
Identificacion-Sistematica-de-Compuestos-Organicos-Ralph-L-Shriner.pdf
440
-
Upload
julian-david-quintero-rendon -
Category
Documents
-
view
4.648 -
download
59
Transcript of Identificacion-Sistematica-de-Compuestos-Organicos-Ralph-L-Shriner.pdf


IDENTIFICACION SISTEMATICA DE
COMPUESTOS ORGÁNICOS
Pro f««O f huéepjd de Q utm iea , U n ive rs id ad Meta- d l*t* 0e1 Sureste , D a lü t , T e x „ E .U .A . E x f . « í « o r y Jef< de l Departam en to Q u i l i c o de la Un)v«r» ldad
. de Sowa. Sowar E .U .A .
R E Y ! f O L D C . F V S O IVP ro feao r huéeped d l ít ln g u ld o d» la U n iv o c id a d d© N evad* . Reno, N evada , E .U .A . E x p / o f e o r de Q u ím ica O rgán ica y m iem bro deJ Cen tró d e E*tu- d l«a Avanzad o i de la U n lv e n ld s d d e m inó le ,
U rban a , I '! ., E .U .A .
D A V I D Y . C V R T l !SP ro fe sa r d t Q u ím ica de la U n ivere ídad de llt ín o la .
U rbana , U t „ E .U .A .
E D I T O R I A L L I M U S AM E X I C OC 0
1 9 7 7

T ítu lo de la obra en ingles:THB SyST&MATlC IlJEKTincATtON of Orgawc COXÍPOVTJDSA lA»02AT0ay ManualO IW 5, 1940, 1948
p o r Ralph I* Shriner y Rcynold C , TFukki. ■V en íó a autorizada en español de la quim a edic:óxjs publicada en inglés p o r Joba Wiley and Sons, Inc.Q 1956, 1% 4 por Jo b * W re y a n d Sons, I n c .
V ertiín cspafíela:Dk. Xofcot A. D ^ k íw je zJefe del D epartam ento de Química y profesor de Química O rgánica del Instituto Tecnológico y de Estudios superiores de M onterrey, N. L , México
Revisión:
D kA. Masía Q u stiKa PsaKíM£.sJ>oaDoctorada en la Universidad de París, profesora de Química Orgánica en la Facultad de Ciencias c investigadora en el Instituto de Química de 3a Universidad Nacional Autónoma de México
Derechos reservados en 3e»gua española;
© 1966, E D IT O R IA L LIM USA, 3. A.Areos de Belén 75, México 1, D. Y.M iembro de la C ám ara Nacional de la In d u stria Editorial. R ec to ro Ntfm. 121
P r im e r* f d iu i ín : lWUi
P r im e ra re im jm W m : J5>72
tíigum ia reimprwiif'Hi: 197-J Ttrcera. reím pm /ín ; 1971
Imprtso ttt tífxiro

PrólogoActualmente sigue predominando el interés en los experimentos de
laboratorio relacionados con la identificación de compuestos orgánicos como método pedagógico, Al mismo tiempo, los métodos para realizar investigaciones en Química Orgánica han experimentado una espectacular revolución con la introducción de los métodos espectroscópicos de análisis, incluyendo la Espectrometría de Masas, y con las técnicas de cromatografía, particularmente la Cromatografía tn Fase Gaseosa, como métodos de separación e identificación. Se lía empezado a usar extensamente la Cristalografía con rayos “X ” cora o un medio definitivo y final para determinar las estructuras de moléculas complejas. El propósito fundamental del curso, tal como se presenta en este libro, ha sido y continúa siendo desarrollar en el estudiante un conocimiento activo, en lugar de uno pasivo, con los conceptos y hechos adquiridos en el curso de química orgánica elemental y proporcionar un nexo entre el trabajo de laboratorio necesariamente muy formal que se efectúa en el curso elemental y el trabajo del laboratorio de investigación. La introducción de los métodos instrumentales antes mencionados presenta la posibilidad de transformar el curso de identificación de compuestos orgánicos en un estudio, preferentemente, de análisis instrumental. Actualmente es posible identificar muchas substancias desconocidas ¿n recurrir a ninguna reacción química, debido a los adelantos logrados en instrumentación.
Aunque se puede desarrollar un excelente curso de este tipo, hemos decidido continuar con el objetivo original de la enseñanza, de la química orgánica fundamental. TJna plena comprensión de los grupos funcionales es básica para la investigación de compuestos orgánicos, ya sea que los problemas impliquen síntesis, reacciones o estudios físicos de las estructuras moleculares. Simultáneamente, se ha modificado el enfoqvj en un esfuerzo para evitar que los métodos aquí utilizados se aparten de las técnicas que el estudiante usará posteriormente al emprender sus investigaciones.
Ya que al designar las clases de solubilidad con las letras Sj, S¿, Ax, etc., se obliga al estudiante a memomar una marcha de trabajo que no usará posteriormente su trabajo de investigación, se ha decidido conservar las ventajas de las pruebas de solubilidad, pero omitiendo su

^ ■ a a s m u i ..m nip*
mu 6 Prólogo
clasificación formal incluida en '‘clases de solubilidad^, Este cambio ocasiona que nos* apartemos aím más de una "marcha analítica” y exige
I J que el estudiante confíe más en su propia iniciativa.^ ' ' El u$o de ia extracción y destilación por arrastre con vapor para, separar mezclas, depende bastante de las diferencias de acidez y basici-J 4k dad de los grupos funcionales y puede proporcionar a este respecto una í W información más digna de confianza que las pruebas de solubilidad.
Por esta razón lia parecido apropiado discutir las separación de mezclas —^ inmediatamente después del estudio de 1a$ solubilidades. Aunque se I W podrían introducir otros métodos de separación tales como destilación,
sublimación cromatografía en fase gaseosa, en columna, en papel y en capa delgada, se ha considerado que su discusión excedería indeseable- menee el contenido y propósito de este libro. Por tanto, se deja al criterio
j * del profesor la presentación de dichos métodos. Se ha aumentado un poco el uso de métodos espectroscópicos y se ha añadido una sección sobre resonancia magnética de protones, la cual tiene actualmente, para
f j W químico orgánico, una importancia general sólo inferior a la Espec-| • troscopia Inírairoja.
Finalmente, se han ampliado las tablas de derivados con 7J.2 anota- ciones nuevas que se han añadido a las 2000 originales. Debe hacerse
I : notar que éstas pueden complementarse con Jas tablas de la ChemicoiRvbber PubJufting; estas tablas son las más completas de este tipo de que se dispone y a las cuales se hace referencia en la explicación del uso
• de la bibliografía.Estamos en deuda con muchos profesores de cursos de identificación,
por las sugerencias y críticas constructivas aportadas. También los cstu- « j ^ P diantes de estos cursos han planteado preguntas y ensayado combina
ciones raras de compuestos y reactivos, mediante las cuales se han hecho correcciones convenientes. A todos ellos patentizamos nuestro aprecio
v j k y agradecimiento.
R .- L . S h w n k r
Marzo, 1965. R . C. Fu s o n
D. Y. C ortini»

Contenido1- I n t r o d u c c i ó n ....................................................................................................... 9
2» I d e n t if ic a c ió n d e s u e s t a n c s a s d e s c o n o c id a s .......................... 15
3 . E x a m e n p r e l im in a r ..................................................................................... 37
4. DETERMINACION DE PROPIEDADES FÍSICAS ............................... 39
5 . A n Xu SIS CUA1JTATIVO E L E M E N T A L ........................................................ 75
6. C omportamiento pe solubilidad .......................................... 81
7 . S e p a r a c ió n d e m e z c l a s ............................................................................. 103
8 . A p l ic a c ió n d e l a s p r u e b a s d e c l a s if ic a c ió n .......................... 123
9. U so »R LOS métodos espectroscópicos para la determinación DE GRUPOS FUNCIONALES ............................................. 197
1 0 . P r e p a r a c ió n d e d e r iv a d o s ........................................................................ 2 4 9
11. T a b l a s d e d e r iv a d o s ..................................................................................... 3 2 9
1 2 . I n t r o d u c c ió n a l a r e s o l u c ió n d e p r o b l e m a s e s t r u c t u
r a l e s ........................................................................................................................... 4 ^ 7
13. P r o b l e m a s ............................................................................................................. 42 1A p é n d i c e ................................................................................................................... 4 3 9
I n d ic e a l f a b é t i c o .............................................................................................. 4 4 9

*
• • .

Introducción
CAPITULO
i
RELACIONES DEL ESTUDIO DE LA IDENTIFICACION DE LOS COM PUESTOS ORGANICOS CON LA INVESTIGACION EN QU IM ICA ORGANICA
L a TüOftÍA y t é c n ic a pa r a , identificar compuestos orgánicos constituye una introducción esencial para la investigación en química orgánica, Este estudio'organiza el conocimiento acumulado sobre las propiedades físicas, las estructuras y las reacciones de miles de compuestos orgánicos dentro de un procedimiento sistemático de identificación lógica. Aunque su objetivo inicial es distinguir los compuestos previamente conocidos, el procedimiento de ataque de los problemas constituye la primera etapa para dilucidar la estructura de nuevos compuestos orgánicos de reciente preparación.
Si, por ejemplo, dos compuestos conocidos A y B se diluyen en un disolvente C3 se añade un catalizador D, y :oda la mezcla se somete a las condiciones de temperatura y presión apropiadas para la reacción, resulta una m eada ds nuevos productos además de las materias primas que no reaccionaron.
Inmediatamente surgen dos problemas: .1. ¿Q ué procedimiento se escogerá para separar la
componentes?

1 0 In tro d u cc ió n
2. ¿Cómo se distinguirán definitivamcntí' loa compuestos individuales (E basta K )? ¿Cuáles son Jos reactivos que no cambiaron? ¿Cuáles de estos compue3íos han sido descritos previamente por otros químico*? Finalmente, ¿cuáles productos son nuevos?
Estos dos problemas están intimamente relacionados. Las separaciones de mételas orgánicas usan tanto procesos físicos como químicos y dependen de Jas estructuras de los constituyentes.
El presente estudio enfoca )a atención, primero, sobre la identificación sistemática de compuestos. Los pasos específicos se dan en el capítulo 2. Después se bosqueja, en el capítulo 7, el uso de estos principios para proyectar procedimientos eficientes para la separación de mezclas. Los métodos prácticos de laboratorio y el examen de principios para los pasos en la identificación se dan en los capítulos 3, 4, 5, 6, B, 9, 10 y II.
SUGERENCIAS A ESTUDIANTES E INSTRUCTORES
La revisión de los planes de estudio para las carreras de química requiere, en muchas escuelas, un creciente número de cursos avanzados durante el último año. Será necesario, por lo tanto, limitar las horas de teoría y laboratorio, especialmente si éstas interfieren con el tiempo disponible para la investigación er¡ este último año. De aquí que ¿ez conveniente planear el trabajo para que no necesite tiempo extra. Los párrafos siguientes con cieñen algunas sugerencias para acelerarlo.
P rogram a. No se puede fijar un horario exacto que sea aplicable a todas las «cuelas debido al uso de diversos calendarios de instrucción tales como semestres, trimestres y cursos de verano. Sin embargo, para un semestre de 16 semanas, además de Jas clases teóricas, son aconsejables dos sesiones de laboratorio de 3 horas cada una, por semana. Se pueden efectuar modificaciones del curso para adaptarlo a una escuela en particular.
M aterial de clase. Los siguientes temas son útiles a Jo* estudiantes: El enfoque sistemático a la identificación; estructura y solubilidad; separación de mezclas; ejemplos selectos de pruebas de clasificación y derivados; estructura y métodos espectroscópicos; polifuncionalidad; deducción de estructuras a partir de datos experimentales; problemas. Es innwursario repetir o intentar abarcar todo el material de los capítulos3 al lü ; los estudiantes deberán leerlos y estudiarlos a medida que avan- J an en su trabajo.
T raba je d e laboratorio r—. Substancias desconocidas. Muchos profesores han comprobado que el estudiante promedio puede analizar cuatro substancias individuales y dos mezclas {conteniendo tres componentes) sin trabajar tiempo extra.
Para lograr un comienzo rápido e ilustrar la m archa' sistemática, puede ser útil dar un ácido valorablc a cada estudiante como primera

S uge ren c ia* o e stu d ian te s € instructor** 11
substancia desconocida. A l estudiante se le dice que la substancia puede valorarse y que debe determinar el análisis elemental,, el punto de fusióno de ebullición y el equivalente de neutralización, y calcular los posibles pea» moleculares. Si Ja substancia desconocida contiene halógenos o nitrógeno, el estudiante deberá seleccionar y ensayar tres o cuatro (pero no más) pruebas de clasificación. Después deberá preparar una lista de los posibles compuestos con sus derivados mediante la consulta de la tabla de ácidos {tabla 20). Se prepara un ju n tocon.el informe ¿ver págs, 24-3S). El análisis de esta primera substancia desconocida oeberá completarse y entregarse en una fecha determinada.
Para las otras tres substancias desconocidas, que separadamente se le entregarán al estudiante, se le puede dar eJ espectro en el infrarrojo de una de todas las substancias y puede usarlas según el plan de trabajo bosquejado en el capítulo 2. También se deberá señalar una fecha definida para el reporte de cada una de las mezclas.
Es posible identificar, totalmente, compuestos orgánicos onocidos sin usar ningún método espectroscópíco. Por lo tanto* la carc d a de curvas espectrales en el infrarrojo, ultravioleta o de resonancia magnética nuclear, no impide el buen éxito en Ja interpretación de los grupos funcionales y de sus reacciones. Si no se usan las curvas espectrales, deberá estudiarse totalmente el material del capítulo 9 y estar preparado para el trabajo futuro. •
Pureza de substancias desconocidas. Aun cuando se hacen todos los esfuerzos posibles para proporcionar muestras de compuestos con un alto, grado de pureza, los estudiantes e instructores deben reconocer que muchos compuestos orgánicos se descomponen o reaccionan con el oxígeno, ]a humedad o el anhídrido carbónico, cuando se les almacena por un tiempo considerable. Tales muestras tendrán un amplio intervalo en el punto de fusión o de ebullición, frecuentemente inferior a los valores reportados en la bibliografía, por lo que, para cada substancia desconocida, el estudiante debe hacer un informe preliminar de los valorea observados para el punto de fusión o de ebullición. £1 instructor debe verificar estos datos y si es necesario indicar al estudiante que purifique la muestra por recristalización O destilación y que repita la determinación de la constante física en cuestión. Esto evita pérdida de tiempo y frustraciones al obtener datos en desacuerdo {leer también pág. 15)-.
Cantidades de las substancias desconocida?. Como guía general se sugieren las cantidades siguientes:
Substancia desconocida N® t , un ácido eitulablc, 4 g de un sólido o 10 mi de un líquido.
Substancia desconocida N* t , 3 g de un sólida u 3 t»«t de un liquido.Substancia desconocida. N® 3> 2 g de un sólido o 5 ral de un líquido.S u s ta n c ia desconocida N 7 4 , 1 g de un sólido o 5 inl de un líquido.

12 Introducción
Las mezclas deberán tener 4 ó 5 g de cada componente, Nota: Si se requiere purificación de una muestra, se deberá proporcionar al estudiante una cantidad adicional.
Se pueden identificar muestra1: aún más pequeñas de ios compuestos, usando cantidades menores de reactivos en las pruebas de clasificación y preparando derivados por medio de técnicas semimicro.1 Las instrucciones pueden modificarse y usar aparatos para escala semimicro. Los aparatos para « te curso pueden adquirirse bajo el nombre de “Bantam Ware”, en equipos fabricados por )a. Komes Glass Co,, Vtne- land, N. Jf., E.U.A.
Itxdicadone* para econom izar tiem p o . Es importante planear por adelantado el trabajo de laboratorio, ¿ato puede hacerse determinando en una sesión de laboratorio los análisis elementales, las constantes físicas, el comportamiento de solubilidad y los espectros en el infrarrojo de varias substancias desconocidas. Esta información deT trá anotaree cuidadosamente en el diario de laboratorio y después repa¿y>rse (junto con la explicación de cada, uno de «tos pasos} la noche anterior a la siguiente sesión de laboratorio. Preparar una Usta seleccionando algunas pruebas de clasificación que deberán ensayarse y efectuarlas en el laboratorio al d ía siguiente. En algunos casos puede prepararse una lista preliminar de compuestos posibles y sus probables derivados. Es importante hacer notar que sólo deberán efectuarse algunas de las treinta pruebas de clasificación para un compuesto dado. Puede no ser necesario preparar más de dos derivados; frecuentemente se encontrará que con un solo derivado es suficiente. El objetivo es utili2ar la secuencia de pasos sistemáticos bosquejados en el capítulo 2, en la forma más eficiente posible.
El instructor deberá guiar a los estudiantes para que la identificación correcta resulte de un proceso de deducciones lógicamente razonadas. U na vez estableada la estructura de la substancia desconocida, la interpretación de las reacciones de prueba y de los espectros en el infrarrojo se hace evidente. La práctica de esta fase del razonamiento desde las observaciones del laboratorio basta llegar a la estructura se facilita con la lectura anticipada del capítulo 10, U n método para desarrollar esta habilidad es aquél en que el instructor escribe una fórmula estructural en el pizarrón y pide a los estudiantes que predigan el comportamiento de solubilidad y seleccionen las pruebas de clasificación apropiadas.
Para relacionar el trabajo de identificación de este curso con la investigación real, el instructor puede seleccionar algunos ejemplos típicos de productos naturales, tales como nicotina, j>ribosa, quinina, penicilina Ci y vitamina Bj, y repasar las reaccione» de identificación
1 Qkk«1i y Entttfcl», StnM tr» Qvaííiarw Orteiie AucJyiü, 2* «4., InteucJcoc* PublUim, N eo» Vori, 1957; CbcrMb. fc a in in g Kxtot'mtKtal Oti«iVC Ck*iftUtryt John DrGlftíf, N*trt York, 19».

S u g t fd t tc ia s o e th id la m * * « in s tn jd o r o i 13
usadas para deducir estas estructuras. La bibliografía rédente proporciona también ejemplos del valor de los espectros en el infrarrojo, ultravioleta y de resonancia magnética nuclear, para establecer estructuras. El estudio de los mecanismos de las reacciones usadas para las pruebas de clasificación y para preparar derivados requiere el conocimiento de los grupos funcionales y sus estructuras electrónicas.
En el transcurso de este libro se citan numerosas referencias a artículos origínales, monografías y obras de referencia. Muchas de éstas no se usarán durante el curso de un semestre. Sin embargo, las referencias se han seleccionado para proporcionar valiosas fuentes bibliográf ¡cas que sirvan de guía en trabajos futuros y son de gran utilidad en la investigación al nivel de licenciatura y de graduados.
El uso de este manual se facilitará enormemente con la preparación de índices marcadores para cada capítulo y parte de los capítulos. El tiempo empleado en la preparación de estos índices marcadores se recupera con creces al acelerar la localización de experimentos para los grupos funcionales, procedimientos para obtener derivados y las tablas de derivados.
En todo momento el instructor y los estudiantes deberán observar las reglas de stguridad. Siempre deberán itsar anteojos en el laboratorio y deberán estar familiarizados con tos tratamientos de emergencia,

99
I*:l-
m
f *m m
H»L»1»í *s
f» -f»

CAPITULO
2Identificación de substancias desconocidasL a s i g u i e n t e s e r i e d e i n s t r u c c i o n e s está destinada a guiar al estudiante en el proceso de identificación de una substancia desconocida. Por supuesto, se espera que él llevará una anotación cuidadosa y sistemática de sus observaciones; la preparación de estas anotaciones se simplificará enormemente aplicando la secuencia de operaciones sugerida.
DETERM INACION B E GRUPOS FUNCIONALES
E xam en p re lim inar. C onsulte el capítulo 3r p¿ff&...32±3&.Observe si la substancia es homogénea, anote su estado físico (só-
lido o líquido), color y olor. Efectúe Ja prueba de Ignición (pág, 38) y anote los resultados.
Constantes físicas. Consulte el capítulo 4, págs. 39-73»Si la substancia, desconocida es un sólido, determine su punto de
fusión (págs, 40-45). Si el intervalo del punto de fusión es mayor de 2.0^0, el compuesto deberá recristalizarse. Si la substancia desconocida es un punto líquido o un sólido de punto de fusión muy bajo, determine su punto de ebullición (págs. 46-49); el intervalo de esta constante no deberá excederse de 5.0cC, exceptuando cuando sean compuestos de punto de ebullición muy elevado. Se recomienda la destilación si el intervalo del punto de ebullición indica una contaminación excesiva, sí el compuesto no es homogéneo o sí debe ser incoloro.
AnálUÍÉ de ios elem ento*. Consulte el capítulo 5, págs. 75-80.Investigue en el compuesto la presencia de nitrógeno, azufre, cloro,
bromo y yodo (págs. 7&-80). Si se observó un residuo en la prueba de ignición, identifique el metal que contiene.
Experimentos de control. Si el estudiante no está familiarizado con el procedimiento para descomponer el compuesto o con las pruebas para identificar los elementos, deberá efectuar experimentos de control sobre un compuesto conocido al mismo tiempo que investiga la subs-

U Jd e n H flc c c ió n d e su b s ta n c ia s d e s co n o c id o *
tan d a desconocida. Por supuesto que la substancia que se use para controlar el experimento deberá contener nitrógeno» azufre y un halógeno.
Prueba* de so lubilidad . Consulte el capítulo 6, págs. 81-102. Determine la solubilidad de la substancia desconocida en todos
aquellos de los siguientes reactivos de ios que puede esperarse proporcionen información útil: agua, ácido clorhídrico diluido, hidróxido de ¿odio diluido, solución de bicarbonato de sodio, áddo sulfúrico concentrado, ácido fosfórico y éter etílico (págs. 83-845). Si la dasificación es dudosa, trate una muestra pesada exactamente con un volumen de disolvente medido con exactitud.
Experimentos de práctica. La deducción exacta sobre la base de pruebas efe solubilidad requiere práctica en compuestos conocidos y con este propósito se proporciona un experimento (págs. 83-65),
Guando se investiga la solubilidad de un compuesto en agua, deberá determinarse la reacción de la solución o suspensión con tornasol y .con feaolftaleína. \
Una vez determinado el comportamiento de solubilidad de la substancia desconocida, escriba una lista de todas las clases de compuestos químicos a las que puede pertenecer la substancia,
inform e preliminar. Para evitar pérdidas de tiempo por observa- dones equivocadas, se recomienda que en este punto el estudiante entregue al instructor sus datos sobre las constantes físicas7 composición elemental y comportamiento de solubilidad.
Prueba* de d e if ic a c ió n . Consulte el capítulo 3, págs. 12ÍM95r A partir de la evidencia que se ha acumulado, deduzca qué grupo o
grupos fuadonales es más probable que estén presentes en la substancia desoonodda e investigúelos mediante reactivos de clasificación que sean apropiados. Treinta de los más importantes se mencionan en el capítulo 8 (pág. 127), en donde se pueden encontrar instrucciones para su empleo. En la lista siguiente se han ordenado estas pruebas de acuerdo con el tipo de compuestos que pueden identificar.
Se aconseja rigurosamente al estudiante que evite realizar pruebas innecesarias, ya que no soló son un desperdicio de tiempo sino, además, aumentan la posibilidad de error, Así, no tiene sentido empezar las pruebas de grupos funcionales de un compuesto básico que contiene nitrógeno con pruebas para el grupo alcohol o cetona. Por oirá parte, están claramente indicadas las pruebas que es de esperarse proporcionen informadón acerca del grupo amino, que es casi seguro esté presente,
TJn buen camino a seguir, cuando el trabajo preliminar no ha dado ningún indicio, es ensayar primero las pruebas más sencillas y confiables. Asi, varias de las pruebas para cetonas y aldehidos son, en general, más fáciles de efectuar y de más precisión que las pruebas pam otras fundones cop oxígeno. Por lo tanto, es aconsejable que en la clasificación de un compuesto neutro que se sospecha contiene oxigeno, se empiece con las pruebas de carbonilo.

D e t e r m i n a c i ó n d e g r u p o i f u n c i o n a l a » 1 7
A c o n tin u a c ió n se. e n lis ta n las p ru e b a s d e g ru p o s fu n c io n a le s , a p ro x im ad «uaente e n e l o rd e n g e n e ra l d e su fa c ilid a d y la c o n fian za q u e s e Ies p u e d e te n e r , a u n q u e , p o r su p u esto , h a y m u c h a s excep cio n es a e ste o rd e n ,
A m inas
a) Cloruro de bencen*ulfonilo (pág- 135).b) Acido nitroso (p% . 15S).c) Cloruro» d e acilo (pág. 128),
Halógenos
a ) Solución etanólica de nitrato de plata (pág. 169).b) Yoduro de sodio en acetona (pig. 1?5),
Aldehidos y « to n a l
c ) Fenilhidracina (pág. 164).b ) 2,4-dínitroíenilhidradna (pág. 142).c ) Solución de bbulfito de jodio (p¿g. Í17 ), é ) Yodo e hidió*ido d e «odio (pAg. 154). é ) Clorhidrato de hidroxilamina (p á j, 150).
Aldehidosd) Solución de Benedict (pág. 132).b ) Reactivo d e TollenS (pág. 192).c) Reactivo de Fuchsina par* aldehido* (pág. I í6 ) .d) Solución de bisulfito de jodio (pág. 191;)-
Fenoles
a) Agua de bromo (pág. 140).b) Solución de cloruro férrico (pág. 143).c) Cloruro* de acilo (pág. 123).
Acidos cerboxilitOt
Ninguna prueba; generalmente se supone que una substancia fuertemente ácida, que no es fenol ni enot, debe ser un ácido carbojulico.
Insaíurcción: •
o ) Solución de permangaflato.de potasio (pág. 16&). b ) Bromo en tctracloruro de Carbono (pag, 137).
Compuestos nitrados
a) Cinc y cloruro de amonio (p¿g> 192).Hidxóxido ferroso (pág. 144).
í ) Solución de hidróaddo de sodio (pág. 1&2),

I J
I
9
£*í/<r«
4,? Solución de hidróxádo de sodio (pág. 182).Clorhidrato de hidroxikmína {pág. 150).
Alcoholes
a) Reactivo d e ácido clorhídrico-cloruro de cinc (pág. 14$).b) Cloruro* de adío (pá£. 128).e) Nitrato cérico ( píg. 140).i ) Sodio metálico (pág. f80),*) Acido peryódico {pág. 162}-f) Anhídrido crómico {pág. 141),
18 Id e n t if ic a c ió n d e su b s ta n c ia * d e s co n o c id o s
Hidrocarburos d tcm itiíd s
a.) Acido i'.ilíúnco fumante (pág, 189),b) Cloniro de aluminio anhidro y azo»benceno o cloroforme (pág. 130),
E teret
a) Acido yodhtdríco (pág. 146),b) Agua de broino {pág. 140),
Indique ios reactivos empicados en las pruebas de clasificación, ios resultados y Ja inferencia hecha a partir de cada una. Anote cualquier observación sobre fa naturaleza de los productos formados en éstas pruebas, que pueda ser utilizada posteriormente para excluir compuestos de la lista de posibilidades; por ejemplo, si la hidrólisis con ácido de un compuesto neutro nitrogenado proporciona un producto que no es soluble en agua, la substancia no puede ser un derivado de un ácido que es insoluble en agua.
Pruebas de control, A menos que el estudiante' tenga experiencia previa con una determinada prueba de clasificación, deberá realizar una prueba de control o testigo con un compuesto conocido, &! mismo tiempo que aplica la prueba a la substancia desconocida.
Datos de especírot. Consulte el capítulo 9, págs. 197-247,Los resultados de las pruebas de clasificación se complementan
generalmente con datos sobre Jos espectros en el inírarroio, resonancia magnética nuclear o ultravioleta. La amplitud con la cual se usarán tales datos dependerá del balance entre el trabajo químico y el instrumental que se ha establecido en un determinado curso. Deberán anotarse todas las frecuencias usadas para sacar conclusiones «obre estructuras, junto con su interpretación. Siempre es deseable interpretar juntos loa datos químtcos e instrumentales.

Examen de la bibliografía 19
EXAMEN DE LA BIBLIOGRAFIA
Buscando en las fuentes bibliográficas, prepare una. lista de compuestos que hiervan o fundan dentro de un margen de 5°C con respecto al valor observado para Ja substancia desconocida y que tengan la misma composición elemental, la misma solubilidad y el mismo comportamiento frente a los reactivos de clasificación de la substancia desconocida. M uchas de estas substancias se pueden encontrar en las tablas del capítulo 11. Sin embargo, hay otras obras de referencia que contienen listas de compuestos en orden creciente de sus puntos de fusión o de ebullición. Se recomiendan las siguientes:
L Frankel, Patai, p*r1cis-Kadmon >• Zijltha, TabU f }or Jdentificatio* af Of£aflic CompQtintfo, Suppiem eni te tlandbóok o f Chemistry and PhysicS, Hodg- m an, Editor-in-Chief, Chemical R ubber Publíihíng Compapy, Cleveland, Ohio, 1960. E*ta obra Contiene la serie más completa de tab ta i de compuestos de que puede dis-poncrFe, ordenadoe según íu punto de fusión y punto de ebullición, con más d e 4380 compuesto* fundamentales.
2. Huntrew y Mullíkett, Identification o} P vrt Orgavie Ccmpounds Order l. (Compounds of carbón, hydrogen, and oxygen), 1&4L
3. HuntreEE, Orgóvie CM ojtnc Compoundi, 1943.
4. Utcnnarf:, $ehniclzpur,i;i Tabellen or¿<jnítektf Verbittdungen, 19M.
Otras obras más antigua^ que también pueden ser útiles, son las • siguientes:
5. Ctarke, liandbaek o f Organic A ualytii, 1931.
M ulliken, Jdentificaiion of Puré Organic Compounds, 19M.\
Una vez que se ha encontrado un posible compuesto, deberá hacerse una búsqueda bibliográfica para encontrar derivados del mismo. El camino más directo para realizar una búsqueda total sobre un compuesto en particular, consiste en buscarlo por su fórmula-molecular en los índices de fórmulas de cada una d¿ las siguientes publicaciones, en el orden en que se citan.
Beüscein'x H andbuth der organitchtn Chcmie, 4th ed., Index to ihc 2nd Supplemecit. Esto cvbre la bibliógrafo hasta 1929- £1 uso del Bcílstcin será discutido más adelante.
C hem iichij Zentralblatt, Colfective Indices; 1922-1924, 1925-1929, 1930-1934.
C h tm vcke t Zenlrclblatt, Annual Formula Indices, 1935*1.939.
Después de 1939, el índice de mayor confianza es el “Subject Index del Chemical Abstracts*. Los índices decenales cubren los años de 193?- 1946 y 1947-1956. Para los años siguientes deberán consultarse los índices

2 0 Id e n t if ic a c ió n d e su b s ta n c ia * d e s co n o c id a s
anuales.* Aunque el Chemical Abstrctts tiene un índice colectivo de fónmúas (Collecáve Formula Index) que cubre ios años de 1920-194$, lo mismo que índices anuales de fórmulas para Ic* años posteriores, no son muy completos y no deberá basarse en ellos cuando se desee realizar una búsqueda exhaustiva.
L a importancia del Hcndbuch de Beilstein es tal que se justifica una explicación más amplia sobre su empleo.1 La obra principal o Havptwcrk cubre exhaustivamente, en 27 volúmenes, la bibliografía hasta 1909. La organización de la obra está basada en la estructura, de tal manera que es posible encontrar con bastante facilidad c) compuesto que se quiera sin usar el índice, una vez que se ha familiarizado con la obra. Asi, los hidrocarburos alifáticos y sus alcoholes, aldehidos y cetonas están en el Vol. 1; los ácidos alifátieos en el Vol. 2; los ácidos alifáticos con grupo hidroxilo» aklo y ceto están en el Vol- 3; los ácidos sulfónicos, las aminas y las foafíoas en el Vol, 4. En el Vol. 5 se tratan los hidrocarburos cíclicos (inclusive Jos aromáticos) y la presentación de Jos compuestos cí* el icos continúa dentro de este lincamiento hasta el Vol. 27, en donde empieza la presentación de los compuestos heterociclícos.
U to vez que un determinado compuesto ha sido localizado en la obra principal, se localiza fácilmente en los suplementos. El primer suplemento (Ersfes Ergánzungíwerk), que cubre la bibliografía desde 1910 hasta 1919, riene, asi como también los suplementos posteriores, la misma distribución que la obra principa!. Asi. un compuesto que se encuentra en el Vol. 1 de la obra principa), está en el Vol. 1 del primer suplemento. Además, una serie auxiliar de números de páginas, colocados en el centro superior de cada página en cada uno de los suplementos, relaciona el material de esa página con la página correspondiente de la obra principa!. El segundo suplemento cubre, en igual forma, el material desde 1920 hasta 1929. El tercer suplemento, que cubre un periodo más extenso (1930-1949), está aún en elaboración.
L a Encydopedia of Organic Chcmistry de Elsevier se planeó inicialmente para cubrir la bibliografía de la Química Orgánica tan exhaustivamente como el Hándbuch de Beilstein, Desafortunadamente, la publicación fue discontinuada después de la aparición de unos cuantos volúmenes, Sin embargo, los volúmenes publicados proporcionan un suplemento valioso para el Hándbuch de Beilstein,
Los siguientes son compendios de menor amplitud:
Htíübron, Dietiottcry of Organic Compounds, 1953.Rodd, Chemistiy of Carien Ccmptmkdi; ccmpleted, 1962.
Para decidir ú un compuesto realmente tiene las constantes físicas observadas, debe permitirse una amplitud razonable respecm al error cx-
* NeJa /W /r§i/w;»r. También k piurfc to(Multar «□ el Endite quinquenal que cubra 1M7 • 1961.
* HuaUa*, A Brirf lnt7Mt*£li*X to ikt Uk */ Brültrin'i iit vffavüchrif Ch mU, W w wiky 4*4 S m , Non» Yoi*. 19»,

Pf<&fXJiadón de derivado* 21
perimental. Así, si el punto de ebullición es muy alto o el punto de fusión muy bajo, et ¡inérvalo debe extenderte un poco más ailá de Otras constantes, tales como !a densidad especifica (pág. 55), cí índice de refracción (pág. 62) y los equivalentes de neutralización (pág. 259), pero no las constantes de Duclaux (pág. 261), pueden usarse, con las concesiones apropiadas a los errores experimentales, para excluir compuestos de la lista de posibilidades. Siempre deberá escribirse una lista completa de Ico compuestos posibles con kw derivados para cada amo, aun cuando algún producto de los obtenidos en Jas pruebas de clasificación, aparen (emente sea un derivado apropiado.
Al mismo tiempo que se prepara una lista de compuestos posibles, deberán tomarse notas sobre los derivados de los compuestos. En esta forma se puede economizar mucho tiempo. Lo mejor es consultar el capítulo 10 sobre derivados y anotar los tipos de compuestos apropiados para la identificación final. Las mismas obras de referencia antes citadas se usan para obtener esta información y, además, deberán consultarse las referencias a la bibliografía original.
El examen de la lista de posibilidades a menudo sugiere otras pruebas de grupos funcionales que deben efectuarse. Por ejemplo, si una lista de compuestos nitrados posibles contiene una nítrocetona, Jas pruebas de carbonilo pueden ser muy valiosas.
PREPARACION DE DERIVADOS
Consulte los capítulos 10 y i 1, págs. 24% y 329.La lista de los compuestos posibles, que resulta de los pasos ante
riores para el examen de una substancia desconocida, ordinariamente contiene compuestos pertenecientes a uno o dos tipos. Él paso siguiente en Ja identificación es la confirmación de la identidad de una de estas posibilidades con la substancia desconocida y la demostración simultánea de que cada una de las posibilidades restantes di/iere en alguna forma de la substancia desconocida. Esta prueba final se consuma con Ja preparación de los derivados.
Para eliminar compuestos de la lista de posibilidades, no se está restringido al uso de derivados; puede emplearse cualquier propiedad suficientemente característica tal como: densidad específica, indico de refracción, rotación óptica o equivalente de neutralización.
Propiedades de u n derivado iatü>)acíoria. a) U n derivado satisfactorio debe poderse preparar y purificar fácil y rápidamente. En general, esto significa que el derivado debe ser sólido debido a que, en el aislamiento y purificación de pequeñas cantidades de materia], los sólidos son más fáciles de manipular y los puntos de fusión se determinan con más precisión y más facilidad que los puntos de ebullición. Los derivados más apropiados funden arriba de 50°C pero ahajo de 250°G. La
\

2 2 kd«nt¡f¡<ac(¿n de su b s ta n c ia s <íéf co n o c id o s
mayoría de los compuestos que funden abajo de son difíciles de cristalizar y un punió de fusión arriba de 250°C es indeseable debido a la poábte descomposición y a que la corrección por el vástago del termómetro a menudo es de varios grados.
b) El derivado debe poderse preparar mediante una reacción que produzca un compuesto con buen rendimiento. Deben evitarse los procesos que estén acompañados de transposiciones y reacciones secundarias.
c) El derivado debe poseer propiedades bastante diferentes de las del compuesto original. Normalmente, esto significa que habrá una marcada diferencia entre su punto de fusión y el de la substancia original.
o) El derivado seleccionado debe ser uno que distinga inequívocamente un compuesto dentro de todas las posibilidades. De aquí que los puntos de fusión de los derivados que se comparen deberán diferir entre sí cuando menos en 5|JC.
Com Ite los capítulos JO y 11 y seleccione un derivado apropiado a partir de 'os que se sugieren. Deberá observarse que los derivados que son satisfactorios para propósitos de identificación son numerosos, pero a menudo de alcances limitados. A continuación se enlistan lew más útiles; éstos deben ser los primeros en sc-r considerados cuando se busca un derivado.
Acidos
a) Amidw y anilida?.b) Estere* de ¿-nitnsbencilo y ¿i'bromoíí vacilo.
Las constantes de Duclaux, los coeficientes de partición y los equivalentes de neutralización son muy útiles en este tipo de trabajo y pueden substituir al derivado. Sin embargo, raras veces « aconsejable depender únicamente de ellos.
Alcohol*!
&) Fenít y a-na/Hlurcutios.b) 3,5-Dmitrobcn2<KUos.
Aldehidos y cetonas
ó) FenílhidrXKmaF y ¿’-nitTofenilhidrwonM.b) 2,4-DiiutrofenitfiidrazonaS.Ó Semícartazonas.d) Oximaj.
Anhídridos d¿ ¿cido
a) Acidos.b) Amidas.C) Aailidas.

*
ftrapcrticíón d« dertvodo* 23
C loturoí de ácido
a) Acidos.b ) Amidw.c ) A n i l id a s .
H aturo i de utqttüo y aviloa) Anilida*.b) Halaros alquilmercúricos.
Am ittat (primarios y fecundarlas)
a) B«ncensuIíojiamfclas.b) /-Toluen5üIíonaTni<)^s.C) Acetanudai:.¿ ) Benzamidas.e ) FemlüourcfU.
Am inas (íetcidiias)Compuesto» de adición con:
«.) Acido cloroplatítuco.b) ^Toluepwj]for>ato de m etib .c) Yoduro <k metilo.d ) A cido plcrico,
H idrcearb iita crCitnitkcs
a) Derivados nitrados.b) Acidos aroilbenzoícos.
iEieret (arom álitós)
a) Derivados nitrados.b ) Derivados bromado?.
f tn o l t i
a ) « 'N a f tilu rtia n o s .b ) Derivados bromados.c ) Acetatos.b ) Benzoatos.
Muchos tipos de compuestos pueden hidrotearse hasta ácidos, aminas, alcoholes, etc., y a menudo se identifican con más facilidad por referencia a tales productos. En este grupo e¿fón los acetales, Jos anhídridos de ácido, los cloruros de acilo, Jas amidas, los ásteres, ciertos ¿teres y los nitrilos.
Los compuestos nitro, nitroso, azo e hídrazo pueden reducirse hasta las correspondientes aminas y muchos compuestos pueden identificarse fácilmente por referencia a tales productos de reducción.
El siguiente paso en la identificación es la preparación del derivado. En el capítulo 10 se encuentran procedimientos para muchas de estas

preparaciones. Sj se preparan otros derivados, deberán buscarse las indicaciones en la bibliografía original-
MEZCLAS
Consulte el capitulo 7, págs. 103-122.En alguna fecha, durante el cureo, se darán al estudiante una o más
mezclas. Se entiende que ninguna de éstas contendrá más de cinco componentes orgánicos (usualmentc sólo tres). Después de recibir la muestra entregada por el instructor, proceda a la separación de acuerdo con los métodos bosquejados en el capítulo 7* Muchas de las mezclas contienen un componente volátil que puede separarse por calentamiento de la mezcla en un baño mana. Al m anejar mezclas de composición desconocida se aconseja no intentar una destilación a temperaturas más elevadas.
Cuando los componentes de la mezcla han sido separados, identifique cada uno de acuerdo con el procedimiento seguido para las substancias simples que sean desconocidas.
INFORM ES DÉ SUBSTANCIAS DESCONOCIDAS
Después que se ha completado la identificación de una substancia desconocida, se informan los resultados en formas especiales proporcionadas por el instructor. Los siguientes ejemplos de informes ilustran el procedimiento correcto.
Cotnpuetfo Alcohol n-bvtlíico Nombre Juan ? /w Sufemncia desconocida N 9 1 F ech a I* á t} v n ¡o de 1963
J, ^ ¡ i in > n £ ¿ c o :a) E itado físico liquido. b ) Color ninguno. c) O lor ^ fó cen te ,d ) Prueba de ignición arde con flam a ru lo ta , no d<)& raiduo.
2. Ooasta&tes íbiea*;a) p .f.: observado 5 corr. t>) d«ns. cep. 0.812f
p .tb .z observado COTT. U 5 -}]8 ', C. c) rJ U¡98ii
3 . A n i í ú ; : c l e n K o t i J :
P -".Cl Br —) I —, N —, S —, Metales ninguno.* 4. Pfuchi* de jolubilidad:
2 4 td s n t if ic o d ó n de n jb íK w c ia » desee noc ida»
H iO NaOH NaHGOj HC1 HjSO*
+
Reacción a l to m a» ! ninguna; a la íenolftalcín*. ninguna.

In fo rm es d e su b s ta n c ia s d e scon oc ida s 25
5, Pruebas de clasificación prclirtu rtar:
Reactivo Resultados Inferencias
2,4’diniirofeniLh idraciw
# 0 hay bpt. S o hay grupo carbonilo
Cloruro de acetilo Reacción*—falo r— olor ti frutal Presencia d t grupo
N iha to cérico Color rejah id ffixib
Reactivo de Lucas P isvflt) en el reactivo; nc hubo jífiarticiJr. de a p a aceitoío
Probablemente un alcohol primario
Grupo funcional indicado por estas pruebas: Alcohol, prababler/iente primario.
6. Resultados eepectroseópieoí:
T ipo de espectro y disolvente
PrecuetKia»significativas Infcrtncisu
¡R en CCl< 3600,3300 cm “ ’ — 0 —-H
1025 (m uy ancha) | — C— 0 —
.
*1•*

26 Identificación de substancia* desconocida*
7. Examen prelí.r.:;,*r de k. bibliografía:
Compucstwi poitbltap.f.0
p.eb.¡ Sugerencia? para pruebas adicionales
!Alcohol itabulUico w r c
M ttilisopropiicerbínol 113 Un fec*m¿tjIcarbinút’¿Jcberia. d e j la prueba del yodoforrr.o
Peniatiúi-3 126
Alcohol n-bulílico 117
Prn(anol-2 119 Un Síc-m tiilcerbinol-dtbería J e t la p rv fb a del yodoformo
Sí < lil-t-but iV carbinol j 120
0. Clasificación adicional y pruebas especíale*:
Reactivo Resoltados Inferencias
Prueba d tl yed e fo tn a
fio hay ppt. S o t i un stc-meúlciirbinoi
Infoon#* da M jbjiondoj desconocidas 27
9. Compuestos probable*:
Nombre
Derivado* írílefi y au* p.f.» E.N., etc-
3^-dinztrobenzoato
tt-nafltl-uretuno Feniluutano V ent. esp.
Alcohol n-buíllico 64° C 7 r c s r c 0JÍ10
Alcohol isobutilico as 104 ee 0.805
PírtíurttM 97 71 49 0.820
10. Preparación de derivados;
Nombre del derivado p.f. observado p.f, reportado
3¿~DimlT¿b¿n¿r2aU> 62*63'C 64°C
iX-Seftiluretano $8-69 71
FfrtiluTttMO 57-59 61
11. Comentario* especiales:

A
l¡¿. B ibliografía utilizada:
Tabia i en eiU Uxlo; H uniréis and M uüiken, Identification oí Pare Organic
Compounds, O rder l.
28 Id en t if ica c ió n d e su b s ta n c ia * d e s co n o c id a s
Compuesto 2-am ino^nifro to lueno Nombre Juan Pérez Substancia desconocida N* 2 Fecha 13 de julio d< !$5'¿
1. Examen físico:a) E itado físico sólido* b) Color amarillo. c) Olor ¿ ) Prueba de ignición flama amarilla, no d t ja raiduo.
2 . C cm tanw i Juicas :t ) p.f.: obiervado I07-I08*C í corr. / Í ÍW /O 'C . b ) Dens.esp.
p.eb.: observado ; corr, c ) n53 . A n i ü j u e l e m e n t a l :
F -,C 2I - , B r - , 1 - , N -f-,S - > E l id e s
4. Prueba» de solubilidad:
H jO N aOK N aH C O i HGI | HiSO<
- - * i
Reacción id tornasol ; a la fenol/taleSna
5. Pruebas de clasificación preliminar:
Reactivo Resaltado! Inferencias
Hiniberg WaOH: sotn% transparente.H C l.p p t Am ina primaria
Acido nitrosa Con naftol-2, ppt. anaranjado Amina aromática primaria
Grupo funcional indicado por « ta* pruebas: amina a io m itic i primaria.

6. Examen prelim inar de la bibliografía:
In fo rm as d * su b s ta n c ia s d e s co n o c id a s 29
Compuestos posiblesp.f.0
peb-Sugerencias para pruebas adídonale»
p-Arninoa«to}<none ¡06>C Es pc*t¡neM* la prueba para metilctiona
2'AmtnQ’J'flitT'atoíveno 107 Son pertinentes ia i pruebai para grupo nitro
a -Saftilantinc i ¡2
m -K itto anilina ¡14 Prueba para grupo nitro
M w inc~3-niirot jlu tno 116 Prueba para p u p o nitro
1
7. Resultado! «pectroscópicw :
T ipo de espectro y disolvente
FrecuenciasSignificativas
Inferencias
Ninguno

30 Identificación <4* svbifancktf d*»eonoci<fa*
8. Clasificación adic>on*l y pruebas especiaba:
Reactivo Retul tado« Inferencias
2 f i 'D mitro j enilhid ratina No hay ppt. No es p-aminoacetofeaona
Yodo e hidróxido de sodio No hay yodoformo S o es una meiilcetona
Cine y cloruro de amonio sobre el derivado bem oi' lado de la substancia datconotida
Espeja de plata Grupo nitro presente
4
9- Compüe*toE probables:
Derivados útiles y «os p.f.k E.N., etc.
Nombrep.f.
BenecttesvLfonamida
Aeeiamída Fenol
2*Amino*4'nitiotolueno 107' 172* J 50* 118a '
m-Nitroanilina 114 136 155 97
4-A m ino^-ttitrotolueno 116 102 96 32
10. Preparación de derivadoi:
Nombre de! derivado p.f- observado p.f. reportado
Beneentulfovamída m - n r 172*
2-Oxi~4-niirotolueno 1 I G U 7 118
Informe» d« jubstonden énttenocldos 3X
11. Comentario» íspecialei:
Se ha informada qtse el í-amino-3-nitrotolitcno se hidrolha hasUi 4*oxi-
3-ttitrGtolvcfío con solución de h'utróxído de todio [N t tille y W intker, Ber.,
15, 2083 (1392)']. La substancia desconocida permaneció inalterada bajo
titos condiciones.
La substancia desconocida se convirtió en e l fenol fo r el m¿tod*) infor
mado por m im e n y F itunham , Bcr., 38, 3790 (1905).
12. Bibliografía utilizada:
Tablas en esté tex to; les referencias especiales citadas arriba.
Compuesto $-naftol Nombre Juan PérezSubstancia desconocida N* 3 Fecha 13 de septiembre de 1 fJ63
1. Examen fia ico:a ) Estado físico sólido. b ) Color blanco. c) O lor sugiere bolas
contra polilla.a) Prueba de ignición flama fuliginosa ■— no hay residuo—• sugiere con-
puesto aromático.
2. Constantes física»:
a) p .f.: observado /2 /- /2 2 .5 ° C ; corr. b ) Dene. esp.p-cb.;.observado 2$4-2B6.*C; corr. e) ttp
3. Análisis clemertlaJ: t
P — i Cl —, Br —,1 —, N —, S —, Metales ninguno.
4. Pruebas de solubilidad:
H jO N aO H NaHCO j ' H a HjSOt
- +■
Reacción a l tornasol ; a la fencfftaíeíiui

7 ": '> \r-* ' " *
5 . P rueba» d e clarificac ión preK uurtar:
3 2 Id e n t if ic a c ió n d e su b stan c io * d e * « o iw id < tt
Reactivo Resultados Inícrenciafi
A gua de broma Precipitado -s frobabltrn tn ie un jenci
Cloruro fh r u o Solución t t r d t -*
G rupo íuncíouil indicado pc>r « t a i pruebas: fenol.
6 . Resultados «pectrcucóptcos:
T ipo de espectro y disolvente
Frecuencias¿gniñeativa*
Inferencias
IR t* CHCl, '3300 em ~' (ancha) Grupo oxhidrilo
3600 ( azuda)
7632 cm~* (mediana) ¿G tvpo cctbonilo? <-C = C?
1605 e m " 1 Aromático
JZOOcm-* ( n v y ancha) 0 1 O
' _
w. *

Informas de tu bsfcinrie* d*iconoc«das 33
7. Exam en prelim inar de I* bibliografía:
Compuesto* poii blesp.f.o
p .th .Sugerencia* para prnebaj adiciónale*
p-H ¡J/oxibt m olde hldo ns9 Deberla dar pruebas d f carbonüc
Etar monobcneUico de i a hidroquinona 122 Debería eicindine con ácidos
Acido (Actlco 122 Contiene nitr¿geno—^no es probable qm¡ sea ácido fuerte
b -N tftc l 122
ToluohidroqUxnona 124 Debería oxidarse fácilmente hasta gúmena amarilla
'
8- Clasificación adicional y pinchas especíale*:
Reactivo R em itidos Inferencia*
2,4-diniirofe n ilhid rocina No h a y ppt. El p’h idroxibemaldihido e t improbable
A g ( M , ) S Espejo de plato No se elimina la tclvohidroquu nona
4

9. Compuestos probables;
24 Id en t if ica c ió n d e su b s ta n c ia s d e s co n o c id a s
NombreDerivados útiles y tu j p.f. ( ‘ O . E. N., ele.
Bromu.ro AcetatoI
1
$-nafla¡ ( b ) 84 70
Tola oh idro quinona (tú -) 204 (d i-) 52
10. Preparación de derivados:
Nojribre dcJ derivado p.f. observad* *C
p.f. reportado "C
M onobrcmo 844 6 84
Acetato 67-68 70
*
1!. Comentarios erpeciales:
E l derívado bromado te preparó por tratamiento con bromo y ¿ d io acético [Smith, / . A»t. Chem. Soc,, 15, 789 (1913)].
La toluohidroqvtnone es improbable por varias rozones. Se ha infor
m ado fácilmente soluble en agua. El cloruro'férrico en solucton concentrada
acuoso da. un color café rojizo y en solución ¿iluida, un color amarillo (Beil.,
V I , 8 74), mientras que la substancia desconocido da Un colar verde (como
lo do el fy-naftol; H u n trttt and M uUiían, p. 234).
Se ha informado <¡ue el $-T¡oftol da positiva la prueba de Tolteñi (Beil.,
V I , €28). Los naftálenos 0> y $-iubstituidos pueden lenef una absorción de
anillo aromático hasta 1650 í m ' 1.

I?. Bibliografía utilizada:
Tablat en este texto; (as r<t{erenc''U ya citadas; BeUamy, T he Iníra-rcd ________ _________ / _______________ _________________________ ___________
in fo rm a s d e su b s ta n c ia s d e s co n o c id a s 35
Spctlra of Ccmplex Xiolccal«s.


Examen preliminar
CAPITULO
3
Estado físico . Se toma nota de la substancia desconocida, si es un líquido o ’*n sólido. Esta, información es útil para consultar las tablas de co m p u es^ (capiculo 11), las cuales están subdívídidas sobre esta base.
Color. He anota el color de la muestra original, así como cualquier cambio de color que pueda ocurrir durante la determinación del punto de fusión o de ebullición (pág. 46).
El color de algunos compuestos se debe a impurezas; éstas frecuentemente se producen debido a la oxidación lenta del compuesto por el oxígeno del aire. Por ejemplo, la anilina ordinariamente es café rojiza, pero una muestra destilada recientemente es casi incolora.
Muchos líquidos y sólidos son realmente coloridos debido a la presencia de grupos cromóforos en la molécula. Muchos compuestos nitrados, quinonas, compuestos azoicos, sales de carbonio (colorantes del triítm l- metano) y compuestos con sistemas conjugados extensos, son coloridos. Si un compuesto desconocido es un líquido estable incoloro o un sólido cristalino blanco, esta información es valiosa porque excluye los grupos funcionales cromóforos* así como muchos grupos que por oxidación podrían convertirse en cromóforos.
O lor. Muchos tipos de compuestos orgánicos tienen olores característicos. No es posible describir olores en unja forma precisa, pero el estudiante deberá familiarizarse con los olores de compuestos usuales. Los alcoholes tienen olores diferentes a Jos de los ésteres; los fenoles, a los de las aminas; los aldehidos, a los de las ectortas. Los mercaptanos, los insonítrilos y la pcntametikndiaauna s e . df:.s¿xihen_ jj.Miatmente^J3>mo_ poseedores.de, jalares--desagradables;, sin embargo, difieren entre sí. ¿ d e más, el olor es más pronunciado, en un grupo determinado, en los miembros de bajo peso molecular, ya que éstos son más volátiles. El benzaldehído, el nitrobenceno y el benzonitrilo tienen olor a aceite de almendras amargas. El eugenol, la cumarina, la vainillina, el salidlato de metilo y el acetato de isoamilo tienen olores característicos que pueden recordarse con facilidad. Los hidrocarburos también difieren en sus

E xo rnen p re lim in a r
olores: tí tolueno, el hexano, «1 isopren^, el indeno, el píneno y el naftaleno poseen olores característicos.
El estudiante deberá observar c u i d a d o ¡ a m e n i e los olores de los compuestos orgánicos usuales empleados en las pruebas y compararlos con los olores de sus substancias desconocidas.
Prueba de ignición. Procedintiúnío, Una muestra de 0.1 g de la substancia se coloca en la tapa de un crisol de porcelana y se acerca a la orilla de una flama para determinar su inflamabilidad. Después se calienta suavemente sobre una flama débil y finalmente se incinera calentando fuertemente. Se hacen observaciones sobre: 1} inflamabilidad y naturaleza de la flama (¿es el compuesto explosivo?) : 2) si el compuesto es un sólido, ver si fundió y la forma de su fusión; 3) olor de los gases o vapores desprendidos ( ¡prcca\iáón!) ; 4) el residuo después de la incineración* ¿se fundirá? Si queda un residuo, se deja eníriar la tapa* se lo añade una gota >. ? agua destilada y la solución se prueba con tornasol. Se le añade una gota de ácido clorhídrico, ¿se desprende un gas? Con un alambre de platino se coma una gota de la solución en ácido clorhídrico para determinar si hay metales.
Ensaye esta prueba con: 1) .etanol, 2) tolueno, 3) benzoato ¿c bario, 4} acetato cúplico, 5) tartrato de sodio y ¡xnasio (Sal de la Ro- ehellc), 6) sacarosa.
Comentarios. Muchos líquidos arden con flama característica que ayuda a determinar la naturaleza del compuesto. Así, un hidrocarburo aromático (que tiene un contenido de carbono relativamente alto), arde con una llama amarillenta y fuliginosa. Los hidrocarburos alifáticos arden con llamas amarillas pero mucho rnenos fuliginosas A medida que aumenta el contenido de oxígeno en el compuesto, la flama se vuelve más y más clara (azul). Sí la substancia es inflamable deberán tomarse las precauciones usuales para la manipulación subsecuente del compuesto, Esta prueba también muestra si deberá determinarse el punto de fusión de un sólido e indica si el sólido es explosivo.
Si después de la incineración queda un residuo, debe examinársele en busca de elementos metálicos. Ya que el numere de elementos metálicos que se encuentran en los compuestos orgánicos no es muy grande- unas cuantas pruebas sencillas determinarán ordinariamente la naturale- 2a del metal presente. Sí la prueba a la flama indica sodio, del>erá incinerarse sobre una lámina de platino en lugar de la tapa de un crisol de porcelana, ¿Por qué?
Pregunta*
J, Nombre algunos residuo) que funden al incinerarse. Nombre algunos que n<> funden.
2. Nombre algunos coinpueítos organometálicos que no dejan residuo a l incinerarse.

II
; CAPITULO
! . 4Determinación
' de propiedades físicasij PUNTOS D E FU SIO N 1
E l p u n to de fu s ió n de un compuesto es el intervalo de temperatura en el cual la fase sólida cambia a liquida. Ya que este proceso se ve acompañado frecuentemente por descomposición, el valor puede no ser la temperatura de equilibrio sino sólo la temperatura de transición de
j sólido A líquido. Si !a prueba de ignición indica que un sólido fundei fácilmente (25 hasta SOO^C), el punto de fusión deberá determinarse por
el procedimiento A. Para puntos de fusión más elevados {300 hasta 500°C) deberá usarse el procedimiento B. Sí !a determinación del punto
| de fusión por el procedimiento A índica una descomposición definida ouna transición de un estado cristalino a otro, se recomienda el procedí-
■ miento C.
Puntos de fusión de mczdas
El método tradicional para probar la identidad de dos sólidos (que deben, por supuesto, tener puntos de fusión idénticos si es que hay una posibilidad de identidad )., ha sido el examen del comportamiento
: v del punto de fusión de una mezcla mecánica de los dos. En general,j una mezcla de muestras de compuestos diferentes presenta una depre
sión del punto de fusión.1 Aunque el uso de puntos de fusión mixtos es valioso en determinados pasos del procedimiento de identificación
I (capítulo 2 ) , el punto de fusión de la mezcla de una substancia desconocida con una muestra conocida de Jas substancias disponibles en
j los anaqueles, no será aceptado en este curso como evidencia para asignarla estructura.
Algunos cuantos pares de substancias, cuando se mezclan, no producen depresión en el punto de fusión^ aunque con mayor frecuencia, la falta de depresión puede observarse sólo en determinadas composiciones de las mezclas. Usando el siguiente método se requiere muy poco
1 Stau, Arthwr y W «kdw», capitula 7 «n fhyritél Jtirthadt oí O rfirñ t Chsmutry, VoJ. T, P iü i I, 3» fd,, «d!»do púr Wtiubcriyer, IpiirKÍcncc Pubfíihcn, Nuevx YftHc. 1959.
39

40 D*t«rmm«icMn d« propíedodo* físicas
esfuerzo adicional para determinar d punto de fusión en mezclas de varías composiciones.
Tome una pequeña porción, aproximadamente iguai, de cada uno de los dos componentes (A y B) que se están examinando. Mezcle la mitad de la porción A con la mitad de B. Ahora, separe la mezcla de A y B en tres partes iguales, A la poniera, añádale el resto del componente A y a Ja tercera, el resto del componente B. Se obtienen así tres meadas con las siguientes composiciones ? 80% de A, 20% de B; 50% de A, 50% de B; y 20% de A, 80% de B. Los puntos de fusión de las tres mezclas deben determinarse simultáneamente por cualquiera de los procedimientos siguientes.
Procedim iento A „ U n tubo de vidrio blando de 45 a 50 cm de longitud y 15 de diámetro, se limpia lavándolo con solución diluida de un detergente neutro, después, con ácido clorhídrico diluido (al 10%) yr finalmente, con agua destilada. El tubo se « c a acercándolo y retirándolo de la parte superior de un mechero Burnen. Este tubo seco y limpio se usa para preparar tubos capilares, de paredes delgadas y de aproxima fia mente 1 mm de diámetro, por calentamiento de una pane del tubo en una llama caliente hasta que el tubo se ablanda y entonces se estira.
El capilar se corta en tramos de unos 6 cm de longitud y el extremo más grande de cada tubo se cierra a la llama. Una cantidad pequeña del compuesto se pulveriza sobre un portaobjetos para microscopio o sobre un vidrio de reloj y parte del polvo se introduce en el tubo capilar! Entontes, el tubo capilar se sostiene verticalmente y se frota suavemente con una Urna, con k> cual el polvo se sedimenta en el fondo. El material te empaca dando golpecitos con la parte inferior del tubo sobre la mesa de laboratorio; después, el tubo se fija al termómetro mediante una liga de bule (caucho) de modo que la muestra quede cérea del bulbo de mercurio (figura, 1), -En el tubo de Thiele se coloca aceite mineral incoloro o acate de semilla de algodón, calentándolo con llama pequeña, Para proteger la llama de las corrientes de aire se coloca un protector cilindrico de metal,2; abierto en las partea superior e inferior» como se muestra en la figura. 1. La velocidad de calentamiento debe ser tal que produzca una elevación en la temperatura ^e 1 ó 2° por minuto. Deberá observarse la temperatura a la cual el compuesto empieza a fundir y aquélla a la cual se licúa. Estos valores se anotan como el intervalo del punto de fusión. También deberá observarse la temperatura registrada por eJ termómetro auxiliar (fa) ; el bulbo de este termómetro deberá colocarse a inedia distancia entre la superficie del aceite y la parte
1 Un* hl* prnwlt j*mc* (1330 g), » 2a que « han quitado bl tsjvil interior yw jw ró r r w » ¿ót o w k h «raode» tü Morrea d t V n 1> y trU iníeriur, ¿r fc la iw nto bkü, Id ata» «juc fctzt m qut x awwtumW» empacar «Utunoaa torao «l p«qí«clgniio de í&íoro.

Pun to s d e fu s ió n 41
superior de la columnilla de mercurio en f3. La. corrección del vástago se calcula por medio de la fórmula;
C o rre c c ió n = + A ; (*i — ¿2) 0 .0 0 0 1 5 4
en donde N = grados de la columnilla de mercurio arriba del nivel del baño de aceite; íi = punto de fusión observado; h = temperatura promedio de la columnilla de mercurio. Esta corrección se añade al punto de fusión observado. Para un compuesto desconocido deberán anotarse canto los puntos de fusión observados como los corregidos.
Frecuentemente x ahorra tiempo haciendo una determinación preliminar del punto de fusión elevando rápidamente la temperatura del
baño. Después de que se averigua el punto de fusión aproximado, se efectúa una segunda determinación elevando rápidamente la temperatura hasta unos 5CC antes del valor aproximado y entonces se continúa lentamente como se describió con anterioridad. Para cada determinación siempre deberá usarse una muestra nueva del compuesto.
Comentarios. La omisión de la limpieza del tubo antes de estirarlo para formar los tubos capilares para fusión, es una de las causas de obtención de puntos de fusión bajos e intervalos amplios. Estos se deben al álcali de la superficie del vidrio, el cual cataliza la co n d es.

J*i?
iJ
i í
ni
r
ír
bs i
42 &?termln«¡6n de propiedades físicas
¿ación aldólica de aldehidos y cetonas, la muiarrotación de les azúcares y sus derivados, ]a descarboxilación de ciertos ácidos y algunas transposiciones'intramoleculares (tales como un desplazamiento de un grupo acüo del oxígeno hacia un grupo oxhidrilo corttiguo). Por ejem* pío, en un tubo <jue no se limpió, la « -g lu c o sa se empezó a reblandecer a 133° y fundió a 14-3-146°, mientras que en tubo dcsalcalinizado el reblandecimiento ocurrió a 142° y la fusión a 146-147°. La 2,3,4fi- tctracetil-<x-D-glucosa.1 se reblandeció a 112° y fundió a 123-126° en un tubo sin limpiar, mientras que el punto de reblandecimiento fue de 135° y el punto de fusión de 137.5-133° en un tubo limpio. La fenacetina, la antipirina y el salol fundieron de 4 a 7o más abajo en tubos sin limpiar que en tubos limpios.1
Una excepción4 fue la sacarosa (p.f. 180-185°), cuyo punto de fusión descendió hasta 150-160° cuando se determinó en un tubo limpiado con ácido clorhídrico. Esto se debió f 'obablemente a la influencia del ácido (silícico) sobre los enlaces ace til ico \
Ftgvru 2. Cvrva «dlbrodAn,
El termómetro siempre deberá calibrarse por observación de los puntos de fusión de varios compuestos puros, tales como:]>.f. (COÍT.) p f. íco rr.)
0 °C hielo 167*0 t o d o hipwifíco3S /Sdielorobcnccno 200 ¡satina90 77í*4initrobí nterw 2 lf> an traccno
114 A cm nüidA 238 carbam licU121 ¿ d d o benzoico 257 oxénilida132 urea 286 ontraquincm *>57 ¿c¡dc* saJiciliro SS2 WJV-d Lacet ílbcncidm a
Si se procura usar el mismo aparato y el mismo termómetro en todas las determinaciones de puntos de fusión, es conveniente y ahorra
1 M orton, I^ iO ra to rj T*ckni<]u< »|t t Chem íítry. McGraw-HilJ Book Co-, N'utvaYork, ¡ m , pí«- 22.
« G «*z. H<\o. CKim. M u , IS, 924 <193».

Pun to s de fu s ió n 4 3
tiempo preparar una curva de calibración co m o la fino se muestra en la. figura 2. El punto de fusión observado para d compuesto usado como referencia, se grafica contra el valor corregido y se traza la curva OA entre estos puntos. En las determinaciones subsecuentes el valor observado, B, se proyecta horizontalmente en la curva, luego se hace descender verticalniente hasta obtener el valor corregido C. Dicha curva de calibración incluye correcciones por las inexactitudes del termómetro y la corrección del vastago.
Rs im p o r ta d atvttar ri in tw a lfl Hri pnntn rf* fmión de un com* puesto desconocido, ya que éste es un valioso índice de pure?a~ Una gian mayoría de los compuestos orgánicos puros funde dentro de un intervalo de 0.5° o funde con descomposición dentro de un pequeño intervalo de temperatura (aproximadamente 1°). Sí el intervalo del punto de fusión,o intervalo de descomposición es amplio, el compuesto deberá recrista- iizarse en un disolvente apropiado y nuevamente se determinará el punto de fusión o de descomposición. Muchos compuestos_or_g¿n_ico£t_talcs
_co$ <^Í!K_aminoácidp.s^Jas_s¿dgsjÍ£_ácidos_0. de_aminas .y los carbohidratos, funden . con descomposición dentro de u n . intervalo dc_ temperatura, considerable.
Presentas
1. ¿Por qué Ja velocidad de calentam iento influye en el punto de fusión?2- ¿Por qué el punto ófi fusión de una m uestra que h a sido ya. fundida a menudo
d a un valor bastante diferente del original?3. ¿Q ué eí un punto mixto d e fuaiSn? ¿C uál t i el valor de tal determinación?
¿H ay algunas excepciones que dejen dudoso c! empleo de puntos mixtos de fusión?
P roced im ien to B. Si un compuesto funde arriba de 250°, su punto de fusión puede deteeminarse mediante la baria de Maquennc. En la figura 3 se muestra un diagrama esquemático de una barra sencilla calentada con gas.
Un termómetro espcü' J (.d ), que registra temperaturas hasta de 500°0, ftc insert^ en la barra de cobre {B), de modo de que el bulbo
Figura 3. Borra «fe Maqvann*.

quede cerca del centro. La parte superior de ia barra de cobre se pule con papel lija. Las llamas que hay debajo de 1* barra se ajustan mediante Jos aditamentos de aire y gas hasta que se produce una flama casi incolora. El compuesto se pulveriza finamente y una cantidad pequeña se esparce sobre ci centro de la barra de Maquenne, a medida que se va elevando la temperatura. Si el compuesto no funde inmediatamente después de haber caído sobre la barra, se le elimina con un pedazo de papel filtro. Después de elevar la temperatura se añade una nueva muestra y este procedimiento de añadir el compuesto pulverizado y quitarlo se repite basta que se alcanza una temperatura a la cual el polvo funde instantáneamente al ponerse en contacto con la barra.
Existen termómetros especiales que registran temperaturas hasta de 50Q°C para usarse con la barra de Maquennc. Por lo tanto, este método es el mejor para todos los compuestos de punto de fusión elevado. Tam bién es muy valioso para compuestos que se descomponen gradualmente a medida que se calientan. La barra de Maquenne da una temperatura de descomposición instantánea, mientras que el método del tubo capilar proporciona un valor que representa el punto de fusión de una mezcla de Ja substancia original junto con cualquiera de sus productos de descomposición, Por esta razón, los puntos de fusión determinados por el método del tubo capilar, frecuentemente difieren de aquellos que íe determinan por el método de la barra de Maqucnne.
P rocedim iento C. Para el estudio de puntos de fusión de compuestos orgánicos se han hecho muchas modificaciones al sencillo aparato
44 D e te rm in a c ió n d e p ro p ie d a d e s f fo ia w
tfpvra 4 , Aporoto para punte» d* t*gún Fithw -Jotat. (Rtpredudtfo por cortttfad« (a F lth tr ScftnKflc C«„ PittiJburgh, Pa,J

Pun to s d e fu s ió n 45
antes mencionada El aparato Fisher-Jobns, mostrado en la. figura 4, tiene una platina de aluminio calentada eléctricamente y provista de un termómetro que registra lecturas hasta de 300°, JUt muestra se coloca entre dos cubreobjetos de 18 min¿ de los usados para microscopio, y los cubreobjetos se colocan en la depresión de la platina de aluminio. La temperatura se regula por medio de un transformador variable y el punto de fusión se observa con ayuda de un foco de iluminación - y un vidrio de aumento. Se debe preparar una curva de calibración para este instrumento, usando como referencia compuestos conocidos tal y como se describió en la pág. 42.
El aparato de Nalge^Axelrod, mostrado en 3a figura 5, consiste en un microscopio de 25 aumentos que tiene insertado un filtro Polaroide,.
Figuro 5 . Aparóle p a ra punte* <i* fo t l ín vtflCn Nale**Jb»lrarf. |l«prad«Kldo j w u¡r~ to iío d* Nalp* Co.. Roch**tíi", N. V.)
□na fuente de iluminación, una platina de aluminio para calentamiento eléctrico controlada mediante un transformador variable, y‘ un termómetro que registra temperaturas hasta de 400°. La raezda se coloca entre ' dos cubreobjetos de 18 mm y estos se ponen dentro de la depresión de la platina de aluminio. La cubierta, que tiene un orificio pequeño, se coloca

J
í»
r»r»
P
:u/—•I
U
¿J
¿ I
r *? •Ce»
46 Determinación de propiedades físicas
en su lugar, se enciende la luz y se enfoca el microscopio- El tubo se gira hasta que casi se cruzan los filtros Polaroide y se observan los cris- , \tales a rpedida que se eleva ia temperatura. Los puntos de fusión de cristales anisótropos se observan fácilmente, ya que los colora de la polarización desaparecen cuando se efectúa la fusión. También se puede observar flciln<ente la transición de una modificación de cristal alotrópico a otra. Debe prepararse., igualmente, una curva de calibración (pág. 42).
PUNTOS DE CONGELACION
Procedimiento* Unos mililitros del líquido se colocan dentro de un tubo de ensayo ordinario que. esté provisto de un termómetro y un alambre agitador (hecho de cobre, níquel o platino). El tubo se sostiene dentro de un tubo de ensayo un ]>oco más grande ^ r medio de un tapón de corcho y se enfria en un baño de hielo o de hielo-sal, o en una mezcla de hielo seco-acetona, y e! liquido se agita vigorosamente (figura 6). Tan pronto como empiezan a formarse cristales, el tubo se saca
del baño y se continúa agitando vigorosamente mientras se lee la temperatura que registra el termómetro. El punto de congelación es la temperatura alcanzada después que ha desaparecido el efecto de sobreenfriamiento inicial.
N o t a . La temperatura deí baño de enfriamiento no deberá estar muy alejada del punto de congelación del compuesto.
Pr«gunuu
Para 3a mayoría de loa compucííOS or- gáfiícos 104 puntos de Cftfigdactón, obtenidos en condiciones ordinariai, no son dignos de confianza, ¿P o r que?
PUNTOS DE EBULLICION
P roced im ien to A . Se prepara un aparato de destilación en «cala peque- ña, como el que se muestra en la figura 7. El aparato consiste en un matraz de destilación de 25 mi, colocado sobre
^ u na placa de asbesto con un orificio de

Pu n to s d e e b u IN c& i 4 7
2 cm en el centro. Se emplean como colectores tubos de ensayo sumergidos en un vaso de precipitados con hielo, para que condense los vapores. Dentro de) matraz se coloca, un tros» de porcelana porosa y se añaden 10 mi del líquido cuyo punto de ebullición se desea determinar. Se inserta el termómetro de tal manera que el bulbo de mercurio quede justamente abajo del tubo lateral. El líquido se calienta hasta ebullición, por medio de una flama pequeña, la cual deberá estar protegida de las corrientes de aire mediante un protector colocado en el lugar adecuado. Se hace todo lo posible para que el líquido destile a una velocidad uniforme. Después que se han recogido 2 ó 3 mi deí destilado, se cambia el colector, sin interrumpir la destilación y tos siguientes 5 6
6 mi se recogen en un tubo de ensayo limpio y seco. Habrá un considerable retardo en la lectura del termómetro, pero ordinariamente se puede determinar el intervalo del punto de ebullición durante la reco- lección de la segunda fracción del destilado. Deberá anotarse crte intervalo del punto de ebullición.
El intervalo del punto de ebullición es un índicc útil de la pureza de la muestra, ivfuchos líquidos orgánicos son higroscópicos y algunos se descomponen con el tiempo. Generalmente los primeros mililitros del destilado contendrán agua o cualquiera de las impurezas volátiles y la segunda fracción contendrá la substancia pura. Si el intervalo del punto de ebullición es grande, el liquido leberá volverse a fraccionar en una columna apropiada.

4 $ D e te rm in a c ió n d e p ro p ie d a d e s f ís ic o s
El punto de ebullición determinado por destilación da una cantidad pequeña de líquido, según se describió antes, cor* frecuencia no es exacto. A menos que se tenga cuidado especial, el vapor puede estar sobrecalentado; también los puntos de ebullición, observados en liquidas de punto de ebullición elevado, pueden se»* demasiado bajos debido al tiempo que se necesita para que el mercurio del bulbo del termómetro alcance la temperatura del vapor. La segunda fracción, recogida en la destilación ya descrita, puede usarse para una determinación más exacta del punto de ebullición por el procedimiento B ó G. Partes de esta misma fracción también deberán usarse para la determinación de la densidad específica, el índice de refracción o la rotación óptica,
P i-o c e d it> ú e n lo Ü , Cuando se dispone de 1 mi de líquido puro y anhidro., se puede determinar su punto de ebullición por medio del aparato mostrado en la figura 8. Este, consiste en un tubo de ensayo ordina
rio, marca Pyrex, de 16 por 150 mm (<4), previsto de un termómetro (Tj) por medio de un corcho (5 ) que tiene una ranura para proporcionar acceso al aire. La copa deí percolador ( C) se construye con un tubo de vidrio de diámetro tal, que haya cerca de 1 mm de claro entre el bulbo del termómetro y Jas paredes de la copa, Usuaimente el tubo de 8 mm resulta apropiado. El fondo de la copa se abre hacia afuera para que cubra la mayor parte del fondo de! tubo, en el cual se han colocado unas cuantas piedras efe ebullición (E ). El tubo de ensayo está aislado por dos capas de papel de asbesto (F ), el cual sobresale unos 5 mm de la altura de la copa. El aparato se prepara en posición vertical, colocando el tubo de. ensayo sobre una placa de asbesto (£>) con un orificio de unos 8 mm de diámetro, Se coloca un segundo termómetro (7*) al lado del primero, para medir la temperatura de la co- lumniüa de mercurio que queda fuera del tubo y así poder calcular la corrección del vástago.
En el tubo de ensayo se coloca un mililitro del liquido y !a llama se ajusta para producir una ebullición moderada. El protector (G) evita el efecto de las corrientes de aíre sobre la llama. El vapor deberá ascender a través de la copa del percolador, arrastrando un poco de líquido, con lo cual el bulbo de
üd r'fWKi mercurio queda en contacto tanto con el líqui-Bltjón. do como con el vapor. El líquido deberá hervir

PWfltOS dft ebullición 49
con fuerea suficiente para que !cs vapores asciendan cerca de 1.0 cm ?.rriba de la envoltura de asbesto ( F ) ; k parte superior del tubo da ensayo actúa como refrigerante de reflujo. L a temperatura (Ti) &c lee después que se ha alcanzado el equilibrio (de 3 a 5 minutos). También se lee el termómetro auxiliar {T2)3 se calcula la corrección del vástago y se añade a la lectura de T j. El punto de ebullición corregido se obtiene con la ecuación siguiente:
p.eb. = Ti + .V (r , - T i) 0.000154
P rocedim iento C . Se prepara un tubo micro para puntos de ebullición, como se muestra en la figura 9. El tubo extemo se hace cerrando un extremo de un tubo de vidrio de 5 mm de diámetro y cortándolo para que quede de 5 cm de largo. U n tubo capilar se cierra a unos Z ó4 mm de uno de sus extren. ^s y se coloca dentro del tubo más grande (figura 9}. Se añaden doa golas del líquido cuyo punto de ebullición se desea determinar y el tubo se fija con una banda de hule (caucho) al termómetro del aparató usado para la determinación de puntos de fusión (ver figura I ) . La temperatura se eleva hasta que, del capilar pequeño, salga una rápida y continua corriente de burbujas que pase a través del liquido. La Dama se retira y se deja enfriar el baño, al mismo tiempo que se continúa agitando vigorosamente. Se anota la temperatura que hay en el instante en que dejan de salir burbujas del capilar y justo antes de que el líquido entre a él. Esta temperatura es el punto de ebullición.
Efecto de la presión sobre el punto de ebullición
Guando se determina un punto de ebullición, deberá anotarse la presión atmosférica que hay en ese momento. Cuando Ja presión está cerca de 760 mm, la corrección causada por una diferencia de presión de 10 mm puede encontrarse dividiendo el punto de ebullición absoluto entre 850, para líquidos no asociados (hidrocarburos, haluroa de alquile, éteres, ásteres), y entre 1020, para líquidos asociados (alcoholes,5 ácidos). La tabla 1 ejemplifica Ja magnitud de tales correcciones barométricas para presiones que no difieran de 760 mm en más de unos 30 mm.
F/gura ful* pora puntal d* «btriH- cí¿rt. Mttedo mlcw.
flfI
n
■i«6
*
<4 l f
< i
n
1 Sañrt» 7 Mctuta» }. Ám. Cirm. $ac., V, 5#7 (1910).

uÍ 9
í i
r»r»t*
r-i
L#
O
É
r»
50 Determinación de propiedades físicos
T a b l a . 1
♦
pj«b.Bhtoluio
Corrección *<* *C paya 16-rom At djfereaci» Mi Jl pTtiiÓn
LIquid» na LÍQUÍdcciKXudg;
50 323* 0.33 0.32100 973 .44 .37150 423 .50 .42200 473 r56 .46300 573 M .56400 673 «79 .665C0 773 .91 .76
Es eviderite que en el trabajo ordinario, las.desviaciones de 760 mm que sean pequeñas, tales como 5 mm, pueden despreciarse
Se han propuesto muchas ecuaciones diferentes para Acular los puntos de ebullición de los líquidos orgánicos a presiones reducidas. Se ha usado la forma integrada de la ecuación de ClausiuS’Clapeyron junto con la regla de Trouton. Se ha encontrado que este método no es satisfactoriOj debido a que la constante de Trouton no es la misma pftra líquidos asociados que para no asociados, además de que el grado de asociación varía con la temperatura. Este hecho ha conducido al uso de las ecuaciones empíricas en las cuales se lian empleado constantes diferentes, dependiendo de la clase de compuestos. Una de las más titiles es la ecuación desarrollada por Hass y Newton 4 y convertida por Bor- denca 7 a una serie simple de ocho gráficas correspondientes a ocho tipos de compuestos.
Los investigadores que trabajan en laboratorios que están en sitios de mucha altitud* y baja presión barométrica, han encontrado conveniente determinar una serie de correcciones empíricas que deberán añadirse a los puntas de ebullición observados para obtener puntos de ebullición a 760 mm. Las correcciones se obtienen destilando varios tipos de diferentes compuestos con distintos puntos de ebullición. La diferencia entre el punto de ebullición registrado en la bibliografía, y el punto de ebullición observado, da la corrección.
Se han hecho una serie de gráficas en coordenadas lineales con los datos de presión de vapor-temperatura de 183 compuestos orgánicos9 y se ha encontrado que estas gráficas son de mucha, utilidad.
Para, dar una idea del cambio del punto de ebullición con la presión,
1 Huí» y N orton, Haadbotk of Ckemíttry anJ P k fjia , Chemical Rvbber Cocipany, Cía- vrh& j, Obi©, I954-19W, pijr, 2119.
T Bortltíica, InJ. Eag. C htm . A*o¡. Jü ., 19» 99 <1946).• Ei\ U cica* del Me. Evnn* <□ C oknids, E.U.A-, «l hierve i 31* a uoa pro»4n
pwmcdio de <60470 eaia y * una «lüjud d« oí.♦ CcmuTUi y Kafcht, U ¿. £ * f 26, « 7 (19M).

