Genética molecular del melanoma familiar. Revisión del ... · Genética molecular del melanoma...
31
102 Rev. Chilena Dermatol. 2011; 27(1):102-108 Genética molecular del melanoma familiar. Revisión del tema y aplicación clínica Pedro Jaramillo Z. 1 , Pablo Uribe G. 1 , Montserrat Molgó N. 1 , Claudia Balestrini D. 3 , Sergio González B. 2 Departamentos de 1 Dermatología y de 2 Anatomía Patológica, Facultad de Medicina, Pontificia Universidad Católica de Chile. 3 Servicio de Dermatología Hospital Dr. Sótero del Río, Santiago, Chile. Correspondencia: Dr. Sergio González B. Correo electrónico: [email protected] DERMATOPATOLOGÍA Resumen El Melanoma maligno cutáneo (MMC) representa el 4% del total de los tumores malignos de la piel, dando cuenta del 80% de las muertes producidas por cáncer cutáneo. La sobrevida a 5 años de individuos portadores de enfermedad metastásica se estima en 14%. Las formas de presentación incluyen una variante esporádica y otra familiar o hereditaria. En ambas el papel de diferentes mutaciones genéticas que otorgan susceptibilidad al desarrollo de MMC es indiscutido, así como la interacción entre las características genéticas del individuo y eventos ambientales. En el MMC familiar se han establecido dos genes de alta susceptibilidad con diferente penetrancia y frecuencia: CDK4 y CDKN2A. CDKN2A (Cyclin- dependent kinase inhibitor 2A) es el más importante gen de susceptibilidad a MMC, cuyas mutaciones se han identificado en aproximadamente un 40% de familias que presentan tres o más casos de MMC. Las características clínicas asociadas a la mutación de CDKN2A son número elevado de individuos afectados por MMC dentro de una familia, MMC primario múltiple y presencia conjunta de MMC y cáncer de páncreas dentro de una familia. En Chile se desconoce la frecuencia y tipos de mutaciones que afectan a CDKN2A en familias con predisposición a MMC familiar. Palabras clave: melanoma familiar, CDKN2A, genética del melanoma, melanoma hereditario Summary Cutaneous malignant melanoma (CMM) represents 4% of all malignant skin tumors and accounts for 80% of deaths related to cutaneous cancer. 5-year survival rates in individuals with metastatic disease is around 14%. An hereditary or familial variant of CMM has been identified. It is related to different mutations of so-called susceptibility genes as well as to interactions between genetic characteristics and environmental factors. Familial CMM is related to two genes of elevated susceptibility, penetrance, and frequency: CDK4 and CDKN2A (Cyclin-dependent kinase inhibitor 2A). CDKN2A is the most important susceptibility gene and its mutations have been identified in approximately 40% of the families bearing three or more members with CMM. Clinical features associated to CDKN2A mutations are elevated number of family members with CMM, multiple primary CMM, and the presence of both CMM and pancreatic cancer in the same family. In Chile, the incidence and mutation variants of CDKN2A in families with CMM is unknown. Key words: familial melanoma, CDKN2A, melanoma genetics, hereditary melanoma
Transcript of Genética molecular del melanoma familiar. Revisión del ... · Genética molecular del melanoma...

102 Rev. Chilena Dermatol. 2011; 27(1):102-108
Genética molecular del melanoma familiar. Revisión del tema y aplicación clínicaPedro Jaramillo Z.1, Pablo Uribe G.1, Montserrat Molgó N.1, Claudia Balestrini D.3, Sergio González B.2
Departamentos de 1Dermatología y de 2Anatomía Patológica, Facultad de Medicina, Pontificia Universidad Católica de Chile. 3Servicio de Dermatología Hospital Dr. Sótero del Río, Santiago, Chile.
Correspondencia: Dr. Sergio González B.Correo electrónico: [email protected]
DERMATOPATOLOGÍA
Resumen
El Melanoma maligno cutáneo (MMC) representa el 4% del total de los tumores malignos de la piel, dando cuenta del 80% de las muertes producidas por cáncer cutáneo. La sobrevida a 5 años de individuos portadores de enfermedad metastásica se estima en 14%. Las formas de presentación incluyen una variante esporádica y otra familiar o hereditaria. En ambas el papel de diferentes mutaciones genéticas que otorgan susceptibilidad al desarrollo de MMC es indiscutido, así como la interacción entre las características genéticas del individuo y eventos ambientales. En el MMC familiar se han establecido dos genes de alta susceptibilidad con diferente penetrancia y frecuencia: CDK4 y CDKN2A. CDKN2A (Cyclin-dependent kinase inhibitor 2A) es el más importante gen de susceptibilidad a MMC, cuyas mutaciones se han identificado en aproximadamente un 40% de familias que presentan tres o más casos de MMC. Las características clínicas asociadas a la mutación de CDKN2A son número elevado de individuos afectados por MMC dentro de una familia, MMC primario múltiple y presencia conjunta de MMC y cáncer de páncreas dentro de una familia. En Chile se desconoce la frecuencia y tipos de mutaciones que afectan a CDKN2A en familias con predisposición a MMC familiar.
Palabras clave: melanoma familiar, CDKN2A, genética del melanoma, melanoma hereditario
Summary
Cutaneous malignant melanoma (CMM) represents 4% of all malignant skin tumors and accounts for 80% of deaths related to cutaneous cancer. 5-year survival rates in individuals with metastatic disease is around 14%. An hereditary or familial variant of CMM has been identified. It is related to different mutations of so-called susceptibility genes as well as to interactions between genetic characteristics and environmental factors. Familial CMM is related to two genes of elevated susceptibility, penetrance, and frequency: CDK4 and CDKN2A (Cyclin-dependent kinase inhibitor 2A). CDKN2A is the most important susceptibility gene and its mutations have been identified in approximately 40% of the families bearing three or more members with CMM. Clinical features associated to CDKN2A mutations are elevated number of family members with CMM, multiple primary CMM, and the presence of both CMM and pancreatic cancer in the same family. In Chile, the incidence and mutation variants of CDKN2A in families with CMM is unknown.
Key words: familial melanoma, CDKN2A, melanoma genetics, hereditary melanoma

103Rev. Chilena Dermatol. 2011; 27(1):102-108
Genética molecular del melanoma familiar. Revisión del tema y aplicación clínica
IntroducciónEl cáncer cutáneo es el más común de todas las neoplasias malignas en seres humanos. El MMC ocupa el tercer lugar en frecuencia, correspondiendo aproximadamente al 4% de todas las neoplasias malignas cutáneas(1,2). Su incidencia continúa aumentando, estimándose que el aumento es mayor que el de cualquier otra neoplasia en hombres, y sólo sobrepasada por el cáncer pulmonar en mujeres(3), por lo que actualmente es considerado un fenómeno de proporciones casi epidémicas. El MMC tiene un comportamiento agresivo reflejado en la alta tasa de metástasis linfáticas regionales y a distancia de las formas invasoras.
Se estiman más de 68.000 nuevos casos y más de 8.000 muertes anualmente en EEUU asociadas a MMC, con un promedio individual de riesgo de desarrollo de enfermedad invasiva a lo largo de la vida cercano a 1 en 39 y 1 en 58 para hombres y mujeres, respectivamente(3). La incidencia más alta de MMC se encuentra en Queensland, Australia, con 41,1 nuevos casos en mujeres y 55,8 en hombres, anualmente por cada 100.000 habitantes. Según la distribución por sexos, el MMC predomina en hombres en EEUU y Australia, mientras que no existen diferencias significativas en Europa(4). En Chile la tasa de mortalidad por MMC aumentó un 14% entre los años 1988 y 1998, mientras que la incidencia aumentó en 105% entre los años 1992 y 1998(5).
En términos económicos, el costo del tratamiento del MMC aumenta significativamente con la etapa de la enfermedad. El año 2009 un estudio realizado en Brasil determinó que más del 95% del costo en tratamiento de MMC fue invertido en pacientes en etapas III y IV, que representaron el 36% de todos los casos de MMC(6).
La mortalidad asociada a las formas invasivas de MMC no se ha modificado en forma significativa a pesar de las diferentes innovaciones terapéuticas, debido fundamentalmente a un diagnóstico tardío de la enfermedad. A pesar de lo anterior, un diagnóstico precoz asociado a un adecuado y oportuno tratamiento transforman al MMC en una condición potencialmente curable, aumentando la sobrevida y disminuyendo los costos asociados a su tratamiento(7).
El MMC es un claro ejemplo de tumor maligno fuertemente influenciado por factores ambientales que se desarrolla en un individuo genéticamente susceptible(8). Los factores de
riesgo individuales más importantes para su desarrollo son el fototipo bajo, color de ojos claro y pelo claro o rojo, la historia familiar de MMC, presencia de múltiples nevos atípicos y el antecedente personal de melanoma. La RUV es el factor de riesgo ambiental más importante, en términos de exposiciones intensas e intermitentes.
La historia familiar de MMC es un potente factor de riesgo, estimándose que al tener un familiar de primer grado afectado, el riesgo de padecer la enfermedad se incrementa en 1,7 veces, mientras que si son dos familiares de primer grado el riesgo aumenta 9 veces(9).
Múltiples estudios se han enfocado en determinar el trasfondo genético del MMC, logrando establecer relación con elementos clínicos y epidemiológicos de la población afectada. Se estima que aproximadamente un 10% de los MMC se produce en un escenario familiar(10), definido como aquellas familias que poseen al menos dos familiares de primer grado afectados por la enfermedad en países con tasas intermedias de incidencia de MMC(11). En estos casos se han establecido dos genes cuyas características en términos de penetrancia y frecuencia de sus mutaciones difieren en forma importante: CDKN2A y CDK4(12). CDKN2A (Cyclin-dependent kinase inhibitor 2A) es el más importante gen de susceptibilidad a MM identificado a la fecha en familias pertenecientes a distintos continentes(12). La frecuencia de las diferentes mutaciones de este gen en familias portadoras de MMC hereditario varía de 5% a 83%, cifra que fluctúa de acuerdo a las tasas locales de incidencia basal de MMC de las diversas áreas geográficas y según los criterios de selección de los diferentes estudios realizados(13-15). El gen CDKN2A codifica para dos distintas proteínas supresoras de tumores: p16INK4a y p14ARF. Ambas actúan en diferentes puntos de control del ciclo celular: p16INK4a lo hace a través de la vía del Retinoblastoma, mientras que p14ARF ejerce su acción a través de la vía de p53. Aunque la mutación de cada una de las proteínas conduce a un mayor riesgo de desarrollar MM, el sector de CDKN2A que codifica para p16INK4 es el que a la fecha ha demostrado mayor frecuencia de mutaciones(12).
Las características asociadas significativamente a la presencia de mutación de CDKN2A son: número elevado de individuos afectados por MMC dentro de la familia, presencia de MM primario múltiple y presencia conjunta de MMC y cáncer de páncreas dentro de la familia(9). Por otra parte,

104 Rev. Chilena Dermatol. 2011; 27(1):102-108
Pedro Jaramillo y cols.
se ha establecido que una edad precoz de presentación de MMC no predice una alta probabilidad de encontrar mutación de CDKN2A. Asimismo, la presencia de nevos atípicos/displásticos en familias con MMC es altamente predictor de MMC, independiente de la existencia de mutación(9).
El denominado modelo de Clark describe los cambios histopatológicos que caracterizan la progresión de un melanocito normal a un MMC. Este modelo consta de cinco etapas, que a su vez se asocian a los diferentes cambios genéticos relacionados con la activación de distintas vías oncogénicas (participación de los genes CDKN2A, CDK4, BRAF, PTEN, NRAS y otros). Por último, en etapas más avanzadas destaca la participación de diversas moléculas de adhesión (β-catenina, E-cadherina, N-cadherina, αVβ3 integrina) que en condiciones normales controlan la migración celular, organización tisular y organogénesis, y cuya descoordinación permite a las células neoplásicas pasar de un crecimiento radial a uno vertical invasor y capaz de originar metástasis(2). Los diferentes genes participan en diferente proporción de acuerdo de acuerdo a si el MMC se presenta en forma esporádica o hereditaria.
Alteraciones genéticas de alta susceptibilidad en MMCDiversos estudios multicéntricos se han enfocado en la determinación de los genes que otorgan mayor susceptibilidad en MMC familiar. Hasta la fecha dos genes de alta susceptibilidad han sido identificados: CDKN2A y CDK4, ambos heredados en patrón autosómico dominante y de penetrancia incompleta(16).
a. CDKN2A: Cyclin-dependent kinase inhibitor 2AEl gen se localiza en el cromosoma 9p21, y es el más importante gen de susceptibilidad para el desarrollo de MMC(17). Corresponde a un gen supresor de tumores que codifica dos transcriptos distintos para las proteínas supresoras de tumores p16INK4a y p14ARF, esta última a partir del mismo segmento de ADN que p16, pero con un marco de lectura diferente (ARF: Alternative Reading Frame). El primer transcrito, p16, se forma a partir de los exones 1α, 2 y 3 de CDKN2A, y su acción natural es bloquear el ciclo celular a nivel del punto de control G1-S, inhibiendo la fosforilación de la proteína del Retinoblastoma (pRb) mediada por quinasas
dependientes de ciclina (CDK4). El segundo transcrito, p14ARF, está codificado en los exones 1β, 2 y 3, y actúa a través de la vía p53 induciendo detención del ciclo celular o apoptosis(12,16)
(Figura 1). Las diferentes mutaciones de CDNK2A están presentes en promedio en un 20% de los casos de MMC hereditario(15). Además del MMC, la predisposición a otros cánceres se ha visto en familias portadoras de MMC en quienes se ha detectado mutación de CDKN2A, como el cáncer de páncreas, aun cuando la relación exacta entre la mutación y la neoplasia no ha sido aclarada(18,19). Datos publicados en diferentes estudios han permitido establecer que la frecuencia de mutaciones de CDKN2A aumenta en forma significativa con el número de casos de MMC en la familia; la presencia de individuos con MMC múltiples y la presencia de cáncer de páncreas en sujetos pertenecientes a familias portadoras de MMC hereditario(13,20). Por otra parte, en ausencia de historia familiar de MMC, la aparición a edad precoz de MMC no predice una mayor probabilidad de encontrar mutación. Por último la presencia de nevos atípicos/displásticos en familias con MMC es un elemento fuertemente predictor de MMC, independientemente de la
Figura 1 Locus CDKN2A y control del ciclo celular12.

