
Gastro cb py cep
58
Colangitis Esclerosante Primaria Colangitis Esclerosante Primaria Cirrosis Biliar Primaria Cirrosis Biliar Primaria Hospital Regional Lic.Adolfo López Mateos Hospital Regional Lic.Adolfo López Mateos Andrés Pérez Barceló RMI Andrés Pérez Barceló RMI
-
Upload
hospital-regional-lic-adolfo-lopez-mateos -
Category
Documents
-
view
168 -
download
2
Transcript of Gastro cb py cep

Colangit is Esclerosante PrimariaColangit is Esclerosante PrimariaCirrosis Bil iar PrimariaCirrosis Bil iar Primaria
Hospital Regional Lic.Adolfo López MateosHospital Regional Lic.Adolfo López MateosAndrés Pérez Barceló RMIAndrés Pérez Barceló RMI

Colangitis Esclerosante Primaria
Enfermedad colestàsica crónica de origen desconocido caracterizada por destrucciòn progresiva de conductos biliares asociada a inflamaciòn difusa y fibrosis que eventualmente desencadena cirrosis y malignidad ...

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Descrita inicialmente a mediados de 1850 con una presentaciòn clinica compleja, 100 años despues relacionada con EII
Incidencia y Prevalencia varia alrededor del mundo. Geoepidemiología
Prevalencia en Norteamèrica 6-16 casos por 100,000 habitantesIncidencia 1-100,000
Media de edad del diagnòstico de CEP es 41 añosMas Comùn en hombres.Pacientes diagnosticados a menudo incidentalmente, alrededor de 50%
son asintomàticos.

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
5 años posterior al diagnòstico de CBP asintomatica 22% muestra sintomas clìnicos, posteiror a 6 años 76% tiene alguna evidencia de progresiòn de la enfermedad (bioquìmica, clìnica o radiogràfica)
Aumento de incidencia de CEP a lo largo del tiempo
Fatiga a menudo presente al inicio de diagnòstico

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Mayor numero de estudios recientes en paises asiàticos a desencadenado nuevo interés en estudio de CEP
Variaciones Geoepidemiològicas--> menor incidencia de asociación con enfermedad Inflamatoria Intestinal → 37% de los pacientes en Asia y en Sur de Europa, en comparación 60-80% en norte de Europa y Norteamèrica
Solo 1-4% de pacientes con CUCI desarrolla colangitis esclerosante de importancia clìnica
“Nacimiento” de una entidad clinica asociada pero diferente de Colangitis Esclerosante con niveles elevados de IgG4
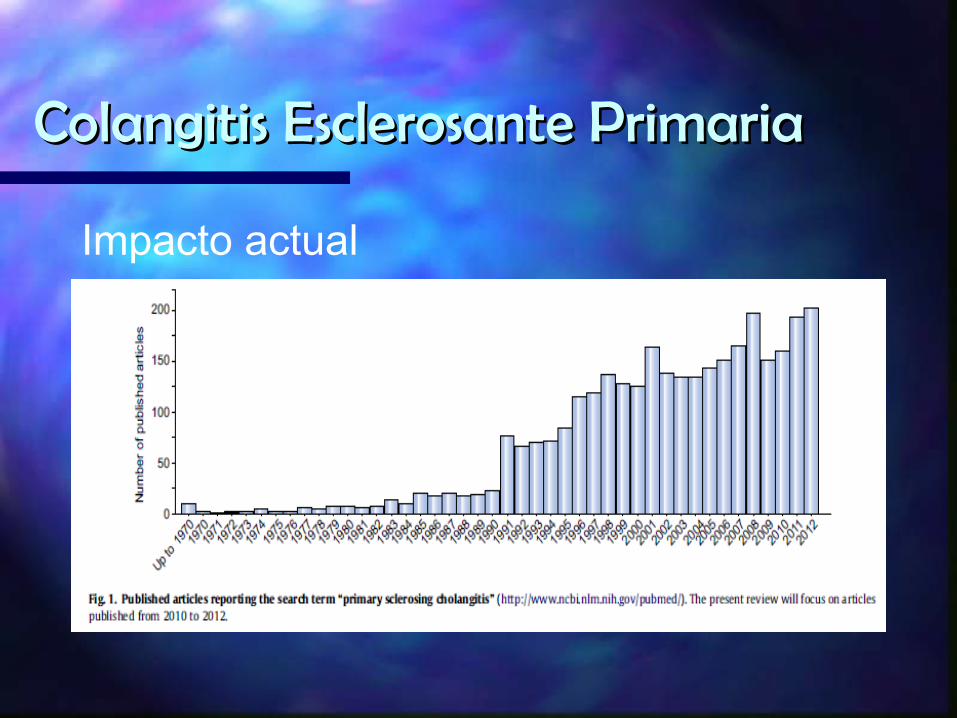
Impacto actual
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

