

Fisiopatologia cardiovascular gelpi_booksmedicos.org
429
-
Upload
antonio-alonso-castillo -
Category
Documents
-
view
619 -
download
11
Transcript of Fisiopatologia cardiovascular gelpi_booksmedicos.org

FISIOPATOLOGÍACARDIOVASCULAR


RICARDO J. GELPI - MARTÍN DONATO
FISIOPATOLOGÍACARDIOVASCULAR
Bases racionales para la terapéutica
www.corpuslibros.com

La presente es una publicación de:
Fisiopatología cardiovascularRicardo J. Gelpi - Martín Donato1º Edición
DERECHOS RESERVADOS© 2010 Corpus Editorial y [email protected]@corpus.comwww.corpuslibros.com.arSuipacha 581 - Tel/Fax: (+54 341) 439 4978 / 437 1327(S2002LRK) Rosario - ArgentinaEditor: Esteban Oscar Mestre
Tirada: 1 000 ejemplaresSe terminó de imprimir en abril de 2010Rosario - Argentina
ISBN: 978-950-9030-02-2
No está permitida la reproducción total o parcial de esta obra, nisu tratamiento o transmisión por cualquier medio o método, sinautorización escrita de la Editorial.
NOTALa medicina es una ciencia en constante desarrollo. Conforme surjan nuevos conocimientos, se requerirán cambios de la
importancia con respecto a fármacos nuevos o de uso no frecuente.
los valores normales y medicamentos permitidos o recomendados.
Gelpi, Ricardo JorgeFisiopatología cardiovascular: bases racionales para la terapéutica / Ricardo Jorge Gelpi y Martín Donato
CDD 616.1

Este libro está dedicado a la memoria del Dr. Carlos A. Bertolasi


Directores coordinadores
Ricardo J. Gelpi: Profesor Titular, Universidad de Buenos
Universidad de Buenos AiresMartín Donato: Jefe de Trabajos Prácticos, Universidad
Cardiovascular, Universidad de Buenos Aires
ColaboradoresErnesto A. Aiello:
Raúl Altman:Nacional de TucumánCharles Antzelevitch: Director del Masonic Medical Research Laboratory, Utica, New York, Estados UnidosNidia Basso:
-
Gabriela Berg: Profesora Adjunta, Universidad de Bue-
Bioquímica Clínica, Laboratorio de Lípidos y Lipoproteí--
dad de Buenos Aires.Carlos A. Bertolasi:
“Dr. Cosme .
Fernando Brites: Profesor Adjunto, Universidad de Bue-
Bioquímica Clínica, Laboratorio de Lípidos y Lipoproteí--
dad de Buenos AiresSusana Cavallero: Facultad de Farmacia y Bioquímica, Universidad de Buenos AiresPablo Chiale:
Horacio E. Cingolani:Cardiovasculares de La Plata, Universidad Nacional de
Christophe Depre:
Verónica D´Annunzio: Jefe de Trabajos Prácticos,
Marcelo Elizari:
Irene L. Ennis: -nes Cardiovasculares de La Plata, Universidad Nacional
Eduardo Escudero: Profesor Adjunto, Universidad
Belisario Fernández: Facultad de Farmacia y Bioquímica,
Marcela Ferreiro:
Germán E. González: Jefe de Trabajos Prácticos del -
-
Diego Grinfeld:
Italiano de Buenos AiresLiliana Grinfeld:
Aires.Marcelo Halac:
Italiano de Buenos AiresDerek J. Hausenloy:
Autores y colaboradores A

Stefan Hein: Médico de planta del Kerckhoff-Clinic,
Ramón Nicasio Herrera: Académico de la Universidad
en Trombosis de la Universidad Nacional de Tucumán.Alejandro Hita:Hospital Universitario Austral, Buenos AiresVadim Kotowicz:
Rubén P. Laguens: -
Jorge Lerman: Profesor Titular, Universidad de Buenos
Guillermo Liniado:
Héctor L. Luciardi:Autorizado de la Facultad de Medicina Universidad
Universidad Nacional de TucumánAlicia Mattiazzi:Cardiovasculares de La Plata, Universidad Nacional de
Patricia Cabeza Meckert:-
ro. Universidad FavaloroCelina Morales: Profesor Titular, Universidad de
Cardiovascular, Universidad de Buenos Aires. Especialista
Juan Muntaner:Nacional de TucumánJosé Navia: Académico de Número, Academia Nacional
Hospital Universitario AustralNéstor G. Pérez: -nes Cardiovasculares de La Plata, Universidad Nacional de
Daniel Piñeiro: Profesor Titular, Universidad de Buenos
Aldo Prado: Universidad Nacional de La PlataAna María Puyó: Facultad de Farmacia y Bioquímica, Universidad de Buenos AiresManuel Rodríguez: Jefe de Trabajos Prácticos, Univer-
-
Ricardo Ronderos:Hospital “ , La Plata
Aurora Ruiz:“Juan A. Fernández
Jutta Schaper:
Nauheim, AlemaniaLaura Schreier: Profesora Titular, Universidad de
y Bioquímica Clínica, Laboratorio de Lípidos y
Universidad de Buenos AiresGuillermo B. Semeniuk: Profesor Consulto de la Uni-versidad de Buenos Aires; Médico de planta del Instituto
de Buenos AiresSerge Sicouri:Research Laboratory, Utica, New York, Estados Unidos.Ana L. Tufare: -nes Cardiovasculares de La Plata, Universidad Nacional de la PlataMarina Vallaza:
“Dr. Ramos MejíaStephen F. Vatner: Director del Departamento de
Martín Vila-Petroff:
Leticia Vittone: -nes Cardiovasculares de La Plata, Universidad Nacional de
Cecilia Mundiña-Weilenmann:
Regina Wikinski: Profesora Emérita, Universidad de
Clínica, Laboratorio de Lípidos y Lipoproteínas; Hospital
Universidad de Buenos AiresDerek Yellon:
Unido.Valeria Zago: Bioquímica, Tesista del Instituto de
Lípidos y Lipoproteínas; Hospital de Clínicas “José de

P Prólogo / 17
1 Fisiopatologia de las dislipemias / 19
Familias de lipoproteínas
Dislipoproteinemias
Dislipoproteinemias secundarias
Modificaciones cualitativas de las lipoproteínas
Fisiopatología de las dislipemias secundarias más frecuentesDiabetes mellitus tipo 2
Síndrome metabólico
Hipotiroidismo
Dislipemia en la posmenopausia
Lipoproteínas e injuria endotelial
Bibliografía
2 Remodelamiento de la placa aterosclerótica / 31Liliana Grinfeld, Marcelo Halac
Unidad lesional: el ateroma
Hallazgos incipientes en la formación de un ateroma
Complicación trombótica de una placa aterosclerótica
Factores de riesgo y vulnerabilidadDislipidemia
Hipertensión arterial
Diabetes
Tabaquismo
Resistencia a la aspirina
Factor tisular y apoptosis
Homocisteína
Proteína C-Reactiva
Índice Í
Otros hallazgos que podrían aumentar el riesgo de un evento coronario
Placa culpable y placa vulnerable
Factores intrínsecos de la vulnerabilidad de una placa ateromatosaCápsula fibrosa
Erosión-Ruptura
Modificaciones estructurales de la cápsula fibrosa
Núcleo lipídico
Factores extrínsecos de la vulnerabilidad de una placa ateromatosaFatiga de la placa
Vasoespasmo
Estrés hemodinámico
Presión arterial
Consideraciones en el desarrollo de nuevas terapéuticas
Identificación diagnóstica
Estudios por Imágenes. Descripción técnica esquemática
Bibliografía
3 Fisiopatología de los síndromes coronarios / 47
Síndromes coronarios agudosSíndromes coronarios agudos sin elevación del segmento ST. Angina inestable e infarto sin elevación del segmento ST
Infarto agudo de miocardio con elevación del segmento ST
Síndromes coronarios crónicosAngina crónica estable
Dolor anginoso
Angina de umbral variable
Espasmo coronario. Angina de PrinzmetalInfluencias neurales
Modulación endotelial
Otros mecanismos

Sindrome X
Isquemia silenteModulación y percepción del dolor
Duración de la isquemia
Magnitud del área isquémica
Casos particulares de isquemia miocárdicaIsquemia miocárdica e hipertrofia ventricular
Isquemia miocárdica y diabetes
Bibliografía
4 Restenosis coronaria / 63
ReestenosisMecanismos tempranos
Mecanismos tardíos
Hiperplasia fibrointimal
Fenómeno inflamatorio
Remodelado arterial crónico
Reestenosis intrastent
Ciclo celular y reestenosisFactores predictivos de la aparición de reestenosis intrastent
Influencia de las variables del procedimiento
Factores genéticos
Stents liberadores de droga (SLD)Stents liberadores de drogas antiproliferativas en escenarios no favorables
Reestenosis de los stents liberadores de droga (SLD)
Prevención farmacológica de la reestenosis intrastent
Clasificación de la reestenosis intrastent
Tratamiento de la reestenosisAngioplastia simple con balón
Aterectomía
Stent intrastent
Radiación intracoronaria
SLD, intrastents convencionales
Conclusión
Bibliografía
5 Miocardio atontado, miocardio hibernado / 79
Fisiología y relevancia clínica del fenómeno de isquemia/reperfusión transitoria: miocardio atontado
Mecanismos moleculares responsables del miocardio atontado
Atontamiento como mecanismo del miocardio hibernado
Genómica de la sobrevida celular en el atontamiento miocárdico
Relación entre miocardio atontado, hibernado y precondicionamiento
Conclusión
Bibliografía
6 Protección del corazón en la injuria por isquemia reperfusión: precondicionamiento y poscondicionamiento isquémico / 89Derek M. Yellon, Derek J. Hausenloy
Protección miocárdicaProtección miocárdica por precondicionamiento isquémico
Características del precondicionamiento isquémico
Indicadores de la protección inducida por precondicionamiento isquémico
Precondicionamiento clásico
La segunda ventana de protección en el precondicionamiento isquémico (precondicionamiento tardío)
Precondicionamiento remoto o de zonas alejadas
Precondicionamiento del miocardio humano
Precondicionamiento en el área clínica
Protección miocárdica por intervenir durante la reperfusión
Injuria letal por reperfusión
La vía de las kinasas de rescate activadas en la isquemia/reperfusión (RISK)
Poscondicionamiento isquémico
Protección del miocardio humano utilizando terapias adyuvantes en la reperfusión
Conclusiones
Bibliografía
7 Fisiopatología de la protección miocárdica en la cirugía cardiaca / 109
Fisiopatología del miocardio isquémico-metabolismo de la isquemia
Autorregulación del flujo coronario. Disfunción endotelial
Proceso de injuria por isquemia/reperfusión
Historia de la protección miocárdica
Protección del miocardio isquémicoTécnicas de administración de cardioplejia
1. Vía anterógrada
2. Vía retrógrada
Consideraciones anatómicas y hemodinámicas (Seno coronario). Efecto de reperfusión retrógrada

Métodos de administración de la solución cardiopléjica
1. Método combinado
2. Método retrógrado
3. Método anterógrado con oclusión del seno coronario
4. Reperfusión con intervenciones adicionales sobre el seno coronario
Efectos clínicos de las intervenciones sobre el seno coronario
Soluciones cardiopléjicas
Temperatura óptima y protección miocárdica
Soluciones enriquecidas
Bibliografía
8 Remodelamiento posinfarto de miocardio / 123
Morales, Guillermo Liniado
Infarto de miocardioEfectos de la isquemia sobre los miocitos cardiacos
Evolución morfológica
Infarto de miocardio con reperfusión
Fisiopatología
Sistema nervioso simpático
Sistema renina-angiotensina-aldosterona
RemodelamientoConcepto de remodelamiento
Remodelamiento posinfarto de miocardio
Expansión del infarto
Remodelamiento tardío posinfarto de miocardio
Remodelamiento de los miocitos cardíacos en el miocardio remanente
Degeneración y muerte de los miocitos cardíacos en el miocardio remanente
Remodelamiento de la matriz extracelular
Remodelamiento temprano/remodelamiento tardío: un continuo hacia la insuficiencia cardíaca
Factores que modifican el Remodelamiento
Aspectos clínicosEpidemiología
Género
Diabetes
Hipertensión arterial
Evaluación clínica del remodelado
Métodos de estudio
Prevención y modulación del remodelamiento posinfarto de miocardio
Terapéutica farmacológica
Inhibidores de la enzima de conversión de angiotensina
Antagonistas de la angiotensina II
Antagonistas de la aldosterona
Bloqueantes β-adrenérgicos
Nitratos
Estatinas
Permeabilidad de la arteria responsable
Terapia celular
Modulación no farmacológica
Cirugía
Métodos de contención pasiva
Soporte circulatorio mecánico
Métodos para restaurar una contracción sincrónica (resincronización cardiaca)
Ejercicio
Bibliografía
9 Sistema renina angiotensina y Remodelamiento cardiaco / 151Germán E. González, Celina Morales, Ricardo J. Gelpi
Remodelamiento ventricularConcepto, generalidades, importancia
Aspectos generales del remodelamiento fisiológico y patogenia del remodelamiento patológico
Sistema renina angiotensina en el remodelamiento ventricular
Rol del sistema renina angiotensina en el infarto de miocardio
Aspectos fisiopatológicos y morfológicos del remodelamiento ventricular posinfarto
Remodelamiento temprano y su relación con la activación del sistema renina angiotensina
Participación del sistema renina angiotensina en el remodelado de zonas alejadas del infarto
Rol del sistema renina angiotensina en el remodelamiento del tejido conectivo
Remodelamiento tardío y su relación con el sistema renina angiotensina
Inhibición farmacológica del sistema renina angiotensina en el remodelamiento ventricular posinfarto
Bibliografía
10 Reparación del corazón / 169
Inducción de los miocitos adultos remanentes a dividirse en células hijasReplicación de los miocitos en el corazón normal
Replicación de los miocitos en el corazón patológico
Estrategias para inducir mitosis y citocinesis en miocitos adultos
Inducción de la replicación de una población de progenitores miocíticos residentes en el corazón y diferenciación en miocitos adultos
Introducción en el corazón de células progenitoras indiferenciadasCélulas tronco embrionarias
Células tronco de la médula ósea del adulto
Conclusión
Bibliografía

11 Señales intracelulares que siguen al estiramiento miocárdico y conducen a la hipertrofia/ 177
Néstor G. Pérez, Irene L. Ennis
Vías intracelulares que conducen a la hipertrofia miocárdicaActivación de calcineurina
Activación de receptores acoplados a proteína G (GPCRs)
Activación de la PI3K (fosfatidilinositol 3-kinasa)
Activación de la vía gp130
Estiramiento miocárdico: señales que inician la hipertrofia
Intercambiador Na+/H+ e hipertrofia miocárdica
Inhibición del intercambiador Na+/H+ y regresión de la hipertrofia cardiaca
Bibliografía
12 Características estructurales del remodelamiento cardiaco en el desarrollo de la insuficiencia cardiaca / 189
Estructura normal del miocito y del intersticio
El miocito en la insuficiencia cardiaca
Cambios nucleares
Pérdida de miocitos por muerte celular
Fibrosis
Correlación estructura/función
Bibliografía
13 Acoplamiento éxcito-contráctil en el corazón normal y patológico / 197
El acoplamiento éxcito-contráctil en el miocardio normalMecanismos subcelulares
Ca2+ vs. respuesta al Ca2+ de las proteínas contráctiles en la determinación de la contractilidad miocárdica
El acoplamiento éxcito-contráctil en diferentes patologías
Insuficiencia cardiaca
Mecanismos subcelulares
El fenómeno de la escalera en el corazón normal e insuficiente
Respuesta β-adrenérgica en el corazón normal e insuficiente
Las proteínas involucradas en el manejo del Ca2+ como blancos de la transferencia génica en la IC
Pérdida de Ca2+ por el RyR2 y muerte súbita
Atontamiento miocárdico (Stunning)
Disfunción contráctil en el atontamiento miocárdico: Ca2+ vs. disminución de la respuesta al Ca2+ de las proteínas contráctiles
Disminución de la respuesta al Ca2+ de los miofilamentos como causa principal del AM
TnI vs. otras proteínas como causa de la menor respuesta al Ca2+ de los miofilamentos
Evidencia que demuestra que la disfunción contráctil del AM puede ser producida por una disminución del Ca2+ disponible para la contracción
Rol de las catecolaminas en el atontamiento miocárdico
Conclusiones
Bibliografía
14 Fisiopatología de la insuficiencia cardiaca / 223Ricardo J. Gelpi, Martín Donato,
Remodelamiento cardiaco
Hipertrofia celular
Apoptosis en la insuficiencia cardiaca
Regeneración y reparación miocárdica
Sistema renina-angiotensina-aldosterona
Sistema adrenérgico
Citoquinas inflamatorias
Rol de la matriz extracelular
Bibliografía
15 Péptidos natriuréticos / 237
Generalidades. Síntesis, secreción, receptores, funcionesPéptidos natriuréticos en situaciones patológicas
Cardiomiopatías. Insuficiencia cardiacaPéptidos natriuréticos como marcadores de insuficiencia cardiaca
Infarto de miocardio
Hipertensión arterial
Arritmias
Valvulopatias
Disnea

Transplante cardiaco
Rol de los péptidos natriuréticos en la regulación de la hipertrofia del miocardio
Aterosclerosis
Patologías endocrinas y renales
Conclusiones
Futuro de los péptidos natriuréticos
Bibliografía
16 Factores protrombóticos en la insuficiencia cardiaca / 251
Raúl Altman, Juan Muntaner
Tríada de Virchow
Cambios hemorreológicos
Disfunción endotelial
Activación de trombina y fibrinólisis
Activación plaquetaria
Efectos del tratamiento médico de la falla cardiaca sobre el estado protrombótico
Agentes antiplaquetarios en insuficiencia cardiaca
Anticoagulación en insuficiencia cardiaca
Conclusiones
Bibliografía
17 Fisiopatología de las valvulopatías / 261Alejandro Hita
Estenosis aórticaDe la estructura a la función: La válvula
Etiología
La respuesta ventricular: Estructura, geometría y función
La estructura: El miocito, el intersticio y la circulación coronaria
La geometría
La función
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica
Circulación pulmonar
La valvulopatía y el ejercicio
Insuficiencia aórticaDe la estructura a la función: La válvula
La respuesta ventricular: Estructura, geometría y función
La estructura: El miocito, el intersticio y la circulación coronaria
La geometría
La función
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica
La valvulopatía y el ejercicio
Insuficiencia mitralDe la estructura a la función: La válvula
La etiología
La respuesta ventricular: estructura, geometría y función
La estructura: El miocito, el intersticio y la circulación coronaria
La geometría
La función
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica
Circulación pulmonar
La valvulopatía y el ejercicio
Estenosis mitralDe la estructura a la función: La válvula
La etiología
La respuesta ventricular: Estructura, geometría y función
Adaptación de la circulación sistémica y pulmonar
La valvulopatía y el ejercicio
Bibliografía
18 Fisiopatología de la hipertensión arterial / 287Nidia Basso
Sistema nervioso autónomo
Sistema renina-angiotensina-aldosterona
Especies reactivas del oxígeno
Factores genéticos
Bibliografía
19 Mecanismos de adaptación fisiológicos y patológicos del corazón en la hipertensión arterial / 301Eduardo Escudero, Ana L. Tufare
Modificaciones estructuralesHipertrofia ventricular
Definición
Significado de la respuesta hipertrófica
El camino hacia la hipertrofia
La hipertrofia ventricular, ¿aparece siempre ante la elevación de la presión arterial?
La respuesta, ¿es igual en todos los pacientes hipertensos?
¿Cómo se regresa de la hipertrofia?
¿Se comporta el ventrículo derecho como órgano blanco?
Remodelamiento concéntrico

Modificaciones funcionalesFunción sistólica
La función en la fibra muscular
La función en el corazón entero
Corazón entero: función medioventricular
Función diastólica
Relajación
Llenado ventricular
El camino a la insuficiencia cardiacaDeterioro de la función ventricular
Alteración en la perfusión miocárdica
Hipertensión arterial y aterosclerosis coronaria
Alteraciones estructurales de la microcirculación coronaria
Alteraciones funcionales de la microcirculación coronaria
ArritmasArritmias supraventriculares
Arritmias ventriculares
Bibliografía
20 Fisiopatología de la hipertensión pulmonar / 321
Epidemiología y factores de riesgo
Fisiopatología
Subtipos de hipertensión pulmonar primaria
Historia clínica
Examen físico
Evaluación del paciente con sospecha de hipertensión pulmonar
Evaluación de enfermedades del tejido conectivo y/u otros factores capaces de producir hipertensión pulmonar
Otras enfermedades y anormalidades asociadasEnfermedad tromboembólica
Enfermedad parenquimatosa pulmonar como causa de hipertensión pulmonar
Hipertensión arterial pulmonar y trastornos respiratorios durante el sueño
Biopsia pulmonar
Severidad y pronóstico de la hipertensión arterial pulmonarPronóstico de la hipertensión arterial pulmonar
Tratamiento quirúrgico
Tratamiento médico
Bibliografía
21 Fisiopatología de las arritmias / 335
Charles Antzelevitch
Historia de la actividad eléctrica del corazón. De los experimentos de Galvani a la heterogeneidad de los potenciales de acción ventriculares
El estudio del potencial de acción
Bases celulares de las manifestaciones eléctricas del corazónLa fase 0 ó de despolarización rápida
La fase 1 ó de repolarización precoz
La fase 2 ó plateau (meseta)
La fase 3 ó de repolarización rápida
La fase 4: El potencial de reposo y la despolarización diastólica espontánea
Potencial de membrana de reposo de las células auriculares y ventriculares
El automatismo normal en el nódulo sinusal y el sistema His Purkinje
Mecanismos de las arritmiasAlteraciones en la formación del impulso
Automatismo normal
El automatismo anormal
Pospotenciales y actividad gatillada
Alteraciones en la conducción del impulso
Mecanismo de reentrada
La heterogeneidad eléctrica celular en el miocardio ventricular
El síndrome de BrugadaBases celulares del síndrome de Brugada
El síndrome de QT prolongado y la torsión de puntas
Bibliografía
22 Fisiopatología del síncope / 361Aurora Ruiz
Causas de síncope
Fisiopatología general del síncope
Síncopes neuromediados
Síndrome vasovagal maligno
Tilt testRespuestas al Tilt test
Respuesta mixta
Cardioinhibidora tipo A
Cardioinhibidora tipo B
Vasodepresora
Excepción 1
Excepción 2

Fisiopatología del síncope vasovagal ante el ortostatismoInterrelación entre la presión arterial, la frecuencia cardiaca y la respiración
Respuesta fisiológica ante el ortostatismo
Teorías sobre la fisiopatología del síncope vasovagal
Teoría de los mecanorreceptores intramiocárdicos
Teoría neurohumoral
Teoría de la disfunción barorrefleja
Sensibilidad barorrefleja espontánea
Teoría de la reducción de volumen
Teoría del desacoplamiento de las señales
Respuestas vasculares paradojales en pacientes con síncope
Alteración de la respuesta venoconstrictora
Reacción paradojal de la vasculatura cerebral
El rol de los receptores 2-adrenérgicos
Síncope vasovagal y respuesta a la emoción
Participación del mecanismo neurocardiogénico en el síncope que ocurre en otras entidadesIsquemia miocárdica
Miocardiopatía hipertrófica
Arritmias supraventriculares y ventriculares
Otros cuadros de intolerancia al ortostatismoHipotensión ortostática
Taquicardia postural
Bibliografía
23 Fisiopatogenia de la Enfermedad de Chagas / 387
EpidemiologíaFactores que favorecen el desarrollo de la endemia
Triatominos. Distribución. Relaciones con el agente y el huésped
Tripanosoma cruzi. Ciclo biológico
Vías de transmisiónVía entomológica o vectorial
Vía transfusional
Vía transplacentaria
Historia natural de la Enfermedad de ChagasEsquema de Rosenbaum
Estadio agudo
Estadio indeterminado
Miocardiopatía chagásica crónica
Bibliografía
24 Remodelamiento en la resincronización del ventrículo izquierdo / 413Ricardo Ronderos, Aldo Prado
Remodelamiento ventricular
Bibliografía
Í Índice analítico / 423


poesía
belleza en sí misma pero con pocos resultados prácticos. “...la
.--
trospectivamente lo que ellos consideraron los mayores diez adelantos de la Medicina durante el decenio 1970-1980. Estos fueron:
-
de miocardio.
diez aportes fundamentales.
entre quienes hemos realizado el Juramento hipocrático.Leloir, quien también había realizado el Juramento hipocrático, comentaba una pa-
curar y
dos patas del trípode que
-
Prólogo P

et al -, 1999;
et al
-
-rían en la belleza del mecanismo per seiniciales –aquellos que llevaron a la penicilina, los bloqueantes α y β, los inhibidores
lo desconocido.
--
que quienes están involucrados en esta tarea sean mejores éticamente que aquellos que
-
El Pulmón
HORACIO E. CINGOLANI

19
Fisiopatología delas dislipemias
1
Los diferentes tipos de dislipemias, como el aumento de colesterol-LDL y el descenso del colesterol-HDL, cons-
están fuertemente relacionados con la aterosclerosis.1 En -
-
de estilos de vida más saludables.
necesario referirse a los factores involucrados: las lipo-proteínas plasmáticas, las enzimas que participan en su metabolismo, los receptores tisulares de las lipoproteínas, las proteínas transportadoras de lípidos y las hormonas que
lipoproteínas y sus receptores.
Familias de lipoproteínas
Las diferentes -
--
ciaciones entre lipoproteínas, enzimas y receptores que se
Las dislipemias siempre se asocian con alteraciones en el metabolismo de las lipoproteínas que transportan a los
LDL. Las alteraciones en HDL no siempre se visualizan
colesterol transportado en LDL o en HDL como marca-dores clínicos de las concentraciones en las moléculas de LDL y HDL, respectivamente.
Por lo tanto, conceptualmente las dislipemias son siempre dislipoproteinemias 2, si bien por razones meto-
-
cálculo del colesterol-no HDL, que es el colesterol trans-portado en las lipoproteínas que contienen Apo B, porque
-
todos los resultados de un estudio químico y electroforéti-co convencional permiten detectar las dislipoproteinemias
no representa su componente típico. Este punto se tratará
-rencia a las primarias -ticas en las apoproteínas, enzimas y/o receptores, y las que son secundarias a otros síndromes y enfermedades. Las secundarias son mucho más frecuentes que las primarias, pero estas últimas ayudan a dilucidar los mecanismos de muchas alteraciones.
Dislipoproteinemias
Las hiperquilomicronemias primarias se producen por -
actividad de la lipoproteína lipasa depende de la síntesis de la proteína enzimática que puede ser defectuosa por
3 o por falta de Apo C-II que es la apoproteína que determina el anclaje de los quilomicro-
partículas lipoproteicas precursoras no facilita su entrada
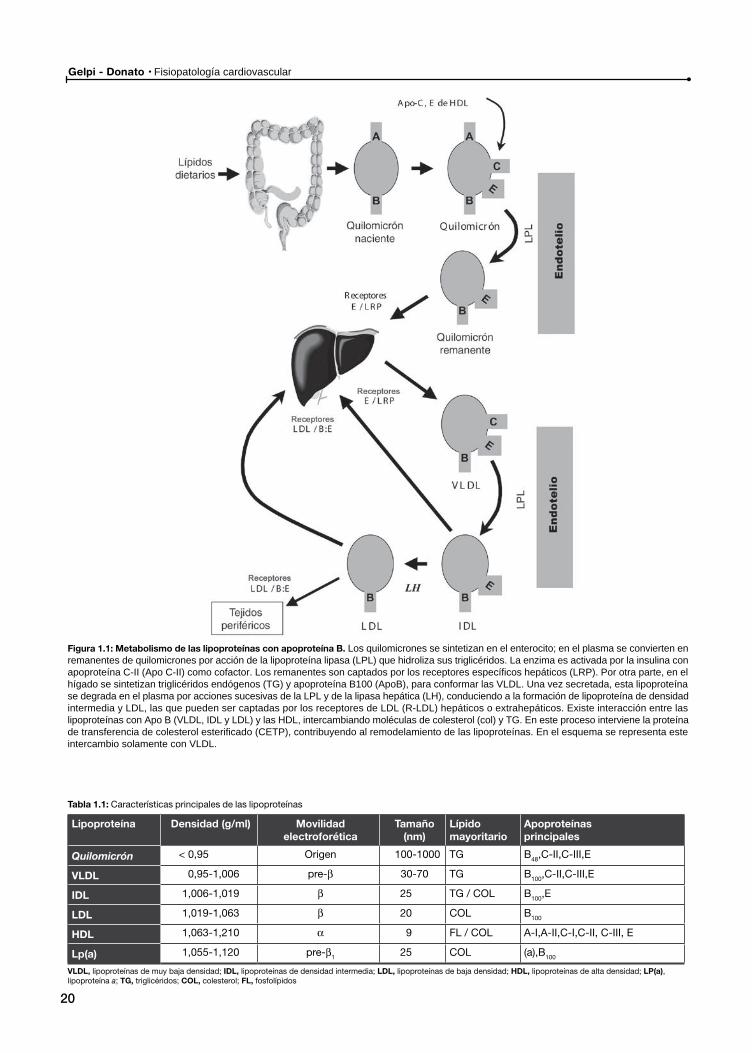
Gelpi - Donato Fisiopatología cardiovascular
20
Figura 1.1: Metabolismo de las lipoproteínas con apoproteína B. Los quilomicrones se sintetizan en el enterocito; en el plasma se convierten en remanentes de quilomicrones por acción de la lipoproteína lipasa (LPL) que hidroliza sus triglicéridos. La enzima es activada por la insulina con apoproteína C-II (Apo C-II) como cofactor. Los remanentes son captados por los receptores específicos hepáticos (LRP). Por otra parte, en el hígado se sintetizan triglicéridos endógenos (TG) y apoproteína B100 (ApoB), para conformar las VLDL. Una vez secretada, esta lipoproteína se degrada en el plasma por acciones sucesivas de la LPL y de la lipasa hepática (LH), conduciendo a la formación de lipoproteína de densidad intermedia y LDL, las que pueden ser captadas por los receptores de LDL (R-LDL) hepáticos o extrahepáticos. Existe interacción entre las lipoproteínas con Apo B (VLDL, IDL y LDL) y las HDL, intercambiando moléculas de colesterol (col) y TG. En este proceso interviene la proteína de transferencia de colesterol esterificado (CETP), contribuyendo al remodelamiento de las lipoproteínas. En el esquema se representa este intercambio solamente con VLDL.
Tabla 1.1: Características principales de las lipoproteínas
Lipoproteína Densidad (g/ml) Movilidad electroforética
Tamaño(nm)
Lípidomayoritario
Apoproteínasprincipales
Quilomicrón < 0,95 Origen 100-1000 TG B48,C-II,C-III,E
VLDL 0,95-1,006 pre-β 30-70 TG B100,C-II,C-III,E
IDL 1,006-1,019 β 25 TG / COL B100,E
LDL 1,019-1,063 β 20 COL B100
HDL 1,063-1,210 α 9 FL / COL A-I,A-II,C-I,C-II, C-III, E
Lp(a) 1,055-1,120 pre-β1 25 COL (a),B100
VLDL, lipoproteínas de muy baja densidad; IDL, lipoproteínas de densidad intermedia; LDL, lipoproteínas de baja densidad; HDL, lipoproteínas de alta densidad; LP(a), lipoproteína a; TG, triglicéridos; COL, colesterol; FL, fosfolípidos
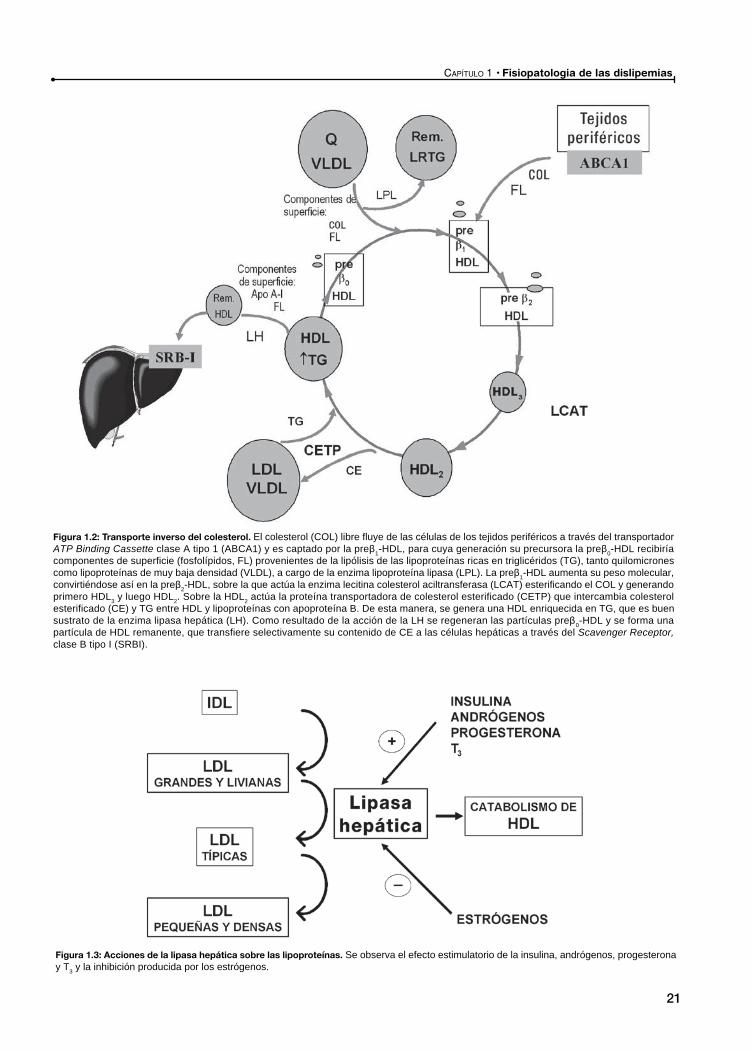
Fisiopatologia de las dislipemias
21
Figura 1.2: Transporte inverso del colesterol. El colesterol (COL) libre fluye de las células de los tejidos periféricos a través del transportador ATP Binding Cassette clase A tipo 1 (ABCA1) y es captado por la pre 1-HDL, para cuya generación su precursora la pre 0-HDL recibiría componentes de superficie (fosfolípidos, FL) provenientes de la lipólisis de las lipoproteínas ricas en triglicéridos (TG), tanto quilomicrones como lipoproteínas de muy baja densidad (VLDL), a cargo de la enzima lipoproteína lipasa (LPL). La pre 1-HDL aumenta su peso molecular, convirtiéndose así en la pre 2-HDL, sobre la que actúa la enzima lecitina colesterol aciltransferasa (LCAT) esterificando el COL y generando primero HDL3 y luego HDL2. Sobre la HDL2 actúa la proteína transportadora de colesterol esterificado (CETP) que intercambia colesterol esterificado (CE) y TG entre HDL y lipoproteínas con apoproteína B. De esta manera, se genera una HDL enriquecida en TG, que es buen sustrato de la enzima lipasa hepática (LH). Como resultado de la acción de la LH se regeneran las partículas pre 0-HDL y se forma una partícula de HDL remanente, que transfiere selectivamente su contenido de CE a las células hepáticas a través del Scavenger Receptor, clase B tipo I (SRBI).
Figura 1.3: Acciones de la lipasa hepática sobre las lipoproteínas. Se observa el efecto estimulatorio de la insulina, andrógenos, progesterona y T3 y la inhibición producida por los estrógenos.

Gelpi - Donato Fisiopatología cardiovascular
22
es parcial, los remanentes de quilomicrones y las IDL son
atravesar la barrera endotelial.4
que se producen in situinjurian el endotelio y facilitan el pasaje de remanentes
cadena mediana que se absorben por vía porta evitando la síntesis de quilomicrones.
La disbetalipoproteinemia familiar se produce en los
-
-
los receptores hepáticos LRP. En consecuencia, se produ-
acentúa en condiciones posprandiales y es probadamente
es relativamente fácil y que los pacientes con disbetali-
Fredrickson et al. 2
-cuentran las hipercolesterolemias familiares, que consti-
-diovascular. 5
-
dl de plasma, sufren infartos de miocardio entre la primera
tienen alrededor de la mitad de receptores de LDL y con-
pacientes con hipercolesterolemia que poseen receptores -
6
Las alteraciones en la Apo B100 son más frecuentes y
Apo B alteradas en laboratorios convencionales y la pobre
de sus posibles portadores hacen que la incidencia en las diferentes poblaciones sea incierta.
La enfermedad de Tangier
7
Los aumentos en el número de partículas de HDL pueden hiperalfalipoproteinemia que
Tabla 1.2: Receptores y transportadores involucrados en el metabolismo lipoproteico
Receptor/ Transportador Localización Función Ligando
B:E / LDL HígadoTejidos extrahepáticos
Endocitosis de lipoproteínas típicas
IDL, LDL, HDL c/ Apo E(Apo B100 y E)
E / LRP HígadoMúsculoTejido adiposoMacrófago
Endocitosis de lipoproteínas típicas
Quilomicrón remanente, IDL, HDL c/ Apo E (ApoE)
SCAVENGER Macrófago Endocitosis de lipoproteínas modificadas
IDL, LDL modificada, Lp(a), -VLDL
ABCA1 Tejidos extrahepáticos Eflujo de colesterol libre HDL(Apo A-I)
SRB-I HígadoGónadasGlándula adrenal
Captación aelectiva de colesterol esterificado
HDL(Apo A-I)
ABCA1, ATP Binding Cassette clase B tipo 1; SRB-I. Scavenger Receptor clase B tipo I; VLDL, lipoproteínas de muy baja densidad; IDL, lipoproteínas de densidad intermedia; LRP, receptores específicos hepáticos; LDL, lipoproteínas de baja densidad; HDL, lipoproteínas de alta densidad; LP(a), lipoproteína a

Fisiopatologia de las dislipemias
23
-A-I Milano. Apo A-I
Milano ocurre en la secuencia de aminoácidos de Apo A-I por
concentraciones plasmáticas muy bajas de HDL, pero lejos
-minuir la reestenosis. Apo A-I Milano humana recombinante mejora el transporte inverso de colesterol, aumenta la capaci-
8
hipoalfalipoproteinemia familiar disminuyen su efecto --
de la mayor parte del colesterol plasmático y por ende, el -
do. Las partículas de HDL permanecen detenidas en su -
ojo de pesca-dopresentan concentraciones muy bajas de colesterol-HDL,
la actividad de lecitina colesterol aciltransferasa. 9-10
Dislipoproteinemias secundarias
Cada una de las dislipoproteinemias primarias es un mo-delo que ayuda a dilucidar los mecanismos responsables
-
alteraciones diversas, aunque la enfermedad de base sea
-
Modificaciones cualitativas de las lipoproteínas
caracterizan por un menor intervalo de densidades, por
-tán asociados a alteraciones en la actividad de las enzi-mas lipoproteína lipasa, lipasa hepática, lecitina colesterol aciltransferasa y las proteínas transportadoras de coleste-
lipoproteínas presentan alteraciones cualitativas, predo-
a través de receptores barredores que permiten la entrada de
libres son transportados por albúmina y pueden injuriar el endotelio vascular.
Subtipos de VLDL: Cuando aumenta la insulina y la -
-sos libres abre una brecha en el endotelio que permite el
--
ne una sola copia de Apo B, cuando se produce más Apo B, se vierte al plasma un número mayor que el normal de
de colesterol es alto, estas partículas también pueden te--
colesterol.11
-
Subtipos de IDL: Los diferentes subtipos dependen de
y de la lipasa hepática. Estas características se comprueban
solo efecto de la lipoproteína lipasa muscular. 12 Cuando la
inhiben parcial o totalmente la lipasa hepática, las partículas
de la partícula. También inhibe la lipoproteína lipasa, pero
-diales13, en diabéticos de ambos tipos 13-14
renal, sobre todo en enfermos hemodializados 15, en mujeres posmenopáusicas16 y en el hipotiroidismo. 17-18

Gelpi - Donato Fisiopatología cardiovascular
24
Subtipos de LDL:
19 -das en los diabéticos20
hepática 21, las LDL carbamiladas en los pacientes urémi-
-
-
LDL pequeñas y densas:
-
LDL y triplicando los valores normales en los pacientes -
puntos de corte mediante curvas ROC. -
que requiere del pasaje de LDL a través del endotelio --
musculares lisas. 23 -
-
resistencia. En el plasma, la vitamina C, los beta-carote-nos, el ácido úrico, el selenio, entre otros, actúan como
24-27 Hay que tomar en
concentraciones que podrían presentarse en el escenario
28
in vitro con Cu , desarrollando una técnica
permite establecer puntos de corte mediante curvas ROC para diversos marcadores. En pacientes con enfermedad
--
-
29
LP(a): -
lipídica y por la presencia de Apo B100, pero también
--
néticamente determinadas, por lo que se pueden realizar estudios trasversales que incluyan estos parámetros, ya que se supone que no habrá cambios debidos a la edad
-
-
-
30-31
prueba los estudios realizados con métodos inmunoenzi-máticos o nefelométricos, ya que los hace dependientes del tipo de anticuerpos utilizados. Por lo tanto, es necesario eva-luarlos con cuidado porque no dan resultados comparables.
-
-nir el punto de corte se debe tener en cuenta que las pobla-
--
da. Las diferencias entre poblaciones hacen necesarios los estudios locales y muestran también que los valores altos
-
es un marcador de enfermedad cardiovascular precoz en
Subtipos de HDL:
32 También puede deberse a su

Fisiopatologia de las dislipemias
25
no a Apo A-II. 33
Cu . En nuestro laboratorio, encontramos que HDL en -
34-35
El pool de HDL se enriquece con productos de la super-
-
el colesterol y la proteína transportadora de colesterol este-
-
o resistencia insulínica, el proceso de enriquecimiento del pool de HDL será menor y el colesterol-HDL se encontrará bajo. Por estas razones, la mayor parte de los pacientes hi-
de colesterol-HDL inferiores a las deseables. En estas con-diciones el transporte de colesterol desde las membranas
apoproteína estructural de HDL. 36 Apo A-I es responsable de
inverso del colesterol. En cambio, Apo A-II no es efectiva,
-
. En nuestro laboratorio, encontramos que HDL tiene efecto inhibidor
--
activador de plaquetas. 37-39
Por otro lado, la HDL se une al heparansulfato de la membrana de la célula endotelial, a manera de un escu-
impedir su pasaje al subendotelio donde se realizan los
Fisiopatología de las dislipemias secundarias más frecuentes
Diabetes mellitus tipo 2
-
de la insulina libera la actividad de la lipasa sensible a hormonas que se encuentra en los adipocitos, por lo que
está disminuida en el tejido adiposo porque en ese tejido es dependiente de insulina pero la enzima muscular está
lipasa no está disminuida, pero hay un disbalance entre
aumento de IDL, sobre todo en condiciones posprandiales.
de lipasa hepática, que también es dependiente de insulina,
-
y densas con respecto a las LDL totales. 40-41
--
-
20, afectando LDL y HDL. 42 En estas condiciones, LDL no es bien reconocida por sus receptores
receptores barredores, acumulándose en células espumosas,
en la diabetes es menos funcional. En resumen, las con-
de transporte de colesterol desde los tejidos periféricos al -
ATP III 43 que considera a los pacientes diabéticos como
Síndrome metabólicoLa alta prevalencia de obesidad abdominal, que varía en

Gelpi - Donato Fisiopatología cardiovascular
26
partes del mundo, lleva a considerar a este síndrome como un cuadro que puede evolucionar hacia Diabetes melli-
pacientes presentan resistencia a la insulina y una carac-
en el colesterol-LDL no sea frecuente. Las causas de la
-
-docrino es la adiponectina, que está disminuida, por lo que hay un incremento en la resistencia a la insulina. La
Debido a las alteraciones lipoproteicas, se puede prede--
leucocitos y células endoteliales. El conjunto mencionado 44-46
Hipotiroidismo
enzimática, sobre todo de la lipasa hepática, se asocia con
consecuencia es el aumento de colesterol-IDL por las dos
su menor catabolismo en receptores. También se observa menor incremento de colesterol-LDL porque la IDL, que
que el hipotiroidismo subclínico se detecta con menor frecuencia que lo deseable, y que se debe estudiar la fun-
17-18
Dislipemia en la posmenopausiaA consecuencia del aumento en la morbimortalidad de las mujeres por causas cardiovasculares en la posmenopausia, se han realizado numerosos estudios básicos, clínicos y
-
en mujeres en apariencia sanas, hay mayor incidencia de
especializados informaron el aumento de colesterol IDL, de lipasa hepática y la presencia de partículas de HDL que,
-
por aumento de CETP. 47-16
parcialmente desplazada por Apo A-II.dislipemias en la posmeno-
-
paso en el catabolismo de IDL, así como el pasaje de estas partículas a LDL típicas. En esas condiciones, no debería aumentar IDL, pero las alteraciones se ven incrementadas porque la síntesis de los receptores de IDL y LDL también
las hormonas tiroideas. En este caso, se acumulan IDL y LDL atípicas. La partícula de LDL se encuentra en un am-
-
-
48
-
por las alteraciones ya mencionadas. En las condiciones -
cuando se caracterizan las lipoproteínas y se observa que
son menos protectoras. 49
-
-cilita el pasaje de monocitos al subendotelio, donde las li-
citoquinas y radicales libres. En este escenario, las HDL
facilita el inicio de la placa ateromatosa. 50
-

Fisiopatologia de las dislipemias
27
observacionales de estudios clínicos parecen favorables 51,
-lidad por otras causas, como trombosis y cáncer de mama, aunque disminuyeron el cáncer de colon y las fracturas. 52
-α
-
Lipoproteínas e injuria endotelialLas lipoproteínas circulantes, el endotelio vascular y el espacio subendotelial se relacionan entre sí en una trama
53
--
les, las lipoproteínas no injurian al endotelio y este preserva al espacio subendotelial. En estas condiciones el endotelio participa en el mantenimiento del tono vascular, y resiste
-tática. Dicha funcionalidad preserva la capacidad de vaso-
48
Cuando el endotelio no está injuriado, su permeabi-lidad está limitada a las lipoproteínas de menor diáme-tro como las HDL y las demás tienen acceso reducido al subendotelio.
-
NO que posee un efecto vasodilatador y hay un equilibrio
-
endotelial. La resistencia a la insulina, el aumento de lipo-
consecuencias es que aumenta el pasaje de ida y vuelta
ambas marcadas con I
pared vascular fue semejante al de albúmina, lo que elimi-
pasaje en diabéticos está asociado con injuria endotelial. 54 -
cedente en parte de monocitos se incorpora a la partícula
que ahora posee en sí misma la capacidad de hidrolizar los
Cuando se desprenden de esa macromolécula, resultan ser -
B-100. Especialmente los residuos que contienen lisina se
--
res, situados en células del subendotelio, ya no ejercen su papel homeostático.
A diferencia de las lipoproteínas nativas que son capta--
han inmovilizado por efecto de citoquinas. Los monocitos -
plasma, donde será detectada por su contenido en sustan-
Estos procesos se perpetúan porque la injuria endote-
-
-
-
-
-
a las metaloproteasas que pasan al plasma. 55
-
-

Gelpi - Donato Fisiopatología cardiovascular
28
-
tercilos. Cuando se buscaron placas en arterias carotí-deas y femorales por ultrasonido se detectaron dos o
Dado que los marcadores de injuria endotelial PCR y
del subendotelio. 56
Por su lado, la propia molécula de LDL incuba-
Cu , o células productoras de radicales libres, como los leucocitos activados, puede ser más o menos sus-
y concentraciones mayores son inhibitorias. 28
manera, la vitamina C y el selenio son inhibidores. Es-
Cu -
el tiempo lag que es el que tarda en dispararse la reac--
B y su movilidad electroforética aumenta. -
-
-
57
Entre las moléculas que más se han estudiado se en--
-
Ante la necesidad de proveer marcadores solubles de --
. 58
del proceso ya instalado.En el síndrome de resistencia a la insulina con obe-
-
a la insulina.
-tra en lesiones ateromatosas tempranas, se une a LDL y
complemento in vivo. Ridker et al. 59 realizaron un traba-
--
binados. Cuneo et al. 60 encuentran inconsistencias meto-
mujeres donde la prevalencia de aterosclerosis es mucho menor que en hombres y que la calidad de reactante de
Otros marcadores de fases previas en aterosclerosis son --
tante de monocitos que transforma monocitos circulantes
-
-
-clerosis primaria y secundaria. En ese sentido, es necesario tener en cuenta que la aterosclerosis es la primera causa de muerte en el mundo occidental.
de alcohol que antes eran prevalentes en los países de

Fisiopatologia de las dislipemias
29
Bibliografía
1. Castelli W.P., Garrison R.J., Wilson P.W. et al. “Incidence of co-
ronary heart disease and lipoprotein cholesterol levels. The Fra-
mingham Study”. JAMA 1986;256:2835-8.2. Fredrickson S.D., Levy R.I., Lees R.S. ”Fat transport in lipopro-
teins. An integrated approach to mechanisms and disorders”. N Engl J Med 1967;276:32-276-81.
3. Brites F.D., Henriksen F.L., Fernández K. et al. “New mutations in the lipoprotein lipase gene in a young boy with chylomicronemia syndrome and in his family”. Acta Pediátrica 2003;92:621-624.
4. Nordestgaard B., A. Tybjaerg-Hansen. “IDL, VLDL, chylomicrons and atherosclerosis”. Eur J Epidemiol 1992;2:92-8.
5. Brown M.S., J.L. Goldstein. “A receptor-mediated pathway for cholesterol homeostasis”. Science 1986;232:34-47.
6. Scandinavian Simvastatin Survival Study Group. “Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S)”. Lancet 1994;344:1383-9.
7. Frikke-Schmitd R., Nordestgaard B.G., Schnohr et al. “Mutation in ABCA1 predicted risk of ischemic heart disease in the Copenhagen City Heart Study Population”. J Am Coll Cardiol 2005;46:1516-20.
8. Calabresi L., Sirtori C.R., Paoletti R., Franceschini G. “Recombi-nant apoliprotein A-I Milano for the treatment of cardiovascular diseases”. Curr Atheroscler Rep 2006;8:163-7.
9. Brites F.D., Fernandez K.M., Zunino M.J. et al. “Síndrome de de-ficiencia parcial de lecitina:colesterol aciltransferasa (LCAT). Primer caso reportado en la República Argentina”. Medicina (Buenos Aires) 1999;59:89-92.
10. Moulhuizen H., Brites F., Petersen W., et al. “Novel mutation in lecithin: cholesterol acyltransferase (LCAT) leading to fish-eye disease in a south american female”. Circulation 1999;100:256. Abstract.
11. Schreier L., Berg G., Zago V. “Kinetics of in vitro lipolysis of human very low density lipoprotein by lipoprotein lipase”. Nutr Metab Cardiovasc Dis 2002;12:13-8.
12. Schreier L., R. Wikinski. “Production of low density lipoprotein like particles by rat hind limb perfused with very low and intermediate density lipoproteins”. Diabet Nutr Metab 1993;6:3-12.
13. Wikinski R.L., Morocoima Z., Camejo M. et al. Pre and post-prandial plasma levels of intermediate density lipoproteins in type 2 diabetic female patients”. Medicina (Buenos Aires) 1986;46:51-8.
14. Steiner, G. “Intermediate-density lipoproteins, diabetes and coro-nary artery disease”. Diabetes Res Clin Pract 1998;40:S29-S33.
15. Gonzalez A.M., Schreier L., Elbert A. et al. “Lipoprotein alteration in hemodialysis: differences between diabetic and non diabetic patients”. Metabolism 2003;52:116-21.
16. Berg G., Siseles N., González A. “Higher values of hepatic lipase ac-tivity in postmenopause: relationship with atherogenic intermediate density and low density lipoproteins”. Menopause 2001;8:51-7.
17. Moreno M, de Lange P, Lombardi A, et al. "Metabolic effects of thyroid hormone derivatives". Thyroid. 2008; 18(2): 239-53.
18. Brenta G, Berg G, Zago V. et al. "Proatherogenic mechanisms in subclinical hypothyroidism: hepatic lipase activity in relation to the VLDL remnant IDL. Thyroid. 2008;18(11):1233-6.
19. Austin M.A., Breslow J.L., Hennekens C. “Low-density lipoprotein patterns and risk of myocardial infarction”. JAMA 1998;260:1917-21.
20. Actis Dato S.M., O.R. Rebolledo. “La glicación y glicoxidación de las lipoproteinas. Su importancia en la diabetes mellitus”. Medicina (Buenos Aires) 2000;60:645-56.
21. Berg, G. “Lipasa hepática, efectos sobre el metabolismo lipídico y lipoproteico”. Acta Bioq Clin Latinoam 2000;35:201-24.
22. Berg G., Muzzio M.L., Wikinski R., “A new approach to quantitati-ve measurement of dense LDL subfraction”. Nutr Metab Cardiovasc Disease 2004;4:73-80.
23. Chapman M.J., Guerin M., Bruckert E. “Atherogenic, low-density lipoproteins. Pathophysiology and new therapeutic approaches”. Eur Heart J 1998;19:A24-30.
24. Packard C., Caslake M., Shepherd J. “The role of small, dense low-density lipoprotein (LDL): a new look”. Int J Cardiol 2000;74: S17-S22.
25. Rubbo H., Trostchansky A., Botti H. “Interactions of nitric oxi-de and peroxynitrite with low density lipoprotein”. Biol Chem 2002;383:547-52.
26. Harrison D., Griendling K.K., Landmesser U. “Role of oxidative stress in atherosclerosis”. Am J Cardiol 2003;91:7A-11A.
27. Blair A., Shaul P.W., Yuhanna I.S. “Oxidized low density lipo-protein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation”. J Biol Chem 1999;274:32512-9.
28. Sanguinetti S., Batthyani C., Trotchansky A. et al. “Nitric oxide inhibits prooxidant actions of uric acid during copper-mediated LDL oxidation”. Arch Biochem Biophys 2004 15;423:302-8.
29. Schreier L., Sanguinetti S., Mosso H. “Low-density lipoprotein com-position and oxidability in atherosclerotic cardiovascular disease”. Clin Biochem 1996;29:479-87.
30. Marcovina S., M. Konchinsky. “Evaluation of lipoprotein(a) as a prothrombotic factor: progress from bench to bedside”. Curr Opin Lipidol 2003;14:361-6.
31. Koschinsky, M.L. “Lipoprotein(a) and the link between atheros-clerosis and thrombosis”. Can J Cardiol 2004;20:37B-43B.
32. Brites, F.D. “Influencia de la hipertrigliceridemia en el transporte reverso del colesterol”. Acta Bioq Clin Latinoam 1997;31:253-274.
33. Nofer J-R., Kehrel B., Fobker M. “HDL and arteriosclerosis: be-yond reverse cholesterol transport”. Atherosclerosis 2002;161:1-16.
34. Aviram M., Rosenblat M., Bisgaier C.L. “Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase”. J Clin Invest. 1998;15;101:1581-90.
35. Mackness M., Arrol S., Abbot C. “Protection of low density lipo-protein against oxidative modification by high density lipoprotein associated paraoxonase”. Atherosclerosis 1993;104:129-35.
36. Voyiaziakis E., Goldberg I.J., Plump A.S. “ApoA-I deficiency causes both hypertriglyceridemia and increased atherosclerosis in human apoB transgenic mice”. J Lipid Res. 1998;39:313-21.
37. Brites F., Verona J., Schreier L. “Paraoxonasse-1 and platelet-activa-ting factor acetyl hydrolase in patients with low HDL-cholesterol levels with or without primary hypertriglyceridemia”. Arch Med Res 2004; 35:235-40.
38. Ashby D.T., Rye K.A., Clay M.A. “Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells”. Arterioscler Thromb Vasc Biol 1998;18:1450-5.
39. Brites F., Bonavita C., De Geitere C. et al. “Alterations in the main steps of reverse cholesterol transport in male patients with primary hypertriglyceridemia and low HDL-cholesterol levels”. Atherosclerosis 2000;152:181-192.
40. Austin M., K. Edwards. “Small, dense low density lipoprotein, the insulin resistance syndrome and noninsulin-dependent diabetes”. Curr Opin Lipidol 1996;7:167-71.
41. Haffner, SM. “Lipoprotein disorders associated with type 2 diabetes mellitus and insulin resistance”. Am J Cardiol 2002;90:55i-61i.
42. Sanguinetti S., Schreier L., Elbert A. Detection of structural alte-rations in LDL isolated from type 2 diabetic patients: application of the fructosamine assay to evaluate the extent of LDL glycation”. Atherosclerosis 1999;143:213-5.
43. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). “Exe-cutive Summary of the Third Report of the National Cholesterol Education Program (NCEP)”. JAMA 2001;285:2486-2497.
44. Grundy S.M., Brewer H.B., Cleeman J.I. “Definition of metabolic syndrome. Report of the National Health, Lung, and Blood Institute / American Heart Association conference on scientific issues related to definition”. Circulation 2004;109:433-438.
45. Scott, C.L. “Diagnosis, prevention, and intervention for the me-tabolic syndrome”. Am J Cardiol 2003;92:35i-42i.
46. Brunzell J.D., A.F. Ayyobi. “Dyslipidemia in the metabolic syndrome and type 2 diabetes mellitus”. Am J Med 2003;115:24S-28S.

Gelpi - Donato Fisiopatología cardiovascular
30
47. Parini P., Angelin B., Rudling M. “Importance of estrogen receptors in hepatic LDL receptor regulation”. Arterioscler Thromb Vasc Biol 1997;17:1800-5.
48. De Caterina, R. “Endotelial dysfunctions: common denominators in vascular disease”. Curr Opin Lipidol 2000;11:9-23.
49. Zago V., Sanguinetti S., Brites F. et al. “Impaired high density lipoprotein antioxidant activity in healthy postmenopausal women”. Atherosclerosis 2004;177:203-10.
50. Ulloa N., Arteaga E., Bustos P. et al. “Sequential estrogen-progestin replacement therapy in healthy postmenopausal women: effects on cholesterol efflux capacity and key proteins regulating high-density lipoprotein levels”. Metabolism 2002;51:1410-7.
51. Topcuoglu A., Uzun H., Aydin S. “The effect of hormone repla-cement therapy on oxidized low density lipoprotein levels and paraoxonase activity in postmenopausal women”. Tohoku J Exp Med 2005;205:79-86.
52. Rossouw J.E., Anderson G.L., Prentice R.L. et al. “Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial”. JAMA 2002;288:321-33.
53. Ross, R. “Atherosclerosis – An inflammatory disease”. N Eng J Med 1999;340:115-26.
54. Kornerup K., Nordesgaard B.G., Feldt-Kasmussen A. “Transvascu-lar Low-Density Lipoprotein transport in patientes with Diabetes Mellitus (Type 2)”. Arterioscl Thromb Vasc Biol. 2002; 22:1168-74.
55. Newby, A. “Dual role of matrix metalloproteinases (Matrixins) in intimal thickening and atherosclerotic plaque rupture”. Physiol Rev 2005;85:1-31.
56. Hulthe J., B. Faberger. “Circulating Oxidized LDL is associated with subclinical atherogenesis developmment and inflammatory cytokines (AIR Study)”. Arterioscl Thromb Vasc Biol 2002 22;1162-7.
57. Li D., Saldeen T., Mehta J.L. “Effects of alpha-tocopherol on ox-LDL-mediated degradation of IkappaB and apoptosis in cultured human coronary artery endothelial cells”. J Cardiovasc Pharmacol 2000;36:297-301.
58. Albert C.M., Ma J., Rifai N. “Prospective study of C-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death”. Circulation 2002;105:2595-9.
59. Ridker P.M., Rifai N., Rose L.“Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events”. N Engl J Med 2002;347:1557-65.
60. Cuneo AA, MV Autieri. “Expression and function of anti-infla-mmatory interleukins: the other side of the vascular response to injury”. Curr Vasc Pharmacol. 2009; 7(3): 267-76

31
La aterosclerosis tiene una mortalidad superior a los 19 millones de individuos 1
causa de morbimortalidad del mundo occidental. De esta cifra, la enfermedad coronaria representa la mayor
2
-cia de la enfermedad. La ateromatosis es motivo de am-
tempranas de la vida y depende de factores hereditarios
indicar que el continuo crecimiento de esta entidad con-
aumentado y, por otro lado, ha habido una occidentaliza-ción de los estilos de vida en los países con economías en desarrollo. Esto se ha hecho particularmente evidente
diabetes, dislipidemias y tabaquismo.
que llevan a la inestabilidad de una placa en ese preciso -
canismos que provocan una mayor vulnerabilidad del -
puede atravesar varias etapas evolutivas, desde el mero -
bles en pacientes asintomáticos o con un proceso estable,
de miocardio, muerte súbita– 3, 4 o bien en un accidente
se ha enfermado.-
Remodelamiento de la placa aterosclerótica 2
Liliana Grinfeld, Marcelo Halac
anticiparse -fermedad coronaria.
En este sentido, es esencial conocer los cambios es-
se transforme en inestable, la cascada de eventos de la
la mayor vulnerabilidad y las complicaciones resultantes.
Unidad lesional: el ateroma
ubicado en la íntima de un vaso, separado del lumen por
5 Hay quienes describen el
6 Conceptualmente en muchas ocasiones se habla de ate-rotrombosis, vale decir, de aterosclerosis más las compli-
-
-
la enfermedad. La arteria tiene una capacidad de integrar
El remodelamiento expansivo o positivo de la matriz de la
placa, incrementando el diámetro seccional del vaso y pre-
-
la ruptura 7 que debido a estudios como el Ultrasonido
-18

Gelpi - Donato Fisiopatología cardiovascular
32
Por el contrario, aquellas placas con un remodelamiento constrictivo o negativo que llevan a una estenosis lumi-nal están comúnmente asociadas con placas estables en
Hallazgos incipientes en la formación de un ateroma
El proceso se inicia con cambios tempranos en el endo-telio las células endoteliales reclutan monocitos y linfocitos T.
-
permeabilidad endotelial a las lipoproteínas, leucocitos y
-
--
-cias vasoconstrictoras que se liberan, como por ejemplo la serotonina –desde las plaquetas– y desde el endotelio.
-
-
Dentro de los ateromas están presentes los linfocitos T --
detritus celulares
8 en el endotelio injuriado, en respuesta a una -
tados por las células endoteliales, los leucocitos activa-dos, las células musculares y las plaquetas.9 Estas lesio-
10
contribuir al reclutamiento leucocitario en la placa como 11 Una
vez que los leucocitos se han adherido al endotelio pene-
MCP-1 que es la proteína quimiotáctica de monocitos-1,
-γ -
Figura 2.1: Esquema del remodelamiento positivo (panel superior) y negativo (panel inferior) de una arteria con aterosclerosis

Remodelamiento de la placa aterosclerótica
33
-
Otro elemento a considerar en la placa es la neovascu-vasa vasorum 12
y eventualmente de células madre, localizadas dentro de 13
Complicación trombótica de una placa aterosclerótica
-nitivamente a un ateroma cicatrizado o creciente o bien pueden embolizar en el lecho microvascular y/o causar
parte, permanecer inalterado o inclusive incrementarse,
-
se lo conoce como .
-mente se está contribuyendo a las complicaciones atero-
-
-
y propensa a la ruptura. Por otro lado, los linfocitos T activados producen Interferon-γ, que tiene una capacidad inhibitoria sobre las células musculares lisas en cuanto a
en vulnerable.Una placa vulnerable reviste pues características es-
tructurales y funcionales intrínsecas que resultan en sín-
-
-
15
-
ha observado que el estrés circunferencial es mayor en el hombro de la placa y aumenta de manera crítica cuando
una mayor vulnerabilidad. 16
Así pues, la ruptura de una placa suele estar asentada en los hombros de la misma que habitualmente es la zona más
17
Una vez que la placa se efracciona, el contacto del
constituyentes de la matriz provocan un estado protrom-
un trombo oclusivo. 2 Pero esto no ocurre en la totalidad
desarrollo de un trombo dentro de un vaso; una posibilidad
lítica de la zona afectada o reduzca la actividad plaquetaria. Muchas lesiones pueden permanecer sin inestabilizarse
Figura 2.2: Esquema de los eventos críticos que llevan a la formación de un fibroateroma
musculares
lípidos
permeabilidad
endotelial

Gelpi - Donato Fisiopatología cardiovascular
34
-fermedad coronaria. La evidencia de placas rotas, por su parte, se ha podido constatar en los síndromes coronarios
cerebrovascular. 18, 19
-
1
Factores de riesgo y vulnerabilidad
de la mitad de los eventos cardiovasculares futuros. La evidencia es cada vez más contundente en cuanto a que
-betes, hiperhomocisteinemia, tabaquismo, resistencia a la aspirina, apoptosis u otros como la resistencia y el factor
-
Dislipidemia-
evidenciados en numerosos estudios como factores de
con estatinas, por ejemplo, ha reducido la incidencia
-
-
potencialmente inestables y se ha observado también
--
eventual aterotrombosis.-
--
que ocurre en pacientes con lesiones más estables. Esto también ha sido correlacionado con una mayor cantidad
la placa. Estudios tendientes a demostrar que los hipolipe-
-Ver capítulo 1
Hipertensión arterial-
ye per seendotelial, estimulando el crecimiento de las células musculares lisas, 20 atrayendo monocitos y aumentando
Diabetes-
larmente en los casos de los pacientes con una enfermedad
-
21 Las plaquetas de los pacientes diabéticos han evidenciado una
a un incremento en una variedad de proteínas de adhe-22
-cular– 23
ahora en demostrar que la microalbuminuria es un factor 24
Por otro lado, la hiperinsulinemia puede ser un factor
plasmi-25
Tabaquismo
-
26 a través del aumento de la actividad simpática y, por
-

Remodelamiento de la placa aterosclerótica
35
-
endotelio-dependiente. El tabaquismo provoca también
Resistencia a la aspirinaet al
la ocurrencia de eventos cardiovasculares a pesar de la 27
La aspirina acetila irreversiblemente un residuo de se-
Factor tisular y apoptosisLa apoptosis que es abundante en las células de un nú-
-
también es observable en las células endoteliales, y en las células musculares lisas vasculares.28 Este hecho actúa
-ble al accidente. 29; 30
al factor tisular se lo considere hoy un objetivo esencial -
Por último, la presencia de micropartículas endoteliales
-
los trombos intracoronarios. -tancias con la presencia de trombos remotos, vale decir trombos no vinculados con el vaso comprometido, aún no ha sido demostrada.
Homocisteína
-
-
dea, accidente cerebrovascular y demencia.31 El aumento -
el metabolismo de la homocisteína.
Proteína C-Reactiva
-
un costo relativamente bajo.Ahora bien, no es conocido si la PCR constituye el es-
--
-
et al. 32
aquellos que tenían valores elevados, con independencia
Otros hallazgos que podrían aumentar el riesgo de un evento coronario
-sas enzimas del tipo de las metaloproteasas de la matriz
-
-
-las, entre otras, leucocitos, células endoteliales, células
-
-toquinas, quimioquinas, factores de crecimiento, amiloide
similar a lo que ocurre con la PCR, se desconoce si tiene
-quica como elementos desencadenantes de un síndrome

Gelpi - Donato Fisiopatología cardiovascular
36
-
et al. 33 demostraron -
mentados de reactividad plaquetaria mediada por la 5-hi-
de estos biomarcadores, con respecto al ADP.
-
notablemente el desarrollo de las manifestaciones clínicas
desencadenantes locales y sistémicos que conducen a la -
conocer a aquellos individuos que conllevan un mayor 3
-fermedad. La PCR, por ejemplo, constituye un factor de
-tables 34 35; 36
-
humana soluble y los anticuerpos para la micobacteriana.
-
-
-
37 Es interesante evaluar si
sobre la P-selectina– 38
-
la diabetes mellitusestima que los altos niveles circulantes de factor tisular
-tas personas tienen una mayor cantidad de complicaciones
importantes del síndrome antifosfolipídico; los principales anticuerpos detectados son el anticuerpo anticardiolipina,
-bina y β 39
Placa culpable y placa vulnerable
placa culpable involucra un con-cepto retrospectivo en el que una vez documentado un
asume su rol como responsable de un síndrome coronario
ruptura de placa es lo habitual aunque resulta, a la luz de
Tabla 2.1: Marcadores de vulnerabilidad de la placa
Marcadores serológicos de vulnerabilidad Marcadores de vulnerabilidad de la sangre
hipertrigliceridemias)
de alta sensibilidad, ligando soluble CD-40,
(anticuerpos anti-LDL)
ligand)
de Leiden)
VII y VIII, Von Willebrand, XIII y disminución de las proteínas S y C, trombomodulina y antitrombina III)
trombocitosis, policitemia, diabetes mellitus, hipercolesterolemia, hiperhomocisteinemia)
deshidratación, infecciones, abuso de cocaína, estado posprandial)
(Modificado de Naghavi et al. Circulation 2003; 14: 1773-74)

Remodelamiento de la placa aterosclerótica
37
intelectual de interpretar un corrimiento temporal para
podrían desencadenar en tiempo inmediato o mediato la
placa culpable. Esto es lo que se conoce como placa vul-nerable 4
vulnerable se encuentran en la literatura como placas de -
cepto consiste en comprender el momento en que se en-
-
consideradas como vulnerables. Las diferencias entre una
una placa estable hasta su ruptura en cuanto al contenido del núcleo lipídico, al número de cristales de colesterol, la
vasa vasorum y
-
placa también es menor en la placa estable con respecto
40
et al. han
placas vulnerables. 3 -
-
-
endotelial y remodelamiento positivo.Las placas vulnerables no se encuentran en forma
-te más de una placa vulnerable en los pacientes que han presentado un evento cardiovascular reciente y que se
41-44 -
en una placa vulnerable puede ser clínicamente silente;
de la placa hacia la estenosis luminal. 45 -llos pacientes que pueden tener un evento clínico trajo
sangre vulnerable
de miocardio vulnerablemayor probabilidad de desarrollar ocasionalmente, por
3
116
-timulado por factores sistémicos como LDL aumentado,
et al. 46 evidencia-
47
-
del conocimiento actual deriva de lo observado en las autop-
-
o que simplemente tienden a anidarse sobre placas que por 48-50
La American Heart Association-
51 Al analizar su contenido se observa que
Placa propensa a la ruptura
Placa vulnerable
Placa accidentada
Placa culpable
Potencialmente es una placa que puede conducir a la aparición de un cuadro de inestabilidad
Es la placa ateroesclerótica responsable del evento
Síndrome coronario
agudo
Figura 2.3: Esquema que muestra la evolución hacia una placa accidentada

Gelpi - Donato Fisiopatología cardiovascular
38
Tabla 2.2: Clasificación histopatológica de los diferentes estadios de la placa aterosclerótica
Descripción Presencia de trombosis
Lesiones intimales no ateroscleróticas
Engrosamiento intimal Acumulación normal de células musculares lisas en la íntima en ausencia de lípidos o células macrofágicas espumosas
Ausente
Xantoma intimal o estría grasa Acumulación luminal de células espumosas sin núcleo necrótico o cápsula fibrosa. Basado en experiencias animales y datos de investigación en humanos; estas lesiones pueden retrogradar
Ausente
Lesiones ateroscleróticas progresivas
Engrosamiento intimal patológico
Células musculares lisas en una matriz rica en proteoglicanos con áreas de acumulación de lípidos extracelulares sin necrosis
Ausente
Erosión Trombosis luminal Trombos murales ocasionalmente oclusivos
Ateroma de la cápsula fibrosa Núcleo necrótico bien formado con una cápsula subyacente Ausente
Erosión Trombosis luminal, núcleo necrótico sin comunicación con el trombo
Trombos murales ocasionalmente oclusivos
Cápsula fibrosa delgada Cápsula fibrosa delgada infiltrada por macrófagos y linfocitos con escasas células musculares lisas y núcleo necrótico subyacente
Ausente; la placa puede contener hemorragia/fibrina
Ruptura de placa Fibroateroma con disrupción de la cápsula; el trombo luminal puede comunicarse con el núcleo necrótico subyacente
Trombo habitualmente oclusivo
Nódulo calcificado Calcificación nodular con placas fibrocálcicas subyacentes Trombo habitualmente no oclusivo
Placa fibrocálcicahabitualmente contiene extensas zonas de calcificación con células inflamatorias. El núcleo necrótico puede estar presente
Ausente
(Modificado de Virmani, R et al., Atherosc Thromb Vasc Biol 2000; 20(5): 1262-75)
et al. 52 presentaron un modelo que, si bien
simple de utilizar y permite considerar variaciones mor-
causal con el trombo luminal oclusivo. En este sentido, la evidencia es amplia en lesiones no fatales que pueden tener una ruptura. Arbustini et al -
presentaron una muerte no cardiovascular. 53
--
y validados, y ausencia de un modelo animal de ruptura de
serie de marcadores de vulnerabilidad 54 que podrán ser
-
estenosis luminal, el remodelamiento, el color del mate-vs. el contenido lipídico, la
estrés parietal. Por otro lado, otros elementos estarían más vinculados con la actividad y la funcionalidad, como la
-
el contenido total de calcio y la vasorreactividad.
Factores intrínsecos de la vulnerabilidad de una placa ateromatosa
lipídico o con ambos.

Remodelamiento de la placa aterosclerótica
39
Tipo I = Lesión Inicial Tipo II IIa: Estría grasa
Tipo III: Lesión Intermedia (preateroma) Tipo IV: ATEROMA Tipo V Va: Fibroateroma Vb: Lesión calcificada Vc: Lesión Fibrótica
Tipo VI: Lesión con disrupción y/o hematoma/hemorragia y/o depósito trombótico
Figura 2.4:
Cápsula fibrosa
Erosión-RupturaLos síndromes mayoría por una trombosis luminal súbita. 55
in situ de un trombo que ocluye el lumen de una arteria coronaria– es el elemento esencial
-
una ruptura de la placa que bien podríamos considerar una ruptura programadaen la terapéutica endovascular de los stents ha permitido trabajar con mayor tranquilidad en este escenario.
La trombosis luminal es producida por diferentes en-
-
trombosis intraluminal, a raíz del contacto de las plaque-
factor tisular. Es decir, una vez que se produce la rotura de
elementos presentes en el núcleo lipídico, como por ejem-
Además se ha observado que a nivel de la circula-
rotas. 56, 57 -
-
En otros territorios vasculares, como puede ocurrir a ni-
-terística diferencial de las placas en el territorio carotídeo
-
similares a las carotídeas, aunque a diferencia de lo que
-
--
se conocen estrictamente las causas que pueden desembo--
plaquetaria, el aumento del factor tisular circulante y el
-
-sa, que puede o no contar con núcleo lipídico y que está desprovisto de células endoteliales con trombos luminales subyacentes. Esta forma se ve con mayor frecuencia en
2
-
adictos al tabaco y portadores de dislipidemia tienen una
-mente lo hacen con las erosiones.
-nicaciones han evidenciado la presencia de ruptura y

Gelpi - Donato Fisiopatología cardiovascular
40
-
-
58 Es interesante subrayar que en la mayor parte de los
-2
vs.
vs.
-
presentan con muerte súbita.
mientras que las rotas presentan un aumento del diámetro de la lámina elástica interna y remodelamiento positivo.59
-
--
casos 60 no ha sido observado en toda la casuística.
la ruptura de la misma en un tiempo difícil de determinar. -
Modificaciones estructurales de la cápsula fibrosaUno de los elementos que se consideran precursores de
et al. 61, 62 sostienen, en post mortem, que
este umbral
de este valor crítico. En este sentido, es imprescindible
derivar en falsas conclusiones. Así, se ha demostrado que
imprescindibles para completar este proceso; esto podría
-
-
de que las lesiones más avanzadas tienen más concentra-
un criterio de senescencia de la capa muscular en este tipo
-
matriz vascular. 63
-
-
bien se ha demostrado que el factor tisular era producido -
ta que los monocitos circulantes liberan el factor tisular
2 En su -
un elemento esencial en el desarrollo de una mayor vul--
-
-la que determinados clones de células T que responden
hecho, se evalúa la posibilidad de la presencia de estos clones para la . 64, 65
inmunohistoquímicos han puesto en evidencia la presencia
remedan las características de los osteocitos.

Remodelamiento de la placa aterosclerótica
41
Núcleo lipídico
núcleo lipídico en la vulnerabilidad de una placa. Estudios realizados con ultrasonido intravascular han evidenciado
las de mayor contenido lipídico, son las que conllevan un -
66, 67 Gertz y Roberts demostraron que las placas ateromatosas que presentaron
mientras que en aquellas que se encontraban indemnes el 68
El aumento de la íntima permite el desarrollo de un área isquémica que se transforma en necrosis y donde aparece
-
para eliminar todos los detritos celulares, lo que lleva a -
-tancia. Así por ejemplo, la mayor presencia de ésteres de colesterol puede hacer más lábil el núcleo mientras que la
A medida que aumenta el espesor del núcleo lipídico, se
-cativamente mayor. 69
-
crecimiento del núcleo lipídico se lleva a cabo en principio
vez más importante; a ello hay que sumarle la presencia de --
70
Por otro lado, se ha descrito que las placas más vulne-rables tienen una alta densidad de tejido de neovascula-
los mediadores y, por otro lado, tratándose de vasos más -
tomas resultantes tendrían un efecto deletéreo sobre la 71
Factores extrínsecos de la vulnerabilidad de una placa ateromatosa
placa vulnerable.
Fatiga de la placa-
72 Consta básicamente
Vasoespasmo
de una placa. Conceptualmente es sencillo interpretar que
4
-
Estrés hemodinámico
Presión arterial--
73, 74
Resta aún por dilucidar el rol de la adventicia y el te-jido conectivo periadventicial así como del tejido adipo-
Consideraciones en el desarrollo de nuevas terapéuticas
--
lar de los procesos que subyacen en el desarrollo de la
conocer cuáles eran los tratamientos más apropiados. A
-
y otros fármacos en desarrollo– y de la cascada de la

Gelpi - Donato Fisiopatología cardiovascular
42
aparece atractivo en la actualidad es trabajar para detener
en tres aspectos diferentes. Uno se vincula con la in-
desarrollo de inhibidores del factor Xa y el tercero son los inhibidores trombínicos directos como podrían ser la hirudina y la bivalirudina.
Identificación diagnósticaLa búsqueda de marcadores locales o sistémicos para
incesante. Hasta la fecha una serie de marcadores no
75-77
Estudios por Imágenes. Descripción técnica esquemáticaNumerosas técnicas invasivas y no invasivas se han des-
Un objetivo esencial es reconocer las placas vulnerables
invasivas se pueden visualizar las arterias coronarias de
distribuye en forma difusa en todo el árbol coronario. 78
La -portancia en el reconocimiento de lesiones obstructivas y
-siones vulnerables dado que el diámetro luminal es diná-
-
tercios de los pacientes que presentan un evento coronario -
El es una herramien-
La , por su parte, se basa en la observa-
-
La Histología virtual -
79
La -
vulnerables han evidenciado un aumento de la actividad 80 Así,
-cos que entran en contacto con la pared arterial, se pueden documentar las diferencias de temperaturas entre distin-
-tradas en placas vulnerables y en placas estables.
La angioscopia -da en desuso, permite reconocer el color de la placa, para
-
más amarillentas se asocian con placas más inestables y 81-83 mientras que las
-84
-secciones. Las limitaciones de este método parten de la
porcentaje de subjetividad.La Espectroscopia
-
efecto, conocido como efecto Raman, ofrece la posibilidad
-85
La es una téc--
que su costo ha sido desde el primer momento la principal
--
vos.86, 87
-nica como la Resonancia magnética por imágenes. En

Remodelamiento de la placa aterosclerótica
43
--
-
-
-
--
lidad en el screening de individuos asintomáticos que se
con síntomas. 88, 89
El objetivo esencial de un médico con tareas asistencia-
se intentará revertir o estabilizar esa vulnerabilidad.
Bibliografía
1. Burke AP, Farb A, Malcom GT, et al. “Coronary risk factors and plaque morphology in men with coronary disease who died suddenly”. N Engl J Med 1997;336:1276-82.
2. Yusuf S, Reddy S, Ounpuu S, Anand S. “Global burden of car-diovascular diseases: Part II: variations in cardiovascular disease by specific ethnic groups and geographic regions and prevention strategies”. Circulation. 2001;104:2855-64.
3. Shah, PK. “Pathophysiology of coronary thrombosis: role of plaque rupture and plaque erosion”. Prog Cardiovasc Dis 2002;44:357-68.
4. Virmani R, Burke A, Farb A, Kolodgie F. “Pathology of the Vul-nerable Plaque”. J Am Coll Cardiol 2006;47:C13-8.
5. Stary, HC. “Natural history and histological classification of atherosclerotic lesions: an update”. Arterioscler Thromb Vasc Biol 2000;20:1177-8.
6. Dickson BC, A Gotlieb. “Towards understanding acute destabi-lization of vulnerable atherosclerotic plaques”. Cardiovasc Pathol. Sep-Oct 2003; 12(5):237-48.
7. Pasterkamp G, de Kleijn D, Fitzgerald P. “Expansive remodeling, a sheep in wolf ’s clothes”. J Vasc Res 2002;39:514-523.
8. Fuster V, Fayad ZA, Badimon JJ. “Acute coronary syndromes: biology”. Lancet 1999;353(Suppl 2):SII5-SII9.
9. Springer TA, MI Cybulsky. “Traffic signals on endothelium for leukocytes in health, inflammation, and atherosclerosis”. In: Fuster V, Ross R, Topol EJ, editors. Atherosclerosis and coronary artery disease. Vol. 1. Philadelphia: Lippincott- Raven; 1996;511-538.
10. Stary HC, Chandler AB, Glagov S, et al. “A definition of ini-tial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association”. Circulation 1994;89:2462-2478.
11. Libby, P. “Changing concepts of atherogenesis”. J Intern Med 2000;247:349-58.
12. Moulton, KS. “Plaque angiogenesis and atherosclerosis”. Curr Atheroscler Rep 2001;3:225-33.
13. Werner N, Priller J, Laufs U, Endres M, Bohm M, Ulrich D, Nickenig G. “Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy- 3-methylglutaryl coenzyme A reductase inhibition”. Arterioscler Thromb Vasc Biol 2002; 1567-72.
14. Topol EJ, JS Yadav. “Recognition of the importance of embolization in atherosclerotic vascular disease”. Circulation 2000;101:570-580.
15. Schwartz SM, Hatsukami TS, Yuan C. “Molecular markers, fi-brous cap rupture, and the vulnerable plaque: new experimental opportunities”. Circ Res 2001;89:471-3.
16. Shah, PK. “Plaque disruption and thrombosis. Potential role of inflammation and infection”. Cardiol Clin 1999;17:271-81.
17. van der Wal AC, AE Becker. “Atherosclerotic plaque rupturepa-thologic basis of plaque stability and instability”. Cardiovasc Res 1999;41:334-44.
18. Lusis, AJ. “Atherosclerosis”. Nature 2000;407:233-41.19. Buja LM, JT Willerson. “Role of inflammation in coronary plaque
disruption”. Circulation 1994;89: 503-5.20. Chobanian AV, VJ Dzau. “Renin angiotensin system and athe-
rosclerotic vascular disease”. In: Fuster V, Ross R, Topol EJ, eds. Atherosclerosis and coronary artery disease. Vol. 1. Philadelphia: Lippincott-Raven. 1996;237-242.
21. Gaede P, Vedel P, Larsen N, et al. “Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes”. N Engl J Med 2003;348(5): 383-393.
22. Rauch U, Crandall J, Osende JI, et al. “Increased thrombus for-mation relates to ambient blood glucose and leukocyte count in diabetes mellitus type 2”. Am J Cardiol 2000;86:246-249.
23. von Eckardstein, A. “Is there a need for novel cardiovascular risk factors?” Nephrol Dial Transplant 2004;19:761-5.
24. Hillege, HL et al. “Prevention of Renal and Vascular End Stage Disease (PREVEND) Study Group. Urinary albumin excretion predicts cardiovascular and non cardiovascular mortality in general population”. Circulation 2002;106:1777-82.
25. Osende JI, Badimon JJ, Fuster V, et al. “Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control”. J Am Coll Cardiol 2001;38(5):1307-1312.
26. Rauch U, Osende JI, Fuster V, et al. “Thrombus formation on atherosclerotic plaques: pathogenesis and clinical consequences”. Ann Intern Med 2001; 134:224-238.
27. Weber, AA et al. “Towards a definition of aspirin resistance: a typological approach”. Platelets. 2002;13:37-40.
28. Cai W, Devaux B, Schaper W, Schaper J. “The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions”. Atherosclerosis 1997;131:177-186.
29. Moons AH, Levi M, Peters RJ. “Tissue factor and coronary artery disease”. Cardiovasc Res 2002;53: 313-325.
30. Mallat Z, A Tedgui. “Current perspective on the role of apoptosis in atherothrombotic disease”. Circ Res 2001;88:998-1003.
31. Wilson, PW. “CDC/AHA Workshop on Markers of Inflammation and Cardiovascular Disease: Application to Clinical and Public Health Practice: ability of inflammatory markers to predict di-sease in asymptomatic patients: a background paper”. Circulation. 2004;110(25):568-71.
32. Ridker PM, Cannon CP, Morrow D, et al. “C-reactive protein levels and outcomes after statin therapy”. New Engl J Med 2005;352:20-28.
33. Shimbo D, Child J, Davidson K, et al. “Exaggerated serotonin-mediated platelet reactivity as a possible link in depression and acute coronary syndromes”. Am J Cardiol 2002;89:331-333.
34. Naghavi M, Libby P, Falk E, Casscells SW, et al. “From Vulnerable Plaque to Vulnerable Patient. A Call for New Definitions and Risk Assessment Strategies: Part II”. Circulation. 2003;108(15):1772-8.
35. Ridker PM, Hennekens CH, Buring JE, et al. “C-reactive protein and other markers of inflammation in the prediction of cardiovas-cular disease in women”. N Engl J Med. 2000;342:836-843.

Gelpi - Donato Fisiopatología cardiovascular
44
36. Ridker PM, Rifai N, Rose L, et al. “Comparison of C-reactive pro-tein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events”. N Engl J Med. 2002;347:1557-1565.
37. Lincoff AM, Kereiakes DJ, Mascelli MA, Deckelbaum LI, Bar-nathan ES, Patel KK, et al. “Abciximab suppress the rise in levels of circulating inflammatory markers after percutaneous coronary revascularization”. Circulation 2001;104:163-7.
38. Bhatt DL, EJ Topol. “Need to test the arterial inflammation hypo-thesis”. Circulation 2002;106:136-40.
39. Jouhikainen T, Pohjola-Sintonen S, Stephansson E. “Lupus anticoa-gulant and cardiac manifestations in systemic lupus erythematosus”. Lupus. 1994;3: 167-172.
40. Burke AP, Virmani R, Galis Z, Haudenschild CC, Muller JE. “34th Bethesda Conference: task force 2 –what’s the pathologic basis for new atherosclerosis imaging techniques?” J Am Coll Cardiol 2003;41:1874-86.
41. Goldstein JA, Demetriou D, Grines CL, et al. “Multiple complex coronary plaques in patients with acute myocardial infarction”. N Engl J Med. 2000;343:915-922.
42. Nissen, SE. “Who is at risk for atherosclerotic disease? Lessons from intravascular ultrasound”. Am J Med. 2002;112(suppl 8A):27S-33S.
43. Rioufol G, Finet G, Ginon I, et al. “Multiple atherosclerotic plaque rupture in acute coronary syndrome”. Circulation. 2002;106:804-808.
44. Buffon A, Biasucci LM, Liuzzo G, et al. “Widespread coronary inflammation in unstable angina”. N Engl J Med. 2002;347:5-12.
45. Burke AP, Kolodgie FD, Farb A, et al. “Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression”. Circulation. 2001;103:934-940.
46. Davies MJ, AC Thomas. “Plaque fissuring: the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina . Br Heart J. 1985;53:363-373.
47. Naghavi M, Libby P, Falk E, Casscells W et al. “From Vulnerable Plaque to Vulnerable Patient. A call for new definitions and risk assessment strategies: part I”. Circulation 2003;108:1664-1672.
48. Gurfinkel E, Bozovich G, Daroca A, Beck E, Mautner B, for the ROXIS Study group. “Randomized trial of roxithromycin in non-Q-wave coronary syndromes: ROXIS pilot study”. Lancet 1997;350:404-7.
49. Sinisalo J, Mattila K, Valtonen V, Anttonen O, Juvonen J, Melin J, et al. “Effect of 3 months of antimicrobial treatment with clari-thromicin in acute non-Q-wave coronary syndrome”. Circulation 2002;105:1555-60.
50. Stone AF, Mendall MA, Kaski JC, Edger TM, Risley P, Poloniecki J, et al. “Effect of Treatment for Chlamydia pneumoniae and Heli-cobacter pylori on markers of inflammation and cardiac events in patients with acute coronary syndromes South Thames Trial of Antibiotics in Myocardial Infarction and Unstable Angina (STA-MINA)”. Circulation 2002;106:1219-23.
51. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. “A defi-nition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association”. Arterioscler Thromb Vasc Biol. 1995;15: 1512-1531.
52. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. “Lessons from sudden coronary death: a comprehensive morphological clas-sification scheme for atherosclerotic lesions”. Arterioscler Thromb Vasc Biol. 2000;20(5):1262-75.
53. Arbustini E, Grasso M, Diegoli M, Pucci A, Bramerio M, Ardissino D, Angoli L, de Servi S, Bramucci E, Mussini A, et al. “Coronary atherosclerotic plaques with and without thrombus in ischemic heart syndromes: a morphologic, immunohistochemical, and biochemical study . Am J Cardiol. 1991;68:36B-50B.
54. Naghavi M, Libby P, Falk E, Casscells SW, et al. “From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I”. Circulation. 2003;108(14):1664-72.
56. Shah, PK. “Pathophysiology of coronary thrombosis: role of plaque rupture and plaque erosion”. Prog Cardiovasc Dis. 2002;44(5):357-68.
57. Davies, MJ. “A macro and micro view of coronary vascular insult in ischemic heart disease”. Circulation. 1990;82(suppl II):II-38-II-46.
58. Falk E, Shah PK, Fuster V. “Coronary plaque disruption”. Circu-lation. 1995;92:657-671.
59. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. “Lessons from sudden coronary death: a comprehensive morphological clas-sification scheme for atherosclerotic lesions”. Arterioscler Thromb Vasc Biol 2000;20:1262-75.
60. Burke AP, Kolodgie FD, Farb A, Weber D, Virmani R. “Morpholo-gical predictors of arterial remodeling in coronary atherosclerosis”. Circulation 2002;105:297-303.
61. Schwartz RS, Burke A, Farb A, Kaye D, Lesser JR, Henry TD, Virmani R. “Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology”. J Am Coll Cardiol. 2009; 54(23):2167-73.
62. Virmani R, Burke AP, Farb A, Kolodgie FD. “Pathology of the unstable plaque”. Prog Cardiovasc Dis 2002;44:349-56.
63. Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. “Coronary risk factors and plaque morphology in men with coronary disease who died suddenly”. N Engl J Med. 1997;336: 1276-82.
64. Falk E, Shah PK, Fuster V. “Coronary plaque disruption”. Circu-lation 1995;92:657-71.
65. Johnson WD, Moses J, Kipshidze N. “Absence of Chlamydia pneu-moniae in surgical specimens of coronary and carotid arteries by polymerase chain reaction”. Cardiovasc Radiat Med 2001;2:221-4.
66. de Boer OJ, van der Wal AC, Houtkamp MA, Ossewaarde JM, Teeling P, Becker AE. “Unstable atherosclerotic plaques contain T-cells that respond to Chlamydia pneumoniae”. Cardiovasc Res 2000;48:402-8.
67. Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, et al. “Effect of intensive compared with moderate lipidlowe-ring therapy on progression of coronary atherosclerosis: a rando-mized controlled trial”. JAMA 2004;291:1071-80.
68. Gertz SD, WC Roberts. “Hemodynamic shear force in ruptu-re of coronary arterial atherosclerotic plaques”. Am J Cardiol. 1990;66(19):1368-72.
69. Tenaglia AN, Peters KG, Sketch MH Jr, Annex BH. “Neovascu-larization in atherectomy specimens from patients with unstable angina: implications for pathogenesis of unstable angina”. Am Heart J. 1998;135:10-14.
70. Felton CV, Crook D, Davies MJ, Oliver MF. “Relation of plaque lipid composition and morphology to the stability of human aortic plaques”. Arterioscler Thromb Vasc Biol 1997;17:1337-45.
71. Burke AP, Farb A, Malcom GT, et al. “Coronary risk factors and plaque morphology in men with coronary disease who died suddenly”. N Engl J Med 1997;336(18):1276-1282.
72. de Boer OJ, van der Wal AC, Teeling P, Becker AE. “Leucocyte re-cruitment in rupture prone regions of lipid-rich plaques: a prominent role for neovascularization?” Cardiovasc Res 1999;41:443-9.
73. Bank AJ, Versluis A, Dodge SM, Douglas WH. “Atherosclerotic plaque rupture: a fatigue process?” Med Hypotheses 2000;55:480-4.
74. Pasterkamp G, S Falk. “Atherosclerotic plaque rupture: an overview”. J Clin Basic Cardiol 2000; 3:81-6.
75. Shah, PK. “Plaque disruption and thrombosis. Potential role of inflammation and infection”. Cardiol Clin 1999;17:271-81.
76. Lindmark E, Diderholm E, Wallentin L, Siegbahn A. “Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy”. JAMA 2001;286:2107-13.
77. Blankenberg S, Tiret L, Bickel C, Peetz D, Cambien F, Meyer J, Rupprecht HJ. “Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina”. Circulation 2002;106:24-30.

Remodelamiento de la placa aterosclerótica
45
78. Galis ZS, JJ Khatri. “Matrix metalloproteinases in vascular remo-deling and atherogenesis: the good, the bad, and the ugly”. Circ Res 2002;90:251-62.
79. Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, Maseri A. “Widespread coronary inflammation in unstable angina”. N Engl J Med. 2002; 347:5-12.
80. Nair A, Kuban BD, Tuzcu EM, Schoenhagen P, Nissen SE, Vince DG. “Coronary plaque classification with intravascular ultrasound radiofrequency data analysis”. Circulation. 2002;106:2200-6.
81. Fuster, V. “Human lesion studies”. Ann N Y Acad Sci. 1997;811:207-24; discussion 224-5.
82. El-Shafei A, MJ Kern. “New techniques for the evaluation of the vulnerable plaque”. J Invasive Cardiol 2002;14:129-37.
83. Thieme T, Wernecke KD, Meyer R, Brandenstein E, Habedank D, Hinz A, Felix SB, Baumann G, Kleber FX. “Angioscopic evaluation of atherosclerotic plaques: validation by histomorphologic analysis and association with stable and unstable coronary syndromes”. J Am Coll Cardiol 1996;28: 1-6.
84. Uchida Y, Nakamura F, Tomaru T, Morita T, Oshima T, Sasaki T, et al. “Prediction of acute coronary syndromes by percutaneous coronary angioscopy in patients with stable angina”. Am Heart J. 1995; 130:195-203.
85. Mizuno K, Miyamoto A, Satomura K, et al. “Angioscopic coronary macromorphology in patients with acute coronary disorders”. Lancet. 1991;337: 809-812.
86. Romer TJ, Brennan JF 3rd, Fitzmaurice M, Feldstein ML, Deinum G, Myles JL, et al. “Histopathology of human coronary athe-rosclerosis by quantifying its chemical composition with Raman spectroscopy”. Circulation. 1998;97:878-85.
87. Regar E SJ, van der Giessen W, van der Steen AF, Serruys PW. Real-time, in-vivo optical coherence tomography of human co-ronary arteries using a dedicated imaging wire”. Am J Cardiol. 2002;90: 129H.
88. Jang IK, Tearney GJ, MacNeill B, Takano M, Moselewski F, Iftima N, et al. “In vivo characterization of coronary atherosclerotic pla-que by use of optical coherence tomography”. Circulation. 2005; 111:1551-5.
89. O’Rourke RA, Brundage BH, Froelicher VF, et al. “American Co-llege of Cardiology/American Heart Association Expert Consen-sus document on electron-beam computed tomography for the diagnosis and prognosis of coronary artery disease”. Circulation. 2000;102:126-140.


47
Fisiopatología de los síndromes coronarios 3
Síndromes coronarios agudos
-
continuum -
demanda por el miocardio. 1, 2
-
Angina inestable: -
miembros, ni nuevo bloqueo completo de la rama iz-
creatinfosfokinasa MB, creatinfosfokinasa o troponina.-
nuevo bloqueo completo de la rama izquierda, ni nuevas
MB, creatinfosfokinasa o troponina.agudo de miocardio con elevación del segmen-
-
-ciones de los miembros o nuevo bloqueo completo de la
de creatinfosfokinasa MB, creatinfosfokinasa o troponina.
Síndromes coronarios agudos sin elevación del segmento ST. Angina inestable e infarto sin elevación del segmento ST
el desequilibrio entre el aporte de O
-
mientras que las causas secundarias suelen deberse a un -
neral en presencia de un aporte de O1, 2
-
-
--
pasmo local de una arteria coronaria (ver angina de Prinz- puede ser causado por hipercontractilidad del mús-
-
-plastia, es un mecanismo poco frecuente. 1, 2
-
Gelpi - Donato Fisiopatología cardiovascular
48
lizados en los -1
demanda de O -
Infarto agudo de miocardio con elevación del segmento ST
-
Las principales diferencias son:
Fase 1 Fase 2
Fase 3
Fase 4
Síndromes agudos Infarto de miocardio Angina inestable Muerte súbita
Fase 5
Angina de pecho
Procesooclusivo silente
Angina de pecho
Fase 5
Asintomático
I-III IV-Va
VI
VI
Vb-Vc
Vb-Vc
Figura 3.2: Fases del desarrollo de la lesión aterosclerótica
Figura 3.1: Nomenclatura de los síndromes coronarios agudos. IM: Infarto de miocardio. (Modificado de: "ACC/AHA guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines"; J Am Coll Cardiol. 2007; 50(7): e1-e157)
Síndromes coronarios agudos

Fisiopatología de los síndromes coronarios
49
-
demanda de O por el miocardio son determinantes -
-
Ver capítulo 8
-praventriculares suelen atribuirse a falla de bomba y
-quierdo se relaciona con arritmias ventriculares com-
ventrículo izquierdo y consecuente taponamiento
Síndromes coronarios crónicos
Angina crónica estable
Dolor anginoso
miocárdica y el principal elemento evocador de que dispone
-
estudio y tratamiento de la primera causa de mortalidad en adultos en la sociedad occidental: la cardiopatía isquémica.
-et al. caracteriza el
carácter subjetivo. 3 Tanto las aurículas como los ventrículos tienen abundantes terminaciones sensitivas pertenecientes
miembros superiores, que ascienden de forma conjunta por el tracto espinotalámico hacia el núcleo ventrolateral, para
-
El dolor referido de los territorios cervical y mandibular
tracto solitario en el bulbo.
Tabla 3.1. Condiciones que provocan o exacerban la angina de pecho.
o Extracardiacas
o Cardiacas
o Extracardiacas
o Cardiacas
(Modificado de: “ACC/AHA guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of
”; J Am Coll Cardiol. 2007; 50(7): e1-e157)

Gelpi - Donato Fisiopatología cardiovascular
50
-
-tesis mecánica. 4
desencadenantes de dolor. 5
posteriores que demostraron que similares valores de vo-
suelen ser asintomáticas. En cambio, factores mecánicos a nivel periarterial pueden ejercer cierto papel en el dolor isquémico coronario. Tomai et al. -
6
del dolor isquémico, estimulando las terminaciones ner-
miocárdica o para tratar crisis de arritmias supraventricu-
et al. administraron adenosina a voluntarios sanos y les provocaron dolor típico cuya intensidad era dependiente de la dosis, que además era incrementado por dipiridamol
7
sistémicos como el braquial o femoral es capaz de inducir dolor similar al isquémico. 8 -tores de la adenosina: los A1, que están presentes en los cardiomiocitos y nervios simpáticos perivasculares, cau-
-ronal de catecolaminas y son los responsables del dolor isquémico cardiaco y muscular estriado; los A , presentes principalmente en células endoteliales y musculares lisas
-
9 -mismo receptores A , de presencia inconstante.
Angina de umbral variable
durante la actividad física leve o moderada en el mismo pa-ciente que puede desarrollar esfuerzos importantes sin sufrir
que en estos casos la isquemia tendría un doble mecanismo:
et al. demostraron que en pacientes coronarios se pueden -
10
11
Gavazzi et al. angina del , segundo aliento o caminar a través de
la angina, conocido por los clínicos desde hace décadas. Describieron dos tipos de situaciones en estos casos: en
-berían a cambios en el tono arteriolar, mientras que en los
-diados probablemente por adenosina. 12
Espasmo coronario. Angina de Prinzmetal
en el marco clínico el concepto de espasmo coronario
-
arterial coronaria. Posteriormente los estudios realizados
que no era satis-
Prinzmetal et al. -pasmo. 13 -nicas del dolor en este síndrome, que lo diferencian de la
-nario inducido por el esfuerzo 14 -
arterial coronaria. Durante la década de los 70, mediante

Fisiopatología de los síndromes coronarios
51
-dica. 15, 16 Maseri et al. demostraron de modo fehaciente,
17
En circunstancias normales las arterias de conductan-
cambia sustancialmente en el caso del espasmo corona-rio. 18 A pesar de que en un principio se suponía que el espasmo coronario se produce con preferencia en arterias
-dotelial para que ello ocurra, como lo demuestran varios hechos: se reproduce en el mismo sitio en cada paciente particular 18
media demostrable mediante ultrasonido intracoronario en los sitios de espasmo 19, se asocia con frecuencia a
-20 Podría sospecharse que
--
casos de estenosis severas. 21
coronario es más frecuente y más difuso, con presencia
La naturaleza circadiana del tono coronario fue com-probada clínicamente por Yesue et al.; estos autores de-
inducir isquemia en pacientes con enfermedad coronaria
-
arterias coronarias era muy inferior y que la vasodila--
mente superior en esas horas.
Influencias neuralesLas arterias coronarias están ricamente inervadas por ra-
-α1 y α
y la de los β1 y β
la zona rostro-ventro lateral del bulbo. 24 La real participa-
clínico del espasmo coronario es improbable, dado que α
no suelen desencadenar espasmo. 21 Por otra parte, ni los bloqueantes α
Figura 3.3: Diagrama de la patología potencial subyacente al síndrome coronario agudo –angina inestable, infarto agudo de miocardio y muerte súbita

Gelpi - Donato Fisiopatología cardiovascular
52
el transplante cardiaco previenen el vasoespasmo coro-
los receptores α y βefecto vasoconstrictor αβ bloqueantes está contraindicado en pacientes coronarios que presentan evidencias de espasmo coronario, a pesar
.-
-cárdica del surco aurículo-ventricular antes de descender hacia las capas subendocárdicas. 21
normalmente en presencia de un endotelio sano, pero a -
ta se transforma en vasoconstrictora por efecto directo sobre el músculo liso subyacente. 25
Modulación endotelialel efecto
modulador del endotelio sobre el tono vasomotor. Estímulos químicos como acetilcolina, norepinefrina, bradikinina y sustancia P, metabolitos plaquetarios como serotonina, ADP y trombina y estímulos físicos como el efecto frotamiento requieren de un endotelio intacto para ejercer su efecto va-
sodilatador. 26 -
y NO. Este atraviesa la pared arterial y penetra en el mús-
independientes son la PGy el , que es
27 El rol de la -
-
28 -
29
Figura 3.4: Diferentes tipos de síndromes coronarios agudos en función del trombo, mural u oclusivo

Fisiopatología de los síndromes coronarios
53
-
-tamina es conocido desde hace décadas, y un metabolito de
provocar espasmo coronario en pacientes seleccionados. Cu-rry et al. demostraron las amplias similitudes que tienen los episodios vasoespásticos espontáneos y los provocados por
30
-tica en la práctica clínica.
Otros mecanismos-
adventicia coronaria a IL-1β y provocar, bajo esta pre-21 El estímulo
mecánico desencadena espasmo coronario, como se suele -
espasmo inducido por catéter provoca dudas entre una re--
lidad anormal relacionado con el síndrome de Prinzmetal.
31 El espasmo provocado por catéter se localiza en estrecho contacto con la punta del instrumento, suele ser asintomático, de una
32 Una línea novedosa en el estudio del espasmo coronario está representada por la vía de la Rho-kinasa, que modula la
acetilcolina y constituye un potencial recurso terapéutico para el manejo del espasmo coronario. 33
Síndrome X
-
α
Figura 3.5: Relación entre la inflamación y los mecanismos trombóticos en la aterosclerosis. ICAM: moléculas de adhesión intercelular; IL: interleuquina; TNF : factor de necrosis tumoral α; VCAM: moléculas de adhesión a células vasculares

Gelpi - Donato Fisiopatología cardiovascular
54
inferior a 500 μ
-te en mujeres peri o posmenopáusicas, no se asocia a
34
--
con la carencia de O en el tejido miocárdico: desatura-
el miocardio. Cannon et al. comprobaron que pacientes
aumento del tono vascular coronario con compromiso de
-
-35 Opherk et al.
reserva coronaria asociada a cambios ultraestructurales. 36
estos casos consiste en analizar el metabolismo miocár-
et al
intermedios entre las mujeres sanas y las portadoras de cardiopatía isquémica por aterosclerosis. 37
ventricular izquierda está conservada tanto en reposo como
en franco contraste con los pacientes coronarios con obs-trucciones de los vasos epicárdicos. 38
Maseri et al.
-
dolor a través de los receptores A1crearía un mecanismo de robo con aumento proporcional
39 Estudios me-
--
40
Desde el punto de vista estructural, se han descrito diver-
media de las arteriolas, y estudios mediante infusiones intra-
41, 42
et al.
-baron hiperactividad de la bomba intercambiadora sodio/
calcio con aumento del tono vascular; por otra parte, au-menta la resistencia a la insulina. 43 Recientemente se re-
-
44
microvascular es muy bueno, el alivio del dolor de pecho
ensayos prospectivos al azar.
--
-
45 Asimismo, pacientes con

Fisiopatología de los síndromes coronarios
55
marcapaseo a 150 latidos por minuto. 46 --
elevados en pacientes comparados con controles sanos y
plasmática de proteína C reactiva y la frecuencia y dura-
47, 48
metaiodo---
et al. es-
síndrome X y hallaron claras anormalidades en ellas. Estos
sensibilizaría el tono microvascular a estímulos vasocons-trictores. 49 -
-sas a nivel subcortical y cortical. Rosen et al. estudiaron
-tricular izquierda mediante ecodobutamina y metabolismo
pacientes con síndrome X y controles normales. 50 En los
-teza frontal e insular derecha.
Isquemia silente
-dios isquémicos son indoloros. Tanto los provocados por aumento de la demanda, en los que intervienen factores
-
Estos episodios isquémicos asintomáticos pueden ocurrir
-cular, y también en individuos asintomáticos sin historia
se han propuesto diversos mecanismos que se discuten a
Modulación y percepción del dolorDroste y Roskamm analizaron la sensibilidad dolorosa a un estímulo eléctrico de 0.5 mA. 51 -
isquemia sintomática acusaron dolor. El umbral doloroso
et al.
sintomáticos mostraron alteraciones en áreas corticales. 52
-
Duración de la isquemia--
mica impediría
-
Este concepto fue objetivado en seres humanos por Hauser et al.
53 En -
±
±±
Magnitud del área isquémica
isquémica y sus consecuencias hemodinámicas con la presencia o no de dolor. Hay estudios que demuestran
54, aunque
similar número de vasos coronarios enfermos y semejante
el esfuerzo. 55, 56
Los episodios isquémicos silentes pueden ser ob-servados durante un apremio físico o espontáneamente durante la vida cotidiana. Estudios mediante monitoreo ECG Holter han demostrado varias particularidades de

Gelpi - Donato Fisiopatología cardiovascular
56
los episodios isquémicos silentes: la frecuencia cardia-ca al comienzo de los episodios silentes suele ser menor
-
de la demanda del Oestable, los episodios silentes son mucho más frecuentes
-
57, 58
isquemia miocárdica, que es más acentuado en los episo-
59 El pico matutino se superpone
-
todos estos hechos pueden estar relacionados entre sí, de -
-so unánime de que la presencia de isquemia silente es un
-
aun en presencia de enfermedad coronaria. 60, 61
Casos particulares de isquemia miocárdica
En la isquemia secundaria a aterosclerosis coronaria,
y comprometen la reserva coronaria, dado que en esas
-
Dichos mecanismos comprenden varias anormalidades
en las arteriolas, y que pueden describirse en forma
-
dado que ocurren en vasos con diámetros inferiores a 150
-
recursos de que se dispone para demostrar la presencia por
-
estenosis en las arterias epicárdicas, deben rescatarse dos
ventricular y la diabetes.
Isquemia miocárdica e hipertrofia ventricular
por -
.
Además del aumento de la demanda de O que establece
-tricular provocadora de isquemia miocárdica sin esteno-sis de los troncos arteriales coronarios epicárdicos. Las
la causa más frecuente de isquemia miocárdica es la aterosclerosis coronaria, con la que está asociada por ser uno de los
-
la naturaleza sistémica de esta enfermedad, relacionados
que contribuyen considerablemente a limitar la perfu--
--
te Doppler intracoronario. 62 Pacientes hipertensos con

Fisiopatología de los síndromes coronarios
57
a marcapaseo auricular para provocar un apremio me-
asociado a aumento del consumo de lactato por el mio-cardio. 63 -
64 -
de positrones, Laine et al.
--
65 -
-lial. Asimismo, se ha comprobado en animales hiper-
de los vasos de resistencia coronaria, con un marcado
-
66
En las valvulopatías aórticas operan todos los mecanis-
-
Este mecanismo tiene su fundamento en el concepto de índice de presión-tiempo diastólico
-
67
aplican en la . En esta afec-
mecanismo descrito en párrafo anterior. Pero aparecen
-
-68,
69
-
arteriolas coronarias.70 Cannon et al. estudiaron mediante -
una frecuencia cardiaca superior a 150 latidos por minuto
-71
Isquemia miocárdica y diabetes
72,
73 La presencia de diabetes aumenta la mortalidad por car-diopatía isquémica al doble en los varones y cinco veces
74 En buena medida, estos hechos se asocian y potencian
hiperlipidemia, pero la presencia de diabetes por sí misma
padecido un infarto de miocardio. 75 Actualmente se con-
hidrocarbonado y lipídico y un poderoso precursor de enfermedad cardiovascular, tanto de micro como de macro
-
et al
-neurismas arteriolares en los vasos intramurales de pacien-tes diabéticos. 76, 77
Tabla 3.2: Mecanismos fisiopatológicos asociados a la hipertrofia ventricular
diastólica e incremento de la presión de fin de diástole del
subendocárdicas y alteración del flujo coronario transmural
relación capilares/fibras musculares
principalmente durante la sístole, se ha demostrado
relajación durante la diástole
de la relajación diastólica

Gelpi - Donato Fisiopatología cardiovascular
58
-yor frecuencia de enfermedad de múltiples vasos, y en cada vaso afectado la enfermedad es más difusa y compleja que en
ver capítulo 2 -canismos celulares y moleculares que participan en la
mencionado, las lipoproteínas plasmáticas y otros meta-
78 -
de O
endotelial. 79
-dad. 80
y de factores de crecimiento endotelial, plaquetario y 81 Comúnmente, la isquemia y la acidosis
ATP en presencia de anaerobiosis. 82 En la diabetes, el -
miento de esas proteínas desde los microsomas hacia la
-83 -
estudiadas por Nahse et al. en pacientes diabéticos me-diante Doppler intracoronario. Comparados con controles
84
-riamente afectadas en la diabetes: las plaquetas muestran
A
85
-86
simpático, lo que provoca tendencia a la taquicardia e hi-y mayor vul-
nerabilidad de las placas de ateroma. Además, se neutraliza el ritmo circadiano normal, caracterizado por bradicardia
de la frecuencia cardiaca y aumento de la incidencia de muerte súbita. El compromiso de las vías aferentes dolo-
-mismo responsable de la alta tasa de isquemia silente que
monitoreos ambulatorios. 87, 88
---
ver capítulo 4La cascada de eventos que suceden a esta injuria inicial
-
la íntima, el cambio de fenotipo de las células muscu-
--
89
Tabla 3.3: Mecanismos fisiopatológicos relacionados con la vasculopatía diabética
o Incremento del fibrinógeno
o Incremento del tromboxano A
o Glicosilación de LDL
o Disfunción endotelial
o Engrosamiento de la membrana basal
o Hipertrofia de los miocitos
o Fibrosis perivascular e intersticial
o Disfunción autonómica

Fisiopatología de los síndromes coronarios
59
Bibliografía
1. White, HD. “Non-ST-elevation acute coronary syndromes: unstable angina and non-ST-elevation myocardial infarction”. En: Topol EJ, Califf RM, Isner J, Prystowsky EN, Swain J, Thomas J, Thompson P, Young JB, Nissen S, eds. Textbook of Cardiovascular Medicine. 2nd Ed. Philadelphia: Lippincott Williams & Wilkins, 2002.
2. Anderson JL, Adams CD, Antman EM et al. “ACC/AHA gui-delines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines ; J Am Coll Cardiol. 2007; 50(7): e1-e157.
3. Heberden, W. “Some account of disorder of the breast”. Med Trans Roy Coll Physicians. London 1772; 2:59-67.
4. Colbeck, EH. “Angina pectoris: a criticism and a hypotesis”. Lancet. 1903;1:793-95.
5. Lewis, T. “Pain in muscular ischemia: its relation to anginal pain”. Arch Intern Med 1932;49:713-27.
6. Tomai F, Crea F, Gaspardone A, Versaci F, Esposito C, Chiariello L, et al. “Mechanisms of cardiac pain during coronary angioplasty”. J Am Coll Cardiol. 1993;22:1892-96.
7. Sylven C, Beermann B, Jonzon B, Brandt R. “Angina pectoris-like pain provoked by intravenous adenosine in healthy volunteers”. BMJ. 1986;293:227-30.
8. Sylven C, Jonzon B, Borg G, Fredholm BB, Kaijser L. “Adenosine injection into the brachial artery produces ischemia-like pain or discomfort in the forearm”. Circulation Res. 1998;22:674-78.
9. Sylven C, Jonzon B, Edlund A. “Angina pectoris like pain provoked by IV bolus of adenosine: relationship to coronary sinus blood flow, heart rate and blood pressure in healthy volunteers”. Eur Heart J. 1989;10:48-54.
10. Chierchia S, Brunelli C, Simonetti I, Lazzari M, Maseri A. “Se-quence of events in angina at rest: Primary reduction in coronary flow”. Circulation. 1980;61:759-65.
11. Maseri A, Chierchia S, Kaski JC. “Mixed angina pectoris”. Am J Cardiol. 1985;56:30E-35E.
12. Gavazzi A, De Servi S, Cornalba C, Falcone C, Scuri PN, Specchia G. “Significance of the walk-through angina phenomenon during exercise testing”. Cardiology. 1986;73:47-53.
13. Prinzmetal M, Kennamer R, Merliss R, Wada T, Bor N. “A variant form of angina pectoris”. Am J Med. 1959;27:375-88.
14. Specchia G, De Servi S, Falcone C, Bramucki E, Angoli L, Mussini GP et al. “Coronary arterial spasm as a cause of exercise-induced ST-segment elevation in patients with variant angina”. Circulation. 1979;59:948-54.
15. Guazzi M, Polese A, Fiorentini C, Magrini F, Olivari MT, Barto-relli C. “Left and right heart haemodynamics during spontaneous angina pectoris. Comparison between angina with ST segment depression and angina with ST segment elevation”. Br Heart J. 1975;37:401-06.
16. Maseri A, Mimmo R, Chierchia S, Marchesi C, Pesola A, L’Abbate A. “Coronary spasm as a cause of acute myocardial ischemia in man”. Chest. 1975;68:625-30.
17. Maseri A, Pesola A, Marzilli M, Severi S, Parodi O, L’Abbate A et al. “Coronary vasospasm in angina pectoris”. Lancet. 1977;i:713-17.
18. Feigl, EO. “Coronary physiology”. Physiol Rev. 1983;63:1-205.19. Reddy KG, Nair RN, Sheehan HM, Hodgson JM. “Evidence
that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis”. J Am Coll Cardiol. 1994;23:833-43.
20. Bertrand ME, Lablanche JM, Tilmant PY, Thieuleux FA, Delforge MR, Carre AG et al. “Frequency of provoked coronary artery spasm in 1089 consecutive patients undergoing coronary arteriography”. Circulation. 1982;65:1299-306.
21. Konidala S, DD Gutterman. “Coronary vasospasm and the regulation of coronary blood flow”. Progr Cardiovasc Dis. 2004;46:349-73.
22. Pristipino C, Beltrame JF, Finocchiaro ML, Hattori R, Fujita M, Mongiardo M et al. “Major racial differences in coronary cons-
trictor response between Japanese and Caucasians with recent myocardial infarction”. Circulation. 2000;101:1102-08.
23. Yasue H, Omote S, Takizawa A, Nagao M, Miwa K, Tanaka S. “Circadian variation of exercise capacity in patients with Prinzmetal variant angina: role of exercise-induced coronary arterial spasm”. Circulation. 1979;59:938-48.
24. Goodson AR, La Master TS, Gutterman DD. “Coronary cons-trictor pathway from anterior hypothalamus includes neurons in rostroventrolaretal medulla”. Am J Physiol. 1993;265:R1311-17.
25. Zeiher AM, Drexler H, Wollschlager H, Just H. “Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis”. Circulation. 1991;83:391-401.
26. Furchgott RF, JV Zawadzki. “The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine”. Nature. 1980;228:373-76.
27. Gauthier KM, Deeter C, Krishna UM, Reedy YK, Bondlela M, Falck JR et al. “Epoxyeicosa-5-enoic acid: A selective epoxyeico-satrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries”. Circ Res. 2002;90:1028-36.
28. Kawachi Y, Tomoike H, Maruoka Y, Kikuchi Y, Araki H, Ishii Y et al. “Selective hypercontraction caused by ergonovine in the canine coronary artery under conditions of induced atherosclerosis”. Circulation. 1984;69:441-50.
29. Miyata K, Shimokawa H, Yamawaki T, Kunihiro I, Zhou X, Higo T et al. “Endothelial vasodilator function is preserved at the spastic/inflammatory coronary lesions in pigs”. Circulation. 1999;100:1432-37.
30. Curry RC Jr, Pepine CJ, Sabom MB, Conti CR. “Similarities of ergonovine-induced and spontaneous attacks of variant angina”. Circulation. 1979;59:307-12.
31. Brum JM, Sufan Q, Lane G, Bove A. “Increased vasoconstrictor activity of proximal coronary arteries with endotelial damage in intact dogs”. Circulation. 1984;70:1066-73.
32. Friedman AC, Spindola-Franco H, Nivatpumin T. “Coronary spasm: Prinzmetal’s variant angina vs. catheter-induced spasm: refractory spasm vs. fixed stenosis”. Am J Roentgenol. 1979;132:897-904.
33. Masumoto A, Morí M, Shimokawa H, Urakami L, Usui M, Takes-hita A. “Suppression of coronary artery spasm by the Rho-kinase inhibitor Fasudil in patients with vasospastic angina”. Circulation. 2002;105:1545-47.
34. Kaski JC, Rosano GMC, Collins P, Nihoyannopoulos P, Maseri A, Poole-Wilson PA. “Cardiac syndrome X: clinical characteristics and left ventricular function. A long-term follow-up study”. J Am Coll Cardiol. 1995;25:807-14.
35. Cannon RO, Watson RM, Rosing DR, Epstein SE. “Angina caused by reduced vasodilator reserve of the small coronary arteries”. J Am Coll Cardiol. 1983;1:1359-73.
36. Opherk D, Zebe H, Weihe E, Mall G, Durr C, Gravert B et al. “Reduced coronary dilatory capacity and ultrastructural changes of the myocardium in patients with angina pectoris but normal coronary arteriograms”. Circulation. 1981;63:817-25.
37. Buchthal SD, den Hollander JA, Merz CN, Rogers WJ, Pepine CJ, Reichek N et al. “Abnormal myocardial phosphorus-31 nuclear mag-netic resonance spectroscopy in women with chest pain but normal coronary angiograms”. N Engl J Med. 2000;342:829-35.
38. Picano E, Lattanzi F, Masini M, Distante A, L’Abbate A. “Usefulness of a hugh-dose dipyridamole-echocardiographic test for diagnosis of syndrome X”. Am J Cardiol. 1987;60:508-12.
39. Maseri A, Crea F, Kaski JC, Crake T. “Mechanisms of angina pectoris in syndrome X”. J Am Coll Cardiol. 1991;17:499-506.
40. Galassi AR, Crea F, Araujo LI, Lammertsma AA, Pupita G, Ya-mamoto Y et al. “Comparison of myocardial blood flow in syn-drome X and one-vessel coronary artery disease”. Am J Cardiol. 1993;72:134-42.
41. Crea F, GA Lanza. “Angina pectoris and norrnal coronary arteries: cardiac syndrome X”. Heart. 2003;90:457-63.

Gelpi - Donato Fisiopatología cardiovascular
60
42. Mohri M, Koyanagi M, Egashira K, Tagawa H, Ichiki T, Shimokawa H et al. “Angina pectoris caused by coronary microvascular spasm”. Lancet. 1998;351:1165-69.
43. Gaspardone A, Ferri C, Crea F, Versachi F, Tomai F, Santucci A. “Enhanced activity of sodium-lithium countertransport in patients with cardiac syndrome X: a potential link between cardiac and metabolic syndrome X”. J Am Coll Cardiol. 1998;32:2031-34.
44. Mohri M, Shimakawa H, Hirokawa Y, Masumoto A, Takeshita A. “Rho-kinase inhibition with intracoronary fasudil prevents myo-cardial ischemia in patients with coronary microvascular spasm”. J Am Coll Cardiol. 2003;41:15-9.
45. Egashira K, Inou T, Hirooka Y, Yamada A, Urabe Y, Takeshita A. “Evidence of impaired endothelium-dependent coronary va-sodilation in patients with angina pectoris and normal coronary angiograms”. N Engl J Med. 1993;328:1659-64.
46. Quyyumi AA, Cannon RO, Panza JA, Diodati JG, Epstein SE. “Endotelial dysfunction in patients with chest pain and normal coronary arteries”. Circulation. 1992;86:1864-71.
47. Tousoulis D, Davies GJ, Asimakopoulos G, Homaei H, Zouri-dakis E, Ahmed N et al. “Vascular cell adhesión molecule-1 and intercellular adhesión molecule-1 serum level in patients with chest pain and normal coronary arteries (syndrome X)”. Clin Cardiol. 2001;24:301-04.
48. Cosin-Sales J, Pizzi C, Brown S. Kaski JC. “C-reactive protein, clinical presentation and ischemict activity in patients with chest pain and normal coronary angiograms”. J Am Coll Cardiol. 2003;41:1468-74.
49. Lanza GA, Giordano AG, Pristipino C, Calcagni ML, Meduri G, Traini C et al. “Abnormal cardiac adrenergic nerve function in patients with síndrome X detected by 131-I metaiodobenzylgua-nidine myocardial scintigraphy”. Circulation. 1997;96:821-26.
50. Rosen SD, Paulesu E, Wise RJ, Camici PG. “Central neural con-tribution to the perception of chest pain in cardiac syndrome X”. Heart. 2002;87:513-19.
51. Droste C, H Roskamm. “Experimental pain measurement in pa-tients with asymptomatic myocardial ischemia”. J Am Coll Cardiol. 1983;1:940-45.
52. Rosen SD, Paulesu E, Nihoyannopoulos P, Tousoulis D, Frackowiak RS, Frith CD et al. “Silent ischemia as a central problem: regional brain activation compared in silent and painful myocardial ische-mia”. Ann Intern Med. 1996;124:939-43.
53. Hauser AM, Gangadharan V, Ramos RG, Gordon S, Timmis GC, Dudlets P. “Sequence of mechanical, electrocardiographic and cli-nical effects of repeated coronary artery occlusion in human beings: Echocardiographic observations during coronary angioplasty”. J Am Coll Cardiol. 1985;5:193-99.
54. Narins CR, Zareba W, Moss AJ, Goldstein RE, Hall WJ. “Cli-nical implications of silent versus symptomatic exercise-induced myocardial ischemia in patients with stable coronary disease”. J Am Coll Cardiol. 1997;29:756-63.
55. Lindsey HE, PF Cohn. “Silent myocardial ischemia during and after exercise testing in patients with coronary artery disease”. Am Heart J. 1978;95:441-46.
56. Hirtzel HO, Leutwyler R, Krayenbuehl HP. “Silent myocardial ischemia: hemodynamic changes during dynamic exercise in proven coronary artery disease despite the absence of angina pectoris”. J Am Coll Cardiol. 1985;2:275-80.
57. Shang SJ, CJ Pepine. “Transient asymptomatic ST-segment de-pression during daily activity”. Am J Cardiol. 1977;39:396-402.
58. Deanfield JE, Maseri A, Selwyn AP, Ribeiro P, Chierchia S, Krikler S, Morgan M. “Myocardial ischemia during daily life in patients with stable angina: Its relation to symptoms and heart rate changes”. Lancet. 1983;2:753-58.
59. Hausmann D, Nikutta P, Trappe HJ, Daniel WG, Wenzlaff P, Lichtlen PR. “Circadian distribution of the characteristics of is-chemic episodes in patients with stable coronary artery disease”. Am J Cardiol. 1990;66:668-72.
60. Weiner DA, Ryan TJ, McCabe CH, Luk S, Chaitman BR, Shef-field LT, et al. “Significance of silent myocardial ischemia during
exercise testing in patients with coronary artery disease”. Am J Cardiol. 1987;59:725-29.
61. Peidro R, Lerman L, Chwojnik A, Chiozza M, Bajraj F, Suarez L. “Desnivel del segmento ST sin dolor en la ergometría. Evalua-ción y seguimiento de pacientes asintomáticos con antecedentes coronarios”. Rev Arg Cardiol. 1993;61:441-48.
62. Houghton JL, Frank MJ, Carr AA, Von Dohlen TW, Prisant M. “Relations among impaired coronary flow reserve, left ventricular hypertrophy and thallium perfusion defects in hypertensive patients without obstructive coronary artery disease”. J Am Coll Cardiol. 1990;15:43-51.
63. Brush JE Jr, Cannon RO, Schenke WH, Bonow RO, Leon MB, Maron BJ, et al. “Angina due to coronary microvascular disease in hypertensive patients without left ventricular hypertrophy”. N Engl J Med. 1988;319:1302-07.
64. Brush GE, Faxon DP, Salmon S, Jakobs AK, Ryan TJ. “Abnormal endothelium-dependent coronary vasomotion in hypertensive patients”. J Am Coll Cardiol. 1992;19:809-15.
65. Laine H, Raitakari OT, Niinikoski H, Pitkanen OP, Iida H, Viikari J, et al. “Early impairment of coronary flow reserve in young men with borderline hypertension”. J Am Coll Cardiol. 1998;32:147-53.
66. Strauer, BE. “Significance of coronary circulation in hypertensive heart disease for development and prevention of heart failure”. Am J Cardiol. 1990;65:34G-41G.
67. Buckberg G, Eber L, Herman M, Gorlin R. “Ischemia in aortic stenosis: hemodynamic prediction”. Am J Cardiol. 1975;35:778-84.
68. Pearlman ES, Weber KT, Janicki JS, Pietra GG, Fishman AP. “Muscle fiber orientation and connective tissue content in the hypertrophied human heart”. Lab Invest. 1982;46:158-64.
69. Schwarztkoopf B, Mundhenke M, Strauer BE. “Alterations of the architecture of subendocardial arterioles in patients with hyper-trophic cardiomyopathy and impaired coronary vasodilator reser-ve: a possible cause for myocardial ischemia”. J Am Coll Cardiol. 1998;31:1089-96.
70. Tanaka M, Fujiwara H, Onodera T. “Quantitative análisis of narrowing of intramyocardial small arteries in normal hearts, hy-pertensive hearts and hearts with hyperterophic cardiomyopathy”. Circulation. 1987;75:1130-39.
71. Cannon RO, Rosing DR, Maron BJ, Leon MB, Bonow RO, Watson RM, et al. “Myocardial ischemia in patients with hyper-trophic cardiomyopathy: contribution of inadequate vasodilator reserve and elevated left ventricular filling pressures”. Circulation. 1985;71:234-43.
72. Kannel WB, DL McGee. “Diabetes and cardiovascular risk factors: The Framingham study”. Circulation. 1979;59:8-13.
73. Stamler J, Vaccaro O, Neaton JD, Wentworth D. “Diabetes, other risk factors and 12 years cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial”. Diabetes Care. 1993;16:434-44.
74. Woodfield S, Lundergan C, Reiner J, Greenhouse S, Thompson M, Rohrbeck S, et al. “Angiographic findings and outcome in diabetic patients treated with thrombolytic therapy for acute myo-cardial infarction: the GUSTO-I experience”. J Am Coll Cardiol. 1996;28:1661-69.
75. Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. “Mor-tality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction”. N Engl J Med. 1998;339:229-34.
76. Blumenthal HT, Alex M, Goldemberg S. “A study of lesions of intramural coronary by branches in diabetes mellitus”. Arch Pathol. 1960;70:27-42.
77. Hamby RI, Zoneraich S, Sherman L. “Diabetic cardiomyopathy”. JAMA. 1974;229:1749-54.
78. Bucala R, Tracey KJ, Cerami A. “Advanced glycosilation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes”. J Clin Invest. 1991;87:432-38.
79. Giugliano D, Ceriello A, Paolisso D. “Diabetes mellitus, hyper-tension and cardiovascular disease: the role of oxidetive stress?” Metabolism. 1995;44:363-68.

Fisiopatología de los síndromes coronarios
61
80. Takahashi K, Ghatei MA, Lam HC, O’Halloran DJ, Bloom SR. “Elevated plasma endothelin in patients with diabetes mellitus”. Diabetología. 1990;33:306-10.
81. Bucala R, Makita Z, Koshinsky T. “Lipid advanced glycosilation induces transendothelial human monocyte chemotaxis and secre-tion of platelet-derived growth factor: role in vascular disease of diabetes and aging”. Proc Natl Acad Sci USA. 1990;87:9010-14.
82. Sun D, Nguyen N, De Grado T, Schwaiger M, Brosius F. “Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocites”. Circulation. 1994;89:793-98.
83. Stanley W, Hall J, Hacker T, Hernandez L, Whitesell L. “Decreased myocardial glucose uptake during ischemia in diabetic swine”. Metabolism. 1997;46:168-72.
84. Nahser P, Brown R, Oskarsson H, Winniford M, Rossen J. “Maxi-mal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus”. Circulation. 1995;91:635-40.
85. Davi G, Catalano I, Averna M, Notarbartolo A, Strano A, Cia-battoni G, et al. “Thromboxane biosynthesis and platelet function in type II diabetes mellitus”. N Engl J Med. 1990;322:1769-74.
86. Toyry J, Niskanen L, Mantysaari M, Lansimies E, Uusitupa M. “Occurrence, predictors and clinical significance of autonomic neuropathy in NIIDM”. Diabetes. 1996;45:308-15.
87. Nesto RW, Phillips RT, Kett KG, Hill T, Perper E, Young E, et al. “Angina and exertional myocardial ischemia in diabetic and nondiabetic patients: assessment by exercise thallium scintigraphy”. Ann Intern Med. 1988;108:170-75.
88. Chiariello M, Indolfi C, Cotecchia MR, Sifola C, Romano M, Con-dorelli M. “Asymptomatic transient ST changes during ambulatory ECG monitoring in diabetic patients”. Am Heart J. 1985;110:529-34.
89. Aronson D, Bloomgarden Z, Rayfield EJ. “Potential mechanisms promoting reestenosis in diabetics patients”. J Am Coll Cardiol. 1996;27:528-35.


63
1 -2 La
ATC ha revolucionado el tratamiento de la enfermedad
uno de los problemas no resueltos.La reestenosis
-
las distintas técnicas, que se realice con o sin stent y que 3-8
mientras que la reestenosis aparece en semanas o meses, pro-
-stent en el momento de la ATC 9, estos
La reestenosis es el resultado de una compleja repara-
10
stent producen factores de crecimiento que
crecimiento neointimal. 11 -
tejido conectivo de la matriz para producir un crecimiento endovascular desde la neoíntima, que puede disminuir la
9-12
de la reestenosis posATC puede ser analizado desde varios aspectos:
-
-
-
A pesar del conocimiento sobre los mecanismos y la respuesta arterial a la injuria, la causa y los mecanismos íntimos de la reestenosis no están completamente deve-
-da por factores de crecimiento y citoquinas, que también participan en la reestenosis vascular, dado que este es un
13-15
-teliales perdiéndose su capacidad protectora; esto determi-
Reestenosis
--
naria; estos se pueden subdividir en tempranos y tardíos.
Reestenosiscoronaria 4
Reestenosis
SLD10% RAVEL17,9% Taxus V ISR19,1% Sirius ISR14% e-Cypher13% Wisdom
Balón32% en BENESTENT42% en STRESS57% en CAVEAT
Stent29% vs. 43% STRESS22% vs. 32% BENESTENT18% vs. 32% REST
Tabla 4.1: Incidencia de reestenosis, según la técnica y comparando diferentes estudios multicéntricos
SLD: Stent liberador de droga
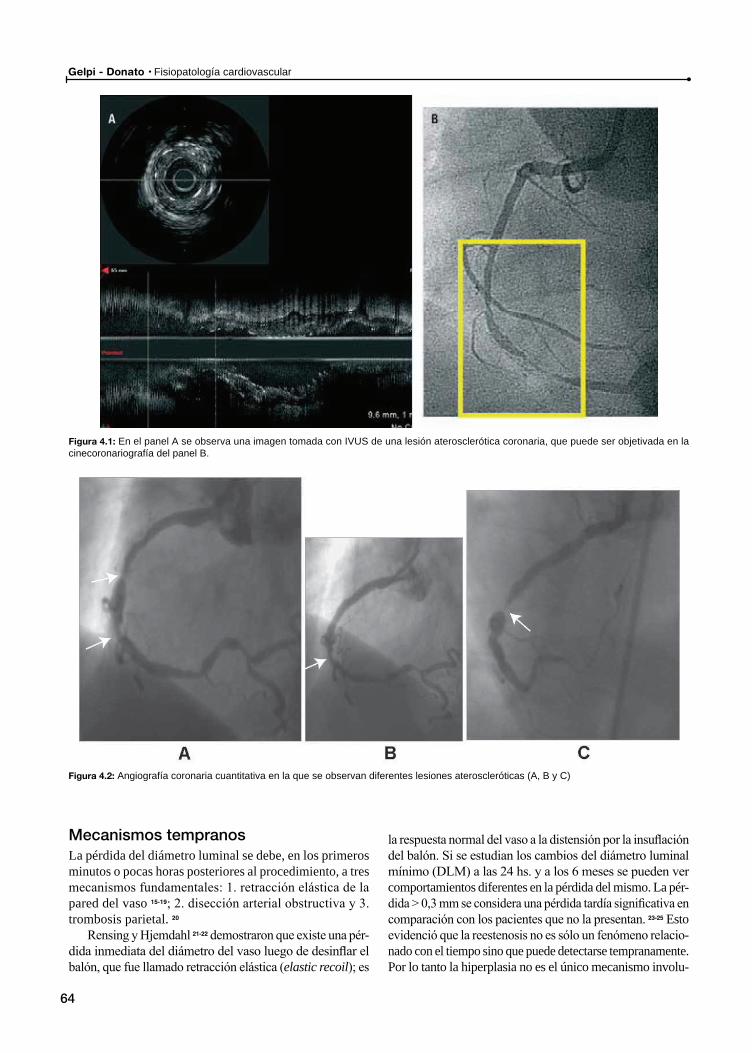
Gelpi - Donato Fisiopatología cardiovascular
64
Figura 4.2: Angiografía coronaria cuantitativa en la que se observan diferentes lesiones ateroscleróticas (A, B y C)
Figura 4.1: En el panel A se observa una imagen tomada con IVUS de una lesión aterosclerótica coronaria, que puede ser objetivada en la cinecoronariografía del panel B.
Mecanismos tempranosLa pérdida del diámetro luminal se debe, en los primeros minutos o pocas horas posteriores al procedimiento, a tres
pared del vaso 15-19
trombosis parietal. 20
21-22 -
elastic recoil
comportamientos diferentes en la pérdida del mismo. La pér-
23-25 Esto -
nado con el tiempo sino que puede detectarse tempranamente. Por lo tanto la hiperplasia no es el único mecanismo involu-

Restenosis coronaria
65
vs. concéntricas,
et alcambios en el DLM, en los primeros minutos u horas pos-
de la reestenosis23 tanto inmediata como a seis meses. 26-27
-
oclusivos 28; como demostraran Jain et al. es también de utilidad para detectar los mecanismos de la reestenosis
29
26
26 -nante importante de la tasa de reestenosis posterior, cuando
30
Por lo tanto, parece claro que los mecanismos princi-
vez, son dependientes del volumen de placa y el remode-lado previo, que permiten obtener un diámetro luminal
-
Mecanismos tardíosLos mecanismos tardíos involucrados en la reestenosis
Hiperplasia fibrointimal-
vancia en la reestenosis coronaria posATC y más aún
y profunda, como sucede en la ATC con stent o aterec-tomía direccional o rotacional 31, 32
-lares comienzan a aumentar la síntesis de ADN y cambian su habitual fenotipo contráctil por uno secretor, pierden
-
hacia la íntima, donde comienzan a proliferar y a secretar
endoteliales sanas para recubrirla. Este proceso de reen-
de las células musculares lisas mediante el factor inhibidor -
intimal. Una vez que las células musculares lisas dejan de proliferar pasan nuevamente al fenotipo contráctil. Este
-
Figura 4.3: Angioscopia que muestra la lesión culpable y el proceso de inestabilidad multifocal

Gelpi - Donato Fisiopatología cardiovascular
66
-timal es el mecanismo de reestenosis más importante en
Fenómeno inflamatorio
et al. realizaron autopsias a pacientes fallecidos en el -
33 Moreno et al. analizaron las muestras obtenidas al realizar aterectomía
con las procedentes de lesiones que habían sido respon-
34
-dola más vulnerable y susceptible a la ruptura, con lo que se pone en contacto el núcleo lipídico, rico en factor
demostrado la presencia de enzimas, como los activadores -
humanas 35, 36, detectándose en las lesiones que se habían inestabilizado mayor cantidad de enzimas en el interior
tenían más síntesis activa.
de la placa ha sido postulada por varios estudios, quienes -
isquémicos. 37-38 Así, Liuzzo et al. 37 encontraron que los
más elevados de proteína C reactiva y amiloide A, evolu-
y/o muerte súbita y tenían más recurrencia de isquemia du-
concordantes en el
varones sin enfermedad coronaria conocida, demostrando
-
para efectuar diversos estudios con vistas a probar nuevas
monocitos a la pared vascular, estaban más elevados en
miocardio. 39 En el caso de la proteína C reactiva, el efecto -
tes que tenían los niveles más elevados de este marcador.
bajas como para alcanzar concentraciones elevadas en el tejido, es posible que actúe sobre los monocitos, mientras
Figura 4.4: En el panel A se observa el proceso de remodelamiento de una placa aterosclerótica y su posterior evolución (panel B).

Restenosis coronaria
67
-
--
matoria. El reclutamiento de leucocitos circulantes hacia -
-40 El anclaje de estas células
-41
42 43
es provocado por stentstents arteriales provoca una
-
44
demostrado que el bloqueo con anticuerpos 45 o la elimina--
citaria y limita el crecimiento neointimal. 46 En la misma
que el bloqueo de la MCP-1 disminuye el crecimiento de
de presentar reestenosis.
Remodelado arterial crónico
en particular, el remodelamiento vascular es el principal mecanismo compensatorio para conservar la luz del vaso. 47 -ta llevando la placa de ateroma hacia la adventicia, sin
-48, 49
Reestenosis intrastent
stent coronario, la incidencia de la reestenosis ha disminuido notablemente, desde un 50-
stent
El stent es un dispositivo utilizado en la revasculariza--
et al.50
-
stent de 51 52 -
cacia del stent
stent
--
53 Al mejorar la técnica de
a la pared arterial, sin dejar soluciones de continuidad que resultaban en bolsillos 53
54, 55, la incidencia del
ha disminuido drásticamente. Estos aspectos han favorecido un uso casi sistemático del stent -
intrastent
la luz del vaso en el interior del stent. La misma puede -
intrastent-
la necesidad de un nuevo procedimiento de revasculari-intrastent
56
-dadas. Así, si se analizan los trabajos que han incluido
57-59
de reestenosis clínica.60
intra-stent
para documentar dos mecanismos básicos. -
intimal que se produce a través y dentro del stent, proce-
--

Gelpi - Donato Fisiopatología cardiovascular
68
stent, documentado por eco intravascular y -
stents, puede ser responsable stent. Debemos
establecer que los stents en uso clínico tienen un espesor -
vulnerable.
Ciclo celular y reestenosis
Ciclo celular es la secuencia de procesos en la vida de una célula eucariota que conserva la capacidad de dividirse. Consiste en interfase, mitosis y citocinesis, lo que se de-
La vida de las células transita por dos etapas que se La interfase se
subdivide en tres periodos g1 gap : en -
S: fase de síntesis o
g2: es el tiempo
la mitosis, durante el que la célula crece, replica su ADN -
m es el estadio más trascendente de la célula,
del núcleo –se separan los cromosomas hijos replicados
dos células hijas. En condiciones normales, las células musculares lisas
-
61,62 El ciclo celular está re-
63-65 -
-
espaciales como temporales, en arterias normales como
no se observa en arterias normales, pero se encuentra ele-
66
-
las kinasas dependientes D, E y A en células quiescentes y
64-65
66
-
61-65 -
un fenotipo vascular con marcado crecimiento neointimal
mecánica.67, 68
69 El ciclo celular, por lo tanto, es un punto en común en el
stents
Figura 4.6: Incidencia global de reestenosis intrastent. Se observa una gran variabilidad, que está influenciada principalmente por el tipo de lesiones (calibre < 3 mm, lesiones con una longitud superior a 15 mm)
Figura 4.5: Tipos de remodelamiento de placa aterosclerótica. El diámetro del vaso se incrementa llevando la placa de ateroma hacia la adventicia, sin provocar una disminución en la luz arterial. En el remodelamiento negativo existe reducción de la luz arterial por una disminución del área de la lámina elástica interna.