105Rev. Chilena Dermatol. 2011; 27(1):102-108
Genética molecular del melanoma familiar. Revisión del tema y aplicación clínica
existencia de mutación de CDKN2A(9). La prevalencia de mutación de CDKN2A en pacientes portadores de síndrome de nevos atípicos ha sido estimada en 3,2%21. Un estudio realizado por de Snoo et al. en 167 individuos portadores de nevos atípicos, determinó que de 136 sujetos que completaron el seguimiento, 10 se convirtieron individuos pertenecientes a familias con melanoma hereditario, de los cuales 4 presentaron mutación de CDKN2A(22). Hasta la fecha, el papel de la presencia de síndrome de nevos atípicos en familias portadoras de melanoma hereditario como predictor de mutación de CDKN2A no ha sido completamente esclarecido.
b. CDK4 (Cyclin-dependent kinase 4)El gen se localiza en el brazo largo del cromosoma 12 y codifica una quinasa que normalmente es bloqueada por p16INK4a, permitiendo la fosforilación de pRB y de esta forma el paso de la fase G1 a S del ciclo celular.
Las mutaciones de CDK4 varían en frecuencia y tipo en las diferentes poblaciones estudiadas, sin embargo solo un escaso número de familias han sido identificadas como portadoras de mutaciones de línea germinal de este gen(13). La frecuencia de mutaciones reportada es extremadamente baja, variando entre un 2% a 3% dependiendo de la localidad geográfica estudiada(13).
Se postula que los casos restantes de MMC hereditario estarían relacionados a mutaciones de áreas no codificantes de CDKN2A o CDK4, mutaciones de otros genes o cambios postraduccionales(12,23). Algunos polimorfismos de importancia discutida se han descrito en CDKN2A, como es el caso de p.A148T(24) en el exón 2 y dos polimorfismos comunes en la región 3´ no traduccional de CDKN2A(17,25).
C. Alteraciones genéticas de baja susceptibilidad en MMC
a. MC1R: Melanocortin-1 receptor
Considerado un gen de baja susceptibilidad, el gen MC1R tiene un papel fundamental en la pigmentación constitutiva del individuo. Con al menos 65 polimorfismos reportados, algunas variantes se asocian estrechamente con el fototipo de piel clara y cabello rojo. La presencia de una variante de MC1R agregada a la mutación de CDKN2A aumenta en forma
significativa la penetrancia global de melanoma, reduciendo la edad de presentación hasta en 20 años, en comparación a los individuos que únicamente presentan mutación de CDKN2A(26).
Mutaciones de CDKN2A en MMC familiar
La expresión aberrante de los diferentes exones que componen CDKN2A se traduce en una alteración en los mecanismos de control del ciclo celular. Los exones más frecuentemente afectados por mutaciones son 1α, 2 y 3, mientras que la mutación del exón 1β ha sido observada en una frecuencia significativamente menor(27).
a. Sitios de las mutaciones
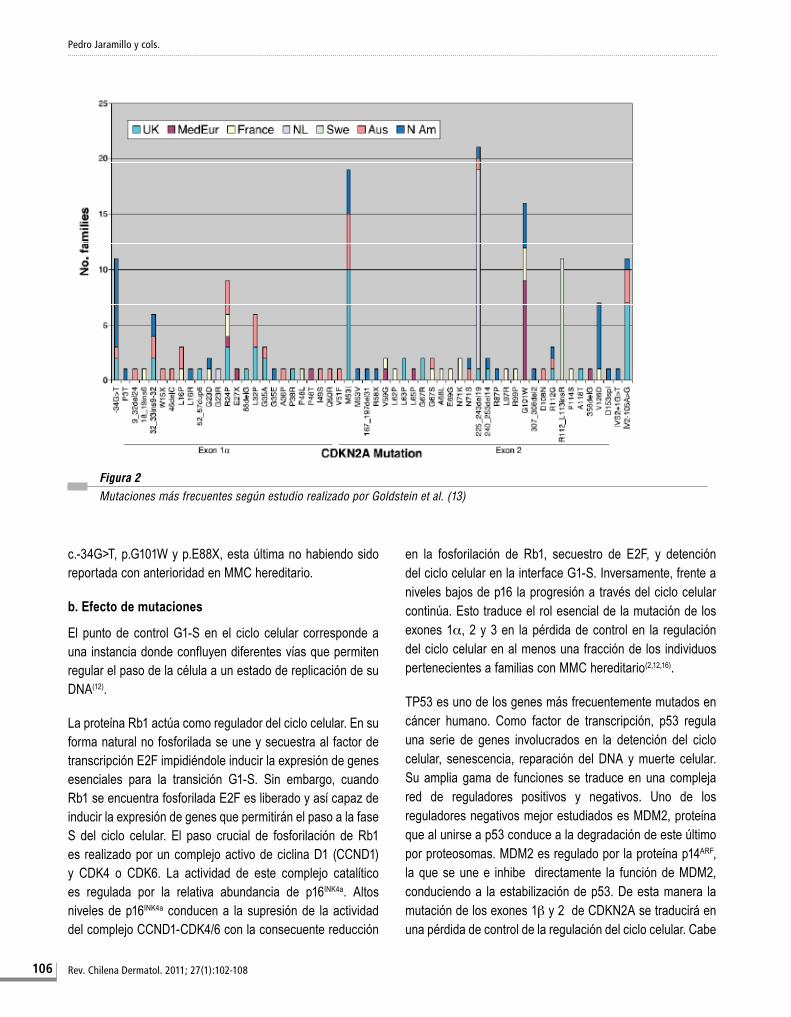
Diversos estudios se han realizado a nivel internacional con el fin de establecer las mutaciones más frecuentes que afectan a CDKN2A. El mayor de ellos corresponde al conducido por Goldstein et al., que el 2006 a través de un estudio multicéntrico intercontinental determinó las mutaciones más prevalentes en un total de 466 familias procedentes de Norteamérica, Europa, Asia y Australia (Fig.2)(13,28). Del total de familias, 41% presentó mutaciones de las cuales el 93,6% comprometieron a CDKN2A (exones 1α, 1β, 2 y 3), con frecuencia variable de acuerdo a las diferentes localidades estudiadas. Un 40% de las mutaciones afectó al exón 1α, mientras que un 53% ocurrió a nivel del exón 2. El 65% correspondieron a mutaciones por cambio de sentido, 16% deleciones, 7% inserciones o duplicaciones, y 5% fueron mutaciones sin sentido o splicing. Las mutaciones del exón 1β fueron observadas sólo en un 2% a 3%. La proporción de familias portadoras de las mutaciones más frecuentes varió significativamente entre las diferentes regiones estudiadas. Esta variación sugiere que factores ambientales, de comportamiento, y la composición étnica local contribuyen en forma importante al desarrollo de MMC aun en presencia de mutaciones(9).
En Sudamérica un estudio realizado por Larre-Borges et al.(15) en un total de 13 individuos pertenecientes a 6 familias uruguayas, utilizando como principal criterio de inclusión la presencia de dos familiares de primer grado afectados, determinó la existencia de mutación de CDKN2A en 5 de las 6 familias estudiadas (83%), detectando además mutación de este gen en el 100% de los individuos afectados por MMC. Las mutaciones más frecuentemente observadas fueron
106 Rev. Chilena Dermatol. 2011; 27(1):102-108
Pedro Jaramillo y cols.
c.-34G>T, p.G101W y p.E88X, esta última no habiendo sido reportada con anterioridad en MMC hereditario.
b. Efecto de mutaciones
El punto de control G1-S en el ciclo celular corresponde a una instancia donde confluyen diferentes vías que permiten regular el paso de la célula a un estado de replicación de su DNA(12).
La proteína Rb1 actúa como regulador del ciclo celular. En su forma natural no fosforilada se une y secuestra al factor de transcripción E2F impidiéndole inducir la expresión de genes esenciales para la transición G1-S. Sin embargo, cuando Rb1 se encuentra fosforilada E2F es liberado y así capaz de inducir la expresión de genes que permitirán el paso a la fase S del ciclo celular. El paso crucial de fosforilación de Rb1 es realizado por un complejo activo de ciclina D1 (CCND1) y CDK4 o CDK6. La actividad de este complejo catalítico es regulada por la relativa abundancia de p16INK4a. Altos niveles de p16INK4a conducen a la supresión de la actividad del complejo CCND1-CDK4/6 con la consecuente reducción
en la fosforilación de Rb1, secuestro de E2F, y detención del ciclo celular en la interface G1-S. Inversamente, frente a niveles bajos de p16 la progresión a través del ciclo celular continúa. Esto traduce el rol esencial de la mutación de los exones 1α, 2 y 3 en la pérdida de control en la regulación del ciclo celular en al menos una fracción de los individuos pertenecientes a familias con MMC hereditario(2,12,16).
TP53 es uno de los genes más frecuentemente mutados en cáncer humano. Como factor de transcripción, p53 regula una serie de genes involucrados en la detención del ciclo celular, senescencia, reparación del DNA y muerte celular. Su amplia gama de funciones se traduce en una compleja red de reguladores positivos y negativos. Uno de los reguladores negativos mejor estudiados es MDM2, proteína que al unirse a p53 conduce a la degradación de este último por proteosomas. MDM2 es regulado por la proteína p14ARF, la que se une e inhibe directamente la función de MDM2, conduciendo a la estabilización de p53. De esta manera la mutación de los exones 1β y 2 de CDKN2A se traducirá en una pérdida de control de la regulación del ciclo celular. Cabe
Figura 2 Mutaciones más frecuentes según estudio realizado por Goldstein et al. (13)

107Rev. Chilena Dermatol. 2011; 27(1):102-108
Genética molecular del melanoma familiar. Revisión del tema y aplicación clínica
destacar que la mutación del exón 1β producirá alteración sólo en la proteína p14ARF, la mutación del exón 2 producirá alteración de la función de p16INK4a y p14ARF simultáneamente, mientras que la mutación exclusiva de 1α afectará solamente a p16INK4a (2,12,16).
Las mutaciones más prevalentes afectan a los exones 1α y 2, lo que es consistente con la inactivación de la proteína p16INK4a como mayor factor predisponente para MMC(29). Se ha estimado que el riesgo acumulado de desarrollar MMC antes de los 80 años en portadores de mutación de CDKN2A varía entre 58% en Europa y 91% en Australia (30,31,32).
c. Utilidad de la pesquisa de mutaciones de CDKN2A
El riesgo de desarrollar MMC en individuos portadores de mutación de CDKN2A se estima en 30% a los 50 años, y en 67% a los 80 años, cifras que son influenciadas fuertemente por factores ambientales(29).
Con anterioridad se ha enfatizado que en localidades donde las tasas de incidencia de MMC son bajas y las mutaciones fundadoras son frecuentes, el testeo genético puede potenciar la adherencia a las recomendaciones clínicas entre los portadores de mutación(33). Por otra parte un estudio de Aspinwall et al. determinó un aumento en screening y comportamiento precautorio tanto en pacientes portadores
como no portadores de la mutación, después del testeo genético(34). Datos obtenidos de estudios que han evaluado el testeo genético en otro tipo de neoplasias indican que los individuos no portadores pertenecientes a familias en que la mutación ha sido identificada tienden a continuar un seguimiento adecuado de acuerdo a su riesgo(35,36).
El estudio clínico y genético de familias con predisposición hereditaria a MMC permitiría una detección precoz de una población de alto riesgo de desarrollar MMC, en la que un estricto seguimiento es mandatorio. Además, contribuir al conocimiento del comportamiento de estas mutaciones en nuestra población, en consideración a la amplia variación con que dichas mutaciones se presentan en diferentes regiones geográficas(37) y en el contexto de una población mayoritariamente descendiente de hombres blancos de origen español y mujeres amerindias(38).
Desde el punto de vista clínico, la identificación de mutaciones en la población estudiada, y luego de un seguimiento razonable, permitirá comprender de mejor manera el comportamiento de las mutaciones de CDKN2A en términos de penetrancia, es decir, el riesgo de los individuos portadores de mutación de desarrollar efectivamente MMC, así como otras neoplasias asociadas a dichas mutaciones.
Bibliografía1. Goodson AG, Grossman D. Strategies for early melanoma detection:
Approaches to the patient with nevi. J Am Acad Dermatol 2009;60(5):719-35; quiz 36-8.
2. Miller AJ, Mihm MC, Jr. Melanoma. N Engl J Med 2006;355(1):51-65.
3. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009;59(4):225-49.
4. MacKie RM, Hauschild A, Eggermont AM. Epidemiology of invasive cutaneous melanoma. Ann Oncol 2009;20 Suppl 6:vi1-7.
5. Zemelman V, Garmendia ML, Kirschbaum A. Malignant melanoma mortality rates in Chile (1988-98). Int J Dermatol 2002;41(2):99-103.
6. Souza RJ, Mattedi AP, Rezende ML, Correa Mde P, Duarte EM. An estimate of the cost of treating melanoma disease in the state of Sao Paulo - Brazil. An Bras Dermatol 2009;84(3):237-43.
7. Kirkwood JM, Eggermont A. Highlights from the perspectives in melanoma XII conference. Pigment Cell Melanoma Res 2009;22(2):156-65.
8. Psaty EL, Scope A, Halpern AC, Marghoob AA. Defining the patient at high risk for melanoma. Int J Dermatol 2010;49(4):362-76.
9. Leachman SA, Carucci J, Kohlmann W, Banks KC, Asgari MM, Bergman W, et al. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol 2009;61(4):677 e1-14.
10. Meyer LJ, Zone JH. Genetics of cutaneous melanoma. J Invest Dermatol 1994;103(5 Suppl):112S-16S.
11. Fraser MC, Goldstein AM, Tucker MA. The genetics of melanoma. Semin Oncol Nurs 1997;13(2):108-14.
12. Sekulic A, Haluska P, Jr., Miller AJ, Genebriera De Lamo J, Ejadi S, Pulido JS, et al. Malignant melanoma in the 21st century: the emerging molecular landscape. Mayo Clin Proc 2008;83(7):825-46.
13. Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res 2006;66(20):9818-28.
14. Bruno W, Ghiorzo P, Battistuzzi L, Ascierto PA, Barile M, Gargiulo S, et al. Clinical genetic testing for familial melanoma in Italy: a cooperative study. J Am Acad Dermatol 2009;61(5):775-82.
15. Larre Borges A, Cuellar F, Puig-Butille JA, Scarone M, Delgado L, Badenas C, et al. CDKN2A mutations in melanoma families from Uruguay. Br J Dermatol 2009;161(3):536-41.

108 Rev. Chilena Dermatol. 2011; 27(1):102-108
Pedro Jaramillo y cols.
16. Bloethner S, Scherer D, Drechsel M, Hemminki K, Kumar R. Malignant melanoma--a genetic overview. Actas Dermosifiliogr 2009;100 Suppl 1:38-51.
17. Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 mutations in familial melanoma. Nat Genet 1994;8(1):15-21.
18. Vasen HF, Gruis NA, Frants RR, van Der Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer 2000;87(6):809-11.
19. Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 1995;333(15):970-4.
20. Begg CB, Orlow I, Hummer AJ, Armstrong BK, Kricker A, Marrett LD, et al. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst 2005;97(20):1507-15.
21. Ung-Juurlink C. American Academy of Dermatology 1999 Awards for Young Investigators in Dermatology. The prevalence of CDKN2A in patients with atypical nevi and malignant melanoma. J Am Acad Dermatol 1999;41(3 Pt 1):461-2.
22. de Snoo FA, Kroon MW, Bergman W, ter Huurne JA, Houwing-Duistermaat JJ, van Mourik L, et al. From sporadic atypical nevi to familial melanoma: risk analysis for melanoma in sporadic atypical nevus patients. J Am Acad Dermatol 2007;56(5):748-52.
23. Goldstein AM, Chidambaram A, Halpern A, Holly EA, Guerry ID, Sagebiel R, et al. Rarity of CDK4 germline mutations in familial melanoma. Melanoma Res 2002;12(1):51-5.
24. Aitken J, Welch J, Duffy D, Milligan A, Green A, Martin N, et al. CDKN2A variants in a population-based sample of Queensland families with melanoma. J Natl Cancer Inst 1999;91(5):446-52.
25. Kumar R, Smeds J, Berggren P, Straume O, Rozell BL, Akslen LA, et al. A single nucleotide polymorphism in the 3’untranslated region of the CDKN2A gene is common in sporadic primary melanomas but mutations in the CDKN2B, CDKN2C, CDK4 and p53 genes are rare. Int J Cancer 2001;95(6):388-93.
26. Goldstein AM, Chaudru V, Ghiorzo P, Badenas C, Malvehy J, Pastorino L, et al. Cutaneous phenotype and MC1R variants as modifying factors for the development of melanoma in CDKN2A G101W mutation carriers from 4 countries. Int J Cancer 2007;121(4):825-31.
27. Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, et al. Features associated with germline CDKN2A
mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet 2007;44(2):99-106.
28. Harland M, Goldstein AM, Kukalizch K, Taylor C, Hogg D, Puig S, et al. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL). Eur J Cancer 2008;44(9):1269-74.
29. Hansson J. Familial melanoma. Surg Clin North Am 2008;88(4):897-916, viii.
30. Landi MT, Goldstein AM, Tsang S, Munroe D, Modi W, Ter-Minassian M, et al. Genetic susceptibility in familial melanoma from northeastern Italy. J Med Genet 2004;41(7):557-66.
31. Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility, Adopted on February 20, 1996. J Clin Oncol 1996;14(5):1730-6; discussion 37-40.
32. Ferrone CR, Ben Porat L, Panageas KS, Berwick M, Halpern AC, Patel A, et al. Clinicopathological features of and risk factors for multiple primary melanomas. JAMA 2005;294(13):1647-54.
33. Kefford R, Bishop JN, Tucker M, Bressac-de Paillerets B, Bianchi-Scarra G, Bergman W, et al. Genetic testing for melanoma. Lancet Oncol 2002;3(11):653-4.
34. Aspinwall LG, Leaf SL, Dola ER, Kohlmann W, Leachman SA. CDKN2A/p16 genetic test reporting improves early detection intentions and practices in high-risk melanoma families. Cancer Epidemiol Biomarkers Prev 2008;17(6):1510-9.
35. Hadley DW, Jenkins JF, Dimond E, de Carvalho M, Kirsch I, Palmer CG. Colon cancer screening practices after genetic counseling and testing for hereditary nonpolyposis colorectal cancer. J Clin Oncol 2004;22(1):39-44.
36. Botkin JR, Smith KR, Croyle RT, Baty BJ, Wylie JE, Dutson D, et al. Genetic testing for a BRCA1 mutation: prophylactic surgery and screening behavior in women 2 years post testing. Am J Med Genet A 2003;118A(3):201-9.
37. Gerstenblith MR, Goldstein AM, Tucker MA, Fraser MC. Genetic testing for melanoma predisposition: current challenges. Cancer Nurs 2007;30(6):452-9; quiz 62-3.
38. Rocco P, Morales C, Moraga M, Miquel JF, Nervi F, Llop E, et al. [Genetic composition of the Chilean population. Analysis of mitochondrial DNA polymorphism]. Rev Med Chil 2002;130(2):125-31.
39. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304(5676):1497-500.
d)1. b) 2. d) 3. a) 4. c) 5.
d) 6. c)7. a)8. d)9. b) 10.
Evaluación (Quiz) (pag. 38)
Respuestas correctas:Dermatosis ParaneoplásicasNélida Raimondo

