
Comparación de los pacientes de un registro español de enfermedad de Fabry en dos periodos de...
6
Original Comparacio ´n de los pacientes de un registro espan ˜ol de enfermedad de Fabry en dos periodos de tiempo Miguel A ´ ngel Barba Romero a, *, Alberto Rivera Gallego b , Guillem Pintos Morell c , en representacio ´n del Grupo Espan ˜ol de Estudio del FOS ^ a Servicio de Medicina Interna, Complejo Hospitalario y Universitario de Albacete, Albacete, Espan ˜a b Servicio de Medicina Interna, Complexo Hospitalario y Universitario de Vigo, Vigo, Pontevedra, Espan ˜a c Servicio de Pediatrı´a, Hospital Universitari Germans Trias i Pujol, Universitat Auto `noma de Barcelona, Badalona, Barcelona, Espan ˜a Med Clin (Barc). 2012;139(9):379–384 I N F O R M A C I O ´ N D E L A R T I ´ C U L O Historia del artı´culo: Recibido el 23 de septiembre de 2011 Aceptado el 29 de noviembre de 2011 On-line el 21 de enero de 2012 Palabras clave: Enfermedad de Fabry Enfermedades por depo ´ sito lisoso ´ mico Alfa-galactosidasa A Estudio familiar Fabry Outcome Survey Espan ˜a R E S U M E N Fundamentos y objetivo: La enfermedad de Fabry (EF) es un raro trastorno lisosomal por de ´ ficit de la enzima alfa-galactosidasa A, con herencia ligada al cromosoma X, que ocasiona disfuncio ´n multiorga ´ nica y muerte temprana. En 2004 se publicaron las caracterı ´sticas de los primeros 24 pacientes espan ˜ oles incluidos en el registro Fabry Outcome Survey (FOS), habiendo aumentado significativamente su nu ´ mero desde entonces. Este trabajo analiza si ha habido cambios en el perfil clı ´nico y diagno ´ stico de los mismos entre los 2 perı ´odos. Pacientes y me ´todo: En 2009 existı ´an en el registro FOS datos de 92 pacientes. Se compararon los pacientes de 2003 con los posteriores (68) respecto a la edad de inicio de los sı ´ntomas y de diagno ´ stico, gravedad y diagno ´ sticos previos erro ´ neos, segu ´n sexo. Similar ana ´ lisis se efectuo ´ entre los casos ı ´ndice (31) y el resto de pacientes. Resultados: En ambos sexos la demora media del diagno ´ stico fue de 10 an ˜os. Los varones presentan un fenotipo cla ´ sico, y hasta un 40% de las mujeres referı ´an sı ´ntomas. En estas, la actividad enzima ´ tica parece condicionar la gravedad de la enfermedad. No hubo diferencias en los para ´ metros analizados entre los pacientes del primer perı ´odo y los posteriores, ni al comparar los casos ı ´ndice con el resto. Conclusiones: Los registros como el FOS poseen valor para ahondar en el conocimiento de las enfermedades minoritarias. Confirmamos que las mujeres no son meras portadoras de la EF. Todavı ´a falta formacio ´n y difusio ´n para incluir la EF en el diagno ´ stico diferencial de otros procesos. Realizar estudios familiares exhaustivos ayudarı ´a a un diagno ´ stico ma ´s precoz. ß 2011 Elsevier Espan ˜a, S.L. Todos los derechos reservados. Comparison of patients from a Spanish Registry of Fabry disease in two periods Keywords: Fabry disease Lysosomal storage diseases Alpha-galactosidase A Pedigree Fabry Outcome Survey Spain A B S T R A C T Background and objective: Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by a deficiency of the enzyme alpha-galactosidase A, that leads to multiorgan dysfunction and premature death. Data from the first 24 Spanish patients enrolled on the Fabry Outcome Survey (FOS) were published in 2004, with a significant increase in the number of patients since then. This manuscript analyzes whether the clinical profile or diagnosis of these patients between the 2 periods has changed. Patients and methods: In 2009 the FOS included data from 92 patients. Patients included up to 2003 and those included after that year (68) were compared by sex, regarding age at onset of symptoms and diagnosis, severity and previous misdiagnoses. Similar analysis was performed between the index cases (31) and the other patients. Results: Mean delay in diagnosis was 10 years for both sexes. Male had a classic phenotype, and up to 40% of the females reported symptoms. In females, the enzyme activity seemed to determine disease severity. No differences were observed in any parameter when comparing the patients included in the first period to those included afterwards, nor when comparing index cases with the rest of the patients. * Autor para correspondencia. Correo electro ´nico: [email protected] (M.A ´ . Barba Romero). ^ Los nombres de los componentes del Grupo Espan ˜ol de Estudio del FOS esta ´n relacionados en el Anexo 1. ww w.els evier.es /med ic in ac lin ic a 0025-7753/$ – see front matter ß 2011 Elsevier Espan ˜a, S.L. Todos los derechos reservados. doi:10.1016/j.medcli.2011.11.026
Transcript of Comparación de los pacientes de un registro español de enfermedad de Fabry en dos periodos de...

