
FENÓMENOS DE SEPARACIÓN DE FASES EN … · FENÓMENOS DE SEPARACIÓN DE FASES EN ALEACIONES DE...
233
FENÓMENOS DE SEPARACIÓN DE FASES EN ALEACIONES DE POLIFLUORURO DE VINILIDENO Y POLIMETACRILATO DE METILO José Carlos Canalda Cámara Director: Dr. Javier Martínez de Salazar Bascuñana Investigador científico del CSIC Memoria presentada para optar al grado de doctor en Ciencias Químicas por la Universidad de Alcalá Alcalá de Henares, 2000
Transcript of FENÓMENOS DE SEPARACIÓN DE FASES EN … · FENÓMENOS DE SEPARACIÓN DE FASES EN ALEACIONES DE...

FENÓMENOS DE SEPARACIÓN DE FASES
EN ALEACIONES DE
POLIFLUORURO DE VINILIDENO
Y POLIMETACRILATO DE METILO
José Carlos Canalda Cámara
Director: Dr. Javier Martínez de Salazar Bascuñana
Investigador científico del CSIC
Memoria presentada para optar al grado de doctor en
Ciencias Químicas por la Universidad de Alcalá
Alcalá de Henares, 2000


A mi padre


AGRADECIMIENTOS
Deseo expresar mi agradecimiento al doctor Javier Martínez de Salazar,
investigador científico del Instituto de Estructura de la Materia (CSIC), por la dirección y
eficaz ayuda en la elaboración de este trabajo de tesis doctoral.
Al doctor Enrique Saiz por su labor de tutoría y la atención prestada durante el
período de realización del presente trabajo.
Al profesor Francisco José Baltá Calleja, director del Centro de Física Miguel
Catalán, por las facilidades prestadas durante la realización de esta tesis doctoral.
A los componentes del departamento de Física Macromolecular, del Instituto de
Estructura de la Materia, por su ayuda y colaboración.
Al departamento de Química Física de la Universidad de Alcalá, del que dependo
académicamente como alumno de doctorado.
A mi madre y a todos mis familiares y amigos.
Y muy especialmente, a Carmen.


ÍNDICE
Prólogo Pág. 1
Capítulo 1. Introducción
1.1 Mezclas compatibles de polímeros Pág. 3
1.2 Determinación de la compatibilidad de una mezcla de polímeros Pág. 6
1.2.1 Parámetro de interacción. Ecuación de Flory Pág. 6
1.2.2 La transición vítrea en polímeros puros y en mezclas compatibles de polímeros Pág. 9
1.3 Polímeros cristalinos Pág. 15
1.3.1 Grado de cristalinidad Pág. 16
1.3.2 Morfología esferulítica Pág. 17
1.3.3 Modelo laminar Pág. 18
1.4 Polifluoruro de vinilideno. Descripción y propiedades Pág. 19
1.4.1 Polimorfismo cristalino del PVDF Pág. 19
1.4.2 Transformaciones de fase en el PVDF cristalino Pág. 21
1.5 Polimetacrilato de metilo. Descripción y propiedades Pág. 23
1.6 Compatibilidad de las mezclas PVDF/PMMA Pág. 23
1.7 Objetivos Pág. 24

Capítulo 2. Materiales y técnicas experimentales
2.1 Materiales utilizados Pág. 27
2.1.1 Preparación de mezclas homogéneas de PVDF y PMMA Pág. 27
2.1.2 Preparación de monocristales de PVDF y dispersión de los mismos en una matriz de PMMA Pág. 28
2.2 Técnicas experimentales Pág. 30
2.2.1 Difracción de rayos X a ángulos altos (WASX) Pág. 31
2.2.2 Difracción de rayos X a ángulos bajos (SAXS) Pág. 32
2.2.3 Calorimetría diferencial de barrido Pág. 35
2.2.3.i Corrección de las medidas experimentales
Pág. 36
2.2.3.ii Endotermas de fusión en polímeros
Pág. 39
2.2.3.iii Transiciones vítreas en polímeros
Pág. 41
2.2.4 Microdureza. Conceptos generales Pág. 41
2.2.5 Medida de densidades por el método de flotación Pág. 43
Capítulo 3. Resultados
3.1 Efecto de la composición en la temperatura de fusión en el equilibrio Pág. 45
3.1.1 Cálculo del parámetro de interacción Pág. 51
3.2 Mezclas amorfas de PVDF y PMMA Pág. 60
3.2.1 Variación de la transición vítrea con la concentración Pág. 60
3.2.2 Incremento de calor específico en función de la concentración Pág. 64
3.2.3 Dependencia de la densidad macroscópica con la concentración Pág. 68
3.2.4 Inverstigación de la microdureza en función la concentración Pág. 71
3.3 Morfología de las mezclas de PVDF y PMMA y transiciones de fase cristalina Pág. 73
3.3.1 Dependencia con la temperatura Pág. 76
3.4 Cinética de segregación de fases en las primeras etapas del proceso. Difusión molecular Pág. 80
3.5 Cinética de segregación de fases en mezclas amorfas Pág. 82
3.5.1 Dependencia temporal Pág. 83
3.5.2 Dependencia con la temperatura Pág. 89
3.5.3 Dependencia de la transición vítrea con la concentración Pág. 94

3.6 Cinética de cristalización en mezclas de PVDF y PMMA Pág. 98
3.6.1 Cristalización isotérmica. Dependencia temporal Pág.104
3.6.2 Cristalización no isotérmica. Dependencia con la temperatura Pág.108
3.6.3 Cristalización no isotérmica. Dependencia de la entalpía de fusión con el tiempo de cambio de régimen Pág.114
3.6.4 Cristalización no isotérmica. Dependencia de los puntos de fusión con la temperatura Pág.118
3.7 Interfase cristalina. Segunda transición vítrea Pág.119
3.7.1 Evolución temporal de la interfase Pág.130
3.7.2 Dependencia con la temperatura Pág.131
3.7.3 Masa total Pág.136
Capítulo 4. Discusión de los resultados
4.1 Consideraciones generales Pág.139
4.2 Compatibilidad del sistema PVDF/PMMA. Parámetro de interacción Pág.139
4.3 Compatibilidad de las mezclas PVDF/PMMA a temperaturas inferiores a la de fusión Pág.145
4.4 Estudio de las mezclas amorfas PVDF/PMMA en estado vítreo. Dependencia con la concentración Pág.148
4.5 Cinética de segregación de fases en las primeras etapas del proceso. Difusión molecular Pág.149
4.5.1 Ecuación de William-Landel-Ferry Pág.153
4.6 Cinética de segregación y cristalización en mezclas PVDF/PMMA Pág.162
4.6.1 Transición vítrea del PMMA Pág.169
4.6.2 Punto de cambio de régimen Pág.171
4.6.3 Cinética para t<tc y Ta<TgPMMA Pág.172
4.6.4 Cinética para t>tc y Ta<TgPMMA Pág.174
4.6.5 Cinética para t<tc y Ta>TgPMMA Pág.176
4.6.6 Cinética para t>tc y Ta>TgPMMA Pág.178
4.6.7 Temperaturas de fusión Pág.179
4.7 Cinética de segregación y cristalización en mezclas con exceso de PVDF Pág.182
4.8 Naturaleza de la interfase cristalina Pág.184
4.8.1 Estudio de la interfase cristalina Pág.185
4.9 Cinética de fusión en monocristales de PVDF Pág.192

4.10 Morfología y fases cristalinas Pág.197
4.10.1 Fases cristalinas presentes en las mezclas segregadas de PVDF y PMMA Pág.198
4.11 Teorías cinéticas de cristalización Pág.203
4.12 Conclusión Pág.209
Capítulo 5. Conclusiones generales Pág.213
Bibliografía Pág.217

1
PRÓLOGO
Desde hace varias décadas, los materiales poliméricos desempeñan un papel
fundamental en la práctica totalidad de las facetas de nuestra vida. En realidad los
polímeros naturales (celulosa, algodón, lana, seda...) han sido utilizados por el hombre
desde los albores mismos de la civilización, e incluso la propia vida se sustenta asimismo
en una compleja bioquímica que tiene su soporte fundamental en polímeros orgánicos tales
como los ácidos nucleicos, las proteínas o los polisacáridos. Sin embargo, en el lenguaje
coloquial solemos identificar a los polímeros con los materiales macromoleculares de
origen artificial, a los que se les denomina habitualmente con el poco apropiado calificativo
de plásticos.
Aunque la síntesis de polímeros artificiales tuvo su origen en la segunda mitad del
siglo XIX con el descubrimiento de la baquelita, no fue sino a partir de la II Guerra
Mundial cuando la industria química comenzó a producir en grandes cantidades distintos
tipos de polímeros uno de los cuales, el polietileno, es probablemente el material
polimérico más extendido y utilizado. Habría que esperar, no obstante, hasta los años
sesenta para que los plásticos comenzaran a ser habituales en la vida cotidiana,
reemplazando en multitud de aplicaciones a otros materiales tradicionales tales como los
metales, la madera, el vidrio o las fibras textiles naturales. Hoy en día, huelga decirlo, es tal
su grado de implantación, que resultaría imposible imaginar nuestro modo de vida actual
sin ellos.
Durante las últimas décadas se han desarrollado multitud de nuevos materiales
poliméricos, cuyas propiedades y aplicaciones mejoran en mucho a las de los más antiguos.
Dentro de ellos cabe reseñar las mezclas compatibles, o aleaciones, de dos o más polímeros
distintos. Aunque las mezclas poliméricas no suelen formar, por lo general, materiales

2homogéneos a nivel molecular, en los sistemas en los que sí ocurre existe la posibilidad de
estudiar la dependencia de las propiedades de las mezclas en función de diferentes
variables, tales como la composición o las condiciones de tratamiento de las mismas, así
como, en su caso, los fenómenos de segregación molecular y cristalización de las especies
involucradas.
El objetivo de este trabajo de investigación es la profundización en el conocimiento
de la estructura y las propiedades específicas de una de las mezclas compatibles más
clásicas en la literatura científica, la formada por polifluoruro de vinilideno (PVDF) y
polimetacrilato de metilo (PMMA). En el capítulo 1 se desarrollan los conceptos generales
manejados en este trabajo, describiéndose asimismo los fundamentos termodinámicos de
las mezclas de polímeros. En el capítulo 2 se detallan los métodos de preparación de las
mezclas de PVDF y PMMA, y se describen las técnicas experimentales utilizadas para la
caracterización de las mismas. En el capítulo 3 se recogen los resultados experimentales y
por último, en el capítulo 4, se realiza la discusión de los mismos. Finalmente, se detallan
las conclusiones generales a las que ha conducido el presente trabajo de tesis doctoral.

3
CAPÍTULO 1
INTRODUCCIÓN
1.1 Mezclas compatibles de polímeros.
En toda mezcla de polímeros existe un factor importante que es preciso tener en
cuenta, la gran complejidad de su estructura molecular, que condiciona su miscibilidad o
compatibilidad. En general las mezclas de polímeros de alto peso molecular suelen
constituir sistemas incompatibles al estar confinada la miscibilidad de los componentes
únicamente a una pequeña región interfacial [Kammer, 1988; Ougizawa, 1986]; esto es
debido al hecho de que la entropía de mezcla de los polímeros es por lo general mucho
menor que la de los compuestos de bajo peso molecular como consecuencia de los elevados
grados de libertad de los que gozan las cadenas poliméricas [Tanaka, 1987].
Así pues, las mezclas compatibles de polímeros se presentan generalmente cuando
existen interacciones específicas entre los monómeros constitutivos de cada una de las
especies macromoleculares. Las propiedades macroscópicas del material resultante de la
mezcla dependerán de la naturaleza de los propios polímeros, de la proporción existente
entre ellos y de la clase de interacciones que tengan lugar entre los mismos.
Por ejemplo, la posibilidad de formación de asociaciones moleculares de tipo
dipolo o enlace de hidrógeno entre ambos polímeros favorecerá la compatibilidad del
sistema al existir una entalpía de mezcla que lo estabiliza termodinámicamente. Existen por
ello algunos pares bien conocidos de polímeros compatibles, los cuales pueden ser los dos
amorfos o uno amorfo y otro semicristalino. Mucho menos frecuente es el caso de la
cocristalización de dos polímeros cristalinos, ya que para que este fenómeno pueda tener
lugar se precisa no sólo una miscibilidad en el fundido, sino también una compatibilidad
entre las estructuras cristalinas de los constituyentes individuales [Tanaka, 1987].

4
Para que dos polímeros puedan ser considerados miscibles es necesario que formen
una mezcla homogénea a nivel molecular en un amplio rango de concentraciones y
temperaturas. Es habitual que estas mezclas tiendan a segregarse en sus dos componentes
iniciales por encima de una temperatura lo suficientemente elevada que constituye el límite
entre la fase homogénea y la región de inmiscibilidad. En esta última, los polímeros están
segregados formando dos fases distintas. A este fenómeno se le ha denominado
temperatura consoluta inferior (LCST en sus siglas inglesas) y es una magnitud física
característica de cada sistema, estando separada la región en la que existe miscibilidad de
aquélla en la que ambas fases están segregadas por una curva de tipo binodal. Dependiendo
de cómo corte la curva binodal a la curva de puntos de fusión, la miscibilidad de la mezcla
en el fundido abarcará un rango mayor o menor de concentraciones [Tanaka, 1989]. Si la
binodal no corta a la curva de puntos de fusión la miscibilidad en el fundido existirá en
todo el rango de concentraciones para temperaturas inferiores a la del mínimo de la curva
de LCST, mientras que si esta binodal se encuentra por encima de la temperatura de
descomposición de alguno de sus componentes la separación de fases no podrá ser
detectada experimentalmente de forma directa, siendo necesario recurrir a algún tipo de
mediciones indirectas que permitan salvar esta dificultad [Bernstein, 1977].
Algunos sistemas binarios muestran también una segregación de fases a bajas
temperaturas, fenómeno que recibe el nombre de temperatura consoluta superior o UCST
en sus siglas inglesas. Al contrario de lo que ocurre en el caso anterior, este
comportamiento es bastante poco común para mezclas homogéneas de polímeros de alto
peso molecular, en especial si se trata de especies muy diferentes químicamente
[Ougizawa, 1986].
En resumen, la condición necesaria para que dos polímeros sean miscibles es, al
igual que ocurre con cualquier otro sistema físico, que las interacciones entre las moléculas
de los dos componentes sean más fuertes que las existentes entre cada uno de ellos por
separado, originándose en estas circunstancias una tranferencia de cadenas poliméricas a la
interfase polímero-polímero que produce un gradiente continuo de concentraciones que
finaliza una vez alcanzada una mezcla homogénea [Freitas Siqueira, 1991].
Desde el punto de vista termodinámico se ha de cumplir la condición general de
que la energía libre de mezcla del sistema:

5
S T - H = G mixmixmix ∆∆∆ [I,1]
Sea negativa [Paul, 1978a; 1980]. Esta condición no es suficiente por sí misma
debido a que, para que exista una verdadera miscibilidad, ésta debe cumplirse en cualquier
rango de proporciones de la mezcla, ya que puede ocurrir que la variación de la entalpía de
mezcla sea negativa tan sólo para unas determinadas concentraciones y positiva para el
resto. Por lo tanto, la condición de miscibilidad total en todo el rango de concentraciones
exigirá que la variación de la entalpía de mezcla con la fracción (en volumen o en cualquier
otra unidad de medida) de alguno de los componentes sea siempre negativa, lo que implica
que la segunda derivada de esta magnitud sea mayor que cero:
0 > ]
G [ PT,2i
mix2
Φ∂∆∂ [I,2]
Tomando Φi como la fracción (normalmente en volumen) de uno cualquiera de los
componentes de la mezcla. Por su parte, podemos desglosar tanto el término entálpico
como el entrópico siguiendo el desarrollo clásico que Flory [Flory, 1941; 1942] y Huggins
[Huggins, 1941] aplicaron inicialmente a las disoluciones de polímeros basándose en la
teoría de red, y que posteriormente Scott [Scott, 1949] extendió a las mezclas. Este modelo
aplica una dependencia cuadrática de la entalpía en función de la composición del tipo de
van Laar:
2112 ννχ ΦΦ∆ 1 T R = H mix [I,3]
Siendo χ12 el parámetro de interacción entre los dos componentes, Φv1 y Φv2 las
respectivas fracciones en volumen de los mismos y R y T la constante de los gases y la
temperatura absoluta. En lo que respecta a la entropía de mezcla, este modelo considera
que la única contribución es la combinatoria, que viene dada por [Flory, 1953]:
) n
+ n
( R- = S vvmix ΦΦ
ΦΦ
∆ 22
21
1
1 lnln νν [I,4]

6
Donde n1 y n2 representan los grados de polimerización de los dos componentes de
la mezcla.
1.2 Determinación de la compatibilidad de una mezcla de polímeros.
Aunque existen diversos métodos que permiten estudiar cualitativa y
cuantitativamente la compatibilidad de una mezcla de polímeros, los más comúnmente
utilizados son la evaluación del parámetro de interacción de Flory χ12 y la determinación de
una única temperatura de transición vítrea en el sistema.
1.2.1 Parámetro de interacción. Ecuación de Flory.
Tal como quedó explicado en el apartado 1.1, las condiciones termodinámicas
necesarias para que exista compatibilidad molecular en una mezcla de polímeros son las
que vienen determinadas por las ecuaciones [I,1], [I,3] y [I,4]. Sustituyendo las dos últimas
en la primera, ésta queda convertida finalmente en:
)+n
+ n
( RT = G vv12vvmix ΦΦΦΦ
ΦΦ
∆ 2122
21
1
1 lnln χνν [I,5]
De la expresión de la ecuación [I,4] se deduce que, excepto para bajos grados de
polimerización o para valores de Φvi cercanos a cero o a uno, ambos cocientes tienden a
cero, y por lo tanto el término entrópico puede ser despreciado en la ecuación [I,5] dejando
a la entalpía de mezcla como única responsable de la variación de la energía libre del
sistema, quedando reducida entonces la ecuación a:
ΦΦ∆ 21 vv12mix RT = G χ [I,6]
Puesto que para que el proceso de mezcla sea termodinámicamente favorable es
preciso que la variación de energía libre sea negativa, y dado que de todos los términos que
aparecen en la ecuación [I,6] el único que puede adoptar valores menores de cero es el
parámetro de interacción χ12, será el signo de éste el que determine la miscibilidad o no de

7
un sistema determinado. El parámetro de interacción, que por esta razón es una de las
magnitudes físicas que permiten estudiar experimentalmente las mezclas compatibles de
polímeros, puede ser definido como el factor que resume el hecho de que la termodinámica
de la mezcla sea favorable a la misma.
El punto de partida es el modelo de Flory-Huggins [Flory, 1953] y Scott [Scott,
1949], planteado inicialmente como ya ha sido comentado para disoluciones de polímeros,
siendo válido únicamente para sistemas en equilibrio termodinámico. Más tarde Nishi y
Wang [Nishi, 1975b] aplicaron este modelo a mezclas compatibles asumiendo que en el
polímero cristalino la diferencia de energía libre por mol entre el polímero puro y la mezcla
era de:
])-(1+)-(1 ) n1-
n1 ( +
n [
VVRT = G 2
v12v122
v
1
2mix ΦΦ
Φ∆ 222ln
χ [I,7]
Donde Vi y ni son respectivamente el volumen molar y el grado de polimerización
del componente i, correspondiendo el subíndice 1 al polímero amorfo y el subíndice 2 al
cristalino. Por otro lado, la diferencia de energía libre entre la fase cristalina y el líquido
subenfriado (es decir, la fase amorfa) del mismo polímero es igual a:
ST-H = G mmmm ∆∆∆ [I,8]
Siendo ∆Hm y ∆Sm la entalpía y la entropía de fusión molar por monómero del
polímero cristalino y Tm la temperatura de fusión del mismo. Suponiendo que el cociente
∆Hm/∆Sm sea independiente de la temperatura e igual a la temperatura de fusión en el
equilibrio Tm0, la ecuación [I,8] queda convertida en:
)TT-(1 H = G 0
m
mmm ∆∆ [I,9]
En el punto de fusión del polímero cristalino la suma de la energía libre de fusión y
la energía libre de mezcla ha de ser cero, por lo que agrupando las ecuaciones [I,7] y [I,9]
se obtiene finalmente:

8
])-(1+)-)(1n1-
n1(+
n [
VHVR- =
T1-
T1 2
v12v122
v
1m
20mm
ΦΦΦ
∆ 222ln
χ [I,10]
Para altos grados de polimerización es posible despreciar los cocientes en cuyo
denominador aparece mi, con lo que la ecuación [I,10] queda reducida a:
)-(1VH
RV-=T1-
T1 2
v121m
20mm
Φ∆ 2χ [I,11]
Donde Tm y Tm0 son las temperaturas de fusión del polímero cristalino en la mezcla
y en el estado puro respectivamente. A su vez, y puesto que χ12 presenta una dependencia
con la temperatura, es conveniente definir un parámetro B independiente de la temperatura
que representa la densidad de energía de interacción característica de cada par polimérico,
de acuerdo con la siguiente expresión:
RTVB= 1
12χ [I,12]
Una vez sustituida en la ecuación [I,11] queda finalmente, tomando T = Tm:
T)-(1
HVB- = )
T1-
T1(
)-(11
m
v
m
20mmv
Φ∆Φ
2
2 [I,13]
Esta ecuación es una recta de cuya pendiente puede calcularse B y de ella, a su vez,
χ12.
La ecuación de Flory, ampliamente utilizada por distintos autores, cuenta con varias
restricciones importantes que condicionan su aplicación. La más inmediata de ellas es de
carácter cinético y viene determinada por el hecho de que las medidas experimentales no
corresponden a una situación de equilibrio termodinámico del sistema. El segundo punto a

9
tener en cuestión es el morfológico, que se debe al hecho de que la estructura y el tamaño
de los cristales poliméricos dependen de las condiciones de obtención de los mismos, al
tiempo que algunos polímeros presentan también un polimorfismo cristalino. En tercer
lugar, las técnicas experimentales utilizadas para la determinación del parámetro de
interacción provocan asimismo una gran disparidad en los valores aportados en la
bibliografía.
Por todo ello, aunque son muy numerosos los estudios publicados sobre la
compatibilidad de polímeros [Bohdanecky, 1990; Chow, 1990; Coleman, 1990; DiPaola-
Baranyi, 1982; Eguiazábal, 1987; Etxeberría, 1994; Goh, 1988; Graessley, 1993;
Hadziioannou, 1984; Ito, 1987; Jo, 1991; Kim, 1993; Lu, 1992; Morra, 1982a; Mumby,
1994; Nishi, 1975b; Paul, 1978b; Plans, 1984; Privalko, 1990; Rield, 1984; Roerdink,
1978; Wendorff, 1980], son muy importantes las discrepancias existentes entre los distintos
autores, tanto entre los que defienden la validez de la ecuación de Flory con o sin
modificaciones, como entre los que la cuestionan proponiendo otras formulaciones
alternativas.
1.2.2 La transición vítrea en polímeros puros y en mezclas compatibles de polímeros.
La transición vítrea es una transición de segundo orden que tiene lugar, en la
fracción amorfa, entre un estado en el que las moléculas tienen cierto grado de libertad de
movimientos y el estado vítreo, y está asociada a la variación de diversas magnitudes tales
como la capacidad calorífica, los coeficientes de expansión o la compresibilidad del
material. Aunque su naturaleza no está totalmente establecida, en el caso de los materiales
poliméricos puede ser asociada a movimientos de segmentos moleculares.
La transición vítrea no es un proceso exclusivamente termodinámico ya que
depende también de una cinética que, en el caso de medidas calorimétricas, se registra
como un salto en la línea base en forma de curva sigmoide [Wunderlich, 1994].
La temperatura de transición vítrea, definida como Tg, corresponde al punto de
inflexión de la sigmoide, y se define físicamente como la temperatura a la cual el cincuenta
por ciento del material ha experimentado el proceso de vitrificación o desvitrificación
[Wunderlich, 1994].
Otra magnitud importante que puede ser estudiada mediante técnicas calorimétricas
es la variación del calor específico (habitualmente a presión constante) entre el estado

10vítreo y el amorfo flexible, ∆cp, la cual corresponde experimentalmente al salto
energético comprendido entre las líneas base anterior y posterior a la transición vítrea.
Desde el punto de vista termodinámico, el calor específico cp puede ser definido como la
variación de entalpía con la temperatura a presión constante [Young, 1987]:
)TH( = c Pp ∂
∂ [I,14]
Siendo ∆cp = cpl - cp
g. En una primera aproximación que suele ser válida
habitualmente, ∆cp puede ser considerado independiente de la temperatura, pero en algunos
casos es preciso asumir una dependencia lineal [Couchman, 1984] del tipo:
T b + a = c p ∆∆∆ [I,15]
Donde ∆a y ∆b son constantes propias de cada polímero. En realidad, la
dependencia de ∆cp con la temperatura sigue un desarrollo polinómico del cual pueden ser
tomados todos los términos que resulten necesarios.
El estudio de la transición vítrea puede ser realizado también en base a la variación
de otros parámetros físicos del sistema tales como el coeficiente de expansión cúbica α,
definido como [Young, 1987]:
)TV(
V1 = P∂
∂α [I,16]
Es decir, la variación de volumen con la temperatura a presión constante. En el caso
particular de la transición vítrea, ∆α = αl - αg de forma análoga a ∆cp.
Un caso particular en el estudio de la transición vítrea son las mezclas compatibles
de polímeros. Experimentalmente se ha podido determinar que cuando estos polímeros son
compatibles a nivel molecular la mezcla presenta una transición vítrea única a una
temperatura intermedia entre las correspondientes a los dos polímeros por separado. Por el
contrario, si la mezcla no es lo suficientemente homogénea de forma que puedan existir
microdominios diferentes en el seno de la misma, el sistema presentará varias transiciones

11
vítreas diferentes pertenecientes cada una de ellas a la correspondiente fase de la mezcla.
Del estudio experimental de la transición o las transiciones vítreas de una muestra se podrá
obtener, pues, información acerca de su estructura microscópica.
En el caso de mezclas homogéneas, la transición vítrea es función de las respectivas
transiciones vítreas de los polímeros en estado puro y de la proporción de ambos en la
mezcla. Sin embargo, la dependencia con la composición de la transición vítrea de la
mezcla no es lineal debido a las interacciones intermoleculares que aparecen al mezclar los
dos polímeros, lo que obliga a formular una expresión que sea capaz de recoger estas
desviaciones de la linealidad, las cuales pueden ser positivas o negativas e incluso originar
curvas asimétricas de variación de la transición vítrea con la composición de las mezclas.
Han sido varios los autores que han estudiado esta cuestión tanto desde el punto de vista
teórico como desde el empírico, por lo que en la bibliografía se encuentran diversas
ecuaciones diferentes para este mismo fenómeno. De entre todas ellas las principales
ecuaciones empíricas, que son también las más utilizadas, son las siguientes:
-Gordon-Taylor [Gordon, 1952]:
ΦΦΦΦ
21
2211
mm
gmgmg k +
T k + T = T [I,17]
En la que Tg1 y Tg2 son las respectivas transiciones vítreas de los polímeros puros,
Φm1 y Φm2 las fracciones en peso de los dos polímeros y k un parámetro ajustable para cada
sistema.
-Jenckel-Heusch [Jenckel, 1953]:
ΦΦΦΦ 21122211 mmgggmgmg )T - T( b + T + T = T [I,18]
El término b es un parámetro ajustable relacionado con la “calidad del solvente”,
mientras el resto de las variables tienen el mismo significado que en la ecuación anterior.

12
-Fox [Fox, 1956]:
T +
T =
T1
g
m
g
m
g 2
2
1
1 ΦΦ [I,19]
Comparando esta ecuación con la anterior de Gordon-Taylor, es fácil comprobar
que se trata de un caso particular de la misma en el que el parámetro k adopta como valor el
cociente entre las transiciones vítreas de los dos polímeros en estado puro, es decir,
k = Tg1/Tg2.
-Kwei [Kwei, 1984]:
ΦΦΦΦΦΦ
2121
2211mm
mm
gmgmg q +
k + T k + T = T [I,20]
Esta última ecuación es simplemente una modificación de la de Gordon-Taylor, a la
que se le ha añadido un término cuadrático en las concentraciones para considerar las
interacciones específicas existentes en la mezcla. Usando fracciones en volumen en vez de
fracciones en peso, la ecuación de Gordon-Taylor queda convertida en una relación aditiva,
mientras la de Kwei se transforma en una expresión de virial de dos términos. Si se
introduce además un término cúbico en esta última ecuación mejora el ajuste de los puntos
experimentales, en especial en los casos en los que la curva presenta una forma asimétrica.
Las ecuaciones teóricas, por su parte, parten de consideraciones termodinámicas
tales como la aproximación termodinámica de continuidad a Tg, entropía producida por los
volúmenes de exceso, etc., y consideran en algunos casos la aproximación de la aditividad
en los volúmenes trabajando con relaciones continuas en volumen en vez de en masa. Han
sido varios los autores que han propuesto distintos tipos de expresiones [Aubin, 1988;
Braun, 1965; Brekner, 1988; Di Marzio, 1981; 1990; Gibbs, 1958; Kanig, 1963; Kelley,
1961; Kwei, 1978; Nishi, 1975a; Pochan, 1979; Riguetti, 1993; Schneider, 1988; 1992],
pero uno de los desarrollos más conocidos es el de Couchman [Couchman, 1978a; 1978b;

13
1979; 1980; 1983; 1984; 1987], que aplica la teoría entrópica a los sistemas de mezclas
poliméricas. Según esta teoría, la entropía total del sistema depende linealmente de las
entropías de los componentes puros mas la entropía de mezcla. En el caso particular de dos
componentes:
S + S + S = S mix2m1m ∆ΦΦ 21 [I,21]
Siendo Si la entropía de cada uno de los componentes, Φmi su fracción en peso y
∆Smix la entropía de mezcla, término que engloba todos los excesos de entropía
(conformacional, térmica, etc.) asociados a la mezcla de los componentes. Si llamamos Sl a
la entropía de la fase líquida y Sg a la de la fase vítrea, tendremos:
S + S + S = S lmix
l2m
l1m
l ∆ΦΦ 21 [I,22]
S + S + S = S gmix
g2m
g1m
g ∆ΦΦ 21 [I,23]
Por otro lado, al ser la transición vítrea una transición termodinámica de segundo
orden, las propiedades extensivas del sistema tales como la entropía, el volumen libre o la
entalpía serán continuas y no experimentarán variación durante el proceso. Así:
S = S gl [I,24]
Por lo que restando miembro a miembro se obtiene:
0 = S - S + )S - S( + )S - S( gmix
lmix
g2
l2m
g1
l1m ∆∆ΦΦ 21 [I,25]
A continuación puede considerarse la equivalencia termodinámica de la entropía
con la temperatura y el calor específico a presión constante:

14
0 = S - S + TdT c +
TdT c g
mixlmix
T
Tpm
T
Tpm
g
g
g
g
∆∆∆Φ∆Φ ∫∫21
2211 [I,26]
Siendo ∆cpi la diferencia de calor específico entre el amorfo flexible y el estado
vítreo para el componente i, y Tg, Tg1 y Tg2 las temperaturas de transición vítrea de la
mezcla y de los dos polímeros respectivamente. En el caso de polímeros de alto peso
molecular se puede asumir que la variación de entropía de mezcla es aproximadamente
cero, lo que simplifica la expresión anterior. Si además se considera a la diferencia de calor
específico independiente de la temperatura, la integración de la ecuación anterior dará
finalmente la expresión de Couchman:
c 2 + c T c + T c
= T pmpm
gpmgpmg
211
222111 lnlnln
∆Φ∆Φ
∆Φ∆Φ [I,27]
Desarrollando en serie los términos logarítmicos de la ecuación de Couchman y
haciendo diferentes tipos de simplificaciones se obtienen finalmente otras ecuaciones,
entre ellas las empíricas de Gordon-Taylor y Fox.
Las desviaciones de la linealidad de la ecuación de Couchman vienen dadas por los
valores relativos entre las dos Tg y las dos ∆cp de los componentes de la mezcla. Si
Tg2 > Tg1 y ∆cp2 < ∆cp1, como ocurre en la mayoría de los pares poliméricos, la curva de
dependencia de la transición vítrea con la concentración será cóncava. Si por el contrario
Tg2 > Tg1 y ∆cp2 > ∆cp1, la curva será convexa. Por último, si ∆cp1 = ∆cp2 la dependencia
será lineal con la concentración.
En lo que respecta al incremento de capacidad calorífica de la mezcla, diversos
autores [Couchman, 1980; 1984; Naito, 1978] han propuesto para la misma una expresión
lineal del tipo:
c + c = c pmpmp 2211 ∆Φ∆Φ∆ [I,28]
La cual se obtiene combinando uno de los posibles desarrollos en serie de la
ecuación logarítmica de Couchman con la primera de las reglas semiempíricas de Simha-

15
Boyer [Boyer, 1973; Simha, 1962], los cuales proponen una relación general entre ∆cp y el
incremento del coeficiente de expansión cúbica (∆α) con la temperatura de transición vítrea
del tipo:
K = T K = T c 2g1gp α∆∆ [I,29]
Correspondiendo a K1 y a K2 unos valores de 27.5 cal/g y 0.113 respectivamente.
Una mejor aproximación de las reglas de Simha-Boyer [Boyer, 1973] considera una
dependencia lineal con la temperatura del tipo:
cal/g T 104 + 15 = T c g-2
gp ⋅∆ [I,30]
T 101.0 + 0.07 = T g-4
g ⋅∆α [I,31]
Que a su vez determina una dependencia lineal de ∆cp con la temperatura:
T b + T b + a + a = c g2mg1m2m1mp ∆Φ∆ΦΦΦ∆ 2121 [I,32]
Las expresiones [I,29] son válidas para numerosos sistemas poliméricos pero no
son totalmente universales, por lo cual será preciso comprobar su validez en cada sistema
determinado antes de utilizar las ecuaciones derivadas de ellas.
1.3 Polímeros cristalinos
Aunque muchos polímeros son susceptibles de cristalizar, las características propias
de los materiales macromoleculares hacen que los cristales poliméricos se diferencien de
los cristales de los materiales no poliméricos. Una cuestión fundamental es que la longitud
de las cadenas moleculares resulta ser en los polímeros muy superior al tamaño de la
celdilla unidad del cristal, lo cual se resuelve mediante el plegado periódico de las
moléculas tal como demostraron varios autores a finales de la década de los cincuenta
[Fischer, 1957; Keller, 1957; Till, 1957].

16En la celdilla unidad de un cristal polimérico existen una o varias secciones de
las cadenas moleculares que pueden pertenecer bien a diferentes tramos de una misma
molécula, bien a moléculas distintas. Las fuerzas de cohesión que mantienen la estructura
de los cristales son interacciones de segundo orden tales como fuerzas de Van der Waals o,
si la composición química del polímero es adecuada, enlaces polares del tipo de los puentes
de hidrógeno, las cuales se rompen cuando un polímero funde o es disuelto.
Conforme a lo comentado anteriormente, la celdilla unidad de un cristal polimérico
estará determinada por los segmentos moleculares que contiene así como por la ordenación
y las posibles conformaciones de los mismos, que dependerán a su vez de la propia
estructura química del polímero y, si existe, de la tacticidad.
1.3.1 Grado de cristalinidad
Los cristales poliméricos se diferencian de otros tipos de materiales cristalinos no
sólo en lo comentado anteriormente sino también en el grado de cristalinidad, definido
como la fracción del polímero presente en estado cristalino. Mientras en las sustancias no
poliméricas la cristalinidad suele ser del 100%, en el caso de los polímeros cristalinos
nunca se llega a rebasar un determinado porcentaje de cristalinidad debido tanto a factores
cinéticos (las moléculas no tienen tiempo suficiente para desentremezclarse) como a
factores morfológicos, ya que la propia estructura molecular, la tacticidad, los
impedimentos estéricos (bucles, finales de cadena) o los defectos de cualquier tipo
(ramificaciones, entrecruzamientos, uniones cabeza-cabeza o cola-cola) impiden que la
totalidad de la cadena molecular pueda entrar en la estructura cristalina. Dependiendo de
estos factores el grado de cristalinidad de un polímero variará entre determinados límites,
razón por la que resulta más preciso hablar de polímeros semicristalinos. Aunque lo más
habitual son cristalinidades del orden de un 50%, en condiciones óptimas se puede llegar a
alcanzar unas cristalinidades máximas situadas en torno a un 85-90%.
La fracción no cristalizada de los polímeros se encuentra en estado amorfo, y desde
un punto de vista físico puede ser considerado como un líquido subenfriado, es decir, un
vidrio. Las cadenas moleculares presentes en la fracción amorfa pueden estar conectadas a
su vez con las laminillas cristalinas ya que, como ha sido comentado, tan sólo una parte de
la longitud total de las moléculas es la que entra a formar parte de los cristales.

17
1.3.2 Morfología esferulítica
Aunque la estructura microscópica de los polímeros semicristalinos puede adoptar
diferentes geometrías (fibrilares, laminares, dendríticas...) la estructura más habitual es la
esferulítica, que es la que se forma comúnmente cuando un polímero cristaliza desde el
fundido [Khoury, 1976]. Tal como indica su nombre se trata de agregados policristalinos de
geometría esférica, es decir, sin direcciones de crecimiento predominantes, formados por
cristales laminares que irradian en todas direcciones a partir de un núcleo central. El
crecimiento de las esferulitas puede tener lugar de dos maneras distintas dependiendo de la
naturaleza del núcleo central, tal como viene representado en la figura 1.1:
Figura 1.1 Morfología esferulítica [Wunderlich, 1973]
Si el núcleo inicial es heterogéneo (A) la esferulita aumenta de tamaño por
radiación de numerosas subestructuras cristalinas que crecen en todas direcciones
ramificándose periódicamente en el espacio libre existente entre las ramas principales. Por
el contrario, si el núcleo inicial es un monocristal (B) todas las ramificaciones tienen lugar
de forma sucesiva a partir de una única estructura inicial, con lo cual la parte central de la
esferulita presentará una morfología no homogénea.
Asimismo, el crecimiento libre de las esferulitas puede ser alterado por
impedimentos estéricos al entrar en contacto los frentes de crecimiento de dos o más

18esferulitas vecinas, por lo cual la geometría esférica tan sólo podrá existir, en un sentido
estricto, durante las primeras etapas del proceso de cristalización, cuando las esferulitas
todavía no han crecido lo suficiente como para chocar entre sí.
La propiedad óptica más característica de las esferulitas es su comportamiento
frente a la luz polarizada, produciendo un patrón típico de interferencia en forma de cruz de
Malta con anillos concéntricos de extinción.
1.3.3 Modelo laminar
Una cuestión importante en el estudio de la morfología de los materiales
poliméricos es la estructura interna de las esferulitas, las cuales están formadas tal como ha
sido comentado anteriormente por un conjunto de cristales laminares entre los cuales se
sitúa el material amorfo no cristalizado, dando como resultado unas estructuras periódicas
en las cuales se alternan, a modo de sandwich, las laminillas cristalinas con las regiones
amorfas. La distancia promedio entre dos laminillas cristalinas consecutivas, característica
de la estructura laminar de los materiales poliméricos, es conocida con el nombre de largo
espaciado.
La siguiente cuestión consiste en establecer la forma en la que las cadenas
moleculares se pliegan para formar las laminillas cristalinas. Puesto que en un material
polimérico existe siempre una gran cantidad de moléculas, al producirse el plegado que da
como resultado la cristalización caben dos posibilidades extremas: Plegado regular
adyacente [Keller, 1957] y plegado irregular no adyacente o al azar [Flory, 1962]. El
plegamiento real de las moléculas suele ser un caso intermedio entre estos dos extremos, y
dependiendo de las condiciones de cristalización el predominio será de uno u otro modelo:
La cristalización a partir de disoluciones diluidas facilitará el plegamiento regular al estar
dispersas las diferentes moléculas en el disolvente, mientras la cristalización a partir del
fundido, al estar las moléculas entremezcladas, conducirá a un predominio del plegamiento
irregular al azar. Por este motivo, aunque la estructura de las laminillas de las esferulitas es
en principio muy similar a la de los monocristales, en las primeras el porcentaje de
plegados al azar es muy superior al existente en estos últimos.

19
1.4 Polifluoruro de vinilideno. Descripción y propiedades.
El polifluoruro de vinilideno, conocido con las siglas PVDF, es el producto de la
polimerización del 1,1-diflúor etileno, comúnmente denominado fluoruro de vinilideno,
siendo su unidad repetitiva:
─(CH2─CF2)n─
El PVDF es un polímero lineal cuyo defecto más frecuente son las uniones cabeza-
cabeza y cola-cola que alteran la secuencia normal dando grupos de tipo -(CF2-CF2)- y
-(CH2-CH2)-, siendo habitual un porcentaje de defectos en torno al 5% del número total de
monómeros sin que las muestras que contienen una cantidad mayor de los mismos lleguen
a rebasar usualmente proporciones del 10% [Chen, 1984; Millich, 1988; Nandi, 1994].
Puesto que este polímero carece de carbonos asimétricos sus moléculas no presentan
tacticidad, pero sí propiedades polares debido a la gran diferencia de electronegatividad
existente entre los átomos de flúor y los de carbono.
1.4.1 Polimorfismo cristalino del PVDF
El PVDF es un polímero semicristalino cuyo grado de cristalinidad oscila
habitualmente en torno a un 50%, si bien dependiendo del historial térmico de la muestra
estudiada pueden llegar a alcanzarse cristalinidades del orden del 70-75% [Broadhurst,
1984; Canalda, 1995 Davis, 1978; Millich, 1988; Nandi, 1994]. Sin embargo, una de las
características más notables del polifluoruro de vinilideno es su polimorfismo cristalino,
habiéndose identificado un total de cinco fases cristalinas diferentes, denominadas
respectivamente con las letras griegas α, β, γ, δ y ε (o, en algunos casos, con ordinales
romanos) [Broadhurst, 1984; Grubb, 1981; Lovinger, 1982b; Marand, 1989; Rashmi,
1987; Yang, 1984], si bien la fase ε sólo es descrita en un caso [Lovinger, 1982b] y los
trabajos más antiguos citan únicamente a las tres primeras [Cortili, 1967a; 1967b; Miller,
1976; Prest, 1975; Takahashi, 1985; Tashiro, 1983b] o incluso solamente a dos [Boerio,
1969; Gal'Perin, 1969]. Las diferencias entre estas fases vienen determinadas por las
distintas conformaciones adoptadas por las cadenas moleculares en el proceso de plegado
de las mismas, así como por la disposición de estas cadenas en la celdilla unidad.

20Las dos estructuras principales son la α y la β o, según otros autores, la forma II y
la forma I respectivamente. Su principal diferencia macroscópica estriba en el hecho de que
la fase β presenta un momento dipolar intrínseco del que carece la α [Broadhurst, 1984;
Marand, 1989; Rashmi; 1987], propiedad que viene determinada por las distintas
estructuras de sus respectivas celdillas unidad, las cuales dependen tanto de la
conformación de las cadenas moleculares como de la disposición de éstas en la red
cristalina.
En lo que respecta a las conformaciones de las cadenas moleculares, éstas son
TGTG' en la fase α y TTTT en la β [Boerio, 1969; Broadhurst, 1984; Grubb, 1981; Prest,
1975; Takahashi, 1985], las cuales son definidas por algunos autores como cis-trans y
trans-trans [Gal'Perin, 1969] por analogía con la isomería producida por los dobles enlaces
C═C, o también como no planar y planar en zig-zag respectivamente [Boerio, 1969; Prest,
1975] en atención al desarrollo geométrico de las mismas [Grubb, 1981].
Estas cadenas se disponen a su vez de diferente manera en la celdilla unidad. Según
Geiss y Ruscher [Geiss, 1989], en la fase α las cadenas moleculares están colocadas de dos
en dos pero alternadas, lo que hace que los momentos dipolares de las dos cadenas
moleculares tengan signos opuestos y se anulen entre ellos, razón por la que esta fase no
presenta momento dipolar neto. En la fase ß, por su parte, son cinco las cadenas
moleculares dispuestas en forma paralela a lo largo del eje z en la celdilla unidad,
conservando todas ellas la misma orientación; los momentos dipolares de las cinco cadenas
moleculares, que ya de por sí son mayores por separado en la conformación TTTT que en
la TGTG', se suman al ser todos del mismo signo, lo que da como resultado el fuerte
momento dipolar neto característico de esta fase cristalina.
La tercera fase, denominada γ o III, fue propuesta por vez primera en los años
sesenta por Cortili y Zerbi [Cortili, 1967a; 1967b; Zerbi, 1965] para explicar unas
pequeñas diferencias surgidas en los espectros infrarrojos de la fase β, siendo
posteriormente estudiada y descrita por numerosos autores [Broadhurst, 1984; Grubb,
1981; Lovinger, 1982b; Marand, 1989; Miller, 1976; Prest, 1975; Rashmi, 1987;
Takahashi, 1985; Tashiro, 1983b; Weinhold, 1980; Yang, 1984]. Su estructura
conformacional, que ha sido definida como TTTGTTTG', es intermedia entre las
correspondientes a las dos anteriores, y al igual que sucede con la fase β presenta polaridad
intrínseca [Broadhurst, 1984; Grubb, 1981; Rashmi, 1987; Tashiro, 1983b] al ser la

21
distribución de las cadenas moleculares en la celdilla unidad similar a la existente en la fase
ß, con cinco cadenas paralelas con idéntica orientación [Geiss, 1989].
Las fases δ y ε, o IV y V, son respectivamente la variante polar de la fase α
[Marand, 1989; Yang, 1984] y la variante antipolar de la fase γ [Lovinger, 1982b], y no han
sido identificadas en el presente trabajo.
En lo que respecta a las constantes cristalográficas de las distintas fases cristalinas,
éstas han sido estudiadas por numerosos autores [Davis, 1978; Grubb, 1981; Lovinger,
1982b; Miller, 1976; Prest, 1975], apareciendo en la bibliografía considerables
discrepancias tanto en la magnitud de los parámetros de las correspondientes celdillas
unidad, como en la asignación de los respectivos sistemas cristalinos, ya que si bien la gran
mayoría de los trabajos publicados coinciden en definir como ortorrómbica a la fase α,
algunos autores la definen como monoclínica seudoortorrómbica, con un ángulo ß de 90°
[Morra, 1982b]. Algo similar ocurre con la fase ß, a la cual los distintos autores la
consideran bien monoclínica [Grubb, 1981], bien ortorrómbica [Gal'Perin, 1969; Miller,
1976; Morra, 1982b; Yang, 1984].
Mucho más controvertidas son las constantes cristalográficas correspondientes a la
fase γ, sobre las que existe una gran disparidad de criterios: Ortorrómbica [Grubb, 1981;
Weinhold, 1979] o monoclínica [Lovinger, 1981; Miller, 1976; Takahashi, 1980; Yang,
1984], discrepando todos estos autores a la hora de asignar no sólo el grupo cristalográfico,
sino incluso los propios parámetros de la celdilla unidad.
La fase δ, por tratarse de una variante de la fase α, cristaliza al igual que ésta en el
sistema ortorrómbico con sus mismos parámetros cristalográficos [Davis, 1978], y lo
mismo ocurre con la fase ε con respecto a la fase γ, aunque debido a su reciente
descubrimiento todavía no existe bibliografía suficiente al respecto.
En un trabajo más reciente [Weinhold, 1982] se explican las discrepancias
anteriormente expuestas con respecto a las estructuras cristalinas de las fases α y γ
postulándose la existencia de dos modificaciones cristalográficas para cada una de ellas,
dependiendo la obtención de una u otra variante del método de preparación utilizado.
1.4.2 Transformaciones de fase en el PVDF cristalino.
Dado el gran polimorfismo que presenta este polímero, la obtención de una u otra
fase cristalina dependerá fundamentalmente de las condiciones de trabajo [Geiss, 1989],

22aunque en determinadas circunstancias es posible la aparición de mezclas de varias de
ellas en distintas proporciones.
De las cinco fases conocidas la más común de todas ellas es la α, al ser la más
estable termodinámicamente. Esta fase cristalina se obtiene cuando se cristaliza el PVDF a
partir del fundido [Gal'Perin, 1969; Geiss, 1989; Grubb, 1981; Marand, 1989; Miller,
1976; Prest, 1975; Rashmi, 1987; Tashiro, 1983b; Yang, 1984] o cuando se precipita de
una disolución en acetona [Geiss, 1989], aunque algunos autores informan también de la
obtención de una mezcla de las fases α y γ en determinados rangos de temperatura de
cristalización debido a la circunstancia de que temperaturas altas cercanas ya al punto de
fusión favorecen el crecimiento de la fase γ en detrimento de la α [Marand, 1989]. Otros
trabajos informan, por último, sobre de la posibilidad de modificar la fase cristalina del
PVDF controlando las condiciones de polimerización que, en un principio, suelen producir
también la fase α [Gal'Perin, 1969; Prest, 1975].
La fase β es la segunda fase cristalina más común del PVDF y también la más
favorecida cinéticamente, y puede ser obtenida en condiciones determinadas de
temperatura o presión, por aplicación de campos eléctricos, por aplicación de altas
presiones [Jawhari, 1992] o por precipitación a partir de soluciones con disolventes
específicos [Geiss, 1989; Marand, 1989; Miller, 1976; Prest, 1975; Tashiro, 1983b],
aunque el método habitual descrito en la bibliografía es la deformación mecánica de la fase
α bien a temperatura ambiente [Grubb, 1981; Marand, 1989; Rashmi, 1987; Takahashi,
1985; Yang, 1984], bien acompañada de un tratamiento térmico simultáneo o posterior
[Gal'Perin, 1969; Takase, 1989; Tashiro, 1983b]. Más recientemente se ha informado de la
obtención de fase β no orientada a partir directamente del fundido mediante enfriamiento
ultrarrápido [Hsu, 1986; Yang, 1987] o por deformación mecánica de la fase γ [Geiss,
1989].
La obtención de la fase γ es descrita en la bibliografía en función de diferentes
métodos según los autores consultados: Por cristalización del fundido a temperatura y
presión elevadas [Geiss, 1989; Grubb, 1981; Miller, 1976; Yang, 1984] o sólo a
temperatura elevada [Marand, 1989; Prest, 1975; Yang, 1984], por evaporación de
soluciones preparadas con disolventes específicos [Geiss, 1989; Grubb, 1981; Rashmi,
1987], por tratamiento térmico a temperaturas cercanas al punto de fusión de otras fases
cristalinas [Geiss, 1989] o mediante aplicación de campos eléctricos externos [Marand,

23
1989]. Otros trabajos de investigación apuntan también que la formación de la fase γ podría
estar favorecida en muestras de PVDF de bajos pesos moleculares [Prest, 1975], o que en
determinadas condiciones lo que se obtiene en realidad no es fase γ pura sino una mezcla
de las fases α y γ.
1.5 Polimetacrilato de metilo. Descripción y propiedades.
El polimetacrilato de metilo, conocido por las siglas PMMA, es el producto de la
polimerización del éster metílico del ácido metacrílico, o metil acrílico, siendo su unidad
repetitiva:
CH3 │
─CH2─C─ │ COOCH3
En este monómero existe un carbono asimétrico (o quiral), lo que implica la
existencia de tacticidad en este polímero. Aunque las propiedades físicas del PMMA varían
mucho en función de la tacticidad, el PMMA atáctico utilizado en este trabajo se presenta
siempre en estado amorfo al no poder cristalizar por no poseer una distribución regular los
sustituyentes del carbono asimétrico.
1.6 Compatibilidad de las mezclas PVDF/PMMA
La compatibilidad a nivel molecular de las mezclas formadas por PVDF y PMMA
fue determinada por vez primera a finales de los años sesenta [Schmidt, 1969], siendo desde
entonces muy numerosos los autores que han estudiado esta compatibilidad determinando
que la misma tiene lugar en un amplio rango de concentraciones y a temperaturas inferiores
a la LCST del sistema, establecida en torno a los 330 °C [Bernstein, 1977].
En general, las mezclas de polímeros suelen ser incompatibles a nivel molecular
debido a que el proceso de mezcla acostumbra a ser termodinámicamente desfavorable en
relación con los dos polímeros por separado. Por esta razón, para que la mezcla sea posible
desde el punto de vista termodinámico es preciso que exista algún tipo de fuerzas atractivas
que puedan contribuir a que el balance energético del proceso de mezcla sea favorable al
mismo. Estas fuerzas no pueden ser las de van der Waals ya que éstas ya existen en los

24polímeros puros, por lo que habrá que buscar algún otro tipo de fuerzas atractivas
intermoleculares tales como las de tipo polar, cuyo ejemplo más frecuente son los enlaces
por puente de hidrógeno.
Si entre las moléculas de dos polímeros distintos pueden establecerse enlaces de
tipo polar, existe una alta probabilidad de que estos dos polímeros sean compatibles
[Wendorff, 1980]. Esto es lo que ocurre en el sistema formado por el PVDF y el PMMA,
siendo bastante fácil de explicar estudiando la composición química de sus respectivos
monómeros.
Por un lado, los enlaces flúor-carbono del PVDF presentan una fuerte polarización
debido a la gran diferencia de electronegatividad que existe entre ambos átomos. Por efecto
inductivo esta polarización se transmite parcialmente al esqueleto de la cadena y de allí a
los enlaces carbono-hidrógeno, dando como resultado final una polarización positiva de los
átomos de hidrógeno existentes en la molécula.
El PMMA posee grupos carbonilo en los que el oxígeno, más electronegativo que
el cabono, se encuentra polarizado negativamente. Puestos ambos polímeros en contacto se
forman puentes de hidrógeno entre los hidrógenos del PVDF y los oxígenos de los grupos
carbonilo del PMMA, como han demostrado varios autores [Coleman, 1977; Léonard,
1985; 1988; Roerdink, 1980] aplicando técnicas de espectroscopía infrarroja, así como más
recientemente [Eijkelenboom, 1992; Maas, 1991] mediante resonancia magnética nuclear,
interpretando todos ellos que la existencia de estas interacciones polares es la responsable
de la compatibilidad molecular de las mezclas de PVDF y PMMA. La magnitud de la
entalpía de enlace de estas interacciones es del orden de 1 kcal/mol [Léonard, 1985].
Varios autores [Roerdink, 1978; Sasaki, 1995] han encontrado una dependencia del
valor del parámetro de interacción con la tacticidad del PMMA, asignando todos ellos un
valor negativo al mismo, para todo el rango de concentraciones, en el caso de mezclas de
PVDF con PMMA atáctico.
1.7 Objetivos
El presente trabajo de investigación pretende contribuir con nuevas aportaciones al
estudio de las mezclas poliméricas compatibles a nivel molecular, a través del sistema
formado por el polifluoruro de vinilideno (PVDF) y el polimetacrilato de metilo (PMMA).
En este sistema hemos evaluado el nivel de compatibilidad molecular del sistema y hemos

25
estudiado la cinética de segregación de fases y su composición, junto con la cristalización
del PVDF segregado, a partir de mezclas homogéneas subenfriadas. El sistema
seleccionado ofrece una gran complejidad debido a las diferencias existentes entre las
propiedades estructurales de los dos polímeros, y su estudio mediante diferentes técnicas ha
permitido aportar nuevos datos para la elaboración del diagrama de fases del mismo.
Dentro de la problemática actual de este campo, en el que quedan todavía
numerosas preguntas por resolver, el presente trabajo de tesis ha ido encaminado a
esclarecer las siguientes cuestiones:
1.- Evaluar la compatibilidad a nivel molecular del sistema PVDF/PMMA
realizando una estimación del parámetro de interacción de Flory por medición de la
depresión del punto de fusión de monocristales de PVDF inmersos en una matriz amorfa de
PMMA.
2.- Investigar la compatibilidad del sistema en función de la dependencia de la
temperatura de transición vítrea y el incremento de la capacidad calorífica con la
concentración, a la luz de diversas aproximaciones.
3.- Examinar la compatibilidad del sistema en función de otras magnitudes físicas
tales como la densidad macroscópica o la microdureza mecánica.
4.- Estudiar la dependencia de la cristalinidad, en muestras parcialmente
cristalizadas, con la temperatura y la concentración, lo que ha permitido establecer, junto
con los estudios de los apartados anteriores, un diagrama de fases para el sistema
PVDF/PMMA.
5.- Investigar las primeras etapas de la cinética de segregación y cristalización de
mezclas inicialmente amorfas de PVDF y PMMA tratadas térmicamente por encima de la
temperatura de transición vítrea.
6.- Finalmente, estudiar la evolución de la cristalinidad del PVDF, en mezclas
inicialmente amorfas de PVDF y PMMA tratadas térmicamente por encima de la
temperatura de transición vítrea, en función de diferentes parámetros físicos tales como la
concentración, la temperatura o el tiempo de tratamiento. Ídem con mezclas con exceso de
PVDF inicialmente segregadas. Ídem una profundización en el estudio para mezclas con
una fracción en peso de PVDF de 0.5, determinadas como las más apropiadas para el
estudio del sistema.

26

27
CAPÍTULO 2
MATERIALES Y TÉCNICAS EXPERIMENTALES
2.1 Materiales utilizados.
El polifluoruro de vinilideno utilizado en este trabajo ha sido Dyflor 2000
producido por Atochem (Mw = 1.2 · 105 g/mol), y el polimetacrilato de metilo atáctico ha
sido sintetizado en el Instituto de Ciencia y Tecnología de Polímeros (CSIC) por los
doctores Madruga y San Román [Madruga, 1984] (Mw = 8.7 · 104 g/mol, Mn = 5.3 · 104
g/mol). El PVDF se encontraba como granza, mientras el PMMA se presentaba en forma
de gránulos finos.
2.1.1 Preparación de mezclas homogéneas de PVDF y PMMA.
Las mezclas homogéneas de PVDF y PMMA han sido preparadas en todos los
casos por disolución y precipitación conjunta de los dos polímeros. Por tal motivo, es
importante elegir un disolvente adecuado para ambos. Aunque son numerosos los
disolventes propuestos en la bibliografía [Bottino, 1988], no todos ellos resultan ser
apropiados para las condiciones de preparación de las muestras utilizadas en el presente
trabajo. Tras realizar varios ensayos previos con distintos disolventes, se ha elegido
finalmente la acetona debido a que es un aceptable disolvente de ambos polímeros, no los
ataca químicamente, la disolución conjunta es fácilmente precipitable evitándose
fraccionamientos indeseados y la posible acetona residual adsorbida en la superficie del
precipitado es fácilmente eliminable por calentamiento en vacío a temperaturas moderadas.
Una vez seleccionada la acetona como disolvente se han preparado disoluciones
diluidas de PVDF con una concentración de 5 g/l llevando la acetona a ebullición
(Tb = 56.2 °C) y manteniéndola a reflujo hasta alcanzar la disolución total del PVDF. Una
vez disuelto este polímero se ha añadido a la disolución el PMMA en las cantidades
necesarias para cada concentración. Tras homogeneizar suficientemente la disolución se ha
vertido ésta en un cristalizador precipitándose los polímeros con agua destilada fría. La

28rapidez y la gran cantidad de la precipitación garantizaron la homogeneidad del
precipitado.
A continuación se ha procedido a evaporar ambos líquidos, acetona y agua, en
vacío y a una temperatura moderada (T ≈ 60 °C), obteniéndose el material precipitado en
forma de un polvo fino completamente seco. De esta manera se evitan las posibles
pérdidas, tanto por filtrado como por falta de precipitación total, asegurándose que la
composición real de las mezclas coincide con la teórica. Por otro lado la volatilidad del
agua y de la acetona garantizan su completa eliminación, evitándose así los posibles
problemas creados por la retención del disolvente por adsorción del mismo en el
precipitado.
Finalmente, el material en polvo así obtenido ha sido moldeado a 190 °C y 150 bar
de presión, enfriándose las muestras en la misma prensa bajo presión de 50 bar. Las
muestras obtenidas en estas condiciones son unas láminas homogéneas de mezclas de
PVDF y PMMA con diferentes proporciones en peso. Para evitar la aparición de burbujas o
de inhomogeneidades se ha moldeado cada muestra dos veces, la primera partiendo del
polvo en forma de láminas muy finas, y la segunda a partir de esas láminas. Se han
obtenido así otras más gruesas, de 0.5 mm de espesor, las cuales han sido utilizadas
directamente en las medidas. Se ha procurado evitar en todo momento la aparición de
procesos de despolimerización del PMMA, razón por la cual no se ha elevado la
temperatura de moldeado más de lo necesario para fundir completamente las muestras y
borrar su historial térmico.
2.1.2 Preparación de monocristales de PVDF y dispersión de los mismos en una
matriz de PMMA.
Para la evaluación del valor del parámetro de interacción de las mezclas de PVDF y
PMMA se ha procedido en primer lugar a la preparación de monocristales de PVDF. En
esta ocasión la técnica utilizada para la obtención de las mezclas homogéneas descrita en el
apartado anterior no es la más apropiada, ya que se trata de PVDF puro y no de mezclas de
los dos polímeros y porque además en lugar de un precipitado rápido se busca obtener
monocristales de PVDF lo más perfectos posibles.
De nuevo es necesario seleccionar cuidadosamente el disolvente dado que la
acetona, por no ser un buen disolvente del PVDF, es ahora inapropiada. Por otro lado,

29
según informa la bibliografía [Geiss, 1989], la naturaleza del disolvente resulta ser
importante para la obtención de una u otra fase cristalina del PVDF. En este caso interesa
obtener monocristales de una única fase cristalina para evitar posibles efectos
morfológicos, y de entre todas las fases es preferible la α por ser la más estable
termodinámicamente y la mejor caracterizada.
Para la obtención de los monocristales de PVDF hemos recurrido a la técnica
descrita por Sakaoku y col. [Sakaoku, 1985] modificando las condiciones de concentración
y temperatura de cristalización, al observarse que las utilizadas por estos autores no eran las
idóneas para nuestros requerimientos. Tras varios ensayos que permitieron encontrar las
condiciones de trabajo más apropiadas, el método de preparación de los monocristales ha
sido finalmente el siguiente:
Se ha preparado una disolución de PVDF, a una concentración del 0.1% en peso, en
una mezcla de disolventes formada por ciclohexanona (Tb = 155.6 °C) y bromobenceno
(Tb = 156.4 °C) [Handbook, 1983-84] en una proporción 3:7 en volumen. Disuelto el
polímero a 130° C se ha trasladado la disolución a un baño de silicona termostatizado a
125° C, donde se ha mantenido durante 24 horas para obtener una cristalización isoterma a
una temperatura lo más cercana posible (∆T ~ 5° C) a la de disolución.
Terminada la cristalización se han separado los monocristales de PVDF del
disolvente mediante centrifugación a 3.500 rpm durante cinco minutos, procediéndose
varias veces a su lavado con acetona a temperatura ambiente hasta la eliminación total del
disolvente. Por último, los monocristales han sido secados por evaporación de la acetona a
vacío y a 50° C hasta alcanzar un peso constante.
Los monocristales de PVDF puros y secos han sido caracterizados por calorimetría
diferencial de barrido, hallándose cristalinidades del orden del 70% en todas las muestras
estudiadas. Asimismo se ha encontrado una única fase cristalina que ha sido identificada
como la α mediante microscopía electrónica. Este porcentaje de cristalinidad es muy
superior al que se obtiene habitualmente en muestras de PVDF cristalizadas desde el
fundido o desde una disolución (χ ~ 50%), y similar al obtenido por Xue y col. [Xue, 1995]
por congelación rápida con nitrógeno líquido de una disolución diluida de PVDF en
dioxano (c = 0.05% en peso) con posterior extracción del disolvente congelado con etanol,
con la ventaja con respecto al método de Xue de poderse utilizar concentraciones de PVDF
más elevadas, lo que no era posible para estos autores.

