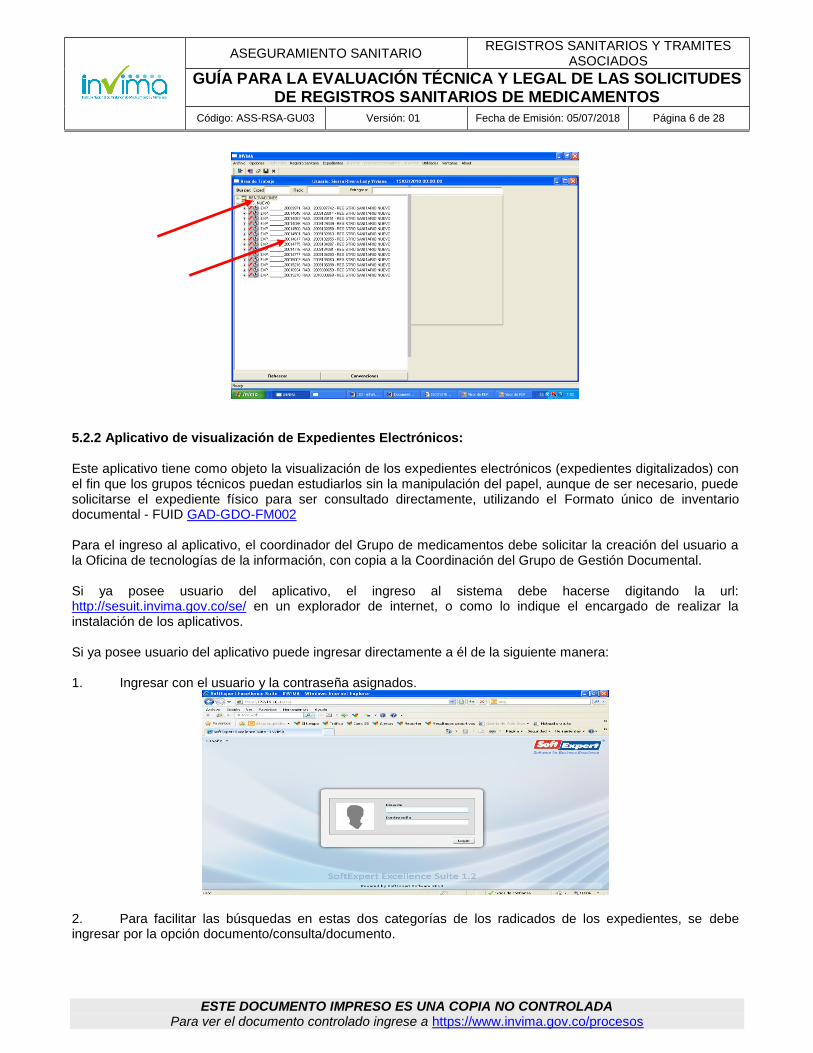
3. En el área de trabajo se presentan los trámites asignados divididos en carpetas por tipos de trámites y en cada carpeta van presentados por orden de radicación, respetando el derecho de turno. Las carpetas son: Autorizaciones, Certificaciones, Modificaciones, R.S. Nuevo, Renovaciones revisiones de Oficio, Correcciones, Otros trámites (desgloses, cancelaciones, etc.) La visualización de la pantalla es la siguiente:
4. Desplegar las carpetas en las cuales va a realizar el estudio, allí aparecerá un listado que relaciona el expediente y el número de radicado, con el historial de los pasos que ha recorrido el trámite, desde el momento de la radicación hasta la asignación en la pantalla con las fechas en que estos pasos se realizaron. Identificar el trámite que tenga la fecha más antigua.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 6 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
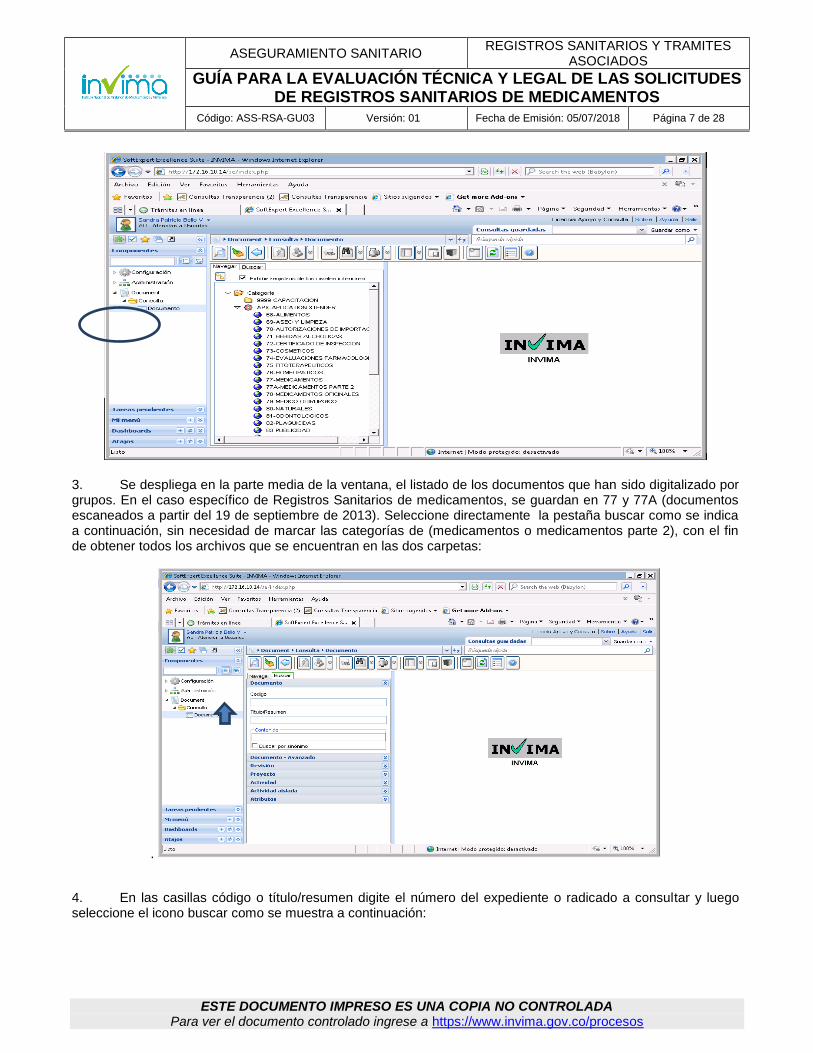
5.2.2 Aplicativo de visualización de Expedientes Electrónicos: Este aplicativo tiene como objeto la visualización de los expedientes electrónicos (expedientes digitalizados) con el fin que los grupos técnicos puedan estudiarlos sin la manipulación del papel, aunque de ser necesario, puede solicitarse el expediente físico para ser consultado directamente, utilizando el Formato único de inventario documental - FUID GAD-GDO-FM002 Para el ingreso al aplicativo, el coordinador del Grupo de medicamentos debe solicitar la creación del usuario a la Oficina de tecnologías de la información, con copia a la Coordinación del Grupo de Gestión Documental. Si ya posee usuario del aplicativo, el ingreso al sistema debe hacerse digitando la url: http://sesuit.invima.gov.co/se/ en un explorador de internet, o como lo indique el encargado de realizar la instalación de los aplicativos. Si ya posee usuario del aplicativo puede ingresar directamente a él de la siguiente manera: 1. Ingresar con el usuario y la contraseña asignados.
2. Para facilitar las búsquedas en estas dos categorías de los radicados de los expedientes, se debe ingresar por la opción documento/consulta/documento.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 7 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
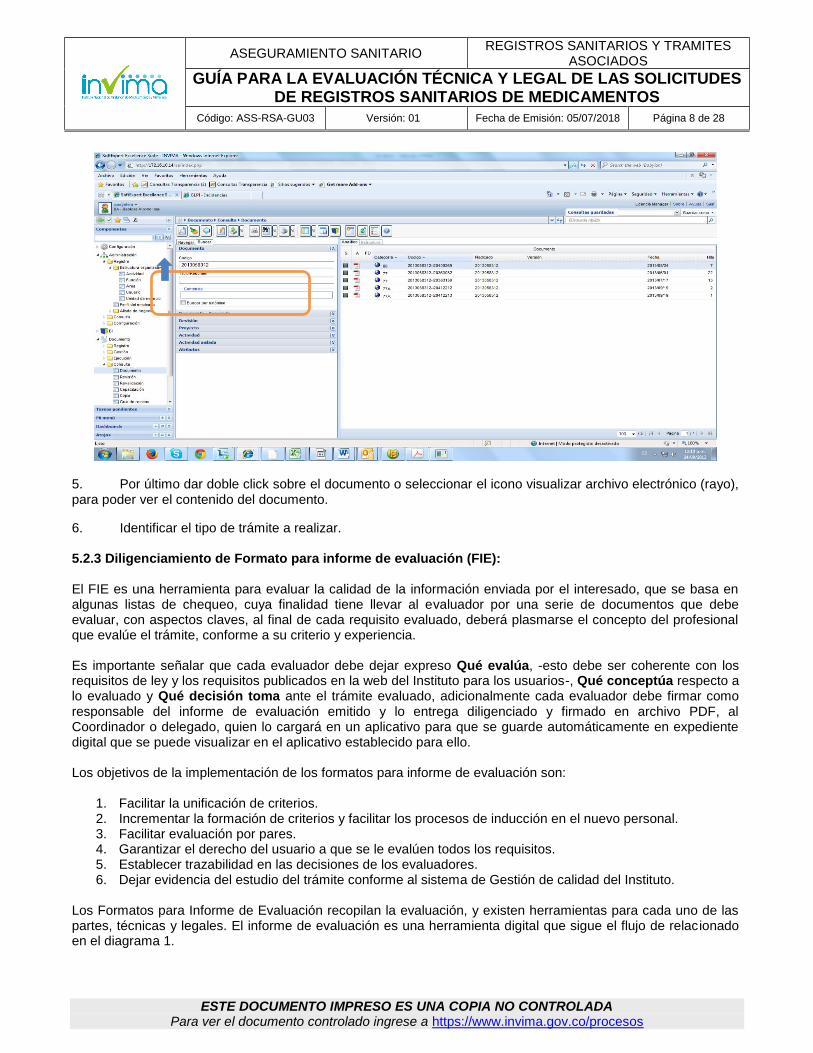
3. Se despliega en la parte media de la ventana, el listado de los documentos que han sido digitalizado por grupos. En el caso específico de Registros Sanitarios de medicamentos, se guardan en 77 y 77A (documentos escaneados a partir del 19 de septiembre de 2013). Seleccione directamente la pestaña buscar como se indica a continuación, sin necesidad de marcar las categorías de (medicamentos o medicamentos parte 2), con el fin de obtener todos los archivos que se encuentran en las dos carpetas:
.