Med Clin (Barc). 2012;139(9):379–384
Original
Comparacion de los pacientes de un registro espanol de enfermedad de Fabryen dos periodos de tiempo
Miguel Angel Barba Romero a,*, Alberto Rivera Gallego b, Guillem Pintos Morell c,en representacion del Grupo Espanol de Estudio del FOS^
a Servicio de Medicina Interna, Complejo Hospitalario y Universitario de Albacete, Albacete, Espanab Servicio de Medicina Interna, Complexo Hospitalario y Universitario de Vigo, Vigo, Pontevedra, Espanac Servicio de Pediatrıa, Hospital Universitari Germans Trias i Pujol, Universitat Autonoma de Barcelona, Badalona, Barcelona, Espana
I N F O R M A C I O N D E L A R T I C U L O
Historia del artıculo:
Recibido el 23 de septiembre de 2011
Aceptado el 29 de noviembre de 2011
On-line el 21 de enero de 2012
Palabras clave:
Enfermedad de Fabry
Enfermedades por deposito lisosomico
Alfa-galactosidasa A
Estudio familiar
Fabry Outcome Survey
Espana
R E S U M E N
Fundamentos y objetivo: La enfermedad de Fabry (EF) es un raro trastorno lisosomal por deficit de la
enzima alfa-galactosidasa A, con herencia ligada al cromosoma X, que ocasiona disfuncion multiorganica
y muerte temprana. En 2004 se publicaron las caracterısticas de los primeros 24 pacientes espanoles
incluidos en el registro Fabry Outcome Survey (FOS), habiendo aumentado significativamente su numero
desde entonces. Este trabajo analiza si ha habido cambios en el perfil clınico y diagnostico de los mismos
entre los 2 perıodos.
Pacientes y metodo: En 2009 existıan en el registro FOS datos de 92 pacientes. Se compararon los
pacientes de 2003 con los posteriores (68) respecto a la edad de inicio de los sıntomas y de diagnostico,
gravedad y diagnosticos previos erroneos, segun sexo. Similar analisis se efectuo entre los casos ındice
(31) y el resto de pacientes.
Resultados: En ambos sexos la demora media del diagnostico fue de 10 anos. Los varones presentan un
fenotipo clasico, y hasta un 40% de las mujeres referıan sıntomas. En estas, la actividad enzimatica parece
condicionar la gravedad de la enfermedad. No hubo diferencias en los parametros analizados entre los
pacientes del primer perıodo y los posteriores, ni al comparar los casos ındice con el resto.
Conclusiones: Los registros como el FOS poseen valor para ahondar en el conocimiento de las
enfermedades minoritarias. Confirmamos que las mujeres no son meras portadoras de la EF. Todavıa
falta formacion y difusion para incluir la EF en el diagnostico diferencial de otros procesos. Realizar
estudios familiares exhaustivos ayudarıa a un diagnostico mas precoz.
� 2011 Elsevier Espana, S.L. Todos los derechos reservados.
Comparison of patients from a Spanish Registry of Fabry disease in two periods
Keywords:
Fabry disease
Lysosomal storage diseases
Alpha-galactosidase A
Pedigree
Fabry Outcome Survey
Spain
A B S T R A C T
Background and objective: Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by a
deficiency of the enzyme alpha-galactosidase A, that leads to multiorgan dysfunction and premature
death. Data from the first 24 Spanish patients enrolled on the Fabry Outcome Survey (FOS) were
published in 2004, with a significant increase in the number of patients since then. This manuscript
analyzes whether the clinical profile or diagnosis of these patients between the 2 periods has changed.
Patients and methods: In 2009 the FOS included data from 92 patients. Patients included up to 2003
and those included after that year (68) were compared by sex, regarding age at onset of symptoms and
diagnosis, severity and previous misdiagnoses. Similar analysis was performed between the index cases
(31) and the other patients.
Results: Mean delay in diagnosis was 10 years for both sexes. Male had a classic phenotype, and up to
40% of the females reported symptoms. In females, the enzyme activity seemed to determine disease
severity. No differences were observed in any parameter when comparing the patients included in the
first period to those included afterwards, nor when comparing index cases with the rest of the patients.
ww w.els evier .es /med i c in ac l in i c a
* Autor para correspondencia.
Correo electronico: [email protected] (M.A. Barba Romero).^ Los nombres de los componentes del Grupo Espanol de Estudio del FOS estan relacionados en el Anexo 1.
0025-7753/$ – see front matter � 2011 Elsevier Espana, S.L. Todos los derechos reservados.
doi:10.1016/j.medcli.2011.11.026

M.A. Barba Romero et al / Med Clin (Barc). 2012;139(9):379–384380
Conclusions: Registries like FOS have a great value to deepen our understanding of rare diseases. We
confirm that women are not just carriers of the disease. There is still a lack of education and awareness in
order to include FD in the differential diagnosis of other processes. Complete family studies would allow
early diagnosis of this disorder.
� 2011 Elsevier Espana, S.L. All rights reserved.
Introduccion
La enfermedad de Fabry (EF) es una rara enfermedad hereditariade deposito lisosomico de esfingolıpidos, con transmision ligada alcromosoma X, originada por la actividad deficiente de la enzimalisosomica alfa-galactosidasa A (GLA)1. Dicha deficiencia conduce ala acumulacion de su principal sustrato, la globotriaosilceramida,en los lisosomas de practicamente todos los tejidos y organos. Losrasgos clınicos principales incluyen lesiones cutaneas (angioque-ratomas), proteinuria, dano renal progresivo, neuropatıa dolorosade predominio en manos y pies (acroparestesias), miocardiopatıahipertrofica (con afectacion valvular y arritmias), ictus, manifes-taciones gastrointestinales, distrofia corneal, hipohidrosis eintolerancia al frıo-calor, ası como al ejercicio2. Todo ello originauna afectacion multisistemica grave y progresiva, con marcadodeterioro de la calidad de vida, y que conlleva una muerte precozde los afectados3.
En 1989 fue identificado el gen que codifica la GLA, quese localiza en posicion Xq 22.114. Debido a su modo de herencia,los varones hemicigotos presentan habitualmente un deficitmarcado de la actividad de la enzima expresando, generalmente,un fenotipo grave de la enfermedad. En cambio, lasmujeres heterocigotas habıan sido consideradas clasicamentemeras «portadoras» del trastorno. Sin embargo, investigacionesrecientes han puesto claramente de manifiesto que la mayorıade las mujeres heterocigotas presentan manifestaciones de laenfermedad, algunas de ellas tan graves como las de los varoneshemicigotos5–10. Esta heterogeneidad en la expresion clınica de laenfermedad en las mujeres se ha atribuido al fenomeno delyonizacion (inactivacion aleatoria de uno de los 2 cromosomas X)que sucede en etapas precoces de la embriogenesis, junto a otrosfactores que originan una actividad enzimatica parcheada dentrode cada organo o tejido, con expresion del gen mutado en unascelulas y el normal en otras11,12.