30Una vez obtenidos los monocristales de PVDF conforme al procedimiento
descrito en el párrafo anterior, han sido dispersados en una matriz de PMMA en
proporciones en peso que oscilaban entre el 10 y el 90%, a intervalos de un 10%. Para ello
se han tomado diez fracciones de monocristales de PVDF secos, a los que se ha añadido la
cantidad de PMMA correspondiente a cada proporción y finalmente se ha añadido acetona
en cantidad suficiente para disolver el PMMA calentando ligeramente para favorecer la
disolución sin llegar a la ebullición, condiciones en las que el PVDF es insoluble en
acetona. Tras dispersar con ultrasonidos los monocristales de PVDF en la disolución de
PMMA, se obtiene una suspensión homogénea. Finalmente, se deposita la
suspensión/disolución en cápsulas de aluminio evaporándose la acetona a vacío y 50° C,
quedando las muestras listas para ser estudiadas en el calorímetro diferencial.
2.2 Técnicas experimentales.
La identificación de las formas cristalinas en las mezclas de PVDF y PMMA ha
sido llevada a cabo mediante técnicas de difracción de rayos X a ángulos altos (WAXS),
mientras las primeras etapas de la cinética de segregación se han estudiado por difracción
de rayos X a ángulos bajos (SAXS). Todas las medidas de rayos X han sido realizadas con
un generador Rigaku de ánodo rotatorio modelo RU-200V y 16 kw de potencia máxima,
siendo las condiciones de trabajo de 40 kV y 160 a 200 mA respectivamente. La radiación
utilizada es la Kα del cobre (λ = 1.5405 Å), eliminándose la Kß con un filtro de níquel.
El estudio de la difracción de los rayos X a ángulos altos ha sido realizado en un
goniómetro vertical Rigaku modelo 2152R1, con geometría Bragg-Bretano, equipado con
un contador de centelleo y rendijas colimadoras de 0.5 / 0.15 / 0.5 mm. Para ángulos bajos
se ha utilizado una cámara Rigaku provista de un detector multicanal y, para medidas a alta
temperatura, una cámara Kratky con celda térmica incorporada.
Los estudios calorimétricos se han realizado con un calorímetro diferencial de
barrido (DSC) Perkin Elmer modelo DSC-4, equipado con una estación de datos Perkin
Elmer modelo 3600 y un baño termostático de circuito cerrado con metanol como fluido
refrigerante.
La microdureza ha sido estudiada con un microdurímetro Leitz modelo Durimet.
Las densidades de las muestras, por último, han sido estimadas por el método de
flotación.

31
2.2.1 Difracción de rayos X a ángulos altos (WAXS).
La difracción de rayos X es una técnica muy utilizada en la investigación de los
materiales poliméricos semicristalinos dado que permite estudiar tanto la parte cristalina de
los mismos mediante los máximos correspondientes a las reflexiones de los planos
cristalinos, como la fracción amorfa causante de la aparición de un halo continuo. De las
reflexiones de Bragg se obtiene información acerca de los parámetros estructurales de la
celdilla unidad, mientras el halo amorfo lo hace con respecto a las distancias medias
interatómicas existentes en la fracción amorfa del polímero.
La utilización de un goniómetro de las características del descrito implica la
existencia de varios factores instrumentales que afectan tanto al máximo como a la anchura
de los perfiles de difracción, por lo cual es preciso corregir los valores experimentales
considerando los siguientes errores:
-Error de absorción. Se debe a la penetración parcial del haz de rayos X en la
muestra, y corresponde a la expresión:
1)] - ( [ R 2t -
R2sen2 = )2(
sent2aθ
µ
θµ
θθexp
cos∆ [II,1]
Siendo µ el coeficiente de absorción lineal de la muestra, t el espesor de la misma y
R el radio del goniómetro.
-Error de planaridad. Corresponde a la diferencia existente entre la superficie plana
de la muestra y una superficie de radio de curvatura correspondiente al radio R del
goniómetro:
R 122 senL = ) 2 (2
2
pθθ∆ [II,2]
Siendo L la superficie iluminada por el haz de rayos X, que a su vez depende de la
apertura de la rendija de divergencia.

32
-Error de divergencia vertical. Aparece cuando se utilizan rendijas del tipo Soller,
y su expresión es:
242 = ) 2 (
2
sθδθ cot
∆ [II,3]
Siendo δ el ángulo de divergencia.
-Error de posición. Aparece cuando la superficie de la muestra no está alineada con
el eje del goniómetro. Su corrección se realiza mediante un calibrado con un material cuyas
reflexiones estén tabuladas con exactitud, empleándose habitualmente silicio.
-Error de anchura. La diferencia entre la semianchura real del pico de difracción y
la experimental puede ser corregida con un calibrado con una muestra patrón, también
normalmente silicio, y corresponde a la expresión:
βδβδβδ 2Si
22r
- = exp [II,4]
Donde δßr, δßexp y δßSi son respectivamente las semianchura real y experimental de
los picos de difracción de la muestra, y del silicio utilizado como patrón.
2.2.2 Difracción de rayos X a ángulos bajos (SAXS).
La distinción entre ángulos bajos y ángulos altos es en principio artificial, puesto
que la resolución obtenida en el detector o en la película fotográfica depende tan sólo de la
distancia existente entre la muestra y el detector; a mayor distancia mayor resolución, es
decir, ángulos más bajos.
Sin embargo, desde un punto de vista físico la información obtenida a ángulos bajos
es diferente y complementaria de la aportada a ángulos altos. La interacción de los rayos X
con materiales poliméricos semicristalinos a ángulos bajos, considerando como tales los
valores de 2θ iguales o menores de 2 - 3°, produce dos fenómenos diferentes y
superpuestos: dispersión continua y difracción discreta [Alexander, 1969]. La difracción
discreta consiste esencialmente en el mismo fenómeno que el producido a ángulos altos, si
bien de acuerdo con la ley de Bragg corresponderá a espaciados mayores que ya no son,

33
como en el caso de los ángulos altos, los planos cristalográficos definidos por la celdilla
unidad, sino que dado su orden de magnitud (decenas de nanometros) se relacionan con el
largo espaciado de los cristales poliméricos correspondiente a la distancia promedio entre
dos cristales laminares consecutivos. La dispersión continua, por el contrario, presenta un
comportamiento monótono con un valor máximo a 0° y un decrecimiento continuo de
intensidad hasta los 6 - 7°, siendo producida por la interacción entre los rayos X
dispersados por las nubes electrónicas que rodean a los núcleos atómicos. En términos
generales puede afirmarse que los fenómenos de interferencia resultan de las variaciones de
densidad electrónica existentes entre los distintos puntos del material, razón por la que los
cristales perfectos o las sustancias completamente homogéneas no dispersarán los rayos X
a ángulos muy bajos [Alexander, 1969]. Por esta razón, para el estudio de este fenómeno se
podrá prescindir en la práctica de las concentraciones electrónicas que definen las redes
cristalinas, sustituyéndolas por una distribución continua de electrones dentro de la celdilla
unidad.
La magnitud experimental relacionada con la dispersión continua es el invariante,
que se define como la integral de la radiación dispersada por la muestra y que corresponde
a las expresiones [Porod, 1951]:
ds I(s) s = Q 2
o∫∞
[II,5]
Para colimación puntual del haz de rayos X, o:
ds I(s) s = Qo∫∞
[II,6]
Para colimación lineal, siendo I la intensidad de la radiación incidente y s el vector
de la red recíproca definido como:
θλ sen2 = s [II,7]

34
Con lo cual, registrándose las variaciones del invariante en función del tiempo se
podrá estudiar la evolución de la densidad electrónica (y por consiguiente de la densidad
macroscópica) de las muestras.
Figura 2.1 Cámara de Kratky [Kakudo, 1972].
(a) Vista general. (b) Posición correcta del diafragma D3 con el borde inferior
situado en el mismo plano que la superficie D2. (c) El borde inferior del diafragma D3 está biselado para evitar la
dispersión parásita.
En el presente trabajo de investigación han sido utilizados dos tipos de cámara de
ángulos bajos. La primera de ellas es la cámara de Kratky [Kakudo, 1972] (Fig. 2.1), que es
una modificación de la cámara de ángulos bajos convencional (una cámara de Laue con la
distancia entre la muestra y la placa fotográfica incrementada) en la cual es posible alcanzar
una resolución superior a la de otras cámaras evitándose el fenómeno de la dispersión
parásita. Esta cámara se diferencia de las convencionales en el diseño de su rendija
colimadora, que tiene forma de U, en cuyos extremos descansa un diafragma. Puesto que la
superficie superior del cuerpo en forma de U, la superficie inferior del diafragma y la

35
superficie superior del “beam stop” están en el mismo plano, cuando el haz incidente
atraviesa la rendija es colimado en forma de un haz muy fino que produce dispersión sólo
por un lado ya que la disposición geométrica de la rendija y el diafragma impiden que la
radiación pase también por el otro lado, evitándose de este modo la aparición de dispersión
parásita. Este diseño permite trabajar con focos de rayos X muy finos (0.1 × 1 mm) siendo
posible alcanzar resoluciones muy altas (100 nm) libres de dispersión parásita.
La segunda cámara de ángulos bajos utilizada ha sido una cámara Rigaku, con una
distancia entre la muestra y el detector de 1 metro, en la cual estaba instalado un contador
lineal multicanal (“leti”) con una resolución de 108 µm por canal.
2.2.3 Calorimetría diferencial de barrido.
Una de las formas más habituales de estudiar los procesos termodinámicos que
tienen lugar en los polímeros es la calorimetría diferencial de barrido (DSC). Esta técnica
realiza una medida de la velocidad de absorción de calor de la muestra objeto de estudio en
relación con una referencia inerte. Ambas cápsulas son calentadas a velocidad constante y
mantenidas en todo momento a idéntica temperatura, registrándose la cantidad de calor que
es preciso suministrar o quitar a la muestra para mantener su temperatura igual a la de la
cápsula de referencia. De esta manera, conociendo la velocidad de calentamiento se obtiene
un registro de la absorción o el desprendimiento de calor de la muestra en función de la
temperatura durante todo el proceso.
El calorímetro ha sido utilizado bajo purga constante de gas inerte, habitualmente
nitrógeno aunque en algunas ocasiones, cuando era preciso subenfriar rápidamente las
muestras, fue sustituido por helio refrigerado en un serpentín con nitrógeno líquido. La
refrigeración del horno del calorímetro, necesaria para trabajar a temperaturas inferiores a
la del ambiente, ha sido realizada con un baño termostático externo utilizando como líquido
refrigerante metanol, lo que nos ha permitido alcanzar temperaturas del orden de
-80 °C. Los tratamientos térmicos de las muestras han sido realizados en el mismo
calorímetro antes de efectuar el barrido. Los márgenes de precisión instrumentales han sido
estimados en ± 0.1 K para la temperatura y en ± 0.1 cal/g para la entalpía.

36
2.2.3.i Corrección de las medidas experimentales
La endoterma de fusión de un material en el DSC es afectada por una serie de
factores tales como la masa de la muestra, la velocidad de barrido, la transferencia de calor
entre los pocillos del horno y la muestra o el flujo del gas inerte utilizado como purga.
Asimismo, a la hora de estudiar los termogramas experimentales es preciso tener en cuenta
un factor importante, el retraso térmico, que puede ser definido en una primera
aproximación como la diferencia existente entre la temperatura de fusión real y la
experimental.
En la práctica, los procesos experimentales de fusión se presentan en forma de pico
endotérmico con una amplitud y una anchura determinadas que dependen tanto de factores
instrumentales como de la naturaleza de la muestra estudiada. Debido a la existencia de un
intervalo de temperaturas en el cual tiene lugar el proceso, es preciso definir con precisión
la temperatura de fusión, que en el caso de una sustancia pura no corresponde al máximo
del pico sino al inicio del mismo (Tin), definido geométricamente por la intersección de la
tangente de la curva de fusión con la línea base.
El valor experimental de Tin es a su vez función de la velocidad de barrido del
calorímetro, conforme a la expresión:
C + v K = T - T rn i ⋅ [II,8]
Siendo Tr la temperatura de fusión real de la muestra, v la velocidad de barrido en
grados Kelvin por minuto y K y C dos constantes instrumentales. El calibrado con indio del
calorímetro nos ha dado el resultado que viene reflejado en la figura 2.2:
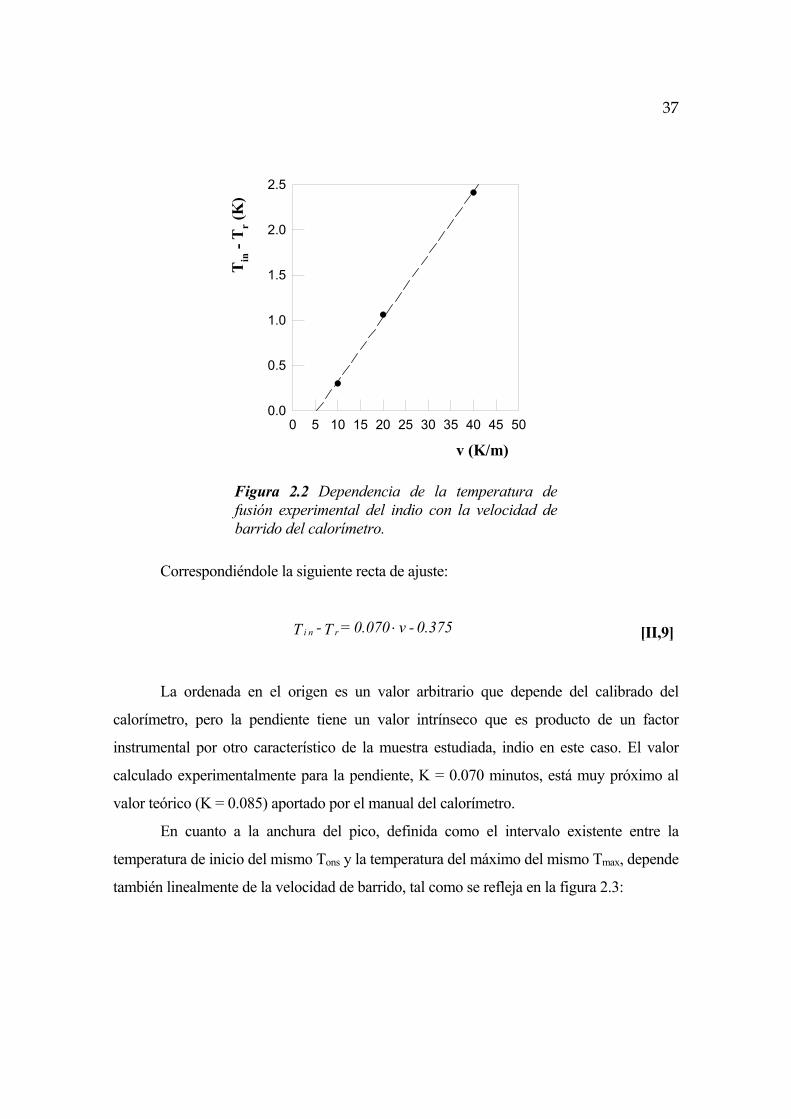
37
Tin
- T
r (K
)
0 5 10 15 20 25 30 35 40 45 500.0
0.5
1.0
1.5
2.0
2.5
v (K/m)
Figura 2.2 Dependencia de la temperatura de fusión experimental del indio con la velocidad de barrido del calorímetro.
Correspondiéndole la siguiente recta de ajuste:
0.375 - v0.070 = T - T rn i ⋅ [II,9]
La ordenada en el origen es un valor arbitrario que depende del calibrado del
calorímetro, pero la pendiente tiene un valor intrínseco que es producto de un factor
instrumental por otro característico de la muestra estudiada, indio en este caso. El valor
calculado experimentalmente para la pendiente, K = 0.070 minutos, está muy próximo al
valor teórico (K = 0.085) aportado por el manual del calorímetro.
En cuanto a la anchura del pico, definida como el intervalo existente entre la
temperatura de inicio del mismo Tons y la temperatura del máximo del mismo Tmax, depende
también linealmente de la velocidad de barrido, tal como se refleja en la figura 2.3:
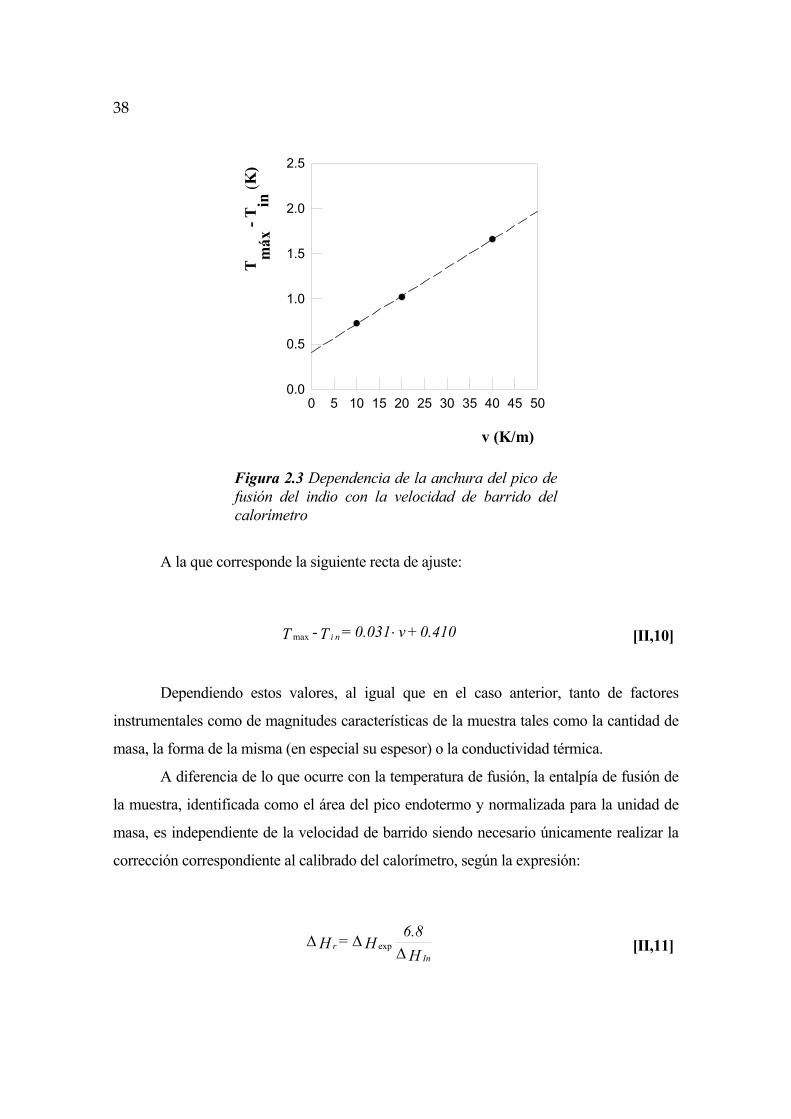
38
Tm
áx -
Tin
(K)
0 5 10 15 20 25 30 35 40 45 500.0
0.5
1.0
1.5
2.0
2.5
v (K/m)
Figura 2.3 Dependencia de la anchura del pico de fusión del indio con la velocidad de barrido del calorímetro
A la que corresponde la siguiente recta de ajuste:
0.410 + v0.031 = T - T n i ⋅max [II,10]
Dependiendo estos valores, al igual que en el caso anterior, tanto de factores
instrumentales como de magnitudes características de la muestra tales como la cantidad de
masa, la forma de la misma (en especial su espesor) o la conductividad térmica.
A diferencia de lo que ocurre con la temperatura de fusión, la entalpía de fusión de
la muestra, identificada como el área del pico endotermo y normalizada para la unidad de
masa, es independiente de la velocidad de barrido siendo necesario únicamente realizar la
corrección correspondiente al calibrado del calorímetro, según la expresión:
H6.8 H = H
Inr ∆
∆∆ exp [II,11]
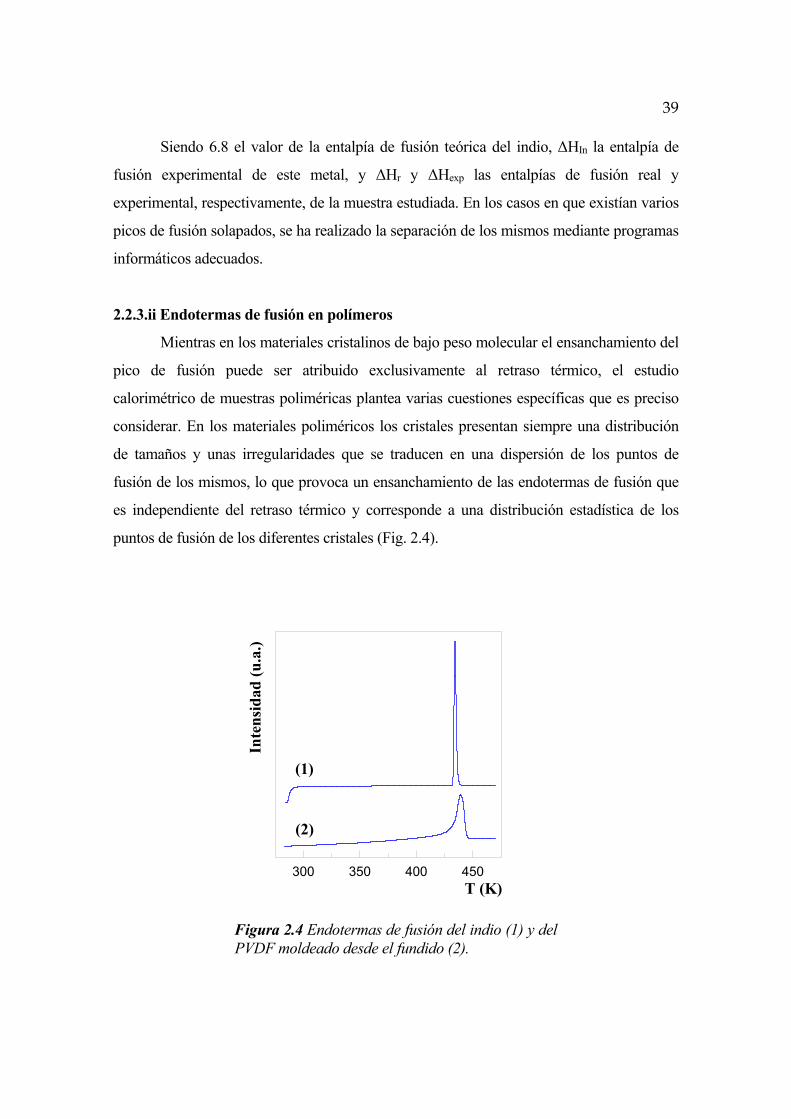
39
Siendo 6.8 el valor de la entalpía de fusión teórica del indio, ∆HIn la entalpía de
fusión experimental de este metal, y ∆Hr y ∆Hexp las entalpías de fusión real y
experimental, respectivamente, de la muestra estudiada. En los casos en que existían varios
picos de fusión solapados, se ha realizado la separación de los mismos mediante programas
informáticos adecuados.
2.2.3.ii Endotermas de fusión en polímeros
Mientras en los materiales cristalinos de bajo peso molecular el ensanchamiento del
pico de fusión puede ser atribuido exclusivamente al retraso térmico, el estudio
calorimétrico de muestras poliméricas plantea varias cuestiones específicas que es preciso
considerar. En los materiales poliméricos los cristales presentan siempre una distribución
de tamaños y unas irregularidades que se traducen en una dispersión de los puntos de
fusión de los mismos, lo que provoca un ensanchamiento de las endotermas de fusión que
es independiente del retraso térmico y corresponde a una distribución estadística de los
puntos de fusión de los diferentes cristales (Fig. 2.4).
300 350 400 450
Inte
nsid
ad (u
.a.)
T (K)
(2)
(1)
Figura 2.4 Endotermas de fusión del indio (1) y del PVDF moldeado desde el fundido (2).

40Estos puntos de fusión dependen del espesor laminar de los cristales conforme a
la conocida expresión de Thomson-Gibbs:
) l H
2- 1 ( T = (l)T eomm ∆
σ [II,12]
Donde Tm(l) es la temperatura de fusión para un cristal de espesor l, Tmo la
temperatura de fusión en el equilibrio para un cristal de espesor infinito, σe la energía
superficial de plegado y ∆H la entalpía de fusión del cristal. Por este motivo los criterios
seguidos para el cálculo de la temperatura de fusión del indio ya no son válidos aquí, ya
que al tratarse de una distribución estadística de los diferentes puntos de fusión resulta más
adecuado tomar el valor promedio de la misma (es decir, el máximo) en lugar del inicio de
la endoterma (“onset”).
Otro factor a tener en cuenta es la baja conductividad térmica de los polímeros, que
hace que la transferencia de calor entre la superficie de la muestra y su interior sea lenta,
tanto más cuanto mayor sea el espesor de la misma. Este gradiente de temperatura hace que
no todo el material funda al mismo tiempo, lo que provoca un ensanchamiento de la
endoterma de fusión. Por esta razón, resulta conveniente utilizar cantidades reducidas de
muestra (no más de unos 5 mg.) y que ésta sea homogénea y tenga el menor espesor
posible.
La velocidad de barrido es otro factor a considerar. Una velocidad baja puede
producir durante el barrido modificaciones morfológicas en la muestra tales como
recristalizaciones o engrosamiento de los cristales preexistentes. Una velocidad demasiado
alta evita estos problemas, pero incrementa el error producido por el retraso térmico y
provoca una pérdida de sensibilidad. Por ello, es necesario un compromiso. En general las
velocidades de barrido habituales suelen ser del orden de los 10 - 20 kelvin por minuto,
pero en el estudio de las mezclas de PVDF y PMMA fue preciso utilizar una velocidad
mayor
(40 K/min.) para evitar que se produjera la segregación de los dos componentes de la
mezcla o una recristalización del PVDF preexistente.
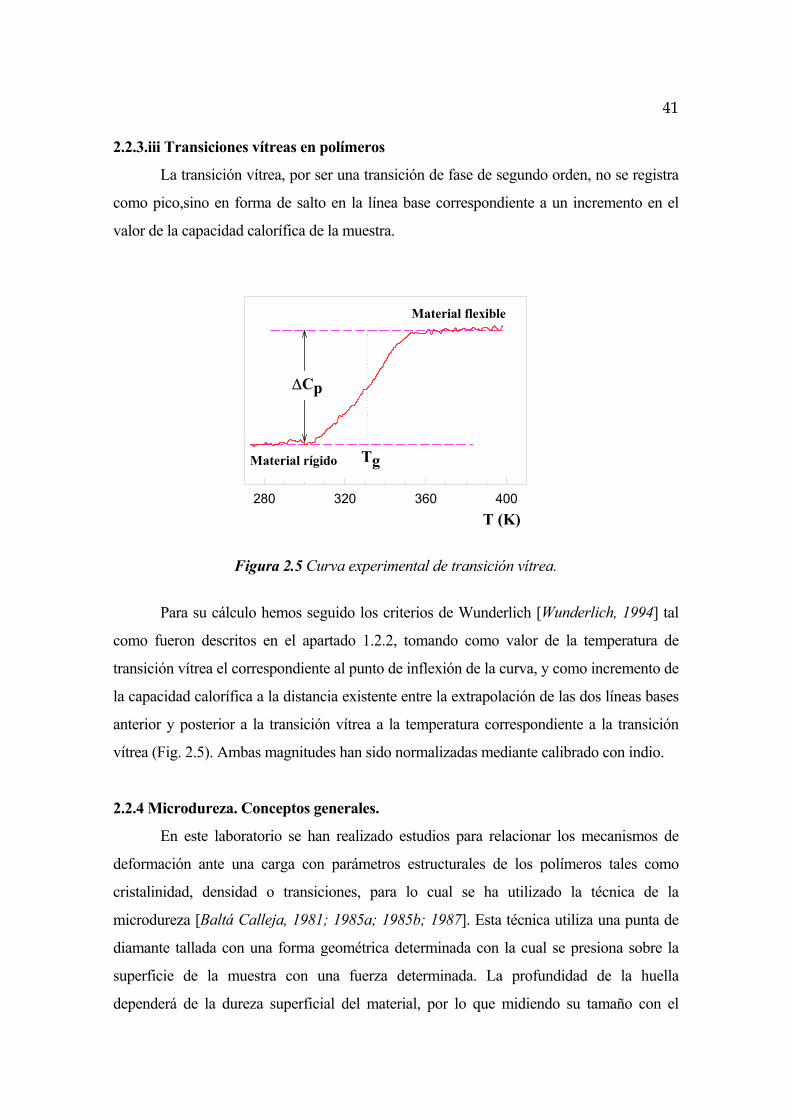
41
2.2.3.iii Transiciones vítreas en polímeros
La transición vítrea, por ser una transición de fase de segundo orden, no se registra
como pico,sino en forma de salto en la línea base correspondiente a un incremento en el
valor de la capacidad calorífica de la muestra.
280 320 360 400
Material flexible
Material rígido Tg
T (K)
∆Cp
Figura 2.5 Curva experimental de transición vítrea.
Para su cálculo hemos seguido los criterios de Wunderlich [Wunderlich, 1994] tal
como fueron descritos en el apartado 1.2.2, tomando como valor de la temperatura de
transición vítrea el correspondiente al punto de inflexión de la curva, y como incremento de
la capacidad calorífica a la distancia existente entre la extrapolación de las dos líneas bases
anterior y posterior a la transición vítrea a la temperatura correspondiente a la transición
vítrea (Fig. 2.5). Ambas magnitudes han sido normalizadas mediante calibrado con indio.
2.2.4 Microdureza. Conceptos generales.
En este laboratorio se han realizado estudios para relacionar los mecanismos de
deformación ante una carga con parámetros estructurales de los polímeros tales como
cristalinidad, densidad o transiciones, para lo cual se ha utilizado la técnica de la
microdureza [Baltá Calleja, 1981; 1985a; 1985b; 1987]. Esta técnica utiliza una punta de
diamante tallada con una forma geométrica determinada con la cual se presiona sobre la
superficie de la muestra con una fuerza determinada. La profundidad de la huella
dependerá de la dureza superficial del material, por lo que midiendo su tamaño con el

42sistema óptico que el microdurímetro lleva incorporado, será posible evaluar la
microdureza de la muestra investigada.
Para que las medidas de microdureza tengan validez ha de cumplirse la condición
de que la penetración máxima del diamante no rebase el límite de 1/10 del espesor total de
la muestra, ya que de ser mayor se producirían desviaciones que harían errónea a la
medida.
Aunque existen varios diseños diferentes de las cabezas del microdurímetro el más
usual es el Vickers, el cual consiste en una pirámide de base cuadrada. Puesto que resulta
más sencillo medir la diagonal de la huella que la profundidad de la misma, el tallado del
diamante se realiza respetando unas proporciones determinadas de forma que permita
establecer una relación matemática sencilla entre la diagonal de la huella y la microdureza
del material objeto de estudio, razón por la que es necesario que la incidencia del diamante
tenga lugar de forma perpendicular a la superficie de la muestra, ya que de no ser así la
huella queda deformada falseándose con ello la medida.
En el caso de una cabeza Vickers, que ha sido la utilizada en el presente trabajo, la
relación entre la diagonal de la huella y la profundidad de la misma es de 7 a 1, lo que en la
práctica limita el tamaño de la huella a las dos terceras partes del espesor de la muestra.
Teniendo en cuenta esta relación geométrica, la microdureza Vickers, que viene expresada
en unidades de presión, corresponde a la fórmula:
dP 1854 = HV 2 [II,13]
Donde P es la fuerza de ensayo en gramos, d la longitud de la diagonal en micras y
HV viene expresada en kg/mm2. Poniendo la expresión anterior en unidades del sistema
internacional:
dP 186 18 = HV 2 [II,14]
Con P y d en las mismas unidades y HV en Mpa.

43
2.2.5 Medida de densidades por el método de flotación.
La gran diferencia existente entre las densidades macroscópicas del PVDF
(ρ ≈ 1.8 g/cm3 para el material semicristalino) y el PMMA (ρ ≈ 1.2 g/cm3) hace que la
densidad de sus mezclas sea muy sensible a la concentración de las mismas, siendo su
medida una técnica complementaria a la calorimetría y a la difracción de rayos X.
El método elegido para la medición de las densidades ha sido el de flotación,
realizado con mezclas de dos líquidos que deben cumplir las siguientes condiciones: Ser
completamente miscibles, no atacar ni física ni químicamente a las muestras, y poseer unas
densidades mayor y menor respectivamente que las de las muestras.
Los líquidos empleados han sido el ciclohexano (ρ = 0.774 g/cm3) y el tetracloruro
de carbono (ρ = 1.584 g/cm3). La alta densidad del PVDF, y por lo tanto de sus mezclas,
supone un límite difícil de rebasar al no ser fácil encontrar líquidos a temperatura ambiente
con densidades superiores a la del PVDF; de hecho, el tetracloruro de carbono es la mejor
opción que hemos podido encontrar. Al ser la densidad de este producto inferior a la del
PVDF no ha sido posible utilizarlo para medir la densidad de este polímero puro ni de las
mezclas muy ricas en el mismo, pero resulta apropiado para el estudio de las densidades de
mezclas de concentraciones medias (ρb = 1.38 g/cm3 para un 50% de riqueza en peso),
pudiéndose extender el intervalo de medida hasta mezclas con un 75% de riqueza en peso
de PVDF
(ρb ≈ 1.55 g/cm3).
Una vez obtenida una mezcla de los dos líquidos de densidad similar a la de la
muestra, hemos aplicado la siguiente ecuación para calcularla:
ΦΦ 21 v2v1b + = ρρρ [II,15]
Siendo Φvi las respectivas fracciones en volumen de los dos líquidos. En esta
expresión no han sido considerados los posibles efectos de los volúmenes de exceso, por
tratarse de una perturbación irrelevante a esta escala.

44

45
CAPÍTULO 3
RESULTADOS
3.1 Influencia de la composición en la temperatura de fusión en el equilibrio.
Las muestras formadas por monocristales de PVDF dispersos en una matriz amorfa
de PMMA, preparadas tal como fue descrito en el capítulo anterior, han sido estudiadas por
calorimetría diferencial en un rango de concentraciones en peso de PVDF (Φm)
comprendido entre el PVDF puro (100%) y un 10% de este polímero, a las velocidades de
barrido de 10, 20 y 40 K/min.
0.0 0.2 0.4 0.6 0.8 1.00
5
10
15
∆H
(cal
/g)
Φm PVDF
40 K/min20 K/min10 K/min
Figura 3.1 Dependencia de la entalpía de fusión de los monocristales de PVDF con la proporción de la mezcla.

46
La figura 3.1 muestra la variación de la entalpía de fusión en las mezclas en función
de la proporción de PVDF, la cual se ajusta a una línea recta. Ello confirma que el método
de preparación de las muestras respeta la proporción correcta de los componentes de la
mezcla. Los valores de Tm han sido calculados, para las tres velocidades de barrido, a partir
de las curvas de DSC.
Por el contrario, la forma de los picos de fusión en el PVDF puro demuestra
depender fuertemente de la velocidad de barrido. Mientras a velocidades altas de barrido
(40 K/min) se aprecia un único pico de fusión, a velocidades más moderadas (10 y 20
K/min) se observa la aparición de un hombro correspondiente a un segundo pico de fusión
de temperatura más elevada (figura 3.2 A).
(B)
(A)PVDF
80:20
90:10
PVDF
10 K/min
cal/g
K (u
nida
des a
rbitr
aria
s)
20 K/min
T (K)
40 K/min
10 K/min
360 380 400 420 440
Figura 3.2 Dependencia de la recristalización de los monocristales de PVDF con la velocidad de barrido (A) y con la proporción de mezcla con el PMMA (B).

47
La adición de PMMA y la consiguiente interacción de sus moléculas con los
monocristales de PVDF produce una importante modificación en la cinética de fusión de
los monocristales de PVDF. Aun a una velocidad de barrido baja (10 K/min) la presencia
de PMMA hace disminuir significativamente la intensidad de este pico endotermo de alta
temperatura, el cual ya apenas se aprecia con una concentración del 20% en peso de
PMMA y desaparece por completo a partir de un 50% en peso de PMMA (figura 3.2 B).
Estos resultados experimentales confirman que las moléculas de PMMA están mezcladas
homogéneamente con los cristales de PVDF, inhibiendo la reordenación de los mismos.
Asimismo, a proporciones medias (0.5 ≤ Φm ≤ 0.7) y a todas las velocidades de
barrido utilizadas se detecta también otro pico endotermo (figura 3.3) que aparece a
temperaturas inferiores a las correspondientes al pico de fusión principal y no superiores,
como ocurría en el caso anterior.
10 K/min
20 K/min
40 K/min
350 375 400 425 450
cal/g
K (u
nida
des a
rbitr
aria
s)
T (K)
Figura 3.3 Endotermas de fusión de los monocristales de PVDF en una mezcla 50:50 con PMMA.
El comportamiento de los máximos de los dos picos secundarios es muy diferente
tal como se refleja en la figura 3.4. Mientras el de alta temperatura mantiene (para v = 10

48K/min) un valor prácticamente constante, el de baja temperatura muestra, a todas las
velocidades estudiadas, una fuerte dependencia con la proporción de PVDF existente en la
mezcla. Extrapolando los valores experimentales de este último a Φm = 1 se obtiene para la
temperatura de fusión un valor de 459.7 K, muy cercano al estimado (como se verá más
adelante) para la temperatura de fusión en el equilibrio de los cristales de PVDF
(Tmo = 458 K) y superior en unos veinte grados kelvin al valor (Tm = 439 K) de los puntos
de fusión del pico de alta temperatura.
Resulta interesante constatar que la proporción a la que tiene lugar el corte de las
dos curvas (Φm ≈ 0.7) es aquélla a la cual los máximos de los dos picos secundarios
coinciden no sólo entre sí, sino también con el valor del punto de fusión principal, que
como se verá más adelante experimenta un suave incremento al aumentar la proporción de
PVDF presente en el sistema.
40 K/min20 K/min10 K/min
0.0 0.2 0.4 0.6 0.8 1.0410
420
430
440
450
Φm PVDF
Tm
(K)
Figura 3.4 Puntos de fusión secundarios de los monocristales de PVDF en función de la cantidad de PMMA presente en la mezcla.
Una información complementaria puede ser obtenida a partir de la proporción de
las áreas de los dos picos secundarios en relación con el área total de la endoterma. El

49
resultado viene reflejado en las figuras 3.5 y 3.6, donde han sido representados los valores
correspondientes a cada uno de los picos secundarios para todas las velocidades de barrido
utilizadas.
Como puede comprobarse en la figura 3.5, el pico de baja temperatura experimenta
un crecimiento muy rápido para Φm = 0.5, tanto mayor cuanto mayor es la velocidad de
barrido, alcanzando un máximo aproximadamente para Φm = 0.6 antes de decrecer de
nuevo, desapareciendo totalmente para concentraciones más elevadas del orden de
Φm = 0.7. A pesar de lo estrecho del intervalo en el que tiene lugar la aparición de este pico,
su porcentaje sobre el área total llega a alcanzar unos valores bastante considerables,
superiores incluso al 40%.
0.2 0.4 0.6 0.8 1.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Φm
PVDF
∆H
/ ∆H
tot
40 K/min20 K/min10 K/min
Figura 3.5 Porcentaje sobre el área total del área del pico secundario de baja temperatura de los monocristales de PVDF en función de la cantidad de PMMA.
La figura 3.6 muestra, por su parte, el comportamiento del pico de alta temperatura
también a diferentes velocidades de barrido, aunque en esta ocasión únicamente se detecta
para v ≤ 20 K/min. Como puede apreciarse, mientras para v = 20 K/min el pico tan sólo
aparece a proporciones muy elevadas de PVDF (v ≥ 0.8) creciendo muy rápidamente en
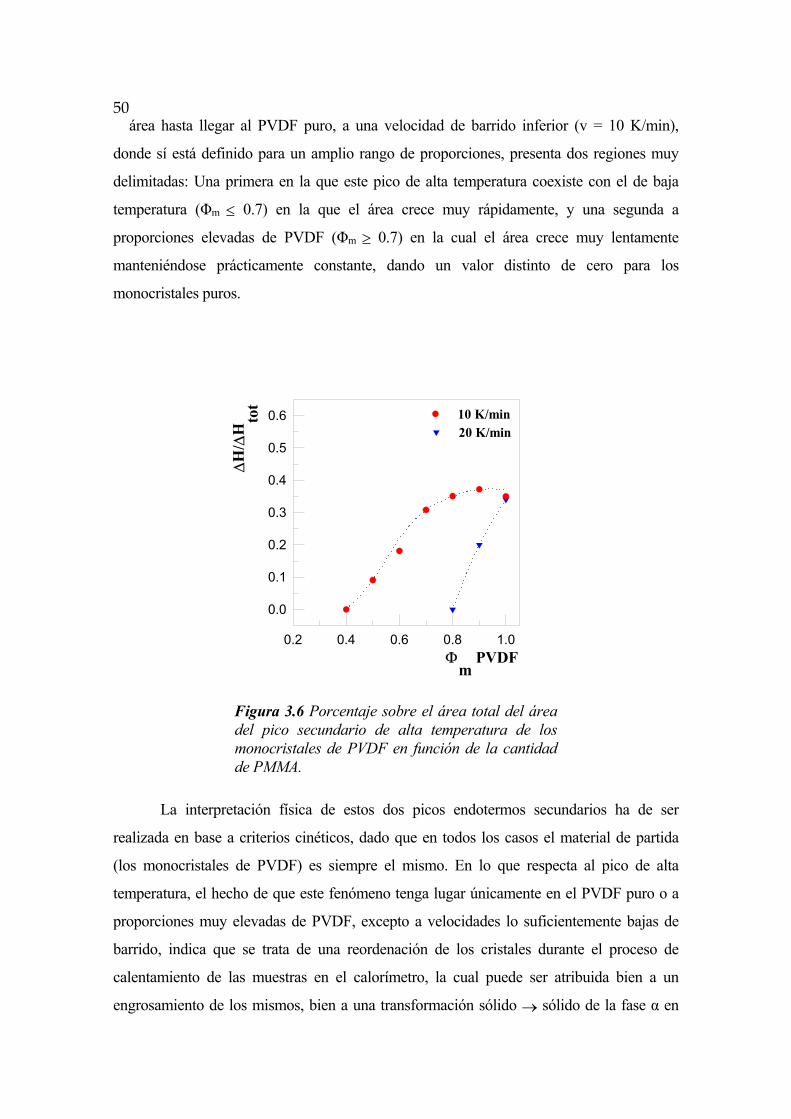
50área hasta llegar al PVDF puro, a una velocidad de barrido inferior (v = 10 K/min),
donde sí está definido para un amplio rango de proporciones, presenta dos regiones muy
delimitadas: Una primera en la que este pico de alta temperatura coexiste con el de baja
temperatura (Φm ≤ 0.7) en la que el área crece muy rápidamente, y una segunda a
proporciones elevadas de PVDF (Φm ≥ 0.7) en la cual el área crece muy lentamente
manteniéndose prácticamente constante, dando un valor distinto de cero para los
monocristales puros.
0.2 0.4 0.6 0.8 1.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Φm
PVDF
∆H
/ ∆H
tot
20 K/min10 K/min
Figura 3.6 Porcentaje sobre el área total del área del pico secundario de alta temperatura de los monocristales de PVDF en función de la cantidad de PMMA.
La interpretación física de estos dos picos endotermos secundarios ha de ser
realizada en base a criterios cinéticos, dado que en todos los casos el material de partida
(los monocristales de PVDF) es siempre el mismo. En lo que respecta al pico de alta
temperatura, el hecho de que este fenómeno tenga lugar únicamente en el PVDF puro o a
proporciones muy elevadas de PVDF, excepto a velocidades lo suficientemente bajas de
barrido, indica que se trata de una reordenación de los cristales durante el proceso de
calentamiento de las muestras en el calorímetro, la cual puede ser atribuida bien a un
engrosamiento de los mismos, bien a una transformación sólido → sólido de la fase α en

51
fase γ, la última de las cuales es estable a altas temperaturas [Lovinger, 1980]. Este
fenómeno de reordenación cristalina es bien conocido en los polímeros semicristalinos
[Wunderlich, 1980], siendo más pronunciado en los monocristales de pequeño tamaño.
Puesto que la utilización de velocidades de calentamiento menores o iguales a 20
K/min provoca la aparición de esta reordenación cristalina, y dado que ésta supone una
dificultad para el estudio de la influencia de la matriz de PMMA en la fusión de los
monocristales al existir factores morfológicos de difícil cuantificación con técnicas de
calorimetría diferencial [Rim, 1984], resulta conveniente seleccionar una velocidad de
barrido lo suficientemente elevada (40 K/min) para evitar recristalizaciones durante el
proceso.
En lo que respecta al pico de baja temperatura, éste no depende de la velocidad de
barrido, existiendo únicamente en muestras con proporciones medias de PVDF. Puesto que
además aparece antes de la fusión de los monocristales, es preciso descartar la existencia de
una transición de fase similar a la del caso anterior, pudiendo asumirse como hipótesis de
partida, que será desarrollada en el siguiente capítulo, que este pico endotermo pueda ser
producido por algún tipo de interacción entre la interfase que rodea a los monocristales de
PVDF (aproximadamente un 30% en peso de la masa total de este polímero) y la matriz de
PMMA en la que éstos están dispersos.
Finalmente, es preciso tener en cuenta también que el comportamiento del sistema
formado por los monocristales de PVDF dispersos en una matriz de PMMA presenta
analogías con el encontrado, como se verá más adelante, en muestras homogéneas
parcialmente segregadas, lo que nos permitirá en su momento realizar comparaciones entre
dos sistemas que en principio son muy diferentes.
3.1.1 Cálculo del parámetro de interacción.
Para el cálculo del parámetro de interacción del sistema PVDF/PMMA es necesario
conocer la temperatura de fusión en el equilibrio de los cristales de PVDF (Tmo). Este valor
se obtiene a partir de la ecuación:
) lH
2 - 1 ( T = Tm
e0mm ∆
σ [III,1]
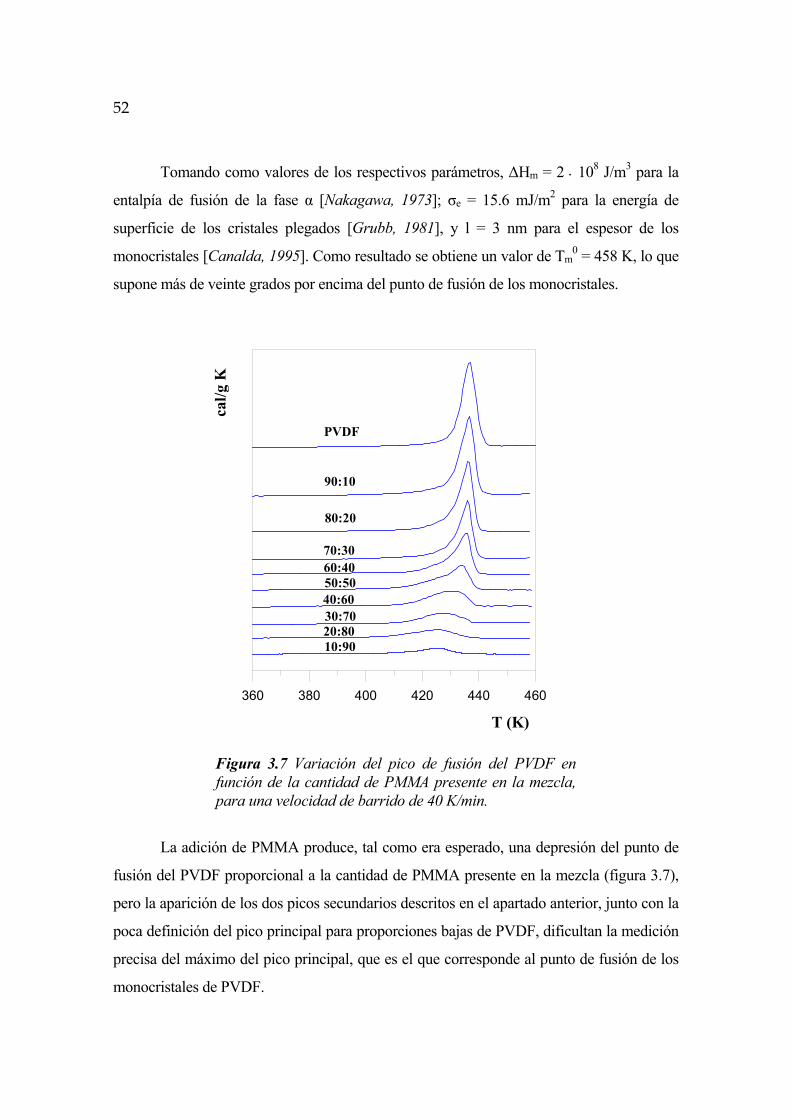
52
Tomando como valores de los respectivos parámetros, ∆Hm = 2 ⋅ 108 J/m3 para la
entalpía de fusión de la fase α [Nakagawa, 1973]; σe = 15.6 mJ/m2 para la energía de
superficie de los cristales plegados [Grubb, 1981], y l = 3 nm para el espesor de los
monocristales [Canalda, 1995]. Como resultado se obtiene un valor de Tm0 = 458 K, lo que
supone más de veinte grados por encima del punto de fusión de los monocristales.
T (K)
360 380 400 420 440 460
cal
/g K
10:9020:8030:7040:6050:5060:4070:30
80:20
90:10
PVDF
Figura 3.7 Variación del pico de fusión del PVDF en función de la cantidad de PMMA presente en la mezcla, para una velocidad de barrido de 40 K/min.
La adición de PMMA produce, tal como era esperado, una depresión del punto de
fusión del PVDF proporcional a la cantidad de PMMA presente en la mezcla (figura 3.7),
pero la aparición de los dos picos secundarios descritos en el apartado anterior, junto con la
poca definición del pico principal para proporciones bajas de PVDF, dificultan la medición
precisa del máximo del pico principal, que es el que corresponde al punto de fusión de los
monocristales de PVDF.

53
Por ello resulta conveniente realizar un estudio no sólo de los valores máximos del
pico de fusión (Tmáx), sino también de otros parámetros relacionados con éste tales como la
temperatura inicial de fusión (Tin), la temperatura final de fusión (Tfin) y el valor promedio
de la distribución estadística del área del pico de fusión, definida como:
c c T = >T<pi
pii
∆Σ∆Σ
[III,2]
Los valores obtenidos para las diversas concentraciones en peso del PVDF, en
función de las distintas velocidades de barrido, están recogidos en las tablas siguientes:
Φm Tin <T> Tmáx Tfin
0.1 415.8 425.0 427.0 437.8
0.2 415.0 425.1 427.4 442.6
0.3 416.8 427.7 428.5 441.9
0.4 418.7 429.9 432.7 443.3
0.5 429.5 430.8 435.1 444.7
0.6 431.4 433.9 437.7 445.5
0.7 433.6 435.4 437.8 446.2
0.8 434.3 435.5 438.4 445.2
0.9 433.8 435.8 437.9 448.5
1 432.4 435.3 436.9 445.6
Tabla 3.1 Parámetros del pico de fusión principal de los monocristales de PVDF en función de la fracción en masa de PVDF, para v = 10 K/min.

54
Φm Tin <T> Tmáx Tfin
0.1 413.4 424.6 426.4 443.4
0.2 413.8 424.9 426.9 441.1
0.3 417.0 427.7 426.2 440.4
0.4 416.2 428.9 426.6 443.7
0.5 427.5 430.6 435.3 443.5
0.6 431.8 433.2 437.4 443.7
0.7 433.4 434.6 437.7 445.7
0.8 433.3 434.3 437.8 444.4
0.9 433.0 434.5 437.1 445.7
1 431.4 434.0 436.0 444.9
Tabla 3.2 Parámetros del pico de fusión principal de los monocristales de PVDF en función de la fracción en masa de PVDF, para v = 20 K/min.
Φm Tin <T> Tmáx Tfin
0.1 416.0 424.2 427.2 439.1
0.2 410.2 424.4 427.4 448.0
0.3 416.1 427.5 429.2 444.7
0.4 416.3 429.1 432.4 443.6
0.5 426.2 430.7 435.4 443.6
0.6 429.2 432.6 437.3 444.4
0.7 431.4 433.5 437.7 445.1
0.8 432.0 434.2 438.0 445.4
0.9 432.0 435.0 438.3 445.7
1 431.5 435.1 437.1 445.9 Tabla 3.3 Parámetros del pico de fusión principal de los monocristales de PVDF en función de la fracción en masa de PVDF, para v = 40 K/min.
Comparando los respectivos valores de cada uno de estos parámetros en función de
las diferentes velocidades de barrido, se aprecia en general una ligera tendencia a la
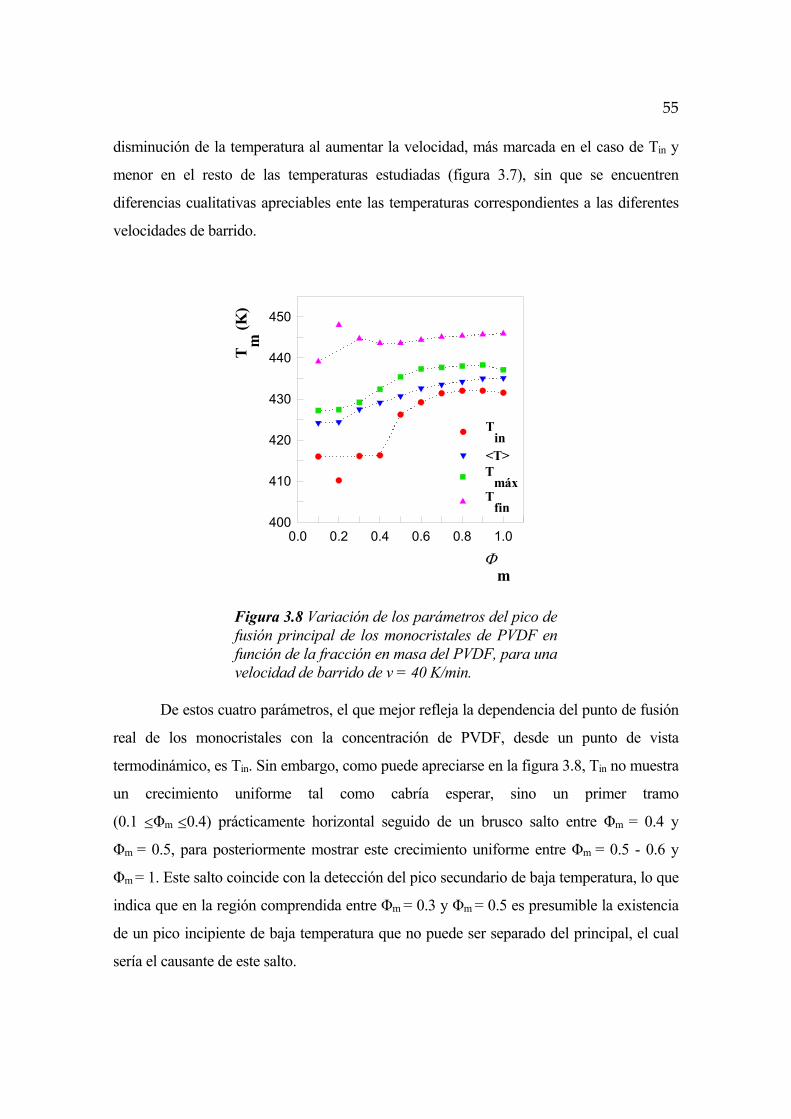
55
disminución de la temperatura al aumentar la velocidad, más marcada en el caso de Tin y
menor en el resto de las temperaturas estudiadas (figura 3.7), sin que se encuentren
diferencias cualitativas apreciables ente las temperaturas correspondientes a las diferentes
velocidades de barrido.
Tmáx
<T>
0.0 0.2 0.4 0.6 0.8 1.0400
410
420
430
440
450
Φm
Tfin
Tin
Tm
(K)
Figura 3.8 Variación de los parámetros del pico de fusión principal de los monocristales de PVDF en función de la fracción en masa del PVDF, para una velocidad de barrido de v = 40 K/min.
De estos cuatro parámetros, el que mejor refleja la dependencia del punto de fusión
real de los monocristales con la concentración de PVDF, desde un punto de vista
termodinámico, es Tin. Sin embargo, como puede apreciarse en la figura 3.8, Tin no muestra
un crecimiento uniforme tal como cabría esperar, sino un primer tramo
(0.1 ≤Φm ≤0.4) prácticamente horizontal seguido de un brusco salto entre Φm = 0.4 y
Φm = 0.5, para posteriormente mostrar este crecimiento uniforme entre Φm = 0.5 - 0.6 y
Φm = 1. Este salto coincide con la detección del pico secundario de baja temperatura, lo que
indica que en la región comprendida entre Φm = 0.3 y Φm = 0.5 es presumible la existencia
de un pico incipiente de baja temperatura que no puede ser separado del principal, el cual
sería el causante de este salto.