FISIOPATOLOGIA
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
CEP se asocia a Antigenos de CMH HLA B-28 y DR3-DR4

– FISIOPATOLOGIA
– Toxicidad o Inmunopatologìa??
Estudios Genèticos de asociaciònToxicidad de àcidos biliaresEje intestino-Hìgado
–
–
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
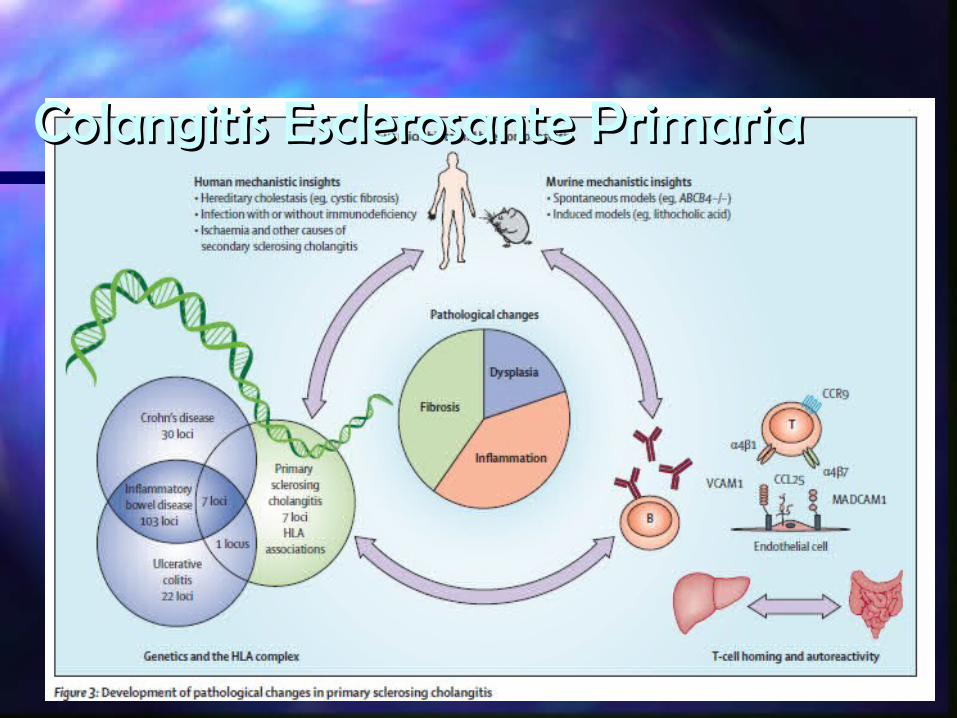
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