El diagnostico de este trastorno se realiza ante la existencia deun cuadro clınico tıpico, o bien el hallazgo de una hipertrofiamiocardica, insuficiencia renal o ictus de origen incierto, determi-nando la actividad enzimatica GLA (en plasma, leucocitos ocultivos celulares) y/o realizando el estudio molecular de su gen. Enlos varones homocigotos, la medicion de la actividad enzimaticarevelara que es baja o ausente. En cambio, muchas mujeresheterocigotas pueden presentar una actividad enzimatica dentrodel intervalo normal. El analisis del gen GLA confirma el diagnosticoen ambos sexos, siendo el unico metodo diagnostico definitivo enlas mujeres heterocigotas2.
Debido a la existencia de formas atıpicas u oligosintomaticas, laincidencia y prevalencia de la enfermedad siguen sin conocerse conexactitud. Publicaciones basadas en nuestro entorno geograficohabıan descrito una incidencia de un caso por cada 476.000habitantes (uno por cada 230.000 varones)13, aunque en algunasotras mas recientes se obtiene una prevalencia de hasta un caso porcada 3.100 recien nacidos varones, con predominio de lasmutaciones responsables de una forma de inicio tardıo de laenfermedad14.
Desde el ano 2001 se dispone de un tratamiento de sustitucionde la enzima deficitaria15,16 que ha demostrado ampliamente sueficacia17,18, con la perspectiva de poder mejorar la calidad de viday alargar la supervivencia de los que sufren dicho trastorno.Paralelamente, y con el fin de mejorar el conocimiento sobre la EF,
se han puesto en marcha registros de practica clınica habitualsobre la misma19,20, con el objetivo de paliar el inconveniente quela baja prevalencia de este trastorno puede suponer en laadquisicion de experiencia en su manejo clınico, ası comoposibilitar el diseno de estudios sobre una cohorte importantede pacientes con la misma.
El Fabry Outcome Survey (FOS) es un registro multicentrico depacientes con diagnostico confirmado de EF que reciben trata-miento de sustitucion enzimatica (TSE) con agalsidasa a (Repla-gal1, Shire Human Genetic Therapies, Cambridge, MA, EE.UU.) obien son candidatos al mismo. Con los datos de los pacientesinicialmente incluidos en dicho registro, en el ano 2004publicamos las caracterısticas clınicas de la serie mas grande depacientes espanoles diagnosticados de dicho padecimiento hastaese momento21. Con el paso de los anos, el numero de pacientesdiagnosticados de EF incluidos en dicho registro ha aumentado demanera progresiva y espectacular. Recientemente hemos publi-cado las caracterısticas clınicas de los pacientes espanoles adultosincluidos en el FOS hasta enero de 2009, comparandolos con elresto de paıses europeos22. En el presente trabajo nos preguntamossi el incremento en el conocimiento de esta enfermedad habıamodificado el perfil clınico o de diagnostico en los pacientesespanoles. Por dicho motivo, decidimos comparar las caracterıs-ticas basales de los nuevos pacientes incluidos en el registro con lasde nuestra primera cohorte. Ası mismo hemos evaluadola influencia que puede suponer el tener un miembro en la familiapreviamente diagnosticado de una EF, en el diagnostico yla gravedad de la misma del resto de miembros afectados.
Metodos
Pacientes
El registro FOS ha sido aprobado por los comites de etica detodos los centros participantes y los pacientes han otorgado suconsentimiento informado escrito para ser incluidos en el mismo.Todos los pacientes incluidos en el FOS tienen un diagnosticoenzimatico y/o genetico de certeza de la enfermedad. Los analisisde actividad enzimatica (plasma y/o leucocitos) y el analisismolecular del gen GLA han sido realizados en centros de referenciacon estandares contrastados de calidad (Instituto de BioquımicaClınica-Corporacion Sanitaria Hospital Clınic de Barcelona y Centrode Investigaciones en Biologıa Molecular-Nanomedicina delHospital Universitario Vall d’Hebron, Barcelona). En el momentode realizar el presente analisis (enero de 2009), existıan datos de92 pacientes espanoles procedentes de 29 hospitales. De ellos,68 habıan sido incluidos desde el ano 2003.