109Rev. Chilena Dermatol. 2011; 27(1):109-115
REVISIÓN DE REVISTAS
The knowledge, awareness, and practice patterns of dermatologists toward psychocutaneous disorders: results of a survey study.Int J Dermatol 2010; 49(7):784-9.Jafferany M, Vander A, Dumitrescu A, Hornung RL.
Coordinación: Perla Calderón H.Servicio de Dermatología, Hospital Clínico, Universidad de Chile.
Introducción: Más de un tercio de los pacientes que consultan por afecciones dermatológicas tienen algún problema psicológico que complica su enfermedad cutánea. Se ha reportado que los pacientes dermatológicos hospitalizados y ambulatorios tienen una mayor prevalencia de enfermedades psiquiátricas que la población general. A pesar de la creciente evidencia de esta relación, pocos centros formadores en Dermatología, Psiquiatría y Medicina Familiar incorporan conocimientos sobre psicodermatología en sus programas de estudio. Este trabajo busca, mediante una encuesta, conocer el interés, conocimiento y experiencia de los dermatólogos sobre la psicodermatología.
Materiales y Métodos: Se enviaron encuestas anónimas por correo a dermatólogos de la Asociación de Dermatología del Estado de Washington (US). Buscaban evaluar el interés, conocimiento, experiencia e interés en el aprendizaje sobre psicodermatología de los encuestados. Se envió una segunda carta a los que no respondieron la encuesta dentro de tres semanas. Los datos fueron analizados estadísticamente usando x2 y el coeficiente de Pearson para correlacionar el entrenamiento previo en psicodermatología y la comodidad del dermatólogo tratando desórdenes psicodermatológicos.
Resultados: se enviaron 237 encuestas y 102 dermatólogos contestaron (43%). El 32% respondió no tener entrenamiento
en psicodermatología, 49% reportó limitado o parcial y el 18% reportó tener un claro conocimiento del tema. Cerca de un 93% respondió tener al menos experiencia ocasional con la psicodermatología y un 83% sentirse cómodo tratando este tipo de pacientes. Se encontró una fuerte correlación entre el entrenamiento que ha recibido el dermatólogo previamente y la comodidad que siente tratando pacientes con trastornos psicodermatológicos. En relación al interés en recibir educación médica continua sobre el tema, 39% respondió afirmativamente, 44% estaba indeciso y 17% no interesado. Los temas de mayor interés en aprender fueron lesiones cutáneas autoinferidas y dismorfofobia.
Discusión: Los resultados sugieren que la mayoría de los dermatólogos encuestados tienen limitados conocimientos sobre psicodermatología, pero la mayoría quiere aprender más. Es interesante que los dermatólogos de esta muestra se sentían cómodos tratando los trastornos psiquiátricos de sus pacientes, no teniendo la necesidad de derivarlos a otro especialista. Además se demostró que los especialistas que tenían algún grado de entrenamiento en el tema se sentían más cómodos tratando este tipo de pacientes. Estos resultados demuestran la necesidad de generar nuevas instancias de aprendizaje sobre esta nueva rama de la dermatología. (CB)
Vitiligo Treatment in Chilhood: A State of the Art Review. Pediatr Dermatol 2010; 27(5):437-4Tamesis ME, Morelli JG
Introducción: Existen múltiples modalidades de tratamiento para el vitiligo, siendo la repigmentación completa raramente obtenida. Un 15 a 30% de los pacientes nunca responden. En la mayoría de los estudios, un 75% de la repigmentación es considerada excelente, obteniéndose esta respuesta en un 60% de los pacientes.
Materiales y Métodos: Este estudio consiste en la revisión de múltiples modalidades de tratamiento utilizadas para tratar vitíligo en los niños. Entre ellos, el uso de terapias tópicas de corticoides, inhibidores de la calcineurina, calcipotriol y combinaciones de éstos, fototerapia UVBnb, PUVA y laser Excimer y finalmente tratamientos quirúrgicos.

110 Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Resultados y Conclusiones: Los corticoides tópicos potentes resultaron tener tasas de repigmentación, que varían desde 45% a 60% en niños, siendo de elección los corticoides potentes más que los ultrapotentes. Los inhibidores de la calcineurina tienen eficacia clínica similar, pero con un menor perfil de efectos secundarios asociados a su uso. Ambos son considerados como terapia de primera línea en vitíligo que compromete menos de un 20% de la superficie corporal total. Mientras que Calcipotriol, como monoterapia, no es tan efectivo como los corticoides tópicos o los inhibidores de calcineurina.
La fototerapia es considerada el tratamiento de elección en pacientes con un compromiso más extenso, mayor al 20%, siendo la nbUVB la más eficaz, con mayores tasas de
repigmentación y mejor perfil de seguridad que PUVA tópica u oral.
El laser Excimer emite una longitud de onda similar a nbUVB, con la ventaja de permitir un tratamiento enfocado en un área localizada y por lo tanto disminuir los efectos adversos.
Para finalizar, en los últimos años se ha creado una nueva tendencia, hacia el tratamiento quirúrgico del vitiligo. Los mejores resultados se han obtenido con el injerto de ampollas de epidermis obtenidas por succión.
Con una adecuada protección solar, la opción de no realizar tratamientos, con o sin camuflaje cosmético, es una alternativa inocua a cualquiera de estas modalidades terapéuticas. (AMK)
Introducción: Los ungüentos antibióticos tópicos son comúnmente usados en heridas postoperatorias superficiales secundarias a procedimientos dermatológicos. Sin embargo, estudios previos no han demostrado superioridad de los antibióticos tópicos por sobre el uso de ungüentos libres de antibióticos, por lo que no aportarían un beneficio en la curación de estas heridas, teniendo un riesgo potencial de causar dermatitis de contacto alérgicas, contribuyendo al desarrollo de resistencia antibiótica.
Objetivos: Comparar la eficacia y seguridad de un ungüento petrolado libre de antibiótico (Aquaphor Healing Ointment de Eucerin) versus el ungüento antibiótico de primeros auxilios Polysporin (Poly-Bac), que combina Polimixina B con Bacitracina, en el tratamiento de heridas creadas tras la remoción quirúrgica de queratosis seborreicas.
Métodos: Es un estudio doble ciego en el que participaron 30 pacientes. A cada uno de ellos se le realizó el shaving de 2 queratosis seborreicas ubicadas en el tronco o abdomen, una a la derecha y la otra a la izquierda. La hemostasia fue conseguida con cloruro de aluminio. Una de las heridas fue tratada con Aquaphor y la otra manejada con ungüento antibiótico Polysporin (Poly-Bac), ambos productos fueron aplicados 2 veces al día, manteniéndose cubiertas por los primeros 7 días. La mejoría clínica de la curación de la herida, así como los síntomas subjetivos de irritación, fueron
evaluados en los días 7, 14 y 28 post procedimiento quirúrgico. Los eventos adversos también fueron registrados.
Resultados: La mejoría clínica no mostró diferencias significativas entre heridas tratadas con Aquaphor versus Polysporin (Poly/Bac) en los parámetros de eritema, edema, reepitelización y formación de costras en ningún momento de la evaluación. La evaluación subjetiva de irritación demostró que las heridas tratadas con Polysporin (Poly-Bac) presentaban un aumento significativo de la sensación de ardor a los 7 días. Se reportó un caso de dermatitis de contacto alérgica tras la aplicación de Polysporin (Poly-Bac). La evaluación del aspecto general de la curación de la herida fue similar para ambos productos.
Conclusión: Este estudio demostró que el ungüento petrolado Aquaphor presenta una eficacia equivalente a la del ungüento antibiótico biasociado Polysporin (Poly-Bac) en la curación de heridas postquirúrgicas. Los ungüentos antibióticos, por lo tanto, no aportarían un beneficio adicional para lograr la curación satisfactoria de las heridas, pudiendo incluso causar dermatitis de contacto alérgica. Los autores señalan como limitante el tamaño pequeño del estudio, además cabe señalar que algunos de los autores fueron o son consultores para Beiersdorf Inc y Johnson & Johnson, fabricantes de Aquaphor y Polysporin, respectivamente. (PCP)
A comparison of postprocedural wound care treatments: Do antibiotics-based ointments improve outcomes?Zoe D. Draelos, Ronald L. Rizer, Nathan S. TrookmanJ Am Acad Dermatol 2011; 64:S23-29.

111Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Infantile Hemangiomas With Minimal or Arrested GrowthA Retrospective Case SeriesKi-Young Suh, MD; Ilona J. Frieden, MDArch Dermatol 2010; 146(9):971-976
Introducción: Una minoría de los hemangiomas de la infancia (HI) presentan un crecimiento mínimo o ausente; éstos se definen como aquellos hemangiomas cuyo componente proliferativo equivale a menos del 25% de su superficie total. Estudios previos confirman que son positivos para el transportador de glucosa tipo 1 (GLUT-1).
Método: Estudio restrospectivo. Se realizó una búsqueda en los archivos fotográficos del Centro de referencia ambulatorio en la Universidad de California, San Francisco. Se revisaron los antecedentes médicos de 42 pacientes que presentaban un total de 47 hemangiomas de la infancia con crecimiento mínimo o ausente (HI-MA).
Resultados: La presentación más frecuente fue en parches con telangectasias finas o gruesas. En el 30% de los casos (14 de 47) se evidenció algún componente proliferativo,
por lo general con pápulas pequeñas en la periferia del hemangioma. 68% (32 de 47) de los HI-MA se presentaron en la parte inferior del cuerpo. Sólo 4 lesiones presentaron ulceración, todas ubicados en la zona anogenital. 17 pacientes presentaban un HI clásico en otra parte del cuerpo.
Conclusiones: Los HI-MA tienen una apariencia clínica distinta a los HI que conocemos. Se presentan preferentemente en la parte inferior del cuerpo, lo que sugiere que factores locales como la densidad de la vasculatura pueden jugar un rol en su crecimiento.
Su diagnóstico puede ser difícil, su reconocimiento permite un diagnóstico más preciso de las anomalías vasculares presentes al nacimiento. La presencia de un HI-MA en un paciente no excluye el potencial proliferativo de otros HI que pueden estar presentes en el mismo paciente. (AC)
Vitamin D inhibits captopril-induced cell detachment and apoptosis in keratinocytes.Zeeli T., Langberg M., Rotem C., David M., Koren R, Ravid A.BJD 2011: 164, 62–7.
Introduccion: El Captopril se ha asociado a aparición de pénfigo y lesiones penfigoide simil. Se han propuesto mecanismos inmunológicos y bioquímicos responsables de la acantolisis mediada por captopril. Por otra parte, la hormona Calcitriol (1alfa,25-dihydroxyvitamina D) protege a los queratinocitos de la apoptosis.
Objetivo: Examinar la influencia del calcitriol sobre la acantolisis de queratinocitos inducida por captopril.
Material y Método: Como modelo de queratinocitos de la capa basal se utilizó queratinocitos de proliferación autónoma, los cuales fueron expuestos in vitro a captopril. Se observó separación de las células mediante microscopía de luz. Se midió respuesta citotóxica mediante tinción Hoechst 33342 y liberación de lactato deshidrogenasa. La apoptosis se verificó mediante la monitorización de la actividad de caspasa 3- simil.
Resultados: Las células expuestas a captopril se separaron de la matriz de cultivo entre sí y se hicieron redondeadas, lo cual fue acompañado de muerte celular. El tratamiento previo de estas células con calcitriol disminuyó significativamente estos eventos, incluso a concentraciones tan bajas como 1 nmol L-1
En este modelo de estudio, la muerte celular programada fue consecuencia de la acantolisis y no al revés.
Conclusiones: Los resultados de este estudio demuestran el rol protector de calcitriol sobre la acantolisis y apoptosis mediada por captopril en queratinocitos. Podría ser que el mecanismo protector de calcitriol se debiera al fortalecimiento de la cohesión intercelular. Esto abriría ventanas terapéuticas con esta hormona en la acantolisis mediada por captopril y tal vez en otras enfermedades ampollares.
Este es el primer trabajo publicado sobre este tema. (PC)

112 Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Effectiveness of the association of cetirizine and topical steroids in lichen planus pilaris – an open-label clinical trial.D´Ovidio R, Rossi A, Di Prima TMDermatol Ther 2010; 23:547-552
Introducción: El liquen plano es considerada una enfermedad inmunológica mediada por células T. Los queratinocitos basales tanto de la piel como del folículo piloso pueden representar el target de la agresión inmune, determinando la destrucción del mismo y la consecuente alopecia cicatricial. Por esto es importante lograr un tratamiento eficaz de esta dermatosis potencialmente desfigurante. Los mastocitos jugarían un rol clave en la fase activa de la enfermedad, lo que provee una oportunidad terapéutica, el uso de antihistamínicos en régimen “anti-inflamatorio”.
Casos: Se trataron 21 pacientes con diagnóstico de liquen plano pilaris histológicamente confirmado. Todos los pacientes recibieron cetirizina 30 mg al día asociado a esteroides tópicos. La duración del tratamiento se determinó caso a caso de acuerdo a la evaluación clínica.
Resultados: La detención de la inflamación (eritema, hiperqueratosis folicular, pérdida de cabello en anágeno) se logró en 17 pacientes. El tratamiento tuvo una duración entre 3 y 24 meses. Aquellos pacientes que concomitantemente presentaron liquen plano cutáneo o mucoso mostraron mejoría sin terapia tópica. La alta dosis de la droga fue bien tolerada, un paciente desarrolló arritmia cardíaca al tercer mes de tratamiento por lo cual la cetirizina fue suspendida.
Discusión: La terapia combinada de esteroides tópicos con cetirizina 30 mg al día es una opción eficaz, de bajo costo y relativamente segura en el tratamiento de liquen plano pilaris moderado a severo, especialmente en aquellos pacientes en quienes el uso de corticoides sistémicos o inmunosupresores está contraindicada. Se requerirá de estudios prospectivos doble ciego para aseverar estas conclusiones (AC)
Iron deficiency in female pattern hair loss, chronic telógeno effluvium, and control groupsOlsen EA, Reed KB, Cacchio BS, Caudill L. J Am Acad Dermatol 2010; 63:991-9
Introducción: La literatura sugiere que la deficiencia de hierro (DH) puede jugar un importante rol en la alopecia de patrón femenino (APF) o en el efluvio telógeno crónico (ETC).
Objetivos: En el presente estudio se buscó determinar si la deficiencia de hierro es más común en mujeres con APF o ETC que en sujetos sin caída del cabello.
Metodología: Se llevó a cabo un estudio controlado en 381 mujeres caucásicas de 18 años o más con APF o ETC vistas en la clínica de desórdenes del cabello de la Universidad de Duke. El grupo control lo formaron 76 mujeres caucásicas de 18 años o más, reclutadas de las cercanías de la universidad y que no presentaban historia o hallazgos físicos de caída del cabello. Todas las participantes tuvieron al menos medición de ferritina sérica y niveles de hemoglobina junto a una historia del estatus menopáusico. Se establecieron 3 criterios distintos de corte según los niveles de ferritina, con diferente sensibilidad y especificidad según publicaciones anteriores.
Resultados: Cuando se utilizó como definición de DH un punto de corte de ferritina menor o igual de 15 ug/L, la DH ocurrió en 12,4%, 12,1% y 29,8% de las mujeres premenopáusicas con APF, ETC y sujetos control, respectivamente. En las mujeres postmenopáusicas la DH ocurrió en el 1,7%, 10,5% y 6,9% de aquellas con APF, ETC y sujetos control, respectivamente. Al utilizar como punto de corte un valor menor o igual de 40 ug/L, la DH ocurrió en el 58,8%, 63,8 y 72,3% de las mujeres premenopáusicas con APF, ETC y sujetos control, respectivamente. En las mujeres postmenopáusicas la DH ocurrió en el 26,1%, 36,8% y en el 20,7% de aquellas con APF, ETC y sujetos control, respectivamente. No existió un incremento significativo en la incidencia de DH en las mujeres premenopáusicas o postmenopáusicas con APF, ETC en comparación al grupo control.
Conclusiones: La DH es común entre las mujeres pero no se encuentra aumentada en mujeres con APF o ETC al compararlas con mujeres sin alopecia.(JL)

113Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Serum prolactin levels in psoriasis and correlation with cutaneous disease activityDilmé-Carreras E, Martin-Ezquerra G, Sánchez-Regaña M, Umbert-Millet P.Clin Exp Dermatol 2011; 36:29-32.
Introducción: La prolactina (PRL) ha sido implicada como un importante inmunomodulador e incluso ejerce efectos proliferativos en cultivos humanos de queratinocitos. Algunos estudios han demostrado incrementos de la PRL sérica en pacientes con psoriasis y exacerbaciones de la misma cuando existe un prolactinoma.
Objetivos: Evaluar la correlación existente entre los niveles de PRL sérico y el índice de severidad y área de psoriasis (PASI).
Métodos: Se midieron los niveles séricos de PRL en 20 pacientes (10 hombres y 10 mujeres, entre 18-88 años) con psoriasis en placas antes y después de un tratamiento tópico
Imiquimod 2.5% and 3.75% for the treatment of Actinic Keratoses: results of two placebo-controlled studies of daily application to the face and balding scalp for 2-week cyclesSwanson N, Abramovits W, Berman B, Kulp J, Rigel D, Levy Sh.J Am Acad Dermatol 2010; 62:582-90.
Introducción: El régimen aprobado imiquimod crema al 5% para tratar la queratosis actínica requiere un largo tiempo de tratamiento y se limita a una pequeña área de piel.
Objetivo: Evaluar imiquimod 2,5% y 3,75% para el tratamiento de corta duración de cara completa o cuero cabelludo calvo.
Métodos: En dos estudios idénticos, adultos con 5 a 20 lesiones fueron distribuidos aleatoriamente para recibir placebo, imiquimod 2,5%, o imiquimod 3,75% (1:1:1). Hasta dos paquetes fueron aplicados por dosis una vez al día dos ciclos de dos semanas de tratamiento, con 2 semanas de intervalo sin tratamiento entre los ciclos. La eficacia fue evaluada en 8 semanas después del tratamiento.
Resultados: Un total de 479 pacientes fueron aleatorizados para recibir placebo, imiquimod 2,5% o 3,75%. Clearence completo y parcial (reducción ≥75% de las lesiones) fueron 6,3% y 22,6% para placebo, 30,6% y 48,1% para imiquimod 2,5%, y 35,6% y 59,4% para imiquimod 3,75%, respectivamente
(P < 0,001 vs placebo, cada uno, P = 0.047, 3,75% vs 2,5% para clearence parcial). La mediana de reducción del valor inicial en el número de lesiones fue de 25% para el placebo, el 71,8% de imiquimod 2,5% y 81,8% para el imiquimod 3,75% (P < 0,001, cada uno frente a placebo, P = 0,048 3,75% vs 2,5%). Hubo pocas interrupciones relacionadas con el tratamiento. Tasas de período de descanso de paciente fueron de 0% para el placebo, 6,9% para imiquimod 2,5%, y 10,6% para imiquimod 3,75%.
Limitaciones: Los efectos farmacológicos locales de imiquimod, incluyendo el eritema, pueden haber ocultado la limitada respuesta al tratamiento en algunos pacientes.
Conclusiones: Tanto imiquimod cremas 2,5% y 3,75% fueron más efectivos que placebo y fueron bien tolerados cuando se administraron diariamente en 2 ciclos de 2 semanas, con un intervalo de 2 semanas de descanso para tratar queratosis actínicas de cara o cuero cabelludo calvo. (FL)
con ungüento de tacalcitol por 6 semanas. Los resultados se compararon con un grupo de 20 voluntarios sanos.
Resultados: Los niveles séricos de PRL fueron significativamente más altos en el grupo psoriático comparado con el grupo control (P< 0,001) y se redujeron significativamente los niveles luego del tratamiento (P=0,001). Existió una correlación entre los niveles séricos de PRL pretratamiento y el PASI. (r=0,33; P=0,02).
Conclusiones: Estos resultados indican que los niveles séricos de PRL podrían servir como marcador biológico de actividad en psoriasis. (JL)

114 Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Recovery of scleroderma-induced atrophic alopecia by autologous fat transplantation. Cho SB, Roh MR, Cheng KY.Dermatol Surg. 2010; 36(12):2061-3
Cerca de la mitad de los pacientes con formas localizadas de esclerodermia presentarán secuelas pigmentarias junto a cicatrices atróficas. En el caso de lesiones ubicadas en cuero cabelludo, se puede observar desarrollo de alopecia secundaria.
Se presenta el caso de una mujer de 26 años con morfea, quien al examen físico presentaba una placa alopécica de 3 a 4 cm en la región frontal del cuero cabelludo, sobre la que se extiende una depresión lineal hacia la frente de 5 cm; que había sido tratada con fototerapia, corticoides sistémicos e intralesionales, sin respuesta satisfactoria. Se decide realizar trasplante de grasa autóloga para corrección del defecto cosmético. El procedimiento se realizó en 2 etapas, hasta obtener la sobrecorreción deseada, desarrollando complicaciones mínimas como edema y equimosis, que se resolvieron espontáneamente. Al tercer mes de realizado el
trasplante se observó crecimiento de pelo terminal, el que se mantuvo hasta el sexto mes de seguimiento.
Existen diversos tratamientos para la esclerodermia locali-zada, pero generalmente se necesitan terapias adicionales para corregir los defectos cosméticos secundarios. Entre estos, se han utilizado rellenos y el trasplante de grasa autólogo con buenos resultados. Hasta la fecha se desconocen los mecanismos por los cuales el tejido adiposo implantado induciría la aparición de pelo. Se piensa que podría deberse a la presencia de células madre, factores angiogénicos, liberación de hormonas, citoquinas y factores de crecimiento. Por lo tanto, los autores sugieren que debiese realizarse seguimiento por algunos meses a pacientes tratadas con esta técnica, antes de decidirse a realizar otras terapias como reducción quirúrgica del defecto o uso de implante de pelo. (CGG)
Clinical differentiation of primary from secondary hyperhidrosisWalling H W.J Am Acad Dermatol 2011; 2:1-6.
Antecedentes: La hiperhidrosis (HH) es la sudoración excesiva que puede ser primaria (idiopática) o secundaria al uso de fármacos o enfermedades. Las características clínicas que apoyan la etiología primaria o secundaria no han sido bien documentadas.
Objetivo: Identificar las características clínicas y demográficas que puedan predecir el diagnóstico de una HH primaria en comparación con una HH secundaria.
Métodos: Se realizó una revisión retrospectiva de todos los pacientes (niños y adultos) , con código de HH, atendidos en el policlínico universitario de pacientes externos de dermatología, basados en la revisión del código de diagnóstico del International Statistical Classificacion of Disease and Related Health Problems ( ICD-9), durante un período de 13 años (1993-2005). (N = 415)
Resultados: 187 pacientes (93,3%) tenían HH primaria (HHP), 28 pacientes (6,7%) tenían HH secundaria (HHS).
Los pacientes con HHS eran mayores (39,0 + 18,6 años v/s 27,3 + 12,3 años). Su edad de presentación era con más frecuencia en mayores de 25 años (55% en HHS v/s 12,1% en HHP, odds ratio [OR] = 8,7; Intervalo de confianza del 95%: 3,5 a 21,4; p < 0,00001).
La HHS fue más frecuente unilateral/asimétrica (OR: 51; IC 95%: 12,6 a 208), generalizada (vs focales, OR: 18, IC 95%: 7,3 a 47,6), y era nocturna (OR: 23,2, IC 95%: 4.3-126, p <0,00001). De los casos de HHS, 57% presentaron una enfermedad endocrina (incluyendo diabetes mellitus, hipertiroidismo e hiperpituitarismo). Las enfermedades neurológicas representaron un 32% (Incluyendo lesión de nervios periféricos, Enfermedad de Parkinson, distrofia simpática refleja, lesiones de columna vertebral y malformación de Arnold-Chiari). Otras causas son: neoplasias (feocromocitoma), enfermedades respiratorias, y la patología psiquiátrica.

115Rev. Chilena Dermatol. 2011; 27(1):109-115
Revisión de Revistas
Colaboradores:
Catalina Bley (CB)Pedro Claveria (PC)Andrea Cortés (AC)Carmen Gloria González (CGG)Ana María Kutz (AMK)Francisco Lama (FL)Jorge Larrondo (JL)Marlene Waissbluth (MW)
En comparación con otras causas secundarias, la HH asimétrica favorece a la patología neurológica (O: 63, IC 95%: 4,9 a 810), p = 0,0002).
Limitaciones: Los resultados fueron obtenidos a partir de una sola población, basada en el policlínico de la universidad.
Conclusiones: En base a estos datos, los criterios diagnósticos de la HHP fueron evaluados estadísticamente. Los criterios incluyen: sudoración excesiva de 6 meses o más de duración, con 4 o más de las siguientes características:
incluyen principalmente sitios con mayor densidad de glándulas ecrinas (axilas, palmas/plantas/región craneofacial), ser bilateral y simétrica; no se presenta en forma nocturna; episodios por lo menos una vez por semana; inicio a los 25 años de edad o menos, historia familiar positiva, y alteración de las actividades diarias. Estos criterios permitirían discriminar bien entre HHP y HHS (sensibilidad: 0,99, especificidad: 0,82) y podrían favorecer un manejo clínico óptimo. (MW)
Ultraviolet light exposure and skin cancer in the city of Arica, Chile.Rivas M, Araya MC, Durán V, Rojas E, Cortes J, Calaf GM Mol Med Rep 2009; 2 : 567-572
Propranolol en el tratamiento de los hemangiomas en la infancia.Kramer D, Muñoz P, Alfaro P, Araníbar L, Cárdenas L, Morovic CG, Vidal C, Whittle C. Rev Chil Pediatr 2010; 81 (6): 523-530
Lumbar association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies.Iacobas I, Burrows P, Frieden Ij, Liang MG, Mulliken JB, Mancini AJ, Kramer D, Paller AS, Silverman R, Wagner AM, Metry DW J Pediatr. 2010 Nov;157(5):795-801.e1-7. Epub 2010 Jul 2.
Publicaciones científicas chilenas en la literatura mundial

116 Rev. Chilena Dermatol. 2011; 27(1):116-124
COMENTARIOS DEL DIRECTOR
Juan Honeyman M.
NOTICIAS a) Treinta años de fundación del Departamento de Dermatología de la Pontificia Universidad Católica de Chile. El 26 de marzo del presente año se realizaron las Jornadas de ex Residentes de Dermatología UC en el marco del aniversario de la creación de la docencia en Dermatología en la Universidad. La reunión fue organizada por el Jefe del Departamento, Dr. Sergio Silva, Dr. Félix Fich, Jefe del programa de postítulo y la Dra. Marianne Kolbach Subjefe.
Con un gran orgullo y muchos deseos de que el Departamento siga incrementando su reconocido prestigio nacional e internacional, saludamos este aniversario.
b) Comité editorial de nuestra revista:Tenemos que lamentar que la Dra. Pilar Valdés ha renunciado recientemente al nuestro Comité. La Dra. Valdés ha hecho una extraordinaria colaboración con nuestra revista. Tiene un largo historial en el Comité ejecutivo, se desempeñó como Secretaria del Comité desde 1996 a 2007. Es mucho lo que la Sociedad Chilena de Dermatología y la Revista en particular debemos agradecer a tan distinguida dama de la dermatología nacional. Nuestro agradecimiento y mejores deseos de éxito.
Las Dras. Irene Araya, Presidenta y Eliana Faúndez, Secretaria General de nuestra Sociedad nos han informado que el Comité Ejecutivo de la Sociedad Chilena de Dermatología, ha tomado conocimiento de la incorporación del Dr. Pablo Uribe G. como nuevo miembro del Comité Editorial de la Revista Chilena de Dermatología.
Para el Comité Ejecutivo es motivo de alegría el ver incorporar a nuevos socios como colaboradores en las grandes tareas de difusión de la dermatología que nuestra institución se ha impuesto, más aún en la Revista Chilena de Dermatología, órgano oficial de nuestra Sociedad.
Desamos todo el éxito al Dr. Pablo Uribe en esta importante labor que está pronto a iniciar.
c) Actividades Científicas programadas para el año 2011
MES DE MAYOViernes 20 3ª REUNIÓN CIENTÍFICA Coordina: Unidad de Dermatología CRS Cordillera OrienteLugar: Hotel Plaza El Bosque
MES DE JUNIOViernes 10 y Sábado 11XXIV SIMPOSIUM “ACTUALIZACION EN DESORDENES PIGMENTARIOS, PATOLOGÍA UNGUEAL Y PELO” Organiza: Unidad de Dermatología, Pontificia Universidad CatólicaDirector: Dr. Sergio Silva. Coordinadores: Dres. Ariel Hasson - Néstor Carreño.Lugar: Hotel Intercontinental MES DE JULIOSábado 22ª SESIÓN DE EDUCACIÓN CONTINUACoordina: Grupo de Trabajo ITS. Lugar: Hotel Plaza El Bosque
Viernes 224ª REUNIÓN CIENTÍFICACoordina: Servicio de Dermatología, Clínica AlemanaLugar: Hotel Plaza El Bosque
MES DE AGOSTOJueves 18, Viernes 19 y Sábado 20XXV CURSO INTERNACIONAL “TERAPÉUTICA DERMATOLÓGICA”Organiza: Departamento de Dermatologia, Hospital Clínico Universidad de ChileCoordinadoras: Dra. Rosamary Soto y Dra. Verónica CatalánLugar: Hotel Intercontinental

117Rev. Chilena Dermatol. 2011; 27(1):116-124
Comentarios del Director
MES DE SEPTIEMBRE
Sábado 33ª SESIÓN DE EDUCACIÓN CONTINUACoordina: Grupo de Trabajo de AcnéLugar: Hotel Plaza El Bosque Viernes 23 II SIMPOSIO INTERNACIONAL DE DERMATOLOGÍA “CORRELACIÓN CLÍNICO-HISTOPATOLÓGICO EN DERMATOLOGÍA”Organiza: Departamento de Dermatología, Clínica Las CondesDirector: Dr. Pedro Lobos. Coordinador: Dr. Rodrigo SchwartzLugar: Auditorio Mauricio Wainer, Clínica Las Condes
MES DE OCTUBRE Viernes 75ª REUNIÓN CIENTÍFICACoordina: Servicio de Dermatología Clínica Las CondesLugar: Hotel Plaza El Bosque
Jueves 20 y Viernes 21 XVII JORNADAS DERMATOLÓGICA DE CLÍNICA ALEMANAOrganiza: Departamento de Dermatología, Clínica AlemanaDirector: Dr. Raúl Cabrera. Coordinador: Dr. Roberto ValdésLugar: Centro de Eventos Manquehue
MES DE NOVIEMBREJueves 24, Viernes 25 y Sábado 26JORNADAS ANUALES PROF. DR. HERNÁN HEVIA P.Lugar: Gran Hotel Pucón - Enjoy
d) Fallecimiento de un maestro de la dermatología mundial.Lamentamos el fallecimiento del Profesor Jean Thivolet. El Profesor Thivolet era Master en Biología Cutánea e inmunodermatólogo. Fue presidente de la Sociedad Francesa de Dermatología, Profesor de Dermatología, Immunología, Alergología en el Hospital de la Universidad de Lyon, Francia.
En 1991 funda el European Journal of Dermatology y fue su editor jefe.
Tuve el honor de compartir con el profesor Thivolet en numerosas ocasiones y de visitar su Departamento en Lyon. También compartí con él en los Estados Unidos, cuando en 1971 junto a otros destacados especialistas como Denny Tuffanelly, Steve Katz, Beno Michel y Ernest Beutner, entre otros, organizamos el Primer Simposio de Inmunodermatología en el Meeting de la Society of Investigative Dermatology en la ciudad de Atlantic City.
COMENTARIOS CIENTÍFICOSNOVEDADES DE PUBLICACIONES RECIENTES
1.- Dermatomiositis
Mammen AL. Dermatomyositis and polymyositis Clinical presentation, autoantibodies, and pathogenesis Ann. N.Y. Acad. Sci. 1184 (2010) 134– 153
2.- Fotoenvejecimiento y vitaminas
Zussman J, Ahdout J, Kim J. Vitamins and photoaging: do scientific data support their use? J Am Acad Dermatol. 2010 Sep;63 (3):507-525.
3.- Hiperhidrosis
Callejas MA, Grimalt R, Cladellas E. Hyperhidrosis Update. Actas Dermosifiliogr. 2010; 101 :110-118
4.- Guía europea para el manejo del linfogranuloma venéreo
de Vries HJ, Morré SA, White JA, Moi H. STI. European guideline for the management of lymphogranuloma venereum, 2010. Int J STD AIDS. 2010 Aug;21(8):533-536.
5.- Sindrome de Sézary.
Olsen EA, Rook AH, Zic J, et al. Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol. 2011 Feb; 64(2):352-404.

118 Rev. Chilena Dermatol. 2011; 27(1):116-124
Juan Honeyman M.
6.- Propranolol en angiomas infantiles ulcerados.Propranolol for treatment of ulcerated infantile hemangiomas. Saint-Jean M, Léauté-Labrèze C, Mazereeuw-Hautier J, et al. J Am Acad Dermatol. 2011 Feb 24. ( En prensa)
7.- Recomendaciones para la evaluación inicial, diagnóstico, tratamiento y seguimiento de los pacientes con melanoma maligno primario de la piel.Mangas C,. Paradelo C, Puig S, et al. Initial evaluation, diagnosis, staging, treatment, and follow-up of patients with primary cutaneous malignant melanoma. Consensus statement of the network of Catalan and Balearic Melanoma Centers. Actas Dermosifiliogr. 2010; 101 :129-142
8.- Guías para monitorizar el manejo de los pacientes con Lupus Eritematoso.- Sally Koch Kubetin. Guidelines Define Optimal Lupus
Monitoring
- January2010•Skin&AllergyNews
http://www.skinandallergynews.com/news/more- news/single-view/guidelines-define-optimal-lupus-monitoring/868645feba.html
- Urowitz MB, Feletar M, Bruce IN, Ibañez D, Gladman DD. Prolonged remission in systemic lupus erythematosus. J Rheumatol.2005 Aug; 32(8):1467-14672.
CONCLUSIONES DE PUBLICACIONES RECIENTES1.- La vitamina D inhibe receptores y controla los genes que se asocian con enfermedades autoinmunes como esclerosis múltiple, diabetes tipo 1, enfermedad de Crohn y cánceres como leucemia y cáncer colorrectal. El suplemento con vitamina D sería importante en estos casos.
Ramagopalan SV, Heger A, Berlanga AJ,et al. A ChIP-seq defined genome- wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010 Oct; 20(10):1352-1360.
2.- El trasplante de células de Stem produce remisión importante de las manifestaciones clínicas en pacientes con Lupus Eritematoso Sistémico severo.
Sun L. Stem-cell transplants can achieve durable. Lupus. 2010; 19(12):1468- 1473.
Burt RK, Traynor A, Statkute L, et al. Nonmyeloablative hematopoietic stem cell transplantation for systemic lupus erythematosus. JAMA. 2006 Feb 1; 295(5):527-535.
3.- El Itraconazol en pulsos es igualmente eficaz que en esquema continuo en el tratamiento de la esporotricosis cutánea.
Song Y, Zhong SX, Yao L, Cai Q, Zhou JF, Liu YY, Huo SS, Li SS. Efficacy and safety of itraconazole pulses vs. continuous regimen in cutaneous sporotrichosis.
J Eur Acad Dermatol Venereol. 2011 Mar;25(3):302-305
4.- La exposición de la madre embarazada a corticoides tópicos de alta potencia se asocia a riesgo para el crecimiento del feto. No ocurre lo mismo con corticoides tópicos de baja potencia.
Chi CC, Mayon-White RT, Wojnarowska FT. Safety of Topical Corticosteroids in Pregnancy: A Population-Based Cohort Study. J Invest Dermatol. 2010 Dec 30. En prensa.
5.- El diagnóstico precoz es esencial para el manejo del DRESS. Lamentablemente es una patología difícil de diagnosticar ya que frecuentemente se presenta como una reacción adversa a drogas morbiliforme, relativamente benigna y con una biopsia inespecífica. El dermatólogo debe sospechar el diagnóstico, hacer los estudios de laboratorio que permitan identificar la droga causal y en especial indagar sobre tratamientos previos y el tiempo de evolución de la enfermedad relacionado con el inicio de dicha terapia.
Owen CE, Stratman EJ. Failure to Recognize and Manage Patients With
DRESS. Comment on “Drug Reaction With Eosinophilia and Systemic Symptoms” Failure to Recognize Patients With DRESS. Arch Dermatol. 2010;146(12):1379.
6.- Dieta y Acné.
Aún existe la controversia sobre la inducción de acné causado por algunos alimentos. A pesar de que existen alimentos que potencian los mecanismos de producción de las lesiones de acné, aún no existen trabajos clínicos que demuestren que dichos alimentos causan acné.

