
VOCALIA TECNICA NACIONAL DIRECCION NACIONAL DEFENSA PERSONAL.

© 2017, www.tecno-med.es 1
EVALUACIÓN CLÍNICA DE PRODUCTOS SANITARIOS
de acuerdo al Reglamento (EU) 2017/745

© 2017, www.tecno-med.es 2
AEFI SECCIÓN CATALANA - VOCALIA PRODUCTOS SANITARIOS
Estudio del Reglamento (UE)
2017/745
del Parlamento Europeo y del
Consejo de 5 de abril de 2017
sobre los productos sanitarios Resumen del Capítulo VI relacionado con la
Evaluación Clínica e Investigaciones Clínicas:
• Artículos 61 al 82 y Anexos XIV y XV.
Pepa Boté, Claire Murphy, Elena Simó, Jeny Villanueva,
Reunión Vocalía 18.05.2017

© 2017, www.tecno-med.es 3
Evaluación Clínica - Resumen
1. Situación actual
– Evaluación clínica según Directiva 93/42/CEE y MEDDEV 2.7/1
– Problemas frecuentes
2. Cambios en los conceptos
3. Transición a MDR
- Evaluación clínica según Reglamento 2017/745
- Obligación / Necesidad de Investigaciones Clínicas
- Esquema de un proceso continuado de EC - Enlace con PMS / PMCF
4. Conclusiones

© 2017, www.tecno-med.es 5
Evaluación Clínica - Resumen
1. Situación actual
– Evaluación clínica según Directiva 93/42/CEE y MEDDEV 2.7/1
– Problemas frecuentes
2. Cambios en los conceptos
3. Transición a MDR
- Evaluación clínica según Reglamento 2017/745
- Obligación / Necesidad de Investigaciones Clínicas
- Esquema de un proceso continuado de EC - Enlace con PMS / PMCF
4. Conclusiones

© 2017, www.tecno-med.es 6

© 2017, www.tecno-med.es 7

© 2017, www.tecno-med.es 8

© 2017, www.tecno-med.es 9

© 2017, www.tecno-med.es 10

© 2017, www.tecno-med.es 11

© 2017, www.tecno-med.es 12
Anexo X – Obliga a - IC para implantes y Clase III
- Inclusión de la EC en TF
- Actualización de la EC con datos PMS y PMCF

© 2017, www.tecno-med.es 13
Anexo X

© 2017, www.tecno-med.es 14
Nueva edición

© 2017, www.tecno-med.es 15
Etapas de Evaluación clínica MEDDEV 2.7/1 rev.4
Q&A pero más reducido
ACTUALIZACIÓN!
CUALIFICACIONES!!
• 0. Alcance
• 1. Identificación datos
• 2. Valoración datos
• 3. Análisis datos
• 4. Informe EC
Anexos
EQUIVALENCIA!!
INVESTIGACIONES!!
Pautas ANÁLISIS!!
EC en el contexto SGC

© 2017, www.tecno-med.es 16
Procedimiento esquemático de Evaluación Clínica
según MEDDEV 2.7/1 rev.4

© 2017, www.tecno-med.es 18
Ejemplo índice de contenido (según MEDDEV
2.7/1 2016)

© 2017, www.tecno-med.es 19
Problemas frecuentes
1. Equivalencia, Equivalencia, EQUIVALENCIA!!!
¿cuándo podemos alegar equivalencia? (equivalencia “clínica”, “técnica” y “biológica”)
(“mismo” v “similar”)
IDEAL: Otro producto único equivalente ya con marcado CE
2. Objetivos “medibles” para la evaluación clínica
3. “Estado del arte”: Situación del producto versus otras
opciones disponibles?
4. Validez científica (y estadística) de los datos
5. Acceso a la información clínica del equivalente
6. Conclusión clara balance beneficio / riesgo – Perfil B/R
7. Planes de PMS / Plan PMCF / EC en el contexto del SGC

