
Enfermedades ligadas a Cromosoma X
65
El cromosoma X presenta una herencia de carácter sexual, por lo cual sus mutaciones son fácilmente identificables. Presenta 1100 genes, el 40% tiene relación con enfermedades.
-
Upload
oswaldo-angeles -
Category
Health & Medicine
-
view
641 -
download
2
Transcript of Enfermedades ligadas a Cromosoma X

El cromosoma X presenta una herencia de carácter sexual, por lo cual sus mutaciones son fácilmente identificables.
Presenta 1100 genes, el 40% tiene relación con enfermedades.

Sus mutaciones pueden tener un comportamiento recesivo o dominante.

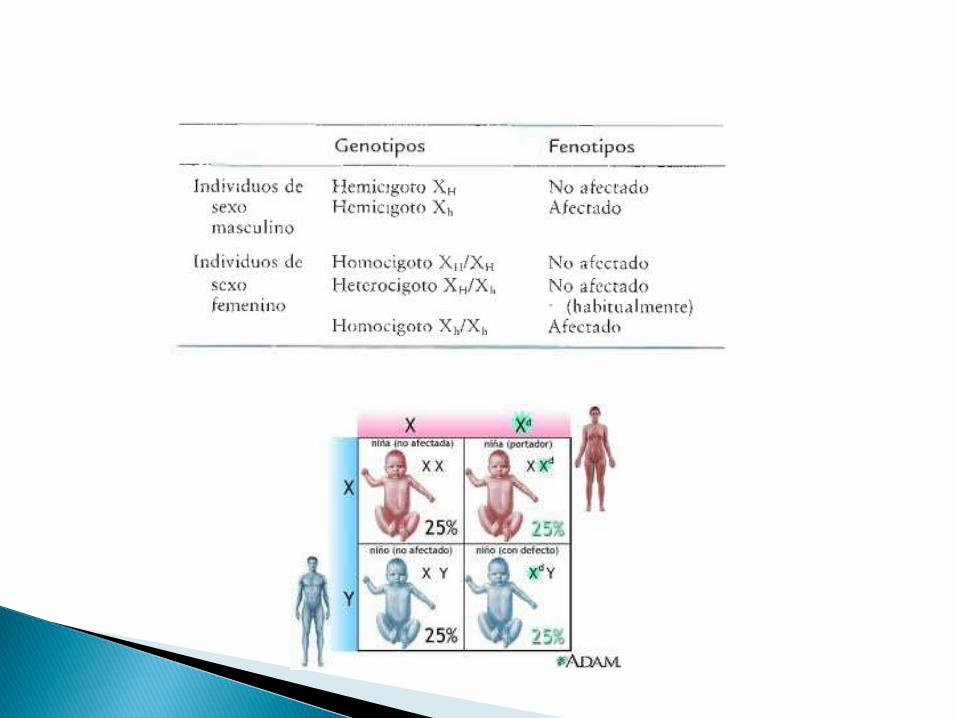
Masculinos con un alelo mutante = HEMICIGOTO
Femeninos con un alelo mutante = HETEROCIGOTOS
Femeninos con doble alelo = HOMOCIGOTOS


En todas las mujeres uno de los dos cromosomas X se inactiva en cada célula, entre los 12-16 días de vida intraembrionaria.
Se inactiva el cromosoma paterno o materno al azar. Lo cual se continua en las descendencias de células.
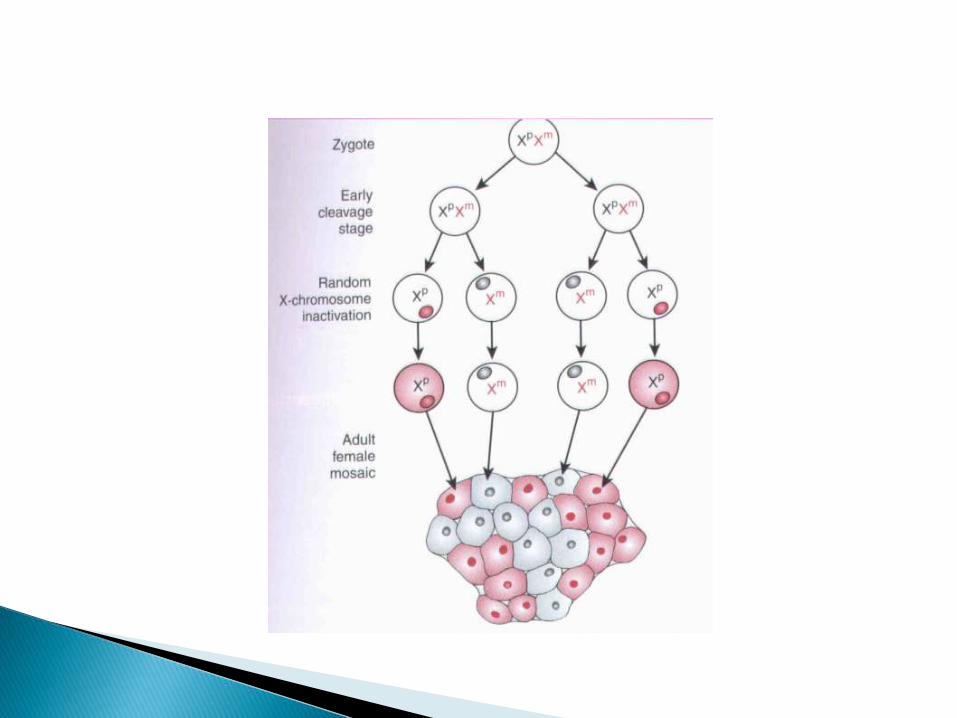

Cromosoma inactivo = corpúsculo de Barr.
Se compensa la dosis de genes entre machos y hembras.
Se inactivan con la metilación del DNA. El 15 % de los genes no se inactiva.


Centro de inactivación del cromosoma X se encuentra en el cromosoma inactivado.
Inactive X- specific transcripts. Es clave para la inactivación.
El gen XIST produce un RNA no codificante.


