
ESTUDIO TEÓRICO DE LA ESTRUCTURA ELECTRÓNICA Y DE LOS...
131
UNIVERSIDAD DE CHILE Facultad de Ciencias Químicas y Farmacéuticas ESTUDIO TEÓRICO DE LA ESTRUCTURA ELECTRÓNICA Y DE LOS MECANISMOS DE TAUTOMERÍA LACTAMA-LACTIMA EN BILINAS DE FITOCROMO Tesis Entregada a la U. de Chile Para Optar al Grado Académico de Doctor en Química POR Ricardo Andrés Matute Morales DIRECTOR: Renato Contreras Ramos CO-DIRECTOR: Leticia González Herrero Santiago, Chile 2010
Transcript of ESTUDIO TEÓRICO DE LA ESTRUCTURA ELECTRÓNICA Y DE LOS...

UNIVERSIDAD DE CHILEFacultad de Ciencias Químicas y Farmacéuticas
ESTUDIO TEÓRICO DE LA ESTRUCTURA ELECTRÓNICA Y DE LOS MECANISMOS DE TAUTOMERÍA LACTAMA-LACTIMA
EN BILINAS DE FITOCROMO
Tesis Entregada a la U. de ChilePara Optar al Grado Académico de
Doctor en Química
POR
Ricardo Andrés Matute Morales
DIRECTOR:Renato Contreras Ramos
CO-DIRECTOR:Leticia González Herrero
Santiago, Chile
2010

UNIVERSIDAD DE CHILEFACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS
INFORME DE APROBACIÓNTESIS DE DOCTORADO
Se informa a la Dirección de Postgrado de la Facultad de Ciencias Químicas y Farmacéuticas que la Tesis de Doctorado presentada por el candidato:
RICARDO ANDRÉS MATUTE MORALES
Ha sido aprobado por la Comisión Informante de Tesis para optar al grado de Doctor en Química, en el examen de defensa de Tesis rendido el día ….........................................................................................................
______________________________________Directores de Tesis:
Dr. Renato Contreras …..........................................
Dr. Leticia González …..........................................
Comisión Informante de Tesis:
Dr. Octavio Vásquez (Presidente) …..........................................
Dr. Boris Weiss …..........................................
Dr. Antonio Zanocco …..........................................
Dr. Eduardo Lissi …...................................
ii

A la memoria de mi padre,
Ricardo Segundo Matute Carvajal
iii

Publicaciones Originadas de esta Tésis
1. Ricardo A. Matute, Renato Contreras, Guillermo Pérez-Hernández, and
Leticia González*. (2008) The Chromophore Structure of the Cyanobacterial
Phytochrome Cph1 As Predicted by Time-Dependent Density Functional
Theory. J. Phys. Chem. B, 112, 16253-16256.
2. Ricardo A. Matute*, Renato Contreras, and Leticia González*. (2010).
Time-Dependent DFT on Phytochrome Chromophores: A Way to the Right
Conformer. J. Phys. Chem. Lett., 1, 796-801.
3. Ricardo A. Matute*, Jorge Soto-Delgado, Leticia González, and Renato
Contreras. (2010). A Lactam-Lactim Tautomerism Involved in the Pr/Pfr
Interconversion of Phytochrome. (Submitted)
* Corresponding Author
iv

Presentaciones en Reuniones Científicas
1. Ricardo A. Matute, Renato Contreras, Guillermo Pérez-Hernández, Leticia
González, “Time-Dependent DFT on Phytochrome Chromophores: A Way to
Find the Right Conformer”, XXIV International Conference on
Photochemistry (ICP2009), 2009, Toledo, Spain. (Oral Presentation)
2. Ricardo A. Matute, Renato Contreras, Guillermo Pérez-Hernández, Leticia
González, “Conformation adopted by Phytochromobilin in the Pr isoform of
plant phytochrome: ZZZssa or ZZZasa?”, 15th International Congress on
Photobiology (ICP2009), 2009, Düsseldorf, Germany. (Poster)
3. Ricardo A. Matute, Renato Contreras, Guillermo Pérez-Hernández, Leticia
González, “Time-Dependent DFT on Phytochrome Chromophores: A Way to Find
the Right Conformer”, 9th International Conference on Tetrapyrrole
Photoreceptors of Photosynthetic Organisms (ICTPPO2009), 2009, Asilomar
Conference Center, Monterey, California, USA. (Poster)
v

AGRADECIMIENTOS
Agradezco a mi director de tesis, Prof. Dr. Renato Contreras, quién supervisó mi
trabajo, brindándome todo el apoyo necesario al momento de proponer y desarrollar mis ideas.
De igual manera, agradezco profundamente a mi co-director de tesis, Prof. Leticia González,
quién me recibió especialmente bien en su laboratorio cuando realicé las pasantías de tesis en
Alemania, inicialemte en Berlin (Freie Universität Berlin) y luego en Jena (Friedrich-Schiller-
Universität Jena), donde pude aprender métodos de fotoquímica computacional.
Toda mi gratitud para ambos profesores, Prof. Contreras y Prof. González, por guiarme
en el diseño de metodologías y estrategias de trabajo, en la discusión y organización de
resultados, y en la redacción de los artículos correspondientes, entre otras muchas tareas que se
ven plasmadas en este trabajo. Además, agradezco al Dr. Gerrit Groenhof, quién me recibió
cordialmente y fue mi supervisor durante la pasantía de tesis que realice en su laboratorio, en el
Instituto Max-Planck de Química Biofísica (Göttingen, Alemania). Con él pude aprender y
deasorrollar todo el trabajo relacionado con simulaciones de dinámica molecular.
Esta tesis contó con el financiamiento de la Beca de Doctorado CONICYT, la Beca
Integrada CONICYT-DAAD para poder realizar mi primera pasantía de tesis en Alemania, los
beneficios de BECAS CHILE de Pasantías Doctorales para la realización de mi segunda
pasantía de tesis, y la Beca de Asistencia a Conferencias Internacionales para poder presentar
mis resultados en la conferencia sobre fitocromos realizada en California. Sin tal financiamiento
esta tesis no hubiese sido posible, por lo que agradezco a CONICYT, BECAS CHILE y DAAD.
Finalmente, agradezco a mi madre y hermanas por su incondicional cariño,
comprensión, y apoyo; y extiendo mi sincero agradecimiento a toda la gente que de uno u otro
modo colaboraron y me apoyaron en este proyecto de tesis, especialmente al Prof. Dr. Lagarias
y al Dr. Rockwell de la UC Davis (California, USA) por sus valiosos consejos y discusión sobre
fitocromo; también a Jorge Soto, Claudio Pérez, Guillermo Pérez, Rodolfo Briones, Mauricio
Sandoval, Julio Matute, Mauricio Ipinza, Michel Agredo, Sebastian Miranda, Carlos Areche,
German Miño, Daniel Kinzel, Hartmut Preuβ, Marteen Wolf, y Sarath Chandra.
vi

TABLA DE CONTENIDOS
Página
AGRADECIMIENTOS ............................................................................................................ vi
TABLA DE CONTENIDOS ….......................................................................................... vii
ÍNDICE DE FIGURAS ….................................................................................................... ix
ÍNDICE DE TABLAS ….................................................................................................... x
ABREVIATURAS Y ACRÓNIMOS …................................................................................ xi
RESUMEN …....................................................................................................................... xii
ABSTRACT …....................................................................................................................... xiv
CAPÍTULO I INTRODUCCIÓN 1
1.1 Estructura y Función del Fitocromo …............................................................. 1
1.2 Hipótesis y Objetivos …...…............................................................................... 11
1.3 Referencias ….................................................................................................... 13
CAPÍTULO II ANÁLISIS ESTRUCTURAL 15
2.1 Aspectos Generales ......................................................................................... 15
2.2 Modelos Moleculares y Metodología Computacional ….................................... 16
2.3 Efecto de Protonación y Entorno …................................................................... 22
2.4 Estructura de la Ficocianobilina en Fitocromo de Cianobacteria ….................. 25
2.5 Estructura de la Fitocromobilina en Fitocromo de Planta …............................. 29
2.6 Modelo Teórico del Comportamiento del Índice Q/S …...................................... 34
2.6 Referencias …..................................................................................................... 37
vii

CAPÍTULO III TAUTOMERÍA EN LA INTERCONVERSIÓN 40
3.1 Aspectos Generales ......................................................................................... 40
3.2 Modelos Moleculares y Metodología Computacional ….................................... 42
3.3 En Busca del Desplazamiento Batocrómico de Pfr …........................................ 44
3.4 Mecanismo de la Tautomería Lactama-Lactima en Bacteriofitocromo …............. 46
3.5 13C-NMR de Ficocianobilina en Fitocromo …....................................................... 52
3.6 Referencias …..................................................................................................... 57
CAPÍTULO IV
CONCLUSIONES 59
APÉNDICE A MARCO TEÓRICO 61
A.1 Teoría del Funcional de la Densidad Dependiente del Tiempo .......................... 62
A.2 Modelo de Continuo Polarlizable …................................................................... 65
A.3 Cálculo de Apantallamiento RMN por el Método GIAO …............................... 69
A.4 Referencias …..................................................................................................... 71
APÉNDICE BTABLAS SUPLEMENTARIAS 72
B.1 Tablas Suplementarias del Capítulo II …........................................................... 73
B.2 Tablas Suplementarias del Capítulo III ….......................................................... 80
APÉNDICE C PUBLICACIONES Y PRESENTACIONES ASOCIADAS 84
viii

ÍNDICE DE FIGURAS
Página
Figura 1.1. Esquema para el fotociclo del fitocromo en planta 2
Figura 1.2. Diagrama de estados electrónicos involucrados en la fotoconversión del fitocromo 3
Figura 1.3. Bilinas con estructuras en discusión 6
Figura 1.4. Espectros electrónicos de absorción para fitocromo de plantas 7
Figura 1.5. Tautomería Lactama-Lactima 10
Figura 1.6. Entorno proteico de la biliverdina en bacteriofitocromo 10
Figura 2.1. Modelos para predecir estructura de PCB en fitocromo Cph1 de cianobacteria 17
Figura 2.2. Estructura del anillo A del cromóforo según el estado de unión a la cisteína 18
Figura 2.3. Modelos para predecir estructura de PФB en fitocromo A de planta 21
Figura 2.4. Espectros TDDFT de absorción UV-Vis para BV y PCB en fitocromo 26
Figura 2.5. Orbitales moleculares involucrados en el espectro de absorción UV-Vis de PCB-ssa 26
Figura 2.6. Espectros TDDFT de absorción UV-Vis para modelos de la fitocromobilina 30
Figura 2.7. Orbitales moleculares involucrados en el espectro de absorción UV-Vis de PФB-ssa 30
Figura 2.8. Estructura del cromóforo dentro de la estructura cristalográfica 31
Figura 3.1. Modelos de tautómeros para la forma Pfr de biliverdina en bacteriofitocromo 44
Figura 3.2. Espectros TDDFT de absorción UV-Vis para tautómeros de BV en bacteriofitocromo 45
Figura 3.3. Mecanismo no disociativo de tautomería con transferencia protónica intramolecular 46
Figura 3.4. Estados de transición para el mecanismo intramolecular de tautomería 48
Figura 3.5. Interacciones de la biliverdina en el cristal y en el TS catalizado por agua 49
Figura 3.6. Mecanismos propuestos para la tautomería de BV en bacteriofitocromo 51
Figura 3.7. Desplazamientos químicos para 13C-RMN por cálculos GIAO DFT 54
ix

ÍNDICE DE TABLAS
Página
Tabla 2.1. Efectos de protonación y entorno sobre la TDDFT para BV y PCB en fitocromo 23
Tabla 2.2. Máximos de absorción e índices Q/S calculados por la TDDFT 33
Tabla 2.3. Relación entre Q/S, F8, FQ 35
Tabla 3.1. Desplazamientos químicos para espectros 13C-RMN de modelos Pfr de PCB 53
Tabla B.1. Tabla suplementaria del espectro TDDFT de la figura 2.4-a 73
Tabla B.2. Tabla suplementaria del espectro TDDFT de la figura 2.4-b 74
Tabla B.3. Tabla suplementaria del espectro TDDFT de la figura 2.4-c 75
Tabla B.4. Tabla suplementaria del espectro TDDFT de la figura 2.6-a 76
Tabla B.5. Tabla suplementaria del espectro TDDFT de la figura 2.6-b 77
Tabla B.6. Tabla suplementaria del espectro TDDFT de la figura 2.6-c 78
Tabla B.7. Tabla suplementaria del espectro TDDFT de la figura 2.6-d 79
Tabla B.8. Tabla suplementaria del espectro TDDFT de la figura 3.2-a 80
Tabla B.9. Tabla suplementaria del espectro TDDFT de la figura 3.2-b 81
Tabla B.10. Tabla suplementaria del espectro TDDFT de la figura 3.2-c 82
Tabla B.11. Tabla suplementaria del espectro TDDFT de la figura 3.2-d 83
x

ABREVIATURAS Y ACRÓNIMOS
Å Angstrom. 10-10 metrosδ Desplazamiento Químico; utilizado en Resonancia Magnética Nuclearε Constante DieléctricaB3LYP Becke-3-parameter-Lee-Yang-ParrBphP BacteriofitocromoBV BiliverdinaCASSCF Complete-Active-Space Self-Consistent-Fieldcols. colaboradores13C-RMN Resonancia Magética Nuclear de 13CDFT Teoría del Funcional de la Densidade.g. “exempli gratia” (en latin), se usa en escritura inglesa: “por ejemplo”f fuerza de osciladorFWHM Full Width at Half Maximum HF Hartree-FockHOMO Orbital Molecular ocupado de mayor energíaLUMO Orbital Molecular desocupado de menor energíaMAS Magic Angle SpinningMPW1PW91 Funcional de intercambio-correlación de Perdew-Wang nm nanómetro. 10-9 metrosPФB FitocromobilinaPBE0 Funcional híbrido no empírico (libre de parámetros)PCB FicocianobilinaPCM Modelo de Continuo PolarizablePDB Protein Data BankPfr Forma fisiológicamente inactiva del fitocromo (absorbe en rojo-lejano)ppm partes por millónPr Forma fisiológicamente activa del fitocromo (absorbe en rojo)Q/S Cuociente de fuerzas de oscilador de la banda Q sobre la banda SoretRef. / Refs. Referencia / ReferenciasRMN Resonancia Magnética NuclearTDDFT Teoría del Funcional de la Densidad dependiente del TiempoTS Estado de Transiciónu.a. unidades arbitrariasZZEssa C5-Z,syn; C10-Z,syn; C15-E,antiZZZasa C5-Z,anti; C10-Z,syn; C15-Z,antiZZZssa C5-Z,syn; C10-Z,syn; C15-Z,antiZZZsss C5-Z,syn; C10-Z,syn; C15-Z,syn
xi

RESUMEN
El fitocromo es un fotoreceptor de plantas que también se encuentra en bacterias
y hongos. En plantas, el fitocromo regula respuestas fotomorfogénicas de la planta
como, por ejemplo, el desarrollo y crecimiento del tallo, regulación en el periodo de
dormancia y de germinación de las semillas, floración, fototaxis, mecanismos de
evasión de la sombra, etc. Todos ellos activados o desactivados en forma diferencial
mediante longitudes de onda específicas en la región UV-Visible del espectro
electromagnético. El receptor se fotoconvierte entre una forma fisiológicamente
inactiva, Pr, y una forma fisológicamente activa, Pfr. La interconversión entre ambas
formas ocurre por mecanismos diferentes que aun se desconocen. Por ello, hay un
interés creciente respecto al mecanismo de fotoconversión del fitocromo debido a
potenciales aplicaciones en biotecnología. Sin embargo, un obstáculo en la
investigación del fitocromo ha sido la escasez de estructuras cristalográficas resueltas en
el sistema.
Con el propósito de predecir las estructuras del cromóforo en fitocromo, hemos
utilizado la teoría del funcional de la densidad dependiente del tiempo (TDDFT). Un
análisis estructural con cálculos teóricos basados en la TDDFT nos permitieron predecir
correctamente una estructura semicíclica ZZZssa para la ficocianobilina en el fitocromo
Cph1 de cianobacteria. Con dicho análisis pudimos evaluar el efecto del estado de
protonación del cromóforo y el entorno proteico sobre las energías de excitación que
xii

entrega la TDDFT, logrando una aproximación cuantitativa cuando se utiliza la
geometría de un molde cristalográfico sin optimizar junto con el modelo de polarizable
continuo (PCM) para simular las interacciones electrostáticas entre el cromóforo y la
apoproteína. Utilizamos un modelo teórico similar para predecir una estructura
semicíclia ZZZssa para la fitocromobilina en el fitocromo A de planta donde, además de
la aproximación cuantitativa del modelo, también pudimos reproducir el índice
espectroscópico Q/S, correspondiente a la razón entre las fuerzas de oscilador de las
bandas Q y Soret, respectivamente. Con el propósito de explicar el comportamiento
general del índice Q/S para bilinas, se propuso un modelo teórico, donde definimos un
término empírico F para el sistema, el cual presenta un componente FS asociado a las
funciones intrínsecas del sistema conjugado (corriente de anillo y aromaticidad) y un
componente FQ asociado a la fotoisomerización del cromóforo. Nuestro modelo teórico
plantea que, cuando se cambia la conformación del sistema, el término F se conserva
(F = FS + FQ), por lo que solamente cambia la distribución entre sus componentes.
Con respecto al fotociclo del fitocromo, estudiamos la fase térmica de la
fotoconversión de Pr a Pfr. Los espectros teóricos de absorción para la forma Pfr, el
análisis de los estados de transición involucrados, y los desplazamientos químicos de
RMN nos sugieren que durante la interconversión Pr/Pfr ocurre una tautomería lactama-
lactima en el anillo A del cromóforo (no así en el anillo D). Además, nuestros
resultados indican que es posible que la tautomería se lleve a cabo a través de un
mecanismo catalizado por cierta molécula de agua cristalográfica localizada en las
proximidades del anillo A de la bilina, , denominada “agua pirrólica”, la que haría de
catalizador bifuncional.
xiii

ABSTRACT
Theoretical Study of the Electronic Structure and of the
Mechanisms of Lactam-Lactim Tautomerism in Bilins of Phytochrome
Phytochrome is a photoreceptor of plants that is also found in bacteria and fungi.
In plants, phytochrome regulates photomorphogenic responses of the plant, e.g., the
development and growth of the stem, regulation in the dormancy period and seed
germination, flowering, phototaxis, shade avoiding mechanisms, etc. All of them are
activated or deactivated in a differential way by means of specific wavelengths on the
UV-Visible region of the electromagnetic spectra. The receptor is photoconverted
between a physiological inactive form, Pr, and a physiological active form, Pfr. The
interconversion between both forms occurs through different mechanisms that are still
unknown. Thus, there is a growing interest concerning the mechanism of
photoconversion of phytochrome since the potential applications in biotechnology.
However, an obstacle in the research on phytochrome has been the scarce of
crystallographic structures resolved in the system.
With the purpose to predict the structures of the chromophore in phytochrome,
we have used the time-dependent density functional theory (TDDFT). A structural
analysis with theoretical calculations based on the TDDFT has allowed to predict
correctly a semicyclic structure ZZZssa for the phycocyanobilin in phytochrome Cph1
of cyanobacteria. With such analysis we could assess the effect of the protonation state
and the protein environment over the excitation energies delivered by the TDDFT,
getting a quantitative approximation when the geometry of a crystallographic template
xiv

is used without optimization, together with the polarizable continuum model (PCM) to
mimic the electrostatic interactions between the chromophore and the apoprotein. We
have used a similar theoretical model to predict a semicyclic structure ZZZssa for the
phytochromobilin in phytochrome A of plants where, besides the quantitative
approximation of the model, we could also predict the spectroscopic index Q/S,
corresponding to the ratio between the oscillator strengths of the Q and Soret bands,
respectively. With the purpose to explain the general behavior of the index Q/S for
bilins, a theoretical model was proposed, where we defined an empirical term F for the
system, which has a component FS associated to the intrinsic functions of the conjugated
system (ring current and aromaticity) and a component FQ associated to the
chromophore photoisomerization. Our theoretical model suggests that, when changing
the conformation of the system, the term F is preserved (F = FS + FQ), so that only
changes the distribution between its components.
With respect to the photocycle of phytochrome, we studied the thermal phase of
Pr to Pfr photoconversion. The theoretical absorption spectra for the Pfr form, the
analysis of the involved transition states, and the NMR chemical shifts suggest that
during the Pr/Pfr interconversion, a lactam-lactim tautomerism occurs in ring A of the
chromophore (not in ring D). In addition, our results indicate that is possible that the
tautomerism is carried out through a mechanism catalysed by a certain crystallographic
water molecule located in the vicinity of ring A of the bilin, called "pyrrole water,"
which would be a bifunctional catalyst .
xv

CAPÍTULO I. INTRODUCCIÓN
CAPÍTULO I
INTRODUCCIÓN
1.1 Estructura y Función del Fitocromo
En plantas se pueden encontrar tres tipos de fotoreceptores: fitocromos,
criptocromos, y fototropinas. En el espectro UV-visible, los fitocromos absorben en la
región del rojo y rojo-lejano, mientras que los criptocromos y fototropinas absorben en
la región del UV-azul. La clorofila y los carotenoides presentes en las hojas de las
plantas absorben fotones en gran parte del espectro visible de la luz solar incidente
correspondiente a la región fotosintéticamente activa, salvo en la región del rojo lejano,
donde hay muy poca fotoabsorción, por lo tanto la luz, al ser transmitida o reflejada por
las hojas, genera una mayor cantidad relativa de luz en el rojo lejano (far red o FR) con
respecto a la luz en el rojo (red o R), y así la razón R:FR se presenta como una señal
clara de la presencia de otras plantas en la vecindad, siendo el fitocromo el responsable
de percibirla, dada su sensibilidad a dichas longitudes de onda. Además, los fitocromos
que se encuentran en plantas responden a la cantidad, calidad, y dirección de la luz
incidente mediante la regulación de procesos de fotomorfogénesis, como la germinación
de semillas, fototropismo, floración, crecimiento, y desarrollo de la planta [1].
1

CAPÍTULO I. INTRODUCCIÓN
El fitocromo puede encontrarse en dos estados metaestables interconvertibles: Pr
y Pfr [1]. La interconversión entre ambos estados depende de la longitud de onda de la
luz incidente [2]; es así como el paso de Pr a Pfr se induce con luz en el rojo, mientras
que el proceso inverso de Pfr a Pr se logra en respuesta a la luz en el rojo lejano. La
forma Pr corresponde al estado fisiológicamente inactivo, mientras que la forma Pfr es
el estado fisiológicamente activo [1]. En el fotociclo de fotoconversión del fitocromo en
planta se han detectado algunos de los intermediarios, tanto para la fotoconversión
directa (desde Pr a Pfr) como para la fotoconversión reversa (desde Pfr a Pr): Lumi-R,
Meta-RA, y Meta-RC para la fotoconversión directa; Lumi-F y Meta-F para la conversión
reversa (ver Figura 1.1).[3;4] Además, cabe señalar que la fotoconversión reversa compite
con una conversión térmica que no requiere la presencia de luz, conocida como
reversión oscura.[1]
Figura 1.1. Esquema para el fotociclo del fitocromo en planta.Intermediarios en la fotoconversión directa: Lumi-R, Meta-RA, Meta-RC. Intermediarios en la fotoconversión reversa: Lumi-F, Meta-F. El esquema también incluye la reversión oscura que ocurre sin la necesidad de luz.
2

CAPÍTULO I. INTRODUCCIÓN
Figura 1.2. Diagrama de estados electrónicos involucrados en la fotoconversión del fitocromo.El diagrama se reproduce tal como lo describe Sineshchekov (Ref. 5) en base a mediciones efectuadas a bajas temperaturas para la fotoconversión y fluorescencia de Pr y Lumi-R. A, estado inicial, Pr; B, el primer fotoproducto estable, Lumi-R; A-B, el intermediario inestable de corta vida, posiblemente, prelumi-R o bato-R; k, constantes de velocidad para la fluorescencia (kf), des-excitación independiente de la temperatura (kd), fotoreacción primaria (kp), fototransformación en producto (kab), y retorno al estado inicial (kba); Ea y Ea', energías de activación de las fotoreacciones primarias dependientes de la temperatura directas y reversas.
En el fotociclo del fitocromo (Figura 1.1), la fotoconversión del fitocromo
presenta una primera fase fotoinducida y una segunda fase térmica (ver Figura 1.2).[5]
El cromóforo presente en el fitocromo es responsable de la detección y fase
inicial en la transducción de la señal luminosa; pertenece al grupo de pigmentos
biológicos conocidos como bilinas, que son tetrapirroles lineales estructuralmente
relacionados a las porfirinas. La estructura del crómoforo depende del tipo de
3

CAPÍTULO I. INTRODUCCIÓN
fitocromo, ya que los fitocromos no sólo se encuentran en plantas, sino que también en
bacterias, cianobacterias, y hongos.[1] En plantas, el cromóforo es la fitocromobilina
(PФB); en algunas cianobacterias, es la ficocianobilina (PCB); mientras que en
bacterias, hongos, y otras cianobacterias, es la biliverdina (BV). Cabe señalar que estos
cromóforos tienen diferencias puntuales en su estructura (Figura 1.3) y, considerando a
la fitocromobilina como referencia de comparación, podemos notar que la
ficocianobilina tiene un enlace reducido en la cadena lateral del anillo D y que la
biliverdina presenta un anillo A más oxidado, con una cadena lateral alílica en vez del
grupo etilideno.[1] Las diferencias y similitudes en el anillo A determinan el proceso de
ensamblaje del cromóforo con la apoproteina, proceso en el cual se genera un enlace
tioeter covalente con una cisteína de la apoproteína a partir del ataque nucleofílico del
grupo sulfidrilo del residuo aminoacídico a la cadena lateral del anillo A. En
fitocromobilina y ficocianobilina, el cromóforo se encuentra unido por el carbono C31 de
la cadena lateral, en tanto que en biliverdina, el ataque nucleofílico sobre el grupo alilo
genera un grupo etilideno además de un carbono quiral en el carbono C2 del anillo,
quedando así el grupo prostético enlazado por el carbono C32. No obstante, el proceso
de ensamblaje del cromóforo aun dista mucho de estar claro, ya que al parecer no sólo
depende del tipo de cromóforo, sino que además depende de la especie, entre otros
factores, y en algunos casos incluso se ha detectado que el cromóforo no se encuentra
unido covalentemente. Algunos estudios sugieren que en dichos casos una histidina
podría estar interaccionando con el cromóforo en forma no covalente. Además, el
proceso de ensamblaje podría estar precedido por la activación del nucleófilo,
posiblemente con la participación de otros aminoácidos contiguos a la cisteína [6-8].
4

CAPÍTULO I. INTRODUCCIÓN
Figura 1.3. Bilinas con estructuras en discusión. BV-sss: Biliverdina en conformación helicoidad cíclica ZZZsss; BV-ssa: Biliverdina en conformación semicíclica ZZZssa; PCB-ssa: Ficocianobilina en conformación semicíclica ZZZssa; PCB-asa: Ficocianobilina en conformación extendida ZZZasa; PФB-ssa: Fitocromobilina en conformación semicíclica ZZZssa; PФB-asa: Fitocromobilina en conformación extendida.
5

CAPÍTULO I. INTRODUCCIÓN
En general, la caracterización conformacional de los fitocromos ha sido una
materia de mucho debate, y sólo recién a fines del 2005 se dio a conocer la primera
estructura cristalizada determinada por difracción de rayos-X para el fitocromo de la
bacteria Deinococcus radiodurans,[9] con lo que se logró un notable avance para poder
determinar las conformaciones que el fitocromo estaría adoptando en las diferentes
especies. Con lo de caracterización conformacional nos estamos refiriendo
puntualmente a la configuración Z o E que podrían tener los dobles enlaces en los
puentes entre anillos, y también a la conformación syn o anti que podrían adoptar los
enlaces simples en esos puentes (ver Figura 1.3). La estructura que se encontró en el
cristal tiene todos los dobles enlaces de los tres puentes con una conformación Z,
mientras que los enlaces simples de los puentes presentan una conformación syn en los
carbonos C5 y C10; y una conformación anti en C15.[9;10] La estructura del cromóforo
suele denotarse en base a las conformaciones de los tres puentes, refiriéndose a la
conformaciones syn o anti con su primera letra, por lo que la estructura del cristal de
fitocromo correspondería a ZZZssa, que es una conformación semicíclica, a diferencia
de la conformación helicoidal cíclica ZZZsss que adoptan los cromóforos libres en
solución,[1] o de la conformación extendida ZZZasa que adopta, por ejemplo, la
ficocianobilina en la C-ficocianina,[11] una proteína antena del complejo fotosintético de
plantas. En base al cristal de fitocromo[9;10] de la bacteria D. radiodurans se podría
generalizar, considerando una estructura ZZZssa para todos los bacteriofitocromos; no
así para cianobacterias, donde hay experimentos de Resonancia Raman que sugieren
una estructura ZZZasa para el cromóforo,[4] aunque últimamente también han aparecido
evidencias experimentales apoyando una estructura ZZZssa[12;13]; en cuanto al fitocromo
de plantas, se presentan dos propuestas para la forma Pr del cromóforo (Figura 1.3):
(i) ZZZssa, apoyada por estudios de homología entre secuencias peptídicas de diferentes
especies;[1] y (ii) ZZZasa, apoyada por experimentos de Resonancia Raman[4].
6

CAPÍTULO I. INTRODUCCIÓN
Figura 1.4. Espectros electrónicos de absorción para fitocromo de plantas. a) Forma Pr de fitocromo; b) Forma Pfr de fitocromo. Los datos experimentales correspondientes fueron puestos a nuestra disposión por gentileza del Prof. Dr. James C. Lagarias (UC Davis, California, EE.UU.). Estos espectros se encuentran publicados en Ref. 14 [Lagarias et al., Photochem. Photobiol. (1987), 46, 5-13].
Nuestra estrategia para abordar la problemática respecto a la estructura del
cromóforo en fitocromo de cianobacterias y de plantas considera el análisis de espectros
teóricos de absorción electrónica, generados mediante la teoría funcional de la densidad
dependiente del tiempo, comúnmente llamada TDDFT[15] (ver marco teórico de la
TDDFT en el Apéndice A). En plantas, la forma Pr presenta su máximo de absorción en
666 nm, y la forma Pfr absorbe con un máximo en 730 nm, ambas con presencia de dos
bandas: la banda Q en la región del rojo y la banda Soret en la región del azul (Figura
1.4); por lo que el primer aspecto a considerar es el que tiene relación con la longitud de
onda de la banda Q del cromóforo, ya que esta banda absorbe en forma dependiente de
la configuración estructural del cromóforo, por lo que a partir de un enfoque teórico
pretendemos asignar los valores experimentales de absorción a determinadas
configuraciones del cromóforo. El segundo aspecto a considerar tiene como base una
propuesta teórica presentada a fines de los años setenta,[16-18] la que señala una estrecha
7

CAPÍTULO I. INTRODUCCIÓN
relación entre la conformación de las bilinas y su propiedades espectroscópicas,
particularmente sobre la razón de fuerzas de oscilador entre la banda roja (o banda Q) y
la banda azul (o banda Soret) del espectro de absorción, de modo que resulta práctico
referirnos a la razón entre estas bandas como índice "Q/S", considerando las iniciales de
dichas bandas.
El análisis del Q/S obtenido mediante métodos semiempíricos[16-18] CNDO
mostraron que los valores de la razón Q/S permitían predecir la “extensión” en que se
encontraban las distintas conformaciones de las bilinas: los valores menores a 1 se
relacionaron con una conformacion “cíclica o cerrada” del tipo ZZZsss, los valores en el
rango aproximado entre 1 y 4 se relacionaron con una conformación “semicíclica o
semiextendida” del tipo ZZZssa o ZZZass, y los valores mayores a 4 se relacionaron
con una conformación “extendida” del tipo ZZZasa. Las predicciones de estos primeros
estudios computacionales semiempíricos fueron corroboradas posteriormente a través
del análisis de datos experimentales obtenidos a partir de espectros de absorción de
aductos sintetizados para las distintas conformaciones.[19;20] Curiosamente, la
determinación de conformaciones para las bilinas en base al parámetro Q/S no ha sido
mayormente investigada con posterioridad a lo ya mencionado, salvo estudios
esporádicos de tipo semiempírico,[21;22] o bien, como mero parámetro de referencia para
datos espectroscópicos experimentales.[23] En el presente trabajo de tesis analizamos el
índice Q/S, pero esta vez en el contexto de la teoría del funcional de la densidad, ya que
al validar el modelo con tal nivel de teoría se estaría proporcionando una base teórica lo
suficientemente consistente como para poder utilizar el análisis de los valores de Q/S
como un criterio importante para la determinación del estado conformacional de las
bilinas, convirtiéndose así en una herramienta de incalculable valor para poder predecir
estructuras para intermediarios y productos formados en el proceso de fotoconversión,
ya que experimentalmente la espectroscopia ultrarápida de femtosegudo desarrollada en
8

CAPÍTULO I. INTRODUCCIÓN
los últimos años logra resolver los espectros de absorción para los intermediarios pero
no resuelve las estructuras que los generan.
Volviendo al fotociclo del fitocromo (Figura 1.1), se dice que la etapa
fotoinducida se caracteriza por una fotoisomerización del doble enlace C15=C16 , que es
la hipótesis que se ha respaldado por muchos años.[24] No obstante, un estudio reciente
sorprendió con la idea de una fotoisomerización en el doble enlace C4=C5 en lugar del
enlace C15=C16.[25] En el contexto de esta controversia, nuestro estudio asume la
fotoisomerización del doble enlace C15=C16 en la etapa fotoinducida, apoyando así el
consenso general que existe al respecto[24]. Pero dejaremos a un lado el proceso de
fotoisomerización, debido a que su estudio a nivel teórico requiere métodos
multiconfiguracionales de alta demanda de cómputo, tales como cálculos CASSCF,[26] y
nos preocuparemos de la fase térmica del fotociclo (ver Figura 1.2), enfocándonos
particularmente en el proceso directo de la fotoconversión desde Pr a Pfr, intentando
dilucidar los mecanismos de reacción involucrados. El grupo de Lagarias propuso que
en dicha etapa se produce intercambio de protones entre el cromóforo y su entorno
proteico;[1] tal propuesta nos lleva a pensar en la posibilidad de que ocurra una
tautomería en la bilina, ya que ésta implica una transferencia protónica, ya sea
intramolecular, o bien, intermolecular cuando hay algun catalizador presente.[27;28] Por lo
tanto, es posible pensar en una tautomería lactama-lactima (Figura 1.5); y,
considerando la presencia de un agua cristalográfica en las inmediaciones del nitrógeno
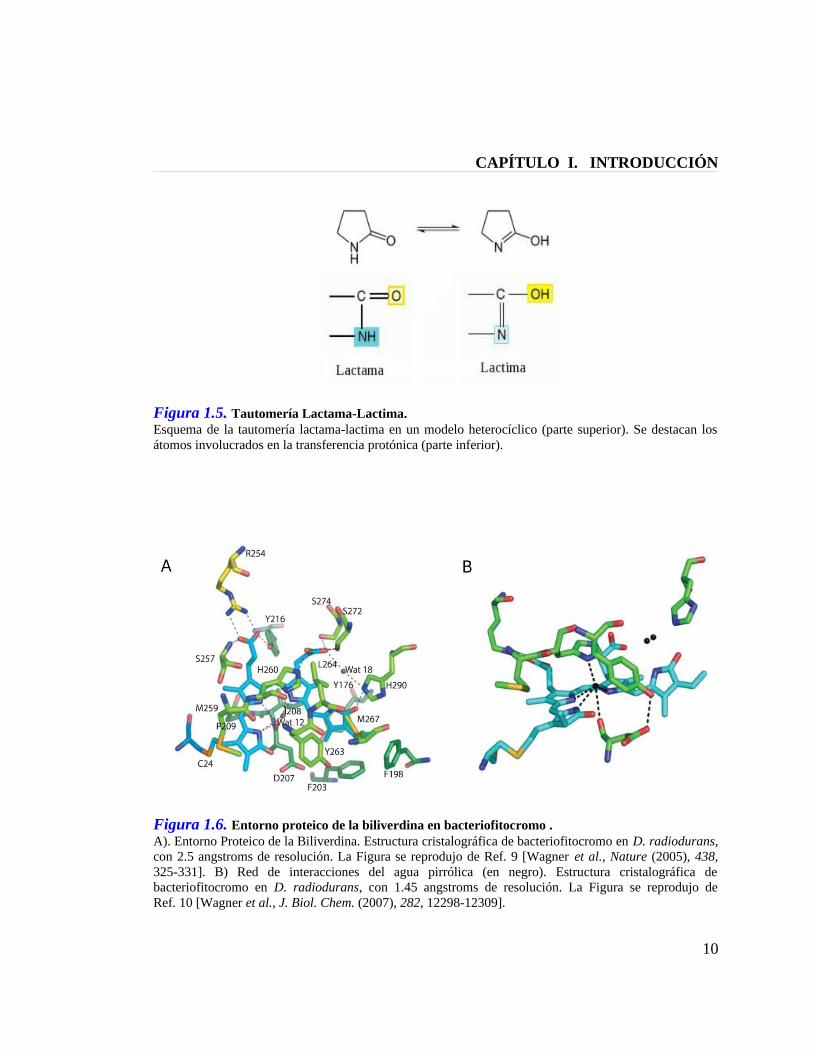
del anillo A de la bilina, denominada “agua pirrólica” (Figura 1.6),[9;10] proponemos que
un mecanismo de tautomería lactama-lactima catalizado por agua sobre el anillo A
podría estar involucrado en la fase térmica de la fotoconversión de Pr a Pfr, pese a que
un estudio reciente haya descartado la posibilidad de tautomería en el sistema.[29]
9
CAPÍTULO I. INTRODUCCIÓN
Figura 1.5. Tautomería Lactama-Lactima. Esquema de la tautomería lactama-lactima en un modelo heterocíclico (parte superior). Se destacan los átomos involucrados en la transferencia protónica (parte inferior).
Figura 1.6. Entorno proteico de la biliverdina en bacteriofitocromo . A). Entorno Proteico de la Biliverdina. Estructura cristalográfica de bacteriofitocromo en D. radiodurans, con 2.5 angstroms de resolución. La Figura se reprodujo de Ref. 9 [Wagner et al., Nature (2005), 438, 325-331]. B) Red de interacciones del agua pirrólica (en negro). Estructura cristalográfica de bacteriofitocromo en D. radiodurans, con 1.45 angstroms de resolución. La Figura se reprodujo de Ref. 10 [Wagner et al., J. Biol. Chem. (2007), 282, 12298-12309].
10