4. En las casillas código o título/resumen digite el número del expediente o radicado a consultar y luego seleccione el icono buscar como se muestra a continuación:
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 8 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
5. Por último dar doble click sobre el documento o seleccionar el icono visualizar archivo electrónico (rayo), para poder ver el contenido del documento.
6. Identificar el tipo de trámite a realizar. 5.2.3 Diligenciamiento de Formato para informe de evaluación (FIE): El FIE es una herramienta para evaluar la calidad de la información enviada por el interesado, que se basa en algunas listas de chequeo, cuya finalidad tiene llevar al evaluador por una serie de documentos que debe evaluar, con aspectos claves, al final de cada requisito evaluado, deberá plasmarse el concepto del profesional que evalúe el trámite, conforme a su criterio y experiencia. Es importante señalar que cada evaluador debe dejar expreso Qué evalúa, -esto debe ser coherente con los requisitos de ley y los requisitos publicados en la web del Instituto para los usuarios-, Qué conceptúa respecto a lo evaluado y Qué decisión toma ante el trámite evaluado, adicionalmente cada evaluador debe firmar como responsable del informe de evaluación emitido y lo entrega diligenciado y firmado en archivo PDF, al Coordinador o delegado, quien lo cargará en un aplicativo para que se guarde automáticamente en expediente digital que se puede visualizar en el aplicativo establecido para ello. Los objetivos de la implementación de los formatos para informe de evaluación son:
1. Facilitar la unificación de criterios. 2. Incrementar la formación de criterios y facilitar los procesos de inducción en el nuevo personal. 3. Facilitar evaluación por pares. 4. Garantizar el derecho del usuario a que se le evalúen todos los requisitos. 5. Establecer trazabilidad en las decisiones de los evaluadores. 6. Dejar evidencia del estudio del trámite conforme al sistema de Gestión de calidad del Instituto.
Los Formatos para Informe de Evaluación recopilan la evaluación, y existen herramientas para cada uno de las partes, técnicas y legales. El informe de evaluación es una herramienta digital que sigue el flujo de relacionado en el diagrama 1.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 9 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
En el caso de la evaluación de un Registro Sanitario nuevo o renovación, debe remitirse a los formatos:
Formato de informe de evaluación -registro sanitario modalidad importar y vender ASS-RSA-FM010. El Informe debe evidenciar claramente la evaluación de todos y cada uno de los requisitos de ley para el trámite, debe plasmar los conceptos técnico científicos y farmacéuticos emitidos por el evaluador en cada punto y debe reflejar claramente que decisión toma el evaluador, respecto a la calidad de la información enviada por el solicitante.
Diagrama 1. Flujo del Formato de Informe de Evaluación
Los Formatos para Informe de Evaluación (FIE), recopilan la evaluación, y existen herramientas para cada uno de las partes, técnicas y legales. El informe de evaluación es una herramienta digital que sigue el flujo de relacionado en el diagrama 1.
6 DESARROLLO DE LA GUIA
6.1 El profesional revisa y verifica por sistemas en el aplicativo de registros sanitarios en el área de trabajo, que el encargado de repartir los trámites le haya hecho la correspondiente asignación de los mismos. 6.2 Se inicia el estudio de un trámite haciendo la selección del mismo en la opción área de trabajo, eligiendo un radicado, por mandato constitucional se debe respetar el derecho a turno, esto se traduce en resolver los trámite más antiguos en estricto orden de fecha de radicación.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 10 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.3 Una vez seleccionado el tramite se busca este mismo radicado en el aplicativo de Sesuite para obtener la visualización del expediente con toda la información que contiene el tramite a estudiar 6.4 En el aplicativo de registros sanitarios en el área de trabajo, completar datos del producto, llenando la base de datos a partir de la información escaneada y visualizada en el aplicativo de Sesuite para el expediente y radicado correspondiente

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 11 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.5 EVALUACIÓN FARMACÉUTICA DE LA INFORMACIÓN TÉCNICA 6.5.1 Observaciones y consideraciones generales sobre la evaluación de la información técnica: En
este paso se inicia propiamente el análisis de la calidad de la información enviada por el interesado y para ello los evaluadores deben tener en cuenta todos los soportes técnico científicos que reporta la literatura y las redes de información internacional por ejemplo: reportes tanto nacionales como internacionales sobre alertas de medicamentos emitidos por ANR –FDA, EMEA, ANVISA, ANMAT, etc-, OPS, OMS, FDA, EMEA etc, las normas farmacológicas vigentes, el manual de normas técnicas colombianas vigente, las farmacopeas oficiales en Colombia: la de Estados Unidos de América (USP), Británica (BP), Alemana (DAB), así mismo las guías ICH, Los criterios de grupo adoptados mediante actas resultantes de las reuniones del grupo de Registros Sanitarios de Medicamentos y Productos Biológicos, los conceptos emitidos por la Sala Especializada de Medicamentos de la Comisión Revisora (SEMPB) y sala de dispositivos médicos si es el caso, los conceptos emitidos por la oficina jurídica o cualquier dependencia del Invima, o del Ministerio Salud y la Protección Social. Así mismo es obligación del evaluador elevar consultas escritas a otras dependencias tales como la SEMPB, a la Oficina de Laboratorios y Control de Calidad de Invima, grupo Técnico de Medicamentos, Grupo de Farmacovigilancia, entre otros, en aras de dar claridad y solventar cualquier duda sobre la calidad, coherencia e integralidad de la información que está evaluando, así mismo tiene el deber de solicitar recolección de muestras o solicitar y de ser posible realizar visitas en caso que el tramite evaluado lo amerite. Para realizar la visita e inspeccionar la validación de procesos se podrá hacer de acuerdo al Instructivo de visita a laboratorios farmacéuticos desde el grupo de registros sanitarios de medicamentos de la dirección de medicamentos y productos biológicos ASS-RSA-IN009
En todo caso el evaluador no puede dejar pasar ninguna inconsistencia sin haber tomado las medidas del caso y documentarlas. Adicionalmente y de manera específica para cada requisito de ley, el evaluador debe tener en cuenta los aspectos referidos a continuación los cuales en su mayoría se explican con más detalle en el numeral 6.5.2: a. Nombre del medicamento: Verificar que no se infrinja el Artículo 78 del Decreto 677/1995 parágrafo 1 que
establece: “No se otorgará registro sanitario a medicamentos de igual composición, pero con diferente nombre, a favor de un mismo titular”, haciendo la respectiva búsqueda en la base de datos de registros sanitarios del Invima. Se debe evaluar si el nombre se ajusta a parámetros de moderación científica, si revisada la base de datos del instituto el nombre puede confundirse con el nombre de otro medicamento previamente aprobado, si el nombre va acompañado de siglas tales como NF, HR, etc, en todo caso debe

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 12 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
haber explicación del significado y si hace referencia a una propiedad de la forma farmacéutica esto debe ser verificado con los datos del expediente.
b. Normas Farmacológicas: Verificar que el principio activo o combinación de principios activos, la concentración y la forma farmacéutica se encuentre en normas farmacológicas. En caso de no encontrarse en el mismo listado, se requerirá al interesado mediante auto para que solicite el estudio de la evaluación farmacológica ante la Comisión Revisora, Sala especializada de medicamentos y envié la evidencia de solicitud del mismo estudio (ésta última facilitará la trazabilidad al trámite de evaluación farmacológica internamente) de igual manera se deben buscar en la red de información internacional y nacional alertas farmacológicas de seguridad del medicamento y en caso que las haya solicitar ante la SEMPB pronunciamiento al respecto, esto de acuerdo a la Resolución No. 2009036096 del 27 de noviembre de 2009.