56Descartados los valores de Tin por este motivo, para calcular χ es preciso
considerar tanto <T> como Tmáx. En la figura 3.8 se aprecia que el valor de Tmáx se ve
asimismo afectado, aunque en menor grado, por la aparición del pico secundario, cuya
coexistencia con el pico principal provoca un cambio de curvatura a concentraciones de
PVDF comprendidas en el intervalo 0.4 ≤ Φm ≤ 0.6. Por el contrario, el valor de <T>
muestra una variación más uniforme debido a que al tratarse de un promedio se minimizan
las desviaciones producidas por los fenómenos anteriormente comentados. Asimismo, los
valores de <T> son sistemáticamente inferiores a los correspondientes a Tmáx, y por lo tanto
más cercanos a los valores de Tin, por lo cual resulta más adecuado utilizar estos valores en
lugar de Tmáx. Para el cálculo de χ12 hemos utilizado la ecuación [Canalda]:
) - (1 VH
RV - T1 =
)( T1 )
lH2 - 1 ( 2
v121m
20mvmm
e Φ∆Φ∆
χσ [III,3]
Referida a la fracción en volumen de PVDF (Φv).
0.0 0.2 0.4 0.6 0.8 1.02.26
2.28
2.30
2.32
2.34
2.36
2.38
Φv2
Tmáx<T>
1/T
m x
103 (K
-1)
Figura 3.9 Dependencia de la temperatura de fusión del PVDF con la concentración para v = 40 K/min.

57
En la figura 3.9 se ha representado el inverso de las temperaturas de fusión del
PVDF (tanto <T> como Tmáx) en las mezclas frente al cuadrado de Φv, para v = 40 K/min,
utilizando los siguientes valores de los parámetros: Hm = 1606 cal/mol [Nakagawa, 1973],
V1 = 84.9 cm3/mol, V2 = 34.6 cm3/mol, R = 1.98 cal/mol K.
El valor de ∆Hm corresponde a la fase α del PVDF, por pertenecer a ésta los
monocristales obtenidos. V1 y V2 han sido calculados a partir de los valores
correspondientes a las densidades de los dos polímeros:
ρ1 = 1.1838 g/cm3 (PMMA)
ρ2 = 1.85 g/cm3 (PVDF)
ρ1 ha sido calculada a partir de medidas de flotación [Martínez de Salazar, 1991a] y
ρ2 como media ponderada de los valores correspondientes a la densidad amorfa (1.692
g/cm3) [Martínez de Salazar, 1994] y a la densidad cristalina de la fase α (1.92 g/cm3)
[Lovinger, 1982a] del PVDF, suponiendo una cristalinidad media de ~ 67-70%.
Como puede apreciarse en la figura las pendientes de ambas rectas son muy
similares, aunque en el caso de Tmáx la desviación de los puntos experimentales con
respecto a la recta de ajuste es superior a la existente en los valores de <T>. La diferencia
entre las dos ordenadas en el origen, correspondiente a unos 3.5 K, viene determinada por
el desfase existente entre ambos parámetros.
De las pendientes de las dos rectas, obtenidas mediante la aplicación de la ecuación:
)-(1 VH
RV- = T1 -
)(T1 2
v121m
20mv
0m
Φ∆Φ
χ [III,4]
Se obtienen los siguientes valores de χ12, para una velocidad de barrido de 40
K/min:

58
Temperatura χ12
<T> -0.140
Tmáx -0.150
Tabla 3.4 Valores del parámetro de interacción para v = 40 K/min.
Lo cual indica que la diferencia en el valor de χ12 tomando uno u otro parámetro es
muy pequeña, por lo que en base a las razones comentadas anteriormente resulta preferible
utilizar los valores de <T> para el cálculo de χ12. En el caso de las otras dos velocidades de
barrido, v = 10 K/min y v = 20 K/min, donde además del pico secundario de baja
temperatura existe también el pico de alta temperatura correspondiente a la transición
α → γ, se repite la misma situación aunque con una mayor desviación de la linealidad de
los inversos de Tmáx en relación con los correspondientes valores de <T>, razón por la que
en estos casos está todavía más justificado optar por este parámetro. Tomando las medidas
experimentales de <T> para todas las velocidades de barrido, se obtienen finalmente los
siguientes valores de χ12:
v (K/min) χ12
0 -0.147
10 -0.149
20 -0.137
40 -0.140
Tabla 3.5 Valores del parámetro de interacción para velocidades de calentamiento de 0 (obtenido por extrapolación), 10, 20 y 40 K/min.
Lo que ofrece un valor medio de χ12 = -0.14, sin que se aprecien diferencias
significativas en función de la velocidad de barrido. Puesto que varios autores postulan una
dependencia de χ12 con la concentración de la mezcla, hemos efectuado también una
estimación de esta posible dependencia considerando al parámetro de interacción como una

59
función cuadrática de la fracción en volumen de los componentes de la mezcla [Chow,
1990]:
) - (1 B + A = 2v12 Φχ [III,5]
La relación entre estos parámetros A y B y las medidas experimentales de la
depresión de los puntos de fusión puede ser obtenida a partir de la expresión de Nishi y
Wang:
)-(1 VH
RV - = T1 -
T1 2
v121m
20mm
Φ∆
χ [III,6]
Despejando χ12:
)T1 -
T1(
) - (1K =
0mm
2v
12Φ
χ [III,7]
Para:
V RV H - = K2
1m∆ [III,8]
E igualando los segundos términos de las ecuaciones [III,5] y [III,7], finalmente
quedará:
) - (1 B + A = )T1 -
T1(
) - (1K 2
v0mmv
2 ΦΦ
[III,9]

60Por lo tanto, representando el primer término de [III,9] frente a (1 - φv)2, se
obtiene una recta cuyos parámetros son A y B. Sustituyendo en esta ecuación los datos
experimentales, para tres velocidades de barrido diferentes, se obtienen los resultados que
vienen recogidos en la tabla siguiente:
v (K/min) A B
0 -0.137 -3.83 ⋅ 10-2
10 -0.139 -2.98 ⋅ 10-2
20 -0.152 -2.51 ⋅ 10-2
40 -0.139 -0.69 ⋅ 10-2
Tabla 3.6 Valores de los parámetros de la ecuación de Chow para velocidades de calentamiento de 0 (obtenidos por extrapolación), 10, 20 y 40 K/min.
Del análisis de los resultados de esta tabla se concluye que la dependencia de χ12
con la concentración, para todas las velocidades de barrido, puede ser considerada
prácticamente nula, dado que en todos los casos el parámetro B presenta un valor muy
próximo a cero.
3.2 Mezclas amorfas de PVDF y PMMA.
3.2.1 Variación de la transición vítrea con la concentración.
Una de las características de las mezclas compatibles de polímeros es la existencia
de una temperatura de transición vítrea única. Dicha temperatura presenta un valor
intermedio entre las correspondientes a los dos polímeros puros, el cual varía en función de
la proporción de ambos polímeros presentes en la mezcla. La variación del calor específico
(∆cp) en la transición ofrece una información adicional en relación a la concentración
relativa de los componentes.
Para el estudio del sistema formado por las mezclas amorfas de PVDF y PMMA se
han preparado muestras homogéneas, fundidas y subenfriadas por debajo de sus respectivas
transiciones vítreas, a distintas concentraciones en peso de PVDF, junto con las
correspondientes a los dos polímeros en estado puro. Las muestras con Φm ≥ 75% de PVDF
han sido obtenidas en forma de mezclas parcialmente segregadas, con una fracción de
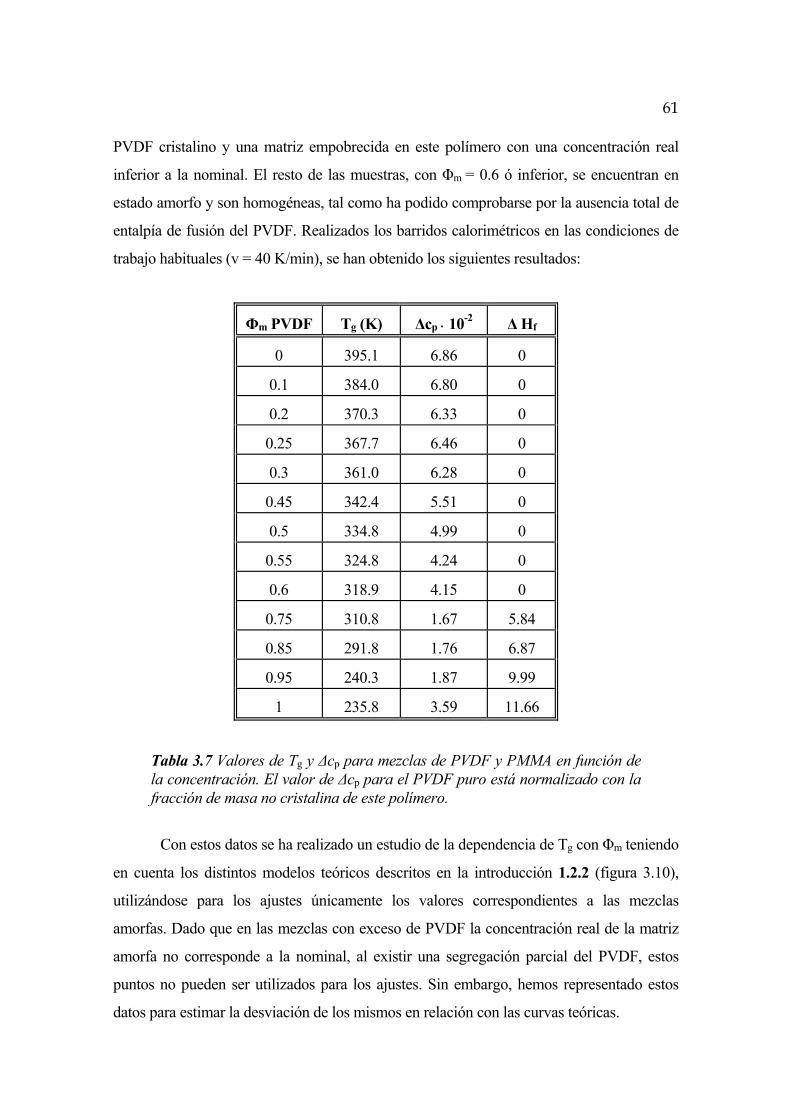
61
PVDF cristalino y una matriz empobrecida en este polímero con una concentración real
inferior a la nominal. El resto de las muestras, con Φm = 0.6 ó inferior, se encuentran en
estado amorfo y son homogéneas, tal como ha podido comprobarse por la ausencia total de
entalpía de fusión del PVDF. Realizados los barridos calorimétricos en las condiciones de
trabajo habituales (v = 40 K/min), se han obtenido los siguientes resultados:
Φm PVDF Tg (K) ∆cp ⋅ 10-2 ∆ Hf
0 395.1 6.86 0
0.1 384.0 6.80 0
0.2 370.3 6.33 0
0.25 367.7 6.46 0
0.3 361.0 6.28 0
0.45 342.4 5.51 0
0.5 334.8 4.99 0
0.55 324.8 4.24 0
0.6 318.9 4.15 0
0.75 310.8 1.67 5.84
0.85 291.8 1.76 6.87
0.95 240.3 1.87 9.99
1 235.8 3.59 11.66
Tabla 3.7 Valores de Tg y ∆cp para mezclas de PVDF y PMMA en función de la concentración. El valor de ∆cp para el PVDF puro está normalizado con la fracción de masa no cristalina de este polímero.
Con estos datos se ha realizado un estudio de la dependencia de Tg con Φm teniendo
en cuenta los distintos modelos teóricos descritos en la introducción 1.2.2 (figura 3.10),
utilizándose para los ajustes únicamente los valores correspondientes a las mezclas
amorfas. Dado que en las mezclas con exceso de PVDF la concentración real de la matriz
amorfa no corresponde a la nominal, al existir una segregación parcial del PVDF, estos
puntos no pueden ser utilizados para los ajustes. Sin embargo, hemos representado estos
datos para estimar la desviación de los mismos en relación con las curvas teóricas.

62
Mezclas parcialmente segregadas
Mezclas homogéneas
0.0 0.2 0.4 0.6 0.8 1.0
240
280
320
360
400 Kwei
0.0 0.2 0.4 0.6 0.8 1.0
240
280
320
360
400 Fox
0.0 0.2 0.4 0.6 0.8 1.0
240
280
320
360
400 Jenckel-Heusch
0.0 0.2 0.4 0.6 0.8 1.0
240
280
320
360
400
Φm
PVDF
Gordon-Taylor
Tg (K
)
Figura 3.10 Dependencia de la transición vítrea con la concentración. Ajuste de varias ecuaciones empíricas.
Como puede apreciarse en la figura 3.10, los valores experimentales se desvían
completamente de la ecuación de Fox. Sin embargo, ofrecen un ajuste satisfactorio con las
tres restantes (Gordon-Taylor, Jenckel-Heusch y Kwei). Como estas tres ecuaciones son
empíricas, el ajuste de las mismas a los puntos experimentales se hace mediante unos
parámetros a los cuales, en nuestro caso, les corresponden los siguientes valores: k = 1.61
para la ecuación de Gordon-Taylor; b = 0.45 para la de Jenckel-Heusch, y k = 1.89 y
q = -24.12 para la de Kwei.
A pesar de que estas tres ecuaciones justifican la dependencia de la transición vítrea
con la concentración, su condición de expresiones empíricas les impide ofrecer un
significado físico a sus respectivos parámetros de ajuste. Por esta razón, resulta más
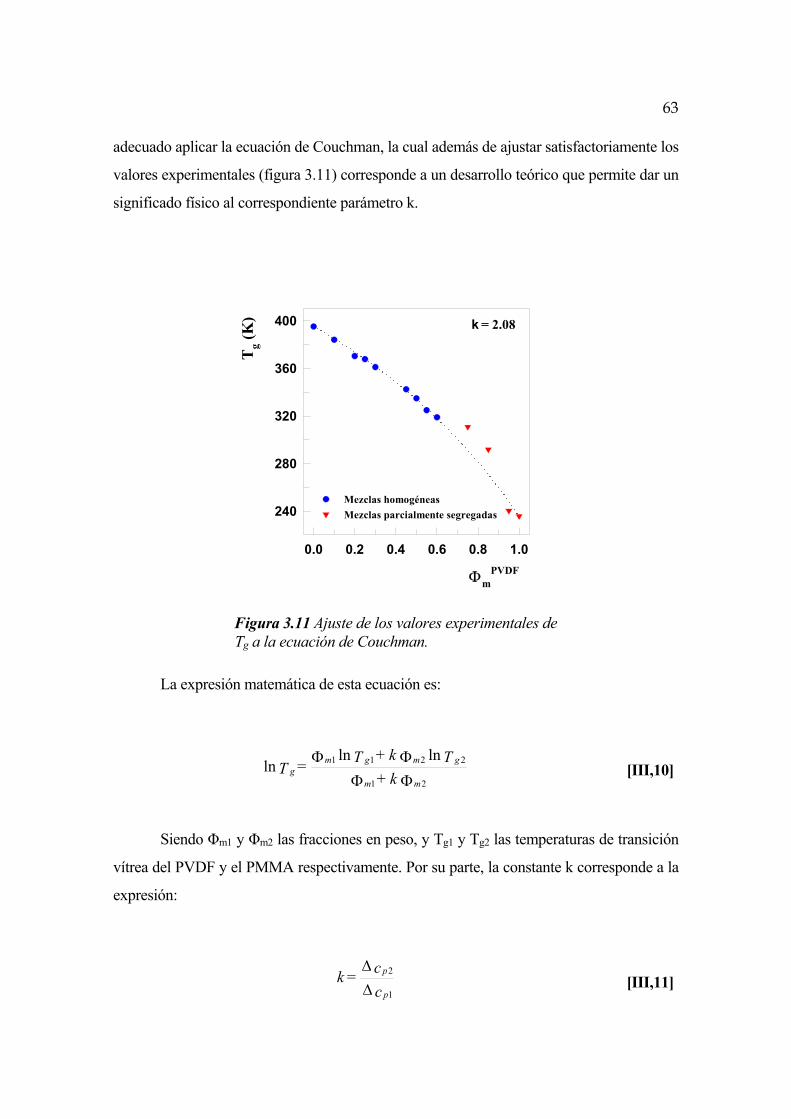
63
adecuado aplicar la ecuación de Couchman, la cual además de ajustar satisfactoriamente los
valores experimentales (figura 3.11) corresponde a un desarrollo teórico que permite dar un
significado físico al correspondiente parámetro k.
0.0 0.2 0.4 0.6 0.8 1.0
240
280
320
360
400 k = 2.08
ΦmPVDF
Tg (
K)
Mezclas parcialmente segregadasMezclas homogéneas
Figura 3.11 Ajuste de los valores experimentales de Tg a la ecuación de Couchman.
La expresión matemática de esta ecuación es:
ΦΦΦΦ
21
2211 lnlnln
mm
gmgmg k +
T k + T = T [III,10]
Siendo Φm1 y Φm2 las fracciones en peso, y Tg1 y Tg2 las temperaturas de transición
vítrea del PVDF y el PMMA respectivamente. Por su parte, la constante k corresponde a la
expresión:
cc = k
p
p
1
2
∆∆
[III,11]

64
Correspondiendo ∆cp1 y ∆cp2 a las variaciones de calor específico del PVDF y el
PMMA.
Ajustando la curva a los puntos experimentales se obtiene un valor para la constante
de k = 2.08, cercano al resultado de k = 1.91 correspondiente al cociente de los incrementos
de calor específico de los dos polímeros en estado puro. Una vez obtenida la relación
existente entre la temperatura de transición vítrea y la concentración, es posible calcular a
partir de la misma la concentración real de la matriz amorfa en las mezclas parcialmente
segregadas. Aplicando la ecuación de Couchman se obtiene:
Φm1 = 64.34% para el 75% en peso de PVDF,
Φm1 = 74.70% para el 85% en peso de PVDF,
Φm1 = 98.21% para el 95% en peso de PVDF.
Como era de esperar, la concentración real de la matriz amorfa resulta ser inferior a
la nominal en todas las muestras excepto en la correspondiente al 95% en peso de PVDF,
lo cual puede ser interpretado en base a la dificultad de obtener una mezcla lo
suficientemente homogénea en los límites de las concentraciones, siendo posible suponer
que parte del PMMA (la diferencia entre el 95 y el 98%) no ha llegado a mezclarse con el
PVDF manteniéndose en estado puro. Sin embargo, no es posible detectar en los
termogramas la transición vítrea del PMMA debido tanto a su escasa cantidad como a su
solapamiento con la cola del pico de fusión del PVDF.
3.2.2 Incremento de calor específico en función de la concentración.
A diferencia de lo que ocurre con las temperaturas de transición vítrea, el cálculo de
los incrementos de calor específico asociados a estas transiciones vítreas resulta ser
extremadamente dificultoso debido tanto a la pequeña magnitud de los mismos, como a su
sensibilidad frente a posibles distorsiones de la línea base.
En lo que respecta a la dependencia de ∆cp con la concentración de las mezclas, es
comúnmente aceptado que esta dependencia es una función lineal con la concentración
[Couchman, 1980] según la ley de aditividad de una mezcla binaria:
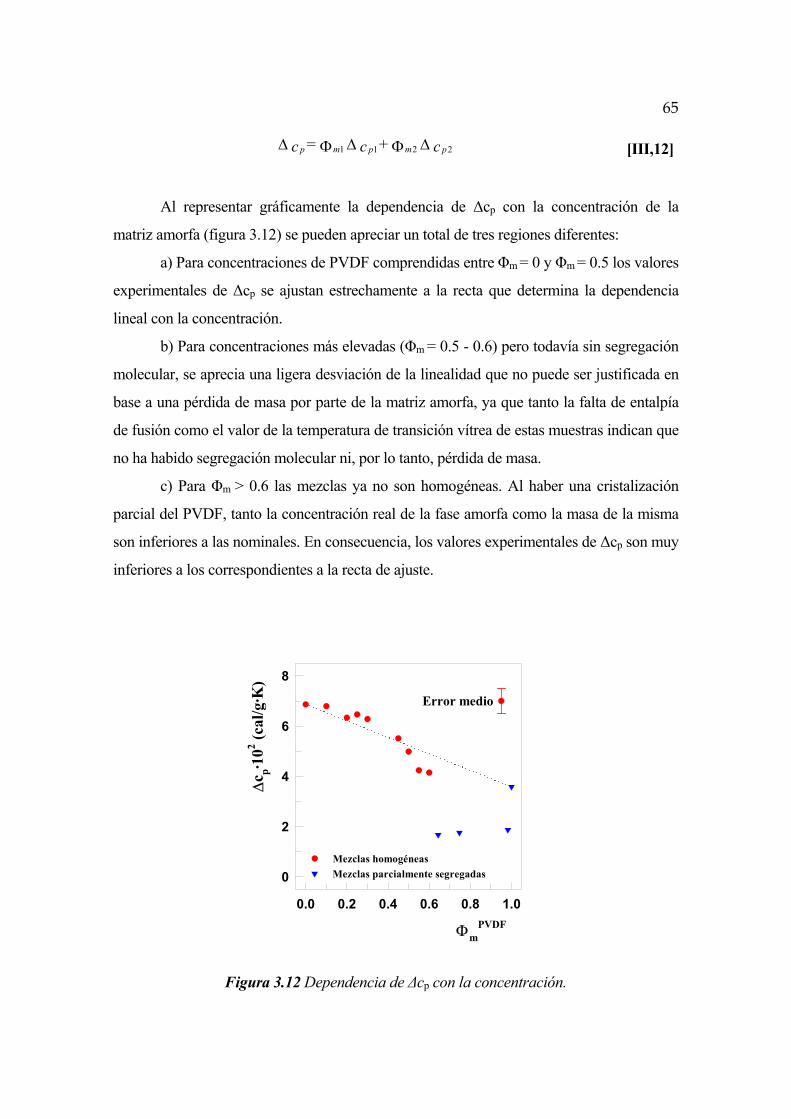
65
c + c = c pmpmp 2211 ∆Φ∆Φ∆ [III,12]
Al representar gráficamente la dependencia de ∆cp con la concentración de la
matriz amorfa (figura 3.12) se pueden apreciar un total de tres regiones diferentes:
a) Para concentraciones de PVDF comprendidas entre Φm = 0 y Φm = 0.5 los valores
experimentales de ∆cp se ajustan estrechamente a la recta que determina la dependencia
lineal con la concentración.
b) Para concentraciones más elevadas (Φm = 0.5 - 0.6) pero todavía sin segregación
molecular, se aprecia una ligera desviación de la linealidad que no puede ser justificada en
base a una pérdida de masa por parte de la matriz amorfa, ya que tanto la falta de entalpía
de fusión como el valor de la temperatura de transición vítrea de estas muestras indican que
no ha habido segregación molecular ni, por lo tanto, pérdida de masa.
c) Para Φm > 0.6 las mezclas ya no son homogéneas. Al haber una cristalización
parcial del PVDF, tanto la concentración real de la fase amorfa como la masa de la misma
son inferiores a las nominales. En consecuencia, los valores experimentales de ∆cp son muy
inferiores a los correspondientes a la recta de ajuste.
0.0 0.2 0.4 0.6 0.8 1.0
0
2
4
6
8
∆c p·1
02 (cal
/g·K
)
ΦmPVDF
Mezclas parcialmente segregadasMezclas homogéneas
Error medio
Figura 3.12 Dependencia de ∆cp con la concentración.

66Un estudio detallado de los termogramas experimentales muestra la existencia en
el caso b) de un pico de cristalización seguido de la posterior fusión de los cristales
anteriormente formados, fenómenos ambos producidos durante el proceso de barrido de la
muestra en el calorímetro. El hecho de que el pico de cristalización tenga su inicio a
temperaturas más altas que la transición vítrea indica asimismo que esta segregación
inducida por el calentamiento de la muestra durante el barrido tiene lugar con posterioridad
al proceso de la transición vítrea, pero la cola que marca el inicio del pico de cristalización
afecta a la línea base provocando la depresión de la misma, lo cual a su vez determina que
las medidas de ∆cp queden subvaloradas con respecto al valor real del salto. Dadas las
características gráficas de los termogramas, no resulta fácil evitar este fenómeno producido
por los picos de cristalización. Sin embargo, debido a lo moderado de su efecto (una
diferencia de aproximadamente un 15% entre los valores experimentales de ∆cp y los
teóricos) esta causa puede ser asumida como la responsable de la desviación observada.
Más compleja resulta la interpretación de los puntos correspondientes a las
muestras parcialmente segregadas, porque aquí sí se ha producido una pérdida de masa en
la matriz amorfa que afecta a los valores de ∆cp, ya que para que éstos puedan ser
comparados con el resto de los datos es necesario referirlos a una misma cantidad de masa.
El cálculo de la cantidad de masa perdida por la matriz amorfa no es sencillo debido a que
las muestras segregadas presentan una compleja distribución de fases:
i) Una matriz amorfa empobrecida en PVDF (como indica la Tg).
ii) Cristales de PVDF.
iii) Una región interlaminar formada por PVDF amorfo, y
iv) Una cuarta región, a la que se puede denominar interfase, constituida por una
población de PVDF y PMMA mezclados de forma probablemente no homogénea
formando un gradiente de concentraciones.
Aunque diversos autores [Wendorff, 1980; Yoon, 1991; Ando, 1992] han postulado
la existencia de una interfase formada exclusivamente por PVDF puro en estado amorfo en
las mezclas parcialmente segregadas de PVDF y PMMA, la aplicación de este modelo a la
normalización de la masa en los puntos correspondientes a estas concentraciones no
conduce a resultados satisfactorios al no ajustarse los valores así normalizados a la recta
teórica de dependencia de ∆cp con la concentración.

67
Procediendo a la inversa, es decir, tomando los valores teóricos de ∆cp y
combinándolos con los valores de la concentración de la matriz amorfa (calculados a partir
de la Tg) y con los de la entalpía de fusión de los cristales de PVDF, se pueden estimar las
cantidades de ambos polímeros que se encuentran tanto fuera de la matriz amorfa como de
los cristales. Para aplicar este modelo es necesario asumir que la interfase ya no está
formada por PVDF puro sino que contiene cierta cantidad de PMMA expulsado de las
regiones cristalinas [Saito, 1994], obteniéndose como resultado, para una masa total de
referencia de un gramo, las siguientes cantidades de PVDF y PMMA:
Φm PVDF mPVDF mPMMA
0.75 0.29 0.13
0.85 0.28 0.05
Tabla 3.8 Masas de PVDF y de PMMA presentes fuera de la matriz amorfa y de la región cristalina, para mezclas parcialmente cristalizadas. Masa total = 1 g.
La muestra con Φm = 0.95 no ofrece resultados físicamente coherentes, por las
razones anteriormente expuestas, pudiéndose asumir que en esta muestra la interfase podría
estar formada por PVDF puro.
Las cantidades de PVDF reflejadas en la tabla anterior se reparten a su vez entre la
interfase propiamente dicha, donde el PVDF estaría mezclado con el PMMA, y la región
adyacente a los cristales, que estaría formada por PVDF puro. No resulta posible
cuantificar la proporción de moléculas de PVDF correspondientes a una u otra región, del
mismo modo que no se puede definir una concentración de la interfase al no estar
mezclados homogéneamente los dos polímeros en ella. Sin embargo, asumiendo que la
proporción de PVDF amorfo sea similar a la existente en los monocristales de PVDF, que
alcanzan unas cristalinidades del orden del 70%, el reparto de las masas de los dos
polímeros en las diferentes fases del material, referido a 1 g de masa total, queda de la
siguiente manera:

68
región laminar interfase matriz amorfa
Φm PVDF PVDFcrist PVDFamf PVDF PMMA PVDF PMMA
0.75 0.23 0.10 0.19 0.13 0.22 0.12
0.85 0.27 0.12 0.17 0.05 0.30 0.10
Tabla 3.9 Distribución de masas del PVDF y el PMMA en las diferentes regiones de las mezclas parcialmente cristalizadas. Masa total = 1 g.
A partir de los datos reflejados en la tabla anterior se puede concluir que una
apreciable cantidad de la masa total se encuentra en la interfase; un 32% en la muestra de
Φm = 0.75 y un 22% en la de Φm = 0.85, correspondiéndoles unas proporciones de PVDF de
un 70 y un 85% respectivamente. Estas concentraciones son más elevadas que las
concentraciones de las matrices amorfas, calculadas en un 64 y un 70% para cada una de
las dos muestras. El hecho de que en los termogramas no se detecte ninguna transición
vítrea a las temperaturas correspondientes a estas proporciones, a pesar de la elevada
cantidad de masa presente en la interfase, confirma la falta de homogeneidad en la
interfase.
3.2.3 Dependencia de la densidad macroscópica con la concentración.
Al igual que ocurre con otros parámetros físicos, la compatibilidad de las mezclas
de PVDF y PMMA puede ser estudiada en relación con la dependencia de la densidad
macroscópica de las mezclas en función de la concentración. Para este fin se prepararon
mezclas amorfas de distintas concentraciones midiéndose sus densidades por el método de
flotación, descrito en el apartado correspondiente. Los resultados obtenidos son los
siguientes:

69
Φm PVDF ρ (g/cm3)
0 1.184
0.25 1.277
0.45 1.370
0.5 1.390
0.55 1.419
0.6 1.444
0.75 1.546
Tabla 3.10 Dependencia de la densidad macroscópica con la concentración de las mezclas PVDF/PMMA. La muestra con Φm = 0.75 está parcialmente cristalizada.
Como puede apreciarse en la figura 3.13, existe una clara dependencia lineal entre
el volumen específico de las mezclas y la concentración de PVDF para las muestras
amorfas. El caso de la muestra con Φm = 0.75, que está parcialmente segregada siendo la
concentración real de la fase amorfa de tan sólo un 64%, resulta difícil de interpretar ya que
no se trata de un material homogéneo sino que está formado por diferentes regiones
(fracción cristalina, interfase y matriz amorfa), las cuales contribuyen de una manera
compleja a la densidad total. Por esta razón no resulta posible obtener experimentalmente
una medida derivada del valor de la densidad de una mezcla amorfa y homogénea de
composición igual a la proporción del 75%, si bien se observa que la densidad
macroscópica de esta muestra no se desvía demasiado de la teórica, probablemente porque
las distintas densidades se compensan entre sí.

70
0.0 0.2 0.4 0.6 0.8 1.00.5
0.6
0.7
0.8
0.9
1.0
Φm PVDF
1/ρ
(cm
3 /g)
Figura 3.13 Dependencia de la densidad macroscópica con la concentración.
La extrapolación de la línea recta para Φm = 1 ofrece un valor para el PVDF amorfo
de 1.692 g/cm3. Comparando este valor y el experimental de la densidad del PMMA de
1.184 g/cm3 con los aportados por la bibliografía, se aprecia una notable concordancia que
permite establecer la dependencia del volumen específico con la concentración conforme la
siguiente expresión:
ρρρ 2
m
1
m
b
+ = 1 21 ΦΦ [III,13]
Siendo ρ1 y ρ2 la densidad amorfa del PVDF y la densidad del PMMA
respectivamente, y Φm1 y Φm2 las fracciones en masa de los dos polímeros. Este modelo
asume implícitamente la existencia de aditividad en los volúmenes, aproximación que se ha
venido considerando en todo este estudio.

71
3.2.4 Investigación de la microdureza en función de la concentración.
La compatibilidad de las mezclas de PVDF y PMMA puede ser también analizada
mediante el estudio de la microdureza, a temperatura ambiente, en función de la
concentración. Esta técnica permite detectar cambios estructurales y morfológicos en
polímeros semicristalinos. La microdureza depende tanto de la componente de la fracción
cristalina (Hc), como de la amorfa (Ha). En el caso de polímeros flexibles a temperaturas
muy superiores a la de transición vítrea, la contribución de la fracción amorfa a la
microdureza total es muy pequeña en relación con la correspondiente a la fracción
cristalina [Baltá Calleja, 1981].
En las mezclas de PVDF y PMMA, al tener el PVDF una temperatura de transición
vitrea (Tg ≈ -40 °C) muy inferior a la temperatura ambiente, será posible esperar una
dependencia lineal de la microdureza con la composición de la mezcla; en muestras
homogéneas y amorfas no existirá aportación de la fracción cristalina del PVDF, por lo que
la microdureza total será función exclusivamente de la correspondiente al PMMA puro al
ser despreciable la aportación del PVDF amorfo. Preparadas mezclas amorfas de
concentraciones correspondientes al 25, 45, 50, 55, 60 y 75% en peso de PVDF (esta
última parcialmente segregada) y de PVDF y PMMA puros, se han obtenido los siguientes
resultados:
Φm PVDF H (MPa) Φm PVDF H (MPa)
0 213.68 0.55 105.35
0.25 163.32 0.6 71.70
0.45 125.98 0.75 54.49
0.5 107.83 1 127.13
Tabla 3.11 Dependencia de la microdureza con la concentración de las mezclas
PVDF/PMMA. La muestra con Φm = 0.75 está parcialmente cristalizada.
Tal como se refleja en la figura 3.14, en el caso de las mezclas homogéneas amorfas
(Φm ≤ 0.6) se aprecia una dependencia lineal de la microdureza con la concentración que
puede ser expresada como:
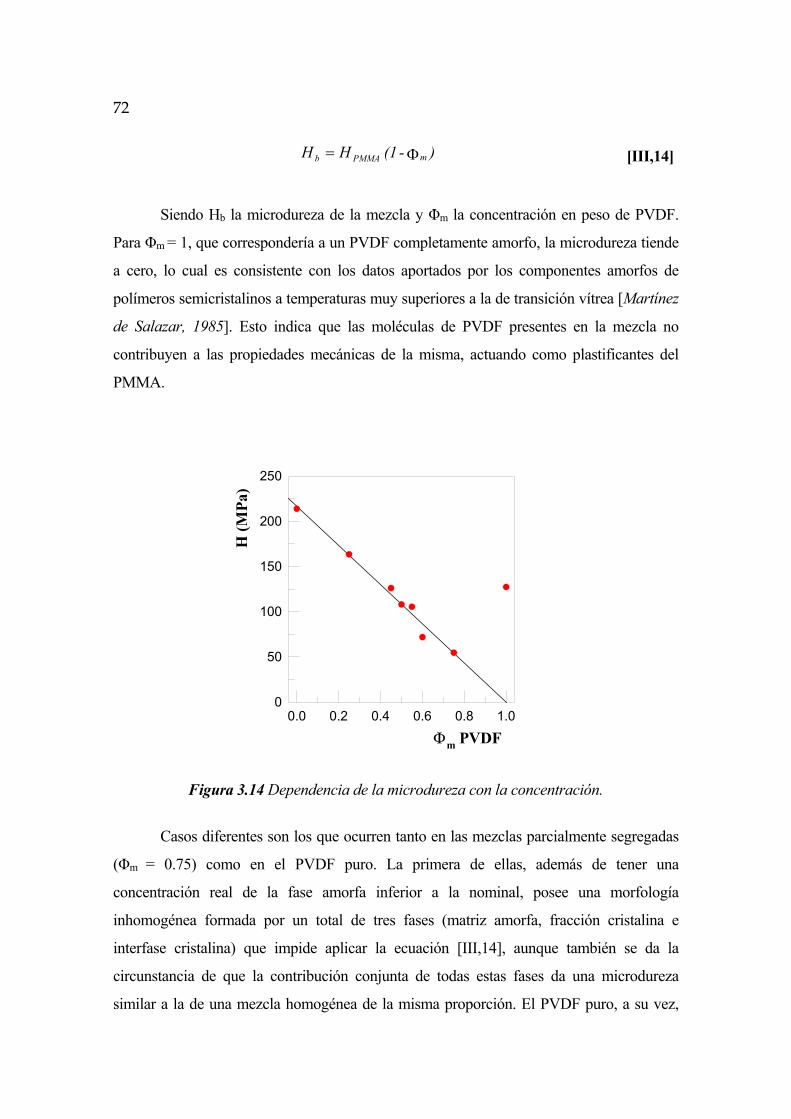
72
) - (1 HH mPMMAb Φ= [III,14]
Siendo Hb la microdureza de la mezcla y Φm la concentración en peso de PVDF.
Para Φm = 1, que correspondería a un PVDF completamente amorfo, la microdureza tiende
a cero, lo cual es consistente con los datos aportados por los componentes amorfos de
polímeros semicristalinos a temperaturas muy superiores a la de transición vítrea [Martínez
de Salazar, 1985]. Esto indica que las moléculas de PVDF presentes en la mezcla no
contribuyen a las propiedades mecánicas de la misma, actuando como plastificantes del
PMMA.
0.0 0.2 0.4 0.6 0.8 1.00
50
100
150
200
250
Φm PVDF
H (M
Pa)
Figura 3.14 Dependencia de la microdureza con la concentración.
Casos diferentes son los que ocurren tanto en las mezclas parcialmente segregadas
(Φm = 0.75) como en el PVDF puro. La primera de ellas, además de tener una
concentración real de la fase amorfa inferior a la nominal, posee una morfología
inhomogénea formada por un total de tres fases (matriz amorfa, fracción cristalina e
interfase cristalina) que impide aplicar la ecuación [III,14], aunque también se da la
circunstancia de que la contribución conjunta de todas estas fases da una microdureza
similar a la de una mezcla homogénea de la misma proporción. El PVDF puro, a su vez,

73
presenta una morfología semicristalina con una fracción cristalina que contribuye a la
microdureza de forma diferente a la correspondiente a las mezclas homogéneas.
3.3 Morfología de las mezclas de PVDF y PMMA y transiciones de fase cristalina.
En esta parte del presente trabajo se ha utilizado principalmente la técnica de
difracción de rayos X para la identificación cualitativa de las diferentes morfologías
presentes en una muestra determinada así como de los posibles procesos de cristalización y
transiciones de fase. Dado que la historia térmica de las muestras resulta ser fundamental a
la hora de establecer la cinética de cristalización de las mismas, siendo necesario realizar la
fusión previa al subenfriado a una temperatura lo suficientemente elevada como para que
los dos polímeros queden mezclados de forma totalmente homogénea, la temperatura
mínima de fusión ha sido estimada en 190/200 °C. En muestras fundidas a temperaturas
inferiores se obtienen unas cinéticas de cristalización totalmente distintas a causa de una
homogeneización incompleta de la mezcla en el fundido.
A continuación se ha procedido a seleccionar muestras cuyos difractogramas
reflejaran con claridad las reflexiones de las diferentes fases cristalinas. Para identificar las
reflexiones de la fase α hemos recurrido a una muestra de PVDF cristalizada desde el
fundido, obteniéndose los siguientes valores experimentales:
Plano 2θ (°) d (nm) I rel.(%)
100 17.55 0.505 53.2
100 17.67 0.502 53.2
020 18.26 0.486 68.1
110 19.83 0.448 100
021 26.62 0.335 38.3
121 32.98 0.272 23.4
200 35.66 0.252 25.5
131 38.32 0.235 29.8
Tabla 3.12 Posición angular, espaciado e intensidad relativa respecto a la reflexión más intensa de los diversos planos cristalográficos de la fase α del PVDF.

74
En el caso de mezclas parcialmente segregadas de PVDF y PMMA, las
reflexiones menos intensas desaparecen y el resto se atenúa, por lo que para identificar
en ellas a la fase α se han seleccionado las cuatro reflexiones más intensas, que son en
la práctica las únicas que aparecen en las mismas. Éstas son el triplete formado por los
tres picos estrechos situados sobre el halo amorfo (2θ = 17.6, 18.3 y 19.8°) así como un
cuarto pico de mayor anchura que los anteriores (en realidad un triplete, aunque no
siempre se resuelve en sus componentes) centrado en torno a 2θ = 26.5°.
Las reflexiones de la fase ß han sido investigadas en mezclas parcialmente
segregadas de PVDF y PMMA debido a que en las condiciones de trabajo habituales no es
posible obtener PVDF puro cristalizado en la fase ß. En concordancia con la bibliografía
[Léonard, 1983] se aprecia la existencia de fase ß prácticamente pura en mezclas con un
75% en peso de PVDF sometidas a las condiciones de tratamiento térmico más adecuadas
para ello. Para identificar la fase ß hemos recurrido a la reflexión principal (planos
200/110), tabulada en torno al valor de 2θ = 20.5°. Esta reflexión, además de ser muy
nítida, no solapa con ninguna de las reflexiones de la fase α, lo que la hace muy apropiada
para la investigación de la existencia de fase ß. El resto de las reflexiones, al tratarse de
mezclas y no de PVDF puro, o no se aprecian o lo son en forma muy débil.
La determinación experimental de la fase γ resulta más compleja debido a que el
escaso grado de cristalinidad total existente en las muestras estudiadas dificulta la
distinción de las reflexiones características de las fases α y γ, detectándose tan sólo los
picos correspondientes a las reflexiones intensas, los cuales coinciden en ambas. Sin
embargo, basándonos en los datos aportados por numerosos autores [Gal'Perin, 1970;
Lovinger, 1980; Osaki, 1975; Prest, 1975; 1978; Tashiro, 1983a] que identifican a la fase γ
en muestras cristalizadas a temperaturas cercanas al punto de fusión del PVDF, hemos
supuesto la existencia de esta fase en muestras ricas en PVDF (w = 0.75) tratadas a
160 °C, en las cuales se aprecia, tanto por difracción de rayos X como por calorimetría
diferencial, una transición de fase que puede ser identificada con la transición ß → γ [Prest,
1978]. Sin embargo, la información proporcionada por los difractogramas no es totalmente
explícita al no poderse diferenciar con claridad las reflexiones pertenecientes a la fase γ de
las correspondientes a alguno de los tipos de fase α descritos en la bibliografía [Gal'Perin,
1970] a causa de la escasa cristalinidad de las muestras estudiadas.

75
En resumen, los difractogramas experimentales correspondientes a las fases α, ß y γ
vienen representados en la figura 3.15:
Fase β
5 10 15 20 25 30
111022
120
110
020100
200/110
021
110
020
100
Fase γ
Fase α
I (u.
a.)
2θ (°)
Figura 3.15 Reflexiones experimentales de las fases α, ß y γ del PVDF.
El siguiente paso consiste en estudiar la cristalinidad presente en mezclas de PVDF
y PMMA fundidas y subenfriadas por debajo de la temperatura de transición vítrea. Como
puede apreciarse en la figura 3.16, el efecto más inmediato de la adición del PMMA es la
ya comentada inhibición de la cristalización del PVDF, obteniéndose mezclas amorfas para
concentraciones de PMMA iguales o mayores a un 35% en peso aproximadamente.
Muestras ricas en PVDF presentan un menor grado de cristalinidad que el correspondiente
al PVDF puro.
Las muestras ricas en PMMA poseen, tal como ha sido comentado, una estructura
amorfa, si bien se aprecia una notable diferencia entre sus difractogramas y el
correspondiente al PMMA puro. El máximo del halo amorfo, que en el PMMA se sitúa en
torno a los 14-15°, experimenta un corrimiento hasta aproximadamente los 16-17° en las

76mezclas amorfas de PVDF y PMMA. Puesto que este máximo está relacionado con la
distancia media intermolecular existente en el material amorfo, y dada la relación inversa
entre los ángulos de reflexión y los espaciados, el incremento en dos grados corresponde
aproximadamente a una disminución de esta distancia media de unos 0.07 nm. Este
corrimiento del máximo, que no se ve afectado significativamente por la concentración de
PVDF, puede ser explicado por la aparición de fuerzas atractivas entre ambos polímeros,
del tipo de puentes de hidrógeno, inexistentes en el PMMA, lo que trae como consecuencia
una disminución en la distancia media entre las moléculas adyacentes al estar éstas más
fuertemente unidas en las mezclas que en el PMMA.
PMMA
5 10 15 20 25 30 35 40
25:75
50:50
75:25
85:15
PVDFI (u.
a.)
2θ (°)
Figura 3.16 Dependencia de la cristalinidad de las mezclas PVDF/PMMA con la concentración.
3.3.1 Dependencia de la temperatura.
Para concentraciones medias (0.4 ≤ Φm ≤ 0.6) se aprecia, incluso a las
concentraciones más bajas de PVDF, una mezcla de fases α y ß. La proporción entre estas
dos fases cristalinas depende fuertemente de diversas variables tales como concentración,
temperatura y tiempo de tratamiento, observándose como tendencia general un predominio
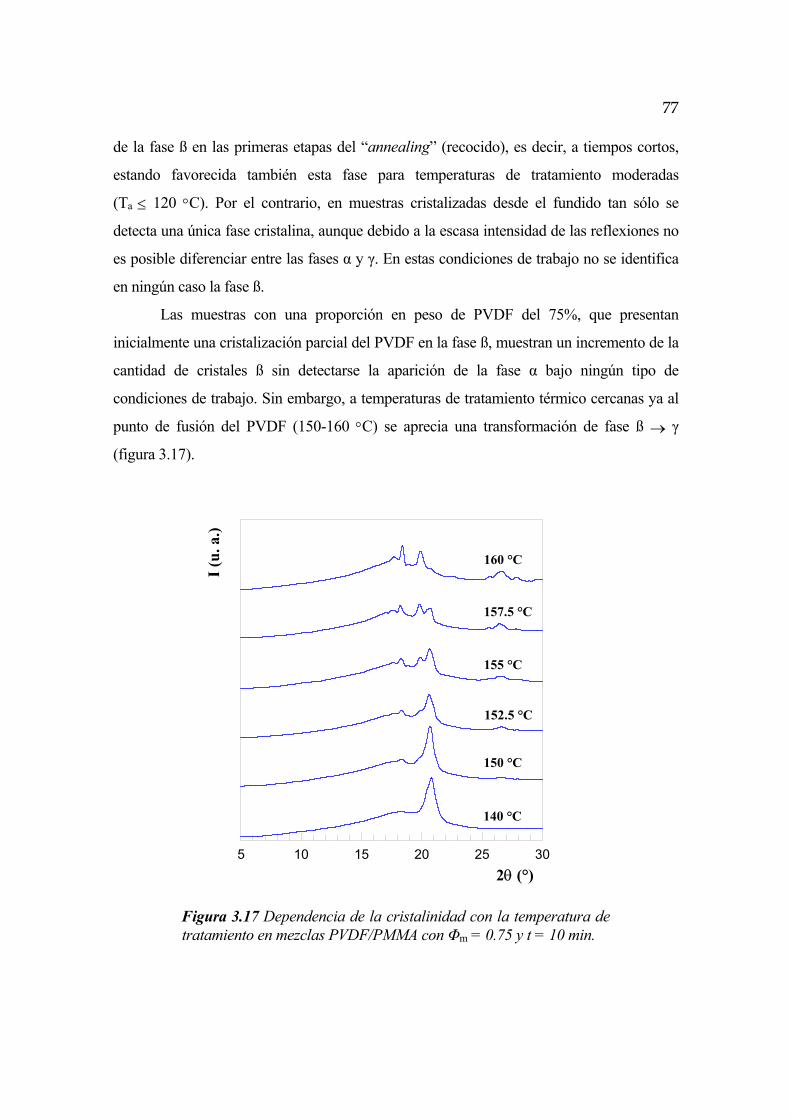
77
de la fase ß en las primeras etapas del “annealing” (recocido), es decir, a tiempos cortos,
estando favorecida también esta fase para temperaturas de tratamiento moderadas
(Ta ≤ 120 °C). Por el contrario, en muestras cristalizadas desde el fundido tan sólo se
detecta una única fase cristalina, aunque debido a la escasa intensidad de las reflexiones no
es posible diferenciar entre las fases α y γ. En estas condiciones de trabajo no se identifica
en ningún caso la fase ß.
Las muestras con una proporción en peso de PVDF del 75%, que presentan
inicialmente una cristalización parcial del PVDF en la fase ß, muestran un incremento de la
cantidad de cristales ß sin detectarse la aparición de la fase α bajo ningún tipo de
condiciones de trabajo. Sin embargo, a temperaturas de tratamiento térmico cercanas ya al
punto de fusión del PVDF (150-160 °C) se aprecia una transformación de fase ß → γ
(figura 3.17).
5 10 15 20 25 30
140 °C
150 °C
152.5 °C
155 °C
157.5 °C
160 °C
I (u.
a.)
2θ (°)
Figura 3.17 Dependencia de la cristalinidad con la temperatura de tratamiento en mezclas PVDF/PMMA con Φm = 0.75 y t = 10 min.
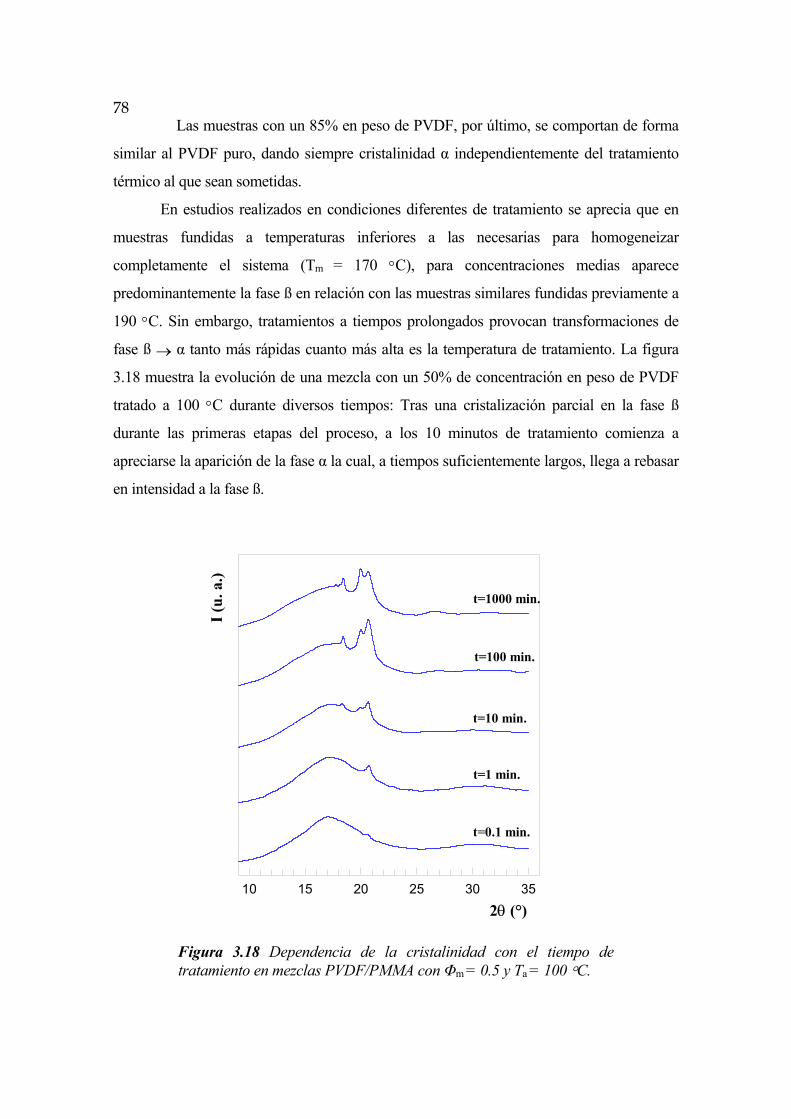
78Las muestras con un 85% en peso de PVDF, por último, se comportan de forma
similar al PVDF puro, dando siempre cristalinidad α independientemente del tratamiento
térmico al que sean sometidas.
En estudios realizados en condiciones diferentes de tratamiento se aprecia que en
muestras fundidas a temperaturas inferiores a las necesarias para homogeneizar
completamente el sistema (Tm = 170 °C), para concentraciones medias aparece
predominantemente la fase ß en relación con las muestras similares fundidas previamente a
190 °C. Sin embargo, tratamientos a tiempos prolongados provocan transformaciones de
fase ß → α tanto más rápidas cuanto más alta es la temperatura de tratamiento. La figura
3.18 muestra la evolución de una mezcla con un 50% de concentración en peso de PVDF
tratado a 100 °C durante diversos tiempos: Tras una cristalización parcial en la fase ß
durante las primeras etapas del proceso, a los 10 minutos de tratamiento comienza a
apreciarse la aparición de la fase α la cual, a tiempos suficientemente largos, llega a rebasar
en intensidad a la fase ß.
10 15 20 25 30 35
t=0.1 min.
t=1 min.
t=10 min.
t=100 min.
t=1000 min.
I (u.
a.)
2θ (°)
Figura 3.18 Dependencia de la cristalinidad con el tiempo de tratamiento en mezclas PVDF/PMMA con Φm = 0.5 y Ta = 100 °C.

79
Paralelamente a este estudio cristalográfico se han medido los volúmenes
específicos de todas estas muestras, observándose un importante incremento en el
crecimiento de la densidad coincidiendo con la aparición de la fase α (Fig. 3.19).
10-2 10-1 100 101 102 103 1040.705
0.710
0.715
0.720
0.725
1/ρ
(cm
3 /g)
t (min)
Figura 3.19 Dependencia de la densidad macroscópica en mezclas PVDF/ PMMA con Φm = 0.5 y Ta = 100 °C en función del tiempo de tratamiento.
Puesto que la densidad de la fase α es inferior a la correspondiente a la fase ß
[Gal'Perin, 1969; Gregorio, 1994; Narula, 1989], resulta una aparente contradicción que
las densidades de las mezclas cristalizadas con una mezcla de las fases α y ß presenten
valores superiores a los que corresponderían a muestras de similar grado de segregación
cristalizadas únicamente en la fase ß, las cuales se habrían de ajustar a la extrapolación de
la recta perteneciente a los puntos experimentales de las primeras etapas del proceso. Este
fenómeno indica que la cristalinidad total aumenta, y que las fases α y ß se originan de
forma independiente, a partir del material amorfo inicial, siguiendo procesos de
segregación molecular y cristalización diferentes, no existiendo una transformación de fase
entre una y otra.

803.4 Cinética de segregación de fases en las primeras etapas del proceso. Difusión
molecular.
El estudio de la segregación de fases en las mezclas amorfas de PVDF y PMMA en
las primeras etapas del proceso ha sido realizado mediante técnicas experimentales de
difracción de rayos X a ángulos bajos (SAXS). Mezclas de PVDF y PMMA, preparadas
según el método habitual, han sido estudiadas con una cámara Rigaku con colimación
lineal y un detector multicanal de 108 µm de resolución. Las muestras han sido calentadas
en la propia cámara, y las medidas de intensidad de difracción acumulada se han realizado
en tiempo real.
A partir de los resultados experimentales se han calculado los valores
correspondientes al invariante en función del tiempo, para una temperatura de tratamiento y
una concentración determinadas. La representación de la raíz cúbica de Q frente a la raíz
cuadrada del tiempo refleja en todos los casos el comportamiento que viene recogido en la
figura 3.20:
0 4 8 12 16 20
0.0
0.5
1.0
1.5
2.0Q1/
3
t1/2 (min1/2)
Ta= 360.5 K
Figura 3.20 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia del invariante con el tiempo.
Como puede apreciarse en la figura, existe una dependencia lineal entre la raíz
cúbica del invariante y la raíz cuadrada del tiempo para los valores correspondientes a
tiempos cortos de tratamiento, produciéndose una desviación de la linealidad a partir de
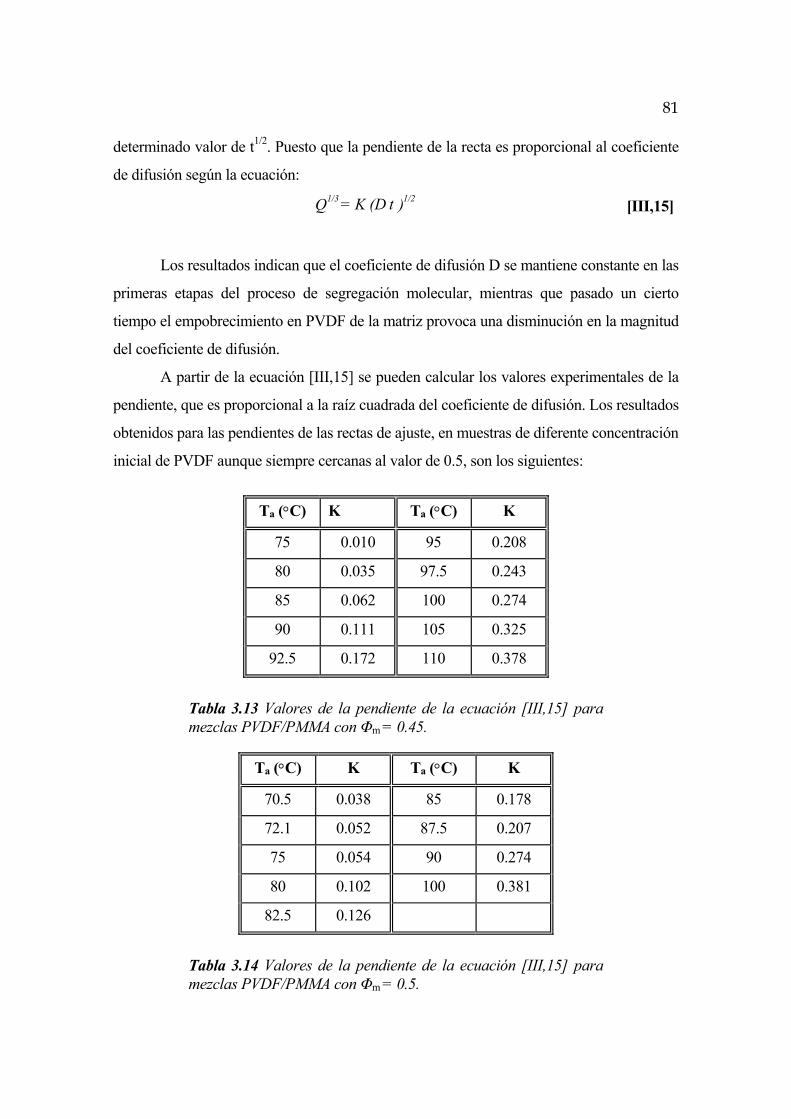
81
determinado valor de t1/2. Puesto que la pendiente de la recta es proporcional al coeficiente
de difusión según la ecuación:
)t (D K = Q 1/21/3 [III,15]
Los resultados indican que el coeficiente de difusión D se mantiene constante en las
primeras etapas del proceso de segregación molecular, mientras que pasado un cierto
tiempo el empobrecimiento en PVDF de la matriz provoca una disminución en la magnitud
del coeficiente de difusión.
A partir de la ecuación [III,15] se pueden calcular los valores experimentales de la
pendiente, que es proporcional a la raíz cuadrada del coeficiente de difusión. Los resultados
obtenidos para las pendientes de las rectas de ajuste, en muestras de diferente concentración
inicial de PVDF aunque siempre cercanas al valor de 0.5, son los siguientes:
Ta (°C) K Ta (°C) K
75 0.010 95 0.208
80 0.035 97.5 0.243
85 0.062 100 0.274
90 0.111 105 0.325
92.5 0.172 110 0.378
Tabla 3.13 Valores de la pendiente de la ecuación [III,15] para mezclas PVDF/PMMA con Φm = 0.45.
Ta (°C) K Ta (°C) K
70.5 0.038 85 0.178
72.1 0.052 87.5 0.207
75 0.054 90 0.274
80 0.102 100 0.381
82.5 0.126
Tabla 3.14 Valores de la pendiente de la ecuación [III,15] para mezclas PVDF/PMMA con Φm = 0.5.

82
Ta (°C) K Ta (°C) K
65 0.148 80 0.423
70 0.165 82.5 0.469
72.5 0.238 85 0.511
75 0.314 87.5 0.478
77.5 0.367 90 0.354
Tabla 3.15 Valores de la pendiente de la ecuación [III,15] para mezclas PVDF/PMMA con Φm = 0.55.
3.5 Cinética de segregación de fases en mezclas amorfas.
Una forma directa de estudiar la cinética de segregación de las mezclas amorfas de
PVDF y PMMA es mediante medidas calorimétricas, tanto en lo referente a la transición
vítrea como a la entalpía de fusión de la fracción cristalina. De esta manera se obtiene
información tanto de la matriz amorfa restante como del PVDF cristalizado, lo cual
permitirá extraer conclusiones acerca de la estructura interna de las muestras parcialmente
segregadas.
La cinética global del proceso depende de diferentes parámetros tales como la
composición inicial de la mezcla, la temperatura de tratamiento o el tiempo de tratamiento.
Puesto que es preciso partir de mezclas inicialmente amorfas pero segregables y
cristalizables, hemos seleccionado una concentración inicial adecuada (50% en peso de
PVDF) que permite controlar en un amplio rango de tiempos y temperaturas la cinética del
proceso.
El primer resultado que se obtiene es la comprobación de que los dos procesos
cinéticos estudiados (la variación de la transición vítrea y la entalpía de fusión) presentan
un máximo en torno a los 120 °C de temperatura de tratamiento para un tiempo
determinado, máximo que demuestra ser asimismo independiente de la concentración
inicial de PVDF en las mezclas. Resultados posteriores permiten fijar este máximo en un
valor coincidente con el de la temperatura de transición vítrea del PMMA, establecida en
torno a los 122 °C. La cinética en ambas regiones anterior y posterior al máximo es
diferente en los dos casos, siendo asumible que a temperaturas próximas al punto de fusión
de las mezclas predominaran los procesos de cristalización, mientras a temperaturas

83
inferiores al máximo era de prever un mayor protagonismo de los fenómenos de difusión
molecular. Por esta razón, el estudio ha sido realizado por separado tanto en la región de
temperaturas inferiores como en la de temperaturas superiores a la transición vítrea del
PMMA.
3.5.1 Dependencia temporal.
Dado que la transición vítrea correspondiente a las mezclas con un 50% en peso de
PVDF se sitúa en torno a los 61 °C, el intervalo de temperaturas utilizado en este estudio ha
sido el comprendido entre este valor y el del punto de fusión del PVDF cristalizado (Tm). A
continuación presentamos los resultados sobre la dependencia temporal de los procesos
isotérmicos en este rango de temperaturas.
t (min)0 40 80 120 160 200
5.80
5.82
5.84
5.86
5.88
5.90
0 40 80 120 160 2005.80
5.82
5.84
5.86
5.88
5.900 40 80 120 160 200
5.80
5.82
5.84
5.86
5.88
5.90
0 40 80 120 160 2005.80
5.82
5.84
5.86
5.88
5.90
ln T
g
Ta=100 ºC
Ta=120 ºC
Ta=110 ºC
Ta=140 ºC
Figura 3.21 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de la transición vítrea con el tiempo para diferentes temperaturas de tratamiento.