FisiopatologìaLesiòn progresiva y crònica se produce en conductos biliares
pequeños, medianos y grandes con una fibrosis inflamatoria concentrica y obliterante
En enfermedad temprana los cambios estan confinados a los espacios porta con infiltrado de linfocitos, celulas plasmàticas y neutròfilos.
Histologicamente el grado de inflamaciòn no corresponde con la fibrosis y su gravedad.
El riesgo de displasia y neoplasia bilair no se correlaciona con la duraciòn o severidad de la fibrosis biliar.
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
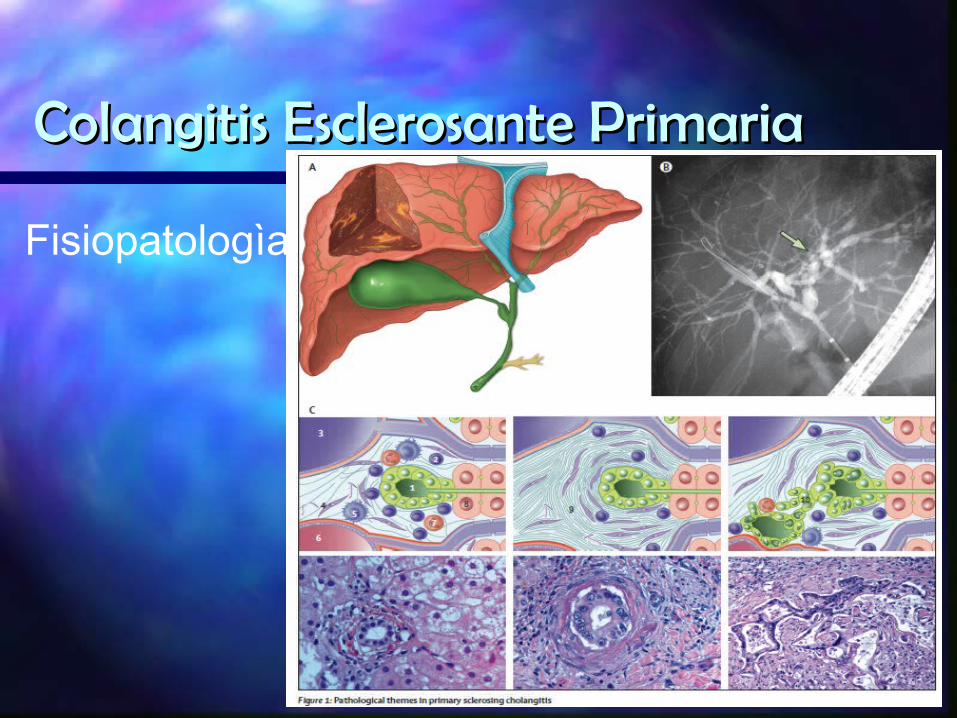
Fisiopatologìa
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

V
Colangitis ¿primaria o secundaria?
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
CX

Colangitis esclerosante – IgG4Enfermedad Multisitèmica Rel IgG4
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Presentaciòn Clínica
50% asintomàticos al inicio de diagnòstico
Sintomas generales, principalmente fatiga
Dolor abdominal (37%). Ictericia (30%) y fiebre (17%)

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Ictericia Obstructiva Progresiva
Fatiga, Prurito (hasta ser incontrolable) Anorexia e Indigestiòn
En etapas avanzadas –> Osteoporosis y malabsorciòn de vitaminas liposolubles
En pacientes con CUCI + CEP Ausencia de afecciòn rectal e ileìtis manifiesta
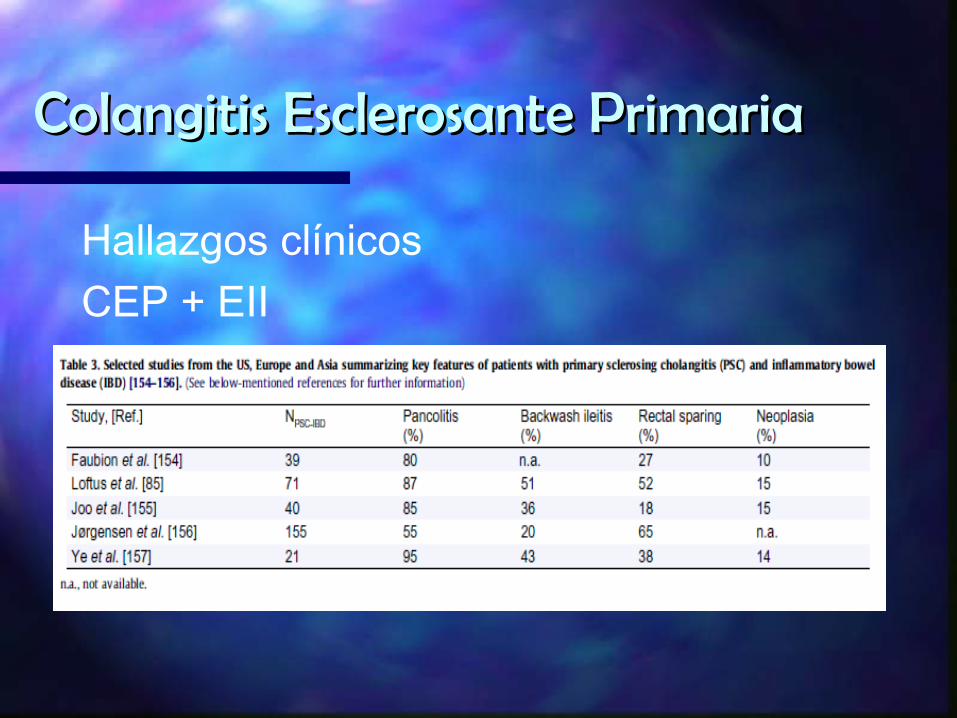
Hallazgos clínicosCEP + EII
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