© 2017, www.tecno-med.es 20
Evaluación Clínica - Resumen
1. Situación actual
– Evaluación clínica según Directiva 93/42/CEE y MEDDEV 2.7/1
– Problemas frecuentes
2. Cambios en los conceptos
3. Transición a MDR
- Evaluación clínica según Reglamento 2017/745
- Obligación / Necesidad de Investigaciones Clínicas
- Esquema de un proceso continuado de EC - Enlace con PMS / PMCF
4. Conclusiones

© 2017, www.tecno-med.es 21
Qué significa <<Evaluación Clínica>>
Proceso sistemático y planificado para: - generar,
- recoger,
- analizar, y
- evaluar de forma continua
los datos clínicos relativos a un producto para - VERIFICAR SU SEGURIDAD Y FUNCIONAMIENTO, INCLUIDOS LOS
BENEFICIOS CLÍNICOS, cuando se utilice conforme a la finalidad prevista por el fabricante.

© 2017, www.tecno-med.es 22
Qué significa <<Datos Clínicos>>
Qué significa <<Beneficio Clínico>>

© 2017, www.tecno-med.es 23
Qué significa <<Pruebas Clínicas>>
… y <<Pruebas Clínicas Suficientes>>
Datos clínicos de una cantidad y calidad que garantizan la validez científica de las conclusiones. (MEDDEV 2.7/1)

© 2017, www.tecno-med.es 24
Qué significa <<Seguimiento Poscomercialización>>
PMS = Post Market Surveillance
Qué significa <<Seguimiento Clínico Poscomercialización>>
PMCF = Post Market Clinical Follow-up

© 2017, www.tecno-med.es 25
Qué significa <<ESTUDIO de Seguimiento Clínico Poscomercialización>>
Un estudio realizado después del marcado CE de un producto con la intención de proporcionar respuestas a preguntas específicas relacionadas con la seguridad o el funcionamiento clínico (es decir, riesgos residuales) de un producto cuándo éste se utiliza de acuerdo con su etiquetado aprobado.

© 2017, www.tecno-med.es 26
Evaluación Clínica - Resumen
1. Situación actual
– Evaluación clínica según Directiva 93/42/CEE y MEDDEV 2.7/1
– Problemas frecuentes
2. Cambios en los conceptos
3. Transición a MDR
- Evaluación clínica según Reglamento 2017/745
- Obligación / Necesidad de Investigaciones Clínicas
- Esquema de un proceso continuado de EC - Enlace con PMS / PMCF
4. Conclusiones

© 2017, www.tecno-med.es 27
MDR – ”Considerandos” enfocados a EC

© 2017, www.tecno-med.es 28
MDR – Obligaciones generales
Artículo 5 Introducción en el mercado y puesta en servicio ...
3. La demostración de la conformidad con los requisitos
generales de seguridad y funcionamiento deberá incluir una
evaluación clínica con arreglo al artículo 61.
…
Artículo 10 Obligaciones generales de los fabricantes ... 3. Los fabricantes llevarán a cabo una evaluación clínica con
arreglo a los requisitos establecidos en el artículo 61 y en el
anexo XIV, incluido el seguimiento clínico
poscomercialización
Aplicable a TODOS
los productos

© 2017, www.tecno-med.es 29
ARTÍCULO 61: EVALUACIÓN CLÍNICA

© 2017, www.tecno-med.es 30
MDR (UE) 2017/745 - Artículo 61
Artículo 61.1 Obligación general para planificar, llevar a cabo y documentar una evaluación clínica Los datos clínicos deben aportar las ”pruebas clínicas suficientes” para confirmar: - La conformidad con los requisitos generales de seguridad y funcionamiento
del Anexo I - La evaluación de los efectos secundarios indeseables - La evaluación de la aceptabilidad de la relación beneficio-riesgo El fabricante especificará y justificará el nivel de las pruebas clínicas necesario para demostrar la conformidad con los correspondientes requisitos generales de seguridad y funcionamiento. Dicho nivel de pruebas clínicas será el adecuado habida cuenta de las características del producto y su finalidad prevista. El fabricante planificará, llevará a cabo y documentará una evaluación clínica de acuerdo a este artículo y a la parte A (Evaluación clínica) del anexo XIV.
¿Has definido la especificación / justificación del nivel “necesario” de pruebas clínicas?