b. Productos nuevos de origen biológico o biotecnológico: (Ej: Hormonas peptídicas, Hemoderivados,
Vacunas, entre otros) deben presentar obligatoriamente evaluación farmacológica como requisito previo para su evaluación aunque estén ya incluidos en normas farmacológicas, por concepto técnico de la SEMPB. En caso de no haber sido evaluado por la SEMPB se redireccionará el trámite a éste órgano asesor, quien conceptuará mediante acta, de manera aprobatoria o no: si el concepto es aprobatorio, el evaluador debe tener presente la Guía de evaluación técnica y legal para concesión de registro sanitario nuevo para productos biológicos y biotecnológicos (ASS-RSA-GU025) y diligenciar el formato de evaluación en ella referido, posteriormente debe continuar con el estudio farmacéutico descrito en este mismo documento, de lo contrario, si el concepto de la SEMPB no aprueba el producto, se requerirá al interesado mediante auto de comisión revisora, en los aspectos determinados. Para el informe de evaluación de la parte específica a Biológicos/biotecnológicos se debe seguir la guía correspondiente (ASS-RSA-GU025) y el formato de evaluación adjunto a la misma.
c. Solicitud de protección de datos( Decreto 2085/2002): el evaluador debe proceder con la evaluación específica y diligenciar el formato de evaluación correspondiente, los aspectos específicos a tener en cuenta en esta evaluación son: consultar las actas de la SEMPB y verificar si existe el concepto de aceptación del nuevo medicamento bajo los lineamientos del decreto 2085 de 2002, revisar si se presenta debidamente diligenciado el formulario de solicitud de protección a la información no divulgada, si incluye la declaración correspondiente a la realización de esfuerzo considerable, si incluye la declaración correspondiente a los títulos y rangos de folios y/o folios específicos de la evaluación farmacológica referentes a la información que desea proteger y si corresponden exactamente con los establecidos en la documentación de Evaluación Farmacológica que suministra a la SEMPB, si en la información que solicitan proteger incluyen capítulos secciones o folios correspondientes a la información farmacéutica (síntesis, caracterización del activo, estructura molecular, control de calidad, método de fabricación, etc). Si La información objeto de protección es coherente con los procedimientos mundialmente aceptados en cuanto a Investigación y Desarrollo de nuevos medicamentos (estudios toxicológicos, farmacocinéticos, farmacodinámicos, estudios preclínicos, estudios clínicos, entre otros), si dentro de la información objeto de protección se encuentran referencias bibliográficas (en el texto, como pie de página o al final de cada capítulo) que nombren revistas de índole científico en donde se haya publicado alguna información relevante sobre la información objeto de protección, el evaluador debe buscar evidencia en cuanto a que la información que se declara como no divulgada se encuentra divulgada, realizando la búsqueda en internet a través de motores de búsqueda, portales reconocidos de información médica y científica, organismos o Agencias Regulatorias Nacionales, Regionales o Continentales, Instituciones Académicas, entre otros. Una vez terminado este estudio específico el evaluador debe continuar con el estudio farmacéutico descrito en este mismo documento y continuar con el diligenciamiento del formato de evaluación para Importar y vender, para el informe de evolución de la parte específica a la protección se debe seguir la guía PM01-RS-G6 y el formato de evaluación adjunto a la misma. Una vez evaluada la parte de la protección continúa el estudio evaluando los demás aspectos referidos en esta guía.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 13 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
d. Indicaciones, contraindicaciones, advertencias y/o posología: Verificar en las actas de la SEMPB y evaluar si al medicamento se le han ampliado o modificado las indicaciones, contraindicaciones, advertencias y/o posología, también si el producto ha tenido llamados a Revisión de oficio, alertas nacionales o internacionales u otros requerimientos u observaciones que pudieran afectar la evaluación de la información para la concesión del registro sanitario.
e. Evaluación del certificado de venta libre o certificado de producto farmacéutico: debe adjuntarse un certificado de producto farmacéutico o certificado de venta libre que cumpla con los requisitos técnicos establecidos en el Decreto 426 de 2009 que entre otros aspectos corresponde a: el (o los) principio(s) activo(s), la forma farmacéutica, la concentración del producto, los fabricantes y los envasadores y los dispositivos si es el caso.
f. Cumplimiento de las BPM: Revisar para el laboratorio fabricante de los lotes del producto, cuya
información debe anexarse, el cumplimiento de las BPM las cuales deberán ser emitidas por el Invima o por un país de referencia (Decreto 162 de 2004, Decreto 549 de 2001). Así mismo se tendrá en cuenta los conceptos generados por el Grupo Técnico de Medicamentos sobre la materia. Es importante tener en cuenta que en el certificado de BPM debe estar claro si el laboratorio tiene capacidad para determinado tipo de moléculas (antineoplásicos, antibióticos, etc), capacidad para determinadas formas farmacéuticas, (inyectables, cápsulas blandas, etc.), capacidad para determinados procesos productivos: (envase pequeño/gran volumen; envase de jeringas prellenados, etc), o procesos que requieren cadena de frió, de igual manera estas características se deben tener en cuenta si se solicita acondicionamiento en planta diferente a la de fabricación. Adicionalmente es importante evaluar la fecha de vigencia del certificado contra la fecha de fabricación de los lotes reportados en los estudios de estabilidad.
g. Presentación comercial y forma farmacéutica: verificar que la presentación comercial esté descrita indicando el color, clase y tipo de material de envase y empaque, en el caso de que el producto se presente en blister cada presentación indicará el número de unidades por blister. En cuanto a la forma farmacéutica, revisar si corresponde con las consignadas en la literatura farmacéutica como farmacopeas oficiales en Colombia u otras formas farmacéuticas nuevas aprobadas en actas de la SEMPB. Tener presente si la presentación comercial incluye dispositivos médicos y los mismos se encuentran legislados por el Decreto 4725 de 2005.
h. Composición cualicuantitativa, formula estructural y fórmula del lote estandarizado: Verificar que la
composición cualicuantitativa sea expresada según lo que se establece en el literal c del artículo 22 del decreto 677/1995.
Confirmar que cada sustancia incluida en la fórmula del medicamento esté identificada con nombre genérico y químico (IUPAC), y no con nombres de marca; para el activo se escribirá su nombre según la Denominación Común Internacional (DCI).
Revisar que se presenten de forma separada los excipientes de los principios activos. El activo debe ser descrito con certeza indicando si corresponde a una sal, éster o solvato. Si es una sal se deberá expresar su contenido como tal, o por el contrario aclarar si corresponde al equivalente de la forma ácida, básica o anhidra de la sustancia activa. En ningún caso los excipientes pueden tener actividad terapéutica.
Verificar que la fórmula cualicuantitativa establecida corresponda con la que se describe en el Certificado de Producto Farmacéutico o Certificado de Venta Libre.
Verificar que la formula estructural y condensada del principio activo, corresponda con el activo solicitado.
Verificar que la fórmula del lote estandarizado de fabricación corresponda proporcionalmente a la del lote industrial proyectado y a la formulación por unidad posológica (ejemplo por tableta).
No se aceptará los excesos en el principio activo para compensar la degradación en los estudios de estabilidad. Si el fabricante agrega excesos para compensar pérdidas en el proceso productivo debe

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 14 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
establecer claramente la magnitud de estos excesos.
i. Proceso de fabricación: Revisar la descripción y/o diagrama de flujo detallado del proceso de fabricación del medicamento, el cual debe estar en orden lógico y estar acorde con el principio activo, excipientes y la forma farmacéutica producida.
j. Controles en proceso: Revisar los controles en proceso y sus especificaciones que se llevan a cabo dentro de la manufactura del medicamento.
k. Especificaciones de calidad de las materias primas: estas se deben verificar (excipientes, principios activos, intermediarios de fabricación y otros).
l. Especificaciones de calidad del producto terminado y metodología de análisis: Verificar las especificaciones de calidad del producto terminado, descripción detallada de la metodología analítica utilizada para el control de calidad del producto y la validación de la metodología analítica para la valoración o cuantificación del principio activo en el medicamento.
m. Estudio de estabilidad: Evaluar el Protocolo, Resultados y conclusiones sobre el estudio de estabilidad de medicamento para el otorgamiento de vida útil.
n. Artes del material de envase y empaque: Evaluar los artes de material de envase y empaque o las muestras físicas de estos con base en los artículos 72 o 74 del decreto 677/1995 (según corresponda).