Punto* d* ebullición 51
en la Tabla 2 se reportan los datos sobre tres pares de compuestos nú asociados y asociados. Las temperaturas están dadas aproximando a grados enteros.
T a b l a 2
Puntos de ebullición a presiones reducidas
OúljwWítoPrttlífl «l KÚJiíOOtM* flHTCW* T»>.
Tn.7*> 700 « 0 cao « o
n-HepCano 93’C 96 °C 94CC 91 °G 86CC 10*Alcohol n-propSlico 97 95 93 91 89 3Yodobenceno IfsH 135 162 179 m 13Acido ^-valeriánico 186 133 180 178 175 11Fluorcno 298 294 290 286 282 16^-Naftol 295 292 288 264 280 15
Los datos indican que, a medida que se reduce la presión, el punto de ebullición de un líquido asociado no disminuye tanto como el punto de ebullición de un líquido no asociado.
Comentarios-. Los puntos de ebullición de los miembros de una determinada señe homóloga aumentan a medida que asciende la serie. Los puntos de ebullición ascienden de manera uniforme como se muestra en la figura 10:
Número <fe átomo* carbonoFigura 10. l*toe(4n *nlí* lo* p u n ta ó* rbuflUtén y «I p««» nwUcvtar.

52 D*tornilnadón de propiedades físicos
pero el incremento por grupo —CH¿ no es constante, áendo mayor al principio de las series que para los miembros superiores (tabla 3).
TA2LA 3
p A , A ’C
n-pentano 36> 32
tt-hexano 68> SO
n-heptano 98>2?
« o c tan o 125> 24
K-ao&ano 149>24
s-decano 173>21
R-undecano 194>21
H-dodecano 215
Si se substituye un átomo de hidrógeno de uno de los hidrocarburos paxaíínicos por otro átomo o grupo, se produce un incremento en el punto de ebullición. Así, los haiuros de alquilo, los alcoholes, los aldehidos, las cetonas, los ácidos, etc., hierven a mayor temperatura que los hidrocarburos con el mismo esqueleto de carbono.
Si el grupo introducido es de ta l naturaleza que provoque asocia- d ó n , se produce una elevación muy notable del punto de ebullición. Este efecto es particularmente pronunciado en los alcoholes (figura 10) y en loa ácidos, ya. que pueden formarse enlaces o puentes de hidrógeno. Por ejemplo, la diferencia en el punto de ebullición entre el propano (no asociado) y el alcohol n-propíiico (asociado) es de 142° •—una diferencia mucho mayor que la que el cambio en el pes¿£ molecular indicaría. A medida que se introducen más grupos oxhidrilo, el punto de ebullición se eleva más, pero el cambio no es tan grande como el causado por el primer grupo oxhidrilo- El incremento por grupo oxhidrilo es mucho mayor que el incremento por grupo metileno (tabla 4 ) .
T a b ía 4CH, CH,
¿ H , CH,
CH, CHjOH- A S * +w*
CHaOH c h 2o h
CHj CHOH
CHjOH|
CH*OH+ Í16'
tíS* 74*

Pun to» d e e b u llic ió n 53
Si el grupo oxhidrilo se convierte a enlace etéreo» se evita la asociación debida a los enlaces de hidrógeno y el punió de ebullición desciende. Las series siguientes ejemplifican este efecto.
0 1 1 ,0 1 1 C H jO G jH s
CHOH CHOH
GHjOH+2S©°
c h 2o h42SD5’
O H jO C jH j
GHOHI
C H íO C jH s + 191*
C H xO C í H ,
C H O Q H 5i
C H jO C jH s
U na comparación de los derivados oxigenados con sus análogos sulfurados, también muestra que la asociación es un factor más potente
■que el peso molecular. Los tioles (tioalcoholes) están sóJo ligeramente asociados y, por Jo tanto, hierven más bajo que sus análogos oxigenados, aunque los primeros tienen pesos moleculares mayores cjue los últimos.
HOH CHjOH CHjOOOH
p.e b., °C
10066
119
HSHCHjSHGH¿C0SH
p.eb., °C
— 62 + 6
93
Los éteres y los tioéteres no están asociados, por lo que los sulfures de alquilo tienen un punto de ebullición más elevado que los éteres, ya que tienen un peso molecular mayor.
p.eb. °G . p*b. *C
(CH3)aO - 2 4 (CH 3),S . + 3 8(C2Hj) ,0 + 3 5 (C^HjJjS + 92
Estos datos sobre los compuestos sulfurados y oxigenados, y sobre los hidrocarburos, los cloruros, bromuros y yoduros de alquilo, ilustran la regla general de que la substitución de un átomo por otro de mayor peso atómico origina una elevación en el punto de ebullición, siempre que d o
ocurra un aumento o disminución en el grado de asociación como resultado de esta substitución. ,
Exactamente en la misma forma que en las relaciones de solubilidad (ver pág. 86), la arborescencia de la cadena y la posición del grupo funcional influyen sobre el punto de ebullición. Los alcoholes alifá ticos saturados (tabla 5) sirven para ¡lustrar las generalizaciones siguientes:

.rj
5 4 D e te rm in a c ió n d e p ro p ie d a d e s
T a b l a 5
r.Alcflfcolej primario* Akefcotd iCcutlíUnM AlcofaolCf ttroífiai
FécnMjb Ip.cb. *C Fórmula p.tb, *C Fúrjmil* p.«b. °C
caüOB 66CHjCHjOH 70CH*CH:CHLOH 9? CHjCHClíi
¿H
$2
CK>Clt>CHíCHjOK
CHjCHCHjOH
116
1W
CHjCHjCHCKj
¿H
59 CH>C H > -¿-C » b
¿ K
BS
CHj
CHjCHiCHjGHjCIGOH
CHjCHCHjCHjOH
138
131
GKiCffcCHiCHCft»
¿K114
GHi
GHCItCHCMtCIIj
¿ H
115
CHjCHjCHCHjOH¿Mí
m
CH»
C H j-i-C K iO H
¿H>
3H CÍLCH—CHCH,
Í h > ó h
CHj
CHiCHí—G—Oftj ¿ H
ifcz
I« Entre alcoholes isómeros, el isómero de cadena lineal tiene el punto de ebullición más elevado.
2. Si se hacen comparaciones de alcoholes del mismo tipo, entre más arborescente es la cadena, menor es el punto de ebullición,
3, Una comparación de los puntos de ebullición de los alcoholes isómeros primarios, secundarios y terciarios, muestra cjue los alcoholes primarios hierven a mayor temperatura que los alcoholes terciarios, siempre y cuando se comparen alcoholes isómeros con 3a misma longitud de Ja cadena más larga.
Frecuentemente es muy valioso el conocimiento de los puntos de ebullición de los compuestos sencillos pará- excluir determinados tipos de compuestos. Las siguientes generalizaciones son de mucha ayuda:
1: Un compuesto orgánico clorado que hierve abajo de 132CC debe ser aiifátíco. Sí hierve arriba de 132CC puede ser alifático o aromático. Esto es consecuencia de que el haluro aromático más sencillo, el cloro- benceno, hierve a I$2°C.
2. En forma similar, un compuesto orgánico bromado que hierve abajo de 157°C, o un compuesto yodado que hierve abajo de 188°C,

D ensidad esp « jfíca 55
debe ser a]ifático. Otros compuestos bromados y yodados pueden ser ali- íáticos o aromáticos.
P rep o n ías
l* ¿Q ué conreccíonfts pueden hacerse al punto de ebullkiÓB determinado por el procedimiento A ? ¿Por el procedimiento 3 ?
2. U n intervalo pequeño en el punto de ebullición no ea índice seguro de pu n an . ¿Por qué?
DENSIDAD ESPECIFICA
La. densidad específica puede determinarse mediante un pienómetro pequeño» o más convenientemente, mediante un densítómetro de Fisher- Davídson.
P roced im ien to para e l uso del den&Uómetro« El principio de este instrumento se funda en. el hecho de que, si dos manómetros conteniendo líquidos de densidades diferentes están conectados a una fuente de succión común, las alturas de los líquidos son inversamente proporcionales a sus densidades (ver figura 11), por lo que, si en un tubo se coloca un líquido de densidad conocida, se puede calcular la densidad específica del liquido en el otro tubo, midiendo las alturas de los dos líquidos.
El densitómetro Fishér-Davidson ha sido proyectado en tal forma, que sólo varía la altura de un líquido en uno de los tubos; asi, puede obtenerse uria sola lectura directa. El instrumento consiste de dos tubos capilares de vidrio, uno en forma de L y el otro en fonna de Z. Los tubos están conectados en su parte superior a un dispositivo de succión que opera pac variación de la longitud de un tubo de hule oprimido entre rodillos (indicados per r en la figura 11). Si se colocan dos líquidos dentro de los tubos y se hace succión hasta que los meniscos del tubo Z queden en la parte horizontal superior y en la parte horizontal inferior, respectivamente, sólo se observará un nivel variable, es decir, el que corresponde al tubo L. El instrumento se puede construir óon una altura fija pará el tubo Z para que, con el etilbenceno (dens. esp.f ss 0,867) como líquido de referencia, se puede calibrar la escala para leer directamente la densidad esp<-Jfíca.
En el uso ordinario, ei etilbenceno se introduce en el tubo L por medio de un gotero medicina), hasta que el nivel del líquido quede entre las dos marcas que hay u Uia9tama c„ ^ arriba del brazo horizontal. No deben quedar «i* Rsfcw-Davídw».

56 0 •term inoción d e p rop iedades físicas
burbujas de aíre en la columna do líquido; $i aparece alguna, debe eliminarse regresando el etilbenceno al bulbo. Esto se hace dando todas lo: vueltas posibles, en sentido opuesto al de las manecillas del reloj, a la perilla que controla la succión, oprimiendo el hule conector que hay entre los tubos L y Z y después girando la perilla en el sentido de las manecillas del reloj. Después que se expulsan las burbujas de aire, se suelta el tubo y se dan a la perilla todas las vueltas posibles en el sentido de las manecillas del reloj. El tubo Z> el cual debe estar bien limpio y seco, se une y prensa con unas pinzas a las ranuras V. Se nivela el instrumento mediante unos tornillos para ajuste que están colocados en las patas y se introduce el líquido cuya densidad específica se desea determinar, dentro de la copa colocada en el extremo del tubo Z. Se necesitan unos 0.3 mi para llenar la copa.
Para succionar el líquido dentro del tubo Z, se da vuelta a la perilla en sentido opuesto al de las manecillas del reloj, hasta que los meniscos queden colocados aproximadamente a la mitad de los brazos horizontales de las partes superior « inferior. El cursor, con su lente de aumento y venúer, se desplaza had a arriba o hacia abajo, sobre la escala opuesta a l tubo L, hasta que la línea índice quede nivelada con el menisco del etilbenceno. La densidad específica se lee hasta la tercera cifra decimal por medio del verníer • este valor representa la densidad específica a 20°C comparada con la del agua a 4°. Para asegurarse del equilibrio, el líquido que hay dentro del tubo Z deberá desplazarse un poco hacia adelante o hada atrás, dando vueltas a la perilla y verificando Id lectura de! menisco del etilbenceno. Después se regresa la muestra a la copa dando vueltas a la perilla y se elimina medíante un gotero. El tubo Z se desconecta y después se lava con etanol o acetona y éter. Se le seca introduciéndole una corriente lenta de aíre seco. Siempre que 1% temperatura ambiente y la de los líquidos sea de 20 ± 5°C, no es necesario hacer correcciones de temperatura, ya que la mayoría de los líquidos orgánicos tienen coeficientes’ de temperatura comprendidos dentro de un intervalo muy pequeño. Los resultados son exactos con aproximación de 0.1%,
El tubo estándar Z, de diámetro interno de 1.9 mm, es el más apn>- piado para la mayoría de los líquidos. Para líquidos viscosos o soluciones acuosas deberá usarse el de 4 mm y para líquidos de muy baja viscosidad deberá usarse el tubo Z de 0.6 mm.
Con el etilbenceno como líquido de referencia, el intervalo de den- sidades específicas que puede medirse es de 0.600 a 2.000, Para líquidos de mayor densidad específica se usa el tetracloruro de carbono (dens. esp.f = 1.595) como líquido de referencia y la escala de lectura? se multiplica por 1.84.
P rocedim iento para e l picr. 'metro* Si no se dispone de un pic- nómetro pequeño^ con capacidad de 1 a 2 mi, puede construirse uno de los dos que se muestran en las figuras 12 y 13. •

D e n s id a d e sp ec ífica 5 7
Figuro 12. Piwi¿ai«<n>.
E1 picnómetro de 1? figura. 12 se hace con un tramo de tubo de vidrio de diámetro inferno de 1 a 2 mm. Se le dobla en la forma indicada, con un bulbo pequeño soplado en la porción medía y un extremo estirado hasta que forme un capilar fino. En el otrg brazo, y a la misma altura que la punta del capilar, se hace una marca. El picnómetro se cuelga mediante un alambre de diámetro muy pequeño de “Nichro- me”, aluminio o platino y so determina su peso.Después, se llena el picnómetro con agua destilada, un poco más arriba de la marca, y se suspende dentro de un vaso de precipitados con agua a 20<3C. Después de unos 1Ü minutos se ajusta )a cantidad de líquido que hay en el tubo mediante un pedazo de papel filtr' que se acerca a la punta del capilar hasta que el menisco del brazo ’'.'bre coincida con la marca. Entonces se saca el picnómetro del vaso de precipitados, se seca y se pesa.
L a figura 13 muestra un pequeño bulbo para la determinación de densidades específicas. Se hace cerrando uno de los extremos de un tubo
de vidrio de 5 mm y soplando hasta formar un bulbo pequeño. El bulbo puede aplanarse por calentamiento del fondo con un sople Le pequeño y aplacado con una espátula. Se pone una marca en el tubo por medio de una lima o con un grabador. Se de* termina el peso del bulbo. Después, se llena el tubo con agua destilada y se suspende con un alambre dentro de un vaso de precipitados conteniendo agua a 20°C. Se ajusta hasta la marca el nivel del agua dentro del bulbo mediante una pipeta capilar. Entonces se 6aca el bulbo del vaso de precipitados, se seca y se pesa.
El peso del picnómetro vacío y su peso cuando está lleno de agua, se anotan y conservan permanentemente. Para determinar Ja densidad específica de un líquido, se llena el bulbo o el picnómetro con el líquido a 20°C y se determina su peso.
peso de la muestraDens. esp." =: -----------r * peso del agua
El aparato debe limpiarse cuidadosamente y secarse inmediatamente después de usarse.
Debe prestarse atención especial a que la muestra usada para esta determinación sea pura. Lo más conveniente es usar una parte de la segunda fracción recogida en el experimento de destilación (pág, 46).
Iml.
Figura 1 3 . Bulbo poro d m iid s d « i w p i -
cíf lea*.
M LM
í
CJ
U
« i
O
*
t e
*3

D eterm tncrc tón d e p ro p la d a d e * f ís ic a s
Algunas veces es necesario determinar la densidad con referencia a la dcí agua a 4ÚC, Esto puede hacerse empleando el factor 0.99823.
Dens. « * » , x 0.998231 peso del agua
Comentarios. A menudo puede u&arse la densidad específica de un liquido para excluir ciertos compuestos de la lista de posibilidades, La densidad varía tanto con la composición como con Ja estructura del compuesto.
r»
Pa»1*
¿j
LB
ÍMi
NCtti«o de átomo* de carta no
figuro 14. RtJatlw»*» do la dentldo'i otp€<1l¡ea y «I p ito molecular.
Los hidrocarburos generalmente son más ligeros que el agua. A medida que se asciende en una determinada serie homologa de hidrocarburos, aumenta la densidad especifica de sus miembros, petxj el incremento por radical mettleno disminuye gradualmente. Las curvas I,II y I I I de )a figura 14 muestran el cambio de la densidad de los alcanos, de los alquenos-1 y de los alquinos-1. Deberá notarse que la densidad específica del hidrocarburo acetil¿níco es mayor que la de la defina correspondiente, la cual, a su vez, es más densa que el hidrocarburo para'

D ensidad espadfica 59
fínico con el mismo número de átomos de carbono. La posición que ocupa el enlace no saturado también influye en Ja densidad. Desplazando el enlace doble hacia la parte media de la molécula, se produce un aumento en la densidad específica. Los datos de la tabla 6 ejemplifican este cambio.
T abla . 6
Nftaibrí Completo1| Detu. r jf.
Centeno- 1 CHb-=CHCH:CH 3C H 1 0.645?Pcnteno-2 CHjCH^CHCHxCH* 0.65 i rPentad ieuo-], 4 CH»= C HCH jCH ~ C H j 0.659?P e n tad ie n o l, 3 C H j= C H C H = C H C H* Q£96?F<ntadiern>2, S C H jC H = C s=CHCHi 0.702?H cxeao-l C H j= C H C H iC H jC H iCH] 0.573”H exenc -2 C H jC H = GHCH jC H jC H j 0.681?Hexeno-3 CHiCHjCH= CHCH íC H j 0.722?
Al examinar las tablas 35, 36 y 37 del capítulo 11, se observa que la mayoría de los hidrocarburos son más ligeros que el agua.
L a substitución de un átomo por otro de mayor peso atómico, ordinariamente aumenta la densidad. Así, la Curva IV , figura 14, qu„e representa las densidades específicas de los cloruros de alquilo normales (lineales), se encuentra arriba de las curvas de los hidrocarburos. Deberá notarse que los cloruros de alquilo son de menor densidad que el agua y que las densidades específicas disminuyen a medida que aumenta el número de átomos de carbono.
Los datos, bastante limitados, sobre los fluoruros de alquilo, se muestran en Ja curva V, figura 14. Esta curva es interesante debido a que revela sólo un cambio pequeño en Ja densidad a medida que aumenta el-número de átomos de carbono. La curva V representa la etapa intermedia entre Jas curvas I y IV.
Las curvas V I y V i l de la figura 14 muestran que la densidad especifica de los bromuros y yoduros de alquilo primarios es mayor que 1.0 y que en estas penes homólogas la densidad específica disminuye' a medida que se aumenta el número de átomos de carbono. La pendiente descendente de las curvas IV, V I y V II se debe a que el átomo de halógeno representa un porcentaje ¿ \da vez i lás y más pequeño de la molécula, a medida que el peso molecular aumenta por los incrementos de los radicales mctileno. L a posición relativa de las curvas en la figura 14 muestra que la densidad específica aumenta en el orden
R H < R F < R C 1 < RBr < RI

siempre que las* comparaciones se hagan con haiuros de alquilo con el mismo esqueleto de carbono y de la misma clase. Los cloruro», bromuros y yoduros, secundarios y terciarios, manifiestan relaciones parecidas.
densidades especificas de los haluros de ajilo también se ordenan según el peso creciente del substituyente (tabla 7 ).
6 0 D e te fm in a c ió n d o p ro p ie d a d * » f í* ic a i
T a b l a 7
Competo p.íb. *C Den», «p.*
Benceno 75.6 o . mFluorobenceri© 36 1.024Clorobcnceno 132 1.107Broinoberxxoo 156 1.497Yodobenccca 188 1.332
U n incremento en el número de átomos de halógeno presentes en la molécula aumenta la densidad especifica. Los compuestos que contienen dós o más átomos de cloro o un átomo de cloro junto con un átomo de oxígeno o un grupo arilo, generalmente tendrán una densidad específica mayor que la unidad (tabla 8).
T a b la 3
JMM-a p . Compítale D«ai. of>.
C loruro de bencilo 1.1026? Tetracloruro de carbono 1.595?C loruro de bensal 1.25571* C lorhidrina del ctüeno 1.213?Btnzotji ck>mro 1.380% doroacetonA 1,162»C loruro de m*tilano 1.336“ C loroacítato ds m etilo 1-235»Cloroformo 1.498+i*
La introducción de grupos funcionales conteniendo oxígeno causa un aumento en la densidad específica. Las curvas de Ja figura 15 representa o el cambio en la densidad específica de algunos de los tipos comunes de compuestos ( - er pág. *51).
Los éteres (curva VTII) son los más ligeros de todos Jos compuestos orgánicos oxigenados. Los alcoholes alifáticos (curva IX ) son más pesados que los éteres, pero más ligeros que el agua. La densidad específica de los alcoholes se hace mayor que í 0 si se introduce un átomo de doro (clorhidrina del etileno), un segundo oxhidrilo (etílenglicol) o un núcleo aromático (alcohol bencílico).

D e n a id ad e sp e c íf ic a 61
1550
1.200
1.150
1 .1 0 0
1,080
1.060
1j04D
s * 1 .0 2 0
I J.ooo £ 0.930
Z 0 .960•V| awo«= 0 .9 2 0
0 -9 0 0
0 .8 S 0
O.BW
0 ^ 4 0
0 .8 2 0
0 ^ 0 0
a 7 8 0
0 .7 6 0
0 .7 4 0
0 .7 2 0
0 .7 0 00 1 2 3 4
f i g v r a 1 5 . ü t ! « ( í « n n d a d t r o f r t a d H f i t c l f k o y p t t o m e l ic u lo r .
La depresión en la curva IX se debe a que el metanol está mucho más asociado que el etanoh Las aminas (curva X) no son tan densas como los alcoholes y están menos asociadas. La asociación también hace que las densidades específicas del ácido fórmico y del ácido acético sean mucho mayores que la unidad; [os ácidos grasos superiores, líquidos, son máé ligeros que el agua (curva X I).
Loa esteres sencillos (curva. X II) son más ligeros que el agua, mientras que los ésteres de los ácidos polibásicos (curva X III), los ¿ateres
*
5 6 ? a 9 1 0 J ] 12 13 Número Nomos <it carbol»

halogenados, Jos cetoésteresi o los oxi ásteres, son más pesados que el agua. La introducción del anillo aromático también puede J.acer que Jos Asieres sean más pesados que el agua. Ejemplos de ásteres de estos tipos, que son más pesados que el agua, son el acetato de fenilo, el benzoato de metilo, el acetato de bencilo, el salicilato de etilo, el o xa lato de n-butílo, la tri- ace ti na, el cartrato de ísopropjlo y eJ citrato de etilo. Ya que los hidrocarburo# w n mÁA ligero* que d agua, es de esperarse que los ásteres que contienen cadenas largas de hidrocarburo m vcilftn Ja corr«pondiente disminución de la densidad específica.
En-general, los compuestos que contienen varios grupos funcionales —especialmente aquellos grupos que favorecen la asociación— tendrán una densidad específica mayor que 1.0. Simplemente, observando si un compuesto es más ligero o más pesado que eí agua, se obtiene una idea de su complejidad. Esto es de considerable valor en el caso de Jos líquidos neutros. Si el compuesto no contiene halógeno y tiene una densidad especifica menor que 1.0, probablemente no contiene más de un grupo funcional sencillo, además de )a parte de hidrocarburo o de éter. Si el compuesto es más pesado que el agua probablemente es polifuncional.
IN D ICE DE REFRACCION DE LIQUIDOS
E! índice de refracción de un líquido es igual a la relación del seno del ángulo de incidencia de un rayo de luz en el aire, con respecto al seno del ángulo de refracción en el líquido (figura 16). El rayo de luz
62 D e te rm in a c ió n d e p ro p ie d a d e s f ís ic o ?
Figura R«frtKCl¿n á* la tot.

Ind ice «fe re fra cc ió n d« lo s U qu ld oe 6 3
sufre cambies eri la velocidad de onda [VúU - ^ P lt,*l(,B) y en dirección, en. el límite de la interfase, y estos cambios dependen de la temperatura y de la longitud de onda de la luz. No es posible medir directamente les ángulos de incidencia y de refracción, por lo que se han desarrollado sistemas ópticos especiales que dependen del ángulo crítico de reflexión en el límite del líquido con un prisma de vidrio de índice de refracción conocido. El refractómetro del tipo de Abbe opera sobre este principio y existe en el mercado una variedad de estos instnjcoentcs-w Las venia/as del reíractómetro del tipo Abbe son: a) para iluminar se puede usar, una fuente de luz blanca, porque-el sistema de prismas da índice de refracción para la línea D del sodio; b) sólo se necesitan unas cuantas gotas de) líquido; c) tiene incorporado un control de temperatura de los prismas y de la muestra; y d) los prismas compem sadores de Araici permiten la determinación de la dispersión específica.
En la figura 17 se muestra un dibujo esquemático del sistema óptico del refractómetro de Abbe 3L de la Bausch & Lomb y en la figura 18> el instrumento. Se deja pasar agua a 20° a través de las chaquetas ( / ) que rodean los prismas (Pj, P2). Sí la muestra líquida fluye libremente, se le introduce mediante una pipeta a través de uno de los canales que están al lado de los prismas (Z>). Si ja muestra es viscosa, se levanta el prisma superior y se extienden unas cuantas gotas sobre el prisma con un aplicador de madera. Después se cierran lentamente los prismas, expulsándose cualquier c*ceso de líquido. Se enciende la lámpara (L) con los lentes de lucita opaca, conectándola a una línea de corriente alterna de 110 voltios. Mientras se observa en el ocular ( i ) , se da vuelta al tomillo macrométrico {y!) y también se ajusta la. posición de la lámpara {Z.) hasta obtener urr campo iluminado uniformemente. Entonces, se enfoca e) ocular (E ) sobre las retículas cruzadas (H ) y se da vuelta al tornillo (j4) hasta que la linea divisoria entre las mitades iluminada y obscura coincida con el centro de las retículas cruzadas O rdinariamente la linea divisoria aparece coloreada y se acromatiza moviendo el tornillo (Z) hasta que en el centro 11 se obtenga una línea divisoria entre blanco y negro muy bien definida. Este tomillo de ajuste (Z ) hace girar los dos prismas de Amíci {C\,C2) , los cuales compensan las diferencias que hay en el grado de refracción de las luces de diferentes longitudes de onda que componen la luz blanca. Después que la líne^ divisoria queda lo mejor definida posible, se da vuelta al tornillo micrometrico (fl) hasta que la línea divisoria queda exactamente en el centro de las rctícu-
® B&u&H I I LoetO O ptica) G o., R o c h a ¡c r 2 . N.' CuH Z*. J jk ,, Nucv* Y ork 17. N . Y-¡ S p f f l^ r LeuF C o ., Buílalo, N. Y ., V*l<ntiiK> Tcchalcal Inftrw raírtt Co.( SkhoK W Í-»<yk, a.
>» CiertW lEquídtt con diíí>cn¡onr« n u u no d u l UJLi Ií<k» b lm definida de *ep«T*d¿n cirnido W u d K a eléctrica cncrto íu tn tc d e ÜonÚMuJó*. F.n l»l** c t» * , bi tt<opar* d eb e rá b i j a n c y co lo cara u n a hoja de ca « 6 n blanco ei» iasedo UJ, co a rc*pec« «1 p r íw » d e 3 y i i - nucíóii, q u e Ia fcu »olar que en tre por. « n * vmUma, *> r r ík ío « a e l i « tí/m u o a lo . E fto ardí*»* ri.im cule propnrtiófi» u c a llri«a d jv tiorii b ií ll definida a t H»ccr «Jurte» c tiid x io sn e l ta rú flo ¿t dbj>ertí f« (Z)•

64 D etentttnodtón d e p fo p le cS ad e i f im o s
FIj«(q 17 , P teftram a « s ^ t n i t l a » <M iW tn a óptico d#l r*fr<iri6n * l te Abfc« 31. (Captodo <M m anual d« o p m K lo n ti po r to rtfiío d» la B««mh A Lomb Oplical Co)
las causadas, como se muestra en F. Entonces se oprime el interruptor que está al lado izquierdo del instrumento para lograr que se ilumine la escala (S ) . En el ocular (ES) se lee el Indice de refracción para la línea D de sodio hasta la tercera decimal y la cuarta se determina apreciativamente. El resultado se anota en la forma siguiente:
- 1.4357
AI mismo tiempo que se lee el índice de refracción deberá hacerse la lectura del tambor (Z ). Después se limpian los prismas con un algo* dón empapado en benceno o en éter de petróleo para eliminar los compuestos insolubles en agua. Los compuestos solubles en agua se eliminan con agua destilada. Se debe ser extremadamente cuidadoso para evitar que te rayen ios prismas. Deberán evitarse los apücadorts de metal o

Ind ice d e re fra cc ión d e los l íq u id o s 65
vidrio y sólo deber? usarse algodón absorbente limpio y libre de polvo para limpiar los prismas. Siempre deberán consultarse los manuales para operar los diferentes instrumentes, ya que hay variaciones en los procedimientos para manipulaciones exactas. U na discusión completa de los principios de la rcfractometría y los refraelómetros ha sido dada por Bauer, Fajans y Lew¡n.c
F ig v ro 1 8 . jt*fr<jtí¿rci6jr<i d * A b h » 3 1 d # t { C o r í« í a d t l a B e u u f i tUjmb CpH cat Co.J
Comentarios. Les valores de (a densidad y el índice de refracción son útiles para excluir determinados compuestos, entre los que se consi* deran probables, durante la identificación de una substancia desconocida. En iodo caso debe tomarse la precaución de que Ja muestra sea pura, por lo que es mejor determinar esras constantes físicas sobre una parte del destilado que se obtiene en la determinación del punto de ebullición.
>* ® * v ír , F a jw n y L ew io , R cík u S o c tim iy , c a p ítu lo 18, p i j v 1140-1281,« a PhysU ci S í n h o ¿i « / O r f u i t C k t m i n y , Vcp|. I . P t t t t I I . 5* «d ., rtliiado por W á l b t t t » , Inlencienoe FubUifcfirr,
Yorfc, 1960.

66 D e te rm in a c ió n d e p ro p ie d a d e s f ís ic a s
n*
Estas dos constante también pueden servir para una verificación final de la substancia desconocida, una. v«s que se ha aclarado su identidad y su esfructura. En el trabajo de investigación son valiosísimas para )a comprobación rápida de estructuras, Esta verificación se acom- pana con el cálculo de las refracciones moleculares teóricas y observadas. La refracción molecular observada se calcula por medio de la ecuación de Lorenz y Lorentz:
"-{ feO ©en donde n ~ índice de refracción, m = peso molecular, d = densidad. La refracción molecular es la sutna de las refracciones atómicas más las exaltaciones debidas a las insaturaciones, formación de anillos o grupos especiales. Los valores usados ordinariamente son los siguientes.12
Elemento Na, Nitrógeno en Na cC 2.42 Oximas alifátic&s 3.93H 1.10 Amidas 2.60O en OH 1.52 Amidas secundarias 2.2?O en íster OR. 1.64 Amidas terciarias 2.71o= 2.21C1 5.96 Grupo nitro enBr 3.86 Nitratos de alquilo 7.59r 13.90 Nitritos de alquila 7.44S en Sil 7.69 Nitroparafinas 6.72S en R.-S 7.97 Compuestas aromáticas nitro 7.30S en RCNS 7.91 Nitroaminas 7.51S en RjSi 8.11
Grupo nitroso en Nitritos 5.91
Nitrógeno en Nitrosoaminas 5.37Hidroxilaminas 2.4BHidrazinas 2.47 Unidades ntrveiuralerAminas primarías ¿filáticas 2.32 Enlace doble ].?:<Aminas secundarias alifáticas 2.49 Enlace triple 2.40Aminas terciarias ali fóticas 2.84 Anillo de tres miembros 0.71Amina* primarias aromáticas 3.21 Anillo de cuatro miembros 0.41JAminas secundarias aromáticas 3.59Aminas terciarias aromática; 4.36 0 tiranoCianuros aiiíá'iit* 3.05 Terminal 2.02Cianuros aromáticos 3.79 No-terminal 5.05
Conj«ig»ó6n— Valores especiare*. Consulte 3a referencia 12.
Dispersión* Con el refractómetro de Abbe puede usarse la lectura del tambor (Z) junto con el índice de refracción para obtener la. dis
persión del líquido. Este valor, v, es igual a —— • en doiule rij< yn? — »c

I n d io d e refracción d« los líquidos 6?
«c son los índices de refracción para la línea D del sodio, la línea azul del hidrogeno V la. línea roia del hidrógeno, í'esptctivamcnte. Loa valores de dispersión, v, se obtienen por medio de la tabla, de dispersiones pn>* perdonada con cada instrumento y son características para cada compuesto. También puede calculara Ja dispersión parcial, «p — u obtenerse de las tablas, y a partir de ésta puede obtenerse la dispersión específica, S, dividiendo la dispersión parcial entre la densidad obtenida a la misma temperatura. Por conveniencia en. su manejo, ordinariamente se multiplican los valores por 10,000.
s» = x 10*
Se ha encontrado que las dispersiones especificas son muy útiles, ya que los valores son característicos para determinadas clases de compues- tOS-,s Por ejemplo, todos los hidrocarburos saturados (pamfinas, mono-o poli-C.iclopara finas) tienen un valor cercano a 99 ± 1. Las dispersiones especificas no son constantes; por ejemplo, los alquilbencenos varían de 174 hasta 190; los alcoholes, de 68 hasta 90.
Las lecturas obtenidas en el tambor (frecuentemente caracterizadas como valores '*2”) varían con los instrumentos de fabricación diferente. Sin embargo, paja un determinado instrumento estas lecturas quedan dentro de ciertos límites, dependiendo del tipo de compuesto. En uno de los refractómetros de Abbe de la Bausch & Lomb, muchos compuestos alífáticos incoloros dieron una lectura aproximadamente de 17, mientras que la mayoría de los compuestos aromáticos incoloros dieron una lectura de unos 24. Así, la magnitud del valor Z proporciona una posibilidad para distinguir entre compuestos alífáticos y aromáticos. Es necesario determinar para cada instrumento los valores de Z de Jos compuestos alífáticos y aromáticos,” ya que estos varían considerablemente con !a naturaleza del vidrio usado en los prismas Pi y Pj y en los prismas compensadores Ci y Cj. Se han observado ciertas excepciones: el bíSulfuro de carbono tiene un valor Z de 40 y los compuestos insaturados muy conjugados también pueden tener valores de Z mayores de lo que debería esjxrrarse. La regla antes citada debe interpretarse, por lo tanto, como cierta para u>i número considerable de compuestos orgánicos usuales pero no como una generalización infalible.
» r u n i i u » . C o m f t . R t n J . , 17* . 9 » 172. l i t e W .r » m * a • Jimt. ¡'tttíJtvni Ttih., Id. 413 0927)í VHulcri WMem»n r Wtiiei», itté., 21, 66t (195J); Fvrtu T ¡•^¿rrvyn, Jnd. tf>t. C kfm ., SKI ( I W > ; WArd y K uft», 1*4, Htg. Ck*m., AKat.Jfl, 5)9 0 9 3 8 ) ; WiUaut, JtoOfc, L i /l t t tf ’jk . 0*w lw l< V SMnUenbírS, B-ti. T fé v . C k tm ., 5*, 3W ( 1 9 » ) ; O ro** T W ackher, ia ¿ . E n , . C h f -n.> ¿ « f . M - , I I . 614 (19»> .
H En «1 UjH> A(íV« de C u ! ZíitJ >«* alifitlOM d»n «■)« lrt<ya«n «1 um lxir «1» mi*«* I I t k i eemputocji »rriniltie«* un <te 35 [JtfiWiw Jné.Ck4m., Anal. EA., Z. 127 (1930)).

6 4 D e l e n n l n o d ó n d e p r o p i f t d o d e s f í s k a i
RO TA CIO N OPTICA
L a rotación óptica y» determina sólo si la lista de compuestos posibles contiene substancias ópticamente activas.
Preparación de Ja solución
P rocedim iento . U na muestra del compuesto, pesada exactamente (cerca de 0.1 a 0.5 g ), se disuelve e r 25 mi del disolvente dentro de un matraz aforado. Los disolventes usados ordinariamente son: agua, etanol y cloroformo. La solución deberá ser transparente; no debe tener en suspensión partículas de polvo o papel filtro. Si es posible, también debe ser incolora. Si la solución no es transparente, deberá recristalizarse el compuesto original o deberá diluirse hasta 50 mi y filtrarse a través de un pequeño papel filtro teco. Se descartan los primeros 25 m i del filtrado; lo» últimos 25 mi se usan para la determinación.
• Llenado d d tubo del polaríraetro
P roced im ien to , Se atornilla el tapón sobre el extremo pequeño del tubo, el tubo se sostiene verticalmente y la solución se vierte en él hasta que esté lleno y el menisco redondeado se extienda sobre el extremo superior del tubo. La placa de vidrio se desliza sobre el extremo del tu l» en tal forma que do queden atrapadas burbujas de aire y «e atornilla entonces la tapa de latón.
Precauciones
1 . Deberá colocare una rondana de hule entre la placa de vidrio y la tapa d e latón . No hay rondana entie la placa do vidrio y el extremo del tubo de vidrio. Este 5* cierra por un contacto d e vidrio a vidrio.
2. Las tapa* no deberán atornillarse demasiado ajustadas. Se k t apretará lo suficiente para que la unión sea firme y rin fugas. St las u p w t t atornillan detnajiado ajustadas, las placas de vidrio se deform an y se observará rotación aunque no re tenga n ada dentro d ti tubo. Pava substancias con rota* cioner muy bajas « aconseja aflojar laa tapai y apretarlas de nuevo entre lectura y lectura,
3. Si las roscas de meia! se desprenden del tubo de vidrio, i c les puede fijar nuevamente en su lugar por medio de cemento litargirio-glicerina; Al colocar de nuevo una de citas renca*, deberá asegurarte que la párte de vidrio »« extienda l mm m il allá del metal.

R o ta c ión ¿ p l ic a 6 9
Usa del polaiímcfcro
U n ejemplo de polariroetro lo constituye un instrumento del tipo de Lippich, de doble campo. En las figuras 19 y 20 se muestra un diagrama de sus partes funcionales.
figuro 19, Diagrama «t^uan^tlca cí«l pokf(m*tw (itcclón lonpi(td!r>d).
r llama de wdio trica de v>dio.
D = filero de dk ro m aw ? ~ nicol polarkador.Q, = a ju jie de Ja penumbra. t í — nicol de pfiDumbia.T = tubo conteniendo la solución.
A = nicol anaiiiador. E — ocular.S = escala.R = in te rru p to r.
X
F ig u ra 2 0 . tg lo r ím v fc a {v í»1b port»rforj.
E — ocular.Z ~ aju»te del cero.S = escala.C — ajuste macrométrico.
N “ tom illo de cierre.Ai — ajuste mierométxico. X — lámpara.

7 0 D e ta rm tnac tón d « p ro p ie d a d e s f ís ic a s
P rocedim ienio> Se enciende la lámpara (L) mediante un interruptor R> Se'enfoca el ocular (E) haciéndolo girar hacía la derechao la izquierda hasta que la linea divisoria de los dos campos quede perfectamente definida.
Para determinar la lectura cero se afloja el seguro ( j V ) y los dos campos se igualan aproximadamente por medio de la palanca de macro- ajuste (C ), Para usar C es necesario aflojar primero N y luego apretar C dando vueltas a la perilla blanca que está en el extremo de C. t>spu¿a de que aproximadamente se igualan los campos, se aprieta N y los campos se igualan exactamente haciendo girar el ajuste micrométrico (M ) a la derecha o a la izquierda. Se enciendo la lámpara {-AT) y se lee la escala (S ). El círculo principal está dividido en grados y 0.25 de grado. £1 vemier, o escala exterior* está dividido en veinticinco divisiones, permitiendo lecturas hasta de 0,01 de grado. Cuando menos deberán hacersc cinco lecturas y promediarse los valores. Para trabajo muy exacto deberán efectuarse las lecturas de aml>a¿ chalas (5} y promediarse jvara corregir cualquier error en el centrado de la escala con respecto a! ocular y a los Nicols,
Para determinar la rotación de la solución, el tubo de! polariroetro se coloca en el portatubos. Se cierra la cubierta y se repite el procedimiento seguido para la determinación del cero. Deberá tomarse el promedio de cuando menos cinco lecturas. La rotación observada es la diferencia entre este valor y la lectura del cero.
Notas y precauciones
1. Ordinariamente el instrumento está acondicionado para usarse con la luz amariHa correspondiente a la línea D det jodio. La luz de cita longitud de onda se obtiene con mayor facilidad mediante lámparas de jodio calentadas eléctricamente, las cuales producen u^a luz amarilla brillante. También puede usarte una llana de sodio, pero no es Un brillante. Una luz blanca, filtrada a través de la •oluctón siguiente, contenida en una celda de 5 cm, da bueno» resultados en compuestos que tienen pequeñas rotaciones experimentales. La solución para el filtro ejtá formada por 6.9 % de sulfato de cobre (J I) hidratado, 9.4 g de diern- mato de potasio y 300 5 de agua. La solución se filtra y se deja reposar para permitir que se sedimenten las partículas de polvo-
Un arco de mercu/io hace posible el irnpleO de la linea verde deí mismo. Para usar la línea verde del mercurio se quita la lámpara (£.) y se desatornilla el nitro de dicromato de *odío. El arco se pone en substitución de L y se repite el procedimiento anterior,
2 . El instrumento se monta y se ajusta para todo trabajo ordinario. La palanca de penumbra ((2) 0 cl tornillo (Z ) no deberán moverse.
3. Al efectuar tai lecturas es mejor empezar con Ja escala ($ ; ajustada casi a cero. Si se mueve demasiado el brazo (C ), habrá una inversión de los campo*.
4. La palanca (C) y el seguro ( N ) no deberán atornillarse demasiado ajustados. Sólo es necesario darles vuelta hasta que queden firmes.

Rotación óptica 71
La rotación especifica de una substancia se calcula con uaa de las fórmulas siguientes:
Para líquidos puros: Para j eluciones:
r a p» _ r -123» _I* Jo - w í.aJD - íe
en donde [c t^ f — rotación específica a 25° a ¡= rotación observadal = longitud del tubo (en decímetros)
d “ densidadc = gramos en 100 mt de solución.
Deberá notarse que la rotación especifica es bastante sensible a la naturaleza del disolvente y, en determinados casos, aun a la concen' tración de la substancia que se examina. La longitud de onda de la luz usada oara la medición puede, también, afectar tanto la magnitud como el signo de la rotación. Por lo tanto, deberá prestarse atención a ias condiciones exactas bajo las cuales se midieron las rotaciones reportadas en 3a bibliografía.
Estudios extensos han demostrado que una gráfica de la rotación óptica contra la longitud de onda en las regiones visible y ultravioleta del espectro, es mucho más valiosa que lo rotación a una soja longitud de onda. Se ha encontrado que dichas curvas do dispersión rotatoria óptica (DRO) son de inmensa utilidad en la determinación de las configuraciones y las conformaciones de compuestos ópticamente activos.15
La determinación de rotaciones ópticas o curvas de DRO está limitada a los compuestos que son ópticamente activos;16 esto es, las formas ópticamente activas deben encontrarse en la naturaleza o poderse obtener por resolución de formas racímicas sintéticas. Sin embargo, en 1846 Faraday observó que cualquier.líquido giraría la luz polarizada si se le colocaba dentro de un campo magnético poderoso cuya dirección fuera paralela al plano de la luz polam ada.17 Esta actividad magneto-óptica fue estudiada extensamente por W. H. Ferian, Sr. Recientemente se han determinado curvas de dispersión rotatoria magneto-óptica mediante el aparato usado para determinar las curvas de DRO, pero colocando la
11 F i n UrtU u l t n u ¿ íK n jio n del UW d e la 4 l ( $ m iá t l r a t i l o r i l ó p tic a tf) Q ttlm ie* t o r t o lea v e r D je n s i i , O p iiítti I to ia ic r y r í í » r , K ícC raw -H iU JBook C o M K v c v a Yorfc, >9® .
* E JItl , S t tr * o (k m ¡ jir > of C o m p o n e r , M tC n w r-H U l Book C o ., N u ív » Y o d t, 1963.* W nrin$ y C s i le r , DéCeerniajidor» <A t b e F w u d i f E /íe c l , M oJfolo 37 t n P k jñ ta i M t t k o é ,
o / O rfsK Íc CA rM ÍJfry. V a l. I , P i i t e I I I , 3» t d . , e d ita d o p o t W e-Luberfcr, ]n te c s» e p c c P u b lh b e n , N u e v a Yo«*, 15*10.
Expresión de los resultados

7 2 D e le rm ln c ic ttm d o p ro p ie d a d e s f ís ic a s
muestra en un campo magnético. £>ia técnica proporciona a la investí • , gación un instrumento adicional para usarse en el estudio de la conformación de las moléculas.
PESO MOLECULAR
Los métodos usuales para la determinación exacta de pesos moleculares son demasiado embarazosos y tardados para ser usados en e! trabajo de identificación rápida. Para los compuestos que han sido descritos previamente, las determinaciones de peso molecular son innecesarias. En ciertos casos especiales, cuando no se ha preparado un derivado o es desconocido, el peso molecular puede determinarse convenientemente por medición de la depresión del punto de fusión del «¿-alcanfor natural (método de R a s ttt) .
P rocedim iento . Se determina el peso de un tubo pequeño (8 por50 m m ), limpio y seco. En el tubo se colocan aproximadamente 50 mg del compuesto y todo « pesa exactamente. Después se añaden unos0.5 g de alcanfor y el tubo se pesa nuevamente. El contenido del tubo se funde rápidamente con una llama muy pequeña hasta que quede un liquido transparente. (Precaución: N o caliente demasiado tiempo). Después que se ha enfriado el contenido del tubo se le saca y coloca sobre un vidrio de reloj limpio. El material se pulveriza y se determian su punto de fusión por el método del tubo capilar. Deberá usarse un termómetro graduado hasta décimas de grado y con un intervalo de 130° hasta 180°. El tubo capilar en el que se determine el punto de fusión deberá contener una columna del material de sólo 1 mm de altura; la columna se comprime por medio de un tubo capilar más angosto. Se toma la temperatura del punto de fusión en el que la disolución queda totalmente transparente y sin sólido. Se determina el punto de fusión del alcanfor original." La diferencia entre estos puntos de fusión da la depresión que en el punto de fusión del alcanfor produjo el compuesto. El peso molecular se calcula con la fórmula:
w _ 39.7 X P X 1000 A T X P
en donde p = peso del compuesto P ~ peso del alcanfor
AT = depresión en el punto de fusión.
Comentarios. El método anterior se basa en que la depresión molar en el punto de fusión del alcanfor es muy grande. Se ha demostrado
u S ia u , A rth u r y « p lh i lo "i eñ « / O rfenít Chemhtry, 1,P*rtt I. 5* od., ediudo *or Weájlxrsrr, XalrrKlcace PwbluHen, Nu«v* York. I9W.
n M e td m ra , S * x er j Jw w j, J . A m . C *<m . S c c ., 6 5 | 5023 (1 9 i9 ) ; R ic o , M J . . W , 65«

P«so m o le cu la r 73
que el valor de 39.7 para la constante de depresión melar K del ¿-alcanfor se cumple en concentraciones de soluto mayores que 0-2 molar. Las soluciones más diluidas originan un aumento en la constante hasta de 50, aproximadamente. Consecuentemente, es necesario hacer una determinación preliminar del peso molecular de una substancia desconocida y calcular un peso molecular aproximado. Después, se efectúa una segunda determinación, usando tal cantidad del soluto que resulte una soluciones (140 a 176^0). En tales casos deberán seleccionarse otros disol-
Este método del alcanfor de Rast, en algunas ocasiones proporciona pesos moleculares incorrectos debido a asociación de las moléculas del soluto 0 a descomposición a Jas temperaturas en que se efectúan Jas diso^ luciones {140 a 176°). En tales casos deberán seleccionarse otros disolventes que tengan una depresión molar elevada del punto de congelación. Algunos ejemplos de ellos son los siguientes:
p¿.Dctjro&n
nioUf*c {*>
C anfina 49.0 31.03Ciclchexano 6.5 20.0Cidopentadecanona ■ 65.6 21.3T etrahíd rotiofeao'diúxído-l ¡ 1 27,0 66.0TríbromofMioJ 96.0 20.4
Se pueden obtener resultados de mayor confianza determinando (a relación de la depresión del punto de congelación de la substancia desconocida con la de una substancia conocida, en el mismo disolvente. El compuesto de referencia deberá tener la misma estructura general y grupos funcionales, y aproximadamente él mismo peso molecular. Ver referencia D, pág. 250, para los procedimientos de determinación de pesos-moleculares y su discusión.
i I
esf i
TP
5

í>
í t
r*r>n
í i
¡>
I

CAPITULO
Análisis cualitativo elementalLos e l e m e n t o s QUE o r d in a w a m r n t e se e n c u e n t r a n j u n t o c o n e l car^ b o n o , e l h id r ó g e n o y c t o x íg e n o , s o n : n i t r ó g e n o . ,S / :u f r c > - f iú o r . - c lo r o y.
La ideo tí:' ación de estos elementos se basa en su conversión en compuestos iónicos solubles en agua y en la aplicación a éstos de pruebas específicas.
FU SIO N DE LOS COMPUESTOS ORGANICOS CON METALES
Precaución. Algunas clases de compuestos orgánicos, tales como los nitroakanos, a2idas orgánicas, dmoésteres, sales de diazonio y algunos polibalucoe alifáticos (cloroformo, tetra cloruro de carbono), reaccionan explosivamente con el sodio o el magnesio calientes. Siempre deberán usarse anteojos de seguridad cuando se realicen estas descomposiciones.
Si se escucha un estallido o se produce una explosión cuando se ha calentado con el sodio la primera porción de la substancia desconocida, se interrumpe la manipulación y aproximadamente unos 0 5 g de la substancia desconocida se reducen, hirviéndolos suavemente con 5 mi de ácido acético glacial y 0.5 g de polvo de cinc. Después que se ha disuelto la mayor parte deí cinc, la mezcla se evapora a sequedad y, entonces, todo el residuo se desco>n|>one por los procedimientos A 6 fí, siguientes;
P roced im ien to A, Sodio} Un tulw de «n#ayo peqiu:ño (50 por 8 mm) *c coloca en posíóón vertical sosteniéndolo con una* pinza*. Se introduce en el tubo de ensayo un trocíto cúbico de sodio metálico limpio,
1 P i r a u j i í b u to » ía M r t 'i f tK u ín d» Ib p ru tb * d e L a n í l^ n * p*W d n c a m p to lc 'i fa c o a to d lo , v í* CkmpHíQ j Cart^bíl], / - C k tn . £dnc.t 27, 2GJ {1950).
.NaCNS

7 6 A n ó l b í s c u a l i t a t i v o * l « n t e n l a l
de unos 4 mrn por lado. La porción inferior del tubo se calienta hasta que ce funde el sodio y sus vapores empiezan a elevarse dentro del tubo. Entonces se añaden 5 rng del compuesto, previamente mezclados con un peso igual de sacarosa, y de nuevo se calienta el tubo. La adición y el calentamiento se repiten por segunda vez y entonces se calienta el fondo del tubo a[ rojo vivo. Se deja enfriar el tubo y se le añade 1 mi de etanol pata disolver el sodio que no hubiera reaccionado. El tubo se calienta de nuevo y, aún caliente, se le deja caer dentro de un vaso de precipitados pequeño que contenga 20 m) de agua dfcstilada (¡precaución!). El tubo se rompe con una varilla de vidrio y la Isolodón se calienta hasta ebullición y se filtra. El filtrado, que deberi ser incoloro* se usa p^.ra las pruebas específicas que se describen a continuación.
P rocedim iento B. Magnesio y carbonato áe potasio.1 La mezcla de fusión se prepara triturando juntas 2 partes de carbonato de potasio an’ fdró y 1 parte de polvo de magnesio. Cerca de 0>l g del compue&to que ; ' va a analizar se coloca en el fondo de un pequeño tubo de ensayo seco (3 X 50 m m ). Si la substancia es un líquido, es mejor añadirla por medio de una pipeta, para evitar que se mojen las paredes del tubo de ensayo. Con unas pinzas el tubo se coloca en un ángulo de 30° con respecto a la horizontal y la mezcla de magnesio-carbonato de potasio se hace descender por la pared del tubo de ensayo. Esta mezcla deberá extenderse por unos 3 cm a lo largo del tubo y quedar a 1 cm de la muestra. Se dejan caer dos gotas de éter sobre la muestra y la boca del tubo se cierra con un tapón de lana de vidrio. ‘ ,
El calentamiento de la mezcla se empieza con el mechero Bunsen cerca de la boca del tubo. Cuando la masa se empieza a poner incandescente., la llama del mechero se desplaza hacia la narte inferior tíel tubo para que destile el compuesto sobre la masa incandescente.
El compuesto que e s e n el Cubo de ensayo no debe calentarse h^sta que no haya empezado la reacción entre el magnesio y el carbonato. La mezcla de magnesk>*carfconato de potasio debe estar al rojo incandescente.
Se continúa el calentamiento y, finalmente, todo el tubo se pone al rojo vivo. Entonces se deja caer el tubo dentro de 20 mi de agua destilada contenidos en un vaso de precipitados. El tubo se rompe, Ea mezcla se agita vigorosamente y después se filtra. El filtrado se usa para las pruebas subsecuentes.
Pruebas específicas para los e lem en tos. Azufre. Unos mililitros de la solución anterior se acidulan con ácido acético y se añaden unas gotas de solución de acetato de plomo. U n precipitado negio de suliuro de plomo indica azufre.
A otro mililitro de la solución se le añaden 2 gotas de una solución de nkroprusiato de sodio. U na intensa coloración violeta rojiza indica azufre.
* Buiej- y BnxVrtituí, W , E * ( . C htm ., A ñ t í . Ed„ i» 135 (19Í7).

. , Nitrógeno, a) Unos 3 ni] de la solución madre ^ acidulaiJ^óS? ¿ado. .acético» s<“. añaden dos gotas de una solución, preparada reciente- n25Ste,,de,ben.cidin^.^I.l%..en.áddo..acético..al.5O%>¡w0^ri3da se agita. L a adición de 1 gota, de una solución al 1% de suBiffp.de cobre f l l ) produce un color azul o un precipitado azul si hay nitrógeno presente. Los cloruros y bromuros no producen colorea, pero los yoduros dan un precipitado verdoso. Si tanto el nitrógeno (p.ej., cianuro) como los yoduros se encuentran presentes, el precipitado es azul.
b) Se ajusta el pK de 1 mi del filtrado a 13, medido con papel Hydrion E. Se añaden dos gotas* tanto de una solución saturada de sulfato de fierro (II) y amonio, como de una solución de fluoruro de potasio al 30% }' Ia solución resultante se hierve cuidadosamente durante vinos 30 segundos, La solución caliente se acidula con precaución añadiéndole ácido sulfúrico al 30% gota a gota justo hasta que se disuelva el hídróxido de hierro, £1 exceso de ácido puede ser perjudicial. La aparición del precipitado característico de a2ul de Prusía indica la presencia de nitrógeno.
Es'e precipitado puede observarse mejor si se recoge y lava sobre un papel filtro blanco. Si no se observa precipitado y se obtiene una solución azul o azul verdosa, es probable que la descomposición inicial con sodio no haya sido completa,
c) Unas 2 gotas de solüción de polisulfuro de amonio se añaden a2 mi de la solución madre y la mezcla se evapora hasta sequedad en baño de vapor. Se añade ácido clorhídrico diluido (5 mi) y la solución se calienta y se filtra. Se añaden unas gotas de solución de cloruro férrico al filtrado. U na coloración roja indica nitrógeno.
d) Unos cuantos cristales de nitrito de sodio se disuelven en 3 mi de la solución madre, se adicionan 2 gotas de solución de cloruro férrico y la $o!ución resultante se acidula con ácido sulfúrico diluido. L a mezcla se calienta hasta ebullición, se alcalinúa con hidróxido de amonio y se filtra* Al adicionar al filtrado 1 gota de agua saturada con ácido sulfhídrico o un sulfuro alcalino, se produce un colpr violeta si hay nitrógeno presente.
En ocasiones la prueba para nitrógeno no es satisfactoria, peno frecuentemente se pueden obtener indicios de su presencia como resultado de las determinaciones de solubilidad o reacciones de clasificación» Es particularmente difícil obtener una prueba positiva de nitrógeno a partir de compuestos nitrados. El grupo nitro puede dilucidarse debido a que la mayoría de ios compuestos nitrados dan un intenso color rojo con los álcalis. Muchos compuestos orgánicos que contienen nitrógeno desprenden amoniaco cuando se calientan en un tubo de ensayo pequeño junto con cal sodada. El amoníaco puede identificarse por su olor o por medio de un pedazo de papel rojo de tornasol, húmedo, colocado en la boca del tubo.
Fu s ión d e lo> co m p u e s to s o rg á n ic o s con m e ta le s

.. /■ íá í » r §
r§
t »
=•
a»i
i
^A nótisis cv>elftotív« ola mental
____ oj, a j Unos 2 mi de la soUicíón se acidulan con ácido ni*VrííoViSuido y se hierven suavemente durante unos minutos para expulsar lo que hub iera^^ácido cianhídrico o ácido sulfhídrico. Se añaden unas gotas de solucwjlsde nitrato ...de...plata. Un precipitado denso indica U_presencia de cloro, bromo o yodo. El cloruro de plata es blanco, d bromuro de plata es amarillo pálido y el yoduro de plata es amarillo. Si sólo se produce una débil turbiedad u opalescencia, es probable que se deba a la presencia de impurezas en los reactivos o en el vidrio del tubo de ensayo usado en la descomposición inicial con sodio.
b) Prueba de Bcihteiix. En el extremo de un alambre dé cobre se forma un anillo pequeño y se le calienta en la llama de un mechero Bunsen hasta que la llama quede incolora. Se en/na el alambre; el anillo se introduce en el compuesto original, tomando un poco y se calienta en la orilla de la llama del mechero. U na llama verde indica halógenos.
Esta prueba es extremadamente sensible y siempre deberá confirmarse con la prueba del nitrato de plata, ya que son suficientes pequeñísimas huellas de ím pum as conteniendo halógeno? para que se produzca una llama verde. Los líquidos muy volátiles pueden evaporarse totalmente antes de que el alambre pueda calentarse lo suficiente para producir la descomposición, por lo que la prueba falla.
Se ha indicado que ciertos compuestos que tío contienen halógenos hacen que la llama adquiera un color verde. Entre estos compuestos están los derivados de la quínoleína y la piridina, los ácidos orgánicos,5 la urea y el cianuro de cobre,*
c) Bromo y yodo. Unos 3 mi de la solución de la descomposición con sodio se acidulan con ácido sulfúrico y se hierven durante algunos minutos. La solución se enfría y se introduce 1 mi de tetracloruro de carbono; entonces se añade una gota de agua, de cloro/ preparada recientemente. La producción de un color púrpura en el jetracloruro de carbono, indica yodo. La adición de agua de cloro se continúa gota a gota, agitando Ja solución después de cada adición. El color púrpura desaparecerá gradualmente y será reemplazado por uno café rojizo, si hay bromo presente,
d) Bromo. A 3 mi de la solución madre, colocados en un tubo de ensayo, se le añaden 3 mi de ácido acÉtko glacial y 0.1 g de dióxido de plomo. Se coloca sobre Ja boca del tubo de ensayo un pedazo de papel filtro humedecido con el reactivo de fucsina-aldehido (pág, 145)^ y el contenido del tubo se calienta hasta ebullición. Si hay bromuro presente en la solución, los vapores de bromo virarán eS color del papc-1 a violeta. Los cloruros y cianuros no producen ningún color. Los yoduros dan un color azul.
5 G iltnm i ) ■ licfcy. / - Á m . C k tm . S o e .. 51, 3575 (1 9 2 9 ),4 V n - v jo A frh cB . R t f . ÍVffi1, CAi’m ,, 5 2 , 567 <3953).'■> Carrx» «l «tfia de duro m<¡ K cohícrv* blfn, pl*«d« i»»rse un b!j>ocV>f»ta ¿ t *od¡0 fK»-
Wlízaik», caoio el ''ChVwox”, jkio (a JO Ilición de «ivj»ya debe mbfllencrx: íclda al papel loraa»!.

Fustán d e lo s com puesto* o rg án ico* co n m eta la* 7 9
E n . lugar del reactivo de fucsina-aldehido puede usarse un peda») de papel filtro empapado en una solución alcohólica de íluoresceín» al1 %. Los vapores de bromo hacen que la fluoresedna amarilla se vuelva rosa debido a la formación de eosina. Los cloruros y cianuros no interfieren con esta prueba. Los yoduros dan un color café.
c) Cloro. Si las pruebas anteriores para bromo y yodo son negativas y e l nitrato de plata produjo un buen precipitado, indica la presencia de doro. Si se ha encontrado que hay bromo y yodo, para identificar cloro deberá usarse uno de los procedimientos siguientes.
f ) Cloro, bromo y yodo. Unos 1.0 mi de la solución madre se ad- . dulan con ácido sulfuríco-diluido v se hierven durante algunos minutos.
La solución se enfria y se investiga si hay yodo añadiendo ,QJi_JE!jCÍe tetraclonixo. de carbono a 1 mi de Ja solución y unas gotas de solución de mtritft <j&_godfa. U n color púrpura indica yodo. Si hay yodo presente, el resto de la soludón se trata con nitrito dé sodio y el yodo se extrae con tetracloruro de cjir. \nq. Finalmente, la solución se hierve durante un minuto y después Se en fr \ A 1 mi de esta solución se le añaden 0.5 mi de tetracloruro de carbono y 2 gotas de a g ^ r o , 5 Una coloradón café indica bromo. El resto de la solución se diluye hasta 60 m i; entonces, se le añaden 2 mi de ácido Jtúíúrfco cQiiccflirídft y 0¿g jl£ j^s.q1fa tp jifi. potasio, y la soludón se hierve durante 5 minutos. Después que la inepcia
■ se íia enfriado, se añade una solución de nitrato de n lata: un precipitado blanco indica doro.
También puede usarse un procedimiento semejante para identificar cloro, en el que el peróxido de plomo y el áddo acético substituyen a!
* persulíato de potasio y al áddo sulfúricOj como agentes oxídan{£S.. . g) Cloro fin presencia de nitrógeno/ azufre, bromo y yodo. Aproximadamente 10 mi de la soludón madre se acidulan con ¿cido nítrico.
, diluido y se hierven para expulsar el ácido cianhídrico y el áddo sulfhídrico . Se añade suficiente nitrato _de plata para precipitar totalmente tocios los halógenos como haiuros de plata y se separa el precipitado. S¡
, ‘ tanto el nitrógeno como el azufre están presentes, el precipitado se hierve durante 10 minutos con 30 mi de ácido nítrico concentrado para destruir Cualquier sulfoci^nuro de plata que pudiera estar presente. Entonces se diluye la mezcla con 30 mi de agua destilada y se filtra. Después, el precipitado de baJuro de plata se hierve con 20 mi de hidróxido de-sodio a l_0,l3fc durante 2 minutos. La soludón se filtra y el filtrado se acidula con ácido nítrico y se añade solución de nitrato de piafa. U n precipitado blanco indica cloro.
h) Flúor. Unos 2 mi de la $oluc¡ón madre se acidulan con ácido acético y la soludón se hierve y se enfría. U na gota de la soludón se coloca sobre un trozo de papel reactivo de arcomo-alisarina. Un color amarillo sobre el papel rojo indica la presencia del fluoruro. El papel reactivo se prepara, empapando un pedazo de papel filtro con una solución compuesta de 3 mi de solución etanólica al 1% de ^lizarin'a y 2 mi

de solución al 0.4%- de clorun> (o nitrato) de zírconio. El papel filtro rojo se seca y, justo antes de usarse, se humedece con una gota de ácido acé tk o _ a lJ4 )^
« » FUSION DE COM PUESTOS ORGANICOS CON CINC-OXIDO DE CALCIO
Cuando se desea comprobar ia fusión con sodio se usa un método alternante para efectuar e! análisis cualitativo de los elementos; el método se basa en que la fusión de un compuesto orgánico que contiene nitrógeno, halógenos y azufre, con una mezcla de óxido de calcio y cinc metálico, convierte el nitrógeno en amoniaco, los halógenos en ion haluro y el azufre en ion sulfuro. Estos se identifican por los métodos ordinarios de la ^ roñica analítica.*
flO A n á t is i» c u a lita t iv o e lem e n ta l
• B r tiM t, G t t íd , Sw iU 7 N ic p w a n . A * d . C W , 1». 1 0 » (1 9 4 7 ),

CAPITULO
Comportamiento de solubilidad
T ré s c l a s e s GENf.ftALP.S df- in f o r v ía c ió k pueden obtenerse acerca de una substancia desconocida por eí estudio de su comportamiento de solubilidad en varios líquidos como: agua, solución de hidróxido de sodio al 5% , solución de bicarbonato de sodio al ácido clorhídrico al 5% y ácido sulfúrico concentrado. Primero, frecuentemente se señala la presencia de un grupo funcional. Por ejemplo, ya que los hidrocarburos son insolubles en agua, el simple hecho de que una substancia desconocida, tal como el éter etílico, sea parcialmente soluble en agua, indica que está presente un grupo funcional. Segundo, la solubilidad en ciertos disolventes a menudo conduce hada información más específica acerca del grupo funcional, Por ejemplo, el ácido benzoico es insoluble en el disolvente polar, agua, pcrí> es convertido por el hidróxido de sodio diluido en una saJ> el benzoato de sodio, que es fácilmente soluble en agua. En este caso, la solubilidad en una solución de hidróxido de sodio al de una substancia desconocida que es ínsoluble en agua, es una poderosa indicación de un grupo funcional ácido. Finalmente, en algunas ocasiones pueden hacerse ciertas deducciones acerca del peso molecular. Por ejemplo, en muchas series homologas de compuestos moaofuncio- nales," los miembros con menos de cinco átomos de carbono son solubles en agua, mientras que los homólogos superiores son. insolubles.
Primero se determina la solubilidad de los compuestos en aguü. -Los compuestos solubles en agua se dividen en tres H as^ principaW- 1) compuestos ácidos que dan soluciones que viran a rojo el papel a?ul de tornasol y 21. edmcuestos básicos que viran a azul el papel roio de tornasol; 3) compuestos neutros y ácidos y bases lo suficientemente débiles para no virar el papel tornasol. Al considerar la solubilidad en agua, se dice, arbitrariamente, que una substancia es “soluble1* si se disuelve cuando menos en la relación de 3 g por 100 mi de disolvente. Éste patrón está dictado por las limitaciones inherentes ai método empleado, el cual se basa en observaciones, visuales semicuantitativaa, burdas, como se verá posteriormente. Es necesario ser cuidadoso en la interpretación de
<4
<4
M
3 1

62 Comportamiento d» solubilidad
A
J»
M
9
B
las cJasi/icaeiones de “solubles” e "iusolublcs” en otros casos, ya que ha menudo se usan definiciones diferentes de « ta s palabras.
Cuando, se está considerando la solubilidad en ácido o base, la observación significativa que debe haccise no es la de si la substancia desconocida es soluble cuando menos en un 3$> o cualquier proporción arbitraria, sino más bien, si es notablemente más soluble en áci¿o o baso acuosa que en agua. Este incremento en la solubilidad es la prueba positiva deseada para un grupo funcional básico o ácido.
’T t t t t i w l ro jf.
Sol. íonuM muí
Agua
InaoJ.
¡Tof a a w l lanK o& do
Sol. NaHCOj
So].
Jnsol.
N aO H Sol.
InsoU
HCl
Jnsol,a p o 4 Sol. ¡
So).Insol. j
HtSO*
Insol.
Acidos solubles en agua (pX 4 < 3)
Bases solubles en agua (pKj, < 9)
Compuesto» neutros solubles enagua, ácido* débiles y bases débiles.
Acido* fuertes
Acidos débiles
Compuestos básicos
.Compuestos neutro* varios
Compuestos neutros
Compuestos inertesF ig u ra 2 1 . R « » u m en d e la s p rw ^ fa a i 4 * l o lu b í ü d e d .
Los compuestos ácidos se reconocen por su solubilidad en hidróyido de so^’o al 5$>. Los ácidos fuertes y los débiles se diferencian por la solubilidad de los primeros, pero no de los segundos, en un disolvente débilmente básico, bicarbonato de sodio z\ 5% , Los compuestos que se comportan como bases en solución acuosa se identifican por su solubilidad en ácido clorhídrico al 5% y, en general, no se hace ningún intento para diferenciar entre b?.:s fue/ccs y débiles1
* Par* un m£to<J<> qut pcroAiui eícctiuí tftj jjií«rtl'ciad6a v«r P c l ía je a , / . O t f - C h tm-, M , 1171 (1 5 5 9 ).

D «fa rm ¿rx jaóo d# to J y b rW o d e s 83
Muchos compuestos que son neutrus aun ea soluciones acuosas fuertemente acidas, se comportan como bases en disolventes más ácidos, tales como el ácido suIflírico concentrado. En general, los compuestos que contienen azufre o nitrógeno tienen un átomo con un par de electrones no compartido y es de esperarse que se disuelvan en esos medios fuertemente ácidos. Por lo tanto, no se obtendrá ninguna información adicional mediante la observación de tales solubilidades y, por esta razón, cuando eí análisis elemental ha demostrado la presencia de sutufre o nitrógeno, no deben efectuarse pruebas de solubilidad más allá de las de acidez y basi- cídad en solución acuosa.
La mayoría de los compuesto» que son neutros en agua y contienen oxígeno en cualquier tipo de función, se comportan romo |>?w* medianamente fuertes en ácido sulfúrico concentrado. La solubilidad en él, o cualquier otra evidencia de una. reacción con este reactivo, indica un átom a.d¿j^eaQ jtL.otx^inníÚ én-reactiva en un hidrocarburo, por-ejem- plO, .un ,enlace _olefínico .o un aniUs aromático Jáciimcnte.sulfonable.
Ya que el comportamiento de solubilidad de los compuestos solubles en agua no proporciona información acerca de la presencia de grupos funcionales ácidos o básicos, esta información debe obtener» por ensayo de jus soluciones acuosas con papel tornasol. En general, el comportamiento de un ácido en ácido clorhídrico al 5% y el de una base en solución de hidróxido de sodio, deberá examinarse rutinariamente, ya que la molécula puede tener grupos funcionales tanto ácidos como básicos.
En las págs, 86-94- se presenta una revisión más detallada del comportamiento de solubilidad.
DETERM INACION DE SOLUBILIDADES
A Experimentos sobre compuestos conocidos-J i • ^
Coloque 0,2 mi (0.1 g de un sólido) del compuesto en un tubo ce ensayo', y añada, en porciones, 3 mí de agua. Agite vigorosamente después de" la adición de cada porción del disolvente, poniendo especial atención en mantener la mezcla a la temperatura ambiente. Si el compuesto se cfisuclvc totalmente, anótelo como soluble.
Ya que los pesos nxdeculares de muchos compuestos orgánicos comunes se encuentran entre 50 y 300, una concentración de 3 g por 100 mi es de 0.1 a 0-6AÍ. Deberá recordarse que cuando se disuelve un ácido orgánico en agua pura, la concentración de iones hídronio de la solución resultante está dada aproximadamente por la ecuación:
[H jO 'l = V K £
en donde K a es la constante de disociación ácida y C la concentración estequiométrica del ácido. El pH de la solución de un ácido soluble,

obtenido en la prueba de solubilidad eu agua, será aproximadamente Igual a pKrf/2. Consecuentemente, los fenoles, con pK a cercano a 10, dan soluciones acuosas muy débilmente ácidas (pH ahededor de 5) para virar el papel a rojo de tornasol.2 Por razones similares, una amma aromática, por ejemplo, la anilina, es una base demasiado débil (p/úi 9.4} para que en solución acuosa vire el papel tornasol a azul. Aunque se pueden emplear procedimientos más refinados usando un papel indicador de pH tal como el papel “Hydríon”, los problemas para obtener agua de pureza adecuada y eliminar el anhídrido carbónico son suficientes para que sea más conveniente, corno regla general para dilucidar fenoles, confiar en pruebas como la del cloruro férrico, que se discutirá en el capítulo 8.
P ara ensayar la solubilidad en soluciones diluidas de hidróxido de sodio, de bicarbonato de sodio o de ácido clorhídrico, agite perfectamente la mezcla, separe (fili,* si es necesario) la solución acuosa para eliminar cualquier substancia desconocida que no se haya disuelto y neutralice con ácido o base. Examine muy cuidadosamente la solución buscando señales de separación de la substancia desconocía original. Aun la aparición de una turbiedad en el filtrado neutralizado constituye una prueba positiva.
M ediante el procedimiento anterior determíne la solubilidad de .c a d a uno de los compuestos siguientes: 1) tolueno, 2) alcohol bencílico,
3) acetato de etilo, 4-) acetanilida, 5) sacarosa, 6) benzoato de etilo, ?) di- oietiíanilina, 8) ítalimida, 9) benzonitrilo, 10) ácido antranílico. Tabule 1» resultados como se muestra en la tabla 9. En esta tabla, + indica solubilidad, — insolubilidad y * caso dudoso o en el límite de solubilidad. Todos los resultados del último tipo deberán verificarse mediante una segunda determinación.
Comentarios. Los sólidos deberán estar pulverizados finamente para aumentar la velocidad de disolución. Si el sólido aparenta ser insoluble tn agua o cter, algunas veces es aconsejable calentar la mezcla moderadamente. Si en estas condiciones hay disolución, el líquido se enfría a temperatura ambiente y se agita para evitar la sobresaturación. Siempre conviene, en tales casos, “sembrar” la solución fría añadiéndole un cristal del sólido. Al pesar la muestra hay que hacerlo con cuidado; deberá pesar 0.10 g con una diferencia no mayor de 0.01 g.
Los líquidos se manejan más convenientemente mediante una pipeta graduada, la cual permite mediciones exactas de la cantidad añadida. Cuando dos fases líquidas están estratificadas, a menudo es posible pasar por alto la interfase y así ver sólo una fase. Generalmente puede evitarse este error agitando vigorosamente el tubo de ensayo, cuando un líquido desconocido se ha dísuelto aparentemente en el disolvente. Si están presentes dos fases, la solución se pondrá opalescente. En los raros casos en que dos fases incoloras tienen el mismo índice de refracción, no se podrá
0 4 C o m p o r ta m ie n to d e s o lu b il id a d
i El tu m i» ! m rejo tt pH á t 4.5 y tz u l, * m ayoría d e 3,3.

Solub
ilidad
«n
Xo
* 0 *
i l i
+ i i
i i
£ as o j j
«i (j¡r' 8 8
•H
■H
■«
0 —1au s
mn
m«
út i
r i
J
2m
-i

99
f *
observar la presencia de una secunda fase aunque se tome la precaución indicada.
• ^Mküñn Vi f a i t h t e ttb¿UcUd &> ácido -o en álcali, ¿>odf!f>erá H¡/ikd(it¡ tnítrí, y» (¡ut pttede rfMiukm hí/a fáátffaá\, h tat*- d a se agita perfectamente, el tiempo requerido para que se realíce Ja disolución no deberá ser mayor de 1 ó 2 minutos.
En el caso de los disolventes de reacción, frecuentemente es más ■j J P cómodo y más económico colocar 3 mi del disolvente en el tubo de ensayo
V añadir el soluto poco a poco. Así, si un compuesto es muy insoluble, se averiguará usando sólo una muy pequeña cantidad de sustancia y no se necesitará pesarla o medirla. En general, en donde no se requieran
, las pruebas cualitativas, el procedimiento prescrito puede simplificarseconsiderablemente; pero siempre que exista cualquier duda, las determinaciones de solubilidad deberán efectuarse con exactitud.
Con frecuencia es posible utilizar una sola, parte del soluto para pruebas con varios disolventes. Así» st se encuentra que el compuesto es insoluble en agua, puede obtenerse una idea bastante exacta de su solví.
r * & bílidad en solución diluida de hidróxido de sodio mediante la adición de j 1 mi de una solución de hidróxido de sodio al 20%. Los 4 mi de disol-! vente que resultan contendrán aproximadamente un 5% de hidtóxida
de sodio. Si la substancia es muy insoluble, a menudo puede recuperarse y usarse subsecuentemente para la prueba con ácido clorhídrico.
Cuando se usa el ácido sulfúrico, es más conveniente colocar los3 mi del disolvente en el tubo de ensayo y entonces añadir el soluto. En algunas ocasiones este reactivo produce reacciones significativas y es importante observar tales manifestaciones como son: producción de calor, cambio de color, formación de un precipitado o desprendimiento de un gas. Deberán tomarse notas cuidadosas de todas estas observaciones ya que pueden ser muy útiles en una etapa posterior de la identificación.
GENERALIZACIONES
Mediante el estudio de los datos de solubilidad ha sido posible establecer ciertas generalizaciones que a menudo permiten predecir el comportamiento de solubilidad de un compuesto, simplemente por el examen de su fórmula estructural. Como introducción, quizá valga la pena discutir brevemente el fenómeno de disolución.
Polaridad y solubilidad. Cuando se disuelve un soluto, sus m oléculas o iones quedan distribuid» más o menos al azar entre las del disolvente. Por ejemplo, en los cristales de cloruro de sodio la distancia promedio entre los iones, sodio y cloruro es de 2 8 Á. En una solución \M , el disolvente se ha intercalado en tal forma que los iones sodio y cloruro quedan separados unos 10 A. La dificultad para separar dichos iones queda indicada por el elevado punto oe fusión (800°C) y punto de ebullición (1413°C) del cloruro de sodio puro.
86 C o m po rtam ien to á& s o lu b il id a d
i - . i

G e n e ra liza c io n e s 87
De ia física elemental deberá recordarse que el trabajo requerido para separar dos placas de cargas opuestas se disminuya con la introducción de materia entre ellas, por un factor m » « l W a*» h, ( ^ é tfr íb * . <&£ m e tf i í . E a iíra c E . ü l t <ss s jíp rem & ntK : ip je eT agua^ (con unú. constante dieléctrica de 30, favorezca la separación de los iones sodio y cloruro y, de hecho, disuelva rápidamente el cloruro de sodio, mientras que el éter (constante dieléctrica 4,4) o el hexano (constante dieléctrica J.»9) sean disolventes excesivamente malos para este tipo de sales. Las moléculas de agua distribuidas entre dos iones (o las placas cargadas de un condensador), son dipoJos pequeños que se autoorientan extremo con extremo, de tal manera, que neturalizan parcialmente las cargas iónicas y así estabilizan el sistema. Por lo tanto, debería esperarse que la capacidad de solvatación y la constante dieléctrica fueran paralelas. Sin embargo, esto no es totalmente cierto. U na elevada constante dieléctrica es necesaria, pero no suficiente, para un disolvente efectivo de iones. Por ejemplo, el áddo cianhídrico, con una constant dieléctrica de 116, es un disolvente muy malo para sales como el clon' -o de sodio. Aunque el fenómeno es complejo, un factor importante, responsable de la eficiencia del agua y otros disolventes oxhidrilados, es su capacidad para formar enlaces de hidrógeno.*
La elevada constante dieléctrica del agua, y su capacidad para form ar enlaces de hidrógeno, que la hacen un buen disolvente para Jas sales, también la hacen un disolvente malo para substancias no polares. En el agua pura las moléculas están orientadas en tal forma que los centros positivos y negativos quedan contiguos. Intentar disolver una substancia no polar, como el benceno, en agua, es intentar la separación de cargas opuestas en un medio de constante dieléctrica baja, Entonces, en general, debe esperarse que un disolvente polar sólo disuelva fácilmente solutos polares y un disolvente no polar, sólo solutos no polares. Esta generalización ha sido resumida brevemente como '‘semejante disuelve a semejante”.
Ya que la mayoría de las moléculas orgánicas tienen tanto una parte polar como una parte ao polar, debería esperarse que la solubilidad dependiera del balance entre ambas paites. A medida que aumenta la parte de hidrocarburo de la molécula, las propiedades de los compuestos se aproximan a las de Jos hidrocarburos de los cuales puede considerarse que derivan estos compuestos. Esto significa que disminuye la solubilidad en agua y aumenta la solubilidad en éter. Un cambio parecido en la solubilidad ocurre a medida que aumenta en la molécula el número de restos hidrocarbonados aromáticos. Así, el a-naftol y el jS-hidroxibi- fentlo son menos solubles que el fenol. Cuando el radical fenilo se en* cuentra presente como substituyente en los ácidos alífáticos, en los alcoholes, en los aldehidos y en compuestos similares, tiene un efecto
? V e r H u n o ic tt , f 'f ty t i ia l O t fa tu c C k tm is l ry, M e C m w H il l B ook C o .t Kce%% Y ú i i , IM G ,; p í¿ , 38 y aijuitnlt».
S

8 3 C o t n p o T t t f t n f e n t a c í e s o l u b i l i d a d
sobre la. solubilidad aproximadamente equivalente al de un radical ali- fático de cuatro átomos de carbono. Por ejemplo, el alcohol bencílico es aproximadamente tan soluble como el alcohol amílico normal, y el ácido hídrocinámko muestra una solubilidad semejante 2 la del ácido «•heptanoico.
E fecto sobre la solubilidad de las fuerza* ¡niermoleculares e n e l so lu to p u ro ♦ La solubilidad de una substancia es una medida del equilibrio entre la substancia pura y su solud6n. Se ha visto que dicho equilibrio se ve afectado, no tan sólo por las interacciones de disolvente- soluto ya discutidas, sino también por las fuerzas intcrmoleculares en el soluto puro. Estas fuerzas son independientes de la polaridad u otras propiedades del disolvente y $e pueden estimar sus magnitudes relativas comparando sus puntos de fusión y de ebullición, ya que los procesos de fusión de un sólido o de ebullición de un líquido, involucran una separación de moléculas, relacionada en alguna forma con la separación que ocurre en la disolución.
Los ácidos dicarboxíHeos ejemplifican la relación inversa entre los puntos de fusión y la solubilidad. Los datos de la tabla 10 muestran que los miembros con número p ar de átomos de carbono funden más alto que el ácido que les antecede o que el que les sigue (ambos con núme-
• ro impar de átomos d? carbono). Evidentemente, las fuerzas rntracristali*• ñas en los miembros con número par de átomos de carbono son mayores
T a b l a . 10
NÚBXTU pard t twroox di
c u ta x i l
&úh;b5Hdwi K/S00g*ffü*
* ‘J t f C
NáaurO impar de £tom>« d<
<arfcoth) P‘CSoJulálidjul
g/lQOgisuna2&pC
CWIÍCO 109 9-5 M alónfc© 135 73.5S u c c ín k o 3S5 6 .8 G lu tá r ic o 97 6+A d íp ico 153 2 P im élic o 103 5S u b é ric o Í4Ü 0 .1 6 A w la íc o 106 0 .2 4S e b á c e o 133 • 0 .1 0
que las de aquéllos con número impar. Ya que el límite de solubilidad para las sólidos ha sido fijado en 3.3 g por 100 mi de agua, es evidente que el ácido adípico (seis átomos de carbono) es insoluble en agua, pero el ácido pimélico {siete átomos de carbono) es soluble en agua.
La correlación entre el punto de fusión elevado y la baja solubilidad está aun mejor ejemplificada por los ácidos isómeros maleíco y fumárico.
' H C -C O jH HOOCCJiil IIHG—COiH HOCOOH
A i ido JTMÜCÍCD (cu) Acida fu in irk o (irme)
E) ácido fumárico sublima a 200*0 y es insoluble en agua. El ácido maleico funde a 130° y es soluble en agua. Entre los isómeros cis-irans.