Laboratorio
Colangitis Esclerosante PrimariaColangitis Esclerosante PrimariaColangitis Esclerosante PrimariaColangitis Esclerosante Primaria

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Marcadores Bioquímicos
Incremento en los niveles séricos de fosfatasa alcalina es la anormalidad bioquìmica mas comùn detectada
En algunos casos es la ùnica alteracion observada y puede variar en el curso de la enfermedad
Los niveles de Aminotransferasas suelen ser normales → Hay reportes de disminuciòn de 3-4 veces el limite normal en pacientes con CEP
Niveles aumentados de Aminotransferasas sugieren obstrucciòn aguda de via biliar o sindrome de sobreposiciòn con HAI

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Marcadores Bioquímicos
Bilirrubinas son tipicamente normalesNiveles de albúmina, INR, Plaquetas son típicamente normales a
menos que se encuentre ya cirrosis hepática e hipertensiòn portal.
Alta sospecha clìnica en alteraciones de funcionamiento hepàtico en pacientes con EII

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Marcadores Bioquímicos
Marcadores bioquìmicosCEP esta asociada en alta proporciòn con autoanticuerpos no
especìficos. No hay un marcador serològico especìfico para el diagnòstico de
CEPSe debe realizar diagnóstico diferencial con marcadores para HAI,
CE IgG4 + PAI

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
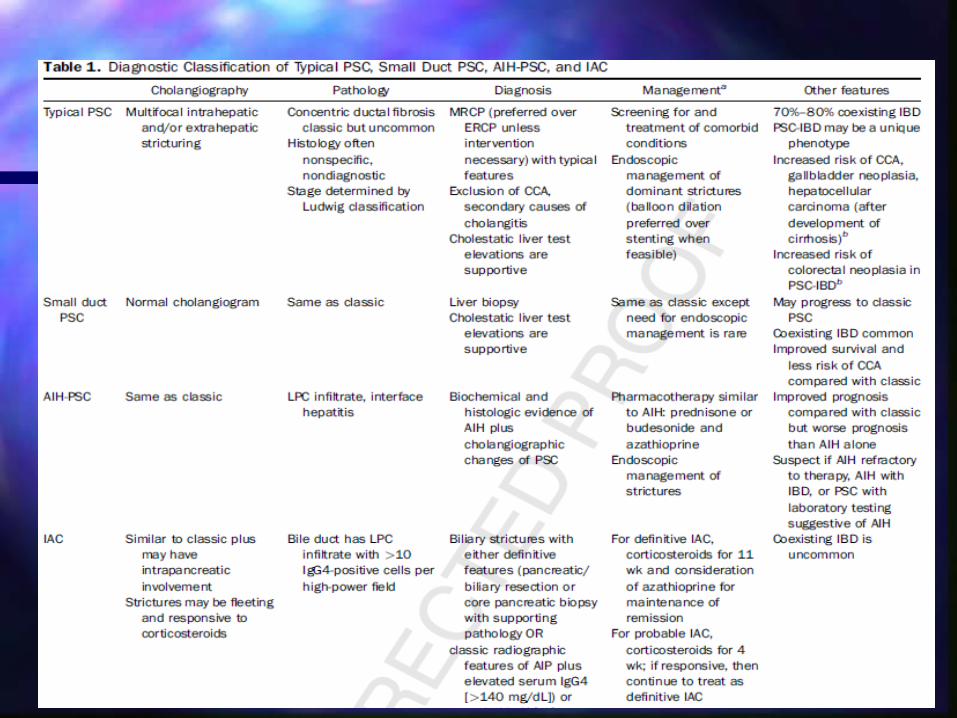
IMAGENColangiografìa es la forma mas conveniente de evaluar a
pacientes con sospecha de CEPLos Hallazgos caracterìsticos incluyen estructuras anulares
multifocales en via biliar intra y extrahepàtica con segmentos alternados normales y dilatados.
Colangiorresonancia a sustituido a CPRE como estudio de elecciónNo invasivaEvita radiaciónMejor costo-efectividad