CAPÍTULO I. INTRODUCCIÓN
1.2 Hipótesis y Objetivos
Sobre la base de los antecedentes expuestos, en esta tesis planteamos como
Hipótesis que tanto la ficocianobilina como la fitocromobilina presentes en fitocromo
de cianobacterias y plantas, respectivamente, adoptan una estructura semicíclica
ZZZssa tal como su símil de biliverdina presente en fitocromo de proteobacterias; y
que, además, la fase térmica del fotociclo del receptor presenta un mecanismo de
tautomería lactama-lactima catalizado por agua sobre el anillo A del cromóforo.
Nuestra Hipótesis nos conduce a los siguientes Objetivos Generales :
I. Analizar mediante modelos teóricos la estructura que adopta la
ficocianobilina y la fitocromobilina en los fitocromos de cianobacterias
y plantas, respectivamente.
II. Evaluar los posibles mecanismos de tautomería lactama-lactima en
la biliverdina presente en bacteriofitocromo.
Con el propósito de abordar apropiadamente los Objetivos Generales se
proponen los siguientes Objetivos Específicos :
1. Predecir la estructura de la ficocianobilina presente en el fitocromo de
cianobacterias mediante espectros electrónicos de absorción calculados
en el contexto de la teoría del funcional de la densidad dependiente del
tiempo.
11

CAPÍTULO I. INTRODUCCIÓN
2. Predecir la estructura de la fitocromobilina presente en el fitocromo de
plantas mediante espectros electrónicos de absorción calculados en el
contexto de la teoría del funcional de la densidad dependiente del tiempo.
3. Analizar la relación entre el índice Q/S y el isomerismo conformacional
de las bilinas; y, en caso de encontrar que exista dependencia entre
ellos, proponer algun modelo teórico que sea capaz de generalizar.
4. Analizar el desplazamiento de la banda Q en espectros teóricos de
absorción de posibles estructuras de biliverdina para la forma Pfr del
fitocromo, asumiendo que ocurre una tautomería lactama-lactima.
5. Encontrar las estructuras para los estados de transición de diferentes
mecanismos de tautomería lactama-lactima para la biliverdina de
bacteriofitocromo, y calcular las energías asociadas.
6. Calcular los desplazamientos químicos de RMN para la ficocianobilina
de fitocromo de cianobacterias, y comparar con valores experimentales
para evaluar la posibilidad de tautomería lactama-lactima.
12

CAPÍTULO I. INTRODUCCIÓN
1.3 Referencias
[1.] N. C. Rockwell, Y. S. Su, J. C. Lagarias, Ann. Rev. Plant Biol. 2006, 57, 837-858.
[2.] Y. G. Chai, B. R. Singh, P. S. Song, J. Lee, G. W. Robinson, Anal. Biochem. 1987, 163, 322-
330.
[3.] K. Schaffner, W. Gärtner, Spectrum 1999, 12, 1-7.
[4.] M. A. Mroginski, D. H. Murgida, P. Hildebrandt, Acc. Chem. Res. 2007, 40, 258-266.
[5.] Sineshchekov, V. A. 1999. Phytochromes: molecular structure, photoreceptor process and
physiological function. In Concepts in Photobiology: Photosynthesis and Photomorphogenesis.
(Edited by G. S. Singhal, G. Renger, S. K. Sopory, K.-D. Irrgang and Govindjee), pp. 755–795.
Kluwer Academic, Boston.
[6.] B. Quest, W. Gärtner, Eur. J. Biochem. 2004, 271, 1117-1126.
[7.] H. Scheer, K. H. Zhao, Mol. Microbiol. 2008, 68, 263-276.
[8.] H. J. Jorissen, B. Quest, I. Lindner, N. Tandeau de Marsac, W. Gärtner, Photochem. Photobiol.
2002, 75, 554-559.
[9.] J. R. Wagner, J. S. Brunzelle, K. T. Forest, R. D. Vierstra, Nature 2005, 438, 325-331.
[10.] J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra, K. T. Forest, J. Biol. Chem. 2007, 282,
12298-12309.
[11.] M. Mimuro, P. Fulglistaller, R. Rimbeli, H. Zuber, Biochim. Biophys. Acta 1986, 848, 155-
166.
[12.] J. Hahn, R. Kühne, P. Schmieder, ChemBioChem 2007, 8, 2249-2255.
[13.] J. J. van Thor, M. Mackeen, I. Kuprov, R. A. Dwek, M. R. Wormald, Biophys. J. 2006, 91,
1811-1822.
[14.] J. C. Lagarias, J. M. Kelly, K. L. Cyr, W. O. Smith Jr., Photochem. Photobiol. 1987, 46, 5-13.
[15.] M. E. Casida, Recent Advances in Density Functional Methods, Part I; World Scientific;
Singapore, 1995.
[16.] Q. Chae, P. S. Song, J. Am. Chem. Soc. 1975, 97, 4176-4179.
[17.] G. Wagnière, G. Blauer, J. Am. Chem. Soc. 1976, 98, 7806-7810.
[18.] W. Pastenak, G. Wagnière, J. Am. Chem. Soc. 1979, 101, 1662-1667.
[19.] P. Nesvadba, A. Gossauer, J. Am. Chem. Soc. 1987, 109, 6545-6546.
13

CAPÍTULO I. INTRODUCCIÓN
[20.] J. B. Iturraspe, S. Bari, B. Frydman, J. Am. Chem. Soc. 1989, 111, 1525-1527.
[21.] C. R. W. Guimaraes, J. D. D. M. Neto, R. B. De Alencastro, Int. J. Quant. Chem. 1998, 70,
1145-1157.
[22.] A. H. Göller, D. Strehlow, G. Hermann, ChemPhysChem 2005, 6, 1259-1268.
[23.] J. R. Wagner, J. Zhang, D. von Stetten, M. Gunther, D. H. Murgida, M. A. Mroginski, J. M.
Walker, K. T. Forest, P. Hildebrandt, J. Biol. Chem. 2008, 283, 12212-12226.
[24.] N. C. Rockwell, J. C. Lagarias, ChemPhysChem 2010, 11, 1172-1180.
[25.] A. T. Ulijasz, G. Cornilescu, C. C. Cornilescu, J. Zhang, M. Rivera, J. L. Markley, R. D.
Vierstra, Nature 2010, 463, 250-254.
[26.] B. O. Roos, Adv. Chem. Phys. 1987, 69, 399.
[27.] J. A. Kereselidze, T. Sh. Zarqua, T. J. Kikalishvili, E. J. Churgulia, M. C. Makaridze, Russ.
Chem. Rev. 2002, 71, 993-2002.
[28.] M. J. Field, I. H. Hillier, J. Chem. Soc., Perkin Trans. 2 1987, 617-622.
[29.] C. Bongards, W. Gärtner, Acc. Chem. Res. 2010, 43, 485-495.
14

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
CAPÍTULO II
ANÁLISIS ESTRUCTURAL
2.1 Aspectos Generales
Este capítulo se dedica al análisis estructural del cromóforo en fitocromo, con el
fin de evaluar las estructuras que estan en discusión. La estructura semicíclica ZZZssa
se propuso en base a comparaciones de homología entre las secuencias de aminoácidos
de las estructuras proteicas de diversas especies, considerando entre ellas
proteobacterias, cianobacterias, hongos, y plantas;[1;2] mientras que la estructura
extendida ZZZasa fue sugerida por estudios con espectroscopia de resonancia Raman,
en los cuales se compararon espectros experimentales y teóricos.[3-6] Nuestro estudio
considera ambas propuestas, ZZZssa y ZZZasa, con el propósito de predecir la
estructura del cromóforo en fitocromo Cph1 de cianobacteria y en fitocromo A de
planta.
La primera parte de este capítulo describe la manera en que el estado de
protonación del cromóforo y la geometría que impone el entorno proteico sobre el
sistema conjugado de la bilina pueden afectar las energías de excitación de la TDDFT
(ver marco téorico de la TDDFT en Apéndice A), las que se utilizan para la obtención
15

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
de los espectros teóricos de absorción. Para ello, se utilizó la estructura cristalográfica
de la biliverdina como un control de estructura, y se utilizaron modelos moleculares
para representar las dos propuestas estructurales de ficocianobilina y fitocromobilina.
Un análisis de los efectos de protonación y entorno nos muestra las condiciones
propicias para realizar los cálculos de la TDDFT y, por lo tanto, en la segunda parte del
capítulo utilizamos tales condiciones de cálculo para predecir la estructura de la
ficocianobilina en fitocromo Cph1 de cianobacteria y la estructura de la fitocromobilina
en fitocromo A de planta.
2.2 Modelos Moleculares y Metodología Computacional
Para discriminar entre las conformaciones propuestas se hizo un análisis
comparativo entre los espectros teóricos provenientes de nuestros cálculos y los
espectros experimentales publicados.
En el caso del fitocromo Cph1 de cianobacteria, se diseñaron modelos
moleculares para las dos estructuras propuestas, ZZZssa y ZZZasa, incluyendo una
estructura control, correspondiente a la biliverina ZZZssa de una de las estructuras
resueltas de bacteriofitocromo.[7;8] La Figura 2.1 muestra los modelos utilizados: BV-ssa
(control), PCB-ssa, y PCB-asa. Los modelos con geometría semicíclica, BV-ssa y
PCB-ssa, se construyeron a partir de la estructura cristalográfica resuelta para el
fitocromo de la proteobacteria Deinococcus radiodurans;[7;8] mientras que el modelo de
geometría extendida se construyó a partir de la estructura cristalográfica de la
C-ficocianina,[9] proteína que tiene cromóforos de ficocianobilina con estructura
ZZZasa, y que por ello suele utilizarse para modelar el cromóforo de fitocromo en dicha
conformación.
16

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.1. Modelos para predecir estructura de PCB en fitocromo Cph1 de cianobacteria. BV-ssa, biliverdina con estructura semicíclica ZZZssa; PCB-ssa, ficocianobilina con estructura semicíclica ZZZssa; y PCB-asa, ficocianobilina con estructura extendida ZZZasa.
Los modelos BV-ssa y PCB-ssa utilizaron como molde la estructura
cristalográfica del dominio de unión a cromóforo del bacteriofitocromo con resolución
de 1.45 Å (PDB:2O9C).[8] El modelo PCB-asa hizo uso de la estructura cristalográfica
de la subunidad α de la C-ficocianina con resolución de 1.45 Å (PDB:1JBO).[9] Las
estructuras cristalográficas utilizadas no tienen resueltos los hidrógenos, por lo que
estos se agregaron apropiadamente segun los requerimientos estructurales de cada uno
de los modelos, haciendo uso de parámetros geométricos internos del programa
GaussView[10]. Es importante señalar que los modelos corresponden a los cromóforos
ensamblados a la apoproteina, en donde el puente de unión a cisteína es sustituido por
un hidrógeno (Figura 2.2). Además, con el fin de minimizar el costo computacional, no
se incluyeron las cadenas laterales de ácido propiónico presentes en los anillos B y C
del cromóforo (compárese los modelos de la Figura 2.1 con las estructuras de los
cromóforos de la Figura 1.3), por considerar que estas no afectan la energías de
absorción al no formar parte del sistema π conjugado, lo que ya ha sido comprobado en
los cálculos teóricos realizados por el grupo de Durbeej y cols.[11]
17

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.2. Estructura del anillo A del cromóforo según el estado de unión a la cisteína. (a) BV con unión a cisteína; (b) BV sin unión a cisteína; (c) PCB con unión a cisteína; (d) PCB sin unión a cisteína.
Con el propósito de evaluar el efecto de la protonación y el entorno para cada
uno de los modelos moleculares de PCB, se definen cuatro condiciones de cálculo:
CONDICIÓN I: Modelo neutro in vacuo.
CONDICIÓN II: Modelo catiónico in vacuo.
CONDICIÓN III: Modelo catiónico en agua.
CONDICIÓN IV: Modelo catiónico en ambiente proteico.
Los modelos catiónicos corresponden a las estructuras que se muestran en la
Figura 2.1, y el efecto de protonación hace referencia al protón unido al nitrógeno del
pirrol C. Para generar la condición III se utilizó un modelo de continuo polarizable
(PCM)[12] (ver marco teórico de PCM en Apéndice A) con la constante dieléctrica del
agua (ε=78,4). La condición IV también se genera con un modelo de continuo
polarizable, pero utilizando una constante dieléctrica igual a 4,0, valor que se ha
sugerido como el apropiado para simular entornos proteicos.[13;14]
18

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
El efecto del entorno sobre la geometría del cromóforo se evaluó al calcular las
energías de excitación, tanto para geometrías cristalográficas sin optimizar como
para geometrías previamente optimizadas por DFT con un nivel de cálculo
B3LYP/6-31G(d).[15;16]
Los estados excitados de las transiciones verticales y las fuerzas de oscilador
asociadas se obtuvieron mediante cálculos de la DFT dependiente del tiempo
(TDDFT),[17] utilizando un nivel de teoría B3LYP/6-31G(d)[15;16] sobre los ocho
primeros singuletes (S1 – S8). Respecto a la elección del funcional de intercambio-
correlación en la TDDFT, hemos optado por B3LYP porque se trata de un funcional
híbrido que posee un 20% proveniente del marco teórico Hartree-Fock (HF), ya que
estudios del efecto del funcional sobre el cálculo TDDFT proponen que precisamente el
grado de contribución de un término de intercambio HF en el funcional juega un papel
importante, con mejores resultados en funcionales con mayor intercambio HF.[18] Por
ello, es común en TDDFT el uso del funcional B3LYP (20% de intercambio HF) o el
funcional PBE0 (25% de intercambio HF). En lo referente al set de base, lo óptimo es
utilizar una base de tamaño mediano con polarización, como por ejemplo lo son las
bases 6-31G(d), 6-31+G(d), y 6-311+G(2d,p).[18] Por lo tanto, un nivel de teoría TD-
B3LYP/6-31G(d), como el que hemos utilizado, es el adecuado para nuestros
requerimientos.
Los espectros teóricos se generaron mediante la convolución de las energías de
excitación con funciones Gaussianas, utilizando arbitrariamente un FWHM (Full Width
at Half Maximum) de 4000 cm-1. Todos los cálculos de DFT y TDDFT se hicieron con
el programa Gaussian03;[10] y la expansión en Gaussianas para generar los espectros se
hizo con el programa GaussSum2.2.[19]
19

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
En el caso de la estructura de la fitocromobilina en el fitocromo A de planta,
se construyeron tres modelos moleculares: PΦB-sss, PΦB-ssa, y PΦB-asa (Figura 2.3).
Cabe destacar que durante el desarrollo de esta tesis el grupo de Essen resolvió la
estructura cristalográfica de un fitocromo de cianobacteria,[20] la que hemos considerado
en el análisis estructural de la fitocromobilina. Por lo tanto, con el propósito de evaluar
el efecto que tiene el uso de diferentes moldes para determinada conformación, se
utilizaron los dos moldes estructurales disponibles para la estructura semicíclica
ZZZssa: el cristal de la forma Pr del bacteriofitocromo de Deinococcus radiodurans con
resolución de 1,45 Å (PDB:2O9C);[6] y el módulo fotosensor completo de la forma Pr
del fitocromo Cph1 de la cianobacteria Synechocystis 6803 con resolución de 2,45 Å
(PDB:2VEA).[6;20] Para modelar la fitocromobilina en la estructura extendida ZZZasa se
utilizó como molde la subunidad α-84 del cristal de C-ficocianina de la cianobacteria
termófila Synechococcus elongatus con resolución de 1,45 Å (PDB:1JBO).[9] Para los
modelos PΦB-ssa y PΦB-asa, los hidrógenos se agregaron con el programa
GaussView[11] de acuerdo al arreglo molecular específico de cada modelo, pero esta vez
los hidrógenos se relajaron con DFT B3LYP/6-31G(d),[15;16] manteniendo fijos los
átomos restantes del cromóforo. Para modelar la situación de la bilina en solución, la
que adopta una conformación helicoidal cíclica ZZZsss,[1] se utilizó la estructura del
modelo ZZZssa proveniente de biliverdina,[6] al cual se le hizo una rotación syn/anti
sobre el enlace C14-C15, para luego optimizar in vacuo la geometría del sistema usando
un nivel de teoría B3LYP/6-31G(d).[15;16]
20

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.3. Modelos para predecir estructura de PΦB en fitocromo A de planta. PΦB-sss, fitocromobilina con estructura helicoidal cíclica ZZZsss; PΦB-ssa, fitocromobilina con estructura semicíclica ZZZssa; y PΦB-asa, fitocromobilina con estructura extendida ZZZasa.
La metodología con las condiciones óptimas de cálculo que fueron utilizadas
para la predicción de la estructura de PCB en fitocromo Cph1 de cianobacteria fueron
las que se utilizaron para la predicción de la estructura de la fitoccromobilina en
fitocromo A de planta. Sin embargo, para el caso particular de la fitocromobilina, se
evaluó también el comportamiento del índice espectroscópico Q/S en los espectros
teóricos y su relación con el isomerismo conformacional del cromóforo, con el
propósito de utilizarlo como un segundo criterio para dicriminar entre las
conformaciones ZZZssa y ZZZasa.
21

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
2.3 Efecto de Protonación y Entorno
La posición de la banda Q presente en el espectro de absorción del fitcromo
depende de la estructura del cromóforo, por lo se puede utilizar para discriminar entre
las dos conformaciones en estudio, ZZZssa y ZZZasa.
Los máximos de absorción de las bandas Q de los modelos de biliverdina
(Figura 2.1) se evalúan para cuatro condiciones específicas (ver Tabla 2.1). Las
condiciones I y II corresponden a cálculos TDDFT en fase gas (in vacuo) para el
cromóforo en su forma neutra y en su forma protonada, respectivamente. La
comparación entre ambas condiciones permite evaluar el efecto de la protonación del
cromóforo sobre el desplazamiento de la banda Q. Luego, las condiciones III y IV
corresponden a cálculos que incluyen el efecto del entorno utilizando un modelo de
continuo polarizable (ver marco teórico en Apéndice A) con constantes dieléctricas
apropiadas para representar un entorno acuoso y un entorno proteico, respectivamente.
Además, se tabulan los resultados de la TDDFT para estructuras cristalográficas sin
relajar y para geometrías cristalográficas optimizadas por DFT con el fin de evaluar el
efecto de la geometría del cromóforo sobre el cálculo de las energías de excitación.
Finalmente, se muestran como referencia los valores experimentales de absorción
(banda Q)[8;21-23] y las energías de excitacion calculadas por otros grupos.[24;25]
22
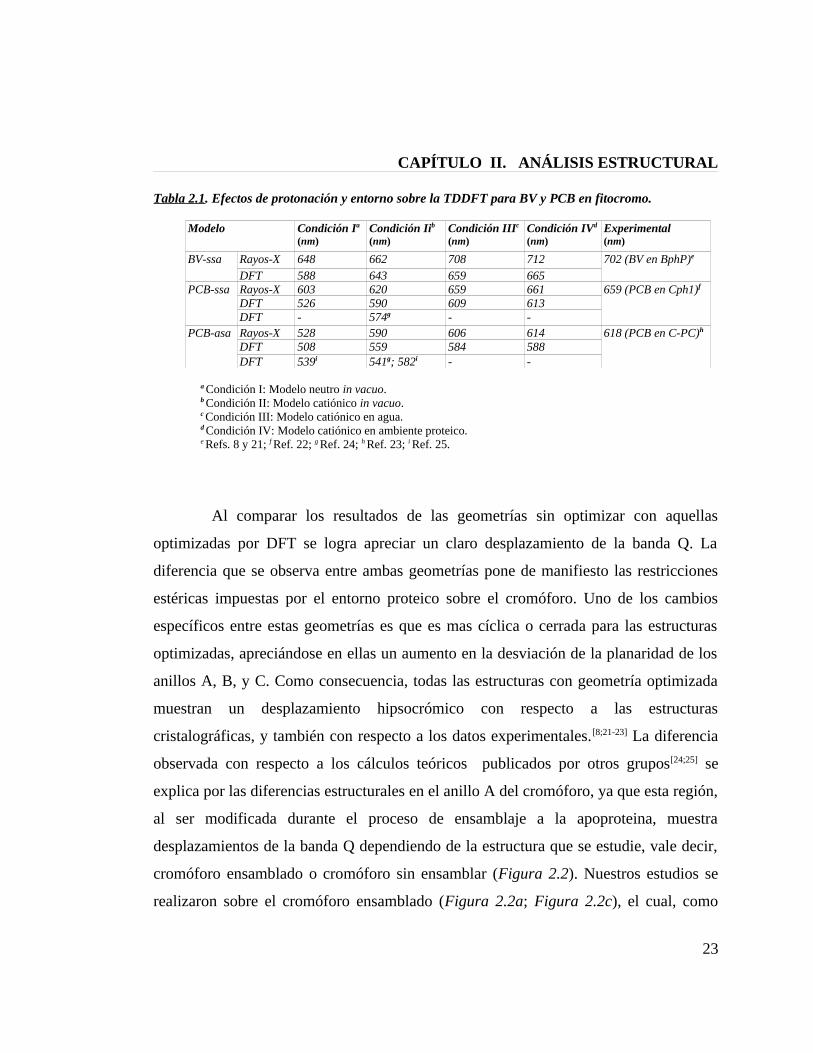
CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Tabla 2.1. Efectos de protonación y entorno sobre la TDDFT para BV y PCB en fitocromo.
Modelo Condición Ia
(nm)Condición Iib
(nm)Condición IIIc
(nm)Condición IVd
(nm)Experimental(nm)
BV-ssa Rayos-X 648 662 708 712 702 (BV en BphP)e
DFT 588 643 659 665PCB-ssa Rayos-X 603 620 659 661 659 (PCB en Cph1)f
DFT 526 590 609 613DFT - 574g - -
PCB-asa Rayos-X 528 590 606 614 618 (PCB en C-PC)h
DFT 508 559 584 588DFT 539i 541g; 582i - -
a Condición I: Modelo neutro in vacuo.b Condición II: Modelo catiónico in vacuo.c Condición III: Modelo catiónico en agua.d Condición IV: Modelo catiónico en ambiente proteico.e Refs. 8 y 21; f Ref. 22; g Ref. 24; h Ref. 23; i Ref. 25.
Al comparar los resultados de las geometrías sin optimizar con aquellas
optimizadas por DFT se logra apreciar un claro desplazamiento de la banda Q. La
diferencia que se observa entre ambas geometrías pone de manifiesto las restricciones
estéricas impuestas por el entorno proteico sobre el cromóforo. Uno de los cambios
específicos entre estas geometrías es que es mas cíclica o cerrada para las estructuras
optimizadas, apreciándose en ellas un aumento en la desviación de la planaridad de los
anillos A, B, y C. Como consecuencia, todas las estructuras con geometría optimizada
muestran un desplazamiento hipsocrómico con respecto a las estructuras
cristalográficas, y también con respecto a los datos experimentales.[8;21-23] La diferencia
observada con respecto a los cálculos teóricos publicados por otros grupos[24;25] se
explica por las diferencias estructurales en el anillo A del cromóforo, ya que esta región,
al ser modificada durante el proceso de ensamblaje a la apoproteina, muestra
desplazamientos de la banda Q dependiendo de la estructura que se estudie, vale decir,
cromóforo ensamblado o cromóforo sin ensamblar (Figura 2.2). Nuestros estudios se
realizaron sobre el cromóforo ensamblado (Figura 2.2a; Figura 2.2c), el cual, como
23

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
producto del proceso de unión a la apoproteina, pierde un doble enlace. Los resultados
de Wan y cols. para geometrías optimizadas por DFT,[25] con valores de 539 nm y
582 nm en las condiciones I y II, respectivamente (ver las entradas correspondientes en
la Tabla 2.1), se obtuvieron a partir de cálculos con la ficocianobilina sin ensamblar
(Figura 2.2d), lo que explica el desplazamiento de alrededeor de 30 nm hacia el azul.
Por otro lado, el grupo de van Thor trabajó con el cromóforo ensamblado,[24] por lo que
atribuimos el desplazamiento de alrededor de 15 nm en sus cálculos a que ellos utilizan
un funcional de intercambio-correlación distinto para la TDDFT (MPW1PW91)
respecto al que utilizamos nosotros (B3LYP).
Una información relevante que muestra la Tabla 2.1 corresponde a la
comparación de los cálculos TDDFT de los cálculos realizados sobre geometrías
cristalográficas sin optimizar con respecto a aquellos realizados sobre geometrías
relajadas. Al comparar los valores respectivos podemos concluir que sólo las estructuras
cristalográficas sin optimizar, al considerar la geometría que impone el entorno proteico
sobre el cromóforo, logran reproducir cuantitativamente los valores
experimentales,[8;21-23] ya que con las geometrías optimizadas se obtienen valores
bastante desplazados hacia el azul y muy alejados del valor experimental, por lo tanto, a
continuación nos referiremos únicamente a los resultados provenientes de cálculos
TDDFT sobre geometrías cristalográficas sin optimizar. Además, en la Tabla 2.1 se
observa que el efecto de la protonación en los tres modelos corresponde a un
desplazamiento batocrómico (hacia el rojo), el cual se aproxima bien a los valores
experimentales.[8;21-23] En la conformación ZZZssa este desplazamiento es de alrededor
de 15 nm, mientras que para la conformación extendida ZZZasa se aprecia un
desplazamiento de más de 60 nm. Por lo tanto, nuestro resultados permiten sugerir que
la forma protonada del cromóforo es la que se encuentra presente en fitocromo y en
C-ficocianina, apoyando así otros estudios realizados al respecto.[25-28]
24

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
El efecto solvatocrómico y el efecto del entorno proteico sobre el
desplazamiento de la banda Q también fueron evaluados (condiciones III y IV en la
Tabla 2.1). Tanto en entorno acuoso (condición III) como en entorno proteico
(condición IV) se observa un desplazamiento batocrómico considerable con respecto a
las condiciones in vacuo (condiciones I y II). Sin embargo, la diferencia entre el entorno
acuoso (condición III) y el entorno proteico (condición IV) es sutil.
2.4 Estructura de la Ficocianobilina en Fitocromo de Cianobacteria
Con el propósito de dilucidar la estructura de la ficocianobilina en fitocromo
Cph1 de Cianobacteria, se calcularon los espectros teóricos que se muestran en la
Figura 2.4 para los modelos BV-ssa, PCB-ssa, PCB-asa (Figura 2.1) en sus formas
protonadas y en entorno proteico (Condición IV), condición que consideramos la
apropiada para generar espectros en este sistema. Para los ocho primeros estados
excitados singuletes, S1 a S8, se calculó por TDDFT la energía de excitación y la fuerza
de oscilador asociada. La expansión en Gaussianas de los picos resultantes del cálculo
TDDFT generan dos bandas para cada espectro: la banda Soret y la banda Q. Los
espectros nos muestran que la banda Soret se genera a partir de múltiples estados (con
distinta longitud de onda), mientras que la banda Q se forma solamente con el primer
singulete excitado, S1, el cual presenta como contribución principal la transición
HOMO ---> LUMO en los tres modelos estudiados. La banda Soret, en cambio,
presenta en su estado más preponderante una superposición de dos transiciones:
HOMO–1 ---> LUMO y HOMO ---> LUMO+1. Los orbitales moleculares involucrados
en las transiciones electrónicas para uno de los modelos de interés, PCB-ssa, se
muestran en la Figura 2.5, los que corresponden a orbitales π,π* delocalizados a lo
largo de toda la molécula.
25
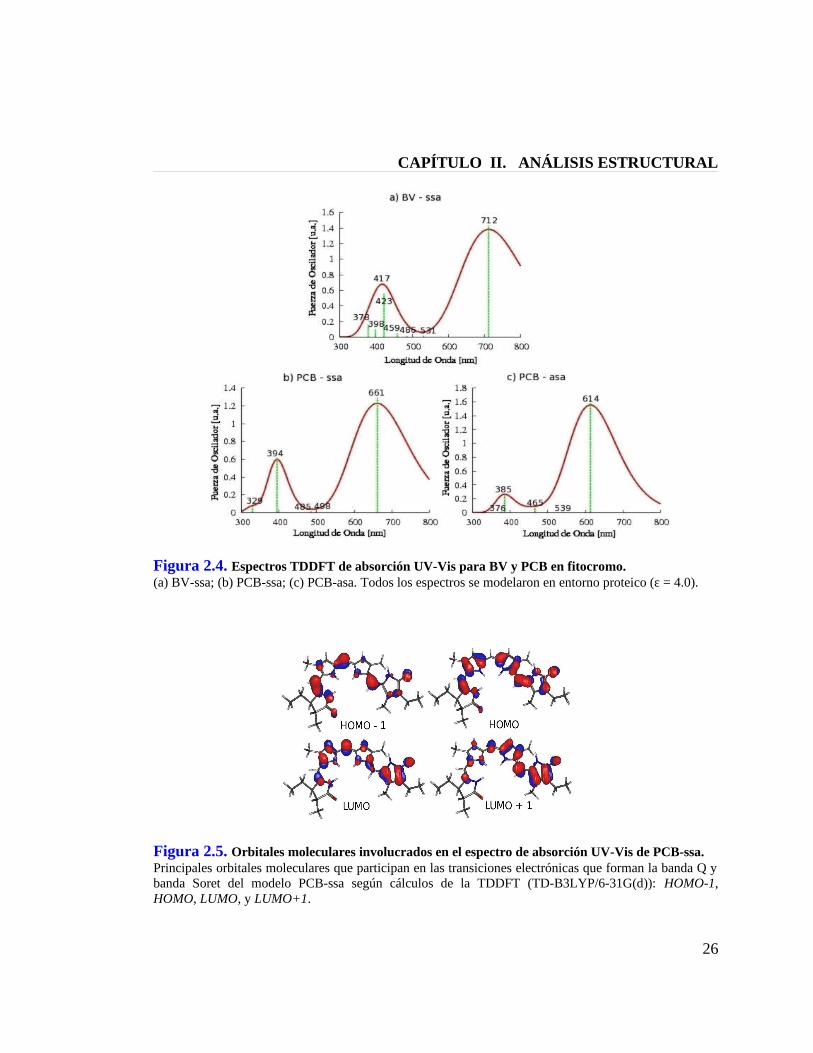
CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.4. Espectros TDDFT de absorción UV-Vis para BV y PCB en fitocromo.(a) BV-ssa; (b) PCB-ssa; (c) PCB-asa. Todos los espectros se modelaron en entorno proteico (ε = 4.0).
Figura 2.5. Orbitales moleculares involucrados en el espectro de absorción UV-Vis de PCB-ssa.Principales orbitales moleculares que participan en las transiciones electrónicas que forman la banda Q y banda Soret del modelo PCB-ssa según cálculos de la TDDFT (TD-B3LYP/6-31G(d)): HOMO-1, HOMO, LUMO, y LUMO+1.
26

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Los máximos de absorción para la banda Q de los espectros teóricos de
absorción (ver Tabla 2.1 y Figura 2.4) nos muestran que los valores experimentales de
702 nm[8;21] para el modelo BV-ssa y de 618 nm[23] para el modelo PCB-asa en
C-ficocianina son cuantitativamente bien reproducidos por los valores teóricos de
708/712 nm y 606/614 nm, respectivamente. En lo que respecta al modelo PCB-ssa, los
659/661 nm de nuestros cálculos reproducen los 659 nm del dato experimental[22] para
ficocianobilina en fitocromo Cph1 de cianobacteria y, considerando que dicho valor
experimental se encuentra lo suficientemente alejado del valor teórico de PCB-asa,
podemos entonces concluir que dicho valor experimental se genera por la
ficocianobilina en conformación ZZZssa y no así por la conformación ZZZasa propuesta
por el grupo de Mroginski.[4-6] Además, en la Tabla 2.1 se puede observar que el cambio
de una conformación semicíclica a una extendida provoca un desplazamiento
hipsocrómico, tal como ha sido descrito en estudios con cálculos semiempíricos.[29]
La conformación ZZZssa propuesta por el grupo de Lagarias se fundamentó en
base a los alineamientos de secuencia para 122 proteínas correspondientes a fitocromo y
proteínas relacionadas,[1;2] donde se observó lo siguiente: (i) todos los fitocromos
mantienen en general un alto grado de conservación en dos de los tres dominios de la
región fotosensora; (ii) los residuos clave que forman el nudo entre dichos dominios se
encuentran altamente conservados; (iii) las diferencias observadas entre las secuencias
se encuentran en regiones que no son importantes en la estructura secundaria de la
proteína. Tales observaciones nos señalan que el cromóforo en fitocromo de
cianobacteria adopta la misma conformación que su par en proteobacteria, Por otro
lado, la conformación ZZZssa ha sido respaldada por experimentos de espectroscopia
RMN sobre fitocromo Cph1 de cianobacteria.[30] Nuestros estudios teórico-
computacionales también apuntan hacia esta misma conformación semicíclica,
descartando la conformación extendida ZZZasa propuesta por Mroginski y cols.,[4-6]
27

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
fundamentada en el análisis con espectroscopia de resonancia Raman. Al respecto,
podemos discutir que los cálculos teóricos utilizados como referencia en dichos
estudios, para asignar las bandas del espectro experimental y discriminar la
conformación del cromóforo, se realizaron en estructuras relajadas in vacuo, y por ello
la conformación ZZZasa que se propone puede no corresponder al cromóforo en su
entorno nativo, tal como lo discute el mismo grupo de Mroginski en estudios teóricos
realizados en la ficocianobilina de C-ficocianina,[31] donde se demuestra que al
considerar el entorno en forma explícita por medio de un método híbrido QM/MM se
logra una mejoría significativa con respecto al cálculo QM del cromóforo aislado.
Durante el proceso de revisión y publicación de los resultados de la TDDFT
expuestos en el presente capítulo, donde respaldamos la propuesta de conformación
semicíclica ZZZssa para la ficocianobilina en fitocromo Cph1 de cianobacteria,
aparecieron publicados dos trabajos importantes en relación directa al tema en
cuestión.[20;32] El primero de ellos presentó la primera estructura en solución para el
fitocromo de cianobacteria, resuelta mediante espectroscopia RMN,[32] donde se
encontró que el cromóforo adopta la conformación ZZZssa. Pero es el trabajo de Essen
y cols. el que definitivamente confirmó la conformación semicíclica ZZZssa al resolver
la estructura cristalográfica de la forma Pr para el módulo fotosensor completo del
fitocromo Cph1 de cianobacteria.[20] En consecuencia, la confirmación de la estructura
ZZZssa de la ficocianobilina nos permite asumir que nuestra metodología de cálculo
para la obtención de espectros teóricos de absorción logra reproducir y predecir en
forma acertada los espectros de absorción experimental en este sistema.
28