Recogida de datos
Los datos incluidos en el FOS se obtienen de las historias clınicasde los pacientes y proceden de las diferentes visitas clınicas de rutinaque realizan. Dichos datos comprenden informacion respecto a:aspectos demograficos, historia familiar, sıntomas y su evoluciontemporal, exploracion fısica, estudios analıticos generales yespecıficos de la enfermedad, exploraciones complementariasnecesarias en funcion de la evolucion, tratamientos concomitantesy aparicion de acontecimientos adversos. Todos los datos se remitenelectronicamente a una base de datos centralizada. Se consideran
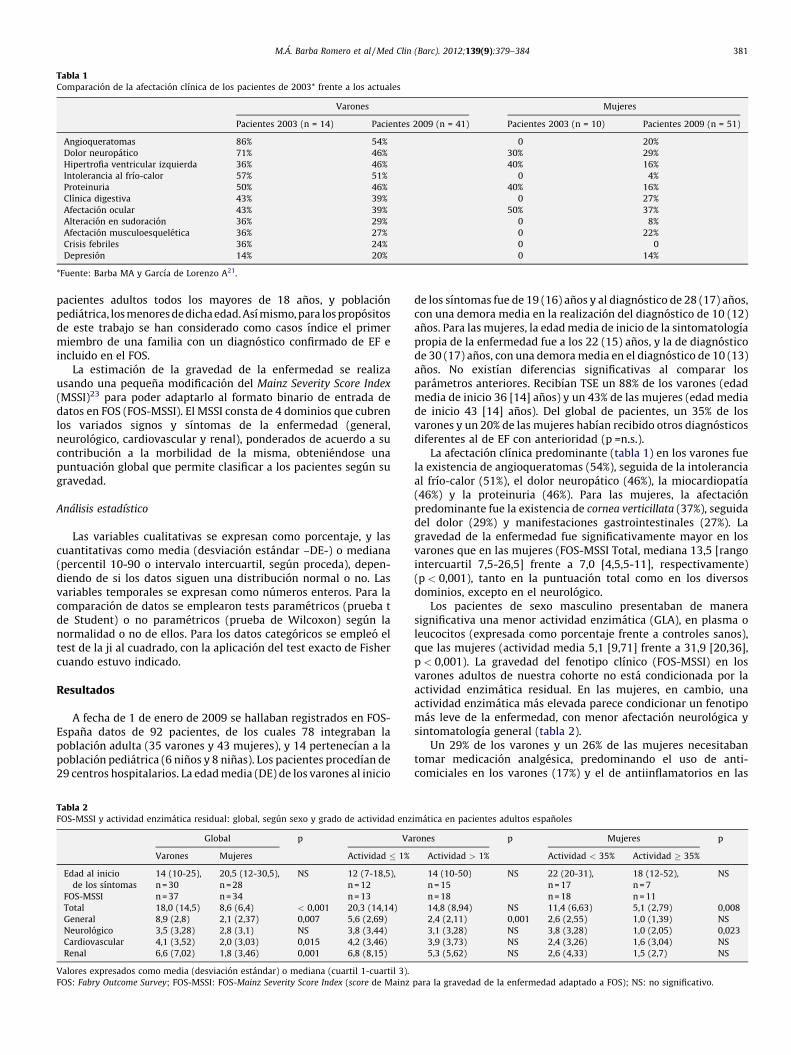
Tabla 1Comparacion de la afectacion clınica de los pacientes de 2003* frente a los actuales
Varones Mujeres
Pacientes 2003 (n = 14) Pacientes 2009 (n = 41) Pacientes 2003 (n = 10) Pacientes 2009 (n = 51)
Angioqueratomas 86% 54% 0 20%
Dolor neuropatico 71% 46% 30% 29%
Hipertrofia ventricular izquierda 36% 46% 40% 16%
Intolerancia al frıo-calor 57% 51% 0 4%
Proteinuria 50% 46% 40% 16%
Clınica digestiva 43% 39% 0 27%
Afectacion ocular 43% 39% 50% 37%
Alteracion en sudoracion 36% 29% 0 8%
Afectacion musculoesqueletica 36% 27% 0 22%
Crisis febriles 36% 24% 0 0
Depresion 14% 20% 0 14%
*Fuente: Barba MA y Garcıa de Lorenzo A21.
M.A. Barba Romero et al / Med Clin (Barc). 2012;139(9):379–384 381
pacientes adultos todos los mayores de 18 anos, y poblacionpediatrica, los menores de dicha edad. Ası mismo, para los propositosde este trabajo se han considerado como casos ındice el primermiembro de una familia con un diagnostico confirmado de EF eincluido en el FOS.
La estimacion de la gravedad de la enfermedad se realizausando una pequena modificacion del Mainz Severity Score Index
(MSSI)23 para poder adaptarlo al formato binario de entrada dedatos en FOS (FOS-MSSI). El MSSI consta de 4 dominios que cubrenlos variados signos y sıntomas de la enfermedad (general,neurologico, cardiovascular y renal), ponderados de acuerdo a sucontribucion a la morbilidad de la misma, obteniendose unapuntuacion global que permite clasificar a los pacientes segun sugravedad.
Analisis estadıstico
Las variables cualitativas se expresan como porcentaje, y lascuantitativas como media (desviacion estandar –DE-) o mediana(percentil 10-90 o intervalo intercuartil, segun proceda), depen-diendo de si los datos siguen una distribucion normal o no. Lasvariables temporales se expresan como numeros enteros. Para lacomparacion de datos se emplearon tests parametricos (prueba tde Student) o no parametricos (prueba de Wilcoxon) segun lanormalidad o no de ellos. Para los datos categoricos se empleo eltest de la ji al cuadrado, con la aplicacion del test exacto de Fishercuando estuvo indicado.
Resultados
A fecha de 1 de enero de 2009 se hallaban registrados en FOS-Espana datos de 92 pacientes, de los cuales 78 integraban lapoblacion adulta (35 varones y 43 mujeres), y 14 pertenecıan a lapoblacion pediatrica (6 ninos y 8 ninas). Los pacientes procedıan de29 centros hospitalarios. La edad media (DE) de los varones al inicio
Tabla 2FOS-MSSI y actividad enzimatica residual: global, segun sexo y grado de actividad enz
Global p Va
Varones Mujeres Actividad � 1%
Edad al inicio
de los sıntomas
14 (10-25),
n = 30
20,5 (12-30,5),
n = 28
NS 12 (7-18,5),
n = 12
FOS-MSSI n = 37 n = 34 n = 13
Total 18,0 (14,5) 8,6 (6,4) < 0,001 20,3 (14,14)
General 8,9 (2,8) 2,1 (2,37) 0,007 5,6 (2,69)
Neurologico 3,5 (3,28) 2,8 (3,1) NS 3,8 (3,44)
Cardiovascular 4,1 (3,52) 2,0 (3,03) 0,015 4,2 (3,46)
Renal 6,6 (7,02) 1,8 (3,46) 0,001 6,8 (8,15)
Valores expresados como media (desviacion estandar) o mediana (cuartil 1-cuartil 3).
