
DINÀMICA MOLECULAR EN ESTATS ELECTRÒNICS...
87
DINÀMICA MOLECULAR EN ESTATS ELECTRÒNICS EXCITATS EXCITATS
Transcript of DINÀMICA MOLECULAR EN ESTATS ELECTRÒNICS...

DINÀMICA MOLECULAR EN ESTATS ELECTRÒNICS
EXCITATSEXCITATS

1. Metodología1.1 Cálculos Electrónicos- Uso de programas “standard” (GAUSSIAN)
- Nivel de cálculo:
- Cálculos semiempíricos
- Cálculos ab initio (HF y post-HF)
- DFT (B3LYP)
- Métodos Híbridos: QM/MM; QM/QM (ONIOM)
E t d it d CIS CASSCF CASPT2 TDDFT- Estados excitados: CIS; CASSCF; CASPT2; TDDFT
1.2 Cálculos Nucleares- Programas de creación propia (“Software”)

É1.2.1 MÉTODOS DE “PUNTOS”(O como ver el mundo a través de agujeros)

G W B hAnsar G W BushG.W. BushAnsar G.W. Bush
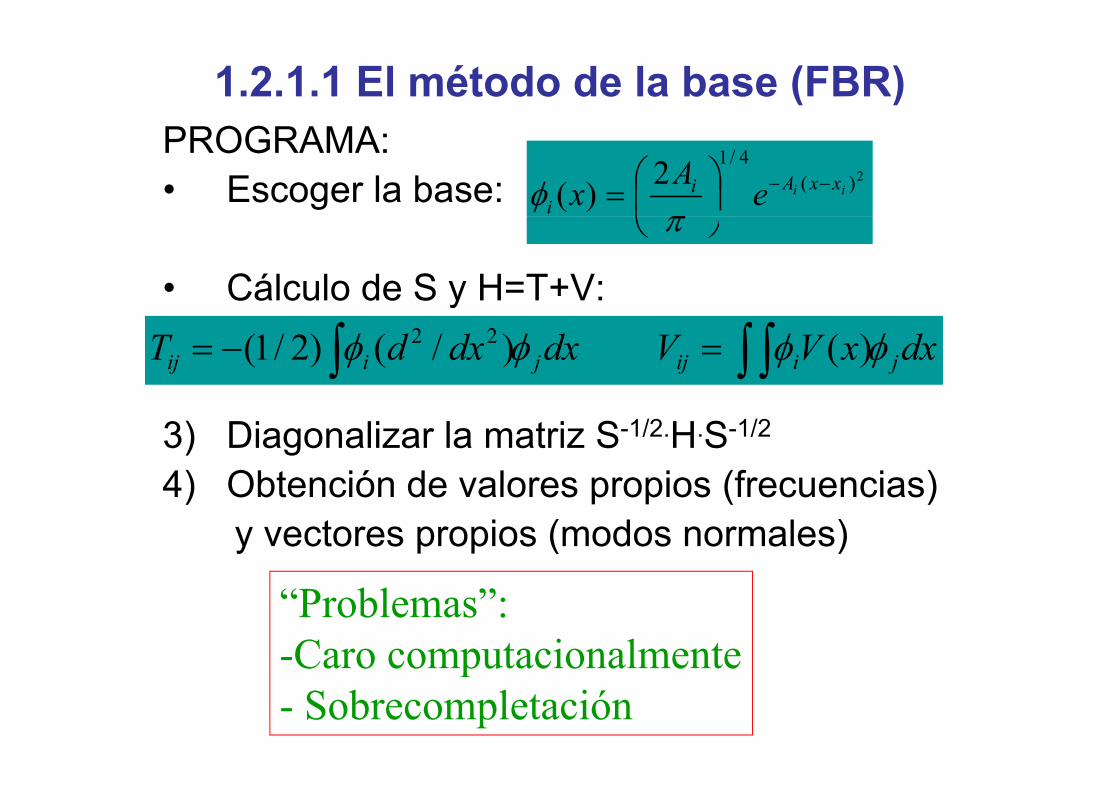
1.2.1.1 El método de la base (FBR)PROGRAMAPROGRAMA:• Escoger la base: 2)(
4/12)( ii xxAii eAx −−⎟
⎠⎞
⎜⎝⎛=φ
• Cálculo de S y H=T+V:
⎠⎝ π
ydxxVVdxdxdT jiijjiij ∫ ∫∫ =−= φφφφ )()/()2/1( 22
3) Diagonalizar la matriz S-1/2.H.S-1/2
4) Obtención de valores propios (frecuencias)4) Obtención de valores propios (frecuencias)y vectores propios (modos normales)
“Problemas”:-Caro computacionalmenteCaro computacionalmente- Sobrecompletación
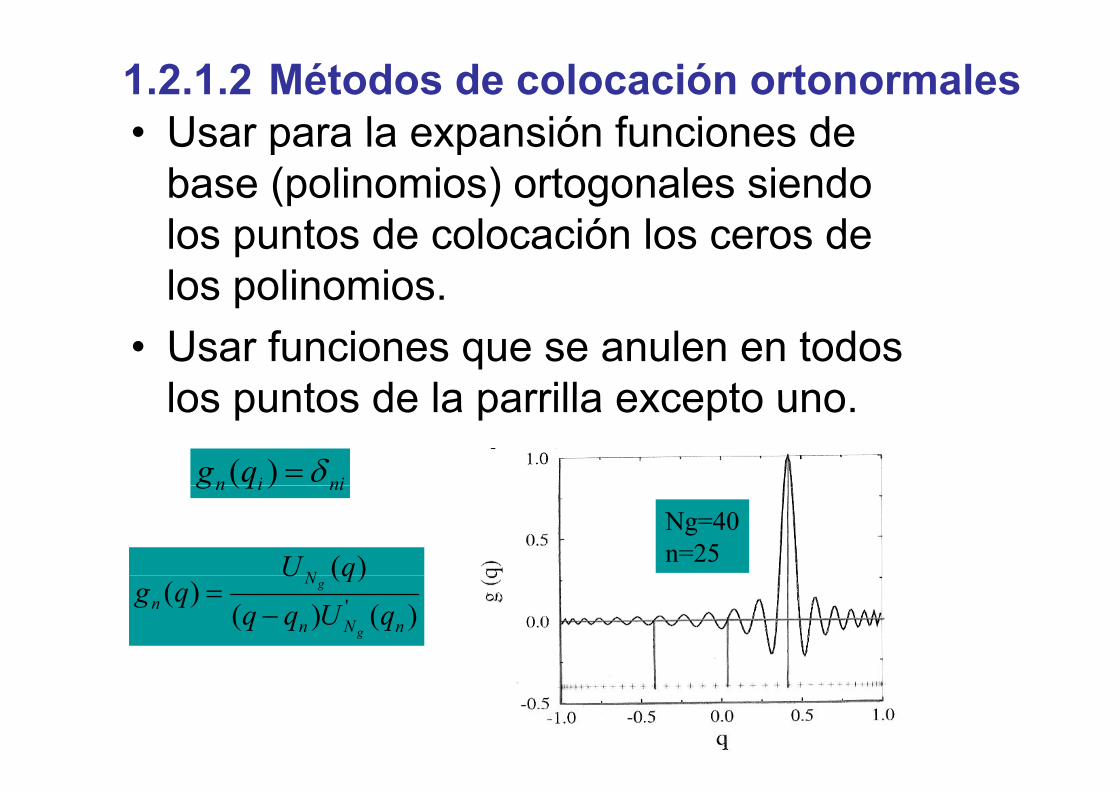
Usar para la expansión funciones de1.2.1.2 Métodos de colocación ortonormales• Usar para la expansión funciones de
base (polinomios) ortogonales siendo los puntos de colocación los ceros de los polinomios.p
• Usar funciones que se anulen en todos los puntos de la parrilla excepto unolos puntos de la parrilla excepto uno.
niin qg δ=)(
)(N qUNg=40n=25
niin qg )(
)()(
)()( '
nNn
Nn qUqq
qUqg
g
g
−=
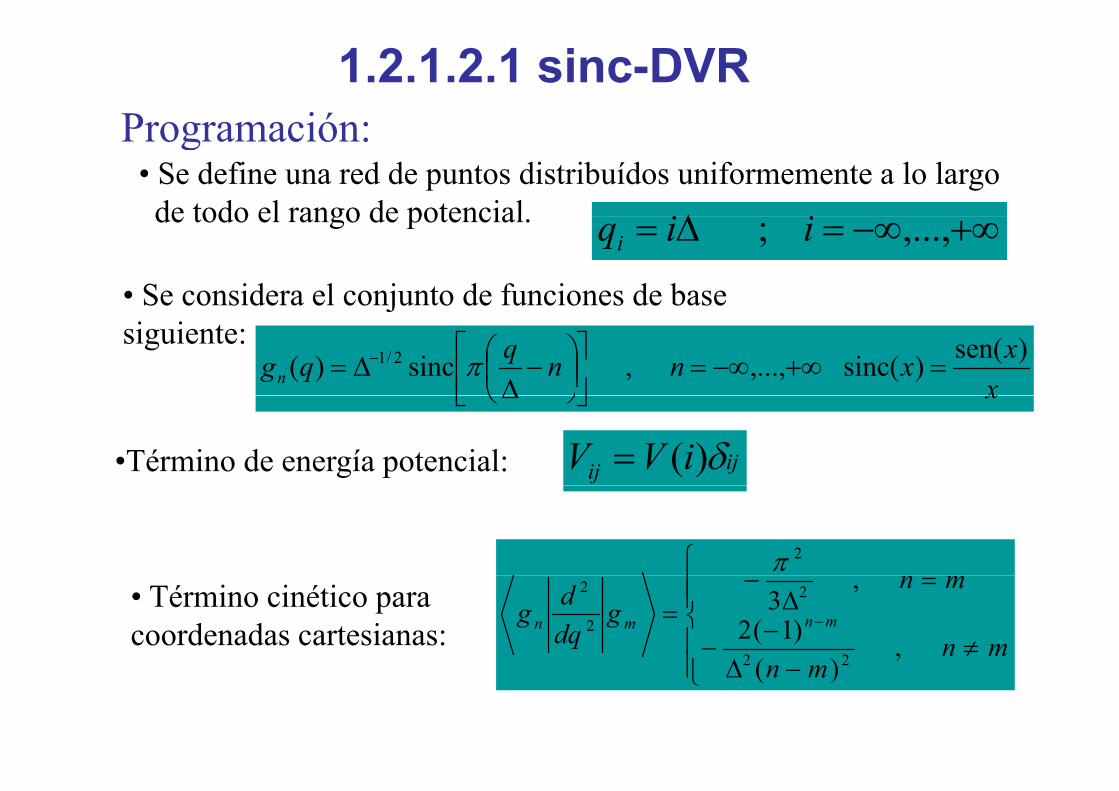
1.2.1.2.1 sinc-DVR Programación:
• Se define una red de puntos distribuídos uniformemente a lo largode todo el rango de potencial Δ ii
Programación:
de todo el rango de potencial.
• Se considera el conjunto de funciones de base
+∞−∞=Δ= ,...,; iiqi
jsiguiente:
xxxnnqqgn)sen()sinc(,...,,sinc)( 2/1 =+∞−∞=⎥
⎦
⎤⎢⎣
⎡⎟⎠⎞
⎜⎝⎛ −Δ
Δ= − πx⎦⎣ ⎠⎝ Δ
•Término de energía potencial: ijij iVV δ)(=
⎪⎧ 2π
• Término cinético para coordenadas cartesianas:
⎪⎪⎩
⎪⎪⎨
≠Δ
−−
=Δ
−= −
mn
mng
dqdg mnmn
,)(
)1(2
,3
22
2
2
2
⎪⎩ −Δ mn )( 22

sinc-DVR (II)• Para coordenades no cartesianas (radiales, angulares,...) las expresiones
anteriores deben ser recalculadas y programadas convenientemente. [D T Colbert y W H Miller JCP 96 (1992) 1982 1991][D.T. Colbert y W.H. Miller, JCP,96 (1992) 1982-1991]
• En la práctica los límites no seran nunca infinito lo que equivale a decirque la función de onda será nula fuera del rango de definición del q gpotencial.
Ventajas del método DVR (sobre el FBR):Ventajas del método DVR (sobre el FBR):• Mucho más barato computacionalmente.• Necesita de muy pocos parámetros• No presenta el problema de la “sobrecompletación”Inconvenientes...
i i i l• No es estrictamente variacional• Puede precisar de cierta algebra previa si se usa una
coordenada “exòtica”coordenada exòtica• Estricta colocación de los puntos (filtraje)

1.2.1.2.2 El método de Fourier (FFT)- Es también un método de colocación ortonormal donde los puntos están
equiespaciados que se basa en la transformada de Fourier:
gggLkqi
k qNLNNkeqg /2 2/,...,0),...,12/(,)( π Δ=−−==
k
Lkqikeaq /2)( π∑=Ψ
Lkqijk
k
jeqa /2)(1 π−∑Ψ=j
jg
k eqN
a )(∑Ψ
• Se trata de un método especialmente útil en dinámica cuántica• Se trata de un método especialmente útil en dinámica cuántica• Puede ser muy eficiente computacionalmente si se implementa el
algoritmo Fast Fourier Transform (FFT) que escala la transformada
39de N puntos en el orden O(Nlog 2N). ¡Esto representa un factor 100 veces más rápido para N=1000 !

1 2 3 Más allá del mundo 1-D1.2.3 Más allá del mundo 1-D
L li ió d l fó l d TODOS l• La generalización de las fórmulas de TODOS los métodos de colocación a 2,3,...n dimensions es trivial.
• El problema es computacional: un cálculo que precisaEl problema es computacional: un cálculo que precisa 20 puntos en una dimensión precisarà 400 en dos, 8000 en tres, ...
• Esto lleva a la necesidad de explorar otros métodos (no necesariamente cuánticos puros) para estudiar problemas químicos en toda su dimensionalidad realproblemas químicos en toda su dimensionalidad real.
• Usando métodos de diagonalización parcial se han llegado a tratar problemas DVR que involucran matrices g p qde más de 500000 x 500000...

Equació de Schrödinger dependent del temps :• Els mètodes estàndard de resolució de l’equació de Schrödingerq g
dependent del temps es basen en expandir la funció d’ona del sistemaen una base de funcions independents del temps, i aplicar el principivariacional per a deduir les equacions de moviment:
Principi variacionalde Dirac-Frenkel
Sistema de N equacionsq(N = N1 x … x Nf)

Dinàmica Molecular Quàntica. El Mètode MCTDH :
• El mètode MCTDH és un algorisme eficient per a resoldre l’equació deSchrödinger dependent del temps de forma aproximada.
• La funció d’onda del sistema s’expandeix com una suma de productes defuncions monopartícula de base dependents del temps:
Principi varacional de Dirac FrenkelPrincipi varacional de Dirac-Frenkel
2n equacions acoblades
∏=
=f
kknn
12n « N

κnI unes poques fòrmules…
∑=
=κ
κκκ ϕϕj
jjP1
)()()(
j 1
Single hole functions:
∑ ∏ΨΨκ κκκ A )'()()( ∑ ∏
≠
=Ψ=Ψκκ
κκ
κκ ϕϕ κJ jJlll
A'
)(´
)()(
)( )()()( ˆ κκκljjl
HH ΨΨ=
∑=ΨΨ=κκκκ
κκρJ JJljjl AA*)()()( ∑J JJljjl
lj
- U Manthe H -D Meyer L S Cederbaum J Chem Phys 97 (1992) 3199- U. Manthe, H. -D. Meyer, L.S. Cederbaum, J. Chem. Phys. 97 (1992) 3199
- M.H. Beck, H. -D. Meyer, Z. Phys. D 42 (1997) 113

2. Aplicaciones2.1 Transferencia protónica en estados electrónicos
excitados2.1.Sistemas estacionarios (doble pozo simétrico)
excitados
BAΔ1 - Δ0}
νAB
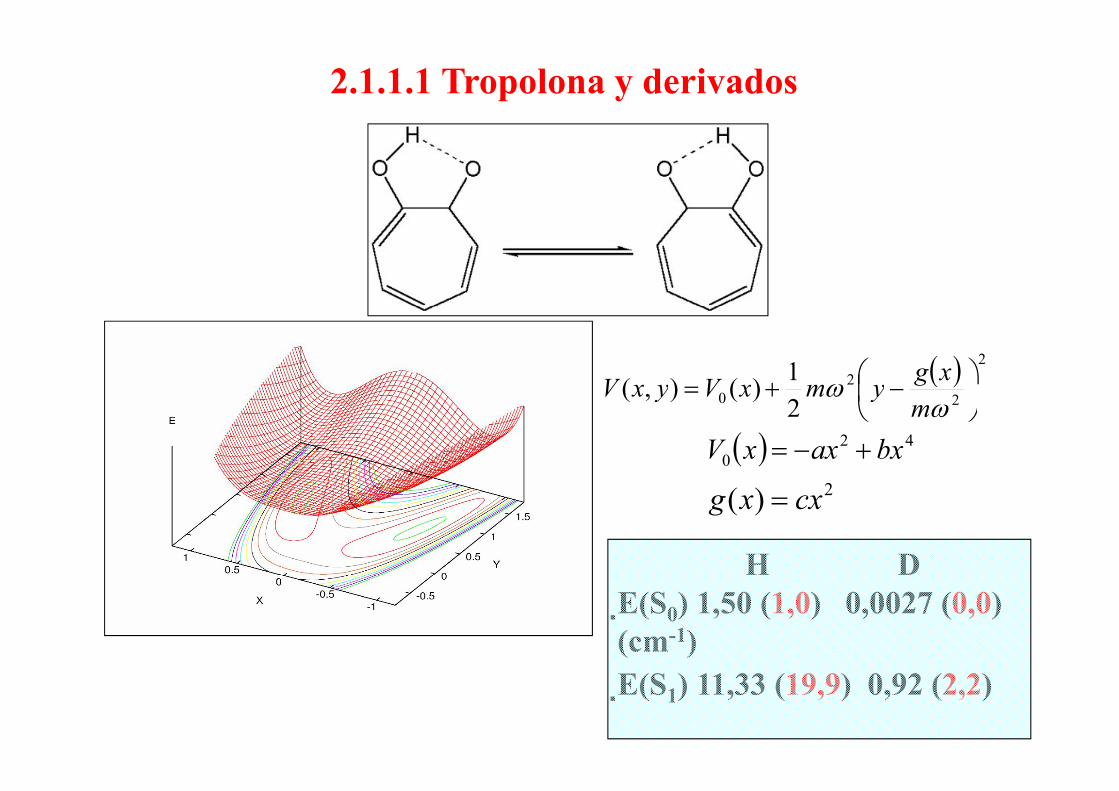
2.1.1.1 Tropolona y derivados
( ) 2
22
01)(),( ⎟
⎠⎞
⎜⎝⎛ −+= ω xgymxVyxV 20 2
)(),( ⎟⎠
⎜⎝
+ω
ωm
ymxVyxV
( ) 420 bxaxxV +−=
22)( cxxg =
H DE(S0) 1,50 (1,0) 0,0027 (0,0)(cm-1)E(S1) 11,33 (19,9) 0,92 (2,2)