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
2.5 Estructura de la Fitocromobilina en Fitocromo de Planta
Para dilucidar la estructura de la fitocrombilina en fitocromo A de plantas, se
calcularon los espectros teóricos que se muestran en la Figura 2.6 para los modelos
PФB-sss, PФB-ssa, PФB-asa (Figura 2.3) en sus formas protonadas y en entorno
proteico (Condición IV), tal como lo hicimos para el caso de la ficocianobilina (sección
2.4). Pero para el caso de fitocromobilina, además del análisis de máximos de absorción
de la banda Q, se hizo un análisis de índices Q/S y, por lo tanto, se realizaron cálculos
para biliverdina y fitocromobilina en las tres conformaciones de estudio: helicoidal
cíclica ZZZsss, semicíclica ZZZssa, y extendida ZZZasa.
La contribución orbitalaria en los espectros se logra apreciar en la Figura 2.7,
donde se muestran los orbitales moleculares involucrados en las transiciones
electrónicas para el modelo PФB-ssa proveniente del molde de biliverdina.[8] En este
modelo, la transición HOMO ---> LUMO que genera la banda Q es π,π* entre orbitales
delocalizados, mientras que la transición principal que compone la banda Soret es una
superposición de dos transiciones: HOMO-1 ---> LUMO y HOMO ---> LUMO+1, que
también son de naturaleza π,π* entre orbitales delocalizados. Estas mismas transiciones
se observaron para ficocianobilina (sección 2.4) y biliverdina. Curiosamente, los cuatro
orbitales que generan la banda Soret de estas bilinas coinciden con aquellos propuestos
por Gouterman para porfirinas en su llamado “modelo de cuatro orbitales”: HOMO-1,
HOMO, LUMO, LUMO+1.[33] Aparentemente la banda Soret podría ser clave en un
intento de relacionar tetrapirroles abiertos, como las bilinas, con tetrapirroles cíclicos,
como las porfirinas; y al respecto podemos preveer que la simetría del sistema podría
estar involucrada: sistemas más simétricos originando una banda Soret más
pronunciada, y sistemas menos simétricos originando una banda Soret más débil.
29
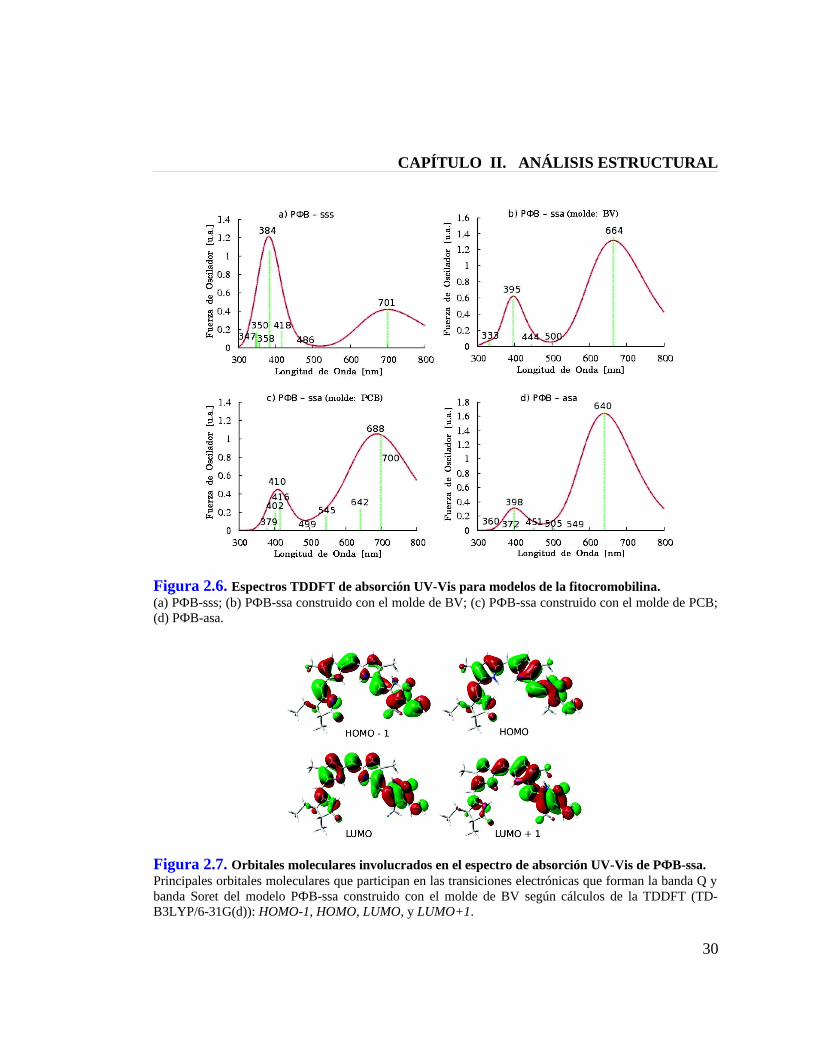
CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.6. Espectros TDDFT de absorción UV-Vis para modelos de la fitocromobilina.(a) PФB-sss; (b) PФB-ssa construido con el molde de BV; (c) PФB-ssa construido con el molde de PCB; (d) PФB-asa.
Figura 2.7. Orbitales moleculares involucrados en el espectro de absorción UV-Vis de PФB-ssa.Principales orbitales moleculares que participan en las transiciones electrónicas que forman la banda Q y banda Soret del modelo PФB-ssa construido con el molde de BV según cálculos de la TDDFT (TD-B3LYP/6-31G(d)): HOMO-1, HOMO, LUMO, y LUMO+1.
30
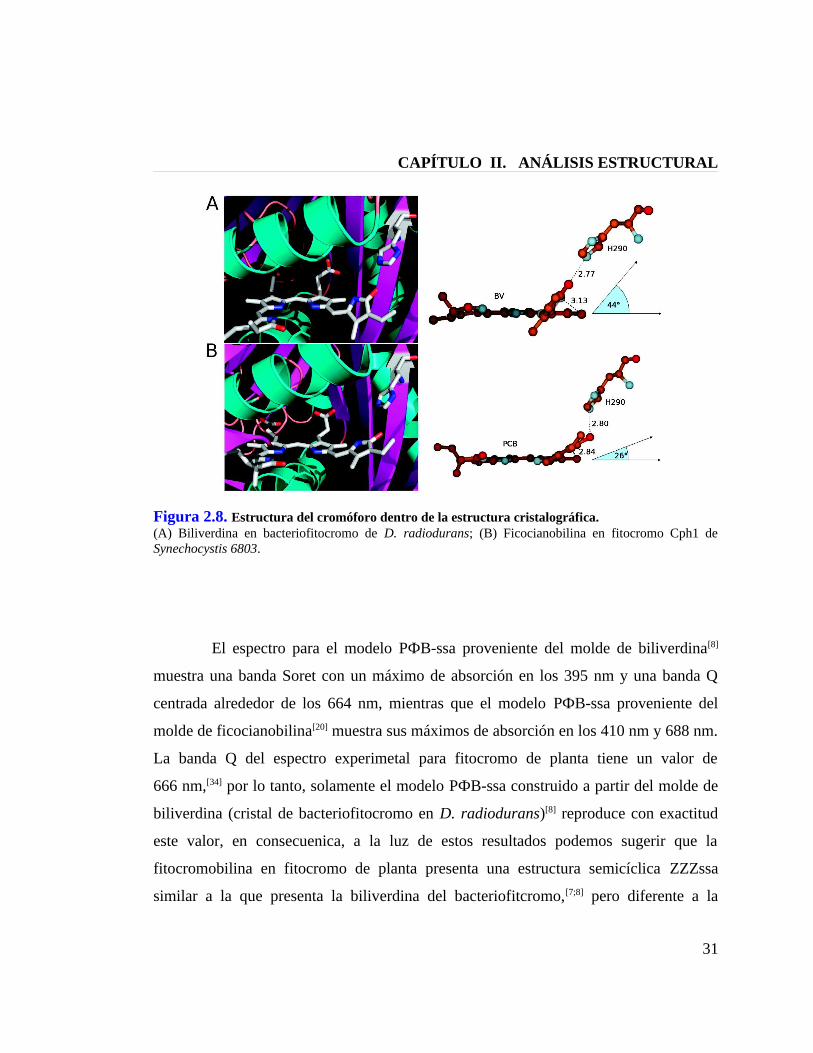
CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Figura 2.8. Estructura del cromóforo dentro de la estructura cristalográfica.(A) Biliverdina en bacteriofitocromo de D. radiodurans; (B) Ficocianobilina en fitocromo Cph1 de Synechocystis 6803.
El espectro para el modelo PФB-ssa proveniente del molde de biliverdina[8]
muestra una banda Soret con un máximo de absorción en los 395 nm y una banda Q
centrada alrededor de los 664 nm, mientras que el modelo PФB-ssa proveniente del
molde de ficocianobilina[20] muestra sus máximos de absorción en los 410 nm y 688 nm.
La banda Q del espectro experimetal para fitocromo de planta tiene un valor de
666 nm,[34] por lo tanto, solamente el modelo PФB-ssa construido a partir del molde de
biliverdina (cristal de bacteriofitocromo en D. radiodurans)[8] reproduce con exactitud
este valor, en consecuenica, a la luz de estos resultados podemos sugerir que la
fitocromobilina en fitocromo de planta presenta una estructura semicíclica ZZZssa
similar a la que presenta la biliverdina del bacteriofitcromo,[7;8] pero diferente a la
31

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
estructura ZZZssa que presenta la ficocianobilina en el cristal de fitocromo de
cianobacteria.[20] Pero si analizamos las causas del desplazamiento espectroscópico para
PФB-ssa en las dos situaciones, podemos intuir que se trata de diferencias estructurales
entre los moldes utilizados,[8;20] y, efectivamente, al comparar los cristales de fitocromo
de proteobacteria y de fitocromo de cianobacteria, podemos apreciar una diferencia en
la torsión del anillo D del cromóforo por sobre el plano formado por los tres anillos
restantes (ver Figura 2.8), ya que en bacteriofitocromo (proteobacteria) esta torsión
tiene un ángulo diedro de aproximadamente 45˚,[8] mientras que en fitocromo de
cianobacteria este ángulo de torsión es de alrededor de 26˚,[20] de modo que el anillo D
del cromóforo de cianobacteria es más coplanar con respecto al anillo D del cromóforo
de proteobacteria. Por lo tanto, el molde de ficocianobilina presenta una mayor
delocalización electrónica en su sistema π conjugado con respecto al molde de
biliverdina, y en consecuencia, un modelo PФB-ssa construido a partir del molde de
ficocianobilina tiene menor energía que aquel construido a patir de proteobacteria, lo
que se refleja en un desplazamiento batocrómico de 664 nm a 688 nm. Estudios de
resonancia Raman en fitocromo de planta sugieren una torsión del anillo D de la
fitocromobilina en fitocromo de planta con similares características a lo que se observa
para biliverdina, segun lo demuestra la fuerte actividad que se encontró en el modo
vibracional correspondiente al puente metilo que une dicho anillo.[35] Alternativamente,
estudios de mutagénesis han sugerido que la torsión del anillo D por sobre el plano del
sistema se produce como respuesta a la ausencia del dominio PHY,[36,37] como es el caso
de los cristales disponibles de bacteriofitocromo.
32
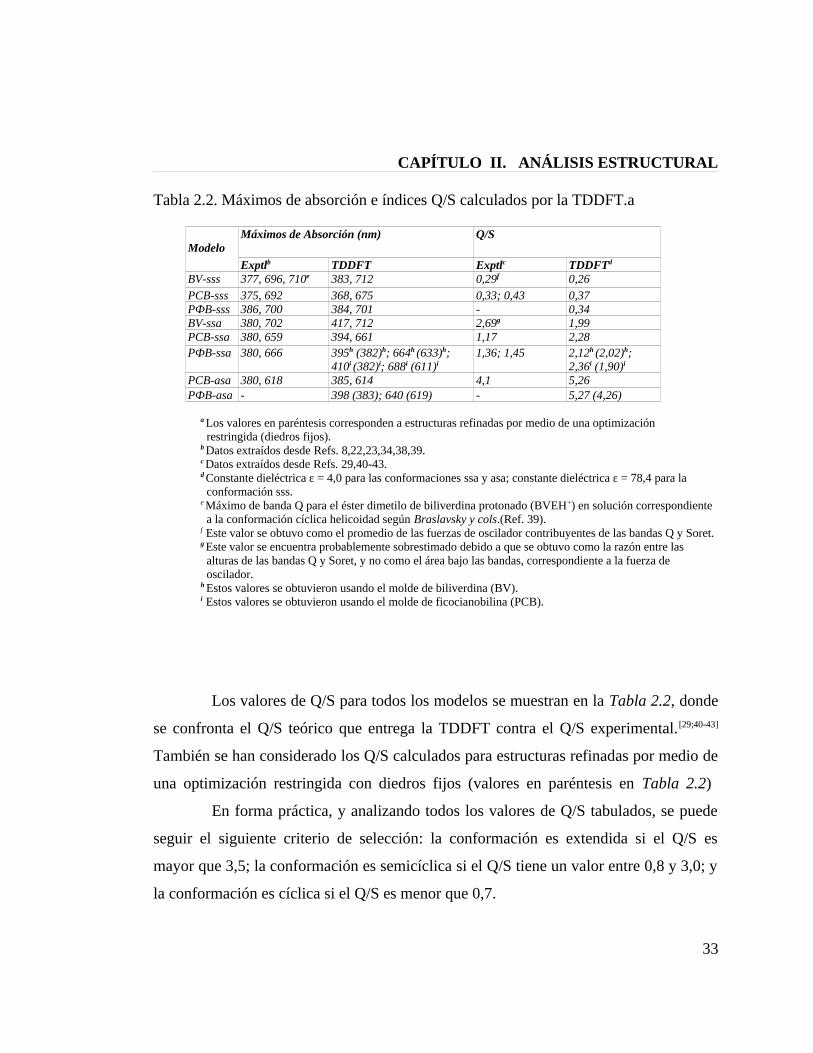
CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Tabla 2.2. Máximos de absorción e índices Q/S calculados por la TDDFT.a
ModeloMáximos de Absorción (nm) Q/S
Exptlb TDDFT Exptlc TDDFTd
BV-sss 377, 696, 710e 383, 712 0,29f 0,26PCB-sss 375, 692 368, 675 0,33; 0,43 0,37PФB-sss 386, 700 384, 701 - 0,34BV-ssa 380, 702 417, 712 2,69g 1,99PCB-ssa 380, 659 394, 661 1,17 2,28PФB-ssa 380, 666 395h (382)h; 664h (633)h;
410i (382)i; 688i (611)i1,36; 1,45 2,12h (2,02)h;
2,36i (1,90)i
PCB-asa 380, 618 385, 614 4,1 5,26PФB-asa - 398 (383); 640 (619) - 5,27 (4,26)
a Los valores en paréntesis corresponden a estructuras refinadas por medio de una optimización restringida (diedros fijos).b Datos extraídos desde Refs. 8,22,23,34,38,39.c Datos extraídos desde Refs. 29,40-43.d Constante dieléctrica ε = 4,0 para las conformaciones ssa y asa; constante dieléctrica ε = 78,4 para la conformación sss.e Máximo de banda Q para el éster dimetilo de biliverdina protonado (BVEH+) en solución correspondiente a la conformación cíclica helicoidad según Braslavsky y cols.(Ref. 39).f Este valor se obtuvo como el promedio de las fuerzas de oscilador contribuyentes de las bandas Q y Soret.g Este valor se encuentra probablemente sobrestimado debido a que se obtuvo como la razón entre las alturas de las bandas Q y Soret, y no como el área bajo las bandas, correspondiente a la fuerza de oscilador.h Estos valores se obtuvieron usando el molde de biliverdina (BV).i Estos valores se obtuvieron usando el molde de ficocianobilina (PCB).
Los valores de Q/S para todos los modelos se muestran en la Tabla 2.2, donde
se confronta el Q/S teórico que entrega la TDDFT contra el Q/S experimental. [29;40-43]
También se han considerado los Q/S calculados para estructuras refinadas por medio de
una optimización restringida con diedros fijos (valores en paréntesis en Tabla 2.2)
En forma práctica, y analizando todos los valores de Q/S tabulados, se puede
seguir el siguiente criterio de selección: la conformación es extendida si el Q/S es
mayor que 3,5; la conformación es semicíclica si el Q/S tiene un valor entre 0,8 y 3,0; y
la conformación es cíclica si el Q/S es menor que 0,7.
33

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
El Q/S teórico para la conformación ZZZssa es 2,12 y para la
conformación ZZZasa es 5,27. Si consideramos que el Q/S experimental para la
fitocromobilina en fitocromo de plantas es 1,36/1,45,[41;42] entonces es posible predecir
una conformación semicíclica ZZZssa para la forma Pr de la fitocromobilina.
2.6 Modelo Teórico del Comportamiento del Índice Q/S
Nuestros resultados muestran la dependencia del índice Q/S con respecto al
isomerismo conformacional de los cromóforos PCB y PФB de fitocromo. Sin embargo,
debido a que dicho comportamiento se encontró específicamente para sólo estos dos
cromóforos, es necesario proponer un modelo teórico que explique la relación entre los
índices Q/S y los confórmeros, de manera de poder generalizar el fenómeno. Con tal
propósito, hacemos uso de las fuerzas de oscilador con las que se construyeron los
espectros TDDFT de absorción (Figura 2.4 y Figura 2.6; Tablas Suplementarias en
Apéndice B).
En el caso particular de las bilinas, contamos con la fuerza de oscilador para
cada uno de los ocho estados excitados calculados (S1 a S8). Además, para nuestro
análisis hemos definido un término empírico “F8” como la sumatoria de las fuerzas de
oscilador de estos ocho estados singuletes:
F8 = ∑i=1
8
S i (1)
Es interesante observar que, para todos los espectros TDDFT de absorción
calculados, F8 presenta un valor relativamente constante para los isómeros estudiados:
sss, ssa, y asa (ver valores de F8 en las Tablas Suplementarias del Apéndice B).
34

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
Tabla 2.3. Relación entre Q/S, F8, FQ para confórmeros de PФB
Modelo Q/S F8 FQ a
PФB-sss 0,34 2,1 0,4354PФB-ssa b 2,12 2,1 1,3611PФB-asa 5,27 2,1 1,6693
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b Se consideró solamente el confórmero construido a partir del molde de BV.
La banda Q se compone mayoritariamente a partir de una única transición
electrónica, a la que llamaremos SQ, que presenta un fuerza de oscilador asociada que
denominaremos FQ. Al tabular cada conformación con sus respectivos valores de Q/S,
F8, y FQ (Tabla 2.3), podemos comprobar que efectivamente F8 se mantiene constante y
que el valor de FQ aumenta a medida que la conformación es más extendida. Por lo
tanto, el aumento del Q/S puede explicarse por una mayor efectividad en la excitación
al estado SQ para las conformaciones extendidas, es decir, en estas estructuras la
transición SQ se encuentra “más permitida” en comparación con estructuras más
cíclicas.
Podemos proponer un modelo téorico que logre explicar satisfactoriamente el
comportamiento de Q/S en bilinas si consideramos que la banda Soret en porfirinas se
asocia tanto a la corriente de anillo[44] como a la aromaticidad[45]. Por otro lado, se
postula que sólo el primer singulete excitado (S1) participa en la fotoisomerización de
bilinas en fitocromo.[46;47] En nuestros resultados encontramos que, en la mayoría de los
casos, el estado SQ corresponde al estado S1, por lo que consideraremos que la transición
electrónica que forma la banda Q corresponde al estado electrónico asociado a la
fotoisomerización del fitocromo. Por lo tanto, es posible particionar la constante
empírica F8, o simplemente F, en dos componentes variables: (i) FS, correspondiente a
35

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
transiciones electrónicas asociadas a la actividad intrínseca del sistema π conjugado
(corriente de anillo y aromaticidad); (ii) FQ, correspondiente a la transición electrónica
asociada a la fotoisomerización del cromóforo. Entonces, proponemos que la base
teórica que logra explicar el comportamiento del índice Q/S es:
F = FS + FQ (F constante; FS y FQ variables) (2)
La aditividad asumida en la ecuación (2) puede asociarse con una
aproximación de osciladores independientes, es decir, despreciando la interacción entre
ellos. Este modelo es ciertamente muy aproximado y requiere de una justificación
formal.
36

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
2.7 Referencias
[1.] N. C. Rockwell, Y. S. Su, J. C. Lagarias, Ann. Rev. Plant Biol. 2006, 57, 837-858.
[2.] N. C. Rockwell, J. C. Lagarias, Plant Cell 2006, 18, 4-14.
[3.] C. Kneip, P. Hildebrandt, W. Schlamann, S. E. Braslavsky, F. Mark, K. Schaffner,
Biochemistry 1999, 38, 15185-15192.
[4.] M. A. Mroginski, D. H. Murgida, D. von Stetten, C. Kneip, F. Mark, P. Hildebrandt, J. Am.
Chem. Soc. 2004, 126, 16734-16735.
[5.] M. A. Mroginski, D. H. Murgida, P. Hildebrandt, Acc. Chem. Res. 2007, 40, 258-266.
[6.] D. H. Murgida, D. von Stetten, P. Hildebrandt, P. Schwinte, F. Siebert, S. Sharda, W. Gartner,
M. A. Mroginski, Biophys. J. 2007, 93, 2410-2417.
[7.] J. R. Wagner, J. S. Brunzelle, K. T. Forest, R. D. Vierstra, Nature 2005, 438, 325-331.
[8.] J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra, K. T. Forest, J. Biol. Chem. 2007, 282,
12298-12309.
[9.] J. Nield, P. J. Rizkallah, J. Barber, N. E. Chayen, J. Struct. Biol. 2003, 141, 149-155.
[10.] Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.
Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M.
Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.
A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M.
Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P.
Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O.
Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G.
A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C.
Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz,
Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P.
Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A.
Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C.
Gonzalez, and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
[11.] B. Durbeej, O. A. Borg, L. A. Eriksson, Phys. Chem. Chem. Phys. 2004, 6, 5066-5073.
[12.] J. Tomasi, B. Menucci, R. Cammi, Chem. Rev. 2005, 105, 2999-3094.
37

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
[13.] M. R. A. Blomberg, P. E. M. Siegbahn, G. T. Babcock, J. Am. Chem. Soc. 1998, 120, 8812-
8824.
[14.] J. Neugebauer, ChemPhysChem 2009, 10, 3148-3173.
[15.] A. D. Becke, Phys. Rev. A 1998, 38, 3098-3100.
[16.] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B, 1988, 37, 785-789.
[17.] M. E. Casida, Recent Advances in Density Functional Methods, Part I; World Scientific;
Singapore, 1995.
[18.] D. Jacquemin, E. A. Perpète, I. Ciofini, C. Adamo, Acc. Chem. Res. 2009, 42, 326-334.
[19.] N. M. O'Boyle, A. L. Tenderholt, K. M. Langner, J. Comput. Chem. 2008, 29, 839-845.
[20.] L. O. Essen, J. Mailliet, J. Hughes, Proc. Natl. Acad. Sci. USA 2008, 105, 14709-14714.
[21.] R. Tasler, T. Moises, N. Frankenberg-Dinkel, FEBS J. 2005, 272, 1927-1936.
[22.] T. Lamparter, B. Esteban, J. Hughes, Eur. J. Biochem. 2001, 268, 4720-4730.
[23.] M. Mimuro, P. Fulglistaller, R. Rimbeli, H. Zuber, Biochim. Biophys. Acta 1986, 848, 155-
166.
[24.] J. J. van Thor, M. Mackeen, I. Kuprov, R. A. Dwek, M. R. Wormald, Biophys. J. 2006, 91,
1811-1822.
[25.] J. Wan, X. Xu, Y. Ren, G. Yang, J. Phys. Chem. B 2005, 109, 11088-11090.
[26.] B. Durbeej, O. A. Borg, L. A. Eriksson, Chem. Phys. Lett. 2005, 416, 83-88.
[27.] F. III. Andel, J. C. Lagarias, R. A. Mathies, Biochemistry 1996, 35, 15997-16008.
[28.] F. III. Andel, J. T. Murphy, J. A. Haas, M. T. McDowell, I. v. d. Hoef, J. Lugtenburg, J. C.
Lagarias, R. A. Mathies, Biochemistry 2000, 39, 2667-2676.
[29.] A. H. Göller, D. Strehlow, G. Hermann, ChemPhysChem 2005, 6, 1259-1268.
[30.] J. Hahn, R. Kühne, P. Schmieder, ChemBioChem 2007, 8, 2249-2255.
[31.] M. A. Mroginski, F. Mark, W. Thiel, P. Hildebrandt, Biophys. J. 2007, 93, 1885-1894.
[32.] G. Cornilescu, A. T. Ulijasz, C. C. Cornilescu, J. L. Markley, R. D. Vierstra, J. Mol. Biol.
2008, 383, 403-413.
[33.] M. Gouterman, G. H. Wagniere, L. C. Snyder, J. Mol. Spect. 1963, 11, 108-127.
[34.] Y. G. Chai, B. R. Singh, P. S. Song, J. Lee, G. W. Robinson, Anal. Biochem. 1987, 163, 322-
330.
[35.] P. Schwinté, W. Gärtner, S. Sharda, M. A. Mroginski, P. Hildebrandt, F. Siebert, Photochem.
Photobiol. 2009, 85, 239-249.
[36.] A. J. Fischer, J. C. Lagarias, Proc. Natl. Acad. Sci. USA 2004, 101, 17334-17339.
38

CAPÍTULO II. ANÁLISIS ESTRUCTURAL
[37.] J. M. Yoon, T. R. Hahn, M. H. Cho, J. S. Jeon, S. H. Bhoo, Y. K. Kwon, Biophys. Res.
Commun. 2008, 369, 1120-1124.
[38.] J. Cornejo, S. I. Beale, M. J. Terry, J. C. Lagarias, J. Biol. Chem. 1992, 267, 14790-14798.
[39.] S. E. Braslavsky, A. R. Holzwarth, K. Schaffner, Angew. Chem. Int. Ed. Engl. 1983, 22, 656-
674.
[40.] Q. Chae, P. S. Song, J. Am. Chem. Soc. 1975, 97, 4176-4179.
[41.] W. Pastenak, G. Wagnière, J. Am. Chem. Soc. 1979, 101, 1662-1667.
[42.] W. Parker, P. Goebel, C. R. Ross, P. S. Song, J. J. Stezowski, Bioconjugate Chem. 1994, 5,
21-30.
[43.] J. R. Wagner, J. Zhang, D. von Stetten, M. Gunther, D. H. Murgida, M. A. Mroginski, J. M.
Walker, K. T. Forest, P. Hildebrandt, J. Biol. Chem. 2008, 283, 12212-12226.
[44.] E. Steiner, P. W. Fowler, ChemPhysChem 2002, 3, 114-116.
[45.] J. A. N. F. Gomes, Chem. Rev. 2001, 101, 1349-1384.
[46.] B. Durbeej, O. A. Borg, L. A. Eriksson, Phys. Chem. Chem. Phys. 2004, 6, 5066-5073.
[47.] B. Durbeej, O. A. Borg, L. A. Eriksson, Chem. Phys. Lett. 2005, 416, 83-88.
39

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
CAPÍTULO III
TAUTOMERÍA EN LA INTERCONVERSIÓN
3.1 Aspectos Generales
El presente capítulo no se enfoca en la etapa fotinducida, sino que en la etapa
térmica que le sigue (Figura 1.2). El estudio de mecanismos involucrados en la etapa
térmica de la fotoconversión del fitocromo es de gran interés dado que se sabe muy
poco al respecto. Por tal motivo, y aprovechando la disponibilidad de estructuras
cristalográfica de la forma Pr de fitocromo, se estudió el mecanismo de la etapa térmica
que describe el proceso de interconversión desde la forma Pr a la forma Pfr,
entendiendo que el mecanismo involucrado puede también formar parte de la etapa
térmica de la interconversión en sentido inverso, es decir, desde Pfr a Pr.
Nuestro estudio se centra en un mecanismo en particular: la tautomería lactama-
lactima,[1;2] que puede ser considerada para los anillos A y D de la bilina, ya sea como
una tautomería favorecida para uno de los anillos, o bien, como una tautomería que
involucre a ambos anillos.
40

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Lo que se sabe acerca de la tautomería lactama-lactima se basa principalmente
en los estudios realizados en el sistema de la 2-piridona,[2-5] los que sugieren que la
tautomería que rige el equilibrio 2-piridona/2-hidroxipiridina puede cursar dos tipos de
mecanismo: disociativo y no disociativo, ambos basados en transferencia de protones.[2]
El mecanismo disociativo de transferencia protónica es típico de reacciones ácido-base,
donde ocurre una protonación y desprotonación en serie del sustrato, que es el
mecanismo dominante para soluciones acuosas no neutras. Por otro lado, el mecanismo
no disociativo de transferencia protónica es importante en fase gaseosa, en medios
apróticos, y en soluciones acuosas neutras. Este mecanismo no disociativo puede ocurrir
de tres maneras: (i) transferencia de protón a través de un mecanismo intramolecular;
(ii) interconversión tautomérica a partir de la formación de un dímero; (iii) un
mecanismo que considere la participación de una o más moléculas de agua como
catalizador bifuncional para la formación de un intermediario de estructura cíclica. [2]
Considerando todos estos antecedentes, se hizo un análisis teórico sobre los
mecanismos plausibles de tautomería para el sistema de la bilina en fitocromo.
La posibilidad de que ocurra una tautomería no es una idea nueva en el sistema
de las bilinas, ya que hay estudios relacionados para bilinas en solución.[6] Sin embargo,
para el caso de las bilinas presentes en fitocromo la posibilidad de tautomería ha sido
abordada solamente, y a un nivel muy superficial, por el grupo de Matysik,[7] el cual en
base a experimentos de espectroscopia 13C-RMN en estado sólido por rotación al ángulo
mágico (MAS) señala que no es posible la ocurrencia de una tautomería. Las
asignaciones en mediciones bidimensionales son usualmente muy seguras, pero
evidentemente los resultados en estado sólido no son extrapolables a una solución. Por
lo tanto, con el propósito de incluir este antecedente en nuestro estudio, hemos realizado
cálculos teóricos de 13C-RMN para el caso de la ficocianobilina de fitocromo.
41

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Por otro lado, mucho se ha especulado acerca del desplazamiento batocrómico
que ocurre tras la fotoconversión directa de Pr a Pfr, y se ha buscado una explicación en
base a la estructura de la forma Pfr.[8] Por lo tanto, también abordamos este aspecto,
utilizando espectros teóricos de absorción.
3.2 Modelos Moleculares y Metodología Computacional
Debido a que la forma Pfr se ha resuelto solamente para el caso de la biliverdina
en fitocromo de proteobacteria,[9] se utilizaron modelos de esta bilina para evaluar la
posibilidad de tautomería.
Los modelos moleculares de biliverdina para la forma Pfr (Figura 3.1) se
construyeron en base a la biliverdina del cristal resuelto por el grupo de Moffat.[9] Las
cadenas laterales de ácidos propiónicos presentes en los anillos B y C no se
consideraron, para así evitar la distorsión indeseada que generan sus cargas negativas
durante el proceso de optimización de geometría. Los hidrógenos, al no estar resueltos,
se agregaron de acuerdo al arreglo molecular de los modelos. Las estructuras con las
que se trabajó para los cálculos de TDDFT fueron geometrías cristalográficas sin
optimizar, en las que solamente los hidrógenos fueron relajados, manteniendo el resto
de los átomos fijos, usando la DFT con el funcional B3LYP[10;11] y la base 6-31G(d).
Para la obtención de los espectros teóricos de absorción de la biliverdina en la
forma Pfr, se calcularon las energías de excitación y fuerzas de oscilador asociadas a
partir de cálculos TDDFT, utilizando un PCM[12] con una constante dieléctrica de 4,0,
tal como fue descrito en el capítulo anterior. La TDDFT se calculó para 8 raíces, con un
nivel de teoría de B3LYP/6-31G(d);[10;11] luego los picos de excitación se
convolucionaron con funciones Gaussianas utilizando un FWHM de 4000 cm-1 para
generar las bandas de absorción.
42

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Se calcularon las barreras de energía para los distintos mecanismos propuestos
de tautomería lactama-lactima, utilizando el funcional híbrido de correlación-
intercambio B3LYP[10;11] en conjunto con el set de base 6-31G(d). Para las
optimizaciones se utilizó el método de optimización por gradiente analítica de
Berny.[13;14] Todos los cálculos se hicieron con el programa Gaussian03.[15] Los puntos
estacionarios se caracterizaron por cálculos de frecuencia.
Para los desplazamientos químicos de RMN, las estructuras se optimizaron in
vacuo por DFT con el funcional híbrido B3LYP[10;11] de la aproximación de gradiente
generalizada (GGA) y el set de base 6-31G(d). Los tensores de apantallamiento químico
para los átomos 13C se calcularon para cada estructura optimizada por DFT usando el
formalismo de la GIAO[16] (ver marco teórico de la GIAO en Apéndice A) junto con el
set de base 6311+(2d,p). Se evaluaron dos funcionales de intercambio-correlación
B3LYP y mPW1PW91. Los desplazamientos químicos anisotrópicos (δ) en ppm se
calcularon para ambos niveles de teoría: B3LYP/6-311+(2d,p)//B3LYP/6-31G(d) y
mPW1PW91/6-311+(2d,p)//B3LYP/6-31G(d). Los valores para δ se calcularon como la
diferencia entre el apantallamiento isotrópico atómico 13C de los modelos moleculares
de la ficocianobilina con respecto a los átomos de carbono en la molécula de referencia,
correspondiente a tetrametilsilano (TMS). La corrección por escalamiento linear
propuesta por Aliev y cols.,[17] implementada para corregir en forma específica los
errores sistemáticos asociados con un nivel de teoría GIAO B3LYP/6-
311+(2d,p)//B3LYP/6-31G(d), se utilizó para los cálculos de los desplazamientos
químicos de 13C-RMN de acuerdo a la siguiente relación: δscalc = 0.95 δcalc + 0.30; en
donde δcalc y δscalc corresponden al valor calculado y al valor corregido,
respectivamente.
43

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.1. Modelos de tautómeros para la forma Pfr de biliverdina en bacteriofitocromo.a) DITAM: Dilactama (anillos A y D); b) TIM-A: Lactima en A, Lactama en D; c) TIM-D: Lactama en A, Lactima en D; d) DITIM: Dilactima (anillos A y D).
3.3 En Busca del Desplazamiento Batocrómico de Pfr
Con el propósito de explicar el desplazamiento batocrómico que se observa con
la interconversión directa desde Pr a Pfr, se calcularon los espectros teóricos que se
muestran en la Figura 3.2 para los modelos DITAM, TIM-A, TIM-D, y DITIM (Figura
3.1); en sus formas protonadas y en entorno proteico, acorde a las condiciones de
cálculo propicias discutidas en detalle en el capítulo II.
Los espectros teóricos de absorción de los modelos tautoméricos de Pfr (Figura
3.2) reproducen dos bandas de absorción. La banda Q, en la región del rojo en el
espectro visible, es descrita principalmente por la transición electrónica
HOMO ----> LUMO, mientras que la banda Soret, en la región del azul en el espectro
visible, está compuesta por varias transiciones electrónicas, con una mayor contribución
por parte de cuatro orbitales: HOMO-1, HOMO, LUMO, y LUMO+1.
44
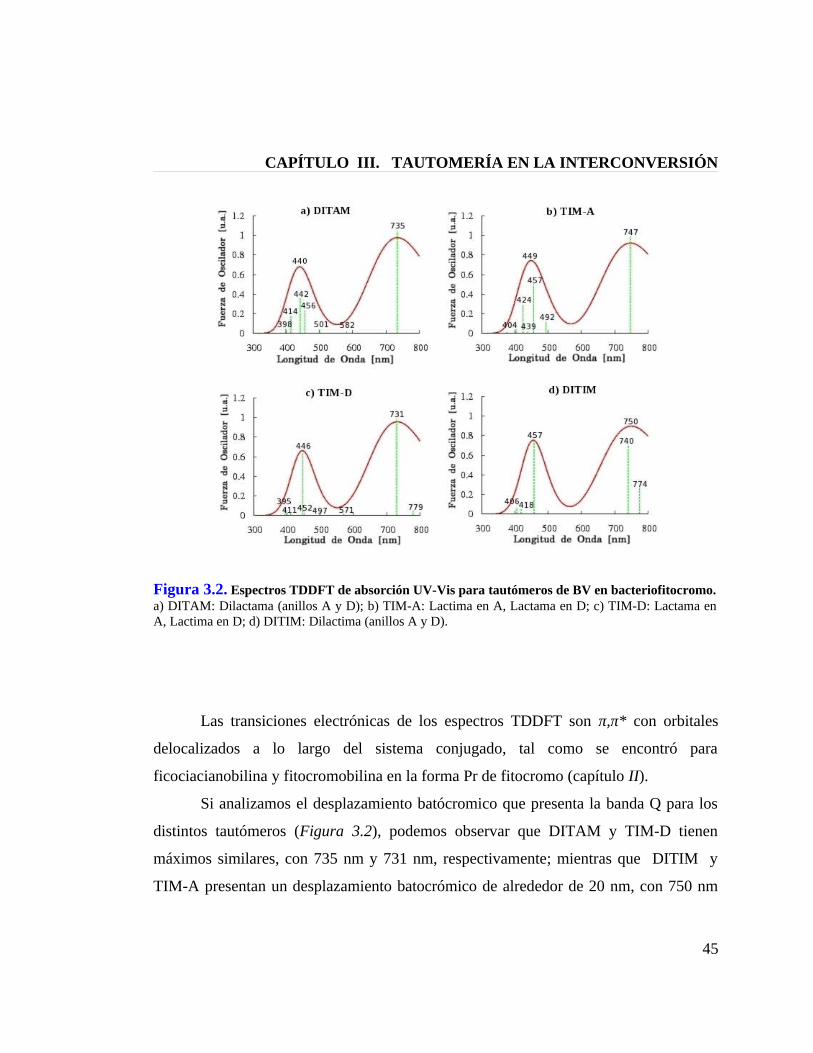
CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.2. Espectros TDDFT de absorción UV-Vis para tautómeros de BV en bacteriofitocromo.a) DITAM: Dilactama (anillos A y D); b) TIM-A: Lactima en A, Lactama en D; c) TIM-D: Lactama en A, Lactima en D; d) DITIM: Dilactima (anillos A y D).
Las transiciones electrónicas de los espectros TDDFT son π,π* con orbitales
delocalizados a lo largo del sistema conjugado, tal como se encontró para
ficociacianobilina y fitocromobilina en la forma Pr de fitocromo (capítulo II).
Si analizamos el desplazamiento batócromico que presenta la banda Q para los
distintos tautómeros (Figura 3.2), podemos observar que DITAM y TIM-D tienen
máximos similares, con 735 nm y 731 nm, respectivamente; mientras que DITIM y
TIM-A presentan un desplazamiento batocrómico de alrededor de 20 nm, con 750 nm
45