6.5.2 Observaciones y consideraciones particulares sobre la evaluación de la información técnica. 6.5.2.1 Información General del Producto Se compone del nombre del producto, principio activo, forma farmacéutica, roles de las empresas registradas con su domicilio (titular, fabricante, envasador, acondicionador, importador, etc). Esta información se diligencia en el formato de informe de evaluación de los registros sanitarios nuevos o renovaciones y tiene la finalidad de resumir los parámetros a tener en cuenta durante la revisión. 6.5.2.2 Información Farmacológica En este numeral, se debe revisar la inclusión en normas farmacológicas, en actas de la SEMPB, alertas internacionales, llamados a revisión de oficio del principio activo, información aprobada para el principio activo de las indicaciones, contraindicaciones y advertencias, dosis y frecuencia de administración, condición de venta, condición del medicamento (esencial, controlado), vía de administración, ATC. Se sugiere la búsqueda de la información en los aplicativos disponibles para su uso: Actas de Comisión Revisora, o el consolidado de las actas de comisión revisora, las cuales se encuentra en el controlador de dominio y en dado caso la información publicada en la web por agencias sanitarias de referencia para Colombia, Ministerio de Salud y Protección social (o quien haga sus veces), bases de datos a las que se tenga acceso, OMS, entre otros. Verificar que el principio activo o combinación de principios activos, la concentración y la forma farmacéutica se encuentre en la norma farmacológica asignada en la evaluación farmacológica, o verificar si hubo alguna actualización o manifestación de la SEMPB respecto al producto o productos similares. En caso de no encontrarse en el mismo listado o incluido mediante acta, se requerirá al interesado mediante auto para que solicite el estudio de la evaluación farmacológica ante la SEMPB y que en la respuesta del requerimiento

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 15 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
adjunte la evidencia de solicitud realizada a la Sala, (ésta última facilitará la trazabilidad al trámite de evaluación farmacológica internamente) En este punto se debe tener en cuenta que coincidan, entre otros, los aspectos relacionados a continuación: forma farmacéutica, vía de administración, dosis y frecuencia de la administración, indicaciones farmacológicas y uso terapéutico, contraindicaciones, efectos secundarios y advertencias y condición de venta, los cuales debe coincidir con lo informado en la solicitud. 6.5.2.3 Llamado a revisión de oficio Verificar en la base de datos de las actas de Comisión Revisora si hay algún llamado a revisión de oficio para el principio activo relacionado con información de seguridad y/o eficacia. De ser así, hay dos opciones: a. Que la evaluación farmacológica tenga una fecha de expedición anterior a la fecha del llamado a revisión de oficio, por lo cual, no hay lugar a una corrección y la información se incluirá en el registro sanitario dejando tres posibilidades para dar continuidad al trámite:
Llamado a revisión de oficio por indicaciones: estas se incluirán directamente en el registro sanitario y sólo en el caso que no soliciten inserto o información para prescribir, no será necesario realizar requerimiento para que se incluyan las nuevas indicaciones.
Llamado a revisión de oficio por contraindicaciones y advertencias: se realizará requerimiento para que se incluyan en los artes envase y empaque y de ser necesario en los insertos, información para prescribir, etc.
Llamado a revisión de oficio por seguridad y eficacia: se realizará requerimiento para que se allegue la documentación de seguridad y sea evaluado por la Sala Especializada.
b. Que la evaluación farmacológica tenga una fecha de expedición posterior a la fecha del llamado a revisión de oficio y la información no se encuentre contenida dentro de la evaluación farmacológica, debe emitirse un requerimiento en donde le solicite al interesado que envíe la corrección a la evaluación farmacológica y se realice la aclaración para continuar con el trámite. 6.5.2.4 Alertas internacionales Verificar las páginas web de otras agencias tales como EMA, FDA, entre otras, para saber si existen alertas farmacológicas de seguridad del medicamento. Si se encuentra una alerta emitida por estas agencias, se debe informar al grupo de Farmacovigilancia al correo [email protected], el grupo se encargará de realizar la gestión de acuerdo con el instructivo para la gestión de la información de seguridad sobre medicamentos y productos biológicos. 6.5.2.5 Biodisponibilidad y/o Equivalencia terapéutica En este numeral se debe revisar si es necesario contar con estudios de biodisponibilidad y/o equivalencia, de acuerdo con la normatividad vigente, siguiendo los lineamientos del Acta 19 de 2002, numeral 2.3.13. Se verifica principalmente si están aprobados dichos estudios por la Sala Especializada de Medicamentos y Productos Biológicos, en qué actas y el concepto de la Sala. Si no se encuentra el acta, deberá requerirse y solicitar al interesado que envíe los estudios farmacocinéticos o de biodisponibilidad y bioequivalencia a la Sala Especializada de Medicamentos y Productos Biológicos de la Comisión Revisora para que sean aprobados y que se envíe una copia de la solicitud para que se de continuidad al trámite una vez se tenga un concepto.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 16 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.5.2.6 Protección de datos Este es un numeral de carácter informativo, el evaluador debe seguir la Guía de evaluación técnica y legal para concesión de registro sanitario nuevo con solicitud de protección Decreto 2085 de 2002 ASS-RSA-GU004 y diligenciar el Formato para informe de evaluación-registro sanitario con solicitud de protección decreto 2085 de 2002 ASS-RSA-FM009. 6.5.2.7 Evaluación del certificado de venta libre o certificado de producto farmacéutico. De acuerdo con la normatividad vigente, decreto 426 de 2009, debe adjuntarse un certificado de producto farmacéutico o certificado de venta libre, el cual debe ser expedido por una autoridad sanitaria que sea catalogada de referencia para Colombia, incluir el (o los) principio(s) activo(s), la forma farmacéutica, la concentración del producto, los fabricantes, los envasadores, indicar si tiene autorización para ser comercializado en el país exportador, las presentaciones comerciales, y si el producto se acompaña de algún dispositivo médico. Este certificado, entre otras cosas, avala que en otro país ya se ha estudiado la información y que se considera pertinente que esté disponible para la comercialización en ese país, en el cual ya ha superado la evaluación de una Agencia Regulatoria. 6.5.2.8 Evaluación del certificado de cumplimiento de las BPM Independientemente del país de origen, el laboratorio fabricante debe contar con certificado de Buenas Prácticas de Manufactura, así como cada uno de los integrantes de la cadena de fabricación (envasadores, acondicionadores) emitido por un país de referencia conforme a lo requerido en el Decreto 162 de 2004. El certificado siempre debe indicar si el producto se realiza en un área común o requiere un área específica, si cuenta con áreas para la fabricación de productos estériles, las formas farmacéuticas autorizadas, si se requieren áreas con cadena de frío, entre otros. El documento debe encontrarse vigente al momento de la expedición del Registro Sanitario, lo cual se indica en la parte considerativa de la Resolución con un párrafo de redacción similar al siguiente:
Que mediante resolución No. XXXXXXXXXX del dd/mm/aaaa (ENTIDAD REGULATORIA QUE AVALA) concedió certificación de Buenas Practicas de Manufactura BPM al laboratorio fabricante XXXXX con domicilio en DIRECCIÓN ubicada en CIUDAD, PAÍS, para fabricar en área de producto (ESTERIL / NO ESTERIL), (PRINCIPIOS ACTIVOS COMUNES/ BETALACTAMICOS / CON ÁREAS ESPECIALES), (FORMA FARMACÉUTICA), con una vigencia hasta el dd/mm/aaaa; por lo tanto puede fabricar el producto de la referencia.
En algunos casos, el Certificado de Producto Farmacéutico puede servir como el soporte para el cumplimiento de las BPM. En este caso, se debe prestar especial atención y al momento de emitir la resolución, la vigencia de las BPM será la fecha de vigencia del CPP o en su defecto un año después de la expedición del mismo, el cual debe estar vigente al momento de la expedición del Registro Sanitario. La leyenda de la parte considerativa deberá ser esta o una similar:
Que como evidencia de la certificación de BPM, el interesado aportó certificado de producto farmacéutico No. XXXXX, emitido por AGENCIA REGULATORIA, en el cual se observa que el establecimiento LABORATORIO FABRICANTE, con domicilio en DIRECCIÓN ubicado en CIUDAD, PAÍS, y las operaciones que realiza, cumplen con lo establecido en las Buenas Prácticas de Manufactura. Que éste documento es considerado válido hasta XXX años después del dd/mm/aaaa como consta en dicha certificación.