84
Representando la variación de Tg en función del tiempo para una temperatura de
tratamiento determinada se aprecia la existencia de dos regiones distintas, una a tiempos
cortos y otra a tiempos largos, en todo el intervalo de temperaturas de tratamiento a partir
de una temperatura de 85 °C. La dependencia de Tg con el tiempo de tratamiento es
aparentemente lineal en ambas regiones tal como se aprecia en la figura 3.21, en la cual los
ejes de ordenadas están representados en escala logarítmica con objeto de resaltar las
diferencias existentes entre las dos regiones.
Con objeto de confirmar esta dependencia lineal hemos procedido a realizar por
separado, para cada una de estas dos regiones, un estudio de su dependencia temporal. Para
la primera región, correspondiente a tiempos cortos de tratamiento, hemos aplicado una
ecuación del tipo:
t K = T - T nogg [III,16]
Siendo K una constante de ajuste, y tomándose como ordenada en el origen para
tiempo cero la temperatura de transición vítrea inicial de la mezcla (Tgo), se buscó el valor
del exponente que mejor se ajustaba a los valores experimentales. Los resultados obtenidos
para el exponente n están recogidos en las tablas 3.16 y 3.17:
Ta (°C) n Ta (°C) n
75 0.91 100 1.33
80 0.89 105 1.12
85 1.20 110 1.04
90 1.06 115 1.05
95 1.03 120 0.83
Tabla 3.16 Valores del parámetro n de la ecuación [III.16] para temperaturas de tratamiento inferiores a la de la Tg del PMMA

85
Ta (°C) n Ta (°C) n
125 1.02 140 1.15
130 0.85 145 0.99
135 1.14 150 1.01
Tabla 3.17 Valores del parámetro n de la ecuación [III.16] para temperaturas de tratamiento superiores a la de la Tg del PMMA
En todos los casos se aprecia que los valores calculados de n son siempre cercanos
a la unidad bien por exceso bien por defecto, con un valor medio de n = 1.04, lo que
excluye una desviación sistemática de los mismos hacia otro valor diferente confirmando
de esta manera la dependencia lineal.
Asumida la dependencia lineal de la transición vítrea con la temperatura, se pueden
recalcular los valores de la pendiente a partir de la ecuación:
t K = T - T ogg [III,17]
Los valores obtenidos para la pendiente son los siguientes:
Ta (°C) K (K/s) Ta (°C) K (K/s)
75 3.28 ⋅ 10-4 100 5.47 ⋅ 10-4
80 7.41 ⋅ 10-4 105 7.18 ⋅ 10-3
85 1.39 ⋅ 10-3 110 9.35 ⋅ 10-3
90 2.11 ⋅ 10-3 115 8.18 ⋅ 10-3
95 4.11 ⋅ 10-3 120 12.27 ⋅ 10-3
Tabla 3.18 Valores de la pendiente de la ecuación [III.17] para temperaturas de tratamiento inferiores a la de la Tg del PMMA

86
Ta (°C) K (K/s) Ta (°C) K (K/s)
125 9.18 ⋅ 10-3 140 4.17 ⋅ 10-3
130 8.17 ⋅ 10-3 145 2.06 ⋅ 10-3
135 6.01 ⋅ 10-3 150 4.00 ⋅ 10-4
Tabla 3.19 Valores de la pendiente de la ecuación [III.17] para temperaturas de tratamiento superiores a la de la Tg del PMMA
Para la segunda región, correspondiente a tiempos de tratamiento largos, se puede
aplicar un tratamiento matemático similar sin más modificación que la de considerar como
origen el punto de intersección de las dos regiones, denominado punto de cambio de
régimen, quedando una expresión del tipo:
) t - t ( K = T - T nc
cgg [III,18]
Siendo Tgc y tc los valores correspondientes a la transición vítrea y el tiempo de
tratamiento en el punto de cambio de régimen.
Una dificultad que existe cuando se realiza este ajuste es que para calcular los
valores de Tgc y tc es preciso suponer previamente una dependencia lineal en la ecuación
[III,18] al obtenerse estos valores por la intersección de las dos rectas, mientras que para
confirmar la dependencia lineal de Tg con el tiempo estimando el valor de n que mejor se
ajusta a los resultados experimentales se necesita conocer previamente Tgc y tc.
Como solución hemos optado por asumir directamente la dependencia lineal de la
temperatura de transición vítrea con el tiempo también para esta segunda región, por
analogía con la primera región y ante la evidencia experimental reflejada en la figura 3.21.
Calculando los puntos de corte de las dos rectas para cada temperatura de tratamiento, se
obtienen finalmente los valores de Tgc y tc para los puntos de cambio de régimen que
vienen recogidos en la tabla 3.20:
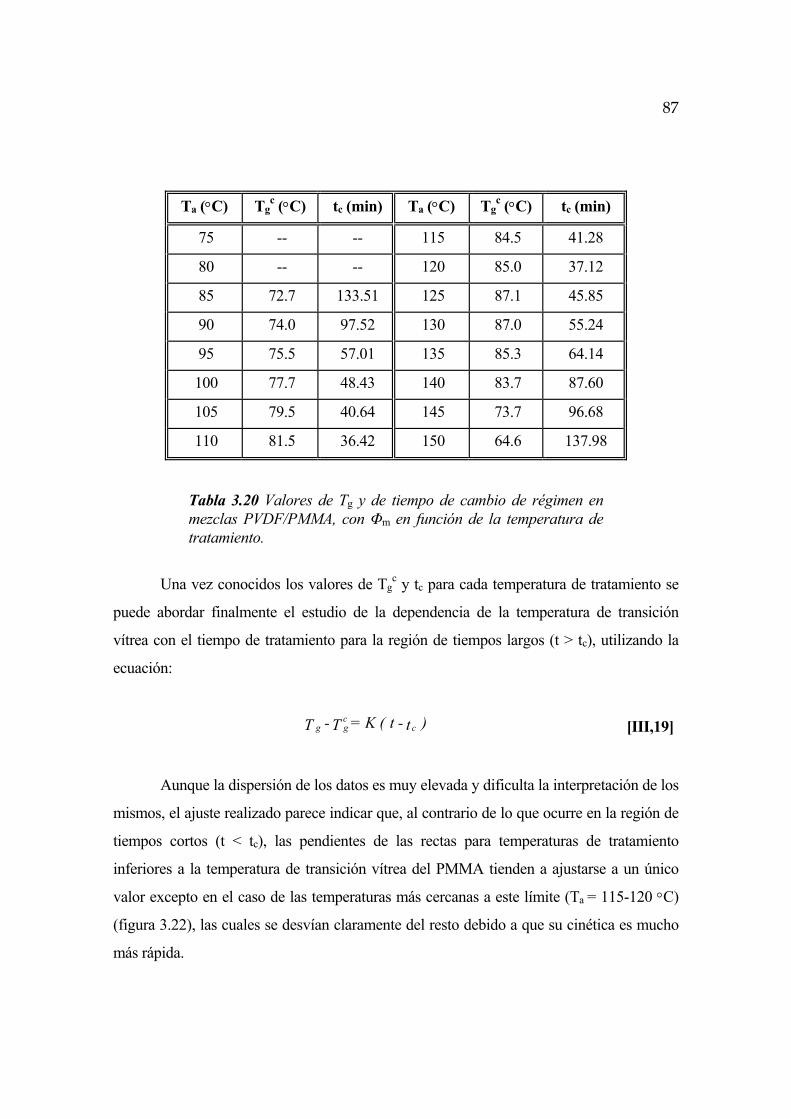
87
T (°C) a T (°C) gc t (min) c T (°C) T (°C) a g
c t (min) c
75 -- -- 115 84.5 41.28
80 -- -- 120 85.0 37.12
85 72.7 133.51 125 87.1 45.85
90 74.0 97.52 130 87.0 55.24
75.5 57.01 135 85.3 64.14
100 77.7 48.43 140 83.7 87.60
105 79.5 40.64 145 73.7 96.68
110 81.5 36.42 150 64.6 137.98
95
Tabla 3.20 Valores de Tg y de tiempo de cambio de régimen en mezclas PVDF/PMMA, con Φm en función de la temperatura de tratamiento.
Una vez conocidos los valores de Tgc y tc para cada temperatura de tratamiento se
puede abordar finalmente el estudio de la dependencia de la temperatura de transición
vítrea con el tiempo de tratamiento para la región de tiempos largos (t > tc), utilizando la
ecuación:
) t - t ( K = T - T ccgg [III,19]
Aunque la dispersión de los datos es muy elevada y dificulta la interpretación de los
mismos, el ajuste realizado parece indicar que, al contrario de lo que ocurre en la región de
tiempos cortos (t < tc), las pendientes de las rectas para temperaturas de tratamiento
inferiores a la temperatura de transición vítrea del PMMA tienden a ajustarse a un único
valor excepto en el caso de las temperaturas más cercanas a este límite (Ta = 115-120 °C)
(figura 3.22), las cuales se desvían claramente del resto debido a que su cinética es mucho
más rápida.
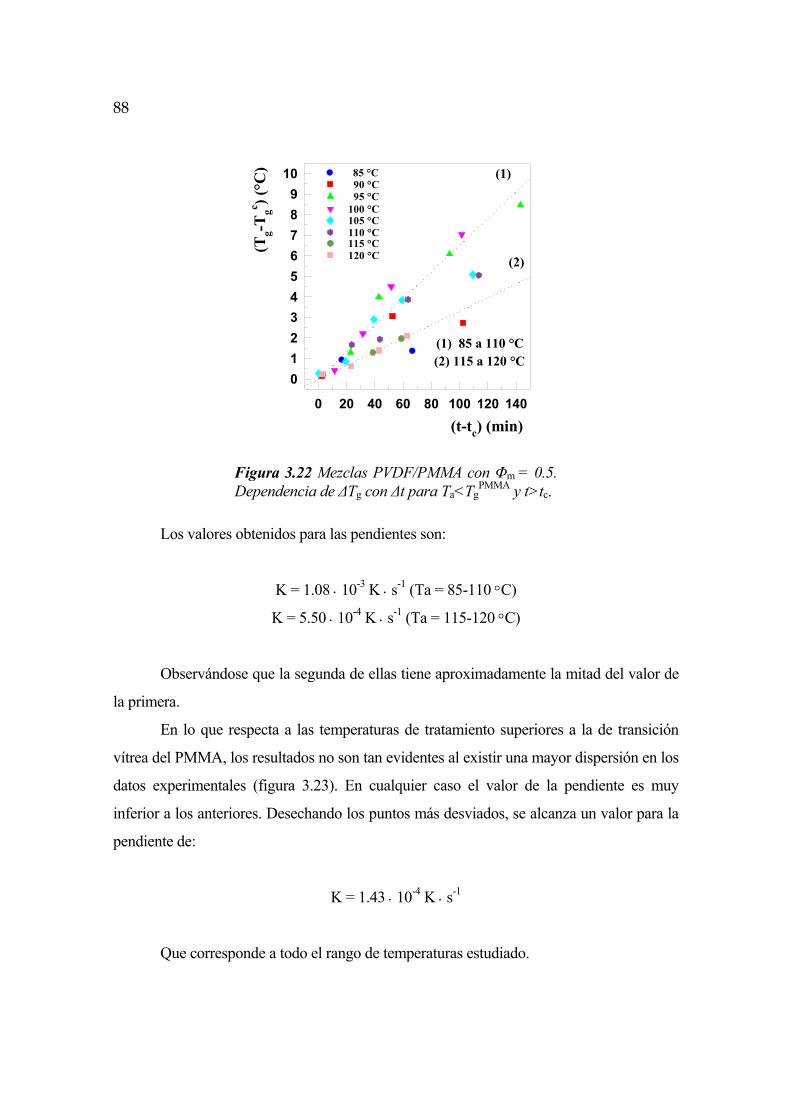
88
0 20 40 60 80 100 120 140
0123456789
10
(Tg-
Tgc ) (
°C)
(t-tc) (min)
(2) 115 a 120 °C(1) 85 a 110 °C
(1)
(2)
90 °C 95 °C100 °C105 °C110 °C115 °C120 °C
85 °C
Figura 3.22 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de ∆Tg con ∆t para Ta<Tg
PMMA y t>tc.
Los valores obtenidos para las pendientes son:
K = 1.08 ⋅ 10-3 K ⋅ s-1 (Ta = 85-110 °C)
K = 5.50 ⋅ 10-4 K ⋅ s-1 (Ta = 115-120 °C)
Observándose que la segunda de ellas tiene aproximadamente la mitad del valor de
la primera.
En lo que respecta a las temperaturas de tratamiento superiores a la de transición
vítrea del PMMA, los resultados no son tan evidentes al existir una mayor dispersión en los
datos experimentales (figura 3.23). En cualquier caso el valor de la pendiente es muy
inferior a los anteriores. Desechando los puntos más desviados, se alcanza un valor para la
pendiente de:
K = 1.43 ⋅ 10-4 K ⋅ s-1
Que corresponde a todo el rango de temperaturas estudiado.
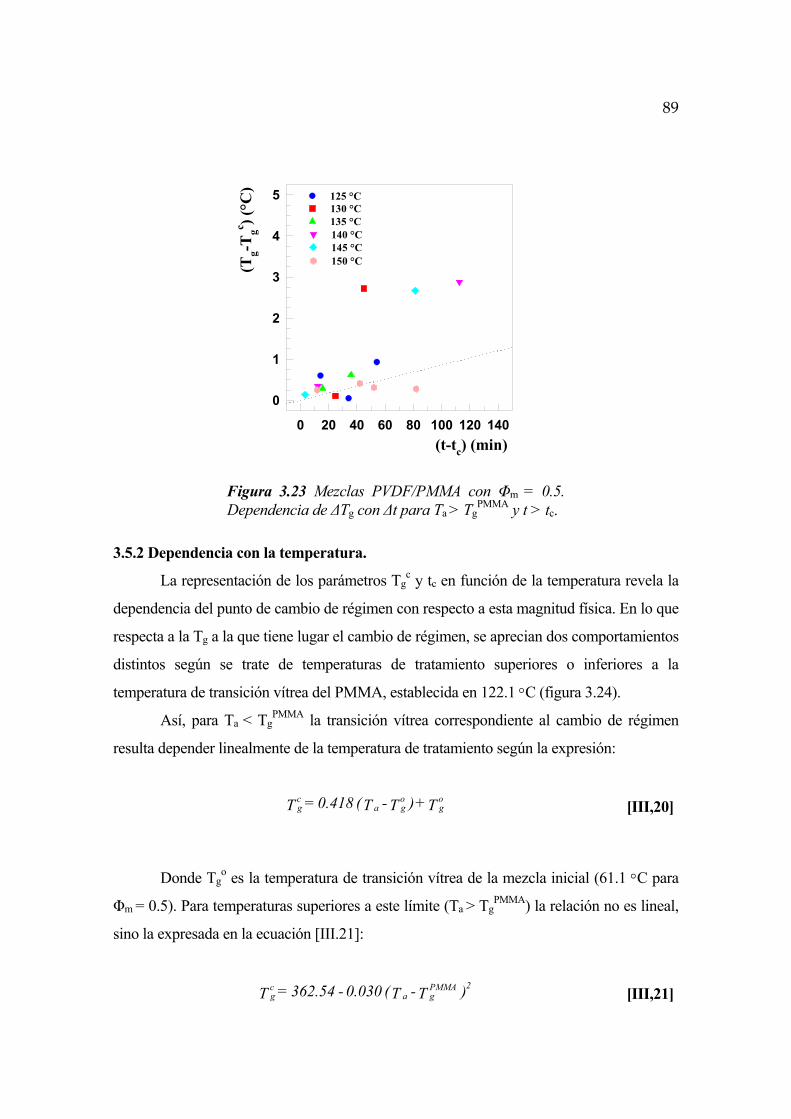
89
0 20 40 60 80 100 120 140
0
1
2
3
4
5
(Tg-
Tgc ) (
°C)
(t-tc) (min)
125 °C130 °C135 °C140 °C145 °C150 °C
Figura 3.23 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de ∆Tg con ∆t para Ta > Tg
PMMA y t > tc. 3.5.2 Dependencia con la temperatura.
La representación de los parámetros Tgc y tc en función de la temperatura revela la
dependencia del punto de cambio de régimen con respecto a esta magnitud física. En lo que
respecta a la Tg a la que tiene lugar el cambio de régimen, se aprecian dos comportamientos
distintos según se trate de temperaturas de tratamiento superiores o inferiores a la
temperatura de transición vítrea del PMMA, establecida en 122.1 °C (figura 3.24).
Así, para Ta < TgPMMA la transición vítrea correspondiente al cambio de régimen
resulta depender linealmente de la temperatura de tratamiento según la expresión:
T + )T - T( 0.418 = T og
oga
cg [III,20]
Donde Tgo es la temperatura de transición vítrea de la mezcla inicial (61.1 °C para
Φm = 0.5). Para temperaturas superiores a este límite (Ta > TgPMMA) la relación no es lineal,
sino la expresada en la ecuación [III.21]:
)T - T( 0.030 - 362.54 = T 2PMMAga
cg [III,21]

90
0 20 40 60 80 100
330
335
340
345
350
355
360
365
Tg
PMMA-T
g
o
Ta-T
g
o (K)
Tgc (K
)
Figura 3.24 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de la transición vítrea de cambio de régimen con la temperatura de tratamiento.
Por su parte, la representación de los tiempos a los que tiene lugar el cambio de
régimen (figura 3.25) muestra asimismo un comportamiento distinto en función de que las
temperaturas de tratamiento sean superiores o inferiores a la de la transición vítrea del
PMMA. Sin embargo, en este caso la divisoria entre una y otra rama no coincide
exactamente con el valor de este parámetro físico. Esta circunstancia puede ser atribuible a
la dificultad de cálculo de los valores de tc en las proximidades de la transición vítrea del
PMMA, al corresponder este rango a la región en la cual la cinética de segregación es más
rápida. Para las temperaturas de tratamiento térmico comprendidas en el intervalo
Ta < TgPMMA se aprecia una dependencia del tipo:
)T - T( 10 2.10 = t1 2o
ga7-
c
⋅ [III,22]

91
Mientras para el intervalo Ta > TgPMMA la ecuación adopta la expresión:
)T - (441.9 10 1.59 + 10 7.40 = t1 2
a7-5-
c
⋅⋅ [III,23]
Según esta ecuación, la inversa del tiempo de cambio de régimen tc depende de la
diferencia existente entre la temperatura de tratamiento térmico y un valor asimilable al del
punto de fusión del PVDF cristalizado.
1/tc ·
104 (s
-1)
0 20 40 60 80 100
0
1
2
3
4
5
Ta-Tgo (K)
TgPMMA-Tg
o
Figura 3.25 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia del tiempo de cambio de régimen con la temperatura de tratamiento.
Para realizar el estudio de la dependencia de la transición vítrea con la temperatura
hemos supuesto, en los valores correspondientes a la región de tiempos cortos (t < tc), una
relación exponencial entre el cociente (Tg-Tgo)/t (las pendientes de las expresiones
isotérmicas, expresadas en grados Kelvin por segundo) y la temperatura de tratamiento Ta,
a partir de los siguientes valores experimentales:

92
Ta (°C) (Tg-Tgo)/t σ Ta (°C) (Tg-Tg
o)/t σ 75 3.28 ⋅ 10-4 4.45 ⋅ 10-5 115 8.18 ⋅ 10-3 2.05 ⋅ 10-3 80 7.41 ⋅ 10-4 9.58 ⋅ 10-5 120 12.27 ⋅ 10-3 6.75 ⋅ 10-4 85 1.39 ⋅ 10-3 2.54 ⋅ 10-4 125 9.18 ⋅ 10-3 1.53 ⋅ 10-3 90 2.11 ⋅ 10-3 1.94 ⋅ 10-4 130 8.17 ⋅ 10-3 2.00 ⋅ 10-4 95 4.11 ⋅ 10-3 4.24 ⋅ 10-4 135 6.01 ⋅ 10-3 3.07 ⋅ 10-4 100 5.47 ⋅ 10-4 6.92 ⋅ 10-4 140 4.17 ⋅ 10-3 4.03 ⋅ 10-4 105 7.18 ⋅ 10-3 4.18 ⋅ 10-4 145 2.06 ⋅ 10-3 2.23 ⋅ 10-4 110 9.35 ⋅ 10-3 7.24 ⋅ 10-4 150 4.00 ⋅ 10-4 9.83 ⋅ 10-4
Tabla 3.21 Pendientes de las rectas isotérmicas en función de la temperatura de tratamiento. σ = desviación típica de las medidas experimentales.
A los cuales se les ha aplicado la ecuación:
)T - T( b + a = t
)T - T( 2M
ga
ogg lnln [III,24]
O lo que es lo mismo:
])T - T( [b t a + T = T 2M
gaogg exp [III,25]
La ecuación [III,25] es la expresión de una gaussiana y, como puede apreciarse en
la figura 3.26, nuevamente el ajuste sólo es posible para los puntos con Ta < TgPMMA por un
lado y Ta > TgPMMA por el otro, ya que en ambos casos la cinética es distinta, aunque en las
dos ocasiones el máximo de la curva coincide con el correspondiente a la temperatura de
transición vítrea del PMMA (Tg = 395.1 K).

93
TgM
340 360 380 400 420 440
0
2
4
6
8
10
12
14
(Tg-
Tgo )/t
· 10
3 (K/s
)
Ta (K)
Ta > TgM
Ta < TgM
Figura 3.26 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de la transición vítrea con el tiempo y la temperatura de tratamiento, para tiempos inferiores al tiempo de cambio de régimen.
Para las temperaturas de tratamiento inferiores a Tg
PMMA se obtienen los siguientes
valores para los parámetros a y b:
a = 1.21 ⋅ 10-2 K s-1
b = -1.62 ⋅ 10-3 K-2
Y para las temperaturas superiores a ella:
a = 9.62 ⋅ 10-3 K s-1
b = -2.89 ⋅ 10-3 K-2
En lo que respecta a la región de tiempos de tratamiento superiores al tiempo de
cambio de régimen (t > tc), la ya comentada independencia de los valores de las pendientes
isotermas con la temperatura hace innecesario realizar un estudio similar al correspondiente
a la región de tiempos cortos, asumiéndose los valores de (Tg-Tgc)/(t-tc) calculados
anteriormente para temperaturas de tratamiento bajas (Ta = 85-110 °C), temperaturas de
tratamiento próximas (aunque inferiores) a la transición vítrea del PMMA (Ta = 115-120
°C) y temperaturas de tratamiento superiores a este valor (Ta > 125 °C).
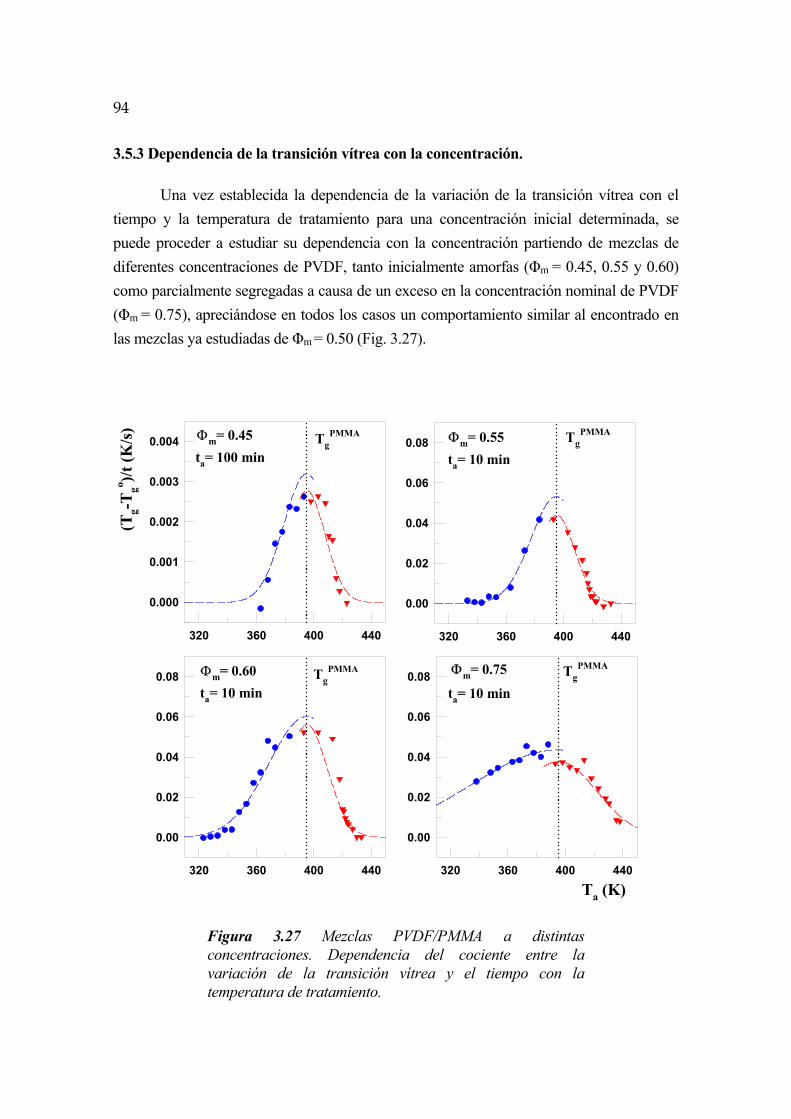
94
3.5.3 Dependencia de la transición vítrea con la concentración.
Una vez establecida la dependencia de la variación de la transición vítrea con el tiempo y la temperatura de tratamiento para una concentración inicial determinada, se puede proceder a estudiar su dependencia con la concentración partiendo de mezclas de diferentes concentraciones de PVDF, tanto inicialmente amorfas (Φm = 0.45, 0.55 y 0.60) como parcialmente segregadas a causa de un exceso en la concentración nominal de PVDF (Φm = 0.75), apreciándose en todos los casos un comportamiento similar al encontrado en las mezclas ya estudiadas de Φm = 0.50 (Fig. 3.27).
Ta (K)320 360 400 440
0.00
0.02
0.04
0.06
0.08
320 360 400 440
0.00
0.02
0.04
0.06
0.08
320 360 400 440
0.00
0.02
0.04
0.06
0.08
320 360 400 440
0.000
0.001
0.002
0.003
0.004
(Tg-
Tgo )/t
(K/s
) Φm= 0.45 Φm= 0.55
Φm= 0.60 Φm= 0.75
ta= 100 min ta= 10 min
ta= 10 min ta= 10 min
TgPMMA Tg
PMMA
TgPMMA Tg
PMMA
Figura 3.27 Mezclas PVDF/PMMA a distintas concentraciones. Dependencia del cociente entre la variación de la transición vítrea y el tiempo con la temperatura de tratamiento.

95
De esta manera se puede diferenciar de nuevo entre dos cinéticas distintas
dependiendo de que la temperatura de tratamiento Ta sea inferior o superior a la
correspondiente a la de la transición vítrea del PMMA, fijada en 395.1 K. También es
posible ajustar cada una de las dos regiones a una gaussiana del tipo:
] ) T - T( b [ a = tT - T 2PMMA
ga
ogg ⋅⋅ exp [III,26]
Utilizando la ecuación [III,26] se encuentran los siguientes parámetros de ajuste a y
b en función de la concentración (tablas 3.22 y 3.23):
Φm 0.45 0.50 0.55 0.60 0.75
a ⋅ 102 0.32 1.21 5.33 6.01 4.35
b ⋅ 103 -1.97 -1.62 -1.60 -0.71 -0.14
Tabla 3.22 Valores de los parámetros a y b de la ecuación [III,26] para Ta < Tg
PMMA.
Φm 0.45 0.50 0.55 0.60 0.75
a ⋅ 102 0.28 0.96 4.40 5.61 3.78
b ⋅ 103 -3.03 -2.89 -3.11 -2.40 -0.69
Tabla 3.23 Valores de los parámetros a y b de la ecuación [III,26] para Ta > Tg
PMMA.
Representados a su vez los valores de los parámetros a y b en función de la
concentración (figuras 3.28 y 3.29), se aprecia una variación monótona de los mismos en
los correspondientes a las mezclas amorfas. El factor preexponencial (el parámetro a), que
determina la amplitud de la cinética de segregación, presenta un comportamiento que se
puede ajustar a una gaussiana sin que se aprecien diferencias significativas entre las dos
regiones. La expresión encontrada, común para ambas series, es:
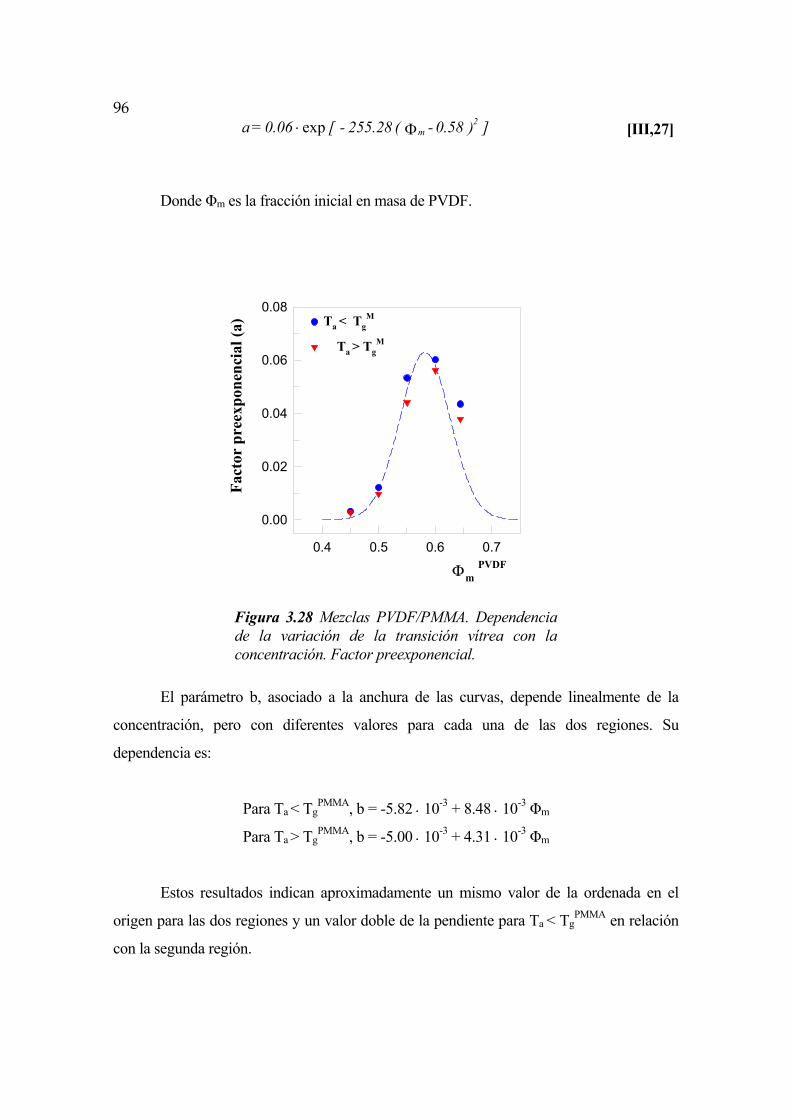
96] )0.58- ( 255.28- [ 0.06 = a 2
mΦ⋅ exp [III,27]
Donde Φm es la fracción inicial en masa de PVDF.
0.4 0.5 0.6 0.7
0.00
0.02
0.04
0.06
0.08
Φm PVDF
Ta > TgM
Ta < TgM
Fact
or p
reex
pone
ncia
l (a)
Figura 3.28 Mezclas PVDF/PMMA. Dependencia de la variación de la transición vítrea con la concentración. Factor preexponencial.
El parámetro b, asociado a la anchura de las curvas, depende linealmente de la
concentración, pero con diferentes valores para cada una de las dos regiones. Su
dependencia es:
Para Ta < TgPMMA, b = -5.82 ⋅ 10-3 + 8.48 ⋅ 10-3 Φm
Para Ta > TgPMMA, b = -5.00 ⋅ 10-3 + 4.31 ⋅ 10-3 Φm
Estos resultados indican aproximadamente un mismo valor de la ordenada en el
origen para las dos regiones y un valor doble de la pendiente para Ta < TgPMMA en relación
con la segunda región.

97
0.4 0.5 0.6 0.7 0.8-4
-3
-2
-1
0
1
Φm PVDF
Fact
or e
xpon
enci
al (b
) · 1
03
Ta> TgM
Ta< TgM
Figura 3.29 Mezclas PVDF/PMMA. Dependencia de la variación de la transición vítrea con la concentración. Factor exponencial.
Un caso especial es el de las muestras con Φm = 0.75 a causa de la ya comentada
segregación parcial existente en las muestras iniciales. Debido a este fenómeno la
concentración inicial de estas muestras no es realmente la nominal, al estar parte del PVDF
cristalizado, sino inferior a ésta. Puesto que la temperatura de transición vítrea inicial de
estas muestras es de 37.8 °C (la correspondiente a un 75% real sería de 18.5 grados
centígrados), la concentración correspondiente a la matriz amorfa ha sido calculada en Φm
= 0.64. Esta corrección en la concentración no resulta suficiente, sin embargo, para explicar
las discrepancias encontradas en estas muestras.
Un estudio de la dependencia del cociente (Tg-TgPMMA)/t frente a Ta revela que, a
diferencia de lo que ocurre en las muestras con concentraciones inferiores de PVDF, en el
caso de Φm = 0.75 el ajuste de la gaussiana no cumple una de las condiciones de contorno:
Para Ta = Tgo el incremento Tg - Tg
o debería ser igual a cero, mientras los valores obtenidos
para ese intervalo son de 9.8 y 0.2 K respectivamente para antes y después de la transición
vítrea del PMMA. Esta discrepancia puede ser atribuida a la diferente morfología existente
en las mezclas de Φm = 0.75 en relación con las inicialmente amorfas, lo que permite
esperar una mayor complejidad en su estructura interna. Por esta razón, hemos realizado un

98estudio por separado de estas muestras al haberse apreciado en ellas otras peculiaridades
en relación con el resto de las concentraciones.
3.6 Cinética de cristalización en mezclas de PVDF y PMMA.
La segunda magnitud termodinámica cuya cinética ha sido estudiada es la entalpía
de fusión de muestras segregadas, tanto en aquéllas inicialmente amorfas (Φm ≤ 0.6) como
en las que poseían un exceso neto de PVDF (Φm = 0.75). Lo primero que se observa al
estudiar los termogramas es la aparición, en todas las concentraciones investigadas, de dos
(y para las concentraciones más altas, tres) picos de fusión, uno de los cuales experimenta
corrimientos en sus máximos en función de la temperatura de tratamiento mientras el otro
mantiene constante este valor. Estos picos pueden deberse, en principio, tanto a la fusión de
cristales de distinta morfología como a dos poblaciones de la misma fase pero de diferente
tamaño, siendo preciso un estudio más detallado para determinarlo.
Como puede apreciarse en las figuras 3.30 a 3.34, un incremento en la
concentración del PVDF se traduce en una mayor definición de los picos de fusión y en la
aparición, para Φm ≥ 0.6, del tercero de ellos a temperaturas elevadas, del orden de
Ta = 150 °C. Este tercer pico, incipiente en las muestras de Φm = 0.6 y muy desarrollado
para Φm = 0.75, corresponde a un proceso de transición de fase similar al registrado
mediante difracción de rayos X.
En lo que respecta a los dos picos principales, el comportamiento de los mismos
resulta ser muy distinto en los procesos isotermos. El primero de ellos aparece en las
primeras etapas del proceso creciendo de una manera uniforme en función del tiempo hasta
alcanzar una saturación. Su punto de fusión es independiente o prácticamente
independiente de la temperatura de tratamiento, lo que sugiere que tiene su origen en un
sistema muy ordenado.
La segunda población se inicia a cierta temperatura y continúa creciendo
significativamente después de que la primera se satura. Su punto de fusión varía
apreciablemente en función de la temperatura de tratamiento, siendo mayor cuanto más
cerca esté de la temperatura de la transición vítrea del PMMA a la cual tiene lugar la
cinética máxima, por lo que cabe admitir que estos cristales se forman a partir de un medio
desordenado.
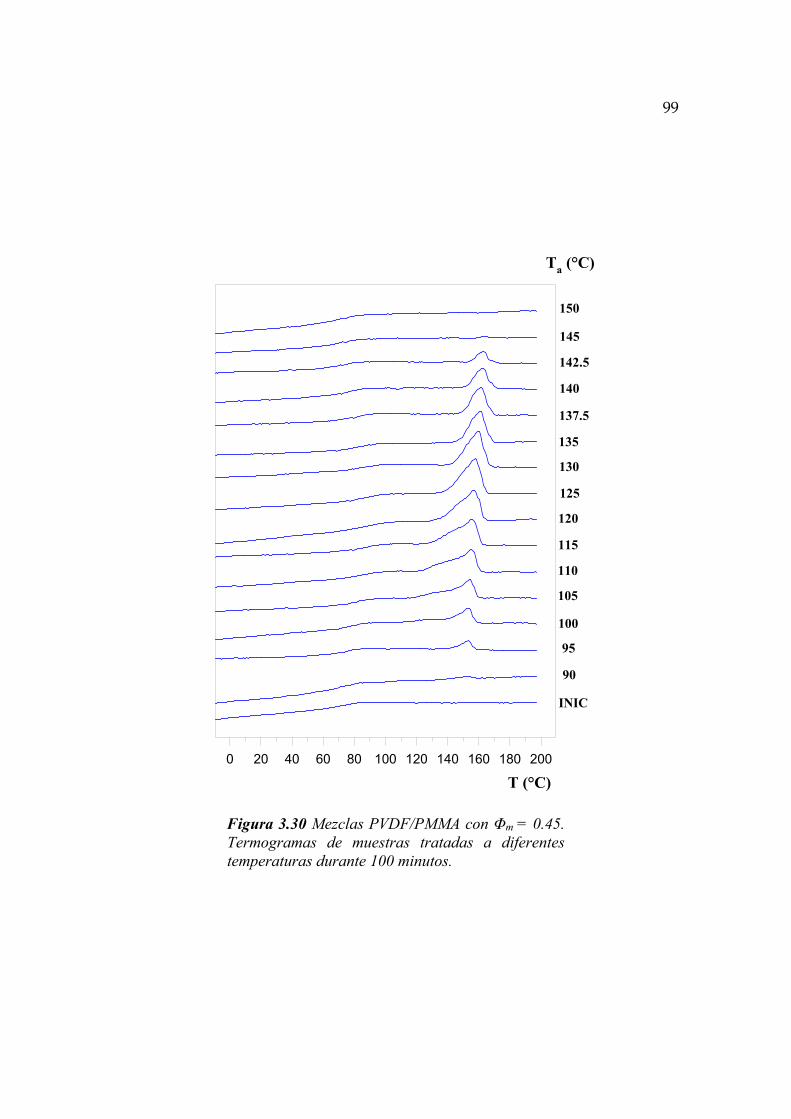
99
130
150
145
142.5
137.5
110
100
95
0 20 40 60 80 100 120 140 160 180 200
INIC
Ta (°C)
90
105
115
120
125
135
140
T (°C)
Figura 3.30 Mezclas PVDF/PMMA con Φm = 0.45. Termogramas de muestras tratadas a diferentes temperaturas durante 100 minutos.

100
70
75
65
8085
150
145
130
110
10095
0 20 40 60 80 100 120 140 160 180 200
90
T (°C)
Ta (°C)
140
135
125120115
105
INIC
Figura 3.31 Mezclas PVDF/PMMA con Φm = 0.5. Termogramas de muestras tratadas a diferentes temperaturas durante 100 minutos.
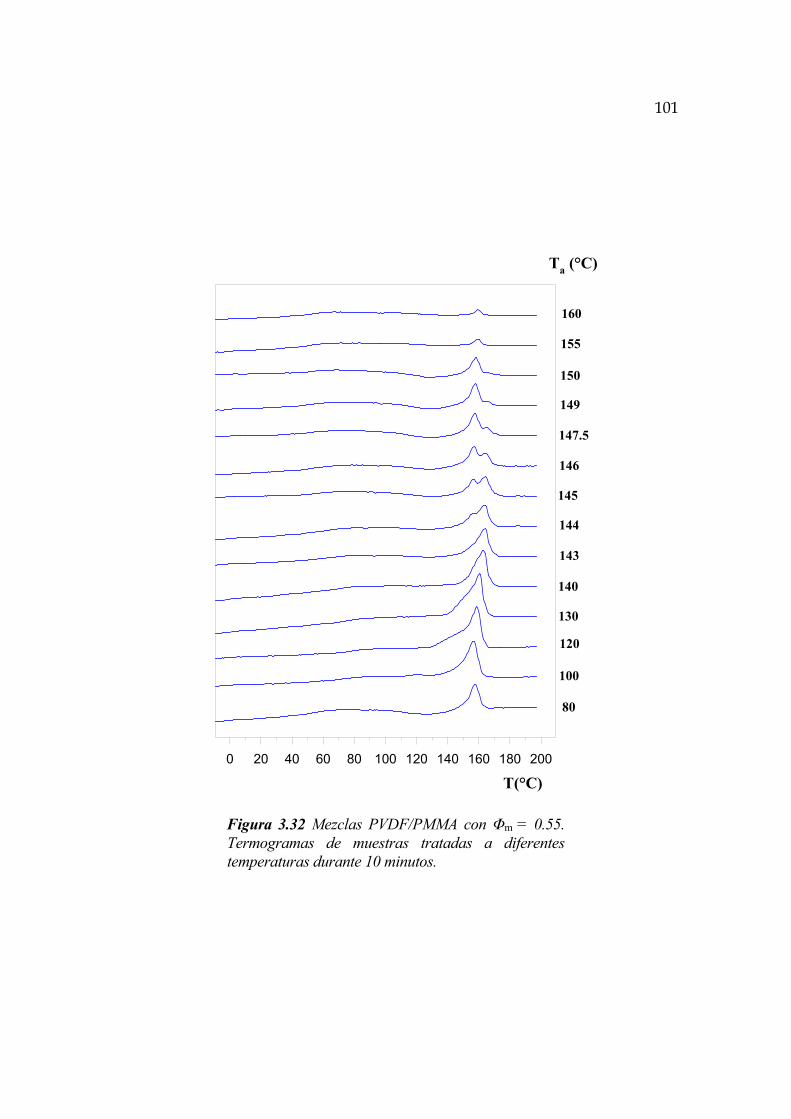
101
80
100
130
145
147.5
150
155
160
0 20 40 60 80 100 120 140 160 180 200
T(°C)
Ta (°C)
146
120
140
143
144
149
Figura 3.32 Mezclas PVDF/PMMA con Φm = 0.55. Termogramas de muestras tratadas a diferentes temperaturas durante 10 minutos.
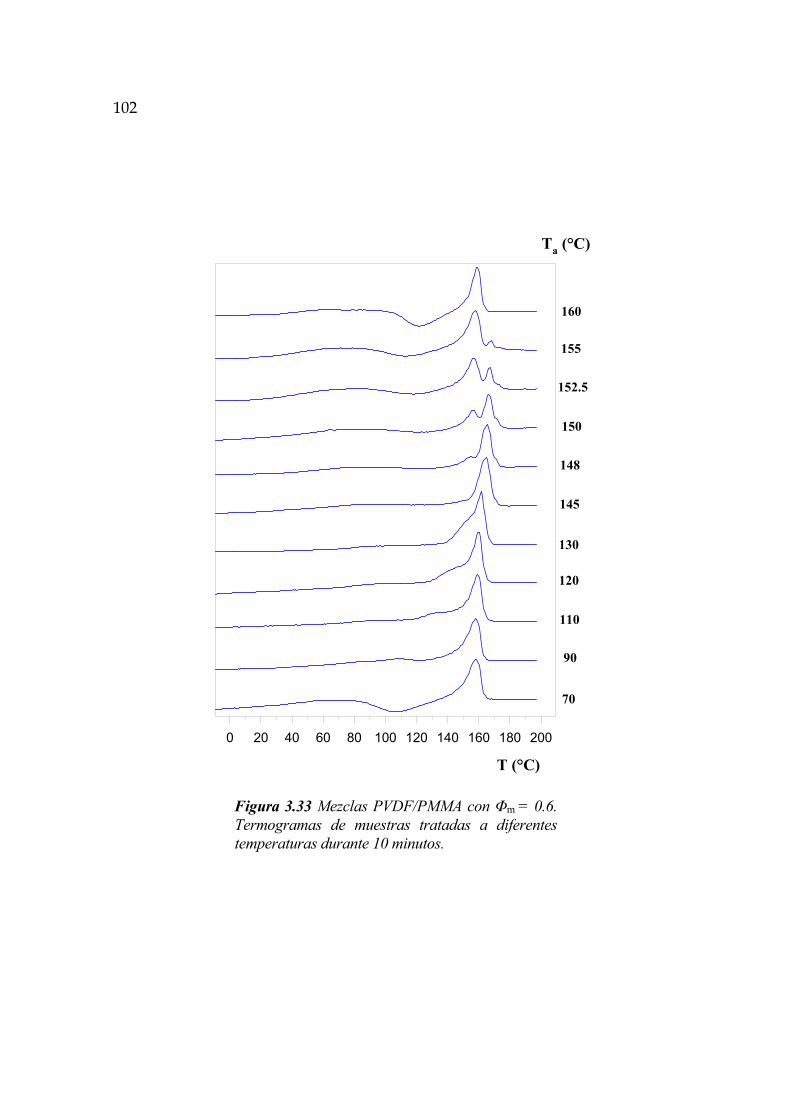
102
160
70
90
130
145
148
150
152.5
155
0 20 40 60 80 100 120 140 160 180 200
Ta (°C)
T (°C)
110
120
Figura 3.33 Mezclas PVDF/PMMA con Φm = 0.6. Termogramas de muestras tratadas a diferentes temperaturas durante 10 minutos.

103
150
145
130
100
80
155
160
0 20 40 60 80 100 120 140 160 180 200
Ta (°C)
T (°C)
INIC
120
140
157.5
162.5
165
Figura 3.34 Mezclas PVDF/PMMA con Φm = 0.75. Termogramas de muestras tratadas a diferentes temperaturas durante 10 minutos.

104
3.6.1 Cristalización isotérmica. Dependencia temporal.
El estudio de la cristalización isotérmica ha sido realizado en muestras de
concentración media (Φm = 0.5), por contar ésta con la cinética más adecuada para ser
investigada experimentalmente. Las mezclas inicialmente amorfas se han tratado
térmicamente a diferentes temperaturas y se ha estudiado la evolución de la cristalinidad en
función del tiempo. Los resultados obtenidos, en lo que respecta a la entalpía de fusión
total, muestran curvas sigmoidales que tienden, después de determinado tiempo, a la
saturación. Este comportamiento es general para todas las temperaturas estudiadas, si bien
la velocidad del proceso demuestra depender fuertemente de la temperatura de
cristalización. La cinética de cristalización es más rápida en torno a unas temperaturas de
tratamiento del orden de Ta = 120-130 °C, y es tanto más lenta cuanto más cercana sea Ta al
valor de la Tg inicial ( ~ 61 °C) o al valor de Tm ( ~ 155 °C) (figuras 3.35 y 3.36).
0 50 100 150 200 250 300
0
1
2
3
4
5
∆H
(cal
/g)
115 °C 110 °C105 °C100 °C
90 °C
85 °C
70 °C 60 °C
t (min)
Figura 3.35 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de muestras cristalizadas a partir del estado amorfo, en función del tiempo de tratamiento (1).
95 °C
80 °C
75 °C

105
0 50 100 150 200 250 300
0
1
2
3
4
5∆
H (c
al/g
)120 °C
130 °C
145 °C
140 °C
t (min)
Figura 3.36 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de muestras cristalizadas a partir del estado amorfo, en función del tiempo de tratamiento (2).
125 °C
150 °C
El siguiente paso ha consistido en separar las contribuciones de los dos picos de
fusión de la entalpía total. La descomposición de las curvas isotérmicas revela un
comportamiento sistemático en todos los casos (figura 3.37): Aparición en las primeras
etapas del proceso del primer pico de fusión, crecimiento rápido del mismo hasta alcanzar
una saturación, y la aparición a tiempos más largos de un segundo pico que continúa
creciendo una vez que el primero se ha saturado. Aunque existe una región intermedia en la
que ambos picos crecen simultáneamente, se puede considerar, como primera
aproximación, que el crecimiento de la entalpía total se debe en su mayor parte a la primera
población de cristales para tiempos cortos, y a la segunda población para tiempos largos. El
tiempo al que tiene lugar el cambio de régimen depende de la temperatura de tratamiento y,
como se verá más adelante, está relacionado con el tiempo de cambio de régimen en la
transición vítrea.
En la figura 3.37 se ha representado la entalpía de fusión (en escala logarítmica para
facilitar la observación del cambio de régimen) en función del tiempo. Este

106comportamiento es similar para todas las temperaturas estudiadas. El segundo pico de
fusión no aparece para temperaturas de tratamiento inferiores a 85 °C, dependiendo de Ta el
tiempo al que tiene lugar la aparición del mismo. En lo que respecta al tercer pico de
fusión, éste no ha sido observado en muestras de estas concentraciones, excepto de forma
incipiente, para temperaturas cercanas al punto de fusión y tiempos de tratamiento largos
(para Φm = 0.5), y más definido y a tiempos menores de tratamiento, pero a temperaturas
también cercanas al punto de fusión para concentraciones más elevadas (Φm = 0.6).
t (min)0 40 80 120 160 200
-3
-2
-1
0
1
2
3
0 40 80 120 160 200
-3
-2
-1
0
1
2
3
0 40 80 120 160 200
-3
-2
-1
0
1
2
3
0 40 80 120 160 200
-3
-2
-1
0
1
2
3
ln ∆
H Ta=90 ºC
Ta=100 ºC
Ta=95 ºC
Ta=120 ºC
Primera población cristalinaSegunda población cristalina
Entalpía total
Figura 3.37 Mezclas PVDF/PMMA con Φm = 0.5. Entalpías de fusión de las dos poblaciones cristalinas en función del tiempo de tratamiento.

107
Otro factor a tener en cuenta es el estudio de los puntos de fusión de las dos
poblaciones cristalinas, pudiéndose apreciar que el pico principal se mantiene constante,
para cada Ta, dentro del margen de error experimental. La determinación del punto de
fusión del segundo pico resulta mucho más dificultosa debido a que en numerosas
ocasiones éste está muy poco definido, no pudiéndose apreciar la existencia de ninguna
dependencia sistemática con el tiempo, por lo que se puede considerar que, al igual que
ocurre con el pico principal, el punto de fusión se mantiene constante (figura 3.38).
Segunda población cristalinaPrimera población cristalina t (min)
0 40 80 120 160 200
120
130
140
150
160
170 Ta=140 °C
0 40 80 120 160 200
120
130
140
150
160Ta=120 °C
0 40 80 120 160 200
120
130
140
150
160
170 Ta=110 °C
0 40 80 120 160 200
120
130
140
150
160
170
Tm
(°C
)
Ta=95 °C
Figura 3.38 Mezclas PVDF/PMMA con Φm = 0.5. Temperaturas de fusión de las dos poblaciones cristalinas en función del tiempo de tratamiento.
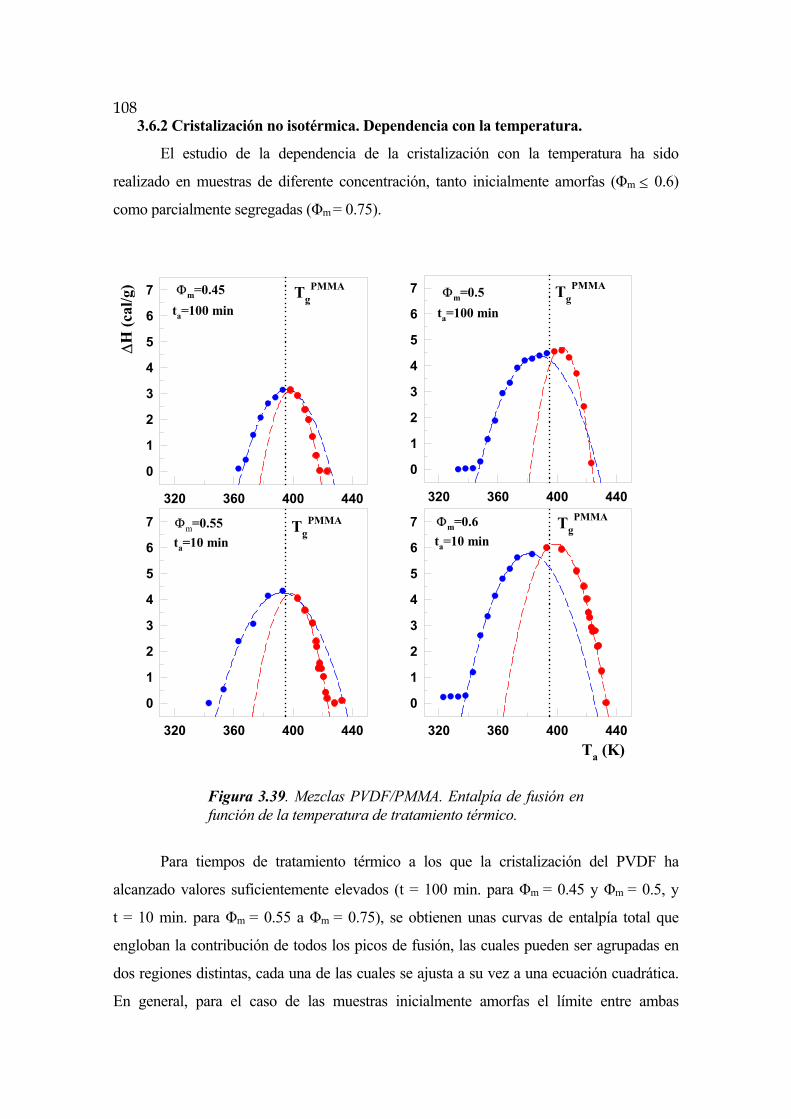
1083.6.2 Cristalización no isotérmica. Dependencia con la temperatura.
El estudio de la dependencia de la cristalización con la temperatura ha sido
realizado en muestras de diferente concentración, tanto inicialmente amorfas (Φm ≤ 0.6)
como parcialmente segregadas (Φm = 0.75).
Φm=0.6Φm=0.55
Φm=0.5Φm=0.45
320 360 400 440
0
1
2
3
4
5
6
7
320 360 400 440
0
1
2
3
4
5
6
7
320 360 400 440
0
1
2
3
4
5
6
7
320 360 400 440
0
1
2
3
4
5
6
7
Ta (K)
∆H
(cal
/g)
ta=10 minta=10 min
ta=100 min ta=100 minTg
PMMA TgPMMA
TgPMMA Tg
PMMA
Figura 3.39. Mezclas PVDF/PMMA. Entalpía de fusión en función de la temperatura de tratamiento térmico.
Para tiempos de tratamiento térmico a los que la cristalización del PVDF ha
alcanzado valores suficientemente elevados (t = 100 min. para Φm = 0.45 y Φm = 0.5, y
t = 10 min. para Φm = 0.55 a Φm = 0.75), se obtienen unas curvas de entalpía total que
engloban la contribución de todos los picos de fusión, las cuales pueden ser agrupadas en
dos regiones distintas, cada una de las cuales se ajusta a su vez a una ecuación cuadrática.
En general, para el caso de las muestras inicialmente amorfas el límite entre ambas

109
regiones se sitúa en torno a los 120 °C (es decir, la temperatura de transición vítrea del
PMMA), aunque la determinación de los límites resulta difícil de precisar en la zona de
transición entre las dos regiones al solaparse sus respectivas contribuciones (figura 3.39).
Las muestras con Φm = 0.75 siguen una vez más un comportamiento totalmente
diferente ya que, aunque también se pueden delimitar dos regiones distintas, el límite entre
ambas regiones queda situado en torno a los 150 °C, estando claramente relacionado con la
transición de fase cristalina que tiene lugar a esa temperatura (figura 3.40).
320 340 360 380 400 420 4403
4
5
6
7
8
9
10
Ta (K)
∆H
(cal
/g) Tg
PMMA
Figura 3.40 Mezclas PVDF/PMMA con Φm = 0.75 y ta = 10 min. Entalpía de fusión en función de la temperatura de tratamiento térmico.
Una información adicional puede ser obtenida por extrapolación de las curvas de
ajuste, cuyas intersecciones con el eje de temperatura corresponden físicamente con el
inicio de la cristalización (limitado a su vez por la transición vítrea en el caso de la primera
población cristalina) el primero, y con el punto de fusión del PVDF segregado el segundo.
En las mezclas inicialmente amorfas el ajuste es aceptable por ambos extremos y para las
dos curvas, correspondiéndose cada una de ellas en primera aproximación con el inicio de
los dos picos de fusión registrados en los termogramas. Las muestras con Φm = 0.75, como
ya era esperado, se desvían también de este comportamiento.