© 2017, www.tecno-med.es 31
MDR (UE) 2017/745 - Artículo 61
Artículo 61.2 Consulta previa para productos de “riesgo”
• Para productos de la Clase III (todos!) y productos activos de la Clase IIb destinados a administrar y/o retirar un medicamento, se establece la posibilidad de consulta previa a un panel de expertos relacionada con la estrategia de desarrollo clínico.
• En su caso, el hecho de que se realizó la consulta se debe incluir en el informe de EC.
• La consulta es informativa – no conlleva derecho a nada.
Habrá un panel de expertos (ad hoc) para estas consultas... ¿criterios? ……COM y MDCG

© 2017, www.tecno-med.es 32
MDR (UE) 2017/745 - Artículo 61
Artículo 61.3 Procedimiento de evaluación clínica (definido y metodológicamente fundado)
• 3 elementos a tener en cuenta – Evaluación crítica de bibliografía científica disponible en ese momento
(en caso de demostrar que el producto, respecto de la finalidad prevista, es equivalente a otro existente Y cuando los datos demuestran adecuadamente
la conformidad), y
– Evaluación crítica de los resultados de todas las investigaciones clínicas disponibles realizadas de acuerdo al Reglamento, y
– Consideración de las opciones alternativas disponibles en ese momento para el mismo fin.
• Atención a que se establece lo disponible “en ese momento”.
Métodos a aplicar de forma ACUMULATIVA… los 3!

© 2017, www.tecno-med.es 33
MDR (UE) 2017/745 - Artículo 61
Artículo 61.4 Obligación de Investigación Clínica para productos de Clase III y productos implantables SALVO determinadas excepciones:
• Cuando se trata de la modificación del diseño por el mismo fabricante, y
• Se demuestra de forma específica la EQUIVALENCIA con el producto comercializado con la aprobación del ON, y
• “suficiencia” de la EC del producto comercializado para demostrar la conformidad del producto modificado
Si se cumplen estas condiciones, el ON debe comprobar que hay un Plan PMCF adecuado que incluye ESTUDIOS poscomercialización.
¿Criterios que aplicarán los ON para determinar si se cumplen estas condiciones?... Consulta previa de la ”estrategia”?

© 2017, www.tecno-med.es 34
MDR (UE) 2017/745 - Artículo 61
Artículo 61.5 Condiciones adicionales (añadir a las del 61.4) aplicables para no tener la obligación de llevar a cabo una evaluación investigación clínica cuando el producto nuevo “equivalente” no es fabricado por el mismo fabricante:
• CONTRATO en vigor para dar pleno acceso a la documentación técnica
• EC inicial de acuerdo al REGLAMENTO (es decir, nueva o actualizada
con los nuevos criterios!)
• Pruebas claras de ello a disposición del ON
“Errata” la palabra evaluación - En la versión en inglés, se utiliza la palabra “investigation”!
¿Viabilidad de este tipo de acuerdos… OEM? Entre competidores?

© 2017, www.tecno-med.es 35
MDR (UE) 2017/745 - Artículo 61
Artículo 61.6 No aplicabilidad de la obligación de realizar Investigación Clínica a los productos Clase III y los productos implantables que cumplen las condiciones indicadas:
• Productos ya en el mercado conforme a Directivas y con evaluación clínica ya basada en datos clínicos suficientes Y que se ajusten a la Especificación Común aplicable de ese tipo de producto (en su caso)
• Lista específica de productos (implantables) para los que no se exigirá IC pero también con la condición de que la evaluación clínica esté “suficientemente avalada” por datos clínicos y que se apliquen las Espec.Comunes, en su caso para ese tipo de producto.
¿”Retro-aplicabilidad” de las Especificaciones Comunes?
¿Implica nueva EC para TODOS los implantables y Clase III aún sin introducir cambios en el diseño?