Colangitis Esclerosante PrimariaColangitis Esclerosante PrimariaBiopsia hepàtica
Tìpicamente no es necesaria para diagnosticar Colagitis Esclerosante Pirimaria
Se requiere si se desea documentar CEP de conductos pequeños o en sospecha de dignòsticos diferenciales como HAI, CE IgG4
Hallazgos en CEP no son específicos debido a la participaciòn heterogenea de la via biliar
El hallazgo clàsico de fibrosis ductal (papel de cebolla) periductales en el espacio porta se encuentra raramente en la pràctica clìnica

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
Variaciones clínicasCEP de pequeños conductos
– Mejor pronòstico, 20% evoluciona a grandes conductos en 7-10 años. Menor riesgo de colangiocarcinoma
Colangiocarcinoma.– 10% de los pacientes. Debe descartarse desde el
diagnòstico de CEP.
Asociaciòn con IgG4
Sindrome de sobreposiciòn (HAI)

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
TRATAMIENTOÀCIDO URSODESOXICÓLICO
AASLD → Contraindica el uso de AUDCEASLD → No lo recomienda ni contraindica basado en falta de estudios

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
TRATAMIENTO
INMUNOSUPRESORESEsteroides, Etanercept, tacrolimus, Ciclosporina, azatioprina,
metotrexate e infliximab No han mostrado beneficios clìnicos
Pacientes con evidencia de HAI-CEP deben ser tratados con terapia inmunosupresora en base a esquemas de tx para HAI

Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
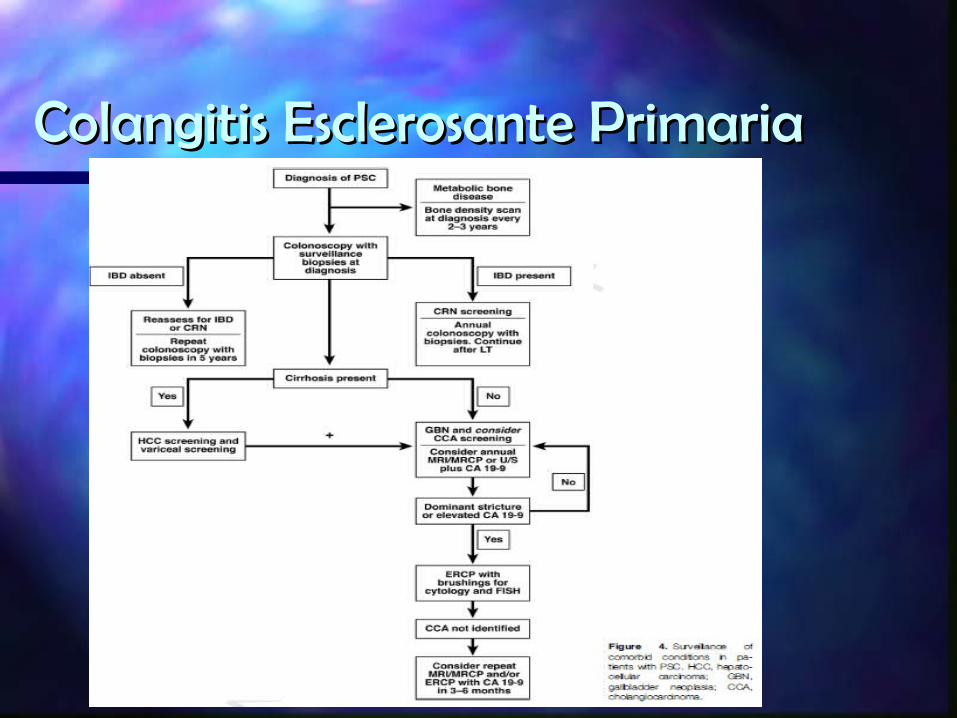
TRATAMIENTO
TERAPIA ENDOSCOPICAVigilancia de lesiones premalignas
TRASPLANTE HEPATICO– Adecuada tolerancia– Indicaciones relacionadas con complicaiones,falta de
respuesta a tratmaiento, prurito incontrolable, colangitis recurrente
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria

Recomendaciones
Colangitis Esclerosante PrimariaColangitis Esclerosante Primaria
CIRROSIS BILIAR PRIMARIA

Cirrosis Biliar Primaria
DEFINICIÓN
Enfermedad hepática granulomatosa crónica de naturaleza inmunitaria caracterizada por destrucciòn de conductos biliares intrahepàticos (pequeños, lobares e interlobares) , colestasis y en algunos casos cirrosis

Cirrosis Biliar Primaria
Antecedentes
1761 → Decrita inicialmente por patólogo Giovanni Battista Morgagni
1851 → Adisson u Gull estudio de misma patología1965 → Reconocimiento de Autoanticuerpos
mitocondriales (AMA`s) por Walker y cols1987 → Antigenos fueron clonados e identif icados como
subunidades de complejo piruvato deshidrogenasa

Epidemiologìa
Asociación con otras enfermedades de origen autoinmuneCaracterística la presencia de anticuerpos antimitocondrialesMujeres son mas afectadas entre la 5ta y 6ta década de la vida en
9 de 10 casosPuede existir afección en personas mas jóvenes, incluso niños
Cirrosis Biliar Primaria

Epidemiología
Progresión lentaIncidencia de 10-12 casos por millón de personas añoPrevalencia 100-300 casos por millón (creciente). Reportes de
hasta 402.AMA en la población general puede ser ta alto como 0.5%Riesgo acumulado de desarrollo de CBP en pacientes con Historia
familiar de la enfermedad, Historia de infecciones vaginales y del tracto urinario, comorbilidades con enfermedades autoinmunes, tabaquismo pasado o presente, uso de esmalte de uñas y tinte en el cabello.
Cirrosis Biliar Primaria

Epidemiologìa
Cirrosis Biliar Primaria

Epidemiología Enfermedades autoinmunes asociadas:
Cirrosis Biliar Primaria

Fisiopatología3 puntos a seguir para el entendimiento de la patogenia:1) Presencia de AMA previa a enfermedad hepatica clìnica sugiere
una perdida de la tolerancia de autoantigenos mitocondriales como un evento temprano independiente para el desarrollo de la enfermedad
Cirrosis Biliar Primaria
Fisiopatología
3 puntos a seguir para el entendimiento de la patogenia:2) A pesar de que el Autoantìgeno se encuentra presente en todas
las celulas nucleadas, la respuesta inmune solo esta restringida a celulas epiteliales de la via biliar intrahepàtica y en cierto grado a glandulas salivales y lacrimales
Cirrosis Biliar Primaria

Fisiopatología
3 puntos a seguir para el entendimiento de la patogenia:3) La recurrencia de Cirrosis Biliar Primaria en pacientes
postrasplantados apoya la idea de que las celulas epiteliales de los conductos biliares es un objetivo genèrico y no el unico
Cirrosis Biliar Primaria

Fisiopatología
Similiar a otros complejos de enfermedades autoinmunitarias la susceptibilidad genètica y la interacciòn con factores ambientales es necesaria para desencadenar la enfermedad
1-6% de pacientes con CBP tiene al menos 1 familiar con enfermedad manifiesta.
63% de concordancia entre gemelos monocigotos
Relación con polimorfismos de Antigenos HLA DBQ B1, IL12A, IL12RB2 y STAT
Cirrosis Biliar Primaria

Fisiopatología
Criterios de Witebsky a favor y en contra de origen autoinmune de CBP
A favor de autoinmunidad:1. Anticuerpos séricos especìficos2. Células T autorreactivas3. Transferencia adaptativa de colangitis usando ceulas T Cdb+ (modelos
murinos)4. Defectos funcionales regulatorios de Celulas T5. Predominio femenino6. Comorbilidades autoinmunes7. Asociaciòn CMH
Cirrosis Biliar Primaria

Fisiopatología
Criterios de Witebsky a favor y en contra de origen autoinmune de CBP
En contra de autoinmunidad:1. Ausencia de enfermedad después de transferencia anticuerpos (en
ratones)2. Ausencia de correlaciòn entre el titulo de anticuerpos antimitocondriales
y la severidad de la enfermedad3. Falla a la respuesta con inmunosupresores
Cirrosis Biliar Primaria