G eneralizaciones 69
la forma cis generalmente es más soluble. En forma parecida, en las substancias polimorfas, tales corno la bemofenona, la forma, de menor punto de fusión es la de mayor solubilidad.
Las diamidas de los ácidos dícarboxílicos constituyen otro grupo de compuestos en los que el punto de fusión es un índice valioso de las fueras presentes en los cristales. La urea (p.f. 132CC) es soluble en agua. Por ocra parte, la oxamida tiene un punto de fusión muy elevado, 420°C y, consecuentemente, una baja solubilidad en agua. La substitución en el grupo amida de los átomos de hidrógeno por grupos metilo disminuye
CONHa CONHCH3 CON(CH5)jI I í
C O N K j . C O N H C H j GON(CM 3) í
p.f. p . r w c p . u i r c p . i .w c
el punto de fusión y aumenta la solubilidad en agua; la iV^'-dímctil- y la ^V^V^Y^tétrametnoxamida son solubles en agua. L a adipamída es insoluble en agua mientras que su ^ jY ^V ^ '-te trae til derivado es soluble en agua
la s amidas del tipo RCONH¿ y RCONHR obedecen la regía general que los compuestos en los límites de solubilidad contienen cerca de cinco átomos de carbono (pág- 94). Sin embargo, las /'/¿V-díalqiiiJ- amidas (RCONR^) funden más bajo que las correspondientes amidas sin substituir y son mucho más solubles en agua, estando el límite dv solubilidad en la vecindad de nueve o diez átomos de carbono. Se ha sugerido que las amidas que tienen el grupo — CONH¿ catán asociada^* debido a que pueden actuar tanto como aceptores o como donadores al formar enlaces de hidrógeno.
O H R R HI* I I f f
R —C - N —H 0 = C —N - H 0 = C —N - I l
S k ' A ¿I,’ I t I
R t- G —N —H Ó = C —N —H R •
Tal asociación es imposible en las amidas A^N-disubstúuidas (RCONR*) y de aquí que su estado de agregación molecular sea bajo, como lo indican su¿ bajos puntos de fusión y elevadas solubilidades.
En general, un aumento en el peso molecular produce uq aumento en las fuerzas íntermoloculares de un sólido. Los polímeros y otros compuestos de peso molecular elevado, generalmente manifiestan bajas
< CK»p|Jrt t Hunlcr» J . CA<*1. S<n., 1837, l i l i ; IW8, 37», 1PM; Cú¡>\r¡t 1á \Iw írr y M atvel, J . Ám . ChvA. Soc., W , 2G66 (>933).
NHj/c=o\
n h 3

■y
solubilidades en agua y en éter. Así, el formaldehído es fácilmente soluble en agua mientras que el paraíormaldehído es insoluble.
C H jO -> H O íC H jO - ) ^ !wluble en **uk iPMfablc «n *4ua
El acríiato de metilo es soluble en agua, pero su polímero es insoluble.
C H ,= ;C H C O O C H 3-> ( - C H jC H - ) ,
¿ O O C H j
La glucosa es soluble en agua, pero sus polímeros —almidón, glucógeno y celulosa— son insolubles. Muchos aminoácidos son solubles en agua, pero sus polímeros de condensación, las proteínas, son insolubles. La. tendencia de algunos tipos tales como las proteínas,, las dextrinas y los almidones, a formar dispersiones coloidales, puede ser engañosa.
Otro método para aumentar el peso molecular de una molécula es el de introducción de halógenos. Ordinariamente, esta introducción sólo produce una. disminución de la solubilidad en agua, dando coma resultado que algunos compuestos que son solubles en agua, cuando se les iniroducen halógenos, pasan a la clase de los insolubles en agua.
Efecto de las arborescencias de la cadena sobre Ut solubilidad. Podría anticiparse, de la consideración del efecto de las arborescencias en las cadenas hidrocarbonadas sobre los puntos de ebullición de los miembros inferiores de las series homólogas, taíes como las de los hidrocarburos y las de los alcoholes, que las arborescencias disminuyen las fuerzas intermoleculares y hacen dec reccr la atracción intermoleeular. Entonces, no es sorprendente que un compuesto que tiene una cadena arborescente sea más soluble que el correspondiente compuesto de cadena lineal. Esta es una regla muy general y es particularmente útil en relación con los compuestos alifátieos sencillos. Por ejemplo, la solubilidad de un compuesto íso difiere ampliamente de la de su isómero normal y está más cercana a Ja del miembro normal que le antecede en la serie homó- loga en estudio. En la tabla 11 se muestran los efectos de las arborescencias en la cadena. En general, el más arborescente de dos compuestos isómetros posee la mayor solubilidad.
T abú* 11
90 Com portamiento d e solubilidad
“T t p o id f .compuíaw» Scloble Limite Josohible
A cido» JPjvAlíco Ii<vyaleríájiicc> « •V ftlír iá n íc oC lo ru ro s de a c ito íscbu tirilo i « -B u tir íloA lcoholes N ec>pem üico M e ti l-2 -b « ta n o l-3 n-A m ilA n id a s Is c b u tíra m id a n -B u tir a m id aE a tírea A c e ta to d e ijo p x o p ilo A c e ta to d e n -p ro p íloC o tonas M etiliso p ro p ll M e ti l-n -p ro p iiN i (rilo* Is o b u tiro n i t r i lo rr-B u tíro n ítrilo

Generdbmclorw*
La posición del grupo funcional en la cadena carbonada también afecta Ja solubilidad. Por ejemplo, el pentanol-3 es más &c! jble que el pentanol-2, el cual a Su vez es más soluble que el pcfltanoM. Cuando el efecto de la» arborescencias se combina con el. desplazamiento del grupo funcional hacia el centro de la molécula, corno lo ejemplifica el meül-2-butanol-2, se observa un marcado aumento en la solubilidad. En general, mientras más compacta es la estructura, mayor es la solubilidad, siempre y cuando las comparaciones se hagan con alcoholes del mismo tipo.
E fecto de la estructura sobre la acidez y la b a ste idad f En ge-neralj el problema de decidir ai una substancia desconocida que es insoló le en agua, d?be disolverse en un ácido diluido O en una base, es esencialmente el caso de evaluar aproximadamente su fuerza como ácido O base. Hay dos efectos principales de la estructura sobre la acidez o la basícidad y éstos, serán discutidos por tumo,
Efectos electrónicos sobre la acide* y la basicid» í. Se han efectuado estudios amplio» sobre la correlación cuantitativa de a estructura con la fuerza ácida o básica de las substancias aromáticas con substituyeme* en mela y en para} Estos efectos son esencialmente electrónicos y se ha logrado disponer de importante evidencia sobre la naturaleza. de la transmisión de dichos efectos eléctricos, desde las posiciones meta- o futra del anillo bencémeo hasta el sitio de reacción. Se ha establecido una correlación cuantitativa de la» constantes de disociación de los ácidos, en las serie* alifáticas, en las cuales no deben tomarse en consideración sólo Jos importantes efectos polares, sino también los efectos esféricos. Sin embargo, tales estudios detallados están más allá del objetivo de esta discusión.
La mayoría de los ácidos orgánicos carboxílicos tienen constantes de disociación en el agua a 25°C de 10-" o mayores y* por lo tanto, son fácilmente solubles en hidróxido de sodio al 5 c/o. Por otra parte, los fenoles, como generalmente son menos ácidos (la constante de disociación del fenol es aproximadamente de 1 0 'lQ), aunque solubles en solución fuertemente básica de hidróxido de sodio, son insolubles en bicarbonato de sodio diluido, (la primera constante de disociación del ácido carbónico es de 4 X 10”T). Sin embargo, la introducción de grupos aubstituyentes puede tener un efecto man:ado sobre la acidez. Así, el o• y el £-nitrofeno! tienen constantes de disociación de aproximadamente 6 x I0~x; en otras palabras, Ja introducción de un grupo nitro orto o para, aumenta la acidez del fenol en un factor de unos 600. Como podría anticiparse,
4 .Por* tM «!írt!C» ro r iu k j « A r e la tt» rreljc*ón d e la íW ru c ia ra c o n U c a o U m lt <k diftK Í*- c3tfn ver biuwn. fcísDonkl y Haíli^w, capltvV» 14 «a D iiím in a tiú n O fg tn h Stm clurts b? Ffcnit-at ¡¿cthoAs, f>4¡U<ÍO ptrr Brande y Ntthorf, Atadcrolc P(t«, Nittv* Yotfc» 1953, • }[im- mead, capitulo' 9 en Strríc E f f t t i l ta. 0'£A*U Chtmíilry, «3»udo pot Nrwnan» John Vi'ílry *ad Soou, K u e v » Y o rk , 19K3.
♦ V e r H in t , Phyáccd O r¡a * ic C & tm lH ry, 2» « t . , M eC raw -H U l B o e t C o .. N u ev x Y a r i , 19C2, npltolo 4.

9 2 C o m p o r ta m ie n to d e s o lu b i l id a d
l a adición de dos grupos nitro, como en el 2,4-diniírcfenol, aumenta tanto la acidez que el compuesto es soíable en solución diluida, de bicar-
H ,0
o- O
I |¡ «;0 —N—O ~ :0 —N—O:
+
bonato de sodio. El efecto de incrementar Ja acide?, mostrado por eJ grupo nitro, se debe a Ja estabilización del anión fenóxido a consecuencia de la distribución parcial de la carga negativa en el grupo nitro.
Un efecto parecido, reforzante de Ja acidez, se observa cuando se introducen halógenos en el fenol. Así, un átomo <.' • bromo orto aumenta la acidez del fenol en un factor de unos 30 y un á\-,mo de bromo para, en un factor de unos 5. N o es sorprendente, pues, que el 2.4,6-tribromo- fenol sea un ácido lo suficientemente fuerte corno para disolverse en una solución de bicarbonato de sodio.
Probablemente el efecto inductivo del bromo es en gran parte responsable de la estabilización del anión tribromofenóxido. Es evidente que no hay la misma posibilidad de estabilización por resonancia como la que tienen los aniones nitrofenóxido.
Influencias electrónicas parecidas afectan Ja basicidad de las aminas, Asi, las aminas aliíáticas en solución acuosa tienen constantes de hidrólisis de unos 10“5 6 10~ , no muy diferentes de la del amoníaco (10"5). Sin embargo la introducción del grupo fenílo disminuye la. basicidad en6 potencias de Í0. Así, la anilina tiene un K* de 5 X 10“ ,D. El efecto del giupo fenílo es de estabilizar la amina líbre por resonancia, corno se ve en el lado izquierdo del equilibrio, lo que es imposible con la base conjugada, que está a la derecha.
HjO +
HU
H—N—H
+ OH"
No es de sorprender el que un segundo substituyeme íenilo disminuya tanto la basicidad, que no sea posible determinar en agua la basi- cidad de la amina. Asi, la difenílamina es insoluble en ácido clorhídrico diluido. La introducción de un grupo nitro en el anillo fenilo de la anilina disminuye su fuerza cómo báse, debido a la estabilización de la ni-

tcoamina por la contribución de 1a estructura, siguiente al híbrido Je resonancia de la amina, pero no al del ácido conjugado.
H—N —H Ü
\
Generctizüríorws 93
- : O ^ N —O :- +
E fectos ettérico* sobre Ja acides y la basicidad. Desde hace tiempo es sabido que los fenoles substituidos en orto tienen una solubilidad muy reducida en álcalis acuosos y se ha usado el término “criptoíe- nol'1 para destacar este comportamiento. Se ha usado el álcali de Claisen (hidróxido de potasio al 35% en metanol-agua) para disolver dichos fenoles impedidos. U n ejemplo extremo es el 2,4,6'tri-í-butilfenol, que no se disuelve ni en hidróxido diluido ni en el álcali de Claisen,7 Se le puede convertir en sal sódica tratándolo con sodio en amoníaco líquido.
La 2,4,6-trí-t'butíIanilina muestra, en igual forma, este insólito comportamiento.5 Es una base tan débil que el pKa de su áddo conjugado es demasiado pequeño para poderse medir en solución acuosa.
La 2,6-di-í-butílpirídina también es una base notablemente más débil que la correspondiente dímctilpiridina’ Se ha sogerido que la díswinudón en la fuerza básica de las aminas se debe al impedimento estético, que impide que el átomo de nitrógeno adicione un protón, j>crc parece más probable que la inestabilidad del ion 2,6-di-í-butilfcnóxida y de !<» iones amonio bloqueados que se mencionó antea, se deba a la interferencia « térica con la solvatación de iones.®
Los impedimentos e&téricos pueden aumentar o disminuir la acidez de los ácidos carboxíiicos. Por ejemplo, ia introducción de grupos alquilo en el átomo de carbono-a del ácido acético tiende a disminuir la acidez. El áddo raetil-t-butiineopentilacético tiene & de la fuerza del ácido acético, probablemente, debido a desestabílizacíón de la base conjugada por inhibición estérica de la solvatación.10 Por otra parte, los áddos benzoicos orío-substituidos son notablemente más fuertes que los correspondientes isómeros para. Es probable que el efecto de los substituyentes sea el de sacar el grupo carboxilo fuera del piano del anillo, con la consiguiente mayor desestabilización del ácido que del anión.11 riste último caso ejempltfira una segunda clase de efecto estérico, la cual es frecucn-
T s t i l lw p , S*w yer y H u n l , / . A m , C h e m . S c c ., « 7 , 303 <1945).
* B*rtLc«, feiiuj y Scíle», / . A tn. C k tm, 7$, Í 349 (19St),
♦ BrftWp 7 Kofiner, / . Atn. C h itn . Soc ., B , 3865
w HíLfljatoBd y Hogla» } . A a . Ch*m . -f *c.j 77, 33S
» I rg o k J , S ln w ta r* (Mii U « k 4 \ d tm !m O r¡9MÜ C k tm ii tr ? , C orecU V ltim a itT P r a j , I tk » c i ,• N. V., 1953, p ip . 74&-750.

r
r*i
n
il ai'
temente importante y se ¡«idíca cotno «toa inhibición cstérica de !a resonancia ,n Como segundo ejemplo, aunque el jD-nitrofenol es unas 2,8 unidades de pK /m á s fuerte que el fenol, el 3,5*dimetil4-nitrofenol tan sólo es más fuerte en l.b unidades de pK aV
9 4 C om po rtam t«n fo d# s o lu b i l id a d
OH OH OH
CH3\ ^ C H a N O , NOj
P*« 1 25* -r.il 3.54
Una parte del efecto de los dos grupos metilo en h reducción de la acidez del nitrofenol es de naturaleza electrónica, pero una gran parte parece deberse a la inhibición estérica de la resonancia en el anión. Así, la estructura de A, que se indica abajo, para contribuir significativamente al híbrido requiere la coplanaridad o la casi coplanaridad del grupo nitro y el anillo aromático. Tal coplanaridad está inhibida por la presencia de los grupos metilo en el ion (B) -
O O
" :0 —N—0 :~
CHaV / C H 3
- . o —ji—o í -
e x a m e n DEL CO M PO RTA M IEN TO DE SOLUBILIDAD
Solubilidad en agua. Como el agua es un compuesto polar, es un mal disolvente de los hidrocarburos. Los enlaces olefínicos y acetilénicos o las estructuras bencenoides, modifican muy poco la polaridad, por lo que los hidrocarburos insaturados y aromáticos no difieren mucho de las parafinas en su salubilídad en agua. La introducción de átomos de haló* geno no altera notablemente la polaridad, pero aumenta el peso molecular y jx>r esta razón siempre disminuye la solubilidad en agua,. Por otra parte, las sales son extremadamente polares; las que se encuentran en esta obra generalmente son solubles en agua.
Otras compuestos están colocados entre estos dos extremos. Aquí se hallan loa alcoholes, los esteres, los éteres, los ácidos, las amtnas, los nitri- los, las amidas, las cetonas, y los aldehidos—por mencionar sólo algunas de las clases más comunes.
1* W hetand , f i e l v w \ee ¡4 Q rg a r \ü C h r lfiii try i J a J in H'ile«r Muí S<Ktí, X u cv a Vori!, 1555.” Wheland, JSrawntll y Mayo, / . Am. Cht«t. 70, 2Í&2 (JÍMB).

£xam « n d d co m p o rta m ie n to d « s o lu b il id a d 95
Como se podría esperar, los ácidos y las a rJnas generalmente son más solubles que los compuestos neutros. i «¡ am y»«¡ ffrhf *
.elevada solubilidad a su tendencia a formar complejos, enlazados por hidr¿geno_ajnoljcula5 de agua. Esta teoría está en armonía con el hecho de que la solubilidad de 1?. amina disminuye a medida que disminuye la basicidad. Esto también explica la observación de que mu- chas aminas terciarías son más solubles en agua fría que en caliente. Aparentemente, a baja temperatura está, involucrada, la solubilidad d d hidrato, mientras que a temperatura elevada el hidrato es inestable y la solubilidad que se mide es la de la amina libre. Los compuestos xnono- fundonalcs, .los .dd.tipo.de los éteres, los éste res, las cetonas, los aldehidos,
_lqs__alcohqles, los nitriloa, las amidas, los ácidos y las aminas, pueden considerarse en .conjunto, por Jo que respecta a su solubilidad en agua. Bn le mayoría de los series komóioeas de este tipo, el límite superior de.
jolubilidad en agua ¿¿encontrará cerca d d miembro o c o n t e n g a f inco átomos de carbono. '
Esta regla resulta de un principio muy general, el de que la cociente semejanza estructural entre el soluto y el disolvente va acompañada de una solubilidad creciente. Debido a la naturaleza polar del agua, los compuestos deben su solubilidad en ella totalmente a los grupos polares que puedan contener, A medida que se asciende en una serie homóloga, la parte de hidrocarburo (no polai) de la molécula aumenta continuamente mientras que la función polar permanece esendalmente sin cambio. De aquí la tendencia hacia una disminución de la solubilidad en disolventes polares como el agua.
El que para muchas series los límites superiores de solubilidad en agua estén en la misma vedndad, se debe a que Jas polaridades de muchos grupos funcionales son similares. L a región particular (aquella del miembro que contiene cinco átomos de carbono) en. la que en estas diversas series se alcanza el límite superior de solubilidad en agua, está enteramente determinada por las proporciones de disolvente y soluto que arbitrariamente se seleccionaron para usarse en esta marcha de separación. Habría sido igualmente fácil y quizá tan satisfactorio el emplear una relación dé soluto a disolvente que colocará el límite en otro lugar.
La tendencia de determinados compuestos que contienen oxigeno a formar hidratos, también contribuye a su solubilidad en agua. La estabilidad de estos hidratos es, por lo tanto, factor determinante de la solubilidad en agua y en éter. Los compuestos como el doral, probablemente deben su gran solubilidad en agua a la formación de hidratos.
Los ¿stores de bajo peso molecular de los áridos fórmico y pirúvko, se hidrolísan en agua a temperatura ambiente, como k> indica el hecho de que la solución se vuelva francamente árida al tornasol.
S o lu b ilid a d e n ácido clorhídrico d ilu id o . Las aminas alifáticas primarias, secxmdarias y terciarias forman sales (compuestos polares)

con el ácido clorhídrico. De aquí que las aminas alífáticas sean fácilmente solubles en ácido clorhídrico diluido.
Los grupos arilo disminuyen la basicidad del átomo de nitrógeno; las aminas aromáticas primarias, aunque san más débilmente básicas que las aminas aJifátácas primarías, son solubles en ácido clorhídrico diluido; las diarílaramas y las triarilaminas no son solubles. Por ejemplo, la difeni&mina, la trifenilamma y el carbazol, son insolubles, Las arilal- quilaminas que contienen tan sólo un grupo arílo, son solubles
Las amidas d[substituidas (RCONR2) que son de peso molecular lo suficientemente alto para ser insolubles en agua, son solubles en ácido clorhídrico diluido. Este comportamiento contrasta con el de las amidas sencillas (RGONHj), que son compuestos neutros» La mayoría de las amidas substituidas (RCONHR) también son neutras. Sin embargo, la A'-bencilacetaraida es básica.
Deberá notarse que las aminas pueden reaccionar con ácido clorhídrico al 5% pata formar clorhidratos insolubles. Los compuestos de este tipo pueden aparentar ser insolubles. Por ejemplo, determinadas aríla- minas, tales como Ja a-naftilamina, forman clorhidratos que son muy poco solubles en ácido cloiWdrico. Calentando Ja mezcla un poco y diluyéndola con agua, algunas veces puede efectuarse la disolución. Usual* m ente el aspecto del sólido mostrará si la amina h a experimentado un cambio. Para decidir en los casos dudosos, deberá separarse el sólido y compararse su punto de fusión con el del compuesto original. Una prueba de halógenos con nitrato de plata indicaría la formación del clorhidrato.
Hay unos cuantos tipos de compuestos que contienen oxígeno y forman sales de oxonio al tratárseles con ácido clorhídrico, por lo que se les clasifica, como básicos.
- Solubilidad en soluciones d ilu idas d e h id ró x id o de $odio y e n soluciones diluidas d e bicarbonato de sodio . Los ácidos carboxí- líeos, áddos sulfónicos, ácidos sulfúricos, fenoles, algunos enoles, imidas, compuestos nitro primarios, secundarios y terciarios, derivados de arilsul- fonilo de las aminas primarias, arilsulfonamidas sin substituir, oximas, tiofenoles y muchos tipos de compuestos menos familiares, son solubles en soluciones diluidas de hidróxido de sodio. De ésto^ sólo los tres grupos mencionados primero son solubles en soluciones diluidas de bicarbonato de sodio.
Los aldehidos y las cetonas son lo suficientemente ácidos para « ac cionar con álcalis acuosos proporcionando amones,'los cuales sirven como intermediarios de reacción en procesos tales corno la condensación aldó- lica; sin embargo, son ácidos demasiado débiles para disolverse en cantidad apredaWe en solución de hidróxido de sodio. Cuando dos grupos carbomlo están unidos al mismo átomo de carbono, como en los ésteres acetoacético y malónico y en las dicetonas-1,3, la acidez aumenta marcadamente debido al incremento en la estabilización del anión, en el
96 Com portom tenío d e solubilidad

mm
n fi'T
i'i'
\
Examen del comportamiento de loíuhlUdcd 97
cual la carga negativa puede distribuirse entre los dos átomos de oxígenn así como en el átomo de carbono,
.6 - O. ; 0 " O. O ’Ó r• \ - • / ' * \ x ■ : \ / *
c — C - C « - C - C —C 4-» c —c - c/ I \ / 5 \ • / I \Las siguientes constantes de disociación ilustran este punto.54
c h 3c o c h 2c o o c 2h , 2 x i o - n G H iC O C H í G tH s) C O Q C sH j 2 X IO- ;11CH2(COOC,H5) 2 5 x 10-14CHsCOCH2COCH3 1 X io -»
Deberá notarse que aunque estas substancias son aproximadamente tan ácidos como los fenoles, la velocidad de separación del protón del carbono puede ser una reacción relativamente lenta.14 y la velocidad dé la disolución de dichas substancias puede ser tan lenta que aparenten ser insolubles en álcali.
Aun un grupo nitro confiere a una substancia la acidez suficiente para hacerla soluble en hidróxido de sodio diluido. Así, el nitroetano15 tiene una K a de unos 3,5 X 10-®.
Es interesante que los compuestos nitro tengan una forma tautó- mera, la forma aci, que es aproximadamente un ácido tan fuerte como los ácidos carboxí líeos. La forma 4t i del nitroetano tiene una K a de 3.6 X 1<H.
Así como el grupoO H O
—C —C —C —I
es ácido, también lo es el grupo imidoO H OII 1 IJ
—C —N —u —y las ¡midas son solubles en solución diluida de hidróxido de sodio, pero no en la de bicarbonato de sodio. Aun el grupo ¿-nitrofemlo o el grupo R S 0 2 hacen la función —CONH— débilmente ácida en salución acuosa. Así, Ja p-nitroacetanilida y Jas sulfonamídas (RSOjNHj} se disuelven en solución de hidróxido. de sodio, pero no ejr bicarbonato de sodio. Las oximas, que tienen un grupo oxhidrilo><mido al átomo de nitrógeno, muestran un comportamiento de solubilidad parecido,
Los esteres con cinco o seis áWmos de carbono, que son totalmente insolubles en agua, pue^éS ser hidrolizados por una prolongada
m V tr y SÍÜtt, J. A ta . Ckam. Soc., 72, (1550),« V ar P « « 0o r D ílloc, / , Am. Cktr*. Sce., 72, 557* (1950),
«ÉJLi é
Ü i i

4 98 Com portam iento d* solubilidad
í i
í *
f »t *
=9
I d
¿ J
e l
1
agitación con soludón diluida de hidróxido de sodio. El álcali no deberá calentarse y la solubilidad o i n s o l u b i l i d a d deberá considerarse para su anotación, después de 1 ó 2 minutos.
Los ácidos grasos que contienen doce o más átomos de carbono, reaccionan lentamente con álcalis formando sales que son jabones. La mezcla no ca transparente, sino forma una dispersión coloidal opalescente que produce espuma cuando se agita. Cuando se ha observado una vez este comportamiento se le puede reconocer fácilmente.
Determinadas sales de sodio de fenoles altamente substituidos son insolubles en hidróxido de sodio. Se puede reconocer esta propiedad probando la solubilidad de cualquier residuo, en agua. Ciertos fenoles que son muy insolubles en agua, pueden precipitar debido a hidrólisis, por lo que aparentan Ser insolubles en álcalis.
Solubüitlad de com puestos anfóferotu Los compuestos que contienen tanto un grupo ácido como uno básico, son anfóteros. Los aminoácidos de bajo peso molecular se encuentran principalmente como sales dipolare*.
NH* NH,-
RCHCOOH ^ R¿HCOO“Son solubles en agua y pueden dar soluciones neutras al tornasol.
Los compuestos anfóteros insolubles en agua actúan como bases y como ácidos fuertes o débiles, dependiendo de basicidad relativa del grupo amino, ya que la basicidad determina la extensión en que el grupo ácido será neutralizado por formación de la sal interna. Si el grupo amino lleva sólo substituyentes alifáticos, ios compuestos se disolverán en ácido clorhídrico e hidróxido de sodio, pero no en bicarbonato de sodio. Este grupo se ilustra con los compuestos de los tipos siguientes:
NH, NHR NRj
LCH<RCHCOiH RGHCO.H RCHCOjHSin embargo, la presencia de un grupo arilo en el átomo de nítró*
geno disminuye su basicidad, por lo que tales compuestos son solubles aun en soludón acuosa de bicarbonato. Esto queda ilustrado con los ccm pucitd siguiente*:
Q H jNHCH jCOjHCflH¿NCHa
RCHCOjHSi hay dos grupos arilo unidos al átomo de nitrógeno, el compuesto
no es básico sino que se comporta simplemente como un ácido fuerte.(C*H3) jNCH jCO jH
Solxibilidad en ácido su lfú r ico concentrado y frío» Este disolvente se usa para compuestos neutros, insolubles en agua, que no con

tienen más elementos que caibono, hidrógeno y oxígeno. Si d compuesto es iraaturado, se sulfona fácilmente o posee un grupo funciona] con oxígeno, se disolverá en ácido sulfúrico concentrado y frío. Frecuentemente la disolución en ácido sulfúrico se ve acompañada por «na reacción tal como: sulfonadón, polimerización, deshidratadla o adición del áddo sulfúrico a enlaces olefínicos o acetílénicos; pero en muchos casos se producen iones * a partir de los cuales puede recuperarse el soluto per diludón con agua helada. En seguida se ejemplifican algunas de las reacciones más comunes:
O S O j H
RCH=CHR-f H7SO<----► [RCHí-CHR] RCHCHRRCHjOH+ ÍHjSOí — -+ RCH20 S0 3H + H,C>*+ HSO«-
(El agua, originada por formación del ester sulfúrico, se convierte en ion hidronio con el ácido sulfúrico concentrado),
(RCH^CHOH + 2H2SO* -* (RCH>),CH0 SO3H + H,0 + + HS<V (RCHj)jCOH + H;SOt -* (RCHj)2C=CHR + H*0 + + HSOt-
C H ,o Q + 2HjS04 -> CH3o/^SO ,H + HaO* + HSO<- ck2 ch3
C h /~ \ + 2H2SO< CH«{ ) s°jH + H30+ + HSOr CHa
(Q H ^C O H + 2H,S0 4 -v (CflH ^C + + H*0 + + 2HS0 4~
{C„H5) , 0 = 0 + HfcSO, (C,H*)86 0 H + H S O r
/ ° / 0 H C€H3COH + HjSO, — C*HaC + HSOr
\OHQ CH, CHj O
COtH + 2HjS04 Ch/ ~ J C + + H,0+ + 2HSO<-
f» ^ h .
/V°CHjCOCCHj + 3H1S04 — 2CH3í(OH)t + HSOá- + HS*OrJ* HiaMactt, Pkyticai Crftnii Ckmúiry, MeGravr-HiH Book Ce., Yftrt, 19443: pÍR*.
45 y d«a.: Burlen y M I , * /« . . 6, SOS (1332); CUtapí# y L«í*«i>, í í í i . . 6. -W ( í9 H ) ;GtBwpte * OcbrMge, J . Ch+m, $<X., 28*4, (1359) 7 reiírcacW» lh l dtad»*. Ver DeiW, R^Ley, Lili, Hodg*. Houta y Wt*otsJc7, }. Am, Cturn. M, 3ÜIS (1&S2J, p*rti rcferepcta iofart la £ócm*t¿vn <ft«t ioa iarfc«a[o.
(braman efe! com portam iento de solubilidad 9 9

Las parafinas, cidriparafinas y sus derivados halogenados, son insolubles en áddo sulfúrico. Los hidrocarburos aromáticos sencillos y sus derivados ha Jovenados no se sulfonan bajo estas condiciones y son insolubles, Sin em balo , la inserción de dos o más grupos alquilo en el núcleo bencénico permite que el compuesto se sulfooe fácilmente y de aquí que los políaJquIlbencenos se disuelvan con bastante facilidad en ácido sulfúrico. Por esta razón el isodureno y el mesitileno son solubles. Ocasionalmente el soluto puede reaccionar en tal forma que produzca un compuesto insoluble.
Algunos éteres de alto peso molecular, como por ejemplo el éter fenllico, se sulfonan tan lentamente a temperatura ambiente que pueden no disolverse.
Muchos alcoholes secundarios y terciarios se deshidratan fácilmente con ácido sulfúrico concentrado formando olefinas, las que entonces se polirjoerizan. Los polímeros resultantes son insolubles en ácido'sulfúrico concentrado y frío y, por lo tanto, forman una capa definida s^bre la parte superior del ácido. El alcohol bencílico y sus productos de substitución se disuelven en ácido sulfúrico concentrado» lo cual provoca su condensación con formación de precipitados de color naranja.
Ejercido*
• I. D isponga en una tabla, ColfiO Se muestra en fa tabla de ejemplo, las fórmulas,nombres y comportamiento de solubilidad de los compoeati» siguientes:
100 - ComfWteml&jito do solubilidad
Fórmula tfU>K<pr.»l Nornto trivial y/o 4i*trmiíki>
Coir'pomoiieDto <fc wtubíJidad aj>o»io
N H ,
0C l
^-Cloroanilinü Insolublo en agua poique úeae seis ¿tomos de carbono y un átomo d e doro , £* básica, sólo un grupo arilo unido al jrrupo amisvo y, p o r lo tanto,
• ei solublr en ácido cloribídri* co diluido.
C H jC O ÍC H jJaC H j Kcptanoná-2 Una m etil ce tona con más de cinco átomos d«3 carbono. Es insoluble en agua (neutra}.

J, Cloruro de «-butilo 9. C*H¿COCH(OH)C»H»
£ x cm « n d« i c o m p o rta m ie n to d e s o lu b i l id a d 101
2. C . H ^ ^ W f ,
3. C ^ C H ^ Q O H
4. (C H ^C H N H C H i5. OH SH SH
I f fCHt—CH—C H 2
6. C ^ o Q . ,
H 0 ,y =J ) S0»H
8. COzKí
<c h 2)s
c o :c , h 5
l’O. Cartazo!
1£. 2 * N itro b u tan o
Cf^CHa/ \
12, C*H6CH CO\ /
CH2CHj
NHNH-
13.
14. Fenilalanina
15. Q u-níJdina
II. Coloque Jos compuestos siguientes en el orden aproximado d¿ «u basicidad en icído clorhídrico acuoso diluido-
1. Bcnzanilida2. n-Amiíataína3. Difenilamina4. 2,S-Dibrorat>-4’rQetIiíimlina
5 . B eu c ilax a in a6. a-Broisoaaiíina7 . ¿ 'T o lu id ín a
I I I . Coloque lo.» compuestos siguientes en el orden de Su solubilidad creciente en agva. Indique Us razónos de la distribución.
I, C H ^C H ^C O K C H jís CHa(CH2)aC 0 2NH<CH^CH^CONHCHa (CjH^CHCONíCH^
CHj(CHt)«CONHt2. C R j
ICH, CH.
i '1CH,
1CH;
1 , CH, CH;
CHa Br CH«OH B r\ I I 1
C H j—COH (CHO* (CH& (C H ^I I / ! ’t (CH,OH CHjCHOH C,H* CHtOH CH,OH CHjOCHj

IV , Agrupe lo* compuesto* ¿gáfente; en el o rá tn aprox ím ídc de ?u acides
102 Com portam iento de íolvbfltdod
decreciente.
1. Acido m ¿íü-tartifico2. E jtearam ida3- Acido ^tolueuiulfóm co4. 2-Brnnw-6-n¡trofenoJ5. Sacarina6. Acido «-nftftoico
7. fl-Naítoí0, Acido beruohidroxárrtk»9. /j-ToluearulfonaniHa
10. Desaxíbeiuolna11. ¿-Tiocresol12. fi-Bcnjoüacetoñc*tato de etilo

Separación de mezclas
CAPITULO
7
L a separación de m ezclas es mi problema que el químico confronta constantemente y para cuya resolución ha desarrollado una amplía variedad de técnicas. Los métodos que aquí se consideren se limitarán a aquellos que aprovechan la naturaleza química de los compuestos que se van a separar y» más específicamente, las diferencias en polaridad y en acidez y basicidad. La extracción y la destilación por arrastre con vapor pertenecen a eate tipo de métodos.
Aunque otros métodos de separación, como destilación, cromatografía en columna, cromatografía en papel, cromatografía en fase gaseosa y electroforeás, tienen una tremenda importancia en cualquier laboratorio moderno de investigación química, son demasiado especializados para ser incluidos en este libro. En las referencias citadas están disponibles buenas discusiones de ellos.
U na excelente referencia general para los métodos de separación aquí mencionados es Ja siguiente:
K . B. W iberg, Laioratary T tch n iq u t in Óigante C h tm ütry , McGraw-Hcll Book Co., Nueva Y ork, 1960.
Destilación
C am ey, Lahoratory Fractional £>istijiatwn, T he MacraiNan Ca., N ueva York, Í949. D ittü la lión , Vql. IV de T tch n iq u t o} Oiganle Chtmisiry, editado por Wtimberger,
IntcncIcacD Publkhen , Nueva York, 1951.
Crom atografía
L ederer y Lederex, Chromalogrcphy. Elsevicr PufeHíhiag Ce., Nueva York, 1953. CaMidy, Adsorption and ChromaUigraphy, Vo1. V de Ttchntqu* c f Organie
Chemistry, editado por Weiüber&er, Intencience Pubü then , Nueva YorV, 1951.
Zechmeister, Progreu in Chramatography, C h ap o n a &. H all, Londrei, 1950. S lra in , Chromalographu; A d w p tfo n A nalytii, Intencience Publóher*, Nu*v* York.
103

BJock, D um m i y Zwcig, P o p /r ChTomc'.ography and Po.ptr Etectroph<rreiii, Aca- demic P rtjj, Nueva York, 1955.
Crom atografía en fase ga*co«
Dal Nogare y Juvet, C&s-Liquid Chromatography, Intcnciencc Publisher», Nueva York, 1962.
• Keuletnans, Gas Cftromatography, 2* cd,, Reinhold Fublíjhm # Corp., Nueva Y ork, 1959.
Pewok, Principia and Practite o[ Gas Chr ornato graphy, Jo h n Wtfcy and Soi>3, Nueva York, 1959.
Purnell, Gas Chromalographj1, John W icy and So»3, Nueva York, 1962.
Exam en general
Berg, Phyricvl and Chemical M elhodt o f Srparation, M cGraw-Hill Book Co., N ueva York, 1962.
Además de las técnicas que acabar» de mencionarse, las cuales con parte fundamental de casi toda la investigación en química orgánica, las siguientes son de gran valor en determinadas áreas:
Distribución a contracorriente
Craiy y Craiy, capítulo IV en T teh n iq u t of Organic Ch¿mii(ry, Vol. I I I . editado por Weissberfcr, Intersc-lence Publisher?, Nueva York, 1950.
Ekctroforecii
Atberty, J . C h tm . U v e . 25, 42$, 619 (1 9 ,8 ) .
Centrifugación
Nidxols y Bailey, capítulo III en Ttchniqv* o f Orgenic C fun tiftry, Vol. I , 2* ed., editado p o r Weis*berger, Intcncience PublUhera, Nueva York, 1949.
Coid, capítulo I I I en T¿(ht¡iqu< o f Otganic C kem iltry, Vol, I I I , editado por Weiwbergcr, Intertcicnce Fublishers, Nueva York, 1950.
104 Separación d o w ew las
SEPARACION DE MEZCLAS BINARIAS
Extracción: separaciones basadas en la formación de sales
Los principios fundamentales pueden aclararse por referencia a ejemplos sencillos. L a separación de anilina del benceno se efectúa por extracción con ácido clorhídrico diluido. La anilina pasa a la fase acuosa

B in a r ía s 105
como su sal, el clorhidrato de anilina- Mientras que la anilina es muy soluble en benceno y casi insoluble en agua, su sai —debido a su naturaleza polar— es soluble en agua e insoluble en benceno. En fomna parecida, el fenol se separa del benceno agitando la mezcla con una solución diluida de hidróxido de sodio. £1 fenol se transforma en su sal, el feuóxido de sodio, cuyo carácter fuertemente polar lo hace insoluble en benceno y soluble en agua. El benzaldehído puede separarse del benceno con un proceso parecido. En este caso, la mezcla se agita con solución acuosa de bisulfito de sodio, el cual convierte el aldehido en su derivado bisulfícico. Este e¿ una sal típica y, por lo tanto, insoluble en benceno, pero soluble en agua. En cada uno de estos ejemplos, el ácido, la baseo el aldehido originales se recuperan fácilmente por descomposición de la sal por los métodos usuales.
Si los compuestos que se van a separar son solubles en agua en grado apreciable, los métodos de extracción tienen un valor limitado. Sin en i' baigo, generalmente pueden ser substituidos por desdíación por arrastre con vapor. Por ejemplo, una mezcla de ácido acético y cic!ohe*anona puede separarse añadiendo suficiente álcali para transformar el ácido acético en acetato de sodio y arrastrando la mezcla con vapor. La cotona será separada en el destilado mientras que !a sal, que no es volátil, se queda en el recipiente. Acidulando oon ácido fosfórico se regenera e; ácido orgánico, el cual, entonces, puede destilarse con vapor.
En fomna semejante puede separarse la dietüamina del alcohol n-butílíeo. Se añade ácido fosfórico en cantidad suficiente para neutralizar la base. Entonces, la destilación con arrastre de vapor separará el alcohol y la amina puede recuperarse por adición de hidróxido de sodio al residuo y repitiendo la destilación con vapor.
Otro recurso útil para establecer una amplía diferencia en el carácter polar de los componentes está ejemplificado por la separación de los componentes de mezclas de aminas primarias con aminas terciarias. La acetilación o la benzoilación convierten la amina primaria en una «•imida ;teuira. La extracción con ácido clorhídrico diluido yparará la amina terciaria y dejará la amida. Por hidrólisis, la amida se convierte en Ja amina original.
Un principio muy semejante está involucrado en el método de Hinsbcrg para la separación de aminas primarias y secundarias. La sul- fonamida de la amina primaria forma una sal con los álcalis y asi puede separarse por extracción con solución diluida de hidróxido de sodio.
Se puede desarrollar un método general par? separar compuestos ácidos que difieran en su grado de acidez. Los ácidos fuertes forman sales cuando se tratan con bicarbonato de sodio y pueden, por lo tanto, ser extraídos con este reactivo. Así, si una mezcla de o-cresol y ácido benzoico se agita con solución diluida de bicarbonato de sodio, el ácido pasa a la fase acuosa como benzoato de sodio, dejando en la í¿se orgánica el cresol, que es débilmente ácido.

10 6 S e p a ra c ió n d e m # z tkn
Una variante útil del tipo de separación anrerior es la conversión de ambo* ácido^ fuerte y débil, en sales de sodio y eJ burbujear de anhídrido carbónico a través de la solución- Así se produce bicarbonato de sodio que hace que ía substancia débilmente ácida se separe. En esta forma podrá separarse cualquier compuesto menos ácido que el ácido carbónico.
Un procedimiento muy ingenioso para separar las cotonas de otros compuestos neutros e insolubles en agua, está ejemplificado por el uso del reactivo de Gírard, clorhidrato de betainhidrazida. Esta, reacciona con el compuesto carbonílico en ía misma forma que las hidrazidas sencillas. Sin embargo, el producto, como es una sal cuaternaria de amonio, es fuertemente polar y, por lo tanto, soluble en agua.
RjCO -f CHj) jNCHjCONHNHj (CHJ)>NCHJCONHN=CRj+HrOa - c i -
La extracción con éter separa los compuestos insolubles en agua, dejando la sal tu la capa acuosa. Por hidrólisis se -regenera el compuesto car- bonílíco originé. El ácido ^-hidrazinbencensulfónico ha sido utilizado en forma parecida,1
Los aldehidos reaccionan con el reactivo de Gira rd en foima análoga, pero la estabilidad de la hidrazida a la hidrólisis, hace que generalmente no sea práctica la recuperación del aldehido después de la separación*
Destilación por arrastre con vapor
Generalmente, un segundo grupo funcional en la molécula proporciona una diferencia en la polaridad, suficiente para permitir una separación por destilación con arrastre de vapor. Así, los alcoholes monoxhidrílicos pueden separarse de los alcoholes dioxhidrflioos y po* Jioxhidrílicos mediante este procedimiento- En forma semejante, los ácidos sencillos, las aminas y muchos otros compuestos volátiles pueden separarse de los correspondientes compuestos di y polifuncionales. Además, el grupo o grupos adicionales no necesitan ser iguales al original. Los aminoácidos, hidroxi ácidos, nitroácidos, cetoácidos, cetoalcoholes y ciano- cetonas, rara vez son arrastrables con vapor. De hecho, es regla general que la presencia en tina molécula de dos o más grupos funcionales (polares) hace a un compuesto no volátil. La tabla 12 muestra qué tipos de compuestos son arrastrables con vapor y cuáles no lo son. El ácido acético y el ácido oxálico, el alcohol etílico y el etilenglicol, el ácido benzoico y el ácido ftálico, son mezclas que ilustran este punto. En cada
l T ic lb » y JU jtiW rt, C h a n, B i t . , M , « 3 (19511.

B l n a r k i i 1 0 7 -
par, el primero que se nombra puede separarse por destilación por arrastre con vapor, mientras que el otro no « ana^t rabie.
T a b l a 12
SolablKW T¡pc* d t coopuotoi VoUiiUdid vil*Volatilidad eon
Juraitre de v*pa*
SoiuMu <>1 Dgua y en í« r .
Alcohole* do buje pe*j malíes Ur, «Mebiika, ectocud, í á do», (*•lera, I im iu . , Bjtrika, clora roí de acjJo ■ Cb"/i Ós0-/¿J .
Omitan fAcilttonte, Jtfucfaot eempueifo» hierven *b» ja de IW C.
A r r t t i n b l t l eon v»por
SoluWw CP *¡po piro IwJubJej íú ítCT.
Atcoholrt poJtcsHidrÜkoí, ¿í»jtjí|xm, dr+hjMdrutoc, ale* de jupinid, Mía nxcAlkü, iti¿o» poliUt(- ca»; bídfoidftWchtdoi, < « 00»» y -ácidoi; axnJnoácidóí
B»jo valni)td»d. Con deru» excepcioneseítOi CCúspuertM PO(C pwaím drttikr x pre¿£« *troorf¿r»ca
Nd u n s t n l ln cao vapor
lruotubltl cd «su» pero Pfllubfc» en KaOK y N*HGOj
Acíden de alto pe*> violtcuhT;fenol:* cor *itoltuyenl« pcí*-( I w
Bí Jjl volatilidad U*t»ihncatc qo »rr»itrjb!í»,H«to hay *1* guau tx ttp - <»«ve»
IwKtUc» m nguk y tn N*HCOi ( « o k>- toile* Hx N*OH
Feoole*, tuIÍH invdii dt MBÍiui primarlas. cdnjnxáiM altto jiñ.manen _ y aecuodaños; ¡mldai, tíofebolei
Pur\Kn de • tbtdlici¿T» eJewfos
Muüha, no puedeú dertibtlt
Uw».lrxnte ttó g n i i n M t t .
lo c to ite i cu i j u pero »óKiUti hi HCt diluido
A n b » <j«e íw lld io » no mSi de un jrupO «tilo HE ido jJ ui(r4- getto; hUfrazinna
Punto» de (4ul1lej¿D titeados
W uehfl» ion arnutrab lcí- « n vapor •
[wohibtet rn ngtu fUO H y HC1 dis id o , j w c oofuíe- aen olí©» eleraeirtoi adíraii de cwt*oeo, Wdrísero, <w(jü«o
j y iuldgetioj.
Nitro eonlpue*tot f l t t ) , brrtíJt». «miau 050 luiwtiüiyemeC afiW* dM», mlfocamldu ¿6 « n ín l aecuadutu; *uo y ajoxlcem. ruerna; «IqulJ o *nl cúaorot,nitrílM, nÍQTkt<a, a lfa (Ai, fotf*- tM
Punta» de ebultfctto e le ftd o t. t íu cb í» no pueden depilar- »c
A lg u n o * ion iiffa»trtili1ep eoa fípor
InwJuVe* en ara», NrOK T HCl diluido*, peto foltfbla
> ta H1SO4
Alcohola» «Ideiídw, cztanak, i*, tere», cocnpuírtOí ÍDBitiirado*
Compiteftoi de pefl-tot de efcaUielún tü n u ka
U ia a l m e n t é arr»»tr*bl<( con vlrpQt
InsoUlbtM <a i jn i , NaOtt diluyo, HO difaído y HiSOt
r e d f o a lW » HDOiiüm y aJifé- tjeoí y ttii derivad™ Jwlejfem- ded
VoJátik» A r r » i t m b l e i con vapor
En las seríes aromáticas se encuentra un grupo muy interesante de excepciones a la regla de la función múltiple. El o-nitrofenol, el salicílal- dehído y muchos otros derivadas o¡ fo-disubstituidos del benceno son arrastradles con vapor. L a explicación a esta aparente pérdida anormal del carácter polar se encuentra en la observación de que todos estos compuestos de excepción son capaces de encontrarse en formas célicas.

IO S S e p a ra c ió n d e m e zc la s
Gomo « bien conocido, las compuestos cíclicos tienden a ser no polares. A continuación se muestran las estructuras con enlaces de hidrógeno del o-nitrofenol y el salicilaidehfdo.
H
01T
H
Se¿id(*lilebjdo
U n ejemplo muy notable de esta disminución de propiedades polares lo constituyen los isómeros 2,4- y 4,6-diacetílresorcinoles. El primero funde a 91° y es arrastrable con vapor; el segundo funde a 132° y no se arrastra con vapor.
Recursos especiales
L a introducción de vario» grupos funcionales polares en una molécula produce una disminución en la solubilidad en disolventes no polares tales como éter, lígroína, tetracloruro de carbono y benceno (pág;. 86). De aquí que frecuentemente sea posible separar un compuesto soluble en éter, de uno insoluble, por tratamiento con éter absoluto. El tratamiento con un disolvente no polar puede también usarse p a% separar compuestos pertenecientes a otras clases de solubilidad. Por ejem5 pío, el ácido benzoico es mucho más soluble en éter que el ácido tereftá- lico. Los fenil- y a naftilurctanos son solubles en ligroína caliente o tetracloruro de carbono, mientras que la ítm-difenilurea y la «m-di-a- naftilurea son insolubles. Por lo tanto, el tratamiento con ligroína proporciona un medio de separación y se aprovecha este hecho para la purificación de los uretanos (pág. 268). A menudo tales separaciones no son tan aproximadamente cuantitativas como aquéllas que dependen de la separación de una substancia neutra de una sal.
En el capítulo 6 se indicó que el enlace de hidrógeno entre las moléculas del soluto y entre soluto y disolvente, es un factor importante que controla la solubilidad. Los compuestos que poseen grupos funció- nales que sólo actúan como donadores en la formación de enlace de hidrógeno, son muy solubles en cloroformo, pero no tanto en hexanoo tetraclomro de carbono. Se puede hacer uso de este hecho en la separación de mezclas.
A menudo, los hidrocarburos aromáticos se separan de los hidrocarburos paraflmcos o al [cíclicos por tratamiento con sulfúrico fumante que convierte el componente aromático en un ácido sulfónico fuertemente polar, el cual puede separara y volverse a transformar en el hidrocarburo por tratamiento con vapor sobrecalentado.
O
a.Niirdírwl

Con Iros o m ás componentes 109
SEPARACION DE MEZCLAS CON TRES O MAS COMPONENTES
Si una mezcla contiene más de dos compuestos, las combinaciones de los métodos anteriores frecuentemente conducen a separaciones satisfactorias. La condición necesaria para tener éxito en una separación es que tos componentes sean de tal nat»ralez<ij que posean una amplia diferencia de polaridad a que ésta pueda inducirse enfre los miembros de tutlquier par de ellos.'
Én la práctica, las mezclas caen dentro de dos categorías, dependiendo de su solubilidad en agua. Estos dos tipos se considerarán separadamente.
Extracción (mezdaá insolubles en agua}
Una rnczcla de clorobcnceno, dimeiüanüina, 0-naftol y anisaidehído ejemplificará el tipo insoluble én agua. Podría separarse de acuerdo con el siguiente proceso. Para hacer mínimas las pérdidas mecánicas y facilitar el manejo' de ia me*cla, primero se Je disuelve en éter,
Solución de Ja mezcla en éter
Extracción con ácido clorhídrico
Capa etérea
!Extracción con
hidróxido de «odio
Capa acuow
I’Adición de hidróxido
de sodio
Capa acucí*|
Aciduladóu
C apa etérea
1 'Extracción con bisulfito
de sodio
Vimitilanüttx*
0-8 afl<A rCapa acuosa
IAcidu] ación
Capa etérea
IDestilación
de! éter
A n u a l ¿«fado C larobeiiccno

1 1 0 S e p a ra c ió n dm m ezc lo»
En la pág. 116 se bosqueja un procedimiento genera! para este tipo de mezclas* Sólo es apropiado para mezclas en las cuales ninguno de loa componentes es*soluble en agua.
Destilación por arrastre con vapor
Si todos los componentes <le la mezcla son soluble» en agua, la extracción debe ser substituida por destilación con vapor, como su indicó en la discusión de mezclas binarías solubles en agua. La mezcla de pipe* rídina, ácido láctico, ácido acético y alcohol n-propíllco proporciona un ejemplo de este tipo de separación- La separación puede efectuarse con el proceso siguiente:
Disolución en Oj¡ru* de !a mezcla
!Arrastre con vapor A
Residuo A ñ ¿ o ¡¿ctico
DcMitacloIAddulación con KiFO«
y a rrau re -con vapor
Residuo D «tilado
Adición d« N aO H y » m it r e con vapor
iSolución xcuoKi de piperkfhd
A dición de N aO H y « ra s tre
con vapor
Residuo Destilado
Adición de H^POi y arrastre
con Vapor Iv
Solución acuosa
de ¿cido ac¿t'tc<>
SaturAción con K 3C 0 j y separación
de capas
Capa superior
alcohol r^propllieo
En la pág. 114 se bosqueja un método general para separar mezclas solubles en agua. Deberá notarse que este método puede resultar muy poco satisfactoria si no se escoge cuidadosamente la mezcla. Cuando los componentes de la mezcla han sido seleccionados en fo tm a inadecuada, reaccionan frecuentemente entre sí o con la solución acuosa de ácido o álcali caliente, durante la destilación por arrastre con vapor.

Procedimiento* d« loboWgri© 111
PROCEDIM IENTOS DE LABORATORIO PARA LA SEPARACION DE MEZCLAS
La identificación de Jos componentes de una inepcia implica, primero, la separación de ellos en compuestos individuales y, segundo, la caracterización de cada uno de estos últimos, de acuerdo con ]« procedimientos de los capitulo! siguiente*, Rara vez es jvosiblc Identificar lo* constituyentes de una m erda ain separarlo! previamente. La separación de los compuestos de una mezcla deberá ser casi tan cuantitativa como sea posible para obtener idea aproximada del porcentaje real de cada componente. Sin embargo, es mucho más importante llevar a cabo la separación de tal manera, que cada compuesto se obtenga totalmente puro, ya que esto hace mucho m ál fácil la identificación individual.
El método de separación seleccionado deberá ser tal, que los compuestos se obtengan tal y como estaban en la mezcla original. Los derivados de los compuestos originales no son muy útiles, a menos que puedan volverse a convertir con facilidad en los compuestos originales. Este criterio de separación es necesario debido a que la identificación de un compuesto se apoya finalmente en la concordancia entre las constantes físicas de la substancia original y las de un derivado de ella con datos similares obtenidos de la bibliografía.
El origen de una mezcla usualmente proporcionará información suficiente para indicar el grupo al cual pertenece la mezcla y, por lo tanto, ei modo de separación que deberá usarse.
Examen preliminar de las mezclas
1. Se otwcrva el estado físico. Si es un sólido suspendido en un líquido, se separa el sólido por filtración y se examina separadamente. Sí catán presentes dos líquidos inmiscibles, también se separan y se examinan individualmente.
2. Se determina la solubilidad o insolubilidad de la mezcla en agua.3. En mezclas liquidas, se evaporan 2 mi de la solución lm ía se
quedad, sobre un vidrio de reloj o la cubierta de porcelana de un crisol, y se observa la presencia o ausencia de residuo. Se hace al residuo la prueba de ignición. En una mezcla sólida se hace directamente la prueba de ignición.
4. En mezclas líquidas se averigua la presencia o ausencia de agua como sigue: a) Determinando la miscibilidad de la solución con éter; h ) sulfato de cobre (II) anhidro-, c) prueba de destilación para agua. L a prueba de destilación es más segura y se realiza en la forma siguiente: En un pequeño matraz de destilación, se colocan cinco mililitros de la mezcla líquida y 5 mi de tolueno anhidro. La mezcla se calienta suavemente para que se efectúe la destilación y se recogen 2 mi de destilado. Se añaden de 5 a 10 m i de tolueno anhidro al destilado. La presencia de

1 1 2 S e jw ro c iá rt m e zc la s
dos capas o gotas características suspendidas en t) tolueno, indican agua. Si la solución cata sólo opalescente^ es indicio de huellas de agua,
5. Sí se ha encontrado que no hay agua, deberá determinarse la presencia o ausencia de un disolvente volátil en la mezcla líquida, para lo cual se colocan 10 mi de la mezcla en un matraz de destilación de 25 mi. El matraz se introduce en un vaso de precipitados con agua iría, la cual después se calienta hasta ebullición. Cualquier liquido que destile bajo estas condiciones se clasifica como un disolvente volátil. El destilado, que puede ser una mezcla de compuestos fácilmente volátiles, y el residuo del matraz, se examinan separadamente.
Frecuentemente sucede que la destilación de una mezcla soluble en agua proporciona un disolvente volátil y un residuo insoluble en agua. Por lo tanto, Ja separación de dicha mezcla se efectúa eliminando todo el disolvente volátil. Entonces, se traía el residuo como mezcla insoluble en agua.
Si después de la destilación el residuo es un líquido soluble en agua, « preferible 'no separar el disolvente en esta etapa, ya que ordinariamente la separación no es cuantitativa.
Sin embargo, si el residuo después de la destilación es un sólido soluble en agua y la separación del disolvente parece ser cuantitativa, entonces es conveniente separar todo el disolvente volátil y examinar separadamente el destilado y el residuo.
Deberá tenerse en cuenta que sí hay agua presente no deberá intentarse dicha separación.
6. Deberá determinarse la reacción de la solución acuosa o suspensión de la mezcla con tornasol y fenolftaleína. Si la mezcla es francamente ááda, 5 mi de ella deberán valorarse con solución 0.1 JY de hidróxido de sodio, para determinar si están presentes cantidades considerables de ácido libre o si la acidez se debe a huellas de ácidos liberados por hidrólisis de ésteres. La titulación deberá realizarse con una solución helada y el punto final corresponderá a la aparición del primer color rosa de la fenolftaleína.
1. Dos mililitros de la mezcla se acidulan con ácido clorhídrico y la solución se enfría. Se observa si hay desprendimiento de gas o formación de precipitado. Después se añade solución diluida de hidróxido de sodio y se observa d resultado.
8. Se alcatinrzan con solución de hidróxido de sodio dos mililitros de la mezcla Se observa si hay separación de un aceite o sólido, liberación de amoníaco o algún cambio de color. La solución deberá calentarse justo hasta ebullición y entonces enfriarse. Después se compara su olor con el de Ja mésela original. A menudo un cambio de olor indica Ja presencia de ésteres. En seguida se añade ácido clorhídrico diluido y se observa el resultado.
9. En el caso de mezclas insolubles en agua deberá hacerse un análisis elemental. Si en una mezcla soluble en agua están presentes

Procedimientos de laboratorio 113
agaa 0 gran cantidad de disolvente volátil, se omite d análisis elemental. Sí la mezcla soluble en agua está compuesta por sólidos, se hace un análisis elemental
10. Si hay ausencia de agua se determina el efecto de Je* siguientes reactivos de clasificación: a) sodtO metálico; b) cloruro de acetilo.
11, Deberá determinarse la acdón de los siguientes reactivos de clasificación sobre una solución o suspensión acuosa de la meada original; a) agua de bromo; b) solución de permanganato de potasio; c) so* ludón de cloruro férrico; d ) solución alcohólica de nitrato de plata;e) reactivo de íucsina-aideJiído, á) fcnilhidrazina.
En esta etapa d d examen se resumen los resultados de las pruebas anteriores y se deduce toda la información posible a partir del comportamiento de la mezcla. El estudio preliminar mostrara el grupo en el que deberá clasificarse la mezcla y, por lo tanto, indicará cuál de los procedimientos siguientes deberá usarse para su separadón.
Mezclas de compuestos solubles .en agua
El diagrama esquemático de la pág. 114 representa mi método para la separación de este tipo de mezclas. Se puede y deberá modificarse para cim as mezclas, cuando el examen preliminar indique que tal modificación es conveniente.
P roced im ien to
1. Se colocan unos 50 mí de la mezcla en un matraz de 500 mi con fondo redondo y acondidonado para destilación por arrastre con vapor» Deberá emplearse una tram pa de vapor y un tubo de seguridad. Por arrestrc con vapor se recogen de 50 a 60 mi de destilado 1. El residuo1 del matraz se coloca en una cápsula para evaporadón y se evapora el agua sobre un baño m ará. Ocasionalmente pueden eliminarse mejor las últimas huellas de agua calentando a presión redudda. El residuo, sólido o líquido, se examina.
2. Se addula el destilado 1 con áddo fosfórico y se arrastra con vapor hasta obtener de 40 a 50 mi del destilado 2, El residuo 2, formado por el fosfato de Ja amina, se alcaliniza y se desfila la amina. Si k amina es muy volátil deberá recogerse en ácido clorhídrico y aislarse como dorhidrato. Las aminas menos volátiles deberán separarse de la solución acuosa por medio de carbonato de potasio.
3. El destilado 2 se alcaüniza ligeramente con h:.’róxido cL sodio diluido y de nuevo se destila por arrastre con vapor, recogiéndose de 30 a 40 mi del destilado 3, El residuo S {en el matraz) Jo constituye la sal de sodio del compuesto áddo, La m itad de ¿1 se acidula con áddo fosfórico y se destila con vapor. En el destilado se determinan los nú* meros de Dudaux. La otra mitad de la solución de la sal de sodio se usa para la preparación del derivado.
2*
üa

rJ
4. El destilado 3 cor tiene compuestos neutro? volátilea-alcoholes, aldehidos y cetonas. Estoí pueden separarse del agua por saturación coi» carbonato de potasio. Mediante loa reactivos de clasificación se determina si están presentes todos o alguno de ellos. Si sólo está presente uno, se destila el líquido, se observa el punto de ebullición y se prepara un derivado.
JJ4 Seperactóii de mételos
m ezcla i>e c o m p u e s t o s s o l u b l e s £N aova
M EZCLA1, Destilación}con vapor________
Residuo 1 Destilado 1 Acidulado coa H 1PO4
2. Destilación con vapor
Destilado 2 (ácido volátil;
cojnpucitos neutro* volátiles)
4
Residuo 2 (Fojfato de am ina)
IAdición de N aO H diluido
Adición d« N aOH y destilación
i3. Dtitilación con vapor Destilado
(Solución de amina)
1 ,Residuo 3
(Sal de N a del ácido volátil)
Dividir e n jd o i parte»
A ñadir H>PO* y destilar
iDeiiilado
iNúmero de Duclavx
para ácido volátil
Evaporar a «qu ed ad
iResiduo
Iir
Derivado
Destilado 3 (coíoj>u«to» neutros volátiles)
iSaturar con K^GOj y « p a ra r las capas
1Capa ruperíor
(Alcoholes, aldehido*, cetonas) Aplicar reactivos de clasificación
Comentarios. Er» la primera destilación por arrestre con vapor deberá recordarse que las sales aminas de ácidos débiles {v. gr. acetato de anilina) pueden experimentar una hidrólisis en tal amplitud, que tanto el ácido como la base se encontrarán en el destilado. Las sales de los ácidos fuertes (v. gr. clorhidrato de anilina) no se afectan con la destilación por arrastre con vapor, quedándose en el matraz de destilación.

Ptecadhnfento* d® laborírtotró 115
Nótese que el procedimiento bosquejado en las págs. 113-114 pro* vacará Ja hidrólisis de cualquier éster que pudiera estar presente* Si la presencia de un éster queda indicada por e l oJor o por el cambio de olor «n la prueba preliminar 8> deberá modificarse el procedimiento como se muestra en el proceso siguiente:
MEZCLAS SOI«UaLRS EN A O VA QUE CONTIENEN ESTERES
MEZCLA1
1. Enfriar, Wturarcon KjCO j
Separar la* capas
Capa acuoia I (Incluyendo cualquier
precipitado en suspensión)
Capa superior 1 Enfriar, neu traü iar exactamente
at anaranjado de atedio con HtSOi diluido y frío
Acidular Con HiPO<, filtrar
í l Precipitado 2 Filtrado 2
.Compuesto* ácidos) ^Destilar con vapor
r ~Destilado 3
(compuestos ácidos) *
Dcstfíar icón vapor
Destilado 5I
Enfriar, tóturar con KjCO j
Capa ¡superior 2
(compuestos neutros)
“ 1Residuo 3
IEvapora?
asequedad
- . . £ x tr« r t& n*««»» 4 J cÓkot HÍ7<TbUH
Residuo 6 i
(Sulfato* de aminai)
Capa «cuosa
Solución alcohólica de los ácidos
Si la mezcla no contiene compuestos ácidos,, 3a separación puede em( ezarse en el mismo punto que el tratamiento de la capa superior 1. En tal caso3 el residuo 6 puede contener compuestos ácidos (Jos cuales originalmente estaban presentes como sales) además de Jos sulfatos de aminas. Pueden separarse por el primer método que se indicó.
Este procedimiento modificado también puede usarse ventajosamente con mezclas que contienen ácidos y alcoholes, ya que en este caso no hay ninguna oportunidad para que ocurra una esterificación.

116 Seporocttn de mexclos
Mezclas de compuestos insohiblcs en agua
Después de la separación de cualquier disolvente volátil, deberá usarse el siguiente procedimiento para la separación de una mezcla insoluble en agua:
MEZCLAS DE COM PUESTOS IKSQLUBLES EN ACtfA
M E Z C L A
R es id u o 1
Añadir éter y filtrar
JS o lu c ió n l
IE x tra e r co n Acido
c lo rh íd r ico
ICapa etérea 2
1E itra e r con
hidróxido I de jodio
íC apa etérea 4
1 ' Evaporar
el é i«
Compílenosneu tn»
iS o lu c ió n 4
1S a tu ra r c o n
a n h íd r id o c a rb ó n ic o
1E x t ra e r c o n é te r
I 1Capa etér«3 5
íSolución 5
11
EvaporarT
Acidularel ¿ter
| 1T
Acidos▼
. Acidoidébilej fuertei
Solución 2
iAñadir hidróxido de tedio
extraer co n ! éter
r r r iCapa etérea 3 Solución S
1 1Evaporar
el éter
Ba»e*
Neutralizar exactamente con ácido
acético
C o iu p u c u o *AJífóterOJ

Proc«dlrotemos do laboratorio 117
P rocedim iento
De 25 a 50 g de la mezcla se agitan con 7 ó mi de éter y los compuestos insolubles {residuo 1) se separan sobre un íütro y se lavan con éter. Los lavados etéreos se añaden a la soludón etérea original y entonces se extrae la solución etérea ( I ) con tres porciones de 30 mi de solución de ácido clorhídrico al 5%. Si durante esta extracción se separa un clorhidrato de amina sólido, deberá diluirse con agua el áddo clorhídrico hasta que se obtenga una solución.
Se reúnen los extractos de ád d o dorhídrico (soludón 2), se alca- línizan con solución de hidróxido de sodio y la mezcla resultante se extrae con vacias porciones de 25 mi de é te r La capa etérea (3} se seca con sulfato de sodio y se destila el éter. El residuo está constituido por ha9?s.
De pués de la extracción la soludón (3) se neutraliza cuidadosamente con ácido acético. Si se separa un sólido puede diminarse por filtradón, pero, es mejor extraer la soludón cuatro o cinco veces con porciones de 25 mi de éter para recuperar cualquier compuesto anfótero.
La capa etérea (2 ) , proveniente de la extraedón con áddo dor- hidríco, se extraen Con tres porciones de 30 mi de soludón de hidróxido de sodio al 5%. Si se forma una emulsión puede añadirse más agua y un poco de alcohol para provocar la separadón de dos capas.
La capa etérea (4) se seca con sulfato de sodio y se destila d éter. El residuo contiene los compuestos neutros. Deberán emplearse reactivos de clasificación, lo mismo que pruebas planeadas, para determinar si el residuo es o no una mezcla. A menudo esta mezcla puede separarse por medio de una destilación oon arrastre de vapor. Si está presente un aldehido, deberá extraerse por medio de una soludón de bisulfito de sodio, con adición de éter para facilitar la separación. Si el residuo es un sólido, frecuentemente se Je puede cristalizar fraccionadamente de alcohol, Si el residuo es un líquido y aparentemente no es posible separarlo por métodos químicos, quizá se le pueda destilar fraccionadamente.
La soludón de hidróxido de sodio (4} se enfría y se satura con anhídrido carbónico. Los ácidos débiles que allí estuvieran se extraen con varias porciones de éter (capa etérea 5). Para separar cualquier éter disuelto, la solución (5) se calienta sobre el baño de vapor durante unos cuantos minutos y se agita. Entonces se le acidula y cnMa y los ácidos más fuertes se separan por filtración o por extracción con éter.

í *
b §’f "
i b
L * i
t
9
h
11 8 S e p a ra c ió n de m é te lo *
Mezclas encontradas en d trabajo de síntesis
Todo estudiante ha separado ya muchas mezclas durante su primer año de trabajo de laboratorio en química orgánica. Los principios usados para planear procedimientos conducentes al aislamiento y purificación de un compuesto orgánico a partir de una mezcla de reacción, son idénticos a los que ya han sido señalados en las secciones precedentes. De hecho, los grandes mejoramientos en los rendimientos de los compuestos orgánicos, que se pueden obtener a partir de una determinada reacción, se han debido a la aplicación de los conocimientos concernientes a la solubilidad y comportamiento frente a Jos reactivos de clasificación, con el fin de planear ct método de separación más eficiente.
Los siguientes procedimiento;- usados en los cursos iniciales de química orgánica, muestran la separación de mezclas encontradas en el trabajo de síntesis.
Preparación de bromuro d i n-h¿xí!o. Se refluja durante 2.5 horas una mezcla de alcohol n-hexilico, áddo brombídrjeo al 46% y ácido sulfúrico concentrado. Después de enfriar, se añade agua y se separa la capa inferior de brumuro de n-hexilo y alcohol n-hexOico que no reaccionó. Este producto crudo se lava varias veces con porciones sucesivas de áddo sulfúrico concentrado y frío. Entonces se lava con agua, se seca con cloruro de calcio anhidro y se destila.
En la etapa de la purificación con ácido sulfúrico concentrado, el bromuro de n-bexilo es insoluble mientras que el alcohol n-hexílico reacciona para formar sulfato ácido de n-hexilo, eJ cual se disuelve en el ácido sulfúrico concentrado y, por lo tanto, se separa. El bromuro de n-hexüo puro no puede obtenerse simplemente secando y destilando el producto crudo de la reacción porque el bromuro de n-hexilo hierve a 157°C y el alcohol «-hexiüco a 156®C.
Preparedón de acetato de n-butilo. Se hierve un exceso de ácido acético con alcohol «-butílíco, en presencia de una pequeña cantidad de ácido sulfúrico concentrado. La mezcla resultante de los compuestos indicados se separa según el proceso de la pág. 119.

P ro ced im ien to» la b o ra to r io 119
MEZCLA
Acido acético, alcohol n-butílico, acetato de «4iutüo, agua, ácido sulfúrico „ i
D e s t ila r ,
-------------------------------JResiduo
ifV m iado
i(Acido acóüco, alcohol n-bu tilico
acetato de n-butiio, »#ua)
l(A ddo *utfí(rico)
aón tAAadir solución |tatut*d?. de NjijOO,
íCapa inferior
I{Solución de
acetato d í fodio)
Capa rjpeñoT
4(Alcohol n*hutílico, acetato
de n-buúio)
Añadir solución saturada de CaClt
lCapa inferior Capa luperior
; i (Solucióc conteniendo (A cétalo de
alcohol n-butílico) 8-buttto)i
Secar con rulfato de rnagneaio y filtrar
rFiltrado
Acetato de n-budío
Destila*-
1Destilado
(Acetato de «t-budio puro)
Sólidoi
(Sulfato de ma#ae»io
h idratado)

120 S*pdrtKl¿n
Preparación d e q u in o ie ín c . La reacción de Skraup produjo una mezcla compuesta principalmente de las substancias que se indican abajo, las cuales se separaron como sigue;
MEZCLA
Sulfato de quínoleína, sulfato de anilina, nítrobenceno, gUccrma, ácido sulfúrico, sulíatos ferroso y férrico, polímeros de la acroleína.
D iluir con agua I y destilar con vapor
i-------- |D ettiltd o 1 Residuo 1
} i(NítrobcrtccTic*)
(Sulfato de quinoleína, »dfato de anilina, gl icen na, ácido sulfúrico, salíate* ferroso y férrico, polímero* de la acroleína)
Alcaliniiar coa N aO H I y destilar con vapor
|----- lDestilado 2 Reiiduo 2
i i (Q u indelna , anilina, agua) (Glicerína, sulfato de jodio,
; sulfato* ferroso y férrico,
D ifo lw r « „ le íd o «u liü rico. I " * » " » * « « M u . ) añ ad ir NaNO» y calentar
4Solución
(Sulfato de quínoleína fenol, ácido sulfúrico,
sulfato de tedio)
íAr.adir NaOH y destilar con vapor
_______ I_______________
D a tila d o R e tiJo o
i *(Quinolcina, agua) .(Fenóxido de sodio, sulfato
j, de sodio)
Separar, tecar con MgSOt y destilar
iDestilado
{Quinoleína pura)
P ro ce d im ie n to s ele la b o ra to r io 121
Procedimiento alterno para la purificación de la quinoUina:
Destilado 2 (Quínoleína, anilina, agua)
Extraer | con benceno_____
Cafia Acuosa
Solución en ácido clorhídrico (clorhidrato
de quinoleína)
A ñadir N aO H y extraer con C«H*
Solución nn benceno (Quinoleína, anilina)
iA ñadir cloruro de /B-toluearulfonílo
7 verter sobre ácido clorhídrico
í ---------------1----------Solución en
benceno ÍKalucnsulfonatnilida
£Solución bencóriiea {Quinoleína}
Secar con MgSO< destilar fraccionadamente
1 .Soluciónacuosa
Primera fraccióni v
(Berree no)
-----Segunda fracción
(Quínoleína pura.)
Ejercicio»
I. En forma de diagram a desarrolle u a procedimiento satisfactorio para !a separación de las mezclas siguientes y la identificacióxi de cada componente.
1. Agua, alcohol isopropílico, sulfato de anilina, lactosa, ácido ¡sobutírko.2. Cloroformo, anilina, ácido benzoico, A^V-dietiJanilina, naítaleno.3. Benceno, d iíen ilam ina, qu ino lelna, nifrobenceno, «i-dinicrobenceno,4. Alcohol m etílico, o -cre jc l, ácido BalícíJico, m etib n ilin a , /j-to lu id ína, es-
tireno , antraceno.5. T etrac lo ru ro de ca rb o n o , benzaldeh ído , «alicilaldetiido, triíerúlcarbino!,
alcohol bencílico.6. E ter etílico, alcohol etílico, acetona, dietilcetuiia, dietilaniina, ácido
acético,7. Dieiilamma, dibromoanilina -2,4, quinaldina, ftalimida, fenol, p-dicloro-
benetno, benzamida.8. Ciclohexano, p-toluidína, quinoleína, ácido 2-<'W>4-amiiwbeQzo¡co, m-
cresol, benceno, S-nilro-S-bromoanisol. «9. U na mezcla sólida d e benzoato de íodio, oxalato de sodio, clorhidrato
de fenilhidracina, ácido heníoico, alcanfor.10- U n instructor mezcló lo> compuesto» siguiente» en el orden indicado y
enlrígó la niévela tom u problema para identificación, podría «i-

122 Sepccrcoé ti d* c e r d a s
pcrar encontrar el esuxl'anie ? Hara. yn diagram a para la posible separación de los componen leí que realmente habría d espuá que la mezclarse hubiera, quedado una sem ana en reposo. {Suponga que las «acciones hubieran ¿ido completa»). Alcohol kr-butíl¡co (7.4 j j ) ; alcohol bencílico (10.3 g ) ; benzaldehído (10.6 g ) ; cloruro de acetilo (15.6 g ) : acctofenona (12.0 g ) ; dimctilanilina (-B4.7 g ).
I I . Los problenm siguiente* ejemplifican la separación de mezclas encontradas realmente durante el cuno de preparación de cocipuestoj simples. Se Indican las m ate ras primas y reactivos esenciales. Primero haga una lista de loe compuestos producidos corno resultado de las reacciones principal y secundarias y cotonees pODga en forma de diagram a un procedimiento apropiado para la separación de lo» compuestos, empezando con la mezcla obtenida al concluirse la reacción. E n seguida com pare su m étodo con el descrito en la bibliografía.
1. Se preparó éter etílico adicionando etanol al 95% a tutfato Acido de etilo calentado a 145*.
2. Se preparó bromuro de n -l tilo por calentamiento a reflujo de alcohol n-tutíÜco con solución acuosa i e ácido bmjnhldrico-Scido sulfúrico.
3. Se preparó b rom ob encino y ¿ * d ibromobenccno por acción del bromo sobre el benceno, en presencia de hierro,
4 . Se preparó p-brcmonUrohcuceno por acción de una m étela de ácidos nítrico y sulfúrico concentrados sobre el bromobenceno.
5. Se preparó etilbenceno p o t reacción entre el bromuro de ¿tilo y el benceno, en presencia de tricloruro de aluminio anhidro.
6. Se preparó anilina por acción de citano y ácido clorhídrico sobre nitro- benceno.
7. Se preparó p-clordolueno por reacción de Sandmcyer, a partir de o-to* luí di T ía.
8. Se preparó alcohol bencílico y ácido benzoico p o r reaección de Canni- ziaro, u partir de bcnzaldehído e hidróxido de potasio en solución acuosa.
9. Se preparó ácido bencílico por acción de una solución acuosa de h idróxido de sodio y brem ato de sodio sobre benzoína.
10. Se preparó el « ‘hutilacetoacetato de etilo p o r akjuilacíón del aceto- acetato de etilo con bromuro de n-butilo, en presencia de etóxido de sodio en etanol absoluto.
11. Se preparó fl*amll-me<íkeiona por hidrólisis del n-bucHacetoacetaio de etilo.

CAPITULO
8Aplicación de las pruebas de clasificaciónEn l l EXAMEN de un c o m p u e s t o orgánico, la aplicación de las pruebas de clasificación viene después de la determinación del punto de ebullición (o de fusión), de la solubilidad y del comportamiento en la ignición. A partir de esto» datos y del aspecto del compuesto (color, estado físico, olor), generalmente es posible descubrir indicios acerca del tipo de grupos funcionales que pueden estar presentes. El siguiente paso en la identificación, es la búsqueda de información específica concerniente a la presencia o ausencia de grupos funcionales comunes. Para este propósito deben Seleccionarse algunos reactivos de clasificación.
En cada uno de los experimentos incluidos en este capítulo, se dan instrucciones para el uso del reactivo que se ha encontrado útil en la identificación de grupos funcionales. Algunos de estos reactivos son específicos para un grupo funcional en particular, ya que cada una de las pruebas posee determinadas limitaciones. Consecuentemente, es necesario ensayar varios de ellos, antes de que pueda hacerse una clasificación satisfactoria. Sin embargo, es indeseable ensayar todos los reactivos sobre cada substancia desconocida. Este modo de ataque no tan sólo es un desperdicio de tiempo sino, además, pue^e ocasionar serias confusiones.
Las sugestiones siguientes proporcionan una forma de iniciar e). ataque del problema estableciendo o excluyendo la presencia de grupos funcionales. 41 mismo tiempo que se efectúa una prueba con la substancia desconocida, deberá realizarse una prueba control con los compuestos conocidos sugeridos en cada experimento, Así, es posible hacer comparaciones directas e inmediatas entre los resultados de los experimentos sobre los compuestos conocidos y el desconocido. Es muy importante observar fielmente como se manifiestan las pruebas positivas y negativas.
SELECCION DE PRUEBAS PARA CLASIFICACION
Para una rápida referencia, las pruebas de clasificación han sido ordenadas alfabéticamente en forma de experimentos numerados. En la discusión siguiente, los números dentro de paréntesis o paréntesis angu-

1 2 4 A p lic a c ió n d e lo s p ru e b a s d e d o s if ic a c ió n
laxes se refieren a estos experimentos. Siempre que sea posible, la prueba de clasificación realizada deberá <er la sugerida por la información obtenida previamente sobre la substancia desconocida.
C om puestas ácido* ¿n so lución ¿fi bicarbonato desodio , Es muy probable que los ácidos carboxílicos, ácidos sulfónicos y ciertos fenoles substituidos, sean las substancias que se encuentren er, esta clase. A menudo no son necesarias otras pruebas de grupo funcional y es muy ventajoso preparar una lista de posibilidades y proceder con la preparación de derivados y la determinación de otras constantes físicas, ta! como se describe en el capítulo siguiente.
Los cloruros de acilo y los anhídridos de ácidos de bajo peso molecular, que pueden comportarse corno ácidos en virtud de su hidrólisis a ácidos durante la prueba de solubilidad, pueden reconocerse per su rápida conversión a anilidas [1{&) J. Los fenoles que se encuentren <n esta clase frecuentemente pueden distinguirse por las pruebas de fenoles que se describen a continuación.
C om puestos déb ilm en te ácidos. Los fenoles son el tipo más común de compuestos en esta clase. Su presencia la indican las pruebas d d cloruro de acetilo [!{ ¿ ) ], del agua de bromo (6 ), del nitrato cérico (7) y del ácido nitroso (19), así como Ja reducción del permanganato <22).
C om puestos básicos. Las aminas básicas primarias, secundarias y terciarias se encuentran en este grupo y se identifican por medio de la prueba de Hinsberg (4) o del ácido nitroso (19). Las arilaminas se bn> m an fácilmente (6).
C om puestos soluble* ett agua. Los compuestos solubles en agua presentan problemas particulares; ya que los ácidos débiles (tales como muchos fenoles) y las bases débiles (tales como las aminas aromáticas) frecuentemente no se identifican fácilmente con pruebas generales, usando indicadores tales como tomasok Cuando estas substancias se encuentran en una mezcla, sü comportamiento durante la separación por destilación con arrastre de vapor a menudo de muestra la presencia del grupo débilmente áddo o básico. De otro modo, es necesario ser particularmente cuidadoso al realizar las pruebas de grupos funcionales. Además de los compuestos de bajo peso molecular, tales como los aldehidos, tetonas, alcohole*, fenoles, éteres, éteres, ácidos, aminas, amidas y otros compuestos monofuncionales con grupos funcionales más complejos, los compuestos solubles en agua pueden incluir otros grupos, por lo que ameritan una discusión especial.
Saies de ácidos carboxlltcos. L a solución de Ja sal se acidula y se libera el áddo carboxllico. Si el ácido es insoluble en agua, se le separa por filtuclón y se le identifica como ya se indicó. Si el ácido es soluble, de la solución acuosa se le puede aislar por extracción con éter o cloroformo y posterior eliminación del disolvente por destilación.

