
Bases genético – moleculares de las proteínas...
122
DEPARTAMENTO ACADÉMICO: Morfología Humana CURSO: Genética Médica TÍTULO: Bases Genético – Moleculares de las Proteínas Estructurales INTEGRANTES: Pérez Ramírez, James Polo Mejía, Juan Julio Joseph Polo Saona, Christian Poma González, Elka Pretell Vargas, Crystel Yasmín Pumamango Córdova, Jimmy Emerson Quiroz Aldave, Juan Eduardo Reyes Florián, Giuliana Rivalles Alvarez, Renzo Renato
Transcript of Bases genético – moleculares de las proteínas...

DEPARTAMENTO ACADÉMICO:
Morfología Humana
CURSO:
Genética Médica
TÍTULO:
Bases Genético – Moleculares de las Proteínas Estructurales
INTEGRANTES:
Pérez Ramírez, James
Polo Mejía, Juan Julio Joseph
Polo Saona, Christian
Poma González, Elka
Pretell Vargas, Crystel Yasmín
Pumamango Córdova, Jimmy Emerson
Quiroz Aldave, Juan Eduardo
Reyes Florián, Giuliana
Rivalles Alvarez, Renzo Renato
TRUJILLO – PERÚ
2010

Seminario II Bases genético – moleculares de las proteínas estructurales
2
Tabla de contenido
Resumen......................................................................................................................................................3
Introducción................................................................................................................................................3
Proteínas Estructurales Intracelulares.........................................................................................................4
Distrofina.................................................................................................................................................4
Queratinas.............................................................................................................................................11
Espectrina..............................................................................................................................................20
Proteínas Estructurales de Membrana......................................................................................................28
Receptor de lipoproteínas de baja densidad.........................................................................................28
CFTR (Regulador de la Conductancia Transmembrana de la Fibrosis Quística).....................................32
Proteínas Estructurales Transportadoras..................................................................................................39
Hemoglobinas........................................................................................................................................39
Proteínas Estructurales Extracelulares......................................................................................................48
Colágeno................................................................................................................................................48
Fibrilina..................................................................................................................................................56
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
3
Resumen
El presente trabajo de investigación bibliográfica trata acerca de las bases genéticas y moleculares del
funcionamiento y estructura de las proteínas estructurales, entre las cuales tenemos a las distrofinas,
queratinas, espectrinas; receptores de LDL, CFTR; hemoglobinas; colágeno, fibrilina, etc.
Se tratarán puntos como la organización génica de cada proteína, considerando la ubicación de los genes que
la codifican dentro del genoma; organización estructural, es decir, la forma de su estructura primaria,
secundaria, terciaria y cuaternaria, y su interacción con otras proteínas y biomoléculas; su función dentro o
fuera de la célula, o como transportadoras de otras moléculas; y finalmente, se consideró a la correlación
entre el genotipo y el fenotipo, normales y alterados
Palabras clave: proteínas estructurales, distrofina, queratina, espectrina, receptores de LDL, CFTR,
hemoglobina, colágeno, fibrilina, correlación genotipo – fenotipo.
Introducción
Dentro de la célula existe una organización bastante compleja, empezando por la membrana (del
eritrocito), donde las proteínas se unen para formar una corteza celular resistente en la que participa la
espectrina, las proteínas de banda, etc. En la célula muscular la distrofina conecta el citoesqueleto de
actina con la matriz extracelular a través de la membrana plasmática. La queratina es una proteína que
constituye el componente principal de las capas más externas de la epidermis de los vertebrados y de
otros órganos derivados del ectodermo.
También tenemos proteínas de membrana, como los receptores de lipoproteínas de baja densidad, que
participan en el metabolismo del colesterol asociado a LDL, y el regulador de conductancia
transmembrana de la Fibrosis Quística (CFTR), que participa de manera decisiva en el proceso de esta
enfermedad.
Entre las proteínas transportadoras, la que más resalta es la hemoglobina, que transporta oxígeno
dentro de los eritrocitos.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
4
Por último, también encontramos proteínas estructurales fuera de la célula, como el colágeno que se
encuentra en la matriz del tejido cartilaginoso, y la fibrilina, esencial para las fibras elásticas del tejido
conectivo
Proteínas Estructurales Intracelulares
Distrofina
Organización génica
El gen DMD que codifica a la distrofinas fue identificado en 1987 en el brazo corto del Cromosoma X.
Dentro del gen DMD existen al menos siete promotores que regulan su expresión de manera tejido
específica y en función del desarrollo. Tres promotores localizados en el extremo 3’ codifican tres mRNA
de 14 kb que constan de un dominio amino-terminal de unión a actina, un dominio central
superenrollado de triple hélice y un dominio carboxilo-terminal con sitios regulatorios y de unión a un
complejo de proteínas membranales conocidas como proteínas asociadas a la distrofina (DAPs).
Hacia el extremo 3’ existen otros cuatro promotores que dan lugar a productos más pequeños.
Alrededor del exón 30 se encuentra el promotor que regula la transcripción de la distrofinas de 260 kDa
(Dp260, que se identificó por primera vez en la retina. El siguiente promotor, característico del sistema
nervioso central da lugar a una proteína de 140kDa.
Otro producto del gen DMD es la Dp116 cuya expresión es característica del sistema nerviosoperiférico.
En el extremo distal del gen, entre los exones 62 y 63 se localiza el promotor de la distrofina Dp71. Esta
distrofina presenta, entre otras variantes generadas por procesamientos alternativos, la remoción del
exón 78 que genera un nuevo extremo hidrofóbico con los últimos 31 aa del COOH-terminal. La Dp71 en
general se expresa ampliamente en tejido no-muscular, principalmente en los diferentes tipos celulares
del sistema nervioso.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
5
El gen DMD y sus productos. Se
muestra la posición del gen DMD en la
región P21 del cromosoma X. La
localización de los diferentes
promotores en el gen y sus productos,
que se nombran de acuerdo a su masa
molecular (Dp por “Dystrophin
protein”). Se representa la estructura
primaria de la distrofina (Dp427) y de los
productos cortos del gen DMD.
Mapa parcial de ligamiento del cromosoma
X que muestra algunos de los genes de
enfermedades, como la distrofia muscular de
Duchene (Alberto Juan Solari, Genética
humana: fundamentos y aplicaciones en
medicina, Ed. Médica Panamericana, 2004
pág. 239)
Organización estructural
La proteína distrofina tiene 3687 aminoácidos y un peso molecular de 427KDa. La distrofina posee
cuatro regiones o dominios característicos, a saber, el dominio amino terminal, el dominio central, un
dominio rico en cisteína y el dominio carboxiterminal. El dominio amino terminal tiene 240 aminoácidos
y puede unirse a la proteína esencial de las miofibrillas y citoesqueleto, la actina; el dominio central
incluye 24 repeticiones imperfectas similares a las de la espectrina y que probablemente constituye el
eje de la molécula de la distrofina ,entre ellas dos secuencias “bisagra” ricas en prolina; el dominio rico
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
6
en cisteína es similar a la región carboxiterminal de la a-actinina .Finalmente el dominio
carboxiterminal, muy especifico, es el que se une a glucoproteínas de la membrana. Por consiguiente la
distrofina es una proteína alargada y con forma de cordón, que por su extremo NH 2 se conecta con la
actina citoplasmática y por su extremo COOH con la membrana (sarcolema) a través de glicoproteínas
de membrana; es una especie de “puente” al exterior .Además es muy probable que se presente en
forma de dímero (con los monómeros con polaridad opuesta)
Estructura de la distrofina, con sus cuatro dominios que ocupan sus casi 3700 aminoácidos (Alberto
Juan Solari, Genética humana: fundamentos y aplicaciones en medicina, Ed. Médica Panamericana, 2004
pág. 239)
Molécula de distrofina. Esta molécula une, por sus extremos carboxilo y aminoterminales, dos MF a dos
moléculas intrínsecas de la membrana. (Marc Maillet, Biología Celular, Elsevier, España, 2002, pág. 147)
Función
Se han sugerido tres funciones principales para las distrofinas:
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
7
1. estabilidad membranal
2. transducción de fuerza
3. organización de especializaciones membranales, donde las distrofinas pueden organizar la
topología membranal o mantener un complejo membranal fijo en un sitio. La distrofina refuerza
y estabiliza el sarcolema durante la tensión de la contracción muscular mediante el mecanismo
de un enlace mecánico entre el citoesqueleto y la matriz extracelular a través del complejo
DAPs. De esta manera participa a la unión sarcómero-membrana y funciona como elemento de
la transducción de fuerza durante la contracción, o puede participar en la formación de
especializaciones como los contactos focales de adhesión. Una de las características de la
distrofina es su presencia en regiones especializadas de la membrana post-sináptica de la unión
neuromuscular donde va a anclar canales de sodio.
El complejo de las proteínas asociadas a la distrofina comprende la distrofina y dos subcomplejos: el
complejo de distroglucano (subunidades y) y el complejo sarcoglucano (Otras
proteínas adicionales son las sintrofinas (subunidad la distrobrevina y el sarcospan. La
distrofina, las sintrofinas y las distrobrevina se localiza en el sarcoplasma, mientras que los
distroglucanos, los sarcoglucanos y el sarcospan son glucoproteínas transmembrana.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
8
Correlación genotipo – fenotipo
Clasificación actualizada de las distrofias musculares progresivas
Enfermedad Localización
genética
Producto del gen PM (kDa) Localización
Duchene - Becker Xp21 Distrofina a 427 Citoesqueleto
LGMD2A 15q15 Calpaína 3 - -
LGMD2B 2p13 Desconocido - -
LGMD2C 13q12 g-sarcoglicano, A4 a 35 Transmembrana
LGMD2D 17q21 a-sarcoglicano, A2 a 50 Transmembrana
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
9
LGMD2E 4q12 b-sarcoglicano, A3b a 43 Transmembrana
LGMD2F 5q33-3s4 d-sarcoglicano a 35 Transmembrana
LGMD1A 5q22-31 Desconocido - -
LGMD1B 1q11-21 Desconocido - -
Distrofia muscular
congénita merosina
negativa
6q22-23 Merosina (cadena a2) a - Matriz extracelular
a Proteínas con anticuerpos monoclonales comercializados
Distrofinopatias: Distrofia muscular de Duchenne y de Becker
Características fenotípicas principales
Edad de inicio: infancia
Debilidad Muscular
Hipertrofia de las pantorrillas
Moderado compromiso intelectual
Valores elevados de creatincinasa sérica degeneración muscular progresiva acabando en silla de
ruedas antes de 5 años.
Muerte frecuente por parada cardio-respiratoria
GEN: DMD Xp21
Distrofias musculares autosómicas recesivas de cintura y miembros
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
10
Distrofia muscular congénita sindrómica
En el músculo, la distrofina une la matriz extracellular (laminina) al citoesqueleto de actina . La
distrofina presenta interacción con un complejo multimérico constituido pot los distroglucanos (DG),
los sarcoglucanos, las sintrofinas y distrobrevina.
(Margaret W. Thompson, Robert L. Nussbaum, James Scott Thompson, Roderick R. McInnes,
Huntington F. Williard Genética en medicinaElsevier España, 2004, pág. 376)
En por lo menos tres, la DMC coexiste con afectación del sistema nervioso central: DMC de Fuku-
yama, enfermedad del músculo, el ojo y el encéfalo y el síndrome de Walkerg-Walburg.
La característica principal es un transtorno de la migración celular hasta la corteza, entre los meses
cuarto y quinto de la gestación, que conduce a la policromigiria, lisencefalia y heterotipia. Otras
anomalías pueden incluir fusión de los lóbulos frontales, hidrocefalia, quistes periventriculares,
atrofia del nervio óptico, hipoplasia de los tractos piramidales, reducción del número de células del
asta anterior e inflamación de las leptomeninges.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
11
Queratinas1
El citoesqueleto de todas las células eucarióticas consiste en tres redes principales: los microfilamentos
(5 a 7 nm de diámetro), los microtúbulos (alrededor de 25 nm de diámetro) y los filamentos intermedios
(de aproximadamente unos 10 nm de espesor). Los microfilamentos y microtúbulos participan en la
división celular, contracción, orientación y polarización, y anclaje. En cambio, la función de los
filamentos intermedios no se comprendió hasta el reconocimiento de mutaciones puntuales en diversos
trastornos de la piel.
Existen más de 50 proteínas de filamentos intermedios que se clasifican en 6 tipos esenciales según la
secuencia de aminoácidos, la especificidad tisular y las características inmunológicas. En el citoplasma de
las células epiteliales los filamentos intermedios de queratina forman una trama compleja desde el
núcleo hasta la membrana celular, a cuyo nivel interactúan con proteínas específicas de unión,
desmosomas intercelulares y hemidesmosomas en la superficie basal de la célula.
Organización estructural
Las queratinas (al menos unas 30 en piel y cabello) se clasifican en dos grupos (tipo I y tipo II) de
proteínas de filamentos intermedios.
Las queratinas tipo I (ácidas, K9 a K20) están codificadas en el cromosoma 17q12-q21 (con excepción de
K18) mientras que las del tipo II (básicas, K1 a K8) lo están por el cromosoma 12q11q-14.
Estructuralmente, las queratinas consisten en un dominio central de ALFA-hélice y dos dominios
laterales no helicoidales (V1 y V2). Las queratinas tipo II tienen además dos subdominios (H1 y H2).
Al inicio y fin del bastón central hay dos regiones cortas: dominios de iniciación y terminación de la
hélice en los extremos amino y carboxiterminal, respectivamente. Las secuencias de estas regiones
están muy conservadas en la familia de las proteínas de filamentos intermedios. Se considera que estas
regiones limítrofes son esenciales durante el ensamblado de los filamentos. Todas las queratinas de un
tipo determinado tienen la misma organización genómica e igual posición de intrones.
1 Solari, Genética Humana: Fundamentos y aplicaciones, 3ra edición
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
12
Los dímeros de queratina son heterodímeros formados por la asociación de una de las queratinas de
tipo I con una de tipo II. Dada la existencia de un total de 30 queratinas entre ambos tipos,la cantidad de
heterodímeros es muy grande. Esta gran variabilidad de filamentos de queratina permite que algunas
células epiteliales tengan queratinas específicas y que aún dentro de un epitelio las células situadas a
diferentes alturas presenten filamentos de diferente composición.
Las queratinas se ordenan con números arábigos en orden decreciente según su peso molecular (la
queratina 1 y la queratina 9, Q1 y Q9, son las mayores en los tipos II y I).En la epidermis general hay una
distribución especifica de las queratinas: las células basales poseen las suyas, los queratinocitos
intermedios poseen otras y la capa córnea otras diferentes.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
13
Función
En cuanto a la función de las queratinas, es por ahora en su mayor parte desconocida. El citoesqueleto
de queratinas se extiende desde la membrana nuclear hasta la plasmática, donde interacciona con los
desmosomas, al parecer con la desmoplaquina . Se ha propuesto que tal distribución puede tener
importancia en la transmisión de información entre la membrana celular y el núcleo, con posibles
implicaciones en el control de la expresión génica. En este contexto, recientemente ha sido descubierto
que la integrina 43 forma parte del complejo hemidesmosómico asociado a la membrana basal y es
posible por tanto que esté asociada con los IF de queratinas de las células epiteliales.
Las integrinas son receptores transmembrana que intercambian señales entre la matriz extracelular y el
interior celular. Normalmente están asociadas al citoesqueleto de actina y actúan recibiendo señales
externas y traduciéndolas en otras que afectan la organización citoesquelética, forma y motilidad
celular. En el sentido contrario, cambios intracelulares pueden modificar la afinidad de la integrina por
ciertos ligandos.
La existencia de queratinas en todos los vertebrados estudiados hasta ahora y la conservación de sus
secuencias entre especies sugieren una importante función para estas proteínas. Del mismo modo, la
delicadamente regulada expresión de las queratinas en pares característicos en cada célula epitelial
hace pensar que las diferentes proteínas son responsables de procesos específicos de cada tipo celular.
Sin embargo, los datos de que se dispone se contradicen con estas suposiciones. In vitro, la combinación
equimolar de cualesquiera proteínas de tipo 1 y 11 en las adecuadas condiciones conduce a la formación
de filamentos, incluso entre queratinas de distintas especies, aunque la fuerza de interacción varía
según el par. Este fenómeno ha recibido el nombre de promiscuidad .In vivo, la microinyección o
transfección de mRNAs de epidermis en líneas celulares epiteliales de la misma o distinta especie (que
expresan las queratinas típicas de epitelio simple K8 y K18) no parece producir ningún efecto. Las
queratinas epidérmicas se integran en el citoesqueleto endógeno y las células crecen con normalidad. Lo
mismo sucede cuando se inyectan estos mRNA en células no epiteliales. Estos datos sugieren que tal vez
la función de las queratinas deba ejercerse al nivel del organismo completo, o al menos en un nivel
superior al celular.
Para determinar la función de las queratinas, se han realizado algunos experimentos en los que se han
eliminado o sustituido los genes de ciertas queratinas en un organismo. La sobreexpresión de formas
mutadas de la K18 de ratón en células de carcinoma embrional (EC) impide la aparición del endodermo
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
14
visceral, comprometiendo el desarrollo del cuerpo embrionario. Sin embargo, la eliminación de ambos
alelos de la K8 en células stem (ES) de ratón no tiene ninguna consecuencia y permite la normal
formación de la capa del endodermo visceral y la formación del cuerpo embrionario aunque la
eliminación de ambos alelos de la K8 en ratones transgénicos es letal en un momento cercano a la
implantación, lo que podría indicar que la función de las queratinas se lleva a cabo a partir de un cierto
estado de desarrollo, siendo tal vez prescindibles en estadios embrionarios más tempranos.
Sin embargo, la utilidad del citoesqueleto de filamentos intermedios está fuera de toda duda: muy
recientemente se han establecido las primeras relaciones entre queratinas mutantes y enfermedad:
ratones transgénicos que expresan diversas formas truncadas de la queratina K14 tienen el
citoesqueleto de IF de la capa basal de la epidermis perturbado en diversos grados y poseen fenotipos
similares a los de varias formas del conjunto de patologías denominadas epidermólisis ampollosa simple,
EBS. Estudios genéticos han demostrado que mutaciones en diversos residuos de la K5 y K14 son la
causa de casos hereditarios de EBS. Una de estas mutaciones se da en la secuencia TYRKLLEGE del final
de la α-hélice, conservada en todas las queratinas. Igualmente, se ha detectado que una cierta forma de
la EBS se debe a mutaciones puntuales que afectan a un determinado aminoácido de la primera parte
del alfa-hélice de la K14. Este residuo (Arg-125) está altamente conservado no sólo en las queratinas,
sino en todos los IF. Mutaciones en este aminoácido en la lamina A afectan seriamente a la formación de
la lámina nuclear. Estos datos sugieren que las queratinas están directamente implicadas en la
resistencia mecánica de la epidermis. Los individuos afectados por distintas formas de EBS tienen una
piel macroscópicamente normal, aunque su citoesqueleto de queratinas esté desorganizado en la capa
basal y la forma de estas células sea distinta. El fenotipo de la EBS, causado por la lisis de las células
afectadas, sólo se manifiesta de forma post-traumática, lo que indicaría que las células que poseen
queratinas defectuosas son capaces de subsistir de forma satisfactoria en condiciones de ausencia de
estrés, sin descartar que otras mutaciones más fuertes sean letales.
Correlación genotipo – fenotipo
Epidermolisis bullosa simple
La epidermolisis bullosa simple (EBS) fue el primer trastorno hereditario identificado. Hasta la fecha
se han reconocido 18 mutaciones en genes de queratina asociadas con patología en el hombre. La
EBS es una de las tres formas principales de epidermolisis bullosa (EB). Las otras dos formas son la EB
de la unión y la EB distrófica.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
15
La formación característica de ampollas intraepidérmicas en la EBS se debe a la citólisis en la región
subnuclear de los queratinocitos basales. La EBS normalmente se hereda en forma autosómica
dominante. Se la considera la forma más leve de EB y tiene una incidencia aproximada de 1 en 50000.
La EBS Dowling-Meara (EBS-DM) es la forma más grave ya que la patología ampollar puede ser
intensa en el momento del nacimiento y puede llevar a la muerte en el transcurso de los primeros
meses de vida. Algunos casos son esporádicos pero la mayoría se heredan en forma autosómica
dominante. Las ampollas surgen con una agrupación herpetiforme en el tronco y extremidades
proximales. Curan con una leve cicatriz. El compromiso bucal es común y también pueden estar
afectados el esófago y los dientes. Hay hiperqueratosis progresiva de palmas y plantas.
Histológicamente se observa un agrupamiento anormal de los filamentos de queratina en los
queratinocitos basales que precede a la citólisis de las células basales y a la formación intradérmica
de ampollas.
En la EBS Weber-Cockayne (EBS-WC) -forma más común de EBS- las ampollas afectan
fundamentalmente manos y pies. Las lesiones aparecen durante la primera infancia o
posteriormente, cuando el niño comienza a caminar. Los traumas mínimos originan la formación de
ampollas aunque las lesiones también pueden formarse espontáneamente. Curan sin dejar cicatriz.
La hiperhidrosis del pie es característica y puede haber hiperqueratosis leve de palmas y plantas. Las
lesiones son más frecuentes en los meses de verano.
La EBS tipo Köbner (EBS-K) es similar a la EBS-WC pero la patología afecta toda la superficie corporal y
la cavidad oral. Las ampollas aparecen en la primera infancia. Aunque curan sin dejar cicatriz, la
infección secundaria es un problema frecuente.
Los estudios histológicos con anticuerpos revelaron tonofilamentos agrupados de queratinas de las
células basales (K5 y K14). La genética molecular confirmó mutaciones en los genes KRT5 y KRT14
como la alteración subyacente en EBS, confirmada posteriormente en experimentos en animales
transgénicos. Estas primeras mutaciones no sólo identificaron la causa de las EBS sino que
permitieron confirmar la teoría de que las queratinas ejercen un papel estructural esencial en las
células epiteliales. Hasta la fecha se han reconocido varias mutaciones en K5 y K14 en la EBS-DM y las
formas leves (EBS-K y EBS-WC). La mayoría son mutaciones heterocigotas y ocurren en la secuencia
de inicio de la hélice de K14.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
16
Los estudios de correlación genética y fenotípica revelaron que las formas más graves obedecen a
mutaciones en las secuencias limítrofes de la ALFA-hélice que parecen cruciales durante el
ensamblado de los filamentos. Las mutaciones en otras regiones de la hélice central se toleran mejor.
Otras formas de EBS incluyen la EBS con pigmentación, atrofia de la piel del tronco y extremidades,
hiperqueratosis de palmas y plantas y uñas distróficas y las EBS de transmisión recesiva.
Eritrodermia bullosa ictiosiforme congénita (EBIC)
También se la conoce como hiperqueratosis epidermolítica. Se presenta con eritrodermia y
formación de ampollas desde el nacimiento, con progresión a hiperqueratosis generalizada grave en
la vida adulta. La citolisis tiene lugar en las capas suprabasales de la epidermis, en forma diferente de
lo que ocurre en las EBS. Estructuralmente, las células basales son normales mientras que en las
células suprabasales hay agrupamiento de tonofilamentos con colapso del citoesqueleto. Debido a
que las células migran hacia la epidermis, la expresión de K5 y K14 se reduce y la de K1 y K10 se torna
predominante.
Se han identificado numerosas mutaciones en K1 y K10, habitualmente heterocigotas. Sobre la base
de las alteraciones genéticas, la EBIC puede dividirse en dos grupos. Una forma se asocia con
hiperqueratosis de palmas y plantas, usualmente por mutaciones en K1, mientras que la otra forma -
sin compromiso palmoplantar- obedece por lo general a mutaciones en K10. La K9 es capaz de
compensar en parte la anormalidad de K10 y por ello los enfermos con este segundo tipo no tienen o
sólo presentan afectación leve de palmas y plantas. Una variante de la EBIC, la ictiosis anular
epidermolítica, es histológicamente y clínicamente similar a la EBIC, con placas hiperqueratósicas
eritematosas en tronco y extremidades superiores.