© 2017, www.tecno-med.es 36
Artículo 61.6.b) – Lista de implantes EXENTOS de obligación IC
- Suturas
- Grapas quirúrgicas
- Productos de obturación dental
- Aparatos de ortodoncia
- Coronas dentales
- Tornillos
- Cuñas
- Placas
- Alambres
- Alfileres
- Clips o conectores

© 2017, www.tecno-med.es 37
MDR (UE) 2017/745 - Artículo 61 Artículo 61.7 La aplicación de la excepción de IC para productos implantables y Clase III se debe explicitar y justificar en el informe de EC del fabricante y en el informe de revisión de la EC por el ON.
¿Puede haber objeciones por parte de las AACC?
Artículo 61.8 Posibilidad de que COM pueda actualizar (mediante actos “DELEGADOS”) la lista de los productos implantables y Clase III a los que se puede aplicar la exención de IC.
Actos ”DELEGADOS” son para modificar el Reglamento. Actos de EJECUCIÓN son para desarrollarlo.
Artículo 61.9 Evaluación clínica de productos sin finalidad sanitaria relacionados en el Anexo XVI – basarla en demostrar su funcionamiento (no “beneficio clínico”).
¿Ética de IC con productos sin finalidad sanitaria? Habrá criterios específicos para estos casos, ej. Para los CEIC?

© 2017, www.tecno-med.es 38
MDR (UE) 2017/745 - Artículo 61
Artículo 61.10 Se explicita la posibilidad de basar la demostración de conformidad en datos “no clínicos” siempre que se justifique en la documentación técnica del producto. Basar la demostración de la conformidad en métodos de ensayo “no clínicos”, e.g. los resultados a la gestión de riesgos, la evaluación del funcionamiento, evaluación pre-clínica.
¿Ejemplos? … ¿Productos históricos?
¿Suficiente con una evaluación clínica bibliográfica para justificar que el producto está en línea con el “estado general de la técnica”?
¿Rasero en el criterio a aplicar por los ON?

© 2017, www.tecno-med.es 39
MDR (UE) 2017/745 - Artículo 61
Artículo 61.11 Obligación de mantener actualizada la EC durante todo el ciclo de vida del producto con los datos PMCF y PMS (aplicando el sistema, el plan y los métodos PMS previstos).
Para productos implantables y los de Clase III, los informes de PMCF y el informe resumen de seguridad PSUR se deberán actualizar al menos 1 vez al año.
¿Directrices / Formatos para informes PMCF / PSUR?
Artículo 61.12 Informe de evaluación clínica con un contenido de acuerdo a la sección 4 del anexo XIV y que debe formar parte de la documentación técnica a la que se refiere el Anexo II.
¿”Formar parte de la documentación técnica” implica estar bajo el control documental establecido por el sistema de calidad?

© 2017, www.tecno-med.es 40
ANEXO XIV: EVALUACIÓN CLÍNICA y PMCF

© 2017, www.tecno-med.es 41
A.1 PASOS para la planificación del la evaluación clínica, la determinación de datos clínicos existentes, la evaluación de la adecuación de los datos existentes para determinar la seguridad y funcionamiento, la generación de nuevos datos clínicos (en caso necesario) y el análisis final con las conclusiones sobre seguridad, funcionamiento y beneficios clínicos logrados.
Atención a la necesidad de describir los beneficios clínicos previstos con parámetros de resultados clínicos significativos y concretos.
Implica cuantificar / parametrizar los resultados clínicos que se esperan.
PLAN DE DESARROLLO CLÍNICO a iniciar en fases TEMPRANAS del proyecto
A.2 Profundidad y amplitud de la EC en consonancia con el riesgo y con las declaraciones (i.e. “alegaciones” = ”claims”) del fabricante respecto al producto.
ANEXO XIV: Parte A - EVALUACIÓN CLÍNICA Parte B - SEGUIMIENTO CLÍNICO POSCOMERCIALZACIÓN