Se le cc ión d e p ru e b e s 125
Sales de ¿cides fu l fónicos. L a sai de un ácido sulfónico se transforma en su correspondiente cloruro tratándola con pentadoruro de fósforo. El cloruro de sulfonílo tratado con hidróxido de amonio produce una solfonamída. Cuando la amida se ha. purificado y se ha determinado su punto de fusión, puede prepararse una lista de compuestos posibles (ver pág. 324).
Sales de aminas La adición de álcali liberará las bases libres, las cuales se tratan según el caso. Ordinariamente se ahorra tiempo y es' más conveniente aplicar directamente las diversas pruebas para aminas [1 (6 ), 4, 6, 1?, 18, 19] a la sal de la amina.
Compuestos polifuncioncUs. Los ácidos polibásicos y Jos hidroxí- ácidos se caracterizan mejor por referencia, a sus equivalentes de neutralización (capítulo 10). Los azúcares, que en la prueba de ignición se carbonizan en forma característica y desprenden un olor acaramelado, deberán tratarse con solución de Bcnedict (3} y reactivo de Tollens (29). Si está presei.:-*. un azúcar reductor, m hace la prueba de la osazo- na (21)* Si no na y reducción, la substancia desconocida deberá hervirse con ácido clorhídrico diluido durante algunos minutos y repetirse las pruebas. Si se obtiene una prueba positiva para azúcares, deberá determinarse la rotación óptica (p<cg. 63), Los alcoholes poiioxhidrílicos y l$s cetoakpholes se c a i^ te ró an por su reacción con h al uros de acüo (1), hidroxílamina (15), fenílhidrazína (21), 2,4-drnitrofenilhidraziru o áddo peryódico (20). Los aminoácidos, pueden investigarse con áddo nitroso (19). Los equivalentes de neutralización {capítulo 10) también pueden determinarse en los derivados benzoilados
El grupo carbonilo de aldehidos y cetonas es el grupo fundonal de esta clase y es para el que se dispone de las pruebas más generales y segura s, Tanto los aldehidos como las ce tonas responden a las pruebas con 2,4-dinítrofenílhicLrazina (9 ), hidroxilamina (15) y fenrlhidrazi- na t21), Una diferenciación más completa se obtiene usando fucsina (12), solución de Benedict (3), reactivo de Tollens (29) o hipoyodito de sodio (16). Los alcoholes pueden investigarse mediante el cloruro de acetilo [1 (</)], cloruro de benzoilo £1 (* )], anhídrido crómico (8) o sodio (24). Si estas pruebas son positivas, se puede obtener información adicional empleando la prueba de Lucas (14), ácido peryódico (20), la prueba del yodoformo o el nitrato cérico (?)» Los ésteres pueden evidenciarse con la prueba del ácido hidroxámico [1 5 (¿ )] o por hidrólisis con solución de hidróxido de sodio £26(a) ] y dan útiles equivalente de saponificación (capítulo 10). Los anhídridos y haluros de acilo reaccionan con la anilina formando anilidas [1(¿0 , 1 (¿ )] y son hidroíizados por álcali (26). El ácido que se produce puede caracterizarse por deter- minadón del equivalente de neutralización, del coeficiente de particióno de las constantes de Dudaux (ver capítulo 30). En ausencia de pruebas positivas para otros grupos funcionales, generalmente deberá pensarse

12 6 A p lic a c ió n d e la s p ru e b a s d e d o s if ic a c ió n
en los éteres; éstos pueden escindirse con ácido ycdhídríco (13) y, si son aromátírro, se irán bnwr.cjido lentamente (5).
C om puesto# neutros que con tienen nitrógeno- La hidrólisis con solución caliente de hidróxido de sodio (26) sirve para convertir las amidas, amidas substituidas, los nitrilos y determinadas aminas aromáticas con subsrituyentes negativos, en los ácidos o fenoles correspondientes, con liberación de amoníaco o aminas. Los grupos nitro pueden dilucidarse mediante hidróxido ferroso (11} o cinc y cloruro de amonio (30).
C om puestos inertes» U n halógeno reactivo se evidencia con la prueba del nitrato de plato (23) o !a del yoduro de sodio (27). El ácido sulfúrico fumante (28) puede usarse pana investigar núcleos aromáticos, haya o no halógeno presente. La condensación con azoxibcnceno [2(cí)]o con cloroformo [2(¿>) ], en presencia de tricloruro de aluminio anhidro, también sirve para averiguar la presencia de anillos aromáticos.
¡m aturación . Las pruebas pa-a enlaces dobles o triples {5, 22) pueden empicarse con todos los com^ 'estos excepto aquéllos que no se afectan con el áddo sulfúrico concentrado.
C om puestos halogenados. Tanto la prueba del nitrato de plata alcohólico (23) como la del yoduro de sodio (27) pueden ensayarse para obtener información concerniente a la reactividad del halógeno.
C^nentarios. No se ha desarrollado ningún “proceso analítico” de rutina que pueda garantizar que conduzca a la identificación rápida de cualquier compuesto orgánico desconocido. Por el contrarío, el camino más eficiente para averiguar ía estructura de una substancia puede ser totalmente diferente del seguido para otra. Sin embargo, algunas sugestiones generales serán útiles.
Efectúe cada prueba con suficiente cuidado como para que esté seguro de que si repite la prueba obtendrá un resultado idéntico. Deberá tomarse en cuenta que las pruebas tienen diferentes grados de contabilidad y que dos pruebas diferentes para el mismo grupo funcional pueden dar resultados discordantes, En dichos casos habrá que juzgarlos críticamente y, cuando'sea posible, dar más importancia a ciertas pruebas que a oteas. Por esta razón, a menudo, es aconsejable colocar juntas todas las pruebas para un determinado grupo funcional, cuando se les anota en las hojas de reporte. Una buena prueba positiva puede tener más valor que muchas negativas, por cjempío, suj>onga que una substancia desconocida da un precipitado con 2,4-dínitrofcniíhidrazina, pero Ja prueba con fenilhidrazina' es negativa. En lugar de repetir cada prueba varias veces (lo que generalmente da los mismos resultados anteriores)o efectuar otras pruebas de grupos funcionales, !o cual a menudo aumenta la confusión, c¿ más ventajoso examinar más dcienidamente la prueba con la 2,4-dinitrofenolhídra2Ína. La posibilidad de que el producto obtenido sea en realidad 2,4-dinitrofcmlhidrazána recuperada o el compuesto original, puede excluirse fácilmente por comparación de los puntos de fusión o por el punto de fusión de la mezcla. La posibilidad de que el

d o p ru e b a* 127.
reactivo dé 2,4^aiitrof«nilliidm iaa fuera demasiado sensible y diera una prueba positiva debido a huellas de alguna impureza de la substancia desconocida, usualmente Se puede excluir por un balance de materia]. Una. vez que se eliminan estas posibilidades, la prueba negativa de fenil- h ídraána y la prueba positiva de )a 2,4-dinitrofenilhidrazina, juntas constituyen una prueba positiva para un aldehido o cetona.
A menudo las substancias desconocidas están contaminadas con impurezas, las cuales se separan con dificultad y pueden interferir seriamente con las pruebas de clasificación. Así, los alcoholes bencílicos pueden estar contaminados con los aldehidos correspondientes (formados por oxidación con el aire) y dar positiva una prueba de carboníio.
A'Cintro ¿ti 4*ptñr>t**tO Pr*t1* Pigiru
1 Cloruro de acílo 1262 Cloruro de aluminio con azoxibenccno o cloroformo ISO3 Solución de Bcnedict 1324- Cloruro da benecnaulfoililo (prueba de Hinsbcrg) 1355 Solución de bromo en telradoruro de carbono 1 3 7 4 r-6 Agua de bromo 1397 R eactivo 'd« nitrato cérico (408 Anhídrido crómico 1419 2,4-dinitroíenilhIdw ina 142
10 Solución de cloruro férrico 1+311 H idróxido ferroso 14412 Reactivo Íucíina-aldehido 14513 Acido yodhldrico (método de Zeísd pa ra alcoxítoe) 14614 Acido dorhidrico-cloiuro d e d n c (prueba de L ucm} 14815 C lorhidrato d e hidroxilamina 15016 Yodo t hidróxido da codío (prueba de yodoíonno} 15417 Cloruro de níquel, blíulforo de carbono e hidróxido de amonio i 56ia C loruro d e níquel y 5-mtroBalidlaIdchfdo 15719 Acido nitro*» 15820 Acido peryódico 16221 Fenilhidrazina y í-nitrojeriiüudrazina 16422 Solución de permanganato de potasio 166 •23 Soludón de nitrato de plata. (etanfilíca) 16924 Sodio 18025 Solución d e bisulfito de sodio l e í26 Soludón de hidrósido de «odio 18227 Yoduro de jodio en acetona 188
. 28 A ddo sulfúrico (fumante) 19029 Reactivo de Tov.cn» 19230 Cinc y cloruro de amonio 192

1 2 8 A |r f k o d ¿ « d * l a » p r u e b a * d e d a « 1 f l< c d ó n
L a rccriitalización de un sólido o la destilación de un líquido ayuda a evitar tales dificultades o, por o tra parte, puede crear &lgunas*nuevas. P or ejemplo, cuando el éter dibencilico se destila a presión atmosférica sufre una descomposición parcial a benzaldehído y tolueno y ciertos alcoholes alQicos, cuando se calientan, se transponen a sus aldehidos saturados isómeros.
El comportamiento detallado, aun de los compuestos relativamente sencillos, es suficientemente complejo, por lo que los conocimientos obtenidos por la sola lectura de libros de texto pueden inducir a muchas equivocaciones. Se aconseja a los estudiantes que contmuamente se re- fieran a ía bibliografía para obtener información acerca de la química de los'compuestos que esten considerando. En más de una ocasión, la identificación del nitrobenccno ha sido retardada porque el investigador 7» sabía que puede desprender amoníaco cuando se calienta con hi- dróxido de sodio concentrado. En forma similar, la falla de una subs- Uncía desconocida, que se pensó era jt-hidroxibenzaldebído, en formar el producto sólido de adición bisulfítíca o el color esperado de la fucsina, es una. desconcertante falla en la identificación hasta que se buscan dalos en la bibliografía; posteriormente, estas observaciones son confirmación valiosa de la estructura postulada.
Para una rápida referencia se ha preparado una lista de las pruebas de grupas funcionales, ordenadas alfabéticamente en la pág, 127.
E xperim ento 1. C loruros de acilo
O O/ /
a ; RC—Cl + H20 — RC—OH 4- HCi\
Con precaución, añada unas cuantas gotas de d o n iro de a£¿liln a 1 mi de agua y toque cl tubo para ver si se ha desprendido calor. En forma similar, ensaye el comportamiento del cloruro d e ’benzoila en agua,1 ¿Qué diferencia observa?
* Tt*g» Ja prcoudón de dctinAf «1 c a c c IO ¿k clonwo An bcnroilo («r lúliicfóo diluid» de h M r& á d o do «uoonk» w t c d e v e rte r io t rtíixlu'7» tv . e l fzCg»zlen>. E « t cóíi> iai«kj •n BJJ p o U n Kiurimfscuo.

E xp e r im e n to 1. C lo ru ro s d e a c iio 129
Repita cada una de las pruebas anteriores usando C.5 mi de anilina en lugar de agua. ¿ Cuáles son los productos de esta reacción? Vierta la mezcla en 5 mi de agua, ¿Qué es el precipitado? ¿Qué hay en la solución?
c)
Trate 0.5 mi de piridina o quinoleína con unas gotas de cloruro de acetilo. Explique por qué se libera calor en esta reacción, tomando en cuenta *^je la amina se recupera después que la mezcla se diluye con agua y enseguida se neutraliza,
/ ° / °d ) RC—C1 + R'OH -h. R C -O R ' + HCl
Añada gota a gota 1 ra] de cloruro de acetilo a (I) 1 mi de etanol; (2) 0.5 g de fenol. En cada caso deje reposar la mésela en reacción durante uno o dos minutos y luego viértala con precaución en 5 mi de agua. En (2 ), separe el líquido y pruebe su solubilidad en soludón diluida y fría de hidróxido de sodio.
e) Reacción de SchoU en-Batw uum ,
/ ° s °RC—C1 + R'OH + NaOH — R C -O R ' + NaCl + HsO
/ °RC—C1 + R JNH2 + NaOH — RC—N H R ' + NaCl + HáO
En xtn frasco pequeño con tapón de vidrio coloque 5 mi de etanoí, 10 mi de agua y 2 mi de cloruro de bertzoílo. A esta solución, añádale, en porciones y con agitación vigorosa, 10 mi de soludón de hidróxido de sodio al 20$>, Agite Ja mezcla durante varios minutos y entonces pni^be la solución con papel tornasol hasta asegurarse que aún está* alcalina. ¿Cuáles son los productos de la reacción? Repita el experimento ante-
.rior usando 2 mi de anilina en lugar del etanol. ¿Qué ventajas tiene este procedimiento sobre la beruoiladón sin el uso de álcali como en (6 )? ¿De qué factores depende el éxito de la reacción de Schotten- Baumann?

§
TB
a©
230
Ib
n
13 0 A p lic a c ió n la s p ru e b a s d e d a j f f ic a d ó n
2Jk. Comentarios. Es evidente que los doraros d* acilo (y probable-W Ynente también los anhídridos) reaccionan a travos de modificaciones de
dos mecanismos limitantes, el primero, análogo a 1 os mecanismos de adición al carbonilo que se observa en la hidrólisis de esteres y, e] segundo, análogo a la hidrólisis de haluros alifáticos terciarios.*
OCH jO O O
Re! ~ X - - * • > RC* - j jg - > RC - O H
La reacción de Schotten-Bauman. . es de especial interés, ya que sería de esperarse que el agua y el ¿o.* oxhidrilo presentes pudieran competir con el alcohol o amina que se van a a c u la r y redujeran fuertemente el rendimiento del producto deseado. El éxito de la reacción se debe a una combinación de circunstancias. Generalmente ocurre en un nwdio heterogéneo con el reactivo orgánico y el cloruro de ben- zoílo en Ja misma fase. Además, especies tales como el ion etóxido y las aminas, pueden competir bastante bien con el ion oxhidrilo por el
J cloruro de acilo, aun en un medio homogéneo.*
«JP
: ba) Coloque 2 mi de benceno seco en un tubo de ensayo limpio y
¿ " f e seco; añada uno o dos cristales de azoxibencéno y aproximadamente m W 0.1 g de cloruro de aluminio anhidro. Observe el color. Si inmediata
mente no so produce color, caliente la mezcla durante unos cuantos minutos. Efectúe la prueba con éter de petróleo, dorobenccno, bromuro
' M de etilo y naftalcno.4\ • Cornentarioj. Esta prueba deberá aplicarse sólo a compuestos que
sean insolubles en ácido sulfúrico. El color producido se debe a un compuesto de adición formado a partir del p^fenitazobenceno y el elo-
£ L" rvro de aluminio.5
E xp erim en to 2 . C loruro de a lum in io con axox¡benceno o c lo ro form o
* Vtr GoU, Killao y Jcfíerton, /. Ch*m. -Sor., 1W, par* rtíer«r»c¡ai.> Vr,r Svf»St» Y Scott, / , Am. Cheto. íce -, 7 í . 141* Si « l hidrocarburo e* VO vbMdo, puede U ia v UO» ♦oloaófi i*. C-5 $ ó* él «o 2 fn' de
YiiíuUntu ¿c catri>vD0 WcO.* V e- P u a u o t r 1 fcuiabfel, B*r., M , 2763 ( J92l> y Pumraerer, Birajlíet, BÍMiWT y
S d * * s ra ít 53, f m <1532).

E xp e r im e n to 2. C lo ru ro d* a lu m in io 131
El cambio a p-íe¡úIazobenreno se puede formular como una substitución aromática del benceno por el Áddo conjugado {o el aducto de doruro de aluminio) del azoxibenceno, seguida por eliminación de los dementas del agua {o HOAlCIj'J.
Azoxíbenceno + H-* -— >
N—N<
^-FeniJarobcricírio
Los hidrocarburos aromáticos derivados del benceno y sus derivados halogenados forman una sojudón cuyo color va del anaranjado intenso hasta un rojo obscuro o forman un predpit&do. Los hidrocarburos polinucleares condensador tales como el naftaleno, el antracenó y el. fenantxeno, dan colores cafes. Los hidrocarburos alifáticos no dan coloro cuando mucho un amarillo pálido.
b) A 2 mi de cloroformo seco, contenidos en un tubo de ensayo, añádales 0.1 mi (o 0.1 g) de benceno. Mézdelos perfectamente e indine el tubo de ensayo hasta humedecer la pared. Entonces añada de Ü.5 a LO g de doruro de aluminio anhidro, procurando que algo del polvo Se pegue a la pared laterat del tubo de ensayo. Observe el color del polvo que está sobre la pared, lo mismo que el color de la soludón. Efectúe.la prueba con éter de petróleo, clorobenceno y bifenilo.
Comentarios. Los colores producidos por Ja reacción de compuestos aromáticas con eJ cloroformo y -d cloruro de aluminio son bastante característicos. Los compuestos alífáticos insolubles en áddo sulfúrico no dan color o sólo un amarillo muy pálido. Los colores típicos producidos son los siguientes:
Compuesto Color
Heneen? y sub homólogos Anaranjado a rojoHaJuros de ¿rilo A naranjado a to jo
N afuJeno • Azuli>jfenilo PúrpuraFenantreno PúrpuraAntracenó V«rde
Con el tiemj>o, los colores cambian a varios tonos de café. CoJores pa- neddos se obtienen cuando se substituye el cloroformo por tetracloruro de carbono.

Ei producto principal de la reacciór. de Friedel y Crafts entre cl cloroformo y el benceno es el trifenilmetano:
S C A + C H C lj ¿SU* (C.H.) ,C H + 3H C i
Probablemente la reacción ocurra con participación del ion C H C l/, como se indica:
1 3 2 A p lic a c ió n d e t a s p r u e b a s ém c la s i f ic a d la
CHCJ3 + AICIj
C,Hg + CHC\t*
HC— Cl <-* H C =C I
k k .
[ A ia , ] -
CHCL
Entonces, el cloruro de berzal se transformaría en trifenilmetano por la secuencia de dos reacciones adicionales de alquilación, según Fríedcl- Crafts.
La desproporción origina especies tales como el ion trífemlmetil- carbonio, el cual queda en solución como sales del AlCLf y son éstas 1a$ responsables de los colores observados. La desproporción es una transferencia de hidrógeno ion carbonio, semejante a la que ocurre -en muchas otras reacciones de jos hidrocarburos saturados.*
(OH*) ,C —H-t- (CíH s) *CH' - > (C íH,) ,€ * + <C*H,)2CH2
El ion (CeiHs)iCH+ podría formarse a partir del cloruro de ben2ohidrito y el cloruro de aluminio.
E xp erim en to S< S o lu c ió n de Bénedict
a) C om puestos que no con tienen azu fre ,
RCHO - » RCO2H + Cu20
RCH G K R ~ C 0 + C u 20I t
RCO R -C O
ArNHNHAr—* ArN=NAr+CujO A r N H N H j A r H + ArOH + Na+ CujO
• Ver St«vmiQn, W«gnfT, y Otvoi, J . Am . Chira. í* t-, 74, 32$9 p*¡^1 T * Í« Z C T X Í1 1 .
2Cu**Complejo de citrato +

E xpe rim en to 3 , S o lu c ió n ám B a n e d k f 133
Sulfato de cobre hidratado (17,3 g ) . C iirato de ¿odio (173,0 %). Carbonata de lodío anhidro (10€ j ) .
• ?« IA una solución o suspensión de C.2 g dej compuesto en 5 nü de
agí’a se le añaden 5 mi de la soludón de Benedict. Observe si se produce un precipitado amarillo o amarillo verdoso^ entonces caliente la = > mezcla hasta ebullición y observe si se formó un precipitado y si lo hu- ^ j» biera, observe su color. Efectúe la prueba con; 1) n-butiraldehído; I f i2) acetoína; $) benzoina; 4) glucosa; 5) sacarosa; 6) glicerina; 7) bu- tanol-2; 8) acetona; 9) fenUhidrazina.
A una solución de 0,2 g de saearosa en 5 ral de agua se le añaden2 gotas de ácido clorhídrico concentrado y se hier/e durante un minuto.Se enfría la solución, se neutraliza el ácido con solución diluida de hidróxido de sodio y se prueba la acción del reactivo de Benedict. Explique el resultado.
Reactivo. Se prepara la solución de Benedict disolviendo las siguientes sales en agua destilada: £
d
« ?
<1El citrato y el carbonato se disuelven calentándolos oon 800 mi de agua. Se adiciona más agua hasta ajustar el volumen de la soludón a 850 mi. El sulfato de cobre se disuelve en 100 m! de agua y la solución resultante se vierte lentamente, agitando, en la soludón de d tra - to y carbonato. La solución final se afora a I It añadiéndole agua.
Comentarios. La soludón de Benedict, que contiene el cobre unido al anión complejo, funciona como un oxidante selectivo. Se introdujo como reactivo para adúcares reductores en substitución de la soludón de Fehling, la cual es fuertemente alcalina. El reactivo de Benedict demues- ^J¡tra la presencia hasta de 0.01%. de glucosa en agúa. El color del prc- J Hcípitado puede ser rojo, amarillo o amarillo verdoso, dependiendo de la naturaleza y cantidad del agente reductor presente.
El reactivo de Benedice es reduddo por g -hidroxiaidehído^ a-hi- droxicetonas y oc-cetoaldehídos. No es un reactivo general para oxidar aldehidos sencillos alifát'tcos o aromáticos,7 Las moléculas que sólo condenen el grupo fundonal alcohol (alcoholes primarios, secundarios, terciarios o glicoles) o sólo el grupo ceto, no se oxidan con la soludón de Benedict. . 1
Los-derivados d e la fridraguja. ejemplificados por la fenilhidrazína y JU .h id m ohm ^n^^ jQ fddaD ^JL ^tjijrg iic jivo . Otras substancias que ■se oxidan con facilidad, tales como la fenilhidroxilamína, el p-amino-
T DfthUJl, Rulb y Bígrr, / . CAfftí. E4vt., ¡ti, 205 (19C0).i
r i
é
« jl •

fenol y reveladores fotográficos afines,- también reducen la solución de Benedict.
134 A p lkadán cíe la* pruabos d e <|afffica<¡¿n
b) C om pueíto t conteniendo azu fre .
RSH + solución de Benedict -» (R S )2Cu
ArSH. ~ solución de Benedict -» j
ii?':
A una solución o suspensión de 0.2 g del compuesto en 3 mi de solución de bicarbonato de sodio al 10%. se le añaden 2 mí de la solución de Benedict. Observe el color y sí se ha formado un precipitado; entonces hierva la mc*cla durante 1 mir -co* y observe el resultado obtenido. Ensaye esta prueba sobre 1) laur; 'lercaptano; 2} dofenol; 3) tiofeno; 4) disulíuro de di-rt^butilo; 5} clorhidrato de t--risteína; 6) tíourea.
Comentarios. Los mercaptanos alifáticer y aromáticos dan un precipitado amarillo que por ebullición se aglomera formando una bola grasos* de color amarillo. Los mcrcaptanos de alquilo terciarios, simplemente dan una solución azul, como tambíír. los compuestos que contienen el grupo sulfuro o dísulfuro. Algunos compuestas que contienen el grupo tiol (ácido tioglicólico, mercaptoetanol y ácido dosaticílíco) no producen precipitados sino que dan soluciones casi incoloras..
La jL-cisteína y L-ctstina sufren una profunda degradación, for-, mando sulfuro de cobre de color negro. Las dialquilsulfonas, diarüsul- íonas y suJíonatos, no reaccionan con el reactivo de Benedict.
Prxieba de FekUrtg. A una solución de 0.2 g de glucosa en 5 mi de agua se le añaden 5 mi de solución de Fehüng y la mezcla se calienta hasta ebullición. Repita el experimento, usando en lugar de glucosa; 1) sacarosa; 2} glícerina; 3) maltosa; 4) lactosa; 3) femlhidrazina. Ensaye esta prueba sobre sacarosa {0.2 g), la cual se ha hervido durante unos minutos con 5 mi de agua que contenga 2 gotas de ácido clorhídrico concentrado. Es necesario que se neutralice el ácido clorhídrico antes que se añada la solución de Fehling. ¿Por qué? La solución de Fehling se prepara mezclando volúmenes iguales de las soluciones siguientes, justo antes de usarlasi
Nc 1. Solución de sulfato de cobre [34.6 g de cristales de sulfato de cobre hidratado en 500 mi de agua].
X? 2. T artrato de sodio y potasio ( l “3 g) c hidróxido de sodio {70 g ) , en 500 mi de agua.

E xp e r im e n to 4 . C lo ru ro d e b e t ice n su lfo n lfo 1 3 5
E xp erim en to 4 . C loruro de bencenstd fon ílo (M étodo de R insberg para d istingu ir en tre «(, amina* prim arias, secundarias y terciaria*)
A 0-3 mi de anilina, contenidos en un tubo de- ensayo, se les añaden5 mi de solución de hidróxido de sodio al 10% y 0,4 mi de cloruro de benccnsulfomlo. Tape el tubo de ensayo y agite la mezcla vigorosamente.6 Haga una prueba para asegurarse de que la solución esté alcalina. Después que haya reaccionado todo el cloruro de bencensul- fonilo, enfríe la soludón y fütteJa o decántela para separarla de cualquier residuo (A ). Observe si eJ residuo A es un sólido o un líquido y si es más ligero o más pesado que Ja solución alcalina. ;Q ué información puede deducir usted a partir de estas observaciones? Pruebe en agua y ácido clorhídrico diluido, la solubilidad del residuo (A). Note que la solubilidad de A en ácido clorhídrico indica que el compuesto original era una amina terciaria. Las sales de sodio de ciertas sulfonamidas de alto peso molecular pueden ser insolubles en solución alcalina.* O rdinariamente son solubles en agua.
Acidule el filtrado transparente. Raspe las paredes del tubo de ensayo para apresurar la cristalización del producto (B ).
Repita esta prueba usando metilanilina y dimetilanílina en lugar de anijina. Algunas aminas secundarías reaccionan lentamente y en ocasiones es necesario calentar la mezcla en reacción.
Mediante un diagrama* muestre cómo el procedimiento anterior diferencia aminas primarias, secundarías y terciarias.
Cuando se usa el método de Hínsberg para separar mezclas de aminas, es necesario recuperar Jas aminas puras en forma individual. Las bencensulfonamidas se pueden hidrolizar como sigue r
* SI la ock Ih <£« raodáp H «aliesrf» cQfiSvcforablcroditt» deberá cnfrUrw. D tftm u u d u jV,V*4[jiEqiáliiúlinM producen un cotoftuotc púrpura ú la rv*tl* *a calient» dwi4¿itJo. £ita pvttdí tfíctauwfo Ir rM tóén entre 15’C y SD^C.
* Dítttrlnuuij.) 4JBÍHA1 primaria* yur-icn producir <kr¡v*d»J <£« diíulíonQo irrtohlbfí* ín £lc*K. j>utdea btirolisM íí hirvitndoJoi SO mtnutoj con *p1oc¡<íb de etáxWn ¿e póáio ni 5% «D ctxco! »b»o[lrt«.
O |S° 2° + R N H j 4- 2NaOH -
O |S° !CI + R 2NH + NaOH - |NR* + NaCl -f HjO
O 1oS 0 2NHR + HaO + HCI —
>S02NR8 + HaO + HCi -
O *O '
iSO^H + RNH,Cl
¡03H -f R2NH8CJ

Para obtener material suficiente, la separación antes descrita se efectúa usando 50 veces más que las cantidades indicadas.
La sulfonamida se hidmliza calentando a reflujo 10 g de ella con J00 m i de ácido clorhídrico al 25%. Las sulfonamidas de las aminas primarias requieren de 24 a 36 toras de reflujo, mientras que las sulfo- namidas de las aminas secundarias se pueden hidrolizar en 10 a 12 horas. Después que se ha completado la disolución, la mezcla se enfría, se akaliniza con solución de hidróxido de sodio al 20% y se extrae con tres porciones de 50 mi de éter, La solución etérea se seca y, después que se ha eliminado el éter» se destila la amina. Con determinadas aminas de puntos de ebullición muy bajos o muy altos, a menudo es más conveniente recuperarlas como clorhidratos burbujeando cloruro de hidrógeno gaseoso y scco en la solución etérea seca.
Muchas sulfonamidas se hidrolizan sólo con gran dificultad; un procedimiento más satisfactorio implica el uso de áddo brornhídneo al 4&% y fenoLM Esta reacción no es una simple hidrólisis sino una degradación reductora en !a que el bromuro de hidrógeno se oxida hasta bromo y la sulfonamida se reduce hasta el disulfuro. El propósito primario d d fenol es eliminar el bromo convirtiéndose en p-bromofcnol.
136 Apílcodén cte k*> prueba* cíe cbiWcadón
2 ArSOjNRi+SHBr+ 5C*H*OH -*ArSS Ar -f- 2R2NH -f SRrC^H^OH + 4H* .
Comentarios. Los cloruros de arilsulfonilo son especialmente útik-í para caracterizar aminas primarias y secundarias. El método de Hins- beig para separar aminas se funda en que las sulfonamidas de Jas aminas primarias son solubles en álcali, mientras que las de las aminas secundarias no lo son. Ya que las aminas terciarias no dan amidas, el método proporciona un medio p a ta clasificar y separar los tres tipos de aminas. Sin embargo, los resultados de la prueza de Hinsberg no deben usarse sólo par* clasificar aminas, también es necesario considerar la solubilidad del compuesto original. Si el compuesto es a n i ó cero, es decir, soluble tanto en ácido como en álcalis, cl método de Hinsberg no ' puede distinguir entre las tres clases. Por ejemplo, el ácido amino)«benzoico reacciona con cloruro de bencensulfonilo y álcali dando una solución de la sal de sodio del derivado de A^bencensulfonilo.
COUH COONa
* Sttricr y BwAírt, f . Am. Cktm, S*c;, 74, 2006 (l$52); Saj-drí y C*1W, ibUC., 74. 4164 (1952).

Expe rim en to 5 . S o lu c ió n d« b ro m o 137
La acidulación precipita el ácido libre; este hecho, considerado aisladamente, indicaría que el compuesto original era una amina primaria y no una am ina secundaria.
Parccc que « ta s reacciones ocurren por un mecanismo semejante al mecanismo de la adición carbonílica de Jos cloruros de acilo, proba- bSemente con expansión del octeto del azufre.
E x p e rim en to S. Solución de b ro m o en tetracloruro de carbono
\ / Br, \ yC^-C - V C—c
/ \ / i ! \Br Br
En esta prueba, 0.1 g (0.2 mi de un líquido) del compuesto que se desea ensayar, se añad n a 2 m) de tetracloruro de carbono y se adiciona gota a go;a {agitando)' ma solución de bromo al 5% en tetracloruro de carbono hasta que persista el color del bromo. Aplique la prueba a: 1) penteno-2; 2) hexano; 3) benceno; 4) fenol; 5) ácido fórmico;6) benzaldehído; 7} etanol; 8} alcohol alílico; 9) acetofenona; 10) anüina.
Cotnenidrios. Este veactiyo se usa extensamente para averiguar 3a presencia de un enlace oleíínico o acetilénico, Deberá emplearse junto con la prueba del permanganato de potasio (experimento 22).
El tetracloruro de carbono es un buen disolvente para el bromo y para muchos compuestos orgánicos, pero no disuelve el bromuro de hidrógeno. Por lo tanto, el desprendimiento de bromuro de hidrógeno se acepta como evidencia de que la reacción es de substitución en lugar de adición. Cuando se emplea para revelar la presencia de insa- tu rae iones, este reactivo puede conducir a conclusiones erróneas por dos razones. La primera es que no todos los compuestos olefírdcos absorben bromo, La presencia de grupos negativos sobre los átomos de carbono de un enlace etílénico hace que la adición sea lenta y en casos extremos inhibe la reacción. Las siguientes reacciones mostrarán este punto:
C*H*CH = CH2-f Br2^ 2 > C6HjCHBiCH2Br
C*H jCH =CH G O jH -f Br, C«H5GHBrCHBiC02H
U na prueba positiva para insaturación es aquélla en la cual el color del bromo desaparece, ¡in que se desprenda bromuro de hidrogeno.
Cuando la bcomación se empica como prueba cualitativa, el tetra- cloruro de carbono es el disolvente preferido; sin embargo, resulta una elección desafortunada para un es'tudio del mecanismo de la reacción, ya que en disolventes no polares la reacción es complicada. Hay una reacción heterogénea que ocurre sobre las paredes deJ recipiente. Ade

L*
9
i »
Ir»jf»
hb
u
í i
n
más, ia reacción es catalizada tuertemente por huellas de agua O de ácidos y puede ser inhibida por el oxígeno." Podría anticiparse que tales efectos dificultarán la obtención de resultados reproducibles y harán incierta ía interpretación de cualquier prueba cualitativa que sea. negativa-
Se ha demostrado que en un disolvente polar, tal como el agua, el metanol, o el ácido acética, la reacción es un proceso en dos pasos.u El primero es la reacción de la defina con el bromo dando un ion bro- monio. El segundo paso es la reacción del ion bromonio con el ion bromuro para dar el producto. Ya que el ion bromonio se abre con inversión de configuración, el curso estereoquímieo total de la reacción conducc a la adición de los dos átomos de bromo en lados opuesto del doble enlacc.
Br Br/ \ ‘ IR jC = C R j *f* Br—Br -*■ RjC------- CR^ . * R^C——CR2
Br" Br
Les sttbstituyentcs que ayudan a estabilizar cl ion bromonio, cargado positivamente, aumentan la velocidad; aquéllos qyc inestabilizan el ion positivo, retardan la reacción. Por ejemplo, la substitución de un grupo alquilo en cualquiera de los dos átomos de carbono que tiene el enlace doble en las oJeíinas del tipo RCH=sCH2, aumenta la velocidad de. adición en solución de ácido acético en un factor de 30 a 40.u ,
La desaparición del color del bromo, acompañada de desprendimiento de bromuro de hidrógeno, indica que ha habido substitución y es característica de muchos compuestos A esta categoría pertenecen los cnoles, muchos fenoles y compuestos enolizablcs. Las metilcetonas son más reactivas que otras cetonas pero, como Jos otros compuestos carbo- níJicos, pueden mostrar un período de inducción, ya que eJ ácido bromhí- drico liberado actúa como catalizador para el paso de enolisación previo a la broraación. Los ásteres sencillos no dan esta prueba. Ei acetoacetato de etilo decolora inmediatamente la solución, mientras que cl malo- nato de etilo puede requerir hasta un minuto. Alguno*: compuestos con motile no activo que no hacen desaparecer el color a temperatura ambiente, dan fácilmente to prueba a 70°C. Entre estas substancias se encuentran el propionaldehído y la ciclopentanona. Los iteres de arilo se comportan en forma semejante. El cianuro de* benciio, aún « 704C, puede requerir varios minutos.
Las aminas aromáticas son una excepción en la que el primer mol de bromuro de hidrógeno formado no se desprende, sino que
1 3 B A p l ic a c ió n d e Jas p ru eba» d » c la s if ic a c ió n
l V*r De Lo Qwúrt. R<ot., 3, 14* (1349).» I)e La Uaj-í, Anm. Rtpts., «7, M> (1050). il K'jSfíUMi, Hr>’c» y Sweálund, / . Clttm . Svc., IM2, (0)4.
P■ B »

I
reacciona para convertir la am¡n? en su sal. Por esta razón a menudo la reacción se confunde con una adición sencilla,
NH¡ NH*Br
0 * ,r- - 0Br
La bencilainina representa un tipo fuera de lo común, que reacciona fácilmente con d bromo. Aparentemente, la substitución de los átomos <le hidrógeno unidos al átomo de nitrógeno es seguida por descomposición hasta benzonitnlo.
3CsH 3CHiN ff ,+ 2Brj -> [CéHsGHjNBrj] + 2 0 fiHsGH2N H 1Br
CfHjCN-f- 2HBrDeterminadas aminas terciarias, por ejemplo, la piridína, cuando se
tratan con bromo forman pcrfcromuros.
E xp e r im e n to é . A g u a d e b ro m o 139
En forma parecida, el color del bromo es eliminado por todos los tipos de aminas ali/átteas.
E xp erim en to 6 . A gua de b ro m o
OH OH
0 +3Bra ^BrO Br+3HBrBr
• Prepare soluciones acuosas al l% d e : 1) fenol, 2) anilina, 3) áddo | salicilico y 4} ¿-nitrofc.nol ¡ a cada solución añádale agua de bromo, gota aí ■ gota, hasta que ya no desaparezca el color del bromo.
Comentarios. Se ha demostrado que en la bromación del b&nceno y del o-nitro. .*iisol con *.gua de bromo, el agente de bromación es el B r—Br en lugar del Br4, Brj" o BrOH.w
Los mercaptanos se transforman rápidamente con el agua de bromo en disulfuros.
2RSH+P.r2 -> RSSR+2H Br
11 W ítton i S«pcr, / . Chtm . !•* ., 1X 9 , 3376.

La ventaja del agua de bromo sobre la solución de bromó en tetra- cloruro de carbono se debe a que el disolvente más polar aumenta la velocidad de bromación debido ai mecanismo iónico. Por supuesto que ' con este disolvente es imposible observar el desprendimiento de bromuro de hidrógeno. Un exceso de agua de bromo convierte el tribromofcnol
-tn u n derivado amarillo tetrabromado, !a 2,4,4,6-tetrabromociclohcxadie- nona,u El compuesto tetrabromado rápidamente $e convierte en el tn- bromofenol cuando se lava con ácido yodhídrico al 2% .
¿ P o r qué e$ Ja tribrccnoanilína insoluble «n ácido brom hidrico diluido? ¿P odría la. decoloración del agua de bromo originarse por la presencia de un compuesto ioorginico? C ite ejemplo*. ¿Se hidroliza e l trem e en &gu&? ¿Q ué efecto Le adría el agua de bromo sobre una lal soluble en ?gua de un ícido im oluble en agua?
E xperim en to 7 . Reactivo d e n itra to cérico w
a) P ara compuesto* to la ble* en agua, Eh un tobo de ensayo diluya 0,5 mi del reactivo de nitrato cérico con 3 mi d e agua destilada y mézclelos perfectamente. Añada de 4 a 5 gotas del compuesto que se va a ensayar, agite la mezcla y observe el cambio de color. Ensaye esta prueba con: 1) etanol; 2) glicerina; 3) ácido tartárico; 4 ) glucosa; 5) fenol. Sí el compuesto es un sólido, disuélvalo en agua y añada al reactivo de 4 a 5 gotas de la solución acuosa,
b ) P ara com puesto* insolubles e n agua, A 0.5 mi de reactivo, ’ contenidos en un tubo de ensayo, añádales 3 mi de dícccano. Si se forma un precipitado, añada agua (de 3 a 4 gotas), agitando, basta que la solución esté transparente. Añada 4 6 5 gotas del compuesto que se va a investigar, agite y observe el color. Ensaye la prueba con: l ) alcohol heptilico; 2) alcohol bencílico; 3) ácido salicliico. Si el compuesto es un sólido, disuélvalo en dioxano y añada de 4 a 5 gotas de la solución de dioxano al reactivo. '
Reactivo, Disuelva 200 g de nitrato cérico amónico [{NH«}2 Ce (NO*)*], en 500 mi de ácido nítrico 2jV. La solución se acelera bastante por calentamiento.
Comentarios. Una prueba positiva para un alcohol queda indicada por un cambio en el color del reactivo de amarillo a rojo. Los fenoles dan en solución acuosa un precipitado que va de café a café verdoso; en dioxauo se forma una coloración que va de rojo intenso a café.
Se obtiene prueba positiva con alcoholes y fenoles que nó contengan más de diez átomos de carbono. Cuando las moléculas son más grandes, el color que se produce es insuficiente para hacer útil la prueba. Los hídrojdácidos, hid^oxi aldehidos y la mayoría de los otros compuestos que contienen un grupo oxhidrilo alcohólico y que están dentro de los
B AJnta *pd UndbtíB, ¿ cta CMrm. itm u i., | i 6 (1952).* Duke y SmiOi, I - U . K * t . C t u m . , E i „ 1 ? , 201 ( íW í) .
| 4 0 A p l ic a c ió n & le s p ru ebo * d a d o s if ic a c ió n

Experimento 8, Anhídrido crómico 141
límites antes mencionados, dan prueba positiva. Les aminoalcoholcs elevan tanto el pH que el ion cérico precipita como hidróxido y la prueba falla.
Los aldehidos, cetonas, ácidos, haJuros de alquilo, ésteres y otros compuestos comunes que sólo contienen carbono, hidrógeno, oxígeno y halógeno, no interfieren. Las aminas aromáticas, los clorhidratos de amina y los compuestos que ordinariamente contienen estructuras oxidadas hasta grupos cromóforos, dan colores o precipitados con el reactivo. Por esta razón, Jas aminas iromáticas algunas veces dan la prueba de fenoles. Los compuestos que se oxidan muy rápidamente algunas veces decoloran !a solución de nitrato cérico antes de que se pueda observar la prueba.
Se ha encontrado que el reactivo de nitrato cérico da reacciones coloridas con derivados del tiofeno y algunos otros compuestos heten>- cíclioos.17
E xperim en to 8 . A nhídrido cróm ico
3RCH2O H + 4 0 t0 ,+ SHiSO* -> 3 R C 0 jH + 9 H 20 + 2 C r z(S0*) , 3R2C H O H + 2CrOj -f -+ SR ^C O +eH jO +C r^SO *),
A 1 mi de acetona, contenido en un pequeño tubo de ensayo, añada 1 gota del líquido o cerca de 10 mg del compuesto sólido. Entonces añada 1 gota del reactivo de áddo crómico-ácido sulfúrico y observe el resultado en 2 segundos. Efcctúc una prueba de control con acetona y compare el resultado. U na prueba positiva para alcoholes primarios o secundarios coasiste en la formación de una suspensión opaca de color verde o azul. Los alcoholes terciarios no dan reacción apreciable en 2 segundos y la solución permanece de color anaranjado. Desprecie cualquier cambio después de 2 segundos.
R eactivo . Se vierte, lentamente y agitando, una suspensión de 25 g de anhídrido crómico en 25 tnl de ácido sulfúrico concentrado en 75 mi de agua. L a solución, de intenso color anaranjado-rojizo, se enfría a temperatura ambiente antes de usarse..
Puede emplearse acetona comercial que sea suficientemente pura. Algunas muestras de acetona se pueden enturbiar en 20 segundos, pero esto no interfiere, siempre y cuando la solución continúe de color amarillo. Si la acetona diera prueba positiva, deberá purificarse añadiéndole una cantidad pequeña de pennanganato de potado y destilando.
Comentarios. Esta prueba es un método rápido para diferenciar los alcoholes primario? y secundarios de los alcoholes terciarios.1* Los alcoholes primarios y secundarías dan positiva la prueba sin restricciones de
» fo rto v g l» , Aw¿d. C írm ., XJ, 1128■ 'O oidwtW t W eU o u n , } . C fc f* . 3 » . 300 (1 9 6 2 ).

> pese- molecular. Aun el colesterol (C^H^O) da prueba positiva. Los aldehidos dan prueba positiva, pero podrán investigarse medíante otros experimentos de clasificación. Las definas, acetilenos, aminas, ¿teres y
f ce tonas, dan pruebas negativas en 2 segundos, siempre y cuando no estén contaminados con cantidades pequeñas de alcoholes. Los enoles pueden dar prueba positiva y los fenoles forman una solución de color
{ obscuro., totalmente diferente del color verde-azuloso de la prueba positiva.
' E xp erim en to 9 . 2 >4~dinxtrúfenUhidraxitxa
N H N H y .......... ^
+ yc O - ) > C - N N k /~ ^ N 0 2 + H,0
NOj
Disuelva 1 6 2 gotas del compuesto que se va a investigar en 2 mi de etanol al 95% y añada la solución a 3 mi de! reactivo de 2,4-dínitro- íenilhídrazma. Agite vigorosamente y si no se. forma inmediatamente un precipitado, deje reposar la solución durante 15 minutos.
En casos difíciles, es aconsejable ensayar la obtención de la dinitro- fenilhidrazona en éter dimetílico del distilenglicol, como se describe en el capítulo 10 (pág- 276).
Reactivo* El reactivo se prepara disolviendo 3 g de 2,4-dinitro- fenühidiarina. en -15 mi de ácido sulfúrico concentrado. Entonces se añade o ta solución» agitando, a 20 mi de agua y 70 mi de etanol al 95%. Se métela perfectamente la solución y se filtra.
Comentarios. La mayoría de los aldehidos y las cotonas producen dinitrofenilhidrazonas que son sólidos insolubles. Al principio, el precipitado puede ser aceitoso y, al reposar, volverse cristalino. Sin embargo, algunas cetonas dan dinitrofenilhidrazonas que son aceites.19 Por ejemplo: la metil-n-octiketona. la di-n-amilcetona y substancial similares, no forman dinitrofenilhidrazonas sólidas.
Una. dificultad adicional de esta prueba la causan ciertos derivados del alcohol aliüco que pueden ser oxidados por el reactivo hasta aldehidos o cetonas, los que entonces dan una prueba positiva. Por ejemplo, las 2,4-dinitrofenilbidrazonas de los correspondientes compuestos carboníceos derivados del alcohol, cinamílico, del fenil-4'-buten-3~ol-2 y de Ja vitamina A h a n sido obtenidas con rendimientos del 10 al 25%. También se ha encontrado que el benzohídrol se convierte, con bajo rendimiento, en la dinitrofemlhidrazona de la benzofenona. Es innece* sario mencionar que siempre hay el peligro adicional de que la muestra
"B ra d A x k 7 WHkrd, ] y ó * . C W , l*, 513 (M M ), a Bc*udc y Fort>cK }. Ck*m.t Soc., US1> -1562.
242 ApHcndón de las pruebo» d* <la«if¡eadÓn

/s
de un alcohol pueda «star lo suficientemente contaminada con su aldehido o cetona, formados por oxidación con el aire, para dar una prueba jjositíva. Si la dinitrofeniihidrazona se forma en cantidwd muy pequeüa, es aconsejable efectuar la reaccdón en la escala empleada para La preparación de un derivado (procedimiento 18, pág. 275) y hacer un cálculo del rendimiento (con base en el peso molecular del compuesto carbonOíco, el cual puede estimarse con bastante aproximación consultando la lista de posibilidades sacadas de las tablas).
El color de una 2,4-dínitrofenilhidrazona puede dar indicación acerca de la estructura de los aldehidos o cetonas de los que deriva. Las dinitrofenilhidrazonas de aldehidos o cetonas en las que el grupo carbonilo no está conjugado con otro grupo funcional, son amarillas. La conjugación con un doble enlace carbono-carbono o con un aniüo bencénico desplaza el máximo de absorción hacia el visible y se revela fácilmente por un examen del espectro ultravioleta.*1 Este desplazamiento también es responsable de un cambio de color del amarillo al rojo anaranjado. Entonces, en general puede suponerse que una dinitro- fenilhidrazona amarilla no esté conjugada. Sin embargo, uo color anaranjado o rojo debe interpretarse con precaución, ya que puede deberse a la contaminación con una impureza (por ejemplo: la 2,4-dinitrofenii- hidrazina es roja-anaranjada).
¿C u¿i es Ja ventaja de este reactivo sobre Ja fcnílhidrazína? ¿Cómo « ¿a te tiza Ja S^-dinitrofenjliudraana? ¿Q ué se forroa Como producto final <i« Ja condensación del aceto ace tato de etilo con: 1) ícniJJiídrazmfl, 2) 2,4-diniCro- *■ fenilhidraztQa?
E xp e r im e n to 10 . So lu c ión d a rfo ru ro fé rr ic o 143
Experimento 10« SaJtofíon Ác cloruro férrico
Prepare soluciones acuosas diluidas (aproximadamente al 0.1%) de I) fenol, 2) acetoacetato de etilo, 3} resorcinol, 4) ácido benzoico, 5) ácido £-hidroxibenzoíco y 6} ácido "saHcíKco; a cada soludón añada una gota de soludón de cloruro fénico al 1%. Compare el-color pro- duddo con el del agua pura y una gota de solución de cloruro férrico. Ocasionalmente, el color producido no es permanentemente, por lo que deberá procurarse observar cuidadosamente la solución en el instante en que se añada la gota de cloruro férrico.
Un' método alterno, que se ha encontrado que es útil para varios fenoles que i.j dan coLr con el procedimiento anterior, es el siguiente: tL A unos 30 mg de sólido o 1 gota de líquido, disuelto o suspendido en í mi de cloroformo, añada 2 gotas de una solución que se preparó disolviendo 1 g de cloruro férrico en 100 mi de cloroformo. Entonces añada 1 gota de piridina y observe cualquier cambio de color que pueda ocurrir*
» P^fcerti y G r Am. $oir., 08, 214 (!$«>).

CoTMiilarioss La producción de color con soludón de cloruro íérri- co** es típica de fenoles y de enoles; sin embarga, muchos de ellos no dan colorea* y, así, una prueba negativa no debe considerarse como significativa a meno$ que se tenga evidencia que la confirme. La mayoría de las oximas y los ácidos hidroxámicos también dan un color rojo con soludón de cloruro fénico. Este hecho se utiliza en la prueba dél ácido hidroxá- mico (pág. 152).
U na prueba para grupos “metileno activo” y fenoles, que h a sido propuesta como, compañera de Ja prueba del cloruro fénico, pero que sólo ha sido aplicada en forma limitada, es la descrita por WarfieW,25
7 4 4 ■ A p lic a c ió n - d« la s p rueba» d e d o s if ic a c ió n
E xperim ento II* H idróxido fe rro so
R K 0 2+ 6 F e (0 H )2+ 4 H 20 ^ R N H 2-f6 F e r0 H )3 .
Añada una cantidad pequeña (unos 10 mg) del compuesto que se desea ensayar a 1 mi de reactivo de sulfato ferroso (A) contenido en un tubo de ensayo y entonces adicione 0.7 mi de solución alcohólica de hidró- xido de potasio (B ), Inserte un tubo de vidrio hasta que llegue al fondo del tubo de ensayo y pase una comente de gas de alumbrado dentro del tubo durante unos 30 segundos para eliminar el aire. Rápidamente tape el tubo y agítelo. Después de 1 minuto observe el color del precipitado. Ensaye la prueba con: 1) njtrobenceno; 2) m-nitroanilina; 3) alcohol etílico; 4 ) alcohol isopropílico.
Reactivos, (A) A 500 mi de agua destilada, recientemente her- / vida, se 1c añaden 25 g de cristales de sulfato ferroso amónico y 2 mi de ád d o sulfúrico concentrado. Se introduce un clavo de hierro para retardar la oxidación producida por el aíre,
(B) Treinta gramos de hidróxido de potasio se disuelven en 30 mi de agua destilada y esta solución se añade a 200 m i de etanol al 95%,.
Comentarios. Una prueba es positiva cuando se. forma un precipitado café rojizo o café. Este es el hidróxido férrico formado por oxidación del hidróxido ferroso con el compuesto nitrado, el que a su vez se reduce hasta la amina. La prueba negativa queda indicada por un precipitado verdoso. En algunos casos la oxidación parcial puede producir un obscu-
Yedmiento d d hidróxido ferroso.Prácticamente todos los compuesto? nitrados14 dan una prueba
posiuva en ui.os 30 segundos. La velocidad con la cual se reduce d compuesto nitrado depende de su solubilidad. El ácido ¿-nitrobenzoíco, que es soluble en el reactivo alcalino, da la prueba casi instantáneamente mientras que el a-nitronaftaleno debe agitarse cerca de 30 segundos.
" Soícm w t 7 W íl* » , A wo¿.‘ C fitm ., V , V n ( IS 5 2 ) ,3» W u f c u . A * mI . C W , HA, 830 (1552).» H ta n m 7 G tm v to e , Iki . C A r»v A m L S , 33? (1957). •

L a píueba es positiva también par» otros compuestos que oxidan cl hidróxido ferroso. Los compuestos nitroso, quínonas, hidroxilaminas, nitratos de alquilo y nitritos de alquilo, pertenecen a esre grupo. Los compuestos fuertemente coloridos no pueden ensayarse.
Experim ftn to 12.-' R e a c tiv o d« fu c s in a 145
E xperim en to 1 2 ► ítemerivo de fu c iin a para aldehidos ^
Ponga 2 mi de reactivo de fucsina pará aldehidos en un tubo de ensayo y añada una gota de butiraldchído. Agite suavemente cl tubo y observe el color desarrollado en 3 ó 4 minutos. Repita la prueba con;1) benzaldehído; 2) solución de formaldehído; 3) acetofenona; 4) acetona.
En esta prueba el reactivo np. deberá calentarse y la solución en- sayada no deberá ser alcalina, C ulñdo la prueba se realiza sobre una substancia desconocida, deberá efectuarse una prueba simultánea sobre un aldehido conocido, para omparación.
Heoclívo, Disuelva 0.5 3 de fuchsina pura {clorhidrato de p*no- saniliña) en 500 mi de agua destilada y filtre la solución. Sature 500 mi de agua destilada 'con anhídrido sulfuroso, mézclela perfectamente con la solución fittrada de fuchsina y déjela reposar una noche. Así se obtiene un reactivo prácticamente incoloro y muy sensible.
Comentarios. La fucsina es un colorante rosa del mfertíjpetano, el cual es convertido por cl ácido sulfuroso en el ácido látifcosulfónico incoloro. Aparentemente,, la reacción involucra Ía adición ácidosulfuroso al núcleo quinoideo del colorante.®
\
| h 2N ^ ^ | - C H 0 - N Í W + 3H*SO» -
(Sqluílóúrc**)
w,*~Q~c (“(f^)>“NHso2H)(a_)+2Ha°s o 3h
(Rcactjvxi d« Sdüíf-irrto low )
Este ácido leucosulfónico w inestable y pierde áddo sulfuroso cuando se trata con un aldehido, produciendo un colorante quinoideo de color violeta-púrpura-
H . Ñ = / ~ V -C
O OH"
O R -
{SóliictC*! viole 18- |¡úrjAtr¿l
(Cl-)
* V er W * 6 Í v Schculng, b tr„ M , 2527 (C 9 ít) ; Schtook r Bergm *nn, ¿/kr6«#A d tr<H(e*tukrw Chemit, Vd!. I , p . 2S2.

1 4 6 A p l ic a c ió n d e lo s p ru e b a s d© d o s if ic a c ió n
Es importante notar que el color de este colorante es diíeic/ite al de la fucsina original. No es un rosa claro ano que tiene un matiz azul muy cercano al violeta o púrpura. Algunas ce tonas y compuestos insatu- rados reaccionan con el ácido sulfuroso regenerando el color rosa de la
a ^ l fucsina. El desarrollo de un color ligeramente rosado en el reactivoi no es, por lo tanto, una prueba positiva para aldehidos.
El hecho de que determinados compuestos causen la regeneración del color rosa de Ja fucsina original, ha servido de base para una prueba. Se ha reportado que, cuando se usa un reactivo preparado especialmente26 y el tiempo de reacción es de 1 hora* las aldosas. produ* cen un color rosa, mientras que las cctosas y disacáridas (exceptuando la maltosa) no lo producen. Esta modificación de la prueba de Schiff
I debe emplearse con precaución, ya que muchos compuestos orgánicos i M k producen un color rosa con et reactivo cuando se agitan al aire; otros
compuestos, tales como las cotonas a^-insaturadas; se cr -obinan con el áddo sulfuroso y asi invierten la primera reacción que se /hdicó antes.
R O R ' 4- 2H I R 'I + R I 4- H jO H O R '+ H I ^ R l + H j O
A rO R ' + H I - » R 'I 4- ArOH R C O O R '+ H Í -> R 'H -R C O O H
R C H (O R ')a+ 2 H r ^ 2RTÍ+RCHO-f HaO
Ponga aproximadamente 0.1 g {o 0.1 mi) del compuesto en un tubo de ensayo de 16 X 150 mm. Añada cuidadosamente, por medio
Í" ^ de una pipeta, 1 mi de ácido acérico glacial y 1 mi de áddo yodhídrico9 al 57% (dens. esp, 1,7). Añada un tiocito de porcelana porosa e in
serte en la boca del tubo de ensayo un tapón de gasa preparado como se describe más adelante. El tapón de gasa se tuerce hasta que ajuste
J I bien y se empuja hasta que descienda unos 4 cm a partir de la boca• del tubo de ensayo. Un pedazo pequeño de algodón no absorbente se
apisona suavemente sobre la parte superior del tapón, por medio de una varilla de vidrio, hasta formar un disco de 2 a 3 mm de espesor. Un
i ” iroso de papel filtro de 2 por 10 cm se dobla longitudinalmente, seI humedece con una solución de nitrato mercúrico y se coloca sobre el disco
4 ^ de algodón. El tubo de ensayo se sumerge hasta una profundidad de f ? 4 a 5 era en un baño de aceite, conservado entre 120 y 130°C, Cuando
hierve la mezcla de reacción, los vapores atraviesan el tapón poroso, el
*í
E xp e r im e n to 13 . A cido yodh ídrico ( m é to d o ¿a Zeital pwrtt a lco x ilo i)J
-i
a » Tobir, M . £■ [. C ktm ., A*él. E¿., 14, 405 (1912).

E x p e r im w ito 13 . A c id o y o d h fd rfc o 1 4 7
cual generalmente íe pore gris. El yoduro de alquilo volátil ascendiendo a través de los tapones, reacciona con el nitrato mercúrico produciendo un color ligeramente anaranjado o bermellón, que se debe a la formación de yoduro mercúrico, U na prueba positiva consiste en la formación de un color anaranjado o bermellón sobre el papel de ensayo dentro de un período de calentamiento de 10 minutos. U n color amarillo constituye una prueba negativa o dudosa.
Ensaye esta prueba sobre: í) anisol; 2) benzoato de metilo; 3) <*- metUglucósido.
Toponeé de ga»a. Se añade una solución de 1 g de acetato de plomo en -10 mi de agua a 60 mi de una solución de hidróxido de sodio 1N y se agita hasta que se disuelva el precipitado. A esta solución de plumbtto de sodio se le añade una solución de 5 g de tiosulfato de sodio hidratado en 10 rol de agua. Se añade aproximadamente 1 mi de glicerina y la solución se diluye hasta 100 mi. Unos 5 mi de esta solución se pipetean sobre tiras de manta de ciclo o ga¿a doble de 2 por 45 cm. Se secan las tiras y se enrollan para que ajusten en el tubo de ensayo.
So lución de n itra to m ercúrico . Se prepara una solución saturada de nitrato mercúrico en 49 mi de agua destilada a la que se le ha añadido 1 mi de ácido nítrico concentrado.
Comentarios. Esta prueba21 está basada en el método clásico de Zcisel para determinar cuantitativamente el porcentaje de grupos metoxilo o etoxilo. Los grupos funcionales que contienen radicales metilo, etilo, n-propilo o isopropilo, unidos al oxígeno, son escindidos por cl ácido yodhJdríco con formación de un haJuio de alquilo volátil. Los derivados alcoxi, en los que el grupo es n-butilo o uno más grande, son difíciles de romper y el yoduro tiene un punta de ebullición demasiado elevado para que se le pueda volatilizar. Algunos compuestos «-bulo» dan prueba positiva, pero el procedimiento no es de confiar {el punto de ebullición del yoduro de «-butilo es 131 "C ).
Bata reacción de clasificación es muy útil para los éteres, ásteres y acetales, en los que los grupos son metilo o etilo. El metanol, el etanol, los dos alcoholes propílicos y aun alcoholes superiores tales como el alcohol ft-butílico y el isoamílico, también dan una prueba positiva. La prueba ha. sido aplicada a numerosos alcaloides y azúcares mediados. La interferencia principal la origina Ja presencia de algúsi grupo funcional quo contenga azufre y que desprenda áddo sulhldrico cuando se calienta con áciú-> yodhíd.ieo.
Algunos éteres pueden requerir un reactivo más potente3* para la escisión. En ocasiones es ventajoso usar 2 m i de áddo yodhidrico, 0.1 g de fenol y 1 mi de anhídrido propiónico por 0.1 g de muestra.
® Tofci*, Ind. C htm ., And. E J ., W» 45 j (JS53); A u d c r w y I>aac»o, Cfum. tn i., ♦57 (15W).
* Skk, faá . Ene. CArm., Antd. E¿., 11, 174 ( l» d ) .

1 4 8 A p l ic a c ió n d e Ico p ru e b a s d e d o s if ic a c ió n
E x p e r im e n to 1 4 . A cido clorhídrico — C loruro d e cinc
{ prueba de tu c a s )
RjCHOH-hHGl- R,CHC1+ H20 RjCOH+ HC2 R3GGI -f H 20
q.) A 1 mi del alcohol, contenido en un tubo de ensayo, anúdale10 mi del reactivo de ácido clorhídrico-cloruro de cinc, que estén a 26-27°C. Tape el tubo y agítelo, después deje reposar la mezcla. Observe el tiempo requerido para la formación del cloruro de alquilo, el cual aparece como una capa insoluble o una emulsión. Efectúe la prueba con cada un o de los alcoholes siguientes y observe, con reloj, el tiempo necesario para que se efectúe la reacción: 1} bu tanoM ; 2) pcntanoí-2; 3? propanol-1; 4} alcohol ter-butílico; b) alcohol ísoa milico; 6) alcohol atílico; 7} alcohol bencílico.
R eactivo . El reactivo de ácido clorhídrico-cloxuro de cinc se prepara disolviendo, con enfriamiento, 136 g (1 mol) de cloruro de cinc anhidro en 105 g (1 mol) de ácido clorhídrico concentrado.
b) A 1 mi del alcohol, contenido en un tubo de ensayo, añádale6 m i de ácido clorhídrico concentrado. Agite la mezcla y déjela reposar. Durante 2 minutos obsérvela cuidadosamente. Pruebe los alcoholes siguientes y anote sus resultados: 1) alcohol K-propilico; 2) pentanoí-2; 3) alcohol fer-butílico; 4) alcohol bencílico.
Comentarios. Ya que la prueba de Lucas1* depende de la aparición del cloruro de alquilo como una segunda fase líquida, evidentemente sólo es aplicable a los alcoholes que son solubles en el reactivo. Esto limita la prueba, en general, a Jos alcoholes monofuncionalca menores que el hexilo y a determinadas moléculas polifundonales.
L a reacción de alcoholes con ácidos halogenhldricos es una reacción de substitución en la cual la especie reactiva es el ácido conjugado del alcohol, R —OHj+, y, como ca de esperarse, es completamente análoga a las reacciones de substitución de los haluros orgánicos y compuestos similares con el nitrato de plata y el ion yoduro (experimentos 23 y 27}, Como es de esperarse en estas reacciones, los efectos de la estructura sobre la reactividad están íntimamente relacionados. Así, los alcoholes primarios no reaccionan aprecíablemente a temperaturas ordinarias con ei ácido clorhídrico, aun en presencia de cloruro de cinc; por una parte, el ion cloruro es un agente nucleófilo demasiado malo para efectuar una reacción de desplazamiento concertado y, por otra parte, el ion carbonio primario es demasiado inestable para servir como intermediario en. el mecanismo de ion carbonio. El bromuio de hidrógeno
“ L «c i4 , / . Chtm. 62, $03 (1950),

E xpe rim en to 1 4 . A c id o c lo rh íd r ico 149
y d yoduro de hidrógeno, que tiene aniones con reactividad nucleófila que aumenta en ese orden, son. en Forma ascendente, más reactivos hacia los alcoholes primarios. Se ha demostrado que la reacción del etanol con el bromuro de hidrógeno es una reacción bimolecular del ion bromuro con el ácido conjugado del etanol,*
Los alcoholes terciarios reaccionan tan rápidamente con el ácido clorhídrico concentrado, que el haluro de alquilo se puede observar después de unos cuantos minutos a temperatura ambiente, al principio como una suspensión lechosa y después como una capa aceitosa. La acidez del medio se aumenta por la adición de) cloruro de cinc anhidro (un ácido fuene de Lcwis) y la velocidad de la reacción aumenta aún más. Esta reacción no es un despla2amíento nucleófilo comparable' al experimentado por los alcoholes primarios, sino más bien ocurre a través de un ion carbonio intermediario. La elevada reactividad de los alcoholes terciarios es.una_oonsr'aiencia de la relativa gran estabilidad del. ion.carbonio .intermediario,. Así, el ion f-butilcarbónio tiene distribuida su carga positiva en parte sobre los átomos de hidrógeno de los grupos metilo y en parce sobre cl átomo de carbono central (pág. 172). El alcohol alílico, aunque es un alcohol primario, también tiene un ion carbonio relativamente estable debido a que su cai^ga está distribuida igualmente sobre los dos átomos de carbono terminales. Como debería
[C H ^C K C H r ^ ’CHiCH = CHj]
espera ríe. reacciona rápidamente con el reactivo de Lucas, desprendiendo calor. Puede lograrse que se separe cl clorura de alílo, diluyendo 1:i mezcla con agua helada.
Los alcoholes secundarios tienen una reactividad intermedia entre los alcoholes primarios y terciarios. Aunque no se ven afectados apre- ciable mente por el ácido clorhídrico concentrado solo, reaccionan bastante rápido en presencia del cloruro de cinc anhidro; la apariencia turbia de la mezcla se observa en unos 5 minutos y en 10 minutos ordinariamente se puede ver una capa bien definida.
Para una discusión más amplia del eiccto de la estructura sobre la reactividad en las reacciones de substitución de este tipo, vea los comen* taños del experimento 23.
Escriba las fórmula* estructurales y nombres de los isómeros de Jos alcoholes saturados de cinco carbonos que no se usaron co este e>3>BriiDemo. ¿Cómo rcac- donarían con este reactivo? ¿Cómo explicaría et comportamiento diferente entre el alcohol edilicoy el alcohol n-propí¡ico? ¿Entre ci alcohol bencílico y el alcohol B-aiailíco? ¿Qué otros métodos pueden usarse para clasificar un alcohol desconocido?
11 Gruo*»tó 1 WiastEiD, / . C ktm . Sóf., 5$, 20M (W 7 ).

l *t i
E xperim en to 15* C lorhidrato de hidroxilrnnitta
A, ALDEHIDOS Y CtTONAS
RCHO -f H.NOH • HG1 - * R C H = N O H + H CI-f H ¿ ) R jCO + HjN O H • HCI - > R :C — N O H - f -H O 4 - H j O
15 0 A p lic a c ió n de la * p ru eba» d e c la s if ic a c ió n
a) Para com puestos neutros. A 1 mi del reactivo añada una gota o unos cristales del compuesto y observe el cambio de color. Sí a temperatura ambiente no hay un cambio marcado, caliente la mezcla hasta ebullición. U n cambio de color de anaranjado a rojo constituye una prueba positiva. Ensaye Ja prueba sobre: 1} n -batiráIdebído; 2) ace- tona; 3) benzofenona; 4) glucosa.
flPr b) P are com puestos ácidos o básicos. A í tnl de 1* solución indicadora añádale aproximadamente 0.2 g del compuesto y iuste el color de la mezcla hasta que iguale a 1 mi del reactivo, contenido en otro tubo de ensayo del mismo tamaño. Esto se hace añadiendo unas cuantas gotas de hidróxido de sodio diluido (1% ) o solución de ácido clorhídrico. Entonces, añada. la solución resultante a 1 mi del reactivo y observe si se produce un color rejo. Ensaye esta prueba sobre: 1} sali- dlaldehído; 2) />-dimetiíaminobenzaldehído. Ensaye las pruebas sobre ácido tartárico por los procedimientos (a ) y ( b).
R eactivo . A una soludón de 5 g de clorhidrato de hidroxilamina en 1 1 de etanol aJ 9 5 # se !e añaden 3 mi del indicador universa) de Bogen o Grammercy. El color de la solución se ajusta a un tinte anaranjado brillante (pH 3,7 a 3.9) añadiendo, gota a gota, una solución
r r j A diluida (5% ) de hidróxido de sodio en etanol. El reactivo es estable durante varios meses.If S o lu c ió n indicadora . Se prepara una solución del indicador aña-diendo cualquiera de los indicadores anteriores a 1 l de etanol al 95%.
Comentarios.*1 El cambio de color en el indicador se debe al ácido clorhídrico literado en la reaedón del compuesto carbonílico con el dorhidrato de hidroxilamina, ya que la oxima no es lo suficientemente básica para formar un clorhidrato. Todos los aldehidos y la mayoría de las cetonas producen un cambio inmediato de color. Algunas cotonas de alto peso molecular, tales como la benzofenona, benzllo, ben-
S k zoína y alcanfor, requieren calentamiento. Los a rca res , quinonas y cetoijas con impedimentos estericos (tales como el ácido o-bervs'oilben- zoíco), dan una prueba negativa.
• Muchos aldehidos experimentan una autoxidadón en el aire y contienen cantidades apredables de ácidos; de aquí que siempre deba determinarse la acción dé una solución o suspensión acuosa sobre el
* Duke, ind . K * t' Chtm ., /UoJ. 18, 110 (IW > .

E xp e r im e n to 1S. C lo rh id ra to d e h ld ro x lla m ln c i 151
tornasol. Si la solución es ácida debe usarse el procedimiento (b ); lo mismo puede decirse para los compuestos cuyo comportamiento de solubilidad muestre que son ácidos o bases.
Aparentemente, el mecanismo de la reacción de los compuestos carbonüícos con la hidroxilamina está muy íntimamente relacionado con loa mecanismos de las reacciones con fenilhidiazina, dinitxofenilhidra- zina y semicarbazida. La formación de semicarbazonas es la reacción de este grupo que ha sido estudiada más ampliamente. Más adelante K se muestra el mecanismo en solución acuosa, cuando es catalizado por el ácido HA-
La conclusión más. importante a que se ha llegado en el estudio de la reacción es que, como debería anticiparse del mecanismo que después se muestra, se ve retardada fuertemente por soluciones que son demasiado acidas o demasiado básicas. Y a que la reacción requiere tanto de la semicarbazida libre como de una fuente de protones {HA}, una solución fuertemente ácida disminuye la velocidad de la reacción por convertir toda Ja semicarbazida en su ácido conjugado inactivo, mientras que una solución fuertemente básica disminuye la velocidad ai convertir el ácido H A en su base conjugada inactiva. Las condiciones m is favorables son aquéllas en las cuales el producto (HA) (NH1NHCONH1) es máximo.
R jC = 0 + HA + H*NNHCONHa ^ R ,0 = < )H A + H^NNHCONHa
1.OH y O R
RjC .+ HA ^ R X + A ~ 'NHNHCONHa '"'NHJNHCONH*
$OH’HA
RjO ^ R4C=N H N H CO N H 2 + H*0 + A-
X’NHNHCONHa 1 RaC =N N H CO N H a + H*0 + HA
La reacción es reversible. Aunque en soluciones ácidas, relativamente débiles, se puede hacer que el equilibrio esté desplazado had a la derecha, el ácido fuerte elimina la semicarbazida convirtiéndola en el ácido con- jugado y desplaza el equilibrio hacia la cetona libre,
** V«r Burílete «n O rtanit CfitmJriry <Jc Gilc*»i>; III, John WUey Sow, N«ev* Vork, 1353, pág. 1J7; Hojee, Btttioi v SmiÜ!, / , l?ff. Ckrm. » , 752 (1558); Hbie, FAyjúW O tfanií C fw náiry, 2* tú ., McGraw-HilJ Co., Nutv» Y oii, pjgi, 254 y pji.

15 2 A p lk a c U n d e Jas p ru# bas d e c la s if ic a c ió n
8 , fc S T £ R ta , C L O R U R O S D E ACIDO Y A N H ÍD R ID O S '
(PRUEBA D E L ACCDO H tD R O X A M IO O )
o oII 1
RCOIV + H , NOH — RCNHOH + R'OH O OI; IRCCI + H2NOH — RCNHOH + HCI
O
(RCO)jO 4- H jN O H - RCNHOH + RCOfH Ol
3RCNHOH + FeCI3/°V
RC
X N -0 /1H .
Fe + 3HCI
P ru eb e pre lim inar, a) Disuelva una gota o unos cuantos cristales del compuesto que se va a examinar en i mi de alcohol al 95% y añada 1 mi de ácido clorhídrico i.W. Observe el color producido cuando se añade a la solución 1 gota de solución de cloruro férrico al 5% . Si se produce una coloración definida anaranjada, roja, azul o violeta, la siguiente prueba para el grupo acilo no es aplicable y deberá omitirse. Sin embargo, note que el ácido clorhídrico adicional evita el desarrollo de los complejos coloridos de muchos fenoles y todos los enoles.
bJ Caliente hasta ebullición una mezcla de 1 gota o cerca de 40 mg del compuesto, I mi de clorhidrato de hidroxilamina 0 .5# en etanoE al 95% y 0.2 mi de hidróxido d<- sodio acuoso HAr. Después de que la solución se ha enfriado ligeramente, añada 2 mi de ácido clorhídrico IA\ Si la solución está turbia, añuda 2 mi de etanol al 95%. Observe el color producido cuando se añade 1 gota de solución de cloruro férrico al 5%, Sí no persiste el color producido por la gota de solución de cloruro férrico, continúe Ja adición del reactivo, gota a gota, hasta que el color observado invada toda la solución problema. Compare el.color con el producido en (a). Una prueba positiva será netamente de color rojo tinto o violáceo, en comparación con el amarillo observado cuando el compuesto original se ensaya con cloruro férrico en presencia de ácido.
Conuntarioi. Todos los v-*ci« de ácidos carfeoxíHros, incluyendo poliésteres y lactonas, dan colores violáceos definidos con diferentes grados

do intensidad. I.os cloruros de acüo, anhídridos de ácido y compuestos trihalogsnados tales como el benzctridorura y el cloroformo, dan' pruebas positivas.
El ácido fórmico produce un color rojo; con todos los otros ácidos la prueba es negativa. Debe mencionarse que el ácido láctico comercial da prueba positiva debido a la presencia de lactida o ácido polílác- tieo. Ordinariamente el ácido itálico contiene anhídrido ftálico y por lo tanto da prueba positiva.
Los compuestos nitrados primarios o secundarios dan prueba positiva debido a [a reacción del cloruro férrico con la forma aci producida por el álcali.
La mayoi'jy de las imidas dan pruebas positivas; alguna* amidas (pero no todas) dan colorea violáceos claros y la mayoría de los nítrilos dan prueba negativa con el procedimiento (b ) anterior.
Los aldehidos sin átomos de hidrógeno en posición a pueden dar pruebas débilmente positivas; COt. ..otros aldehidos y cetonas Ja prueba ea negativa,
C . N IT R IC O S Y A M ID A S
NHlí
RCN+HjNOH -» ROIHOHO Oíl 11 RCNB2+H,NOH RGNHOH+NH*
. A 2 iiil de una solución de clorhidrato de hidroxilamina M en pro- pilcngticol se les a naden 1 gota o unos 30 mg del compuesto, también dtíuelto en propilenglicol (mínima cantidad), y 1 mi de hidróxido de potasio liV. La mezcla se hierve moderadamente durante 2 minutos y se deja enfriar a temperatura, ambiente; entonces se le añaden de 0,5 a 1.0 mJ de solución alcohólica de cloruro férrico (5% ). U na coloración de rojo a violeta constituye una prueba positiva. Los colores amarillos indican pruebas negativas y los' colores o precipitados cafes son indeterminados.
Comentarios. 'Iodos los nítrilos y amidas comunes dan pruebas positivas, La Jffiflyannifo, díacetilbencidina y determinadas amidas con
-impedimentos este ricos, no dan prueba positiva.Para la* uiodcíicaviourc dr prut-bas y los comentarios sobre el
comportamiento de los tipos especiales de compuestos orgánicos del nitrógeno, los estudiantes deberán consultar la bibliografía original.21
» C htrn. E d u t., 17, «1 ílíM fl); Buctlr* y T h* lfn , AtuJ. C l* m ., 22, ¿76ÍI9 S 0 ]; Solow nr y L lpjrK iir, i b i i , , 24, GS8
E xp e r im e n to 15 . C lo rh id ra to d e h id ro x ila m in a 153

- t E xp erim en to 16 . Y o d o e h id róx ido de sodio0
(p ru e b a del yo d o fo r m o )
1 5 4 A p l ic a c ió n d e la s p ru e b a s d * d o s if ic a c ió n
fO
^ RCH O H CH 3+I,-f-2N aO H R - ^ - C H , + 2N al+213*0
f £ RCOCH3+ 31: + 3NaOH -> R - C - CI3+ 3N aI+ 3H20 | n *o h
RCO jN a-f C’H Íj
r »
i *
:
í s
i
Aplique la prueba siguiente a : 1) alcohol isopropílico; 2) ac';*ona;3) acetato de etilo; 4) acetoaectato de etilo; 5) acetofenona; 6) me- tanol puro*
Coloque 4 gotas det líquido (0.1 g del sólido) que se va a probar en un tubo de ensayo (de 15 mm de ancho). Añada 5 mi de díoxano y agite hasta que toda la muestra se haya disuelto. Añada 1 mi de solución de hidróxido de sodio al 10% y después la solución de yodo-yoduro de potasio (lugol), con agitación, hasta que un pequeño exceso produzca d característico color obscuro del yodo.-'5 Si se decoloran, menos de 2 mi de la soludón de yodo, coloque el tubo de ensayo en un baño maría que se mantiene a 60CC. Si se decolora el pequeño exceso de yodo presente, continúe la adición de la solución de yodo (conservando la soludón de dioxano a 60°C ), con agitación, hasta que nuevamente un pequeño cxceso de la soludón de yodo produzca el carácter color obscuro. Se continúa añadiendo yodo hasta que el color obscuro no desaparezca en 2 minutos de calentamiento a 60 "C. Él cxceso de yodo se elimina añadiendo con agitadón unas cuantas gotas de solución de hidróxido de sodio al 10%. Después, llene el tubo con agua y déjelo reposar durante 15 minutos. Si el compuesto eá sólido, es conveniente filtrar el yodoformo y determinar su punto de íusión- que deberá ser l t iM 2 l0C. Si el yodoformo está rojizo, disuélvalo en 3 ó 4 mi de dioxano, añádale 1 mi de solución de hidróxido de sodio al 10% y agite hasta que sólo quede un color ligeramente verdoso. Diluya con agua y filtre.
* £1 n3cl»tK>l c«aerci»l treeuwitcMCJrte d a . pniebw p«düvm dffcido & la p ram cl» ¿ 1 ímpv/ci**.
W 1.a lolucióft de yodi>-y(xíuro de poiajio debe ad¡eiorwr»e iiaat» que on % «« twr*o produxza im color (tacuro, detído í que «rjuno* <>>mpcesto* <t*a wo cfcW am»Tt1l° »t tratm« coa htooyodiia. Si Ja diUwÜft c«p «su» te eíettúa rM, tjo te ebiieo* ycxMornio. Deberé twune qoc «c ha rtjx/rtvdo qu« b dftbkítor.» <U yodofomo, pero a un* velocidad aueho m epu que 1» aaetUjropilcetooi [ver Pr¡f, OcJnnú, Karabauoi, Cratuin y Y»ne, J. O fi- C ktm . 37, 1914

Reactivo. La solución de yodo-yoduro de potasio se prepara añadiendo 200 g de yoduro de potasio y 100 g de yodo a 300 mi de agua destilada y agitando hasta que la disolución sea total.
Comentarios. Esta prueba es positiva para Jos compuestos que contienen el grupo C H iC O —> C H jlC O — o C H I2C O — unido a un átomo de hidrógeno o a un átomo de carbono que no tenga átomos de hidrógeno activo o grupos que produzcan un exceso de impedimento estéríco. Por supuesto que la prueba será también positiva para cualquier coro* puesto que reaccione con el reactivo para dar un derivado que contenga uno de los grupos requeridos, Inversamente, los compuestos que contienen uno de los grupos requeridos, no dará la prueba del vodoformo si se destruye ese grupo por la acción bidrolítica det reactivo antes de que se termine la yodación.
A continuación están los tipos principales de compuestos que dan la prueba:
CH3CHO CH3CH2OH CHjOOR CH¿CHOHR
O H OH
RCOCHjCOR RCHCHjGHR
(R cualquier radical alquilo o arila excepto un radical arilo di-orto- substituido).
La prueba es negativa para los compuestos de los tipos siguientes:
GHjCOCH iCO jR CHiCOCHjGN CH3ÜOOH,NOa etc.
En dichos compuestos ei grupo acetílo se fragmenta con el reactivo hasta áddo acético, el cual resiste la yodación.36
Se ha propuesto un reactivo modificado37 para diferenciar mctíl- cetonas de metilcarbinoles, el cual consiste en una solución de 1 g de cianuro de potasio, 4 g de yodo y 6 mi de solución concentrada de hidróxido de amonio en 50 mi de agua. Este reactivo produce yodo- formo a partir de metilcetonas, pero no de metücárbinoles.
La ruptura de las trihalocetonas con una base, ejemplificada por el segundo paso de la prueba del yodoformo, cscá relacionada con la inversa de la condensación de Clai*eo“ En estos casos la reacción puede proseguir dehído a la estabiüdi.' del aniCn resultante.
* P i r t Wl# «JiicuáAtí general de ttftl p iuc tm VcA FuWV 7 BulJ, C * im . R tv i. , 15, 275 (19*),
w IRothlui, A u k . E¡au!t¡ PatfA. Fac ci. M<¿. CárAaiá (R .A -J Srcf. t í . , I f t » , N» 10, j>*B. J ; C A . . » ( 5W 1 ( 1 W I) .
» A icatH kr, F ñ n t ip l t t ú f Icaie O rfanie ÍU tu iú ia t, J o b a ' rfíley aü£ Son», Nucy* York, ¡9M, |> i*. 106.
E xp e r im e n to 16- Y o d o • h id ró x id o d e so d io 155

1 5 6 A p l ic a c ió n d e la s p rueba» d o c la s if ic a c ió n
/ 0R C - C la + O H -
s ° / °RC~~CHSCR + OH’
r y : ✓ °RC—CIj
. OH .
RC + -CI,
OH
Q" Ol o OY X / /
RC—CHjCR -v RC +\
OH
-CH^CR
. OH .
E xp erim en to 1 7 . C lorura de n íq u e l, b isu lfu ro de carbono e h idróx ido de am onio *
R 2N H + CSs + NH4OH -> R 2N—C—S NHg + H20jwci«
S
.R jN —C—S“ -_ ‘Ni
Prepare una solución acuosa de la amina añadiendo 1 6 2 gotas a 5 m i de agua. Si es necesario pueden añadirse una o dos gotas de ácido clorhídrico concentrada para disolver la ainina. A 1 rnl del reactivo, contenido en un tubo de- ensaye, añádale de 0.5 a 1 mi de hidróxido de amonio concentrado, seguido por 0.5 a 1 mi de la solución de la amina. U n precipitado definido indica una amina secundaria. U na turbidez ligera indica huellas de amina secundaria, seguramente como impureza. Ensaye esta prueba sobre: 1) anilina; 2) Ar-metilanümaj 3) trietilamina.
R eactivo . A 0.5 g de cloruro de níquel hexahidratado en 100 mi de agua se les añade tal cantidad de bisulfuro de carbono que, después de agitar la mezcla, quede un glóbulo de bisulfuro de carbono en el fondo del frasco. Si se guarda en un frasco Upado herméticamente, el reactivo es estable durante largos períodos. Guando se evapora el bisulfuro de carbono que no se disolvió, debe añadirse más.
Comentarios, Esta prueba la dan todas las aminas secundarias, pero no las aminas primarias. Es .,ivy sensible y muchas muestras comerciales de aminas terciarias producen turbidez debido a la presencia de pequeñas cantidades de aminas secundarias.. Esto ocurre en las piridmas, cjuiftoleinas t isoquinoleínas, substituidas, aisladas de los destilados del alquitrán de hulla.
» r h * u 1*d. Eag. C W , 4nt¡. BJ., 17, 1 » (1 9 « ) .