Restenosis coronaria
69
Factores predictivos de la apariciónde reestenosis intrastent
-
quienes se les ha implantado un stent. Diversos estudios -
mo responsable de la reestenosis intrastent es la hiperplasia neointimal. Por el contrario, el mecanismo predominante
intrastent, tanto clínicos y an-
clínicos, el más consistente es la presencia de diabetes. Otros factores clínicos relacionados han sido una edad avanzada o
70-72, 10, 12 -
71, 73, 74, la
70, 71 y la localiza-
72, 73
-dimiento. Los factores de procedimiento relacionados han sido un diámetro luminal mínimo no adecuado al diámetro
múltiples stents en una misma arteria. 70, 71, 74 Kastrati et al. 75
-pendientes de reestenosis intrastent que hallaron en su serie. Así, un paciente no diabético, con un solo stent implantado
de múltiples stents y un diámetro intraluminal posproce-
Influencia de las variables del procedimiento
que tras el procedimiento de ATC quedan con una este--
Fase SReproducción delDNA
Fase G2Preparación para la división celular
Fase MDivisión celular(mitosis)
Fase G1Aumento del citoplasmaincorporación de nuevasproteínas
Fase G0Células funcionantes
inactiva para la
reproducción
Antisense
Syrolimusciclinas/cdksp27
Agentecitostático(interrupción delciclo celular)
Punto de control(no hay retorno delciclo celular)
Radiaciónactinomicina DirinotecanADN
Agentecitotóxico(muerte celular)
Paclitaxelmicrotúbulos
Figura 4.7: Esquema representativo del ciclo celular, en el que se indican los puntos en los que actúan algunos de los fármacos inhibidores de la reestenosis

Gelpi - Donato Fisiopatología cardiovascular
70
intrastent tendrá una mayor
-
pero la injuria vascular provocada es también mayor, lo
factores del procedimiento relacionados han sido un menor
stents en la misma arteria y la injuria arterial amplia y profunda.76
Factores genéticosLa reestenosis no es un problema del azar, y ciertos pa-
-
no se ha podido relacionar con estos procesos o factores.-
son los marcadores más utilizados en los estudios de la
-
Estudios recientes han detectado una serie de factores
intrastent. 77-82
Stents liberadores de droga (SLD)
El uso rutinario del stent
que lo transporta hasta lesiones distales con muy baja incidencia de complicaciones, hicieron del método de
-
A pesar de esta evidencia clínica, la reestenosis del stent -
stents
crecimiento intrastent
--
stent
un importante avance para disminuir la reestenosis intra-stent
A partir de este concepto de stentstent,
-
Los primeros ensayos clínicos fueron realizados en un stent -
stent
-
-83
con lesiones más complejas, en pacientes más complejos,
-stent convencional.
-
resultados positivos o neutros en los estudios realizados en
Stents liberadores de drogas antiproliferativas en escenarios no favorables
--
cundarios, sino también por la elevada tasa de reestenosis,
fueron aleatorizados para recibir un stent con sirolimus en
un stent con sirolimus en ambos vasos. 84
-
series clásicas de stent con el stent

Restenosis coronaria
71
-
todas las trombosis ocurrieron en pacientes tratados con stent con sirolimus en ambos vasos.
similar a la de los stents recientes han mostrado que los resultados del tratamiento con stent con sirolimus en estos pacientes, en cuanto a reesteno-sis y trombosis del stent, son comparables a los obtenidos en pacientes sin infarto. 85
Los datos sobre los stents en puentes de safena están basados en estudios con pocos pa-cientes, en los que se ha obtenido una tasa de nuevos proce-
se ha obtenido una menor tasa de reestenosis y de nuevos -
control con stents 86
que recibieron stent con sirolimus en puentes de safena fue
-
87
Clásicamente, la enfermedad del tronco ha sido una in-stent puede ser una
-
pacientes con enfermedad del tronco tratados con stent con 88, 89
implantar un stent, la tasa de reestenosis es muy elevada. En
favorables respecto de las series con stents convencionales. 90
Reestenosis de los stents liberadores de droga (SLD)
son los mismos que en los stents
fracturas de los puentes o struts-
mites del stent mostrado tener más incidencia en el desarrollo de la rees-
stents convencionales. En el estudio
cobertura de la placa fue la causa de esta reestenosis en struts
estuvieron presentes en un paciente. Las características de la hiperplasia neointimal tal vez
stents
Figura 4.8: Esquema representativo de una arteria en la que se observa un stent y las nanopartículas de droga liberadas hacia la pared arterial.
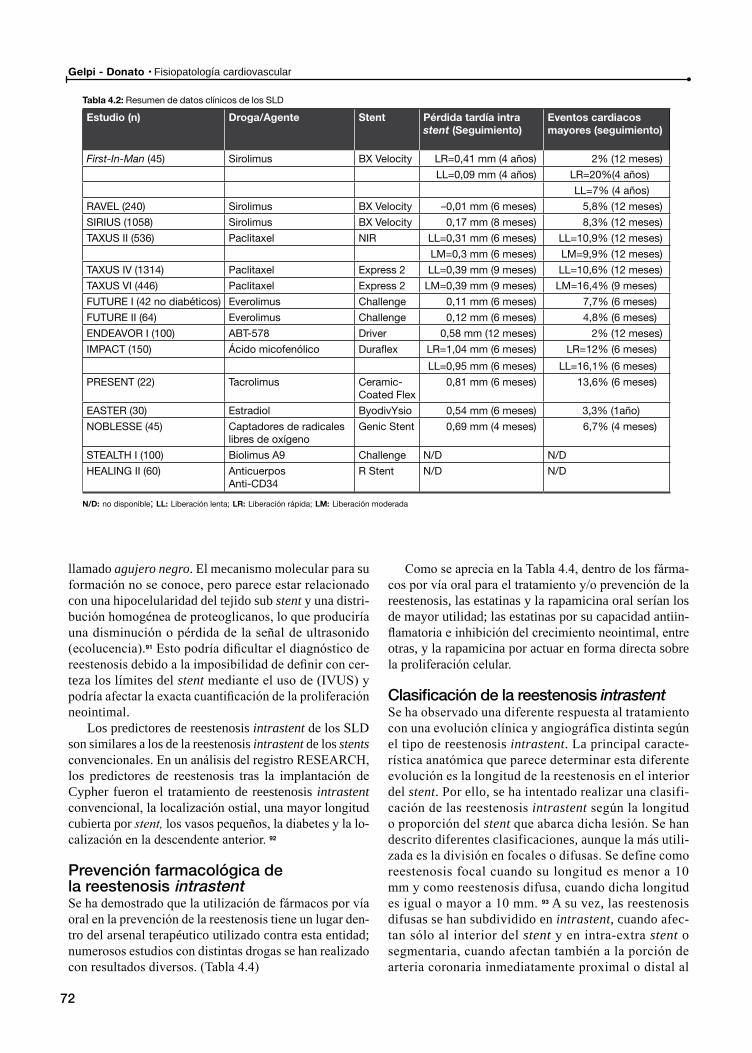
Gelpi - Donato Fisiopatología cardiovascular
72
Tabla 4.2: Resumen de datos clínicos de los SLD
Estudio (n) Droga/Agente Stent Pérdida tardía intra stent (Seguimiento)
Eventos cardiacos mayores (seguimiento)
First-In-Man (45) Sirolimus BX Velocity 2% (12 meses)
RAVEL (240) Sirolimus BX Velocity –0,01 mm (6 meses) 5,8% (12 meses)
SIRIUS (1058) Sirolimus BX Velocity 0,17 mm (8 meses) 8,3% (12 meses)
TAXUS II (536) NIR LL=0,31 mm (6 meses) LL=10,9% (12 meses)
LM=0,3 mm (6 meses) LM=9,9% (12 meses)
TAXUS IV (1314) Express 2 LL=0,39 mm (9 meses) LL=10,6% (12 meses)
TAXUS VI (446) Express 2 LM=0,39 mm (9 meses) LM=16,4% (9 meses)
FUTURE I (42 no diabéticos) Everolimus Challenge 0,11 mm (6 meses) 7,7% (6 meses)
FUTURE II (64) Everolimus Challenge 0,12 mm (6 meses) 4,8% (6 meses)
ENDEAVOR I (100) ABT-578 Driver 0,58 mm (12 meses) 2% (12 meses)
Duraflex LR=1,04 mm (6 meses) LR=12% (6 meses)
LL=0,95 mm (6 meses) LL=16,1% (6 meses)
Tacrolimus Ceramic-Coated Flex
0,81 mm (6 meses) 13,6% (6 meses)
EASTER (30) Estradiol ByodivYsio 0,54 mm (6 meses)
NOBLESSE (45) Captadores de radicales libres de oxígeno
Genic Stent 0,69 mm (4 meses) 6,7% (4 meses)
STEALTH I (100) Biolimus A9 Challenge N/D N/D
HEALING II (60) AnticuerposAnti-CD34
R Stent N/D N/D
N/D: no disponible; LL: Liberación lenta; LR: Liberación rápida; LM: Liberación moderada
llamado agujero negro. El mecanismo molecular para su
con una hipocelularidad del tejido sub stent y una distri-
91
-teza los límites del stent
neointimal. Los predictores de reestenosis intrastent
son similares a los de la reestenosis intrastent de los stents
Cypher fueron el tratamiento de reestenosis intrastent
cubierta por -92
Prevención farmacológica de la reestenosis intrastent
-tro del arsenal terapéutico utilizado contra esta entidad;
-
reestenosis, las estatinas y la rapamicina oral serían los de mayor utilidad; las estatinas por su capacidad antiin-
otras, y la rapamicina por actuar en forma directa sobre
Clasificación de la reestenosis intrastent
el tipo de reestenosis intrastent. La principal caracte-
del stent. Por ello, se ha intentado realizar una clasifi-intrastent
stentdescrito diferentes clasificaciones, aunque la más utili-
93 A su vez, las reestenosis difusas se han subdividido en intrastent, cuando afec-
stent stent o

Restenosis coronaria
73
stentque las reestenosis se subdividen en focales o difusas
stent94; así, se clasifican en focales si afectan a menos del
stent y en difusas si afectan a
et al., con 95, divide a las lesiones
stent intrastent focalizada. Lesiones
stents o en los gapsstent stent;
Reestenosis intrastent difusa. Lesiones stent
reestenosis intrastent proliferativa. Lesio--
stent Reestenosis intrastent
focal. Aunque la reestenosis intrastent -
la reestenosis situada
stents. La incidencia de stent puede reducirse
stentfármaco. 92, 96
de reestenosis de los stents convencionales, sobre todo en lo que respecta a la frecuencia de cada una de las locali-zaciones.
Tratamiento de la reestenosis
97-101
Angioplastia simple con balónFue el primer tipo de tratamiento empleado. 102 Los resul-
-
cuando se analizan las series que presentan un porcentaje elevado de reestenosis difusas, la tasa de necesidad de
98, 103
en el tratamiento de las reestenosis intrastent debido a las escasas complicaciones asociadas al procedimiento y
trata de reestenosis de tipo difuso, la elevada incidencia -
cionado el estudio de otras alternativas.
AterectomíaInvariablemente, esta técnica se complementa con la reali-
-
del stent 93
intraprocedimiento. 98 Cuando se analizan los resultados a
104
Stent intrastentstent adicional en el in-
terior del primer stent stent adicional intrastent -
Tabla 4.3: Mecanismos de reestenosis de SLD
Causas biológicas Causas mecánicas
ausencia o insuficiente inhibición de la drogaDistribución y cobertura asimétrica del stent
Ausencia o retraso en la reendotelización Mala aposición del stent
Depósitos de fibrina Infra expansión del stent
Inflamación crónica inducida por el polímero Fracturas de los struts
Toxicidad de la droga hacia el vaso Ruptura o irrupción del polímero

Gelpi - Donato Fisiopatología cardiovascular
74
aumentar la luz del vaso hasta un diámetro superior al stent
93 stent intrastent es un procedi-
Radiación intracoronaria
de la reestenosis intrastent
en el tratamiento de la reestenosis intrastent. Los resul-tados son diversos, Teirstein et al. 105 reportaron una tasa
vs.
SLD, intrastents convencionales
el tratamiento de la reestenosis intrastent se asocia con una tasa de reestenosis más elevada que la de las lesiones
-currencia de reestenosis intrastent
-
106
-cientes con reestenosis intrastent para recibir tratamien-
107 Los resultados provisionales han mostrado una recurrencia de reestenosis intrastent
--
intrastent fueron distri-
stent Cypher y stentla reestenosis intrastentrespectivamente. 108
109 -
110
Por los resultados obtenidos en los estudios menciona-
el tratamiento de la reestenosis intrastent es, hasta ahora, el mejor método.
Conclusión
El descubrimiento de los mecanismos celulares y molecula-
stent
importante de pacientes, como los diabéticos o quienes
etc., que presentan cifras altas de reestenosis.
Figura 4.9: Esquema de los 4 patrones de la clasificación de reestenosis, considerando la reestenosis como focal o difusa. Obsérvese que el patrón 1 presenta 4 subtipos.
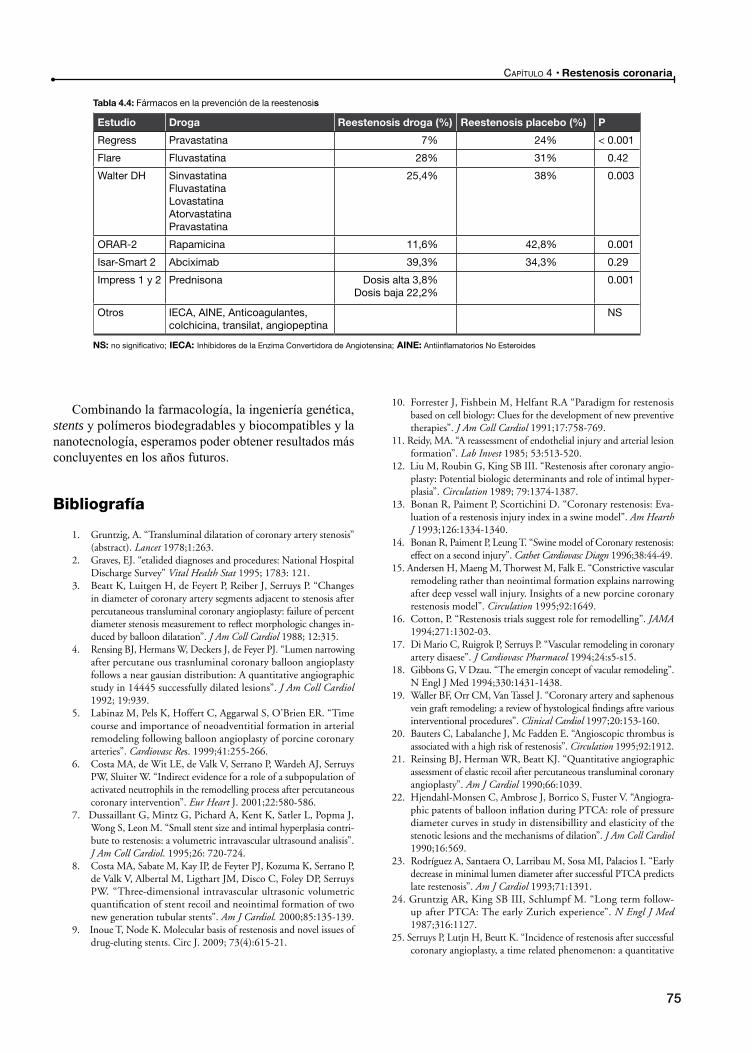
Restenosis coronaria
75
stents
Bibliografía
1. Gruntzig, A. “Transluminal dilatation of coronary artery stenosis” (abstract). Lancet 1978;1:263.
2. Graves, EJ. “etalided diagnoses and procedures: National Hospital Discharge Survey” Vital Health Stat 1995; 1783: 121.
3. Beatt K, Luitgen H, de Feyert P, Reiber J, Serruys P. “Changes in diameter of coronary artery segments adjacent to stenosis after percutaneous transluminal coronary angioplasty: failure of percent diameter stenosis measurement to reflect morphologic changes in-duced by balloon dilatation”. J Am Coll Cardiol 1988; 12:315.
4. Rensing BJ, Hermans W, Deckers J, de Feyer PJ. “Lumen narrowing after percutane ous trasnluminal coronary balloon angioplasty follows a near gausian distribution: A quantitative angiographic study in 14445 successfully dilated lesions”. J Am Coll Cardiol 1992; 19:939.
5. Labinaz M, Pels K, Hoffert C, Aggarwal S, O’Brien ER. “Time course and importance of neoadventitial formation in arterial remodeling following balloon angioplasty of porcine coronary arteries”. Cardiovasc Res. 1999;41:255-266.
6. Costa MA, de Wit LE, de Valk V, Serrano P, Wardeh AJ, Serruys PW, Sluiter W. “Indirect evidence for a role of a subpopulation of activated neutrophils in the remodelling process after percutaneous coronary intervention”. Eur Heart J. 2001;22:580-586.
7. Dussaillant G, Mintz G, Pichard A, Kent K, Satler L, Popma J, Wong S, Leon M. “Small stent size and intimal hyperplasia contri-bute to restenosis: a volumetric intravascular ultrasound analisis”. J Am Coll Cardiol. 1995;26: 720-724.
8. Costa MA, Sabate M, Kay IP, de Feyter PJ, Kozuma K, Serrano P, de Valk V, Albertal M, Ligthart JM, Disco C, Foley DP, Serruys PW. “Three-dimensional intravascular ultrasonic volumetric quantification of stent recoil and neointimal formation of two new generation tubular stents”. Am J Cardiol. 2000;85:135-139.
9. Inoue T, Node K. Molecular basis of restenosis and novel issues of drug-eluting stents. Circ J. 2009; 73(4):615-21.
10. Forrester J, Fishbein M, Helfant R.A “Paradigm for restenosis based on cell biology: Clues for the development of new preventive therapies”. J Am Coll Cardiol 1991;17:758-769.
11. Reidy, MA. “A reassessment of endothelial injury and arterial lesion formation”. Lab Invest 1985; 53:513-520.
12. Liu M, Roubin G, King SB III. “Restenosis after coronary angio-plasty: Potential biologic determinants and role of intimal hyper-plasia”. Circulation 1989; 79:1374-1387.
13. Bonan R, Paiment P, Scortichini D. “Coronary restenosis: Eva-luation of a restenosis injury index in a swine model”. Am Hearth J 1993;126:1334-1340.
14. Bonan R, Paiment P, Leung T. “Swine model of Coronary restenosis: effect on a second injury”. Cathet Cardiovasc Diagn 1996;38:44-49.
15. Andersen H, Maeng M, Thorwest M, Falk E. “Constrictive vascular remodeling rather than neointimal formation explains narrowing after deep vessel wall injury. Insights of a new porcine coronary restenosis model”. Circulation 1995;92:1649.
16. Cotton, P. “Restenosis trials suggest role for remodelling”. JAMA 1994;271:1302-03.
17. Di Mario C, Ruigrok P, Serruys P. “Vascular remodeling in coronary artery disaese”. J Cardiovasc Pharmacol 1994;24:s5-s15.
18. Gibbons G, V Dzau. “The emergin concept of vacular remodeling”. N Engl J Med 1994;330:1431-1438.
19. Waller BF, Orr CM, Van Tassel J. “Coronary artery and saphenous vein graft remodeling: a review of hystological findings aftre various interventional procedures”. Clinical Cardiol 1997;20:153-160.
20. Bauters C, Labalanche J, Mc Fadden E. “Angioscopic thrombus is associated with a high risk of restenosis”. Circulation 1995;92:1912.
21. Reinsing BJ, Herman WR, Beatt KJ. “Quantitative angiographic assessment of elastic recoil after percutaneous transluminal coronary angioplasty”. Am J Cardiol 1990;66:1039.
22. Hjendahl-Monsen C, Ambrose J, Borrico S, Fuster V. “Angiogra-phic patents of balloon inflation during PTCA: role of pressure diameter curves in study in distensibillity and elasticity of the stenotic lesions and the mechanisms of dilation”. J Am Coll Cardiol 1990;16:569.
23. Rodríguez A, Santaera O, Larribau M, Sosa MI, Palacios I. “Early decrease in minimal lumen diameter after successful PTCA predicts late restenosis”. Am J Cardiol 1993;71:1391.
24. Gruntzig AR, King SB III, Schlumpf M. “Long term follow-up after PTCA: The early Zurich experience”. N Engl J Med 1987;316:1127.
25. Serruys P, Lutjn H, Beutt K. “Incidence of restenosis after successful coronary angioplasty, a time related phenomenon: a quantitative
Tabla 4.4: Fármacos en la prevención de la reestenosis
Estudio Droga Reestenosis droga (%) Reestenosis placebo (%) P
Regress 7% 24% < 0.001
Flare Fluvastatina 28% 31% 0.42
Walter DH SinvastatinaFluvastatinaLovastatinaAtorvastatina
25,4% 38% 0.003
ORAR-2 Rapamicina 11,6% 42,8% 0.001
Isar-Smart 2 Abciximab 39,3% 34,3% 0.29
Impress 1 y 2 Dosis alta 3,8%Dosis baja 22,2%
0.001
Otros IECA, AINE, Anticoagulantes, colchicina, transilat, angiopeptina
NS
NS: no significativo; IECA: Inhibidores de la Enzima Convertidora de Angiotensina; AINE: Antiinflamatorios No Esteroides

Gelpi - Donato Fisiopatología cardiovascular
76
angiographic study in 342 consecutive patients at 1, 2, 3 and 4 months”. Circulation 1988;77:361.
26. Rodríguez A, Palacios I, Fernández M, Brose J. “Time course and mechanisms of early luminal diameter loss after PTCA”. Am J Cardiol 1995;76:1131.
27. La Blanche, JM. “On Behalf of the FACT investigators. Recoil twenty-four hours after coronary angioplasty: a computerized angiographyc study”. J Am Coll Cardiol 1993;21:34A.
28. Escobar A, With CJ, Ramee SR. “Angioscopic vs. angiographic coronary lesions morphology in unstable angina”. Circulation 1993;88:I-586.
29. Feld S, Ganim M, Carell E. “Comparison of angioscopy, intravas-cular ultrasound imaging and quantitative coronary angiography in predicting clinical outcomes after coronary intervention in high risk patients”. JACC 1996;28(1):97.
30. Malekianpour M, L. Chen. “Restensosi or recoil? The role of sub acute recoil in perceived restenosis”. JACC 1997;29:1081.
31. Foley D, P. Serruys. “Influence of coronary vessel size on renarrowing process and late angiographic outcome after successful balloon angioplasty”. Circulation 1994;90(3):1239.
32. Leguizamon J, Chambre D, Torresani E, Fernández A. “Aterecto-mía rotacional coronaria de alta velocidad: ¿Predicen los factores angiográficos? fracasos, complicaciones mayores o reestenosis. Análisis estadístico multivariado”. Rev Argen Cardiol 1994;62:165.
33. Van der Wal AC, Becker AE, Van der Loos CM, Das PK. “Site of Intimal Rupture or Erosion of Thrombosed Coronary Athe-rosclerotic Plaques Is Characterized by an Inflammatory Process Irrespective of the Dominant Plaque Morphology”. Circulation 1994;89:36-44.
34. Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT et al. “Macrophage Infiltration in Acute Coronary Syndromes: Implication for Plaque Rupture”. Circulation 1994;90:775-778.
35. Henney AM, Wakeley PR, Davies MJ, Foster K, Hembry T, Mur-phy G et al. “Localization of Stromelysin Gene Expression in Atherosclerotic Plaques by in situ Hybridazation”. Proc Natl Acad Sci USA 1991;88:8154-8158.
36. Li L, Zielke HR, Cheng L, Xiao R, Crow MT et al. “Increased Expression of 72-kd Type IV Collagenase (MMP-2) in Human Aortic Atherosclerotic Lesions”. Am J Pathol. 1996;148:121-128.
37. Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A et al. “The Prognostic Value of C-Reactive Protein and Serum Amyloid. A Protein in Severe Unstable Angina”. N Eng J Med. 1994;331:417-424.
38. Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J et al. “Plasma Concentration of Soluble Intercellular Ad-hesion Molecule 1 and Risks of Future Myocardial Infarction in Apparently Healthy Men”. Lancet 1998;351:88-92.
39. Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. “Pros-pective Study of C-Reactive Protein and the Risk of Future Cardio-vascular Events Among Apparently Healthy Women”. Circulation 1998;98:731-733.
40. Springer, TA. “Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm”. Cell. 1994;76:301-314.
41. Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. “Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendo-thelial migration of human neutrophils in vitro”. J Clin Invest. 1989;83:2008-2017.
42. Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. “Regulation of leukocyte-endothelium interaction and leukocyte trans-endothelial migration by intercellular adhesion molecule 1-fibrinogen recognition”. Proc Natl Acad Sci U S A. 1995;92:1505-1509.
43. Welt FG, Tso C, Edelman ER, Kjelsberg MA, Paolini JF, Seifert P, Rogers C. “Leukocyte recruitment and expression of chemokines following different forms of vascular injury”. Vasc Med 2003;8:1-7.
44. Rogers C, Edelman ER, Simon DI. “A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after
angioplasty or stent implantation in rabbits”. Proc Natl Acad Sci U S A. 1998;95:10134-10139.
45. Cipollone F, Marini M, Fazia M, Pini B, Iezzi A, Reale M, Paloscia L, Materazzo G, D’Annunzio E, Conti P, Chiarelli F, Cuccurullo F, Mezzetti A. “Elevated circulating levels of monocyte chemoat-tractant protein-1 in patients with restenosis after coronary an-gioplasty”. Arterioscler Thromb Vasc Biol. 2001;21:327-334.
46. Danenberg HD, Golomb G, Groothuis A, Gao J, Epstein H, Swaminathan RV, Seifert P, Edelman ER. “Liposomal alendronate inhibits systemic innate immunity and reduces in-stent neointimal hyperplasia in rabbits”. Circulation 2003;108:2798-2804.
47. Glagov S, Weisemberg E, Zarins CK, Koletttis GJ. “Compensatory enlargement of human atherosclerotic coronary arteries”. N Eng J Med 1987; 316: 1371-1375.
48. Fuster, V. “Remodelado del trombo: punto clave en la progresión de la aterosclerosis coronaria”. Rev Esp Cardiol 2000;53(sup1)2-7.
49. Nagavi M, Libby P, Falk E, Serruys P, Virmani R, Ridker P. “From Vulnerable Plaque to Vulnerable Patient: A Call for New Defini-tions and Risk Assessment”. Circulation 2003;108:1664-1672.
50. Sigwart U, Puel J, Mirkovitch V, Joffre F, Kappenberger L. “Intra-vascular stents to prevent occlusion and restenosis after transluminal angioplasty”. N Engl J Med 1987;316: 701-706.
51. Serruys P, De Jaeger P, Kemeneiji F, Macaya C, Rutsch W, Materne P et al. “A comparision of ballon-expandable-stent implantation with ballon angioplassty in patients with coronary artery disease”. N Engl J Med. 1994;331:489-495.
52. Fischman D, Leon M, Baim D, Schatz R, Savage M, Nobushoshi M et al. “A randomized comparision of coronary stent placement and balloon angioplasty in the treatment of coronary artery disease”. N Engl. J Med. 1994;331:496-501.
53. Colombo A, Hall P, Nakamura S, Almagor Y, Maiello L, Tobis JM et al. “Intracoronary stenting without anticoagulation accomplished with Intravascular guidance”. Circulation 1995;91:1676-1688.
54. Bertrand ME, Legrand V, Boland J, Fleck E, Bonnier J, McFadden EP et al. “Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elec-tive coronary stenting”. Circulation 1998;98:1.597-1.603.
55. Leon MB, Baim DS, Popma JJ, Gordon PC, Cutlip DE, Kunz M et al. “A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting”. N Engl J Med 1998;339:1.665-1.671.
56. Kereiakes D et al. “Usefulness of stent length in predicting in-stent restenosis (the MULTI-LINK stent trials)”. Am J Cardiol 2000 Aug 1;86(3):336-41.
57. Akiyama T, Moussa I, Reimbers B, Ferraro M, Kobayashi Y, Colom-bo A et al. “Angiographic and clinical outcome following coronary stenting of small vessels. A comparison with coronary stenting of large vessels”. J Am Coll Cardiol 1998;32;1.610-1.618.
58. Savage MP, Fischman DL, Rake R, Leon MB, Schatz RA, Penn I et al. “Efficacy of coronary stenting versus balloon angioplasty in small coronary arteries. Stent restenosis study (STRESS) investigators”. J Am Coll Cardiol 1998;31:307-311.
59. Lau KW, He Q, Ding ZP, Johan A. “Safety and efficacy of angio-graphy-guided stent placement in small native coronary arteries of < 3.0 mm in diameter”. Clin Cardiol 1997;20:711-716.
60. Mintz GS, Hoffmann R, Mehran R, Pichard AD, Kent KM, Leon MB et al. “In-stent restenosis: the Washington Hospital Center experience”. Am J Cardiol 1998;81:7E-13E.
61. Tanner FC, Yang ZY, Duckers E, Gordon D, Nabel GJ, Nabel EG. “Expression of cyclin dependent kinase inhibitors in vascular disease”. Circ Res. 1998;82:396-403.
62. Nabel, EG. “CDKs and CKIs: molecular targets for tissue remo-deling”. Nat Rev Drug Discov. 2002;1:587-598.
63. Morgan, DO. “Principles of CDK regulation”. Nature. 1995;374:131-134.
64. Ser, CJ. “Cancer cell cycles”. Science. 1996;274:1672-1677.65. Ekholm SV, SI Reed. “Regulation of G(1) cyclin-dependent kinases
in the mammalian cell cycle”. Curr Opin Cell Biol. 2000;12:676-684.

Restenosis coronaria
77
66. Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massague J. “Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimi-togenic signals”. Cell. 1994;78:59-66.
67. Sherr CJ, JM Roberts. “CDK inhibitors: positive and negative regulators of G1-phase progression”. Genes Dev. 1999;13:1501-1512.
68. Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. “p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest”. Genes Dev. 1994;8:9-22.
69. Boehm M, Olive M, True AL, Crook MF, San H, Qu X, Nabel EG. “Bone marrow-derived immune cells regulate vascular disea-se through a p27(Kip1)-dependent mechanism”. J Clin Invest. 2004;114:419-426.
70. Kimura T, Yokoi H, Nakagawa Y, Tamura T, Kaburagi S, Sawa-da Y et al. “Three-year follow-up after implantation of metallic coronary artery stents”. N Engl J Med 1996;334:561-566.
71. Asakura M, Ueda Y, Nanto S, Hirayama A, Adachi T, Kitakaze et al. “Remodeling of in-stent neointima, wich became thinner and transparent over 3 years: serial angiographic and angioscopic follow-up”. Circulation 1998;97:2003-2006.
72. Klugherz BD, DeAngelo DL, Kim BK, Herrmann HC, Hirshfeld JW, Kolansky DM. “Three-year clinical follow-up after Palmaz-Schatz Stenting”. J Am Coll Cardiol 1996;27:1185-1191.
73. Serruys PW, Kootstra J, Melkert R, de Jaegere P, van den Brand M, Morek M. “Periprocedural quantitative coronary Angiography following Palmaz-Schatz stent implantation predicts reestenosis rate at 6 months: result of a meta-analysis of Benestent-I, Benestent- II and Music [resumen]”. J Am Coll Cardiol 1998;31(Supl A):64. 1997;80:711-715.
74. Kastrati A, Schömig A, Elezi S, Schühlen H, Wilhelm M, Dirs-chinger J et al. “Interlesion dependence of the risk for restenosis in patients with coronary stent placement in multiple lesions”. Circulation 1998;97:2396-2401.
75. Kastrati A, Schömig A, Elezi S, Schühlen H, Dirschinger J, Ha-damitzky M et al. “Predictive factors of restenosis after coronary stent placement”. J Am Coll Cardiol 1997;30:1428-1436.
76. Kasaoka S, Tobis J, Mauri J, Akiyama T, Reimers B, Di Mario C, Colombo A. et al. “Angiographic and intravascular ultrasound predictors of in-stent restenosis”. J Am Coll Cardiol 1998;32:1630-1635.
77. Amant C, Bauters C, Bodart JC, Lablanche JM, Grollier G, Dan-chin N et al. “Allele of the angiotensin I-converting enzyme is a major risk factor for restenosis after coronary stenting”. Circulation 1997;96:56-60.
78. Jefferson BK, Foster JH, McCarthy JJ, et al. “Aspirin resistance and a single gene . Am J Cardiol. 2005; 15;95: 805-8.
79. Leone AM, De Stefano V, Burzotta F, et al. “Glycoprotein Ia C807T gene polymorphism and increased risk of recurrent acute coronary syndromes: a five year follow up . Heart. 2004; 90: 567-9.
80. Nguyen TA, Diodati JG, Pharand C. “Resistance to clopidogrel: a review of the evidence . J Am Coll Cardiol. 2005; 19: 1157-64.
81. Isordia-Salas I, Leaños-Miranda A, Sainz I. “Asociación entre el polimorfismo 4G/5G en el gen del inhibidor del activador del plas-minógeno-1 (PAI-1) y el infarto agudo de miocardio con elevación del ST en pacientes jóvenes . Rev Esp Cardiol. 2009; 62: 365-72.
82. Grinfeld D, Sarmiento R, Dizeo C, et al. “Estudio del polimorfismo leu33/pro33 del receptor glicoproteico plaquetario IIIa (PLA) y su relación con la reestenosis postangioplastia coronaria con stent en una población argentina . Rev Argent Cardiol 2003; 71: 425-33.
83. Mourice MG, Serruys P, Fajade JJ, Sousa J. “Ravel study group. Randomized study with the sirolimus coated Bx Velocity balloon expandable stent in the tratement of patients with the novo native coronary lesions. A randomized comparison of a sirolimus eluting stent with a standrt stent for coronary revascularization”. N Eng J Med 2002;346:1773-80.
84. Colombo A, Moses JW, Morice MC, Ludwig J, Holmes DR. Jr., Spanos V et al. “Randomized study to evaluate sirolimus-eluting
stents implanted at coronary bifurcation lesions”. Circulation. 2004;109:1244-9.
85. Lemos PA, Saia F, Hofma SH, Daemen J, Ong AT, Arampatzis CA et al. “Short- and long-term clinical benefit of sirolimus-eluting stents compared to conventional bare stents for patients with acute myocardial infarction”. J Am Coll Cardiol 2004;43:704-8.
86. Hoye A, Lemos PA, Arampatzis CA, Saia F, Tanabe K, Deger-tekin M et al. “Effectiveness of the sirolimus-eluting stent in the treatment of saphenous vein graft disease”. J Invasive Cardiol 2004;16:230-3.
87. Ge L, Iakovou I, Sangiorgi GM, Chieffo A, Melzi G, Matteo M et al. “Treatment of saphenous vein grafts lesions with drug-eluting stents: immediate and mid-term outcome”. J Am Coll Cardiol 2005;45:A25.
88. De Lezo JS, Medina A, Pan M, Delgado A, Segura J, Pavlovic D et al. “Rapamycin-eluting stents for the treatment of unprotected left main coronary disease”. Am Heart J 2004;148:481-5.
89. Arampatzis CA, Lemos PA, Tanabe K, Hoye A, Degertekin M, Saia F et al. “Effectiveness of sirolimus-eluting stent for treatment of left main coronary artery disease”. Am J Cardiol 2003;92:327-9.
90. Hoye A, Tanabe K, Lemos PA, Aoki J, Saia F, Arampatzis C et al. “Significant reduction in restenosis after the use of sirolimus-eluting stents in the treatment of chronic total occlusions”. J Am Coll Cardiol 2004;43:1954-8.
91. Costa MA, Sabate M, Angiolillo DJ, Jimenez-Quevedo P, et al. “Intravascular ultrasound characterization of the ‘black hole’ phe-nomenon after drug-eluting stent implantation . Am J Cardiol. 2006; 97: 203-6.
92. Lemos PA, Hoye A, Goedhart D, Arampatzis CA, Saia F, Van der Giessen WJ et al. “Clinical, angiographic, and procedural predictors of angiographic restenosis after sirolimus-eluting stent implantation in complex patients: an evaluation from the Rapamycin-Eluting Stent Evaluated At Rotterdam Cardiology Hospital (RESEARCH) study”. Circulation 2004;109:1366-70.
93. Mintz GS, Hoffmann R, Mehran R, Pichard AD, Kent KM, Leon MB et al. “In-stent restenosis: the Washington Hospital Center experience”. Am J Cardiol 1998;81:7E-13E.
94. Kasaoka S, Tobis J, Mauri J, Akiyama T, Reimers B, Di Mario C, Colombo A. et al. “Angiographic and intravascular ultrasound predictors of in-stent restenosis”. J Am Coll Cardiol 1998;32:1630-1635.
95. Mehran R, Dangas G, Abizaid A, Mintz G, Lansky A, Satler L, Pichard A, Leon M et al. “Angiographic patterns of in-stent res-tenosis. Classification and implications for long-term outcome”. Circulation 1999;100:1872-1878.
96. Colombo A, Orlic D, Stankovic G, Corvaja N, Spanos V, Mon-torfano M et al. “Preliminary observations regarding angiographic pattern of restenosis after rapamycin-eluting stent implantation”. Circulation 2003;107:2178-80.
97. Reimbers B, Moussa I, Akiyama T, Tucci G, Di Mario C, Colom-bo A et al. “Long-term clinical follow-up after successful repeat percutaneous intervention for stent restenosis”. J Am Coll Cardiol 1997;30:186-192.
98. Dauerman HL, Baim DS, Cutlip DE, Sparano AM, Gibson CM, Cohen DJ et al. “Mechanical debulking versus balloon angioplas-ty for the treatment of diffuse in-stent restenosis”. Am J Cardiol 1998;82:277-284.
99. Bauters C, Banos JL, Van Belle E, Mc Fadden EP, Lablanche JM, Bertrand ME. “Six-month angiographic outcome after suc-cessful repeat percutaneous intervention for in-stent restenosis”. Circulation 1998;97:318-321.
100. Mehran R, Mintz GS, Satler LF, Pichard AD, Kent KM, Leon MB et al. “Treatment of in-stent restenosis with excimer laser coronary angioplasty: mechanisms and results compared with PTCA alone”. Circulation 1997;96:2.183-2.189.
101. Waksman R, White L, Chan R, Porrazo M, Bass B, Leon MB et al. “Intracoronary radiation theraphy for patients with in-stent res-tenosis: 6 month follow-up of a randomized clinical study”. Circulation 1998;98 (Supl 1):651:1.005-1.010.

Gelpi - Donato Fisiopatología cardiovascular
78
102. Mehran R, Mintz GS, Popma JJ, Pichard AD, Satler LF, Leon MB et al. “Mechanisms and results of balloon angioplasty for the treatment of in-stent restenosis”. Am J Cardiol 1996;78:618-622.
103. Mehran R, Dangas G, Minz G, Waksman R, Hong M, Leon M et al. “In-stent restenosis: ‘The great equalizer’. Disappointing clini-cal outcomes with all interventional strategies” [resumen]. J Am Coll Cardiol 1999;33(Supl A):63A.
104. Lee SG, Lee CW, Cheong SS, Hong MK, Kim JJ, Park SJ et al. “Immediate and long-term outcomes of rotational atherectomy versus balloon angioplasty alone for treatment of diffuse in-stent restenosis”. Am J Cardiol 1998;82:140-143.
105. Terstein PS, Massullo V, Jani S, Popma JJ, Minz GS, Leon MB et al. “A double-blinded randomized trial of catheter based radio-therapy to inhibit restenosis following coronary stenting”. N Engl J Med 1997;336:1.697-1.703.
106. Tanabe K, Serruys PW, Grube E, Smits PC, Selbach G, Van der Giessen WJ et al. “TAXUS III Trial: in-stent restenosis treated with
stent-based delivery of paclitaxel incorporated in a slow-release polymer formulation”. Circulation 2003;107:559-64.
107. Alfonso F, Ángel J, Cequier A, Augé JM, López Mínguez JR, Bethencourt A et al. “Results of the restenosis intrastent: balloon angioplasty versus rapamycin-eluting stent implantation rando-mized study”. J Am Coll Cardiol 2005;45:A83.
108. Kastrati A, Mehilli J, Von Beckerath N, Dibra A, Hausleiter J, Pache J et al. “ISAR-DESIRE Study investigators. Sirolimus-eluting stent or paclitaxel-eluting stent vs balloon angioplasty for preven-tion of recurrences in patients with coronary in-stent restenosis: a randomized controlled trial”. JAMA. 2005;293:165-71.
109. Stone GW, Ellis SG, O'Shaughnessy CD, Martin SL, et al. “Pacli-taxel-eluting stents vs vascular brachytherapy for in-stent restenosis within bare-metal stents: the TAXUS V ISR randomized trial . JAMA. 2006; 295: 1253-63.
110. Moses JW, Leon MB, Popma JJ, et al. “Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery . N Engl J Med. 2003; 349: 1315-23.
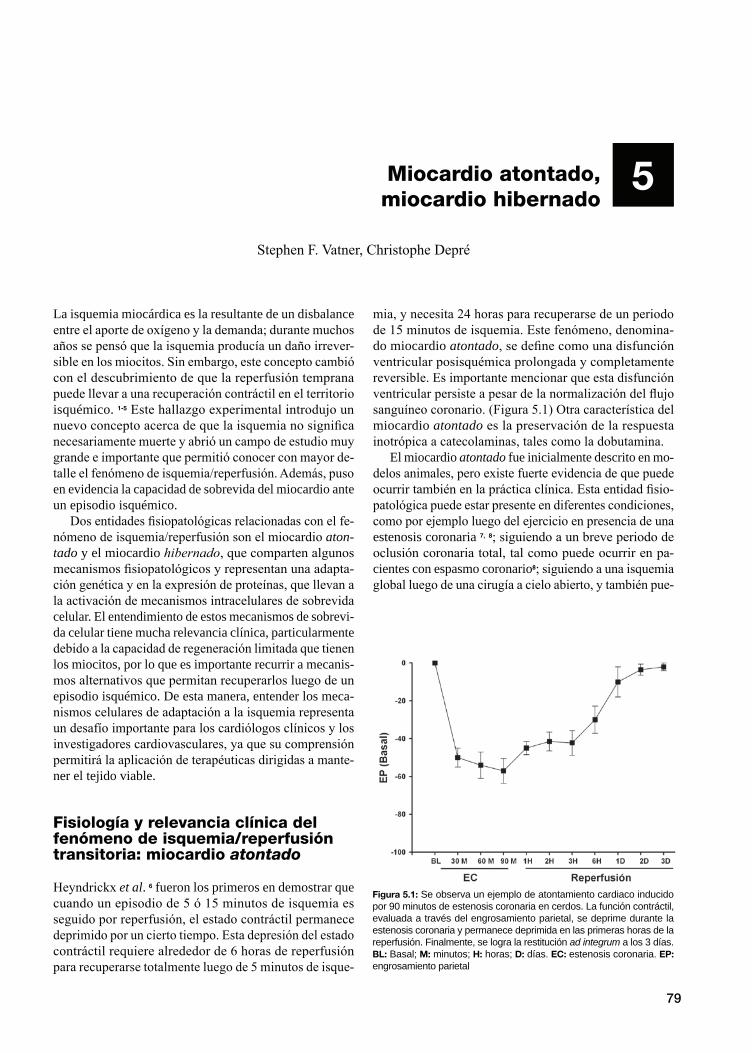
79
Miocardio atontado, miocardio hibernado
5
La isquemia miocárdica es la resultante de un disbalance
-
isquémico. 1-5
-
en evidencia la capacidad de sobrevida del miocardio ante un episodio isquémico.
-aton-
tado y el miocardio -
celular. El entendimiento de estos mecanismos de sobrevi-da celular tiene mucha relevancia clínica, particularmente
los miocitos, por lo que es importante recurrir a mecanis-
episodio isquémico. De esta manera, entender los meca-
-ner el tejido viable.
Fisiología y relevancia clínica del fenómeno de isquemia/reperfusión transitoria: miocardio atontado
et al. 6 fueron los primeros en demostrar que
-
-do miocardio atontado
miocardio atontado
El miocardio atontado fue inicialmente descrito en mo-
-
estenosis coronaria 7, 8
-cientes con espasmo coronario8
-
Figura 5.1: Se observa un ejemplo de atontamiento cardiaco inducido por 90 minutos de estenosis coronaria en cerdos. La función contráctil, evaluada a través del engrosamiento parietal, se deprime durante la estenosis coronaria y permanece deprimida en las primeras horas de la reperfusión. Finalmente, se logra la restitución ad integrum a los 3 días. BL: Basal; M: minutos; H: horas; D: días. EC: estenosis coronaria. EP: engrosamiento parietal

Gelpi - Donato Fisiopatología cardiovascular
80
de ocurrir en el subepicardio de un individuo con infarto subendocárdico. 9 Una de las características del miocardio atontado -
en el área atontadaatontado pue-
durante los episodios isquémicos intermitentes inducidos 9
las otras causas de atontamiento mencionadas que inducen
último, una causa frecuente de atontamiento miocárdico
de miocardio.
Mecanismos moleculares responsables del miocardio atontado
-mentales y especies animales para estudiar el miocardio atontado -
-
del calcio. Esta última postula que el miocardio atontado
atontado. 10-13
-atontado fue
probada por Kim et al. 14 en un modelo de atontamiento en
la arteria descendente anterior durante 90 minutos, con una
-nes atontadas -
in vivocuando los miocitos fueron aislados. Tanto los movimientos del calcio como la corriente de calcio a través de los canales tipo L estuvieron disminuidas en los miocitos atontados, lo
la troponina I en el modelo de atontamiento en cerdo 14, 15, lo que se opone a los datos obtenidos en modelos de rata. Este
el atontamiento tiene un comportamiento diferente de acuer-
en los miocitos atontados de rata 16
miocitos aislados de cerdo, lo que resulta consistente con lo observado in vivo en mamíferos, incluyendo estudios en
-
en el modelo de miocardio atontadoLas diferencias entre especies en el manejo del calcio
intracelular no son sorprendentes, considerando que los co-razones de ratas pueden contraerse y relajarse a frecuencias
-
-celulares del retículo sarcoplásmico. 17 En contraste, en
-
entra a la célula a través de los canales tipo L de calcio;
frecuencia lleva a un
-
fraccional a frecuencias cardiacas elevadas, lo que lleva a 17 Por el contrario,
positivo, que puede ser asociado con un aumento en las corrientes de calcio a través de los
ver capítulo 13
-mas de las cadenas pesadas de miosina, ya que la isoforma α
18 Por lo tanto, es posible
fosfolamban, en el manejo del calcio y en las alteraciones potenciales de otras proteínas, tales como la troponina C
atontado fue
6
Considerando las diferencias entre especies ya mencionadas,
troponina I. 19-22
al calcio intracelular. Recientemente, Murphy et al. 23 descri-bieron un fenotipo de miocardio atontado, caracterizado por
de la troponina I inducida por calpaína I.

Miocardio atontado, miocardio hibernado
81
En consecuencia, el miocardio atontado puede ser des--
tos
15, 24 Además, se ha
25 En este -
tran que la proteolisis de la troponina I ocurre en pacientes 19 No
-
19
-cales libres. Desde principios de la década del 80, muchos
--
26-33 Es conocido que los radicales libres son producidos, principalmente,
34, 35 El incremento de estas especies reactivas 36, 37
37 y la dis-
-38 La
38 Aunque el
se han evaluado varias posibilidades. 9, 36 El mecanismo más
alterar la bomba de calcio del retículo sarcoplásmico. Otros -
cambiador Na /Ca y la bomba Na /K ATPasa. Una desre-
a través del sarcolema y junto con la actividad alterada del
39
40
Atontamiento como mecanismo del miocardio hibernado
El miocardio -
41
-
-tada del miocardio. El término , que se usa
por primera vez por Diamond et al. en 197842, pero fue
41
El miocardio hibernado fue propuesto como una con-
--
sis del corazón inteligente 41
y aton-tado
-
-
-bajos clínicos y, de esta forma, comienza a aparecer un
no puede aumentar apropiadamente debido a la presencia de estenosis coronaria y a la limitada reserva vasodilata-
presencia de episodios repetitivos de isquemia y reper-
atontamiento que en
atontamiento repetitivo 43
de la reserva vasodilatadora coronaria, que representa la
44 En los
-45
De esta manera, cuando la estenosis coronaria se vuelve crítica, la reserva vasodilatadora se encuentra totalmente reclutada, y por lo tanto cualquier estímulo que aumente
inducirá isquemia. 46 Estos breves pero repetidos episo-dios de isquemia, que pueden no ser sintomáticos, son
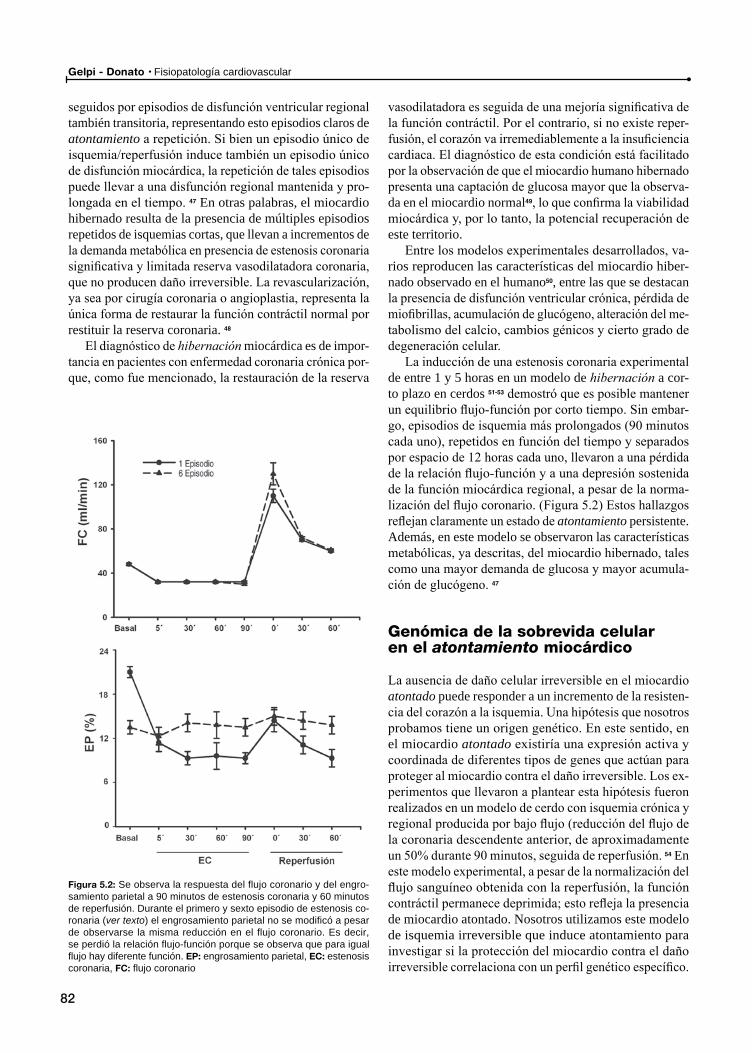
Gelpi - Donato Fisiopatología cardiovascular
82
también transitoria, representando esto episodios claros de atontamiento
-47 En otras palabras, el miocardio
hibernado resulta de la presencia de múltiples episodios repetidos de isquemias cortas, que llevan a incrementos de
restituir la reserva coronaria. 48
miocárdica es de impor--
-
-da en el miocardio normal49
este territorio.-
rios reproducen las características del miocardio hiber-nado observado en el humano50, entre las que se destacan
-
de entre 1 y 5 horas en un modelo de a cor-to plazo en cerdos 51-53
-
-
atontamiento persistente. Además, en este modelo se observaron las características
-47
Genómica de la sobrevida celular en el atontamiento miocárdico
atontado puede responder a un incremento de la resisten-
el miocardio atontado
-
54 En
de miocardio atontado. Nosotros utilizamos este modelo de isquemia irreversible que induce atontamiento para
Figura 5.2: Se observa la respuesta del flujo coronario y del engro-samiento parietal a 90 minutos de estenosis coronaria y 60 minutos de reperfusión. Durante el primero y sexto episodio de estenosis co-ronaria (ver texto) el engrosamiento parietal no se modificó a pesar de observarse la misma reducción en el flujo coronario. Es decir, se perdió la relación flujo-función porque se observa que para igual flujo hay diferente función. EP: engrosamiento parietal, EC: estenosis coronaria, FC: flujo coronario
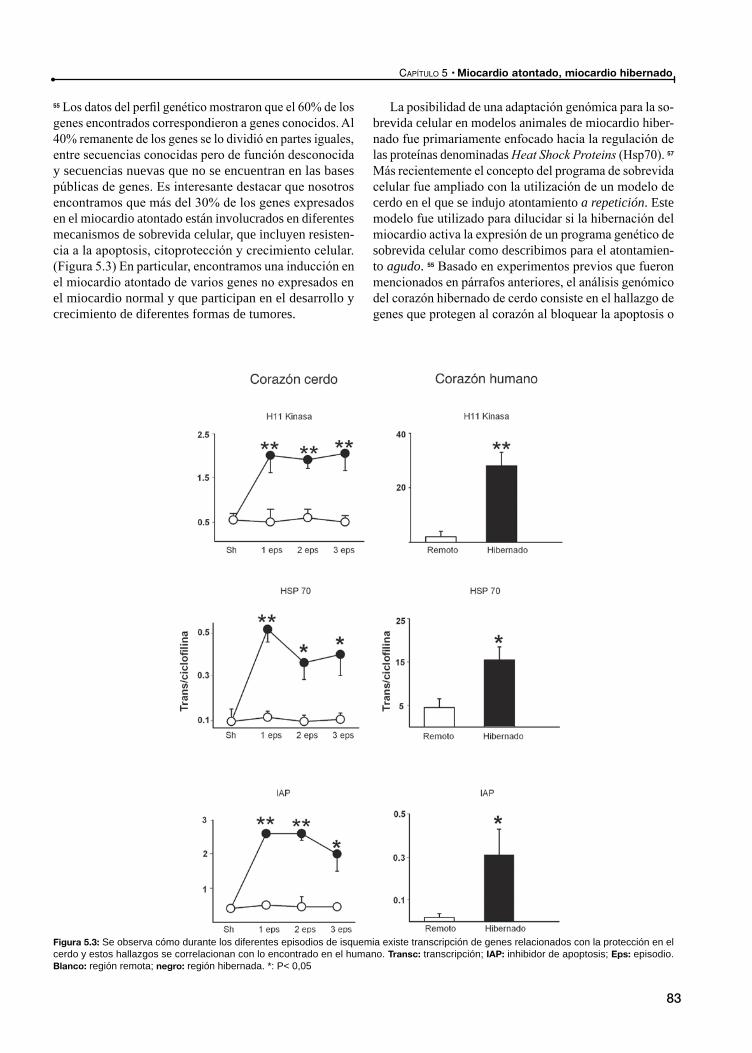
Miocardio atontado, miocardio hibernado
83
55
y secuencias nuevas que no se encuentran en las bases
en el miocardio atontado están involucrados en diferentes mecanismos de sobrevida celular, que incluyen resisten-
el miocardio normal y que participan en el desarrollo y crecimiento de diferentes formas de tumores.
-brevida celular en modelos animales de miocardio hiber-
las proteínas denominadas 57
cerdo en el que se indujo atontamiento a repetición. Este
sobrevida celular como describimos para el atontamien-to agudo. 55
Figura 5.3: Se observa cómo durante los diferentes episodios de isquemia existe transcripción de genes relacionados con la protección en el cerdo y estos hallazgos se correlacionan con lo encontrado en el humano. Transc: transcripción; IAP: inhibidor de apoptosis; Eps: episodio. Blanco: región remota; negro: región hibernada. *: P< 0,05

Gelpi - Donato Fisiopatología cardiovascular
84
55
como también tiene un efecto citoprotector por estimular
por isquemia tanto in vivo como in vitro. 58-60
-mulan el crecimiento celular y la sobrevida, tales como el H11-kinasa 61
62
-
Dado que en el miocardio hibernado humano ocurren
-
en pacientes. 47 -co del área hibernada y también del territorio normal del
estuvieron aumentados después del primer episodio de -
citoprotectores y promotores del crecimiento, es inducido
-
-
el crecimiento celular, independientemente de cualquier 63
-
muestras de tejido del área hibernada y del área normal
-
comparado con el miocardio normal. Las proteínas corres-
celular, lo que probablemente representa el mecanismo molecular de viabilidad celular en el territorio isquémico. Por lo tanto, a partir de este mecanismo podremos conocer la forma de recuperar funcionalmente a los miocitos que
-
y hemodinámicos que ocurren en el miocardio hibernado
sobrevida celular.En otras palabras, el miocardio humano hibernado está
-
-cular del miocardio hibernado. Es importante mencionar que en el modelo de cerdo con episodios repetidos de is-quemia hubo un aumento de la apoptosis después del pri-
sobrevida celular mencionados para el miocardio atonta-do e hibernado también podrían bloquear los mecanismos de apoptosis.
De esta manera, hemos detectado, en nuestro modelo
-cia no se encuentra en bases públicas de datos mientras
-
nuestro conocimiento de los mecanismos involucrados en la sobrevida celular, que son activados por la isquemia.
-atontado
como del , lo que redundará en una terapéutica más apropiada.
La sensibilidad de la técnica de PCR permite medir y evaluar, en forma separada, el endocardio y el epicardio, lo
-
55 Por lo tanto, -

Miocardio atontado, miocardio hibernado
85
todo o nada sino que es proporcional a la intensidad del estímulo inicial.
Relación entre miocardio atontado, hibernado y precondicionamiento
puede desarrollar mecanismos -
tido, la a los mecanismos involucrados, con el precondiciona-
64 El precondiciona-miento isquémico
infarto; por ello sus mecanismos moleculares son aún
Una primera ventana del precondicionamiento isqué--
sodio de isquemia65
de precondicionamiento66, 67
horas a partir del estímulo precondicionante. Los meca-nismos involucrados en la primera ventana consisten en
-
Figura 5.4: Se observa esquematizada la hipótesis de protección celular endógena en la que se propone que diferentes genes son activados en respuesta a una isquemia de corta duración (miocardio atontado) o una más prolongada (miocardio hibernado). Esta activación induce la síntesis de proteínas protectoras y podría estar en la misma dirección que el precondicionamiento isquémico.
¿3era ventana del precondicionamiento isquémico?

Gelpi - Donato Fisiopatología cardiovascular
86
68 Mientras que la relevancia clínica del precondicionamiento isquémico todavía es materia de debate, la posibilidad de que tanto el miocardio atontado como el equivalente a la observada con el precondicionamiento
et al. demostraron que episodios repetitivos de isquemia inducen una nueva ventana para el precondicionamiento
era
diferentes al precondicionamiento clásico. 69
El miocardio atontado, y el precondicio-namiento isquémico presentan dos importantes caracte-rísticas en común. 70
Conclusión
son caracterizadas por una pérdida del equilibrio entre el
En el miocardio atontado, este presenta alteraciones de la
la ausencia de necrosis. En forma similar, en el miocardio
que ocurre en el miocardio es con frecuencia -
parte, cuando el miocardio está -
--
atontado
-
Bibliografía
11. Ginks WR, Sybers HD, Maroko PR, Covell JW, Sobel BE, Ross J Jr. “Coronary artery reperfusion. II. Reduction of myocardial infarct size at 1 week after the coronary occlusion”. J Clin Invest 1972; 51:2717-2723.
12. Maroko PR, Kjekshus JK, Sobel BE, Watanabe T, Covell JW, Ross J Jr et al. “Factors influencing infarct size following experimental coronary artery occlusions”. Circulation 1971; 43:67-82.
13. Maroko PR, Libby P, Ginks WR, Bloor CM, Shell WE, Sobel BE et al. “Coronary artery reperfusion. I. Early effects on local myocardial function and the extent of myocardial necrosis”. J Clin Invest 1972; 51:2710-2716.
14. Matsuzaki M, Gallagher KP, Kemper WS, White F, Ross J Jr. “Sustained regional dysfunction produced by prolonged coronary stenosis: gradual recovery after reperfusion”. Circulation 1983; 68:170-182.
15. Theroux P, Ross J Jr, Franklin D, Kemper WS, Sasayama S. “Co-ronary arterial reperfusion. III. Early and late effects on regional myocardial function and dimensions in conscious dogs”. Am J Cardiol 1976; 38:599-606.
16. Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF. “Regional myocardial functional and electrophysiological al-terations after brief coronary artery occlusion in conscious dogs”. J Clin Invest 1975; 56:978-985.
17. Ambrosio G, Betocchi S, Pace L, Losi MA, Perrone-Filardi P, Soricelli A et al. “Prolonged impairment of regional contractile function after resolution of exercise-induced angina. Evidence of myocardial stunning in patients with coronary artery disease”. Circulation 1996; 94:2455-2464.
18. Ambrosio G, I Tritto. “Clinical manifestations of myocardial stun-ning”. Coron Artery Dis 2001; 12:357-361.
19. Bolli, R. “Myocardial ‘stunning’ in man”. Circulation 1992; 86:1671-1691.
10. Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF. “Regional myocardial functional and electrophysiological al-terations after brief coronary artery occlusion in conscious dogs”. J Clin Invest 1975; 56:978-985.
11. Braunwald E, RA Kloner. “The stunned myocardium: Prolon-ged, postischemic ventricular dysfunction”. Circulation 1982; 66:1146-1149.
12. Kusuoka H, Porterfield J, Weisman H, Weisfeldt M, Marban E. “Pathophysiology and pathogenesis of stunned myocardium. De-pressed Ca activation of contraction as a consequence of reper-fusion-induced cellular calcium overload in ferret hearts”. J Clin Invest 1987; 79:950-961.
13. Marban, E. “Myocardial stunning and hibernation. The physiology behind the colloquialisms”. Circulation 1991; 83:681-688.
14. Kim SJ, Kudej RK, Yatani A, Kim Y, Takagi G, Honda R, Colantonio D, VanEyk J, Vatner D, Rasmusson R, Vatner SA. “Novel mechanism for myocardial stunning involving impaired Ca2+ handling”. Circ Res 2001; 89:821-837.
15. Thomas S, Fallavollita J, Lee T, Feng J, Canty J. “Absence of tro-ponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine”. Circ Res 1999; 85:446-456.
16. Gao W, Atar D, Backx P, Marban E. “Relationship between in-tracellular calcium and contractile force in stunned myocardium. Direct evidence for decreased myofilament responsiveness and altered diastolic function in intact ventricular muscle”. Circ Res 1995; 76:1036-1048.
17. Bers, D. “Calcium fluxes involved in control of cardiac myocyte contraction”. Circ Res 2000; 87:275-281.
18. Schwartz K, Lompre AM, Bouveret P, Wisnesky C, Whalen RG. “Comparison of rat cardiac myosins at fetal stages, in young animals and in hyptohyroid adults”. J Biol Chem 1982; 257:14412-14417.

Miocardio atontado, miocardio hibernado
87
19. McDonough J, Arrell D, Van Eyk J. “Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury”. Circ Res 1999; 84:9-20.
20. Gao WD, Liu Y, Mellgren R, Marban E. “Intrinsic myofilament alterations underlying the decreased contractility of stunned myo-cardium. A consequence of Ca2+-dependent proteolysis?” Circ Res 1996; 78:455-465.
21. Gao W, Atar D, Liu Y, Perez N, Murphy A, Marban E. “Role of troponin I proteolysis in the pathogenesis of stunned myocardium”. Circ Res 1997; 80:393-399.
22. Van Eyk J, Powers F, Law W, Larue C, Hodges R, Solaro R. “Break-down and release of myofilament proteins during ischemia and ische-mia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation”. Circ Res 1998; 82:261-271.
23. Murphy A, Kogler H, Georgakopoulos D, McDonough J, Kass D, Van Eyk JEM. “Transgenic mouse model of stunned myocardium”. Science 2000; 287:488-491.
24. Luss H, Boknik P, Heusch G, Muller F, Neumann J, Schmitz W, Schulz R. “Expression of calcium regulatory proteins in short-term hibernation and stunning in the in-situ porcine heart”. Cardiovasc Res 1998; 37:606-617.
25. Luss H, Boknik P, Heusch G, Muller F, Neumann J, Schmitz W, Schulz R. “Expression of calcium regulatory proteins in short-term hibernation and stunning in the in-situ porcine heart”. Cardiovasc Res 1998; 37:606-617.
26. Feng J, Schaus BJ, Fallavollita JA, Lee T-C, Canty JM, Jr. “Preload Induces Troponin I Degradation Independently of Myocardial Ischemia”. Circulation 2001; 103:2035-2037.
27. Myers M, Bolli R, Lekich R, Hartley C, Michael L, Roberts B. “Enhancement of recovery of myocardial function by oxygen free-radical scavengers after reversible regional ischemia”. Circulation 1985; 72:915-921.
28. Bolli R, Jeroudi M, Patel B, Aruoma O, Halliwell B, Lai E, McCay P. “Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reper-fusion. Evidence that myocardial ‘stunning’ is a manifestation of reperfusion injury”. Circ Res 1989; 65:607-622.
29. Bolli R, Patel B, Jeroudi M, Lai E, McCay P. “Demonstration of free radical generation in a ‘stunned’ myocardium of intact dogs with the use of spin trap alpha-phenyl N-Tertbutyl nitrone”. J Clin Invest 1988; 82:476-485.
30. Gross G, Farber N, Haroman H, Waltier D. “Beneficial actoins of superoxide dismutase and catalase in stunned myocardium of dogs”. Am J Physiol 1986; 250:H372-H377.
31. Koerner J, Anderson B, Dage R. “Protection against postischemic myocardial dysfunction in anesthetized rabbits with scavengers of oxygen-derived free radicals”. J Cardiovasc Pharmacol 1991; 17:185-191.
32. Przyklenk K, R Kloner. “Reperfusion-injury by oxygen-derived free radicals”. Circ Res 1989; 64:86-96.
33. Przylenk K, R Kloner. “Superoxide dismutase plus catalase im-proves contractile function in the canine model of the ‘stunned’ myocardium”. Circ Res 1986; 58:148-156.
34. Sekili S, McCay P, Li X, Zughaib M, Sumn J, Tang X, Thornby J, Bolli R. “Direct evidence that the hydroxyl radical plays a patho-genic role in myocardial ‘stunning’ in the conscious dog and that stunning can be markedly attenuated without subsequent adverse effects”. Circ Res 1993; 73:705-723.
35. Li X, McCay P, Zughaib M, Jeroudi M, Triana J, Bolli R. “Demons-tration of free radical generation in the ‘stunned’ myocardium in the conscious dog and identification of major differences between conscious and open-chest dogs”. J Clin Invest 1993; 92:1025-1041.
36. Zughaib M, Abd-Eltfattah A, Jeroudi M, Sun J-Z, Sekili S, Tang X-L, Bolli R. “Augmentation of endogenous adenosine attenutates myocardial stunning independently of coronary flow or hemody-namic effects”. Circulation 1993; 88:2359-2369.
37. Sun JZ, Tang XL, Knowlton AA, Park SW, Qiu Y, Bolli R. “Late preconditioning against myocardial stunning. An endogenous protective mechanism that confers resistance to postischemic dys-
function 24 h after brief ischemia in conscious pigs”. J Clin Invest 1995; 95:388-403.
38. Huang C, Vatner S, Peppas A, Yang G, Kudej R. “Cardiac Nerves Affect Myocardial Stunning Through Reactive Oxygen and Nitric Oxide Mechanisms”. Circ Res 2003; 93:866-873.
39. Corretti MC, Koretsune Y, Kusuoka H, Chacko VP, Zweier JL, Marban E. “Glycolytic inhibition and calcium overload as conse-quences of exogenously generated free radicals in rabbit hearts”. J Clin Invest 1991; 88:1014-1025.
40. Macfarlane N, D Miller. “Depression of peak force without altering calcium sensitivity by the superoxide anion in chemically skinned cardiac muscle of rat”. Circ Res 1992; 70:1217-1224.
41. Rahimtoola, SH. “A perspective on the three large multicenter randomized clinical trials of coronary bypass surgery for chronic stable angina”. Circulation 1985; 72:V123–V135.
42. Diamond GA, Forrester JS, de Luz PL, Wyatt HL, Swan HJ. “Post-extrasystolic potentiation of ischemic myocardium by atrial sti-mulation”. Am Heart J 1978; 95:204-209.
43. Vanoverschelde JL, Wijns W, Depre C, Essamri B, Heyndrickx GR, Borgers M et al. “Mechanisms of chronic regional postis-chemic dysfunction in humans. New insights from the study of noninfarcted collateral-dependent myocardium”. Circulation 1993; 87:1513-1523.
44. Feigl, EO. “Coronary physiology”. Physiol Rev 1983; 63:1-205.45. Uren NG, Melin JA, De Bruyne B, Wijns W, Baudhuin T, Camici
PG. “Relation between myocardial blood flow and the severity of coronary-artery stenosis”. N Engl J Med 1994; 330:1782-1788.
46. Berne, RM. “The role of adenosine in the regulation of coronary blood flow”. Circ Res 1980; 47:807-813.
47. Kim SJ, Peppas A, Hong SK, Yang G, Huang Y, Diaz G et al. “Persistent stunning induces myocardial hibernation and protec-tion: flow/function and metabolic mechanisms”. Circ Res 2003; 92:1233-1239.
48. Wijns W, Vatner SF, Camici PG. “Hibernating myocardium”. N Engl J Med 1998; 339:173-181.
49. Tamaki N, Yonekura Y, Yamashita K, Saji H, Magata Y, Senda M et al. “Positron emission tomography using fluorine-18 deoxyglucose in evaluation of coronary artery bypass grafting”. Am J Cardiol 1989; 64:860-865.
50. Borgers M, J Ausma. “Structural aspects of the chronic hibernating myocardium in man”. Basic Res Cardiol 1995; 90:44-46.
51. Arai AE, Pantely GA, Anselone CG, Bristow J, Bristow JD. “Active downregulation of myocardial energy requirements during prolon-ged moderate ischemia in swine”. Circ Res 1991; 69:1458-1469.
52. Schulz R, Guth BD, Pieper K, Martin C, Heusch G. “Recruitment of an inotropic reserve in moderately ischemic myocardium at the expense of metabolic recovery. A model of short-term hibernation”. Circ Res 1992; 70:1282-1295.
53. Schulz R, Rose J, Martin C, Brodde OE, Heusch G. “Development of short-term myocardial hibernation. Its limitation by the severity of ischemia and inotropic stimulation”. Circulation 1993; 88:684-695.
54. Kudej RK, Kim SJ, Shen YT, Jackson JB, Kudej AB, Yang GP, et al. “Nitric oxide, an important regulator of perfusioncontraction matching in conscious pigs”. Am J Physiol Heart Circ Physiol 2000; 279:H451-H456.
55. Depre C, Tomlinson JE, Kudej RK, Gaussin V, Thompson E, Kim SJ et al. “Gene program for cardiac cell survival induced by transient ischemia in conscious pigs”. Proc Natl Acad Sci USA 2001; 98:9336-9341.
56. Van Eyk J, M Dunn. “Proteomic and genomic analysis of cardio-vascular disease”. Wiley 2002; 85-98.
57. Fallavollita JA, Jacob S, Young RF, Canty JM Jr. “Regional al-terations in SR Ca(2+)-ATPase, phospholamban, and HSP-70 expression in chronic hibernating myocardium”. Am J Physiol 1999; 277:H1418-H1428.
58. Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. “Small heat shock proteins and protection against ischemic injury in cardiac myocytes”. Circulation 1997; 96:4343-4348.

59. Radford NB, Fina M, Benjamin IJ, Moreadith RW, Graves KH, Zhao P et al. “Cardioprotective effects of 70-kDa heat shock protein in transgenic mice”. Proc Natl Acad Sci USA 1996; 93:2339-2342.
60. Trost SU, Omens JH, Karlon WJ, Meyer M, Mestril R, Covell JW, et al. “Protection against myocardial dysfunction after a brief ischemic period in transgenic mice expressing inducible heat shock protein 70”. J Clin Invest 1998; 101:855-862.
61. Depre C, Hase M, Gaussin V, Zajac A, Wang L, Hittinger L et al. “H11 kinase is a novel mediator of myocardial hypertrophy in vivo”. Circ Res 2002; 91:1007-1014.
62. Williams RS, IJ Benjamin. “Protective responses in the ischemic myocardium”. J Clin Invest 2000; 106:813-818.
63. Suzuki G, Lee T-C, Fallavollita JA, Canty JM Jr. “Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle”. Circ Res 2005; 96:767-775.
64. Murry CE, Jennings RB, Reimer KA. ”Preconditioning with is-chemia: a delay of lethal cell injury in ischemic myocardium”. Circulation 1986; 74:1124-1136.
65. Murry CE, Richard VJ, Jennings RB, Reimer KA. “Myocardial protection is lost before contractile function recovers from ischemic preconditioning”. Am J Physiol 1991; 260:H796-H804.
66. Kuzuya T, Hoshida S, Yamashita N, Fuji H, Oe H, Hori M et al. “Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia”. Circ Res 1993; 72:1293-1299.
67. Marber MS, Latchman DS, Walker JM, Yellon DM. “Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction”. Circulation 1993; 88:1264-1272.
68. Bolli, R. “Cardioprotective function of inducible nitric oxide syn-thase and role of nitric oxide in myocardial ischemia and precon-ditioning: an overview of a decade of research”. J Mol Cell Cardiol 2001; 33:1897-1918.
69. Shen Y, Depre C, Yan L. et al. Repetitive ischemia by coronary stenosis induces a novel window of Ischemic preconditioning circulation 2008; 118: 1961-1969.
70. Heusch G, Schulz R, Rahimtoola SH. “Myocardial hibernation: a delicate balance”. Am J Physiol 2005; 288:H984-H999.

89
Protección miocárdica
La enfermedad arterial coronaria es la mayor causa de mortalidad y morbilidad en el mundo occidental, y para
el mundo. 1 Representa un continuo de diferentes estados -
de estos estadios de la enfermedad representa un objetivo
como en el área básica. Es importante tener en cuenta que
-rejadas consecuencias importantes y con frecuencia fatales,
que ocluye las arterias coronarias mayores y lleva indefec-tiblemente al infarto de miocardio. 2 En este escenario, la
--
Por lo tanto, para disminuir la morbimortalidad es ne-
protejan al miocardio de la injuria por isquemia/reperfu--
tratamiento que es necesario administrar antes de que el evento isquémico ocurra, lo que resulta difícil de aplicar en la práctica clínica dado lo impredecible del comienzo de
-
Protección del corazón en la injuria por isquemia/reperfusión: precondicionamiento y
poscondicionamiento isquémico6
Derek M. Yellon, Derek J. Hausenloy
-no de precondicionamiento isquémico que fue descrito por primera vez por Murry et al 3; esta maniobra
-
debe ser realizada antes de que el evento isquémico ocu-
---
4
del miocardio contra este tipo de injuria, que resulta de la apertura de una arteria coronaria ocluida, ofrece por sí
-mico5 describe un efecto protector inducido por múltiples
-
Protección miocárdica por precondicionamiento isquémico
et al. 3 demostraron, utilizando un modelo
que cuatro episodios alternativos de cinco minutos de
-
3
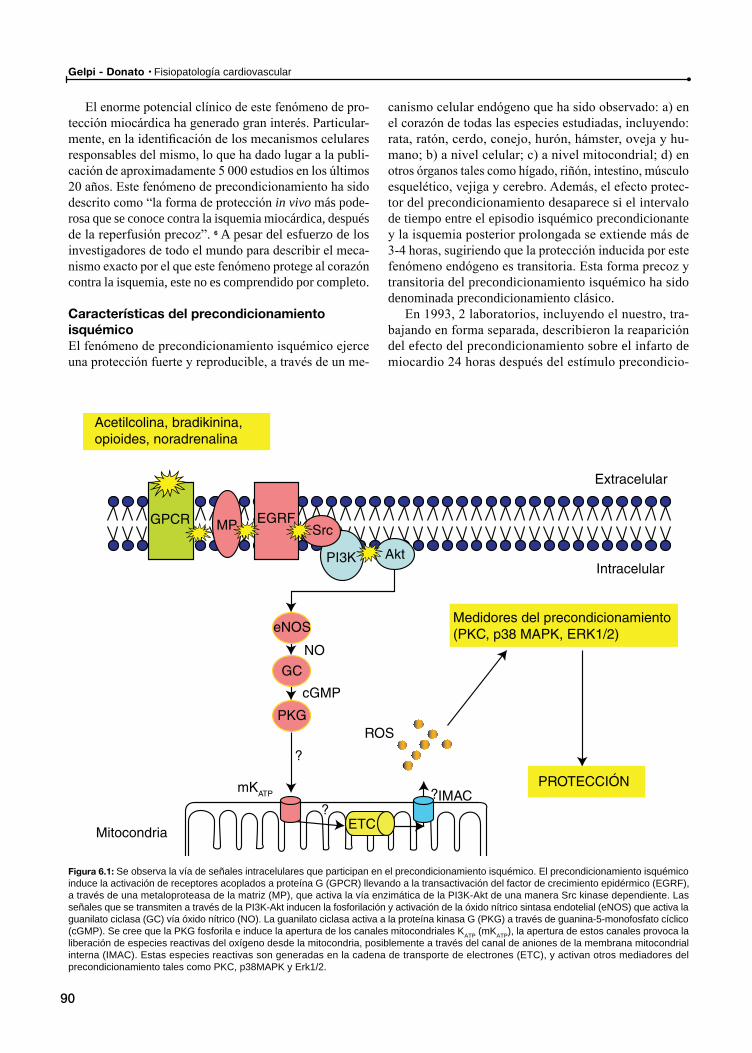
Gelpi - Donato Fisiopatología cardiovascular
90
--
-
in vivo más pode-rosa que se conoce contra la isquemia miocárdica, después
6 A pesar del esfuerzo de los -
contra la isquemia, este no es comprendido por completo.
Características del precondicionamiento isquémico
-
-
-tor del precondicionamiento desaparece si el intervalo de tiempo entre el episodio isquémico precondicionante
transitoria del precondicionamiento isquémico ha sido denominada precondicionamiento clásico.
-
del efecto del precondicionamiento sobre el infarto de -
Figura 6.1: Se observa la vía de señales intracelulares que participan en el precondicionamiento isquémico. El precondicionamiento isquémico induce la activación de receptores acoplados a proteína G (GPCR) llevando a la transactivación del factor de crecimiento epidérmico (EGRF), a través de una metaloproteasa de la matriz (MP), que activa la vía enzimática de la PI3K-Akt de una manera Src kinase dependiente. Las señales que se transmiten a través de la PI3K-Akt inducen la fosforilación y activación de la óxido nítrico sintasa endotelial (eNOS) que activa la guanilato ciclasa (GC) vía óxido nítrico (NO). La guanilato ciclasa activa a la proteína kinasa G (PKG) a través de guanina-5-monofosfato cíclico (cGMP). Se cree que la PKG fosforila e induce la apertura de los canales mitocondriales KATP (mKATP), la apertura de estos canales provoca la liberación de especies reactivas del oxígeno desde la mitocondria, posiblemente a través del canal de aniones de la membrana mitocondrial interna (IMAC). Estas especies reactivas son generadas en la cadena de transporte de electrones (ETC), y activan otros mediadores del precondicionamiento tales como PKC, p38MAPK y Erk1/2.
Acetilcolina, bradikinina,opioides, noradrenalina
GPCR MP EGRF
PI3K Akt
eNOS
GC
PKG
ETC
Src
Medidores del precondicionamiento(PKC, p38 MAPK, ERK1/2)
PROTECCIÓN
Extracelular
Intracelular
ROS
IMAC
NO
cGMP
mKATP
Mitocondria
?
??

Protección del corazón en la injuria por isquemia/reperfusión...
91
-nejos 7 y perros. 8
denominada segunda ventana de protección o precon-dicionamiento tardío.
Indicadores de la protección inducida por precondicionamiento isquémico
-
ventriculares 9
ventricular posisquémica en corazones aislados. 10
-
producido por un episodio no letal de isquemia, es motivo 11
-
12 -
Precondicionamiento clásico
el precondicionamiento isquémico no han podido ser di--
estimulados por estos componentes intracelulares.
--
ya que ciertos componentes pueden actuar como estimu-ladores o como mediadores/efectores. Recientemente, han sido descritas las diferentes vías intracelulares que
-
-
Estimuladores del precondicionamiento clásicoLos estimuladores del precondicionamiento isquémico son factores que actúan antes del episodio isquémico
-tracelulares, y son los que tienen la memoria del precon-dicionamiento isquémico.a) Ligandos de receptores acoplados a proteína G
-
estímulo del precondicionamiento isquémico. Demos--
miento isquémico podía ser abolida con el pretratamiento
Figura 6.2: Representación esquemática de las 4 clases más importantes de familias de MAPK. Cada clase mayor de MAPK, Erk1/2, BMK1, JNK1/2 y p38 comprenden 3 módulos de un MKKK-MKK-MAPK. Estas MAPK’s son activadas en respuesta a la isquemia/reperfusión y pueden ser activadas también en respuesta al precondicionamiento isquémico (con receptores acoplados a proteína G, GPCR’s o receptores con actividad de tirosina kinasa, RTyrK’s). Estas MAPK’s fosforilan sus sustratos (en verde, ver texto para detalles), que median la protección a través de mecanismos anti-apoptóticos o regulando la expresión de genes.
Isquemia/Reperfusión
GPCR, RTyrK
MKKK: Raf1/2MKK: MEK1/2MAPK: ERK1/2
MEKK1-3MEK5
BMK1(ERK5)
MEKK1-3MKK 3/6p38a/
cfos/elk-1p90RSK, BAD
c-junMEF2A/MEF2C
c-jun
MAPKAPK2
HSP 27
Antiapoptóticoestabilizacióncitoesqueleto
Antiapoptótico Expresión, genes
MEKK1-3MKK4/7JNK1/2

Gelpi - Donato Fisiopatología cardiovascular
92
-
-
que actuarían como estímulos del precondicionamiento -
cionamiento activa un receptor acoplado a proteína G,
la teoría del receptor acoplado a proteína G y describieron -
II y endotelina 1.
a proteína G, durante periodos cortos de isquemia/reper-
membrana celular a la mitocondria.b) La cascada de la enzima PI3K-Akt kinasa
-
et al. 13 fueron los primeros en demostrar que el precondi-
al abolir la actividad de la Akt, utilizando un inhibidor de
isquémico desaparecía. Los mismos autores mostraron la
-
del precondicionamiento.Por otro lado, Downey et al. 14-16 han demostrado que
--
16 mostraron -
namiento isquémico a la mitocondria, a través de una vía 17
c) El óxido nítrico como estimulador del precondicionamiento
17
ciclasa, a través del GMPc, activa la proteína kinasa G 17 Finalmente se cree que la PKG fosforila y abre
los canales KATP de la mitocondria.d) Los canales KATP y la protección miocárdicaEl rol de los canales KATPprecondicionamiento isquémico es una historia interesante
et alATP -
triculares aislados de hámster. 18 En este trabajo pionero, Noma et al. 18 postulan que la apertura de este canal,
a través de una corriente repolarizante de K . El acorta-
Posteriormente, Cole et al. 19 -
ATP
KATP -et
al. 20 describen que la apertura de los canales KATP está -
namiento isquémico, en el mismo estudio demostraron que
Como se ha mencionado, los primeros trabajos pos--
miento isquémico y por los canales KATP se debía al acor-
con la apertura de los canales KATP era independiente del
canales KATP del sarcolema podían no ser los responsables -
-ATP, ubicado en la membrana
mitocondrial interna, que estaría directamente involucrado
e) Los canales KATP mitocondriales y la protección miocárdicaEn 1991 21, Inoue et al. -
ATP. En
rol para los canales KATP de la mitocondria en la protec-22
En este estudio, se reconstituyeron proteínas de la mem-brana interna de la mitocondria conteniendo canales KATP

Protección del corazón en la injuria por isquemia/reperfusión...
93
que fueron reconstituidos dentro de proteoliposomas, y -
ATP del sarcolema para inducir una corriente de K 22
canal K ATP del sarcolema ni acortaban el potencial de -
ATP, y por el 22
f) El canal KATP mitocondrial como estimulador del precondicionamiento isquémico
23
ATP de la mitocon-dria indujo un estado de precondicionamiento de hasta
ATP
inducida por el precondicionamiento isquémico.g) Relación entre los canales KATP de la mitocondria y las especies reactivas del oxígeno (ROS)Diferentes estudios consideraron a las especies reactivas
-dicionamiento 24-25
ser el complejo III de la cadena de transporte de electrones mitocondrial, pues se ha demostrado que son liberados en respuesta al estímulo de precondicionamiento. 12, 17 En este sentido, Forbes et al. 26 fueron los primeros en relacionar
los canales KATP la apertura de los canales KATP
son liberados dentro del citosol y activan mediadores del precondicionamiento tales como la PKC. 12
21 et
al. 22ATP de la
mitocondria permanece desconocida. Como consecuencia de esto, muchas de sus acciones han sido inferidas reali-
de estos canales.h) Especies reactivas del oxígeno
-
27 Por
et al. 24 demostraron
por diversos estudios. 28-29
En este sentido, Tritto et al.25 fueron los primeros en
-
et al.12 mostraron, en miocitos de pollo neonato, que el precon-
-O
la cadena de transporte de electrones de la mitocondria. El mecanismo por el que el precondicionamiento indu-
ATP. 16, 23 Una
mitocondria podrían activar kinasas, tales como PKC29,
precondicionamiento.
Mediadores del precondicionamiento clásicoLos mediadores del precondicionamiento isquémico son factores que actúan durante o después del episodio isqué-mico y median el efecto protector.a) Proteína kinasa CYtrehus et al. fueron los primeros en mostrar en el cora-
30
-condicionamiento. Estudios posteriores demostraron que
estimuladores del precondicionamiento, tales como bra-
por inhibidores de la PKC.b) Tirosina kinasas
-ceptor con actividad intrínseca de tirosina kinasa, que puede actuar como un estímulo del precondicionamiento
puede actuar como mediador del precondicionamiento,
et al. 31
-
32
el periodo de isquemia inicial y estaba probablemente -
nos en el miocardio de conejo. Estudios realizados en
puede ser fosforilada al mismo tiempo que la PKC,
mediante el que el receptor de tirosina kinasa induce -

Gelpi - Donato Fisiopatología cardiovascular
94
c) Proteínas kinasas activadas por mitógenos (MAPK´s)
-
Estas cascadas de kinasas son activadas en respuesta a
tirosina kinasa, receptores asociados a proteína G y PKC.
asociada con el precondicionamiento clásico no es claro, ya que estudios previos demuestran que es activada antes
-
33 -
precondicionamiento es también controvertido. 34 La dis-crepancia en estos resultados puede ser atribuida a las
α
35
Takeishi et al. 33
la JNK durante el periodo isquémico inicial, aunque la
En este sentido, Iliodromitis et al. 34 demostraron que el
-
la PKC durante el precondicionamiento en conejos. A pe-
corazones de conejo fueron sometidos al protocolo de precondicionamiento.
Un nuevo miembro de la familia de las MAPK fue
77 et al. 36 mostraron
y reportaron que el precondicionamiento aumenta la ac-tividad de la BMK133, aunque la importancia de esta vía
-
disparidad entre los diferentes estudios puede ser en parte debido a la presencia de diversas isoformas con acciones
-les y especies.
d) La vía intracelular JAK-STAT--
la membrana celular al núcleo, donde se produce la mo-et
al. 37 encontraron que el precondicionamiento isquémico
corazones aislados de rata.
Efectores finales del precondicionamiento clásico: mecanismo de protección
-da por precondicionamiento isquémico es desconocido,
Función mitocondrial
-nica, es crucial para la sobrevida de los miocitos. Murry et al.3,
-
-mentar la síntesis de ATP.Protección contra la apoptosis
muerte celular: la necrosis y la apoptosis. La apoptosis es -
-ver capítulo 12
ruptura de la membrana celular, masiva de los
-mente, debido a que tampoco son claros cuáles son los estímulos necesarios para que se desarrolle la necrosis
et al.39 fueron los primeros en

Protección del corazón en la injuria por isquemia/reperfusión...
95
informar que el precondicionamiento isquémico reduce
-
-
canales KATP mitocondriales. 40
El canal KATP de la mitocondria
ATP de la mi-
22 --
condicionamiento. La apertura de este canal produciría:
-
Reducción de la generación de especies reactivas del oxígeno
-
Uniones de hendidura (GAP)--
terconecta los miocitos con el sincisio funcional. El rol de las uniones GAP en el precondicionamiento es con-
Figura 6.3: Representación esquemática del poro de transición de la permeabilidad mitocondrial (mPTP). El mPTP comprende el canal de aniones voltaje dependiente (CAVD) de la membrana mitocondrial externa (MME), el translocador de nucleótidos de adenina de la membrana mitocondrial interna (MMI) y la ciclofilina D (CY-D) de la matriz mitocondrial. El mPTP puede ser regulado por componentes adicionales tales como la creatinkinasas (CK) ubicada en el espacio intermembrana mitocondrial, el receptor para benzodiazepinas (RB) y la hexokinasa (HQ). La apertura del mPTP puede ser inducida por miembros proapoptóticos de la familia Bcl-2, BAX y BAK. Además, BAX BID truncado (t-BID) podría formar poros en la MME y causar la liberación de factores proapoptóticos tales como citocromo C, pro-caspasa 2, 3 y 9, Factor de inducción de apoptosis (AIF), Smac/DIABLO y la endonucleasa G (Endo G), independientemente de la apertura del mPTP. Los miembros anti-apoptóticos Bcl-2, y Bcl-XL inhibirían la apertura del mPTP y estabilizarían la MME. Los inductores de la apertura del mPTP incluyen al Ca2+, Pi, estrés oxidativo y agentes tioles y desacopladores. Los inhibidores de la apertura del mPTP incluyen la ciclosporina-A, cationes divalentes, Mg2+, Sr2+, Mn2+, Ba2+, protones ADP/ATP y NADH.
Citosol
Membranamitocondrialexterna (MME)
Membranamitocondrialexterna (MMi)
Matriz Mitocondrial
Inhibidor CAVD
RB
CK
H+, Mg2, Sr2+, Mn2+,Ba2+
ADP/ATP/NAD(P)HCa2+
PiEstrésoxidativo+ Tioles
Ciclosporina-A
Citocromo Cprocaspasa 2, 3, 9AIF, Endo GSMAC-DIABLO
BAKBcl-2
Canalanionesvoltaje
dependiente(CAVD)
Translocadornucléotidos
adenina
Translocadornucléotidos
adenina
Canalanionesvoltaje
dependiente(CAVD)
BAX
BAX t-BID
Desacopladores
Espaciointermembrana
HQ
CY-D

Gelpi - Donato Fisiopatología cardiovascular
96
es necesaria su presencia para que se pueda inducir la
estas uniones puede mimetizar el precondicionamiento
necrosis celular.El intercambiador Na+-H+ (NHE)
-H -puesta a un descenso del pH intracelular durante el periodo
-
-
intercambiador se produce antes de la isquemia o después
puede ser mayor que la inducida por el precondiciona-miento. 41
La vía de las kinasas de rescate activadas en la isquemia/reperfusión (RISK)Estudios de nuestro laboratorio 42 -lle los eventos más relevantes que ocurren en los primeros
isquémico. A partir de estos estudios, hemos demostrado
42, lo que hemos denominado la vía de las kinasas de rescate activadas en la isquemia/
42
-
celular.El poro de transición mitocondrial
-
43 El mPTP lleva este nombre por la similitud que tiene con los poros proteicos
la permeabilidad mitocondrial en respuesta a diferentes factores. 43 -da pero se cree que tiene tres componentes principales: el canal de aniones voltaje dependiente de la membrana
adenina de la membrana mitocondrial interna y la ciclo-
Figura 6.4: Representación esquemática de los componentes involucrados en el precondicionamiento tardío. El precondicionamiento tardío puede ser mimetizado por agentes farmacológicos tales como especies reactivas del oxígeno (ROS), donantes de óxido nítrico (NO). Estos estimuladores o agentes activan mediadores del precondicionamiento tardío que incluyen a la proteína kinasa C- (PKC ), tirosina kinasa (TK Src/Lck), kinasas activadas por mitógenos (MAPK’S). Estas kinasas activan la transcripción de factores tales como NF- B y JAK-STAT, que colaboran en la protección inducida en los días 2-4. Estos mediadores distales incluyen proteínas del shock de calor (HSP’s), óxido nítrico sintasa inducible (iNOS), superóxido dismutasa manganeso (MnSOD), los canales KATP.
Estímulo precondicionanteIsquemia/Reperfusión subletalestrés, marcapaso, ejercicio
Estímulo NO, ROS, adenosinabradikinina, opioides
Mediadoreskinasas: PKC, TK, MAPK
Efectores medidores alejadosHSP, INOS, MnSOD, KATP, Cox-2
Aldosa reductasa
Protección celular
Días 2-4
Día 1
Factores transcripciónNF-KB-JAK-STAT
Transcripción genes
Precondicionamiento farmacológicoEndotoxina, citoquinas, ROSdonantes NO, adenosinaopioides, abridores KATP

Protección del corazón en la injuria por isquemia/reperfusión...
97
La apertura del mPTP induce la muerte celular por
volumen de la mitocondria con ruptura de la membra--
de esta forma el proceso de apoptosis. La apertura del
es un determinante crítico de la muerte celular; esta -
del ATP mitocondrial. Estas tres alteraciones ocurren en
-closporina A40
sobre el miocardio.Nosotros fuimos los primeros en proponer y demostrar
-dio inhibiendo la apertura de los mPTP en el momento de
40 -
44
La segunda ventana de protección en el precondicionamiento isquémico (precondicionamiento tardío)
-quémico presenta una respuesta bifásica, con una fase
horas 3
7-8
-
observado también en cerdos, ratones, ratas, y reproducido
-
atontado--
manos y tejido auricular humano.
Mecanismo de protección del precondicionamiento tardío
-7-8
triggers-
Estimuladores del precondicionamiento tardíoa) Adenosina
La adenosina liberada como respuesta a un episodio sub-
la isquemia precondicionante, puede actuar como estimu-lador iniciando un fenotipo miocárdico más resistente a
en demostrar el rol de la adenosina como estimulador del precondicionamiento tardío. 45
b) Especies reactivas del oxígenoet al.46
-cardio
estudios posteriores.c) Óxido nítrico
et al. fue el primero en demostrar
ejercida por el precondicionamiento tardío contra el miocardio atontado47 y posteriormente contra el infarto del miocardio. 48
d) Bradikinina Yellon et al.49 demostraron que la bradikinina puede ac-tuar como un estimulador del precondicionamiento tardío. Además, en nuestro laboratorio encontramos que la ad-
estímulo por debajo del umbral de precondicionamiento puede ser incrementado por la presencia de un inhibidor
se sabe, aumenta los niveles de bradikinina induciendo un precondicionamiento tardío en un modelo de cerdo
e) Opioides
Mediadores del precondicionamiento tardíoa) Proteína kinasa CYamashita et al.50 fueron los primeros en demostrar que
b) Tirosina kinasa

Gelpi - Donato Fisiopatología cardiovascular
98
inducida por el precondicionamiento tardío contra el mio-cardio atontado y el infarto de miocardio.c) Proteínas kinasas activadas por mitógenos (MAPK´s)Un protocolo de precondicionamiento isquémico que
puede activar tres miembros mayores de la familia de las
d) La vía de la PI3K-AktNuestro laboratorio 51 ha demostrado que la cascada de
-condicionamiento tardío en el modelo in vivode conejo.e) Canales KATP cardiacos
ATP sarcole-males y mitocondriales como mediadores del precondicio-namiento tardío inducido por isquemia, estrés por calor,
los canales KATP
f) Factores de transcripción en el precondicionamiento tardío
-titutivamente y activado por el precondicionamiento. Es
Xuan et al.52
ocurre como respuesta a un estímulo precondicionante o a un dador de NO. Estos autores mostraron también que
-
52
g) La vía intracelular JAK-STAT
a un estímulo precondicionante y ha sido demostrado up-regulation
efectores distales del precondicionamiento tardío. De las cuatro isoformas de la JAK que han sido descritas,
Mediadores distales y efectores finales del precondicionamiento tardío
Las características temporales del precondicionamien-to tardío requieren la síntesis de proteínas que actúen como mediadores/efectores distales, posiblemente bajo
-
el efecto protector del precondicionamiento tardío in vivo. a) Óxido nítrico sintasa
et al. 53
-
contra el miocardio atontado y el infarto de miocardio. b) Ciclo-oxigenasa-2 (COX2)
et al. 54
PGE y/o PGI
up-regulation
c) Aldosa-reductasa
familia de las aldo-keto-reductasas, puede metabolizar los
-
el contrario, el efecto protector se bloqueaba si la aldosa-reductasa era inhibida. d) Enzimas antioxidantes (manganeso super óxido dismutasa – MnSOD)
--
sea que este fuera realizado por isquemia, estrés por calor,
e) Proteínas de shock por calor (HSP)Dillman et al.55 -
et al.56
-
-cionante. 7requerida para que ocurra el precondicionamiento tardío inducido por adenosina.
Precondicionamiento remoto o de zonas alejadas
menos reconocido, por el que el precondicionamiento de

Protección del corazón en la injuria por isquemia/reperfusión...
99
Przyklenk et al.57 en un estudio en el que mostraron que -
descendente anterior. Posteriormente, otros estudios mos-
-miento clásico e involucran a la adenosina, bradikinina,
Precondicionamiento del miocardio humanoPara poder trasladar los conceptos de los párrafos ante-riores sobre el precondicionamiento isquémico miocár-dico en animales a la práctica clínica, especialmente en
-
-
-
bien controlados.
Estudios in vitro
Ikonomidis et al.58 fueron los primeros en demostrar que el
mecanismo de precondicionamiento de miocitos humanos. et al.59 demostraron que un periodo
miocitos auriculares de humanos adultos, recolectados o
A1 y A
de la PKC y de los canales KATP de la mitocondria como mediadores del precondicionamiento.
et al.61 demostraron la presencia de precon-
de necrosis, mostraron que la misma disminuye después del estímulo precondicionante.
Estudios in vivo
--
efecto similar al precondicionamiento, y no a un reclu-
con el precondicionamiento, tales como adenosina, bra-dikinina, y los canales KATP no parecen estar implicados
del precondicionamiento. Por el contrario, otros estudios ATP en el
Otra entidad clínica en la que podría estar involucrado el precondicionamiento isquémico es en los pacientes con
-tricular, menor cantidad de arritmias, reducida inciden-
aparente efecto protector se debe al precondicionamiento.
previas al infarto de miocardio62, lo que resulta interesante porque ese periodo de tiempo está dentro de la ventana de
Un modelo clínico de precondicionamiento isquémi-
--
jorar los indicadores de isquemia miocárdica, en forma independiente del reclutamiento de vasos colaterales. Los índices de isquemia miocárdica más utilizados incluyen la presencia de dolor de pecho, anormalidades del movi-
-
de CK. 63
corazones pudieron haber sido preacondicionados duran-63;
los mecanismos podrían ser los que actúan en el precon-dicionamiento clásico: canales KATP, adenosina, opioides, y bradikinina.
Con respecto al precondicionamiento tardío, Leesar et al. 64 -
-plastia coronaria, atenúa los síntomas del dolor de pecho,
et al. 65 han mos-trado que un test de tolerancia al ejercicio puede conferir

Gelpi - Donato Fisiopatología cardiovascular
100
Otro modelo de precondicionamiento en el miocardio
coronario. Yellon y otros autores demostraron que el clam-
consumo de ATP 66 -na T. 67
precondicionamiento durante la hipotermia o paro cardio---
-
en un trabajo 68 y en otro el precondicionamiento isquémico 1 de la ade-
Por otro lado, el precondicionamiento realizado duran-
-
morbilidad. Además, el precondicionamiento isquémico no
-68
-
Precondicionamiento en el área clínica
precondicionado en diferentes situaciones de isquemia/re--
--
punto importante a considerar es que a veces, en cuadros
Figura 6.5: Esquema que representa la vía de las kinasas de rescate activadas en la reperfusión (RISK). Los compuestos que protegen durante la reperfusión se unen a receptores acoplados a proteína G, RAS, activan la enzima PI3K-Akt y la cascada de kinasas Raf1-MEK1/2-Erk1/2, las que en conjunto comprenden las kinasas de rescate denominadas RISK. La protección contra la injuria por reperfusión es mediada por: 1) La fosforilación e inactivación de factores proapoptóticos, caspasas 3 y 9, BIM, BAX, BAD y p53, los que previenen la liberación del citocromo C desde la mitocondria; 2) la fosforilación y activación de la eNOS (óxido nítrico sintasa endotelial), lo que produce óxido nítrico y podría proteger inhibiendo la apertura del poro de transición de la permeabilidad mitocondrial (mPTP); 3) La fosforilación y activación de las proteínas p70S6K y p90S6K que protegen por inactivar BAD; y 4) la regulación de la expresión de genes que intervienen en la sobrevida celular.
Unión del ligando al receptor
PI3K RAS Raf-1
BIMMOM2
BAX
BAD
p70S6K
p53p90S6K
Traducciónproteica
Inhibición de laliberación del citocromo C
Expresióngenes
Núcleo
Caspasa 3Caspasa 9
Mitocondria
eNOS
?
mPTP
Sobrevida celular
MEK 1/2
ErK 1/2Akt/PKB

Protección del corazón en la injuria por isquemia/reperfusión...
101
-
porque obviamente no es posible saber cuándo ese paciente va a tener el infarto de miocardio. En este sentido, se ha
-69
A1 de adenosina, puede mantener al miocardio en un estado de precondicionamiento por varios días.
El nicorandil, un abridor de los canales KATP, ha sido
inestable. Estos estudios demostraron que puede atenuar
-rios mayores. 70 -piedades vasodilatadoras de este fármaco no pudieron ser descartadas y pudieron ser la causa del efecto favorable.
-minar el efecto positivo o no del precondicionamiento en
-
-
el concepto de poscondicionamiento isquémico, que se
Protección miocárdica por intervenir durante la reperfusión
Injuria letal por reperfusión--
plastia coronaria primaria, es un requisito fundamental
-
resultar en muerte celular, lo que se conoce como injuria
-rio después de un episodio isquémico, que lleva a la muer-te a células que presentaban solamente injuria reversible.
entidad independiente de la injuria isquémica es motivo
Figura 6.6: La vía de señales RISK y el mPTP como un mecanismo de cardioprotección universal. La activación de la Akt y Erk1/2 y la subsecuente inhibición de la apertura del mPTP en el momento de la reperfusión protegen al corazón contra la injuria por isquemia/reperfusión. La vía de señales RISK puede ser activada farmacológicamente o a través de maniobras cardioprotectoras como el precondicionamiento y poscondicionamiento isquémico.
Precondicionamientoisquémico
Adenosina
Protocoloprecondicionamiento
Adenosina
Activación deRISK
Poscondicionamientoisquémico
Períodoisquémico Fase reperfusión
Fosforilación de:1. BAD/BAX2. eNOS3. GSK3
Agentes farmacológicos(estatinas, EPO)
Inhibición de laapertura del mPTPen la reperfusión
Protección cardíaca

Gelpi - Donato Fisiopatología cardiovascular
102
injuria celular, aun en ausencia de isquemia.
-
-
La vía de las kinasas de rescate activadas en la isquemia/reperfusión (RISK)
-
71
71, -
En diferentes estudios fue mostrado que diversos tipos -
-
-milar a la insulina, el factor de crecimiento transformante
adenosina tales como AMP 579 y NECA, eritropoyeti-
por una variedad de mecanismos que incluirían efectos
Poscondicionamiento isquémico
72
por primera vez el concepto de poscondicionamiento
isquémico. 5
-
5 Utilizando un modelo canino de injuria por isquemia/in vivo et al.5 demostraron
-
-
este un efecto comparable al del precondicionamiento -
males sometidos al protocolo de poscondicionamiento
5
Poscondicionamiento isquémico como una forma de reperfusión modificada
Una posibilidad es que el concepto de poscondicionamien-
que podría simplemente representar una forma de reper-
estudios previos como una manera de atenuar la injuria 73 et al.73 utilizando
Posteriormente, Kin et al.74 demostraron que el pos-condicionamiento también es efectivo en ratas, y en el
En este estudio el poscondicionamiento fue inducido uti--
-cionante puede ser especie dependiente. Este hecho está, además, avalado por trabajos de nuestro laboratorio en los
el trabajo de Kin et al.74
que la cantidad de los mismos.
han mostrado ser útiles para limitar la injuria miocárdi-

Protección del corazón en la injuria por isquemia/reperfusión...
103
que nos permite especular que el poscondicionamiento isquémico ejerce su efecto protector por los mismos me-canismos. 75
La protección inducida por poscondicionamiento es mediada a través de la señalización RISK
La primera evidencia de que la cascada de kinasas de rescate era responsable del efecto protector del poscondicionamiento
75 Encontramos
corazones aislados de ratas, era abolida cuando se adminis-
et al. 76 mostraron -
nejos fue atenuada en presencia de un inhibidor de la MEK ATP
-
de los canales KATP mitocondriales. Por último, en un trabajo posterior, Downey et al.77
por el poscondicionamiento isquémico.
Mediadores que activan la vía de señalización RISK
activada por el poscondicionamiento no está claro. El --
-nista del receptor A . 78 En el mismo sentido, Downey et al.77
estos estudios muestran que la adenosina actúa como un -
namiento. De esta manera, nosotros especulamos que la
Mediadores que actúan después de la activación de la vía de señalización RISK
75 et al. han mostrado que tanto el poscondicionamiento isquémico 79 como la reperfu-
de la permeabilidad mitocondrial en el momento de la
La vía de señalización RISK y el poro de transición mitocondrial como vías universales de protección
-
el hecho de que el reclutamiento de esta vía intracelu-
cardioprotectoras, tales como el precondicionamiento
del mPTP podrían ser administrados en el momento de la
Protección del miocardio humano utilizando terapias adyuvantes en la reperfusión
-
cardioprotectores como terapia adyuvante a las estrate-
Insulinaet al. introdujeron por primera
-na-potasio podía ofrecer un efecto protector en respuesta
80 Posteriormente,
-
-81; además, el estudio multicén-
82
83 y un
podía ser protectora.Desafortunadamente el estudio CREATE, que incor-
-84, aunque
fueron administrados tardíamente. En la actualidad, la
85

Gelpi - Donato Fisiopatología cardiovascular
104
-
-86 Además, diferentes trabajos mostraron
-
Magnesio-
87
88
89 -
como terapia adyuvante a la trombolisis. La disparidad en-tre estos trabajos parece estar relacionada con el momento
Adenosina
adenosina como terapia adyuvante a la trombolisis induce
90
Inhibidores del intercambiador sodio/hidrógenoLos estudios iniciales que utilizaron inhibidores del inter-
-farto de miocardio, mostraron un efecto protector por
miocardio. 91
protector, administraron el inhibidor del intercambiador antes o al comienzo del episodio de isquemia. En este sentido, el estudio GUARDIAN 92
--
Canales KATP mitocondriales
ATP mitocon-
et al., 93
de los canales KATP -
94
Barredores de las especies reactivas del oxígeno
-
-
de los mismos. 95
contradictorios. 96
Inhibición de la apertura del poro de transición mitocondrial (mPTP)
El hecho de que la apertura del mPTP en los primeros
que pueda ser administrada como terapia adyuvante a la
inhibir transitoriamente la apertura del mPTP. Nosotros hemos mostrado, en tejido auricular humano sometido
ciclosporina-A, tiene un efecto protector.
Conclusiones
-mente isquémico, el precondicionamiento isquémico es
actualidad. Desafortunadamente, el precondicionamiento
su efecto deben ser aplicados antes de que el episodio de -
siblemente, podrían ser utilizados en procedimientos tales

Protección del corazón en la injuria por isquemia/reperfusión...
105
puede ser anticipado.
-lisis, la morbimortalidad del infarto de miocardio continúa siendo elevada; en consecuencia, son necesarias nuevas es-
del poscondicionamiento isquémico durante la trombolisis
Una posibilidad más factible para alcanzar cardiopro-
son utilizadas para el tratamiento de otras condiciones pa-
Bibliografía
1. Murray CJ, AD López. “Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study”. Lancet 1997; 349(9064):1498-1504.
2. Naghavi M, Libby P, Falk E, et al. “From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I”. Circulation 2003; 108(14):1664-1672.
3. Murry CE, Jennings RB, Reimer KA. “Preconditioning with is-chemia: a delay of lethal cell injury in ischemic myocardium”. Circulation 1986; 74(5):1124-1136.
4. Braunwald E, RA Kloner. “Myocardial reperfusion: a double-edged sword?” J Clin Invest 1985; 76(5):1713-1719.
5. Zhao ZQ, Corvera JS, Halkos ME et al. “Inhibition of myocardial injury by ischemic postconditioning during reperfusion: compa-rison with ischemic preconditioning”. Am J Physiol Heart Circ Physiol 2003; 285(2):H579-H588.
6. Kloner RA, Bolli R, Marban E et al. “Medical and cellular impli-cations of stunning, hibernation, and preconditioning: an NHLBI workshop”. Circulation 1998; 97(18):1848-1867.
7. Marber MS, Latchman DS, Walker JM et al. “Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associa-ted with resistance to myocardial infarction”. Circulation 1993; 88(3):1264-1272.
8. Kuzuya T, Hoshida S, Yamashita N et al. “Delayed effects of su-blethal ischemia on the acquisition of tolerance to ischemia”. Circ Res 1993; 72(6):1293-1299.
9. Shiki K, DJ Hearse. “Preconditioning of ischemic myocardium: reperfusion-induced arrhythmias”. Am J Physiol 1987; 253(6 Pt 2):H1470-H1476.
10. Asimakis GK, Inners-McBride K, Medellin G et al. “Ischemic preconditioning attenuates acidosis and postischemic dysfunction in isolated rat heart”. Am J Physiol 1992; 263(3 Pt 2):H887-H894.
11. Bolli R, E Marban. “Molecular and cellular mechanisms of myo-cardial stunning”. Physiol Rev 1999; 79(2): 609-634.
12. Vanden Hoek TL, Becker LB, Shao Z et al. “Reactive oxygen species released from mitochondria during brief hypoxia induce precon-ditioning in cardiomyocytes”. J Biol Chem 1998; 273(29):18092-18098.
13. Tong H, Chen W, Steenbergen C et al. “Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C”. Circ Res 2000; 87(4):309-315.
14. Krieg T, Qin Q, McIntosh EC et al. “ACh and adenosine acti-vate PI3-kinase in rabbit hearts through transactivation of re-ceptor tyrosine kinases”. Am J Physiol Heart Circ Physiol 2002; 283(6):H2322-H2330.
15. Oldenburg O, Critz SD, Cohen MV et al. “Acetylcholine-induced production of reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol 3- and Src-kinase activation and mitochondrial K(ATP) channel opening”. J Mol Cell Cardiol 2003; 35(6):653-660.
16. Oldenburg O, Qin Q, Sharma AR et al. “Acetylcholine leads to free radical production dependent on K(ATP) channels, G(i) proteins, phosphatidylinositol 3-kinase and tyrosine kinase”. Cardiovasc Res 2002; 55(3):544-552.
17. Oldenburg O, Qin Q, Krieg T et al. “Bradykinin induces mito-chondrial ROS generation via NO, cGMP, PKG, and mKATP channel opening and leads to cardioprotection”. Am J Physiol Heart Circ Physiol 2003.
18. Noma, A. “ATP-regulated K+ channels in cardiac muscle”. Nature 1983; 305(5930):147-148.
19. Cole WC, McPherson CD, Sontag D. “ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage”. Circ Res 1991; 69(3):571-581.
20. Gross GJ, JA Auchampach. “Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs”. Circ Res 1992; 70(2):223-233.
21. Inoue I, Nagase H, Kishi K et al. “ATP-sensitive K+ channel in the mitochondrial inner membrane”. Nature 1991; 352(6332):244-247.
22. Garlid KD, Paucek P, Yarov-Yarovoy V et al. “Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection”. Circ Res 1997; 81(6):1072-1082.
23. Pain T, Yang XM, Critz SD et al. “Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals”. Circ Res 2000; 87(6):460-466.
24. Murry CE, Richard V, Jennings RB et al. “Preconditioning with ischemia: is the protective effect mediated by free radical-induced myocardial stunning?” Circulation 78. 1988.
25. Tritto I, D’Andrea D, Eramo N et al. “Oxygen radicals can induce preconditioning in rabbit hearts”. Circ Res 1997; 80(5):743-748.
26. Forbes RA, Steenbergen C, Murphy E. “Diazoxide-induced cardio-protection requires signaling through a redox-sensitive mechanism”. Circ Res 2001; 88(8):802-809.
27. Droge, W. “Free radicals in the physiological control of cell function”. Physiol Rev 2002; 82(1):47-95.
28. Tanaka M, Fujiwara H, Yamasaki K et al. “Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit”. Cardiovasc Res 1994; 28(7):980-986.
29. Baines CP, Goto M, Downey JM. “Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium”. J Mol Cell Cardiol 1997; 29(1):207-216.
30. Ytrehus K, Liu Y, Downey JM. “Preconditioning protects ischemic rabbit heart by protein kinase C activation”. Am J Physiol 1994; 266(3 Pt 2):H1145-H1152.
31. Maulik N, Watanabe M, Zu YL et al. “Ischemic preconditioning triggers the activation of MAP kinases and MAPKAP kinase 2 in rat hearts”. FEBS Lett 1996; 396(2-3):233-237.
32. Baines CP, Wang L, Cohen MV et al. “Protein tyrosine kinase is downstream of protein kinase C for ischemic preconditioning’s anti-infarct effect in the rabbit heart”. J Mol Cell Cardiol 1998; 30(2):383-392.

Gelpi - Donato Fisiopatología cardiovascular
106
33. Takeishi Y, Huang Q, Wang T et al. “Src family kinase and ade-nosine differentially regulate multiple MAP kinases in ischemic myocardium: modulation of MAP kinases activation by ischemic preconditioning”. J Mol Cell Cardiol 2001; 33(11):1989-2005.
34. Iliodromitis EK, Gaitanaki C, Lazou A et al. “Dissociation of stress-activated protein kinase (p38-MAPK and JNKs) phosphorylation from the protective effect of preconditioning in vivo”. J Mol Cell Cardiol 2002; 34(8):1019-1028.
35. Wang Y, Huang S, Sah VP et al. “Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family”. J Biol Chem 1998; 273(4):2161-2168.
36. Takeishi Y, Abe J, Lee JD et al. “Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts”. Circ Res 1999; 85(12):1164-1172.
37. Hattori R, Maulik N, Otani H et al. “Role of STAT3 in ischemic preconditioning”. J Mol Cell Cardiol 2001; 33(11):1929-1936.
38. Jennings RB, Murry CE, Reimer KA. “Preconditioning myocar-dium with ischemia”. Cardiovasc Drugs Ther 1991; 5(5):933-938.
39. Piot CA, Padmanaban D, Ursell PC et al. “Ischemic preconditio-ning decreases apoptosis in rat hearts in vivo”. Circulation 1997; 96(5):1598-1604.
40. Hausenloy DJ, Maddock HL, Baxter GF et al. “Inhibiting mito-chondrial permeability transition pore opening: a new paradigm for myocardial preconditioning?” Cardiovasc Res 2002; 55(3):534-543.
41. Gumina RJ, Buerger E, Eickmeier C et al. “Inhibition of the Na(+)/H(+) exchanger confers greater cardioprotection against 90 minutes of myocardial ischemia than ischemic preconditioning in dogs”. Circulation 1999; 100(25):2519-2526.
42. Hausenloy DJ, Tsang A, Mocanu M et al. “Ischemic Preconditioning Protects by Activating Pro-Survival Kinases at Reperfusion”. Am J Physiol Heart Circ Physiol 2005; 288:H971-H976.
43. Hunter DR, RA Haworth. “The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms”. Arch Biochem Biophys 1979; 195(2):453-459.
44. Juhaszova M, Zorov DB, Kim SH et al. “Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore”. J Clin Invest 2004; 113(11):1535-1549.
45. Baxter GF, Marber MS, Patel VC et al. “Adenosine receptor invol-vement in a delayed phase of myocardial protection 24 hours after ischemic preconditioning”. Circulation 1994; 90(6):2993-3000.
46. Sun JZ, Tang XL, Park SW et al. “Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs”. J Clin Invest 1996; 97(2):562-576.
47. Bolli R, Bhatti ZA, Tang XL et al. “Evidence that late preconditio-ning against myocardial stunning in conscious rabbits is triggered by the generation of nitric oxide”. Circ Res 1997; 81(1):42-52.
48. Qiu Y, Rizvi A, Tang XL et al. “Nitric oxide triggers late precondi-tioning against myocardial infarction in conscious rabbits”. Am J Physiol 1997; 273(6 Pt 2):H2931-H2936.
49. Ebrahim Z, Yellon DM, Baxter GF. “Bradykinin elicits ‘second window’ myocardial protection in rat heart through an NO-dependent mechanism”. Am J Physiol Heart Circ Physiol 2001; 281(3):H1458-H1464.
50. Yamashita N, Nishida M, Hoshida S et al. “Induction of manganese superoxide dismutase in rat cardiac myocytes increases tolerance to hypoxia 24 hours after preconditioning”. J Clin Invest 1994; 94(6):2193-2199.
51. Kis A, Yellon DM, Baxter GF. “Second window of protection fo-llowing myocardial preconditioning: an essential role for PI3 kinase and p70S6 kinase”. J Mol Cell Cardiol 2003; 35(9):1063-1071.
52. Xuan YT, Tang XL, Banerjee S et al. “Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits”. Circ Res 1999; 84(9):1095-1109.
53. Takano H, Manchikalapudi S, Tang XL et al. “Nitric oxide syn-thase is the mediator of late preconditioning against myocardial infarction in conscious rabbits”. Circulation 1998; 98(5):441-449.
54. Shinmura K, Tang XL, Wang Y et al. “Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic precon-ditioning in conscious rabbits”. Proc Natl Acad Sci USA 2000; 97(18):10197-10202.
55. Dillmann WH, Mehta HB, Barrieux A et al. “Ischemia of the dog heart induces the appearance of a cardiac mRNA coding for a protein with migration characteristics similar to heat-shock/stress protein 71”. Circ Res 1986; 59(1):110-114.
56. Knowlton AA, Brecher P, Apstein CS. “Rapid expression of heat shock protein in the rabbit after brief cardiac ischemia”. J Clin Invest 1991; 87(1):139-147.
57. Przyklenk K, Bauer B, Ovize M et al. “Regional ischemic‚ precon-ditioning‘ protects remote virgin myocardium from subsequent sustained coronary occlusion”. Circulation 1993; 87(3):893-899.
58. Ikonomidis JS, Tumiati LC, Weisel RD et al. “Preconditioning human ventricular cardiomyocytes with brief periods of simulated ischaemia”. Cardiovasc Res 1994; 28(8):1285-1291.
59. Arstall MA, Zhao YZ, Hornberger L et al. “Human ventricular myocytes in vitro exhibit both early and delayed preconditio-ning responses to simulated ischemia”. J Mol Cell Cardiol 1998; 30(5):1019-1025.
60. Walker DM, Walker JM, Pugsley WB et al. “Preconditioning in isolated superfused human muscle”. J Mol Cell Cardiol 1995; 27(6):1349-1357.
61. Loubani M, Ghosh S, Galinanes M. “The aging human myo-cardium: tolerance to ischemia and responsiveness to ischemic preconditioning”. J Thorac Cardiovasc Surg 2003; 126(1):143-147.
62. Kloner RA, Shook T, Antman EM et al. “Prospective temporal analysis of the onset of preinfarction angina versus outcome: an ancillary study in TIMI-9B”. Circulation 1998; 97(11):1042-1045.
63. Laskey WK, D Beach. “Frequency and clinical significance of ische-mic preconditioning during percutaneous coronary intervention”. J Am Coll Cardiol 2003; 42(6):998-1003.
64. Leesar MA, Stoddard MF, Dawn B et al. “Delayed preconditioning-mimetic action of nitroglycerin in patients undergoing coronary angioplasty”. Circulation 2001; 103(24):2935-2941.
65. Lambiase PD, Edwards RJ, Cusack MR et al. “Exercise-induced ischemia initiates the second window of protection in humans independent of collateral recruitment”. J Am Coll Cardiol 2003; 41(7):1174-1182.
66. Yellon DM, Alkhulaifi AM, Pugsley WB. “Preconditioning the human myocardium”. Lancet 1993; 342(8866):276-277.
67. Jenkins DP, Pugsley WB, Alkhulaifi AM et al. “Ischaemic precondi-tioning reduces troponin T release in patients undergoing coronary artery bypass surgery”. Heart 1997; 77(4):314-318.
68. Belhomme D, Peynet J, Louzy M et al. “Evidence for precondi-tioning by isoflurane in coronary artery bypass graft surgery”. Circulation 1999; 100(19 Suppl):II340-II344.
69. Dana A, Baxter GF, Walker JM et al. “Prolonging the delayed phase of myocardial protection: repetitive adenosine A1 receptor activation maintains rabbit myocardium in a preconditioned state”. J Am Coll Cardiol 1998; 31(5):1142-1149.
70. “Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial”. Lancet 2002; 359(9314):1269-1275.
71. Yellon DM, GF Baxter. “Reperfusion injury revisited: is there a role for growth factor signaling in limiting lethal reperfusion injury?” Trends Cardiovasc Med 1999; 9(8):245-249.
72. Baxter GF, DM Yellon. “Current trends and controversies in ische-mia-reperfusion research--meeting report of the Hatter Institute 3rd International Workshop on Cardioprotection”. Basic Res Cardiol 2003; 98(2):133-136.
73. Okamoto F, Allen BS, Buckberg GD et al. “Reperfusion conditions: importance of ensuring gentle versus sudden reperfusion during relief of coronary occlusion”. J Thorac Cardiovasc Surg 1986; 92(3 Pt 2):613-620.
74. Kin H, Zhao ZQ, Sun HY et al. “Postconditioning attenuates myo-cardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion”. Cardiovasc Res 2004; 62(1):74-85.

Protección del corazón en la injuria por isquemia/reperfusión...
107
75. Tsang A, Hausenloy DJ, Mocanu MM et al. “Postconditioning: a form of ‘modified reperfusion’ protects the myocardium by acti-vating the phosphatidylinositol 3-kinase-Akt pathway”. Circ Res 2004; 95(3):230-232.
76. Yang XM, Proctor JB, Cui L et al. “Multiple, brief coronary occlu-sions during early reperfusion protect rabbit hearts by targeting cell signaling pathways”. J Am Coll Cardiol 2004; 44(5):1103-1110.
77. Yang XM, Philipp S, Downey JM et al. “Postconditioning’s pro-tection is not dependent on circulatin blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation”. Basic Res Cardiol 2005; 100(1):57-63.
78. Kin H, Lofye MT, Amerson BS, Zatta AJ et al. “Cardioprotection by ‘Postconditioning’ is Mediated by Increased Retention of Endo-genous Intravascular Adenosine and Activation of A2A Receptors During Reperfusion”. Circulation 100[17], 803. 2004.
79. Argaud L, Gateau-Roesch O, Raisky O et al. “Postconditioning inhibits mitochondrial permeability transition”. Circulation 2005; 111(2):194-197.
80. Sodi-Pallares D, Testelli MR, Fishleder BL et al. “Effects of an intravenous infusion of potassium-insulin-glucose solution on the electrocardiographic signs of myocardial infarction. A preliminary clinical report”. Am J Cardiol 9, 166-181. 1962.
81. Fath-Ordoubadi F, KJ Beatt. “Glucose-insulin-potassium therapy for treatment of acute myocardial infarction: an overview of randomi-zed placebo-controlled trials”. Circulation 1997; 96(4):1152-1156.
82. Malmberg K, Ryden L, Efendic S et al. “Randomized trial of insu-lin-glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): effects on mortality at 1 year”. J Am Coll Cardiol 1995; 26(1):57-65.
83. Diaz R, Paolasso EA, Piegas LS et al. “Metabolic modulation of acute myocardial infarction. The ECLA (Estudios Cardioló-gicos Latinoamérica) Collaborative Group”. Circulation 1998; 98(21):2227-2234.
84. Mehta SR, Yusuf S, Diaz R et al. “Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial”. JAMA 2005; 293(4):437-446.
85. Janiger JL, JW Cheng. “Glucose-insulin-potassium solution for acute myocardial infarction”. Ann Pharmacother 2002; 36(6):1080-1084.
86. Nikolaidis LA, Elahi D, Hentosz T et al. “Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy”. Circulation 2004; 110(8):955-961.
87. Herzog WR, Schlossberg ML, MacMurdy KS et al. “Timing of magnesium therapy affects experimental infarct size”. Circulation 1995; 92(9):2622-2626.
88. Woods KL, Fletcher S, Roffe C et al. “Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LI-MIT-2) ”. Lancet 1992; 339(8809):1553-1558.
89. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group”. Lancet 1995; 345(8951):669-685.
90. Mahaffey KW, Puma JA, Barbagelata NA et al. “Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial”. J Am Coll Cardiol 1999; 34(6):1711-1720.
91. Zeymer U, Suryapranata H, Monassier JP, et al. “The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial”. J Am Coll Cardiol 2001; 38(6):1644-1650.
92. Theroux P, Chaitman BR, Danchin N et al. “Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocar-dial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators”. Circulation 2000; 102(25):3032-3038.
93. Sugimoto K, Ito H, Iwakura K, Ikushima M et al. “Intravenous nicorandil in conjunction with coronary reperfusion therapy is associated with better clinical and functional outcomes in patients with acute myocardial infarction”. Circ J 2003; 67(4):295-300.
94. Mizuo N, Iwao I, Yoshiya M, Yoshiaki S et al. “Comparison between nicorandil and magnesium as an adjunct cardioprotective agent to percutaneous coronary intervention in acute anterior myocardial infarction”. Circ J 2004; 68(3):192-197.
95. Flaherty JT, Pitt B, Gruber JW et al. “Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction”. Circulation 1994; 89(5):1982-1991.
96. Salonen RM, Nyyssonen K, Kaikkonen J et al. “Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: the Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study”. Circulation 2003; 107(7):947-953.


109
Fisiopatología de la protección miocárdica en la cirugía cardiaca
7
-do al cirujano a tratar de conocer e interpretar, al momento
y molecular. Un mayor conocimiento del problema de la isquemia miocárdica y los posibles intentos de minimizar o disminuir sus efectos deletéreos sobre el músculo car-diaco redundarán en mejores resultados sobre la calidad y cantidad de vida de los pacientes.
Fisiopatología del miocardio isquémico-metabolismo de la isquemia
-
1, 2
-
-dores y constrictores.
por el endotelio-
control, momento a momento, del tono coronario impuesto por un apropiado equilibrio sobre los vasos de resistencia
-mente a través de la membrana celular, concentrándose en el compartimiento intersticial. La adenosina actúa directa-mente en el músculo liso de las arteriolas causando relaja-
. La adenosina
satisfecha, aun temporalmente, el disbalance entre oferta y
del ADP y más adenosina es formada y difundida al parén-quima y a los microvasos. Este incremento en la concen-
-
-
canales de Cl-. 2
sistema nervioso simpático actúa a través de los
-
--
-creta el neuropéptido Y, que podría contribuir a la vaso-
2
-
-
representados principalmente por el péptido vasoconstric-
-mero de factores entre los que se encuentran la adenosina, acetilcolina y el estrés de rozamiento endotelial secundario

Gelpi - Donato Fisiopatología cardiovascular
110
. El NO representa un potente vasodilatador en los vasos de resisten-
simpáticos, o el
liso vascular. A su vez, el NO es un potente inhibidor directo -
-noácidos derivado de la preproendotelina-1 en el endote-
-
coronario, tanto normal como en la enfermedad. Es rápi-damente sintetizada por el endotelio vascular, en particular
-
ET-1, que puede disminuir los efectos vasodilatadores de los autacoides como la adenosina y el NO. También con la edad avanzada se observa un aumento de los efectos de la
2
3
Autorregulación del flujo coronario. Disfunción endotelial
-
2
está comprometida y así también su capacidad de pro-
isquemia temporal, se produce un círculo vicioso entre
predominio de los vasoconstrictores. 2
-
-
adenosina, ambos con potentes efectos antiplaquetarios --
efectos deletéreos, aumentando la permeabilidad vascu-
---
de ataque de membrana, como así también los radicales
E-selectina e ICAM-1. 2
--
-
de Na /K
2
Las manifestaciones y mecanismos de la injuria mio-
Las diferentes evidencias documentan las alteraciones ce--
4 Los cambios que caracterizan a los patro-nes de injuria celular isquémica comprenden alteraciones
, acu-

Fisiopatología de la protección miocárdica en la cirugía cardíaca
111
, Cl- y H O, como así también de Ca .
-
celular. 4
caracterizada por el severo aumento de la permeabilidad
cationes bivalentes y trivalentes, incluyendo el Ca . 4 -secuentemente, el edema celular desarrolla soluciones de
ruptura.
Proceso de injuria por isquemia/reperfusión
-ver capítulo
5
5
prevenir la injuria irreversible que lleva a la necrosis mio---
recuperable.
iniciados por ella. 5
La injuria --
espectro de manifestaciones clínicas, que abarcan desde el atontamiento ver capítulo 6 -
6
muerte a células que sufrían un proceso reversible. También
-
--
5
-
miocárdico. 5
-
-
-
-
5
También diversos mecanismos enzimáticos provocan
-
-
-
y ácido hipocloroso. 5
de los radicales libres son efectivos durante la reperfu--
scavengers. Estos
en su capacidad o bien afectados por la injuria provocada por la isquemia. 5
-scavenger de los radicales libres o
-tes, y la imposibilidad de la célula de producir equivalentes
reductasa y altera la homeostasis celular. De esta manera,
importante para el control de los radicales libres. 7 -
-
7
-7

Gelpi - Donato Fisiopatología cardiovascular
112
-
-5
5, resultan en un efecto de car--
-
por los factores C5a, IL-8 y el factor de crecimiento trans-formante -α α
-
-
5
contactar con el endotelio, que pierden su capacidad de
-
posicionarse por sí mismos y activarse durante el proceso
del endotelio cumplen un rol importante orientando los
proceso de varias etapas, observado en las vénulas posca-
5
-
--
los y el reclutamiento, durante la isquemia; el factor C5a
5
-
con profundo efecto sobre la vasculatura y sobre los acti-5
está asociada a una secuencia de eventos en la que el com-
el ensamblado del complejo de ataque de membrana con C5b-8 y C9, a través de la membrana plasmática, provoca
electrolitos. Así también el ataque directo lítico, mediante
-
5
--
se adhieren a las células endoteliales y a los miocitos. 6
-6
6
-
-
El factor tisular inicia
6 --
continuidad en la misma. 6 El endotelio, bajo condiciones
-
6 De todas ma-neras, ambas células poseen la capacidad de estimular la salida del factor tisular en respuesta a una variedad de es-
-tor de necrosis tumoral-α, Interferon- , interleuquina-1 y
de trombos intravasculares.6
del factor tisular en el monocito y las células endoteliales.

Fisiopatología de la protección miocárdica en la cirugía cardíaca
113
positivo en el que la trombina estimula los monocitos y células endoteliales y, a su vez, estos producen fac-
-
intravasculares. 6
-
--
PARs. 6
zonas del miocardio, a pesar de contar nuevamente con el 8 Este
. Cuando
capilar y la estenosis residual en la zona de la arteria afec-tada. La zona del cambia dinámica-mente con las horas a causa del vasoespasmo, edema del
-8
-
libres. 8
la primera etapa, en la zona de ya se observan microtromboembolias. Las aminas vasoactivas, producto de las plaquetas activadas, causan espasmo microvascular
-8
microvasculatura lesionada.
-fía de contraste. En corazones normales, la capacitancia
-res, es perfundida durante la diástole sin incremento de la
8 --
la funcional del miocito, etc. 8
normales, la impedancia miocárdica se incrementa durante
-cuentra obstruida y no puede pasar a las vénulas, resultando
8
puede no observarse de inmediato una vez iniciado el fe-
-ciada la isquemia. 8 En aquellos casos en que se presenta un
8
una ayuda multidisciplinaria en busca de disminuir o mini-
Historia de la protección miocárdica
-nalmente mediante el uso de la hipotermia. 9 Más tarde, Melrose et al.10 describieron el paro cardiaco electrome-cánico mediante el uso de infusiones ricas en potasio,
-
11 et al. 12 de--
tricamente causaba injuria isquémica subendocárdica,
ventricu-
-
11
drenaje de las cavidades ventriculares distendidas, que
-traciones de adenosina trifosfato y creatinfosfato, que
-cardio. 11
y de su capacidad contráctil al retomar su actividad mio-cárdica normal. 11

Gelpi - Donato Fisiopatología cardiovascular
114
-cárdica con soluciones cardiopléjicas durante el mismo,
--
estable.
et al.9, quienes demostraron que la hipotermia --
El paro cardiaco mediante hipercalemia fue descrito por Melrose et al.10 -
--
El precepto fundamental de cualquier técnica de pro-
-
-
-
-13
Protección del miocardio isquémico
Técnicas de administración de cardioplejia
-
-
una vez confeccionados, que sería un complemento de la
1. Vía anterógradaLa cardioplejia se administra en forma tradicional a través
-14
-
en forma intermitente durante todo el tiempo de clampeo o bien en forma continua, pero esta última forma ocasio-
En los pacientes con enfermedad coronaria severa
-
-
de los ostium -
-ostium coronario.
También durante el tratamiento de la válvula mitral
2. Vía retrógrada
seno coronario fue descrita por Gott et al. 15 en 1957. -
con una sutura, o indirecta, sin la apertura de la aurícula, aunque la presencia de válvulas en el seno muchas veces
et al.16 demostraron que
-cular posterior. También se desarrollaron diferentes cánulas

Fisiopatología de la protección miocárdica en la cirugía cardíaca
115
-
desplazamiento. 17
-
puede desplazar hacia la aurícula derecha produciendo la
distal como pueden en el seno, mientras que otros, lo ostium de la vena
coronaria derecha. -
interrumpen para terminar las anastomosis distales. Un
18
Consideraciones anatómicas y hemodinámicas (Seno coronario).Efecto de reperfusión retrógrada
seno coronario, la vena cardiaca anterior y la vena cordis mínima. 19
El seno coronario se abre dentro de la aurícula dere-
el seno coronario. 19
derecho, terminando en la aurícula derecha a nivel de la
-19
La vena cordis minimaldirectamente dentro de la aurícula derecha y del ventrículo
y, ocasionalmente, al ventrículo izquierdo. 19
-ventricular, Hammond y Austen 20 -
que el septum es un territorio pobremente cubierto por la
-
seno coronario muy cerca de su ostium. La distancia me-dia entre el ostium del seno coronario y la vena cardiaca media es de 1,7 ±
de dicha vena. 19, 21
está muy profundo dejan de perfundirse el septum y el ventrículo derecho; si en cambio está colocado al inicio, puede sufrir su desplazamiento hacia la aurícula derecha.
-te el lecho capilar total, aun en pacientes valvulares con
suplan diferentes lechos capilares. 19
Thebesio actúan como conductos de baja resistencia, que
-
-forme de las soluciones cardiopléjicas. 19
et al. 22 -
-pléjica aun en presencia de enfermedad coronaria severa. 19
El sistema venoso del seno coronario es una red vascular densa que no resulta afectada por la arteriosclerosis coronaria y está desarrollada en forma más abundante y uniforme en la zona subendocárdica de ambos ventrículos. 19
En ciertas situaciones clínicas, tales como reoperacio--
de soluciones cardiopléjicas puede tener un efecto de la-
posibles émbolos ateromatosos. 19
o desplazamiento del embolismo aéreo, además de poder retroperfundir las soluciones en forma continua durante
19

Gelpi - Donato Fisiopatología cardiovascular
116
--
manece controversial, especialmente, en lo relacionado a 19
-
selectiva de los ostium --
-traventricular. 23
Aronson et al.24
minuto después del cardiopulmonar, mientras que
-
-
Métodos de administración de la solución cardiopléjica
-
1. Método combinado
rewarming
El paro cardiaco es iniciado con el uso de la cardio--
-
para el recalentamiento miocárdico a temperatura nor-mal.
2. Método retrógrado
3. Método anterógrado con oclusión del seno coronario
-
4. Reperfusión con intervenciones adicionales sobre el seno coronarioEs, básicamente, un método para aumentar la recupe-
coronario.Durante el recalentamiento miocárdico por medio de
-
-
-
25
Figura 7.1: Método de administración de cardioplejiaFigura 7.2: Cánula balón para infusión de cardioplejia retrógrada por seno coronario

Fisiopatología de la protección miocárdica en la cirugía cardíaca
117
Efectos clínicos de las intervenciones sobre el seno coronario
-
Talwaker et al.26 demostraron que la cardioplejia adminis-
et al. 27
randomizado retrospectivo. Determinaron que la cardio-
Jasinski et al.28
-
--
lo derecho. De esta manera es lícito pensar que la combina-
-
et al.29
-
y el septum interventricular posterior no se perfunden en forma ideal.
Soluciones cardiopléjicas
Las soluciones cardiopléjicas cristaloides se basan en -
das con diferentes complementos, como por ejemplo β-bloqueantes. Feindel et al.30 demostraron que las solucio-
cuanto a la reversibilidad de la injuria miocárdica en un
En un estudio prospectivo y randomizado, Fremes et al.31
-
32
Temperatura óptima y protección miocárdica
-
-13 Hayashida et
al.29, en un estudio randomizado, compararon los efectos
--
Figura 7.3: Anatomía del seno coronario y venas de drenaje en aurícula derecha

Gelpi - Donato Fisiopatología cardiovascular
118
-
Las técnicas de cardioplejia fría intermitente provocan hipotermia miocárdica, isquemia y retraso en la recupe-
13
hipotermia en los sistemas enzimáticos y bioquímicos,
como la cardiaca. 13
et al.33
enriquecida, con el objeto de preservar los niveles de ATP -
-
de ATP en diferentes estudios clínicos. 13
El Warm Heart Trial es un estudio randomizado y pros--
-sentaron menor índice de bajo volumen minuto posopera-
mortalidad e infarto perioperatorio. 13
-tados clínicos con el uso de la cardioplejia cristaloide fría en
--
13
Soluciones enriquecidas-
diopléjicas enriquecidas, se intenta atenuar la injuria miocárdica.
β -
simpático y estabilizando las membranas. 13
-frina se elevan cerca de 150 veces en el sistema coronario,
reservas de ATP se encuentran disminuidas, el bloqueo β-blo-
queantes administrados al sistema coronario contribuiría 34
Aunque la mayoría de los β-bloqueantes provocan un
corta y cardioselectiva como el esmolol, que tiene una vida
-
31 El uso del
13
-mún en el tratamiento del miocardio isquémico. El reciente estudio
-13
denominados scavengersel compartimiento intravascular e intersticial durante la
-13
anticuerpos monoclonales y acadesina y también con la in-
13
-
-
miocardio posisquémico. 13
Figura 7.4: En la figura de la izquierda se observa el catéter balón en correcta posición. Puede ocluir los ostiums de las venas cardiacas medianas y pequeñas. El área clara del corazón indica áreas perfundidas. La zona oscura indica zonas sin perfusión. En la figura de la derecha se esquematiza la cánula balón ocluyendo las venas 5 y 6 y también las venas cardiacas medianas y pequeñas. El área de no perfusión es mayor. 2. Seno coronario. 3. Gran vena cardiaca. 5. Ventrículo izquierdo (posterior). 6. Vena interventricular posterior.