FOS: Fabry Outcome Survey; FOS-MSSI: FOS-Mainz Severity Score Index (score de Mainz
de los sıntomas fue de 19 (16) anos y al diagnostico de 28 (17) anos,con una demora media en la realizacion del diagnostico de 10 (12)anos. Para las mujeres, la edad media de inicio de la sintomatologıapropia de la enfermedad fue a los 22 (15) anos, y la de diagnosticode 30 (17) anos, con una demora media en el diagnostico de 10 (13)anos. No existıan diferencias significativas al comparar losparametros anteriores. Recibıan TSE un 88% de los varones (edadmedia de inicio 36 [14] anos) y un 43% de las mujeres (edad mediade inicio 43 [14] anos). Del global de pacientes, un 35% de losvarones y un 20% de las mujeres habıan recibido otros diagnosticosdiferentes al de EF con anterioridad (p =n.s.).
La afectacion clınica predominante (tabla 1) en los varones fuela existencia de angioqueratomas (54%), seguida de la intoleranciaal frıo-calor (51%), el dolor neuropatico (46%), la miocardiopatıa(46%) y la proteinuria (46%). Para las mujeres, la afectacionpredominante fue la existencia de cornea verticillata (37%), seguidadel dolor (29%) y manifestaciones gastrointestinales (27%). Lagravedad de la enfermedad fue significativamente mayor en losvarones que en las mujeres (FOS-MSSI Total, mediana 13,5 [rangointercuartil 7,5-26,5] frente a 7,0 [4,5,5-11], respectivamente)(p < 0,001), tanto en la puntuacion total como en los diversosdominios, excepto en el neurologico.
Los pacientes de sexo masculino presentaban de manerasignificativa una menor actividad enzimatica (GLA), en plasma oleucocitos (expresada como porcentaje frente a controles sanos),que las mujeres (actividad media 5,1 [9,71] frente a 31,9 [20,36],p < 0,001). La gravedad del fenotipo clınico (FOS-MSSI) en losvarones adultos de nuestra cohorte no esta condicionada por laactividad enzimatica residual. En las mujeres, en cambio, unaactividad enzimatica mas elevada parece condicionar un fenotipomas leve de la enfermedad, con menor afectacion neurologica ysintomatologıa general (tabla 2).
Un 29% de los varones y un 26% de las mujeres necesitabantomar medicacion analgesica, predominando el uso de anti-comiciales en los varones (17%) y el de antiinflamatorios en las
imatica en pacientes adultos espanoles
rones p Mujeres p
Actividad > 1% Actividad < 35% Actividad � 35%
14 (10-50)
n = 15
NS 22 (20-31),
n = 17
18 (12-52),
n = 7
NS
n = 18 n = 18 n = 11
14,8 (8,94) NS 11,4 (6,63) 5,1 (2,79) 0,008
2,4 (2,11) 0,001 2,6 (2,55) 1,0 (1,39) NS
3,1 (3,28) NS 3,8 (3,28) 1,0 (2,05) 0,023
3,9 (3,73) NS 2,4 (3,26) 1,6 (3,04) NS
5,3 (5,62) NS 2,6 (4,33) 1,5 (2,7) NS
para la gravedad de la enfermedad adaptado a FOS); NS: no significativo.

Tabla 3Pacientes espanoles de 2003 frente a pacientes nuevos de los anos 2003-2008, segun sexo
Varones Mujeres
Pacientes 2003
(n = 14)
Pacientes nuevos
2003-2008 (n = 27)
p Pacientes 2003
(n = 10)
Pacientes nuevos
2003-2008 (n = 41)
p
Edad al inicio de los sıntomas 13,1 (6,89), n = 12 20,5 (17,49), n = 20 NS 20,6 (13,78), n = 7 22,1 (16,20), n = 24 NS
Edad al diagnostico 25,4 (11,75), n = 12 29,3 (19,13), n = 25 NS 32,9 (15,91), n = 8 29,9 (17,19), n = 37 NS
Demora entre el diagnostico
y el comienzo de los sıntomas (anos)
12,3 (8,71), n = 12 10,4 (13,10), n = 20 NS 9,4 (13,5), n = 7 10,7 (12,87), n = 23 NS
Edad al inicio del TSE 31,4 (11,39), n = 12 38 (15,14), n = 23 NS 40 (19,16), n = 3 41,6 (13,82), n = = 17 NS
Edad de entrada a FOS 34,6 (12,62) 34,3 (17,52) NS 34,5 (16,77) 34 (18,10) NS
Actividad GLA (% normal) 8,7 (10,39), n = 11 4,7 (11,24), n = 24 NS 34,0 (16,0), n = 8 30 (21,66), n = 29 NS
Diagnostico previo 50% (6/12) 35% (9/26) NS 12,5% (1/8) 23,5% (8/34) NS
FOS-MSSI n = 14 n = 25 n = 10 n = 31
Total 13,3 (9,5-26,5) 13,5 (7-23,5) NS 5 (1-9) 8 (5-12) NS
General 4,5 (1,5-5,5) 3,5 (0-6) NS 0 (0-1) 1 (0-6) NS
Neurologico 4,5 (1-6) 4 (0-6) NS 0 (0-4) 3 (0-6) NS
Cardiovascular 2 (0-6) 6 (0-7) NS 0 (0-1) 0 (0-6) NS
Renal 4 (0-8) 4 (0-8) NS 0 (0-4) 0 (0-4) NS
Valores expresados como media (desviacion estandar) o mediana (cuartil 1-cuartil 3) (porcentaje para diagnosticos previos).
FOS: Fabry Outcome Survey; FOS-MSSI: FOS-Mainz Severity Score Index (score de Mainz para la gravedad de la enfermedad adaptado a FOS); GLA: alfa-galactosidasa A; NS: no
significativo; TSE: tratamiento de sustitucion enzimatica.
M.A. Barba Romero et al / Med Clin (Barc). 2012;139(9):379–384382
mujeres (12%). Un 31% de los varones tomaban inhibidores de laenzima conversiva de la angiotensina (IECA) o antagonistas delreceptor de la angiotensina II (ARA II), frente a un 14% de lasmujeres (p = 0,03 para los IECA).