Ictiosis bullosa de Siemens
Es una forma de hiperqueratosis epidermolítica con engrosamiento epidérmico y formación de
ampollas superficiales, principalmente en las zonas de flexión. Se ha observado agregación de los
tonofilamentos y citolisis que se limitan a las capas superficiales espinosa y granular de la epidermis.
Los estudios de ligamiento sugieren la participación del gen KRT2e en esta forma de patología. La
enfermedad leve puede simular clínicamente la ictiosis bullosa de Siemens. Aunque las técnicas de
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
17
genética molecular contribuyen con el diagnóstico diferencial en casos dudosos, la variación
fenotípica intrafamiliar complica la identificación precisa del trastorno.
Paquioniquia congénita (PC)
Incluye un espectro de displasias ectodérmicas con distrofia hipertrófica ungueal como hallazgo
principal. Se hereda en forma autosómica dominante y se clasifica en dos tipos: PC-1 y PC-2.
En la PC-1 hay además queratodermia no epidermolítica de palmas y plantas y leucoqueratosis oral.
La microscopia electrónica revela tonofilamentos anormales en los queratinocitos suprabasales pero
diferentes de los que se observan en EBS-DM y EBIC. La primera mutación identificada fue en los
genes K6a y K16, habitualmente en las secuencias de iniciación de la hélice. Otras mutaciones en K16
explican fenotipos inusuales de PC-1 en los cuales el compromiso de la piel y de uñas no se hace
evidente hasta los 6 años de vida.
En la PC-2 se observan quistes pilosebáceos múltiples que aparecen en la pubertad. Durante la
primera infancia puede ser difícil la distinción entre PC-1 y PC-2.
Las alteraciones genéticas incluyen mutaciones en el gen KRT17; todas las identificadas hasta ahora
afectan la secuencia de inicio de la hélice.
El esteatocistoma múltiple es una variante de PC-2 que se caracteriza por múltiples quistes
pilosebáceos semejantes a los que se observan en la PC-2. La misma mutación puede originar
distintos fenotipos, lo cual indica que la expresión clínica puede depender de la combinación de otros
factores genéticos y ambientales.
Queratoderma palmoplantar
Abarca un grupo de patologías que pueden ser clínicamente difíciles de distinguir. La hiperqueratosis
de palmas y plantas ocurre en forma difusa o focal y puede asociarse con otros hallazgos
ectodérmicos. Obedece a mutaciones en K9 y K16.
La forma epidermolítica (EPPK) es una enfermedad autosómica dominante que se presenta en las
primeras etapas de la vida. Hay citolisis histológica en las capas suprabasales de la epidermis gruesa;
con microscopia electrónica se observan agregados de filamentos de queratina.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
18
La hiperqueratosis difusa amarillenta de palmas y plantas sin otras manifestaciones clínicas orientó la
búsqueda a alteraciones genéticas en el gen KRT9 por su expresión específica en las células
suprabasales de la piel plamoplantar. El análisis de tres familias con EPPK leve reveló mutaciones en
el gen KRT1. En la forma Unna-Thost se identificó una mutación inusual en el gen KRT1.
Otros tipos de queratodermia palmoplantar incluyen la variante Curth-Macklin, detectada en varias
generaciones de una familia afroamericana, se caracteriza por filamentos de queratina anormales en
la capa espinosa y granular. La alteración genética es en el cromosoma 12 (donde se codifican las
queratinas tipo II).
La forma focal no afecta o sólo compromete en forma leve las uñas y no hay compromiso de la
cavidad oral. Se identificaron mutaciones en la secuencia de iniciación de la hélice de K16.
Trastornos de la queratina no epidérmica
Otros tejidos expresan ciertas queratinas, cuyas mutaciones se asocian con fenotipos particulares de
patología. El nevus blanco es un trastorno benigno autosómico dominante, por mutaciones en K4 o
K13.
La distrofia corneal epitelial de Meesmann se hereda en igual forma. Se caracteriza por la presencia
de quistes intraepiteliales en la córnea anterior, llenos de restos intracelulares y, probablemente,
agregados de queratina. Se inicia durante la infancia pero puede comenzar más tardíamente. El
trastorno habitualmente es asintomático y no hay alteraciones visuales. Los enfermos, por la
fragilidad de la córnea, pueden no tolerar lentes de contacto. Las queratinas K12 y K13 se expresan
específicamente en las células epiteliales de la córnea; esto permitió identificar mutaciones en los
respectivos genes.
Las queratinas epiteliales K8 y K18 se expresan en hígado, páncreas y epitelio intestinal y se han
encontrado mutaciones en K18 en pacientes con cirrosis criptogénica.
Los resultados de estudios en animales sugieren que mutaciones en K8 o K18 pueden predisponer a
los enfermos a patología hepática de comienzo tardío.
Trastornos de la queratina del cabello
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
19
Los genes de las queratinas del pelo tipo I y tipo II se localizan en los cromosomas 17 y 12,
respectivamente. El moniletrix es una patología autosómica dominante infrecuente con expresión
fenotípica variable, desde pérdida leve del cabello hasta alopecía casi completa. La microscopia
electrónica revela defectos en la estructura de los microfilamentos del cabello. Puede haber
queratosis folicular y anormalidades de las uñas.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
20
Espectrina
La espectrina es una de las proteínas más importantes del eritrocito. Su nombre se debe a que si
introducimos glóbulos rojos en un medio hipotónico éstos absorben agua y se acaban lisando
(estallando). Al lisarse, la hemoglobina sale hacia el exterior de la célula quedando sólo la membrana, lo
que se denomina como fantasma de eritrocito. Una de las proteínas del fantasma es la espectrina, y de
ahí le viene el nombre.
Organización estructural 2
La formación y la integridad de la membrana asociada a la red de apoyo que las moléculas espectrina
pueden formar dependen de las asociaciones intermoleculares e intermoleculares en dos puntos clave
2 William N. Kelley, Medicina interna, Pág. 1376
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
21
de la molécula de espectrina. En la “cabeza” terminal, unión intercatenaria entre las cadenas y que
componen la molécula de espectrina, asociando estas cadenas en heterodímeros o tetrámeros. Al final
de la “cola”, los sitios de unión intercatenario integran los tetrámeros de espectrina en una red por
medio de asociaciones con proteína 4.1, la actina y otras proteínas. Entre estos sitios extremos, gran
parte de la longitud de espectrina corresponde a secuencias repetidas en tándem o "segmentos", cada
uno de los cuales contiene 100 a 120 aminoácidos.
La eliminación de estas repeticiones tándem en términos de las unidades de conformacionales a las que
dan lugar, han sido establecidas sin ambigüedad en la estructura cristalina de una de estas repeticiones.
Históricamente, manera de mostrar y referirse a los segmentos de repetición se basó en secuencias de
alineamientos y colocar al principio y al final de cada uno de los motivos de la fase (aproximadamente
30 residuos) con los segmentos que ya se han definido bioquímica y estructuralmente. Debido a que la
función depende de la estructura pertinente.
Estructura de los segmentos de repetición
La espectrina es una proteína periférica de membrana, por lo que su unión con ésta será relativamente
débil. Se encuentra en la cara citoplasmática y supone el 25% de las proteínas periféricas. Cada hematíe
contiene unas 250 mil copias de espectrina.
La espectrina se encuentra siempre dimerizada formando una doble cadena, en la que la cadena alfa es
ligeramente más pesada y grande que la beta.
El análisis del equilibrio de sedimentación, además de la electroforesis en gel, ha demostrado que, en
solución, un segmento de espectrina utilizado para la cristalografía se somete a análisis de dimerización
reversible. El equilibrio entre las poblaciones de monómero y dímero se produce rápidamente por
encima de 20 ° C, pero a una tasa insignificante a -5 ° C. El equilibrio muy lento a bajas temperaturas
permite caracterizar las propiedades hidrodinámicas tanto de las especies de monómeros y dímeros.
Unión intercatenaria al extremo de la cabeza de la cadena
El estudio de los sitios de espectrina que sean accesibles a la degradación proteolítica han sido
importantes en el avance de nuestro conocimiento de la espectrina. En investigaciones sobre
mutaciones en β -espectrina que afectó la susceptibilidad a la proteólisis de la α- espectrina existe la
hipótesis de que la relación entre la cabeza y la cola entre la α y β espectrina podrían explicar la
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
22
susceptibilidad de la espectrina a proteólisis. Esta hipótesis plantea que las hélices A y B contienen
alrededor de 60 residuos del segmento parcial que se encuentran cerca del extremo carboxilo de β
espectrina, forman un haz de tres hélices con el ensamblaje de la hélice C alrededor del 30 residuos del
segmento parcial que se encuentra en el extremo amino de la espectrina. Esta hipótesis ha sido
ampliamente comprobada por genética y estudios bioquímicos que han identificado con exactitud la
ubicación de los residuos y los motivos necesarios para la unión a la cabeza de la cadena.
A nivel genético se ha encontrado que muchas consecuencias funcionales importantes surgen de
eliminaciones y mutaciones en la región de espectrina que forma la hélice C de un segmento parcial, y
dentro de las regiones, de β de espectrina que forman las hélices A y B de un segmento parci al. Estas
consecuencias incluyen efectos que van desde los de leves a graves, tanto en las moscas (En el que se
produce la detención del desarrollo) y en los seres humanos (en los que los efectos son eliptocitosis
hereditaria y la no inmunidad a hidropesía fetal). Estos efectos pueden, en general, atribuirse a un
debilitamiento o ruptura de la red de espectrina como resultado de un defecto en la unión entre cabeza
y cola de la cadena.
A nivel bioquímico, los ensayos de unión directa y ensayos de huella de proteasa, que utilizan
fragmentos peptídicos de especies nativas y recombinantes de α y β espectrina, confirmó que los
aminoácidos, cerca del carboxilo terminal de de espectrina (aminoácidos que son homólogas a las
hélices A y B) y aminoácidos cerca del amino terminal (que son homólogas a las de la hélice C) son los
residuos esenciales requeridos para la unión intercatenaria entre cabeza y cola.
Un notable descubrimiento fue que el sitio de unión dentro de la β espectrina de la hélice C solitaria
cerca del extremo amino terminal de α espectrina se puede recrear posiblemente en cualquier
segmento de repetición de β espectrina con sólo eliminar un segmento de la hélice C. Este
descubrimiento apoya la idea de que la unión intercatenaria depende de la interaccion hélice-helice que
produce la unión de tres hélice de otras repeticiones de α espectrina y β espectrina. Los demás
residuos, que se encuentran fuera de las regiones que son los homólogos de las hélices A, B y C de
otros segmentos de repetición también se requieren
Dominio similar a calmodulina
La secuencia del segmento no repetitivas en el carboxilo terminal de α espectrina (a22) es a la vez
homóloga al segmento 4 de actinina y calmodulina.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
23
Un análisis de secuencias predice la presencia de cuatro EF-hands (EFs 1 a 4) dentro de estas secuencias
no repetitivas del extremo carboxi-terminal de la α espectrina. Estudios demostraron que, en ausencia
de calcio, las hélices EF-1 están estrechamente unidas, mientras que la hélice EF-2 son menos compactas
y están involucrados en las interacciones laterales la hélice EF-1. La unión de Ca+2 causa una
redistribución de las interacciones hidrofóbicas en EF-1, resultando en una apertura de la estructura de
hélice-giro-hélice que, a su vez, es propagado a EF-2.
Estos cambios conformacionales pueden modificar la interfaz entre los segmentos α22 y β1, y puede, en
particular, modificar la estructura en bucle entre EF y EF-1-2, que desempeña un papel importante en la
unión intercatenaria en el extremo de la cola de las subunidades de espectrina . Esto puede explicar
cómo Ca+2 regula la interacción entre los filamentos (F)-actina y espectrina.
Unión intercatenaria al extremo de la cola de la cadena
Además de una interacción entre la α22 y β1, la unión intercatenario final de la cola también incluye
segmentos α20 y α21, y β2 y β3, que comparten una similitud en secuencias limitadas con otros
segmentos, donde un segmento está unido al siguiente por una inserción octámero que también se
encuentran entre α19 y α20. La supresión o duplicación de octámeros encontrados entre α20 y α21 y
entre β2 y β3 da como resultado una pérdida de la unión intercatenaria. Curiosamente, las sustituciones
no conservativas de estos residuos (por ejemplo, la sustitución de Arg por Gly) no afectan vinculante.
Esto sugiere que los octameros no son por sí mismos sitios de unión intercatenaria, sino que son
fundamentales en la definición del registro, o de la posición relativa de los segmentos de las cadenas de
α - espectrina y β -espectrina que contienen los sitios de unión verdaderos.
Una variante de α espectrina eritroide humana, α LELY (α low expression allele), es muy común entre los
caucásicos. Provoca efectos calamitosos sólo si están acompañados de una mutación del gen que se
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
24
asocia generalmente a eliptocitosis hereditaria (HE), un defecto que suele ser atribuibles a alteraciones
o supresiones en el extremo de sitios de unión intercatenaria de la cabeza. La mutación implica la
supresión de seis residuos en el extremo carboxilo de una hélice en el segmento α 21, esta supresión
debe afectar a la unión intercatenaria de la cola, pero en los seres humanos la presencia del alelo α LELY
por sí mismo es asintomáticos.
Esquema de cómo la mutación de α LELY afecta la estabilidad de la red de espectrina
Sitios de asociación con la membrana celular
La región final de espectrina incluye varios sitios de unión que se superponen o se yuxtaponen cerca
entre sí. Por ejemplo, la asociación del extremo amino de β espectrina con la proteína 4.1 y, a través de
proteína de 4.1, con la membrana celular.
Desde la posición de la mutación Kissimmee en β 1, una mutación que disminuye la capacidad de
unión de la espectrina a la proteína 4.1, podemos entender que la región de unión 4,1 es
inmediatamente adyacentes al dominio de unión a actina y podría superponerse al sitio de unión
intercatenario. Esta superposición puede explicar la acción estabilizadora de la proteína 4.1 en el
complejo binario espectrina-actina El extremo amino terminal de la cabeza de β espectrina también se
asocia directamente con las proteínas integrales de membrana, pero no está claro cuáles son los
residuos de espectrina.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
25
Los sitios de unión de β espectrina a la membrana está contenida en el segmento β 2, y sugieren que
una secuencia conservada de cinco residuos (Gly-Lys-Pro-Pro - Lys) de β 2 constituiria un sitio de unión a
la membrana.
Por otro lado, también se puede concluir que los sitios de unión a la membrana se encuentran en una
mayor región que incluye los segmentos de β 3-5 y β 7-8.
De hecho, en ambas posiciones, estos polipéptidos sintéticos con una alta afinidad por la membrana
contienen el octámero conservado entre β 2 y β 3 que es necesarios para la unión intercatenaria. Este
octámero también puede constituir un sitio de unión a la membrana. Si es así, puede explicar por qué
las sustituciones no-conservativas en este octámero conservado no afectan la unión.
El residuo conservado en el octámeros puede ser requerido de modo dependiente de la secuencia como
un motivo de unión a la membrana, y de manera independiente de la secuencia para definir el registro
de segmentos alternos que contienen los sitios de unión. La presencia de un octámero similar entre α 20
y α 21 implica que las cadenas de α -espectrina pueden también estar independientemente asociados
con la membrana celular.
La presencia de este octámero en α Espectrina podría explicar la asociación de de cadenas con la
periferia de las células epiteliales que carecen de β subunidades. La dificultad de definir el sitio de unión
a la membrana en espectrina se podría explicar por la multiplicidad de los sitios espectrina en la
membrana celular utilizada en estos experimentos.
El dominio de homologo a plectrina
Los extremos carboxilo de isoformas de espectrina β II y β 1∑2 contienen dominios homologos a
plectrina (PH). Aunque la similitud de secuencia entre los diferentes dominios PH es débil, su estructura
terciaria se conserva. El dominio PH de la espectrina participa en el anclaje de espectrina a la
membrana.
En la plectrina, el fosfatidilinositol 4,5-bisfosfato (PIP2) es un ligando potencial de el dominio PH y puede
ser responsable de la orientación de la membrana.
IP 3 se une a la con carga positiva de la hendidura entre los lazos de conexión de las cadenas β 1 y 2, y 5
y 6. Considerando que los grupos 4-fosfato y 5-fosfato interactuar a través de puentes salinos y enlaces
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
26
de hidrógeno y están rodeados por varios residuos de carga positiva, el anillo de inositol y los grupos 1-
fosfato apenas participan en la interacción.
Tomando en cuenta la orientación de la molécula de IP 3 y la relativamente débil constante de
disociación para el complejo de dominio IP3-PH, es probable que PIP2, en lugar de IP3, sea el ligando
natural para el dominio PH. Como la unión implica residuos de carga positiva que se conservan en
muchos dominios PH, la interacción con el PIP 2 puede ser una característica general de otras proteínas
que contienen dominios PH.
Correlación genotipo – fenotipo
Esferocitosis
La esferocitosis hereditaria es una enfermedad genética (75% de los casos son autosómicos dominante y
el otro 25% corresponde a un patrón autosómico recesivo),1 que forma parte de las llamadas anemias
hemolíticas, caracterizada por la producción de hematíes de forma esferoidal, por un defecto en la
membrana del mismo, lo cual hace que se destruya con facilidad en el bazo.
Causas
La esferocitosis hereditaria está causada por una variedad de mutaciones en los genes que
transcriben para la espectrina4 (el defecto más frecuente),5 ankirina (o anquirina), y otras proteínas
de la membrana del hematíe.6 Estas proteínas son necesarias para mantener la forma normal del
hematíe, que es la de un disco bicóncavo. Estos defectos de membrana disminuye en el glóbulo rojo
su elasticidad y deformabilidad características y aumenta la concentración corpuscular media de
hemoglobina 7 (CCMH). Como una de las funciones del bazo es eliminar aquellos hematíes de
formas anormales (que generalmente son antiguos), también destruye los esferocitos.
Síntomas
Generalmente comienza a los 5-6 años de edad, de forma lenta, como una anemia crónica con
ictericia (hiperbilirubinemia no conjugada por hemolisis intravascular) y esplenomegalia (aumento
del tamaño del bazo). También puede cursar con palidez, fatiga, debilidad, dolor abdominal, fiebre,
cálculos biliares. Las infecciones pueden desencadenar crisis hemolíticas y aplásicas.
Prevalencia
Se estima que la prevalencia es de 2.2 de cada 10,000 nacidos vivos, la mayoría heredados de sus
padres. Un 25% de los casos es debido a una neomutación.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
27
Eliptocitosis3
Es un trastorno hereditario, en el cual los glóbulos rojos sanguíneos tienen una forma anormal (elíptica).
Es más común en personas de ascendencia africana y mediterránea. Existe una mayor probabilidad de
desarrollar esta enfermedad si alguien en la familia la ha padecido.
La eliptocitosis con frecuencia es inofensiva. En los casos leves, menos del 15% de los glóbulos rojos son de
forma elíptica. Sin embargo, algunas personas pueden tener crisis en las cuales los glóbulos rojos se rompen,
especialmente si tienen una infección viral. Las personas con esta enfermedad pueden padecer anemia,
ictericia y cálculos biliares.
3 Bernadette F. Rodak, Hematología: fundamentos y aplicaciones clínicas, Pág. 274
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
28
Proteínas Estructurales de Membrana
Receptor de lipoproteínas de baja densidad
La mayor parte del colesterol se transporta en la sangre, unido a proteínas, formando unas partículas
conocidas como lipoproteínas de baja densidad o LDL (de Low Density Lipoproteins).
Cuando la célula necesita colesterol para la síntesis de membrana, produce proteínas receptoras de LDL
y las inserta en su membrana plasmática. Cuando el colesterol es captado pasa a los lisosomas donde se
hidrolizan los ésteres de colesterol dando lugar a colesterol libre, que de esta forma queda a disposición
de la célula para la biosíntesis de las membranas. Si se acumula demasiado colesterol libre en la célula,
ésta detiene tanto la síntesis de colesterol como la síntesis de proteínas receptoras de LDL, con lo que la
célula produce y absorbe menos colesterol.
Esta vía regulada para la absorción del colesterol está perturbada en algunos individuos que heredan
unos genes defectuosos para la producción de proteínas receptoras de LDL y, por consiguiente, sus
células no pueden captar LDL de la sangre. Los niveles elevados de colesterol en sangre resultantes
predisponen a estos individuos a una aterosclerosis prematura, y la mayoría de ellos mueren a una edad
temprana de un infarto de miocardio como consecuencia de alteraciones de las arterias coronarias. La
anomalía se puede atribuir al receptor de LDL el cual puede estar ausente o ser defectuoso.
El receptor de LDL es una proteína mosaico que media la endocitosis de LDL rico en colesterol. Es un
receptor de la superficie celular que reconoce la apo-proteína B100 que se encaja en la capa
fosfolipídica externa de partículas LDL. El receptor también reconoce la proteína apoE, encontrada en
los remanentes de quiolomicrones y de VLDL (IDL). Brown y Goldstein ganaron un Premio Nobel para su
identificación del receptor de la lipoproteína de la baja densidad (LDL) en 1985 mientras que estudiaban
hipercolesterolemia familiarl.
Organización génica
Se ubica en el cromosoma 19, 19p1.06-1.1
El gen que codifica al receptor de LDL tiene 18 exones. El exón 1 contiene una secuencia de señal que
ubica al receptor en el retículo endoplasmic para el transporte a la superficie de la célula. Más allá, los
exones 2 al 6 codifican la región de unión del ligando; del 7 al 14 codifican el dominio EGFP; el 15
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
29
codifica la región rica en oligosacáridos, el 16 (y parte del 17) codifican la región que atraviesa la
membrana, y el 18 (con el resto del 17) codifica el dominio citosólico.
Organización estructural
El receptor de LDL se puede describir como una proteína quimérica. Está compuesto de un número de
dominios funcionalmente distintos que puedan funcionar independientemente el uno del otro.
El extremo amino del receptor de LDL contiene siete repeticiones de secuencia (~50% idénticas) cada
aproximadamente 40 aminoácidos, con 6 residuos de cisteína. Estas regiones de unión del ligando (LB)
se doblan automáticamente cuando son sintetizadas como péptidos individuales. Los residuos de
cisteína forman enlaces disulfuro, formando un enrejado octaédrico, coordinado a un ion del calcio, en
cada repetición. El mecanismo exacto de la interacción entre las repeticiones LB y ligando (LDL) es
desconocido, pero se piensa que las repeticiones actúan como “abrazaderas” para sostener el LDL.
Al lado del dominio de unión al ligando está un factor epidérmico del crecimiento (EGF) precursor del
dominio de homología (dominio EGFP). Esto demuestra la homología del aproximadamente 30% con el
gen del precursor de EGF. Hay tres repeticiones de “factor del crecimiento”; A, B y C. A y B están
cercanamente de cerca mientras que C es separada por un motivo propulsor-beta. El dominio EGFP ha
estado implicado en el lanzamiento de los ligandos al receptor. Se piensa que un cambio conformacional
ocurre en las condiciones ácidas (pH5.0) del endosome, trayendo al propulsor-beta en contacto con las
repeticiones de unión al ligando 4 y 5.
Un tercer dominio de la proteína es rico en oligosacáridos unidos por enlace O-glucosídico pero parece
demostrar poca función.
Un dominio que atraviesa la membrana que contiene una cadena de residuos de aminoácidos
hidrofóbicos cruza la membrana plasmática de la célula. Dentro de la célula, el dominio C-terminal
contiene una secuencia de señal que es necesaria para la entrada del receptor.