Fisiopatología de la protección miocárdica en la cirugía cardíaca
119
--
13
42 También se
13
--
través del bloqueo del complejo CD11/CD18, mediante el uso de anticuerpos anti CD18, o bien mediante anticuerpos
13
--
13
reportada por Fitch et al. 35 que administraron anticuerpo
-
El factor de necrosis tumoral-α
13
Los pacientes tratados con adenosina previa a la isque-mia demostraron tener menores niveles enzimáticos, espe-
índice cardiaco. 13 También la adenosina puede activar la vía de precondicionamiento a través de la proteína kinasa
13 ver capítulo 6
ATPasa. 13
Los canales de potasio sensibles al ATP se ven impli--
mitocondrial. 13
los opioides ofrecen al tejido mayor tolerancia a la isque-13
la isquemia. 13
hibernado, llamados , tie-nen la propiedad, similar a los opioides, de aumentar la
-13
-13, pues parecen
imitar el precondicionamiento a través de un mecanismo no aclarado aún; aparentemente actúan mediante la apertura de los canales de potasio sensibles al ATP. 13
de sodio no puede ser manejada adecuadamente por la bomba de Na /K
intracelular activa su intercambio con el Ca / Ca , y
intracelular, responsable
-
-
la isquemia. 36 La injuria endotelial puede ser disminuida
NO. Básicamente se puede optar por dos caminos para es-
36
et al.37
contráctil posisquémica y, además, reduce la
13
Mizuno et al.38
totales de nitrato/nitrito. Además, la acetilcolina estimula 38 Aun-
de NO, no contribuye, aparentemente, en forma directa a 38, a pesar de que
adenosina en las soluciones cardiopléjicas podría tener po-tentes efectos cardioprotectores en la isquemia severa. La

Gelpi - Donato Fisiopatología cardiovascular
120
infarto, la injuria microvascular posisquémica y el tiempo de contractura isquémica. 36 El bloqueo no selectivo de los receptores de adenosina A1 y Ainfarto cuando es dada antes de la isquemia, y se observa
-36 -
, con menos efecto durante la isquemia con el receptor A1. 36
-
niveles de adenosina intersticial. A pesar de ello se de-
36
-
-
Bibliografía
1. Kassab, GS. “The coronary vasculature and its reconstruction . Ann Biomed Eng, 2000; 28(8): 903-15.
2. Vinten-Johansen J, Zhao Z.Q, Guyton RA. “Cardiac Surgical Physiology . Card. Surg. Adult, 2003; 2: 53-84.
3. Ramasamy R, Hwang YC, Liu Y, et al. “Metabolic and functional protection by selective inhibition of nitric oxide synthase 2 during ischemia-reperfusion in isolated perfused hearts . Circulation, 2004; 109(13): 1668-1673.
4. Buja LM, ML Entman. “Modes of myocardial cell injury and cell death in ischemic heart disease . Circulation, 1998; 98(14): 1355-1357.
5. Park JL, BR Lucchesi . “Mechanisms of myocardial reperfusion injury . Ann Thorac Surg, 1999; 68(5): 1905-12.
6. Steffens S, Montecucco F, Mach F. “The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury . Thromb Haemost. 2009; 102(2): 240-7.
7. De Vecchi E, Paroni R, Pala MG, et al. “Role of leucocytes in free radical production during myocardial revascularisation . Heart, 1997; 77(5): 449-55.
8. Kaul S, H Ito. “Microvasculature in acute myocardial ischemia: part II: evolving concepts in pathophysiology, diagnosis, and treatment . Circulation, 2004; 109(3): 310-5.
9. Bigelow WG, Lindsay WK, Greenwood WF. “Hypothermia; its possible role in cardiac surgery: an investigation of factors gover-ning survival in dogs at low body temperatures . Ann Surg, 1950; 132(5): 849-66.
10. Melrose DG, Dreyer B, Bentall HH, et al. “Elective cardiac arrest . Lancet, 1955; 269(6879): 21-2.
11. Bessho R, DJ Chambers. “Experimental study of intermittent crossclamping with fibrillation and myocardial protection: reduced
injury from shorter cumulative ischemia or intrinsic protective effect? J Thorac Cardiovasc Surg, 2000; 120(3): 528-537.
12. Buckberg GD, CE Hottenrott. “Ventricular fibrillation. Its effect on myocardial flow, distribution, and performance . Ann Thorac Surg, 1975; 20(1): 76-85.
13. Nicolini F, Beghi C, Muscari C, et al. “Myocardial protection in adult cardiac surgery: current options and future challenges . Eur J Cardiothorac Surg, 2003; 24(6): 986-93.
14. Buckberg GD. FOREWORD-Development of Blood Cardio-plegia and Retrograde Techniques, The Experimenter/Observer Complex, En: Mohl, W. Editorin Coronary Sinus Interventions in Cardiac Surgery. Second edition, Vienna, Austria. Eurekah.com/ Landes Bioscience 2000.
15. Gott VL, Gonzalez JL., Paneth M, et al. “Cardiac retroperfusion with induced asystole for open surgery upon the aortic valve or coronary arteries . Proc Soc Exp Biol Med, 1957; 94(4): p. 689-92.
16. Rudis E, Gates RN, Laks H, et al. “Coronary sinus ostial occlusion during retrograde delivery of cardioplegic solution significantly improves cardioplegic distribution and efficacy . J Thorac Cardiovasc Surg, 1995; 109(5): 941-6; discussion 946-7.
17. Castella M, GD Buckberg. “Reduction of systolic and diastolic dysfunction by retrograde coronary sinus perfusion during off-pump coronary surgery . J Thorac Cardiovasc Surg, 2004; 127(4): 1018-25.
18. Ikonomidis JS, Yau TM, Weisel RD, et al. “Optimal flow rates for retrograde warm cardioplegia . J Thorac Cardiovasc Surg, 1994; 107(2): 510-9.
19. Ruengsakulrach P, BF Buxton. “Anatomic and hemodynamic con-siderations influencing the efficiency of retrograde cardioplegia . Ann Thorac Surg, 2001; 71(4): 1389-95.
20. Hammond GL, WG Austen. “Drainage patterns of coronary arterial flow as determined from the isolated heart . Am J Physiol, 1967; 212(6): 1435-40.
21. von Lüdinghausen M, S Chiba, “The Venous Drainage of the Myo-cardium in the Human Heart , En: Mohl, W., Editorin Coronary Sinus Interventions in Cardiac Surgery. Second edition, Vienna, Austria. Eurekah.com/ Landes Bioscience 2000, p. 32.
22. Lillehei CW, Gott VL, Varco RL. “The direct vision correction of cal-cification of calcific aortic stenosis by means of pump-oxygenator and retrograde coronary sinus perfusion . Dis Chest, 1965; 30: 123-132.
23. Farge A, Mousseaux E, Acar C, et al. “Angiographic and electron-beam computed tomography studies of retrograde cardioplegia via the coronary sinus . J Thorac Cardiovasc Surg, 1996; 112(4): 1046-53.
24. Aronson, S. “Cardioplegia delivery: more (or less) than what you see . Ann Thorac Surg, 1999; 68(3): 797-8.
25. Mohl, W. “Basic Considerations and Techniques in Coronary Sinus Interventions. En: Mohl, W., Editorin Coronary Sinus Interventions in Cardiac Surgery. Second edition, Georgetown, Texas Eurekah.com/ Landes Bioscience 2000, p. 1-17.
26. Talwalkar NG, Lawrie GM, Earle N, et al. “Can retrograde cardio-plegia alone provide adequate protection for cardiac valve surgery? Chest, 1999; 115(1): 135-9.
27. Arom KV, Emery RW, Petersen RJ, et al. “Evaluation of 7,000+ patients with two different routes of cardioplegia . Ann Thorac Surg, 1997; 63(6): 1619-24.
28. Jasinski M, Kadziola Z, Bachowski R, Domaradzki, W, et al. “Com-parison of retrograde versus antegrade cold blood cardioplegia: randomized trial in elective coronary artery bypass patients . Eur J Cardiothorac Surg, 1997; 12(4): 620-6.
29. Hayashida N, Ikonomidis JS, Weisel RD, et al. “Adequate distri-bution of warm cardioplegic solution . J Thorac Cardiovasc Surg, 1995; 110(3): 800-12.
30. Feindel CM, Tait GA, Wilson GJ, et al. “Multidose blood versus crystalloid cardioplegia. Comparison by quantitative assessment of irreversible myocardial injury . J Thorac Cardiovasc Surg, 1984; 87(4): 585-95.
31. Fremes SE, Christakis GT, Weisel RD, et al. “A clinical trial of blood and crystalloid cardioplegia . J Thorac Cardiovasc Surg, 1984; 88: 726-41.

Fisiopatología de la protección miocárdica en la cirugía cardíaca
121
32. Tulner SA, Klautz RJ, Engbers FH, et al. “Left ventricular function and chronotropic responses after normothermic cardiopulmonary bypass with intermittent antegrade warm blood cardioplegia in patients undergoing coronary artery bypass grafting . Eur J Car-diothorac Surg, 2005; 27(4): 599-605.
33. Buckberg, GD. “A proposed ‘solution’ to the cardioplegic contro-versy . J Thorac Cardiovasc Surg, 1979; 77(6): p. 803-15.
34. Killingsworth CR, Wei CC, Dell’Italia LJ, et al. “Short-acting beta-adrenergic antagonist esmolol given at reperfusion improves survival after prolonged ventricular fibrillation . Circulation, 2004; 109(20): 2469-74.
35. Fitch JC, Rollins S, Matis L, et al. “Pharmacology and biological efficacy of a recombinant, humanized, single-chain antibody C5 complement inhibitor in patients undergoing coronary artery
bypass graft surgery with cardiopulmonary bypass . Circulation, 1999; 100(25): 2499-506.
36. Vinten-Johansen J, Zhao ZQ, Sato H. “Reduction in surgical ischemic-reperfusion injury with adenosine and nitric oxide the-rapy . Ann Thorac Surg, 1995; 60(3): p. 852-7.
37. Nakanishi K, Zhao ZQ, Vinten-Johansen J, et al. “Blood cardio-plegia enhanced with nitric oxide donor SPM-5185 counteracts postischemic endothelial and ventricular dysfunction . J Thorac Cardiovasc Surg, 1995; 109(6): 1146-54.
38. Mizuno A, Baretti R, Buckberg GD., et al. “Endothelial stunning and myocyte recovery after reperfusion of jeopardized muscle: a role of L-arginine blood cardioplegia . J Thorac Cardiovasc Surg, 1997; 113(2): 379-89.


123
Remodelamiento posinfarto de miocardio
8
Infarto de miocardio
miocardio es un sector de músculo cardiaco que ha sufrido
-tantes:
-tensa.
y de la estructura normales de la célula, causando ini-
de manera que la muerte celular representa el resul-
-juria celular subletal. Dado que no todas las células
-
celular en un sector del músculo cardiaco.--
de los miocitos cardiacos en un sector del miocardio. En un infarto, todos los tipos celulares que compo-
--
de muerte celular en la mayoría de las células paren-quimatosas presentes en ese sector de tejido.
El infarto de miocardio es la forma más importante de
bien diversas lesiones que provocan estrechamiento de la luz de una arteria coronaria pueden causar isquemia mio-cárdica, el infarto de miocardio es producido, en más del
puede decirse que el infarto de miocardio resulta de una
coronaria epicárdica, trombosis intraluminal superpuesta -
soespasmo. No obstante, pueden ocurrir infartos en en-
--
-
ocurra o no infarto depende, a su vez, de dos factores: de
-res: un sector central
,
no participan primariamente del evento isquémico.
En la zona central la necrosis comienza en el miocardio
-1

Gelpi - Donato Fisiopatología cardiovascular
124
músculo cardiaco comprometido pertenece al territorio de
miocardio es en el ventrículo izquierdo, pudiendo afectar un sector de la pared libre y/o del septum interventricular. Los infartos del ventrículo derecho y los auriculares son
parte de un infarto ventricular izquierdo.
-
-cardio subendocárdico adyacente y el epicardio pueden quedar indemnes.
-
transmurales y subendocárdicos. Los infartos transmurales
desde el endocardio hacia el epicardio. Los infartos suben-
de la pared. Así, la ocurrencia de un infarto transmural o
--
pared ventricular izquierda causan frecuentemente shock -
-
Efectos de la isquemia sobre los miocitos cardiacos
complejos en diversos sistemas celulares. Con el trans-curso del tiempo estas alteraciones empeoran, compro-metiendo en última instancia componentes estructurales y bioquímicos vitales, causando la muerte celular. No obs-tante, hasta cierto nivel y durante un periodo que tiene una
puede reparar y las células afectadas pueden recuperarse
-
2
Figura 8.1: Progresión de la necrosis tras la oclusión de una arteria coronaria epicárdica. A los 40 minutos era sólo subendocárdica; a las 3 horas era más extensa; a los 4 días presentaba su máxima extensión.
Figura 8.2: Determinantes del tamaño de infarto

Remodelamiento posInfarto de miocardio
125
-
están compuestas casi enteramente por creatina-fosfato,
--
-
-
isquemia, dándole al tejido un color pálido.Hasta los 15 minutos de isquemia total los cambios son
--
-dan lesionados de manera irreparable. En este sentido, todos los procesos isquémicos alcanzan un punto en el
cuando se le da la oportunidad de hacerlo, de manera que se puede considerar que alcanza un punto de no retorno,
-
ATP y los niveles de lactato se encuentran muy elevados, -
te injuriante, en este caso la isquemia, la célula no puede
irreversible conduce a la muerte celular.-
versibilidad de la injuria isquémica es la microscopía elec-3
- Los miocitos con injuria reversible se encuentran au-
--
cromatina, por debajo de la membrana nuclear. La mem-
densos y un aclaramiento incompleto de la matriz.- Los miocitos con injuria irreversible presentan el nú-
No obstante, los dos sellos distintivos de injuria isquémica -
mática asociadas con vacuolas subsarcolemales y presen-
Evolución morfológica
infarto es la presencia de una zona de palidez en la su-
horas se va delimitando cada vez más del tejido vecino
de infarto va tornándose blanquecino-amarillenta, y a su alrededor se hace presente un borde rojizo que indica
-mente, podemos dividir en tres etapas que se superponen
-
la muerte celular de los miocitos cardiacos en el área de
-
Figura 8.3: Evolución temporal y regional de la necrosis de coagulación. En la ordenada se representa una escala de medición semicuantitativa de 0 (ausente) a 3 (máxima o completa). La necrosis de coagulación estuvo presente durante las primeras 2 semanas, y alcanzó su máxima expresión en el miocardio medial.
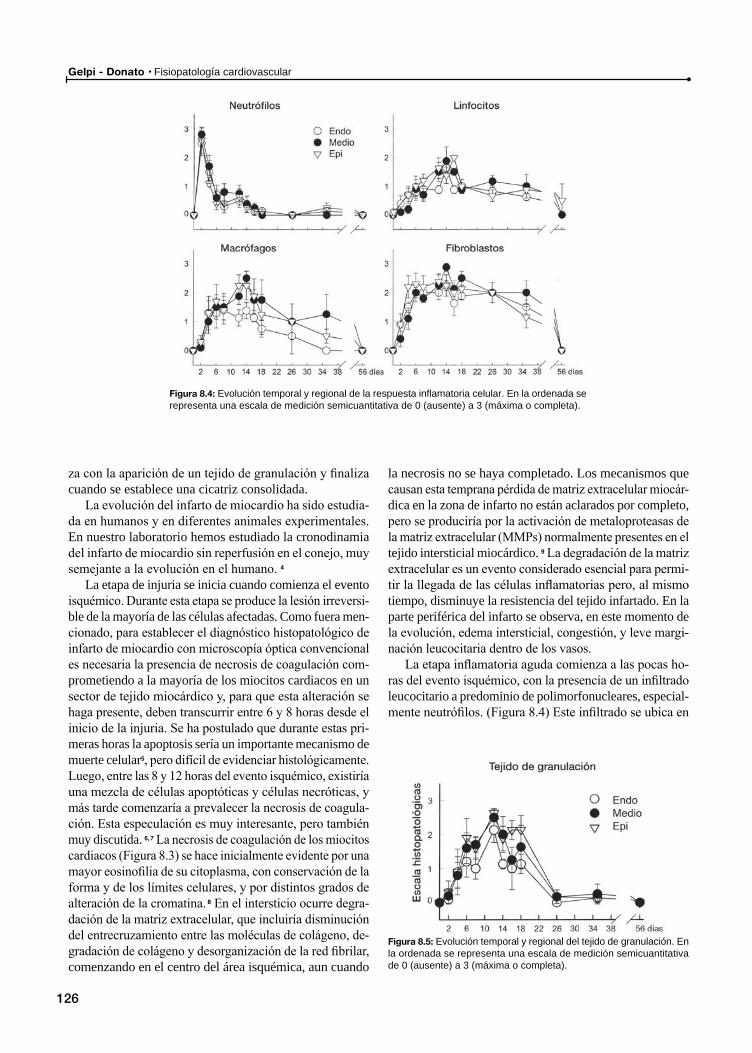
Gelpi - Donato Fisiopatología cardiovascular
126
cuando se establece una cicatriz consolidada.-
En nuestro laboratorio hemos estudiado la cronodinamia
4
La etapa de injuria se inicia cuando comienza el evento -
ble de la mayoría de las células afectadas. Como fuera men-
-prometiendo a la mayoría de los miocitos cardiacos en un
-meras horas la apoptosis sería un importante mecanismo de muerte celular5
-
muy discutida. 6, 7
8 -
-
comenzando en el centro del área isquémica, aun cuando
la necrosis no se haya completado. Los mecanismos que -
dica en la zona de infarto no están aclarados por completo,
tejido intersticial miocárdico. 9-
tiempo, disminuye la resistencia del tejido infartado. En la parte periférica del infarto se observa, en este momento de
-
-
leucocitario a predominio de polimorfonucleares, especial-
Figura 8.4: Evolución temporal y regional de la respuesta inflamatoria celular. En la ordenada se representa una escala de medición semicuantitativa de 0 (ausente) a 3 (máxima o completa).
Figura 8.5: Evolución temporal y regional del tejido de granulación. En la ordenada se representa una escala de medición semicuantitativa de 0 (ausente) a 3 (máxima o completa).
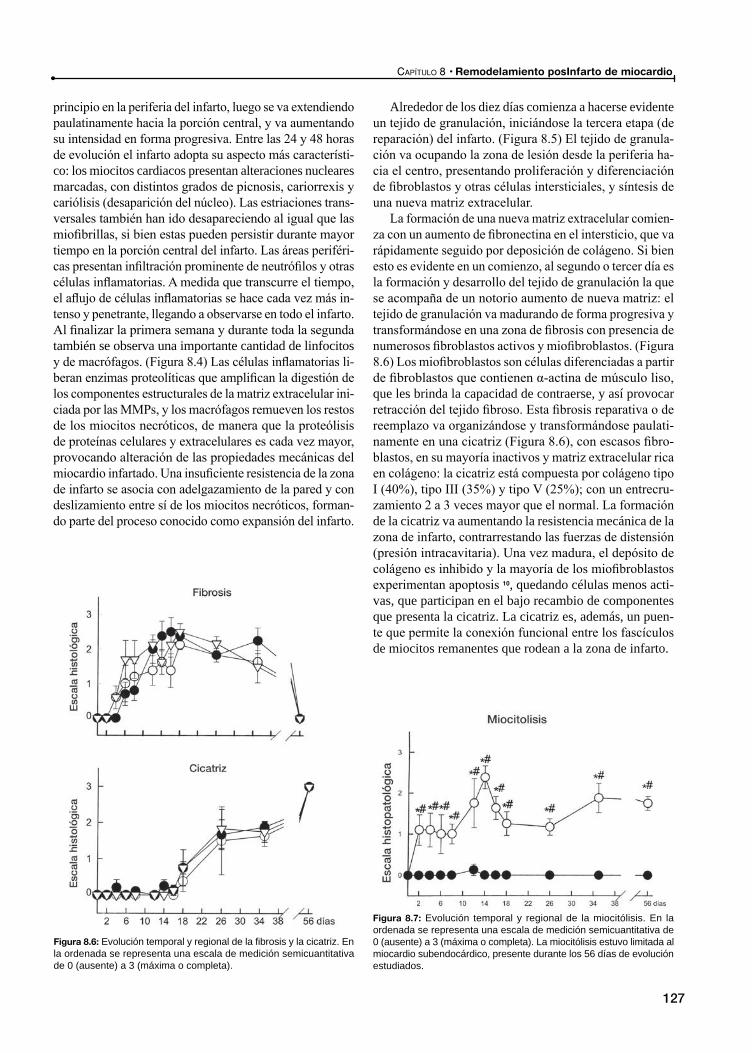
Remodelamiento posInfarto de miocardio
127
-co: los miocitos cardiacos presentan alteraciones nucleares
-
-
-
también se observa una importante cantidad de linfocitos -
-
-
Alrededor de los diez días comienza a hacerse evidente
--
-
que les brinda la capacidad de contraerse, y así provocar
--
-
de la cicatriz va aumentando la resistencia mecánica de la
10, quedando células menos acti-vas, que participan en el bajo recambio de componentes que presenta la cicatriz. La cicatriz es, además, un puen-
de miocitos remanentes que rodean a la zona de infarto.
Figura 8.6: Evolución temporal y regional de la fibrosis y la cicatriz. En la ordenada se representa una escala de medición semicuantitativa de 0 (ausente) a 3 (máxima o completa).
Figura 8.7: Evolución temporal y regional de la miocitólisis. En la ordenada se representa una escala de medición semicuantitativa de 0 (ausente) a 3 (máxima o completa). La miocitólisis estuvo limitada al miocardio subendocárdico, presente durante los 56 días de evolución estudiados.

Gelpi - Donato Fisiopatología cardiovascular
128
-
-nos de la mitad del volumen del miocardio al comienzo del infarto. 11
En el subendocardio suele observarse otro tipo de muerte -
Figura 8.7
-
-do como células vacías, con aspecto alveolar, delimitadas
-
postulado que en las áreas periféricas también puede pro-ducirse muerte celular mediante apoptosis. 12
Infarto de miocardio con reperfusión
adopta características peculiares. El primer factor que mo-
antes de que se complete el frente de onda, células que habían sufrido isquemia pero que no habían muerto son rescatadas
subendocárdico o transmural que no ocupa todo el espesor -
-
--
capilares de la zona de infarto: a mayor necrosis de capi-13 Los miocitos también pueden
mostrar un tipo particular de necrosis: necrosis en bandas 14
En los infartos reperfundidos es habitual que el tejido
rescatados -
-
-
-
-
.
-
15
Fisiopatología
mensurable es una pérdida funcional en un sector de tejido
durante un cierto tiempo. La persistencia de la isquemia, la
hacen que la pérdida funcional sea permanente.La pérdida funcional se hace evidente por una dismi-
-
-
y del volumen minuto.--
cida, se sabe que la isquemia y el infarto afectan ambos
-.
-námica implica un estiramiento del tejido miocárdico. En

Remodelamiento posInfarto de miocardio
129
Como fuera mencionado, durante los primeros días -
-
-
-to produce estiramiento del miocardio remanente, donde
--
sectores remanentes del ventrículo infartado. Adaptaciones
miocitos conlleva un aumento del volumen de la cámara
-
que se autoperpetúa.El mayor estrés parietal actúa sobre todos los compo-
citoquinas 16
17, quienes a su vez estimulan la apop-
mecanismos pueden ser estimulados por el incremento
como un mecanismo compensador, tiende a restablecer el -
mos neurohumorales pueden resultar inicialmente bene--
neurohumoral sistémica media cambios compensatorios
de remodelamiento.
Sistema nervioso simpático
representa un mecanismo compensador. 18 No obstante, -
tos deletéreos. 19
-
nalina. Estos cambios pueden inducir apoptosis de los
-
así el círculo vicioso.
Sistema renina-angiotensina-aldosteronaOtro importante sistema que presenta actividad aumentada
ver capítulo 9 20 La
incrementa la vasopresina. Todos estos efectos sirven para
de sodio, e incrementar el tono vascular. Además, dentro
un infarto de miocardio.-
ral en pacientes que han sufrido un infarto de miocardio
--
nozcamos más acerca de otros sistemas seremos capaces
Como fuera mencionado, como resultado de la dilata-
-21
22
Remodelamiento
Concepto de remodelamiento
23 -
-formar su esencia. Con el mismo sentido, el concepto de remodelamiento fue introducido en la década de 1980 para describir los cambios estructurales que se producen en la pared ventricular y en la cavidad ventricular izquierda como
infarto de miocardio. 24

Gelpi - Donato Fisiopatología cardiovascular
130
utilizándolo también para referirse a cambios en las paredes y en las cámaras cardiacas que ocurren en otras enfermeda-
-patías y las miocarditis. El término también es aplicado,
de volumen y a los mecanismos moleculares involucrados. En determinadas circunstancias es utilizado, además, para referirse a cambios en el crecimiento normal y en situacio-
En cualquier caso, el concepto de remodelado es un con-cepto estructural: ha ocurrido remodelado cuando en el co-
-
La importancia del remodelamiento ventricular se ha
-
lo ha transformado en un importante blanco terapéutico.
Remodelamiento posinfarto de miocardio
-tructurales que se producen como consecuencia de un
--
Ello obedece, en principio, a dos hechos fundamentales -
los sectores remanentes. De esta manera, el infarto de
-lamiento tardío.
Expansión del infarto
25
Los cambios estructurales que se producen en la zona
de miocitos, deslizamiento de los miocitos entre sí, rup-
resultando todo ello en un menor número de miocitos a través del espesor de la pared. 26 A los cambios miocita-
27
--
resultado de una nueva injuria isquémica y necrosis mio-28
-nicamente inaparente hasta la ruptura ventricular. 29 Los
-
el espesor del miocardio infartado y las condiciones de
actúa como factor protector.
Figura 8.8: Esquema de infarto sin y con expansión. El infarto con expansión presenta adelgazamiento de la pared, aumento de la longitud epicárdica correspondiente al infarto, y dilatación regional de la cavidad ventricular.

Remodelamiento posInfarto de miocardio
131
A los cambios mencionados en la zona de infarto se -
manente, con deslizamiento de los haces musculares entre
través de la pared ventricular y alteraciones de los mioci-
incrementa de 7 a 8 veces.
-
-
-
-
-
y/o rotura del ventrículo.El deterioro funcional que caracteriza a estos pacientes,
es posible que relacionado con un incremento en el estrés parietal sobre el miocardio no infartado, y la mortalidad
-conocimiento temprano de esta forma de remodelamiento.
-
-
-lar, eventos isquémicos miocárdicos adicionales o muer-
infarto es vista como el comienzo de un proceso tiempo-
del infarto de miocardio. 30
Remodelamiento tardío posinfarto de miocardio
después del evento isquémico, como resultado de una
31
-delado tardío es el mecanismo por el que aparece la falla de bomba tardía en los pacientes que han padecido un infarto de miocardio, hecho que ocurre en una elevada
32
El remodelado tardío obedece a cambios estructurales en la pared del ventrículo, tanto en la zona de infarto como
33
Los cambios en la zona de infarto, que forman parte del remodelamiento tardío, se deben al proceso de repa-
--
vo que reemplaza al tejido miocárdico irreversiblemente
Este proceso puede llevar semanas o aun meses, depen-
moduladores y de la especie considerada, pero siempre
El volumen del tejido cicatrizal siempre es menor que el volumen del tejido lesionado al que reemplaza, y adi-
-
en la zona de infarto. La cicatriz es menos distensible que
de la zona de infarto no es un componente importante del remodelamiento tardío. Así, el remodelado tardío en la zona de infarto está compuesto principalmente por un
-
Al contrario de los cambios en la zona de infarto, son --
--
Los miocitos cardiacos remanentes se ven envueltos en -
Remodelamiento de los miocitos cardiacos en el miocardio remanenteEl remodelamiento de los miocitos remanentes se mani-

Gelpi - Donato Fisiopatología cardiovascular
132
es considerada, al menos en principio, como una res-
trabajo no puede ser compensada a través de la forma-
hecho habitual en otros tipos de celulares. El mecanismo
de los miocitos cardiacos los componentes estructurales
-
de un aumento en el diámetro transversal del miocito y, por ende, en el espesor de la pared del ventrículo,
-
-pectivamente .
En el infarto de miocardio, los miocitos de los sectores -
esta manera, los miocitos presentan un marcado aumento
34,
35 Como consecuencia, el espesor de la pared ventricular
Una mayor demanda funcional para el tejido muscular
--
-
-ciada por desencadenantes mecánicos y/o por desencade-
El desencadenante mecánico es el estiramiento de los miocitos causado por un mayor estrés parietal. El estira-
-
-
las proteínas contráctiles, que pasan de las formas adultas
-36 -
37 En forma adicional, también
constitutivas, aumentando la síntesis de cadena liviana de 38 Por último,
39
---
-
forma paracrina o autocrina. En cualquiera de estos casos, -
ET-1 son similares entre sí y actúan acoplados a proteínas Gq. 40-42
43 Los factores de crecimiento
de crecimiento epidérmico, factor de crecimiento derivado
MAP kinasa. 44 -

Remodelamiento posInfarto de miocardio
133
transmitidos a través de un mecanismo común que involucra
miocardio, la masa ventricular izquierda y el peso del co-
tales parámetros pueden estar aumentados, encontrarse dentro de los valores normales, o estar disminuidos. Ello
-tar la pérdida producida. Por el contrario, si la pérdida es
-lizar dichos parámetros.
Degeneración y muerte de los miocitos cardiacos en el miocardio remanente
los miocitos, van apareciendo paulatinamente diversos
de miocitos por muerte de los mismos. Los cambios de-
-
que provocan la muerte de los miocitos remanentes y la forma de muerte que provocan son poco conocidos. En la actualidad se considera que la muerte de los mioci-tos puede producirse por necrosis, por apoptosis45, o por
46
-
47
El remodelado del tejido intersticial en los sectores
-
básica y clínica.
Remodelamiento de la matriz extracelular
miocárdica es un complejo microambiente para los mio-citos cardiacos. 48 Consiste en una red macromolecular
sostén a los miocitos cardiacos y, junto con ellos, determi-
Tabla 8.1: Componentes de la matriz extracelular cardiaca

Gelpi - Donato Fisiopatología cardiovascular
134
formada por componentes estructurales y no estructurales:
-
la abundancia relativa de uno u otro componente podrían
-nas remotas es diferente en etapas tempranas y tardías.
-
49 Como
En etapas tardías se produce un aumento de la matriz
-parativa como en la zona de infarto, sino que es reactiva.
-
tejido intersticial. Como fuera mencionado, la pérdida de
manera que en el miocardio remanente va apareciendo
reactiva y reparativa. 50
El remodelamiento vascular en la zona remota, eva-
-mostrando un retraso en el crecimiento adaptativo de la microvasculatura con respecto a los miocitos. 51 Debido a ello, podría esperarse que el ventrículo izquierdo infartado
El remodelamiento tardío se caracteriza, entonces, por -
-
-diovasculares adversos, de forma que el remodelamiento
-dinámica y consecuencias clínicas desfavorables durante
52-54
Remodelamiento temprano/remodelamiento tardío: un continuo hacia la insuficiencia cardiaca
-
-
-cipal condicionante de este espectro. Como dijéramos,
mínimamente deformada. En los corazones con infarto,
-
miocardio residual.-
duce un desplazamiento de la curva hacia la derecha: para
es lo mismo que decir que se necesita un mayor volumen
-
55
-tos moderados tal incremento de volumen fue encontrado
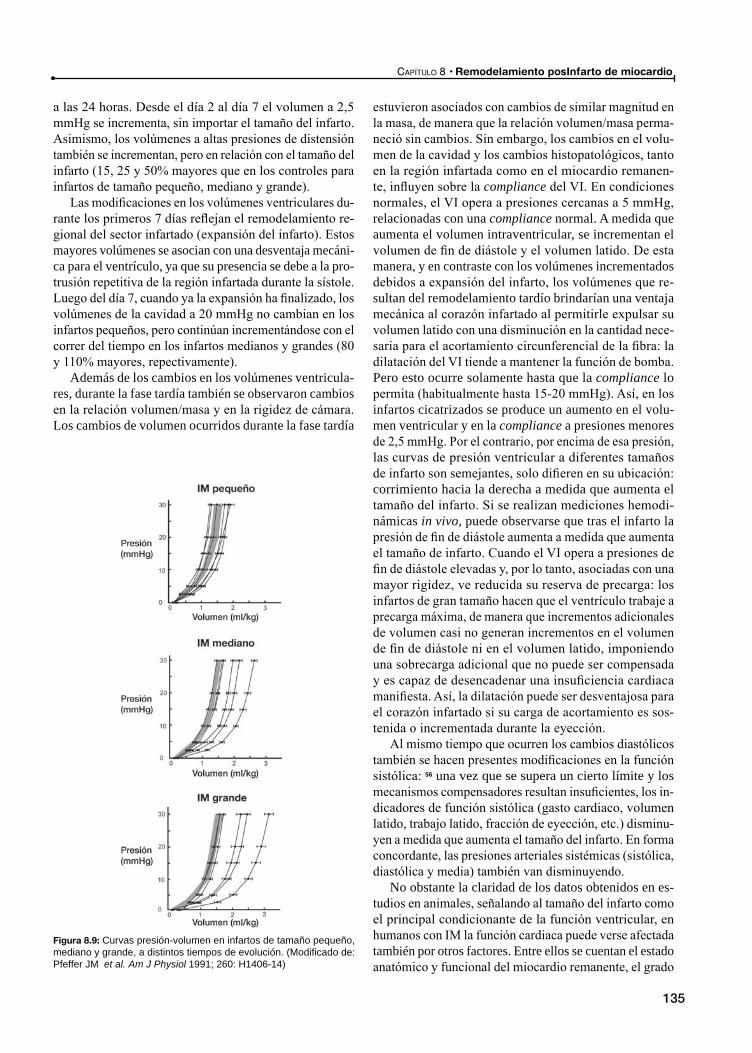
Remodelamiento posInfarto de miocardio
135
--
mayores volúmenes se asocian con una desventaja mecáni-ca para el ventrículo, ya que su presencia se debe a la pro-
Además de los cambios en los volúmenes ventricula-res, durante la fase tardía también se observaron cambios
Los cambios de volumen ocurridos durante la fase tardía
--
-compliance
relacionadas con una compliance normal. A medida que aumenta el volumen intraventricular, se incrementan el
manera, y en contraste con los volúmenes incrementados -
sultan del remodelamiento tardío brindarían una ventaja
-
Pero esto ocurre solamente hasta que la compliance lo
infartos cicatrizados se produce un aumento en el volu-men ventricular y en la compliance a presiones menores
corrimiento hacia la derecha a medida que aumenta el -
námicas in vivo, puede observarse que tras el infarto la
-
56 una vez que se supera un cierto límite y los -
-
No obstante la claridad de los datos obtenidos en es-
también por otros factores. Entre ellos se cuentan el estado Figura 8.9: Curvas presión-volumen en infartos de tamaño pequeño, mediano y grande, a distintos tiempos de evolución. (Modificado de: Pfeffer JM et al. Am J Physiol 1991; 260: H1406-14)

Gelpi - Donato Fisiopatología cardiovascular
136
Factores que modifican el remodelamiento
-
parietal y de la permeabilidad de la arteria relacionada con el infarto. Los dos primeros ya han sido considerados
ahora en la permeabilidad de la arteria relacionada con el infarto. 57-60
--
de eventos ha traído aparejados esfuerzos para limitar el
dolor torácico, intentando prevenir el desarrollo completo -
lar izquierda. En tal sentido, el pilar principal de la reper-
-lular miocárdica que avanza desde el endocardio hacia el
la terapéutica trombolítica a todos los pacientes con infarto
Cuando la terapéutica trombolítica se utiliza rápida-
-
debe a una mejoría en la motilidad parietal de la zona de
-
la mejoría en la sobrevida es mayor que la esperable por
Por otra parte, en un estudio en pacientes con infarto 58,
En concordancia, otros estudios 59, 60
arteria relacionada con el infarto se encontraba permeable
-
ocurriría demasiado tarde como para rescatar al miocardio isquémico, evitando la muerte celular.
-cionada con el infarto se encuentra permeable, este hecho
-
trombolisis tardía brinda una mejoría en la sobrevida. Por
-te el periodo inicial sino por otro mecanismo.
Los mecanismos mediante los que la trombolisis tar-
días pos infarto la arteria relacionada con el infarto per-
-
-
-canismo adicional sería una mayor estabilidad eléctrica
Por lo tanto, la reapertura de una arteria responsable de infarto produce una mayor sobrevida que no está en
-
de una mayor estabilidad eléctrica y de una mejor perfu-
infarto. 61
Aspectos clínicos
Epidemiología
miocardio es uno de los principales mecanismos invo-

Remodelamiento posInfarto de miocardio
137
-
62
estar relacionado, como hemos visto, con un importante número de factores que incluyen la permeabilidad de la
-
-
-
Género-
-
63
--
cardiaca.-
64
-
65
--
un subanálisis del estudio DIG66
que las mujeres tienen una mayor mortalidad con el uso
Diabetes
presentar enfermedad cardiovascular, ya que la diabetes se asocia a una variedad de alteraciones cardiacas, inclu-
67
de un infarto de miocardio los diabéticos tienen mayor
cardiaca. 68
-serva en los pacientes que desarrollan más remodelado
-
-lar que los no diabéticos. De manera que, aun cuando en
infarto duplica a la de los no diabéticos, esta no se asocia
69 no ha sido observado en otros estudios más pe-70 o mostraron
71 La diabetes se asocia a una variedad de cambios ultraestruc-turales en el miocardio, caracterizada por un aumento de
72 67 y aumento 73 Este ha sido asociado, en el
-74 Aún cuando en los pacientes con diabetes se pre-
75, lo que puede
--
celular76, cambios que serían responsables del aumento -
ventricular izquierda podría ser una forma de adapta-
-
de eventos cardiovasculares y de muerte. El hecho de
tendría menor capacidad de adaptarse a la pérdida de
efectivo. Así, el único recurso para sostener el volu-

Gelpi - Donato Fisiopatología cardiovascular
138
-
-
77
-
infarto, este no parecería involucrar un mayor remodela-do ventricular.
Hipertensión arterial
--
78
-
79; por otra, el aumento de la apoptosis
-do. 80
aumento del estrés parietal, un conocido estímulo para el desarrollo de remodelado. El aumento de la actividad neu-
81 Por último, la -
ventrículo izquierdo, y que también ha demostrado pre-
Evaluación clínica del remodelado
Métodos de estudio
-
se encontraba asintomática. 82 Por tal motivo, debería in-
ventricular izquierda.
-
-
-
métodos básicos, que forman parte de la rutina de la eva-
los cuales el médico puede sospechar remodelado y, por lo tanto, podrán servir como pesquisa del remodelado ventricular posinfarto.
Electrocardiograma
--
infartado y de las anormalidades de la motilidad parietal.
ventricular en las zonas adyacentes al área de necrosis, -
de la onda T puede indicar la presencia de miocardio
---
la presencia de miocardio viable aunque disfuncionante y el tejido normal circundante pueden ser los responsables de crear diferentes campos eléctricos localizados, que a su vez pueden ser responsables de los cambios electrocardio-
-lados 83 o asociados a la clínica 84
III 85 -
-
-

Remodelamiento posInfarto de miocardio
139
-
ventrículo izquierdo.
la presencia de infarto.--
mente importante y de una utilidad clínica mayor, no es un
ventricular. En el caso particular de los infartos que com-prometen la punta del ventrículo izquierdo pueden aparecer
-
Radiografía de tórax-
Ambas medidas se suman y el resultado se divide por la distancia entre la línea media y el borde interno del radio
cardiotorácica supera los 0,5.
-
costo, su alta accesibilidad y su calidad de no invasiva,
-
-
diámetro transverso cardiaco durante el ciclo cardiaco:
puede caer en cualquier parte del mismo y producir varia-
-fías que se hacen en la cama del paciente no respetan la
-
ventrículo izquierdo, de manera que el crecimiento anor-
aneurismática, puede ocupar el borde derecho de la silueta, o el crecimiento anormal del ventrículo derecho lo hace ocupar el lado izquierdo de la misma. A estas alteraciones se le pueden sumar las que involucran al pericardio, que
-
-
86
-
--
la severidad del síndrome.Resultados semejantes se obtuvieron del análisis del
estudio DIG87
han encontrado resultados similares.
-
-
Ecocardiografía--

Gelpi - Donato Fisiopatología cardiovascular
140
temprano y sumamente sensible de la presencia de is-
a la morbimortalidad.
-
--
clínica y hemodinámica, con las complicaciones del infarto
del infarto mayores las complicaciones y mortalidad. Los -
además, permiten evaluar muchas de sus complicaciones,
su capacidad para evaluar en forma seriada la evolutividad
todos y cada uno de los estadios de la enfermedad coro-
-
de la masa ventricular izquierda, de los volúmenes ven--
88 -riencia del operador es también un factor crucial, y su uso
de obesidad o de enfermedad pulmonar.Como dijéramos, durante el infarto se producen altera-
desplazamiento espacial respecto de los adyacentes, sino -
-
-rritorio verdaderamente comprometido.
-cados en la motilidad parietal que se traducen por aqui-
-temente, por hipoquinesia marcada. En cambio, en el in-farto no transmural, el trastorno de la motilidad es menos
-
-
plazo y mortalidad. 89
Otras técnicas de evaluación
ventrículo izquierdo en forma adecuada y no tienen las
-
-ventaja, estos estudios no pueden hacerse en la cabecera
--
Figura 8.10: Evolución de los volúmenes ventriculares durante el remodelado posinfarto de miocardio; antes del infarto (A), durante su periodo agudo (B), y a dos diferentes tiempos de evolución más tardíos (C y D)
Fi 8 10 E ol ción de los olúmenes entric lares d rante el

Remodelamiento posInfarto de miocardio
141
-
-
probablemente el mejor método para el cálculo de volú-
y reproducibilidad, pero su costo es aún mayor y también hay que trasladar al enfermo.
-
-ca, varios estudios han demostrado que es también una de
-
-
-
Prevención y modulación del remodelamiento posinfarto de miocardioEl tratamiento del infarto de miocardio incluye las te-
--
-queantes tiene su fundamento en el hecho de que la ac-tividad neurohumoral es un componente importante del proceso de remodelamiento. Por lo tanto, intervenciones
-
-
-
90 91 y la diabetes. 92
-
de ocurrido el infarto, antes de que se produzcan cambios duraderos. En la actualidad no se dispone de un único
necesario utilizar terapéuticas combinadas.Dado que los cambios producidos durante el remode-
lamiento posinfarto tienen una elevada concordancia con
cardiaco puede ser considerado como un objetivo prima-rio del tratamiento y asimismo servir como un indicador
en poblaciones menos numerosas.-
no son ideales ya que, a pesar del tratamiento, en muchos
-
del estrés parietal actúan sobre la zona de infarto y sobre
Así, el objetivo de la terapéutica antirremodelamien-
revertir el remodelamiento estructural desfavorable, inte--
-res métodos para revertir este proceso hacia la normalidad.
-
-

Gelpi - Donato Fisiopatología cardiovascular
142
en la zona de infarto como en la zona remota. Pero am-
--
tiva en las zonas remotas sería, por el contrario, un evento
indiscriminadamente sobre ambas zonas, podría presentar,
-
--
mente compensada, y el proceso contendría, en sí mismo,
-
Terapéutica farmacológica-
hemodinámica podría ayudar a disminuir las fuerzas de
-vo del ventrículo izquierdo. Por otra parte, si se evitara la
Por último, considerando que el remodelamiento pos-
-mento en los valores plasmáticos de los mediadores que
-
los mediadores que presentan propiedades vasodilatadoras,
disminución de la actividad neurohormonal
En la actualidad, los fármacos utilizados para modular el remodelamiento posinfarto de miocardio son los inhi-
Inhibidores de la enzima de conversión de angiotensina
-
en dos situaciones clínicas diferentes. Durante el periodo
cardiaca. En el primer escenario, los IECA fueron evaluados 62, AIRE93 y TRACE. 94
95 96 y tratamiento 97
-
-
pueden ser deletéreos durante el infarto de miocardio, tales -
-
-
IECA han demostrado ser muy útiles en el infarto de mio-cardio, por diversos mecanismos: previenen el desarrollo
--
emanada de estos estudios indica que los IECA deben ad-
-
hubiera historia de estenosis bilateral de la arteria renal; o
-
-
Antagonistas de la angiotensina II

Remodelamiento posInfarto de miocardio
143
brindan un bloqueo más selectivo de los efectos de la -
de los efectos hemodinámicos de estos, todavía necesitan más estudios preclínicos y clínicos que apoyen su utiliza-
en pacientes con intolerancia a los IECA.
Antagonistas de la aldosterona-
-
compliance arterial
El bloqueo de la aldosterona previene el remodelado -
miocardio98 -
El bloqueo de la aldosterona es otra forma de inhi-
-99 -
100 Eple-
-
talidad y forma parte del tratamiento estándar de ambas
-dosterona sobre el remodelado ventricular en pacientes
-
101
Bloqueantes adrenérgicos
--
-
muchos de estos efectos adversos producto de la sobres-
un doble rol en el tratamiento del remodelado posinfarto.
102 y timolol103, que demostraron -
pio, los médicos eran remisos a su empleo en el infarto por
-nían mayor deterioro clínico y hemodinámico.
-
Tabla 8.2: Efectos del enalapril y del carvedilol sobre los volúmenes ventriculares y la fracción de eyección del ventrículo izquierdo
Basal 4 meses 6 meses 12 meses Valor de p
Volumen de fin de diástole (ml)
SOLVD placebo 200 ± 42 208 ± 43 210 ± 46 p = 0.025
SOLVD enalapril 196 ± 41 198 ± 37 197 ± 39
ANZ placebo 175 ± 52 - 185 ± 58 194 ± 54 p = 0.0015
ANZ carvedilol 187 ± 72 - 179 ± 63 178 ± 63
Volumen de fin de sístole (ml)
SOLVD placebo 148 ± 38 155 ± 43 156 ± 42 p = 0.019
SOLVD enalapril 146 ± 38 147 ± 36 145 ± 38
ANZ placebo - 133 ± 52 139 ± 47 p = 0.0001
ANZ carvedilol 136 ± 64 - 121 ± 56 121 ± 57
Fracción de eyección (%)
SOLVD placebo 26 ± 11 26 ± 11 26 ± 11 p = 0.612
SOLVD enalapril 25 ± 11 26 ± 11 26 ± 11
ANZ placebo 30.4 ± 9.1 - 29.3 ± 8.2 29.2 ± 7.8
ANZ carvedilol 28.6 ± 7.1 - 33.5 ± 8.3 34.1 ± 9.7

Gelpi - Donato Fisiopatología cardiovascular
144
-104
ya tratados con trombolíticos y IECA. En este estudio, el
mortalidad total, de la mortalidad cardiovascular y de la incidencia de infarto no fatal. Un subestudio ecocardio-
105
-
De manera que el carvedilol produce efectos claramente
parece ser aditivos a los de los IECA. --
106, metoprolol 107 o bisoprolol 108
la supervivencia, disminuyen la morbilidad, aumentan la
calidad de vida, y disminuyen el número de internaciones y
-
hidrosalina se recomienda el ajuste del tratamiento diuréti-
NitratosLos nitratos han sido ampliamente utilizados en el infarto de miocardio durante varias décadas. Tienen varios efectos
-
-
ventricular. Pese a estos resultados positivos, los estudios 109 110 -
-tos no se recomiendan rutinariamente salvo ante la persis-
-
111
sobre la mortalidad fue inferior al del enalapril cuando ambos fármacos fueron evaluados en forma comparativa.
Estatinas-
una enzima limitante en la vía de la biosíntesis del co-
niveles plasmáticos de colesterol. Pero, además, estos -
Tales efectos dependen de las acciones sobre una cadena ---
ventricular con mejoramiento de los volúmenes y de la 112 En humanos
-113 mientras otros
114 También se observaron 115, de
manera que hasta el presente es prematuro recomendar
de la enfermedad coronaria.
Permeabilidad de la arteria responsableLa permeabilidad de la arteria responsable del infarto
-
stent-
del futuro remodelado ventricular, la permeabilidad de la -
La permeabilidad de la arteria responsable se corre-
116, a tal punto que,
--
117
salvar tejido endocárdico y restaurar el miocardio atontado

Remodelamiento posInfarto de miocardio
145
en los bordes del infarto. Los infartos reperfundidos tienen,
-118
del infarto, dentro del mes de producido el evento, con --
119
Terapia celularLas stem cellsque se desarrollan y se mantienen los distintos tejidos del
-
---
--
mecanismo íntimo a través del que se produce dicho bene-
-nismo diferente al implante de células latiendo en forma
una mejoría de las propiedades mecánicas pasivas, con 120, y con un
efecto paracrino121 -
Por varias razones, la terapéutica con células madre es un área controversial e intensamente debatida. Por un
-
células madre podría tener un impacto favorable en pa-cientes con infarto de miocardio. En el mismo sentido, estudios en pacientes indican que la terapéutica con cé-lulas madre es factible, y que las células madre tendrían
No obstante, es importante resaltar que tales ensayos clí-
y sus resultados pueden considerarse francamente preli-minares; resulta notable el hecho de que esta terapéutica haya sido introducida en ensayos clínicos cuando no se han elucidado mecanismos básicos. Actualmente se están
-jor controlados, para obtener más y mejores resultados en
-
-
aspectos fundamentales, de manera que la terapéutica con células madre en pacientes con infarto de miocardio, hoy
-tiva de tratamiento.
Modulación no farmacológica
pueden interrumpir el remodelamiento adverso posinfarto -
delado posinfarto de miocardio puede hacerse mediante --
Cirugía
aneurismas ventriculares sintomáticos han sido tratados
cierre directo lineal del ventrículo izquierdo 122 o con im-123 De acuerdo
con la ley de Laplace, los ventrículos dilatados tienen
Batista et al.124 han reducido el volumen de los ventrícu-
pared libre del ventrículo izquierdo. Aun cuando ha sido reportado un efecto favorable en pacientes seleccionados, este método no es recomendado debido a la alta tasa de
-
125,

Gelpi - Donato Fisiopatología cardiovascular
146
-
-
rescate en pacientes críticamente enfermos.
Métodos de contención pasiva
abandonadas, pero fundaron las bases para el desarrollo
la forma del ventrículo izquierdo mediante una conten-
pacientes con insuficiencia cardiaca. En lo que respecta
el remodelado.
Soporte circulatorio mecánicoLos dispositivos de asistencia circulatoria mecánica brin-dan un soporte circulatorio en forma parcial o total en
-
-
solamente el trasplante cardiaco y los dispositivos de asis-tencia circulatoria mecánica pueden permitir mejorar la
-
El uso de dispositivos de asistencia circulatoria me-
pacientes en espera de trasplante como para el tratamiento
-cia ventricular ha provisto conocimientos únicos acerca del proceso de remodelamiento. En efecto, observar que
--
mecanismos celulares que subyacen en el remodelamiento
de remodelamiento se han vuelto el objeto de intensas
nuevos blancos terapéuticos pueden proveer nuevos cami-nos para producir un remodelamiento inverso, mejorando
Métodos para restaurar una contracción sincrónica (resincronización cardiaca)Uno de los más recientes avances en tratamiento de la
-lar en el que ambos ventrículos son resincronizados para contraerse en forma simultánea. Este proceso es conocido
ver capítulo 24 -
-lar e intraventricular, y así mejorar la contractilidad del
-
el índice cardiaco. 126
127
Ejercicio
-maneciendo en reposo en cama durante, por lo menos, dos o tres meses. Entre las bases racionales de dicha conducta se encontraba el objetivo de mantener un bajo consumo
-ca suele iniciarse en fases tempranas de la convalecencia
-
indica el entrenamiento físico. No obstante, los efectos del ejercicio sobre el remodelamiento y sus implicancias
-
remodelamiento con efectos a favor y en contra.Por una parte, el aumento de la actividad física en eta-
-

Remodelamiento posInfarto de miocardio
147
-
-
-cuencia cardiaca, un efecto favorable sobre la resistencia
-
-ciencia cardiaca el entrenamiento mejora el metabolismo
Al evaluar el impacto del ejercicio sobre el remodelado posinfarto de miocardio deberían tenerse en cuenta dis-
mantener el , momento en la evolu-
-
En los estudios realizados en pacientes se presentan varias
-
un alto número de pacientes. 128 -
perjudicial. 129 De esta manera, los datos disponibles son limitados y, a menudo, contradictorios.
Bibliografía
1. Reimer KA, RB Jennings. “The ‘wavefront phenomenon’ of myo-cardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow”. Lab Invest 1979;40:633-44.
2. Jennings RB, Steenbergen C Jr, Reimer KA. “Myocardial ischemia and reperfusion”. Monogr Pathol 1995;37:47-80.
3. Jennings RB, CE Ganote. “Structural changes in myocardium during acute ischemia”. Circ Res 1974;35 Suppl 3:156-72.
4. Morales C, González GE, Rodríguez M, Bertolasi CA, Gelpi RJ. “Histopathologic time course of myocardial infarct in rabbit hearts”. Cardiovasc Pathol 2002;11:339-45.
5. Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. “Apop-tosis and myocardial infarction”. Basic Res Cardiol 1998; 93 Suppl 3:8-12.
6. Rodríguez M, Lucchesi BR, Schaper J. “Apoptosis in myocardial infarction”. Ann Med. 2002;34(6):470-9.
7. Rodríguez M, J Schaper. “Apoptosis: measurement and technical issues”. J Mol Cell Cardiol 2005;38(1):15-20.
8. Fujiwara H, Ashraf M, Sato S, Millard RW. “Transmural cellular damage and blood flow distribution in early ischemia in pig hearts”. Circ Res. 1982;51(6):683-93.
9. Takahashi S, Barry AC, Factor SM. “Collagen degradation in is-chaemic rat hearts”. Biochem J 1990;265: 233-241.
10. Zhao W, Lu L, Chen SS, Sun Y. “Temporal and spatial characte-ristics of apoptosis in the infarcted rat heart”. Biochem Biophys Res Commun 2004;325(2):605-11.
11. Fishbein MC, Maclean D, Maroko PR. “Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution”. Am J Pathol. 1978;90(1):57-70.
12. Abbate A, Morales C, De Falco M, Fedele V, Biondi Zoccai GG, Santini D, Palleiro J, Vasaturo F, Scarpa S, Liuzzo G, Severino A, Baldi F, Crea F, Biasucci LM, Vetrovec GW, Gelpi RJ, Baldi A. “Ischemia and apoptosis in an animal model of permanent infarct-related artery occlusion”. Int J Cardiol 2007;121:109-11.
13. García-Dorado D, Théroux P, Solares J, Alonso J, Fernández-Avilés F, Elizaga J, Soriano J, Botas J, Muñoz R. “Determinants of hemo-rrhagic infarcts. Histologic observations from experiments involving coronary occlusion, coronary reperfusion, and reocclusion”. Am J Pathol 1990;137(2):301-11.
14. Baroldi G, Mittleman RE, Parolini M, Silver MD, Fineschi V. “Myo-cardial contraction bands. Definition, quantification and signifi-cance in forensic pathology”. Int J Legal Med 2001;115(3):142-51.
15. Reffelmann T, RA Kloner. “The no-reflow phenomenon: A basic mechanism of myocardial ischemia and reperfusion”. Basic Res Cardiol 2006;101(5):359-72.
16. Nian M, Lee P, Khaper N, Liu P. “Inflammatory cytokines and post-myocardial infarction remodeling”. Circ Res 2004;94(12):1543-53.
17. Sun, Y. “Oxidative stress and cardiac repair/remodeling following infarction”. Am J Med Sci 2007;334(3):197-205.
18. Sigurdsson A, K Swedberg. “The role of neurohormonal activation in chronic heart failure and postmyocardial infarction”. Am Heart J 1996;132(1 Pt 2 Suppl):229-34.
19. Remes, J. “Neuroendocrine activation after myocardial infarction”. Br Heart J 1994;72(3 Suppl):S65-9.
20. Ferrario, CM. “Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research”. J Renin Angiotensin Aldosterone Syst 2006;7:3-14.
21. Rumberger, JA. “Ventricular dilatation and remodeling after myo-cardial infarction”. Mayo Clin Proc 1994;69:664-74.
22. Grossman W, Jones D, McLaurin LP. “Wall stress and patterns of hy-pertrophy in the human left ventricle”. J Clin Invest 1975;56:56-64.
23. Diccionario de la lengua española, Vigésima segunda edición, Real Academia Española (http://www.rae.es/rae.html)
24. Roberts CS, Maclean D, Maroko P, Kloner RA. “Early and late remodeling of the left ventricle after acute myocardial infarction”. Am J Cardiol 1984;54:407-10.
25. Hochman JS, BH Bulkley. “Expansion of acute myocardial infarc-tion: an experimental study”. Circulation 1982;65(7):1446-50.
26. Weisman HF, Bush DE, Mannisi JA, Weisfeldt ML, Healy B. “Cellular mechanisms of myocardial infarct expansion”. Circulation 1988;78(1):186-201.
27. Whittaker P, Boughner DR, Kloner RA. “Role of collagen in acute myocardial infarct expansion”. Circulation 1991;84(5):2123-34.
28. Hutchins GM, BH Bulkley. “Infarct expansion versus extension: Two different complications of acute myocardial infarction”. Am J Cardiol 1978;41:1127-32.
29. Schuster EH, BH Bulkley. “Expansion of transmural myocardial infarction: a pathophysiologic factor in cardiac rupture”. Circulation 1979;60(7):1532-8.
30. McKay RG, Pfeffer MA, Pasternak RC, Markis JE, Come PC, Nakao S, Alderman JD, Ferguson JJ, Safian RD, Grossman W. “Left ventricular remodeling after myocardial infarction: a corollary to infarct expansion”. Circulation 1986;74(4):693-702.

Gelpi - Donato Fisiopatología cardiovascular
148
31. Pfeffer MA, JM Pfeffer. “Ventricular enlargement and reduced survival after myocardial infarction”. Circulation 1987;75(5 Pt 2):IV93-7.
32. Gaudron P, Eilles C, Kugler I, Ertl G. “Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors”. Circulation 1993;87(3):755-63.
33. Capasso JM, Li P, Zhang X, Anversa P. “Heterogeneity of ventricular remodeling after acute myocardial infarction in rats”. Am J Physiol 1992;262(2 Pt 2):H486-95.
34. Anversa P, JM Capasso. “Cardiac hypertrophy and ventricular remodeling”. Lab Invest 1991;64(4):441-5.
35. Sadoshima J, Jahn L, Takahashi T, Kulik TJ, Izumo S. “Molecular characterization of the stretch-induced adaptation of cultured car-diac cells. An in vitro model of load-induced cardiac hypertrophy”. J Biol Chem 1992;267:10551-60.
36. Gupta MP, Gupta M, Zak R, Sukhatme VP. “Egr-1, a serum-indu-cible zinc finger protein, regulates transcription of the rat cardiac alpha-myosin heavy chain gene”. J Biol Chem 1991;266:12813-6.
37. Yamazaki T, Komuro I, Yazaki Y. “Molecular mechanism of cardiac cellular hypertrophy by mechanical stress”. J Mol Cell Cardiol 1995;27:133-40.
38. Schwartz K, Boheler KR, de la Bastie D, Lompre AM, Mercadier JJ. “Switches in cardiac muscle gene expression as a result of pressure and volume overload”. Am J Physiol 1992;262(3 Pt 2):R364-9.
39. Yamazaki T, Shiojima I, Komuro I, Nagai R, Yazaki Y. “Invol-vement of the renin-angiotensin system in the development of left ventricular hypertrophy and dysfunction”. J Hypertens Suppl 1994;12:S23-7.
40. Vahebi S, RJ Solaro. “Cardiac sarcomeric function, small G-protein signaling, and heart failure”. Panminerva Med 2005;47:133-42.
41. Nicol RL, Frey N, Olson EN. “From the sarcomere to the nucleus: role of genetics and signaling in structural heart disease”. Annu Rev Genomics Hum Genet 2000;1:179-223.
42. Goldspink, G. “Changes in muscle mass and phenotype and the expression of autocrine and systemic growth factors by muscle in response to stretch and overload”. J Anat 1999;194 (Pt 3):323-34.
43. Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarai-nen S, Sugden PH. “Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses”. J Cell Physiol 2007;212:311-22.
44. Schneider MD, McLellan WR, Black FM, Parker TG. “Growth factors, growth factor response elements, and the cardiac pheno-type”. Basic Res Cardiol 1992;87 Suppl 2:33-48.
45. Sabbah HN, VG Sharov. “Apoptosis in heart failure”. Prog Cardiovasc Dis 1998;40:549-62.
46. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA et al. “Cardiac autophagy is a maladaptive response to hemo-dynamic stress”. J Clin Invest 2007;117:1782-93.
47. Sorescu D, KK Griendling. “Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure”. Congest Heart Fail 2002;8:132-40.
48. Weber KT, Sun Y, Tyagi SC, Cleutjens JP. “Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms”. J Mol Cell Cardiol 1994;26:279-92.
49. Wilson EM, Moainie SL, Baskin JM, Lowry AS, Deschamps AM, Mukherjee R et al. “Region-and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling”. Circulation 2003;107:2857-63.
50. Spinale, FG. “Myocardial matrix remodeling and the matrix me-talloproteinases: influence on cardiac form and function”. Physiol Rev 2007;87:1285-342.
51. Anversa P, Loud AV, Levicky V, Guideri G. “Left ventricular failure induced by myocardial infarction. II. Tissue morphometry”. Am J Physiol 1985;248(6 Pt 2):H883-9.
52. Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA et al. “Myocardial infarct size and ventricular function in rats”. Circ Res 1979;44:503-12.
53. Gaudron P, Eilles C, Ertl G, Kochsiek K. “Compensatory and noncompensatory left ventricular dilatation after myocardial in-
farction: Time course and hemodynamic consequences at rest and during exercise”. Am. Heart J 1992;123:377-85.
54. Pfeffer MA, E Braunwald. “Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications”. Circulation 1990,81:1161-72.
55. Pfeffer JM, Pfeffer MA, Fletcher PJ, Braunwald E. “Progressive ventricular remodeling in rat with myocardial infarction”. Am J Physiol 1991;260:H1406-14.
56. Fletcher PJ, Pfeffer JM, Pfeffer MA, Braunwald E. “Left ventricular diastolic pressure-volume relations in rats with healed myocardial infarction. Effects on systolic function”. Circ Res 1981;49(3):618-26.
57. Bolognese L, G Cerisano. “Early predictors of left ventricular remo-deling after acute myocardial infarction”. Am Heart J 1999;138(2 Pt 2):S79-83.
58. Lamas GA, Pfeffer MA, Braunwald E. “Patency of the infarct-related coronary artery and ventricular geometry”. Am J Cardiol 1991;68:41D-51D.
59. Bonaduce D, Petretta M, Villari B, Breglio R, Conforti G, Mon-temurro MV et al. “Effects of late administration of tissue-type plasminogen activator on left ventricular remodeling and function after myocardial infarction”. J Am Coll Cardiol 1990;16:1561-8.
60. Topol EJ, Califf RM, Vandormael M, Grines CL, George BS, Sanz ML et al. “A randomized trial of late reperfusion therapy for acute myocardial infarction. Thrombolysis and Angioplasty in Myocardial Infarction-6 Study Group”. Circulation 1992;85:2090-9.
61. Zarrabi A, Eftekhari H, Casscells SW, Madjid M. “The open-artery hypothesis revisited”. Tex Heart Inst J 2006;33(3):345-52.
62. St John Sutton M, Pfeffer MA, Moye L, Plappert T, Rouleau JL, Lamas G et al. “Cardiovascular Death and Left Ventricular Remo-deling Two Years After Myocardial Infarction: Baseline Predictors and Impact of Long-term Use of Captopril: Information From the Survival and Ventricular Enlargement (SAVE) Trial”. Circulation 1997;96:3294-9.
63. Ho KK, Pinsky JL, Kannel WB, Levy D. “The epidemiology of heart failure: the Framingham Study”. J Am Coll Cardiol 1993;22:6A–13A.
64. Cavasin MA, Tao Z, Menon S, Yang XP. “Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice”. Life Sci. 2004;75:2181-92.
65. Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. “Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population-based cohort”. J Am Coll Cardiol 1999;33:1948-55.
66. Rathore SS, Wang Y, Krumholz HM. “Sex-based differences in the effect of digoxin for the treatment of heart failure”. N Engl J Med 2002;347:1403-11.
67. Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK et al. “Impact of diabetes on cardiac structure and function: the strong heart study”. Circulation. 2000;101:2271-6.
68. Melchior T, Kober L, Madsen CR, Seibaek M, Jensen GV, Hil-debrandt P et al. “Accelerating impact of diabetes mellitus on mortality in the years following an acute myocardial infarction: TRACE Study Group Trandolapril Cardiac Evaluation”. Eur Heart J 1999;20:973-8.
69. Solomon SD, St John Sutton M, Lamas GA, Plappert T, Rouleau JL, Skali H et al. “Survival and Ventricular Enlargement (SAVE) Investigators. Ventricular Remodeling Does not Accompany the Development of Heart Failure in Diabetic Patients after Myocardial Infarction”. Circulation 2002;106:1251-5.
70. Iwasaka T, Takahashi N, Nakamura S, Sugiura T, Tarumi N, Kimura Y et al. “Residual left ventricular pump function after acute myocardial infarction in NIDDM patients”. Diabetes Care 1992;15:1522-6.
71. Dini FL, Volterrani C, Azzarelli A, Lanciani A, Lunardi M, Ber-nardi D et al. “Left ventricular size and function in patients with noninsulin-dependent diabetes and postinfarction total or subtotal coronary occlusions”. Angiology 1998;49:967-73.
72. Kawaguchi M, Techigawara M, Ishihata T, Asakura T, Saito F, Maehara K et al. “A comparison of ultrastructural changes on

Remodelamiento posInfarto de miocardio
149
endomyocardial biopsy specimens obtained from patients with diabetes mellitus with and without hypertension”. Heart Vessels 1997;12:267-74.
73. Di Bello V, Talarico L, Picano E, Di Muro C, Landini L, Paterni M et al. “Increased echodensity of myocardial wall in the diabe-tic heart: an ultrasound tissue characterization study”. J Am Coll Cardiol 1995;25:1408-15.
74. Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N et al. “Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model”. Circulation 2000;101:899-907.
75. Zarich SW, Arbuckle BE, Cohen LR, Roberts M, Nesto RW et al. “Diastolic abnormalities in young asymptomatic diabetic patients assessed by pulsed Doppler echocardiography”. J Am Coll Cardiol 1988;12:114-20.
76. Schaffer SW, M Mozaffari. “Abnormal mechanical function in diabetes: relation to myocardial calcium handling”. Coron Artery Dis 1996;7:109-15.
77. Aaronson KD, Schwartz JS, Chen TM, Wong KL, Goin JE, Man-cini DM. “Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation”. Circulation 1997;95:2660-7.
78. Gradman AH, F Alfayoumi. “From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease”. Prog Cardiovasc Dis 2006;48:326-41.
79. Jilaihawi H, Greaves S, Rouleau JL, Pfeffer MA, Solomon SD; Healing and Early Afterload Reducing Therapy Trial Investiga-tors. “Left ventricular hypertrophy and the risk of subsequent left ventricular remodeling following myocardial infarction”. Am J Cardiol 2003;91:723-26.
80. Díez J, Panizo A, Hernández M, Pardo J. “Is the regulation of apoptosis altered in smooth muscle cells of adult spontaneously hypertensive rats?” Hypertension 1997;29:776-80.
81. Muscholl MW, Schunkert H, Muders F, Elsner D, Kuch B, Hense HW et al. “Neurohormonal activity and left ventricular geome-try in patients with essential arterial hypertension”. Am Heart J 1998;135:58-66.
82. McDonagh TA, Morrison CE, Lawrence A, Ford I, Tunstall-Pedoe H, McMurray JJ et al. “Symptomatic and asymptomatic left-ventricular systolic dysfunction in an urban population”. Lancet 1997;350:829-33.
83. Palmeri ST, Harrison DG, Cobb FR, Morris KG, Harrell FE, Ideker RE et al. “A QRS scoring system for assessing left ventricular function after myocardial infarction”. N Engl J Med 1982;306:4-9.
84. Silver MT, Rose GA, Paul SD, O’Donnell CJ, O’Gara PT, Eagle KA. “A clinical rule to predict preserved left ventricular ejection fraction in patients after myocardial infarction”. Ann Intern Med 1994;121:750-6.
85. Bosimini E, Giannuzzi P, Temporelli PL, Gentile F, Lucci D, Mag-gioni AP et al. “Electrocardiographic evolutionary changes and left ventricular remodeling after acute myocardial infarction: results of the GISSI-3 Echo substudy”. J Am Coll Cardiol 2000;35:127-35.
86. Cohn JN, Johnson GR, Shabetai R, Loeb H, Tristani F, Rector T et al. “Ejection fraction, peak exercise oxygen consumption, cardiothoracic ratio, ventricular arrhythmias, and plasma norepinephrine as determinants of prognosis in heart failure. The V-HeFT VA Cooperative Studies Group”. Circulation 1993;87(Suppl VI):V15-6.
87. Philbin EF, Garg R, Danisa K, Denny M, Gosselin G, Hassapo-yannes C et al. “The relationship between cardiothoracic ratio and left ventricular ejection fraction in congestive heart failure. Digitalis Investigation Group”. Arch Intern Med 1998;158:501-6.
88. Gottdiener, JS. “Left ventricular mass, diastolic dysfunction and hypertension”. Ann Intern Med 1993;38:31-56.
89. Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ Jr, Cuddy TE et al. “Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the Survival and Ventricular Enlargement Trial”. N Engl J Med 1992;327:669-77.
90. Casaclang-Verzosa G, Gersh BJ, Tsang TS. “Structural and functional remodeling of the left atrium: clinical and therapeutic implications for atrial fibrillation”. J Am Coll Cardiol 2008;51:1-11.
91. Casas JP, Chua W, Loukogeorgakis S, Vallance P, Smeeth L, Hin-gorani AD et al. “Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis”. Lancet 2005;366:2026-33.
92. Ostergren, J. “Renin-angiotensin-system blockade in the prevention of diabetes”. Diabetes Res Clin Pract 2007;76 Suppl 1:S13-21.
93. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. “Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure”. Lancet 1993;342:821-8.
94. Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyng-borg K et al. “A clinical trial of the angiotensin-converting enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction”. N Engl J Med 1995;333:1670-6.
95. The CONSENSUS Trial Study Group. “Effects of enalapril on mor-tality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS)”. N Engl J Med 1987;316:1429-35.
96. Konstam MA, Kronenberg MW, Rousseau MF, Udelson JE, Melin J, Stewart D et al. “Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dilatation in patients with asymptomatic systolic dysfunction”. Circulation 1993;88:2277-83.
97. Konstam MA, Rousseau MF, Kronenberg MW, Udelson JE, Melin J, Stewart D et al. “Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure”. Circulation 1992;86:431-8.
98. Rodríguez JA, Godoy I, Castro P, Quintana JC, Chávez E, Yovano-vich J et al. “Ramipril vs. espironolactona en el remodelamiento ventricular izquierdo post-infarto: randomizado y dobleciego”. Rev Med Chile 1997;125:643-52.
99. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A et al. “The effect of spironolactone on morbidity and mortality in pa-tients with severe heart failure. Randomized Aldactone Evaluation Study Investigators”. N Engl J Med 1999;341:709-17.
100. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B et al. “Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction”. N Engl J Med 2003;348:1309-21.
101. Chan AK, Sanderson JE, Wang T, Lam W, Yip G, Wang M et al. “Aldosterone receptor antagonism induces reverse remodeling when added to angiotensin receptor blockade in chronic heart failure”. J Am Coll Cardiol 2007;50:591-6.
102. Beta-Blocker Heart Attack Trial Research Group. “A Randomized trial of propranolol in patients with acute myocardial infarction”. J Am Med Assoc 1982;247:1707-14.
103. The Norwegian Multicenter Study Group. “Timolol-induced re-duction in mortality and reinfarction in patients surviving acute myocardial infarction”. N Engl J Med 1981;304:801-7.
104. CAPRICORN Investigators. “Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomized trial”. Lancet 2001;357:1385-90.
105. Doughty RN, Whalley GA, Walsh HA, Gamble GD, López-Sendon J, Sharpe N, on behalf of the CAPRICORN Echo Substudy investi-gators. “Effects of carvedilol on left ventricular remodeling following acute myocardial infarction: the CAPRICORN Echo Substudy”. Circulation 2004;109:201-6.
106. Packer M, Bristow MR, Cohn JN, for the US Carvedilol Heart Failure Study Group. “The effect of carvedilol on morbidity and mortality in patients with chronic heart failure”. N Engl J Med 1996;334:1349-55.
107. MERIT-HF Study Group. “Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomized intervention trial in congestive heart failure”. Lancet 1999;353:2001-7.

Gelpi - Donato Fisiopatología cardiovascular
150
108. CIBIS II Investigators and Committees. “The Cardiac Insuffi-ciency Bisoprolol Study II (CIBIS II): a randomized trial”. Lancet 1999;353:9-13.
109. GISSI-3. “Effects of lisinopril and transdermal glyceryl trinitrate singly and together on 6 week mortality and ventricular function af-ter acute myocardial infarction: Gruppo Italiano per lo Studio della Sopravvivenza nell’infarto Miocardico”. Lancet 1994;343:1115-22.
110. ISIS-4 (Fourth International Study of Infarct Survival) Collabora-tive Group. “ISIS-4: a randomized factorial trial assessing early oral captopril, oral mononitrate and intravenous magnesium sulphate in 58 050 patients with suspected acute myocardial infarction”. Lancet 1995;345:669-82.
111. Cohn JN, Archibald DG, Ziesche S, Franciosa JA, Harston WE, Tristani FE et al. “Effect of vasodilator therapy on mortality in chronic congestive heart failure: results of a Veterans Administration Cooperative Study”. N Engl J Med 1986;314:1547-52.
112. Choi EY, Chang W, Lim S, et al. “Rosuvastatin inhibits norepinephrine-induced cardiac hypertrophy via suppression of Gh . Eur J Pharmacol. 2010;627(1-3):56-62.
113. Sola S, Mir MQ, Lerakis S, Tandon N, Khan BV. “Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure”. J Am Coll Cardiol 2006;47:332-7.
114. Krum H, A Tonkin. “The Rosuvastatin Impact on Ventricular Remodeling, Cytokines and Neurohormones (UNIVERSE) Study (abstr)”. J Am Coll Cardiol 2006;47:61A-62A.
115. Kjekshus J, Apetrei E, Barrios V, Böhm M, Cleland JG, Cornel JH et al. “CORONA Group. Rosuvastatin in older patients with systolic heart failure”. N Engl J Med 2007;357:2248-61.
116. White HD, Cross DB, Elliott JM, Norris RM, Yee TW. “Long-term prognostic importance of patency of the infarct-related co-ronary artery after thrombolytic therapy for acute myocardial infarction”. Circulation 1994;89:61-7.
117. Jeremy RW, Hackworthy RA, Bautovich G, Hutton BF, Harris PJ. “Infarct artery perfusion and changes in left ventricular volume in the month after acute myocardial infarction”. J Am Coll Cardiol 1987;9:989-95.
118. Marino P, Zanolla L, Zardini P (GISSI). “Effect of streptokinase on left ventricular modeling and function after myocardial infarction: the GISSI trial”. J Am Coll Cardiol 1989;14:1149-58.
119. Hochman JS, Lamas GA, Buller CE, Dzavik V, Reynolds HR, Abramsky SJ et al. “Coronary intervention for persistent occlusion after myocardial infarction”. N Engl J Med 2006;355:2395-407.
120. Jain M, DerSimonian H, Brenner DA, Ngoy S, Teller P, Edge AS et al. “Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction”. Circulation 2001;103:1920-7.
121. Reffelmann T, Dow JS, Dai W, Hale SL, Simkhovich BZ, Kloner RA. “Transplantation of neonatal cardiomyocytes after permanent coronary artery occlusion increases regional blood flow of infarcted myocardium”. J Mol Cell Cardiol 2003;35:607-13.
122. Cooley DA, GL Hallman. “Surgical treatment of left ventricular aneurysm: experience with excision of postinfarction lesions in 80 patients”. Prog Cardiovasc Dis 1968;11:222-8.
123. Dor, V. “The endoventricular circular patch plasty (‘Dor Pro-cedure’) in ischemic akinetic dilated ventricles”. Heart Fail Rev 2001;6:187-93.
124. Batista RJ, Verde J, Nery P, Bocchino L, Takeshita N, Bhayana JN et al. “Partial left ventriculectomy to treat end-stage heart disease”. Ann Thorac Surg 1997;64:634-8.
125. Athanasuleas CL, Buckberg GD, Stanley AW, Siler W, Dor V, Di Donato M et al. “RESTORE group. Surgical ventricular restoration in the treatment of congestive heart failure due to post-infarction ventricular dilation”. J Am Coll Cardiol 2004;44:1439-45.
126. Kass DA, Chen CH, Curry C, Talbot M, Berger R, Fetics B et al. “Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay”. Circulation 1999;99:1567-73.
127. Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L et al. “Cardiac Resynchronization-Heart Failure (CARE-HF) Study Investigators. The effect of cardiac resynchro-nization on morbidity and mortality in heart failure”. N Engl J Med 2005;352:1539-49.
128. Dubach P, Myers J, Dziekan G, Goebbels U, Reinhart W, Vogt P et al. “Effects of exercise training on myocardial remodeling in patients with reduced left ventricular function after myocardial infarction”. Circulation 1997;95:2060-7.
129. Jugdutt BI, Michorowski BL, Tissa Kappagoda C. “Exercise training after anterior Q wave myocardial infarction: importance of regional left ventricular function and topography”. J Am Coll Cardiol 1988,12:362-372.

151
proporciona las herramientas que permiten interpretar -
damentos en que se basan las elecciones terapéuticas
consecuencias clínicas.En este sentido, el estudio del remodelamiento ven-
-
1-3 Un
remodelamiento ventricular posinfarto de miocardio, es -
Entre dichos sistemas hormonales, el sistema renina an--
el remodelamiento de zonas alejadas a él. En el presente -
farto de miocardio, el remodelamiento ventricular y su
--
mentales y clínicos en los que el bloqueo del sistema renina
miocardio y el remodelamiento ventricular.
Remodelamiento ventricular
Concepto, generalidades, importancia
proceso de remodelamiento ventricular puede desencade-
deportistas de alto rendimiento, en la mujer embarazada,
se hace referencia a él cuando subyace a una serie de pa-
de un proceso a través del que se desencadenan una serie
Ver Tabla 9.1 1
-
1 De esta manera, considerando al -
-
re-modelamiento
-
4
Linzbach et al. 5-
cardiaca, considerando un peso cardiaco crítico de 500 -
Más tarde, Kostuk et al.6 et al. 7 mostraron que
respecto, Pfeffer et al. 8, Ertl et al. 9 y Gaudron et al. 10 -rrelaciona en forma directa con la mortalidad.
Aspectos generales del remodelamiento fisiológico y patogenia del remodelamiento patológico
Sistema renina angiotensina y remodelamiento cardiaco 9
Germán E. González, Ricardo J. Gelpi, Celina Morales

Gelpi - Donato Fisiopatología cardiovascular
152
de las enfermedades cardiovasculares primarias, como -
patías, la miocarditis, y las malformaciones cardiacas.
en marcha el remodelamiento cardiaco, en la mayoría de
-
cada uno de los componentes de la pared miocárdica,
pared vascular. 11 Por ello, considerar al remodelamiento
factores funcionales, humorales, bioquímicos, hormona-
proceso de remodelamiento cardiaco habría una secuencia
y funcionales en común, que pondrían en marcha el cambio
secuencia terminal de eventos, en conjunto con los cam-
-
remodelamiento compensatorio -
Tabla 9.1: ventricular
Patologías adquiridas
1. Infarto de miocardio2. Miocardiopatía hipertensiva, valvular o
congénita3. Miocarditis, Enfermedad de Chagas-Maza
Patologías genéticas
A) Cardiopatías hereditarias1. Miocardiopatía hipertrófica familiar2. Miocardiopatía dilatada3. Enfermedad de Marfán4. HemocromatosisB) Modelos experimentales transgénicos1. Cardiopatía hipertrófica2. Cardiopatía dilatada
Misceláneas 1. Remodelamiento de la senescencia2. Remodelamiento por frecuencia cardiaca3. Remodelamiento por activación neuro-
hormonal4. Diabetes5. Déficit de Vitamina B6
6. Atrofia postransplante heterotópico
(Modificado de Swynghedauw, B. Physiol Rev 1999; 79(1):215-62)
se perpetúan en el tiempo o el remodelamiento no alcanza -
4
de remodelamiento.
Sistema renina angiotensina en el remodelamiento ventricular
-
-12
involucrado en la homeostasis cardiovascular, tanto en
-lamiento ventricular. 13-15
La
13, 16-20
-
aspartil proteasa que se sintetiza y localiza principalmente -
ser producida en otros tejidos. 21
fl
fi
Figura 9.1: Aspectos etiopatogénicos y evolutivos vinculados al remodelamiento ventricular. Un conjunto de cambios bioquímicos, morfológicos y funcionales da origen al remodelamiento ventricular. Este se considera favorable cuando de un estado de descompensación inicial se logra una compensación intermedia, y desfavorable cuando se llega a insuficiencia cardiaca terminal.

Remodelamiento cardíaco y sistema renina angiotensina
153
-dasa que se encuentra principalmente unida a la membrana de células endoteliales de la mayoría de los lechos vas-
-
1-4
22
-
1 -
-23
-mente caracterizados. 24, 25
El AT1 pertenece a la familia de receptores acoplados a proteína G y es el efector de la mayoría de las acciones
que el receptor ATfetal, también pertenece a la familia de los receptores
-tor AT1. 12, 15, 17, 26
y AT27
28
-ma hormonal forma parte del tejido cardiaco y, aunque su
21, 29, 30
Akasu et al. 28
7 especies de mamíferos incluyendo humanos. Los resulta-
aorta de humanos, a diferencia del conejo y el cerdo, la ma-
enzima quimasa, que también se encontraría en la mayoría de los mastocitos y células endoteliales.
Otros estudios también mostraron que en la aurícula de
-
ventrículo es similar en las dos especies. 21
-
Figura 9.2: Esquema representativo del sistema renina angiotensina clásico. El angiotensinógeno, formado en el hígado, es hidrolizado por la renina para formar angiotensina I. A partir de la angiotensina I se forma angiotensina II, una acción catalizada por la enzima convertidora de angiotensina. Esta enzima también cataliza la formación, a partir de la angiotensina II de otros componentes menos activos como la angiotensina III y IV.
ECA: Enzima convertidora de angotensina; EPs: endopeptidasas. (Modificado de Barlucchi, L et al. Circ Res. 2001;88:298-304.)

Gelpi - Donato Fisiopatología cardiovascular
154
-
-
II, también podría ser variable en las diferentes especies. 31
1
17, 18 Por otro lado, tiene efectos sobre el crecimiento car-diaco, activando la síntesis de proteínas tanto en miocitos
32 -sina II también han sido descritos en los cardiomiocitos
-cardio humano. 13
et al. 33 estudiaron el efecto del bloqueo del re-ceptor AT1 y ATpapilar de conejos. Estos autores mostraron que el efecto
por el receptor AT1 . Ishihata et al. 17, 18
-ferentes especies de mamíferos, mostrando que dicho efecto
densidad de receptores presentes en el sarcolema. En cone-
17
-
34, 35
cardiaco fue demostrado tanto in vivo como in vitro -doshima et al. 36
la síntesis de proteínas en miocitos, incluso en ausencia 37 En
36 Estudios recientes mostraron que la respuesta celular producida por la acti-
1 también podría producirse por la
proteína G. 38, 39
et al. 26 mostra--
tor AT
del receptor AT1.-
-
40
-
-
la ECA, su actividad no es inhibida por los inhibidores de esta. 22, 41
--
-to posinfarto.
Rol del sistema renina angiotensina en el infarto de miocardioEl infarto de miocardio pone en marcha una respuesta
--
químicos y neurohormonales, que dan como resultado la
et al. 42
así como también se incrementa la densidad de los recep-tores AT1
-
nuestro laboratorio mostraron que en corazones de co-
receptores AT1, tanto en la zona de infarto como en zonas remotas, es similar al control durante los
43
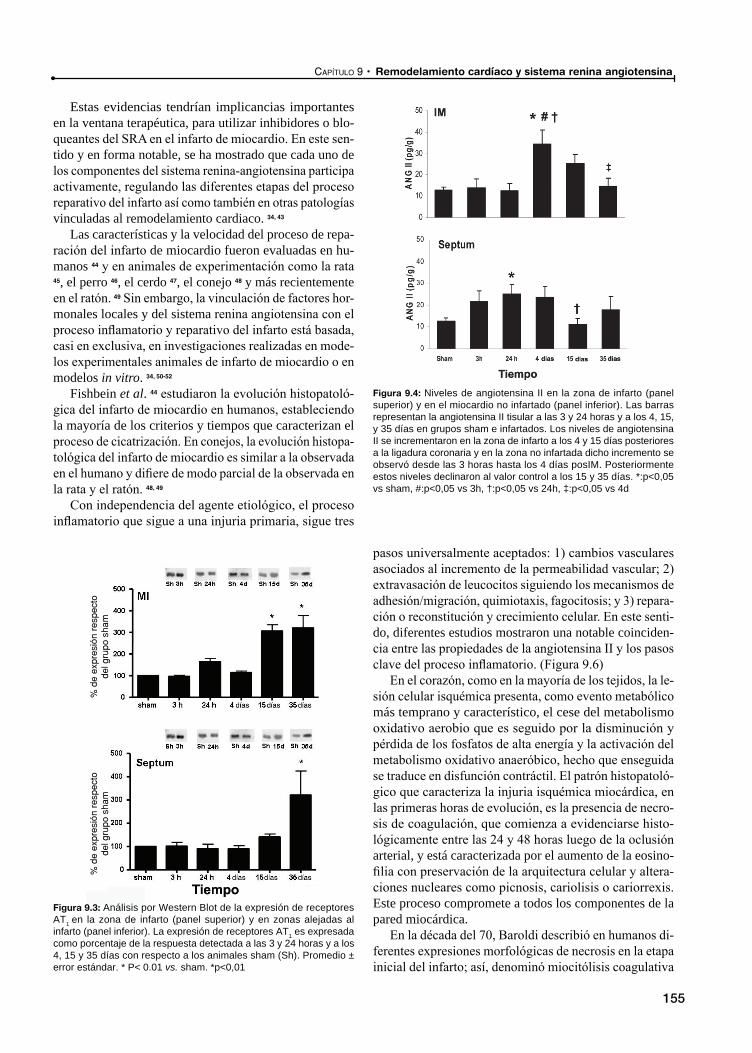
Remodelamiento cardíaco y sistema renina angiotensina
155
Estas evidencias tendrían implicancias importantes en la ventana terapéutica, para utilizar inhibidores o blo-
-tido y en forma notable, se ha mostrado que cada uno de
vinculadas al remodelamiento cardiaco. 34, 43
Las características y la velocidad del proceso de repa-de miocardio fueron evaluadas en hu-
manos 44
45, el perro 46, el cerdo 47, el conejo 48 y más recientemente 49 -
monales locales y del sistema
-
modelos in vitro. 34, 50-52
Fishbein et al. 44 -
la mayoría de los criterios y tiempos que caracterizan el -
48, 49
--
do, diferentes estudios mostraron una notable coinciden-
-
más temprano y característico, el cese del metabolismo
-
--
arterial, y está caracterizada por el aumento de la eosino--
Este proceso compromete a todos los componentes de la pared miocárdica.
-
% d
e ex
pres
ión
resp
ecto
del g
rupo
sha
m%
de
expr
esió
n re
spec
tode
l gru
po s
ham
Figura 9.4: Niveles de angiotensina II en la zona de infarto (panel superior) y en el miocardio no infartado (panel inferior). Las barras representan la angiotensina II tisular a las 3 y 24 horas y a los 4, 15, y 35 días en grupos sham e infartados. Los niveles de angiotensina II se incrementaron en la zona de infarto a los 4 y 15 días posteriores a la ligadura coronaria y en la zona no infartada dicho incremento se observó desde las 3 horas hasta los 4 días posIM. Posteriormente estos niveles declinaron al valor control a los 15 y 35 días. *:p<0,05 vs sham, #:p<0,05 vs 3h, †:p<0,05 vs 24h, ‡:p<0,05 vs 4d
Figura 9.3: Análisis por Western Blot de la expresión de receptores AT1 en la zona de infarto (panel superior) y en zonas alejadas al infarto (panel inferior). La expresión de receptores AT1 es expresada como porcentaje de la respuesta detectada a las 3 y 24 horas y a los 4, 15 y 35 días con respecto a los animales sham (Sh). Promedio ± error estándar. * P< 0.01 vs. sham. *p<0,01

Gelpi - Donato Fisiopatología cardiovascular
156
-
autor, en forma de focos subendocárdicos y perivasculares. -
48
-
-
48
En la mayoría de los mamíferos, el evento celular que
-
-cos, como los componentes activos del complemento, las
51 Estos fac-
de estos leucocitos a las células endoteliales. 51 Además,
52, 55, 56
en zonas alejadas al infarto, contribuyen al proceso de re-
Asimismo, se ha mostrado que la enzima convertidora -
participa activamente, en forma directa o indirecta, en el
-57, 58
Asimismo se ha mostrado que la
paracrina, contribuyendo al proceso reparativo de la zona de infarto. En este sentido, también estimularía el estrés
-parativa. Lu et al. 59 encontraron en la zona de infarto un
-
de la zona de infarto.
metaloproteasas 8 y 9, en particular en la zona de infarto, 56 Estas enzimas
Figura 9.5: Muestra la distribución de áreas positivas para angiotensina II en la zona de infarto y zonas remotas, en función del tiempo. La inmunomarcación para angiotensina II se incrementó en el septum a las 3 hs., 24 hs. y 4 días postMI, mientras que en la zona de infarto dichas áreas positivas fueron más evidentes a los 4 y 15 días posligadura.
Figura 9.6: Participación de la angiotensina II en cada una de las etapas claves de la respuesta inflamatoria. PGs: Prostaglandinas; VEGF: Factor de crecimiento del endotelio vascular; MEC: Matriz extracelular. (Modificado de Suzuki et al. Int J Biochem Cell Biol. 2003;35:881-900).
PGs, VEGFReagrupamiento de
proteínas del citoesqueleto
Ang IIActivación de
células inmunes
↑ medidoresproinflamatorios
↑ Permeabilidadvascular
↑ Infiltrado de celulasinflamatorias
Reparación tisulary remodelamiento
Resolución Fibrosis
↑ crecimiento celular
↑ MEC, angiogénesis

Remodelamiento cardíaco y sistema renina angiotensina
157
-ponentes del tejido conectivo en la zona de infarto, lo que
-56 Diferentes evidencias
42, 58, 60, 61 Esta -
20, 62 y de metaloproteasas de matriz.De forma concomitante con el incremento en la síntesis
56, 63
--
tracelular. Las MMPs median una amplia variedad de pro--
Ha sido mostrado que todas las células encontradas en
-
tipos de MMPs y sus inhibidores.
--
64
terapéuticos como los inhibidores de la enzima converti-
AT1 -lance MMP/TIMP. En este sentido, varios autores mos-
la pared miocárdica. 65, 66
polimorfonucleares
42 -
-citoquinas y factores de crecimiento necesarios
--
que las proteínas de matriz a depositarse desde la zona borde del infarto, reemplazan
-ma y reblandecimiento que presenta el tejido durante esta etapa, la zona de infarto es muy vulnerable al aumento del
-portancia durante el proceso de remodelamiento.
et al. 60 mostraron en ratas un aumento en la -
del infarto. Al respecto, Cleutjens et al. 69, utilizando in situ y
de inmunohistoquímica mostraron un incremento en la tipo I y III, en célu-
de necrosis desde los 7 días del infarto. Deten et al. 56 -
el aumento de las citoquinas e interleuquinas descritas en el apartado anterior.
-
--
bido a su capacidad de brindar resistencia a la pared in-
--
las metaloproteasas. 55, 56
reemplazará a la zona de necrosis.
Aspectos fisiopatológicos y morfológicos del remodelamiento ventricular posinfartoCleutjens et al. 70 consideran al remodelamiento posinfar-to de miocardio como un proceso activo y bien controlado
al infarto de miocardio como el factor desencadenante
adoptará el ventrículo y que le permitirá, en un principio, adaptarse a los cambios hemodinámicos producidos por la
71, 72
involucrada en el proceso reparativo. 4
-

Gelpi - Donato Fisiopatología cardiovascular
158
proceso reparativo de la zona de infarto, los cambios he--
monal, tanto local como sistémica, produciendo como con-
-vinientes en cada etapa del remodelamiento redundaría en
-car que, a pesar de que el remodelamiento ventricular y la
lo constituye el incremento de los volúmenes ventricu-lares que, poniendo en marcha el mecanismo de Frank-
4, 10, aumenta la fuerza de los miocitos viables para
-
zona de infarto y favorecer la contractilidad dado el efecto
et al. 73
-
superpuestas. Una primera fase, denominada temprana, que se produciría dentro de los tres primeros días de evo-
en particular limitados a la zona de infarto y la cavidad -
día, en donde adquieren relevancia el inicio, el desarrollo
-
Remodelamiento temprano y su relación con la activación del sistema renina angiotensina
como consecuencia de la pérdida de miocitos, se traduce -
que ocurre de inmediato después de producido el infarto, -
Estrés parietal
Figura 9.7: Representación esquemática de los aspectos fisiopatológicos que participan en el remodelamiento ventricular posinfarto de miocardio. La disminución de la fracción de eyección producida por el infarto de miocardio aumenta el volumen de fin de sístole (VFS) produciendo dilatación. Esta pone en marcha, en forma temprana, factores fisiológicos (ley de Frank-Starling), para intentar normalizar el volumen sistólico, junto con la activación de factores neurohormonales locales y sistémicos. Por otro lado, esta dilatación incrementa el estrés parietal que, actuando sobre la zona de infarto, contribuye al proceso de expansión y sobre la zona no infartada, produce hipertrofia. Si esta logra compensar en forma completa el aumento del estrés, se puede considerar que el remodelamiento fue favorable, mientras que si la reducción es incompleta, el remodelamiento es desfavorable, conduciendo a mayor dilatación y, progresivamente a insuficiencia cardiaca.

Remodelamiento cardíaco y sistema renina angiotensina
159
responder a los cambios hemodinámicos intraventricula-res, producidos por la pérdida de masa contráctil. Además,
resultan directamente de los trastornos hemodinámicos,
Gaudron et al. 10, 74 estudiaron en pacientes con infarto -
-
pondría en marcha el primer mecanismo compensatorio,
recurriendo en el músculo no infartado al mecanismo de ---
73 -
ley de Laplace, este aumento -
tal en toda la circunferencia de la pared ventricular, cons-
de infarto como en el músculo no infartado. Por lo tanto, así como la muerte de las células miocárdicas constituye
el remodelamiento posinfarto, dado que marca un punto de
en el remodelado de la zona de infarto y de la pared no isquémica. 75
-
Pfeffer et al. 71, utilizando parámetros funcionales,
-
como también el incremento del estrés parietal sobre dicha
--
siderada como una de las primeras manifestaciones mor-
-torias, la zona de necrosis es particularmente vulnerable al incremento del estrés parietal, desarrollándose un proceso conocido como Ver capítulo 8
La por Hutchins y Bulkey, en un estudio de autopsia de pa-
infarto, y constituye una de las primeras manifestaciones
La vulnerabilidad de la zona de infarto en esta etapa,
de los
y a la pérdida del andamiaje que brindan
55, 56
-portante la metaloproteasas de matriz. Es por eso que, el
-
Figura 9.8: Representación esquemática de las diferentes etapas que caracterizan al remodelamiento ventricular en el infarto de miocardio. Los cambios morfológicos y geométricos representativos de cada etapa se asocian a trastornos hemodinámicos y disfunción ventricular (modificada de Pfeffer JM et al., Annu Rev Physiol 1995; 57: 805-826)
VI normal temprano tardío
Expansión delinfarto
Trastornomecánico
Recuperaciónmecánico
Trastornomecánico
Paredlibre
Septum
PDF VIPDF VI PDF VI
Adelgazamiento+ dilatación VI
Aumento dedilatación VI
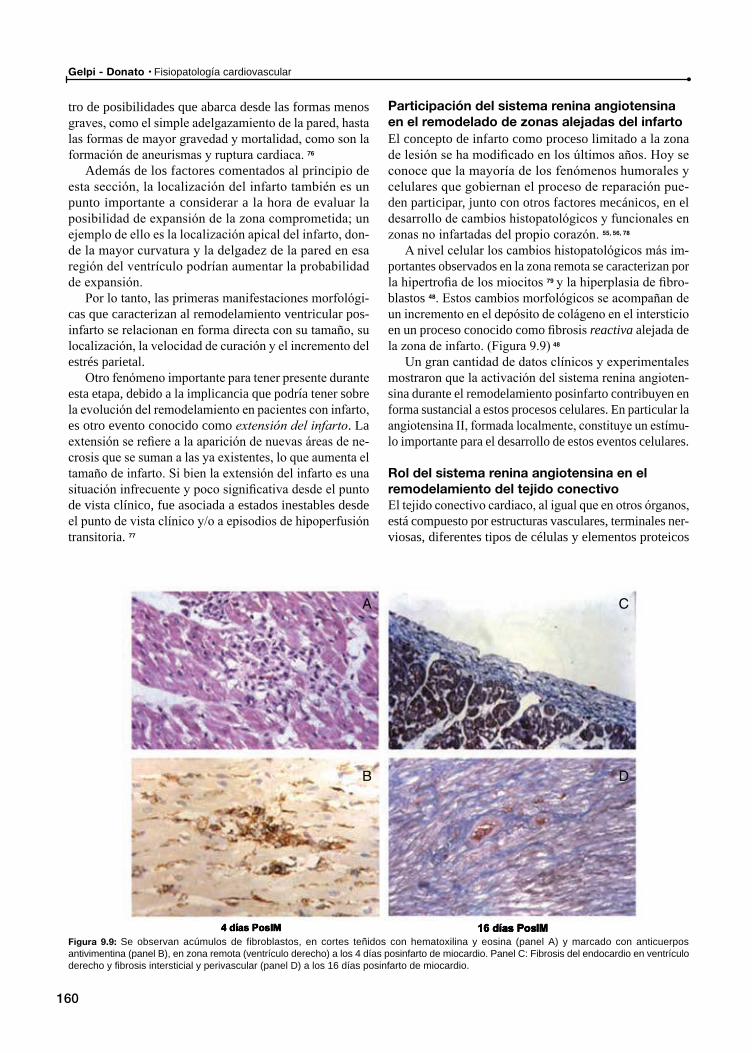
Gelpi - Donato Fisiopatología cardiovascular
160
tro de posibilidades que abarca desde las formas menos
76
Además de los factores comentados al principio de
punto importante a considerar a la hora de evaluar la
-
-cas que caracterizan al remodelamiento ventricular pos-
estrés parietal.
esta etapa, debido a la implicancia que podría tener sobre
es otro evento conocido como . La -
de vista clínico, fue asociada a estados inestables desde
transitoria. 77
Participación del sistema renina angiotensina en el remodelado de zonas alejadas del infartoEl concepto de infarto como proceso limitado a la zona
-den participar, junto con otros factores mecánicos, en el
55, 56, 78
-portantes observados en la zona remota se caracterizan por
79 -blastos 48
en un proceso conocido como reactiva alejada de 48
-sina durante el remodelamiento posinfarto contribuyen en forma sustancial a estos procesos celulares. En particular la
-lo importante para el desarrollo de estos eventos celulares.
Rol del sistema renina angiotensina en el remodelamiento del tejido conectivo
está compuesto por estructuras vasculares, terminales ner-viosas, diferentes tipos de células y elementos proteicos
C
D
A
B
Figura 9.9: Se observan acúmulos de fibroblastos, en cortes teñidos con hematoxilina y eosina (panel A) y marcado con anticuerpos antivimentina (panel B), en zona remota (ventrículo derecho) a los 4 días posinfarto de miocardio. Panel C: Fibrosis del endocardio en ventrículo derecho y fibrosis intersticial y perivascular (panel D) a los 16 días posinfarto de miocardio.

Remodelamiento cardíaco y sistema renina angiotensina
161
y no proteicos que, en condiciones normales, cumplen funciones especializadas. Cada uno de sus componentes rodea las células contráctiles y provee el medio adecuado
cardiaca. 80
-
-
-
78
Los componentes proteicos que forman parte de la
encuentran en menores cantidades formando parte de las -
et al. 81 -
-
Ante la pérdida de la homeostasis tisular, como ocurre -
tracelular del tejido conectivo de la zona isquémica se ve
-
51
-lar de la zona de infarto cumple un rol reparativo. En este
-rativa, cicatrizal o de reemplazo. 80, 82
puramente mecánica.Durante el proceso de remodelamiento ventricular, la
-se afectada por factores mecánicos y hormonales. Estos
dichas zonas, la mayoría de los componentes celulares de la matriz reaccionan en forma independiente al proceso reparativo de la zona de infarto. 62, 83
et al. 84 et al. 85 mostraron en
-nas remotas incrementan la síntesis de ADN, a la vez que
-nas no infartadas.
Cleutjens et al. 69 mostraron, en zonas remotas al in--
-
-48
después por otros autores en la misma especie 86, 87 y en otras especies. 35, 48
80 llamaron a estos cambios histopato--
adventicia de las arterias coronarias intramiocárdicas y del intersticio cardiaco, que aparece en ausencia de ne-
rodeando a los miocitos.
zona de infarto previene la ruptura cardiaca al oponerse al 88
de la pared ventricular que, indefectiblemente, conducirán 66, 89
-
66, 90
Los -yor importancia en el proceso reparativo y reactivo de la
-
82
78, 83 mostraron que la
por factores mecánicos, como el estiramiento, y en res-puesta a factores neurohormonales locales y sistémicos 83
sobre todo en forma perivascular, intersticial y endocár-dica. 82 -
35, 82
II, actuando a través del receptor AT1 y el factor de creci-

Gelpi - Donato Fisiopatología cardiovascular
162
horas en zonas remotas al infarto. 56 Este aumento de los
I y III en dichas zonas.
de TGF-ß1, constituye uno de los principales factores que
McEwan et al. 91 mostraron en ratas que la adminis-
las arteriolas intracoronarias. Paralelamente a la prolifera-
intersticial y perivascular.
Remodelamiento tardío y su relación con el sistema renina angiotensinaLa pérdida de masa contráctil demanda, en el miocardio
al mismo tiempo que se desarrolla el proceso reparativo -
en marcha, en el miocardio no isquémico, un conjunto de
una etapa avanzada del proceso de remodelamiento; re-presentan la respuesta celular que implica un crecimiento
Por lo tanto, en esta etapa del proceso de remodela-miento ventricular, se van a manifestar los cambios morfo-
a hacerse evidentes hasta una etapa avanzada del proceso.
los cambios en la arquitectura ventricular permitirían una
-
88
-
fundamental durante el proceso de remodelamiento; este
parte de los miocitos. 4
--
denomina índice de masa y presenta como límite estable- 4
hemodinámica, también se van a ver involucrados todos los componentes del intersticio miocárdico. Este hecho
las células del endotelio, así como del remodelado de la
65, 78
-
4
Cuando el aumento del índice de masa cardiaca no se ventricular, se con-
Tal es el caso del infarto de miocardio que, por tratarse
--
-
síntesis de proteínas estructurales y contráctiles a través de diferentes vías.
Rubin et al. 92 mostraron, en ratas con infarto de miocar-
compensadora, el área de miocardio no infartado y el diáme-tro de los miocitos de la pared no infartada se relacionan en
-
-dora desarrollado por los miocitos de la zona no infartada
crecimiento celular podría limitar la respuesta funcional. Al respecto, Olivetti y col.93 mostraron, en ratas con infarto del
tanto, a pesar de que el crecimiento de los miocitos se tra-dujo en un aumento de la masa, fue incapaz de reducir la

Remodelamiento cardíaco y sistema renina angiotensina
163
Anversa et al. -
dio viable y el volumen de los miocitos en forma propor-cional. Asimismo, estos autores vincularon el incremento
-
29, 37
-
mecánicos, como el estiramiento, y por factores neuro-
no acopladas a receptores. Estudios realizados en ratas
-
los miocitos. 94
-nas y factores de crecimiento, así como también la acti-
--
95 -
–como el péptido natriurético auricular y cerebral. Asimis-
IGF-I en miocitos viables de la zona adyacente al infarto, que están sometidos a un alto estrés parietal. 96 -
-
97 Por lo tanto, este factor de crecimiento cumpliría un rol importante en la respuesta
-
de vías intracelulares, relacionadas con el receptor de este Ver capítulo 11
mediado por el estiramiento de los miocitos y por la ac---
in vivo como in vitro
-cos relacionados con un incremento en las condiciones de
20, 37 et al. mostraron un -
mecánico del miocardio, y también que el incremento en la síntesis proteica en los miocitos puede ser bloqueado
32
-mentales de infarto, estudiaron el efecto de fármacos inhi-
no infartadas. Así, se ha observado que los inhibidores de
comparables a los animales sham. Resultados similares
Estudios recientes de nuestro laboratorio muestran que,
-
endocárdica en septum y ventrículo derecho.La aldosterona es otro componente importante del siste-
del remodelamiento ventricular en forma independiente de
los niveles de sodio, potasio, hormona antidiurética, pépti-do natriurético auricular y endotelina también contribuyen
-blastos dentro del miocardio. Estudios in vitro mostraron
tratamiento con espironolactona inhibe el incremento de
-traron que la mayoría de las citoquinas, que incrementan
-nas, factor de necrosis tumoral, y el factor de crecimien-
infartadas. 55, 56 Estas citoquinas, actuando en forma auto-
de miocitos. 98
Inhibición farmacológica del sistema renina angiotensina en el remodelamiento ventricular posinfarto
-
determina que este sistema hormonal cumpla funciones
el proceso de remodelamiento ventricular. 34, 35, 60
-
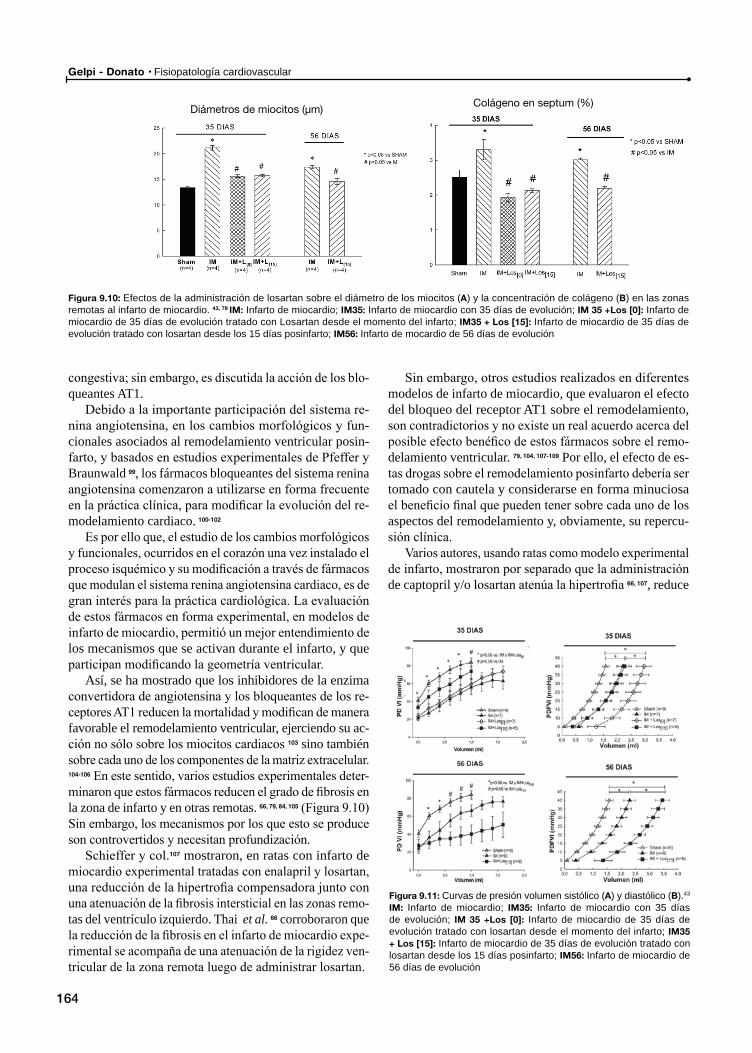
Gelpi - Donato Fisiopatología cardiovascular
164
Figura 9.11: Curvas de presión volumen sistólico (A) y diastólico (B).43
IM: Infarto de miocardio; IM35: Infarto de miocardio con 35 días de evolución; IM 35 +Los [0]: Infarto de miocardio de 35 días de evolución tratado con losartan desde el momento del infarto; IM35 + Los [15]: Infarto de miocardio de 35 días de evolución tratado con losartan desde los 15 días posinfarto; IM56: Infarto de miocardio de 56 días de evolución
-queantes AT1.
--
cionales asociados al remodelamiento ventricular posin-
Braunwald 99, los fármacos bloqueantes del sistema renina
-modelamiento cardiaco. 100-102
los mecanismos que se activan durante el infarto, y que
Así, se ha mostrado que los inhibidores de la enzima -
favorable el remodelamiento ventricular, ejerciendo su ac-103 sino también
104-106 -
la zona de infarto y en otras remotas. 66, 79, 84, 105
107 mostraron, en ratas con infarto de
-tas del ventrículo izquierdo. Thai et al. 66 corroboraron que
--
modelos de infarto de miocardio, que evaluaron el efecto del bloqueo del receptor AT1 sobre el remodelamiento,
-delamiento ventricular. 79, 104, 107-109 Por ello, el efecto de es-
tomado con cautela y considerarse en forma minuciosa
aspectos del remodelamiento y, obviamente, su repercu-
de captopril y/o 66, 107, reduce
Figura 9.10: Efectos de la administración de losartan sobre el diámetro de los miocitos (A) y la concentración de colágeno (B) en las zonas remotas al infarto de miocardio. 43, 79 IM: Infarto de miocardio; IM35: Infarto de miocardio con 35 días de evolución; IM 35 +Los [0]: Infarto de miocardio de 35 días de evolución tratado con Losartan desde el momento del infarto; IM35 + Los [15]: Infarto de miocardio de 35 días de evolución tratado con losartan desde los 15 días posinfarto; IM56: Infarto de mocardio de 56 días de evolución
Diámetros de miocitos (μm)Colágeno en septum (%)

Remodelamiento cardíaco y sistema renina angiotensina
165
99, 109 y la 66, 87, tanto en la
y col.104 -
y col.110
mejoraría la --
-
-tante tener en cuenta la especie que será utilizada para el estudio del remodelamiento ventricular y el bloqueo del
-
sobre el remodelamiento ventricular. Los resultados de es-
también podría producir un efecto desfavorable sobre el
del tratamiento y no del momento de su inicio. Este efecto desfavorable estaría dado por el incremento de la dilata-
contractilidad fue atenuada y no se observo un incremento
importancia de hallar una ventana terapéutica adecuada para comenzar el tratamiento con bloqueantes AT1, y de considerar la especie animal utilizada a la hora de evaluar
-modelamiento ventricular posinfarto.
--
-na sobre muchos aspectos del remodelamiento. Como se
estudios clínicos que muestran mayoritariamente efectos
variables que los clínicos, analizan el remodelamiento
Bibliografía
11. Swynghedauw, B. “Molecular mechanisms of myocardial remodeling”. Physiol Rev. 1999;79:215-262.
12. Fedak PW, Verma S, Weisel RD, Li RK. „Cardiac remodeling and failure From molecules to man (Part II)”. Cardiovasc Pathol. 2005;14:49-60.
13. Fedak PW, Verma S, Weisel RD, Skrtic M, Li RK. “Cardiac remodeling and failure: from molecules to man (Part III)”. Cardiovasc Pathol. 2005;14:109-119.
14. Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. “Controversies in ventricular remodeling”. Lancet. 2006;367:356-367.
15. Linzbach, AJ. “Heart failure from the point of view of quantitative anatomy”. Am J Cardiol. 1960;5:370-382.
16. Kostuk WJ, Kazamias TM, Gander MP, Simon AL, Ross J Jr. “Left ventricular size after acute myocardial infarction. Serial changes and their prognostic significance”. Circulation. 1973;47:1174-1179.
17. White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. “Left ventricular end-systolic volume as the major de-terminant of survival after recovery from myocardial infarction”. Circulation. 1987;76:44-51.
Figura 9.12: Concentración de colágeno en cicatriz, en animales tratados a diferentes tiempos de evolución del infarto. Microfotografías representativas de la zona de cicatriz, en cortes teñidos con la técnica de picrosirius red, pertenecientes a los grupos de la figura (Modificado de González, GE et al. Am J Physiol Heart Circ Physiol. 2009; 297: H375-H386).

Gelpi - Donato Fisiopatología cardiovascular
166
18. Pfeffer MA, E Braunwald. “Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications”. Circulation. 1990;81:1161-1172.
19. Ertl G, Gaudron P, Neubauer S, Bauer B, Horn M, Hu K, Tian R. “Cardiac dysfunction and development of heart failure”. Eur Heart J. 1993;14 Suppl A:33-37.
10. Gaudron P, Eilles C, Kugler I, Ertl G. “Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors”. Circulation. 1993;87:755-763.
11. Turhan H, Aksoy Y, Yetkin E, Kosar F. “Ventricular remodeling following acute myocardial infarction: The role of altered collagen homeostasis”. Int J Cardiol. 2006.
12. de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. “Interna-tional union of pharmacology. XXIII. The angiotensin II receptors”. Pharmacol Rev. 2000;52:415-472.
13. Baker KM, Booz GW, Dostal DE. “Cardiac actions of angiotensin II: Role of an intracardiac renin-angiotensin system”. Annu Rev Physiol. 1992;54:227-241.
14. Gavras H, HR Brunner. “Role of angiotensin and its inhibition in hypertension, ischemic heart disease, and heart failure”. Hyperten-sion. 2001;37:342-345.
15. Oishi Y, Ozono R, Yoshizumi M, Akishita M, Horiuchi M, Oshima T. “AT2 receptor mediates the cardioprotective effects of AT1 receptor antagonist in post-myocardial infarction remodeling”. Life Sci. 2006;80:82-88.
16. Crawford DC, Chobanian AV, Brecher P. “Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat”. Circ Res. 1994;74:727-739.
17. Ishihata A, M Endoh. “Pharmacological characteristics of the posi-tive inotropic effect of angiotensin II in the rabbit ventricular myo-cardium”. Br J Pharmacol. 1993;108:999-1005.
18. Ishihata A, M Endoh. “Species-related differences in inotropic effects of angiotensin II in mammalian ventricular muscle: recep-tors, subtypes and phosphoinositide hydrolysis”. Br J Pharmacol. 1995;114:447-453.
19. Booz GW, KM Baker. “Molecular signalling mechanisms control-ling growth and function of cardiac fibroblasts”. Cardiovasc Res. 1995;30:537-543.
20. Dostal DE, Hunt RA, Kule CE, Bhat GJ, Karoor V, McWhin-ney CD, Baker KM. “Molecular mechanisms of angiotensin II in modulating cardiac function: intracardiac effects and signal transduction pathways”. J Mol Cell Cardiol. 1997;29:2893-2902.
21. Dostal DE, KM Baker. “The cardiac renin-angiotensin system: concep-tual, or a regulator of cardiac function?” Circ Res. 1999;85:643-650.
22. Ferrario CM, Trask AJ, Jessup JA. “Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angio-tensin-(1-7) in regulation of cardiovascular function”. Am J Physiol Heart Circ Physiol. 2005;289:H2281-H2290.
23. Berry C, Touyz R, Dominiczak AF, Webb RC, Johns DG. “An-giotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide”. Am J Physiol Heart Circ Physiol. 2001;281:H2337-H2365.
24. Tallant EA, Ferrario CM, Gallagher PE. “Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas recep-tor”. Am J Physiol Heart Circ Physiol. 2005;289:H1560-H1566.
25. Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. “Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice”. Hypertension. 2006;47:996-1002.
26. Masaki H, Kurihara T, Yamaki A, Inomata N, Nozawa Y, Mori Y, Murasawa S, Kizima K, Maruyama K, Horiuchi M, Dzau VJ, Takahashi H, Iwasaka T, Inada M, Matsubara H. “Cardiac-specific overexpression of angiotensin II AT2 receptor causes attenuated response to AT1 receptor-mediated pressor and chronotropic effects”. J Clin Invest. 1998;101:527-535.
27. Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD. “Angiotensin
II receptors and angiotensin II receptor antagonists”. Pharmacol Rev. 1993;45:205-251.
28. Akasu M, Urata H, Kinoshita A, Sasaguri M, Ideishi M, Arakawa K. “Differences in tissue angiotensin II-forming pathways by species and organs in vitro”. Hypertension. 1998;32:514-520.
29. Barlucchi L, Leri A, Dostal DE, Fiordaliso F, Tada H, Hintze TH, Kajstura J, Nadal-Ginard B, Anversa P. “Canine ventricular myocytes possess a renin-angiotensin system that is upregulated with heart failure”. Circ Res. 2001;88:298-304.
30. de Boer RA, Pinto YM, Suurmeijer AJ, Pokharel S, Scholtens E, Humler M, Saavedra JM, Boomsma F, van Gilst WH, van Veldhuisen DJ. “Increased expression of cardiac angiotensin II type 1 (AT(1)) receptors decreases myocardial microvessel den-sity after experimental myocardial infarction”. Cardiovasc Res. 2003;57:434-442.
31. Griendling KK, Lassegue B, Alexander RW. “Angiotensin receptors and their therapeutic implications”. Annu Rev Pharmacol Toxicol. 1996;36:281-306.
32. Malhotra R, Sadoshima J, Brosius FC, III, Izumo S. “Mecha-nical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro”. Circ Res. 1999;85:137-146.
33. Scott AL, Chang RS, Lotti VJ, Siegl PK. “Cardiac angiotensin receptors: effects of selective angiotensin II receptor antagonists, DUP 753 and PD 121981, in rabbit heart”. J Pharmacol Exp Ther. 1992;261:931-935.
34. Zhao W, Ahokas RA, Weber KT, Sun Y. “ANG II-induced cardiac molecular and cellular events: role of aldosterone”. Am J Physiol Heart Circ Physiol. 2006;291:H336-H343.
35. Toko H, Zou Y, Minamino T, Masaya M, Harada M, Nagai T, Sugaya T, Terasaki F, Kitaura Y, Komuro I. “Angiotensin II type 1a receptor is involved in cell infiltration, cytokine production, and neovascularization in infarcted myocardium”. Arterioscler Thromb Vasc Biol. 2004;24:664-670.
36. Sadoshima J, S Izumo. “Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype”. Circ Res. 1993;73:413-423.
37. Liu Y, Leri A, Li B, Wang X, Cheng W, Kajstura J, Anversa P. “Angiotensin II stimulation in vitro induces hypertrophy of normal and postinfarcted ventricular myocytes”. Circ Res. 1998;82:1145-1159.
38. Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, Irie K, Holle E, Yu X, Kupershmidt S, Roden DM, Wagner T, Yatani A, Vatner DE, Vatner SF, Sadoshima J. “Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice”. J Clin Invest. 2005;115:3045-3056.
39. Rajagopal K, Lefkowitz RJ, Rockman HA. “When 7 transmembra-ne receptors are not G protein-coupled receptors”. J Clin Invest. 2005;115:2971-2974.
40. Silva DM, Vianna HR, Cortes SF, Campagnole-Santos MJ, Santos RA, Lemos VS. “Evidence for a new angiotensin-(1-7) receptor subtype in the aorta of Sprague-Dawley rats”. Peptides. 2006.
41. Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. “An-giotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats”. Am J Physiol Heart Circ Physiol. 2005;289:H1013-H1019.
42. Sun Y, Zhang J, Zhang JQ, Weber KT. “Renin expression at sites of repair in the infarcted rat heart”. J Mol Cell Cardiol. 2001;33:995-1003.
43. Gonzalez GE, Palleiro J, Seropian I, Lopez Verilli M.A., Gironacci MM, Krieger L, Cavallero S, Tomasi VH, Gelpi RJ, Morales C. “Effect of Early vs. Late AT1 Receptor Blockade with Losartan on Post-Myocardial Infarction Ventricular Remodeling in Rabbits”. Am J Physiol Heart Circ Physiol. 2009; 297: H375-H386
44. Fishbein MC, Maclean D, Maroko PR. “The histopathologic evo-lution of myocardial infarction”. Chest. 1978;73:843-849.

Remodelamiento cardíaco y sistema renina angiotensina
167
45. Fishbein MC, Maclean D, Maroko PR. “Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution”. Am J Pathol. 1978;90:57-70.
46. Richard V, Murry CE, Reimer KA. “Healing of myocardial infarcts in dogs. Effects of late reperfusion”. Circulation. 1995;92:1891-1901.
47. Fujiwara H, Ashraf M, Sato S, Millard RW. “Transmural cellular damage and blood flow distribution in early ischemia in pig hearts”. Circ Res. 1982;51:683-693.
48. Morales C, Gonzalez GE, Rodriguez M, Bertolasi CA, Gelpi RJ. “Histopathologic time course of myocardial infarct in rabbit hearts”. Cardiovasc Pathol. 2002;11:339-345.
49. Vandervelde S, van Amerongen MJ, Tio RA, Petersen AH, van Luyn MJ, Harmsen MC. “Increased inflammatory response and neovascularization in reperfused vs. non-reperfused murine myo-cardial infarction”. Cardiovasc Pathol. 2006;15:83-90.
50. Sandmann S, Li J, Fritzenkotter C, Spormann J, Tiede K, Fischer JW, Unger T. “Differential effects of olmesartan and ramipril on inflammatory response after myocardial infarction in rats”. Blood Press. 2006;15:116-128.
51. Frangogiannis, NG. “The mechanistic basis of infarct healing”. Antioxid Redox Signal. 2006;8:1907-1939.
52. Bujak M, NG Frangogiannis. “The role of TGF-beta signaling in myocardial infarction and cardiac remodeling”. Cardiovasc Res. 2006.
53. Bouchardy B, G Majno. “Histopathology of early myocardial in-farcts. A new approach”. Am J Pathol. 1974;74:301-330.
54. Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. “Inflammation and angiotensin II”. Int J Biochem Cell Biol. 2003;35:881-900.
55. Deten A, Volz HC, Briest W, Zimmer HG. “Cardiac cytokine expres-sion is upregulated in the acute phase after myocardial infarction. Experimental studies in rats”. Cardiovasc Res. 2002;55:329-340.
56. Deten A, Holzl A, Leicht M, Barth W, Zimmer HG. “Changes in extracellular matrix and in transforming growth factor beta isoforms after coronary artery ligation in rats”. J Mol Cell Cardiol. 2001;33:1191-1207.
57. Berthonneche C, Sulpice T, Tanguy S, O’Connor S, Herbert JM, Janiak P, de Leiris J, Boucher F. “AT1 receptor blockade prevents cardiac dysfunction after myocardial infarction in rats”. Cardiovasc Drugs Ther. 2005;19:251-259.
58. Sun Y, Zhang JQ, Zhang J, Ramires FJ. “Angiotensin II, transfor-ming growth factor-beta1 and repair in the infarcted heart”. J Mol Cell Cardiol. 1998;30:1559-1569.
59. Lu L, Quinn MT, Sun Y. “Oxidative stress in the infarcted heart: role of de novo angiotensin II production”. Biochem Biophys Res Commun. 2004;325:943-951.
60. Sun Y, KT Weber. “Angiotensin II receptor binding following myo-cardial infarction in the rat”. Cardiovasc Res. 1994;28:1623-1628.
61. De Carvalho FC, Sun Y, Weber KT. “Angiotensin II receptor bloc-kade and myocardial fibrosis of the infarcted rat heart”. J Lab Clin Med. 1997;129:439-446.
62. Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. “Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis”. Circ Res. 2002;91:1119-1126.
63. Etoh T, Joffs C, Deschamps AM, Davis J, Dowdy K, Hendrick J, Baicu S, Mukherjee R, Manhaini M, Spinale FG. “Myocar-dial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs”. Am J Physiol Heart Circ Physiol. 2001;281:H987-H994.
64. Laguens RP, Castagnino HE, Jorg ME, Hamamura S. “Reduced injury and scar in acute myocardial infarctions treated with human growth hormone”. Jpn Heart J. 1998;39:809-817.
65. Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, Crawford J, Black S, Libby P, Mitchell PG, Lee RT. “Selective matrix metalloproteinase inhibition reduces left ventri-cular remodeling but does not inhibit angiogenesis after myocardial infarction”. Circulation. 2002;105:753-758.
66. Thai HM, Van HT, Gaballa MA, Goldman S, Raya TE. “Effects of AT1 receptor blockade after myocardial infarct on myocardial fibrosis, stiffness, and contractility”. Am J Physiol. 1999;276:H873-H880.
67. Vanhoutte D, Schellings M, Pinto Y, Heymans S. “Relevance of matrix metalloproteinases and their inhibitors after myocar-dial infarction: a temporal and spatial window”. Cardiovasc Res. 2006;69:604-613.
68. Xu X, Wan W, Ji L, Lao S, Powers AS, Zhao W, Erikson JM, Zhang JQ. “Exercise training combined with angiotensin II recep-tor blockade limits post-infarct ventricular remodelling in rats”. Cardiovasc Res. 2008;78:523-532.
69. Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. “Collagen remodeling after myocardial infarction in the rat heart”. Am J Pathol. 1995;147:325-338.
70. Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. “The in-farcted myocardium: simply dead tissue, or a lively target for thera-peutic interventions”. Cardiovasc Res. 1999;44:232-241.
71. Pfeffer JM, Pfeffer MA, Fletcher PJ, Braunwald E. “Progressive ventricular remodeling in rat with myocardial infarction”. Am J Physiol. 1991;260:H1406-H1414.
72. Chareonthaitawee P, Christian TF, Hirose K, Gibbons RJ, Rum-berger JA. “Relation of initial infarct size to extent of left ventricular remodeling in the year after acute myocardial infarction”. J Am Coll Cardiol. 1995;25:567-573.
73. Sutton MG, N Sharpe. “Left ventricular remodeling after myo-cardial infarction: pathophysiology and therapy”. Circulation. 2000;101:2981-2988.
74. Gaudron P, Eilles C, Ertl G, Kochsiek K. “Compensatory and noncompensatory left ventricular dilatation after myocardial in-farction: time course and hemodynamic consequences at rest and during exercise”. Am Heart J. 1992;123:377-385.
75. Bowen FW, Jones SC, Narula N, John Sutton MG, Plappert T, Edmunds LH Jr, Dixon IM. “Restraining acute infarct expansion decreases collagenase activity in borderzone myocardium”. Ann Thorac Surg. 2001;72:1950-1956.
76. Hochman JS, BH Bulkley. “Expansion of acute myocardial infarc-tion: an experimental study”. Circulation. 1982;65:1446-1450.
77. Hutchins GM, BH Bulkley. “Infarct expansion versus extension: two different complications of acute myocardial infarction”. Am J Cardiol. 1978;41:1127-1132.
78. MacKenna D, Summerour SR, Villarreal FJ. “Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis”. Cardiovasc Res. 2000;46:257-263.
79. Gonzalez GE, Palleiro J, Monroy S, Perez S, Rodriguez M, Masucci A, Gelpi RJ, Morales C. “Effects of the early administration of losartan on the functional and morphological aspects of postmyo-cardial infarction ventricular remodeling in rabbits”. Cardiovasc Pathol. 2005;14:88-95.
80. Weber KT, CG Brilla. “Myocardial fibrosis and the renin-angioten-sin-aldosterone system”. J Cardiovasc Pharmacol. 1992;20 Suppl 1:S48-S54.
81. Robinson TF, Cohen-Gould L, Factor SM. “Skeletal framework of mammalian heart muscle. Arrangement of inter- and pericel-lular connective tissue structures”. Lab Invest. 1983;49:482-498.
82. Weber KT, Anversa P, Armstrong PW, Brilla CG, Burnett JC, Jr., Cruickshank JM, Devereux RB, Giles TD, Korsgaard N, Leier CV. “Remodeling and reparation of the cardiovascular system”. J Am Coll Cardiol. 1992;20:3-16.
83. Gurantz D, Cowling RT, Villarreal FJ, Greenberg BH. “Tumor necrosis factor-alpha upregulates angiotensin II type 1 receptors on cardiac fibroblasts”. Circ Res. 1999;85:272-279.
84. Smits JF, van Krimpen C, Schoemaker RG, Cleutjens JP, Daemen MJ. “Angiotensin II receptor blockade after myocardial infarc-tion in rats: effects on hemodynamics, myocardial DNA synthe-sis, and interstitial collagen content”. J Cardiovasc Pharmacol. 1992;20:772-778.
85. van Krimpen C, Smits JF, Cleutjens JP, Debets JJ, Schoemaker RG, Struyker Boudier HA, Bosman FT, Daemen MJ. “DNA synthe-

Gelpi - Donato Fisiopatología cardiovascular
168
sis in the non-infarcted cardiac interstitium after left coronary artery ligation in the rat: effects of captopril”. J Mol Cell Cardiol. 1991;23:1245-1253.
86. Dixon IM, Ju H, Jassal DS, Peterson DJ. “Effect of ramipril and losartan on collagen expression in right and left heart after myo-cardial infarction”. Mol Cell Biochem. 1996;165:31-45.
87. Ju H, Zhao S, Jassal DS, Dixon IM. “Effect of AT1 receptor block-ade on cardiac collagen remodeling after myocardial infarction”. Cardiovasc Res. 1997;35:223-232.
88. Holmes JW, Borg TK, Covell JW. “Structure and mechanics of healing myocardial infarcts”. Annu Rev Biomed Eng. 2005;7:223-253.
89. Van Kerckhoven R, Kalkman EA, Saxena PR, Schoemaker RG. “Altered cardiac collagen and associated changes in diastolic func-tion of infarcted rat hearts”. Cardiovasc Res. 2000;46:316-323.
90. Matsubara LS, Matsubara BB, Okoshi MP, Cicogna AC, Janicki JS. “Alterations in myocardial collagen content affect rat papillary muscle function”. Am J Physiol Heart Circ Physiol. 2000;279:H1534-H1539.
91. McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ. “Dif-ferential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo”. Circulation. 1998;98:2765-2773.
92. Rubin SA, Fishbein MC, Swan HJ. “Compensatory hypertrophy in the heart after myocardial infarction in the rat”. J Am Coll Cardiol. 1983;1:1435-1441.
93. Olivetti G, Capasso JM, Meggs LG, Sonnenblick EH, Anversa P. “Cellular basis of chronic ventricular remodeling after myocardial infarction in rats”. Circ Res. 1991;68:856-869.
94. Thienelt CD, Weinberg EO, Bartunek J, Lorell BH. “Load-induced growth responses in isolated adult rat hearts. Role of the AT1 receptor”. Circulation. 1997;95:2677-2683.
95. Ju H, Zhao S, Tappia PS, Panagia V, Dixon IM. “Expression of Gq alpha and PLC-beta in scar and border tissue in heart failure due to myocardial infarction”. Circulation. 1998;97:892-899.
96. Cittadini A, Grossman JD, Stromer H, Katz SE, Morgan JP, Douglas PS. “Importance of an intact growth hormone/insulin-like growth factor 1 axis for normal post-infarction healing: studies in dwarf rats”. Endocrinology. 2001;142:332-338.
97. Donohue TJ, Dworkin LD, Lango MN, Fliegner K, Lango RP, Benstein JA, Slater WR, Catanese VM. “Induction of myocardial insulin-like growth factor-I gene expression in left ventricular hypertrophy”. Circulation. 1994;89:799-809.
98. Burger A, Benicke M, Deten A, Zimmer HG. “Catecholamines stimulate interleukin-6 synthesis in rat cardiac fibroblasts”. Am J Physiol Heart Circ Physiol. 2001;281:H14-H21.
99. Pfeffer JM, Pfeffer MA, Braunwald E. “Influence of chronic cap-topril therapy on the infarcted left ventricle of the rat”. Circ Res. 1985;57:84-95.
100. Chowdhary S, JN Townend. “The OPTIMMAL trial: losartan or captopril after acute myocardial infarction”. Lancet. 2002;360:1886.
101. Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, Deedwania PC, Ney DE, Snavely DB, Chang PI. “Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE)”. Lancet. 1997;349:747-752.
102. Witte K, Thackray S, Banerjee T, Clark AL, Cleland JG. “Update of ELITE-II, BEST, CHAMP, and IMPRESS clinical trials in heart failure”. Eur J Heart Fail. 2000;2:107-112.
103. Guo X, Wang J, Elimban V, Dhalla NS. “Both enalapril and losar-tan attenuate sarcolemmal Na+-K+-ATPase remodeling in failing rat heart due to myocardial infarction”. Can J Physiol Pharmacol. 2008;86:139-147.
104. Hu K, Gaudron P, Anders HJ, Weidemann F, Turschner O, Nahrendorf M, Ertl G. “Chronic effects of early started angio-tensin converting enzyme inhibition and angiotensin AT1-receptor subtype blockade in rats with myocardial infarction: role of bra-dykinin”. Cardiovasc Res. 1998;39:401-412.
105. Patten RD, Aronovitz MJ, Einstein M, Lambert M, Pandian NG, Mendelsohn ME, Konstam MA. “Effects of angiotensin II receptor blockade versus angiotensin-converting-enzyme inhibition on ventricular remodelling following myocardial infarction in the mouse”. Clin Sci (Lond). 2003;104:109-118.
106. Hao J, Wang B, Jones SC, Jassal DS, Dixon IM. “Interac-tion between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro”. Am J Physiol Heart Circ Physiol. 2000;279:H3020-H3030.
107. Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN, Drexler H. “Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat”. Circula-tion. 1994;89:2273-2282.
108. Tanimura M, Sharov VG, Shimoyama H, Mishima T, Levine TB, Goldstein S, Sabbah HN. “Effects of AT1-receptor blockade on progression of left ventricular dysfunction in dogs with heart failure”. Am J Physiol. 1999;276:H1385-H1392.
109. Jain M, Liao R, Ngoy S, Whittaker P, Apstein CS, Eberli FR. “Angiotensin II receptor blockade attenuates the deleterious effects of exercise training on post-MI ventricular remodelling in rats”. Cardiovasc Res. 2000;46:66-72.
110. Xia QG, Chung O, Spitznagel H, Illner S, Janichen G, Rossius B, Gohlke P, Unger T. “Significance of timing of angiotensin AT1 receptor blockade in rats with myocardial infarction-induced heart failure”. Cardiovasc Res. 2001;49:110-117.

169
-yores causas de mortalidad. 1, 2 Los mecanismos involucra-dos en su desarrollo son poco conocidos. En el curso del tiempo, a medida que se adquirían nuevos conocimientos, se postularon diferentes factores que podrían estar involu-
-
-
fue posible discriminar, entre los diferentes mecanismos
Más recientemente, el conocimiento de los mecanis--
--
3, 4
Este enfoque cuantitativo permite suponer que la pre--
vas células contráctiles, pueden ser alternativas terapéuti-
--
stem cells
de la masa de miocardio faltante con nuevas células con-
-
consideradas alternativas terapéuticas para el tratamiento, -
terapéuticos recientes han mejorado la sobrevida, no han
Inducción de los miocitos adultos remanentes a dividirse en células hijas
Replicación de los miocitos en el corazón normal
de todos los eucariotes. En los metazoarios, el número
-
hiper-
De esta manera, los cardiomiocitos adultos están en
del individuo; vale decir que los cardiomiocitos con que nacemos son los mismos con los que morimos. Por el con-trario, las células de los vasos y del tejido conectivo, en respuesta a estímulos apropiados, son capaces de entrar en
que implican una pérdida de miocitos. Una de las razones
Reparación del corazón
10

Gelpi - Donato Fisiopatología cardiovascular
170
D, que se unen con la kinasa correspondiente, formando
es la llave que decide la entrada en el ciclo celular. Cuando
bajo el control de dos ciclinas, la A, en la fase temprana, y la B, en la fase tardía. A diferencia de las otras ciclinas, que están ubicadas en el interior nuclear, la ciclina B es
que la célula inicie la fase M. En esta etapa de la mitosis,
se disuelve la membrana nuclear y se cumplen las etapas
-lulas hijas mononucleadas o células bi o multinucleadas. Cada una de esas etapas está bajo el control de sus corres-pondientes inhibidores. Los de los complejos CDK son
de inhibidores son proteínas de la familia CIP/KIP, cuyos CIP1 KIP1 y P57 , que
de ciclinas sería la responsable de la ausencia de replica-
de las divisiones sea un mal necesario para mantener la estabilidad de las funciones eléctricas y mecánicas del miocardio.
El concepto de que los miocitos adultos normales son células incapaces de dividirse está basado en dos tipos de observaciones. Una de ellas es que no es posible inducir
ex vivo. Mientras que las células cardiacas de fetos o de animales recién nacidos son fácilmente cultivables, las de los adultos mantienen su via-bilidad durante breve tiempo, sin reproducirse, a menos que reviertan a un estado indiferenciado, similar a un miocito
todas las especies de mamíferos estudiadas hasta la fecha, cuando se usan las técnicas convencionales de microscopía
se realizan durante la vida fetal. Unos pocos días después
-nucleadas en vez de formar dos células hijas. Desde ese
a partir del destete, antes del mes de vida, la entrada en el ciclo celular se encuentra bloqueada, aparentemente por el resto de la vida del animal. No solo no se observan mitosis,
que la síntesis de ADN por los núcleos miocíticos está casi
-
están en ciclo celular. 5
Los factores involucrados en ese bloqueo de la repli--
6 Las cuatro fases del mismo –G1, preparadora de la síntesis del ADN; S, de síntesis del ADN; G2, etapa que transcurre
M, o mitosis, en la que los cromo-
el mismo contenido de ADN, en dos núcleos hijos– deben cumplirse en forma ordenada y secuencial. Cada una de
points
Las células que se encuentran en el estadio de diferen-
de entrar en el ciclo celular, por lo que se las denomina célu-
induce una variedad especial de ciclinas, llamadas ciclinas
Figura 10.1: Esquema del ciclo celular

Reparación del corazón
171
7, 8
incapacidad de los miocitos murinos para entrar en el ci-
9, 10
inhibidores del ciclo celular, como por ejemplo la proteína 11
de los inhibidores del ciclo celular, y que es posible actuar -
canismos responsables de la incapacidad replicativa de los miocitos.
-
durante la vida intrauterina y la época temprana posnatal,
la de los roedores. En los corazones embrionarios y feta-les estas células son uninucleadas, diploides, y se dividen por citocinesis y cariocinesis. Después del nacimiento es
-ciales humanos, no se sabe en qué momento las células dejan de dividirse o de entrar en el ciclo celular. A dife-rencia de lo que ocurre en la mayoría de los mamíferos,
mayoría tetraploides, poseen el doble del contenido de ADN de los miocitos diploides fetales o del recién na-cido. 12 Ello implica que, a diferencia de los roedores y de otras especies como el cerdo, cuyas células son po-linucleadas pero con núcleos diploides, en el desarrollo
fundamental para comprender el funcionamiento del ciclo -
-rían responsables de un recambio continuo, pero lento, de células que se pierden en el curso del tiempo, mante-
de los miocitos, para preservar la masa contráctil. Este -
del anciano comparado con el joven. 13
Replicación de los miocitos en el corazón patológicoDe la misma manera que durante el desarrollo normal
-to sostenido del trabajo, la respuesta de estas células es
el remodelamiento consecutivo a la pérdida de miocitos que ocurre en las miocarditis o en el infarto de miocardio. En esas circunstancias las células aumentan de volumen,
volumen celular y el nuclear, los núcleos también se hi-
de ADN, los miocitos deben mantener la capacidad de entrar en el ciclo celular, pues de otra manera no podrían
de ADN puede ser de hasta treinta y dos veces el de una 14, 15
-cremento del contenido de ADN y las razones por las que
que se realizan todas las etapas de la mitosis pero dentro de una membrana nuclear intacta, sería la responsable de
16
-cos, realizados en su mayor parte en corazones humanos,
-citos por unidad de volumen cardiaco, es decir, una ver-dadera hiperplasia. Linzbach 17
18
Figura 10.2: Imágenes de núcleos de miocitos normales (A) e hipertróficos (B)
A B

Gelpi - Donato Fisiopatología cardiovascular
172
miocitos faltantes, basta recordar que en los infartos de
Estrategias para inducir mitosis y citocinesis en miocitos adultos
--
del ciclo celular, si bien son de utilidad para conocer los
-
factores de crecimiento, tales como el
del
en el ciclo celular a miocitos inmaduros en sistemas in vitro 22, 23 -tos in vivo. Los primeros estudios realizados en humanos y
isquémicas, a los que se administraron estos factores como -
de esos productos durante mayor tiempo. Recientemente
y una hiperplasia de los miocitos. 24, 25
infarto y entre otros efectos, induce un aumento del número de mitosis de los miocitos. 26
-
-
Inducción de la replicación de una población de progenitores miocíticos residentes en el corazón y diferenciación en miocitos adultos
que, a semejanza de lo que ocurre en el tejido epitelial,
-paces de replicar y diferenciarse en miocitos adultos. 27,
28 Dado lo reciente de esas publicaciones, todavía no han
-lular es necesario que se cumplan todas las etapas de la
humano en diferentes circunstancias, principalmente en la 19
miocardio 20
documentado la presencia de citocinesis convencionales.
hiperplasia importante o que ese mecanismo sea capaz de
Ello ha motivado que se plantee la posibilidad de
-cinesis clásicas. 17
21 De estos estudios
vida posnatal no se pierde la capacidad de los miocitos -
-
descarta la posibilidad de que operen otros mecanismos
para reemplazar con células funcionalmente útiles los
Figura 10.3: Metafase de un miocito adulto. Las flechas señalan la inserción del huso

Reparación del corazón
173
de la posibilidad de inducir estas células a replicar, o de in vitro y después
a adquirir el fenotipo de un miocito adulto normal y no
-pandidas ex vivo y reinoculadas en animales con infarto de
Introducción en el corazón de células progenitoras indiferenciadas
Células tronco embrionarias -
diferenciarse indistintamente en elementos del endo, ecto y mesodermo. Estas células totipotentes, llamadas tronco embrionarias, o stem cells, una vez ubicadas
-nar elementos más diferenciados, como miocardiocitos
dado que, si se dispusiera de un número adecuado de embriones humanos, hecho posibilitado por las técnicas
in vitro, se podría desarrollar, tanto en el
diferenciados de tejidos humanos y emplearlos para re-
29 Además, por medio
obtener embriones humanos clonados y desarrollar cual-quier estirpe celular del individuo dador, eliminando de
se las transplanta. Por último, si se emplearan moldes in vitro
-
para desarrollar la mayoría, o todas, las posibilidades descri-
humana está prohibida en la mayoría de las naciones.stem
Por el contrario, esos dilemas éticos están ausentes cuando las células capaces de diferenciarse en otras estir-
Los primeros, si bien fueron implantados con relativo -
data. 30
-ladores implantables, hizo decaer el entusiasmo inicial. En
el mayor número de ensayos clínicos y publicaciones.
Células tronco de la médula ósea del adulto
-bro–, cuando han sufrido una pérdida celular. Incluso si
eso, se comprende el entusiasmo que produjo la observa--
células que eran capaces de diferenciarse en otros tejidos.-
ban por vía endovenosa neuroblastos inmaduros cultivados in vitro -
31
32-34
-
stem cellsy del transplante celular. Aparecieron nuevas palabras en el
y casi la inmortalidad.
mamíferos adultos, entre ellos el hombre, contiene una
a los elementos de estirpe hemopoyética, sino que, cuando
35 Los -
-

Gelpi - Donato Fisiopatología cardiovascular
174
-
del dador. 36
transplantadas colonizaban en casi todos los tejidos del receptor, donde se diferenciaban en células endoteliales, osteocitos, hepatocitos, células epiteliales, miocitos, e in-
discute si es real la plasticidad de estas células y actual-mente es motivo de controversia si en realidad se trans-
núcleos entre las células tronco y las residentes en el tejido 37, 38, o si persisten como elementos hemopoyéticos. 39, 40
Una de las razones de la controversia acerca de la di-stem cells adultas en otras células radica
una estirpe celular. Por ejemplo, un miocito cardiaco pue--
-
su capacidad de contraerse y de conectarse eléctricamente con otros miocitos.
Lamentablemente, la mayoría de los estudios emplean
-citos adultos funcionalmente competentes.
--
espesor de la pared, como consecuencia de la incorpora-
miocitos, células endoteliales y músculo liso. 41
Pese a que este estudio fue realizado con pocos anima-
pocos meses más tarde, en lo que es probablemente un ejemplo único en la medicina contemporánea, aparecieron publicados los primeros estudios humanos en los que se inyectaron por vía intracoronaria células mononucleares
de miocardio. 42, 43 -dores publicaron los resultados de un ensayo de fase 1 con
de los cuales recibieron por vía intracoronaria células de
44
En el momento actual, ya se han publicado los resul-
y otro no aleatorizado, en el que se comparan el efecto
sido la falta de reacciones adversas, por lo menos durante
ventricular. 45, 46 Recientemente se ha intentado transplan-
-meable, con resultados contradictorios. En tanto que en
51, el
52
-
-
trasplante celular se debe a que las células se incorporan al sincicio funcional del miocardio, o si ejercen un efecto indirecto.
Esta última posibilidad está apoyada por el hecho de -
ramente no contribuyen al ciclo cardiaco, como los mio-
-
la cercanía del infarto, ya sea evitando la muerte celular -
nadas. Por otra parte, en el caso del infarto de miocardio,
-
Por otra parte, a medida que aumenta el número de ensayos clínicos, no se observa un control adecuado de la
-cado efectos adversos, estos se han producido en el corto
-
cuarta parte de los casos, con los posibles efectos perju-
Además, se ha planteado la posibilidad de que el trans-
aterosclerosis, induciendo el crecimiento de la placa, como se ha observado en modelos de roedores.
de los posibles problemas que pueden aparecer, reciente-
en el que se administraron células asociadas con factores

Reparación del corazón
175
stents, porque ese proce-dimiento inducía restenosis coronaria. 48
Pese a estos efectos adversos posibles, lo más proba-
-
-lizada en cualquier centro médico que cuente con un ser-
plantea problemas éticos, especialmente cuando en mu-chos casos se trata de pacientes en los que ya no es posible
-mos por los que las células transplantadas inducen una mejoría de la contractilidad. Pese a que de acuerdo con
mismas se transformen en miocitos adultos, la posibilidad de que produzcan factores de crecimiento, que a su vez
-rada. Por otra parte, de contarse con un mayor número de
-
estudios en animales, especialmente en mamíferos de
las mejores células a trasplantar, si es o no necesaria
quizás es más importante, cuáles son las enfermedades
isquemia cardiaca.--
estos fracasaran y tuvieran que suspender ensayos en curso -
Conclusión
de la terapia celular, como por ejemplo el desconocer si
permitan aumentar la masa de cardiomiocitos adultos fun-
49
Bibliografía
1. Leeder, SR. “The economic impact of cardiovascular disease . Heart Beat: The World Heart Federation Newsletter. Available at: <htp://www.worldheart.org/pdf/publications.heartbeat.2004>
2. Murray CJL, AD López (eds). The global burden of disease: a com-prehensive assessment of mortality and disability from diseases, injuries and risk factors in 1990 and projected to 2020. Cambridge, Harvard School of Public Health on behalf of the World Health Organization and The World Bank, 1996 (Global Burden of Disease and Injury Series, Vol. I).
3. Chimenti C, Kajstura J, Torella D, et al. “Senescence and Death of Primitive Cells and Myocytes Lead to Premature Cardiac Aging and Heart Failure . Circ Res. 2003;93:604-13.
4. Sarkar S, Chawla-Sarkar M, Young D. “Myocardial Cell Death and Regeneration during Progression of Cardiac Hypertrophy to Heart Failure . J Biol Chem 2004;279:52630-42.
5. Soonpaa MH, LJ Field. “Survey of Studies Examining Mammalian Cardiomyocyte DNA Synthesis . Circ Res. 1998;83:15-26.
6. Li JM, G Brooks. “Cell cycle regulatory molecules (cyclins, cyclin-dependent kinases and cyclin-dependent kinase inhibitors) and the cardiovascular system: potential targets for therapy? . Eur Heart J 20:406-20; 1999.
7. Pasumarthi KBS, L Field. “Cardiomyocyte cell cycle regulation . Circulation 90:1044-54; 2002
8. Mc Lellan WR, MD Schneider. “Genetic dissection of cardiac growth control pathways . Annu Rev Physiol 62:289-319; 2000.
9. Soonpaa MH, Koh GY, Pajak L. “Cyclin D1 Overexpression Promotes Cardiomyocyte DNA Synthesis and Multinucleation in Transgenic Mice . J Clin. Invest 99:2644-54; 1997.
10. Akli S, Zhan S, Abdellatif M. “E1A Can Provoke G1 Exit That Is Refractory to p21 and Independent of Activating Cdk2 . Circ Res. 85:319-328; 1999.
11. Poolman RA,Li JM, Durand B. “Altered Expression of Cell Cycle Proteins and Prolonged Duration of Cardiac Myocyte Hyperplasia in p27 KIP1 Knockout Mice . Circ Res 85:117-127; 1999.
12. Pfitzer, P. “Nuclear DNA content of human myocardial cells . Curr Top Pathol 54:125-68; 1971.
13. Anversa P, J Kajstura. “Ventricular myocytes are not terminally diffe-rentiated in the adult mammalian heart . Circ. Res; 83:1-14; 1998.
14. Sandritter W, CP Adler. “Polyploidization of heart muscle nuclei as a prerequisite for heart growth and numerical hyperplasia in heart hypertrophy . Recent Adv Stud Cardiac Struct Metab 12:115-27; 1976.
15. Vliegen HW, Eulderink F, Bruschke AV. “Polyploidy of myocyte nuclei in pressure overloaded hearts: a flow cytometric study in left and right ventricular myocardium . Am J Cardiovasc Pathol 5:27-319; 1995.
16. Cabeza Meckert P, Garcia Rivello H, Vigliano C. “Endomitosis and polyploidization of cardiac myocytes in the periphery of acute myocardial infarction . Cardiovasc Res 67:116-123; 2005.
17. Linzbach AJ. “Heart failure from the point of view of quantitative anatomy . Am J Cardiol 5:370-382; 1960.