Tropolonas substituídas:5-hidroxitropolona y 5-aminotropolonap y p
ABC D
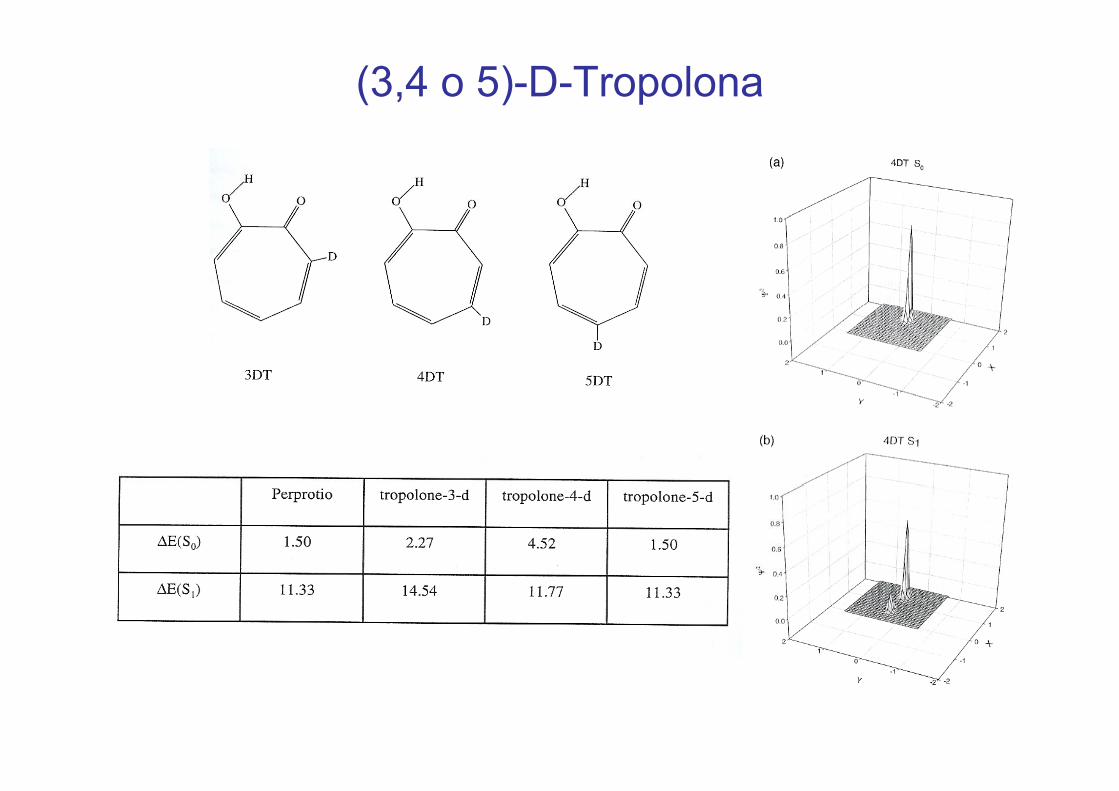
(3,4 o 5)-D-Tropolona
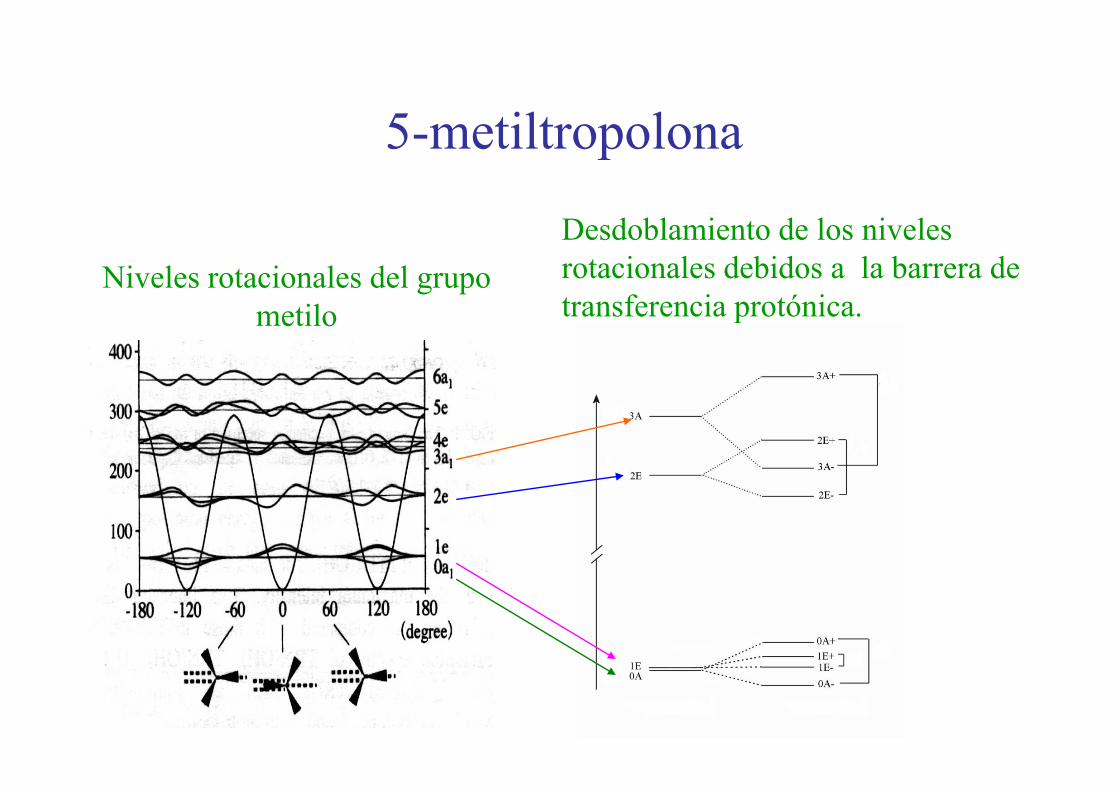
5 metiltropolona5-metiltropolona
Desdoblamiento de los niveles rotacionales debidos a la barrera de transferencia protónica
Niveles rotacionales del grupo il transferencia protónica.metilo

Superficie de energía potencial analíticap g p( ) ( ) ( )θθ ;,,, xVyxVyxVtot +=
( ) 2
22
0 21)(),( ⎟
⎠⎞
⎜⎝⎛ −+=
ωω
mxgymxVyxV
( ) ( ) ( ) ⎟⎟⎠
⎞⎜⎜⎝
⎛ +⎥⎦⎤
⎢⎣⎡ −+−=
0
063
26cos1
23cos1
2;
xxxVVxV θθθ
⎠⎝
( ) ( )
( ) ⎟⎞
⎜⎛ −⎤⎡ −
⎟⎟⎠
⎞⎜⎜⎝
⎛ −⎥⎦⎤
⎢⎣⎡ −++−
20
263
0
063
61
26cos1
23cos1
2
xxVVV
xxxVV
t θ
θθ
( ) ⎟⎟⎠
⎞⎜⎜⎝
⎛⎥⎦⎤
⎢⎣⎡ −−+ 2
0
063 6cos122 x
xxVVV ts θ
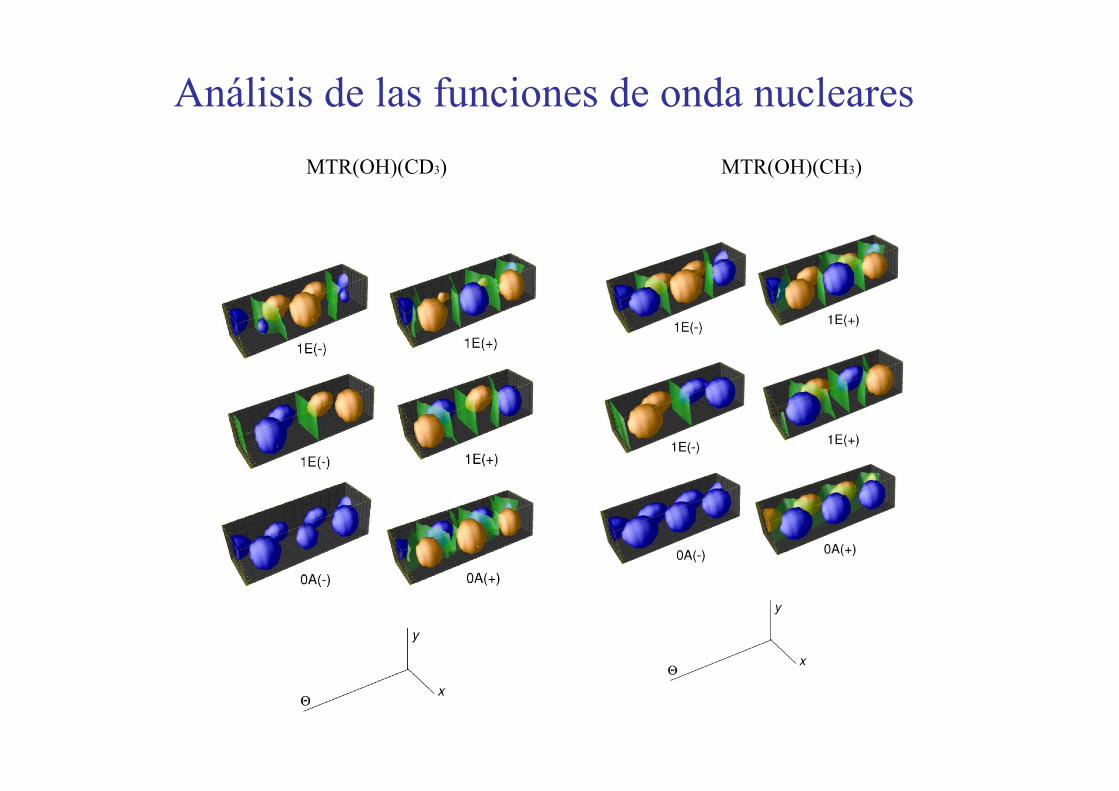
Análisis de las funciones de onda nuclearesMTR(OH)(CH3)MTR(OH)(CD3)
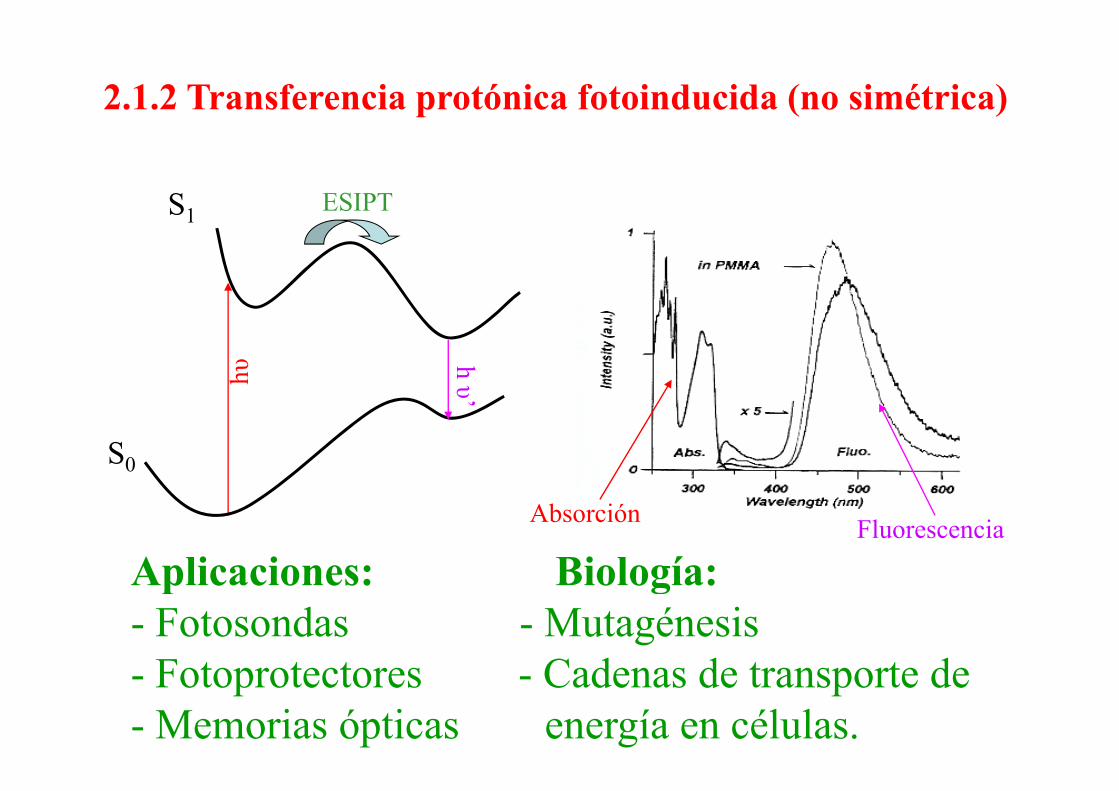
2.1.2 Transferencia protónica fotoinducida (no simétrica)
ESIPTS11hυ
h υυ’
S0
Absorción Fluorescencia
Aplicaciones: Biología:Aplicaciones: Biología:- Fotosondas - Mutagénesis
F t t t C d d t t d- Fotoprotectores - Cadenas de transporte de- Memorias ópticas energía en células.
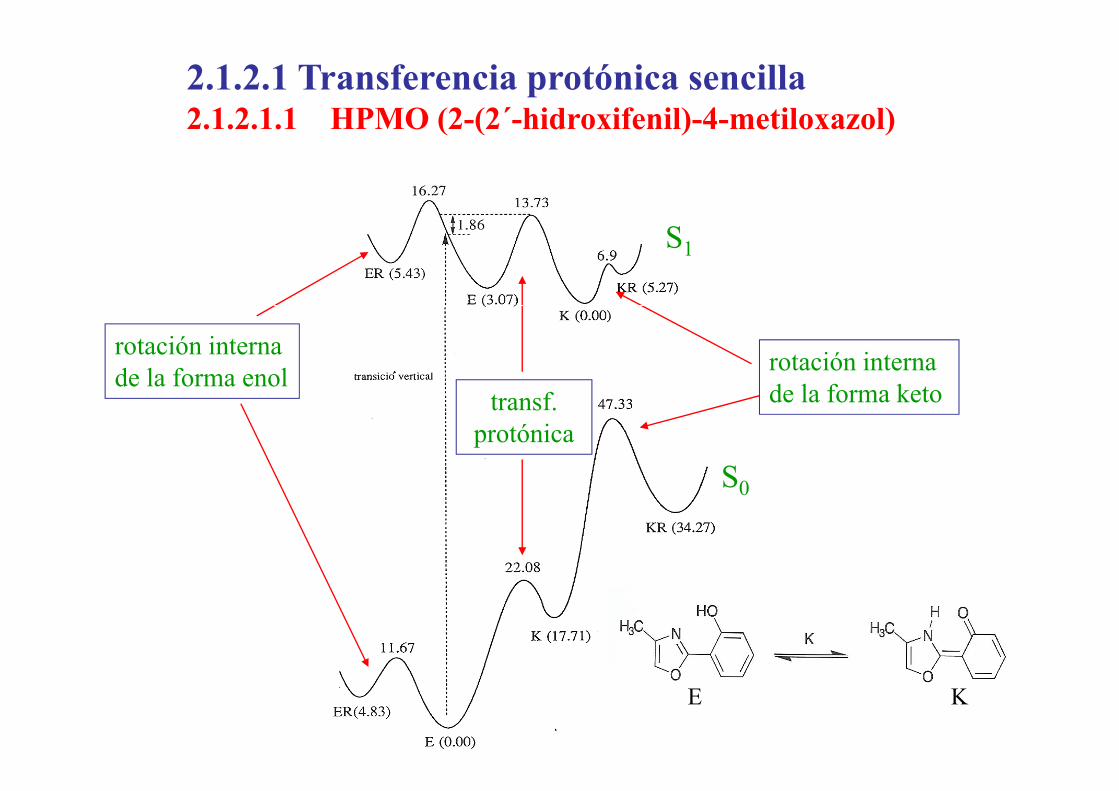
2.1.2.1 Transferencia protónica sencilla2.1.2.1.1 HPMO (2-(2´-hidroxifenil)-4-metiloxazol)2.1.2.1.1 HPMO (2 (2 hidroxifenil) 4 metiloxazol)
S1
rotación internade la forma keto
rotación interna de la forma enol
t f
S0
de la forma ketotransf.protónica
S0
E K
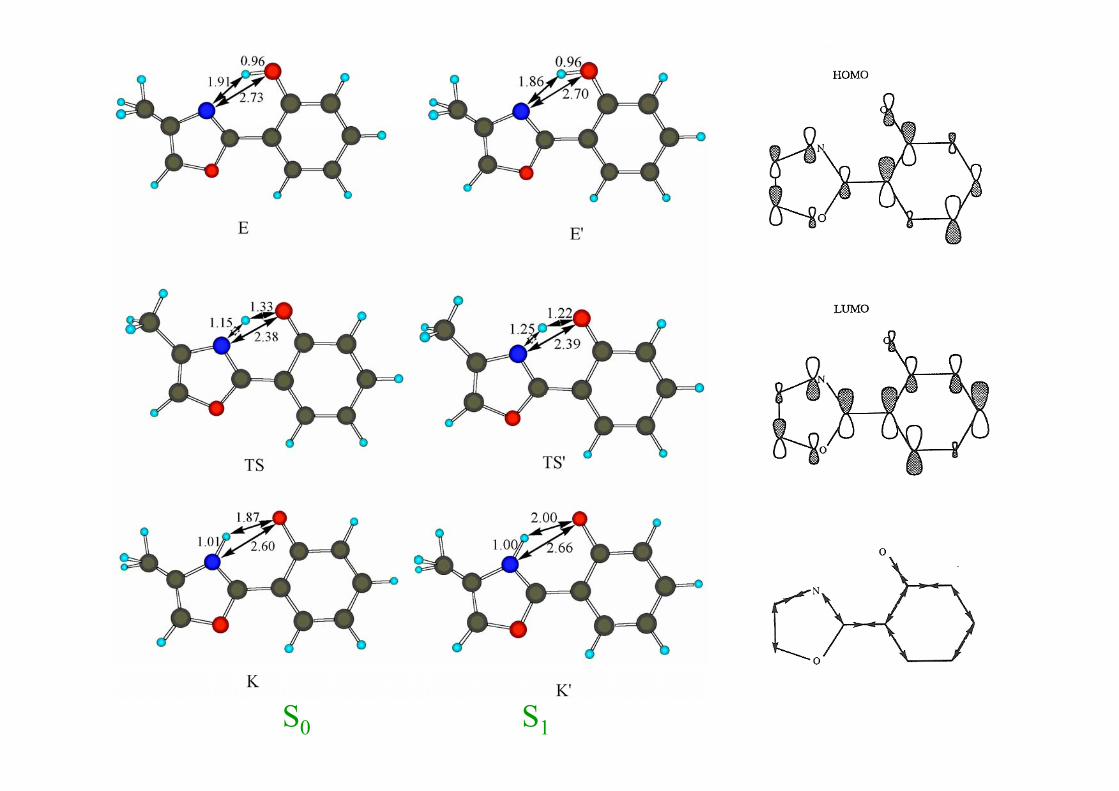
S1S0