E x p e rim & n to 1 6 . C lo ru ro d e n iq u e l 1 5 7
E xperim en to 1 8 . C loruro de tú g u e l y S -nhfosalicila ldeh’do *
A 5 mi de agua añádales 1 6 2 golas de la amina que se va a investigar, Si es necesario puede añadirse una gota o dos de áddo clorhi- drlco concentrada para disolver la amina. Añada 0,5 mi de la soludón de amina resultante a 3 mi del reactivo. U na amina aiifática primaria produce inmediatamente un precipitado abundante, mientras que las aminas aromáticas primarias ordinariamente requieren 2 ó 3 minutos para dar un predpitado definido. La aparición de una ligera turgidez no es una prueba positiva, índica que pueden estar presentes huellas de aminas primarias como impurezas. Ensaye sobre: 1) n-butílamina;2) dietilamina; 3) anilina; 4) dimetilanilina.
R eactiva . A 15 m! de trietanolamina se les añaden 0.5 g de 5-nitra- salicilaldeMdo (p.f, 124-125’C) y aproximadamente '25 mi de agua y se disuelve el aldehido. Entonces se añaden 0.5 g de cloruro de níquel hexahidratado, disueltos en unos cuantos mililitros de agua, y el volumen total se ajusta a 100 mi. Si la trietanolamina contiene etanolamina, puede necesitarse añadir otros 0.5 g del aldehido y eliminar cl precipitado resultante por filtración.
.Comentarios, Esta prueba <*v tan sensible que debe fentrse cuidado al interpretarla. Sólo cuando se forma un precipitado en gran cantidad se puede considerar indicativo de una amina primaria; una turbidez ligera es meramente señal de impurezas. Debe tenerse cuidado de usar las cantidades antes especificadas, ya que la adidón de grandes canti-
Muchas muestras comerciales de aminas secundarias y terciarias condene» huellas de aminas primarias y producen turbidez.
L a prueba la dan todas las aminas primarias capaces de formar la base de Schiff con ct 5-nitrosalicilaldehído. L a hidroxiLamina y las hidra-
CHO C »-= N Rr ^ > O H
C H = NR R
N = C H
Ni/ \
dad es de soludones de aminas secundarias también darán predpitado.
» Dubr, l*¿. S n t. C fiv» .' A«*l. Ed.t 17, J96

w r
2 9J- 156 A plkodón de las p ro e jas de clasificación
* J |z in a s que sólo tienen substituyentes en un átomo de nitrógeno, dan s- " p r u e b a s positivas. Las amidas no dan precipitado. Esta prueba, no se
puede aplicar a los aminoácidos.
E xperim en to 1 9 . Acido nitroso
(a) ( ) N H * + HONO + HCI ^ / X n^CI + 2HsO
r»r»f * ^
mOH + N2 + HCJ
Diazooción, Disuelva 1 mi de anilina en 3 mi'de ácido clorhídrL concentrado, diluido con 5 mi de agua, y enfríe la solución a 0°C en un ivaso de precipitados que contenga hielo picado» Disuelva 1 g de nitrito de sodio en 5 m i de agua y añada lentamente la solución, con agitación, a la soludón helada del clorhidrato de anilina. Continúe la adición hasta que la mezcla dé prueba positiva para áddo nitroso. Esta prueba se efectúa colocando una gota de la solución sobre pape^ de almidón- yodo j un color azul indica la presencia de ácido nitroso. Pa¿« 2 ó 3 miSé.t -----„ — ------- — ----------------— I----------------- — ---------— -------------- --- - —de Ja solución a otro tubo de ensayo, caliente moderadamente y observe
jj^^fcel desprendimiento de gas. ¿Qué se formó? ¿Cuál es el término aplicado esta última reacción?
j ^ j j N j C l + ^ ' ^ ' ^ * | O N a -f NaOH
i *
q »
Prnrnm7 *
N=N^__
+ NaCl + HtO
C opulación . Añada una segunda porción de la soludón helada de diazonio —unos 2 mi— a una solución de 0.1 de 0-naftol en 2 mi de solución de hidróxido de sodio al 10% y 5 mi de agua. Observe la formación del colorante anaranjado rojizo.
fc) RJM H +H O N O - » R 2NN = 0 + H ¿>
Disuelva 2 mi de: metálanilina en 5 mi de áddo clorhídrico con- ntrado, diluya con 5 mi de agua y enfríe en un baño de hielo. Añada

E xp e r im e n to 19 , A d d o ir it r o io 159
lentamente y con agitación 1.5 g de nitrito de sodio disueltos en 5 mi de agua y observe el resultado. ¿Cuáí es el producto? ¿Por lo que concierne a esta reacción, hay alguna diferencia entre las aminas secundarias aromáticas y alifaticas?
m » H N R|Cl-
(d) r j + HONO + HCI — I j J + H*0
NORepita ( c) usando dímetilanílina. Observe el color y carácter del
producto de reacción. ¿Se comportan las aminas aiifátícas terciarias en forma semejante?
<<?)
+ HONO ------* + HsO
NO NOKA írtan lfo
cíh*oh| hjso4
o - O - h - o » -*■ K X HRojo A m l tHSO<}-
| niOH
K > n- < > 5 N °Ami
A ñada 'un cristal de nitrito de sodio a 2 mi de ácido sulfúrico concentrado y agite hasta que se disuelva. Añada 0.1 g de fenol y observe loi cambios de color,. Vierta la solución en 20 mi de agua, helada y observe el color. Añada solución de hidróxido de sodio basta que la mezcla est¿ alcalina y de nuevo observe el color.
El color azul observado en esta reacción se déte al fenolindoícno!, formado a partir del p-nitrosofenol (moaoxima de quínona) producido inicialmente en la reacción con c* exceso de fenol. Esta reacción se conoce como “nitroso de Liebenxiann1’ y es característica de los fenoles en los que hay una posición orto o para sin substituir. Puede usarse para averiguar si hay grupo nitroso mezclando cantidades iguales del compuesto nitroso y fenol, añadiendo la mezcla a ?cido sulfúrico y procediendo como en la prueba anterior.

1 6 0 A p l ic a c ió n d e la s p ru e b e s de» d o s if ic a c ió n
Comentarios. Reacción d< <rn?r*!<r< primarias. Tanto las aminas primarias aromáticas como las alifá ticas, reaccionan con ácido nitroso, dando ínicíalmente el correspondiente ion dia2onio. Como se podría anticipar, los compuestos alifáticos de diazomo son mucho más inesta* bles que los compuestos aromáticos y se descomponen rápidamente, aun a 0°C, produciendo nitrógeno gaseoso y el alcohol, olefina y productos de otros desplazamientos y reacciones del ion carbonio R'* representa el ion caitonio formado por transposición 1, 2.
R- --------> ROM + Olefina + RC1 + ROR
R N K ¿ ^ > R - N , ^ + Nj •
' ' V * -------- ► R 'O H + d e f in a + R 'C l + R'OIV
Por otra parte, las sales aromáticas de díazonio generalmente son «•wahles t-r> solución a ÜC‘C. Cuando se calientan en solución acuosa pier- den nitrógeno dando el ion ariiearbonio/1 A r\ Este ion reacciona rápidamente con el agua para dar el fenol.
Se ha demostrado que la reacción de copulación entre determinadas sales de díazonio y fenoles implica la reacción entre el ion diazonío y el ion fenóxído.<l Si la solución es demasiado acida, el ion fenórtído so convierte en el íenol y así ae retarda 3a reacción; si la solución es demasiado alcalina el ion díazonio reacciona con el ion oxhidrilo para dar díazo- hidróxido, el cual no se copula. Por lo tanto, la soludón debe estar convenientemente amortiguada para obtener una reacción de copulación satisfactoria.
La diazoacíón del ácido antranílico produce un ion híbrido (zwit- teríon) o un acildiazoato cíclico. Este compuesto es inestable y pierde anhídrido carbónico y nitrógeno para formar un intermediario extrema*
OI1
NaNOj r^ \ C O a-' 1 r \ > C >o
N
11 Ver lngold, a n d MttkanUm in O tisnic CktmiUrt, GurftelJ tfnlvtuíty Frctt.l ü v t a , N . V . , 1953. pfe. 7 » y » 7 .

Expe rim en to 19 . A c id o n itro so 161
d amenté inactivo llamado "bendno” y simbolizado por Ja fórmula A {pág. 160). El bencíno reactivo se combina con eumol para formar fcnetol} con maleato <lc dietilo para formar un benzociclobuteno substituido y con antraceno para formar tripticeno. Los mejores rendimientos de los producto» se obtienen realizando la díazoación con nitrito de amílo y la reacción de desplazamiento, en un disolvente aprótico tal como cloruro de metileno, tetrahidrofurano o acetonitrilo, Los intermediarios bencino 41 también se forman por la acción de bases muy fuertes, como el feml-litio y la amida potásica, sobre los haluros de arílo.
Reacción con aminas secundarias. Tanto las aminas secundarias aromáticas como las alifáticas, reaccionan con el áddo nitroso para formar compuestos A’-nitrosos, que son aceites o sólidos amarillos.
Reacción con aminas terciarias. Como el ácido nítrico, el ácido nitroso es un agente substituyeme para núcleos aromáticos. Sin embargo, como es considerablemente menos reactivo que el agente fútrante, el ácido nitroso ataca sólo anillos aromáticos que -eaccionan fácilmente por substitución tales como ios fenoles y aminas te' "iarías. La reacción con una amina terciaría puede verse acompañada por una desalquiladón de Ja amina con formación del derivado nitroso de la amina secundaria; la oxidación subsecuente del grupo nitroso p ro p o ^o n a una nitroamina.^
Las aminas terciarias alifaticas no experimentan ninguna rearxión de transcendencia con el áddo nitroso, pero algunas veces forman nitritos insolubles (o dorhidratos cuando se usa ácido clorhídrico para preparar el ácido nitroso). Estas sales pueden, por supuesto, reconocerse por su reacción con bases para regenerar la amina.
Aunque el ácido nitroso es más útil para caracterizar aminas, otros grupos funcionales también reaedonan con cl. Un grupo metileno adyacente a un grupo celo es convertido en una oxixmnocetonau y los alquilmercaptanos producen compuestos tionitrosos rojos,
GHjCOCH2GH, -H~ q > CHjGO - C - CHj+ HjOI!
N —OH
RCHZSH - ^ 2 * RCHjSMO +11,0
Se obtienen rendimientos muy superiores usando un nitrito de alquilo44 y ácido clorhídrico en lugar de nitrito de sodio y áddo clorhídrico. Para investigar mercaptanos,*5 añada unos 10 mg del compuesto que contiene azufre a l m] de éter. Entonces, añada 1 gota de ácido clorhídrico 6jV y 2 gotas de .nitrito de n -butilo (o nitrito de etilo). Agite
*1 KMAY, CfirtH. R«w.. w , SI ÍMfcJí S-il« y Miller, / . A«i. CÍ4n . S ti .¡ (1960);Stllfk Mlllw y B w xk W », &M-, « . m ¿ U&S3); Friednwa y Lojullu, IbU., W, <1963?.
** Crowley, Milton, Reade y TwJi, / . S«¿.r 1 W , 1286,u Scithm y DamereJI, Ot¿. Sjuthests, 2, 204 {1W JJ,
: « H íd c T J e r d v , C k tm . B er., B * 929 11952); L « b er y SScfktn, to r - , W . ( ) « « .

16 2 A p lic a c ió n d e la s p ru e b a* d a c ta s lf ic a d ó n
suavemente la mezcla y observe d color producido- Loa alquilmercap*
Los alquílmercaptanos terciarios, R jC —SH, tioéteres y disulfuros dan un color que va de verde pálido a angarillo, en la capa etérea. Los sul- fóxidos, sulfonas, sulfmatos y sultanatos dan prueba negativa.
En todas estas reacciones el reactivo nitrosante es un derivado del áddo nitroso y .no el ácido nitroso en s í Asíj Jos O s= N —NOj, O a rN — CU y 0 = N —Br, han demostrado ser especies efectivas en la día2oarión de aminas. Lo mismo que el +N = 0 > NjO< y H*NCV* que también pueden ser en otros casos los agentes nitrosantes.44
R C = 0 + H IO ,+H ..O -> 2RCOOH + H IO ,
R C = 0
RCHOHCHNHjR’+ H 1 0 , -> R C H O -f R 'C H O -I-NHj+ H IO í
Ponga 2 mi del reactivo de ácido peryódico en un pequeño tubo de ensayo, añada 1 gota {no más) de ácido nítrico concentrado y agite vigorosamente. Entonces, añada 1 gota o un cmtalito del compuesto que se va a ensayar. Agite la me*c)a de 10 a 15 segundos y añada de1 a 2 gotas de solución acuosa de nitrato de plata (5<&). La formación instantánea de un precipitado blanco (yodato de plata) indica que el compuesto orgánico ha sido Oxidado por el peryodato, el Cual, por consiguiente, se reduce a yodato. Esta reacción constituye una prueba positiva. Guando no hay formación de precipitado o aparece un precipitado café que se redisuelve al agitar, se considera que la prueba es negativa.
Aplique la prueba a las substancias siguientes: *7 alcohol isopropí- lico, acetona, etilenglico!, gjicerina, glucosa, formaíina, ácido tartárico, ácido Jáctíco.
M I r ío M j StrVrnrra anJ Mtckaa iim ¡a Qrgtmic C^cmifUy, CcwFiíll U ltivcn llY P ttU . Jtbaca, N. Y., 1SW, p¿p.w Por QoavtfiiefKb, írtM pnletcy d íb irÍD ccaíizarje jíroultóneameo«e.
E xp erim en to 2 0 ♦ A d d o peryódico
RCHOH + H IO « —» 2 R G H 0 -f-H ¡0 + H l0 ¿
RCHOHRCHOH
-f- HIOt —» RCHO -f R COOH+ HIOj

Experimento 20. Addo p©ry6dko 163
R& tCtítiO, El reactivo de áddo peryódko se prepara disolviendo0.5 g de áddc paraperyódico (H nIO r.) en 100 mi de agua destilada.
Cotnetilarios. El áddo peryódico tiene urja.acción. oxidante muy selectiva* sobre los._gÍicolefcl)2, a~hidro:rialdehidos, ct-hidroxicetonas, 1£~ 8icetona|^a;-hidrojdácidor,-y. «-anúooalcohQles. La velocidad de la reacción disminuye en el orden mendonado, Bajo las condiciones arriba especificadas, algunas veces los a-hidro»ácidos dan prueba negativa. También reaccionan los compuestos (3-dicarbonflicos y otros compuestos con metilenos activos.45
Es impórtente que se usen las cantidades exactas de reactivo y ácido nítrico. La prueba se funda en que el yodato de pJata es muy poco soluble en áddo nítrico diluido mientras que el peryodato de plata es muy soluble. Sin embargo, si hay presente demasiado ácido nítrico, el yodato de plata no predpitará.
Bajo las condiciones anteriores, las olefinas, los alcoholes secundarios, los gIicoles-lj3, las « to n as y los aldehidos no se ven afectados por el ácido peryódico. La prueba del áddo peryódico es más apropiada para compuestos.jwlubfo en agua. Para compuestos insolubles en agua pueden usarse soluciones en dioxano,
También,se. puede diluddar la presencia de un grupo cpoxi por medio del ácido^ peryódico, siempre que se añada suíidente áddo para cataJizar la hidrólisis hasta el glícol* Para investigar la presenda d d enlace.epoxi se disuelven 2 gotas de Ja subst&nda desconodda en 2 mi de ácido acético glacial y la solución se añade a 2 mi de la soludón de áddo peryódico al 0.5% que ha sido acidulada con 2 gotas de áddo
r c h - c h r + h 2o R G H -C H R
V ¿ h ¿ h
nítrico concentrado. L a mezcla se agita perfectamente y se le añaden de 2 a 3 gotas de solución de nitrato de plata al 3fó. U n a jiru e l^ p^sitiya queda indicada por un precipitado blanco de_ypdato_de.plataí._,
Se ha efectuado un estudio de la cinética de la reacción del ácido peryódico con el etilenglicol y sus análogos con substituyentes metilo. El primer paso es la formación reversible del éster cíclico del peryodato por reacción del ion ICV (o su dihidrato octahédrico, H J O fi") con el glicol. Después, el ester del peryodato se rompe por un mecanismo
“ Ver Jadean, cajetilla fl de Orgáníe Rtaciíen* d? A<tams, Vol. II. JoHr. WSlty aíid Sooi, Kvív» V«{í, 1944.
* Wollrom t Bobbltt, / . Ám. C he*. Soc., 78. 24S9 <195G); S « > Fcatcr y St<rpb«i, / . Chin i. So*., 33M (J959).
J» Fucb», Water* y V*oiJ£rL»'etfJ Anal. Cb*m.t W» 1514 (1952).>l Buiat y Bonton, / . Ch*tn. So*. 1954. 1406; Pck« y BuJgrín, / . Ch¿m. S « - , M,
3S03 C39W); Bailusr Cf\<m. TnJ.t W7 (19J9) y rcfírtfleifn citad» ¿bS.

1 6 4 A p lic o c ió n d » la s p ru e b a » d e d o s if ic a c ió n
molecular, dando directamente 2 motes de) aldehido y el ion yodato h «dilatado ( HiiO*’ —IO 3" -f-Hj(J).
E xp erim en to 21 ♦ Fenilhidrasinct y i>nUrofenUhidra»ina
A 5 mi de agua añádales 0.5 mi de fenilhidrazina o 0.5 g de p-rútro- fenilhidrazána y después, gota a gota, ácido acético hasta que justamente se disuelva la fenilhidrazina. Entonces, añada 0.5 mi de acetona y agite. Sí no hay precipitación, caliente moderadamente durante.un minuto con flama baja, añada 2 mi de agua y enfríe.
Disuelva 0.5 mi de acetofenona en 2 mi de etanol y añada agua, gota a gota, hasta que justamente desaparezca la turbídes al agitar. Si se añade demasiada agua deberá agregarse un poco de alcohol para ad a ra r la solución, A esta solución transparente se le añaden 0.2 mi de fenilhidrazina pura o 0.2 g de p-nitrofenilhidrazina. Si la solución permanece transparente durante varias minutos, se cataliza la reacción añadiendo una gota de ácido acético; caliente moderadamente durante algunos minutos y entonces enfríe. ¿Q ué ventaja tiene este procedimiento sobre el dado para la acetona?
Comentarios. Aunque se puede esperar que la mayoría de los aldehidos y cetonas formen fenilhidrazonas bajo las condiciones empleadas aquí, el uso de la fenilhidrazina tiene la seria desventaja de que muchas fcnilhidrazonas son Aceites y, por lo tanto, difíciles de apreciar vitual- mente. La />-nitrofeniIhidr¿ ín a evití. esta dificultad en muchos casos y se recomienda su empleo. Los mecanismos de estas reacciones probablemente son semejantes aJ de formación de la semicarhazona, el cual se discute en la pág. 152.
Se ha determinado al menos una de las frentes de dificultad en el uso de la fenilhidrazina. Las fettiihidrazonas producidas inicialmente
(a) y c = 0 + H jN N H Q H j ) f e N N H Q H 5 + HeO

Exp«rím«nío 21. Fenilhidrazina y p-n¡frofenllb¡drcztno*>Í65
no tan sólo pueden existir en dos formas estereoquímicas,53 sino pueden tautomerizarae en solución ha*& el compuesto azo isóme*u.- uw #* grupos nitro en un anillo ayudan a estabilizar la hidrazona y así evitan ' la tautomerizacíón.*
b) Osaiona* ■
Prepare muestras de 0.2 g de glucosa, maltosa, sacarosa y galactosa. Pruebe simultáneamente los cuatro azúcares en la forma siguiente: Coloque las muestras en tubos de ensayo, por separado, y en cada tubo de ensayo añada 0.4 g de clorhidrato de fenilhidrazina, 0.6 g de acetato de sodio cristalino y 0.4 mi de agua destilada. Tape los tubos de ensayo con tapones de corcho monohoradados y póngalos todos en un vaso de precipitados con agua a ebullición. Tome ñora del tiempo en que se hizo la inmersión y deí tiempo de precipitación de cada osazona.® Es necesario agitar Jos tubos ocasionalmente para evitar sobresaturación.
Después de 20 minutos, saque los tubos del baño Ue agua caliente y déjelos enfriar. Después que ae han enfriado, vierta uria cantidad pequeña del líquido y cristales sobre un vidrio de reloj y absorba aígo de Laa agTjas madre? con un trozo de pape! filtro, teniendo cuidado de no triturar o romper los conglomerados de cristales. Examíne loa cristales con un microscopio simple o de poco alimento (aproximadamente 80 a 100 X ) y compárelos Con fotomicrografías.56
Comentarios. £1 tiempo requerido para la formación de la osazona es frecuentemente una ayuda valiosa para diferenciar varios azúcares. Mulliken da los siguientes valores para el tiempo requerido para que
n Fm*krvWrg, SttrtoeKcrtie, Vol. S, Prao* D*in(eJí% Utipas, 19S2, pá«p. 1WG j *¡$1.
k O’ Coqoor, / . Ore. Chtzn., 25, 437S (láfil)j xn etolxiJíO, vcft KanlMn y Talkr, / . Ah , Ckem. Soc., Sí, 3624 {1 9 9 }.
» O’ConBflr y Rocen t iv o i , J . Orf . Ch*r»., M, 5269 (1961), '” L» formación d r producto* m ln o m i, drfcíóo * i* OXÍdariaq de U fcpífoldrM iai, perde
ev ita r» aondiÁndo 0.5 n i de tolnci¿B btum cU de bóutfito <k ted io , lo eu»í deberí W a re e ó k dcica «jtbr h otazúo» 7 dete rro itu r xu ponto do Im ito .
RCHOH
:HOR C -N N H Q H j
RCOCK =N N H C*H 5
+ C6HíNH2-hNH, + 2HíO

LÍ
>
í »f ti
*9
14
P t■ C .
1 6 6 A p l it a c ió n (Jé la s p ru e b a s d« « ¡o s if ica c ió n
f lk precipite la osazona de una solución caliente: fructosa, 2 minutos;* glucosa, de 4 a 5 minutos; idiosa, 7 minutos; arabinosa, 10 minutos; ga
lactosa, de 15 a 19 minutos; rafinosa, 60 minutos; lactosa, osatona. soluble en agua caliente: maltosa, osazona soluble en agua calióme; mañosa, % minuto (hidrazona); sacarosa, 30 minutos, debido a la hidrólisis y formación de giucosazona.
E xp erim en to 2 2 . S o luc ión de p erm angana to de potm io
2K M n04 + H20 ^ 2KOH *f 2M n02 + 3(0)
> c = o ( - f (O) + h 2o - > C — c <
o h ó yOiídna'óíi | tnib*Ccu«Díe
> c = o o==c<^
A 2 mi de agua o etanol añada 0,1 g (o 0.2 mi} del compuesto que se va a examinar. Entonces, añada una solución de permanganato de potasio al 2<fc, gota a gota con agitación, hasta que persista e í color púrpura del permanganato de potasio. Aplique esta prueba a : 1) pen-
¿ teno-2; 2) tolueno; 3) fenol; 4) berualdehído; 5) anilina; 6) ácidoitd 0 F fórmico; 7) áddo cinámico; 8) glucosa. Si el color no cambia entre
0.5 y 1 minuto, deje reposar los tubos durante 5 minutos con ocasional | agitación vigorosa. No se engañe con una reacción débil, ya que ésta
■ ™ puede deberse a la presencia de impurezas.Aunque para algunos compuestos insolubles en agua se ha usado
f ocasionalmente acetona como disolvente, en lugar de etanol, se ha en-J ^ contrado que determinadas parafinas, purificadas cuidadosamente, dan fcL jp piueba negativa en acetona, pero la dan positiva en etanol. El etanol
no reacciona a temperatura ambiente con el permanganato de potasio en U ^ los primeros 5 minutos.57ji| B El permanganato de potasio en ácido acético acuoso ha sido usado| para diferenciar alcoholes sencillos de tipo primario, secundario y ter
ciario, En las condiciones de la prueba, reaccionan los alcoholes prima* k rio» y secundarios, pero los alcoholes terciarios no reaccionan.5*
Comentarios. Una solución de permanganato de potasio se decolora con compuestos que tienen enlaces ecilémcos acetilénieos. Este ensayo
¿w flk se conoce como la prueba de Baeyer -para insatuiaciones. En soluciones acuosas diluidas y frias, el producto principal de la acción def perman-
55 Ijrttídt, TfcnmptOa y P ica , / , Am. CA cra. S « t . , í » , 1658■ R irter, } . Ck*m. 90, m (J353>,

E xp e r im e n ta 2 2 . So lu c ión d e p a n n cu ig a n o to d e p o ta s io 167
gaaato de potasio sobre una oleíína es el pilco!. Si se callenta la-aiezcla- de reacción se ecUiá._VL>:»á oxidación adicional - que -conduce-finaimente—. a la ruptür'aTde la cadena de carbono,
R C H ^ C H R -» R G tí—C H R -» R.—C2=iO + 0 = C - R
¿ H ¿ H ¿ H OH
Los enlaces acetilénicos usualmente se escinden por oxidación y producen ácidos
R - C a C - R '+ H * 0 + 3 { O J -* R C O O H + R 'C O O H
Esta prueba es general; en particular es útil en relación con compuestos tales como el tetrafeniletileno (G4H5)íC = G (C íHs)¿, y <4 dibro- muro de tolano, CíHsCBr^CBrC^Hj, que no decoloran la solución de bromo en tetracloruro de carbono.
La velocidad a la cual ios compuestos insaturados decoloran el permanganato depende de la solubilidad del compuesto orgánico. Si el compuesto es muy insoluble, es necesario pulverizarlo y agitar la mezcla vigorosamente durante varios minutos o disolver la substancia en un disolvente no afectado por el permanganato.
El examen de la ecuación siguiente muestra que, a medida que la reacción prosigue, la solución se vuelve alcalina.
3> C =C < ( 4- 2KMnO< + 4 H .0 - 3^>C— C<( + 2MnOa + 2KOH '
O H OH
No obstante, es necesario evitar el uso de una solución fuertemente alcalina, ya que esto cambia la naturaleza de la prueba Por ejemplo, en solución de carbonato de sodio, aun la acetona da prueba positiva. Con frecuencia no se observa la formación del precipitado de bióxido, de manganeso; el color púrpura cambia gradualmente a un café rojizo.
Sin embargo, es factible el uso del pormanganato de potasio en medio neutro. Así, con el permanganato de cinc, el hidró»do de cinc producido sólo es soluble ligeramente y la solución permanece prácticamente neutra. También es posible usar permanganato de potasio en presencia de sulfato de magnesio para alcanzar este objetivo. En este caso, el ion oxhidrilo se precipita en forma de hidróxido de magnesio insoluble.
La prueba de Baeyer, aunque es superior a la prueba de bromo paja compufstos insaturados, a su vez presenta complicaciones. Todas las substancias fácilmente oxidables dan esta prueba. Los compuestos carbonilicos que decoloran las soluciones de bromo, generaluir te dan negativa la prueba de Baeyer, La acetona es un buen ejemplo, aunque

T 66 A p ííc o a ó r t d e ( o í p ru e b a s d e d o s if ic a c ió n
decolora rápidamente las soluciones de bromo, se le puede usar como disolvente en la prueba de Baeyer. Los aldehidos dan prueba de liaeyer positiva; sin embargo, muchos de ellos, tales como d bertzaldchído y ei formafclehído, no decoloran las soluciones de bromo. El ácido fórmico
y sus ésteres, Jos cuales tienen cl grupo 0 = d lH , también reducen el permanganato.
Los alcoholes forman otra clase importante de compuestos que ■decoloran las soluciones de permanganato de potasio, pero no Jas soluciones de bromo. Los alcoholes puros no dan esta, prueba fácilmente; sin embargo, a menudo contienen impurezas que son oxidadas fácilmente. Otros tipos de compuestos también pueden contener cantidades pequeñas de impurtzas que pueden decolorar las soluciones de permanganato. Por esta razón, ia decoloración de sólo una o dos gotas de la solución de permanganato no siempre puede aceptarse como prueba positiva.
Los fenoles y las arilaminas también reducen la solución de per- manganato y sufren una oxidación a quinonas; éstas pueden oxidarse aún más con un exceso de reactivo, dando una serie de productos de oxidación entre los cuales están cl ácido raaleico, el ácido oxálico y el anhídrido carbónico.
OH O■ I i!
Los compuestos orgánicos azufrados en los que el azufre está pre* senté en estado reducido, también reducen el permanganato y se oxidan. Los mercaptanos se oxidan a disulfuros y los tioóteres-y los tioacetales hasta sulfonas.
2RSH + {O) — » R $ S R + H 20
RSR -£ !+ R ¿ 0 R S 0 2R
R*C(SR), -Í21* R¿C(SO¿R)2
Esta oxidación de los tiocompuestos ocurre con mayor facilidad en un medio áddo, por lo que, bí el análisis elemental muestra la presencia de azufre, es conveniente disolver 0.1 g del compuesto en 2 mi de ácido acético glacial y añadir la solución diluida de permanganato de potasio gota a gota. Si el color púrpura desaparece, es jyosible que « té presente un grupo funcional que contenga azufre oxidable. Guando el azufre está
c h c o 2h w c o 2h <01— I > COj
c h c o 2h c o *h

E xp e r im e n to 2 3 , So lu c ión d e n itra to d o p ía t e 169
presente en forma de sulfonas, sulfatos de alquilo o ácidos iulfónícos no substituidos, la solución de perra anganato de potasio no se reduce. Sin embargo, determinados sulfonatos substituidos, tales como los cora- puestos de adición bisulfítica a aJdehídos y cotonas y los ácidos fenol- sulfónicos, reducen el pennanganato.
El hecho de que los glicoles formados a partir de olefinas, que pueden encontrarse como isómeros geométricos, sean loa productos de adición cis de las dos funciones oxhidrilo sobre el mismo lado del enlace doble ha sugerido fa posibilidad de que baya un ester intermediario de manganeso cíclico,w
¿Qué grupos funcionales responden tanto a las pruebas de bromo como a las de permanganato? ¿Cuál de estas pruebas es mejor para investigar la presencia de enlaces múltiples? ¿En qué casos es provechoso el uso de ambos reactivos?
E xperim en to 2 3 . S o luc ión de n itra to de p í^ta ( etanáíiea)
R X + AgNOj —> AgX-f- RONOa
Este reactivo es útil para clasificar compuestos que se sabe contienen halógenos, Añada una gota del compuesto halogenado a 2 mi de una solución etanólica de nitrato de plata al 2%. Si después de 5 minutos de reposar a temperatura ambiente no se observa ninguna reacción, la solución se calienta hasta ebullición y se observa si se ha formado un precipitado. Si hay precipitado, observe su color. Añada 2 gotas de ácido nítrico diluido (5%) y observe si se disuelve el precipitado. Los haluros de plata son insolubles en ácido nítrico diluida; las sales de plata de los ácidos orgánicos son solubles.
Ya que los haluros de alquilo a menudo contienen como impurezas cantidades pequeñas de isómeros, puede ser aconsejable en determinados casos extremos, recoger y pesar, el haluro de plata seco obtenido a partir de una muestra pesada de Ja substancia desconocida. Ordinariamente se puede llegar a un valor aproximado del peso molecular a partir de una consideración de sus constantes físicas y de un examen de la lista de posibilidades; el rendimiento teórico calculado del haluro de plata puede así compararse con la cantidad obtenida. Sí las cantidades del rendimiento obtenido son sólo un porcentaje pequeño, la prueba es negativa, Un haluro de alquilo que sólo da una cantidad pequeña de haluro de plata porque reacciona lentamente, puede diferenciarse d d d c una mezcla de un haluro inerte con una cantidad pequeña de impureza reactiva, mediante filtración del haluro precipitado ¡nieta Imcmc y pro- bando después el filtrado con má's nitrato de plata,
M Vfir 'Wfiien, «apítulo 12 de O r & C i t / m u t r y do Cilrtt»o, Vot. JV, John WíleT Sano, Nueva York, 14i3, p ij* . l2 ll j agí.

9
i*i •
170 A p lic a c ió n d e la s p ru e b a s d e c la s if ic a c ió n
jj ^ Aplique la prueba a: l) cloruro de benzoílo; 2} cloruro ae bencilo;3) bromuro de etilo; 4) bromobenceno; 5) cloroformo; 6) ácido clor- ac ético, *
Í * Comentarios, Muchas substancias que contienen halógenos neaccio-P nan con nitrato de plata dando un ha luto de plata insduble y la velocidad
de esta reacción es un índice del grado de reactividad del átomo de halógeno que se estudia. Esta información es valiosa, ya que permite
g ■ hacer determinadas deducciones acerca de la estructura de la molécula.I Ixis halaros más reactivos son aquéllo* que son iónicos. Entre los
compuestos orgánicos, las safes de aminas de los ácidos halógena dos constituyen los ejemplos más comunes.
[RNHj]*X-
Se encuentran con menor frecuencia las saies -de oxooio y las sales de carbonio, las cuales contienen halógeno iónico.
R:O rRH
x- C+Q"
c rin » ] vScilft*
Las soluciones acuosas de estas sales dan inmediatamente un precipitado de haluro de plata con solución acuosa de nitrato de plata.
TEORIA DE LAS REACCIONES DE SUBSTITUCION .
Debido a que la prueba con nitrato de plata alcohólico y la substitución del ion haluro con yoduro de sodio (experimento 27) están tan Intimamente relacionadas, se ha encontrado prudente discutirlas juntas en esta sección. Así como es conveniente el clasificar los tipos de enlace, definiendo los dos extremos como co valen le y iónico, aunque la mayoría de los enlaces son intermedios entre los dos. es conveniente definir los dos mecanismos extremos por los que los balurós y substancias similares experimentan substituciones nuclcóíilas. El primero de éscos ha sido llamado por varios investigadores “S,.-2V (substitución nudeófíta bimolecular), “desplazamiento''’ o “IN"’ (nucleófilo) y el segundo, “Syl”, “solvólisis" o “Lím.” (lim itante). Ya que los dos mecanismos tienen requisitos estructurales muy diferentes, cualquier consideración del efecto de la estructura sobre la reactividad de los haluros debe tomar en cuenta el mecanismo involucrado.
La discusión detallada de este tema está más allá del pian de este libro, pero existen excelentes revisiones críticas.*0 Sin embargo, una
• S n íM i o r , Chem. R<9!., W, 571 (ÍS5C), y M v tly i ie Dí¡pUc*n>t<u‘ R«<trtitn¡, McGravr» HlH Pook Co-, Nurva Yorfc, S%2. Birllrtt. cu Organls ChimiUry Cilman, V’oJ. 151. Jófart
Son?, Nueva Y«rk, i9i3, p ig . Irg<>W, Stnitiur* and M r t >'* Qtgaiüi Cktmíüry, ComeD tsaranit? Pn:«* lH u n , N. Y., 1953.

T eo r ía d e lo s re a cc ion e s d e su b s t itu c ió n 171
mención breve de los dos mecanismos aludidos será una ayuda considerable para comprender Ja prueba del nitrato de plata y 'tam bién Ja prueba que usa el yoduro de sodio en acetona (experimento 27).
El mecanismo de desplazamiento SN2, es una reacción concertada en Ja que un reactivo nuclcófilo, como el ion yoduro u oxhidrilo, choca con el haluro en reacción y la colisión ocurre en la parte posterior del átomo de carbono del cual se está separando el ion haluro,
\ R / R I- + H-C—X !-*..« ¿..-X-H I—c ú a + x -
S » ' \ \lEuftdo de
Se ha visco que el estado de transición tiene la, carga negativa distribuida preferentemente sobre los grupos entrante y saliente. Ya que es muy pequeña la carga positiva o negativa inducida sobre el átomo de carbono central, un grupo R que puede estabilizar una carga positivao negativa ayuda muy poco a la reacción. Por otra parte, ya que la reacción se inicia con la formación del enlace co va lente con I“, la parte posterior del carbono en el que se está efectuando eí desplazamiento debe estar libre de impedimento para permitir la entrada fácíl del átomoo grupo atacante. Así, es de esperarse que esta reacción sea sensible al bloqueo estérico en el sitio de reacción, pero relativamente insensible a los efectos electrónicos. El S,vl, o mecanismo de solvóllsis, involucra dos etapas de las cuales la primera ea la ionización del haluro reac^ donante.
R - X - + R - + X -
R+ + Y --* R - Y
Es evidente que este mecanismo no debería retardarse por el bloqueo estérico en Ja parce posterior'por R y, por otra parte, debería verse muy favorecido por un cambio de R que ayudara a la distribución de la carga positiva. En general, los haluros que reaccionan con el yoduro de sodio en acetona deben hacerlo por el mecanismo S,v2 y aquellos que reaccionan con el nitrato de plata alcohólico deben hacerlo por un tipo S.vl- JBstas conclusiones se deducen de la naturaleza de los reactivos. El yoduro de sodio en acetona tiene c! ion yoduro que es un agente desplanante muy bueno (se dice qu^ es fuertemente nucleófilo) y la. acetona que es un disolvente muy malo para la ionización, por lo tanto, este reactivo favorece fuertemente el mecanismo N. Por otra parte, el. ion nitrato es un agente nucleófilo malo, el etanol es un disolvente ionizante intermedio y la plata; por su poder de coordinación con el halógeno que se está separando, es excelente para ayudar ¿ la ionización, consecuentemente, este reactivo favorece el mecanismo Lim.

*■
Coa estos principios generales en mente, es posible correlarionar un gi*n número de dates. Desafortunadamente, en el medio semipolar en donde Jos químicos orgánicos a menudo prefieren trabajar, las salea están presentes no íólo corno iones disociados, sino también (y principalmente) como pares iónicos, iones triples y grupos iónicos. Así, una solurióa diluida de yoduro de sodio en acetona contiene iones sodio, iones yoduro y pares iónicos de yoduro de sodio. Ya que el ion disociado es normalmente la especie más reactiva, la velocidad neta de la reacción dependerá de la cantidad de yoduro que se encuentre presente como el ion menos reactivo, es decir, de la constante de disociación del par-iónico así como de la reactividad del ion yoduro disociado. Tales consideraciones tienen implicaciones importantes cuando es necesario correlacionar la estructura con la reactividad. Así, el orden de reacción de los haluros de litio en acetona es i L il > Liflr > LiCl, orden inverso al de reactividad de los iones disociados M en ese disolvente.
Los disolventes dipolares apróticos, tales como el dimctilsulfóüádo, A^W-KtímetíIforTn&mida y sulfolano, han demostrado ser particularmente efectivos como disolventes para reacciones de substitución nucleófila.^
H aluros saturados . Los iones carbonio terciarios son más estables que los secundarios, los cuales, a su vez, son más estables que los primarios. U na explicación de estas diferencias es que el ion terciario tiene una manera de distribuir su carga positiva que es más eficiente que la de los iones secundarios y la de éstos es más eficiente que la de los primarios. Esta distribución de la carga puede ser discutida en el lenguaje de la teoría de enlace-valencia, mediante la descripción de un ion, por' ejemplo; el ion f-butilcarbonio, como un híbrido de resonancia de las estructuras que se muestran a continuación,*3 Dicha distribución de la carga sobre Jos átomos de hidrógeno del alquilo ha sido llamada "hiper- conjugacíón’V
H CHj H CH,
H — i — <!> v> H *C =C «-» Otras estructuras semejantesl i l i
H CH, H CH,
El segundo factor que favorece la formación de iones carbonio ter- líanos a partir de sus haluros, en comparación con la formación correspondiente de los iones primarios o secundarios, es 3a tensión estéríca |ue puede estar presente en jo Iialur^ terciario (con ángulos de 109° ilrededor del átomo de carbono central), la cual es aminorada pardal- neme al pasar al ion plano (con ángulos de 120°C entre los grupos unidos
M Wuidrin, SlVefofí. SllliOl, Stcvtna y G*llf TtfraheJron Lrttiri, 9, 24 '1960}. »* Páffcír, Q ^ n . I», 163 {1962).« WbífaTwJ. A d a a \tt¿ Orgati* C ktm itíry, > Jofee Wiíujr pjwt Son», N u tv a Y ojÍ,
¥M, ms. 4W.
1 7 2 A p l ic a c ió n d e la * p ru e b a s d e d c s r f íc o c ió n

T eo r ía d e io s re a cc io n e s d e substituc ión i 73
al átomo central).M Entonces, es de esperarse que el ondea de reactividad de los haluros de alquilo con el nitrato de plata, sea el que se ha encontrado expcrimcntalmente:
Terciarios > Secundarios > Primarios
Es interesante que la reactividad solvolítica sea muy glande en moléculas tan excesivamente arborescentes como el cloruro de tri-í-butil« metilo, el cual puede reaccionar hasta 40,000 veces más rápido que el cloruro de fcr-butilo.*5
Como podría anticiparse, el orden de reactividad de los haluros sencillas se invierte en las reacciones de desplazamiento con yoduros alcalinos, ya que el orden de accesibilidad estérica al ion yoduro es: primario > secundario > terciario.
Con el yoduro de sodio, los bromuros primarios forman, a 25C>C, un precipitado de bromuro de sodio en 3 minutos •, menos, mientras que los d o m o s no forman precipitado y deben calenurse hasta 50°C para que haya rcacción. Los bromuros secundarios y terciarios reaccionan a 50CC, pera los cloruros terciarios no reaccionan dentro del tiempo especificado, Los cloruros terciarios reaccionarán si las soluciones de prueba se dejan reposar durante un día o dos.
Haluro* alicícíicos. En la reacción con nitrato de plata, los haluros de cidohexilo presentan una reactividad menor que los haluros secundarios de cadena abierta. El cloruro de cidoheNÍlo es inactivo y el bromuro de cidohexilo es menos reactivo que el 2 -bromohexano, aunque da precipitado con el nitrato de plata alcohólico, Símilarmerite, el cloruro de i-metilcíclohexilo es considerablemente menos reactivo que los cloruros terciarios acíclicos, Sin embargo, tanto el cloruro de l-metil- dclopentilo como el doruro de I-mctílcicloheptilo, son más reactivos que los análogos de cadena abierta,*6 Se ha discutido el efecto del tamaño del anillo sobre la velocidad de solvólisis en los anillos de 4 a 17 miembros.*1
En, la rcacción con el yoduro de sodio en acetona, el cloruro ac ciclopcntilo reacciona a una velocidad comparable con la de los cloruros secundarios acíclicos,w mientras que la rcacción con cloruro de ciclo- hexilo es considerablemente más lenta.w Así, el cloruro y el bromuro de ciclohcxiío, el cloruro de bomílo y los compuestos similares, no reaccionan apreciablemente con el yoduro de sodio a 50°C en un tiempo r.o
'«H ÜMlleU, CT1 Qrfanic Chzmiltry de CilíiVan, Vol, í l l , Jijfio Wiler and Son», Nuevo Yojl, p í$» . y s ip .M Bpnlett, en Qrgavie Chamiliry d* GiUnin, Vol. III, Jota Wi|<y and Swi», Xuevt Yuti.
1M3, pí*. ¿9.* firvwn, Fletdiír y / . Am. Ckeot. Soe„ 71, 2J2 (1951).» browti y BorfcowiW. / . Ám. Ck*m. So*., 74, 1894 (3442); B im ii, Br*w*er y Sfeechwr,
ríM., 7*. «7 (19H).** flwdwiit y Coúfxr, / . Am. Ck*m, Scf.j J4*S (1851).

m a y o r d«* 6 m in u to s . E s ta d ism in u c ió n d e l a v e lo c id a d en el s istem a d e l ñ c lo h e x ü o se d e b e a la g e o m e tría e sp ec ia l im p u e s ta a l e s ta d o d e tran s ic ió n p o r c l a p illo d e l c ic lo h ex a n o .
H a lu r o s b ic íc l ic o s « U n h a ló g e n o e n el á to m o d e c a rb o n o , p u e n te d e c ie rto s s istem as b ic íd ic o s , es m u y in a c t iv o t a n to e n las reacc io n es d e solvóliste co m o en las d e d e sp la z a m ie n to .^ P o r e jem p lo , e l d o r u r o d e a p o c a n fü o ( I ) n o se a fe c ta p o r t r a ta m ie n to c o n so lu c ió n ca lien te de h id ró x id o d e p o ta s io a l 3 0 ^ e n e ta n o l a l 8 0 % d u r a n te 24 h o ra s , n i co n n i tra to d e p la ta en e ta n o l acu o so y ca lien te d u r a n te 4 8 horas.™
E l a ta q u e d e l io n o x h id rilo es b lo q u e a d o p o r e! e sq u e le to en fo rm a d e ja u la y la d is tr ib u c ió n d e la c a rg a d e l io n c a rb o n io te rc ia r io , p o r re so n an c ia d e l t ip o q u e se m u e s tra , e s tá im p e d id a e s to ic a m e n te ( re c o r d a r la reg la d e B re d t) .
174 A p lic a c ió n d a la s p ru e b a s d e d o s if ic a c ió n
E l l-b ro m o h ic5 .c lo [2 .2 .1 ]H ep tan o se c o n v ie r te en el carb in o l c u a n d o se traca d u ra n te 2 d ía s co n n i tr a to de* p la ta a cu o so a 1 5 0 °C . C u a n d o se t ie n e la j a u la del 2 .2 .2 -b íd c io o c ta n o , c o m o e n e l b ro m u ro ( I I ) . el n i tr a to d e p la ta , acuoso fo rm a el a lc o h o l c o rre sp o n d ie n te d esp u és d e 4 h o ra s a te m p e ra tu ra a m b ie n te .11 L a m a y o r re a c tiv id a d d e l s istem a 2 .2 .2- e n co m p a ra c ió n c o n e l 2 .2 .1- es p o sib le d e b id o a las e s tru c tu ra s d e l p rim ero , q u e e s té tic a m e n te son u n p o c o m ás fa v o ra b le s p a r a la re so n an c ia p o r h ip e rc o n ju g a c ió n .
H aluros de crilo y v in iic «n L o a h a lu ro s d e v in ilo y a q u e llo s ha* lu ro s d e a rilo en. los q u e el h a ló g e n o e s tá u n id o d ire c ta m e n te a l an illo a ro m á tic o son in e r te s ta n to a l d e sp la z a m ie n to lin ea l co m o a l m ecan ism o d e solvólisis, y a q u e h a y u n b lo q u e o csté rico q u e e v ita et p r im e ro y la re la tiv a in e s ta b ilid ad d e loa io n e s c a rb o n io c o n c a rg a p o sitiv a sobre u n á to m o c o n e n la c e d o b le o u n á to m o d e c a rb o n o d e a rilo , im p id e e l segundo .
Sin em b arg o , se d isp o n e d e u n n u ev o m e c a n ism o p a ra los h a lu ro s d e a rilo c o n su b s titu y en te s o r to O p a n 2, ta le s c o m o g ru p o s n itro , en p resen c ia d e bases fu e rte s , v . g r., o x h id r ilo o a lcox¡lo .w Así, el c lo ru ro
14 Apj>k<*uiít y Roben*, C ítun . Rain., W , 10&5» Bnrtldt r Kaac, / . Am. CKtm. .?<*., «1, 3184 (1933),n D oerins, X*vi»j S iyigti, Spr«d»cr y W heUB, J. Am. C ktm . S tc-, 75, locen P v n una díicirffiD ¿ t t a «ac tiv idades de loi haluto» de an ta , con ctfcrcittiaJ, Vea
K am nw id y P * rb , i . Am. Chrm. Sot., 77, 340 (1935).15 Buimett y Zshkr, CJLrm. Rtvr>, 49, 273 (3951); ta^nld, S<n>e¡itrt and Mtchmun i>
OrgM it CktMÜtty, Coroell IJo tvcnity P rcrt, J tb a ia , N . Y.» 1953. capitu lo X V .
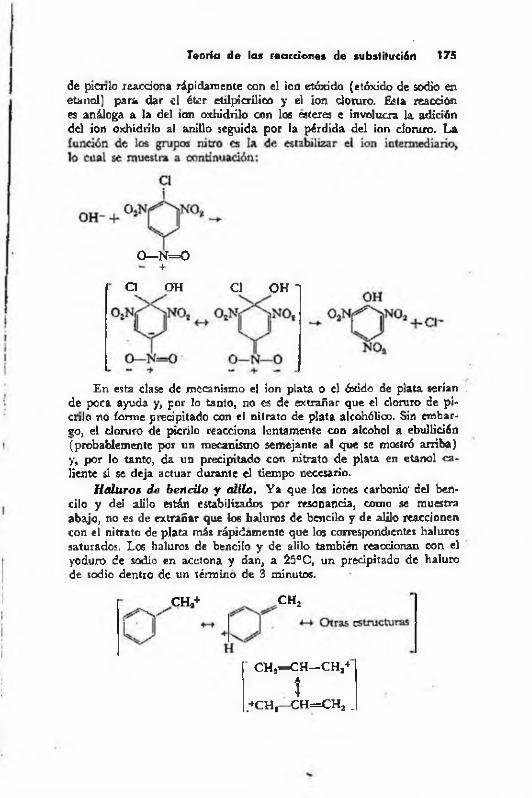
T eo r ía d e la s re a cc io n e s d e su b s t itu c ió n 175
d e p íe n lo re a c c io n a r á p id a m e n te c o n e l ion e tó x id o (e tó x id o d e sodio e ne ta n o l) p a r a d a r e l é te r e tilp ic rílico y c\ io n c lo ru ro . É s ta reacc ió n es a n á lo g a a la d e l io n o x h id r ilo c o n los esteres e in v o lu c ra k a d ic ió n d e l io » o x h id r ilo a l a n illo s e g u id a p o r l a p é rd id a d e l io n c lo ru ro . L a
d e p o c a a y u d a y , p o r lo ta n to , n o e s d e e x tr a ñ a r q u e e l c lo ru ro d e p í e n lo n o fo rm e p re c ip ita d o c o n e l n i tr a to d e p la ta a lcohó lioo . Sin e m b a rg o , e l c lo ru ro d e p ic ó lo re ac c io n a le n ta m e n te c o n a lco h o l a eb u llic ió n (p ro b a b le m e n te p o r u n m e c a n ism o se m e ja n te a l q u e se m o stró a r r ib a ) y , p o r lo ta n to , d a u n p re c ip i ta d o c o n n i tr a to d e p la ta en e ta n o l c a lie n te si se d e ja a c tu a r d u r a n te e l t ie m p o n ecesa rio .
H a l u r o s d e b e n c ü o y a i l l o . Y a q u e lo s io n es carb o n io 1 de l b en - c ilo y d e l a ü lo e s tán estab iliz ad o s p o r re so n an c ia , co m o se m u e s tra a b a jo , n o es d e e x tr a ñ a r q u e los h a lu ro s d e b e n c ílo y d e a li lo re a c c io n e n
y o d u ro d e sod io e n a c e to n a y d a n , a 2 5*G , u n p re c ip ita d o d e h a lu ro d e sod io d e n tro d e u n té rm in o d e 3 m in u to s .
O—N= 0+
C1 O H C I O H i
E n e s ta c lase d e m e c a n ism o el io n p la ta o e l ó x ido d e p la ta s e r ía n .
c o n e l n i tra to d e p la ta m ás rá p id a m e n te q u e los c o rre sp o n d ien te s h a lu ro s sa tu ra d o s . L o s h a lu ro s d e b e n c ilo y d e a lilo ta m b ié n re ac c io n a n c o n el
C H 2+ CH j
' c h 2= c h — c h 3+
t,+ C H ,— c h = c h 2 .

1 7 6 A p l ic a c ió n d e la s p ru e b a s d e d o s if ic a c ió n
E l e fe c to d e im a su b s titu c ió n a d ic io n a l e n e l a r ilo , g e n e ra lm e n te es e l d e a u m e n ta r l a v e lo c id ad d e l a reacc ió n d e solvóüsis. Asi, el c lo ru ro d e b e n sh id r ilo re ac c io n a r á p id a m e n te c o n e tan o l c a lien te , a u n sin a d ic ió n d e n i tr a to d e p la ta , y el c lo ru ro d e tr ife n ilm e tü o en d ió x id o d e a z u fre l íq u id o (d iso lv en te e sco g id o p o rq u e e s e fec tiv o e n la so lva- ta c ió n d e io n es y n o re a c c io n a ) s e io n iza e x te n sa m e n te a l c lo ru ro de tr ife m lm e tílc a rb o n io (C¡¿Hi) jC C l"
A u n q u e se ría d e e sp e ra rse q u e e l c lo ru ro d e t r ífe n i lrn e tib tu v ie ra d e m a s ia d o im p e d im e n to esté rico p a ra e x p e r im e n ta r u n a reacc ió n d e d e sp lazam ien to con e! io n y o d u ro , se h a e n c o n tra d o q u e re ac c io n a m u c h o m ás r á p id a m e n te q u e el c lo ru ro d e b e n e ilo co n e l re a c tiv o d e y o d u r o 'd e so d io -ace to n a . S in e m b a rg o , la reacc ió n n o es u n a sim ple su b stitu c ió n p a ra fo rm a r y o d u ro d e tr if i lo , com o lo d e m u e s tra el h e ch o d e q u e se ob se rv e e l c o lo r de l y o d o . E s te e je m p lo h a c e re sa l ta r la a te n c ió n q u e es n e c e sa r io p o n e r a l in te rp re ta r p ru e b a s c u a lita tiv a s d e e ste t ip o . D eb e re c o rd a rse q u e los m iem b ro s d e u n a c la se d e com p u esto s e n e s tu d io p u e d e n re a c c io n a r co n el m ism o re a c tiv o e n v a r ia s fo rm a s y q u e p a r a c a d a c lase d e reacc ió n p u e d e s e r d ife re n te el e fe c to d e l a e s tru c tu ra so b re la re a c tiv id a d .
P a r t i c ip a c ió n d e g r u p o s v e c in o s . E s d e e sp e ra r q u e el e fec to de c u a lq u ie r á to m o o g ru p o su b s tra c to r d e e lec tro n es, a d y a c e n te a u n á to m o d e c a rb o n o en e l q u e p u e d a e fe c tu a rse u n a re a c c ió n d e solvóüsis, s e a d d e d ism in u ir la v e lo c id ad , y a q u e es d e sfav o rab le la p resen c ia de u n a c a rg a p o s itiv a p a rc ia l a d y a c e n te a l s itio en el c u a l se v a a in d u c ir u n a c a rg a p o sitiv a . Este e fe c to e s tá i lu s tra d o p o r la ca re n c ia d e re a c tiv id a d d e los h a lu ro s d e fi-alcox ilo , fc -h a locc tonas ( ta le s c o m o la c t-d o ro a o e to - f e n o n a ) , fl-c lo rocsteres d e ác id o s to lu e n su lfó n ic o s ÍJ y i ,2 ’d ic lo ru ro s .7! S in e m b a rg o , d e b e rá n o ta rs e q u e las a -h a lo c e to n a s , ésteres, a m id a s y n itrilo s , son fu e r te m e n te reac tiv o s h a c ia e l io n y o d u ro y c u a n d o «c t r a ta n co n y o d u ro d e sod io en a c e to n a a 25 °C , d a n u n p re c ip ita d o d e h a lu ro d e so d io .d en tro d e i té rm in o d e 3 m in u to s .
S e ha aportado evidencia de que l a aceleración de Jas reacc io o es de desplazamiento por u n a función c a rb o n ilo y x se d e b e a la e stab ilizac ió n d e i e s ta d o d e tran s ic ió n p o r u n a d is tr ib u c ió n p a rc ia l d e la c a rg a n e g a tiv a so b re e l á to m o d e o x íg e n o c a rb o n ílic o . T a l e stad o d e tran s ic ió n te n d r ía la e s tru c tu ra q u e se m u e s tra e n seguida.™ L a e x ag e rad a in a c tiv id a d de la d o r o m e t í l^ to l i l s u l f o n a e s tá d e a c u e rd o c o n este p u n to d e v ista .17
» Wtuki*. 4 f , Am. Cfum. $0Í-, 7Í, MI (I9 tf|; OwamUJbM .r 73, « 5 8
O Qjvx7t. f le o í., 5 , 254 (1951).
y TífcitcnbteK , } . Am. Cktm. S«e., S i, 5606
i» R w w , IfcCiíítry y Kfcutr, 1 . Am. Ch<n. Soc., 19, 2280 (1954).

\ r
i / \ lL o s g ru p o s R jN — , R S — , I — 7 b e ta a l h a ló g e n o , a d ife ren c ia del
a lco x ilo o c lo ro , a u m e n ta n la v e lo c id a d d e so lv ó lis is” L o s li: lluros que c o n tie n e n estos g ru p o s re a c c io n a n p o r d e sp laz am ien to interi»o, <_omo se m u e s tra a co n tin u a c ió n :
T e o r ía d « la » reacc ión t i d é « ob stilv c ló n 177
1+ O H
E n e l caso d e o tro s g ru p o s v e c in o s , ta le s co m o «I b ro m o y c l ace- to x ilo , la d ism in u c ió n d o l a v e lo c id a d de solvólisis (a n á lo g a a la d ism in u c ió n d e la v e lo c id a d p o r u n c lo ro v e c in o ) a c au sa d e l e fec to in ductivo es c o m p e n sa d a en g r a n p a ite , a u n q u e n o to ta lm e n te , p o r la acción d e sp lan a n te in te rn a , p o r lo q u e las v e lo c id ad e s d e solvóüás d e las su b s tan c ia s d e los t ip o s q u e se in d ic a n a b a jo , só lo so n ligeram en te in fe r io re s a las d e l a n á lo g o sin su b s ti tu y e m e ^
B r C H jC O O H
' _ U _ , -L L < -L L 1 i ' i ' í
Ya que para desplazar u n átomo saliente» X, el grupo vecino debe ser capaz de participar en una inversión de Waldcn con ataque p o r la parte poftteríor, Jo» grupo* c\i d e lo* «Utema* cíclicos no (on efectivas. Así, cuando e l p-brornobencenosulfonato d e ciclohexilo está siiUútuíto con un acetoxflo c is e n la posición-2, su velocidad de solvólbis en ácido acético glacial se disminuye en un factor de casi 5000 (efecto inductivo),
71 V tt Wiomio y Gnmw»ld, ) . Am- Chin* &*<., 70» 82S P « r * &n rtxumo.

176 A p lic a c ió n d * la s p ru e b a s d e d a s íf ie a d ó n
poro u n g ru p o acetox i(o -2 lra n iy l a d ism in u y e sólo en u n ía c to r d e 5 ,14 ya. q u e d e fec to in d u c tiv o es c o m p e n sa d o e n o rm e m e n te p o r e l h e c h o d e q u e e l a ce to x ilo es u n b u e n a g e n te d e sp la z a n te in te rn o y e s tá en l a p o sic ió n eatérica a p ro p ia d a p a r a u n d e sp la z a m ie n to trans.
L o s g ru p o s a r ilo ta m b ié n p u e d e n p a r t ic ip a r c o m o g ru p o s v ecin o s.” Asi* e l c lo ru ro d e 2 ,2^2-trifenU etilo se so lvo liza e n so lu c ió n d e á c id o fó rm ic o íc o n m ig ra c ió n d e l fe n ilo ) 10,000 veces m ás r á p id o q u e e d o r u r o d e ceo p e n tilo .31 E n e s ta tran sp o s ic ió n d e W a g n e r-M e e rw e in e l fen iio desp laza in te rn a m e n te a l io n c lo ru ro , e n u n a r e a e d ó n q u e es a n á lo g a , p o r u n a p a r te , a lo s d e sp la z a m ie n to s in te rn o s a n te rio re s p ro d u c id o s p o r o tro s g ru p o s v ec in o s y , p o r o t r a p a r te , a la a lq u i la d ó n a ro m á tic a d e i t ip o F rie d e l-C ra f ts .
O j 5 p" H r *
O tr o * c o m p u e s t o s k a lo g e t ia d o s . Y a q u e la m ay o ría d e lo s d o - ituos^ y b ro m u ro s d e a d ío re a c c io n a n r á p id a m e n te c o n e l e ta n o l, a u n sin n i tr a to d e p la ta , éstos; fo rm a n in m e d ia ta m e n te u n p re c ip ita d o c o n el n i tr a to d e p la ta a lcohó lico . T a m b ié n r e a e d o n a n r á p id a m e n te c o n e l yo* d u ro d e so d io e n a c e to n a , p ro b a b le m e n te p o r Un m e c a n ism o d e ad ic ió n d e carbonita*
U n é te r a -h a lo g e n a d o se so lv o liza r á p id a m e n te y e l io n c a rb o n io re s u lta n te e s re la tiv a m e n te e s tab le ( e l io n p u e d e se r c o n s id e ra d o co m o e l é te r d e l á d d o c o n ju g a d o d e u n a c e to n a ) . A sí, lo s c o m p u esto s d e este
[ R - O - C - Ó = C —1■ i • I
t ip o a m e n u d o fo rm ar, in m e d ia ta m e n te u n p re c ip ita d o c o n n i tr a to de p la ta ?.1cohólico .
L a a c u m u la c ió n d e á to m o s d e d o r o so b re e l m ism o á to m o d e car* b o n o e n lo s com p u esto s a lifá tíco s se n d llo s , p ro v o c a u n n o ta b le g ra d o d e in a c t iv id a d c o n e l n i tra to d e p la ta , ta l c o m o se o b se rv a en e l c lo ro
** W ioutóu , G ru tw iJ d C In s v th ír t , / . A m . C hrm . So*., 70 , 821 (1$4S),■ Wiortán, M m h, Cniov/zíá, Sdlrtiber f GtTH, J. A m . C fum . S o t,, 74, l lt3 (IS52);
W üUtiú E í o s n J u a i , ib U l., 7 7 , 1 7 3 8 ( 1 5 5 5 ) .
a Injold, StmttMTá attJ k ttdw n itte ín Ow¡ar¿t C kim ítttf, ComcU ü rJw ttic r Prew, ítlwK*, N . Y ,, 1953. p ig . 514.

Iftcrfa da le*» reaccione» do subrtftuetón 179
fo rm o , te tra c lo ru ro d e c a rb o n o , í - te t r a d o r o e ta n o y ác id o tr id o ro a c é tic o . E s to n c o c u rre e n los com p u esto s b ro m a d o s ; e l ic tra b ro m u ro d e c a rb o n o re a c c io n a c o n el n i tra to d e p la ta a 2 5 ° C y e l b r o m o f o m o y e t > te t r a - b ro o io e ta n o f o r m a r u n p re c ip ita d o , h irv ié n d o lo s c o n n i tr a to d e p l a t a a lco h ó lico .
En los compuestos del tipo del cloruro de aliío, la acumulación de átomos de cloro sobre un solo átomo de carbono, no disminuye su reao tívidad, sino que, en realidad, parece que la incrementa. El benzotri- cloruro se hidroliza con más facilidad que el cloruro de benzal, el cual a su vez es más reactivo que cl cloruro de bencilo.
C C i3 C H C la O H jC l
0>0>0L o s com p u esto s p o lic lo rad o s sencillos c o n los h a ló g en o s so b re u n
so lo á to m o d e c a rb o n o , ta le s co m o e l c loroform o* e l te tra c lo ru ro d e car* b o n o y e l á c id o tr id o ro a c é t ic o , n o re a c c io n a n c o n y o d u ro d e sod io e n a c e to n a . S in em b arg o , di d o r u r o d e b en za l y e l b e rrzotri d o r u r o son re a c t iv o s d e b id o a l s is tem a a iíiic o p re sen te .
A c o n tin u a c ió n se e x p o n e u n re su m en d e los re su ltad o s q u e so n d e e sp e ra rse en la p ru e b a c o n n i t r a to d e p la ta a lcohó lico .
L Loa siguientes com puestos íohtbicB e n ag u a d a n inm ediatam ente u n p recip ita d o c o a n itra to d e p la ta &cuo&o.I. S a ta de amina* de hidrácidc» halogenados,
(R N H j) *X "+Ag*NCV -*■ A gX -f (R N H i)*N O r
2> Sales de oxotuo.$. Hftlnn* de catbooio.4« Claniíoí de icálo de bajo peso molecular. Mudioa de ¿atos *e hidroUiaa
con agua y &*i proporciona el ion haluro.RCOC3+ H ¿> - * R C O O K + HCI
I I , L 05 com puestos inxolubles e n ag u a k co locan d e w o d o genera l den tro" d etre i jru p o íj con respecto a comportam iento frente a «olucionei de nitratod e p l a t a sJcohó lico .1 . L o ? C om piie ito* d e l p r im a r g ru p o d a n in m e d ia ta m e n te a te m p e ra tu ra
a m b ie n te u n p re c ip ita d o .
RCOC1 RCHClOR R»CC1B.CH—CH CH jX RCHBrCHjBr R I
% E l segundo grupo Incluye compraeítol. que re ac ro n an lentam ente © no reaccionan a tem peratura ambiente, pero con facilidad dan u n precipitado a temperaturas m is elevadas.
R C EíjC l R jC K C l R C H B rj

3, £1 últíux? está formado por compueito» inerte frente 3 lolucioocj cí- teeote» de nitrato de plats íücchólico.
ArX R CH =C H X HCCI, AiCOGHtCI RO CH O
Comer ya se hizo resaltar antes, la reactividad con el nitrato de plata alcohólico es totalmente independíente de la reactividad con el yoduro de sodio en acetona, por lo que ambos reactivos deberán probarse con cualquier compuesto halogenado (ver experimento 27),
E x p e r i m e n t o 2 4 . S o d i o
2R O H + 2Na -*■ 2R O N a+ H 20
A 1 mi de alcohol n-butílico añádale trozos delgados de sodio metálico hasta que ya no se disuelva más.- Enfríe la solución y observe. Añada un volumen igual de éter. ¿Qué es el precipitado? Aplique la prueba a acetona, éter n-buálico y tolueno,42
Comentarios. Este reactivo es de los más valiosos para investigar compuestos neutros que contengan átomos de hidrógeno que se puedan jubstituir con facilidad. Les grupos funcionales que contienen un átomo de hidrógeno unido a oxígeno, nitrógeno o azufre, pueden reaccionar con sodio desprendiendo hidrógeno,
2 R O H + 2Na - » 2R O N a+ H>
2R jN H +2N a - » 2R3N N a+H ¿
2 R S H + 2 N a-> 2R SN a+ H,
Esta prueba es de mucha utilidad en alcoholes de peso molecular intermedio, es decir, aquéllos que contienen de tres hasta unos ocho átomos de carbono. Los alcoholes más sencillos sólo con dificultad pueden obtenerse en condiciones anhidras. La presencia de huella* de humedad hace que la prueba sea positiva. Los alcoholes de peso molecular elevado reaccionan lentamente con el sodio, y a menudo el desprendimiento de gas es tan lento que hace que la prueba sea de poco valor. Cuando se corta sodio metálico en aire húmedo, adsorbe agua sobre su superficie, por lo que cuando se coloca en un disolvente perfectamente seco, como el benceno, desprende el hidrógeno producido por la interacción del metal con la humedad adsorbida.
Los átomos de hidrógeno unidos al carbono no son desplazados por los metales a menos que haya grupos funcionales adyacentes que activen los átomos de hidrógeno. Los compuestos con grupos merino
b E « * p ru c b » put<i«i a p ro a r te * t x m f n t t i t t %#M <u o I tip ú d o f a u if t& coto*, dliD Jvjíndt>l« en m dúotvmte C») ««na o bcrw*»o, jwhídn».
1 8 0 A p l ic a c ió n cíe io s p ru eba» d e c la s if ic a c ió n

activos, tales como el acetileno o acetilenos monosubstituidos, reaccionan con sodio.
HC ® G íi + 2Na -> NaC a GNa+ H j
2RC - CH + 2Na -► 2RC . C N a+ H,
Un grupo metí leño adyacente a un grupo activante, particularmente uno entre dos de dichos grupos, posee átomos de hidrógeno que pueden ser desplazados por ¡odio.
2ClliOOGH2GOOC2Hj-f-2N a -> 2[CHjGOGHCOOC:H j]'N a^-f Hj
Los grupos metilo reactivos se encuentran en ciertos compuestos, particularmente en metilcetonas tales como la acetona y la acetofenona. Estas reaccionan con sodio para dar el derivado sódico de la cetona y la- acetofenona. Estas reaccionan con sodio para dar rl derivado sódico de la cetona y una mezcla de productos formados por /aducción y condensación. Por ejemplo, la acetona produce acetona sódica, isopropóxido de sodio, pinacoJato de sodio, óxido de mesitilo y forona.
Asi, el sodio metálico es un reactivo útil para investigar tipos de compuestos con hidrógenos reactivos, que no son lo suficientemente activos para producir ion35 hidrógeno en un disolvente ionizante. Obviamente, es innecesario probar la acción del sodio sobre compuestos que se sabe son ácidos.
Prediga la acción del sodio lobrc cl fenol, ef ácido benzoico, 3aa ojomíu, el nitiom etano y la bencenjulfonamída. ¿Por qué « t a prueba aunca je usa con «nos compuestos? ¿ Q u í efecto tendría la preBcnda de hum edad en erta prueba? ¿C uál ej el principal defecto d*l jodio metálico como reactivo de clasificación?
E xp e r im e n to 2 5 . S o ív d ó n d e b is u lf ito d e so d io 181
E xp erim en to 25* S o luc ión de b isu lfito da jod io
RCHO + NaHSOa
OHR --C
H SO,Na
Prepare una solución alcohólica de bisulfito de sodio añadiendo 3 mi de etanol a 12 mi de una solución acuosa de la sal al 40%. £! alcohol precipitará una cantidad pequeña de la sal y deberá eliminarse por decantación o filtración antes de que el reactivo quede listo para usarse.
Ponga 1 nd del reactivo en un tubo de ensayo y añádale 0,3 mi de benzaldehído. Tape el tubo de ensayo y agite vigorosamente. Repita la prueba con 1) hcptaldehido, 2) acetofenona.
a« *
M
3
J!
I
« i

18 2 A p t ie a c fó u d o fas p ru e b a s d e d o s if ic a c ió n
C o n u n ta r io i. L a fo rm ac ió n d e c o m p a c to s d e a d ic ió n b isu lf itk a , lo* c u a le s se h a d e m o s tra d o q u e so n < *-h id ro x ia lq u ilsu !fo n ato s“ e s u n a re a c c ió n general* de a ld eh id o s. L a m a y o ría d e las m etilce to n as , c e to n as cicli* c a s d e b a jo p e so m o le c u la r (b a s ta la c ic lo o c ta n o n a ) y c ie rto s com puestos q u e tien en g ru p o s ca rb o n ita m u y re ac tiv o s , se c o m p o r ta n en fo rm a sim ila r . S in em b arg o , a lg u n as m e tilc e to n a s sólo fo rm a n le n ta m e n te los com p u esto s d e ad ic ió n o n o los fo rm a n . E je m p lo s d e e llo lo io n las a r il- m e tilc e to n a s , la p in aco lo tia y e l ó x id o d e m esitifo . P o r o t r a p a r te , e[ c in a m a ld c h íd o fo rm a un co m p u esto d e ad ic ió n q u e co n tien e dos m olécu la s d e b isu lfito .
E l b isu lfito e stá en e q u ilib r io c o n e l c o m p u e s to carb o n llico , y com o e l b isu lfito d e so d io se d esco m p o n e t a n to c o n á c id o s c o m o c o n á lcalis, ios co m p u esto s d e a d ic ió n sólo so n e s tab les e n so lu c io n es n e u tra s . L os co m p u e s to s d e r iv a d o s d e com p u esto s c a rb o n ílic o s d e b a jo peso m o lecu la r w n solubles e n ag u a . Su u t i l id a d se d e b e a q u e so n sólidos p u riñ ca b le s c o n fa c ilid a d y st q u e « d e sc o m p o n e n fá c i lm e n te c o n ácidos y á lca lis re g e n e ra n d o lo s com puestos o rig in a les .
L o s a n á lo g o s n itro g e n ad o s d e los a ld e h id o s , las a ld im in a s o bases d e S ch iff, ta m b ié n a d ic io n a n b isu lf ito d e so d io ; e l p r o d u c to es id én tico al
R C K = N — R ' + N a H S 0 2 -> R — C H — N H R '
/ SOaNaR C H O H + R 'N H 2
S O jN a
fo rm a d o p o r l a a cc ió n d e u n a a m in a p r im a r ia so b re e l co m p u esto b isu lf ito -a ld e h id o .* E l e n la c e c a A o n o -a ^ u f re d e e s to s com p u esto s es re a c tiv o , s ien d o desp lan ad o e l g ru p o su lfo n a to p o r re acc io n es c o n a n io n e s tales c o m o e l CNf“.
Sugiera u n a explicación de por qué la ciclohexanona reacciona • fácilmente con el bisulfito de «¿dio m ien tra que Ja dietilcetona no. ¿CuSl es la explicación d e la falta de reactividad de Ja pm acolona? Com pan: Este caso con el de la aceto/cnona, Explique el comportam iento del cinam aldchído, ¿Por que je usa u n a solución alcohólica de bisulfito de sodio? Ensaye esta prueba sobre acetona usando una solución acuosa.
E x p e r i m e n t o 2 6 . S o l u c i ó n d e h i d r ó x i d o d e s o d io
a ) A c id o s . E n e l c a p ítu lo 6 se d isc u tió la c o n v ers ió n d e Ja m a y o ría d e las su b stan c ias o rg á n ic as q u e so n a c id as en so lu c ió n acu o sa a
H Laaer 7 tdin£fcimoi^rcr, / . Am. C kta t. Ít»í„ S7, 2360 {1935); Shrincr y X rxá, / . Otr. chtm.. %. eaj <mi)-
M N eítitiflU n y H*rtaus, / . O t t , C k t a t ; 2*, 1ÍH3 {1959),

E xp e r im e n to 2 6 . S o lu c ió n d o h id ró x id o do s o d io 183
sales de sodio solubles en agua carao prueba para, ácidos. Esta reacción puede modificarse para determinaciones cuantitativas de peses molecu* lares por función ácida, cantidad conocida como equivalente de neutralización. el cual se discute en el capítulo 10 (pág. 259).
b) Satas de am onio y ¿aíes de amina*
RCO O N H ,+ NaOH -> R CO O N a+ N H ,+ H 20 RNH3C1+ NaOH -vR N H j-f- NaCl+ HjO
Ponga .5 mi de solución, de hidróxido de sodio al 10% en un tubo de ensayo, añada 0.2 g de benzoato de amonio y .agite la mezcla vigorosamente, Note el olor del amoníaco,
A 0,4 % de clorhidrato de anilina, contenidos en un tubo de ensayo, añádales 5 mi de solución de hidróxido de sodio al 10%. Agite la. mezcla y déjela reposar unos cuantos minutos. Observe la separación de la capa aceitosa de la amina.
c ) E s te r e *
RC—OR' + NaOH -»■ RC~O N a + R'OH
M étodo A . En un matraz de fondo redondo, provisto de un eficiente refrigerante a reflujo, coloque 40 mi de una solución de hidróxido de sodio al 25%. Añada 5 mi de benzoato de etilo y caliente hasta ebullición. Deberá ponerse dentro del matra2 un trozo de placa porosao un tubo de ebullición para evitar la ebullición descontrolada. Continúe el calentamiento hasta que desaparezca la capa del éster o el olor característico {cerca de Yt hora). Cambie la posición del refrigerante, destile unos 5 mi y sature el destilado con carbonato de potasio. Observe la formación de dos capas. Explique. Obviamente, la cantidad de muestra requerida dependerá del peso molecular del alcohol que se va a aislar así como del peso molecular del éster original.
Enfríe el residuo del matraz y acidúlelo con ácido fosfórico diluido. ¿Qué es el sólido que se separa? ¿Cómo puede usted estar seguro de que no es fosfato de sodio? Si el ácido fuera un líquido volátil ¿ je le podría separar por destilación? 115
Comentarios. Los ésieres varían considerablemente en la facilidad con la que pueden ser saponificados. La mayoLía de los esteres sencillos que hierven a menos de 110°C se saponificarán totalmente por reflujo con solución de hidróxido de sodio al 25% durante % hora, como se‘describió anteriormente en el Método A, Los ésteres que hierven entre 110
» Ver tí m¿to4o para <ktcrraui*r h í n&merOt de D*c1mu* dtKcjto ea 1* pig. 261-

J 8 4 A p l ic a c ió n d o f o t p ru e b a s t U d o s if ic a c ió n
y 200°C requieren un tiempo mayor ( de 1 a 2 horas} para al saponificación totaL
La hidrólisis do ésteres insolubles en agua puede acelerarse marcadamente por adición de 0.1 % de sulfato de laurilo sódico (Gardinol) al álcali y al éster. La mezcla se agita vigorosamente para emulsificar el éster y entonces se calienta a reflujo. Debe usarse un matraz grande, ya que el agente emunificante produce una gran cantidad de espuma.
Los ésteres de punto de ebullición muy elevado (arriba de 200°C) que son insolubles en agua, se hídrolizan lentamente y el reflujo prolongado puede ocasionar la pérdida de un alcohol volátil. El procedimiento siguiente u utiliza una solución de hidróxido de potasio en dietil- englicol (p. eb. 244°C), El díetiienglicol no tan sólo es un disolvente excelente para esteres ¿no que también permite una temperatura de reacción más elevada y todos los alcoholes, con excepción de los de punto de ebullición muy elevado, pueden obtenerse en íorzna pura por destilación de la mezcla.
M étodo B . En un matraz de destilación de 10 ó 25 mi ponga 3 mi de dietíienglicol, 0.5 g de pastillas de hidróxido de potasio y 0,5 mi d e agua. Caliente la m acla con Llama pequeña hasta que se haya di- suelto el álcali y después enfrfe; añada 1 ó 2 g del éster y mezcle perfectamente. Por. medio de un corcho ponga un termómetro y coloque como receptor un tubo de ensayo enfriado en un vaso de precipitados lleno de hielo. E l matraz se calienta ínicialraente con llama pequeña y su contenido se agita para mezclado. Cuando sólo haya una fase líquidao una fase líquida y una sólida, la mezcla se calienta 'vigorosamente para que destile el alcohol. El destilado se usa para la preparación de un derivado sólido como el 3,5-dinitrobenzoato,
El residuo del matraz es la solución o la suspensión de la sal potásica d d ácido derivado de! éster. Añada al residuo 10 tnl de agua y 10 mi de etanol y agite perfectamente, Añada áddo sulfúrico diluido (6A/) hasta que la solución esté ligeramente ácida a la íenolftaleína. Deje reposar la mezcla durante unos 5 minutos y entonces fíltrela. El filtrado se usa directamente para la preparación de un derivado. Puede tratarse con bromuro de ¿-nitrobencilo o bromuro de ^-fenilfenad- lo para obtener el correspondiente éster sólido. Sí el cster original tuviera un punto de ebullición tan elevado que no se pudiera determinar un buen punto de ebullición, puede resultar conveniente el que se divida el filtrado en dos porciones y se preparen dos derivados.
Comentarios. La saponificado., representa el procedimiento más útil para caracterizar los ésteres. Sin embargo, deberá recordarse que el álcali concentrado y caliente también afecta otros grupos funcionales. Los ftldefiídos que tienen hidrógenos-a sufren condensación aldol y re$ini-
* Redraiwn y Luco, W . £ « /. C ktm ., Anal* E l , 9, 321 {[937).

fícacion, Los aldehidos que no tienen hidrógenos-a sufren una reacción de Cannizzaro y forman el alcohol y la sal de sodio deJ ¿oído,
2R,CCHO + NaOH -> RjCCH jO H + R5CCOONa
Los compuestos pol ¡funcionales, tales como las [3~<Ucetonas y los £-ccto- ásteres, sufren una escisión bajo la acción de los álcalis calientes. La posibilidad de que ocurran tales reacciones ¡nterferentes puede investigarse por medio de los otros reactivos de clasificación mencionados en este capítulo, y esto enfatiza el hecho de que un solo reactivo de clasxji- cación no puede tomarse corno prueba de ¡a presencia de uti determinado grupo funcional. Es importante correlacionar todas las pruebas al intenta r deducir conclusiones concernientes a la estructura de un compuesto desconocido.
Los esteres de los ácidos con impedimento estérico, tales como los Z^fi-trialquilbenzoatos de alquilo, son muy difíciles de hidrolizar con álcalis. Sin embargo, estos ésteres pueden hidrolizarae ráp. 'ámente disolviéndolos en áddo sulfúrico al 100% y diluyendo la solución con agua helada." Este resultado se debe a que los ésteres de ácidos con impedimento es ¡¿rico experimentan la siguiente reacción cuando «e les disuelve en ácido sulfúrico al 100#,*
CÜOR CO
« > X T > + W ■-* H3C0 ° Hí + O W + 2Hsor o í , CH,
Cuando se añade agua, el ion carbonio forma el ácido,
CO COOH
!’cQ ch*+(wCH j
Inversamente, el ácido con impedimento esterico puede convertirse fácilmente en un éáter disolviéndolo en ácido sulfúrico al 100% y tratando la solución con el alcohol.
Los esteres sin impedimento no experimentan estas reacciones. Se disuelven en ácido sulfúrico al 100% y se recuperan sin alteración cuando se vierte la solución en agua helada.
Los esteres de ácidos y alcoholes con impedimento estérico, tales como el 2,4,6-trimetilbenzoato de ter-butilo, aunque son extremada
* NcKin&n, / . A m . Chtm . Soc., S3, 2431 (1M1),, “ * 5 “ «5ít t » W yn '«*f OrfflPifr M c C m v f H il ) B o o i C e . , Y « * , 1 * 0 ,
p4« l. + W 9,
E xpe rim en to 2 6 . S o lu c ió n d e h id r ó x id o do s o d io 165

1 8 6 A p l ic a c ió n tf* Jas p ro » b a * d e d o s if ic a c ió n ¡
mente resistentes a la hidrólisis con álcalis, pueden hidrolizane fácilmente hirviéndolos durante 1 hora con ácido clorhídrico al I85&.*5
p o r medio de ecuaciones ]oí producto» fo rm ad?: por la hidrólisis alca- Una de: í ) acetato de ¿.fcnilfcnacito: 2) dibenzoato de etilenghcol; 3) oxalato de n-butilo; 4 ) el poliéster de giicerina y ácido (tilico ; sugiera procedimientos apropiados pa ra investigar los productos formados en estas reaecic-iies.
d ) A m idas, amidas substituida* y n itrtto s.
RCONHj + N aO H -> RGOONa + NH3
RCONHR' 4 . N aO H —> RCO O N a + R'NH2
RCONR'j 4- N aO H -*• R CO O N a 4- R 'jNK RCN + NaOH 4 H jO - * RCOONa + NH,
O í N / ^ N H j 4 NaOH — ^O N a 4 NHá
N O , “ N O ,Trate 5 mi de solución de hidróxido de sodio al 10%, contenida
en un tubo de ensayo, con 0.2 g de urca. Agíte la mezcla y observe si se desprende amoníaco- Repita la prueba con: I) benzamida; 2) aceta- nilída; 3) benzonitrilo; 4) 2,4-dinitroanilina. Después, caliente hasta ebullición cada una de las mezclas y observe el olor. Pruebe la acción de los vapores sobre el papel tornasol rojo, humedecido.
Enfríe las soluciones anteriores y acidule cada una de ellas con ácido clorhídrico. Observe el resultado. ¿Q ué origina el que se formeo no un precipitado?
Comentarios. E] amoníaco o la amina que se produzcan en esta hidrólisis alcalina pueden caracterizarse por la prueba de Hinsberg o usando los reactivos de los experimentos 17 y 18. Para este propósito lo mejor es usar una muestra grande (1 g) y destilar el amoníaco o la amina volátil, de la solución alcalina, en un receptor que contenga ácido clorhídrico diluido. Entonces se puede neutralizar esta solución y hacer el ensayo como se indica en el experimento 4*
U na amina insoluble en agua puede separarse por extracción con óter. Entonces se puede destilar el óter y la. am ina se caracteriza por las pruebas usuales. U na amina no volátil, soluble en agua, también puede convertirse en un derivado de arilsulfonilo y separarse del ácido orgánico (que es el otro producto de la hidrólisis) aprovechando el comportamiento de solubilidad o las diferencias en volatilidad.
Muchas amidas substituidas se hidrolízan con mayor facilidad calentándolas b. reflujo con ácido sulfúrico al 20%.
2R C O N H R '4 H £ 0 * 4-2H2O 2 R C O O H 4 (R 'NHjijSO, 2RCO N R':4 'H¿SO*+ 2H¿0 -> 2RCOOH + (R 'jN H ^S O ,
** Ceben y ScHncider» / • Chem. SciC., 6 3 , 3352 (1943).