Cuando se trate de productos de origen Biológico o Biotecnológico se deberá documentar el fabricante del principio

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 17 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
activo y del producto terminado. En el certificado de fabricante del principio activo no debe incluirse los productos en investigación. En caso que el certificado de BPM no coincida o no estén vigentes las buenas prácticas de manufactura, o no se allegue con la documentación se deberá realizar un requerimiento de solicitud del certificado que coincida con lo indicado por la normatividad vigente. Una vez emitido el registro sanitario, si el fabricante, envasador o acondicionador ha sido aprobado con un certificado de BPM o Certificado de Producto Farmacéutico de país de referencia, deberá diligenciar una base de datos, en el siguiente link https://docs.google.com/forms/d/1-dxiyAdyvf4KgwIrEeEJ9gtkGE2iAKJm--vTYInLO2Q/edit?usp=drive_web, con el fin de tener una base de datos de dichos fabricantes. 6.5.2.9 Presentaciones comerciales y forma farmacéutica: Confirmar que la presentación comercial esté descrita, por el interesado, con precisión indicando el color, clase y tipo de material de envase y empaque. En el caso de que el producto se presente en forma de blister cada presentación indicará el número de unidades por blister. Además se debe revisar si la forma farmacéutica solicitada (teniendo en cuenta su composición y descripción) por el interesado corresponde con las consignadas en la literatura farmacéutica como farmacopeas oficiales en Colombia u otras formas farmacéuticas nuevas aprobadas en actas de la SEMPB. Si se evidencia el uso de dispositivos médicos deberá incluirlos formalmente en la presentación comercial y describirlos completamente; si dichos dispositivos se encuentran legislados por el Decreto 4725 de 2005 y el interesado no allega copia del Registro Sanitario otorgado por la Dirección de Dispositivos, deberá complementar la información de los mismos allegando nombre genérico o marca del dispositivo médico, clasificación de acuerdo al riesgo, indicaciones y uso, indicar el código internacional (ECRTI, GMDN u otro de igual reconocimiento internacional), advertencias, precauciones y contraindicaciones; descripción del dispositivo médico: listado de principales partes, funcionamiento, información descriptiva, estudios técnicos y comprobaciones analíticas. Resumen de los documentos de verificación y validación (informe de pruebas) de diseño o certificado de análisis. Para el sistema de infusión estéril por ser catalogado como dispositivo médico clase IIa debe allegar adicionalmente la información científica necesaria que respalde la seguridad del producto y un análisis de riesgos del dispositivo médico según sus indicaciones, descripción de soluciones adoptadas para cumplir con los requisitos esenciales de seguridad y funcionamiento, los respectivos estudios de estabilidad que sustenten el tiempo de vida útil. La información presentada en este punto, servirá como soporte para evaluar los certificados de calidad del material de envase, empaque y los dispositivos que acompañan el producto, lo cual deberá coincidir en su totalidad. 6.5.2.10 Composición cualicuantitativa, formula estructural y fórmula del lote estandarizado: Esta información hace referencia al principio activo, su estructura y la fórmula cualicuantitativa, y para su evaluación deberá tener en cuenta los siguientes aspectos: Verificar que la composición cualicuantitativa sea expresada según lo que se establece en el literal c del artículo 22 del decreto 677/1995. Se debe verificar que cada sustancia incluida en la fórmula del medicamento esté identificada con nombre genérico y químico (IUPAC), y no con nombres de marca; para el activo se escribirá su nombre según la Denominación Común Internacional (DCI). Un resumen de la interpretación de la evidencia de la estructura e isomerismo, información tal como el potencial

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 18 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
de isomerismo, la identificación de estereoquímica o el potencial para formar polimorfos debe ser incluido. Cuando un principio activo es quiral, debe indicarse si se trata de un estéreo-isómero específico o una mezcla de estéreo-isómeros que fueron utilizados en los estudios clínicos y no clínicos, por lo tanto, si es necesario, deberá compararse la información farmacológica allegada, principalmente en el caso de los productos nuevos. A continuación se señala la letra con la que se identifica la composición y será la completada en la base de datos en la pestaña otros, en el campo “Concentr”:
a. Por unidad en formas de presentación dosificada en caso de tabletas, grageas, cápsulas, óvulos, supositorios, inyectables y similares.
b. Por 100 mL en caso de composiciones líquidas no inyectables. c. Por cada mililitro en líquidos para administración por gotas en inyectables en multidosis. d. Por cada 100 gramos en caso de productos presentados en forma de polvos, cremas, ungüentos,
geles, emulgeles, etc e. Por gramos de polvo para reconstituir a 100 mL, en caso de polvos para reconstituir a solución o
suspensión. f. En porcentaje de peso o volumen en el caso de aerosoles.
Si la expresión de la fórmula cuali-cuantitativa no coincide, deberá emitirse un requerimiento señalando que debe corregirse y expresarse como lo indica el decreto, dado que será la formulación que figurará en la base de datos, esto también evita que ingresen correcciones al evaluador por este aspecto. Revisar que se presenten de forma separada los excipientes de los principios activos. El activo debe ser descrito con certeza indicando si corresponde a una sal, éster o solvato. Si es una sal se deberá expresar su contenido como tal, o por el contrario aclarar si corresponde al equivalente de la forma ácida, básica o anhidra de la sustancia activa. En ningún caso los excipientes pueden tener actividad terapéutica. El incumplimiento de estos parámetros dará lugar a un requerimiento. Verificar que la sal, éster o solvato, sea la aprobada en normas farmacológicas, si no es así, deberá requerirse al interesado indicando que no es posible continuar con el estudio hasta que sea incluido en normas farmacológicas por la Sala Especializada de Medicamentos y Productos Biológicos En la fórmula cuali-cuantitativa también se describen las cápsulas empleadas y los recubrimientos en caso de ser empleados. Si no se encuentran, deberá emitirse un requerimiento para que las incluyan Verificar que la fórmula cualicuantitativa establecida corresponda con la que se describe en el Certificado de Producto Farmacéutico o Certificado de Venta Libre. En cuanto a la formula estructural y condensada del principio activo revisar que corresponda con el activo solicitado. Confirmar que la fórmula del lote estandarizado de fabricación corresponda proporcionalmente a la del lote industrial proyectado y a la formulación por unidad posológica (ejemplo por tableta).No se aceptará los excesos en el principio activo para compensar la degradación en los estudios de estabilidad. Si el fabricante agrega excesos para compensar pérdidas en el proceso productivo debe establecer claramente la magnitud de estos excesos y justificarlo debidamente. 6.5.2.11 Proceso de fabricación Revisar la descripción detallada del proceso de fabricación del medicamento, el cual debe estar en orden lógico y estar acorde con el principio activo, excipientes y la forma farmacéutica producida. Es suficiente un diagrama de flujo detallado del proceso de fabricación del medicamento, el cual debe estar en orden lógico y estar acorde con el principio activo, excipientes y la forma farmacéutica producida.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 19 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.5.2.12 Especificaciones de calidad de las materias primas: 6.5.2.12.1 PRINCIPIOS ACTIVOS Y EXCIPIENTES FARMACOPÉICOS: El interesado puede únicamente relacionar la farmacopea oficial y su edición, de donde se toma la monografía para el análisis del principio activo o excipiente. También podrá allegar fotocopia de las monografías o transcripciones literales de las mismas. El fabricante debe realizar todas las pruebas descritas en la farmacopea oficial escogida. No será necesario que el interesado allegue soportes de estos ensayos o certificados de análisis del fabricante del medicamento o del principio activo o excipiente. 6.5.2.12.2 PRINCIPIOS ACTIVOS Y EXCIPIENTES NO FARMACOPÉICOS: Se aceptará la metodología analítica establecida por el fabricante del medicamento, en donde se incluya la descripción detallada de todos los ensayos que se realicen y sus respectivos límites o especificaciones. En casos particulares se solicitará toda la información proveniente del fabricante de la materia prima, referente a perfil de impurezas, descripción de la materia prima con sus pruebas de identificación; todo debe ser presentado con los respectivos soportes. En el caso de que el interesado manifieste que los resultados fueron obtenidos aplicando el método del fabricante del activo se solicitará toda la información proveniente de éste. De estar consignado en la literatura científica de acceso público que de la materia prima en evaluación existen varias formas polimórficas y/o isómeros (que podrían afectar la biodisponibilidad y farmacodinamia) se solicitará al interesado que especifique y demuestre el tipo de cristal e isómero utilizado, el cual debe ser el adecuado. En general se consideran como excipientes a las sustancias inertes dentro de la formulación, ayudantes de formulación o intermediarios de fabricación. En todo caso se debe verificar que ningún excipiente tenga actividad terapéutica, verificar si algún excipiente corresponde a la categoría de Material especifico de riesgo (MER) o cualquier otra situación establecida en actas de la SEMPB para solicitar certificados de seguridad, debe verificar las especificaciones de sustancias relacionadas, solventes residuales y/o impurezas orgánicas. En el caso de que el producto contenga ciertos excipientes como: Alcohol bencílico, Metabisulfito, Tartrazina (Amarillo FD&C No. 5) y Aspartame, se debe tener precaución ya que estos requieren la inclusión de leyendas especiales en las contraindicaciones y advertencias. (Ver casos especiales referidos en actas de Comisión Revisora en el numeral 6.8.11). En el caso de materias primas (activos, excipientes e intermediarios de fabricación) de origen animal, se debe establecer claramente el origen de estas y de ser necesario debe presentar certificado del proveedor o de la autoridad sanitaria del país, en donde se establezca la ausencia agentes infecciosos y/o patógenos como la Encefalitis Espongiforme Bovina. En el caso de productos biológico hemoderivados se debe allegar certificado de ausencia del virus del Nilo y priones de Encefalitis Espongiforme Bovina y todos aquellos que sean incluidos en las actas de comisión revisora. Teniendo en cuenta que existen varios casos especiales y particulares, además de las que se exponen anteriormente, con respecto a los excipientes o materias primas usadas en los medicamentos, el evaluador deberá informarse y actualizarse constantemente sobre nuevas disposiciones al respecto y comunicarlo al resto del grupo, solicitando al coordinador un espacio en las reuniones de comité primario y dejando constancia de la socialización en las actas de dichas reuniones.