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
para DITIM y 747 nm para TIM-A, valores que concuerdan muy bien con el valor
experimental de referencia de 749 nm[18] o 750 nm[19;20], correspondiente a la forma Pfr
de bacteriofitocromo. El desplazamiento batocrómico para el modelo dilactima
(DITIM) es consecuencia de una tautomería en el anillo A, ya que no se observa
desplazamiento para TIM-D cuando se compara con el modelo dilactama (DITAM)
como referencia. Por lo tanto, estos resultados sugieren que una tautomería lactama-
lactima puede explicar el desplazamiento batocrómico que se observa con la formación
de Pfr, pero en forma dependiente del anillo, es decir, presentándose sólo para el
tautómero lactima en el anillo A.
3.4 Mecanismos de la Tautomería Lactama-Lactima en Bacteriofitocromo
La tautomería lactama-lactima puede ocurrir en el grupo funcional imida
presente en la bilina, por lo tanto, puede afectar tanto al anillo A como al anillo D del
cromóforo. Con el propósito de saber si la tautomería es favorecida en uno de los anillos
por sobre el otro se calcularon las barreras de energía considerando, por motivos
prácticos, solamente el mecanismo no disociativo de tautomería con transferencia
protónica intramolecular (Figura 3.3). De esta manera, pretendemos obtener la
estructura de los TS asociados para ambos casos, a los que nos referirermos como TSA
y TSD para la tautomería en el anillo A y en el anillo D, respectivamente.
Figura 3.3. Mecanismo no disociativo de tautomería con transferencia protónica intramolecular.La interconversión de lactama a lactima puede ocurrir mediante la transferencia de un protón desde el nitrógeno al oxígeno con formación de un estado de transición.
46

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
La coordenada de reacción para la transferencia intramolecular no disociativa
corre desde el reactivo, correspondiente al tautómero dilactama DITAM (Figura 3.1),
hasta la formación del producto TIM-A en el caso de una tautomería en el anillo A, o
bien, hasta la formación del producto TIM-D en el caso de una tautomería en el anillo
D. Ambas reacciones compiten, y deben pasar por sus respectivos TS antes de formar
cada producto, de modo que la barreras energéticas asociadas al TSA y al TSD nos
pueden mostrar, en principio, si la tautomería lactama-lactima se encuentra
energéticamente favorecida en uno de lo anillos. La Figura 3.4 muestra la estructura de
los estados de transición TSA y TSD, sus respectivas barreras de energías, y la
frecuencia imaginaria asociada a la transferencia del protón. El estado de transición
TSA (Figura 3.4A) presenta una diferencia de 7 kcal/mol con respecto al estado de
transición TSD (Figura 3.4B), es decir, la tautomería en A se encuentra
energéticamente favorecida con respecto a la tautomería en D, lo que concuerda
perfectamente con los resultados de la TDDFT para Pfr discutidos en la sección anterior
(sección 3.3). Sin embargo, las barreras de energía para la formación de los estados de
transición TSA y TSD aun son bastante altas para que la tautomería ocurra por este
mecanismo en condiciones normales de temperatura y, por lo tanto, un mecanismo de
transferencia protónica intramolecular es inviable para la tautomería lactama-lactima de
biliverdina en bacteriofitocromo. En consecuencia, la tautomería en el anillo A,
energéticamente favorecida, podría ser viable solamente en presencia de un catalizador,
y si consideramos la presencia inequívoca de una molécula de agua cristalográfica en la
vecindad del anillo A,[9;20-23] podemos intuir que dicha molécula de agua podría actuar
como catalizador.
47

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.4. Estados de transición para el mecanismo intramolecular de tautomería.(A) TSA: Estado de transición para tautomería intramolecular en el anillo A. (B) TSB: Estado de transición para tautomería intramolecular en el anillo B. Las estructuras de los estados de transición se muestran junto a la barrera de energía y a la frecuencia imaginaria asociada a la transferencia protónica.
En todos los cristales resueltos para el fitocromo se ha encontrado una molécula
de agua en la vecindad del grupo carbonilo del anillo A del cromóforo, [9;20-23] la que
estaría formando parte de una red de interacción electrostática estabilizada por una
histidina en la parte superior, el carbonilo del enlace petídico de un aspartato en la parte
inferior, y los nitrogénos pirrólicos de la bilina en los costados (Figura 3.5A, Figura
3.5B). Como la forma Pfr ha sido resuelta solamente para el fitocromo de
proteobacteria,[9] nos preocupamos sólo de la situación de la biliverdina en
bacteriofitocromo. En dicho contexto, la biliverdina presenta un entorno cercano similar
al que se muestra en la Figura 3.5A para la forma Pr[22] y como el que se muestra en la
Figura 3.5B para la forma Pfr.[9] En estas figuras se logra apreciar que la histidina y el
aspartato forman parte de la red de interacción del agua, residuos presentados como
His260 y Asp207, respectivamente.
48
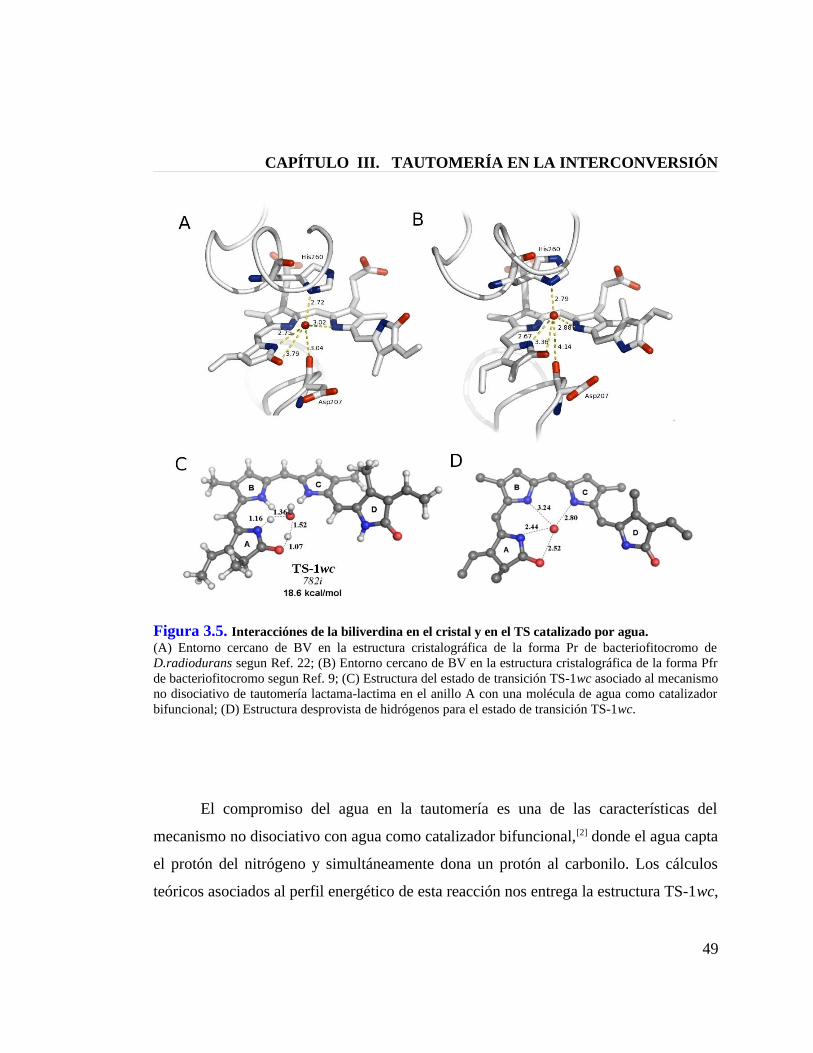
CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.5. Interacciónes de la biliverdina en el cristal y en el TS catalizado por agua.(A) Entorno cercano de BV en la estructura cristalográfica de la forma Pr de bacteriofitocromo de D.radiodurans segun Ref. 22; (B) Entorno cercano de BV en la estructura cristalográfica de la forma Pfr de bacteriofitocromo segun Ref. 9; (C) Estructura del estado de transición TS-1wc asociado al mecanismo no disociativo de tautomería lactama-lactima en el anillo A con una molécula de agua como catalizador bifuncional; (D) Estructura desprovista de hidrógenos para el estado de transición TS-1wc.
El compromiso del agua en la tautomería es una de las características del
mecanismo no disociativo con agua como catalizador bifuncional,[2] donde el agua capta
el protón del nitrógeno y simultáneamente dona un protón al carbonilo. Los cálculos
teóricos asociados al perfil energético de esta reacción nos entrega la estructura TS-1wc,
49

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
correspondiente al estado de transición asociado a dicho mecanismo (Figura 3.5C). El
estado de transición TS-1wc nos muestra que efectivamente la molécula de agua actúa
como catalizador bifuncional en la reacción, con el protón del nitrógeno localizado a
1,36 Å del oxígeno del agua, mientras que el protón que el agua dona a la bilina se
encuentra a 1,07 Å del oxígeno del carbonilo del anillo A.
Al comparar la estructura de TS-1wc (Figura 3.5C) con el entorno cercano de la
biliverdina en los cristales para Pr y Pfr (Figura 3.5A; Figura 3.5B), podemos observar
que la posición del agua en TS-1wc no dista mucho de la posición que adopta el agua
cristalográfica; por ello hemos incluido la estructura del TS sin los hidrógenos (Figura
3.5D) para hacer más directa la comparación de las distancias interatómicas importantes
con respecto a las del cristal. Podemos así observar que la similitud en el
posicionamiento del agua entre TS-1wc y el cristal es evidente. Además, la barrera
energética de 18,6 kcal/mol asociada al estado de transición TS-1wc se puede superar
en condiciones normales de temperatura, por lo tanto, estos resultados indican que la
tautomería lactama-lactima por medio de un mecanismo no discociativo con agua es
viable.
El mecanismo de tautomería que involucra un estado de transición TS-1wc
presenta una catálisis concertada por parte del agua, pero es importante considerar que
existe una segunda forma en que puede presentarse la catálisis: el mecanismo
disociativo. La catálisis disociativa consiste en un mecanismo por pasos con formación
de intermediarios iónicos (Figura 3.6). Este mecanismo no se ha considerado en los
cálculos de este trabajo, sin embargo, pensamos que puede ser perfectamente viable.
Por lo tanto, en la Figura 3.6 se resumen los mecanismos de tautomería lactama-lactima
que proponemos: (i) mecanismo no disociativo de transferencia protónica catalizada por
agua, y (ii) mecanismo disociativo de transferencia protónica catalizada por agua.
50
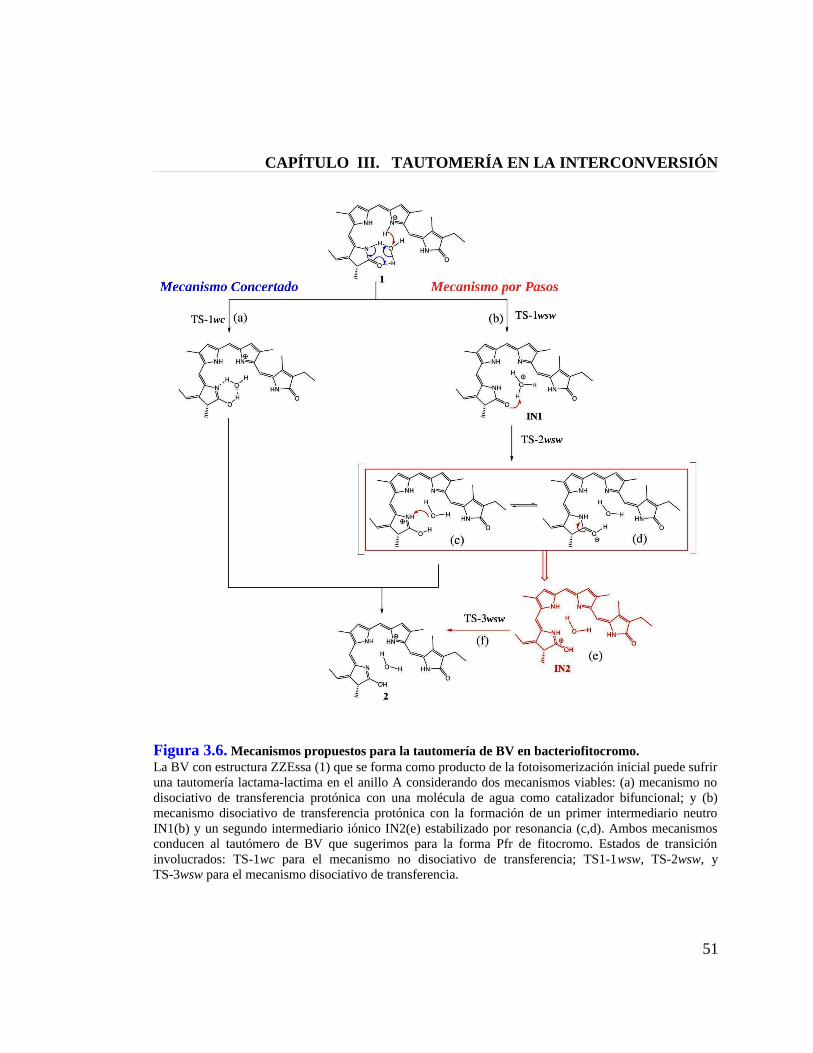
CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.6. Mecanismos propuestos para la tautomería de BV en bacteriofitocromo.La BV con estructura ZZEssa (1) que se forma como producto de la fotoisomerización inicial puede sufrir una tautomería lactama-lactima en el anillo A considerando dos mecanismos viables: (a) mecanismo no disociativo de transferencia protónica con una molécula de agua como catalizador bifuncional; y (b) mecanismo disociativo de transferencia protónica con la formación de un primer intermediario neutro IN1(b) y un segundo intermediario iónico IN2(e) estabilizado por resonancia (c,d). Ambos mecanismos conducen al tautómero de BV que sugerimos para la forma Pfr de fitocromo. Estados de transición involucrados: TS-1wc para el mecanismo no disociativo de transferencia; TS1-1wsw, TS-2wsw, y TS-3wsw para el mecanismo disociativo de transferencia.
51

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
3.5 13C-RMN de la Ficocianobilina en Fitocromo
La tautomería lactama-lactima puede ocurrir en el grupo funcional imida
presente en la bilina, por lo tanto, el cromóforo puede tautomerizar en el anillo A, en el
anillo D, o en ambos. Sin embargo, el grupo de Matysik descarta la posibilidad de
tautomería en base a estudios experimentales con espectroscopia 13C-RMN,[7] donde
encuentran que en Pfr los grupos carbonilos de los anillos A y D no se presentan en la
forma enólica sino que en la forma ceto, por lo que señalan que la bilina en Pfr es
dilactama como en Pr.[7] Por consiguiente, resulta de gran utilidad realizar los cálculos
teóricos respectivos para corroborar que las bandas experimentales se asignaron
correctamente, sin ambigüedades, y es por ello que nuestros resultados teóricos los
comparamos con los resultados experimentales del grupo de Matysik.[7]
Los cálculos para RMN se hicieron con la GIAO DFT (ver marco teórico en el
Apéndice A),[19] pero utilizando dos funcionales de correlación-intercambio, B3LYP y
mPW1PW91, que son los que se utilizan con mayor frecuencia en este tipo de cálculos
(ver Tabla 3.1). Además, para el caso de la GIAO B3LYP hemos utilizado la corrección
por escalamiento lineal propuesta específicamente para el nivel de teoría B3LYP/6-
311+G(2d,p)//B3LYP/6-31G(d) en base a estudios sistemáticos realizados en una serie
de moléculas orgánicas.[20]
52
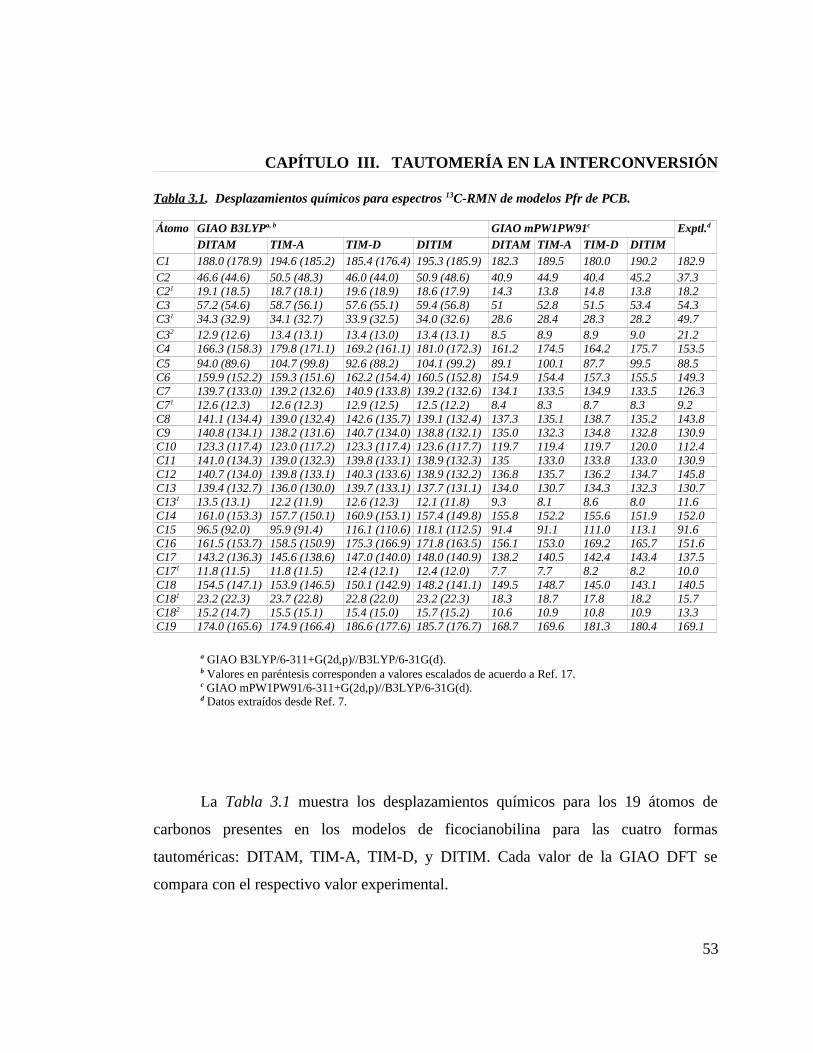
CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Tabla 3.1. Desplazamientos químicos para espectros 13C-RMN de modelos Pfr de PCB.
Átomo GIAO B3LYPa, b GIAO mPW1PW91c Exptl.d
DITAM TIM-A TIM-D DITIM DITAM TIM-A TIM-D DITIMC1 188.0 (178.9) 194.6 (185.2) 185.4 (176.4) 195.3 (185.9) 182.3 189.5 180.0 190.2 182.9C2 46.6 (44.6) 50.5 (48.3) 46.0 (44.0) 50.9 (48.6) 40.9 44.9 40.4 45.2 37.3C21 19.1 (18.5) 18.7 (18.1) 19.6 (18.9) 18.6 (17.9) 14.3 13.8 14.8 13.8 18.2C3 57.2 (54.6) 58.7 (56.1) 57.6 (55.1) 59.4 (56.8) 51 52.8 51.5 53.4 54.3C31 34.3 (32.9) 34.1 (32.7) 33.9 (32.5) 34.0 (32.6) 28.6 28.4 28.3 28.2 49.7C32 12.9 (12.6) 13.4 (13.1) 13.4 (13.0) 13.4 (13.1) 8.5 8.9 8.9 9.0 21.2C4 166.3 (158.3) 179.8 (171.1) 169.2 (161.1) 181.0 (172.3) 161.2 174.5 164.2 175.7 153.5C5 94.0 (89.6) 104.7 (99.8) 92.6 (88.2) 104.1 (99.2) 89.1 100.1 87.7 99.5 88.5C6 159.9 (152.2) 159.3 (151.6) 162.2 (154.4) 160.5 (152.8) 154.9 154.4 157.3 155.5 149.3C7 139.7 (133.0) 139.2 (132.6) 140.9 (133.8) 139.2 (132.6) 134.1 133.5 134.9 133.5 126.3C71 12.6 (12.3) 12.6 (12.3) 12.9 (12.5) 12.5 (12.2) 8.4 8.3 8.7 8.3 9.2C8 141.1 (134.4) 139.0 (132.4) 142.6 (135.7) 139.1 (132.4) 137.3 135.1 138.7 135.2 143.8C9 140.8 (134.1) 138.2 (131.6) 140.7 (134.0) 138.8 (132.1) 135.0 132.3 134.8 132.8 130.9C10 123.3 (117.4) 123.0 (117.2) 123.3 (117.4) 123.6 (117.7) 119.7 119.4 119.7 120.0 112.4C11 141.0 (134.3) 139.0 (132.3) 139.8 (133.1) 138.9 (132.3) 135 133.0 133.8 133.0 130.9C12 140.7 (134.0) 139.8 (133.1) 140.3 (133.6) 138.9 (132.2) 136.8 135.7 136.2 134.7 145.8C13 139.4 (132.7) 136.0 (130.0) 139.7 (133.1) 137.7 (131.1) 134.0 130.7 134.3 132.3 130.7C131 13.5 (13.1) 12.2 (11.9) 12.6 (12.3) 12.1 (11.8) 9.3 8.1 8.6 8.0 11.6C14 161.0 (153.3) 157.7 (150.1) 160.9 (153.1) 157.4 (149.8) 155.8 152.2 155.6 151.9 152.0C15 96.5 (92.0) 95.9 (91.4) 116.1 (110.6) 118.1 (112.5) 91.4 91.1 111.0 113.1 91.6C16 161.5 (153.7) 158.5 (150.9) 175.3 (166.9) 171.8 (163.5) 156.1 153.0 169.2 165.7 151.6C17 143.2 (136.3) 145.6 (138.6) 147.0 (140.0) 148.0 (140.9) 138.2 140.5 142.4 143.4 137.5C171 11.8 (11.5) 11.8 (11.5) 12.4 (12.1) 12.4 (12.0) 7.7 7.7 8.2 8.2 10.0C18 154.5 (147.1) 153.9 (146.5) 150.1 (142.9) 148.2 (141.1) 149.5 148.7 145.0 143.1 140.5C181 23.2 (22.3) 23.7 (22.8) 22.8 (22.0) 23.2 (22.3) 18.3 18.7 17.8 18.2 15.7C182 15.2 (14.7) 15.5 (15.1) 15.4 (15.0) 15.7 (15.2) 10.6 10.9 10.8 10.9 13.3C19 174.0 (165.6) 174.9 (166.4) 186.6 (177.6) 185.7 (176.7) 168.7 169.6 181.3 180.4 169.1
a GIAO B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d).b Valores en paréntesis corresponden a valores escalados de acuerdo a Ref. 17.
c GIAO mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G(d).d Datos extraídos desde Ref. 7.
La Tabla 3.1 muestra los desplazamientos químicos para los 19 átomos de
carbonos presentes en los modelos de ficocianobilina para las cuatro formas
tautoméricas: DITAM, TIM-A, TIM-D, y DITIM. Cada valor de la GIAO DFT se
compara con el respectivo valor experimental.
53

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Figura 3.7. Desplazamientos químicos para 13C-RMN por cálculos GIAO DFT.a) DITAM: Dilactama (anillos A y D); b) TIM-A: Lactima en A, Lactama en D; c) TIM-D: Lactama en A, Lactima en D; d) DITIM: Dilactima (anillos A y D). En Azul: cálculo GIAO B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d) con valores escalados de acuerdo a Ref. 17. En Rojo: cálculo GIAO mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G(d). En Negro: valores experimentales extraídos de Ref. 7.
Para mayor claridad en la presentación de nuestros resultados, los
desplazamientos químicos de la Tabla 3.1 que se consideraron significativos para el
análisis se muestran en la Figura 3.7.
En la Tabla 3.1, al fijar nuestra atención en el carbono C1 que forma el carbonilo
en el anillo A, y comparando el valor teórico de cada tautómero con respecto al valor
de referencia experimental,[7] podemos ver que el par DITAM/TIM-D presenta un valor
de alrdedor de 185 ppm para el caso de la GIAO B3LYP sin corregir, mientras que el
par DITIM/TIM-A presenta un valor cercano a los 195 ppm, y en comparación con el
valor experimental[7] de 182.9 ppm podemos pensar que la asignación de la forma ceto
en A es la correcta; aunque esta apreciación se vuelve ambigua cuando consideramos
54

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
los valores corregidos (valores en azul en la Figura 3.7 y en paréntesis en la Tabla 3.1),
ya que en ese caso el par DITAM/TIM-D marca en alrededor de 178 ppm y el par
DITIM/TIM-A marca en alrededor de 185 ppm, quedando el valor de referencia en el
medio de ambos pares. No obstante, los cálculos por GIAO mPW1PW91 nos terminan
confirmando que la asignación correcta para el C1 es ceto, ya que el par DITAM/TIM-D
se encuentra muy cercano al valor experimental (compárese los 182.3 ppm de DITAM
con respecto a los 189.9 ppm del valor de referencia[7]), en tanto el par DITIM/TIM-A
se encuentra desplazado por cerca de 10 ppm.
Análogamente, en el caso del carbono C19, que forma el carbonilo del anillo D,
podemos observar en la Tabla 3.1 y en la Figura 3.7 que, en el caso de los cálculos
GIAO B3LYP, tanto los valores corregidos como los sin corregir se ajustan al valor
experimental en el caso del par en DITAM/TIM-A, y no así para el par DITIM/TIM-D,
que se encuentra desplazado. Además, los valores de 168.7 ppm y 169.6 ppm
calculados por GIAO mPW9PW91 para DITAM y TIM-A, respectivamente,
concuerdan con los 169.1 ppm de la referencia experimental,[7] en tanto el par
DITIM/TIM-D se ve desplazado por alrededor de 10 ppm. En consecuencia, estos
resultados nos indican que el anillo D se encuentra como tautómero lactama.
De lo anterior, confirmamos que los espectros 13C-RMN obtenidos por el grupo
de Matysik[7] para la forma Pfr de ficocianobilina efectivamente corresponden a una
estructura dilactama, con ambos carbonilos en su forma ceto. Este resultado parece
contradecir nuestra hipótesis acerca de la formación del tautómero lactima en A
(modelo TIM-A) durante la interconversión del fitocromo de Pr a Pfr. Incluso, la
revisión reciente que hizo Gärtner[8] sobre estructuras del cromóforo en fitocromo
también discute sobre aquello, señalando que la participación de una tautomería en la
fotoconversión se puede descartar, ya que es un mecanismo incompatible con los
espectros 13C-RMN para la forma Pfr (referencia experimental de la Tabla 3.1). Sin
55

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
embargo, nuestra posición al respecto es que el argumento de la incompatibilidad de los
espectros RMN con una tautomería es discutible, ya que se sabe que la tautomería es un
mecanismo dependiente de las condiciones experimentales. En un medio polar, por
ejemplo, se favorece la forma lactama, y lo contrario sucede en un medio apolar, donde
se favorece la forma lactima.[2] También se ha obsevado que en fase sólida se favorece el
tautómero lactama[24-27] y, por lo tanto, es común encontrar ese tautómero en las
estructuras cristalográficas; y, precisamente, esa podría ser la situación para el
fitocromo, donde las bilinas en los cristales se encontrarían como tautómeros lactama.
Pero el argumento que consideramos de mayor utilidad en favor de nuestra hipótesis de
tautomería es que los estudios experimentales a los que hemos hecho referencia[7] en
esta sección (referencia experimental en Tabla 3.1 y en Figura 3.7) corresponden a
resultados obtenidos con espectroscopia RMN en fase sólida,[7;8] lo que explicaría la
presencia de una estructura dilactama (modelo DITAM), con sus dos carbonilos en la
forma ceto. Por lo tanto, en virtud de nuestros espectros TDDFT de absorción, la
barrera de energía para la formación del estado de transición TS-1wc, y la comparación
de la estructura de TS-1wc con respecto a las estructuras cristalográficas de Pr y
Pfr,[9;21] podemos concluir que nuestra hipótesis de tautomería es válida, asumiendo que
los espectros de 13C-RMN de la Ref. 7 fueron condicionados experimentalmente al
realizarse en fase sólida.
56

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
3.6 Referencias
[1.] J. A. Kereselidze, T. Sh. Zarqua, T. J. Kikalishvili, E. J. Churgulia, M. C. Makaridze, Russ.
Chem. Rev. 2002, 71, 993-2002.
[2.] M. J. Field, I. H. Hillier, J. Chem. Soc., Perkin Trans. 2 1987, 617-622.
[3.] A. Lledós, J. Bertrán, Tetrahedron Lett. 1981,22, 775-778.
[4.] A. Lledós, J. Bertrán, J. Mol. Stuct. 1985, 120, 73-78.
[5.] M. Moreno, W. H. Miller, Chem. Phys. Lett. 1990, 171, 475-479.
[6.] S. E. Braslavsky, A. R. Holzwarth, K. Schaffner, Angew. Chem. Int. Ed. Engl. 1983, 22, 656-
674.
[7.] T. Rohmer, C. Lang, J. Hughes, L. O. Essen, W. Gärtner, J. Matysik, Proc. Natl. Acad. Sci.
USA 2008, 105, 15229-15234.
[8.] C. Bongards, W. Gärtner, Acc. Chem. Res. 2010, 43, 485-495.
[9.] X. Yang, J. Kuk, K. Moffat, Proc. Natl. Acad. Sci. USA 2008, 105, 14715-14720.
[10.] A. D. Becke, Phys. Rev. A 1998, 38, 3098-3100.
[11.] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B, 1988, 37, 785-789.
[12.] J. Tomasi, B. Menucci, R. Cammi, Chem. Rev. 2005, 105, 2999-3094.
[13.] H. Schlegel, J. Comput. Chem. 1982, 3, 214-218.
[14.] H. Schlegel, Geometry Optimization on Potential Energy Surface In Modern Electronic
Structure Theory, (Ed.: D. R. Yarkony) World Scientific Publishing: Singapore, 1994.
[15.] Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.
Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M.
Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.
A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M.
Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P.
Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O.
Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G.
A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C.
Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz,
Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P.
57

CAPÍTULO III. TAUTOMERÍA EN LA INTERCONVERSIÓN
Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A.
Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C.
Gonzalez, and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004.
[16.] F. A. A. Mulder, M. Filatov, Chem. Soc. Rev. 2010, 39, 578-590.
[17.] A. E. Aliev, D. Courtier-Murias, S. Zhou, J. Mol. Struct. 2009, 893, 1-5.
[18.] B. Karniol, R. D. Vierstra, Proc. Natl. Acad. Sci. USA 2003, 100, 2807-2812.
[19.] B. Borucki, D. v. Stetten, S. Seibeck, T. Lamparter, N. Michael, M. A. Mroginski, H. Otto, D.
H. Murgida, M. P. Heyn, P. Hildebrandt, J. Biol. Chem. 2005, 280, 34358-34364.
[20.] X. Yang, E. A. Stojkovic, J. Kuk, K. Moffat, Proc. Natl. Acad. Sci. USA 2007, 104, 12571-
12576.
[21.] J. R. Wagner, J. S. Brunzelle, K. T. Forest, R. D. Vierstra, Nature 2005, 438, 325-331.
[22.] J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra, K. T. Forest, J. Biol. Chem. 2007, 282,
12298-12309.
[23.] L. O. Essen, J. Mailliet, J. Hughes, Proc. Natl. Acad. Sci. USA 2008, 105, 14709-14714.
[24.] H. W. Yang, B. M. Craven, Acta Crystallogr. B 1998, 54, 912–920.
[25.] B. R. Penfold, Acta Crystallogr. 1953, 6, 591–600.
[26.] U. Ohms, H. Guth, E. Heller, H. Dannöhl, A. Schweig, Z. Kristallogr. 1984, 169, 185–200.
[27.] J. Almlöf, A. Kvick, I. Olovsson, Acta Crystallogr. B 1971, 27, 1201–1208.
58

CAPÍTULO IV. CONCLUSIONES
CAPÍTULO IV
CONCLUSIONES
1. Los espectros teóricos de absorción calculados en el contexto de la teoría
del funcional de la densidad dependiente del tiempo sugieren que la
ficocianobilina presente en el fitocromo Cph1 de cianobacteria adopta
una estructura semicíclica ZZZssa.
2. Los espectros teóricos de absorción calculados en el contexto de la teoría
del funcional de la densidad dependiente del tiempo sugieren que la
fitocromobilina presente en el fitocromo A de planta adopta una
estructura semicíclica ZZZssa.
3. En el contexto de la TDDFT, el índice Q/S y el isomerismo
conformacional se encuentran relacionados: valores menores de Q/S para
la estructura helicoidal cíclica ZZZsss, valores medios de Q/S para la
estructura semicíclica ZZZssa, y valores mayores de Q/S para la
estructura extendida ZZZasa. Proponemos un modelo teórico para el
comportamiento general de Q/S en bilinas: F = FS + FQ.
59

CAPÍTULO IV. CONCLUSIONES
4. Los tautómeros lactima de biliverdina inducen un desplazamiento
batocrómico en forma dependiente del anillo pirrólico. La tautomería en
el anillo A induce un desplazamiento batocrómico con 20 nm adicionales
por sobre la tautomería en el anillo D, y reproduce cuantitativamente el
valor experimental de la forma Pfr de bacteriofitocromo. Estos
resultados sugieren que en la interconversión de Pr a Pfr ocurre una
tautomería lactama-lactima en el anillo A de la biliverdina.
5. Los estados de transición para el mecanismo intramolecular de
tautomería lactama-lactima en los anillos A y D muestran que la
tautomería se encuentra favorecida energéticamente para el anillo A, pero
las altas barreras de energía asociadas descartan la posibilidad
de un mecanismo intramolecular. Por otro lado, el estado de transición
para el mecanismo de tautomería catalizado por agua tiene una barrera de
energía de 19 kcal/mol, por lo tanto, la tautomería lactama-lactima es
viable con este mecanismo.
6. Los cálculos RMN corroboran que, en los espectros 13C-RMN del grupo
de Matysik, la estructura de la ficocianobilina en la forma Pfr de
fitocromo es dilactama, lo que sugiere que en la interconversión de Pr a
Pfr no ocurre tautomería lactama-lactima. Sin embargo, los experimentos
de referencia se realizaron en estado sólido, donde se favorece la
presencia del tautómero lactama, por lo tanto, una tautomería lactama-
lactima no se puede descartar en el sistema real.
60

__________________________________________________________________________________________________________________________
APÉNDICE A
Marco Teórico __________________________________________________________________________________________________________________________
61

APÉNDICE A. MARCO TEÓRICO
A.1 Teoría del Funcional de la Densidad Dependiente del Tiempo
La teoría del funcional de la densidad dependiente del tiempo (TDDFT),
formulada a partir de los teoremas de Runge-Gross[1] y sus extensiones,[2;3] utiliza las
ecuaciones de Kohn-Sham dependientes del tiempo,[3-6]
i∂
∂ tir ,t = FKS
r ,t i r ,t (1)
Además, la densidad dependiente del tiempo (r,t) se encuentra definida como
r , t =∑i
nocc
∣i r , t ∣² (2)
En las ecuaciones (1) y (2), (r,t) representa los orbitales moleculares ocupados
dependientes del tiempo y FKS(r,t) es el operador Kohn-Sham dependiente del tiempo.
Existen dos formas de aplicar la TDDFT:
(1) Método de Yabana o TDDFT en tiempo real
(2) Método de Casida o TDDFT de respuesta lineal
En el método de Yabana, la función de onda Kohn-Sham tiempo-dependiente se
propaga en el tiempo.[7;8] En el método de Casida, se analiza la respuesta lineal de las
ecuaciones Kohn-Sham dependientes del tiempo cuando se perturban con un campo
eléctrico externo.[4] En el trabajo de esta tésis se utilizó la TDDFT según el método de
Casida, cuyo formalismo se explica a continuación.
62