Al comparar los pacientes considerados casos ındice conaquellos en los que algun miembro de la familia ya habıa recibidoun diagnostico de EF, no se observaron en los varones diferenciassignificativas en la edad al diagnostico, demora en el mismo, recibirun diagnostico diferente al de EF, ni en la gravedad de laenfermedad (medida por el FOS-MSSI). En las mujeres tampocose apreciaron diferencias significativas en los parametros ante-riores, aunque existıa una tendencia en ellas a recibir menosdiagnosticos erroneos en los casos con antecedentes familiares(44,4% en los casos ındice frente a 14,8% en las demas, p = 0,06) yexhibir una menor gravedad (FOS-MSSI Total, mediana 11,3 [rangointercuartil 8-17] para los casos ındice frente a 6 [4,0-9,5] para lasdemas, p = 0,08). El inicio del TSE en los varones fue significati-vamente mas temprano en los casos ındice frente a los demas(edad media de 32 [13] anos frente a 41 [14] anos, p = 0,04), pero noası en las mujeres.
Al comparar los pacientes incluidos en FOS en nuestra primerapublicacion con los nuevos pacientes incluidos desde entonces(tabla 3), tampoco se encontraron diferencias estadısticamentesignificativas en la edad al inicio de los sıntomas, edad aldiagnostico, demora en el mismo, recibir un diagnostico diferenteal de la EF ni en la gravedad de la enfermedad (FOS-MSSI), alanalizar dichos datos en funcion del sexo. Tampoco se observarondiferencias en la edad en que se inicio la TSE.
Discusion
La EF pertenece al grupo de las denominadas enfermedadesraras o minoritarias, para las cuales los registros multicentricoscomo FOS aportan informacion extremadamente valiosa, que serıadifıcil de obtener con la practica clınica individual. El numero depacientes espanoles incluidos en FOS ha aumentado de manerasostenida desde nuestra primera publicacion en el ano 200421. Almismo tiempo, el numero de publicaciones en relacion con la EF haaumentado de manera significativa desde la introduccion del TSE.Ası, desde 1966 hasta el ano 2001 (35 anos) existen 692 entradasen PubMed (MeSH –Medical Subject Heading- Major Topic «Fabry
disease») para dicha enfermedad, en comparacion con las mas de1.100 referencias en el perıodo 2001 a 2011 (10 anos)24.
A pesar de ello, en los resultados que mostramos en este trabajosigue llamando la atencion que la demora media para llegar aldiagnostico de la enfermedad aun se encuentra alrededor de los 10anos para los pacientes de ambos sexos, ası como la elevadaproporcion de diagnosticos previos erroneos o distintos al de dichotrastorno. Aunque pudiera pensarse que en los pacientes inicial-mente incluidos en el registro existiera un teorico sesgo de gravedad,tambien los nuevos casos incluidos en FOS desde la publicacion de2004 presentan una afectacion, al ser incluidos en el registro,de similar gravedad que los de la cohorte inicial. Estos hallazgos, ennuestra opinion, constituyen un indicador de que todavıa esnecesario formar e informar a la comunidad cientıfica para que seincluya la EF en el diagnostico diferencial de otros procesos25.
Al igual que en nuestra publicacion previa, la mayorıa de losvarones presentan un fenotipo clasico de la enfermedad, conpredominio de la afectacion cutanea, neurologica, cardiaca y renal.Pero, a diferencia de nuestra primera publicacion, donde habıa mascasos registrados de pacientes del sexo masculino, en la actualidadcomienzan a predominar los del sexo femenino. Dicho hallazgo escongruente con el modo de transmision de esta enfermedad,aunque algo lejos todavıa de la teorica proporcion 2:1 que se esperaen las entidades con transmision ligada al cromosoma X. Por otrolado, en el momento actual es incuestionable que las mujeres noson meramente «portadoras» asintomaticas de la EF5–10. Ası, hastaun 40% de nuestras pacientes presentan sintomatologıa propia dela enfermedad. En ellas, la existencia de cornea verticillata y el dolorneuropatico siguen siendo los sıntomas mas prevalentes, siendomenos frecuente la existencia de miocardiopatıa. El mayor numerode mujeres registradas en la actualidad, con inclusion de formasmas leves o de evolucion mas lenta, explicarıa dichas diferencias.Por tanto, como ya observamos en nuestra cohorte inicial,podemos confirmar que las mujeres no son meras espectadorasen este trastorno de herencia ligada al cromosoma X.
Del mismo modo que en nuestra primera publicacion, noencontramos diferencias en la gravedad de la afectacion clınica enlos varones en relacion con la actividad enzimatica residual.En cambio, en las mujeres sı parece haber menor gravedadclınica en relacion a la existencia de una actividad enzimaticaresidual significativa. Este hallazgo, desde nuestro punto de vista,es extremadamente interesante, aunque contrasta con los cono-cimientos que existen al respecto en la actualidad. Puede estarcondicionado por el tamano de nuestra poblacion, ya que en otrapublicacion previa con mas pacientes no se obtuvo una correlacionentre la gravedad de la enfermedad y la actividad enzimatica

M.A. Barba Romero et al / Med Clin (Barc). 2012;139(9):379–384 383
medida en plasma y/o leucocitos7. Por otro lado, la actividadenzimatica medida en sangre se sabe que no es un buen reflejo dela actividad tisular en los diferentes tejidos y organos (determi-nante princeps de la diferente expresividad clınica del deficitenzimatico), que puede estar condicionada por el fenomeno delyonizacion11,26. Es mas, en una publicacion reciente con datosprocedentes de FOS se ha observado que puede haber diferenciasen la afectacion clınica de las mujeres europeas con EF segun lazona geografica de residencia, pero sin relacion con la actividadenzimatica en sangre27.