Gelpi - Donato Fisiopatología cardiovascular
176
18. Grajek S, Lesiak M, Pyda M. “Hypertrophy or hyperplasia in cardiac muscle. Post-mortem human morphometric study . Eur Heart J 14: 40-47; 1993.
19. Kajstura J, Leri AS, Finato N. “Myocyte proliferation in end-stage cardiac failure in humans Proc. Natl. Acad. Sci USA 95: 8801-05; 1998.
20. Beltrami AP, Urbanek K, Kajstura J. et al. “Evidence that hu-man myocytes divide after myocardial infarction . N Engl J Med 344:1750-1757; 2001.
21. Moravec M, Turek Z, Moravec J. “Persistence of neoangiogenesis and cardiomyocyte divisions in right ventricular myocardium of rats born and raised in hypoxic conditions . Basic Res Cardiol 97:153-60; 2002.
22. Claycomb WC, RL Moses. “Growth factors and TPA stimulate DNA synthesis and alter the morphology of cultured terminally differentiated adult rat cardiac muscle cells . Dev Biol 127:257-65; 1988.
23. Decker RS, Cook MG, Behnke-Barclay M. “Some growth factors stimulate cultured adult rabbit ventricular myocyte hypertrophy in the absence of mechanical loading . Circ Res 77:544-55.; 1995
24. Laguens R, Cabeza Meckert P, Vera Janavel G. et al. “Entrance in Mitosis of Adult Cardiomyocytes in Ischemic Pig Hearts After Plasmid-Mediated rhVEGF165 Gene Transfer . Gene Therapy 2002; 9: 1676-81; 2002.
25. Laguens R, Cabeza Meckert P, Vera Janavel G, et al. “Cardiomyocyte hyperplasia after plasmid-mediated vascular endothelial growth factor gene transfer in pigs with chronic myocardial ischemia . J Gene Med. 6:222-27; 2004.
26. Vera Janavel G, Cabeza Meckert P, Crottogini A. et al. “Reducción del tamaño de infarto de miocardio mediante la transferencia gé-nica de VEGF en ovejas . Rev Argent Cardiol 72:197-202; 2004.
27. Beltrami AP, Barlucchi L, Torella D. et al. “Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration . Cell 114:763-76; 2003.
28. Laugwitz KL, Moretti A, Lam J, et al. “Postnatal isl1+ cardio-blasts enter fully differentiated cardiomyocyte lineages . Nature 433:647-53; 2005.
29. Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Sil-vestri F, et al.“Miocardial regeneration by activation of totipotent cardiac stem cells ischemic heart failure . Proc Natl Acad Sci USA 102:8692-97; 2005.
30. Oh H, Bradfute SB, Gallardo TD, et al. “Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction . Proc Natl Acad Sci USA 100:12313-18; 2003.
31. Doss MX, Koehler CI, Giss G. “Embryonic stem cells: a promising tool for cell replacement therapy . J Cell. Mol. Med 8:465-73; 2004.
32. Menasche P, Hagege AA, Scorsin M, et al. “Myoblast transplantation for heart failure . Lancet 357: 279-280; 2001.
33. Menard C, Hagege AA, Agbulut O, et al. “Transplantation of cardiac-committed mouse embryonic stem cells to infarcted sheep myocardium: a preclinical study . Lancet 2005;366:1005-12.
34. Bjornson CR, Rietze RL, Reynolds BA. “Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo . Science 283:534-37; 1999.
35. Eglitis MA, E Mezey. “Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice . Proc Natl Acad Sci USA 94: 4080-5; 1997.
36. Azizi SA, Stokes D, Augelli BJ. “Engraftment and migration of human bone marrow stromal cells implanted in the brains of al-bino rats-similarities to astrocyte grafts . Proc Natl Acad Sci USA 95: 3908-13; 1998.
37. Ferrari G, Cusella-De Angelis G, et al. “Muscle regeneration by bone marrow-derived myogenic progenitors . Science 279: 1528-30; 1998.
38. Jiang Y, Jahagirdar BN, Reinhardt RL, et al. “Pluripotency of mesenchymal stem cells derived from adult marrow . Nature 418:41-49; 2002.
39. Hruban RH, Long PP, Perlman EJ, et al. “Fluorescence in situ hybridization for the Y-chromosome can be used to detect cells of recipient origin in allografted hearts following cardiac transplan-tation . Am J Pathol 142:975-80; 1993
40. Dolado MA, Pardal R, García-Verdugo JM, et al. “Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes . Nature 425:968-73; 2003.
41. Nygren JM, Jovinge S, Breitbach M, et al. “Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequen-cy through cell fusion, but not transdifferentiation . Nat Med 10:494-501; 2004.
42. Balsam LB, Wagers AJ, Christensen JL. “Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium . Nature 428:668-73; 2004.
43. Murry CE, Soonpaa MH, Reinecke H, et al. “Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts . 428:664-68; 2004.
44. Planat-Bénard V, Menard C, André M, et al. “Spontaneous Car-diomyocyte Differentiation From Adipose Tissue Stroma Cells . Circ Res. 94:223-29; 2004.
45. Orlic D, Kajstura J, Chimenti S, et al. “Bone marrow cells regenerate infarcted myocardium . Nature 410:701-05; 2001.
46. Hamano K, Nishida M, Hirata K, et al. “Local implantation of autologous bone marrow cells for therapeutic angiogenesis in pa-tients with ischemic heart disease: clinical trial and preliminary results . Jpn Circ J. 65:845-47; 2001.
47. Strauer BE, Brehm M, Zeus T, et al. “Myocardial regeneration after intracoronary transplantation of human autologous stem cells following acute myocardial infarction . Dtsch med Wschr. 126:932-38; 2001.
48. Strauer BE, Brehm M, Zeus T, et al. “Repair of infarcted myocar-dium by autologous intracoronary mononuclear bone marrow cell transplantation in humans . Circulation 106:1913-18; 2003.
49. Schachinger V, Assmus B, Britten MB, et al. “Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction: final one-year results of the TOPCARE-AMI Trial . J Am Coll Cardiol 44:1690-99; 2004.
50. Wollert KC, Meyer GP, Lotz J, et al. “Intracoronary autologous bone-marrow cell transfer after myocardial Infarction: the BOOST rando-mised controlled clinical trial . Lancet 364:141-48; 2004.
51. Kuethe F, Richartz BM, Kasper C. “Autologous intracoronary mononuclear bone marrow cell transplantation in chronic ischemic cardiomyopathy in humans . Int J Cardiol 100:485-91; 2005.
52. Strauer BE, Brehm, M, Zeus T. “Regeneration of human infarcted heart muscle by intracoronary autologous bone marrow cell trans-plantation in chronic coronary artery disease, The IACT study . J Am Coll Cardiol 46:1651–58; 2005.
53. Yoon YS, Park JS, Tkebuchava T. “Unexpected severe calcification after transplantation of bone marrow cells in acute myocardial infarction . Circulation 109:3154-57; 2004.
54. Kang HJ, Kim HS, Zhang SY, et al. “Effects of intracoro-nary infusion of peripheral blood stem-cells mobilised with granulocyte-colony stimulating factor on left ventricular systolic function and restenosis after coronary stenting in myocardial infarction: the MAGIC cell randomised clinical trial . Lancet 363:751-56; 2004.
55. Melo LG, Pachori AS, Kong D, et al. “Molecular and cell-based the-rapies for protection, rescue, and repair of ischemic myocardium: reasons for cautious optimism . Circulation 109: 2386-93; 2004.

177
Señales intracelulares que siguen al estiramiento miocárdico y conducen a la hipertrofia 11
Vías intracelulares que conducen a la hipertrofia miocárdica
numerosas enfermedades cardiovasculares como la hiper-
al disminuir el estrés miocárdico frente al aumento de la
intraventricular; r: radio del ventrículo y h: espesor de la
cardiovascular. 1
2, 3
-res de crecimiento, y principalmente incremento del estrés
miocitos, consecuencia del aumento en la síntesis proteica,
4-7
-los que promueven el crecimiento celular lo hacen me-
.
in-i -
tracelular dependientes de este ion. En lo que respecta a
proteína kinasa dependiente de calmodulina: la CaMKII, y la de la proteína fosfatasa dependiente de calmodulina: la calcineurina.
i
i8
de Ca 9 10, -
miocitos in vitro -
i, aunque dependiente de la calmodulina. 11
Activación de calcineurinaEsta proteína es una serina/treonina fosfatasa depen-diente de calmodulina que ante aumentos sostenidos en
i es activada por el complejo Ca /calmodulina. La calcineurina activada desfosforila a miembros de la
-mitiendo que estos se trasloquen al núcleo y faciliten la
-tamente necesaria. 12, 13
-
• -cineurina activa o de NFAT activo y que desarrollan HC. 14
• Εl tratamiento con inhibidores de la calcineurina, -
15-17
•calcineurina.
• del miocardio. 18, 19
Activación de receptores acoplados a proteína G (GPCRs)Tres clases de proteínas G son las involucradas en la res-
α

Gelpi - Donato Fisiopatología cardiovascular
178
siete dominios transmembrana activa a la proteína Gq des-20-22
-i,
-
β
-te vinculada al control de la contractilidad miocárdica y de
i
23, de PKC 24 25
Activación de la PI3K (fosfatidilinositol 3-kinasa)
transmembrana, incluyendo los receptores con actividad -
ejemplo α y β -
Figura 11.1: Representación esquemática de vías intracelulares que conducen a la hipertrofia. Una amplia variedad de condiciones fisiológicas o patológicas son capaces de inducir hipertrofia cardiaca al activar diferentes receptores de membrana y vías de señalización intracelular. Como puede apreciarse, existen diversos puntos de contacto cruzado entre las vías intracelulares activadas por los receptores acoplados a proteína G (GPCR), receptores de tirosina kinasa (RTyrK) y receptores de citoquinas, y todos ellos comparten la estimulación de la cascada de la MAP kinasa (MAPK). Las tres familias de MAPK (ERK 1/2, p38MAPK y JNK) han sido involucradas en la génesis de hipertrofia cardiaca de diferentes modelos experimentales. Estas kinasas estimulan factores de transcripción nuclear y otras proteínas involucradas en la señalización intracelular. En el caso particular de las ERK1/2, estas también han sido implicadas en la estimulación del intercambiador Na+/H+ (NHE) a través de la kinasa p90rsk. La amplia mayoría, si no todas, de las vías intracelulares que participan en el desarrollo de hipertrofia cardiaca confluyen en la producción de un incremento de la [Ca2+]i. El aumento de la [Ca2+]i es una señal hipertrofiante ampliamente reconocida que puede actuar a través de al menos dos caminos de señalización intracelular distintos luego de interactuar con la calmodulina: la activación de la proteína kinasa II (CaMKII) y/o la activación de la fosfatasa calcineurina. Ambas vías esencialmente conducen a la activación y traslocación al núcleo de factores de transcripción nuclear y estimulación de la transcripción génica. De este esquema se desprende que ninguna cascada de señalización intracelular regula la hipertrofia de los cardiomiocitos de manera aislada. Por el contrario, daría la impresión de que cada vía intracelular opera como un componente integrado de una respuesta orquestada.
Ca2+
IGF-1
PI3K
Ca2+/Calmodulina
GPCR
PLC
RTyrK Citoquinas
JAK
gp130 CT-1R
IP3 DAG
PKC Ras
Raf
MEKsMEK1/2
Rac1 STAT
Src
βγ α
Akt/PKB
p70S6k
GSK3β
Transcripción
Calcineurina CaMKII ERK 1/2
GATA 4 MEF-2
Elk1
p53BaxBNPANF βMHC
AP-1NFAT
GATA 4
MEF-2 NFκB
NFAT
p38MAPK JNK

Señales intracelulares que siguen al estiramiento miocárdico...
179
β favorecien-β en su estado
activo fosforila e inhibe a varias moléculas vinculadas
Akt promueve la sobrevida celular al inhibir en múltiples β también se
fosforilan en respuesta a estímulos mecánicos a partir de -
β1. 26 La melusina ha sido
26
fracasan en el desarrollo normal del crecimiento de los
26
Activación de la vía gp130-
-
evidencia que sustenta un papel citoprotector para la vía
el desarrollo de HC. 27-29
Estiramiento miocárdico: señales que inician la hipertrofia
--
mediante los que es posible adaptar el volumen minuto cardiaco a las variaciones en las condiciones hemodi-
-
30, en
inicial. Esto constituye el bien conocido mecanismo de
o, para eyectar el mismo volumen latido contra una mayor
-
aumento en la
-
y sus colaboradores 31
efecto efec-
-recho aislado de perro. Estos autores denominaron a este mecanismo colaboradores 32 autorregu-
33 reprodujeron por primera vez el efecto contráctil del estiramiento en tiras aisladas de ventrículo miocárdico. Estos autores mostraron que
-tos correspondientes rápidos y lentos de la fuerza desa-rrollada. El cambio rápido en la fuerza se piensa que es la base del mecanismo de
Ca
de cada -
estaba presente en músculos aislados de animales reser-
Figura 11.2: Cuando cardiomiocitos aislados de ratas neonatas son cultivados sobre placas de silicona y sometidos a estiramiento puede recuperarse del medio en que están cultivados angiotensina II y endotelina, siendo ambas moléculas importantes eslabones de la cadena de eventos autocrina/paracrina, que culmina con el aumento de la síntesis proteica, la reexpresión de genes del patrón fetal y el desarrollo de hipertrofia cardiaca.
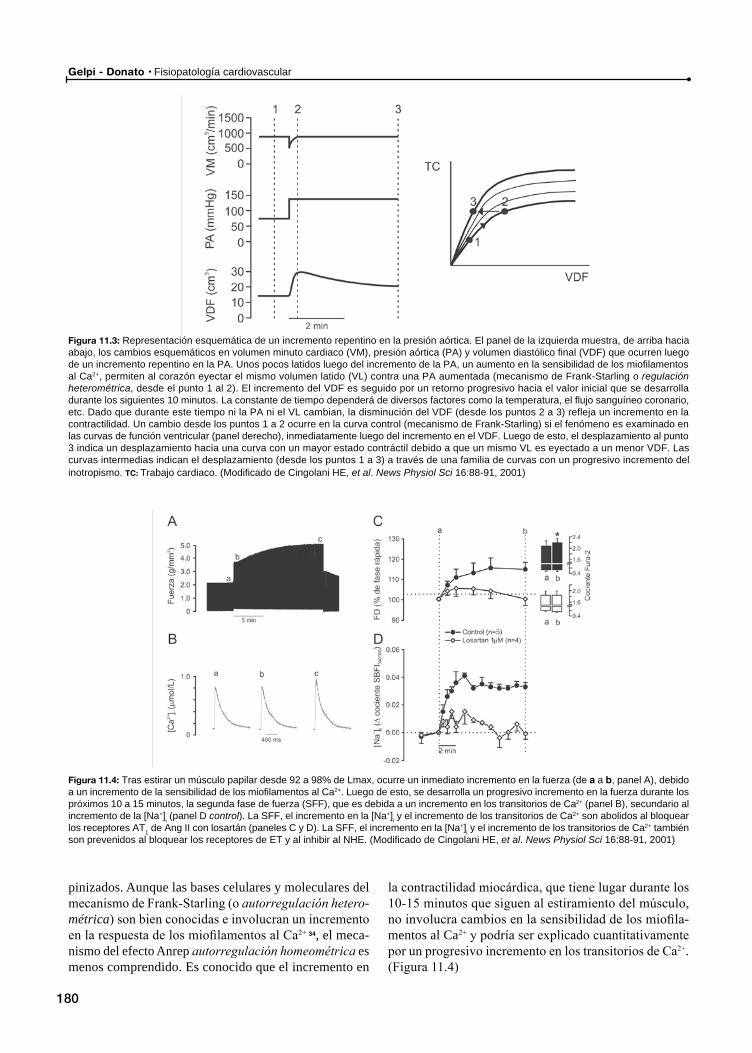
Gelpi - Donato Fisiopatología cardiovascular
180
Figura 11.3: Representación esquemática de un incremento repentino en la presión aórtica. El panel de la izquierda muestra, de arriba hacia abajo, los cambios esquemáticos en volumen minuto cardiaco (VM), presión aórtica (PA) y volumen diastólico final (VDF) que ocurren luego de un incremento repentino en la PA. Unos pocos latidos luego del incremento de la PA, un aumento en la sensibilidad de los miofilamentos al Ca2+, permiten al corazón eyectar el mismo volumen latido (VL) contra una PA aumentada (mecanismo de Frank-Starling o regulación heterométrica, desde el punto 1 al 2). El incremento del VDF es seguido por un retorno progresivo hacia el valor inicial que se desarrolla durante los siguientes 10 minutos. La constante de tiempo dependerá de diversos factores como la temperatura, el flujo sanguíneo coronario, etc. Dado que durante este tiempo ni la PA ni el VL cambian, la disminución del VDF (desde los puntos 2 a 3) refleja un incremento en la contractilidad. Un cambio desde los puntos 1 a 2 ocurre en la curva control (mecanismo de Frank-Starling) si el fenómeno es examinado en las curvas de función ventricular (panel derecho), inmediatamente luego del incremento en el VDF. Luego de esto, el desplazamiento al punto 3 indica un desplazamiento hacia una curva con un mayor estado contráctil debido a que un mismo VL es eyectado a un menor VDF. Las curvas intermedias indican el desplazamiento (desde los puntos 1 a 3) a través de una familia de curvas con un progresivo incremento del inotropismo. TC: Trabajo cardiaco. (Modificado de Cingolani HE, et al. News Physiol Sci 16:88-91, 2001)
Figura 11.4: Tras estirar un músculo papilar desde 92 a 98% de Lmax, ocurre un inmediato incremento en la fuerza (de a a b, panel A), debido a un incremento de la sensibilidad de los miofilamentos al Ca2+. Luego de esto, se desarrolla un progresivo incremento en la fuerza durante los próximos 10 a 15 minutos, la segunda fase de fuerza (SFF), que es debida a un incremento en los transitorios de Ca2+ (panel B), secundario al incremento de la [Na+]i (panel D control). La SFF, el incremento en la [Na+]i y el incremento de los transitorios de Ca2+ son abolidos al bloquear los receptores AT1 de Ang II con losartán (paneles C y D). La SFF, el incremento en la [Na+]i y el incremento de los transitorios de Ca2+ también son prevenidos al bloquear los receptores de ET y al inhibir al NHE. (Modificado de Cingolani HE, et al. News Physiol Sci 16:88-91, 2001)
pinizados. Aunque las bases celulares y moleculares del -
métrica34, el meca-
nismo del efecto Anrep es menos comprendido. Es conocido que el incremento en
-mentos al Ca
.

Señales intracelulares que siguen al estiramiento miocárdico...
181
Ca
de Ca mediado por el intercambiador Na /Ca
Na /H 35-41 Trabajos
incremento en el transitorio de Ca , ni por un incremento desde el retículo sarcoplásmico42,
ni por un incremento en la corriente de Ca del tipo L. 43
-
-carbonato. 35
la respuesta contráctil provocada por el estiramiento, pero se pudo de detectar y caracterizar una cascada de eventos autocrinos/paracrinos que conducen al incremento de la
-20 así como la de
ET 44 por los cardiomiocitos de ratas neonatas sometidos a estiramiento, ya había sido descrita. La mayor contri-
-ciones multicelulares de corazones de animales adultos la
por el estiramiento. 35 -
lo referido a los receptores de membrana y a las vías de
relevancia para conocer los mecanismos involucrados en el desarrollo de la HC.
iun H intracelular por un Nadel NHE inducida por estiramiento podría potencialmente incrementar la fuerza por dos mecanismos: el incremen-to en el pHi –lo que aumenta la sensibilidad de los mio-
i –que a través del NCX operando en
de estiramiento se realizan en presencia de medios que -
cativos en el pHi. 35,37
cambios en el pHi se puede encontrar en el hecho de que
del pHiintercambiador Cl--HCO - Na 44, 45 La
al NHE y al AE, por lo tanto minimiza los cambios en el pHi -
ipuede ser detectada por un incremento en el pHibicarbonato está ausente del medio, pero un aumento en
i
i puede inducir un aumento en los niveles intracelulares de Ca a través del
de Cala entrada de Ca
37, 42
de Ca debiera ser mediado por el modo inverso del NCX i ,
del NHE.37i desplaza
NCX
a la célula, determinando el incremento i
46 Te-i y considerando que
inM, el cálculo de ENCX NCX Na Ca
-brana positivos al ENCX -verso de este transportador. Debido a que la célula cardiaca normalmente alcanza aquel valor de ENCX
-
NCX participaría muy poco o nada de la contractilidad basal.
NCX modo inverso a la contractilidad basal de músculos pa-36, 46, 47
i
36, 46 ,47 llevan al ENCXesta manera el funcionamiento del modo inverso del NCX
i el único mecanismo responsable
i inducido por estiramiento y por
NHE. 36, 37, 46, 47 -ría que este mecanismo es el único determinante del efecto
-i -
a través del NCX en el modo inverso. Acerca de este punto, evidencias recientes demuestran que, a pesar de que el bloqueo del modo inverso
i/K ATPasa, para un nivel
i, el aumento en la fuerza de-
del NCX fue inducido por ET-1, que cuando la bomba de
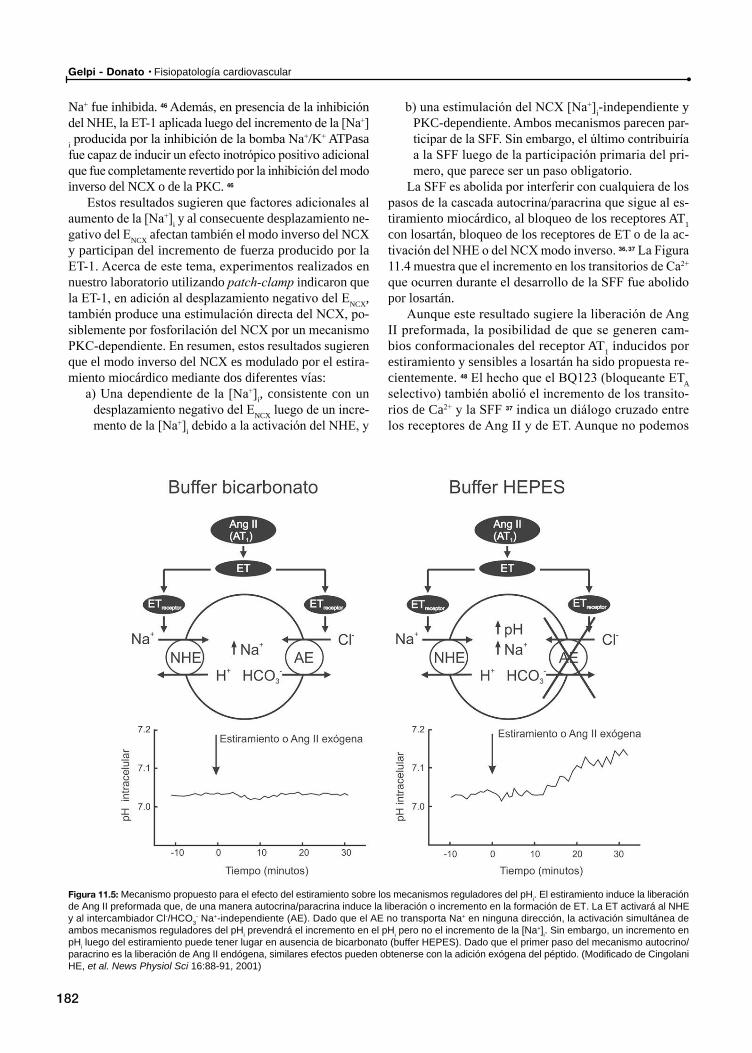
Gelpi - Donato Fisiopatología cardiovascular
182
Na fue inhibida. 46
i /K ATPasa
inverso del NCX o de la PKC. 46
i y al consecuente desplazamiento ne-NCX afectan también el modo inverso del NCX
y participan del incremento de fuerza producido por la
nuestro laboratorio utilizando indicaron que NCX,
-
que el modo inverso del NCX es modulado por el estira-miento miocárdico mediante dos diferentes vías:
i, consistente con un NCX -
i
i-independiente y PKC-dependiente. Ambos mecanismos parecen par-
-
-tiramiento miocárdico, al bloqueo de los receptores AT1 con losartán, bloqueo de los receptores de ET o de la ac-
36, 37
por losartán.
-bios conformacionales del receptor AT1 inducidos por estiramiento y sensibles a losartán ha sido propuesta re-cientemente. 48
A -
rios de Ca 37
Figura 11.5: Mecanismo propuesto para el efecto del estiramiento sobre los mecanismos reguladores del pHi. El estiramiento induce la liberación de Ang II preformada que, de una manera autocrina/paracrina induce la liberación o incremento en la formación de ET. La ET activará al NHE y al intercambiador Cl-/HCO3
- Na+-independiente (AE). Dado que el AE no transporta Na+ en ninguna dirección, la activación simultánea de ambos mecanismos reguladores del pHi prevendrá el incremento en el pHi pero no el incremento de la [Na+]i. Sin embargo, un incremento en pHi luego del estiramiento puede tener lugar en ausencia de bicarbonato (buffer HEPES). Dado que el primer paso del mecanismo autocrino/paracrino es la liberación de Ang II endógena, similares efectos pueden obtenerse con la adición exógena del péptido. (Modificado de Cingolani HE, et al. News Physiol Sci 16:88-91, 2001)

Señales intracelulares que siguen al estiramiento miocárdico...
183
-perimentos de 49
-
-cárdico.
Los resultados descritos hasta aquí enfatizan el hecho
-
Caun lento incremento en la cantidad de Ca disponible en el
potencialmente importante desde el punto de vista clínico,
El papel que cumple el NHE en el mecanismo que
trabajando en distintas especies. 37, 39-41, 50
es controvertida. El papel de la ET ha sido parcialmen-38
1, aunque descrito en miocitos aislados 20, 51
38 no pudieron demostrar la
-
39
41, 50
Intercambiador Na+/H+ e hipertrofia miocárdica
Aunque el estiramiento miocárdico ha sido ampliamente
-1 y los
del NHE. 52 esquemática de nuestra propuesta, basada principalmente en resultados obtenidos en preparaciones multicelulares cardiacas sometidas a estiramiento.
-
53-57i es bien reconocido como
-cineurina o de la CaMKII. En cualquier caso, el aumento
i i podría ser la consecuencia de la -
Figura 11.6: Representación esquemática de la cascada de eventos propuesta luego del estiramiento miocárdico. La Ang II endógena es liberada desde los miocitos activando de manera autocrina los receptores AT1. La estimulación de los receptores AT1 induce la liberación/formación de ET, que activará simultáneamente al NHE y al AE a través de los receptores de ET. La estimulación del AE previene el esperado incremento en el pHi mediado por el NHE, pero no previene el incremento en la [Na+]i. El incremento en la [Na+]i posiblemente llevará al intercambiador Na+/Ca2+ (NCX) a operar en su modo inverso, determinando el incremento en el transitorio de Ca2+. Como se describió en el texto, una estimulación directa [Na+]i-independiente del NCX también puede contribuir al incremento de fuerza. Nótese que aunque en este esquema se identifica al mecanismo inducido por estiramiento como autocrino, no podemos descartar la posibilidad de que las células endoteliales o los fibroblastos contribuyan de un modo paracrino. (Modificado de Cingolani HE, et al. News Physiol Sci 16:88-91, 2001)

Gelpi - Donato Fisiopatología cardiovascular
184
Nosotros queremos enfatizar que no proponemos que el
i y 58-59 Incluso, se ha descrito recientemente que
i. 60 -
-
-actividad del NHE en el miocardio de ratas espontáneamente
i. 61
El hecho de que una actividad elevada del NHE no fue i fue difícil de inter-
pretar, porque la actividad de este intercambiador ha sido i que a otros
eventos celulares. Más aún, el estudio seminal realizado en
i62,
-i puede incrementar-
se o disminuir con factores de crecimiento, dependiendo de la ausencia o presencia de bicarbonato en el medio63,
64
i son o no paralelos a los cambios en el pHiincremento en la actividad del NHE, los mecanismos de-
Na /Ki
i no detectados por la bomba Na /K ATPasa son su-65
En resumen hemos descrito:
en el pHi61
i pero no en el pHi44, 45
-44 y del NHE. 35
i en presencia de bicarbonato. 36, 37
-tiramiento miocárdico, cuando la actividad del AE
-66
-67, 68 Todas estas
i
y muestran qué factores de crecimiento pueden ac-i aun cuando el
pHiPutney y Barber 69
i en
/M siendo por lo tanto
i induzca un incremento en la síntesis de proteínas, con
i i.
Inhibición del intercambiador Na+/H+ y regresión de la hipertrofia cardiaca
Dado que la actividad del NHE está incrementada en el 61, se
-
70
--
Figura 11.7: Vías intracelulares hipertróficas hipotéticas disparadas por el estiramiento miocárdico. Ver texto para las abreviaturas y referencias.

Señales intracelulares que siguen al estiramiento miocárdico...
185
miocitos sobrevivientes. Kusumoto et al. demostraron que
del infarto. 58 et al. demostraron
el tratamiento con cariporide. 53 -
con losartán, enalapril o nifedipina 71, y también con el 53 Este último estudio
-
72, aunque este
a un efecto relacionado con la apoptosis. 73, 74 Además, la
β54, 55
-diaca ha sido establecido también para diversos modelos
Ennis et al. 55
hipertiroideas poseen actividad del NHE incrementada 75
--
76
ventricular izquierda y la actividad de la hormona parathor-mona ha sido descrito en pacientes con enfermedad renal terminal e hiperparatiroidismo secundario. 77 Dado que la actividad del NHE puede ser modulada por la parathormona 78
del NHE en cardiomiocitos de ratas neonatas. 56 Es inte-
-tividad de la MAPK se mantuvo elevada.56
-i o el pHi
i i induce di-
79 Diversos factores de crecimiento
-
NHE. 80, 81
estudiado, la leptina, la proteína plasmática secretada por -
82
i y la i
83, su potencial uso --
fueron oscurecidos por una elevada incidencia de acciden-
fue interrumpido. 84 No se conoce si el retiro abrupto del -
-poride. 85 Es desconocido para nosotros, por el momento,
por otros inhibidores del intercambiador.
Bibliografía
1. Levy D, Garrison RJ, Savage DD. “Prognostic implications of echocardiographically determined left ventricular mass in the Framingham heart study . N Engl J Med. 1990;322:1561-1566.
2. Mathew J, Sleight P, Lonn E, “Heart Outcomes Prevention Eva-luation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ra-mipril . Circulation 2001;104:1615-1621.
3. Verdecchia P, Schillaci G, Borgioni C. “Prognostic significance of serial changes in left ventricular mass in essential hypertension . Circulation 1998;97:48-54.
4. McKinsey TA, EN Olson. “Cardiac hypertrophy: sorting out the circuitry . Curr Opin Genet Dev. 1999;9:267-274.
5. Sadoshima J, S Izumo. “The cellular and molecular respon-se cardiac myocytes to mechanical stress . Annu Rev Physiol. 1997;59:551-571.
6. Chien, KR. “Stress pathways and heart failure . Cell. 1999;98:555-558.
7. MacLellan WR, MD Schneider. “Success in failure: mode-ling cardiac decompensation in transgenic mice . Circulation 1998;97:1433-1435.
8. Sei CA, Irons CE, Sprenkle AB. “The α-adrenergic stimulation of atrial natriuretic factor expression in cardiac myocytes requires calcium influx, protein kinase C, and calmodulin-regulated pathways . J Biol Chem. 1991;266:15910-15916.
9. Sonnenberg, H. “Mechanism of release and renal tubular action of atrial natriuretic factor . Fed Proc. 1986;45:2106-2110.
10. LaPointe MC, Deschepper CF, Wu JP, Gardner DG. “Extracellular calcium regulates expression of the gene for atrial natriuretic factor . Hypertension 1990;15:20-28.

Gelpi - Donato Fisiopatología cardiovascular
186
11. Shen M, Zhao Q, Solaro J. “A novel calcium-independent pathway mediates α1-adrenergic receptor-stimulated hypertrophy in adult rat cardiac myocytes . Circulation 110:III-92 (Abstract), 2004.
12. Zhang W, Kowal RC, Rusnak F. “Failure of calcineurin inhibitors to prevent pressure-overload left ventricular hypertrophy in rats . Circ Res. 84:722-728, 1999.
13. Ding B, Price RL, Borg TK. “Pressure overload induces severe hypertrophy in mice treated with cyclosporine, an inhibitor of calcineurin . Circ Res. 84:729-734, 1999.
14. Molkentin JD, Lu JR, Antos CL. “A calcineurin-dependent trans-criptional pathway for cardiac hypertrophy . Cell. 93:215-228, 1998.
15. Sakata Y, Masuyama T, Yamamoto K. “Calcineurin inhibitor atte-nuates left ventricular hypertrophy, leading to prevention of heart failure in hypertensive rats . Circulation. 102:2269-2275, 2000.
16. Meguro T, Hong C, Asai K. “Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure . Circ Res. 84:735-740, 1999.
17. Oie E, Bjornerheim R, Clausen OP. “A inhibits cardiac hypertrophy and enhances cardiac dysfunction during postinfarction failure in rats . Am J Physiol. 278:H2115-H2123, 2000.
18. Rothermel BA, McKinsey TA, Vega RB. “Myocyte-enriched calci-neurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo . Proc Natl Acad Sci USA 98:3328-3333, 2001
19. Hill JA, Rothermel B, Yoo KD.“Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function . J Biol Chem 277:10251-10255, 2002.
20. Sadoshima J, Xu Y, Slayter. “Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro . Cell. 1993;75:977-984.
21. Shubeita HE, McDonough PM, Harris AN. “Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy . J Biol Chem. 265:20555-20562, 1990.
22. Maruyama Y, Nishida M, Sugimoto Y. “Galpha(12/13) mediates alpha(1)-adrenergic receptor-induced cardiac hypertrophy . Circ Res. 91:961-969, 2002.
23. Zou Y, Yao A, Zhu W. “Isoproterenol activates extracellular signal-regulated protein kinases in cardiomyocytes through calcineurin . Circulation 104:102-108, 2001.
24. Braun M, Simonis G, Birkner K. “Regulation of protein kinase C isozyme and calcineurin expression in isoproterenol induced cardiac hypertrophy . J Cardiovasc Pharmacol. 41:946-954, 2003.
25. Oudit GY, Crackower MA, Eriksson U. “Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure . Circulation 108:2147-2152, 2003.
26. Brancaccio M, Fratta L, Notte A. “Melusin, a muscle specific- integrin b1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload . Nature Medicine 2003;9:68-75.
27. Yoshida K, Taga T, Saito M. “Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders . Proc Natl Acad Sci USA 93:407-411, 1996.
28. Kunisada K, Negoro S, Tone E. “Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced car-diomyopathy . Proc Natl Acad Sci USA 97:315-319, 2000.
29. Jacoby JJ, Kalinowski A, Liu MG. “Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age . Proc Natl Acad Sci USA 100:12929-12934, 2003.
30. Von Anrep, G. “On the part played by the suprarenals in the normal vascular reactions on the body . J Physiol. 1912;45:307-317.
31. Rosenblueth A, Alanais J, Lopez E. “The adaptation of ventricular muscle to different circulatory conditions . Arch Int Physiol Bio-chem. 1959;67:358-373.
32. Sarnoff SJ, Mitchell JH, Gilmore JP. “Homeometric autoregulation in the heart . Circ Res. 1960;8:1077-1091.
33. Parmley WW, L Chuck. “Length-dependent changes in myocardial contractile state . Am J Physiol. 1973;224:1195-1199.
34. Solaro, J. “Mechanism regulating cardiac myofilament response to calcium . En: Heart Physiology and Pathophysiology. Fourth Edition. Eds. Speralakis, Kurachi, Terzic, Cohen. Academic Press, San Diego, 2001 519-526.
35. Cingolani HE, Alvarez BV, Ennis IL. “Stretch-induced alkalini-zation of feline papillary muscle. An autocrine-paracrine system . Circ Res. 1998;83:775-780.
36. Pérez NG, Camilión de Hurtado MC, Cingolani HE. “Reverse mode of the Na+/Ca2+ exchange following myocardial stretch. Underlying mechanism of the slow force response . Circ Res. 2001;88:376-382.
37. Alvarez BV, Pérez NG, Ennis IL. “Mechanisms underlying the increase in force and calcium transient that follows stretch of car-diac muscle: A possible explanation of the Anrep effect . Circ Res. 1999;85:716-722.
38. Calaghan SC, E White. “Contribution of angiotensin II, endothelin 1 and the endothelium to the slow inotropic response to stretch in ferret papillary muscle . Pflügers Arch. 2001;441:514-520.
39. Calaghan S, E White. “Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart . J Physiol. 2004;559:205-214.
40. von Lewinski D, Stumme B, Fialka F. “Functional relevance of the stretch-dependent slow force response in failing human myo-cardium . Circ Res. 2004;94:1392-1398.
41. von Lewinski D, Stumme B, Maier LS. “Stretch-dependent slow force response in isolated rabbit myocardium is Na+ dependent . Cardiovasc Res. 2003;57:1052-1061.
42. Kentish JC, A Wrzosek. “Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular tra-beculae . J Physiol. 1998; 506:431-444.
43. Hongo K, White E, Le Guennec J-Y. “Changes in [Ca2+]i, [Na+]
i
and Ca2+ current in isolated rat ventricular myocytes following an increase in cell length . J Physiol. 1996;491:609-619.
44. Camilión de Hurtado MC, Alvarez BV, Ennis IL. “Stimulation of myocardial Na+-independent Cl-/HCO
3- exchanger by angiotensin
II is mediated by endogenous endothelin . Circ Res. 2000;86:622-627.
45. Camilión de Hurtado MC, Alvarez BV, Pérez NG. “Angiotensin II activates Na+-independent Cl-/HCO
3- exchange in ventricular
myocardium . Circ Res. 1998;82:473-481. 46. Aiello EA, Villa-Abrille MC, Dulce RA. “Endothelin-1 stimulates
the reverse mode of the Na+/Ca2+ exchanger through Na+i-depen-
dent and Na+i-independent pathways . Hypertension 2005;45:1-7.
47. Pérez NG, Villa-Abrille MC, Aiello EA, Camilión de Hurtado MC. “A low dose of angiotensin II increases inotropism through activation of reverse Na+/Ca2+ exchange by endothelin release . Cardiovasc Res. 2003;60:589-597.
48. Zou Y, Akazawa H, Qin Y. “Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II . Nature Cell Biol. 2004;6:499-506.
49. Aiello EA, Villa-Abrille MC, Cingolani HE. “Autocrine stimulation of cardiac Na+/Ca2+ exchanger currents by endogenous endothelin released by angiotensin II . Circ Res. 2002;90:374-376.
50. Yamazaki T, Komuro I, Kudoh S. “Role of ion channels and exchan-gers in mechanical stretch-induced cardiomyocyte hypertrophy . Circ Res. 1998;82:430-437.
51. Leri A, Claudio PP, Li Q. “Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell . J Clin Invest. 1998;101:1326-1342.
52. Cingolani, HE. “Na+/H+ exchange hyperactivity and myocar-dial hypertrophy: are they linked phenomena? Cardiovasc Res. 1999;44:462-467.

Señales intracelulares que siguen al estiramiento miocárdico...
187
53. Camilion de Hurtado MC, Portiansky EL, Perez NG. “Regression of cardiomyocyte hypertrophy in SHR following chronic inhibition of the Na+/H+ exchanger . Cardiovasc Res. 2002;53:862-868.
54. Engelhardt S, Hein L, Keller U. “Inhibition of Na+/H+ exchange prevents hypertrophy, fibrosis, and heart failure in beta 1-adrenergic receptor transgenic mice . Circ Res. 2002;90:814-819.
55. Ennis IL, Escudero EM, Console GM. “Regression of isoproterenol-induced cardiac hypertrophy by Na+/H+ exchanger inhibition . Hypertension 2003;41:1324-1329.
56. Karmazyn M, Liu Q, Gan XT. “Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes . Hypertension 2003;42:1171-1176.
57. Kusumoto K, Haist JV, Karmazyn M. “Na+ /H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats . Am J Physiol. 2001;280:H738-H745.
58. Hefti MA, Harder BA, Eppenberger HM. “Signaling pathways in cardiac myocyte hypertrophy . J Mol Cell Cardiol. 1997;29:2873-2892.
59. Schluter RD, HM Piper. “Regulation of growth in the adult car-diomyocytes . FASEB J. 1999;13:S17-S22.
60. Shen M, Zhao Q, Solaro J. “A novel calcium-independent pathway mediates α
1-adrenergic receptor-stimulated hypertrophy in adult
rat cardiac myocytes . Circulation 2004;110:III-92 (Abstract). 61. Pérez NG, Alvarez BV, Camilión de Hurtado MC. “pH
i regula-
tion in myocardium of the spontaneously hypertensive rat. Com-pensated enhanced activity of the Na+-H+ exchanger . Circ Res. 1995;77:1192-1200.
62. Epel, D. “The role of Na+/H+ exchange and intracellular pH chan-ges in fertilization . En: Grinstein S, editor. Na+/H+ exchange. Florida: CRC Press 209-223; 1998.
63. Boron WF, Boyarski G, Ganz M. “Regulation of intracellular pH in renal mesangial cells . Ann N Y Acad Sci. 1989;574:321-332.
64. Thomas, RC. “Bicarbonate and pHi response . Nature
1989;337:601. 65. Bluhm WF, Lew WY, Garfinkel A. “Mechanism of length history-
dependent tension in an ionic model of the cardiac myocyte . Am J Physiol. 1998;274:H1032-H1040.
66. Cingolani HE, Chiappe de Cingolani GE, Ennis IL. “Influence of Na+-independent Cl-/HCO
3- exchange on the slow force response
to myocardial stretch . Circ Res. 2003;93:1082–1088. 67. Chiappe de Cingolani G, Morgan P, Mundina-Weilenmann C.
“Hyperactivity and altered mRNA isoform expression of the Cl-/HCO
3- anion-exchanger in the hypertrophied myocardium . Car-
diovasc Res. 2001;51:71-79. 68. Farias F, Morgan P, Chiappe de Cingolani GE. “Involvement of
the AE3 isoform of the Na+-independent Cl-/HCO3- exchange in
the compensation of myocardial NHE-1 hyperactivity in SHR . Can J Physiol Pharmacol 83:397-404;2005.
69. Putney LK, DL Barber. “Na-H exchange-dependent increase in intracellular pH times G
2/M entry and transition . J Biol Chem.
2003;278:44645-44649. 70. Cingolani HE, MC Camilion de Hurtado. “Na+-H+ exchanger inhi-
bition: a new antihypertrophic tool . Circ Res. 2002;90:751-753.
71. Alvarez BV, Ennis IL, Camilión de Hurtado MC. “Effects of anti-hypertensive therapy on cardiac sodium/hydrogen ion exchanger activity and hypertrophy in spontaneously hypertensive rats . Can J Cardiol. 2002;18:667-672.
72. Cingolani HE, Rebolledo OR, Portiansky EL. “Regression of hypertensive myocardial fibrosis by Na+/H+ exchange inhibition . Hypertension 2003;41:373-377.
73. Teshima Y, Akao M, Jones SP. “Cariporide (HOE 642), a selecti-ve Na+-H+ Exchange inhibitor, inhibits the mitochondrial death pathway . Circulation 2003;108: 2275-2281.
74. Chakrabarti S, Hoque AN, Karmazyn M. “A rapid ischemia-induced apoptosis in isolated rat hearts and its attenuation by the sodium-hydrogen exchange inhibitor HOE 642 (cariporide) . J Mol Cell Cardiol. 1997;29:3169-3174.
75. Bak MI, JC Ingwall. “Contribution of Na+/H+ exchange to Na+ overload in the ischemic hypertrophied hyperthyroid rat heart . Cardiovasc Res. 2003;57:1004-1014.
76. Shohet RV, Kisanuki YY, Zhao XS. “Mice with cardiomyocy-te-specific disruption of the endothelin-1 gene are resistant to hyperthyroid cardiac hypertrophy . Proc Natl Acad Sci USA 2004;101:2088-2093.
77. Saleh FN, Schirmer H, Sundsfjord J. “Parathyroid hormone and left ventricular hypertrophy . Eur Heart J. 2003;24:2054-2060.
78. Helmle-Kolb C, Counillon L, Roux D. “Na/H exchange activi-ties in NHE1-transfected OK-cells: cell polarity and regulation . Pflugers Arch. 1993;425:34-40.
79. Pastukh VM, Wu S, Ricci C. “Reversal of Hyperglycemic pre-conditionng by Angiotensin II: role of calcium transport . Am J Physiol. Am J. Physiol 288:H1965-1975; 2004
80. Rothstein EC, Byron KL, Reed RE. “H2O
2-induced Ca2+ overload
in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway . Am J Physiol. 2002;283:H598-H605.
81. Snabaitis AK, Hearse DJ, Avkiran M. “Regulation of sarcolemmal Na+/H+ exchange by hydrogen peroxide in adult rat ventricular myocytes . Cardiovasc Res. 2002;53:470-480.
82. Xu FP, Chen MS, Wang YZ. “Leptin induces hypertrophy via Endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes . Circulation 2004;110:1269-1275.
83. An J, Varadarajan SG, Camara A. “Blocking Na+/H+ exchange re-duces [Na+]
i and [Ca2+]
i load after ischemia and improves function
in intact hearts . Am J Physiol. 2001;281:H2398-H2409. 84. Theroux P, Chaitman BR, Danchin N. “Inhibition of the so-
dium-hydrogen exchanger with cariporide to prevent myocardial infarction in high -risk ischemic situations. Main results of the GUARDIAN trial . Circulation 2000;102:3032-3038.
85. Camilión de Hurtado MC, Ennis IL, Pérez NG. “Upregulation of myocardial Na+/H+ exchanger induced by chronic treatment with a selective inhibitor . J Mol Cell Cardiol. 2002;34:1539-1547.
86. Rugale C, Delbose S, Cristol J. “Sodium restriction prevents cardiac hypertrophy and oxidative stress in angiotensin II hypertension . Am J Physiol. 2003;284:H1744-H1750.


189
cardiaca son discutidos desde hace mucho tiempo. En el
-te de la del miocardio normal. 1
en tejidos obtenidos post mortem
confocal y con la capacidad de preservar viable el tejido -
2-4 -
-
de los β
hipotetizamos que estos cambios funcionales son cau-
los túbulos T del sarcolema y sistema reticuloendotelial, mitocondria, discos intercalares, citoplasma y microtú-
como tres factores importantes responsables de la re-
El remodelamiento es un término utilizado en la litera-
Cohn et al
5 Nosotros, respetaremos este término en
Estructura normal del miocito y del intersticio
-
μ μm de ,
μm . 6 El tejido inters-ticial representa el esqueleto, que provee estabilidad a los miocitos. Además, contiene a la
-
7-9
--
tos son mononucleados.10 El núcleo muestra una forma
un nucleolo prominente. Los miocitos están constituidos
endoplásmico.11, 12
su actividad continua durante toda la vida del individuo.
El miocito en la insuficiencia cardiaca
miocitos presentan
μmμm
μm.
μm es característica de un cardiomiocito normal, μm
miocitos. 6
Características estructurales del remodelamiento cardiaco en el desarrollo
de la insuficiencia cardiaca12
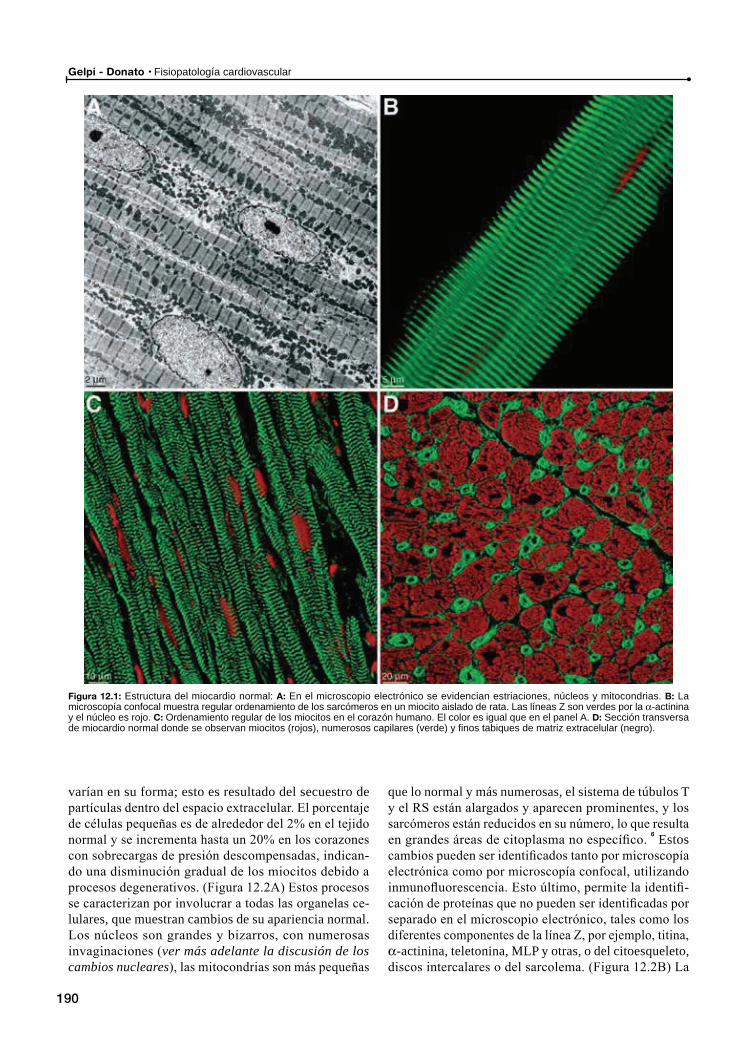
Gelpi - Donato Fisiopatología cardiovascular
190
varían en su forma; esto es resultado del secuestro de
-
-lulares, que muestran cambios de su apariencia normal.
ver más adelante la discusión de los cambios nucleares
que lo normal y más numerosas, el sistema de túbulos T
6 Estos
-
α-actinina, teletonina, MLP y otras, o del citoesqueleto,
Figura 12.1: Estructura del miocardio normal: A: En el microscopio electrónico se evidencian estriaciones, núcleos y mitocondrias. B: La microscopía confocal muestra regular ordenamiento de los sarcómeros en un miocito aislado de rata. Las líneas Z son verdes por la α-actinina y el núcleo es rojo. C: Ordenamiento regular de los miocitos en el corazón humano. El color es igual que en el panel A. D: Sección transversa de miocardio normal donde se observan miocitos (rojos), numerosos capilares (verde) y finos tabiques de matriz extracelular (negro).

Características estructurales del remodelamiento cardíaco...
191
celular y del intersticio. 13
El término degeneración ha sido casi borrado de la lite-14
cambios estructurales en los miocitos que indican deterioro
--
6
-
ambas alternativas son posibles
Cambios nucleares
El núcleo es especialmente importante para la sobrevida de la célula, por esto, sus alteraciones son discutidas en
Figura 12.2: Degeneración de los miocitos y fibrosis. A: Miocitos (rojo) de diferentes tamaños y formas (flechas), mucho más pequeños que los normales, se encuentran rodeados por grandes áreas de matriz extracelular (negro) indicando la presencia de fibrosis. B: α-actinina (verde, los núcleos son rojos) muestra importante desorganización de la estructura sarcomérica.
Figura 12.3: Degeneración de los miocitos en el microscopio electrónico. A: Pérdida de material contráctil y numerosos fragmentos nucleares Barra=5μm. B: Los núcleos presentan múltiples invaginaciones. Barra=2μm.
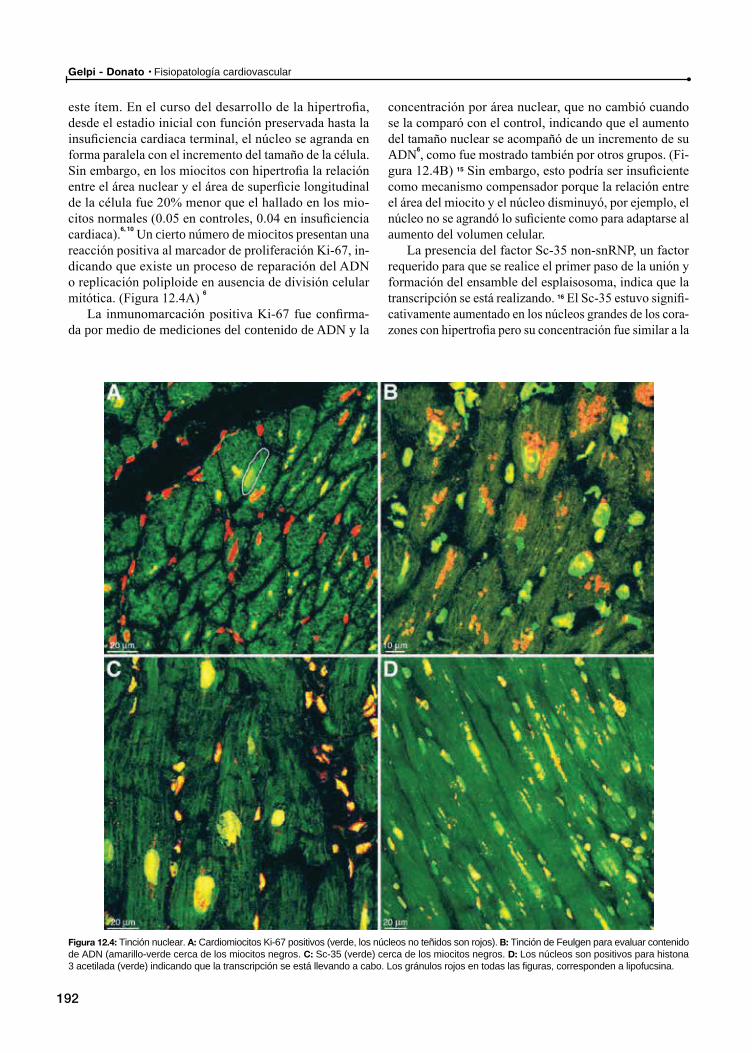
Gelpi - Donato Fisiopatología cardiovascular
192
-
6, 10 Un cierto número de miocitos presentan una -
6
-da por medio de mediciones del contenido de ADN y la
ADN6 -15
aumento del volumen celular.
16 --
Figura 12.4: Tinción nuclear. A: Cardiomiocitos Ki-67 positivos (verde, los núcleos no teñidos son rojos). B: Tinción de Feulgen para evaluar contenido de ADN (amarillo-verde cerca de los miocitos negros. C: Sc-35 (verde) cerca de los miocitos negros. D: Los núcleos son positivos para histona 3 acetilada (verde) indicando que la transcripción se está llevando a cabo. Los gránulos rojos en todas las figuras, corresponden a lipofucsina.

Características estructurales del remodelamiento cardíaco...
193
6
-
ocurre en los cardiomiocitos puesto que contienen un nú-
puede asumir que en los estadios precoces de la enfer-medad, probablemente en la primera década, el conteni-
-
de los miocitos.
Pérdida de miocitos por muerte celular
6, 17 -μm , mientras que en corazones
ver más adelante 6, 13 Tres formas diferentes de muerte celular deben ser con-
sideradas: apoptosis, oncosis, y la muerte celular autofá-
Apoptosis: es un tipo de muerte celular suicida pro-
método del TUNEL y otras técnicas. Una característica
las mitocondrias y el sarcolema, mientras que el núcleo
que murieron por apoptosis se realiza a través de los ma-18
Por el contrario, en la oncosis las células muertas son eliminadas por los leucocitos y linfocitos durante un pro-
Oncosis: es un tipo de muerte celular accidental, en la
-meabilidad, la célula se presenta edematosa. La oncosis
injuria isquémica, realizando un análisis ultraestructural.19,
20 Además, dado que la membrana celular permite el paso
-cencia con un anticuerpo anti-C9. 21 El término necrosis es utilizado con frecuencia para describir la muerte celular por isquemia. Nosotros y otros autores, preferimos el término oncosis y reservamos la palabra necrosis para describir los
cuando la célula ha pasado el punto de no retorno.Muerte celular autofágica: también ha sido denomi-
nada . Es depen-diente de ATP como la apoptosis, pero es independiente
-
-
Figura 12.5: Muerte celular en el miocardio insuficiente. A. Núcleos apoptóticos (verde) en cardiomiocitos (rojo). B: Muerte celular oncótica identificada por deposición de C9 (verde, miocitos son rojos, núcleos azules). C: Muerte celular autofágica identificada por marcación de ubiquitina (verde, cardiomiocitos son rojos, núcleos azules)

Gelpi - Donato Fisiopatología cardiovascular
194
proteínas está involucrado en este tipo de muerte celular.
proteínas en los tejidos.22, 23 Normalmente, las proteínas se acoplan a la ubiquitina a través de una serie de enzimas
-tos complejos son transportados a través de la célula, para que isopeptidasas y otros sistemas hidrolíticos eliminen la
Este tiene una estructura cilíndrica que cliva proteínas en péptidos y aminoácidos. La ubiquitina es reciclada para ser nuevamente utilizada.
-cionada con ubiquitina ocurre en condiciones neurode-
o Alzheimer 24, 25 y ha sido recientemente descrita en co-razones humanos. 26, 27 El estadio preliminar consiste en
-cleo y, por último, de la célula completa. 28, 29 Las vacuolas
27 Por otro lado, los complejos ubiquitina son positivos
27 En un análisis
-
proteína y muerte celular. 27 El contenido y la actividad de
de su valor control. Por lo tanto, podemos concluir que
complejos ubiquitina/proteína llevan a la muerte de los
es causado por un incremento de la actividad de la enzi-
capacidad de eliminar la ubiquitina.
-
corazones normales. Antes de que sea determinada la im-portancia real de estas tasas de muerte celular, se deben
-ble asumir que los mismos tiempos se respetan en el mio-
-
decir que los diferentes tipos de muerte celular ocurren a
a la pérdida de un tercio de todos los miocitos y a la insu-
Figura 12.6: Identificación de fibrosis (miocitos son rojos, material fibrótico es verde). A: Fibrosis moderada. B: Fibrosis severa, fibrosis de reemplazo y presencia de miocitos de diferente tamaño.

Características estructurales del remodelamiento cardíaco...
195
Fibrosis
conectivo 14
la MEC tienen la capacidad de producir citoquinas, que estimulan la síntesis de proteínas de la matriz, metalopro-
7, 30, 31 -
-
32
reactiva y de reemplazo. La -
-
en las cardiomiopatias.14 Usualmente se localiza en el área perivascular y, en ocasiones, se puede encontrar en el intersticio. La es focalizada y causada por la pérdida de miocitos. El aumento de la
elementos microvasculares, lo que incrementa la dis-
33
el factor de crecimiento transformante β1 β1 -34, 35
-
como consecuencia, la injuria de los miocitos remanentes.
Correlación estructura/función
-
pero comienza a incrementarse la muerte celular. Esto y el número elevado de miocitos con pérdida de elementos
-
10, 13 -
ducida densidad capilar. Esto se considera el equivalente -
6
-
-
densidad capilar. La isquemia subendocárdica contribuye al
-
-
Bibliografía
1. Aschoff L, S Tawara. Die heutige Lehre von den pathologisch-ana-tomischen Grundlagen der Herzschwäche. Jena: Fischer; 1906.
2. Schaper J, Meis M, Froede R. Morphologische Untersuchungen am Myokard von Patienten mit koronarer Herzkrankheit. Berlin-Heidelberg-New York-Tokyo: Springer Verlag; 1985.
3. Schaper J, Froede R, Buck A, et al. “Impaired myocardial ultrastructure and cytoskeleton in cardiomyopathic human myocardium”. In: Schultheiß HP, ed. New Concepts in Viral Heart Disease. Berlin, Heidelberg, New York: Springer; 1988:295-302.
4. Schwartz K, JJ Mercadier. “Molecular and cellular biology of heart failure”. Current Opinion in Cardiology. 1996;11:227-236.
5. Cohn JN, Ferrari R, Sharpe N. “Cardiac remodeling-concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling”. J Am Coll Cardiol. 2000;35:569-582.
6. Hein S, Arnon E, Kostin S, et al. “Progression from compensated hypertrophy to failure in the pressure-overloaded human heart - Structural deterioration and compensatory mechanisms”. Circulation. 2003;107:984-991.
7. Weber, KT. “Cardiac interstitium in health and disease: the fibrillar collagen network”. J Am Coll Cardiol. 1989;13:1637-1652.
8. Speiser B, Riess CF, Schaper J. “The extracellular matrix in human myocardium: Part I: Collagen I, III, VI, and IV”. Cardioscience. 1991;2:225-232.
9. Speiser B, Weihrauch D, Riess CF, et al. “The extracellular matrix in human cardiac tissue. Part II: vimentin, laminin, and fibronectin”. Cardioscience. 1992;3:41-49.
10. Scholz D, Diener W, Schaper J. “Altered nucleus/cytoplasm re-lationship and degenerative structural changes in human dilated cardiomyopathy”. Cardioscience. 1994;5:127-138.

Gelpi - Donato Fisiopatología cardiovascular
196
11. Schaper J, Meiser E, Stämmler G. “Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice and from human hearts”. Circ Res. 1985;56:377-391.
12. Barth E, Stämmler G, Speiser B, et al. “Ultrastructural quantita-tion of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man”. J Mol Cell Cardiol. 1992;24:669-681.
13. Schaper J, Froede R, Hein S, et al. “Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy”. Circulation. 1991;83:504-514.
14. Majno G, Joris I. Cells, Tissues, and Disease. Principles of general pathology. Cambridge, Mass.: Blackwell Science; 1996.
15. Soonpaa MH, Field I. “Survey of studies examining mammalian cardio myocyte DNA synthesis”. Circ Res. 1998;83:15-26.
16. Fu XD, Maniatis T, T. “The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3’ splice site”. Proc Natl Acad Sci U S A. 1992;89:1725-1729.
17. Olivetti G, Melissari M, Capasso JM, et al. “Cardiomyopathy of the aging human heart (myocyte loss and reactive cellular hyper-trophy)”. Circ Res. 1991;68:1560-1568.
18. Majno G, Joris I. “Apoptosis, oncosis, and necrosis. An overview of cell death”. Am J Pathol. 1995;146:3-15.
19. Jennings RB, Sommers HM, Herdson PB, et al. “Ischemic injury of myocardium”. Ann N Y Acad Sci. 1969;156:61.
20. Schaper J, Mulch J, B. W, Schaper W. “Ultrastructural, functional, and biochemical criteria for estimation of reversibility of ischemic injury: A study on the effects of global ischemia on the isolated dog heart”. J Mol Cell Cardiol. 1979;11:521-541.
21. Ferreira MAS, Owen HE, Howie AJ. “High prevalence of acute myocardial damage in a hospital necropsy series, shown by C9 immunohistology”. J Clin Pathol. 1998;51:548-551.
22. Ciechanover A, Orian A, Schwartz AL. “The ubiquitin-mediated proteolytic pathway: Mode of action and clinical implications”. J Cell Biochem, suppl. 2000;34:40-51.
23. Glickman MH, Ciechanover A. “The ubiquitin-proteasome pro-teolytic pathway: destruction for the sake of construction”. Physiol Rev. 2002;82:373-428.
24. Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. “Ubiquitin, cellular inclusions and their role in neurodegeneration”. Trends Neurosci. 1998;21:516-520.
25. Jellinger KA. “Cell death mechanisms in Parkinson’s disease”. J Neural Transm. 2000;107:1-29.
26. Knaapen MW, Davies MJ, De Bie M, et al. “Apoptotic ver-sus autophagic cell death in heart failure». Cardiovasc Res. 2001;51:304-12.
27. Kostin S, Pool L, Elsässer H, et al. “Myocytes die by multiple mechanisms in failing human hearts”. Circ Res. 2003;92:715-724.
28. Klionsky DJ, Emr SD. “Autophagy as a regulated pathway of cellular degradation”. Science. 2000;290:1717-1720.
29. Bursch W, Hochegger K, Török L, et al. “Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments”. J Cell Sci. 2000;113:1189-1198.
30. Weber KT, Sun Y, Tyagi SC, et al. “Collagen network of the myocar-dium: function, structural remodeling and regulatory mechanisms”. J Mol Cell Cardiol. 1994;26:279-92.
31. Brilla CG, Maisch B, Zhou G, et al. “Hormonal regulation of cardiac fibroblast function”. Eur Heart J. 1995;16:45-50.
32. Hein S, Schaper J. “The extracellular matrix in normal and diseased myocardium”. J Nucl Cardiol. 2001;8:188-196.
33. Sabbah HN, Sharov VG, Lesch M, et al. “Progression of heart failure: a role for interstitial fibrosis”. Mol Cell Biochem. 1995;147:29-34.
34. Weber KT. “Angiotensin II and connective tissue: homeostasis and reciprocal regulation”. Regul Pept. 1999;82:1-17.
35. Schultz J, Witt SA, Glascock BJ, et al. “TGF-ß1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II”. J Clin Invest. 2002;109:787-796.

197
Acoplamiento éxcito-contráctil enel corazón normal y patológico 13
El acoplamiento éxcito-contráctil en el miocardio normal
Mecanismos subcelulares
que comienzan en la membrana celular o sarcolema, con
, se denomina aco- El ciclo se completa
es un esquema de los procesos celula--
despolariza por la entrada de Na a través de los canales rápidos de Na -nales lentos de Ca de tipo L y el Ca entra a la célula a
Ca plateau o meseta del potencial libera una mayor cantidad
de Ca o receptores de rianodina
inducida por el Ca –, prove-yendo así el Ca -croscopía confocal ha revelado que el Ca es liberado en cuantos que se denominan , a
Ca control local de Ca ocurren estocásticamente durante la diástole, a muy baja frecuencia y en ausencia de ICa que las dispare, varios miles de pueden ser sincronizadas por la ICa
-liberado se suma
Ca transient de Ca 1 El Ca
de las dos proteínas contráctiles fundamentales, la actina
no actúa
un desrepresor. Finalmente, el Ca es retomado en su ma-
el intercambiador Na /Caver más adelante
En condiciones normales, virtualmente todo el Ca que entra a la célula lo hace a través de los canales de tipo L del sarcolema. Estos canales, concentrados en su mayor parte en los túbulos T sarcolemales, son complejos proteicos multimé-
α1, α , β, γ, y δα1 contiene
el poro por el que entra el Ca
proteína kinasa dependiente de Ca2-4
del canal. Las subunidades α ,β, γ, y δ -
. Durante el AEC, el Ca
cercana al canal. Este au-mento tiene un efecto dual: interactúa directamente con el
Figura 13.1: Secuencia de eventos en el acoplamiento éxcito-contráctil. El potencial de acción (PA), que ocurre a nivel de la membrana celular, culmina en la contracción. El nexo es el aumento de calcio intracelular ([Ca2+]i).

Gelpi - Donato Fisiopatología cardiovascular
198
Figura 13.2: Esquema del acoplamiento éxcito-contráctil. La entrada de Ca2+ por los canales L (receptor de dihidropiridinas, DHPR) del sarcolema (SL) produce la liberación de Ca2+ del retículo sarcoplasmático (RS), a través del canal de liberación de Ca2+ (receptor de rianodina, RyR2). El Ca2+ liberado se une a la troponina C de los miofilamentos (MF) y produce la contracción. Parte del Ca2+ es extruido de la célula por el intercambiador Na+/ Ca2+ (NCX), pero la mayor parte es retomado por el RS a través de la Ca2+-ATPasa del RS (SERCA2a). Esta enzima está regulada por la fosfolamban (PLN). La Ca2+-ATPasa del sarcolema participa en el mantenimiento del Ca2+ diastólico.
activa a la CaMKII, que fosforila al canal y facilita la ICa.
-dades polipeptídicas, con un dominio citoplasmático y un dominio de transmembrana, que forma el canal de Ca .
-ladores intrínsecos 5
--
de Ca inducida por Ca
calsecuestrina se une al Ca , constituyendo un amortigua-dor del Ca amor-tiguador del Ca intraluminal, la calsecuestrina modula la
juntina-calsecuestrina sería el sensor, a nivel molecular, del Ca intraluminal y estaría involucrado directamente en la
6
El Caa través de la Ca -ATPasa o bomba de Ca -bién denominada
Sarcoplasmic (E Reticulum Calcium A activamente desde el cito-
responsable del descenso del Ca -
libre intraluminal, también favorece 7
-nico8
a instante, durante el ciclo cardiaco, depende del potencial de membrana y además de las concentraciones de Na y Ca a ambos lados de la misma. En condiciones normales,
, que introduce a la célu-
también puede funcionar en sentido inverso, introduciendo Ca rever-se 9 Esto ocurriría, aunque por un periodo
-nes especiales como, por ejemplo, de alto Na intracelular,
-riable de acuerdo a las especies. En la rata, por ejemplo,
caída del Ca
Ca

Acoplamiento éxcito-contráctil en el corazón normal y patológico
199
10
secuestro de Ca será mayor y el Ca secuestrado por el -
del Ca 11
fosfolamban
in vivo 16 -
17 12
-
bomba por el Ca y, como consecuencia, la recaptura de Ca
-
funcional de la PLN fue dilucidado a través del uso de
models -
stem cells
13 Estudios
por el Ca -
son compatibles con los descritos previamente en mem-branas in vitro
13 Por otro -
el Ca . Las propiedades contráctiles de estos animales
-nor contractilidad y una menor amplitud y velocidad de caída del transient de Ca . 14
-das en las mismas. Por otra parte, es de suma importancia
hace fundamentalmente a través del sistema β
β -tractilidad miocárdicas, como se verá más adelante. 11
Ca2+ vs. respuesta al Ca2+ de las proteínas contráctiles en la determinación de la contractilidad miocárdica
-den ocurrir cambios en la contractilidad miocárdica: A.
transient de Ca intracelular y
al Caaumenta el Ca intracelular, aumenta la contractilidad.
respuesta de los 2+ tiene,
. Un aumento de la sensibilidad de
dado del Cade producir mayor fuerza o acortamiento. Una disminu-
Ca intracelular se produce menor fuerza o acortamiento.
-veles saturantes de Ca
intracelular o en la -
-
β
15
El acoplamiento éxcito-contráctil en diferentes patologías
Insuficiencia cardiaca-
-
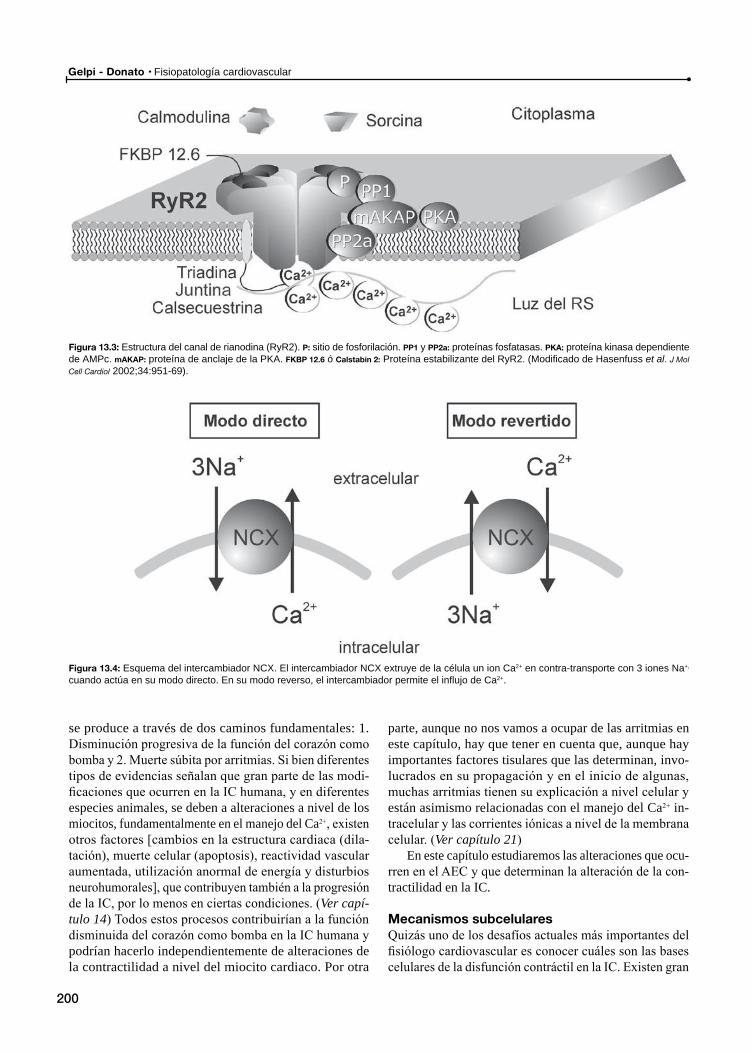
Gelpi - Donato Fisiopatología cardiovascular
200
Figura 13.3: Estructura del canal de rianodina (RyR2). P: sitio de fosforilación. PP1 y PP2a: proteínas fosfatasas. PKA: proteína kinasa dependiente de AMPc. mAKAP: proteína de anclaje de la PKA. FKBP 12.6 ó Calstabin 2: Proteína estabilizante del RyR2. (Modificado de Hasenfuss et al. J Mol
Cell Cardiol 2002;34:951-69).
Figura 13.4: Esquema del intercambiador NCX. El intercambiador NCX extruye de la célula un ion Ca2+ en contra-transporte con 3 iones Na+, cuando actúa en su modo directo. En su modo reverso, el intercambiador permite el influjo de Ca2+.
se produce a través de dos caminos fundamentales: 1.
-
especies animales, se deben a alteraciones a nivel de los miocitos, fundamentalmente en el manejo del Ca
-
Ver capí-tulo 14
podrían hacerlo independientemente de alteraciones de la contractilidad a nivel del miocito cardiaco. Por otra
parte, aunque no nos vamos a ocupar de las arritmias en este capítulo, hay que tener en cuenta que, aunque hay importantes factores tisulares que las determinan, invo-
están asimismo relacionadas con el manejo del Ca in-
Ver capítulo 21 En este capítulo estudiaremos las alteraciones que ocu-
-tractilidad en la IC.
Mecanismos subcelulares
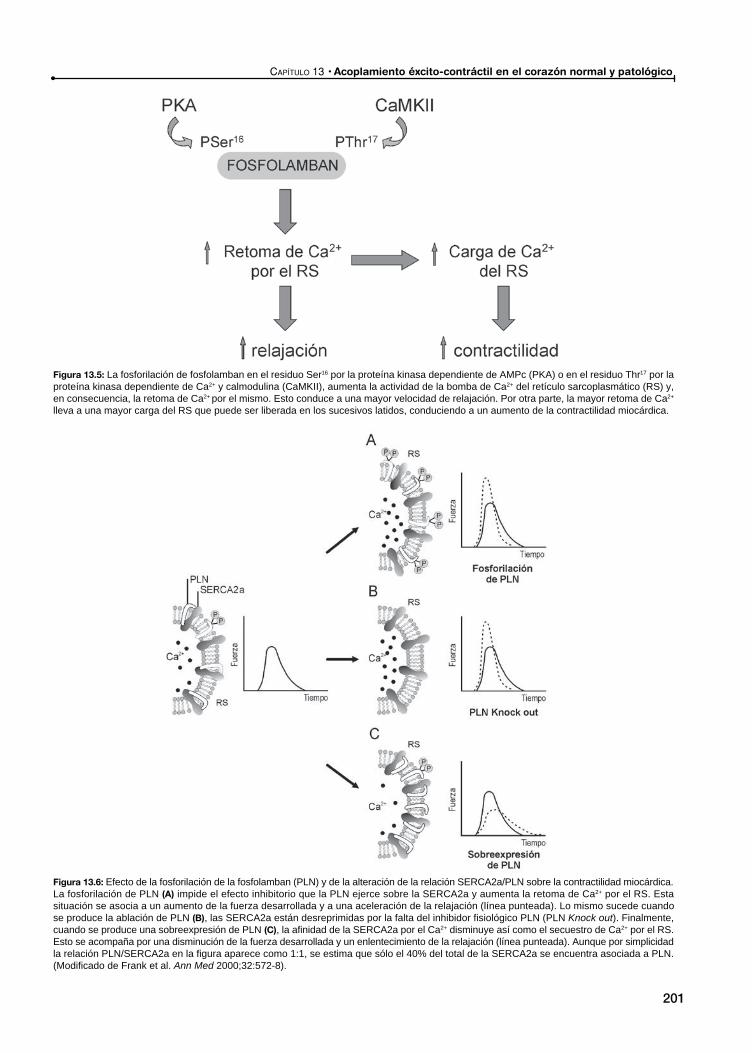
Acoplamiento éxcito-contráctil en el corazón normal y patológico
201
Figura 13.5: La fosforilación de fosfolamban en el residuo Ser16 por la proteína kinasa dependiente de AMPc (PKA) o en el residuo Thr17 por la proteína kinasa dependiente de Ca2+ y calmodulina (CaMKII), aumenta la actividad de la bomba de Ca2+ del retículo sarcoplasmático (RS) y, en consecuencia, la retoma de Ca2+ por el mismo. Esto conduce a una mayor velocidad de relajación. Por otra parte, la mayor retoma de Ca2+ lleva a una mayor carga del RS que puede ser liberada en los sucesivos latidos, conduciendo a un aumento de la contractilidad miocárdica.
Figura 13.6: Efecto de la fosforilación de la fosfolamban (PLN) y de la alteración de la relación SERCA2a/PLN sobre la contractilidad miocárdica. La fosforilación de PLN (A) impide el efecto inhibitorio que la PLN ejerce sobre la SERCA2a y aumenta la retoma de Ca2+ por el RS. Esta situación se asocia a un aumento de la fuerza desarrollada y a una aceleración de la relajación (línea punteada). Lo mismo sucede cuando se produce la ablación de PLN (B), las SERCA2a están desreprimidas por la falta del inhibidor fisiológico PLN (PLN Knock out). Finalmente, cuando se produce una sobreexpresión de PLN (C), la afinidad de la SERCA2a por el Ca2+ disminuye así como el secuestro de Ca2+ por el RS. Esto se acompaña por una disminución de la fuerza desarrollada y un enlentecimiento de la relajación (línea punteada). Aunque por simplicidad la relación PLN/SERCA2a en la figura aparece como 1:1, se estima que sólo el 40% del total de la SERCA2a se encuentra asociada a PLN. (Modificado de Frank et al. Ann Med 2000;32:572-8).
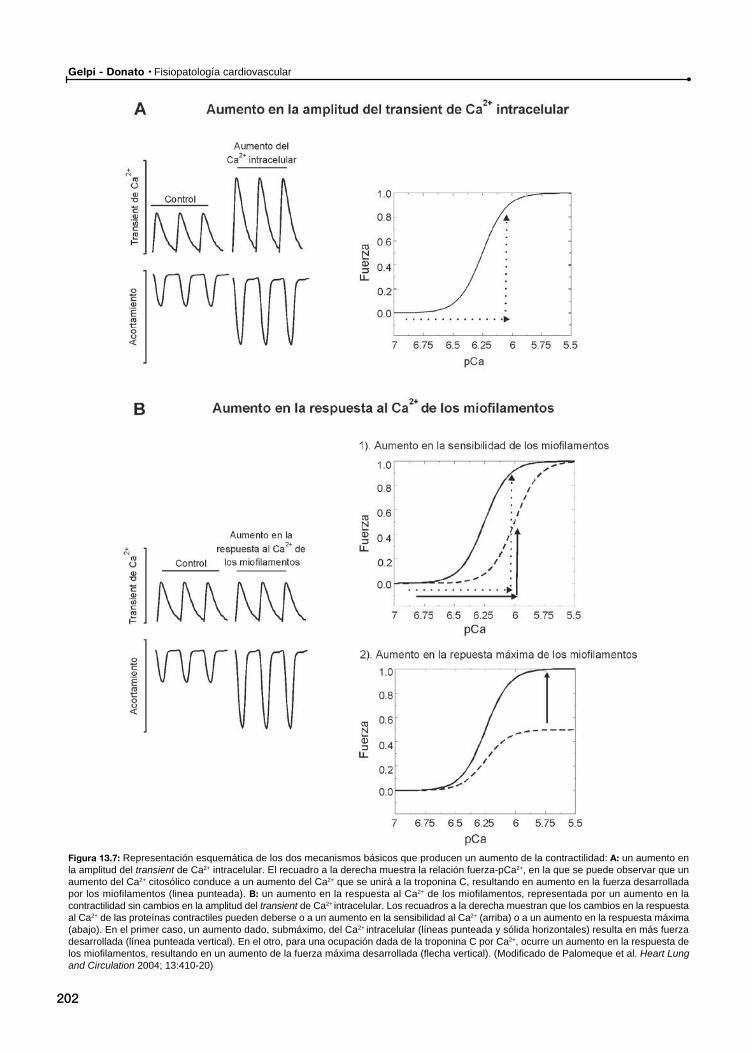
Gelpi - Donato Fisiopatología cardiovascular
202
Figura 13.7: Representación esquemática de los dos mecanismos básicos que producen un aumento de la contractilidad: A: un aumento en la amplitud del transient de Ca2+ intracelular. El recuadro a la derecha muestra la relación fuerza-pCa2+, en la que se puede observar que un aumento del Ca2+ citosólico conduce a un aumento del Ca2+ que se unirá a la troponina C, resultando en aumento en la fuerza desarrollada por los miofilamentos (linea punteada). B: un aumento en la respuesta al Ca2+ de los miofilamentos, representada por un aumento en la contractilidad sin cambios en la amplitud del transient de Ca2+ intracelular. Los recuadros a la derecha muestran que los cambios en la respuesta al Ca2+ de las proteínas contractiles pueden deberse o a un aumento en la sensibilidad al Ca2+ (arriba) o a un aumento en la respuesta máxima (abajo). En el primer caso, un aumento dado, submáximo, del Ca2+ intracelular (líneas punteada y sólida horizontales) resulta en más fuerza desarrollada (línea punteada vertical). En el otro, para una ocupación dada de la troponina C por Ca2+, ocurre un aumento en la respuesta de los miofilamentos, resultando en un aumento de la fuerza máxima desarrollada (flecha vertical). (Modificado de Palomeque et al. Heart Lung and Circulation 2004; 13:410-20)

Acoplamiento éxcito-contráctil en el corazón normal y patológico
203
Figura 13.8: Cambios en las proteínas relacionadas con el manejo del Ca2+ intracelular en el miocardio normal e insuficiente. En el miocardio insuficiente, se describe una expresión aumentada del NCX y una expresión disminuida de la SERCA2a y la PLN en forma aproximadamente proporcional, respecto del miocardio normal. La fosforilación del RyR2 podría ser el mecanismo responsable de una pérdida de Ca2+ del RS en diástole, que normalmente es muy pequeña. En conjunto, estos 3 factores contribuirían a disminuir la carga del RS en el miocardio insuficiente. Se describe además una disminución de la expresión de la bomba Na+/K+-ATPasa, que podría colaborar a la alteración del manejo del Ca2+, perjudicando fundamentalmente la función diastólica (ver texto).
cantidad de modelos de IC en donde se han estudiado las bases celulares de la misma. Los resultados obtenidos,
todos los modelos de IC, de lo que ocurre a nivel celular en la IC. Describiremos aquí las alteraciones encontradas
tran-sient de Ca intracelular. 16, 17
un manejo de Ca anormal en la IC fue hecha hace casi transient de Ca con
el indicador de Ca acuarina, en trabéculas de corazones miopáticos humanos, obtenidos en el momento del trans-
-transient de Ca
21 Esto fue corroborado posteriormente en nume-
que el transient de Ca en la IC se caracterizaba por tener elevado Ca
respuesta al Ca de las proteínas contráctiles18, este meca-
la contractilidad en la mayor parte de los modelos de IC.transient de Ca
transient de Ca en la IC es la
-delos de IC, se describen alteraciones de distintos compo-
17 En la mayoría de los estudios, esta dismi-
proporcional de la PLN estaría conservada. De esta manera, la dependencia del Ca del transporte de Ca
pero el transporte de Ca estaría disminuido a todas las concentraciones de Ca
la menor respuesta al sistema β
de la , reduciendo aún más el trans-porte de Ca -
del
caída del transient de Caobservada en muchos modelos de IC. Asociado a la dis-
-tente en los diferentes modelos de IC estudiados, inclui-do el humano. 17
fuera de la célula, un aumento en

Gelpi - Donato Fisiopatología cardiovascular
204
-
-
circulante, que en condiciones normales sería recuperado
del NCX, trabajando en su modo directo. Por otra parte, la
un efecto adicional sobre la cinética del canal sarcolemal de Ca
de Ca -secuencia puede aumentar la entrada de Ca a la célula y
transient de Ca , 19
la IC. En cuanto al número, se ha descrito una disminu-
17 -
- por parte de los
mismos frente al estímulo, como se dijo anteriormente. -
dida de Ca en diástole, contribuyendo a la menor car-20
ocurrir a pesar de la menor respuesta βver más adelante
-20 -
-
normales, Otros estudios revelan por otra parte, que en
-
que aumenta la actividad del canal, llevando a una pér-dida de Ca
la IC. 29 También se ha descrito un aumento en la fos-
desconoce su posible incidencia en la menor contracti-β
-
Menor captura de Ca de
- en diástole, a través de
y, en consecuencia, en el transient de Ca .
intracelular, es la dismi-/K -ATPasa, descrita en
muchos de los modelos estudiados. 21
llevaría a un aumento del Na intracelular que podría demorar el modo directo del NCX o favorecer su modo revertido, lo que tendería a producir un aumento del Ca
-
-
a un nivel que promueve el modo revertido del NCX, y por lo tanto la entrada de Ca , como fue mencionado. La entrada de Ca por el modo revertido del NCX po-dría resultar perjudicial si ocurre en forma importante,
la escaleralenta del Ca
22 Finalmente, es necesario mencionar que la ICa, disparadora del AEC, no se ha encontrado disminui-da en la mayoría de los modelos estudiados, por lo que
transient de Ca de la IC.
contractilidad anormal de la IC es la causa o la consecuen-
-
-, como aca-
bamos de describir. 23
24
está en línea con evidencia obtenida en modelos animales
se pudo demorar25, revertir 26 o prevenir 27 por tratamientos

Acoplamiento éxcito-contráctil en el corazón normal y patológico
205
- y con
contractilidad normal o aun aumentada. Al respecto, se ha
-
Para resumir, la evidencia hasta el momento indica que la
siempre necesaria.
El fenómeno de la escalera en el corazón normal e insuficienteEl aumento de contractilidad, que ocurre con indepen-dencia del control neurohumoral, en respuesta al aumento
-mente conservado entre las distintas especies, incluido
positivo, producido por el aumento en la frecuencia de
transient de Ca 28 -mos subcelulares que determinan dicho aumento no han
indican que el aumento del transient de Ca , que ocu-
es consecuencia de un aumento en el contenido y en la por el 5 A su vez, el aumento de
entrada de Ca a la célula a través de los canales de Catipo L, como resultado del incremento del número de despolarizaciones por unidad de tiempo 51, 2 y un aumento en el Catravés del NCX. 28
el que el
-,
-28 Por otra parte, el aumento del Na intracelular,
que resulta del mayor número de despolarizaciones por uni-
podría retardar al modo directo del NCX y así disminuir o favorecer la entrada de Ca por el
aumento del Na intracelular y el aumento de contractilidad provocado por el incremento en la frecuencia de estimu-
Ca a través de un mecanismo Na -dependiente. 28 -
al aumento del Na-
y el incremento de fuerza resultante del aumento de la frecuen-
especies, el NCX podría contribuir a la escalera positiva a .
por los canales tipo L
a través de los mecanismos antes descritos, promueven un de Ca -
-
se presentan en forma de una ausencia de incremento de contractilidad en respuesta al aumento de la frecuencia de
-
29, 30 La
preparaciones enteras como en miocitos, aislados, y clí-
in vitro
alteraciones subcelulares y moleculares que podrían ser -
Alteraciones subcelulares-
-transient de Ca
por el mismo. 31
muestra el efecto del aumento de la fre-
evaluado a través de la contractura inducida por una brusca
el vaciamiento instantáneo del Ca 32 La altura
por un aumento paralelo del contenido de Ca
-
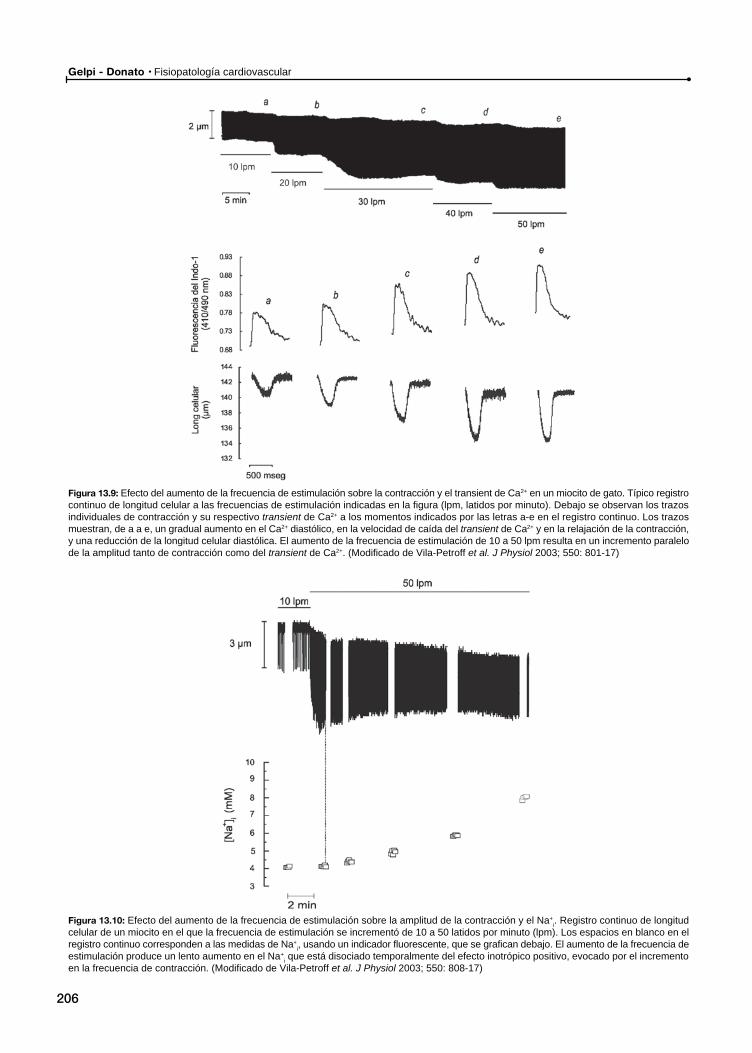
Gelpi - Donato Fisiopatología cardiovascular
206
Figura 13.9: Efecto del aumento de la frecuencia de estimulación sobre la contracción y el transient de Ca2+ en un miocito de gato. Típico registro continuo de longitud celular a las frecuencias de estimulación indicadas en la figura (lpm, latidos por minuto). Debajo se observan los trazos individuales de contracción y su respectivo transient de Ca2+ a los momentos indicados por las letras a-e en el registro continuo. Los trazos muestran, de a a e, un gradual aumento en el Ca2+ diastólico, en la velocidad de caída del transient de Ca2+ y en la relajación de la contracción, y una reducción de la longitud celular diastólica. El aumento de la frecuencia de estimulación de 10 a 50 lpm resulta en un incremento paralelo de la amplitud tanto de contracción como del transient de Ca2+. (Modificado de Vila-Petroff et al. J Physiol 2003; 550: 801-17)
Figura 13.10: Efecto del aumento de la frecuencia de estimulación sobre la amplitud de la contracción y el Na+i. Registro continuo de longitud
celular de un miocito en el que la frecuencia de estimulación se incrementó de 10 a 50 latidos por minuto (lpm). Los espacios en blanco en el registro continuo corresponden a las medidas de Na+
i, usando un indicador fluorescente, que se grafican debajo. El aumento de la frecuencia de estimulación produce un lento aumento en el Na+
i que está disociado temporalmente del efecto inotrópico positivo, evocado por el incremento en la frecuencia de contracción. (Modificado de Vila-Petroff et al. J Physiol 2003; 550: 808-17)
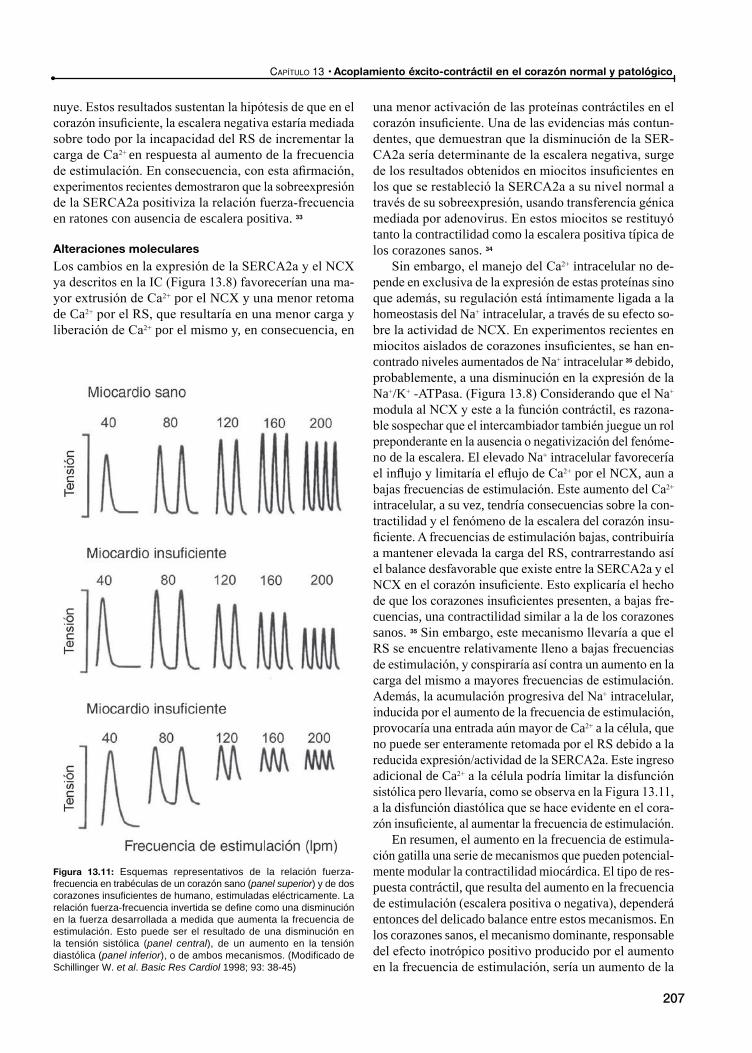
Acoplamiento éxcito-contráctil en el corazón normal y patológico
207
en respuesta al aumento de la frecuencia
en ratones con ausencia de escalera positiva. 33
Alteraciones moleculares
- por el NCX y una menor retoma
de Ca por el mismo y, en consecuencia, en
--
tanto la contractilidad como la escalera positiva típica de los corazones sanos. 34
intracelular no de-
homeostasis del Na intracelular, a través de su efecto so-
-contrado niveles aumentados de Na intracelular 35 debido,
Na /K -
-no de la escalera. El elevado Na intracelular favorecería
por el NCX, aun a
intracelular, a su vez, tendría consecuencias sobre la con--
-cuencias, una contractilidad similar a la de los corazones sanos. 35
intracelular,
provocaría una entrada aún mayor de Ca a la célula, que no puede ser
adicional de Ca
-
En resumen, el aumento en la frecuencia de estimula--
mente modular la contractilidad miocárdica. El tipo de res-puesta contráctil, que resulta del aumento en la frecuencia
entonces del delicado balance entre estos mecanismos. En los corazones sanos, el mecanismo dominante, responsable
Figura 13.11: Esquemas representativos de la relación fuerza-frecuencia en trabéculas de un corazón sano (panel superior) y de dos corazones insuficientes de humano, estimuladas eléctricamente. La relación fuerza-frecuencia invertida se define como una disminución en la fuerza desarrollada a medida que aumenta la frecuencia de estimulación. Esto puede ser el resultado de una disminución en la tensión sistólica (panel central), de un aumento en la tensión diastólica (panel inferior), o de ambos mecanismos. (Modificado de Schillinger W. et al. Basic Res Cardiol 1998; 93: 38-45)
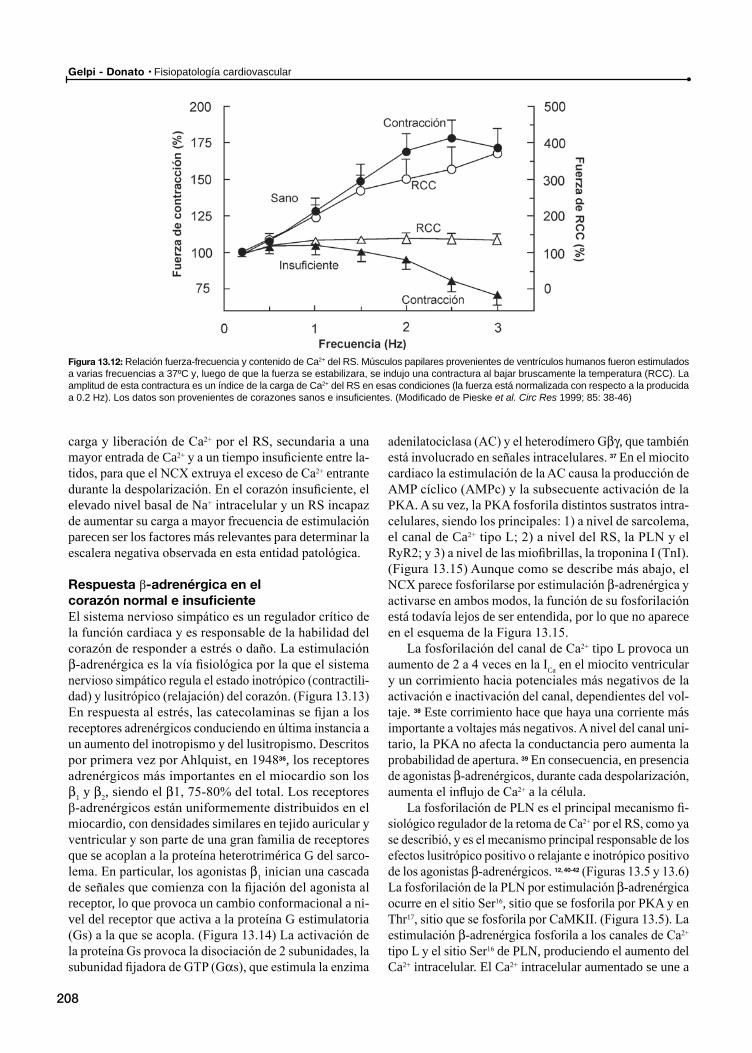
Gelpi - Donato Fisiopatología cardiovascular
208
mayor entrada de Ca - entrante
elevado nivel basal de Na
parecen ser los factores más relevantes para determinar la
Respuesta -adrenérgica en el corazón normal e insuficienteEl
βcontractili-
un aumento del inotropismo y del lusitropismo. Descritos 36, los receptores
β1 y β , siendo el β
miocardio, con densidades similares en tejido auricular y
que se acoplan a la proteína heterotrimérica G del sarco-β1 inician una cascada
receptor, lo que provoca un cambio conformacional a ni-vel del receptor que activa a la proteína G estimulatoria
α
βγ, que también 37 En el miocito
PKA. A su vez, la PKA fosforila distintos sustratos intra-
el canal de Ca
β
está todavía lejos de ser entendida, por lo que no aparece
tipo L provoca un Ca en el miocito ventricular
-taje. 38 Este corrimiento hace que haya una corriente más
-tario, la PKA no afecta la conductancia pero aumenta la probabilidad de apertura. 39 En consecuencia, en presencia
β a la célula.
-
β 12, 40-42
β, sitio que se fosforila por PKA y en
Thr17
β de PLN, produciendo el aumento del
Ca intracelular. El Ca intracelular aumentado se une a
Figura 13.12: Relación fuerza-frecuencia y contenido de Ca2+ del RS. Músculos papilares provenientes de ventrículos humanos fueron estimulados a varias frecuencias a 37ºC y, luego de que la fuerza se estabilizara, se indujo una contractura al bajar bruscamente la temperatura (RCC). La amplitud de esta contractura es un índice de la carga de Ca2+ del RS en esas condiciones (la fuerza está normalizada con respecto a la producida a 0.2 Hz). Los datos son provenientes de corazones sanos e insuficientes. (Modificado de Pieske et al. Circ Res 1999; 85: 38-46)
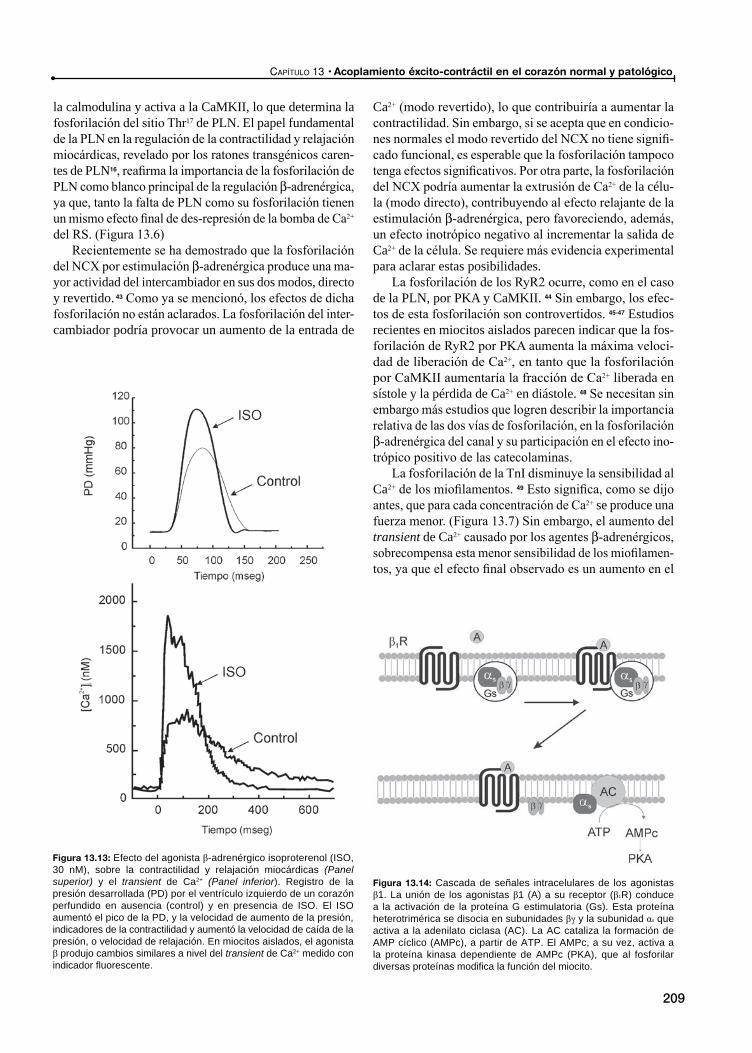
Acoplamiento éxcito-contráctil en el corazón normal y patológico
209
Ca--
de la célu-
β
Capara aclarar estas posibilidades.
de la PLN, por PKA y CaMKII. 44 -45-47 Estudios
recientes en miocitos aislados parecen indicar que la fos--
liberada en sístole y la pérdida de Ca en diástole. 48
β -
Ca 49
se produce una
transient de Ca β-
Figura 13.13: Efecto del agonista -adrenérgico isoproterenol (ISO, 30 nM), sobre la contractilidad y relajación miocárdicas (Panel superior) y el transient de Ca2+ (Panel inferior). Registro de la presión desarrollada (PD) por el ventrículo izquierdo de un corazón perfundido en ausencia (control) y en presencia de ISO. El ISO aumentó el pico de la PD, y la velocidad de aumento de la presión, indicadores de la contractilidad y aumentó la velocidad de caída de la presión, o velocidad de relajación. En miocitos aislados, el agonista produjo cambios similares a nivel del transient de Ca2+ medido con
indicador fluorescente.
Figura 13.14: Cascada de señales intracelulares de los agonistas 1. La unión de los agonistas 1 (A) a su receptor ( 1R) conduce
a la activación de la proteína G estimulatoria (Gs). Esta proteína heterotrimérica se disocia en subunidades y la subunidad s que activa a la adenilato ciclasa (AC). La AC cataliza la formación de AMP cíclico (AMPc), a partir de ATP. El AMPc, a su vez, activa a la proteína kinasa dependiente de AMPc (PKA), que al fosforilar diversas proteínas modifica la función del miocito.
la calmodulina y activa a la CaMKII, lo que determina la 17 de PLN. El papel fundamental
-tes de PLN16
β
β -yor actividad del intercambiador en sus dos modos, directo y revertido. 43
-cambiador podría provocar un aumento de la entrada de

Gelpi - Donato Fisiopatología cardiovascular
210
inotropismo. La menor sensibilidad al Ca -
con que el Ca se desprende de la TnC. En principio esto podría contribuir al efecto relajante de las catecolaminas, aunque hay resultados contradictorios al respecto. En tanto
de la sensibilidad al Ca contribuye al efecto relajante de β 40
de los receptores β1 -β . Evidencias
β1 y β-
cas y terapéuticas. 50
que los receptores β también están acoplados a la proteína 51 Este
PKA más localizada, que puede fosforilar los canales de Ca de tipo L del sarcolema pero no moléculas distantes como la PLN y la TnI. En consecuencia, los efectos ino-
de los receptores β-
tores β
β
ββ
β -
de Gs-adenilato ciclasa-AMPc-PKA.
β a sus receptores, así como ocurre con cualquier otro receptor acoplado a proteína G, puede
52
-
-
-
βlos receptores acoplados a proteína G, también requiere la
β-
β--
sibilizados. Una vez internalizados, los receptores pueden downregulation -
Clásicamente βARK1 se ha descrito como la kinasa β.
βpara fosforilar al receptor. β -
βγβ
es un efectivo inhibidor in vitro e in vivo de la actividad de la βARK1, ya que secuestra a la subunidad Gβγ. En el
β1 como β βARK1, y una actividad aumentada de la βARK1 lleva a una pérdida
Figura 13.15: Esquema de los efectos de la estimulación β-adrenérgica de proteínas fundamentales del AEC en el miocardio. La unión del agonista -adrenérgico a su receptor aumenta el AMPc que, a través de la PKA, fosforila proteínas. En el sarcolema (SL), la fosforilación del canal de Ca2+ aumenta la entrada de Ca2+ a la célula y contribuye al efecto inotrópico positivo. A nivel del retículo sarcoplasmático (RS), la fosforilación de la fosfolamban (PLN) aumenta la retoma de Ca2+ por el RS, lo que conduce a un efecto relajante, pero también, al aumentar la carga de Ca2+ del RS, lleva a un aumento de la contractilidad. La fosforilación de los receptores de rianodina (RyR2), también a nivel del RS, podría contribuir al efecto inotrópico positivo, aunque esta contribución es controvertida. Finalmente, la fosforilación de la troponina I (TnI) de los miofilamentos (MF) disminuye la sensibilidad al Ca2+ de los mismos, contribuyendo al efecto relajante de los agonistas β-adrenérgicos.

Acoplamiento éxcito-contráctil en el corazón normal y patológico
211
βde los niveles aumentados de catecolaminas. 57 La dismi-
-β, incluso la dobutamina,
-
miocárdica. A pesar de su utilidad a corto plazo, la admi-
-
sobrevida. 58 -
transplantados. Estos dispositivos han llevado aparente--
res βpor lo común llamados remodelamiento reverso, que ocu-
de receptores β se restaura a valores normales, llevando a
Figura 13.16: Desensibilización del receptor β. La ocupación del receptor por el agonista (A) conduce a la fosforilación del mismo por la kinasa del receptor acoplada a proteína G (β-ARK1). Al receptor fosforilado se acopla la proteína β-arrestina, que media la internalización de los receptores. El receptor internalizado puede activar nuevas señales intracelulares, degradarse o volver a la membrana plasmática, para lo que se requiere su desfosforilación, llevada a cabo por fosfatasas que trabajan a pH ácido. (Modificado de Lefkowitz, R. J Biol Chem 1998; 273: 18677-80)
Figura 13.17: Alteraciones moleculares del sistema -adrenérgico en la insuficiencia, con respecto al miocardio normal. En el miocardio insuficiente, la densidad de los receptores 1 está disminuida y hay un aumento en la expresión de la -ARK1, enzima que fosforila al receptor y lo desensibiliza, conduciendo a una menor formación de AMPc y activación de PKA.
β. 50 βARKct aumenta
β 50
que el sistema β -
nivel aumentado de catecolaminas que está inversamente relacionado con la sobrevida. 53 Por otro lado, Bristow et al. 54, demostraron que tanto la densidad de re-ceptores β
humano. La pérdida de los receptores β es selectiva al tipo β1, y entonces hay un mayor porcentaje relativo de receptores β que enfatizan sus propiedades diferenciales. 50 Los receptores β1 remanentes y los β están desensibi-lizados, presumiblemente por aumento de la actividad de las GRKs. Además, se han encontrado aumentos de hasta
αi, lo que llevaría a una me-nor respuesta de los sistemas acoplados a Gs. En el último estadio de la IC humana, así como en modelos animales de IC, los niveles cardiacos y la actividad de βARK1 apare-
también se ha encontrado aumentada. 55 Lo importante es que los niveles elevados de βARK1 suelen preceder a la
estadio de la IC. 56
aumentada de la βARK1 como otros cambios en el sistema

Gelpi - Donato Fisiopatología cardiovascular
212
β. Ver capítulo 24
-
Entre los factores neurohumorales, es importante notar que, además de los niveles circulantes aumentados de cate-
sistémicas, que a su vez provocan alteraciones miocárdicas, son contribuyentes fundamentales del círculo vicioso de la
mejorar y aun aumentar la sobrevida en la IC.
Las proteínas involucradas en el manejo del Ca2+ como blancos de la transferencia génica en la IC
-
como una herramienta útil que permite describir la im-portancia de las diferentes alteraciones en la IC, y clínico, porque permite evaluar las consecuencias terapéuticas de la misma. Hay tres áreas principales que han sido blanco
el manejo del Ca durante el AEC, los canales de K y su -
βproteínas que tienen que ver con el manejo del Ca , Hajjar et al
-59
validando en conjunto con otros estudios similares, la
ofrecer una nueva modalidad terapéutica en pacientes con IC y comprobando la importancia de esta proteína en la
IC en rata. 60
antisense contra PLN, es capaz de
-
mejorar el manejo de Ca alterado en la IC. 17
Pérdida de Ca2+ por el RyR2 y muerte súbita
del RyR1, la isoforma del RyR del músculo esquelético, que se asocian a una homeostasis alterada del Ca y son la causa, por ejemplo, de la y la miopatía
de cardiomiopatías que se caracterizan por muerte súbita temprana: la cardiomiopatía arritmogénica ventricular
, y
Es interesante que las mutaciones asociadas con estas
núcleo central, en los RyR1. Las alteraciones de los RyR1 del músculo esquelético se asocian con un aumento del Ca del
-
aumento del Ca ondas de calcio potencial-
Los posibles mecanismos de -se han estudiado re-
cientemente. 61
ejercicio intenso o en el estrés emocional y puede produ-cir, como dijimos, muerte súbita. Uno de los mecanismos
diástole. 61 El aumento de la pérdida de Ca --
yendo Ca
Ca -
hacia el umbral, favoreciendo las posdespolarizaciones -
por los canales L, debido a
de Ca
este ciclo del Ca alterado puede resultar en taquicardia
focos de células ventriculares.
Atontamiento miocárdico (Stunning)-
Ver capítulo 5 62
al periodo pre-isquémico, que se recupera con lentitud. Eventualmente, al cabo de horas, días o semanas, la recu-
63
entender los mecanismos íntimos del AM, ya que aunque

Acoplamiento éxcito-contráctil en el corazón normal y patológico
213
entidad de relevancia clínica, es decir, el atontamiento
cardiopulmonar. 64
Disfunción contráctil en el atontamiento miocárdico: Ca2+ vs. disminución de la respuesta al Ca2+ de las proteínas contráctiles
-
los mecanismos íntimos que llevan al AM se ha realizado -
resultados a veces no coincidentes, lo que ha complicado
modelos es que, en todos ellos, el miocardio es sometido a
aporte de O, como ocurre
63, el mecanismo responsable para esta recupera-
-
---
65
-
dis--
-
mecanismo de la menor contractilidad en el AM es una
65
y en el Ca disponible 66
Disminución de la respuesta al Ca2+ de los miofilamentos como causa principal del AM
Ca está disminuida en el AM. Kusuoka et al. 116 fueron
-
en corazones atontados de hurones. Estudios posteriores,
transients de Ca no estaban alterados. 68 Estos estudios demostraron en
se debe a una menor sensibilidad al Ca , a una menor
67
AEC afectado por el AM es distal a la disponibilidad de Ca
El mecanismo por el que se produce la menor respuesta al Ca -blecido todavía, pero la evidencia disponible apunta a mo-
-69 Este es
y de los radicales
la respuesta al Ca-
vés del Ca70
. 65
desarrollada por Marbán et al. -
65 -
de proteasas dependientes de Ca-
al comienzo de la re--
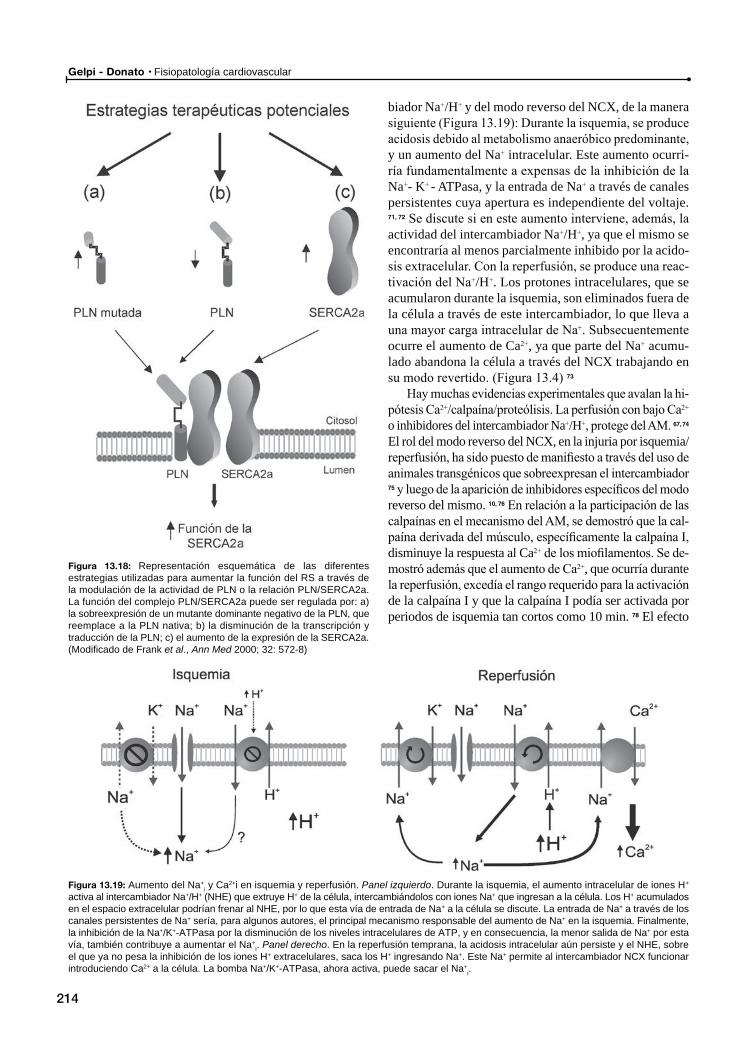
Gelpi - Donato Fisiopatología cardiovascular
214
Figura 13.19: Aumento del Na+i y Ca2+i en isquemia y reperfusión. Panel izquierdo. Durante la isquemia, el aumento intracelular de iones H+
activa al intercambiador Na+/H+ (NHE) que extruye H+ de la célula, intercambiándolos con iones Na+ que ingresan a la célula. Los H+ acumulados en el espacio extracelular podrían frenar al NHE, por lo que esta vía de entrada de Na+ a la célula se discute. La entrada de Na+ a través de los canales persistentes de Na+ sería, para algunos autores, el principal mecanismo responsable del aumento de Na+ en la isquemia. Finalmente, la inhibición de la Na+/K+-ATPasa por la disminución de los niveles intracelulares de ATP, y en consecuencia, la menor salida de Na+ por esta vía, también contribuye a aumentar el Na+
i. Panel derecho. En la reperfusión temprana, la acidosis intracelular aún persiste y el NHE, sobre el que ya no pesa la inhibición de los iones H+ extracelulares, saca los H+ ingresando Na+. Este Na+ permite al intercambiador NCX funcionar introduciendo Ca2+ a la célula. La bomba Na+/K+-ATPasa, ahora activa, puede sacar el Na+
i.
biador Na /H y del modo reverso del NCX, de la manera
y un aumento del Na intracelular. Este aumento ocurri-
Na - K - ATPasa, y la entrada de Na a través de canales persistentes cuya apertura es independiente del voltaje. 71, 72
actividad del intercambiador Na /H , ya que el mismo se encontraría al menos parcialmente inhibido por la acido-
-/H . Los protones intracelulares, que se
acumularon durante la isquemia, son eliminados fuera de la célula a través de este intercambiador, lo que lleva a
ocurre el aumento de Ca , ya que parte del Na acumu-lado abandona la célula a través del NCX trabajando en
73
-
o inhibidores del intercambiador Na /H 67, 74 El rol del modo reverso del NCX, en la injuria por isquemia/
75
reverso del mismo. 10, 76
-
disminuye la respuesta al Ca -, que ocurría durante
de la calpaína I y que la calpaína I podía ser activada por periodos de isquemia tan cortos como 10 min. 78 El efecto
Figura 13.18: Representación esquemática de las diferentes estrategias utilizadas para aumentar la función del RS a través de la modulación de la actividad de PLN o la relación PLN/SERCA2a. La función del complejo PLN/SERCA2a puede ser regulada por: a) la sobreexpresión de un mutante dominante negativo de la PLN, que reemplace a la PLN nativa; b) la disminución de la transcripción y traducción de la PLN; c) el aumento de la expresión de la SERCA2a. (Modificado de Frank et al., Ann Med 2000; 32: 572-8)
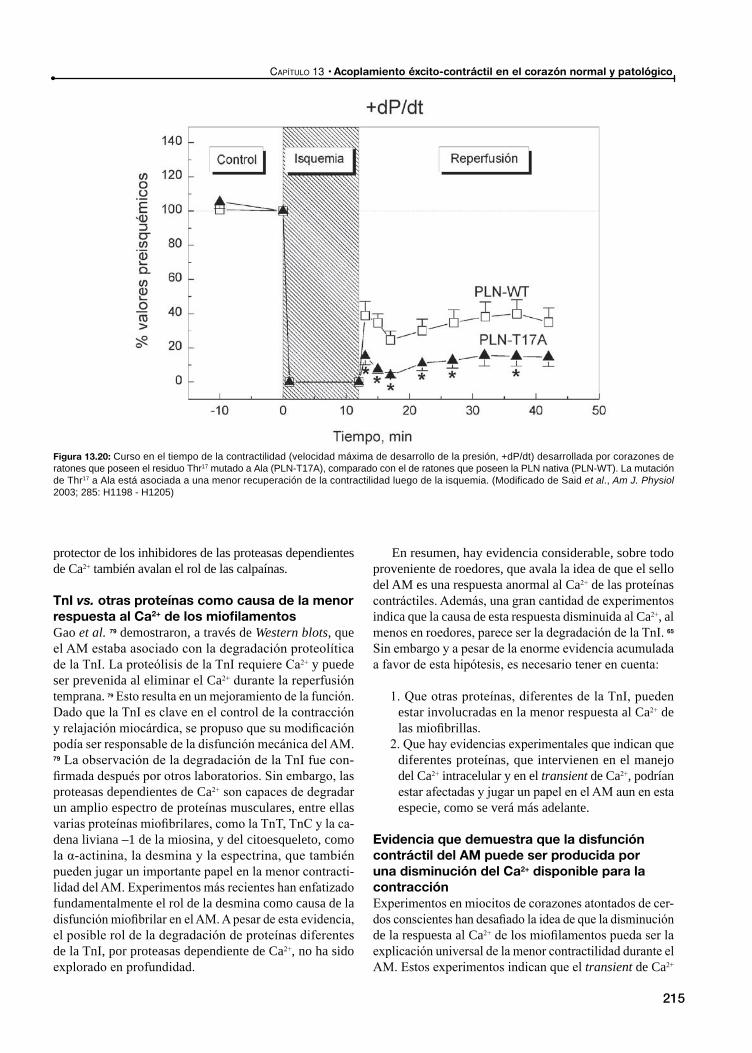
Acoplamiento éxcito-contráctil en el corazón normal y patológico
215
protector de los inhibidores de las proteasas dependientes de Ca también avalan el rol de las calpaínas.
TnI vs. otras proteínas como causa de la menor respuesta al Ca2+ de los miofilamentosGao et al. 79 demostraron, a través de Western blots, que
y puede ser prevenida al eliminar el Catemprana. 79
79 -
proteasas dependientes de Caun amplio espectro de proteínas musculares, entre ellas
-dena liviana –1 de la miosina, y del citoesqueleto, como
-
fundamentalmente el rol de la desmina como causa de la
de la TnI, por proteasas dependiente de Ca , no ha sido
En resumen, hay evidencia considerable, sobre todo proveniente de roedores, que avala la idea de que el sello del AM es una respuesta anormal al Ca de las proteínas
indica que la causa de esta respuesta disminuida al Ca , al 65
estar involucradas en la menor respuesta al Ca de
diferentes proteínas, que intervienen en el manejo del Ca intracelular y en el transient de Ca , podrían
especie, como se verá más adelante.
Evidencia que demuestra que la disfunción contráctil del AM puede ser producida por una disminución del Ca2+ disponible para la contracción
-
de la respuesta al Ca
transient de Ca
Figura 13.20: Curso en el tiempo de la contractilidad (velocidad máxima de desarrollo de la presión, +dP/dt) desarrollada por corazones de ratones que poseen el residuo Thr17 mutado a Ala (PLN-T17A), comparado con el de ratones que poseen la PLN nativa (PLN-WT). La mutación de Thr17 a Ala está asociada a una menor recuperación de la contractilidad luego de la isquemia. (Modificado de Said et al., Am J. Physiol 2003; 285: H1198 - H1205)
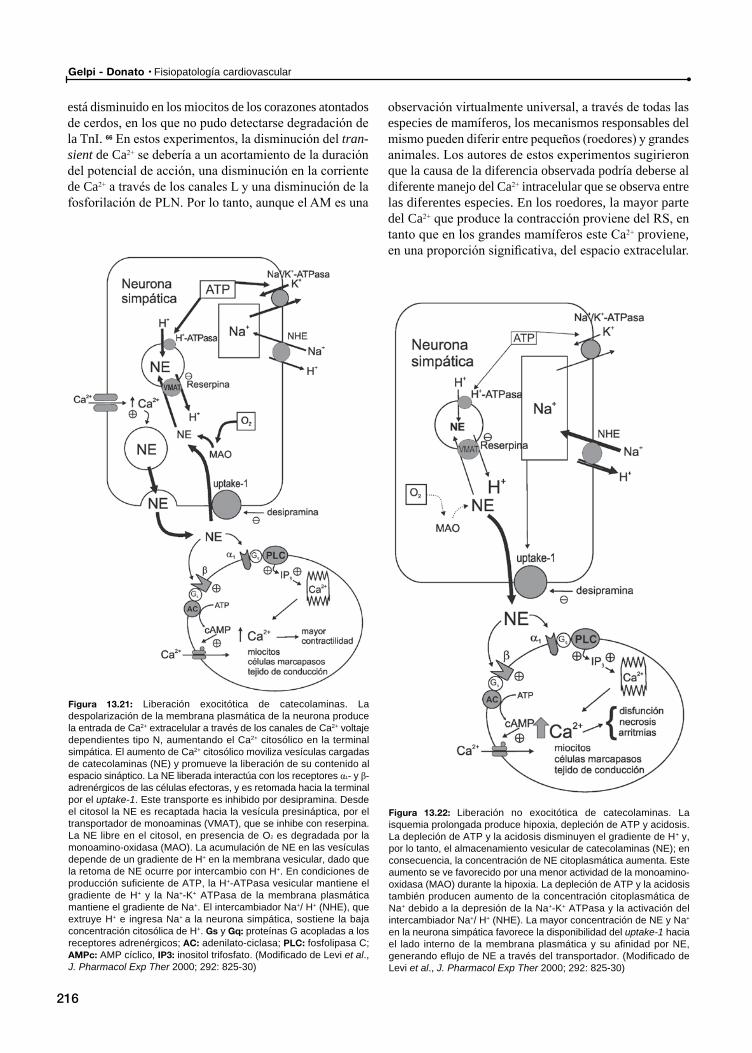
Gelpi - Donato Fisiopatología cardiovascular
216
está disminuido en los miocitos de los corazones atontados
la TnI. 66 tran-sient de Ca
de Ca
especies de mamíferos, los mecanismos responsables del
que la causa de la diferencia observada podría deberse al diferente manejo del Ca intracelular que se observa entre las diferentes especies. En los roedores, la mayor parte del Ca
proviene,
Figura 13.21: Liberación exocitótica de catecolaminas. La despolarización de la membrana plasmática de la neurona produce la entrada de Ca2+ extracelular a través de los canales de Ca2+ voltaje dependientes tipo N, aumentando el Ca2+ citosólico en la terminal simpática. El aumento de Ca2+ citosólico moviliza vesículas cargadas de catecolaminas (NE) y promueve la liberación de su contenido al espacio sináptico. La NE liberada interactúa con los receptores 1- y - adrenérgicos de las células efectoras, y es retomada hacia la terminal por el uptake-1. Este transporte es inhibido por desipramina. Desde el citosol la NE es recaptada hacia la vesícula presináptica, por el transportador de monoaminas (VMAT), que se inhibe con reserpina. La NE libre en el citosol, en presencia de O2 es degradada por la monoamino-oxidasa (MAO). La acumulación de NE en las vesículas depende de un gradiente de H+ en la membrana vesicular, dado que la retoma de NE ocurre por intercambio con H+. En condiciones de producción suficiente de ATP, la H+-ATPasa vesicular mantiene el gradiente de H+ y la Na+-K+ ATPasa de la membrana plasmática mantiene el gradiente de Na+. El intercambiador Na+/ H+ (NHE), que extruye H+ e ingresa Na+ a la neurona simpática, sostiene la baja concentración citosólica de H+. Gs y Gq: proteínas G acopladas a los receptores adrenérgicos; AC: adenilato-ciclasa; PLC: fosfolipasa C; AMPc: AMP cíclico, IP3: inositol trifosfato. (Modificado de Levi et al., J. Pharmacol Exp Ther 2000; 292: 825-30)
Figura 13.22: Liberación no exocitótica de catecolaminas. La isquemia prolongada produce hipoxia, depleción de ATP y acidosis. La depleción de ATP y la acidosis disminuyen el gradiente de H+ y, por lo tanto, el almacenamiento vesicular de catecolaminas (NE); en consecuencia, la concentración de NE citoplasmática aumenta. Este aumento se ve favorecido por una menor actividad de la monoamino-oxidasa (MAO) durante la hipoxia. La depleción de ATP y la acidosis también producen aumento de la concentración citoplasmática de Na+ debido a la depresión de la Na+-K+ ATPasa y la activación del intercambiador Na+/ H+ (NHE). La mayor concentración de NE y Na+ en la neurona simpática favorece la disponibilidad del uptake-1 hacia el lado interno de la membrana plasmática y su afinidad por NE, generando eflujo de NE a través del transportador. (Modificado de Levi et al., J. Pharmacol Exp Ther 2000; 292: 825-30)

Acoplamiento éxcito-contráctil en el corazón normal y patológico
217
66
las proteínas contráctiles están involucradas en el meca--
males, incluido el hombre. 80 Pero, por otra parte, se ha demostrado que las proteínas que manejan el Ca intra-celular están involucradas en el atontamiento miocárdico, aun en roedores. En estos animales se ha encontrado que
81 Además, el
la bomba de Caen corazones atontados de ratas. 82
residuos no fosforilables, indicaron que ambos residuos
mecánica como del Ca intracelular durante el AM83, 84, lo
del Ca podrían participar si no en la causa, en la recupe-
-
respuesta al Ca de las proteínas contráctiles, que parece predominar en los roedores y que posiblemente se deba a
Finalmente, estudios en roedores determinaron que en la -
teínas que, como la PLN, cumplen un papel en el manejo del Ca intracelular.
Rol de las catecolaminas en el atontamiento miocárdico
-quemia. Durante la isquemia miocárdica, las terminales nerviosas simpáticas sufren cambios que ocurren con
de catecolaminas desde la terminal simpática y, en conse-
Figura 13.23: Efecto de la depleción de catecolaminas sobre la recuperación contráctil posisquémica y la producción de radicales libres en el corazón atontado. Paneles superiores: Transcurso en el tiempo de la presión desarrollada (PD) y la presión diastólica final (PDF), de corazones aislados y perfundidos de rata, sometidos a 20 minutos de isquemia seguidos de 30 minutos de reperfusión. Los corazones se obtuvieron de animales no tratados (control) o tratados con reserpina (5 mg/kg, 24 horas antes del sacrificio). La reserpina, que vacía las terminales simpáticas de catecolaminas, mejoró significativamente la recuperación contráctil posisquémica. Panel inferior: El grado de peroxidación lipídica, secundaria a la formación de radicales libres, se evaluó por la producción de sustancias reactivas al ácido tiobarbitúrico (TBARS) al finalizar el periodo de reperfusión, en corazones de rata sometidos al protocolo de isquemia/reperfusión descrito más arriba. El tratamiento con reserpina disminuyó significativamente la producción de TBARS, indicando menor formación de radicales libres en el atontamiento. (Modificado de Vittone et al., Naunyn Schmiedeberg´s Arch Pharmacol 2006; 373: 60-70)

Gelpi - Donato Fisiopatología cardiovascular
218
85, 86
Fase 1 --
tema nervioso simpático central en respuesta al dolor, la
--
de sus aferentes en el miocardio isquémico, provocada
---
en la terminal
está limitada por mecanismos presinápticos de la termi-
los receptores presinápticos α , A1 y H por noradrenali-na, adenosina e histamina, respectivamente, sustancias liberadas al intersticio miocárdico durante la isquemia;
llamado . Fase 2 -
de catecolaminas en el miocardio isquémico cuya libera--
Ca -nápticos y puede ser suprimida por diferentes inhibidores del Estas propiedades en su conjunto indican que, bajo con-diciones isquémicas, el transporte y opera como un conductor que lleva catecola-
Fase 3
con el desarrollo de defectos estructurales de la membrana plasmática y no puede ser ya bloqueada por inhibidores del neuronal.
-
coronario. Es probable que, durante la fase temprana de -
-
ronal a través del , indicando que este mecanismo
87
Las catecolaminas liberadas desde las terminales ner-viosas simpáticas, en respuesta a la isquemia miocárdica,
-tecolaminas liberadas pueden favorecer mecanismos de
-nas inducida por la isquemia está involucrada en la cardio-
-88-91
e isoproterenol, mimetizan los efectos del precondicio-namiento isquémico. 92
en presencia de una limitada reserva coronaria, inclina el
-
85 -
sobre los efectos de las catecolaminas liberadas durante
otros laboratorios93-95 -tráctil posisquémica aumenta en corazones deplecionados de catecolaminas por tratamiento previo con reserpina, lo que corrobora que las catecolaminas, perjudican la recu-
α- y β -rante la isquemia159, 168, o por mecanismos independientes
65
-α y
AMPc, PKA, para el β en el miocito car- durante la
α- y βel precondicionamiento isquémico o en la necrosis tisular

Acoplamiento éxcito-contráctil en el corazón normal y patológico
219
-
en músculos papilares y corazones aislados y perfundidos,
de los receptores βcontráctil en el atontamiento 94 o la mejora. 97 Más aún, la
ciclasa que aumenta los niveles intracelulares de AMPc, 94
βdel miocardio podría activar, al menos en parte, mecanis-mos celulares que llevan al atontamiento.
de los receptores β1- y β
95 resulta-dos de nuestro laboratorio, obtenidos en corazones ais-
α1-, β1- y β -
β1β -
-
β-adre-
β1 no contrarresta
ββ
dependiente de la vía receptor β -lla dependiente de la vía receptor β1- y β -Gs-adenila-tociclasa. 99, 99
β-
β195 Ade-
durante la isquemia activan mecanismos celulares protec-tores a través de la vía α1
95 En resumen, en
posisquémica, efectos opuestos que se sobreponen.
isquémico. 100, 101 En un trabajo101 se demuestra que la de--
desarrollo de necrosis subendocárdica en el área de ries-
efecto protector de la actividad simpática sostenida en la -
-
-
no está aclarado todavía el efecto de las catecolaminas en el AM. Aunque es difícil establecer comparaciones
sobre el miocardio isquémico, dependerían, fundamen-talmente, de la cantidad de catecolaminas acumuladas en
α o β
Conclusiones
En este capítulo se ha realizado una apretada síntesis de los mecanismos involucrados en el AEC en el miocardio
-dico. Los actores principales fueron el manejo celular del ion Ca y las proteínas contráctiles que responden al Ca
-
sano y el enfermo. Basados en esto, la idea rectora del capítulo fue que el entendimiento de estos mecanismos celulares es el camino necesario para comprender las bases
Bigliografía
11. Guatimosin S, Dilly K, Santana LF, et al. “Local Ca2+ signaling and EC coupling in heart: Ca2+ sparks and the regulation of the [Ca2+]
i transient”. J Mol Cell Cardiol. 2002;34:941-50.
12. Hosey MM, Borsotto M, Lazdunski M. “Phosphorylation and dephosphorylation of dihydropyridine-sensitive voltage-dependent Ca2+ channel in skeletal muscle membranes by cAMP- and Ca2+-dependent processes”. Proc Natl Acad Sci USA. 1986;83:3733-37.
13. Sharp AH, Imagawa T, Leung AT, et al. “Identification and charac-terization of the dihydropyridine-binding subunit of the skeletal muscle dihydropyrine receptor”. J Biol Chem. 1987;262:12309-15.