Función
Los complejos del receptor de LDL están presentes en invaginaciones revestidas de clatrina en la
superficie de la célula, que cuando están unidas al LDL-colesterol vía adaptina, se pellizcan para formar
vesículas revestidas de clatrina dentro de la célula. Esto permite que el LDL-colesterol sea unido e
interiorizado en un proceso conocido como endocitosis y previene que el LDL se difunda alrededor de la
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
30
superficie de la membrana. Esto ocurre en todas las células nucleadas (no eritrocitos), pero
principalmente en el hígado que quita el 70% de LDL de la circulación. LDL está implicado directamente
en el desarrollo de arterioesclerosis, debido a la acumulación del LDL-colesterol en la sangre. El La
arterioesclerosis es el proceso responsable de la mayoría de enfermedades cardiovasculares.
Una vez que se interne la vesícula revestida se despojará de su capa de clatrina y se fusionará con un
endosome ácido tardío. El cambio en el pH causa un cambio conformacional en el receptor, que lanza la
partícula de LDL unida. Los receptores entonces o se destruyen o pueden ser reciclados vía ciclo
endocítico de nuevo a la superficie de la célula, donde el pH neutro hará que el receptor se invierta a su
conformación nativa lista recibir otra partícula de LDL.
La síntesis de receptores en la célula es regulada por el nivel del colesterol intracelular libre; si es
superior a las necesidades de la célula entonces la transcripción del gen del receptor será inhibida. Los
receptores de LDL son traducidos por los ribosomas en el retículo endoplasmático y son modificados por
el aparato de Golgi antes de viajar en vesículas a la superficie de la célula.
Correlación genotipo – fenotipo
En la arteriosclerosis
Niveles elevados de colesterol en la fracción LDL ("colesterol LDL" o "colesterol malo") se asocian
fuertemente al desarrollo de enfermedad arteriosclerótica. Diversos modelos experimentales y
observaciones epidemiológicas sistemáticas apoyan, de hecho, un papel causal del colesterol LDL en
la iniciación y progresión de la arteriosclerosis. Sin embargo, debe tenerse en mente que éste no es
el único factor de riesgo asociado a esta enfermedad, y que su manejo médico debe ser planificado
sobre la base de la evaluación del riesgo cardiovascular global individual de cada paciente.
El transporte reverso de colesterol y las células espumosas
Como se mencionó al principio, las LDL no están fisiológicamente involucradas en un influjo neto de
colesterol hacia los tejidos. Sin embargo, en determinadas circunstancias patológicas, como la
hipercolesterolemia LDL, la hipertensión arterial, la diabetes mellitus o el tabaquismo, se desarrolla
una entrega exagerada y no regulada de colesterol desde LDL químicamente modificadas (oxidadas)
a células macrofágicas subendoteliales, que cuando son sobrepasadas en su capacidad de
depuración, en un proceso conocido como "transporte reverso de colesterol" y mediado por las
lipoproteínas de alta densidad (HDL), degeneran en células inestables, propensas a la inflamación y a
la muerte celular patológica (necrosis). La acumulación de estos macrófagos sobrecargados de
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
31
colesterol, conocidos como células espumosas, determina el desarrollo de placas de ateroma en la
pared arterial, hecho anatomopatológico definitorio de la enfermedad aterosclerótica.
Mutaciones del receptor de LDL
Hay 5 clases de mutación del receptor de LDL.
Clase 1: las mutaciones afectan la síntesis del receptor en el retículo endoplasmático (ER).
Clase 2: las mutaciones impiden el transporte apropiado al aparato de Golgi, necesario para las
modificaciones del receptor.
- Un truncamiento de la proteína del receptor en el residuo número 660 conduce a los dominios
3, 4 y 5 del dominio del precursor de EGF que falta. Esto imposibilita el movimiento del receptor
del ER al Golgi, y conduce a la degradación de la proteína del receptor.
Clase 3: las mutaciones detienen la unión de LDL al receptor.
- La repetición 6 del dominio de unión al ligando (N-terminal, líquido extracelular) se suprime.
Clase 4: las mutaciones inhiben la interiorización del complejo receptor-ligando.
- El mutante “JD” resulta de una sola mutación del punto en el dominio de NPVY (C-terminal,
citosólico; residuo de Y convertido a C, residuo número 807). Este dominio recluta a la clatrina y
otras proteínas responsables de la endocitosis de LDL, por lo tanto esta mutación inhibe el
interiorización de LDL.
Clase 5: las mutaciones dan lugar a receptores que no pueden reciclar correctamente. Esto conduce
a un fenotipo relativamente suave, pues los receptores todavía están presentes en la superficie de la
célula (pero deben ser sintetizado nuevamente).
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
32
CFTR (Regulador de la Conductancia Transmembrana de la Fibrosis Quística)
Es el miembro 7 de la subfamilia C de los transportadores ABC (o ATPasas de tráfico)
Organización génica
En 1989 se aisló el gen CFTR, implicado en la FQ. Este gen se encuentra localizado en 7q31 y tiene 27
exones. Se extiende desde el par de bases número 116 907 253 hasta el par 117 095 955. La proteína
que codifica está compuesta por 1480 aminoácidos.
Ubicación génica del gen del CFTR
Organización estructural
CFTR es una proteína de 170000 daltons anclada a la membrana por dos dominios transmembrana (TM-
1 t TM-2) y cada dominio transmembrana atraviesa 6 veces la bicapa lipídica. Tiene dos sitios de unión al
ATP NFB1 y NFB2) y un dominio regulador (R) de alto contenido en aminoácidos como ácidos glutámico
y aspártico, glutamina y lisina. Los lugares de unión al ATP presentan una gran homología con dominios
similares de una familia de proteínas llamadas ABC o ATPasas de tráfico (traffic ATPases), entre las que
se encuentran las proteínas de resistencia a fármacos (MDR) del ratón y humanos, el transportador del
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
33
factor sexual a de Saccharomyces cerevisiae (STE6) y el factor de resistencia a cadmio de levadura
(YCF1).4
Esquema de la proteína CFTR.
TM1 y TM2, dominios transmembrana
NFB1 y NFB2, dominios de unión al ATP
R, dominio regulador
El primer dominio transmembrana (TM1) del CFTR es el soporte físico del poro del canal. Se supone que
determinados residuos de aminoácidos básicos crean dentro del poro las condiciones para dotarle de las
características de un canal de cloro: las cargas positivas de la arginina interaccionarían con algunas de las
moléculas de cloro y crearían un poro por el que fluirían los iones cloruro embebidos entre moléculas de
agua.
El cambio de algunos aminoácidos de la región transmembrana de la proteína modifica su especificidad,
haciéndose más permeable a los iones I que a los iones cloruro. Este hecho avaló experimentalmente,
que la proteína CFTR puede funcionar como un canal de cloruros. Otra evidencia de que la proteína
CFTR es un canal de iones cloruro es que las células que no tienen canal CFTR (de aproximadamente 8-
10 pS estimulable por epinefrina y AMPc y sus análogos) lo adquieren cuando se les introduce el gen y
éste se expresa.
CFTR es un canal cuya apertura y cierre está controlado por estímulos hormonales, cuyo efecto se ejerce
elevando la concentración intracelular de AMPc. El AMPc es un segundo mensajero que activa una
proteína quinasa A (PKA), la cual a su vez fosforila a otras proteínas que son activadas o inactivadas por
dicha fosforilación. El isoproterenol, la epinefrina, las prostaglandinas E1 y E2, la adenosina y el péptido
intestinal vasoactivo son algunas de las sustancias que estimulan el flujo de iones cloruro por este
mecanismo. En todos estos casos, la fosforilación de la CFTR por la proteína quinasa dependiente de
AMPc provoca la apertura del canal y la salida de iones cloruro a favor de la gradiente.
4 Salcedo A. Fibrosis Quística. Ediciones Díaz de Santos, 1998. Págs. 29-30
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
34
Existen 9 sitios “consenso” para fosforilación por PKA en el dominio R de la proteína CFTR. Cuatro de
estos sitios se fosforilan in vivo. Mediante mutagénesis dirigida se pueden cambiar estas serinas por
alaninas y con ello se consigue que la CFTR sea menos sensible al AMPc.
Fosforilación in vivo del dominio R de la proteína
CFTR
La deleción de una gran parte del dominio R (aminoácido 708 hasta el 835) hace que el CFTR se haga casi
insensible a la acción del AMPc (permanece abierto en ausencia de AMPc). En este mutante de CFTR, la
sustitución por alanina de la única serina fosforilable por la PKA que aún conserva (serina 660) provoca
la apertura total y la independencia del AMPc, pero aún necesita de la presencia de ATP para abrirse.
Los análogos no hidrolizables del ATP no pueden sustituirle en provocar este efecto, lo que hace
suponer que es necesaria la hidrólisis del ATP. En este sentido, se ha comprobado que la región NBF1
aislada tiene actividad ATPasa: su constante de Michaells (Km) para ATP es de 0,1 mM y su actividad
molecular es inferior a la de la proteína MDR. La actividad se inhibe por inhibidores de las ATPasas como
el AMP-PNP y el sulfito sódico. Mutantes en aminoácidos muy conservados a lo largo de la evolución de
este dominio pierden la actividad hidrolítica sobre ATP. Sobre el CFRT fosforilado en su dominio R, la
hidrólisis consigue así una apertura transitoria que se puede estabilizar por la presencia de análogos del
ATP no hidrolizables
En base a los datos experimentales actuales, la apertura del canal CFTR se activa por fosforilación del
dominio R por la PKA. Además, es necesaria la hidrólisis de ATP por el NBF1 y que otra molécula de ATP
(o algún análogo no hidrolizable de este) se una al NBF2 para estabilizar la apertura del canal.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
35
Regulación del canal CFTR. De Fibrosis Quística.
(A. Salcedo Posadas)
Función
CFTR es un canal de cloruro regulado por AMPc, que regula otros canales de iones. El CFTR mantiene la
hidratación de las secreciones en los conductos y las vías aéreas liberando cloruro e inhibiendo la
capación de sodio.5
La disfunción de CFTR puede afectar a muchos órganos, especialmente a los que segregan moco, como
las vías aéreas altas y bajas, páncreas, sistema biliar, genitales masculinos, intestino y glándulas
sudoríparas.
Las secreciones deshidratadas y viscosas de los pulmones de los pacientes con fibrosis quística
interfieren con la limpieza mucociliar, inhiben la función de los péptidos antimicrobianos naturales,
proporcionan un medio de cultivo a los gérmenes patógenos y obstruyen el flujo de aire. Durante los
primeros meses de vida, estas secreciones y las bacterias que las colonizan inician una reacción
inflamatoria. La liberación de citocinas inflamatorias, enzimas antibacterianas del huésped y enzimas
bacterianas dañan los bronquiolos. La repetición de los ciclos de infección, inflamación y destrucción
tisular reduce la cantidad de tejido pulmonar funcional y, finalmente, producen un fallo pulmonar.
5 Thompson M, Nussbaum R, Thompson J, McInnes R, Williard H. Genética en medicina. España, Elsevier, 2004. Pag 188
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
36
Fotografía de una sección transversal media del
pulmón de un paciente con fibrosis quística.
Nótense los tapones mucosos y las secreciones
purulentas dentro de las vías aéreas. De “Genética
en medicina” Thompson M, Nussbaum R,
Thompson J, McInnes R, Williard H.
La pérdida de transporte de cloruro del CFTR en los conductos pancreáticos altera la hidratación de las
secreciones y produce retención de enzimas exocrinas en el páncreas. El daño producido por la
retención de estas enzimas causa finalmente fibrosis del páncreas.
CFTR regula también la captación del sodio y cloruro del sudor a medida que éste avanza a lo largo del
conducto sudoríparo. En ausencia de CFTR funcional, el sudor tiene un contenido mayor de cloruro
sódico; ésta es la base del “síndrome del bebé salado” y de la prueba diagnóstica del cloruro en sudor.
Además de con la fibrosis quística, algunas mutaciones en CFTR se asocian con un espectro de
enfermedades, entre las que se incluyen la azoospermia obstructiva, la pancreatitis idiopática, la
bronquiectasia diseminada, la aspergilosis broncopulmonar alérgica, la enfermedad senopulmonar
atípica y el asma. Algunos de estos trastornos se asocian con mutaciones en un solo alelo CFTR, mientras
que otros, como la fibrosis quística se producen cuando existen mutaciones en ambos alelos. En algunos
casos se ha determinado el papel causal de estas mutaciones, pero no en otros.
Sólo existe correlación entre determinados alelos CFTR mutantes y la gravedad de la enfermedad o
insuficiencia pancreática. Algunas mutaciones secundarias o polimorfismos en un alelo CFTR pueden
alterar la eficacia del ensamblaje o de la maduración de la proteína, extendiendo así el espectro de la
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
37
enfermedad asociada con algunas mutaciones. Además, algunas mutaciones se expresan de forma
predominante en ciertos tejidos. Por ejemplo, ciertas mutaciones que afectan la eficacia del ensamblaje
tienen un mayor efecto sobre la exposición del CFTR en los derivados de los conductos de Wolff que en
otros tejidos. Algunos factores ambientales, como exposición al humo del tabaco, empeoran la gravedad
de la enfermedad pulmonar en los pacientes con fibrosis quística.
Correlación genotipo – fenotipo
La Fibrosis Quística es un enfermedad autosómica recesiva causada por mutaciones en el gen regulador
de la conductancia transmembránica (CFTR) que clínicamente se trata de una enfermedad
multisistémica que presenta implicación pulmonar, digestiva y de aparato reproductor.6
Actividad de la proteína CFTR en la membrana celular del humano (Pediatra Broncopulmonar Dr. Luis
E. Vega-Briceño, CFTR: Más que un canal de cloro)
Este gen abarca, aproximadamente 250 kb y está formado por 27 exones, con tamaños comprendidos
entre 38 y 274 pares de bases. Codifica para un ARNm de 6,5 kb detectable mediante northern blot en
varios tejidos, sobre todo en aquellos afectados por la FIBROSIS QUÍSTICA: pulmón, páncreas, glándulas
sudoríparas, hígado, pólipo nasal, glándulas salivales y colon. Este ARNm se traduce en una proteína de
1480 aminoácidos que funciona como un canal de cloro regulado por AMPc. CFTR se ha considerado
miembro de la familia de proteínas ABC (ATP-binding cassete).
6 Salcedo A. Fibrosis Quística. Ediciones Díaz de Santos, 1998. Págs.35-39
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
38
La expresión de la Fibrosis Quística es muy heterogénea en los diferentes pacientes. Dado el amplio
espectro de las mutaciones encontradas y las subsiguientes consecuencias moleculares, debería existir
correlación entre diferentes genotipos y sus fenotipos. Se ha realizado un gran número de estudios
sistemáticos agrupando pacientes que presentaban características clínicas comunes entre individuos con
Fibrosis Quística con el mismo genotipo.
De entre todos los parámetros analizados, sólo la función pancreática se correlaciona bien con los
fenotipos clínicos y parece correlacionar con diferentes mutaciones en CFTR: en este sentido, los
fenotipos que presentan suficiencia pancreática se asocian con pacientes que tienen una o dos
mutaciones leves, la mayoría de ellas de cambio de aminoácido. Aproximadamente el 15% de los
pacientes con Fibrosis Quística presentan suficiencia pancreática, el resto tienen insuficiencia.
Los fenotipos pancreáticos insuficientes se asocian a pacientes que son portadores de dos alelos
severos, tales como F508 u otras mutaciones de parada, de cambio de pauta de lectura, etc. El alelo
leve parece conferir un fenotipo dominante sobre el grave.
En un intento por correlacionar las diferentes mutaciones con el problema funcional que ocasionan, el
sistema original propuesto por Tsui fui redefinido por Welsh y Smith, se establecen 5 clases nombradas
de la I a la V. La clase I corresponde a las mutaciones en las que no se produce proteína CFRT normal,
afectan la biosíntesis. La clase II son mutaciones que afectan la maduración de la proteína, dentro de
este grupo se encuentra la F508. La clase III corresponde a mutaciones que afectan la regulación del
canal de Cl: la CFTR alcanza la membrana celular pero es incapaz de responder a los estímulos con
AMPc. La clase IV agrupa a las mutaciones que afectan la conducción del cloro, la CFTR actúa como un
canal de cloro alterado. Por último, las de clase V son aquellas que dan lugar a una síntesis reducida de
proteína o un procesado defectuoso del CFTR normal. En este caso las propiedades del canal son
normales.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
39
Proteínas Estructurales Transportadoras
Hemoglobinas
Es una heteroproteína de la sangre, de peso molecular 68.000 (68 kD). Transporta el oxígeno desde los
órganos respiratorios hasta los tejidos. La forman cuatro cadenas polipeptídicas (globinas) a cada una de las
cuales se une un grupo hemo, cuyo átomo de hierro es capaz de unirse de forma reversible al oxígeno.
Organización génica
Cada una de las cadenas polipeptídicas de la Hb cuenta con genes propios: a, b, d, g, e. Los genes a y b son
independientes y se ubican en cromosomas distintos. El grupo a, se localiza en el brazo corto del cromosoma
16 (región 16p13.3) y contiene además los codificadores de la cadena z. El grupo b se localiza en el brazo
corto del cromosoma 11 (región 11p15.15) e incluye a los genes de las cadenas g, d y e. Todos los genes
funcionales de la globina comparten una estructura general que consiste en 3 exones (secuencias
codificadoras) y 2 intrones o sectores interpuestos (secuencias que no se traducen). Existen dos secuencias
claves en la iniciación de la transcripción: TATA y CAT. La porción distal del tercer exón (AATAAA) finaliza la
transcripción. La transcripción primaria del ARNm incluye copias de toda la secuencia del ADN genómico
(intrones y exones). Antes de su transporte al citoplasma se procesa por clivaje del extremo 5’, hay
separación de las secuencias transcriptas de los intrones y poliadenilación del extremo 3’. Los puntos de
consenso son secuencias de nucleótidos adyacentes que perfeccionan la síntesis del ARNm.
Síntesis de la Hb
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
40
En el feto humano, en un principio, no se sintetizan cadenas alfa ni beta, sino zeta (z) y epsilon (e) (Hb Gower
I). Al final del primer trimestre la subunidades a han reemplazado a las subunidades z (Hb Gower II) y las
subunidades g a los péptidos e. Por esto, la HbF tiene la composición a2g2. Las subunidades b comienzan su
síntesis en el tercer trimestre y no reemplazan a g en su totalidad hasta algunas semanas después del
nacimiento.
Organización estructural
La hemoglobina es una proteína con estructura cuaternaria, es decir, está constituida por cuatro cadenas
polipeptídicas: dos a y dos b (hemoglobina adulta- HbA); dos a y dos d (forma minoritaria de hemoglobina
adulta- HbA2- normal 2%); dos a y dos g (hemoglobina fetal- HbF).. Las cadenas polipeptídicas alfa contienen
141 aminoácidos, las no alfa 146 (b, g, d) y difieren en la secuencia de aminoácidos.
La estructura secundaria es muy similar: cada una exhibe 8 segmentos helicoidales designados con las letras
A a la H. Entre ellos se encuentran 7 segmentos no helicoidales. Cada cadena a esta en contacto con las
cadenas b, sin embargo, existen pocas interacciones entre las dos cadenas a o entre las dos cadenas b entre
sí.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
41
Las cuatro cadenas polipeptídicas de la Hb contienen cada una un grupo prostético, el Hem, un
tetrapirrol cíclico, que les proporciona el color rojo a los hematíes. Un grupo prostético es una porción
no polipeptídica que forma parte de una proteína en su estado funcional.
Cuando una proteína esta con su grupo prostético se denomina holoproteina, y cuando esta sin este, se
lo denomina apoproteina. Además por poseer un grupo prostético se dice que la Hb es una proteína
conjugada, es una hemoproteina.
Correlación genotipo – fenotipo
Alteraciones de la Hb
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
42
ESTRUCTURALES
Falciforme o Drepanocítica Hb S. En 1949, Pauling descubrió que en la anemia falciforme había
alteración en la molécula de Hb. Es una enfermedad hereditaria, autosómica recesiva, ya que es
necesario que el individuo sea homocigoto (HbSHbS) para tener la enfermedad. Sin embargo, también
puede presentarse como heterocigoto, es decir HbA y HbS produciendo tan sólo el rasgo falciforme y
una resistencia a la malaria. En esta patología se produce un cambio de aminoácido en la posición 6 de
beta globina normal, cambiando ácido glutámico por valina, lo que disminuye la solubilidad de la
proteína, de tal manera que la hemoglobina S forma polímeros produciendo un glóbulo rojo en forma de
hoz, cuando han liberado el oxigeno.
Eritrocitos en forma de hoz
Estos glóbulos rojos falciformes no son flexibles y forman tapones en los vasos sanguíneos pequeños,
produciendo una interrupción de la circulación de la sangre que puede dañar los órganos de cualquier
parte del cuerpo. En un estudio realizado por Robert Hebbel y sus colaboradores, demostraron que el
componente hemo de la hemoglobina tiende a liberarse de la proteína debido a episodios repetidos de
la polimerización de la hemoglobina S. Algunos de estos grupos hemo libres tienden a alojarse en la
membrana de los hematíes, el hierro de este grupo promueve la formación de componentes muy
peligrosos llamados especies reactivas de oxígeno. Estas moléculas dañan los componentes lipídicos y
proteicos de la membrana de los glóbulos rojos, produciendo su destrucción (hemólisis). Por lo tanto, en
la anemia falciforme se incrementa la hemólisis y desciende el valor de la hemoglobina y el hematocrito,
es decir que se presenta como una anemia hemolítica crónica, donde las manifestaciones clínicas se
inician a los seis meses de eda. El balance entre vasoconstrictores y vasodilatadores se altera a favor de
los primeros y el flujo de la sangre se hace lento. También se observan trastornos en el crecimiento y
desarrollo del niño. Generalmente se observan retardos en la maduración sexual. La vasooclusión posee
manifestaciones diversas: isquemia dolorosa, microinfartos, crisis de secuestro esplénico (que puede ser
causa de muerte súbita en niños), neovascularización, necrosis de órganos isquémicos afectados, entre
otras.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
43
Metahemoglobinemias
- Hb M: La metahemoglobina (ferrihemoglobina) es un derivado de la hemoglobina en que el hierro
ferroso se oxida a su forma férrica, lo que origina un color azulado pardo, similar a la cianosis de la piel.
La metahemoglobina forma parte de la hemoglobina "inactiva"; es incapaz de combinarse de modo
reversible con el oxígeno y monóxido de carbono, además desvía la curva de disociación oxígeno en el
sentido de un aumento de su afinidad por este y entorpece por tanto su transporte desde la sangre a los
tejidos. Así, cantidades anormales de metahemoglobinemia, causarán una "anemia" funcional con
cianosis (debido a la capacidad reducida de la sangre para transportar oxígeno). La
metahemoglobinemia congénita se hereda como rasgo autosómico dominante; es consecuencia de
mutaciones de la globina que estabiliza al Fe en el estado férrico, o por mutaciones que merman las
enzimas que reducen la metahemoglobina a Hb. La Metahemoglobinemia adquirida se debe a toxinas
que oxidan el Fe del hemo, en particular los compuestos que tienen nitratos y nitritos. Algunos pueden
presentar moderada policitemia que intenta compensar las necesidades de transportación de oxígeno
tisular. En otros pacientes un ligero retardo mental puede acompañar su evolución. La
metahemoglobinemia tóxica si aparece rápidamente produce síntomas de anoxia.Con cifras de 20 a 30
% aparece fatiga, disnea, taquicardia, cefalea, lipotimia, naúseas y vómitos, aunque algunos de estos
síntomas son propios del agente causal. Con concentraciones mayores de 55 % se presentan estupor y
letargo. Por encima de 70 % de metahemoglobinemia es mortal.