Debido a la elevada prevalencia del dolor neuropatico, tanto envarones como en mujeres, no es de extranar que mas de la cuartaparte de los pacientes tengan necesidad de tomar analgesicos demanera continuada. El hecho de que en nuestra primerapublicacion dichos farmacos fueran tomados por mas de la mitadde los pacientes puede facilmente explicarse por el predominio delos varones en la cohorte inicial, con mayor prevalencia en ellos dela neuropatıa dolorosa. En cambio, sı es llamativa, en nuestraopinion, la baja proporcion del uso de IECA-ARA II en nuestrospacientes, sobre todo si tenemos en cuenta la prevalencia en ellosde hipertrofia ventricular izquierda o proteinuria, susceptibles desu uso. Creemos que ese deberıa ser un aspecto a optimizar en sutratamiento, dada la amplia evidencia del beneficio de dichosfarmacos en las circunstancias comentadas28,29.
Un hecho a tener en cuenta, y que en nuestra opinion siguereflejando una carencia de formacion e informacion en esta y otrasenfermedades de baja prevalencia, es la practica ausencia dediferencias entre los denominados casos ındice de nuestra serie y elresto de casos diagnosticados de una EF. El inicio del TSE mastemprano en los varones ındice puede ser indicativo de quelos medicos que cuidan de estos pacientes son cada vez masconscientes de los beneficios que aporta un inicio temprano delmismo30, aunque dicho enfoque no se haya trasladado aun a lasmujeres. Aun ası, las sutiles diferencias halladas en las mujeres defamilias con casos ındice ya diagnosticados (menor proporcion dediagnosticos erroneos y la tendencia a tener una menor gravedad aldiagnostico) son, en nuestra opinion, un buen exponente de lanecesidad de insistir en la realizacion de estudios familiarescompletos tras el diagnostico de un caso ındice de EF31, explicandoclaramente cual es la realidad de la afectacion clınica de lasmujeres. Al mismo tiempo, creemos que deberıa potenciarse elpapel de las familias de los pacientes como vectores activos en labusqueda del diagnostico entre los miembros de las mismas.
En la mejora de los aspectos anteriormente comentados,consideramos que las guıas o documentos de consenso basadosen nuestra realidad asistencial32 suponen una inestimable ayudapara todos aquellos profesionales de la salud que pueden tenercontacto con esta patologıa.
En resumen, presentamos la extension de nuestra serie depacientes espanoles diagnosticados de EF, la cual sigue siendola mas numerosa publicada hasta la fecha. Ello es posible gracias ala existencia del FOS, un registro de practica clınica habitual de estaenfermedad, de indudable valor y utilidad para ahondar en elconocimiento de la misma. La EF es una enfermedad multisiste-mica, mimetizadora de otros procesos, que origina una morbi-mortalidad significativa. Debemos aun seguir trabajando paraexpandir el conocimiento de la misma, posibilitando el beneficio deun diagnostico precoz que limite el sufrimiento de los pacientes ysus familias, y al mismo tiempo les permita beneficiarse deltratamiento especıfico de que disponemos en la actualidad.
Financiacion
La base de datos del FOS se halla bajo el control independientedel Comite Internacional del Fabry Outcome Survey. La recogidade datos y su analisis estadıstico estan esponsorizados por Shire
Human Genetic Therapies (HGT), Cambridge, MA, EE.UU. Elesponsor no tuvo ningun papel en la interpretacion de los datosde este trabajo ni en la redaccion del manuscrito. Shire HGTfinancio la traduccion del resumen al ingles y la asistencia editorialrealizada por ReSearch Pharmaceutical Services.
Conflicto de intereses
El Dr. Barba Romero ha recibido honorarios por conferencias yayudas para viajes por asistencia a reuniones cientıficas de ShireHGT y Genzyme. El Dr. Rivera Gallego ha recibido honorarios porconferencias de Shire HGT y ayudas para viajes de Shire HGT yGenzyme. El Dr. Pintos Morell ha recibido honorarios y/o ayudaspara viajes de Shire HGT, Genzyme y BioMarin.
Anexo 1
Los siguientes investigadores, miembros del Grupo Espanol deestudio del FOS, han aportado datos de sus pacientes para elpresente trabajo:
Hospital Universitario de Albacete: M.A. Barba; HospitalGeneral de Alicante: A. Franco, V. Climent, M. Arenas; HospitalGeneral de Asturias: J. Herrera; Hospital Central de Asturias: E.Gomez; Hospital Universitario Germans Trias i Pujol (Badalona,Barcelona): G. Pintos, M. Ibernon, J. Ara; Fundacion Puigvert(Barcelona): R. Torra; Hospital Clınic i Provincial de Barcelona: V.Torregrosa; Hospital Puerta del Mar (Cadiz): J. Gonzalez; HospitalUniversitario de Elche (Alicante): A. Mora; Hospital de Elda(Alicante): V. Valverde, J. Martin, E. del Bosque; Hospital Sant Joande Deu (Esplugues de Llobregat, Barcelona): M. Pineda; HospitalClınico de San Cecilio (Granada): M.D. Prados; Hospital de Bellvitge(Hospitalet de Llobregat, Barcelona): J. Torras; Hospital JuanRamon Jimenez (Huelva): I. Martin; Hospital San Agustın (Linares,Jaen): S. Hernandez; Hospital Universitario La Paz (Madrid): F.J.Barbado, M. Lopez; Complejo Hospitalario de Ourense: J.M. deToro; Hospital Son Dureta (Palma de Mallorca): T. Bosch; Hospitaldel Bierzo (Ponferrada, Leon): J. Paniagua; Hospital de Ronda(Malaga): L. Tamargo; Hospital Universitario de Santiago (Santiagode Compostela, A Coruna): V. Fernandez; Hospital de Torrecarde-nas (Almerıa): M.D. del Pino; Hospital General de Valencia: A.Perez, M.I. Febrer; Hospital Universitario de Vigo (Pontevedra): A.Rivera; Hospital de Vinaroz (Castellon de la Plana): F. Cabades.