© 2017, www.tecno-med.es 42
A.3 DEMOSTRACIÓN DE EQUIVALENCIA sobre las siguientes características:
• TÉCNICAS: diseño similar, condiciones similares de uso, especificaciones y propiedades similares, métodos similares de implantación, principios de operación y requisitos críticos de funcionamiento similares
• BIOLÓGICAS: mismos materiales en contacto con los mismos tejidos humanos para un tipo y duración similares de contacto y con unas carácterísticas similares de liberación de sustancias.
• CLÍNICAS: se utiliza para las mismas condiciones o finalidad clínicas, para gravedad y fases similares de la enfermedad, en la misma parte del cuerpo, en una población similar (edad, anatomía, fisiología), mismo tipo de usuario, funcionamiento crítico similar habida cuenta del efecto clínico esperado para una finalidad específica prevista.
SERÁN SIMILARES EN LA MEDIDA EN QUE NO HUBIERA DIFERENCIA CLÍNICA IMPORTANTE EN LA SEGURIDAD Y FUNCIONAMIENTO CLÍNICOS DEL PRODUCTO.
DEMOSTRACIÓN DE QUE EL FABRICANTE DISPONE DE NIVELES SUFICIENTES DE ACCESO A LOS DATOS RESPECTO DE LOS CUALES ALEGA LA EQUIVALENCIA
ANEXO XIV: Parte A - EVALUACIÓN CLÍNICA
Parte B - SEGUIMIENTO CLÍNICO POSCOMERCIALZACIÓN

© 2017, www.tecno-med.es 43
A.4 INFORME DE EVALUACIÓN CLÍNICA:
• Con los resultados de la evaluación clínica y las pruebas clínicas
• Apoyo a la evaluación de la conformidad
• Formará parte de la documentación técnica del producto
• Incluir datos tanto favorables como desfavorables en la documentación técnica.
¿Habrá nueva MEDDEV 2.7.1… con el detalle del contenido?
ANEXO XIV: Parte A - EVALUACIÓN CLÍNICA
Parte B - SEGUIMIENTO CLÍNICO POSCOMERCIALZACIÓN

© 2017, www.tecno-med.es 44
B.5 Proceso CONTINUO para ACTUALIZAR la evaluación clínica mediante el proceso PMCF y Plan de Seguimiento Poscomercialización con la finalidad específica de garantizar la aceptabilidad continua de los riesgos y detectar riesgos emergentes.
B.6 Método documentado de acuerdo al Plan PMCF
• Atención al uso de “pruebas objetivas” (por ejemplo, no vale afirmar que funciona pq no hay quejas o feedback de lo contrario…), durante toda la vida útil
• Atención a garantizar la aceptabilidad continua de la relación beneficio-riesgo
• Atención también a “detectar posibles usos indebidos o no previstos” (off-label use).
B.7 Análisis de los hallazgos del PMCF – documentar resultados en un INFORME.
B.8 ”Feedback” de las conclusiones a la Evaluación Clínica y a la Gestión de Riesgos – ATENCIÓN A POSIBLES MEDIDAS PREVENTIVAS Y/O CORRECTIVAS
ANEXO XIV: Parte A - EVALUACIÓN CLÍNICA
Parte B - SEGUIMIENTO CLÍNICO POSCOMERCIALZACIÓN

© 2017, www.tecno-med.es 45
Necesidad / Obligación de Investigación Clínica
• Producto implantable / Producto Clase III – NO HAY producto ”equivalente” (o no se cumplen las condiciones!)
– La evaluación clínica NO ES suficiente para demostrar la conformidad
– La evalauación clínica NO SE AJUSTA a la especificación común
Art 62: Requisitos generales relativos a las investigaciones clínicas realizadas para
demostrar la conformidad de los producto