Anemia hemolítica Congénita de Cuerpos de Heinz (AHCCH):
Los trabajos de Carrel y Lehmann en 1966 demostraron la patología molecular en este tipo de anemia y
que la inestabilidad de la molécula de Hb era debida a la sustitución de aminoácidos. Las mutaciones
representativas son las que interfieren en los puntos de contactos entre las subunidades alfa y beta, por
ejemplo Hb Philly (b35 Tyr--->Phe); alteran los segmentos helicoidales, como la Hb Génova (b28 Leu---
>Pro); o alteran las interacciones de la bolsas hidrófobas de las subunidades de globina con el hem,
como Hb Koln (b98 Val--->Met). El grado de hemólisis se correlaciona con el grado de mutación de la Hb.
Los hallazgos clínicos como ictericia, palidez, esplenomegalia, orinas oscuras con pigmenturia se
correlacionan con la hemólisis. El valor de la Hb puede ser normal o bajo. En caso de sospecha clínica es
importante establecer la presencia de cuerpos de Heinz, que se forman dentro de las células por
oxidación de la Hb pasando a sulfaHb que se precipita en forma de cuerpos de Heinz insolubles. Estas
inclusiones son eliminadas por el Bazo dando lugar a células “picoteadas”, rígidas de supervivencia
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
44
abreviada, lo que produce anemia hemolítica de gravedad variable, que a veces requiere transfusiones.
Estigmas frecuentes son las ulceras en las piernas y la vesiculopatia prematura, por el alto recambio de
bilirrubina.
Hemoglobinas con alteración de la afinidad por el oxígeno.
Se producen por mutaciones situadas a nivel de las áreas de contacto entre las subunidades a y b de la
molécula de Hb o de la zona de unión del 2,3 DPG a la cadena b. De acuerdo con la naturaleza de la
mutación pueden observarse aumentos o disminuciones de la afinidad de la hemoglobina por el
oxígeno, siendo las primeros mucho más frecuentes que las segundos. Se heredan con carácter
autosómico dominante, siendo la forma homocigota, al igual que otras hemoglobinopatías
estructurales, incompatibles con la vida.
a) Alta afinidad por el Oxígeno
Policitemia
Es un trastorno en el cual hay demasiados glóbulos rojos en la circulación sanguínea. Es el opuesto de
la anemia, que ocurre cuando hay escasez de glóbulos rojos en la circulación. La policitemia también
se denomina plétora (aumento excesivo de sangre). Esto hace que halla mucho oxígeno en la sangre.
b) Baja afinidad por el Oxígeno
Cianosis
Es la coloración azulada de la piel mucosas y lechos ungueales, usualmente debida a la existencia de
por lo menos, 5 g% de hemoglobina reducida en la sangre circulante o de pigmentos hemoglobínicos
anómalos (metahemoglubina o sulfohemoglobina) en los glóbulos rojos.
La cianosis se clasifica como central o periférica:
La cianosis central resulta de la hipoxemia arterial causada por alteración de la función pulmonar
(hipoventilación alveolar, alteraciones de la ventilación-perfusión, transtornos de difusión de
oxígeno) o por la existencia de cortocircuitos intracardiacos derecha-izquierda (defectos septales),
entre los grandes vasos (conducto arterioso) o en los pulmones. También puede observarse en la
policitemia vera en ausencia de insaturación arterial de oxígeno, debido al incremento de
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
45
hemoglobina reducida en la sangre. En la cianosis central tanto la piel como las mucosas tienen el
color azulado.
La cianosis periférica aparece como resultado de la disminución del flujo sanguíneo periférico y de
vasoconstricción. El flujo sanguíneo lento permite que cada hematín dure en contacto con los tejidos
durante más tiempo; en consecuencia, se extrae más oxígeno de la sangre arterial con el posterior
incremento de hemoglobina reducida en la sangre venosa. Se observa habitualmente en los tejidos
periféricos (manos, orejas, nariz y pies), pudiendo ser generalizada o localizada. Las causas que la
originan son múltiples, siendo las principales la exposición al frío, la insuficiencia cardiaca y la
obstrucción venosa.
TALASEMIAS
Los dos tipos principales de talasemia se denominan talasemia alfa y talasemia beta. Los individuos
afectados por el primer tipo no producen suficiente cantidad de globina alfa y los afectados por el
segundo, de globina beta. A su vez, cada uno de estos tipos de talasemia puede adoptar formas
diferentes, con síntomas que van de leves a severos. Los términos “mayor, menor, intermedia y
mínima”, utilizadas para indicar la gravedad de las manifestaciones clínicas, no necesariamente indican
Heterocigota u Homocigota. Las talasemias más importantes se heredan por genes autosómicos
recesivos. Tanto la alfa como la beta talasemia ocasionan disminución de la Hb dentro del eritrocito, lo
que da lugar a una disminución del color (hipocromia) y del tamaño (microcitica) del hematíe. Otras
talasemias descriptas son la delta y la gamma, de escasa frecuencia.
Alfa talasemias:
Son cuatro los genes que controlan la producción de la globina alfa y la cantidad de genes faltantes o
anormales determina la severidad de la enfermedad. El principal mecanismo por el que se producen las
alfa talasemias es la deleción o pérdida total de un gen. Las formas no delecionales son menos
frecuentes y obedecen a mutaciones, alteraciones en la transcripción del ARN o producción de ARN
anómalo.
Pérdida en un solo gen alfa: En este caso no existe manifestación clínica. Solo se diagnostica
mediante técnicas complejas de análisis de ADN.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
46
Pérdida de dos genes alfa: Produce un cuadro denominado talasemia menor o rasgo talasémico; no
suele provocar problemas de salud importantes pero los individuos afectados pueden padecer una
ligera anemia y transmitir la enfermedad a sus descendientes.
Pérdida de tres genes alfa: Constituye la denominada enfermedad de Hb H, esto produce
anormalidades en los glóbulos rojos que derivan en su destrucción rápida. En esta enfermedad la
producción de Hb A va de 25 a 30%.en el adulto, la cadenas beta sin pareja se acumulan y forman
tetrámeros b4, denominadas Hb H. Es frecuente en China e Indonesia y se han descrito también
algunos casos en Italia y Sudamérica y en España. Cursan con un cuadro clínico de anemia hemolítica
de intensidad moderada exacerbada por infecciones o por la ingesta de algunos medicamentos
oxidantes, y moderada esplenomegalia. Es frecuente la supervivencia hasta la etapa media de la
edad adulta, sin transfusiones.
Pérdida de cuatro genes alfa: Es la denominada talasemia grave o mayor en la cual se produce la
muerte del niño durante la gestación o en el periodo que sigue al parto. Esta enfermedad es
incompatible con la vida del niño. Como la síntesis de la cadena alfa falla la HbA y la HbF disminuyen
y en su lugar aumentan la Hb de Bart (cuatro cadenas gamma), que tiene una extraordinaria afinidad
por el O2, y casi no lo suministra a los tejidos, causando asfixia y muerte; o la Hb H (cuatro cadenas
beta).
Beta Talasemias
Las beta talasemias son el resultado de la falta de síntesis de las cadenas beta de globina.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
47
Beta talasemia heterocigota o menor (rasgo talasemico): Aparece cuando sólo está afectada una de las
copias del gen que codifica la cadena. Es la mutación del gen beta, caracterizada por una hematíes
elevada, con concentración de hemoglobina normal o disminuida y generalmente presenta un aumento
de la Hb A2. Las personas portadoras de talasemia menor, no presentan manifestaciones clínicas,
aunque en ocasiones pueden tener una ligera anemia que se pone de manifiesto al realizar un análisis.
Los glóbulos rojos de los portadores del rasgo talasémico son más pequeños de lo normal. La talasemia
menor está presente desde el nacimiento, permanece durante toda la vida y puede transmitirse de los
padres a los hijos.
Beta Talasemia Homocigota O Mayor (Anemia De Cooley): Es la forma mas grave anemia congénita. La
talasemia homocigótica, es en la que las dos copias del gen para una cadena de la hemoglobina son
defectuosas, ocurre cuando no se sintetizan cadenas. Dependiendo de las mutaciones genéticas beta, se
producirá una cantidad nula o muy escasa de cadenas beta, y un menor o mayor número de cadenas
alfa.
La talasemia mayor es una anemia hereditaria grave. Los pacientes afectados con esta anomalía no
pueden fabricar suficientes glóbulos rojos y requieren frecuentes transfusiones de sangre. La
enfermedad se manifiesta durante los primeros meses de vida, habitualmente entre el tercer y octavo
mes. Estos pacientes presentan palidez, alteraciones del sueño, rechazo de los alimentos y vómitos.
Desarrollan hemosiderosis (depósito en todos los tejidos del hierro liberado tras la hemólisis). Es
frecuente la presencia de cálculos biliares por la hemólisis crónica. Adquieren un color pardo-verdoso
por la anemia, la ictericia (la hemólisis libera bilirrubina que produce un color amarillo en la piel y
mucosas) y la hemosiderosis. Se detiene el crecimiento, se retrasa la pubertad. Y finalmente se produce
un fallo cardíaco. Actualmente algunos pacientes pueden también ser tratados, e incluso curados,
mediante un transplante de médula ósea.
Beta Talasemia Intermedia: Se designa así al síndrome talasémico de moderada intensidad, que
condiciona la aparición de una anemia leve y alteraciones óseas. Presentan sintomatología clínica y
requieren transfusiones de sangre durante alguna época de su vida, pueden desarrollar hemosiderosis.
Sus manifestaciones no son tan graves como en los pacientes afectados de la forma mayor de la
enfermedad.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
48
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
49
Proteínas Estructurales Extracelulares
Colágeno
Los colágenos están compuestos por tres cadenas polipeptídicas α, que pueden ser idénticas o
diferentes, cada cadena diferente se designa con un numero arábigo y como las cadenas α son distintos
en cada tipo de colágeno , la nomenclatura de cada una incluye, además de su número arábigo, el
numero romano de su tipo de colágeno ; así por ejemplo, para el colágeno I, el más general del tejido
conectivo, las cadenas α1(I) y α2(I).cada cadena tiene un gen diferente , designado “COL”, seguido del
tipo de colágeno ( en números arábigos ) y la cadena (designada ”A”)y su número. Los genes COL están
dispersos por el genoma humano.
Los colágenos se clasifican ( según formen o no fibrillas) en fibrilares ( tipos I,II,III,V Y IX) y no fibrilares
( tipos IV, VII,VIII,IX,X,XII,XIII Y XIV)y un colágeno fibrilar pero de fibrilla no escalonada (el VI). La fibrilla
típica ( de moléculas escalonadas) tiene la periodicidad característica de 67nm ( periodicidad D), y la
longitud de la molécula formada por las tres cadenas , el tropocolágeno, es 4,4veces D ( alrededor de
300nm) esta fibrilla elemental de colágeno , tan regular es el producto de la triple hélice , muy regular,
de las tres cadenas α ; y esta triple hélice a su vez, es resultado de la secuencia regular de aminoácidos
(aa), en unidades repetidas de a tres, todas las cuales comienzan por glicina y siguen con dos
aminoácidos prefijados ( unidades Gli-X-Y) . las mutaciones que alteran esta secuencia tienen profundas
repercusiones sobre el colágeno ; por otra parte como las tres cadenas empiezan a unirse por la región
terminal o carboxiloterminal (para la formación de la triple hélice) esta región es particularmente
sensible a los cambios mutacionales.
Organización génica
Los genes “COL” están dispersos por muchas regiones DEL GENOMA HUMANO , sin formar un cumulo
definido ; sin embargo, poseen una organización con muchas semejanzas, que son constantes en los
exones correspondientes al dominio helicoidal central , donde estos son siempre pequeños , de un
múltiplo de 9 pb, que puede ser 54pb ( el mas frecuente), 45, 99, 108, y 162pb . Estos exones codifican
respectivamente en unidades petidicas de tripletes gli-x-y) 6 unidades , 5,11,12 y 18 unidades. Lo mas
llamativo es que todos los exones centrales comienzan exactamente por el codón de glicina y terminan
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
50
por el aminoácido “y” ; este patrón de organización es muy antiguo en la evolución poruqe incluso se
halla en erizos de mar , por lo cual es previo a la divergencia de vertebrados e invertebrados en la
evolución de los organismos.
El gen de la cadena pro-α(1)( del colágeno mas común ) ,localizado en el cromosoma 17 ( 17q21.3),
posee 51 exones de los cuales los primeros cinco corresponden al péptido señal(1) y al dominio globular
propetido-N(4) , del 6 al 47 corresponden al dominio helicoidal y los cuatro últimos corresponden al
propeptido-C.
Organización estructural
Tipos de cadenas de colágeno
Tipo Composición de la
Cadena
Símbolo(s) del
Gen
Detalles Estructurales Localización
I [α1(I)]2[α(I)] COL1A1,
COL1A2
300nm, 67nm fibrillas en
banda
piel, tendón, hueso, etc.
II [α1(II)]3 COL2A1 300nm, pequeñas 67nm
fibrillas
cartílago, humor vítreo
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
51
III [α1(III)]3 COL3A1 300nm, pequeñas 67nm
fibrillas
piel, músculo, frecuentemente
con tipo I
IV [α1(IV)2[α2(IV)] COL4A1 por
COL4A6
390nm dominio globular C-
terminal, no fibrilar
toda la lámina basal
V [α1(V)][α2(V)][α3(V)] COL5A1,
COL5A2,
COL5A3
390nm dominio globular N-
terminal, fibras pequeñas
mayoría de tejido intersticial,
assoc. con tipo I
VI [α1(VI)][α2(VI)][α3(VI)] COL6A1,
COL6A2,
COL6A3
150nm, N+C term.
Dominios globulares,
microfibrillas, 100nm
fibrillas en banda
mayoría de tejido intersticial,
assoc. con tipo I
VII [α1(VII)]3 COL7A1 450nm, dímero epitelio
VIII [α1(VIII)]3 COL8A1,
COL8A2
algunas células endoteliales
IX [α1(IX)][α2(IX)][α3(IX)] COL9A1,
COL9A2,
COL9A3
200nm, dominio globular
N-term. unido a
proteoglicano
cartílago, assoc. con tipo II
X [α1(X)]3 COL10A1 150nm, dominio globular
C-term.
cartílago hipertrófico y
mineralizante
XI [α1(XI)][α2(XI)][α3(XI)] COL11A1,
COL11A2
300nm, fibras pequeñas cartílago
XII α1(XII) COL12A1 interactúa con tipo I y III
Correlación genotipo – fenotipo
Osteogénesis imperfecta
Es una afección (también llamada huesos de cristal) que ocasiona huesos extremadamente frágiles.
Debido a las frecuentes fracturas que padecen, en muchas ocasiones se confunde la enfermedad con
maltrato infantil.
Etiología
La osteogénesis u osteogenia imperfecta (OI) es una enfermedad congénita, lo que quiere decir que
está presente al nacer. Con frecuencia es causada por un defecto en un gen que produce el colágeno
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
52
tipo 1, un pilar fundamental del hueso. Existen muchos defectos diferentes que pueden afectar este
gen y la gravedad de esta enfermedad depende del defecto específico de dicho gen.
Existen varios tipos de mutaciones en el COL1A1 gen causa la formas más graves de la osteogénesis
imperfecta, incluyendo los tipos II, III y IV. Algunas de estas mutaciones eliminar los segmentos de
ADN de la COL1A1 gen, dando lugar a una anormalmente reducido, a menudo no funcional pro-α1 (I)
de la cadena. Otros cambios genéticos que alteran la secuencia de aminoácidos en el pro-α1 (I) de la
cadena, por lo general sustituye el aminoácido glicina con un aminoácido diferente. En algunos casos,
el ácido amino sustituciones alterar uno de los extremos de la cadena de proteína (llamada C-
terminal), que interfiere con el ensamblaje de las moléculas de colágeno. Estos COL1A1 mutaciones
conducen a la producción de versiones anormales de colágeno tipo I.Cuando este colágeno
anormales se incorpora en el desarrollo de huesos y otros tejidos conectivos, hace que el médico los
problemas serios asociados con formas graves de osteogénesis imperfecta.
Algunos COL1A2 mutaciones eliminar piezas del gen, lo que lleva a un pro-α2 (I) de la cadena que
falta regiones críticas. Otros cambios genéticos que alteran la secuencia de bloques estructurales de
las proteínas (aminoácidos) en el pro-α2 (I) de la cadena, por lo general sustituye el aminoácido
glicina con un aminoácido diferente. En algunos casos, el ácido amino sustituciones alterar uno de los
extremos de la cadena de proteína (llamada C-terminal), que interfiere con el ensamblaje de las
moléculas de colágeno. Estos COL1A2 mutaciones prevenir la producción normal de colágeno tipo I.
Cuando el colágeno anormales se incorpora en el desarrollo de huesos y otros tejidos conectivos,
hace que el médico los problemas serios asociados con formas graves de osteogénesis imperfecta.
La osteogénesis imperfecta es una enfermedad autosómica dominante, lo que quiere decir que usted
la padecerá si tiene una copia del gen. La mayoría de los casos de OI se heredan de uno de los padres,
aunque algunos casos son el resultado de nuevas mutaciones genéticas.
Una persona con osteogénesis imperfecta tiene un 50% de posibilidades de transmitirle el gen y la
enfermedad a sus hijos.
Clasificación
Tipo I u osteogénesis imperfecta leve: Es el más común y las personas pueden tener una
expectativa de vida normal.
Tipo II: Es una forma severa que generalmente lleva a la muerte en el primer año de vida.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
53
Tipo III También llamada osteogénesis imperfecta severa: Las personas con este tipo presentan
muchas fracturas en el comienzo de su vida y pueden sufrir graves deformidades óseas. Muchos
quedan limitados a una silla de ruedas y generalmente tienen una expectativa de vida algo más
corta.
Tipo IV u osteogénesis imperfecta moderadamente severa: Es similar al tipo I, aunque las
personas necesitan muletas o dispositivos ortopédicos para caminar. La expectativa de vida es
normal o cerca de lo normal.
Existen otros tipos de osteogénesis imperfecta, pero ocurren con muy poca frecuencia y la
mayoría se consideran subtipos de la forma moderadamente severa (tipo IV).
Síntomas
Todas las personas con osteogénesis imperfecta (OI) tienen huesos débiles, lo cual las hace
susceptibles a sufrir fracturas. Las personas con Osteogénesis imperfecta generalmente tienen una
estatura por debajo del promedio (estatura baja). Sin embargo, la gravedad de la enfermedad varía
enormemente.
Radiografías de varios pacientes, niños con osteogénesis imperfecta
Los síntomas clásicos abarcan:
Tinte azul en la parte blanca de los ojos (esclerótica azul)
Fracturas óseas múltiples.
Pérdida temprana de la audición (sordera)
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
54
Debido a que el colágeno tipo 1 también se encuentra en los ligamentos, las personas con
Osteogénesis imperfecta a menudo tienen articulaciones flexibles (hipermovilidad) y pies planos.
Algunos tipo de OI también llevan al desarrollo deficiente de los dientes.
Los síntomas de las formas más severas de Osteogénesis imperfecta pueden abarcar:
Brazos y piernas arqueadas.
Cifosis.
Escoliosis (curvatura de la columna en forma de "S")
Síndrome de Ehlers-Danlos
El síndrome de Ehlers-Danlos es un grupo heterogéneo de trastornos hereditarios del tejido
conectivo caracterizado por hiperlaxitud articular, fragilidad cutánea, e hiperextensibilidad. El defecto
del colágeno se ha identificado en sólo 6 de los 11 tipos del síndrome de Ehlers-Danlos. El tipo IV se
caracteriza por una disminución en la cantidad de colágeno tipo III. Tipos V y VI se caracterizan por
deficiencias en la lisil oxidasa y hidroxilasa, una enzima importante en la modificación
postraduccional de la biosíntesis de colágeno. Tipo VII, tiene un déficit de procolágeno peptidasa
amino-terminal. Tipo IX ha metabolismo del cobre anormal. Tipo X tiene no funcionales fibronectina
plasmática.
Autosómica recesiva VI de Ehlers-Danlos tipo, también conocido como el tipo cifoescoliosis, se
caracteriza por cifoescoliosis neonatal, laxitud articular generalizada, la fragilidad de la piel, e
hipotonía muscular severa al nacer.Bioquímicamente, este tipo se atribuye a una deficiencia de lisil
hidroxilasa (LH), la enzima que hidroxila residuos específicos de lisina en la molécula de colágeno
para formar hidroxilisinas con dos funciones importantes. Los residuos son los sitios de fijación de
galactosa y glucosylgalactose, y actúan como precursores del proceso de reticulación del colágeno
que le da su fuerza de tensión.
Tenascina-X es una gran matriz extracelular de la proteína, la deficiencia de lo que provoca una forma
distinta recesiva clínica de este síndrome. Por lo tanto, otros factores distintos de colágenos o
transformación enzimas colágeno puede causar este síndrome. Esta nueva forma descrita se puede
asociar con anomalías adicionales
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
55
Alteración de la cicatrización de heridas es un rasgo típico de síndrome de Ehlers-Danlos,
probablemente por un defecto de fibroblastos. La reparación de la herida se puede lograr utilizando
el tipo exógeno colágeno V.
Danlos Síndrome de Ehlers pacientes pediátricos se ha demostrado que tienen deficiencias en tres
genes de la familia S-transferasa de glutatión (GSTM1, GSTT1, GSTP1 ). Esto conduce a la generación
de especies reactivas de oxígeno.
Caso clínico 1
Mujer de 37 años de edad, casada, procedente de la provincia del Cotopaxi, con antecedentes
familiares de padre y abuelo paterno portadores de enfermedad cutánea similar la que cursa la
paciente. Nacida por parto normal de un embarazo a término. Desde la infancia presenta fragilidad e
hiperextensibilidad cutáneas, aumento de la flexibilidad articular sobre todo a nivel de articulaciones
interfalángicas. Al examen físico se puede observar pseudotumoraciones de aproximadamente 0,5 a
1 cm de diámetro, movibles, con superficie cutánea normal. Múltiples cicatrices atróficas con la
característica en "papel de cigarrillo", localizadas sobre todo en miembros inferiores y áreas de roce.
Síndromes asociados a alteración del colágeno tipo IV: síndrome de Alport y Liquen escleroso y atrófico.
El colágeno IV es una macromolécula que se localiza específicamente en las membranasbasales. Está
compuesto por tres sub-unidades de cadenas α, de las cuales han sido identificadas seis tipos
diferentes (α1 - α6). Este colágeno constituye un heterotrímero, con dos cadenas α genéticamente
iguales y una cadena α diferente. Estas sub-unidades varían de acuerdo a la localización de la
membrana basal.
Se reconocen dos formas de transmisión del síndrome de Alport: ligada al X en un 85% de los casos y
autosómica recesiva en un 15%.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
56
En la forma ligada al X el defecto radica en el gen COL4A5, ubicado en Xq22-q23 (como consecuencia
de mutaciones sin sentido y delecciones). Dicho gen codifica la cadena α5 del colágeno IV, por lo
tanto su mutación da como resultado ausencia de la misma a nivel de las membranas basales de
epidermis, cóclea, glomérulo renal y ojo, hecho que se correlaciona con las alteraciones clínicas de
este síndrome (sordera, nefropatía y alteraciones oculares).
En la forma autosómica recesiva (poco frecuente) existen dos genes implicados, el COL4A3 y COL4A4,
localizados en el cromosoma 22, que codifican las cadenas α3 y α4 del colágeno IV respectivamente
(éstas se hallan en los mismos tejidos que la cadena α5).
La frecuencia del síndrome de Alport se estima de 2.7% en niños con insuficiencia renal. La alteración
renal comienza con hematuria persistente en la infancia, que progresa con el tiempo a insuficiencia
renal. También puede observarse hipertensión arterial y síndrome nefrótico.