110
Aunque la representación de la entalpía de fusión total apunta de forma evidente a
la existencia de al menos dos cinéticas distintas solapadas entre sí, es conveniente
profundizar en este estudio descomponiendo la entalpía en las contribuciones
correspondientes a los respectivos picos. El estudio se ha realizado para distintas
concentraciones: Muestras de concentración media (Φm = 0.5) inicialmente amorfas y con
una cinética de cristalización lenta, tanto a tiempos de tratamiento cortos (t = 10 min.)
como largos (t = 100 min.); muestras de concentración elevada (Φm = 0.6) inicialmente
amorfas pero con cinética de cristalización rápida, y por último muestras con exceso de
PVDF (Φm = 0.75) parcialmente segregadas.
En el primero de estos casos, una mezcla de Φm = 0.5 tratada térmicamente durante
10 minutos, todos los puntos experimentales se encuentran en la primera región de
segregación con ta < tc (ver tabla 3.20), existiendo una única población cristalina (la
primera, de Tm constante) en todo el intervalo de temperaturas excepto en las proximidades
del máximo (Ta ≈ 120 °C). Aquí aparece una pequeña aportación de la segunda población
cristalina. La curva de entalpías muestra la forma de campana característica de estas
cinéticas, con un máximo coincidente con la transición vítrea del PMMA (Ta = 122 °C) y
una asimetría en sus dos ramas (figura 3.41). La escasa relevancia de la incipiente segunda
población cristalina, tanto en el rango de temperaturas como en los valores absolutos de
entalpías, hace innecesaria la descomposición de la curva en las componentes
correspondientes a las dos poblaciones cristalinas.
Muestras de la misma concentración (Φm = 0.5) pero con tiempos de tratamiento lo
suficientemente elevados (ta = 100 min.) muestran un crecimiento significativo y paralelo
de las dos poblaciones cristalinas, aunque la coexistencia de puntos experimentales
pertenecientes tanto a la primera región de segregación (ta < tc) como a la segunda (ta > tc)
marca unas diferencias notables en la forma de las respectivas curvas. Como puede
apreciarse en la figura 3.42, la curva de entalpías de la primera población cristalina presenta
una forma de campana asimétrica en todo el intervalo de temperaturas excepto en la región
comprendida entre los 90 y los 120 °C, en la cual queda interrumpida. Esta región
corresponde a los puntos experimentales que cumplen simultáneamente la doble condición
de encontrarse en la segunda región de segregación (ta > tc) y tener temperaturas de

111
tratamiento térmico inferiores a la de la transición vítrea del PMMA. La desviación de
estos puntos se debe al hecho de ser la cinética de segregación en esta región diferente de la
correspondiente a temperaturas de tratamiento inferiores, por lo que estos valores no
pueden por ello ser incluidos en el ajuste del resto de la curva.
340 360 380 400 420
0.0
0.5
1.0
1.5
2.0
Ta (K)
∆H
(cal
/g)
Figura 3.41 Mezclas PVDF/PMMA con Φm = 0.5 y ta = 10 min. Entalpía de fusión en función de la temperatura de tratamiento térmico.
En lo que se refiere a la segunda población cristalina, se observa un brusco corte
coincidiendo con la temperatura de transición vítrea del PMMA, correspondiendo a los
valores de temperaturas superiores a este límite unas entalpías de fusión muy inferiores a
las simétricas del rango inferior de temperaturas. Estudios realizados a otros tiempos
distintos de tratamiento térmico (40, 60 y 80 minutos) en los que también coexisten las dos
regiones de segregación en todo el intervalo de temperaturas, ofrecen resultados
equivalentes para ambas poblaciones cristalinas. Asimismo, se aprecia un pico incipiente
perteneciente a una tercera población cristalina a temperaturas de tratamiento próximas a la
de fusión (Ta = 150 °C) y tiempos de tratamiento relativamente elevados iguales o
superiores a los 80 minutos.
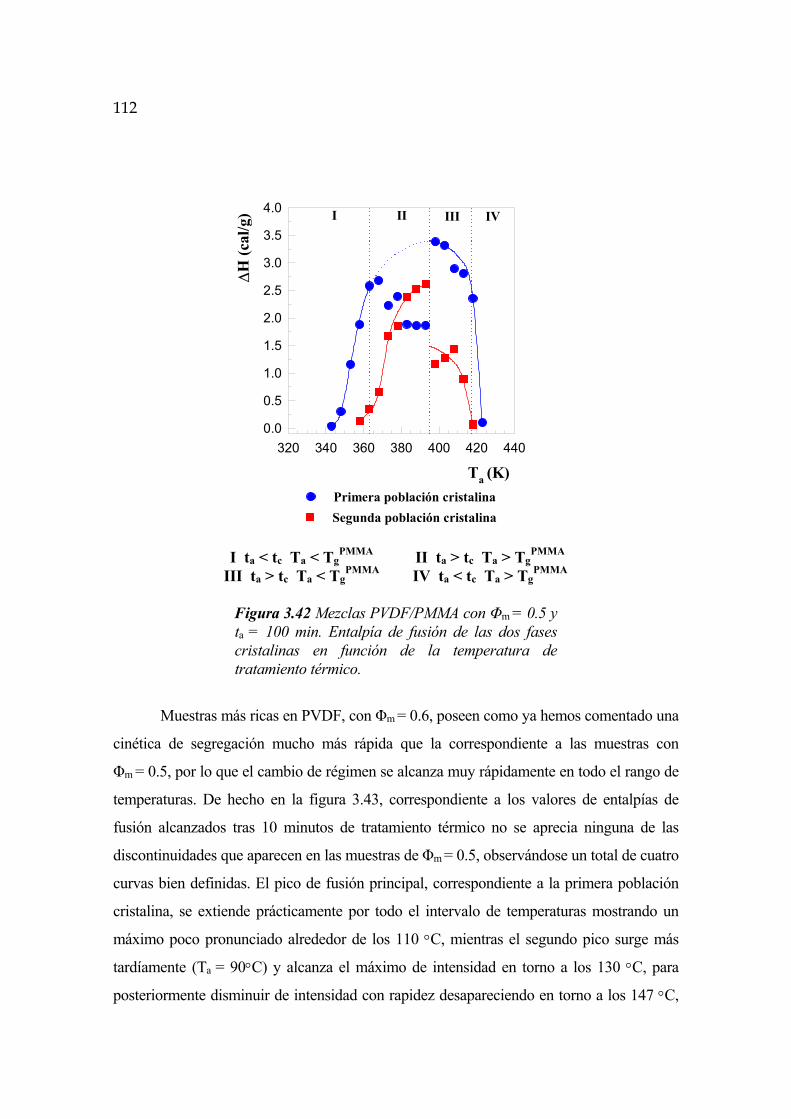
112
320 340 360 380 400 420 4400.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
Segunda población cristalinaPrimera población cristalina
Ta (K)
∆H
(cal
/g) I II III IV
I ta < tc Ta < TgPMMA II ta > tc Ta > Tg
PMMA III ta > tc Ta < Tg
PMMA IV ta < tc Ta > TgPMMA
Figura 3.42 Mezclas PVDF/PMMA con Φm = 0.5 y ta = 100 min. Entalpía de fusión de las dos fases cristalinas en función de la temperatura de tratamiento térmico.
Muestras más ricas en PVDF, con Φm = 0.6, poseen como ya hemos comentado una
cinética de segregación mucho más rápida que la correspondiente a las muestras con
Φm = 0.5, por lo que el cambio de régimen se alcanza muy rápidamente en todo el rango de
temperaturas. De hecho en la figura 3.43, correspondiente a los valores de entalpías de
fusión alcanzados tras 10 minutos de tratamiento térmico no se aprecia ninguna de las
discontinuidades que aparecen en las muestras de Φm = 0.5, observándose un total de cuatro
curvas bien definidas. El pico de fusión principal, correspondiente a la primera población
cristalina, se extiende prácticamente por todo el intervalo de temperaturas mostrando un
máximo poco pronunciado alrededor de los 110 °C, mientras el segundo pico surge más
tardíamente (Ta = 90°C) y alcanza el máximo de intensidad en torno a los 130 °C, para
posteriormente disminuir de intensidad con rapidez desapareciendo en torno a los 147 °C,

113
unos 15 °C por debajo de la temperatura de fusión de la totalidad de los cristales presentes
en la muestra, establecida en torno a los 160 °C.
320 340 360 380 400 420 4400
1
2
3
4
5
6 Segunda población cristalina.Primera población cristalina.
Ta (K)
∆H
(cal
/g)
Fases de alta temperatura.
Figura 3.43 Mezclas PVDF/PMMA con Φm = 0.6 y ta = 10 min. Entalpía de fusión de las distintas fases cristalinas en función de la temperatura de tratamiento térmico.
En el intervalo comprendido entre estos dos límites (147 ≤ Ta ≤ 160 °C) se aprecia
la aparición de tres picos de fusión; el primero de ellos puede ser asignado a la primera
población cristalina, y los restantes a dos formas cristalinas de alta temperatura asociadas
respectivamente a la recristalización de parte del PVDF durante el proceso de barrido en el
calorímetro y a una transición de fase que afecta a la primera población cristalina,
produciendo una especie cristalina diferente.
Las entalpías de fusión de las muestras con Φm = 0.75 revelan, como cabía esperar,
un comportamiento completamente distinto en relación al resto de las series de
concentraciones inferiores (Fig. 3.44). La entalpía de fusión inicial de la primera población
cristalina no es cero debido a la segregación previa existente en estas muestras,
incrementándose su valor inicial muy suavemente hasta llegar a la transición registrada

114entre los 155 y los 165 °C, es decir, unos 5 ó 10 °C por encima de los valores
correspondientes a la concentración anterior. La segunda población cristalina, aunque
existe en un amplio rango de temperaturas, está presente en muy poca cantidad mostrando
un comportamiento similar al de la población anterior, con un ligero incremento seguido de
una brusca caída coincidiendo con el inicio de la transición de fase. La recristalización
durante el proceso de barrido en el calorímetro en la región de altas temperaturas (155 ≤ Ta
≤ 165 °C) es en esta ocasión mucho menos significativa que para las muestras con Φm =
0.6, pero su comportamiento es similar. Por último, los cristales producidos por la
transición de fase experimentada por la primera población cristalina muestran también un
comportamiento similar al de las muestras con Φm = 0.6, aunque a temperaturas superiores
en el intervalo descrito.
320 340 360 380 400 420 440
0
2
4
6
8
10
Ta (K)
∆H
(cal
/g) Primera población cristalina
Fases de alta temperatura
Segunda población cristalina
Figura 3.44 Mezclas PVDF/PMMA con Φm = 0.75 y ta = 10 min. Entalpía de fusión de las distintas fases cristalinas en función de la temperatura de tratamiento térmico.
3.6.3 Cristalización no isotérmica. Dependencia de la entalpía de fusión con el tiempo
de cambio de régimen.
De todos los casos estudiados en el apartado anterior, el más complejo de ellos es
aquél en el que coexisten entalpías de fusión pertenecientes al primer régimen de
cristalización (ta < tc) con otras obtenidas en el segundo régimen de cristalización (ta > tc).
Por esta razón es preciso realizar un estudio más detallado de estas muestras diferenciando

115
en cada caso entre las cinéticas correspondientes a las regiones anterior y posterior al punto
de cambio de régimen halladas en el estudio de la variación de la transición vítrea, las
cuales indican un cambio en la evolución del sistema. Dado que los parámetros que definen
este cambio de régimen (Tgc y tc) dependen de la temperatura de tratamiento, para un
tiempo determinado parte de las entalpías medidas en función de la temperatura de
tratamiento pueden pertenecer a la primera región (t < tc) mientras el resto corresponden a
la segunda (t > tc).
Por tal motivo hemos estudiado la dependencia con la temperatura de las entalpías
de fusión de las dos poblaciones cristalinas para distintos tiempos de tratamiento,
seleccionando una concentración media (Φm = 0.5) en la cual la cinética de cristalización es
lo suficientemente rápida como para poder ser estudiada a tiempos cortos, pero no tanto
como para obtener saturaciones a tiempos más elevados. Tal como ha sido determinado
experimentalmente (tabla 3.20), para muestras de esta concentración el tiempo mínimo de
cambio de régimen resulta ser del orden de los 35-40 minutos para temperaturas de
tratamiento próximas a la correspondiente a la transición vítrea del PMMA, mientras en los
dos extremos del rango de temperaturas estudiados los tiempos críticos máximos están
situados en torno a los 130-140 minutos. Por último, para temperaturas de tratamiento
térmico cercanas a la transición vítrea inicial de las mezclas (Ta ≤ 85 °C) no se ha
encontrado ningún cambio de régimen en el intervalo temporal investigado.
Para tiempos de tratamiento inferiores a los 40 minutos todos los valores
experimentales de las entalpías de fusión corresponden a la primera región
independientemente de la temperatura, existiendo como única división el límite
correspondiente a la transición vítrea del PMMA.
Por el contrario, a partir de los 40 minutos de tratamiento las gráficas de entalpía
pueden ser divididas en cuatro zonas distintas las cuales, en orden creciente de
temperaturas, son:
t < tc y Ta < TgPMMA (región I)
t > tc y T a< TgPMMA (región II)
t > tc y Ta > TgPMMA (región III) y
t < tc y Ta > TgPMMA (región IV)

116Correspondiendo respectivamente a: Crecimiento de la primera población
cristalina por debajo de TgPMMA (región I); crecimiento de la segunda población cristalina
por debajo de TgPMMA (región II); crecimiento de la segunda población cristalina por
encima de TgPMMA (región III), y crecimiento de la primera población cristalina por encima
de TgPMMA (región IV).
Al incrementarse el valor de ta se incrementa también el intervalo correspondiente a
un tiempo de tratamiento superior al tiempo de cambio de régimen tc (es decir, las regiones
II y III), disminuyendo paralelamente las regiones I y IV en las cuales el tiempo de
tratamiento es inferior a tc. Para tiempos de tratamiento suficientemente elevados, todo el
intervalo de temperatura corresponde a las regiones de tiempos de tratamiento superiores a
tc.
Un estudio sistemático de la variación de la entalpía de fusión con la temperatura de
tratamiento, para un tiempo fijo, ofrece unos resultados que es conveniente analizar en la
figura 3.45. En lo que respecta a la primera población cristalina, se aprecia que ésta
experimenta un rápido crecimiento en la región I, un decrecimiento moderado en la región
II, un brusco salto al atravesar el límite entre las regiones II y III (la transición vítrea del
PMMA) que le lleva a alcanzar unas entalpías de fusión superiores al máximo alcanzando
en la región I, y un decrecimiento continuo en las regiones III y IV sin distinción apreciable
en el límite entre ambas y sin que los valores de las entalpías de fusión en estas dos
regiones muestran una dependencia apreciable con el tiempo de tratamiento térmico.
La segunda población cristalina presenta un comportamiento muy distinto.
Incipiente en la región I, crece en la región II hasta alcanzar valores similares al
correspondiente al máximo de la primera población en la región I y superiores a los de esta
misma población al final de la región II. El paso de la región II a la región III supone una
caída brusca en las entalpías seguida de un decrecimiento lento en las regiones III y IV, sin
que tampoco aquí se pueda apreciar ninguna diferencia significativa entre ambas regiones,
aunque sí se detecta una moderada dependencia con el tiempo de tratamiento.

117
(A)
Segunda población cristalina Primera población cristalina
IV IIIIII IVIIIIII
Ta (K)
(D)(C)
340 360 380 400 420
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
340 360 380 400 420
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
(B)
IVIIIIII
340 360 380 400 420
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
340 360 380 400 420
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5III
∆H
(cal
/g)
I II IV
(A) ta = 40 minutos (B) ta = 60 minutos (C) ta = 80 minutos (D) ta = 100 minutos
Región I, t < tc, Ta < TgPMMA Región II, t > tc, Ta < Tg
PMMA Región III, t > tc, Ta > Tg
PMMA Región IV, t < tc, Ta > TgPMMA
Figura 3.45 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de las dos fases cristalinas en función de la temperatura de tratamiento térmico.

1183.6.4 Cristalización no isotérmica. Dependencia de los puntos de fusión con la
temperatura.
El estudio de los puntos de fusión de los diferentes picos nos ha permitido obtener
resultados adicionales (figura 3.46).
(D)(C)
(A)
Fases de alta temperaturaPrimera población cristalina Ta (K)
(B)
320 360 400 440
340
360
380
400
420
440
320 360 400 440
340
360
380
400
420
440
320 360 400 440
340
360
380
400
420
440
320 360 400 440
340
360
380
400
420
440
Tm
(K)
(A) Φm = 0.5 ta = 60 minutos (B) Φm = 0.5 ta = 100 minutos (C) Φm = 0.6 ta = 10 minutos (D) Φm = 0.75 ta = 10 minutos
Figura 3.46 Mezclas PVDF/PMMA. Temperaturas de fusión de las diferentes fases cristalinas en función de la temperatura de tratamiento térmico.
A una concentración (Φm = 0.5) a la que sólo aparecen los dos picos principales, se
aprecia que el primero de ellos mantiene su temperatura aproximadamente constante, o sólo
con un ligero incremento en función de la temperatura, mientras el segundo experimenta un

119
fuerte incremento de aproximadamente unos 50 K a lo largo de todo el intervalo de
temperaturas de tratamiento. Para Φm = 0.6 el comportamiento de estos dos picos es similar
hasta el inicio de los procesos de alta temperatura (recristalización y transición de fase)
descritos en el capítulo 3.6.2, los cuales tienen su inicio, como ya ha sido comentado, a una
temperatura de tratamiento térmico de aproximadamente unos 147 °C. En este intervalo los
puntos de fusión de la primera población cristalina experimentan un ligero incremento
(∆T ~ 5 °C) en sus valores, mientras que la segunda población cristalina desaparece por
completo. Los dos procesos de alta temperatura muestran puntos de fusión superiores e
inferiores respectivamente a los correspondientes a la primera población cristalina, con una
fuerte dependencia de la temperatura de tratamiento térmico.
Por último, en las muestras con Φm = 0.75 se aprecia un comportamiento similar,
aunque más marcado, y a temperaturas superiores en unos 5 ó 10 °C a las anteriores, el cual
es similar al existente entre los puntos de fusión de cristales similares del PVDF para estas
dos concentraciones iniciales.
3.7 Interfase cristalina. Segunda transición vítrea
La siguiente etapa del presente trabajo de investigación ha consistido en estudiar
experimentalmente la presencia de una interfase cristalina ubicada entre los cristales y la
fracción amorfa del polímero. El primer paso consiste en aplicar el modelo más sencillo
posible, el bifásico, formado exclusivamente por una fracción cristalina y una matriz
amorfa. Calculando la cantidad de masa existente en la fracción cristalina a partir de la
entalpía de fusión, y la correspondiente a la fracción amorfa con el valor de la transición
vítrea, se aprecia en todos los casos un defecto sistemático de masa, de forma que la suma
de las masas calculadas para la matriz amorfa y para la fracción cristalina siempre da un
valor inferior al de la masa inicial. Este fenómeno, general para todas las concentraciones y
más acusado en las regiones donde la cinética es más rápida, alcanza su mayor magnitud en
las mezclas de Φm = 0.75, donde como se puede apreciar en la figura 3.47 la pérdida de
masa alcanza unos valores situados entre un 15 y un 20% del total.

120
340 360 380 400 420 4400.0
0.2
0.4
0.6
0.8
1.0
Masa cristalinaMasa amorfa
Masa total
Ta (K)
Frac
ción
de
mas
a
Figura 3.47 Mezclas PVDF/PMMA con Φm = 0.75. Fracciones de masa calculadas según el modelo de dos fases.
Esta pérdida de masa justifica la existencia de una tercera fase, identificada como la
interfase cristalina. Puesto que la concentración de PVDF es mayor en la interfase que en la
matriz amorfa, la determinación experimental de la interfase debe realizarse a través de una
segunda transición vítrea de temperatura inferior a la de la matriz amorfa. La identificación
de esta transición vítrea asociada a la interfase no resulta inmediata a causa de la pequeña
cantidad de material involucrado en la misma, lo que se traduce en unos valores de ∆cp de
un orden de magnitud similar en muchos casos al del error instrumental. Un segundo
inconveniente es el posible solapamiento entre ambas transiciones vítreas, dificultando e
incluso impidiendo la separación de ambas contribuciones. En la práctica tan sólo ha sido
posible apreciar de forma diferenciada ambas transiciones vítreas en mezclas ricas en
PVDF (figura 3.48), mientras para concentraciones menores el solapamiento entre las dos
curvas conduce a un único escalón asimétrico que se prolonga, debido a la contribución de
la transición vítrea de la interfase, por el tramo de temperaturas bajas.
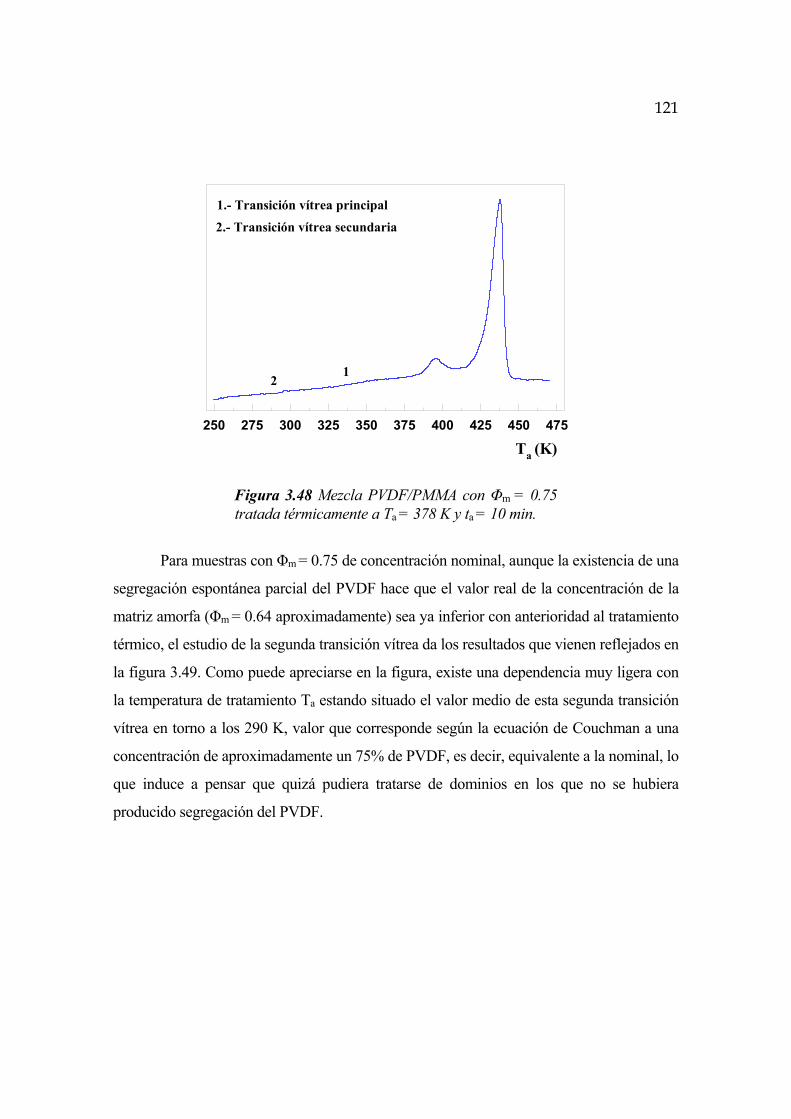
121
250 275 300 325 350 375 400 425 450 475
12
Ta (K)
1.- Transición vítrea principal
2.- Transición vítrea secundaria
Figura 3.48 Mezcla PVDF/PMMA con Φm = 0.75 tratada térmicamente a Ta = 378 K y ta = 10 min.
Para muestras con Φm = 0.75 de concentración nominal, aunque la existencia de una
segregación espontánea parcial del PVDF hace que el valor real de la concentración de la
matriz amorfa (Φm = 0.64 aproximadamente) sea ya inferior con anterioridad al tratamiento
térmico, el estudio de la segunda transición vítrea da los resultados que vienen reflejados en
la figura 3.49. Como puede apreciarse en la figura, existe una dependencia muy ligera con
la temperatura de tratamiento Ta estando situado el valor medio de esta segunda transición
vítrea en torno a los 290 K, valor que corresponde según la ecuación de Couchman a una
concentración de aproximadamente un 75% de PVDF, es decir, equivalente a la nominal, lo
que induce a pensar que quizá pudiera tratarse de dominios en los que no se hubiera
producido segregación del PVDF.

122
320 340 360 380 400 420 440280
285
290
295
300
Ta (K)
Tg
(K)
Figura 3.49 Mezclas PVDF/PMMA con Φm = 0.75. Segunda transición vítrea.
En mezclas amorfas de concentración media (Φm = 0.5) tratadas térmicamente
(figura 3.50), la transición vítrea asociada a la interfase no aparece diferenciada en el
termograma, sino que contribuye al tramo de baja temperatura de la transición vítrea
principal. Asimismo se aprecia que la asimetría de las curvas de transición vítrea producida
por estas colas es dependiente de las condiciones de tratamiento térmico (tiempo y
temperatura), aunque en el conjunto de todos los casos estudiados la temperatura
correspondiente al inicio de la curva de transición vítrea (Tb) alcanza siempre un valor
situado en torno a los 300 K. Esta temperatura corresponde a una concentración de PVDF
de aproximadamente un 70% en peso, valor próximo al encontrado en las muestras ricas en
PVDF y coincidente, de forma aproximada, con la concentración máxima posible en
mezclas homogéneas de PVDF y PMMA.
Los resultados similares alcanzados en ambos casos parecen indicar la aparición de
unos importantes reordenamientos moleculares en el seno de las fracciones no cristalinas
del sistema, los cuales pueden ser interpretados como núcleos de precristalización
enriquecidos en PVDF con respecto a la concentración media de la matriz, la cual va
disminuyendo paulatinamente conforme se desarrolla la segregación, mientras la
concentración de estos núcleos se mantiene aproximadamente constante.

123
280 300 320 340 360 380 400
ta=60 min
Ta=388 K
T (K)
TbT
e
Te
Figura 3.50 Mezclas PVDF/PMMA con Φm = 0.5. Curvas de transición vítrea de una muestra amorfa (inicial) y una muestra parcialmente segregada tras un tratamiento térmico.
Para poder identificar esta asimetría con una segunda transición vítrea asociada a la
interfase es necesario descartar que la misma pueda estar producida por otro fenómeno
diferente. Por ello es preciso considerar la asimetría intrínseca de las curvas
correspondientes a las mezclas amorfas iniciales, así como las de los polímeros puros. Esta
asimetría intrínseca es máxima en el PMMA y mínima en el PVDF, con valores
intermedios en las mezclas de ambos. Puesto que al tratar térmicamente una mezcla de
PVDF y PMMA se produce una segregación parcial del PVDF que implica un
enriquecimiento en PMMA de la matriz amorfa, cabe esperar un incremento en el valor de
la diferencia Tg - Tb, como efectivamente ocurre. Sin embargo, una comparación de la
curva de la transición vítrea del PMMA puro con la correspondiente a una mezcla
parcialmente segregada de PVDF y PMMA (figura 3.51) revela inmediatamente la gran
diferencia existente entre las dos curvas.
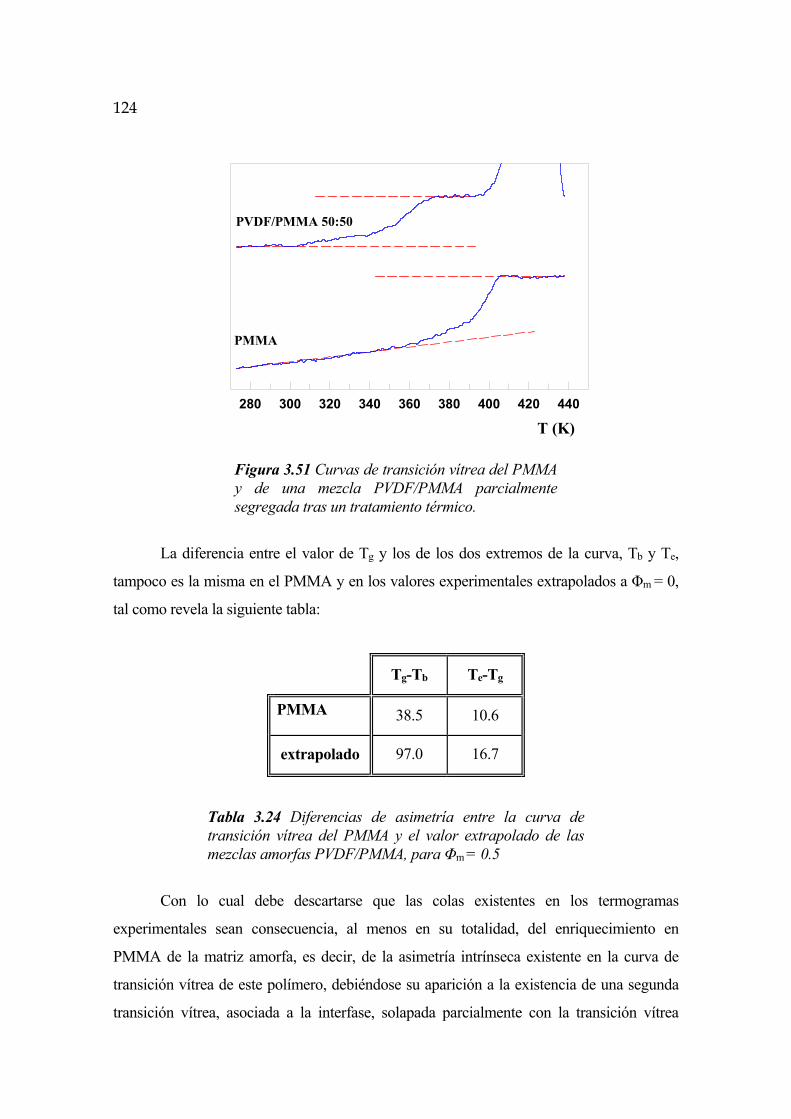
124
280 300 320 340 360 380 400 420 440
T (K)
PMMA
PVDF/PMMA 50:50
Figura 3.51 Curvas de transición vítrea del PMMA y de una mezcla PVDF/PMMA parcialmente segregada tras un tratamiento térmico.
La diferencia entre el valor de Tg y los de los dos extremos de la curva, Tb y Te,
tampoco es la misma en el PMMA y en los valores experimentales extrapolados a Φm = 0,
tal como revela la siguiente tabla:
Tg-Tb Te-Tg
PMMA 38.5 10.6
extrapolado 97.0 16.7
Tabla 3.24 Diferencias de asimetría entre la curva de transición vítrea del PMMA y el valor extrapolado de las mezclas amorfas PVDF/PMMA, para Φm = 0.5
Con lo cual debe descartarse que las colas existentes en los termogramas
experimentales sean consecuencia, al menos en su totalidad, del enriquecimiento en
PMMA de la matriz amorfa, es decir, de la asimetría intrínseca existente en la curva de
transición vítrea de este polímero, debiéndose su aparición a la existencia de una segunda
transición vítrea, asociada a la interfase, solapada parcialmente con la transición vítrea

125
principal. Por esta razón resulta necesario separar ambas transiciones, con objeto de poder
estudiar la segunda de ellas.
El tratamiento matemático realizado consiste en generar la curva correspondiente a
la transición vítrea principal y restársela al termograma experimental, ajustando la cola
restante a una sigmoide. La curva asignada a la transición vítrea principal es creada a partir
de las sigmoides correspondientes a los dos polímeros puros, PVDF y PMMA,
desplazándolas en la escala de temperaturas de modo que sus puntos de inflexión coincidan
entre sí y con el valor experimental de la transición vítrea de la matriz amorfa y sumándolas
ponderadamente en función de la concentración de la misma. Este método tiene la ventaja
de que permite sustraer la asimetría intrínseca de la transición vítrea del PMMA, con lo que
la cola restante corresponde exclusivamente a la contribución de la transición vítrea de la
interfase.
El primer paso consiste en ajustar las curvas experimentales de los dos polímeros
puros a una sigmoide de expresión:
] a
) a - (x - [ + 1
a =y
2
1
o
exp
[III,28]
En el caso del PVDF la curva se ajustó a una única sigmoide, mientras que para el
PMMA ha sido necesario utilizar la suma de dos sigmoides debido a la asimetría de la
curva. Los valores obtenidos para los parámetros de estas curvas son los siguientes:
PVDF: ao = 0.02 a1 = 233.90 a2 = 6.34
PMMA: ao = 0.03 a1 = 339.82 a2 = 31.88 (primera sigmoide)
ao = 0.02 a1 = 393.37 a2 = 4.14 (segunda sigmoide)
Los resultados de ambos ajustes vienen reflejados en la figura 3.52. En el PVDF
parcialmente cristalino, la fracción de masa amorfa ha sido normalizada a la unidad para
poderla comparar con el PMMA.

126
200 225 250 275 300 325 350 375 400 425
PVDF
PMMA
T (K)
Figura 3.52 Ajuste de las curvas patrón de transición vítrea del PVDF y el PMMA a partir de los termogramas experimentales.
Una vez obtenidas las dos curvas patrón, se ha procedido a comprobar la validez
del modelo comparando la suma de las mismas con curvas experimentales de mezclas
amorfas y homogéneas de distinta concentración de PVDF. Para ello, tras desplazar los
puntos de inflexión de las dos curvas patrón hasta hacerlos coincidir con la transición vítrea
de la curva experimental, hemos hecho la suma ponderada de las ordenadas de las curvas
patrón conforme a la expresión:
Φ∆Φ∆∆ 2211 mpmpp c + c = c [III,29]
Siendo Φm1 y Φm2 las concentraciones en peso de los dos polímeros y ∆cp1 y ∆cp2
los respectivos incrementos de capacidad calorífica de los mismos. En la mayor parte de las
muestras estudiadas el ajuste entre el modelo matemático y la curva experimental ha
resultado ser lo suficientemente bueno como para considerar aceptable el modelo (figura
3.53).

127
275 300 325 350 375 400 425 450 475
T (K)
Figura 3.53 Ajuste de la curva patrón de transición vítrea al termograma experimental de una mezcla de PVDF y PMMA con Φm = 0.2.
El último paso consiste en aplicar el modelo a las mezclas de PVDF y PMMA
tratadas térmicamente y parcialmente segregadas, en las cuales aparece la cola
correspondiente a la segunda transición vítrea a temperaturas inferiores (es decir, a
concentraciones superiores) a las de la transición vítrea principal. Tras generar la curva
patrón correspondiente a la concentración final de la matriz amorfa, se le ha restado al
termograma experimental tal como se aprecia en la figura 3.54, ajustándose el resultado de
la resta a una nueva sigmoide (figura 3.55) correspondiente a la transición vítrea de la
interfase.

128
275 300 325 350 375 400 425 450 475
T (K)
Figura 3.54 Ajuste de la curva patrón de transición vítrea al termograma experimental de una mezcla de PVDF y PMMA con Φm = 0.5 tratada a 388 K durante 60 minutos.
225 250 275 300 325 350 375 400
T (K)
Figura 3.55 Ajuste a una sigmoide de la transición vítrea secundaria de una mezcla de PVDF y PMMA con Φm = 0.5 tratada a 388 K durante 60 minutos.

129
Este método matemático se ha revelado adecuado para el ajuste y cálculo de las
transiciones vítreas secundarias, aunque la existencia de errores instrumentales (en especial
las irregularidades de las líneas bases en algunos termogramas) unida a la escasa magnitud
de las transiciones introduce en los resultados un factor de incertidumbre superior al
existente en el estudio de las transiciones vítreas principales, en especial en lo referente al
cálculo de los incrementos de capacidad calorífica.
La sigmoide obtenida proporciona información, a partir de sus diferentes
parámetros, sobre la naturaleza de la interfase. La concentración media puede ser calculada,
mediante la ecuación de Couchman, a partir del valor del punto de inflexión, identificado
con la temperatura de transición vítrea, y de ella puede obtenerse la diferencia existente
entre las concentraciones de la interfase y la matriz amorfa. La amplitud de la sigmoide es
proporcional al incremento de capacidad calorífica asociado a la transición vítrea, siendo
preciso establecer el valor de la constante de proporcionalidad para poder manejar valores
absolutos de ∆cp. Realizados en el calorímetro cálculos de este parámetro para diferentes
termogramas experimentales, y ajustes de sigmoides para estos mismos termogramas,
siempre referidos a la curva correspondiente a la transición vítrea principal, hemos
establecido para la constante de proporcionalidad un valor medio de k = 2.
Otra cuestión a tener en cuenta es el hecho de que los termogramas experimentales
no corresponden a un conjunto homogéneo de datos debido a la existencia de varias
poblaciones cristalinas diferentes, cada una de las cuales presenta una cinética de
cristalización propia que afecta de distinta manera a la cinética de segregación de las fases
amorfas. Dado que a la concentración estudiada (Φm = 0.5) las fases cristalinas de alta
temperatura no aparecen prácticamente en ningún caso, tan sólo es preciso considerar las
dos poblaciones cristalinas principales, separando los termogramas en tres grupos. El
primero de ellos corresponde a las muestras en las que existe un crecimiento exclusivo de
la primera población cristalina (la que aparece a tiempos de tratamiento cortos en las
muestras sometidas a tratamiento térmico), bien por tratarse de temperaturas a las que la
segunda población cristalina no aparece, bien por haberse realizado el tratamiento térmico a
tiempos inferiores a los necesarios para que tenga lugar el crecimiento de la segunda
población cristalina. El segundo grupo reúne los termogramas de las muestras en las que se
aprecia un crecimiento conjunto de las dos poblaciones cristalinas. El tercero agrupa las

130muestras con tiempos de tratamiento largos en las que sólo existe crecimiento de la
segunda población cristalina, por haber llegado a la saturación el crecimiento de la primera.
Con objeto de estudiar la dependencia de la interfase con la cinética del crecimiento
cristalino, hemos desechado los termogramas correspondientes a las muestras con
crecimiento conjunto de las dos poblaciones cristalinas dado que, por tratarse de una región
en la que están solapadas ambas cinéticas, resulta difícil interpretar los resultados. Los otros
dos grupos se han estudiado por separado al tratarse de muestras en las que la cristalización
del PVDF tiene lugar de forma homogénea.
3.7.1 Evolución temporal de la interfase
La dependencia temporal de la concentración de la interfase, obtenida a partir de los
valores de la transición vítrea secundaria, para varias temperaturas de tratamiento, viene
reflejada en la figura 3.56.
Como puede apreciarse en la figura, esta dependencia temporal es reducida en
todos los casos aunque, en general, se observa un ligero incremento de la concentración
para tiempos de tratamiento largos. Sin embargo, el estudio sistemático de todas las series
está dificultado por el hecho de que, para una temperatura de tratamiento determinada, el
intervalo de tiempo correspondiente al crecimiento exclusivo de una cualquiera de las dos
poblaciones cristalinas es por lo general bastante limitado. Debido también a la fuerte
variación que experimentan con la temperatura de tratamiento tanto los tiempos a los que
se inicia la cristalización de la segunda población cristalina, como los tiempos de saturación
del crecimiento de la primera, estas series tampoco son coincidentes unas con otras en sus
intervalos temporales, lo que dificulta una comparación directa de las mismas.

131
Interfase asociada al crecimiento de la primera población cristalinaInterfase asociada al crecimiento de la segunda población cristalina
ta (min)0 50 100 150 200 250
0.0
0.2
0.4
0.6
0.8
1.0 Ta= 393 K
0 50 100 150 200 250
0.0
0.2
0.4
0.6
0.8
1.0 Ta= 363 K
0 50 100 150 200 250
0.0
0.2
0.4
0.6
0.8
1.0 Ta= 363 K
0 50 100 150 200 250
0.0
0.2
0.4
0.6
0.8
1.0
Φm
i Ta= 353 K
Figura 3.56 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia temporal de la concentración de la interfase para diferentes temperaturas de tratamiento.
3.7.2 Dependencia con la temperatura
Al igual que ocurre con la dependencia temporal, las curvas correspondientes a la
variación de la concentración con la temperatura, para un tiempo de tratamiento
determinado existen en un intervalo muy corto en la mayor parte de los casos y tampoco
coinciden en muchas ocasiones los intervalos de temperatura en los que se desarrollan. Para
poder disponer de un estudio completo en todo el intervalo de temperaturas comprendido
entre la transición vítrea inicial de la mezcla (Tg ~ 335 K) y la fusión del PVDF cristalizado
(Tm ~ 435 K), hemos realizado una primera aproximación consistente en tomar la media de
los valores para cada temperatura, lo cual es aceptablemente válido considerando que la

132dependencia temporal es por lo general reducida. Una segunda aproximación más
ajustada a la realidad experimental es la de suponer una dependencia lineal de la
concentración con el tiempo, tomando como valor de la concentración con la temperatura
la extrapolación a tiempo cero en el caso de la interfase asociada al crecimiento de la
primera población cristalina, y la extrapolación a tiempo reducido cero (considerando como
tal el inicio de la cristalización) para la interfase asociada al crecimiento de la segunda
población cristalina.
Los resultados obtenidos en ambos casos son muy similares, lo que indica que
ambas aproximaciones son equivalentes, aunque finalmente hemos elegido la segunda por
estar más justificada desde un punto de vista físico. Entre las curvas asociadas al
crecimiento de la primera y la segunda poblaciones cristalinas aparecen diferencias
significativas, lo que indica que a cada una de las dos poblaciones cristalinas le
corresponde una cinética distinta, tanto en la matriz amorfa tal como se mostró en el
capítulo correspondiente, como también en la interfase cristalina.
a) Concentración
320 340 360 380 400 420 440
0.0
0.2
0.4
0.6
0.8
1.0 (4)(3)(2)(1)
∆Φm
Φmi
Ta (K)320 340 360 380 400 420 440
0.0
0.2
0.4
0.6
0.8
1.0 (4)(3)(2)(1)
∆Φm
Φmi
Ta (K)
Figuras 3.57 y 3.58 Mezclas con Φm = 0.5. Dependencia con Ta de Φmi y ∆Φm asociadas al
crecimiento de la primera población cristalina (izquierda) y la segunda población cristalina (derecha).

133
La dependencia con la temperatura de tratamiento de la concentración de la
interfase (Φmi) y del incremento de concentraciones entre la interfase y la matriz amorfa
(∆Φm = Φmi - Φm), para las interfases asociadas al crecimiento de cada una de las dos
poblaciones cristalinas, viene reflejada en las figuras 3.57 y 3.58.
Correspondiendo las cuatro líneas verticales a los siguientes parámetros:
(1) Temperatura de transición vítrea inicial de las muestras (Tg ~ 335 K).
(2) Temperatura mínima a la cual tiene lugar la aparición de la segunda población
cristalina (Ta ~ 360 K).
(3) Temperatura de transición vítrea del PMMA (Tg ~ 393 K).
(4) Temperatura de fusión del PVDF cristalizado (Tm ~ 435 K).
En el caso de la primera población cristalina se ha realizado un ajuste lineal,
mientras en la segunda ha sido necesario hacerlo cuadrático.
Lo primero que se aprecia en la gráfica correspondiente a la interfase asociada al
crecimiento de la primera población cristalina (figura 3.57) es la existencia de tres regiones
netamente diferenciadas, separadas entre sí por la temperatura mínima a la cual tiene lugar
la aparición de la segunda población cristalina (Ta ~ 360 K) y por la temperatura de
transición vítrea del PMMA (Tg ~ 393 K). Este comportamiento, que es similar para Φmi y
para ∆Φm, marca unos mínimos cercanos al límite teórico (Φmi ~ 0.5 y ∆Φm ~ 0) para estas
dos temperaturas, mientras los máximos de las tres regiones muestran unos valores que
crecen linealmente con la temperatura de tratamiento, correspondiendo a la temperatura de
transición vítrea inicial de las muestras (Tg ~ 335 K) un valor de Φmi = 0.65, lo que supone
un enriquecimiento inicial de la interfase en aproximadamente ∆Φm = 0.15 en relación con
la matriz amorfa. Extrapolando estos valores hasta el punto de fusión del PVDF segregado
y cristalizado (Tm ~ 435 K), los dos parámetros estudiados se incrementan hasta Φmi = 0.74
y ∆Φm = 0.24.
El comportamiento de la interfase asociada al crecimiento de la segunda población
cristalina (figura 3.58) es, como ya ha sido comentado, completamente distinto. En primer
lugar, y a diferencia del caso anterior, no existen temperaturas intermedias que hagan de
frontera entre las distintas regiones, ya que los puntos experimentales describen una curva
suave con el máximo situado en torno a los 385 K aproximadamente, temperatura a la cual
la cinética de cristalización alcanza su valor más elevado.

134Otra diferencia notable entre este caso y el anterior estriba en el hecho de que la
extrapolación de las curvas de ajuste de los puntos experimentales alcanza, dentro de los
límites del error experimental, el valor mínimo teórico (Φmi = 0.5 y ∆Φm = 0) tanto en las
proximidades de la temperatura de transición vítrea inicial de las muestras (Tg ~ 335 K)
como en las cercanías del punto de fusión del PVDF segregado y cristalizado (Tm ~ 435 K),
al contrario de lo que ocurría con la interfase asociada al crecimiento de la primera
población cristalina.
b) Masa
Multiplicando la amplitud de la sigmoide de ajuste (ao) por el factor de
proporcionalidad calculado previamente (k = 2), se obtienen los valores del incremento de
la capacidad calorífica correspondiente a la transición vítrea asociada a la interfase, la cual
a su vez es función de la cantidad de masa existente en la misma. Puesto que los errores
experimentales en la determinación de este parámetro son muy superiores a los existentes
en la medida de las concentraciones, la información obtenida es mucho más limitada.
El criterio seguido para agrupar los puntos experimentales ha sido el mismo que el
empleado para las concentraciones, la separación en tres regiones de la interfase asociada al
crecimiento de la primera población cristalina y la consideración de una única región, en
todo el intervalo de temperaturas, para la interfase asociada al crecimiento de la segunda
población cristalina. En esta ocasión, a diferencia de lo que ocurría con la concentración,
los valores medios dieron mejores resultados que las extrapolaciones a tiempo o tiempo
reducido cero.
En el estudio de la ∆cp de la interfase asociada al crecimiento de la primera
población cristalina (figura 3.59), donde las cuatro temperaturas marcadas con líneas
verticales tienen los mismos significados que en los casos anteriores (figuras 3.57 y 3.58),
resulta difícil cuantificar los resultados, debido a la ya comentada existencia de un error
experimental de magnitud considerable, aunque parece apreciarse una variación de la
tendencia de esta magnitud en función de la temperatura, con una distribución trimodal
cuyos máximos relativos coincidirían respectivamente con las zonas centrales de la región
de crecimiento exclusivo de esta primera población cristalina (Ta < 360 K), del intervalo de
temperaturas comprendido entre la temperatura mínima de aparición de la segunda
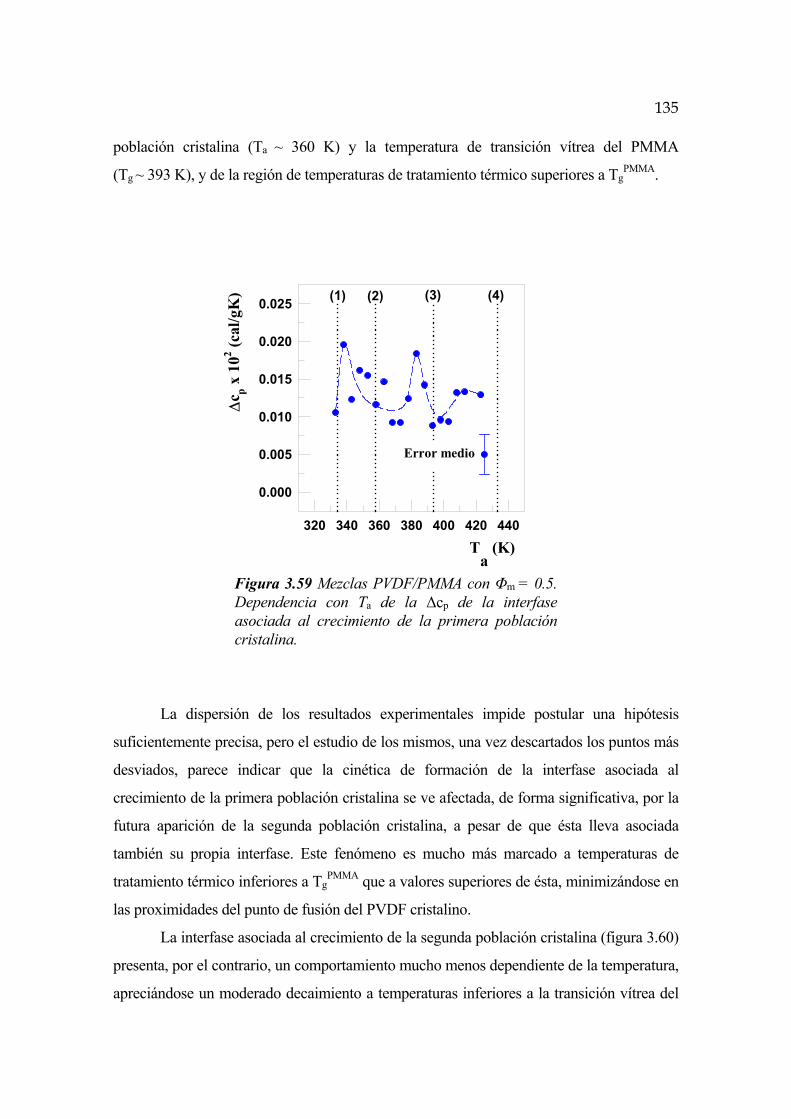
135
población cristalina (Ta ~ 360 K) y la temperatura de transición vítrea del PMMA
(Tg ~ 393 K), y de la región de temperaturas de tratamiento térmico superiores a TgPMMA.
320 340 360 380 400 420 440
0.000
0.005
0.010
0.015
0.020
0.025 (4)(3)(2)(1)
Ta (K)
∆c p x
102 (c
al/g
K)
Error medio
Figura 3.59 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia con Ta de la ∆cp de la interfase asociada al crecimiento de la primera población cristalina.
La dispersión de los resultados experimentales impide postular una hipótesis
suficientemente precisa, pero el estudio de los mismos, una vez descartados los puntos más
desviados, parece indicar que la cinética de formación de la interfase asociada al
crecimiento de la primera población cristalina se ve afectada, de forma significativa, por la
futura aparición de la segunda población cristalina, a pesar de que ésta lleva asociada
también su propia interfase. Este fenómeno es mucho más marcado a temperaturas de
tratamiento térmico inferiores a TgPMMA que a valores superiores de ésta, minimizándose en
las proximidades del punto de fusión del PVDF cristalino.
La interfase asociada al crecimiento de la segunda población cristalina (figura 3.60)
presenta, por el contrario, un comportamiento mucho menos dependiente de la temperatura,
apreciándose un moderado decaimiento a temperaturas inferiores a la transición vítrea del
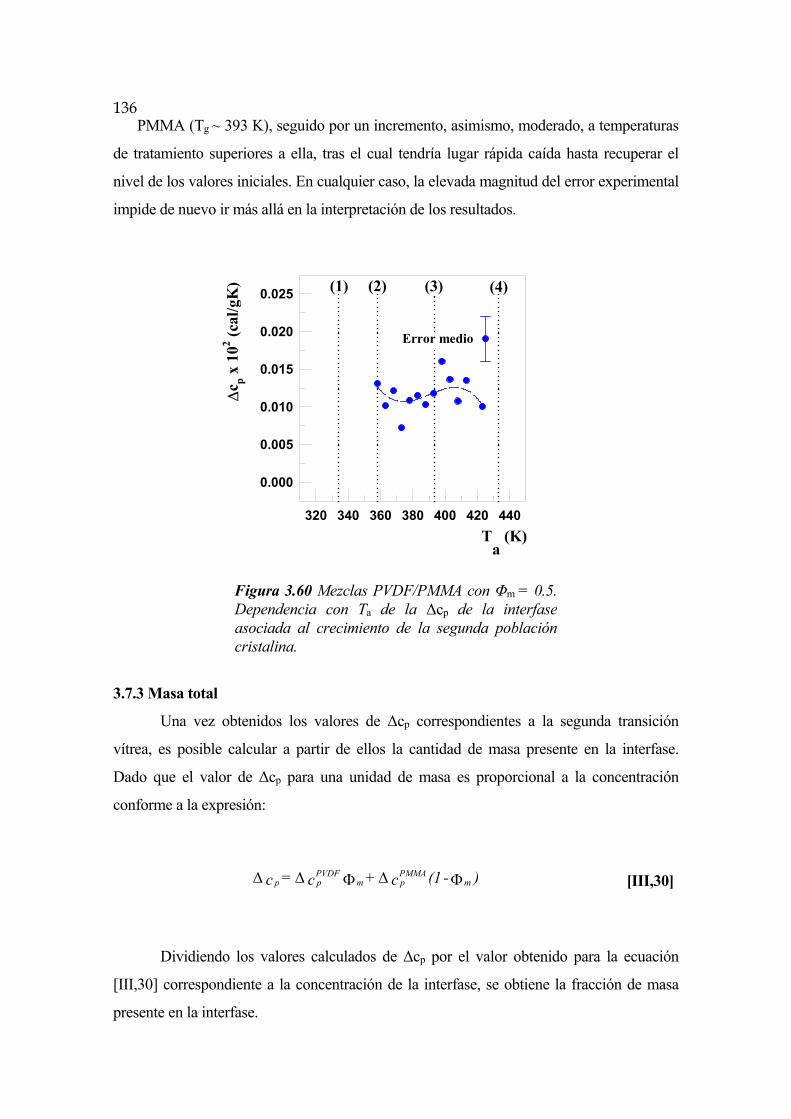
136PMMA (Tg ~ 393 K), seguido por un incremento, asimismo, moderado, a temperaturas
de tratamiento superiores a ella, tras el cual tendría lugar rápida caída hasta recuperar el
nivel de los valores iniciales. En cualquier caso, la elevada magnitud del error experimental
impide de nuevo ir más allá en la interpretación de los resultados.
320 340 360 380 400 420 440
0.000
0.005
0.010
0.015
0.020
0.025
Ta (K)
(4)(3)(2)(1)
∆c p x
102 (c
al/g
K)
Error medio
Figura 3.60 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia con Ta de la ∆cp de la interfase asociada al crecimiento de la segunda población cristalina.
3.7.3 Masa total
Una vez obtenidos los valores de ∆cp correspondientes a la segunda transición
vítrea, es posible calcular a partir de ellos la cantidad de masa presente en la interfase.
Dado que el valor de ∆cp para una unidad de masa es proporcional a la concentración
conforme a la expresión:
)- (1 c + c = c mPMMApm
PVDFpp Φ∆Φ∆∆ [III,30]
Dividiendo los valores calculados de ∆cp por el valor obtenido para la ecuación
[III,30] correspondiente a la concentración de la interfase, se obtiene la fracción de masa
presente en la interfase.

137
La aplicación de la misma ecuación para los valores de ∆cp de la transición vítrea
principal permite calcular la cantidad de masa presente en la fase amorfa, y a partir de la
entalpía de fusión del PVDF cristalino se obtiene la masa de PVDF cristalizado.
Asumiendo un modelo trifásico formado por el PVDF cristalino, la fracción amorfa
y la interfase, la suma de las tres contribuciones debería equivaler a la masa total del
sistema. Los resultados obtenidos vienen reflejados en las figuras 3.61 y 3.62.
320 340 360 380 400 420 440
0.6
0.8
1.0
1.2
1.4
1.6
1.8
m (g
)
Ta (K)
(4)(3)(2)(1)
Error medio
320 340 360 380 400 420 440
0.4
0.6
0.8
1.0
1.2
1.4
1.6
m (g
)
Ta (K)
(4)(3)(2)(1)
Error medio
Figuras 3.61 y 3.62 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia con Ta de la masa total en muestras con crecimiento exclusivo de la primera población cristalina (izquierda) y de la segunda población cristalina (derecha).
Como puede apreciarse en la figura 3.61, en las muestras correspondientes al
crecimiento exclusivo de la primera población cristalina puede asumirse, dentro del margen
de error, que el modelo aplicado es aceptablemente válido excepto para temperaturas
cercanas a la de transición vítrea inicial de las mezclas amorfas (1), en las cuales la escasa
magnitud de la segregación hace que el margen de error sea muy superior, midiéndose
valores superiores al máximo posible de 1 gramo. No resulta fácil apreciar cambios
significativos coincidiendo con la temperatura mínima de cristalización de la segunda
población cristalina (2), ni tampoco con la temperatura de transición vítrea del PMMA (3),
aunque quizá pudiera existir un mínimo local coincidiendo aproximadamente con esta
última.

138En las muestras correspondientes al crecimiento exclusivo de la segunda
población cristalina, por el contrario, (figura 3.62) parece existir una desviación sistemática
hacia valores inferiores a la unidad, es decir, un defecto de masa, excepto para temperaturas
de tratamiento térmico próximas a la transición vítrea del PMMA (3), en las cuales la curva
parece mostrar un máximo local tendente a la unidad. En el capítulo siguiente se realizará
un análisis más detallado de estos resultados.

139
CAPÍTULO 4
DISCUSIÓN DE LOS RESULTADOS
4.1 Consideraciones generales.
El objetivo del presente trabajo de investigación es profundizar en el estudio del
sistema formado por mezclas compatibles de PVDF y PMMA. Aunque este sistema ha sido
previamente investigado por otros autores (véase la introducción), las discrepancias
existentes en ciertos aspectos del mismo hacían necesario abordarlo desde un nuevo
enfoque que permitiera clarificarlas, al tiempo que la aplicación de los métodos de análisis
utilizados ha permitido el estudio controlado de diversas características, tanto
termodinámicas como cinéticas, del sistema objeto de estudio.
Este trabajo está estructurado en tres apartados principales: Compatibilidad
termodinámica del sistema en el estado fundido, propiedades físicas de las mezclas en el
estado vítreo, y cinética de los procesos de segregación y cristalización en muestras tratadas
térmicamente.
4.2 Compatibilidad del sistema PVDF/PMMA. Parámetro de interacción.
La compatibilidad molecular de las mezclas de PVDF y PMMA ha sido
determinada por diversos autores (véase la introducción), recogiendo las tablas 4.1 y 4.2 los
valores aportados para el parámetro de interacción de Flory (χ12), tanto los obtenidos a
partir de la depresión del punto de fusión de los cristales de PVDF en mezclas
PVDF/PMMA parcialmente segregadas, técnica utilizada por nosotros, como los
calculados mediante otras técnicas experimentales diferentes.

140
Autor χ12
Nishi, 1975b -0.295 (160 °C)
Roerdink, 1978 -0.10 (177 °C)
Paul, 1978b -0.375 (165 °C)
Wendorff, 1980 -0.12 (160 °C)
Morra, 1982a -0.2 (170 °C)
Goh, 1988 -0.42
Jo, 1991 -0.11 (170 °C)
Moussaif, 1999 -0.32
Tabla 4.1 Valores de χ12 determinados por distintos autores, mediante medidas de depresión del punto de fusión del PVDF. El valor aportado por Wendorff corresponde a una concentración de PVDF de 0.5.
Autor χ12 Técnica
Wendorff, 1980 -0.07 para w = 0.5 SAXS
Hadziioannou, 1984 -0.226 / -0.059 SANS
Di Paola, 1982 -0.03 para w = 0.5 Cromat. inversa
Privalko, 1990 -0.6 / -0.7 (220 °C) Dilatometría
Tabla 4.2 Valores de χ12 determinados por distintos autores mediante diversas técnicas experimentales.
La comparación de estos valores nos permite concluir que el valor calculado de χ12
depende de la técnica de análisis empleada. Incluso dentro del grupo de medidas de
depresión del punto de fusión la concordancia entre los valores aportados por los distintos
autores es relativa, con un valor medio de χ12 en torno a -0.2, habiéndose observado una
ligera dependencia de este valor con el incremento del peso molecular y con la
concentración de la mezcla.