MUTACION DEL GEN DMD de codifica la Distrofina
•Duplicación•Delecion (ovogenesis)•Inserción•Cambio de Nucleótido (espermatogenesis)
INCIDENCIA 1 EN 3,500 VARONESHETEROGENEIDAD ALELICA
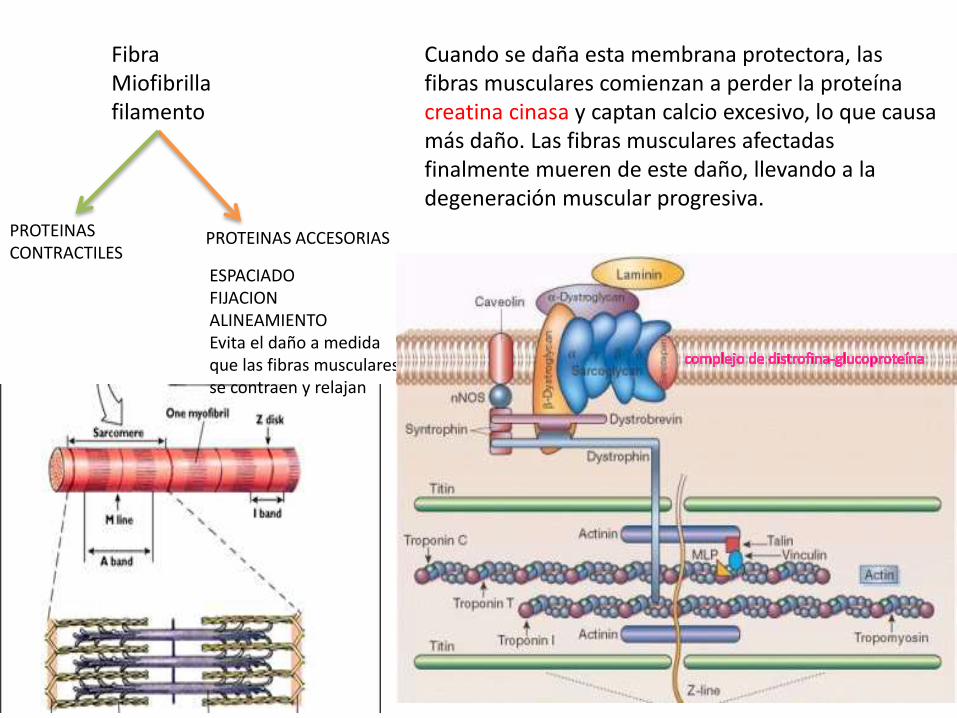
PROTEINAS ACCESORIASPROTEINAS CONTRACTILES
ESPACIADOFIJACIONALINEAMIENTOEvita el daño a medida que las fibras musculares se contraen y relajan
Cuando se daña esta membrana protectora, las fibras musculares comienzan a perder la proteína creatina cinasa y captan calcio excesivo, lo que causa más daño. Las fibras musculares afectadas finalmente mueren de este daño, llevando a la degeneración muscular progresiva.
FibraMiofibrillafilamento
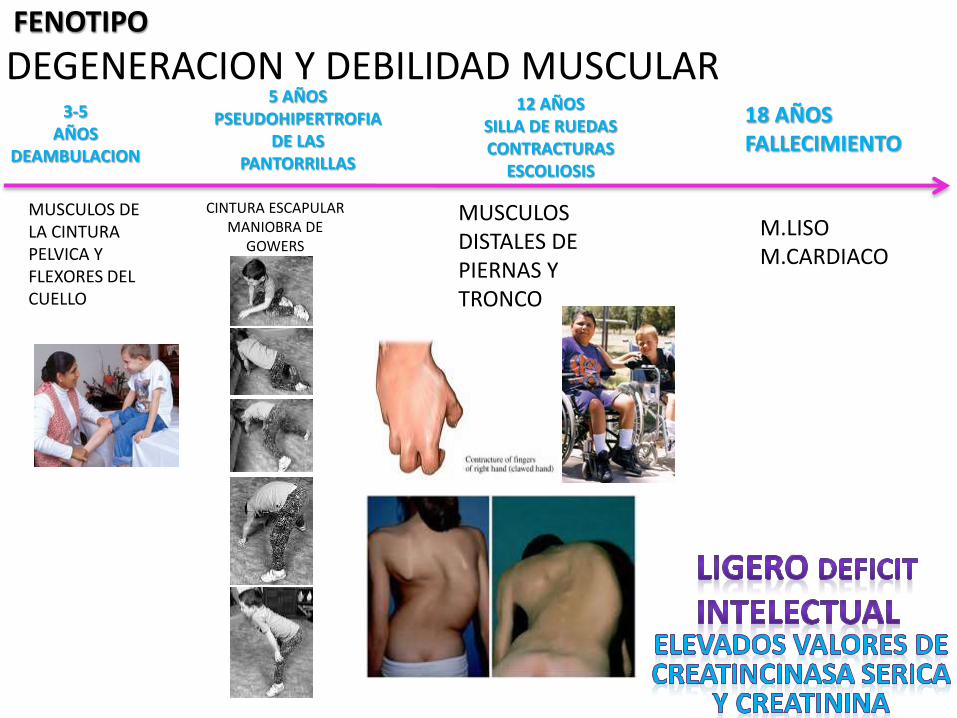
DEGENERACION Y DEBILIDAD MUSCULARFENOTIPO
MUSCULOS DE LA CINTURA PELVICA Y FLEXORES DEL CUELLO
CINTURA ESCAPULARMANIOBRA DE
GOWERS
MUSCULOS DISTALES DEPIERNAS Y TRONCO
3-5AÑOS
DEAMBULACION
5 AÑOSPSEUDOHIPERTROFIA
DE LASPANTORRILLAS
12 AÑOSSILLA DE RUEDASCONTRACTURAS
ESCOLIOSIS
18 AÑOSFALLECIMIENTO
M.LISOM.CARDIACO