119Rev. Chilena Dermatol. 2011; 27(1):116-124
Comentarios del Director
Davidovici BB, Wolf R. The role of diet in acne: facts and controversies. Clinics in Dermatology (2010) 28, 12–16
COMENTARIOS SOBRE PUBLICACIONES RECIENTES1.- Importancia del fenómeno de Raynaud para el diagnóstico de la esclerosis sistémica. (SS)
Un estudio canadiense analiza los criterios para establecer el diagnóstico de una esclerosis sistémica.
Se estudiaron 1.048 pacientes con edad promedio de 55 años (+/- 12 años), 87% mujeres con una evolución de 11 años (+/- 10 años). El 38% tenía compromiso cutáneo generalizado.
El fenómeno de Raynaud (98%) y el compromiso cutáneo (esclerodactilia de los dedos en un 92% o por encima de los dedos en el 58%) fueron las características clínicas más comunes. La afectación visceral incluyó fibrosis pulmonar (35%), hipertensión pulmonar (15%). El aparato digestivo indica que se afecta con síntomas como promedio un 4,2 (± 3.1) de un máximo de 14. Un escaso número de pacientes relatan esclerodermia renal (4,4%). Otras manifestaciones incluyen telangiectasias clínicamente visibles (78%), contracturas de los dedos (37%), y úlceras de la punta de los dedos (17%).
La gravedad de la enfermedad fue moderada. En un rango de 0 a 10, las evaluaciones médicas de la gravedad fueron 2,8 (± 2.3), y las evaluaciones del paciente fueron de 3,5 (± 2.5).
El 71% de los pacientes tuvieron al menos un anticuerpo relacionado a SS. Los anticuerpos anticentrómero fueron los más comunes (34%), seguidos por PM/Scl (17%), RNA Pol III (16%), y topoisomerasa I (16%). La mayoría de los pacientes (85%) tenía sólo uno de estos autoanticuerpos.
La discapacidad funcional fue moderada. En una escala de 0 a 3, fue de 0,8 (± 0.7). Un 38% de los pacientes presentaban dolor moderado a severo.
Los síntomas depresivos fueron altos, con una puntuación promedio de 14 (±10) de un máximo de 16.
La calidad de vida relacionada con la salud se ve afectada considerablemente, los pacientes con esclerosis sistémica se encuentran entre el 15% de la población con la peor calidad de vida.
Los autores concluyen que la gran mayoría de los pacientes con esclerosis sistémica tienen fenómeno de Raynaud. Por lo tanto, de aquellas personas con fenómeno de Raynaud, se debe plantear la sospecha diagnóstica de SS. La sensibilidad de Raynaud y el engrosamiento cutáneo proximal del dedo fue sólo del 57%. La asociación de telangiectasias clínicamente visibles y la detección de los anticuerpos relacionados con esclerosis sistémica (anticuerpos anticentrómero y antitopoisomerasa) mejoran la sensibilidad para el diagnóstico en un 97%.
Baron M, Pope J, Markland J, Robinson D, Jones N, Khalidi N, Docherty P, Kaminska E, Masetto A, Smith D, Sutton E, Mathieu JP, Hudson M, Ligier S, Grodzicky T, Mittoo S, Fritzler M. Systemic sclerosis: establishing diagnostic criteria. Medicine (Baltimore). 2010 May;89(3):159-165.
2.- Guías para el tratamiento de la tiña del cuero cabelludo.
La tinea capitis es una infección por dermatofitos en cuero cabelludo y folículos pilosos y piel circundante, causada por especies del género Trichofitum y Microsporum. El Microsporum canis sigue siendo el agente causal predominante. La Sociedad Europea de Dermatología Pediátrica hace recomendaciones para el tratamiento de esta patología.
Tratamiento Oral:
La tinea capitis requiere de tratamiento sistémico porque los antifúngicos tópicos no penetran en el folículo piloso. El tratamiento tópico sólo se utiliza como terapia adyuvante.
La griseofulvina ha sido la droga de elección desde 1950. Es activa contra dermatofitos y segura a largo plazo. Su desventaja es que se requiere un tratamiento de larga duración (6-12 semanas o más) lo cual puede ocasionar disminución de la adherencia. Otros agentes antifúngicos orales son terbinafina, itraconazol y fluconazol, con eficacia y efectos adversos similares a la griseofulvina en niños con tiña por Tricofitum spp. Con estos fármacos la duración del tratamiento es más corta.

120 Rev. Chilena Dermatol. 2011; 27(1):116-124
Juan Honeyman M.
La decisión de elegir tratamiento con griseofulvina o con los nuevos agentes antifúngicos en niños con tiña por Tricofitum spp depende del tipo de paciente y del balance entre duración de tratamiento y las consideraciones económicas.
En los casos en los cuales el agente es Microsporum, la griseofulvina es el tratamiento de elección. Su eficacia es superior a terbinafina.
Tratamiento Tópico:
La terapia adyuvante tópica como el selenio o ketoconazol shampoo, cremas fungicidas o lociones disminuyen las esporas viables responsables del contagio de la enfermedad y reinfección. Las cremas o lociones fungicidas tópicas deberían aplicarse a las lesiones una vez al día por una semana.
El shampoo debería aplicarse en cuero cabelludo y pelo por 5 minutos 2 veces al día por 2-4 semanas o 3 veces por semana hasta que el paciente esté clínica y micológicamente curado.
Duración del tratamiento:
La duración del tratamiento depende de la evolución del cuadro. Se recomienda realizar exámenes clínicos y micológicos con intervalos regulares (2-4 semanas). El tratamiento puede suspenderse si hay cultivos negativos o si el recrecimiento del pelo normal es evidente.
Kakourou T, Uksal U Guidelines for the management of tinea capitis in children.; European Society for Pediatric Dermatology. Pediatr Dermatol. 2010 May-Jun;27(3):226-228.
3.- Novedades en el tratamiento de la pediculosis.
La pediculosis capitis es una ectoparasitosis altamente transmisible, prevalente a nivel mundial. Es un importante problema de salud que afecta principalmente a niños. El efecto social, económico y educacional es considerable.
El tratamiento de la pediculosis capitis ha consistido en la aplicación de insecticidas, algunas veces combinados con la remoción mecánica de liendres peinando el cabello mojado. Se han usado lindano, permetrina, piretrinas y malatión. El uso de peine para liendres puede asegurar la remoción de los huevos, mejorando la eliminación de liendres viables(1).
El lindano, con propiedades similares al DDT, se absorbe por el tejido adiposo y neural causando neurotoxicidad y anemia. Actualmente se utiliza raramente debido a la disponibilidad de tratamientos menos tóxicos.
Estos tratamientos son de eficacia variable, tienen riesgo de toxicidad y problemas de resistencia, lo que contribuye al incremento de incidencia de pediculosis capitis en países desarrollados y no desarrollados.
Un agente no neurotóxico tópico es el isopropilo miristato al 50%, que actúa disecando el exoesqueleto del piojo y la loción benzil alcohol al 5% que asfixia al piojo. Dado su mecanismo de acción mecánicos es poco probable que se desarrolle resistencia.
Otros tratamientos tópicos son las piretrinas, permetrina y el malatión, los cuales tienen bajo riesgo de absorción percutánea, pero pueden asociarse con irritación, rash, y reacciones alérgicas. El malatión debe usarse con precaución, ya que tiene una base inflamable.
Estos agentes tienen eficacia reducida y pueden fallar en el tratamiento por el desarrollo de resistencia. Los mecanismos de resistencia se atribuyen al compromiso del sistema citocromo monoxigenasa P450, resistencia basada en la glutatión-S- transferasa y mutaciones en el gen del canal de sodio. También existe resistencia cruzada entre los pediculicidas la cual puede ocasionarse por la resistencia de los nervios periféricos(1, 2).
En general la resistencia a pediculicidas está relacionada con la formulación tópica. Esto ha motivado la búsqueda de agentes sistémicos efectivos con bajos riesgos de toxicidad, lo cual representaría una alternativa terapéutica mejor. Por otra parte, en pacientes con excoriaciones severas, los tratamientos tópicos pueden ser dolorosos. La terapia sistémica es preferible si existe infección bacteriana secundaria.
El albendazol, un agente para el tratamiento de infestaciones por helmintos, se ha probado en pediculosis capitis pediátrica. Una dosis simple (400 mg) produce sólo un 60% de éxito comparado con el 80% con permetrina tópica al 1%. El tratamiento por 10 días con levamisol, otro antihelmíntico, erradica la pediculosis capitis en el 70% de los casos(1, 2).

121Rev. Chilena Dermatol. 2011; 27(1):116-124
Comentarios del Director
La trimetropina/sulfametoxazol, que interfiere con la síntesis de vitamina B del piojo, muestra resultados controversiales. Un estudio demostró un 83% de éxito administrada en cursos de 3 días de tratamiento separado por una semana de intervalo. Hay otros trabajos que por el contrario no evidencian ningún beneficio(3).
La resistencia a las drogas y el alto porcentaje de falla de tratamientos con varios agentes tópicos hace que la ivermectina sea una opción terapéutica atractiva para el control de los piojos. Recientemente se ha empleado la ivermectina tópica y sistémica para el tratamiento de la pediculosis(4).
La ivermectina (22, 23-dihidroavermectin) es un análogo semisintético de la ivermectina B1, que se aisló del Streptomyces avermitilis. Es un potente antihelmíntico efectivo y seguro contra helmintos intestinales, y otras ectoparasitosis cutáneas como casos severos de escabiosis, larva migrans cutánea, miasis. Actúa al unirse selectivamente uniendo a un canal de cloro abierto por glutamato que se encuentra en los nervios de los invertebrados y en las células musculares, ocasionando el movimiento libre del cloro, que causa parálisis y muerte de los parásitos. No es neurotóxica a dosis habituales y a esas dosis no atraviesa la barrera hematoencefálica.
La ivermectina es una buena opción para el tratamiento de la pediculosis capitis. Su seguridad se ha demostrado por más de una década en el control de la oncocercosis. La eficacia de la ivermectina tópica ha sido confirmada recientemente en un estudio in vitro. La aplicación de solución tópica al 0,8% en 25 pacientes con pediculosis capitis ha demostrado ser eficaz, aunque no ofrece beneficios adicionales con respecto a los demás pediculicidas tópicos(4,5).
Hay estudios recientes con ivermectina sistémica. Con una sola dosis se han observado efectos potentes en el tratamiento de escabiosis en niños y menores de 5 años. Un estudio abierto de Glaziou y cols. demuestra la eficacia y tolerabilidad de ivermectina en el tratamiento de la pediculosis capitis en niños. Se trataron 26 pacientes (edad 5-17 años) con una dosis de ivermectina oral (200 g/kg),observándose una cura clínica en el 27% de los pacientes y respuesta parcial en el 77%. Este estudio demostró que
una dosis de ivermectina es suficiente para matar todos los parásitos adultos, pero se requirió una segunda dosis en el 41% de los niños por la presencia de liendres viables. Una semana después de la segunda dosis se observó una marcada reducción en la densidad de liendres. Actualmente la resistencia a la ivermectina no se ha reportado en piojos de la cabeza, por lo que es una opción muy útil en el manejo de casos resistentes a los pediculicidas estándares.
Un interesante aporte es un estudio realizado en niños mejicanos realizado con el objetivo de evaluar la eficacia y seguridad de la ivermectina oral en el tratamiento de la pediculosis capitis pediátrica. Se trataron 44 niños entre 6 a 15 años con una dosis de ivermectina oral de 200 ug/kg erradicándose los piojos adultos en todos los niños. El 41% (n =18) requirió una segunda dosis una semana después por la presencia de liendres viables. En la tercera visita, dos semanas luego del comienzo del tratamiento no había evidencia de liendres viables, y había resolución completa de las escoriaciones en todos los niños y sin prurito o mínimo reportado en el 93% (n = 41). No hubo efectos adversos significativos por la administración de ivermectina.
Los autores concluyen que la ivermectina demuestra la alta eficacia y tolerabilidad en el tratamiento de la pediculosis capitis en niños. Un número significativo de niños requirieron una segunda dosis para asegurar la erradicación completa(5).
1.- Diamantis SA, Morrell DS, Burkhart CN. Treatment of head lice.
Dermatol Ther. 2009 Jul-Aug;22(4):273-278.
2.- Review of common therapeutic options in the United States for
the treatment of pediculosis capitis. Clin Infect Dis. 2003 Jun
1;36(11):1355-1361.
3.- Morsy TA, Ramadan NI, Mahmoud MS, Lashen AH. On the efficacy
of Co-trimoxazole as an oral treatment for pediculosis capitis
infestation. J Egypt Soc Parasitol. 1996 Apr;26(1):73-77
4.- Glaziou P, Nyguyen LN, Moulia-Pelat JP, Cartel JL, Martin PM.
Efficacy of ivermectin for the treatment of head lice (Pediculosis
capitis). Trop Med Parasitol. 1994 Sep;45(3):253-4.
5.- Ameen, M, Arenas R.Villanueva-Reyes J. y cols. Oral Ivermectin
for Treatment of Pediculosis Capitis. Pediatr Infect Dis J 2010;29:
991–993.