E xp e r im e n to 2 6 . S o lu c tó n -t ta h id ró x id o d e «o d io 1 8 7
Si el ád d o es volátil^ puede separara por destilación o por fdtra- d ó n « es Insoluble en agua» L a amina puede liberarse por adidón de álcali y caracterizarse por conversión a su derivado de arüsulfoniJo.
Los nitriIos3 particularmente las cianhidrinas, frecuentemente se hidrolizan con ácidos. El tratamiento con ácido sulfúrico de 90 a 95%o ácido clorhídrico concentrado, a temperaturas que van de 10 a 50^0, convierte los nitritos en amidas. Las amidas pueden hidrolizarse aún más diluyendo la mezcla con agua y calentando a reflujo, de K hora a 2 horas.
RCN -7§Sr> RCONH, r c o o h + n h j í s o .HiO
Las arilaminas con grupos nitro orto o para con respecto al grupo amino, se hídrolizaü hasta los correspondientes nitrofenoles y amoníacoo aminas bajo la acdóa de álcalis calientes. El grupo nitroso se asemeja al grupo nitro en su efecto labilizantc sobre k* grupos orto o para respecto a él. Por ejemplo, las p-nitrosodialquilanilinas se hidrolizan con álcalis hasta las aminas secundarias y la sal de sodio del /-nitrosofenol (monoxima de la benzoqumona).
N R,
N bOKR jN H +
NO
e) C om puesto* nitrados
L N = o
A 5 mi de solución de hidróxido de sodio t>\ 20% añada 3 mi de etanol y una gota de nitrobenceno y agíte vigorosamente. Compare el color de la solución con el producido por una gota de nitrobenceno y 5 mi de agua más 3 m i de etanol. Repita la prueba con jj-nitrofenol y f-nitroanilina.
La cau¿W exacta del cambio de color de un compuesto nitrado no ha sido establecida definitivamente, pero la tautomerízadón antertc: a un anillo quinoide ha sido propuesta como una explicación.

Disuelva 0.1 § de j/i-dínitrobencenv en 10 rrx! de acetona y añada de 2 a 3 m i de solución de hidróxido de sodio al 10%, con agitación. Observe el color producido. Ensaye la prueba c o d nitrobenceno. -
Comentarios. Loa compuestos mononitrados no dan color (o un amarillo muy pálido) con estos reactivos. Si están presentes dos grupos nitro se desarrolla un color púrpura azuloso; la presencia de tres grupos nitro produce un color rojo sangre. La presencia de un grupo amino, tunino substituido u oxhidrilo en la molécula, inhibe la formación de los colores característicos rojo y púrpura.”
E xperim en to 2 7 , Y o d u ro de sodio en acetona
RCI + N a l—> R I-fN aC l
R B r-f N s l R I -f-NaBr
A 1 mi de 3a solución de yoduro de sodio en acetona, contenido en un tubo de ensayo, añádale 2 gotas del compuesto cuyo análisis elemental ha mostrado la presencia de cloro o bromo. Si el compuesto es un sólido, disuelva cerca de 0,1 g en el mínímo volumen posible de acetona y añada la solución al reactivo. Agite cl tubo de ensayo y deje reposar la solución a temperatura ambiente durante 3 minutos. Observe si se ha formado un precipitado y también si la solución se pone café rojiza (debido a la liberación de yodo libre). Si no ocurre ningún cambio a temperatura ambiente, coloque el tubo de ensayo dentro de un vaso de precipitados, con agua, a 50°C. Al término de 6 minutos déjelo enfriar a temperatura ambiente y observe sí ha habido reacción. Ensaye esta prueba sobre: l ) bromuro de n-butilo; 2 ) bromuro de «c-butilo; 3) cloruro de ter-buti!o; 4) bromuro de etileno; 5) cloruro de ben- d io ; 6 ) cloruro de beraoílo; 7) cloruro de bencensulfonilo; 8) K-cloro- acetofenona.
R eactivo , Disuelva quince gramos de yoduro de sodio en 100 mi de acetona pura. La solución, inicialmente incolora, adquiere un color amarillo limón. Deberá conservarse en frasco obscuro y desecharse tan pronto como se desarrolle un color café-rojizo definido.
Comentarios. Esta prueba se basa en que el doruro y el bromuro de sodio sólo son muy ligeramente solubles en acetona. El efecto de Ja estructura sobre la reactividad, en esta reacción, ha sido ya discutido en Ja sección sobre el reactivo de nitrato de plata etanólíco (experimento 2S)* Se aconseja ensayar la- reactividad de cualquier haluro desconoddo frente a cada uno de estos reactivos, ya que las reactividades en ambas pruebas no son paralelas.
" Bo« 7 SkhoboB, Eat . C ktm ., Anal. E¿.y 7> 190 (1 9 » ).
1 6 8 A p lic a c ió n d a Ja* p ru e b a* d t c la s if ic a c ió n

Experimento 27, Yoduro d» «odio «n aceiMia 1&9
El ion yoduro puede participar en otro tipo de reacción, la cual se puede reconocer porque origina no tan sólo cloruro bromuro de sodio sino también yodo. Ejemplo de ello es la ya mencionada formación de yodo a partir de cloruro de trifenilmetilo y yoduro de sodio.
Los compuestos 1,2-dícloro y 1,2-dibromo no tao sólo dan un precipitado de cloruro o bromuro de sodio, sino que también desprenden yodo libre.
RCH - CHR+ 2N&I - * RCH - CH R+ 2NaBr
L l i í\\
R C H = C H R + I2
La comparación de los haluros de etilcno dio los resultados siguientes:
Ppt, a 25°C BrCHiCHjBr 1.5 roin.ErCK ^H rC! 3 min.ClCHjCI^Cl Ninguno (Ppt. a 50°C en 2.5 min.)
Aunque so ha reportado que el cloruro de píenlo produce yoduro de picrílo cuax.do se trata con yoduro de potasio en etanol, d tratamiento con yoduro de potasio en medio ácido, por ejemplo: en ácido acético b aviente o aun en acetona que contenga algo de ácido acético, a temperatura ambiente, produce trinitrobenceno y yodo.ÍJ
Los compuestos polibromados, tales como el bvornoformo y el >tctra- bromoetano, reaccionan con el yoduro de sodio a 5ücC dando un precipU tado y libcrandp yodo. El tetrabromuro de carbono reacciona a 25°C.
Los cloruros 'de sulfonílo forman inmediatamente un precipitado y también liberan yodo libre. Probablemente el yodo se forma por la acción del yoduro de sodio sobre el yoduro de sulfonilo.
ArSO¿Cl+NaI ArSO.I + NaCljlVal
ArSOjNa4-Ií
El cloruro de bencemulfonilo forma bencensulfinato de sodio con un rendimiento de 6 0 $ ;;92 Los otros productos que se obtienen son la difenihuifona {21%) y el tiosulfinato de fenilo ( 10%}»
Los sulfonatos de alquilo también reaccionan produciendo los correspondientes sulfonatos de sodio como precipitados.
ArSOiOR+ N al ArSOjONa 4- RI
♦i Blitt y Trútroo, J . Am- CA*p». Soc.> 74, 6272 (1952}.** y B o m , Arck. P h a m - , 3S 7 , M i

!
89
9
fe»L»
í t
N
La reaedón debe tenerse presente para el caso de que uno de los grupos de un íster sulfónico contenga un halógeno.
T90 Aplicocfón d e ío» pruebo? d e «lenificación
Experimento 28. Acido sulfúrico ( fu m ím te )
r . r > +s o j a * . n s0aHPrecaución, Use este reactivo sólo con compuestos inertes.Coloque 2 mi de ácido sulfúrico fumante al 20% en un tubo de
ensayo seco y limpio y añada 1 m) de dclohexano. Agite la mezcla vigorosamente y déjela reposar durante algunos minutos. Observe si se h;i efectuado la solución. Repíta e] experimento usando: 1) benceno; 2) bromobenceno; 3) bromuro de etiieno.
El áddo sulfúrico concentrado es un disolvente extraordinario en dos aspectos* Su comíante dieléctrica es mucho mayor que Ja de cualquier otro disolvente en el que se ha medido esta propiedad.** Así, las fuerzas de atraedón entre cualquiera de los iones disueltos son tan pequeñas en soludón diluida, que los coefidentes de actividad pueden tomarse como la unidad. La segunda propiedad extraordinaria es que, además de ia autoprotólisis, análoga a la encontrada en otros disolventes oxhidrilicos tales como el agus, hay una autodisociadóa que se traduce inidalmente
O
2HOSOiOH — H O - 5 -O H * + H O —S —O-
¿H ¿en la formación de anhídrido sulfúrico y agua. Sin embargo, a las concentraciones consideradas, cada uno de éstos reacciona esencialmente en forma total con d áddo sulfúrico, por lo que el equilibrio completo es el siguiente:
O O
2HOSOjOH ^ H jO : + - 0 - 1 - O - S - OH
O O(A xú¿n tW le n ta d é r i l í t r í c o )
(H S iO /-)
Indudablemente que estarán presentes muchas otras especies, tales como los ácidos polisulfúrícos o sus aniones, pero en concentraciones demasiado pequeñas para que sea posible demostrarlo experimentalmente,
13 Gillítpií, Hugbca j Xnffoldj } . Chtut. S*c., 1950, 2473.

E xp e r im e n to 2 8 . A c id o su lfú r ic o 191
El ácido sulfúrico concentrado reacciona con el etíleno formando sulfato ácida de etilo, pepo cl áddo sulfúrico que contiene trióxido de azufre produce, en lugar del compuesto anterior, áddo edénico. La razón para el comportamiento diferente en las dos condídones se conv
especies sulfonantes, tales como SOj ó HSOj+, en concentración pequeña y, por lo tanto, el reactivo sulfonante fracasa en su. competenda con la adición de protones, primer paso en la formación del éster sulfato.
Sin embargo, en ácido sulfúrico fumante, la adición del KSO j*j SOj o algún otro agente sulfonante se vuelve importante, por lo que la reaedón prindpal es la siguiente:
En cada caso el segundo paso es el mismo; la reaedón del ion carbonio formado inidalmentc con cl ion bisulfato.
El prim er paso en la forroadón del áddo etíónico « , cuando - menos teóricamente, semejante al primer paso en la sulfonadón aromática, La naturaleza exacta del ageüte sulfonante en el áddo sulfúrico fumante no ba sido determinada, aunque se ha encontrado evidencia de que es el HSOj* o un diroero* del S O j(S A )>
La acción del ácido sulfúrico fumante sobre los compuestos dihalo- genados en 1,2 es compleja. La mezcla se pone obscura y se libera algo del halógeno. Es probable que la pérdida del haluro de hidrógeno sea seguida por la polimerización del haluro de vínilo. Por ejemplo: el bromuro de etileno probablemente origina los cambios siguientes:
Note que esta prueba sólo es 6tll p a ra loa compuestos inwluble* en Scldo • tulíúrico. ¿Por qué? Escriba las ecuaciones de las reacciones iovolucractaj. Com-
* Bwiod T Horrura*, / . Ck<m. S e t., ISMt 3522. SÍ6 «piarlo, tc t IngoW. StnirttU4 tr*¿ Mttkmvm ÍK 0>f«K|\r Chta»Tíry, CocdíU V n im p iy P»"e«, ltli»ca« K. y n 1953» r*S*- 299 * llguicnUM.
prende cuando se considera que el áddo sulfúrico al 100% contiene
CHj=s CH*+ KjSO, _» C H jC H /+ HSO*~
CH2rrC H 2-f H SC y -*• "C H !- CHjSOíH
CHjCHj*+HSQf —> GH3CH3—O—SOjH •CHiCKj—SOjH-t-HSOr -» HOjSOCHjCH, - SO.<H
CH2— C Hj = C HB r + HB r
nCH2= C H B r ^ - ( C H j - C H - ) ,
Br
2HBr -f HjSO^ —V Brj+ S O j+ 2HjO

192 A plkecfán «Je / « pruebo* <fe dasifkectón
p u « loa productou de o t a rtaccúSn con aquéllos fo n a a d o i e n j u p ru eb a de solub ilidad con t i Acido tu tfürico concen trad o y ur>x olefina com o el L«em >-1,
Experim enta 29. Reactive de Tóilent
RCHO -f 2Ag (NHj) ¿OH -► 2Ag-r RGOONH4+ H2O + 3NH*
Prepare el reactivo de Tollens de acuerdo con las indicaciones que se dan a continuación y ensaye su reacción con los siguientes compuestos; 1} formaldehido; 2) acetona; 3) benzaldehído; 4) glucosa; 5) hidroquinona; 6} £-aroinofcnol.
Reactivo. En un tubo completamente limpio coloque 2 mi de una solución de nitrato de plata al 5% y añada una gota de soludón diluida de hidróxido de sodio (aproximadamente al 10%). Añada, gota a gota, una solución muy diluida de hidróxido de amonio (aproximadamente al 2% ), agitando constantemente hasta que justamente se disuelva el óxido de plata precipitado. Para obtener un reactivo sensible es necesario evitar un exceso grande de hidróxido de amonio.
Este reactivo se deberá preparar justo antes de usarse y no se deberá conservar, ya que la solución se descompone rápidamente y deposita un precipitado poderosamente explosivo. Si no ocurriera ninguna reacción en frío, la solución deberá calentarse un poco.
¿ I * presencia de un átomo de halógeno remetívo interferiría con esta prueba?
Comentarios. Deberá tomarse en cuenta que las adloínas, la dífenil- amina y otras aminas aromáticas, lo mismo que el ct-naftol y determinados fenoles, dan positiva la prueba de Tollens. También se ha encontrado que la» a-alcoxi y a-dialquilaminocetonas reducen el nitrato de plata amoniacal.9*
E xperim ento 30» Cinc y c lo ru ro d e am onio
O r - ^ o r " * *Disuelva 0,5 mi de nitrobcnceno en 10 m í de etanol al 50% y
añada 0.5 g de cloruro de amonio y 0.5. ^ de cinc en polvo. Agite y caliente hasta ebullición. Deje reposar 5 minutos, filtre y pruebe la acción del filtrado sobre el reactivo de Tollens.
Esta prueba se basa en la reducción del compuesto desconocido hasta, una hidrazina, una hidroxilamma o un aminofenol; todos estos
" L w pnrd y C d f« jd t / . Am. C k ,m . So t., 77r S272 ( » » > .

Expe rim en to 30» C in c y c io rv ro á* am o n io 193
compuestos se oxidan con el «activo de Toilens, el cual se reduce a plata metálica.
CíHsN H 0H + 2Ag(NHi ) í0 H ^ C ^ M 5N O + 2 H 1O -t-2 ^ + 4 N H >
Esta prueba no puede emplearse si el compuesto original reduce eí reactivo de Toilens.
L a reducción de los compuestos nitrados basta compuestos azoicos con hidruro de Jítio y aluminio ha sido también propuesta como una prueba cualitativa de la función nitro.96
1, Escriba la* ecuaciones para ¡4i reacciones que ocurren en las p rn b M siguientes. Suponga que te realizan bajo la¿ condicione» cKperirneixiales especificadas para loj reactivos de clarificación, Cuando sea conveniente, las reacciones pueden escribirse como ji ocurrieran por «capas.
1. Anisol + ácido yodbSdríco.2. Acido crotónico + solución de permanganato de potasio.$. Acewfenona + clorhidrato de hidroxilamina.4. Dibenzoilmctano + hidróxido de sodio diluido y caliente.5. JV-etilftalimid» + solución caliente de hidróxido de sodio.6. Benzoato de e tilo .+ hidroxilam ina.y a continuación cloruro Mrrico.7. Etanolamína + n itrito de Bcdio -h ácido clorhídrico. f¡. ú-diclorobenccnp -+■ ácido sulfúrico fumante-9. D D T + solución caliente da hidróxido de sodio.
10. 2,3-dibromopcntano -f solución de yoduro de sodio.11. Octar.ol-2 + solución de hipoyodito de sodio.12. Yoduro da hencüo *f- nitrato de p lata alcohólico.1$, a-naftol + agua de broma.14. A lanina + cloruro de .bencewulfoailo 4- hidróxido de sodio.15. 0-díetilamínoctanol *f cloruro de acetilo.
I I , Coloque los compuestos siguiente* en el orden aproximado de 1% reactividad decreciente del átomo do halógeno hacia el nitrato de plata alcohólico.
3. CgH.COCHjCI, CaW£COCI, CjHsCOCHjCHtCHjQ, C$HANHaCl.4. B rC H /ÍC H a, CHjCH aC H X I, C H ,C H jC H íBr.
Ejercicios
Cl
j . ( C H j^ a . O2, (CH^CHCl. CH3CHtCHCIC«H4, O O Cl.
* Gilmin y Gojxnv, / . Am. C Ktn. See.t 73, 2939 (1951).

¿ i
Í )
V
í )
V
rr9
L
I I I . ¿Cuál *ería. «1 orden aprwdraado de reactividad J e los átomos de halógena de loe com puesta s ig u ie n te si el reactiva fuer a «na solución de yoduro de sodio en acetona?
1. CÍLCHiCHiBr, CH iCH íCHíCI, (CHi)iCHCl, <CH-.),CC?.2. C .HLCOCH^l, CHiCHsGHjCl. (CHi)*CCfLCl.
IV . Seleccione una prueba de clasificación que pudiera us-a/se pa ra diferenciar las ¿guíente» pares de compuestos.
1. <*)CIC,H.COCH, j CLHiCOCHjCI.2. (C*H»)jC O H y íi-C A O H .3. C jH jCHO y C U G O C H ,.4. H O (C H i ) 4O H y C H ,C H iC H —CH/.
] (O H O H
5. Sacarosa y lactosa.6. CHjO CH zCOjH y H O C H íG O jCHj .7. Ciclohexana y aniso!.8. (CHOiC{CO?CIíi)j y C H j(C O A H ;h . *9. C » tt^H :N H s y <*) CHjCJLNH».
V . Suponga que se perdieron las éciquoias de j í í í irascos Que se sabía contenían los leía compuestos íiguientes:
H O C H jCH zGH iO H [ <C>Ht) jNH7] 2S a
C H (O H )C O jH . C H (N H j)C O ¿H
CH jGO iFI GH iCO jH r 1
O H -C O iN . H O C H iC H (C H O H ) jC H O H '
1--------O -------- 1
Seleccione u n a secuencia de prueba* (no use puntos de fusión o puntos de ebullición) que pudieran usarse para volver a e tiquetar los irasco; rápida y correctamente. ¿C uál es el mínimo núm ero de pruebas que serían suficientes?
VJt. Un compuesto puede K r uno de los siguientes:
1. HeptanoI-2, ó. E ter etilfenilico.2. M etilbendlcetona. 7. Cinaniato de etilo.3. n-vakriauato de etilo. 8. Benzílo.4 . Anhídrido n-butirico. 9. n-hexaldehído.5. 1,3,-dietwipropano. 10. Bemalacetona,
A- Se ensayó la acción de los reactivo» '¡guíenles sobre cada uno de los compuestos anteriore*. Ind ique quó reactivos d an pruebas positivas y cuáles las dan negativas. Escriba las ecuaciones pa ra la* ppjebas positivas.
a) Solución diluida y caliente de hidróxido de sodio-b) Clorhidrato de hidroxilamina.c ) Hipoyodito de sodio.d ) Pcnnanganata de potasio diluido.
t 9 4 A p lk c d ¿ n de la s p ru e b a s do d e i f i c a c ió n

E xp e iim a n lo 30 , C in c y c lo ru ro d e am o n io 195
B. Suponga que cada uno de los reactivos anterior»; diera y r» prueba negativa. ¿O ué posibilidades ouedarían por considerarse y como se podría difcrenciari» en trt st?
V IL Las observaciones siguientes ce efectúan frecuentemente «n cl laboratorio.£ n cada caso exprese las deducciones que pueden hacerse «obre la n a tu raleza del compuesto en estudio.
1. U na solución acuosa de un compuesto insoluble en í te r d a reacción ácida al tornasol.
2. U n compuesto soluble «n solución de bicarbonato de sodio, pero no en agua, decolora en frío una solución diluida de perm anganato de potasio.
3. U n compuesto neutro que contiene nitrógeno se disuelve totalmente en una solución, diluida y caliente, de bidróxido de sodio.
4. U n compuesto neuuro forma un precipitado Citando se agita con lución saturada de bisulfito de Sodio.
5. Un compuesto neutro es insoluble en ácido sulfúrico fumant*.6- U n compuesto soluble en agua reacciona con cloruro de acetilo y
da un precipitado amarillo cuando *e trata con solución de hipoyo* dito da sodio.
7. La acción del ácido nitroso ¿obre un compuesto básico lo convierte en un sólido verde. Sin embargo, cl compuesto original no reacciona con el cloruro de acetitc.
3. U n hidrocarburo ío m a un derivado con sodio.9. U n compuesto que sólo contiene carbono, hidrógeno y oxígeno.fonna
dos productos cuando je tra ta con álcali caliente.30. Por calentamiento un compuesto ácido se transforma en un com
puesto neutro.11. U n compuesto débilmente ácido se convierte por reducción en un
compuesto básico y por hidrólisis en un compuesto neutro.12. U n compuesto débilmente básico d a un compuesto débilmente áddo
cuando se tra ta con cloruro de /McJuensulfonilo y ttn compuesto soluble en bicarbonato acuoso cuando se tra ta con ácido nitroso.
13. Un compuesto soluble en bicarbonato de sodio y solución de ¿cido clorhídrico d a una jotución transparente con cloruro de benceniul* fonílo y álcaii. L a adición de un ácido d a un precipitado. T ratando el compuesto original con ácido clorhídrico y nitrito de «odio, no se desprende nitrógeno aun después de calentar Ja mezcla de reacción.
14. U n compuesto soluble c n 'ag u a dejó un residuo en la prueba de ignición. La solución, ácuo&a de este residuo resultó alcalina al tornasol.
15. U n compuesto soluble en agua fue ácido al tornasol: húm edo. El compuesto redujo el perm anganato, pero.nO decoloró el bromo en tetracloruro de carbono y sí decoloró el agua de bromo.
16. Un compuesto básico reaccionó cor. cloruro de bencensulfonilo y álcali formando una solución transparente. L a neutralización cuida- dou. de esta, solución produjo un precipitado que no era d compuesto origina?, peto que se disolvía en un exceso d e ácido clorhídrico diluido.


CAPITULO
9Uso de los métodosespectroscópicos
ara la determinacióne grupos funcionales
L k i d e n t i f i c a c i ó n d e l o s c o m p u e s t o s orgánicos ha experimentado unü revolución con la introducción de instrumentos que permiten determinar rápida y convenientemente, aun con cantidades pequeñas de material, los espectros en el infrarrojo, de resonancia magnética nuclear y en el ultravioleta de las substancias. La espectroscopia de masas se ha vuelto cada vez más importante debido a la perfección de los espectrómetros, que los hace dignos de confianza, y también a ios adelantos en la interpretación de los espectros. Sin embargo, este capítulo se limitará a una discusión breve sobre el uso de la espectroscopia en el infrarrojo, de resonancia magnética nuclear y en «1 ultravioleta para la determinación de grupos funcionales.
Es evidente que una discusión detallada de la espectroscopia infrarroja está más allá del propósito de este libro y para ello el lector podrá consultar las siguientes referencias generales:
A ) Bcllam^, T k / In fra -itd Sp<clra c f :CompUx M olecuU t, 2* ed-, Mcthueti& Co., Londres; John Wüey and Sons, N ueva York, 1958. U n a introducción m uy buena al tema de la espectroscopia en el infrarrojo coma es UMida por el químico orgánico; « t e libro es m is prolijo que las otras referencia» que te enlistan a continuación.
B) Jones y Sandorfy, capítulo IV ch Chemical Applicatioru c f Sp¿ctroscopy, ed itado por W. W tjt, In te n d e n te Publishers, Nueva York, 1956. ¿ t e ' capitulo contiene una valiosa discusión de la espectroscopia en el infrarrojo con éníasií lobre técnicas,
C ) N&Larúshí, Infrared Abforption Spactroscopy-Practícd, Holden-Day, San Francisco, Calif., 1962. Este libro es de valor especial debido al $ran
: núm ero de problema!» incluyendo espectros, y, en muchos ca*o«, pregunta! «pe-
ESPECTROSCOPIA INFRARROJA
197

199 Uso <fe Fot métodos ©xpectroscópieoi
cíiicaa acerca de vario* aspectos de ellos. Los espectros se discuten posteriormente en uní, lección abarte en donde se din las respuestas.
C) Herthcnson, Infrared Abtorption SpecJta, Index for 1945*195?, Academia Press, fíueva York, 1959. Este trabaja proporciona el medio más Conveniente para localizar en la bibliografía Ios espectros en el infrarrojo publicados en los años cubiertos en ¿1.
£J Sítvenirin y Bassler, SptdtomtUic Identification of Orgcnic Compourids,Jobri Wiley and Sons, Nueva York* 1963. Este libro de referencia proporciona una buena introducción al uso de las espectroscopias en el infrarrojo, de resonancia m ajn ítíca nuclear, espectroscopia de masas y en el ultravioleta pa ta el trabajo de identificación.*
Las moléculas orgánicas no son conjuntos rígidos do átomos, como lo implican los modelos de “bola y palo1’, sino que pueden representarse mejor como unidos entre sí por resortes y experimentando continuos movimientos longitudinales, de flexión y de rotación. Cuando la frecuencia de la radiación infrarroja que entra á una solución o un cristal de un compuesto orgánico corresponde a la frecuencia de un movimiento molecular, se absorbe una radiación. Trazando una curva dd porcentaje de transmisión contra la frecuencia es posible obtener una gráfica de las cantidades relativas de los movimientos moleculares longitudinales y de flexión de los diferentes átomos de la molécula. Las frecuencias de rotación están fuera del intervalo examinado ordinariamente.Debe mencionarse que, para originar una absorción en el infrarrojo, un movimiento debe involucrar un cambio en el momento dipolar de la molécula. Se han realizado detallados análisis teóricos de los espectros de varias motéculas sencillas.1 Sin embargo, el uso de la espectroscopia en el infrarrojo en eí estudio de moléculas- complejas, por los químicos orgánicos, es en gran parte empírico.
Para las mediciones en el infrarrojo ordinariamente sólo se usan como disolventes compuestos no oxhidrílados^ El agua y los alcoholes no tan sólo absorben fuertemente en determinadas regiones en donde el disolvente debe ser transparente, sino que también afectan el espectro causando desplazamientos imposibles de predecir, los que se atribuyen a enlaces de hidrógeno y otras interacciones con el soluto. Debe enfatizarse que las celdas en las que se miden los espectros en el infrarrojo con mucha frecuencia son de cloruro de sodio y, por lo tanto, pueden deteriorarse con una muestra húmeda. El tetracloruro de carbono y el ¿ t v 4 bisulfuro de carbono están entre los disolventes generalmente más usados. ^ Tienen poca tendencia a interactuar con el soluto mediante enlaces do hidrógeno u otra acción de solvatación y, aunque cada uno de ellas absor
* (N. del *r.}^-Mortítlo J MudroRete “A piicaáomtí de Í4 ttfettrcicopia inS'á-r/a/a" Facultad d i Ciencia, Univtttidad de iMadcid, Madrid, 1?02. Libro muy útil p>Jr los '^ m e r o s o t p ro b íem o j c an tó m e y * u i tfttn U sd ai e x p K c a á o n a .
1 K «ttb*nj, and Ram a* $ j» t fhf> D- V an Noitmn<£ Cq., PrirtCíton, N . J ., IWÜr

E spe c tro scop io in fra r ro ja 1 9 9
be fuertemente en determinadas repones del espectro, en conjunto constituyen un disolvente transparente para tedo cü intervalo utilizado. La celda de 0.1 rom, que es la más usada, requiere cerca de 0.1 mi de uno solución de 5 al 10% o cerca de 5 a 15 mg de muestra. Con una “micro* celda” es posible usar muestras tan pequeñas corno 0,02 mg. Otros disolventes que han sido empleados ordinariamente son el cloroformo, el brornof^uooj y otros policlorometanos o etanos. El espectro de laa muestras líquidas puede determinarse sin disolvente y los sólidos insolubles pueden suspenderse en “NujoT* o “Perfluorolube” . £1 sólido también puede pulverizarse con brbnuñodc potasio (transparente en el infrarrojo) y la m eada resultante se comprime a presión elevada para formar una pastilla transpaiente, la cual se usa en lugar de la celda.
Desafortunadamente, diferentes unidades han sido adoptadas por diversos investigadores por lo que es necesario familiarizarse con cada convención^La cantidad de Iu2 absorbida algunas veces se graflca como “pox\ nja\e de_absordóot£t pero es más común graficar “porcentaje de transm:. ion” , lo cual significa que una repión de máxima absorcS n aparece como un valle en. la mayoría de las curvas en el infrarrojo-
ÍL1 problema de la determinación cuantitativa de la radiación infrarroja no ha sido resuelto totalmente. Por lo tanto, la mayoría de los instrumentos comerciales dan lecturas en porcentaje de transmisión, las cuales no son reproducibles de un instrumento a otro. Aun un mismo instrumento puede dar lecturas algo diferentes de una semana a la siguiente. La escala de porcentaje de transmisión no puede usarse para cálculos cuantitativos en forma análoga a la del espectrofotórnetro de ultravioleta y, a menos que se hagan esfuerzos especiales para realizar una calibración, debe considerarse tan sólo aproximada» Sin embargo, a pesar de dichas dificultades, la espectroscopia en el infrarrojo se usa extensamente como un método analítico cuantitativo.2
La frecuencia se gráfica preferentemente en ondas por centímetro (generalmente llamadas redprocaT^Jc centímetros W 1), aunque la longitud de onda en mieras (jj.) se usa Ordinariamente como una alternativa. Las mieras se convierten fácilmente a números de onda sacando su mcíproca y multiplicándola por 10,000.
e n r 1 - 10,000/y.
La tremenda importancia del método de espectroscopia en el infrarrojo se debe a que muchos movimientos .longitudinales y le_ílexiór son esencialmente independiente*.. d e .. los. cambios de estructura en ófras_ partí» ~de,.la molécula. Así, la absorción máxima causada por la vibración longitudinal carbono-oxígeno del grttpo C = 0 en la cielohexa-
* Ver (Urruuj-j Kcir y tJobrintr. / . Cfitm. Stt-x 74, 80 (1562) í■ jfrM., 74, 72 (1SM).

nona (I) es casi exactamente de la misma frecuencia queenr 3a pregnan'3- ona ( I I ) , .
2 0 0 U so d e lo» m étodo* « tp# ctros tó p ico »
Es de gran ayuda tener presente les efectos que sobre Ja absorción deben esperarse al efectuar determinados cambios en la estructura, Porlo general, el enlace doble entre dos átomos tiene una constante de fuerza mayor (es.decir, actúa como un resorte más rígido) que un solo Enlace entre loa mismos dos átomos- La mayoría de Jos enlaces sencillos quedan dentro de Ja región de frecuencias menores de 1GQ0 cm"1, la mayoría de los enlaces dobles, en la región de 1600 a 2000 cm' 1 y la mayoría de ios enlaces triples entre 2000 y 2500 on"1. Entre 2500 y 3700 cm -1 se encuentran Jas frecuencias de vibración longitudinal de los enlaces entre hidrógeno y oxigeno, azufre y nitrógeno.
El conocimiento de la teoría estructural oi^ánica es esencial en cualquier uso de Ja espectroscopia en el infrarrojo. Así, el saber que la conjugación de un enlace doble carbono-carbono con un grupo carbonilo disminuye la cantidad de carácter de enlace doble, tanto en el catbonilo como en el enlace doble carbono-carbono, conduce a esperar que cada una de estas frecuencias se desplace hacía la región del enlace sencillo, es decir, que disminuya. E«o se ejemplifica a continuación:
/ °CHjCCHjCHa C H jC I^ C H ^C H ,C—O 1720 n o - 1 O - C i * J a n . - 1
/ ° / °(CHjC—CH=CH* CH3C==CH~CH^]
0 = 0 16*5 cm ."1C—C W3 m.-'
Generalizaciones
Determinadas generalizaciones del efecto de la estructura sobre los espectros en el infrarrojo pueden ser ¿tiles. Estas se ilustran en la íi- gura 22.
L a conjugación, como se mencionó antes, disminuye la frecuencia de ambos enlaces dobles,

Espedraacopícr infrarroja 201
7.a acumulación de enlace^ dobles, por otra parte, tiene el efecto opuesto. Así, los alanos muestran absorción de enlace doble carbono- carbono aproximadamente a 1950 cm"1 en lagar de a 1650 cm’1, posición de un enlace doble aislado. El enlace doble carbono-oxígeno en las ce- tonas y los ¡sodanatos se incrementa de su posición normal de aproximadamente 1710 cm"1 hasta 2100-2300 crn-1.
El enlace de hidrógeno disminuye tanto la frecuencia de vibración longitudinal del hidrógeno como la del carbomJo cuando está implicado un grupo carbonita. Asi, la frecuencia del carbonilo de la Ay^-dieolace- tamida, en solución de dioxano, es de 1647 ern-1 (sin enlaces de hidrógeno), pero en metano!., en donde hay enlaces de hidrógeno con el disolvente, es de 1615 cm '1.
Los substituyanles negativos pueden incrementar la frecuencia del carbonilo en los aldehidos, cetonia o ácidos y sus derivados. Así, el acetato de etilo tiene su absorción de carbordJo a 1745 cm’1, pero el tri- cloroacetato de etilo absorbe a 1763 cm '1. Dichos desplazamientos deper - den del ángulo entre los enlaces carbono-oxígeno y los carbono-X, como se demostró por primera vea en las «,-bromociclohexanonas complejas. Así, la a-doroacetona en fase líquida muestra dos bandas de carbonilo: una a 1722 y Ja segunda a 1745 cm-1. Cada una de éstas corresponda a una diferente conformación rotacional.
La tensión del anillo aumenta la frecuencia de la vibración longitudinal de un enlace doble exocíclico. Esto se ilustra con las series siguientes:
Las frecuencias de vibración longitudinal carbono-hidrógeno también se aumentan por la tensión del anillo. Así, !a frecuencia de vibración longitudinal carbono-hidrógeno se presenta a 3030, 3007 y 3015 cm-1 en el ciclopropano, óxido de etileno y etilenimina,* en comparación con 2960 cmJ en el cíclohexano.4 En efecto, la contracción del ánguloentre los átomos de carbono unidos al enlace doble ^>C=rCHz, de su ángulo normal de casi 120° basta valores de. 109. 90 y 60° (que debe adquirir en los ar.illos de cinco, cuatro y tres miembros, respectivamente}, conduce a aumentos sucesivos en la frecuencia de vibración longitudinal. .,
La um ión esUrica, que actúa en dirección opuesta, puede presentarse en moléculas con la fórmula general R;:C = C H ¿) si los grupos R son suficientemente grandes. La ampliación por esta causa del ángulo
i«i cm.-' iwnn».' JW D C rt.-1 J750<m .-1
* HoKreo*, Evan* y Gíocklcr, / . A n . C*/m. S*e., 73. 3D2& Í1951). 4 Rjummcn, / , C htm . Phyt., II, 249 (IMS).

§T>
i r<
f f j t f l j Ijj) f f f t f i j m i
T55
SQO ™
- íw
~
¿6----
------
-----
&-ts-
j'i-ito
------
»5----
-io /i
¿ ¿
¿ j'y

2 0 4 U j o d « lo s m é to do s fs p e c iro s e ó p ic o l
^ G = rC H 2 disminuya la frecuencia de vibración longitudinal del enlace doble carbono<at'bono, como lo ilustran los ejemplos siguientes: 5
(CH3C H ,)jC = C H 216*2 cm
En ]a figura 22 se resumen las posiciones de Jos máximos de absorción de los principales grupos funcionales, (%'er págs. 202-203,)
Idealmente, ios espectros deberían determinarse en fase vapor, en donde las interacciones entre las moléculas son mínimas. Sin embargo, por razones prácticas, las detejininacíones en solución, en un disolvente no polar con la menor absorción posible, como el tetracloruro de carbono o el disulfuro de carbono, son muy satisfactorias. Las interacciones moleculares en solución o en ej líquido puro producen normalmente desplazamientos de las bandas con respecto a las posiciones determinadas en fase vapor. Por lo tanto, los espectros de líquidos y soluciones no son comparables con los datos obtenidos a partir de sus vapores,
- Los sólidos insolubles en disolventes apropiados para determina- clones en el infrarrojo, ordinariamente se determinan como suspensiones en aceite de parafina. Los espectros de los “mulla” a menudo son mucho menos satisfactorios que aquéllos que se obtienen eñ solución, ya que las bandas pueden mostrar desplazamientos inesperados, cuando los espectros se determinan en “muLT (o película pulverulenta), debido a las interacciones entre las moléculas que conservan su posición compacta dentro de la xed cristalina.
U n ejemplo de la utilidad de la espectroscopia en el infrarrojo para el esclarecimiento de estructuras nos lo proporciona el espectro mostrado en la figura 23 (pág. 206), de una substancia de fórmula molecular CuKuO. De la relación entre los átomos de hidrógeno y los de carbono puede verse que al compuesto le faltan nueve pares de átomos de hidrógeno para ser saturado (ver capítulo 12 para la interpretación de fórmulas moleculares). L a absorción a más de 3000 cm-1 (vibración longitudinal carbono-hidrógeno aromático u olefínico) junto con las bandas a 1500 y 1600 (anülo bencénico substituido) y la fuerte banda a 590 era-1 (fenilo monosubstituído), indican que cuando menos bay un anillo aromático. La ausencia de absorción en^5 2900 y 3000 cra-< muestra que hay muy pocos, si es que los hay, átomos de hidrógeno unidos a carbonos alifáricos (como también puede verse en la fórmula molecular). La .ausencia de una fuerte absorción a más de 3100 e n r 1 indica que no hay grupo oxhidrilo y la fuerte absorción a 1725 cm"1, que
I Bwtkit y Srik». / . Azu, C ftt**■ S*t.t 17, 2
(CH3),CH—C(CH ,)2 (C H ^ C -C íC H a k
C = C H a C = C H 8
( C T U c / (CH.),C1639 < m - ' f 6 2 | d n - J

lU fra c tro tto p ta In fra rro ja 2 0 5
d único átomo de oxígeno es, caá con corteza, parte de un grupo car be- « lo. Las dos Ixmdas a 2720 y 2800 craul (hidrógeno unido al carbono de una función aldehido) indican que el grupo carbonita es una fundón aldehido. L a posición de la absordón del carbonita del aldehido señala que no está unido directamente a un anillo benccníco (en donde estaría conjugado). Todo esto lleva a la fórmula parcial
C < \
SC H C H = 0
c , h /
«o donde una de las unidades C{Hj probablemente es- un anillo bencé- »co, como lo indica expresivamente el espectro y la otra puede pensarse que también lo sea, aunque este último punto no se deduce de la interpretación del espectro hecha hasta aquí. Deberá notarse que en este, como en general en todos los espectros en el infrarrojo, hay evidencia de huellas de una impureza. La absorción a 1660 y 1275 ero-* sugiere la presenda de un pequeño porcentaje de beraofenona, que es casi siempre ona impureza normal del düenilacctaldehido.
En las págs, 207-214 hay una lista de algunos de los grupos funcionales más comunes con sus bandas en el infrarrojo más características. Para mayores detalles deben consultarse las referencia* mencionadas.

É< .
frI
*
¡>
>
£
v
2 0 6 U so d e fas m é to do s e»pectro$cój&k<»
i »
f»
r>
* o
i*
3 w 2 v
E § U 2 <o
2 £ ü_ —
ÉJ]c
£
|tu
a
(eMjd? pepKinsp} epueqjosqv
.f-Ü*
'i

Alcoholas 207
Acetileno*,*1 Los acetilenos monasubstituidos sencillos del tipo K C m C li tienen una banda moderadamente fuerte debida a la vibración longitudinal del enlace triple carbono-carbono a 2105-2150 cm '1. También hay una banda caracterísdca <le la vibración longitudinal carbono-hidrógeno a 3300 axr* aproximadamente (las oleíinas y los hidrocarburos saturados presentan sus bandas de absorción de vibración longitudinal carbono-hidrógeno a frecuencias inferiores a 3200 cm"'). Como para que una vibración longitudinal sea activa en el infrarrojo debe producir un cambio en el momento dipolar, los acetilenos disubstituidos, RC CR. en donde los R son grupos alquilo similares, no muestran absorción en la región del enlace triple.
A nh ídrid o s de ácido. El grupo anhídrido es una de las pocas funciones carbonita relativamente sencillas que muestra dos frecuencias de absorción correspondientes a la vibración longitudinal carbono-oxí- geno. Así, los anhídridos de ácido aiifáticos saturados tienen una banda aproximadas:: "ite a 1800*1825 cm-1 y una segunda a l?40-t?70 cm-1. La conjugación y tc..sión del anillo tienen cualitativamente los mismos efectos sobre las frecuencias que en las otras funciones carbonilo. Por ejemplo, las bandas de los anhídridos benzoico y crotónico están desplazadas unos 20 a 40 cm-1. El anhídrido succínico* con sus grupos carbonilo unidos a un anillo de cinco miembros, tiene bandas a 1865 y 1782 cm '1. Se ha hecho una correlación del efecto de la estructura sobre la distancia entre los dos máximos del carbonilo.7
H aluros d e aciio, lo s cloruros de acilo saturados tienen el máximo de absorción del carbonilo a 1760-1810 cm-1. De nuevo la conjugación probablemente desplace la posición del máximo, pero se han estudiado relativamente pocos ejemplos de haluros conjugados.
Acido» ca rb o x tlico s/ Aunque a menudo 1a función ácido-cas bo.vtlo se investiga fácilmente por la solubilidad de la substancia desconocida en álcalis, el análisis en el infrarrojo también puede ser ótil. Los fuertes enlaces de hidrógeno afectan las posiciones tanto de las frecuencias de vibración longitudinal del enlace sencillo oxígeno-hidrógeno como las del enlace doble carbono-oxigeno, haciéndolas un poco inciertas. El OH de los ácidos- absorbe a 2500-3000 cm"1. En los ácidos saturados simples la frecuencia de absorción del carbonilo aparece a 1700 cm"'. En general, el efecto de conjugación y de substituyen tes negativos es el misino que para los ésteres (ver pág. 200).
A lcoholes. Las limitaciones bastante severas de las pruebas de grupo funcional para alcoholes hacen que la espectroscopia en el infrarrojo sea especialmente valiosa para su investigación. La absoreióti característica de la función —O H que se aprecia con mayor facilidad es la frecuencia de vibración longitudinal oxígeno-hidrógeno en la región
4 S(i'pj>jrd y S»nij»soji, iluarí. R tci., 6, 10 (1952).7 Cbok, C ktm . G }*¿.t 1155, 142.* M. S t. C. Ftctt, / - S tt., IK I, Htt.

entre 3100-3600 cm-1. Los grupos oxhidrilo sin enlaces de hidrógeno absorben aproximadamente a 3500 ern-1 y los alcoholes terciarios con mucho impedimento pueden mostrar una sola absorción bien precisa a esa frecuencia. Sin embargo, Ja mayoría de loa alcoholes en solución existen como una mezcla en equilibrio de una pequeña parte de mo* féculas sin enlaces de hidrógeno (banda pequeña a 3600 cm-') junto con una gran parte de moléculas que presentan enlaces de hidrógeno en diversos grados y originan una banda ancha a unos 3400 cm-1. Ya que el enlace de hidrógeno puede hacer que la frecuencia de vibración longitudinal hidrógeno-oxígeno se confunda con la de hidrógeno-nitró- geno, deberán determ inara Jos espectros en soluciones más diluidas para evitar confusiones. La dilución disminuye la cantidad de enlaces de hidrógeno intermolecuJares y tiende a desplazar Ja absorción hidrógeno- oxigeno, si está pre$cnte, hacia Ja región de oxhidrilos libres a 3600 cm"1. En ciertos casos se ha encontrado un sobretono de Ja absorción de car* L milo (aproximadamente 2 X 1700 cm’1) a 3400 cm' 1 y deberá tenerse cu; 'ado cuando haya grupos carbonilo presentes para no confundir esta banda con Ja absorción del oxhidrilo.
El efecto que producen las diferencias en el enlace de hidrógeno interno de los glicoles cis y trans se ha usado para asignar configuraciones a dichos compuestos.* La frecuencia de vibración longitudinal carbono-oxígeno a unos 1100 cmw| también se encuentra en el espectro de los alcoholes y se ha realizado una considerable cantidad de trabajo para correlacionar la posición de esta banda con la distribución de los átomos que rodean el grupo oxhidrilo (ver referencias en la pág. 197).
A ldehidos. Los aldehidos saturados sencillos muestran la absorción atribuida al enlace doble carbono-oxígeno a unos 1725 cm '1. La conjugación con ün enlace doble carbono-carbono o un anillo aromático disminuye este valor hasta unos 1700 cm-1. Se puede obtener una ayuda adicional en la identificación de una función aldehido con la frecuencia de vibración longitudinal carbono-hidrógeno correspondiente a l C —H del grupo formilo. Ésta absorción aparece en la región entre 2695-2720 cm"J en los aldehidos alifádeos,. a unos 2730 cm '1 en muchos aldehidos aromáticos y a 2760 cm -1 en determinados aldehidos aromáticos substituidos en orto.K A menudo se observan31 dos bandas cerca de 2720 y 2820 cm"J«
A m ida*. Las amidas saturadas (en solución en tetracloruro de carbono o en cloroformo) tienen una absorción del carbonilo entre 1645 y 1680 cm_í. Lo mismo que en otros compuestos carbonilicqs, la conjugación del grupo carbonilo disminuye la frecuencia un poco y la inclusión del grupo amida en un anillo con tensión, la aumenta. Así, las lactamas de cinco miembros tienen la absorción del cart>onilo a unoa 1700 y las lacta-
» V«T KuIid, J, AnT. « c m . Soc.t 7«, (155*); Wafoonky, ¡ M ., 7 t , 5395lfl Ftoclu,», ¿mí. C4*»».. 27, 2 (IMi).11 PtitUkj r OOttwJatl, Amal. Chrm., 23. 1611 (]»!).
208 Uso de los m étodos «pectro icópicos

Ester*» 209
rnas de cuatro miembros, a unos 1730*1760 oír*. Las amidas con hidrógeno unido ¿1 ótomv de nitrógeno presentan, además, la frecuencia de vibración longitudinal’ nitrógeno-hidrógeno aproximadamente a 3200 cm-1 y a menudo es posible diferenciar entre sí las amidas primarias, secundarias y terciarias. Se dispone1’ de explicaciones detalladas atvjca de los espectros de amidas al estado sólido y en solución, y deben (■««- saltarse.
A m inas. La absorción de la vibración longitudinal nitrógeno.Iii.. árógeno aparece en la región entre 3150-3400 cm '3. Aunque tamban puede aparecer en esta región la absorción de vibración longituditv.il hidrógeno-oxigeno debida a los enlaces de hidrógeno, frecuentemente puede distinguirse determinando el espectro de la substancia desconocida en soluciones más diluidas. La dilución tiende a destruir los enlaces de hidrógeno intermoleculares y, así, la banda de hidrógeno-oxígeno se desplana hacia la región de absorción del enlace sin puentes de hidrógeno (36(X) cm '1). Las aminas primarias también muestran una banda correspondiente al grupo funciona] H —N — H a 1600-1625 era4 .
E steresr. Los esteres alifádcos saturados sencillos absorben, aproximadamente, a 1740 cm-1. La conjugación del grupo carbonilo con un enlace doble carbono-carbono o con el anillo bencénico disminuye la frecuencia de absorción del carbonilo a 1520-1725 cm-’, aproximadamente, La substitución de grupos substractores de electrones en el carbono-a puede originar grandes desplazamientos hacia frecuencias mayores. Así, el cianoacetato de etilo absorbe a 1751 y el tricloroacetato de etilo a 1768 cm4 . Los enolcs de los {3-cetoésteres muestran un insólito desplazamiento en la absorción det carbonilo, la cual aparece como una banda muy fuerte aproximadamente a 1650 Cm'1, Probablemente este desplazamiento se deba a la contribución de la siguiente estructura de
resonancia.53 La contribución de dicha estructura ea reforzada por la existencia de enlaces de hidrógeno. Análogamente, el o-hidroxibenzoato de butilo tiene su absorción de carbonilo a 1675 cmul.
Aunque las Jactonas de seis miembros muestran una absorción'normal para el carbonilo del éster, las lactonas de cinco miembros muestran
u Ver ReírrcníU Círciml A en la pá*. 1J®.u Riumutteo y Braduia, )■ 4m . CKem. Soc,, 7 i , 1073 (IÍMÍ); Haxlwtll, Riciiiudí y
TiKwnpwn* }■ Ch*m. See., HH¡8, 1436. Ver Usebién L w iird , Cvlovaky, M*3<üe<Oft 7 Po teoiit, / . Am. Ctfzs. S ie ., 74, 4G70 (1952); Befljuoy y B etdcr, J. CA*m. Ssc., 1954. 44í?.
H

V
I ^ 2 T0 Uno da lo« métodos espectro*cápteos*■ W
un desplazamiento hasta unos 1770 c™ -1 y las lactonas de cuatro miem- bros Iiasta 1320 cm-1 o más.
( ■ L a frecuencia del carbonilo del éster se ve influida fuertemente pori ™ substitución eo 3a parte alcohólica de la molécula. Así, los «teres de
fenilo y vinilo tienen su absorción de vibración longitudinal de carbonilo a 1750-1770 cm"1. Los substituyentes en el anillo bencénico pueden des-
* W plaza*- la absorción axin más. Así, el acetato de o-nitrofcnilo absorbe! a 1786 cm '1.
Eteret* La presencia de oxígeno en una substancia desconocida, junto con la evidencia sobre la ausencia de otros grupos funcionales
I que contengan oxigeno, constituye evidencia para un éter. Sin embargo,la vibración longitudinal del enlace sencillo carbono-oxígeno produce una absorción en el infrarrojo a 1050-1150 c n rJ en los éteres saturados
*j ^ sencillos y a 1200-1260 era-1 en los éteres de ariio y vinilo.i H idrocarburos saturados. Los compuestos alifátícos con enlaces
m ttk carbono-hidrógeno muestran máximos de absorción, debido a la vibra- ■ P P cíón longitudinal carbono-hidrógeno, a 2850-2970 cmJ y también lasi deformaciones del grupo — CH2— originan absorción a 1465 o n ’ 1 y' las del grupo CUy, absorción a 1450 cm’1. Además, hay un máximo
a 1375 cm-1, debido al C H r, que es bastante característico, Pueden consultarse referencias más detalladas.'*' n La localización precisa de
J estas bandas puede ser de considerable valor en la determinación de es-^ tructuras, ya que se ven influidas por los grupos carbonilo o enlaces
dobles carbono-carbono.1* El hidrógeno unido a un carbono enlace rnúl- <•• • tiple origina absorción a mayores frecuencias de la vibración longitudinal* carbono-hidrógeno. Asi, R2C = C H j muestra absorción a unos 3080 cm-1,
R C « C —I í a 3300 cm“J y el hidrógeno aromático a unos 3050 cm-1. En los anillos con tensión, la frecuencia de vibración longitudinal carbono- hidrógeno aumenta.” Así, los ciclopropanos presentan absorción a 3024- 3058, los ciclobutanos a unos 2970, 2995, los ciclopentanos a 2940-2950 y los cíciohexan.os a unos 2915-2924 cm"1. Además, se ha encontrado que la posición de la banda de absorción del deuterio (y, por lo tanto, es de suponerse que también la del hidrógeno), cuando está en un anillo de ciclohexano, depende de sí es axial o ecuatorial.1*
H idrocarburos arom ático*. A menudo un anillo bencénico se caracteriza no tan sólo por la banda de vibración longitudinal carbono- hidrógeno a 3000-3090 cm-1, sino también por las bandas cercanas a 1500 y 1600 cm-1, aunque una o ambas de éstas dos últimas pueden
v
b
í >
í '- r*
1
1* Shíppxr<l y SlmpMB, Q,Mari. A w „ *1, 19 51 V<r r<ftrcitei» ptfnewil en Ja p4f. 197; pije*. 19-20.M RcfrfCiXM Ceritrai E, p ij . Í97, También ttr HoUn J /oarj / . Au\, C tifm . S e t., 75,
562S (1953) par» cofrrUeiotiM <fet*Da<Ut y U» rebroto*? alUir Ri>l)«rts y dMmbtrt, } . Am. C \ t m, Sos., 72, 5030 (1031JJ Col?, / . C ium . Sót.,
¿ ■ k I6SÍ, MO?.Cetnpi. R.end,, Í35. 154 (1^42); Coit f , Sní^a, Danahcf, Ywins y Kctfcdg*,
C/«» i. ( f ¡* 4 . l*M , 12H- Ver Efntdo y WnJght, P rese n í« S u ttc ck tn th ify , e jítído per Ktyoc, Aoidtaic Prca, Nuw» Y«A, 1954, p ig . 151,

Ce tomas 2 T 1
faltar. Loa bencenos monosubstituidos tienen una banda, intensa cerca, de 700 cm-1.
C e to w u . Las cetonas aKfáticas saturadas tienen la banda de carbonilo a unos 1710 cm-1* Como en el caso de otros compuestos carboní- licos, esta ba.ida es desplazada a 1670-1690 cm4 cuando el grupo carbonilo está conjugado con un enlace doble carbono-carbono o un anillo aromático. Los enoles de las {3-dícetonas muestran la absorción de carbonilo como una banda ancha e intensa a 1540-1640 cm*4 por la razón indicada en la discusión de los enoles de los £-cetoésteres {pág. 209)* El efecto de la tensión de un anillo sobre la absorción carboníiica es paralelo al que corresponde ai enlace doble olefínico. Aunque las cetonas cíclicas de seis miembros presentan la absorción normal del carbonilo alifático, las cetonas de cinco miembros tienen su absorción carbonÜica desplazada a 1740 cm"1 y las cetonas de cuatro miembros a 1780 cm '1. Gomo en los enlaces dobles olefinicos, la ampliación del ángu. ) de enlace por tensión estéric» entre substituyentes voluminosos, diamir. ye 3a frecuencia de absorción del carbonilo.” Así, la di-f-butilcelona. tiene su absorción de carbonilo a 1636 cm4 y la í-butíltriptilcctona (hexametii-hexanona-3) a 1675 cm '1*
Los substituyeme* pueden tener Un efecto considerable sobre la . posición de la banda del carbonilo. El efecto de un átomo de cloro en un átomo de carbono-a ha sido correlacionado con su orientación en el espacio con respecto al enlace carbono-oxígeno.20
Análogamente, los substituyentes en el anillo bencénico pueden originar desplazamientos de la frecuencia del carbonilo en las cetonas aromáticas (conjugadas)
Otro efecto estructura] que puede conducir a desplazamientos significativos de la frecuencia del carbonilo de una cetona se ilustra por la interacción transanular del átomo de nitrógeno de un grupo amino. Así, la aminocetona que se muestra a continuación tiene su frecuencia de vibración longitudinal de carbonilo a 1681 cm-J. En solución ácida
w B sr tlt i t y S ú to , / . A m , C kem . S«e., 77, SáOS [1955].& Jones, Rujnwiy, Herlin* y Efcjbriner,, / , Am. Chrm. ía c ., 7-4, 232fl <1952); Mcuutúma,
SbímanMudvi, Idiuhiou, KumflJiJ, Níkoprna y Sbklo, / , CA*M. Phys,, 2 t , 1515(195?); Cor*?, ; . J im . C ktm . So*., 71, 230», 329? (1953).
11 Selaw iy j Fttóm , / . ¿ m . Ckctn. Soc ., 73 , (1951): Fuion , Jc*¡bh y sk*l<©n, n , 25J$ (1954}.
MO
CÜJ CH* CHa

2 1 2 U so d% lo s m é to d o s a*pactrosc¿p i< os
o en el pcrclorato sólido no hay absorción en la región del carbonilo, p€iC se obsÉjYa absorción de oxhidrilo, Ic cual conduce a Ja asignación d e la. estructura con el oxígeno protonado, indicado arriba ,22
En la discusión de los espectros de anhídridos se señaló que la interacción de dos grupos carbonilo equivalentes o casi equivalentes, unidos mediante un átomo de oxígeno, provoca la separación en dos bandas de la absorción de vibración longitudinal del carbonilo. Dicha separación se dice que se debe a la resonancia de Fermi y su presencia es bien conocida en moléculas sencillas*3 También pueden ocurrir interacciones entre la vibración longitudinal de un carbonilo y el sobretono de alguna otra vibración que por casualidad sea de la frecuencia correcta, Por ejemplo, la difeni3-3,4-c¡cIopemcn-2-ona-i muestra dos bandas de intensidad comparable en la región de carbonilo a 1695 y 1718 cm '1. Se ha demostrado que esta separación se debe a una banda que
hay á 660 cm’1, la cual tiene su primer sobretono cercano a la frecuencia normal del carbonilo- El que la interacción de estas dos, ocasionen vma separación de la frecuencia del carbonilo, lo sugiere el hecho de que en una serie de compuestos afínes, sólo aquéllos con la absorción más baja mostraron la separación de dicha banda. I-a hipótesis de que la banda a fl60 cm’ 1 (y su sobretono) en « te compuesto se deba a una flexión fuera del plano del hidrógeno vinílico en el átomo de carbono alfa con respecto a l grupo carbonilo, condujo a la síntesis y determinación del espectro del compuesto análogo con los protones del anillo substituidos por deuterio para cambiar la frecuencia de la flexión. Se encontró que la separación de la banda de carbonilo desaparecía en el compuesto deuterado.*4
Casi es superfluo señalar el peligro de una interpretación incorrecta de los espectros por no tom ar en cuenta dichas interacciones. L a cíclo- pentanona1 muestra un comportamiento análogo: 55 se observa un segundo pico que desaparece con la deuteracíón de los átomos de carbono-a. Es probable que tales separaciones, originadas por una resonancia
^ Le<>tMr<3t Ole! y Cbivarelfi, ) . Am. C ktm . S tc .. 77, 67H (1555). EÍ«toe slcuibrc» u. hiA encontrado «UCM epmpuMM». Para reviiioriet criticas tconard, R g ío ti Chem. Profr. (K u í í *-Ho9U t So . Lih.fj 17, W (1956); C hima, H , 231 (ISfiOl.
** Heribers:, ¿*¡ier¿4 om4 Harria» Sptttra , X>. V»ti KcUrJtid Co., Pttoceion, Jf, 1945j . p it* . 213 y t¿g«.
» YaW y Williun», / , Am , Ck<m. S«c., «0, 5m** H m b e r r , ttyfrarrd 4nd jh S p tt l ia , D . V »n «fOftaitid C o ., P ñ n c a o n , N , J .
H P$H|
110
pi*. « 7 .

Funciones n itro g en a d a s 2 )3
de Fermi, sean bastante comunes entro los compuestos carbonílicos21 y, como probablemente las dos frecuencias que están interactuando dependen en forma diferente del disolvente, las formas de las curvas pueden depender enteramente del disolvente.2*
ÍYílriío*.” Los mtrílos saturados sencillos muestran la absorción C a K como una banda mediana a 2250 cm"1. La conjugación disminuye la frecuencia hasta 2225 cm-1.
C om puestos n itrados. Tanto los compuestos nitrados aliláticos como los aromáticos2* tienen las dos bandas características del grupo nitro a 1350 y 1520 cm '1.
O lefinai. Las olefinas no conjugadas sencillas del tipo R2C = C iiJ? muestran absorción aproximadamente a 1650 cm-1. Sin embargo, las ole- finas mucho más substituidas pueden estar constituidas en tal forma que la vibración longitudinal carbono-carbono origine pocos cambios en el momento dipolar. En este caso no hay absorción en el infrarrojo en la región dei enlace doble. Shcppard y Simpson han discutido con más detalle los espectros de las definas.*
La conjugación con un segundo enlace doble carbono-carbono, un anillo aromático o un grupo carbonilo puede desplazar la absorción del enlace doble carbono-carbono hasta 1600 cm'1 o un poco menos. Como es de esperar, la polarización de un enlace doble carbono-carbono por conjugación, con un grupo carbonilo o substitución ssímétrica con otros grupos polares, aumenta la intensidad de la absorción. Los efectos por variación de los ángulos de enlace debido a tensión de los anillos o tensión estética, se discutieron en la pág. 201,
A menudo puede determinarse el grado de substitución de una olefina y aun la configuración. '-Por ejemplo, la banda de flexión carbono-hidrógeno de las definas del tipo R jC ^C H * en donde R es un grupo alquilo, aparece a unos 910 cm’1, mientras que Ja absorción correspondiente de las definas del tipo R C H —CHR depende de si la olefina tiene configuración cis o truns; la banda franr está a unos 960 cm*1 y la cis (que es rnenos precisa) a 680-715 cm”1. Las posiciones de estas bandas pueden alterarse apreciablemente por substitución de R en Jas estructuras anteriores por otros grupos funcionales.53
Funciones nitrogenadas diversas. Los estudios de determinados grupos funcionales que contienen nitrógeno han proporcionado datos que no aparecen en las referencias citadas previamente. Algunos de estos datos se mencionan aquí. Se: han examinado algunos ésteres de nitrito (bandas intensas a 1725, 1640 y 1610 cm"1) y nitrosaminas (bandas a 1350-1410 cm' 1),31 Las oximas muestran banda de enlace doble car-
* Btltamjr, Ttant, Ptreday S<x., 63, 34 {1959}.*1 Kltsno y Grif filfa, Anal. Chtrx. 24, 334 (1952).« R ítale y Wlliíítn, / . Ckent. Soc., 1W2. 4153; HantUin*. aW-, 1B53T 262** Slirppard y SbnpíOP, Qm*tI. R ta t., 17 (1952).* KilttS, Anaf. Cbtrx., 23, 14» (1953).W HiocWin* y ) . C W . S.><., 1Í54, 491.

-9 2 1 4 U so tfo tos m é to do s e sp octa rtCÓ p íto s
»
bono-nitrógeno aproximadamente a 1640 cm.-1 y determinadas eximas | ^ Qt y 3 pueden diferenciarse por la posición de la banda.de vibraciónL longitudinal oxígeno-hidrógeno.3* Las cetiminas absorben en la misma
región/” Las dialquílcctiminas muestran la absorción de enlace doble carbono-nitrógeno a 1640-1645 a r r J y Jas de alquiladlo aproximadamente a 1620 cm-1. La dífenilcetimma da una banda a 1602 c n r1.
Los isocianatos, tanto aromáticos como alifáticos, muestran 3a absorción de vibración longitudinal carbono-oxígeno a 2269 ±i 6 cm '1.3* k 05 compuestos diazO muestran una absorción intensa en ía región entre
9 2000-2200 cmwl. Así, el diazometano35 muestra fuerte absorción nitrógeno-nitrógeno a 2101 cm-1. El difenildiazometano tiene su absorción correspondiente a 2045 era"1. Determinadas diazocíclohexadienonas44 también muestran absorción a 2015-2150 cm-1. El fluoroborato de ben-
I cendiazonio, que aparentemente es Ja Gnica sal de diazonio estudiada,34tiene una absorción moderadamente intensa a 2296 cm-1.
i *
i »a
í*
r»
ESPECTROSCOPIA DE RESONANCIA MAGNETICA DE B O T O N E S
El objetivo de esta sección es ilustrar la utilidad de la resonancia magnética de protones en la determinación de estructuras. Para aquellos, a quienes les gustaría aprender más sobre la teoría y práctica del método se les recomiendan las referencias siguientes:
J j k A ) Jacfcman, ApfrtiCatiotJ o / Nuclear 'Magrie tic Resononcc Spectiosccpy ir.Or¿a.n¡c Ctiemixtry, Pergamott Press, Nueva York, 1959,
Robertt, NacUar M a& netu Resonaace, Applicaiinn: to Orgánic C htmistry, McGra.w-HUI Boolc Company, Nueva York, 155$.
I _ R-oberts, A tí fn fro d u e th n io Spin-tphi Spliiling in. H igh ResolntionSuelear Magneiic R tse n M c t Specira, W . A. Benjamín, Nueva York, 1961.
Estos tres libro} proporcionan información príníipaJmerue p3ra ct químico orgánico en ejercicio y eqostituyen una buena introducción al tema.
B ) Pople, Schndder y Beenstein, High^rescluiion iNuclear Aíagnetic Re- loñance, McGravr-Hill Book Company, Nueva York, 1959. t i t a es ía obra de referencia tipo sobre resonancia magnética nuclear y es u n a fuente valiosa
j ¿e información tanto p a ra el experto como para quien vse ocasionalmente la RMN.
i . C ) M artin , “N M R Spccürosccpy as an Analyúcal Tool in Orgam c Chenus-tr , '\ J. Chem. EJue., 38, 266 (1961).
Conroy, "N uclear M agnetic Resonance in Organie S iructural Eluci- datioj. ', en Advances in Orgamc ChtmUfry, Vol. II> editado p o r RaphaeJj Taylor y W ynbsrg, Interscícnce Publishers, Nueva York, 1960,
* PjJm r Wtíbin, Can. J. Chcr*., 31, ]D(H (1 «3 ).» Pickfl^ >• Pollr, 1. A m . Chcm. So í., 76, 5I«9 {1934).* Davina, / . Cko,m,, Soí., Itól. 3? 12.15 C»HÍo«I, tJeWbCT y Ranw»y, J. Chim. 19, <06 (J&l). 11 U F o k , Só«jsi y Wcnwr, ) . Cherti, Soc., 1854, 46áf>.

E jpedro icopfo d e resonando m agnética 21$
Cfela ano d e ¿Atoj, es un « u m e n crítico corto del tem a y proporcionan una. buena introducción, al campo de 3a R m N.
D ) Bhacóa, Johrtton y Shoolery, NM R Speetra CaUlog, Variati Aiaociates, Palo Aito, Calif., J.962, U n católogo de 339 espectros de a lia resolución, de los cuales varios se osan en ía subsecuente discusión de este capítulo.
■Wibcrg y Nist, JnUrpretadon c f N M R S ftc tra , W , A. Benjamín, Nu<va YorV, 1562. E»ta obra es una colección de patrones de frajoieotaccíón teórica 'aJciilados por una computadora IBM-709, con los resultados impresos por el equipo de salida, de cal m anera que Se parezcan a los espectros reales.
Guando una molécula orgánica, tal como la del metano (que tiene átomos de hidrógeno unidos covalentementc), se coloca en un fuerte campo magnético e irradiada con una radiación de radiofrecuencia, hay una absorción de la radiación a una frecuencia que es característica del medio magnético que rodea, a l núcleo del hidrógeno. La absorción está relacionada con el cambio de los spines nucleares de los hidrógenos de uno de los dos estados cuánticos posibles al otro. A diferencia de Ja absorciói. .en el ultravioleta y en el infrarrojo, la frecuencia de la absorción en Ja ri..:>nancía magnética nuclear (RM N) depende no tan sólo deJ medio estructural que rodea al núcleo sino también del campo magnético externo aplicado con el propósito de efectuar la determinación. L/>s espectros de resonancia magnética nuclear han sido determinados con diversas fuerzas de campo. En Ja actualidad, en Estados Unidos, ordinariamente son m ás usados los campos de 14,000 a 23,500 gauss con sus frecuencias respectivas de 60 y 100 megaciclos.
Para obtener la homogeneidad del campo magnético requerido para el trabajo de alta resolución, la muestra debe ser usualmente un liquido móvil o estar disuelta en un disolvente como eJ tetracloruro de carbono, deuterocloroíarmo, bisulfuro de carbono o anhídrido sulfuroso. Ordinariamente es aconsejable una soludón del 5 al 30%, por lo general se emplea aproximadamente I mi de la solución, pero cuando se tiene muy poca muestra se pueden determinar los espectros mediante técnicas especiales. Aproximadamente se añade 1 °/o o menos de tetrame- tilsilano corno deferencia interna, por las razones que en seguida se explican.
En la figura 24 se muestra el espectro del metoxiacetonitrilo. Puede verse que hay dos señales de absorción, Jas cuales están relacionadas con los dos grupos de núcleos de hidrógeno: los del grupo metoxilo y los átomos de hidrógeno del metileno. Las dificultades instrumentales y de otros tipos hacen imposible la determinación de Ja frecuencia y del campo magnético en el que ocurren estas absorciones con exactitud suficiente para calibrar el espectro en unidades absolutas. En lugar de ello se introduce una substancia de referencia, ya sea dentro de la solución que contiene la muestra (“referencia interna") o en un tubo capilar por separado ("referencia externa” ), y la señal de la absorción de esta substancia proporciona el punto de referencia para la calibración del .espectro. Se han usado como substancias de referencia muchos tipos,

216 Uso d« toe métodos ospedroscópicoi
incluyendo el agua, el cloruro <Ie metileno, el cloroformo y el benceno, todos como referencias externas. En la actualidad, la práctica más extensamente empleada es la de usar el tetrametiisilano como referencia interna y asignar a la señal del tetrametiisilano el valor ya sea de 0 6 de 10. L a diminuta sena] de absorción en la m arca cero de la figura 24 se debe al tetrametiisilano añadido como referencia interna.
Debido a que la posición de la absorción depende tanto del carnpo magnético aplicado como de 3a radiofrecuencia, un espectro idéntico al de al figura 24 podría obtenerse, en principio, en dos formas diferentes. El campo podría mantenerse constante y variar continuamente la frecuencia con un generador de barrido lineal, o mantenerse constante ia frecuencia y variar el campo magnético. Por razones prácticas, el espectro mostrado se obtuvo por el último método y podría pensarse en la necesidad de emplear una escala horizontal en miligauss. Sin embargo, experimentalmente es más sencillo calibrar el espectro en ciclos por segando y, por esta razón, aun cuando el espectro se obtenga por barrido del O’mpo magnético, frecuentemente la «ca la horizontal está representada en estas unidades.
Es esta característica de la RMN, única entre los métodos espec- troscópicos, lá que conduce a expresiones como ‘los hidrógenos del metoxilo en el metoxíacetonitrílo absorben a 208 c.p.s. a campo más bajo a partir del tetrametiisilano” . Debido a que las distancias entre las señales son proporcionales a la fuerza del campo aplicado, la posición de esta' señal de metoxüos en un espectro determinado a 40 Me. y 9400 gauss sería. 208 X (9400/14,100) ó 208 X (40/60) ¿s 139 c.p.s. a partir del tetrametiisilano. Para tener unidades que sean independientes del campo extemo al cual se efectuaron las determinaciones, es conveniente dividir 1a diferencia de la frecuencia en ciclos por segundo entre la radiofrecuencia empleada, en este caso, 60 Me. Las diferencias en tales unidades han sido denominadas ' ‘desplazamientos químicos”* Entonces, las unidades son partes por millón. En este caso la absorción del grupo metoxiio a 208 c.p.s. a partir del tetrametiisilano representa un desplazamiento químico de 3.47 p.p.m. Debido a la tendencia de los químicos a representar gráficamente los espectros con la fuerza del campo aumentando, hacia la derecha y su inconformidad con una escala en partes por millón que aumenta hacia la Júqu¡erda, como en la figura 24, se ha usado extensamente una segunda escala en paites por millón en la que la frecuencia del tetrametiisilano se toma como 10 en lugar de 0. Esta convención origina las unidades que han sido llamadas unidadü' -v (tau).
Así, la absorción del metileno en la figura 24 es a 4.20 p.p.in, a partir del tetrametiisilano o a t = 10.00 — 4.20 ó 5.80. En esta discusión usaremos partes por millón a partir del tetrametiisilano, convención que se ha usado en el Varían A'M R Spcctra Catalog al que se hizo referencia en la pág. 215.

E spectroscopia d« reso n an d o m agnética
43-oa5 1 i '
t
' !¡ i i£§3KS r
i
i ; J ;*
§ §*8
iV i
¡m l
■1 s 3 4
i;
. f1 i ;
§ *9yr
i
81£-SíÍH
¡i ¿ S.2 .
1E
x»zim
•i

m
l * *
r *n tj
3 »
a »
uuü i
í í
n
218 Uso de los métodos espectroscopios
Desplazamientos químicos
Quienquiera que esté familiarizado con el uso de los espectros en el infrarrojo,en la química orgánica estructural, inmediatamente supondría que las posiciones de las señales en la RM N estarían, como los máximos en el infrarrojo, relacionadas con la estructura en tal forma que el desplazamiento químico podría proporcionar una información valiosa sobre la estructura. Así, en cJ ejemplo considerado en la figura 24 parecería razonable que se pudiera considerar que la absorción de los protones del metoxiio a 3.47 p.p.m. y Ja de los protones del metileno a 4-20 estuvieran desplazadas respecto a alguna posición característica típica de los protones de un hidrocarburo aíifátíco.
En la tabta 13 se dan ios desplazamientos químicos de varios tipos de compuestos. So verá que los protones del. metoxiio del metoxiaceto- nitrilo, a 3.4?, originan una absorción en la misma posición que el grupo metoxiio del metano!. Además, podría ar. *ciparse la posición del grupo metileno del metoxiacetonitrilo tomando romo punto de partida la absorción del metitcno en un hidrocarburo, 1 .2?, y suponiendo que los desplazamientos debidos a las funciones ciano y oxígeno fueran aditivos. Así, la absorción del rnetileno-a en el 'r-bromobutíronítrilo aparece a 2.58 (supondremos que el álomo de bromo está lo suficientemente alejado como para que pueda ignorársele), lo que sugiere que el grujió ciano es responsable de un desplazamiento a campo más bajo de 2.53 — 1.27 ó 1.31 p.p.rn. El grupo metileno-a del tetrahidrof urano origina una absorción a 3.75, lo cual significa que el grupo éter ha ocasionado un desplazamiento de 3.75 — 1.27 ó 2.48. El efecto combinado de los dos grupos sería entonces 2,48 + 1.31 ó 3.79, lo cual está en concordancia aceptable con el valor de 4.20 observado.
Las complicaciones que se discutirán hacen imposible predecir los desplazamientos químicos con gran exactitud, excepto en casos muy especiales, pero el método esquematizado, aunque burdo, puede proporcionar una valiosa comprobación de lo razonable que puede ser !a asignación de una estructura. U n método un poco más. elegante ha sido desarrollado por Shoolery y se discute en el libro de Jackman, págs. 59 y sigs. y en Ja pág. 130 de la tcferencía A de la pág. 214,
La interpretación y prodición de los desplazamientos químicos no han sido fáciles y aún continúan siendo investigadas extensamente, tanto teórica como experímcntalmente. Aunque se han hecho algunas correlaciones entre los desplazamientos químicos y parámetros químicos tales como elcctronegatividades,*7 momentos dipolaics de grupo * y constantes de substituyentes,* es claro que, en general, deben considerarse otros
* Q a w f t v Dúl«r. J. Ch*m. P kp., M, IMS (I ttJ ) .* Rcddy, Rw *er y GoUstci», / . C htm . P kyt., >CO (líW tJ." F n s e r , Cm*. ¡ . ?226 (IM O).

D e sp la za m íe n lo s q u ím ic o s 2 1 9
T a b la . 13
Gnijxi íuneiotul| D'VpbdonicoLo quCaitCQ,*
EJ'taplo j p .f r t . a partir del | (tieajftcciliiUtvo
OH 0
Oxhidrilo enólico ^p-BrC.H 4C = CH—<Xt U4 B tir ) O ü O1 ü
1 í ¿ J t
CarboxiloC H ,C = C H -C C H 1 c«. 15 A*CH»COOH 11.37?
Hidrógeno de aldehido C H ,C H = 0 9.80
Hidrógeno AromáticoC .H .C H .-0 10.00c , h , 7.37Dur^no (hidrógenos anulare*) <>.SCHCb 7.27
Trihalometanos CHBi, e.«5C H ^ C ÍC H ^ 5.34
Protonca definidos CHjC HjCH.CHi NOj 4.38Compuestos nitrados c»Hto a 4.24?FenolesDiarilmetanos
5.92
3.75Eteres CU, CHX
\ /O
Alcoholes CH,OH 3 A lCH3CH,CB,Ol{ 3.5S
Acetileno* C,H*CüeCH 3.05CH,0=cCtí l.SO
KaJuro* de alquilo CHjCHjCI 3.57CH,CܻBr 3.43CH,CHiT 3.20
Aminas CH,CHtCH,CH,NH, 2.70Nitritos BrCH,CH.CB.CN 2.58Aldehidos y tetonas CH.COíUiCH, 2,4*
CHjCH.Qf.CHO 2.42C adena lateral aromática C.HgCÍJ, 2.32Alcoholes CH.CHjCÜjOH 2.28?
(C H ^ H O Ü “ 1.60?CH,OjJ M 3 t
Amida* CHjCjijCONHü 2.23Hidrocarburos CH,(CH,\CH, 1.27Aminas CH1CH,CHtCH*NHl ‘ 1.10Hidrocarbnrofl CHííCH^CH, 0.88Anillo de ciclopropano ÍCH.li 0.22T ctm netilsilano (CHj^Sl 0.0Q
* En doHc Ia *td6 po Wff, ioi vJm«í ton fot íH Vürii/i NMR Sprcltá Cítalo¡¡ y fueron CVtXenMos « i >1 l ; c m dcUlcrocJofOÍorcoO. D*f>>:ha» regírti^dos n°» Auocíatt*f te «proikoe con te ivmtjfc».
t ProUatfcnicnt« U potíci¿a d« f)U ataorció» depelHtt íuertemenít dfJ cfijolvírtte y líe ta coocmtrtci'jn, debí<¿> a M<KÍ«!<5n e jntCTWínMfi«.

factores. En particular, las anisotropías de enlace ■“ son demasiado importantes para ser ignoradas y el can p o magnético que resulta de Ja corriente anular inducida es responsable en gran parte de las posiciones de las «sonancias de los protones aceti tónicos y aromáticos/ 1
El empleo del tetrametiisilano como referencia interna ha sido favorecido por el hecha de que el campo magnético efectivo en una solución depende de la magnitud de la susceptibilidad magnética del disolvente. Permitiendo que este efecto actúe simultáneamente sobre la muestra y el tetratmtfilsiíano usado como referencia (suponiendo que cada uno se vea afectado en igual extensión) > a menudo se puede cancelar el efecto del disolvente y generalmente es sobre esta base que se efectúan'las comparaciones de los desplazamientos químicos. Alternamente, se pueden comparar substancias diferentes en el mismo medio por extrapolación hasta dilución infinita. Sin embargo, debe notarse que interacciones específicas entre el compuesto en estudio y el disolvente pueden ocasionar que el desplazamiento químico dependa considerablemente del disolvente, aún con la referencia interna de tetrametiisilano.'12
220 U so d e los e n g o d o s e s p e c t r o s c o p ia s
Areas de las señales de absorción
Si la utilidad de la RM N dependiera únicamente de la información derivada de los desplazamientos químicos, sería bastante limitada. Afortunadamente, de los espectros de R M N se pueden obtener varias clases de información. Quizá es ágnifícativo el que en loa estudios iniciales de los espectros de RM N a menudo se Ies interpretó sin tomar en consideración los desplazamientos químicos de las diversas señales, pero como no $c conocía, la importancia d d efecto del disolvente sobre los desplazamientos químicos, probablemente esto fue suficiente. Una de las características de la absorción en RM N que hizo esta forma de interpretación posible fue d hecho de que, a diferencia de las intensidades de absorción en el infrarrojo, la intensidad de la absorción debida a un protón es independiente del medio que lo rodea. Así, en el espectro del CHjOCH’CNJ mostrado en la figura 24, la relación de las ¿reas de las señales de los átomos de hidrógeno del metoxiio y de los átomos de hidrógeno del metileno es de 3/2; como puede verse, sus alturas se encuentran aproximadamente en esta misma relación. La medición de las áreas es muy fácil con un instrumento como el espectrofotómetro Varían A60, que está equipado con un mtegrador.
A coplam iento de ipin^épin. O tro aspecto importante de la espectroscopia de RMN de alta resolución que ha sido de lo más valioso
* A<fer*¿s t e fau T<ícm»dtu z tu m lc t tu lis £1*215, ver Worilz y Sticufwré, AM. P ¡V u, i . 36J (1962)
M Ver R r f t ' . 'c b A y E cr> U p ij . 214.«* V«r Kllflck y Stothcr», Con. ) . C h u z ., + í, Z323 (IÜ€2), y HaltoJi y ItjcíW'ái, Af^f.
M y/,, &é J53 ÍI&52J» pir» tc(cr«ieúí.