141
Estas discrepancias tienen su origen en las limitaciones de la ecuación de Flory. La
primera de ellas, y la más inmediata, es de carácter cinético y viene dada por el hecho de
que las medidas experimentales no corresponden a una situación de equilibrio
termodinámico del sistema al no poderse obtener en la práctica cristales
termodinámicamente perfectos. El recurso habitual para resolver este problema consiste en
utilizar la teoría de Hoffman-Weeks [Hoffman, 1962]. Según este método, se representan
las temperaturas de fusión fuera del equilibrio (Tm) de diferentes muestras frente a las
correspondientes temperaturas de cristalización (Tc) de las mismas muestras. Por
extrapolación de la recta que agrupa a estos puntos experimentales hasta su intersección
con la recta Tm = Tc, se obtiene el punto de fusión en el equilibrio termodinámico (Tm0).
Esta extrapolación es válida únicamente para cristalizaciones isotérmicas a temperaturas
próximas al equilibrio, es decir, para muestras que han sido cristalizadas a temperaturas
cercanas a la de fusión, con lo cual ha de ser cuestionada su aplicación en casos como el del
presente trabajo de investigación en los cuales no se cumple esta condición.
La segunda limitación a la hora de aplicar esta teoría es la morfológica, y viene
dada por el hecho de que la estructura y el tamaño de los cristales poliméricos dependen
notablemente de las condiciones experimentales en que se han originado los mismos,
mientras algunos polímeros como el PVDF presentan también polimorfismo cristalino.
Este punto afecta al parámetro V2 de la ecuación de Flory:
)-(1VH
RV-=T1-
T1 2
v121m
20mm
Φ∆
χ [IV,1]
Es decir, al volumen molar del polímero cristalino, ya que al haber una mezcla de
varias fases cristalinas y al fundir cada una de ellas por separado a distintas temperaturas, el
volumen implicado en el proceso no es el total sino tan sólo la fracción correspondiente a la
fase cristalina fundida a cada temperatura determinada. Otra cuestión importante a tener en
cuenta es el hecho de que la ecuación de Flory solamente es válida para el caso de cristales
de espesor infinito en los que se ha alcanzado el equilibrio termodinámico, por lo que esta
ecuación debe ser escrita según la expresión:

142
)-(1VH
RV-=T1-
)(T1 2
v121m
20mv
0m
Φ∆Φ
χ [IV,2]
Donde Tm0 y Tm
0(Φv) son, respectivamente, las temperaturas de fusión en el
equilibrio de cristales de espesor infinito del polímero aislado, y del polímero en el seno de
una mezcla homogénea de concentración Φv. En el caso de un cristal de espesor finito (l),
la dependencia del punto de fusión en función de l viene determinada por la conocida
ecuación de Thompson-Gibbs [Hoffman, 1976]:
)lH
2-(1 T = Tm
e0mm ∆
σ [IV,3]
Siendo σe la energía superficial de los plegados moleculares en la superficie del
cristal. Si en lugar de un polímero puro se tiene una mezcla compatible de concentración
Φv, la ecuación anterior se puede expresar como:
)lH
2-(1 )(T = )(Tm
ev
0mvm ∆
ΦΦσ [IV,4]
Combinando las ecuaciones [IV,2] y [IV,4] se obtiene finalmente la expresión:
)-(1VH
RV - T1 =
)(T1 )
lH2-(1 2
v121m
20mvmm
e Φ∆Φ∆
χσ [IV,5]
Que relaciona los valores experimentales de los puntos de fusión de cristales
poliméricos en mezclas compatibles de composición Φv, con la temperatura de fusión en el
equilibrio de los cristales de espesor infinito del polímero puro. Este planteamiento es

143
válido sólo en el caso de que la energía libre de superficie σe dependa principalmente del
plegado de las cadenas moleculares [Hoffman, 1976] y no sea modificada
significativamente por la presencia del segundo polímero. Una ecuación similar, que
considera también el efecto del espesor en la depresión del punto de fusión, ha sido
propuesta más recientemente por Chow [Chow, 1990].
Otro factor a tener en cuenta, en el sistema PVDF/PMMA, es que cuando la
concentración de PVDF es inferior a un 25-30% en peso, este polímero no cristaliza bajo
ninguna condición. Por lo tanto, al no poderse obtener PVDF cristalino a partir de mezclas
amorfas en este rango de concentraciones, no es posible medir la depresión del punto de
fusión en las mismas. En el extremo opuesto, para muestras ricas en PVDF existe también
el ya comentado límite, situado en torno a un 60-65% de riqueza en peso de PVDF, por
encima del cual no se pueden obtener mezclas homogéneas al cristalizar el exceso de
PVDF.
Por esta razón, el cálculo de χ12 mediante medidas de la depresión del punto de
fusión, en mezclas parcialmente segregadas de PVDF/PMMA, está limitado en la práctica
a la región de concentraciones medias, no siendo posible ampliarlo a ninguno de los dos
extremos. Otro inconveniente añadido [Rim, 1984] es que la presencia del polímero amorfo
afecta a la temperatura de transición vítrea de la fase amorfa del sistema, y por lo tanto al
subenfriamiento experimentado por los cristales, lo que determina la dependencia del
espesor de los mismos con la concentración y en consecuencia la magnitud de su punto de
fusión, incurriéndose por ello en una subestimación del valor del parámetro de χ12.
Tal como ha sido comentado en el capítulo anterior, hemos conseguido evitar estas
limitaciones recurriendo a un sistema formado por un conjunto de monocristales de PVDF
dispersos en el seno de una matriz de PMMA, lo cual permite controlar la morfología de
los monocristales de PVDF (fase α pura), ampliando asimismo el rango de concentraciones
de trabajo a la totalidad del intervalo. Esta idea ha surgido en nuestro laboratorio y se ha
ensayado con éxito en varios sistemas [Canalda, 1995; Martínez de Salazar, 1991b;
Martínez de Salazar, 1996], permitiéndonos un avance en relación con los trabajos de
investigación anteriores.
Al no partirse de una mezcla homogénea se evitan los efectos cinéticos producidos
por la segregación de fases necesaria para la cristalización del PVDF, y al estar dispersos
los monocristales del mismo en la matriz de PMMA se obtiene un estado de cuasiequilibrio

144equivalente al que se alcanzaría tras la segregación y la cristalización del PVDF
procedente de una mezcla homogénea y amorfa una vez terminado el proceso cinético.
La utilización de los monocristales permite evitar también las posibles
perturbaciones morfológicas comentadas anteriormente, al obtenerse monocristales
homogéneos correspondientes además a una única fase cristalina, la α. Asimismo, dadas las
características del sistema formado por los monocristales y la matriz de PMMA, es posible
medir depresiones del punto de fusión del PVDF en todo el rango de concentraciones,
desde el 10 al 90% de riqueza en peso de este polímero, ampliándose así por ambos
extremos los límites existentes en el caso de las mezclas parcialmente segregadas, ya que al
estar físicamente separados los dos polímeros y el PVDF cristalizado independientemente
de su proporción, la fusión de los cristales no resulta afectada en ningún caso por la cinética
de segregación.
El valor medio obtenido para el parámetro de interacción, para la temperatura
correspondiente al punto de fusión en el equilibrio, es de χ12=-0.14. Dentro del rango de
valores aportados para el parámetro de interacción en la bibliografía, el valor obtenido en
este trabajo se sitúa en la zona baja (en valor absoluto) del mismo, muy alejado de los
propuestos por autores como Nishi y Wang [Nishi, 1975b], Paul y Barlow [Paul, 1978b] o
Goh y Slow [Goh, 1988], y más cercano a los propuestos por Roerdink y Challa [Roerdink,
1978], Wendorff [Wendorff, 1980] o Jo y Kwon [Jo, 1991].
Otra conclusión importante del presente trabajo es la constatación de la práctica
independencia del parámetro de interacción con la concentración, en contraste con los
resultados de estos autores, los cuales encuentran una marcada dependencia de χ12 con esta
variable, principalmente a partir de concentraciones superiores a un 50% en peso de PVDF.
La razón de esta discrepancia parece radicar en el polimorfismo cristalino del PVDF, tal
como proponen Morra y Stein [Morra, 1982a]. Estos autores concluyen que el valor del
parámetro de interacción varía en función de la proporción existente entre las distintas fases
cristalinas del PVDF presentes en el sistema, ya que cada una de las cuales posee un punto
de fusión diferente a los de las demás. Posteriormente Plans y col. [Plans, 1984] han
apuntado que al tener en cuenta la coexistencia de varias fases cristalinas del PVDF en las
mezclas, renormalizando las fracciones en volumen de los diferentes cristales, se puede
conseguir un valor de χ12 independiente de la concentración. Esto hace que los resultados
de Nishi y Wang concuerden con los de Morra y Stein.

145
El hecho de que los monocristales de PVDF utilizados en el presente trabajo fueran
morfológicamente homogéneos, permite explicar la independencia de χ12 con la
concentración de las mezclas. Por otra parte, el valor negativo de χ12 nos lleva a concluir
que, desde un punto de vista termodinámico, la mezcla de PVDF y PMMA es compatible
en el fundido, en todo el rango de concentraciones, por debajo de la LCST, situada en torno
a los 330 °C.
4.3 Compatibilidad de las mezclas de PVDF y PMMA a temperaturas inferiores a la
de fusión.
Una forma sencilla de calcular los límites termodinámicos de la compatibilidad
molecular de dos polímeros consiste en aplicar la ecuación de Flory descrita en el apartado
anterior, la cual al ser representada describe una curva que define la temperatura de fusión
en el equilibrio del PVDF segregado en función de la concentración de la mezcla. Sin
embargo, al no estar los cristales de PVDF en equilibrio termodinámico su temperatura de
fusión real Tm(Φv) es inferior a la correspondiente temperatura de fusión en el equilibrio
Tmo(Φv), lo que provoca un descenso del límite real entre el material fundido y el cristalino
o, lo que es lo mismo, entre la mezcla homogénea y la cristalizada.
La temperatura de transición vítrea se puede interpretar como aquélla a la que las
moléculas del material detienen sus movimientos anulándose con ello la difusión
molecular. Para que un polímero cristalino presente en una mezcla amorfa pueda cristalizar
es necesario que sus moléculas estén segregadas de la mezcla, para lo cual es preciso que la
temperatura a la que se encuentra el sistema sea igual o superior a la correspondiente a la
transición vítrea de la mezcla en esas condiciones. En la práctica, y al igual que ocurre con
los puntos de fusión, los factores cinéticos provocan que el material necesite una
temperatura superior a la de la transición vítrea para comenzar a segregarse. Por
extrapolación de las curvas experimentales de cristalización a entalpía cero, se han podido
determinar las temperaturas mínimas de segregación para varias mezclas correspondientes
al tramo central de concentraciones. De esta manera se obtiene como resultado un
incremento medio en las temperaturas mínimas de segregación del orden de unos veinte
grados Kelvin por encima de Tg.
Estos resultados vienen resumidos en la figura 4.1, en la cual han sido
representados tanto las curvas de Tmo y Tg como los valores experimentales de los puntos

146de fusión Tm(Φv) y de la temperatura de inicio de la cristalización (Tc
min), para distintas
concentraciones de PVDF. Dado que estos puntos experimentales corresponden
únicamente a la región de concentraciones medias, ha sido necesario extrapolar los
resultados a la totalidad del intervalo de concentraciones.
0.0 0.2 0.4 0.6 0.8 1.0240
280
320
360
400
440
480
Φm PVDF
T (K
)
Tg
Tcmin
Tm
Tmo
Figura 4.19 Mezclas PVDF/PMMA. Curvas de puntos de fusión y de transición vítrea en función de la concentración.
Las curvas Tmo y Tm no se cortan para ningún valor de Φm, lo que indica que desde
un punto de vista termodinámico la compatibilidad en el fundido entre el PVDF y el
PMMA se extiende por todo el intervalo de concentraciones. Sin embargo, era de esperar
que la curva de los puntos de fusión experimentales y la curva de la temperatura de inicio
de la cristalización se cortaran conforme a la evidencia experimental de que las mezclas de
PVDF y PMMA no cristalizan bajo ningún tipo de condiciones para concentraciones de
PVDF inferiores a aproximadamente un 25-30% en peso. En la extrapolación de los
valores experimentales de Tcmin se ha tenido en cuenta este factor, razón por la que es
preciso descartar una primera aproximación consistente en asumir para la curva de Tcmin
una dependencia con la concentración similar a la existente para la transición vítrea aunque
con los valores de temperatura incrementados en un factor constante ∆T, ya que en estas

147
condiciones el corte entre las dos curvas tenía lugar para un valor de la concentración de
PVDF de aproximadamente un 10% en peso, muy inferior al determinado
experimentalmente.
Una posible explicación de esta discrepancia es que el incremento de temperatura
existente entre la transición vítrea y la temperatura mínima de cristalización no es
constante, sino que va aumentando al disminuir la concentración de PVDF presente en las
mezclas al ser mayor la dificultad del PVDF para segregarse de las mismas. Este fenómeno
ha sido expuesto por Léonard y col. [Léonard, 1983], que encuentran, por un lado, un
fuerte aumento en el valor de este incremento de temperatura para concentraciones de
PVDF inferiores a las estudiadas en este trabajo (∆T ≈ 30 °C para Φm = 0.4), y por otro la
desaparición de este incremento para concentraciones en las que ya existe segregación
previa al tratamiento térmico (Φm ~ 0.7). Tomando estos dos límites y ajustándolos con
nuestros propios valores experimentales se alcanza una notable concordancia entre este
último ajuste y los datos experimentales.
En el intervalo comprendido entre la temperatura de fusión real y la
correspondiente al inicio de la cristalización, siempre para una concentración dada, se
encuentra una región en la que al sistema le resulta posible segregarse y cristalizar. En la
práctica, para pasar de la fase termodinámicamente estable del fundido a la metaestable del
estado vítreo, es preciso atravesar la región cristalizable, por lo cual el hecho de que la
segregación molecular tenga lugar o no durante este proceso dependerá básicamente de la
competencia entre la cinética de difusión molecular y la velocidad de enfriamiento de la
muestra. Si la segunda de ellas es más rápida que la primera se podrán obtener vidrios
amorfos en los que no haya tenido lugar la segregación, mientras que cuando suceda lo
contrario la muestra se segregará y cristalizará parcialmente durante el proceso de enfriado
del fundido.
La velocidad de enfriamiento del fundido está limitada por las características
técnicas del calorímetro empleado. En el caso del presente estudio tiene un valor máximo
teórico de 320 grados Kelvin por minuto. La difusión molecular, por el contrario, es
función de la concentración de PVDF en la mezcla al ser la movilidad molecular de este
polímero mucho mayor que la del PMMA. Como consecuencia de ello, existe una
concentración límite por encima de la cual no es posible obtener mezclas amorfas en estado
vítreo, debido a que su cinética de segregación es tan rápida que el proceso tiene lugar
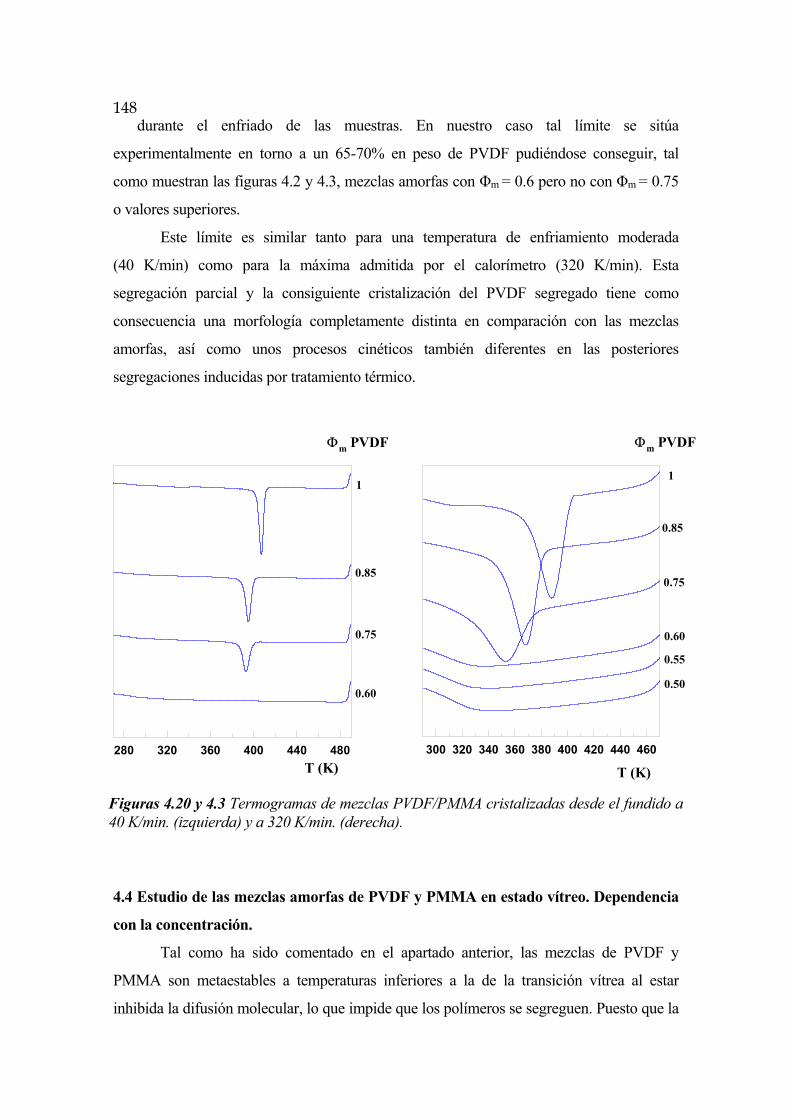
148durante el enfriado de las muestras. En nuestro caso tal límite se sitúa
experimentalmente en torno a un 65-70% en peso de PVDF pudiéndose conseguir, tal
como muestran las figuras 4.2 y 4.3, mezclas amorfas con Φm = 0.6 pero no con Φm = 0.75
o valores superiores.
Este límite es similar tanto para una temperatura de enfriamiento moderada
(40 K/min) como para la máxima admitida por el calorímetro (320 K/min). Esta
segregación parcial y la consiguiente cristalización del PVDF segregado tiene como
consecuencia una morfología completamente distinta en comparación con las mezclas
amorfas, así como unos procesos cinéticos también diferentes en las posteriores
segregaciones inducidas por tratamiento térmico.
T (K)280 320 360 400 440 480
Φm PVDF
0.60
0.75
0.85
1
300 320 340 360 380 400 420 440 460
Φm PVDF
0.60
1
0.85
0.75
T (K)
0.50
0.55
Figuras 4.20 y 4.3 Termogramas de mezclas PVDF/PMMA cristalizadas desde el fundido a 40 K/min. (izquierda) y a 320 K/min. (derecha).
4.4 Estudio de las mezclas amorfas de PVDF y PMMA en estado vítreo. Dependencia
con la concentración.
Tal como ha sido comentado en el apartado anterior, las mezclas de PVDF y
PMMA son metaestables a temperaturas inferiores a la de la transición vítrea al estar
inhibida la difusión molecular, lo que impide que los polímeros se segreguen. Puesto que la

149
transición vítrea de las mezclas de PVDF y PMMA cubre un rango de aproximadamente
160 K entre la correspondiente al PMMA (Tg ~ 393 K) y al PVDF (Tg ~ 233 K), será
necesario considerar la temperatura de transición vítrea de la mezcla en función de la
concentración. Esta temperatura, para una concentración de Φm = 0.5, está situada en torno
a los 334 K.
Manteniendo estas muestras en estado vítreo por debajo de sus respectivas
transiciones vítreas es posible estudiar la dependencia de distintas propiedades físicas de las
mismas en función de la concentración. Las medidas de transición vítrea (temperatura y
calor específico), densidad macroscópica y microdureza (ver figuras 3.11 a 3.14) poseen
una dependencia aditiva con la concentración en el intervalo en el cual las mezclas son
amorfas y homogéneas, comportándose como una especie física única. Por el contrario, en
las mezclas parcialmente segregadas (Φm ≥ 0.65) se aprecia que el comportamiento de las
mezclas se desvía de la aditividad tal como corresponde a sistemas físicos heterogéneos.
4.5 Cinética de segregación de fases en las primeras etapas del proceso. Difusión
molecular.
La dispersión de rayos X, tal como ha sido comentado, se debe a la existencia de
regiones con diferentes fluctuaciones de densidad electrónica en las muestras amorfas. Este
fenómeno hace posible estudiar mediante SAXS la separación de fases en las mezclas
compatibles de PVDF y PMMA gracias a las grandes diferencias de densidad (electrónica
y macroscópica) existentes entre el PVDF cristalino (ρ = 1.92 g/cm3 para la fase α)
[Narula, 1989], el PVDF amorfo (ρ = 1.692 g/cm3) [Martínez de Salazar, 1994], el PMMA
(ρ = 1.1838 g/cm3) [Martínez de Salazar, 1991a] y las mezclas de ambos (ρ = 1.380 g/cm3
para una mezcla al 50% en peso) [Martínez de Salazar, 1991a]. Dado que para los átomos
ligeros la relación entre la masa atómica y el número atómico es aproximadamente igual a
2, resulta válida la suposición de que la relación entre las densidades macroscópicas es
equivalente a la existente entre las densidades electrónicas de los mismos materiales.
Estudiando estas diferencias de densidad electrónica será posible seguir la evolución de la
segregación de fases y la cristalización de muestras tratadas térmicamente por encima de
las correspondientes temperaturas de transición vítrea.

150La magnitud medida experimentalmente es el invariante, definido como la
integral de la intensidad de la radiación dispersada según las expresiones [Porod, 1951]:
ds I(s) s=Q 2∫∞
°
[IV,6]
Para colimación puntual del haz de rayos X, o:
ds I(s) s=Q ∫∞
°
[IV,7]
Para colimación lineal. La dependencia temporal del invariante ha sido estudiada
para distintas condiciones de concentración y temperatura, haciéndose para ello las
oportunas normalizaciones. Para conocer la relación entre el invariante Q y el volumen
segregado de polímero φ en las primeras etapas del proceso, se puede partir de la
expresión:
ρφρφρ Mv
Fav
B ) - (1 + = [IV,8]
Donde ρB, ρaF y ρM son respectivamente las densidades de la mezcla inicial no
segregada, de la fase amorfa del PVDF y del PMMA, mientras Φv representa la fracción
inicial en volumen de PVDF en la mezcla. Pasado un tiempo t la mezcla estará
parcialmente segregada, por lo que la expresión [IV,8] queda transformada en:
) - (1 ) - (1 + ) - (
= M
vFavB
t ϕρφρϕφ
ρ [IV,9]

151
Definiéndose φ como la fracción segregada de PVDF en el tiempo t. Por otro lado,
para un sistema bifásico el invariante será proporcional a la diferencia de densidad
electrónica entre estas dos fases (PVDF segregado y mezcla PVDF/PMMA restante, en
nuestro caso) según la expresión [Alexander, 1969]:
) - (1 ) - ( K = >)(< K = Q 2Bt
F2 ϕϕρρρ∆ [IV,10]
Donde <(∆ρ)2> es el valor cuadrático medio de las fluctuaciones de densidad
electrónica y K agrupa las constantes experimentales y de normalización. Los valores de
densidad electrónica, expresados en moles de electrones por centímetro cúbico, han sido
transformados en densidades macroscópicas expresadas en g/cm3. Experimentalmente
hemos comprobado que para tiempos cortos de tratamiento térmico existe un aumento del
invariante (figura 3.20), lo que indica una segregación de fases con la consiguiente
modificación de las densidades electrónica y macroscópica, sin que sea posible detectar
simultáneamente una cristalización del PVDF segregado. Por lo tanto, puede asumirse que
el material segregado se encuentra en estado amorfo sin cristalizar, por lo que ρF = ρaF,
quedando la ecuación [IV,10] convertida en:
) - (1 ) - ( K = Q 2Bt
Fa ϕϕρρ [IV,11]
Agrupando, por último, las ecuaciones [IV,9] y [IV,11], obtenemos finalmente:
) - (1 ])-(1 ) - [( K = Q 2
vMF
a ϕϕ
φρρ [IV,12]
Puesto que las dos densidades son constantes, la expresión anterior puede resumirse
en:

152
ϕϕ
φ-1
) - (1 K = Q 2v′ [IV,13]
Siendo K' = K(ρaF-ρM)2
Como puede apreciarse en la ecuación [IV,13], para una concentración inicial dada
el invariante depende de la fracción de volumen transformado según la expresión φ/(1- φ).
Por esta razón, y puesto que a tiempos cortos la segregación todavía es incipiente, se puede
despreciar el valor de φ frente a 1, quedando de esta manera la ecuación [IV,13] reducida a:
ϕφ ) - (1 K = Q 2v′ [IV,14]
Con lo que el invariante depende linealmente del valor del volumen segregado φ.
Puesto que en estas primeras etapas del proceso todavía no se ha iniciado la cristalización
del PVDF segregado, puede asumirse que la separación de fases está controlada
básicamente por la difusión intermolecular. Suponiendo una geometría esférica de
dominios, la relación entre el crecimiento radial de los mismos y el tiempo viene dada por
la expresión [Aaron, 1970]:
t D = R λ [IV,15]
Que es un caso particular de la ecuación de Fick [Jost, 1960] donde λ es un
parámetro que depende de la concentración [Aaron, 1970] y D representa el coeficiente de
difusión del PVDF en la mezcla.
Al ser el invariante proporcional al volumen segregado, que se supone esférico,
asumiendo que el número de dominios de PVDF puro permanece constante con el tiempo,

153
tanto el invariante como el volumen segregado serán asimismo proporcionales al cubo del
radio de estos dominios. Agrupando las ecuaciones [IV,14] y [IV,15] conforme a estos
criterios, se obtiene finalmente la expresión:
)(Dt )-(1 K" = Q 1/22/3v
1/3 φλ [IV,16]
Para K" = (4/3 π K')1/3
Esta ecuación determina el valor del coeficiente de difusión en función de la
concentración inicial de la muestra, lo cual en la práctica se cumple únicamente, tal como
quedó reflejado en el capítulo anterior (figura 3.20), en las primeras etapas de la
segregación, produciéndose a tiempos suficientemente largos una desviación de la
linealidad y una disminución del coeficiente de difusión a causa del empobrecimiento en
PVDF de la muestra, con la consiguiente variación de la densidad de la fracción amorfa de
la misma. Por consiguiente, la pendiente m del tramo lineal de la recta experimental
corresponderá a la expresión:
D ) - (1 K" = m 1/22/3φλ [IV,17]
4.5.1 Ecuación de William-Landel-Ferry
La ecuación de William-Landel-Ferry [Arridge, 1975] agrupa en una misma
expresión la dependencia de la cinética de segregación de fases con la temperatura y el
tiempo, pudiendo ser expresada como:
)T-(T + K)T-(T
K = g2
g1gT expνν [IV,18]
Donde vT es la frecuencia de relajación a una temperatura T superior a la
temperatura de transición vítrea Tg y vg la frecuencia de relajación en la transición vítrea,

154medidas ambas a una frecuencia de 0.1 Hz. [Bueche, 1962]. Los valores de las
constantes han sido estimados para el PVDF, mediante medidas de relajación dieléctrica,
en K1 = 33 y K2 = 52 respectivamente [Ezquerra]. La ecuación de William-Landel-Ferry
determina la movilidad de las moléculas del polímero en función de la diferencia de
temperatura existente en el sistema en relación con la transición vítrea. Puesto que la
magnitud v está relacionada con los coeficientes de difusión, la ecuación de William-
Landel-Ferry puede ser transformada de manera que relacione los coeficientes de difusión
con la temperatura. Para ello, se puede utilizar la expresión propuesta por Bueche:
6a = D
2
mν
[IV,19]
Siendo el parámetro a la longitud del segmento polimérico, v la frecuencia de salto
y Dm el coeficiente de difusión del monómero. Por otro lado, admitiendo que la movilidad
de los monómeros de PVDF es mucho mayor que la correspondiente a los monómeros de
PMMA debido a la gran diferencia que existe entre sus respectivos valores de Tg
(∆Tg >150 K), es posible prescindir de la aportación del PMMA al coeficiente de
interdifusión de la mezcla. Así pues, se puede establecer una relación entre el coeficiente de
difusión del monómero de PVDF (Dm) y el coeficiente de interdifusión D. Según el modelo
propuesto por Kramer y col. [Kramer, 1984], desarrollado por Garbella y Wendorff
[Garbella, 1988], se obtiene la expresión siguiente:
φφχ v2
vF
e12m ) - (1
NN | | 2 D = D [IV,20]
Donde χ12 es el parámetro de interacción entre los dos polímeros, Ne la longitud de
entrecruzamiento, NF el grado de polimerización y Φv la fracción en volumen, todos ellos
referidos al PVDF. Dado que según nuestros resultados χ12 puede ser considerado constante
con la concentración, todos los factores de la ecuación [IV,20] son constantes a excepción
de Φv. El valor de Φv puede ser aproximado a la concentración inicial despreciando, como

155
se hizo anteriormente, el volumen segregado φ frente a la misma. Por último, agrupando las
ecuaciones [IV,18], [IV,19] y [IV,20] se llega a la siguiente expresión:
)T-(T + K)T-(T
K ) - (1 D = Dg2
g1v
2vo expφφ [IV,21]
Siendo Do un factor que agrupa a los distintos parámetros constantes según:
NN
3| | a
= DF
e122
go ⋅
χν [IV,22]
Lo cual permite calcular en la ecuación WLF las frecuencias de vibración a partir
de los coeficientes de difusión. En el presente trabajo de investigación no disponemos de
valores absolutos de D sino de valores proporcionales a esta magnitud. Elevando al
cuadrado la expresión de la ecuación [IV,17] y agrupándola con la [IV,21] se alcanza
finalmente:
)T-(T + K)T-(T
K ) - (1 K = mg2
g1v
10/3v
2D
2 expφφλ [IV,23]
Siendo KD = (K")2 ⋅ Do
Para estudiar el comportamiento de la ecuación WLF al utilizarse los valores
experimentales obtenidos a distintas concentraciones iniciales, es necesario evitar el
inconveniente que supone no disponer de valores absolutos de D, así como también es
deseable eliminar su dependencia con la concentración. Por ello es necesario suprimir el
factor preexponencial de la ecuación WLF, lo que permite eliminar tanto el factor que
multiplica a las medidas experimentales de D, como la dependencia con la concentración
inicial. Es necesario recordar que el término exponencial de la ecuación WLF no
corresponde exactamente al coeficiente de difusión en valores absolutos, pero su utilización

156permite la comparación tanto de los valores experimentales entre sí, como la de éstos
con la ecuación teórica en la que, obviamente, se ha suprimido también su correspondiente
factor preexponencial.
El método matemático empleado en este trabajo se basa en el ajuste de los valores
experimentales según la ecuación:
)T-(T + K)T-(T
K A = mg2
g1
2 exp⋅ [IV,24]
Siendo m la pendiente de la recta de ajuste de la parte lineal de la representación de
Q1/3 frente a t1/2 cuyo cuadrado, tal como ha sido comentado, es proporcional al coeficiente
de difusión D. El factor preexponencial A, por su parte, queda definido como:
φφλ v10/3
v2
D )-(1 K = A [IV,25]
Donde K1 y K2 han sido fijados respectivamente en los valores K1 = 33 y K2 = 52,
calculándose el valor del factor A que mejor se ajusta a los resultados experimentales de la
ecuación WLF. Finalmente, con objeto de eliminar la dependencia de todos los factores
constantes y de la concentración inicial hemos normalizado los valores experimentales
dividiendo, en la ecuación [IV,24], el parámetro m2 por el valor preexponencial A.
Un factor a tener en cuenta son los valores de Tg utilizados en el ajuste de la
ecuación WLF. La primera aproximación consiste en tomar los valores de Tg
correspondientes a la concentración inicial, que calculados mediante la ecuación de
Couchman resultan ser:

157
ΦmPVDF Tg (K)
0.45 341.5
0.50 334.1
0.55 326.4
Tabla 4.3 Valores de Tg de las mezclas PVDF/PMMA en función de la concentración de PVDF.
Sin embargo, esta aproximación es válida tan sólo para temperaturas de tratamiento
térmico cercanas a la transición vítrea inicial en las cuales, por ser lenta la cinética de
difusión molecular, el empobrecimiento en PVDF de la matriz amorfa puede considerarse
despreciable. Para temperaturas de tratamiento térmico más elevadas este empobrecimiento
es significativo y no se puede ignorar, ya que se refleja en una variación de la temperatura
de transición vítrea en relación con la inicial. En consecuencia, en estas regiones ha sido
preciso considerar los valores reales de Tg para cada temperatura de tratamiento, que han
sido calculados a partir de los estudios realizados por calorimetría diferencial de barrido.
Teniendo en cuenta esta corrección en los valores de Tg, se han representado los
valores normalizados del cociente m2/A para las tres concentraciones estudiadas,
comparándolos con la curva teórica descrita por la ecuación:
)T-(T + K)T-(T
K = T)( fg2
g1exp∆ [IV,26]
Como puede apreciarse en la figura 4.4, la concordancia de los valores
experimentales con la curva teórica es excelente en los tres casos hasta un valor de (T-Tg)
situado en torno a los 30 K. A partir de este intervalo, y a pesar de las correcciones, los
puntos experimentales se desvían de los valores previstos, debido probablemente a que
para incrementos de temperatura más elevados la cinética es demasiado rápida sucediendo
en la matriz amorfa unos fenómenos para los cuales ya no es válido el modelo propuesto.

158
0 10 20 30 40 50
Φm = 0.55 Φm = 0.50 Φm = 0.45
curva teóricam2 /A
(u.a
.)
Ta-Tg (K)
Figura 4.4 Ecuación WLF para mezclas PVDF/PMMA con distintas concentraciones.
A continuación hemos estudiado la dependencia con la concentración del factor
preexponencial A, que presenta los siguientes valores experimentales:
ΦmPVDF A
0.45 7.50 ⋅ 10-7 0.50 6.09 ⋅ 10-7 0.55 1.66 ⋅ 10-6
Tabla 4.4 Valores del parámetro A de la ecuación WLF para mezclas PVDF/PMMA en función de la concentración de PVDF.
Como hemos visto, este factor preexponencial está constituido por tres términos
diferentes. El primero de ellos (KD) engloba todas las constantes involucradas en el
desarrollo matemático del modelo. El segundo es el cuadrado del parámetro λ de Aaron,
dependiente de la concentración, y el último es un factor f(Φv) dependiente de la
concentración que recoge la dependencia de los coeficientes de difusión con la
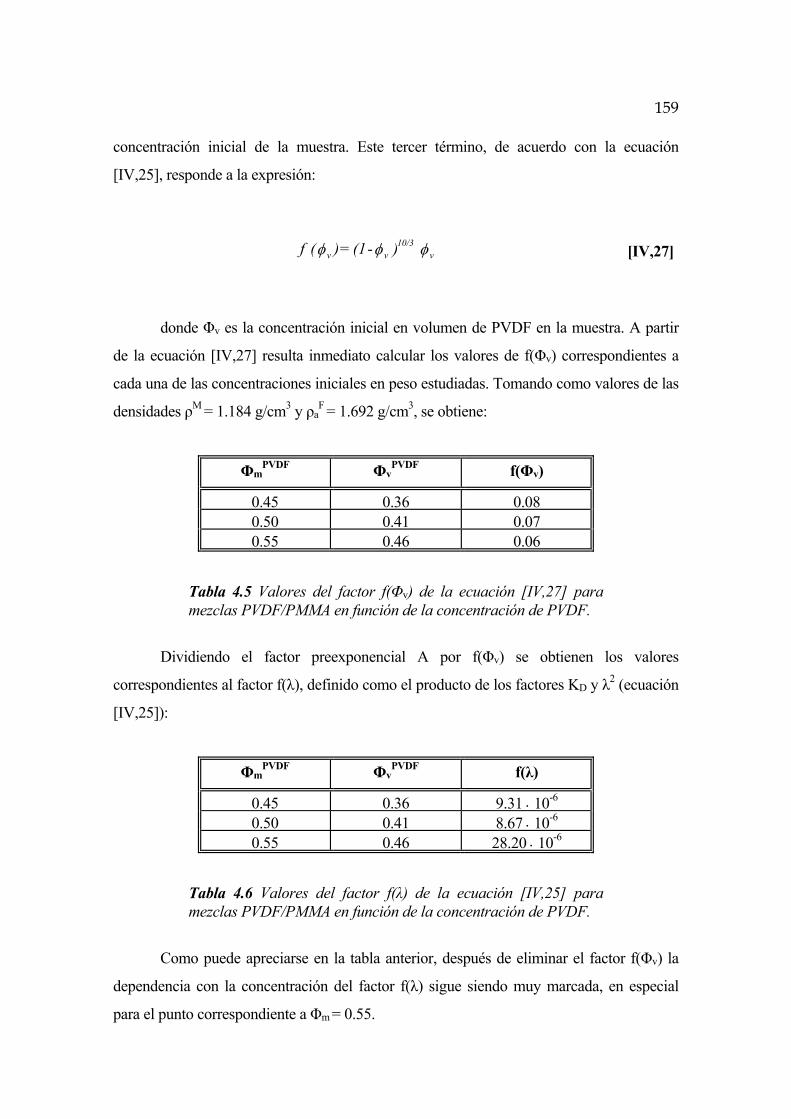
159
concentración inicial de la muestra. Este tercer término, de acuerdo con la ecuación
[IV,25], responde a la expresión:
φφφ v10/3
vv )-(1 = )( f [IV,27]
donde Φv es la concentración inicial en volumen de PVDF en la muestra. A partir
de la ecuación [IV,27] resulta inmediato calcular los valores de f(Φv) correspondientes a
cada una de las concentraciones iniciales en peso estudiadas. Tomando como valores de las
densidades ρM = 1.184 g/cm3 y ρaF = 1.692 g/cm3, se obtiene:
ΦmPVDF Φv
PVDF f(Φv)
0.45 0.36 0.08 0.50 0.41 0.07 0.55 0.46 0.06
Tabla 4.5 Valores del factor f(Φv) de la ecuación [IV,27] para mezclas PVDF/PMMA en función de la concentración de PVDF.
Dividiendo el factor preexponencial A por f(Φv) se obtienen los valores
correspondientes al factor f(λ), definido como el producto de los factores KD y λ2 (ecuación
[IV,25]):
ΦmPVDF Φv
PVDF f(λ)
0.45 0.36 9.31 ⋅ 10-6 0.50 0.41 8.67 ⋅ 10-6 0.55 0.46 28.20 ⋅ 10-6
Tabla 4.6 Valores del factor f(λ) de la ecuación [IV,25] para mezclas PVDF/PMMA en función de la concentración de PVDF.
Como puede apreciarse en la tabla anterior, después de eliminar el factor f(Φv) la
dependencia con la concentración del factor f(λ) sigue siendo muy marcada, en especial
para el punto correspondiente a Φm = 0.55.

160Para estudiar la dependencia del parámetro λ con la concentración hemos tenido
en cuenta el trabajo de Aaron y col. [Aaron, 1970], en el cual se hace un estudio muy
completo de este tema desarrollando varias aproximaciones distintas. En general, y para el
caso de crecimiento cristalino con geometría esférica asumido en el presente estudio, Aaron
define un parámetro k cuya expresión matemática es la siguiente:
C - CC - C 2 = k
IP
MI [IV,28]
Donde CM, CI y CP son respectivamente las concentraciones de la matriz amorfa, de
la interfase y de la fase cristalina. Aaron propone tres diferentes relaciones entre λ y k en
función de la aproximación utilizada:
) k - 4k ( +
2k - = 1/2
2
1/2 ππλ [IV,29]
) k - ( = 1/2λ [IV,30]
]) k 4 + 1 ( + ) k 4 (
) k 4 ( [ = 1/21/31/3
1/34
λ [IV,31]
De estas tres expresiones es preciso considerar cual de ellas es la más apropiada
para representar nuestros resultados experimentales, teniendo en cuenta que los valores de
f(λ) son proporcionales a λ2 y no a λ, por serlo linealmente a los coeficientes de difusión D.
Elevando al cuadrado las expresiones [IV,29], [IV,30] y [IV,31], se aprecia que en la
primera de ellas existe una dependencia cuadrática con k (y por lo tanto con CM, asimilable
a la concentración Φv), en la segunda una dependencia lineal y en la tercera una
dependencia aproximadamente lineal, dado que el cociente de los exponentes máximos de
k en el numerador y en el denominador es uno. En consecuencia, aunque las tres
aproximaciones contemplan un incremento en el valor de λ en función de la concentración,
la única de ellas capaz de explicar el fuerte incremento experimentado por f(λ) para las

161
concentraciones más elevadas es la ecuación [IV,29], por corresponderle una dependencia
cuadrática.
Una vez elegida esta aproximación puede hacerse la simplificación de considerar
inexistente la interfase, al tratarse de las primeras etapas del proceso. Asumiendo un
modelo en dos fases, la expresión [IV,28] queda reducida a:
CC 2 - = kP
M [IV,32]
O, utilizando la nomenclatura empleada en este trabajo:
C 2 -
= kP
vφ [IV,33]
Por último, también es necesario considerar que la constante KD presenta un valor
que no es posible cuantificar. Elevando al cuadrado la expresión [IV,29] y sustituyendo k
por su valor en [IV,33], se llega finalmente a:
} ] )C / (2 - 4
)C / (2[
)C / (2 - )C / (2 -
2)C / (2
{ K = )f( 1/2Pv
2Pv
1/2Pv
Pv
2Pv
D φπ
φπ
φφ
πφλ [IV,34]
Siendo KD y CP dos parámetros ajustables. Al encontrarse el parámetro CP en todos
los sumandos de la ecuación [IV,34] elevado a diferentes potencias, se puede despejar
parcialmente agrupándolo con KD. Por ello son posibles múltiples soluciones que conducen
a curvas similares con diferentes valores de KD y CP. Al no poderse cuantificar en valores
absolutos ninguno de estos dos parámetros, el estudio realizado permite únicamente
constatar de modo cualitativo la validez del modelo aplicado.

162Los resultados del ajuste vienen reflejados en la figura 4.5, donde por
simplicidad se ha asignado a KD el valor de 1, lo que conduce a un valor de CP igual a
0.167. Como puede apreciarse, la existencia de sólo tres puntos experimentales no permite
realizar un ajuste demasiado preciso, aunque sí se detecta un incremento monótono de f(λ)
con la concentración tal como reflejan los resultados experimentales.
0.0 0.2 0.4 0.6 0.8 1.00
10
20
30
40
50
60
70
Φv
f(λ)
Figura 4.5 Dependencia de la constante de Aaron con la concentración.
4.6 Cinética de segregación y cristalización en mezclas de PVDF y PMMA.
El tratamiento térmico a temperaturas superiores a la de transición vítrea de
mezclas inicialmente amorfas de PVDF y PMMA nos ha permitido realizar un estudio de
la segregación molecular y de la cristalización del PVDF, pudiéndose determinar las leyes
cinéticas que controlan estos procesos. Como era de esperar, para muestras inicialmente
segregadas se encuentran desviaciones importantes del comportamiento general de las
mezclas amorfas, razón por la cual es necesario estudiarlas por separado. En el otro
extremo del intervalo de concentraciones, para muestras con cantidades de PVDF inferiores
aproximadamente a un 25% en peso, no es posible inducir la segregación del PVDF en
ninguna de las muestras tratadas.

163
Trabajando a concentraciones medias de PVDF se aprecia que, al igual que ocurre
en otros muchos procesos físicos, la cinética global del sistema resulta ser la combinación
de varias cinéticas sencillas, principalmente las correspondientes a la difusión molecular y
a la cristalización del PVDF segregado. En los casos en los que las velocidades de los
distintos procesos implicados son muy diferentes, se puede asumir que la cinética global
está controlada por la correspondiente al proceso más lento, criterio que permite realizar
por separado el estudio de las distintas regiones del sistema en función de la cinética
particular que gobierna a cada uno de ellos.
En general, los límites entre los distintos procesos han podido ser establecidos con
bastante precisión en función de las tres variables principales consideradas, la
concentración, la temperatura y el tiempo de tratamiento, encontrándose que estos límites
resultan ser coincidentes para las diferentes magnitudes estudiadas.
a) Dependencia con la concentración.
Tal como se ha comentado, en las mezclas en las que se produce segregación
(Φm ≥ 0.3) es preciso diferenciar entre las que forman mezclas amorfas en estado vítreo
(Φm ≤ 0.65) y aquéllas en las que se parte de un material inhomogéneo parcialmente
cristalizado (Φm > 0.65). En ambos casos se puede inducir una cristalización del PVDF que
inicialmente se hallaba en estado amorfo, pero la distinta naturaleza del sustrato inicial
conduce a cinéticas de cristalización completamente distintas.
b) Dependencia con la temperatura.
Tanto en las gráficas que representan la dependencia de la transición vítrea en
función de la temperatura como en las que recogen la relación con este parámetro de la
entalpía de cristalización (figura 4.6) se aprecian curvas en forma de campana que pueden
ser descompuestas en dos ramas distintas, cada una de las cuales cuenta con parámetros
propios. El límite entre ambas cinéticas ha sido establecido, independientemente de la
concentración, en la temperatura de la transición vítrea del PMMA.
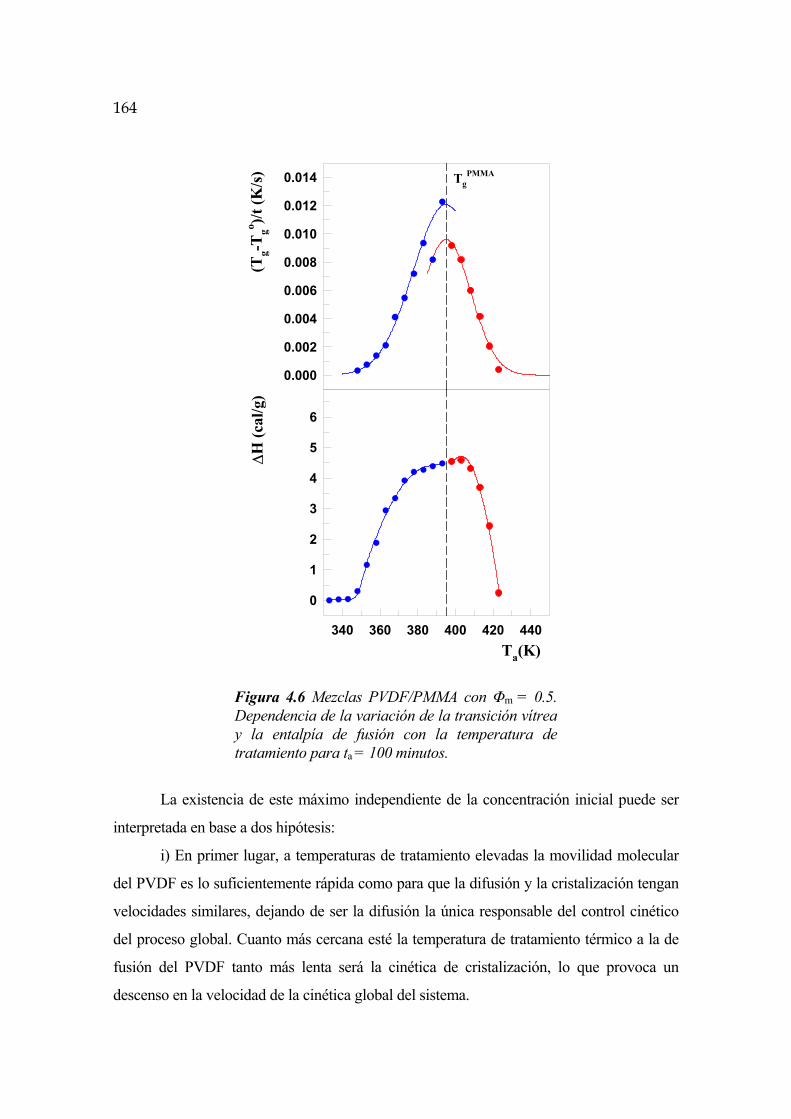
164
340 360 380 400 420 440
0
1
2
3
4
5
6
0.000
0.002
0.004
0.006
0.008
0.010
0.012
0.014 TgPMMA
(Tg-
Tgo )/t
(K/s
)∆
H (c
al/g
)
Ta(K)
Figura 4.6 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de la variación de la transición vítrea y la entalpía de fusión con la temperatura de tratamiento para ta = 100 minutos.
La existencia de este máximo independiente de la concentración inicial puede ser
interpretada en base a dos hipótesis:
i) En primer lugar, a temperaturas de tratamiento elevadas la movilidad molecular
del PVDF es lo suficientemente rápida como para que la difusión y la cristalización tengan
velocidades similares, dejando de ser la difusión la única responsable del control cinético
del proceso global. Cuanto más cercana esté la temperatura de tratamiento térmico a la de
fusión del PVDF tanto más lenta será la cinética de cristalización, lo que provoca un
descenso en la velocidad de la cinética global del sistema.

165
ii) La segunda interpretación de este fenómeno parte de considerar la movilidad de
las moléculas de PMMA presentes en la mezcla. A temperaturas de tratamiento inferiores a
la correspondiente a la Tg del PMMA puede considerarse que las moléculas de este
polímero permanecen inmóviles frente a las del PVDF, únicas responsables en estas
condiciones de la difusión molecular. Sin embargo, esta situación deja de ser válida cuando
la temperatura de tratamiento térmico es superior a la de la citada transición vítrea, ya que
es preciso asumir que en el rango de temperaturas comprendido entre la transición vítrea
del PMMA y la fusión de los cristales de PVDF segregado, las moléculas de PMMA se
difunden simultáneamente con las de PVDF. Para el PVDF la segregación es
termodinámicamente favorable al estar compensada la energía absorbida por este proceso
con la desprendida en la subsiguiente cristalización. Esto no ocurre en el caso del PMMA
atáctico, en el cual la segregación es un proceso energéticamente desfavorable dado que
este polímero no puede cristalizar. Por esta razón las moléculas difundidas de este último
polímero tenderán a oponerse a la salida del PVDF de la matriz amorfa, lo que provoca el
remezclado parcial de los polímeros segregados, dando como resultado una disminución de
la cinética global del proceso.
Entre estas dos hipótesis parece más acertado optar por la segunda de ellas, dado
que en todos los casos estudiados de mezclas inicialmente amorfas la temperatura
correspondiente a la cinética máxima no varía con la concentración a pesar de que sí lo
hacen, tanto la amplitud de este máximo, como la temperatura de transición vítrea inicial de
la mezcla, es decir, el rango de temperaturas en el que tiene lugar el proceso. Por otro lado,
si bien a temperaturas de tratamiento superiores a este máximo se encuentra un
comportamiento cinético similar de sentido opuesto, no es posible ajustar todo el conjunto
de los valores experimentales a una misma curva, lo que indica la existencia de unas leyes
cinéticas diferentes antes y después de la transición vítrea del PMMA. Esta evidencia
experimental resulta difícil de justificar en base únicamente a una competencia entre la
cinética de segregación molecular y la de cristalización, pero sí puede ser interpretada
considerando la movilidad de las moléculas de PMMA, relacionada a su vez con la
transición vítrea de este polímero.
c) Dependencia temporal.
Al igual que ocurre con la dependencia con la temperatura, en las curvas isotermas
se aprecia un comportamiento diferente antes y después de un valor determinado de tiempo

166(tc), definido como tiempo de cambio de régimen, el cual resulta ser dependiente de la
temperatura de tratamiento de la muestra (figura 4.7).
tc
0 50 100 150 200-3
-2
-1
0
1
2
Segunda población crist.
5.80
5.82
5.84
5.86
5.88
5.90
Primera población crist.
t (min)
ln ∆
Hln
Tg
Entalpía total
Figura 4.7 Mezclas PVDF/PMMA con Φm = 0.5. Dependencia de la variación de la transición vítrea y la entalpía de fusión con el tiempo de tratamiento para Ta = 100 °C.
Tanto a tiempos inferiores como superiores a los correspondientes puntos de
cambio de régimen, se aprecia de nuevo una estrecha correlación entre las curvas de
variación de la Tg y las correspondientes a las entalpías de fusión, pudiendo asociarse las
dos regiones de la transición vítrea con las dos poblaciones cristalinas registradas en los
termogramas. Comparando estos resultados con los obtenidos del estudio de la variación de
la densidad electrónica del material en las primeras etapas del proceso, hemos efectuado
una interpretación del significado físico del punto de cambio de régimen, siguiendo el

167
criterio de considerar un control cinético por la difusión molecular en una primera etapa del
proceso y un control conjunto por la difusión y la cristalización a tiempos más largos.
Según esta hipótesis, el punto de cambio de régimen constituiría el límite entre estas dos
regiones. A tiempos inferiores al tiempo de cambio de régimen tendría lugar una variación
rápida de la transición vítrea dependiendo fundamentalmente de la difusión, mientras a
tiempos superiores al tiempo crítico se obtendría un crecimiento lento de la transición
vítrea debido a que la cristalización del PVDF segregado sería ya lenta al estar entorpecido
el crecimiento de los cristales por las colisiones entre ellos, de lo que resultaría un retraso
en la cinética global del proceso.
d) Cristalinidad.
En las mezclas amorfas sometidas a tratamiento térmico no ha sido posible
establecer una identificación total entre los picos de fusión registrados en los termogramas
y las fases cristalinas detectadas por difracción de rayos X, debido a la imposibilidad de
obtener muestras sometidas a tratamientos térmicos completamente equivalentes con las
distintas técnicas experimentales utilizadas. Aunque después de comparar los resultados de
los estudios calorimétricos con los obtenidos por difracción de rayos X se aprecia una cierta
relación entre la primera población cristalina, de punto de fusión constante, con la fase ß, y
entre la segunda población cristalina, de punto de fusión dependiente de la temperatura, con
la fase α, esta relación no es lo suficientemente explícita como para permitirnos confirmar
la citada identificación. Por ello, no es posible descartar en un principio que las diferencias
existentes entre las dos poblaciones cristalinas detectadas por calorimetría puedan deberse a
la estructura interna y al historial térmico de sus respectivos cristales (tamaño, grado de
perfección, dependencia de su crecimiento con la cinética de segregación de la matriz
amorfa) y no a su pertenencia a una fase cristalina determinada.
El estudio de muestras inicialmente segregadas con Φm = 0.75 no presenta
dificultades en lo referente al pico de fusión principal, asignado claramente a la fase ß. Más
difícil resulta la identificación del segundo pico, detectado por calorimetría pero no por
difracción de rayos X, para la que existen dos posibles explicaciones: Fase α presente en
una cantidad insuficiente para ser detectada por difracción de rayos X aunque sí por
calorimetría, conforme al modelo propuesto por Léonard y col. [Léonard, 1983], o una

168segunda población de cristales ß formada inicialmente por cristales de pequeño tamaño
y bajo punto de fusión, los cuales irían engrosándose con la temperatura de tratamiento
incrementando su punto de fusión hasta confundirse con los de la primera población.
Puesto que las medidas de DSC determinan que la proporción de esta segunda población es
aproximadamente un 10% del total, cantidad demasiado elevada para no ser detectada por
rayos X, cabe descartar la primera de las dos hipótesis.
En muestras con concentraciones elevadas de PVDF (Φm ≥ 0.6) sometidas a altas
temperaturas de tratamiento (Ta ~ 150-160 °C), además de las dos poblaciones principales
se detectan otras dos poblaciones cristalinas con puntos de fusión más elevados, tal como
se aprecia en la figura 3.43, correspondiendo la primera de ellas a un proceso de
recristalización durante el barrido en el calorímetro. La identificación de estas dos
poblaciones cristalinas de alta temperatura no resulta evidente ya que sólo en contados
casos pudieron ser estudiadas por difracción de rayos X debido a su pequeña proporción
sobre el total de la masa de las muestras. Sin embargo, en mezclas con Φm = 0.75 se ha
identificado la transición de fase como una transformación cristalina ß → γ. En mezclas
con Φm = 0.6 la transición de fase no ha podido ser registrada por difracción de rayos X,
aunque por analogía hemos asumido que pudiera tratarse también de una transición de fase
similar.
Las fases cristalinas de alta temperatura del PVDF han sido estudiadas por Osaki e
Ishida [Osaki, 1975], Prest y Luca [Prest, 1978] y Okabe y col. [Okabe, 1998], los cuales
las atribuyen al resultado de transformaciones tanto α → γ como ß → γ. En todos los casos
el resultado final es PVDF cristalizado en fase γ, pero dependiendo de la naturaleza del
material inicial y de las condiciones de tratamiento térmico los procesos serán diferentes,
pudiendo tratarse bien de una tranformación en estado sólido (la α → γ según Prest y Luca)
o una fusión lenta seguida de una recristalización del material fundido en el caso de la
transformación ß → γ, también según estos mismos autores.
El trabajo de Osaki e Ishida nos permite interpretar la existencia de dos poblaciones
diferentes de alta temperatura. En las condiciones de trabajo aplicadas a sus muestras
(utilizan PVDF puro en lugar de mezclas PVDF/PMMA como en el presente trabajo) estos
autores encuentran tres picos de fusión que identifican respectivamente con la fase α (el
principal, de más bajo punto de fusión) y con la fase γ los dos restantes. Estas dos
poblaciones γ tendrían puntos de fusión diferentes por haberse formado en condiciones

169
distintas, pero la extrapolación por el método de Hoffmann-Weeks de sus respectivos
puntos de fusión a las condiciones de equilibrio termodinámico conduce a un mismo valor
de Tmo para ambas.
En mezclas de concentraciones medias (Φm = 0.5) no se han apreciado en
prácticamente ningún caso estas poblaciones de alta temperatura. Por el contrario, sí han
resultado ser más adecuadas que las mezclas más ricas en PVDF para realizar el
seguimiento de los procesos cinéticos de segregación molecular y cristalización de forma
controlada, dado que para concentraciones superiores estos procesos son ya demasiado
rápidos y difíciles de estudiar. Tal como ha sido expuesto en el capítulo anterior, la
existencia de varios puntos singulares en la cinética de segregación y cristalización de las
muestras hizo necesario tenerlos en cuenta a la hora de realizar un estudio completo de este
sistema físico.
4.6.1 Transición vítrea del PMMA
La transición vítrea del PMMA (TgPMMA ≈ 395 K) es el principal límite a tener en
cuenta en todas las magnitudes experimentales estudiadas en este trabajo, ya que en todos
los casos se muestra como una frontera que diferencia claramente los valores
correspondientes a temperaturas de tratamiento inferiores a ella de los obtenidos a
temperaturas superiores.
Las discontinuidades observadas tanto en los valores de las entalpías de fusión
como en las transiciones vítreas justo en ese límite de temperatura, han de ser interpretadas
en base a la difusión molecular del PMMA. Dado que los coeficientes de difusión del
PVDF son mayores en varios órdenes de magnitud que los correspondientes al PMMA,
para temperaturas de tratamiento térmico inferiores a TgPMMA, aunque mayores que la
transición vítrea de la mezcla, puede asumirse que la difusión molecular responsable de los
procesos de segregación corre a cargo exclusivamente del PVDF. Por el contrario, rebasado
este límite las moléculas de PMMA están por encima de su transición vítrea, lo que
provoca que la movilidad molecular tenga lugar en ambos polímeros.
A consecuencia de ello, en este segundo caso cabe esperar una oposición de las
moléculas de PMMA a la segregación del PVDF, al ser termodinámicamente favorable la
mezcla de ambos polímeros para el primero de ellos, lo que hace que las moléculas de
ambos polímeros se vuelvan a mezclar de nuevo. Este fenómeno provoca una disminución
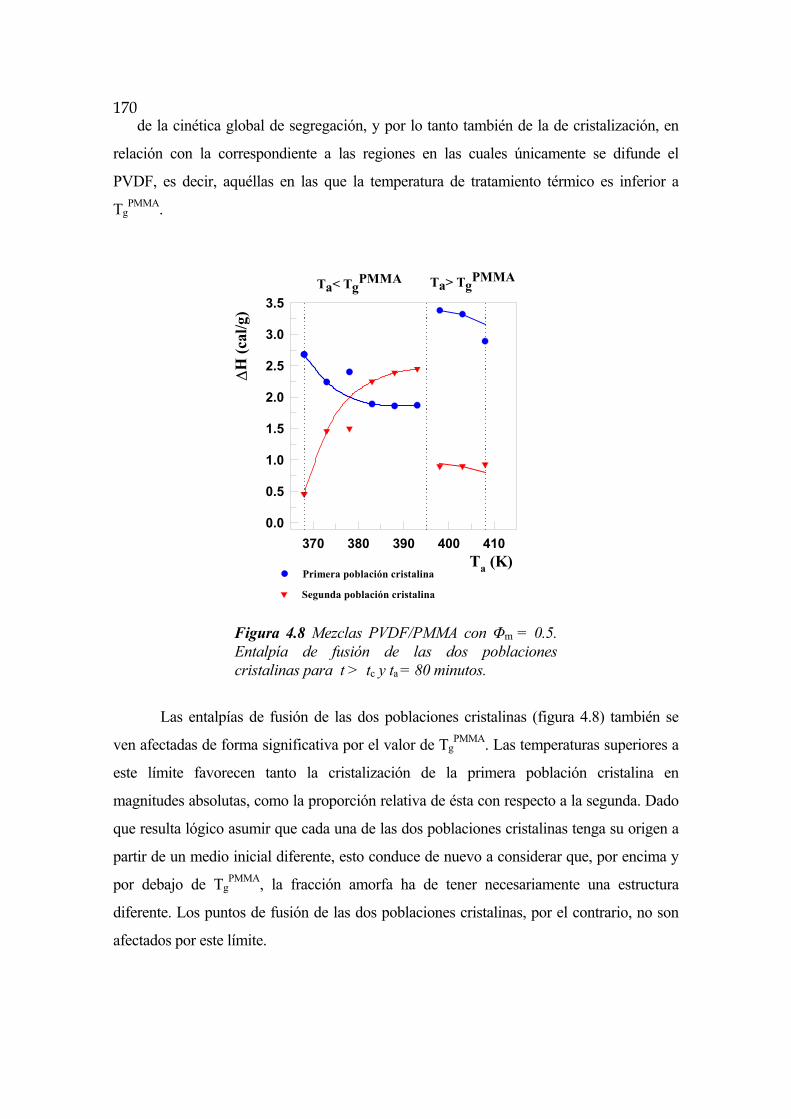
170de la cinética global de segregación, y por lo tanto también de la de cristalización, en
relación con la correspondiente a las regiones en las cuales únicamente se difunde el
PVDF, es decir, aquéllas en las que la temperatura de tratamiento térmico es inferior a
TgPMMA.
370 380 390 400 4100.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Primera población cristalina
Segunda población cristalina
Ta< TgPMMA Ta> TgPMMA
Ta (K)
∆H
(cal
/g)
Figura 4.8 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de las dos poblaciones cristalinas para t > tc y ta = 80 minutos.
Las entalpías de fusión de las dos poblaciones cristalinas (figura 4.8) también se
ven afectadas de forma significativa por el valor de TgPMMA. Las temperaturas superiores a
este límite favorecen tanto la cristalización de la primera población cristalina en
magnitudes absolutas, como la proporción relativa de ésta con respecto a la segunda. Dado
que resulta lógico asumir que cada una de las dos poblaciones cristalinas tenga su origen a
partir de un medio inicial diferente, esto conduce de nuevo a considerar que, por encima y
por debajo de TgPMMA, la fracción amorfa ha de tener necesariamente una estructura
diferente. Los puntos de fusión de las dos poblaciones cristalinas, por el contrario, no son
afectados por este límite.
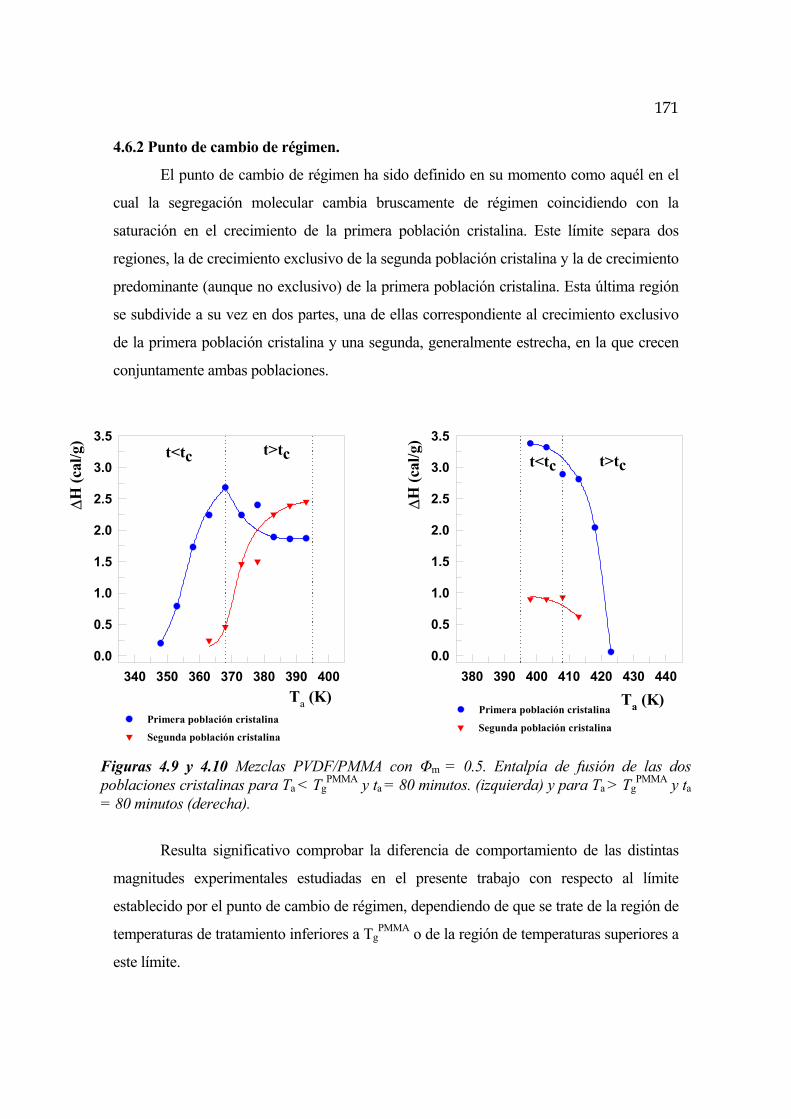
171
4.6.2 Punto de cambio de régimen.
El punto de cambio de régimen ha sido definido en su momento como aquél en el
cual la segregación molecular cambia bruscamente de régimen coincidiendo con la
saturación en el crecimiento de la primera población cristalina. Este límite separa dos
regiones, la de crecimiento exclusivo de la segunda población cristalina y la de crecimiento
predominante (aunque no exclusivo) de la primera población cristalina. Esta última región
se subdivide a su vez en dos partes, una de ellas correspondiente al crecimiento exclusivo
de la primera población cristalina y una segunda, generalmente estrecha, en la que crecen
conjuntamente ambas poblaciones.
340 350 360 370 380 390 4000.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Segunda población cristalina
Primera población cristalina
t>tct<tc
∆H
(cal
/g)
Ta (K)380 390 400 410 420 430 440
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5∆
H (c
al/g
)
Ta (K)
Segunda población cristalina
Primera población cristalina
t<tc t>tc
Figuras 4.9 y 4.10 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de las dos poblaciones cristalinas para Ta < Tg
PMMA y ta = 80 minutos. (izquierda) y para Ta > TgPMMA y ta
= 80 minutos (derecha).
Resulta significativo comprobar la diferencia de comportamiento de las distintas
magnitudes experimentales estudiadas en el presente trabajo con respecto al límite
establecido por el punto de cambio de régimen, dependiendo de que se trate de la región de
temperaturas de tratamiento inferiores a TgPMMA o de la región de temperaturas superiores a
este límite.