La alteración ocular patognomónica es el lenticono anterior que provoca trastornos visuales, la cual
sólo está presente en un 25% de los casos. La pérdida auditiva sensorial es progresiva, iniciándose
con pérdida para las altas frecuencias (2 000 a 8 000 HZ) y luego para las bajas
El diagnóstico se realiza por la historia de hematuria, asociada a déficit auditivo. En la biopsia renal
puede observarse o no engrosamiento de la membrana basal y marcación con anticuerpos
monoclonales de los tipos de cadenas α faltantes.
El estudio inmunohistoquímico de la piel se considera el procedimiento diagnóstico no invasivo en
pacientes con microhematuria con sospecha de síndrome de Alport ligado al X, observando la
ausencia de α5 en la zona de la membrana basal en el 80% de los casos homocigotos.
El tratamiento es sintomático, se realiza con inhibidores de la enzima convertidora de angiotensina y
diálisis en caso de insuficiencia renal.
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
57
Fibrilina
INTRODUCCIÓN
La identificación de un producto génico alterado (FIBRILINA) ha permitido aclarar la Etiología del
Síndrome de Marfan, descrito hace un siglo. Se trata de una enfermedad hereditaria autosómica
dominante, esto significa que dicha enfermedad tiene la misma probabilidad de aparecer en un sexo
que en otro y ser éstos capaces de transmitirlo a la descendencia. Siempre que haya un individuo
afectado, significara que ha recibido al menos un alelo dominante de uno de sus padres. Puede
ocurrir también, que a pesar de que los padres no sean portadores, el nuevo individuo si padezca la
enfermedad, debido a una nueva mutación o por un fenómeno de penetrancia reducida.
Esta enfermedad se asocia al gen FBN1 del cromosoma 15. El FBN1 codifica una proteína
llamada fibrilina, que es esencial para la formación de fibras elásticas del tejido conectivo. Sin el
soporte estructural de las fibras elásticas, muchos tejidos presentan una debilidad que puede
conducir a distintas consecuencias como rotura de paredes arteriales, formación de
aneurismas),megalocórnea, aracnodactilia, etc.
Esta enfermedad es causada por un defecto (mutación) en el gen que determina la estructura de
la fibrilina, ésta es la responsable del ensamblaje de las redes de microfibrillas que, junto con la
elastina, forman parte de la matriz extracelular de los tejidos, es una proteína que constituye una
parte importante del tejido conectivo.Una alteración en esta proteína provocara una destrucción del
ensamblaje de las microfibrillas normales y la producción de fibras elásticas anormales.
Se piensa que la fibrilina normal actuaría inhibiendo la formación de huesos largos y que las
fibras elásticas serian las responsables, mediante su tensión, de controlar dicho crecimiento, por
tanto, al existir alteraciones en estas estructuras, se produciría un aumento exagerado de los huesos.
Se nace con el Síndrome de Marfan, aunque puede ser que no se le diagnostique hasta más
tarde. Aún cuando todas las personas con el Síndrome de Marfan tienen un defecto en el mismo gen,
la mutación es diferente en cada familia; no todas las personas experimentan las mismas
manifestaciones clínicas o con la misma severidad. Esto se conoce como expresión variable, lo que
implica que el gen defectuoso se manifiesta de manera diferente en las personas afectadas. Los
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
58
científicos aún no logran entender por qué ocurre esta expresión variable en las personas con
Marfan.
Esta variabilidad es llamada "heterogeneidad alélica", y es la responsable de que un mismo gen
produzca diferentes mutaciones, y por tanto, variaciones en las manifestaciones clínicas, o incluso
dan lugar a cuadros clínicos diferentes.
Los pacientes afectados por este Síndrome presentan, como ya dijimos, talla elevada,
extremidades largas y finas (Aracnodactilia), cifoescoliosis, hipotonía muscular, subluxación del
cristalino 7(Ectopia Lenticular 8 )y síntomas cardiovasculares: insuficiencia de las válvulas cardiacas y
degeneración de la túnica media de las grandes arterias.
El Gen correspondiente a la Fibrilina – 1 (FBN-1) se localiza en 15q21, cubre 110 kb y posee 65
exones; los dominios correspondientes en la proteína muestran ciertas repeticiones. En especial hay
siete dominios muy ricos en cisteína (posee 8 cisteina), que necesariamente deben estar en registro
cuando se asocian varios monómeros para formar un filamento.
Además, La FIBRILINA, es una glucoproteína de gran tamaño (320kDa) que contiene 42
dominios similares a un precursor del Factor de Crecimiento Epidérmico (EGF). Estos dominios,
contienen(en la mayor parte de casos) un “motivo” o secuencia de aa ligante de Ca++ .
Y como ya mencionamos, el gen también contiene 7 “ motivos “ de 70 aa (con 8 Cisteína cada
uno) similares a una parte de la proteína ligadora al Factor de Crecimiento y Transformación β1
(TGF-β1).
7 Solari. GENÉTICA HUMANA: Fundamentos y Aplicaciones en Medicina. Tercera Edición. 2003 : 333 – 3348 Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 284
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
59
ORGANIZACIÓN GENICA:
La Familia de las Fibrilinas (Fibrillin Family), involucra a varias proteínas, dentro de ellas:
FIBRILINA – 1 FIBRILINA – 2 FIBRILINA – 3
1. FIBRILINA – 1 : Nombre del gen: FBN1 9
Sinónimos: FBN, MASS, MFS1, OCTD, SGS, WMS 10
Símbolos Previos: FBN, MFS1, WMS 11
Alias: MASS, OCTD, SGS 12
Descripción: Fibrillin 1 (HGNC Symbol) 13
Cromosoma: 15 14
Citobanda: q21.1 15
Localización: 15q21.1 16 Ubicación de Cromosoma: 48700505 – 48937918 17 NCBI Gene ID: 2200 18
9 HUGO - Human Genome Organisation - Organización del Genoma Humano HGNC – COMITÉ DE NOMENCLATURA - http://www.genenames.org/ 10 THE HUMAN PROTEIN ATLAS - http://www.proteinatlas.org 11 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 12 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 13,6,7 http://www.proteinatlas.org/ENSG00000166147/gene 14
15
16 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 17 http://www.proteinatlas.org/ENSG00000166147/gene 18 Domain Mapping of Disease Mutations - http://bioinf.umbc.edu/DMDM/gene_prot_page.php?id=2200
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
60
Gen Tipo: Proteína Codificante 19
Resumen: 20
o Este gen codifica un miembro de la Familia de las Fibrilinas (Fibrillin Family). o La proteína codificada es grande, Glicoproteina de la Matriz Extracelular que sirve
como un componente estructural de Microfibrillas de 10 – 12 nm. ligadas al Calcio. Estas Microfibrillas, proporcionan una fuerza que produce soporte estructural en Tejido Conectivo Elástico y No Elástico en todo el cuerpo.
o Mutación en este gen está asociado con Síndrome de Marfan, Ectopia Aislada del Cristalino, Síndrome Autosómico Dominante Weill-Marchesani, Síndrome MASS, Síndrome de Craneosinostosis de Shprintzen-Goldberg.
Visor de Mapa: 21
19 NCBI - http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=2200 20 NCBI - http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=Graphics&list_uids=2200 21 http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?taxid=9606&chr=15&query=uid%28-1811869849,-2146581795%29&QSTR=2200[gene_id]&maps=gene_set&cmd=focus
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
61
Locus : X63556 / 9940 bp 22
Origen : 23
1 agtatttctc tcgcgagaaa ccgctgcgcg gacgatactt gaagaggtgg ggaaaggagg 61 gggctgcggg agccgcggca gagactgtgg gtgccacaag cggacaggag ccacagctgg 121 gacagctgcg agcggagccg agcagtggct gtagcggcca cgactgggag cagccgccgc 181 cgcctcctcg ggagtcggag ccgccgcttc tccactggca ggggccgcct gaagtgggag 241 cagcgcctgg agaaggcggg aggagcccgg cccgggggac gggcggcggg atagcgggac 301 cccggcggcg cggtgcgctt cagggcgcag cggcggccgc agaccgagcc ccgggcgcgg 361 caagaggcgg cgggagccgg tggcggctcg gcatcatgcg tcgagggcgt ctgctggaga 421 tcgccctggg atttaccgtg cttttagcgt cctacacgag ccatggggcg gacgccaatt 481 tggaggctgg gaacgtgaag gaaaccagag ccagtcgggc caagagaaga ggcggtggag 541 gacacgacgc gcttaaagga cccaatgtct gtggatcacg ttataatgct tactgttgcc 601 ctggatggaa aaccttacct ggcggaaatc agtgtattgt ccccatttgc cggcattcct 661 gtggggatgg attttgttcg aggccaaata tgtgcacttg cccatctggt cagatagctc 721 cttcctgtgg ctccagatcc atacaacact gcaatattcg ctgtatgaat ggaggtagct 781 gcagtgacga tcactgtcta tgccagaaag gatacatagg gactcactgt ggacaacctg 841 tttgtgaaag tggctgtctc aatggaggaa ggtgtgtggc cccaaatcga tgtgcatgca 901 cttacggatt tactggaccc cagtgtgaaa gagattacag gacaggccca tgttttactg 961 tgatcagcaa ccagatgtgc cagggacaac tcagcgggat tgtctgcaca aaaacgctct 1021 gctgtgccac agtcggccga gcctggggcc acccctgtga gatgtgtcct gcccagcctc 1081 acccctgccg ccgtggcttc attccaaata tccgcacggg agcttgtcaa gatgtggatg 1141 aatgccaggc catccccggg ctctgtcagg gaggaaattg cattaatact gttgggtctt 1201 ttgagtgcaa atgccctgct ggacacaaac ttaatgaagt gtcacaaaaa tgtgaagata 1261 ttgatgaatg cagcaccatt cctggaatct gtgaaggggg tgaatgtaca aacacagtca 1321 gcagttactt ttgcaaatgt ccccctggtt tttacacctc tccagatggt accagatgca 1381 tagatgttcg cccaggatac tgttacacag ctctgacaaa cgggcgctgc tctaaccagc 1441 tgccacagtc cataaccaaa atgcagtgct gctgtgatgc cggccgatgc tggtctccag 1501 gggtcactgt cgcccctgag atgtgtccca tcagagcaac cgaggatttc aacaagctgt 1561 gctctgttcc tatggtaatt cctgggagac cagaatatcc tcccccaccc cttggcccca 1621 ttcctccagt tctccctgtt cctcctggct ttcctcctgg acctcaaatt ccggtccctc 1681 gaccaccagt ggaatatctg tatccatctc gggagccacc aagggtgctg ccagtaaacg 1741 ttactgatta ctgccagttg gtccgctatc tctgtcaaaa tggacgctgc attccaactc 1801 ctgggagtta ccggtgtgag tgcaacaaag ggttccagct ggacctccgt ggggagtgta 1861 ttgatgttga tgaatgtgag aaaaacccct gtgctggtgg tgagtgtatt aacaaccagg 1921 gttcgtacac ctgtcagtgc cgagctggat atcagagcac actcacgcgg acagaatgcc 1981 gagacattga tgagtgttta cagaatggcc ggatctgcaa taatggacgc tgcatcaaca 2041 cagatggcag ttttcattgc gtgtgtaatg cgggctttca tgttacacga gatgggaaga 2101 actgtgaaga tatggatgaa tgcagcataa ggaacatgtg ccttaatgga atgtgtatca 2161 atgaagatgg cagttttaaa tgtatttgca aacctggatt ccagctggca tcagatggac 2221 gttattgcaa agacattaac gagtgtgaaa cccctgggat ctgcatgaac gggcgttgcg 2281 tcaacactga tggctcctac agatgtgaat gcttccctgg actggctgtg ggtctggatg 2341 gccgtgtgtg tgttgacaca cacatgcgga gcacatgcta tggtggatac aagagaggcc 2401 agtgtatcaa acctttgttt ggtgctgtca ctaaatctga atgctgttgc gccagcactg 2461 agtatgcatt tggggaacct tgccagccgt gtcctgcaca gaattcagcg gaatatcagg 2521 cactctgcag cagtgggcca ggaatgacgt cagcaggcag tgatataaat gaatgtgcac 2581 tagatcctga tatttgccca aatggaatct gtgaaaacct tcgtgggacc tataaatgta 2641 tatgcaattc aggatatgaa gtggattcaa ctgggaaaaa ctgcgttgat attaatgaat 2701 gtgtactgaa cagtctcctt tgtgacaatg gacaatgtag aaatactcct ggaagttttg 2761 tctgtacctg ccccaaggga tttatctaca aacctgatct aaaaacatgt gaagacattg 2821 atgaatgcga atcaagtcct tgcattaatg gagtctgcaa gaacagccca ggctctttta 2881 tttgtgaatg ttcttctgaa agtactttgg atccaacaaa aaccatctgc atagaaacca 2941 tcaagggcac ttgctggcag actgtcattg atgggcgatg tgagatcaac atcaatggag 3001 ccaccttaaa gtcccagtgc tgctcctccc tcggtgctgc gtggggaagc ccgtgcaccc
22 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/X63556? 23 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/X63556?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
62
3061 tatgccaagt tgatcccata tgtggtaaag ggtactcaag aattaaagga acacaatgtg 3121 aagatataga tgaatgtgaa gtgttcccag gagtgtgtaa aaatggcctg tgtgttaaca 3181 ctagggggtc attcaagtgt cagtgtccca gtggaatgac tttggatgcc acaggaagga 3241 tctgtcttga tatccgcctg gaaacctgct tcctgaggta cgaggacgag gagtgcaccc 3301 tgcctattgc tggccgccac cgcatggacg cctgctgctg ctccgtcggg gcagcctggg 3361 gtactgagga atgcgaggag tgtcccatga gaaatactcc tgagtacgag gagctgtgtc 3421 cgagaggacc cggatttgcc acaaaagaaa ttacaaatgg aaagcctttc ttcaaagata 3481 tcaatgagtg caagatgata cccagcctct gcacccacgg caagtgcaga aacaccattg 3541 gcagctttaa gtgcaggtgt gacagcggct ttgctcttga ttctgaagaa aggaactgca 3601 cagacattga cgaatgccgc atatctcctg acctctgtgg cagaggccag tgtgtgaaca 3661 cccctgggga ctttgaatgc aagtgtgacg aaggctatga aagtggattc atgatgatga 3721 agaactgcat ggatattgat gagtgtcaga gagatcctct cctatgccga ggtggtgttt 3781 gccataacac agagggaagt taccgctgtg aatgcccgcc tggccatcag ctgtccccca 3841 acatctccgc gtgtatcgac atcaatgaat gtgagctgag tgcacacctg tgccccaatg 3901 gccgttgcgt gaacctcata gggaagtatc agtgtgcctg caaccctggc taccattcaa 3961 ctcccgatag gctattttgt gttgacattg atgaatgcag cataatgaat ggtggttgtg 4021 aaaccttctg cacaaactct gaaggcagct atgaatgtag ctgtcagccg ggatttgcac 4081 taatgcctga ccagagatca tgcaccgaca tcgatgagtg tgaagataat cccaatatct 4141 gtgatggtgg tcagtgcaca aatatccctg gagagtacag gtgcttgtgt tatgatggat 4201 tcatggcatc tgaagacatg aagacttgtg tagatgtcaa tgagtgtgac ctgaatccaa 4261 atatctgcct aagtgggacc tgtgaaaaca cgaaaggctc atttatctgc cactgtgata 4321 tgggctactc cggcaaaaaa ggaaaaactg gctgtacaga catcaatgaa tgtgaaattg 4381 gagcacacaa ctgtggcaaa catgctgtat gtaccaatac agcaggaagc ttcaaatgta 4441 gctgcagtcc cgggtggatt ggagatggca ttaagtgcac tgatctggac gaatgttcca 4501 atggaaccca tatgtgcagc cagcatgcag actgcaagaa taccatggga tcttaccgct 4561 gtctgtgcaa ggaaggatac acaggtgatg gcttcacttg tacagacctt gatgagtgct 4621 ctgagaacct gaatctctgt ggcaatggcc agtgcctcaa tgcaccagga ggataccgct 4681 gtgaatgcga catgggcttc gtgcccagtg ctgacgggaa agcctgtgaa gatattgatg 4741 agtgctccct tccgaacatc tgtgtctttg gaacttgcca caacctccct ggcctgttcc 4801 gctgtgagtg tgagataggc tacgaactgg acagaagcgg cgggaactgc acagatgtga 4861 atgaatgcct ggatccaacc acgtgcatca gtgggaactg tgtcaacact ccaggcagct 4921 atatctgtga ctgcccacct gattttgaac tgaacccaac tcgagttggc tgtgttgata 4981 cccgctctgg aaattgctat ttggatattc gacctcgagg agacaatgga gatacagcct 5041 gcagcaatga aattggagtt ggtgtttcca aagcttcctg ctgctgttct ctgggtaaag 5101 cctggggtac tccttgtgag atgtgtcctg ctgtgaacac atccgagtac aaaattcttt 5161 gtcctggagg ggaaggtttc cgaccaaatc ctatcaccgt tatattggaa gatattgatg 5221 agtgccagga gctaccaggg ctgtgccaag gaggaaaatg tatcaacacc tttgggagtt 5281 tccagtgccg ctgtccaacc ggctactacc tgaatgaaga tacacgagtg tgtgatgatg 5341 tgaatgaatg tgagactcct ggaatctgtg gtccagggac atgttacaac accgttggca 5401 actacacctg tatctgtcct ccagactaca tgcaagtgaa tgggggaaat aattgcatgg 5461 atatgagaag aagtttgtgc tacagaaact actatgctga caaccagacc tgtgatggag 5521 aattgttatt caacatgacc aagaagatgt gctgctgttc ctacaacatt ggccgggcgt 5581 ggaacaagcc ctgtgaacag tgtcccatcc caagtacaga tgagtttgct acactctgtg 5641 gaagtcaaag gccaggcttt gtcatcgaca tttataccgg tttacccgtt gatattgatg 5701 agtgccggga gatcccaggg gtctgtgaaa atggagtgtg tatcaacatg gttggcagct 5761 tccgatgtga atgtccagtg ggattcttct ataatgacaa gttgttggtt tgtgaagata 5821 ttgacgagtg tcagaacggc ccagtgtgcc agcgcaacgc cgaatgcatc aacactgcag 5881 gcagctaccg ctgtgactgt aagcccggct accgcttcac ctccacagga cagtgcaatg 5941 atcgtaatga atgtcaagaa atccccaata tatgcagtca tgggcagtgc attgacacag 6001 ttggaagctt ttattgcctt tgccacactg gttttaaaac aaatgatgac caaaccatgt 6061 gcttggacat aaatgaatgt gaaagagatg cctgtgggaa tggaacttgc cggaacacaa 6121 ttggttcctt caactgccgc tgcaatcatg gtttcatcct ttctcacaac aatgactgta 6181 tagatgttga tgaatgtgca agtggaaatg ggaatctttg cagaaatggc caatgcatta 6241 atacagtggg gtctttccag tgccagtgca atgaaggcta tgaggtggct ccagatggga 6301 ggacctgtgt ggatatcaat gaatgtcttc tagaacccag aaaatgtgca ccaggtacct 6361 gtcaaaactt ggatgggtcc tacagatgca tttgcccacc tggatacagt cttcaaaatg
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
63
6421 agaagtgtga agatattgat gagtgtgtcg aagagccaga aatttgtgcc ctgggcacat 6481 gcagtaacac tgaaggcagc ttcaaatgtc tgtgtccaga agggttttcc ttgtcctcca 6541 gtggaagaag gtgccaagat ttgcgaatga gctactgtta tgcgaagttt gaaggaggaa 6601 agtgttcatc acccaaatcc agaaatcact ccaagcagga atgctgctgt gccttgaagg 6661 gagaaggctg gggagacccc tgcgagctct gccccacgga acctgatgag gccttccgcc 6721 agatatgtcc ttatggaagt gggatcatcg tgggacctga tgattcagca gttgatatgg 6781 acgaatgcaa agaacccgat gtctgtaaac atggacagtg catcaataca gatggttcct 6841 atcgctgcga gtgtcccttt ggttatactc tagcagggaa tgaatgtgta gatactgatg 6901 aatgttctgt tggcaatcct tgtggaaatg gaacctgcaa gaatgtgatt ggaggttttg 6961 aatgcacctg cgaggaggga tttgagcccg gtccaatgat gacatgtgaa gatataaatg 7021 aatgtgccca gaatcctctg ctctgtgcct tccgatgtgt gaacacttat gggtcatatg 7081 aatgcaaatg tcccgtggga tatgtgctca gagaagaccg taggatgtgc aaagatgagg 7141 atgagtgtga agagggaaaa catgactgta ctgaaaaaca aatggaatgc aagaacctca 7201 ttggcacata tatgtgcatc tgtggacccg ggtatcagcg gagacctgat ggagaaggct 7261 gtgtagatga gaatgaatgt cagacgaagc cagggatctg tgagaatggg cgctgcctca 7321 acacccgtgg gagctacacc tgtgagtgta atgatgggtt taccgccagc cccaaccagg 7381 acgagtgcct tgacaatcgg gaagggtact gcttcacaga ggtgctacaa aacatgtgtc 7441 agatcggctc cagcaacagg aaccccgtca ccaaatcgga atgctgctgt gacggaggga 7501 gaggctgggg tccccactgt gagatctgcc ctttccaggg gactgtggct ttcaagaaac 7561 tctgtcccca tggccgagga ttcatgacca atggagcaga tatcgatgaa tgcaaggtta 7621 ttcacgatgt ttgccgaaat ggggaatgtg tcaatgacag aggatcatat cattgcattt 7681 gtaaaactgg gtacactcca gatataactg ggacttcctg tgtagatctg aacgagtgca 7741 accaggctcc caaaccctgc aattttatct gcaaaaacac agaagggagt taccagtgtt 7801 catgcccgaa aggctacatt ctgcaagagg atggaaggag ctgcaaagat cttgatgagt 7861 gtgcaaccaa gcaacacaac tgccagttcc tatgtgttaa caccattggc ggcttcacat 7921 gcaaatgtcc tcccggattt acccaacacc atacgtcctg cattgataac aatgaatgca 7981 cctctgacat caatctgtgc gggtctaagg gcatttgcca gaacactcct ggaagcttca 8041 cctgtgaatg ccagcgggga ttctcacttg atcagaccgg ctccagctgt gaagacgtgg 8101 acgagtgtga gggtaaccac cgctgccagc atggctgcca gaacatcatt gggggctaca 8161 ggtgcagctg cccccagggc tacctccagc actaccagtg gaaccagtgt gttgatgaaa 8221 acgaatgcct cagcgctcac atctgcggag gagcctcctg tcacaacacc ctggggagct 8281 acaagtgcat gtgtcccgcc ggcttccagt atgaacagtt cagtggagga tgccaagaca 8341 tcaatgaatg tggctctgcg caggccccct gcagctatgg ctgttccaat accgagggcg 8401 gttacctgtg tggctgtcca cctggttact tccgcatagg ccaagggcac tgtgtttctg 8461 gaatgggcat gggccgagga aacccagagc cacctgtcag tggtgaaatg gatgacaatt 8521 cactctcccc agaggcttgt tacgagtgta agatcaatgg ctaccccaaa cggggcagga 8581 aacggagaag cacaaacgaa actgatgcct ccaatatcga ggatcagtct gagacagaag 8641 ccaatgtgag tcttgcaagt tgggatgttg agaagacagc catctttgct ttcaatattt 8701 cccacgtcag taacaaggtt cgaatcctag aactccttcc agctcttaca actctgacga 8761 atcacaacag atacttgatc gaatctggaa atgaagatgg cttctttaaa atcaaccaaa 8821 aggaagggat cagctacctc cacttcacaa agaagaagcc agtggctgga acctattcat 8881 tacaaatcag tagtactcca ctttataaaa agaaagaact taaccaacta gaagacaaat 8941 atgacaaaga ctacctcagt ggtgaactgg gtgataatct gaagatgaaa atccaggttt 9001 tgcttcatta attcaccatc cagaaaccaa ataattaaaa gaaaaacaaa tatagatagg 9061 tagaactata ttttccccca atcagaatca tcatatcata ggtacaatct ttcaccaagt 9121 aaatttgtat aaataagcac tattctttgt attaccaaag caaggtacag gtgactaccc 9181 tagttcaaaa caaccacttt ctcaggcttc tcatgtgtgt agctaagcta ccttgtcata 9241 tgtgttgatt cttgaaaact gggacgtgta tttccattgg gggttggcca tttatgctga 9301 catgccatcc ttccagcaaa cgtaygggaa tgtgctttca attgatggac tactctattt 9361 tttgcaaatt tgtaaacttt gcttctccaa atacaagtac taggttgtcc atttatggta 9421 cctatttggt gctagtaaat tttcaaacta gatttataaa tgcactgtaa tatgtacaca 9481 acttagaaac caaattacaa gtattcagtt ccaatacttc attaatttca atcaaccaaa 9541 gttagttcag tagcttatct cagttatgag tataatacat tacatgtaaa ttaagtgtgt 9601 gtatactgta atcgtgctat tttttatcat tgaaacattt ataaactaga ataataatgc 9661 ccttaatgtg agggtttgta atggtgctta ttaagaccaa agacttgtta aatgtataca 9721 ccaagtggta atgaaatttc kgtgactggc ccacacgtgc atagaggtct gggaggacca
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
64
9781 ggaaacagcc tcagtggcca gaggatcacc agtgcatcct tcatcacagc atgtgcaata 9841 tgccaagatt accctcggtc attcctgtca acaaggggtc aatgtcataa atgtcacaat 9901 aaaacaatct cttctttttt ttagtttaaa aaaaaaaaaa //
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
65
2. FIBRILINA – 2 : Nombre del gen: FBN2 24
Sinónimos: CCA, DA9 25
Símbolos Previos: CCA 26
Alias: DA9 27
Descripción: Fibrillin 2 (HGNC Symbol) 28
Cromosoma: 5 29
Citobanda: q23.3 30
Localización: 5q23-q31 31 Ubicación de Cromosoma: 127593601 – 127994878 32
NCBI Gene ID: 2201 33
Gen Tipo: Proteína Codificante 34
24 HUGO - Human Genome Organisation - Organización del Genoma Humano HGNC – COMITÉ DE NOMENCLATURA - http://www.genenames.org/ 25 THE HUMAN PROTEIN ATLAS - http://www.proteinatlas.org 26 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 27 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 28,21,22 http://www.proteinatlas.org/ENSG00000138829/gene 29
30
31 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 32 http://www.proteinatlas.org/ENSG00000138829/gene 33 http://www.proteinatlas.org/ENSG00000138829/gene 34 NCBI - http://www.ncbi.nlm.nih.gov/gene/2201
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
66
Resumen: 35
o La proteína codificada por este gen es un componente del tejido conectivo y Microfibrillas.