Fisiopatología
Cirrosis Biliar Primaria

Manifestaciones clínicas
Los hallazgos clínicos y la historia natural de la enfermedad varia ampliamente entre pacientes, desde asintomática y lentamente progresiva hasta sintomática y deterioro rapido.
Aumento de detecciòn de enfermedad asintomàtica por laboratorios de rut ina.
2/3 de los pacientes inicialmente asintomáticos desarolla enfermedad hepatica sintomática a los 5 años del diagnóstico.
1/3 se mantiene l ibre de síntomas en muchos años
Cirrosis Biliar Primaria

Manifestaciones clínicas
Fatiga aunque poco específica es el síntoma mas común en CBP → en 80% de los pacientes y mas de 40% de los síntomas en enfermedad moderada-severa.
Síntomas relacionados a colestasis crónica → prurito es el mas típico 20-70% de pacientes, usualmente precedido de ictericia
– Colestasis → Componentes pruritogénicos:– 1. Acidos biliares– 2. Agentes opioides endogenos– 3. Lisofosfolipasa autotaxina (neoplasias e inmunidad)
Cirrosis Biliar Primaria

Manifestaciones clínicasDisminuciòn de la densidad mineral òsea es comùn--> Osteopenia 33%,
Osteoporosis 11%Hipercolesterolemia → Aumento de HDL. No incrementa el riesgo
cardiovascular o ateroesclerosis temprana
Complicaciones de Cirrosisi hepatica por CBP solo difieren en presentaciòn clìnica en la presetnaciòn temprana de vàrices esofàgicas antes de otras manifestaciones de hipertensiòn portal
La incidencia de CHC es similar que en otras enfermedades en estadios avanzados.
Cirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar Primaria

DiagnósticoDebe ser sospechado de acuerdo a Historia clínica y hallazgos de
laboratorio correspondientes con enfermedad colestàsica.
Hallazgos de elevaciòn de fosfatasa alcalina
Elevaciòn de Aminotransferasas e Inmunoglobulinas (principalmente IgM)
Cirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar Primaria

Diagnóstico2 de los siguientes 3 crierios establecen el diagnóstico:
– 1. Anticuerpos antimitocondriales séricos con titulos 1:40 o mayor
– 2. Aumento de la cifra de fostasa alcalina al menos 1.5 por arriba del limite normal por mas de 24 semanas
– 3. Hallazgos histológicos caracetrìsticos, especialmentecolangitis no supurativa o lesion de conductos biliares interlobulares
Cirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar Primaria
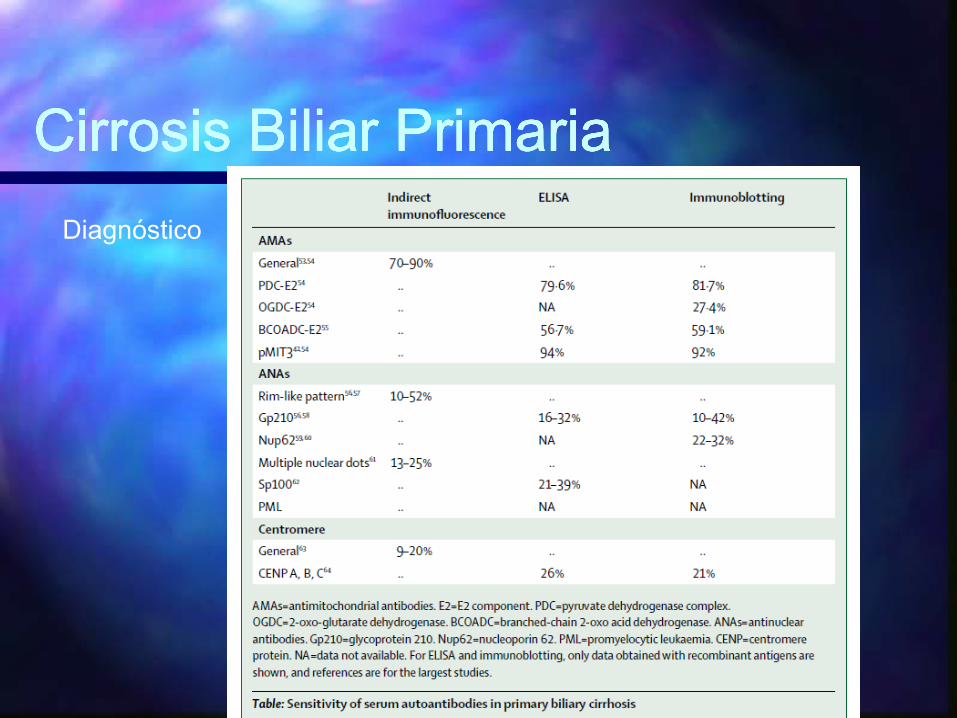
Diagnóstico
Cirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar PrimariaCirrosis Biliar Primaria