© 2017, www.tecno-med.es 46
1. FORMULARIO DE SOLICITUD
2. MANUAL DEL INVESTIGADOR
3. PLAN DE INSVESTIGACIÓN CLÍNICA
4. OTRA INFORMACIÓN
ANEXO XV, Capitulo 2: Documentación relativa a la solicitud de IC
MDR (UE) 2017/745 - Artículo 62
62.1 Requisito general que establece que las investigaciones clínicas deberán estar diseñadas, autorizadas, realizadas, registradas y notificadas con arreglo a los artículo 62 al 81 y ANEXO XV.

© 2017, www.tecno-med.es 47
• 2.2 MANUAL DEL INVESTIGADOR :
Es un documento que contiene un resumen de la información clínica y no clínica disponible hasta el momento. Debe actualizarse.
- Identificación y descripción del producto (finalidad prevista, clase riesgo, regla clasificación)
- Instrucciones del fabricante para instalación, mantenimiento de normas de higiene y uso, requisitos almacenamiento y manipulación
- Información a incluir en etiquetado e instrucciones de uso (IU), cuando vaya al mercado (ojo con representante EU!)
- Info adicional según el producto
- Evaluación preclínica:
a. Cálculo del diseño, ensayos in vitro e in vivo, con animales, mecánicos y/o eléctricos, de fiabilidad.
b. Validación esterilización, verificación programas informáticos, ensayos de funcionamiento
c. Evaluación de la biocompatibilidad y seguridad biológica
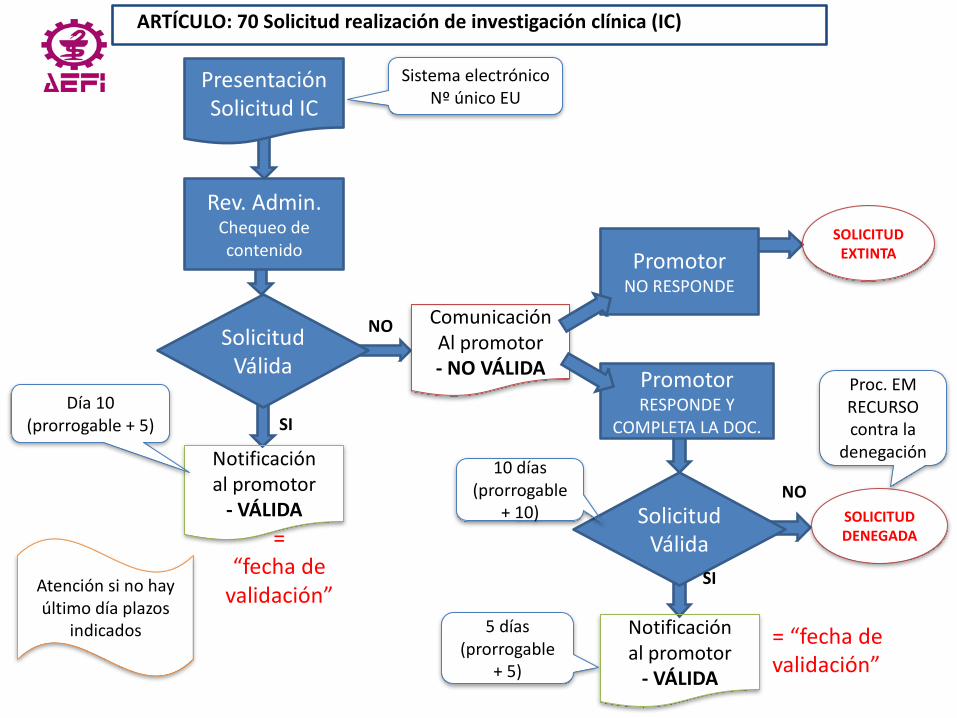
AEFI SECCIÓN CATALANA - VOCALIA PRODUCTOS SANITARIOS ESTUDIO REGLAMENTO PRODUCTOS SANITARIOS
CAPÍTULO VI
ARTÍCULO: 70 Solicitud realización de investigación clínica