Gelpi - Donato Fisiopatología cardiovascular
220
14. O’Callahan CM, Ptasienski J, Hosey MM.. “Phosphorylation of the 165-kDa dihydro-pirydine/phenylalkylamine receptor from skeletal muscle by protein kinase C”. J Biol Chem. 1988;263:17342-49.
15. Zucchi R, Ronca-Testoni S. “The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: Modulation by endogenous effectors, drugs and disease states”. Pharmacol Rev. 1997;49:1-51.
16. Wehrens XHT, Marks AR. “Altered function and regulation of car-diac ryanodine receptors in cardiac disease”. Trends in Biochemical Sci. 2003;28:671-8.
17. MacLennan DH, Abu-Abed M, Kang C. “Structure-Function relationships in Ca2+ cycling proteins”. J Mol Cell Cardiol. 2002;34:897-918.
18. Miura Y, Kimura J. “Sodium-calcium exchange current”. J Gen Physiol. 1989;93:1129-45.
19. Bridge JHB, Smolley JR, Spitzer KW. “The relationship between charge movements associated with I
Ca and I
Na-Ca in cardiac myo-
cytes”. Science. 1990;248:376-8.10. Bers DM, Bassani JW, Bassani RA. Na-Ca exchange and Ca fluxes
during contraction and relaxation in mammalian ventricular mus-cle”. Ann N Y Acad Sci. 1996;779:430-42.
11. Frank K, Kranias EG. “Phospholamban and cardiac contractility”. Ann Med. 2000;32:572-8.
12. Mundiña-Weilenmann C, Vittone L, et al. “Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart”. J Biol Chem. 1996;271:33561-67.
13. Luo W, Grupp IL, Harrer J, et al. “Targeted ablation of the phos-pholamban gene is associated with markedly enhanced myocar-dial contractility and loss of β-agonist stimulation”. Circ Res. 1994;75:401-9.
14. Kadambi VJ, Ponniah S, Harrer JM, et al. “Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice”. J Clin Invest. 1996;97:533-9.
15. Palomeque J, Vila Petroff MG, Mattiazzi A. “Pacing staircase phe-nomenon in the heart: From Bodwitch to the XXI century”. Heart Lung and Circulation. 2004;13:410-20.
16. Gwathmey JK, Copelas L, MacKinnon R, et al. “Abnormal in-tracellular calcium handling in myocardium from patients with end-stage heart failure”. Circ Res. 1987;61:70-6.
17. Hasenfuss G, Pieske B. “Calcium cycling in congestive heart failure”. J Mol Cell Cardiol. 2002;34:951-69.
18. Perez NG, Hashimoto K, McCune S, et al. “Origin of contractile dysfunction in heart failure: calcium cycling versus myofilaments”. Circulation. 1999;99:1077-83.
19. Reuter H, Pott C, Goldhaber JI, et al. “Na+-Ca2+ exchange in the regulation of cardiac excitation-contraction coupling”. Cardiovasc Res. 2005;67:198-207.
20. Reiken S, Gaburjakova M, Guatimosim S, et al. “PKA phosphoryla-tion of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts: role of phoshatases and response to isoproterenol”. J Biol Chem. 2003;278:444-53.
21. Schwinger RHG, Wang J, Frank K, et al. “Reduced sodium pump α
1, α
3, and β
1-isoform protein levels and Na+, K+-ATPase activity
but unchanged Na+-Ca2+ exchanger protein levels in human heart failure”. Circulation. 1999;99:2105-12.
22. Weber CR, Piacentino V 3rd, Houser SR, et al. “Dynamic regulation of sodium/calcium exchange function in heart failure”. Circulation. 2003;108:2224-29.
23. Ito K, Yan X, Tajima M, et al. “Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts”. Cir Res. 2000;87:588-95.
24. DiPaola NR, Sweet WE, et al. “β-adrenergic receptors and calcium cycling proteins in non-failing hypertrophied and failing human hearts: transition from hypertrophy to failure”. J Mol Cell Cardiol. 2001;33:1283-95.
25. del Monte F, Williams E, Lebeche D, et al. “Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic
reticulum Ca2+-ATPase in a rat model of heart failure”. Circulation. 2001;104:1424-9.
26. Lim HW, De Windt LJ, Mante J, et al. “Reversal of cardiac hy-pertrophy in transgenic disease models by calcineurin inhibition”. J Mol Cell Cardiol. 2000;32:697-709.
27. Olson EN, Molkentin JD. “Prevention of cardiac hypertrophy by calcineurin inhibition: hope or hype?” Circ Res. 1999;84:623-32.
28. Vila Petroff MG, Palomeque J, Mattiazzi A. “Na+/Ca2+ exchange function underlying contraction frequency inotropy in cat myo-cardium”. J Physiol. 2003;550:801-17.
29. Feldman MD, Gwathmey JK, Phillips P, et al. “Reversal of the force-frequency relationship in working myocardium from patients with end-stage heart failure”. J Appl Cardiol. 1988;3:273-83.
30. Gwathmey JK, Slawsky MT, Hajjar RJ, et al. “Role of intracellular calcium handling in force-interval relationships of human ventri-cular myocardium”. J Clin Invest. 1990;85:1599-613.
31. Schillinger W, Lehnart SE, Prestle J, et al. “Influence of SR Ca2+ATPase and Na+-Ca2+-exchanger on the force-frequency rela-tion”. Basic Res Cardiol. 1998;93:38-45.
32. Pieske B, Maier LS, Bers DM, et al. “Ca2+ handling and sarco-plasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium”. Circ Res. 1999;85:38-46.
33. Maier LS, Wahl-Schott C, Horn W, et al. “Increased SR Ca2+ cycling contributes to improved contractile performance in SERCA2a-overexpressing transgenic rats”. Cardiovasc Res. 2005;67:636-46.
34. del Monte F, Harding SE, et al. “Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a”. Circulation. 1999;100:2308-11.
35. Pieske B, Maier LS, Piacentino V III, et al. “Rate dependence of (Na+)
i and contractility in nonfailing and failing human myocar-
dium”. Circulation. 2002;106:447-53.36. Ahlquist RP. “A study of the adrenergic receptors”. Am J Physiol.
1948;153:586-600.37. Dohlman HG, Thorner J, Caron MG, et al. “Model systems for
the study of seven-transmembrane-segment receptors”. Ann Rev Biochem. 1991;60:653-88.
38. Tsien RW, Bean BO, Hess P, et al. “Mechanisms of calcium channel modulation by β-adrenergic agents and dihydropyridine calcium agonists”. J Mol Cell Cardiol. 1986;18:691-710.
39. Yue DT, Herzig S, Marbán E. “β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes”. Proc Natl Acad Sci USA. 1990;87:753-7.
40. McIvor ME, Orchard CH, Lakatta EG. “Dissociation of changes in apparent myofibrillar Ca2+ sensitivity and twitch relaxation induced by adrenergic and cholinergic stimulation in isolated ferret cardiac muscle”. J Gen Physiol. 1988;92:509-29.
41. Li L, DeSantiago J, Chu G, Kranias EG, Bers DM. “Phosphorylation of phospholamban and troponin I in β-adrenergic-induced accelera-tion of cardiac relaxation”. Am J Physiol. 2000;278:H769-79.
42. Said M, Mundiña-Weilenmann C, Vittone L, et al. “The relative relevance of phosphorylation of the Thr17 residue of phospholam-ban is different at different levels of β-adrenergic stimulation”. Pflügers Arch. 2002;444:801-9.
43. Perchenet L, Hinde AK, Patel KC, et al. “Stimulation of Na/Ca exchange by the β-adrenergic/protein kinase A pathway in guinea-pig ventricular myocytes at 37ºC”. Pflügers Arch. 2000;439:822-8.
44. Benkusky NA, Jiang MT, Farrell EF, et al. “PKA and CaMKII phosphorylation of ryanodine receptors in canine heart failure”. J Mol Cell Cardiol. 2004;37:225 (Resumen).
45. Valdivia HH, Kaplan JH, Ellis-Davies GC, et al. “Rapid adap-tation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation”. Science. 1995;267:1997-2000.
46. Marx SO, Reiken S, Hisamatsu Y, et al. “PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (rya-nodine receptor): defective regulation in failing hearts”. Cell. 2000;101:365-76.
47. Terentyev D, Viatchenko-Karpinski S, Gyorke I, et al. “Protein phosphatases decrease sarcoplasmic reticulum calcium content

Acoplamiento éxcito-contráctil en el corazón normal y patológico
221
by stimulating calcium release in cardiac myocytes”. J Physiol. 2003;552:109-18.
48. Li L, Satoh H, Ginsburg KS, et al. “The effects of CaMKII on cardiac excitation-contraction coupling in ferret ventricular myo-cytes”. J Physiol. 1997;501:17-32.
49. Okazaki O, Suda N, Hongo K, Konishi M, Kurihara S. “Modula-tion of Ca2+ transients and contractile properties by β adrenoceptor stimulation in ferret ventricular myocytes”. J Physiol. 1990;423:221-40.
50. Rockman HA, Koch WJ, Lefkowitz RJ. “Seven-transmembrane-spanning receptors and heart function”. Nature. 2002;415:206-12.
51. Xiao RP, Ji X, Lakatta EG. “Functional coupling of the β2-adreno-
ceptor to a pertussis toxin-sensitive G protein in cardiac myocytes”. Mol Pharmacol. 1995;47:322-9.
52. Lefkowitz RJ. “G protein-coupled receptors. III. New roles for receptor kinases and β-arrestins in receptor signaling and desen-sitization”. J Biol Chem. 1998;273:18677-80.
53. Packer M. “Neurohormonal interactions and adaptations in con-gestive heart failure”. Circulation. 1988;77:721-30.
54. Bristow MR, Ginsburg R, Minobe W, et al. “Decreased catecho-lamine sensitivity and β-adrenergic-receptor density in failing human hearts”. N Engl J Med. 1982;307:205-11.
55. Yi XP, Gerdes AM, Li F. “Myocyte redistribution of GRK2 and GRK5 in hypertensive, heart-failure-prone rats”. Hypertension. 2002;39:1058-63.
56. Iaccarino G, Keys JR, Rapacciuolo A, et al. “Regulation of myocar-dial βARK1 expression in catecholamine-induced cardiac hypertro-phy in transgenic mice overexpressing α
1B-adrenergic receptors”. J
Am Coll Cardiol. 2001;38:534-40.57. Iaccarino G, Dolber PC, Lefkowitz RJ, Koch WJ. “β-adrenergic
receptor kinase-1 levels in catecholamine-induced myocardial hypertrophy: regulation by β- but not α
1-adrenergic stimulation”.
Hypertension. 1999;33:396-401.58. Packer M. “The development of positive inotropic agents for chro-
nic heart failure: how have we gone astray?” J Am Coll Cardiol. 1993;22(4 Suppl A):119A-26A.
59. Hajjar RJ, Kang JX, Gwathmey JK, et al. “Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes”. Circulation. 1997;95:423-9.
60. del Monte F, Williams E, Lebeche D, et al. “Improvement in sur-vival and cardiac metabolism following gene transfer of SERCA2a in a rat model of heart failure”. Circulation. 2001;104:1424-9.
61. Jiang D, Xiao B, Zhang L, et al. “Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant asso-ciated with ventricular tachycardia and sudden death”. Circ Res. 2002;91:218-225.
62. Braunwald E, Kloner RA. “The stunned myocardium: prolonged postischemic ventricular dysfunction”. Circulation. 1982;66:1146-9.
63. Heyndrickx GR, Millard RW, McRitchie RJ, et al. “Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs”. J Clin Inves. 1975;56:978-85.
64. Nixon JV, Brown CN, Smitherman TC. “Identification of transient and persistent segmental wall motion abnormalities in patients with unstable angina by two-dimensional echocardiography”. Circulation. 1982;65:1497-1503.
65. Bolli R, Marbán E. “Molecular and Cellular Mechanisms of Myo-cardial Stunning”. Physiol Rev. 1999;79:609-34.
66. Kim SJ, Kudej RK, Yatani A, et al. “A novel mechanism for myo-cardial stunning involving impaired Ca2+ handling”. Circ Res. 2001;89:831-7.
67. Kusuoka H, Porterfield JK, Weisman HF, et al. “Pathophysiology and pathogenesis of stunned myocardium. Depressed Ca2+ activa-tion of contraction as a consequence of reperfusion-induced cellular calcium overload in ferret hearts”. J Clin Invest. 1987;79:950-61.
68. Marbán E, Kitakaze M, Chacko VP, et al. “Ca2+ transients in per-fused ferret hearts revealed by gated 19F NMR spectroscopy”. Circ Res. 1988:93:673-8.
69. McDonald KS, Mammen PA, Strang KT, et al. “Isometric and dynamic contractile properties of porcine skinned cardiac myocytes after stunning”. Circ Res. 1995;77:964-72.
70. Bolli R. “Myocardial stunning in man”. Circulation. 1992;86:1671-91.
71. Cross HR, Radda GK, Clarke K. “The role of Na+/K+ ATPase activity during low flow ischemia in preventing myocardial injury: a 31P, 23Na and 87Rb NMR spectroscopic study”. Magnetic Resonance in Medicine. 1995;34:673-85.
72. Van Emous J, Nederhoff MG, Ruigrok TJ, et al. “The role of the Na+ channel in the accumulation of intracellular Na+ during myocardial ischemia: consequences for post-ischemic recovery”. J Mol Cell Cardiol. 1997;29:85-96.
73. Xiao XH, Allen DG. “Role of Na+/H+ exchanger during ische-mia and preconditioning in the isolated rat heart”. Circ Res. 1999;85:723-30.
74. Mosca SM, Cingolani HE. “Comparison of the protective effects of ischemic preconditioning and the Na+/H+ exchanger blockade”. Naunym-Schmiedeberg’s Arch Pharmacol.2000; 362:7-13.
75. Cross H, Lu L, Steenbergen C, Phillipson K, et al. “Overexpression of the Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion in male but not in female transgenic mice”. Circ Res. 1998;83:1215-23.
76 . Ladilov Y, Haffner S, Balser-Schäfer C, et al. “Cardioprotective effects of KB-R7943: a novel inhibitor of the reverse mode of Na+/Ca2+ exchanger”. Am J Physiol. 1999;276:H1868-76.
77. Matsumura Y, Seki E, Otsu K, et al. “Intracellular calcium level required for calpain activation in a single myocardial cell”. J Mol Cell Cardiol. 2001;33:1133-42.
78. Yoshida K, Sorimachi Y, Fujiwara M, et al. “Calpain is implicated in rat myocardial injury after ischemia or reperfusion”. Jpn Circ J. 1995;59:40-8.
79. Gao WD, Atar D, Liu Y, et al. “Role of troponin I proteolysis in the pathogenesis of stunned myocardium”. Circ Res. 1997;80:393-9.
80. Andres J, Moczarska A, Stepkowski D, et al. “Contractile pro-teins in globally stunned rabbit myocardium”. Basic Res Cardiol. 1991;86:219-26.
81. Wu QY, Feher JJ. “Ryanodine perfusion decreases cardiac me-chanical function without affecting homogenate sarcoplasmic reticulum Ca2+ uptake: comparison with the stunned heart”. J Mol Cell Cardiol. 1996;28:943-55.
82. Vittone L, Mundiña-Weilenmann C, Said M, et al. “Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways”. J Mol Cell Cardiol. 2002;34:39-50.
83.
Said M, Vittone L, Mundiña-Weilenmann C, et al. “Role of dual-site phospholamban phosphorylation in the stunned heart: insights from phospholamban site-specific mutants”. Am J Physiol. 2003;285:H1198-H1205.
84. Valverde CA, Mundiña-Weilenmann C, Reyes M, et al. “Phospho-lamban phosphorylation sites enhace the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart . Cardivasc Res 70:335-45; 2006.
85. Schömig, A. “Catecholamines in myocardial ischemia. Systemic and cardiac release . Circulation 82(suppl II):II-13-II-22; 1990.
86. Levi R, Smith NC. “Histamine H3-receptors: a new frontier in
myocardial ischemia”. J Pharmacol Exp Ther. 2000;292:825-30.87. Lameris TW, de Zeeuw S, Alberts G, et al. “Time course and mecha-
nism of myocardial catecholamine release during transient ischemia in vivo”. Circulation. 2000;101:2645-50.
88. Hu K, Nattel S. “Mechanisms of ischemic preconditioning in rat hearts. Involvement of α
1B-adrenoceptors, pertussis toxin-sensitive
G proteins, and protein kinase C”. Circulation. 1995;92:2259-65.89. Vatner DE, Sato S, Vatner SF, et al. “Sympathetic signal trans-
duction in myocardial ischemia”. In: Heyndrickx GR, Vatner SF, Wijns W, editors. Stunning, Hibernation and Preconditioning: Clinical Pathophysiology of Myocardial Ischemia. Lippincott-Raven Publishers, Philadelphia. 1997; p. 31-48.

Gelpi - Donato Fisiopatología cardiovascular
222
90. Sharma A, Singh M. “The possible role of adrenergic component in ischemic preconditioning”. Methods Find Exp Clin Pharmacol. 1997;19:493-9.
91. Lochner A, Genade S, Tromp E, Podzuweit T, Moolman JA. “Is-chemic preconditioning and the β-adrenergic signal transduction pathway”. Circulation. 1999;100:958-66.
92. Miyawaki H, Ashraf M. “Isoproterenol mimics calcium precon-ditioning-induced protection against ischemia”. Am J Physiol. 1997;272:H927-36.
93. Hirata M, Fukui H, Shimamoto N, Goto N. “Inhibition of myocardial calcium accumulation during ischemia and reperfu-sion by reserpine in isolated guinea pig hearts”. Jpn J Pharmacol. 1982;32:573-76.
94. Ishiguro Y, Morgan JP. “Effect of endogenous catecholamine on myocardial stunning in a simulated ischemia model”. Fundam Clin Pharmacol. 2001;15:111-6.
95. Vittone L, Said M, Mattiazzi A. “β2-adrenergic stimulation is
involved in the contractile dysfunction of the stunned heart . Naunyn-Schmiedeberg’s Arch Pharmacol 2006; 373: 60-70.
96. Salvi, S. “Protecting the myocardium from ischemic injury. A critical role for α
1-adrenoreceptors?” Chest. 2001;119:1242-9.
97. Temsah RM, Dyck C, Netticadan T, et al. “Effect of β-adre-noceptor blockers on sarcoplasmic reticular function and gene expression in the ischemic-reperfused heart . J Pharmacol Exp Ther 293:15-23; 2000.
98. Daaka Y, Luttrell LM, Lefkowitz RJ. “Switching of the coupling of the β
2-adrenergic receptor to different G proteins by protein
kinase A . Nature 390:88-91; 1997.99. Bartels LA, Clifton GD, Szabo TS. “Influence of myocardial
ischemia and reperfusion on β-adrenoceptor subtype expression . J Cardiovasc Pharmacol 31:484-7; 1998.
100. Lavallee M, Amano J, Vatner SF, et al. “Adverse effects of chronic cardiac denervation in conscious dogs with myocardial ischemia”. Circ Res. 1985;57:383-92.
101. Huang CH, Vatner SF, Peppas AP, et al. “Cardiac nerves affect myocardial stunning through reactive oxygen and nitric oxide mechanisms”. Circ Res. 2003;93:866-73.

223
Fisiopatología de la insuficiencia cardiaca 14
del ventrículo izquierdo– es la variable que determina si
-
conocidas, los procesos responsables –tanto considerando el ventrículo intacto como los mecanismos moleculares–
-canismos más importantes que se activan en las distintas
-
La --
-ro. 1
-
1 Otro dato no menor es el hecho de que
-
de este concepto simplista también se consideraba a la
-
los problemas circulatorios, consecuencia de una dismi--
-secuencia del mismo. Con cierta cautela se podría decir
-
Citoprotección
Hipertrofia ReparaciónInjuria tisular
Figura 14.1: El esquema muestra los tres mecanismos más importantes que permiten al corazón adaptarse a los distintos tipos de injuria, ya sea isquémica o por cambios severos en las condiciones de carga. La integración y coordinación de estos mecanismos responsables son condiciones críticas para mantener la homeostasis miocárdica que llevará a un adecuado funcionamiento cardiaco.

Gelpi - Donato Fisiopatología cardiovascular
224
-nen volumen minuto normal, o incluso, en lo que sería el caso opuesto, el ventrículo izquierdo puede tener una
ventrículo, además de otras variables como la frecuencia
-
-
tres estadios.El primer estadio comienza en el momento en que el
-
-mos compensadores que permiten mantener el volumen
-
esta etapa, las manifestaciones clínicas están ausentes y
y difícil de predecir.
-miento de los mecanismos compensadores conspiran para
esta etapa, la estructura y la forma del ventrículo izquierdo
y la son acti-
con independencia de las condiciones hemodinámicas del
estos eventos resultarán en manifestaciones clínicas mo-deradas que requerirán tratamiento médico. En este punto,
-2
morbilidad y mortalidad, siendo este estadio terminal, a -
--
-
-canismos involucrados en el proceso de remodelamiento.
Remodelamiento cardiaco
tratada en detalle en el Capítulo 9, aquí mencionaremos
El término remodelamiento cardiaco incluye cambios --
3 Desde un punto --
mitantes que ocurren en las aurículas, válvulas, vasos san-4 Así, los términos remodelamien-
to cardiaco y miocárdico intentan describir los diferentes procesos adaptativos que se producen en el miocardio, y
-portante mencionar que es el remodelamiento cardiaco el determinante más importante de las manifestaciones clí-
-mento de la masa del ventrículo izquierdo pero sin in-
hipertro-
normal.5
de los miocitos que compensa la pérdida de miocardio.
a nivel del miocito, aunque esto no siempre se traduce en
---
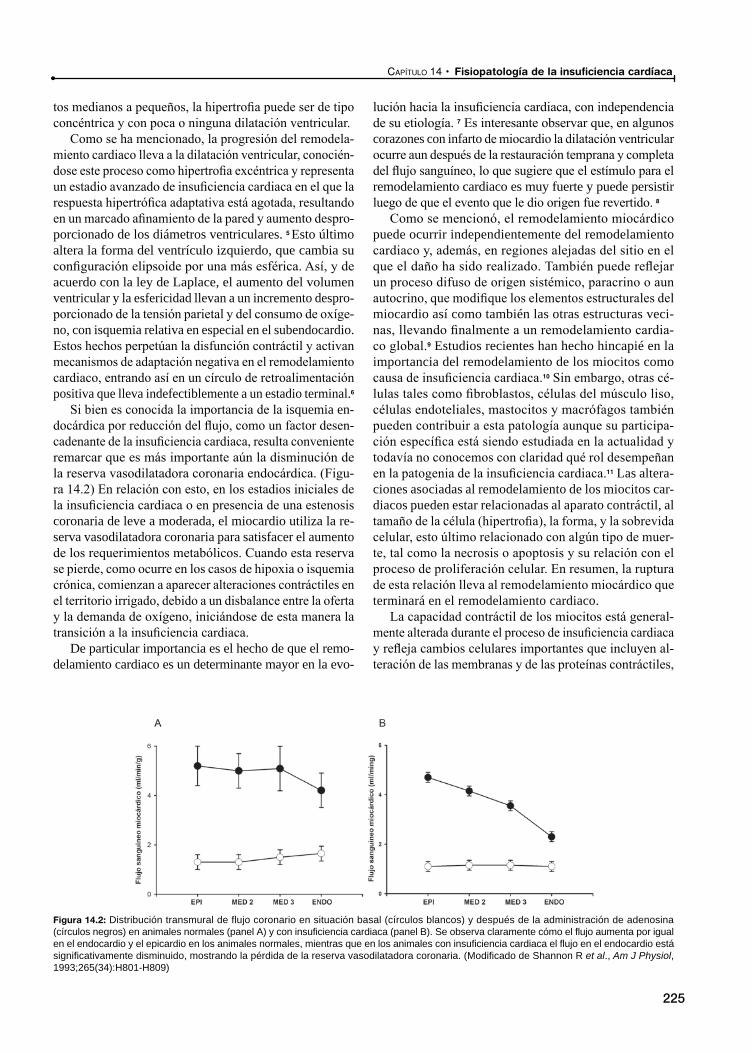
Fisiopatología de la insuficiencia cardíaca
225
--
-porcionado de los diámetros ventriculares. 5 Esto último altera la forma del ventrículo izquierdo, que cambia su
acuerdo con la ley de Laplace, el aumento del volumen ventricular y la esfericidad llevan a un incremento despro-
-no, con isquemia relativa en especial en el subendocardio.
positiva que lleva indefectiblemente a un estadio terminal.6
isquemia en--
-
coronaria de leve a moderada, el miocardio utiliza la re-serva vasodilatadora coronaria para satisfacer el aumento
De particular importancia es el hecho de que el remo-delamiento cardiaco es un determinante mayor en la evo-
7
corazones con infarto
remodelamiento cardiaco es muy fuerte y puede persistir 8
puede ocurrir independientemente del remodelamiento
miocardio así como también las otras estructuras veci--
9 Estudios recientes han hecho hincapié en la importancia del remodelamiento de los miocitos como
10 -
-
11 Las altera-ciones asociadas al remodelamiento de los miocitos car-diacos pueden estar relacionadas al aparato contráctil, al
-
terminará en el remodelamiento cardiaco.-
-
Figura 14.2: Distribución transmural de flujo coronario en situación basal (círculos blancos) y después de la administración de adenosina (círculos negros) en animales normales (panel A) y con insuficiencia cardiaca (panel B). Se observa claramente cómo el flujo aumenta por igual en el endocardio y el epicardio en los animales normales, mientras que en los animales con insuficiencia cardiaca el flujo en el endocardio está significativamente disminuido, mostrando la pérdida de la reserva vasodilatadora coronaria. (Modificado de Shannon R et al., Am J Physiol, 1993;265(34):H801-H809)

Gelpi - Donato Fisiopatología cardiovascular
226
así como también en el metabolismo y el acoplamiento 3, 12 Estudios realizados en células
contráctil de estas células no es simplemente una con-
-
-dad de los miocitos. 12, 13 De esta manera, mientras que la
ventricular, la ausencia de alteraciones contráctiles en el
característicos del remodelamiento cardiaco y serán men-cionados más adelante.
Hipertrofia celular
más importantes a la injuria, ya que está presente en cualquiera de sus formas en prácticamente todas las
-cas, podríamos decir que prácticamente no hay pato-
-ventricular, en lo que se
descompensada14, 15, que transforma a
16 Podría decirse, sin temor a equivocarse, que
actual. Prueba de esto es que aún desconocemos si la
-
de los miocitos sería difícil para el ventrículo mantener
-
es una respuesta muy conocida que ocurre frente al au-
remodelamiento celular.3, 17
miocárdica es probablemente un mecanismo adaptativo,
por aumentar el número de unidades contráctiles dentro de cada miocito, lo que lleva de forma simultánea a una
-
tiempo, se transforma en una respuesta mal adaptativa. En
intersticial y arritmias severas.18
-
claro, así como tampoco son conocidos los mecanismos
-raleza del estímulo el miocito puede sobrevivir, alcanzando
-letéreos tales como la apoptosis, que podría llevar a la in-
Una de las vías que se activan en respuesta a un incremen-
-tracelulares del complejo enzimático de las MAP kinasas
que componen la familia de las MAP kinasas promueven -
tativa, mientras que otras estimulan la -
tricular. Otras vías intracelulares no adaptativas incluyen la
II, a la aldosterona o al factor de crecimiento transforma-
factor bidireccionales, ya que a bajas concentraciones tienen pro-piedades protectoras, mientras que a altas concentraciones son deletéreas. Otras vías intracelulares que promueven un
19
-
-
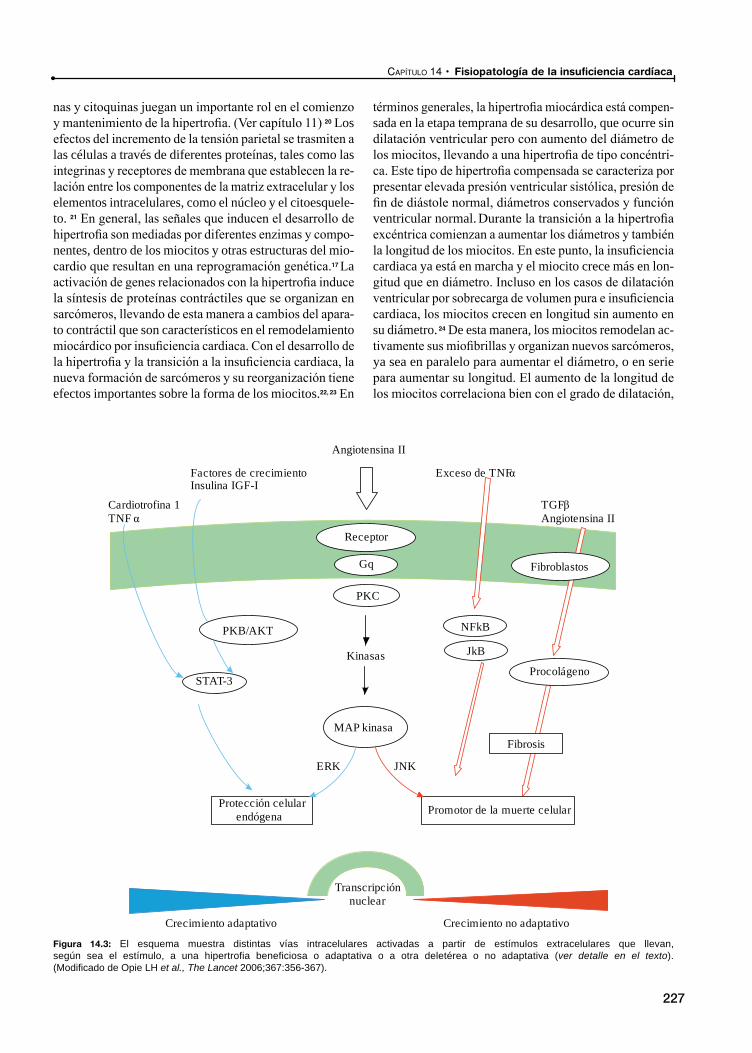
Fisiopatología de la insuficiencia cardíaca
227
20 Los
las células a través de diferentes proteínas, tales como las -
elementos intracelulares, como el núcleo y el citoesquele-to. 21
-nentes, dentro de los miocitos y otras estructuras del mio-
17 La
-to contráctil que son característicos en el remodelamiento
efectos importantes sobre la forma de los miocitos.22, 23 En
-sada en la etapa temprana de su desarrollo, que ocurre sin
-
ventricular normal.
cardiaca ya está en marcha y el miocito crece más en lon-
su diámetro. 24 De esta manera, los miocitos remodelan ac-
ya sea en paralelo para aumentar el diámetro, o en serie
Cardiotrofina 1TNF
Factores de crecimientoInsulina IGF-I
Angiotensina II
PKC
Kinasas
STAT-3
MAP kinasa
ERK JNK
ovitatpada on otneimicerCovitatpada otneimicerC
Transcripción nuclear
NFkB
JkB
Exceso de TNF
TGFAngiotensina II
Receptor
Gq Fibroblastos
PKB/AKT
Procolágeno
Protección celular endógena
Fibrosis
Promotor de la muerte celular
Figura 14.3: El esquema muestra distintas vías intracelulares activadas a partir de estímulos extracelulares que llevan, según sea el estímulo, a una hipertrofia beneficiosa o adaptativa o a otra deletérea o no adaptativa (ver detalle en el texto). (Modificado de Opie LH et al., The Lancet 2006;367:356-367).

Gelpi - Donato Fisiopatología cardiovascular
228
16
realineamiento y desplazamiento de los miocitos en rela-slipage
-
25
Apoptosis en la insuficiencia cardiaca
cardiaca se caracteriza, como fue mencionado, por cambios estructurales adversos y
cierto punto de no retorno, continúa a pesar de que la in-
número de miocitos.26
miocitos se pierde, lo que contribuye al aumento de la
por isquemia relativa, debido al aumento del consumo de
la necrosis, pareciera ser que la apoptosis es el mecanismo 27-29
in vitro están asociados con un aumento del número de células muertas por apoptosis. 30 Esta forma de muerte celular en
31, mientras que en los corazones normales la tasa de apoptosis es de alrededor
32
que la pérdida de miocitos por apoptosis contribuye a la
de si un nivel de apoptosis tan bajo puede contribuir real-
podría ser que una pérdida de miocitos mantenida en el
cascada de enzimas proteolíticas denominadas caspasas. Cuando estas enzimas son activadas, las caspasas termi-
caspasas ocurre a través de dos mecanismos diferentes.
-
-
9. Aunque estos dos mecanismos son distintos en sus orí-
desde la mitocondria.28 Un aspecto interesante y novedoso es que si bien la apoptosis está incrementada en los corazo-
miocitos, en los que el Citocromo C es liberado de la mito-
28, en un proceso conocido como apoptosis interruptus.33, 34 Aunque el mecanismo por
-mento de la actividad de la proteína XIAP, la que a su vez
33 Otra posibilidad -
mitocondria.35 Otro factor adicional para limitar la apop-
del metabolismo de los miocitos. Dado que la apoptosis
estas condiciones de vida muera por apoptosis. 36 De esta
disminuyendo el número de células por pérdida de los mio-
marcado de apoptosis en miocitos y otros tipos celulares
activan bajo ciertas circunstancias.
Regeneración y reparación miocárdica
la sobrevida no son los únicos aspectos involucrados
El concepto, aunque muy controvertido, de la pérdida

Fisiopatología de la insuficiencia cardíaca
229
de miocitos por diferentes tipos de muerte celular y su
en duda la creencia de que estas células no se dividen
miocárdica. Estos conceptos están basados en que la muerte de los
-
Así, estimaciones de la tasa de que un mecanismo de reemplazo de los miocitos podría estar presente para el mantenimiento del músculo cardia-
37 -
-citos, tal como ocurre en el infarto de miocardio, y esto
et al.38-40
miocardio. Estas observaciones se realizaron utilizan-
miocitos estuvo presente en el miocardio normal y par-ticularmente en la zona borde del infarto. Estos inves-
los miocitos cardiacos adultos son células terminalmente
-
-tores mencionan que el ventrículo izquierdo podría ser
41
celular no parecen estar activos en las zonas infartadas, donde la pérdida de miocitos es masiva, y estarían limi-
viable remanente.
-madre cardiacas, que estarían presentes
-
-
miocárdico. 37
Sistema renina-angiotensina-aldosterona
aldosterona es activado desde los estadios iniciales de la
mecánico. En miocitos cardiacos aislados sometidos a
-tos.42, 43
-
-contramos cierta discrepancia, ya que si bien la mayoría demuestra un efecto favorable sobre el remodelamien-to44-46 47 e
48-49 Por otro lado, cuando se analizan las causas de estas diferencias y los mecanismos involucrados, encontramos
-
50
de la ECA puede tener efectos importantes sobre otras vías intracelulares como las activadas por bradikinina,
-
síntesis de
todavía no se conocen.
Sistema adrenérgico
-
-51 Incluso en las etapas iniciales de la
cuando el ventrículo izquierdo está bien compensado, ya -
52
que todavía no conocemos en detalle las consecuencias -
mecanismos compensatorios tales como el incremento de la frecuencia cardiaca y la contractilidad, el avance
-
en deletérea. 53

Gelpi - Donato Fisiopatología cardiovascular
230
54
de los
55 Por otro lado, la estimula-56 -
pática el aumento de masa ventricular. Este efecto dele-
57
que apoya la como causante -
actúan como mecanismos compensadores en las etapas -
Citoquinas inflamatorias
injuria celular y representa uno de los mecanismos por los
rol de la -ponsables de la enfermedad cardiovascular, ha merecido
58 -diada por la
más importantes es el -
cos, tales como el cáncer, el shock séptico y alteraciones
de citoquinas puede tener, bajo ciertas circunstancias, un efecto protector contra las enfermedades, mientras que
citoquinas puede ser deletérea. Levine et al., en 199059, fueron los primeros en mostrar que el -
-
han llevado a plantear la como 60
contribuyen al proceso de remodelamiento y posterior
-
también puede ser sintetizado por los miocitos cardiacos.
61, 62, así como
volumen. 63
--
64
65 -
-
observaron efectos favorables sobre el remodelamiento
cardiaca. 66
los estudios que evaluaron el tratamiento de pacientes
que ser suspendido ante la falta de un efecto favorable en los pacientes. 67 -
con la complejidad del funcionamiento del los tiempos apropiados para la terapia anticitoquina y, por último, a la falta de conocimiento que tenemos sobre
citoquinas.
Rol de la matriz extracelular
La matriz -
-
-
-ciente. El concepto clásico de que la MEC es solamente un mecanismo o una estructura pasiva de soporte de los miocitos ya ha sido reemplazado por completo, pues la MEC constituye por sí misma un tejido dinámico que alte-
68 Un metabolismo activo y continuo de la MEC está pre-

Fisiopatología de la insuficiencia cardíaca
231
miocardio y los 69
los procesos de remodelamiento
tercio de todas las células miocárdicas, aunque ocupen más de dos tercios del volumen total miocárdico. 70 Los
71
de la MEC consiste sobre todo en una compleja red de 72 La elastina es menos abundante en el
miocardio que en la vasculatura y, dado que el músculo cardiaco tiene una elasticidad limitada, el rol de la elastina
intervienen en los mecanismos intracelulares, sin embar-
-
cardiaca están asociadas con alteraciones en la cantidad, 73
y tiene como objetivo dar soporte estructural a un área
necrosis sería un mecanismo
ruptura cardiaca74
---
tica75
-
-sis y cicatriz es que en la primera se observa un proceso
cicatriz se observan -
-
-
--
-
causa o consecuencia. 76
los miocitos es un proceso que puede estar asociado a la -
torio. 77
también en el miocardio viable de zonas alejadas al infarto 78, 79
alejadas del infarto. 80
-
--
-
-
dilatada, inducido por marcapaseo a alta frecuencia car--
80, 81
-
-
miocardiopatía inducida por isoproterenol. 80 Estos datos
Aunque la miocardiopatía inducida por alta frecuencia -
ca en humanos, estos estudios proveen fuerte evidencia -
modelamiento ventricular. Por otro lado, este modelo de miocardiopatía

Gelpi - Donato Fisiopatología cardiovascular
232
con la miocardiopatía dilatada observada en pacientes.
con este tipo de miocardiopatía presentan un aumento 82 -
mentales y la miocardiopatía en pacientes puede radicar
-endocárdica, que es uno de los principales estímulos para
-portante, se deben considerar también su calidad y com-
-
83
-
84
autores 97 mostraron que alteraciones en el entrecruzamien--
cambios ocurren en forma independiente de la cantidad de
del entrecruzamiento está asociado también a un aumen-85
controversia.86-88
-
-
89 De esta manera, los com-ponentes de la MEC no son estructuras estables sino que
-dad coordinada de cuatro familias de enzimas proteolíticas que incluyen las proteínas aspárticas, proteasas de cisteí-na, proteasas de serina y proteasas relacionadas a iones metálicos como son las últimas son las que están principalmente involucradas en
81
Las MMP representan una familia importante de enzi-
-
-
dos, como ocurre en el cáncer, la artritis y la mayoría de las enfermedades cardiovasculares. 89
-teínas de la MEC, aunque se diferencian en que cada una
y coordinada durante el remodelamiento. Las MMP se cla-
-
-
y simultáneamente activan péptidos intersticiales como
-car la arquitectura y el remodelamiento ventricular. En el
-tizan y secretan la mayoría de las MMP y las ADAMs.89, 90
-
remodelamiento de la matriz por las MMP es una respuesta común del miocardio a la injuria tisular. 91-100
un efecto deletéreo importante. Por ejemplo, la MMP-9 aparece como una de las enzimas más importantes en el
101-106 Una prueba del efecto deletéreo de las MMP lo han aporta-
de MMP-1; estos desarrollan un remodelamiento ventri-107
infarto de miocardio. 108 -
ruptura miocárdica después del infarto de miocardio. 109 De
pone a las MMP-9 como una de las enzimas más activas
--
110
de las MMP, en ratones con infarto de miocardio, atenúa

Fisiopatología de la insuficiencia cardíaca
233
y otros factores de crecimiento que están incrementados
como inductores importantes de las síntesis de las MMP, perpetuando de esta manera un círculo vicioso deletéreo.
-
Bibliografía
11. Thom T, Haase N, Rosamond W, et al. American Heart Associa-tion Statistics Committee and Stroke Statistics Subcommittee. “Heart disease and stroke statistics--2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee”. Circulation 2006; 113(6): e85-e151.
12. Hoshijima M, KR Chien. “Mixed signals in heart failure: cancer rules”. J Clin Invest 2002; 109 (7): 849-855.
13. Swynghedauw, B. “Molecular mechanisms of myocardial remodeling”. Physiol Rev 1999; 79:849-855.
14. Cohn JN, Ferrari R, Sharpe N. “Cardiac remodeling – concepts and clinical implications: a consensus paper from and international forum on cardiac remodeling Behalf of and International Forum on Cardiac Remodeling”. J Am Coll Cardiol 2000; 35(3): 569-582.
15. Gelpi RJ, O Schwint. “Hipertrofia cardiaca”. En: Bertolasi, Car-diología. Buenos Aires, Ed. Panamericana, 2000; 111-134.
16. Hittinger L, Mirsky I, Shen YT, et al. “Hemodynamic mechanisms responsible for reduced subendocardial coronary reserve in dogs with severe left ventricular hypertrophy”. Circulation 1995; 92(4): 978-86.
17. Udelson JE, MA Konstam. “Relation between left ventricular remode-ling and clinical outcomes in heart failure patients with left ventricular systolic dysfunction”. J Card Fail 2002; 8(6 suppl):S465-471.
18. Bolognese L, Neskovic AN, Parodi G, et al. “Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications”. Cir-culation 2002; 106(18):2351-2357.
19. Paul M, Poyan Mehr A, Kreutz R. “Physiology of local renin-angiotensin systems”. Physiol Rev. 2006; 86(3):747-803.
10. Li F, Wang X, Yi XP, et al. “Structural basis of ventricular remo-deling: role of the myocyte”. Curr Heart Fail Rep. 2004; 1(1):5-8.
11. Ushikoshi H, Takahashi T, Chen X, et al. “Local overexpression of HB-EGF exacerbates remodeling following myocardial infarction by activating noncardiomyocytes”. Lab Invest. 2005; 85(7):862-73.
12. Houser SR, KB Margulies. “Is depressed myocyte contractility centrally involved in heart failure?” Circ Res 2003; 92(4):350-358.
13. Baicu CF, Stroud JD, Livesay VA, et al. “Changes in extracellular collagen matrix alter myocardial systolic performance”. Am J Physiol 2003; 284(1):H122-132.
14. Wright JW, Mizutani S, Harding JW. “Pathways involved in the transition from hypertension to hypertrophy to heart failure. Treatment strategies”. Heart Fail Rev. 2008; 13(3): 367-75.
15. Morisco C, Sadoshima J, Trimarco B, et al. “Is treating cardiac hypertrophy salutary or detrimental: the two faces of Janus”. Am J Physiol 2003; 284(4): H1043-1047.
16. Galderisi M, O Divitiis. “Risk Factor-induced Cardiovascular Re-modeling and the Effects of Angiotensin-Converting Enzyme Inhibitors”. J Cardiovasc Pharmacol. 2008; 51(6):523-531.
17. Sugden, PH. “Signaling in myocardial hypertrophy: Life after cal-cineurin?” Circ Res 1999; 84(6):633-646.
18. Weber KT, Jalil JE, Janicki JS, et al. “Myocardial collagen remode-ling in pressure overload hypertrophy. A case for interstitial heart disease”. Am J Hypertens 1989; 2(12 pt 1): 931-940.
19. Gelpi RJ, Gao S, Zhai P et al. Genetic inhibition of calcineurin induces diastolic dysfunction in mice with chronic pressure over-load. Am J Physiol 2009; 297(5): H1814-H1819.
20. Cingolani HE, IL Ennis. “Sodium-hydrogen exchanger, car-diac overload, and myocardial hypertrophy”. Circulation 2007; 6;115(9):1090-100.
21. Ross RS, TK Borg. “Integrins and the myocardium”. Circ Res 2001; 88(11):1112-1119.
22. Gerdes AM, JM Capasso. “Structural remodeling and mechanical dysfunction of cardiac myocytes in heart failure”. J Mol Cell Cardiol 1995; 27(3):849-856.
23. Gerdes, AM. “Cardiac myocyte remodeling in hypertrophy and progression to failure”. J Card Fail 2002; 8(6 suppl):S264-268.
24. Spinale FG, Ishihra K, Zile M, et al. “Structural basis for changes in left ventricular function and geometry because of chronic mitral regurgitation and after correction of volume overload”. J Thorac Cardiovasc Surg 1993; 106(6):1147-1157.
25. Olivetti G, Capasso JM, Sonnenblick EH, et al. “Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats”. Circ Res 1990; 67(1):23-34.
26. Beltrami CA, Finato N, Rocco M, et al. “The cellular basis of dilated cardiomyopathy in humans”. J Mol Cell Cardiol 1995; 27(1):291-305.
27. Chandrashekhar Y, J Narula. “Death hath a thousand doors to let out life”. Circ Res 2003; 92(7):710-714.
28. Narula J, Pandey P, Arbustini E, et al. “Apoptosis in heart failure: release of cytochrome from mitochondria and activation of cas-pase-3 in human cardiomyopathy”. Proc Natl Acad Sci USA 1999; 96(14):8144-8149.
29. Garg S, Narula J, Chandrashekhar Y. “Apoptosis and heart failure: clinical relevance and therapeutic target”. J Mol Cell Cardiol 2005; 38(1):73-79.
30. Leri A, Fiordaliso F, Setoguchi M, et al. “Inhibition of p53 function prevents renin-angiotensin system activation and stretch-mediated myocyte apoptosis”. Am J Pathol 2000; 157(3):843-857.
31. Olivetti G, Abbi R, Quaini F, et al. “Apoptosis in the failing human heart”. N Eng J Med 1997; 336(16):1131-1141.
32. Rodriguez M, J Schaper. “Apoptosis: measurement and technical issues”. J Mol Cell Cardiol. 2005; 38(1):15-20.
33. Haider N, Narula N, Narula J. “Apoptosis in heart failure represents programmed cell survival, not death, of cardiomyocytes and likelihood of reverse remodeling”. J Card Fail 2002; 8(Suppl):S512-S517.
34. Morissette MR, A Rosenzweig. “Targeting survival signaling in heart failure”. Curr Opin Pharmacol 2005; 5(2):165-170.
35. Narula N, Narula J, Zhang PJ, et al. “Is the myofibrillarlytic myo-cyte a forme fruste apoptotic myocyte?” Ann Thorac Surg 2005; 794(4):1333-1337.
36. Leist M, Single B, Castoldi AF, et al. “Intracellular adenosine tri-phosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis”. J Exp Med 1997; 185(8):1481-1486.
37. Anversa P, B Nadal-Ginard. “Myocyte renewal and ventricular remodeling”. Nature 2002; 415(6868):240-243.
38. Anversa P, J Kajstura. “Ventricular myocytes are not terminally diffe-rentiated in the adult mammalian heart”. Circ Res 1998; 83(1):1-14.
39. Beltrami AP, Urbanek K, Kajstura J, et al. “Evidence that human cardiac myocytes divide after myocardial infarction”. N Engl J Med 2001; 344(23):1750-1757.
40. Kajstura J, Leri A, Beltrami CA, et al. “Myocyte proliferation in end-stage cardiac failure in humans”. Proc Natl Acad Sci USA 1998; 95(23):1750-1757.
41. Nadal-Ginard B, Kajstura J, Leri A, Anversa P. “Myocyte death, growth, and failure”. Circ Res 2003; 92(2):139-150.
42. Sadoshima J, Xu Y, Slayter HS, et al. “Autocrine release of angioten-sin II mediates stretch-induced hypertrophy of cardiac myocytes in Vitro”. Cell 1993; 75(5):977-984.

Gelpi - Donato Fisiopatología cardiovascular
234
43. Sadoshima J, S Izumo. “Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts Critical role of the AT1 receptor subtype”. Circ Res 1993; 73(3):413-423.
44. Jain M, Liao R, Ngoy S, et al. “Angiotensin II receptor blockade attenuates the deleterious effects of exercise training on post-MI ventricular remodelling in rats”. Cardiovasc Res 2000; 46: 66-72.
45. Patten RD, Aronovitz MJ, Einstein M, et al. “Effects of angiotensin II receptor blockade versus angiotensin-converting-enzyme inhi-bition on ventricular remodelling following myocardial infarction in the mouse”. Clin Sci (Lond) 2003; 104: 109-118.
46. Thai HM, Van HT, Gaballa MA, et al. “Effects of AT1 receptor blockade after myocardial infarct on myocardial fibrosis, stiffness, and contractility”. Am J Physiol 1999; 276: H873-H880.
47. Zdrojewski T, Gaudron P, Whittaker P, et al. “Ventricular remo-deling after myocardial infarction and effects of ACE inhibition on hemodynamics and scar formation in SHR”. Cardiovasc Pathol 2002; 11: 88-93.
48. González GE, Palleiro J, Monroy S, et al. “Effects of the early administration of losartan on the functional and morphological aspects of postmyocardial infarction ventricular remodeling in rabbits”. Cardiovasc Pathol 2005; 14: 88-95.
49. Pourdjabbar A, Parker TG, Nguyen QT, et al. “Effects of pre-, peri-, and postmyocardial infarction treatment with losartan in rats: effect of dose on survival, ventricular arrhythmias, function, and remodeling”. Am J Physiol 2005; 288: H1997-H2005.
50. González G, Seropian I, Krieger L, et al. “Effect of early vs late AT1 receptor blockade with losartan on Post-Myocardial infarc-tion ventricular remodeling in rabbits . Am J Physiol 2009; 297: H375-H386.
51. Hasking GJ, Esler MD, Jennings GL, et al. “Norepinephrine spi-llover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity”. Circulation 1986; 73(4):615-621.
52. Gelpi RJ, Hittinger L, Fujii AM, et al. “Sympathetic augmentation of cardiac function in developing hypertension in conscious dogs”. Am J Physiol 1988; 255:H1525-H1534.
53. Hardt SE, Geng YJ, Montagne O, et al. “Accelerated cardiomyopathy in mice with overexpression of cardiac G(s)alpha and a missense mutation in the alpha-myosin heavy chain”. Circulation 2002; 105(5):614-20.
54. Mann DI, Kent RL, Parsons B, et al. “Adrenergic effects on the biology of the adult mammalian cardiocyte”. Circulation 1992; 85(2):790-804.
55. Port JD, MR Bristow. “Altered beta-adrenergic receptor gene regu-lation and signaling in chronic heart failure”. J Mol Cell Cardiol 2001; 33(5):887-905.
56. Simpson, P. “Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha I adrenergic response”. J Clin Invest 1983; 72(2):732-738.
57. Engelhardt S, Hein L, Wiesmann F, et al. “Progressive hypertrophy and heart failure in beta I -adrenergic receptor transgenic mice”. Proc Natl Acad Sci USA 1999; 96(12):7059-7064.
58. Libby, P. “Inflammation in atherosclerosis”. Nature 2002; 420(6917):868-874.
59. Levine B, Kalman J, Mayer L, et al. “Elevated circulating levels of tumor necrosis factor in severe chronic heart failure”. N Engl J Med 1990; 323(4):236-241.
60. Seta Y, Shan K, Bozkurt B, et al. “Basic mechanisms in heart failure: the cytokine hypothesis”. J Card Fail 1996; 2(3):243-249.
61. Yokoyama T, Nakano M, Bednarczyk JL, et al. “Tumor necrosis factor-α provokes a hypertrophic growth response in adult cardiac myocytes”. Circulation 1997; 95(5):1247-1252.
62. Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. “Expression and functional significance of tumor necro-sis factor receptors in human myocardium”. Circulation 1995; 92(6):1487-1493.
63. Oral H, Sivasubramanian N, Dyke DB, et al. “Myocardial proin-flammatory cytokine expression and left ventricular remodeling
in patients with chronic mitral regurgitation”. Circulation 2003; 107(6):831-837.
64. Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al. “Left ventricular remodeling in transgenic mice with cardiac restric-ted overexpression of tumor necrosis factor”. Circulation 2001; 104(7):826-831.
65. Bozkurt B, Kribbs SB, Clubb FJ, et al. “Pathophysiologically progressive left ventricular dysfunction and remodeling in rats”. Circulation 1998; 97(14):1382-1391.
66. Bradham WS, Moe G, Wendt KA, et al. “TNF- and myocardial matrix metalloproteinases in heart failure: relationship to LV re-modeling”. Am J Physiol 2002; 282(4):H1288-1295.
67. Krum, H. “Tumor necrosis factor-alpha blockade as a therapeutic strategy in heart failure (RENEWAL and ATTACH): Unsuccessful, to be specific”. J Card Fail 2002; 8(6):365-368.
68. Libby P, RT Lee. “Matrix matters”. Circulation 2000; 102(16):1874-1876.
69. Weber, KT. “Cardiac interstitium in health and disease: the fibrillar collagen network”. J Am Coll Cardiol 1989; 13(7):1637-1652.
70. Zak, R. “Cell proliferation during cardiac growth”. Am J Cardiol 1973; 31(2):211-219.
71. Eghbali M, Czaja MJ, Zeydel M, et al. “Collagen chain mRNAs in insolated heart cells from young and adult rats”. J Mol Cell Cardiol 1988; 20(3):267-276.
72. Caulfield JB, TK Borg. “The collagen network of the heart”. Lab Invest 1979; 40(3):364-372.
73. Ju H, IM Dixon. “Extracellular matrix and cardiovascular diseases”. Can J Cardiol 1996; 12(12):1259-1267.
74. Sun Y, KT Weber. “Infarct scar: a dynamic tissue”. Cardiovasc Res 2000; 46(2):250-256.
75. Weber KT, Jalil JE, Janicki JS, et al. “Myocardial collagen remode-ling in pressure overload hypertrophy Acase for interstitial heart disease”. Am J Hypertens 1989; 2(12 Pt 1):931-940.
76. Silver MA, Pick R, Brilla CG, et al. “Reactive and reparative fibrillar collagen remodeling in the hypertrophied rat left ventricle: two experimental models of myocardial fibrosis”. Cardiovasc Res 1990; 24(9):741-747.
77. Swynghedauw, B. “Molecular mechanisms of myocardial remode-ling”. Physiol Rev 1999; 24(9):741-747.
78. Beltrami CA, Finato N, Rocco M, et al. “Structural basis of end-stage failure in ischemic cardiomyopathy in humans”. Circulation 1994; 89(1):151-163.
79. Rossi, MA. “Patterns of myocardial fibrosis in idiopathic cardiomyopathies and chronic chagasic cardiopathy”. Can J Cardiol 1991; 7(7):287-294.
80. Marijianowski MM, Teeling P, Becker AE. “Remodeling after myocardial infarction in humans is not associated with inters-titial fibrosis of noninfarcted myocardium”. J Am Coll Cardiol 1997; 30(1):76-82.
81. Woodiwiss AJ, Tsotetsi OJ, Sprott S, et al. “Reduction in myocar-dial collagen cross linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction”. Circulation 2001; 103(1):155-160.
82. Gunja-Smith Z, Morales AR, Romanelli R, et al. “Remodeling of human myocardial collagen in idiopathic dilated cardiomyopathy. Role of metalloproteinase’s and pyridinoline cross-links”. An J Pathol 1996; 148(5):1639-1648.
83. Marijianowski MM, Teelin P, Mann J, Becker AE. “Dilated car-diomyophathy is associated with an increase in the type I/type III collagen ratio: a quantitative assessment”. J Am Coll Cardiol 1995; 25(6):1263-1272.
84. Buegess ML, Bueggy J, Price RL, et al. “Exercise- and hypertension-induced collagen changes are related to left ventricular function in rat geart”. Am J Physiol 1996; 270:H151-159.
85. Norton GR, Tsotetsi J, Trifunovic B, et al. “Myocardial stiffness is attributed to alterations in cross – linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats”. Circulation 1997; 96(6):1991-1998.

Fisiopatología de la insuficiencia cardíaca
235
86. Spinale FG, Zellner JL, Johnson WS, et al. “Cellular and extra-cellular remodeling with the development and recovery from ta-chycardial- induced cardiomyopathy: changes in fibrillar collagen myocyte adhesion capacity and proteoglycans”. J Mol Cell Cardiol 1996; 28(8):1591-1608.
87. Herrmann KL, McCulloch AD, Omens JH. “Glycated collagen crosslinking alters cardiac mechanics in volume-overload hyper-trophy”. Am J Physiol 2003; 284(4):H1277-1284.
88. Iiomoto DS, Covell JW, Harper E. “Increase in cross-linking of type I and type III collagens associated with volume-overload hypertrophy”. Circ Res 1988; 63(2):399-408.
89. Chancey AL, Broqer GL, Janicki JS. “Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function”. Am J Physiol Heart Circ Physiol 2002; 282(6):H2152-2158.
90. Brower GL, Chancey AL, Thanigaraj S, et al. “Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity”. Am J Physiol 2002; 83(2):H518-525.
91. Spinale FG, Coker ML, Thomas CV, et al. “Time-dependent chan-ges in matrix metalloproteinase activity and expression during the progression of congestive heartfailure: relation to ventricular and myocyte function”. Circ Res 1998; 82(4):482-495.
92. Brower GL, JS Janicki. “Contribution of ventricular remode-ling to pathogenesis og heart failure in rat”. Am J Physiol 2001; 280(2):H674-683.
93. Coker ML, Thomas CV, Clair MJ, et al. “Myocardial matrix metalloproteinase activity and abundance with congestive heart failure”. Am J Physiol 1998; 274(5 Pt 2):H1516-1523.
94. Dixon IM, Ju H, Reid NL, et al. “Cardiac collagen remodeling in the cardiomyopathic Syriam hamster and the affec of losartan”. J Mol Cell Cardiol 1997; 29(7):1837-1850.
95. Feldman AM, Li YY, McTiernan CF. “Matrix metalloproteinanses in pathophysiology and treatment of heart treatment of heart failure”. Lancet 2001; 357(9257):654-655.
96. Lee RT, P Libbt. “Matrix metalloproteinases: not-so-innocent bystanders in heart failure”. J Clin Invent 2000; 106(7):827-828.
97. Li YY, McTiernan CF, Feldman AM. “Interplay of matrix meta-lloproteinase, tissue inhibitors of metalloproteinases and their regulator in cardiac matrix remodeling”. Cardiovasc Res 2000; 46(2):214-224.
98. Li YY, AM Feldman. “Matrix metalloproteinases of heart failure: potential therapeutic implications”. Drugs 2001; 61(9):1239-1252.
99. Mann DL, FG Spinale. “Activation of matrix metalloproteinases in the failing human heart: breaking the tie that binds”. Circulation 1998; 98(17):1699-1702.
100. Nagatomo Y, Carabello BA, Coker ML, et al. “Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control”. Am J Physiol 2000; 278(1):H151-161.
101. Romanic AM, Burns-Kurtis CL, Gout B, et al. “Ohlstein following myocardial onfarction in the rabbit”. Life Sci 2001; 68(7):799-814.
102. Spinale FG, Coker ML, Bond BR, et al. “Myocardial matrix de-gradation and metalloproteinase activation in the failing heart: a potential therapeutic target”. Cardiovasc Res 2000; 46(2):225-238.
103. Thomas CV, Coker ML, Zellner JL, et al. “Increased matrix meta-lloproteinase activity and selective upregulation in LV myocardial from patients with end-stage dilated cardiomyopathy”. Circulation 1998; 97:1708-1715.
104. Yokoseki O, Yazaki Y, Suzuki J, et al. “Association of matrix meta-lloproteinase expression and left ventricular function in idiopathic dilated cardiomyopathy”. Jpn Circ J 2000; 64:352-357.
105. Masutomo K, Makino N, Sugano M, et al. “Extracellular matrix regulation in the development of Syrian cardiomyopathic Bio 146 and Bio 5358 hamsters”. J Mol Cell Cardiol 1999; 31:1607-1615.
106. Thompson MM, IB Squire. “Matrix metalloproteinase-9 expres-sion after myocardial infarction: Physiological or Pathological?” Cardiovasc Res 2002; 54: 495-498.
107. Kim HE, Dalal SS, Young E, et al. “Disruption of the myocardial extracellular matrix leads to cardiac dysfunction”. J Clin Invest 2000; 106(7):857-866.
108. Ducharme A, Frantz S, Aikawa M, et al. “Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement an collagen accumulation after experimental myocardial infarction”. J Clin Invest 2000; 106(1):55-62.
109. King Mk, Coker ML, Goldberg A, et al. “Selective matrix metallo-proteinase inhibition with developing heart failure: effects on lefts ventricular function and structure”. Circ Res 2003; 92(2):177-185.
110. Rohde LE, Ducharme A, Arroyo LH, et al. “Matrix metallopro-teinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice”. Circulation 1999; 99(23):3063-3070.


237
Generalidades. Síntesis, secreción, receptores, funciones
El descubrimiento de los péptidos os por dos líneas de investi-
del retorno venoso producían un incremento de la natriu-
ratas, inyectados a otros animales, producían una intensa natriuresis y diuresis. A partir del aislamiento del factor
una potente actividad diurética, natriurética y vasorrela-
1 Actualmente, se conoce que los péptidos natriuréticos
-puesta por el factor -
péptido péptido
-
de 17 aa con un puente disulfuro. Los tres primeros poseen el puente disulfuro entre dos residuos de cisteína, que es
y el CNP están altamente conservados, mientras que el 2-5
El ANF deriva de una pre-pro-hormona, el pre-pro-ANF -
Péptidos natriuréticos 15
doplásmico produce la pro-hormona el pro-ANF des-
por una proteasa de membrana, la corina6, forman una por-1-98 y la verdadera
hormona ANF pro-ANF1-98 forman el pro-ANF79-98 o péptido kaliurético; el pro-ANFy el pro-ANF o vasodilatador. 2, 7
-
clivado por la furina formando el N-terminal pro-BNP 77-108. El CNP de-
2,8
de ANF se encuentra en la aurícula izquierda, hallándo-se en concentraciones menores en la aurícula derecha, el
-
-trointestinal, el timo y el ojo, aunque las concentraciones
9
El BNP se encuentra en los adultos, principalmente en -
de ANF es mayor que la de BNP, tanto en las aurículas -
circulante. 10 El BNP, a diferencia del ANF, varía de forma considerable entre las distintas especies, por ejemplo en
El CNP es el principal miembro de esta familia de pép-tidos natriuréticos producido por el sistema nervioso cen-tral, en donde actúa como un neuropéptido, con funciones
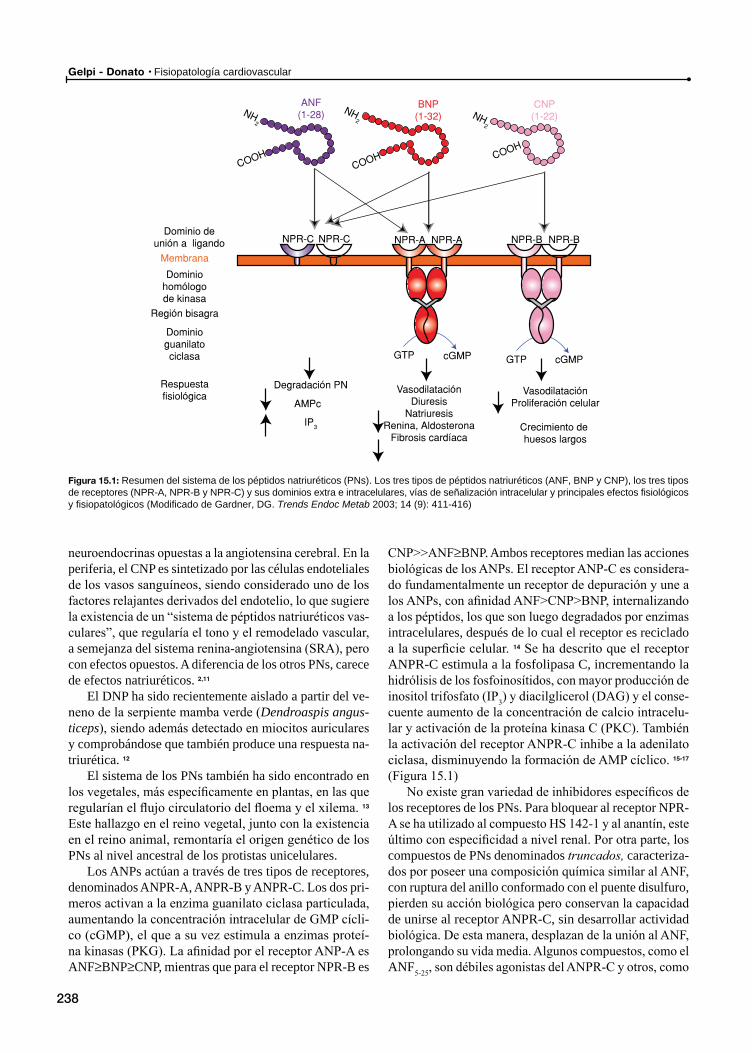
Gelpi - Donato Fisiopatología cardiovascular
238
periferia, el CNP es sintetizado por las células endoteliales
-
a semejanza del sistemacon efectos opuestos. A diferencia de los otros PNs, carece de efectos natriuréticos. 2,11
El DNP ha sido recientemente aislado a partir del ve-Dendroaspis angus-
ticepsy comprobándose que también produce una respuesta na-triurética. 12
El sistema de los PNs también ha sido encontrado en
13
PNs al nivel ancestral de los protistas unicelulares. Los ANPs actúan a través de tres tipos de receptores,
denominados ANPR-A, ANPR-B y ANPR-C. Los dos pri-
--
ANF≥BNP≥CNP, mientras que para el receptor NPR-B es
≥BNP. Ambos receptores median las acciones -
intracelulares, después de lo cual el receptor es reciclado 14
ANPR-C estimula a la fosfolipasa C, incrementando la
--
15-17
los receptores de los PNs. Para bloquear al receptor NPR-
compuestos de PNs denominados caracteriza-
con ruptura del anillo conformado con el puente disulfuro,
de unirse al receptor ANPR-C, sin desarrollar actividad
ANF
Figura 15.1: Resumen del sistema de los péptidos natriuréticos (PNs). Los tres tipos de péptidos natriuréticos (ANF, BNP y CNP), los tres tipos de receptores (NPR-A, NPR-B y NPR-C) y sus dominios extra e intracelulares, vías de señalización intracelular y principales efectos fisiológicos y fisiopatológicos (Modificado de Gardner, DG. Trends Endoc Metab 2003; 14 (9): 411-416)

Péptidos natriuréticos
239
el ANF amida, que une al ANPR-C, se comportan como 2
A diferencia del ANF, verdadera hormona endocrina con
es decir, se liberan a medida que se procesan de la prohormo-na, encontrándose en el tejido cardiaco solo formas maduras de BNP77-108. Además, se cree que no cumplirían funciones endocrinas sino paracrinas y autocrinas controlando la hi-
-
estímulos mecánicos y neuroendocrinos aumentan su libera-
el estiramiento del cardiomiocito de la pared auricular, que involucra un mecanismo en el que interviene una proteína
pertussis18 -
α, IL-1β19
Por otra parte, el estiramiento de la pared ventricular sería el 20
El sistema de los
un sistema vasodepresor para contrarrestar estadios hiper-tensivos. Estos objetivos se cumplen mediante acciones
directos abarcan:
vascular,
el movimiento de plasma desde los capilares al tejido intersticial, y
-
-ciones de:
-merular de la corteza adrenal
-
-lámico
noradrenalina en el sistema nervioso central e in-
células tubulares renales, lo que disminuye la re-
-
-
2,21-30
-
descrito sus efectos sobre los capilares, la mucosa intestinal,
15-31
Todos estos efectos en su conjunto promueven una caída de la volemia y por lo tanto del retorno venoso, y una dis-
--
sobre todo las actividades neuroendocrinas y neurotransmi-
el endotelio vascular y en tejidos tubulares renales. Los niveles circulantes de los PNs pueden aumentar
-
-nas tiroideas, los diuréticos, los inhibidores de la enzima
BNP plasmáticos también están aumentados en el último trimestre del embarazo, pero sus niveles no varían con el ciclo menstrual. 2, 32
-
junto con los PNs como marcadores bioquímicos.
Péptidos natriuréticos en situaciones patológicas
-
-crementan el ANF y el BNP, se pueden mencionar el infar-
-
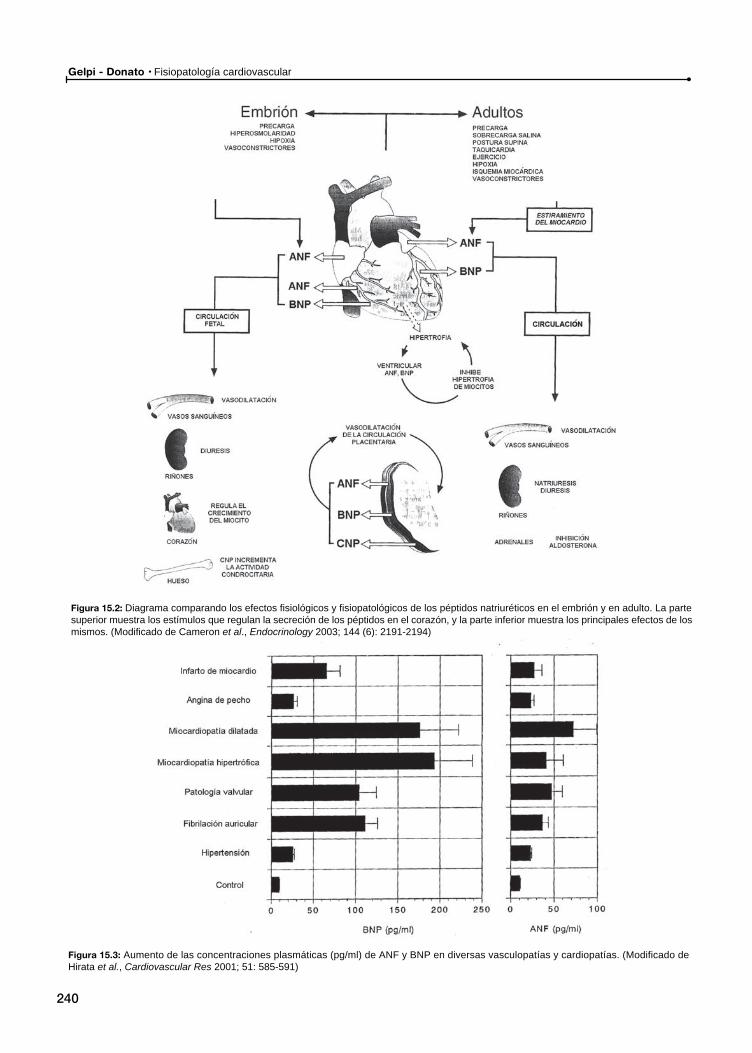
Gelpi - Donato Fisiopatología cardiovascular
240
Figura 15.3: Aumento de las concentraciones plasmáticas (pg/ml) de ANF y BNP en diversas vasculopatías y cardiopatías. (Modificado de Hirata et al., Cardiovascular Res 2001; 51: 585-591)
Figura 15.2: Diagrama comparando los efectos fisiológicos y fisiopatológicos de los péptidos natriuréticos en el embrión y en adulto. La parte superior muestra los estímulos que regulan la secreción de los péptidos en el corazón, y la parte inferior muestra los principales efectos de los mismos. (Modificado de Cameron et al., Endocrinology 2003; 144 (6): 2191-2194)
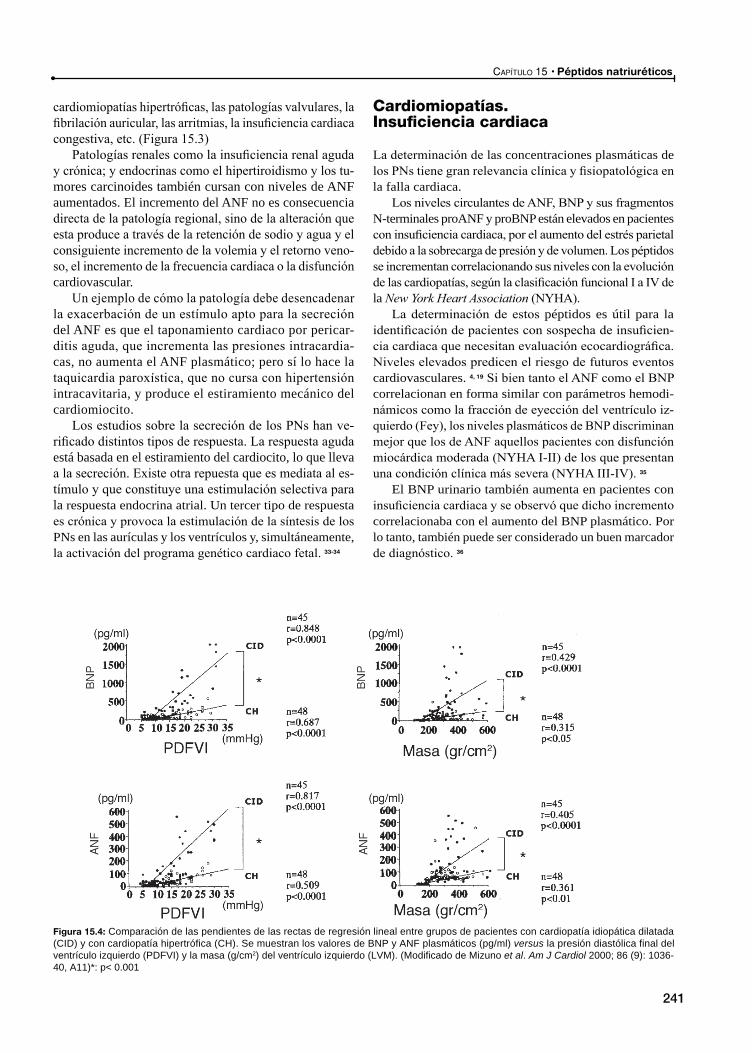
Péptidos natriuréticos
241
-mores carcinoides también cursan con niveles de ANF aumentados. El incremento del ANF no es consecuencia
-
cardiovascular.
del ANF es que el taponamiento cardiaco por pericar--
cas, no aumenta el ANF plasmático; pero sí lo hace la
intracavitaria, y produce el estiramiento mecánico del cardiomiocito.
-
está basada en el estiramiento del cardiocito, lo que lleva -
la respuesta endocrina atrial. Un tercer tipo de respuesta
PNs en las aurículas y los ventrículos y, simultáneamente, 33-34
Cardiomiopatías. Insuficiencia cardiaca
concentraciones plasmáticas de
la falla cardiaca.
N-terminales proANF y proBNP están elevados en pacientes
la péptidos es útil para la
-
cardiovasculares. 4, 19
correlacionan en forma similar con parámetros hemodi--
35
El BNP urinario también aumenta en pacientes con
correlacionaba con el aumento del BNP plasmático. Por lo tanto, también puede ser considerado un buen marcador
36
Figura 15.4: Comparación de las pendientes de las rectas de regresión lineal entre grupos de pacientes con cardiopatía idiopática dilatada (CID) y con cardiopatía hipertrófica (CH). Se muestran los valores de BNP y ANF plasmáticos (pg/ml) versus la presión diastólica final del ventrículo izquierdo (PDFVI) y la masa (g/cm2) del ventrículo izquierdo (LVM). (Modificado de Mizuno et al. Am J Cardiol 2000; 86 (9): 1036-40, A11)*: p< 0.001
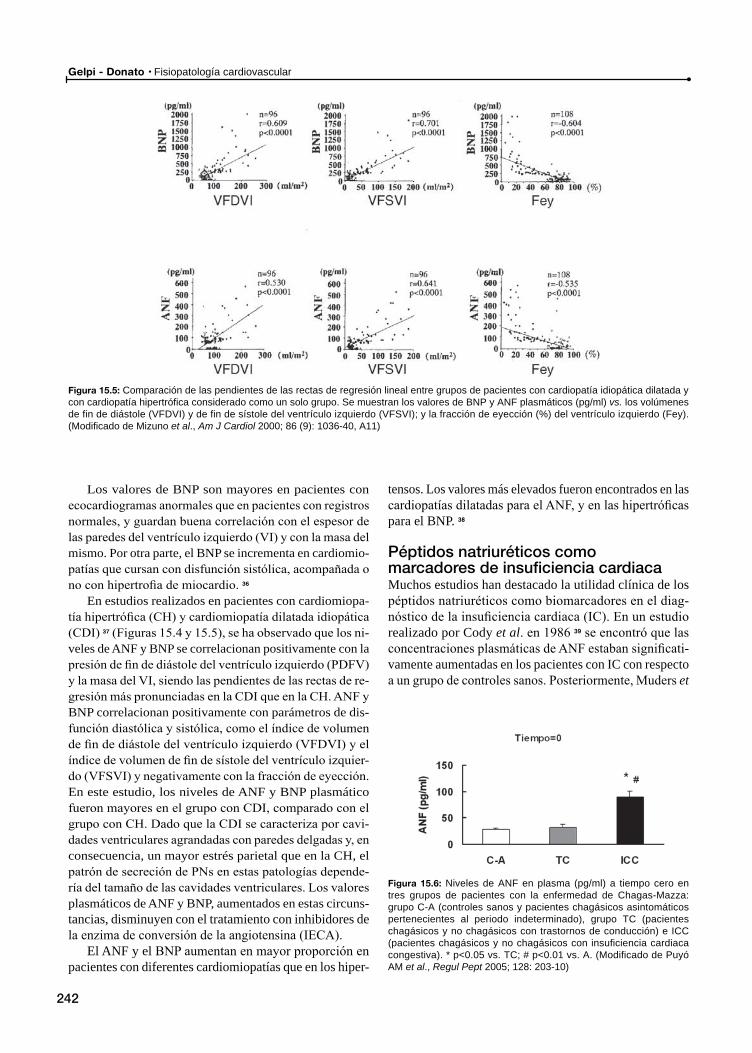
Gelpi - Donato Fisiopatología cardiovascular
242
Los valores de BNP son mayores en pacientes con
mismo. Por otra parte, el BNP se incrementa en cardiomio-
36
En estudios realizados en pacientes con cardiomiopa-
37 -veles de ANF y BNP se correlacionan positivamente con la
-
BNP correlacionan positivamente con parámetros de dis-
-
En este estudio, los niveles de ANF y BNP plasmático
-
consecuencia, un mayor estrés parietal que en la CH, el -
plasmáticos de ANF y BNP, aumentados en estas circuns-tancias, disminuyen con el tratamiento con inhibidores de
El ANF y el pacientes con diferentes cardiomiopatías que en los hiper-
tensos. Los valores más elevados fueron encontrados en las
para el BNP. 38
Péptidos natriuréticos como marcadores de insuficiencia cardiacaMuchos estudios han destacado la utilidad clínica de los
-
realizado por Cody et al 39
-vamente aumentadas en los pacientes con IC con respecto
et
Figura 15.5: Comparación de las pendientes de las rectas de regresión lineal entre grupos de pacientes con cardiopatía idiopática dilatada y con cardiopatía hipertrófica considerado como un solo grupo. Se muestran los valores de BNP y ANF plasmáticos (pg/ml) vs. los volúmenes de fin de diástole (VFDVI) y de fin de sístole del ventrículo izquierdo (VFSVI); y la fracción de eyección (%) del ventrículo izquierdo (Fey). (Modificado de Mizuno et al., Am J Cardiol 2000; 86 (9): 1036-40, A11)
Figura 15.6: Niveles de ANF en plasma (pg/ml) a tiempo cero en tres grupos de pacientes con la enfermedad de Chagas-Mazza: grupo C-A (controles sanos y pacientes chagásicos asintomáticos pertenecientes al periodo indeterminado), grupo TC (pacientes chagásicos y no chagásicos con trastornos de conducción) e ICC (pacientes chagásicos y no chagásicos con insuficiencia cardiaca congestiva). * p<0.05 vs. TC; # p<0.01 vs. A. (Modificado de Puyó AM et al., Regul Pept 2005; 128: 203-10)
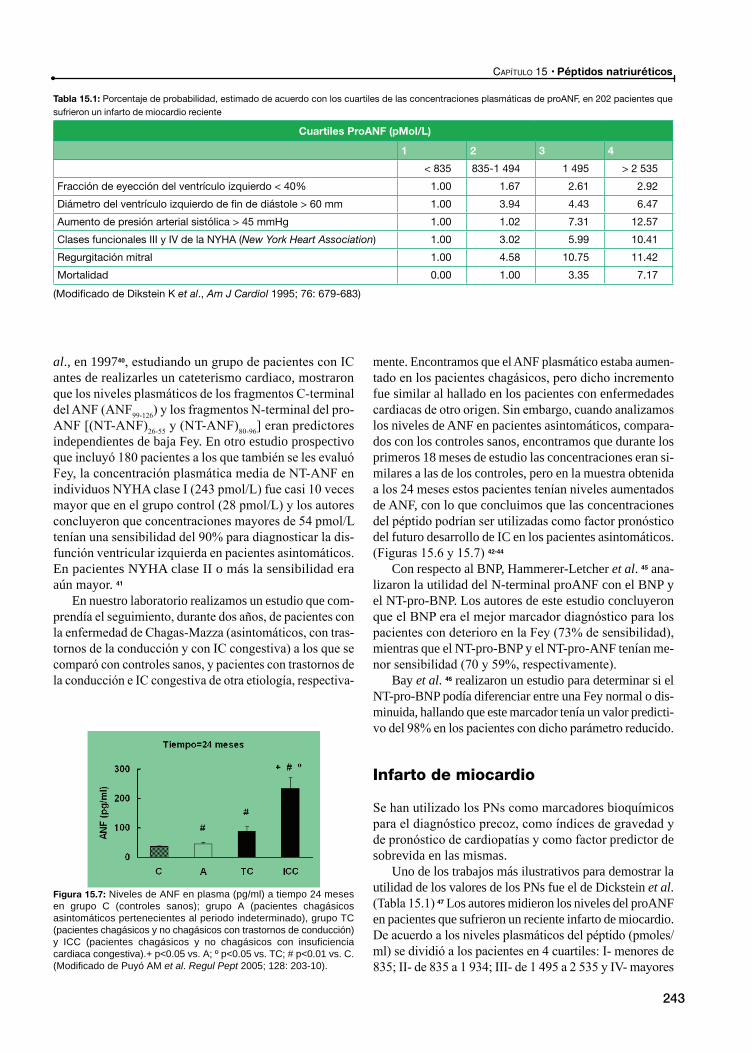
Péptidos natriuréticos
243
al., en 199740
antes de realizarles un cateterismo cardiaco, mostraron
independientes de baja Fey. En otro estudio prospectivo
-
En pacientes NYHA clase II o más la sensibilidad era aún mayor. 41
En nuestro laboratorio realizamos un estudio que com-
-
-
mente. Encontramos que el ANF plasmático estaba aumen-
fue similar al hallado en los pacientes con enfermedades
los niveles de ANF en pacientes asintomáticos, compara-dos con los controles sanos, encontramos que durante los primeros 18 meses de estudio las concentraciones eran si-milares a las de los controles, pero en la muestra obtenida
de ANF, con lo que concluimos que las concentraciones
del futuro desarrollo de IC en los pacientes asintomáticos. 42-44
Con respecto al BNP, Hammerer-Letcher et al. 45 ana-lizaron la utilidad del N-terminal proANF con el BNP y el NT-pro-BNP. Los autores de este estudio concluyeron
mientras que el NT-pro-BNP y el NT-pro-ANF tenían me-
Bay et al. 46 realizaron un estudio para determinar si el NT-pro-BNP podía diferenciar entre una Fey normal o dis-minuida, hallando que este marcador tenía un valor predicti-
Infarto de miocardio
marcadores bioquímicos
predictor de sobrevida en las mismas.
Uno de los trabajos más ilustrativos para demostrar la utilidad de los valores de los PNs fue el de Dickstein et al.
47 Los autores midieron los niveles del proANF en pacientes que sufrieron un reciente infarto de miocardio.
Tabla 15.1: sufrieron un infarto de miocardio reciente
Cuartiles ProANF (pMol/L)
1 2 3 4
< 835 835-1 494 1 495 > 2 535
1.00 1.67 2.61 02.92
1.00 3.94 4.43 06.47
Aumento de presión arterial sistólica > 45 mmHg 1.00 1.02 7.31 12.57
Clases funcionales III y IV de la NYHA (New York Heart Association) 1.00 3.02 5.99 10.41
Regurgitación mitral 1.00 4.58 10.75 11.42
Mortalidad 0.00 1.00 3.35 07.17
(Modificado de Dikstein K et al., Am J Cardiol 1995; 76: 679-683)
Figura 15.7: Niveles de ANF en plasma (pg/ml) a tiempo 24 meses en grupo C (controles sanos); grupo A (pacientes chagásicos asintomáticos pertenecientes al periodo indeterminado), grupo TC (pacientes chagásicos y no chagásicos con trastornos de conducción) y ICC (pacientes chagásicos y no chagásicos con insuficiencia cardiaca congestiva).+ p<0.05 vs. A; º p<0.05 vs. TC; # p<0.01 vs. C. (Modificado de Puyó AM et al. Regul Pept 2005; 128: 203-10).

Gelpi - Donato Fisiopatología cardiovascular
244
-
-
-
-
pacientes con valores más altos de proANF plasmático, los parámetros estudiados empeoraron casi el doble para la frac-
que el número de decesos fue siete veces mayor. El BNP es un marcador predictivo del remodelado ven-
38, 48, 49 Las con-centraciones aumentadas de BNP en la etapa temprana, des-pués del infarto, han sido asociadas con un remodelamiento adverso del ventrículo izquierdo y valores aumentados del mismo también se correlacionaron con la mortalidad de los
En un estudio en el que se analizaron las concentra--
50
De acuerdo con lo descrito, tanto el ANF como el BNP
-pervivencia en pacientes con infarto de miocardio e IC.
Hipertensión arterial
-
-riendo que tiene también un efecto simpatolítico. Debido a estas observaciones se han realizado muchos estudios
Los PNs se encuentran aumentados en diversos tipos -
los PNs no se alteran durante la etapa de remodelamien-to concéntrico, pero sí aumentan cuando se alcanza la
Ver capítulo 18
-
-
un BNP elevado es un marcador en pacientes hipertensos
aumento del BNP en pacientes con cardiomiopatías y en hipertensos. Este efecto no sería directo, sino que estaría
38
En la HTA esencial en el hombre, los valores plasmá--
dos con respecto a los controles normales, aunque hay que
ventrículos puede producir una respuesta diferencial de la -
mental de anastomosis aorta-cava, que produce hipertro-
51
están aumentados en el plasma de los modelos de hiper--
así como en ratas hipertensas DOCA-sal52
de la aorta53
54 -pecto a las ratas control. 55
estos modelos. En estudios realizados con cepas de ratones que son
defecto en la síntesis de ANF puede causar HTA. 56-57 Por otra parte, en otros trabajos, encontraron que la HTA sal

Péptidos natriuréticos
245
sensible, que se desarrolla en ratones para el
1 II, losartán. 58 -
59
Arritmias
El ANF se eleva en la
ARNm y también el ARNm del pre-pro-BNP. Por lo tan-to, sus valores pueden ser predictores del embolismo de
60 Los valores elevados de ANF, hallados en pacientes portadores de arritmias, son inter-medios con respecto a los encontrados en pacientes con cardiopatías dilatadas. 43-44
Valvulopatías
El BNP y el proBNP plasmáticos son considerados mar-cadores de
61 -vado asimismo que el BNP se correlaciona con el cociente
62
Disnea
Otro importante uso de los PNs, ANF y BNP como marca-
disneas. Ambos se incrementan notoriamente en la disnea
aquellos con disnea, pero sin falla cardiaca. 63
Transplante cardiaco
-plantados. Previo al transplante, los niveles de ANF se
-plante, era de esperar que los niveles cayeran bruscamente
los niveles normales. De esta manera, el ANF se consti-
del transplante, pero permanecen elevados durante un tiem-
a dos factores:-
múnmente dejada para efectuar la anastomosis y que continuaría secretando altas cantidades de ANF, y
-no del transplante. 64
Rol de los péptidos natriuréticos en la regulación de la hipertrofia del miocardio
La ventricular se caracteriza por un incremento
, c-jun y Egr-1
etapas tempranas del desarrollo, como la isoforma β de la
tipo III, ANF y BNP. 34, 65 Ver capítulo 11 El ANF y el BNP están elevados en las cardiomiopa-
cardiaca. Los niveles de ANF son mayores en la circula-
ventrículos adultos. En estudios del desarrollo embrionario
un rol de ANF y BNP como ordenadores del crecimiento 66-67 Durante la re-
α actina, y el cambio de las isoformas α y β de las cadenas
efectos de la ANG II local y la endotelina. 52
hemodinámica y, en forma independiente, por el proceso 68 El bloqueo de receptores
A de la

Gelpi - Donato Fisiopatología cardiovascular
246
factores de crecimiento, como la ANG II y la endotelina 69
-
la resistencia vascular periférica.
por lo que las células cardiacas se encuentran en un entorno altamente enriquecido en estos péptidos en el miocardio hi-
63 Los receptores para los PNs se encuentran tanto
69
células de músculo liso vascular 70
cardiomiocitos neonatales en cultivo. 71 Más recientemente,
compartida por el BNP y el CNP. 72
proteína kinasa G. El ANF también puede inducir la apop-tosis de los cardiomiocitos. 73
74
El tratamiento de
-tor de crecimiento insulina simil. 75 Por otra parte, estudios
ANF. 76
-taloproteasas de la matriz que intervienen en la remodela-
77 BNP podría, además, comportarse como un α
78
de la enzima aldosterona sintasa en cardiocitos neonata-les79 -
-
crecimiento, controlando el crecimiento de los miocitos y
-
--
cimiento descontrolado del miocardio. 63
-Nppa-/-
57
Nppb-/-
80 Los ratones Npr1-/- que carecen del receptor común para ANF y BNP desarrollan HTA resistente a la sal, con una desproporcio-
81
-
82
elementos cis que contienen motivos GATA; estas secuen-cias son esenciales para la actividad de los promotores en
-
y para β 83, 84
--
-tas α
82
-miocitos inducida por el estiramiento mecánico. 85
-cial para la supervivencia de los cardiomiocitos embrio-narios, para el mantenimiento del estado diferenciado del
Aterosclerosis
Los PNs se encuentran aumentados en placas ateroscle--

Péptidos natriuréticos
247
--
y transplantes cardiacos. Los PNs son dosados habitualmente mediante radioin-
-
paciente cardiaco.
propende a aumentar sus efectos. Limitados al uso paren--
se comportan como inhibidores de la ECA y la kininasa II. Así refuerzan, por este triple mecanismo, sus efectos sobre los PNs, la ANG II y las cininas. El uso terapéutico en la HTA esencial, de los inhibidores de endopeptidasa
sintéticos del ANF, como el carperitide, y del BNP, como
y del sistema simpático. Asimismo, se abre la posibili-
-
calcio voltaje sensitivos y abridores de canales de pota-sio calcio sensitivos. Contemporáneamente, la industria farmacéutica busca métodos alternativos como el uso de nasales, parches cutáneos y preparados orales
89
Agradecimiento
Bibliografía
11. de Bold AJ, Borenstein HB, Veress AT, et al. “A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats”. Life Sci. 1981;28:89-94.
12. Ruskohajo H. “Atrial natriuretic peptide: Synthesis, release and metabolism”. Pharmacol. Rev. 1992;44(4):479-602.
13. Gutkowska J, Antunes Rodrígues J, Mc Cann SM. “Atrial natriuretic peptide in brain and pituitary gland”. Physiol Rev.;77(2):465-515.
14. Venogupal J. “Cardiac natriuretic peptides - hope or hype?” J Clin Pharm Ther. 2001;26:15-31.
15. Jortani SA, Prabhu SD, Valdes R. “Strategies for developing bio-markers of heart failure”. Clin Chem. 2004;50(2):265-78.
86
Patologías endocrinas y renales
El -les como el aldosteronismo primario y los feocromocitomas. 87 El ANF es responsable del escape mineralocorticoide. 88 Cuando se instala un tratamiento con mineralocorticoi-
incremento de la natriuresis y la diuresis que es producto
-mento de la volemia. Asimismo, el ANF se incrementa
consecuencia del aumento de la volemia y sus niveles descienden después de la hemodiálisis. 31
Conclusiones
tres tipos de acciones: endocrinas, paracrinas y autocrinas. Para el ANF están reservadas principalmente las fun-
-
Para el BNP, ejercer las funciones paracrinas y auto-crinas, fundamentalmente a nivel cardiaco, por sus pro-
-miento cardiaco.
Para el CNP, las acciones locales paracrinas y autocri---
crinas a nivel del sistema nervioso central de la hormona
endocrino, productor de
-
--
no que menor índice de neoplasias presenta en los seres
Futuro de los péptidos natriuréticos
-

Gelpi - Donato Fisiopatología cardiovascular
248
16. Jan W, Wu F, Morser J, Wu Q. “Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme”. Proc Natl Acad Sci USA. 2000;97:8525-29.
17. Yandle TG. “Biochemistry of natriuretic peptides”. J Intern Med. 1994;235:561-76.
18. Sawada Y, Suda M, Yokoyama H, Kanda T, Sakamaki T, Tanaka S, et al. “Stretch-induced hypertrophic growth of cardiocytes and proces-sing of brain-type natriuretic peptide are controlled by proprotein-processing endoprotease furin”. J Biol Chem. 1997;272:20545-54.
19. Levin ER, Gardner DG, Samson WK, Natriuretic peptides”. N Engl J Med. 1998;339(5):321-28.
10. Yoshibayashi M, Yoshihiko S, Nakao K. “Brain natriuretic peptide versus atrial natriuretic peptide--physiological and pathophysiologi-cal significance in children and adults: a review”. Eur J Endocrinol. 1996:135:265-68.
11. Itoh H, Suga S, Ogawa Y, Komatsu Y, et al. “Significance of vascular natriuretic peptide system in vascular remodeling in humans and its application to gene therapy”. Ann N Y Acad Sci. 1997;811:533-41.
12. Lisy O, Jougasaki M, Heublein DM, et al. “Renal actions of syn-thetic dendroaspis peptide”. Kidney Int. 1999;56:502-08.
13. Vessely DL, Giordano AT. “Atrial natriuretic peptide hormonal síntesis in plants”. Bioch Biophys Res Comm. 1991;179:695-700.
14. Pandey NK. “Biology of natriuretic peptides and their receptors”. Peptides. 2005;26:901-32.
15. Bianciotti LG, Vatta MS, Elverdin JC, et al. “Atrial natriuretic factor-induced amylase output in the rat parotid gland appears to be mediated by the inositol phospahte pathway”. Biochem Biophis Res Comm. 1998;247:123-28.
16. Sabbatini ME, Vatta MS, Vescina C, et al. “NPR-C receptors are involvd in C-type natriuretic peptide response on bile secretion”. Regul Pept. 2003;116:13-20.
17. Sabbatini ME, Villagra A, Davio CA, et al. “Atrial natriuretic factor stimulates exocrine pancreatic secretion in the rat through NPR-C receptors”. Am J Physiol Liver Physiol, 2003;285:G929-37.
18. Bensimosn M, Chang A, Kuroski et al. “Participation of G pro-teins in natriuretic peptide hormone secretion from heart atria”. Endocrinology. 2004;145:5313-21.
19. Puyó AM, Vatta MS, Donoso AS, et al. “Central natriuretic peptides regulation of peripheral atrial natriuretic factor release”. Regul Pept. 2000;90:93-99.
20. Ma KK, Ogawa T, de Bold AJ. “Selective upregulation of cardiac brain natriuretic peptide at the transcriptional and translational levels by pro-inflammatory cytokines and by conditioned medium derived from mixed lymphocytes reactions via p38 MAP kinase”. J Mol Cell Cardiol. 2004;36:505-13.
21. Fernández BE, Domínguez AE, Vatta MS, et al. “Atrial natriuretic peptide increases norepinephrine uptake in the central nervous system”. Arch Int Physiol Biochim. 1990;98:127-30.
22. Fernández BE, Domínguez AE, González MA, et al. “Role of atrial natriuretic peptide on calcium channel mechanisms involved in catecholamine release from bovine adrenal medulla”. Arch Int Pharmacodyn Ther. 1992;316:105-13.
23. Vatta MS, Travaglianti M, Bianciotti LG, et al. “Atrial natriuretic factor effects on norepinephrine uptake in discrete telencephalic and diencephalic nuclei of the rat”. Brain Res. 1994;646:324-26.
24. Vatta MS, Rodríguez Fermepín M, et al. “Atrial natriuretic factor enhances norepinephrine uptake in circumventricular organs, locus coeruleus and nucleus tractus solitarii of the rat”. Neurosci Lett. 1995;197(1):29-32.
25. Vatta MS, Presas M, Bianciotti LG, et al. “B and C types natriuretic peptides modulate norepinephrine uptake and release in the rat hypothalamus”. Regul Pept. 1996;65:175-84.
26. Fernández BE, Leder M, Fernández G, et al. “Atrial natriuretic fac-tor modifies the biosynthesis and turn over of norepinephrine in the rat adrenal medulla”. Biochem Biophys Res Comm. 1997;238:343-46.
27. Vatta MS, Presas MF, Bianciotti LG, et al. “B and C types natriu-retic peptides modulate norepinephrine uptake and release in the rat adrenal medulla”. Peptides. 1997;18(10):1843-49.
28. Vatta, MS, Rodríguez Fermepín, M, et al. “Atrial natriuretic fac-tor inhibits norepinephrine biosynthesis and turn-over in the rat hypothalamus”. Regul Pept. 1999;85:101-07.
29. Rodríguez Fermepín, M, Vatta, S, Bianciotti, LG, et al. “B-type and C-type natriuretic peptides modify norepinephrine uptake in discrete encephalic nuclei of the rat”. Cell Mol Neurobiol. 2000;20(6):763-71.
30. Fernández BE, Correa AH, Choi MR. “Atrial natriuretic factor stimulates dopamine uptake mediated by natriuretic peptide-type A receptor”. Regul Pept. 2005;124:137-44.
31. Bianciotti LG, Vatta MS, Bengochea LA, et al. “Atrial natriuretic factor increases peritoneal dialysis efficiency in nephrectomized rats”. Peptides. 1996;17:87-92.
32. Sagnella GA. “Measurement and significance of circulating natriuretic peptides in cardiovascular diseases”. Clin Sci. 1998;95:519-29.
33. Clerico A, Iervasi G, Mariani G. “Pathophysiologic relevante of measuring the plasma levels of cardiac natriuretic peptide hormones in humans”. Horm Metab Res. 1999;31:487-98.
34. de Bold AJ, Bruneau BG, Kuroski de Bold ML. “Mechanical and neuroendocrine regulation of the endocrine heart”. Cardiovasc Res. 1996;31:7-18.
35. Luchner A, Burnett JC Jr, Jougasaki M, et al. “Evaluation of brain natriuretic peptide as marker of left ventricular dysfunction and hy-pertrophy in the population”. J Hypertens. 2000;18(8):1121-28.
36. Ng LL, Geeranavar S, Jennings SC, et al. “Diagnosis of heart failure using urinary natriuretic peptides”. Clin Sci (London). 2004;106(2):129-33.
37. Mizuno Y, Yoshimura M, Harada E, et al. “Plasma levels of A- and B-type natriuretic peptides in patients with hypertrophic cardiom-yopathy or idiopathic dilated cardiomyopathy”. Am J Cardiol. 2000;86(9):1036-40,A11.
38. Yasumoto K, Takata M, Ueno H, et al. “Relation of plasma brain and atrial natriuretic peptides to left ventricular geometric patterns in essential hypertension”. Am J Hypertens. 1999;12:921-24.
39. Cody RJ, Atlas SA,Laragh JH, Kubo SH, Covit AB, Rayman KS, et al. “Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal and hemodynamic response to peptide infusion”. J Clin Invest. 1986;78:1362-74.
40. Muders F, Komer EP, Griese DP, et al. “Elevation of plasma na-triuretic peptides as markers for left ventricular dysfunction”. Am Heart J, 1997;134:442-49.
41. Lerman A, Gibbons RJ, Rodeheffer RJ, et al. “Circulating N-terminal arterial natriuretic peptide as a marker for symptomless left-ventricular dysfunction”. Lancet. 1993;341:1105-09.
42. Scaglione J, Puyó AM, Dupuy A, et al. “Behaviour of atrial na-triuretic factor in an experimental model of trypanosoma cruzi infection in rats”. J. Parasitol. 2001;87:923-26.
43. Puyó AM, Scaglione J, Auger S, et al. “Atrial natriuretic factor as a marker of myocardial compromise in Chagas’ disese”. Regul Pept. 2002;105:139-42.
44. Puyó AM, Scaglione J, Auger S, et al. “Natriuretic peptides as prognostic and diagnostic markers in Chagas´ disease”. Regul Pept. 2005;128:203-10.
45. Hammerer-Lercher A, Neubauer E, Muller S, et al. “Head-to-head comparison of N-terminal pro-brain natriuretic peptide, brain natriuretic peptide and N-terminal pro-atrial natriuretic peptide in diagnosing left ventricular dysfunction”. Clin Chim Acta. 2001;310:193-97.
46. Bay M, Kirk V, Parner J, et al. „NT-proBNP: a new diagnostic scree-ning tool to differentiate between patients with normal and reduced left ventricular systolic function”. Heart. 2003;89:150-54.
47. Dikstein K, Larsen AI, Bonargee V, et al. “Plasma proatrial natriu-retic factor is predictive of clinical status in patients with congestive heart failure”. Am J Cardiol. 1995;76:679-83.
48. Nagaya N, Nishikimi T, Goto Y, et al. “Plasma brain natriuretic peptide is a biochemical marker for the prediction of progressive ventricular remodeling after acute myocardial infarction. Am Heart J. 1998;135(1):21-28.

Péptidos natriuréticos
249
49. Crilley JG, Ferrer M. “Left ventricular remodeling and brain natriuretic peptide after first myocardial infarction”. Heart. 2001;86:638-42.
50. De Lemos JA, Morrow DA, Bentley JH, et al. “The prognostic value of B-type natriuretic peptide in patients with acute coronary syndromes”. N Engl J Med. 2001;345:1014-21.
51. Su X, Brower G, Janicki JS, et al. “Differential expression of natriuretic peptides and their receptors in volume overload cardiac hypertrophy in the rat”. J Mol Cell Cardiol. 1999;31(10):1927-36.
52. Cavallero S, González GE, Puyó AM, et al. “Atrial natriuretic factor behavior and myocyte hypertrophic profile in combined pressure and volume-induced cardiac hypertrophy . J Hypertens, 2007; 25:1940-50
53. Ogawa Y, Tamura N, Chusho H, et al. “Brain natriuretic peptide appears to act locally as an antifibrotic factor in the heart”. Can J Physiol Pharmacol. 2001;79:723-29.
54. Puyo AM, Vega G, Pellegrino de Iraldi A, et al. “Increased circulating levels of atrial natriuretic factor in two kidney-two clip renovascular hypertension”. Medicina (Buenos Aires). 1998;58:165-70.
55. Damiano PF, Cavallero S, Mayer MA, et al. “Impaired response to insulin associated to protein kinase C in chronic fructose-induced hypertension”. Blood Pres. 2002;11:1-7.
56. John SW, Krege JH, Oliver PM, et al. “Genetic decreases in atrial natriuretic peptide and SALT-sensitive hipertension”. Science. 1995;267:679-81.
57. Hill O, Kuhn M. Zucht HD, et al. “Analysis of the human guanylin gene and the processing and cellular localization of the peptide”. Proc Soc Ntl Acad Sci USA. 1995;92:2046-50.
58. Manning P, Schwartz PT, Katsuba NC, et al. “Vasopressin-sti-mulated release of atriopeptin: endocrine antagonists in fluid homeostasis”. Science. 1985;229:395-97.
59. Melo LG, Steinhelper ME, Pang SC, et al. “ANP in regulation of arterial pressure and fluid-electrolyte balance: lessons from genetic mouse models”. Physiol Genom. 2000;3:45-58.
60. Hirata Y, Matsumoto A, Aoyagi T, et al. “Measurement of plasma brain natriuretic peptide level as a guide for cardiac overload”. Cardiovasc Res. 2001;51(3):585-91.
61. Qi W, Mathisen P, Kjekshus J, et al. “Natriuretic peptides in patients with aortic stenosis”. Am Heart J. 2001;142(4):725-32.
62. Uusima P, Tokola H, Ylitalo A, et al. “Plasma B-type natriuretic peptide reflects hypertrophy and diastolic function in hypertension. Anglo-Scandinavian Cardiac Outcomes Trial Investigtors”. Int J Cardiol. 2004;(2):251-56.
63. Gardner DG. “Natriuretic peptides: markers or modulators of cardiac hypertrophy?” Trends Endocrinol Metab. 2003;14(9):411-16.
64. Masters RG, Davies RA, Keon WJ, et al. “Neuroendocrine response to cardiac transplantation”. Can J Cardiol. 1993;9:609-17.
65. Chien KR, Zhu H, Knowlton KU, et al. “Transcriptional regula-tion during cardiac growth and development”. Annu Rev Physiol. 1993;55:77-95.
66. Cameron VA, Aitken GD, Ellmers LJ, et al. “The sites of gene expression of atrial, brain, and C-type natriuretic peptide in mouse fetal development: temporal changes in embryos and placenta”. Endocrinology. 1996;137:817-24.
67. Cameron VA, Ellmers LJ. “Minireview: natriuretic peptides during development of the fetal heart and circulation”. Endocrinology. 2003;144(6):2191-94.
68. Ogawa T, Linz W, Stevenson M, et al. « Evidence for load-depen-dent and load-independent determinants of cardiac natriuretic peptide production”. Circulation. 1996;93:2059-67.
69. Bianciotti LG, de Bold AJ. “Modulation of cardiac natriuretic pep-tide gene expression following endothelin type. A receptor blockade in renovascular hypertension”. Cardiovasc Res. 2001;49:808-16.
70. Lin X, Hänze J, Heese F, et al. “Gene expression of natriuretic peptide receptors in myocardial cells”. Circ Res. 1995;77:750-58.
71. Calderone A, Thaik CM, Takahashi N, et al. “Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts”. J Clin Invest. 1998;101:812-18.
72. Horio T, Nishikimi T, Yoshihara F, et al. “Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes”. Hypertension. 2000;35:19-24.
73. Rosenkranz AC, Woods RL, Dusting GJ, et al. “Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocites: importance of cyclic GMP”. Cardiovasc Res. 2003;57:515-22.
74. Wu CF, Bishopric NH, Pratt RE. “Atrial natriuretic peptide in-duces apoptosis in neonatal rat cardiac myocytes”. J Biol Chem. 1997;272:14860-66.
75. Tokudome T, Horio T, Soeki T, et al. “Inhibitory effect of C-type natriuretic peptide (CNP) on cultures cardiac myocyte hyper-trophy: interference between CNP and endothelin-1 signaling pathways”. Endocrinology. 2004;145:2131-40.
76. Cao L, Gardner DG. “Natriuretic peptides inhibit DNA synthesis in cardiac fibroblasts”. Hypertension. 1995;25:227-34.
77. Cameron VA, Rademaker MT, Ellmers LJ, et al. “Atrial (ANP) and brain natriuretic peptide (BNP) expression after myocardial infarction in sheep: ANP is synthesized by fibroblasts infiltrating the infarct”. Endocrinology. 2000;141:4690-97.
78. Tsuruda T, Boerrigter G, Huntley BK, et al. “Brain natriuretic peptide is produced in cardiac fibroblasts and induces matrix me-talloproteinases”. Circ Res. 2002;9:1127-34.
79. Kapoun AM, Liang F, O´Young G, et al. “B-type natriuretic pep-tide exerts broad functional opposition to transforming growth factor beta in primary human cardiac fibroblasts. Fibrosis, myo-fibroblast conversion, proliferation, and inflammation”. Circ Res. 2004;94:453-61.
80. Ito T, Yoshimura M, Nakamura S, et al. “Inhibitory effect of natriu-retic peptides on aldosterone synthase gene expression in cultured neonatal rat cardiocytes”. Circulation. 2003;107(6):807-10.
81. Oliver PM, Fox JE, Kim R, et al. “Hypertension, cardiac hypertro-phy and sudden death in mice lacking natriuretic peptide receptor A”. Proc Natl Acad Sci USA. 1997;94:14730-35.
82. Temsah R, Nemer M. “GATA factors and transcriptional regulation of cardiac natriuretic peptide genes”. Regul Pept. 2005;128:177-85.
83. Herzig TC, Jobe SM, Aoki H, et al. “Angiotensin II type 1a re-ceptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload”. Proc Natl Acad Sci USA. 1997;94:7543-48.
84. Hasegawa K, Lee SJ, Jobe SM, Kitsis RN. “cis-acting sequences that mediate induction of beta-myosis heavy chain gene expression during left ventricular hypertrophy due to aortic constriction”. Circulation. 1997;96:3943-53.
85. Pikkarainen S, Tokola H, Majalahti-Palviainen T, et al. “GATA-4 is a nuclear mediator of mechanical stretch-activated hypertrophic program”. J Biol Chem. 2003;278(26):23807-16.
86. Alexander MR, Knowles JW, Nishikimi, T, et al. “Increased atheros-clerosis and smooth muscle cell hypertrophy in natriuretic peptide receptor A-/-apolipoprotein E-/- mice”. Arterioscler Thromb Vasc Biol. 2003;23(6):1077-82.
87. Puyó AM, Levín GM, Armando I, et al. “Increased plasma atrial natriuretic factor in catecholamine-producing tumor patients”. Clin. Exp Hypertens. 1999;21(7):1129-44.
88. Yokota N, Bruneau BG, Kuroski de Bold ML, et al. “Atrial natriu-retic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway”. J Clin Invest. 1994;94(5):1938-46.
89. Cha YM, Redfield MM, Shah S, Shen WK, Fishbein MC, Chen Ps. “Effects of omapatrilat on cardiac nerve sprouting and struc-tural remodeling in experimental congestive heart failure”. Heart Rhythm. 2005;2(9):984-90.


251
En términos de incidencia, prevalencia, morbimortali-
problemática creciente de la salud pública. A pesar de los
-1, 2, 3
4 Estudios multicéntricos de IC mostraron, en los pa-
3
-
--
do una causa de elevada morbimortalidad en el mundo occidental15, particularmente en aquellos con severa dis-
6
-
de los mecanismos neurohormonales y mecánicos com-
alterada, aunque aún no es bien conocido si este estado
la enfermedad7
5
acci-tromboembolismo hasta
-caciones la causa de muerte. 8 La incidencia reportada de
-
9, mientras
10, 11 El estudio 12
-tudios observacionales, en portadores de miocardiopatía
13
Así, el y la isquemia o infarto de miocardio con-tribuyen a la elevada mortalidad en IC, con la trombosis
12
tromboembolismo es compleja
-8
Aunque la ICCr per se-
embolismo14
auricular y ventricular izquierda, en particular con severa 15 -
trombos, proceso en el que también están implicados factores hemodinámicos. Las células endoteliales y subendoteliales contribuyen a
-
mastocitos. Estas células son una rica fuente de mediado-16
-17 Heit
et al. 18
-
19, la incidencia de estos eventos vasculares con 20
que lo actualmente reconocido.
Factores protrombóticos en la insuficiencia cardiaca 16

Gelpi - Donato Fisiopatología cardiovascular
252
-
de P selectina soluble que representan, respectivamente,
14 Consecuentemente, los pacien--
8
podría ser la
21
células endoteliales. 22 Los niveles elevados de FT proveen
8 -23 Otro
importante estímulo para aumentar el FT es la citoquina -
lar 24 Am-
posiblemente mediado por el aumento de los niveles de
Tríada de Virchow
tríada de 8
de la contractilidad pueden todos producir anormal
y al subsecuente tromboembolismo. El deterioro de la fun-
en la cámara ventricular izquierda dilatada; a su vez, la
--
25
En la falla cardiaca también se ha demostrado la presen-cia de ,
26 La -
trombosis in situ y al tromboembolismo. -
27, así podrían estar alteradas las mediciones de marcadores
28 las anor--
presentes en la falla cardiaca29, probablemente asociadas
30
-
-
-
-
medios de aneurismas ventriculares izquierdos, independientemente
-
La presencia de los diferentes componentes de la tríada -
nesis. Las anormalidades plaquetarias, de la hemostasia,
mecanismos del tromboembolismo no son simplemente mecánicos, sino que también se relaciona con un estado
Cambios hemorreológicos
en mujeres y en aquellos en clase funcional más severa. 14

Factores protrombóticos en la insuficiencia cardíaca
253
Hoffmeister et al. 31
entre viscosidad plasmática y Fey en IC. Aunque Gibbs et al. 14
de la clase funcional de la IC.
-
-
-
intracardiacos, trombosis in situ
severa podría, por lo menos parcialmente, ser responsable
en estos pacientes. 32
Baker et al. 33
la viscosidad plasmática predicen fuerte y positivamente -
turos eventos isquémicos cerebrales es débil.Los -
-
aumentado en particular en pacientes con ICCr, con corre-
-so debería evitarse en pacientes con ICCr, en vista de su
34 En contraste, en sujetos
efecto muy leve y rápidamente transitorio en los índices 35
Disfunción endotelial
de la
a la pared vascular. Cuando es liberado por las plaquetas inhibe su reclutamiento para ser incorporadas al trombo
bioactivo. 36
-
diferentes estímulos, lo que contribuye a la vasoconstric-26
La endotelina 37 es un potente péptido vasoconstric-tor venoso y arterial, derivado del endotelio, que cumple funciones tanto de hormona circulante como de factor
-
38, particular-
en sitios de injuria vascular y en el plasma de pacientes con falla cardiaca. 39 La ET podría ser responsable del
-liales, y ambos podrían ser marcadores independientes
se describe en la falla cardiaca como parte de la respuesta compensatoria al desorden hemodinámico. Como un po-
endotelial, la -lina de las células endoteliales. Más aún, la AII incrementa
40, 41 Todos estos
pacientes con falla cardiaca.En el estudio 42
los hombres, lo que podría contribuir parcialmente a la -
menino con falla cardiaca, un predictor independiente -
-
endotelio. 27
ventricular izquierdo y muerte pos infarto. 43
Activación de trombina y fibrinólisis
Aunque la trombina-
formándose el complejo trombina-antitrombina III -

Gelpi - Donato Fisiopatología cardiovascular
254
altamente sensibles de la actividad de trombina in vivo, en pacientes con IC. 39
debido a que su vida media es muy corta. Las mediciones de la actividad de trombina en pacientes con falla cardiaca
-
la falla cardiaca. 39
-
El dímero D-
-mentarse en otras enfermedades cardiovasculares como
44 y la estenosis mitral45,
El factor de necrosis tumoral (FNT , una citoquina --
do la posibilidad de trombosis microvascular, de injuria 46
-
no es claro al presente.
con niveles elevados de citoquinas47, que se correlacionan con la seve-
48 Ciertamente, la
-
-
pueden ser mediadas por vía del FT, un potente activador -
dores, así como los valores de la viscosidad plasmática, del
predecir la morbilidad y la mortalidad en la ICCr. Chin et al. 49 encontraron que los niveles plasmáticos
pero no otros índices, fueron mayores en quienes sufrieron eventos que en aquellos que no los presentaron, con una
-
-
plaquetaria corresponden a la ICCr, con muy limitada 50 Chin et al. 51 se-
-
Los niveles basales de
pueden ser una vía de inicio de la trombosis, en un
Activación plaquetaria
cardiaca no ha sido bien establecido, el incremento de la
-
52
-traron incrementados en la falla cardiaca, incluyendo la
-
-tarios circulantes.
leucocitos y
son de particular importancia por las posibles respuestas -
53
del clearanceque puede contribuir a la mayor incidencia de eventos
-
-quetaria en la IC.54
-55 Diversos estudios han mostrado que
los pacientes con falla cardiaca presentan concentraciones 56
-

Factores protrombóticos en la insuficiencia cardíaca
255
mujeres en clase funcional más severa.P-selectina
las plaquetas activadas, con un rol activo en las interac-ciones plaqueta-plaqueta. Además la P-selectina promue-ve el rodamiento de las plaquetas y leucocitos sobre el endotelio activado. 57
La P-selectina es también un componente crucial de la -
-
plaque-58
Los datos referidos en la literatura revelan que los pacientes con IC descompensada presentan P-selectina
59
-
IIb-IIIa. 60
mayores en pacientes con falla cardiaca que en controles. 61
-
idiopática vs.La osteonectina
plaquetaria. Cuando las plaquetas son activadas la osteo-
-vados en pacientes con falla cardiaca. 52
-taria, como el -
-
- desde
-62
Efectos del tratamiento médico de la falla cardíaca sobre el estado protrombótico
--
vasculares futuros63, son pocos los estudios in vivo que
falla cardiaca. 8 Datos acumulativos indican que los IECA 64,
para reducir la frecuencia de reinfarto puede relacionarse 14
La incidencia de manifestaciones clínicas de trombo-embolismo, en pacientes con falla cardiaca, se estima en
65 El tratamiento médico de la falla cardiaca 8
cardiaca proveen un modesto efecto antiplaquetario, con
-ceptores de serotonina plaquetarios. 7
Los diuréticos, como hidroclorotiazida, furosemida, es-pironolactona e indapamida aumentan la síntesis de pros-
-26
son antiplaquetarios menos po-
en este aspecto. , el propanolol y el carvedilol reducen la -frina y ADP. 66
--
en pacientes con falla cardiaca. 14
Los -
trombosis en los estudios clínicos. 67
-nado con los efectos de los IECA sobre el metabolismo de
14
Los receptores de 68, aunque su rol no ha sido aún
-69

Gelpi - Donato Fisiopatología cardiovascular
256
-
70 La -
71
La actividad antiplaquetaria es probablemente un he-cho común a todos los IECA. Los niveles aumentados de
-sis de
cardiaca7
la falla cardiaca. 72
73 74, EPILOG75, y BIP76, y es un importante aspecto clínico aún no dilucidado.
presentan propiedades antiplaquetarias, es probable que
Losartan 77 e irbesartan 78 -
afectan las concentraciones plasmáticas del inhibidor del -
tos hipertensos.
79 80
Agentes antiplaquetarios en insuficiencia cardiaca
-
-
Los inhibidores de los receptores de ADP podrían en
cardiaca tratados con IECA y aspirina. 7
El -
-81 y acoplados a la adenil-
bloqueantes de los receptores de ADP, actúa a través de un metabolito, sobre el receptor plaquetario de ADP sub-
82
, pero inde-
-83
84
tanto el uso de warfarina como de aspirina se asociaba
85
falla cardiaca leve a moderada, y que no disminuía con el tratamiento con warfarina.
86 y CURE. 87 Clopi-, isquemia mio-
con el implante de stents.-
tes
-
-lidad de los datos de los otros estudios analizados. 9 Esta
apropiados, establece que los datos no sostienen con clari-
pacientes con falla cardiaca que presentan ritmo sinusal. 9
con aspirina en bajas dosis, como alternativa a la mono-terapia con aspirina en estos pacientes.
Anticoagulación en insuficiencia cardiaca
En la ICCr, la -
miocardiopatía periparto puede estar indicada la anticoa--
88-89

Factores protrombóticos en la insuficiencia cardíaca
257
90 ---
91
estudios observacionales no randomizados, y el análisis de los efectos de warfarina en comparaciones no
92 93 94 95
Los datos disponibles no sostienen el uso rutinario de -
manecen en ritmo sinusal. 90
randomizados. La terapia
-farina en el tratamiento de la falla cardiaca con ritmo sinusal.
96
-
los que recibieron -dos a warfarina, respectivamente. Hubo más hospitaliza-ciones por razones cardiovasculares, especialmente em-
aspirina.
97
-
-
-
-
por el bajo ritmo de enrolamiento, con la resultante dis-
-hibidores directos de la
es el ximelagatrán -
98 Dos aspectos que deben considerarse son
se dispone de un antídoto para 99
Conclusiones
-sia alterada, aunque no es bien conocido si este estado
7
Los estudios clínicos randomizados al presente no apor-tan evidencia concluyente para recomendar el uso de as-
en pacientes con falla cardiaca y ritmo sinusal. 9
En presencia de falla cardiaca y ritmo sinusal, los estu-dios clínicos randomizados y los observacionales aportan
-
con los controles. 90 Los datos disponibles no sostienen su
-
ritmo sinusal.
Bibliografía
1. Sytkowski PA, Kannel WB, D’Agostino RB. “Changes in risk factors and the decline in mortality from cardiovascular disease. The Framingham Heart Study . New Engl J Med 1990; 322 (23): 1635-1641.
2. Ghali JK, Cooper R, Ford E. “Trends in hospitalization rates for heart failure in the United States, 1973–1986: evidence for increa-sing population prevalence . Arch Intern Med 1990; 150: 769-773.
3. Gheorghiade M, RO Bonow. “Chronic heart failure in the United States: a manifestation of coronary artery disease . Circulation 1998; 97 (3): 282-289.
Figura 16.1: Esquema que interrelaciona los factores protrombóticos con las alteraciones neurohumorales y mecánicas en la insuficiencia cardiaca. Ang II: Angiotensina II; FNT: Factor de necrosis tumoral; TAT: complejo trombina- antitrombina III

Gelpi - Donato Fisiopatología cardiovascular
258
4. Garg RK, Gheorghiade M, Jafri SM. “Antiplatelet and anticoagu-lant therapy in the prevention of thromboemboli in chronic heart failure . Prog Cardiovasc Dis 1998; 41 (3): 225-236.
5. Davis CJ, Gurbel PA, Gattis WA, et al. “Hemostatic abnormalities in patients with congestive heart failure: diagnostic significance and clinical challenge . International Journal of Cardiology 2000; 75, (1): 15-21.
6. Cleland JG, Tygesen K, Uretsky BF, et al. “On behalf of the AT-LAS investigators. Cardiovascular critical event pathways for the progression of heart failure; a report from the ATLAS Study . Eur Heart J 2001; 22:1601-12.
7. Malinin AI, O’Connor CM, Dzhanashvili AI, et al. “Platelet ac-tivation in patients with congestive heart failure: ¿Do we have enough evidence to consider clopidogrel? Am Heart J 2003; 145 (3): 397-403.
8. Lip GY, CR Gibbs. “Does Heart Failure Confer a Hypercoagulable State? Virchow´s Triad Revisited . J Am Coll Cardiol 1999; 44: 1424-6.
9. Lip GY, CR Gibbs. “Antiplatelet agents versus control or anticoa-gulation for heart failure in sinus rhythm: a Cochrane systematic review . Q J Med 2002; 95:461-468.
10. Dunkman WB, Johnson CR, Carson PT, et al., for the V-HeFT VA Cooperative Studies Group. “Incidence of thromboembolic events in congestive heart failure . Circulation. 1993; 87(suppl VI):VI.94–VI.101.
11. Brown A, JGF Cleland. “Influence of concomitant disease on patterns of hospitalisation in patients with heart failure discharged from Scottish hospitals in 1995 . Eur Heart J 1998;19:1063-9.
12. Loh E, Sutton MS, Wun CC, et al. “Ventricular dysfunction and the risk of stroke after myocardial infarction . N Engl J Med 1997; 336 :251-7.
13. Fuster V, Gersh BJ, Giuliani ER, et al. “The natural history of idio-pathic dilated cardiomyopathy . Am J Cardiol. 1981;47:525-531.
14. Gibbs CR, Blann AD, Watson RD, et al. “Abnormalities of He-morheological, Endothelial, and Platelet Function in Patients With Chronic Heart Failure in Sinus Rhythm. Effects of Angiotensing-Converting Enzyme Inhibitor and B-blocker Therapy . Circulation 2001; 103: 1746-1751.
15. Gibbs CR, Davies MK, Lip GY. “ABC of heart failure. Antithrom-botic treatment . BMJ 2000; 320: 495-498.
16. Bankl HC, Radaszkiewiicz T, Klappacher GW, et al. “Increase and redistribution of cardiac mast cells in auricular thrombosis . Circulation 1995; 91: 275-283.
17. Heit JA, Silverstein M, Mohr D, et al. “The epidemiology of venous thromboembolism in the community . Thromb Haemost 2001; 86: 452-463.
18. Heit JA, Silverstein M, Mohr D, et al. “Risk factors for deep vein thrombosis and pulmonary embolism a population-based case-control study . Arch Intern Med 2000: 160: 809-815.
19. Narang R, Cleland J, Erhardt L, et al. “Mode of death in chronic heart failure: a request for more accurate classification . Eur Heart J 1996; 17: 1390-1403.
20. Cleland JG. “Anticoagulation and antiplatelet therapy in heart failure . Curr Opin Cardiol 1997; 12: 276-287
21. Rauchhaus M, Doehner W, Francis DP, et al. “Plasma cytokine parameters and mortality in patients with chronic heart failure”. Circulation 2000; 102:3060–3067.
22. Baillieres Best Pract Res Clin Haematol
23. Ng TM, Cheang KI, Munger MA, et al. “Neurohormonal activation does not explain elevated tissue factor expression in heart failure . Thromb Haemost 2002; 87:176–7.
24. Chin SP, Chung NA, Gibbs CR, et al. “Vascular endothelial growth factor and soluble P-selectin in acute and chronic heart failure . Am J Cardiol 2002; 90:1258–60
25. Falk RH, Foster E, Coats MH. “Ventricular thrombi and throm-boembolism in dilated cardiomyopathy: a prospective follow up study . Am Heart J 1992; 123: 136-142.
26. Kubo SH, Rector TS, Bank AJ, et al. “Endothelium-dependent vasodilation is attenuated in patients with heart failure . Circulation 1991; 84: 1589-1596.
27. Lip GY, AD Blann. “von Willebrand factor and its relevance to cardiovascular disorders . Br Heart J. 1995; 74:580-583
28. Lip GY, Lowe GD, Metcalfe MJ, et al. “Effects of warfarin therapy on plasma fibrinogen, von Willebrand factor and fibrin d-dimer in left ventricular dysfunction secondary to coronary artery disease with and without aneurysms . Am J Cardiol 1995; 76:453-458.
29. Jafri S, Mammen EF, Levine TB, et al. “Effects of warfarin on markers of hypercoagulability in patients with heart failure . Am Heart J 1997; 134:27-36.
30. Lip GY, Lowe GD, Metcalfe MJ, et al. “Is diastolic dysfunction associated with thrombogenesis ? . Int. J Cardiol 1995; 50: 31-40.
31. Hoffmeister A, Hetzel J, Sander S, et al. “Plasma viscosity and fibrinogen in relation to haemodynamic findings in chronic con-gestive heart failure . Eur J Heart Failure. 1999;1: 293-295.
32. Lip, GY. “Fibrinogen and cardiovascular disease . QJM. 1995; 88:155-165
33. Baker IA, Pickering J, Elwood P, et al. “Fibrinogen, viscosity and white blood cell count predict myocardial but not cerebral in-farction: Evidence from the Caerphilly and Speedwell cohort . Thromb Haemost 2002; 87: 421-425.
34. Gibbs CR, Blann AD, Edmunds E, et al. “Effects of acute exercise on hemorheological, endothelial, and platelet markers in patients with chronic heart failure in sinus rhythm . Clin Cardiol 2001; 24 (11): 724-729.
35. Blann, AD. “The effect of running a marathon on routine and research vascular, hematology and biochemistry indices . Journal of Thrombosis and Haemostasis 2003; 1:398.
36. Freedman JE, J Loscalzo. “Nitric oxide and its relationship to thrombotic disorders . J Thromb Haemost 2003; 1:1183-1188.
37. Levin, ER. “Endothelins . New Engl J Med 1995; 333: 356-363.38. Wei CM, Lerman A, Rodeheffer RJ, et al. “Endothelin in human
congestive heart failure . Circulation 1994; 89 (4) 1580-1586.39. Sbarouni E, Bradshaw A, Andreotti F, Tuddenham E, Oakley CM
Cleland JG. “Relationship between hemostatic abnormalities and neuroendocrine activity in heart failure . Am Heart J 1994; 27 (3): 607-612.
40. Ding YA, MacIntyre DE, Kenyon CJ, et al. “Potentiation of adre-naline-induced platelet aggregation by angiotensin II . Thromb Haemost 1985; 54 3: 717-720.
41. Swartz SL, TJ Moore. “Effect of angiotensin II on collagen-induced platelet activation in normotensive subjects . Thromb Haemost 1990; 63 (1): 87-90.
42. The SOLVD Investigators. “Effect of enalapril on survival in pa-tients with reduced left ventricular ejection fractions and congestive heart failure . N Engl J Med 1991; 325: 293-302.
43. Lip GY, Lowe GDO, Metcalfe MJ. “Effects of warfarin ther-apy on plasma fibrinogen, von Willebrand factor and fibrin D-dimer in left ventricular dysfunction secondary to coronary artery disease with and without aneurysms . Am J Cardiol 1995; 76: 453-458.
44. Kumagai k, Fukunami M, Ohmori M, et al. “Increased intracar-diovascular clotting in patients with chronic atrial fibrillation . J Am Coll Cardiol 1990; 16(2): 377-380.
45. Yasaka M, Miyatake K, Mitani M, et al. “Intracardiac mobile thrombus and D-dimer fragment of fibrin in patients with mitral stenosis . Br Heart J 1991; 66 (1): 22-25.
46. van der Poll T, Buller HR, ten Cate H, et al. “Activation of coa-gulation after administration of tumor necrosis factor to normal subjects . New Engl J Med 1990; 322 (23): 1622-1627.
47. Raymond RJ, Dehmer GJ, Theoharides TC, et al. “Elevated in-terleukin-6 levels in patients with asymptomatic left ventricular systolic dysfunction . Am Heart J 2001; 141:435-438.
48. Deswal A, Petersen NJ, Feldman AM, et al. “Cytokines and cyto-kine receptors in advanced heart failure: an analysis of the cytokine database from the vesnarinone trial (VEST) . Circulation 2001; 103:2055–2059

Factores protrombóticos en la insuficiencia cardíaca
259
49. Chin BS, Blann AD, Gibbs CR, et al. “Prognostic value of inter-leukin-6, plasma viscosity, fibrinogen, von Willebrand factor, tissue factor and vascular endothelial growth factor levels in congestive heart failure . Eur J Clin Invest 2003; 33(11): 941-948.
50. Krumholz H, Parent E, Tu N, Vaccarino V, et al. “Readmission after hospitalisation for congestive heart failure among Medicare beneficiaries . Arch Intern Med 1997; 157:99-104.
51. Chin BS, Conway DS, Chung, NA, et al. “Interleukin-6, tissue factor and von Willebrand factor in acute decompensated heart failure: relationship to treatment and prognosis . Blood Coagul Fibrinolysis 2003; 14:515-521.
52. Anfossi G, M Trovati. “Role of catecholamines in platelet function: pathophysiological and clinical significance . Eur J Clin Invest. 1996; 26: 353-370.
53. Devaux B, Scholz D, Hirche A, et al. “Upregulation of cell adhesion molecules and the presence of low grade inflammation in human chronic heart failure . Eur Heart J 1997; 18:470-9.
54. Mehta J, P Mehta. “Platelet function studies in heart disease: enhanced platelet aggregate formation in congestive heart failure . Circulation 1979; 60:497-503
55. Gershlick AH. “Are there markers of the blood–vessel wall interaction and of thrombus formation that can be used clinically? . Circulation 1990; 81 (Suppl 1): I28-I34
56. Jafri SM, Ozawa T, Mammen E, et al. “Platelet function, thrombin and fibrinolytic activity in patients with heart failure . Eur Heart J 1993; 14: 205-212.
57. Boukerche H, Ruchaud Sparagano MH, Rouen C, et al. “A mono-clonal antibody directed against a granule membrane glycoprotein (GMP-140/PADGEM, P-selectin, CD62P) inhibits ristocetin-induced platelet aggregation . Br J Haematol 1996; 92: 442-451.
58. Diacovo TG, Puri KD, Warnock RA, et al. “Platelet-mediated lymphocyte delivery to high endothelial venules . Science 1996; 273: 252-255.
59. O’Connor CM, Gurbel PA, Serebruany VL. “Usefulness of soluble and surface-bound P-selectin in detecting heightened platelet activity in patients with congestive heart failure . Am J Cardiol 1999; 83: 1345-1349.
60. Lasters N, Almendro T, Bellon, JA, et al. “Functional regulation of Platelet/Endothelial Cell Adhesion Molecule-1 by TGF-β1 in promonocytic U-937 cells . J Immunol 1994; 153: 4206-4218.
61. Serebruany VL, Murugesan SR, Pothula A, et al. “Increased soluble platelet/endothelial cellular adhesion molecule-1 (PECAM-1) and osteonectin levels in patients with severe congestive heart failure: independence of disease etiology, and antecedent aspirin therapy . Eur J Heart Fail 1999; 1: 243-249.
62. Cox D. “Methods for monitoring platelet function . Am Heart J 1998; 135 (5): S160–S169.
63. Thompson SG, Kienast J, Pyke SD, et al. “Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris . N Engl J Med. 1995; 332: 635-641.
64. Garg R, S Yusuf . “Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in pa-tients with heart failure . JAMA. 1995; 273:1450-1456
65. Garg RK, Gheorghiade M, Jafri SM. “Antiplatelet and anticoagu-lant therapy in the prevention of thromboemboli in chronic heart failure . Prog Cardiovasc Dis 1998; 41:225-236.
66. Gasser JA, DJ Betterridge. “Comparison of the effects of carve-dilol, propranolol, and verapamil on in vitro platelet function in healthy volunteers . J Cardiovasc Pharmacol 1991; 18(4 Suppl):S29-34.
67. Garg R, S Yusuf. “Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in pa-tients with heart failure . JAMA. 1995; 273:1450-1456.
68. Moore TJ, GH Williams. “Angiotensin II receptors on human platelets . Circ Res 1982;5 1:314-320.
69. Ding YA, MacIntyre DE, Kenyon CJ, et al. “Angiotensin II effects on platelet function . J Hypertens 1985; (33 Suppl):S251-253.
70. Brown NJ, DE Vaughan. “Prothrombotic effects of angiotensin . Adv Intern Med 2000; 45:419-29.
71. Gomi T, Ikeda T, Shibuya Y, et al. “Effects of antihypertensive treat-ment on platelet function in essential hypertension . Hypertens Res 2000; 23:567-72.
72. Moskowitz, R. “The angiotensin-converting enzyme inhibitor and aspirin interaction in congestive heart failure: fear or reality? Curr Cardiol Rep 2001; 3:247-53
73. Nguyen KN, Aursnes I, Kjekshus J. “Interaction between enalapril and aspirin on mortality after acute myocardial infarction: sub-group analysis of the Cooperative New Scandinavian Enalapril Survival Study II (CONSENSUS II) . Am J Cardiol 1997; 79:115-9.
74. The GUSTO Investigators. “An international randomized trial comparing 4 thrombolytic strategies for AMI . N Engl J Med 1993; 329: 673-682.
75. Peterson JG, Topol EJ, Sapp SK, et al. “Evaluation of the effects of aspirin combined with angiotensin-converting enzyme inhibi-tors in patients with coronary artery disease . Am J Med 2000; 109:371-377.
76. Leor J, Reicher-Reiss H, Goldbourt U, et al. “Aspirin and mortality in patients treated with angiotensin-converting enzyme inhibitors: a cohort study of 11,575 patients with coronary artery disease . J Am Coll Cardiol 1999; 33:1920-5.
77. Levy PJ, Yunis C, Owen J, et al. “Inhibition of platelet aggrega-bility by losartan in essential hypertension . Am J Cardiol 2000; 86:1188-92.
78. Li P, Fukuhara M, Diz DI, et al. “Novel angiotensin II AT(1) receptor antagonist irbesartan prevents thromboxane A(2)-induced vasoconstriction in canine coronary arteries and human platelet aggregation . J Pharmacol Exp Ther 2000; 292:238-46.
79. Cohn JN, Tognoni G, Glazer RD, et al. “Rationale and design of the Valsartan Heart Failure trial (Val-HeFT) a large multinatio-nal trial to assess the effects of valsartan, an angiotensin-receptor blocker, on morbidity and mortality in chronic congestive heart failure . J Card Fail 1999; 5:155-60.
80. Pfeffer MA, McMurray J, Leizorovicz A, et al. “Valsartan in acute myocardial infarction trial (VALIANT): rationale and design . Am Heart J 2000; 140:727-50.
81. Coukell AJ, Markham A. “Clopidogrel (review) . Drugs 1997; 54: 745-50.
82. Moshfegh K, Redondo M, Julmy F, et al. “Antiplatelet effects of clo-pidogrel compared with aspirin after myocardial infarction:enhanced inhibitory effects of combination therapy . J Am Coll Cardiol 2000; 36:699-705.
83. Yusuf S. “Effect of enalapril on survival in patients with reduced left ventricular ejection fractions . N Engl J Med 1991; 325:293-302.
84. Loh E, St John Sutton M, Wun CC, et al. “Ventricular dysfunction and the risk of stroke after myocardial infarction . N Engl J Med 1997; 336:251-257.
85. Dunkman WB, Johnson GR, Carson PE et al. “Incidence of throm-boembolic events in congestive heart failure . Circulation 1993; 87 Suppl VI:94 -101.
86. CAPRIE Steering Committee. “A randomized, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE) . Lancet 1996; 348:1329-39.
87. CURE Trial Investigators. “Effects of clopidogrel in addition to aspirin in patients with ACS without ST-segment elevation . N Engl J Med 2001; 345: 494-502.
88. Gonzalez Maqueda I, Armada Romero E, Díaz Recasens J, Gallego García P, García Moll M, González Garcia A, Fernández Burgos C, Iñíguez Romo A, Rayo Llerena I. “Guías de práctica clínica de la Sociedad Española de Cardiología en la gestante con cardiopatía . Rev Esp Cardiol 2000; 53: 1474-1495.
89. Abbas AE, Lester SJ, Connolly H. “Pregnancy and the cardiovas-cular system . International Journal of Cardiology 2003
90. Lip GY, CR Gibbs. “Anticoagulation for heart failure in sinus rhythm: a Cochrane systematic review . Q J Med 2002; 95:451-459.
91. Jones CG, JG Cleland. “Meeting report: the LIDO, HOPE, MOX-CON and WASH studies . Eur J Heart Failure 1999; 1:425-431.
92. Swedberg K. for the CONSENSUS trial study group. “Effects of enalapril on mortality in severe congestive heart failure: results of

Gelpi - Donato Fisiopatología cardiovascular
260
the Cooperative North Scandanavian Enalapril Survival Study (CONSENSUS) . N Engl J Med 1987; 316:1429-435.
93. Dunkman WB, Johnson GR, Carson PE, et al, for the V-HeFT VA Cooperative Studies Group. “Incidence of thromboembolic events in congestive heart failure . Circulation 1993; 87 (Suppl.VI):94 -101.
94. Al-Khadra AS, Salem DN, Rand WM, Udelson et al. “Warfarin anticoagulation and survival: a cohort analysis from the Studies of Left Ventricular Dysfunction . (SOLVD). J Am Coll Cardiol 1998; 31 :749-753.
95. Loh E, Sutton MS, Wun CC, et al. “Ventricular dysfunction and the risk of stroke after myocardial infarction . N Engl J Med 1997; 336:251-257.
96. Cleland JG, Findlay I, Jafri S, Sutton G, Falk R, Bulpitt C, Prentice C, Ford I, Trainer A, Poole-Wilson PA. “The Warfarin /Aspirin Study in Heart failure (WASH): a randomized trial comparing antithrombotic strategies for patients with heart failure . Am Heart J 2004; 148 (1): 157-164.
97. Massie BM, Krol WF, Ammon SE, Armstrong PW, et al. “The Warfarin and Antiplatelet Therapy in Heart Failure trial (WATCH): rationale, design, and baseline patient characteristics . J Card Fail. 2004; (2):101-12
98. Verheugt FW, EM Antman. “Anticoagulation in cardiac disease: New data-New options . Eur Heart J 2004; 6 (2): B1.
99. Crowther MA, JI Weitz. “Ximelagatran: the first oral direct throm-bin inhibitor . Expert Opin Investig Drugs 2004 13 (4): 403-413.