Diferencial vs SobreposiciónRazòn Fosfatasa alcalina/TGO <1.5Niveles de IgG elevadosAnticuerpos antimusculo liso >1:80
Cirrosis Biliar Primaria

Historia Natural de la Enfermedad
Cirrosis Biliar Primaria

Tratamiento
Muchos agentes en estudio, mismos limitados por enfermedad poco comùn, variabilidad de presentación clínica y sx de sobreposición (se necesitan estudios grandes, multicentricos y por largo tiempo)
Àcido ursodesoxicòlicoSal biliar polar hidrofìlica, normalmente <3% de sales biliares
totales.13-15 mg/kg/dìa dividido en 3 dosis
Cirrosis Biliar Primaria

Tratamiento
Àcido UrsodesoxicólicoEl mecanismo de acciòn en CBP permanece poco claro
Reducciòn en cantidad de àcidos biliares primarios y puede regular la señalización celular y evitar la apoptosis
Ensayos clinicos aleatorizados controlados → todos muestran mejoría sustancial de los marcadores sericos de colestasis.
Efecto en la supervivencia solo evidenciado en 1 solo estudio
Tiene el potencial de prevenir la hipertension portal y varices esofàgicas, retardando el tiempo para trasplante de hìgado.
Mejor resultado en Nivels de Bilirrubina mayor a 24 mmol/L
Cirrosis Biliar Primaria

Tratamiento Alternativas cuando el tratmiento con AUD no logrò una respuesta
completa estan siendo estudiados.
Agonista del receptor Farnesoid X
No esta establecido beneficio con uso de esteroides. Solo en sindromes de sobreposiciòn con HAI
Tratamiento de sintomas y comorbilidades
Cirrosis Biliar Primaria

Tratamiento
Cirrosis Biliar Primaria

Es una indicaciòn común de trasplante hepático
El procedimiento es solo efectivo en pacientes con hepatipatìa en etapa terminal, Indicaciones que no difieren con otras causas.
Cirrosis descompensada
Ascitis refractaria a diuréticos
Peritonitis bacteriana espontanea
Hemorragia variceal recurrente
Encefalopatìa
Carcinoma hepatocelular
Sobrevida postrasplante 92% a 1 año y 85% a 5 años.Tasa de recurrencia 30% a 10 años
Cirrosis Biliar Primaria

BIBLIOGRAFIA1. JOHN E. EATON,1 JAYANT A. TALWALKAR. Pathogenesis of Primary Sclerosing
Cholangitis and Advances in Diagnosis and Management. GASTROENTEROLOGY 2013;-:116
2. Tom H. Karlsen1,2, Kirsten Muri Boberg. Update on primary sclerosing cholangitis. Journal of Hepatology 2013 vol. 59 j 571–582
3. Gideon M Hirschfi eld, Tom H Karlsen, Keith D Lindor, David H Adams. Primary sclerosing cholangitis. www.thelancet.com Published online June 28, 2013
4. Atsushi Tanaka*, Hajime Takikawa. Geoepidemiology of primary sclerosing cholangitis: A critical review.Journal of Autoimmunity xxx (2013) 1-6.
5. Carlo Selmi, Christopher L Bowlus, M Eric Gershwin, Ross L Coppel. Primary biliary cirrhosis. Lancet 2011; 377: 1600–09
6. James M Neuberger. Primary biliary cirrhosis. Autoinmune Liver disease. 2011 Elsevier Ltd. All rights reserved
7. Li Wang, Feng-Chun Zhang. Connective tissue diseases in primary biliary cirrhosis: A population-based cohort study. World J Gastroenterol 2013 August 21; 19(31): 5131-5137


