o Puede estar envuelta en las ensambladas fibras elásticas.o Mutación en este gen causa Aracnodactília Contractural Congénita (CCA) (Un
trastorno que es similar o una variante del síndrome de Marfan se hereda como un rasgo autosómico dominante, y se caracteriza especialmente por aracnodactilia, contractura de las articulaciones y escoliosis 36 )
Visor de Mapa: 37
Locus : HSU03272 / 10172 bp 38
35 NCBI - http://www.ncbi.nlm.nih.gov/gene/2201 36 GENETICS HOME REFERENCE - http://ghr.nlm.nih.gov/condition/congenital-contractural-arachnodactyly 37 http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?TAXID=9606&CHR=5&MAPS=model,ugHs,ensgenes,rna,genes[127593601.00%3A127768726.98]-r&QSTR=2201[gene_id]&QUERY=uid%28-1811869421,-2146581794%29&ovr= 38 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/U03272?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
67
Origen : 39
1 atggggagaa gacggaggct gtgtctccag ctctacttcc tgtggctggg ctgtgtggtg 61 ctctgggcgc agggcacggc cggccagcct cagcctcctc cgcccaagcc gccccggccc 121 cagccgccgc cgcaacaggt tcggtccgct acagcaggct ctgaaggcgg gtttctagcg 181 cccgagtatc gcgaggaggg tgccgcagtg gccagccgcg tccgccggcg aggacagcag 241 gacgtgctcc gagggcccaa cgtgtgcggc tccagattcc actcctactg ctgccctgga 301 tggaagacgc tccctggagg aaaccagtgc attgtcccga tttgtagaaa tagttgtgga 361 gatggatttt gttcccgtcc taacatgtgt acttgttcca gtgggcaaat atcatcaacc 421 tgtggatcaa aatcaattca gcagtgcagt gtgagatgca tgaatggtgg gacctgtgca 481 gatgaccact gccagtgcca gaaaggatat attggaactt attgtggaca acctgtctgt 541 gaaaatggat gtcagaatgg tggacgttgc atcgcccaac cgtgtgcttg tgtttatggg 601 ttcactggtc cacagtgtga aagagattac aggacaggcc cgtgtttcac tcaggtcaac 661 aaccagatgt gccaagggca gctgacaggc attgtctgca cgaagactct gtgctgtgcc 721 accactggac gggcgtgggg ccatccctgt gagatgtgtc cagcccagcc tcagccctgc 781 cgacggggtt tcatccccaa catccgcact ggagcttgcc aagatgttga tgaatgccag 841 gctatcccag ggatatgcca aggaggaaac tgtatcaata cagtgggctc ttttgaatgc 901 agatgccctg ctggtcacaa acagagtgaa actactcaga aatgtgaaga cattgatgag 961 tgcagcatca ttcctgggat atgtgaaact ggtgaatgtt ccaacaccgt gggaagctat 1021 ttttgtgttt gtccacgtgg atatgtaacc tcaacagatg gctctcgatg catcgatcag 1081 agaacaggca tgtgtttctc gggcctggtg aatggccgct gtgcacaaga gctcccgggg 1141 agaatgacga aaatgcagtg ctgctgtgag cctggccgct gctggggcat cggaaccatt 1201 cctgaagcct gtcctgtcag aggttctgag gaatatcgca gactttgcat ggatggactt 1261 ccaatgggag gaattccagg gagtgctggt tccagacctg gaggcactgg gggaaatggc 1321 tttgccccaa gtggcaatgg caatggctat ggcccaggag ggacaggctt catccccatc 1381 cctggaggca atggcttttc tcctggcgtt gggggagccg gtgtgggggc cgggggacag 1441 ggacctatca tcactggact aacaattctg aaccagacaa tagatatctg taagcatcat 1501 gctaaccttt gtttaaatgg acgctgtata ccaactgtct caagctaccg atgtgaatgc 1561 aacatgggtt ataagcagga tgcaaatgga gattgtatag atgttgatga atgcacatca 1621 aatccctgca ctaatggaga ttgtgttaac acacctggtt cctattattg taaatgtcat 1681 gctggattcc agaggactcc taccaagcaa gcatgcattg atattgatga gtgcatccag 1741 aatggggttc tttgtaaaaa cggtcgatgc gtgaactcag atggaagttt ccagtgcatt 1801 tgcaatgccg gctttgaatt aactacagat ggaaaaaact gtgttgatca tgatgaatgt 1861 acaactacca acatgtgttt gaatggaatg tgcatcaatg aagatggcag cttcaagtgc 1921 atctgcaaac caggatttgt cttggctcca aatgggcgtt actgtactga tgttgatgaa 1981 tgccagaccc caggaatctg catgaatggg cactgcatca acagtgaagg gtccttccgc 2041 tgtgactgtc ccccaggcct ggctgtgggc atggatggac gtgtgtgtgt tgatactcac 2101 atgcgcagta cctgctatgg aggaatcaag aaaggagtgt gtgtgcgtcc tttccccggt 2161 gcagtgacca agtccgaatg ctgctgtgcc aatccagact atggttttgg agaaccctgc 2221 cagccatgcc ctgcaaaaaa ttcagctgaa ttccacggcc tttgtagtag tggagtaggt 2281 atcactgtgg atggaagaga tatcaatgaa tgtgctttgg atcctgatat atgtgccaat 2341 gggatttgtg aaaacttacg tggtagttac cgttgtaatt gcaacagtgg ctatgaacca 2401 gatgcctctg gaagaaactg tattgacatt gatgaatgtt tagtaaacag actgctttgt 2461 gataacggat tgtgccgaaa cacgccagga agttacagct gtacgtgccc accagggtat 2521 gtgttcagga ctgagacaga gacctgtgaa gatataaatg aatgtgaaag caacccatgt 2581 gtcaatgggg cctgcagaaa caaccttgga tctttcaatt gtgaatgttc gcccggcagc 2641 aaactcagct ccacaggatt gatctgtatt gacagcctga aggggacctg ttggctcaac 2701 atccaggaca gccgctgtga ggtgaatatt aatggagcca ctctgaaatc tgaatgctgt 2761 gccaccctcg gagccgcctg ggggagcccc tgtgagcggt gtgaactaga tacagcttgc 2821 ccaagagggc ttgccaggat taaaggtgtt acgtgtgaag atgttaatga gtgtgaggtg 2881 ttccctggcg tttgtccaaa tggacgctgt gtcaacagta agggatcttt tcattgcgag 2941 tgccctgaag gccttacgtt ggatgggact ggccgtgtat gtttggatat tcgcatggag 3001 cagtgttact tgaagtggga tgaagatgaa tgcatccacc ccgttcctgg aaagttccgc 3061 atggatgcct gctgctgtgc tgtcggggcg gcttggggca ccgagtgtga ggagtgcccc
39 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/U03272?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
68
3121 aaacctggca ccaaggaata cgagacactg tgcccccgcg gggctggctt tgctaaccga 3181 ggggatgttc ttactgggcg gccattttac aaagacatca atgaatgcaa agcatttcct 3241 gggatgtgca cttatgggaa gtgcagaaat acaatcggaa gcttcaaatg ccgttgcaat 3301 agtggctttg ctctagacat ggaggaaaga aactgcacgg acatcgacga gtgcaggatt 3361 tctcctgacc tctgtggcag tggaatctgc gtcaatacac cgggcagctt tgagtgcgag 3421 tgcttcgaag gctatgaaag tggcttcatg atgatgaaga actgcatgga cattgacgga 3481 tgtgaacgta accctctcct ttgtaggggt ggcacctgtg tgaacactga gggcagcttt 3541 cagtgtgact gcccactggg acacgagctg tcaccatccc gtgaggactg tgtggatatt 3601 aatgaatgct ccctgagtga caatctctgc agaaatggaa aatgtgtgaa catgattgga 3661 acctatcagt gctcttgcaa tcctggatat caggctacgc cagaccgcca gggctgtaca 3721 gatattgatg aatgtatgat aatgaacgga ggctgtgaca cccagtgcac aaattcagag 3781 ggaagctacg aatgcagctg cagtgagggt tatgccctga tgccagatgg gagatcgtgt 3841 gcagacattg atgaatgtga aaacaatcct gatatctgtg atggcggcca gtgtaccaac 3901 attcctggag agtatcgctg cctctgctat gatggcttca tggcttccat ggacatgaaa 3961 acatgcattg atgtcaatga atgtgaccta aattcaaata tctgcatgtt tggggaatgt 4021 gagaacacaa agggatcctt catttgccac tgtcagctgg gttactcagt gaagaagggg 4081 accacaggat gtacagatgt ggatgagtgt gaaattggtg ctcataactg cgacatgcat 4141 gcctcatgtc tgaatatccc aggaagcttc aagtgtagct gcagagaagg ctggattgga 4201 aacggcatca agtgtattga tctggacgaa tgttctaatg gaacccacca gtgtagcatc 4261 aatgctcagt gtgtaaatac cccgggctca taccgctgtg cctgctccga aggtttcact 4321 ggtgatggct ttacctgctc agatgttgat gagtgtgcag aaaacataaa cctctgtgag 4381 aacggacagt gccttaatgt cccgggtgca tatcgctgcg agtgtgagat gggcttcact 4441 ccagcctcag acagcagatc ctgccaagat attgatgaat gctccttcca aaacatttgt 4501 gtctctggaa catgtaataa cctgcctgga atgtttcatt gcatctgcga tgatggttat 4561 gaattggaca gaacaggagg gaactgtaca gatattgatg agtgtgcaga tcctataaac 4621 tgtgtcaatg gcctatgtgt caacacgcct ggtcgctatg agtgtaactg cccacccgat 4681 tttcagttga acccaactgg tgtgggttgt gttgacaacc gtgtgggcaa ctgctacctg 4741 aagtttggac ctcgaggaga tgggagtctg tcttgcaaca ccgagatcgg ggtgggcgtc 4801 agtcgctctt catgctgctg ctctctggga aaggcctggg gaaacccctg tgagacatgc 4861 ccccctgtca atagcactga atattacacc ctgtgtcccg gaggtgaagg cttcagacct 4921 aaccccatca caatcatttt agaagacatt gacgaatgcc aggagttacc aggtctctgc 4981 cagggtggaa actgcatcaa cacttttggg agcttccagt gtgagtgccc acaaggctac 5041 tacctcagcg aggatacccg catctgtgag gatattgatg agtgttttgc acatcctggt 5101 gtgtgtgggc ctgggacctg ctataacacc ctgggaaatt acacctgcat ttgcccacct 5161 gagtacatgc aggtcaatgg aggccacaac tgcatggaca tgagaaaaag cttttgctac 5221 cgaagctata atggaaccac ttgtgagaat gagttgcctt tcaatgtgac aaaaaggatg 5281 tgctgctgca catataatgt gggcaaagct gggaacaaac cttgtgaacc atgcccaact 5341 ccaggaacag ctgactttaa aaccatatgt ggaaatattc ctggattcac ctttgacatt 5401 cacacaggaa aagctgttga cattgatgaa tgtaaagaga ttccaggcat ttgtgcaaat 5461 ggtgtgtgca ttaaccagat tggcagtttc cgctgtgaat gccctacagg attcagttac 5521 aatgacctgc tgttggtttg tgaagatata gatgagtgca gcaatggtga taatctctgc 5581 cagcggaatg cagactgcat caatagtcct ggtagttacc gctgtgaatg tgccgcgggt 5641 ttcaaacttt cacccaatgg ggcctgtgta gatcgcaatg aatgtttaga aattcctaac 5701 gtttgcagtc atggcttgtg tgttgatctg caaggaagtt accagtgcat ctgccacaat 5761 ggctttaagg cttctcagga ccagaccatg tgcatggatg ttgatgagtg cgagcggcac 5821 ccatgtggaa atggaacttg taaaaacacc gttggatcct ataactgtct gtgctaccca 5881 gggtttgaac tcactcataa taatgattgc ctggacatag atgagtgcag ttcctttttt 5941 ggtcaggtgt gcagaaatgg acgttgtttt aatgaaattg gttctttcaa gtgtctatgt 6001 aacgaaggtt atgaacttac cccagatggc aaaaactgta tagacactaa tgagtgtgtc 6061 gcccttcccg gctcttgctc tcctggtacc tgtcagaatt tggagggatc cttcagatgc 6121 atctgtcccc cagggtatga agtaaaaagc gagaactgca ttgatataaa tgaatgtgat 6181 gaagatccca acatttgtct ttttggttcc tgtactaata ctccaggggg cttccagtgc 6241 ctctgccccc ctggctttgt actatctgat aatggacgga gatgctttga tactcgccag 6301 agcttctgct tcacaaattt tgaaaatgga aagtgttctg tacccaaagc tttcaacacc 6361 acaaaagcaa aatgctgctg tagtaagatg ccaggagagg gctgggggga cccctgtgag 6421 ctgtgcccca aagacgatga agttgcattt caggatttgt gtccatatgg ccatggaact
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
69
6481 gtccctagtc ttcatgatac acgtgaagat gtcaatgagt gtcttgagag cccaggcatt 6541 tgttcaaatg gtcaatgtat caacaccgac ggatcttttc gctgtgaatg tccaatgggc 6601 tacaaccttg actacactgg agtacgctgt gtggatactg atgagtgttc aatcggcaat 6661 ccgtgtggaa atggtacatg caccaatgtt attgggagtt ttgaatgcaa ttgcaatgaa 6721 ggctttgagc cagggcccat gatgaattgt gaagatatca acgaatgtgc ccagaaccca 6781 ctgctgtgtg ctttacgctg catgaacact tttgggtcct atgaatgcac gtgcccgatt 6841 ggctatgccc tcagggaaga tcaaaagatg tgcaaagatc tggatgaatg tgctgaaggg 6901 ttacacgact gtgaatctag gggcatgatg tgtaagaatc taatcggcac cttcatgtgc 6961 atctgccctc ctggaatggc ccgaaggccc gatggagaag gctgtgtaga tgaaaatgaa 7021 tgcaggacca agccaggaat ctgtgaaaat ggacgttgtg ttaacattat tggaagctat 7081 agatgtgagt gtaatgaagg attccagtca agttcttcag gcactgaatg ccttgacaat 7141 cgacagggtc tctgctttgc agaggtactg cagacaatat gtcaaatggc atccagtagt 7201 cgcaatctcg tcactaagtc agaatgctgc tgtgatggtg ggcgaggctg gggccaccag 7261 tgcgagcttt gcccacttcc tggaactgcc cagtacaaaa agatatgtcc tcatggccca 7321 ggatatacaa ctgatggaag agatattgat gaatgtaagg taatgccaaa cctctgcacc 7381 aatggtcagt gcatcaatac catgggctca ttccgatgct tctgcaaggt tggctacacc 7441 acagacatca gtggaacctc ttgtatagac cttgatgaat gctcccagtc cccgaaacca 7501 tgcaactaca tctgcaagaa cactgagggg agttatcagt gttcatgtcc gagggggtat 7561 gtcctgcaag aggatggaaa gacatgcaaa gaccttgatg aatgtcaaac aaagcagcat 7621 aactgccagt tcctctgtgt caacaccctg ggggggttta cctgtaaatg tccacctggt 7681 ttcacacagc atcacactgc ttgtatcgac aacaacgaat gtgggtctca acctttgctt 7741 tgtggaggaa agggaatctg tcaaaacact ccaggcagtt tcagctgtga atgccaaaga 7801 gggttctctc ttgatgccac cggactgaac tgtgaagatg ttgatgaatg tgatgggaac 7861 cacaggtgcc aacacggctg ccagaacatc ctgggtggct acagatgtgg ctgcccccaa 7921 ggctacatcc agcactacca gtggaatcag tgtgtcgatg agaatgaatg ctccaatccc 7981 aatgcctgtg gctctgcttc ctgctacaac accctgggga gttacaagtg cgcctgcccc 8041 tcggggttct ccttcgacca gttctccagt gcctgccacg acgtgaatga gtgctcgtcc 8101 tccaagaacc cctgcaatta cggctgctct aacacggagg ggggctacct ctgtggctgc 8161 ccccctgggt attacagagt gggacaaggc cactgtgtct caggaatggg atttaacaag 8221 gggcagtacc tgtcactgga tacagaggtc gatgaggaaa atgctctgtc cccagaagca 8281 tgctacgagt gcaaaatcaa cggctatcct aagaaagaca gcaggcagaa gagaagtatt 8341 catgaacctg atcccactgc tgttgaacag atcagcctag agagtgtcga catggacagc 8401 cccgtcaaca tgaagttcaa cctctcccac ctcggctcta aggagcacat cctggaacta 8461 aggcccgcca tccagcccct caacaaccac atccgttatg tcatctctca agggaacgat 8521 gacagcgtct tccgcatcca ccaaaggaat gggctcagct acttgcacac ggccaagaag 8581 aagctcatgc ccggcacata cacactggaa atcactagca tccctctcta caagaagaag 8641 gagcttaaga aactggaaga gagcaatgag gatgactacc tcctagggga gcttggggag 8701 gctctcagaa tgaggctgca gattcagctc tattaaccgt tcacagactt gggcccaggc 8761 tcaaatccta gcacagccag tctgcagaag catttgaaaa gtcaaggact aattttaaag 8821 aggaaaaata ataataactc ttgtttcttt cctccctgtc ttagactttg aatgttgacc 8881 ctcacaggga gggataattt agactctggt atggccaaag atttgagctc aaaggcaacc 8941 gtggttactg tattttttat ataacttcat tttaaaatat attaaaagaa acctaaatgt 9001 tcaagatatc agcatatggc actaaatgca caaaaataat gtgagctttt tttttttttt 9061 cctgttagca gtctgtaaca ctttgggtat tttgctatag ttgctaatta aaaaaatata 9121 gatgtttatt tatttttaat gcagtaatat atggagaaat gaacaaacta tgtaaacaaa 9181 aagggaaact cacttgtttt tctttagatt tataaatttg agctattttt tttagaggtg 9241 ctttttaaaa atccaataga tacaagagat gtttcctttg gttttctgcc agtcatccag 9301 ctgatacaca cctgatcgat tttaaagaaa gccacacaga gctgaatcgg gcagtgctaa 9361 tcaataattt aaaagacatg aatgtcatta gatcctttat aacgtagatc gaagccaaag 9421 cagctcattt gtgacaacat ttcatatcac cagacacacc aggcaacaga agttgaagca 9481 caaccactgt agcaaaatac cttgactgct tgtgagacca ttagcattgc aggccaaacc 9541 gtactgtatt tccttctcat aacctcaagg aaccatatgt gctacccaca acacctcatt 9601 cttacccagg gtgcgctgcg tcctcatggt actgtaggca gctgaagaac cgccgttccc 9661 ttgaaaggga acacctggca ttctgtggtg tttcgtgctg tcttaaataa tggtgcattt 9721 attatgttca agttatttca ggattgccat atgtgcaaac aaatcatgca atgcagccaa 9781 ggaatatatg ttgttgttgt tgttttaaac ccattttttt tttagaattt tcattaatac
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
70
9841 tgtagttata caccatatgc ctcattttat catagcctat tgtgtatgaa agatgtttgt 9901 acaatgaatt gatgtttagt ttgctttagt catttaaaaa gatattgtac caggatgtgc 9961 tattaagagc acgtatccat tattcttctc aacccaagaa cctgtttcct ggaccagtga 10021 ccaaacctca tatgtgaaat ggccaaagca catgcaggct cctggttgtt cctctcaaac 10081 ctgtgctgac caaagattag taaccagtta tacccagtat tttgaggttt tattgttttt 10141 ttaataacta aaaaaaaact cgtgccgaat tc
3. FIBRILINA – 3 :
Genética Médica
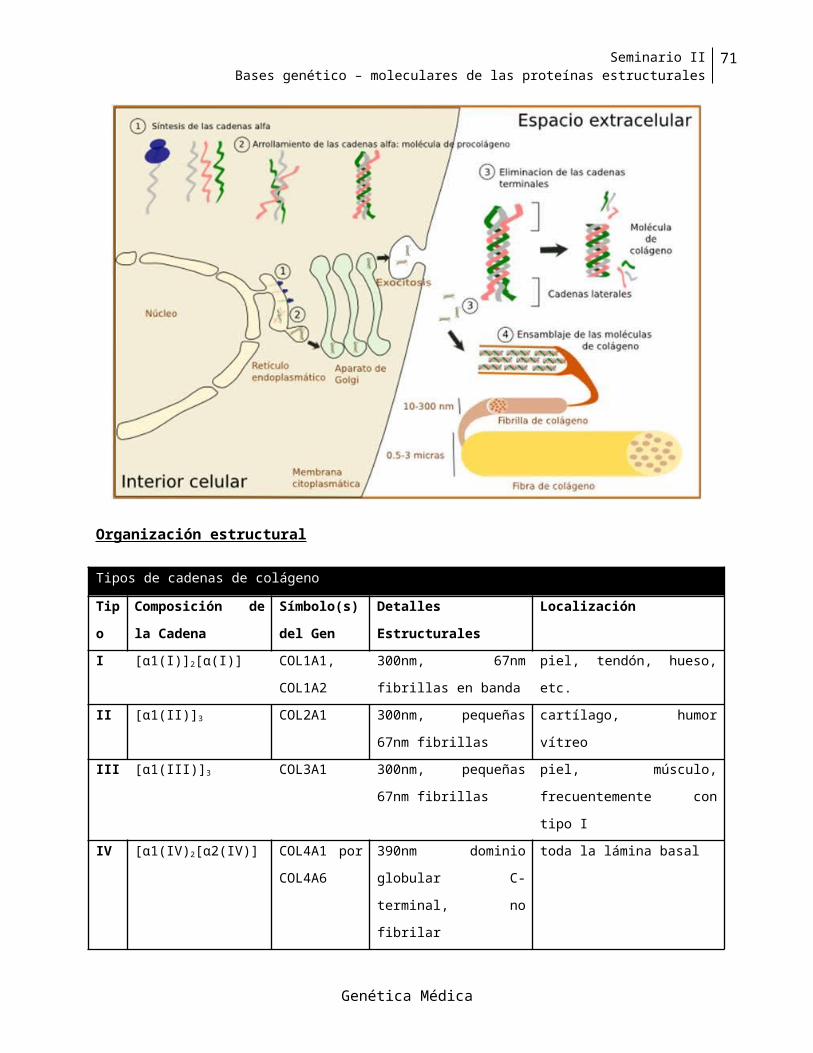
Seminario II Bases genético – moleculares de las proteínas estructurales
71
Nombre del gen: FBN3 40
Sinónimos: --- 41
Símbolos Previos: --- 42
Alias: --- 43
Descripción: Fibrillin 3 (HGNC Symbol) 44
Cromosoma: 19 45
Citobanda: p13.2 46
Localización: 19p13 47 Ubicación de Cromosoma: 8130287 - 8212650 48
NCBI Gene ID: 84467 49
Gen Tipo: Proteína Codificante 50
40 HUGO - Human Genome Organisation - Organización del Genoma Humano HGNC – COMITÉ DE NOMENCLATURA - http://www.genenames.org/ 41 THE HUMAN PROTEIN ATLAS - http://www.proteinatlas.org 42 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 43 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 44, 37, 38 http://www.proteinatlas.org/ENSG00000142449/gene 45
46
47 HGNC - http://www.genenames.org/cgi-bin/hgnc_search.pl 48 http://www.proteinatlas.org/ENSG00000142449/gene 49 http://www.proteinatlas.org/ENSG00000142449/gene 50 NCBI - http://www.ncbi.nlm.nih.gov/gene/84467
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
72
Resumen: 51
o Este gen codifica una proteína que pertenece a la Familia de las Fibrilinas (Fibrillin Family).
o Las fibrilina son moléculas de la matriz extracelular que se ensamblan dentro de las Microfibrillas en el Tejido Conectivo.
o Este gen es más altamente expresado en Tejidos Fetales o Su producto de la proteína se localiza en microfibrillas extracelulares de desarrollo
de elementos esqueletales, piel, pulmón, riñón y músculo esqueléticoo Este gen es potencialmente implicados en el síndrome de Weill-Marchesani.