122 Rev. Chilena Dermatol. 2011; 27(1):116-124
Juan Honeyman M.
TEMA DE REVISIÓNAutoanticuerpos en Dermatomiositis y sus características clínicas asociadasExiste asociación de autoanticuerpos con diferentes fenotipos clínicos en pacientes con miositis. Existen auto anticuerpos asociados a miositis que también se pueden encontrar en pacientes con otras enfermedades del tejido conectivo. Hay otro grupo de autoanticuerpos que son más específicos, ya que se encuentran principalmente en pacientes con miositis, no se encuentran en otras enfermedades del tejido conectivo y están prácticamente ausentes en los pacientes con distrofias musculares inflamatorias y no inflamatorias.
ANTICUERPOS ESPECÍFICOSa) Anticuerpos anti-Jo-1 y otros anti-tRNA sintetasa (1-12)
La sintetasa aminoacil-ARNt se expresa en las enzimas citoplasmáticas que catalizan la esterificación de un aminoácido específico a su ARNt para formar un aminoacil-ARNt. Hay un ARNt único para cada uno de los 20 aminoácidos.
Los auto anticuerpos contra la histidil-ARNt sintetasa (anti-Jo-1) son los más comunes. Hay un grupo de pacientes con el Síndrome Antisintetasa, un síndrome clínico caracterizado por miositis, enfermedad pulmonar intersticial, artritis no erosiva, fiebre y lesiones hiperqueratósicas características de los dedos (manos de mecánico).
Se ha identificado una serie de nuevos anticuerpos aminoacil-ARNt sintetasas (ARS), entre los cuales figuran los treonil-ARNt sintetasa (anti-PL-7), alanil-ARNt sintetasa (anti-PL-12), glicil-ARNt sintetasa (anti-EJ), isoleucil-ARNt sintetasa (anti-OJ), asparaginil-ARNt sintetasa (anti-KS), antitirosilo-ARNt sintetasa, y más recientemente, antifenilalanil sintetasa (anti-Zo).
Los anticuerpos Anti-Jo-1 se encuentran en aproximadamente el 25%-30% de los pacientes con miositis, y los otros autoanticuerpos anti-ARS se presentan en aproximadamente del 1%-5% de los enfermos.
Cada paciente individual no produce más de un anticuerpos antisintetasa. Existen diferencias entre los pacientes con las
diferentes antisintesas. En pacientes con anti-PL-12 el 90% de ellos tenía enfermedad pulmonar intersticial (EPI) y el 65% consultó inicialmente a un neumólogo. En comparación, sólo el 50%-75% de los pacientes con anticuerpos anti-Jo-1 tienen EPI. Aunque el 90% de los pacientes anti-PL-12 tenía una enfermedad subyacente del tejido conectivo, sólo el 32% tenía Polimiositis (PM) y el 19% presentó dermatomiositis (el resto tenía el diagnóstico de esclerosis sistémica, enfermedad del tejido conectivo indiferenciado, lupus eritematoso sistémico y artritis reumatoide).
Por otra parte, el 90% de los pacientes con Jo-1, tenía compromiso muscular. Al compararlos con los pacientes Jo-1 positivos, los pacientes PL-12 tienen tasas mucho más bajas de artritis (58% versus 94%), manos de mecánico (16% versus 71%) y fiebre (45% versus 87%). El compromiso pulmonar se produce en aproximadamente el 30% de los pacientes con miositis en ausencia de anticuerpos antisintetasas conocidos.
En general los pacientes con anti-PL-12 y algunos otros antisintetasa son más propensos a tener enfermedades pulmonares sin enfermedad muscular clínicamente detectable. Un estudio en pacientes japoneses con anticuerpos antisintetasas, reveló que siete de 88 pacientes tenían compromiso pulmonar y no desarrollaron miositis clínica, después de más de 6 años de observación. Estos pacientes tenían anticuerpos anti-KS, anti-PL-7, anti-PL-12, anti-EJ, y anti-OJ, pero no anti-Jo-1.
Los pacientes con Dermatomiositis (DM) amiopática también pueden desarrollar compromiso pulmonar. No está claro si los pacientes con la enfermedad amiopática y compromiso pulmonar constituyen una entidad separada o “atípica” de la dermatomiositis o del síndrome antisintetasa.
Los autoanticuerpos antisintetasas se pueden encontrar en los pacientes con DM ó PM, y algunos antisintetasas pueden estar fuertemente asociados con una de las otras enfermedades. Diferentes estudios de la misma antisintetasa arrojan muy diferentes resultados.
La respuesta de anticuerpos a la proteína de Jo-1 (es decir, la histidil-ARNt sintetasa) representa un proceso dependiente de célula T, inducido por un antígeno contra la proteína Jo-1. Los títulos del anticuerpo anti-Jo-1 se correlacionan con actividad de la enfermedad. La asociación de los niveles anti-

123Rev. Chilena Dermatol. 2011; 27(1):116-124
Comentarios del Director
Jo-1 en las manifestaciones musculares y extramusculares de actividad de la enfermedad sugiere que puede haber un vínculo entre el antígeno Jo-1 y la inflamación en diversos tejidos.
Los autoantígenos Jo-1 se escinden por granzima B, una enzima proteolítica que se encuentra en los gránulos de las células T citolíticas. Se ha postulado que los linfocitos que reconocen epítopos crípticos en Jo-1 no se eliminarían durante la maduración, deberían permanecer en la circulación, y podrían activarse para impulsar una respuesta autoinmune. Se ha demostrado que en los pulmones se produce un microambiente para la generación de fragmentos crípticos Jo-1 por la granzima B y la subsecuente iniciación de una respuesta inmune anti-Jo-1.
La respuesta inmune contra la proteína Jo-1 parece ser un evento importante en la iniciación de la miositis. Esto se ha demostrado en modelos experimentales con ratones, pero sigue por establecerse si ocurre lo mismo en la enfermedad humana. Estudios en ratón sugieren que la respuesta inmune contra Jo-1 puede jugar un papel importante en la patogénesis del síndrome antisintetasa.
El antígeno Jo-1 puede tener propiedades pro inflamatorias, además de su papel en la síntesis de proteínas. La proteína Jo-1 puede atraer a linfocitos, monocitos y células dendríticas inmaduras por medio de su interacción con el receptor 5 de quimoquinas. Se ha propuesto que las células musculares dañadas podrían liberar Jo-1, lo que conduce al reclutamiento de células inflamatorias que podrían, a su vez, perpetuar la destrucción muscular mediada por autoinmunidad.
b) Anticuerpos anti-Mi-2 (13-16)
El 20%-30% de pacientes con dermatomiositis tienen anticuerpos contra la proteína nuclear Mi-2. Con el método de ELISA hay un número significativo de pacientes con PM e incluso distrofia muscular Mi-2 positivos.
El Mi-2 es un componente de la deacetilasa que remodela al nucleosoma, o complejo NuRD. Este complejo nuclear consta de hasta ocho subunidades distintas y regula la trascripción a nivel cromosómico por desacetilación de histonas y remodelamiento del nucleosoma dependiente de ATP. Específicamente, Mi-2 modifica la estructura de la cromatina por medio de su actividad como una ATPasa dependiente de
ADN, estimulada por el nucleosoma. Originalmente, se pensó que Mi-2 funcionaba exclusivamente como un supresor de la trascripción, sin embargo, trabajos recientes indican que el Mi-2 también interactúa con proteínas de transactivación mediante su dominio amino terminal.
Pacientes con DM con anticuerpos anti-Mi-2 tienden a tener manifestaciones cutáneas más severas, como erupciones en heliotropo, erupciones en chal sobre la espalda y el cuello, y crecimiento cuticular excesivo. Sin embargo, los pacientes con anticuerpos Mi-2 tienen un pronóstico más favorable, con una mejor respuesta al tratamiento con esteroides, y una disminución de la incidencia de tumores malignos en comparación con otros con DM. Estas observaciones sugieren que, los pacientes de DM con anticuerpos anti-Mi-2 pueden representar un grupo distinto.
La exposición solar con aumento de la intensidad de radiación de UV aumenta las probabilidades de desarrollar una respuesta inmune contra el Mi-2. Se ha observado que el anticuerpo anti Mi-2 es mas frecuente en latitudes con mayor radiación solar.
Los niveles de proteína Mi-2 niveles son bajos en músculo normal y en muestras de biopsias musculares de PM. En cambio, las biopsias musculares de pacientes con DM tienen un aumento significativo de la expresión de Mi-2. Esto indicaría que la expresión aumentada de Mi-2 en los tejidos de DM impulsa la respuesta inmune anti-Mi-2.
c) Autoanticuerpos de partículas de reconocimiento de antiseñal (17- 21)
Las biopsias de pacientes con DM y PM se caracterizan por la presencia de células inflamatorias. Sin embargo, aproximadamente el 10% de los pacientes que aparentemente tienen enfermedad del músculo autoinmune tienen biopsias que revelan degeneración, necrosis y regeneración de fibras musculares con poca o ninguna infiltración de linfocitos. Algunos de estos pacientes con una “miopatía necrosante” tienen autoanticuerpos dirigidos a los componentes de la partícula de reconocimiento de señal (PRS).
La PRS es un complejo de seis polipéptidos (72, 68, 54, 19, 14, y 9 kDa) y una sola molécula ARN 7SL. Estos autoanticuerpos pueden dirigirse contra uno o más de los seis polipéptidos, así como a ARN 7SL. Un 4% de pacientes

124 Rev. Chilena Dermatol. 2011; 27(1):116-124
Juan Honeyman M.
con PM ó DM tienen autoanticuerpos anti-PRS, en ellos la enfermedad muscular es severa, con los niveles muy elevados de CPK y responden inicialmente a los esteroides aunque con frecuencia se requieren múltiples medicamentos inmunosupresores para su control. Hay escasos pacientes que tienen este anticuerpo que no tienen enfermedad muscular. (Dos tenían esclerosis sistémica y uno síndrome antisintetasa), lo que sugiere que estos anticuerpos pueden no ser específicos para las PM.
Algunos pacientes con miopatías necróticas autoinmunes no tienen anticuerpos anti-PRS. Falta por determinar si estas personas tienen anticuerpos no identificados.
d) Anti-155/140 asociado a cáncer y Dermatomiositis(22- 24)
Recientemente se han detectado anticuerpos antiproteínas de 155 y 140 kDa. Estos anticuerpos anti-155/140 son altamente específicos para DM, y se encuentran en 13%-21% de los pacientes. Los pacientes con dermatomiositis anti-155/140 positivos tienen una tasa mayor de tumores malignos que los pacientes negativos.
Curiosamente, los anticuerpos anti-155/140 también se encuentran en pacientes con la forma juvenil de DM. Esto es especialmente notable porque es rara la presencia de otros anticuerpos en la forma juvenil. Serán necesarios estudios futuros para confirmar la identidad de los auto antígenos reconocidos por los autoanticuerpos anti-155/140 y evaluar su potencial papel patológico.
1.- Marguerie, C., C.C. Bunn, H.L. Beynon, et al . 1990. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q. J. Med. 77: 1019–1038.
2.- Betteridge, Z., H. Gunawardena, J. North, et al . 2007. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 46: 1005–1008.
3.- Kalluri, M., S.A. Sahn, C.V. Oddis, et al . 2009. Clinical profile of anti-PL-12 autoantibody: Cohort study and review of the literature. Chest 135: 1550–1556.
4.- Targoff, I.N. 2008. Autoantibodies and their significance in myositis. Curr. Rheumatol. Rep. 10: 333–340.
5.- Fathi, M., J. Vikgren, M. Boijsen, et al . 2008. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 59: 677–685.
6.- Spath, M., M. Schroder, B. Schlotter-Weigel, et al . 2004. The long-term outcome of anti-Jo-1-positive inflammatory myopathies. J. Neurol. 251: 859–864.
7.- Martin, A., M.J. Shulman & F.W. Tsui. 1995. Epitope studies indicate that histidyl-tRNA synthetase is a stimulating antigen in idiopathic myositis. FASEB J. 9: 1226–1233.
8.- Miller, F.W., K.A. Waite, T. Biswas, et al . 1990. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc. Natl. Acad. Sci. USA 87: 9933–9937.
9.- Stone, K.B., C.V. Oddis, N. Fertig, et al . 2007. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 56: 3125–3131.
10.- Casciola-Rosen, L., F. Andrade, D. Ulanet, et al . 1999. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J. Exp. Med. 190: 815–826.
11.- Katsumata, Y., W.M. Ridgway, T. Oriss, et al . 2007. Species-specific immune responses generated by histidyl-tRNA synthetase immunization are associated with muscle and lung inflammation. J. Autoimmun. 29: 174–186.
12.- Howard, O.M., H.F. Dong, D. Yang, et al . 2002. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J. Exp. Med. 196: 781–791.
13.- Nilasena, D.S., E.P. Trieu & I.N. Targoff. 1995. Analysis of the Mi-2 autoantigen of dermatomyositis. Arthritis Rheum. 38: 123–128.
14.- Wang, H.B. & Y. Zhang. 2001. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res. 29: 2517–2521.
15.- Hengstman, G.J., W.T. Vree Egberts, H.P. Seelig, et al. 2006. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann. Rheum. Dis. 65: 242–245.
16.- Burd, C.J., H.K. Kinyamu, F.W. Miller, et al. 2008. UV radiation regulates Mi-2 through protein translation and stability. J. Biol. Chem. 283: 34976–34982.
17.- Satoh, T., T. Okano, T. Matsui, et al . 2005. Novel autoantibodies against 7SL RNA in patients with polymyositis/dermatomyositis. J. Rheumatol. 32: 1727–1733.
18.- Targoff, I.N., A.E. Johnson & F.W. Miller. 1990. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 33: 1361–1370.
19.- Miller, T., M.T. Al-Lozi, G. Lopate, et al . 2002. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J. Neurol. Neurosurg. Psychiatry 73: 420–428.
20.- Kao, A.H., D. Lacomis, M. Lucas, et al . 2004. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 50: 209–215.
21.- Hengstman, G.J., H.J. Ter Laak, W.T. Vree Egberts, et al. 2006. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann. Rheum. Dis. 65: 1635–1638.
22.- Kaji, K., M. Fujimoto, M. Hasegawa, et al . 2007. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 46: 25–28.
23.- Targoff, I.N., G. Mamyrova, E.P. Trieu, et al . 2006. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 54: 3682–3689.
24.-Gunawardena, H., L.R. Wedderburn, J. North, et al . 2008. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford) 47: 324–328.

IN MEMORIAN
E l pasado 2 de Marzo del presente nos ha dejado el Dr. Marcos Pereira Díaz, quien fuera Médico Dermatólogo del Hospital Barros Luco-Trudeau por
más de 30 años. El doctor Marcos Pereira cursó sus estudios primarios y secundarios en el Liceo Celedón en la ciudad de Santa Marta, Colombia, en donde fue contemporáneo del famoso novelista colombiano Gabriel García Márquez.
Llega a nuestro país en el año 1950 para cursar sus estudios de medicina, obteniendo el título de Médico Cirujano en Junio de 1958. Una vez recibido de médico, el Dr. Marcos Pereira se traslada a la sexta región en donde permanece 10 años desarrollando, en esta provincia, su profunda vocación social, aportando al mejoramiento de políticas de salud pública en los distintos pueblos en que ejerció su profesión. Esto le significó el reconocimiento como hijo ilustre en las localidades de Chépica y Nancagua.
En el año 1971 vuelve a Santiago a realizar sus estudios de Post-Grado en la especialidad de Dermatología y Venereología dirigido por el profesor Dr. Ignacio Gonzalez Díaz, en la sede sur de la Facultad de Medicina de la Universidad de Chile. Posteriormente, el Dr. Pereira participó activamente en la docencia de Pre-Grado junto a su colega y amigo el profesor Dr. Jaime Ruiz Román así como también en la formación de numerosas generaciones de dermatólogo, quienes lo recuerdan como un docente cálido, muy alegre y entregado a sus pacientes. Ahondar en destacar más de sus virtudes y facetas de su personalidad, hubiese herido su auténtica modestia.
En relación a su vida familiar, contrajo matrimonio con doña Maria Graciela Moya Pottstock en Junio de 1958 y de esta unión nacen sus tres hijos a quienes legó su vocación por la ciencia: Susana (Médico Veterinario), Marco (Médico Dermatólogo) y Paulina (Médico Pediatra). Sólo queda recordar su vocación por la medicina, su gran alegría de vivir y su querida Santa Marta, Colombia.
Dr. Juan Pedro Lonza Médico Dermatólogo
Dr. Marcos Pereira Díaz
Rev. Chilena Dermatol. 2011; 27(1):125

126
MES DE MAYO
Viernes 20 11.00 hrs.
3ª REUNIÓN CIENTÍFICA Coordina: Unidad de Dermatología CRS Cordillera Oriente Lugar: Hotel Plaza El Bosque Patrocina: Laboratorio Stiefel a GSK Company
MES DE JUNIO
Viernes 10 y Sábado 11 XXIV SIMPOSIUM “ACTUALIZACIONES EN DESÓRDENES
PIGMENTARIOS Y PATOLOGÍA UNGUEAL Y DE PELO” Organiza: Unidad de Dermatología Pontificia Universidad Católica
Director: Dr. Sergio Silva Coordinadores: Dres. Ariel Hasson y Néstor Carreño
Lugar: Hotel Intercontinental
MES DE JULIO
Sábado 2 10.00 hrs.
2ª SESIÓN DE EDUCACIÓN CONTINUA Coordina: Grupo de Trabajo ITS Lugar: Hotel Plaza El Bosque
Viernes 22 11.00 hrs. 4ª REUNIÓN CIENTÍFICA Coordina: Servicio de Dermatología Clínica Alemana Lugar: Hotel Plaza El Bosque Patrocina: Laboratorio Beiersdorf S.A.
MES DE AGOSTO
Jueves 18, Viernes 19 y Sábado 20 XXV CURSO INTERNACIONAL “TERAPÉUTICA
DERMATOLÓGICA” Organiza: Departamento de Dermatología - Hospital Clínico
Universidad de Chile Coordinadoras: Dra. Rosamary Soto y Dra. Verónica Catalán Lugar: Hotel Intercontinental
CALENDARIO DE ACTIVIDADES CIENTÍFICAS SOCHIDERM
MES DE SEPTIEMBRE
Sábado 3
10.00 hrs.
3ª SESIÓN DE EDUCACIÓN CONTINUA
Coordina: Grupo de Trabajo de Acné
Lugar: Hotel Plaza El Bosque
Patrocina: Laboratorio La Roche Posay
Viernes 23
II SIMPOSIO INTERNACIONAL DE DERMATOLOGÍA
“CORRELACIÓN CLÍNICO-HISTOPATOLÓGICO EN
DERMATOLOGÍA”
Organiza: Departamento de Dermatología Clínica Las Condes
Director: Dr. Pedro Lobos
Coordinador: Dr. Rodrigo Schwartz
Lugar: Auditorio Mauricio Wainer Clínica Las Condes
MES DE OCTUBRE
Viernes 7
11.00 hrs.
5ª REUNIÓN CIENTÍFICA
Coordina: Servicio de Dermatología Clínica Las Condes
Lugar: Hotel Plaza El Bosque
Patrocina: Laboratorio Stiefel a GSK Company
Jueves 20 y Viernes 21
XVII JORNADAS DERMATOLÓGICAS DE CLÍNICA ALEMANA
Organiza: Departamento de Dermatología Clínica Alemana
Director: Dr. Raúl Cabrera
Coordinador: Dr. Roberto Valdés
Lugar: Centro de Eventos Manquehue
MES DE NOVIEMBRE
Jueves 24, Viernes 25 y Sábado 26
JORNADAS ANUALES PROF. DR. HERNÁN HEVIA P.
Lugar: Gran Hotel Pucón - Enjoy
Año 2011
XI CONGRESO CHILENO DE DERMATOLOGÍA Y VENEREOLOGÍA
19 – 21 DE ABRIL 2012
Hotel Sheraton Miramar – Viña Del Mar
Rev. Chilena Dermatol. 2011; 27(1):126