APÉNDICE A. MARCO TEÓRICO
La idea central del método de Casida es aplicar la teoría de perturbación
dependiente del tiempo a primer orden para así describir la respuesta lineal dependiente
del tiempo de la densidad frente a un campo electrico oscilante tiempo-dependiente.
Antes de perturbar el sistema con el campo eléctrico, se asume que el sistema molecular
se encuentra en su estado electrónico fundamental, por lo que se cumple la ecuación
Kohn-Sham para el estado fundamental,
FKS ,0r 0KS=E0
KS0KS (3)
con densidad electrónica igual a
0r =∑
i
nocc
∣ir ∣² (4)
donde FKS,0(r) es el operador Kohn-Sham independiente del tiempo, Φ0KS es el
determinante de Slater Kohn-Sham del estado fundamental, (r) son los orbitales Kohn-
Sham ocupados, y E0KS es la energía total para el estado fundamental.
Al aplicar un campo eléctrico oscilante tiempo-dependiente,
Et =fei t f ∗ e−it (5)
se produce un cambio en los orbitales Kohn-Sham, en la densidad electrónica, y en el
operador Kohn-Sham.
63

APÉNDICE A. MARCO TEÓRICO
Dado que la aplicación del campo externo puede considerarse como una
pequeña perturbación del sistema, la nueva densidad incluye una perturbación de primer
orden,
r , t =0r r , t (6)
y así el operador Kohn-Sham dependiente queda definido como
FKS r ,t = FKS ,0r [ FKS ,0
r ] t=t 0
r , t Et (7)
dónde el segundo término de la derecha corresponde al cambio lineal del operador
Kohn-Sham del estado fundamental FKS,0 al momento de activar la perturbación.
Por otro lado, tenemos que la energía del estado singulete más bajo es
Esing=2 ESsing−ES
trip (8)
Esing=2 ⟨Ssing∣ FKS ∣S
sing⟩ − ⟨Strip∣ FKS ∣S
trip⟩ (9)
por lo tanto, al sustituir las ecuaciones (6) y (7) en la ecuación (9); el posterior análisis
de las restricciones de ortogonalidad; y luego de la transformación de Fourier al espacio
de energía; se obtiene la ecuación de Casida para la TDDFT, [4;5;9;10] con la que se llega a
las energías de excitación y vectores de transición a través de una ecuación de
autovalores. En notación matricial, las ecuación de Casida es
A BB∗ A∗ X
Y = 1 00 −1X
Y (10)
64

APÉNDICE A. MARCO TEÓRICO
Los elementos de la matriz A y B en la ecuación (10) corresponden a
funcionales híbridos de correlación-intercambio,
Aia , jb = ijaba−i ja | ib−CHF ji |ab1−CHF ja | f xc | ib (11)
B ia, jb = ja |bi −CHF jb | ai1−CHF ja | f xc |bi (12)
La ecuación de Casida (10) es una ecuación de autovalores no hermítica, cuya
solución entrega las energías de excitación y los vectores de transición |XY> que
determinan el cambio a primer orden de la densidad electrónica y, por lo tanto,
determinan también la función de onda del estado excitado.
Aun cuando el kernel de correlación-intercambio xc, en las ecuaciones (11) y
(12), es formalmente dependiente de la energía, en la práctica se utilizan funcionales de
correlación-intercambio del estado fundamental (tales como SVWN,[11;12] BLYP,[13;14]
PBE,[15;16] o B3LYP[17]) para evaluar tales términos, lo que es consecuencia de la
aplicación de la aproximación de densidad local adiabática (ALDA),
f xc ,r , r ' = f xcr r−r ' (13)
A.2 Modelo de Continuo Polarizable
En general, un modelo continuo se define como un modelo en el cual los grados
de libertad de las partículas constituyentes se describen mediante un continuo,
usualmente a través de una función de distribución.
En el caso particular del modelo de continuo polarizable (PCM), el solvente se
representa como un dieléctrico continuo infinito, mientras que el soluto se localiza en
65

APÉNDICE A. MARCO TEÓRICO
una cavidad molecular que se genera con esferas entrelazadas localizadas alrededor de
cada átomo del soluto, de manera de poder conservar la forma real de la molécula.[18]
En la interacción eletrostática soluto-solvente, la distribución de carga M del
soluto, dentro de la cavidad, polariza el dieléctrico continuo, el que luego polariza la
distribución de carga del soluto. Esta manera de tratar la interacción electrostática
corresponde a un proceso autoconsistente, el cual se resuelve numéricamente en forma
iterativa.[18]
El potencial de reacción del solvente se define como el potencial de interacción
para modelos continuos, donde el soluto se modela como un dipolo puntual polarizable,
mientras que la interacción electrostática entre un medio polarizable y un dipolo se
expresa en términos de un campo electrostático, el cual tiene su origen en la
polarización del dieléctrico.[18;19] Sin embargo, el modelo básico requiere la solución de
un problema electrostático clásico: el problema de Poisson. Para tratar este problema, es
necesario definir las condiciones de la superficie de la cavidad ,
[V ] = 0 en (14)
[∂V ] = 0 en (15)
La primera condición (14) expresa la continuidad del potencial en la superficie,
donde también se cumple que
[V ] = V i n − V out = 0 (16)
66

APÉNDICE A. MARCO TEÓRICO
La segunda condición (15) involucra la discontinuidad del componente del
campo que es perpendicular a la superficie de la cavidad. Si consideramos una cavidad
con una constante dieléctrica igual a 1 tenemos que
[∂V ] = ∂V∂n
i n
− ∂V∂n
out
= 0 (17)
donde n corresponde al vector que apunta al exterior que es perpendicular a la superficie
de la cavidad.
En el modelo de continuo polarizable se utiliza un método de carga aparente de
superfice denominado método ASC (Aparent Surface Charge), en el cual, a partir de la
condición (17), se deriva una carga aparente de superficie (s), donde s es la variable de
posición en la superficie .[18;20]
El método ASC define un potencial sobre todo el sistema,
V r = ∫
ss
∣r−s∣d2 s (18)
Al considerar que la fuente del potencial de reacción proviene de una
distribución de carga limitada a una superficie cerrada, se simplifica considerablemente
el problema electrostático en comparación con otras formulaciones en las cuales el
medio dieléctrico completo se toma como la fuente del potencial de reacción.
Debido al gran costo computacional que se requiere para la resolución de la
integral de la ecuación (18), la superficie de cavidad se aproxima en términos de
elementos finitos denominados tesserae, que se hacen lo suficientemente pequeños para
considerar (s) prácticamente como una constante dentro de cada tessera.[18] Al
67

APÉNDICE A. MARCO TEÓRICO
describir (s) en forma discreta, es posible definir un set de cargas puntuales, qk, en
términos del valor local de (s) de cada tessera por el área correspondiente, Ak. Por lo
tanto,
V r ∝ ∑k
ssk Ak
∣r−sk∣= ∑
k
qk
∣r−s k∣(19)
Luego, el vector de polarización lo entrega el gradiente del potencial total V(r),
Pir = −i−1
4∇ V r (20)
donde i es la constante dieléctrica de la región i. Además, en la frontera entre dos
regiones i y j, existe una distribución ASC definida como
ij = − P j − Pi⋅nij (21)
donde nij es el vector unitario de la superficie fronteriza que apunta desde el medio i al
medio j.
La definición básica del modelo PCM se deriva de la ecuación (20), tomando en
consideración que el caso más básico es cuando i = 1 y j = 1, lo que nos permite
calcular el gradiente sobre la parte interna de la superficie,
s = − 14
∂
∂ nV M V i n (22)
donde VM es el potencial electrostático generado por una distribución de carga M.
68

APÉNDICE A. MARCO TEÓRICO
A.3 Cálculo del Apantallamiento RMN por el Método GIAO
Como punto de partida se utiliza la aproximación de Hartree-Fock, en la cual la
función del estado fundamental se construye con un determinante de Slater,
0 = N !−1/2∥1 1... j j ...N /2 N /2∥ (23)
Comúnmente, los orbitales moleculares se representan como una combinación
lineal de orbitales atómicos al utilizar la aproximación MO-LCAO,
j =∑
C j (24)
Además, sabemos que la matriz de densidad de una molécula de capa cerrada se
define como
D = 2∑i=1
N /2
Ci∗Ci (25)
La primera derivada de la matriz de densidad con respecto al campo magnético
aplicado, ∂D /∂B0 , se puede obtener a partir de la resolución de las ecuaciones
Hartree-Fock con pertubación acoplada (coupled-perturbed Hartree-Fock o CPHF) en
las cuales la pertubación generada por el campo magnético externo, 12
B0 × r iA−R0, se
incluye en el operador de Fock.[21]
69

APÉNDICE A. MARCO TEÓRICO
El campo magnético externo es independiente del origen de gauge R0, y se
puede demostrar que el tensor de apantallamiento también es independiente de la
elección de R0, es decir,
AR0 − A
0 = 0 (26)
para cualquier valor de R0.[22] La ecuación (26) se cumple solamente si se satisface la
relación de completitud y si las funciones de onda se obtienen variacionalmente. Esta
última condición, en la práctica, se puede satisfacer fácilmente; sin embargo, la primera
condición, al requerir el uso de un set completo de bases, no se puede satisfacer. Por lo
tanto, en la práctica, existe cierta dependencia respecto a la elección del origen de
gauge R0, la que debe ser eliminada por algún método. Uno de estos métodos
corresponde a la aproximación por GIAO (Gauge-Including Atomic Orbitals).[22;23] En el
método GIAO se trabaja con funciones de base complejas que dependen del campo
magnético externo B0,
GIAO
B0 = e−iB0×R −R 0⋅r /2c (27)
las cuales se utilizan en la aproximación MO-LCAO de la ecuación (24). En la ecuación
(27), es un orbital atómico real independiente del campo que se encuentra centrado
en la posición R. Las funciones GIAO tienen la propiedad de cambiar el origen R0 por
R cuando interactúan con el operador de momento, por lo que son muy convenientes
para el cálculo de apantallamiento. Por lo tanto, el uso de GIAOs en el cálculo de los
elementos de matriz para el Hamiltoniano molecular conduce a la cancelación de la
dependecia con en el origen de gauge R0.[22]
70

APÉNDICE A. MARCO TEÓRICO
A.4 Referencias
[1.] E. Runge, E. K. U. Gross, Phys. Rev. Lett. 1984, 52, 997.
[2.] R. van Leeuwen, Phys. Rev. Lett. 1999, 82, 3863.
[3.] R. van Leeuwen, Int. J. Mod. Phys. B 2001, 15, 1969.
[4.] M. E. Casida in Recent Advances in Density Functional Methods, Part I (Ed.: D. P. Chong),
World Scientific, Singapore, 1995, p. 155.
[5.] A. Drew, M. Head-Gordon, Chem. Rev. 2005, 105, 4009.
[6.] E. K. U. Gross, W. Kohn, Adv. Quantum Chem. 1990, 21, 255.
[7.] K. Yabana, G. F. Bertsch, Phys. Rev. B 1996, 54, 4484.
[8.] M. A. L. Marquez, A. Castro, G. F. Bertsch, A. Rubio, Comput. Phys. Commun. 2003, 151, 60.
[9.] S. Hirata, M. Head-Gordon, Chem. Phys. Lett. 1999, 314, 291.
[10.] F. Furche, J. Chem. Phys. 2001, 114, 5982.
[11.] P. A. M. Dirac, Proc. Cambridge Philos. Soc. 1930, 26, 376.
[12.] S. H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 1980, 58, 1200.
[13.] A. D. Becke, Phys. Rev. A 1988, 38, 3098.
[14.] C. Lee, W. Wang, R. G. Parr, Phys. Rev. B 1988, 37, 785.
[15.] J. P. Perdew, K. Burke, M. Enzerhof, Phys. Rev. Lett. 1996, 77, 3865.
[16.] J. P. Perdew, K. Burke, M. Enzerhof, Phys. Rev. Lett. 1997, 78, 1396.
[17.] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
[18.] J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 2005, 105, 2999.
[19.] L. Onsager, J. Am. Chem. Soc. 1936, 58, 1486.
[20.] J. Tomasi, M. Persico, Chem. Rev. 1994, 94, 2027.
[21.] R. M. Stevens, R. M. Pitzer, W. N. Lipscomb, J. Chem. Phys. 1963, 38, 550.
[22.] R. Ditchfield, Mol. Phys. 1974, 27, 789.
[23.] K. Wolinski, J. F. Hinton, P. Pulay, J. Am. Chem. Soc. 1990, 112, 8251.
71

__________________________________________________________________________________________________________________________
APÉNDICE B
Tablas Suplementarias __________________________________________________________________________________________________________________________
72
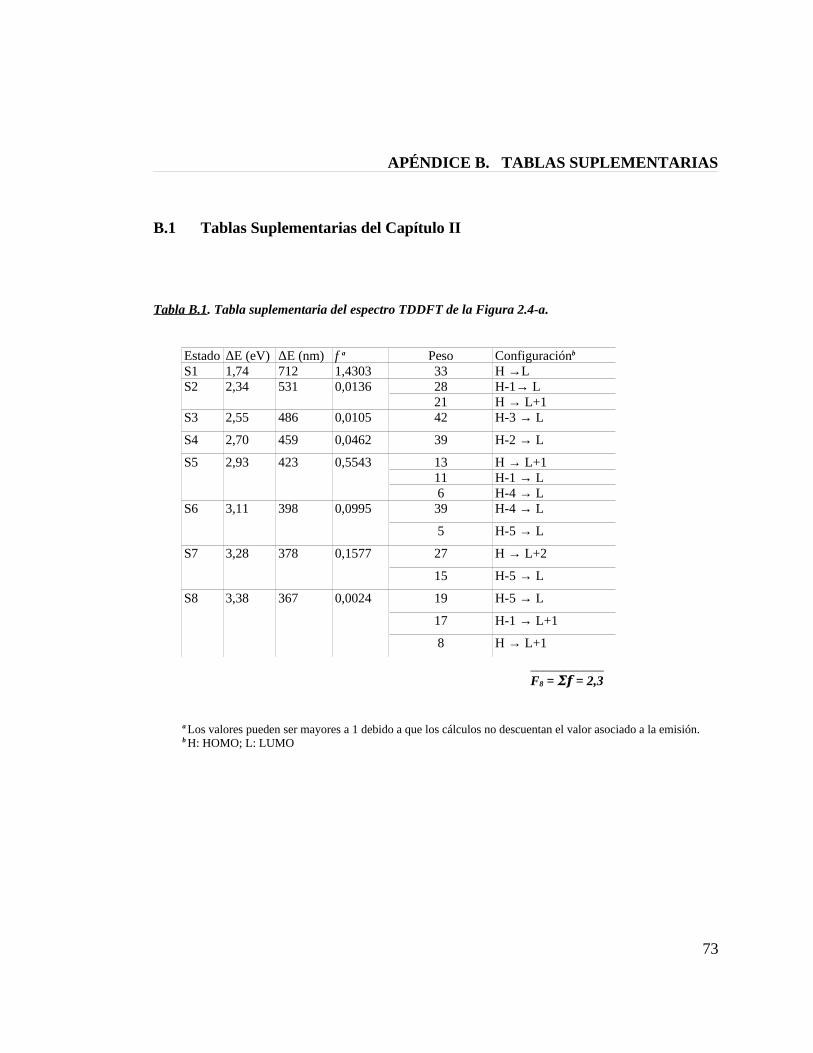
APÉNDICE B. TABLAS SUPLEMENTARIAS
B.1 Tablas Suplementarias del Capítulo II
Tabla B.1. Tabla suplementaria del espectro TDDFT de la Figura 2.4-a.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb
S1 1,74 712 1,4303 33 H →LS2 2,34 531 0,0136 28 H-1→ L
21 H → L+1S3 2,55 486 0,0105 42 H-3 → L
S4 2,70 459 0,0462 39 H-2 → L
S5 2,93 423 0,5543 13 H → L+111 H-1 → L6 H-4 → L
S6 3,11 398 0,0995 39 H-4 → L
5 H-5 → L
S7 3,28 378 0,1577 27 H → L+2
15 H-5 → L
S8 3,38 367 0,0024 19 H-5 → L
17 H-1 → L+1
8 H → L+1
___________F8 = Σf = 2,3
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
73
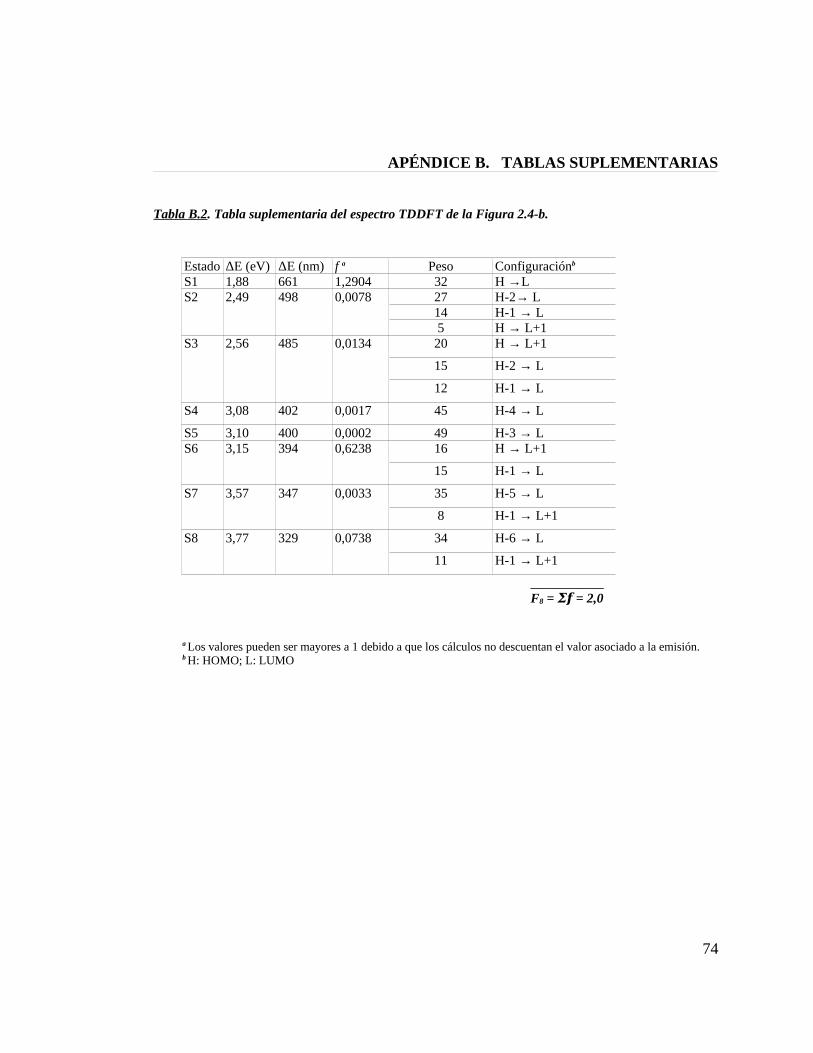
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.2. Tabla suplementaria del espectro TDDFT de la Figura 2.4-b.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb
S1 1,88 661 1,2904 32 H →LS2 2,49 498 0,0078 27 H-2→ L
14 H-1 → L5 H → L+1
S3 2,56 485 0,0134 20 H → L+1
15 H-2 → L
12 H-1 → L
S4 3,08 402 0,0017 45 H-4 → L
S5 3,10 400 0,0002 49 H-3 → LS6 3,15 394 0,6238 16 H → L+1
15 H-1 → L
S7 3,57 347 0,0033 35 H-5 → L
8 H-1 → L+1
S8 3,77 329 0,0738 34 H-6 → L
11 H-1 → L+1
___________F8 = Σf = 2,0
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
74

APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.3. Tabla suplementaria del espectro TDDFT de la Figura 2.4-c.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb
S1 2,02 614 1,5852 35 H →LS2 2,30 539 0,0068 33 H-2→ L
9 H-1 → LS3 2,67 465 0,0682 27 H-1 → L
13 H → L+1
7 H-2 → L
S4 2,81 441 0,0002 49 H-3 → L
S5 3,11 398 0,0016 43 H-4 → LS6 3,22 385 0,2576 26 H → L+1
5 H-6 → L
S7 3,30 376 0,0101 5 H-1 → L
42 H-5 → L
S8 3,42 362 0,0005 24 H-6 → L
16 H-7 → L
___________F8 = Σf = 1,9
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
75
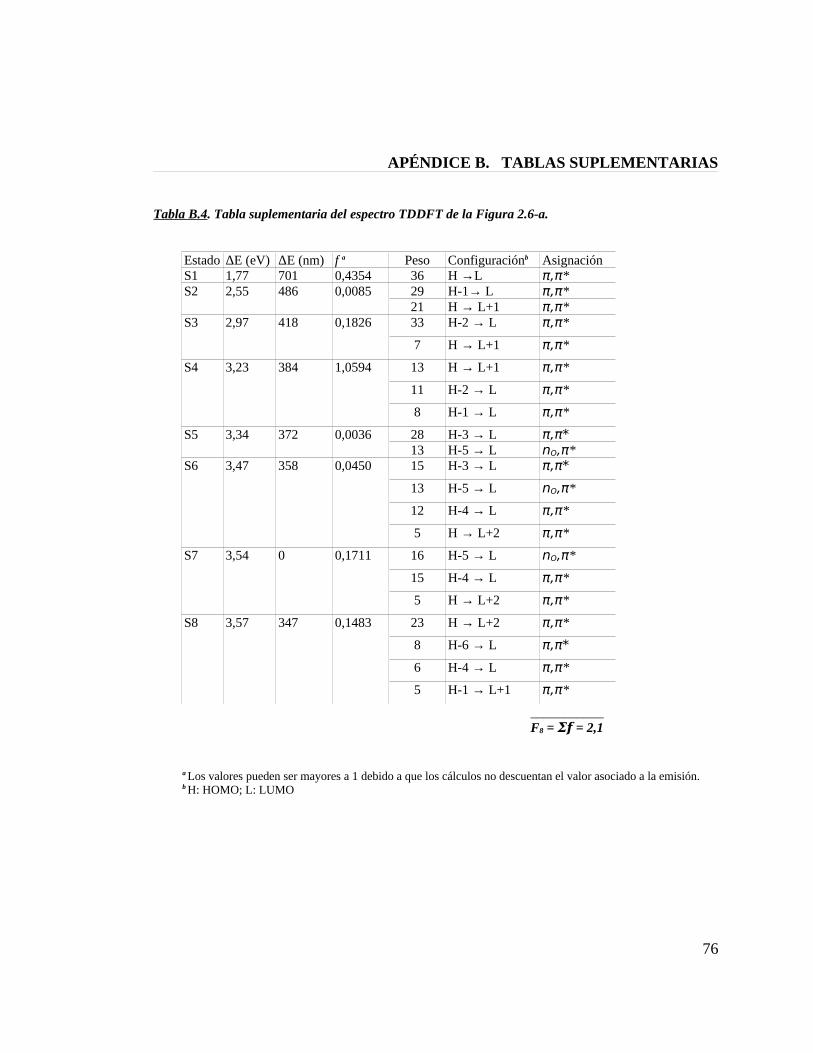
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.4. Tabla suplementaria del espectro TDDFT de la Figura 2.6-a.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,77 701 0,4354 36 H →L π,π*S2 2,55 486 0,0085 29 H-1→ L π,π*
21 H → L+1 π,π*S3 2,97 418 0,1826 33 H-2 → L π,π*
7 H → L+1 π,π*
S4 3,23 384 1,0594 13 H → L+1 π,π*
11 H-2 → L π,π*
8 H-1 → L π,π*
S5 3,34 372 0,0036 28 H-3 → L π,π*13 H-5 → L nO,π*
S6 3,47 358 0,0450 15 H-3 → L π,π*
13 H-5 → L nO,π*
12 H-4 → L π,π*
5 H → L+2 π,π*
S7 3,54 0 0,1711 16 H-5 → L nO,π*
15 H-4 → L π,π*
5 H → L+2 π,π*
S8 3,57 347 0,1483 23 H → L+2 π,π*
8 H-6 → L π,π*
6 H-4 → L π,π*
5 H-1 → L+1 π,π*
___________F8 = Σf = 2,1
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
76
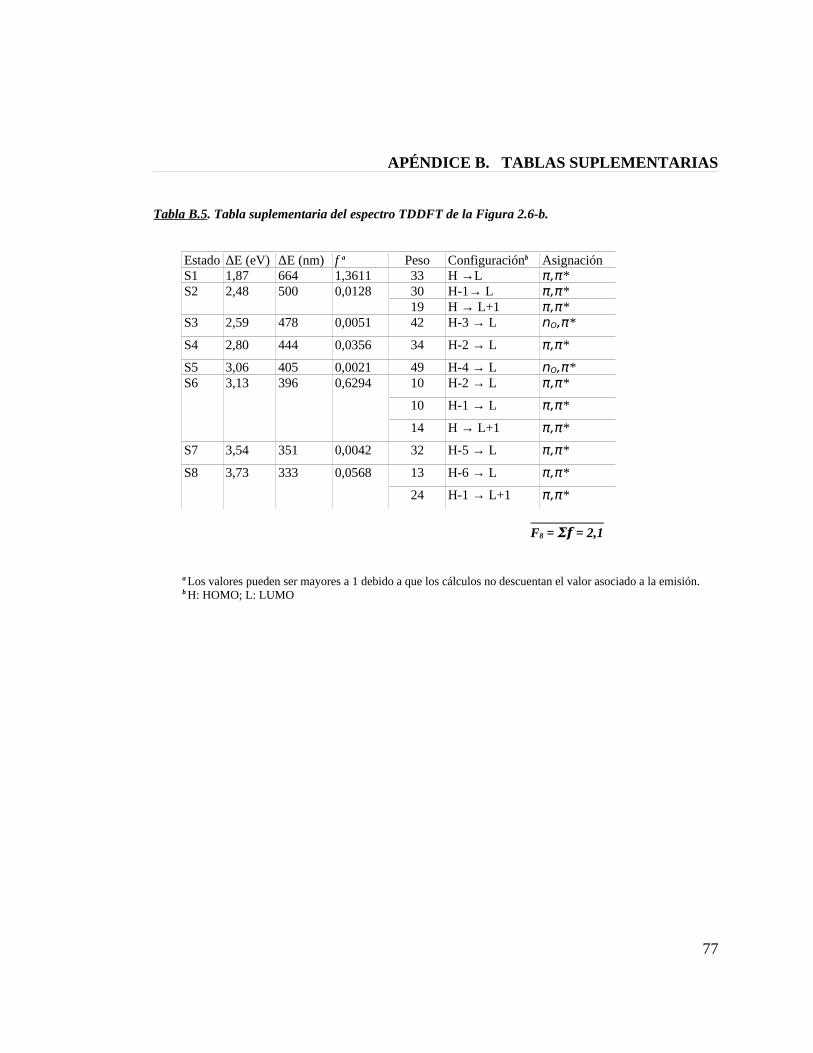
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.5. Tabla suplementaria del espectro TDDFT de la Figura 2.6-b.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,87 664 1,3611 33 H →L π,π*S2 2,48 500 0,0128 30 H-1→ L π,π*
19 H → L+1 π,π*S3 2,59 478 0,0051 42 H-3 → L nO,π*
S4 2,80 444 0,0356 34 H-2 → L π,π*
S5 3,06 405 0,0021 49 H-4 → L nO,π*S6 3,13 396 0,6294 10 H-2 → L π,π*
10 H-1 → L π,π*
14 H → L+1 π,π*
S7 3,54 351 0,0042 32 H-5 → L π,π*
S8 3,73 333 0,0568 13 H-6 → L π,π*
24 H-1 → L+1 π,π*
___________F8 = Σf = 2,1
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
77

APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.6. Tabla suplementaria del espectro TDDFT de la Figura 2.6-c.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,77 700 1,0417 30 H →L π,π*S2 1,93 642 0,2391 23 H-2→ L nO,π*
13 H-1 → L π,π*S3 2,28 545 0,1538 25 H-1 → L π,π*
13 H-2 → L nO,π*
7 H → L+3 π,π*
S4 2,49 499 0,0085 31 H-3 → L π,π*
15 H → L+1 π,π*
S5 2,85 434 0,0031 48 H-4 → L nO,π*S6 2,98 416 0,3111 12 H → L+1 π,π*
8 H-1 → L+1 π,π*
6 H-3 → L π,π*
6 H-2 → L nO,π*
5 H-2 → L+1 nO,π*
S7 3,08 402 0,2100 18 H-1 → L+1 π,π*
12 H-2 → L+1 nO,π*
5 H → L+1 π,π*
S8 3,27 379 0,0352 22 H-2 → L+1 nO,π*
16 H-1 → L+1 π,π*
___________F8 = Σf = 2,0
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
78

APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.7. Tabla suplementaria del espectro TDDFT de la Figura 2.6-d.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,94 640 1,6693 35 H →L π,π*S2 2,26 549 0,0090 36 H-2→ L nO,π*
7 H-1 → L π,π*S3 2,45 505 0,0421 32 H-1 → L π,π*
6 H → L+1 π,π*
5 H-2 → L nO,π*
S4 2,74 453 0,0028 48 H-3 → L nO,π*
S5 2,75 451 0,0348 33 H-4 → L π,π*12 H → L+1 π,π*
S6 3,11 398 0,3042 23 H → L+1 π,π*
7 H-4 → L π,π*
S7 3,33 373 0,0056 46 H-5 → L π,π*
S8 3,45 360 0,0059 18 H-6 → L π,π*
16 H-7 → L π,π*
10 H-1 → L+1 π,π*
___________F8 = Σf = 2,1
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
79

APÉNDICE B. TABLAS SUPLEMENTARIAS
B.2 Tablas Suplementarias del Capítulo III
Tabla B.8. Tabla suplementaria del espectro TDDFT de la Figura 3.2-a.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,69 735 1,0462 34 H → L π,π*S2 2,13 582 0,0162 30 H-1 → L π,π*
20 H → L+1 π,π*S3 2,48 501 0,0381 34 H-2 → L π,π*
7 H → L+1 π,π*
S4 2,72 456 0,2306 23 H-3 → L nO,π*
7 H → L+1 π,π*
S5 2,81 442 0,3665 17 H-3 → L nO,π*6 H → L+1 π,π*6 H-2 → L π,π*5 H-1 → L π,π*
S6 2,99 414 0,1819 32 H → L+2 π,π*
8 H-1 → L+1 π,π*
S7 3,12 398 0,0453 23 H-1 → L+1 π,π*
16 H-4 → L π,π*
S8 3,31 375 0,0058 30 H-2 → L+1 π,π*
13 H-4 → L π,π*
___________F8 = Σf = 1,9
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
80
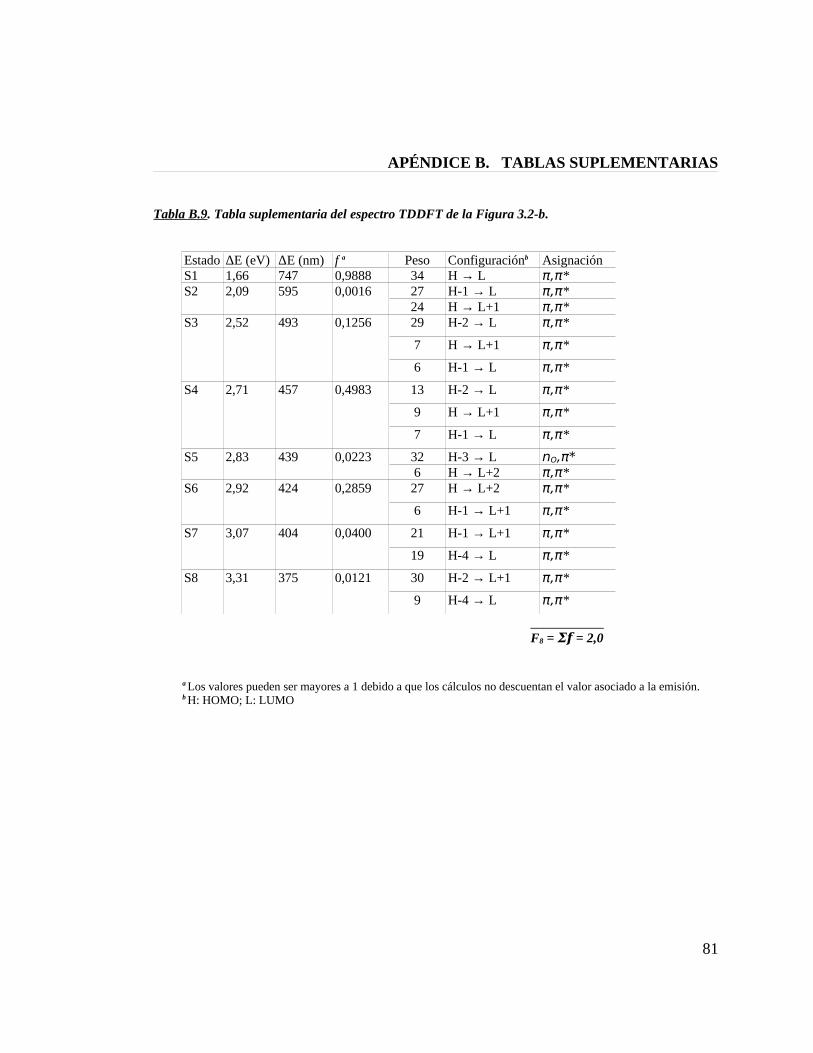
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.9. Tabla suplementaria del espectro TDDFT de la Figura 3.2-b.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,66 747 0,9888 34 H → L π,π*S2 2,09 595 0,0016 27 H-1 → L π,π*
24 H → L+1 π,π*S3 2,52 493 0,1256 29 H-2 → L π,π*
7 H → L+1 π,π*
6 H-1 → L π,π*
S4 2,71 457 0,4983 13 H-2 → L π,π*
9 H → L+1 π,π*
7 H-1 → L π,π*
S5 2,83 439 0,0223 32 H-3 → L nO,π*6 H → L+2 π,π*
S6 2,92 424 0,2859 27 H → L+2 π,π*
6 H-1 → L+1 π,π*
S7 3,07 404 0,0400 21 H-1 → L+1 π,π*
19 H-4 → L π,π*
S8 3,31 375 0,0121 30 H-2 → L+1 π,π*
9 H-4 → L π,π*
___________F8 = Σf = 2,0
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
81
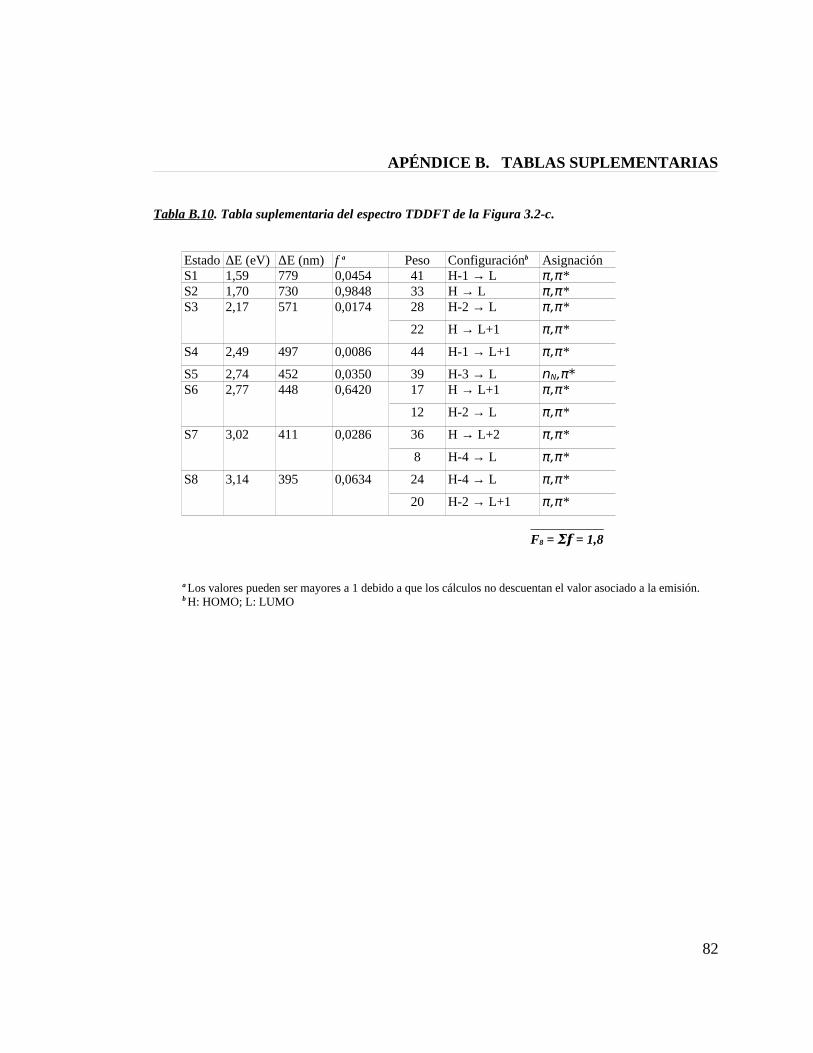
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.10. Tabla suplementaria del espectro TDDFT de la Figura 3.2-c.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,59 779 0,0454 41 H-1 → L π,π*S2 1,70 730 0,9848 33 H → L π,π*S3 2,17 571 0,0174 28 H-2 → L π,π*
22 H → L+1 π,π*
S4 2,49 497 0,0086 44 H-1 → L+1 π,π*
S5 2,74 452 0,0350 39 H-3 → L nN,π*S6 2,77 448 0,6420 17 H → L+1 π,π*
12 H-2 → L π,π*
S7 3,02 411 0,0286 36 H → L+2 π,π*
8 H-4 → L π,π*
S8 3,14 395 0,0634 24 H-4 → L π,π*
20 H-2 → L+1 π,π*
___________F8 = Σf = 1,8
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
82
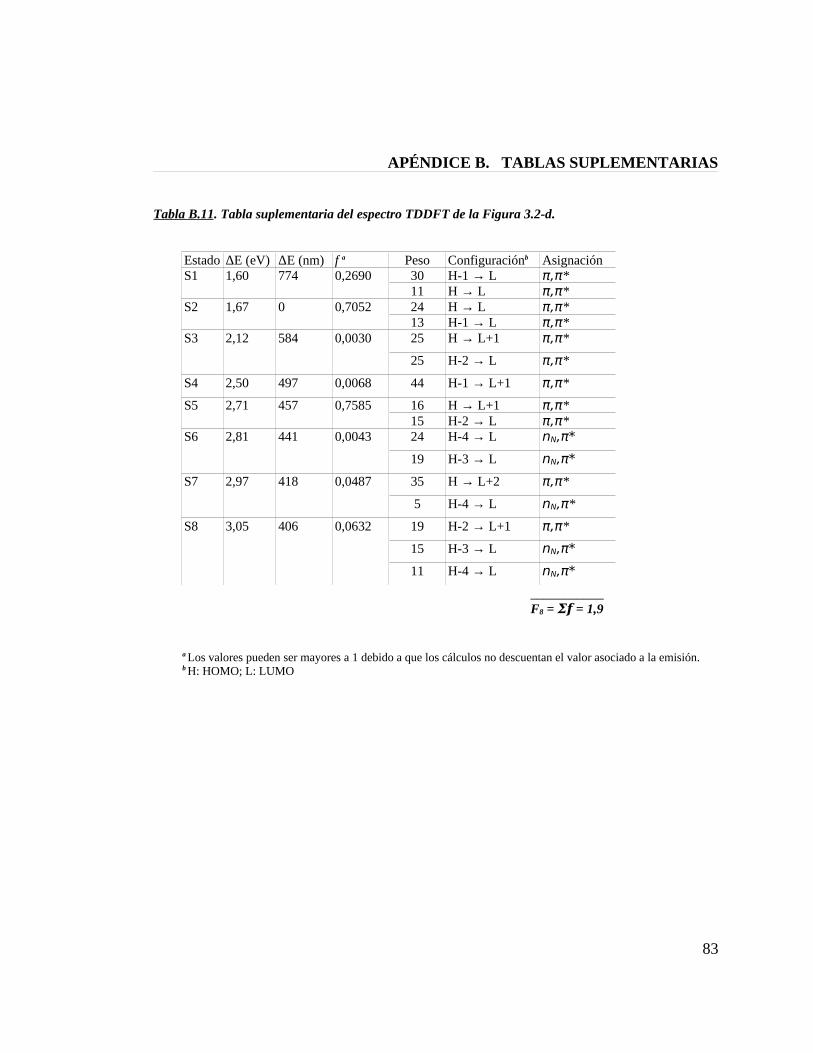
APÉNDICE B. TABLAS SUPLEMENTARIAS
Tabla B.11. Tabla suplementaria del espectro TDDFT de la Figura 3.2-d.
Estado ΔE (eV) ΔE (nm) f a Peso Configuraciónb AsignaciónS1 1,60 774 0,2690 30 H-1 → L π,π*
11 H → L π,π*S2 1,67 0 0,7052 24 H → L π,π*
13 H-1 → L π,π*S3 2,12 584 0,0030 25 H → L+1 π,π*
25 H-2 → L π,π*
S4 2,50 497 0,0068 44 H-1 → L+1 π,π*
S5 2,71 457 0,7585 16 H → L+1 π,π*15 H-2 → L π,π*
S6 2,81 441 0,0043 24 H-4 → L nN,π*
19 H-3 → L nN,π*
S7 2,97 418 0,0487 35 H → L+2 π,π*
5 H-4 → L nN,π*
S8 3,05 406 0,0632 19 H-2 → L+1 π,π*
15 H-3 → L nN,π*
11 H-4 → L nN,π*
___________F8 = Σf = 1,9
a Los valores pueden ser mayores a 1 debido a que los cálculos no descuentan el valor asociado a la emisión.b H: HOMO; L: LUMO
83