Reacción en el interior de la β ciclodextrinaβ-ciclodextrina
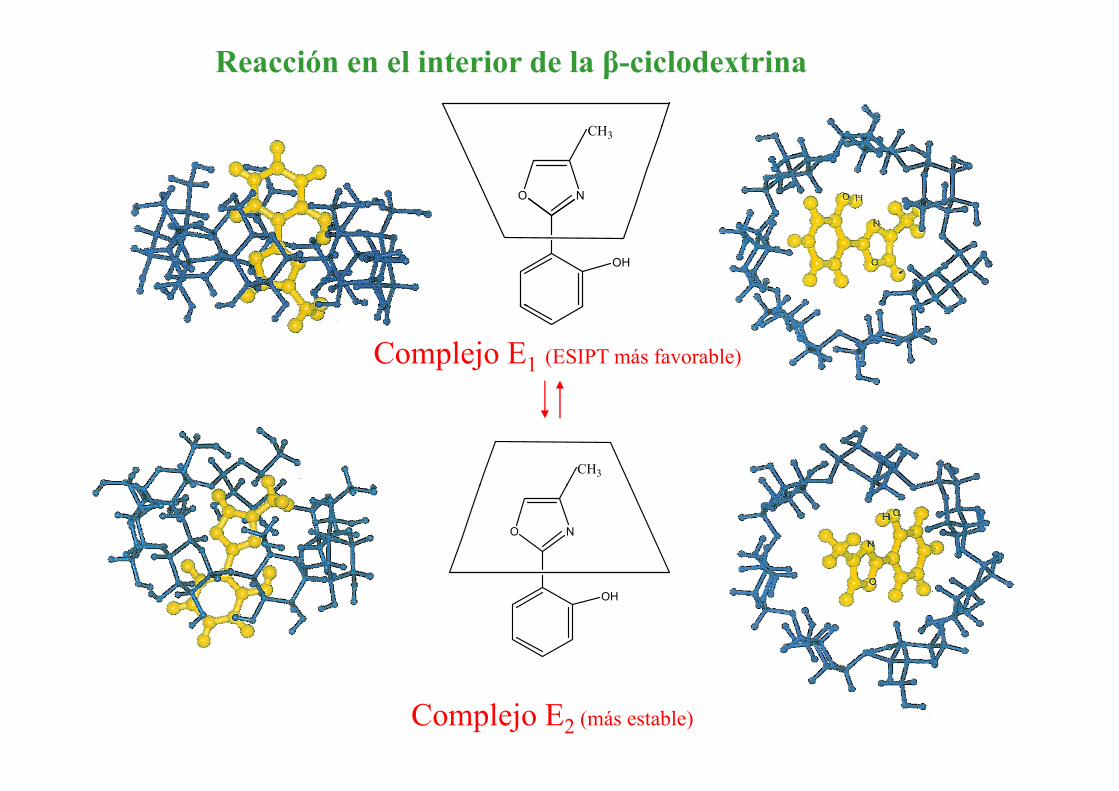
Reacción en el interior de la β-ciclodextrina
CH
O N
CH3
OH
Complejo E1 (ESIPT más favorable)
CH3
O N
OH
Complejo E2 (más estable)
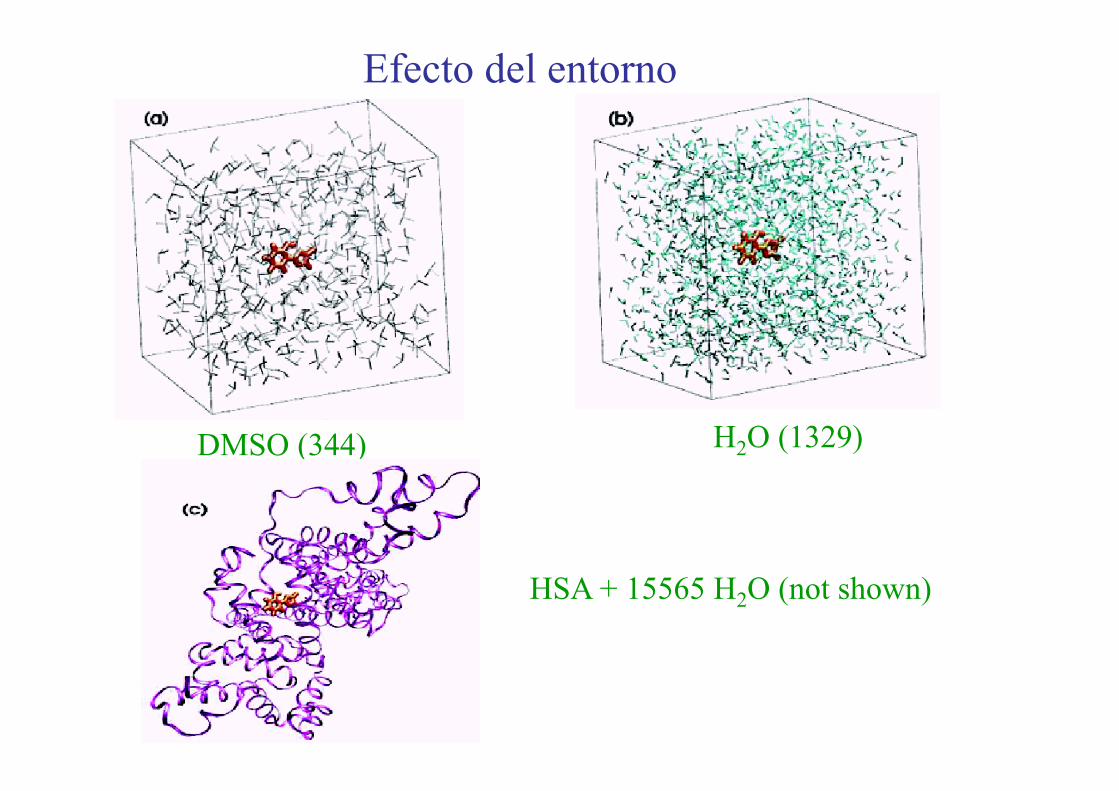
Efecto del entorno
H2O (1329)DMSO (344)
HSA + 15565 H2O (not shown)
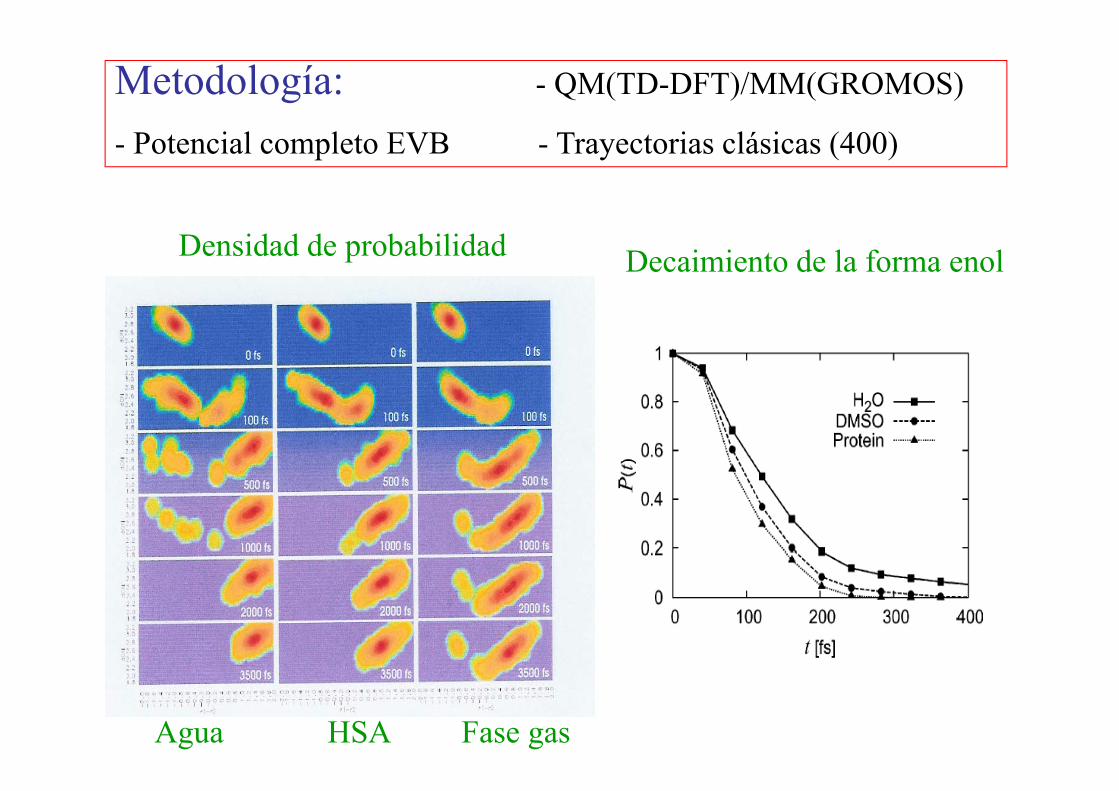
Metodología: - QM(TD-DFT)/MM(GROMOS)
- Potencial completo EVB - Trayectorias clásicas (400)
Densidad de probabilidad Decaimiento de la forma enol
Agua HSA Fase gas
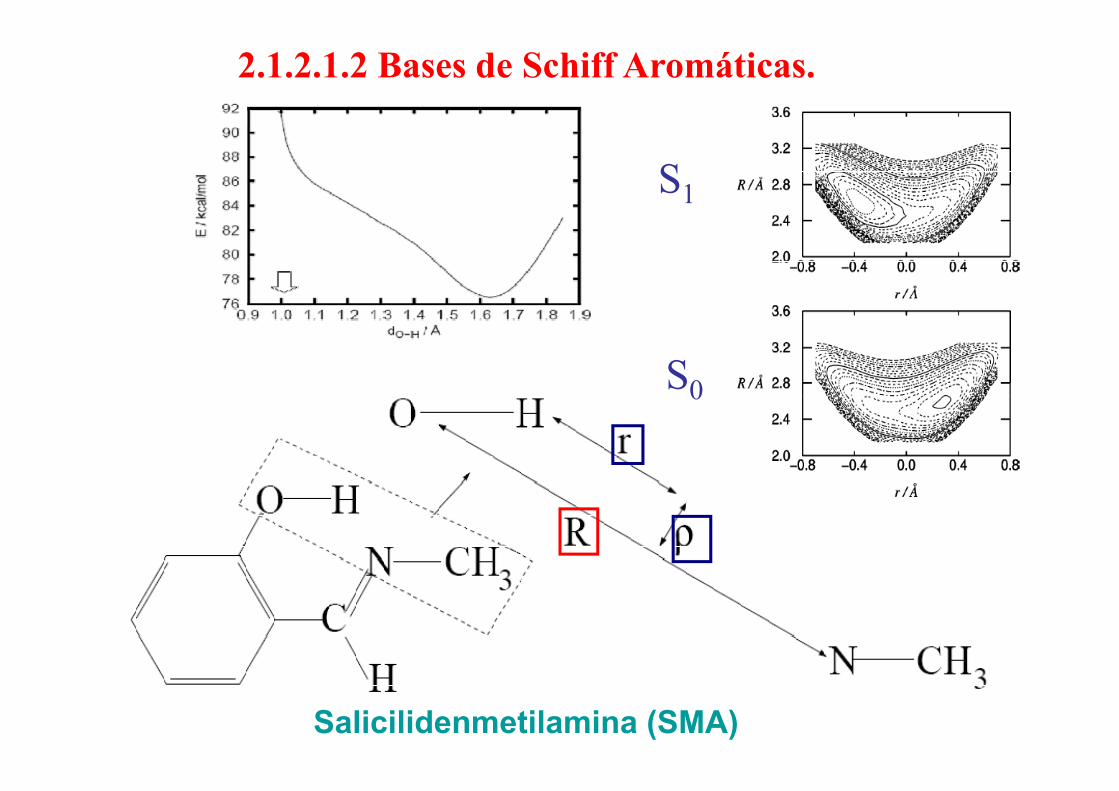
2.1.2.1.2 Bases de Schiff Aromáticas.
S1
S00
Salicilidenmetilamina (SMA)

Término cinético:
Término potencial:∑ ∏= =
=s
r
f
rr hcH1 1
)(ˆκ
κ
Término potencial:12 puntos de R15 puntos de r4 puntos de ρ
720 cálculos electrónicos

Evolución temporal de la función de onda (MCTDH)p ( )

Densidades de probabilidad unidimensionales
H D
~11 fs ~25 fs
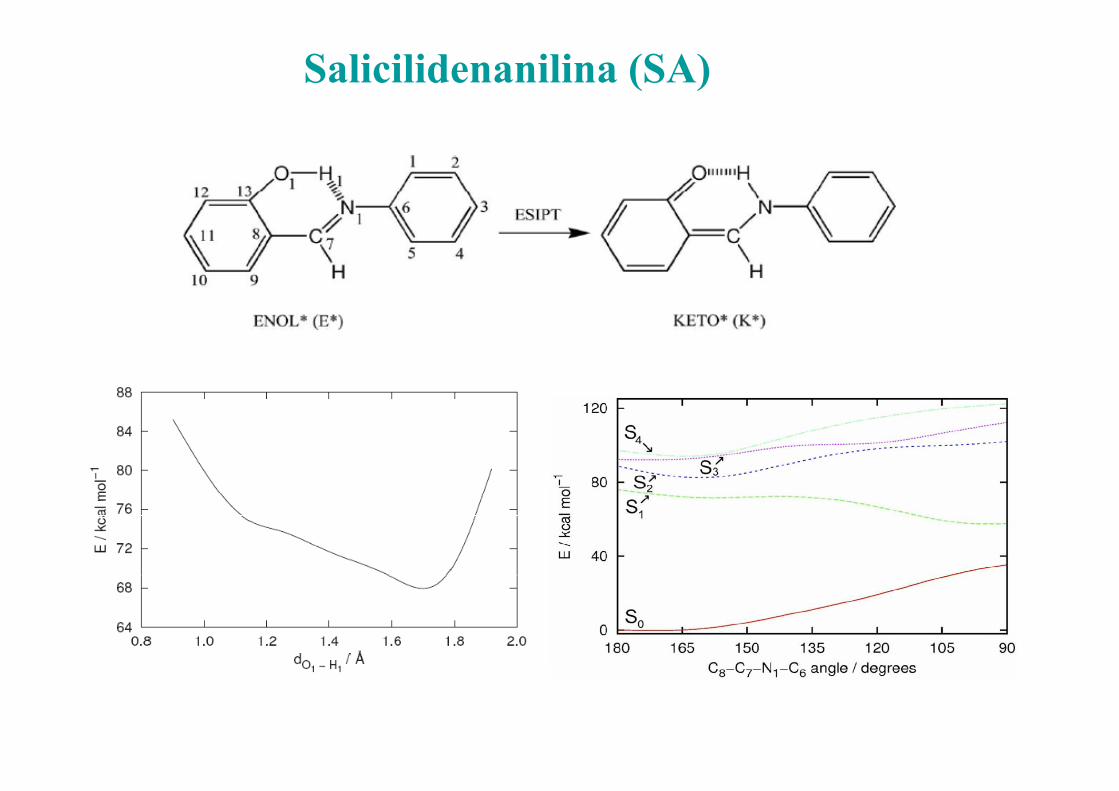
Salicilidenanilina (SA)

474 n
350 nm
nm
m

2.1.2.1.3: HAN (1-hidroxi-2-acetonaftona)
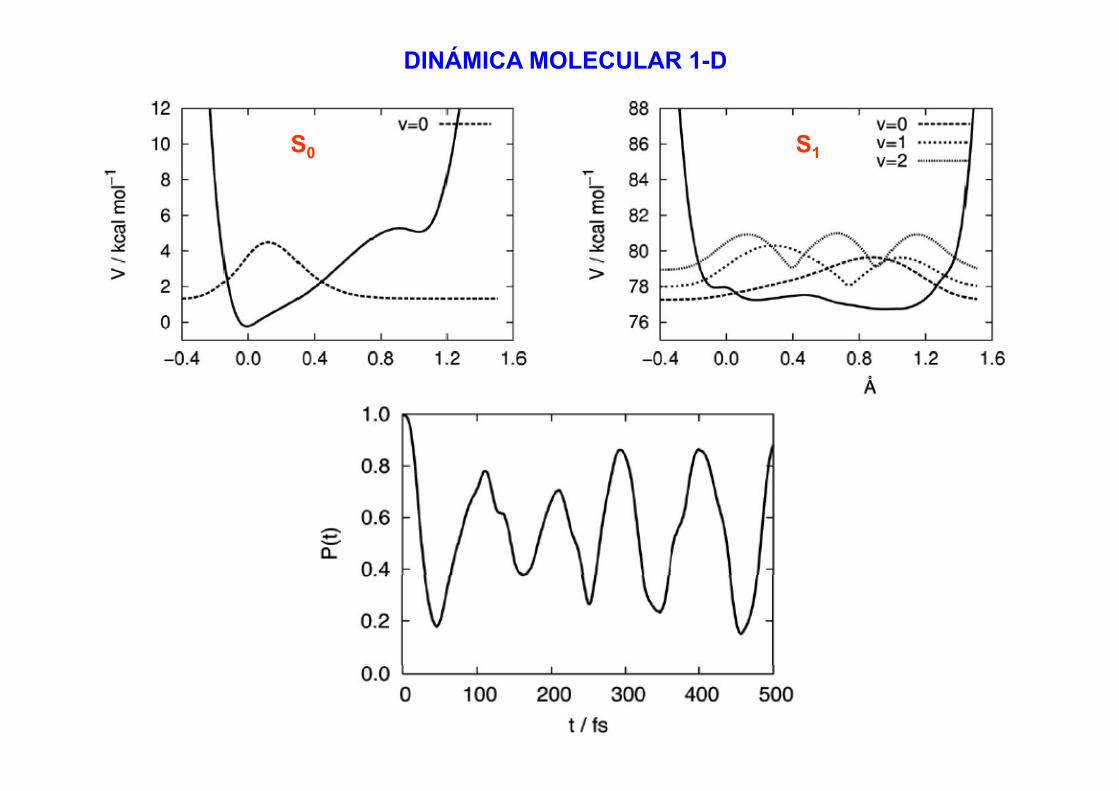
DINÁMICA MOLECULAR 1-D
S0 S1

NN
H1.291.26
2.1.2.2 Transferencia protónica múltiple
NNN
1 191 03
N N
HH
1.261.29protónica múltiple
2 1 2 2 1 Dímero del 7 azaindol yN N
NN
HH1.03
N N 1.83
1.16
1.43
HH
1.19
1.38
1.03
1.83SP2'
DM=3.63
2.1.2.2.1 Dímero del 7-azaindol y bases del ADN
TS1'
SP2
N N
NN
1.04
1.75HH
1.05
1.69
TS1'TS2'DM=7.69
DM=8.33
TS2'
NN NN
2 041 01 2 03 1.01
INT(Z)'DM=8.98
de S0
INT(Z)'SCT N N
N N
HH
1.01
2.041.01
2.02HH
2.03
1.01
1.01
2.06
SL
N N
NN
HH0.99
2.36
0.99
2.44
BP' T'DM=2.63 DM=3.80
BP'T'INT(C)'
SL
INT(C)'DM=10.46