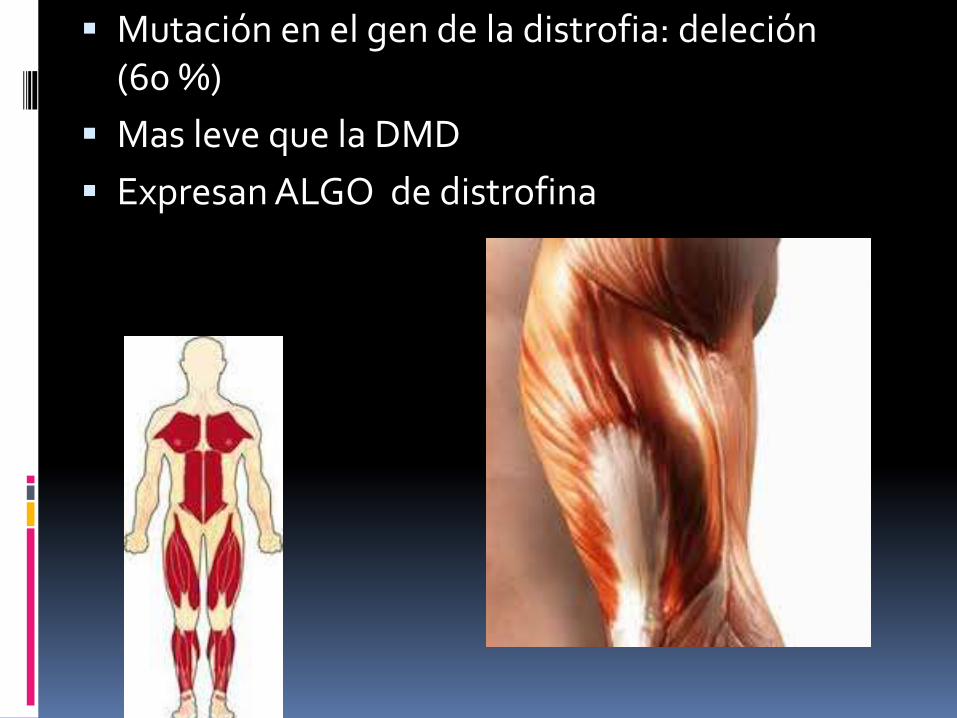
Mutación en el gen de la distrofia: deleción (60 %)
Mas leve que la DMD
Expresan ALGO de distrofina


CARACTERISTICAS
Hemorragias en tejidos blandos, músculos y articulaciones que soportan peso.
Sangrados prolongados
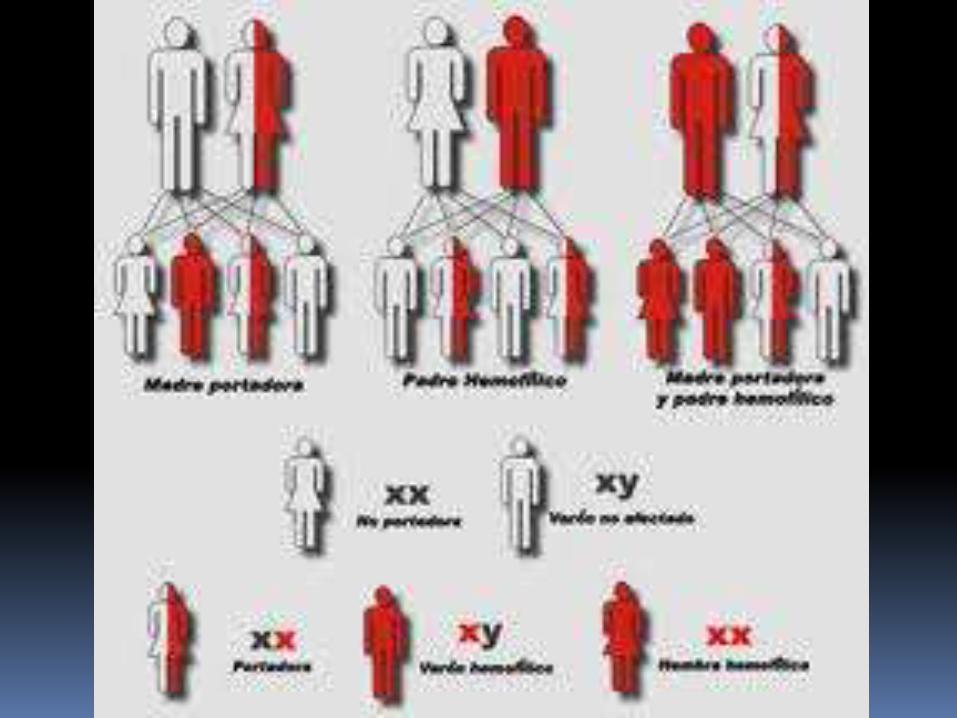
Enfermedad de varones ( raramente la presenta una mujer )

DOS TIPOS:
A B Gen F8 : inversión
(25%)
Deficiencia o disfunción del factor VIII (factor anti hemofílico)
Incidencia: 1 en cada 5 000 a 10 000.
Gen F9
Deficiencia o disfunción del factor IX
Incidencia : 1 en 100 000
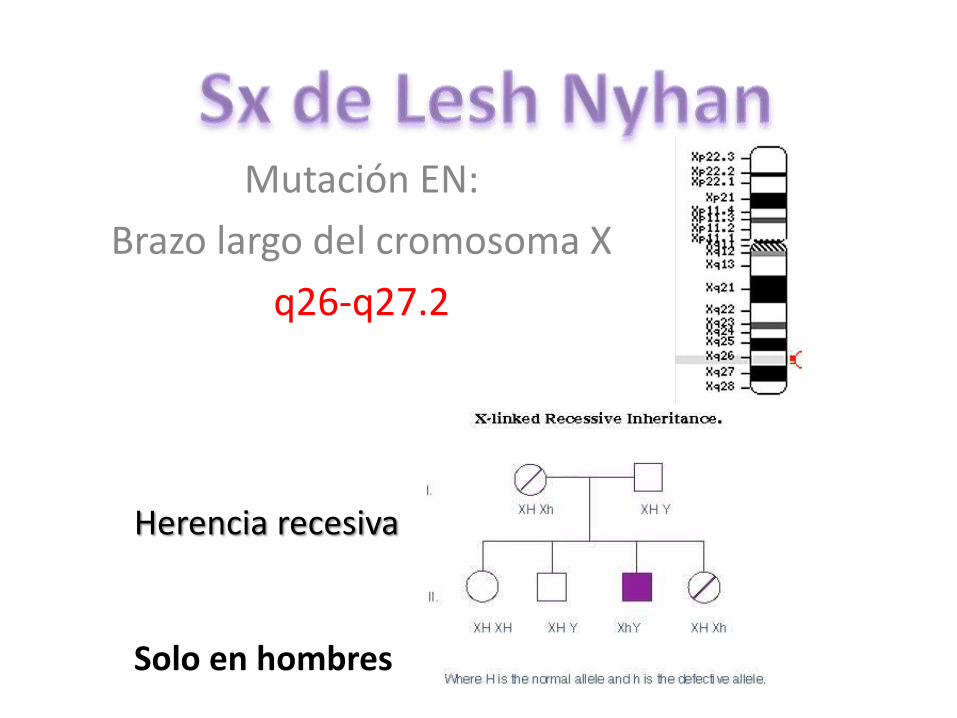
Mutación EN:
Brazo largo del cromosoma X
q26-q27.2
Herencia recesiva
Solo en hombres