127Rev. Chilena Dermatol. 2011; 27(1):127
Año 2011
XIX Jornadas Mediterráneas de Confrontaciones Terapéuticas en Medicina y Cirugía Cosmética
Mayo 13 – 15, 2011 Barcelona, España www.semcc.com
22nd World Congress of Dermatology
Mayo 24-29 Seoul, Korea www.wcd2011.org
16th Annual Clinical Symposium Dermatologic Surgery: Focus on Skin cancer
Mayo 26-29, 2011 Orlando, Florida. USA. www.mohssurgery.org
2nd Summer School of Pediatric Dermatology
Junio 3-6, 2011. Aegean Sea Cruise. Greece. www.espdsummerschool2011.org
Summer Academy Meeting
Agosto 3-7, 2011. New York. USA. www.aad.org
XX CONGRESO ARGENTINO DE DERMATOLOGÍA
Agosto 11-14, 2012 Córdoba - Argentina www.sad2011.com.ar
IX Congreso Colombiano de Dermatología Pediátrica
Agosto 12-15, 2011 Centro de Convenciones Las Américas Cartagena, Colombia [email protected]
EVENTOS INTERNACIONALES
IV Congreso Latinoamericano de Dermatología Pediátrica
Septiembre 21-24, 2011 Guadalajara. México. www.sladp.org www.congressmexico.com/fmdp2011/
5th Continent Congress Lasers and Aesthetic Medicine
Septiembre 15-17, 2011 Cannes. France.
XX AEDV Congress
Octubre 20-24, 2011 Lisboa. Portugal
www.eadv.org
XXVII Congreso Centroamericano y del Caribe de Dermatología
Noviembre 15-19, 2011. San José. Costa Rica. www.dermatologiacr.com/congreso [email protected]
Año 2012
8th World Congress of the International Academy of Cosmetic Dermatology Cancun 2012
31 de Enero al 4 Febrero, 2012. Quintana Roo, México. Hilton Cancún Golf & Spa Resort
www.wcocd2012.com / [email protected]
XIX Congreso Ibero Latinoamericano de Dermatología CILAD 2012 Septiembre 19-22, 2012 Sevilla – España www.cilad.org

128
Acné y Enfermedades Afines. Acné y Dieta Occidental. 2010;26(1):76-78Acné y Enfermedades Afines. Rosácea Infantil. 2010;26(3):303-308Algunos aspectos de compromiso cutáneo por el cáncer de mama. Revisión de la literatura a propósito de tres casos. 2010;26(4):404-411Calendario de Actividades Científicas. 2010;26(2):218-219Calendario de Actividades Científicas. 2010;26(3):345Calendario de Actividades Científicas. 2010;26(4):460Calendario de Actividades Científicas. 2010;26(1):111Caracterización Epidemiológica de los Lipomas Subcutáneos. 2010;26(1):30-34Carcinoma de Células de Merkel: A propósito de dos casos. 2010;26(4):391-394Casos Clínicos. Acroangiodermatitis de Mali. 2010;26(3):309-317Casos Clínicos. Criptocosis diseminada. 2010;26(3):309-317Casos Clínicos. Dacriocistocele congético como diagnóstico diferencial de hemangioma. 2010;26(3):309-317Casos Clínicos. Dermatitis crónica actínica: a propósito de un caso. 2010;26(1):92-99Casos Clínicos. Dermatitis rosácea -like corticoinducida. 2010;26(1):92-99Casos Clínicos. Enfermedad de Hashimoto - Pritzker. 2010;26(1):92-99Casos Clínicos. Enfermedad de Mondor. 2010;26(1):92-99Casos Clínicos. Eritrodisestesia palmoplantar por capecitabina. 2010;26(3):309-317Casos Clínicos. Escleredema diabeticorum. 2010;26(1):92-99Casos Clínicos. Hidradenitis supurativa o acné inversa tratada con terapia fotodinámica (TFD -MAL). 2010;26(2):178-185Casos Clínicos. Hipertricosis cervical anterior esporádica. 2010;26(3):309-317Casos Clínicos. Histiocitosis de células de Langerhans. 2010;26(2):178-185Casos Clínicos. La alopecia universal responde a tratamiento tras 13 años de evolución. 2010;26(2):178-185Casos Clínicos. Linfangioma circunscrito. 2010;26(1):92-99Casos Clínicos. Liquen escleroso. 2010;26(3):309-317Casos Clínicos. Metástasis cutánea atípica de tumor pulmonar. 2010;26(2):178-185Casos Clínicos. Migración de silicona. 2010;26(4):427-434Casos Clínicos. Papulosis fibrosa blanquecina del cuero. 2010;26(2):178-185Casos Clínicos. Pénfigo eritematoso o síndrome de Senear - Usher. A propósito de un caso. 2010;26(3):309-317Casos Clínicos. Porocarcinoma ecrino: tumor maligno infrecuente. 2010;26(1):92-99
ÍNDICE POR TEMAS - 2010
Rev. Chilena Dermatol. 2011; 27(1):128-129
Casos Clínicos. Poroma ecrino, presentación atípica: a propósito de un caso. 2010;26(2):178-185Casos Clínicos. Prúrigo ampollar en adulto por picadura de chinche. 2010;26(4):427-434Casos Clínicos. Prúrigo provocado por mordedura de ácaro de rata. 2010;26(4):427-434Casos Clínicos. Psoriasis pustular desencadenada por antifactor de necrosis tumoral alfa. 2010;26(2):178-185Casos Clínicos. Queratoacantoma: Presentación como Cuerno cutáneo. 2010;26(4):427-434Casos Clínicos. Quiste triquilemal proliferante. 2010;26(2):178-185Casos Clínicos. Signo de Leser-Trélat. 2010;26(4):427-434Casos Clínicos. Síndrome de Goltz. 2010;26(4):427-434Casos Clínicos. Síndrome de Peutz-Jeghers. 2010;26(4):427-434Casos Clínicos. Síndrome mano - pie por capecitabina: a propósito de un caso clínico. 2010;26(3):309-317Casos Clínicos. Tricoblastoma. 2010;26(1):92-99Casos Clínicos. Xantogranuloma Juvenil gigante: presentación de un caso. 2010;26(4):427-434Comentarios del Editor. 2010;26(1):105-109Comentarios del Editor. 2010;26(2):210-216Comentarios del Editor. 2010;26(3):338-344Comentarios del Editor. 2010;26(4):451-458Cuál es su diagnóstico. Granuloma Facial. 2010;26(3):321-324Cuál es su diagnóstico. Liquen Plano Vesiculoampollar. 2010;26(1):79-83Cuál es su diagnóstico. Prúrigo Nodular. 2010;26(2):191-193Cuál es su diagnóstico. Xantogranuloma Juvenil. 2010;26(4):437-440Cuál es su diangóstico. Tumor de células granulares. 2010;26(3):325-327Dermatofibroma Subungueal. Estudio Epidemiológico de 100 casos. 2010;26(1):22-24Dermatopatología. Diagnóstico Diferencial Histológico de los Granulomas Cutáneos. 2010;26(1):88-91Dermatopatología. Inmunohistoquímica en el Diagnóstico Diferencial de las Proliferaciones Basaloides Cutáneas. 2010;26(2):194-201Dermatopatología. Marcadores de Proliferación en Dermatopatología. 2010;26(4):442-443Dermatopatología. Paniculitis y Eritema Indurado de Bazin. 2010;26(3):318-320Dermatosis Asociada a Cáncer Testicular Mixto en paciente Joven: Reporte de un caso y revisión de la literatura. 2010;26(4):399-403Dermatosis Fúngica Invasiva en RN de Peso Extremadamente Bajo. 2010;26(4):412-418

129Rev. Chilena Dermatol. 2011; 27(1):128-129
Dermatosis IgA Lineal: Revisión y Presentación de caso clínico. 2010;26(4):396-398Dermatosis Purpúrica Pigmentada por Actividad Física. 2010;26(4):387-390Diagnóstico Dermatológico: Correlación entre Médicos de Atención Primaria de Salud y Médicos Dermatólogos. 2010;26(1):264-270Editorial Científico. Actualización del Uso de Ivermectina Oral en niños y propanolol en Hemangiomas de la Infancia. 2010;26(1):5-8Editorial Cientifico. Avances en Terapéutica Dermatológica. 2010;26(2):118-120Editorial Cientifico. Rol del Estrés Oxidativo en el Envejecimiento de la Piel. 2010;26(4):351-357Editorial. Asociación Gremial en el camino del Bienestar de sus Asociados. 2010;26(1):4Editorial. Elección de Directorio SOCHIDERM 2010-2012. 2010;26(4):350Editorial. Terremoto y Piel. 2010;26(2):116-117Editorial. Una etapa que finaliza. Informe de Gestión 2008-2010. 2010;26(3):224-226Editorial. Veinticinco años de Historia. 2010;26(3):228-230Educación Médica Continua. Ivermectina: Sus múltiples usos, seguridad y toxicidad. 2010;26(4):358-368Educación Médica Continua. Malformaciones Vasculares. 2010;26(1):10-19Educación Médica Continua. Melasma: Patogénesis y Tratamiento. 2010;26(3):232-248Educación Medica Continua. Ultrasonografia Dopler-color en el diagnóstico de las Anomalías Vasculares de las Partes Blandas superficiales en Pediatría. 2010;26(2):122-130Elevada Proporción de Psoriasis entre pacientes afectados de Onicomicosis. 2010;26(4):370-374Eritema Elevatum Diutinum. Reporte de dos casos y revisión de la literatura. 2010;26(2):164-168Esporotricosis Cutánea: Revisión a propósito de un caso clínico. 2010;26(2):154-158Estudio comparativo sobre las conductas alimentarias entre pacientes con Diagnóstico de Acné Vulgar y pacientes sanos. 2010;26(4):375-378Estudios del efecto inmunomodulador de la Equinácea purpúrea en pacientes con Herpes Labial recurrente. 2010;26(4):379-384Facomatosis Pigmentovascularis: Revisión y Nueva Clasificación. 2010;26(1):36-40Información a nuestros colaboradores para el envío de trabajos. 2010;26(1):112Información a nuestros colaboradores para el envío de trabajos. 2010;26(2):220Información a nuestros colaboradores para el envío de trabajos. 2010;26(3):346Información a nuestros colaboradores para el envío de trabajos. 2010;26(4):462Micosis Fungoides Hipopigmentada: Reporte de Siete Casos. 2010;26(2):144-147Mucormicosis en una paciente diabética. Caso clínico y Revisión de la Literatura. 2010;26(2):148-153Obituario. 2010;26(2):209PLEC. Anticoagulantes y Antiplaquetarios en cirugía Dermatológica. 2010;26(3):328-332
Índice por tema - 2010
PLEC. Peeling combinado en Arrugas Peribucales. 2010;26(1):85-87PLEC. Plasma Rico en Factores de Crecimiento (PRFC). 2010;26(4):435-436PLEC. Tratamiento de Nevo de Ota con Láser Nd: YAG Q-switched. 2010;26(2):188-190Porocarcinoma Ecrino Pigmentado en cuero cabelludo: A propósito de un caso. 2010;26(2):161-163Premiación Revista Chilena de Dermatología 2009. 2010;26(3):337Prevalencia de Patologías Cutáneas en el adulto Mayor de 80 años. Analísis de dos comunas del Sector Norte de Santiago, Chile. 2010;26(2):138-143Protocolo de Farmacovigilancia a Reacciones de Hipersensibilidad a Medicamentos: Resultados de su aplicación en 5 centros de Santiago. 2010;26(2):131-137Psoriasis Ungueal: Experiencia con el tratamiento combinado de Clobetasol al 8% en Laca Ungueal y Calcipotriol. 2010;26(1):42-45Psoriasis. Psoriasis Infantil: ¿Por qué es diferente? 2010;26(4):419-426Publicaciones Científicas Chilenas en la Literatura Mundial. 2010;26(1):104Publicaciones Científicas Chilenas en la Literatura Mundial. 2010;26(4):449Queratodermias Palmoplantares Adquiridas. Revisión Bibliográfica. 2010;26(1):272-278Queratosis Liquenoide Benigna. 2010;26(1):280-283Recomendaciones para el Uso de Agentes Biológicos en Psoriasis. 2010;26(1):55-71Revisión de Revistas. 2010;26(1):100-104Revisión de Revistas. 2010;26(2):202-208Revisión de Revistas. 2010;26(3):333-337Revisión de Revistas. 2010;26(4):444-449Sarcoidosis Cutánea. 2010;26(1):46-54Sección Pediatría. Fibrolipoma Neural de Extremidad Superior. 2010;26(2):170-172Sección Pediátria. Mastocitosis en Edad Pediátrica. 2010;26(3):295-302Sección Pediatría. Telangiectasia Macularis Eruptiva Perstans en la Infancia: Reporte de dos casos. 2010;26(1):72-75Sección Psoriasis. Nutrición y Estilos de vida: Un enfoque complementario para el tratamiento de la Psoriasis. 2010;26(2):173-176Síndrome de Rendu - Osler - Weber en paciente adulto: Reporte de un caso y Revisión de la Literatura. 2010;26(3):285-289Síndrome Metabólico en pacientes Psoriáticos en el Hospital Clínico de la Universidad de Chile. 2010;26(3):259-263Terapia Fotodinámica con Metil Aminolevulinato y Ácido 5-Aminolevulínico en Acné Inflamatorio Leve y Moderado: Experiencia Clínica. 2010;26(1):25-29Terapia Fotodinámica en pacientes con Acné Inflamatorio Facial Leve a Moderadamente Severo: Estudio Abierto en 30 pacientes chilenos. 2010;26(3):250-256Vasculopatía Livedoide: Reporte de dos casos. 2010;26(3):290-294

130
ÍNDICE POR AUTOR - 2010
Ahumada B. Orlando, Córdova B. Ana, Hono P. José, Carvallo T. Rina. 2010;26(3):272-278Alarcón C. Rosario, Martínez D. Catalina, Triviño F. Paula, Muller H. Natalia, Alarcón A. Francisco, Delgado Sch. Carolina. 2010;26(4):412-418Alarcón C. Rosario, Solís H. Felipe, Alarcón A. Francisco, Delgado Sch. Carolina. 2010;26(1):79-93Alarcón P. Daniela, Alarcón C. Rosario, Alarcón A. Francisco. 2010;26(3):309-317Araníbar D. Ligia, Giacaman S. Paula, Sazunic Y. Ivo. 2010;26(1):46-54Araníbar D. Ligia, Hasbún Z. Trinidad, Gamé H. Anne Marie, Sazunic Y. Ivo, Wortsman C. Ximena. 2010;26(2):170-172Araya B. Irene. 2010;26(4):350Arellano L. Javier, Antoniazzi O. Cristina, Navea D. Oscar. 2010;26(4):427-434Avello G. Esteban, Olguín N. Thiare, Cossio T. María Laura. 2010;26(4):427-434Awad R. Perla, Saffie A. Paula. 2010;26(4):387-390Barria S. Katherine, Correa G. Hernán, García-Huidobro R. Isidora, Madrid S. Patricio, González B. Sergio. 2010;26(1):72-75Bello C. Cristina, Cossio T. María Laura, Cárdenas D. Consuelo. 2010;26(1):92-99Benedetto E. Juanita, Cabrera M. Raúl, Britzman Joana, Castro Alex. 2010;26(3):290-294Benedetto E. Juanita, Pinto J. María Paz, Saavedra N. Javiera, Cabrera M. Raúl, Castro Alex. 2010;26(2):164-168Briceño R. Gastón, Guzmán G. Pablo, Anguita H. Rodrigo, Momberg S. Carlos. 2010;26(1):92-99Briceño R. Gastón, Guzmán G. Pablo. 2010;26(3):309-317Briceño R. Gastón, Villaseca H. Miguel, P. Lilia Antonio. 2010;26(2):178-185Calderón H. Perla. 2010;26(1):100-104Calderon H. Perla. 2010;26(3):333-337Cárdenas D. Consuelo, Arellano L. Javier, Alfaro C. Patricia, Faúndez L. Eliana, Le-Bert Z. Marcela, Calderón M. Perla, Molgó N. Montserrat, Chávez C. Pablo. 2010;26(2):131-137Cárdenas V. Rodrigo, Reyes D. Diego. 2010;26(2):178-185Corradini K. Eduardo, Andino N. Romina, Mandiola B. Carlos, Enberg G. Margarita, Corredoria S. Yamile. 2010;26(4):404-411Correa G. Hernán, Jara A. Daniela, Mora G. Carolina. 2010;26(2):144-147De la Sotta F. Pilar, Romero G. William, Kramer H. Daniela, Cárdenas D. Consuelo, González B. Sergio. 2010;26(3):295-302Echeverría P. Ximena. 2010;26(2):202-208
Rev. Chilena Dermatol. 2011; 27(1):130-131
Echeverría P. Ximena. 2010;26(4):444-449Escaffi F. María José, Benedetto B. Antonietta, Podlipnik C. Sebastián, Díaz J. María Cristina, Misad S. Carlos. 2010;26(2):154-158Fajre W. Ximena, Saavedra N. Javiera, Salas A. Camila, Canals C. Magdalena. 2010;26(1):92-99Fajre W. Ximena. 2010;26(1):10-19Faúndez L. Eliana. 2010;26(2):116-117Figueroa B. Andrés, Barassi I. Claudia, Tapia M. Maria Loreto. 2010;26(3):325-327Fischer L. Cristián, Iglesias-Sancho Maribel, Umbert-Millet Pablo. 2010;26(3):309-317Fuenzalida C. Héctor, Cardemil B. Alfredo, Cardemil B. Sebastián, Misad S. Carlos. 2010;26(3):321-324García B. Cristian, Arce V. José D, Méndez C. José Ignacio, Parra R. Rodrigo, Villanueva A. Eduardo, Otero O. Johana. 2010;26(2):122-130Gatica M. José, Morales C. Enrique, Bertoló P. Soledad. 2010;26(2):178-185Gómez H. Orietta, Valenzuela A. Fernando. 2010;26(2):118-120Gómez Z. María Susana, Danielo Cristián, Ragazzini Luciana. 2010;26(4):427-434González B. Sergio. 2010;26(4):442-443Gosch C. Marianne, Larrondo G. Jorge, Araya L. Héctor, Honeyman M. Juan. 2010;26(4):351-357Guerrero A. Robinson, Hono P. José. 2010;26(1):4Guglielmetti V. Antonio, Conlledo V. Rodrigo, Rodríguez C. Álvaro, Pizarro T. Pablo, MacMillan K. Norman, González A. Raúl. 2010;26(4):399-403Guglielmetti V. Antonio, Rodríguez C. Álvaro, Conlledo V. Rodrigo, Vial P. Iván. 2010;26(3):285-289Hasbún Z. Trinidad, Charlín F. Raúl, Bobadilla B. Francisco, Vaswani R. Varsha, Ríos V. Marco, Pinilla P. Jorge. 2010;26(2):161-163Hasson N. Ariel, Nicklas D. Claudia, Burgos C. Susana, Silva V. Sergio. 2010;26(2):188-190Hasson N. Ariel, Pérez R. Mario, Montoya D. Javier. 2010;26(3):232-248Herane H. María Isabel, Orlandi J. Cecilia. 2010;26(3):250-256Herane H. María Isabel. 2010;26(3):303-308Hernández R. Esteban, Leiva V. Josué, Morales H. Claudia. 2010;26(2):178-185Honeyman M. Juan, Gaete G. Marcela. 2010;26(1):76-78Honeyman M. Juan. 2010;26(1):105-109Honeyman M. Juan. 2010;26(2):210-216Honeyman M. Juan. 2010;26(3):228-230