Corrección Híbrida CIS/TDDFT para estados de transferencia de carga:[A. Drew et al. J. Am. Chem. Soc. 126 (2004) 4007][ ( ) ]

CIS TDDFT (corrected)
TDDFT (uncorrected)TDDFT (uncorrected)
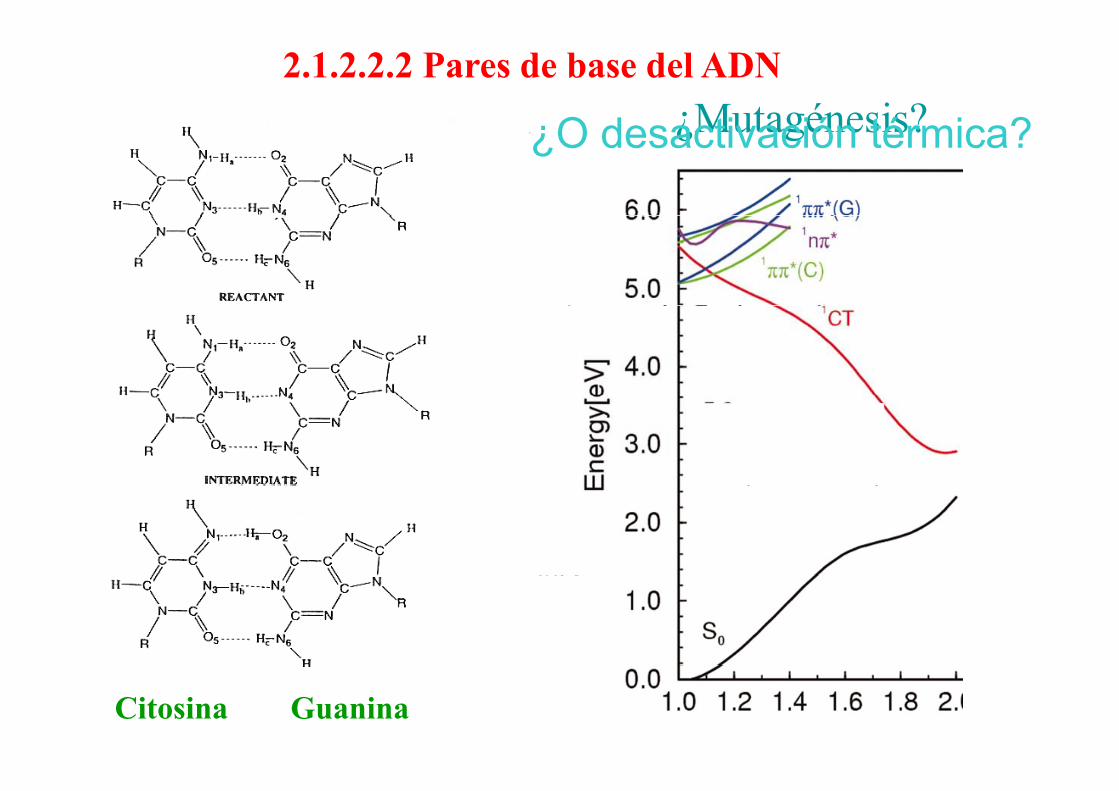
2.1.2.2.2 Pares de base del ADN¿Mutagénesis?¿O desactivación térmica?¿Mutagénesis?¿O desactivación térmica?
-15.67 C–-G+
Citosina Guanina

10.612.6
3.42 A+-T –
T i i ti l
3.45
––– Transiciones verticales
––– Estado de transferencia de carga
Energías en kcal/mol
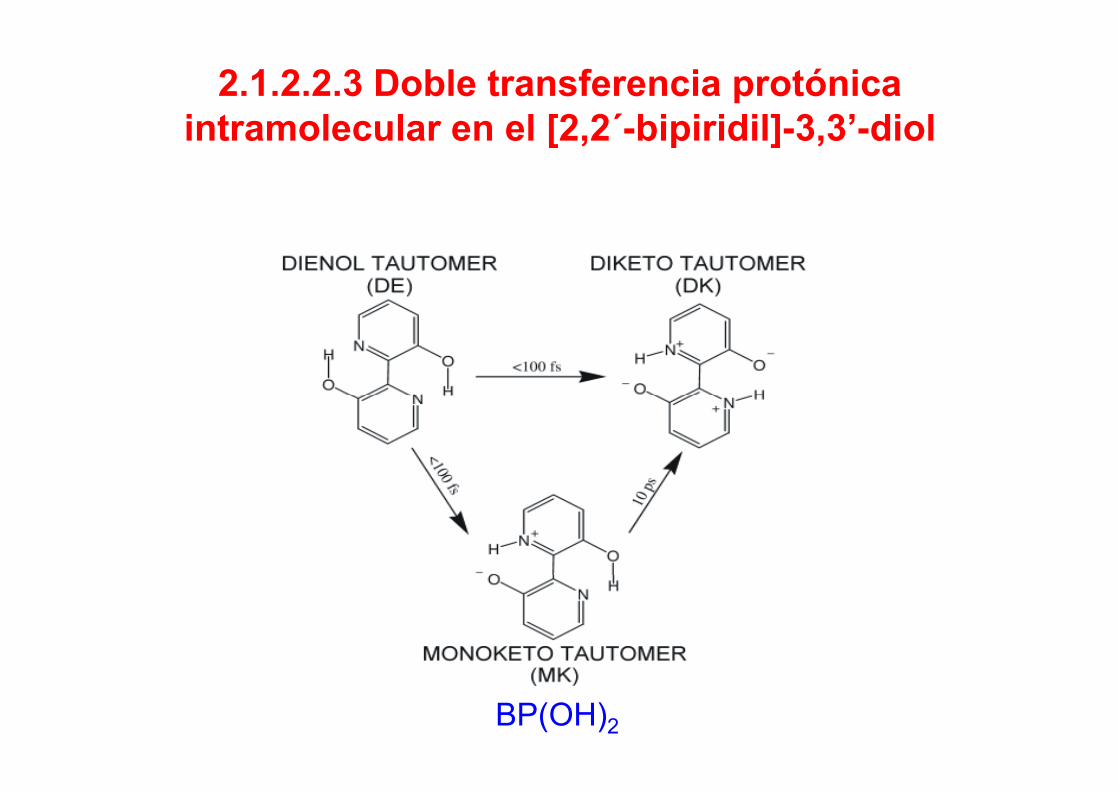
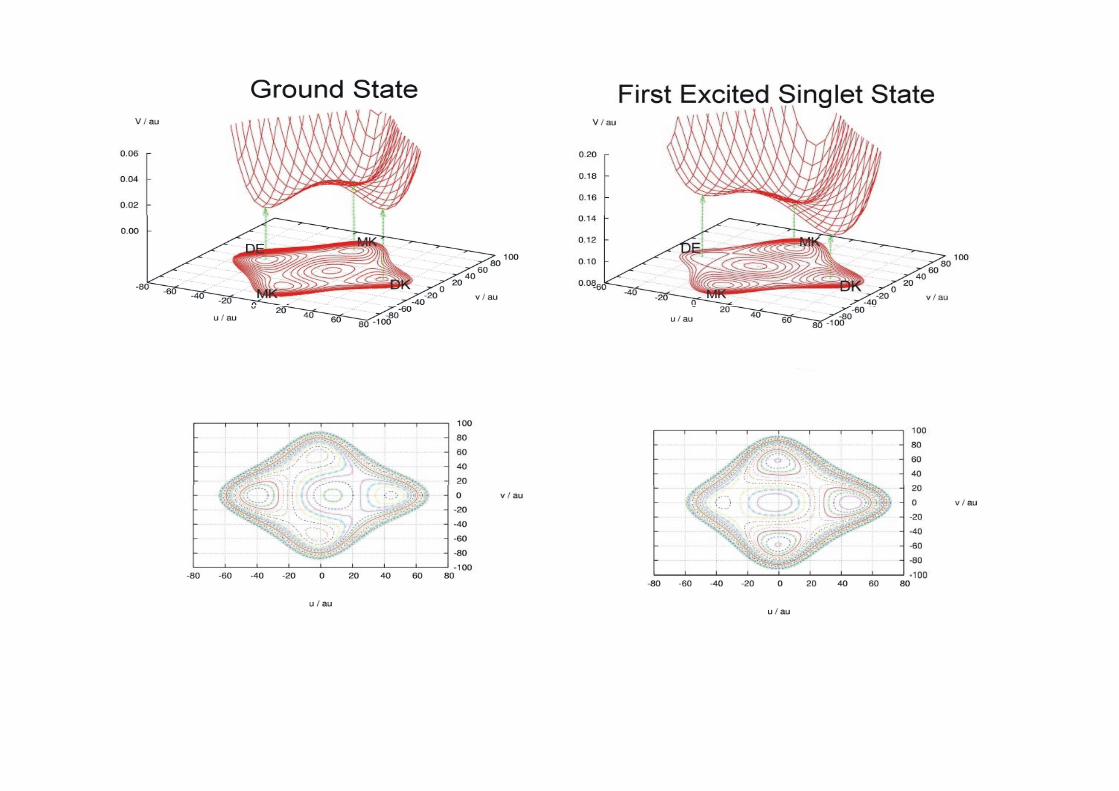
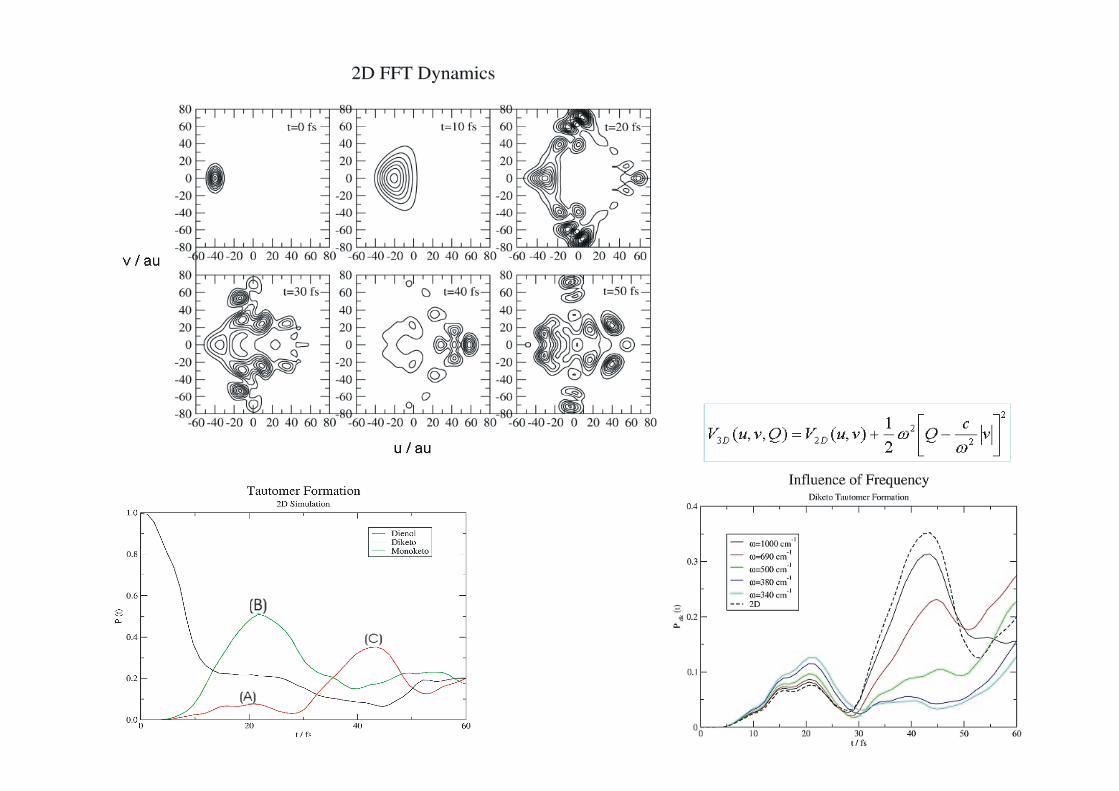
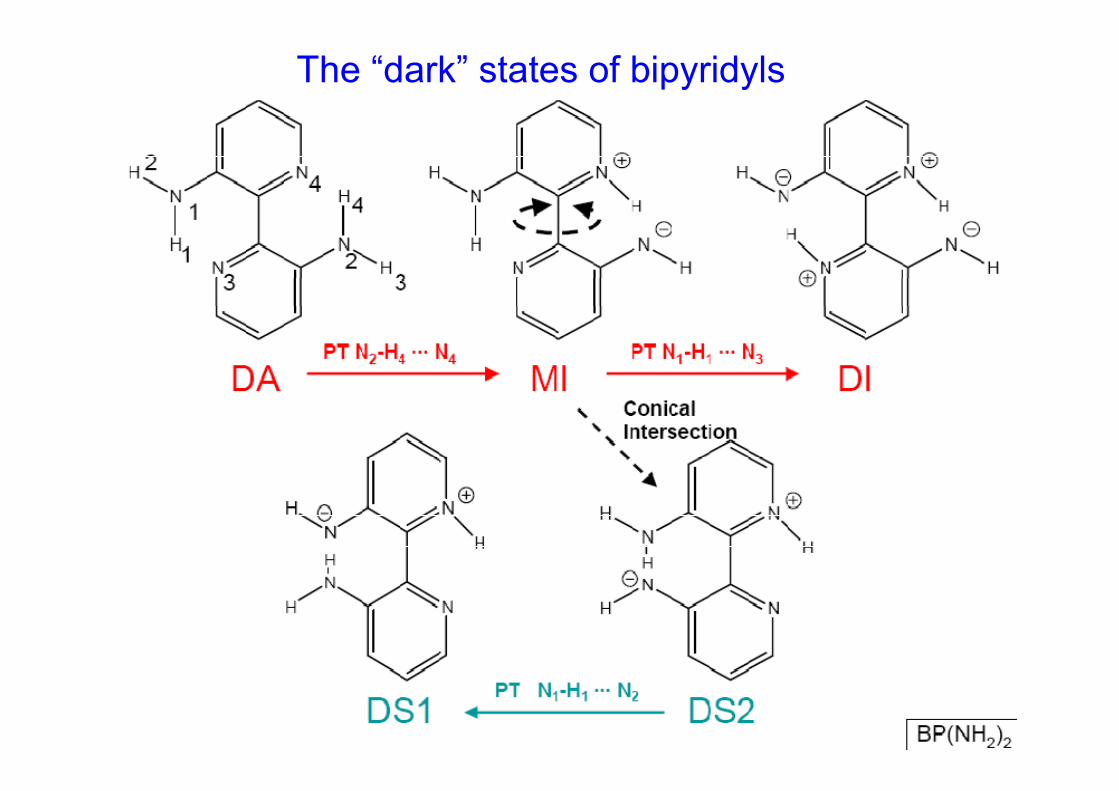
2.1.2.2.3 Doble transferencia protónica intramolecular en el [2 2´ bipiridil] 3 3’ diolintramolecular en el [2,2 -bipiridil]-3,3 -diol
BP(OH)2