__________________________________________________________________________________________________________________________
APÉNDICE C
Publicaciones y Presentaciones Asociadas __________________________________________________________________________________________________________________________
84

The Chromophore Structure of the Cyanobacterial Phytochrome Cph1 As Predicted byTime-Dependent Density Functional Theory
Ricardo A. Matute and Renato ContrerasDepartamento de Química, Facultad de Ciencias, UniVersidad de Chile, Casilla 653, Santiago, Chile
Guillermo Perez-Hernandez and Leticia Gonzalez*Institut fur Physikalische Chemie, Friedrich-Schiller-UniVersitat Jena, Helmholtzweg 4, 07743 Jena, Germany
ReceiVed: August 20, 2008; ReVised Manuscript ReceiVed: September 25, 2008
The UV-vis absorption spectra of the photoreceptor chromophores biliverdin (BV) in the ZZZssa conformationand the phycocyanobilin (PCB) with conformations ZZZssa and ZZZasa have been investigated by means oftime-dependent density functional theory (TD-DFT) with a polarized continuum model. The three systemsare studied in different conditions to include protonation, solvation- and protein-environmental effects on gasphase and available X-ray structures. The crystal structures of BV in bacteriophytochrome of Deinococcusradiodurans and PCB in C-Phycocyanin serve to calibrate the performance of the TD-DFT method and allowestimating the spectral shifts created when gas phase structures instead of a proper environment are used. Incontrast, the structure of PCB in the cyanobacterial phytochrome Cph1 is unknown. The excellent agreementof the theoretical spectrum with experimentally recorded data for the PCB in the cyanobacterial phytochromeCph1 strongly supports a semicyclic ZZZssa structure, similar to that found for the BV chromophore.
Phytochromes are a family of photoreceptors present in allflowering plants (Phy family) and cryptophytes, but also incyanobacteria (phytochromes Cph1 and Cph2), nonoxygenicbacteria (bacteriophytochromes or BphPs), and even fungi(fungal phytochromes or Fphs). Each photoreceptor has achromophore whose function depends on the nature of thephytochrome. In most plant phytochromes the chromophore isthe phytochromobilin (PΦB), while phycocyanobilin (PCB) isin general related to cyanobacteria and biliverdin (BV) tononoxygenic bacteria.1 All these chromophores are bilins andshow a covalently linked open-chain tetrapyrrole (see Figure1), which is structurally related with the macrocyclic tetrapyrrolestructures of the well-known porphyrins. As such, they show asimilar absorption spectrum, governed by the so-called Soretand Q bands.2 In the visible absorption spectra of porphyrinsthe intense absorption band located between 350 and 450 nmis known as the Soret band and the typically weak absorptionbands between 450 and 700 nm are called Q bands.3,4 Likewise,bilins show a Soret band in the visible absorption region butonly one Q-band. The chromophore of plant phytochromescontrols many photomorphogenic processes regulating themetabolic response of the organism to its light environment.Upon light irradiation, phytochromes can switch from theinactive Pr, red-absorbing (R) form with a Q-band peakingaround 666 nm, to the active Pfr, far-red (FR) absorbing formwith a peak around 730 nm.5 This interconversion is lightreversible and it is well known6 that the difference between Pr
and Pfr involves a Z-E photoisomerization at the double bondC15dC16, (see Figure 1).
While numerous spectra have been available for phyto-chromes for several decades, crystal structures are rather scarce.Only a few proteins have been resolved in the last several years,namely, the one corresponding to the chromophore-bindingdomain of the bacterial phytochrome of Deinococcus radiodu-rans (DrBphP) with a BV chromophore in the Pr state7,8 andthe chromophore-binding domain of an unusual bacterialphytochrome RpBphP3 from Rhodompseudomonas palustris,also with a BV chromophore.9 In the X-ray structure of DrBphP,the Pr form of BV adopts a semicyclic ZZZssa (C5-Z, C10-Z,C15-Z, C5-syn, C10-syn, C15-anti) conformation (see Figure 1)and is covalently attached to a cysteine residue near theN-terminal domain by a thioether bond.7 Unfortunately, thecrystal structure of the cyanobacterial phytochrome has not yetbeen obtained. Thus, very often theoretical calculations ormechanistic studies on phytochromes are done using the PCBchromophore of the C-phycocyanin (C-PC), which is a light-harvesting pigment present in photosynthetic cyanobacteria forwhich the crystal structure was resolved long ago.10 In contrastto the semicyclic structure of BV, PCB in C-PC shows anextended ZZZasa conformation.
Nowadays, there is no consensus about the paramountquestion of chromophore conformation in cyanobacterial andplant phytochromes.1,11,12 On one hand, a ZZZssa structure, asin the BV chromophore of DrBphP, is put forward for the Pr
forms of PCB and PΦB with arguments such as the highsequence identity between bacterial, cyanobacterial, and plantphytochromes.1,11 Furthermore, two-dimensional nuclear Over-hauser effect13 as well as 15N NMR spectroscopy14 experimentsare consistent with a ZZZssa conformation for cyanobacterialphytochrome Cph1. On the other hand, theoretical and experi-mental resonance Raman (RR) spectra suggest a ZZZasa
* To whom correspondence should be addressed. E-mail: [email protected].
16253
10.1021/jp807471e CCC: $40.75 2008 American Chemical Society
Published on Web 11/20/2008
2008, 112, 16253–16256

structure.12,15,16 In the present study, we shed light on thisconformational controversy through quantum chemical calcula-tions on PCB in Cph1. First, we investigate the UV-visabsorption spectra of the chromophores BV in BphP and PCBin C-PC for which three-dimensional structures are available.Then we use different models to probe the spectra of PCB inCph1, and we discuss the structure which would be consistentwith the available experimental data.
The calculations are done on the three structures shown inFigure 1. As template the crystal structure of the bacteriophy-tochrome chromophore binding domain at 1.45 Å resolution(PDB: 2O9C) is used for the BV and PCB chromophores inthe ZZZssa conformation,7 while the X-ray structure at 1.45 Åresolution of the R-84 subunit of C-PC from the thermophiliccyanobacterium Synechococcus elongatus (PDB: 1JBO) is usedfor PCB in the ZZZasa conformation.17 The hydrogen atomsare added according to the molecular arrangement of BV andPCB after the assembly with the apoprotein (see Figure 2 andlater in text). In all cases the cysteine linkage was replaced withhydrogen. The propionic-acid sidechains on the rings B and C(cf. Figure 1) are not included because they are not part of theconjugated system of the chromophore and as such they do notaffect the excitations energies (data not shown), which is inagreement with previous studies.18 For each system, four modelsof different complexity have been considered to account forprotonation, solvation, and protein surroundings. Model I is the
unprotonated chromphore in vacuo. Model II is the protonatedform also in vacuo. Model III and IV are protonated formssimulated in water and in a protein environment, respectively. Thespectra is calculated not only on X-ray structures, but also onchromophores previously optimized using density functional theoryDFT with the B3LYP/6-31G(d) protocol,19 as implemented in theGaussian03 set of programs.20 A calculation of the Hessian ensuredthat the obtained geometries are true minima, which can be foundin the Supporting Information. Vertical excited states and corre-sponding oscillator strengths are obtained using the time-depend-ent21 version of B3LYP/6-31G(d) over eight roots. The so-obtainedspectra are then convoluted with Gaussian functions with fullwidths of 4000 cm-1 at half-maximum using the GaussSum 2.1program.22 The environment is modeled with the polarizablecontinuum model (PCM).23 A dielectric constant of ε ) 78.4 isused for water, and ε ) 4.0 is used to represent the surroundingprotein moiety, as first suggested by Blomberg et al.24 and lateron by others.25-27
Experimentally, the absorption spectra of BV in BphP,8,28 PCBin Cph1,29 and PCB in C-PC10 show a UV Soret band centered inall cases at 380 nm and a Q-band around 700 nm. Since the exactposition of the Q-band depends on the specific chromophore andprotein, the accuracy describing this part of the spectrum will beused as a criterion to discern a semicyclic (ssa) from a extended(asa) conformation of the unknown PCB phytochrome in Cph1.Table 1 collects the experimental absorption Q peaks recorded forthe plant (oat) and cyanobacteria chromophores, as well as thecalculated values for the Models I-IV in both the X-ray and DFToptimized structures. Other theoretical values from the literatureare also compiled in Table 1.
First we compare the absorption peaks calculated on therelaxed and on the X-ray geometries with each other. We cansee that the Q bands are rather different, giving account of thesteric constraints imposed by the protein moiety. Specificobserved changes are that the optimized structures are moreclosed (or cyclic) than the X-ray ones and the rings A, B, andC show deviations from planarity. As a consequence, everyrelaxed structure, regardless of the conditions I-IV, exhibits amarkedly blue shift with respect to the crystal structure and the
Figure 1. Protonated biliverdin (BV) and phycocyanin (PCB) chromophores with conformations as indicated. Propionic side chains in rings B andC are not considered.
Figure 2. Possible conformations which can be considered for thering A of the chromophores BV (a and b) and PCB (c and d). In panelsa and c, the chromophores are assembled to the apoprotein, while inpanels b an d they are not.
TABLE 1: TD-B3LYP/6-31G(d) Q-bands (in nm) Contributing to the UV-Vis Absorption Spectra of the Chromophores BVand PCB Calculated on X-ray Structures and DFT Optimized Ones in Different Conformations in Different Environments;Experimental and Other DFT Values Are Given for Comparison
model I(unprotonated/in vacuo)
model II(protonated/in vacuo)
model III(protonated/water)
model IV(protonated/protein)
experimental
BV-ssa X-ray 648 662 708 712 702 (BV in BphP)a
DFT 588 643 659 665PCB-ssa X-ray 603 620 659 661 659 (PCB in Cph1)b
DFT 526 590 609 613DFT 574d
PCB-asa X-ray 528 590 606 614 618 (PCB in C-PC)c
DFT 508 559 584 588DFT 539e 541d, 582e
a References 8 and 28. b Reference 29. c Reference 10. d Reference 13. e Reference 25.
16254 J. Phys. Chem. B, Vol. 112, No. 51, 2008 Letters

experimental values. The difference between the DFT resultsfrom the literature13,25 and our values is related to the structureof the ring A (Figure 2) adopted in the calculations. This aspectcan be fiddly since the structure of the A ring depends, amongother reasons, on whether the chromophore is assembled or notto the apoprotein. Our calculations for BV follow the most recentX-ray structure resolved in 2007 which undoubtedly revealeda chiral center at the carbon C2 after ligation of the cysteineresidue to the C32 carbon8 (Figure 2a). Caution should beexercised in not considering the unassembled structure (Figure2b), since this contains one double bond more in the conjugatedπ system, which typically results in a red shift of 30 nm.8
Analogously to BV, for PCB we have used the structure shownin Figure 2c. In contrast, the calculations of Wan and co-workers25 adopt the chromophore before the apoprotein is linkedvia the cysteine residue at the C31 carbon (see Figure 2d); thisstructure implies an additional C3dC31 double bond, with theconcomitant red shift of ca. 30 nm (compare 508 and 559 nmwith 539 and 582 nm, respectively, in Table 1). The explanationfor the difference between the Q peaks obtained by van Thor13
et al. and ours is more subtle. They used the appropriateassembled chromophores for BV and PCB (Figure 2a,c,respectively), but a different density functional (MPW1PW91),leading to a blue shift of ca. 15-20 nm with respect to ourB3LYP values (see Table 1).
From all these considerations and our results, we concludethat calculated spectra based on relaxed geometries should betreated with caution, and henceforth we shall only discuss thechanges on the spectra calculated on the crystal models.
Since the crystal structure cannot evidence protonation, it isimportant to evaluate its effect on the absorption spectra. As itcan be seen, upon protonation the three chromophores suffer abathocromic shift; taking this into account, the neutral formshows a larger deviation from the experimental values. Thedifference between the neutral and the protonated forms is about15 nm for the conformation ZZZssa and larger for the ZZZasaconformation with a difference of more than 60 nm. Hence,our calculations support the fact that the chromophores areprotonated in agreement with refs 25 and 30-34.
Aqueous solution induces a variable solvatochromic red shiftin all chromophores. Conspicuously, a similar effect is obtainedin the protein environment. These values come very close tothe experimental ones with an accuracy seldom achieved withTD-DFT. The experimental values of 702 nm for BV-ssa and618 nm for PCB-asa in C-PC are quantitatively reproduced bythe theoretical 708/712 nm and 606/614 nm ones, respectively(see Table 1). We now turn to PCB in Cph1. Since the calculatedpeaks at 659 and 661 nm for water and protein environmentsare in excellent agreement with the experimental Q-bandmeasured at 659 nm and far away from 618 nm, which wouldcorrespond to a ZZZasa conformation, we are left to concludethat PCB in Cph1 must adopt a ZZZssa conformation. It is alsogratifying to realize that our calculations are also consistent witha hypsochromic shift which takes place when going to moreextended conformations (in this case from ssa to asa), as it hasbeen observed with semiempirical AM1 calculations on the PCBsystem.35
To uncover more details of the UV-vis spectra of the threechromophores, we show in Figure 3 the calculated absorption bandsin protein media, which we consider the most accurate ones. As itcan be seen the Soret band comprises several states with differentoscillator strengths. In contrast, the Q-band is described by a singlestate which upon inspection of the contributing orbitals can beattributed to an highest occupied molecular orbital (HOMO)flowest
unoccupied molecular orbital (LUMO) transition in the threechromophores. The strongest peak contributing to the Soret bandis a state with a superposition of HOMO-1fLUMO andHOMOfLUMO+1 excitations. The corresponding orbitals forPCB-ssa are shown in Figure 4; all of them correspond to π,π*orbitals delocalized between the four pyrrole rings.
In conclusion, the calculated spectra suggest that the Pr formof PCB very likely adopts a ZZZssa conformation in thecyanobacterial phytochrome. This conclusion fits into the pictureproposed by Lagarias et al. who analyzed a sequence alignmentof 122 known (or suspected) phytochromes and phytochrome-related proteins finding that (i) all phytochromes exhibitsequence conservation in two of the three domains of the
Figure 3. TD-DFT absorption UV-vis spectra of the protonated BV-ssa (a), PCB-ssa (b), and PCB-asa (c) chromophores modeled in aprotein environment (e ) 4).
Letters J. Phys. Chem. B, Vol. 112, No. 51, 2008 16255

photosensory region, (ii) key residues in the knot between thesetwo domains are also conserved, and (iii) differences in thesequences are not within the secondary structure.1,11 These factsindicate that the architecture of the photosensory core is verylikely to be conserved in all phytochromes, implying then thatthe chromophores should all have the same ZZZssa conforma-tion as found in bacteriophytochromes. This proposal is backedup by the recent 13C- and 15N NMR spectroscopic experimentsof refs 13 and 14 respectively, performed in the cyanobacterialphytochrome Cph1. Moreover, taking into account that in bothplants and cyanobacteria the chromophore links in the sameway to the apoprotein (by a cysteine at the 31 carbon, see Figure1), it is very plausible that the plant phytochrome PΦB alsopresents a ZZZssa conformation. Lagarias and co-workers havegone even further on proposing a photoconversion mechanismwhich involves a semicyclic ssa conformation for the Pfr formtoo, with no net charge transport over the full path.1
Contrary to the appealing idea of a unified ssa conformationfor all the phytochrome species, the RR data of Mroginski etal.15,11 suggest an asa structure for both the Pr and Pfr states, aswell as for the intermediate Lumi-R of the photocycle. However,one should note that these calculations have been done in vacuo,that is, without the protein environment, even when it has beendemonstrated that both the experimental36 and theoretical37 RRspectra are very sensitive to the protein environment. Indeed,the recent hybrid quantum mechanics/molecular mechanics(QM/MM) study on the PCB chromophore explicitly bound tothe R-subunit of C-PC clearly shows significant improvementswith respect to the pure QM calculations of the isolatedchromophore indicating that the comparison of experimentalRR spectra of the protein-bound chromophore with calculatedRR spectra of the isolated cofactor may not always beunambiguous.37
Summarizing, according to our results protonation and theconformational change from asa to ssa induce a bathochromicshift, whereas deprotonation and the isomerization from ssa toasa induce a hypsochromic shift. Most importantly, the excellentagreement between the experimental absorption spectra and theherein calculated one provides additional support that the Cph1phytochrome adopts a ZZZssa conformation as in the bacte-riophytochrome.
Acknowledgment. We thank early discussions with Dr. M.A.Mroginski (TU Berlin) and financial support from the DeutscheForschungsgemeinschaft (GO 1059/2-1) as well as CONICYT(Chile) and DAAD (Germany).
Supporting Information Available: This material is avail-able free of charge via the Internet at http://pubs.acs.org.
Note Added in Proof. At the time of processing this article,a ZZZssa confirmation was found in two cyanobacterialchromophores by NMR38 and diffraction39 experiments.
References and Notes
(1) Rockwell, N. C.; Su, Y. S.; Lagarias, J. C. Ann. ReV. Plant Biol.2006, 57, 837.
(2) Gouterman, M. The Porphyrins; Dolphin, D, Ed.; Academic Press:New York, 1978; Vol. III, p 1.
(3) Soret, J. L. C. R. Acad. Sci. 1983, 97, 1267.(4) Weiss, C. J. J. Mol. Spectrosc. 1972, 44, 37.(5) Chai, Y. G.; Singh, B. R.; Song, P. S.; Lee, J.; Robinson, G. W.
Anal. Biochem. 1987, 163, 322.(6) Braslavsky, S. E.; Gartner, W.; Schaffner, K. Plant Cell EnViron.
1997, 20, 700.(7) Wagner, J. R.; Brunzelle, J. S.; Forest, K. T.; Vierstra, R. D. Nature
2005, 438, 325.(8) Wagner, J. R.; Zhang, J.; Brunzelle, J. S.; Vierstra, R. D.; Forest,
K. T. J. Biol. Chem. 2007, 282, 12298.(9) Yang, X.; Stojkovic, E. A.; Kuk, J.; Moffat, K. Proc Natl. Acad.
Sci. U.S.A. 2007, 104, 12571.(10) Mimuro, M.; Fulglistaller, P.; Riimbeli, R.; Zuber, H. Biochim.
Biophys. Acta 1986, 848, 155.(11) Rockwell, N. C.; Lagarias, J. C. Plant Cell 2006, 18, 4.(12) Murgida, D. H.; von Stetten, D.; Hildebrandt, P.; Schwinte, P.;
Siebert, F.; Sharda, S.; Gartner, W.; Mroginski, M. A. Biophys. J. 2007,93, 2410.
(13) van Thor, J. J.; Mackeen, M.; Kuprov, I.; Dwek, R. A.; Wormald,M. R. Biophys. J. 2006, 91, 1811.
(14) Hahn, J.; Kuhne, R.; Schmieder, P. ChemBioChem 2007, 8, 2249.(15) Mroginski, M. A.; Murgida, D. H.; Hildebrandt, P. Acc. Chem. Res.
2007, 40, 258.(16) Mroginski, M. A.; Murgida, D. H.; von Stetten, D.; Kneip, C.; Mark,
F.; Hildebrandt, P. J. Am. Chem. Soc. 2004, 126, 16734.(17) Nield, J.; Rizkallah, P. J.; Barber, J.; Chayen, N. E. J. Struct. Biol.
2003, 141, 149.(18) Durbeej, B.; Borg, O. A.; Eriksson, L. A. Phys. Chem. Chem. Phys.
2004, 6, 5066.(19) (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Becke, A. D.
Phys. ReV. A 1988, 38, 3098. (c) Slater, J. C. Quantum Theory of Molecularand Solids: The Self-Consistent Field for Molecular and Solids; McGraw-Hill: New York, 1974, Vol 4. (d) Vosko, S. H.; Wilk, L.; Nusair, M. Can.J. Phys. 1980, 58, 1200. (e) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. B1988, 37, 785.
(20) Frisch, M. J. et al. Gaussian 03, rev. C.02.; Gaussian, Inc.:Wallingford, CT, 2004.
(21) Casida, M. E. In Recent AdVances in Density Functional Methods;Chong, D. P., Ed.; World Scientific: Singapore, 1995; Part 1, p 155.
(22) O’Boyle, N. M.; Tenderholt, A. L.; Langner, K. M. J. Comput.Chem. 2008, 29, 839.
(23) Tomasi, J.; Mennucci, B.; Cammi, R. Chem. ReV. 2005, 105, 2999.(24) Blomberg, M. R. A.; Siegbahn, P. E. M.; Babcock, G. T. J. Am.
Chem. Soc. 1998, 120, 8812.(25) Wan, J.; Xu, X.; Ren, Y.; Yang, G. J. Phys. Chem. B 2005, 109,
11088.(26) Ren, Y.; Wan, J.; Xu, X.; Zhang, Q.; Yang, G. J. Phys. Chem. B
2006, 110, 18665.(27) Borg, O. A.; Durbeej, B. J. Phys. Chem. B 2007, 111, 11554.(28) Tasler, R.; Moises, T.; Frankenberg-Dinkel, N. FEBS J. 2005, 272,
1927.(29) Lamparter, T.; Esteban, B.; Hughes, J. Eur. J. Biochem. 2001, 268,
4720.(30) Durbeej, B.; Borg, O. A.; Eriksson, L. A. Chem. Phys. Lett. 2005,
416, 83.(31) Andel, F., III.; Lagarias, J. C.; Mathies, R. A. Biochemistry 1996,
35, 15997.(32) Andel, F., III.; Murphy, J. T.; Haas, J. A.; McDowell, M. T.; Hoef,
I. v. d.; Lugtenburg, J.; Lagarias, J. C.; Mathies, R. A. Biochemistry 2000,39, 2667.
(33) Kneip, C.; Hildebrandt, P.; Schlamann, W.; Braslavsky, S. E.; Mark,F.; Schaffner, K. Biochemistry 1999, 15185, 38.
(34) Hasegawa, J.; Isshiki, M.; Fujimoto, K.; Nakatsuji, H. Chem. Phys.Lett. 2005, 410, 90.
(35) Goller, A. H.; Strehlow, D.; Hermann, G. ChemPhysChem 2005,6, 1259.
(36) von Stetten, D.; Seibeck, S.; Michael, N.; Scheerer, P.; Mroginski,M. A.; Murgida, D. H.; Krauss, N.; Heyn, M. P.; Hildebrandt, P.; Borucki,B.; Lamparter, T. J. Biol. Chem. 2007, 282, 2116.
(37) Mroginski, M. A.; Mark, F.; Thiel, W.; Hildebrandt, P. Biophys. J.2007, 93, 1885.
(38) Cornilescu, G.; Ulijasz, A. T.; Cornilescu, C. C.; Markley, J. L.;Vierstra, R. D. J. Mol. Biol. 2008, 383, 403.
(39) Essen, L.-O.; Mailliet, J.; Hughes, J. Proc. Natl. Acad. Sci. U.S.A.2008, 105, 14709.
JP807471E
Figure 4. Selected molecular orbitals of PCB-ssa involved in theabsorption spectra given in Figure 2b.
16256 J. Phys. Chem. B, Vol. 112, No. 51, 2008 Letters

Published on Web Date: February 02, 2010
r 2010 American Chemical Society 796 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
Time-Dependent DFT on Phytochrome Chromophores:AWay to the Right ConformerRicardo A. Matute,*,† Renato Contreras,† and Leticia Gonz�alez*,‡
†Departamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile, and ‡Institut f€urPhysikalische Chemie, Friedrich-Schiller-Universit€at Jena, Helmholtzweg 4, 07743 Jena, Germany
ABSTRACT A theoretical approach based on time-dependent density functionaltheory (TDDFT) togetherwith a polarizable continuummodel for incorporating thebulk effect of the surrounding environment is used to estimate the excitationenergies for the phytochromobilin chromophore of plant phytochrome. TheTDDFT results reproduce well the experimental values of the absorption maximaand those of the ratio of the spectroscopic bands (Qbandover the Soret band orQ/Sindex). Our results suggest that the phytochromobilin within the Pr form of thephytochrome holoprotein should adopt a semicyclic ZZZssa structure as it does inbacteria.
SECTION Biophysical Chemistry
P hytochromes are photoreceptors in plants and are alsofound in nonoxygenic bacteria, cyanobacteria, andfungi. In plants, phytochromes regulate photomorpho-
genic responses as germination, de-elotation, shade avoid-ance, phototropism, chloroplast movement, stomatal open-ing, and flowering. The photocycle of the receptor involves aZ/E photoisomerization around a double bond of its phyto-chromobilin (PΦB) chromophore (Figure 1), directing thephotoconversion between a physiological inactive red formcalled Pr to a physiological active far-red form called Pfr.
1
PhytochromeAabsorbs in theUV-visible range, showing twoprominent bands, the Soret band in the blue region and the Qband in the red region.2,3 The Soret band normally absorbsnear the 380 nm, but the Q band is variant and specific foreach chromophore, for example, in the Pr of the plant phyto-chrome, this band has its maximum at around 666 nm.3,4
According to the pioneering work ofWagner et al. showingthe crystallographic structure in proteobacteria,5 the Pr formof the biliverdin (BV) chromophore adopts a ZZZssa confor-mation (C5-Z, C10-Z, C15-Z, C5-syn, C10-syn, C15-anti). In thecase of the cyanobacterial phytochrome, theoretical calcula-tions based on time-dependent density functional theory6
(TDDFT) predicted satisfactorily a ZZZssa structure for itsphycocyanobilin (PCB) chromophore conformation,7 whichwas then confirmed by NMR solution experiments8 andcrystallographic9 structures. However, no X-ray or NMR struc-tural information is available for plant phytochrome, and thus,there is no consensus about which structure is adopted by itsPΦB chromophore in the holoprotein. On the one hand, asemicyclic ZZZssa structure, as in proteobacteria, is supportedby sequence identity;1a,10 on the other hand, a stretchedZZZasa conformation is proposedbasedon resonanceRaman(RR) evidence11-14 (Figure 1), and it has been adopted in themost recent computational studies of the phytochrome sys-tem.15,16 In this Letter, we elucidate which conformer should
be found in plant phytochromes by means of calculating theelectronic absorption spectra of both possible conformations,as well as that of the cyclic solution structure. The apoproteinismostly transparent, not absorbing in the UV-vis, except forsome aromatic residues such as tyrosine and tryptophan,which together generate the so-called protein band at 280nm, that is, at around 100 nm to the UV side with respect theSoret band of the chromophore. Therefore, an efficient pro-cedure consists of treating the chromophore alone with a fullquantumdescription (TDDFT in our case)while the surround-ing environment can be modeled in a much simpler way, forinstance, with continuum models.7 Such an approach is, forexample, suggested in a recent review on the theoreticaldescription of biomolecular spectroscopy by Neugebauer,17
who points out the necessity to keep fixed parts of thechromophore at the crystal structure geometry, that is, con-sidering implicitly the geometry imposed by the surroundingapoprotein. This treatment is a good shortcut to the moredemanding one of sampling the ensemble of structuresbelonging to the conformational space;18 moreover, it ispreferred over the typical relaxation of the chromophore invacuo since the lattermethod converges to a geometry, whichbeing out of the conformations accessed by the system,delivers overestimated excitation energies.7
As a template for PΦB in the semicyclic ZZZssa model, weuse either the X-ray structure of the BV chromophore found inthe Pr form of the bacteriophytochrome ofDeinococcus radio-durans (PDB: 2O9C)19 or the Pr formof the PCB chromophorefound in the cyanobacterial Cph1 phytochrome from thecyanobacterium Synechocystis 6803 (PDB: 2VEA).9 For thestretched ZZZasa model, we use the PCB found in the R-84
Received Date: December 19, 2009Accepted Date: January 28, 2010
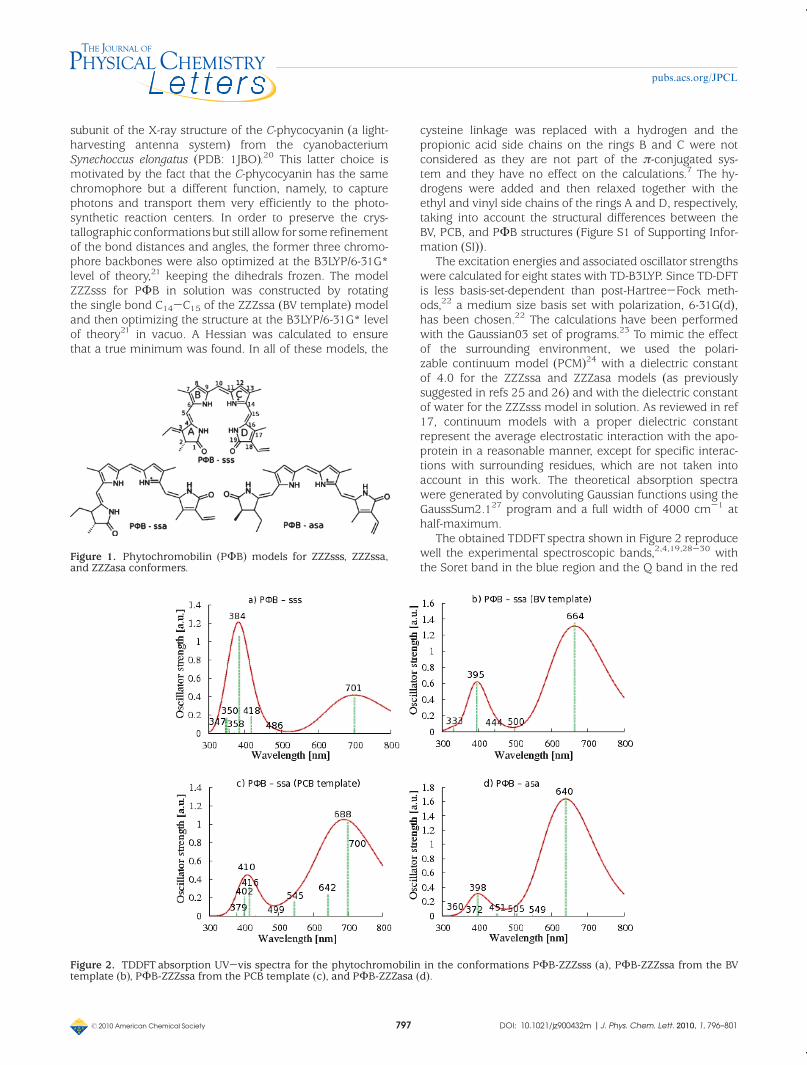
r 2010 American Chemical Society 797 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
subunit of the X-ray structure of the C-phycocyanin (a light-harvesting antenna system) from the cyanobacteriumSynechoccus elongatus (PDB: 1JBO).20 This latter choice ismotivated by the fact that the C-phycocyanin has the samechromophore but a different function, namely, to capturephotons and transport them very efficiently to the photo-synthetic reaction centers. In order to preserve the crys-tallographic conformations but still allow for some refinementof the bond distances and angles, the former three chromo-phore backbones were also optimized at the B3LYP/6-31G*level of theory,21 keeping the dihedrals frozen. The modelZZZsss for PΦB in solution was constructed by rotatingthe single bond C14-C15 of the ZZZssa (BV template) modeland then optimizing the structure at the B3LYP/6-31G* levelof theory21 in vacuo. A Hessian was calculated to ensurethat a true minimum was found. In all of these models, the
cysteine linkage was replaced with a hydrogen and thepropionic acid side chains on the rings B and C were notconsidered as they are not part of the π-conjugated sys-tem and they have no effect on the calculations.7 The hy-drogens were added and then relaxed together with theethyl and vinyl side chains of the rings A and D, respectively,taking into account the structural differences between theBV, PCB, and PΦB structures (Figure S1 of Supporting Infor-mation (SI)).
The excitation energies and associated oscillator strengthswere calculated for eight states with TD-B3LYP. Since TD-DFTis less basis-set-dependent than post-Hartree-Fock meth-ods,22 a medium size basis set with polarization, 6-31G(d),has been chosen.22 The calculations have been performedwith the Gaussian03 set of programs.23 To mimic the effectof the surrounding environment, we used the polari-zable continuum model (PCM)24 with a dielectric constantof 4.0 for the ZZZssa and ZZZasa models (as previouslysuggested in refs 25 and 26) and with the dielectric constantof water for the ZZZsss model in solution. As reviewed in ref17, continuum models with a proper dielectric constantrepresent the average electrostatic interaction with the apo-protein in a reasonable manner, except for specific interac-tions with surrounding residues, which are not taken intoaccount in this work. The theoretical absorption spectrawere generated by convoluting Gaussian functions using theGaussSum2.127 program and a full width of 4000 cm-1 athalf-maximum.
The obtained TDDFT spectra shown in Figure 2 reproducewell the experimental spectroscopic bands,2,4,19,28-30 withthe Soret band in the blue region and the Q band in the red
Figure 1. Phytochromobilin (PΦB) models for ZZZsss, ZZZssa,and ZZZasa conformers.
Figure 2. TDDFT absorption UV-vis spectra for the phytochromobilin in the conformations PΦB-ZZZsss (a), PΦB-ZZZssa from the BVtemplate (b), PΦB-ZZZssa from the PCB template (c), and PΦB-ZZZasa (d).