261
Fisiopatología de las valvulopatías 17
Alejandro Hita
La enfermedad valvular continúa siendo una causa mayor de morbimortalidad. En los Estados Unidos, las valvulopa-
-
1
Estenosis aórtica
De la estructura a la función: La válvulaLa -
-
-
la zona posterior, a la valva anterior mitral que, junto con -
dicionan una parte importante del mecanismo funcional
claridad las frecuentes alteraciones obstructivas del tracto de salida del ventrículo izquierdo en la 2
factores, como el envejecimiento, la presencia de anillo -
-
al desarrollo de un componente obstructivo subvalvular
-
volumen minuto, con incremento de la morbimortalidad
3-4
anillo, y está constituida por las valvas, las comisuras,
-
-
5
-
-tor responsable de las discrepancias en las determinacio-
mediciones realizadas por manometría en cateterismo y las -
-
-
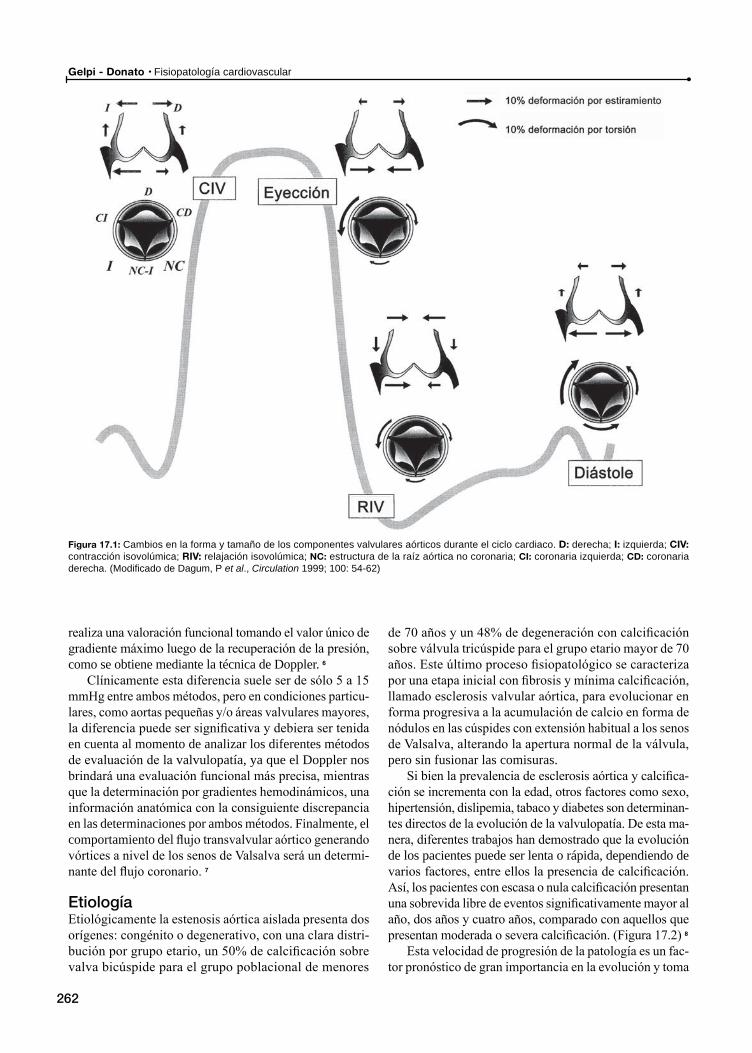
Gelpi - Donato Fisiopatología cardiovascular
262
como se obtiene mediante la técnica de Doppler. 6
-
en cuenta al momento de analizar los diferentes métodos valvulopatía, ya que el Doppler nos
en las determinaciones por ambos métodos. Finalmente, el comportamiento del
-7
Etiología
-
pero sin fusionar las comisuras.-
--
de los pacientes puede ser lenta o rápida, dependiendo de
8
-
Figura 17.1: Cambios en la forma y tamaño de los componentes valvulares aórticos durante el ciclo cardiaco. D: derecha; I: izquierda; CIV: contracción isovolúmica; RIV: relajación isovolúmica; NC: estructura de la raíz aórtica no coronaria; CI: coronaria izquierda; CD: coronaria derecha. (Modificado de Dagum, P et al., Circulation 1999; 100: 54-62)
Fisiopatología de las valvulopatías
263
-
La respuesta ventricular: Estructura, geometría y función
-
-nes celulares, moleculares e intersticiales que se mani-
proceso participan todos y cada uno de los constituyentes cardiacos: miocito, intersticio y estructura vascular, y su
procesos, permitirá eliminar el estímulo hemodinámico y
En este capítulo analizaremos los cambios básicos que -
tricular, que son determinantes de la respuesta ventricular.
La estructura: El miocito, el intersticio y la circulación coronaria
que los mismos serán tratados en profundidad en otros pasajes de la obra y escapan al objetivo del presente ca-
El miocitoEl
ventricular; esta se inicia con una
9
que el miocito presente un incremento de su volumen pero
en el número de mitocondrias, que convierten a esta célula 10
El intersticio
matriz -
-
-11 Particu-
-12, mientras que en
-
acuerdo al estímulo interviniente. 13
Estas diferencias presentan su correlato anatomopa-
--
et al. 14 evaluaron
biopsia endomiocárdica. Estos autores describieron que para incrementos similares de masa miocárdica se obser-vaban dos poblaciones por completo diferentes desde el punto de vista cualitativo o estructural, con diferencias
-
En consecuencia, es claro que un análisis cuantitativo de la -cuada de este proceso adaptativo y se debe avanzar sobre
-
Figura 17.2: Evolución libre de eventos en dos grupos de pacientes, sin calcificación o muy escasa y con moderada o severa calcificación (Modificado de N Engl J Med 2000; 343: 611-7)

Gelpi - Donato Fisiopatología cardiovascular
264
-
15 Esto plantea la importancia de evaluar no solo el ventrículo sino las características del
Doppler tisular, ha
En los pacientes portadores de estenosis -
-
16
-
en esta valvulopatía se relaciona con el remodelamiento inverso de los diferentes constituyentes de la pared mio-
-nando un incremento porcentual relativo de esta última en
de la 17
La circulación coronariade pecho con coronarias nor-
18 y su presencia 19 -
coronario son determi-
-remo-
delamiento vascular, como respuesta adaptativa del sistema -
cia, ya que sus alteraciones serán un determinante directo en -
-
--
-
ambos mecanismos determinará un remodelamiento vas-20
ventrículo izquierdo. 21
-
subendocárdica, con evidencia de re-
Interacción dinámicaSeñales externas
Procesamientoextracelular
Químicos
Factores de crecimento
Metaloproteasa
Hormonas
Mecánicos
Estiramiento
Fibroblastos
Síntesis de colágeno
Síntesis de metaloproteasa
Miocitos
Síntesis de miofibrillas
Integrinas
Receptores hormonales
Receptores parafactores de crecimiento
Receptores detransmembrana
Procesamientointracelular
Respuesta celular
Figura 17.3: Interacción dinámica entre los miocitos y el intersticio frente a diferentes señales externas. (Modificado de Kanekar et al. Cardiovasc Pathol 7:127-133; 1998)
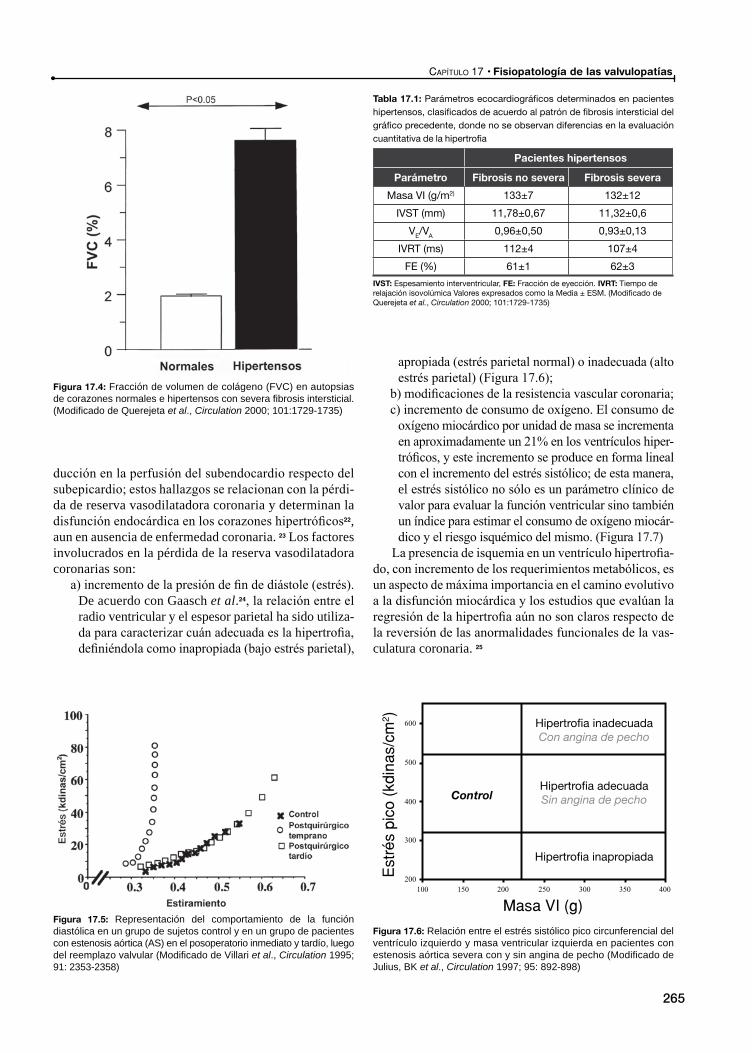
Fisiopatología de las valvulopatías
265
Figura 17.4: Fracción de volumen de colágeno (FVC) en autopsias de corazones normales e hipertensos con severa fibrosis intersticial. (Modificado de Querejeta et al., Circulation 2000; 101:1729-1735)
Tabla 17.1:hipertensos, clasificados de acuerdo al patrón de fibrosis intersticial del gráfico precedente, donde no se observan diferencias en la evaluación cuantitativa de la hipertrofia
Pacientes hipertensos
Parámetro Fibrosis no severa Fibrosis severa
Masa VI (g/m2) 133±7 132±12
IVST (mm) 11,78±0,67 11,32±0,6
VE/VA 0,96±0,50 0,93±0,13
IVRT (ms) 112±4 107±4
FE (%) 61±1 62±3
IVST: Espesamiento interventricular, FE: Fracción de eyección. IVRT: Tiempo de relajación isovolúmica Valores expresados como la Media ± ESM. (Modificado de Querejeta et al., Circulation 2000; 101:1729-1735)
-da de reserva vasodilatadora coronaria y determinan la
22, aun en ausencia de enfermedad coronaria. 23 Los factores involucrados en la pérdida de la reserva vasodilatadora coronarias son:
De acuerdo con Gaasch et al.24
radio ventricular y el espesor parietal ha sido utiliza-
-
el
-
-
-culatura coronaria. 25
Figura 17.5: Representación del comportamiento de la función diastólica en un grupo de sujetos control y en un grupo de pacientes con estenosis aórtica (AS) en el posoperatorio inmediato y tardío, luego del reemplazo valvular (Modificado de Villari et al., Circulation 1995; 91: 2353-2358)
Control
Hipertrofia inadecuadaCon angina de pecho
Hipertrofia adecuadaSin angina de pecho
Hipertrofia inapropiada
Figura 17.6: Relación entre el estrés sistólico pico circunferencial del ventrículo izquierdo y masa ventricular izquierda en pacientes con estenosis aórtica severa con y sin angina de pecho (Modificado de Julius, BK et al., Circulation 1997; 95: 892-898)

Gelpi - Donato Fisiopatología cardiovascular
266
Figura 17.8: Representación esquemática de la secuencia de eventos en la hipertrofia cardiaca y su progresión a la insuficiencia cardiaca
La geometría
Como se ha mencionado, el miocardio responde a los incrementos de la hipertrofia, la que ha sido clasificada desde un punto de vista mor-
ventricular.
-lar, este no es el único factor. Así, varios estudios demos-
26 estenosis -
y con menor clase funcional. 27
bajo estrés parietal circunferencial, para un determinado femenino. Estas
-ferencias funcionales y de comportamiento posoperatorio;
realizado el reemplazo valvular; por otra parte, la mujer
-dola con el hombre, aunque la mortalidad es similar para
28
La función
Función ventricular sistólicaEn los pacientes con enfermedad valvular será la respues-
cardiaca. El miocardio pone en marcha diferentes respuestas para intentar compensar el defecto primario y para evitar la falla miocárdica. En primer lu-
mecanismos neurohormonales con el objeto de incremen-tar la contractilidad. El primer mecanismo es limitado y el tercero es deletéreo cuando se sostiene en el tiempo, es por ello que el incremento de la masa y el proceso de
Figura 17.7: Mecanismos fisiopatológicos involucrados en la angina de pecho en pacientes portadores de estenosis aórtica severa. (Modificado de Julius, BK et al. Circulation 1997; 95: 892-898)

Fisiopatología de las valvulopatías
267
remodelamiento adquieren un rol central en la compen-
estímulo hemodinámico presenta una secuencia evolutiva
29 Como fue descrito, la determina cambios en los miocitos, caracterizados por
-
-30 Dado que
el incremento de la
31
pacientes presenta un comportamiento caracterizado por
-
-
32 Por otra parte, los síntomas en una valvulopatía no se correlacionan en forma
los síntomas y la severidad de la valvulopatía; se puede
como pérdida en la clase funcional y la capacidad de
un rol fundamental en el estudio de una valvulopatía. Pero
-rabello et al.33, las características que se describen en la
-masa, son fac-
-
-
-
de cambios en el estado contráctil; este comportamiento de la -
de bomba del ventrículo más que su estado contráctil. 34
-
y el mismo podrá ser analizado en sístole o en diástole.
-vidad del ventrículo izquierdo y Th el espesor parietal. De
cálculos del estrés parietal son más complejos que esta sim-
ventrículo izquierdo. Dentro de los diferentes tipos de estrés
Tabla 17.2:
1. Sensible a los cambios del inotropismo
2. Independiente de la carga
3.
4. Fácil y seguro de aplicar
5. De probada utilidad en la práctica clínica
(Modificado de Carabello B. et al., Circulation 2002; 105:2701-2703)
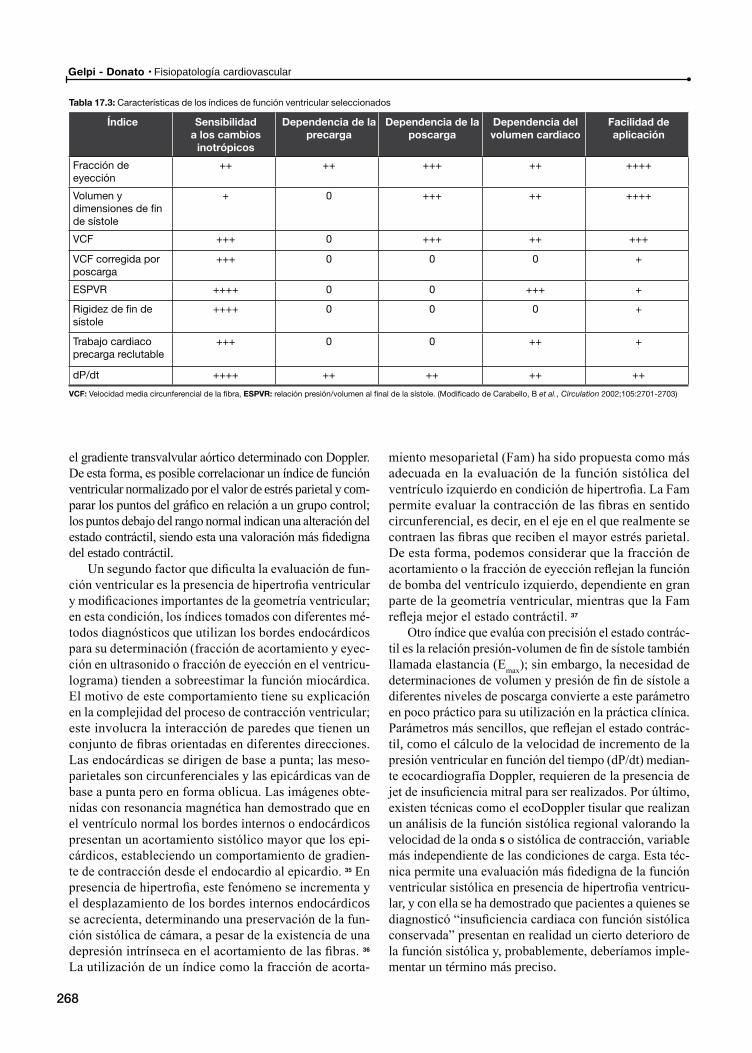
Gelpi - Donato Fisiopatología cardiovascular
268
ventricular normalizado por el valor de estrés parietal y com-
del estado contráctil.-
-
--
-parietales son circunferenciales y las epicárdicas van de
-
el ventrículo normal los bordes internos o endocárdicos --
35 En
el desplazamiento de los bordes internos endocárdicos -
36 -
circunferencial, es decir, en el eje en el que realmente se
parte de la 37
-
llamada
-til, como el cálculo de la velocidad de incremento de la
-
velocidad de la onda s-
-lar, y con ella se ha demostrado que pacientes a quienes se
-mentar un término más preciso.
Tabla 17.3: Características de los índices de función ventricular seleccionados
Índice Sensibilidad a los cambios
inotrópicos
Dependencia de la precarga
Dependencia de la poscarga
Dependencia del volumen cardiaco
Facilidad de aplicación
Fracción de eyección
Volumen y dimensiones de fin de sístole
0
VCF 0
VCF corregida por poscarga
0 0 0
0 0
Rigidez de fin de sístole
0 0 0
Trabajo cardiaco precarga reclutable
0 0
VCF: Velocidad media circunferencial de la fibra, ESPVR: relación presión/volumen al final de la sístole. (Modificado de Carabello, B et al., Circulation 2002;105:2701-2703)
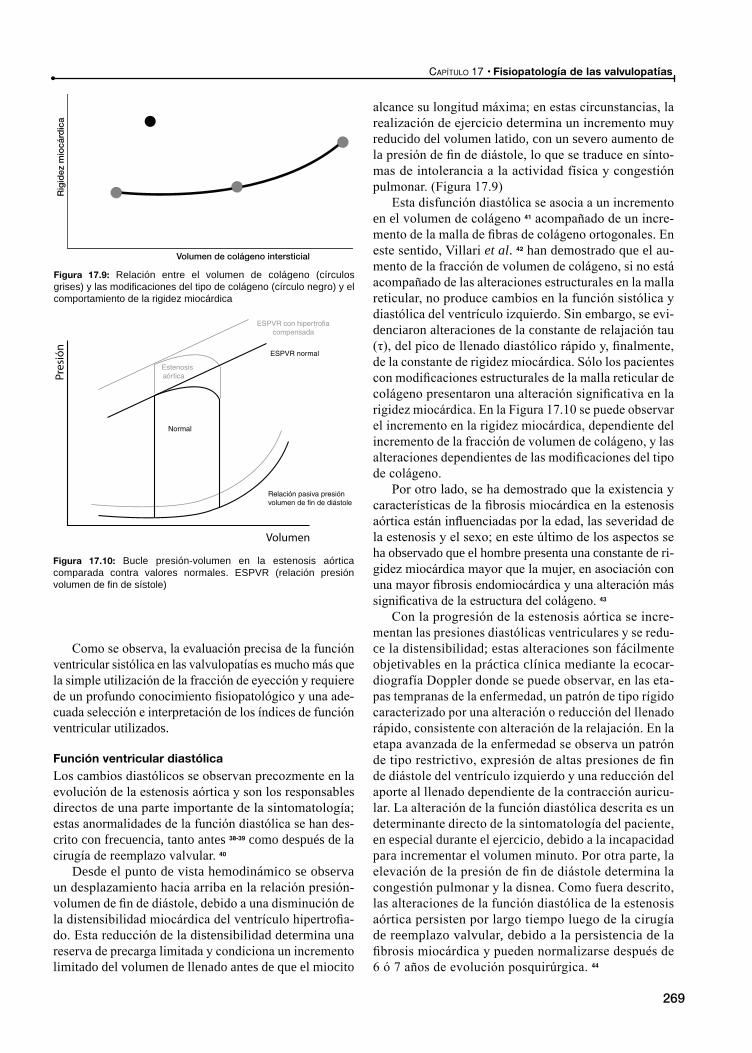
Fisiopatología de las valvulopatías
269
-
ventricular utilizados.
Función ventricular diastólica
-crito con frecuencia, tanto antes 38-39 como después de la
40
Desde el punto de vista hemodinámico se observa
-distensibilidad determina una
limitado del volumen de llenado antes de que el miocito
reducido del volumen latido, con un severo aumento de la -
en el 41 -
et al. 42 han demostrado que el au-
-denciaron alteraciones de la constante de
ha observado que el hombre presenta una constante de ri-
43
--
ce la distensibilidad; estas alteraciones son fácilmente objetivables en la práctica clínica mediante la ecocar-
-
-
en especial durante el ejercicio, debido a la incapacidad para incrementar el volumen minuto. Por otra parte, la
de reemplazo valvular, debido a la persistencia de la
44
Figura 17.10: Bucle presión-volumen en la estenosis aórtica comparada contra valores normales. ESPVR (relación presión volumen de fin de sístole)
Volumen de colágeno intersticial
Rig
idez
mio
cárd
ica
Figura 17.9: Relación entre el volumen de colágeno (círculos grises) y las modificaciones del tipo de colágeno (círculo negro) y el comportamiento de la rigidez miocárdica

Gelpi - Donato Fisiopatología cardiovascular
270
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica-
tica depende fundamentalmente de la severidad de la
-
es la sumatoria del componente valvular y vascular; la importancia de este último factor es evidente cuando
-prusiato, 45
-curso terapéutico permite demostrar la importancia del
Circulación pulmonar
46 En la estenosis sintomática, diferentes trabajos han de-mostrado una frecuencia de
47 -
48; aunque una
a retornar a la normalidad.
La valvulopatía y el ejercicioEl
-
-
la toma de decisiones de esta valvulopatía. Como fuera
de las resistencias periféricas; en forma equivalente, la
49 La respuesta al ejercicio en los pacientes portadores de
frecuencia cardiaca, con un aumento del volumen minuto,
proporcional. 50
durante el esfuerzo. 51 Finalmente, el área valvular tiende a con el ejercicio,
permitiendo un cierto incremento del volumen minuto; a medida que la enfermedad evoluciona, las valvas se
aumentar el volumen minuto y el trabajo latido durante
durante el esfuerzo.
-
-siones en estos pacientes. 52
---
-dobuta-
contráctil conservada, responde incrementando el volumen -
transvalvulares sin cambios en el área valvular, constituyen-
valvular. Por el contrario, los pacientes con pseudoesteno-
Insuficiencia aórtica
De la estructura a la función: La válvula-

Fisiopatología de las valvulopatías
271
et al. 53 La anatomía funcional -
tes: la zona sinotubular, los senos y la base; la zona
básicamente por tejido elástico y soportan la apertura de las arterias coronarias. Además, son el punto para la
-
-
intervalvares, determinantes directos de una adecuada
actúen en forma independiente. Por último, determinan
54 Funcionalmente la base de la aorta
por músculo y se establece, como ya fuera descrito, una
-tría del tracto de salida durante las diferentes fases del
--
-55
56
--
cia y así sucesivamente. Este compromiso asociado de la
-
-
La respuesta ventricular: Estructura, geometría y función
-
La estructura: El miocito, el intersticio y la circulación coronaria
El miocitoEn la
-
-
-
-
57
deslizamiento o slippage de los miocitos permitiendo un aumento del diámetro de la cavidad ventricular con incre-
-
-
et al.; el es--
58 Este proceso de remodelado

Gelpi - Donato Fisiopatología cardiovascular
272
El intersticio
La -
con esto, trabajos en humanos, así como observaciones
-larmente marcada cuando la misma se ha instalado. Tam-bién se ha descrito en esta valvulopatía una up-regulation
de 59 y un incremento de
-
no están totalmente aclaradas, pero este comportamiento
-54 del estrés
La circulación coronaria
la
reserva vasodilatadora corona-ria y las alteraciones estructurales de arterias y arteriolas
-
-
-
determina con frecuencia isquemia subendocárdica 60-61
vasodilatadora coronaria. 62
La geometríaEl comportamiento de la
-
ventrículo42
este volumen latido incrementado determina el aumen-
ventricular. Los cambios de la
que puede presentar diferencias relacionadas a distintos
-
de sístole superiores a 55 mm; por el contrario, solo un
La función
Función ventricular sistólicaComo se ha mencionado, la característica hemodinámica
-
Frank-
este volumen en una cámara de alta impedancia como la
63, que
-
-le, lo que determina un desplazamiento del bucle estrés-
ventrículo mantener un elevado volumen latido con un
-
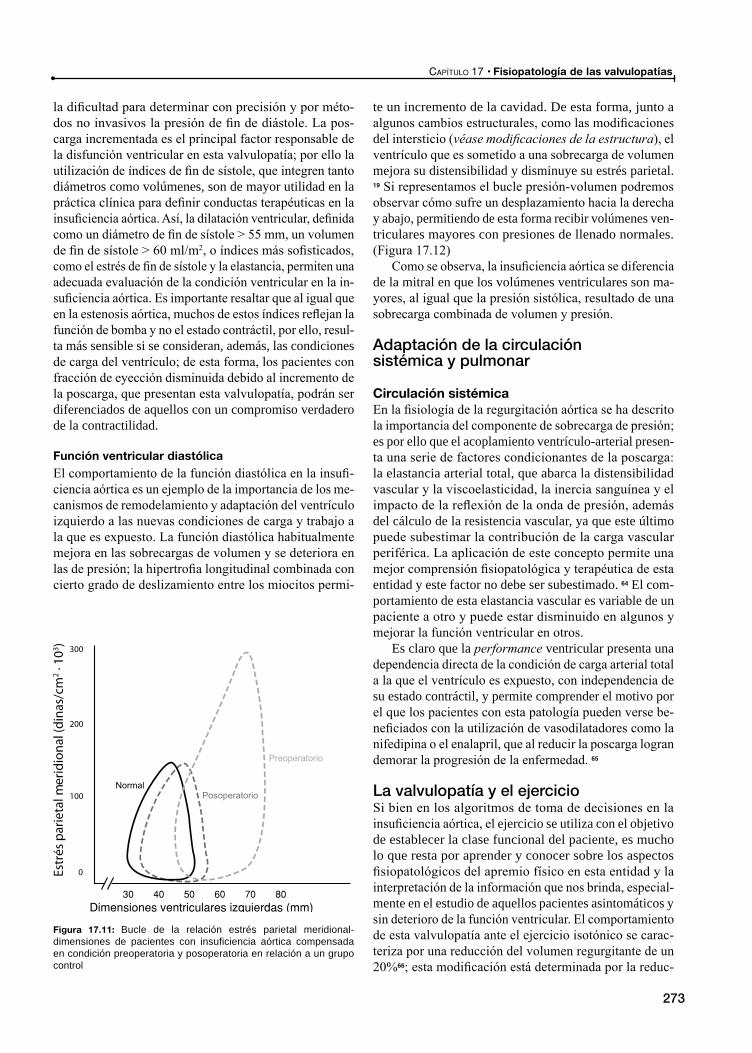
Fisiopatología de las valvulopatías
273
--
diámetros como volúmenes, son de mayor utilidad en la
-
-ta más sensible si se consideran, además, las condiciones
diferenciados de aquellos con un compromiso verdadero de la contractilidad.
Función ventricular diastólica--
-
te un incremento de la cavidad. De esta forma, junto a
del
mejora su distensibilidad y disminuye su estrés parietal. 19
y abajo, permitiendo de esta forma recibir volúmenes ven-triculares mayores con presiones de llenado normales.
de la mitral en que los volúmenes ventriculares son ma-
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica
es por ello que el acoplamiento ventrículo-arterial presen-
la elastancia arterial total, que abarca la distensibilidad
del cálculo de la resistencia vascular, ya que este último
entidad y este factor no debe ser subestimado. 64 El com-portamiento de esta elastancia vascular es variable de un
Es claro que la ventricular presenta una
su estado contráctil, y permite comprender el motivo por -
65
La valvulopatía y el ejercicio
ejercicio se utiliza con el objetivo de establecer la clase funcional del paciente, es mucho lo que resta por aprender y conocer sobre los aspectos
-mente en el estudio de aquellos pacientes asintomáticos y
-
66 -
Figura 17.11: Bucle de la relación estrés parietal meridional-dimensiones de pacientes con insuficiencia aórtica compensada en condición preoperatoria y posoperatoria en relación a un grupo control

Gelpi - Donato Fisiopatología cardiovascular
274
resistencias vasculares sistémicas. Así, en pacientes con
67 El comportamiento inverso,
y deterioro del estado contráctil sometidos a este estrés. Borer et al.60 analizaron el porcentaje de cambio de
la
-
conservada en condiciones basales y observaron que la --
en condiciones basales.
Insuficiencia mitral
De la estructura a la función: La válvulamitral es una valvulopatía de alta preva-
-tico severo, constituyéndose en un importante problema de
determina la necesidad de un profundo conocimiento de
involucrada y tomar las decisiones terapéuticas correctas. El cierre mitral normal depende de una compleja interac-
aurícula izquierda, anillo mitral, valvas, cuerdas, múscu-los papilares y la pared ventricular propiamente dicha; el conocimiento de la estructura anátomo-funcional de esta válvula es esencial, ya que los mecanismos responsables
alteraciones estructurales o funcionales de esta válvula aurículo-ventricular. 68
Como se aprecia, el anillo mitral tiene forma de silla de montar; su estructura anterior no constituye un verdadero
-
-ca y la sístole ventricular, constituyéndose de esta forma en uno de los factores importantes para mantener la com-petencia valvar; este anillo normalmente tiene un diámetro
69
--
reduce de forma ostensible cuando el intervalo PR es corto
auricular o marcapaseo ventricular; estos factores determi-
demostrando la jerarquía del comportamiento auricular en la competencia valvar. La importancia clínica de este fac-
-
La válvula mitral está constituida por dos valvas, la anterior y la posterior, con sus respectivas comisuras me-dial, central y lateral. La valva anterior es la de mayor ESPVR
Figura 17.12: Se observa el bucle presión-volumen en pacientes normales, con insuficiencia mitral y con insuficiencia aórtica. ESPVR: Relación presión volumen de fin sístole Figura 17.13: Esquema ilustrativo de los festones valvares

Fisiopatología de las valvulopatías
275
cmcm; por el contrario, la valva posterior es de menor tama-
cmEstas valvas presentan festones lateral, central y medial;
En la sístole, las valvas contactan inicialmente sus bor--
-micos o funcionales que alteran este proceso son factores
Trabajos como el de Carpentier et al. han demostra-do que una distancia muy incrementada de 10 mm o más
70
lo determina el aparato subvalvular mitral. Los músculos
-
similar para ambas valvas, el músculo anterolateral con una preponderancia a los festones laterales y el músculo pos-teromedial a los festones mediales. Las cuerdas laterales y
previene el prolapso valvar, mientras que las comisurales
-
Los músculos papilares están posicionados a unos dos
descendente posterior–, sino que, al mismo tiempo, nece-
motilidad miocárdica subyacentes a los músculos papilares
-
Así, resulta claro que es necesaria una adecuada inter-
-
-
mitral será totalmente diferente, ya que aquí la válvula se encuentra indemne.
La etiología-
prolapso de válvula mitral
-
Figura 17.14: Esquema ilustrativo de las zonas de coaptación y aposición
Figura 17.15: Esquema ilustrativo del patrón de fijación de las cuerdas a las valvas mitrales. A: Anterior; M: Medio: L: Lateral; MPP: Músculo pailar posterior; MPA: Músculo papilar anterior

Gelpi - Donato Fisiopatología cardiovascular
276
Figura 17.16: Clasificación funcional de los tipos de movimiento cordal y valvar asociados a insuficiencia mitral tipo I movimiento valvar normal, tipo II insuficiencia mitral secundaria a prolapso valvar o movimiento excesivo y tipo III es un movimiento valvar restrictivo que podría ser IIIa de restricción diastólica o IIIb sistólica. En líneas punteadas se esquematiza el movimiento de las valvas durante la sístole. (Modificado de Carpentier, A. “Cardiac valve surgery: the French correction”. J Thorac Cardiovasc Surg 1983; 86:323)
La importancia clínica de este concepto radica en que
--
-
punto de controversia. Por el contrario, en los pacientes
--
nes71
realizarla.
La respuesta ventricular: Estructura, geometría y función
La estructura: El miocito, el intersticio y la circulación coronaria
El miocito
-modinámica. En la
-trículo incrementar su volumen latido para compensar el
-
-
72, probablemente debido a que el
mitral no es muy potente comparado, por ejemplo, con el
-
miocitos se encuentran incrementados en la
reemplazo valvular.-
tativo en esta valvulopatía y el camino a la falla miocárdica
El intersticioEl comportamiento del

Fisiopatología de las valvulopatías
277
estimularía un incremento de la síntesis de -
abajo y a la derecha formando parte de los mecanismos compensadores de la
esfericidad del ventrículo. Nuevamente se observa la importancia del intersticio
y su comportamiento particular en las diferentes valvulo--
La circulación coronaria-
ver valvulopatía aórtica
73
su reserva vasodilatadora, tal como ocurre en toda valvulo-
-
mitral. 74 La falta de evidencia de este componente circu-
con-
consumo de -
75; y un
severa, que es relativamente modesto comparado con el hallado en otras valvulopatías. 76
en perros han demostrado incrementos tan modestos como
isque-
coronariopatía demostrada.
La geometríaEl remodelamiento del ventrículo izquierdo en la insu-
--
El ventrículo adquiere una forma más esférica, tanto en diástole como en sístole77, con las consecuentes alteracio-
-
-
los pacientes con miocardiopatía dilatada o cardiopatía isquémica. En contrapartida, diferentes trabajos han de-
-78
importancia en esta valvulopatía es la aurícula izquierda;
aurícula izquierda se encuentra mar-cadamente dilatada, su distensibilidad está incrementa-
muestra presiones prácticamente normales o levemente incrementadas, lo que permite que las presiones del cir-
79
-lúmenes sean considerados para la toma de decisiones en esta valvulopatía; así, pacientes con parámetros interme-
La función
Función ventricular sistólicaLa
-lumen se realiza a una cavidad de baja impedancia como es

Gelpi - Donato Fisiopatología cardiovascular
278
las diferencias de impedancia entre la aurícula izquierda
caracterizada por una -
trículo que maneja la
descenso del estrés pico y persistencia de un estrés de
mitral descompensada, el estrés pico se encuentra incre-
secundario al cierre de la cámara de baja impedancia. 79
-
El comportamiento hemodinámico descrito le permite al
su trabajo cardiaco con solo un leve incremento del con-
-
-
79
-
-
del estado contráctil; en otras palabras, representan lo que
et al
contráctil. 80 Otros parámetros mucho más simples, como
-
-
de vida de estos pacientes. 81, 82, 83 -
--
lla contráctil ventricular izquierda, sumados al importante -
planteado la importancia de realizar la toma de decisiones en estos pacientes en forma más precoz y antes del inicio
84
-
-vulopatía suelen ser más tardíos y menos precisos en la
de la de bomba ventricular izquierda en la
--
las fases iniciales de la valvulopatía, el estímulo simpáti-
el rol de los betabloqueantes en el manejo terapéutico de esta entidad.
Función ventricular diastólicaEn las -
el slippage
incremento del -
-

Fisiopatología de las valvulopatías
279
--
cardiaca en esta valvulopatía85, especialmente asociadas
-
muestras de biopsia epicárdica, han demostrado prolon-
Adaptación de la circulación sistémica y pulmonar
Circulación sistémica
la posibilidad de utilizar terapéutica vasodilatadora en el tratamiento médico de esta valvulopatía, con el objetivo
comportamiento hemodinámico de la -
del estrés parietal, aun en presencia de un deterioro de la
-
--
ellos clínicos, han demostrado, utilizando esta terapéuti--
ciencia mitral86
-
sintomáticos, menos útil en los asintomáticos y práctica-mente de nula utilidad en asintomático con deterioro leve
87 -
res han planteado que la terapéutica vasodilatadora puede -
ciencia mitral primaria y debería reservarse para aquellos pacientes con
Circulación pulmonar
por una aurícula izquierda complaciente con escasa eleva-
resistencias pulmonares; a pesar de ello, un escaso número de pacientes terminará presentando una -monar reactiva similar a la estenosis mitral. Esta hiper-
y plantea la importancia de evaluar el ventrículo derecho
del ventrículo derecho. Este comportamiento del circuito
vs.reposo o en ejercicio podría aclarar en un alto porcentaje
-
del ventrículo derecho disminuida, aun en presencia de -
88
-rante el ejercicio; la falla de este comportamiento se aso-
89
La valvulopatía y el ejercicioTal como ocurre en todas las valvulopatías, el ejercicio físico puede ser utilizado con el objetivo de aclarar aspec-tos relacionados con la clase funcional del paciente. El comportamiento de esta valvulopatía con el ejercicio está
-
utilizado en esta valvulopatía, pero son escasos los trabajos

Gelpi - Donato Fisiopatología cardiovascular
280
sido parcialmente resuelta con el advenimiento de nuevas -
importancia objetivar diferentes parámetros ventriculares
otro determinante del comportamiento de esta valvulopa-et al.
-
posoperatoria. 90 -
válvula mitral demostrando que muchos de estos pacientes,
91
-nados de pacientes.
Estenosis mitral
De la estructura a la función: La válvulaLos aspectos anatomofuncionales de la válvula mitral ya
en este punto solo se remarcarán los aspectos diferenciales estenosis mitral. La
válvula mitral se produce en la diástole temprana y depende
movimiento del anillo mitral, la
durante el ejercicio, ya que la taquicardia acorta el tiempo
92 El in-
Tabla 17.4: Marcadores de mal pronóstico en la Insuficiencia mitral
Parámetros Conservados Intermedios Disfunción Ventricular
Fracción de eyección 70% 60-70% < 60%
Diám. fin de sístole < 40 mm 40-45 mm > 45 mm
Diám. fin de sístole/superf. corporal < 22 mm/m2 22-26 mm/m2 > 26 mm/m2
Vol. fin de sístole/superf. corporal < 35 ml/m2 30-50 ml/m2 > 50 ml/m2
Diám: diámetro; Vol: volumen
Figura 17.17: A, y
A B
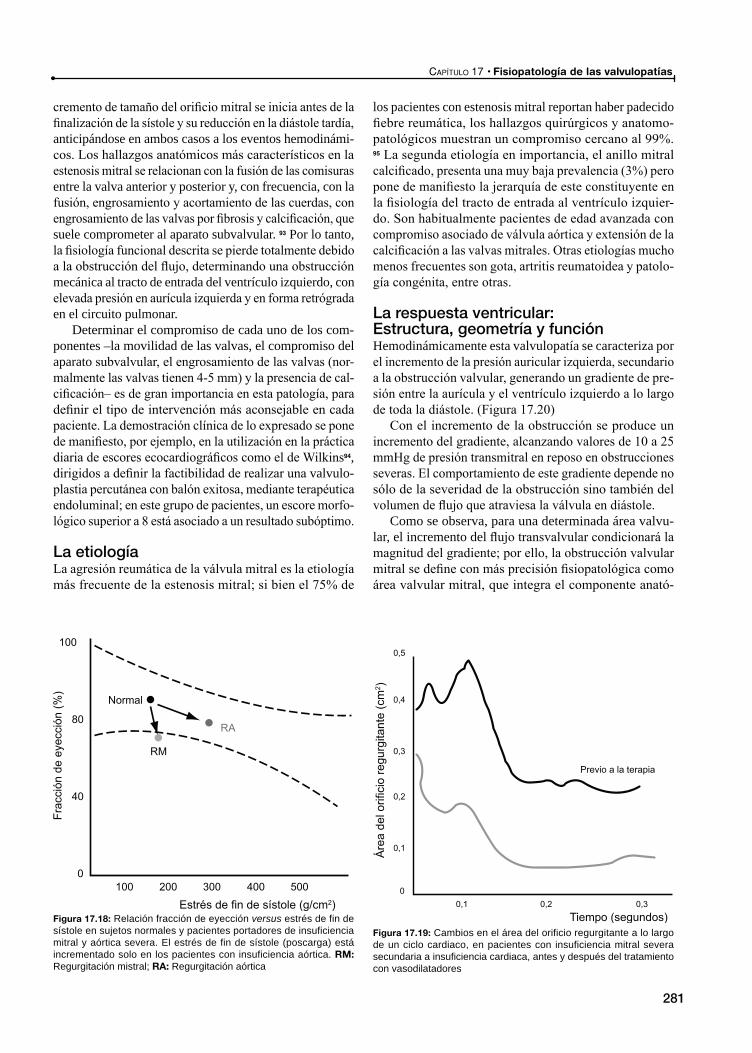
Fisiopatología de las valvulopatías
281
anticipándose en ambos casos a los eventos hemodinámi-
entre la valva anterior y posterior y, con frecuencia, con la
suele comprometer al aparato subvalvular. 93 Por lo tanto,
mecánica al tracto de entrada del ventrículo izquierdo, con
en el circuito pulmonar. Determinar el compromiso de cada uno de los com-
ponentes –la movilidad de las valvas, el compromiso del aparato -
-
94, valvulo-
-
La etiología
los pacientes con estenosis mitral reportan haber padecido -
95
-
-
La respuesta ventricular: Estructura, geometría y funciónHemodinámicamente esta valvulopatía se caracteriza por
-
Como se observa, para una determinada área valvu-lar, el incremento del
-
Figura 17.18: Relación fracción de eyección versus estrés de fin de sístole en sujetos normales y pacientes portadores de insuficiencia mitral y aórtica severa. El estrés de fin de sístole (poscarga) está incrementado solo en los pacientes con insuficiencia aórtica. RM: Regurgitación mistral; RA: Regurgitación aórtica
Figura 17.19: Cambios en el área del orificio regurgitante a lo largo de un ciclo cardiaco, en pacientes con insuficiencia mitral severa secundaria a insuficiencia cardiaca, antes y después del tratamiento con vasodilatadores

Gelpi - Donato Fisiopatología cardiovascular
282
la aurícula izquierda. A diferencia del resto de las valvulopatías, el ven-
-
Los volúmenes ventriculares en la estenosis mitral ais--
mal sin evidencias de impedimento en la velocidad de
-96
-
-carditis o miocarditis reumática, así como presencia de
ventrículo izquierdo suelen ser escasos y la mayor parte de las reducciones del volumen minuto son el resulta-
izquierdo, junto a alteraciones del llenado en un ventrí-culo cuya masa suele encontrarse en valores normales o subnormales. 97
llenado temprano y un desplazamiento de la curva pre--
-tría ventricular izquierda secundarias al incremento de los volúmenes y presiones derechas que provocan una
98
-
la estenosis mitral, la aurícula izquierda es la cámara cardia-
-
de la aurícula también muestra características particulares relacionadas a una variable más, que es la complacencia
-
sal, libres de síntomas en su vida diaria que presentan, ante -
pacientes suelen tener una estenosis mitral cerrada con una
no complaciente. 99 En estos pacientes, la importancia de las
que la del mantenimiento del ritmo sinusal; un incremento de frecuencias de 75 a 150 latidos determina un incremento
-100
-mos, con técnicas de estrés como el ecoestrés, nos permite
Finalmente, es importante remarcar que, aun en ritmo -
en su interior, que predisponen al desarrollo de trombos, en particular en el apéndice de la aurícula izquierda, presen-
observar, el comportamiento de la aurícula izquierda es un
Adaptación de la circulación sistémica y pulmonar
-
sistémica. En la etapa inicial de esta valvulopatía, el incremento
pasivo en las presiones pulmonares venosas, capilares y ar-teriales. Estos cambios hemodinámicos son intermitentes,
-culatura pulmonar; a pesar de ello, estos pacientes pueden
El
-cia de un incremento de las resistencias vasculares pulmo-nares; esta -
severidad de la estenosis y el incremento de las resisten-cias vasculares pulmonares, distintos factores condicionan

Fisiopatología de las valvulopatías
283
-den ser reversibles, pero el mantenimiento en el tiempo
-
en dilataciones aneurismáticas de las paredes arteriales ver capítulo 20
-
-
del ventrículo izquierdo, el área valvular mitral y el ante-101 -
hiperten-
102
está relacionada con la presencia de factores vasoactivos como la endotelina, o un péptido derivado del endotelio con propiedades vasoconstrictoras y proliferativas sobre las células del músculo liso. Estos factores también per-
-
El desarrollo de los cambios referidos en la vascula-
-bre el
está preparado para el manejo de altos volúmenes a baja
o más durante un ejercicio importante, manejando sus pre-siones en valores prácticamente normales. El incremento
-
sus funciones, el ventrículo derecho sufre un proceso de
izquierdo.
-cular en la estenosis mitral; quizá otros factores, como la
-
-
radica en comprender que el acoplamiento ventrículo-vas-
cálculo de las presiones y las resistencias pulmonares, de
mitral, puede ser subestimado al ser evaluado con estas técnicas.
La valvulopatía y el ejercicio
importante en la estenosis mitral se relaciona con el llena-do ventricular izquierdo; como resultado de ello, el volu-men minuto en reposo puede estar reducido o preservado, pero básicamente falla en elevarse de forma apropiada con el al tracto de entrada del ventrículo izquierdo que, si es
-
manifestaciones clínicas durante la actividad física. La
pulmonar y, con frecuencia, a los trastornos del ritmo,
-
situaciones puntuales donde el apremio físico contribuye a Figura 17.20: Representación esquemática del gradiente de presión diastólico entre la aurícula izquierda y el ventrículo izquierdo

Gelpi - Donato Fisiopatología cardiovascular
284
-co de la valvulopatía en condiciones de reposo. Como se
-miento de la distensibilidad auricular no es el habitual y en
el incremento de las presiones pulmonares, por ejemplo mediante técnica de ecoDoppler intraesfuerzo, a partir del
-
Bibliografía
11. Borre JS, OW Isom. “Pathophsiology, evaluation and management of valvular heart disease. The epidemiology of valvular heart disease: An emerging public health problem”. Adv Cardiol 2002;39:1-6.
12. Yacoub M, Onuzo O, Reidel B, et al. “Mobilization of the left and right fibrous trigones for relief of severe left ventricular outflow obstruction”. J Thorac Cardiovasc Surg. 1999;117:126-132.
13. Aurigemma G, Battista S, Orsinelli D, et al. “Abnormal left ventricular intracavitary flow acceleration in patients undergoing aortic valve replacement for aortic stenosis: a marker for high posoperative morbidity and mortality”. Circulation 1992;86:926-936.
14. Bartunek J, Sys SU, Rodrigues AC, et al. “Abnormal systolic in-traventricular flow velocities after valve replacemente for aortic stenosis. Mechanisms, predictive factor, and prognostic signifi-cance”. Circulation 1996;93(4):712-719.
15. Thubrikar MJ, Heckman JL, Nolan SP. “Higt-speed cine-radio-graphic study of aortic valve leaflet motion”. J Heart Valve Dis. 1933;2:653-661.
16. Levine RA, Cape EG, Yoganathan AP. “Pressure recovery distal to stenoses: Expanding clinical aplications of engineering principles”. J Am Coll Cardiol 1993;21:1026-1028.
17. Bellhouse BJ, FH Bellhouse. “Fluid mechanics of the aortic root with application to coronary flow”. Nature 1968;219:1059-1061.
18. Osenhek R, Binder T, Porenta G, et al. “Predictor of outco-me in severe; asymptomatic aortic stenosis”. N Engl J Med. 2000;343:611-7.
19. Iizumo S, Nadal GB, Mahdavi V. “Protooncogenes induction and reprogramming of cardiac gene expresion produced by presure overload”. Proc Nat Acad Sci USA 1988;85:339-343.
10. “Genomics of Cardiovascular development, adaptation, and remode-ling”. NHLBI program for genomic applications, Beth Israel Deaconess Medical Center. <http://cardiogenomics.med.harvard.edu>
11. Matsuo T, Carabello BA, Nagatomo, et al. “Mechanisms of cardiac hypertrophy in canine volume overload”. Am J Physiol. 1998;275:H65-H74.
12. Weber KT, Sun Y, Tyagi SC, et al. “Collagen network of the myocar-dium: function, structural remodeling and regulatory mechanisms”. J Mol Cell Cardiol. 1994;26:279-272.
13. Weber KT, Brilla CG, Campbell SE, et al. “Pathologic hypertrophy with fibrosis: the structural basis for myocardial failure”. Blood pressure. 1992;1:75-85.
14. Querejeta R, Varo N, Begoña Lopez BS, et al. “Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive Heart disease”. Circulation 2000;101:1729-1735.
15. Weber, KT. “Cardioreparation in hipertensive heart disease”. Hy-pertension 2001;38:588-591.
16. Krayenbuehl HP, OM Hess, et al. “Left ventricular myocardial structure in aortic valve disease before, intermediate, and late after aortic valve replacement”. Circulation 1989;79:744-755.
17. Villari B, Vassalli G, Betocchi S, et al. “Normalization of left ventricular nonuniformity late after valve replacement for aortic stenosis”. Am J Cardiol 1996;78:66-71.
18. Basta LL, Raines D, Najjar S, et al. “Clinical, hemodynamic and coronary angiographic correlates of angina pectoris in patients with severe aortic valve disease”. Br Heart J. 1975;37:150-157.
19. Ross J Jr, E Braunwald. “Aortic stenosis”. Circulation 1968;38:V-61-V-68.
20. Sheridan, D. Left Ventricular Hypertrophy. First Edition. Chapter 5: “Coronary vasculature in hypertrophy”. Churchill-Livingstone, London 1998;29-35.
21. Garcia D, Camici PG, Durand LG et al. “Impairment of coronary flow reserve in aortic stenosis.”. J Appl Physiol. 2009;106(1):113-21
22. Ito N, Isoyama S, Takahashi T, et al. “Coronary dilator reserve and morphological changes after relief of pressure overload in rats”. J Mol Cell Cardiol 1993;25:3-14.
23. Hittinger L, Shen Y-T, Patrick TA, et al. “Mechanism of suben-docardial dysfunction in response to excercise in dogs with severe left ventricular hypertrophy”. Circ Res 1992;71:423-434.
24. Gaasch, WH. “Left ventricular radius to wall thickness ratio”. Am J Cardiol. 1979;43:1189-1194.
25. Brilla C, Janicki J, Weber K. “Impaired diastolic function and coronary reserve in genetic hipertensión. Role of intersticial fibrosis and medial thickening of intramyocardial coronary arteries”. Circ Res 1991;69:107-115.
26. Carroll JD, Carroll EP, Feldman T, et al. “Sex-associated differen-ces in left ventricular function in aortic stenosis in the elderly”. Circulation 1992;86:1099-1107.
27. Douglas PS, Otto CM, Mickel MC, et al. “Gender differences in left ventricular geometry and function in patients undergoing balloon dilation of the aortic valve for isolated aortic stenosis”. Br Heart J 1995;73:548-554.
28. Morris JJ, Scaff HV, Mullany CJ, et al. “Gender differences in left ventricular functional response to aortic valve replacement”. Circulation 1994;90:83-89.
29. Gunther S, W Grossman. “Determinants of ventricular function in pressure-overload hypertrophy in man”. Circulation 1979;59:679-688.24.
30. Anversa P, Olivetti G, Melissari M, et al. “Stereological measurement of cellular and subcellular hypertrophy and hyperplasia in the papi-llary muscle of adult rat”. J Moll Cell Cardiol. 1980;12:781-795.
31. Harpole HD, RH Jones. “Serial Assesment of ventricular perfor-mance after valve replacement for aortic stenosis”. J Thor Cardiovas Surg 1990;99:645-650.
32. Hwang MH, Hammermeister KE, Oprian C. “Pre-operative iden-tification of patients likely to have left ventricular dysfunction after aortic valve replacement. Participants in the veterans Adminis-tration Cooperative Study on valvular heart disease”. Circulation 1989;80:65-76.
33. Carabello, BA. “Evolution of the study of left ventricular function. Everything old is new again”. Circulation 2002;105:2701-2703.
34. Otto, C. Valvular Heart Disease. Second edition. Chapter 7: “Left ventricular response to chronic pressure and volume overload”. Philadelphia, Pennsylvania: Elsevier 2004; 158-177.
35. Clark NR, Reicheck N, Bergey P, et al. “Circumferential myocar-dial shortening in the normal human left ventricule. Assement by magnetic resonance imaging using spatial modulation of magne-tization”. Circulation 1991;84:67-74.
36. Palmon LC, Reicheck N, Yeon et al. “Intramural myocardial shor-tening in hypertensive left ventricular hypertrophy with normal pump function”. Circulation 1994;89:122-131.
37. Ballo P, Mondillo S, Guerrini F, et al. “Midwall mechanics in physiologic and hypertensive concentric hypertrophy”. J Am Soc Echocardiog 2004;17:418-427.

Fisiopatología de las valvulopatías
285
38. Cheuk-Man Yu, Hong Ling BM, Hua Yang BM, et al. “Progression of systolic abnormalities in patients with Isolated diastolic heart failure and diastolic dysfunction”. Circulation 2002;105:1195-1201.
39. Galema TW, Yap SC, Gelejinse ML, et al. Early detection of left ventricular dysfunction by Doppler tissue imaging and N-terminal pro-B-type natriuretic peptide in patients with symptomatic severe aortic stenosis. J Am Soc Echocardiog. 21:257-61.
40. Hess OM, Ritter M, Schneider J, et al. “Diastolic stiffness and myocardial structure in aortic valve disease before and after valve replacement”. Circulation 1984;69:855-865.
41. Douglas PS, Berko B, Lesh M, et al. “Alterations in diastolic function in response to progressive left ventricular hypertrophy”. J Am Coll Cardiol 1989;13:461-467.
42. Villari B, Campbell SE, Hess OM, et al. “Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease”. J Am Coll Cardiol. 1 1993;22:1477-84.
43. Villari B, Campbell SE, Schneider J, et al. “Sex-dependent diffe-rences in left ventricular function and structure in chronic pressure overload”. Eur Heart J 1995;16(10):1410-1419.
44. Gjertsson P, Caidhl K, Bech-Hanssen O. Left ventricular diastolic dysfunction late after aortic valve replacement in patients with aortic stenosis. Am J Cardiol. 2005, 96: 722-7
45. Umesh N. Khot, Gian M. Novaro, Zoran B. Popovi, et al. “Nitro-prusside in Critically Ill Patients with Left Ventricular Dysfunction and Aortic Stenosis”. N Eng J Med 2003;348:1756-63.
46. Otto CM, Burwash IG, Legget ME, et al. “A prospective study of asymptomatic valvular aortic stenosis: clinical, echocardiographic and excercise predictors of outcome”. Circulation 1997;95:2262-2270.
47. Faggiano P, Antonini-Canterin F, Ribichini F, et al. “Pulmonary artery hipertension in adult patients with symptomatic valvular aortic stenosis”. Am J Cardiol 2000;85:204-208.
48. Aragam JR, Folland ED, Lapsley D, et al. “Cause and impact of pulmonary hipertension in isolated aortic stenosis on operati-ve mortality for aortic valve replacement in men”. Am J Cardiol 1992;69(16):1365-1367.
49. Archer SI, Mike DK, Hetland MB, et al. “Usefulness of mean aortic valve gradient and left ventricular diastolic filling pattern for distinguishing the symptomatic from the asymptomatic patients”. Am J Cardiol. 1994;73:275-81.
50. Gabay J, Donato M, Pascua A et al. Effects of isometric exercise on the diastolic function in patients with severe aortic stenosis with or without coronary lesion. Int J Cardiol. 2005;104(1):52-8.
51. Movsowitz C, Kussmaul WG, Laskey WK. “Left ventricular dias-tolic response to excercise in valvular aortic stenosis”. Am J Cardiol 1996;77:275-28.
52. Amato, PJ MoVa, Werner KE, Ramires JAF. “Treatment decision in asymptomatic aortic valve stenosis: role of exercise testing”. Heart 2001;86:381-386.
53. Zimmerman, J. “The functional and surgical anatomy of the aortic valve”. Isr J Med Sci 1969;5:862-6.
54. Gross L, MA Kugel. “Topographic anatomy and histology of the valves of the heart”. Am J Pathol 1931;7:445-73.
55. Dare AJ, Veinot JP, Edwards WD. “New observations on the etio-logy of aortic valve disease”. Hum Pathol 1993;24:1330.
56. Keane MG, Wiegers SE, Plappert T, et al. “Bicuspid aortic valves are associated with aortic dilatation out of proportion to coexistent valvular lesions”. Circulation 2000;102:III-35-III-39.
57. Wang X, Li F, Gerdes AM. “Chronic pressure overload cardiac hy-pertrophy and failure in guinea pigs: I. Regional hemodynamics and myocyte remodelling”. J Mol Cell Cardiol 1999;31:307-317.
58. Grossman WD, LP Mc Laurin. “Wall stress and patterns of hypertrophy in the human left ventricle”. J Clin Invest. 1975;56:56-64.
59. Truter S, Kolesar J, Dumlao T, et al. “Abnormal gene expression of cardiac fibroblasts in experimental aortic regurgitation”. Am J Ther. 2000;7:237-243.
60. Borer JS, Truter S, Herrold EM, et al. “Myocardial fibrosis in Chornic aortic regurgitation molecular and cellular responses to volume overload”. Circulation 2002;105:1837-1842.71.
61. Miyahara K, Sonoda M, Kukihara T. “Relationships between left ventricular mass, left ventricular work and coronary artery size in aortic regurgitation, posible mechanism of myocardial ischemia”. Jpn Circ J 1993;57:263-271.
62. Nitemberg A, Foult JM, Anthony I, et al. “Coronary flow and resistanse reserve in patients with chronic aortic regurgitation, angina pectoris and normal coronary arteries”. J Am Coll Cardiol 1988;11:478-486.
63. Wisenbaugh T, Spann JF, Carabello BA. “Differences in myocardial performance and load between patients with similars amounts of chronic aortic versus chronic mitral regurgitation”. J Am Coll Cardiol 1984;3:916-923.
64. Slordahl SA, H Piene. “Haemodynamic effects of arterial complian-ce, total peripheral resistanse, and glyceryl trinitrate on regurgitant volume in aortic regurgitation”. Cardiovasc Res 1991;25:869-874.
65. Scognamiglio R, Rahimtoola SH, Fasoli G, et al. “Nifedipine in asymptomatic patients with severe aortic regurgitation and normal left ventricular function”. N Eng J Med 1994;331(11):689-694.
66. Thompson R, Ross I, Leslie P, et al. “Haemodinamyc adaptation to excercise in asymptomatic patients with severe aortic regurgitation”. Cardiovasc Res 1985;19:212-218.
67. Massie BM, Kramer BL, Loge D. “Ejection fraction response to supine excercise in asymptomatic aortic regurgitation: relation to simultaneous hemodynamic measurement”. J Am Coll Cardiol 1985;5:847-855.
68. Fontaine HS, Schwammenthal E, Yoganathan AP, et al. “Integrated mechanism for functional mitral regurgitation-leaflet restriction versus coapting force: in vitro studies”. Circulation 1997;96:1826-1834.
69. Westaby S, Karp RB, Blackstone EH, et al. “Adult human val-ve dimensions and their surgical significance”. Am J Cardiol 1984;53:552-556.
70. Carpentier, A. “Cardiac valve surgery: the French correction”. J Thorac Cardiovasc Surg 1983; 86:323.
71. Enriquez-Sarano M, Avierinos JF, Messika-Zeitoun D, et al. “Quan-titative determinants of the outcome of asymptomatic mitral re-gurgitation”. N Eng J Med 2005;352:875-83.
72. Carabello BA, Zile MR, Tanaka R, et al. “Left ventricular hyper-trophy due to volume overload versus pressure overload”. Am J Physiol. 1992;263 (32):H1137-H1144.
73. Vassalli G, Hess OM, Krogmann ON, et al. “Coronary artery size in mitral regurgitation and its regression after mitral valve surgery”. Am Heart J. 1993;126:1091-1098.
74. Carabello BA, Nakano K, Ishihara K, et al. “Coronary blood flow in dogs with contractile dysfunction due to experimental volume overload”. Circulation 1991;83:1063-1075.
75. Urschel CW, Covell JM, Sonnenblick EH, et al. “Myocardial me-chanics in aortic and mitral valvular regurgitation: the concept of instantaneous impedance as a determinant of the performance of the intact heart”. J Clin Invest 1968;47:867-883.
76. Kleaveland, JR, Kussmaul WG, Vinciguerra T, Diters R, Carabello BA. “Volume overload hypertrophy in a closed-chest model of mitral regurgitation”. Am J Physiol. 1988;254:H1034-H1041.
77. Boconas P, Gorlin R, Cohn P, et al. “Dynamic geometry of the left ventricle in mitral regurgitation”. Circulation 1973;48:786-796.
78. Hung J, Guerrero L, Handschumacher MD, Suple G, Sullivan S, Levine R. “Reverse ventricular remodeling reduces ischemic mitral regurgitation. Echo-guided device application in the beating heart”. Circulation 2002;106:2594-2600.
79. Braunwald E, WC Awe. “The syndrome of severe mitral regurgi-tation with normal left atrial pressure”. Circulation 1963;27:29.
80. Zile M, Gaasch WH, Levine HJ. “Left ventricular stress-dimension-shortening relations before and after correction of chronic aortic and mitral regurgitation”. Am J Cardiol 1985;56:99-105.
81. Starling MR, Kirsh MM, Montgomery DG, Grass MD. “Impaired left ventricular contractile function in patients with long-term mi-tral regurgitation and normal eyection fraction”. J Am Coll Cardiol 1993;22:239-250.
82. Zile MR, Gaasch WH, Carroll JD, et al. “Chronic mitral regurgi-tation: predictive value of preoperative echocardiographic indexes

Gelpi - Donato Fisiopatología cardiovascular
286
of left ventricular function and wall stress”. J Am Coll Cardiol. 1984;3:235-242.
83. Hammermeister KE, Fischer L, Kennedy JW. “Predictions of late survival in patients with mitral valve disease from clinical, hae-modinamic and quantitative angiographic variables”. Circulation 1978;57:341-349.
84. Consenso de Valvulopatías de la Sociedad Argentina de Cardiología. Rev. Arg Cardiol 2006; 75(4): 304-323.
85. Corin WJ, Murakami T, Monrad ES, et al. “Left ventricular passi-ve diastolic properties in chronic mitral regurgitation”. Circulation 1991;83:797-807.
86. Rosario LB, Stevenson LW, Solomon SD, et al. “The mechanism of decrease in dynamic mitral regurgitation during heart failure treatment: importance of reduction in the regurgitant orifice size”. J Am Coll Cardiol 1998;32(7):1819-1824.
87. Levine HJ, WH Gaasch. “Vasoactive drugs in chronic regurgi-tant lesions of the mitral and aortic valves”. J Am Coll Cardiol 1996;28:1083-1091.
88. Hochreiter C, Niles N, Devereaux RB, et al. “Mitral regurgitation: relationship of noninvasive descriptors of right and left ventricu-lar performance to clinical and hemodynamic findings and to prognosis in medically and surgical treated patients”. Circulation 1986;73,5:900-912.
89. Borer JS, Hochreiter CA, Supino PG. “The importance of right ventricular performance measurement in selecting asymptomatic patients with mitral regurgitation for valve surgery”. Adv Cardiol. 2002;39:144-152.
90. Leung DY, Griffin BP, Haluska B. “Left ventricular function after valve repair for chronic mitral regurgitation: Predictive value of preoperative assessement of contractile reserve by excercise echo-cardiography”. J Am Coll Cardiol. 1996;28:1198.
91. Tischler MD, Battle RW, Saha M, et al. “Observations suggesting a high incidence of excercise –induced severe mitral regurgitation in patients with mild rheumatic mitral valve disease at rest”. J Am Coll Cardiol 1995;25(1):128-133.
92. Tsakiris AG, Gordon DA, Padiyar R. “Relation of mitral valve opening and closure to left atrial pressures in the intact dog”. Am J Physiol 1978;234:H146-H151.
93. Roberts WC, R Virmani. “Aschoff bodies at necropsy in valvular heart disease. Evience from an analysis of 543 patients over 14 years of age that rheumatic heart disease, at least anatomically, is a disease of the mitral valve”. Circulation 1978;57(4):803-807.
94. Wilkins GT, Weyman AE, Abascal VM, et al. “Percutaneous balloon dilatation of the mitral valve: an analysis of echocardiographyc variables related to outcome and the mechanism of dilatation”. Br Heart J 1988;60:299-308.
95. Olson LJ, Subramanian R, Ackerman DM, Edwars WD. “Surgical pathology of the mitral valve: a study of 712 cases spanning 21 years”. Mayo Clinic Proc 1987;62:22-34.
96. Zinder II R, Lange R, Willard J et al. “Frecuency cause and effect on operative outcome of depressed left ventricular ejection fraction in mitral stenosis”. Am J Cardiol 1994;73:65.
97. Kennedy JW, Yarnall SR, Murray JA, et al. “Quantitative angio-cardiography: relationships of left atrial and ventricular pressure and volume in mitral valve disease”. Circulation 1970;41:817.
98. Mittal SR, RS Goozar. “Echocardiographic evaluation of left ventricular function in pure mitral stenosis . Int J Card Imaging. 2000; 16:29-33.
99. Wod, P. “An appreciation of mitral stenosis, I: clinical features”. BMJ 1954;1:1051-1063.
100. Duran M, Valencia E, Bejarano R. “Contribución auricular iz-quierda al llenado ventricular en la estenosis mitral. Evaluación por ecocardiografía Doppler”. Rev Esp Cardiol 1991;44(1):18.
101. Otto CM, Davis KB, Reid CL. “Relation between pulmonary artery pressure and mitral stenosis severity in patients undergoing balloon mitral commissurotomy”. Am J Cardiol 1993;71:874-878.
102. Remetz M, Cleman M, Cabin H. “Pulmonary and pleural compli-cations of cardiac disease”. Clinic in Chest Medicine 1989;10(4):545.

287
Fisiopatología de la hipertensión arterial 18
La del volumen minuto cardiaco y de la resistencia ofrecida por el sistema vascular, es decir, la resistencia periférica
-
dependiente de cualquier factor que altere el equilibrio entre las dos variables mencionadas.
-rial implica el conocimiento de los mecanismos de control
-
-
vinculada al equilibrio cardiovascular y renal, y cambia a través de las distintas etapas de la vida. Los mamífe-ros tienen un sistema circulatorio muy semejante y la PA
-do que forma parte de un complejo sistema, adaptado en
-tos indispensables para el normal funcionamiento tisular.
izquierdo y la hiperplasia del músculo liso vascular. La PA aumenta con la edad durante todo el desarrollo
hombre. El aumento es rápido inmediatamente después
-rante toda la vida. A partir de la mitad de esta comienza a
desarrollado modelos de animales hipertensos selecciona-
-
adultos. Por otra parte, en el desarrollo evolutivo de la rata normal, la PA también aumenta durante toda la vida,
-
-1
de la pared arterial mostrando alteraciones similares en
2 debida a un aumento del espesor de
3 Otros autores
4
-
dependiente de endotelio. 5 En este sentido, en el ser hu-
vasodilatadora, como consecuencia de la menor relaja-
resistencia del antebrazo6; además en pacientes, tanto la -
y de las arteriolas de resistencia. 7
8 Por -
titutiva disminuye con la edad en las ratas normotensas
9
En el ser humano, se ha observado que la PA del adulto -
Nidia Basso

Gelpi - Donato Fisiopatología cardiovascular
288
cencia. El estudio seriado de la PA en los mismos sujetos -
dida que avanza la edad. 10 -
-
-lados entre sí.
-latorio a través de la contractilidad y la frecuencia car-diaca, la resistencia periférica y la capacidad venosa, en consecuencia, es determinante del nivel de ajuste de la
-
La homeostasis del -
-
la cascada enzimática de la renina, que constituye un factor -
-terminan el desarrollo de esta característica y provocan
Sistema nervioso autónomo
El
los cambios posturales y también en forma sostenida en
-
-
los procesos multifactoriales, incluyendo anormalidades -
arterial esencial.
-centro de control va-
somotor
las neuronas cardiovasculares estimulatorias eferentes del -
vía proyecciones neuronales desde y hacia otras áreas del
los mecanorreceptores, sensibles al estiramiento, ubicados -
encuentran en áreas superiores, inclusive el área postrema
-
El --
11
de las columnas intermedio-laterales de la médula a los
--
-
-

Fisiopatología de la hipertensión arterial
289
-do de control hemodinámico. Los cambios varían entre
-ras como la
terminaciones simpáticas periféricas al espacio sináptico es
postsinápticas donde actúa y es metabolizada, o difunde al -
12
modulada por una cantidad de 13 El receptor más importante es el
α a la terminal la presencia del neurotransmisor en el espacio
-
a estos receptores presinápticos inhibitorios, se encuentran los receptores βque aumentan la cantidad de NA liberada por cada impul-so nervioso. La A liberada por la médula adrenal actuaría a nivel de los β tono simpático.
a la NA aumentando el efecto del neurotransmisor. 14
-tar involucrado en el aumento sostenido de la PA. No
efecto hipotensor de los bloqueantes β 15 Los
-cados básicamente en dos subtipos: α y β, cada uno de los cuales tiene varios subtipos con distintos patrones
16 Los efectos de la NA son predominantemente α; los receptores α1 se encuentran sobre todo en las membranas postsinápticas de la capa
ser estimulados. Los receptores β1 se encuentran por so--
dos preferentemente por la A que tiende a aumentar el volumen minuto.
dinámica única de interacciones que permiten aumentar la 17 La principal consecuencia
acciones para aumentar aún más el efecto simpático:1. Actúa a través del AP que carece de barrera hemato-
encefálica y aumenta el tono simpático.
la NA liberada por cada impulso nervioso.
que, a su vez, potencia el efecto de la NA.
intracelular.
Figura 18.1: Esquema del componente aferente y eferente e integración central del sistema nervioso autónomo en la regulación de la presión arterial. NA: noradrenalina; Ach: acetilcolina; NTS: núcleo del tracto solitario

Gelpi - Donato Fisiopatología cardiovascular
290
-
--
te, junto a la NA promueve cambios estructurales como
sodio.
otro de los mecanismos fundamentales en la homeostasis de
-
hemodinámicos renales. Asimismo, pudo observarse en pa-
-
-
-
misma. 18 -
-
-tensos por tratamiento con L-NAME, que en los controles,
-dad. La leptina sería uno de los intermediarios involucrados en el aumento de la actividad simpática. 19
-
-
de la PA, el -
nerando aumento de la frecuencia cardiaca, contractilidad -
y en el sistema de capacitancia, que detectan cambios en -
-
-
--
19 El aumento
-
-
-
avalan esta posibilidad:
a menudo revelan mecanismos hiperquinéticos en la -
cuencia cardiaca y del volumen minuto, vinculados
aumento de la actividad simpática.-
tomada como marcador indirecto del tono nervioso
-mente aumentada en los pacientes hipertensos esen-ciales, cuando se los compara con normotensos de
--
terminales nerviosas está aumentada, en especial a
importancia en la homeostasis de la PA.
por medida directa, a nivel del músculo esquelético, -

Fisiopatología de la hipertensión arterial
291
Los barorreceptores arteriales ejercen un papel permi--
do a la pérdida de capacidad intrínseca para responder al aumento permanente de la PA, mecanismo denominado reajuste de los barorreceptores. Estos receptores son ca-
pueden normalizarla. Dado que representan el mayor me-
del aumento de la PA. Los resultados no han demostrado
que modula la actividad cardiaca. El reajuste estaría vin-
Sistema renina-angiotensina-aldosterona
-
las mismas conclusiones, en forma casi simultánea. Uno
de Medicina de la Universidad de Buenos Aires, bajo la
-
-canismos involucrados en el aumento de la PA. Utilizando
recientemente nefrectomizado. La PA del receptor aumen-taba y se mantenía elevada aun después de retirar el injer-
mantenidos en isquemia total provocaba un ascenso muy
-
soluble en acetona y tenía un marcado efecto vasoconstrictor
capaz de actuar sobre una proteína denominada hipertensi-
-
un tipo de enzima inactiva activada por otra enzima pre-
-nina.
contenía la mitad de cada nombre inicial. En forma simi--
compuesto activo. 20
El sistema -
-
-
-
en el metabolismo de otros polipéptidos como la bradi-
menor peso molecular.
Las

Gelpi - Donato Fisiopatología cardiovascular
292
hombre y deducir la estructura primaria de la enzima y de su precursor.
-plasmáticos en las células epitelioides de la pared arterio-
-
El paso inicial para la síntesis de renina renal es la forma--
diente. Esta forma intermedia es transportada al retículo endoplásmico donde es clivada, liberando la prorrenina,
renal. La prorrenina o renina inactiva también es libera-
de la prorrenina plasmática.
por muchas variables, que actúan a través de uno o más
-ceptor renal, la mácula densa, las terminaciones nerviosas renales, y los factores humorales. El barorreceptor renal es,
21 Es un receptor vascular, situado en la arteriola aferente,
misma puede llevar a un aumento permanente de la libera-
-
la parte distal del asa de Henle y adyacentes a las células
delimitado por las arteriolas aferente y eferente, la má-cula densa y el retículo conjuntivo constituye el llamado
-
renina a través de un mecanismo β 22 La -
pérdida de volumen, etc. Una serie de factores humorales intervienen en el pro-
-β 23 y las
24
Los mecanismos inhibitorios estarían mediados por un aumento del calcio intracelular, calmodulina depen-
-
renina. Los factores humorales inhibitorios incluyen la an-α
1
mensajero del efecto inhibitorio, dado que los factores que
25 El -
como en los tejidos, parece ser un factor limitante de la actividad del sistema enzimático.
-26, mien-
tras que puede ser también sintetizado en el sistema ner-
27
tal forma que resulta dependiente de la especie y del te-
-
-
de la prohormona en plasma. 28
-
---
cripcional y de la vida media tanto del ARN mensajero 26
-cocorticoides de la corteza suprarrenal o la ACTH au-

Fisiopatología de la hipertensión arterial
293
29-30 --
can una dramática caída de la prohormona. Asimismo, 31, las hormonas tiroideas32 -
receptor AT1. 33-34 Recientemente, se ha postulado que -
por varias hormonas, localizado en el ADN nuclear más -
estabilizar el ARNm resultante. 35
-vidad del sistema enzimático y, por lo tanto, en la actividad
36
-
37 que no es
otros polipéptidos incluyendo la bradiquinina, encefalinas y sustancia P. La -nasa o encefalinasa, debido al valor muy reducido de su Km al hidrolizar la bradiquinina. La ECA intervendría en
-do un poderoso sistema vasoconstrictor e inhibiendo un potente sistema vasodilatador. Los inhibidores de la ECA
su estructura aminoacídica derivada de la secuencia del 38 La mayor parte de la ECA se encuentra
-
sucede normalmente y la ECA puede ser detectada en
encuentra en la membrana de las células endoteliales,
son ricos en ECA. El -
La cascada enzimática tiene un papel fundamental en -
-
Figura 18.2: Representación esquemática de los componentes y efectos del sistema renina-angiotensina. Ang: Angiotensina; ECA: Enzima convertidora de angiotensina

Gelpi - Donato Fisiopatología cardiovascular
294
mediata, cuyo objetivo es mantener la homeostasis circula-
sector arterial;-
-mentar el volumen minuto;
-
39
-mente potente sobre el músculo liso vascular. De forma simultánea, a través de la homeostasis hidrosalina, modu-la el volumen plasmático y el volumen minuto, actuando
directo sobre el músculo liso vascular, un efecto presor
40
-
encuentra la hiperplasia del músculo liso vascular, hiper-
y mayor repuesta contráctil vascular a dosis mínimas de --
41
-bre receptores ubicados en la membrana celular. Básica-mente, se han descrito dos tipos principales de receptores
polipéptido se ejercen a través de los receptores AT1. El receptor AT1 del músculo liso de la aorta de la rata y del tejido adrenal bovino ha sido aislado y clonado y se ha
42-43 Los receptores AT1 pertenecen a la familia de los receptores de hormonas peptídicas con
proteína G. 44-45 -
-
46-47
calcio intracelular y activan la proteína kinasa C. 48 La -
cidad intrínseca de tirosina kinasa similar a los receptores
de insulina, aumenta su actividad. En este sentido, se de-
de la rata estaban constitutivamente fosforilados y que la 49
en la adultez. 50 -51 Este receptor tiene una estructura similar al
52
53
ha postulado que estos receptores estarían vinculados al
efecto presor y promotor del crecimiento celular. También se los ha relacionado con los mecanismos de apoptosis. En
--
-
en forma intracrina, paracrina o autocrina.54 Las técnicas de
55 Asimismo, el
56 En el cere-bro, se han detectado todos los componentes del sistema
-péptido que puede actuar como neurotransmisor o neuro-modulador. 40
-
sus metabolitos pueden tener otros efectos además de los -
timo ha sido razonablemente bien analizada mientras que los mecanismos moleculares de los sistemas tisulares aún
-
40 El polipéptido administrado por la vía intracerebroventricular
-
57-58
central parece ser independiente de la circulante ya que

Fisiopatología de la hipertensión arterial
295
59
60 Con
se encontraría en las terminaciones nerviosas, en áreas vinculadas al control cardiovascular y a la homeostasis hidrosalina. 56 La ECA está ampliamente distribuida en el
60 La pared de los vasos contiene la enzima y se la ha localizado intraneuronalmente; su rol funcional parece
que cuando se administran inhibidores de la ECA por la vía intracerebroventricular en animales espontáneamente
-nohistoquímicos en la rata, los primates y el hombre. 72 El octapéptido se encuentra sobre todo en el hipotálamo, el sistema límbico, el bulbo y la médula espinal. Terminales
subforniano. Esta presencia es importante por la capacidad del polipéptido para liberar ADH y ACTH. Por último, se
periventriculaes del tercer ventrículo, en las neuronas del
el locus ceruleus y en el núcleo de la oliva inferior.
-
.
--
sistema presor y potencian un importante sistema vasodi-
65-67
-nen un amplio espectro de uso clínico; son muy efectivos
resultante de las enfermedades cardiovasculares 68 y se han utilizado en el tratamiento de la nefropatía diabética. 69
la última década como consecuencia de un mejor cono-70-71
-
este tipo fue el losartán, un derivado del imidazol que se comporta como un activo bloqueador del receptor AT1. 72-73
y la sed. 72 -
de renina y aumenta la actividad renínica plasmática con
65
-73
74 En
-75-76 Asimismo, ate-
músculo liso vascular. 77 Parece tener efectos protectores
renal. El -merular en las ratas diabéticas por estreptozotocina. Un
IECA en pacientes con enfermedad renal, diabéticos y no diabéticos. 78
-
-
La capacidad del losartán para disminuir la PA se ha 79
-
-nista se mantiene por siete días, el efecto hipotensor se potencia. 80 En estudios comparativos, el efecto sobre la
enalapril o -
vera. 81 Además, estos compuestos presentan efectos adi-tivos cuando se los administra con hidroclorotiazida, sin hipocalemia o hiperuricemia asociada. En pacientes con
la proteinuria en forma similar al enalapril78, también dis-minuye la PA en otras enfermedades renales sin cambios
82 En varios estudios realizados
-ta redujo la resistencia vascular sistémica y la PA media,

Gelpi - Donato Fisiopatología cardiovascular
296
con aumento del volumen minuto cardiaco. 83 La admi-
produjo efectos similares. -
parte, el mismo tratamiento con enalapril o losartán, sumi-nistrado a ratas normales, impide el aumento de la PA que aparece con la edad. Además, tiene un efecto protector sobre
memoria y aprendizaje que se pierde con la edad. 84
-
Especies reactivas del oxígeno
incluye moléculas muy reactivas y de vida corta, deriva- molecular. Múltiples sistemas
enzimáticos usan diferentes sustratos como fuente de elec-
- -
O
-
85
-
86 Requiere tetrahidrobiopterina -
-
87-88
--
en su centro catalítico y transforma el O - en H O
--
-
intersticio vascular. 87
Olipídicos a H
--
el citosol metaboliza el H O a H O y O . También tiene
88
La -
-
-- .
y de la 89
Entre los factores de crecimiento, el factor derivado de las plaquetas, el factor epidermal, el factor transfor-mador β
de necrosis tumoral α,
90
aumenta -
O
99
Entre los lípidos, el aumento del colesterol está asocia-
88

Fisiopatología de la hipertensión arterial
297
-
O - y del H O -
91 -tes antihipertensivos, como los bloqueantes de canales de calcio, los β-bloqueantes, los inhibidores de la enzima de
92 En este
93
94-95
-tivo. Tanto en células endoteliales como vasculares lisas
-
-
96
Factores genéticos
-ma humano que incluyen
-
dado que el control de esta variable depende de los sis-temas cardiovascular, renal, endocrino y nervioso. Por
masa corporal, el tabaquismo y otros factores contribuyen
el mejor tratamiento para cada paciente, adecuado a las
Bibliografía
1. González Bosc LV, Kurnjek ML, Müller A, Terragno NA, Basso N. “Effect of angiotensin II inhibition on the nitric oxide synthase in the normal rat during aging”. J Hypertens. 2001;19:1403-1409.
2. Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B, Goldman S. “Large artery remodeling during aging. Biaxial passive and active stiffness”. Hypertension. 1998;32:437-443.
3. Michel JB, Azizi M, Salzmann JL, Levy B, Ménard J. “Effect of vasodilators on the structure of the aorta in normotensive ageing rats”. J Hypertension. 1987;5:S165-S168.
4. Küng CF, TF Lüscher. “Different mechanisms of endothelial dis-function with aging and hypertension in rat aorta”. Hypertension. 1995;25:194-200.
5. Panza, JA. “Nitric Oxide in Hypertension”. En: Oparil S, Weber MA, editores. Hypertension: A companion to Brenner and Rector´s The Kidney. Philadelphia: Saunders WB Company, 2000; p. 158-165.
6. Gerhard M, Roddy MA, Creager SJ, Creager MA. “Aging pro-gressively impairs endothelium-dependent vasodilation in forearm resistance vessels in humans”. Hypertension. 1996;27:849-853.
7. Panza JA, Quyyumi AA, Brush JE Jr, Esptein SE. « Abnormal endothelium-dependent vascular relaxation in patients with es-sential hypertension”. N Engl J Med. 1990;323:22-27.
8. Hayakawa H, Hirata Y, Suzuki E, Matsuoka H, Kikuchi K, Na-gano T, et al. “Mechanisms of altered endothelium-dependent vasorelaxation in isolated kidneys from experimental hypertensive rats”. Am J Physiol. 1993;264:H1535-H1541.
9. Chou TC, Yen MH, Li CY, Ding YA. “Alterations of nitric oxide synthase expression with aging and hypertension in rats”. Hyper-tension. 1998;31:643-648.
10. Lever AF, SB Harrap. “Essential hypertension. A dissorder of growth with origins in childhood?” J Hypertens. 1992;10:101-120.
11. Allen K, Shykoff BE, Izzo JL Jr. “Cognitive appraisal of threat or challenge predicts hemodynamic responses to mental arithmetic and speechs tasks”. Am J Hypertens. 1998;11:134A.
12. Eisenhofer G, Esler MD, Goldstein DS, Kopin IJ. “Neuronal uptake, metabolism and release of tritium-labeled norepine-phrine during assessment of its plasma kinetics”. Am J Physiol. 1991;261:E505-E515.
13. Langer, SZ. “Pre-synaptic regulation of the release of catechola-mines”. Pharmacol Rev. 1980;32:337-362.
14. Rand MJ, H Majewski. “Adrenaline mediates a positive feedback loop in noradrenergic transmission: Its possible role in development of hypertension”. Clin Exp Hypertens. 1984;A6:347-370.
15. Chang PC, Kriek E, van Brummelen P. “Sympathetic activity and presynaptic adrenoceptor function in patients with long-standing essential hypertension”. J Hypertens. 1994;12:179-190.
16. Insel, PA. “Seminars in medicine of the Beth Israel Hospital, Bos-ton. Adrenergic receptors-Evolving concepts and clinical implica-tions”. N Engl J Med. 1996;334:580-585.
17. Marsden PA, Brenner BM, Ballerman BJ. “Mechanisms of an-giotensin action on vascular smooth muscle, the adrenal and the kidney”. En Laragh JH, Brenner BM editores. Hypertension: Pa-thophysiology, Diagnosis and Treatment. New York: Raven, 1990; 1247-1272.

Gelpi - Donato Fisiopatología cardiovascular
298
18. DiBona, GF. “Neural control of the kidney. Past, present and future”. Hypertension. 2003;41[part 2]:621-624.
19. DiBona, GF. “The sympathetic nervous system and hypertension. Recent developments”. Hypertension. 2004;43:147-150.
20. Basso N, NA Terragno, “History about the discovery of the renin-angiotensin system”. Hypertension. 2001;38:1246-1249.
21. Freeman RH, JO Davis. “The control of renin secretion and meta-bolism”. En: Genest J, Koiw E y Kuchel O, editores. Hypertension. New York: McGraw-Hill. 1977; p. 210-240.
22. Johnson JA, Davis JO, Gotshall RW, Lohmeier TE, Davis JL, Braverman B, Tempel GE. “Evidence for an intrarenal beta re-ceptor in control of renin release”. Am J Physiol. 1976;230:410-418.
23. Johnson JA, Davis JO, Witty RT. “Effect of catecholamines and renal nerve stimulation on renin release in the non-filtering kidney”. Circ Res. 1971;29:646-653.
24. Dunn MJ, VL Hood. “Prostaglandins and the kidney”. Am J Physiol. 1977;233:F169-F184.
25. Churchill, PC. “Second messengers in renin secretion”. Am J Phy-siol. 1985;249:F175-F184.
26. Brasier AR, J Li. “Mechanisms for inducible control of angiotensi-nogen gene transcription”. Hypertension. 1996;27(Part 2):465-475.
27. Campbell DJ, JF Habener. “Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat”. J Clin Invest. 1986;78:31-39.
28. Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, et al. “Genetic control of blood pressure and the angio-tensinogen locus”. Proc Natl Acad Sci USA. 1995;92:2735-2739.
29. Klett C, Hellmann W, Hackenthal E, Ganten D. “Modulation of tissue angiotensinogen gene expression by glucocorticoids, estro-gens and androgens in SHR and WKY rats”. Clin Exp Hypertens. 1993;15:683-708.
30. Brasier AR, Phillipe J, Campbell DJ, Habener JF. “Novel ex-pression of the angiotensinogen gene in a rat pancreatic islet cell line: transcriptional regulation by glucocorticoids”. J Biol Chem. 1986;261:16148-16154.
31. Krattenmacher R, Knauthe R, Parczyk K, Walker A, Hilgenfeldt U, Fritzemeier KH. “Estrogen action on hepatic synthesis of an-giotensinogen and IGFI: direct and indirect estrogen effects”. J Steroid Biochem. 1994;48:207-214.
32. Hong Brown LQ, CF Deschepper. “Effect of thyroid hormones on angiotensinogen gene expression in rat liver brain and cultured cells”. Endocrinology. 1992;130:1231-1237.
33. Kohara K, Brosnihan KB, Ferrario CM, Milsted A. “Peripheral and central angiotensin II regulates expression of genes of the renin-angiotensin system”. Am J Physiol. 1992;262:E651-E657.
34. Klett C, Nobiling R, Gierschik P, Hackenthal E. “Angiotensin II stimulates the synthesis of angiotensinogen in hepatocytes by inhibiting adenylcyclase activity and stabilizing angiotensinogen mRNA”. J Biol Chem. 1993;268:25095-25107.
35. Brasier AR, Li J, Copland A. “Transcription factors modulating angiotensinogen gene expression in hepatocytes”. Kidney Int. 1994;46:1564-1566.
36. Reid IA, Morris BJ, Ganong WG. “The renin-angiotensin system”. Annu Rev Physiol. 1978;40:377-410.
37. Erdos EG, RA Skidgel. “The angiotensin I converting enzyme”. Labor Invest. 1987;56:345-348.
38. Bernstein KE, Martins BM, Bernstein EA, Linton J, Striker L. “The isolation of angiotensin converting enzyme cDNA”. J Biol Chem. 1988;263:11021-11024.
39. Haber, E. “Defining the physiologic and pathophysiologic roles of renin: the role of specific inhibitors”. Am J Kidney Dis. 1985;5:A14-A22.
40. Bunnemann B, Fuxe K, Ganten D. “The renin-angiotensin system in the brain: an update”. Regul Peptides. 1993;46:487-509.
41. Griendling KK, Ushio Fukai M, Lassègue B, Alexander RW. “An-giotensin II signaling in vascular smooth muscle. New Concepts”. Hypertension. 1997;29(Part 2):366-373.
42. Murphy TI, Alexander RW, Griendling KK, Runge MS, Bernstein KE. “Isolation of a cDNA encoding the vascular type 1 angiotensin II receptor”. Nature. 1991;351:233-236.
43. Desarnaud F, Marie J, Lombard C. “Deglycosylation and frag-mentation of purified rat liver angiotensin II receptor. Applica-tion to the mapping of hormone binding domains”. Biochem J. 1993;289:289-297.
44. Hjorth SA, Schambye HT, Greenlee WJ, Schwartz TW. “Identi-fication of peptide binding residues in the extracellular domains of the AT1 receptor”. J Biol Chem. 1994;269:30953-30959.
45. Ohyama K, Yamano Y, Chaki S, Kondo T, Inagami T. “Domains for G protein coupling in angiotensin II receptor type 1: Stu-dies by site directed mutagenesis”. Biochem Biophys Res Commun. 1992;189:677-683.
46. Nabika T, Velletri PA, Lovenberg W, Beaven MA. “Increase in cytosolic calcium and phosphoinositide metabolism induced by angiotensin II and [Arg]vasopressin in vascular smooth muscle cells”. J Biol Chem. 1985;260:4661-4670.
47. Brock TA, Alexander RW, Ekstein LS, Atkinson WJ, Gimbrone MA Jr. “Angiotensin increases cytosolic free calcium in cultured vascular smooth muscle cells”. Hypertension. 1985;7:1105-1109.
48. Griendling KK, Berk BC, Ganz P, Gimbrone MA Jr, Alexander RW. “Angiotensin II stimulation of vascular smooth muscle phos-phoinositide metabolism. State of the Art Lecture”. Hypertension. 1987;9:III181-III185.
49. Kai H, Griendling KK, Lassègue B, Ollerenshaw JD, Runge MS, Alexander RW. “Agonist induced phosphorilation of the vascular type 1 angiotensin II receptor”. Hypertension. 1994;24:523-527.
50. Inagami T, Guo DF, Kitami Y. “Molecular biology of angiotensin II receptors: An overview”. J Hypertens. 1994;12(Suppl 10):S83-S94.
51. Mukoyama M, Nakajima M, Horiuchi M, Sasamura H, Pratt RE, Dzau VJ. “Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven transmembrane receptors”. J Biol Chem. 1993;268:24539-24542.
52. Kambayashi Y, Bardham S, Takahashi K. “Molecular cloning of a novel angiotensin II isoform involved in phosphotyrosin phospha-tase inhibition”. J Biol Chem. 1993;268:24543-24546.
53. DeGasparo M, Bottari S, Leven NR. “Characteristics of angioten-sin II receptors and their role in cell and organ physiology”. En: Laragh JH, BM Brenner, editores. Hypertension: Pathophysiology Diagnosis and Management. 2nd ed. New York: Raven Press. 1995; p. 1695-1720.
54. Campbell, DJ. “Circulating and tissue angiotensin systems”. J Clin Invest. 1987;79:1-6.
55. Dzau VJ, Ellison KE, Brody T, Ingelfinger J, Pratt RE. “A com-parative study of the distributions of renin and angiotensin mes-senger ribonucleic acids in rat and mouse tissues”. Endocrinology. 1987;120:2334-2338.
56. Taugner R, Hackenthal E, Rix E, Nobiling R, Poulsen K. “Im-munocytochemistry of the renin-angiotensin system: renin, an-giotensinogen, angiotensin I, angiotensin II, and converting en-zyme in the kidneys of mice, rats and tree shrews”. Kidney Intern. 1982;22:S33-S43.
57. Bunnemann B, Fuxe K, Metzger R, Bjelke B, Ganten D. “The semi-quantitative distribution and cellular localization of angiotensinogen mRNA in the rat brain”. J Chem Neuroanat. 1992;5:245-262.
58. Stornetta RL, Hawelu Johnson CL, Guyenet PG, Lynch KR. “Astro-cytes synthesize angiotensinogen in brain”. Science. 1988;242:1444-1446.
59. Ruiz P, Basso N, Grinspon D. “Angiotensinogen concentration in the cerebrospinal fluid in different experimental conditions in the rat”. Hypertension. 1983;5(Suppl V):V29-V33.
60. Chai SY, Mendelsohn FAO, Paxinos G. “Angiotensin converting enzyme in rat brain visualized by quantitative in vitro autoradio-graphy”. Neuroscience. 1987;20:615-627.
61. Fitzsimons, JT. “Angiotensin: Thirst and Sodium Appetite. Physiol Rev. 1998;78:583-686.

Fisiopatología de la hipertensión arterial
299
62. Gehlert DR, Gackenheimer SL, Schober DA. “Autoradiographic localization of subtypes of angiotensin II antagonist binding in the rat brain”. Neuroscience. 1991;44:501-514.
63. Leung KH, Smith RD, Timmermanns PBMWM, Chiu AT. “Re-gional distribution of the two subtypes of angiotensin II receptors in rat brain using selective nonpeptide antagonists”. Neurosci Lett. 1991;123:95-98.
64. Rowe BP, Grove KL, Saylor DL, Speth RC. “Discrimination of angiotensin II receptor subtype distribution in the rat brain using nonpeptide receptor antagonists”. Regul Pept. 1991;33:45-53.
65. Schalekamp MA, Derkx FH, van den Meiracker AH. “Renin inhi-bitors, angiotensin converting enzyme inhibitors and angiotensin II receptors antagonists: Relationship between blood pressure responses and effects on the renin-angiotensin system”. J Hypertens. 1992;10:S157-S164.
66. Swales, JD. “The renin angiotensin system as a target for therapeutic intervention”. J Cardiovasc Pharmacol. 1994;24(Suppl 2):S1-S5.
67. Waeber B, Nussberger J, Brunner HR. “Angiotensin converting enzyme inhibitors in hypertension”. En: Laragh JH, BM Brenner, editors. Hypertension: pathophysiology diagnosis and management. New York: Raven Press, 1990; 2209-2232.
68. Kjekshus J, Swedberg K, Snappin S. “Effects of enalapril on long term mortality in severe congestive heart failure”. Am J Cardiol. 1992;69:103-107.
69. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (Collaborative Study Group). “The effect of angiotensin converting enzyme inhibition on diabetic nephropathy”. N Engl J Med. 1993;329:1456-1462.
70. Bernstein KE, BC Berk. “The biology of angiotensin II receptors”. Am J Kidney Dis. 1993;22:745-754.
71. Inagami T, Guo DF, Kitami Y. “Molecular biology of angiotensin II receptors: An overview”. J Hypertens. 1994;12(Suppl 10):S83-S94.
72. Timmermanns PB, Carini DJ, Chiu AT, Duncia JV, Price WA Jr, Wells GJ, et al. “The discovery of a new class of highly specific nonpeptide angiotensin II receptor antagonists”. Am J Hypertens. 1991;4(Part2):275S-281S.
73. Timmermanns PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, et al. “Angiotensin II receptors and angiotensin II receptors antagonists”. Pharmacol Rev. 1993;45:205-251.
74. Azuma H, Hamasaki H, Niimi Y. “Prevention of intimal thickening after endothelial removal by a nonpeptide angiotensin II receptor antagonist: losartan”. Br J Pharmacol. 1992;106:665-671.
75. Murakami H, Liu JL, Zucker IH. “Blockade of AT1 receptors enhances baroreflex control of heart rate in conscious rabbits with heart failure”. Am J Physiol. 1996;271:R303-309.
76. Sladek T, Sladkova J, Kolar F, Papousek F, Cicutti N, Korecky B, Rakusan K. “The effect of AT1 receptor antagonist on chronic cardiac response to coronary artery ligation in rats”. Cardiovasc Res. 1996;31:568-576.
77. Kauffman RJ, Bean JS, Zimmerman KM, Brown RF, Steinberg MI. “Losartan a non peptide angiotensin II (ANGII) receptor antagonist inhibits neointima formation following balloon injury to rat carotid arteries”. Life Sci. 1991;49:PL223-PL228.
78. Gansevoort RT, PE de Jong. “Is the antiproteinuric effect of ACE inhibition mediated by interference in the renin angiotensin sys-tem?” Kidney Int. 1994;45:861-867.
79. Weber MA. “Clinical experience with the angiotensin II recep-tor antagonist losartan: A preliminary report”. Am J Hypertens. 1992;5:247S-251S.
80. Gradman AH, Arcuri KE, Goldberg AI, Ikeda LS, Nelson EB, Snavely DB, Sweet CS. “A randomized placebo controlled double blind parallel study of various doses of losartan potassium compared with enalapril maleate in patients with essential hypertension”. Hypertension. 1995;25:1345-1350.
81. Ruff D, Gazdick LP, Berman R, Goldberg AI, Sweet CS. “Compa-rative effects of combination drug therapy regimens commencing with either losartan potassium an angiotensin II receptor antagonist or enalapril maleate for the treatment of severe hypertension”. J Hypertens. 1996;14:263-270.
82. Gansevoort RT, de Zeeuw D, Shahinfar S, Redfield A, de Jong PE. “Effect of the angiotensin II antagonist losartan in hypertensive patients with renal disease”. J Hypertens. 1994;12(Suppl 2):S37-S42.
83. Dickstein K, Gottlieb S, Fleck E, Kostis J, Levine B, De Kock M, Le Jemtel T. “Hemodynamic and neurohumoral effects of the angiotensin II antagonist losartan in patients with heart failure”. J Hypertens. 1994;12(Suppl 2):S31-S35.
84. Ferder L, Inserra F, Basso N. “Advances in our understanding of aging: role of the renin-amgiotensin system”. Curr Opin Pharmacol. 2002;2:189-194.
85. Thannickal VJ, BL Fanburg. “Reactive oxygen species in cell signa-ling”. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005-L1028.
86. Moncada S, Palmer RM, Higgs EA. “The discovery of nitric oxide as the endogenous nitrovasodilator”. Hypertension. 1988;12:365-372.
87. Wassmann S, Wassmann K, Nickening G. “Modulation of oxidant and antioxidant enzyme expression and function in vascular cells”. Hypertension. 2004;44:381-386.
88. Landmesser U, Dikalov S, Price SR, Mc Cann L, Fukai T, Holland SM, Mitch WE, Harrison DG. “Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension”. J Clin Invest. 2003;111:1201-1209.
89. Taniyama Y, KK Griendling. “Reactive oxygen species in the vasculature. Molecular and cellular mechanisms”. Hypertension. 2003;42:1075-1081.
90. Stralin P, SL Marklund. “Vasoactive factors and growth factors alter vascular smooth muscle cell ec-SOD expression”. Am J Physiol Heart Circ Physiol. 2001;281:H1621-H1629.
91. Redon J, Oliva MR, Tormos C, Giner V, Chaves J, Iradi A, Saez GT. “Antioxidant activities and oxidative stress byproducts in human hypertension”. Hypertension. 2003;41:1096-1101.
92. Ghiadoni L, Magagna A, Versari D, Kardasz I, Huang Y, Taddei S, Salvetti A. “Different effect of antihypertensive drugs on conduit artery endothelial function”. Hypertension. 2003;41:1281-1286.
93. Münzel T, JF Jr Keaney. “Are ACE inhibitors a magic bullet against oxidative stress?” Circulation. 2001;104:1571-1574.
94. Costa LE, La-Padula P, Lores Arnaiz S, D’Amico G, Boveris A, Kurnjek ML, Basso N. “Long-term angiotensin II inhibition increases mito-chondrial nitric oxide synthase and not antioxidant enzyme activities in rat heart”. J Hypertens. 2002;20:2487-2494.
95. Cavanagh EMV de, Piotrkowski B, Basso N, Stella I, Inserta F, Ferder L, Fraga C. “Enalapril and losartan attenuate mitochondrial dysfunction in aged rats”. FASEB J. 2003;17:1096-1098.
96. Lalouel, JM. “Large scale search for genes predisposing to essential hypertension”. Amer J Hypert. 2003;16:163-166.


301
Mecanismos de adaptación fisiológicos y patológicos del corazón en la hipertensión arterial 19
-
trastornos del ritmo cardiaco o provocando episodios de isquemia miocárdica. 1- 3
como enfermedad cardiaca hipertensiva, son la base inicial
1, 2,
4-5, constituyéndose en los principales determinantes pro-6
-
funcionales, principalmente del ventrículo izquierdo, así --
Modificaciones estructurales
Hipertrofia ventricular
Definición
contráctil como consecuencia de un infarto de miocardio
-dientemente del factor causal, se caracteriza sobre todo por el aumento de la masa muscular. 7
Cuando se mantiene la estructura con un aumento
intersticiales y de la red vascular, como en el atleta, la 8
-
desarrollo de la red vascular, se produce un remodela-
distensibilidad de la cámara ventricular y la reserva va-
9 En -
-
precisos no siempre consensuados por la mayoría de los 10-11
ha tomado como referencia el valor de masa ventricular -
pertensa ni obesa, 10 -
11-12 En
tomados como puntos de corte por distintos autores. 10, 13-15
masa ventricular izquierda y probabilidad de eventos, se hace más difícil aceptar un punto único de corte, ya que a menor masa ventricular, menores posibilidades de com-plicaciones.15-16
Eduardo Escudero, Ana L Tufare
Figura 19.1: Se puede observar la relación entre los miocitos y las fibras intersticiales en el miocardio normal, en la hipertrofia fisiológica y en la hipertrofia patológica. En la hipertrofia fisiológica, si bien hay un aumento en el tamaño de los miocitos, esta modificación se acompaña de un incremento proporcional de las fibras intersticiales. En la patológica, ante similar aumento del tamaño de las células musculares, hay un mayor incremento de las fibras intersticiales.

Gelpi - Donato Fisiopatología cardiovascular
302
Significado de la respuesta hipertrófica
-
marcha para normalizar el estrés parietal, con el objetivo -
no miocárdico dentro de límites normales, previniendo 17-18
de masa e incremento de eventos cardiovasculares. 15-16
-
19 Por otra parte, en observaciones llevadas a cabo en nuestro laboratorio, trabajando con ratas
-
por inhibir el intercambiador Na /H --
20,
21
El camino hacia la hipertrofia
incremento en la síntesis proteica, produciendo además -
17
Los cambios a nivel molecular, responsables de las
alternativas, involucrando receptores de la membrana ce-
de estiramiento y mensajeros intracelulares, que terminan
Yamazaki et al.22 han demostrado que el estiramiento
--
23 en el interior celular. El DAG,
24 La endotelina li-berada se une al receptor ETA de membrana del mismo
-
través de una serie de kinasas, fosforilan al intercambiador Na /H , aumentando los niveles de sodio intracelular. Este incremento del sodio activa al intercambiador Na /Ca -nerando como consecuencia un aumento del Ca en el ci-tosol. 25 Los niveles elevados del calcio inician una cadena
Tabla 19.1: por ecocardiograma
Autor Límites para definir HVI
Levy D et al.10 2 en el hombre2 en la mujer
Ganau A et al.13 2 en el hombre2 en la mujer
Schillaci G et al.15 2 en el hombre2 en la mujer
Levy D et al.10
de Simone et al.14 157 g/m2.7 en ambos sexos
g/m2
g/m2.7
Figura 19.2: El gráfico de barras de la izquierda representa los valores del índice de masa ventricular izquierda, antes y a los 30 días de recibir HOE (inhibidor del intercambiador Na+/H+) en un grupo de ratas espontáneamente hipertensas. A la derecha, el estado de la función sistólica del ventrículo izquierdo en similar situación. Se puede comprobar cómo, ante la regresión de la hipertrofia, se encuentra una mejoría de la función ventricular. Los datos fueron obtenidos por ecocardiograma.
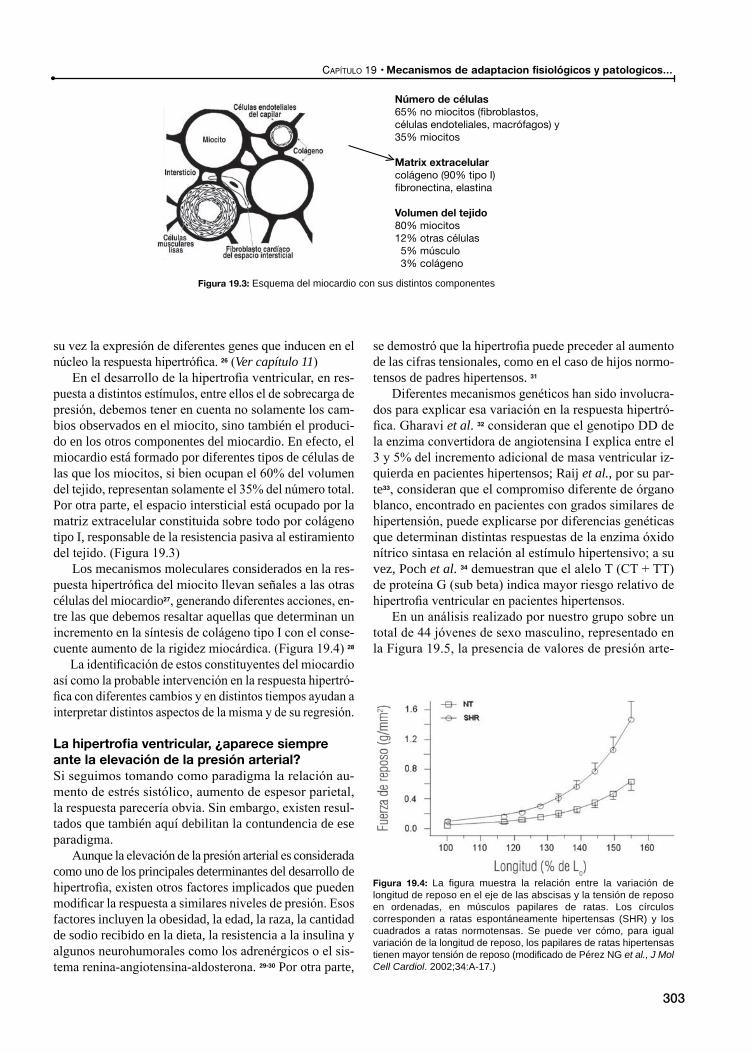
Mecanismos de adaptacion fisiológicos y patologicos...
303
26 Ver capítulo 11-
-bios observados en el miocito, sino también el produci-do en los otros componentes del miocardio. En efecto, el miocardio está formado por diferentes tipos de células de
Por otra parte, el espacio intersticial está ocupado por la
tipo I, responsable de la resistencia pasiva al estiramiento
Los mecanismos moleculares considerados en la res-
células del miocardio27 -tre las que debemos resaltar aquellas que determinan un
-28
-
La hipertrofia ventricular, ¿aparece siempre ante la elevación de la presión arterial?
-
-tados que también aquí debilitan la contundencia de ese
como uno de los principales determinantes del desarrollo de
factores incluyen la obesidad, la edad, la raza, la cantidad de sodio recibido en la dieta, la resistencia a la insulina y
-29-30 Por otra parte,
de las cifras tensionales, como en el caso de hijos normo-tensos de padres hipertensos. 31
--
et al. 32
-quierda en pacientes hipertensos; Raij et al., por su par-te33
vez, Poch et al. 34
-
Figura 19.3: Esquema del miocardio con sus distintos componentes
Número de células65% no miocitos (fibroblastos, células endoteliales, macrófagos) y35% miocitos
Matrix extracelularcolágeno (90% tipo I)fibronectina, elastina
Volumen del tejido80% miocitos12% otras células15% músculo13% colágeno
Figura 19.4: La figura muestra la relación entre la variación de longitud de reposo en el eje de las abscisas y la tensión de reposo en ordenadas, en músculos papilares de ratas. Los círculos corresponden a ratas espontáneamente hipertensas (SHR) y los cuadrados a ratas normotensas. Se puede ver cómo, para igual variación de la longitud de reposo, los papilares de ratas hipertensas tienen mayor tensión de reposo (modificado de Pérez NG et al., J Mol Cell Cardiol. 2002;34:A-17.)
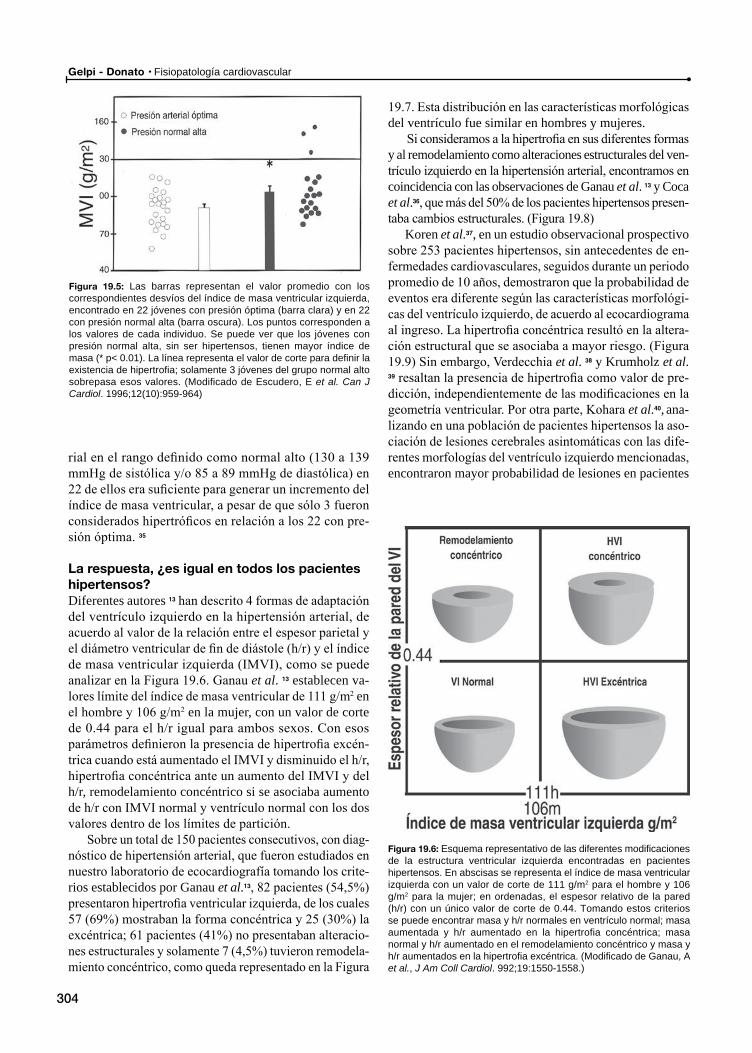
Gelpi - Donato Fisiopatología cardiovascular
304
-35
La respuesta, ¿es igual en todos los pacientes hipertensos?Diferentes autores 13
et al. 13 establecen va- en
en la mujer, con un valor de corte
-
h/r, remodelamiento concéntrico si se asociaba aumento
-
-rios establecidos por Ganau et al.13
--
del ventrículo fue similar en hombres y mujeres.
y al remodelamiento como alteraciones estructurales del ven-
coincidencia con las observaciones de Ganau et al. 13 y Coca et al.36 -
Koren et al.37, en un estudio observacional prospectivo -
-
-
et al. 38 y Krumholz et al. 39 -
et al.40, ana---
encontraron mayor probabilidad de lesiones en pacientes
Figura 19.5: Las barras representan el valor promedio con los correspondientes desvíos del índice de masa ventricular izquierda, encontrado en 22 jóvenes con presión óptima (barra clara) y en 22 con presión normal alta (barra oscura). Los puntos corresponden a los valores de cada individuo. Se puede ver que los jóvenes con presión normal alta, sin ser hipertensos, tienen mayor índice de masa (* p< 0.01). La línea representa el valor de corte para definir la existencia de hipertrofia; solamente 3 jóvenes del grupo normal alto sobrepasa esos valores. (Modificado de Escudero, E et al. Can J Cardiol. 1996;12(10):959-964)
Figura 19.6: Esquema representativo de las diferentes modificaciones de la estructura ventricular izquierda encontradas en pacientes hipertensos. En abscisas se representa el índice de masa ventricular izquierda con un valor de corte de 111 g/m2 para el hombre y 106 g/m2 para la mujer; en ordenadas, el espesor relativo de la pared (h/r) con un único valor de corte de 0.44. Tomando estos criterios se puede encontrar masa y h/r normales en ventrículo normal; masa aumentada y h/r aumentado en la hipertrofia concéntrica; masa normal y h/r aumentado en el remodelamiento concéntrico y masa y h/r aumentados en la hipertrofia excéntrica. (Modificado de Ganau, A et al., J Am Coll Cardiol. 992;19:1550-1558.)
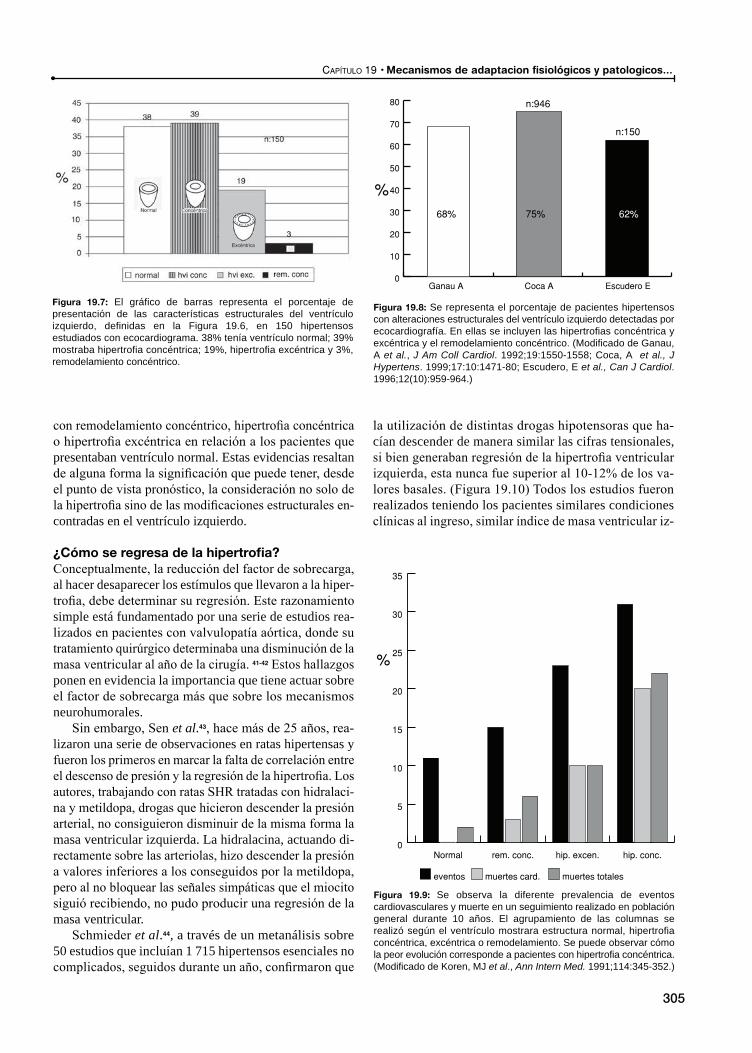
Mecanismos de adaptacion fisiológicos y patologicos...
305
presentaban ventrículo normal. Estas evidencias resaltan
-contradas en el ventrículo izquierdo.
¿Cómo se regresa de la hipertrofia?
al hacer desaparecer los estímulos que llevaron a la hiper-
simple está fundamentado por una serie de estudios rea-
41-42
ponen en evidencia la importancia que tiene actuar sobre
neurohumorales.et al.43 -
lizaron una serie de observaciones en ratas hipertensas y
-
masa ventricular izquierda. La hidralacina, actuando di-
masa ventricular.et al.44, a través de un metanálisis sobre
50 estudios que incluían 1 715 hipertensos esenciales no
-cían descender de manera similar las cifras tensionales,
-
realizados teniendo los pacientes similares condiciones -
Figura 19.8: Se representa el porcentaje de pacientes hipertensos con alteraciones estructurales del ventrículo izquierdo detectadas por ecocardiografía. En ellas se incluyen las hipertrofias concéntrica y excéntrica y el remodelamiento concéntrico. (Modificado de Ganau, A et al., J Am Coll Cardiol. 1992;19:1550-1558; Coca, A et al., J Hypertens. 1999;17:10:1471-80; Escudero, E et al., Can J Cardiol. 1996;12(10):959-964.)
Figura 19.7: El gráfico de barras representa el porcentaje de presentación de las características estructurales del ventrículo izquierdo, definidas en la Figura 19.6, en 150 hipertensos estudiados con ecocardiograma. 38% tenía ventrículo normal; 39% mostraba hipertrofia concéntrica; 19%, hipertrofia excéntrica y 3%, remodelamiento concéntrico.
Figura 19.9: Se observa la diferente prevalencia de eventos cardiovasculares y muerte en un seguimiento realizado en población general durante 10 años. El agrupamiento de las columnas se realizó según el ventrículo mostrara estructura normal, hipertrofia concéntrica, excéntrica o remodelamiento. Se puede observar cómo la peor evolución corresponde a pacientes con hipertrofia concéntrica. (Modificado de Koren, MJ et al., Ann Intern Med. 1991;114:345-352.)
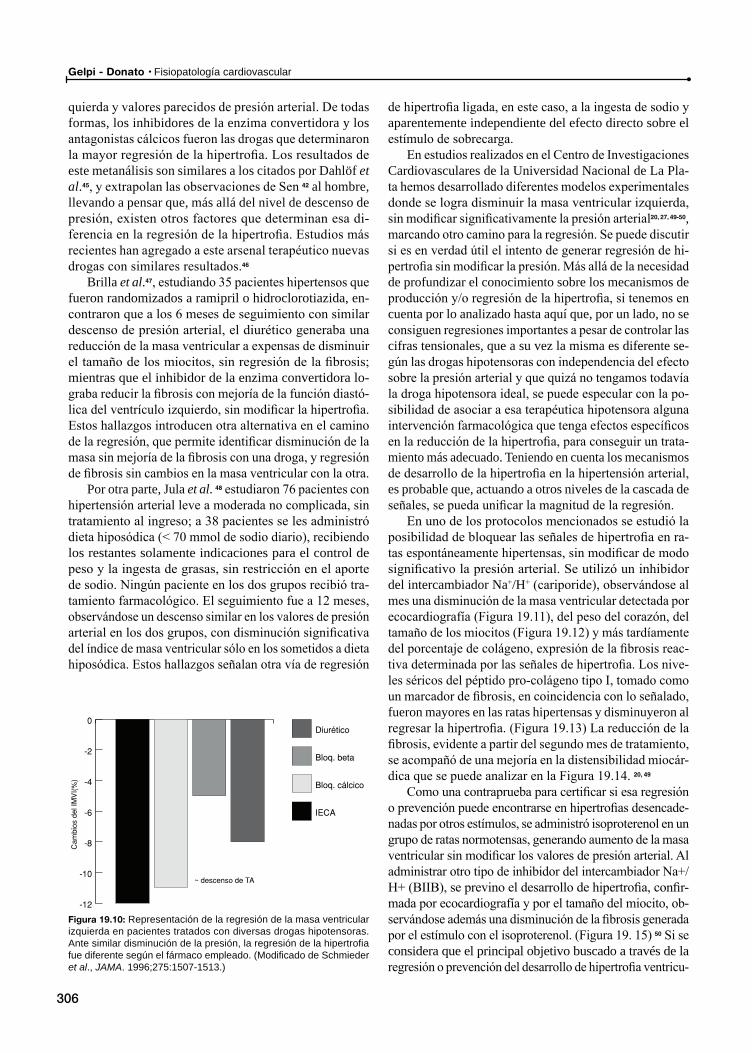
Gelpi - Donato Fisiopatología cardiovascular
306
formas, los inhibidores de la enzima convertidora y los
este metanálisis son similares a los citados por Dahlöf et al.45 42 al hombre, llevando a pensar que, más allá del nivel de descenso de
-
46
Brilla et al.47
fueron randomizados a ramipril o hidroclorotiazida, en-
mientras que el inhibidor de la enzima convertidora lo--
Por otra parte, Jula et al. 48
los restantes solamente indicaciones para el control de
-
aparentemente independiente del efecto directo sobre el
Cardiovasculares de la Universidad Nacional de La Pla-
20, 27, 49-50,
-
de profundizar el conocimiento sobre los mecanismos de
cuenta por lo analizado hasta aquí que, por un lado, no se
cifras tensionales, que a su vez la misma es diferente se-
-
-miento más adecuado. Teniendo en cuenta los mecanismos
es probable que, actuando a otros niveles de la cascada de
-
del intercambiador Na /H
--
fueron mayores en las ratas hipertensas y disminuyeron al
- 20, 49
-
--
50
considera que el principal objetivo buscado a través de la -
Figura 19.10: Representación de la regresión de la masa ventricular izquierda en pacientes tratados con diversas drogas hipotensoras. Ante similar disminución de la presión, la regresión de la hipertrofia fue diferente según el fármaco empleado. (Modificado de Schmieder et al., JAMA. 1996;275:1507-1513.)

Mecanismos de adaptacion fisiológicos y patologicos...
307
Figura 19.11 A: Imagen correspondiente a un registro ecocardiográfico en modo M del ventrículo izquierdo de una rata hipertensa. B: Comparación de los índices de masa ventricular izquierda, entre ratas normotensas (N), ratas espontáneamente hipertensas sin tratamiento (SHR) y SHR luego de un mes de tratamiento con 30 mg de cariporide (SHR-HOE): El índice de masa es significativamente mayor en SHR no tratadas respecto de las normotanesas (N), notándose una significativa regresión del mismo a los 30 días de tratamiento con cariporide.
A
B
Figura 19.12: En estas imágenes se observan, arriba, los corazones de una rata del grupo control (NT), a la izquierda, de una rata SHR sin tratamiento en el centro y de una rata SHR al mes del tratamiento. En la parte inferior se muestran cortes histológicos de corazones de ratas representativas de cada grupo. La hipertensión aumenta el tamaño del miocito y el volumen del espacio intersticial, que disminuyen en la regresión hasta alcanzar una situación similar a la observada en ratas normotensas. (Modificado de Camilión de Hurtado, MC et al., Cardiovasc Res. 2002;53:862-868.)
Tratamiento
PP
(μg
/L)
Frac
ció
n d
eco
lág
eno
(%)
A
B
*
*
*
*
Figura 19.14: Estas curvas representan la resistencia al estiramiento de músculos papilares. Se puede ver cómo el papilar se hace progresivamente más distensible al prolongar el tratamiento con cariporide, como consecuencia de una mayor regresión de la fibrosis. (Modificado de Camilión de Hurtado, MC et al., Cardiovasc Res. 2002;53:862-868.)
Figura 19.13: Las barras representan en la parte superior A: el porcentaje del volumen del miocardio ocupado por colágeno en ratas normotensas y en SHR sin tratamiento y al mes, 2 y 3 meses del mismo. En la parte inferior, B: se pueden analizar los niveles en sangre del PIP (péptido procolágeno tipo I) en los grupos de ratas descritos arriba, NT: normotensos. (Modificado de Camilión de Hurtado, MC et al., Cardiovasc Res. 2002;53:862-868.) respecto a NT

Gelpi - Donato Fisiopatología cardiovascular
308
Pfeffer et al.54, trabajando con ratas espontáneamente hipertensas, Brilla et al.55, utilizando modelos de hiperten-
et al. 56,
demostraron aumento de peso del ventrículo derecho y/o
prueba del compromiso de ese ventrículo en diferentes ti-
-motensos y 79 hipertensos esenciales, midiendo por eco-
derecho, Paolasso et al.57
-et al.54 habían reportado un aumento
ratas espontáneamente hipertensas. Lo analizado muestra claras evidencias del compromi-
so del ventrículo derecho en animales con diferentes tipos
arterial esencial, con independencia de factores hemodi-
Remodelamiento concéntricoComo fue analizado con anterioridad la presencia de un
Figura 19.15: A la izquierda se pueden analizar parámetros que caracterizan la estructura y función sistólica ventricular izquierda, obtenidos por ecocardiograma en ratas normotensas controles (barra clara), luego de recibir isoproterenol (barra gris) y a los 30 días de asociar isoproterenol BIIB (barra rayada). En el centro se ven las imágenes ecocardiográficas del ventrículo izquierdo en control (arriba), isoproterenol (centro) y BIIB (abajo). A la derecha, los cortes histológicos del ventrículo izquierdo en igual situación (Modificado de: Ennis, I et al., Hypertension. 2003;41:1324-1329.) DD: diámetro diastólico; FS: fracción de acortamiento; ED: espesor diastólico de pared anterior; MVI: masa ventricular izquierda
consecuencia de la misma, encontramos evidencias en po-
En efecto, trabajos de Levy et al.51 et al. 52 y recientemente de Mathew et al.53
¿Se comporta el ventrículo derecho como órgano blanco?Analizando los mecanismos que determinan la respuesta hi-
-
-
lado, que el estímulo hemodinámico secundario al aumento
capilar pulmonar, pareciera factible.
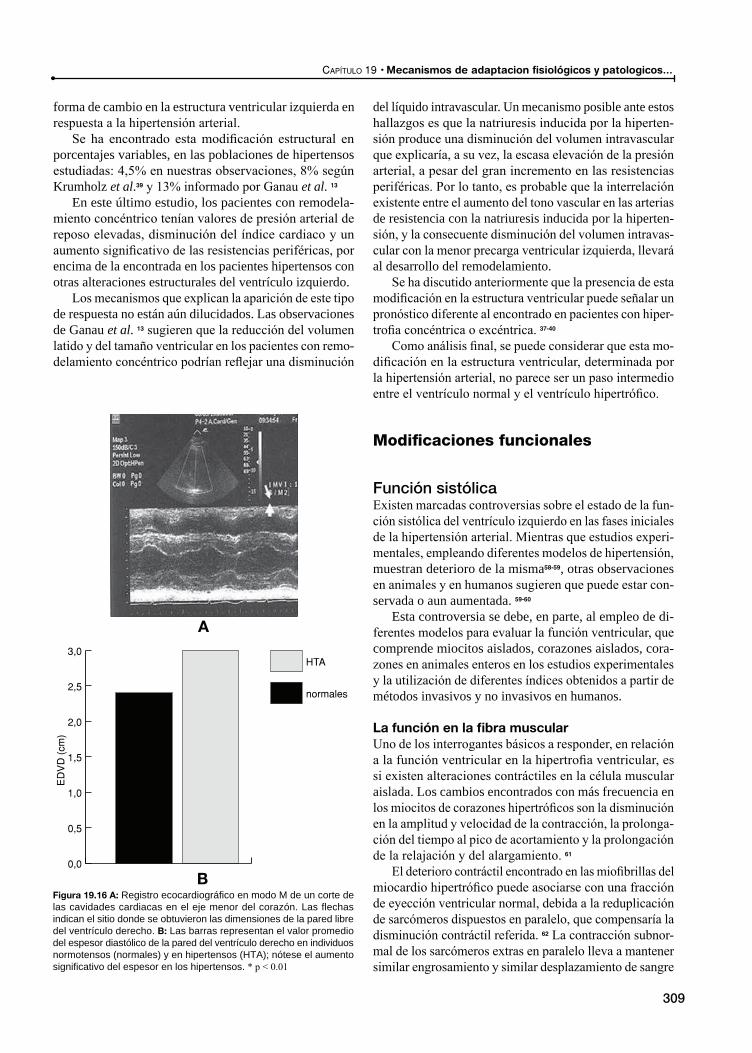
Mecanismos de adaptacion fisiológicos y patologicos...
309
del líquido intravascular. Un mecanismo posible ante estos -
de resistencia con la natriuresis inducida por la hiperten--
al desarrollo del remodelamiento.
-37-40
-
Modificaciones funcionales
Función sistólica-
-
muestran deterioro de la misma58-59, otras observaciones -
servada o aun aumentada. 59-60
Esta controversia se debe, en parte, al empleo de di-
comprende miocitos aislados, corazones aislados, cora-
métodos invasivos y no invasivos en humanos.
La función en la fibra muscular
aislada. Los cambios encontrados con más frecuencia en
-
61
62 -
forma de cambio en la estructura ventricular izquierda en
porcentajes variables, en las poblaciones de hipertensos
Krumholz et al.39 et al. 13
En este último estudio, los pacientes con remodela-
encima de la encontrada en los pacientes hipertensos con otras alteraciones estructurales del ventrículo izquierdo.
de respuesta no están aún dilucidados. Las observaciones de Ganau et al. 13
-
Figura 19.16 A: Registro ecocardiográfico en modo M de un corte de las cavidades cardiacas en el eje menor del corazón. Las flechas indican el sitio donde se obtuvieron las dimensiones de la pared libre del ventrículo derecho. B: Las barras representan el valor promedio del espesor diastólico de la pared del ventrículo derecho en individuos normotensos (normales) y en hipertensos (HTA); nótese el aumento significativo del espesor en los hipertensos.
A
B

Gelpi - Donato Fisiopatología cardiovascular
310
63
La función en el corazón entero-
hipertensos, un aumento del espesor de la pared ventricular
-
64-65 Teniendo en
ventricular izquierda, es uno de los determinantes de la
dentro de límites normales.
masculino de la Facultad de Ciencias Médicas de la Uni-versidad Nacional de La Plata, con edad promedio de
19.17, en los alumnos hipertensos. 60 Estas observaciones
-ferido por otros autores. 59 Es interesante considerar que
-
-céntrico. 66
--
Corazón entero: función medioventricular
-
67
-sultante de utilizar las variaciones endocárdicas. 68
-
la sístole, el acortamiento en el subendocardio es mayor -
crepancia entre el acortamiento del endocardio y el de la
ventricular, a diferencia de las subendocárdicas y subepi-
-
ventricular.-
-
Figura 19.17: A la izquierda se representa el índice de masa ventricular izquierda (IMVI) en los jóvenes con presión óptima y con hipertensión arterial (la barra muestra el valor promedio y los puntos, los datos de cada uno de los jóvenes estudiados). A la derecha se representan los valores del volumen latido y el índice cardiaco en esos grupos. Barras y puntos blancos: presión óptima. Barras y puntos negros: hipertensos

Mecanismos de adaptacion fisiológicos y patologicos...
311
endocardio hacia la cavidad, comparado a lo que ocurre
mantiene un acortamiento endocárdico normal a pesar de -
ne en cuenta que la mitad interna de la pared ventricular
-59-60
--
tamiento medioventricular se asociaba a menor capacidad funcional 69
-
-docárdico con valores similares en el acortamiento me-
- 70 -
nes refuerzan lo analizado con anterioridad, permitiendo considerar que, probablemente en estadios tempranos de
estaría compensada por el aumento en el número de sar-
Función diastólica
características del llenado del ventrículo izquierdo, son las primeras en aparecer como consecuencia de los efectos
izquierdo. 35, 60, 71
borderline y ante
56 -
35, 72, 73
ventricular a partir de diferentes mecanismos. 74
Relajación
impuesta al músculo75 -
el citosol y por aquellos factores como el pH intracelu-
al calcio. 75-76
La rápida caída de los niveles del calcio intracelular es determinada principalmente por la actividad de una bomba del retículo sarcoplásmico ATP-dependiente, co-
Ver capítulo 13-
-
-
-
-
aumento de los niveles del intercambiador Na /Ca en pacientes con miocardiopatía dilatada, aunque no se lo
-
Además de estos mecanismos, se pueden encontrar otros procesos moleculares capaces de comprometer la
Figura 19.18: Esquema de un corte del ventrículo izquierdo perpendicular al eje mayor, que muestra las características del eje menor en un corazón normal (arriba) y en corazón hipertrófico (abajo). Se considera que la pared está constituida por dos capas concéntricas. La mayor velocidad de acortamiento de las fibras de la capa interna determina que el espesor a fin de sístole de esta capa sea mayor que el de la capa externa. Esto explica la migración a fin de diástole de las capas internas hacia afuera, determinando un mayor acortamiento a nivel del endocardio. Estos cambios son más evidentes en un corazón hipertrófico. D: diástole. S: sístole. HVI: Hipertrofia ventricular izquierda
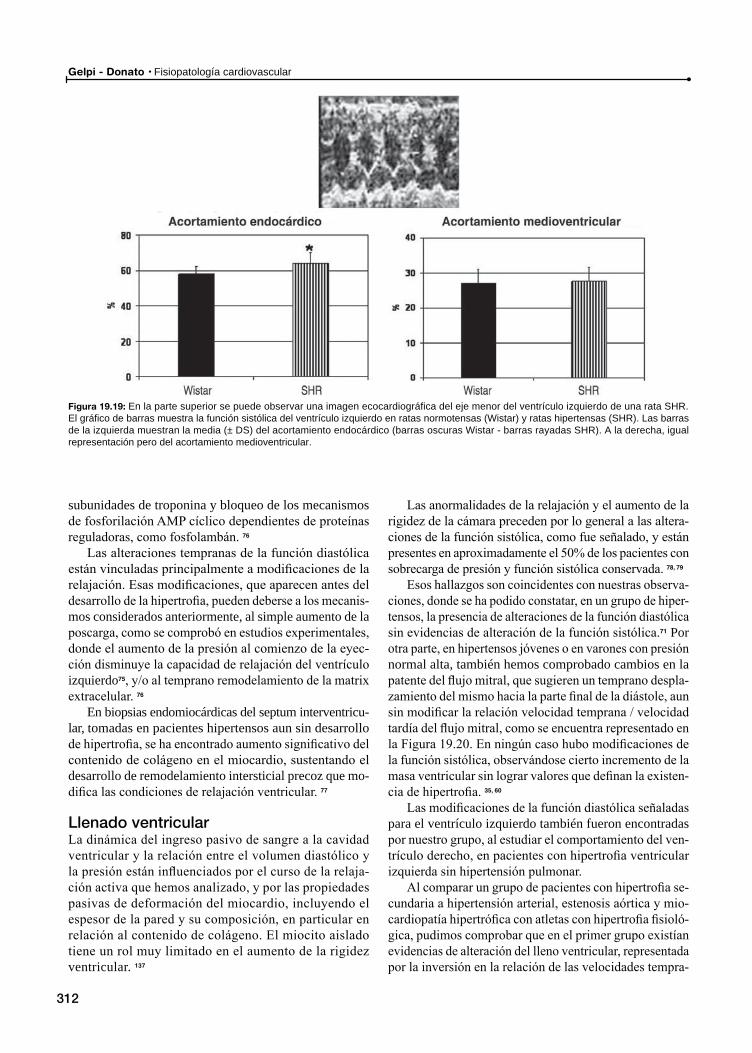
Gelpi - Donato Fisiopatología cardiovascular
312
subunidades de troponina y bloqueo de los mecanismos
76
-mos considerados anteriormente, al simple aumento de la
-
izquierdo75
76
En biopsias endomiocárdicas del septum interventricu-lar, tomadas en pacientes hipertensos aun sin desarrollo
desarrollo de remodelamiento intersticial precoz que mo-77
Llenado ventricular
-
ventricular. 137
-
78, 79
--
71 Por
normal alta, también hemos comprobado cambios en la -
-35, 60
para el ventrículo izquierdo también fueron encontradas -
---
-
Figura 19.19: En la parte superior se puede observar una imagen ecocardiográfica del eje menor del ventrículo izquierdo de una rata SHR. El gráfico de barras muestra la función sistólica del ventrículo izquierdo en ratas normotensas (Wistar) y ratas hipertensas (SHR). Las barras de la izquierda muestran la media (± DS) del acortamiento endocárdico (barras oscuras Wistar - barras rayadas SHR). A la derecha, igual representación pero del acortamiento medioventricular.

Mecanismos de adaptacion fisiológicos y patologicos...
313
80
El camino a la insuficiencia cardiaca
-81 El estudio de
-
82 Al realizar un ajuste por edad y por otros
doble que la que tenían los varones normotensos y el triple
-tivamente, mientras que el infarto de miocardio, con una
82
-
-
Deterioro de la función ventricular
-
de cadena pesada y la densidad de los microtúbulos dentro del miocito, que aumenta la viscosidad intracelular afec-
Figura 19.20 A: Registro ecocardiográfico de las velocidades de los flujos en el tracto de entrada del ventrículo izquierdo en diástole, tomado con Doppler pulsado. Se ven dos picos de velocidades, uno temprano correspondiente al lleno rápido (onda E) y otro tardío, en relación al flujo generado por la contracción auricular (onda A). B: Gráfico de barras representativo del análisis de esas velocidades en jóvenes con presión óptima (barras blancas) y jóvenes hipertensos (barras oscuras). Las barras superiores muestran el valor promedio (± DS) de las áreas bajo la curva de la onda E (izquierda) y A (derecha) y las inferiores, la relación entre esas áreas a la izquierda y el porcentaje que el área bajo la onda E tiene en el área total. En todos los casos se puede deducir un corrimiento del llenado hacia la parte final de la diástole. *p<0,01
BA
Figura 19.21: En la el panel (A) las barras representan el valor medio (± DS) del espesor de la pared libre del ventrículo derecho tomada por ecocardiografía, en individuos normales (C), atletas (A), con hipertensión arterial (HA), con estenosis aórtica severa (AS) y con miocardiopatía hipertrófica primaria (MH). En B se puede analizar la relación entre la velocidad diastólica temprana (E) y tardía (A) del desplazamiento del anillo tricuspídeo en los mismos grupos. Se puede observar un mayor espesor del ventrículo derecho con alteración de la función diastólica en AH, AS y MH respecto a C; mientras que los atletas con espesor mayor de ese ventrículo muestran mejoría de la relación E/A tricuspídea. *

Gelpi - Donato Fisiopatología cardiovascular
314
Tabla 19.2: Factores predisponentes para el desarrollo de insuficiencia cardiaca
Factor analizado
Sexo Riesgo Prevalencia (%)
% Atribuible
Hipertensión arterial
m f
2.07 3.35
60 62
39 59
Infarto de miocardio
m f
6.34 6.01
10 3
34 13
Angina m f
1.43 1.68
11 9
5 5
Diabetes m f
1.82 3.73
8 5
6 12
HVI m f
2.19 2.85
4 3
4 5
Valvulopatía m f
2.47 2.13
5 8
7 8
HVI: m: masculino. f: femeninoEl análisis está basado en la población de Framingham con individuos entre 40 y 89
83-84
no se tiene certeza sobre si la apoptosis participa en la -
rida o, por el contrario, resulta un proceso homeostático
contractilidad en forma ordenada. 74
-
izquierdo. 85
-
aldosterona, acelerando de esa forma el deterioro funcio-nal y conduciendo a un remodelamiento de la cavidad. La
asocia a pérdida de miocitos y a un deslizamiento de los 86, aunque mo-
-
del aumento del volumen ventricular, se produce un incre-
87
-
-ral y funcional de esta red de elementos contráctiles y no
-
Distintos autores 88
-tales, utilizando ratas espontáneamente hipertensas, con
estudio realizado en nuestro laboratorio, donde se ana-
de ratas espontáneamente hipertensas89, se pudo compro-
Alteración en la perfusión miocárdica-
como consecuencia de lesiones coronarias oclusivas y/o alteraciones estructurales y funcionales de la microcir-
de esos factores. -
es dos veces superior a la de los individuos normotensos, -
de padecer un infarto. 90-91
-
isquemia miocárdica, entre los que se incluyen la ace-92, el aumento de la
resistencia coronaria microvascular 93
de la reserva coronaria.94 Estas alteraciones determinan que los individuos hipertensos puedan padecer isquemia
-
Hipertensión arterial y aterosclerosis coronaria
presor más importante, tiene un efecto estimulante sobre el crecimiento del músculo liso vascular. Este péptido
-lo liso, activando a la fosfolipasa C, que incrementa la
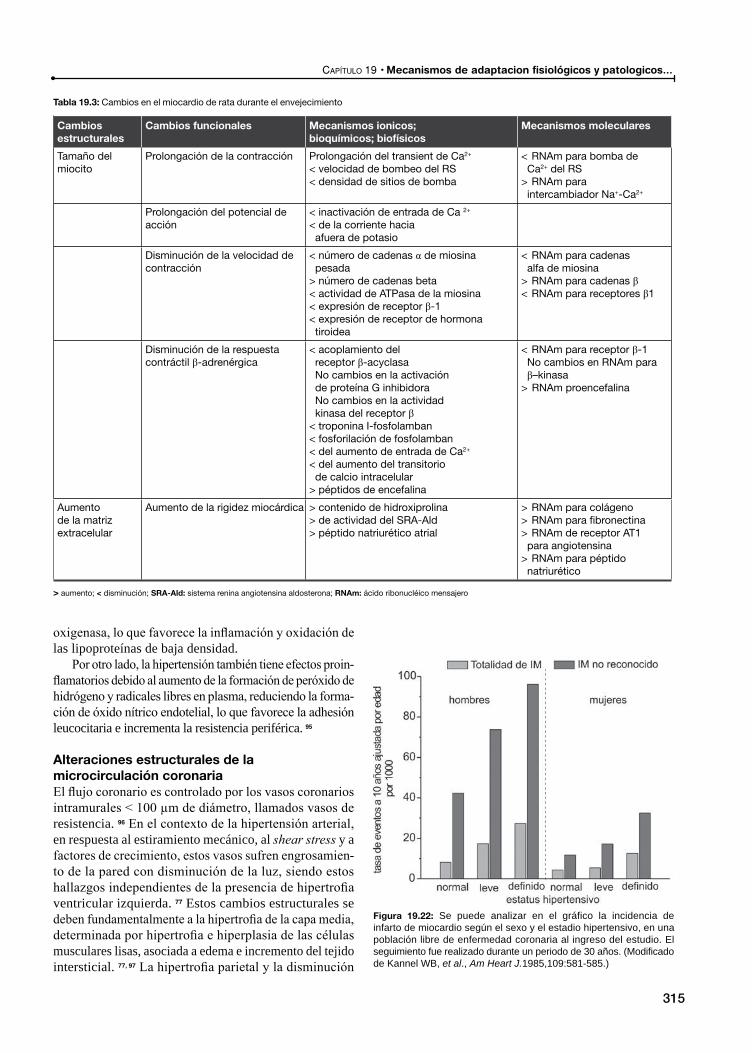
Mecanismos de adaptacion fisiológicos y patologicos...
315
las lipoproteínas de baja densidad.-
-
leucocitaria e incrementa la resistencia periférica. 95
Alteraciones estructurales de la microcirculación coronaria
resistencia. 96
en respuesta al estiramiento mecánico, al y a -
ventricular izquierda. 77 Estos cambios estructurales se
musculares lisas, asociada a edema e incremento del tejido intersticial. 77, 97
Cambios estructurales
Cambios funcionales Mecanismos ionicos;bioquímicos; biofísicos
Mecanismos moleculares
miocito < velocidad de bombeo del RS< densidad de sitios de bomba
< RNAm para bomba de Ca del RS> RNAm para intercambiador Na -Ca
acción< inactivación de entrada de Ca < de la corriente hacia afuera de potasio
Disminución de la velocidad de contracción
< número de cadenas de miosina pesada> número de cadenas beta
< expresión de receptor -1< expresión de receptor de hormona tiroidea
< RNAm para cadenas alfa de miosina> RNAm para cadenas < RNAm para receptores 1
Disminución de la respuesta contráctil -adrenérgica
< acoplamiento del receptor -acyclasa No cambios en la activación de proteína G inhibidora No cambios en la actividad kinasa del receptor < troponina I-fosfolamban< fosforilación de fosfolamban< del aumento de entrada de Ca< del aumento del transitorio de calcio intracelular> péptidos de encefalina
< RNAm para receptor -1 No cambios en RNAm para –kinasa> RNAm proencefalina
Aumento de la matriz extracelular
Aumento de la rigidez miocárdica > contenido de hidroxiprolina> de actividad del SRA-Ald> péptido natriurético atrial
> RNAm para colágeno> RNAm para fibronectina> RNAm de receptor AT1 para angiotensina> RNAm para péptido natriurético
Tabla 19.3: Cambios en el miocardio de rata durante el envejecimiento
> aumento; < disminución; SRA-Ald: sistema renina angiotensina aldosterona; RNAm: ácido ribonucléico mensajero
Figura 19.22: Se puede analizar en el gráfico la incidencia de infarto de miocardio según el sexo y el estadio hipertensivo, en una población libre de enfermedad coronaria al ingreso del estudio. El seguimiento fue realizado durante un periodo de 30 años. (Modificado de Kannel WB, et al., Am Heart J.1985,109:581-585.)