BP(NH2)2
C.I.
BP(OH)2
C.I.
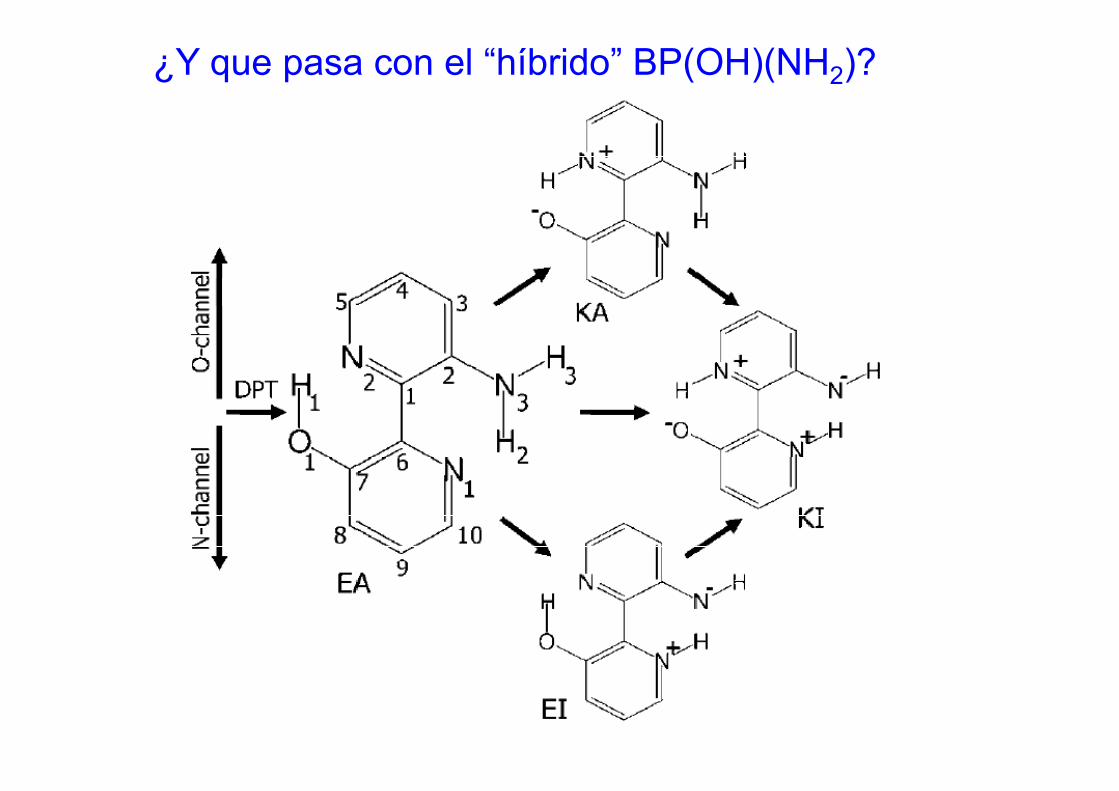
¿Y que pasa con el “híbrido” BP(OH)(NH2)?
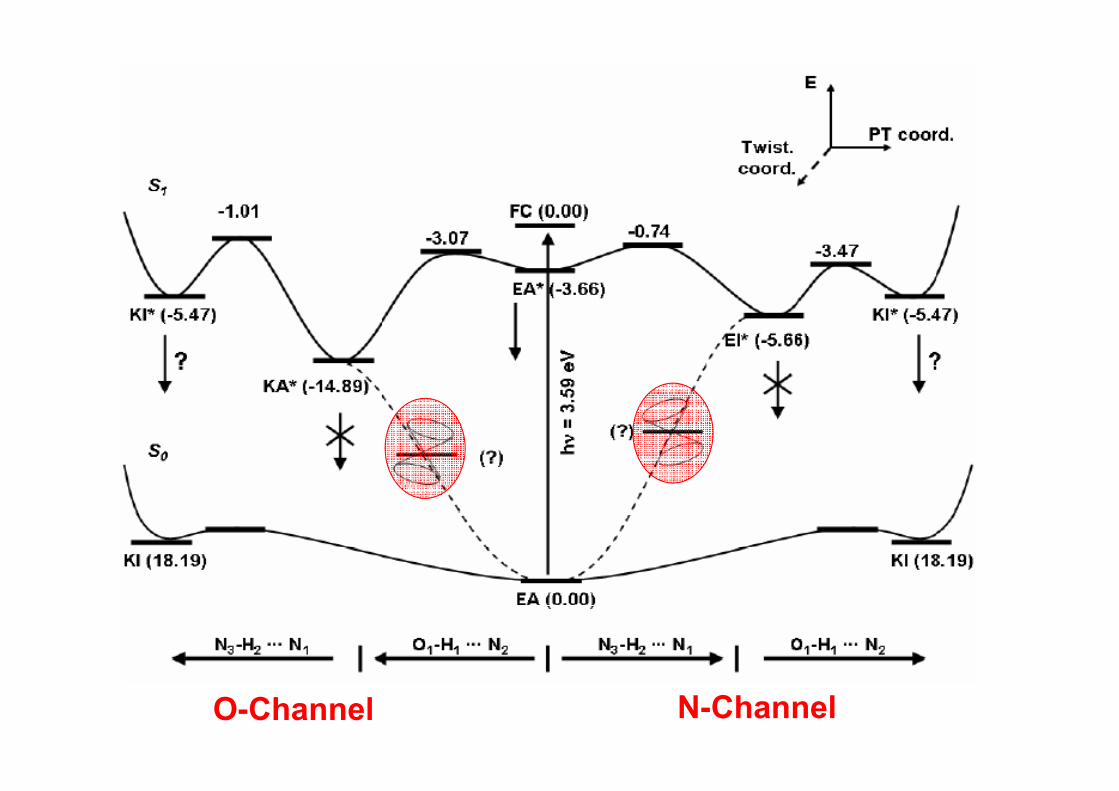
O-Channel N-Channel
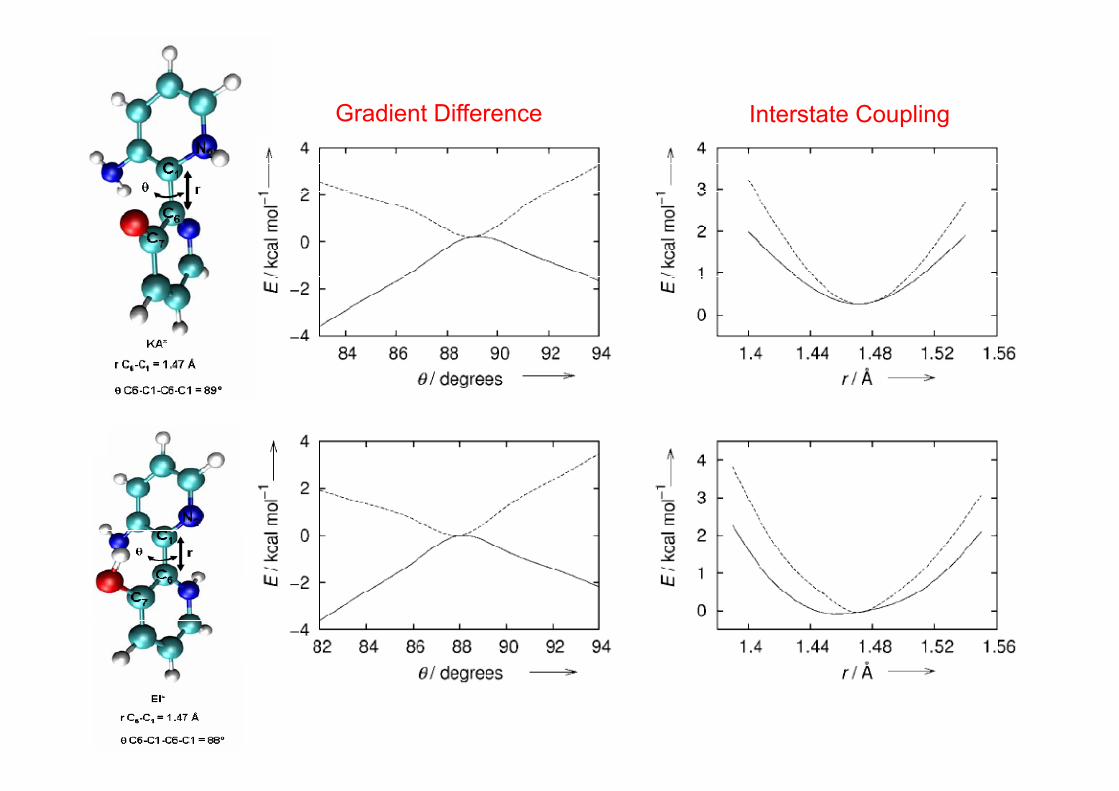
Gradient Difference Interstate Couplingp g
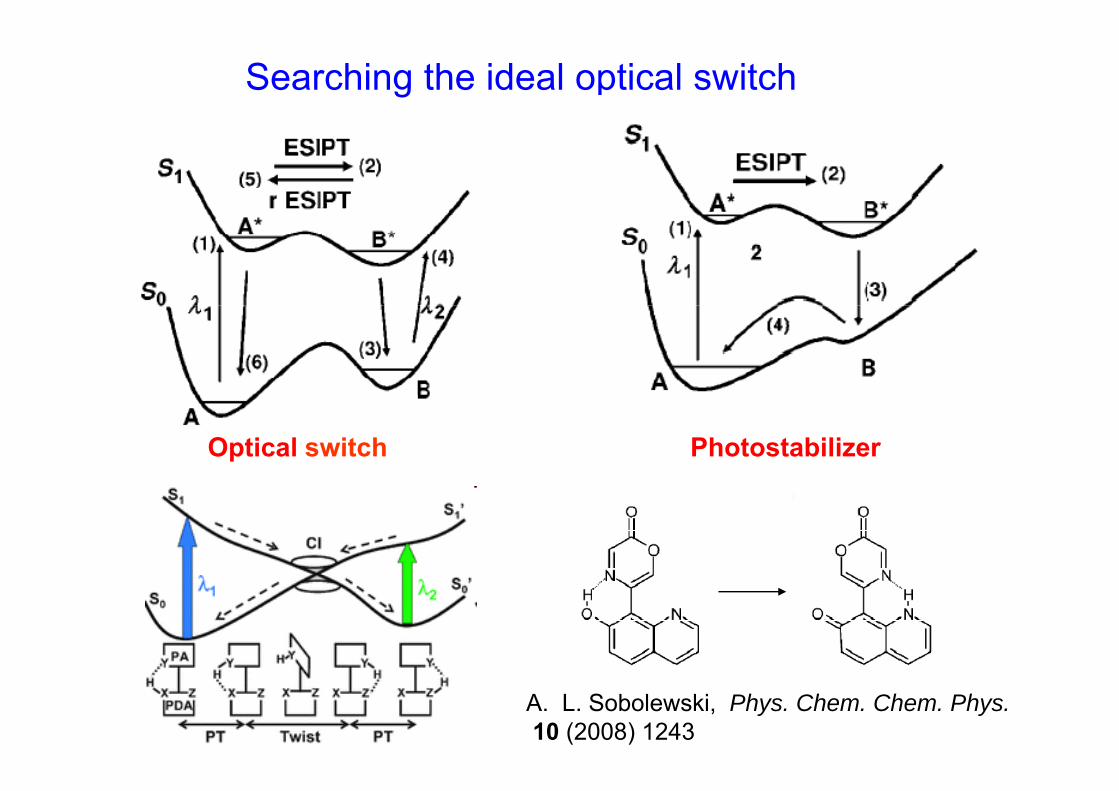
Searching the ideal optical switch
Optical switch Photostabilizer
A. L. Sobolewski, Phys. Chem. Chem. Phys.10 (2008) 1243
The “dark” states of bipyridyls
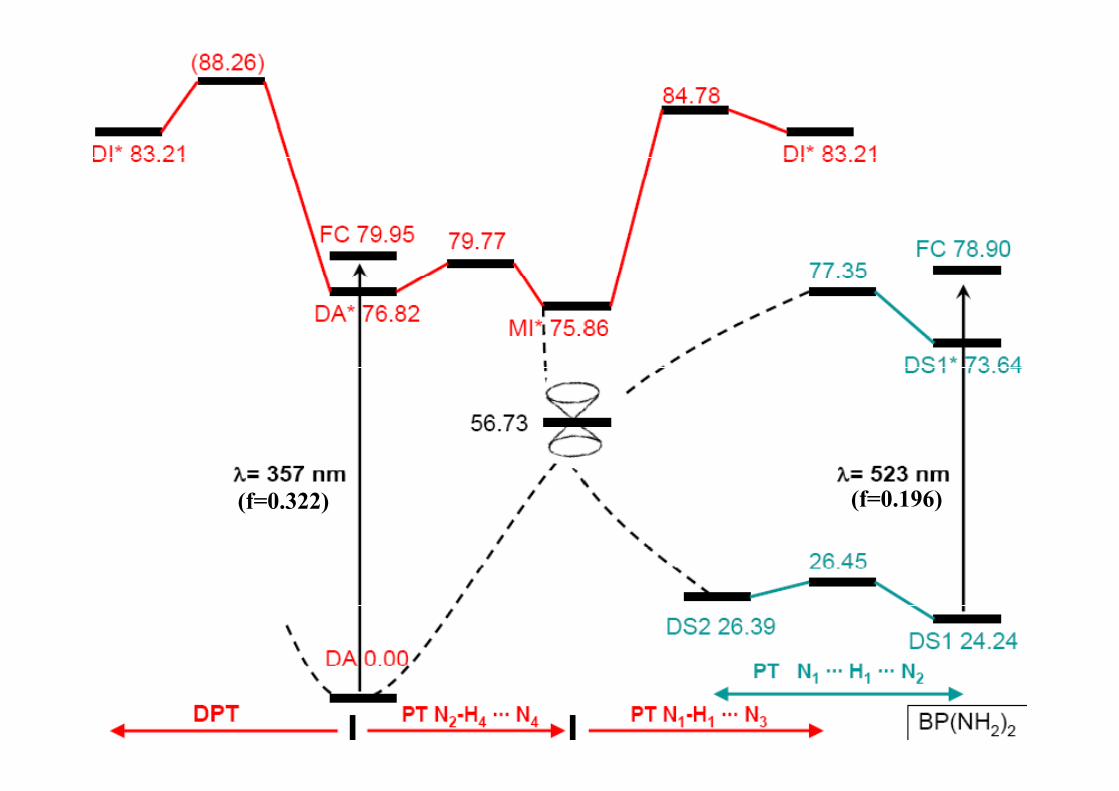
(f=0.322) (f=0.196)

Tropolona y derivados:REFERENCIAS
p y- J. J. Paz et al. J. Chem. Phys. 103 (1995) 353-359- J. J. Paz et al. J. Chem. Phys. 107 (1997) 6275-6282- J. J. Paz et al. J. Chem. Phys. 108 (1998) 8114-8122- J. J. Paz et al. Chem. Phys. 246 (1999) 103-113- O. Vendrell et al. J. Chem. Phys. 117 (2002) 7525-7533
HPMO:- V. Guallar et al. J. Phys. Chem. 100 (1996) 19789-19794- R. Casadesús et al. Chem. Phys. Lett. 356 (2002) 423-430y ( )- O. Vendrell et al. J. Phys. Chem. B 108 (2004) 6616-6623- R. Casadesús et al. J. Photochem. Photobiol. A 173 (2005) 365-374
Bases de Schiff aromáticas:- J. M. Ortiz-Sánchez et al. J. Phys. Chem. A 110 (2006) 4649-4656- J. M. Ortiz-Sánchez et al. J. Chem. Phys. A 129 (2008) 214308y ( )- C. Randino et al. PCCP. En revisión
HAN:HAN:- J. A. Organero et al. Chem. Phys. Lett. 328 (2000) 83-89- J. A. Organero et al. J. Phys. Chem. A 104 (2000) 8424-8431- J. M. Ortiz-Sánchez et al. J. Chem. Phys. 127 (2007) 084318

Antronas y suberonas:REFERENCIAS (2)
y- R. Casadesús et al. J. Phys. Chem. A 108 (2004) 6616-6623 - R. Casadesús et al. Chem. Phys. 328 (2006) 410-420- M. Moreno et al. J. Phys. Chem. A 111 (2007) 10090-10097
Dímero del 7-AI y bases del ADN:- A. Douhal et al. Chem. Phys. Lett. 256 (1996) 370-376A. Douhal et al. Chem. Phys. Lett. 256 (1996) 370 376- V. Guallar et al. Chem. Phys. 228 (1998) 1-7- V. Guallar et al. J. Phys. Chem. A 103 (1999) 6251-6256- M. Moreno et al. J. Phys. Chem. A 105 (2001) 3887-3893y ( )- R. Casadesús et al. Chem. Phys. 290 (2003) 319-336- R. Gelabert et al. J. Phys Chem A 110 (2006) 1145-1151
Bipiridilos:- R. Gelabert et al. ChemPhysChem 5 (2004) 1372-1378- J. M. Ortiz-Sánchez et al. ChemPhysChem 8 (2007) 1199-1206y ( )- J. M. Ortiz-Sánchez et al. ChemPhysChem 9 (2008) 2068-2076- J. M. Ortiz-Sánchez et al. Chem. Eur. J. 16 (2010) 6693-6703- J. M. Ortiz-Sánchez et al. ChemPhysChem 11 (2010) 3696-3703y ( )
TRANSFERÈNCIA PROTÒNICA MÚLTIPLE FOTOINDUÏDA EN LA PROTEÏNA VERDA FLUORESCENT.
ESTUDIS D’ESTRUCTURA ELECTRÒNICA I DE DINÀMICAESTUDIS D’ESTRUCTURA ELECTRÒNICA I DE DINÀMICA MOLECULAR.Miquel MorenoMiquel Moreno

Esquema de la presentació• Introducció
– Que són les Green Fluorescent Proteins (GFP)?P i l GFP?– Per a que serveixen les GFP?
– Quina és l’estructura de les GFP?– Per que són fluorescents les GFP?q
• Mètode de càlcul– Càlculs electrònics– Dinàmica molecular clàssica– Dinàmica molecular quàntica
Res ltats• Resultats– Resultats de dinàmica molecular clàssica– Resultats dels càlculs electrònicsResultats dels càlculs electrònics– Resultats de dinàmica quàntica
• Conclusions• Perspectives de futur

Introducció. Que són les GFP?
Aequorea Victoria
O. Shimomura et al.

Introducció Per a que serveixen les GFP?Introducció. Per a que serveixen les GFP?
Marcador Biològic UniversalMarcador Biològic Universal • Estable
Altament fluorescent• Altament fluorescent• El cromòfor es forma en una reacció
autocatalíticaautocatalítica• Es fusiona amb altres proteïnes sense alterar el
seu funcionamentseu funcionament.• Pot ser usat en sistemes vius (cèl·lules) per a
seguir la seva genètica i/o el seu metabolismeseguir la seva genètica i/o el seu metabolisme mitjançant espectroscòpia de fluorescència