Completa deficiencia de la enzima de recuperación hipoxantina guanina fosforribosil transferasa
HGPRT
Cataliza la adición de la 5-fosforribosil-1-pirofosfato PRPP a las bases de purina GUANINA-HIPOXANTINA en la VIA DE RECUPERACION DE BASES LIBRES
Adenina(base libre)
GuaninaO
hipoxantina
AMP
GMPO
IMPhipoxantina guanina fosforribosiltransferasa
HGPRT
The conversions of hypoxanthine and guanine plus PRPP to IMP and GMP respectively, and PPi by the action of HGPRT.
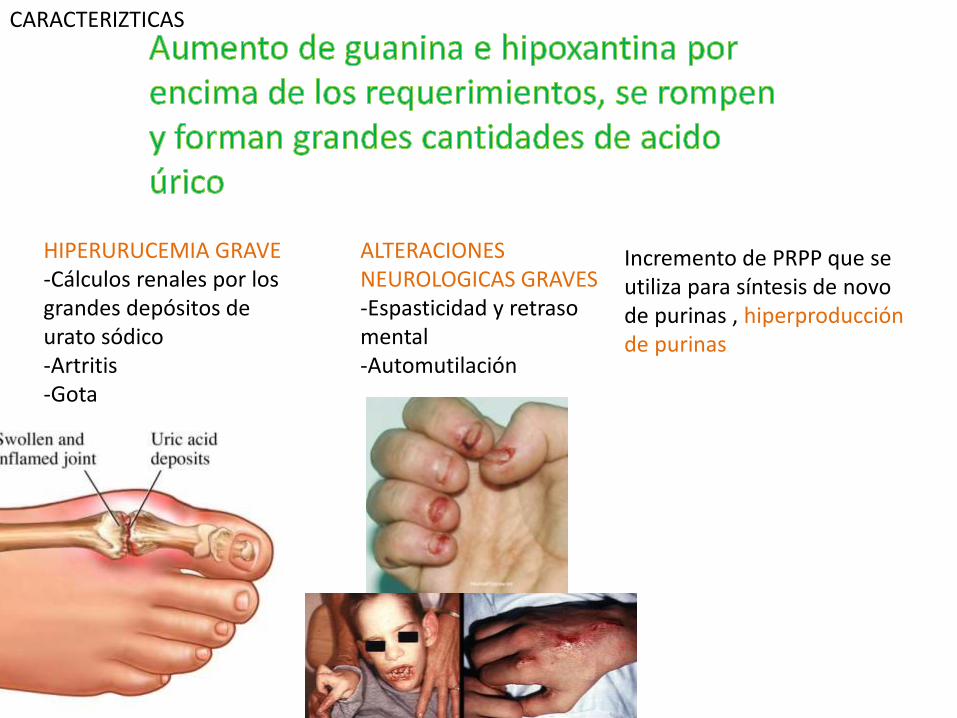
HIPERURUCEMIA GRAVE-Cálculos renales por los grandes depósitos de urato sódico-Artritis-Gota
Incremento de PRPP que se utiliza para síntesis de novode purinas , hiperproducciónde purinas
ALTERACIONES NEUROLOGICAS GRAVES-Espasticidad y retraso mental-Automutilación
CARACTERIZTICAS

Dx
1. Pañal naranja2. Actividad hipoxantina guanina fosforribosil
transferasa3. Síntomas
Tx sintomático• Alopurinol (inhibidor de xantina oxidasa)para disminuir niveles de acido úrico• AINES• DIASEPAM

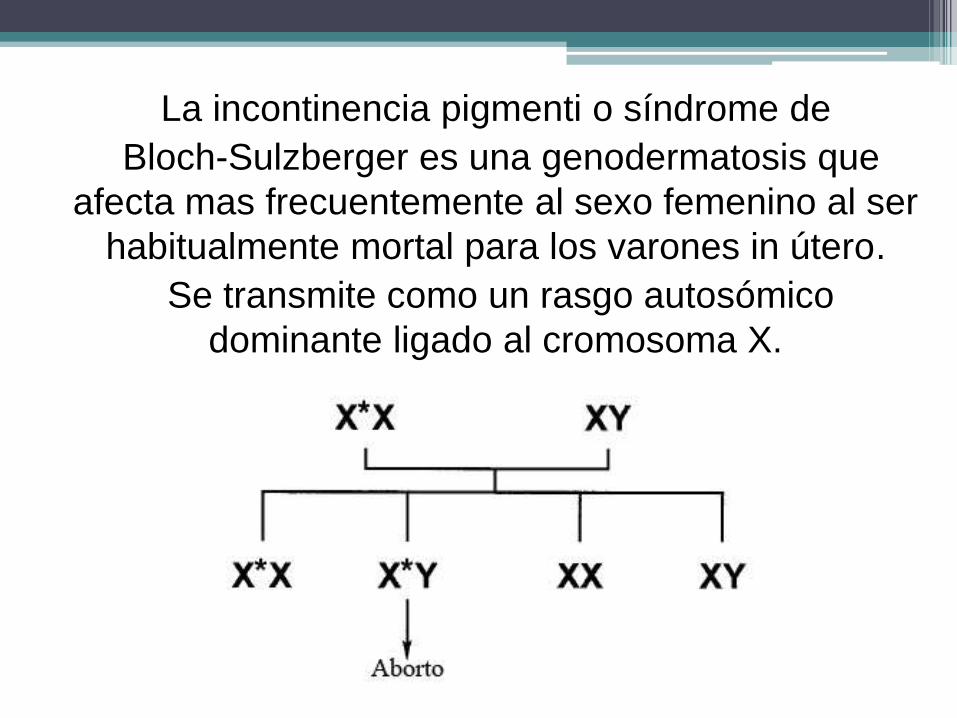
La incontinencia pigmenti o síndrome de
Bloch-Sulzberger es una genodermatosis que
afecta mas frecuentemente al sexo femenino al ser
habitualmente mortal para los varones in útero.
Se transmite como un rasgo autosómico
dominante ligado al cromosoma X.