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 20 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.5.2.12.3 MATERIALES DE ENVASE: Se verificará las especificaciones de calidad del material de envase primario. Las especificaciones de calidad deben ser adecuadas para el tipo de envase y material del mismo. Por ejemplo: de ser plástico se solicitará que el interesado haya confirmado lo indicado por el fabricante como es el tipo de plástico, color, dimensiones, indicar el volumen de rebose, determinación de permeabilidad, Extractables (según capítulo <661> USP) o farmacopeas BP y Europea. Para los goteros la determinación de número de gotas por mL se debe hacer con producto, por tanto si lo han realizado con agua el evaluador debe solicitar el resultado del ensayo con el producto. 6.5.2.12.4 DISPOSITIVOS COMPLEMENTARIOS DEL MEDICAMENTO: Se considera como dispositivo complementario todo implemento de preparación o administración que se comercializara conjuntamente con el medicamento, ejemplos de dichos dispositivos: cucharas y jeringas para administración de productos orales, sets o kits de transferencia y preparación de soluciones y polvos estériles, entre otros. El interesado debe allegar la descripción detallada del dispositivo, si es posible allegar diagramas o fotos y las especificaciones de calidad adecuadas para el uso del dispositivo. En caso de alguna inquietud el evaluador deberá consultar de manera escrita (email o carta) al Grupo de Insumos para la Salud, sobre si el dispositivo requiere o no registro sanitario de acuerdo al decreto 4725 de 2005 Los dispositivos médicos que acompañan un producto, pueden tener registro sanitario o no. Si cuentan con él, es necesario verificar que se encuentre vigente. En el caso que no cuente con registro sanitario verifique con base los requisitos establecidos en el Decreto 4725 de 2005 (los dispositivos son de clase I y Ila) 6.5.2.13 Especificaciones de calidad del producto en proceso:
Los controles en proceso que el fabricante defina deben ser adecuados para el tipo de proceso y de producto farmacéutico. Los controles en proceso pueden incluir para sólidos: dureza, peso, friabilidad, dimensiones, humedad, aspecto; para líquidos y semisólidos se incluyen pH del producto, aspecto, viscosidad, olor y el completar a volumen final, estos ensayos se determinan solamente hasta cuando se observe el cumplimiento de la especificación. El fabricante debe definir claramente la magnitud de los límites de tolerancia para cada control en proceso realizado y establecer la periodicidad de su evaluación. 6.5.2.14 Especificaciones de calidad para el producto terminado y metodología de análisis: 6.5.2.14.1 PRODUCTOS FARMACOPEICOS El interesado puede únicamente relacionar la farmacopea oficial y su edición, de donde se toma la monografía para el análisis del producto terminado. También podrá allegar fotocopia de las monografías o transcripciones literales de las mismas. El fabricante debe realizar todas las pruebas descritas en la farmacopea oficial escogida. No será necesario que el interesado allegue soportes de estos ensayos o certificados de análisis del fabricante del medicamento. Es importante que las pruebas realizadas sean adecuadas para la forma farmacéutica del medicamento y para el principio activo. Para la VALIDACIÓN DEL METODO DE CUANTIFICACION O VALORACIÓN DEL ACTIVO: No será necesario realizar una validación completa del método de cuantificación o valoración del activo si este es farmacopéico. Sin embargo se exigirá el parámetro de adecuabilidad del sistema y especificidad para efectos de estabilidad frente al blanco y al placebo allegando soportes debidamente identificados. 6.5.2.14.2 PRODUCTOS NO FARMACOPEICOS Se aceptará la metodología analítica establecida por el fabricante del medicamento, en donde se incluya la descripción detallada de todos los ensayos que se realicen y sus respectivos límites o especificaciones. Es

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 21 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
importante que las pruebas realizadas sean adecuadas para la forma farmacéutica del medicamento y para el principio activo. Es importante tener en cuenta los análisis y especificaciones referidos en el manual de normas técnicas colombianas vigente. Para los principios activos que se utilicen en formas farmacéuticas como tabletas, suspensiones, polvo para reconstituir a suspensión oral o inyectable, semisólidos donde el activo está suspendido se solicitará el tamaño y distribución de partícula. Para la VALIDACIÓN DEL METODO DE CUANTIFICACION O VALORACIÓN DEL ACTIVO: Los métodos no farmacopeícos de cuantificación del principio activo deben venir totalmente validados de acuerdo a los lineamientos de la Guía ICH Q2 (R1) o la vigente y guías complementarias y la norma compendial <1225> de la USP vigente. En el caso que la metodología para producto terminado sea la misma utilizada en los estudios de estabilidad esta debe estar validada en el parámetro de especificidad con fines de estabilidad, es decir en donde se garantice que los productos de degradación del activo, obtenidos mediante degradación forzada o por enriquecimiento del medicamento con sustancias de degradación y/o impurezas conocidas, no deben interferir con la cuantificación de este activo, tal y como se establece en la Norma Compendial USP <1225> y la Guía ICH Q2 (R1). En el caso que el interesado manifieste que utiliza dos o más métodos de cuantificación del activo, este debe establecer cual utiliza para el estudio de estabilidad y deberá cumplir con lo expuesto anteriormente allegando soportes debidamente identificados.
Para el ensayo de disolución, el cual es requerido por tabletas con y sin cubierta, tabletas masticables, tabletas dispensables, cápsulas (duras y blandas), suspensiones (a la fecha las que sí lo requieren según la monografía), si la farmacopea escogida por el interesado incluye una metodología analítica diferente a la allegada, deberá validarla.
6.5.2.15 Resumen de la información farmacológica Se verifica que el interesado incluya vía de administración, dosis y frecuencia de administración, indicaciones farmacológicas y usos terapéuticos, contraindicaciones, efectos secundarios y advertencias, evaluando la coherencia con la forma farmacéutica y la composición del medicamento. 6.5.2.15 Estudios de estabilidad y periodo de vida útil del producto: 6.5.2.15.1 PROTOCOLOS DE ESTABILIDAD El evaluador deberá tener en cuenta que en el protocolo del estudio de estabilidad el interesado deberá indicar: Una descripción completa del medicamento objeto del estudio (nombre, concentración o potencia, formulación, material de envase, presentación comercial, fecha de inicio del estudio, aspecto del producto, entre otros). Los parámetros y métodos de análisis completos de las pruebas de estabilidad (si utilizan más de una metodología para el análisis del producto indicar cual se utiliza en el estudio de estabilidad) los cuales deben corresponder con los establecidos en las especificaciones de producto terminado, si se omite alguna prueba deberán sustentar suficientemente la causa de esto. Cronograma de tiempos de muestreo del estudio de estabilidad en el cual se indiquen los códigos o números de los lotes utilizados en el estudio, su fecha de fabricación, donde se establezca la razón social y domicilio del lugar donde se realizó el proceso de fabricación de cada lote. Se deben presentar mínimo las tablas de resultados y la conclusión del estudio de la estabilidad, indicando la vida útil sugerida y las condiciones de almacenamiento.