172Como puede apreciarse en las figuras 4.9 y 4.10, en el primer caso la entalpía de
fusión de la primera población cristalina experimenta una clara depresión en la zona
correspondiente a tiempos de tratamiento superiores al tiempo de cambio de régimen (tc) y
temperaturas de tratamiento inferiores a TgPMMA. Este fenómeno no ocurre, ni para la
segunda población cristalina en la región de Ta < TgPMMA, ni para ninguna de las dos en la
región
Ta > TgPMMA.
En realidad esta depresión no indica un descenso de la cristalinidad sino una
disminución en el nivel de saturación del crecimiento de la primera población cristalina,
que en esta región está desfavorecida con respecto a la segunda al contrario de lo que
ocurre en las tres regiones restantes.
En resumen, considerando los límites impuestos tanto por el cambio en la cinética
de segregación molecular (punto de cambio de régimen) como por la temperatura de
transición vítrea del PMMA, se puede realizar un estudio por separado de cada una de las
cuatro regiones en las que quedan divididas las curvas.
4.6.3 Cinética para t < tc y Ta < TgPMMA.
En las primeras etapas del proceso de segregación es de esperar un predominio de
la difusión molecular sobre la cristalización e incluso, a tiempos suficientemente cortos,
una presencia exclusiva del primero de estos dos fenómenos. Por otro lado, a temperaturas
de tratamiento inferiores a TgPMMA también puede asumirse que la difusión molecular, y por
lo tanto la consiguiente segregación de fases, tiene lugar únicamente en el PVDF. Esta
hipótesis se ha visto confirmada por los resultados obtenidos mediante difracción de rayos
X a ángulos bajos y por el estudio de la variación de la transición vítrea y la cinética de
cristalización.
La comparación de los resultados realizados en condiciones isotermas (figura 4.7)
permite comprobar que la segregación tiene lugar linealmente con el tiempo de forma muy
rápida y está asociada básicamente con la primera de las dos poblaciones cristalinas, la cual
crece de forma simultánea hasta alcanzar un punto de saturación coincidente con el punto
crítico de la transición vítrea. Únicamente a tiempos cercanos al punto crítico comienza a
cristalizar la segunda población cristalina; aunque la saturación del crecimiento de la
primera población y la aparición de la segunda población no son exactamente coincidentes,
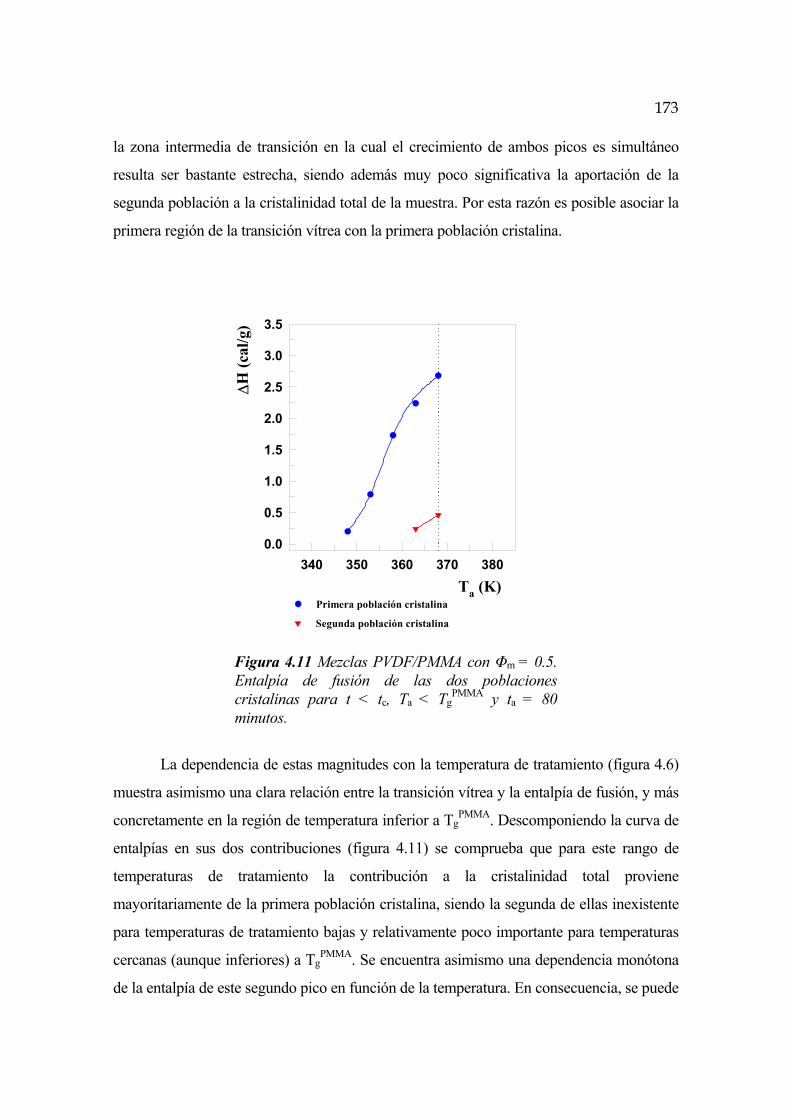
173
la zona intermedia de transición en la cual el crecimiento de ambos picos es simultáneo
resulta ser bastante estrecha, siendo además muy poco significativa la aportación de la
segunda población a la cristalinidad total de la muestra. Por esta razón es posible asociar la
primera región de la transición vítrea con la primera población cristalina.
340 350 360 370 3800.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Segunda población cristalina
Primera población cristalina
∆H
(cal
/g)
Ta (K)
Figura 4.11 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de las dos poblaciones cristalinas para t < tc, Ta < Tg
PMMA y ta = 80 minutos.
La dependencia de estas magnitudes con la temperatura de tratamiento (figura 4.6)
muestra asimismo una clara relación entre la transición vítrea y la entalpía de fusión, y más
concretamente en la región de temperatura inferior a TgPMMA. Descomponiendo la curva de
entalpías en sus dos contribuciones (figura 4.11) se comprueba que para este rango de
temperaturas de tratamiento la contribución a la cristalinidad total proviene
mayoritariamente de la primera población cristalina, siendo la segunda de ellas inexistente
para temperaturas de tratamiento bajas y relativamente poco importante para temperaturas
cercanas (aunque inferiores) a TgPMMA. Se encuentra asimismo una dependencia monótona
de la entalpía de este segundo pico en función de la temperatura. En consecuencia, se puede

174considerar válido el modelo que asocia la variación de la transición vítrea con el
crecimiento de la primera población cristalina.
El estudio de la dependencia del pico de fusión de la primera población cristalina
con la temperatura y el tiempo de tratamiento sirve para complementar lo anteriormente
expuesto. Esta población cristalina cuenta con unos puntos de fusión elevados
(Tm ~ 150-155 °C para muestras con Φm = 0.5) que son prácticamente independientes tanto
del tiempo como de la temperatura de tratamiento, lo cual indica que se trata de unos
cristales homogéneos y de tamaño uniforme, con un punto de fusión elevado e
independiente de la temperatura de tratamiento. Es de esperar que el crecimiento de estos
cristales tenga lugar de forma simultánea a partir de núcleos formados en el material
segregado, sin que las condiciones de cristalización afecten significativamente a su
cinética. Este crecimiento cristalino debería detenerse una vez que los cristales de PVDF
alcanzaran un tamaño suficiente para entrar en contacto entre ellos, al dificultarse la
continuación de su crecimiento.
4.6.4 Cinética para t > tc y Ta < TgPMMA.
Al estudiar la variación de los valores de Tg en las regiones de tiempos superiores al
correspondiente al punto crítico, tomando como tiempo cero el tiempo crítico (figura 3.22),
se observa que estos valores de Tg dependen linealmente con el tiempo al igual que ocurre
en el caso anterior, pero sin ser función aparentemente de la temperatura de tratamiento,
apreciándose desviaciones significativas únicamente para los valores correspondientes a
temperaturas (Ta = 115-120 °C) cercanas al límite, en las cuales la variación de Tg es
inferior a la correspondiente al resto de las temperaturas.
Esta segunda segregación de la fase amorfa se asocia exclusivamente al crecimiento
de la segunda población cristalina, la cual por el contrario sí demuestra depender en forma
sistemática de la temperatura de tratamiento. Esta aparente contradicción ha de ser
explicada suponiendo que, para una misma cantidad de PVDF segregado, la fracción
cristalizada del mismo depende de la temperatura de tratamiento térmico a la que se somete
la muestra en mucha mayor medida que en la primera población cristalina.
El estudio de las entalpías de fusión de ambas poblaciones cristalinas en este
intervalo (figura 4.12) exhibe un fuerte crecimiento de la segunda población mientras la
primera población cristalina presenta una fuerte depresión de los valores de su entalpía de
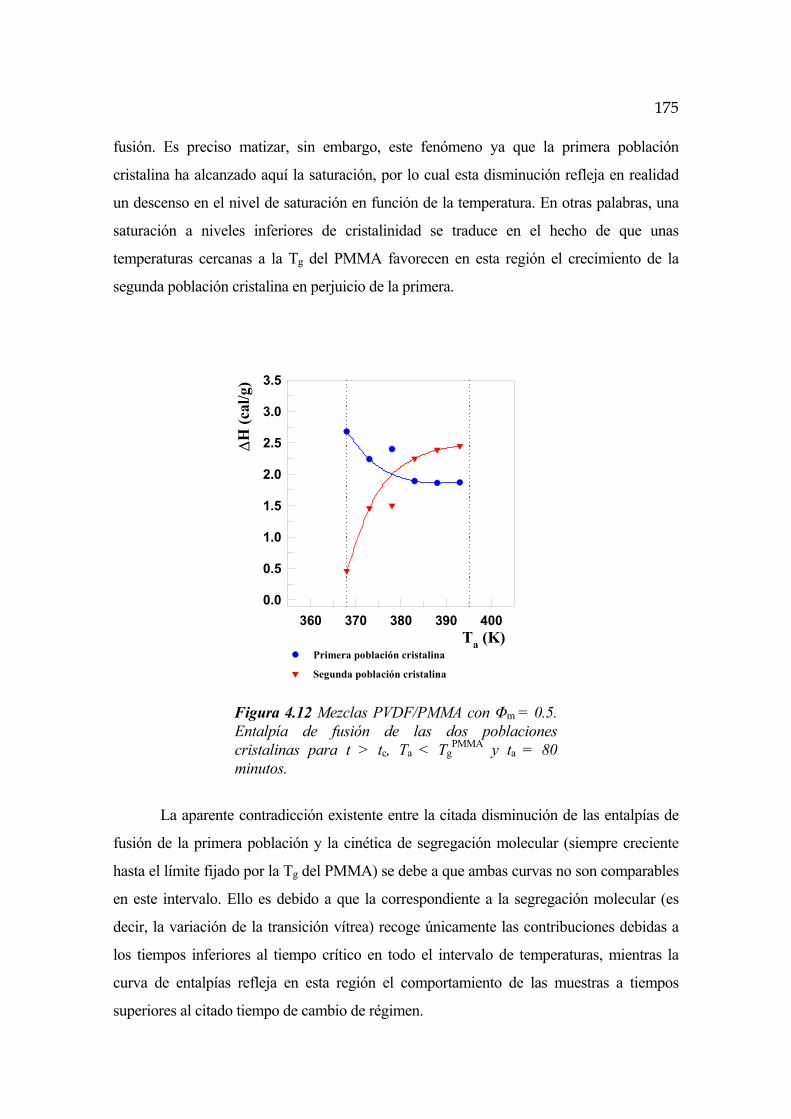
175
fusión. Es preciso matizar, sin embargo, este fenómeno ya que la primera población
cristalina ha alcanzado aquí la saturación, por lo cual esta disminución refleja en realidad
un descenso en el nivel de saturación en función de la temperatura. En otras palabras, una
saturación a niveles inferiores de cristalinidad se traduce en el hecho de que unas
temperaturas cercanas a la Tg del PMMA favorecen en esta región el crecimiento de la
segunda población cristalina en perjuicio de la primera.
360 370 380 390 4000.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Segunda población cristalina
Primera población cristalina
∆H
(cal
/g)
Ta (K)
Figura 4.12 Mezclas PVDF/PMMA con Φm = 0.5. Entalpía de fusión de las dos poblaciones cristalinas para t > tc, Ta < Tg
PMMA y ta = 80 minutos.
La aparente contradicción existente entre la citada disminución de las entalpías de
fusión de la primera población y la cinética de segregación molecular (siempre creciente
hasta el límite fijado por la Tg del PMMA) se debe a que ambas curvas no son comparables
en este intervalo. Ello es debido a que la correspondiente a la segregación molecular (es
decir, la variación de la transición vítrea) recoge únicamente las contribuciones debidas a
los tiempos inferiores al tiempo crítico en todo el intervalo de temperaturas, mientras la
curva de entalpías refleja en esta región el comportamiento de las muestras a tiempos
superiores al citado tiempo de cambio de régimen.

176En lo que respecta a los puntos de fusión de la segunda población cristalina,
también se aprecia una marcada diferencia en relación con los correspondientes a la
primera. Estos puntos de fusión, siempre inferiores en magnitud a los correspondientes a la
primera población cristalina, muestran un comportamiento similar a éstos en lo que
respecta a su dependencia con el tiempo de tratamiento, siendo prácticamente invariables.
Las diferencias que aparecen a tiempos cortos se atribuyen a la falta de definición de los
máximos cuando los picos son todavía incipientes. Sin embargo, su dependencia con la
temperatura de tratamiento es completamente distinta, apreciándose para todas las
concentraciones estudiadas un incremento aproximadamente lineal en función de Ta.
En resumen, los resultados obtenidos indican con claridad que la cristalización de
esta segunda población cristalina estaría vinculada a procesos tardíos de segregación
molecular ocurridos con posterioridad a la saturación de la población anterior o iniciados
poco antes de que tuviera lugar ésta. La cristalinidad de la segunda población cristalina
crece muy lentamente y siempre en pequeña cantidad. El control cinético del proceso
global es de esperar que esté regulado conjuntamente por la difusión molecular y el
crecimiento cristalino o quizá incluso sólo por este último, dado que la difusión molecular
(es decir, la variación de la transición vítrea en la matriz amorfa) demuestra no depender de
la temperatura de tratamiento para tiempos superiores al tiempo crítico (excepto para
valores muy cercanos a la transición vítrea del PMMA), aunque sí del tiempo transcurrido
desde este tiempo crítico. En consecuencia, en estas condiciones se producirían unos
cristales muy imperfectos y de bajo punto de fusión, dependientes además de la
temperatura de tratamiento térmico.
4.6.5 Cinética para t < tc y Ta > TgPMMA.
Las teorías cinéticas de cristalización predicen para el crecimiento cristalino en
función de la temperatura una curva en forma de campana cuyo máximo marca un límite
pasado el cual la velocidad de cristalización decrece conforme disminuye la diferencia
Tc-Tf en el material cristalizado. Puesto que la difusión molecular crece a su vez
exponencialmente con la temperatura, para temperaturas de tratamiento térmico lo
suficientemente elevadas el control cinético del proceso global ya no puede ser atribuido a
la difusión molecular, como ocurría a temperaturas de tratamiento bajas y tiempos cortos,

177
sino que ahora es la cristalización, como proceso más lento, la que controla la cinética
global de la segregación molecular.
Por otro lado, a temperaturas de tratamiento mayores a la correspondiente a la Tg
del PMMA también se ha de tener en cuenta la difusión molecular del PMMA. La
movilidad de las moléculas de este polímero se opone a la difusión molecular del PVDF,
produciéndose una mezcla parcial de las moléculas de PVDF y PMMA segregadas que dan
como resultado una disminución de la cinética global de segregación en relación con la
correspondiente a la difusión del PVDF en solitario.
El estudio de varios parámetros experimentales confirma estas hipótesis, mostrando
una disminución de la segregación molecular en función de la temperatura. Ajustados los
valores experimentales de variación de la Tg en forma lineal con el tiempo y como una
función exponencial cuadrática (gaussiana) con la temperatura, se observa que para un
mismo incremento de temperatura (en valor absoluto) con respecto a la Tg del PMMA, los
valores correspondientes a la región de temperaturas altas (Ta > TgPMMA) son
sistemáticamente inferiores a los equivalentes de la otra región de temperaturas (Ta <
TgPMMA), fenómeno que ha sido interpretado en base a la ya explicada mezcla parcial
producida por la difusión molecular del PMMA en el seno de la matriz amorfa.
En lo que a la entalpía de fusión respecta, el comportamiento de las curvas
isotermas es similar, desde un punto de vista cualitativo, al observado en la primera región
de temperaturas, con la primera población cristalina vinculada estrechamente a la
segregación molecular para tiempos inferiores al tiempo de cambio de régimen y una
saturación de su crecimiento a partir de este valor, junto con una pequeña aportación de la
incipiente segunda población para tiempos cercanos al tiempo de cambio de régimen.
Al observar la dependencia de las entalpías de fusión con la temperatura de
tratamiento (figura 4.13) se aprecia una situación simétrica a la anterior, con una primera
población cristalina decreciente con la temperatura y una segunda población incipiente o
inexistente. La entalpía de fusión de la primera población tiende a cero a temperaturas
cercanas a la de fusión del PVDF (Ta = 150 °C para Φm = 0.5), mientras la segunda
población lo hace a valores ligeramente menores coincidiendo, para concentraciones
suficientemente ricas de PVDF, con el inicio de la transformación de fase que daba como
resultado la aparición de la fase γ.

178
400 410 420 430
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Segunda población cristalina
Primera población cristalina
∆H
(cal
/g)
Ta (K)
Figura 4.13 Mezclas PVDF/PMMA con Φm =0.5. Entalpía de fusión de las dos poblaciones cristalinas para t < tc, Ta > Tg
PMMA y ta = 80 minutos.
Los puntos de fusión siguen la tendencia general ya comentada, aunque no siempre
se detecta la segunda población cristalina debido a su ya citada inexistencia a temperaturas
de tratamiento próximas a la temperatura de fusión del material cristalino.
4.6.6 Cinética para t > tc y Ta > TgPMMA.
En esta región se sigue apreciando una variación de la Tg similar a la registrada en
la región equivalente de temperaturas inferiores a la Tg del PMMA. En este caso hemos
encontrado una dependencia aproximadamente lineal con el tiempo t-tc (tomando como
cero los respectivos tiempos de cambio de régimen) y una independencia con la
temperatura. Puesto que en esta región el número de puntos experimentales es inferior al
existente en el caso anterior la precisión de ajuste es menor. A pesar de ello, se aprecia
claramente que la cinética de segregación molecular es, en cualquier caso, muy inferior en
magnitud a la correspondiente a temperaturas de tratamiento inferiores a la transición vítrea

179
del PMMA, incluso para aquellos valores bajos correspondientes a las temperaturas
cercanas a este límite (Ta = 115-120 °C).
El estudio de las entalpías de fusión de las dos poblaciones cristalinas muestra
resultados diferentes a los obtenidos en la región equivalente de temperaturas inferiores a la
Tg del PMMA. Sin embargo, se sigue observando la saturación del crecimiento de la
primera población cristalina en todo el rango de temperaturas por encima del punto de
cambio de régimen. Comparando los valores de las dos entalpías de fusión con las
correspondientes a un mismo tiempo y un mismo incremento de temperatura con respecto a
la región de temperaturas bajas (Ta < TgPMMA) (figura 4.8), se observa que la primera
población de cristales alcanza de forma sistemática unos límites de saturación de ∆H más
elevados que en el caso anterior. Por el contrario, la segunda población cristalina tiene
valores inferiores a los registrados en el otro rango de temperaturas.
4.6.7 Temperaturas de fusión.
El estudio de la dependencia de los puntos de fusión con la temperatura, para un
tiempo de tratamiento dado, muestra un comportamiento notablemente diferente para cada
una de las dos poblaciones cristalinas.
En general, los valores correspondientes a la primera población cristalina son
elevados y experimentan una leve variación con la temperatura de tratamiento, mientras los
pertenecientes a la segunda población tienen su inicio a temperaturas situadas
inmediatamente por encima de la transición vítrea de la mezcla y experimentan un fuerte
incremento al aumentar la temperatura de tratamiento, alcanzando magnitudes equivalentes
a las de la primera población para temperaturas de tratamiento cercanas ya a la temperatura
de fusión del PVDF en estas muestras (figura 4.14).
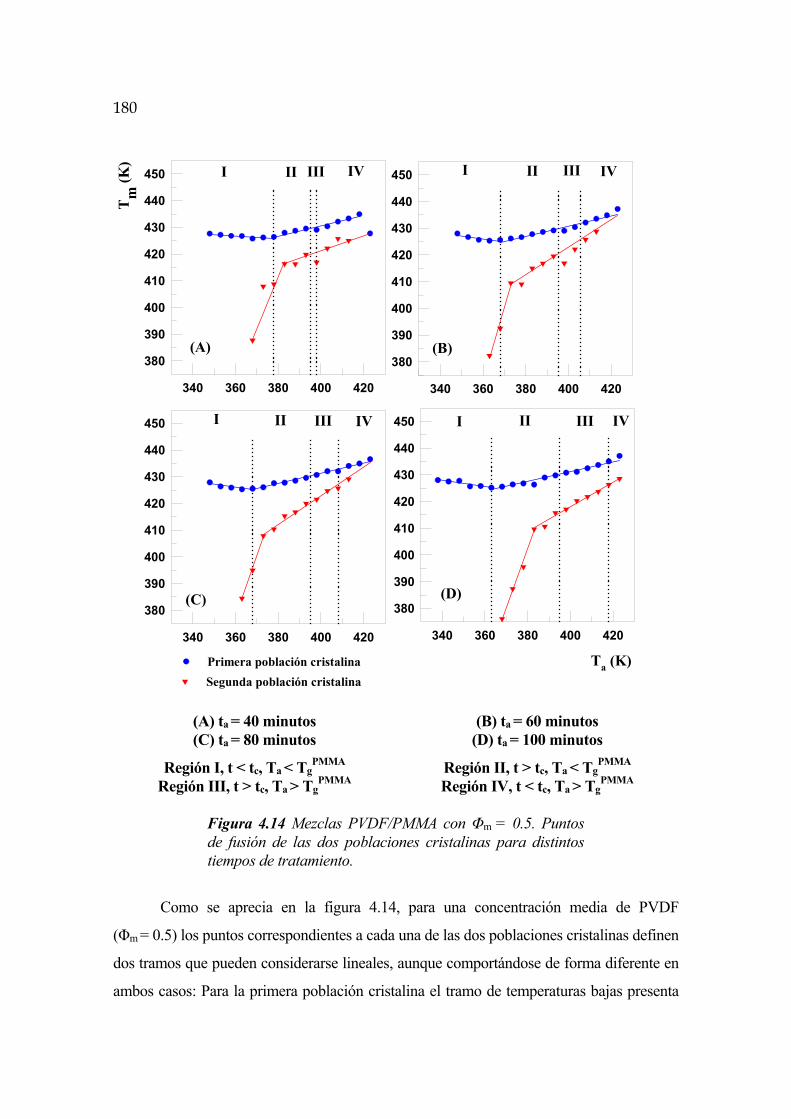
180
Segunda población cristalinaPrimera población cristalina Ta (K)
(D)(C)
(B)(A)
IVIIIIIIIVIIIIII
IVIIIIII
340 360 380 400 420
380
390
400
410
420
430
440
450
340 360 380 400 420
380
390
400
410
420
430
440
450
340 360 380 400 420
380
390
400
410
420
430
440
450
340 360 380 400 420
380
390
400
410
420
430
440
450
Tm
(K) III IVIII
(A) ta = 40 minutos (B) ta = 60 minutos (C) ta = 80 minutos (D) ta = 100 minutos
Región I, t < tc, Ta < TgPMMA Región II, t > tc, Ta < Tg
PMMA Región III, t > tc, Ta > Tg
PMMA Región IV, t < tc, Ta > TgPMMA
Figura 4.14 Mezclas PVDF/PMMA con Φm = 0.5. Puntos de fusión de las dos poblaciones cristalinas para distintos tiempos de tratamiento.
Como se aprecia en la figura 4.14, para una concentración media de PVDF
(Φm = 0.5) los puntos correspondientes a cada una de las dos poblaciones cristalinas definen
dos tramos que pueden considerarse lineales, aunque comportándose de forma diferente en
ambos casos: Para la primera población cristalina el tramo de temperaturas bajas presenta

181
una pendiente ligeramente negativa, mientras para la segunda población cristalina la
pendiente es positiva y de valor elevado. Los tramos de temperaturas altas muestran para
las dos poblaciones cristalinas unas pendientes positivas, mayor la segunda que la primera,
tendiendo a cortarse ambas rectas en valores de temperatura cercanos al punto de fusión en
el equilibrio de los cristales de PVDF. El cambio de tendencia corresponde, para la primera
población cristalina, al inicio del crecimiento de la segunda población cristalina, mientras
para la segunda población cristalina este cambio de tendencia tiene lugar a una temperatura
situada en torno a los 370-380 K.
Para concentraciones mayores de PVDF se hace menor la diferencia entre ambos
tramos en las curvas correspondientes a cada una de las dos poblaciones cristalinas (figura
4.15), apareciendo también las formas de alta temperatura asociadas a la fase γ, con
temperaturas de fusión crecientes con la temperatura.
320 340 360 380 400 420 440340
360
380
400
420
440
460
Tm
(K)
Ta (K)
Fases de alta temperatura
Segunda población cristalinaPrimera población cristalina
Figura 4.15 Mezclas PVDF/PMMA con Φm = 0.6. Puntos de fusión de las diferentes poblaciones cristalinas para un tiempo de tratamiento de 10 minutos.

182Una observación importante es el hecho de que ninguno de los puntos de fusión
de las dos principales poblaciones cristalinas ve afectada su tendencia ni por la Tg del
PMMA, ni por ninguno de los dos puntos de cambio de régimen (fronteras entre las
regiones I a IV), en contraste con lo que ocurre tanto con la variación de la Tg como con las
respectivas entalpías de fusión. En resumen, puede afirmarse que estas tres líneas de
cambio de régimen, y en especial la primera de ellas, afectan fuertemente al crecimiento de
los cristales de las dos poblaciones, pero no a su morfología ni al tamaño de los mismos.
La evolución de los puntos de fusión refleja el tipo de morfología de los cristales
formados: En la primera población de cristales se puede predecir un predominio de la
nucleación sobre el plegado molecular tal como indica el punto de fusión constante,
formándose cristales de tamaño homogéneo de alto punto de fusión. La segunda población,
por el contrario, parece estar formada por cristales imperfectos de bajo punto de fusión,
creados a partir de un medio desordenado, cuyo tamaño depende en gran medida de la
temperatura de tratamiento. En este caso se precisan valores cercanos a los de la
temperatura de fusión para poder alcanzar puntos de fusión similares a los de la primera
población. La morfología y el tamaño promedio de los cristales, determinados a partir del
punto de fusión, son independientes de la cantidad de material cristalizado, determinada a
su vez por las entalpías de fusión, y no se ven afectados por las variaciones de la
segregación molecular en función de la temperatura y el tiempo de tratamiento.
4.7 Cinética de segregación y cristalización en mezclas con exceso de PVDF.
En el caso de mezclas de PVDF y PMMA con exceso de PVDF (Φm ≥ 0.7) la
presencia de PVDF segregado previamente al tratamiento térmico impone unas
condiciones previas que se traducen en una cinética de segregación y cristalización muy
diferente a la encontrada en mezclas inicialmente amorfas. En principio, y en concordancia
con lo publicado por Léonard y col. [Léonard, 1983], se aprecia que tanto el PVDF
segregado previamente al tratamiento térmico como el que lo hace durante este proceso,
pertenecen fundamentalmente a la fase ß, pudiéndose suponer que la existencia previa de
unos núcleos de cristalización condiciona el crecimiento de los cristales de PVDF en esta
fase cristalina. A temperaturas cercanas a la de fusión del PVDF, tal como ha sido
comentado, tiene lugar una transición de fase ß → γ.

183
Los estudios calorimétricos han revelado la existencia, en muestras tratadas
térmicamente, de dos poblaciones cristalinas perfectamente diferenciadas además de la fase
γ, las cuales muestran en sus temperaturas de fusión el mismo comportamiento que el
observado en muestras de menor concentración de PVDF (figura 4.16):
a) Una primera población cristalina, identificable como fase ß, de punto de fusión
elevado cuya temperatura de fusión permanece constante (Tm ~ 438 K).
b) Una segunda población cristalina cuyo punto de fusión varía fuertemente con la
temperatura de tratamiento.
c) Las dos formas de alta temperatura asociadas a la fase γ detectadas también en la
muestras amorfas con Φm = 0.6.
320 340 360 380 400 420 440
340
360
380
400
420
440
460
Tm
(K)
Ta (K)
Fases de alta temperatura
Segunda población cristalinaPrimera población cristalina
Figura 4.16 Mezclas PVDF/PMMA con Φm = 0.75. Puntos de fusión de las diferentes poblaciones cristalinas para un tiempo de tratamiento de 10 minutos.
Como puede apreciarse, la única diferencia cualitativa entre el gráfico que refleja la
variación de los puntos de fusión en función de Ta para las muestras con Φm = 0.75 y las
correspondientes a mezclas con menor contenido en PVDF e inicialmente amorfas, es que

184la curvatura de las dos poblaciones principales prácticamente ha desaparecido para
Φm = 0.75, a excepción de la primera población cristalina en el tramo correspondiente a
temperaturas de tratamiento cercanas al punto de fusión del PVDF cristalizado.
Las entalpías de fusión, por el contrario, muestran en las mezclas con Φm = 0.75 un
comportamiento distinto al correspondiente a las mezclas inicialmente amorfas, existiendo
un predominio de la fase principal (ß) en todo el intervalo de temperaturas a excepción de
aquéllas a las que tiene lugar la transición de fase ß → γ.
La segregación molecular, estudiada a partir de la variación de la transición vítrea
con la temperatura de tratamiento, ofrece unos resultados cualitativamente similares a los
obtenidos en las mezclas amorfas, con unos valores inferiores para Ta > TgPMMA a los
correspondientes a Ta < TgPMMA, pero con una distribución de los puntos experimentales
completamente distinta.
En conjunto, puede afirmarse que el material previamente segregado actúa como
núcleos de cristalización a partir de los cuales crecen los cristales. Esta diferencia explica el
distinto comportamiento tanto de la Tg como de las entalpías de fusión aunque, al igual que
ocurre con las mezclas amorfas, los puntos de fusión no se ven afectados por ello.
Otra singularidad encontrada en las mezclas con Φm = 0.75 consiste en la detección
de una segunda Tg, prácticamente independiente de la temperatura de tratamiento, situada
en torno a los 291 K. A esta temperatura le corresponde una concentración en peso de
aproximadamente un 75% de PVDF, que es superior a la inicial de la matriz amorfa,
calculada en torno a un 65% en función de la Tg inicial.
4.8 Naturaleza de la interfase cristalina
Tal como ha sido explicado en el capítulo anterior, las mezclas PVDF/PMMA
parcialmente segregadas constituyen un sistema físico formado por, al menos, tres fases:
PVDF cristalino, una matriz amorfa enriquecida en PMMA y una interfase. Aunque la
existencia de la interfase es un hecho habitual en estos sistemas físicos y está perfectamente
determinada en las mezclas de estos dos polímeros por los trabajos de numerosos autores,
su naturaleza exacta es objeto de controversia.
Básicamente, se pueden considerar dos modelos límite, el de una interfase
compuesta exclusivamente por PVDF amorfo, o el de una interfase constituida por una
mezcla también amorfa de PVDF y PMMA de concentración diferente a la de la matriz

185
amorfa. La bibliografía existente, tanto relativa al sistema PVDF/PMMA como a otros
similares, no es demasiado extensa, discrepando los distintos autores sobre la naturaleza de
esta región. Así, mientras unos [Hahn, 1985; Yoon, 1991; Ando, 1994; Mijovic, 1997;
Wing, 2000] proponen una interfase cristalina formada exclusivamente por PVDF en estado
amorfo, puro o prácticamente puro, un segundo grupo de autores [Maas, 1994; Saito,
1994b; Bessonova, 1998; Crämer, 1998; Maiti, 1998; Okabe, 1998] describen regiones
intercristalinas en las que existirían moléculas del polímero amorfo mezcladas con el
cristalino en determinada proporción. La cuestión está en determinar si estas regiones
corresponden en sentido estricto a la interfase cristalina o si, por el contrario, se trataría de
un sistema más complejo formado por un total de cuatro fases: PVDF cristalino, interfase
amorfa de PVDF puro, región intercristalina formada por una mezcla de ambos polímeros
y, finalmente, fracción amorfa empobrecida en PVDF en relación con la concentración
inicial. Este sistema conciliaría ambas posturas, a la par que explicaría las diferencias
morfológicas detectadas en el PVDF cristalizado, en relación con las esferulitas de
polímero puro, descritas en varios trabajos recientes [Saito, 1994b; Crämer, 1998; Maiti,
1998; Gregorio, 2000].
El presente trabajo de investigación, por su parte, confirma esta última hipótesis.
Por un lado, como quedó reflejado en el capítulo anterior, queda demostrada la existencia
de una fase enriquecida en PVDF (con valores en torno al 70% de este polímero) en
relación con la fase amorfa. Por otro, el balance de masa de las tres fases estudiadas (PVDF
cristalino, fase enriquecida en PVDF y matriz amorfa) resulta, por lo general, inferior a la
unidad, lo que parece indicar la existencia de una fracción de masa (¿la interfase de
PVDF?) que no ha sido detectada en nuestros experimentos. Partiendo de esta hipótesis, las
conclusiones generales son las siguientes.
4.8.1 Estudio de la interfase cristalina
La primera conclusión que se obtiene del estudio de las transiciones vítreas
secundarias relacionadas con la interfase cristalina, es que cada una de las dos poblaciones
cristalinas principales identificadas en el sistema posee asociada una interfase de cinética, y
presumiblemente de morfología, diferente de la otra, razón por lo que está justificado un
estudio por separado de las interfases correspondientes a cada una de estas dos poblaciones
cristalinas.

186
a) Interfase asociada a la primera población cristalina.
Tal como ha sido expuesto en el capítulo anterior, definimos a la primera población
cristalina como aquélla que aparece en primer lugar cuando las muestras homogéneas son
sometidas a tratamiento térmico, correspondiendo a tiempos de tratamiento cortos e
incrementos altos de cristalinidad tras los cuales su crecimiento se satura. Esta población
cristalina está asociada a la primera etapa de la cinética de segregación de la matriz amorfa.
Tanto la concentración media de la interfase como la cantidad de masa presente en
la misma (figuras 3.57 y 3.59) muestran en todo el rango de temperaturas de tratamiento
dos discontinuidades que modifican sensiblemente la tendencia de las mismas. Las
temperaturas a las que tienen lugar estas discontinuidades son, respectivamente, la
temperatura mínima a la que se ha registrado la aparición de la segunda población cristalina
(Ta ~ 360 K) y la transición vítrea del PMMA (Ta ~ 393 K).
320 340 360 380 400 420 440
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Ta (K)
∆H
m (c
al/g
)
Figura 4.17 Dependencia con la temperatura de la entalpía de fusión de la primera población cristalina, para Φm = 0.5.
El siguiente paso consiste en asociar el comportamiento de la interfase con la
evolución de la población cristalina. En la figura 4.17 hemos reflejado las entalpías de
fusión medidas experimentalmente (es decir, la cantidad de material cristalizado) en
función de la temperatura, para diferentes tiempos de tratamiento. A diferencia de las

187
magnitudes estudiadas en la interfase (concentración media y ∆cp), la cristalinidad es
acumulativa, lo que impide promediar o extrapolar sus valores tal como se hizo en el caso
anterior. Por esta razón, la interpretación física ha de hacerse a partir de los valores
máximos alcanzados por la entalpía de fusión para cada temperatura de tratamiento,
obteniéndose de nuevo una distribución trimodal separada por fronteras similares a las
encontradas en los casos anteriores.
La existencia de estas dos fronteras provoca la aparición de tres regiones netamente
diferenciadas. La primera de ellas corresponde al intervalo en el cual el PVDF segregado
cristaliza exclusivamente en la primera población cristalina aun a tiempos suficientemente
largos de tratamiento. La segunda región agrupa a las muestras en las que también se
produce la aparición de la segunda población cristalina, si bien a tiempos de tratamiento
superiores a los aplicados a estas muestras. La tercera región, por último, se encuentra a
temperaturas de tratamiento superiores a la temperatura de transición vítrea del PMMA.
La naturaleza de estas fronteras ha de ser investigada con precaución debido a que
la saturación en el crecimiento de la primera población cristalina no coincide con el inicio
de la cristalización de la segunda población cristalina, al existir un intervalo de crecimiento
común de extensión variable en función de la temperatura de tratamiento. En esta región
intermedia no resulta posible separar las dos contribuciones individuales correspondientes a
la ∆cp de cada interfase, aunque sí se puede hacer con las entalpías de fusión de las dos
poblaciones cristalinas. Por esta razón, si en lugar de representar los valores máximos de
las entalpías de fusión anteriores a la aparición de la segunda población cristalina, se hace
con valores correspondientes a la saturación de la entalpía de fusión de la primera
población cristalina, se obtienen los resultados que vienen reflejados en la figura 4.18,
correspondiendo la diferencia de ordenadas entre los puntos de esta curva y los de la
anterior, al intervalo de crecimiento conjunto de ambas poblaciones cristalinas.
Tal como puede apreciarse en la figura, la curva de las entalpías de saturación de la
primera población cristalina presenta una forma bastante similar a la curva de las entalpías
máximas coincidentes con la aparición de la segunda población cristalina, con una
depresión muy marcada en las cercanías de la transición vítrea del PMMA y unos valores
más elevados a temperaturas superiores a TgPMMA que a temperaturas inferiores a la misma.
Mucho menos evidente resulta ahora la existencia de la otra frontera, ya que es difícil
apreciar si la depresión existe realmente o si, por el contrario, se trata de un artificio

188matemático provocado por la supresión, en la curva anterior, de los puntos
experimentales intermedios, correspondientes a tiempos de tratamiento térmico en los
cuales tiene lugar un crecimiento conjunto de ambas poblaciones cristalinas. Sin embargo,
esta depresión sí aparece marcada con nitidez en los parámetros asociados a la interfase, lo
que parece indicar que la frontera propuesta en torno a los 360 K existe realmente.
320 340 360 380 400 420 440
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
Ta (K)
∆H
m (c
al/g
)
Figura 4.18 Dependencia con la temperatura de la entalpía de fusión (valores de saturación) de la primera población cristalina, para Φm = 0.5.
En ambos casos el PVDF cristalizado pertenece de forma exclusiva a la primera
población cristalina, pero mientras en la primera región (Ta < 360 K) la segunda población
cristalina no llega a aparecer en todo el proceso, en la segunda región (Ta > 360 K) todavía
no ha aparecido para los tiempos de tratamiento térmico aplicados (figura 4.17) o bien ya
ha aparecido, habiendo alcanzado la saturación el crecimiento de la primera población
cristalina (figura 4.18). En el caso de la cristalinidad, la explicación puede ser que la rápida
aparición de la segunda población cristalina impide contar con los valores correspondientes
a tiempos de tratamiento más elevados, pero este criterio no es aplicable a la interfase dado
que aquí hemos trabajado con valores promediados o extrapolados en los que se ha
minimizado la dependencia temporal. La conclusión es que, aun a tiempos de tratamiento

189
térmico inferiores a los necesarios para que la segunda población cristalina pueda ser
detectada, la interfase evoluciona de forma aparentemente distinta según la aparición de
esta segunda población cristalina vaya a tener lugar o no. En resumen, puede afirmarse que
la futura aparición de la segunda población cristalina provoca una disminución, tanto de la
cristalinidad, como de la cantidad de material involucrado en la interfase, así como un
menor gradiente de concentraciones respecto a la matriz amorfa, lo que puede ser atribuido
a una posible disminución de los núcleos de precristalización.
La depresión provocada por la transición vítrea del PMMA tiene la misma
interpretación que la dada en el estudio de otros parámetros de las muestras, tales como la
cristalinidad o la segregación molecular de la matriz amorfa: La movilidad de las moléculas
de PMMA por encima de la temperatura de transición vítrea de este polímero dificulta la
segregación del PVDF y, por lo tanto, la cristalización del mismo.
b) Interfase asociada a la segunda población cristalina
La segunda población cristalina es aquélla que aparece tardíamente a tiempos de
tratamiento térmico largos. Esta población crece más lentamente que la anterior, y tras un
período inicial de solapamiento de ambas cinéticas, queda como única responsable del
incremento de la cristalinidad total con posterioridad a la saturación del crecimiento de la
primera población cristalina.
La cinética de la interfase asociada al crecimiento de la segunda población
cristalina es muy diferente a la encontrada en el caso anterior. En primer lugar, se aprecia
una dependencia mucho menor con la temperatura de tratamiento, así como un
comportamiento mucho más homogéneo de los dos parámetros estudiados, la
concentración y la ∆cp (figuras 3.58 y 3.60) que el existente en la interfase asociada a la
primera población cristalina. En cuanto a las dos fronteras descritas, la primera de ellas no
ha sido considerada, al no existir valores experimentales a temperaturas inferiores a la
misma. La segunda frontera, correspondiente a la temperatura de transición vítrea del
PMMA, no parece afectar significativamente a la concentración de la interfase, que alcanza
en sus proximidades su valor máximo antes de volver a decrecer a temperaturas de
tratamiento superiores. Los valores que representan la ∆cp de la masa involucrada en la
interfase, por su parte, se muestran aproximadamente constanstes en todo el intervalo de
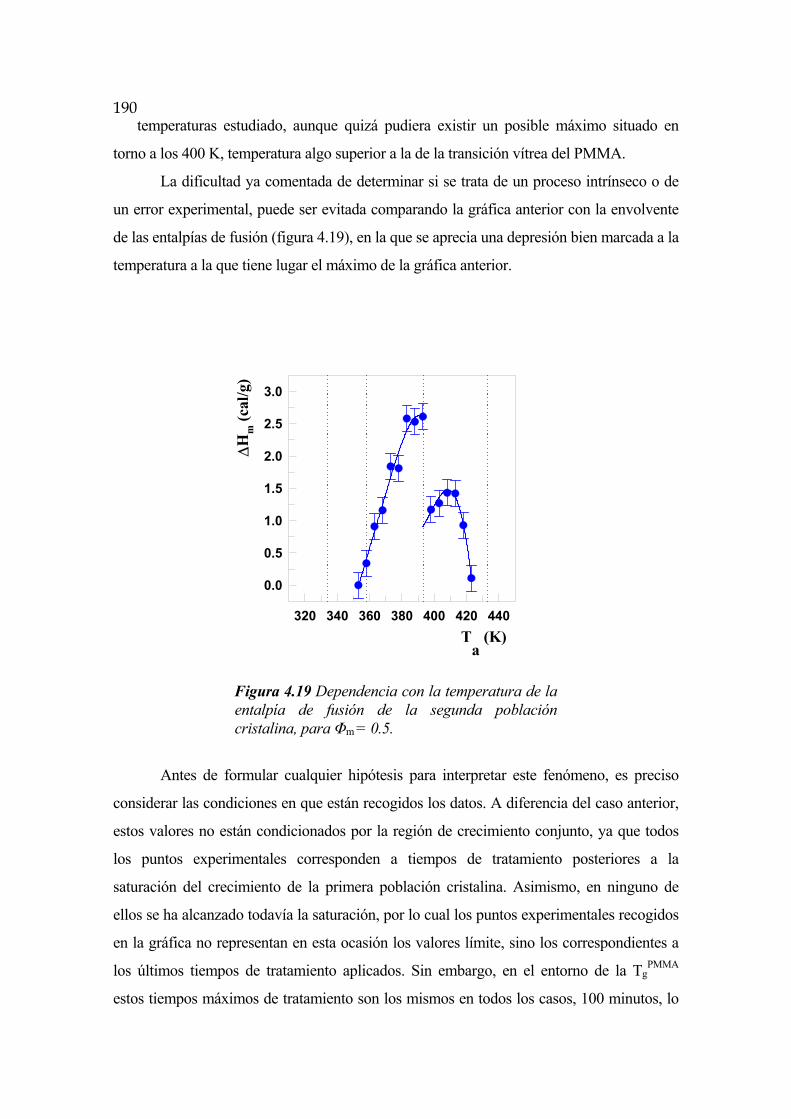
190temperaturas estudiado, aunque quizá pudiera existir un posible máximo situado en
torno a los 400 K, temperatura algo superior a la de la transición vítrea del PMMA.
La dificultad ya comentada de determinar si se trata de un proceso intrínseco o de
un error experimental, puede ser evitada comparando la gráfica anterior con la envolvente
de las entalpías de fusión (figura 4.19), en la que se aprecia una depresión bien marcada a la
temperatura a la que tiene lugar el máximo de la gráfica anterior.
320 340 360 380 400 420 440
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Ta (K)
∆H
m (c
al/g
)
Figura 4.19 Dependencia con la temperatura de la entalpía de fusión de la segunda población cristalina, para Φm = 0.5.
Antes de formular cualquier hipótesis para interpretar este fenómeno, es preciso
considerar las condiciones en que están recogidos los datos. A diferencia del caso anterior,
estos valores no están condicionados por la región de crecimiento conjunto, ya que todos
los puntos experimentales corresponden a tiempos de tratamiento posteriores a la
saturación del crecimiento de la primera población cristalina. Asimismo, en ninguno de
ellos se ha alcanzado todavía la saturación, por lo cual los puntos experimentales recogidos
en la gráfica no representan en esta ocasión los valores límite, sino los correspondientes a
los últimos tiempos de tratamiento aplicados. Sin embargo, en el entorno de la TgPMMA
estos tiempos máximos de tratamiento son los mismos en todos los casos, 100 minutos, lo
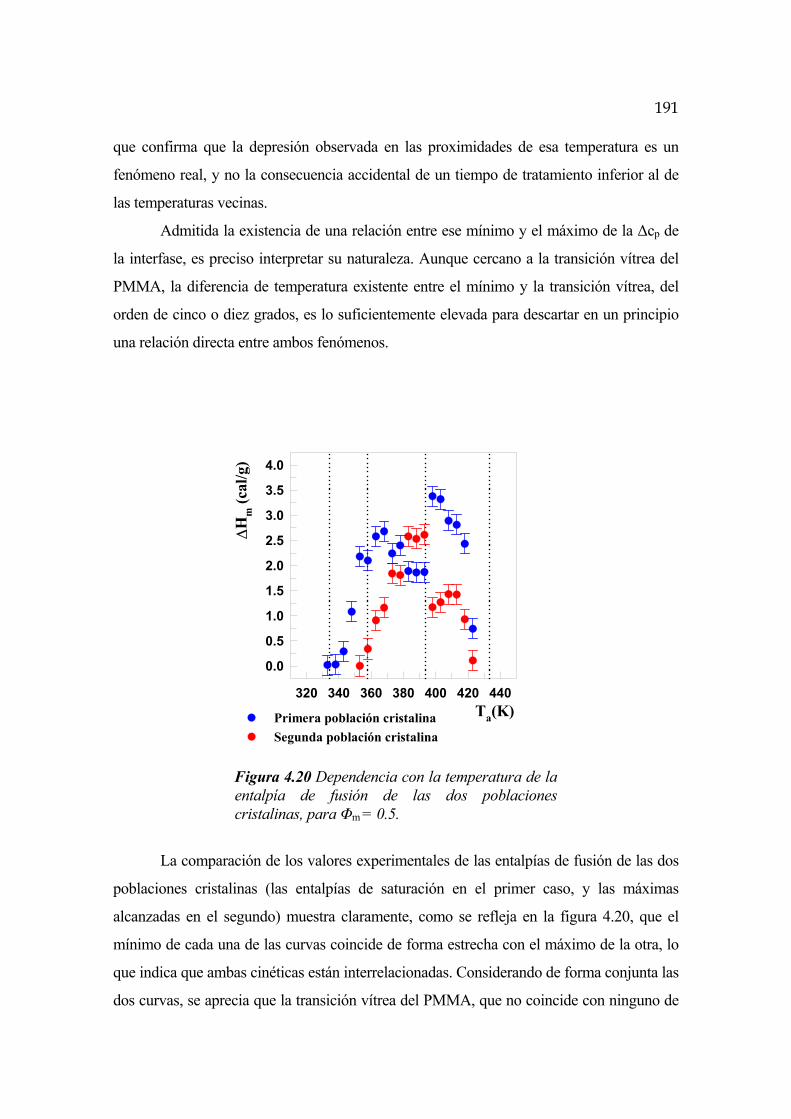
191
que confirma que la depresión observada en las proximidades de esa temperatura es un
fenómeno real, y no la consecuencia accidental de un tiempo de tratamiento inferior al de
las temperaturas vecinas.
Admitida la existencia de una relación entre ese mínimo y el máximo de la ∆cp de
la interfase, es preciso interpretar su naturaleza. Aunque cercano a la transición vítrea del
PMMA, la diferencia de temperatura existente entre el mínimo y la transición vítrea, del
orden de cinco o diez grados, es lo suficientemente elevada para descartar en un principio
una relación directa entre ambos fenómenos.
320 340 360 380 400 420 440
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
Ta(K)
∆H
m (c
al/g
)
Primera población cristalinaSegunda población cristalina
Figura 4.20 Dependencia con la temperatura de la entalpía de fusión de las dos poblaciones cristalinas, para Φm = 0.5.
La comparación de los valores experimentales de las entalpías de fusión de las dos
poblaciones cristalinas (las entalpías de saturación en el primer caso, y las máximas
alcanzadas en el segundo) muestra claramente, como se refleja en la figura 4.20, que el
mínimo de cada una de las curvas coincide de forma estrecha con el máximo de la otra, lo
que indica que ambas cinéticas están interrelacionadas. Considerando de forma conjunta las
dos curvas, se aprecia que la transición vítrea del PMMA, que no coincide con ninguno de

192los dos máximos ni con ninguno de los dos mínimos, es la que determina el cambio de
tendencia de las dos cinéticas.
La intersección entre el máximo de la ∆cp de la interfase y el mínimo de la curva de
entalpías de fusión, para la segunda población cristalina, puede ser explicada ahora de
forma sencilla, ya que la segregación de la matriz amorfa inducida por el tratamiento
térmico produce un incremento de la interfase que no es compensado por la subsiguiente
cristalización del PVDF. La falta de fluctuaciones importantes en la concentración de la
interfase indica, por otro lado, que el fenómeno afecta por igual a los dos polímeros (PVDF
y PMMA), como ocurriría si el aumento de masa se debiera mayoritariamente a uno de
ellos.
4.9 Cinética de fusión en monocristales de PVDF
El estudio calorimétrico del sistema físico formado por monocristales de PVDF
dispersos en una matriz de PMMA ha rendido resultados interesantes no sólo en lo
referente a la determinación del valor del parámetro de interacción, principal objetivo del
experimento, sino también en lo relativo a la cinética de fusión de los monocristales. La
principal ventaja de este estudio en relación con los realizados en muestras con
cristalinidad inducida mediante tratamiento térmico, consiste en que permite determinar la
dependencia de los parámetros investigados (Tm, ∆H) con la proporción existente entre los
monocristales de PVDF y la matriz de PMMA para todo el intervalo de proporciones, lo
cual no es posible en las muestras segregadas.
Resulta interesante recordar que, aunque los monocristales utilizados en el estudio
corresponden en todos los casos a una única fase cristalina perfectamente caracterizada
(fase α), tanto una velocidad de barrido suficientemente baja en el calorímetro como la
presencia de cantidades suficientemente elevadas de PMMA en el entorno de los cristales
de PVDF provocan la aparición de picos de fusión secundarios que muestran unas cinéticas
completamente distintas tanto a la correspondiente al pico principal asociado a la fase α,
como entre sí, como se aprecia en la figura 4.21.
Tal como se vio en el capítulo anterior, los dos picos secundarios muestran unos
comportamientos característicos. El primero de ellos, al que hemos denominado de baja
temperatura por aparecer sus máximos a temperaturas inferiores a las correspondientes al
pico de fusión principal, ha sido detectado únicamente en muestras con proporciones

193
medias de monocristales, independientemente de la velocidad de barrido utilizada en el
calorímetro. Es importante resaltar el hecho de que este intervalo de concentraciones
coincide con la zona en la que las mezclas homogéneas experimentan segregación
mediante tratamiento térmico (0.3 ≤Φm ≤0.7), así como la fuerte dependencia de los valores
de Tm con la proporción de monocristales de PVDF.
Pico principalPico secundario de baja temperatura
0.0 0.2 0.4 0.6 0.8 1.0
410
420
430
440
450
ΦmPVDF
Tm
(K)
Pico secundario de alta temperatura
Figura 4.21 Puntos de fusión principal y secundarios de los monocristales de PVDF en función de la cantidad de PMMA presente en la muestra.
El segundo pico secundario, denominado de alta temperatura por razones similares,
se diferencia del anterior por varias cuestiones:
a) En primer lugar, y al contrario de lo que ocurre con el primero, su presencia
depende en gran medida de la velocidad de barrido del calorímetro, estando favorecida su
aparición por velocidades de barrido bajas.