Per a que serveixen les GFP?
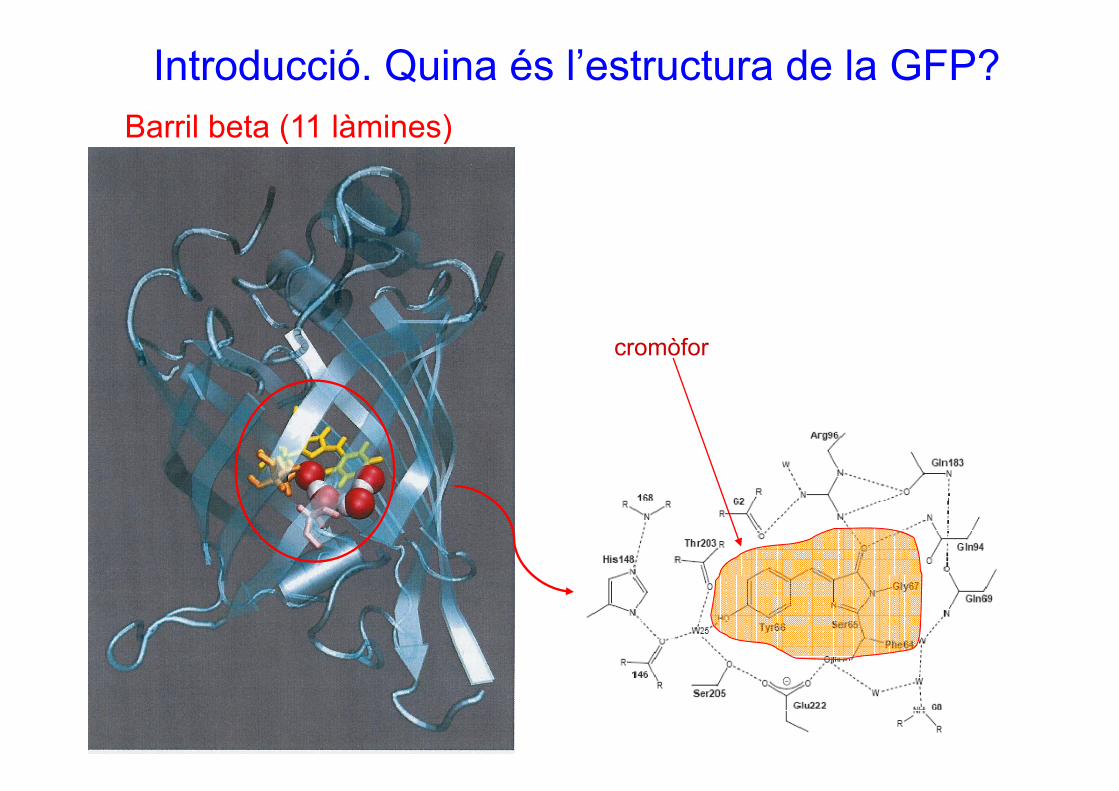
Introducció. Quina és l’estructura de la GFP?Barril beta (11 làmines)Barril beta (11 làmines)
cromòfor
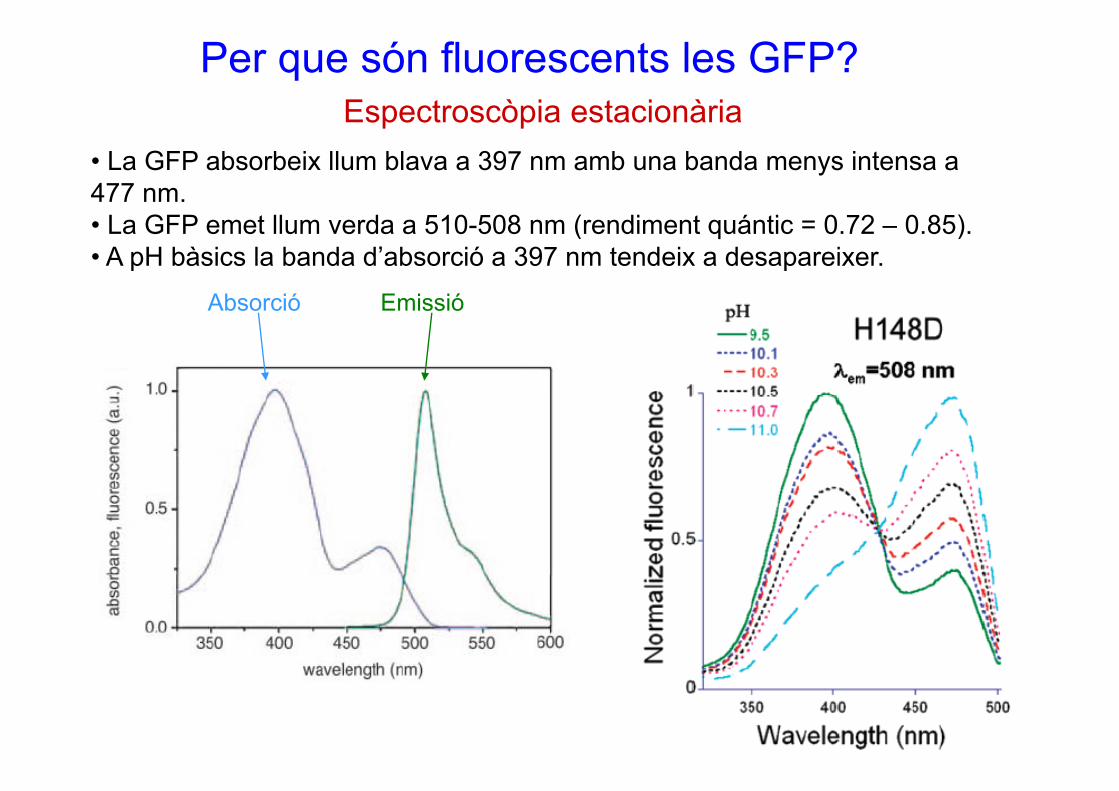
Per que són fluorescents les GFP?Espectroscòpia estacionària
• La GFP absorbeix llum blava a 397 nm amb una banda menys intensa a 477 nm.
L GFP t ll d 510 508 ( di t á ti 0 72 0 85)
p p
• La GFP emet llum verda a 510-508 nm (rendiment quántic = 0.72 – 0.85). • A pH bàsics la banda d’absorció a 397 nm tendeix a desapareixer.
Absorció EmissióAbsorció Emissió
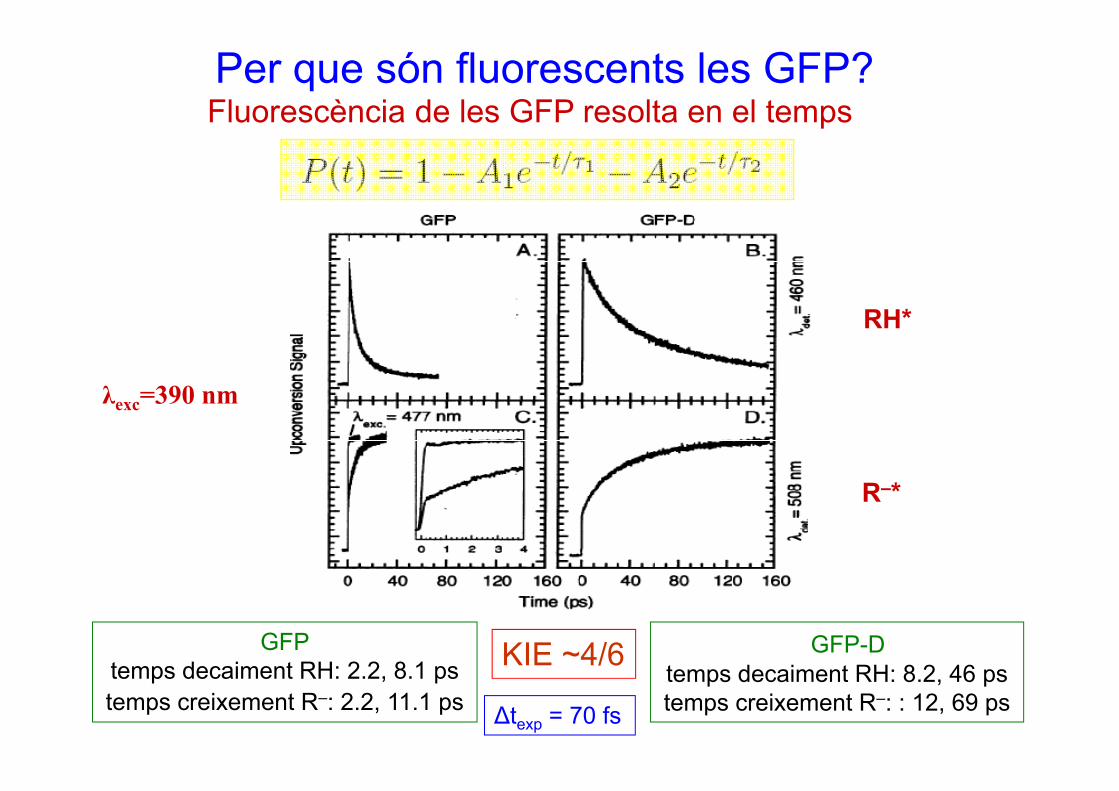
Fluorescència de les GFP resolta en el tempsPer que són fluorescents les GFP?
p
RH*
λ =390 nm
R *
λexc=390 nm
R–*
KIE ~4/6GFPt d i t RH 2 2 8 1
GFP-D d i RH 8 2 46
KIE 4/6temps decaiment RH: 2.2, 8.1 pstemps creixement R–: 2.2, 11.1 ps
temps decaiment RH: 8.2, 46 pstemps creixement R–: : 12, 69 psΔtexp = 70 fs

Transferència protònica múltiple
Estat responsable de la fluorescència

Mètodes de càlcul electrònics
- CASSCF/CASPT2
- Es consideren l’estat fonamental (S0) i els estats excitats electrònics ππ * i πσ * d’energia més baixa.
- S’imposa planarietat del cromòfor i de la cadena de transferència protònicatransferència protònica.
- Es consideren dos espais actius diferents: (6,6) (6 electrons en 6 orbitals) i (9,8) (8 electrons en 9 orbitals). L’espai gran s’usa per “calibrar” el mètode i analitzar els punts estacionaris mentre l’espai petit és usat per generar la superfície de potencial completa.p p p

Métode de càlcul. Dinàmica molecular clàssica• Usada per a analitzar la configuració del cromòfor i les molècules circundants enUsada per a analitzar la configuració del cromòfor i les molècules circundants en
l’entorn proteic complet.• Programa CHARMM (versió 28b2) amb el camp de forces CHARMM 22.
• La proteïna està solvatada en una esfera de 37 Å de radi formada per 5531 molècules d’aigua del tipus TIP35531 molècules d aigua del tipus TIP3.
• Nombre total d’àtoms: 20236
• Temperatura fixada a 300Kp
• Pas de rosca del temps:1fs
• Temps total de propagació: 0.5 nsp p p g

Mètode de Càlcul. Dinàmica molecular quàntica
• Usada per a estudiar el desplaçament dels protons al llarg de la cadena.• La naturalesa quàntica del sistema de triple transferència protònica no ens
permet fer un tractament clàssic de la dinàmica dels nuclis.• Considerant vàlida l’aproximació de Born-Oppenheimer, la dinàmica nuclear
del sistema vindrà descrita per l’equació de Schrödinger dependent delp q g ptemps:
Per resoldre aquesta equació usaremPer resoldre aquesta equació usarem el mètode MCTDH
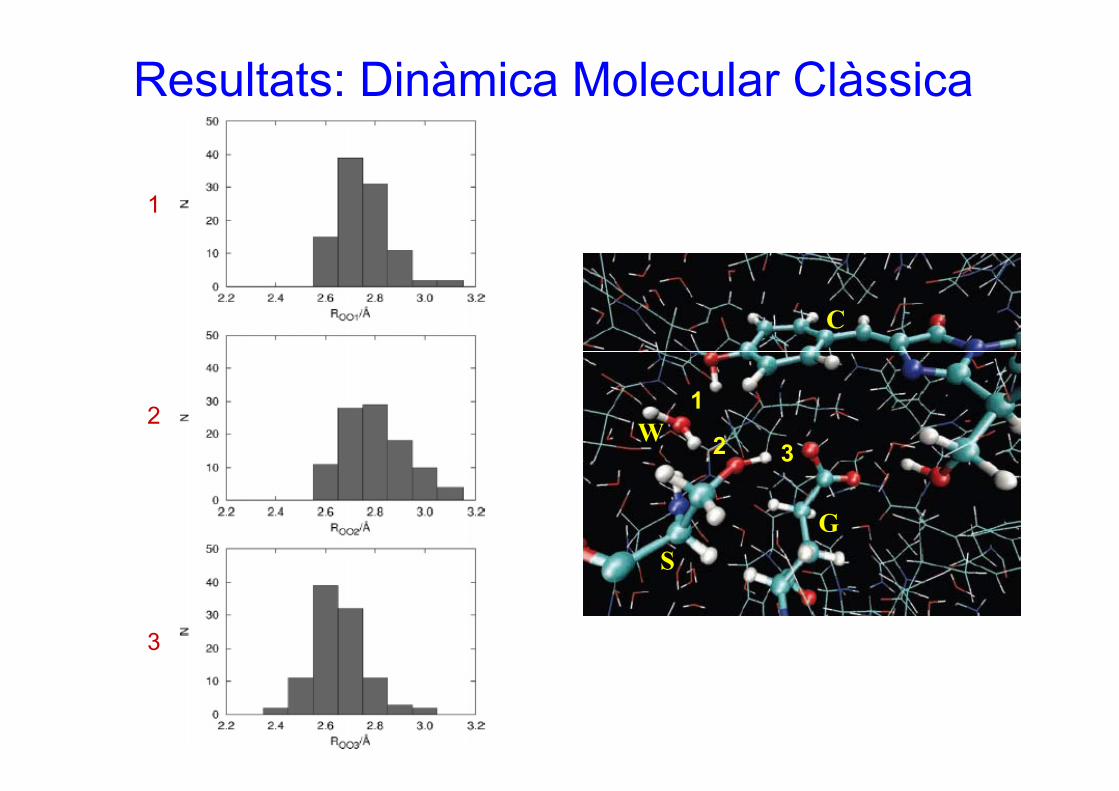
Resultats: Dinàmica Molecular Clàssica
1
2
1
3
1
C
12 1
2 3W
SG
3
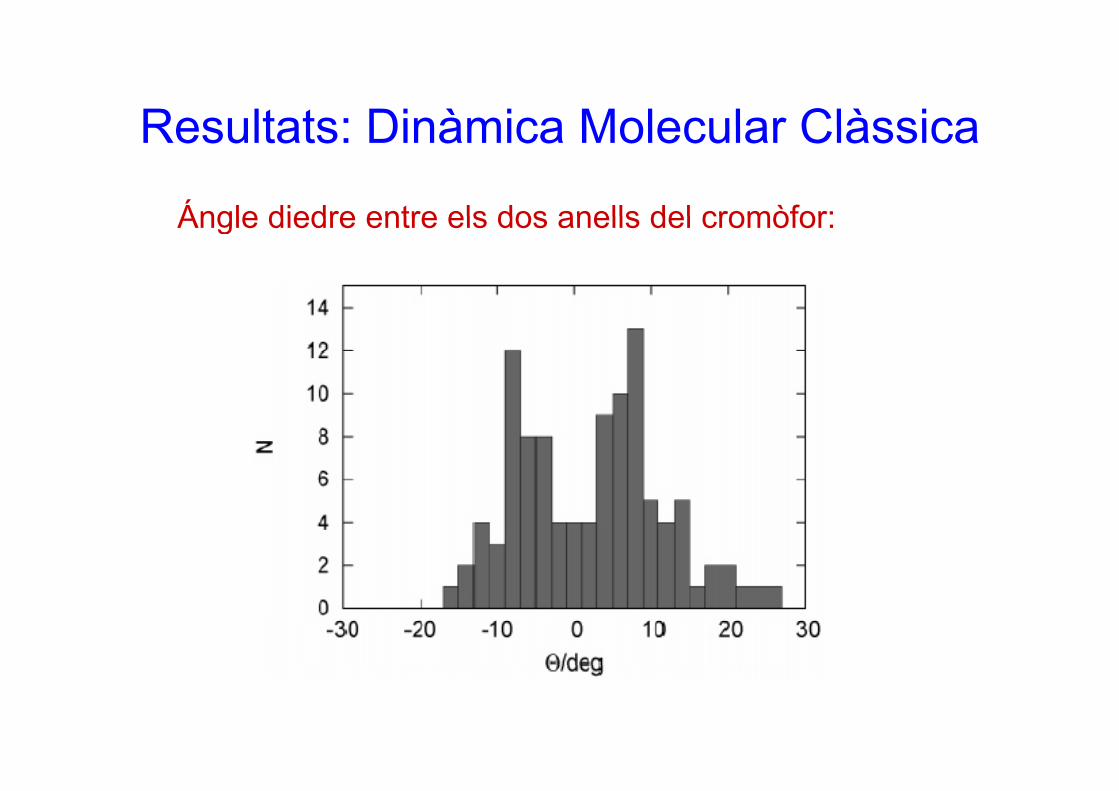
Resultats: Dinàmica Molecular ClàssicaResultats: Dinàmica Molecular Clàssica
Ángle diedre entre els dos anells del cromòfor:Ángle diedre entre els dos anells del cromòfor:
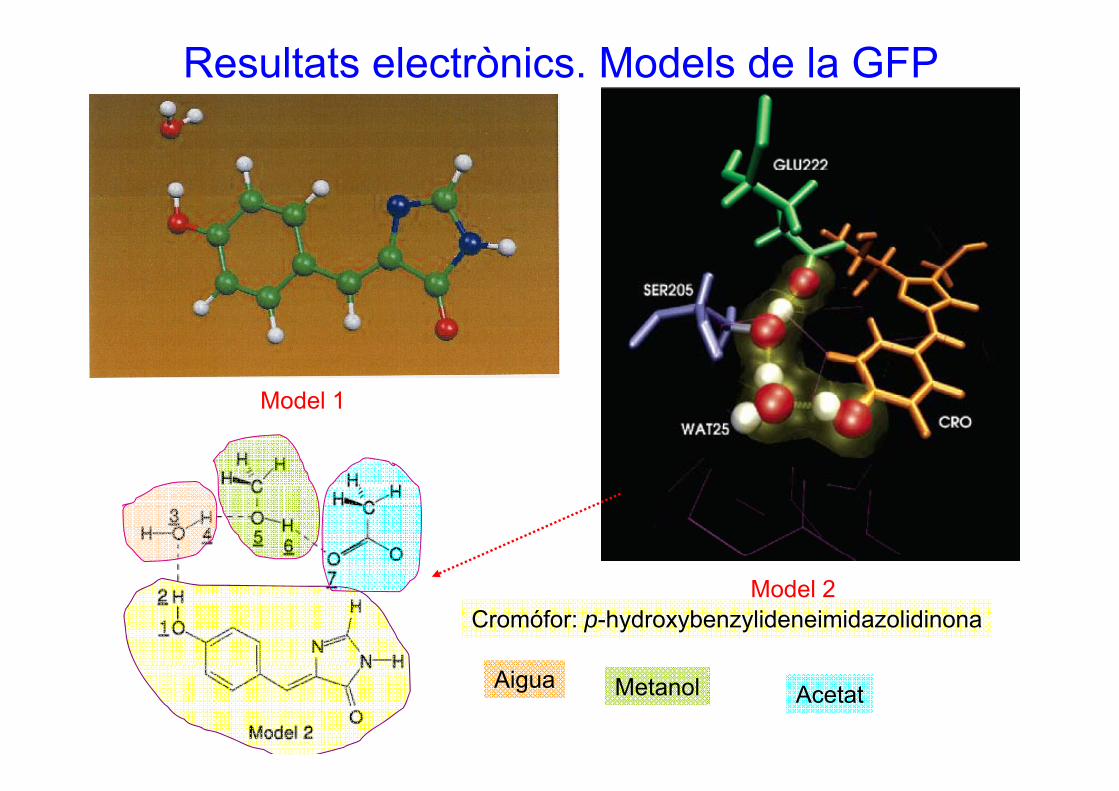
Resultats electrònics. Models de la GFP
M d l 1Model 1
Model 2Cromófor: p-hydroxybenzylideneimidazolidinona
Aigua Metanol Acetat
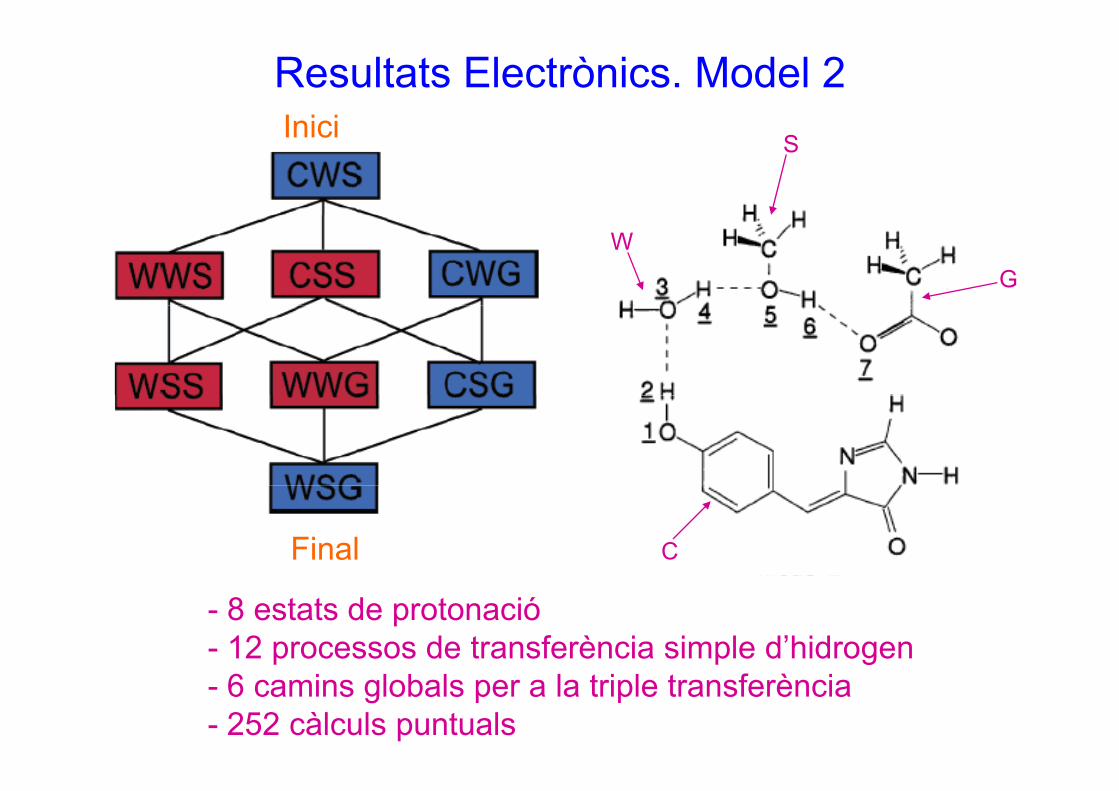
Resultats Electrònics. Model 2Inici SInici
WG
CFinal
- 8 estats de protonació - 12 processos de transferència simple d’hidrogeng- 6 camins globals per a la triple transferència- 252 càlculs puntuals