131Rev. Chilena Dermatol. 2011; 27(1):130-131
Índice por autor - 2010
Honeyman M. Juan. 2010;26(3):338-344Honeyman M. Juan. 2010;26(4):451-458Klein G. Rodolfo, Klein Sh. Paula, Glasinovich P. Andrea. 2010;26(4):427-434Kramer H. Daniela, Alfaro C. Patricia, López Juan Pablo, Whittle P. Carolina. 2010;26(3):309-317Leiva B. Innty, Navarrete M. Washington. 2010;26(2):178-185Lezaeta C. Carlos, Valenzuela L. Karen, Zemelman D. Viviana, Valdés A. María del Pilar. 2010;26(2):138-143Madrid S. Patricio. 2010;26(1):88-91Magliano L. Julio, Larre Borges G. Alejandra, Nicoletti R. Sofía, Martínez A. Miguel. 2010;26(3):280-283Maira P. María Elsa, Aranibar D. Ligia, Hasbún Z. Trinidad, Saavedra U. Tirza, Brant G. Stewart, Morales H. Claudia, Sazunic Y. Ivo. 2010;26(4):391-394Maira P. María Elsa. 2010;26(2):209Martínez Erick, Alas Roberto, Escalante Karen, Miller Karen, Arenas Roberto. 2010;26(1):22-24Molgó N. Montserrat, Burgos C. Susana, Sazunic Y. Ivo. 2010;26(1):92-99Montoya D. Javier, Hasson N. Ariel, Montoya D. Lorena. 2010;26(3):309-317Morales H. Claudia, Carreño T. Laura, Sanhueza L. Verónica. 2010;26(3):318-320Mullins L. Enrique. 2010;26(3):224-226Navea D. Oscar, Arellano L. Javier, Vargas R. Ximena. 2010;26(1):92-99Orlandi J. Cecilia, Bustos O. Freddy, Mujica R. Maximiliano, Fernández Z. Carina, Díaz R. Elisa, Loubies M. Rodrigo. 2010;26(1):30-34Pascualini María Florencia, Reyes María Verónica, Kurpis María, Ruiz Lascano Alejandro. 2010;26(2):178-185Pascualini María Florencia, Sureda Nadia Carolina, Kurpis María, Ruiz Lascano Alejandro. 2010;26(1):92-99Pérez W. Jaime, Aspillaga V. María Soledad, Bentjerodt R. Rose Marie. 2010;26(2):178-185Pérez W. Jaime, Aspillaga V. Soledad, Bentjerodt R. Rosemarie. 2010;26(1):92-99Pérez W. Jaime, Bruning V. Carmen. 2010;26(2):178-185Pinto S. Cristián, García-Huidobro R. Isidora, Hasson N. Ariel. 2010;26(1):25-29Prado A. Bernardita, Le-Bert Z. Marcela, Carrión A. Flavio, Martínez G. María José, Honeyman M. Juan. 2010;26(4):379-384Prado A. Bernardita, Sotomayor S. Claudio, Le-Bert Z. Marcela, Avendaño Ana Isabel. 2010;26(4):427-434
Quezada G. Natacha, Cifuentes M. Mirtha. 2010;26(1):85-87Quezada G. Natacha, Hasson N. Ariel. 2010;26(4):435-436Quiroz P. Claudia, Silva A. Sofia. 2010;26(1):36-40Recavarren A. Marcia, Zemelman D. Viviana, Valenzuela A. Fernando, Valdés A. Pilar, Espinoza P. Miguel. 2010;26(3):259-263Reyes M. Verónica, Marín E. Eric, Garay Iliana, Ruiz L. Alejandro. 2010;26(3):309-317Romero G. William A, De la Cruz F. Claudia. 2010;26(1):55-71Romero G. William A, De la Cruz F. Claudia. 2010;26(4):419-426Ruiz A. Héctor, Oddó B. David, Valls G. Gonzalo, González B. Dante, Prado S. Arturo. 2010;26(2):148-153Ruiz A. Héctor, Olivares S. Ramiro, González B. Sergio. 2010;26(2):191-193Saavedra U. Tirza, Espinoza P. Miguel, Hojman C. Lía, Carreño T. Laura, Morales H. Claudia. 2010;26(3):309-317Saavedra U. Tirza, Espinoza P. Miguel, Hojman C. Lía, Carreño T. Laura, Morales H. Claudia. 2010;26(4):427-434Saavedra U. Tirza, Larrondo G. Jorge, Zapata M. Solange, Clavería P. Pedro, Ruiz O. Marcelo. 2010;26(4):427-434Saavedra U. Tirza, Schrag Beatriz, Hojman C. Lía. 2010;26(4):427-434Salomone B. Claudia, Nicklas D. Claudia, Droppelmann D. Katherine, Pérez Cotapos S. María Luisa. 2010;26(4):375-378Sánchez R. Manuel, Sola O. Joaquín, Fischer L. Cristián, Marti G. Agusti, Umbert M. Pablo. 2010;26(4):370-374Sandoval O. Mauricio, Araya C. Gabriela, Andre G. Tamara, Misad S. Carlos. 2010;26(4):396-398Sandoval O. Mauricio, Droppelmann D. Katherine. 2010;26(3):328-332Sandoval O. Mauricio, González B. Sergio, Farías N. Magdalena. 2010;26(4):437-440Schafer V. Fabiola, Rubio L. Rocio, Pérez Cotapos S. María Luisa, González B. Sergio. 2010;26(1):92-99Sotomayor S. Claudia, Barrios J. Ximena, Espinoza P. Miguel, Soto P. Rosamary, Zemelman D. Viviana. 2010;26(3):264-270Valenzuela V. Yesenia, Sazunic Y. Ivo, Ramirez Constanza. 2010;26(2):194-201Vera K. Cristián. 2010;26(2):176-176Victoria Ch. Jairo. 2010;26(4):358-368Zegpi T. María Soledad, Cárdenas D. Consuelo, Sáenz de Santa María P. María Luisa. 2010;26(1):42-45Zegpi T. María Soledad, Salomone B. Claudia, Silva V. Sergio, Nicklas D. Claudia. 2010;26(1):5-8

132 Rev. Chilena Dermatol. 2011; 27(1):132
Revista Chilena de Dermatología, acepta para su publicación en idioma castellano, siempre y cuando reúnan los requisitos que se mencionan a continuación y sean aprobados por el Comité Editorial. El Contenido del artículo será de exclusiva responsabilidad del autor.
Las contribuciones deben referirse a alguna de las áreas de la Dermatología y salud pública en Dermatología, con referencia a aspectos de investigación básica, investigación clínica, experiencia clínica, revisiones y actualizaciones diagnósticas y terapéuticas, comentarios bibliográficos, entre otros.
Los trabajos deben enviarse directamente a Sociedad Chilena de Dermatología y Venereología, Av. Vitacura 5250 Of. 202, Vitacura. Santiago ó vía e-mail a [email protected]
Instrucciones GeneralesLos trabajos enviados deberán ser originales e inéditos y no podrán ser sometidos 1. a consideración de otras publicaciones hasta que el Comité Editorial haga saber al autor que el artículo no ha sido aceptado. Todos los artículos deberán acompañarse de una declaración simple, firmada por el autor responsable, que indique que el trabajo es inédito y original.
El Comité Editorial se reserva el derecho de rechazar total o parcialmente los 2. artículos o de sugerir al autor las correcciones pertinentes antes de su publicación.
Los artículos deben estar escritos en procesador de texto Windows 2003 o 3. superior.
En la primera página deben anotarse: El titulo del trabajo, que debe ser breve y 4. representativo del contenido del artículo; Nombre y título del autor(es), identificándolo con su nombre de pila, apellido paterno e inicial del materno, al término de cada nombre de autor debe identificarse con número de “superíndice”; Nombre de la(s) Secciones, Departamentos, Servicios e Instituciones a las que perteneció dicho autor durante la ejecución del trabajo; Nombre, dirección, correo electrónico y número de fax del autor principal con quien establecer correspondencia; Fuente de apoyo financiero si lo hubo; Señale con letras minúsculas en “superíndice” a los autores que no sean médicos y use dicho superíndice para identificar su título profesional o su calidad de alumno de una determinada escuela universitaria.
En la segunda página repita el título y comience con el texto que no debe exceder de 5. 10 páginas o 5.000 palabras. Debe incluirse un resumen en castellano e inglés con un máximo de 150 palabras cada uno y tres palabras claves en castellano e inglés que orienten sobre el contenido del artículo.
Las referencias bibliográficas deben escribirse al final del artículo, ordenándolas 6. numéricamente de acuerdo con la secuencia en que aparecen en el texto, en el cual han de señalarse solamente con el número arábigo progresivo correspondiente, entre paréntesis y a nivel de la línea. Su formato debe ser el siguiente: a) Para artículos en revistas: Apellido e inicial del nombre del o los autores. Limite la puntuación a comas que separen los autores entre sí. Sigue el título completo del artículo, en su idioma original. Luego, el nombre de la revista en que apareció, abreviado según el estilo usado por el Index Medicus, año de publicación; volumen de la revista: página inicial y final del artículo. B) Para capítulos en libros. Ejemplo: I Anderson L Human Parvovirus B19. Richman D, Whitley R, Hayden F. Clinical Virology. Ed. Churchill Livington Inc. 1997, Chapter 28, pgs. 613-631. C) Para artículos en formato electrónico: citar autores, título del artículo y Revista de origen tal como para su publicación en papel indicando a continuación el sitio electrónico donde se obtuvo la cita y la fecha en que se hizo la consulta. Ej: Rev Méd Chile 2003; 131:473-782. Disponible en: www.scielo.cl (consultado el 14 de Julio de 2003).
Los autores son responsables de la exactitud de sus referencias.7.
Los cuadros y tablas se deben entregar por separado, acompañados de su título y 8. explicación.
Las fotografías, ilustraciones, gráficos, dibujos y reproducciones fotográficas deben 9. entregarse en imágenes digitales indicando posición correcta de publicación. Las ilustraciones, gráficos y dibujos deben ser claros y legibles e indicar la referencia cuando corresponda. Los archivos digitales deben enviarse en formato jpg, de preferencia separados del texto, no pegados, y deben tener un tamaño 8 x 5,5 cm. con una resolución de 300 dpi o bien 72 dpi si el tamaño es sobre 50 x 34,5 cm.
Toda foto clínica que incluya la cara de un paciente deberá ser alterada o modificada 10. con la finalidad de que la persona no sea reconocida y se resguarde su identidad y deberá ir acompañada de una autorización escrita del paciente para su publicación ya sea en revista formato papel o revista electrónica.
Las fotos deberán ser de primera fuente, sin haber sido trucadas ni retocadas; si 11. necesita hacer cualquier modificación, agregar flechas o tapar ojos, envíe una copia modificada que acompañe a la foto original. Los trabajos deben acompañarse de una declaración simple afirmando la autoría de las fotos y que éstas no han sido alteradas ni modificadas, salvo en el caso de las fotos en que aparezca la cara del paciente, en cuyo caso ésta debe ser alterada o modificada en original a efectos de que no sea reconocida su identidad, resguardar su privacidad y dar cumplimiento a la autorización escrita del paciente. Si las fotos han sido tomadas de otra fuente, debe acompañarse de una autorización para su publicación del autor original. La publicación de figuras en colores debe ser consultada con la revista; su costo es fijado por los impresores y deberá ser financiado por los autores, excepto secciones: Sección Educación Médica Continua, casos clínicos. Cuál es su diagnóstico y Sección Quirúrgica.
Los editores de la Revista Chilena de Dermatología se reservan el derecho a realizar 12. en todos los trabajos sometidos a su consideración y aprobados para su publicación, las correcciones ortográficas y gramaticales que pudiesen resultar necesarias y las adecuadas requeridas para uniformar el estilo y forma de la revista, siempre que estos cambios no signifiquen una modificación de la materia expuesta.
La autorización de reimpresos debe ser solicitada directamente a los autores, 13. quienes informarán a la Sociedad Chilena de Dermatología para que ésta resuelva las condiciones de comercialización.
Todos los aspectos no previstos en esta información deberán ser consultados 14. directamente con el editor de la Revista.
Condiciones especiales para la Sección ¿Cuál es su diagnóstico?Las contribuciones deben referirse a casos clínicos de interés.1.
No debe mencionarse hospital o clínica o lugar de trabajo del autor donde fue visto 2. el paciente ni datos que permitan su localización.
En la primera página debe anotarse: 3.
a) ¿Cuál es su diagnóstico?
b) Nombre y título del(os) autor(es), especialidad, lugar de trabajo, mail de contacto, teléfono y fax del autor responsable.
La primera parte, donde se expone el cuadro clínico, debe tener un máximo de 370 palabras y las páginas siguientes deben tener una extensión máxima de 1.500 palabras.
Deben incluirse dos fotos a color; una de tipo clínica y otra del estudio histopatológico, 4. las fotos deben cumplir los requisitos indicados en el punto 8.
Con la discusión del caso, debe hacerse una revisión del cuadro clínico, que incluya 5. diagnóstico diferencial, tratamientos, etc. Debe incluirse también un comentario final.
Las referencias bibliográficas (mínimo 10) deben cumplir con los requisitos generales 6. indicados en el punto 6.
Condiciones especiales para la Sección Casos Clínicos.Se publicarán casos clínicos de interés particular para la especialidad, ya sea por su 1. baja incidencia en nuestro medio o por su forma clínica poco habitual.
No debe mencionarse hospital o clínica o lugar de trabajo del autor donde fue visto 2. el paciente ni datos que permitan su localización.
Los casos enviados no deben contener más de 350 palabras en total. Los nombres 3. de los autores y sus lugares de trabajo no se consideran para esta cifra.
El título debe ser claro, breve y debe incluir el nombre del autor o autores (no más de 4. cuatro) y su(s) lugar(es) de trabajo. También agregar dirección de E-mail y número de fax del autor responsable.
Se debe incluir una sola foto que ilustre el caso clínico o la histología del mismo, que 5. cumpla con las condiciones indicadas en el punto 8.
En la redacción del caso clínico debe agregarse un breve comentario sobre el 6. mismo.
Deben incluirse dos referencias bibliográficas.7.
Los autores se comprometen, por el solo acto de enviarlo para su publicación, a 8. referir siempre cada publicación, agregando a su título las palabras “Caso clínico” encerradas entre paréntesis.
Información a nuestros colaboradores para el envío de trabajos