Aunque afecta primariamente a la piel, existen
diferentes trastornos asociados, incluyendo
defectos dentales, trastornos convulsivos, retraso
mental, anomalías oculares y neoplasias
infantiles.
Dado que suele ser letal en varones, se
observa casi exclusivamente en
niñas.
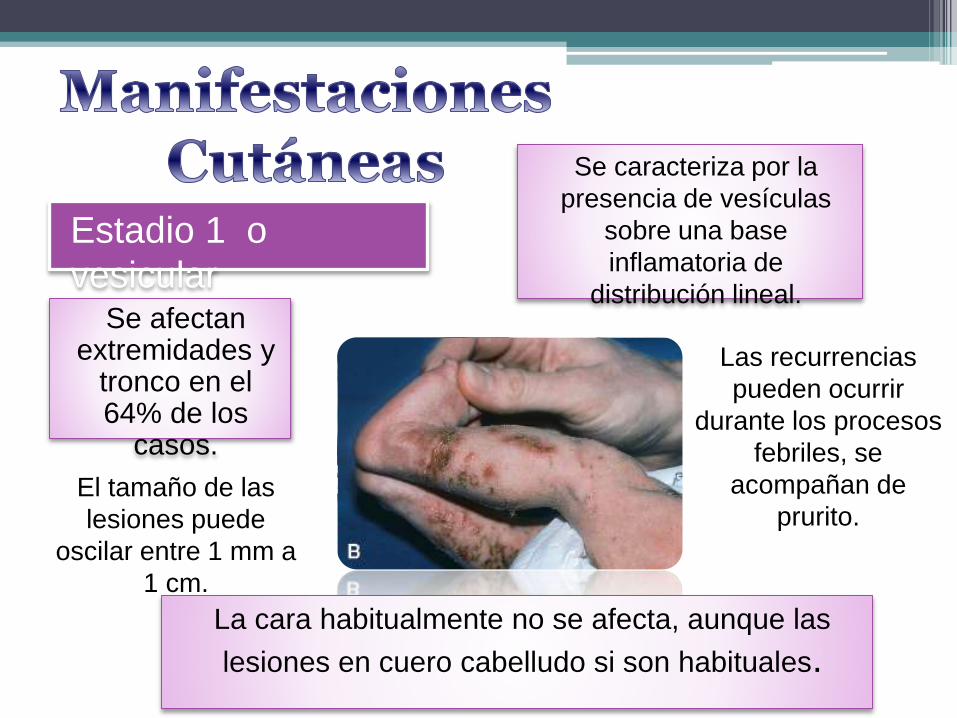
Estadio 1 o
vesicular
Se caracteriza por la
presencia de vesículas
sobre una base
inflamatoria de
distribución lineal.Se afectan
extremidades y tronco en el 64% de los
casos.
La cara habitualmente no se afecta, aunque las
lesiones en cuero cabelludo si son habituales.
El tamaño de las
lesiones puede
oscilar entre 1 mm a
1 cm.
Las recurrencias
pueden ocurrir
durante los procesos
febriles, se
acompañan de
prurito.
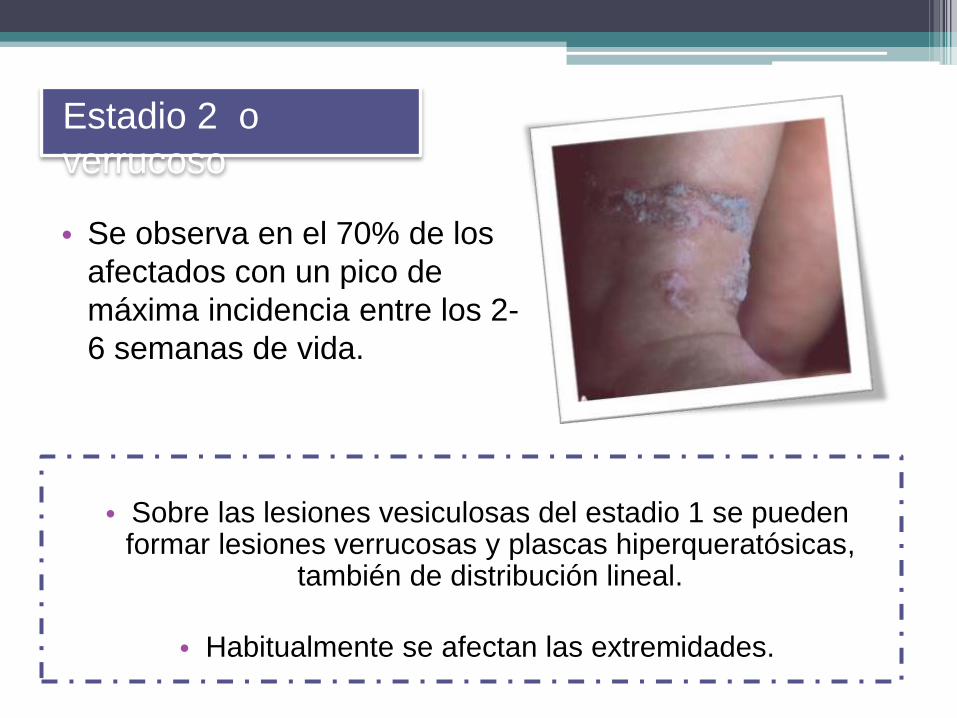
• Se observa en el 70% de los
afectados con un pico de
máxima incidencia entre los 2-
6 semanas de vida.
Estadio 2 o
verrucoso
• Sobre las lesiones vesiculosas del estadio 1 se pueden formar lesiones verrucosas y plascas hiperqueratósicas,
también de distribución lineal.
• Habitualmente se afectan las extremidades.