Resultats Electrònics. Model 2CASPT2 (9 8) πσ*CASPT2 (9,8) πσ*
ππ*ππ
S0
πσ*πσ
ππ*
S0
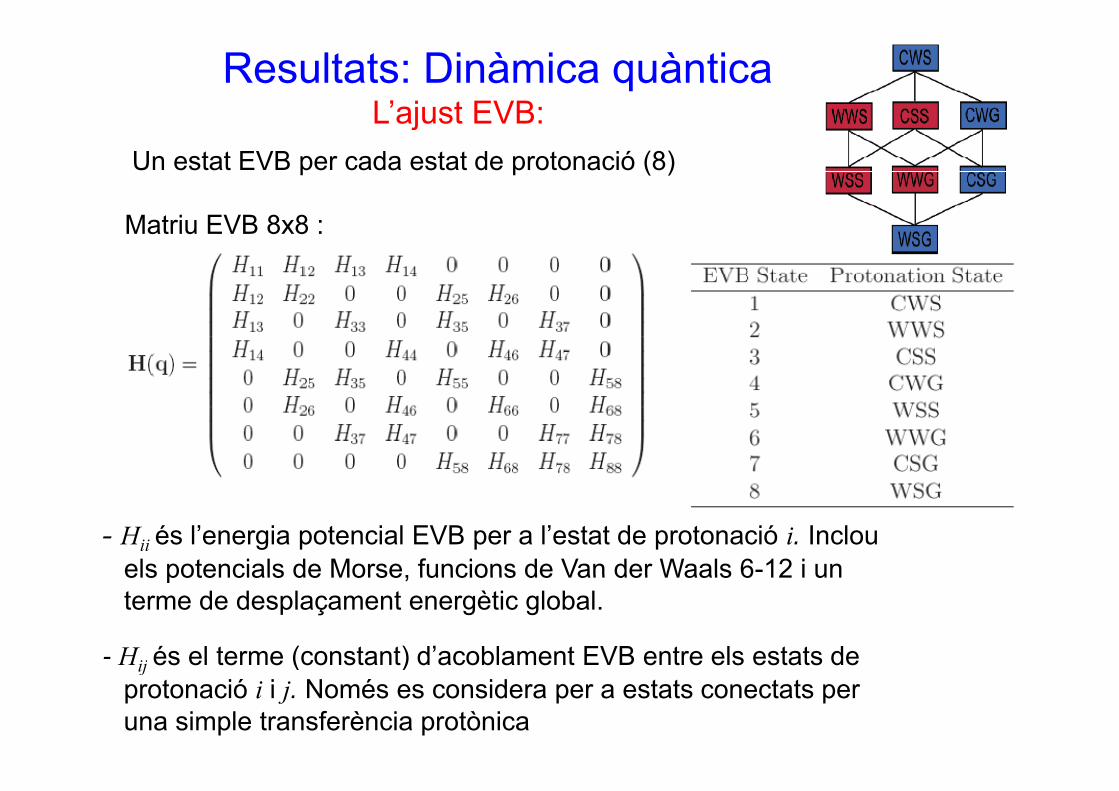
Resultats: Dinàmica quànticaL’ajust EVB:
Un estat EVB per cada estat de protonació (8)
M t i EVB 8 8
j
Matriu EVB 8x8 :
- Hii és l’energia potencial EVB per a l’estat de protonació i. Inclouels potencials de Morse, funcions de Van der Waals 6-12 i unels potencials de Morse, funcions de Van der Waals 6 12 i unterme de desplaçament energètic global.
- Hij és el terme (constant) d’acoblament EVB entre els estats de ij és e te e (co sta t) d acob a e t e t e e s estats deprotonació i i j. Només es considera per a estats conectats per una simple transferència protònica
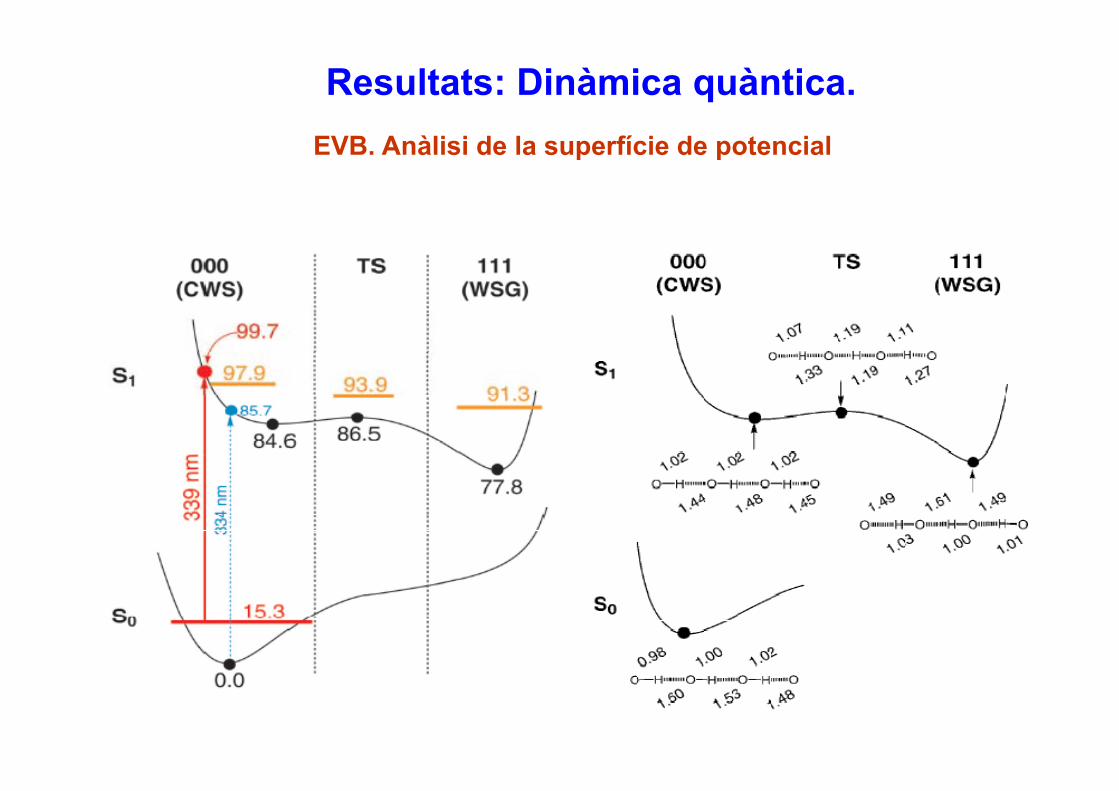
Resultats: Dinàmica quàntica.EVB. Anàlisi de la superfície de potencial
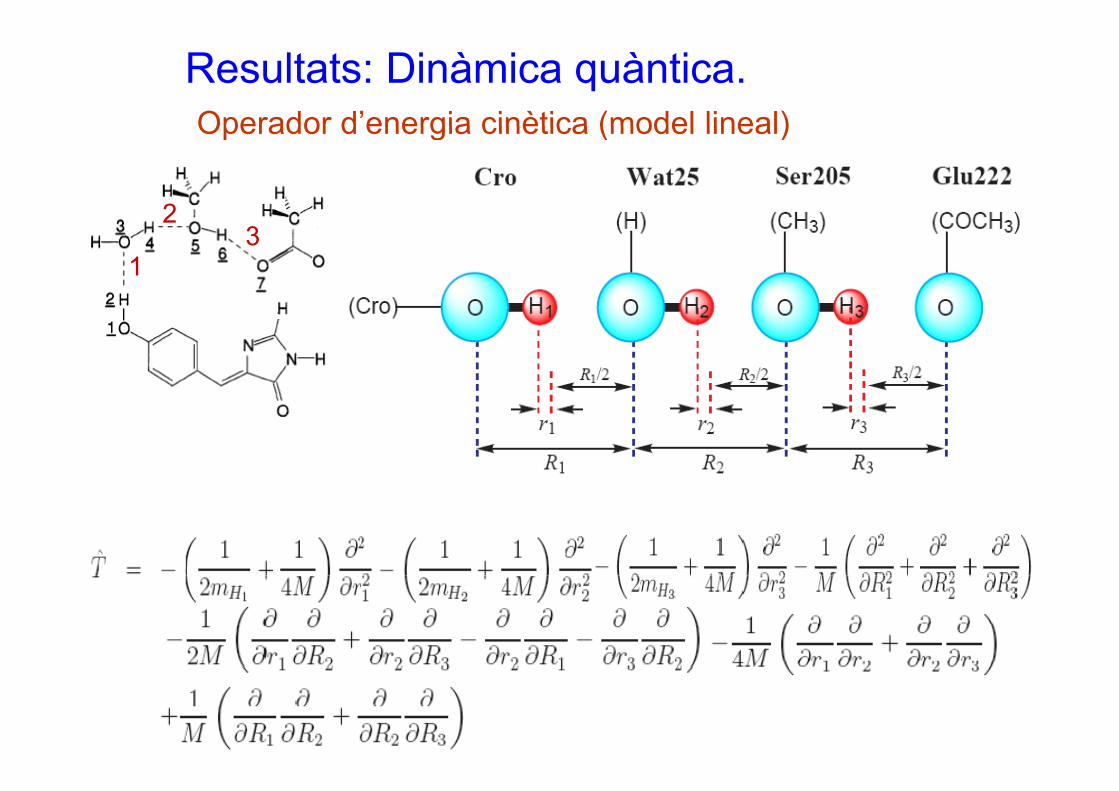
Resultats: Dinàmica quàntica.Operador d’energia cinètica (model lineal)Operador d energia cinètica (model lineal)
2
31
23

Resultats: Dinàmica quàntica.Procediment
1) L’estat vibracional fonamental en S0 es genera mitjançant una llargapropagació en temps imaginari del paquet d’ones (donat que l’operador
Procediment
propagació en temps imaginari del paquet d ones (donat que l operador de Boltzmann i l’operador d’evolució temporal són formalment equivalents).
hrr
tiTkH ee B Hr,p ˆ)( −− ≈ Tkit
B
h−=
2) Es simula la fotoexcitació Franck-Condon col·locant el paquet d’ones *
TkB
resultant en l’estat electrònic excitat ππ*.
3) Seguidament es propaga el paquet d’ones en la PES de l’estat excitat ππ*.3) Seguidament es propaga el paquet d ones en la PES de l estat excitat ππ .
Els 6 graus de llibertat s’han contragut a 3 (ri,Ri).En total s’han usat 27000 productes de Hartree dependents del temps per a definir el paquet d’ondes MCTDH.
132
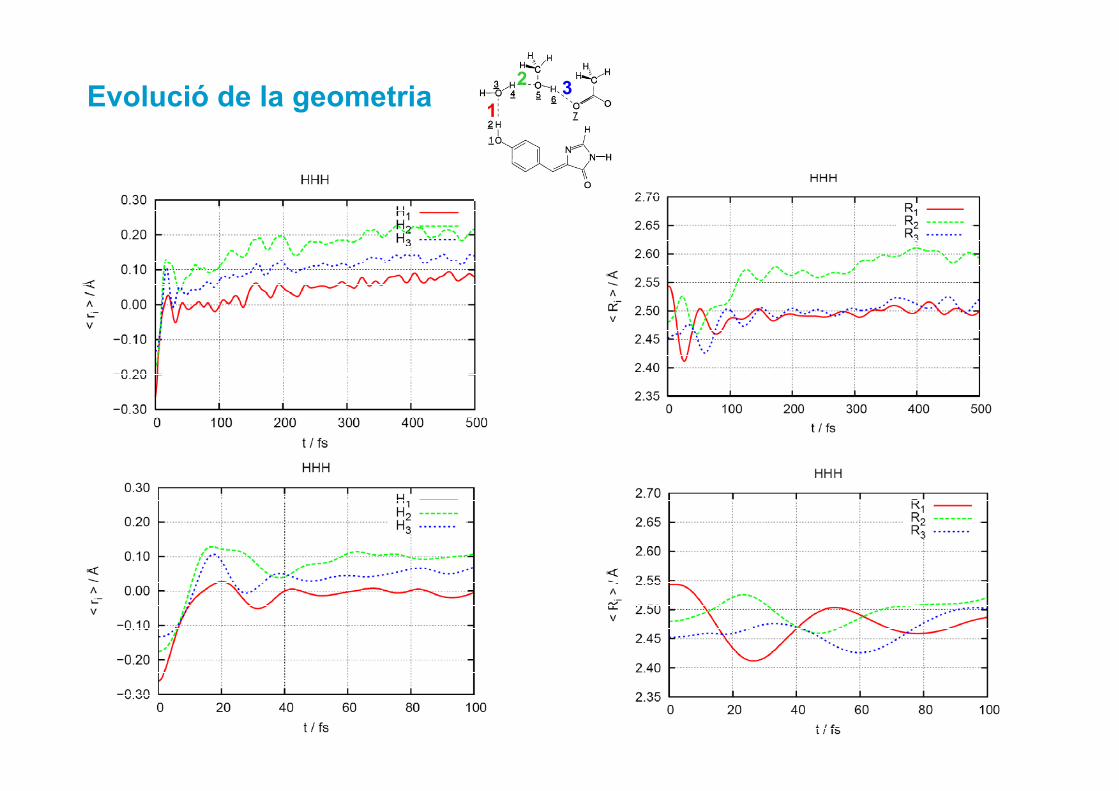
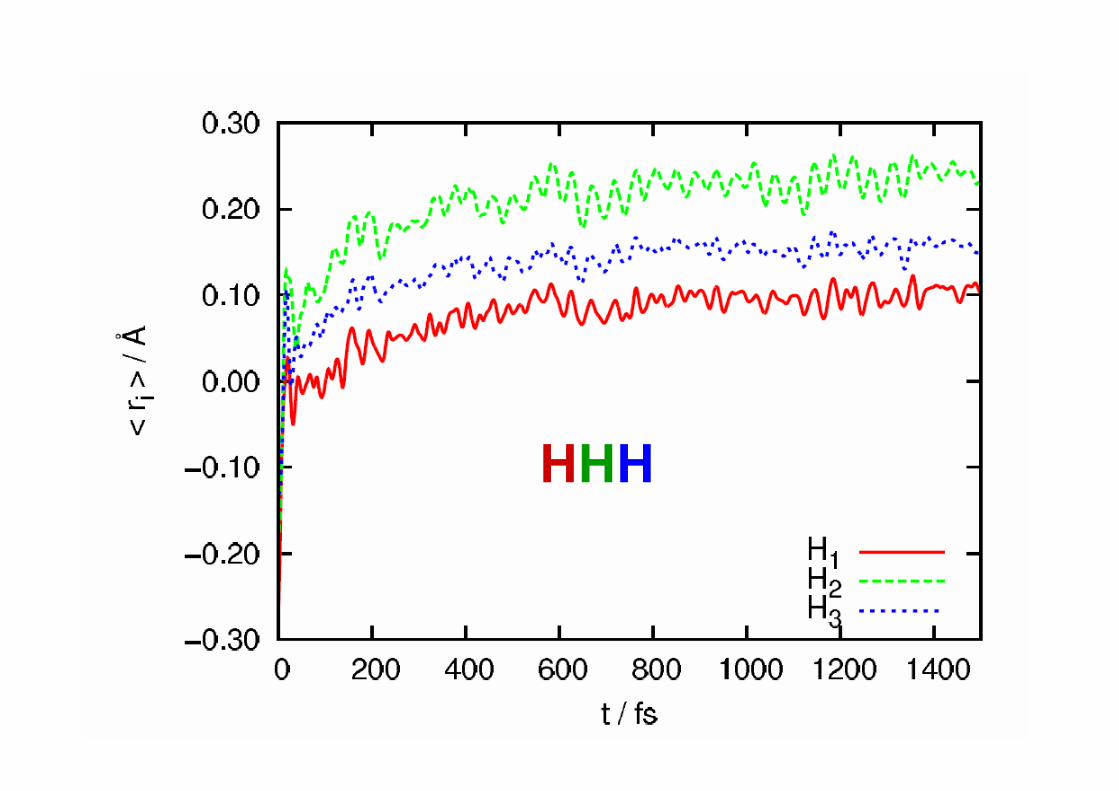
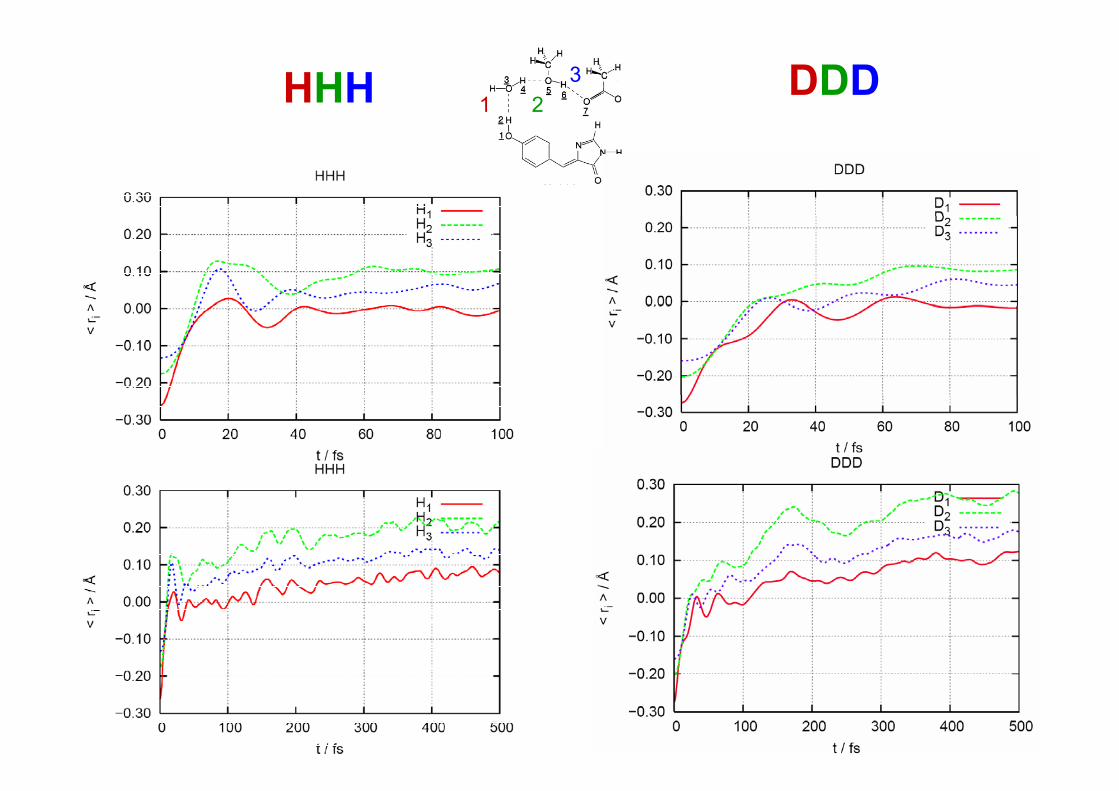
Evolució de la geometria
HHH
13
2HHH DDD
DDDHHH
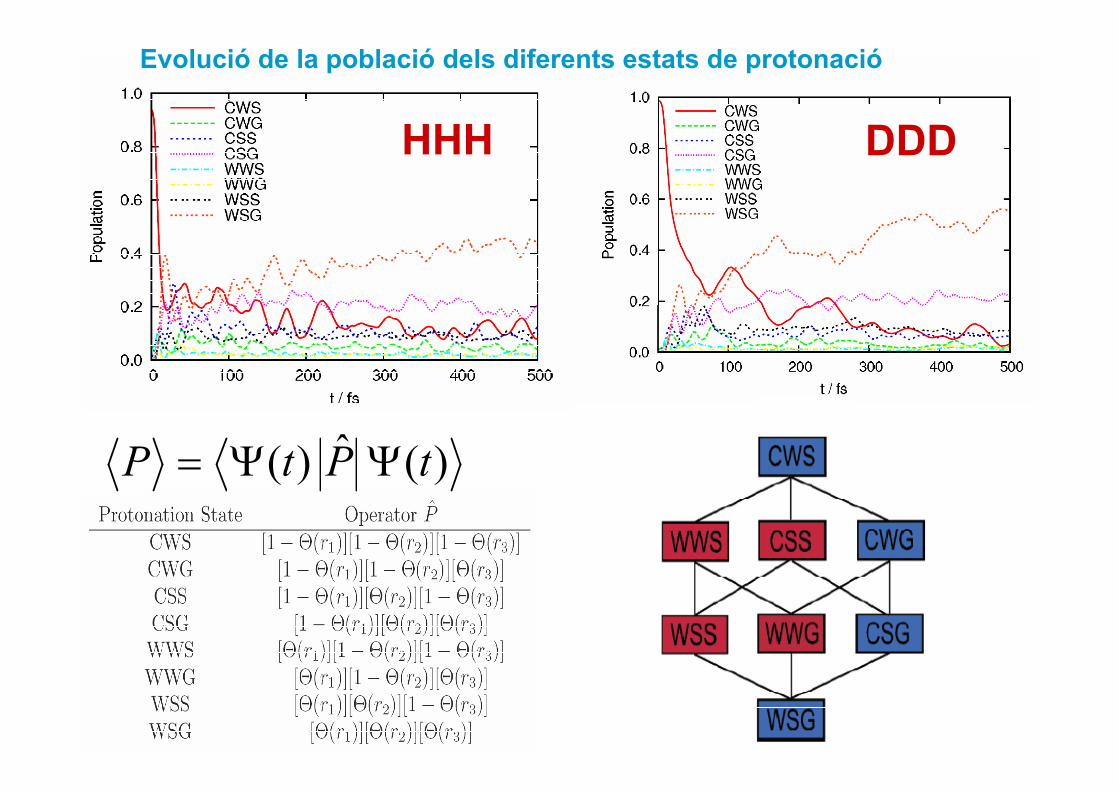
Evolució de la població dels diferents estats de protonació
DDDHHH
)(ˆ)( tPtP ΨΨ=