© 2017, www.tecno-med.es 48
• 2.2 MANUAL DEL INVESTIGADOR cont :
- Datos clínicos existentes:
a. Procedentes de la literatura científica
b. Otros sobre seguridad, funcionamiento, beneficios, características de diseño y finalidad prevista de los productos equivalentes o similares del = fabricante. Incluido el tiempo en el mercado y una revisión del funcionamiento, beneficio, seguridad y acciones correctivas adoptadas.
c. Resumen del análisis beneficio-riesgo y de la gestión de riesgos (riesgos conocidos o previsibles, efectos indeseables, contraindicaciones y advertencias).
d. Si llevan med/sangre/plasma...información detallada y valor añadido incorporación.
e. Lista que detalle el cumplimiento de los requisitos de seguridad.
f. Descripción detallada de los procedimientos clínicos y pruebas de diagnóstico utilizadas en la IC y sobretodo DESVIACIÓN respecto a la práctica clínica habitual.
AEFI SECCIÓN CATALANA - VOCALIA PRODUCTOS SANITARIOS ESTUDIO REGLAMENTO PRODUCTOS SANITARIOS
CAPÍTULO VI
ARTÍCULO: 70 Solicitud realización de investigación clínica

© 2017, www.tecno-med.es 49
= Manual del Investigador Documentación
Técnica
Technical File

© 2017, www.tecno-med.es 50
Documentación Técnica – Technical File Estructura según reglamento y guias
Parte A Resumen
Parte B TODO lo demás
Parte C Plan PMS
(PMCF o PMPF) + Informes anuales
20
17
20
18
…
2 / 3
3 / 3
© 2017, www.tecno-med.es 51
ARTÍCULO:70Solicitudrealizacióndeinvestigaciónclínica(IC)
PresentaciónSolicitudIC
SistemaelectrónicoNºúnicoEU
Rev.Admin.Chequeodecontenido
SolicitudVálida
ComunicaciónAlpromotor- NOVÁLIDA
Notificaciónalpromotor- VÁLIDA
Día10(prorrogable+5)
=“fechadevalidación”
PromotorNORESPONDE
SOLICITUDEXTINTA
PromotorRESPONDEY
COMPLETALADOC.
SolicitudVálida
SOLICITUDDENEGADA
Notificaciónalpromotor- VÁLIDA
=“fechadevalidación”
5días(prorrogable
+5)
10días(prorrogable
+10)
NO
SI
Atenciónsinohayúltimodíaplazos
indicados
Proc.EMRECURSOcontrala
denegación
NO
SI

© 2017, www.tecno-med.es 52
ARTÍCULO:70Solicitudrealizacióndeinvestigaciónclínica(IC)
EvaluaciónEM
¿TodoOK?
Solicitudinformación
complementaria
AUTORIZACIÓN
PuedehaberActosDELEGADOSy
ActosdeEJECUCIÓNpararesolverdivergenciasde
interpretaciónydeaplicación
ClockSTOP
NO
SI
Art.70EVALUACIÓN45días
Prorrogables+20paraconsultaa
expertos
Art.71EVALUADORES• Noconflictodeintereses• Independientes• NºAdecuado• Experiencia• Cualificación

© 2017, www.tecno-med.es 53
Proceso de EC CONTINUADA

© 2017, www.tecno-med.es 54
Evaluación clínica – Modelo habitual de “ahora” …

© 2017, www.tecno-med.es 55
… y ACORDAOS –> La Evaluación clínica debe ser continuada en el ciclo de vida del producto…