Las lesiones típicas de este
estadio aparecen como
lesiones pigmentadas de
color gris oscuro con una
distribución lineal o espiral.
Se pueden afectar tanto
tronco como extremidades;
siendo las axilas y región
pectoral la mas
frecuentemente afectada.
El inicio de las lesiones se
observa entre 12 - 26
meses de edad y persisten
durante años.
Estadio 3 o
verrucoso
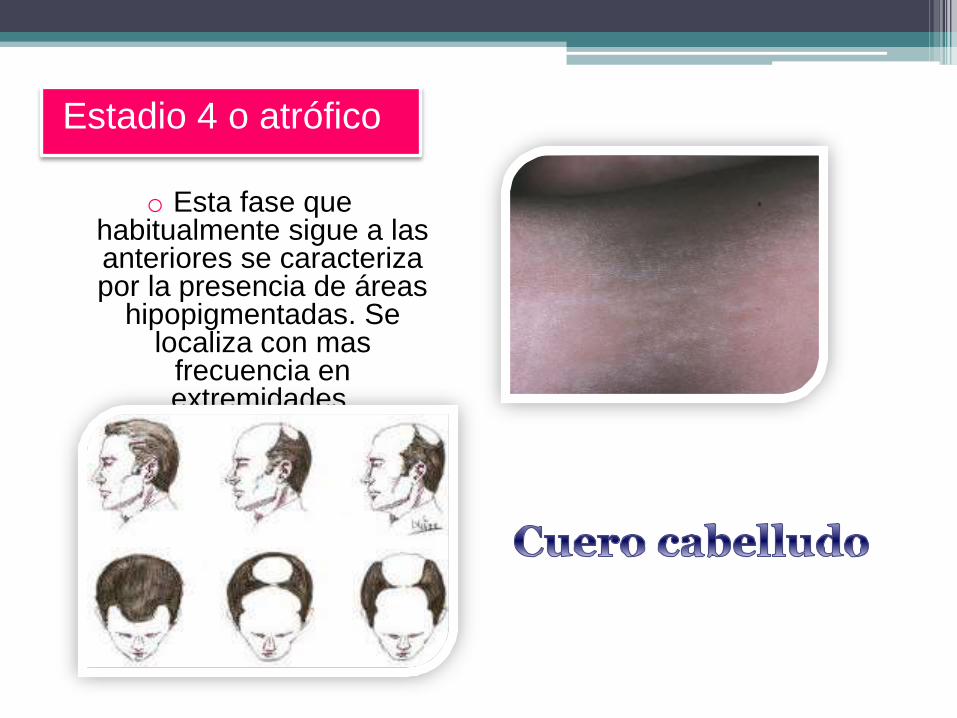
o Esta fase que habitualmente sigue a las anteriores se caracteriza por la presencia de áreas
hipopigmentadas. Se localiza con mas
frecuencia en extremidades.
Estadio 4 o atrófico
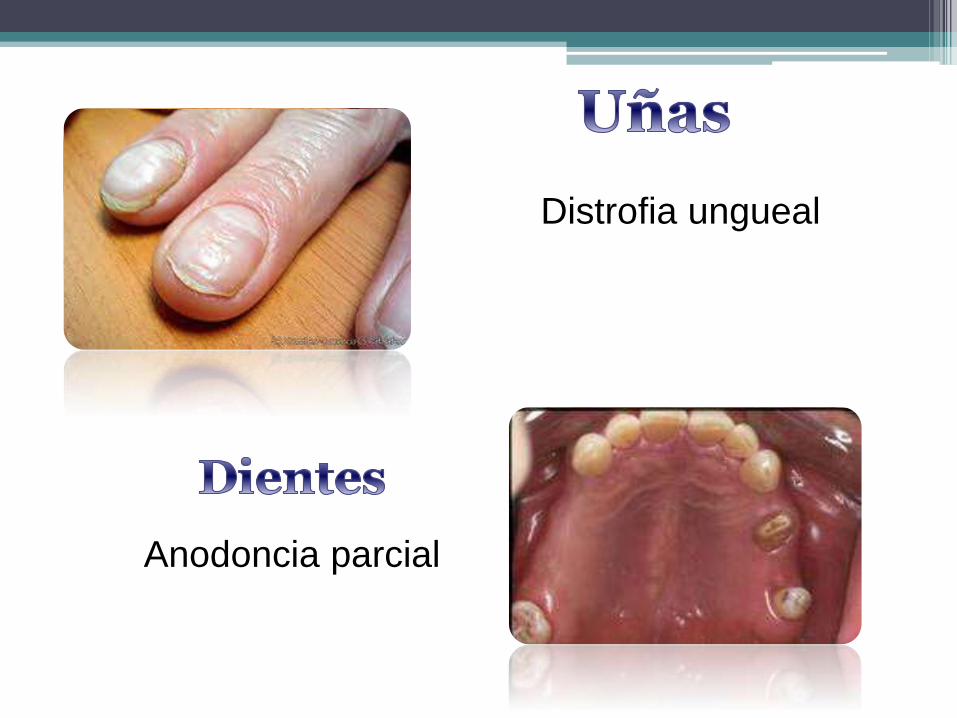
Distrofia ungueal
Anodoncia parcial

No retinales:
estrabismo, ptosis.
Retinales: afectación de
la retina periférica o la
mácula.
Retraso mental,
hidrocefalea.

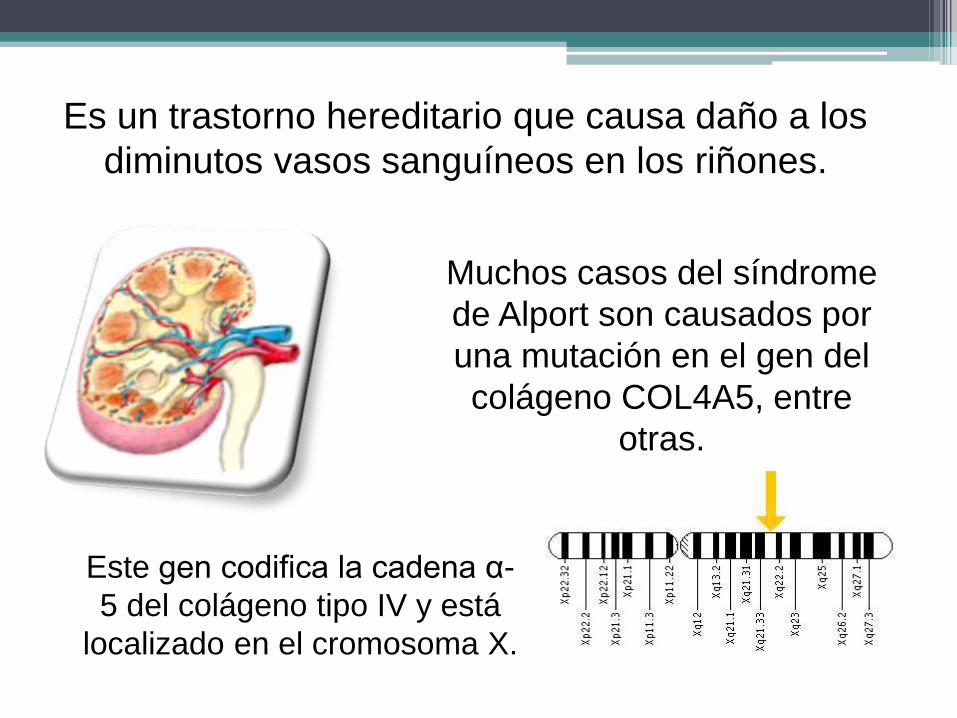
Es un trastorno hereditario que causa daño a los
diminutos vasos sanguíneos en los riñones.
Muchos casos del síndrome
de Alport son causados por
una mutación en el gen del
colágeno COL4A5, entre
otras.
Este gen codifica la cadena α-
5 del colágeno tipo IV y está
localizado en el cromosoma X.