13
2Evolució temporal de la població dels estats de
protonació CXY
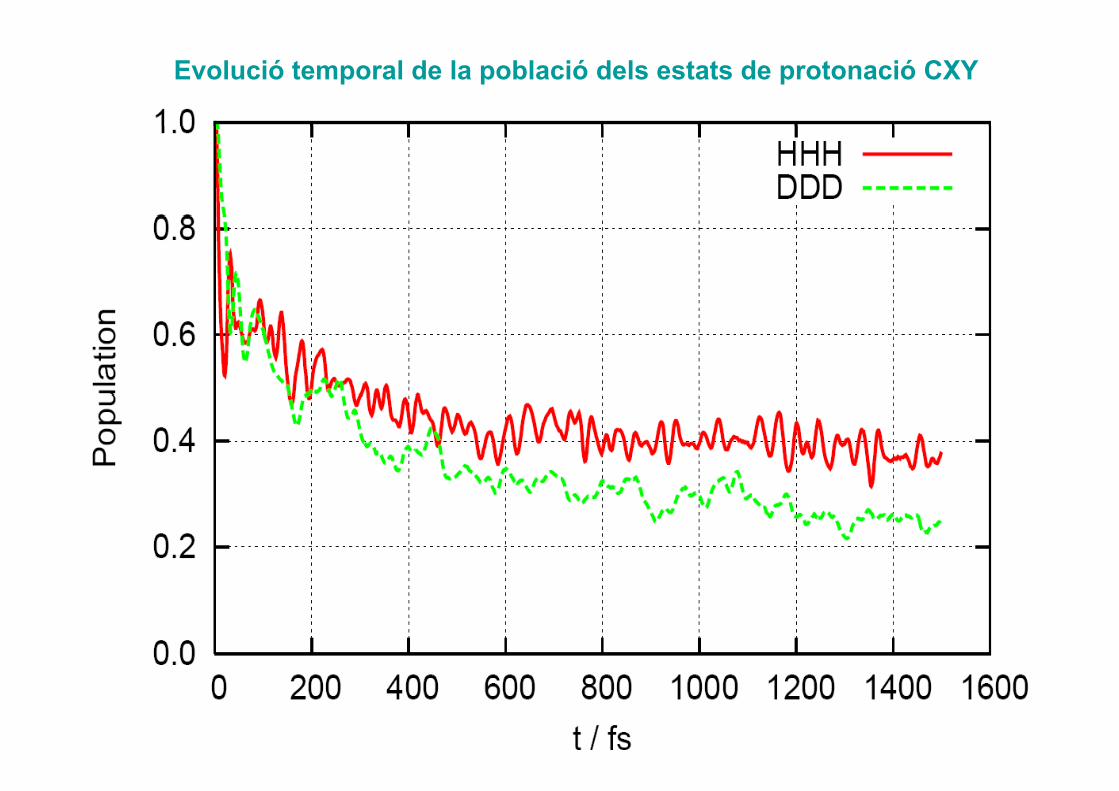
Evolució temporal de la població dels estats de protonació CXY
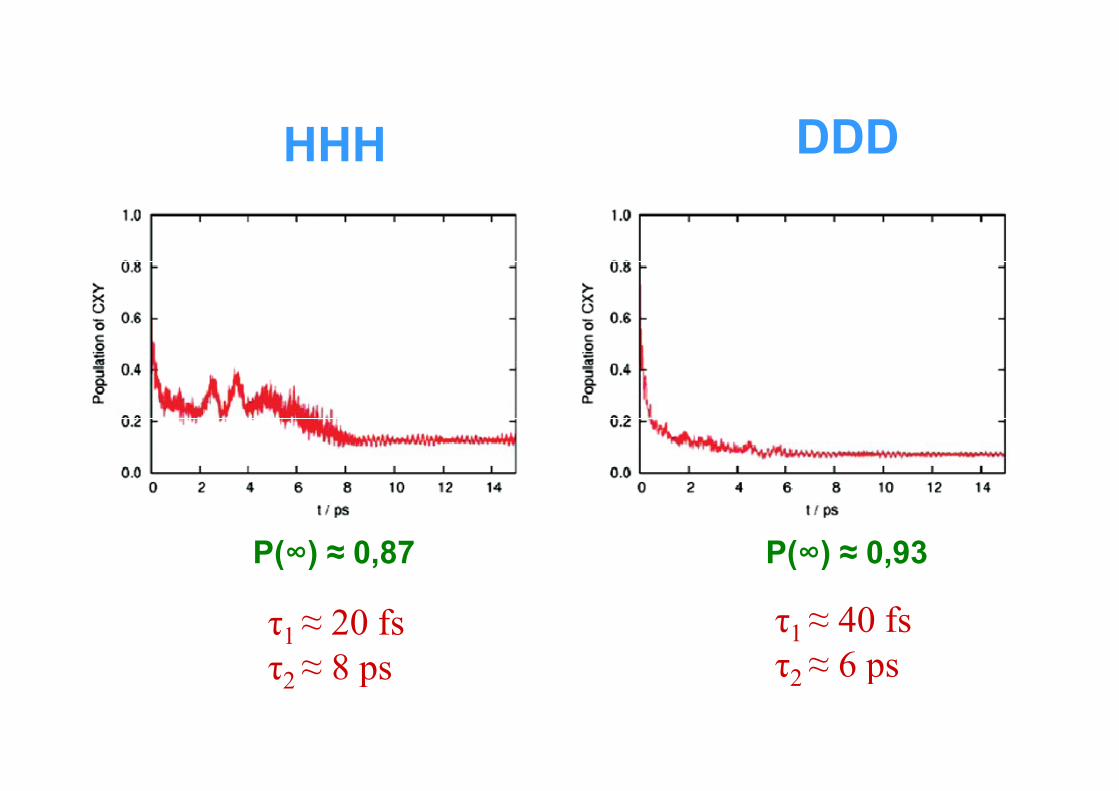
DDDHHH DDD
P(∞) ≈ 0,87 P(∞) ≈ 0,93
τ1 ≈ 20 fsτ ≈ 8 ps
τ1 ≈ 40 fsτ ≈ 6 psτ2 ≈ 8 ps τ2 ≈ 6 ps
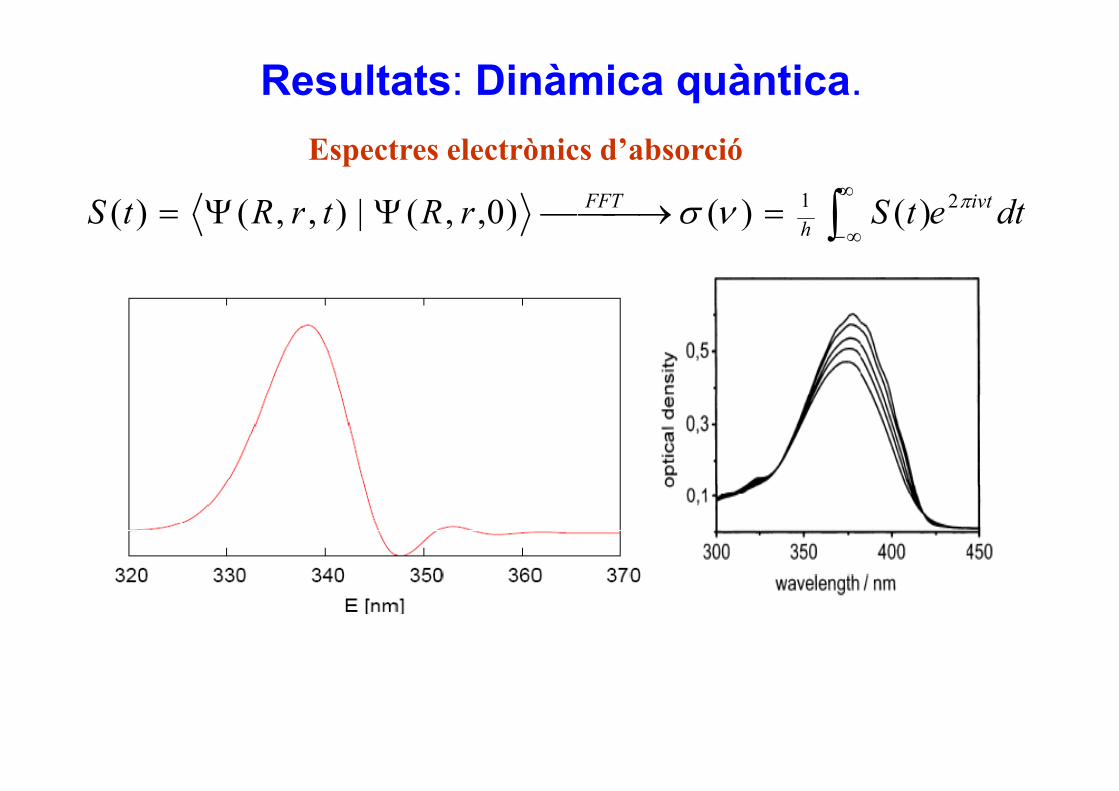
Resultats: Dinàmica quàntica.
dtetSrRtrRtS ivtFFT πνσ 21 )()()0(|)()( ∫∞
=⎯⎯ →⎯ΨΨ=
Espectres electrònics d’absorció
dtetSrRtrRtS hνσ )()()0,,(|),,()( ∫ ∞−=→ΨΨ=

Conclusions• El procés de triple transferència protónica en la GFP mitjançant fotoexcitació a l’ estat
excitat singlet ππ* de menor energia es concertat i pràcticament sincrònic.• El moviment dels protons és molt ràpid i en 20 fs (clarament per sota de la resolució
temporal experimental), tots els protons estan més propers dels seus respectius àtomsacceptors.
• El moviment global dels protons està fortament acoblat mostrant efectes isotòpicssignificatius: un KIE normal per a l’estadi inicial de la reacció (< 100fs) però un KIE inversen el límit asimptòtic.
• Mentre es manté la cadena protònica Cro-Wat25-Ser205-Glu222 la triple transferènciaprotònica no s’arriba a completar. Al contrari, al cap de ~8 ps el sistema entra enressonància. (A ↔ I).
• La introducció d’un bany d’oscil·ladors acoblats al sistema mostra que la relaxacióvibracional interna (IVR) pot augmentar notablement el temps de formació de l’espèciefluorescent tot i que aquest efecte no pot ser l’única causa del valor mesuratexperimentalment en l’escala de temps dels picosegons.
• És molt probable que per a trencar la cadena protònica es produeixi una reorganitzaciód’alguns residus per a permetre una sortida als protons. Aquesta reorganització seria laresponsable del KIE normal que ha estat mesurat experimentalment.

Perspectives de futur”T t l t ” d l d d t f è i tò i”Tancant la porta” de la cadena de transferències protònicas
(Procés I B)

Continuació de la cadena de transferències protòniques en la GFPp q
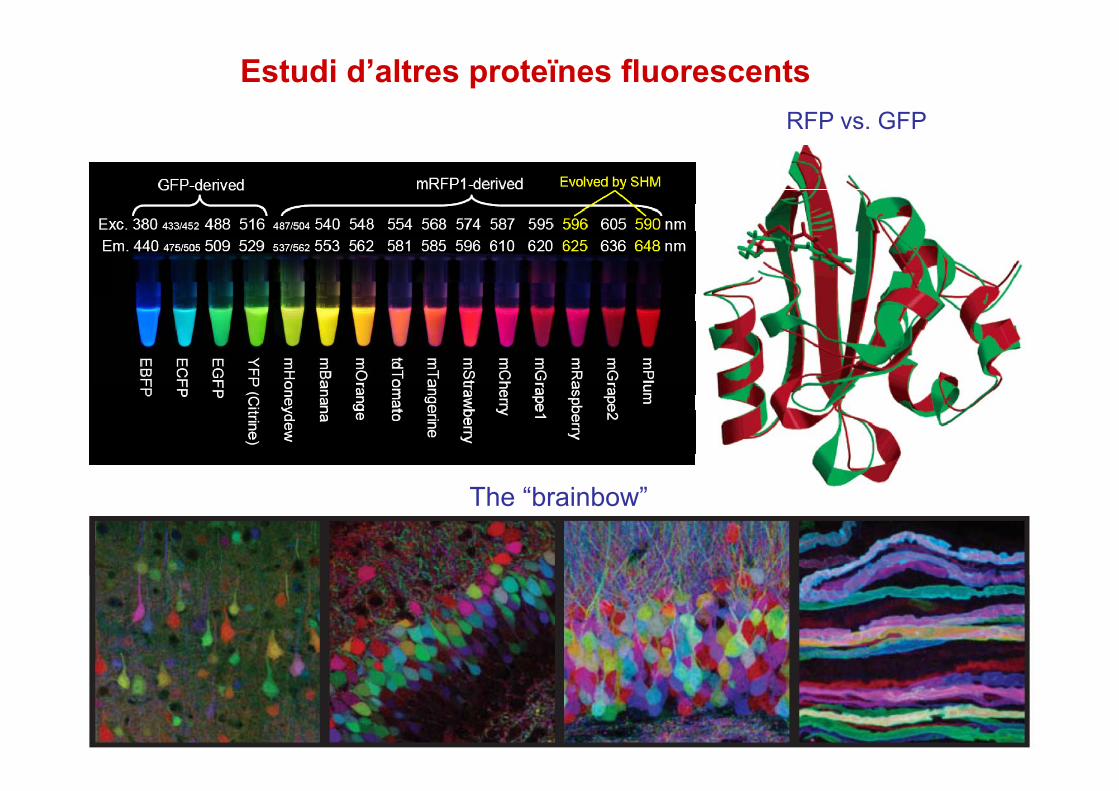
Estudi d’altres proteïnes fluorescentsRFP vs GFPRFP vs. GFP
The “brainbow”

%
Kindling Fluorescent Protein KFP
100% trans50% trans50% cis
100% cis
Llum taronja (568 nm)Llum
vermella
Llum blava (450 nm) (595 nm)

REFERÈNCIES GFPs1 O Vendrell R Gelabert M Moreno y J M Lluch Photoinduced Proton Transfer1. O. Vendrell, R. Gelabert, M. Moreno y J.M. Lluch. Photoinduced Proton Transfer
from the Green Fluorescent protein Chromophore to a Water Molecule: Analysisof the Transfer Coordinate. Chem. Phys. Lett. 396 (2004) 202-207.
2. O. Vendrell, R. Gelabert, M. Moreno y J.M. Lluch. Potential Energy Landscape ofthe Photoinduced Multiple Proton-Transfer Process in the Green FluorescentProtein: Classical Molecular Dynamics and Multiconfigurational ElectronicStructure Calculations. J. Am. Chem. Soc. 128 (2006) 3564-3574.
3. O. Vendrell, R. Gelabert, M. Moreno y J.M. Lluch. Operation of the Proton-Wire inGreen Fluorescent Protein. A Quantum Dynamics Simulation. J. Phys. Chem. B112 (2008) 5500-5511.112 (2008) 5500 5511.
4. O. Vendrell, R. Gelabert, M. Moreno y J.M. Lluch. A Potential Energy Function forHeterogeneous Proton-Wires. Ground and Photoactive States of the Proton-Wiregin the Green Fluorescent Protein. J. Chem. Theory Comput. 4 (2008) 1138-1150.
5. O. Vendrell, R. Gelabert, M. Moreno y J.M. Lluch. Exploring the Effects ofIntramolecular Vibrational Energy Redistribution on the Operation of the ProtonWire in Green Fluorescent Protein. J. Phys. Chem. B 112 (2008) 13443-13452.


















![Bloc 5 Cartografia [Modo de compatibilidad] · Coordenades Geogràfiques Coordenades UTM (Universal Transvers Mercator) Els meridians i els paral·lels no són rectes, no tenen una](https://static.fdocuments.es/doc/165x107/5fa61b438d5c1065141b4547/bloc-5-cartografia-modo-de-compatibilidad-coordenades-geogrfiques-coordenades.jpg)