194b) En segundo lugar, su presencia no se reduce a la zona central de
proporciones ya que se extiende hasta los monocristales puros (Φm = 1) con una entalpía de
fusión siempre creciente, aunque no a la región pobre en PVDF.
c) Por último, los valores registrados para los puntos de fusión muestran ser no sólo
altos, sino también prácticamente independientes con la proporción de los monocristales
presentes en la mezcla.
Es interesante asimismo reseñar que el pico de fusión de baja temperatura
desaparece para una proporción de monocristales de PVDF (Φm ≈ 0.7) en la cual los puntos
de fusión de los dos picos secundarios coinciden entre sí.
En contraste con las dos poblaciones secundarias, la población principal crece con
la proporción de monocristales de forma moderada, con una pendiente que viene
determinada por el parámetro de interacción del sistema. Conviene recordar que, mientras
el comportamiento del pico de fusión principal está regido por factores termodinámicos
(parámetro de interacción), los dos picos secundarios deben su existencia exclusivamente a
orígenes cinéticos.
Comparando las gráficas de entalpías y puntos de fusión de los monocristales de
PVDF en función de la proporción de la mezcla con las equivalentes de las mezclas
homogéneas parcialmente segregadas en función de la temperatura y el tiempo de
tratamiento (figuras 4.22 a 4.24), se aprecian unas analogías que es preciso investigar
teniendo en cuenta la disparidad de ambos sistemas físicos y las diferentes magnitudes
físicas con las que se relacionan la variación de la entalpía y la de los puntos de fusión:
Proporción de monocristales de PVDF en un caso, y temperatura o tiempo de tratamiento
térmico en el otro.
Así, en la figura 4.22 se observa una correlación similar entre los puntos de fusión
del pico secundario de alta temperatura (en los monocristales) y el correspondiente a la
primera población cristalina (en las mezclas segregadas) por un lado, y entre el pico
secundario de baja temperatura y el de la segunda población cristalina por el otro, para
mezclas PVDF/PMMA con una concentración inicial de PVDF igual a 0.6 y un tiempo de
tratamiento de 10 minutos.
Pasando ahora a las entalpías de fusión es fácil descubrir que el comportamiento de
las correspondientes a los dos picos secundarios es muy diferente en ambos asemejándose
cada una de ellas, respectivamente, a las curvas de entalpía obtenidas en las mezclas

195
segregadas para tiempos y temperaturas constantes. Paralelamente aparece una
diferenciación en función de la velocidad de barrido en el calorímetro, inexistente en el
caso de las temperaturas de fusión.
Ta (K)ΦmPVDF
Primera población cristalina
320 360 400 440
380390
400410
420
430440
450460
Segunda población cristalina
0.0 0.2 0.4 0.6 0.8 1.0
380390
400410420
430440
450460
Pico de alta temperaturaPico de baja temperatura
Tm
(K)
(A) (B)
Figura 4.22 Puntos de fusión secundarios de los monocristales de PVDF (A) y de las dos poblaciones cristalinas principales para mezclas parcialmente segregadas (B).
En la figura 4.23 el porcentaje sobre el área total de la entalpía del pico secundario
de baja temperatura, en función de la proporción de monocristales, se compara con las
curvas de entalpía de fusión de las dos poblaciones cristalinas principales en función de la
temperatura de tratamiento, en mezclas segregadas con Φm = 0.6 y ta = 10 min.
Aunque en este caso no sólo es diferente el parámetro que se representa en el eje de
abcisas (proporción en peso y temperatura de tratamiento, respectivamente) sino también
en el del eje de ordenadas (entalpía de fusión en ambos, pero proporción sobre el total y
valor absoluto, respectivamente), es posible apreciar aquí una cierta similitud cualitativa
con curvas en forma de campana con el máximo situado en la zona central del intervalo
estudiado.
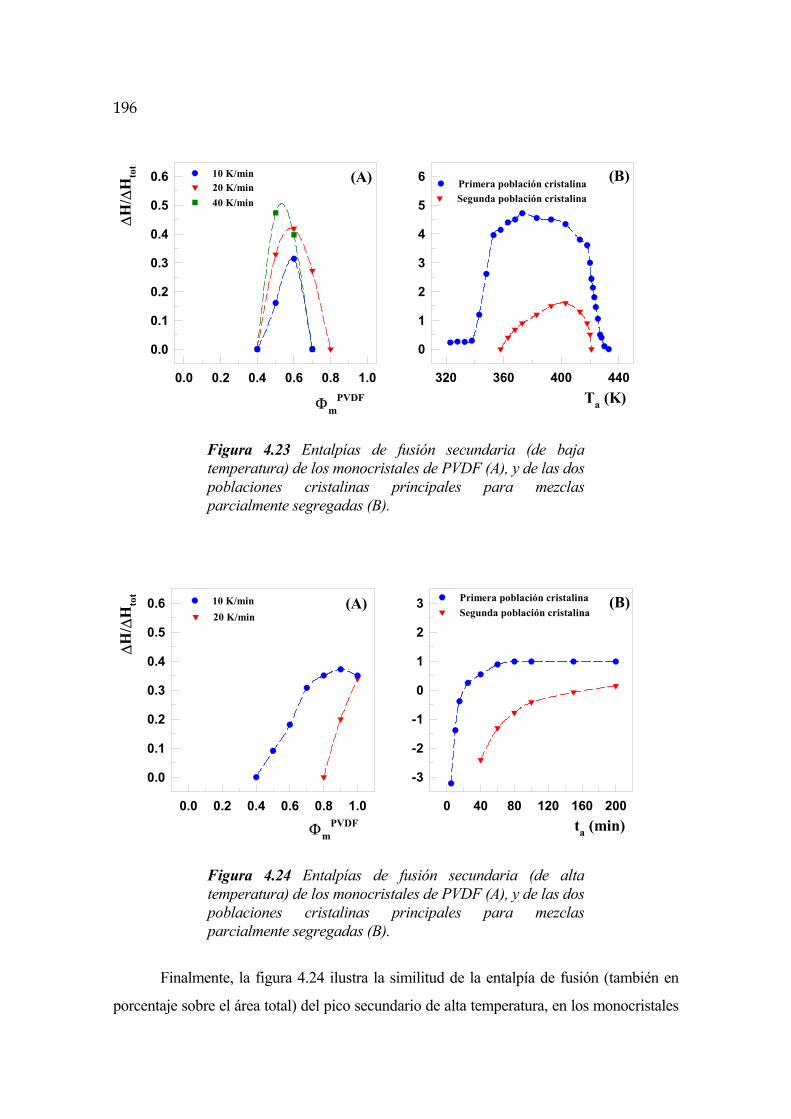
196
Primera población cristalina
Ta (K)ΦmPVDF
320 360 400 440
0
1
2
3
4
5
6Segunda población cristalina
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
∆H
/ ∆H
tot
40 K/min20 K/min10 K/min (A) (B)
Figura 4.23 Entalpías de fusión secundaria (de baja temperatura) de los monocristales de PVDF (A), y de las dos poblaciones cristalinas principales para mezclas parcialmente segregadas (B).
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
ta (min)ΦmPVDF
Primera población cristalina
0 40 80 120 160 200
-3
-2
-1
0
1
2
3Segunda población cristalina
10 K/min20 K/min
∆H
/ ∆H
tot
(A) (B)
Figura 4.24 Entalpías de fusión secundaria (de alta temperatura) de los monocristales de PVDF (A), y de las dos poblaciones cristalinas principales para mezclas parcialmente segregadas (B).
Finalmente, la figura 4.24 ilustra la similitud de la entalpía de fusión (también en
porcentaje sobre el área total) del pico secundario de alta temperatura, en los monocristales

197
de PVDF, con las curvas isotermas de la entalpía de fusión en mezclas segregadas, en este
caso con Φm = 0.5 y Ta = 368 K.
A la hora de establecer conclusiones es preciso obrar, por las razones expuestas,
con cautela. En el sistema formado por monocristales de PVDF dispersos en una matriz de
PMMA existe la evidencia de que las dos endotermas de fusión secundarias se deben
exclusivamente a factores cinéticos y no termodinámicos, pero no resulta inmediata su
equivalencia con el sistema formado por mezclas amorfas parcialmente cristalizadas tras un
tratamiento térmico en unas condiciones de temperatura y tiempo determinadas. En
principio siempre se debería poder llegar a una situación final equivalente actuando sobre
distintos parámetros (temperatura, tiempo, concentración) ya que los procesos físicos que
regulan la segregación molecular y la cristalización del PVDF dependen conjuntamente de
todos ellos. Sin embargo, en la práctica no resulta fácil establecer la concordancia entre
estos parámetros y principalmente entre los dos sistemas físicos diferentes que hemos
comparado.
Obviamente en el sistema formado por monocristales de PVDF y la matriz de
PMMA no existe ninguna dependencia ni con la temperatura ni con el tiempo, al no haber
segregación previa producida por un tratamiento térmico. Por otro lado, en el caso de las
mezclas homogéneas tratadas térmicamente lo que no resulta posible es extender el estudio
fuera de un estrecho intervalo de concentraciones.
No obstante, la conclusión que se puede alcanzar es que la cinética de cristalización
del sistema es sumamente sensible a los distintos parámetros que influyen sobre ella, y que
incluso en unas condiciones similares se pueden formar dos o más poblaciones cristalinas
con comportamientos muy diferentes entre sí.
4.10 Morfología y fases cristalinas
El estudio de la morfología de las muestras parcialmente cristalizadas y la
identificación de las fases cristalinas presentes en las mismas constituye el siguiente
objetivo de este trabajo de investigación. En las muestras inicialmente amorfas puede
asumirse que la única fase existente, la matriz amorfa, presenta una composición
homogénea en todo su volumen. Al iniciarse el tratamiento térmico comienzan unos
procesos de difusión molecular (PVDF en el caso de temperaturas de tratamiento inferiores
a la transición vítrea del PMMA, y ambos polímeros para temperaturas superiores a la

198misma) que provocan la aparición de un gradiente de concentraciones detectado tanto
por las fluctuaciones de la densidad electrónica como por la aparición de colas en las
curvas de las transiciones vítreas, las cuales se extienden a temperaturas inferiores a la
correspondiente al punto de inflexión de la curva. El hecho de que el máximo de estos
gradientes de concentración (Φm = 0.7) coincida con el valor a partir del cual no es posible
inhibir la cristalización del PVDF, sugiere que el mecanismo de segregación molecular
actúa iniciándose en forma de un gradiente de concentraciones que al alcanzar el valor
límite acaba provocando la migración de las moléculas de PVDF (o la expulsión de las de
PMMA) y la cristalización de estas primeras. Este fenómeno ha sido estudiado
recientemente por Wing y Mijovic [Wing, 2000], que describen la existencia de
heterogeneidades microscópicas del orden de magnitud de los nanometros, razón por la
cual las denominan nanoheterogeneidades.
No obstante es preciso considerar que, dada la aparición de dos poblaciones
cristalinas muy diferentes, ha de asumirse la existencia de, al menos, dos mecanismo
distintos de segregación molecular que actúan de forma paralela y cuyas cinéticas
respectivas son extremadamente sensibles a las condiciones de trabajo.
4.10.1 Fases cristalinas presentes en las mezclas segregadas de PVDF y PMMA.
En el presente trabajo de investigación no ha sido posible asignar sin ambigüedad
en todos los casos los picos de fusión detectados en los termogramas a fases cristalinas
concretas, a excepción de la fase γ registrada para temperaturas de tratamiento térmico
cercanas al punto de fusión del PVDF y de la fase ß, identificada en muestras con
concentraciones iniciales de PVDF situadas en torno al 75% en peso. No obstante, existe
cierta relación entre la que hemos denominado primera población cristalina (de punto de
fusión constante) y la fase ß, así como entre la segunda población cristalina (de punto de
fusión dependiente de la temperatura de tratamiento) y la fase α.
Esta relación se apoya fundamentalmente en la comparación cualitativa de los
diagramas de difracción de rayos X con los termogramas obtenidos por calorimetría
diferencial. En los primeros se aprecia la aparición inicial de fase ß seguida por una
aparición más tardía de la fase α, la cual a tiempos de tratamiento suficientemente largos
puede llegar, si la temperatura de tratamiento es suficientemente elevada, a superar en
proporción a la fase ß.

199
Varias comprobaciones indirectas ayudan a confirmar esta relación. En un estudio
simultáneo de difracción de rayos X y medidas de densidad para una muestra con Φm = 0.5
tratada a 100 °C durante distintos tiempos, se aprecia que el crecimiento de la fase α
provoca un aumento en la densidad macroscópica mayor que el correspondiente a la fase ß,
a pesar de que la densidad de la fase α es menor que la de la fase ß. Esta aparente
contradicción puede ser interpretada asumiendo que la fase ß se produce por cristalización
del PVDF segregado de la matriz amorfa; en estas condiciones la densidad de la matriz
amorfa restante disminuye al quedar empobrecida en PVDF, mientras parte del PVDF
segregado no cristaliza pasando a formar parte de la interfase amorfa con una densidad
inferior a la de cualquiera de las fases cristalinas. Por este motivo el aumento de densidad
correspondiente a la cristalización del PVDF en la fase ß estaría compensado en parte tanto
por la menor densidad del PVDF amorfo presente en la interfase, como por la disminución
de la densidad de la matriz amorfa, siendo el incremento de la densidad macroscópica el
resultado conjunto de todos estos fenómenos.
La fase α, por el contrario, se produciría en un medio distinto de la matriz amorfa
principal, enriquecido en PVDF y de mayor densidad que ésta, lo que conduce a un
incremento de la densidad macroscópica. En consecuencia, a pesar de que la densidad del
PVDF α es inferior a la correspondiente al PVDF ß, los distintos procesos de formación de
los mismos provocarían incrementos netos de las densidades macroscópicas mayores en el
caso de la fase α que en el de la fase ß.
Esta identificación de la fase cristalina ß del PVDF con la segregación de la matriz
amorfa, y de la fase α con la reordenación molecular del PVDF amorfo, lleva a asociar a la
fase ß con la primera población cristalina detectada por calorimetría y a la fase α con la
segunda población cristalina, dado que la cinética de cristalización de ambas poblaciones
conduce a conclusiones similares con respecto a su formación tal como ha sido comentado
en los capítulos anteriores.
La identificación de las dos poblaciones cristalinas de alta temperatura tampoco se
ha podido realizar de forma directa debido a las razones expuestas en la introducción.
Aunque son varios los autores [Osaki, 1975; Prest, 1978; Gal'Perin, 1970; Lovinger, 1980;
Tashiro, 1983a; Gregorio, 2000] que informan de la aparición de la fase γ a temperaturas
próximas a la de fusión del PVDF, sus estudios han sido realizados con PVDF puro y no
con mezclas cristalizadas de PVDF y PMMA, por lo que la extrapolación de sus

200conclusiones a nuestro sistema debe ser tomada con precaución. Por otro lado, los
difractogramas correspondientes a la fase de alta temperatura, registrados en muestras con
Φm = 0.75 tratadas a 160 °C, no coinciden tampoco con los asignados por varios autores a
la fase γ [Prest, 1978; Gal'Perin, 1970; Tashiro, 1983a], hecho que puede ser atribuido a la
presencia del PMMA dado que las reflexiones producidas por los cristales de PVDF son
muy dependientes del grado de cristalinidad de las muestras tal como demuestran Gal'Perin
y col. [Gal'Perin, 1970]. Para muestras poco cristalinas, como ocurre con las estudiadas en
el presente trabajo, no resulta fácil distinguir entre las reflexiones de las fases cristalinas α y
γ, ni comparar cada una de ellas con las correspondientes al PVDF puro.
Descartada por ello la identificación por difracción de rayos X de las fases
obtenidas a altas temperaturas de tratamiento, hemos procedido a estudiar más
detenidamente esta transición por calorimetría diferencial, aunque las características de esta
técnica experimental no permiten por sí solas discriminar entre fases cristalinas
determinadas tal como apuntan, entre otros, Prest y Luca [Prest, 1978]. Por otro lado, la ya
comentada diferencia existente entre los sistemas estudiados en la bibliografía (PVDF
puro) y las mezclas de PVDF y PMMA investigadas en esta tesis dificulta en los estudios
calorimétricos, al igual que lo hace en los de difracción de rayos X la interpretación de los
resultados de estos investigadores. Sin embargo, algunos conceptos importantes aportados
por estos trabajos se pueden tener en cuenta:
-Las muestras de PVDF cristalino tratadas térmicamente a temperaturas cercanas al
punto de fusión se transforman siempre en fase γ con independencia de que inicialmente
hubieran estado cristalizadas en fase α [Osaki, 1975; Prest, 1978; Lovinger, 1980;
Gregorio, 2000] o en fase ß [Osaki, 1975; Prest, 1978; Tashiro, 1983a; Okabe, 1998].
-Una misma fase cristalina puede presentar más de un único punto de fusión
dependiendo de las condiciones experimentales [Osaki, 1975; Prest, 1978; Marand, 1988].
La fase γ, en particular, presenta cristales de bajo y alto punto de fusión en diversos
estudios [Osaki, 1975; Prest, 1978; Marand, 1988; Iwamoto, 1988; Gregorio, 2000]. Este
último autor encuentra que el punto de fusión de la fase γ obtenida por una transición de
fase α → γ es superior, en unos 8 °C, al punto de fusión correspondiente a la fase γ
cristalizada directamente desde el fundido.
-Las transformaciones α → γ y ß → γ parecen seguir procesos completamente
diferentes. Según Prest y Luca [Prest, 1978] la primera de estas dos transformaciones es

201
una transición de fase sólido→sólido provocada por cambios conformacionales en las
cadenas de PVDF sin que los cristales α lleguen a fundir. La segunda transformación, por
el contrario, tiene lugar por una fusión lenta (0.31 K/min.) de la fase ß con una posterior
recristalización del PVDF en fase γ. Aunque Prest y Luca no trabajan con mezclas
compatibles de PVDF y PMMA sino con PVDF puro, la utilización de un agente
tensoactivo facilita la formación de la fase γ independientemente de la fase en la que
estuviera cristalizado inicialmente el material de partida (α o ß). Esta transformación se es
justifica por estos autores asumiendo que el agente tensoactivo dificulta la difusión
molecular del PVDF fundido permitiendo que las cadenas adopten una conformación γ que
resulta ser más estable en este caso que en el PVDF puro sin agente tensioactivo. Con todas
las reservas posibles, puesto que el sistema PVDF más agente tensioactivo no es similar al
formado por una mezcla parcialmente segregada de PVDF y PMMA, se puede asumir una
cierta similitud en la cinética de cristalización existente en ambos casos.
La comparación de nuestros resultados experimentales con los datos recogidos en la
bibliografía demuestra que en el caso de mezclas con Φm = 0.75 la transición de alta
temperatura registrada por calorimetría puede ser identificada de forma bastante explícita
con la transición ß → γ descrita por Prest y Luca [Prest, 1978], coincidiendo además los
difractogramas de difracción de rayos X en lo referente a la fase ß existente con
anterioridad a la transición. En lo que respecta a las poblaciones de alta temperatura, éstas
han sido asignadas a la fase γ.
En muestras con menor cantidad de PVDF (Φm = 0.6) la identificación es más
problemática debido tanto a la menor resolución de los picos de fusión, como a la
existencia de una mezcla de fases α y ß con anterioridad a la transformación de alta
temperatura. Por analogía, hemos asumido que estas poblaciones cristalinas pertenecen
también a la fase γ.
La comparación de nuestros experimentos con los publicados por Léonard y col.
[Léonard, 1983] para mezclas de PVDF y PMMA cuenta con el inconveniente de que ni
las condiciones de trabajo ni las técnicas experimentales empleadas son las mismas en
ambos casos, lo que impide una comparación cuantitativa de los resultados. Sin embargo, sí
se aprecian unas discrepancias significativas que no pueden ser atribuidas a las diferencias
instrumentales. La razón de estas discrepancias se explica al comparar las condiciones de
tratamiento térmico (tiempo y temperatura) aplicadas en uno y otro trabajo: Mientras

202Léonard y col. investigan la dependencia de la cristalinidad con la concentración para
un único tiempo y una única temperatura de tratamiento, así como la dependencia de la
cristalinidad con la temperatura también para un único tiempo, nuestros estudios han sido
realizados en un amplio rango de temperaturas y tiempos de tratamiento. Por ello, mientras
estos autores encuentran una única población cristalina (la α) para concentraciones situadas
en torno a Φm = 0.5, nosotros detectamos para estas mismas concentraciones la existencia
de dos poblaciones cristalinas en los termogramas y una mezcla de las fases α y ß en los
difractogramas de rayos X, con una proporción entre ambas en los dos casos que depende
fuertemente de los dos parámetros (tiempo y temperatura) considerados.
Por otro lado, estos autores tampoco tienen en cuenta la existencia de un cambio de
régimen tanto en la cinética de segregación de la matriz amorfa como en la cristalización
del PVDF segregado (punto de cambio de régimen). Para el tiempo de tratamiento
empleado por estos autores (ta = 60 min.) al menos una de las concentraciones (Φm = 0.5)
debería agrupar valores experimentales correspondientes a los regímenes cinéticos anterior
y posterior al punto de cambio de régimen, los cuales experimentan discontinuidades en las
curvas de entalpías de cristalización registradas por nosotros.
Por último los citados autores recogen, para concentraciones elevadas de PVDF
(Φm ≥ 0.6), la existencia de dos únicas fases cristalinas, la α y la ß, lo que contrasta con
nuestros resultados experimentales en los cuales, para estas mismas concentraciones y
temperaturas de tratamiento próximas al punto de fusión de los cristales de PVDF, han sido
detectadas también dos fases de alta temperatura así como una transformación de fase
ß → γ.
Salvando estas diferencias y evitando las comparaciones cuantitativas, no obstante
se aprecia una notable coincidencia entre nuestros resultados y los de estos autores. Así,
para concentraciones elevadas de PVDF (Φm ≥ 0.6) se encuentran similitudes entre la forma
de la curva de la primera población cristalina (Tm = cte.) y la atribuida por estos autores a la
fase ß, coincidiendo también esta apreciación, en el caso de Φm = 0.75, con los resultados
obtenidos por difracción de rayos X. La segunda población cristalina (Tm = f (Ta)) presenta
también un comportamiento similar al correspondiente a la fase α de Léonard, a excepción
del tramo final de temperaturas donde el pico registrado por estos autores como
perteneciente también a esta misma fase α ha sido asignado por nosotros a la fase γ.

203
Más compleja resulta la comparación de las curvas correspondientes a una
concentración de Φm = 0.5, debido a los ya comentados cambios existentes tanto en los
puntos de cambio de régimen como en la transición vítrea del PMMA. Dado que Léonard
no contempla estas transiciones, hamos elegido una curva de entalpías correspondiente a un
tiempo de tratamiento inferior (ta = 10 min.) en el cual todos los puntos experimentales
corresponden a tiempos inferiores a los correspondientes al punto de cambio de régimen,
pudiéndose despreciar la contribución de la segunda población cristalina al aparecer ésta
únicamente, y sólo de forma incipiente, a temperaturas próximas al máximo de la cinética
de cristalización (Ta ~ 120 °C). Considerando, pues, que la totalidad de la entalpía de fusión
corresponde a la primera población cristalina, se encuentra una notable similitud entre
nuestra curva y la de Léonard, aunque en este caso la correspondencia es entre la primera
población cristalina y la fase α de Léonard, es decir, justo lo opuesto a lo encontrado para
concentraciones superiores de PVDF.
Estos resultados confirman la dificultad existente para establecer una correlación
entre las entalpías de fusión registradas por calorimetría y las fase cristalinas investigadas
por difracción de rayos X (nuestro caso) o por espectroscopía infrarroja (Léonard), a la vez
que pone de relieve la gran dependencia de la cinética de cristalización con parámetros
tales como la concentración, la temperatura y el tiempo de tratamiento térmico de las
muestras.
4.11 Teorías cinéticas de cristalización.
Las teorías cinéticas de cristalización consideran que el proceso de cristalización de
un polímero puro desde el estado fundido es el resultado de la suma de dos procesos
elementales, la nucleación y el crecimiento posterior en forma de plegado molecular. De
acuerdo con la teoría de Lauritzen-Hoffman [Lauritzen, 1973], en el crecimiento de un
cristal polimérico pueden definirse tres regímenes distintos en función de que predomine
uno u otro de los dos procesos. Llamando G al crecimiento radial de las esferulitas
cristalinas, i a la velocidad de nucleación y g a la velocidad de plegado de las moléculas
que ya han nucleado, se obtiene:

204Régimen I: G ∝ i para g >> i (nucleación simple).
Régimen II: G ∝ (ig)1/2 para g ≈ i (nucleación múltiple).
Régimen III: G ∝ i para g < i (nucleación múltiple).
Los regímenes I y III están gobernados por la nucleación mientras que el régimen II
depende conjuntamente de la nucleación y el plegado molecular. A su vez, según esta
teoría la expresión general de G será:
) f T T
K - ( G c
gg ∆
∝ expβ [IV,35]
Donde ßg es el término de movilidad que describe la velocidad de transporte de las
moléculas cristalizables al frente de crecimiento y el exponencial recoge el término
responsable de la nucleación, donde Kg es una constante de nucleación cuyo valor depende
del régimen de crecimiento (doble para los regímenes I y III que para el II), Tc la
temperatura de cristalización, ∆T el incremento de temperatura existente entre la
temperatura de fusión en el equilibrio (Tmo) y la temperatura de cristalización y f un factor
corrector dado por la expresión:
T + TT 2 = f
com
c [IV,36]
Partiendo de esta base Saito y col. [Saito, 1991] proponen una modificación de la
teoría de Lauritzen-Hoffman definiendo un modelo de difusión en dos fases cuyo
desarrollo matemático se inicia considerando por separado los términos i y g
relacionándolos respectivamente con los coeficientes de difusión DM y DS según las
expresiones:

205
) f T T
K - ( D ic
gM ∆
∝ exp [IV,37]
Dg S∝ [IV,38]
Con lo que la expresión de G quedará como:
) f T T
K - ( D G c
gM ∆
∝ exp [IV,39]
Para los regímenes de cristalización I y III y:
) f T T
K - ( ) D D ( G c
g1/2SM ∆
∝ exp [IV,40]
Para el régimen II. Si en lugar de un polímero puro se tiene una mezcla compatible
de dos polímeros, será necesario introducir algunas modificaciones en estas ecuaciones. En
primer lugar DM y DS, que son indistinguibles en un polímero puro, aquí ya tienen
significados físicos distintos; se define DM como el coeficiente de difusión mutua y DS
como el coeficiente de autodifusión en la mezcla, correspondiéndoles respectivamente las
expresiones propuestas por Brochard [Brochard, 1988]:
) n / D
+ n / D
( D 1-
2o2
2
1o1
1M
φφ∝ [IV,41]
Siendo Φi la fracción en volumen, Dio el coeficiente de difusión del monómero y ni
el grado de polimerización del polímero i. Y Skolnick [Skolnick, 1988]:

206
] n / n + n ) - (1 +
n / n ) - (1 + [ nD D
e11
e1
1
o1
s γγγγ
∝ [IV,42]
Donde γ es una constante y ne el grado de polimerización entre los puntos de
entrecruzamiento. Como se puede apreciar en estas dos expresiones, mientras DM depende
de los coeficientes de difusión de los dos polímeros y de la proporción existente entre ellos,
DS es función únicamente del polímero cristalizable y, por lo tanto, independiente de la
concentración.
Relacionando esto último con las expresiones [IV,37] y [IV,38], puede concluirse
que en la nucleación intervienen los coeficientes de difusión de los dos polímeros ya que
ambos tienen que separarse previamente para crear un núcleo de cristalización, aunque si
uno de los dos polímeros se difunde mucho más rápidamente que el otro, como ocurre con
el PVDF con respecto al PMMA en determinadas condiciones de tratamiento, podrá
considerarse que la difusión mutua depende únicamente del polímero con mayor movilidad
molecular, asumiéndose que las moléculas del más lento permanecen inmóviles. Sin
embargo, la velocidad de nucleación sí dependerá de la concentración de PVDF existente
en la matriz amorfa.
En el plegado, por el contrario, intervendrá tan sólo el coeficiente de autodifusión
del polímero cristalizable, dado que al estar ya segregados los dos polímeros las moléculas
amorfas del polímero cristalizable tan sólo tendrán que reordenarse entre ellas
(autodifundirse) para poderse plegar formando el cristal.
En el caso de mezclas de polímeros la concentración del polímero cristalizable ha
de ser incluida no sólo en la expresión del coeficiente de difusión DM, sino también en la
propia expresión del término de nucleación i, con lo que los términos i, g y G quedan
finalmente como:
) f T T
K - ( D ic
gM1 ∆
∝ expφ [IV,43]
Dg S∝ [IV,44]
) f T T
K - ( D G c
gM1 ∆
∝ expφ [IV,45]

207
Para los regímenes de cristalización I y III y:
) f T T
K - ( ) D D ( G c
g1/2SM
1/21 ∆
∝ expφ [IV,46]
Para el régimen II.
Dependiendo a su vez DM de Φ1 tal como viene reflejado en la ecuación [IV,41].
Dividiendo entre sí las expresiones [IV,43] y [IV,44], se obtiene:
) f T T
K - ( ) DD ( i/g
c
g
S
M1 ∆
∝ expφ [IV,47]
Lo que significa que el cociente i/g decrece cuando Φ1 decrece, hecho que se
traduce en que las transiciones III → II → I tienen lugar cuando Φ2 (la concentración del
polímero no cristalizable) aumenta.
Saito y col. [Saito, 1991] indican en su trabajo sobre las mezclas de PVDF y
PMMA, que el régimen III únicamente aparece en el caso del PVDF puro, mientras en las
mezclas sólo se registran los regímenes I y II. Sin embargo, el trabajo de estos autores
cuenta con la limitación de estar realizado en muestras cristalizadas desde el fundido con
subenfriamientos muy bajos, por lo que el intervalo de temperaturas estudiado por ellos es
muy inferior al utilizado en el presente trabajo de investigación correspondiendo
únicamente a la región más cercana a la temperatura de fusión. Por otro lado, el desarrollo
matemático realizado por Saito no considera la aparición de ningún punto singular en las
curvas isotermas, mientras la expresión propuesta para la dependencia exponencial de G
con la temperatura no explica ni el máximo alcanzado en el valor correspondiente a la
transición vítrea del PMMA, ni los saltos en las curvas experimentales asociados a ese
valor. Sí considera el cambio de régimen II a régimen I, que sitúa en torno a los 155 °C

208para mezclas con una concentración de PVDF del 55% y un peso molecular del
PMMA similar al del utilizado por nosotros, y considera también la dependencia de los
cambios de régimen con la concentración de la mezcla, que para una temperatura de
cristalización de Tc = 150 °C sitúa en Φ2 = 0.3 (78% en peso de PVDF) para la transición
III → II y en Φ2 = 0.42 (68% en peso de PVDF) para la transición II → I.
Sin embargo, al estudiar la dependencia de G con la temperatura de cristalización
Saito no considera el efecto de la cristalización al ajustar linealmente la expresión
logarítmica de las ecuaciones [IV-45] y [IV-46], lo cual es cierto únicamente para las
etapas iniciales del proceso en las cuales la segregación molecular es todavía incipiente y la
concentración de la matriz amorfa puede ser considerada similar a la inicial. Por
consiguiente, esta aproximación no es válida para la totalidad del proceso de cristalización
ya que la segregación molecular que tiene lugar en la matriz amorfa produce un
empobrecimiento en la concentración de polímero cristalizable en la matriz amorfa que no
ha sido considerada por Saito.
Por estas razones el estudio de Saito es válido únicamente para unas condiciones de
trabajo mucho más limitadas que las utilizadas en el presente estudio, dándose además la
circunstancia de que los límites de concentraciones dados por este autor para las
transiciones de fase III → II (Φm1 = 0.78) y II → I (Φm1 = 0.68) corresponden al rango en el
cual no es posible obtener mezclas homogéneas en estado vítreo, por lo cual este estudio
sólo puede ser realizado en muestras cristalizadas desde el fundido y no en mezclas
homogéneas en estado vítreo sometidas a tratamiento térmico. A consecuencia de ello no es
posible establecer una correlación directa entre los resultados obtenidos por Saito y los de
este trabajo, salvo en lo referente a la aplicación de las teorías cinéticas de cristalización
empleadas por estos autores; a la primera población cristalina le correspondería un
predominio de la nucleación sobre el crecimiento de los cristales (régimen I), dependiendo
exclusivamente del coeficiente de difusión del PVDF para temperaturas de tratamiento
inferiores a la de la transición vítrea del PMMA y de los de ambos polímeros para
temperaturas superiores a ésta. Para la segunda población cristalina, por el contrario, el
control cinético sería conjunto de la nucleación y el plegado molecular (régimen II) o
incluso únicamente de este último, siendo independiente de i.
Otro desarrollo teórico importante es el publicado por Alfonso y Russell [Alfonso,
1986], aunque en este estudio no se investiga el sistema PVDF/PMMA sino el formado por

209
el polióxido de etileno (PEO) y el PMMA. A diferencia de Saito estos autores consideran
una dependencia de la velocidad de crecimiento cristalino G frente a la temperatura,
estando controlado el crecimiento cristalino por la nucleación para subenfriamientos bajos
y por la difusión molecular para subenfriamientos altos.
En el caso de mezclas compatibles de un polímero semicristalino con un segundo
amorfo la modificación del potencial químico en la mezcla hace que la energía de
nucleación y la movilidad molecular se vean alteradas, mientras que para que ocurra la
cristalización es preciso que exista previamente una segregación de los dos polímeros. En
consecuencia, surge una competencia entre la cristalización y la segregación molecular.
El desarrollo matemático realizado por Alfonso y Russell presenta la ventaja, con
respecto al equivalente de Saito, de ser aplicable en un amplio rango de pesos moleculares,
composición y temperaturas de transición, al tiempo que contempla la existencia de un
máximo en la velocidad de cristalización en función de la temperatura. Sin embargo, este
desarrollo no permite explicar ni la existencia de dos poblaciones cristalinas diferenciadas,
cada una de ellas con su propia cinética, ni las discontinuidades observadas
experimentalmente tanto a la temperatura de transición vítrea del PMMA como a los
tiempos críticos correspondientes al cambio en la cristalización de la primera a la segunda
poblaciones cristalinas. Por esta razón, estos resultados tampoco son equiparables a los
nuestros.
4.12 Conclusión.
La aplicación de estas teorías cinéticas al estudio del sistema PVDF/PMMA en las
condiciones aplicadas en el presente trabajo no resulta inmediata, y por ello no se pueden
determinar con precisión los regímenes de crecimiento cristalino presentes en cada una de
las regiones en las que hemos dividido las curvas cinéticas. No obstante, y en base a todo lo
anteriormente expuesto, puede concluirse lo siguiente:
-La primera población cristalina parece estar relacionada directamente con los
procesos de segregación molecular ocurridos cuando la difusión molecular produce en la
matriz amorfa máximos locales de concentración con Φm ≈ 0.7. Esta población, asimilable a
la fase cristalina ß, al menos en alguno de los casos estudiados, tiene su cinética controlada
básicamente por la difusión molecular. Para temperaturas de tratamiento inferiores a la de
la transición vítrea del PMMA este control se debe a la difusión exclusiva del PDVF,

210mientras para temperaturas superiores a la citada el control cinético es ejercido
conjuntamente por la difusión del PVDF y el PMMA. En la cristalización de esta población
cristalina existe un predominio de la nucleación sobre el crecimiento de los cristales
asociada a las primeras etapas del proceso, alcanzando el nivel de saturación en un punto
de cambio de régimen pasado el cual su crecimiento está detenido, probablemente a causa
de factores estéricos.
-La segunda población cristalina tiene su origen en un sustrato diferente del
conjunto de la matriz amorfa, surgido probablemente a raíz de las heterogeneidades
producidas en la misma al hacer su aparición el gradiente de concentraciones provocado
por el tratamiento térmico. Por analogía con el estudio del sistema formado por
monocristales de PVDF dispersos en una matriz de PMMA, se deduce que son factores
exclusivamente cinéticos, y no termodinámicos, los que producen la diferenciación entre
las dos poblaciones cristalinas estudiadas. La cinética de cristalización de esta segunda
población cristalina está controlada principalmente por la cristalización o conjuntamente
por la cristalización y la difusión molecular, y en ella predomina el crecimiento cristalino
sobre la nucleación.
-Las poblaciones de alta temperatura, identificadas como pertenecientes a la fase γ,
se producen únicamente a temperaturas próximas a la de fusión del PVDF en muestras con
concentraciones iniciales de PVDF relativamente elevadas (Φm > 0.5), y son producto,
respectivamente, de la recristalización del PVDF durante el barrido en el calorímetro y de
una transición de fase entre la primera población cristalina y la fase γ.
-Las muestras con exceso de PVDF, y por lo tanto parcialmente segregadas con
anterioridad al tratamiento térmico, poseen una cristalinidad inicial asociada en su mayor
parte a la fase ß para concentraciones de PVDF del orden de Φm ≈ 0.75, comportándose de
forma similar a la del PVDF puro para concentraciones más elevadas (Φm ≥ 0.8). El estudio
de la cinética de segregación molecular de las muestras con Φm = 0.75 sugiere que su
comportamiento puede ser similar, a nivel macroscópico, al que tiene lugar para muestras
de concentraciones inferiores en aquellas regiones de la matriz amorfa en la que los
máximos locales de concentración alcanzan valores del orden de Φm ≈ 0.7.
En resumen, puede afirmarse que las moléculas de PMMA mezcladas con PVDF
inhiben la cristalización de este último induciendo la aparición de la primera población
cristalina (presumiblemente ß) cuando la concentración de este polímero alcanza un valor

211
suficientemente elevado (Φm ≈ 0.7). Cuando las moléculas de PMMA no están presentes, la
reordenación molecular de la interfase producirá la segunda población cristalina
(presumiblemente α).

212

213
CAPÍTULO 5
CONCLUSIONES GENERALES
1.- La compatibilidad a nivel molecular, en el estado fundido, del polifluoruro de
vinilideno y el polimetacrilato de metilo atáctico ha sido estudiada por aplicación
de la ecuación de Flory a las medidas de depresión del punto de fusión del PVDF.
La utilización por primera vez de un sistema formado por monocristales de PVDF
dispersos en una matriz de PMMA permite minimizar los efectos morfológicos que
han dificultado las medidas del parámetro de interacción realizadas anteriormente
por diversos autores. Simultáneamente se ha conseguido una mayor aproximación
al estado ideal de equilibrio termodinámico. El valor obtenido para el parámetro de
interacción de este sistema demuestra ser prácticamente independiente de la
concentración.
2.- La compatibilidad del sistema en estado vítreo ha resultado ser posible, a
temperaturas inferiores a la de la correspondiente transición vítrea, para
concentraciones de PVDF inferiores al 65-70% en peso. En el rango de
concentraciones inferiores a este límite el sistema se comporta como una única
especie física totalmente homogénea y amorfa, tal como ha sido comprobado por
medidas de cristalinidad, temperatura y variación del calor específico de la
transición vítrea, densidad macroscópica y microdureza. Para concentraciones de
PVDF más elevadas la segregación molecular es más rápida que la mayor
velocidad posible de enfriamiento del fundido, por lo que las muestras presentan
cristalización parcial y se comportan en todos los casos como especies físicas
heterogéneas.

214 3.- El tratamiento térmico de las muestras en estado vítreo induce una segregación
molecular que viene acompañada de una cristalización parcial del PVDF segregado.
Esta segregación molecular tiene lugar para concentraciones de PVDF iguales o
superiores aproximadamente a Φm = 0.3, límite por debajo del cual la cristalización
no ocurre al ser la temperatura mínima necesaria para que se inicie la cristalización
superior a la temperatura de fusión del PVDF en esas mismas condiciones. En las
muestras tratadas térmicamente se aprecia, utilizando técnicas de dispersión de
rayos X a ángulos bajos (SAXS), la existencia de una segregación molecular en las
primeras etapas del proceso previa a la cristalización parcial del PVDF segregado.
4.- El estudio de las curvas de transición vítrea de estas muestras revela la aparición de
una interfase enriquecida en PVDF, en relación con la matriz amorfa, posiblemente
vinculada a los núcleos de precristalización de este polímero. Este fenómeno
provoca la aparición de un gradiente de concentraciones cuyo límite superior está
situado en torno a una concentración del 70% en peso de PVDF, valor por encima
del cual el PVDF cristaliza segregándose de la mezcla. La evolución de esta
interfase está directamente relacionada con la cinética de las diferentes especies
cristalinas detectadas en el sistema objeto del presente trabajo de investigación.
5.- En muestras sometidas a un tratamiento isotérmico se ha puesto de manifiesto la
existencia de dos regiones distintas de segregación molecular de la matriz amorfa
así como de dos poblaciones cristalinas diferentes. Cada región de segregación
molecular está asociada a una población cristalina determinada. El límite entre
ambas regiones, denominado punto de cambio de régimen, tiene unos parámetros
que dependen de la temperatura de tratamiento. Para tiempos de tratamiento
inferiores al correspondiente al punto crítico, el control cinético es producido por la
difusión molecular, mientras a tiempos superiores a este límite la cinética pasa a
estar controlada principalmente por la cristalización.

215
6.- Se ha comprobado la existencia de dos regiones de cinéticas diferentes separadas
por la temperatura de transición vítrea del PMMA. Este límite es independiente de
la concentración y del tiempo de tratamiento, y marca la división entre la región de
temperaturas (Ta < TgPMMA) en la cual la difusión molecular es producida
exclusivamente por el PVDF, y aquella otra región (Ta > TgPMMA) en la que se
difunden simultáneamente las moléculas de los dos polímeros.
7.- El estudio de la cinética de cristalización de las dos poblaciones cristalinas revela la
existencia de factores cinéticos inducidos por las inhomogeneidades existentes en
las muestras tratadas. La primera población cristalina, asociada a la primera región
de variación de la transición vítrea de la matriz amorfa, se produce a partir de la
segregación molecular inducida en la matriz amorfa, estando constituida por
cristales de punto de fusión constante. Los resultados experimentales indican que la
cinética global que gobierna el crecimiento de esta población cristalina está
controlada por la difusión molecular, y en la cristalización de la misma
predominaba la nucleación sobre el crecimiento de los cristales.
La segunda población cristalina está asociada a procesos tardíos de segregación
molecular (tiempos posteriores al tiempo de cambio de régimen), estando
constituida por cristales de punto de fusión dependiente de la temperatura de
tratamiento. De los resultados experimentales se desprende que la cinética global
que gobierna el crecimiento de esta población cristalina está controlada por la
cristalización o conjuntamente por la cristalización y la difusión molecular,
predominando el crecimiento de los cristales sobre la nucleación de los mismos.
Por último, y para concentraciones suficientemente elevadas de PVDF, se aprecia la
aparición de dos nuevas poblaciones cristalinas a temperaturas de tratamiento
térmico próximas al punto de fusión del PVDF. Estas poblaciones cristalinas se
producen respectivamente por una transformación de fase de la primera población
cristalina y por procesos de recristalización durante el barrido de las muestras en el
calorímetro, siendo identificadas como pertenecientes en los dos casos a la fase γ, la
cual es termodinámicamente más estable en ese rango de temperaturas.

216
8.- Aunque no ha sido posible realizar con total certeza la identificación de las dos
poblaciones cristalinas principales con las correspondientes fases cristalinas del
PVDF, tanto la relación apreciada en muestras con Φm = 0.75 entre la primera
población cristalina y la fase ß, así como otros indicios indirectos, nos permiten
sugerir la posibilidad de que la primera población cristalina corresponde a la fase ß,
mientras la segunda población cristalina está formada por cristales pertenecientes a
la fase α. Las poblaciones cristalinas de alta temperatura, tal como ha sido
comentado, pertenecen a la fase γ.
9.- Asumiendo los puntos propuestos en el apartado anterior, se concluye que la fase ß
está asociada a mezclas de PVDF y PMMA las cuales comienzan a segregar
cuando la concentración de la matriz amorfa alcanza máximos locales con
Φm ≈ 0.7, cristalizando parcialmente el PVDF segregado en la fase ß. La fase α, por
el contrario, se forma por reordenación molecular de otras regiones de mezcla
heterogénea. Por último, la fase γ está producida por una transformación de fase
ß → γ ocurrida a temperaturas cercanas a la correspondiente al punto de fusión del
PVDF, así como por fenómenos de recristalización de las muestras.

217
BIBLIOGRAFÍA
- Aaron, H.B.; Fainstein, D.; Kotler, G.R. J. Appl. Phys., 41, 4404 (1970).
- Alexander, L. X-Ray diffraction methods in polymer science. Wiley Interscience. New York
(1969).
- Alfonso, G.C.; Russell, T.P. Macromolecules 19, 1143 (1986).
- Ando, Y.; Yoon, D.Y. Polymer J. 24, 1329 (1992).
- Ando, Y.; Hanada, T.; Saitoh, K. J. Polym. Sci. Polym. Phys. 32, 179 (1994).
- Arridge, R.G.C. Mechanics of Polymers, Clarendon Press, Oxford (1975).
- Aubin, M.; Prud'Homme, R.E. Polym. Eng. Sci. 28, 21 (1988).
- Baltá Calleja, F.J.; Martínez de Salazar, J.; Cackovic, H.; Loboda-Cackovic, J. J. Mater.
Sci. 16, 739 (1981).
- Baltá Calleja, F.J. Adv. Polym. Sci. 66, 117 (1985a).
- Baltá Calleja, F.J.; Kilian, H.G. Colloid Polym. Sci. 263, 697 (1985b).
- Baltá Calleja, F.J.; Martínez de Salazar, J.; Rueda Bravo, D. Encycl. Polym. Sci. Eng. 7,
614 (1987).
- Bernstein, R.E.; Cruz, C.A.; Paul, D.R.; Barlow, J.W. Macromolecules 10, 681 (1977).
- Bessonova, N.P.; Danchinov, S.K.; Shibanov, Y.D. Polymer Science, Ser. A 40, 689
(1998).
- Boerio, F.J.; Koenig, J.L. J. Polym. Sci. A-2, 7, 1489 (1969).
- Bohdanecky, M.; Simek, L.; Petrik, S. Polymer Comm. 31, 137 (1990).
- Bottino, A.; Capannelli, G.; Munari, S.; Turturro, A. J. Polym. Sci. Polym. Phys. 26, 785
(1988).
- Boyer, R.F. J. Macromol. Sci. - Phys. B7, 487 (1973).
- Braun, G.; Kovacks, A.J. Physics of Non-Crystaline Solids. J.A. Prins Ed., North Holland,
Amsterdam (1965).
- Brekner, M.J.; Schneider, H.A.; Cantow, H.J. Polymer 29, 78 (1988).
- Broadhurst, M.G.; Davis, G.T. Ferroelectrics 60, 3 (1984).
- Brochard, F.; Jouffroy, J.; Levinson, P. Macromolecules 16, 1638 (1983).
- Bueche, F. Phisical Properties of Polymers, Interscience, N.Y. (1962).

218- Canalda, J.C.; Hoffmann, T.; Martínez de Salazar, J. Polymer 36, 981 (1995).
- Chen, L.T.; Frank, C.W. Ferroelectrics 57, 51 (1984).
- Chow, T.S. Macromolecules 23, 333 (1990).
- Coleman, M.M.; Zarian, J.; Varnell, D.F.; Painter, P.C. J. Polym. Sci. Polym. Lett. 15, 745
(1977).
- Coleman, M.M.; Serman, C.J.; Bhagwagar, D.E.; Painter, P.C. Polymer 31, 1187 (1990).
- Cortili, G.; Zerbi, G. Spectrochim. Acta 23A, 285 (1967a).
- Cortili, G.; Zerbi, G. Spectrochim. Acta 23A, 2216 (1967b).
- Couchman, P.R.; Karasz, F.E. Macromolecules 11, 117 (1978a).
- Couchman, P.R. Macromolecules 11, 1157 (1978b).
- Couchman, P.R. Physics Letters 70A, 155 (1979).
- Couchman, P.R. Macromolecules 13, 1272 (1980).
- Couchman, P.R. Macromolecules 16, 1924 (1983).
- Couchman, P.R. Polym. Eng. Sci. 24, 135 (1984).
- Couchman, P.R. Polym. Eng. Sci. 27, 618 (1987).
- Crämer, K.; Fogliato Santos Lima, M.; Magonov, S.N.; Hellmann, E.H.; Jacobs, M.;
Hellmann, G.P. J. Mat. Sci. 33, 2305 (1998).
- Davis, G.T.; McKinney, J.E.; Broadhurst, M.G.; Roth, S.C. J. Appl. Phys. 49, 4998 (1978).
- Di Marzio, E.A. Ann. NY Acad. Sci. 371, 1 (1981).
- Di Marzio, E.A. Polymer 31, 2294 (1990).
- DiPaola-Baranyi, G.; Fletcher, S.J.; Degré, P. Macromolecules 15, 885 (1982).
- Eguiazábal, J.I.; Elorza, J.M.; Iruin, J.J.; Guzmán, G.M. Anales de Química 83, 159 (1987).
- Eijkelenboom, A.P.A.; Maas, W.E.J.R.; Veeman, W.S.; Werumeus Buning, G.H.; Vankan,
J.M.J. Macromolecules 25, 4511 (1992).
- Enns, J.B.; Simha, R. J. Macromol. Sci. Phys. B13, 11 (1977).
- Etxeberría, A.; Uriarte, C.; Fernández-Berridi, M.J.; Iruin, J.J. Macromolecules 27, 1245
(1994).
- Ezquerra, T. Comunicación privada.
- Fischer, E.W. Z. Naturforsch., bf. 12a, 753 (1957).
- Flory, P.J. J. Chem. Phys. 9, 660 (1941).
- Flory, P.J. J. Chem. Phys. 10, 51 (1942).

219
- Flory, P.J. Principles of Polymer Chemistry. Cornell University Press, Ithaca, New York
(1953).
- Flory, P.J. J. Am. Chem. Soc. 84, 2857 (1962).
- Fox, T.G. Bull. Am. Phys. Soc. 1, 123 (1956).
- Freitas Siqueira, D.; Galembeck, F.; Pereira Nunes, S. Polymer 32, 990 (1991).
- Frensch, H.; Wendorff, J.H. Polymer 27, 1332 (1986).
- Gal'Perin, Y.L.; Kosmynin, B.P. Polym. Sci. USSR 11, 1624 (1969).
- Gal'Perin, Y.L.; Kosmynin, B.P.; Smirnov, V.K. Polym. Sci. USSR 12, 2133 (1970).
- Garbella, R.W.; Wendorff, J.H. Makromol. Chem. 189, 2459 (1988).
- Geiss, D.; Ruscher, C. Progress in Colloid & Polymer Science 80, 119 (1989).
- Gibbs, J.H.; DiMarzio, E.A. J. Chem. Phys. 28, 373 (1958).
- Goh, S.H.; Slow, K.S. Polym. Bull. 20, 393 (1988).
- Gordon, M.; Taylor, J.S. J. Appl. Chem. 2, 493 (1952).
- Graessley, W.; Krishnamoorti, R.; Balsara, N.P.; Fetters, L.J.; Lohse, D.J.; Schulz, D.N.;
Sissano, J.A. Macromolecules 26, 1137 (1993).
- Gregorio, R. Jr.; Cestari, M. J. Polym. Sci. Polym. Phys. 32, 859 (1994).
- Gregorio, R. Jr.; Capitao, R.C. J. Mat. Sci. 35, 299 (2000).
- Grubb, D.T.; Choi, K.W. J. Appl. Phys. 52, 5908 (1981).
- Hadziioannou, G.; Stein, R.S. Macromolecules 17, 567 (1984).
- Hahn, B.R.; Herrmann-Schönherr, O.; Wendorff, J.H. Polymer 28 201 (1987).
- Handbook of chemistry and physics. 64TH ed. C.R.C. Press. Boca Ratón (Florida, USA)
(1983-84).
- Hoffman, J.D.; Weeks, J.J. J. Res. Nat. Bur. Std. 66A, 13 (1962).
- Hoffman, J.D.; Lauritzen, J.I. Treatise on solid state chemistry vol. 3. N.B. Hannay, Plenum
Press, N.Y. (1976).
- Hsu, C.C.; Geil, P.H. Polym. Commun. 27 105 (1986).
- Huggins, M.L. J. Chem. Phys. 9, 440 (1941).
- Ito, H.; Russell, T.P.; Wignall, G.D. Macromolecules 20, 2213 (1987).
- Iwamoto, T.; Itoh, T.; Ando, Y.; Miyata, S.; Konishi, T. Japan. J. Applied Physics Letters
27 L1969 (1988).
- Jawhari, T.; Merino, J.C.; Pastor, J.M. J. Mat. Sci. 27, 2237 (1992).

220- Jenckel, E.; Heusch, R. Kolloid Z. 130, 89 (1953).
- Jo, W.H.; Kwon, I.H. Macromolecules 24, 3368 (1991).
- Jost, W. Diffusion in Solids, Liquids, Gases, Academic Press, N.Y. (1960).
- Kakudo, M.; Kasai, N. X-Ray Diffraction by Polymers. Kudansha Ltd. Tokyo (1972).
- Kammer, H.W. Plastic and Rubber Processing and Applications 9, 23 (1988).
- Kanig, G. Kolloid Z.Z. Polym. 190, 1 (1963).
- Kanig, G. Kolloid Z.Z. Polym. 233, 829 (1969).
- Keller, A. Phil. Mag. 2, 1171 (1957).
- Kelley, F.N.; Bueche, F. J. Polymer Sci. 50, 549 (1961).
- Khoury, F.; Pasaglia, E. Treatise on Solid State Chemistry vol. 9: Crystalline and
Noncrystalline Solids. Ed. Plenum Press (1976).
- Kim, K.J.; Kim, G.B.; Han, S.H. J. Appl. Pol. Sci. 49, 7 (1993).
- Kramer, E.J.; Green, P.; Palmstrom, C.J. Polymer 25, 473 (1984).
- Kwei, T.K.; Frisch, H.L. Macromolecules 11, 1267 (1978).
- Kwei, T.K. J. Polym. Sci. Polym. Lett. Edn. 22, 307 (1984).
- Lauritzen, J.I.; Hoffman, J.D. J. Appl. Phys. 44, 4340 (1973).
- Léonard, C.; Halary, J.L.; Monnerie, L.; Broussoux, D.; Servet, B.; Micheron, F. Polymer
Comm. 24, 110 (1983).
- Léonard, C.; Halary, J.L.; Monnerie, L. Polymer 26, 1507 (1985).
- Léonard, C.; Halary, J.L.; Monnerie, L. Macromolecules 21, 2988 (1988).
- Lovinger, A.J. Polymer 21, 1317 (1980).
- Lovinger, A.J. Macromolecules 14, 322 (1981).
- Lovinger, A.J. Developments in crystalline Polymers 1. Ed. D.C. Bassett. App. Sc.
Publischers, London (1982a).
- Lovinger, A.J. Macromolecules 15, 40 (1982b).
- Lu, X.; Weiss, R.A. Macromolecules 25, 3242 (1992).
- Maas, W.E.J.R.; van der Heikden, W.A.C.; Veeman, W.S.; Vankan, J.M.J.; Werumeus
Buning, G.H. J. Chem. Phys. 95, 4698 (1991).
- Maas, W.E.J.R.; Papavoine, C.H.M.; Veeman, W.S.; Werumeus Buning, G.H.; Vankan,
J.M.J. J. Polym. Sci. Polym. Phys. 32, 785 (1994).
- Madruga, E.L.; San Román, J. J. Macrom. Sci., Chem. A21, 167 (1984).
- Maiti, P.; Nandi, A.K. Macromol. Chem. Phys. 199, 1479 (1998).

221
- Plans, J.; Macknight, W.J.; Karesz, F.E. Macromolecules 17, 1100 (1984).
- Marand, H.L.; Stein, R.S. J. Polym. Sci. Polym. Phys. 26, 1361 (1988).
- Marand, H.L.; Stein, R.S. J. Polym. Sci. Polym. Phys. 27, 1089 (1989).
- Martínez de Salazar, J.; Baltá Calleja, F.J. J. Mater. Sci. Lett. 4, 324 (1985).
- Martínez de Salazar, J.; Canalda Cámara, J.C.; Baltá Calleja, F.J. J. Mater. Sci. 26, 2579
(1991a).
- Martínez de Salazar, J.; Sánchez Cuesta, M.; Plans, J. Polymer 32, 2984 (1991b).
- Martínez de Salazar, J.; Canalda Cámara, J.C.; Vallejo, B. Macromol. Symp. 78, 95 (1994).
- Martínez de Salazar, J.; Alizadeh, A.; Jiménez, J.J.; Plans, J. Polymer 37, 2367 (1996).
- Mijovic, J.; Sy, J-W.; Kwei, T.K. Macromolecules 30, 3042 (1997).
- Miller, R.L.; Raisoni, J. J. Polym. Sci. Polym. Phys. 14, 2325 (1976).
- Millich, F.; Hauber, B.F. J. Polym. Sci. Polym. Phys. 26, 1217 (1988).
- Morra, B.S.; Stein, R.S. J. Polym. Sci. Polym. Phys. 20, 2243 (1982a).
- Morra, B.S.; Stein, R.S. J. Polym. Sci. Polym. Phys. 20, 2261 (1982b).
- Moussaif, R.; Jérôme, R. Polymer 40, 6831 (1999).
- Mumby, S.J.; Sher, P. Macromolecules 27, 689 (1994).
- Naito, G.; Johnson, G.E.; Allara, D.L.; Kwei, T.K. Macromolecules 11, 1260 (1978).
- Nakagawa, K.; Ishida, Y. J. Polym. Sci. Polym. Phys. 11, 2153 (1973).
- Nandi, A.K. Polymer 35, 5202 (1994).
- Narula, G.K.; Pillai, P.K.C. J. Mater. Sci. Lett. 8, 627 (1989).
- Nishi, T.; Kwei, T.K.; Wang, T.T. J. Appl. Phys. 46, 4157 (1975a).
- Nishi, T.; Wang, T.T. Macromolecules 8, 909 (1975b).
- Okabe, Y.; Kyu, T.; Saito, H.; Inoue, T. Macromolecules 31, 5823 (1998).
- Osaki, S.; Ishida, Y. J. Polym. Sci. Polym. Phys. 13, 1071 (1975).
- Ougizawa, T.; Inoue, T. Polymer Journal 18, 521 (1986).
- Paul, D.R. Polymer Blends. S. Newman eds. Academic Press, New York (1978a).
- Paul, D.R.; Barlow, J.W.; Bernstein, R.E.; Wahrmund, D.C. Polym. Eng. Sci. 18, 1225
(1978b).
- Paul, D.R.; Barlow, J.W. J. Macromol. Sci. Rev. Macromol. Chem. C(18), 109 (1980).
- Pochan, J.M.; Beatty, C.L.; Pochan, D.F. Polymer 20, 879 (1979).
- Porod, G. Kolloid Z 124, 83 (1951).

222- Prest, W.M. Jr.; Luca, D.J. J. Appl. Phys 46, 4136 (1975).
- Prest, W.M. Jr.; Luca, D.J. J. Appl. Phys. 49, 5042 (1978).
- Privalko, V.P.; Petrenko, K.D.; Lipatov, Y.S. Polymer 31, 1277 (1990).
- Rashmi, Narula, G.K.; Pillai, P.K.C. J. Mat. Sci. 22, 2006 (1987).
- Rield, B.; Prud'Homme, R.E. Pol. Engineering and Science 24, 1291 (1984).
- Righetti, M.C.; Ajroldi, G.; Marchionni, G.; Pezzin, G. Polymer 34, 4307 (1993).
- Rim, P.B.; Runt, J.P. Macromolecules 17, 1520 (1984).
- Roerdink, E.; Challa, G. Polymer 19, 173 (1978).
- Roerdink, E.; Challa, G. Polymer 21, 509 (1980).
- Saito, H.; Okada, T.; Hamane, T.; Inoue, T. Macromolecules 24, 4446 (1991).
- Saito, H.; Stühn, B. Polymer 35, 475 (1994).
- Saito, H.; Stühn, B. Macromolecules 27, 216 (1994b).
- Sakaoku, K.; Itoh, T.; Kunimura, S. Jap. J. Applied Physics Letters 24, L175 (1985).
- Sasaki, H.; Bala, P.K.; Yoshida, H.; Ito, E. Polymer 36, 4805 (1995).
- Schmidt, J.M. U.S. Patent. 3, 459, 834 (1969).
- Schneider, H.A. Makromol. Chem. 189, 1941 (1988).
- Schneider, H.A.; Di Marzio, E.A. Polymer 33, 3453 (1992).
- Scott, R.L. J. Chem. Phys. 17, 279 (1949).
- Skolnick, J.; Yaris, R.; Kolinski, A. J. Chem. Phys. 88, 1407 (1988).
- Simha, R.; Boyer, R.F. J. Chem. Phys. 37, 1003 (1962).
- Takahashi, Y.; Tadokoro, H. Macromolecules 13, 1317 (1980).
- Takahashi, Y.; Miyamoto, N. J. Polym. Sci. Polym. Phys. 23, 2505 (1985).
- Takahashi, Y. Macromolecules 26, 1471 (1993).
- Takase, Y.; Scheinbeim, J.I.; Newman, B.A. J. Polym. Sci. Polym. Phys. 27, 2347 (1989).
- Tanaka, H.; Lovinger, A.J. Macromolecules 20 2640 (1987).
- Tanaka, H.; Nishi, T. Phys. Rev. A 39, 783 (1989).
- Tashiro, K.; Takano, K.; Kobayashi, M.; Chatani, Y.; Tadokoro, H. Polym. Bull. 10, 464
(1983a).
- Tashiro, K.; Takano, K.; Kobayashi, M.; Chatani, Y.; Takodoro, H. Polymer 24, 199
(1983b).
- Till, P.H. J. Polym. Sci. 24, 301 (1957).
- Weinhold, S.; Litt, M.H.; Lando, J.B. J. Polym. Sci. Polym. Lett. 17, 585 (1979).

223
- Weinhold, S.; Litt, M.H.; Lando, J.B. Macromolecules 13, 1178 (1980).
- Weinhold, S.; Bachmann, M.A.; Litt, M.H.; Lando, J.B. Macromolecules 15, 1631 (1982).
- Wendorff, J.H. J. Polym. Sci. Polym Lett. Ed. 18, 439 (1980).
- Wing Sy, J.; Mijovic, J. Macromolecules 33, 933 (2000).
- Wunderlich, B. Macromolecular Physics vol. 1. Academic Press, N.Y. (1973).
- Wunderlich, B. Macromolecular Physics vol. 3. Academic Press, N.Y. (1980).
- Wunderlich, B. Progr. Colloid Polym. Sci. 96, 22 (1994).
- Xue, G.; Wang, Y.; Liu, S. Macromolecules 28, 3476 (1995).
- Yang, D.C.; Thomas, E.L. J. Mat. Sci. Lett. 3, 929 (1984).
- Yang, D.C.; Chen, Y. J. Mater. Sci. Lett. 6, 599 (1987).
- Yoon, D.Y.; Ando, Y.; Rojstaczer, S.; Kumar, S.K.; Alfonso, G.C. Makromol. Chem.,
Macromol. Symp. 50, 183 (1991).
- Young, R.J. Introduction to polymers. Ed. Chapman & Hall. N.Y. (1987).
- Zerbi, G.; Cortili, G. Chem. Comm. 13, 295 (1965).


