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 22 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Presentar un documento en el cual el responsable del desarrollo de los estudios de estabilidad avale la información que se establece en el protocolo, las tablas de resultados y las conclusiones del estudio. En el caso que se presenten estudios utilizando un modelo tipo BRACKETING o MATRIXING justificar el uso de dichos modelos de acuerdo a la normatividad ICH 6.5.2.15.2 REQUISITOS, METODOS Y TIEMPOS En el caso general de estudios de estabilidad y partiendo de lo referido en el parágrafo 2 del Articulo 22 del Decreto 677/95 se deberán presentar mínimo datos de 3 lotes de tamaño piloto industrial, cuando se trate de un principio activo que lleve menos de cinco (5) años de aprobación por parte de la Comisión Revisora de Medicamentos y cuando el principio activo es conocido, es decir que ya se ha registrado y existen en el mercado productos comercializándose y se le reconoce su estabilidad, los estudios de estabilidad pueden ser desarrollados en tan sólo dos (2) lotes de tamaño piloto industrial. Las pruebas de análisis presentadas para estos deben cubrir la evaluación de parámetros físicos, químicos biológicos y microbiológicos propios de la forma farmacéutica y del activo. Los intervalos de muestreo que deben presentarse serán los definidos en la guía ICH Q1A sección 2.2.6 Testing Frecuency, de la siguiente manera: Los muestreos deben realizarse cada 3 meses durante el primer año, cada 6 meses durante el segundo año y cada 12 meses luego del segundo año. Para el caso general, los interesados idealmente deben allegar datos que soporten todo el tiempo de vida útil solicitada dentro del estudio de estabilidad a largo plazo. Si el interesado no los ha completado y solicita 24 meses de vida útil deberá allegar mínimo la siguiente información se tendrá en cuenta los lineamientos definidos en la guía ICH Q1E (Apéndice A, árbol de decisión) y la guía ICH Q1A. Para medicamentos con formas farmacéuticas como polvos estériles para reconstituir a soluciones inyectables, polvos liofilizados para reconstituir a soluciones inyectables y similares, será obligatorio la presentación de estudios de estabilidad para la solución reconstituida en el solvente recomendado (administración inmediata) o si este se administra por infusión intravenosa, en líquidos parenterales de amplio uso en entidades de salud como agua para inyección, solución salina normal NaCl 0.9 %, dextrosa al 5%. Dichos estudios deben soportar los tiempos máximos de uso y las condiciones de almacenamiento para la solución reconstituida que se establezca en las cajas, etiquetas o insertos del producto. Si no se define explícitamente un tiempo máximo de uso y las condiciones de almacenamiento, el interesado debe establecerlos con base en los estudios de estabilidad realizados. Los estudios de estabilidad para la solución reconstituida deben realizarse con el medicamento objeto de estudio y no pueden ser extrapolados a otras potencias o pesos de llenado. Para medicamentos con formas farmacéuticas como polvo para reconstituir a suspensión oral o similares, se debe presentar estudios de estabilidad de la suspensión o solución reconstituida que soporte el tiempo máximo de uso y las condiciones de almacenamiento propuestas en cajas, etiquetas e insertos. Si no se define explícitamente un tiempo máximo de uso y las condiciones de almacenamiento, el interesado debe establecerlos con base en los estudios de estabilidad realizados. Los estudios de estabilidad para la solución o suspensión reconstituida deben realizarse con el medicamento objeto de estudio y no pueden ser extrapolados a otras potencias o pesos de llenado. 6.5.2.15.3 ACLARACIONES Si el peticionario presenta estudios de estabilidad realizados en zona climática II y teniendo en cuenta que el producto se va a comercializar en zona IV, debe allegar los estudios correspondientes a esta zona. Es importante aclarar que la obligatoriedad de presentar estudios para zona climática IV, no será aplicable a

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 23 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
productos que requieran refrigeración o congelación (ejemplo: vacunas, productos biológicos y productos biotecnológicos) o cuando el interesado pueda sustentar mediante bibliografía científica que el principio activo no soporta las condiciones de estudio para zona climática IV. Para producto que se envase en un contenedor semipermeable deberá presentar los estudios de acuerdo a las condiciones establecidas en la guía ICH Q1A. Sin embargo, el peticionario podrá presentar estudios de estabilidad a condiciones de zona IV para el caso general, siempre y cuando se hagan pruebas específicas que determinen que la permeabilidad del envase se encuentre dentro de límites que no afecten al producto. Como contenedores semipermeables se pueden considerar: bolsas plásticas o semirígidas en polietileno de baja densidad, ampollas, viales o frascos de polietileno de baja densidad, además de los que considere el fabricante o la literatura científica. En el caso de modificaciones a registro sanitario de medicamentos en la modalidad Importar y Vender, que contemplen ampliación de vida útil, solamente se aceptará los estudios de estabilidad a largo plazo que cubran toda el tiempo de vida útil solicitada. No se aceptarán, para este tipo de trámites, estudios de estabilidad acelerada o prospectiva. 6.5.2.15.4 OTORGAMIENTO DE VIDA UTIL Como norma general para el establecer la vida útil de medicamentos en la modalidad importar y vender, se otorgará el tiempo de vida útil que establezca el fabricante como conclusión de los estudios de estabilidad y/o el Certificado de Venta Libre o Certificado de Producto Farmacéutico que se allegue (si incluye este dato), siempre y cuando los datos allegados se consideren adecuados previa evaluación técnica de los mismos, pudiéndose otorgar por parte del Invima vidas útiles menores a las solicitadas. Si el interesado no sugiere o no concluye un tiempo de vida útil el Invima asignará este atributo del medicamento siguiendo los lineamientos de la ICH. 6.5.2.16 Estudios Farmacocinéticos (biodisponibilidad) o similares.
Este requisito aplica únicamente si el producto está incluido, en los conceptos emitidos por la SEMPB en el acta 19 de 2002 numeral 2.3.13, acta 4 de 2004 numeral 2.3.35 u otras actas posteriores. Los estudios de biodisponibilidad, farmacocinéticos, clínicos o farmacológicos son de evaluación exclusiva por parte de la Comisión Revisora. Tener en cuenta el marco normativo que para el caso son las resoluciones 1890 de 2001 y la resolución 1400 de 2001. En caso que el interesado presente estudios de íntercambiabilidad estos deben ser igualmente aprobados por Comisión Revisora. Esto último teniendo en cuenta el Artículo 4°. De la resolución 1400/2001 que dice: Estudios de Bioequivalencia. Se exigirán estudios de Bioequivalencia para los medicamentos que se comercializan en Colombia bajo denominación genérica o de marca, cuando el productor interesado solicite la certificación de intercambiabilidad con el innovador en el mercado. Si el medicamento objeto de estudio no esta contemplado dentro de los grupos obligatorios para presentar biodisponibilidad pero el interesado los allega, estos se enviarán para evaluación de la Comisión Revisora, a no ser que el interesado específicamente declare prescindir de esta evaluación. Todos los productos nuevos de origen biológico o biotecnológico deben presentar evaluación farmacológica ante la Comisión Revisora tanto en la solicitud de registro sanitario nuevo como la de renovación. 6.5.2.17 Artes de material de envase y empaque Si el solicitante utilizará los materiales de empaque y envase aprobados en el país de origen, deberán incluir como mínimo la información en idioma español que se establece en el artículo 74 del decreto 677/95. El solicitante podrá hacer uso de autoadhesivos, impresión directa en la caja u otros métodos para dar cumplimiento al artículo 74 del decreto 677/1995 y podrá realizar esta labor en empresas en el territorio nacional que estén certificadas en BPM por el Invima y que tengan la capacidad técnica de acondicionamiento específico y para la manipulación el medicamento (según caso especifico). De cualquier manera el uso de stikers se hará

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 24 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
de manera tal que no oculte información de la etiqueta original. El solicitante deberá allegar copia del contrato de acondicionamiento en donde se indiquen los procesos a realizar y los productos a acondicionar. Si se van a utilizar diseños de etiquetas y material de empaque exclusivos para el territorio colombiano, el interesado podrá allegar artes o bocetos a color y por duplicado de los mismos. Estos deberán contener la información comprendida en el artículo 72 del decreto 677/95, además si el medicamento lo requiere una franja violeta y la leyenda respectiva, cuando el producto lo requiera (artículo 73 del decreto 677/95). Al momento de la evaluación es importante que el evaluador tenga en cuenta los aspectos que refiere el FIE (ASS-RSA-FM010).
CASOS ESPECIALES REFERIDOS EN ACTAS DE COMISION REVISORA
TEMA DE INTERÉS Nº DE ACTA Y NUMERAL
Sibutramina Acta 04 de 2008 numeral 2.1.4
Aines tópicos acta 08 de 2009 numeral 2,4,15
Manejo regulatorio de AINES Acta 22 de 2006 numeral 2.10.15
Alcohol bencÍlico Acta 35 de 2007 numeral 2.8.29
Aspartame/ Fenilcetoúrea Acta 03 de 2004 numeral 2.8.8
Biodisponibilidad de moléculas nuevas Acta 01 de 04 de 1998 numeral 2.4
Biodisponibilidad y equivalencia Acta 51 de 1997 numeral 2.16
Glusosamina Acta 01 del 2009 numeral 2.7.23
Glusosamina Acta 23 de 2009 numeral 2.5.20
Ibuprofeno Acta 42 de 2007 numeral 2.4.32
Insertos Acta 2 de 2007 numeral 2.2.5.
Medicamento contra los síntomas de la gripa Acta 35 del 2007 numeral 2.8.2
Metabisulfito Acta 35 de 2007 numeral 2.8.28
Sucralosa Acta 8 de 2008 numeral 2.8.1
Precaución uso alcohol con aines y analgésicos Acta 23 de junio de 1999 numeral 2.7.1.
Tartrazina Acta 13 de 2007 numeral 2.7.28
Tartrazina Acta 35 de 2007 numeral 2.8.31
6.5 Una vez el Químico Farmacéutico evalúa la información, y ha diligenciado el FIE de evaluación dará el paso en el aplicativo de registros para evaluación por parte del Abogado. 6.6 EVALUACION LEGAL DE LA INFORMACION: El profesional abogado revisa y verifica por sistemas en el aplicativo de registros sanitarios en el área de trabajo, que el químico farmacéutico le haya pasado el trámite. Se inicia el estudio de un trámite haciendo la selección del mismo en la opción área de trabajo, eligiendo el radicado que le llego del Químico farmacéutico, por mandato constitucional se debe respetar el derecho a turno, esto se traduce en resolver los trámite más antiguos en estricto orden de fecha de radicación.