r 2010 American Chemical Society 798 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
region (Table 1). In all cases, the Q band is mainly composedof a HOMO to LUMO transition, whereas in the Soret band,there are fourmain orbitals involved in the transitions, that is,the HOMO-1, HOMO, LUMO, and LUMOþ1 (see Figure 3and Table S1 of SI). These same four orbitalswere found in theSoret band of the cyanobacterial phytochrome,7 in analogywith the four-orbitalmodel ofGouterman for the Soret band inporphyrins.31
Two spectra are obtained for the semicyclic ZZZssa con-former, depending on the underlying template, either BV(Figure 2b) or PCB (Figure 2c). In the spectrum based onBV, theQ band absorbing at 664 nm is in excellent agreementwith the experimental Q band absorption value of 666 nm. Inthe spectrum based on PCB, the Q band absorbs at 688 nm,that is, it shows a bathochromic shift of 24 nm, which canbe explained by looking at the template structures. The ringD of BV is tilted by 44� over the plane formed by the otherthree pyrrol rings19 (Figure 4A), while in the PCB, it is tiltedonly 26� (Figure 4B).9 The latter arrangement achievesbetter conjugation within the π system, thus lowering theexcitation energy and inducing the concomitant bathochro-mic shift in the spectrum. It has been suggested that the
difference in the torsional angle of the ring D of both crystalscan be explained by the lack of one domain (PHY domain)in the X-ray structure of the phytochrome with BV, whichcould destabilize the ring D in BV.32 Hypothetically, bothpositions of the ringD could be accessed at room temperatureby torsional movement in solution. However, if we assumea different torsion of the ring D between proteobacteriaand cyanobacteria, in view of the better approximation of664 versus 688 nm compared to the experimental value of666 nm, our results would suggest that PΦB adopts a con-formation similar to that of BV in proteobacteria and not likePCB in cyanobacteria. This conclusion is not so clear if oneanalyzes the excitations obtained using partially optimizedgeometries. As seen from the values in parentheses in Table 1,the excitation energies of the constraint geometries for PΦB-ssa (BV template) and PΦB-asa are blue-shifted with respectto those of the unrefined crystal structures. With values of633 and 619 nm, it is not straightforward to assign the Q bandof PΦB to the ssa or asa conformer. Moreover, taking intoaccount that the Q band obtained for the crystal of the ZZZasaconformer (Figure 2d) is blue-shifted only by 24 nm withrespect to that of the ZZZssa structure, a stretched structurecannot be unequivocally discarded.
To get additional information, we shall use the ratio of theoscillator strengths of theQbandover the Soret one, or the so-called Q/S index, to discriminate between both conformers.Q/S indexes have been invoked in the literature for a longtime, first, using semiempirical methods, as in the work ofChae and Song33 andWagni�ere et al.34,35 (see also the reviewby Scheer36), and later experimentally in vitro using synthe-sized adducts of tetrapyrrole locked in different conforma-tions.37,38 Afterward,Q/S indexeswere revisited throughAM1semiempirical studies.39,40 In our calculation of theQ/S ratios,we have also included the cyclic conformer (Figure 2a) be-sides the semicyclic and stretched ones.
Table 1 collects the calculated and experimental2,4,19,28-30
absorptionmaxima togetherwith the experimental33,35,40-42
and TDDFTQ/S ratios. Aswe can see, the calculatedQ/S ratiosreproduce qualitatively the experimental ones, keeping the
Table 1. Calculated TDDFTAbsorption Maxima (in nm) and Q/S Ratiosa
absorption maxima Q/S
model exptl.b TDDFT exptl.c TDDFTd
BV-sss 377, 696,710e 383, 712 0.29f 0.26
PCB-sss 375, 692 368, 675 0.33, 0.43 0.37
PΦB-sss 386, 700 384, 701 0.34
BV-ssa 380, 702 417, 712 2.69g 1.99
PCB-ssa 380, 659 394, 661 1.17 2.28
PΦB-ssa 380, 666 395h (382)h, 664h (633)h, 410i (382)i, 688i (611)i 1.36, 1.45 2.12h (2.02)h, 2.36i (1.90)i
PCB-asa 380, 618 385, 614 4.1 5.26
PΦB-asa 398 (383), 640 (619) 5.27 (4.26)aValues in parentheses correspond to structures refined with a constraint optimization (frozen dihedrals). b Taken from refs 2, 4, 19, and 28-30.
cTaken from refs 33, 35, and 40-42. dDielectric constant ε=4.0 for ssa and asa conformations and ε=78.4 for sss conformation. eQbandmaximumfor the protonated Biverdin dimethyl ester (BVEHþ) in solution corresponding to the helically coiled conformation according to Braslavsky et al.28 fThisvalue is obtained as an average of the oscillator strengths of the two peaks contributing to the Q and S bands, respectively.33 gThis value is likely to beoverestimated since it is obtained42 as the ratio between the heights of the Q and S bands and not the areas below the bands, that is, the oscillatorstrength. hThese values have been obtained using the BV template. i These values have been obtained using the PCB template.
Figure 3. HOMO-1, HOMO, LUMO, and LUMOþ1 orbitals in-volved in the TDDFTabsorption spectrum given in Figure 2b.
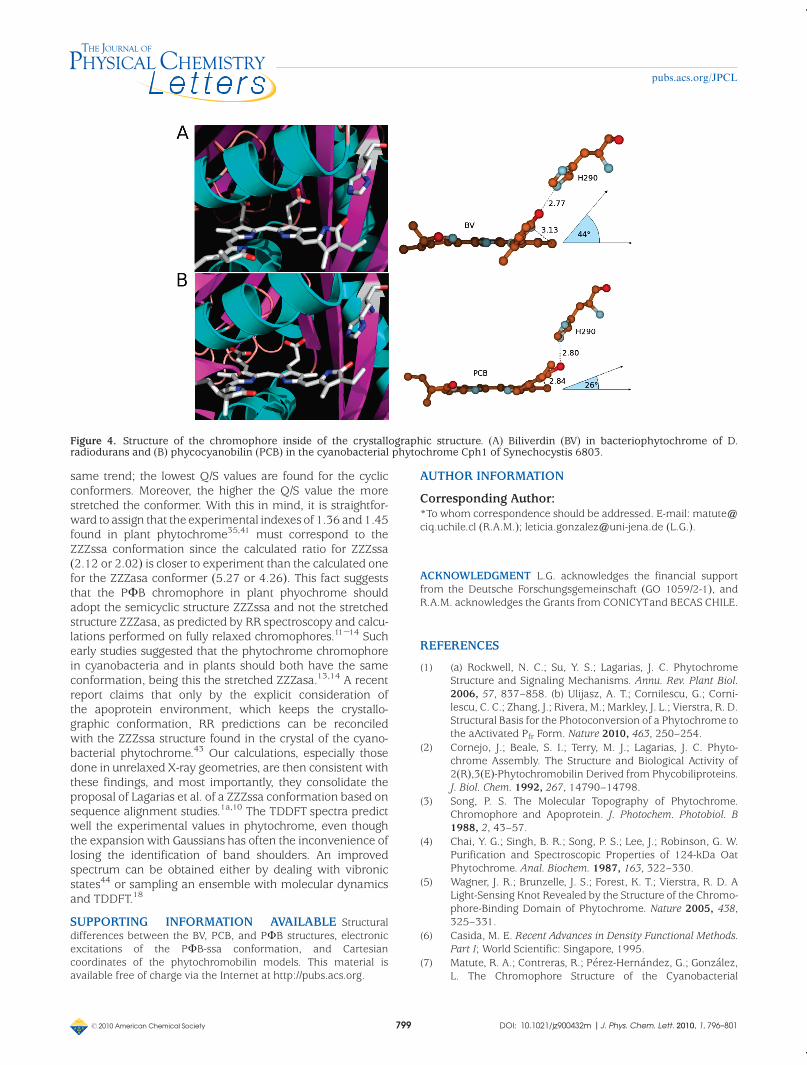
r 2010 American Chemical Society 799 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
same trend; the lowest Q/S values are found for the cyclicconformers. Moreover, the higher the Q/S value the morestretched the conformer. With this in mind, it is straightfor-ward to assign that the experimental indexes of 1.36 and 1.45found in plant phytochrome35,41 must correspond to theZZZssa conformation since the calculated ratio for ZZZssa(2.12 or 2.02) is closer to experiment than the calculated onefor the ZZZasa conformer (5.27 or 4.26). This fact suggeststhat the PΦB chromophore in plant phyochrome shouldadopt the semicyclic structure ZZZssa and not the stretchedstructure ZZZasa, as predicted by RR spectroscopy and calcu-lations performed on fully relaxed chromophores.11-14 Suchearly studies suggested that the phytochrome chromophorein cyanobacteria and in plants should both have the sameconformation, being this the stretched ZZZasa.13,14 A recentreport claims that only by the explicit consideration ofthe apoprotein environment, which keeps the crystallo-graphic conformation, RR predictions can be reconciledwith the ZZZssa structure found in the crystal of the cyano-bacterial phytochrome.43 Our calculations, especially thosedone in unrelaxed X-ray geometries, are then consistent withthese findings, and most importantly, they consolidate theproposal of Lagarias et al. of a ZZZssa conformation based onsequence alignment studies.1a,10 The TDDFT spectra predictwell the experimental values in phytochrome, even thoughthe expansion with Gaussians has often the inconvenience oflosing the identification of band shoulders. An improvedspectrum can be obtained either by dealing with vibronicstates44 or sampling an ensemble with molecular dynamicsand TDDFT.18
SUPPORTING INFORMATION AVAILABLE Structuraldifferences between the BV, PCB, and PΦB structures, electronicexcitations of the PΦB-ssa conformation, and Cartesiancoordinates of the phytochromobilin models. This material isavailable free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author:*To whom correspondence should be addressed. E-mail: [email protected] (R.A.M.); [email protected] (L.G.).
ACKNOWLEDGMENT L.G. acknowledges the financial supportfrom the Deutsche Forschungsgemeinschaft (GO 1059/2-1), andR.A.M. acknowledges the Grants from CONICYTand BECAS CHILE.
REFERENCES
(1) (a) Rockwell, N. C.; Su, Y. S.; Lagarias, J. C. PhytochromeStructure and Signaling Mechanisms. Annu. Rev. Plant Biol.2006, 57, 837–858. (b) Ulijasz, A. T.; Cornilescu, G.; Corni-lescu, C. C.; Zhang, J.; Rivera, M.; Markley, J. L.; Vierstra, R. D.Structural Basis for the Photoconversion of a Phytochrome tothe aActivated Pfr Form. Nature 2010, 463, 250–254.
(2) Cornejo, J.; Beale, S. I.; Terry, M. J.; Lagarias, J. C. Phyto-chrome Assembly. The Structure and Biological Activity of2(R),3(E)-Phytochromobilin Derived from Phycobiliproteins.J. Biol. Chem. 1992, 267, 14790–14798.
(3) Song, P. S. The Molecular Topography of Phytochrome.Chromophore and Apoprotein. J. Photochem. Photobiol. B1988, 2, 43–57.
(4) Chai, Y. G.; Singh, B. R.; Song, P. S.; Lee, J.; Robinson, G. W.Purification and Spectroscopic Properties of 124-kDa OatPhytochrome. Anal. Biochem. 1987, 163, 322–330.
(5) Wagner, J. R.; Brunzelle, J. S.; Forest, K. T.; Vierstra, R. D. ALight-Sensing Knot Revealed by the Structure of the Chromo-phore-Binding Domain of Phytochrome. Nature 2005, 438,325–331.
(6) Casida, M. E. Recent Advances in Density Functional Methods.Part I; World Scientific: Singapore, 1995.
(7) Matute, R. A.; Contreras, R.; P�erez-Hern�andez, G.; Gonz�alez,L. The Chromophore Structure of the Cyanobacterial
Figure 4. Structure of the chromophore inside of the crystallographic structure. (A) Biliverdin (BV) in bacteriophytochrome of D.radiodurans and (B) phycocyanobilin (PCB) in the cyanobacterial phytochrome Cph1 of Synechocystis 6803.

r 2010 American Chemical Society 800 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
Phytochrome Cph1 As Predicted by Time-Dependent Den-sity Functional Theory. J. Phys. Chem. B 2008, 112, 16253–16256.
(8) Cornilescu, G.; Ulijasz, A. T.; Cornilescu, C. C.; Markley, J. L.;Vierstra, R. D. Solution Structure of a Cyanobacterial Phyto-chrome GAF Domain in the Red-Light-Absorbing GroundState. J. Mol. Biol. 2008, 383, 403–413.
(9) Essen, L.-O.; Mailliet, J.; Hughes, J. The Structure of a Com-plete Phytochrome Sensory Module in the Pr Ground State.Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 14709–14714.
(10) Rockwell, N. C.; Lagarias, J. C. The Structure of Phytochrome:A Picture is Worth a Thousand Spectra. Plant Cell 2006, 18,4–14.
(11) Kneip, C.; Hildebrandt, P.; Schlamann, W.; Braslavsky, S. E.;Mark, F.; Schaffner, K. Protonation State and StructuralChanges of the Tetrapyrrole Chromophore During the Pr fPfr Phototransformation of Phytochrome: A Resonance Ra-man Spectroscopic Study. Biochemistry 1999, 38, 15185–15192.
(12) Mroginski, M. A.; Murgida, D. H.; von Stetten, D.; Kneip, C.;Mark, F.; Hildebrandt, P. Determination of the ChromophoreStructures in the Photoinduced Reaction Cycle of Phyto-chrome. J. Am. Chem. Soc. 2004, 126, 16734–16735.
(13) Mroginski, M. A.; Murgida, D. H.; Hildebrandt, P. The Chro-mophore Structural Changes During the Photocycle of Phy-tochrome: A Combined Resonance Raman and QuantumChemical Approach. Acc. Chem. Res. 2007, 40, 258–266.
(14) Murgida, D. H.; von Stetten, D.; Hildebrandt, P.; Schwinte, P.;Siebert, F.; Sharda, S.; Gartner, W.; Mroginski, M. A. TheChromophore Structures of the Pr States in Plant and Bacter-ial Phytochromes. Biophys. J. 2007, 93, 2410–2417.
(15) Durbeej, B. On the Primary Event of Phytochrome: QuantumChemical Comparison of Photoreactions at C4, C10 and C15.Phys. Chem. Chem. Phys. 2009, 11, 1354–1361.
(16) Alto�e, P.; Climent, T.; De Fusco, G. C.; Stenta, M.; Bottoni, A.;Serrano-Andr�es, L.; Merch�an, M.; Orlandi, G.; Garavelli, M.Deciphering Intrinsic Deactivation/Isomerization Routes in aPhytochrome Chromophore Model. J. Phys. Chem. B 2009,113, 15067–15073.
(17) Neugebauer, J. Subsystem-Based Theoretical Spectroscopy ofBiomolecules and Biomolecular Assemblies. ChemPhysChem2009, 10, 3148–3173.
(18) Schaffer, L. V.; Groenhof, G.; Klingen, A. R.; Ullman, G. M.;Boggio-Pasqua, M.; Robb, M. A.; Grubmuller, H. Photoswitch-ing of the Fluorescent Protein asFP595: Mechanism, ProtonPathways, and Absorption Spectra. Angew. Chem., Int. Ed.2007, 46, 530–536.
(19) Wagner, J. R.; Zhang, J.; Brunzelle, J. S.; Vierstra, R. D.; Forest,K. T. High Resolution Structure of Deinococcus Bacteriophy-tochromeYields New Insights into PhytochromeArchitectureand Evolution. J. Biol. Chem. 2007, 282, 12298–12309.
(20) Nield, J.; Rizkallah, P. J.; Barber, J.; Chayen, N. E. The 1.45AThree-Dimensional Structure of C-Phycocyanin from theThermophilic Cyanobacterium Synechococcus elongatus.J. Struct. Biol. 2003, 141, 149–155.
(21) (a) Becke, A. D. Density-Functional Exchange-Energy Ap-proximation with Correct Asymptotic Behavior. Phys. Rev. A1998, 38, 3098–3100. (b) Lee, C.; Yang, W.; Parr, R. G.Development of the Colle-Salvetti Correlation-Energy For-mula into a Functional of the Electron Density. Phys. Rev. B1988, 37, 785–789.
(22) Jacquemin, D.; Perp�ete, E. A.; Ciofini, I.; Adamo, C. AccurateSimulation of Optical Properties in Dyes. Acc. Chem. Res.2009, 42, 326–334.
(23) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Montgomery, J., J. A.; Vreven,T.; Kudin, K. N.; Burant, J. C.; et al.Gaussian 03, revision C.02.;Gaussian, Inc.: Wallingford, CT, 2004.
(24) Tomasi, J.; Menucci, B.; Cammi, R. Quantum MechanicalContinuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094.
(25) Blomberg, M. R. A.; Siegbahn, P. E. M.; Babcock, G. T.Modeling Electron Transfer in Biochemistry: A QuantumChemical Study of Charge Separation in Rhodobacter sphaer-oides and Photosystem II. J. Am. Chem. Soc. 1998, 120, 8812–8824.
(26) Borg, O. A.; Durbeej, B. Relative Ground and Excited-StatepKa Values of Phytochromobilin in the Photoactivation ofPhytochrome: AComputational Study. J. Phys. Chem. B 2007,111, 11554–11565.
(27) O'Boyle, N. M.; Tenderholt, A. L.; Langner, K. M. cclib: ALibrary for Package-Independent Computational ChemistryAlgorithms. J. Comput. Chem. 2008, 29, 839–845.
(28) Braslavsky, S. E.; Holzwarth, A. R.; Schaffner, K. SolutionConformations, Photophysics, and Photochemistry of BilePigments; Bilirubin and Biliverdin, Dimethyl Esters andRelated Linear Tetrapyrroles. Angew. Chem., Int. Ed. Engl.1983, 22, 656–674.
(29) Lamparter, T; Esteban, B.; Hughes, J. Phytochrome Cph1from the Cyanobacterium Synechocystis PCC6803. Purifica-tion, Assembly, and Quaternary Structure. Eur. J. Biochem.2001, 268, 4720–4730.
(30) Mimuro,M.; Fulglistaller, P.; Rumbeli, R.; Zuber, H. FunctionalAssignment of Chromophores and Energy Transfer in C-Phycocyanin Isolated from the Thermophilic Cyanobacter-ium Mastigocladus laminosus. Biochim. Biophys. Acta 1986,848, 155–166.
(31) Gouterman, M.; Wagni�ere, G. H.; Snyder, L. C. Spectra ofPorphyrins: Part II. Four Orbital Model. J. Mol. Spectrosc.1963, 11, 108–127.
(32) Fischer, A. J.; Lagarias, J. C. Harnessing Phytochrome'sGlowing Potential. Proc. Natl. Acad. Sci. U.S.A. 2004, 101,17334–17339.
(33) Chae, Q.; Song, P. S. Linear Dichroic Spectra and Fluores-cence Polarization of Biliverdin. J. Am. Chem. Soc. 1975, 97,4176–4179.
(34) Wagni�ere, G.; Blauer, G. Calculations of Optical Properties ofBiliverdin in Various Conformations. J. Am. Chem. Soc. 1976,98, 7806–7810.
(35) Pasternak, W.; Wagni�ere, G. Possible Interpretation of Long-Wavelength Spectral Shifts in Phytochrome Pr and Pfr Forms.J. Am. Chem. Soc. 1979, 101, 1662–1667.
(36) Scheer, H. Biliproteins. Angew. Chem., Int. Ed. Engl. 1981, 20,241–261.
(37) Nesvadba, P.; Gossauer, A. Synthesis of Bile Pigments. 14.Synthesis of a Bilindionostilbenoparacyclophane as a Modelfor Stretched Bile Pigment Chromophores of Biliproteins.J. Am. Chem. Soc. 1987, 109, 6545–6546.
(38) Iturraspe, J. B.; Bari, S.; Frydman, B. Total Synthesis of“Extended” Biliverdins: The Relation between their Confor-mation and their Spectroscopic Properties. J. Am. Chem. Soc.1989, 111, 1525–1527.
(39) Guimaraes, C. R. W.; Neto, J. D. D.; De Alencastro, R. B.Phytochrome Structure: A New Methodological Approach.Int. J. Quantum Chem. 1998, 70, 1145–1157.
(40) Goller, A. H.; Strehlow, D.; Hermann, G. The Excited-StateChemistry of Phycocyanobilin: A Semiempirical Study. Chem-PhysChem 2005, 6, 1259–1268.

r 2010 American Chemical Society 801 DOI: 10.1021/jz900432m |J. Phys. Chem. Lett. 2010, 1, 796–801
pubs.acs.org/JPCL
(41) Parker, W.; Goebel, P.; Ross, C. R.; Song, P. S.; Stezowski, J. J.Molecular Modeling of Phytochrome Using Constitutive C-Phycocyanin from Fremyella diplosiphon as a Putative Struc-tural Template. Bioconjugate Chem. 1994, 5, 21–30.
(42) Wagner, J. R.; Zhang, J.; von Stetten, D.; Gunther,M.;Murgida,D. H.; Mroginski, M. A.; Walker, J. M.; Forest, K. T.; Hildeb-randt, P. Mutational Analysis of Deinococcus radioduransBacteriophytochrome Reveals Key Amino Acids Necessaryfor the Photochromicity and Proton Exchange Cycle ofPhytochrome. J. Biol. Chem. 2008, 283, 12212–12226.
(43) Mroginski,M. A.; von Stetten, D.; Escobar, F. V.; Strauss, H.M.;Kaminski, S.; Scheerer, P.; Gunther, M.; Murgida, D. H.;Schmieder, P.; Bongards, C.; Gartner, W.; Mailliet, J.; Hughes,J.; Essen, L. O.; Hildebrandt, P. Chromophore Structure ofCyanobacterial Phytochrome Cph1 in the Pr State: Reconcil-ing Structural and Spectroscopic Data by QM/MM Calcula-tions. Biophys. J. 2009, 96, 4153–4163.
(44) Spillane, K. M.; Dasgupta, J.; Lagarias, J. C.; Mathies, R. A.Homogeneity of Phytochrome Cph1 Vibronic AbsorptionRevealed by Resonance Raman Intensity Analysis. J. Am.Chem. Soc. 2009, 131, 13946–13948.

A Lactam-Lactim Tautomerism Involved in the Pr/Pfr
Interconversion of Phytochrome
Journal: The Journal of Physical Chemistry Letters
Manuscript ID: jz-2010-013467
Manuscript Type: Letter
Date Submitted by the Author:
28-Sep-2010
Complete List of Authors: Matute, Ricardo; Universidad de Chile, Facultad de Ciencias, Departamento de Quimica Soto-Delgado, Jorge; Universidad Técnica Federico Santa María, Departamento de Química González, Leticia; Friedrich-Schiller-Universität Jena, Institut für Physikalische Chemie Contreras, Renato; Universidad de Chile, Departamento de Química
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters

A Lactam-Lactim Tautomerism Involved in
the Pr/Pfr Interconversion of Phytochrome
Ricardo A. Matute,*,† Jorge Soto-Delgado,‡ Leticia González,§ and Renato Contreras†
† Departamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile;
‡ Departamento de Química, Universidad Técnica Federico Santa María, Casilla 110-V, Valparaíso,
Chile; and § Institut für Physikalische Chemie, Friedrich-Schiller-Universität Jena, Helmholtzweg 4,
07743 Jena, Germany
E-mail: [email protected]
Title Running Head: Tautomerism in the Phytochrome Interconversion
1
Page 1 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

Abstract
Phytochromes show a strong bathochromic shift in the forward phtotoconversion process from Pr to Pfr,
which could be explained by a lactam-lactim tautomerism occurring after the initial photoisomerization.
Theoretical calculations using time-dependent density functional theory on molecular models for the
biliverdin chromophore of bacteriophytochrome allow to assign such bathochromic shift to a lactam-
lactim tautomerism on ring A but not on ring D of their open tetrapyrrole structure. Moreover, the
predicted Q-band absorption peak for the ZZEssa structure of Pfr with lactim tautomer in ring A is in
excellent agreement with the experimental value. In addition, a mechanistic analysis delivers the
transition states for the intramolecular non-dissociative mechanism of lactam-lactim tautomerism on
ring A and on ring D, being energetically favoured the tautomerization on ring A, but with an energy
barrier still high enough to be overpass. In contrast, the transition state for the lactam-lactim
tautomerism on ring A through a mechanism catalyzed by one water molecule is energetically viable.
Keywords: Phytochrome, Biliverdin, Phycocyanobilin, Tautomerism, TDDFT.
2
Page 2 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

In plants, there are different photoreceptors regulating its development and metabolism, and
between them the phytochrome has acquired particular interest since it is involved in
photomorphogenic responses triggered by specific-wavelength incident light, for instance, the shade
avoidance as a sensing mechanism for the growth process directed by the presence of neighbor plants
competing for the sunlight.1 Another important photomorphogenic responses regulated by phytochrome
include germination, phototropism, and flowering.2 Thus, this photoreceptor is a suitable target for
biotechnology; in fact, there are several studies in that direction, among them the work by the group of
Quail about a light-switchable phtyochrome-based promotor system3 and the recent published work
concerning phytochrome-mediated protein translocation to the nucleus.4 Despite these advances on
phytochrome biotechnology, the photoconversion mechanism of phytochrome is still unclear.
The phytochrome photocycle (Figure S1 of Supporting Information (SI)) is a switching process
between the two photoconvertible forms, namely Pr and Pfr, which absorb the red and far-region region
of the UV-Visible spectrum, respectively.2 Since the resolution of the first crystallographic structure for
the Pr form of phytochrome,5 some other X-ray structures have been reported6-8, including a Pfr
structure for a bacterial phytochrome,8 which together have shed light on the photoreceptor structure,
motivating new proposals for the underlying photoconversion mechanism. In this work, we assess from
a theoretical perspective the possible participation of a lactam-lactim tautomerism in such mechanism.
The lactim-lactam tautomerism is the cyclic equivalent to the amide-imidic acid tautomerism
(Figure 1A), where the lactam tautomer has the keto form next to a protonated nitrogen, whereas the
lactim tautomer has the enol form next to an unprotonated nitrogen.9 A quick revision of the structure
for the Pr form of biliverdin (Figure 1B) is enough for determining the two locations where the
tautomerism could be achieved, i.e., either in the pyrrol ring A or in the pyrrol ring D. Assuming that a
tautomerism is occurring thermally after the primary photoinduced process, we could consider the
plausible combination of the tautomers for both rings in the Pfr product. Thus, the Figure 1C shows the
models selected as the hypothetical Pfr form for the biliverdin in bacteriophytochrome: di-lactam model,
lactim in ring A model, lactim in ring B model, and di-lactim model; which henceforth in the text will
3
Page 3 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

be referred as DITAM, TIM-A, TIM-B, and DITIM, respectively. We worked on these models to build
up their theoretical electronic absorption spectra predicted by the time-dependent density functional
theory (TDDFT),10 looking for the structural basis that could explain the bathochromic shift suffered by
the phytochrome upon the interconversion from the Pr to the Pfr form, clearly focusing on the carbonyl
group.
Another issue that we consider pertinent to address is related to the studies conducted by the
group of Matysik,11,12 in which they used 13C-NMR spectroscopy to resolve the situation of the carbons
in the Phycocyanobilin (PCB) of cyanobacterial phytochrome in its Pfr form. In one of such studies,
they found that both carbonyls of the chromophore (ring A / ring D) should be in the keto form, 11 thus
discarding the possibility of tautomerism occurrence, what was also pointed out in the recent review by
Gärtner.13 With this in mind, we performed NMR shielding calculations on PCB to compare the
chemical shifts with the ones reported by the mentioned reference.11
Figure 1. Selected molecular structures. A. Representation of a Lactam-Lactim tautomerism for a simple model B. Biliverdin in the Pr form of bacteriophytochrome. C. Biliverdin models for the Pfr form: (a) DITAM: di-lactam tautomer of biliverdin. (b) TIM-A: biliverdin with lactim in ring A and lactam in ring D. (c) TIM-D: biliverdin with lactim in ring D and lactam in ring A. (d) DITIM: di-lactim tautomer of biliverdin.
4
Page 4 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
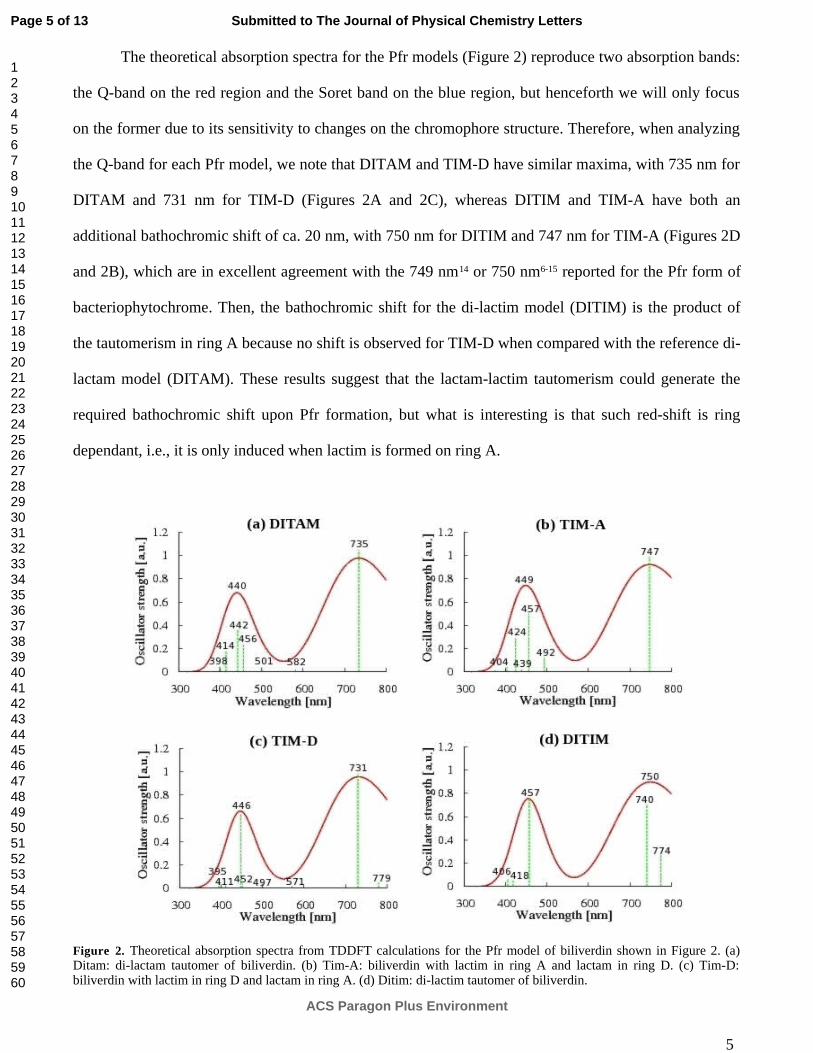
The theoretical absorption spectra for the Pfr models (Figure 2) reproduce two absorption bands:
the Q-band on the red region and the Soret band on the blue region, but henceforth we will only focus
on the former due to its sensitivity to changes on the chromophore structure. Therefore, when analyzing
the Q-band for each Pfr model, we note that DITAM and TIM-D have similar maxima, with 735 nm for
DITAM and 731 nm for TIM-D (Figures 2A and 2C), whereas DITIM and TIM-A have both an
additional bathochromic shift of ca. 20 nm, with 750 nm for DITIM and 747 nm for TIM-A (Figures 2D
and 2B), which are in excellent agreement with the 749 nm14 or 750 nm6-15 reported for the Pfr form of
bacteriophytochrome. Then, the bathochromic shift for the di-lactim model (DITIM) is the product of
the tautomerism in ring A because no shift is observed for TIM-D when compared with the reference di-
lactam model (DITAM). These results suggest that the lactam-lactim tautomerism could generate the
required bathochromic shift upon Pfr formation, but what is interesting is that such red-shift is ring
dependant, i.e., it is only induced when lactim is formed on ring A.
Figure 2. Theoretical absorption spectra from TDDFT calculations for the Pfr model of biliverdin shown in Figure 2. (a) Ditam: di-lactam tautomer of biliverdin. (b) Tim-A: biliverdin with lactim in ring A and lactam in ring D. (c) Tim-D: biliverdin with lactim in ring D and lactam in ring A. (d) Ditim: di-lactim tautomer of biliverdin.
5
Page 5 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