Gelpi - Donato Fisiopatología cardiovascular
316
Figura 19.23: Mecanismos etiopatogénicos de arritmias ventriculares en la hipertensión arterial
en la distensibilidad del vaso que deteriora la conductan-
98
ventrículo izquierdo, que se produce en los pacientes hi-
-cial alrededor de los miocitos, como fue analizado ante-riormente, y de las arterias y arteriolas intramiocárdicas93, alterando aún más la capacidad vasodilatadora de la mi-
99
-quierda, que contribuye a la isquemia miocárdica, es el
crecimiento del compartimiento muscular e intersticial, provocando un aumento de la distancia entre los capilares,
100
Alteraciones funcionales de la microcirculación coronaria
arterias coronarias de aumentar el diámetro de su luz ante un estímulo vasodilatador, como por ejemplo el resultante de la isquemia miocárdica. La hiperemia reactiva es el consi-
luz del vaso. Esta respuesta hiperémica se debe al acúmulo local de sustancias vasodilatadoras como la adenosina, las
101 El análisis de la reserva
102
--
menta la resistencia coronaria dependiente del miocardio. Por otro lado, la capacidad vasodilatadora coronaria puede
un incremento del estrés parietal. 103
Además de estos factores miocárdicos y vasculares,
endotelio. Las células endoteliales sintetizan, bajo condicio-
en respuesta a la bradiquinina, la acetilcolina y a factores físicos como el 104 En los individuos hiperten-sos esta respuesta se ve alterada, disminuyendo los factores vasodilatadores dependientes del endotelio, resultando en un aumento relativo y también probablemente absoluto de los factores vasoconstrictores como la endotelina.
-
pacientes hipertensos con coronarias epicárdicas sin le-
Arritmias
padecer arritmias supraventriculares y ventriculares, siendo
Arritmias supraventricularesLa mayor frecuencia de arritmias supraventriculares, como
-
datos publicados, los pacientes hipertensos que desarrollan
105 La
HTA Arritmias ventriculares

Mecanismos de adaptacion fisiológicos y patologicos...
317
Arritmias ventriculares
arritmias ventriculares, se han postulado varios mecanis-
-
-
Es importante tener en cuenta que, sumado a los me-
-
las arterias coronarias epicárdicas.
Bibliografía
1. Frolich, ED. “The first Irving H.Page lecture: The mosaico of hypertension: past, present and future”. J Hypertens. 1988;6(suppl 4 ):S 2-11.
2. Frolich, ED. “The heart in hypertension: unresolved conceptual challenges”. Hypertension. 1988;1(supplI)I19-24.
3. Strauer, BE. “Ventricular function and coronary hemodynamics in hypertensive heart disease”. Am J Cardiol. 1979;44:999-1006.
4. Kannel WB, AJ Belanger. “Epidemiology of heart failure”. Am Heart J. 1991;121:951-957.
5. McLenachan JM, Henderson A, Morris KL, Dergie HJ. “Ventri-cular arrhythmias in patients with hypertensive LVH”. N Engl J Med. 1987;317:787-792.
6. Kannel, WB. “Role of blood pressure in cardiovascular morbidity and mortality”. Prog Cardiovasc Dis. 1974;7:5-24.
7. Frey N, Katus HA, Olson EN, Hill JA. “Hypertrophy of the Herat. A new therapeutic target?” Circulation. 2004;109:1580-1589.
8. Escudero EM, Tufare AL, Rebolledo O, Pellegrini L, Lobrutto C. “Serum carboxyl terminal propeptide of procollagen type I in exercise induced left ventricular hypertrophy”. Clin Cardiol. 2004;27:471-474.
9. Querejeta R, Varo N, López B, Larman M, Artiñano E, Etayo JC, Martínez Ubago JL, Gutiérrez-Sampa M, Emparanza JI, Gil MJ, Monreal I, Pardo Mindán J, Díez J. “Serum carboxyl-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease”. Circulation. 2000;101:1729-1735.
10. Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP. “Echocardiographic criteria for left ventricular hy-pertrophy: the Framingham Heart Study”. Am J Cardiol. 1987; 58:1072-1083.
11. Devereux RB, Dahlöf B, Levy D. “Comparison of enalapril versus nifedipine to decrease left ventricular hypertrophy in systemic hy-pertension (the PRESERVE trial)”. Am J Cardiol. 1996;78-61-65.
12. de Simone G, Daniels SR, Devereux RB, Meyer RA, Roman MJ, de Devitis O, Alderman MH. “Left ventricular mass and body size in normotensive children and adults: assessment of allometric relations and impact of overwight”. J Am Coll Cardiol.1992;20:1251-1260.
13. Ganau A, Devereux RB, Roman MJ, de Simone G, Pickering TG, Saba PS, Vargiu P, Simognini I, Laragh JH. “Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension”. J Am Coll Cardiol. 1992;19:1550-1558.
14. Devereux RB, Koren MJ, de Simone G, Roman MJ, Laragh JH. “Left ventricular mass as a measure of preclinical hypertensive disease . Am J Hypertens. 1992;5(6 Pt 2):175S-181S.
15. Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. “Continuous relation between left ventricular mass and cardiovascular risk in essential hipertension”. Hypertension. 2000;35:580-586.
16. Casale PN, Devereux RB, Milner M, Zullo G, Harshfield GA, Pickering TG, Laragh JH. “Value of echocardiographic measure-ment of left ventricular mass in predicting cardiovascular morbid events in hypertensive men”. Ann Intern Med. 1986;105:173-178.
17. Grossman W, Jones D, McLaurin LP. “Wall stress and patterns of hy-pertrophy in the human left ventricle”. J Clin Invest. 1975;56:56–64.
18. Chien, KR. “Genomic circuits and the integrative biology of cardiac diseases”. Nature. 2000;407:227–232.
19. Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA “Genetic alterations that inhibit in vivo pressure overload hypertrophy prevent cardiac dysfunction despite increased wall stress”. Circulation. 2002;105:85-92.
20. Escudero EM, Pérez NG, Camilión de Hurtado MC, Cingolani HE. “Left ventricular performance after regression of myocardial hypertrophy in spontaneously hypertensive rats (SHR)”. J Mol Cell Cardiol. 2003;35:A9.
21. Tufare AL, Garciarena C, Escudero EM. “Efectos de la regresión de la hipertrofia ventricular izquierda sobre la función sistólica del ventrículo izquierdo en ratas”. Rev Fed Arg Cardiol. 2004;33:48.
22. Yamazaki T, Komuro I, Yazaki Y. “Role of the rennin - angiotensin system in cardiac hypertrophy”. Am J Cardiol. 1999;83:53H-57H.
23. Sadoshima J, Izumo S. “Signal transduction pathways of angio-tensin II--induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers”. Circ Res. 1993;73:424-438.
24. Camilión de Hurtado MC, Álvarez BV, Ennis IL, Cingolani HE. “Stimulation of myocardial Na+-independent Cl--HCO
3- exchanger
by angiotensin II is mediated by endogenous endothelin”. Circ Res. 2000;86:622-627.
25. Pérez NG, Camilión de Hurtado MC, Cingolani HE. “Reverse mode of the Na+-Ca2+ exchange following myocardial stretch: underlying mechanism of the slow force response”. Circ Res. 2001;88:376-782.
26. Marbán E, Y Koretsune. “Cell calcium, oncogenes, and hypertro-phy”. Hypertension. 1990;15:652–658.
27. Sadoshima J, S Izumo. “Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts”. Circ Res. 1993;73:413-423.
28. Pérez NG, Escudero EM, Portiansky EL, Camilión de Hurtado MC, Cingolani HE. “Regresion and prevention of myocardial hypertrophy and fibrosis by Na+/H+ exchange inhibition”. J Mol Cell Cardiol. 2002;34:A-17.
29. Hammond IW, Devereux RB, Aldermann MH, Laragh JH. “Re-lation of blood pressure and body build of left ventricular mass in normotensive and hypertensive employed adults”. J Am Coll Cardiol. 1988;12:996-1004.
30. Brilla, CG. “Renin-angiotensin-aldosterone system and myocardial fibrosis (editorial)”. Cardiovasc Research. 2000;47:1-3.
31. de Leonardis V, De Scalzi M, Falchetti A, Cinelli P, Croppi E, Livi R, Scarpelli L, Scarpelli PT. “Echocardiographic evaluation of

Gelpi - Donato Fisiopatología cardiovascular
318
children with and without family history of essential hypertension”. Am J Hypertens. 1988;1:3 Pt 1:305-8.
32. Gharavi AG, Lipkowitz MS, Diamond JA, Jhang JS, Phillips RA. “Deletion polymorphism of the angiotensin-converting enzyme gene is independently associated with left ventricular mass and geometric remodeling in systemic hypertension”. Am J Cardiol. 1996;15-77:15-1315-9.
33. Raij, L. “Nitric oxide in hypertension: relationship with re-nal injury and left ventricular hypertrophy”. Hypertension. 1998;31:189-193.
34. Poch E, González D, Gómez Angelats E, Enjuto M, Paré JC, Rivera F, de la Sierra A. “G-protein beta 3 sub-unit gene variant and left ventricular hypertrophy in essential hipertensión”. Hipertension. 2000;35:214-218.
35. Escudero E, DeLena S, Graff.Iversen S, Almiron M, Cingolani H. “Left ventricular diastolic function in young men with high normal blood pressure”. Can J Cardiol. 1996;12(10):959-964.
36. Coca A, Gabriel R, de la Figuera M, López-Sendon JL, Fernández R, Sagastagoitia JD, García JJ, Barajas R. “The impact of different echocardiographic diagnostic criteria on the prevalence of left ventricular hypertrophy in essential hypertension: the VITAE study. Ventrículo Izquierdo Tensión Arterial España”. J Hypertens. 1999;17:10:1471-80.
37. Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. “Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension”. Ann Intern Med. 1991;114:345-352.
38. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Santucci A, Santucci C, Reboldi G, Porcellati C. “Prog-nostic value of left ventricular mass and geometry in systemic hypertension with left ventricular hypertrophy”. Am J Cardiol. 1996;78:197-202.
39. Krumholz HM, Larson M, Levy D. “Porgnosis of left ventricular geometric patterns in the Framingham Heart Study”. J Am Coll Cardiol. 1995;25:879-884.
40. Kohara K, Zhao B, Jiang Y, Takata Y, Fukuoka T, Igase M, Miki T, Hiwada K. “Relation of left ventricular hypertrophy and geometry to asymptomatic cerebrovascular damage in essential hypertension”. Am J Cardiol. 1999;83:3367-70.
41. Vilari BM, Campbell SE, Hess OM. “Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease”. J Am Coll Cardiol. 1993;22:1477-1484.
42. Vilari B, Vassalli G, Monrad ES. “Normalization of diastolic dys-function in aortic stenosis late after valve replacement”. Circulation. 1995;88;2353-2358.
43. Sen S, Tarazi RC, Bumpus FM. “Cardiac hypertrophy and anti-hypertensive therapy”. Cardiovascular Res. 1977;11;427-433.
44. Schmieder RE, Martus P, Klingbeil A. “Reversal of left ventricular hypertrophy in essential hypertension: meta-analysis of randomized double-blind studies”. JAMA. 1996;275:1507-1513.
45. Dahlöf B, Pennert K, Hansson L. “Reversal of left ventricular hy-pertrophy in hypertensive patients: a meta-analysis of 109 treatment studies”. Am J Hypertens. 1992;5:95-110.
46. Gosse P, Sheridan DJ, Zannad F, Dubourg O, Guéret P, Karpov J de Leeuw PW, Palma-Gamiz JL, Pessina A, Motz W, Degaute JP, Chastang C. “Left ventricular regression in hypertensive patients, comparison between indapamida LP 1.5 mg vs enalapril 20 mg: LIVE study”. J Hypertens. 2000;18:1465-1475.
47. Brilla CG, Funck R, Rupp H. “Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease”. Circulation. 2000;102:1388-1393.
48. Jula AM, HM Karanko. “Effects on left ventricular hypertrophy of long-term nonpharmacological treatment with sodium retric-tion in mild-to-moderate essential hypertension”. Circulation. 1994;89:1023-1031.
49. Camilión de Hurtado MC, Portiansky EL, Pérez NG, Rebolledo OR, Cingolani HE. “Regression of cardiomyocyte hypertrophy in SHR following chronic inhibition of the Na+-H+ exchanger”. Cardiovasc Res. 2002;53:862-868.
50. Ennis I, Escudero EM, Cónsole G, Camihort G, Gómez Dumm C, Sidler RW, Camilión de Hurtado MC, Cingolani HE. “Re-gression of isoproterenol-induced cardiac hypertrophy by Na +/ H+ exchanger inhibition”. Hypertension. 2003;41:1324-1329.
51. Levy D, Salomón M, D’Agostino RB, Belanger AJ, Kannel WB. “Prognostic significance of serial changes in left ventricular mass in essential hypertension”. Circulation. 1994;90:1786-1793.
52. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zam-pi I, Reboldi G, Porcellati C. “Prognostic implications of baseline electrocardiographic features and their serial changes in subjects with left ventricular hypertrophy”. Circulation. 1998;97:48-54.
53. Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, Bosch J, Sussex B, Probstfield J, Yusuf S. “Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricu-lar hypertrophy by the angiotenisn-converting enzyme inhibitor ramipril”. Circulation. 2001;104:1615-1621.
54. Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED. “Cardiac function and morphology with aging in the spontaneously hy-pertensive rat”. Am J Physiol. 1979 Oct;237:4H461-8.
55. Brilla, CG. “Regression of myocardial fibrosis in hypertensive heart disease: diverse effects of various antihypertensive drugs”. Cardiovasc Res. 2000;46:324-331.
56. Frolov VA, Drozdoeva GA, Kazanskaia TA. “The right heart ven-tricle during hemodynamic overload of the left ventricle”. Patol Fiziol Eksp Ter. 1995;132(3):26-31.
57. Paolasso J, Escudero E, Ronderos R, Corneli D. “Características estructurales del ventrículo derecho en pacientes con hipertensión arterial”. Rev Fed Arg Cardiol. 2001;30:45.
58. Bing OHL, Matsushita S, Fanburg BL, Levine BL, Levine HJ. “Mechanical properties of rat cardiac muscle during experimental hypertrophy”. Circ Res. 1971;28:234-245.
59. Shorofsky SR, Aggarwal R, Corretti M, Baffa JM, Strum JM, Al-Seikan BA, Kobayashi YM, Jones LR, Wier WG, Balke CW. “Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks”. Circ Res. 1999;84:424-434.
60. Escudero EM, De Lena S, Cingolani HE. “Estructura y función del ventrículo izquierdo en jóvenes estudiantes varones de la Uni-versidad Nacional de La Plata con hipertensión arterial en estadio I”. Medicina. 1997;57:181-190.
61. Nuss H, SR Houser. “Voltage dependence of contraction and calcium current in severely hypertrophied feline ventricular myo-cytes”. J Mol Cell Cardiol. 1991;23:717-726.
62. Carabello, B. “Evolution of the study of left ventricular function. Everything old is new again”. Circulation. 2002;105:2701-2703.
63. de Simone G, Devereux RB, Celentano A. “Left ventricular cham-ber and wall mechanics in the presence of concentric geometry”. J Hypertens. 1999;17:1001-1006.
64. Ford, EF. “Heart size”. Circ Res. 1985;39:297-303.65. Grossman W, Jones D, Mc Lauri LP. “Wall stress and patterns of
hypertrophy in the human left ventricle”. J Clin Invets. 1975;56:56-64.66. Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler
RJ, Barzilai B, Dávila-Román V. “Alterations in left ventricular structure and function in young healthy obese women”. J Am Coll Cardiol. 2004;43:1399-1404.
67. de Simone G, Devereux RB, Vople M, Camargo MJF, Wallerston DC, Laragh JH. “Midwall LV mechanics in rats with or without renovascular hypertension: effect of different Na + intakes”. Am J Phyisiol. 1996;270:H628-637.
68. Reichek N, Wilson J, St John Sutton M, Plappert TA, Goldberg S, Hirshfeld JW. “Noninvasive determination of left ventricular end-systolic stress: validation of the method and initial application”. Circulation. 1982;65:99-108.
69. Schussheim AE, Devereux RB, de Simone G, Borer JS, Herrold EMcM, Laragh JH. “Usefulness of subnormal midwall fractional shortening in predicting left ventricular exercise dysfunction in asymptomatic patients with systemic hipertension”. Am J Cardiol. 1997;79:1070-1074.
70. Escudero EM, Camilión de Hurtado MC, Pérez NG, Tufare AL. “Echocardiographic assessment of left ventricular midwall mecha-

Mecanismos de adaptacion fisiológicos y patologicos...
319
nics in spontaneosuly hypertensive rats”. Eur J Echocardiography. 2004;5:169-175.
71. Escudero EM, Plastino JA, Pedroni P. “Estudio de los patrones de flujo trasnmitral en pacientes hipertensos”. Rev Chilena de Cardiol. 199;9:131-136.
72. Sagie A, Benjamín E, Galderisi M, Larson MG, Evans JC, Fu-ller DL, Lehman B, Levy D. “Echocardiografic assessment of left ventricular structure and diastolic filling in elderly subjects with borderline isolated systolic hypertension (the Framingham Heart Study)”. Am J Cardiol. 1993;662-665.
73. Kapuku GK, Seto S, Mori H, Mori M, Utsunomia T, Suzuki S, Oku Y, Yano K, Hashiba K. “Impaired left ventricular filling in borderline hypertensive patients without cardiac structural chan-ges”. Am Heart J. 1993;125:1710-1716.
74. Lorell BH, BA Carabello. “Left ventricular hypertrophy. Pathoge-nesis, detection and prognosis”. Circulation. 2000;102:470-479.
75. Zile MR, WH Gaasch. “Mechanical loads and the isovolumic and filling indices of left ventricular relaxation”. Prog Cardiovasc Dis. 1990;32:333-346.
76. Solaro JR, HM Rarick. “Troponin and tropomyosin. Proteins that switch on and tune the activity of cardiac myofilaments”. Circ Res. 1998;83:471-480.
77. Schwartzkopff B, Motz W, Knauer S, Frenzel H, Struer BE. “Mor-phometric investigations of intramyocardial arterioles in right septal endomyocardial biopsy of patientes with arterial hypertension and left ventricular hypertrophy”. J Cardiovasc Pharmacol. 1992;20:(su-ppl 1).12-17.
78. Villari B, Hess OM, Kaufmann P. “Effect of aortic valve steno-sis (pressure overload) and regurgitation (volume overload) on left ventricular systolic and diastolic function”. Am J Cardiol. 1992;62:927-934.
79. Hess OM, Villari B, Krayenbuehl HP. “Diastolic dysfunction in aortic stenosis”. Circulation. 1993;87(suppl 5)73-76.
80. Tufare AL, E Escudero. “¿Es el ventrículo derecho órgano blanco en la hipertensión arterial?” Comunicación Oral. XI Congreso Argentino de Hipertensión Arterial, Buenos Aires, 2004.
81. Ericksson H, Svardddsudd K, Larsson B. “Risk factors for heart failure in the general population : the study of men born in 1913”. Eur Heart J. 1989;10:657-666.
82. Levy D, Larson MG, Vasan RS, Kannel WB, Ho KKL. “The progression form hypertension to congestive heart failure”. JAMA. 1996;275:1557-1562.
83. Lamb HJ, Beyerbacht HP van der Laarse A. “Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism”. Circulation. 1999;99:2261-2267.
84. Neubauer S, Horn M, Cramer M. “Myocardial phosphocreatine-to ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy”. Circulation. 1997;96:2190-2196.
85. Serizawa T, Mirsky J, Carabello BA, Grossman W. “Diastolic myo-cardial stiffness in gradually left ventricular hypertrophy in dog”. Am J Physiol. 1982;242:H633-H637.
86. Katz, A. “Cadiomyopathy of overload. A major determinant of prog-nosis in congestive heart failure”. N Engl J Med. 1990;322:100-110.
87. Strauer, BE. “Myocardial oxygen consumption in chronic heart disease. Role of wall stress, hypertrophy and coronary reserve”. Am J Cardiol. 1979;44:999-1006.
88. Slama M, Ahn J, Varagic J, Susic D, Frohlich. “Long-term left ventricular echocardiographic follow-up of SHR and WKY rats: effects of hypertension age”. Am J Physiol Herat. 2004;286:H181-H185.
89. Tufare A, Escudero E, Camilión de Hurtado MC. “Hipertrofia ventricular izquierda y progresión a la insuficiencia cardiaca en ratas espontáneamente hipertensas”. XI Congreso Argentino de Hipertensión Arterial. Buenos Aires, 2004.
90. Kannel WB, Dannenberg AL, Abbott RD. “Unrecognized myo-cardial infarction and hypertension: the Framingham study”. Am Heart J. 1985,109:581-585.
91. Rabkin SW, Mathewson FA, Tate RB. “Prognosis after acute myocardial infarction: relation to blood pressure values before infarction in a prospective cardiovascular study”. Am J Cardiol. 1977 Oct;40(4):604-10.
92. Ross, R. “Atherosclerosis: an inflamatory disease”. N Engl J Med. 1999;340(2):115-126.
93. Bishop JE, G Lindahl. “Regulation of cardiovascular collagen synthesis by mechanical load”. Cardiovasc Res. 1999;42:27-44.
94. Dellsperger KC, ML Marcus. “Effects of left ventricular hypertrophy on the coronary circulation”. Am J Cardiol. 1990;65:1505-1510.
95. Swei A, Lacy F, De Lano FA, Schmid-Schönbein GW. “Oxidative stress in the Dahl hypertensive rat”. Hypertension. 1997;30:1628-1633.
96. Tillmanns H, Steinhausen M, Leinberger H, Tedherau H, Kübler W. “Pressure measurement in the terminal vascular bed of the epimyocardium of rats and cats”. Circ Res. 1981;49:1202-1211.
97. Amann K, Gareghbagli H, Stephan S, Mall G. “Hypertrophy and hyperplasia of smooth muscle cells of small intramyocardial arteries in spontaneously hypertensive rats”. Hypertension. 1995;25:124-131.
98. James, JN. “Morphologic characteristics and functional significance of focal fibromuscular dysplasia of small coronary arteries”. Am J Cardiol. 1990;65:12G-22G.
99. Frohlich, E. “Fibrosis and ischemia: the real risks in hypertensive heart disease”. Am J Hypertens. 2001;14:194S-199S.
100. Tomanek RJ, Schalk KA, Marcus ML, Harrison DG. “Coronary angiogenesis during long-term hypertrophy and left ventricular hypertrophy in dogs”. Circ Res. 1989;65:352-359.
101. Canty JM Jr, TP Jr Smith. “Adenosine-recruitable flow reserve is absent during myocardial ischemia in unanesthetized dogs studied in the basal state”. Circ Res. 1995;76:1079–1087.
102. Hamouda MS, Kassem HK, Salama M, El Masry M, Shaaban N, Sadek E, Khandheria BK, Seward JB, Elhendy A. “Evaluation of coronary flow reserve in hypertensive patients by dipyrida-mol transesophageal Doppler echocardiography”. Am J Cardiol. 2000;86:305-308.
103. Kozakova M, Palombo C, Pratali L, Pittela G, Galetta F, L’Abbate A. “Mechanisms of coronary flow reserve impairment in human hypertensión. An integrated approach by transthoracic and transeophageal echocardiography”. Hypertension. 1997;29:551-559.
104. Lüscher TF, Wenzel RR, Noll G. “Local regulation of the coronary circulation in health and disease. Role of nitric oxide and endo-thelin”. Eur Heart J. Suppl C. 1995;16:51-58.
105. Braunwald, E ed. Heart disease: a textbook of cardiovascular medicine. Capítulo XXIII. 6th ed. Philadelphia: Saunders, 2001.


321
Fisiopatología de la hipertensión pulmonar 20
ventrículo izquierdo, se desarrolla a partir de cuatro perso-
-
-
-
Andre Cournand et al -
en las unidades de terapia intensiva.-
1
pulmonares. El término hipertensión pulmonar primaria 2
-hipertensión arterial
pulmonar idiopáticael de hipertensión arterial pulmonar familiar. El término
la idea sobre la temporalidad y relatividad de las sucesivas verdades.
3
-
en el circuito menor.
---
una propiedad independiente de la velocidad del mismo.
F 8 l
neointimales 4 --
principio una injuria endotelial que puede ser secundaria -
toinmunes. Los vasos sistémicos desarrollan lesiones in-timales en respuesta a injurias tales como edad avanzada,
se sumen los factores antes mencionados.
-
pulmones denervados, pudiendo aumentar el volumen de
-

Gelpi - Donato Fisiopatología cardiovascular
322
duos normales. Respirar concentraciones elevadas de CO o la acidosis aumentan la resistencia vascular pulmonar.
-
-
-bios estructurales:
septum interventricular o
-maria o secundaria
-
y en las arteriolas, que se producen, fundamentalmente, en la media y en la íntima. Las arterias pulmonares mus-culares tienen un diámetro que va desde los 100 a 1000 micrones. La media de estas arterias tiene músculo liso y
Epidemiología y factores de riesgo
5
puede presentarse en cualquier etapa de la vida, la mayor incidencia ocurre en la tercera década en las mujeres y en la cuarta en los varones, con una edad promedio de
étnica y el promedio de tiempo entre el inicio de los sín-
-
--
missense
de receptores heteroméricos, la respuesta celular podrá
smads -
-
--
-
- hay mutaciones
abundan en los vasos pulmonares.--
-6 -
-
-
-
derecha y en la estructura cardiaca ocurren tardíamente, por lo que se deben utilizar métodos que permitan una
El cateterismo cardiaco fue el primer método para
-
las presiones y resistencia vascular pulmonar permite

Fisiopatología de la hipertensión pulmonar
323
-7 -
tes con enfermedad parenquimatosa pulmonar avanzada y -
-
-
antigen 1
al que ocurre en el sarcoma de Kaposi. Un revolucionario
como un proceso vasoproliferativo que como un proceso
tratamiento de esta enfermedad, sumando a los vasodila-
remodelado vascular. 8 El bloqueo de las endotelinas pa-
músculo liso, se produce también en el endotelio vascu-
es producido por el endotelio vascular, catalizado por la
Normalmente hay un balance entre factores relajantes
-
Tabla 19.1: Clasificación de Venecia 2003 de la Hipertensión arterial pulmonar
Hipertensión arterial pulmonar idiopática
Hipertensión arterial pulmonar familiar
Enfermedad vascular del tejido conectivo
Congénita secundaria a shunts
Hipertensión portal
Infección por virus de inmunodeficiencia humana
Drogas y tóxicos
Otras (enfermedad por depósito de glucógeno, enfermedad de Gaucher, teleangiectasia hemorrágica hereditaria, hemoglobinopatías, enfermedades mieloproliferativas, esplenectomía)
Asociada con compromiso venoso o capilar significativo
Enfermedad pulmonar venooclusiva
Hemangiomatosis capilar pulmonar
Hipertensión venosa pulmonar
Hipertensión pulmonar asociada con hipoxemia
Enfermedad pulmonar obstructiva crónica
Enfermedad pulmonar intersticial
Hipoventilación alveolar
Exposición crónica a grandes alturas
Secundaria a enfermedad embólica o trombótica crónica
Obstrucción tromboembólica de arterias pulmonares proximales
Obstrucción tromboembólica de arterias pulmonares distales
Misceláneas
Sarcoidosis, histiocitosis X, linfangioleiomiomatosis, compresión de vasos pulmonares (adenopatías, tumores, mediastinitis fibrosa)

Gelpi - Donato Fisiopatología cardiovascular
324
9 Un disbalance de estos factores eleva el tono vasomotor, promueve la pro-
aumenta la trombosis. Podemos concebir así la enferme--
constrictora, lo que nos lleva a enfatizar el tratamiento con
Inhibir el efecto de la endotelina bloqueando el receptor es un tratamiento nuevo y efectivo que puede efectuarse con bosentan, un no-péptido activo administrado por vía oral,
los receptores ET-A y ET-B. Este tratamiento reduce tanto
--8
-
-
-te de una superfamilia de factores transformadores de cre-
fenotipo permisivo y estímulos del ambiente. Compromete todos los elementos de los vasos y ofrece como blanco te-
canales de potasio y la BMPRII.
Fisiopatología
con el descubrimiento de un posible mecanismo de la
mediadores vasodilatadores producidos por el endotelio vascular pulmonar.
--
-rios; por ejemplo, el uso de estudios no efectuados, como
--
académica, pues muchos prestadores no reembolsan me-
volumen minuto cardiaco y la resistencia vascular pulmonar. -
cular se duplica. El aumento de cualquiera de estos factores -
voltaje dependientes y canales de potasio calcio dependientes --
Tabla 19.2: Clasificación funcional
A. Asociación Cardiológica de Nueva York
Clase 1: sin síntomas con la actividad física común
Clase 2: síntomas con actividades ordinarias. Leve limitación de actividad
Clase 3: síntomas con actividades menores a las habituales. Limitación marcada de actividad
B. Organización Mundial de la Salud
Clase I: pacientes con hipertensión pulmonar pero sin limitación de la actividad física ni síntomas
Clase II: pacientes con hipertensión pulmonar con limitación leve de la actividad física. Confortables en reposo. Actividad física habitual disnea o fatiga, causa no justificada, dolor torácico o casi síncope
Clase III: paciente con hipertensión pulmonar con marcada limitación de la actividad física. Confortable en reposo. Actividad menor a la habitual causa disnea o fatiga, dolor torácico o casi síncope
Clase IV: pacientes con hipertensión pulmonar con imposibilidad de realizar actividad física sin síntomas. Signos de insuficiencia cardiaca derecha. Disnea y/o fatiga aun en reposo. La actividad física aumenta el disconfort.
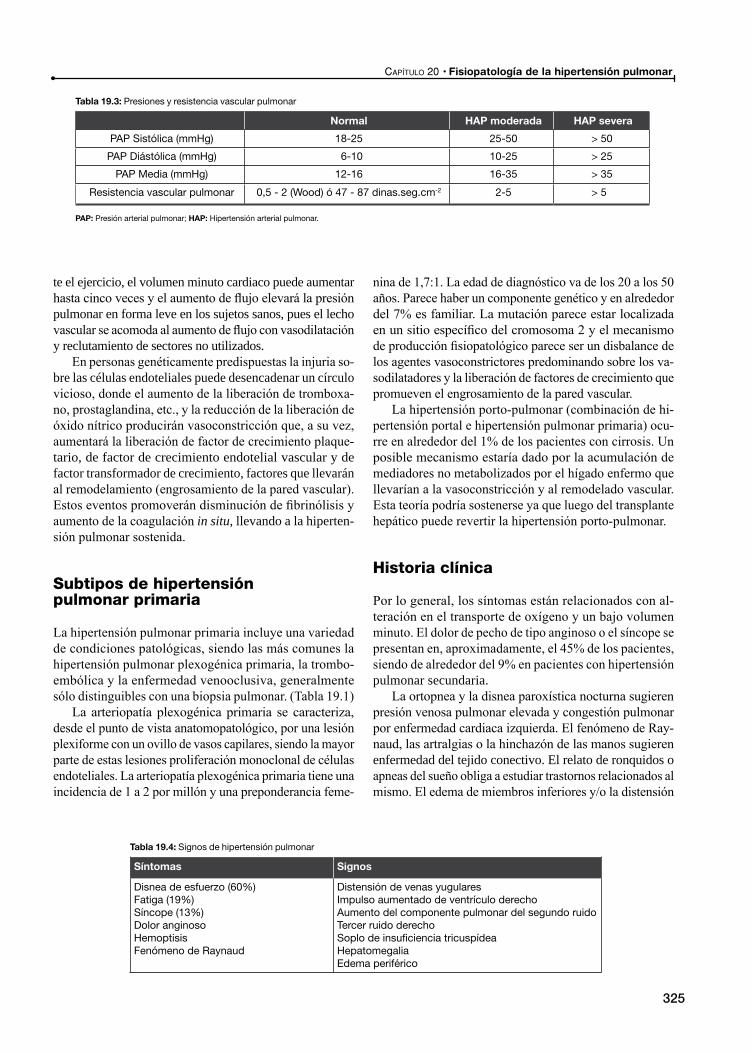
Fisiopatología de la hipertensión pulmonar
325
Tabla 19.3:
Normal HAP moderada HAP severa
18-25 25-50 > 50
6-10 10-25 > 25
12-16 16-35 > 35
Resistencia vascular pulmonar 0,5 - 2 (Wood) ó 47 - 87 dinas.seg.cm-2 2-5 > 5
PAP: HAP: Hipertensión arterial pulmonar.
te el ejercicio, el volumen minuto cardiaco puede aumentar
pulmonar en forma leve en los sujetos sanos, pues el lecho
y reclutamiento de sectores no utilizados.-
bre las células endoteliales puede desencadenar un círculo -
-tario, de factor de crecimiento endotelial vascular y de factor transformador de crecimiento, factores que llevarán
in situ, llevando a la hiperten-
Subtipos de hipertensión pulmonar primaria
-
-
-
--
Historia clínica
-
pulmonar secundaria.
-
enfermedad del tejido conectivo. El relato de ronquidos o
Tabla 19.4: Signos de hipertensión pulmonar
Síntomas Signos
Disnea de esfuerzo (60%)Fatiga (19%)Síncope (13%)Dolor anginosoHemoptisisFenómeno de Raynaud
Distensión de venas yugularesImpulso aumentado de ventrículo derechoAumento del componente pulmonar del segundo ruidoTercer ruido derechoSoplo de insuficiencia tricuspídea Hepatomegalia Edema periférico

Gelpi - Donato Fisiopatología cardiovascular
326
--
Examen físico
-a -v
prominente. La disnea de esfuerzo y el cansancio son los primeros síntomas, sumándose dolor torácico con el es-fuerzo, mareos y eventualmente síncope, en los casos más
izquierdo por la arteria pulmonar dilatada. Los princi-
-
con onda v prominente, edema de miembros inferiores, -
también un clic eyectivo temprano producido por la brusca
un latido en la zona paraesternal izquierda producido por
-
-
-
de derecha a izquierda, severa
de tambor hacen sospechar la posibilidad de enfermedad
--
1 1 -5 . El ventrículo derecho distendido
-
--
mento de la amplitud de la onda P en DII y BIRD todos estos
-monar son: aumento de las sombras de las arterias pulmo-
árbol de invierno -teria pulmonar alcanza diámetros mayores a los 18 mm. Aparecen también líneas de Kerley B y C, con valores
Figura 19.1: Se observa aumento de las sombras de las arterias pulmonares mayores hiliares y atenuación vascular pulmonar periférica (árbol de invierno). Además, se evidencia el aumento del tamaño del ventrículo derecho en la posición de frente como aplanamiento del borde inferior izquierdo y separación de la punta cardiaca de la cúpula diafragmática (indicado por las flechas).

Fisiopatología de la hipertensión pulmonar
327
-
-10
-
Evaluación del paciente con sospecha de hipertensión pulmonar
-
-
, donde v es la velocidad de jet
-
y del ventrículo derechos, con dimensiones ventriculares
del septum interventricular con motilidad anormal. La pared septal y posterior del ventrículo izquierdo está aumentada.
-
screeningdel método utilizado sino también de la prevalencia de la
eco stress
-
Evaluación de enfermedades del tejido conectivo y/u otros factores capaces de producir hipertensión pulmonar
idiopática tiene anticuerpos antinúcleo positivos y alrededor
observarse en cualquier enfermedad del tejido conectivo es más frecuente en la esclerosis sistémica, variando la preva-
-tes en estos pacientes. Los pacientes con esclerodermia con
-cuerpos, comienzo de la enfermedad en la posmenopausia
En otras enfermedades del tejido conectivo, tales como -
-
anticardiolipina y antifosfolípidos.-
Otras enfermedades y anormalidades asociadas
-11 12
-
Figura 19.2:septum
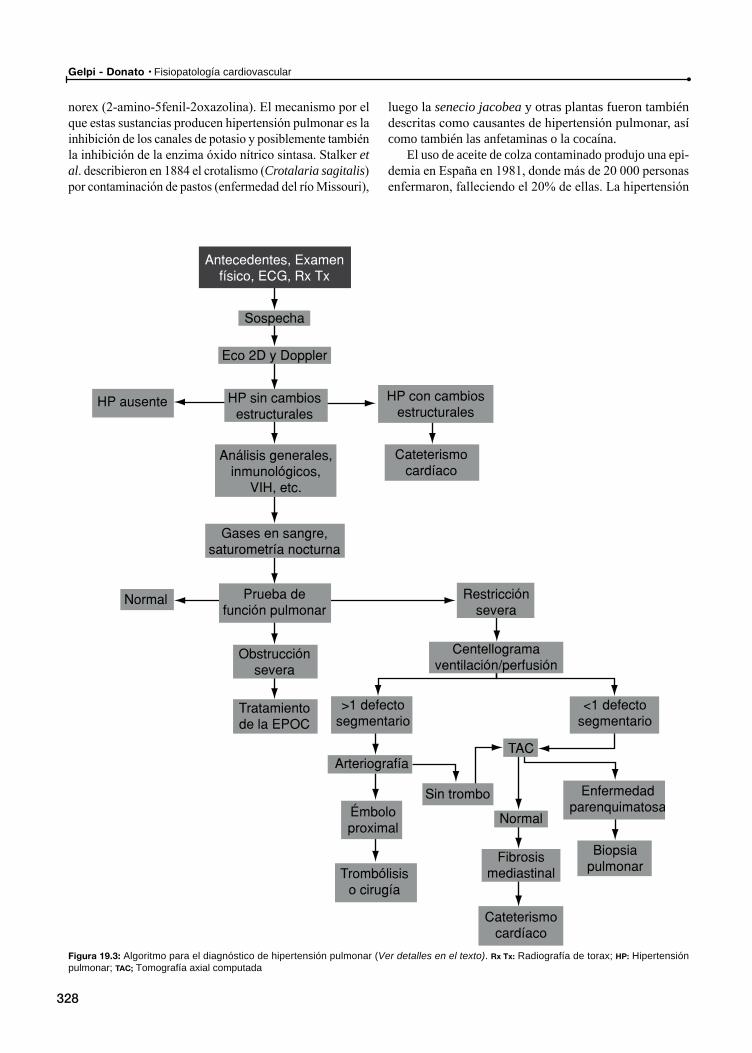
Gelpi - Donato Fisiopatología cardiovascular
328
et al Crotalaria sagitalis
senecio jacobea y otras plantas fueron también
como también las anfetaminas o la cocaína. El uso de aceite de colza contaminado produjo una epi-
Figura 19.3: Algoritmo para el diagnóstico de hipertensión pulmonar (Ver detalles en el texto). Rx Tx: Radiografía de torax; HP: Hipertensión pulmonar; TAC; Tomografía axial computada

Fisiopatología de la hipertensión pulmonar
329
pulmonar se observa también en la enfermedad de células -
enfermedades tiroideas.
Enfermedad tromboembólica
13 -
Para indicar la endarterectomía, los trombos deben co-menzar en el tronco o en las arterias lobares pulmonares. La
ventricular izquierdo, arterias pulmonares centrales dilata-das y, en ocasiones, trombos dentro de ellas. La ausencia de
14 permite evaluar anomalías --
15 permite en ocasiones --
Enfermedad parenquimatosa pulmonar como causa de hipertensión pulmonar
16 --
cardiaco. Otro factor a tener en cuenta es la policitemia secundaria, que aumentando la viscosidad, incide en el
17 se ha -
el remodelado de los vasos pulmonares se observa en
-
no se indican en la EPOC por lo antes mencionado. La
-
-
del lecho vascular por injuria de las células endoteliales.
Hipertensión arterial pulmonar y trastornos respiratorios durante el sueño
-
-
-
-
-
-Figura 19.4: Se observa agrandamiento ventricular izquierdo, arterias pulmonares centrales dilatadas. La ausencia de estos signos no excluye la presencia de enfermedad.

Gelpi - Donato Fisiopatología cardiovascular
330
contractilidad miocárdica, al tiempo que aumenta el con--
-
leve o moderado.
-monar son ampliamente conocidos aunque la prevalencia
cardiaca, enfermedad parenquimatosa pulmonar y obesi-
-
tratamiento con CPAP en caso de encontrarse apneas que
Biopsia pulmonar
lo que no se recomienda efectuarla rutinariamente.
Severidad y pronóstico de la hipertensión arterial pulmonar
-rias perspectivas:
-
-brevida
Con respecto al punto 1, si bien el ecoDoppler es útil,
sintomática correlaciona bien con la sobrevida. La prueba de la caminata de seis minutos 18 provee, en forma sencilla,
-
Pronóstico de la hipertensión arterial pulmonar
pocos meses mientras que otros viven decenios. La causa
-plejo:
tricuspídea
-se en forma no invasiva con nuevas técnicas de resonancia
ventrículo derecho, ya que esta técnica no se ve afectada
-
-dal19
el tiempo de sobrevida. Los niveles de péptido cerebral se relacionan con las alteraciones hemodinámicas en los
Tratamiento quirúrgico

Fisiopatología de la hipertensión pulmonar
331
stents -
colocados en la aorta descendente. La endarterectomía está indicada en aquellas situaciones en las que los trom-bos se encuentran en el tronco o en las arterias lobares pulmonares. En situaciones muy especiales el transplante pulmonar es una terapia a considerar. Las indicaciones
-
Tratamiento médico
-
20
-
y los efectos secundarios frecuentes. Por todo esto, el trata-21
Las pruebas de vasorreactividad son parte importante de
-
-
-terias pulmonares, que sumadas a su corta vida media la
-
vía oral, con warfarina o acenocumarol, y el suplemento
Otros medicamentos22
el ambrisentan.-
-
enfermedad hepática.
-zem durante el cateterismo derecho, se correlaciona con
bloqueantes cálcicos.--
--
co. Esto se debe a la presencia de remodelado del lecho vascular y trombosis microvascular, siendo estos factores
-
ha sido aprobado su uso en forma inhalatoria. El uso de
-
24 -
25, como la esclerosis sistémica
-matorias perivasculares del tipo de linfocitos del tipo T y
-mes. La fractalquina es una quemoquina especial que jue-
la pared vascular.26
-27
--
aumentada del factor de crecimiento plaquetario y RANTE
-
este transportador que controla la hiperplasia muscular

Gelpi - Donato Fisiopatología cardiovascular
332
vías de tratamiento. El uso de microalineamientos permi-
sendas en ese sentido. Además, en diversos trabajos y consensos se proponen
Primera Línea: Bloqueantes de los canales de calcio
y/o edemas. El remodelado vascular y las microtrombosis pueden ser causa de no respuesta en muchos casos, por lo
como se ha probado, mejora la sobrevida. Otros tratamientos pueden ser: prostaciclina intrave-
nosa, que tiene como efectos secundarios diarrea, rubor,
-
-
-
son básicamente tres:
canal de potasio dependiente. Como ya mencio-
-constrictores derivados de la célula endotelial, las
-
de la vasculatura pulmonar cuando la enfermedad
-
pulmonar, por tromboembolismo pulmonar, en la es-
Las enfer- de iz-
quierda a derecha, como los que se encuentran en las comunicaciones interauriculares o interventriculares,
-fermedad de la válvula mitral, la pericarditis cons-
son las causas habituales en estos casos. La obstruc--
venoclusiva son causas poco frecuentes. En base a lo antedicho podría bosquejarse un esquema de tra-
pulmonar
-ricular, transplante pulmonar
Las terapias futuras son:
Bibliografía
1. Romberg, E. “Ueber Sklerose der Lungen Arterie”. Dsch Arch Klin Med 1891;48:197-206.
2. Dresdale DT, Shultz M, Mitchom RJ. “Primary Pulmonary Hypertension: Clinical and Hemodynamic Study”. Am J Med 1951;11:686-705.
3. Krogh, A. “The supply of oxygen to the tissues and the regulation of the capillary circulation”. J Physiol. 1919;20;52:457-74.
4. Wagenvoort, CA. “Morphological substrate for the reversibility and irreversibility of pulmonary hypertension”. Eur Heart J. 1988;9:7-12.
5. Robles AM, D Shure. “Gender issues in pulmonary vascular di-sease”. Clin Chest Med 2004;25(2):373-377.
6. Simonneau G, Robbins IM, Beghetti M et al. “Updated Clinical Classification of Pulmonary Hypertension . J Am Coll Cardiol, 2009; 54:43-54.
7. Raymond RJ, Hinderliter AL, Willis PW, Ralph D, Caldwell EJ, Williams W, Ettinger NA, Hill NS, Summer WR, de Boisblanc B, Schwartz T, Koch G, Clayton LM, Jobsis MM, Crow JW, Long W. “Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension”. J Am Coll Cardiol. 2002;39:1214-9.
8. Gibbons GH, VJ Dzau. “The emerging concept of vascular re-modelling”. N Engl J Med. 1994;330(20):1431-8.
9. Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, Badesch D, Voelkel NF. “Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hyper-tension”. Am J Respir Crit Care Med. 1999;159(6):1925-32.

Fisiopatología de la hipertensión pulmonar
333
10. Fedullo PF, Auger WR, Kerr KM, Rubin LJ. “Chronic thromboembo-lic pulmonary hypertension”. N Engl J Med. 2001;345(20):1465-72.
11. Fishman, AP. “Aminorex to FEN/Phen. An epidemic foretold”. Circulation 1999;99(1):156-61.
12. Archer SL, Djaballah K, Humbert M, Weir KE, Fartoukh M, Dall’ava-Santucci J, Mercier JC, Simonneau G, Dinh-Xuan AT. “Nitric oxide deficiency in fenfluramine- and dexfenfluramine-induced pulmonary hypertension”. Am J Respir Crit Care Med. 1998;158(4):1061-7.
13. Bailey CL, Channick RN, Auger WR, Fedullo PF, Kerr KM, Yung GL, Rubin LJ. “High probability perfusion lung scans in pulmonary venoocclusive disease”. Am J Respir Crit Care Med. 2000;162(5):1974-8.
14. Tardivon AA, Mousseaux E, Brenot F, Bittoun J, Jolivet O, Bourroul E, Duroux P. “Quantification of Hemodynamics in primary pul-monary hypertension with magnetic resonant imaging”. Respir Crit. Care Med 1994;150(4):1075-1080.
15. Selim M, Arcasoy J D, Christie V A, Ferrari M, St Jhon S, Zisman D, Blumenthal N, Pochettino A, Kotloff R. “Echocardiographic As-sessment of Pulmonary Hypertension in Patients with Advanced Lung Disease”. Am J Respir Crit Care Med. 2003;167:735-740.
16. Rafanan AL, Golish JA, Dinner DS, Hague LK, Arroliga AC. “Noc-turnal hypoxemia is common in primary pulmonary hypertension”. Chest 2001;120(3):894-9.
17. Riley MS, Porszasz J, Engelen MP, Brundage BH, Wasserman K. “Gas exchange responses to continuous incremental cycle ergometry exercise in primary pulmonary hypertension in humans”. Eur J Appl Physiol. 2000;83(1):63-70.
18. Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, Nakanishi N, Miyatake K. “Clinical correlates and prognostic significance of six-minute walk test in patients with primary pul-monary hypertension. Comparison with cardiopulmonary exercise testing”. Am J Respir Crit Care Med. 2000;161:487-92.
19. Jones AT, Hansell DM, Evans TW. “Quantifyng pulmonary per-fusion in primary pulmonary hypertension using electron-beam computed tomography”. Eur Resp J. 2004;23(2):202-207.
20. Kessler R, Chaouat A, Weitzenblum E, Oswald M, Ehrhart M, Apprill M, Krieger J. “Pulmonary hypertension in the obstructive sleep apnoea syndrome: prevalence, causes and therapeutic conse-quences”. Eur Respir J. 1996;9(4):787-94.
21. Nagaya, N. “Drug therapy of primary pulmonary hipertension”. Am J Cardiovasc Drugs. 2004;2:75-85.
22. Sanjay Mehta. “Drug therapy for pulmonary arterial hypertension. What’s on the menu today?” Chest 2003; 124:2045-2049.
23. Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, Wang SH, Modry D, Archer SL. “Long-term treatment with oral sildenafil is safe and improves functional capacity and he-modynamics in patients with pulmonary arterial hypertension”. Circulation Oct 28 2003;108(17):2066-9.
24. Adnot S, S Eddahibi. “Genetic and physiopathology or pri-mary and secondary artery hypertension”. Bull Acad Natl Med 2003;187(8):1529-42.
25. Voelkel NF, Cool C, Lee SD, Wright L, Geraci MW, Tuder RM. “Primary pulmonary hypertension between inflammation and cancer”. Chest. 1998;114:225S-230S.
26. Dorfmuller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, Capron F, Coulomb-Lhermine A, Marfaing-Koka A, Simonneau G, Emilie D, Humbert M. “Chemokine RANTES in severe pulmonary arterial hypertension”. Am J Respir Crit Care Med. 2002;165(4):534-9.
27. Opravil M, Pechere M, Speich R, Joller-Jemelka HI, Jenni R, Russi EW, Hirschel B, Luthy R. “HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study”. Am J Respir Crit Care Med. 1997;155(3):990-5.
28. McGoon, MD. “The assessment of pulmonary hypertension”. Clin Chest Med. 2001;22(3):493-508.
29. Cool CD, Rai PR, Yeager ME, Hernández-Saavedra D, Serls AE, Bull TM, Geraci MW, Brown KK, Routes JM, Tuder RM, Voelkel NF. “Expression of human herpesvirus 8 in primary pulmonary hypertension”. N Engl J Med. 2003;349(12):1113-22.


335
Fisiopatología de las arritmias 21
Historia de la actividad eléctrica del corazón. De los experimentos de Galvani a la heterogeneidad de los potenciales de acción ventriculares
-mo de acoplamiento eléctrico-mecánico. El estudio de la
-cia. 1
los primeros estudios sobre bioelectricidad en músculo de rana.2
electricidad animal. -
3
-
estimular el nervio, pero que ésta no estaba involucrada con su funcionamiento normal y apoyaba la doctrina de una fuerza vital desconocida. Cuando obtuvo una copia
-mino potencial de acción para relacionar las variaciones
4
-5
reotomo
-electrómetro capilar desarrollado
6-7
-
8
cuerda y describe en sus publicaciones los primeros re-
9-10
y eran necesarios 5 operadores.11
--
boratorio de Thomas Lewis en Londres y a Alfred Cohn en 12
descubrimiento. Las arritmias cardiacas eran pobremente entendidas has-
-
conocemos, es el resultado de una serie de descubrimientos
rutinariamente utilizadas incluye a las tres de los miembros, --
13
desarrollan una nueva técnica, con microelectrodos intra-celulares. Los microelectrodos eran de vidrio con una pun-
injuria. 14 La técnica fue subsecuentemente perfeccionada --

Gelpi - Donato Fisiopatología cardiovascular
336
15-16
de Purkinje de cabrito, con la técnica del microelectrodo intracelular. 17
membrana de reposo; el aumento de la permeabilidad al
1 Además, -
bre el cocaína. 18
-
solo las corrientes de sodio y potasio.1 A partir de esta época comienza a desarrollarse el estudio de los canales
el primer estudio con la técnica de clampeo de voltaje, y -
19
En 1988, Litovsky y Antzelevich introducen el concep--
cias entre las células endocárdicas y epicárdicas del cora-20 Hasta entonces se creía que los ventrículos
estaban formados por dos tipos celulares: los del sistema
--
en la profundidad del subepicardio del ventrículo derecho e izquierdo canino. Estas células presentan características
-
M21, posteriormente encontradas en ventrículos de cerdos, cobayo, conejo, felino y humanos. 22-26 -
las células endocárdicas, las células epicárdicas y las cé-
lulas M, con características distintivas respecto de la fase
el devenir de la historia y de la ciencia básica abren una
-
El estudio del potencial de acción
El
-menos, la diferente sensibilidad a fármacos y el estudio de
-
Figura 21.1: Luigi Galvani en su laboratorio (1791): los efectos de la electricidad sobre el movimiento muscular
Figura 21.2: El electrómetro capilar de Lippmann (1873), utilizado por Waller. Estaba formado por un delgado capilar de vidrio que contenía una columna de mercurio y ácido sulfúrico. El potencial eléctrico era censado por dos alambres conectados al ácido sulfúrico y al mercurio. Con los cambios en la diferencia del potencial eléctrico se producían movimientos del menisco de la columna de mercurio. La posición del menisco podía ser observada en un microscopio o proyectada en una pantalla. Si esta pantalla era cubierta por un material fotosensible y además se le impartía movimiento perpendicular al eje del capilar, se obtenían las variaciones del potencial eléctrico en función del tiempo. En el panel inferior se observa que el movimiento estaba dado por un particular dispositivo, armado con un vagón de tren de juguete; por detrás se observa el primer registro del potencial eléctrico desde la superficie corporal humana; en su parte superior se registra el tiempo, en la parte media, el trazado del pulso y abajo, en blanco, el electrograma.
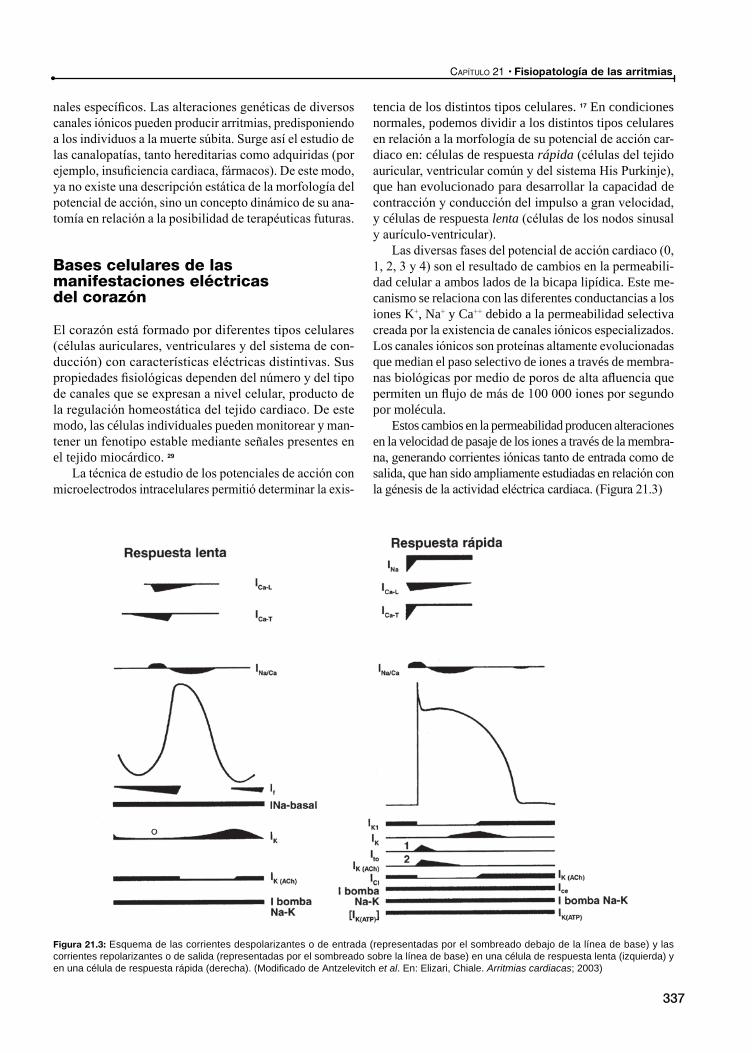
Fisiopatología de las arritmias
337
-
Bases celulares de las manifestaciones eléctricas del corazón
-
modo, las células individuales pueden monitorear y man-
el tejido miocárdico. 29
-
tencia de los distintos tipos celulares. 17 En condiciones normales, podemos dividir a los distintos tipos celulares
-diaco en: células de respuesta rápida
que han evolucionado para desarrollar la capacidad de
y células de respuesta lenta
-dad celular a ambos lados de la bicapa lipídica. Este me-canismo se relaciona con las diferentes conductancias a los iones K , Na y Ca debido a la permeabilidad selectiva
Los que median el paso selectivo de iones a través de membra-
por molécula. Estos cambios en la permeabilidad producen alteraciones
en la velocidad de pasaje de los iones a través de la membra-
Figura 21.3: Esquema de las corrientes despolarizantes o de entrada (representadas por el sombreado debajo de la línea de base) y las corrientes repolarizantes o de salida (representadas por el sombreado sobre la línea de base) en una célula de respuesta lenta (izquierda) y en una célula de respuesta rápida (derecha). (Modificado de Antzelevitch et al. En: Elizari, Chiale. Arritmias cardiacas; 2003)

Gelpi - Donato Fisiopatología cardiovascular
338
La fase 0 ó de despolarización rápida-
se produce un rápido ascenso del potencial transmem-
se alcanza el potencial umbral, los canales de Na voltaje
abren en forma abrupta, de modo que la membrana pasa de ser permeable al K a ser altamente permeable al Na
CaT, ICaLpotencial
potencial umbral
La fase 1 ó de repolarización precoz
de salida de potasio denominada Ito. 20 La corriente Ito
to1--
nopiridina.-
to1 contribuiría en forma -
corriente I
taquiarritmias. La corriente Ito, estudiada mediante técnicas de clampeo de voltaje, es cinco veces mayor en el epicar-
-
Ito--
ventriculares por mecanismo de reentrada denominadas .
La fase 2 ó plateau (meseta)-
diacos es notoriamente mayor si se la compara con las
28
-
Durante este periodo la conductancia a los iones decrece
-
la frecuencia cardiaca. -
-Ca -
K
Na
Capor dos tipos de canales, denominados T y L. La corriente ICa-L
-Ca-T está involucrada
-
Ca-L, que depende del -
lar, es la determinante principal del comienzo de la repo-
rápido y lento. Es así como el aumento del calcio intrace-
confundida con la corriente lenta de sodio denominada co-
y los anestésicos locales.
La fase 3 ó de repolarización rápida

Fisiopatología de las arritmias
339
Kr e IKs
K1
K
Kr Ksson abundantes en las aurículas, los ventrículos y el sis-tema His-Purkinje, pero escasos en las células nodales. 29
Kr e IKs-
tes antiarrítmicos ejercen efectos sobre las corrientes IKr e IKs kr Kr y Iks
30
En el proceso de -
La corriente I ) es sensible a los niveles citoplasmá-ticos de ATP. Los canales correspondientes están cerrados a niveles de ATP citoplasmático normales pero se abren
31
La corriente I se activa cuando la acetilcolina se une al receptor muscarínico de la membrana plasmática, con el resultado previsible de la salida de potasio, el acor-
potencial de membrana en reposo hacia valores más ne-
nivel de las aurículas y del sistema especializado. 32
La corriente Ik que se activa en presencia de con-
-
cálcica. 33
La fase 4: El potencial de reposo y la despolarización diastólica espontánea
lados de la membrana celular, debido a su permeabilidad selectiva, como consecuencia se establece una diferen-cia de voltaje denominada potencial de membrana o de reposo celular.
Como hemos mencionado previamente, en las células cardiacas el valor del potencial de membrana de reposo es variable dependiente del tipo celular: alrededor de -50
-lulas His-Purkinje y de -80 a -90 a nivel las células del
-
dulos sinusal y aurículo-ventricular, del haz de His y de las
-
Potencial de membrana de reposo de las células auriculares y ventriculares
-
a alta permeabilidad selectiva de la membrana al potasio,
k1, -
-je correspondiente y al potencial de membrana de reposo. Del mismo modo se describe una baja permeabilidad al Na y Ca ≈dentro de la célula, tales como las proteínas, no pueden difundir junto al
es 0 podemos
EK i 0 El término de la izquierda Ek representa la diferen-
El término de la derecha representa la diferencia de po-i 0
. Para , Na ,
Cl- potencial de membrana, se debe considerar la permeabilidad relativa de cada uno de ellos, además de
iones:
k i / Pk 0 Na i / PNa 0 Cl
-i / PCl
-0

Gelpi - Donato Fisiopatología cardiovascular
340
-34
La bomba Na –K -traciones intracelulares de ambos iones constantes, impi-
que ine-vitablemente ocurrirían como consecuencia de la continua
, con
El automatismo normal en el nódulo sinusal y el sistema His Purkinje
de His y el sistema His Purkinje, para activar al músculo ventricular.
-tricular y las células de Purkinje tienen actividad espontánea llamada actividad de marcapasos. Estas células se carac-terizan por poseer un potencial de membrana no constante
-nea lleva el potencial de membrana desde los valores más
-
--
-
-
potencial de mar-capasos
b
k -
a través de canales voltaje-dependiente no selectivos, que
f
por la y previene el cese de la actividad nodal durante la estimu-
-lenta -
lulas de respuesta rápida
Mecanismos de las arritmias
Las
Figura 21.4: Clasificación de los mecanismos de las arritmias cardiacas. (Modificado de Antzelevitch et al. En: Elizari, Chiale. Arritmias cardiacas; 2003)
gatillada
anillo
en fase 2

Fisiopatología de las arritmias
341
-
arritmias, a veces potencialmente letales. De la posibilidad -
cio actuar, tendremos la respuesta hacia una terapéutica
Los mecanismos responsables de las arritmias cardia-cas se dividen en:
Alteraciones en la formación del impulso-
por comenzar en forma espontánea, mientras que un ritmo -
te. Las alteraciones en los ritmos automáticos pueden ser
deberse a posdespolarizaciones precoces o tardías.
Automatismo normalEl automatismo es la propiedad de las células cardiacas de
-sal como el aurículoventricular o el sistema His-Purkinje presentan actividad automática, correspondiente a la fase
-
El automatismo normal es el responsable del ritmo si-
presentes ante una falla en la salida del estímulo desde centros de comando superiores a los del latido de escape.
-lares de potasio modulan la pendiente de la DDE: el estí-
El aumento del automatismo normal es la causa de al-
taquicardia sinusal, atribuido al aumento del automatismo sinusal y al ritmo idioventricular acelerado, atribuido al aumento del automatismo del sistema de His-Purkinje. 35
El automatismo anormalOcurre en condiciones que reducen el potencial de mem-
-
se puede observar en la isquemia miocárdica y el infar-
-pontánea en el automatismo anormal es similar al del au-
-diente del tiempo, de la corriente Ik junto con un aumento
Ca. 36
observado también en células humanas aisladas de ven-37
--
-licos o quizás aquellas provenientes de las venas pulmona-
38
Pospotenciales y actividad gatilladaLos pospotenciales o posdespolarizaciones fueron de-
39 Los pospotenciales dependen
en forma espontánea y por lo tanto no son automáticos. -
nivel del potencial umbral, se produce un nuevo potencial gatillada. Por
-
Posdespolarizaciones precocesLos pospotenciales o posdespolarizaciones precoces
40, si bien dependen de la especie animal con-siderada, del tipo de tejido así como del método uti-lizado, tienen como característica común una crítica
todos los casos. 41
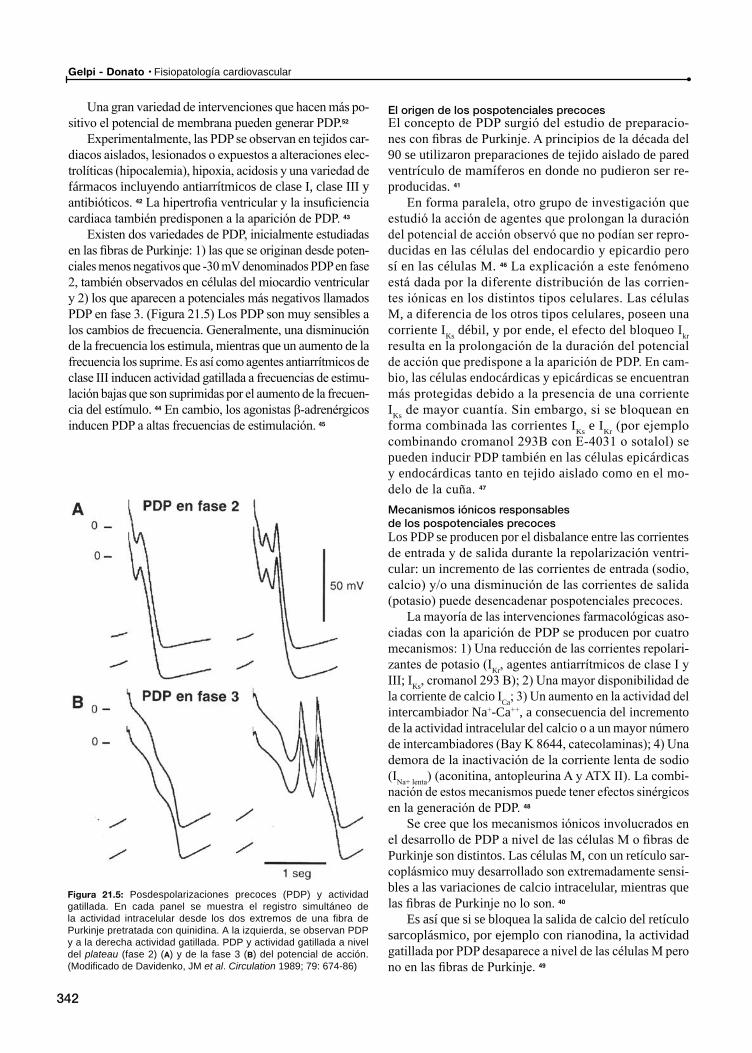
Gelpi - Donato Fisiopatología cardiovascular
342
-52
--
fármacos incluyendo antiarrítmicos de clase I, clase III y 42
43
-
de la frecuencia los estimula, mientras que un aumento de la
--
cia del estímulo. 44
45
El origen de los pospotenciales precoces-
90 se utilizaron preparaciones de tejido aislado de pared ventrículo de mamíferos en donde no pudieron ser re-producidas. 41
-ducidas en las células del endocardio y epicardio pero sí en las células M. 46
-
M, a diferencia de los otros tipos celulares, poseen una corriente IKs débil, y por ende, el efecto del bloqueo Ikr
-bio, las células endocárdicas y epicárdicas se encuentran
IKsforma combinada las corrientes IKs e IKr
pueden inducir PDP también en las células epicárdicas y endocárdicas tanto en tejido aislado como en el mo-
47
Mecanismos iónicos responsables de los pospotenciales precocesLos PDP se producen por el disbalance entre las corrientes
-cular: un incremento de las
-
-Kr
III; IKsla corriente de calcio ICaintercambiador Na -Ca , a consecuencia del incremento de la actividad intracelular del calcio o a un mayor número
-
48
Purkinje son distintos. Las células M, con un retículo sar--
bles a las variaciones de calcio intracelular, mientras que 40
Es así que si se bloquea la salida de calcio del retículo sarcoplásmico, por ejemplo con rianodina, la actividad
49
Figura 21.5: Posdespolarizaciones precoces (PDP) y actividad gatillada. En cada panel se muestra el registro simultáneo de la actividad intracelular desde los dos extremos de una fibra de Purkinje pretratada con quinidina. A la izquierda, se observan PDP y a la derecha actividad gatillada. PDP y actividad gatillada a nivel del plateau (fase 2) (A) y de la fase 3 (B) del potencial de acción. (Modificado de Davidenko, JM et al. Circulation 1989; 79: 674-86)

Fisiopatología de las arritmias
343
El rol de las posdespolarizaciones precoces en las arritmias cardiacas
estaría involucrada en el mecanismo de inicio de las taqui-cardias ventriculares polimorfas del tipo torsión de puntas
-tario y adquirido. Las PDP serían, además, responsables
-
-Na.
Posdespolarizaciones tardías-
-
frecuencias cardiacas relativamente rápidas. Toda vez
como para alcanzar el valor del potencial umbral, se activan los canales rápidos de sodio y se desencadena
50
-centraciones crecientes de calcio, se producía una serie de poscontracciones espontáneas. 51
-
51 Estudios posteriores, realizados
alcanzar el potencial umbral e iniciar actividad automática aun cuando la actividad del marcapaso normal estuviera deprimida. 53
Causas y mecanismos subyacentes de la actividad gatillada por posdespolarizaciones tardías
-
o ausente en condiciones de normalidad. 38 Cuando la -
momento oportuno del ciclo cardiaco, es decir, cuando la -
cular, se transforma en un evento disparador de diversos
derivar en La corriente de entrada
Na /Ca. 54
Ejemplos clásicos de pospotenciales tardíos son los -
laminas en la aurícula, ventrículo, válvula mitral y tejido del seno coronario; así como también en corazones insu-
del área de miocardio infartado. 55
En la pared ventricular, las PDT han sido observadas a -
46, 50, 56
densidad de la corriente IKs presente en el endocardio y el
29 Más aún, se -
vida por el isoproterenol induce PDT en perros y cobayos a nivel del endocardio y epicardio. 57 Así queda estableci-
ventricular. También las PDT han sido descritas en miocitos ais-
lados de las venas pulmonares y podrían ser responsables de ciertos tipos de 58
-
/K , cuya
-dor Na - Ca , que se traduce como un aumento en la con-
fármaco.-
-
--
lina, en concentraciones similares a las que se observan
-lar potencia las PDT inducidas por isoproterenol. 59 Las
calcio desde el retículo sarcoplásmico, con lo que contri-buyen a manifestar las PDT, mientras que las concentra-

Gelpi - Donato Fisiopatología cardiovascular
344
por el retículo sarcoplásmico suprimiéndolas. También los -
amiodarona suprimen las PDT
lo tanto, del calcio intracelular. 60 El pinacidil, activador
K-ATP56 Los fármacos que reducen
-tura, como la rianodina, pueden suprimir las PDT. 38
Arritmias cardiacas y actividad gatillada por posdespolarizaciones tardías
-
evidencias indirectas del vínculo. El escenario clínico
-
-
-
objeto de bloquear la corriente de calcio transarcolémica 61
Dentro de las arritmias que podrían ser desencadenadas -
rritmias ventriculares idiopáticas; los ritmos idioventri-
62
Figura 21.6: Posdespolarizaciones tardías (PDT) inducidas por digital en células M. Efectos de la acetilestrofantidina (AcS) sobre la actividad transmembrana de preparado multicelular de una célula epicárdica (Epi), endocárdica (Endo) y M. La concentración extracelular de potasio es de 4mM. A. Control. B. Después de 90 minutos de exposición a AcS (10-7g/ml). Cada panel muestra los últimos tres latidos de un tren de 10 latidos estimulados a una longitud de ciclo constante (LCB) de 250 mseg. Después de cada tren de estimulación basal hay una pausa de tres segundos. La AcS indujo PDT tardías en la célula M, pero no en la célula endocárdica o epicárdica. Clin Electrophysiol 1991; 14: 1714-20)

Fisiopatología de las arritmias
345
acoplamientos cortos o el marcapaseo, a frecuencias más
frecuencia de escape de la PDT. Los estímulos prematuros causan un efecto similar. Puede inferirse con estos hallaz-
inducidas por este mecanismo no son suprimibles fácil-mente y pueden ser desencadenadas en forma espontánea o inducidas por marcapaseo. 38
-trantes son: la taquicardia ventricular sensible a la adeno-sina e inducida por el ejercicio; la taquicardia ventricular uniforme repetitiva mediada por el AMP cíclico; las ta-
-diaca. 63
Alteraciones en la conducción del impulso
-
-do un impulso atraviesa con lentitud la zona de bloqueo;
acuerdo al sitio en que ocurren. Así, estos pueden lo-
en el fascículo de His o en las ramas del haz de His. Los -
bradiarritmias por bloqueos, sino que
un mecanismo denominado de reentrada.
Mecanismo de reentrada-
hacia los ventrículos a través de los haces internodales, nodo aurículoventricular, tronco común del haz de His,
-tribuida en el seno del miocardio ventricular. Una vez
-
de despolarizarse. El concepto de reentrada implica que
35 El mecanismo de reentrada es el responsable de la mayoría de las arritmias supra-ventriculares y ventriculares.
Mecanismo clásico de reentradareentrada, el del anillo repre-
senta su forma más simple. Mayer, en sus publicaciones a
-
64
circular y lo correlaciona con casos clínicos de taquia-rritmia. 65
-
del movimiento circular por el corte del circuito en cual-
insertan en el miocardio ventricular. A partir de este mode-
acopladas de corazones de mamíferos. 66
De tal forma, las condiciones necesarias para que se --
-
-
unidireccional
una zona de conducción lenta
de no ser así, el impulso encontrará tejido refractario por delante y el ritmo circular se interrumpirá. En la clínica,
-
ventriculares. 38
-
apropiado para crear un obstáculo anátomo-funcional con

Gelpi - Donato Fisiopatología cardiovascular
346
anató-mica
anisotrópica:-
unidireccional. En el mecanismo de el impul-
opuestas. 67
El circuito puede presentar diferentes localizaciones:-
trada auricular.
a. En el músculo. -miento circular. El circuito de reentrada es muy
-
y de los ritmos hiperactivos del ventrículo por re-entrada.
b. -
c. En un circuito con participación de la unión A-V. -
-
parte de lo esencial del circuito de reentrada. Un ejemplo clásico lo proporcionan las taquicardias
-
ejemplo típico del circuito sistema His-Purkinje – -
paralelo con la vía normal. Habitualmente, la vía anormal conduce con rapidez, pero al mismo tiem-po posee periodos refractarios relativamente lar-
-lar durante el periodo refractario efectivo de la vía
través de la vía normal hacia los ventrículos. Lle-
-rículas, estableciéndose entonces un movimiento
-68
tipo de mecanismo llamado de reentrada intramural.
el ventrículo determina las condiciones favorables para el desarrollo de reentrada. 69
-
no es necesaria en este tipo de reentrada debido a que la
70
Reentrada en fase 2
de isquemia, en el subepicardio del ventrículo derecho. 71 gatillo
de movimiento circular de reentrada. Es producido por la
to es más prominente. 20
-
el domo. Esa pérdida se produce por el predominio de las Figura 21.7: Modelos anulares de reentrada. A: Esquema anular de reentrada. B: Mecanismo de reentrada en el síndrome de Wolff Parkinson White que involucra al nódulo aurículoventricular y una vía accesoria aurículoventricular. C: El mecanismo de reentrada en un asa Purkinje–músculo propuesto por Schmitt y Erlanger. (Modificado de: Schmitt et al. Am J Physiol 1928; 87: 326-47)

Fisiopatología de las arritmias
347
la muesca
to
Ca71 Bajo estas circunstancias el
plateau del
que pueden iniciar uno o más ciclos de movimientos cir-culares de reentrada. 71
de potasio como el pinacidil, los bloqueantes de los canales -
-72 A su vez, los bloqueantes de los
canales Ito -
neidad eléctrica e interrumpen la reentrada. La arritmia
aspecto de una taquicardia ventricular multiforme, que se
-
ventricular observada en el 35 Las bases celulares de este síndrome serán objeto de otra sec-
La heterogeneidad eléctrica celular en el miocardio ventricular
La pared ventricular está formada por distintos tipos ce-lulares: las células endocárdicas, epicárdicas y M, que
-
las células epicárdicas llamada 20 Más aún,
-las M se debe a la presencia de una corriente IKs atenuada,
Nacorriente de intercambio sodio/calcio aumentada. Esas di-ferencias en las intensidades de las corrientes determinan
-29, 73
-lulas epicárdicas con las endocárdicas, se observa que las
20, 73 El potencial de ac-
típica de en el endocardio y más acentuada en el ventrículo derecho que en el izquierdo. 69, 74
más pro-minente en el epicardio que en el endocardio ha sido descrita en los modelos canino in vitro e in vivo, miocitos aislados de
35 En el ventrículo derecho,
pronunciada que en el ventrículo izquierdo, tanto a bajas -
Figura 21.8: Reentrada en fase 2 en un modelo de isquemia en una preparación multicelular canina del epicardio ventricular derecho. Registros obtenidos de cuatro sitios señalados arriba a la derecha. Luego de 35 minutos de isquemia, el domo del potencial de acción se desarrolla normalmente en el sitio 4 pero no en los sitios 1, 2, 3. El domo se propaga en el sentido de las agujas del reloj y reexcita los sitios 3 y 2 con la consecuente generación de una extrasístole en el sitio 1. Longitud de ciclo de base LCB 700 mseg. (Modificado de Lukas et al. Cardiovasc Res 1996; 32: 593-603)

Gelpi - Donato Fisiopatología cardiovascular
348
-les de Ito Ks en el
canales de IKr e IK1 -74 Como se discutirá más adelante, la diferencia
de la corriente Ito entre ambos ventrículos constituye la
es esencialmente una enfermedad del ventrículo derecho.
Ito75
-
la pared libre de los ventrículos sino también en estructu-ras endocárdicas profundas. 75
subendocardio profundo hasta el miocardio medio en la pared anterior del ventrículo izquierdo, desde el subepicar-dio profundo hasta el miocardio medio en la pared lateral del mismo y en los tractos de salida así como en las capas profundas de estructuras endocárdicas, como los músculos papilares, las trabéculas y el tabique interventricular. 21, 75
diferencia respecto de las células endocárdicas y epicárdi--
y ventriculares. A diferencia de las células de Purkinje, las
-
-
42, 44,
50, 76 -tes bloqueantes IKr, comportamiento que no se observa en el epicardio ni en el endocardio. Desarrollan PDT, como
-
los canales IKsy endocárdicas.
---
gap junctions
-
--
Es así como muchos fármacos, sean de uso corriente en
como torsión de puntas.
por diversos mecanismos que ya hemos mencionado, como --
las condiciones apropiadas para la reentrada intramural. Diversos fármacos se encuentran en la lista, entre ellos
-
de puntas. Esto se debe a que, además del efecto de clase III, la amiodarona posee diversas acciones:
77 En animales y en el ---
cas, mientras que los de las células o se abrevian. 76, 78
-
Torsade de pointes
-
con amiodarona. 78 La lista de fármacos que han demos-
-
-queantes de la corriente IKr. Es así como la incidencia de
-79
-neidad eléctrica del miocardio ventricular. Es así como la

Fisiopatología de las arritmias
349
Figura 21.9: Dependencia de la frecuencia de los potenciales de acción del epicardio, región M y endocardio de los ventrículos derechos e izquierdos caninos. Se muestra la actividad transmembrana a longitudes de ciclo de base (LCB) de 300, 1000, 2000, y 5000 mseg. (Modificado de Sicouri et al. Circ Res 1991; 68: 1729-41)
Figura 21.10: Diferencias iónicas entre las células de las regiones epicárdica (Epi), M y endocárdica (Endo). A: Potenciales de acción de miocitos aislados de las respectivas regiones del ventrículo izquierdo canino. B: Relaciones intensidad/voltaje para la corriente IK1 en los miocitos de Epi, Endo y M. Los valores son los promedios ± del desvío estándar. C: Corriente transitoria de salida (Ito) en los tres tipos celulares. Los trazados de la corriente se registraron durante la aplicación de pulsos despolarizantes desde un potencial que se mantuvo constante a - 80 mV para explorar los potenciales de membrana entre -20 y -70 mV. D: Relación corriente/voltaje pico promedio para la corriente Ito en los tres tipos celulares (promedio ± del desvío estándar). E: Activación dependiente del voltaje del componente lento de la corriente rectificadora de potasio (IKs). Las corrientes se obtuvieron por el protocolo de pulsos de voltaje que se muestra en el inserto; solución desprovista de sodio, potasio y calcio. F: Activación dependiente del voltaje de las corrientes IKs (remanente después de la exposición al E-4031) e IKr (sensible al E-4031). Los valores se expresan en promedio ± el desvío estándar.*p < 0.05 en comparación con las células del epicardio o del endocardio. G: Modalidad reversa de las corrientes de intercambio sodio-calcio registradas en soluciones desprovistas de potasio y cloro, a un voltaje de -80 mV. La corriente INa-Ca se activó hasta un valor máximo al cambiar la perfusión por una solución externa desprovista de sodio, en el momento que indica la flecha. H: La densidad del intercambiador sodio-calcio del las células M es 30% mayor que la de las células del Endo, calculada como la corriente INa-Ca saliente pico normalizada para la
capacitancia de la célula. La densidad del intercambiador sodio-calcio fue similar en Endo y Epi. I: La corriente tardía de sodio sensible a la tetrodotoxina (TTX). El potencial de membrana se mantuvo a -80 mV y se aplicaron pulsos despolarizantes breves para llevarlo a -45 mV y así inactivar la corriente rápida de sodio antes de pasar a -10 mV. J: La corriente tardía de sodio normalizada medida a los 300 mseg dentro del pulso de prueba se relaciona con el potencial de este último. (Modificado de Zygmunt et al. Am J Physiol 2000; 278: H1671-78)
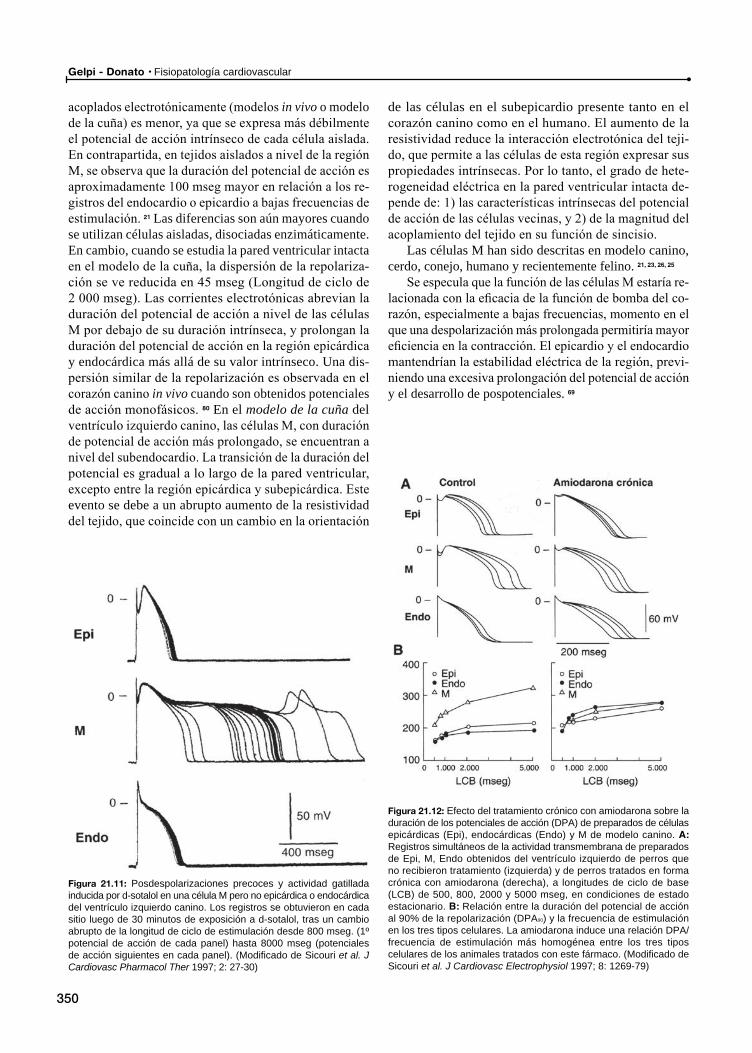
Gelpi - Donato Fisiopatología cardiovascular
350
in vivo o modelo
-
21 Las diferencias son aún mayores cuando se utilizan células aisladas, disociadas enzimáticamente. En cambio, cuando se estudia la pared ventricular intacta
-
y endocárdica más allá de su valor intrínseco. Una dis-
in vivo cuando son obtenidos potenciales 80 En el modelo de la cuña del
evento se debe a un abrupto aumento de la resistividad
Figura 21.11: Posdespolarizaciones precoces y actividad gatillada inducida por d-sotalol en una célula M pero no epicárdica o endocárdica del ventrículo izquierdo canino. Los registros se obtuvieron en cada sitio luego de 30 minutos de exposición a d-sotalol, tras un cambio abrupto de la longitud de ciclo de estimulación desde 800 mseg. (1º potencial de acción de cada panel) hasta 8000 mseg (potenciales de acción siguientes en cada panel). (Modificado de Sicouri et al. J Cardiovasc Pharmacol Ther 1997; 2: 27-30)
Figura 21.12: Efecto del tratamiento crónico con amiodarona sobre la duración de los potenciales de acción (DPA) de preparados de células epicárdicas (Epi), endocárdicas (Endo) y M de modelo canino. A: Registros simultáneos de la actividad transmembrana de preparados de Epi, M, Endo obtenidos del ventrículo izquierdo de perros que no recibieron tratamiento (izquierda) y de perros tratados en forma crónica con amiodarona (derecha), a longitudes de ciclo de base (LCB) de 500, 800, 2000 y 5000 mseg, en condiciones de estado estacionario. B: Relación entre la duración del potencial de acción al 90% de la repolarización (DPA90) y la frecuencia de estimulación en los tres tipos celulares. La amiodarona induce una relación DPA/frecuencia de estimulación más homogénea entre los tres tipos celulares de los animales tratados con este fármaco. (Modificado de Sicouri et al. J Cardiovasc Electrophysiol 1997; 8: 1269-79)
de las células en el subepicardio presente tanto en el
-
--
Las células M han sido descritas en modelo canino, cerdo, conejo, humano y recientemente felino. 21, 23, 26, 25
--
-
y el desarrollo de pospotenciales. 69

Fisiopatología de las arritmias
351
-planteado conceptos clásicos y ha planteado nuevos con-ceptos.
-cardio-endocardio incluyen:
1. La onda J de la hipotermia;
-
M incluyen:-
preferencialmente en las células M;
-
-tricular, con la posibilidad del desarrollo de nuevas tera-
en su conjunto.
El síndrome de Brugada
-
-cordiales derechas, con y sin bloqueo de rama derecha en tres hombres sanos. 81
la normalidad. 82
Recién en 1989, Martini et al -ra et al
83, 84
de edad fue llevado por sus padres a una consulta médica con los doctores Josep y Pedro episodios de síncope y muerte súbita recuperada, con un
-
caracterizada por una alta incidencia de síncope o muerte -
-
rama derecha en las precordiales derechas. 85
El término síndrome de Brugada fue introducido duran-
en julio de 1995, y la primera referencia en la literatura 86
-tarse fenotípicamente en la edad adulta. La muerte súbita
87
se estima en 5 por cada 10 000 habitantes y, fuera de los accidentes de tránsito, es la causa principal de muerte en
en los países donde la enfermedad es endémica. 88 El área considerada como endémica se encuentra en el sureste
132 La -
do en Laos, en la frontera con Tailandia y Camboya, la
89
El folclore de estos países le ha dado múltiples deno-minaciones, que aluden a un tipo particular de muerte que
Figura 21.13: Desenmascaramiento del síndrome de Brugada con ajmalina (1 mg.kg-1). Paciente de sexo masculino (37 años) con historia de síncope recurrente. El registro electrocardiográfico basal (izquierda) no presenta alteraciones. La ajmalina induce supradesnivel del segmento ST en precordiales derechas (V1 a V3), y un patrón electrocardiográfico del síndrome de Brugada (tipo 1). (Modificado de Sangenis P, et al. Rev Arg Cardiol 2005; 73:20-26)

Gelpi - Donato Fisiopatología cardiovascular
352
Los episodios de muerte súbita o síncope son causados por
Antes del episodio, la mayoría de los pacientes presentan
-cediendo a la arritmia, se han observado secuencias de
90
-valencia real de la enfermedad debido a las características
-
veces se encuentran ocultas, pero pueden ser puestas de
ajmalina, procainamida y la 86, 91
-
-cúpula
85, 92
≥-
en silla de montar. 92
del tipo silla de montar o cúpula o ambos. 92
Estos patrones pueden ser observados secuencialmente
-
-
la misma forma pueden observarse anormalidades en la
93
-
con baja penetrancia en particular en mujeres. Chen y sus colaboradores, en 1998, fueron los primeros en relacio-
α de los canales 94 Cerca de cinco docenas de
95 Estas mutaciones se han relaciona-
Na por un desplazamiento en la dependencia del voltaje y del tiempo de sus procesos
--
93 Otro locus
-
-
-
96
Bases celulares del síndrome de Brugada
corriente de salida transitoria de potasio denominada Ito, más notoria a nivel de la
, característica de las
INa e ICa
nada, llevando a una pérdida del domodomo resulta
domo-
sístoles con intervalo de acoplamiento breve que pueden iniciar uno o más ciclos de movimientos circulares de
-
71
se observan los cambios celulares
plateau-

Fisiopatología de las arritmias
353
en silla de montar
-
cúpula, manifestaciones fenotípicas características del sín-97
El
la rama derecha. El panel B muestra una de las morfolo-
el sustrato necesario para que el mecanismo de reentrada
corta. 97
--
86 En los pa-
-mulos se aplican en el sitio de la refractariedad más breve, situado casi siempre en el lado epicardio. En pacientes,
se realiza desde el endocardio del ventrículo derecho; sin
través del seno coronario. En tejidos epicárdicos aislados pertenecientes a ventrículo derecho se provoca con mayor
to más -
98 -nibilidad de la corriente de Ito
--
-97
en tejido aislado, los bloqueantes de los canales de so-
del epicardio del ventrículo derecho, que coincide con -
-
e intraventricular del ventrículo derecho, comentados en apartados anteriores. 99
Finalmente, la corriente de Ito tiene un rol fundamental -to
-primiendo así la actividad arrítmica. 69
El síndrome de QT prolongado y la torsión de puntas
Los síndromes de -
T y, a menudo, una taquicardia multiforme específica,
100
calcio o sodio a través de la membrana celular. La

Gelpi - Donato Fisiopatología cardiovascular
354
Figura 21.14: Esquema comparativo de los mecanismos responsables de la génesis de las arritmias en los síndromes de Brugada y QT prolongado. (Modificado de Antzelevitch C, et al. En: Zipes, DP. Cardiac Electrophysiology: From cell to bedside; 2000)
Figura 21.15: Esquema de los posibles mecanismos responsables de los cambios electrocardiográficos en el síndrome de Brugada. Endo: célula endocárdica, M: célula M; Epi: célula epicárdica. (Modificado de Dumaine R, et al. Circ Res 1999; 85: 803-9) (Ver explicaciones en el texto.)

Fisiopatología de las arritmias
355
-
98
reciben tratamiento.
-
-ciado a síncope, convulsiones, arritmias ventriculares y
-
101 -
Ks --
en la homeostasis del oído interno, se asocia con sordera -
--
-nante. 102, 103
tendrían manifestaciones clínicas y la posibilidad de trans-mitir el
10 000 personas, aunque representa la variante más común.
et al. describieron la primera forma recesiva del síndrome 104
fundamentalmente de sodio y potasio a nivel de los cro--
nocen siete variedades.-
Ks -
Kr-
Na-
K1 -
-
al desarrollo del
--
más adecuada. 69
-
allí la importancia del estudio, sobre todo de estas formas más frecuentes en la práctica clínica.
-
Figura 21.16: Efecto diferencial de la ajmalina sobre las células epicárdicas del ventrículo derecho (Epi VD) e izquierdo (EpiVI) sometidas a un cambio brusco en la frecuencia de estimulación. La longitud del ciclo (LC) se modificó de 2000 a 1000 mseg. En la célula Epi VD, el número 1 indica el último potencial de acción estimulado a la longitud del ciclo de 2000 mseg, y los números 2 a 5 corresponden a potenciales de acción registrados durante la estimulación a la LC de 1000 mseg. El domo del potencial de acción, ausente a LC de 2000 mseg (1), se restablece en forma progresiva a la vez que el potencial de acción se prolonga cuando LC se abrevia a 1000 mseg (2 a 5). La ajmalina no modificó la duración del potencial de acción en Epi VI. (Modificado de Miyazaki, T et al. J Am Coll Cardiol 1996; 27: 1061-70)

Gelpi - Donato Fisiopatología cardiovascular
356
otra parte, con la sensibilidad aumentada de estos pacientes
en presencia del bloqueante IKs abrevia los potenciales
-
de este síndrome. -
Kr
-
Na
células M que en los otros tipos celulares,
de la pared ventricular. El ATX II también produce un
de las células endocárdicas y epicárdicas, que resulta en un marcado retraso en el comienzo de la onda T, como
M que el del resto de las células de la pared ventricular, y
105
-
-
La
como mecanismo responsable del mantenimiento de la arritmia se considera a la reentrada como el más probable.
la corriente neta repolarizante a través de diversos meca-Kr o IKs,
el incremento de las corrientes tardías ICa o IKs, mutacio-
son responsables de las diversas variantes de los síndro-69 Los bloqueantes de la
corriente IKrpromotores de la corriente tardía INa y las mutaciones del
-
del miocardio ventricular, induciendo de esta manera un
-dominantemente en este tipo celular. Los bloqueantes IKr
a nivel de las células M que en las células endocárdicas y
Ca
corriente tardía de sodio como el ATX-II. Con respecto a la quinidina, el efecto sobre la disper-
dosis. A concentraciones terapéuticas relativamente bajas,
Figura 21.17: QT prolongado y Torsade de pointes posinfarto de miocardio. Registro electrocardiográfico de una paciente del sexo femenino (65 años) dentro de las primeras 24 horas del curso de un infarto agudo de miocardio inferodorsal y que desarrolla prolongación del intervalo QT y torsión de puntas. Na+: 145 mEq/l, K+: 4.5 mEq/l, Mg: 3mg/100ml, Ca++ (iónico): 4.25 mg/dl

Fisiopatología de las arritmias
357
predomina el bloqueo de la corriente IKr
en forma importante, pero no en las células epicárdicas ni
de puntas que no ocurre a altas dosis terapéuticas o frente
El --
A pesar de que ambos, dl-sotalol y d-sotalol, han mos--
mias supraventriculares y ventriculares, estudios clínicos
en pacientes posinfarto fue prematuramente interrumpido 30
se han detectado posdespolarizaciones precoces, mientras
et al. mostraron que el
en forma preferencial respecto de las células endocárdicas y epicárdicas. 42
comportamiento se basa en que el bloqueo por el d-sotalol de la corriente IKr
-
especial en las células M donde la corriente IKs
notoria en este tipo celular. 42
dependencia reversa del uso. Este comportamiento es común a los antiarrítmi-cos de clase III. El d-sotalol constituye así un ejemplo de aumento
-
III, lo constituye la amiodarona que, como previamente mencionamos, induce una disminución
Bibliografía
1. Surawicz, B. “Brief history of cardiac arrhythmias since the end of the nineteenth century . Part I. J Cardiovasc Electrophysiol 2003; 14(12):1365–71.
2. Galvani LA. De viribus electricitatis in motu musculari: communtarius comment. Bologna: Bonon. Scient. et Art Inst 1791; 7: 363-418.
3. Matteucci, C. “Sur un Phénomène physiologique produit par les muscles en contraction . Ann Chim Phys 1842; 6:339-341.
4. Du Bois-Reymond, E. Untersuchungen über thierische Elektricität. Bd. I u II Berlin, Reimer 1848-60.
5. Kolliker A, H Muller. “Nachweis der negativen schwankung des muskelstronmes am naturlich sich contrahieden muskel verhandl .J Phys Med Gesellsch 1856 ; 6 :528.
6. Waller, AD. “A demonstration on man of electromotive changes accompanying the heart’s beat . J Physiol 1887; 8:229-34.
7. Lippmann, G. “Relations entre les phenomenes electriques et capillaires . Ann Chim Phys 1875; 5 (11): 494-549.
8. Einthoven, W. “Ueber die Form des Menschlichen Electrocardio-gramms . Arch f d Ges Physiol 1895; 60:101-23.
9. Einthoven, W. “Un nouveau galvanometre . Arch Neerl Sci Exactes Nat 1901;6:625-33.
10. Einthoven, W. “Galvanometrische registerung des menschlichen electrocardiogramms, zugleich eine beurtheilung der anwedung des kapillar-electrometers in physiologie . Pflugers Arch 1903; 99:472-80.
11. Fisch, C. “Centennial of the string galvanometer and the electro-cardiogram . J Am Coll Cardiol 2000; 36:1737-45.
12. Fye, WB. “A History of the origin, evolution and impact of elec-trocardiography . Am J Cardiol 1994; 73:937-49.
13. Goldberger, E. “A simple, indifferent, electrocardiographic elec-trode of zero potential and a technique of obtaining augmented, unipolar, extremity leads . Am Heart J 1942; 23: 483-92.
14. Ling G, RW Gerard. “The normal membrane potential of frog sartorius fibers . J Cell Comp Physiol 1949; 34: 383-96.
15. Nastuk WL, AL Hodgkin. “The electrical activity of single muscle fibres . J Cell Comp Physiol 1950; 35: 39-73.
16. Hodgkin AL, Huxley AF, Katz B. “Ionic current underlying activity in the giant axon of the squid . Arch. Sci Physiol 1949; 3:129-50.
17. Weidmann, S. “Effect of current flow on the membrane potential of cardiac muscle . J Physiol 1951; 115: 227-36.
18. Weidmann, S. Elektrophysiologie der Herzmuskelfaser. Bern und Stuttgart: Verlag Hans Huber, 1956.
19. Neher E, B Sakmann. “Single-channel currents recorded from membrane of denervated frog muscle fibers . Nature 1976; 260: 799-802.
20. Litovsky SH, Antzelevitch C. “Transient outward current promi-nent in canine ventricular epicardium but not endocardium . Circ Res. 1988;62(1):116-26.
21. Sicouri S, C Antzelevich. “A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle . The M cell. Circ Res 1991; 68:1729-41.
22. Stankovicova T, Szilard M, De Scheerder I, Sipido KR. “M cells and transmural heterogeneity of action potential configuration in myocytes from the left ventricular wall of the pig heart . Cardiovasc Res 2000; 45: 952 – 60.
23. Sicouri S, Quist M, Antzelevitch C. “Evidence for the presence of M cells in the guinea pig ventricle . J Cardiovasc Electrophysiol 1996, 7:503- 11.
24. Mcintosh MA, Cobbe SM, Smith GL. “Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure . Cardiovasc Res 2000; 45(2): 397-409.
25. Aiba T, Shimizu W, Inagaki M, Hidaka I, Tatewaki T, Sunagawa K. “Transmural heterogeneity of the action potential configuration in the feline left ventricle . Circ J 2003; 67: 449 – 54.
26. Drouin E, Charpentier F, Gauthier C, Laurent K, Marec HL. “Electrophysiologic characteristics of cells spanning the left ven-tricular wall of human heart: Evidence for presence of M cells . J Am Coll Cardiol 1995;26:185-92.
27. Lukas A, C Antzelevitch. “Differences in electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role of the transient outward current . Circulation 1993; 88: 2903-15.
28. Hübner C, Jentsch T. “Ion channel diseases . Hum Mol Genet 2002; 11(20): 2435- 45.
29. Liu DW, C Antzelevitch. “Characteristics of the delayed rectifier current (I
Kr and I
Ks) in canine ventricular epicardial, midmyocardial,

Gelpi - Donato Fisiopatología cardiovascular
358
and endocardial myocytes a weaker IKs
contributes to the longer action potential of the M cell . Circ Res 1995; 76: 351-65.
30. Waldo AL, Camm AJ, deRuyter H, Friedman PL, MacNeil DJ, Pauls JF et al. “Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction . The SWORD Investigators. Survival With Oral d-Sotalol. Lancet 1996;348(9019):7- 12.
31. Noma, A. “ATP-regulated K+ channels in cardiac muscle . Nature 1983; 305(5930):147- 48.
32. Carmeliet E, K Mubagwa. “Characterization of the acetylcholine-induced potassium current in rabbit cardiac Purkinje fibres . J Physiol 1986; 371(1): 219- 37.
33. Zygmunt, AC. “Intracellular calcium activates a chloride current in canine ventricular myocytes . Am J Physiol 1994v;267(5 Pt 2):H1984-95.
34. Carmeliet, E. “Cardiac ionic currents and acute ischemia: from channels to arrhythmias . Physiol Rev; 79(3):917-1017.
35. Antzelevitch, C. “Los mecanismos arritmogénicos . En: Elizari MV, Chiale P, eds. Arritmias Cardiacas. Buenos Aires, Ed. Panamericana, 2003; 81-112.
36. Katzung BG, JA Morgenstern. “Effects of extracellular potassium on ventricular automaticity and evidence for a pacemaker current in mammalian ventricular myocardium . Circ Res 1977; 40: 105-11.
37. Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. “Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure . Am J Physiol 1999; 277(1 Pt 2):H80- 91.
38. Zipes, D. “Mechanisms of clinical arrhythmias . J Cardiovasc Electrophysiol 2003;14:902-12.
39. Cranefield, P. “Action potentials, afterpotentials, and arrhythmias . Circ Res 1977;41:415-23.
40. Akar FG, DS Rosenbaum. “Transmural Electrophysiological Hete-rogeneities Underlying Arrhythmogenesis in Heart Failure . Circ Res 2003; 93:638-45.
41. Boutjdir M, N el-Sherif. “Pharmacological evaluation of early afterdepolarisations induced by sea anemone toxin (ATXII) in dog heart . Cardiovasc Res 1991;25(10):815-9.
42. Sicouri S, Moro S, Elizari MV. “D-sotalol induces marked action potential prolongation and early afterdepolarizations in M but not epicardial or endocardial cells of the canine ventricle . J Cardiovasc Pharmacol Ther 1997;2:27-30.
43. January CT, Riddle JM, Salata JJ. “A model for early afterdepola-rizations: induction with the Ca2+ channel agonist Bay K 8644 . Circ Res 1988; 62(3):563-71.
44. Davidenko JM, Cohen L, Goodrow R, Antzelevitch C. “Quini-dine-induced action potential prolongation, early afterdepolari-zations, and triggered activity in canine Purkinje fibers. Effects of stimulation rate, potassium, and magnesium . Circulation 1989;79:674-86.
45. Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R et al. “Similarities between early and delayed afterdepolariza-tions induced by isoproterenol in canine ventricular myocytes . Cardiovasc Res 1997; 34(2):348-59.57.
46. Sicouri S, C Antzelevitch. “Afterdepolarizations and triggered activity develop in a select population of cells (M cells) in canine ventricular myocardium: the effects of acetylstrophanthidin and Bay K 8644 . Pacing Clin Electrophysiol 1991;14(11 Pt 2):1714-20.
47. Burashnikov A, C Antzelevitch. “Failure of canine ventricular epicardial and endocardial cells to develop early afterdepolarization activity is due to the presence of prominent IKs . Circulation 1997; 96: I -292 (Resumen).
48. Patterson E, Scherlag BJ, Szabo B, Lazzara R. “Facilitation of epinephrine-induced afterdepolarizations by class III antiarrhythmic drugs . J Electrocardiol 1997;30 (3):217-24.
49. Burashnikov A, C Antzelevitch. “Mechanisms underlying early afterdepolarization activity are different in canine Purkinje and M cell preparations. Role of intracellular calcium . Circulation 1996; 94:1 (Resumen).
50. Antzelevitch C, S Sicouri. “Clinical relevance of cardiac arrhythmias generated by afterdepolarizations. Role of M cells in the generation
of U waves, triggered activity and torsade de pointes . J Am Coll Cardiol 1994;23(1):259-77.
51. Reiter, M: “Die Entstehung von ‘Nachkontraktionen’ im Herz-muskel unter Einwirkung von Calcium un don Digitalisglykosiden in Abhaengigkeit von der Reizfrequenz .Arch Exp Path U Pharmk 1992;242:497-507.
52. Ferrier, GR. “The effects of tension on acetylstrophanthidin-induced transient depolarizations and aftercontractions in canine myocardial and Purkinje tissues . Circ Res 1976;38(3):156-62.
53. Rosen MR, Gelband H, Merker C, Hoffman BF. “Mechanisms of digitalis toxicity. Effects of ouabain on phase four of canine Purkinje fiber transmembrane potentials . Circulation 1973;47(4): 681-9.
54. Zygmunt AC, Goodrow RJ, Weigel CM. “INaCa and ICl(Ca) contribute to isoproterenol-induced delayed after depolarizations in midmyocardial cells . Am J Physiol 1998;275(6 Pt 2):H1979-92.
55. Rozanski GJ, SL Lipsius. “Electrophysiology of functional subsi-diary pacemakers in canine right atrium . Am J Physiol 1985;249(3 Pt 2):H594-603.
56. Sicouri S, C Antzelevitch. “Drug-induced afterdepolarizations and triggered activity occur in a discrete subpopulation of ventricular muscle cells (M cells) in the canine heart: quinidine and digitalis . J Cardiovasc Electrophysiol 1993;4(1):48-58.
57. Burashnikov A, C Antzelevitch. “Block of I(Ks)
does not induce early afterdepolarization activity but promotes beta-adrenergic agonist-induced delayed afterdepolarization activity . J Cardiovasc Electrophysiol 2000;11(4):458-65.
58. Chen YJ, Chen SA, Chen YC, Yeh HI, Chan P, Chang MS, Lin CI. “Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: implication in initiation of atrial fibrillation . Circulation 2001; 104 : 2849-54.
59. Song Y, L Belardinelli. “ATP promotes development of afterdepola-rizations and triggered activity in cardiac myocytes . Am J Physiol 1994; 267: H2005-11.
60. Rosen MR, P Jr Danilo. “Effects of tetrodotoxin, lidocaine, vera-pamil, and AHR-2666 on Ouabain-induced delayed afterdepola-rizations in canine Purkinje fibers . Circ Res 1980;46(1):117-24.
61. Zaugg, C. “Current concepts on ventricular fibrillation: a vicious circle of cardiomyocyte calcium overload in the initiation, main-tenance and termination of ventricular fibrillation . Indian Pacing Electrophysiol J 2004;4(2):85-92.
62. Wit AL, MR Rosen. “Afterdepolarizations and triggered activity: Distinction from automaticity as an arrhythmogenic mechanism . En: Fozzard HA y col (Ed). The Heart and Cardiovascular System. New York, Raven Press;1992; 2113.
63. Vermeulen, JT. “Mechanisms of arrhythmias in heart failure . J Cardiovasc Electrophysiol 1998; 9:208–21.
64. Mayer, AG. “Rhythmical pulsations is scyphomedusae . Publication 47 of the Carnegie Institute 1906;1.
65. Mines, GR. “On dynamic equilibrium in the heart . J Physiol 1913; 46: 349-82.
66. Schmitt FO, J Erlanger. “Directional differences in the conduction of the impulse through heart muscle and their possible relation to extrasystolic and fibrillary contractions . Am J Physiol 1928:87: 326-47.
67. Elizari, MV. “Bases Fisiopatológicas de las Arritmias Cardiacas . En: Bertolasi C, Barredo C, Gimeno G, Liniado G, Mauro V, editores. Cardiología 2000. Buenos Aires, Ed Panamericana, 2000; 135-90.
68. Bayés de Luna, A. “Arritmias . En: Bayés de Luna A, editor. Tra-tado de Electrocardiografía Clínica. Barcelona Editorial Científico médica, 1992; : 351-451.
69. Antzelevitch C, Shimizu W, Yan G, Sicouri S, Weissenburger J, Nesterenko VV et al. “The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart . J Cardiovasc Electrophysiol 1999;10 (8):1124-52.
70. Akar FG, Yan GX, C Antzelevitch, Rosenbaum DS. “Unique topo-graphical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome . Circulation 2002; 105(10):1247-53.

Fisiopatología de las arritmias
359
71. Lukas A, C Antzelevitch. “Phase 2 reentry as a mechanism of ini-tiation of circus movement reentry in canine epicardium exposed to simulated ischemia . Cardiovasc Res 1996;32(3):593-603.
72. Di Diego JM, C Antzelevitch. “High [Ca2+]o-induced electrical
heterogeneity and extrasystolic activity in isolated canine ventricular epicardium . Phase 2 reentry. Circulation 1994;89:1839 –50.
73. Zygmunt AC, Goodrow RJ, Antzelevitch C. “INa-Ca
contributes to electrical heterogeneity within the canine ventricule . Am J Physiol 2000;278:H1671-78.
74. Volders P, Sipido K, Carmeliet E, Spatjens R, Wellens H, Vos M. “Repolarizing K
1 Currents I
to1 and I
Ks are larger in right than left
canine ventricular midmyocardium . Circulation 1999;99:206-10.75. Sicouri S, Fish J, Antzelevitch C. “Distribution of M cells in the
canine ventricle . J Cardiovasc Electrophysiol 1994;5:824-37.76. Sicouri S, Belardinelli L, Carlsson L, Antzelevitch C. “Potent antia-
rrhythmic effects of chronic amiodarone in canine pulmonary vein sleeve preparations . J Cardiovasc Electrophysiol. 2009; 20(7): 803-10.
77. Chezalviel-Guilbert F, Davy JM, Poirier JM, Weissenburger J. “Mexiletine antagonizes effects of sotalol on QT interval duration and its proarrhythmic effects in a canine model of torsade de pointes . Am Coll Cardiol 1995;26 (3):787- 92.
78. Drouin E, Lande G, Charpentier F. “Amiodarone reduces transmural heterogeneity of repolarization in the human heart . J Am Coll Cardiol.1998;32:1063-7.
79. Freedman, R. “Schizophrenia . N Engl J Med 2003;349:1738-49.80. Antzelevitch C, Yan GX, Shimizu W, Burashnikov A. “Electrical
heterogeneity, the ECG and cardiac arrhythmias . En: Zipes DP, Jalife J, Eds. In Cardiac Electrophysiology: From Cell to Bedside, 3rd edition. Philadelphia, W: B. Saunders Company, 2000; 222-38.
81. Osher HL, L Wolff. “Electrocardiographic pattern simulating acute myocardial injury . Am J Med Sci 1953;226:541-5.
82. Edeiken, J. “Elevation of RS-T segment, apparent or real in right precordial leads as probably normal variant . Am Heart J 1954;48:331-9.
83. Martini B, Nava A, Thiene G, et al. “Ventricular fibrillation wi-thout apparent heart disease: description of six cases . Am Heart J 1989; 118:1203–9.
84. Aihara N, Ohe T, Kamakura S, Matsuhisa M, Takagi H, Shimomura K. “Clinical and electrophysiologic characteristics of idiopathic ventricular fibrillation . Shinzo 1990;22:80-6.
85. Brugada P, J Brugada. “Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report . J Am Coll Cardiol 1992;20:1391-6.
86. Yan GX, C Antzelevitch. “Cellular basis for the electrocardiographic J wave . Circulation 1996;93: 372-9.
87. Antzelevitch C, Brugada P, Brugada J, Brugada R. “Brugada Syndrome:From Cell to Bedside . Curr Probl Cardiol 2005;30:9-50.
88. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuri-panyo K, Likittanasombat K et al. “Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men . Circulation 1997;96:2595-600.
89. Tungsanga K, P Sriboonlue. “Sudden unexplained death syndrome in north-east Thailand . Int J Epidemiol. 1993;22:81-7.
90. Priori SG, Dile L, Schwartz PJ. “Torsade de Points. En: Podrid PJ, Kowey PR (eds) . Cardiac arrhythmia. Williams and Wilkins Baltimore, 1995; p. 951
91. Sangenis P, Vitagliano L, Ferreiro M, Ansaldi SM, Sicouri S. “El chequeo médico-deportivo en individuos asintomáticos: anomalías cardiológicas . Rev Arg Cardiol 2005; 73:20-26)
92. Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin J for the Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. “Proposed Diagnostic Criteria for the Brugada Syndrome . Eur Heart J 2002; 23:1648-54.
93. Smits JP, Eckardt L, Probst V, Bezzina CR, Schott JJ, Remme CA et al. “Genotype-phenotype relationship in Brugada syndrome: elec-trocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients . Am Coll Cardiol 2002;40(2):350-6.
94. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P et al. “Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation . Nature 1998;392:293-6.
95. Tan HL, Bezzina C, Smits JP, Verkerk AO, Wilde AA. “Genetic con-trol of sodium channel function . Cardiovasc Res 2003;57:961–973.
96. Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV et al. “Ionic mechanisms responsible for the electro-cardiographic phenotype of the Brugada syndrome are temperature dependent . Circ Res 1999;85:803-9.
97. Antzelevitch, C. “The Brugada syndrome: ionic basis and arrhyth-mia mechanisms . 2001;12:268-72.
98. Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. “Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome . J Am Coll Cardiol 1996;27:1061-70.
99. Civetta MM, Moro S, Chiale P, Elizari MV, Escobar J, Sicouri S. “Efecto preferencial de la ajmalina sobre el epicardio ventricular derecho canino puede explicar las manifestaciones electrocardiográ-ficas del síndrome de Brugada . Rev Argent Cardiol 2002;70:118-126.
100. Shimizu, W. “Genotype-specific clinical manifestation in long QT syndrome . Expert Rev Cardiovasc Therapy 2003;1(3):401-9.
101. Jervell A, F Lange-Nielsen. “Congenital deaf-mutism, functional heart disease with prolongation of the QT interval, and sudden death . Am Heart J 1957; 54: 103-6.
102. Chiang CE, DM Roden. “The long QT syndromes: genetic basis and clinical implication . J Am Coll Cardiol 2000;36:1-12.
103. Ward, OC. “A new familial cardiac syndrome in children . J Irish Med Assoc 1964; 64: 103-6.
104. Priori SG, Schwartz PJ, Napolitano C, Bianchi L, Dennis A, De Fusco M et al. “A recessive variant of the Romano-Ward long-QT syndrome? Circulation 1998;97:2420-2425.
105. Shimizu W, C Antzelevitch. “Cellular basis for the ECG featuresof the LQT1 form of the Long-QT syndrome: effects of -adrenergic agonists and antagonists and sodium channel blockerson transmural dispersion of repolarization and torsade de pointes”. Circulation 1998; 98:2314-22


361
Fisiopatología del síncope 22
Aurora Ruiz
-ciencia y el tono postural que revierte espontáneamente, sin secuelas posteriores, es una entidad de alta prevalencia
que abarca tanto aquellos cuadros severos que pueden preceder a la muerte como aquellos menores comúnmente denominados lipotimia o
de las causas predisponentes y determinantes del cuadro, -
otras circunstancias no representa más que una intercu-
Independientemente de la importancia con respecto al
severo deterioro en la calidad de vida del paciente. Linzer et al. han comparado dicho deterioro al causado por otras
1
-
en forma retrospectiva y basándose en el valor predictivo de diferentes métodos. Muchas de las causas de síncope han
-
u otras enfermedades cardiacas estructurales o del sistema
--
el cuadro de síncope que lo provocan. De las observaciones realizadas durante la
un solo mecanismo pueda estar involucrado en todos ellos.
-
Causas de síncope
-
en los valores predictivos de los distintos métodos por
una sospecha. Por último, contribuyen a la complejidad
concomitancia de más de una de ellas en un mismo pa-ciente. Dos tipos de factores deben ser tenidos en cuenta: los predisponentes y los desencadenantes. En los ancianos es claro el rol de los factores predisponentes tal como
un estado infeccioso intercurrente, provocando la pérdida
Unidos, en donde se indica que el costo por paciente fue 2
conocimiento del valor de los distintos métodos. Habiendo hecho estas salvedades, las causas más fre-
-

Gelpi - Donato Fisiopatología cardiovascular
362
-
no solamente la ventaja de su sencillez sino también im-
a los mecanismos subyacentes. Pacientes con una misma
pueden presentar síncope por mecanismo hemodinámico,
certeza, entendiendo por tal a los mecanismos que pro-vocan la pérdida de conocimiento, es alcanzada en muy
contadas ocasiones ya que para ello sería necesario contar
el episodio. En su ausencia, podemos a lo sumo sospechar,
ocurre tiene un rol causal.-
3-7
Fisiopatología general del síncope
-
clínica, es decir, a la pérdida transitoria de la conciencia -
-
cerebral para provocar la pérdida de la conciencia. Para que ocurra pérdida del conocimiento y del tono postural
-misferios o a toda la parte superior del tronco encefálico.
La -
Causa no aclarada ................................Neurocardiogénica ...............................Cardíaca ................................................ arrítmica .................................... Taquiarritmia ................ Bradiarritmia ................. No arrítmica ..............................Neurológica ..........................................Otros ......................................................
% Tabla 22.2:
(Modificado de Barcroft H, et al. Lancet 1944;I:489-490 y Oberg B, et al. Acta Physiol Scand. 1973;87:121-32.)
Tabla 22.1: Causas de síncope de acuerdo con el mecanismo fisiopatológico
Alteraciones vasculares
Ateromatosis de vasos de cuello
Artrosis cervical
Oclusión proximal de la subclavia
Disminución del volumen minuto
Obstrucciones al flujo de salida ventricular
Obstrucciones al tracto de entrada ventricular Insuficiencia cardíaca
Hipovolemia
Rémora de sangre en los miembros inferiores
Disminución de la resistencia periférica
Situacional
Neurocardiogénico o vasovagal
Hipersensibilidad del seno carotídeo
Drogas simpaticolíticas
Simpaticectomía
Disautonomías primarias o secundarias
Reposo prolongado
Drogas vasodilatadoras
Exposición al calor
Estados febriles
Hipoxemia
Asfixia
Enfermedades pulmonares
Anemia
Déficit metabólico
Hipoglucemia
Vasoconstricción cerebral
Hipocapnia
Psiquiátrico
Depresión
Histeria
Reacción de pánico

Fisiopatología del síncope
363
--
--
la coronaria, responde a la necesarios para observar dicha
Por el contrario, el CO
hipercapnia es rápida y puede deberse al incremento conco-mitante de los iones HpH en el líquido cefalorraquídeo. No se descarta tampoco un efecto directo del CO sobre los vasos ya que, en el caso de
o de lesiones focalizadas en la vasculatura cerebral, la hi-
-rrecto aporte es el mecanismo más frecuente de síncope.
Síncopes neuromediados
-
un determinado receptor con su correspondiente vía aferente,
efectora. Tal es el caso de los síncopes situacionales como el -
tales como síncope miccional o síndrome del seno carotídeo. En cambio, el síncope más estrictamente denominado
como vasovagal o neurocardiogénico, que ocurre frente --
puede ser considerado como una de las causas bradiarrít-
o demostrada más frecuente de síncope. Así, Day et al.
-8
-
-mentarios. Los desencadenantes clásicos son la permanencia
-
sudoroso. El pulso es difícilmente palpable, la frecuencia car-diaca puede ser baja o elevada de acuerdo a la variedad de la
-
-
-
-
Tabla 22.3: Cuadros de intolerancia al ortostatismo
Taquicardia postural sin hipotensión (POTS)
Hipotensión con estado normoadrenérgico o hiperadrenérgico
S. Neurocardiogénico o vasovagal
Hipovolemia
Hipotensión con estado hipoadrenérgico
Insuficiencia autonómica primaria
Enfermedades debilitantes
Reposo prolongado en cama
Viajes espaciales
Tabla 22.4: Síncope de causa bradiarrítmica
Alteraciones intrínsecas del nódulo sinusal, nódulo aurículo-ventricular o del sistema de conducción distal
Alteraciones secundarias a:
Drogas
Trastornos electrolíticos
Enfermedades metabólicas
Neuromediadas

Gelpi - Donato Fisiopatología cardiovascular
364
siones, no se puede detectar un claro desencadenante
-
-cuencia interpretados como paro cardiorrespiratorio o crisis epilépticas, respectivamente.
-
-nismo del síncope en individuos portadores de diferentes
Síndrome vasovagal maligno
sincopales recurrentes, en particular en ancianos, reprodu-
y con una alta incidencia de traumatismos.9
circunstancias consideradas como malignas impliquen un 10
-
este tipo de respuestas, una mayor incidencia de trastornos -
persensibilidad del seno carotídeo que en los pacientes con
En estudios retrospectivos y prospectivos no se ha detectado un mayor índice de recurrencias que en los pacientes con
sin recurrir al implante de un marcapaso.
bueno, no está del todo aclarado en pacientes con una en-fermedad subyacente.
Uno de los clásicos desencadenantes del cuadro va-
parte de los conocimientos que poseemos en la actualidad
observaciones realizadas durante el TT o prueba de ortos-
en qué consiste la prueba.
Tilt test
La prueba se realiza en una camilla basculante de con-
primera etapa consiste en un periodo de reposo de 15
-
-
frecuencia cardiaca. El TT debe realizarse en un am-
Los individuos susceptibles reproducen el episodio de
-
más utilizada fue el isoproterenol y, en la actualidad, son los nitritos.
Respuestas al Tilt testEl TT es positivo cuando la prueba provoca síncope o presíncope. Durante el síncope es posible observar:
elevada;
arterial y la frecuencia cardiaca. En esta última circunstancia, más frecuente en los
--
28
-
hemodinámicas periféricas. Una nomenclatura ampliamente difundida en la ac-
11
Respuesta mixta
Cardioinhibidora tipo A
Fisiopatología del síncope
365
la frecuencia cardiaca.
Cardioinhibidora tipo B
-
Vasodepresora
Excepción 1
Excepción 2
--
námica en el momento en que el síncope ocurre sino ob-
-
-damente en los momentos previos a la pérdida de cono-
Figura 22.2: Registro continuo de electrocardiograma (en rojo), presión arterial (en azul) y frecuencia cardiaca (en verde) durante el Tilt test en un paciente de 27 años. A los 24 minutos se observa disminución de la presión arterial y de la frecuencia cardiaca, conformando una respuesta mixta.
Figura 22.1: Camilla basculante para realización de Tilt test
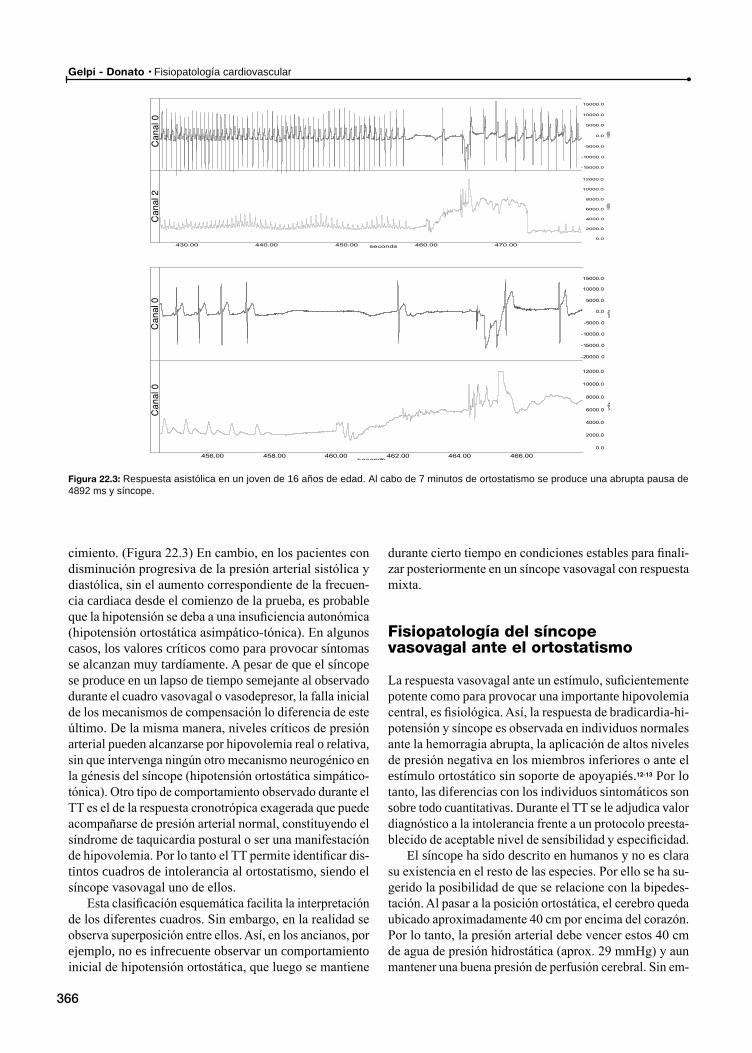
Gelpi - Donato Fisiopatología cardiovascular
366
-cia cardiaca desde el comienzo de la prueba, es probable
casos, los valores críticos como para provocar síntomas se alcanzan muy tardíamente. A pesar de que el síncope se produce en un lapso de tiempo semejante al observado
arterial pueden alcanzarse por hipovolemia real o relativa,
-tintos cuadros de intolerancia al ortostatismo, siendo el
ejemplo, no es infrecuente observar un comportamiento
-
Fisiopatología del síncope vasovagal ante el ortostatismo
potente como para provocar una importante hipovolemia bradicardia-hi-
estímulo ortostático sin soporte de apoyapiés.12-13 Por lo tanto, las diferencias con los individuos sintomáticos son sobre todo cuantitativas. Durante el TT se le adjudica valor
-
El síncope ha sido descrito en humanos y no es clara --
-
Figura 22.3: Respuesta asistólica en un joven de 16 años de edad. Al cabo de 7 minutos de ortostatismo se produce una abrupta pausa de 4892 ms y síncope.