Bibliografıa
1. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymaticdefect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med.1967;276:1163–7.
2. Rodrıguez-Palmero Seuma A, Esteban Oliva D, Pintos Morell G. Enfermedad deFabry. En: Sanjurjo P, Baldellou A, editores. Diagnostico y tratamiento de lasenfermedades metabolicas hereditarias. 3.a ed, Madrid: Ergon; 2010. p. 847–58.
3. Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, et al. Natural courseof Fabry disease: changing pattern of causes of death in FOS - Fabry OutcomeSurvey. J Med Genet. 2009;46:548–52.
4. Konreich R, Bishop DF, Desnick RJ. The gene encoding alpha-galactosidase A andgene rearrangements causing Fabry disease. Trans Assoc Am Physicians.1989;102:30–43.
5. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical man-ifestations and impact of disease in a cohort of 60 obligate carrier females. J MedGenet. 2001;38:769–75.
6. Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, et al.Anderson-Fabry disease: clinical manifestations of disease in female hetero-zygotes. J Inherit Metab Dis. 2001;24:715–24.
7. Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M,et al. Natural History of Fabry disease in females in the Fabry Outcome Survey. JMed Genet. 2006;43:347–52.
8. Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are notjust carriers, but have a significant burden of disease and impaired quality oflife. Genet Med. 2007;9:34–45.
9. Street NJ, Yi MS, Bailey LA, Hopkin RJ. Comparison of health-related quality oflife between heterozygous women with Fabry disease, a healthy control popu-lation and patients with other chronic disease. Genet Med. 2006;8:346–53.

M.A. Barba Romero et al / Med Clin (Barc). 2012;139(9):379–384384
10. Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U,et al. Females with Fabry disease frequently have major organ involvement:lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–28.
11. Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.).Nature. 1961;190:372–3.
12. Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT, et al. Inheritanceof most X-linked traits is not dominant or recessive, just X-linked. Am J MedGenet A. 2004;129A:136–43.
13. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al.The frequency of lysosomal storage diseases in The Netherlands. Hum Gen.1999;105:151–6.
14. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Saturaba H, et al. Highincidence of later-onset Fabry disease revealed by newborn screening. Am JHum Genet. 2006;79:31–40.
15. Schiffmann R, Kopp JB, Austin 3rd HA, Sabnis S, Moore DF, Weibel T, et al.Enzyme replacement therapy in Fabry disease: a randomized controlled trial.JAMA. 2001;285:2743–9.
16. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety andefficacy of recombinant human a-galactosidase A-replacement therapy inFabry’s disease. N Engl J Med. 2001;345:9–16.
17. Lidove O, West ML, Pintos-Morell G, Reisin R, Nicholls K, Figuera LE, et al. Effectsof enzyme replacement therapy in Fabry disease–a comprehensive review ofthe medical literature. Genet Med. 2010;12:668–79.
18. Rivera Gallego A, Lopez Rodrıguez M, Barbado Hernandez FJ, Barba Romero MA,Garcıa de Lorenzo y Mateos A, Pintos Morell G, en representacion del GrupoEspanol de Estudio del Fabry Outcome Survey. Enfermedad de Fabry en Espana:primer analisis de la respuesta al tratamiento de sustitucion enzimatica. MedClin (Barc). 2006;127:481–4.
19. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al.Fabry disease defined: baseline clinical manifestations of 366 patients in theFabry Outcome Survey. Eur J Clin Invest. 2004;34:236–42.
20. Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabrydisease: baseline medical characteristics of a cohort of 1756 males and femalesin the Fabry Registry. J Inherit Metab Dis. 2007;30:184–92.
21. Barba MA, Garcıa de Lorenzo A, en representacion del Grupo Espanol de Estudiode FOS. Enfermedad de Fabry en Espana. Analisis de 24 casos. Med Clin (Barc).2004;123:57–60.
22. Barba-Romero MA, Rivera Gallego A, Pintos Morell G, on behalf of the SpanishFOS study group. Fabry disease in Spain. Description of Spanish patients and acomparison with other European countries using data from the Fabry OutcomeSurvey (FOS). Int J Clin Pract. 2011;65:903–10.
23. Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E, et al.The Mainz Severity Score Index: a new instrument for quantifying the Ander-son-Fabry disease phenotype, and the response of patients to enzyme replace-ment therapy. Clin Genet. 2004;65:299–307.
24. PubMed [consultado el 20 Nov 2011]. Disponible en: http://www.ncbi.nlm.nih.gov/sites/pubmed
25. Barbado Hernandez FJ. La enfermedad de Fabry como modelo de enfermedadmultisistemica. Rev Clin Esp. 2006;206 Supl 3:1–4.
26. Hughes DA. Early therapeutic intervention in females with Fabry disease. ActaPaediatrica. 2008;97:41–7.
27. Barba-Romero MA, Deegan P, Giugliani R, Hughes D. Does geographical locationinfluence de phenotype of Fabry disease in women in Europe? Clin Genet.2010;77:131–40.
28. Remuzzi G, Ruggenenti P, Perico N. Chronic renal diseases: renoprotectivebenefits of renin-angiotensin system inhibition. Ann Intern Med. 2002;136:604–10.
29. Kjeldsen SE, Julius S. Hypertension mega-trials with cardiovascular end points:effect of angiotensin-converting enzyme inhibitors and angiotensin receptorblockers. Am Heart J. 2004;148:747–54.
30. Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M, onbehalf of the FOS Investigators. Fabry disease in children and response to enzymereplacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2011Apr 1. doi:10.1111/j.1399-0004.2011.01671.x. [Epub ahead of print].
31. Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history.J Genet Couns. 2008;17:79–83.
32. Garcıa de Lorenzo A. Consenso para el estudio y tratamiento de la enfermedadde Fabry. Fundacion GETER. Med Clin (Barc). 2011;137:178–83.


