From above, we have TDDFT spectra suggesting that the Pfr form could be either lactim in ring
A (TIM-A model) or lactim in rings A/D (DITIM models). With this in mind, we turn now to
complementary information provided afer assessing different mechanisms of tautomerism, i.e., we look
for the associated transition states (TS) through DFT calculations.
First, we run the analysis for the lactam-lactim tautomerism involving either ring A or ring D,
separately. Thus, we can assess whether the tautomerism is energetically favoured in one ring over the
other, considering for that purpose the intramolecular non-dissociative mechanism described by Lledós
and Bertrán16 (see Scheme 1).
The barriers that have to be overpass during the intramolecular non-dissociative mechanism
from lactam to lactim were calculated through a single point calculation for each TS structure: TSA for
the tautomerism on ring A and TSD for the tautomerism on ring D (Figure 3). Hence, when comparing
the structures and energies, we get an energy difference of 7 kcal/mol between them. Such analysis
indicates unambiguously that TSA is energetically favoured (Figure 3A). Therefore, these results are in
agreement with the description of the absorption spectra from the models with the lactim tautomer on
ring A (TIM-A and DITIM), suggesting altogether that the photoconversion of phytochrome is likely to
occur via a lactim-lactam tautomerism on ring A and not on ring D of the bilin. Nevertheless, the energy
barrier of 48 kcal/mol for TSA is still high enough to be overpass, indicating that an intramolecular
process is not viable; rather, a catalyzed mechanism should take place.
Scheme 1. Reaction mechanism for the intramolecular non-dissociative tautomerization.
6
Page 6 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
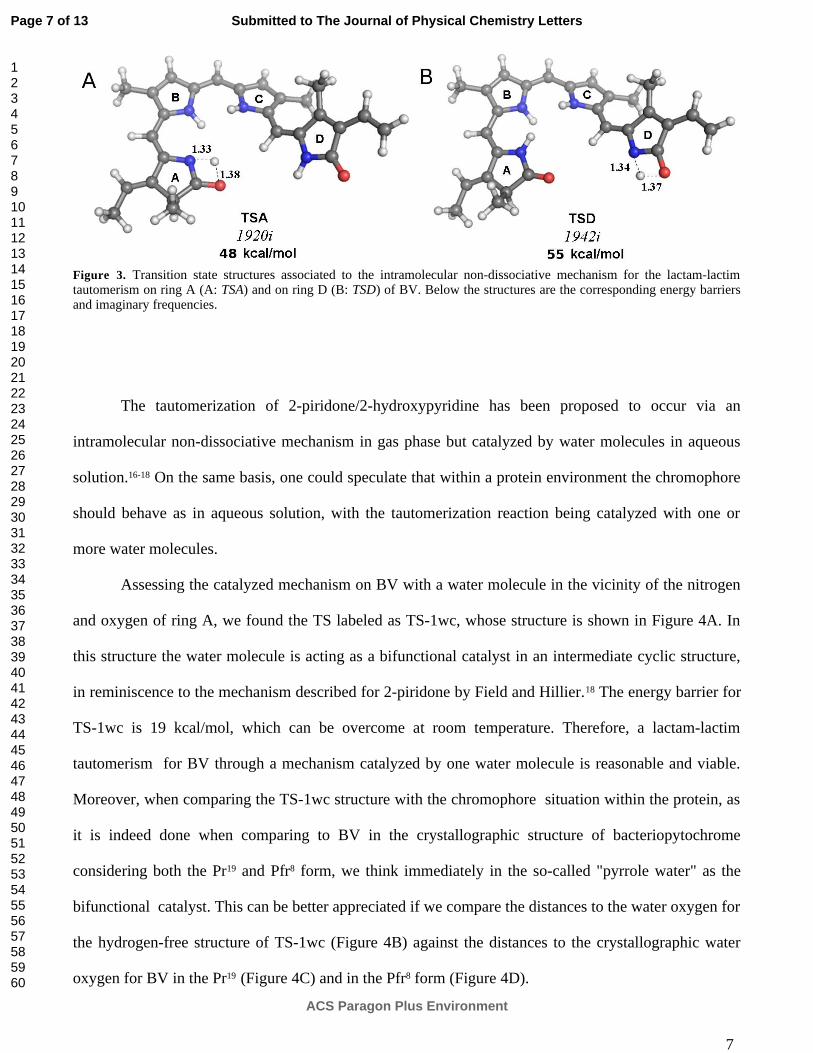
Figure 3. Transition state structures associated to the intramolecular non-dissociative mechanism for the lactam-lactim tautomerism on ring A (A: TSA) and on ring D (B: TSD) of BV. Below the structures are the corresponding energy barriers and imaginary frequencies.
The tautomerization of 2-piridone/2-hydroxypyridine has been proposed to occur via an
intramolecular non-dissociative mechanism in gas phase but catalyzed by water molecules in aqueous
solution.16-18 On the same basis, one could speculate that within a protein environment the chromophore
should behave as in aqueous solution, with the tautomerization reaction being catalyzed with one or
more water molecules.
Assessing the catalyzed mechanism on BV with a water molecule in the vicinity of the nitrogen
and oxygen of ring A, we found the TS labeled as TS-1wc, whose structure is shown in Figure 4A. In
this structure the water molecule is acting as a bifunctional catalyst in an intermediate cyclic structure,
in reminiscence to the mechanism described for 2-piridone by Field and Hillier.18 The energy barrier for
TS-1wc is 19 kcal/mol, which can be overcome at room temperature. Therefore, a lactam-lactim
tautomerism for BV through a mechanism catalyzed by one water molecule is reasonable and viable.
Moreover, when comparing the TS-1wc structure with the chromophore situation within the protein, as
it is indeed done when comparing to BV in the crystallographic structure of bacteriopytochrome
considering both the Pr19 and Pfr8 form, we think immediately in the so-called "pyrrole water" as the
bifunctional catalyst. This can be better appreciated if we compare the distances to the water oxygen for
the hydrogen-free structure of TS-1wc (Figure 4B) against the distances to the crystallographic water
oxygen for BV in the Pr19 (Figure 4C) and in the Pfr8 form (Figure 4D).
7
Page 7 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
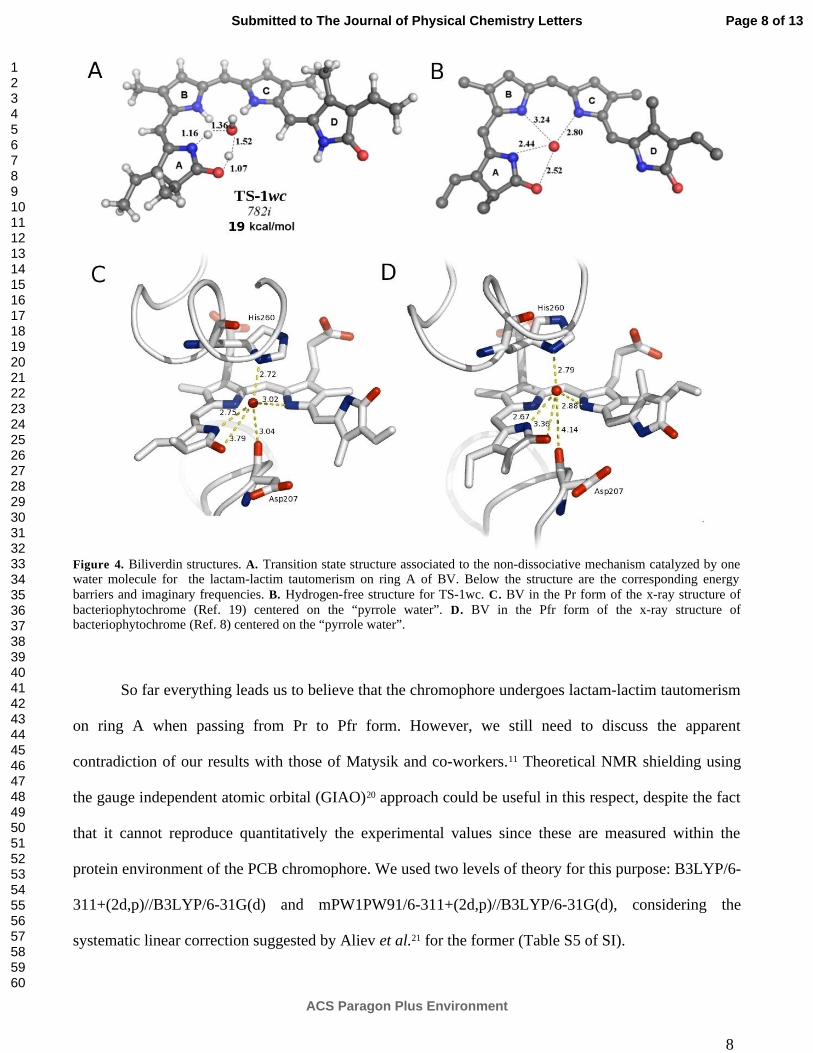
Figure 4. Biliverdin structures. A. Transition state structure associated to the non-dissociative mechanism catalyzed by one water molecule for the lactam-lactim tautomerism on ring A of BV. Below the structure are the corresponding energy barriers and imaginary frequencies. B. Hydrogen-free structure for TS-1wc. C. BV in the Pr form of the x-ray structure of bacteriophytochrome (Ref. 19) centered on the “pyrrole water”. D. BV in the Pfr form of the x-ray structure of bacteriophytochrome (Ref. 8) centered on the “pyrrole water”.
So far everything leads us to believe that the chromophore undergoes lactam-lactim tautomerism
on ring A when passing from Pr to Pfr form. However, we still need to discuss the apparent
contradiction of our results with those of Matysik and co-workers.11 Theoretical NMR shielding using
the gauge independent atomic orbital (GIAO)20 approach could be useful in this respect, despite the fact
that it cannot reproduce quantitatively the experimental values since these are measured within the
protein environment of the PCB chromophore. We used two levels of theory for this purpose: B3LYP/6-
311+(2d,p)//B3LYP/6-31G(d) and mPW1PW91/6-311+(2d,p)//B3LYP/6-31G(d), considering the
systematic linear correction suggested by Aliev et al.21 for the former (Table S5 of SI).
8
Page 8 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

Figure 5. GIAO-based NMR shieldings for selected carbons of the phycocyanobilin chromophore of cyanobacterial phytochrome. Blue values: B3LYP/6-311+(2d,p)//B3LYP/6-31G(d). Red values: mPW1PW91/6-311+(2d,p)//B3LYP/6-31G(d). Values in parenthesis: experimental NMR shifts form Ref. 11. (a) DITAM: di-lactam tautomer of biliverdin. (b) TIM-A: biliverdin with lactim in ring A and lactam in ring D. (c) TIM-D: biliverdin with lactim in ring D and lactam in ring A. (d) DITIM: di-lactim tautomer of biliverdin.
The Figure 5 shows the Pfr models for the PCB chromophore together with the 13C-NMR shifts
predicted by the GIAO for the two levels of theory mentioned above compared with the experimental
values in parenethesis.11 Despite the results with the B3LYP22 functional seems to be more ambiguous
than those using the mPW1PW91,23 it is possible to support the findings of the group of Matysik about a
lactam tautomer for the Pfr form of the chromophore.11 Nevertheless, it is known that the nature of the
tautomerism is quite sensitive to the environment conditions, e.g., in gas phase and in non-polar solvents
the lactim form predominates, whereas in aqueous solution and in polar solvents the lactam tautomer
predominates,18 but what is more interesting is that in solid state the lactam form is favoured. 24-27 Hence,
we suggest that the lactam form found by the group of Matysik11 is a consequence of the experimental
conditions, i.e., NMR in solid state.
In summary, the lactam-lactim tautomerism on ring A of the chromophore induces a
bathochromic shift consistent with the Pr/Pfr interconversion; the mechanism catalyzed by one water
molecule is energetically viable; and the NMR in solid state should not discard the occurrence of
tautomerism.
9
Page 9 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

Computational Methods
The biliverdin models for Pfr were constructed using as template the crystal structure for the Pfr
reported by the group of Moffat.8 The hydrogens are not resolved, so they were placed according to the
molecular structures depicted in Figure 1C. For the TDDFT10 calculations we have used the unrelaxed
crystal geometry, since it has been previously demonstrated that it predicts properly the experimental
values.28,29 Hence, for these calculations the added hydrogens were previously optimized at B3LYP/6-
31G(d) level of theory22 in vacuo keeping frozen the rest of the atoms. The BV models (Figure 1C) do
not consider the propionic side-chains of the pyrrol rings B and C because i) they do not affect the
excitation calculations as they are not part of the conjugated system, and ii) because their interaction
with the pyrrole nitrogens during full geometry optimization cause an undesirable distortion in the
resulting structure.
We have used the hybrid functional B3LYP22 with a 6-31G(d) basis set for TDDFT calculation.
The excitation energies and associated oscillator strengths were calculated using the Linear Response
formulation of the TDDFT as implemented10 in the Gaussian03 package30. The calculations have been
done with TD-B3LYP/6-31G(d) over eight roots, where the surrounding environment has been
represented with a polarizable continuum model (PCM)31 using a dielectric constant of 4.0 to account
for the average electrostatic interaction between the bilin chromophore and the apoprotein, as it has been
recommended in the review by Neugebauer.32 The vertical excitations were convoluted with Gaussian
functions considering a full width of 4000 cm-1 at half-maximum for the generation of the theoretical
absorption bands using the GaussSum2.2 program.33
The mechanistic analysis was carried out with the B3LYP exchange-correlational functional,22
together with the standard 6-31G(d) basis set. The optimizations used the Berny analytical gradient
optimization method.34,35 All calculations were achieved with the Gaussian03 suite of programs.30 The
stationary points were characterized by frequency in order to verify that the TS structures had an only
one imaginary frequency. The intrinsic reaction coordinate (IRC)36 path was traced to check the energy
10
Page 10 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

profiles connecting each transition structure to the two associated minima corresponding to reactants
and products.
In the NMR Shielding calculations, the model structures were fully optimized in vacuo using
DFT with the hybrid generalized gradient approximation (GGA) functional B3LYP22 and the 6-31G(d)
basis set. The 13C atomic chemical shielding tensors (σ) were computed for each optimized structure at
the density functional level using the gauge independent atomic orbital (GIAO)20 formalism with the
6-311+(2d,p) basis set. Two exchange-correlation functionals were assessed, namely, B3LYP22 and
mPW1PW91.23 Isotropic atomic chemical shifts (δ) in units of ppm for both levels of theory, i.e.,
B3LYP/6-311+(2d,p)//B3LYP/6-31G(d) and mPW1PW91/6-311+(2d,p)//B3LYP/6-31G(d), were
computed as differences between the 13C atomic isotropic shieldings of the models and the carbon atoms
of the reference tetramethylsilane (TMS). The linear scaling correction of Aliev et al.,21 which is
implemented to correct specifically systematic errors associated with the GIAO B3LYP/6-
311+(2d,p)//B3LYP/6-31G(d) level of theory, was used on the computed 13C-NMR chemical shifts
according to the following relationship: δscalc = 0.95 δcalc + 0.30, where δcalc and δscalc are the
calculated and the linearly scaled values, respectively.
Acknowledgment. We thank to Dr. N.C. Rockwell and Prof. J.C. Lagarias (Department of Molecular
and Cell Biology at University of California, Davis) for the interesting discussion on some of the ideas
of this work; L.G. acknowledges the financial support from the Deutsche Forschungsgemeinschaft (GO
1059/2-1); J.S.D. acknowledges project 130922 from USM.; and R.A.M. acknowledges the Grants from
CONICYT and BECAS CHILE.
Supporting Information Available: Tables for the theoretical absorption spectra and NMR
shielding calculations; and cartesian coordinates for reactants and TS structures. This material is
available free of charge via the Internet at http://pubs.acs.org.
11
Page 11 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

References
1 Franklin, K. A.; Whitelam, G. C. Annals of Botany 2005, 96, 169.
2 Rockwell, N. C.; Su, Y. S.; Lagarias, J. C. Annu. Rev. Plant Biol. 2006, 57, 837.
3 Shimizu-Sato, S.; Huq, E., Tepperman, J. M.; Quail, P. H. Nat. Biotechnol. 2002, 20, 1041.
4 Levskaya, A.; Weiner, O. D.; Lim, W. A.; Voigt, C. A. Nature 2009, 461, 997.
5 Wagner, J. R.; Brunzelle, J. S.; Forest, K. T.; Vierstra, R. D. Nature 2005, 438, 325.
6 Yang, X.; Stojkovic, E. A.; Kuk, J.; Moffat, K. Proc. Natl. Acad. Sci. USA 2007, 104, 12571.
7 Essen, L.-O.; Mailliet, J.; Hughes, J. Proc. Natl. Acad. Sci. USA 2008, 105, 14709.
8 Yang, X.; Kuk, J.; Moffat, K. Proc. Natl. Acad. Sci. USA 2008, 105, 14715.
9 Kereselidze, J. A.; Zarqua, T. Sh.; Kikalishvili, T. J.; Churgulia, E. J.; Makaridze, M. C. Russ. Chem. Rev. 2002, 71, 993.
10 Casida, M. E. In Recent Advances in Density Functional Methods; Chong, D. P., Ed.; World Scientific: Singapore, 1995; Part 1, p 155.
11 Rohmer, T.; Lang, C.; Hughes, J.; Essen, L. O.; Gärtner, W.; Matysik, J. Proc. Natl. Acad. Sci. USA 2008, 105, 15229.
12 Rohmer T, Lang C, Bongards C, Gupta KB, Neugebauer J, Hughes J, Gärtner W, Matysik J. J. Am. Chem. Soc. 2010, 132, 4431.
13 Bongards C, Gärtner W. Acc. Chem. Res. 2010, 43, 485.
14 Karniol, B.; Vierstra, R. D. Proc. Natl. Acad. Sci. USA 2003, 100, 2807.
15 Borucki, B.; v. Stetten, D.; Seibeck, S.; Lamparter, T.; Michael, N.; Mroginski, M. A.; Otto, H.; Murgida, D. H.; Heyn, M. P.; Hildebrandt, P. J. Biol. Chem. 2005, 280, 34358.
16 Lledós, A.; Bertrán, J. Tetrahedron Lett. 1981, 22, 775.
17 Lledós, A.; Bertrán, J. J. Mol. Struct. 1985, 120, 73.
18 Field, M. J.; Hillier, I. H. J. Chem. Soc., Perkin Trans. 2 1987, 617.
19 Wagner, J. R.; Zhang, J.; Brunzelle, J. S.; Vierstra, R. D.; Forest, K. T. J. Biol. Chem. 2007, 282, 12298.
20 Mulder, F. A. A.; Filatov, M. Chem. Soc. Rev. 2010, 39, 578.
21 Aliev, A. E.; Courtier-Murias, D.; Zhou, S. J. Mol. Struct. 2009, 893, 1.
22 (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Becke, A. D. Phys. Rev. A 1988, 38, 3098. (c ) Slater, J. C. Quantum Theory of Molecular and Solids: The Self-Consistent Field for Molecular and Solids; McGraw-Hill: New York, 1974, Vol. 4. (d) Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys. 1980, 58, 1200. (e) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
12
Page 12 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

23 (a) Perdew, J. P. In Electronic Structure of Solids ’91; Ziesche, P., Eschig, H., Eds.; Akademie Verlag: Berlin, 1991; p 11 (b) Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664 (c) Lynch, B. J.; Zhao, Y.; Truhlar, D. G. J. Phys. Chem. A 2003, 107, 1384.
24 Yang, H. W.; Craven, B. M. Acta Crystallogr. B 1998, 54, 912.
25 Penfold, B. R. Acta Crystallogr. 1953, 6, 591.
26 Ohms, U.; Guth, H.; Heller, E.; Dannöhl, H.; Schweig, A. Z. Kristallogr. 1984, 169, 185.
27 Almlöf, J.; Kvick, A.; Olovsson, I. Acta Crystallogr. B 1971, 27, 1201.
28 Matute, R. A.; Contreras, R.; Pérez-Hernández, G.; González, L. J. Phys. Chem. B 2008, 112, 16253.
29 Matute, R. A.; Contreras, R.; González, L. J. Phys. Chem. Lett. 2010, 1, 796.
30 Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; et al. Gaussian 03, revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.
31 Tomasi, J.; Menucci, B.; Cammi, R. Chem. Rev. 2005, 105, 2999.
32 Neugebauer, J. ChemPhysChem 2009, 10, 3148.
33 O`Boyle, N. M.; Tenderholt, A. L.; Langner, K. M. J. Comp. Chem. 2008, 29, 839.
34 Schlegel, H. J. Comput. Chem. 1982, 3, 214.
35 Schlegel, H. Geometry Optimization on Potential Energy Surface In Modern Electronic Structure Theory, (Ed.: D. R. Yarkony) World Scientific Publishing: Singapore, 1994.
36 Fukui, K. J. Phys. Chem. 1970, 74, 4161
13
Page 13 of 13
ACS Paragon Plus Environment
Submitted to The Journal of Physical Chemistry Letters
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960



Monday20,07
Room A Room B Room C
Opening
Keynote Lecture, Ahmed H. Zewail, USA
COFFEE BREAK
(S1) Photonics and Imaging (I) (S2) Photochromic Materials and Molecular Switches (S3) Gas Phase and Theory
IL1 (Javier Solis, Spain) IL2 ( Keitaro Nakatani, Japan) IL3 (Luis Bañares, Spain)
OC1 - Hiroshi Uji-i OC4 - Li-Zhu Wu OC7 - Alexey V. Baklanov
OC2 - Tsuyoshi Asahi OC5 - B. Seefeldt OC8 - Tatiana Domratcheva
OC3 - Thorben Cordes OC6 - A. Jorge Parola OC9 - Ricardo A. Matute
LUNCH
PL1, Michel Orrit, The Netherlands
(S4) Single Molecule Spectroscopy (S5) Photoinduced Electron and Charge Transfer (S6) Femto(bio)chemistry
IL4 (Maarten B.J. Roeffaers, Belgium) IL7 (Eric Vauthey, Switzerland) IL9 (Pascal Plaza, France)
OC10 - Dominik Wöll OC14 - Johanna Brazard OC19 - A. Weigel
OC11 - Sadahiro Masuo OC15 - Luis G. Arnaut OC20 - François-Alexandre Miannay
COFFEE BREAK
IL5 (Markus Sauer, Germany) IL8 (Franco Scandola, Italy) IL10 (Dongping Zhong, USA)
OC12 - Mike Heilemann OC16 - Anita Bruckner OC21 - P. Gilch
OC13 - Kei Murakoshi OC17 - S. Haacke OC22 - Michel Sliwg
IL6 (Boiko Cohen, Spain) OC18 - Eléna Ishow OC23 - Marcin Ziolek
Poster Session I
Reception
9:00
9:30
10:3011:00
11:30
11:50
12:10
12:30
14:00
14:45
14:55
15:25
15:45
16:0516:30
17:00
17:20
17:40
18:00
20:00

TIME-DEPENDENT DFT ON PHYTOCHROME CHROMOPHORES:
A WAY TO FIND THE RIGHT CONFORMER
Ricardo A. Matute,1 Renato Contreras,
1
Guillermo Pérez-Hernández,2 Leticia González
2
1Departmento de Química, Facultad de Ciencias, Universidad de Chile,
Casilla 653, Santiago, Chile 2Institut für Physikalische Chemie, Friedrich-Schiller-Universität Jena,
Helmholtzweg 4, 07743 Jena, Germany
E-mail: [email protected] or [email protected]
A theoretical approach based on the time dependent density functional theory
(TDDFT) is implemented together with a polarizable continuum model for incorporating the
bulk effect of the surrounding environment to estimate the excitation energies for different
chromophores found in phytochrome. We realized that a crystallographic structure without
geometry optimization as template for the calculations is critical to account for the steric
effect of the apoprotein over the chromophore. Through an analysis of the theoretical
spectra we predicted that phycocyanobilin inside the cyanobacterial phytochrome Cph1
likely adopts a semicyclic conformation ZZZssa (C5-Z,syn;C10-Z,syn;C15-Z,anti) [1], which
was then confirmed experimentally from a crystallographic structure by x-ray
diffraction [2]. Analogously, we implemented the method to phytochromobilin, i.e. the
chromophore of plant phytochrome, with results that suggest a ZZZssa structure too.
Therefore, we assume that theoretical spectra based on TDDFT seems to give us a good way
to predict the right conformer. Furthermore, we assess the ratio of the spectral bands on the
theoretical spectra (Q-band over the Soret band or Q/S ratio) finding that the spectroscopic
Q/S index indeed is directly related to the conformational isomerism of the chromophore.
Acknowledgements: FONDECYT Grant Nº 1070715, the Doctoral Fellowships from
CONICYT, and the Deutsche Forschungsgemeinschaft (GO 1059/2-1).
References [1]. R. A. Matute, R. Contreras, G. Pérez-Hernández, and L. González, J. Phys. Chem. B
2008, 112, 16253.
[2]. L. O. Essen, J. Mailliet, and J. Hughes, Proc. Natl. Acad. Sci. USA 2008, 105, 14709.

��
����������
������ ��������������� ������������������

����
�F,>�"�������������������������� ��� #% ��������0���
%���� $����-L� ��!��������5������ $������� ��
�������� ����1 � ���# �
�"�($,-2�,':"��,$4�('#!�!� �$��$"$�!��(��"�!5����� �!��"$��"���!�$* �8��&�!�##,�!!�(� �,'���� ���$* �8� ��!�'+� ��� =2�4�D�� �"����$!������������) �����.����� ������?����@�$"(� �������� ���������������. ���?���@2#,' ��"!����������#,' ��"�+$/��&��$!�+'�,�/�/4�,!�����5�����5����=�$"(����������!��#,' ��"!�+�"* �'"�$!�,�#,�!!',!�4&� $,�� �"��$* �8$ ',!�'+� ������� �,�!#'"!��+',�(��,$($ �'"������+'�"(� �$ �$���+'�,�����#,' ��"!�#�&!�*$��&��" �,$* �:� ���������'�� ��,�:� ��4�'*��/�*$��$"(���"� �*��8�(�"*�5� ��!��,�!�� !��"(�*$ �� �$ �����#,' ��"!�$* � '�� ��,�:� �� ����4�D�� �"����$!�������������4�'*��/�*$��+�"* �'"!�'+� �������#,' ��"!��"�$����;�����*'/#��A5��':�8�,5�$,�� ��!�+$,��"-"':"���&�$"$�&>�"��8$,�'�!�/�� �#���!#$�/� $" !�:��+'�"(� �$ �����#,' ��"!��$8��'8�,�$##�"��4� �$�!'�(�! �"* �+�"* �'"!������(�++�,�" �+�"* �'"!�*'��(�4��(��� '� ���(�++�,�" �$���A#,�!!�'"�'+� ���������"�!�$"(;',� '�(�++�,�"*�!��"� ���,�#,' ��"�!�D��"*�!����++�,�"*�!��"�#,' ��"�!�D��"*�!�*'��(���$(� '�(�++�,�" ��" �,$* �'"�#$, "�,!�',�(�++�,�" �/�� �2#,' ��"�*'/#��A�!�*'" $�"�"������#,' ��"!���"�',(�,� '�$"$�&!���+� �������#,' ��"!�*$"��" �,$* �:� ���$*��' ��,5�,��$,(�"��$��$,��,�#,' ��"�*'/#��A5�:��! $, �(�*$,,&�"��'� ��'2�//�"'#,�*�#� $ �'"�?�'��@��A#�,�/�" !��!�"��$"��"�8� ,'� ,$"!*,�# �'"�$"(� ,$"!�$ �'"�!&! �/�����$�!'�!�':��"� ��!��A#�,�/�" !� �$ � ����2 �,/�"�!��!�/�(�$ �"�� ����" �,$* �'"�$/'"�� ������!�����, ��,� ,$"!��" �*'2�'*$��>$ �'"��A#�,�/�" !��"��,$4�('#!�!��#�(�,/$��*���!��!�"��+��',�!*�" �#,' ��"�+�!�'"!�$"(���2.'��*��$,����',�!*�"*���'/#��/�" $ �'"��?����@�$!!$&!��"�'"�'"�*���!�*'"+�,/�(� ����" �,$* �'"!��"�#�$" $���"�',(�,� '��"8�! ��$ �����;�����*'/#��A�!�/',���"�(� $��5�:��,$�!�(�$"�$" �4'(&�$�$�"! � ��������#,' ��"��
�F2>�%��������������������&������������&������������������������� �������������������MMM�������MMM���;�
6�%������� $�6������ ������� �#�#�# ���� $�"�������# ���� �$����0 :1- ���# :$�&�����:�� :����� �������� ��$�� ����
����������� ��7����� ����
��& '*�,'/���!�$�#�' ',�*�# ',�+'�"(��"�#,' '4$* �,�$5�*&$"'4$* �,�$5�+�"��5�$"(�#�$" !���"�#�$" !5�#�& '*�,'/��$* !���-��$�4�'�'��*$��!:� *�� �$ �*'" ,'�!� ���#�' '/',#�'��"�*�,�!#'"!�!�?#�$" ��,': �5�+�':�,�"�� �/�5�!�$(��$8'�(�"�5�!��(�(',/$"*&5�#�' ' ,'#�!/5�� *�@�4$!�(�'"� ���:$8���"� ��'+� ����"*�(�" ����� ����!#� �� ����" �,�! � '�(�!*,�4�� ���/�*�$"�!/�'+�#�' '*'"8�,!�'"�'+� ���#�& '*�,'/�5� ��,���!�$�,��$ �(�#,'4��/� �$ ��$!� '�4��!'�8�(�+�,! %� ���*'"+',/$ �'"�$('# �(�4&� ���*�,'/'#�',�5������#�& '*�,'/'4���"5��"!�(��#�$" �#�& '*�,'/������,���!�"'�*'"!�!�!��"� ��!�,�!#�* �&� ���:'�$� �,"$ �8��! ,�* �,�!��$8��4��"�#,'#'!�(�+',� ����"$* �8��+',/��,�'+� ���!:� *�%�$"��A �"(�(�*'"+',/$ �'"�KKK$!$�?K5$" �HK5!&"HK5$" �@�',�$�!�/�*&*��*�*'"+',/$ �'"�KKK!!$�?K5!&"HK5!&"HK5$" �@������,'����$�*'/#� $ �'"$��! �(&�4$!�(�'"� ��� �/�2(�#�"(�" �(�"!� &�+�"* �'"$�� ��',&�?�����@�:��*'��(�#,�(�* � ���! ,�* �,��+',� ���*�,'/'#�',��'+� ���*&$"'4$* �,�$��#�& '*�,'/�5�!�##', �"��$�!�/�*&*��*�KKK!!$�! ,� �,��N�O�����"5� ���*,&! $��! ,�* �,��'+� ���#�& '*�,'/��!�"!',&�/'(����'+�$�*&$"'4$* �,�$��#�& '*�,'/��:$!�,�#', �(�4&� !!�"�� �$���N�O5�*'"+�,/�"��'�,� ��',� �*$�2*'/#� $ �'"$��#,�(�* �'"����"� ���/� �'(5�:���!�� ����������"�*'"C�"* �'"�:� �� ���#'�$,�>$4���*'" �"��/�/'(���?��.@�'8�,� ���$8$��$4���A2,$&�! ,�* �,�!�$!� �/#�$ �!�����',� �*$��!#�* ,$�*'"!�(�,�"�� ���#,' '"$ �'"5�!'�8$ �'"5�$"(�#,' ��"��"8�,'"/�" ��++�* !5�$,��*'"! ,�* �(�4$!�(�'"� ������* ,'"�*� ,$"!� �'"!�+',����� ��A*� �(�! $ �!���**',(�"�� '�'�,�,�!� �!5�#,' '"$ �'"�$"(� ���*'"+',/$ �'"$��*�$"���+,'/�KKK$!$� '�KKK!!$��"(�*��$�4$ �'*�,'/�*�!��+ 5�:��,�$!�(�#,' '"$ �'"�$"(� ����!'/�,�>$ �'"�+,'/�KKK!!$� '�KKK$!$��"(�*��$��&#!'*�,'/�*�!��+ ����!�(�!5�*'/#$,�!'"�

��=�
4� :��"� ��������2��.�*$�*��$ �'"!�'"�A2,$&�! ,�* �,�!�:� ��$"(�:� �'� ���'/� ,&�'# �/�>$ �'"��"(�*$ �!� �$ � ���/� �'(��!�D�$" � $ �8��'"�&�:��"� ���/'(��!�$,��*'"! ,�* �(�+,'/�*,&! $�� �/#�$ �!�:� �'� ���'/� ,&�'# �/�>$ �'"5� �$ �,�#,�!�" !�4� �,� ���!#$ �$��!� �$ �'"�'+� ���*�,'/'#�',���"� ���#,' ��"��"8�,'"/�" ���#$, �'+� ���D�$" � $ �8��#,�(�* �'"�'+� ����A*� $ �'"��"�,�&5� ��� ��',� �*$��!#�* ,$�!�':�#,'#�,�&� ���(�#�"(�*��'+� ���,$ �'!�4� :��"� ���4$"(!�?J24$"(�'8�,��',� �4$"(5�',�J;���"(�A@�$"(� ���*'"+',/$ �'"�'+� ���*�,'/'#�',��?�A �"(�(5�!�/�*&*��*5�',�*&*��*@���'�4' �5� ���$4!',# �'"��"�,�&�$"(� ���J;���"(�A5�:��"�*'"+,'" �(�:� ���A#�,�/�" $��($ $5�$,���!�(�$!�*,� �,�$�+',�(� �,/�"�"�� ���*'"+',/$ �'"�$('# �(�4&�#�& '*�,'/'4���"��"�#�$" �#�& '*�,'/�����& '*�,'/'4���"��"�� !��,�+',/�$4!',4!�$ �111�"/�?J24$"(@5�:��,�$!�'�,� ��',� �*$��!#�* ,�/�!�':!�$�#�$-�$ �11��"/�+',� ���KKK!!$�/'(���$"(�1�9�"/�+',� ���KKK$!$�/'(������!'5�:���� �$�J;��'+������+',�KKK!!$�$"(���3�+',�KKK$!$5�*'"+,'" �(�:� �� ����A#�,�/�" $��J;��'+���=12����+',�#�$" �#�& '*�,'/����'�� ��,5� ��!��,�!�� !�$��':��!�� '�#,�(�* �$�!�/�*&*��*�KKK!!$�*'"+',/$ �'"�+',� ���*�,'/'#�',���"�#�$" �#�& '*�,'/�5�$!��$8��4��"�+'�"(��"� ���A2,$&�! ,�* �,��'+�4$* �,�'#�& '*�,'/��$"(�*&$"'4$* �,�$��#�& '*�,'/�5�!'�!�##', �"�� ����(�$�'+�$��"�+��(�KKK!!$�*'"+',/$ �'"�+',� ����,��!'+',/�'+�$��� ���#�& '*�,'/��!#�*��!��� �N�O������.$ � �5�����'" ,�,$!5������,�>2�,"$"(�>5�$"(�����'">$��>5�0����&!�����/���5����5��1�=�?�996@��N�O������ !!�"5�0��.$����� 5�$"(�0������!5��,'*���$ ����*$(���*������5��95���397�?�996@�
�F4>���������������1�����#����������������� � ��&������������������� ����������������������������.�������������&#�������������&������� ������������������
6���A�� ��%�� �1&�#���1��� ��� ��2 ���$�5��������� �!���� �<�1�!���� $�2 ����$�%�0��2 ���# :���� ����� ���$�, (������0�����0��������$��� ��$����"��?� $�%��-�#����� $�*��' ������%�� �1&�#���1��� ��� ��2 ���$�5��������� �!���� �<�1�!���� $�2 �����
������������� ���������� ���1� ����# �
��& '*�,'/����?#�&�@��!� ���#,�/$,&�#�' ',�*�# ',�+',�!�"!�"���A ,�/��&��':�$/'�" !�'+����� �$"(�+',�/�(�$ �"��8$,�'�!�+$,2,�(����� 2�"(�*�(�,�!#'"!�!��"������,�#�$" !���,$"!�'*$ �'"�+,'/� ���*& '!'�� '� ���"�*���!��!�$"��!!�" �$��! �#��"�#�&��!��"$�� ,$"!(�* �'"�� �����!�$"��24'A�#,' ��"� �$ �+�"* �'"!�$!�$�"��$ �8��,����$ ',��"�+$,2,�(����� �!��"$��"��(':"! ,�$/�'+� ���#�&���"��,$4�('#!�!� �$��$"$���'��(�" �+&�+$* ',!��"8'�8�(��"� ���2(�#�"(�" ����� �!��"$�� ,$"!(�* �'"5�#''�!�'+�� �&�/� �&�!��+'"$ �2 ,�$ �(���(�2=�!��(!�:�,��!*,��"�(�+',�!��(��"�!� �$ �!�##,�!!� ����&#�,!�"!� �8��#��"' &#��'+� ���/� $" ������#��"' &#��'+� ���!�##,�!!',�/� $" �#,�!�" �(���,���!�*$�!�(�4&�$�/�!!�"!��/� $ �'"��"� ���������"�� �$ ���$(!� '�$"�$/�"'�$*�(� ,$"!� �'"��"�� !���! �(�"��-�"$!�2,��$ �(�('/$�"������"'8���#�&�2�9��$������$� �,!� ���#�$" Y!�!�"!� �8� &� '�(�++�,�" ����� �:$8���"� �!��"(�*�"��+$,2,�(�������,,$(�$"*��,�!#'"!�!�$"(�(�*,�$!�!� ���#�,!�! �"*��'+� ���+$,2,�(2���� 2�"(�*�(�!��"$��"���"#� ������! ,'"����(�2=�!�##,�!!',�#��"' &#��'+�#�&�2�9��*'" ,$! !�:� �� ���/'(�,$ ��#��"' &#��'4!�,8�(��"� ���:��(2 &#��4$*-�,'�"(5�:��*���"(�*$ �!� �$ � ���/� $ �'"�/$�"�&�$� �,!�+�"* �'"!��"�$"� ���2(�#�"(�" �!��"$��"��*$!*$(�������/� $ �'"�!#�*�+�*$��&��"��4� !�"�*��$,�$**�/��$ �'"�'+� ���#�' ',�*�# ',�/'��*�����#'"�,�(2���� ��,,$(�$ �'"��8�"� �'����� �! �����" �,$* !�:� ������?+$,2,�(��'"���&#'*' &���@�$"(����?���2��-��#,' ��"@5� :'�+$* ',!� �$ �$,���!!�" �$��+',�"�*��$,�$**�/��$ �'"�'+�#�&�������/� $ �(�#�&���A��4� !��"$� �,�(�(��,$($ �'"�-�"� �*!��8�"��"(�,����� �*'"(� �'"!� �$ ��"��4� �� !�"�*��$,�$**�/��$ �'"�����!�+�"(�"���"(�*$ �!� �$ ��� ��,�#�&��(��,$($ �'"� $-�!�#�$*��"�$,�&��A*��!�8��&��"� ���*& '#�$!/�',� �$ �*& '#�$!/�*�$"(�"�*��$,�(��,$($ �'"�*'�" �,4$�$"*���$*��' ��,��

August 8, 2009 Ricardo Matute, PhD Candidate Universidad de Chile Facultad de Ciencias Departamento de Quimica Casilla 653 Santiago, Chile Dear Mr. Matute, I thank you for your poster presentation at the 9th International Conference on Tetrapyrrole Photoreceptors of Photosynthetic Organisms (ICTPPO) held at Asilomar Conference Center in Monterey, CA from July 26 to July 31, 2009. Your contribution to the meeting was most welcome and I am happy that we will be able to develop a research cooperation of mutual interest. Sincerely yours,
J. Clark Lagarias Paul K. and Ruth R. Stumpf Professor of Plant Biochemistry















![MECANISMOS TRANSDUCCIONALES DE INSULINA EN EL ...repositorio.uchile.cl/tesis/uchile/2010/qf-contreras_ae/pdfAmont/qf... · 11.3 La primera fase del incremento del [Ca2+] i depende](https://static.fdocuments.es/doc/165x107/5e3de3906b213d3075414b06/mecanismos-transduccionales-de-insulina-en-el-113-la-primera-fase-del-incremento.jpg)