A re a d e to s se ñ a le s d e a b so rc ió n 221
%*'4V*
“ I i I"1
o O -í
1 1
<
i 1 l '
g § s * 5 ! 1
1—
t»
2 8 3 s* r
1i
11
i ..
3 8 ? 3 s ! i :
* § 3 S T ' ¡
,
S ■ft
Figu
ra
93,
p»p«
<ha
RMN
d*í
ysdv
ro
d«
«tifa
«n
¿«ul
erot
loro
foT
ino.
(D
«r«<
ha*
r«g]
tfío
d©t
por
Vor
tao
Ait
oila
Ui
jr n
pw
ívd
do
«
n*w
p*rr
n!to
.)

2 2 2 U to d e lo s m é to d o s e sp e d ro s cá p fe o s
cr. I* dilucidación de estructuras es el acoplamiento de spin-spin. El problema se ilustra con el espectro del yoduro de etilo, mostrado en la figura 25. Aunque es de esperarse que el grupo etilo tenga dos señales, una de los tres protones del metilo y una de los dos protones del me* tileno (coYi áreas en relación de 3 /2 ), puede verse que estas dos absorcio- n « están subdivididas; la señal del metilo se ha convertido en un triplete y la del metileno en un cuadruplete, Lo que es más, otros grupos etilo, en el éter diétílico o en el etilbenceno, por ejemplo, muestran un tri- píete y un cuadruplete similares, U na propiedad importante de la subdivisión observada aquí es que las distancias entre las señales del triplcteo del cuadruplete son independientes del campo magnético empleado (aunque, por supuesto, la distancia entre los centros del cuadruplete y tripíete son proporcionales al campo apJicado). U na forma sencilla, pero generalmente muy útil, de explicar la subdivisión es la observación de que cada uno de los protones equivalentes del grupo metilo puede tener sus spínes orientados con el campo magnético externo u opuestos a él. Así son posibles las siguientes combinact nes de los spines:
•+-+ 4- +■ -f* — — — + — ------— 4 -4 - 4 - - - 4------f' — -\—
Estas orientaciones pueden afectar el campo en la región de los protones del metileno en tal forma que se originen posiciones de resonancia ligeramente diferentes de los protones del metileno en moléculas que, por ejemplo, tengan el grupo metilo ( -f- 4- - f ) Y «l grupo metilo (.4- + —}. Ya que los tres protones del grupo metilo son equivalentes, cada una de las combinaciones centrales ( —), ( + ^ + ) y ( — + + }> tiene un efecto equivalente y si la proporción de las moléculas con cada una de las posibles combinaciones de spínes es la misma, la relación de intensidad de las cuatro señales debería- ser 1, 3> 3, 1, o sea, casi Ja observada, Análogamente, los dos protones del metile.no pueden tener las orienta* dones indicadas a continuación'.
+ + - ++ -
Así se tendrían tres clases de moléculas con sus absorciones de protones de metilo a tres posiciones ligeramente diferentes y eri la. relación de intensidad 1,2,1. Se deberá notar que ios miembros de grupos equiva- lentes de protones no originan subdivisión. Esto es la manifestación de un principio general que se adoptará al aplicar esta interpretación de primera aproximación del acoplamiento de spin-spin. Así, los seis protones del benceno, siendo equivalentes, producen una sola línea de absorción.
De una ampliación del anterior análisis de la absorción d d etilo se deduce un segundo principio general. Si en un grupo equivalente hay

A re a d e la s señ a le s d e ab so rc ió n 2 2 3
n protones, deberán esperarse n -f 1 lineas en la absorción, de un segundo grupo que esté lo suficientemente cercano para ser afectado.
En genera], el acoplamiento entre los hidrógenos de átomos adyacentes es Jo suficientemente grande como para producir el tipo de áub- división que se ha encontrado aquí, pero si ios protones están separados por tres átomos de carbono saturados, la subdivisión es demasiado pequeña para ser observada, En los sistemas insaturados es común el acoplamiento a mayores distancias. La aproximación de primer orden mencionada aquí debe usarse con precaución, particularmente en moléculas rígidas* debido a que frecuentemente predice muchas menos líneas de las que han de esperarse por aplicación de una teoría más rigurosa. Por ejemplo, para el benceno 1,2,3-trisubstituido con un solo protón para rodeado de dos protones equivalentes, simplificadamente debería esperarse que tuviera un tríplete para & absorción del protón para y un doblete para los dos protones meia. De hecho, una aproximación más rigurosa da un máximo teórico d«- nueve líneas y ordinariamente se observan más de cinco.
U na interpretación, más exacta de los acoplamientos de spin-spín se encuentra en las referencias de las págt. 214-215.
Existe una situación que requiere una especial advertencia debido a que ocasiona complicaciones espectrales que pueden desorientar a quien esté descuidado. Esto se ilustra con el espectro de la dienoná siguiente. La región del espectro correspondiente a los protones del
del metileno del grupo bencilo podrían parecer a primera vista como
equivalentes y así originar una absorción única. Pero si se hace un dibujo de la molécula que muestre detalladamente su estereoquímica, se verá que estos dos protones no son equivalentes. Indudablemente, la substancia existe como una mezcla de isómeros conformacionates con varios grados de rotación del enL^e sencillo entre d grupo bencilo y el ?nilio dienona. El estudio cuidadoso de ellas muestra que en ninguna
Y
H
anillo aromático y los ghipos metilo es fácil de interpretar. Los protones
OCH3 II CM XjHj
CHa

conformación la posición de uno de lo* protones del bcncilo es equivalente a Ja del otro protón en cualquiera Je sus posiciones posibles. Porlo tanto, estos dos protones no deben ser equivalentes desde el punto de vísta de Jos átomos que los rodean. La absorción debida a estos dos protones es un cuadruplete; cada uno separa al otro en un doblete. En casos análogos es muy difícil decidir por adelantado ai la no equivalencia originará un desplazamiento químico lo suficientemente grande para hacer evidente su separación. En este ejemplo, el desplazamiento químico a 60 Me es de 25 c.p.s,
No sólo es valioso el conocimiento del número de señales que resultan del acoplamiento de spin-spin, sino también la ‘'constante de acoplamiento”, parámetro que está asociado con la distancia que los sepára (como la constante de acoplamiento es independiente del campo. se le reporta en ciclos por segundo en lugar de partes por millón). En el espectro del yoduro de etilo que se muestra en la figura 25, la constante de acoplamiento (representada por / ó A ) es aproximadamente de 7 c.p.jL, valor característico del grupo etilo en diversos compuestos.
En la tabla 14 se resumen algunos de los valores absolutos de las constantes de acoplamiento de protones unidos a átomos de carbono contiguos que forman parte de moléculas cuya rigidez relativa permite una estimación del ángulo diedro que se forma entre los protones. Puede verse que, tanto en los derivados del ciclohexano como en Jas definas, los protones trans (separados por el máximo ángulo diedro posible) generalmente tienen las constantes de acoplamiento mayores» Se ha observado una variación similar con respecto al ángulo y al acoplamiento de los protones gemínales, como se muestra en los datos de la tabla 14, De la teoría se ha derivado una relación entre el ángulo y la constante de acoplamiento.4* La teoría todavía requiere mayor refinamiento, sin embargo, se ha demostrado que los protones unidos al mismo átomo de carbono tienen constantes de acoplamiento de signo opuesto a las de los protones que están eo átomos de carbono contiguos,44 Las constantes Se acoplamiento de muchos, si no es que de la mayoría de los protones gemínales, probablemente son negativas,46 En Ja interpretación. de los espectros más sencillos el signo del acoplamiento no es de interés práctico, ya que no puede deducirse del espectro y tampoco afecta su aspecto.
2 2 4 U so d e lo* m é todo s o s p e d ro s c ó p ic o t
^ ^ « K í i p l u s , / . Ckrrx. n y t - , » , U (19WJ; Kan>lu* * Gcmt, ¡bid.t SJ, 1278
41 Ver Fitíiec, ¿Y». !■ Ckcm-, 43, I48J (1062), p»r* roeícot**,‘S M, KwtJux, / . A a . Cfcrm, S « c , 84, SUtt (1962)5 U r t tr W y Kurlrnd. ífr¡¿» « ,
SMS (!* « > ; Anrt, M i. . S4, 37É? (1962),

Espectros <U RMN 225
T abla 14
ACgunOS valorea absolutos de Las constantes de acoplamiento (A 6 ]■) de las protonel unidos & átomos de carbono contiguos.
/ , c.p,j. Anjuln <3¡«ir»
Derivados del ciclohexanoH hhj—H juui 8.12* ÉSO*
3-*5* 60Hidrógenos olefinicoi
trans 11-1Sf 130m 6-14f 0
Hidrógenos aromáticosorto 7-lOfrntta 2-31para H
Protones gcminaleu
Andalo ( H - C —H )—C H j— (tetraédneo) =C}í(okfía»ccO,
Í2-15 105.5°0-4 120
* Ver Im reícundi* mi tn piff*. 2H-ÍS y tawbvín Anct, J. Am . C ktm . Svc., 84, lt&4
t L. Mr JackoD3Ti, Rrfírvflcia A en J* p íj . 211, píg*. 93 y >¡5 1 .
Espectros de RM N y velocidades de rcacción
Determinados procesos experimentados por las substancias orgánicas comunes ocurren a tal velocidad que los hace especialmente apropiados para estudios mediante RM N. Reciprocamente, y esle es el punto de vista que nos interesa aquí, la apariencia del espectro puede verse afectada por estos procesos lo suficiente como para dar lugar a importantes deducciones estructurales. Los fenómenos de intercambio de protones y el equilibrio conformacional son dos de los más importantes^ de dichos procesos. Asi, en el espectro del etanol puro V seco, el protón del oxhidrilo aparece como un triplete debido a la subdivisión provocada por ios protones del grupo metileno contiguo. Análogamente, el espectro de los protones del metileno se complica por el acoplamiento con el protón del oxhidrilo.
Si se añade aunque sea una cantidad pequeña de ácido clorhídrico, la absorción del oxhidrilo origina una banda única y la del metileno, un cuadruplete de! tipo observado en el yoduro de etilo. La raión de esto es que el ácido clorhídrico (lo mismo que muchas otras substancias) actúa como catalizador para el intercambio de protones de una molécula de etanol a otra. En el intercalo de tiempo que es importante para las mediciones de RMN, cualquier protón se mueve tan rápidamente que

su campo magnético es simplemente el promedio de los campos de los muchos sitios que ocupa. Como cada molécuía tiene ei mismo medio
^ circundante atómico promedio, hay una sola posición para la absorcióndel protón del oxhidrilo.
Si á una solución de etanol se le añade una pequeña cantidad de ácido acético, no so presentan dos señales, una en la posición caracteris-i tica del ácido y otra en ia posición del etanol; en lugar de ellas hay una
sola señal ligeramente desplazada de La posición del etanol original hada la posición del ácido acético puro. A medida que se va añadiendo más ácido acético hay un desplazamiento gradual que continúa hasta un punto en el que la solución es casi ácido acético puro y )a absorción es esencialmente la del propio ácido acético. Por supuesto que la razón de este comportamiento es que cada protón pasa una fracción de itu e?ds- tenda unido a las moléculas de etanol y el resto unido a las moléculas de ácido acético. Las fracciones dependen de las cantidades relativas de
S p P ambas clases de moléculas presentes. Como > redistribución es muyrápida, el campo responsable de la posición de la absorción de RMN os confuso y su intensidad es un promedio que depende de la composi- ción de la mezcla.
Es evidente la posibilidad de usar este comportamiento para el análisis cuantitativo. También es de considerable valor en la determinación
t ^ de estructuras, ya que a menudo hay cierta indecisión acerca del lugar deaparición de la absorción del oxhidrilo en el espectro de un alcohol.
b La adición de una cantidad pequeña de áddo bencensulfónico, o algún otro ácido, origina un desplazamiento de la absorción del oxhidrilo que conduce a su rápida identificación. O tra apücadón posible, es la de desplazar la absorción del oxhidrilo para alejarla de algún otro grupo de señales que puedan estarla ocultando. ;
¡ | Los efectos conformacionales pueden desorientar bastante la interpretación de los espectros de RM N si no se sospecha su presenda. Así, a temperatura ambiente la dimetilformamida muestra como doblete la
i fc absordón debida a los grupos metilo; a temperatura ambiente o menor,1 " las dos señales son de igual intensidad y están separadas por unas
0.25 p.p.m, A temperaturas más elevadas ios picos del metilo se van j - v acercando y, finalmente, a I50°C forman un solo pico. Aquí, la explica-i f ción es que en el intervalo de tiempo de la RM N, la suposición usual
de los químicos orgánicos de que la rotación alrededor del enlace “sencillo”
n carbono-nílrógeno de la dimetilformamida es lo suficientemente rápida como para que los dos grupos metilo estén en medios circundantes atómi- | eos equivalentes, no es correcta a temperatura ambiente. A temperaturasmás elevadas aumenta la velocidad de rotadón y los medios circundantes atómicos de los metilos llegan a promediarse. La situación se complica
* aún más por el hecho de que la distancia entre las dos absordones demetilo, a temperatura ambiente, depende fuertemente del disolvente y
j 2 2 6 U so d» lo s m é to do s aspoc tro scép fco s
PiI
i

E spe c tro scop ia e n «1 u líro v to le ta 2 2 7
con el cambio apropiado de disolvente el desplazamiento quimirr» entre los grupea metilo puede llegar a cero o aun puede cambiar de signo.**
O tro ejemplo del efecto del equilibrio conformacional es el comportamiento del espectro del cidohexano a diferentes temperaturas.47 A tem peratura ambiente, el cidohexano está en rápido equilibrio entre Jas dos formas de silla, por lo que cualquier protón absorbe a un campo intermedio entre el de una posición axial y una ecuatorial. Sin embargo, al enfriarse la absorción se amplía y cuando la muestra se ha enfriado a - J 0 0 ° C el espectro del protón aparece corno dos señales complejas resueltas parcialmente, una correspondiente a los protones axiales y otra a los ecuatoriales. Puede anticiparse que con substituyeme* en el anillo del cidohexano, la ‘'congeladón” de la molécula en una sola posición puede ocurrir a temperatura ambiente o mayor. Asi, muchos anillos de cidohexano substituido están “congelados” en una sola conformación de silla o se iuterconvierten tan lentamente que sus protones axiales y ecuatoriales muestran absordones separadas.
Absorción de protones unidos al nitrógeno
El nitrógeno'14 tiene dos características que ocasionan complica- dones adicionales al interpretar los espectros con grupas N —H. El núdeo del N u, a diferenda del .carbono, tiene un momento cuadripolar eléctrico, el cual es de esperarse que amplíe la señal de absorción de los protones unidos a él. Además, a diferenda del H 1, <jue tiene un spin de Yi, o como el Cti o el O14 con spínes de 0, el N1* tiene un spin de I, lo cual significa que hay tres orientadones posibles para el spin nuclear (con una constante de acoplamiento nitrógeno-hidrógeno de unos 50 c.p.s.). Por ejemplo, el amoníaco muy puro muestra el espectro de protones como tres «fíales bastante amplias separadas por 50 c.p.s. Las aminas en solución fuertemente ácida también muestran estos efectos del núcleo del nitrógeno debido a que bajo estas condidones el intercambio de protones es muy lento. Sin embargo, en la forma en que normalmente se examinan, muchas aminas experimentan cambio de protones lo sufí- dentem ente rápido como para 'que ios protones unidos al nitrógeno den una absorción bastante ddinida. L a absorción del N —H de las amidas es, igualmente, ancha y puede llegar a ser tan ancha que llegue a ser difícil o imposible localizar en los espectros determinados en condicio^ nes ordinarias.
ESPECTROSCOPIA LN EL ULTRAVIOLETA
Aunque una discusión amplia de la espectroscopia en el ultravioleta está, más allá de los límites de este libro, se intentara indicar brevemente
* H ttt tio y RJclunl*. Mci. P h p ., S , 139 (2992}.* Jemco, Ntfyci, Sedertiolm y Eortc, / . A m . Ck*m. 5 * t., 385 (I9Í21.

2 2 $ (Jko é% lo s m étodo s espectros c ó p k o s
los tipos de problemas estructurales que se espera pueda resolver. Se recocdendan las siguientes referencias generales y deberán ser consultadas por cualquier persona interesada en usar el método para la de-
.terminación de estructuras,
A ) J a i f é y O rchin, Thttory a n d A p p lic a iio n s o f U i tr a c iu l t i S p e c t io tc o p y , JoKtt W üey and Soru, Nueva York, 1962.
Giltam y Stcrn, A n ín tr o d u c t io n te E le c tr o n ic A b s o r fi í io n S p t c t r o x o p y ín O rg a n ic C h tm i í i r y , 2* ed., Edward Aroold, Ltd., London, 1957,
Estos libro? proporcionan la¿ más amplias fuentes de inform ación de utilidad general p 3 ra el químico orgánico.
B ) Braude, D e tsrm in a tityn o ¡ O rg a n ie S t r v c t u n s b y P k y s k a i M e th td s , editado p o r Braude y N ichod, Academia Press, N ueva York, 1955, capítulo 4.
C) Majen, Quart, Rsot., 15t 287 (1961),Strritwicfccr, M o h e c í Orbital Tktory for Organie Chtnmts, John
Wxtey and Sons, Nueva Y o it , 1961,
. Eita» dos revisiones criticas proporcionan una buena introducción a ]& leoría de Ja aborción de la luí visible y ultravioleta,
t>) Hcrshemon, VltrevioU t and Visible Absorpticm, Aeademic Pres», Nueva York, 1956. Esta obra proporciona una guía relativam ente «endite, de lo* espectros publicados entre 1930 y 1954.
■ • E) Fiiedel y Orchin, U U ravioU t S p « t r a o f A r o r n t í ie C o m p o u n d i , John Yfíley and Sonsa Nueva Y ori, 1951. E íte libro es principalmente una colección de espectros, como lo indica su titulo, aunque en las página» iniciales también hay ais© de discusión de técnicas y principios generales.
Por razones prácticas, la región ultravioleta se divide ordinariamente en la ultravioleta “cercana.” o de “cuarzo” (región en la cual el aire y el cuarzo son transparentes), que incluye la región de 200-750 ray* (2000- 7500 A ), y el ultra violeta ‘‘lejano” o de “vacío*. Los espectrofotómctros Cary y Beckmxtnn determinan convenientemente los espectros en la región cercana, aunque las determinaciones a longitudes de onda de 200- 220 jnp. sólo son de fiar bajo condiciones especiales. Se dispone de instrumentos que pueden utilizarse hasta 175 m ji. El ultravioleta lejano requiere técnicas especiales, ya que el aire que hay en el trayecto de la luz absorbe intensamente en esta región y no son fácilmente asequibles a los químicos orgánicos.
El espectro en el ultravioleta, a diferencia deí espectro en c! infra-• rrojo o ¿el Raman, se origina por excitación electrónica de la molécula co.i la luz irradiante. La consecuencia importante de este hecho, desde el punto de vista del químico interesado en estructuras, es que los espectros en la región ultravioleta señalan la presencia de insatur aciones en la molécula absorbente. Esto se debe a que, con pocas excepciones, sólo las moléculas que contienen enlaces múltiples tienen suficientes estados excitados estables para originar absorciones en el ultravioleta cercano.

E spec tro scop ia en «1 u lt ra v io le ta 229
Así, las hidrocarburos saturados, los alcoholes y los éteres son tianspa’ rentes en esta región. Además, las oí afinas mottofuncionales» Jos acetilenos, los ácidos carboxí líeos, 'los esteres* las anudas y las eximas tienen máximos de absorción justo fuera de la región cercana (a unas 200 mjx) y, en general, sólo muestran el fmal de la absorción.
U na tercera clase de compuestos que comprende Jos aldehidos, ce- tonas, compuestos nitrados alifáticos y los ésteres de nitrato 3c caracteriza por máximos de absorción en el ultravioleta cercano, pero éstos son de intensidades tan pequeñas que sólo son útiles bajo circunstancias especiales. Por cjempio, la acetona tiene un coeficiente de extinción molecular {s, medida de la intensidad de absorción) de 16 en su máximo a 270 mfi. Con fines de comparación, una banda moderadamente fuerte tiene un € de 1000-10,000 y no son raros los valores inri elevados como 100,000. A pesar de ello, la espectroscopia ultravioleta en algunas oca-
T a b l a , 15
. Grvpv {uocUmo) Xatc my> t
R C H = C H R 185 8,000R ¿ ¡ = 0 270 1 6
R.COOH 204 41R.COC1 235 5 3R N O , 275 J2R S 210 1,200R jC ^ s N N H C O N H j m 10,&00R N = N R 35 0 140
R N = 0 (300(6 7 0
10020
siones ha sido muy útil para estudiar dichos grupos funcionales con absorción débil. En la tabla 15 se tiene una lista de algunos datos representativos que dan las posiciones de los máximos de absorción y los coeficientes de extinción molecular de algunos grupos funcionales aislados (no conjugados).
Puede verse que si el uso de la espectroscopia en el ultravioleta estuviera restringido a moléculas rnonofuncionaíes, sería de utilidad muy limitada. Sin embargo, afortunadamente, aun grupos funcionales tales corno los olefímcos. acctilcuicos y carboxtlicos (y, en general, cualquier función que contenga un enlace doble o triple) originan uria fuerte absorción en el ultravioleta cercano cuando están conjugados entre sí. Así, la espectroscopia en e! ultravioleta ha resultado ser principalmente un arma en el estudio de sistemas conjugados. De estos comentarios resulla obvio que el uso de los espectros en el ultravioleta en determinaciones de estructuras, es algo diferente ¿I de las técnicas en el infrarrojo; de hecho, a menudo los dos métodos se complementan.

2 3 0 U so d e lo s m é to do s e s p e c t ro s c o p io s
Los modernos espectro fotómetros de ultravioleta de doble haz, generalmente trazan gráficas de la absorbanciú (densidad óptica) contra la longitud de onda en milimicras. Ya que Ja absorbancia (A ) de una solución tiene un significado cuantitativo en la espectroscopia en el ultravioleta y como las soluciones empleadas son lo .suficientemente diluidas como para que se cumpla la. ley de Becr, casi siempre es conveniente convertir la absorbancia en coeficiente de extinción molecular (c) en cualquier curva que se vaya, a publicar, Estas cantidades bc relacionan entre sí por medio de la ecuación siguiente:
£ = Á /bC
en donde £> • longitud de Ja celda en centímetros fú silm en te 1 cm} y (■ = Concentración en moles por litro. Se lia visto que z es una medida de la absorbencia de una solución a un;i concentración de I mol pur litro en una celda de 1 cm, p o r lo que Jo* valores de f en espectros diferentes son comparables aunque se hubit. .in usado concentraciones tm p o c o distintas para iraz a r h * dos curvas.
Los disolventes m is usados en la. espectroscopia en el ultravioleta son: cidohexano, etanol al §b%, etanol absoluto que no contenga benceno y dioxano. La concentración aproximada que se debe usar puede estimarse, si se dispone de alguna información sobre el valor esperado para c) máximo valor de e, A sí, Ja escala de absorbancia de Cary va de 0.0 a 2.4 unidades de absorbancia y para una curva en Ja que un máximo con £ = 10* tenga una absorbancia de 2.0? se ha visto que la concentración correcta es 2/JÜ1 ó 2 X JO-* molar cuando se utiliza una celda de 1 cm.
Identificación de grupos funcionales partiendo del conocU m iento de la» posiciones de lo$ m á xim os d e absorción. Este uso de la espectroscopia, que es tina técnica común en el infrarrojo, es rara en el ultravioleta por dos razones. Primero, Ja mayoría de los grupos funcionales sencillos absorben débilmente o no absorben y, segundo, Jos espectros de la mayoría de Jas moléculas son relativamente sencillos, es decir, sólo tienen uno o dos máximos en lugar de los die2 o veinte que son usuales en las curvas en el infrarrojo. Por Jo tanto, necesariamente muchos grupos funcionales absorben en la misma región. A pesar de ello, el examen del espectro en el ultravioleta por un observador con experiencia ocasionalmente ha revelado la presencia de un grupo funcional que previamente no se habia sospechado. Estos exámenes, por lo general, se Jiacen para investigar la clase de anillos aromáticos o hetero- cíclicos que están presentes en un producto natural de estructura des- conodda. Por ejemplo, la presencia de un grupo nitro fenilo en la cloromicetina fue propuesta por Doub y Vandenbelt basándose en el espectro en el ultravioleta,48 Sin embargo, por lo general se requiere
** V<r RcbiLock, CrooVi, Ceatreuli* j Surtí, / . Am, Cftim . íc f - , 7X, 24íé <1949}.

/
alguna, información previa acerca de loa posibles grupos funcionales para que el espectro en el ultravioleta sea útil. Por supuesto que la ausencia de los grupos funcionales que se sabe absorben en el ultravioleta puede a menudo inferirse sin Jugar a duda.
Uso <íe com puestos m odelo . Aunque a primera vista podría parecer una desventaja que únicamente grupos funcionales muy específicos originen una absorción en el ultravioleta y que el espectrofotómetro sea ciego a los cambios que no afecten la unidad absorbente, estas limitaciones son» en gran parte, responsables de la utilidad de la espectroscopia en el ultravioleta en la investigación de estructuras, porque) aunque casi nunca es posible predecir sobre bases teóricas el espectro de una rao- leeula, con frecuencia puede obtenerse el espectro de un "modelo4*, es decir, el <le una substancia sencilla que difiera de Ja substancia des- conocida en tal forma, que no se afecte la unidad absorbente.
U n ejemplo de dicho uso de Ja espectroscopia en eJ ultravioleta lo constituye el problema estructural presentado por el fS-aminocrotonato de etilo. Se sabe que esta substancia es uno u otro de Jos tautómeros IA, IB, Ya que IA tiene dos cromóforos aislados, es de esperarse que
NH O MHj O!l II I «
CHjC ~ C H jC O aH j CKjC = CHCOCáHsi a ra
sólo muestre una pequeña absorción en 3a región del ultravioleta cercano. Para conocer el espectro aproximado de Ja estructura de la vinilamina (IB) se preparó el (J-diinetilaminocrotonato de etilo I I y se determinó su espectro. Como se puede ver en la figura 26, el espectro de la substancia desconocida concuerda muy bien con el del modelo de vinilarrúna, y así
(CHj)jN O1 H.. CHjC= CHGOC2H5
n
se confirmó la estructura IB/* También puede verse en la figura 26 que el espectro del (^-metilaminocjotonato de etilo es análogo al de las am inas prim aria y terciaria afines.
U n segundo ejemplo del uso de substancias modelo para tener una idea de los probables espectros en el ultravioleta, lo proporciona la demostración de la estructura de los dicianuros de quinoleína.31 La quino- lcína reacciona con el bromuro de cianógeno y el ácido cianhídrico formando dos dicianuros que se creía tenían las estructuras I I I y IV .
« OíídcnajEt. y Coc*. / . Am. Chsnt. 4<X.> f7 . 10! 7 <19+5).* Setter. Y t i a f ÑolJer, J. A m . CK*m* So*., 73, T72 (1951).
Uso <f« com puestos m edafa 231

J 3 2 U so d« lo> m é to do s e $ p e d ro ic ó p íc o »
Loflgiiurf de Dndi. A
F ijara 26. Curva 1, «ip«clro da obiorcMn d»l rf« «tíku corvo 2.^m tH lpnlm avlQ iK ito d* «l)k>; curra 3 . p-dlm«Mcimlnocrol«nfl1o d a «llk>. Tcnám lm eM innfciaclanw *on en □ (la h n c ra , {t«pn>ducídc dt< Jc-vrnoE o f the Amalean Cbemlcal
Sociary por cortttlú cfal pr^faior Arthut C. Cop* y <f* la So<Jednd.|
Se necesitaba, confirmar ias estructuras y, además, aclarar cuál de los d¡cianuros de quinoleina era I I I y cuál era IV . Como no se disponía de ningún método de síntesis o de degradación que no fuera ambiguo,
« H
n CH,
^ - n - c h ,
CNVI VW
fue necesario recurrir al estudio de los espectros en el ultravioleta. Como substancias r¿-x3elo para el di cianuro de quinoleina I I I se escogieron la fenilciauamida (V) y la N-rnetilfenilcianamida (V I) , En la figura 27

Principio d« odttivldad . 233
se puede ver que los contornos generales de las curvas, las posiciones y las alturas de ios raá-'inx* son bastante concordantes. El dicianuro de quinoleina I I I tiene una unidad de nítrüo a^ in sa tu rad o que no aparece en las substancias modelo. Este grupo pudo ignorarse debido a que su absorción es mucho más pequeña que la de la otra unidad absorbente.
Corno modelo para el otro dicianuro de quinoleina (IV) se escogió la W-metü-o-estirilciariamida (V II) . Los espectros de IV y V II se muestran juntos en la figura 23 y puede verse que concuerdan bastante bien entre sí y son diferentes de los espectros de Ja figura 27. Así, los espectros en el ultravioleta de los dicianuros de quinoleina proporcionan utia fuerte evidencia de que tienen las estructuras indicadas.
Figuro 17 . E ip « c fr« t d« o b t o r d ó n de: 1} tt¡<lgnvrv d« qulncUtaa d e b a ^ punió d* fuilén, 2j fraltácuvamída, 31 N-«*Hlíwi1lcípnpnifrfa. ^ p ro d u c id a del J quitioI «{ tha Amciican Chemical $txi«ty per eortwío dft ki So<í«dpd y d«l protaior CoH I . Naltor.)
P rincip io de uditiuicUid. El aspecto de la espectroscopia en el ultravioleta que le da su mayor flexibilidad en los problemas estructu-
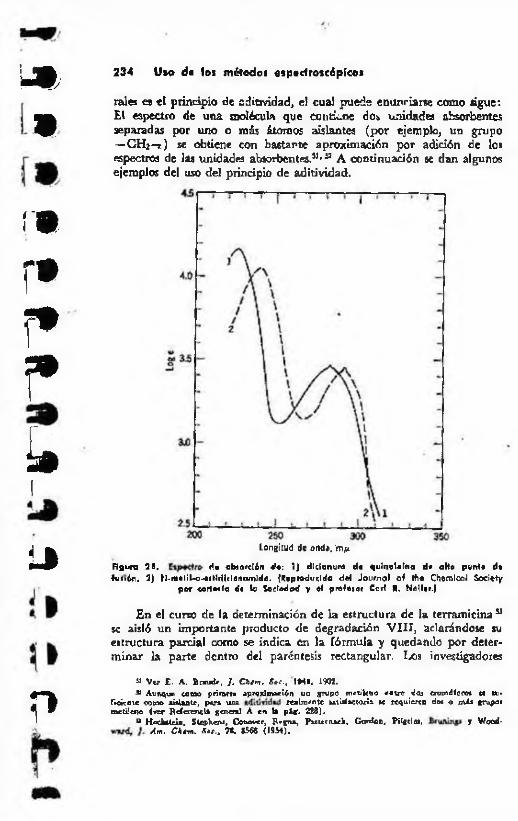
\M
9
m
r »
Pf
Ui
U
r>
rales e$ «I principio de cdittvidad, el cual puede enunriarse como sigue*. El espectro de una molécula que coutLoe do* unidades absorbentes separadas por uno o más ¿tomos aislantes (por ejemplo, un grupo - C H í - t.) se obtiene con bastante aproximación por adición de los espectros de las unidades absorbentes.55'*2 A continuación se dan algunos ejemplos del uso del principio de aditividad.
2 3 4 U w d i Io í m é fodo s e sp« cfro se ¿p íco*
Longitud de 6itd»,'mp
fifluro 29. H.» abiardón <í*: 1) dtrfanura d« qulnoUrtia d» alto pvní® d»foj»¿n, 2) t-J-itttlíUoHUtMIclancunJda. {Kfproduttdo dM Joufnol o í th« ChomlceJ Soci^ty
pet «ortti(« ¿ i la Seefodorf y «I profttot CüH I . NalUr.|
£ n el curso de la determinación de la estructura de la terramicina 53 se aisló un importante producto de degradación V III , aclarándose su estructura parcial como se índica en la fórmula y quedando por determinar la parte dentro del paréntesis rectangular. Ixis investigadores
M V « E. A. Bcrofc, b C hfhT, Soc., I « » , 1902.** Aunque coDtt pñriHi* iprcjdmaciír* üo arwpo metilotiiJ entre daj CrronáíOfOi c* ttJ*-
ííeicDle cotüo jüíUtttc, P*íl una «atinente tatirf»csoria K K<iuiej<L de* o míi crupaloeüleno (ver RdTeraicLt gowmt A tu b píy, 228).
O Hociultla, Sitiaban, CcinAtr, Ríjiul, Puteriu^k, Cardón, PiCgílúli j V w ¿ -Am. C htm . S e t., 7t* S 5 « <J9M).

Efeefoi de fo geometría molecular 235
sospecharon que el resto [C*Hj(OCHa}j] era uno de Jos trimetoxibea- cenos. Como se ve, los do? cromóforos, el sistema de!, naftaleno y el
CH3
V V C W i O HCH^O OCHa
viusistema del benceno, están aislados (no conjugados) por el — C H O H — y, por lo tanto, el espectro de la molécula puede obtenerse aproximadamente sumando los espectros de las dos unidades. Consecuentemente, Jos autores sumaron el espectro de la substancia IX , del cual disponían, respectivamente con el del 1,2,3, el del 1,2,4 y el del 1,3^-trimetoxiben- ceoo. Sólo i>ara el derivado 1,2,4 la curva de adición coincidió razonablemente bien con el espectro de V III .
CH,H*OH
IX
MacKenzie,* en un estudio de la estructura dsl ácido úsnico, examinó el espectro de los productos de degradación X y XI- Observó que si las estructuras propuestas son correctas, la estructura X tiene el crotnófora, X I, unido al cromóforo aislado de la acetilacetona (forma enólica) (X II) . Por lo tanto, substrajo el espectro de X I del X y
OH ^ OH
X T Í 5CH3CO °
y xiO
. IICHaC—CHCCHj
IOH
xir
obtuvo una curva que esencialmente se podía sobreponer a la de la acetilacetona. Asi se confirmaron las estructuras propuestas.
Efecto* de la geom etría m olecu lar ¿obre los espectro* d e absorción en e l ultravio leta . Una dificultad que se encuentra siempre
« MacICíbcdc, / . Am . C W io r ., 74, 405?

presente al seleccionar compuestos modelo para comparar sus espectros de absorción con los espectro» de substancias desconocidas es Ja posibilidad de cambios inesperados en el espectro a causa de efectos estcricos específicos en uno u otro de 3os compuestos que van a compararse. El origen más usual de dichas dificultades es Ja inhibición estéríca de la resonancia. Por ejemplo, el espectro del dífenilo no corresponde a la suma de los espectros de dos anillos bencénicos. Esto es debido a que Ja interacción de Ja resonancia de los dos anillos bencénicos conjugados del difeaOo estabiliza ios estados excitados que resultan de la absorción de luz. Por otra parte, se ha demostrado que tal interacción de resonancia requiere que los dos anillos del difenilo sean aproximadamente copla- nares. El dimesitilo (X III) , que tiene un grupo metilo en cada una de las cuatro posiciones arto, es incapaz de adoptar la conformación en la que los dos anillos bencénicos sean coplanares, Por lo tanto, se inhibe
¿ 3 6 U so d * lo s m é todo s e sp é tfro s có p U o s
la interacción de resonancia y, en consecuencia, el espectro del dimesi- tilo es muy similar a la suma de los espectros de dos anillos aislados de niesitjleno y diferente de! que posee el difenilo.55
U n examen de Jos espectros de los nitrobencenos ¿rtg-substituidos ha mostrado que, a medida que aumenta el tamaño del substituyeme orto y el grupo nitro es forzado más y más fuera del plano del anillo del benceno, el espectro cambia gradualmente; finalmente, el máximo a 265 mfi, que tiene un r de rnás de 10,000 en los ¿-/-alquilnitrobencenos, casi desaparece totalmente en el o-nitro-í-butübenceno*
En Jos estudios sobre el efecto de los substituyeles orto en los espectros de los derivados de las anilinas se ha observado una análoga Inhibición estéríca de la resonancia.57 La benzoquinuclidina (X IV ) proporciona 'un ejemplo extremo en el que el átomo de nitrógeno está colocado en un es-
xiv
* PicIccU, W altw r J . A m . Chtm. S o t . , I» , Z2% ( t 'J X ); O'ShMWl.ncny yR odcbujfc, 8 2 290$ (l!M 0).
» B co*n 7 R iig a n , / . A m . C h tm . Ü et., « . m ' i (134?). n R cra insion , / , Am. C ktm . Scc„ €7, ie38

C o rre lo c io n e s m á* p e r fe c c io n a d a s 237
queleto rígido de carbono a un ángulo que inhibe efectivamente la interacción de resonancia entre el grupo amino y el anillo aromática Como es de esperar, el espectro ultravioleta de la bcnzoquinuclidina se pareoc mucho más al del benceno que al de la dimetilanilina5*
Además de Ja inhibición estérica de la resonancia, los espectros en el ultravioleta pueden cambiar notablemente por factores estéricos que alteren mutuamente las posiciones de los dipolos. Posteriormente se citará un ejemplo en la discusión (pág. 239) del efecto de la posiúón del átomo de cloro, en b s <r-clorocrtonas sobre la posición del espectro en el ultravioleta de la función carbonilo. Un estudio de las c-dicetonas M ha mostrado que sus espectros se ven influidos drásticameute por la orientación de tos grupos carbonilo, uno con respecto al otro. Entonces, por lo general deberá tenerse cuidado en la selección de las substanc ias modelo que se usen en la espectroscopia en el ultravioleta.
Otro efecto que en gran parte depende de la geometría molecular es la interacción de los c*omóforos no conjugados que están situados convenientemente en el espurio. Por ejemplo, la dicetoolefina siguiente
presenta un máximo de absorción a 223 mjji { lag t = 3.36), así como máximos de absorción a 296 y 307 m^i (log t = 2.41). El espectro en el infrarrojo muestra que el compuesto no está cnolkado y Ja absorción inusitadamente intensa lia sido atribuida a la interacción transanular del enlace doble olefínico con los grupos carbonilo.*1
Se han revisado críticamente e interpretado otras interacciones de los cromóforos no conjugados.61
Correlaciones m ás perfeccionadas <U la estructura con los espectros en e l ultravioleta. Se han realizado numerosos estudios dei efecto sobre el espectro en el ultravioleta cuando se aumenta el número de enlaces dobles o triples en sistemas del tipo X — (C H = C H )„ —Y y X — (C )„—Y. Como estos han sido resumidos por Cillam y Sterr,*2 no se discutirán más aquí.
Para el químico que investiga productos naturales la correlación cuantitativa más importante de los espectros es la de Woodward,M quien
» W cp tíír, R t f . rnr<-. C*í|ti. 71. tlM> (1 S « ) .v‘ Lcuiuird y M*dcr, J. Am. Ch*m. Soe.► 72, S38fl*> C w b v U V ilt, i t t x v - Cfctm . Ac<q, 4 3 , J3W« Wilcox, Wioiteln y / . Am. Chrtn. Soc., « , (1950).** Ver Rrífrtfltift geatnf A fti la 22».♦1 V er F iitc r y F íe x r , SteroUIi, RciirttoM l’otiliibíivr , K ncvi Y ork, 13W. p iy i 15
7 ág?. o o ia lq u ie ra de b t lYÍcrfHeioi jne»iiio«u*lfl» r» U pA*. 228.

*
! •
r *
1 *
f
formuló una regla empírica para determinar la posición de los máximos de absorción d«*. los dienos conjugados y de ias tetonas a,íS-insaíuradas¡. En la discusión precedente hemos supuesto que la substitución de los átomos dé hidrógeno, aun de aquéllos unidos directamente a un cromó- foro, por grupos alquilo, deja inalterado el espectro; sin embargo, esto es derto sólo como primera aproximación. En el sistema XV, el máximo de absorción del compuesto fundamental sin substituyenos, etanol como disolvente, puede tomarse como 215 Se pueden predecir los máximos de una extensa'Variedad de compuestos afines con subsdtuyentcs
23 3 U so d e io s m é todo s e sp o c tto scóp ie o s
P A J - C = C - C = 0
¡ 1 k xv
alquilo a y 3 usando las generalizaciones de qu^ por cada substituyeme-* se deben añadir 10 triji al valor de 215 mjXj por cada substituyeme-^ se añaden 12 y por cada anillo al que sea exociclico el enlace doble carbono-carbono se añaden 15 mp. El ejemplo siguiente ilustra )a aplicación del método.
Problema
Prediga Ja posición del máximo en <3 ultravioleta de la lubstancía ¿¡guíente feo solución e tañó lica):
« O
r T t r ^c u ,
Un eolaca doble exociclico está unido a un Átomo de carbono de vn anillo como 6c muestra ea la fórmula a 3a izquierda. El calacc doble del segundo
ejemplo es exocíclíco a l anillo A pero «ndocidico a3 anillo B. No hay rubitjtu- yentes en el átom o de carbono-tz pero hay do* en et p, por lo que deben añadirte 24 m u (2 X 12) al valor b a « d e 215 r>n*. Además, «3 enlace doble e& exocíclica a l anillo A p o r lo que debe añadirse un total de 5 + 24 = 29 m u a las 215 mji para dar el valor de 244 n^i que Je predijo íería la posición del máximo.

Compuestos earijonfllco* 239
La regla para los dienos conjugados es similar. Para detalle: deberán consultarse Ia3 referencias citadas. En vísta de la simplicidad del método, es notable su extensa concordancia con Jos datos experimentales, habiendo sido causa de Ja revisión de las estructuras incorrectas de varios productos naturales.
U na segunda correlación importante para la química aromática, es Ja de Douh y Vandenbelt,64 quienes encontraron que el efecto de los. substituyentes sobre el espectro del benceno se podía correlacionar con las constantes de Hammet para Jos suí*tituyentes.
En el libro de Gillam y Stern, aJ que se hizo referencia en Ja pág. 228, se discuten otros estudios sistemáticos sobre la variación del espectro con el cambio estructural en tipos de compuestos como los polie- nos y acetilenos conjugados, Jas scraícarbazoms, las 2,4-dinitrofenilhi- drazonas y las oximas-conjugadas o no conjugadas. Se ha reportadow adicionalmente un amplio estudio sobre las 2,4-dinitrofeniJh ¡drazonas.
A bsorción de com puestos m ono funcionales. Algunos grupos, tales como el azo y el nitroso, tienen una intensa absorción en el ultravioleta cercano. Sin embargo, los máximos de absorción de otros compuestos, tales como aldehidos, cetonas y aminas, en la región del ultravioleta cercano, son de muy baja intensidad. El peligro inherente al uso de dichas bandas se pone en evidencia cuando se considera que a una concentración de 0.1% una impureza con una z de 15,000 originaría un máximo de absorción con la misma intensidad que una cetona pura, por ejemplo con una £ de 15. A pesar de ello, se ha obtenido información valiosa de tales espectros.
Va que la región del ultravioleta accesible al químico orgánico, J>a sido ampliada hasta incluir la absorción debida a muchos grupos funcionales aislados, aquí el tema se revisa brevemente,
O lefina» n o con fugad tu . Aunque el principal máximo de absorción de las definas no conjugadas está en el ultravioleta al vacío (160- 200 my,)j el número , de substituyentes alquilo sobre los átomos de carbono del enlace doble puede ser coirelacionado con el espectro; u tales correlaciones son valiosas, ya que esta región generalmente es más accesible al químico orgánico® Además, ha sido posible vsar el final de la absorción en el ultravioleta cercano (205-225 roy.) para determinar el grado de substitución de los enlaces dobles carbono-carbono.61
C om puestos carboníllcoa. L a débil absorción del carbonilo a 260-300 nm bien podría ser, en determinados casos, complemento útil del espectro en el infrarrojo, ya que puede usarse para distinguir entre cetonas o akk-nídos y esteres. Por ejemplo, tanto las cetonas cíclicas de
W D m * y V a n d tn fe tk , J . A «u C k t m . S < x . « . 2714 <1947); 71, 2*14 (1X9).m Johnson, / , Am. CJupf*. 5ee„ T t, 2720 (ÍS53),* V e Scdkmw, Hamm) y C u t, / . Chtnt. P Íy t, tZ, 538 (1954); o T urnc r. / . Cktm.
■S*t.j 30 (1»S), ifiicuiMn y rrfcrttidí»« Mkhrtí y Appkwhiit, Ort . Cke,o.t 77 , 5*5 (1362).** Bludan, Ha»Ví! y Wcxxi, / . O u m . $*<., IM2. 2737.

2 4 0 Uso de los métodos espec troscop ios
c'uko miembros como los &cercs alifáticos absorben en el infrarrojo a 1740 cm '1, mientras , que sólo Jas primeras tienen una apreciable absorción en el ultravioleta a más de 210 mjx. Se ha encontrado w urv uso adicional para eJ máximo del carbonilo: en una ciclohexanona substituida su posición se desplaza por la presencia de un átomo-a de cloio o bromo y la magnitud del desplazamiento depende de si el átomo de halógeno es ecuatorial o axial. Se han observado desplazamientos similares en los espectros de las a-hidroxi y ¿t-acetoxicetonas.,'> Por lo tanto, Ja técnica complementa al infrarrojo corno medio para asignar configuraciones a dichas cetonas substituidas.
A m inas. El examen de los espectros de aminas sencillas en e! ultravioleta a} vacio7* indica que ia extensión de la región práctica puede ser un instrumento valioso para distinguir entre aminas alifátícas primarias, secundarias y terciarias. Ya que las aminas terciarias saturadas tienen un máximo de absorción justo dentro de la región del ultravioleta cercano (aproximadamente a 214 m ^ ), el cual se desplaza a mayony longitudes de onda en las vinílaminas con estructura RjN —C H = C H R (228-238 m tt), la espectroscopia en el ultravioleta ha demostrado ser valiosa para diferenciar este grupo funcional del de las aminas saturadas o el de las aminas no conjugadas, en las cuales la amina y el enlace doble olefínico están Aislados.7”'
C ru p o í funcionales conten iendo a zu fre . Los mcrcaptanos, tío- éteres y di- y polisulíuros tienen espectros característicos en la región del ultravioleta cercano.71 Las sulfonas y los sulfóxidos también tienen una absorción característica,*
Ejercicios
Si el lector n o está familiarizado con la interpretación d e la información estructural a p a r tir de fám ulas moleculares, debe referirse para este punto al capitulo 12. En cada uno d e los ejemplos « c rib a una estructura (o estructuras) concordante Con les hecho* presentado*.
I . El espectro en el infrarrojo de la p % 241 es el de un compuesto (en solución al 2 % en tetracloruro de carbono) con fórmula molecular C »H *0.
II . El espectro de RMN de la pS$. 242 es el de una substancia con fórmula CtH»0.
I I I . El espectro do RMN de ia páx. 243 es el d e un» substancia con fórmulaCdffcO*.
IV, El espectro de la pág. 244 es el de un compuesto neutro con fórmula C.H,>NO.
C & cban, / . C li m . l$$4, 292.74 Coofaoa y DuKtcgAMkcr, / , Cker*. 1WÍ, Si?,n T«nne*t>»uro, CuUin y Harrium, / . C4*m. Pky¿.t 21, 311 (1253).™ L coaanl 7 l*>cke, J. - In . O t»ra. $o<-„ 77, 437 (líf tS ),n F c W r r . n u cí, J. Am. Ck*n*. So*., 71, M (14*9); Hmt y Carm*fc, W 4 ., 11,
1215 (1 * 9 ).H ítiH -t 1 C m K t , / . Am. V k tm . Soc.t 71, 232 <tW9); 7J, 1242 11950}.

E¡eic¡do& 241
J
i ti
i
í

U so d o io s m étodos « p « e fro * c ¿ p íc o j E je rc id o s 2 4 3
3 [ 1s 1
Í3
1
?
S8
’
« s s
<
8 u
Ctp
v'lro
d*
*M
N po
ro
•! *{*
rc4H
i>
llf,
p6
g.
240.
{P
ropi
edad
O
UraH
a d»
Vc
Korv
A
íirxJ
pi*»
y
npro
duilt
íe
con
u»
p*rm
Ho.
)

244 U to <í« los m é todo s e s p e c t ro s c o p io sii
"2 |í>
1 "T* 1 ‘ ¡ I -o o -
[
si
5
■8o5 S R 1
* f 1í
>1
j .
1 i >¡ 1i
§ 3 8 * B ;- ■
i i 1l
- 4
§8 s s *
: /
8 § 8 s s \
s
Effw
fre
d« RM
N po
ra »!
ÍV,
p¿g.
5W
, (P
ropl
ídod
lit
erar
ia
do V
aria
d A
jwtla
m
y líp
icif
utilí
í »n
1W
p«rr
r1ja
.J

£*pr
<trg
de
RMN
pata
•í •[■
mid
o V,
pó
g,
247,
[p
ropi
ciad
1j)
«ro<
f«r
d* V
arón
A*
*gcr
ox>t
y
r*pr
ocíi/
cfdo
con
**»
P*
rmJi
0.J .

5
r »
f #
246 Uso do los métodos «spodroscéplcos
i® f * [ 0 h©
= • u *
í * »
»
§ *€»
p«<t
ro
*n «1
Infr
arro
jo
paro
al
«]er
ifcTo
V!
, p£
{|.
14
?.

V. El espectro d e U p ig . 245 e» «I de un» * ubi u n cía neutra, C*H»*0. Las peque¿í«i ta n d a s a 1.15 j 1.27 p.p.m . se deben a impureza».
V r. E l espectro d e la ptg.«24<S es e l de una substancia con fórmula C*H»Oj. El tratam iento con cloruro de deuterío seco produce el intercambio de aólo do» átomos da hidrógeno por deuterío.
V II. U n com pueíto Con un máximo en el ultravioleta a 216 (e 8300) se redu jo catalíticam ente a CHiCH O H G K (CH >)t
V IH , U a hidrocarburo ( I ) , CiHu, 242 nq j, por oronólisií d a formaldeMdo adem ás de otros producto*. Reacciona con /tf-bromosuccinimida (1 mol) pa ra d a r un bromuro, el cual, por calentamiento, d a l í , G»Hu, que tiene absorción en el infrarrojo a 1500 j 1600 cm-1.
IX. ¿Cómo distinguiría usted los compíteseos siguientes u c o d o lo» métodos «spoccroscópkos en tantas formas coroo sea posible? (Presente una discusión breve sobre las diferencias entre los espectro» que uited espera para cada uno de los métodos escogidos).
í . CHjCHzCH= C H —C át= C H C H jCH^
y OH >CH jCH ~CH O H iCH CH CH*.
Ejercicios 247


CAPITULO
1 0
Preparación de derivados
U k a v e z q u e S £ e s t a b l e c e l a naturaleza de uno o más de los grupa6 funcionales presentís en la substancia desconocida y se ha preparado una lista de cor*puestos posibles, es necesario eliminar todas las posibilidades menos uu.-'; para ello se obtienen constantes físicas adicionales de la substancia desconocida, las cuales pueden compararse con las de las posibilidades. Las constantes físicas empleadas tradícionalmente para este propósito sido los puntos de fusión de las substancias sólidas derivadas de la substancia desconocida por alguna reacción química. Sin embargo, cuando no se puede preparar ningún derivado deben usaric- otras constantes, tales tomo rl índice de refracción y la densidad. El arrastre de los ácidos con vapor {constantes de Üuclau*, pág. 261} y los coeficientes de partición éter-agua también pueden ser valiosos.
Determinados grupos funcionales, tales como los grupos carbonita, el carboaJcoxilo, el olefinico, el metoxilo y el C*-metilo, se prestan fácilmente para determinaciones cuantitativas {por ejemplo: *‘equivalen tt* de neutralización”, "equivalente de saponificación” , ''número de yodo” y otros más). Dichas constantes, que efectivamente dan el peso molecular gramo por grupo funcional, son de un valor especia), ya que pueden calcularse a partir de la fórmula molecular y no requieren ser comparadas con v-alores de la bibliografía. Se ha logrado un progreso considerable en el desarrollo de métodos cuantitativos para la determinación de otros grupos funcionales orgánicos y dichos métodos se usan en la identificación, determinación de pureza y análisis cuantitativo de meadas. Estos métodos están más allá del propósito de la presente obra y no necesitan usarse en los cursos ordinarios de identificación de compuestos orgánicos. En seguida se dan algunas referencias útiles:
A ) S igila , Qvanfitatioe Organie Analjuis oía Functiorwl G rcvpf, 3* ed., John Wilcy and Son3, Nueva York, 1963.
B) Múchcll, KoUhoFÍ, Proskaucr y W elubcrgcr, Orgattic A na!ysis,'\'oJí. I, l í , I I l y IV , IntcrscienCe Fublishers, Nueva York, 1953-1960.
C ) Frita y Hacmmond, (¿uoniiiatiue Orgoníc A ttalyth, John Wilcy and Son», Nueva York, 1957.

i 9
J J
m .
. W
EW
TM
id
#
Ü1
D ) Snúth y Shriner, T h t Exam ination f>{ N ew Otgdtile ComJ>ovnds: M aetc y Se mintiere A naiyiicd A ftihóds, Jobr. W iley Art . Sons, Nueva York, 1956.
Este capítulo trata principalmente la selección y preparación de derivados,, tema que se esbozó brevemente en el capítulo 2. Los derivados se agrupan de acuerdo con las clases de compuestos para los que se usan y, a su vez, k s clases se han ordenado alfabéticamente. Las instruc- dones para preparar derivados se designan en este capítulo como procedimientos. Frecuentemente pueden usarse las reacciones de clasificación discutidas en el capitulo 8 para preparar derivados; Jas instrucciones para éstas se mencionan como experimentes. También se incluyen determinados métodos para obtener constantes físicas, tales como los equivalentes de neutralización de los ácidos o los equivalentes de saponificación de los esteres. Para propósitos de identificación, dichas constantes pueden ser tan significativas o aún más que c! punto de fusión de un derivado.
A menudo surge la pregunta <u- qué tantos derivados se requieren. Sólo hay una respuesta lógica: se deberán preparar tantos derivados como sean necesarios para completar la identificación, Algunas substancias desconocidas pueden identificarse con ayuda del punto de fusión de un solo derivado. Otras pueden requerir tres o cuatro. Aún otras, tales k.omo determinados hidrocarburos saturados, pueden fallar en la formación de un derivado satisfactorio y tienen que identificarse mediante constantes físicas tales como punto de fusión, punto de ebullición, índtce de refracción, densidad, peso molecular y espectro en el infrarrojo.
Un error común es recuperar el material inicial y fracasar en identificarlo corro tal. El principiante a menudo cree que podría reconocer el material inicial examinándolo visualmente, pero las propiedades que considera como características (huellas de color debido a una impureza, forma cristalina, y tamaño del cristal) pueden cambiar notablemente cuando la substancia se recristaiiza o se recupera después de algún intento de reacción. Por esta razón es indispensable, cuando se está realizando trabajo de síntesis, conocer el punto de fusión del material inicial y también los puntos de fusión de cualquiera de los productos que se esperan como resultado de las reacciones colaterales. Par ejemplo, al intentar preparar derivados de alcoholes con isodanato de fcnilo, es de esperarse que, en presencia de cantidades pequeñas de agua de contaminación, se produzca W,N'-d ¡fon ¡1 urea y el investigador debe estar en guardia para no confundir este producto con un derivado de su alcohol. Sin embargo, puede suceder que, por coincidencia, el punto de fusión del derivado sea el mismo que el de uno de los reactivos. Por lo tanto, siempre que x sospecha que se ha recuperado el material inicial deberá tomarse un punto de fusión mixto (ver pág. 39).
El siguiente registro muestra que los procedimientos están numerados de acuerdo con un orden alfabético de las clases de compuestos
250 Preparación d e d«r|vetdos

de los cuales deben prepararse derivado*. A continuación se dan consejos para seleccionar el procedimiento adecuado para un compuesto determinado.
R sg is t ío d e Ja s n ú m e ro s 251
R e g i s t r o d e l o s n ú m e r o s d e l o s p r o c e d i m i e n t o s
N (¿antro del procedimiento Derivado» para TÍJM det dtriixló Pi*.
l Acido* Ettcrcs de /enacilo y bcndlo substituidos 257
2 Acidos Amidas 25?3 Acidos Aníftdasj ¿ 'to luididas, ¿-bromoaoiJidas
Fenilhidrazídas, sales de fenilhid racoma
2584 Acidos
2585 Acide* Sales de tiouronÍQ f-íubstituídas 2596 Acidos Equivalentes de neutralización 2597 Acidos Constante* de D udaux 2616 Acidos Coeficiente» de partición íter-a^ua 2629 Alcoholcj U re taño* substituidos 268
10 Alcohole* Benzoato», ^-nitrobenzoatoj 268U Alcoholes Acetatos 26912 Alcoholes 3,5-diaitfubenxoato» 26913 Alcoholes 3 -ni tróvala tos 27014 Alcoholes Sulfatos ácidos de alquilbericilúouronío 27015 Aldehidos Acido» 27416 Aldehidos $e.wcarbazonas 27517 Aldehidos ¿-nitrofenilhíd razonas 27518 \1dehidos 2,4dinitroíentlbidrftronaa 27519 Aldeídos Derivado» de metona 27620 Amidas 9-aciUmidoxaatendj¡ 27821 Aminas ( I y I I ) Amida» ribslituldas 28122 Aminas (1 y H ) Sulfonamidas 28323 Aminas ( I y I I ) FcniltiourcBs 28324 Aminas ( I I I ) S a líi cuatcm sria j de amonio 28425 Amina» ( I I I ) Compue»tos nitroso 28426 Aminas ( I I I ) Picratcn 28527 Aminas ( I I I ) ClorcplatinatoB 28528 Aminoácidos Derivado* de ¿-tolueneulfonilo 28829 Aoíínoicidos Derivados de 2/í-áinitrofenilo 2A930 Eitere* Equivalente de lapomíícacíón 29231 Esteres iV-tendlamídaJ 29532 Esteres Hidratadas de Ácido 29533 E» teres 3,5-dínitrobenioatos 29534 Eteres (aJifá<íco$) 3,5-d¡niirebea2oato* . 29735 Eteres (aromáticos) Producto» de biomación 298**6 p.terea (aromático») Cloruros de aulfonüo 298■¿7 Eteres (aromático») Sulfonamidas 29938 Eteres (aromático») Pícratos 29999 HaluroJ de alquilo Haluroa de atquil mercurio 30140 Haluro» de alquiló Anilidas, (oluididae, naftalidas 30141 H alaros de alquilo Eteres de alquil-ji-naftílo 30242 Halaros di. alquilo Picratos de ¿-alquiliiotiourea 30243 Haluros de aiílo Cloruros de sulfomlu 30344 Haluroa de a rilo Sultana midas 304

/
252 Preparación de derivados
¡(ilMfO (U procedimiento Derivado» pan Tipo det dwivudo W *
45 Hidrocarbuiofl aromático* Nitraciáu 306
46 Hidrocarburos aromáticos Oxidación de cadenas b tera les 307
47 Hidrocarburos aro-«áticos Acidos aroilbenioícw - 307
« Hidrocarburos aro-xn&rieos McfUarilcetoiias 308
49 Getonas Oxxmw 311¿0 Nícnlos Acidos 31451 M tiilos Aminas, fcniltiourcas sub$C. 31552 Nitrilo» HGI del ácido im’moalQuilmercapla-
acédeo 31653 Compuestas nitro Aminas 31854 _ Fenoles A ddej_anloxiacéticos 320
3 5 --------- Fenoles Bromación "320-56 Fenol« Fenílurettnos 32157 Sulfoo amidas jY-xantUsuifonamidas 32358 Acidos * afónico» Sulfonato» de bcnalriogronia 32559 Salea de eulfonaUX C laruroi de sulfowlo y julíonsaoidas 32560 Acidos suIÍÓdícoj Sale» de /R oluidina 326
.. 61 Acido® íuifóaicoj Cloroarílsulfonaniidaí, -anilidxs 326
ACETALES
(Enlistados en la pág. 330)
Los acétales se identifican por referencia al alcohol y al aldehido o cetona que producen cuando se hidroüzan. La hidrólisis se logra por calentamiento de los acetales con ácidos diluidos; en estas condiciones ocurre la reacción siguiente:
R O R ' R\ / \
C + H ,0 ^ 2R'OH + C—O/ \ /
(R)H OR' (R)H
Los acétales de bajo peso molecular se hidrolizan fácilmente por calentamiento a reflujo, con ácido clorhídrico al I ó 2°/c, durante 3 a 5 minutos. Los ace tales de peso molecular más elevado requieren tiempos mayores (SO a 60 minutos), L a hidrólisis de los acetales de alcoholes irtsolubles en agua y aldehidos o cetbnas puede facilitarse usando como disolvente una solución de dioxano al 50%.
?!• procedimiento exacto que deberá seguirse depende de los productos de hidrólisis. Si tanto el albohol como el compuesto carbonílíco

H a lu ro s d e a d ío y a n h íd r id o s 2 5 3
son solubles en agua., la mezcla de hidrólisis se neutraliza al tornasol y se divide a la mitad. Se usa una parte para la c2racterización del aldehidoo la cetona medíante la preparación de un derivado conveniente. Son útiles laa semicarbazonas (procedimiento 16), las fenilhidrazonas (experimento 21, pág, 164) y las ¿vnítrofcnílhldrabonas (procedímíento 17). La segunda parte de la mezcla de hidrólisis se trata con cloruro de benzoílo de acuerdo con el método de Schotten-Baumann [experímeri- to 1(<0, pág, 129J. Así se convierte el alcohol en el correspondiente benzoato de alquilo, el cual se aísla y se hídrolíza por el método A del experimento 26, pág. 183. Entonces se caracteriza el alcohol por uno de los métodos descritos en ia pág. 263.
Si uno de los productos de la hidrólisis es insofuble en la mezcla de reacción, se le aísla y caracteriza. Entonces se trata el producto que está en la solución acuosa por cualquiera de los métodos arriba mencionados. Si tanto el alcohol como el compuesto carbonílíco son insolubles, se les separa de k ’t>a acuosa. En caso de que uno de los productos sea un aldehido o una metilcetona aJifátíca, se le puede separar del alcohol por medio de su compuesto bisulfítico (experimento 25, pág, 161). En algunas ocasiones se puede recurrir a la destilación fraccionada para separar el alcohol y el compuesto carbonílíco,
HALUROS DE ACILO Y ANHIDRIDOS
(Enlistados en las págs. 331-333)
Muy a menudo los haluros de acílo y anhídridos se identifican por referencia a las anílidas que forman cuando se les trata con anilina [experimento L(&), P^g- 128]* Si la amida correspondiente es ínsoluble en agua, el haluro de acílo o el anhídrido pueden tratarse con hidró- xído de amonio concentrado para producir la amida,
Los cloruros de acilo de los ácidos díbásícos reaccionan con exceso de anilina, a temperatura ambiente, produciendo díanilidas. Si el anhídrido de un árido dibásico se calienta con anilina, puede producirse la jV-fenílaaida. Esta, frecuentemente sirve como derivado.
Por hidrólisis los haluros de,.acilo o los anhídridos se convierten en los ácidos correspondientes. Si el ácido es sólido, frecuentemente puede servir como derivado. En otros casos se puede hidrolÍ2ar el halurn de acílo o el anhídrido con álcali diluido y la solución neutralizarse con ácido clorhídrico (fenotf calcina) y evaporarse. La mezcla de Ja sal ae sodio del ácido y el cloruro de sodio puede usarse para preparar ésteres sólido» tales como los que se describen bajo el encabezado de ácidos en la pág. 254.
Los haluros de acilo y los anhídridos reaccionan con los alcoholes y . los fenoles produciendo ésteres [experimento I ( á ) , pág. 129], los

2 5 4 P re p a ra c ió n d® d e r iv a d o *
cuales pueden servir para identificación. F1 alcohol o el fenol deben seleccionarse en forma tal que ¿.1 producto sea un bólido.
Loí alcoholes reaccionan con los cloruros de acilo de los ácidos dibásicos produciendo esteres normales, pero con un anhídrido de un ácido dibásico, el producto principal es el éster ácido. El anhídrido itálico 5c combina con ios alcoholes dando ftalatos ácidos de alquilo.
Se produce e( dicater sólo si se usa un catalizador y hay exceso del alcohol.
ACIDOS
(Enlistados en las págs. 334- 38)
L a constante de Duclaux (procedimiento 7) proporciona un medio para la identificación de los ácidos monobásicos saturados que tienen menos de siete átomos de carbono. No es apropiada para Jos ácidos valeriánico t isobutlrico debido a que Jas constantes para estos dos ácidos están muy cercanas. Los equivalentes de neutralización de los ácidos (procedimiento 6) y los coeficientes de partición (procedimiento 8) son particularmente valiosos, Usualroentc es aconsejable, y frecuentemente es necesario, que se confirme la identificación de Jos ácidos por medio de derivados sólidos.
Los ésteres sólidos proporcionan un medio útil para caracterizar ácidos. Algunos ésteres metílicos son sólidos^ pero en Ja mayoría de los casos se deben usar éste res de ¿►nítrobencilo> feriadlo, /?-cbrofenacik>, /j-bromofenadlo o ¿-fenilfenaciío. Estos se preparan tratando Jas sales de los ácidos con el correspondiente haluro (procedimiento 1),
R C O O N a-f C)CHaCfiH<NO2(p) -^R C O O C H 3CtH4NO¿(^) + NaCl
RCOONa+BrCHzCOAr -> RCOOCH:C O A r+N aB r
Este método es particularmente ventajoso debido a que no requiere una muestra anhidra del ¿cádo.
También pueden prepararse los ésteres de p-bromo y ¿-fcnilfenacilo tratando el ácido con Ja a-diaíjoacetofenona apropiada.
RCOOH+NíCHCOAr-» RCOOCHiCOAr+Ni
Los productos de reacción de Ja fenilhidrazina y los ácidos también son buenos derivados. A la temperatura de ebullición (243°C) de La

Seles d e ó a do* 255
íemlhidrazma, lo» ácidos mooo y di básicos aliíiticos, sencillos y sin substituyen tes, forman fenilhidraadas (procedimiento 4 ) .
R C O a H + H 2N N H C < H s - * R C O N H N H C jH s 4-H^O
Los ácidos sulfónicos, ácidos aiifátícos «-clorados y Jos ácidos d ¿básicos aiifátícos forman salea de fenUhidrazina cuando se calientan con una solución bencénica de fenilhidrazma.
Xx» ácidos pueden convertirse en amidas por tratamiento con cloruro de tionilo y luego con amoníaco.
RCOOH+ SOCIj RCOC1 + SOj -f- HCl
RCOC1+2NH3 - * RCONHj -f NH^CI
Este método es particularmente apropiado si la amida es insoluble en agua; puede efectuarse añadiendo el cloruro de acilo aJ hidróxido de amonio concentrado (procedimiento 2).
Las añil idas, toluididas y p-bromoanilidas son derivados excelentes debido a la facilidad con la cual pueden prepararse y purificarse. Se pueden preparar a partir del ácido libre o de su sal (procedimiento 3).
Las dimetilamidas y las diamidas derivadas del £,pr-d¡aminodifenil- metano también han encontrado aplicación como derivados. Los ácidos se condensan con la o-fcnilendiamina produciendo 2>alquil1>endmida- zoles, Jos cuales son derivados útiles.
Las sales derivadas de los ácidos orgánicos y las aminas fuertemente básicas» tales como la benolamina, a-femletilamina y la piperazina, también son buenos derivados’.
Sales de ácidos
La adición de un ácido minera) fuerte a la sal metálica de un ácido carboxílico deja en libertad el ácido orgánico.
2R C O O N a+H 3SO* -> 2R C O O H + N aíSO<
El áddo orgánico libre puede aislarse por destilación, extraedón o filtración. Gomo las sales metálicas funden sólo a temperaturas muy elevadas y ordinariasL^-nte se descomponen, es necesario usar las cocs- taates físicas del áddo para preparar la lista de posibilidades. Entonces

256 Preparación de derivados
e l á c id o p u t t te c o a v e r t ín c e n u n d e r iv a d o a p ro p ia d o o la sa l p u e d e u sa rse d ire c ta m e n te p a r a l a p re p a ra c ió n d e la a n i l id a , to lu id a {procedí* m ie n to 3 ) o c s te r d e ¿ -n itro b e n c ilo o ¿ -b ro m o fe n a c ilo (p ro c e d im ie n to 1 ) .
E l b ro m u ro d e i '- ( /> -b ro m o b e n e il) 't io u rc n io > p re p a ra d o p o r c a len ta m ie n to d e l b ro m u ro d e /j-b ro m o b en c ilo c o n t ío u re a , re ac c io n a con Jas sales d e sodio o p o ta s io d e los á c id o s ca rb o x ílico s p ro d u c ie n d o Jas sales d e S - (¿ -b ro m o b e n á l) - tio u ro n io (p ro c e d im ie n to 5 ) ;
B r^ ^ C H aB r + S Q N H ?) 2 [■ O CH2SC(NH2 Br-
Br/ ^ ' C H 2SC<NH2)2
(RCOO)- B r / )CH ,SC{N H 2), + NaBr
L a s sales d e S -bencil- y $ » { jD -c lo ro b cn cil)-tio u ro m o se p re p a ra n e n fo rm a a n á lo g a . L a s saJes se o h ü e n e n cas i p u ra s , p e ro t ie n e n p u n to s d e fusión b a s ta n te s cercanos.
Referencias
Amidas y dimct&uxúdás. M itchíl) y Rcid, J. Am. Che**. S&c., 53» 1979 (19SS). Anllidas, j^-brojnoanilidas y p'tohxidldas. Kuehn y McEtvairt, / . A m . Chtm.. So<-,
53, 1173 (1931); Biyant y M ilcbe», ibV ., 60, 2746 (1938), íV-ac»I»cjiria*v Siepben y Stephen, ] . Chem. Soc., 492 (1&57),E neres d i fctucOo y p^lordfeuatílo . H aaa , Reíd y Jaroieícm, / . Am . Chem. Soe.,
52, 818 (1930); R atijcr y Reíd, ibld., 41, 75 11919); M os« y Reíd, ibíd.,54, 2101 (1932); Luiidqulrt, ibíd., 60, 2000 (1938).
E s te r» de p-brtwnoíetuciJo. Reíd y Jodcíind, / . A m . Chem. Soc., 42, 1043 (1920); Powell, M d ., 53, U ?2 (1931); KcHy y KJcfí, ¡bld., 54, 4444 (1932); Chcn y SSúb., Trans. S ti. Soc. Chuta, 7 , 81 (1 9 3 1 ); ErickBon, Dechary y Kc»lin.j, / . Am . Chem. Soe., 73, 5301 (1951).
Esíetta de p-feoilfejtMcHo. D ía le y Bronitaky, / . Am . Chem. Soc.t 52, S715 (1930); D ía le y Sweeney, ibl¿., 54, 2059 (1932); Kelly y Morisani, ibid., 5$, 1502(1936).
E neres de p ’bnMnobepcílo. Fiekers y D iG ercm w ?, / . A m . Chem. $oc., 70. 3854 (1948).
Esteres de p-nltzt>l*acno. Reíd, / , A m . Chem- Soe., 39, 124 (1917); í.yroan y Reíd, ibid., 89, 701 (1917); Lyons y Reíd, ibid., 39, 1727 (1917); Kelly y Segura, i b i d 56, 2497 (1934).
Sales de S 'boiultiQuroiúo. Donleavy, / . A m . Chem. Soc., 56, 1004 (1936); Cham berí y Scberer, Ind . Ett¿. Chem., 16, 1272 (1 9 2 4 ); Veibet y Lillelund, Bvll. Scc. Chxm., [5], 5, 1153 (1938).
Sales de S-{p-bromo y 5-{p-eIorob«aciltwuronte. Dewey y Spcrry, / . Am . Chem.Soc., 61, 3251 (1939); Dewey y Shasky, ibid., 63, 3526 (194-1),
Sales de S-t'&aftümctíltúniraDti), Booncr, /? A m . Chem. Soc., 70, 3503 (1948),

Proc«djm ¡tnto 2, Amida» 257
Frniin id reídas y wk* de feD¡lbtdta¿jra¡o. Stcrapcl y S chaííel, / . A m . C hem . Soc., 64, 470 (194?).
Carbozilatoé de bcpálamouio y caitoxiJatoi de «'fcnilstiUmoulo. Buehler, Cartón y Eddi, / . Am. Chtm. Soc., 57, 2181 (1335),
Sales de ptperaxonio. PoHaid, Adclson y Baiti, / . A m . Chem. Soc., 56, 1759(1934); Prigot y Pdktd, 70, 275B (1943).
2-aJquílb«adüi{daiQlcs, Pool, Harwood y Ralston, /. Am. Chevt. Soc., 59, 170(1937); Brown y Cwnpbeíl, J. Chtm. Soc., 1937, 1699.
Dinnidú* de p^-diatoiaodifcnilmetiBiX Ralston y McCorklc, /. Ant. Chcm. Soc., 61, 1604 (1939).
Derivados de ácidos grase* superiores. Ford, lowa State Coüege }. Sel., 12, 121 (193.7); C. A., 32, 4943 (1938).
P roced im ien to i , Esteres de p-nitrobencilo , fenacilo , p•clor</fenacilo9 \*brom ofenccilo y p -fenilfunacilo
U n gramo <k ácido se añade a. 5 mi de agua contenidos en un matraz pequeño y Se neutralizan cuidadosamente con solución de hidró^ xido de sodio al 10%. Se añade un poco del ácido y se continúa la adición hasta que la solución esté justo ád d a al tornasol. Si el áddo original se obtuvo como sal de sodio, se disuelve 1 g de la sal en 5 a 10 mi de agua. Si esta solución es alcalina, añada una o dos gotas de ácido clorhídrico diluido. Se añaden diez mililitros de alcohol y 1 g de bromuro de fen ac ilo en to n ces se calienta a reflujo la mezcla durante 1 hora si el ácido es monobásico, 2 horas si es dtbásico y 3 horas si es tribásico. Ocasionalmente puede ser necesaria la adición de unos cuantos mililitros más de alcohol si durante e! reflujo precipita un sólido. Se deja enfriar la polución y el éster precipitado se purifica por reeri*- tali2ación de alcohol.
Al preparar derivados con estos reactivos debe tenerse cuidado de que la mezcla original no esté alcalina. Los álcalis provocan la hidrólisis de los haluros de fenacilo hasta alcoholes fenacüicos. Además, el bro* muro de f-bromofcnacilo no deberá usarse si hay cantidades considerables de cloruro de sodio en la sal de sodio del ácido,2 Debe seguirse un procedimiento especial para obtener los formiatos de fenacilo substituido.*
P roced írm ^nto 2 , A m idas a partir de ácidos
Durante 15 a 30 minutos se calienta a reflujo un grazno del ácido con 5 rnl de cloruro de tionilo. La mezcla se vierte con precaución en 15 mi de hidróxido de amonio concentrado, helado. La amida prtfcipi' tada se filtra y se purifica por rccristalización de agua o de etanol diluido.
1 ] Predación 1 Lox W urtt 4f {«tr*cik> ton l*Chtn¿e«nM.1 Paira» y BcruaUm, } . j4m. C htm . S ie . t 15, 2096 (1943). y
. i Kubws* y MnUuyr», / . JaíI Fáíytitk- Om ¡4, 1, 4S (I9S0), /

kb
! * t
- a
La
La
í i
l i
r»*
5Ü—
2 5 8 P re p a ra c ió n d a d e r iv a d o s
P r o c c á iw ie n i^ 3 . /ánüitfíW , p-foíuí<ííá<xy y p-fcrom c£;«iiírfo.í
a) En un tubo de ensayo se mezcla un gramo del ácido o la sal de jodio con 2 mi de cloruro de tionilo y Ja mezcla se calienta a reflujo, usando un refrigerante pequeño, durante 30 minutos. La mezcla se enfría, se añade una solución de 1 a 2 g de la amina (anilina, /Molui- dina, o ^-bromoanilina) en 30 mi de benceno y la mezcla se calienta moderadamente, en uft baño de vapor, durante 2 minutos. La solución bencénica se pasa a un embudo de separación y se lava sucesivamente con 2 mi de agua, 5 mi de ácido clorhídrico al 5%, 5 mi de solución de hidróxido de sodio al 5% y 2 mi de agua. Se evapora el benceno y la amida se recristaliza de agua o etanol.
b) En un-tubo de ensayo se coloca una mezcla de 0.4 g de la sal de sodio del ácido, seca y pulvcrinda, 1 m; de la arilamina y 0.3 mi de ácido clorhídrico concentrado. El u ^ o de ensayo se coloca en un baño de aceite que entonces se calienta: la temperjftura se mantiene entre 150°C y 160°C de 45 a 60 minutos. Al finalizar este tiempo se saca el tubo de ensayo y el producto se purifica por uno de los métodos siguientes:
1. Si el áado que se está considerando tiene menos de seis átomos de carbono, se hierve el producto crudo con 5 mi de etanol al 95% y la solución se decanta en 50 mi de agua caliente. La solución resultante se evapora hasta un volumen de 10 a 12 mi y se enfría en un baño de hielo. Los cristales $e filtran y se rtcristalízan de ur.a pequeña cantidad de agua o etanol diluido.
2. Si el ácido contiene seis o más átomos de carbono, el producto crudo de la reacción se pulveriza y se lava con 15 mi de ácido clorhídrico al 5% y después con 15 mi de agua helada. El residuo se hierve con 30 a 40 tnl de etanol al 50% y se filtra la solución. El filtrado se enfría dentro de un baño de hielo y los cristales de la amida substituida se* filtran. El producto puede recristalizarse de etanol diluido.
Procedim iento 4 . F en ilh id rábidos y sales de fe.nilhidrazonio
a) Un gramo del ácido se disuelve en 2 mi de fcnilhidrawna y la solución se hierve suavemente durante 30 minutos. El producto crista* lino, que precipita cuando se enfría la sofvci&i, se filtra a presión reducida y se lava con pequeñas porciones de l>cnceno o éter hasta que los cristales queden blancos. Cuando se usa urt gran exceso de íenilhidra- 2ina, algunas veces es necesario diluir la mezcla con benceno para que ocurra la precipitación del producto. Los derivados de los ácidos monobásicos inferiores se rec riscal izan ce benceno caliente, mientras que los de los ácidos superiores y de los ácidos dibásicos se recristalízan mejor de

Procedim iento 6. Equivalentes «fe ftout#oílzoerírt 259
etanol o mezclas de etanol-agua. Los derivados cbícnidos de los ácidos dibásieos por este método son las bís-{J-feni{hídr asidas.
b) Se mezcla, un gramo del ácido con 2 mi de fe nilhid razana di- sueltos en 5 m( de benceno. Algunas veces precipita inmediatamente un sólido blanco; se le recri&taliza de etanol. Si no precipita ningún sólido, la mezcla se calienta a reflujo durante 30 minutos y el producto que precipita al enfriar se filtra, se lava en éter y se recristaliza de benceno o etanol. Los ácidos sultánicos, los ácidos aiifátícos substituidos con halógenos y los ácidos dibásieos aiifátícos producen sales por este procedimiento, mientras que los ácidos aiifátícos sencillos y sin substitu- yentes forman fenílhid rasdas.
P roced im ien to 5 . Sales de bpncil-, p>clorobencii y *>bromobencilt¡ouronio
Aproximadamente 0.3 g del ácido (o 0.5 g de la sal de sodio o otasio) se adicionan a 3 ó 4 mi de agua, se añade una gota de una
solución del indicador fenolftalelna y la solución se neutraliza por adición, gota a cota, de una solución de hidróxido de sodio al 5% . Debe evitarse un exceso de álcali, Si se adiciona, demasiado, se añade ácido clorhídrico diluido hasta que la solución adquiera un color rosa pálido. A esta solución acuosa de Ja sal de sodio se le añade una solución caliente de1 g de cloruro o bromuro de arilalquihiouronio en 10 mi de etanol al 95%. Se enfría la mezcla y la sal se filtra. Algunos ácidos (v. gr., el fórmico) no forman precipitados y parte del etanol debe evaporarse para obtener la sal.
Estas sales de tiouronio de los ácidos orgánicos se obtienen en un estado de alta pureza y ordinariamente no requieren ser rccristalittdas. Si es necesario pueden recristalizarse de una cantidad pequeña de dioxano.
Los puntos de fu&ión de muchas de estas sales están bastante cercanos y un número considerable de ellas funden dentro de un pequeño intervalo de temperaturas. De aquí que siempre sea mejor confirmar la identificación con algún dato adicional tal como la constante de Duclaux, el coeficiente de partición o el equivalente de neutralización.
P r e n d im ie n to 6* Equivalentes de neutralización
R C O jH +N aO H -» R C 0 2N a + H/D
Se pesa exactamente una muestra del ácido (aproximadamente0.2 g) y se disuelve en 50 a 100 mi de agua o etanol. ¿ a mezcla puede
r calentarse, si es necesario, para disolver todo el compuesto. Esta solución

se titula con una solución de hidróxido de sodio previamente valorada y que tenga un factor de normalidad (F. N.) cercano a 0.1; como indicador se usa fenolftaleína* El equivalente de neutralización del ácido se calcula de acuerdo con la fórxnuls.:
_ , , , .. peso de ia muestra x 1000Equivalente de neutralización = v0^ en dd ilc a |¡ (m]) x F , N,
Comentarios. El peso molecular do un ácido puede determinarte a partir del equivalente de neutralización, multiplicando este valor por el número de grupos ácidos que haya en la molécula.
El cambio de medio, aun de agua pura a etanol puro, ¿ifecía el pK canto deí ácido orgánico como del indicador/’ Por esta razón se obtienen mejores resultados en agua o en etanol acuoso con sólo suficiente etanol para disolver el ácido orgánico. En etanol absoluto o al 9 5 frecuen^ tómente es imposible obtener un punto final bien definido con fenolfta- Jeína. En tales casos, deberá emplearse azul de bromotimol como indicador, Los ácidos también pueden titularse en una mezcla de disolventes compuesta de etanol y benceno o tolueno.
Siempre debo correree un testigo con el disolvente; la misma cantidad de fenolftaleíoa que se usó en la titulación debe emplearte *n el testigo. En el trabajo ordinario, los equivalentes de neutralización deberán concordar con los valores calculados dentro de un margen de ± 1 % . Usando muestras cuidadosamente purificadas y secadas y una buena técnica, el error puede reducirse basta ±0 .3% .
Para dar un equivalente de neutralización exacto, la substancia titulada debe ser pura y anhidra. Si el valor obtenido para el equivalente de neutralización no concuerda con el valor teórico, el compuesto de* berá recristalitarsc en un disolvente apropiado y secarse cuidadosamente.
Las sales de aminas de áddos fuertes pueden titularse por el mismo procedimiento.
Preguntas
1. S i un ácido no es*í perfeci amenic «eco, ¿su equivalente de neutralización será alto o bajo?
2. ¿Interferirá la p/csencia de un grupo amíno aromático en )a determinación del equivalente de neutralización? ¿C uál sería el efecto de un grupo amíno alifitico?
3. ¿Q ué tipo* de fenoles pueden titularse cuantitativam ente?De*de el punto de villa teórico, ¿cuál debería ser la constante de ionización de un ¿cido para que el ¿ tid o se pudiera titu lar con fendfiaW ua?
* Friti 7 Hkmmond, HaantUttivs OrganU Ana¿yñi, }i>hn W lty »ad Soa»., Ntaev* Yerfc, dpienfe 3; Kotlfccrff, A¿t4'8íil* la&calarj, The ÍÚínúlW C*„ Nu«v» Volt, 1537;
Grutwild v B c r W u , / . .<«*. CA*«u. Sot.. 73, « J 9 Gnmmld, C -W , 26,1696 (1354),
260 Preparación d e derivados

Pro<edírolent© 7. Arrostre con vapor 261
P r o c e d im ie n to 7 , A r r a s t r e c o a v a p o r
c o n s t a n res d e d u c l a u x p a r a á c i d o s a l i f a t t c o s v o l á t i l e s
Ésta prueba es útil para ácido* aiifátícos sin substituyenos y de bajo peso molecular que contienen de uno a seis; ¿tomos de carbono. También es valiosa para la caracterización de los ésteres de tales ácidos, ya que los ácidos pueden obtenerse de elíos por hidrólisis.
Se disuelven tres gramos del ácido en 150 mi de agua destilada y la solución se mezcla perfectamente. Se toman diez mililitros de la solución por medio de una pipeta y se titulan con solución de hidróxido de eodio 0.1 usando fenolftaleína como indicador. Entonces se colocan 100 mi de la solución en un matraz de destilación de 250 a 300 mi y se añaden unos trozos de placa porosa. No hay necesidad de usar termómetro, pero la parte superior de! matraz de destilación deberá taparse con un tapón de hule lú. pió. El matraz se conecta mediante un tapón de hule a un refrigerante uorto enfriado con agua, en tal forma que el brazo lateral dd matraz de destilación penetre bastante dentro ¿e la paite angosta del tubo del refrigerante. El aparato deberá montarse de tal manera que el refrigerante forme un ángulo de 45° con la parte superior de la m e». El matraz de destilación se calienta hasta que del extremo de salida del refrigerante salgan gotas a una velocidad constante. La destilación no deberá ser tan rápida que haya un flujo continuo de destilado o tan lenta que haya un intervalo apreciable entre gota y gota. Se recogen tres fracciones de 10 mi (A, B, C) usando una probeta de 10 mi. Cada fracción se titula separadamente cor; la solución de hidróxido de sodio0.1.V usada inicíalmcnte, Los resultados se expresan como porcentaje de acidez de los 100 mi usados.Número de Duclaux ^
volumen de ¿Icali (mi) para La fracción de 10 mi x 100 volumen de álcali (m i) para los 10 mi iniciales X 10
Los números de Duclaux para los ácidos aiifátícos comunes se reportad en la tabla 16.
Comentarios. La solución de hidróxido de sodio no necesita valorarse con exactitud, pero deberá ser muy aproximadamente 0 .1N. Este procedimiento es empírico y los valores sólo son relativos, siendo un índice de la volatilidad relativa del ácido y el agua a la concentración señalada. El volumen de la probeta deberá verificarse con el de una pipeta de 10 mi. ’
Preguntas
1. ¿Qué ei una mésela o trópica?2. El ácido fórmico (?7.5%) y el agua (22.5%) coonituyen w u n*ac!a &ao-
. trópica. Si ésta fuera la concentración usada en la destilación, ¿cuál» «rían

L tí «r«
n
f t
3»
*
262 Preparación de derivado*
1 a » u\ 16 Números de Duciaux
I Fríi«16ri
A ¡ B C
Fórmico 3.95 4.40 4.55Acético 6.8 7.1 7.4Prop iónico n .9 t i . 7 11.3a-butírico ) 7.9 13.» 14.6Isobutíríco 25.0 20.9 16.0« v a le rián ico ?4.S 20 6 17.0IsovalcriániCo 20.7 23-1 16.3«•C&proico 33.0 24.0 19.Q
los rió me rus dé Oucli -x? Suponga que se usó una. solución que contiene 90% de ácido fórmico.- ¿Q ué d iría usted con respecto a ]t>s números de Duciaux Correspondientes a las fraccione* A. B y C ?
3- El ácido acético (2&%) y el tolueno (7 2 % ) constituyen una mezcla azeotró. pica. ¿Cómo podría separarse dicha mezcla?-
4- i Puede usted sugerir alguna explicación, desde el punto de vfcta estructural, al hechí* de <|üc Ua ácidos superiores no formen mezclas ¿ 2eOtrópicai con el agua? «Por qyr los número} de Duclaux para la fracción A de estos icidos es mayor que los de las fraccione* B y C?
5, El acerato de etilo w saponificó por trata miento con solución de hidróxido de sodio. Se añadió ácido fosfórico y se destiló una parte de 3a Solución. C uando sr determinaron en este destilado los números de Duclaux, no dieron.los vá!or«s esperados partí el á d d o acético. Explique eíto . ¿Cómo debería haberse m odificado el procedimiento?
P rocedim iento 8 . C oefic ien tes de partic ión éter+agua p ara ácidos orgánico* *
Unos 2 g <teS ácido desconocido se disuelven en 50 mi de agua destilada y exactamente 10 mi de la solución {medidos con pipeta o bureta) se titulan con una solución de hidróxido de sodio previamente valorada (aproximadamente O.hV), usando íenolítaleina como indicador. Se calcula la normalidad exacta de la solución ácida original y una parte de ella se diluye con suficiente agua desellada para, que la solución sea 0.1 A? (±0.003).
Por medio de una pi[>eta se transfieren exactamente 50 mi de esta solución O .ltt ^ un embudo de separación de 200 mi, limpio y seco, y so añaden, también mediante una pipctaf 50 mi de. éter saturado con agua (pero libre de etanol). La mezcla se agita durante 5 minutos y se deja reposar duran ce algunos minutos para permitir que se separen las capas. La capa acuosa inferior se pasa a un matraz limpio, tornando la precau*
5 Derroer y Dcrrcicr» J- ¿r*- Cke*t. Sor., 6S, IOS <19*3).