Visor de Mapa: 52
Locus : NM_032447 / 8967 bp 53
51 NCBI - http://www.ncbi.nlm.nih.gov/gene/84467 52 http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?taxid=9606&chr=19&query=uid%28-2146542617%29&QSTR=84467[gene_id]&maps=gene_set&cmd=focus 53 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/NM_032447?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
73
Origen : 54
1 gcagcctcca ggggacacgc catgactctg gagggtctgt atttggcaag gggccccctg 61 gcccggctcc tgctggcctg gtcggccctg ttgtgcatgg caggtggcca aggccgctgg 121 gacggggcct tggaggctgc aggtcctgga cgtgtgcgga ggcggggcag cccaggcatc 181 ttgcaggggc cgaatgtgtg cggctcccgg ttccatgcct actgctgtcc aggctggagg 241 acattccctg gcaggagcca gtgtgtcgta cccatctgta ggcgcgcctg cggtgaaggc 301 ttctgctccc agcccaacct gtgcacctgt gcggatggga cgctggctcc cagctgcggg 361 gtgagccgag ggtcagggtg cagtgtgagc tgtatgaatg ggggcacctg ccggggggcg 421 tcctgtctgt gtcagaaggg ctacacaggc accgtgtgtg ggcagcccat ctgtgaccgc 481 ggctgccaca atgggggtcg ctgcattggg cccaaccgct gcgcctgtgt gtatggcttc 541 atgggacctc aatgtgagag agattaccgg acgggaccct gctttggcca agtaggcccc 601 gaggggtgcc agcatcagct gacgggcctc gtgtgcacca aggcactttg ctgtgccact 661 gtgggccgtg cctggggcct tccatgtgaa ctttgccctg cacagccaca cccctgccgc 721 cgcggcttca tccccaatat ccacacgggg gcctgccaag atgtggatga gtgccaggct 781 gtgccaggcc tgtgccaggg aggcagctgc gtcaacatgg tgggctcctt ccattgccgc 841 tgtccagttg gacaccggct cagtgacagc agcgccgcat gtgaagacta ccgggccggc 901 gcctgcttct cagtgctttt cgggggccgc tgtgctggag acctcgccgg ccactacact 961 cgcaggcagt gctgctgtga caggggcagg tgctgggcag ctggcccggt ccctgagctg 1021 tgtcctcctc ggggctccaa tgaattccag caactgtgcg cccagcggct gccgctgcta 1081 cccggccacc ctggcctctt ccctggcctc ctgggcttcg gatccaatgg catgggtccc 1141 cctcttgggc cagcgcgact caacccccat ggctctgatg cgcgtgggat ccccagcctg 1201 ggccctggca actctaatat tggcactgct accctgaacc agaccattga catctgccga 1261 cacttcacca acctgtgtct gaatggccgc tgcctgccca cgccttccag ctaccgctgc 1321 gagtgtaacg tgggctacac ccaggacgtg cgcggcgagt gcattgatgt agacgaatgc 1381 accagcagcc cctgccacca cggtgactgc gtcaacatcc ccggcaccta ccactgccgg 1441 tgctacccgg gcttccaggc cacgcccacc aggcaggcat gcgtggatgt ggacgagtgc 1501 attgtcagtg gtggcctttg tcacctgggc cgctgtgtca acacagaggg cagcttccag 1561 tgtgtctgca atgcaggctt cgagctcagc cctgacggca agaactgtgt ggaccacaac 1621 gagtgtgcca ccagcaccat gtgcgtcaac ggcgtgtgtc tcaacgagga tggcagcttc 1681 tcctgcctct gcaaacccgg cttcctgctg gcgcctggcg gccactactg catggacatt 1741 gacgagtgcc agacgcccgg catctgcgtg aacggccact gtaccaacac cgagggctcc 1801 ttccgctgcc agtgcctggg ggggctggcg gtaggcacgg atggccgcgt gtgcgtggac 1861 acccacgtgc gcagcacctg ctatggggcc atcgagaagg gctcctgtgc ccgccccttc 1921 cctggcactg tcaccaagtc cgagtgctgc tgtgccaatc cggaccacgg ttttggggag 1981 ccctgccagc tttgtcctgc caaagactcc gctgagttcc aggcactgtg cagcagtggg 2041 cttggcatta ccacggatgg tcgagacatc aacgagtgtg ctctggatcc tgaggtttgt 2101 gccaatggcg tgtgcgagaa ccttcggggc agctaccgct gtgtctgcaa cctgggttat 2161 gaggcaggtg cctcaggcaa ggactgcaca gacgtggatg agtgtgccct caacagcctc 2221 ctgtgtgaca acgggtggtg ccagaatagc cctggcagct acagctgctc ctgccccccc 2281 ggcttccact tctggcagga cacggagatc tgcaaagatg tcgacgaatg cctgtccagc 2341 ccgtgtgtga gtggcgtctg tcggaacctg gccggctcct acacctgcaa atgtggccct 2401 ggcagccggc tggacccctc tggtaccttc tgtctagaca gcaccaaggg cacctgctgg 2461 ctgaagatcc aggagagccg ctgtgaggtg aaccttcagg gagccagcct gcggtctgag 2521 tgctgcgcca ccctcggggc agcctggggg agcccctgcg aacgctgcga gatcgaccct 2581 gcctgtgccc ggggctttgc ccggatgacg ggtgtcacct gcgatgatgt gaacgagtgt 2641 gagtccttcc cgggagtctg tcccaacggg cgttgcgtca acactgctgg gtctttccgc 2701 tgtgagtgtc cagagggcct gatgctggac gcctcaggcc ggctgtgcgt ggatgtgaga 2761 ttggaaccat gtttcctgcg atgggatgag gatgagtgtg gggtcaccct gcctggcaag 2821 taccggatgg acgtctgctg ctgctccatc ggggccgtgt ggggagtcga gtgcgaggcc 2881 tgcccggatc ccgagtctct ggagttcgcc agcctgtgcc cgcgggggct gggcttcgcc 2941 agccgggact tcctgtctgg ccgaccattc tataaagatg tgaatgaatg caaggtgttc 3001 cctggcctct gcacgcacgg tacctgcaga aacacggtgg gcagcttcca ctgcgcctgt 3061 gcggggggct tcgccctgga tgcccaggaa cggaactgca cagatatcga cgagtgtcgc 3121 atctctcctg acctctgcgg ccagggcacc tgtgtcaaca cgccgggcag ctttgagtgc54 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/NM_032447?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
74
3181 gagtgttttc ccggctacga gagtggcttc atgctgatga agaactgcat ggacgtggac 3241 gagtgtgcaa gggacccgct gctctgccgg ggaggcactt gcaccaacac ggatgggagc 3301 tacaagtgcc agtgtccccc tgggcatgag ctgacggcca agggcactgc ctgtgaggac 3361 atcgatgagt gctccctgag tgatggcctg tgtccccatg gccagtgtgt caatgtcatc 3421 ggtgccttcc agtgctcctg ccatgccggc ttccagagca cacctgaccg ccagggctgc 3481 gtggacatca acgaatgccg ggtccagaat ggtgggtgtg acgtgcactg tattaacact 3541 gagggcagct accggtgcag ctgtgggcag ggctactcgc tgatgcccga cggaagggca 3601 tgtgcagacg tggacgagtg tgaagagaac ccccgcgttt gtgaccaagg ccactgcacc 3661 aacatgccag ggggtcaccg ctgcctgtgc tatgatggct tcatggccac gccagacatg 3721 aggacatgtg ttgatgtgga tgagtgtgac ctgaaccctc acatctgcct ccatggggac 3781 tgcgagaaca cgaagggttc ctttgtctgc cactgtcagc tgggctacat ggtcaggaag 3841 ggggccacag gctgctctga tgtggatgaa tgcgaggttg gaggacacaa ctgtgacagt 3901 cacgcctcct gtctcaacat cccggggagt ttcagctgta ggtgcctgcc aggctgggtg 3961 ggggatggct tcgaatgtca cgacctggat gaatgcgtct cccaggagca ccggtgcagc 4021 ccaagaggtg actgtctcaa tgtccctggc tcctaccgct gcacctgccg ccagggcttt 4081 gccggggatg gcttcttctg cgaagacagg gatgaatgtg ccgagaacgt ggacctctgt 4141 gacaacgggc agtgcctcaa tgcgcccggc gggtaccgct gtgaatgtga gatgggcttt 4201 gaccccaccg aggaccaccg ggcctgccag gatgtggacg agtgtgcgca agggaacctc 4261 tgtgcatttg ggagctgtga gaacctgcct ggaatgttcc gctgcatctg caatggtggc 4321 tacgaactgg accgaggggg tggcaactgc acagacatca acgagtgtgc agacccagta 4381 aactgcatca acggcgtgtg cattaacacc cccggcagct acctctgcag ctgcccccag 4441 gattttgagc tgaaccccag cggagtgggc tgcgtggaca ctcgggccgg gaactgtttc 4501 ctggagacgc atgaccgagg ggacagtggc atttcctgca gtgccgagat cggagttggt 4561 gtcacccgag cttcctgctg ttgctccctg ggccgggctt ggggcaatcc ctgtgagctg 4621 tgccctatgg ccaacaccac tgagtacaga accctgtgcc cgggtggtga gggcttccag 4681 cctaaccgca tcactgtcat tctggaagac atcgacgagt gccaagagct gccagggctg 4741 tgtcaggggg gtgactgcgt caacacgttt ggcagtttcc agtgtgagtg cccacctggc 4801 taccacctca gtgagcacac ccgcatctgt gaggatattg acgaatgctc cacacactcc 4861 ggcatctgtg gccctggcac ctgctacaac accctgggga actacacctg tgtctgccct 4921 gcagagtacc tccaagtcaa tggtggcaac aactgcatgg atatgaggaa gagtgtctgc 4981 ttccggcact ataacggcac atgtcaaaat gagctggcct tcaacgtgac ccggaaaatg 5041 tgttgctgct cctacaacat tggccaggcc tggaatagac cctgtgaggc ctgccccact 5101 cccatcagtc ctgactacca gatcctgtgt ggaaatcagg ccccgggatt cctcactgac 5161 atccacacgg ggaagcccct tgacattgat gagtgtgggg agatccccgc catctgtgcc 5221 aatggcatct gcataaacca gatcgggagt ttccgctgcg agtgccccgc aggcttcaac 5281 tacaacagca tcctgctggc ttgtgaagat gtcgatgagt gtggcagcag ggagagtccc 5341 tgccagcaga atgctgactg catcaacatc cccggtagct accgctgcaa gtgcacccga 5401 gggtacaaac tgtcgccagg cggggcttgt gtgggacgga atgagtgtcg ggagatcccg 5461 aatgtctgta gccatggtga ctgcatggac acagaaggca gctacatgtg tctgtgtcac 5521 cgtggattcc aggcctctgc agaccagacc ctgtgcatgg acattgacga gtgtgaccgg 5581 cagccttgtg gaaatgggac ctgcaagaac atcattggct cctacaactg cctctgcttc 5641 cctggctttg tggtgacaca caatggggat tgtgtggatt ttgatgagtg tactaccctg 5701 gtggggcagg tgtgccgatt tggccattgc ctcaacacag ctggttcctt ccactgcctc 5761 tgccaggatg gctttgagct cacagctgat gggaagaact gtgtggacac caatgagtgc 5821 ctcagccttg caggaacctg cctacccggc acttgccaga acctcgaggg ctccttccgc 5881 tgcatctgtc cccctggctt ccaggtgcag agtgaccact gcattgatat cgacgagtgc 5941 tcagaggagc ccaacctctg cctctttggc acctgtacca acagccctgg gagcttccag 6001 tgcctctgcc cacctggctt tgtcctctct gacaatgggc accgttgctt tgacacacgg 6061 cagagtttct gcttcacccg ttttgaggct gggaagtgct cggtgcccaa agctttcaac 6121 accaccaaga cccgctgctg ctgcagtaag aggcctgggg agggctgggg agacccctgc 6181 gaactgtgtc cccaggaggg cagcgctgcc tttcaggagc tctgcccctt tggccacggg 6241 gcagtcccag gcccggatga ctcccgagaa gacgtgaatg agtgtgcaga gaaccctggc 6301 gtctgcacta acggcgtctg tgtcaacacc gatggatcct tccgctgtga gtgtcccttt 6361 ggctacagcc tggacttcac tggcatcaac tgtgtggaca cagacgagtg ctctgtcggc 6421 cacccctgtg ggcaagggac atgcaccaat gtcatcggag gcttcgaatg tgcctgtgct 6481 gacggctttg agcctggcct catgatgacc tgcgaggaca tcgacgaatg ctccctgaac
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
75
6541 ccgctgctct gtgccttccg ctgccacaat accgagggct cctacctgtg cacctgtcca 6601 gccggctaca ccctgcggga ggatggggcc atgtgtcgag atgtggacga gtgtgcagat 6661 ggtcagcagg actgccacgc ccggggcatg gagtgcaaga acctcatcgg taccttcgcg 6721 tgcgtctgtc ccccaggcat gcggcccctg cctggctctg gggagggctg cacagatgac 6781 aatgaatgcc acgctcagcc tgacctctgt gtcaacggcc gctgtgtcaa caccgcgggc 6841 agcttccggt gcgactgtga tgagggattc cagcccagcc ccacccttac cgagtgccac 6901 gacatccggc aggggccctg ctttgccgag gtgctgcaga ccatgtgccg gtctctgtcc 6961 agcagcagtg aggctgtcac cagggccgag tgctgctgtg ggggtggccg gggctggggg 7021 ccccgctgcg agctctgtcc cctgcccggc acctctgcct acaggaagct gtgcccccat 7081 ggctcaggct acactgctga gggccgagat gtagatgaat gccgtatgct tgctcacctg 7141 tgtgctcatg gggagtgcat caacagcctt ggctccttcc gctgccactg tcaggccggg 7201 tacacaccgg atgctactgc tactacctgc ctggatatgg atgagtgcag ccaggtcccc 7261 aagccatgta ccttcctctg caaaaacacg aagggcagtt tcctgtgcag ctgtccccga 7321 ggctacctgc tggaggagga tggcaggacc tgcaaagacc tggacgaatg cacctcccgg 7381 cagcacaact gtcagttcct ctgtgtcaac actgtgggcg ccttcacctg ccgctgtccg 7441 cccggcttca cccagcacca ccaggcctgc ttcgacaatg atgagtgctc agcccagcct 7501 ggcccatgtg gtgcccacgg gcactgccac aacaccccgg gcagcttccg ctgtgaatgc 7561 caccaaggct tcaccctggt cagctcaggc catggctgtg aagatgtgaa tgaatgtgat 7621 gggccccacc gctgccagca tggctgtcag aaccagctag ggggctaccg ctgcagctgc 7681 ccccagggtt tcacccagca ctcccagtgg gcccagtgtg tggatgagaa tgagtgtgcc 7741 ctgtcgcccc ccacctgcgg gagcgcctcc tgtcgcaaca ctcttggtgg cttccgctgc 7801 gtctgcccct ctggctttga ctttgatcag gccctcgggg gctgccagga ggtggatgag 7861 tgcgccggac ggcgtggccc ctgtagctac agctgtgcca acacgcctgg tggcttcctg 7921 tgcggctgtc ctcaaggcta cttccgggct gggcaagggc actgtgtctc cggcctgggc 7981 ttcagccccg gaccccagga caccccggac aaagaggagc tgctctcgtc tgaagcctgc 8041 tacgaatgca agatcaatgg cctctcccct cgggaccggc cacgacgcag tgcccacagg 8101 gaccaccagg tgaacctggc cacccttgac tccgaggccc tgctgacctt gggcctgaac 8161 ctctcacacc tgggccgggc cgagcgcatc ctggagctcc ggccggccct ggagggtcta 8221 gagggccgga tccgctacgt catcgtccgc ggaaacgagc aaggtttctt tcgcatgcat 8281 cacctccgtg gcgtcagctc cctgcagctg gggcggaggc ggccggggcc tggaacctac 8341 cggctggagg tggtgagcca catggcagga ccctggggtg tccagccaga ggggcagcca 8401 gggccatggg gccaggcctt gaggctgaag gtgcagctgc agttgcttta gttgggagga 8461 gcctcagtgg gccccagctg tccagagaag ggggattctg gaactgggaa ggactgatcc 8521 ccagaagcga tggctgacca gattgaaccc cgaaactcag gaagagtgaa atgctacacg 8581 acaacctcag gcaagcccgg cctctgcctg ggcctctgtg ccagccccgg gggcccccca 8641 gttactcagt ctttcctgga gacagcaaga agctgcaatg tgcaatcccc ctgcccccac 8701 agccaaggtc aggaagaggc cctgtggtca ccgtgtctgg ccaatctcag gctttcactt 8761 ctgtactgca ctgtggcttg ccctggcggg gggcaggggg ttggcaggac atggcaatgg 8821 gcaactgggg tgggcacagg gcttattcct cggagtagaa gggtgtacag ggggcccaga 8881 ctccacagtg acttgccaca tttgcccccc atttggagaa tgcttttata tcaaaagtgg 8941 agacgataat aaagttattt tgggtta
ORGANIZACIÓN ESTRUCTURAL:
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
76
1. FIBRILINA – 1 : 55
YFSREKPLRGRYLKRWGKEGAAGAAAETVGATSGQEPQLGQLRA EPSSGCSGHDWEQPPPPPRESEPPLLHWQGPPEVGAAPGEGGRSPARGTGGGIAGPRR RGALQGAAAAADRAPGAARGGGSRWRLGIMRRGRLLEIALGFTVLLASYTSHGADANL EAGNVKETRASRAKRRGGGGHDALKGPNVCGSRYNAYCCPGWKTLPGGNQCIVPICRH SCGDGFCSRPNMCTCPSGQIAPSCGSRSIQHCNIRCMNGGSCSDDHCLCQKGYIGTHC GQPVCESGCLNGGRCVAPNRCACTYGFTGPQCERDYRTGPCFTVISNQMCQGQLSGIV CTKTLCCATVGRAWGHPCEMCPAQPHPCRRGFIPNIRTGACQDVDECQAIPGLCQGGN CINTVGSFECKCPAGHKLNEVSQKCEDIDECSTIPGICEGGECTNTVSSYFCKCPPGF YTSPDGTRCIDVRPGYCYTALTNGRCSNQLPQSITKMQCCCDAGRCWSPGVTVAPEMC PIRATEDFNKLCSVPMVIPGRPEYPPPPLGPIPPVLPVPPGFPPGPQIPVPRPPVEYL YPSREPPRVLPVNVTDYCQLVRYLCQNGRCIPTPGSYRCECNKGFQLDLRGECIDVDE CEKNPCAGGECINNQGSYTCQCRAGYQSTLTRTECRDIDECLQNGRICNNGRCINTDG SFHCVCNAGFHVTRDGKNCEDMDECSIRNMCLNGMCINEDGSFKCICKPGFQLASDGR YCKDINECETPGICMNGRCVNTDGSYRCECFPGLAVGLDGRVCVDTHMRSTCYGGYKR GQCIKPLFGAVTKSECCCASTEYAFGEPCQPCPAQNSAEYQALCSSGPGMTSAGSDIN ECALDPDICPNGICENLRGTYKCICNSGYEVDSTGKNCVDINECVLNSLLCDNGQCRN TPGSFVCTCPKGFIYKPDLKTCEDIDECESSPCINGVCKNSPGSFICECSSESTLDPT KTICIETIKGTCWQTVIDGRCEININGATLKSQCCSSLGAAWGSPCTLCQVDPICGKG YSRIKGTQCEDIDECEVFPGVCKNGLCVNTRGSFKCQCPSGMTLDATGRICLDIRLET CFLRYEDEECTLPIAGRHRMDACCCSVGAAWGTEECEECPMRNTPEYEELCPRGPGFA TKEITNGKPFFKDINECKMIPSLCTHGKCRNTIGSFKCRCDSGFALDSEERNCTDIDE CRISPDLCGRGQCVNTPGDFECKCDEGYESGFMMMKNCMDIDECQRDPLLCRGGVCHN TEGSYRCECPPGHQLSPNISACIDINECELSAHLCPNGRCVNLIGKYQCACNPGYHST PDRLFCVDIDECSIMNGGCETFCTNSEGSYECSCQPGFALMPDQRSCTDIDECEDNPN
55 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/X63556?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
77
ICDGGQCTNIPGEYRCLCYDGFMASEDMKTCVDVNECDLNPNICLSGTCENTKGSFIC HCDMGYSGKKGKTGCTDINECEIGAHNCGKHAVCTNTAGSFKCSCSPGWIGDGIKCTD LDECSNGTHMCSQHADCKNTMGSYRCLCKEGYTGDGFTCTDLDECSENLNLCGNGQCL NAPGGYRCECDMGFVPSADGKACEDIDECSLPNICVFGTCHNLPGLFRCECEIGYELD RSGGNCTDVNECLDPTTCISGNCVNTPGSYICDCPPDFELNPTRVGCVDTRSGNCYLD IRPRGDNGDTACSNEIGVGVSKASCCCSLGKAWGTPCEMCPAVNTSEYKILCPGGEGF RPNPITVILEDIDECQELPGLCQGGKCINTFGSFQCRCPTGYYLNEDTRVCDDVNECE TPGICGPGTCYNTVGNYTCICPPDYMQVNGGNNCMDMRRSLCYRNYYADNQTCDGELL FNMTKKMCCCSYNIGRAWNKPCEQCPIPSTDEFATLCGSQRPGFVIDIYTGLPVDIDE CREIPGVCENGVCINMVGSFRCECPVGFFYNDKLLVCEDIDECQNGPVCQRNAECINT AGSYRCDCKPGYRFTSTGQCNDRNECQEIPNICSHGQCIDTVGSFYCLCHTGFKTNDD QTMCLDINECERDACGNGTCRNTIGSFNCRCNHGFILSHNNDCIDVDECASGNGNLCR NGQCINTVGSFQCQCNEGYEVAPDGRTCVDINECLLEPRKCAPGTCQNLDGSYRCICP PGYSLQNEKCEDIDECVEEPEICALGTCSNTEGSFKCLCPEGFSLSSSGRRCQDLRMS YCYAKFEGGKCSSPKSRNHSKQECCCALKGEGWGDPCELCPTEPDEAFRQICPYGSGI IVGPDDSAVDMDECKEPDVCKHGQCINTDGSYRCECPFGYTLAGNECVDTDECSVGNP CGNGTCKNVIGGFECTCEEGFEPGPMMTCEDINECAQNPLLCAFRCVNTYGSYECKCP VGYVLREDRRMCKDEDECEEGKHDCTEKQMECKNLIGTYMCICGPGYQRRPDGEGCVD ENECQTKPGICENGRCLNTRGSYTCECNDGFTASPNQDECLDNREGYCFTEVLQNMCQ IGSSNRNPVTKSECCCDGGRGWGPHCEICPFQGTVAFKKLCPHGRGFMTNGADIDECK VIHDVCRNGECVNDRGSYHCICKTGYTPDITGTSCVDLNECNQAPKPCNFICKNTEGS YQCSCPKGYILQEDGRSCKDLDECATKQHNCQFLCVNTIGGFTCKCPPGFTQHHTSCI DNNECTSDINLCGSKGICQNTPGSFTCECQRGFSLDQTGSSCEDVDECEGNHRCQHGC QNIIGGYRCSCPQGYLQHYQWNQCVDENECLSAHICGGASCHNTLGSYKCMCPAGFQY EQFSGGCQDINECGSAQAPCSYGCSNTEGGYLCGCPPGYFRIGQGHCVSGMGMGRGNP