• Afección renal
• Coclear
• Ocular
La principal señal de este síndrome, es la hematuria
microscópica (microhematuria).
Los hombres con el síndrome Alport ligado al cromosoma X (XLAS)
padecen micro hematuria desde una edad muy temprana.
Alrededor del 90% de mujeres con XLAS también la tienen.

SÍNDROME DE HUNTER

Enfermedad por acumulación lisosómica debida adeficiencia de Iduronato sulfatasa.
Recesivo ligado al cromosoma X.
Mucopolisacaridosis. (mas de 12 enfermedadespor acumulación lisosomal)
Los mucopolisacáridos se acumulan en el interiorde los lisosomas por deficiencia de enzimasnecesarias para su degradación.
Aparecen en la orina, se pueden detectarmediante pruebas de cribado.

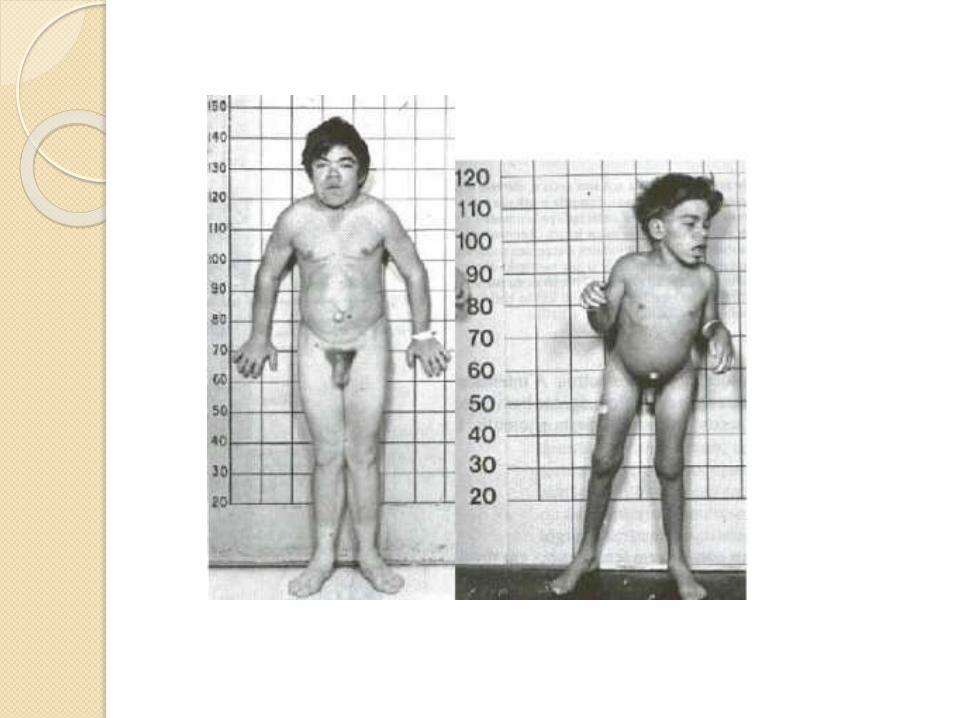
CLINICA
Dx. Fase temprana <18 meses
Opacación corneal
Alteraciones esqueléticas
Hepatoesplenomegalia
Rasgos faciales toscos
Fallecimiento antes de los diez años
de edad.

DEFECTO ENZIMÁTICO,
GENÉTICA Iduronato sulfatasa
XR
Debido a alelos que alteran la
actividad enzimática, fenotipo más
leve que el Sx. De Hurler, con
afectación variable del SNC.

Los diferentes patrones de transmisión
hereditaria del Sx. De Hurler autosómico y
el Sx. De Hunter ligado al cromosoma X
indican que se deben a mutaciones en
genes distintos.
Los fibroblastos deficientes en α-L-
iduronidasa del Sx. De Hurler captan la α-
L- iduronidasa normal liberada por los
fibroblastos del Sx. De Hunter y viceversa.

Afectan a proteínas distintas.
La demostración de que un producto
procedente de un mutante de un mutante
del genoma puede corregir el defecto
bioquímico de otro mutante se denomina
complementación genética.
Los estudios para ver si es posible ésta
son los análisis de complementación.

La capacidad de una célula para captar a
partir del líquido extracelular la enzima
lisosomal de la que carece explica el
éxito en trasplantes de células normales.
Se ha visto éxito en el tratamiento de
trasplante de médula ósea.


SÍNDROME DE RETT

Afecta casi exclusivamente a mujeres. (1:10000,1:15000)
Trastorno dominante ligado a X con letalidad delos varones hemicigotos.
Mutaciones espontáneas de un gen delcromosoma X, MECP2, que codifica una proteínade unión al DNA, proteína de unión a metil-Cp G2 causando su alteración una pérdida de funciónde factores neurotróficos por disrupción enmetilación de unión a ADN y alteración enrepresores transcripcionales.