© 2017, www.tecno-med.es 56
REPETIR

© 2017, www.tecno-med.es 57
Sistema de seguimiento poscomercialización
Plan PMS / PMCF – Métodos y Procesos a ENLAZAR
Gestión de Riesgos - Nuevos riesgos / Emergente - Cambios en perfil de riesgo - Datos que impactan BBR?
Evaluación Clínica / PMCF - Actualización CER en base a nuevos datos (frecuencia??)
PMS - PSUR - Cambios en “estado del arte” - Actualización PMS Plan?
VIGILANCIA - Notificar incidentes graves - Seguir y analizar tendencias - FSCA / FSN
Trazabilidad / UDI - Alcance definido - Proveedores / Distribuidores - Mantenimiento BBDD
RECLAMACIONES - Recepción y evaluación - Investigación - Comunicación
CAPA - Acciones correctivas / preventivas - Análisis de causas - Determinación de eficacia
Análisis de datos - Indicadores / Umbrales - Métodos estadísticos - ¿Aumento significativo?
COMUNICACIÓN - Con AASS y ON - Proveedores / Distribuidores - Usuarios

© 2017, www.tecno-med.es 58
Evaluación Clínica - Resumen
1. Situación actual
– Evaluación clínica según Directiva 93/42/CEE y MEDDEV 2.7/1
– Problemas frecuentes
2. Cambios en los conceptos
3. Transición a MDR
- Evaluación clínica según Reglamento 2017/745
- Obligación / Necesidad de Investigaciones Clínicas
- Esquema de un proceso continuado de EC - Enlace con PMS / PMCF
4. Conclusiones

© 2017, www.tecno-med.es 59
EC vs PMCF vs PMCFS vs IC Evaluación clínica vs PMCF vs Estudios PMCF vs Investigación
clínica
• Evaluación clínica EC
• Seguimiento clínico poscomercialización PMCF general
• Estudios de seguimiento clínico poscomercialización PMCF Studies
• Investigación clínica IC
TODOS LOS
PRODUCTOS
SANITARIOS
PRODUCTOS
SANITARIOS
NOVEDOSOS
IMPLANTES Y
CLASE III

© 2017, www.tecno-med.es 60
Conclusiones
Evaluación clínica: Obligación para todos los productos sanitarios AHORA.
MEDDEV 2.7/1 rev. 4 da otro salto cualitativo – La EC se ha convertido en
un proceso transversal / estratégico.
MEDDEV 2.7/1 rev.4 es el marco de referencia actual pero viene el
Reglamento y las exigencias van aún más en aumento. La revisión 4 se
publica con cierta voluntad de preparar el mercado para las exigencias
obligatorias que se aplicarán cuando se aplique el reglamento.
Las empresas se tendrán que adaptar para incorporar y sistematizar la
evaluación clínica a lo largo del ciclo de vida del producto. Los
mecanismos deberán abordar una estrategia de desarrollo clínico y una
evaluación clínica continuada en el tiempo.
¿Procedimientos y competencia para ello?
* Implicará más colaboración Empresa / Universidad / Hospitales

© 2017, www.tecno-med.es 61

© 2017, www.tecno-med.es 62
• Claire Murphy [email protected] Ingeniero Electrónico MIEMBRO SOCIEDADES
ASQ CQA – Biomedical Auditor
Socio Consultor Tecno-med Ingenieros Experto en auditorias Organismos Notificados
Experto en auditorias FDA y CMDCAS
• Xavier Canals-Riera [email protected] Euroingeniero, Ingeniero Telecom col 2912 MIEMBRO SOCIEDADES
Director Tecno-med Ingenieros Presidente Subcomité Normalización Electromedicina SC62/209
Presidente Capítulo Español Sociedad Ingeniería Biomédica IEEE EMBS
Vice presidente SEEIC Sociedad Esp.Electromedicina e Ing.Clínica
Evaluador científico de la ANEP – Ministerio de Ciencia e Innovación
Experto en auditorias de Organismos Notificados
Profesor del Master Ingeniería Biomédica de UPC-UB, UPV, UAM,
Suerte…. y GRACIAS!

© 2017, www.tecno-med.es 63
Preguntas …… ?


