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
78
EPPVSGEMDDNSLSPEACYECKINGYPKRGRKRRSTNETDASNIEDQSETEANVSLAS WDVEKTAIFAFNISHVSNKVRILELLPALTTLTNHNRYLIESGNEDGFFKINQKEGIS YLHFTKKKPVAGTYSLQISSTPLYKKKELNQLEDKYDKDYLSGELGDNLKMKIQVLLH
2. FIBRILINA – 2 : 56
MGRRRRLCLQLYFLWLGCVVLWAQGTAGQPQPPPPKPPRPQPPP QQVRSATAGSEGGFLAPEYREEGAAVASRVRRRGQQDVLRGPNVCGSRFHSYCCPGWK TLPGGNQCIVPICRNSCGDGFCSRPNMCTCSSGQISSTCGSKSIQQCSVRCMNGGTCA DDHCQCQKGYIGTYCGQPVCENGCQNGGRCIAQPCACVYGFTGPQCERDYRTGPCFTQ VNNQMCQGQLTGIVCTKTLCCATTGRAWGHPCEMCPAQPQPCRRGFIPNIRTGACQDV DECQAIPGICQGGNCINTVGSFECRCPAGHKQSETTQKCEDIDECSIIPGICETGECS NTVGSYFCVCPRGYVTSTDGSRCIDQRTGMCFSGLVNGRCAQELPGRMTKMQCCCEPG RCWGIGTIPEACPVRGSEEYRRLCMDGLPMGGIPGSAGSRPGGTGGNGFAPSGNGNGY GPGGTGFIPIPGGNGFSPGVGGAGVGAGGQGPIITGLTILNQTIDICKHHANLCLNGR CIPTVSSYRCECNMGYKQDANGDCIDVDECTSNPCTNGDCVNTPGSYYCKCHAGFQRT PTKQACIDIDECIQNGVLCKNGRCVNSDGSFQCICNAGFELTTDGKNCVDHDECTTTN MCLNGMCINEDGSFKCICKPGFVLAPNGRYCTDVDECQTPGICMNGHCINSEGSFRCD CPPGLAVGMDGRVCVDTHMRSTCYGGIKKGVCVRPFPGAVTKSECCCANPDYGFGEPC QPCPAKNSAEFHGLCSSGVGITVDGRDINECALDPDICANGICENLRGSYRCNCNSGY EPDASGRNCIDIDECLVNRLLCDNGLCRNTPGSYSCTCPPGYVFRTETETCEDINECE SNPCVNGACRNNLGSFNCECSPGSKLSSTGLICIDSLKGTCWLNIQDSRCEVNINGAT LKSECCATLGAAWGSPCERCELDTACPRGLARIKGVTCEDVNECEVFPGVCPNGRCVN SKGSFHCECPEGLTLDGTGRVCLDIRMEQCYLKWDEDECIHPVPGKFRMDACCCAVGA AWGTECEECPKPGTKEYETLCPRGAGFANRGDVLTGRPFYKDINECKAFPGMCTYGKC RNTIGSFKCRCNSGFALDMEERNCTDIDECRISPDLCGSGICVNTPGSFECECFEGYE SGFMMMKNCMDIDGCERNPLLCRGGTCVNTEGSFQCDCPLGHELSPSREDCVDINECS
56NCBI - http://www.ncbi.nlm.nih.gov/nuccore/U03272?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
79
LSDNLCRNGKCVNMIGTYQCSCNPGYQATPDRQGCTDIDECMIMNGGCDTQCTNSEGS YECSCSEGYALMPDGRSCADIDECENNPDICDGGQCTNIPGEYRCLCYDGFMASMDMK TCIDVNECDLNSNICMFGECENTKGSFICHCQLGYSVKKGTTGCTDVDECEIGAHNCD MHASCLNIPGSFKCSCREGWIGNGIKCIDLDECSNGTHQCSINAQCVNTPGSYRCACS EGFTGDGFTCSDVDECAENINLCENGQCLNVPGAYRCECEMGFTPASDSRSCQDIDEC SFQNICVSGTCNNLPGMFHCICDDGYELDRTGGNCTDIDECADPINCVNGLCVNTPGR YECNCPPDFQLNPTGVGCVDNRVGNCYLKFGPRGDGSLSCNTEIGVGVSRSSCCCSLG KAWGNPCETCPPVNSTEYYTLCPGGEGFRPNPITIILEDIDECQELPGLCQGGNCINT FGSFQCECPQGYYLSEDTRICEDIDECFAHPGVCGPGTCYNTLGNYTCICPPEYMQVN GGHNCMDMRKSFCYRSYNGTTCENELPFNVTKRMCCCTYNVGKAGNKPCEPCPTPGTA DFKTICGNIPGFTFDIHTGKAVDIDECKEIPGICANGVCINQIGSFRCECPTGFSYND LLLVCEDIDECSNGDNLCQRNADCINSPGSYRCECAAGFKLSPNGACVDRNECLEIPN VCSHGLCVDLQGSYQCICHNGFKASQDQTMCMDVDECERHPCGNGTCKNTVGSYNCLC YPGFELTHNNDCLDIDECSSFFGQVCRNGRCFNEIGSFKCLCNEGYELTPDGKNCIDT NECVALPGSCSPGTCQNLEGSFRCICPPGYEVKSENCIDINECDEDPNICLFGSCTNT PGGFQCLCPPGFVLSDNGRRCFDTRQSFCFTNFENGKCSVPKAFNTTKAKCCCSKMPG EGWGDPCELCPKDDEVAFQDLCPYGHGTVPSLHDTREDVNECLESPGICSNGQCINTD GSFRCECPMGYNLDYTGVRCVDTDECSIGNPCGNGTCTNVIGSFECNCNEGFEPGPMM NCEDINECAQNPLLCALRCMNTFGSYECTCPIGYALREDQKMCKDLDECAEGLHDCES RGMMCKNLIGTFMCICPPGMARRPDGEGCVDENECRTKPGICENGRCVNIIGSYRCEC NEGFQSSSSGTECLDNRQGLCFAEVLQTICQMASSSRNLVTKSECCCDGGRGWGHQCE LCPLPGTAQYKKICPHGPGYTTDGRDIDECKVMPNLCTNGQCINTMGSFRCFCKVGYT TDISGTSCIDLDECSQSPKPCNYICKNTEGSYQCSCPRGYVLQEDGKTCKDLDECQTK QHNCQFLCVNTLGGFTCKCPPGFTQHHTACIDNNECGSQPLLCGGKGICQNTPGSFSC ECQRGFSLDATGLNCEDVDECDGNHRCQHGCQNILGGYRCGCPQGYIQHYQWNQCVDE
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
80
NECSNPNACGSASCYNTLGSYKCACPSGFSFDQFSSACHDVNECSSSKNPCNYGCSNT EGGYLCGCPPGYYRVGQGHCVSGMGFNKGQYLSLDTEVDEENALSPEACYECKINGYP KKDSRQKRSIHEPDPTAVEQISLESVDMDSPVNMKFNLSHLGSKEHILELRPAIQPLN NHIRYVISQGNDDSVFRIHQRNGLSYLHTAKKKLMPGTYTLEITSIPLYKKKELKKLE ESNEDDYLLGELGEALRMRLQIQLY
3. FIBRILINA – 3 : 57
MTLEGLYLARGPLARLLLAWSALLCMAGGQGRWDGALEAAGPGR VRRRGSPGILQGPNVCGSRFHAYCCPGWRTFPGRSQCVVPICRRACGEGFCSQPNLCT CADGTLAPSCGVSRGSGCSVSCMNGGTCRGASCLCQKGYTGTVCGQPICDRGCHNGGR CIGPNRCACVYGFMGPQCERDYRTGPCFGQVGPEGCQHQLTGLVCTKALCCATVGRAW GLPCELCPAQPHPCRRGFIPNIHTGACQDVDECQAVPGLCQGGSCVNMVGSFHCRCPV GHRLSDSSAACEDYRAGACFSVLFGGRCAGDLAGHYTRRQCCCDRGRCWAAGPVPELC PPRGSNEFQQLCAQRLPLLPGHPGLFPGLLGFGSNGMGPPLGPARLNPHGSDARGIPS LGPGNSNIGTATLNQTIDICRHFTNLCLNGRCLPTPSSYRCECNVGYTQDVRGECIDV DECTSSPCHHGDCVNIPGTYHCRCYPGFQATPTRQACVDVDECIVSGGLCHLGRCVNT EGSFQCVCNAGFELSPDGKNCVDHNECATSTMCVNGVCLNEDGSFSCLCKPGFLLAPG GHYCMDIDECQTPGICVNGHCTNTEGSFRCQCLGGLAVGTDGRVCVDTHVRSTCYGAI EKGSCARPFPGTVTKSECCCANPDHGFGEPCQLCPAKDSAEFQALCSSGLGITTDGRD INECALDPEVCANGVCENLRGSYRCVCNLGYEAGASGKDCTDVDECALNSLLCDNGWC QNSPGSYSCSCPPGFHFWQDTEICKDVDECLSSPCVSGVCRNLAGSYTCKCGPGSRLD PSGTFCLDSTKGTCWLKIQESRCEVNLQGASLRSECCATLGAAWGSPCERCEIDPACA
57 NCBI - http://www.ncbi.nlm.nih.gov/nuccore/NM_032447?
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
81
RGFARMTGVTCDDVNECESFPGVCPNGRCVNTAGSFRCECPEGLMLDASGRLCVDVRL EPCFLRWDEDECGVTLPGKYRMDVCCCSIGAVWGVECEACPDPESLEFASLCPRGLGF ASRDFLSGRPFYKDVNECKVFPGLCTHGTCRNTVGSFHCACAGGFALDAQERNCTDID ECRISPDLCGQGTCVNTPGSFECECFPGYESGFMLMKNCMDVDECARDPLLCRGGTCT NTDGSYKCQCPPGHELTAKGTACEDIDECSLSDGLCPHGQCVNVIGAFQCSCHAGFQS TPDRQGCVDINECRVQNGGCDVHCINTEGSYRCSCGQGYSLMPDGRACADVDECEENP RVCDQGHCTNMPGGHRCLCYDGFMATPDMRTCVDVDECDLNPHICLHGDCENTKGSFV CHCQLGYMVRKGATGCSDVDECEVGGHNCDSHASCLNIPGSFSCRCLPGWVGDGFECH DLDECVSQEHRCSPRGDCLNVPGSYRCTCRQGFAGDGFFCEDRDECAENVDLCDNGQC LNAPGGYRCECEMGFDPTEDHRACQDVDECAQGNLCAFGSCENLPGMFRCICNGGYEL DRGGGNCTDINECADPVNCINGVCINTPGSYLCSCPQDFELNPSGVGCVDTRAGNCFL ETHDRGDSGISCSAEIGVGVTRASCCCSLGRAWGNPCELCPMANTTEYRTLCPGGEGF QPNRITVILEDIDECQELPGLCQGGDCVNTFGSFQCECPPGYHLSEHTRICEDIDECS THSGICGPGTCYNTLGNYTCVCPAEYLQVNGGNNCMDMRKSVCFRHYNGTCQNELAFN VTRKMCCCSYNIGQAWNRPCEACPTPISPDYQILCGNQAPGFLTDIHTGKPLDIDECG EIPAICANGICINQIGSFRCECPAGFNYNSILLACEDVDECGSRESPCQQNADCINIP GSYRCKCTRGYKLSPGGACVGRNECREIPNVCSHGDCMDTEGSYMCLCHRGFQASADQ TLCMDIDECDRQPCGNGTCKNIIGSYNCLCFPGFVVTHNGDCVDFDECTTLVGQVCRF GHCLNTAGSFHCLCQDGFELTADGKNCVDTNECLSLAGTCLPGTCQNLEGSFRCICPP GFQVQSDHCIDIDECSEEPNLCLFGTCTNSPGSFQCLCPPGFVLSDNGHRCFDTRQSF CFTRFEAGKCSVPKAFNTTKTRCCCSKRPGEGWGDPCELCPQEGSAAFQELCPFGHGA VPGPDDSREDVNECAENPGVCTNGVCVNTDGSFRCECPFGYSLDFTGINCVDTDECSV GHPCGQGTCTNVIGGFECACADGFEPGLMMTCEDIDECSLNPLLCAFRCHNTEGSYLC TCPAGYTLREDGAMCRDVDECADGQQDCHARGMECKNLIGTFACVCPPGMRPLPGSGE GCTDDNECHAQPDLCVNGRCVNTAGSFRCDCDEGFQPSPTLTECHDIRQGPCFAEVLQ
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
82
TMCRSLSSSSEAVTRAECCCGGGRGWGPRCELCPLPGTSAYRKLCPHGSGYTAEGRDV DECRMLAHLCAHGECINSLGSFRCHCQAGYTPDATATTCLDMDECSQVPKPCTFLCKN TKGSFLCSCPRGYLLEEDGRTCKDLDECTSRQHNCQFLCVNTVGAFTCRCPPGFTQHH QACFDNDECSAQPGPCGAHGHCHNTPGSFRCECHQGFTLVSSGHGCEDVNECDGPHRC QHGCQNQLGGYRCSCPQGFTQHSQWAQCVDENECALSPPTCGSASCRNTLGGFRCVCP SGFDFDQALGGCQEVDECAGRRGPCSYSCANTPGGFLCGCPQGYFRAGQGHCVSGLGF SPGPQDTPDKEELLSSEACYECKINGLSPRDRPRRSAHRDHQVNLATLDSEALLTLGL NLSHLGRAERILELRPALEGLEGRIRYVIVRGNEQGFFRMHHLRGVSSLQLGRRRPGP GTYRLEVVSHMAGPWGVQPEGQPGPWGQALRLKVQLQLL
FUNCIÓN:
1. FIBRILINA – 1 : La proteína codificada es grande, Glicoproteina de la Matriz Extracelular que sirve como un componente estructural de Microfibrillas de 10 – 12 nm. ligadas al Calcio. Estas Microfibrillas, proporcionan una fuerza que produce soporte estructural en Tejido Conectivo Elástico y No Elástico en todo el cuerpo.
2. FIBRILINA – 2 :
Componente del tejido conectivo y Microfibrillas. Puede estar envuelta en las ensambladas fibras elásticas.
3. FIBRILINA – 3 :Las fibrilina son moléculas de la matriz extracelular que se ensamblan dentro de las Microfibrillas en el Tejido Conectivo.
Este gen es más altamente expresado en Tejidos Fetales
Su producto de la proteína se localiza en microfibrillas extracelulares de desarrollo de elementos esqueletales, piel, pulmón, riñón y músculo esquelético
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
83
CORRELACIÓN GENOTIPO - FENOTIPO:
1. FIBRILINA – 1 : Mutación en este gen está asociado con SÍNDROME DE MARFAN, Ectopia Aislada del
Cristalino, Síndrome Autosómico Dominante Weill-Marchesani, Síndrome MASS, Síndrome de Craneosinostosis de Shprintzen-Goldberg
2. FIBRILINA – 2 :
Mutación en este gen causa ARACNODACTÍLIA CONTRACTURAL CONGÉNITA (CCA). (Un trastorno que es similar o una variante del síndrome de Marfan se hereda como un rasgo autosómico dominante, y se caracteriza especialmente por aracnodactilia, contractura de las articulaciones y escoliosis 58 )
3. FIBRILINA – 3 :Este gen es potencialmente implicados en el SÍNDROME DE WEILL-MARCHESANI.
58 GENETICS HOME REFERENCE - http://ghr.nlm.nih.gov/condition/congenital-contractural-arachnodactyly
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
84
SÍNDROME DE MARFANANTECEDENTES:
Este padecimiento, descrito en 1896 por Antoine Marfan, se clasifica entre las enfermedades del tejido conjuntivo (conectivo); Sus manifestaciones son muy variables y, por tanto, a veces es difícil diagnosticarlo cuando los síntomas son leves y no hay antecedentes familiares. 59
PRINCIPIOS: 60
Mutaciones Dominantes Negativas 61
Expresividad Variable
59 Guizar Vásquez. GENÉTICA CLÍNICA: Diagnóstico y Manejo de Enfermedades Hereditarias. Tercera Edición. 2001 : 202 -20360 Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 28461 Acción Perjudicial de una Proteína Mutante que impide, en mayor o en menor grado, la organización de la Proteína Normal con la cual coexiste. Solari. GENÉTICA HUMANA: Fundamentos y Aplicaciones en Medicina. Tercera Edición. 2003 : 333 - 334
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
85
CARACTERÍSTICAS FENOTÍPICAS PRINCIPALES:
Edad de inicio: Temprana Infancia.
Estatura Desproporcionada
Genética Médica
Fig. 1. Lactante con Síndrome de Marfan. Se observa facies estrechas y Aracnodactília (Tomado de Guizar Vásquez. GENÉTICA CLÍNICA: Diagnóstico y Manejo de Enfermedades Hereditarias. Tercera Edición. 2001: 202 -203

Seminario II Bases genético – moleculares de las proteínas estructurales
86
Anomalías Esqueléticas
Genética Médica
Fig. 2. Paciente en hospital con Síndrome de Marfan. Se observa gran altura (Tomado de Páez, Haack, Bemergui CÁTEDRA EN MEDICINA III . http://www.revistas.luz.edu.ve/index.php/ic/article/viewFile/2321/2250 )
Fig. 3. Paciente en hospital con Síndrome de Marfan. Se observa anormalidades en extremidades inferiores (Tomado de Páez, Haack, Bemergui CÁTEDRA EN MEDICINA III . http://www.revistas.luz.edu.ve/index.php/ic/article/viewFile/2321/2250 )

Seminario II Bases genético – moleculares de las proteínas estructurales
87
Ectopía Lenticular
Genética Médica
Fig. 4. Paciente en hospital con Síndrome de Marfan. Se observa en manos: Aracnodactilia (Tomado de Páez, Haack, Bemergui CÁTEDRA EN MEDICINA III . http://www.revistas.luz.edu.ve/index.php/ic/article/viewFile/2321/2250 )
Fig. 5. Paciente con Síndrome de Marfan. Se observa: Aracnodactilia . (Tomado de Bobrow CRISTALINO y CATARATAS – Google Books

Seminario II Bases genético – moleculares de las proteínas estructurales
88
Prolapso de la Válvula Mitral
Genética Médica
Fig. 6. Ectopía Lenticular. Aspecto bajo la lámpara de hendidura del ojo izquierdo de un paciente con Síndrome de Marfan. El asteriscoindica el centro del cristalino, que está desplazado superomedialmente; por lo general el Cristalino está en el centro de la pupila. Las Flechas indican el borde del Cristalino que está visible en la pupila de forma anormal (Tomado de Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 285 )
Fig. 7. Prolapso de la Válvula Mitral. Casaca (Tomado de http://www.nlm.nih.gov/medlineplus/spanish/ )

Seminario II Bases genético – moleculares de las proteínas estructurales
89
Dilatacion y Ruptura de la Aorta
CROMOSOMA 15: 62
62 Guizar Vásquez. GENÉTICA CLÍNICA: Diagnóstico y Manejo de Enfermedades Hereditarias. Tercera Edición. 2001 : 917
Genética Médica
Fig. 8. Dilatación de la Aorta. Casaca (Tomado de http://www.nlm.nih.gov/medlineplus/spanish/ )

Seminario II Bases genético – moleculares de las proteínas estructurales
90
FRECUENCIA: 63
1 en 10 000
INCIDENCIA DE MUTACION IN NOVO : 64
25 - 35 %
CASO CLÍNICO
63 Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 284 64 Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 284
Genética Médica

Seminario II Bases genético – moleculares de las proteínas estructurales
91
HISTORIA Y HALLAZGOS FÍSICOS: 65
65 Nussbaum, Mcinnes, Willard. THOMPSON & THOMPSON. GENÉTICA EN MEDICINA. Séptima Edicion. 2005 : 284
Genética Médica
Un chico de 16 años sano y que es una estrella del baloncesto en su instituto, es remitido a la clínica de genética por un posible Síndrome de Marfan. Se parece físicamente a su padre, un hombre alto y delgado, que murió mientras realizaba su sesión de ejercicio matinal. Ningún otro familiar presenta historia de anomalías esqueléticas, muerte súbita, perdida de visión o anomalías congénitas. En el examen físico, se aprecia que tiene un hábito asténico, paladar ojival, un ligero pectus carinatum, aracnodactilia, una relación envergadura: altura de 1,1, un soplo diastólico y marcas de estiramiento en los hombros y muslos. Se le solicita una ecocardiografía, que muestra dilatación de la raíz de la aorta con regurgitación aórtica. El examen oftalmológico evidencia iridodonesis bilateral y ligero desplazamiento superior de los cristalinos. Basándose en el examen físico y en los resultados de las pruebas, el genetista le explica a él y a su madre, que él tiene el Síndrome de Marfan.

Seminario II Bases genético – moleculares de las proteínas estructurales
92
Genética Médica


