PRINCIPIOS
Mutaciones de pérdida de función
Penetrancia incompleta
Expresividad variable
Fenotipo dependiente del sexo

CARACTERÍSTICAS
FENOTÍPICAS PRINCIPALES.
Inicia: neonatal o infancia temprana
Regresión en el desarrollo neurológico

Mecanismo desconocido, alteraciones
en la regulación de un conjunto de
genes en el cerebro en desarrollo.
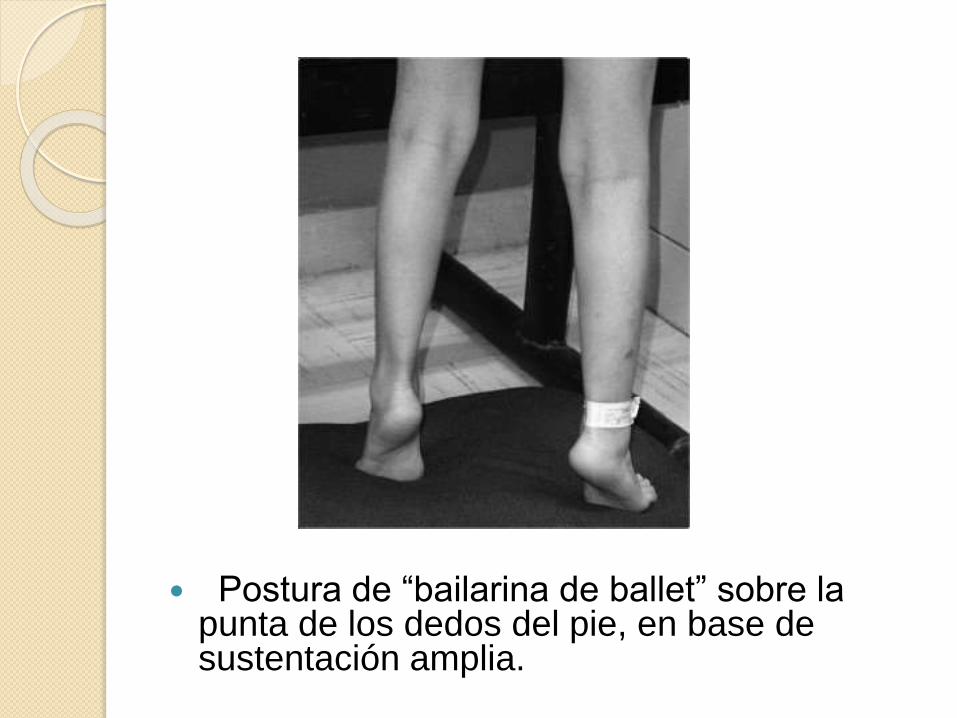
Postura de “bailarina de ballet” sobre la punta de los dedos del pie, en base de sustentación amplia.

Crecimiento y desarrollo prenatal y
neonatal normales, seguido de la
aparición rápida de sintomatología
neurológica con pérdida entre los 6 y
los 18 meses de edad de los hitos del
desarrollo ya alcanzados.

CLÍNICA
Espasticidad
Ataxia
Rasgos autistas
Comportamiento irritable con arrebatosde llanto.
Movimientos de retorcimiento o aleteo nodeliberados en la manos y brazos.
Reducción del crecimiento de la cabeza
Microcefalia
Convulsiones (50%)

Cerebro pequeño, atrofia cortical y
cerebelar sin pérdida de neuronas.
(no es degenerativa típica)
Córtex e hipocampo neuronas mas
pequeñas y dispuestas en forma mas
densa, patrón de ramificaciones
dendríticas simplificado.
MeCP2 establece y mantiene
interacciones neuronales.
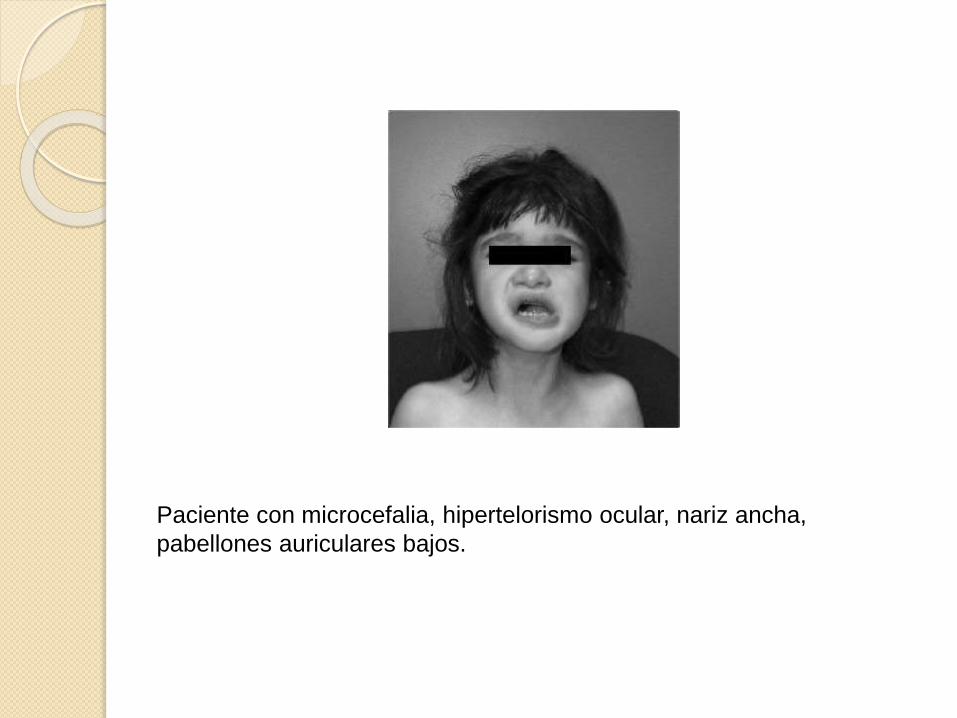
Paciente con microcefalia, hipertelorismo ocular, nariz ancha,
pabellones auriculares bajos.


NIÑAS
Afectación grave: no habla, darse
vuelta o andar. Epilepsia grave.
Ligera.


NIÑOS
Muerte intrauterina
Encefalopatía congénita
Retraso mental
clásico: sólo en niños con mosaicismo
somático para una mutación en
MECP2 o un cromosoma X extra.

El progreso del deterioro mental se
interrumpe después de unos años.
Pacientes sobreviven con una
discapacidad neurológica grave pero
estable.



CONTROL Y TRATAMIENTO
Dx. Clínico y por pruebas de DNA
Tx. Sintomático y de apoyo:
anticonvulsivantes, carbidopa o
levodopa para la rigidez, melatonina
para trastornos del sueño

HERENCIA
99% esporádicos
Raros casos heredado de una madre
no afectada o poco afectada con un
desequilibrio en la inactivación del
cromosoma X.
70% mutaciones de Novo se originan
de línea germinal paterna.
Hijo afectado y padres no: riesgo bajo
para futuros hermanos.

Madre portadora de una mutación en
MECP2: hijo o hija con 50% de
probabilidad.


