Fisiopatología del síncope
367
factor determinante, ya que muchos animales poseen una
Interrelación entre la presión arterial, la frecuencia cardiaca y la respiración
--
nales presentan continuas oscilaciones. Dichas oscilaciones
-
Este complejo sistema funciona en base a múltiples sensores,
el sistema nervioso, provocando un efecto cuya resultante
La pérdida del -
frecuencias, el análisis espectral de las oscilaciones de un
los distintos componentes de dicho control.
-
arterial y la frecuencia cardiaca.-
que provocan alteraciones hemodinámicas, repercutiendo
por los
-
perfundido de animales que el estiramiento auricular pro-voca taquicardia. A favor de la importancia de los efectos
-co, no desaparece por completo14 y persiste en pacientes con
centrales. El acoplamiento entre el centro respiratorio y los -
arterial. Tanto en animales anestesiados como conscientes y en seres humanos, la arritmia sinusal respiratoria per-
Oscilaciones dela presión arterial
Oscilaciones dela frecuencia cardíaca
Vía mecánica Vía receptores periféricos Vía central
Respiración
Distensión del NS Cambios en el retorno venoso
Distensión de AD, venas pulmonares,
pulmón
Acoplamientode centro respiratorio centros
vasomotores
Alteraciones en el llenado del VD y VI
Barorreflejo
Figura 22.4: Relación entre la respiración, la presión arterial y la frecuencia cardiaca (ver explicación en el texto). NS: nódulo sinusal; AD: aurícula derecha; VD: ventrículo izquierdo; VI: ventrículo izquierdo

Gelpi - Donato Fisiopatología cardiovascular
368
siste aún en ausencia de movimientos inspiratorios y de
15
-pal mecanismo a través del que se relacionan la arterial y la frecuencia cardiaca.
-
-
-
-
cardiaca. Mullen et al.16
componentes de un circuito cerrado que relaciona a la
1: Mecánica circulatoria
frecuencia determinada. Está determinada por las propie-
-diaca es la arritmia sinusal respiratoria.
--
son principalmente mecánicas y no se alteran de forma
-mico.
Respuesta fisiológica ante el ortostatismo
-miten adaptar el comportamiento hemodinámico a la
17 En la
inicial-
pulmonares. Estos, al inhibirse, provocan un aumento del tono simpático, de la frecuencia cardiaca, del inotro-pismo y de la resistencia periférica. Todos estos efectos
-
en el músculo, el aumento de la frecuencia cardiaca y la
por los barorreceptores carotídeos. 18
El comportamiento hemodinámico inicial durante el
En el proceso de pararse se contraen los músculos de los miembros inferiores y del abdomen, provocando un au-
-
y dura has-
frecuencia cardiaca continúa elevándose. La de
-
Teorías sobre la fisiopatología del síncope vasovagal
la asistolia. Esta última se
del simpático. El mecanismo por el que se produce la
19 propusieron que la
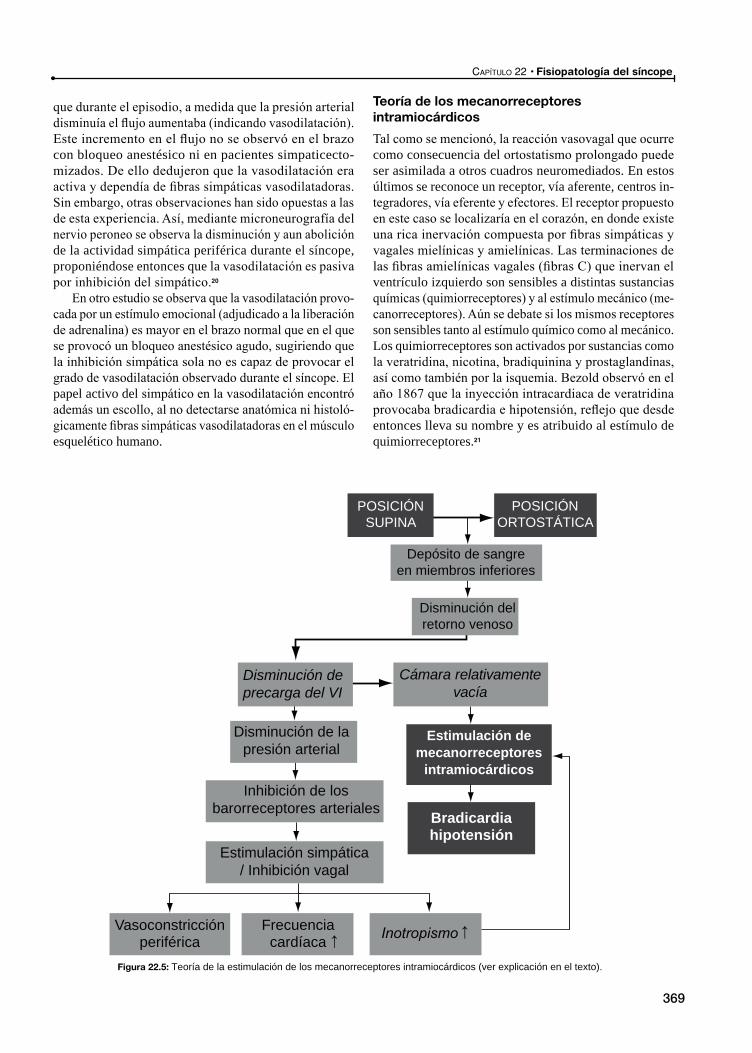
Fisiopatología del síncope
369
con bloqueo anestésico ni en pacientes simpaticecto-
de la actividad simpática periférica durante el síncope, proponiéndose entonces que la
20
-
papel activo del -
esquelético humano.
Teoría de los mecanorreceptores intramiocárdicos
como consecuencia del ser asimilada a otros cuadros neuromediados. En estos últimos se reconoce un receptor, vía aferente, centros in-
ventrículo izquierdo son sensibles a distintas sustancias -
son sensibles tanto al estímulo químico como al mecánico. Los quimiorreceptores son activados por sustancias como
entonces lleva su nombre y es atribuido al estímulo de quimiorreceptores.21
POSICIÓN SUPINA
Depósito de sangre en miembros inferiores
Disminución del retorno venoso
Disminución de precarga del VI
Disminución de la presión arterial
Inhibición de los barorreceptores arteriales
Estimulación simpática / Inhibición vagal
Vasoconstricción periférica
Frecuencia cardíaca
Cámara relativamente vacía
POSICIÓN ORTOSTÁTICA
Bradicardiahipotensión
Estimulación de mecanorreceptores
intramiocárdicos
Figura 22.5: Teoría de la estimulación de los mecanorreceptores intramiocárdicos (ver explicación en el texto).
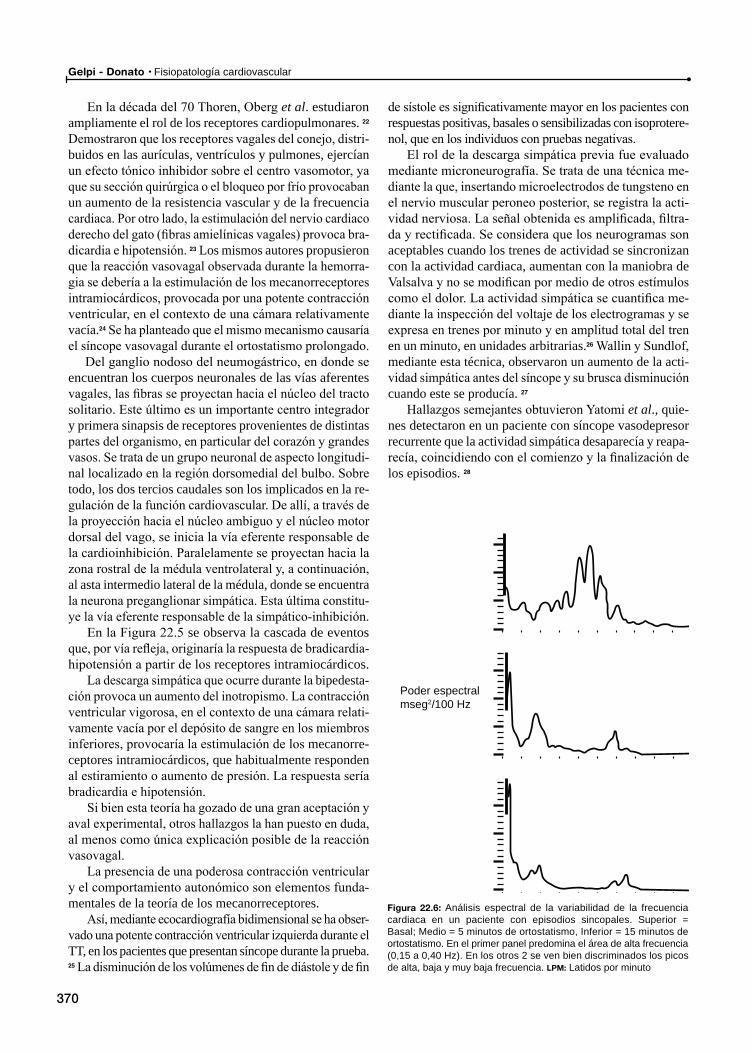
Gelpi - Donato Fisiopatología cardiovascular
370
et al. estudiaron ampliamente el rol de los receptores cardiopulmonares. 22
-buidos en las aurículas, ventrículos y pulmones, ejercían
un aumento de la resistencia vascular y de la frecuencia
-23 Los mismos autores propusieron
-
vacía.24
encuentran los cuerpos neuronales de las vías aferentes
y primera sinapsis de receptores provenientes de distintas
-
todo, los dos tercios caudales son los implicados en la re-
al asta intermedio lateral de la médula, donde se encuentra -
se observa la cascada de eventos
receptores intramiocárdicos.bipedesta-
-
mecanorre-ceptores intramiocárdicos, que habitualmente responden
-mentales de la teoría de los mecanorreceptores.
-
TT, en los pacientes que presentan síncope durante la prueba. 25
respuestas positivas, basales o sensibilizadas con isoprotere-
-
--
aceptables cuando los trenes de actividad se sincronizan con la actividad cardiaca, aumentan con la maniobra de
-
en un minuto, en unidades arbitrarias.26
mediante esta técnica, observaron un aumento de la acti-
cuando este se producía. 27
et al., quie-nes detectaron en un paciente con síncope vasodepresor recurrente que la actividad simpática desaparecía y reapa-
los episodios. 28
Poder espectralmseg2/100 Hz
Figura 22.6: Análisis espectral de la variabilidad de la frecuencia cardiaca en un paciente con episodios sincopales. Superior = Basal; Medio = 5 minutos de ortostatismo, Inferior = 15 minutos de ortostatismo. En el primer panel predomina el área de alta frecuencia (0,15 a 0,40 Hz). En los otros 2 se ven bien discriminados los picos de alta, baja y muy baja frecuencia. LPM: Latidos por minuto

Fisiopatología del síncope
371
El dosaje de catecolaminas es otro de los métodos me-diante el que se evalúa al simpático. Chosy y Graham
29
que en los pacientes que no pierden el conocimiento. El comportamiento de la adrenalina y la noradrenalina fue evaluado durante el TT. En los individuos normales, la
-mentan a medida que avanza la prueba. La adrenalina se mantiene sin cambios durante el TT. En los pacientes con respuestas positivas, el comportamiento de la noradrena-
la actividad simpática neuronal.30
-
-
-
durante el síncope. Un método más reciente, que permite evaluar de ma-
la variabilidad de la frecuencia cardiaca. La descompo-
permite observar un pico de alta frecuencia relacionado -
entre las áreas de baja y alta frecuencia es considerada -
simpático durante el TT en los pacientes con respues-
que precede al síncope. -
mecanorreceptores pone el énfasis en el -
bién es posible que, en los pacientes susceptibles, dichos receptores sean particularmente sensibles, aun ante estí-mulos de intensidad normal.
et al. probaron esta posibilidad evaluando la respuesta de los de pacientes con síncope, la mitad de ellos con TT positivo
31
--
miembros inferiores. La respuesta normal es el aumento del tono simpático, el aumento de las catecolaminas plas-
-mente más la resistencia vascular, ante distintos niveles
-multáneas, se desconoce si el estímulo fue semejante en
-
Las observaciones realizadas constatan que la respuesta se produce en dos etapas. En la primera de ellas se observa un
-
la actividad simpática renal como la frecuencia cardiaca disminuyen.32
-res arteriales.33 La última etapa puede ser bloqueada por
-miocárdicos.34
Ante el reconocimiento del rol causal de las catecola--
son los estudios realizados en forma controlada.
las condiciones para que se estimulen los mecanorrecep-tores están dadas en los pacientes con síncope neurocar-
poner en duda esta teoría.---
es abolida por completo por la et al
-
de la frecuencia de la aurícula remanente, mientras que la frecuencia del ventrículo denervado permanecía estable y aun aumentaba levemente.35 Otras respuestas positivas
-critas después.36 -

Gelpi - Donato Fisiopatología cardiovascular
372
de la frecuencia, tanto de la aurícula remanente como del -
depresor como, por ejemplo, los barorreceptores arteria-les, o el sistema nervioso central, o bien un mecanismo
se encuentra relativamente vacía durante el ortostatismo.
-tricular no es unánime. En un estudio, por ejemplo, se ha
una hipercontractilidad.37
-
-
mecanorreceptores intramiocárdicos son activados predominantemente durante la sístole, la fre-
el aumento de la contractilidad ventricular en los momen-tos previos al síncope no es un desencadenante claro del
-
respuesta vasoconstrictora a nivel de los miembros supe-riores en los primeros minutos de ortostatismo, mucho antes de que aparezcan los síntomas. 38-39
cardiaca. En un estudio reciente se ha observado que los pa-
--
40
la prueba, otros autores han realizado observaciones dife-rentes. Así, se ha descrito que la principal característica
-
pacientes con síncope, observamos que el único predictor independiente del comportamiento de los diferentes pará-metros de la variabilidad de la frecuencia cardiaca fue la edad.41
-
--
-
42
Por último, la inefectividad de los bloqueantes -
-
-
-
-
un estímulo de los mecanorreceptores intramiocárdicos.
Teoría neurohumoral
Opioides endógenosLos
-43 -
prevenir el colapso circulatorio causado por hipovolemia et al
44
-
-
que intervinieran los mecanorreceptores intramiocárdicos.
reacciones vasodepresoras inducidas por el ejercicio, re-
que en los controles sometidos al mismo nivel de ejercicio

Fisiopatología del síncope
373
y que no desarrollaron síncope.45
únicas hormonas pituitarias que se incrementan durante la
cortisol plasmático y la prolactina también se encuentran incrementados.46
Arginina-vasopresina, renina-
y de renina plasmáticas, en dos pacientes que en el
47
-
a la hipovolemia central, y está mediada por los receptores arteriales y cardiopulmonares, respectivamente, que ejercen
-
-
la respuesta hipotensora.-
-
precoz durante la etapa hemodinámicamente compensada.
llevado a postular que en los pacientes susceptibles, los
-ceptores intramiocárdicos y los barorreceptores.
Serotoninaserotonina sería el mediador de la
-
-
-48
-nos presentes durante el síncope. Los receptores seroto-
cardiovascular y el sistema nervioso central en donde la -
--
- 49 Estas últimas se encuentran altamente incrementadas durante el síncope. Lo mismo ocurre con el cortisol plasmático, por lo que la serotonina parece ser una sustancia involucrada en poten-
-tral, tal como lo es la clomipramina, es mayor en los pa-
aferentes.
Óxido nítrico (NO)
-
encuentra en forma constitutiva en la pared endotelial y
se encuentra en las neuronas tanto centrales como peri-
intracelular y produce una escasa -
san. Finalmente una isoforma inducible interviene en la
-
distintos estímulos dependientes de receptores como la acetilcolina, serotonina, bradiquinina y noradrenalina o no mediados por receptores, tales como el estiramiento
trabajos de Mellander et al. 50
51 En condiciones de
-
resistencia. En cambio, en preparaciones denervadas la respuesta mediada por las catecolaminas circulantes se

Gelpi - Donato Fisiopatología cardiovascular
374
mayores de resistencia a este control. En ellos, el efecto vasodilatador estaría mediado por el NO.
Kaufmann et al
respuestas positivas.52 Por otro lado, Dietz et al. han -
tal también depende del NO.53 En el caso del síncope
Otra sustancia cuyo rol queda por dilucidar es la ade--
54
que se han medido, falta el establecimiento claro de una
-
de la vasopresina o la endotelina pueden ser la respuesta
Teoría de la disfunción barorrefleja
-
resultados altamente contradictorios. Esta discrepancia
-
Barorreceptoresbarorreceptores: los de
-monares, también conocidos como mecanorreceptores ya nombrados en párrafos anteriores.
Los barorreceptores arteriales son estimulados por el -
cardiaca, de la contractilidad, de la resistencia periférica y
-bién actúan en condiciones de reposo contrabalanceando
El
-
--
es muy dependiente de la distensibilidad del vaso. Los re-
-
La curva de respuesta de los barorreceptores tiene una
-
intramiocárdicos, la primera sinapsis ocurre en el núcleo del tracto solitario, desde donde se conecta con el núcleo
-lateral de la médula. El principal neurotransmisor de las
ácido 55 aunque también se ha propuesto a la sustancia
-
se suele evaluar a través de la actividad del nervio peroneo -
-
56, 57
-do adicional se produce a nivel del efector en la respuesta
-
provenientes de los mecanorreceptores, quimiorrecepto-
--

Fisiopatología del síncope
375
Evaluación de la sensibilidad barorrefleja (SBR) El estudio de la sensibilidad de los barorreceptores ha des-
-do de miocardio 58 59 El estudio
Diversos métodos han sido utilizados para medir la -
-
nervio peroneo o la actividad simpática renal.
de los receptores como de las vías aferentes, los centros
-
en el animal in vivoen la respuesta.
-
del cambio en la frecuencia cardiaca ante un determinado -
mostrado tener valor clínico.
Maniobras fisiológicasManiobra de Valsalva
La maniobra de
60
-
arterias periféricas y al aumento transitorio del trabajo
por el simpático, ya que ocurre aun en pacientes con sec--
-
la taquicardia y del aumento de la resistencia periférica.
y tiende a aumentar hasta los valores basales, aun cuan-
en los barorreceptores arteriales, cardiopulmonares y en los quimiorreceptores. La tercera fase corresponde a los
-
de la TA. Por último, en la cuarta fase, constituida por los
de los barorreceptores arteriales es característica de esta
no parece ser muy importante ya que la bradicardia no
-
et al.61
-
Por lo tanto, la bradicardia alcanzada en esta fase indica
Figura 22.7: Etapas de la maniobra de Valsalva. TAS: Presión arterial sistólica; FC: Frecuencia Cardiaca. (Ver texto)

Gelpi - Donato Fisiopatología cardiovascular
376
-
-servado durante la cuarta fase y el RR más corto durante la
--
Durante la cuarta fase es posible medir la sensibilidad -
-te el análisis de la primera fase tiene la ventaja de que el
-
Ortostatismo
de la respuesta simpática que se observa ante el ortosta-tismo, no son los únicos. Ya se han nombrado los meca-
62 Este último mecanismo ha sido claramente demostrado en ani-
el ser humano. Una forma clásica de evaluar la respuesta
El valor normal es superior a 1.-
-canorreceptores, observándose bradicardia mediada por
-
aunque los valores hallados son inferiores.63
-
índices, la multiplicidad de factores que la determinan
Estimulación farmacológicaet al
64
versus los cambios de
la frecuencia cardiaca o del intervalo RR, obteniéndose la
ambas variables. Un valor elevado indica una fuerte res-
respuesta simpática.--
de los barorreceptores arteriales, ya que no ocurre ante la
--
Mientras que en la bradicardia provocada por un au-mento de la -
et al-
-
65
que los cambios en ambas variables no son aleatorios, se
los barorreceptores, alterando el parámetro que se desea medir.Succión a través de un collar en el cuello
-
66 Posteriormente se -
sentidos. Mediante esta técnica es posible estudiar no solo los cambios en la frecuencia cardiaca provocados por la
-
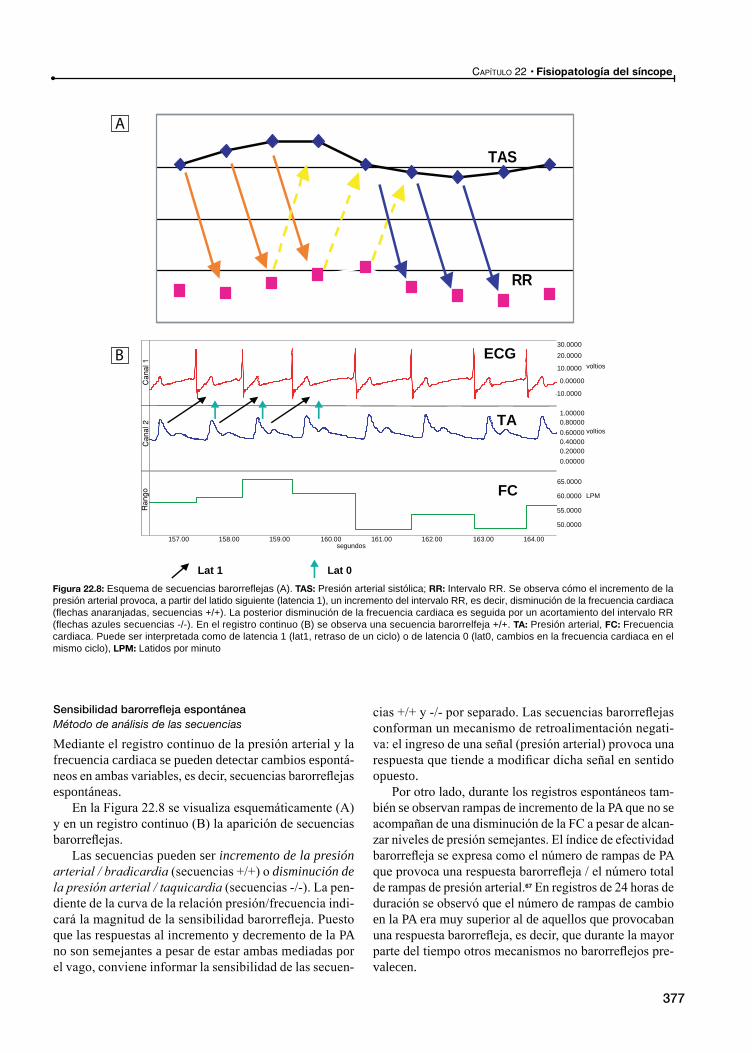
Fisiopatología del síncope
377
Sensibilidad barorrefleja espontáneaMétodo de análisis de las secuencias
frecuencia cardiaca se pueden detectar cambios espontá-
espontáneas.
Las secuencias pueden ser incremento de la presión disminución de
--
que las respuestas al incremento y decremento de la PA no son semejantes a pesar de estar ambas mediadas por
-
-
opuesto.-
bién se observan rampas de incremento de la PA que no se -
67
en la PA era muy superior al de aquellos que provocaban
-valecen.
157.00 158.00 159.00 160.00 161.00 162.00 163.00 164.00 segundos
-10.0000
0.00000
10.0000
20.000030.0000
voltios
0.00000 0.200000.400000.600000.800001.00000
voltios
50.0000
55.0000
60.0000
65.0000
LPM
ECG
TA
FC
Lat 1 Lat 0
TAS
RR
B
A
Figura 22.8: Esquema de secuencias barorreflejas (A). TAS: Presión arterial sistólica; RR: Intervalo RR. Se observa cómo el incremento de la presión arterial provoca, a partir del latido siguiente (latencia 1), un incremento del intervalo RR, es decir, disminución de la frecuencia cardiaca (flechas anaranjadas, secuencias +/+). La posterior disminución de la frecuencia cardiaca es seguida por un acortamiento del intervalo RR (flechas azules secuencias -/-). En el registro continuo (B) se observa una secuencia barorrelfeja +/+. TA: Presión arterial, FC: Frecuencia cardiaca. Puede ser interpretada como de latencia 1 (lat1, retraso de un ciclo) o de latencia 0 (lat0, cambios en la frecuencia cardiaca en el mismo ciclo), LPM: Latidos por minuto

Gelpi - Donato Fisiopatología cardiovascular
378
-
-
rápida.14
-
análisis secuencial tiene la ventaja de permitir una cuanti-
Análisis espectral: puede modular a la frecuencia cardiaca en determinada banda de frecuencia, es decir, que a determinada oscila-
-jante en la frecuencia cardiaca mediada por la actividad
et al 69
como coherencia. Esta debe ser superior a 0.50 para que dicha frecuencia sea tenida en cuenta para el análisis.
estaría determinado por otros de los múltiples mecanis--
baja frecuencia.
validados comparándolos con los métodos más tradiciona--
70
Medición de la rama simpática del barorreflejo (MNSA): Los --
díamente la respuesta simpática.
La respuesta simpática mensurable a través de la mi--
diástole.71
--
tividad de las motoneuronas simpáticas. Cuando la acti-
-
total del tren medida en unidades arbitrarias. En indivi-duos sanos, la actividad simpática es muy variable entre individuos, pero reproducible para un mismo sujeto aun
Sensibilidad barorrefleja durante el ortostatismo-
sino que también se acorta la latencia con respecto al intervalo RR previo. Ello puede deberse a una dismi-
de motoneuronas simpáticas con mayor velocidad de 72
-
mismo ocurre en otras circunstancias en las que está ele-
el ejercicio192
-73
Sensibilidad barorrefleja en los pacientes con síncope vasovagal
-
son variados los métodos utilizados y los criterios de se-74-83
Tabla 22.5: Sensibilidad barorrefleja (SBR) mediante diferentes métodos
negativo o controles
Autor Edad Método SBR
Julu et al; 1981 30-55 Secuencial
Jardine et al; 1976 52±17 Secuencial >
Freitas et al; 1977Secuencial =Espectral =
et al; 1978 30±14 Secuencial
Morillo et al; 1979 48±3 <et al; 1974 21-41 <
Thomson et al; 1975 44±17 Collar-FNE =El-Sayed et al; 1980 Collar
Cooper et al; 1982 37 Collar =Béchir et al; 1983 30±7 Espectral <
FNE: Fenilefrina, NTP: Nitroprusiato

Fisiopatología del síncope
379
-
-
tienen un efecto directo sobre el
-
Cambios no detectados por la primera pueden ponerse de
los pacientes y controles reclutados puede representar otra fuente de discrepancia.
se ha informado una menor74 o semejante75
El método de análisis de secuencias también arroja resultados contradictorios de una mayor o semejante76-77
Ante el estímulo -
cia con los controles como un valor inferior o una mayor caída en los pacientes con síncope.
-
--
nes basales. Durante el ortostatismo, los pacientes con ante-
En vista de las discrepancias halladas en los diferentes
-
hormonales, que predisponen al cuadro
-
durante el TT.
Teoría de la reducción de volumenEn vista de que con frecuencia el síncope puede ser pre-venido por medidas que incrementen el volumen vascular,
evidencia parece indicar que, más que el volumen plas-
el desarrollo del síncope.
et al., midiendo la impedancia torácica, esplác-nica y pélvica durante el Tilt test, han observado que los adolescentes con síncope y TT positivo presentan un me-
-
pélvico ni de los miembros inferiores. El lecho esplácnico representa individualmente el mayor reservorio de volu-
84
Teoría del desacoplamiento de las señalesLipsitz et al. han descrito la presencia de una desincroni-
frecuencia cardiaca en los momentos previos al síncope. 85 -
amplitud respiratoria aumenta. Un minuto y medio antes
espectral de la variabilidad de la frecuencia cardiaca en las áreas de alta frecuencia y de baja frecuencia. Por lo
-toria pero no lo hacen el intervalo RR ni la variabilidad.
Respuestas vasculares paradojales en pacientes con síncope
el síncope.
Alteración de la respuesta venoconstrictoraEl comportamiento de los vasos de capacitancia en los pacientes con TT positivo es debatido. Por un lado, se ha observado que no se producen los cambios normales del tono venoso del muslo durante el ortostatismo 86 y que -
--
et al. observaron que no hay diferencias en la respuesta venoconstrictora de los miembros superiores, ante la
-

Gelpi - Donato Fisiopatología cardiovascular
380
en presencia de una respuesta venoconstrictora adecuada, no pudiendo ser esta última la causa primera del síncope
-
capacitancia esplénicos durante el ejercicio. 87
Reacción paradojal de la vasculatura cerebral-
88 mediante Doppler transcraneano, lo que se interpreta como una
de conocimiento en los pacientes con síncope neurocar-
la respuesta de la vasculatura cerebral no es paradojal sino --
más de un minuto. 89
--
El rol de los receptores 2-adrenérgicos-
-tico a nivel del nervio peroneo durante el síncope, otra
-
-
1 1 -
-
iniciada la prueba con respecto a las basales, siendo dicho
TT positivo.90
-
-
, no im-91
Síncope vasovagal y respuesta a la emoción
Un intenso estímulo emocional puede producir dos tipos
cuando lo que ocurre es temor o un intenso malestar, se
92
-
hipotalámica en zonas cercanas a las responsables de la
Participación del mecanismo neurocardiogénico en el síncope que ocurre en otras entidades
Nos hemos referido hasta ahora principalmente al cua-
síncope que se presenta en forma aislada, en ausencia de -
-
Isquemia miocárdicaLa isquemia y el infarto de miocardio desencadenan re-
-
Los mecanorreceptores intramiocárdicos están prin-cipalmente distribuidos en las paredes inferior y poste-rior del ventrículo izquierdo. En los infartos que afectan esta área, es frecuente observar episodios de bradicardia-
pared anterior es más frecuente que ocurra taquicardia
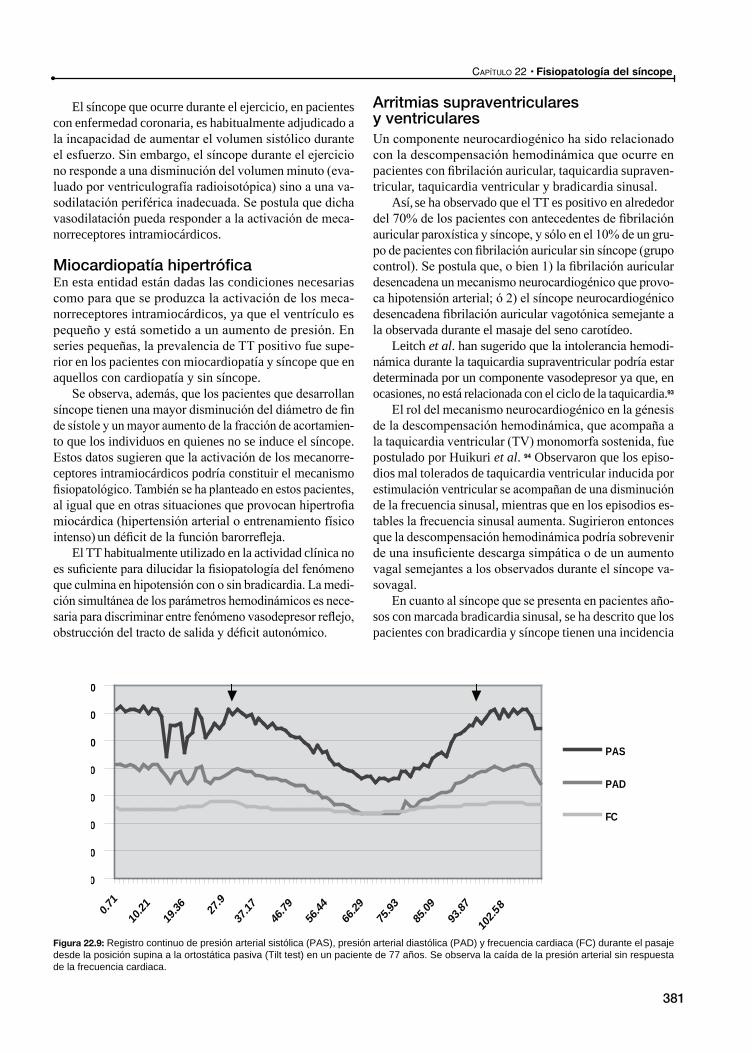
Fisiopatología del síncope
381
El síncope que ocurre durante el ejercicio, en pacientes con enfermedad coronaria, es habitualmente adjudicado a
--
-norreceptores intramiocárdicos.
Miocardiopatía hipertróficaEn esta entidad están dadas las condiciones necesarias
-norreceptores intramiocárdicos, ya que el ventrículo es
-rior en los pacientes con miocardiopatía y síncope que en aquellos con cardiopatía y sin síncope.
-to que los individuos en quienes no se induce el síncope.
mecanorre-ceptores intramiocárdicos podría constituir el mecanismo
El TT habitualmente utilizado en la actividad clínica no
--
Arritmias supraventriculares y ventriculares
pacientes con auricular, taquicardia supraven-tricular, taquicardia ventricular y bradicardia sinusal.
Así, se ha observado que el TT es positivo en alrededor
-
-
la observada durante el masaje del seno carotídeo. Leitch et al -
námica durante la taquicardia supraventricular podría estar determinada por un componente vasodepresor ya que, en ocasiones, no está relacionada con el ciclo de la taquicardia.93
postulado por Huikuri et al. 94 Observaron que los episo-dios mal tolerados de taquicardia ventricular inducida por
de la frecuencia sinusal, mientras que en los episodios es-
-
-sos con marcada bradicardia sinusal, se ha descrito que los pacientes con bradicardia y síncope tienen una incidencia
0
0
0
0
0
0
0
0
0.71
10.21
19.36 27
.9
37.17
46.79
56.44
66.29
75.93
85.09
93.87
102.5
8
PAS
PAD
FC
Figura 22.9: Registro continuo de presión arterial sistólica (PAS), presión arterial diastólica (PAD) y frecuencia cardiaca (FC) durante el pasaje desde la posición supina a la ortostática pasiva (Tilt test) en un paciente de 77 años. Se observa la caída de la presión arterial sin respuesta de la frecuencia cardiaca.
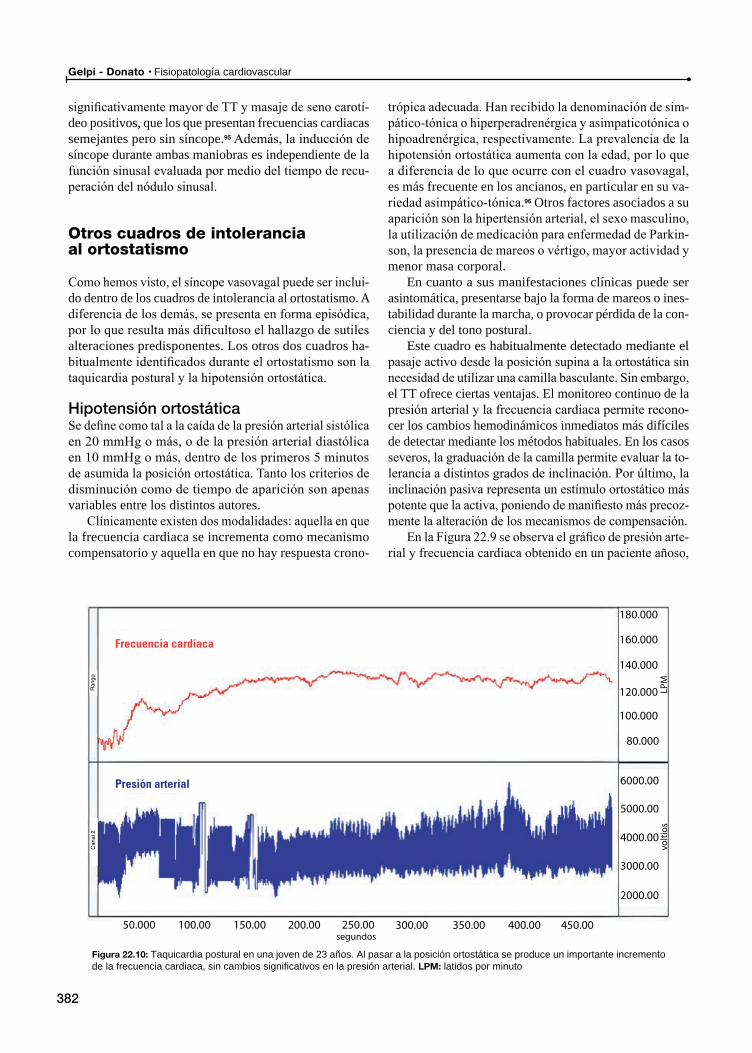
Gelpi - Donato Fisiopatología cardiovascular
382
-deo positivos, que los que presentan frecuencias cardiacas semejantes pero sin síncope.95
síncope durante ambas maniobras es independiente de la -
Otros cuadros de intolerancia al ortostatismo
-do dentro de los cuadros de intolerancia al ortostatismo. A
alteraciones predisponentes. Los otros dos cuadros ha-
Hipotensión ortostática
variables entre los distintos autores.
la frecuencia cardiaca se incrementa como mecanismo compensatorio y aquella en que no hay respuesta crono-
-
es más frecuente en los ancianos, en particular en su va-96 Otros factores asociados a su
-
menor masa corporal. En cuanto a sus manifestaciones clínicas puede ser
asintomática, presentarse bajo la forma de mareos o ines-tabilidad durante la marcha, o provocar pérdida de la con-ciencia y del tono postural.
Este cuadro es habitualmente detectado mediante el
el TT ofrece ciertas ventajas. El monitoreo continuo de la -
cer los cambios hemodinámicos inmediatos más difíciles de detectar mediante los métodos habituales. En los casos
-
-
-
Frecuencia cardíaca
Presión arterial
50.000 100.00 150.00 200.00 250.00segundos
300.00 350.00 400.00 450.00
180.000
160.000
140.000
120.000
100.000
80.000
6000.00
5000.00
4000.00
3000.00
2000.00
volt
ios
LPM
Figura 22.10: Taquicardia postural en una joven de 23 años. Al pasar a la posición ortostática se produce un importante incremento de la frecuencia cardiaca, sin cambios significativos en la presión arterial. LPM: latidos por minuto

Fisiopatología del síncope
383
-
-
Ello puede suceder por alteraciones en el funciona-
--
ton afecta a las terminaciones simpáticas y se caracteriza
forma asociada a otros trastornos centrales como enfer--
dos veces más frecuente en el hombre que en la mujer, se -
relacionados con el equilibrio y la marcha. Cuando afecta
a los del Parkinson, con el que suele confundírselo. Lo
-
los cuerpos de Lewis que predominan más en las células
están preservadas, los niveles basales de NA son normales pero no aumentan con el ortostatismo.
neuropatía periférica presente en múltiples enfermedades -
mo. En ellas los niveles de NA se encuentran basalmente disminuidos.
-
-
Taquicardia postural-
-
el ortostatismo tales como mareos, palpitaciones, sensa-
disconfort torácico, presíncope y síncope. Este cuadro ha recibido otros nombres tales como síndrome de taquicardia
asociado con el prolapso de válvula mitral, el síndrome de corazón de soldado.97
-98 y/o de la masa eritroci-
taria.99
inferiores es otro de los mecanismos más propuestos en
volumen minuto observada durante el ortostatismo es un
-
inferiores. Así, a la hipersensibilidad venosa localizada en
sudomotora en las piernas100 -hidrosis101
102
-
103
104 taquicar-dia postural, un incremento de la
la complianceel umbral de edema durante el ortostatismo estuvo reduci-
-
et al.105
familia con síndrome de taquicardia postural refractaria a
del transportador y se condice con el incremento en la con-
familiar. Golstein et al. encuentran que, en los pacientes con
-
simpático. En otro estudio se ha detectado la presencia de un factor circulante, que inhibe selectivamente a los recep-
1

Gelpi - Donato Fisiopatología cardiovascular
384
la falla primaria podría encontrarse en los receptores del 106
-dros de intolerancia al ortostatismo tiene sus caracterís-
-
-
-
central. Ella, a su vez, hipotéticamente, estaría determina-
--
terapéutica también muestra similitudes. En todos estos cuadros, el incremento de la volemia y su correcta redis-
demostrado ser de utilidad.et al -
-
pacientes que solo presentaron taquicardia aislada inicial 107
del volumen minuto, o de la resistencia periférica, no cuen--
presentadas, más allá de su relevancia clínica, ponen de
al medio.
Bibliografía
11. Linzer M, Gold DT, Potinen M. “Impairment of physical and psychosocial function in recurrent syncope”. J Clin Epidemiol 1991;44:1037-43.
12. Krahn AD, Klein GJ, Yee R, Manda. “The high cost of syncope: cost implications of a new insertable loop recorder in the investi-gation of recurrent syncope”. Am Heart J. 1999;137:870-7.
13. Eagle KA, HR Black. “The impact of diagnostic tests in evaluating patients with syncope”. Yale J Biol Med. 1983;56:1-8.
14. Martin GJ, Adams SL, Martin HG, Mathews J, Zull D, Scanlon PJ. “Prospective evaluation of syncope”. Ann Emerg Med 1984;13:499-504.
15. Silverstein MD, Singer DE, Mulley AG, Thibault GE, Barnett GO. “Patients with syncope admitted to medical intensive care units”. JAMA 1982;248: 1185-9.
16. Chang-Sing P, CT Peter. “Syncope: evaluation and management. A review of current approaches to this multifaceted and complex clinical problem”. Cardiol Clin 1991;9:641-51.
17. Dubner SJ, Roel J, Sokn FJ. “Estudio de los pacientes con sín-cope. Evaluación diagnóstica y seguimiento”. Rev Arg Cardiol 1992;60:147.
18. Day SC, Cook EF, Funkenstein H, Goldman L. “Evaluation and outcome of emergency room patients with transient loss of cons-ciousness”. Am J Med 1982;73:15-23.
19. Fitzpatrick A, Theodorakis G, Vardas P. “The incidence of ma-lignant vasovagal syndrome in patients with recurrent syncope”. Eur Heart J 1991;12: 389-94.
10. Maloney JD, Jaeger FJ, Fouad-Tarazi FM, Morris HH. “Malignant vasovagal syncope: prolonged asystole provoked by head-up tilt. Case report and review of diagnosis, pathophysiology and therapy”. Clevel Clin J Med 1988;55:542-8.
11. Brignole M, Menozzi C, Del Rosso A, Costa S, Gaggioli G, Bottoni N, Bartoli P, Sutton R. “New classification of haemodynamics of vasovagal syncope: beyond the VASIS classification”. Europace 2000;2:66-76.
12. Wasmund SL, Smith ML, Takata TS et al. “Sympathoexcitation is attenuated during low level lower body negative pressure in subjects who develop pre-syncope”. Clin Auton Res. 2003;13:208-13.
13. Sander-Jensen K, Mehlsen J, Secher NH et al. “Progressive central hypovolaemia in man- resulting in a vasovagal syncope? Haemo-dynamic and endocrine variables during venous tourniquets of the thighs”. Clin Physiol. 1987;7:231-42.
14. Saul JP, Berger RD, Albrecht P, Stein SP, Chen MH, Cohen RJ. „Transfer function analisis of the circulation: unique insights into cardiovascular regulation”. Am J Physiol 1991;261:H1231-1245.
15. Horner RL, Brooks D, Kozar LF, Gan K, Phillipson EA. “Respira-tory-related heart variability persists during central apnea in dogs: mechanisms and implications”. J Appl Physiol 1995;78:2003-2013.
16. Mullen TJ, Appel ML, Mukkamala R, Mathias JM, Cohen RJ. “System identification of closed-loop cardiovascular control: effects of posture and autonomic blockade”. Am J Physiol 1997;272 (1 Pt 2): H448-61.
17. Wieling W, JJ van Lieshout. “Maintenance of postural Normo-tension in humans”. In: P. Low (ed). Clinical Autonomic Disorders. Little Brown Co. Boston 1993;69-75.
18. Abboud, FM. “Integration of reflex responses in the control of blood pressure and vascular resistance”. Am J Cardiol 1979;44:903-911.
19. Barcroft H, Edholm OG, McMichael J. “Posthaemorrhagic fainting: Study by cardiac out-put and forearm flow”. Lancet 1944;I:489-490.
20. Morillo CA, Eckberg DL, Ellenbogen KA, Beightol LA, Hoag JB, Tahvanainen KU, Kuusela TA, Diedrich AM. “Vagal and sympa-thetic mechanisms in patients with orthostatic vasovagal syncope”. Circulation 1997;96:2509-13.
21. Bezold A, L Hirt. “Ueber die physiologischen Wirkungen des Essigsauren Veratrins”. Physiol Lab Wurzburg 1867;1:73-156.
22. Oberg B, P Thoren. “Studies on left ventricular receptors, signalling in non-medullated vagal afferents”. Acta Physiol Scand 1972;85:145-163.
23. Oberg B, P Thoren. “Circulatory responses to stimulation of me-dullated and non medullated afferents in the cardiac nerve in the cat”. Acta Physiol Scand. 1973;87:121-32.
24. Oberg B, P Thoren. “Increased activity in left ventricular receptors during hemorrhage or occlusion of caval veins in the cat: a possible

Fisiopatología del síncope
385
cause of the vaso-vagal reaction”. Acta Physiol Scand 1972;85: 164-173.
25. Shalev Y, Gal R, Tchou PJ, Anderson AJ, Avitall B, Akhtar M, Jazayeri MR. “Echocardiographic demonstration of decreased left ventricular dimensions and vigorous myocardial contrac-tion during syncope induced by head-up Tilt”. J Am Cardiol 1991;18:746-51.
26. Mark AL, Victor RG, Nerhed C, Wallin BG. “Microneurographic studies of the mechanisms of sympathetic nerve resposes to static exercise in humans”. Circ Res 1985;57:461-9.
27. Wallin BG, G Sundlof. “Sympathetic outflow in muscles during vasovagal syncope”. J Autonom Nerv Syst 1982;6:287-291.
28. Yatomi A, Iguchi A, Uemura K, Sakamoto N, Iwase S, Mano T. “A rare case of recurrent vasodepressive attacks of 2-hours duration: Analysis of the mechanism by muscle sympathetic nerve activity recording”. Clin Cardiol 1989;12: 164-8.
29. Chosy J, D Graham. “Catecholamines in vasovagal fainting”. J Psichosom Res 1965;9:189.
30. Sra JS, Murthy V, Natale A, Jazayeri MR, Dhala A, Deshpande S, Sheth M, Akhtar M. “Circulatory and catecholamine change during head-up tilt testing in neurocardiogenic (vasovagal) syncope”. Am J Cardiol 1994;73:33-7.
31. Sneddon JF, Counihan PJ, Bashir Y, Haywood GA, Ward DE, Camm AJ. “Assessment of autonomic function in patients with neurally mediated syncope: augmented cardiopulmonary barore-ceptor responses to graded orthostatic stress”. J Am Coll Cardiol 1993;21:1193-8.
32. Evans RG, Ludbrook J, Ventura S. “Role of vagal afferents in the haemodinamic response to acute central hypovolaemia in una-naesthetiz rabbits”. J Auton Nerv Syst 1993;46:251-60.
33. Morita H, SF Vatner. “Effects of hemorrhage on renal nerve activity in conscious dogs”. Circ Res 1985;57:788-793.
34. Evans RG, Hayes IP, Ludbrook J, Ventura S. “Factors confoun-ding blockade of cardiac afferents by intrapericardial procaine in conscious rabbits”. Am J Physiol 1993;264:H1861-1870.
35. Scherrer U, Vissing S, Morgan B, Hanson P, Victor RG. “Vasovagal syncope after infusion of vasodilator in a heart transplant recipient”. New Engl J Med 1990;322:602-4.
36. Fitzpatrick AP, Banner N, Cheng A, Yacoub M, Sutton R. “Vaso-vagal reactions may occur after orthotopic heart transplantation”. J Am Coll Cardiol 1993;21:1132-37.
37. Yamanouchi Y, Jaalouk S, Shehadeh AA, Jaeger F, Goren H, Fouad-Tarazi FM. “Changes in left ventricular volume during head up tilt in patients with vasovagal syncope: An echocardiographic study”. Am Heart J 1996;131:73-80.
38. Sneddon JF, Counihan PJ, Bashir Y, Haywood GA, Ward DE, Camm AJ. “Impaires immediate vasoconstrictor responses in patients with recurrent neurally mediated syncope”. Am J Cardiol 1993; 71:72-76.
39. Thomson HL, Atherton JJ, Khafagi FA, Frenneaux MP. “Failure of reflex venoconstriction during exercise in patients with vasovagal syncope”. Circulation 1996;93:953-59.
40. Goldstein DS, Holmes C, Frank SM, Dendi R, Cannon RO 3rd, Sharabi Y, Esler MD, Eisenhofer G. “Cardiac sympathetic dysauto-nomia in chronic orthostatic intolerance syndromes”. Circulation. 2002;106:2358-65.
41. Ruiz GA, Madoery C, Arnaldo F, Menéndez C, Tentori MC. “Frequency-domain analysis of heart rate variability during positive and negative head-up tilt test: importance of age”. Pacing Clin Electrophysiol. 2000;23:325-32.
42. Calkins H, Kadish A, Sousa, J Rosenheck S, Morady F. “Compa-rison of responses to isoproterenol and epinephrine during head up tilt in suspected vasodepressor syncope”. Am J Cardiol 1991;67: 207-9.
43. Burke SL, PK Dorward. “Influence of endogenous opiates and cardiac afferents on renal nerve activity during hemorrhage in conciencious rabbits”. J Physiol 1988;402:9-27.
44. Smith ML, Carlson MD, Sheehan HM. “Naloxone does not pre-vent vasovagal syncope during simulated orthostasis in humans”. Physiologist 1991;34: 238-239.
45. Perna G, Ficola U, Salvatori MP, Stanislao M, Vigna C, Villella A, Russo A, Fanelli R, Paleani Vettori PG, Loperfido F. “Increase of plasma beta endorphins in vasodepressor syncope”. Am J Cardiol 1990;1:929-30.
46. Jardine DL, Melton IC, Crozier IG, Bennett SI, Donald RA, Ikram H. “Neurohormonal response to head-up tilt and its role in vasovagal syncope”. Am J Cardiol 1997;79:1302-1306.
47. Gunter A, A Wagner. “Excessive secretion of vasopressin during vasovagal reaction”. Am Heart J 1991;121:602.
48. Abboud, FM. “Neurocardiogenic syncope”. New Engl J Med 1993;328:1117-1120.
49. Theodorakis GN, Markianos M, Livanis EG. Zarvalis E, Flevari P, Kremastinos DT. “Central serotoninergic responsiveness in neurocardiogenic syncope”. Circulation 1998;98:2724-2730.
50. Mellander S, Bjornberg J, Ekelund U, Alm P. “Cardiovascular regulation by endogenous nitric oxide is essential for survival after acute haemorrhaga”. Acta Physiol Scand 1997;160:57-65.
51. Maspers M, Bjornberg J, Grande PO, Mellander S. “Sympathetic an adrenergic control of large bore arterial vessels, arterioles and veins, and of capillary pressure and fluid exchange in whole organ cat skeletal muscle”. Acta Physiol Scand 1990;38: 509-521.
52. Kaufmann H, Berman J, Oribe E, Oliver J. “Possible increase in the synthesis of endothelial derived relaxing factor (EDRF) during vasovagal syncope (abstr)”. Clin Autonom Res 1993;3:69.
53. Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. “Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans”. J Physiol Lond 1994;480:361-8.
54. Nelson SD, Stanley M, Love ChJ, Coyne KS, Schaal SF. “The au-tonomic and hemodynamic effects of oral theophylline in patients with vasodepressor syncope”. Arch Intern Med 1991;151:2425-29.
55. Reis DJ, Granata AR, Perrone MH, Talman WT. “Evidence that glutamic acid is the neurotransmitter of baroreceptor afferent terminating in the nucleus tractus solitarius (NTS)”. Auton Nerv Syst 1981;3:321-34.
56. Kunze, DL. “Reflex discharge patterns of cardiac vagal efferent fibres”. J Physiol 1972;222:1-15.
57. Fagius J, BG Wallin. “Sympathetic reflex latencies and conduction velocities in normal man”. J Neurol Sci 1980;47:433:448.
58. La Rovere MT, Bigger TJ, Marcus FI, Mortara A, Schwartz PJ for the ATRAMI (Autonomic Tone and Reflexes After Myocardial Infaction) investigators. “Baroreflex sensitivity and heart rate va-riability in prediction of total cardiac mortality after myocardial infaction”. Lancet 1998;351:478-484.
59. Ostersiel KJ, Hanlein D, Willenbrook R, Eichhorn C, Luft F, Dietz R. “Baroreflex sensitivity and cardiovascular mortality in patients with mild to moderate heart failure”. Br Heart J 1995;73:517-522.
60. Eckberg, DL “Parasympathetic cardiovascular control in human disease: a critical review of methods and results”. Am J Physiol 1980;239:H581-93.
61. Smith ML, Beihtol LA, Fritsch-Yelle JM, Ellenbogen KA, Porter TR, Eckberg DL. “Valsalva´s maneuver revisited: a quantitative meted yielding insights into human autonomic control”. Am J Physiol 1996;271:H1240-H1249.
62. Doba N, DJ Reis. “Role of the cerebellum and the vestibular apparatus in regulation of orthostatic reflexes in the cat”. Circ Res 1974;34: 9-18.
63. Takahashi N, Nakagawa M, Saikawa T, Ooie T, Akimitsu T, Kaneda K, Hara M, Iwao T, Yonemochi H, Ito M, Sakata T. “Noninvasive assessment of the cardiac baroreflex: Response to downward til-ting and comparison with the phenylephrine method”. J Am Coll Cardiol 1999;34:211-5.
64. Smyth HS, Sleight P, Pickering GW. “Reflex regulation of arterial pressure during sleep in man”. Circ Res 1969;24:109-121.
65. Chen RY, Fan FC, Schuessler GB, Chien S. “Baroreflex control of heart rate in humans during nitroprusside-induced hypotension”. Am J Physiol 1982;243:R18-R24.
66. Ernsting J, DJ Parry. “Some observations on the effect of stimu-lating the carotid arterial stretch receptors in the carotid artery of man”. J Physiol 1957;137:45P.

Gelpi - Donato Fisiopatología cardiovascular
386
67. Mancia G, Groppelli A, Di Rienzo M, Castiglione P, Parati G. “Smoking impairs baroreflex sensitivity in humans”. Am J Physiol 1997;273(part 2):H1555-H1560.
68. Di Rienzo M, Castiglione P, Parati G, Mancia G, Pedoti A. “Ba-rorreflex modulation of the cardiovascular system: new insights from the joint analysis of blood pressure and heart signals”. Technol Health Care 1996;4:121-128.
69. De Boer RW, Karemaker JM, Strackee J. “Relationships between short-term blood pressure fluctuations and heart rate variability in resting subjects. II a simple model”. Med Biol Eng Comput 1985; 23:359-364.
70. Parati G, Di Rienzo M, Castiglione P, Frattola S, Omboni A, Pedotti A, Mancia G. “Daily-life baroreflex modulation: new perspectives from computer analysis of blood pressure and heart rate variability”. En: Di Rienzo M, Mancia G, Parati G, Pedotti A, Zanchetti A (eds). Computer analysis of cardiovascular signals. Amsterdam: IOS Press 1995;209-218.
71. Sundlof G, G Wallin. “Human muscle nerve sympathetic acti-vity at rest. Relationship to blood pressure and age”. J Physiol 1978;274:621-637.
72. Furlan R, Jacob G, Palazzolo L, Rimoldi A, Diedrich A, Harris PA, Porta A, Malliani A, Mosqueda Garcia R, Robertson D. “Se-quential modulation of cardiac autonomic control induced by cardiopulmonary and arterial baroreflex mechanisms”. Circulation 2001;104: 2932-7.
73. Wasmund SL, Smith ML, Takata TS, Joglar JA, Li JM, Kowal RC, Page RL, Hamdan MH. “Sympathoexcitation is attenuated during low level lower body negative pressure in subjects who develop pre-syncope”. Clin Auton Res 2003;13:208-13.
74. Mosqueda-Garcia R, Furlan R, Fernandez-Violante R, Desai T, Snell M, Jarai Z, Ananthram V, Robertson RM, Robertson DJ. “Sympathetic and baroreceptor reflex function in neurally mediated syncope evoked by tilt”. Clin Invest 1997; 99:2736-44.
75. Thomson HL, Wright K, Frenneaux M. “Baroreflex sensitivity in patients with vasovagal syncope”. Circulation 1997;21;95:395-400.
76. Jardine DL, Ikram H, Frampton CM, Frethey R, Bennett SI, Crozier IG. “Autonomic control of vasovagal syncope”. Am J Physiol 1998;274(6 Pt 2):H2110-5.
77. Freitas J, Pereira S, Lago P, Costa O, Carvalho MJ, Falcao de Freitas A. “Impaired arterial baroreceptor sensitivity before tilt-induced syncope”. Europace 1999;1:258-65.
78. Pitzalis M, Parati G, Massari F, Guida P, Di Rienzo M, Rizzon B, Castiglioni P, Iacoviello M, Mastropasqua F, Rizzon P. “Enhanced reflex response to baroreceptor deactivation in subjects with tilt-induced syncope”. J Am Coll Cardiol 2003;41:1167-73.
79. Morillo CA, Eckberg DL, Ellenbogen KA, Beightol LA, Hoag JB, Tahvanainen KU, Kuusela TA, Diedrich AM. “Vagal and sympa-thetic mechanisms in patients with orthostatic vasovagal syncope”. Circulation 1997;96:2509-13.
80. El-Sayed H, R Hainsworth. “Relationship between plasma volume, carotid baroreceptor sensitivity and orthostatic tolerance”. Clin Sci (Lond) 1995; 88:463-70.
81. Julu PO, Cooper VL, Hansen S, Hainsworth R. “Cardiovascular regulation in the period preceding vasovagal syncope in conscious humans”. J Physiol May 15 2003;549(Pt 1):299-311.
182. Cooper VL, R Hainsworth. “Effects of head-up tilting on barore-ceptor control in subjects with different tolerances to orthostatic stress”. Clin Sci (Lond) 2002;103:221-6.
183. Bechir M, Binggeli C, Corti R, Chenevard R, Spieker L, Ruschitzka F, Luscher TF, Noll G. “Dysfunctional baroreflex regulation of sympathetic nerve activity in patients with vasovagal syncope”. Circulation 2003;107:1620-5.
184. Stewart JM, McLeod KJ, Sanyal S, Herzberg G, Montgomery LD. “Relation of postural vasovagal syncope to splanchnic hypervolemia in adolescents”. Circulation 2004;110:2575-81.
185. Lipsitz LA, Hayano J, Sakata S, Okada A, Morin RJ. “Complex demodulation of cardiorespiratory dynamics preceding vasovagal syncope”. Circulation 1998;98:977-83.
186. Hargreaves AD, AL Muir. “Lack of variation venous tone poten-tiates vasovagal syncope”. Br Heart J 1992;67:486-90.
187. Thomson HL, Atherton JJ, Khafagi FA, Frenneaux MP. “Failure of reflex venoconstriction during exercise in patients with vasovagal syncope”. Circulation 1996;93:953-59.
188. Grubb BP, Gerard G, Roush K, Temesy-Armos P, Montford P, Elliott L, Hahn H, Brewster P. “Cerebral vasoconstriction during head up tilt test induced vasovagal syncope. A paradoxic and unex-pected response”. Circulation 1991;84:1157-1164.
189. Lagi A, Cencetti S, Corsoni V, Georgiadis D, Bacalli S. “Cerebral vasoconstriction in vasovagal syncope: any link with symptoms? A transcranial Doppler study”. Circulation 2001;(27)104:2694-8.
190. Smith, ML. “Mechanisms of vasovagal syncope: Relevance to postflight orthostatic intolerance”. J Clin Pharmacol 1994;34:460-65.
191. Dietz NM, Halliwill JR, Spielmann JM. “Sympathetic withdrawal and forearm vasodilatation during vasovagal syncope in humans (Abstr)”. Circulation 1996;94:I-544.
192. Goldstein, D. “Stress response patterns”. In: Stress, catecholamines, and cardiovascular disease. New York: Oxford University Press 1995;287:328.
193. Leitch JW, Klein GJ, Yee R et al. “Syncope associated with su-praventricular tachycardia: An expression of tachycardia rate or vasomotor response?” Circulation 1992;85:1064.
194. Huikuri HV, Zaman L, Castellanos A et al. “Changes in spon-taneous sinus node rate as an estimate of cardiac autonomic tone during stable and unstable ventricular tachycardia”. J Am Coll Cardiol 1989;13:646.
195. Gaggioli G, Brignole M, Menozzi C et al. “A positive response to head-up tilt testing predicts syncopal recurrence in carotid sinus syndrome patients with permanent pacemakers”. Am J Cardiol 1995;76:720.
196. Ooi WL, Barrett S, Hossain M, Kelley-Gagnon M, Lipsitz LA. “Pat-terns of orthostatic blood pressure change and their clínical correlates in a frail, elderly population”. JAMA 1997;277:1299-304.
197. Jacob G, I Biaggioni. “Idiopathic orthostatic intolerance and postural tachycardia syndromes”. Am J Med Sci 1999;317:88-101.
198. Abi-Samra F, Maloney JD, Fouad FM, Castle LW. “The usefulness of head up tilt testing and haemodynamic investigation in the work -up of syncope of unknown origin”. PACE 1988;11:1202-14.
199. Hoeldtke RD, KM Davis. “The orthostatic tachycardia syndrome: evaluation of autonomic function and treatment with octreotide and ergot alkaloids”. J Clin Endocrin Metab 1991;73:132-9.
100. Schondorf R, PA Low. “Idiopathic postural orthostatic tachycardia syndrome (POTS): an attenuated form of acute pandysautonomia?” Neurology 1993;43:132-7.
101. Khurana, RK. “Orthostatic intolerance and orthostatic tachycardia: a heterogeneous disorder”. Clin Auton Res 1995;5:12-8.
102. Hoeldtke RD, Gworkin GE, Gaspar SR, Israel BC. “Sympatho-tonic orthostatic hypotension: a repost of four cases”. Neurology 1989;39:34-40.
103. Jacob G, Ertl AC, Shannon JR et al. “The effect of standing on neurohumoral responses and plasma volume in healthy subjects”. J Appl Physiol 1998; 84:914-21.
104. Stewart JM, A. Weldon. “Vascular perturbations in the chronic orthostatic tolerance of the postural orthostatic tachycardia syn-drome”. J Appl Physiol 2000;89:1505-1512.
105. Shannon JR, Flattem NL, Jordan J et al. “Orthostatic intolerance and tachycardia associated with norepinephine-transporter defi-ciency”. N Engl J Med 2000;342:541-9.
106. Shapiro RE, Winters B, Hales M et al. “Endogenous circulating sympatholytic factor in orthostatic intolerance”. Hypertension 2000;36: 553-560.
107. Streeten DHP, TF Scullard. “Excessive gravitational blood pooling caused by impaired venous tone is the predominant non-cardiac mechanism of orthostatic intolerance”. Clin Sci 1996;90:277-285.

387
Fisiopatogenia de la Enfermedad de Chagas 23
Epidemiología
El descubrimiento de la -cho sin precedentes en la historia de la medicina mundial,
de Mina Geraes, Brasil, comisionado para efectuar una
Pans-, conocido en Brasil como el barbei-
ro, por su tendencia a picar a sus víctimas en la cara. Al
tripanosoma cruzi en honor a su maestro, Osvaldo Cruz. 1
denominada también Tripanosomiasis americana, que se -
los principales problemas médico-sanitarios de América
de personas infectadas y alrededor de 100 millones de 2
En países donde la enfermedad es endémica, la prevalen-
3
la familia Tripanosomidae y la clase -
es transmitido a los huéspedes mamíferos, vehiculizado
hemípteros, familia de los reduvídeos, subfamilia triato-minae, del que han sido descritas más de 100 especies. De las 17 especies de nuestro país, el vector más importante
-méstico, es el . 4
Estados Unidos, donde fue encontrado el triatoma sangui-suga
.
Factores que favorecen el desarrollo de la endemiaLa tripanosomiasis americana era primitivamente una
-les silvestres y era transmitida por triatomíneos también
-tres naturalmente infectados, ya que estos no presentan
Estudios de ADN realizados en momias humanas deseca-das en forma natural, en la zona costera andina sur, han demostrado que el ciclo selvático de la enfermedad estaba probablemente bien establecido cuando los primeros hu-
esta zona se unieron, en forma inadvertida, a los mamí-feros huéspedes de la enfermedad. Los resultados de este
-5
susceptibilidad del hombre y de ciertos animales domés-
importantes reservorios, así como los roedores. Los fac-tores sociales que favorecen la endemia empiezan por la vivienda: los

Gelpi - Donato Fisiopatología cardiovascular
388
las condiciones de oscuridad, transitoriedad, analfabetismo
del hombre y de los animales suceptibles se disemina. La prevalencia de la endemia está condicionada por
área, en particular el tipo precario de la vivienda y condi-
-
parasitosis a países no endémicos. Estudios recientes co-
--
infectados residen en ese país. 6
Triatominos. Distribución. Relaciones con el agente y el huésped
-
cruzi. Dos tercios de las especies descritas presentaron la --
liados en América se superpone al mapa de la enfermedad humana en el continente.
Las especies mejor adaptadas a la vivienda humana
-barbeiro, ,
parede; en Estados Unidos como , arizona-bedbug; que prevalece en el sur del continente, desde
predomina al norte del Ecuador y vive, entre sus focos naturales, en las palmeras
Pas-, el Triatoma dimidiata y el Triatoma
sordidacomo en América Central.
Los triatomas infectados transmiten el parásito en un ciclo silvestre a una variedad de animales huéspedes, los reservorios naturales del . Los más importan-
acercamiento a la vivienda humana, representan la princi-
al ciclo domiciliario. Otros animales reservorios, con índi-
Los animales que son reservorios domiciliarios de la
-das, ya que disminuyen cuando los triatomas domésticos
en las viviendas humanas se han hallado infectados por el Tripanosoma cruzi
peridomiciliario
--
tras se alimentan y habilidad para adaptarse a la vivienda humana, características que son determinantes de su capa-cidad para transmitir el al ser humano.
Tripanosoma cruzi. Ciclo biológico
El tripanosoma cruzi, durante su ciclo de vida, presenta una serie de transformaciones dentro del insecto vector, del huésped mamífero y de los reservorios silvestres, pa-
-
El reduvídeo vector se infecta mientras se alimenta
en el intestino posterior del insecto, transformándose en
parásitos alcanzan el recto del insecto, transformándose
al huésped animal y que es eliminada con las heces del insecto. Los parásitos penetran a las células del huésped por endocitocis, a través de microescoriaciones produci-das por rascado, o directamente por las mucosas, como la conjuntiva del mamífero.
-
Con la lisis del -

Fisiopatogenia de la enfermedad de chagas
389
especial el tejido muscular esquelético y el cardiaco, por
intracelulares. Asimismo, las formas circulantes pueden
del parásito.
Vías de transmisión
Vía entomológica o vectorial
-
de la presencia de los triatomíneos. El Tripanosoma cruzi es transmitido en el momento en
hués-ped humano. El hábito del triatomíneo de defecar mientras
-
metacíclicas infectantes del parásito. El termotropismo es el estímulo más poderoso al que
obedece el reduvídeo en procura de alimento. Atraído por el cuerpo caliente de la víctima, el reduvídeo pica a la persona mientras duerme. Las picaduras, sobre todo del
, son principalmente en la cara, pero
-
estas escoriaciones.
Vía transfusional
Tripanosoma cruzi -
-
12 Esta -
los países latinoamericanos. La mayoría de los infectados desarrolla una enferme-
--
-
la zona que, a su vez, sería directamente proporcional a la
Vía transplacentariaLa tasa de pasaje transplacentaria del T. cruzi va desde el
-
-7 En
8
parasitemia materna; el feto puede ser afectado en su crecimiento y
-
de los casos, no se observa compromiso fetal, como tam-poco alteraciones en el crecimiento ni en la viabilidad del
-
No obstante se han descrito abortos, muertes fetales, recién nacidos prematuros y desnutridos fetales. 9
-
son de baja prevalencia y sin importancia desde el punto
Historia natural de la Enfermedad de Chagas
Esquema de Rosenbaum
indeterminado o indiferenciado
-
-
enfermedad. El área sombreada sobre este umbral simbo-liza el porcentaje de los pacientes con evidencias clínicas ostensibles, mientras que en la zona por debajo se ilustran los casos descubiertos cuando se utilizan procedimientos
-senta los casos con evidencias de compromiso miocárdico
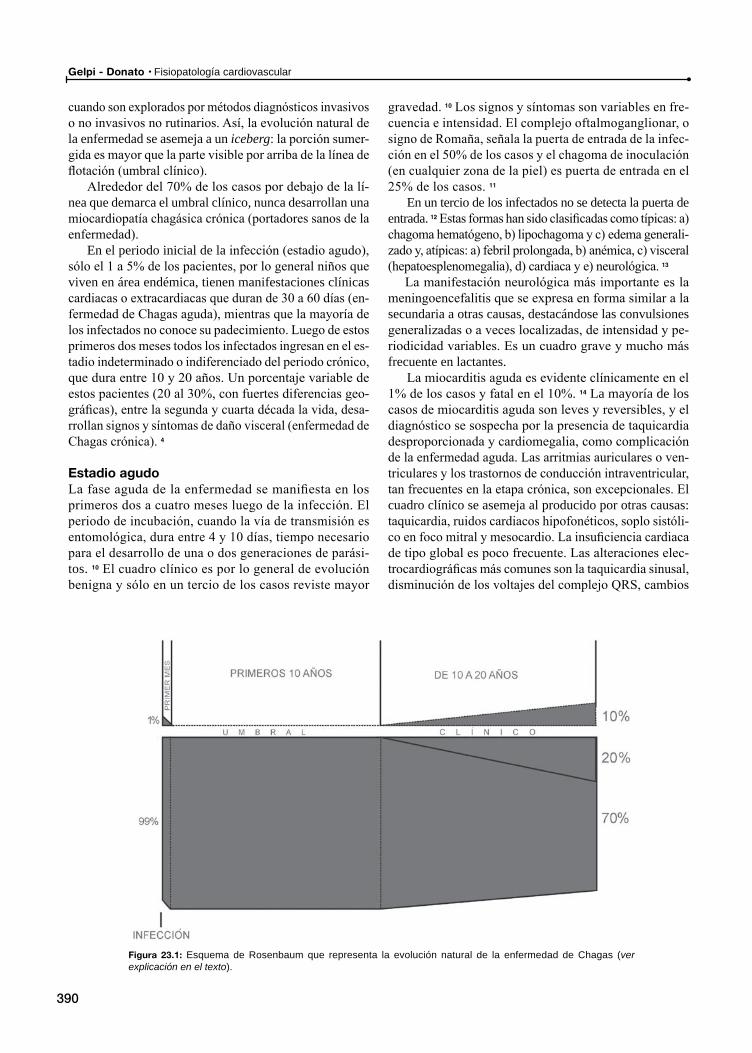
Gelpi - Donato Fisiopatología cardiovascular
390
la enfermedad se asemeja a un iceberg -
-nea que demarca el umbral clínico, nunca desarrollan una
En el periodo inicial de la
viven en área endémica, tienen manifestaciones clínicas -
-
--
4
Estadio agudo
-tos. 10
10 -
-
11
En un tercio de los infectados no se detecta la puerta de entrada. 12
-
13
secundaria a otras causas, destacándose las convulsiones -
frecuente en lactantes.
14 La mayoría de los
-
cuadro clínico se asemeja al producido por otras causas: -
-
Figura 23.1: Esquema de Rosenbaum que representa la evolución natural de la enfermedad de Chagas (ver explicación en el texto).

Fisiopatogenia de la enfermedad de chagas
391
-
El ---
15
Las escasas autopsias realizadas en pacientes con mio-
---
parásitos, aunque esto está relacionado con el número de 25 -
-
taquicardia ventricular e incluso el aleteo ventricular pueden -
de miocardio por cardiomiopatía isquémica. La muerte se produce como consecuencia de taquiarritmias ventriculares
-bolismo pulmonar o sistémico.
-área en-
démica, contacto con los triatominos, tipo de vivienda,
-
paciente y del tipo e intensidad de las manifestaciones.
en el plazo de semanas o meses.-
yor el índice de mortalidad en la primera infancia, debida miocarditis. Las cifras
de mortalidad, comunicadas por diferentes autores, están -
16
Estadio indeterminado
primeras manifestaciones clínicas de la miocardiopatía -
rentes denominaciones: preclínico, indeterminado, indi-ferenciado, latente. Es un periodo clínicamente silente, de
-yoría nunca presentará evidencias clínicas de enfermedad.
-
-17
-
-
--
-
asintomáticos con buena capacidad funcional. 18
Figura 23.2: Bloqueo completo de rama derecha. Onda monofásica semejante a un infarto agudo de miocardio. Electrocardiograma obtenido en el curso de una miocarditis chagásica aguda necrotizante y fulminante. (Modificado de Rosenbaum, MB. Prog Cardiovasc Dis. 1964; 7: 199-225.)

Gelpi - Donato Fisiopatología cardiovascular
392
Durante este periodo, clínicamente silencioso, el pro-
-
19
de muestras de biopsias endomiocárdicas, método de ele-
-
20
-
-
21
Estos porcentajes concuerdan más con el hecho de que
-4
-mas congeladas: pacientes que presentan en las biopsias
-
--
-miocárdica o la prueba de ajmalina.
El detector de lesiones que
--
todos de estudio no invasivos e invasivos más sensibles
que evolucionarán a la miocardiopatía.
--
tes series. 22
-23
Fueron descritos potenciales ventriculares tardíos con 24
Han sido comunicadas diferentes alteraciones ecocar-et al.
izquierdo aumentado. 25 También se detectaron anorma-
pacientes, con un predominio de la hipocinesia apical. 26
et al.
de los pacientes presentaba transtornos en la motilidad. 27
-
marcapaseo auricular. 28 -
29
La disparidad observada en la prevalencia de anorma-lidades, comunicadas con los diferentes procedimientos
-dría atribuirse a que las muestras no fueron semejantes en
periodo intermedio, así como también a que los diferentes -
miocárdico subclínico.
de ajmalina, por su intenso y efímero efecto depresor de -
30 Es destacable que esta prueba
personas infectadas sin otras manifestaciones de compro-miso miocárdico. 31
-
prueba de ajmalina es un marcador clínico más sensible de
por otra parte, permite detectar el compromiso miocárdico -
-
la presencia de compromiso miocárdico leve, que pudo ser
32
activa en las biopsias endomiocárdicas de pacientes con 33 En un es-
-
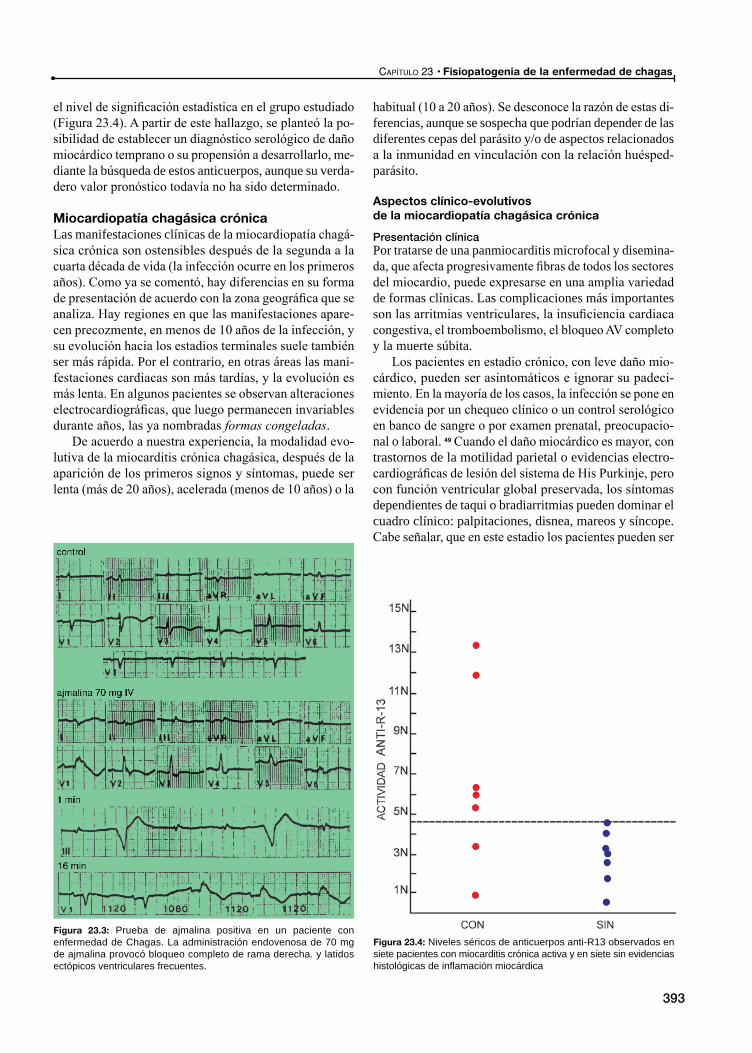
Fisiopatogenia de la enfermedad de chagas
393
-
-diante la búsqueda de estos anticuerpos, aunque su verda-
Miocardiopatía chagásica crónicaLas manifestaciones clínicas de la -
-
ser más rápida. Por el contrario, en otras áreas las mani-
.-
-ferencias, aunque se sospecha que podrían depender de las diferentes cepas del parásito y/o de aspectos relacionados
parásito.
Aspectos clínico-evolutivos de la miocardiopatía chagásica crónica
Presentación clínicaPor tratarse de una panmiocarditis microfocal y disemina-
de formas clínicas. Las complicaciones más importantes
y la muerte súbita.--
-nal o laboral. 49
trastornos de la motilidad parietal o evidencias electro-
dependientes de taqui o bradiarritmias pueden dominar el cuadro clínico: palpitaciones, disnea, mareos y síncope.
Figura 23.3: Prueba de ajmalina positiva en un paciente con enfermedad de Chagas. La administración endovenosa de 70 mg de ajmalina provocó bloqueo completo de rama derecha. y latidos ectópicos ventriculares frecuentes.
Figura 23.4: Niveles séricos de anticuerpos anti-R13 observados en siete pacientes con miocarditis crónica activa y en siete sin evidencias histológicas de inflamación miocárdica

Gelpi - Donato Fisiopatología cardiovascular
394
-
el ejercicio y atípico para isquemia miocárdica. Constituida la cardiomiopatía dilatada, con deterioro de
completo, que caracterizan a los estados avanzados de la en-
cardiopatías, la severidad de los síntomas no siempre se co-
-
--
-cos. Es considerado como característico de la miocarditis
-dica. A menudo contiene trombos murales adheridos al
-nadas por el ventrículo izquierdo, reveladas por la trans-
--
trado celular difuso, compuesto por células mononucleares
-
-emplazan al tejido muscular y son el sustrato de las anor-
-
y constituye el sustrato de las arritmias ventriculares y supraventriculares.
En otras áreas se encuentra
-
-
en la cardiomiopatía dilatada idiopática.
de métodos inmunocitoquímicos ha permitido demostrar más casos parásito-positivos. 34 Mediante la técnica de
Tripanosoma cruzi
35
-
desarrolla en forma temprana y preferencial, pero las ra-
anterior de la rama izquierda del haz de His y la rama dere-
-
el hemibloqueo anterior izquierdo y el bloqueo completo de rama derecha. Estas anormalidades tan características
-
rama izquierda son las estructuras menos comprometidas
A nivel auricular, se han descrito lesiones miocárdicas que
Anormalidades electrocardiográficas
Bloqueos intraventriculares y auriculoventriculares
-
intraventricular, como el bloqueo de rama derecha con o sin hemibloqueo anterior izquierdo, en personas menores
frecuente y, por ello, ha sido utilizado como evidencia de
su prevalencia es baja en pacientes asintomáticos con 36 El bloqueo de rama de-
recha aislado o asociado a hemibloqueo anterior izquierdo
bien indican la presencia de compromiso del sistema de
variable.
-
bloqueo de rama izquierda, mucho menos frecuente, impli-
del bloqueo de rama izquierda y del hemibloqueo posterior -
lidad de la rama derecha y el fascículo anterior, de manera
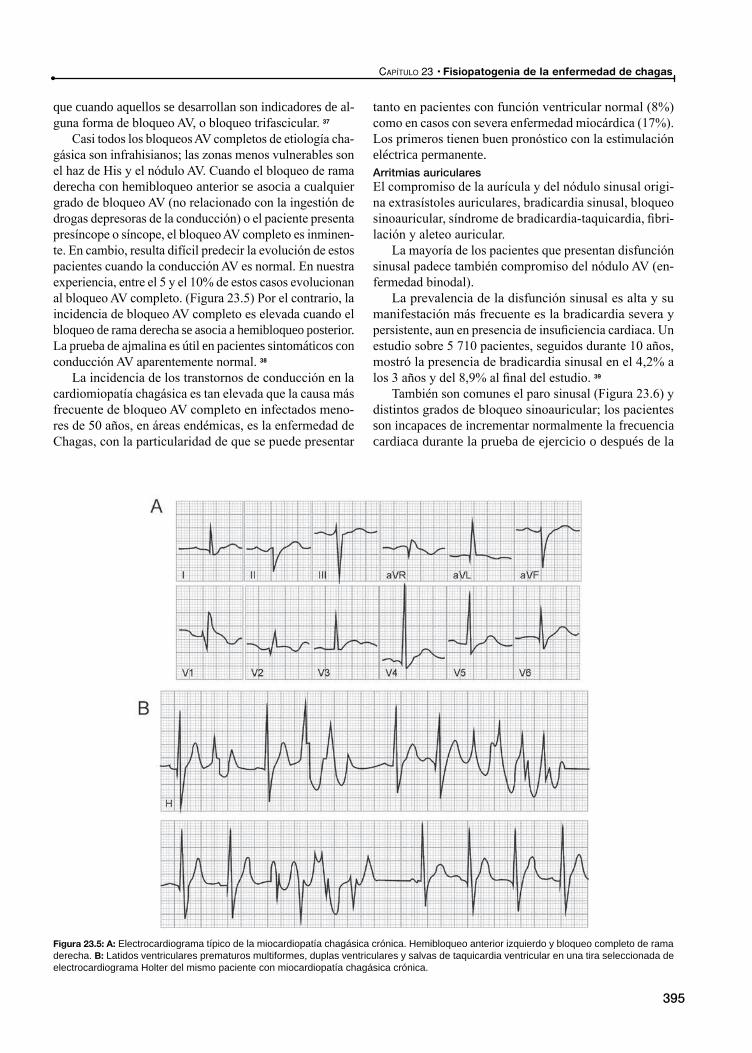
Fisiopatogenia de la enfermedad de chagas
395
que cuando aquellos se desarrollan son indicadores de al-37
-
derecha con hemibloqueo anterior se asocia a cualquier
-
bloqueo de rama derecha se asocia a hemibloqueo posterior. La prueba de ajmalina es útil en pacientes sintomáticos con
38
-
eléctrica permanente. Arritmias auriculares
-
-
-
39
son incapaces de incrementar normalmente la frecuencia cardiaca durante la prueba de ejercicio o después de la
Figura 23.5: A: Electrocardiograma típico de la miocardiopatía chagásica crónica. Hemibloqueo anterior izquierdo y bloqueo completo de rama derecha. B: Latidos ventriculares prematuros multiformes, duplas ventriculares y salvas de taquicardia ventricular en una tira seleccionada de electrocardiograma Holter del mismo paciente con miocardiopatía chagásica crónica.
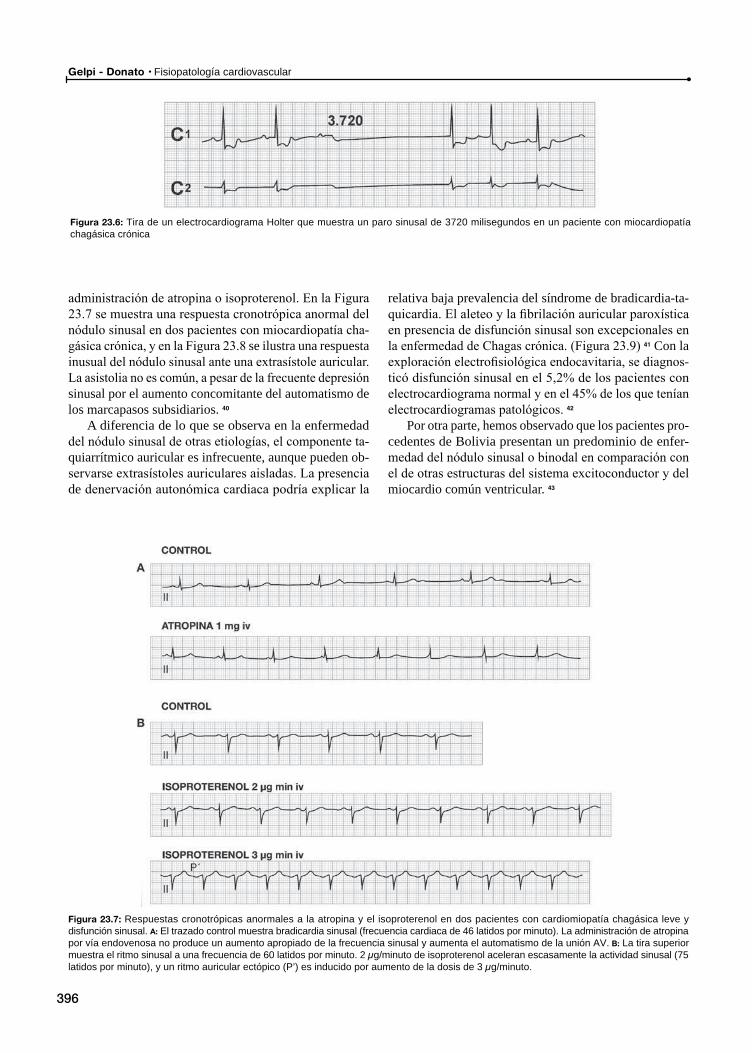
Gelpi - Donato Fisiopatología cardiovascular
396
Figura 23.6: Tira de un electrocardiograma Holter que muestra un paro sinusal de 3720 milisegundos en un paciente con miocardiopatía chagásica crónica
Figura 23.7: Respuestas cronotrópicas anormales a la atropina y el isoproterenol en dos pacientes con cardiomiopatía chagásica leve y disfunción sinusal. A: El trazado control muestra bradicardia sinusal (frecuencia cardiaca de 46 latidos por minuto). La administración de atropina por vía endovenosa no produce un aumento apropiado de la frecuencia sinusal y aumenta el automatismo de la unión AV. B: La tira superior muestra el ritmo sinusal a una frecuencia de 60 latidos por minuto. 2 μg/minuto de isoproterenol aceleran escasamente la actividad sinusal (75 latidos por minuto), y un ritmo auricular ectópico (P’) es inducido por aumento de la dosis de 3 μg/minuto.
-
se ilustra una respuesta
sinusal por el aumento concomitante del automatismo de los marcapasos subsidiarios. 40
A diferencia de lo que se observa en la enfermedad -
quiarrítmico auricular es infrecuente, aunque pueden ob-
de
relativa baja prevalencia del síndrome de bradicardia-ta-
41 Con la -
42
Por otra parte, hemos observado que los pacientes pro-cedentes de Bolivia presentan un predominio de enfer-
miocardio común ventricular. 43

Fisiopatogenia de la enfermedad de chagas
397
En los estadios más avanzados de la enfermedad, en
44
Arritmias ventriculares
-lencia y complejidad de las arritmias ventriculares se
presentan con más frecuencia en pacientes con aneurisma
-arritmias ma-
Figura 23.8: Manifestación poco común de disfunción sinusal en una mujer de 51 años con cardiomiopatía chagásica crónica de severidad moderada. A: La frecuencia sinusal es de 68 latidos por minuto y existe bloqueo de rama derecha.B: Registro simultáneo en DI, DII y V1. Una extrasístole auricular (P’), con una ligadura de 480 mseg inicia un episodio prolongado de paro sinusal. El ritmo sinusal reaparece después de dos pausas de 2880 y 1840 mseg, respectivamente, que terminan con escapes auriculares (Pe). Al final de la tira, otra extrasístole auricular con ligadura de 500 mseg deprime nuevamente la actividad sinusal.
Figura 23.9: Electrocardiograma Holter que muestra episodios de fibrilación auricular paroxística que revierten espontáneamente y ponen de manifiesto la depresión del nódulo sinusal por sobreestimulación con pausas de hasta 6132 segundos, en un paciente con miocardiopatía chagásica crónica.
Tabla 23.1: Hallazgos clínicos, radiológicos y electrocardiográficos en 41 pacientes chagásicos crónicos con arritmias ventriculares potencialmente malignas
Síntomas Pacientes (n= 41)
Hallazgos clínicosSíncope recurrenteMareos
ICC
CARDIOMEGALIAModeradaSevera
TRANSTORNOS DE CONDUCCIÓN
HAI
181622 11
1716
1816111131
ICC: Insuficiencia cardiaca congestiva. BRD:HAI: HPI:

Gelpi - Donato Fisiopatología cardiovascular
398
Tabla 23.2: Extrasístoles ventriculares en la miocarditis crónica chagásica
54%
Cardiomegalia e ICC
Silueta cardiaca normal
Variabilidad espontánea
Incidencia posejercicio
presentan MS8 de 10
presentan TV3 de 7
ECG con BRD y onda T anormal
ICC: Insuficiencia cardiaca congestiva. EV: Extrasistolia ventricular.MS: +++: Alta frecuencia.++: Frecuencia intermedia. +: Baja frecuencia.
Figura 23.10: Extrasistolia ventricular multiforme en un paciente con miocardiopatía chagásica crónica e insuficiencia cardiaca
síntomas severos que podían ser atribuidos a arritmias ventriculares sostenidas: 8 de ellos padecieron sínco-
-
--
ventriculares ocurren a menudo en presencia de severo -
monitoreo ambulatorio. 23, 24
-
súbita es precedida por arritmias ventriculares potencial-46
Las -
característica más frecuente y permanente, aun cuando -
una remarcable persistencia y muy escasa variabilidad
repetidos. La persistencia y estabilidad de las arritmias -
muerte súbita.
--
el ritmo cardiaco y las arritmias ventriculares, mostraron
ventriculares, tanto simples como complejas, era homo-
-minuían con el reposo. 45
-cardia ventricular polimorfa. Otras anormalidadesNo es infrecuente observar, en estadios avanzados de la
--
-
muestra la frecuencia 47
Anormalidades miocárdicas
Aneurisma ventricular-
nica con respecto a otras miocardiopatías es la presencia -
lar, que predisponen a las complicaciones más temibles de la enfermedad: el tromboembolismo y las arritmias

Fisiopatogenia de la enfermedad de chagas
399
estadio indeterminado es posible hallar áreas de acinesia -
sional, así como alteraciones leves en la motilidad apical 29 Con la
El aneurisma ventricular apical izquierdo de la enfer--
en la cardiopatía isquémica. 48 Ambas entidades comparten
relacionadas con la presencia del aneurisma ventricular: -
embolismo pulmonar y sistémico. 49
--
podría llevar al debilitamiento focal de la pared ventricular.
la vulnerabilidad de esta zona, particularmente por su es-
--
50
-
-
-
--
El monitoreo ambulatorio con sistema Holter revela el --
risma ventricular o severas alteraciones de la motilidad
51
-
predictor independiente de muerte súbita cardiaca. 52
Disfunción autonómica cardiovascular-
-
53
Las evidencias de disautonomía detectadas por dife--
determinado de la enfermedad, son: a. respuesta cardiovascular alterada al esfuerzo, in-
-cuencia cardiaca
-
En todos los estadios de la enfermedad se han obser-
Los niveles de noradrenalina plasmática se correlacionan
Las alteraciones en las pruebas de los pacientes que se encuentran en el periodo indeterminado, demuestran que
relacionada con la presencia de enfermedad cardiaca clíni-
-ral. 54
podrían contribuir a la muerte súbita cardiaca arrítmica en 55
Insuficiencia cardiaca congestiva. Tromboembolismo
La
Tabla 23.3:
Alteraciones electrocardiográficas % de pacientes
Zonas inactivas y trastornos de repolarización 57,0
55,7
Extrasistolia ventricular 53,9
23,0
Extrasistolia auricular 12,6
Fibrilación auricular 12,3
17,0
16,4
14,4
Aleteo auricular 14,4
12,6
10,9

Gelpi - Donato Fisiopatología cardiovascular
400
Tabla 23.4: Clasificación clínica de la enfermedad de Chagas
Chagas agudo Parasitemia positiva
Chagas indeterminado Serología positivaExamen físico normalEstudios cardiológicos normales
Chagas con cardiopatía Grupo A Sin cardiomegaliaGrupo BCon cardiomegalia
Arritmias y/o trastornos de la conducciónArritmias y/o trastornos de la conducciónCon insuficiencia cardiacaSin insuficiencia cardiaca
-56
La incidencia de tromboembolismo arterial y venoso
-
-56 Las
autopsias han demostrado la presencia de trombos mura-
57
Criterios diagnósticos
basa en la tríada:
-
--
Clasificación
variedad de formas clínico-evolutivas y las innumerables
-
58
Muerte súbita. Pronóstico. Factores de riesgoLa incidencia de muerte súbita en la enfermedad de Cha-
sido subestimada, sobre todo en las áreas rurales.
-
-
-69
-ciencia cardiaca, y también presentan arritmias ventricu-
dinámico ambulatorio. 59
-padeiro en personas que sufrieron accidentes de tránsito
enfermedad es endémica, revelan que la cardiomiopatía 60
que la causa de la muerte en tales casos es producida por -
Alrededor de la mitad de tales pacientes eran asintomáti-cos antes de la muerte y casi todos tenían anormalidades
En un estudio de Mendoza et al. sobre diez pacientes
-
una taquicardia ventricular sostenida. 61
arritmias y bloqueos intraventriculares y auriculoventri-
décadas en forma lenta desde la miocarditis subclínica, a --
-tener conclusiones al respecto, debido a la necesidad de

Fisiopatogenia de la enfermedad de chagas
401
et al.62, utilizando un análisis mul-
que demanden mayor esfuerzo físico, padecer enfermeda-des asociadas.
Tripanosoma cruzi -
ha sido referida en varios estudios. Después del desarrollo
-sitivos, en estadio indeterminado, tuvieron una sobrevida
-
Los hechos más fuertemente asociados con un desen-
-
predictores independientes de mortalidad y de arritmias --
muerte súbita cardiaca y los episodios de taquicardia ven-tricular sostenida no fatal, en los pacientes con aneurisma
fuerte de mortalidad, en esta y en otras series. 62
La presencia de -
marcapasos endocavitarios. Por otro lado, los pacientes
-trículo izquierdo y el aneurisma ventricular constituyen los sustratos presentes más frecuentes que conducen a la muerte, como consecuencia de las arritmias ventriculares
52
Fisiopatogenia de la miocardiopatía chagásica crónica
distintas formas clínico-evolutivas de la cardiomiopatía -
A pesar de los importantes avances en la última déca-
-
-
Hipótesis no autoinmunesDenervación autonómica
-ronas parasimpáticas de los -
por el tripanosoma. 63
-
-
Menos probable aún es que las lesiones neuronales puedan producir las propias de la
las alteraciones que se observan en la enfermedad, sobre -
nos autores consideran que las anormalidades neuronales
su causa. 64
Anomalías microvascularesMorris et al.endoteliales y miocárdicas con Tripanosoma cruzi de-
-

Gelpi - Donato Fisiopatología cardiovascular
402
-
65
Los autores han demostrado que el endotelio se afec-
--
endoteliales.
-66
en la actividad de la adenilciclasa. Teniendo en cuenta que
estaría relacionada con la presencia del parásito, sino con 67
arterias coronarias epicárdicas son infrecuentes. Por el contrario, las arterias
68
--
69 Por otra parte, Andrade y Castro Filho describieron alteraciones vasculares locales e ines-
70 Torres et al. obser--
dependiente del endotelio de los vasos coronarios, podía
la contractilidad. 71
-
72
Efectos tisulares directos del parasitismo celularparásito cuya con-
-
tripanosoma en las células requiere el reconocimiento de
-
-
-duce en forma temprana durante el curso de la enfermedad,
acetilcolina transferasa producida por el parasitismo de las
Las variaciones en la enzima neuraminidasa, elaborada -
minar tanto la virulencia del parásito como la respuesta del huésped. Esta enzima remueve el ácido siálico de los
el parásito y, al mismo tiempo, aumenta la respuesta in-
en cultivos de células con Tripanosoma cruzi. El papel directo de la presencia del parásito en los mecanismos
-
ADN 73 74 en los miocitos y leucocitos de las lesiones tisulares. Hipótesis autoinmune
--
-
inmunidad celular ha sido considerado en todas las fases de la enfermedad. En el
-
con predominio de células mononucleares. Los linfocitos
-dos afectados. Este proceso es semejante al rechazo de transplante cardiaco. La transferencia de linfocitos CD
-
-pos humorales. Asimismo, también se hallaron anticuer-
Tripanosoma cruzi. 75
Otros estudios han demostrado que los linfocitos sen-sibilizados con Tripanosoma cruzi imitan varios epitopes
76 Estos datos indican una respuesta coordinada con un componente autoinmune,

Fisiopatogenia de la enfermedad de chagas
403
que compromete tanto a la inmunidad mediada por células como a la inmunidad humoral.
-puesta mediada por células, que comienza en una fase tan
--
el torrente circulatorio, desde nidos latentes en diversos
-
leishmanias provocaría reacciones inmunes y autoinmu-
-mo de mimetismo molecular. 77
-
celular antitejido en seres humanos y animales infectados Tripanosoma cruzi.
-
T. cruzidel huésped, por el mecanismo de mimetismo molecular.
el parásito y el huésped mamífero, que determinarían que -
-
-
-nistas. 78
la diabetes insulinorresistente son ejemplos que han sido bien documentados. Del mismo modo, este mecanismo
de cardiopatías diversas, como la miocardiopatía dilatada
Evidencias de mimetismo molecular en la cardiomiopatía crónica chagásica
-
periféricos, ribosomales P, tubulina, miosina, laminina y
Anticuerpos antimiosinaCunha-Neto et al
anticuerpos que β de
la cadena pesada de la miosina cardiaca humana. 79
y los asintomáticos presentan reactividad contra la misma subunidad de la miosina. Otros autores no encontraron
80 y atribuyen la presencia de los anticuerpos antimiosina en el suero de pacientes con car-
81
cardiopatías, incluyendo las miocarditis, el infarto mio-
de anticuerpos antimiosina cardiaca. 80
pueda atribuirse a estos anticuerpos. En estos casos se
anticuerpos antimiosina. 82
Anticuerpos antiglicoproteínas - lamininaLos anticuerpos antilaminina presentes en el suero de humanos y de monos infectados con T. cruzi fueron de-
et al. Dichos anticuerpos se pro-
83 La pre-sencia de estos anticuerpos no pudo correlacionarse con la
84
Anticuerpos antiproteínas del tejido nerviosoet al. describieron, en el suero de
-cia de anticuerpos antineuronas y Khoury et al., la de anticuerpos antinervios periféricos. 85, 86 A pesar de una
T. cruzitejido nervioso, la reactividad antinervios periféricos en el
87
Anticuerpos antitejido endotelial, vasos sanguíneosy estructuras intersticiales (EVI)Cossio et al.
anticuerpo que se unía al endotelio, venas y estructuras
-
88 Las biopsias de músculo esquelético y
-

Gelpi - Donato Fisiopatología cardiovascular
404
Figura 23.11: Modelo del receptor -adrenérgico humano. Tm1 a t7: siete segmentos transmembrana. e2, e3, e4: tres asas extracelulares. i1, i2, i3: tres asas intracelulares. NH2: extremo terminal extracelular y COOH: extremo terminal intracelular.
miocárdica. 89 Al incubar el suero con lisado de epimasti--
T. cruzi
90
-
91
Los títulos de anticuerpos antinervios periféricos, así
86
Anticuerpos antirreceptores autonómicos-
-receptores β1
cardiacos, aumentando el AMPc, del mismo modo que 92
pacientes con cardiomiopatía dilatada, puesta en contacto con cultivos de cardiomiocitos de rata neonata, aumentaba la frecuencia de latidos de estas células.
de los receptores cardiovasculares β1 y β
93
-muestran que los anticuerpos que reconocen y estimulan
Asimismo, estos anticuerpos estarían vinculados con las arritmias que presentan dichas cardiopatías.
Por otra parte, se ha demostrado en varias enferme-
los receptores hormonales, interactuando con ellos como
de las células. 78
La estructura molecular y propiedades inmunogénicas de los receptores β-adrenérgicos: La estructura molecular del receptor β
-
los epítopes de las células T y B necesarios para inducir una respuesta autoinmune.
Couraud et al. 94 demostraron que los receptores β -
-et
al95
--
los anticuerpos en el suero de los pacientes y de animales
Figura 23.12: Efectos de los anticuerpos antirreceptor β1 adrenérgico (Ac anti β1) purificados sobre el cronotropismo de los miocardiocitos de ratas neonatas (ver detalles en el texto)
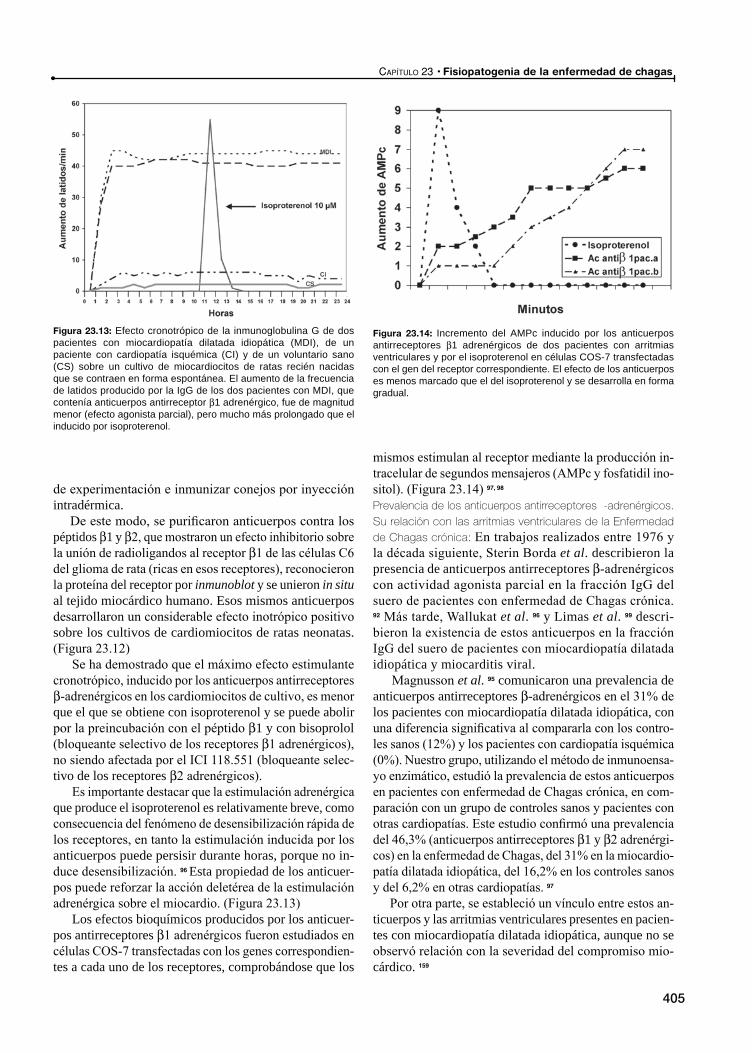
Fisiopatogenia de la enfermedad de chagas
405
intradérmica.
péptidos β1 y ββ
la proteína del receptor por inmunoblot y se unieron in situ al tejido miocárdico humano. Esos mismos anticuerpos
sobre los cultivos de cardiomiocitos de ratas neonatas.
βque el que se obtiene con isoproterenol y se puede abolir
β1 y con bisoprolol β
-tivo de los receptores β
que produce el isoproterenol es relativamente breve, como
anticuerpos puede persisir durante horas, porque no in-96 Esta propiedad de los anticuer-
Los efectos bioquímicos producidos por los anticuer-pos antirreceptores β
-tes a cada uno de los receptores, comprobándose que los
--
97, 98
Prevalencia de los anticuerpos antirreceptores -adrenérgicos. Su relación con las arritmias ventriculares de la Enfermedad de Chagas crónica:
et al. describieron la presencia de anticuerpos antirreceptores β
92 et al. 96 y Limas et al. 99 descri-
idiopática y miocarditis viral.et al. 95 comunicaron una prevalencia de
anticuerpos antirreceptores βlos pacientes con miocardiopatía dilatada idiopática, con
-
-
-
β1 y β --
97
-ticuerpos y las arritmias ventriculares presentes en pacien-tes con miocardiopatía dilatada idiopática, aunque no se
-cárdico. 159
Figura 23.14: Incremento del AMPc inducido por los anticuerpos antirreceptores β1 adrenérgicos de dos pacientes con arritmias ventriculares y por el isoproterenol en células COS-7 transfectadas con el gen del receptor correspondiente. El efecto de los anticuerpos es menos marcado que el del isoproterenol y se desarrolla en forma gradual.
Figura 23.13: Efecto cronotrópico de la inmunoglobulina G de dos pacientes con miocardiopatía dilatada idiopática (MDI), de un paciente con cardiopatía isquémica (CI) y de un voluntario sano (CS) sobre un cultivo de miocardiocitos de ratas recién nacidas que se contraen en forma espontánea. El aumento de la frecuencia de latidos producido por la IgG de los dos pacientes con MDI, que contenía anticuerpos antirreceptor β1 adrenérgico, fue de magnitud menor (efecto agonista parcial), pero mucho más prolongado que el inducido por isoproterenol.
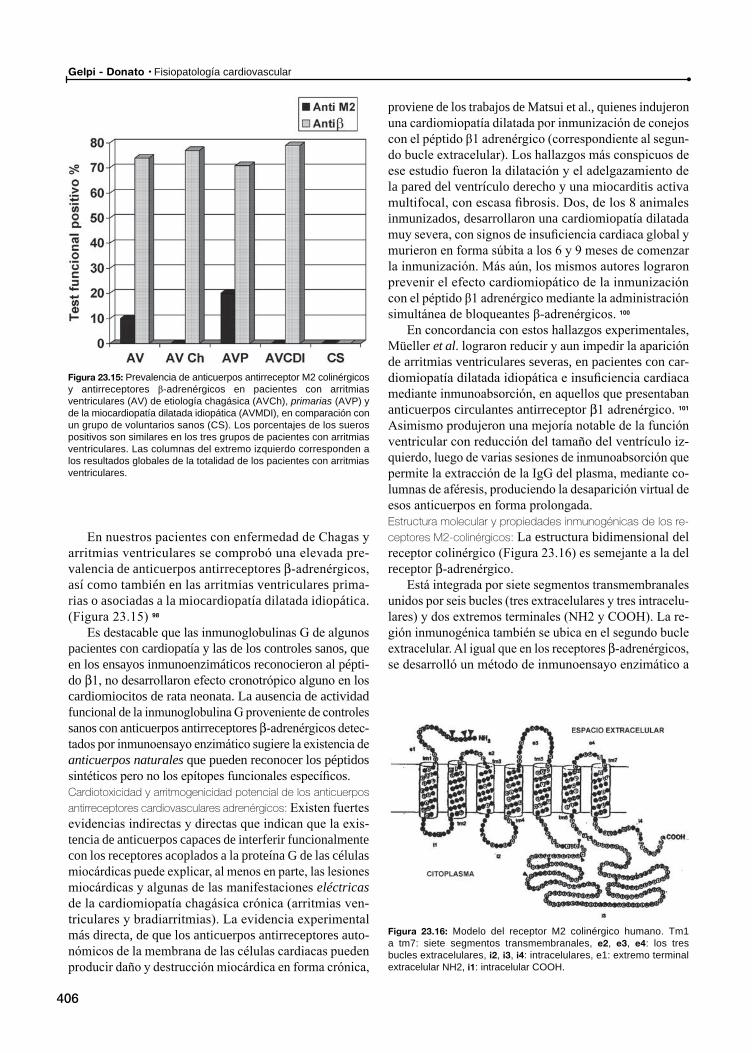
Gelpi - Donato Fisiopatología cardiovascular
406
-valencia de anticuerpos antirreceptores βasí como también en las arritmias ventriculares prima-rias o asociadas a la miocardiopatía dilatada idiopática.
98
pacientes con cardiopatía y las de los controles sanos, que en los ensayos inmunoenzimáticos reconocieron al pépti-do βcardiomiocitos de rata neonata. La ausencia de actividad funcional de la sanos con anticuerpos antirreceptores β -
anticuerpos naturales que pueden reconocer los péptidos
Cardiotoxicidad y arritmogenicidad potencial de los anticuerpos antirreceptores cardiovasculares adrenérgicos:
-tencia de anticuerpos capaces de interferir funcionalmente con los receptores acoplados a la proteína G de las células
eléctricas -
más directa, de que los anticuerpos antirreceptores auto-
proviene de los trabajos de Matsui et al., quienes indujeron
-
la pared del ventrículo derecho y una miocarditis activa
inmunizados, desarrollaron una cardiomiopatía dilatada
100
et alde arritmias ventriculares severas, en pacientes con car-
anticuerpos circulantes antirreceptor β 101
-
-
Estructura molecular y propiedades inmunogénicas de los re-ceptores M2-colinérgicos: La estructura bidimensional del
receptor β
--
β
Figura 23.15: Prevalencia de anticuerpos antirreceptor M2 colinérgicos y antirreceptores -adrenérgicos en pacientes con arritmias ventriculares (AV) de etiología chagásica (AVCh), primarias (AVP) y de la miocardiopatía dilatada idiopática (AVMDI), en comparación con un grupo de voluntarios sanos (CS). Los porcentajes de los sueros positivos son similares en los tres grupos de pacientes con arritmias ventriculares. Las columnas del extremo izquierdo corresponden a los resultados globales de la totalidad de los pacientes con arritmias ventriculares.
Figura 23.16: Modelo del receptor M2 colinérgico humano. Tm1 a tm7: siete segmentos transmembranales, e2, e3, e4: los tres bucles extracelulares, i2, i3, i4: intracelulares, e1: extremo terminal extracelular NH2, i1: intracelular COOH.
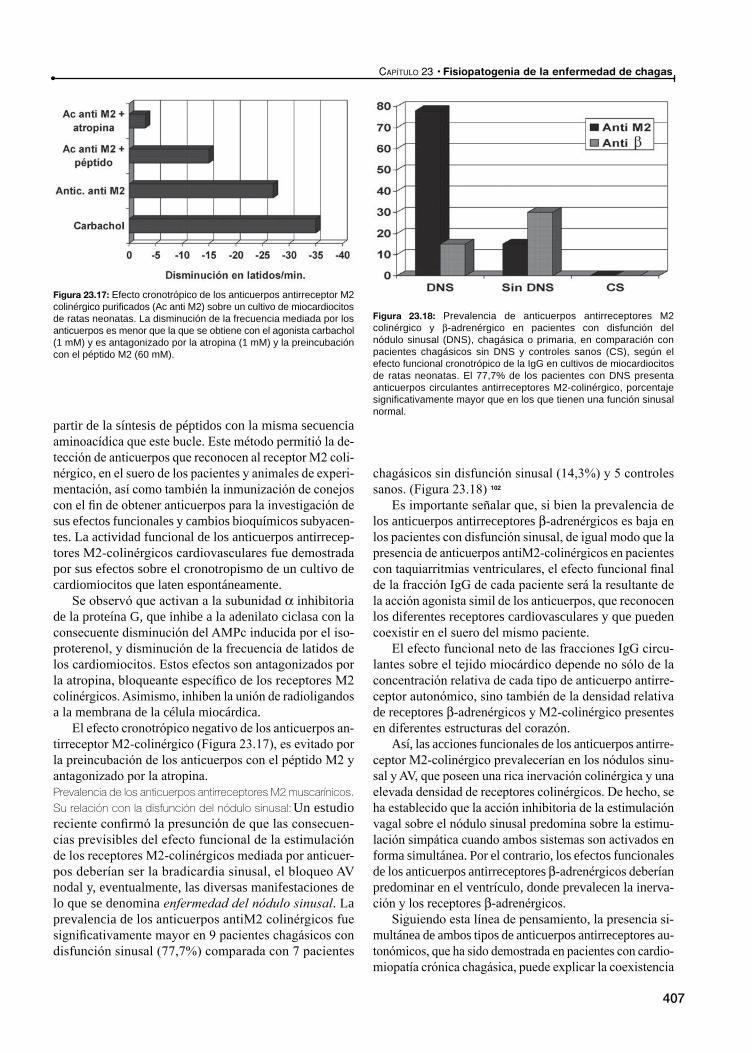
Fisiopatogenia de la enfermedad de chagas
407
partir de la síntesis de péptidos con la misma secuencia ---
sus efectos funcionales y cambios bioquímicos subyacen-tes. La actividad funcional de los anticuerpos antirrecep-
por sus efectos sobre el cronotropismo de un cultivo de cardiomiocitos que laten espontáneamente.
α inhibitoria de la proteína G, que inhibe a la adenilato ciclasa con la
-
a la membrana de la célula miocárdica.-
Prevalencia de los anticuerpos antirreceptores M2 muscarínicos. Su relación con la disfunción del nódulo sinusal: Un estudio
-
-
nodal y, eventualmente, las diversas manifestaciones de lo que se denomina . La prevalencia de los
102
los anticuerpos antirreceptores β
los diferentes receptores cardiovasculares y que pueden
-
-
de receptores β
Así, las acciones funcionales de los anticuerpos antirre--
-
forma simultánea. Por el contrario, los efectos funcionales de los anticuerpos antirreceptores βpredominar en el ventrículo, donde prevalecen la inerva-
β--
multánea de ambos tipos de anticuerpos antirreceptores au--
Figura 23.17: Efecto cronotrópico de los anticuerpos antirreceptor M2 colinérgico purificados (Ac anti M2) sobre un cultivo de miocardiocitos de ratas neonatas. La disminución de la frecuencia mediada por los anticuerpos es menor que la que se obtiene con el agonista carbachol (1 mM) y es antagonizado por la atropina (1 mM) y la preincubación con el péptido M2 (60 mM).
Figura 23.18: Prevalencia de anticuerpos antirreceptores M2 colinérgico y β-adrenérgico en pacientes con disfunción del nódulo sinusal (DNS), chagásica o primaria, en comparación con pacientes chagásicos sin DNS y controles sanos (CS), según el efecto funcional cronotrópico de la IgG en cultivos de miocardiocitos de ratas neonatas. El 77,7% de los pacientes con DNS presenta anticuerpos circulantes antirreceptores M2-colinérgico, porcentaje significativamente mayor que en los que tienen una función sinusal normal.

Gelpi - Donato Fisiopatología cardiovascular
408
la frecuencia cardiaca suele ser relativamente baja en los
Cardiotoxicidad y arritmogenicidad potencial de los anticuerpos antirreceptor M2-colinérgico: Otros estudios también han permitido demostrar que los anticuerpos antirreceptores
-
-
-
100
-
eran contrarrestados por la atropina. 103
efecto molecular de los anticuerpos antirreceptores β
--
diovasculares. 98
primarioscon los anticuerpos antirreceptores β
primaria -
de esas anormalidades. 97, 97, 102
--
pechadas perspectivas para el tratamiento de las arritmias
Origen de los anticuerpos antirreceptores cardiovasculares en la enfermedad de Chagas: anticuerpos anti-
--
parásito, que condicionara la pérdida de tolerancia por
En el laboratorio del Dr. M. J. Levin104
ribosomales P del Tripanosoma cruzi que son tres: P0, P1
Figura 23.19: La generación de los anticuerpos que reconocen y reaccionan con los receptores autonómicos miocárdicos en la miocardiopatía chagásica crónica estarían dirigidos contra antígenos del parásito y reaccionarían en forma cruzada contra epítopes del segundo bucle extracelular de los receptores.

Fisiopatogenia de la enfermedad de chagas
409
-
-
-mente más altos que los sueros de pacientes sin evidencias
de las proteínas ribosomales P de bajo peso molecular de T. cruzi
-tas autoinmunes en el lupus eritematoso sistémico. Kaplan et al. 105
EEEDDD, y en particular el tercer residuo Glu del péptido
por T. Cruzi -
β El análisis de las secuencias de los péptidos correspon-
-diovasculares βdemostrar que, el reconocimiento por el suero de pacientes
-
β-
tídicas de las proteínas P ribosomales de T. cruzi y las receptores β
-
anticuerpos anti-P y los anticuerpos funcionalmente ac-tivos antirreceptores cardiovasculares β
-cen los receptores β
T. cruzi. -105-106
--
Tripanosoma cruzi y que esta -
rasitario. Además, los resultados del estudio indican que
-dad para reaccionar en forma cruzada con proteínas de la membrana celular cardiaca.
De acuerdo a los estudios descritos, en la miocardio-
-
del parásito, es muy probable que el reconocimiento de las proteínas ribosomales P parasitarias ocurra en esos sitios.
--
-
la presencia del parásito en las células miocárdicas es el -
responsable de la respuesta inmune antiparasitaria incre-
107
-bre receptores de la membrana celular depende en forma
estructura citoplasmática del Tripanosoma cruzi, junto a
Estas observaciones y los estudios recientes ya comen-et al.100 y los de
Mueller et al.101 -
muchas de las manifestaciones clínicas de la enfermedad, y
a impedir los efectos funcionales de los anticuerpos so-β
de T. cruzicruzada contra los receptores cardiovasculares humanos β
-
del un tratamiento. Asimismo, estos péptidos podrán uti--
Bibliografía
1. Elizari, M. “La miocardiopatía chagásica. Perspectiva histórica”. Medicina (Buenos Aires). 1999;59:25-40.
2. WHO: “Control of Chagas disease. Report of a WHO Expert Com-mittee”. WHO Technical Report Series Ginebra. 1991.; N 811.
3. Hayes R, C Schofield. “Estimación de las tasas de incidencia de infecciones y parasitosis crónica a partir de la prevalencia: la enfer-medad de Chagas en América Latina”. Bol Sanit Panam. 1991;108.
4. Rosenbaum, MB. “Chagasic cardiomyopathy”. Prog Cardiovasc Dis. 1964;7:199-225.

Gelpi - Donato Fisiopatología cardiovascular
410
5. Aufderheide A, Salo W, Madden M, Streitz J, Buikstra J, Guhi F, Arriaza B, et al. “A 9000 year record of Chagas’disease”. PNAS 101. 1994;(7):2034-2039.
6. Dias, J. “Epidemiology of Ghagas’disease”. En: Wendel S, Brenner Z y col. Chagas’disease (American tripanosomiasis): It’s impact on transfusion and clinical medicine. San Pablo. ISBT. 1992;49-80.
7. Bittencourt AL, Barbosa H, Rocha T y col. “Incidencia da trans-missao congenita da Doença de Chagas em partos prematuros em a maternidade Tsyla Balbino (Salvador, Bahía)”. Rev Inst Med Trop Sao Paulo. 1972;14:31.
8. Barousse AP, Esposto M, Mandel S y col. “Enfermedad de Chagas congénita en área no endémica”. Medicina. 1978;38:611.
9. Howard, JE. “La enfermedad de Chagas congénita”. Colección de monografías biológicas. De Standler. Universidad de Chile, 1962;16.
10. Chagas, C. “Tripanosomiase americana: forma, aguda da molestia”. Mem Inst Oswaldo Cruz. 1916;8(2):5.
11. Romaña, C. Enfermedad de Chagas. Buenos Aires, López Libreros. 1963.
12. Cançado Romeo, J. “Forma aguda de la enfermedad de Chagas en Brasil”. Rev Ass Med Brasil. 1980;26.
13. Lugones H. “Consideraciones acerca de los síntomas del perio-do agudo de la enfermedad de Chagas en la infancia”. Anais do Congreso Internacional da Doença de Chagas, Río de Janeiro, Brasil. 1959;3:861.
14. Pan American Health Organization. Health conditions in the Americas Scientific Publications. Washington DC: PAHO.1990;524(1)160.
15. Mazza, S. “La enfermedad de Chagas en la República Argentina”. Mem Ins Oswaldo Cruz. 1949;47:273.
16. Consejo de Enfermedad de Chagas y Miocardiopatías Infecciosas "Dr. Salvador Mazza". Consenso de Enfermedad de Chagas. Rev Arg Cardiol 2002;70:1-87.
17. Andrade, Z. “Aspectos de la patogenia de la enfermedad de Chagas”. Rev Fed Arg de Cardiol. 1988;17:155.
18. Prata A, Lopes E, Chapadeiro F. “Caracteristicas da morte subita como nao esperada na doença de Chagas”. Rev Soc Bras Med Trop. 1986;19:9.
19. Reis López E, Chapadeiro E, Andrada Z y col. “Anatomía patoló-gica de corazones chagásicos asintomáticos fallecidos de un modo violento”. Mem Inst Osw Cruz. 1981;76:189.
20. Pereira Barreto AC, Mady C, Arteaga Fernandez E y col. “Right ventricular endomyocardial biopsy in chronic Chagas’disease”. Am Heart J. 1986;111:307.
21. Madoery RJ, A Dománico. “Enfermedad de Chagas: periodo intermedio”. Rev Arg Cardiol. 1987;55:217.
22. Madoery RJ, Dománico A, Marcelino A y col. “Alteraciones elec-trocardiográficas durante el periodo intermedio de la enfermedad de Chagas: consideraciones evolutivas”. Rev Lat Cardiol.1992;13:55.
23. Prata, A. “Natural history of chagasic cardiomyopathy”. En: Pan American Health Organization.Washington DC 1976; Pub n 318:191.
24. Madoery C, Guindo J, Esparza E y col. “ECG de señal promediada en la enfermedad de Chagas”. Rev Arg de Cardiol. 1992;60:93.
25. Storino R, J Milei. Miocardiopatía chagásica crónica. Un enfoque para el clínico general. Bs As, Ed. Club de Estudio, 1986.
26. Pereira Barreto, A, Serro Azul L, Mady C y col. “Forma indeter-minada da doença de Chagas: uma doença polimórfica”. Arq Bras Cardiol. 1990;55:347.
27. Arreaga N, Puigbó J, Acquatella H. y col. “Radionucleide evaluation of left ventricular function in chronic Chagas cardiomyopathy”. J Nucl Med. 1983;24:563.
28. Pimenta J, Miranda M, Brito Pereyra C. “Electrophysiologic fin-dings in long term asyntomatic chagasic individuals”. Am Heart J. 1983;106: 374.
29. Carrasco H, Barbosa J, Inglessis G y col. “Left ventricular cinean-giography in Chagas’disease: detection of early myocardial damage”. Am Heart J. 1982;104(3):595.
30. Chiale P, Przybylski J, Laiño R y col. “Usefulness of the ajmaline test in patients with latent bundle branch block”. Am J of Cardiol. 1982;49:21.
31. Chiale P, Przybylski J, Halpern M y col. “Comparative effects of ajmaline on intermittent bundle branch block and the Wolff-Parkinson White Syndrome”. Am J of Cardiol. 1977;39:651.
32. Chiale P, Galperín J, Groppa J, Vallazza M, Guerchi J, Sanchez R, Franco D, Von Wulffen MA, Elizari M. “Detección no invasiva de lesiones miocárdicas subclínicas en pacientes con infección chagásica crónica”. Rev Arg de Cardiol. 1997;65(3):311.
33. Levin M, Levitus G, Kerner N y col. “Autoantibodies in Chagas’ Heart Disease: posible markers of severe Chagas’ heart complaint”. Mem Ins Osw Cruz. 1990; 85:539.
34. Chandler W, J Watts. “Inmunofluorescence as an adyuvant to the histopathologic diagnosis of Chagas’disease”. J Clin Microbiol. 1988;26:567.
35. Anez N, Carrasco H, Parada H y col. “Myocardial parasite persistence in chronic chagasic patients”. Am J Trop Med Hyg. 1999;60(5):726.
36. Maguirre J, Mott K, Lehman J y col. “Relationship of electro-cardiographic abnormalities and seropositivity ti Trypanosoma cruzi within a rural comunity in northeast Brazil”. Am Heart J. 1983;94:105.
37. Rosenbaum M, Elizari M, Lazzari J, Halpern M, Ryba D. “QRS patterns heralding the development of complete heart block with particular emphasis on right bundle branch block with left posterior hemiblock”. En: Sandoe E, FlentedJensen E, Olesen K: Symposium on Cardiac Arrhythmias, Elsinore, Denmark. 1970;149.
38. Moleiro F, Mendoza I, Octavio J y col. “Los transtornos de con-ducción intraventricular y la prueba de ajmalina en pacientes con infección chagásica”. Rev Lat Cardiol. 1982;3:414.
39. Chiale P, Laiño R, Pastori J, Sanchez R, Elizari M, Rosenbaum M. “Normal and abnormal ventricular automaticity in the human heart”. En: Rosenbaum M, Eliari M: Frontiers of cardiac electrophy-siology. La Haya. Martinus Nihjoff Publishers. 1983;159.
40. Elizari M, Nau G, Levi R, Lazzari J, Halpern M, Rosenbaum M. “Experimental production of rate-dependent bundle branch block in the canine heart”. Circ Res. 1974;34 730.
41. Marin Neto J, Maciel B, Gallo L y col. “Effects of parasympa-thetic impairment on the hemodynamic response to handgrip in Chagas’heart disease”. Brit. Heart J. 1986;55:204.
42. Carrasco Guerra, HA. Diagnóstico de daño miocárdico en la enfer-medad de Chagas. Mérida, Universidad de los Andes. Consejo de Publicaciones. 1983.
43. Elizari, MV. “Enfermedad del nódulo sinusal en la Enfermedad de Chagas”. Simposio latinoamericano sobre enfermedad de Chagas. Rev Fed Arg de Cardiol. 1988;17:205-12.
44. Pinto Dias J, K Kroetzel. “The prognosis value of the electrocar-diographic features of chronic Chagas’disease”. Rev Ins Med Trop São Paulo. 1968;10(3):158-162.
45. Chiale P, M Rosenbaum. “Clinical and pharmacological characterization and treatment of potentially malignant arrhytmias of Chronic chagasic cardiomyopathy”. En: Williams, EM: Antiarrhythmic drugs. Handbook of Experimental Pharmacology. Heidelberg, Springer Verlag Berling. 1989;601.
46. Chiale P, Halpern M, Nau G y col. “Malignant ventricular arrhyth-mias in chronic chagasic myocarditis . Pace 1982;5:162.
47. Rosenbaum M, A Alvarez. “The electrocardiogram in chronic Chagas’myocarditis”. Am Heart J. 1955;50:492.
48. Borges Pereira J, Xavier S, Pirmez C, Coura J.“Enfermedad de Chagas en Virgem da Lapa, Mina Gerais, Brasil. Aspectos cínicos y epidemiológicos del aneurisma ventricular izquierdo”. Rev Soc Med Trop. 1998;31(5):457.
49. Castagnino H, F Toranzos. “Correlación histológica de los aneu-rismas ventriculares chagásicos e isquémicos. Relación con la fi-siopatología”. Arch Ins Cardiol Mex. 1988;58(5):425.
50. Hagar J, M Rahimtoola. “Chagas’heart disease”. En: Current Pro-blems in Cardiology. 1995; Vol XX,12:825.78.

Fisiopatogenia de la enfermedad de chagas
411
51. Milei J, Pesce R,Valero E y col. “Electrophysiologic-structural correlations in chagasic aneurysm causing malignant arrhythmias”. Int J Cardiol. 1991;32(1):65-73
52. Bestetti RB, Dalbo C, Arruda C y col. “Predictors of sudden cardiac death for patients with Chagas’disease: a hospital-derived cohort study”. Cardiology. 1996;87(6):481.
53. Palmero H, Caeiro T, Iosa D. “Características distintivas de la cardiopatía en la enfermedad de Chagas”. Medicina. 1980;40 (Supl 1):234.
54. Almeida H, Brandao M da C, dos Reis M y col. “Denervacion e doença de coraçao em pacientes chagasicos crónicos”. Arq Bras Cardiol. 1987;48:43.
55. Junqueira, LF. “Posible rol de la disfunción cardiaca autonómica en la muerte súbita asociada con la enfermedad de Chagas”. Arq Bras Cardio. 1991;56:429.
56. Araujo R, Bestetti R, Godoy R y col. “Chronic Chagas’heart disease in children and in adolescents: a clinicopathologic study”. Int J Cardiol. 1985;9:439.
57. Arteaga Fernandez E, Barretto A, Ianni B, y col. “Trombose cardiaca e embolia em pacientes falecidos de cardiopatía chagásica crónica”. Arq Bras Cardiol. 1989;52:189.
58. Barisani JL, Storino R y col. “Consenso de Enfermedad de Chagas”. SAC Rev Arg de Cardiol. 2002;Sup 1, 70.
59. Espinosa R, Pericchi L, Carrasco H y col. “Prognostic indicators of chronic chagasic cardiopathy”. Int J Cardiol. 1991;30:195.
60. Lopes E, Chapadeiro E, Almeida H y col. “Contribuiçao ao es-tudo da anatomia patologica dos coraçoes de chagasicos falecidos subitamente”. Rev Soc Bras Med Trop. 1975;9:269.
61. Mendoza I, Moleiro F, Marques J. “Morte subita na doença de Chagas”. Arq Bras Cardiol. 1992;59:3.
62. Pugliese C, Lessa I, Santos Filho A. “Sobrevida de la miocardiopatía chagásica crónica descompensada”.Rev Inst Med Trop San Pablo. 1976;18:191.
63. Koberle, F. “Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis”. Adv Parasitol. 1968; 6:63.
64. Davila DF, Rossel RO y Donis JH. “Cardiac parasympathic ab-normalities: cause or consequence of Chagas disease”. Parasitol Today. 1989;5:327
65. Morris SA, Tanowitz HB, Wittner M, Bilezikian JP. “Pathophy-siological insights into the cardiomyopathy of Chagas’ disease”. Circulation. 1992; 82:1900.
66. Morris SA, Weiss I M, Factor SM, Bilezikian JP, Tanowitz HB, Wittner M. “Verapamil ameliorates clinical, pathological and bio-chemical manifestetions of experimental Chagasic cardomyopathy in mice”. J A Col. Cardiol. 1989;14:782-789.
67. Morris SA, Tanowitz HB, Factor SM, Bilezikian JP, Wittner M. “Myocardial adenylate cyclase activity in acute murine Chagas’ disease”. Circ. Res. 1988; 62: 800-810.
68. Oliveira JS, dos Santos JC, Muccillo G, Ferreira AL. “Increased capacity of the coronary arteries in cronic Chagas´ heart disease: further support for the neurogenic pathogenesis concept”. Am heart J. 1985;109:304-408.
69. Torres, CM. “Arterioloesclerose das finas ramificacoes das arteriais do miocardio (coronarite chagásica) e miocitolise focal do miocár-dio na cardiopatía chagásica crónica”. Hospital (Río do Janeiro). 1958;54:597-610.
70. Andrade ZA, BG Castro Filho. “As lesoes vasculares na miocardite crónica chagásica”. Gaz Med Bahía. 1970;70:105-12.
71. Torres FW, Acquatella H, Condado J y col. “Coronary vascular reactivity is abnormal in patients with Chagas’ heart disease”. Am Heart J. 1995;129:995-1001.
72. Milei, J. “Patogenia”. En: Storino R, J Milei. Enfermedad de Chagas. Buenos Aires: Mosby. Doyma. 1994,103-28.
73. Brandariz S, Schijman A, Vigliano C y col. “Detection of parasite DNA in Chagas’ heart disease”. Lancet. 1995; 346:1370-1371. 1995;346:1370.
74. Bellotti G, Alcides Bocchi E, Villela de Moraes A y col. “In vivo detection of Trypanosoma cruzi antigens in hearts of patients with chronic Chagas’ heart disease”. Am Heart J. 1996;131:301.
75. Laguens RP, Cabeza Meckert PM, Chambo JG. “Immunologic studies on a murine model of Chagas’disease”. Medicina (Bs. As). 1989;49:197- 202.
76. Felix JC, von Kreuter BF, Santos-Buch CA. “Mimicry of heart cell surface epítopes in primary anti-Trypanosoma cruzi Lyt 2+ T lynfocytes”. Clin Immunol Immunopathol. 1993;68:141-6.
77. Ferrari I, Levin M, Wallukat G, et al. “Molecular mimicry between the immunodominant ribosomal protein PO et Trypanosoma cruzi and a funtional epitope on the human beta 1 adrenergic receptor”. J Exp Med. 1995;182:59-65.
78. Davies, JM. “Molecular mimicry: can epitope mimicry induce autoimmue disease?” Immunol Cell Biol. 1997;75:113.
79. Cunha-Neto E, Duranti M, Gruber A y col. “Autoimmunity in Cha-gas disease cardiopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen”. Proc Natl Acad Sci USA. 1995;(11); 92:3541.
80. De Scheerder IK, de Buyzere ML, Delanghe JR y Wieme RJ. “Anti myosin humoral immune response following cardiac injury”. Autoimmunity. 1989;4:51.
81. Levin, MJ. “In chronic Chagas heart disease, don´t forget the parasite”. Parasitol Today. 1996;12:415.
82. Neu N, Ploier B y Ofner C. “Cardiac myosin induced myocarditis. Heart autoantibodies are not involved in the induction of the disease”. J Immunol. 1990;145:4094.
83. Szarfman A, Terranova VP, Rennard SI y col. “Antibodies to laminin in Chagas’ disease”. J Exp Med. 1982;155:1161.
84. Sanchez JA, Milei J, Yu ZX, Storino R, Wenthold R y Ferrans VJ. “Immunohistochemical localization of laminin in the heart of patients with chronic chagasic cardiomyopathy. Relationship to thickening of basement membranes”. Am Heart J. 1993;126:1392.
85. Khoury EL, Ritacco V, Cossio PM y col. “Circulating antibodies to peripheral nerve in American trypanosomiasis (Chagas’ disease)”. Clin Exp Immunol. 1979;36:8.
86. Ribeiro dos Santos R, Marquez JO y Koberle F. “Antibodies against neurons in chronic Chagas’ disease”. Trop Med Parasit. 1979;30:19.
87. Szarfman A, Luquetti A, Rassi A y col. “Tissuereacting immunoglo-bulins in patients with different clinical forms of Chagas’ disease”. Am J Trop Med Hyg. 1981;30:43.
88. Cossio PM, Diez C, Szarfman A y col. “Chagasic Cardiopathy: Demostration of a serum gamma globulinn factor which reacts with endocardium and vascular structres”. Circulation. 1974;49:13.
89. Laguens RP, Cossio PM, Diez C y col. “Immunopathologic and morphologic studies of skeletal muscle in Chagas’ disease”. Am J Pathol. 1975;80:153.
90. Sterin-Borda L, Cossio PM, Gimeno MF y col. “Effect of chagasic sera on the rat isolated atrial preparation: immunological, morpho-logical and functional aspects”. Cardiovasc Research. 1976;19:613.
91. Khoury EL, Diez C, Cossio PM, Arana RM. “Heterophil nature of EVI antibody in Trypanosoma cruzi infection”. Clin Immunol Immunopathol. 1983;27:283-8.
92. Sterin-Borda L, Pascual J y Borda E. “Interaction of antibodies with cardiac neurotransmitterreceptors”. Medicina. 1989;49:181.
93. Medei EH, Nascimento JH, Pedrosa RC, et al. “Antibodies with beta-adrenergic activity from chronic chagasic patients modulate the QT interval and M cell action potential duration . Europace. 2008; 10(7):868-76.
94. Sterin-Borda L, Pascual J y Borda E. “Interaction of antibodies with cardiac neurotransmitterreceptors”. Medicina. 1989;49:181.
95. Chiale PA, Elizari MV, Rosenbaum MB y col. “Sera of chagasic patients recognize functional autoimmune epitopes on G protein coupled cardiovascular receptors”. XXth Annual Meeting on
96. Wallukat G, Fu ML, Magnusson Y. y col. “Agonistic effects of anti-peptide antibodies and autoantibodies directed against adre-nergic and cholinergic receptors: absence of desensitization”. Blood Press. 1996;3:31.
97. Chiale PA, Ferrari I, Mahler E, Vallazza M y col. “Prevalencia y efectos funcionales y bioquímicos de los anticuerpos antirreceptores autonómicos cardiacos en pacientes con arritmias ventriculares y disfunción sinusal”. Rev Arg Cardiol. 2000;68(3):399.

Gelpi - Donato Fisiopatología cardiovascular
412
98. Chiale PA, Ferrari I, Vallazza M, Mahler E. y col. “Differential pro-file and biochemical effects of antiautonomic membrane receptor antibodies in ventricular arrhythmias and sinus node dysfunction”.Circulation. 2001;103:1765.
99. Limas CJ, Goldemberg IF, Limas C. “Autoantibodies against b adre-noceptors in human dilated cardiomyopathy”. Circ Res. 1989;64:97.
100. Matsui S, Fu MLX, Katsuda S y col. “Peptides derived from cardiovascular G-protein-coupled receptors induce morphologi-cal cardiomyophathic changes in immunized rabbits”. J Mol Cell Cardiol. 1997;29:641.
101. Müeller J, Wallukat G, Spiegelsberger S, et al. “Autoantibodies directed against b adrenoceptors in patiens with severe arrhythmia and an implantable cardioverter-defibrillator” (Resumen). Pacing Clin Electrophysiol. 1997;20:1073.
102. Ferrari I, Levin MJ, Elizari MV, et al. “Cholinergic autoantibodies in sinus node dysfunction”. Lancet. 1997;350:262.
103. Farias de Oliveira S, Coury Pedrosa R, Nascimiento JM y col. “Sera from chronic chagasic patients with complex cardiac arrhythmias
depress electrogenesis and conduction in isolated rabbit hearts”. Circulation. 1997;96:2031.
105. Levin MJ, Levitus G, Kerner N y col. “Autoantibodies in Chagas’ heart disease: possible markers of severe Chagas’ heart complaint”. Mem Inst Osw Cruz. 1990;85:539.
106. Kaplan D, Ferrari I, Lopez Bergami P y col. “Antibodies to ribo-somal P proteins of T. Cruzi in Chagas’ disease possess functional autoreactivity with heart tissue and different from anti-P autoan-tibodies in Lupus”. Proc Natl Acad Sci USA. 1997;94:10301.
107. Kaplan D, Ferrari I, Lopez Bergami P y col. “Anticuerpos contra las proteinas ribosomales P estimulan al receptor adrenérgico en enfermedad de Chagas pero no en Lupus Eritematoso Sistémico”. Rev Arg Cardiol. 1998;66:127.
108. Levin, M. “Molecular mimmicry and Chagas heart disease: High anti R13 autoantibodies levels are markers of severe Chagas heart complaint”. Res Immunol. 1991;142:157.

413
Remodelamiento en la resincronización del ventrículo izquierdo 24
bomba -
--
mo simple, por lo que en principio fue considerado como
-mente complejos e interrelacionados.
-didad, debemos conocer ciertos detalles acerca de la ana-
-
en cada cavidad auricular y haces comunes, que envuel-
anillos auriculoventriculares. De acuerdo a los clásicos tratados de anatomía, a nivel
--
y una capa más interna, que forman los pilares y trabéculas
-dico y de los músculos papilares.
-
cavidad torácica, mientras que los anillos auriculoventri-
No podemos dejar de mencionar al sistema cardionec-
los fascículos auriculoventriculares, el haz de His y la red de Purkinje.
-da, para lo que, de ser necesario, el lector deberá remitirse a un tratado de anatomía, debemos mencionar que la rama
del tabique interventricular, alcanzando los pilares ante-rior y posterior del ventrículo derecho, donde termina su
La rama izquierda, de mayor diámetro, se divide en
su recorrido alcanzando los pilares anterior y posterior del
-
El haz de His se encuentra cubierto por una capa de tejido conectivo que lo aísla de las células miocárdicas
-
-
Cabe recordar que la respuesta evidenciada por el músculo cardiaco se sustenta en la ley del todo o nada,
Ricardo Ronderos, Aldo Prado

Gelpi - Donato Fisiopatología cardiovascular
414
-
desarrollo de las funciones de cada elemento. Las aurículas deben contraerse antes que los ventrícu-
los, y estos deben efectuar este proceso de modo tal que se realice una actividad uniforme y coordinada en todas
-terística particular de sus células de presentar potenciales de membrana más bajos que las células miocárdicas y
-
el iniciador de la actividad cardiaca. De allí, el impulso es transmitido a ambas aurículas, completándose su des-
impulso es retardado. Este mecanismo estaría determina-do por el menor número de uniones intercalares comuni-
al llenado total de ambos ventrículos que se encuentran
son diferentes en todas las estructuras, rondando los 0.05 --
eléctrico. Estas diferencias hacen que el estímulo, una vez
hacia el territorio distal o, dicho de otra manera, alcance
rama izquierda. En base a estos datos, se establece que la despola-
-
nivel del septum interventricular, en su cara izquier-
ventrículo izquierdo, y a la altura del tracto de salida del ventrículo derecho.
-ciales, lo que llevaría a pensar que durante la sístole, al
-cular.
ser tan sencillo. Al inicio de la sístole, durante el periodo
precede a la circunferencial, pero ambas alcanzan el pico -
cunferencial o menor del ventrículo izquierdo se reduce
-
las primeras es notoria. Las velocidades de desplazamiento miocárdico se in-
Durante la diástole, las principales deformaciones ocu-rren en las etapas tempranas y tardíamente, junto al llena-
Recientemente, a través de estudios en animales, con la
-
transitorio de las
-
-mentos ventriculares, permitiendo un adecuado llenado
--
sincronía re-lacionando los tiempos de desplazamiento de ventrículo
-
Todas estas manifestaciones eléctricas comenzaron a
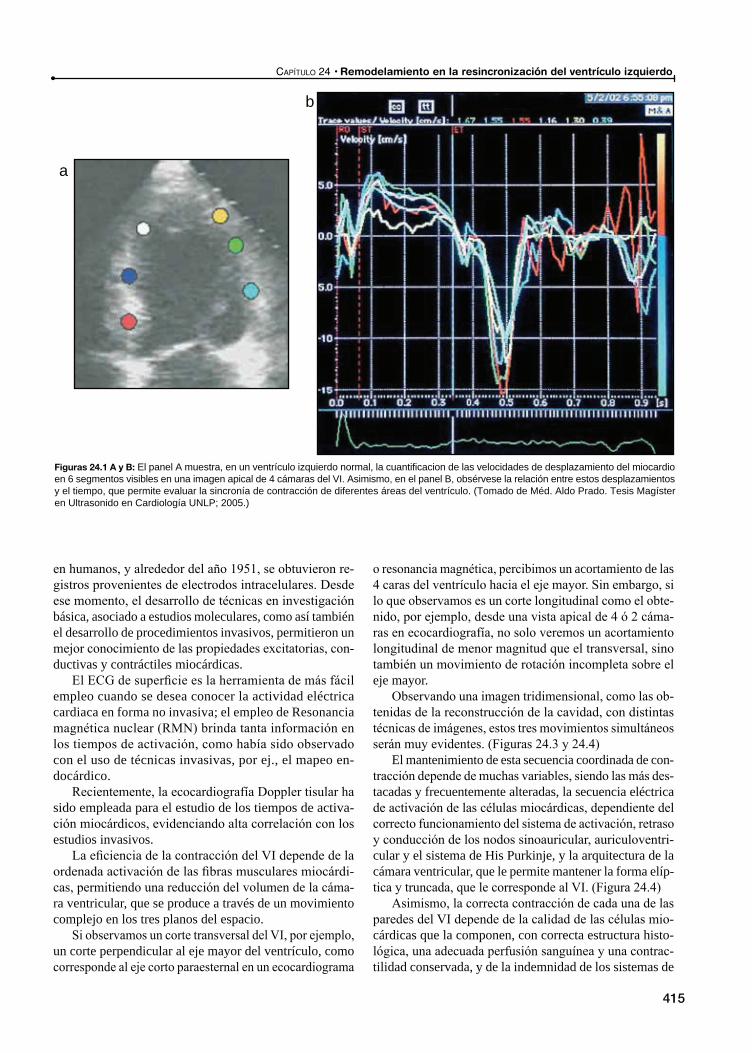
Remodelamiento en la resincronización del ventrículo izquierdo
415
-
básica, asociado a estudios moleculares, como así también el desarrollo de procedimientos invasivos, permitieron un
-ductivas y contráctiles miocárdicas.
empleo cuando se desea conocer la actividad eléctrica cardiaca en forma no invasiva; el empleo de Resonancia
con el uso de técnicas invasivas, por ej., el mapeo en-docárdico.
sido empleada para el estudio de los tiempos de activa-
estudios invasivos.
--
ra ventricular, que se produce a través de un movimiento complejo en los tres planos del espacio.
un corte perpendicular al eje mayor del ventrículo, como
acortamiento de las
--
eje mayor.-
El mantenimiento de esta secuencia coordinada de con--
tacadas y frecuentemente alteradas, la secuencia eléctrica
-cular y el sistema de His Purkinje, y la arquitectura de la cámara ventricular, que le permite mantener la forma elíp-
-cárdicas que la componen, con correcta estructura histo-
-tilidad conservada, y de la indemnidad de los sistemas de
Figuras 24.1 A y B: El panel A muestra, en un ventrículo izquierdo normal, la cuantificacion de las velocidades de desplazamiento del miocardio en 6 segmentos visibles en una imagen apical de 4 cámaras del VI. Asimismo, en el panel B, obsérvese la relación entre estos desplazamientos y el tiempo, que permite evaluar la sincronía de contracción de diferentes áreas del ventrículo. (Tomado de Méd. Aldo Prado. Tesis Magíster en Ultrasonido en Cardiología UNLP; 2005.)
a
b
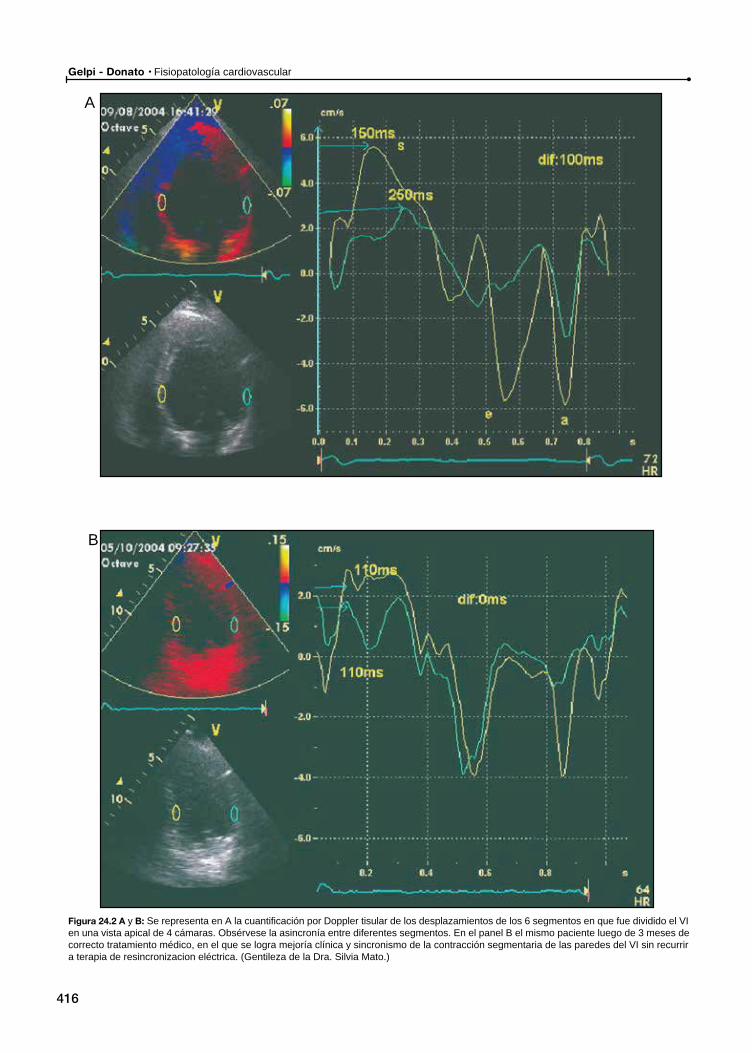
Gelpi - Donato Fisiopatología cardiovascular
416
Figura 24.2 A y B: Se representa en A la cuantificación por Doppler tisular de los desplazamientos de los 6 segmentos en que fue dividido el VI en una vista apical de 4 cámaras. Obsérvese la asincronía entre diferentes segmentos. En el panel B el mismo paciente luego de 3 meses de correcto tratamiento médico, en el que se logra mejoría clínica y sincronismo de la contracción segmentaria de las paredes del VI sin recurrir a terapia de resincronizacion eléctrica. (Gentileza de la Dra. Silvia Mato.)
A
B

Remodelamiento en la resincronización del ventrículo izquierdo
417
Figura 24.4: Muestra el modelo en 3D obtenido por ecocardiografía del ventrículo izquierdo, donde es posible apreciar la geometría normal de elipse truncada de esta cámara. Los segmentos representados en diferentes colores sobre la superficie de la cámara representan, de acuerdo a un modelo de 17 segmentos, los distintos segmentos que componen las paredes del VI.
Figura 24.5: Representa en la parte superior el modelo 3D del VI, y en la parte inferior las curvas de volumen (ml) y tiempo (mseg) de cada uno de los 17 segmentos que componen las paredes del VI durante un ciclo cardiaco, representados en diferentes colores.
Figura 24.3: Muestra el modelo 3D del ventrículo izquierdo, obtenido con ecocardiografía en tiempo real a final de la diástole

Gelpi - Donato Fisiopatología cardiovascular
418
que constituyen dos sistemas musculares coordinados. Las -
la cámara en sentido horario.
indefectiblemente una pérdida de la calidad y sincronía de
la -
-
de marcapasos, colocados en principio en el ventrículo de-recho, a través de catéteres epicárdicos o endocavitarios.
secuencia de contrac-
efectos hemodinámicos, por la pérdida de sincronía y de -
-tencia de ritmo sinusal.
Desde comienzos de la década del 80, el implante de marcapasos secuenciales aurículoventriculares, con la ca-pacidad de detectar y coordinar la actividad eléctrica au-
intervalo aurícula-ventrículo, ha demostrado una impor-
tipo de asistencia. 1
-cas de las paredes de la cámara ventricular, es conocida la importancia de la presencia de cicatrices, secundarias
ante la presencia de discinesias secundarias a aneurismas
-
-jidos de esta cámara. Técnicas como la de Dor, o la simple plicatura de los aneurismas ventriculares izquierdos, se han
-
-
Las el normal funcionamiento del aparato subvalvular mitral,
-2, 3
-rales que reemplacen tejido miocárdico normal por tejido
-
contráctil, con remodelamiento ventricular, pérdida de la -
Remodelamiento ventricular
-metría y volúmenes del ventrículo izquierdo, por ejemplo,
tejido miocárdico y ha disminuido la capacidad contráctil del conjunto de esta cámara.
Un buen ejemplo de este proceso es lo que sucede
miocardio. La necrosis de parte del tejido que compone las paredes de esta cámara, disminuye la masa de células musculares capaces de contraerse en cada sístole. 5
-miento del volumen latido.
--
de la mortalidad en enfermedades cardiovasculares. El -
cisa de la de esta cámara, en especial en 1, 4

Remodelamiento en la resincronización del ventrículo izquierdo
419
sincronía de
-
de las velocidades y sentido de desplazamiento de las pa-
La técnica conocida como Doppler tisular ha tenido
Poder medir el en forma cualitativa y cuantitativa, permite evaluar con
--
-
2, 6
-
o -tajes de desplazamiento de las paredes del miocardio, y
las velocidades del desplazamiento de las paredes del ven-trículo izquierdo permite, asimismo, evaluar la sincronía de
-
Asimismo, pueden reconocerse dentro de las ondas de
--
po al pico de estas dos ondas puede compararse entre los asincronía y la
-
Trabajos multicéntricos internacionales, basados so-
de los estímulos eléctricos intraventriculares en el ECG,
y para producir un remodelamiento inverso en las cámaras -
6-10
De los resultados de los estudios multicéntricos antes -
-
-
-
-
clínica o invasiva.En temas como la isquemia miocárdica, es indispen-
-nico-dietética y medicamentosa. La adecuada terapéutica
izquierdo, así como el remodelamiento inverso de esta cámara, sin necesidad de recurrir a terapias de estimula-
3, 5, 8
décadas con los marcapasos bicamerales.
el marcapaseo bicameral como en el marcapaseo aislado -
-

Gelpi - Donato Fisiopatología cardiovascular
420
sincronía intraventricular depende asimismo de
-mercialmente disponibles, mapeos tridimensionales de la
las distintas caras en el bloqueo de la rama izquierda, es-pontáneo o producido durante el marcapaseo unicameral
Estas situaciones clínicas, en pacientes con clase fun-cional III, terapéutica medicamentosa completa y sin is-quemia miocárdica, constituirían las indicaciones aproba-
1
-centaje de pacientes con estas características, no responden con adecuada mejoría de la clase funcional, capacidad de
-
Es fácil comprender los fracasos de la terapéutica de
contráctiles inferoposteriores y/o lateral, dada la incapa--
Pero aun en ausencia de estas, los resultados no son siempre adecuados, a pesar de la presencia de un retardo
-tricular. 7
Pero en el análisis de la sincronía intraventricular, la
-tricular.
Asimismo, es indispensable conocer si la anatomía venosa de la vaso venoso para acceder, a través de él, a la zona de la
para mejorar la sincronía. El uso solo del seno coronario, como vía para la colo-
-
lateral de esta cámara. Las venas coronarias izquierdas son inconstantes en
puntualizan las limitaciones de esta anatomía, particular--
parece coincidir con los porcentajes más elevados de fraca-
-
en términos de ser seleccionados para este tipo de terapia.
la -
-
herramientas de estudio de la actividad eléctrica ventri-
-lucrados y posibilidad de acceso venoso de los catéteres a las áreas necesarias de calidad de los resultados.
Cuando pueden obtenerse datos precisos del área disin--
auriculoventriculares y biventriculares, con la secuencia
-
7, 12, 13
En estos casos, se obtiene una de la cámara ventricular izquierda, que reduce los volú-menes ventriculares y optimiza los parámetros que, como se comentaba más arriba, son de utilidad para predecir la
--
dos a los normales, aunque no se recupere la secuencia de
miocárdicas. 2, 3, 5, 8, 9, 10
Bibliografía
1. St John Sutton MG, Plappert T, Abraham WT, Smith AL, DeLur-gio DB, Leon AR, Loh E, Kocovic DZ, Fisher WG, Ellestad M, Messenger J, Kruger K, Hilpisch KE, Hill MR; Multicenter InSync Randomized Clinical Evaluation (MIRACLE) Study Group. “Effect of cardiac resynchronization therapy on left ventricular size and function in chronic heart failure”. Circulation. 2003;30;108(13).
2. Stellbrink C, Breithardt OA, Franke A, Sack S, Bakker P, Auricchio A, Pochet T, Salo R, Kramer A, Spinelli J; PATH-CHF (PAcing THerapies in Congestive Heart Failure) Investigators; CPI Guidant Congestive Heart Failure Research Group. “Impact of cardiac resynchronization therapy using hemodynamically optimized pacing on left ventricular remodeling in patients with congestive heart failure and ventricular conduction disturbances”. J Am Coll Cardiol. 2001; 38(7):1971-3.

Remodelamiento en la resincronización del ventrículo izquierdo
421
3. Yu CM, Fung JW, Chan CK, Chan YS, Zhang Q, Lin H, Yip GW, Kum LC, Kong SL, Zhang Y, Sanderson JE. “Comparison of efficacy of reverse remodeling and clinical improvement for relatively narrow and wide QRS complexes after cardiac resyn-chronization therapy for heart failure”. J Cardiovasc Electrophysiol. 2004;15(9):1058-65.
4. Erol-Yilmaz A, Verberne HJ, Schrama TA, Hrudova J, De Winter RJ, Van Eck-Smit BL, De Bruin R, Bax JJ, Schalij MJ, Wilde AA, Tukkie R. “Cardiac resynchronization induces favorable neurohu-moral changes”. Pacing Clin Electrophysiol. 2005;28(4):304-10.
5. Yu CM, Fung WH, Lin H, Zhang Q, Sanderson JE, Lau CP. “Predictors of left ventricular reverse remodeling after cardiac resynchronization therapy for heart failure secondary to idiopa-thic dilated or ischemic cardiomyopathy”. Am J Cardiol. 2003; 15;91(6):684.
6. Yu CM, Fung JW, Zhang Q, Chan CK, Chan I, Chan YS, Kong SL, Sanderson JE, Lam CW. “Improvement of Serum NT-ProBNP Predicts Improvement in Cardiac Function and Favorable Prognosis After Cardiac Resynchronization Therapy for Heart Failure”. J Card Fail. 2005;(5 Suppl):42-6.
7. Yu CM, Abraham WT, Bax J, Chung E, Fedewa M, Ghio S, Le-clercq C, Leon AR, Merlino J, Nihoyannopoulos P, Notabartolo D, Sun JP, Tavazzi L; PROSPECT Investigators. “Predictors of response to cardiac resynchronization therapy (PROSPECT)--study design”. Am Heart J. 2005;149(4):600-5.
8. Woo GW, Petersen-Stejskal S, Johnson JW, Conti JB, Aranda JA Jr, Curtis AB. “Ventricular reverse remodeling and 6-month outcomes in patients receiving cardiac resynchronization therapy:
analysis of the MIRACLE study”. J Interv Card Electrophysiol. 2005;12(2):107-13.
9. Yu CM, Fung JW, Chan CK, Chan YS, Zhang Q, Lin H, Yip GW, Kum LC, Kong SL, Zhang Y, Sanderson JE. “Comparison of efficacy of reverse remodeling and clinical improvement for relatively narrow and wide QRS complexes after cardiac resyn-chronization therapy for heart failure”. J Cardiovasc Electrophysiol. 2004;15(9):1058-65.
10. Achilli A, Sassara M, Ficili S, Pontillo D, Achilli P, Alessi C, De Spirito S, Guerra R, Patruno N, Serra F. “Long-term effectiveness of cardiac resynchronization therapy in patients with refractory heart failure and narrow QRS”. J Am Coll Cardiol. 2004 16;44(10):2096.
11. Abraham, WT. “Rationale and design of a randomized clinical trial to assess the safety and efficacy of cardiac resynchronization therapy in patients with advanced heart failure: the Multicenter InSync Randomized Clinical Evaluation (MIRACLE)”. J Card Fail. 2000; 6(4):369-80.
12. Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J; MIRACLE Study Group. “Multicenter InSync Randomized Clinical Evaluation. Cardiac resynchronization in chronic heart failure”. N Engl J Med. 2002; 346(24):1845-53.
13. Kron J, Aranda JM Jr, Miles WM, et al. “Benefit of cardiac resynchronization in elderly patients: results from the Multicenter InSync Randomized Clinical Evaluation (MIRACLE) and Multicenter InSync ICD Randomized Clinical Evaluation (MIRACLE-ICD) trials . J Interv Card Electrophysiol. 2009; 25(2):91-6.


423
A
actividad,
adenosina 91, 97
, ,
, ,
de Prinzmetal 50de umbral variable 50
, , , 177, , , , ,
,
,
, , , ,
, , , ,
,
, ,
B
,
, ,
bloqueantes,
bloqueo
, ,
C
calcineurina 177calcio 80, 197, ,
calmodulina 198
Índice analítico Í

Gelpi - Donato Fisiopatología Cardiovascular
424
células
, ,
stem cells
, , , ,
citoquinas 157, ,
, ,
tipo I 157tipo III 157
colesterolHDL 19LDL 19
, ,
,
corrientes
crecimientocelular 158
D
slippage
, 57
ventricular 159,
, 110,
dislipemia 19,
dislipoproteinemias 19
,
E
efectoAnrep 179Bowditch 179
, , ,
, , ,
,
enfermedad
estatinas 11,
, , ,
estiramiento 179,
estrés
,
, ,

425
F
,
familia de lipoproteína 19
, ,
, ,
, , ,
,
fosfolamban 199, , ,
,
frecuencia, ,
, , ,
,
G
,
,
,
H
hiperquilomicronemia 19
, ,
, , ,
, 177, 189, , , , , ,
, ,
miocárdica 177,
, ,
I
infarto
, 155, ,
con reperfusión 128
inhibidores,
injuria
, ,
cardíaca 189, , , , , , , , ,
, , , ,
intercambiadorNa /Ca 197Na -H

Gelpi - Donato Fisiopatología Cardiovascular
426
Na /H ,
, ,
,
,
isquemia 55, 109, , , ,
miocárdica 79,
L
ley de Laplace 159lipoproteínas 19
,
M
, ,
matriz, , ,
, , ,
de matriz 157
miocardio 79atontado 79hibernado 79, 119
miocardiopatía
muerte,
N
nitratos 11,
,
O
, ,
, 97,
P
,
placa
,
,
poscondicionamiento 89isquémico 89,
,
,
precondicionamiento 85, 89remoto 98tardío 97

427
, , ,
, , 117proteína
,
, 97
Q
R
radicales libres 80
acoplados proteína G 177
, ,
, , , 189, ,
,
,
cardíaca 11,
,
S
,
, ,
, ,
síndrome/s,
,
sin stent sistema
,
nervioso simpático 109,
, ,
,
,
,
, ,
,
stem cells , ,
stents
,
subtipo/s

Gelpi - Donato Fisiopatología Cardiovascular
428
T
tono
,
tripanosoma cruzi
,
,
V
,
, , ,
, ,
vía de las kinasas de rescate activadas en la isquemia/
volumen
,
W
,
X


