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 25 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
El abogado inicia el diligenciamiento del FIE correspondiente a la evaluación legal código ASS-RSA-FM034, teniendo en cuenta que cuando considere pertinente diligenciará el FIE-l-, en caso de aprobar el trámite, pero diligenciará siempre el informe FIE cuando se haga un requerimiento, cuando sea una corrección o cuando haya respuesta a un requerimiento legal o recurso de reposición, en todo caso si a consideración del abogado no requiere diligenciar el FIE en caso de aprobaciones no se exime al evaluador legal de realizar una evaluación concienzuda de toda la documentación legal que le permita soportar lo aprobado y dejará plasmado en la base de RS el concepto legal. el Informe debe evidenciar claramente la evaluación de todos y cada uno de los requisitos de ley para el tramite, debe plasmar los conceptos legales emitidos por el evaluador en cada punto y debe reflejar claramente que decisión toma el evaluador, respecto a la calidad de la información enviada por el solicitante. – esto es el requerimiento y si el abogado lo considera la aprobación del trámite- Al final el informe debe quedar debidamente diligenciado por los evaluadores Abogados. En este paso se inicia propiamente el análisis legal de la calidad de la información enviada por el interesado El profesional Abogado será encargado de verificar los documentos correspondientes a: REQUISITOS LEGALES, Art. 24 y 31 decreto 677 de 1995, decreto 426 de 2009: Formulario de solicitud suscrito por el representante legal de la empresa titular o un apoderado legalmente constituido. Poder debidamente otorgado a un abogado si el titular actúa mediante apoderado. Certificado de existencia y representación legal de la empresa titular. Certificado de existencia y representación legal de la empresa fabricante. Certificado de existencia y representación legal de la empresa importadora. Certificado de venta libre, expedido por autoridad sanitaria del país de origen, en el cual conste que el producto está autorizados para ser puesto en el mercado del país exportador. Certificado de B.P.M., del laboratorio fabricante, expedido por autoridad sanitaria. (Tener en cuenta decreto 549 de 2001 y decreto 162 de 2004). Autorización del fabricante al importador para importar, distribuir, comercializar y ser el titular del registro sanitario, del producto, en la Republica de Colombia. Certificado expedido por la Superintendencia de Industria y Comercio en el cual conste que la marca está registrada o en trámite de registro a nombre del titular del Registro Sanitario. Si la marca está registrada a nombre de un tercero, adicionalmente debe adjuntar autorización de uso de marca, acreditando la calidad en la que actúa quien otorga la autorización. Recibo de Pago en original por la tarifa legal correspondiente. NOTA: Todos los documentos públicos, provenientes del exterior deben venir con sello de apostille o con sello de consularización. Todos los documentos de carácter legal, provenientes del exterior deben tener una vigencia no mayor de un (1) año, a la fecha de radicación del trámite. Si los documentos legales, provenientes del exterior se encuentran en idioma diferente al español adicionalmente se debe adjuntar traducción oficial al español.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 26 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
Si de acuerdo a los hallazgos y conceptos planteados en el informe de evaluación los evaluadores emiten una aprobación, generan una resolución la cual pasa por sistema al Coordinador o a la persona encargada por éste para revisar el concepto emitido por el profesional. Si el Coordinador o persona responsable no está de acuerdo con el concepto, este lo devuelve al profesional que lo estudió para que realice la corrección correspondiente y deja la observación en el formato de evaluación en la casilla correspondiente.
6.8 Si el Químico Farmacéutico o el abogado evaluador, dentro del estudio necesitan elevar una consulta a otra dependencia como Comisión Revisora, debe dar paso a esta opción dentro del sistema y diligenciar la consulta a través de la aplicativa base de datos salas de comisión revisora. Imprimir una copia de dicha consulta y entregarla al coordinador del grupo –ver diagrama árbol de decisión y direccionamiento a Comisión Revisora al final de esta guía.
Si requiere elevar una consulta a una dependencia diferente por ejemplo Grupo Técnico de Medicamentos o grupo de Fármacovigilancia, realizar la consulta como correspondencia del Grupo de Registros de la Dirección de Medicamentos y dejar constancia de esto en el Informe de evaluación.
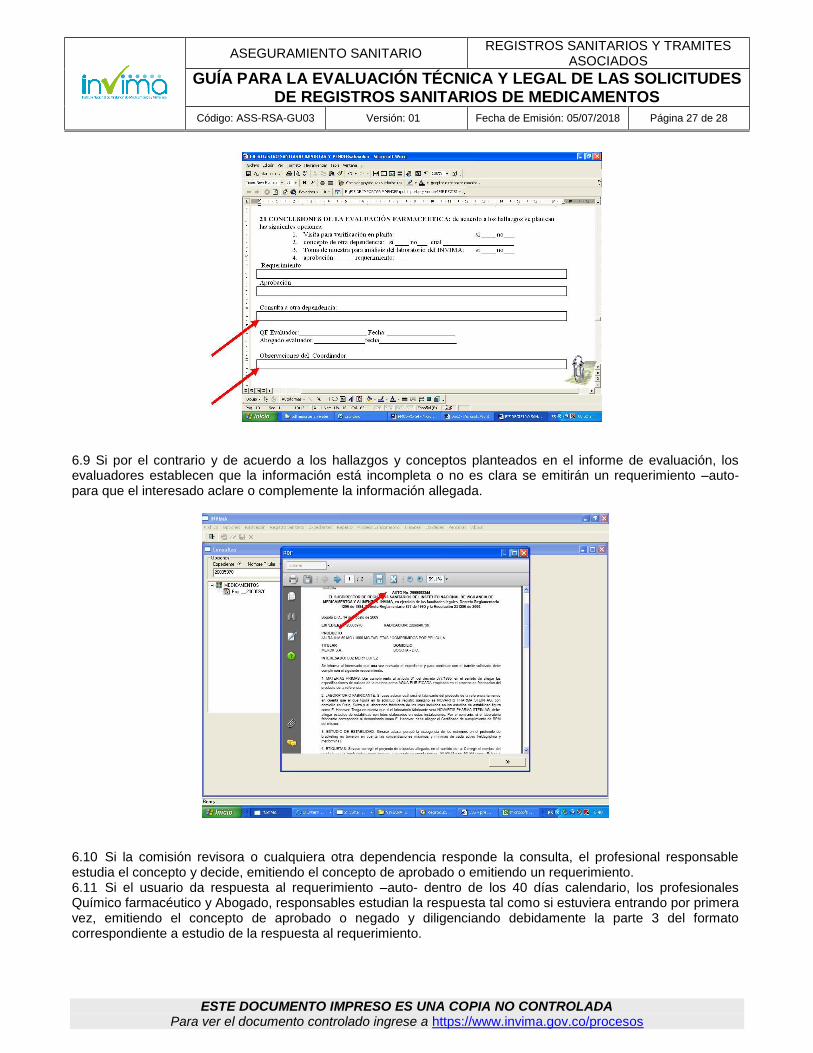
ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 27 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.9 Si por el contrario y de acuerdo a los hallazgos y conceptos planteados en el informe de evaluación, los evaluadores establecen que la información está incompleta o no es clara se emitirán un requerimiento –auto- para que el interesado aclare o complemente la información allegada.
6.10 Si la comisión revisora o cualquiera otra dependencia responde la consulta, el profesional responsable estudia el concepto y decide, emitiendo el concepto de aprobado o emitiendo un requerimiento. 6.11 Si el usuario da respuesta al requerimiento –auto- dentro de los 40 días calendario, los profesionales Químico farmacéutico y Abogado, responsables estudian la respuesta tal como si estuviera entrando por primera vez, emitiendo el concepto de aprobado o negado y diligenciando debidamente la parte 3 del formato correspondiente a estudio de la respuesta al requerimiento.

ASEGURAMIENTO SANITARIO REGISTROS SANITARIOS Y TRAMITES
ASOCIADOS
GUÍA PARA LA EVALUACIÓN TÉCNICA Y LEGAL DE LAS SOLICITUDES DE REGISTROS SANITARIOS DE MEDICAMENTOS
Código: ASS-RSA-GU03 Versión: 01 Fecha de Emisión: 05/07/2018 Página 28 de 28
ESTE DOCUMENTO IMPRESO ES UNA COPIA NO CONTROLADA Para ver el documento controlado ingrese a https://www.invima.gov.co/procesos
6.12 Si el usuario pasados 40 días no ha respondido el requerimiento se da por negada la solicitud de acuerdo a lo establecido en el código contencioso administrativo artículo 13, o en caso de que el mismo usuario cierre el tramite sin haber contestado el requerimiento, el abogado será el encargado de generar la resolución de abandono de la solicitud de acuerdo con el artículo 8 del mismo código. 6.13 Documento final 7. PERIODICIDAD DE REVISIÓN Y ACTUALIZACIÓN DEL PRESENTE DOCUMENTO: Este documento se revisará y actualizará cada vez que surja un avance en el campo de los medicamentos, que modifique y obligue a solicitar nuevo documento no incluido en el presente documento. Cuando alguno de los decretos o resoluciones reglamentarias de la legislación sanitaria de medicamentos sea modificada. 8. ANEXOS: Guía de evaluación técnica y legal para concesión de registro sanitario nuevo para productos biológicos y biotecnológicos ASS-RSA-GU025 Instructivo para la industria farmacéutica: visitas con propósito de obtención de registro sanitario ASS-RSA-IN045 Instructivo de Visita a Laboratorios Farmacéuticos desde el Grupo de Registros Sanitarios de Medicamentos y Productos Biológicos ASS-RSA-IN009 Instructivo para la toma de decisiones y direccionamiento de trámites a Comisión Revisora ASS-RSA-IN014 Instructivo para el manejo de la comunicación al interior del Grupo de Medicamentos y Productos Biológicos ASS-RSA-IN015 Instructivo para la aplicación de Evaluación por pares ASS-RSA-IN016 Formato para Informe de Evaluación registro sanitario modalidad importar y vender ASS-RSA-FM010 Formato para Informe de Evaluación Técnica-Registro Sanitario Productos Biológicos ASS-RSA-FM014 Formato de Informe de Evaluación correcciones a resoluciones de registro sanitario de medicamentos y trámites asociados ASS-RSA-FM015 Formato de informe de evaluación Legal ASS-RSA-FM034 Formato para informe de Evaluación respuesta a requerimiento o Evaluación de Recurso de Reposición Medicamentos ASS-RSA-FM035


