Alcoholes 263
ción de dejar unos cuantos mililitros en el embudo de separación. Per medio de una pipeta se toman veinte mililitros de la capa acuosa, y se titulan con el álcali 0.1 N , usando fenolftaleína como indicador. Entonces se transfieren, mediante vina pipeta, 20 mi de la capa etérea a un m a tm limpio, Se elimina ia mayor parte del éter por destilación cuidadosa, se añaden 20 mi de agua destilada y la solución se titula con álcali O.LV. A partir de estos valores se calcula la relación de la concentración en el a^ua con respecto a la del éter (Cc/C ¿).
Los valores de Ca/C K para algunos ácidos comunes son los siguientes: ácido bencenstilfónico, 300; rf-canforsulfónico, 300; d-tartárico, 280; acetílglicina, !50; cítrico, 150; glicina. 120: mesotartáríco, 120; /-malicio, 75; ¿ft-málico» 52; sulfosalicílico, 36; díglicólico, 35; glicóKco, 35; oxámico, 31; oxálico, 25; tricarbalílico, 20: láctico, 14; malónico, 12,0; maleico, 10.9; hidracrílico; 10.0; ftalato ácido de potasio, 8,1.; succínico, 7.8; mctoxíacétioo, 5.7; levulínico, 4.4; tx-hidroxüsobutcríco, 4.4; aconftico, 4.2; glutárico, 3,7; eionoacético, 3.3: ítacónico, 3.0; fórmico, 2.8; eto- xiacético, 2.2; acético, 2.1; adipíco, 1.9; fumárico, 0.80; ftáüco, 0.79; pimélico, 0.73; £-bromopropiónico, 0.?0; propiónico, 0.63; <tf-mand6- lico, 0.50; aerílicc, 0.49: cloroacético, 0.46; bromoaofitico, 0.31; furoi- co, 0.26; croeóníco, 0.25; a,(J-dibromopropiónico, 0,24; tricloroacético, 0.24; (3-cloropropiónico, 0.24; butírico, 0.21; valeriánico, 0.20; ísobutt- rico, 0-19; dicloroacético, 0,18; beruoij fórmico, 0.16; a-cloropropiónico, D,í4; a-bromopropiónico, 0.110; ¡sovaleriánko. 0,108; ^-yodopropió^ nicOj 0.103; ¿ft-me ciletilacétíco, 0.092; 0-clorobutirico, 0,087; tiimetil- acctico, 0.077; isocaproico, 0.053; <**bromobutí¡'íco. 0,056; fenilacéiico. 0.047.
Comentarios, Este método de partición entre disolventes tiene algunas ventajas sobre el método de Duclaux, En especial, que no está limitado a los ácidoa volátiles y de aquí que tenga una aplicación más amplia. Sin embargo, loa valores para los ácidos que son muy intimamente afines están demasiado cercanos para que sean útiles como criterio de identificación [c f . butirico (0.21), valeriánico (0.20) e isobutírico (0.19],
La precisión en la medición de estas relaciones es de aproximadamente ± .2#>, con respecto a sus valores propios, cuando C4¡C t — 1, pero disminuye hasta ± 4 % cuando Ca/C t es mayor que 35 o tan pequeña como 0.05. Para valores de la relación aún mayores, la precisión
. es mucho menor ( ± 10 a 20 & ).
ALCOHOLES
>. (Enlistados en las págs. 339-342}
Los derly.dos más generales de los alcoholes primados y secundarios son los a-naftil-, £-nitrofenil- y fenüuretanos, Estos. se forman

2 6 4 Pr«para<l¿<i d e d e r iv a d o s
cuando se tratan con los iaocianatos de ft-naftilo, ¿-r.iírofenilo y fenilo (procedimiento 9 ) , respectivamente.
G»H7N =5 C = O + R O H C»H7NHCOOR ( ^ ) 0 jN C y H 4 N = C = 0 - f - R 0 H f ^ ) OjNOíH.NHCOOR
Q H sN ^ C = O + ROH - y C6H5NHCOOR
También se producen uretanos tratando los alcoholes con azidas de ácidos [procedimiento 9 (¿} ].
AfOON3+ ROH -> ArNHCOOR+N2
También se recomienda usar los 4-yododiferiiluretanos, los o- y w-nitro- feniluretanos» loa 3,5-diuitrofenUuretanos, Jos 4-xeniluretanos, los £-aní~ áluretanos y ios ^-bromofeniluretanos para caracterizar los alcoholes.
La presencia de agua, como impureza en el akohol origina dificultades en la obtención de urétanos por cualquiera de estos métodos. El agua hidroliza los isocianatas dando arilaminas, las cuales se combinan ron el exceso de reactivo produciendo ureas disubstítuidas.
A rN = C = O + H a0 -*• ArNHCOOH -+ ArNHi+OO*ArN= C O + ArNHj - * ArNHCONHAr
Las ureas tienen puntos de fusión más elevados y son menos solubles que los uretanos correspondientes y> las ureas, aun en cantidades pequeñas, dificultan considerablemente el aislamiento y purificación de los uretanos. Por esta razón, este procedimiento es más útil para alcoholes que son ínsolubles en agua y, por lo tanto, que se obtienen con más faoÜdad en condiciones anhidras.
S6k> con grandes dificultades pueden obtenerse uretanos a partir de alcoholes terciarios. Los isocianatos provocan su deshidrataran con fo rmación de la defina y la diarilurea.
- Para los alcoholes solubles en agua, que es probable contengan huellas de humedad, los 3,5-dinítrobenzoatas son derivados más satisfactorios que los uretanos. Los 3,5-dinitrobenzoatos se preparan tratando los alcoholes con cloruro de 3,5-dinitrobenzoiIo (procedimiento 12).
NO,
COCI + ROH
Los alofanatos, los cuales k preparan por reacción del ácido ciánico con los alcoholes, han sido usados ampliamente para determinados tipos
. de alcoholes. Generalmente el ácido ciánico se genera calentando ácido ciaDÚríco y se burbujea directamente, en forma gaseosa, en la solución
COOR + HCl

A lco h o le s 265
alcohólica. Probablemente el reactivo es un dímero del ácido ciánico, como indica en seguida:
Este procedimiento tiene la ventaja de poderte aplicar extensamente. Es útil Jo mismo para alcoholes terciarios que para primarios y secundarios.
Se ha encontrado que también son útiles los compuestos dobles de ios 3,5-dinitrobenzoatos de alquilo con la a-naftilamina,
Ox.?s tipos de esteres, tales como los />-nitrobenzoatos [procedimientos i ?(a), 1 0 (¿ )J , ftalatos de ^nitrobencilo y nitro-l-amraquúion- earboxilatos-2, han .encontrado uso como derivados. En el caso de los g] i col es y compuestos polioxhidrílicos, pueden usarse los benzoatos (procedimiento JO) y los acetatos [procedimiento 11 (a) u 11(6)]. Los 3‘nitroítalatos (procedimiento 13) son valiosos no tan sólo para los alcoholes simples íino. también para los alquiléteres del etilenglicol (celo- solves) y dictilenglicol (carbitoles), Los xantatos también son apropiados p ara la identificación de alcoholes primarios y secundarios.
Se les ha usado para, identificar los celosolves y carbitoles, ya que los derivados no tan sólo tienen puntos de descomposición definidos, sino que también pueden ser caracterizados por titulación con una solución valorada de yodo.
Guando (os alcoholes se tratan con cloruro de tritilo, en presencia de piridina, producen éteres de tritilo.
celosolves, carbitoles y glicoles afines. Estos alcoholes suhstituidos también producen 3,4,fMriyodobrnzoaios cristalinos.
de fósforo sobre la sacarina, reacciona con los alcoholes produciendo
NH
CO COJ 1 -*• H N = C = 0 -*■ NH2C 0N > =C = 0
NH NH
NH2C 0N = C = 0 + ROH — NHjCONHCO^R
• SROH + GSj + KOH R O ¿S K + H20
S S
El cloruro de pseudosacarina, preparado por acción del peatacloruro

la identificación
HCl
Numerosos alcoholes primarios y secundario* se identifican fácil-. mente por conversión a suliatos ácidos ce a-quilo, seguid:’ de formación de u n a sal con cloruro de S-bencilHouronio (procedimiento 1 4 ) .
ROH + CISOjH -» ROSOjH+ HCl
ROSO,H + C ,H iCH2SC{NHJ) ,Cl[CíHjCHzSC (KH¿) 2y< ROSO;) - + lIC l
Lo* alcoholes tem arios se convierten en cloruros bajo la acción de cloruros de acilo y ac deshidratan bajo la acción de anhídridos. Sin embargo, los ésteres puede;i prepararsc con rendimientos satisfactorios si se deja que la reacción ocurra en presencia de una base o un metal que reaccione con cl ácido clorhídrico a la misma velocidad con que se forma. Los trialouilcarbitxolcs sencillos pueden convertirse en tetracloroftalaco?. según las reacciones siguientes:
RaCOH + C?HsMg,Br -> R3COMgBr + C4H6
-|COOMgBrJC O O C R ,
1,0
^jCOOH ¿COOCR,
Estos ítalatos substituidos pueden caracterizarse por sus puntos de des» composición y equivalentes de neutralización.
Referencias
Cl
3
3
Ci
i*Cl
Cl
derivados de O-alquilsacarina, los cuales son útiles en de alcoholes primarios y secundarios.
266 Preparación d e derivados
CC1 COR
N + ROH — I N +. y w k /SO. SOj
fenüuretanos. Dewey y W itt, Ind . Eng. Chem., Anal. F.d., 12> 459 (1940) j 14, 648 (1942).
2»4,G-triyodoí<niluretanw, Johnson, / . C htm . Soc., 3322 (19ü5}.

A lc o h o le s 2 6 7
ix-naftílurcunoi. Bickel y Frend», / . Am. Ch<m. Soc., 48, 747 (1926); Frt*fch y W irtel, ibid., 48, 1736 (I926>-
3.5-dinjtrobenzoato?. Bry&nt, / . A m . Chem. Soc., 54, 3758 (1932).3.5-diniirobenrostos y sus compuesto? 6e edición « m l-nsitllam ina. Benfey,
Staitmyer, M jjljjan y Vr'esthead, / . Org. Cht,m.. 20, 1777 (1955); Sutter, Hetv. Chim. Acta, 21, 1266 (1938).
SuHatos ácido? de alquilbcnciltiouronio, Baír v Swtft, / . A m . Chem. Soc., 64, 197a (1 9 + 2 ) .
¿¿í-carbamatos a p a rtir de toluen-2,4.diisociansto. Boisert, } . Or*. Chem., 23,_ 906 (1958).
o-nitrobenzoatcs. Lowc, } . Am. C htm . Soc.. 74, 841 (1952). p-nitrobenzoatos. Henslock, / . Chem. Soc., 1933, 236; Armstrong y Copenhavcr,
J. Am. Chem. Soc., 65, 2252 (1943). p-aitrofenilacetatos. ty'ard y Jcnkirw, / . Org. Chem., 10, 371 (1945).Alofauatos. BehaJ, Compt. Rand., i 68, 945 (1 9 1 9 ): Spíelman, Barnci y Cióse.
] . A m . Chem. Soc.. 12, 2520 (1950); Lañe, ']■ C htm ., Soc., 1951, 2 7 64 . firomometilftaJimidas. Maneera y Lcrntargcr, ] . Org. C hem ., 15, 1253 (1950). p-nítrofcnilurcttaos. H om e y Shriner, / . A m , C htm . Soc., 53, 3166 (1 9 3 1 ) ;
ShnDcr y Cox, ibid., 53, 1601 (E 9 3 1 ) ; v a n H oojjs t r a te n , Rec. Trdv. Chim.. 51, 414- (1 9 3 2 ) .
2,4-dinItrofcniluretano9. Van Glinklo, R tt . Trav. Chim., 61, 149 (1942).Esteres del Ácido J*aitroftálim . Nicolei y Sacks, J. A m . Ckcm. Soc., 4?, 2340
(1925); Dickinson, Crosson y Copcnhaver. ibid., 59, 1094 ({937); Veraguih y D iehl, ibid., 62, 233 (1940).
S^dtbldroxibenzoatos de alquilo. Suter y W cston, / . A m . Chem. Soc,, 61, 531 (1939).
Esteres d d ácido itálico. Go^jaitE y Copenhavcr. / . Am . Chem. Soc., 61, 2909 (1939).
4r-yoáodtíeni3ureíaooj. K&wai y Tam ura, / . Chem. Soc. ¡apon, 52, 77 (3931). Ftalatos d< p-nitrobcncilo. Reíd, } . A m . C hem . Soc.. 39, 1249 (193 7).XanUtOS. W hitm ore y Liebcr, fnd . Eng. C httñ ., Anal F.d., 7, 127 (1935); Shupe,
J. Assoe. Offic. Agr. Chemiits, 25, 495 (1942). p^toraofenifu reíanos de alquilo. Sah y T ao , R íe . Trau. Chim., 58, 12 (1939), N¡tro-l-antraqu¡Doncajboxi3atos>2 de alquilo. ¡Sah y M a, / . Chínese Chem. Soc.,
1 ,5 1 (1933).o-tlltro-, m -nilro- y 3,5-dinitroíeniluretanos. H oeie, R<c. Trav. Chim.. 54, 505
(1935).4-xeniIurttanos. Morgan y Pettct, / . Chem. Soc., 1931, 1124. p-ankiluretano«. Brutine y Wohrl, idonatsh., 63 , 374 (1933). p-trifcoilm etilfenii. y 2-fluoreniluretanos. W itlcn y Rcid, J. A m . Chem. Soc.,
69, 2470 (1947).Sales de p-toluidina de ácidos alquílsulfúricos. Barlon y Yoiing, J. A m . Chem.
Soc., 65, 294 ( 1S43)2J4-dinhrOÍcnilhidl,aíOnas de compuestos carhonilicos a p a rtir de alcoholes. Duke
y W itm an, Anal. Chem., 20, 4 9 0 ( ! 9 4 8 ) .Tetracloroft¿latos ácidos de Certndqutto. Ff^sler y Shriner, / . Am. Chem. Soc.,
5 8 , 1334 (1 9 3 6 ) .Derivados de éteres monoalquíticos del eitlco glicol y dictUen glieol. Masón y
M anninfi, / . A m . Chem , Soc., 62, 1$35 (1910); M anning y Maso», ibid., 62 , 3136 (1940).
Eteres tnfilieos de celoaolv«, carbitole* y glícole? afines. Scikcl y Hunlrcas, / . A m .Chem. Soc., 63, 593 (1941); C reen y C reen , ibid., 66, 1610 (1944).
Derivados de O-álquiUacarina. Mcadoe y Rcjjí, J. Am . Chem. Soc., 65, 457 (1943). 3,4r5'triyodobv..'.i!Oatos< O ’Donnell y Carey, / . A m . Chem. Soc., 68, 1865 (1946);
O*í>onnell, Kellcy, O ’M atlcy y UpUam. ibid., 70, 1657 (1948).

r<o
268 P re p a ra c ió n d o d e r iv a d o *
а) Fenil y cA-nafiiluretWtOs. En un tubo de eusayo se coloca un gramo del alcohol o fenol anhidro y se añaden 0.5 mi de fenilisocianato ®o «-naítilisocianato. Si el reactante es un fenol, la reacción deberá ser catalizada por adición de 2 a 3 gotas de pirídina o trietilamína anhidras. Si no ocurre una reacción espontánea, deberá, calentara la solución en baño m ana durante 5 minutos» Entonces se enfria en un vaso de precipitados con hielo y las paredes del tubo se raspan con una varilla de vidrio para inducir la cristalización. Se purifica el urctano disolviéndolo en 5 mi de éter de petróleo o tetracloruro de carbono, filtrando la solución caliente y enfriando el filtrado en un baño de hielo. Los cristales se filtran y se ¡secan sobre una placa porosa; entonces se determina el punto de fusión»
б) D lfenüuretanos. Se mezcla medio gramo del fenol con 0.5 g de cloruro de difenilcarbamilo y la mezcla se calienta durante algunos minutos con llama baja. El producto solidifica al enfriar la mezcla dentro de un bario con hielo y se purifica por recristal&ación de éter de petróleo, tetracloruro de carbono o benceno.
c) p-introfenllvrfi¿ano*. Medio gramo de £-nitrofenilusoc¡anato en 5 mi de benceno se trata con 0.5 g del alcohol. <£e produce una reacción espontánea que se completa en unos 5 minutos! Se evapora el benceno en un baño m ana y el residuo se calienta con 10 mi de tetracloruro de carbono. La solución se filtra en caliente y el tetracloruro de carbono se evapora a baño maxía hasta un volumen de 1 a 2 mi. Guando se enfría, esta solución precipita\v\ uretano, el cual se filtra.
d) p -n itro fen ilu re tarw f a p a r tir de oxidas de ácido. Durante2 horas se calienta a reflujo una mezcla de 0.5 g de ¿'mtrobenzazida, 1 g del fenol o alcohol y 10 a 20 mi de ligroína seca (p. eb. 80-125°; después se deja enfriar y los cristales del uretano se filtran. Frecuentemente el derivado se aísla en estado puro, pero sí es necesario puede recristalizarec de lígroína, benceno o acetato de etilo.
U n procedimiento análogo puede seguirse para la preparación de uretanos a partir de otras benzazidas substituidas.
P roced im íen to 1 0 , B enzoatos y p-nitrobenzoaios
a) Se disuelve un mililitro del alcohol en 3 mi de pirídina anhidra y se añaden 0.5 g de cloruro de benzoílo o ¿-mtrobenzoUo. Después que ha cesado la reacción inicial, la mezcla se calienta durante un minuto con flama baja y se vierte, agitando vigorosamente, en 10 mí de
Procedimiento 9. UreUmoi
* | Precaución! Loi botUnattrt soci bcnntó^onw.

agua. Se deja que el precipitado se asiente y se decanta el líquido sobrenadante. El residuo se agita perfectamente con 5 mi de .solución de carbonato de sodio al 5%, se fDtra y se purifica por recrhtalízación de alcohol.
b) Un mililitro de] alcohol se meacía con C.5 g de cloruro de ben- zoilo o de p-nítrobenzoílo y se hierve con llama baja durante algunos minutos. La mezcla sé vierte en agua y se purifica como en (a).
P roced im ien to 1 1 . Acetatos
a) Tres gramos del compuesto políoxhidrilado anhidro se mezclan con 1.5 g de acetato de sodio fundido y pulverizado y 15 mi de anhídrido acético. La mezcla se calienta, en baño mana, agitándola ocasionalmente, durante 2 horas. Al finalizar este tiempo la solución caliente se vierte, con agíf-'cíón vigorosa, en 100 mi de agua helada. Se deja reposar la mezcla, a lta n d o ocasionalmente, hasta que el exceso de anhídrido acético se ha hidrolizado. Los cristales se filtran, se lavan perfectamente con agua, y se purifican por recristaíización de alcohol*
b) JDos gramos del compuesto políoxhidrilado se añaden a 20 mi de pirídina anhidra. Se añaden ocho gramos de anhídrido acético, con agitación y después que ha cesado la reacción inicial, )a solución se hierve a reflujo durante]# V 5 minutos. La mezcla se enfría y se vierte en 50 a 75 ral de aguamelada. El derivado acetUado se filtra, se lava con ácido clorhídrico al 2 ^ , frío, y después se Iasra con agua. Se purifica por recristalización de alcohol.
Procedim iento 12, 3,5-D Ínitrobenzoakrt 269
P rocedim iento 1 2 . ’’ 3 ^-d in ílro b en zo a to s
Aproximadamente 0,5 g de cloruro de 3,5-dinitrobenzoílo se mezclan con 2 mi del alcohol en un tubo de ensayo y la mezcla se hierve suave-
. mente durante 5 minutos. Entonces se añaden 10 mi de agua destilada y la solución se enfría en un baño de hielo hasta que solidifica el pro- ducto. El precipitado se filtra, se lava con 10 mi de solución de carbo- nato de sodio al 2% y se recristalíza eti 5 a 10 mi de una. mezcla etanol - agua de una composición cal que cl ester se disuelva en la solución caliente, pero que se precipite cuando Se enfríe la solución. Después qye los cristales se han filtrado y se han secado sobre una placa porosa, se les determina el punto de fusión.
Si no se disjxme de cloruro de 3,5-dinitrobenzoílo se le puede preparar mezclando en ur> tubo de ensayo 0.5 g de ácido 3,5-dinitro- benzoico con 1 g de pentaclorvro de fósforo. La mezcla se calienta suavemente para iniciar la reacción. Después que ha cesado la rápida reacción inicial, la mezcla se calienta durante unos 4 minutos a la tcm-
■ peratura necesaria para que se produzca un burbujeo vigoroso. La mez-

2 7 0 P rep a ra c ió n d e d e r iv a d o s
cía, aún líquida, se vierte soorc u» vidrio d i reloj y se deja que la masa solidifique. El material se pasa a una placa porosa limpia y se comprime con una espátula para eliminar el oxicloruro de fósforo. El cloru/o de acilo residual s£ usa inmediatamente para la preparación del derivado como se describió antes.
Los 3,5-dimtxobenzoatos también pueden prepararse por el método de la piridina descrito en el procedimiento 10(a).
P rocedim iento 13* 3<^xilrof¿alalox
o) -A partir do alcoholes <ju* h ierven a m enos de 150*. Unamezcla de 0,4 g de anhídrido 3-nitroftálico y 0.5 mi do alcohol se hierve suavemente en un tubo de ensayo provisto de un tubo de vidrio que sirve como refrigerante. R) calentamiento se continúa durante 5 a 10 mi* ñutos más después de que ’a mezcla se licúa. La mezcla se enfria, se diluye con 5 mi de agua y _ calienta hasta ebullición. Si la disotucíón no es total se añaden otros 5 a 10 mi de agua caliente. La solución se enfría y el etier te deja cristalizar. Algunas -veces el derivada se separa como un aceite y debe dejársele reposar hasta el día siguiente para que cristalíce. El producto se rccristaiLza una o dos veces de agua caliento,
b) A parlir de, alcoholes que h ierven a tn¿8¿4e¿150\ Una mezcla de 0,4 g de anhídrido 3-mtroftáüco, 0.5 g det -áicohol_y 5 mi de tolueno scco se hierve hasta que se ha disuelto todo el ar®clrido y la ebullición se prolonga 15 minutos más. Después se eliminad :el- tolueno a] vacio, usando una trompa. El residuo se extrae dos veces- con '5' m i de agua caliente, el aceite residual se disuelve en 10 mi de alcohol a} 95% y la solución se callenta hasta ebullición. Si la solución caliente no es transparente deberá filtrarse. Se añade a$oa a la solución caliente hasta que se produzca una turbíde* que desaparezca al añadir una o doa gotas d« alcohol. La solución se deja enfriar lentamente y finalmente se deja en ripioso. Muchos da los 3-nitroftalatos de alquilo superiores, derivados de los éteres monoalquílicos del ctilenglícol y del dietilenglicol, se separan como aceites y deben dujarse reposar durante varios días para que solidifiquen ► Ocasional mente el tolueno puede substituir a la mezcla alcohol^agua para la recmtalización. Algunas veces es aconsejable determinar el equivalente tic neutralización del ft^lato ácido de alquilo así como su punto du fusión.
P rocedim iento 14 . Su lfa to* á c ido t do a lquilhenciltiouronio
Unas 5 gotas del alcohol se añaden a una mezcla de 4/gotas de ácido clorosulfónico y ,r> m>tas de dio\<mo. Si no se desprende inmedia- ta/ricnie cloruro d<: hidrógeno, la mezcla resultante se calienta suavemente, con agitación, y sy deja reposar durante 5 ó 10 minutos. Entonces,

Aldehidos 271
después de la adición de 1 mi de agua, se añade 1 mi de una solución acuosa saturada (o una. solución alcohólica al 15%) de cloruro de S-benciltiouronio. Si en unos cuanto» minutos no se forman cristales, la solución se enfría en un baño con hielo. Los derivados de los alcoholes de bajo peso molecular (hasta el rc-hexilo, inclusive) pueden rtcristalújarse de etanol al 10# y los derivados de los alcoholes superiores, de etanol al 50$r.
ALDEHIDOS
(Enlistados en las págs. 343-344)Los derivados más útiles de los aldehidos son las íenil-, p-bromo-
fenil-, />-nitrofenil- y 2-4-dinitrofcnilh id rabonas (procedimientos 17 y 18). Si en la prueba de clasificación con la fenilhidm ina (experimento 21, pág. 164) se obtiene una fe ni Ih id razona sólida, por supuesto que se usará como derivado. Sin embarg^a-lá^ fenilhidrazona es líquida, se puede recurrir a una de las fcn ilh id r^ ^ ^ s iib ^ jttiid a s ; de éstas, las 2,4-dmitro- íenilhid razonas son recomeñdábte^ebijdo a 'quc hay mayor probabilidad de que sean sólidas (|xnocodtim ^ a@ B)j
R^> C = 0 + HaN N H í ^ ) K p 4 ^ ^ > C ^ N N H ^ ^ N O ? 4- H.O
■3.. \ . 2,4*dirtitíO[«nil}l!dF»«>na.El reactivo de dimtrofentlhidrazina, generalmente consiste en una
solución de dinitrofenilhidrazína en .etanol acuoso y ácido sulfúrico. So. ha reportado que en algunos 'Cásós la dinitrofenilhidm ina disuelea en el éter dimetílico del dierilengliroljícon ácido clorhídrico como catalizador, es superior al reactivo con ácido sulfúrico.
Para los aldehidos .solubles eñ agua» de bajo peso molecular, a menudo es ventajoso emplear corno reactivo !a semicarbazida [procedí' miento l6 (a ) ó 1 6 .(6 )]. Todas las scrnicarbazonas son sólidas y generalmente pueden obtenerse casi puras sin recristaíización. Algunas veccs estos derivados se forman lentamente y debp. tenerse cuidado de dejar la reacción el tiempo suficiente para que sC3yCompletar ;
R R *C=0 4- H,NNHCONH2 -* \c=NNHCOKH? 4- H20
ScimcWUttoaa
A menudo pueden usarse eximas {procedimiento 49) i>ero, en general, es probable que estos derivados fundan má¿ bajo que las corres- pbndíentea 2,4*¿ vútrofeni lltidrazónas y semicarbazonas. Una ventaja de las oximas como derivados es que, por lo general, se forman rápida

272 Preparación d e d«rivodo*
mente. L a reacción entre loes compuestos carboníceos y la hidroxilamina es reversible y debe tenerse cuidado para evitar t\ contacto innecesario con soluciones fuertemente ácidas; bajo estas condiciones la oxima puede regenerar el compuesto original.
Los aldehidos de alto peso molecular a menudo se identifican convenientemente por referencia a los ácidos correspondientes, los cuales se forman cuando el aldehido se oxida [procedimientos 15 (a) y 15(i>)].
Otros derivados que también han sido recomendados para los compuestos carbonílicos son Jas fcnilsemtcarbazonas, las a - y (3-naftílhidra- zonas» las o*, m- y p-tolilsemicarfeazonas, «- y P-naftilsemicarfeazonas, f-xeoilsemicarbazonas, 3j5-dinitroFeml$emicarbazonas, o- y p-clorüben- zoh id razonas, 3-nitroberwhídiazonas, nitroguanilhidrazonas, difenílhidra- zonas, benzotiazoles, bcnzotiasoluia¿ yck* productos de condensación con la metona (dimedona) {proce<
Tanto los aldehidos como las ce ton as reaccionan fácilmente con carbonato de amonio y cianuro de potasio o sodio para producir hidan- ioínas substituidas. Estas, son sólidos cristalinos y sirven como derivados útiles.
C H ^-C O\
i/
CH2 + RCHO
CHj—CO
H (R )
HN NH

Aldehidos 273
2,4-dinhrofenUW naorLaí, Alie*», J. A m . Chem. Soc., 32, 2955 (1930); Brady> / . Chem. Soc., 1931 756; S train, J. A m . Chem. S a e 57, 758 (1935}; Campbell, Analyst, 61, 391 (1936) ¡ Íddleí, Low, Roicn y Kart, In d Eng. Chem., Anal. Ed., 11, 102 (1 9 3 9 ); Alien y Rrchmond, / . Org. Chem., 2, 222 (1937); Johnaoo, / . A m . Chem. Soc., 75, 2720 (1953); Johmon, ¡bU.,73, 5838 (1951); Brady y J a n e » , / . Chem. Soc., 1950, 1021.
2,4*dihítrofednhi<!r3zcnas en ¿ter dim ei3íco del ctiicnglicol como dkolvente. Shinc.J . Org. Chem., 24, 252 (1959).
B eitioúazolo y benzotiazdtnas, L an idnva y Sharnoff, / . Atn. Chem. Soc., 53, 2654- (1931),
P'O éftílhiiim onas. Lee, Sah y Kao, $ci. R cp lt. Nati. Tsing Hua Unto., (A) 2, 335 (19S4),
p-clorobcnzofaMmonas, SJjlh y Salí, 5c/. Reptr. N ati. T i in t tiu a Univ., (A) 2, 353 (1934).
«Nclorol 'teohidraronaí, Sun y Sah, Sci. Reptr. N ati. Tsing Hua Univ., (A) 2, 359 1934).
m-yodob«itzphiártjonas, 5ah y Li, J . Chínete Chem. Soe., 14, 24 (1946) [{?. A-, 43, 6973 (1949)].
j7*yadobeaxohidTíuoníL£. Sah y HiU, Rec. Trav. Chin:., 59, 349 (1940); DeGaudt* marU y Duhoií, Bult. Soc. Chim. Pranee, 1950, 63.
í-mcntl& idraronas. Wcodwand, K o h m an ,y HarrLj, / . A m . Chem. Soe., 63, 120 (1941). ■■
K ítrnguanilhidm oiias, Whítmorc, Rírvukas v Smith, / . Am. Chem. Soc., 5?, 706(1935).
DiíenjlHidrazónm. Mauixnbrcchcr, Ber., 39, 3563 (1906).Scmlgarbíaoiias. Shríncr y T urncr, J. A m . Chem. Soc., 52, 1267 (1930). Derivados de metona. W einbergcr, Ind . Eng.. Chem., A n a l . Kd., 3, 365 (1931);
H om ing y R om ing, J, Org. Chem., 11, 95 (1946).P'toliíseoucaibazoa&s. Sah y Leí, J. Chínese Chem. Soe., 2, 167 (1934). S-oztrobegxohídrazonaLS. Strain, / . A m . Chem. Soc., 57, 758 (1935); M en$ y Sah,
Sci, Repts. N etl. Tsíng Uva. Univ., (A) 2, 347 (1934).M crcap taks con ácidos aurcaptocazboxf Ileos. Stfcier y Lover, / . Am . Chem. Soe.,
74, 5576 (1952).2-nhrú-4'm etilbcnzohIdm ona2, Sah y W ang. / , Chínese Chem. Soc., 14, 31
(1946) (C , A ., 43, 6972 (1949)].S^^raetB^-xütrotK& zohídrazona*. Sah y Ltu, / . Chínese Chem. Soc., 14, 94
(1946) íC . A ., 43, 6972 (1949)1.2,6-diiútiü*4<iu«tübczizohidra2DrLa5. Sah, J . Chínese Chem. Soc., 14, 45 (1946)
fC. A., 43, 6972 (1949)].o-tojüscmícarbazon&s. Lci, Sah y Shih, / . Chine¡e Chem. Soc,, J f 246 (1935). m -nitrobcncensulíonhídm onas. Clamaron y S torne, J. Chem. Soc., 1934; .1330. Fcnllscqúcartmonafc Sah y M a, / . Chínete Chem. Soc., 2, 32 (1934). ' iK-tolUsemicarbazotm. S>h, Wang y Kao, / , Chínese Chem. Soe., 4 , 187 (1936)'. a-uaítílwroicar& ajonis. Sah y Chiang, /, Chínete Chem. S o c 4, 496 (1936). f3*iuítQ$eisfcartazona$' Sah y Tao, J. Chínese Chem. Soc., 4, 501 (1936), JjínlinítrofenlIscQiicsrijwoajií, Sah y T ao , / . Chínese Chem. Soc., 4» 506 (1936). ¿Ntenikemicarhiuonas. Sah y Kao, Rec, Trav. Chim., 58, 459 (1939).
Refe reacia*
\
ftft
ft
j» .>
-

Hidanloír** substituidas. Hcnze y Specr, / . / Í th. CAtfnt. 64, 522 (1942); también Am. Doc. Ir.it.. Doc. N* 1603.
Sales de bcDciltioaionio de ios compuestos bisulfíticos de aldehidos y cetou&s. Von Vr'acdt y K ra t2l, B t r 76, 1209 (1943).
o-cirboxifcTi2bidnzotias. Zcltner, M onaisk., 80, 330 {1551),p-carboxifenSUúdrarcnas. Veibcl, Blaaberg y Siesos, D antk Tids. P<xim., 14, 3fi4
(1940) ÍC. A., 36, 2495 (19*2); Veibel, MonaUh, 81, 330 (1951)].AnKdro 4.-amiooiaorfolÍB&s y 4-(¿>-aminofcol]) m&rídmas. Dugan y Haendfer,
/ . Am. Chem. Soc., 64, 2502 (1942),2,4^ÜoitrofeniUeniicacbaaon3s. M cV tigh y Rose; / . Ckem. Soc., 1945, 713,a - o ^oaítíUf.tnio*am]zídas» Sah, Yang, Chin y Wang, t. Chínese Chttn. Soc.,
14, 101 (1946) [C. A v 43, 6973 (1949)].5-ícniIamjDosemioxafljazi<b. Sah y W ang, / , Chínese Chem. Soc., 14, 39 (1946)
[C . A., 43, 69? 1 (1 9 4 9 )].5-{a-{enüetn)ttmiox4aii2Íd3. Leonard y Boyer, ] . Org. Chem.., 15, 42 (1950).G ira rá , Reactivos-de. G irard y Sandulesco, H e h . Chim. Ac(a, 19, 1095 (1936).AminobLsulfíticos productos de adición. Adams y GaHjcr, J. A m . Ckern. Soc.,
' 71, 522 (1949).p-ditnetilanúüoanílos de aldehiv "'s, U tzm ger y RegenasB, He!v. Chim. Acto, 37,
1901 (1954),Carbón»tcná* y caíbcloxihíd!razonas. Rabjohn y Barnstorff, / . Am . Ckem. Soc.,
75, 2259 (1953).p-tolueasutfom to de JY-nKtK-S-carbohídnuidofiridmk), Derivados d t, A lkn y
G atet, / . Org. Chem., 6, 596 (1941).. .Tioscmicarbazonas. Sah y Daniels, R íe . T tqv. C kim ,, 69, 1545 (1950),
274 Preparación de derivados
P rocedim iento 1 5 . O x idac ión hasta ácidos
a) M étodo del perm cm ganato. A una solución o suspensión de1 g del aldehido en 10 a 20 mi de agua a la cual se le han adicionado unas gotas de solución de hidróxido de sodio a] 10%, se le añade una solución saturada de pentianganato de potasio en agua. La mezcla se agita vigorosamente y se añade suficiente permanganato para impartirle un color púrpura definido. La mezcla se acidula con ácido’ sulfúrico diluido y se añade solución de bisulfito de sodio hasta que el perman- ganato y e! dióxido de manganeso se hayan convertido en sulfato de manganeso. El ácido se filtra y se recristaliza de acetona-agua. Si el ácido no precipita se puede extraer con cloroformo o éter.
b) M étodo del p eróx ido de h idrógeno (a g u a oxigenada). En un matraz de 500 mi se colocan 20 mi de solución de hidróxido de sodio al 5% y 30 mi de solución de peróxido de hidrógeno sil 3%- La solución se calienta hasta 65*70°C y se añade 1 g del aldehido. La mezcla se agita y se mantiene a 65°C durante 15 minutos. Sí <1 aldehido no se ha disuelto $c pueden añadir unos mililitros de etanol. Se adicionan otros 10 mi de peróxido de hidrógeno y la mezcla se calienta durante 10 minutos mas. La solución se acidula al rojo congo y el ácido que precipita se filtra.

Procedim iento 18. 2 r4-Dín¡trofetilthidrazona$ 275
SI el ácido es wluble en agua ea mejor neutralizar Ja solución a la fenolftaleína y evaporar a sequedad. Entonces, la sai de sodio se conviene ert el derivado apropiado por el procediruiento 1, 2 ó 3.
P rocedim iento 16 . Sem icarbasonas
a) Para com puestos solubles en agua. U n mililitro del aldehido o de la cetona, 1 g de clorhidrato de semicarbazida y 1.5 g de acetato de sodio se disuelven en 10 mi de agua contenida en un tubo de ensayo. La mezcla so agita vigorosamente y el tubo de ensayo se introduce en un vaso de precipitados con agua caliente y se deja en éí hasta que se enfríe el agua. Entonces, se coloca en un vaso de precipitados con hielo y se raspan las paredes del tubo con una varilla de vidrio. Los cristales de la semicarbazona se filtran v se recristalizan de agua o etanol del 25 al 50%.
b) Para com puestos ínsolubles en agua. U n mililitro del aldehido o de la tetona se disuelven en 10 mi de etanol. Se añade agua hasta que la solución esté ligeramente turbia; la turbiedad ss elimina con unas gotas de etanol. Entonces je adicionan 1 g de clorhidrato de semi- carbazáda y 1.5 g de acetato de sodio y de aquí en adelante se sigue el procedimiento (a).
Procedim iento 17. p-nilro fenilh idraxonas
U na mezcla de 0,5 g de ¿-nitrofenilbidrazina, 0.5 g del aldehido o de la cetona y 10 a 15 mi de etanol se calientan hasta ebullición y se añade una gota de ácido acético glacial. L a mezcla, se mantiene caliente durante unos cuantos minutos, y si es necesario se añade más etanol hasta obtener una solución transparente. La solución se enfría y la p-nitrofenil- hidrazona se filtra. Puede remstalizársele en una pequeña cantidad de etanol.
Si el derivado no precipita al enfriar la solución, la mezcla se calienta hasta el punto de ebullición, se añade agua hasta que Ja solución esté turbia y entonces se añaden una o dos gotas de etanol para clarificarla. La hidrazona que precipita al enfriar se rern'stalíza de etar.cl-agua.
P rocedim iento 18. 2 t4-dini¿rofeniJhidrasonas
Se prepara una solución de 2,4-dinitrofenilhidrazina de la manera siguiente: A 0.4 g de la 2,4-dinitrofonilhidra2Ína, contenidos en un matraz Erlenmeyc'r de 25 mi, se le añaden 2 mi de ácido sulfúrico concentrado. So adiciona agua (3 m i), gota a gota con agitación, haata

que la disol ación sea total. A esta solución caliente se le avaden 10 mi de etanol al 95% .
Se prepara, una solución del compuesto carbonilico en etanol disolviendo 0.5 g deí compuesto en 20 mi de etanol al 95%. Se adiciona ía solución recientemente preparada de 2,4-dinitrofenilhidrazina y la mezcla resultante se deja reposar a temperatura ambiente# Ordinariamente la cristalización de la 2,4-dinitrofenilhid razona ocurre a los 5 ó 10 minutos. Si no se foima precipitado, la mezcla se deja reposar hasta el día siguiente»
Ordinariamente, la recristalización puede efectuarse en la forma siguiente: La 2-4-dinitrofenílhídrazona en 30 m i de etanol al 95% se calienta sobre el baño maría. Si se disuelve inmediatamente, se añade agua Lentamente hasta que se llega al punto de turbidez o hasta que se ha adicionado un máximo de 5 mi de agua. Si la dinitrofenilhidrazona no se disuelve, se añade lentamente acetato de etilo a la mezcla caliente hasta que se logra la disolución. Se filtra la solución caliente a través de un filtro de pliegw y se deja reposar a temperatura ambiente hasta que la cristalización sea total (aproximadamente 12 horas).
La reacción en éter dimetflico del dietilenglicol se efectúa con una solución de 4 g de 2,4-dimtrofenilhidrazina disuelta con calentamiento en 120 m i del éter y dejándola en reposo durante varios dias. A 5 mi de esta solución, a tem peratura ambiente, se le añaden 0.1 g del compuesto carbonilico en 1 mi de etanol al 95% o en 1 mi de éter dimetílico del dietilenglicol y después tres gotas de ácido clorhídrico concentrado. Si no hay precipitación inmediata de la hidrazona, la solución deberá diluirse con agua y dejarte reposar.
P roced im ien to 19 . D erivados d e m e tona con aldehido*
a) P roducto$ de condensación de bism etona-aldehído. A una solución de 0.1 g del aldehido en 4 in! de etanol al 50% se le añaden .0.4- g de metona, ú el aldehido es alífático. Sí es un aldehido aromático sólo se usan 0.3 g de metona. Se añade una gota de piperidina y la nmestxa se hierve suavemente durante 5 minutos. Si al final de este tiempo la solución caliente es transparente, se añade agua, gota a gota, hasta el ounto de turbiedad. Entonces se enfría la solución v el derivado.
776 Pr«p«m<(6n d» derivados
derivado se ^cristaliza do mezclas de metanól y agua,b) C iclitación « octakidroxan tenos substitu idos. Si es conve
niente preparar un derivado adicional, el producto de condensación de metona anterior puede convertirse en un octahidroxanteno substituido hirviendo una solución de 0,1 g del derivado de metona en etanol al 80% (3 a 6 m i) al cual se íe ha añadido t gota de ácido clorhídrico concentrado. La cidisadón es total en 5 minutos, después de este tiempo

A m idas e ímídas 7.77
se añade agua hasta cl punío de turbiedad. La mezcla se enfría y el xart- teño substituido S2 filtra, Este producto ordinariamente es puro pero puede ^cristalizarse de roetanol acuoso.
AMIDAS E IMIDAS
{Enlistadas en las págs. 345-348)
El método más general para caracterizar amidas consiste en hídro- lizadas con ácidos o álcalis [experimento 26(4), pág, 186].
RGOKH*+ H20 + HCl -► R C O O H + NH,CI R C O N H j+N aO H -► RCO O N a+N H ¿
La hidrólisis de las amidas substituidas produce aminas primarias o secunda ;as en lugar de amoníaco.
R C O N H R '-f NaOH -> R C O Q N a+ R 'N H a R C O N H j'-f NaOH RC O O N a-f R /N H
La identificación se realiza por caracterización de los productos, es decir, el ácido, el amoníaco y la amina primaria o secundaria. En las tablas 20 y 24 Be dan los punto* de fusión de algunos de estos productos de hidrólisis y de sus derivados.
El cloruro de ftaiilo reacciona con amidas no substituidas para producir W-acilftalimidas, las cuales son derivados sólidos útiles.
C O
+ RCONH2 ^ N C O R + 2HCI
COEl xanthidrol reacciona fácilmente con amidas no substituidas e
imidas formando ^acilamidoxantenco, los cuates son buenos derivados (procedimiento 20).
NHCOR
k c o n h ^ Q ^ Ü + H i 0
Las amidas ^-substituidas no se condensan.£1 óxido de mercurio reacciona con determinadas amidas ‘for
mando ■N^'-mercuribísacilamidas, las cuales son derivados sólidos de punto de fusión elevado.
H gO + 2RCONH, Hg (NHCOR) 3+ H 20

276 Preparación de derivados
Algunas amidas forman sales estables con el áddo oxálico3 que sirven para su identificación.
Los barfjituratos pueden convertirse en derivados de IVJV'-p-ni.tro- bencilo por reacción con bromuro de /j-nitrobencilo mediante un procedimiento análogo al usado para la preparación de los ésterfó ¿-nitro- bencílicos de k>s ácidos {procedimiento 1).
JV-Adtftalímidas. Eviwu y l 'eh n , / . A m . Chem. Soc., 51, 3653 (1929).Oxalatcs de amidas primaria*' MacKenüie y R aw ltt, InJ . Eng. Chem., Anal E4„
12, 737 (1940). i.9-AcilamjdoraíU<n<*s. Phillips y P itl, J. A m . Chem. Sac., 65, 1355 {1943); Phillíp.<¡
y Praaik, / . Org. Cherri., 9, 9 (1944).NA-McrcuribisacilámidLu. Williami, FLainev v Leopold, / . A m . Chem. Soc., 64,
1736 (J942).B.irbiluratos de jV,/V'-jvDÍtrobci»ciio. Caalle y Poe, J. Am. Chem. Soc., 66, 14*0
(1944).
Aproximadamente 0.5 g de xanthidrol se disuelven en 7 mi de ácido acético glacial. Si la solución no es transparente (debido a los productos de desproporción del xanthidrol), se deja reposar unos minutos o se centrifuga y la solución transparente se decanta en un tubo de ensayo limpio, A esta solución se le añaden 0.5 g de la amida y la mezcla se calienta a 85°C en un vaso de precipitados con agua durante 20 a 30 minutos. Después de enfriar, el acilamídoxantcno se filtra y se recrista- liía de una mezcla de 2 panes de dio>cano y 1 parte de agua.
Algunas amidas no se disuelven en ácido acético, y para convertirlas . en el derivado hay que usar una mezcla de 5 mi de etanol, 2 mi de ácido acético glacial y 3 mi de agua como disolvente para la reacción.
\ / CONH
+• R&C
CON—CH2QH<NQ í(/>)
: co
VC 0 N ^ C H 2Q H 4N 02í/3)
Referencias
P r o c e d im ie n to 2 0 . 9*A cil<im idoxanXeno3
AMINAS — PRIMARIAS Y SECUNDARIAS
(Enlistadas en las págs. 349-357).
Los tipos más útiles de derivados de las aminas primarias y secundarias son las correspondientes acetamidas y benzamidas, las feníltíoureas y fenil y ¿-toluilsulfonamidas. De éstas, probablemente las bencensul-

Amina» — Primaría* y secundarías 279
fonamidas se usan con más frecuencia debido a que la preparación de estos derivados está involucrada en el método de Hinsberg para la clasificación de aminas [experimento 4, pág. 135, procedimiento 22(a)]> Cuando se encuentra que las bencensulfonarotdas son inapropiadas, se puede recurrir a numerosos derivados similares, cada uno de los cuales presenta ventajas en la identificación de ciertos tipos de aminas. Se recomiendan Jas p»tolucn~ [procedimiento 22(a}], ¿-bromobencen- [procedimiento 22(¿>J, ?n-nitrobencen~ [procedimiento 2 2 (¿ )] f metan- [ procedimiento 22 (6 )} y a-naftilsulfonamidas [procedimiento 22(6)].
Las feniltioureas son especialmente valiosas para caracterizar aminas solubles en agua y de bajo peso molecular. Estas se forman por reacción con c! fcnilisotiocianato (procedimiento 23),
RNH2 4- CíHsNCS —► CdHjNHCSNHR R^NH 4- CUHjNCS CéH^IHCSNRj
El reactivo de fenilsotáocianato no es sensible al agua; esta reacción puede llevarse a cabo con soluciones acuosas diluidas de las aminas.
Las amidas de los ácidos acéteico [procedimiento 21 {<*)], benzoico [procedimiento 2 l(fe )] y ¿-nitrobenzoico [procedimiento 21(¿)] Se preparan convenientemente tratando la amina respectivamente con anhídrido acética, cloruro de benzoílo y cloruro de ¿-nitrobenzoilo.
RNH2+ (G H jC O íiO -^ RNHCOCH3+ C H 3COOH 2RNH¿-J- C*H5COCl RNHCOC íH s+ R N H jCI
2RNHt + O ^ Q C O C I -v O jN / V oNHR + RNH,CI
Para casi todas las aminas, primarias y secundarias se conocen los derivados acetilados y benzoilados, por esta razón estoa derivados son muy útiles.
El anhídrido 3-nitroftálico reacciona con las aminas primarias y secundarias produciendo ácidos f talármeos. El ácido ftalámíco producido por una amina primaria sufre úna deshidratadón cuando se calienta a 145°C y forma la aquil>3-nitraftalimida. El de la amina secundaria es estable al calor.
" " 'N e o
U )0n o 2
KNHrp r 00" a s . p f Vk ^ C O N H R
NO,

Por ío tanto, este reactivo proporciona un medio adicional para diferencia* aminas primarias, sccrund arias y terciarias y proporciona derivados para las aminas primarias y secundarias»
Otros tipos de derivados que « ha encontrado son valiosos en ia caracterización de las aminas son los derivados del 2,4-dinitrobenzal, los azocompuesv» (experimento Í9, pág. 158), las sales del ácido p-to- luensulfónico, los compuestos moleculares con fenol, los compuestos con haíuros de ^nítrotHEncilo y los picratos (procedimiento 26),
Las am bas primarias y secundarias se combinan con los isocianatos de arilo o azídaj de ácido para producir ureas substituidas.
A rN = C = 0 + R N H j------ > ArNHCX>NHRAri\r= C = 0 4 -R » N H -------► ArNHCONRj
ArCONj-------► A rN = C = O ArNHGONHR
Se han descrito las íenit-, a - y (S-naftil, m- y />-bromofenil-, m- y /r-cloro- fenil-, m- y ¿-nitrofenil- y 3>5-dinitrofenilureas.
Varios clorhidratos de aminas se pueden obtener burbujeando cloruro de hidrógeno seco en una solución de la anima, en éter o benceno. Algunos de los clorhidratos tienen puntos de fusión o descomposición satisfactorios y, por lo tanto, pueden servir como derivados.
U na forma más apropiada para preparar los clorhidratos de los amina-alcoholes consiste en neutralizar las soluciones en alcohol propílico de las akanolamínas con cloruro de hidrógeno seco,
2 8 0 P r v p o r c K l o n d e d e r i v c d o j
Referen cím
P^neDObiakxaouffqiiamidííi, Marvel. y Smíth, j. Am. Chem. Soc., 45, 2696 (1923),
m-NitrobccccESU liona mí das. Marve, Kingsbury y Smilh, ]. Am. Chem, $QC„ 47, 166 41925).
|>-Benc«lUUObcDccnsuiÍQflaa]idis, Wooltolk, Réynold» y M&JOft, J. Org. Chem., 24, 1445 (1959).
Bendbulfooanitóai. Marvel y Gil3esj>ie, / , Am. Chem, Soc., 48, 2943 (1926),Mctamugcaiaoitdat, Marvel, Helírick y Belsley, J. Am, Chem. Soc,, 51, 1272
<1929),Fenil- y o-toiíltioureM. Octerbacher y Whícmore, / . Am. Chem. Soc., 51, 1909
(1929).«t-NitroíeaiiuoFaneafl, Sah y Lci, /♦ Chinen Chem, Soc., 2, 153 (1934-),ir-Yodcítuiluicaa. Sah y Waixg, Rec. Trav. Chim., 59, 364 (1940),Gorbidr&tc* de amjiKekcbaks. Jon«. 7, Assoe. Offic. Agr, Chemtttf, 27. 467
(1944).Axüwlfojuipida» de imÚMHÜcohofe*. Shupc, 7, Ame. Offic. Agr. Chemiils. 25,
227 (1942).2 {-Diiütnibcnzal, Derivados del» Lowy y Wescott, /. Am. Chem, Soc., 42, 849
(1920); Lowy y Dc*vney, ibid., 43, 346 (1921); 45, 1060 {1923}; Bennett y WillU, ). Cktm. Soc., 1928, 1962; Bennett y Pont, ibid., 1929, 1465.

Procedimiento 21* Aratdo* substituida* 281
Sale» del ««ido p~toluczLsulfónico. N oller y Lian#, / . A m . Chem, Soc., 54, 670 (1932).
Compuestos tnolccularc* coa fenol. Buehler, W ood, H u ll y Erwin, ] . A m . Chem.S o c 54, 2598 <1^32),
CofnpuMttra con haluroe de p-mfcrobencilü. Lyons, / . Am . Pharm.- Asioc., 21, 224 (1932).
Sale* d d ácido 3,5-<üflitrúb«ttJsOÍcó. Buehlcr, C urrier y L a^rence, JW . En*. Chem., Anal. E ¿.. 5 , 277 (1933).
Ar-(ArT3iait>amctilJ(taliiiuda5. VYitUtead y Hcine, / . A m . Chem.. Soc., 77, 3913 (1955).
NUfO-2-indaúdioaa'I,3, Salía de, C rktem cn, W ang, Davies y H am s, Ana!. Chem., 21, 1573 (1949).
m*Yodobcxua2¡da. Sah y Chen, } . Chínese Chem. Soc., 14,74 (1946) [C. A., 43* 7447 {1949)]
nt-Nitrobenzaaida. Karm um , Svcnsk Kem. T id ., 50, 61 (1948).4-M etü‘3-mtrol>cnz6zida. Sah, Lí, Tsung y W ang, J, C him sc Chem. Soc., I4T
84 (194$).Acidas de ácidos iY-Axilcnámicoí, Sah, Yu, C hía, Chcn y Chao, / . Chinase Chem.
¿oc., i '- . 52 (1946).2-Ison¡trosocii ’-^hcíÍlamÍaai. Birch, ] . Chem. S<x., 1944, 314.2,G‘Díxnetil-4'piricloi»s-'l-substitnidas. Chanvelíer, BtiU. Soc. Chim. France, 1949,
611.5-NitrobRriHtaratoi (dllU aniías). Dewey y Plein , Ind . Eng. Cheto., A n d , Ed.,
18, 515 (1946).Bisulfito». A dañu y Garbcr, / , Am . Chem. Soc., 71, 622 (1949).Uso de quiuonas halógena das. Buu-hoT, Royer y Ecfcert, Rec. Trau. Chim., 71,
1059 (1952) [tí. A.. 47, 0008 (1953)],¿Iquilam inam ctilcnm alonalci de etilo. Lappen, J. Chem. Edite., 28, 126 (1951), Sales del ácido 2,4'dinUrobenEtíica. Buchler y Catíce, In d . Eng. Chem., Am L Ed.,
6, 351 (1934).Sales de ácidos amraquIaoittulíónkoS' PeHtin y ScweJl, ] . Soe. C íen j. J71A., 42*
27T (1923).Sdíonbkacetam ictas. Aldea y Houston, J. A m . Chem. Soc., 56, 413 (3934). tt-Naíciitioürtaá, Suter y Mofíeíc, / . Am- Chem. Soc., 55, 2497 (1933).P'NaítíJ- y p-xeniltianrcat, B roun y Campbell, J. Chem. Soc., 1937. 1699. m -B rom ofeailurr» , Sah y Chang, Rec, Trat1, C him ., 58, 8 (1939), 3^-DiOitrofrmluTeas. Sah y M a, J t Chínese Chem. S o c 2, 159 (1934). j>-Clorofcn5IqrcM. Kao, Fai\ y Sah, / , Chínese C hem , Soc., 3, 137 (1935). tti-Nitrofenilurcai. M en# y Sah, / . Chínete Chem. Soc., 4, 75 (1936). j^Broraofcniluccas. Sah, KaO y W an$, }. Chínese Chem. $oc., 4, 193 (1936), hi-Cloroícnüurcas. Sah y "Wu, / . Chine se Chem, Soc., 4 , 53 3 (1936). (J.jSiítíJiireaj. Sah, / . Chínese Chem. S o c 5 , 100 (1937).Aminas y anhídrido S-nítroftálico, Alexartdcr y M cElvain, ] . Am . Chem. Soc.,
60, 2285 (1938).l-í2,4*Dim troícnU )3-alqm t(a arilju rcas, McVeígh y Rose, / . Chem. S*c, 1945,
621.
P roced im ien to 21* Anudas substitu ida* a partir de amináj
a) Acelam tdas substituidas a p a r tir de am inas insolubies en agua* Se prepara una solución de Ja amina disolviendo 0.5 g del compuesto en 25 mi de ácido clorhídrico al 5%. Se añaden cantidades

2 f l2 P rep a ra c ió n d e d e r iv a d o s
pequeñas de solución de hidróxido de sodio al 5%. hasta que la mezcla se enturbie y se elimina Ja turbiedad mediante la adiHón de 2 a 3 mi de ácido clorhídrico al 5%. Se añaden unos trocitos de hielo y en seguida 5 mi de anhídrido acético. La mezcla se agita vigorosamente y se adiciona, en una sola vez, una solución, preparada previamente, de 5 g de acetato dc'sodio (trihidratado) en 5 mi de agua. Si el producto no cris» talúa, la. mezcla se deja en el refrigerador durante Ja noche.
La recrista]«ación puede efectuarse en ciciohexano o en una mezcla de ciciohexano y Onceno. La acetamida debe estar perfectamente seca antes de que se intente recnstalizarla en estos disolventes. También puede usarse una mezcla de etanol-agua para Ja «cristalización.
b ) Benaamidas substitu idas. M étodo de la p irid ina . A una solución de 0.5 g del compuesto en 5 mi de píridina seca y 10 mi de benceno seco se le añaden, gota a gota, 0.50 mi de cloruro de benzoílo. La mezcla resultante se calienta en un baño maría a 6G-70°C durante 30 minutos y entonces se viertt "n 100 mi. de agua. Se separa la capa beneénica y la solución acuosa se- lava una .vez con 100 mi de benceno. Las soluciones bencénicas se reúnen, se lavan con agua y con solución de carbonato de sodio al 5% y se secan con un poco de sulfato de magnesio anhidro. Se elimina el agente secante por filtración a través de un filtro de pliegues y el benceno se evapora hasta un volumen pequeño {3 a 4 m i). La mezcla se agita con hexano (unos 20 mi) y los cristales de) derivado benzoilado se filtran y se lavan con hexano. Usualmcnte la reeristalización puede efectuarse en una mezcla de ciciohexano y hexano o en una mezcla de hexano y acetato de etilo. El etanol o el etanol acuoso también pueden usarse para muchos compuestos.
c) Benito y p-n itrobenzam idas substituidas. Puede emplearse el procedimiento usual para la reacción de Schotten-Eaumann descrito en el experimento l(e ) , pág. 129. Los siguientes son dos procedimientos modificados:
1. Una mezcla de 20 mi de solución de hidróxido de sodio al 5%, 5 mi de cloroformo, 0.5 g del compuesto y 0.5 mi de cloruro de fcenzoSlo se agita durante unos 20 minutos y se deja reposar durante 12 horas. Se separa la capa clorofórmíca y la capa acuosa se lava con 10 mi de cloroformo. Las soluciones cloroformices reunidas se lavan con agua, se secan con sulfato de magnesio anhidro y se evaporan hasta un volumen pequeño (2 a 3 m i). La solución se mezcla con hexano (20 a 25 mi) y se agita; el derivado se filtra y se lava con hexano.
2. Aproximadamente 1 mt de la amina se añade a una solución de 1 g de cloruro de bcnzoilo o clorvro de ^nitrobenzoílo en 20 mi de benceno seco. La solución resultante se hierve con un refrigerante de reflujo durante 15 minutos y se deja enfriar. La solución se filtra y el precipitado se lava con 10 mi de benceno caliente, los lavados se añaden al filtrado original. En seguida se lava la solución benccnica con 10 mi

Amincts — Terciario* 283
de solución de carbonato de sodio al después con 10 mi de ácido clorhídrico ai %% y finalmente con 10 ral d>. agua destilada. Entonces se evapora el benceno y el residuo se recristaliza de alcohol diluido.
P roced im ien to 2 2 . Sulfonamxda* a p a r tir de aminas
a) B ene en y p-to iuentu lfonam idas. Estos derivados se preparan por el procedimiento descrito en el experimento 4, usando cantidades suficientes de material para permitir la recristalizacíón del producto final de etanol a! 95%.
b) B encil, p-bromobenc¿n*t m*nitrobencén~, a-no/ui» y me- ta n su ífo n a m id a t. Se prepara una solución de 1 g del cloruro de sulfonilo en 25 mi de benceno seco y se añaden 2 mi de la amina. La soIucíód se agita y se deja reposar durante 10 minutos. El clorhidrato de la amina se elimina por filtración y el filtrado se evapora El residuo se recristaliza una o dos veces de etanol diluido.
ProcedímumU> 23 . F enikxou reas
En un tubo de ensayo se mezclan cantidades iguales de la amina y det fenilisotiodanaio y se agita durante 2 minutos. Si la reacción no ocurre espontáneamente la mezcla se calienta durante 3 minutos con llama d¿bi!. Ordinariamente las aminas aiifáticas reaccionan inmediatamente mientras que las aminas aromáticas requieren calentamiento. Después se mantiene ía mezcla dentro de un vaso de precipitados con hielo hasta que la masa solidifica. El sólido se pulveriza y se lava con li- groína y etanol al 5% para eliminar cualquier exceso de alguno de los reactivos. Entonces se recristaliza de etanol al 95%.
AMINAS — TERCIARIAS
(Enlistadas en las págs. 358-361)
Las aminas terciarias son de naturaleza tan variada que no se ha encontrado un tipo de derivado que sea de aplicación general. Quizá Jos derivados más útiles son fas sales cuaternarias de amonio formadas por combinación de la amina con el yoduro de metilo [procedimíen-
24( a ) ] el cloruro de bencilo [procedimiento 2 4 (6 )] o el /J-toluen- suífonato de metilo {^procedimiento 24( t ) ] .
R,N +CH 31 (RjNCHj)*I_
RjN+C HsCHp-* (RjNCWAH,)*CI-RjN - f - C H jC í H íS O jC H s (RiNCH3)<(CHiQÍI»SOJ)~

También se emplean con frecuencia salea de ácidos halogenados, ácido plcríco (procedimiento 26), ácido p-toluensulfónico y ácido cloro- platínico, Los picrolonatos (sales de 3 -metíl-4-ní tro- 1 -p-nitioíeni 1-5-pira- zclona) pueden prepararse por el mismo procedimiento señalado para los picratos {procedimiento 26). Los picrolonatos figuran entre los derivados de las aminas terciarias que generalmente son más satisfactorios y a menudo se forman con facilidad en casos en que otros derivados presentan dificultades. También se han empleado los reineck&tos [sales del ácido H *C r(N H ,)j(SC N )<l
Determinadas .N^-dialquilanilinas reaccionan con el ácido nitroso formando ¿>-mtrosoderivados y éstos pueden algunas veces servir como derivados.
+ HONO R jN / )N O + H,0
264 Preparación d t dw ivados
Rtfcreacüu
•• jhN itroiodialkjlaiiniiiai, M arvcl, Scott y Amscutt, / . A m Ch*m. S o e ., 51, 363B <1929).
R elneckate . Ayrock, Eisenbniun y S c i r a d c r , ] . A m . Ckem. Scc., 73, 1351 (1931).
P rocedim iento 2 4 . Sales cuaternarias de am onio de am inas terciaria*
a) U na mezcla de 0.5 g de la amina y 0.5 mi de yoduro de metilo se calienta durante algunos minutos en un tubo de ensayo con llama débü y se enfria en un baño de hielo. El tubo se raspa con una varilla de vidrio para acelerar la cristalización. El producto se purifica por recmtaikación de alcohol metílico o etílico absoluto o de acetato de etilo.
b) A una solución de 2 a 3 g de cloruro de bencilo o ¿-toluensul- fonato de metilo en 10 mi de benceno seco se le añade un gramo de la amina. La solución se hierve durante 10 a 20 minutos y se enfria. Los productos se recristalizan disolviéndolos en la mínima cantidad posible de etanol a ebullición, se añade acetato de etilo basta que comienza la precipitación y la mezcla se enfria. £1 producto se filtra y se seca rápi* damente comprimiéndolo sobre una placa porosa; el punto de fusión se determina inmediatamente.
P rocedim iento 25* C om puesto* n itroso
¿ ¿ disuelven dos gramos de la amina en 20 mi de ácido clorhídrico al 10% y la solución se enfría en una mezcla congelante. Se le añade

A m ín o é « l< f« 285
lentamente y con agitación vigorosa una solución de 13 g de nitrito de sodio en 2 ral d i agua. L* mezcla se deja reposar 15 minutos en el baño de hielo y entonces se filtra el clorhidrato del compuesto nitroso. Los cristales amarillos se mezclan con 3 mi de agua y se colocan en un baoo de hielo, Se adiciona una solución diluida de hidróxido de sodio hasta que la solución esté alcalina. El compuesto nitroso, de color verde, se extrae con éter y se deja evaporar cl éter. Se obtienen cristales de color verde brillante que se filtran y se secan sobre una placa porosa.
P rocedim iento 2 6 . P lcra lo j
a) Se añade una muestra del compuesto (0.3 a 0.5 g) a 10 ral de etanol al 95%, Si la disolución no es total, la mezcla se agita hasta que se obtenga una solución saturada y entonces se filtra. El filtrado se añade a 10 mi de solución saturada de ácido pícrico en etanol al 95% y entonces la soi -ráón se calienta hasta ebullición. La solución se deja enfriar lentam ente se filtran los cristales amarillos del picrato y se re- cristalizan de etanol. Determinados picratos, especialmente los de los hidrocarburos (tabla 35), se disocian cuando se calientan y, consecuentemente, no pueden «cristalizarse. En tales casos,, el precipitado originat deberá lavarse con una cantidad muy pequeña de éter y secarse sobre una placa porosa para la determinación del punto de fusión. (Precaución: Algunos picratos explotan cuando se calienta).
ó ) En un tubo de ensayo se mezclan cantidades iguales del compuesto y ácido pícrico y se calienta en el baño de vapor durante JO minutos o con llama muy débil hasta, que Ja mezcla se funda. El sólido se deja enfriar y, si es suficientemente estable, se recristaliza de etanol.
P rocedim iento 27- C loro platínalo*
A una solución de 0.5 g de la amina en 10 mi de ácido clorhídrico at 10% se le añaden, lentamente y agitando, 10 mi de una solución acuosa de ácido cloroplatínico (HjPtCVGHjO) al 25%, El cloroplati- nato cristalino que precipita deberá filtrarse y lavarse con ácido clorhídrico al 10%. Puede wcristalaaree de etanol que contenga una gota de ácido clorhídrico concentrado para evitar la hidrólisis. Debe evitarse el contacto de los cloroplatinatos con espátulas de metal, ya que provoca la formación de platino metálico.
AMINOACIDOS
(Enlistados en las págs. 362*363)
* Los puntos de fusión o puntos de descomposición de los aminoácidos no son exactos. Los valores dependen de la velocidad de calen»

2 8 6 Prflj»ar*neión d e d a r ív o d o s
tamicnto. B e aquí que al usar estas constantes para preparar una lista de posibilidades debe darse un margen de tolerancia para compensar au inexactitud.
Lo* «^aminoácidos naturales, de origen, vegetal y anima!, o los que se obtienen por hidrólisis acida o enzimática' 'de las proteínas y■„ los pepti- dos. son ópticamente activos (excepto la glicina)'y por su configuración pertenecen a l a serie L. La5 rotaciones específicas son constantes valiosas para identificarlos. En la tabla 17 se da una lista. Las rotaciones específicas en otros disolventes y referencias a ía bibliografía pueden encontrarse en la referencia G de la pág. 288.
Una prueba colorida usada ampliamente es la 0reacción de la ninhídrina” Esta prueba se realiza añadiendo unos 2 ing del a-amino- ácido a 1 mi de una solución de 0.2 g de ninhidrina (monohidrato de indantríona-1,2,3) en 50 mi de agua. La mezcla Se calienta a ebullición durante 15 a 30 segundos; una ‘ '•ueba positiva la constituye Ja aparición de un color que va de azul a a z u l. -íoleta que es el que dan loa examino- ácidos y Jos ^^aminoácidos. Otros colores (amarillo, anaranjado, rojo) son prueba negativa.
CO R
^C (O H )2 + H*N—CHCO¡>H -
COCO R
" \ íC = N —CHCO^H
COjn to
CO
I NCHNH2 + RCHO + co ,
Nco.coCO CO
\ / I 1 y*01*1*C H N = C ^ N í o
S / \ /co coAzul
La prolína, hidroxiprolina y los ác>do& o-, m- y -fr-aminobenzoicos no' dan color azul sino amarillo. Las sales de amonio dan prueba positiva y algunas aminas (anilina) dan colores que van de anaranjado a rojo, siendo la prueba considerada coma negativa, Esta prueba de color es particularmente ..valiosa en la caracterización de los «-amino ácidos por

A m in o á c id o * 2 8 7
cromatografía de partición en papel. Esta técnica !'.2 sido estudiada exten- saínente y se han desarrollado procedimientos dignos de confianza para identificar y separar los aminoácidos naturales más comunes. Las referencias mencionadas en la pág. 283, describen las investigaciones sobre cromatografía de partición en papel de aminoácidos. En las referencias D y F se encuentran procedimientos experimentales específicos y la referencia G enlista los valores de Rp, En la cromatografía de partición en papel y en el cálculo de los valores de R? es importante realizar experimentos de control con aminoácidos conocidos y en condiciones experimentales exactamente iguales.
T abla 17Rotaciones tspeefficas ¿s lot a-^minodcídvt
Amiiwí6dú« OootveGte í & T^ P í ± , |rmxa
L-alanina LO# HCI 5.8 15 + 1 4 .7L-ar£¡nína 6-Oí/ HCI 1.6 23 + 26 .9A d d a i>a$pirtfco 6 .0 # HCI 2.0 24 + 24 .6L-eistina 1 .0# HCI 1.0 24 —2144Acido L-gíutámico 6 ,0 # HCI 1.0 22 + 31.2v h istíd m a 6 .0# HCI 1.5 22 + 13.0Hidroxí-i^prolinaAloh3droxi-L-p!*ítJna
l.OiV HCI 1.3 20 -4 7 .3Agua 2.6 13 -3 8 .1
k -jid e u d n a $ .0 # HCÍ 5.1 20 + 40 .6w lo iw k 'ü c in a 6 .0 # HCI 3.9 20 +38.1L-leucina 6 .0 # HCI 2.0 26 + 15.1I^lisána 6 .0 tf HCI 2.0 23 +25.9x.-metionina 0 .2 # HC) 0.6 25 +21.2L-íenilalinína 5 .4 # HCI 3.8 20 - 7 .1L-prollna 0 .5 # HC! 0.6 20 - 5 2 .6v-seriaa 1 .0# HCI 9.3 25 + 14.4L - tre o n in a Agua 1.3 26 + 28 .4L-alotr-ooina Agua 1.6 26 + 9 .6L-triptófano Agua 1.0 22 —31.5L-tlrosina 6 .3 # HCI 4.4 20 - 8 .6L-valina • 6 .0 # HCI 3.4 20 + 28.8
Los derivados sólidos de los aminoácidos que se preparan, ordinariamente son derivados del tipo usado para las aminas más que los descritos para los ácidos. El método de Schotten-Baumann [experimento. (1«)> páí!\ 129] proporciona derivados de benzoilo y el mismo procedimiento general, usando anhídrido acético en lugar de cloruro de benzoilo, da los derivados aceitados [procedimiento 21 (<*)]. El feniliso- cianato reacciona con los aminoácidos para producir las correspondientes feniluteas substituida [procedimiento 9(<t)]. La reacción de Hinsbcrjí, usando cloruro de / toluensulfonilo (procedimiento 28), proporciona buenos derivados para un número considerable de aminoácidos» Las 2J4-di»

V
mtíoanilinas ¿/-substituidas, obtenidas por acción del 2,4'dimtirofluoro- benceno (procedimiento 29) sobre los aminoácidos, han encontrado un
2 8 8 P r e p a r a c i ó n d * d e r i v a d o *
amplio uso como derivados de los aminoácidos, péptidos y proteínas. Estos derivados de dinitroarilo responden a las pruebas coloridas con álcali [experimento 26(í ) , pág. 187].
Refe renda*
A ) C rccn í« in y WiníU, C hsm utry o¡ (he A mino A á d s, 3 vola., John Wiley and Son», Nueva Yor*, 1961.
B ) Mewter, Biochcmiitry of íhe Amino A cid t, Acaderoic Prcw, Nueva York,1957.
C ) GreeobcrB, Am ino Aeids and Proietns, C harles C. Thomai, Springfield^I H . , 1 9 5 1 .
D ) Litu-ack, Exp<u¡mml&l BioeUemitiry, John Wilcy and Sons, Nueva York,
E ) Wock, Durrum y Zwclg, A M a n a d of Paper Chromatogjophy and Paper Eltctrophoresif, Academíc Pre-M, Nueva York, 1955.
F ) The Biochemiitry S u íí , In lroductcry B ieckem iiity Labore io iy Manual, Univ. of lowa Prinling Service, íow a City, Icwa, 1962-1E^63,
C ) Tablti for Id tnú jueüon o f Organie Compounds, Chemical Rubber Co.» Cleveland 14, Ohio, 1960, p i& . 148-154.
Derivado* de p-toíueasalfoalio de am inaiddos. McChecney y Swann, / , Am.C h m . Soe., 59, 1116 (1937).
Q oroform iato de al Lio como reactivo. Scevens y W atanabc, / , A m . Chem. Soe., 72, 625 (1950).
CiaiXttdlaciÓB d d grupo amino. M cKinney, U bíng , Setzkora y Cowan, / , Am .Clutn. Soc., 72, 2599 (1950).
A'ATDÍCaiboxídíanhídrído de cía tin a . Baüey, ¿tature, 164, 809 (1949).Sales coa aItro-2-in<iandk>n*-l ,3. Laríen, W ítt y Poc, M ikrocfw nU ver, Mikroehim.
Act*, 14, 351 (1949) [C. A .f 43, 69g (1949)].5-Nitrobari)itHtai« (d ílicun tos). Lorien, W itt y Poe M ifaochcm 'u ver. M ikrockim.
Acta, 34, 351 (1949) [C. A .., 44, I855h (1950)].Derivados de S^-dijútrobemoOo de aminoácidos. Saundcrs, Biochem. / . , 28, 560
(1934); Town, iblá., 35, 578 (1941).
Procedimiento 28. Derivados de p^olueruulfonilo
R -C H C O zH
FI
NH
1960.
de aminoácidos
Aproximadamente 1 g del aminoácido se disuelve en 20 mi de solución 1N de hidróxido de sodio, se añade una solución de 2 g de do-

C a rb o h id ra to s y g lu có s id o » 2 6 9
ruro de ¿-toluensulfonilo en 23 mi de éter y la mezcla se agita mecánicamente o se agita v iro sam en te durante 3 a 4 horas. Se separa la capa etérea y la capa acuosa se acidula, hasta el viraje del rojo congo, con ácido clorhídrico diluido. Ordinariamente el derivado es sólido; se filtra y se recristaliza en 4 a 5 m) de etanol al 6 0 # , Si al 3£Ídular se obtiene un aceite, la mezcla se deja en el refrigerador durante la noche para inducir !a cristalización,
Las sales de sodio de los derivados de la fenilalanina y la tirosina son poco, solubles en agua y precipitan duraníe la reacción inicial. La suspensión resaltante se acidula y las sales se redisuelven. Entonces, los derivados de p-toluemulfonilo cristalizan de la capa etérea y se filtran.
Los derivados de los ácidos glutámico y aspártico, de la arginina, la lisina, el triptófano y la prollna cristalizan con dificultad; se deberán ensayar otros derivados en caso de que se produzcan aceites,
P ro c e d im ^n io *29. D erivados de 2 ,4 -d in itro fen ilo de aminoácido»
A una solución ó suspensión de 0.5 g del aminoácido en 10 mi de agua y 1,0 g de bicarbonato de sodio se le añade una solución de 0,8 g de 2,4'dinítrofluorobenccno en 5 mi de etanol. La mezcla se agita vigorosamente y sé deja reposar a temperatura ambiente durante una hora, con agitación vigorosa ocasional. Después, se añaden 5 mi de una solución saturada de sal y la mezcla se extrae dos veces con 10 mi de éter para eliminar los reactivos sin tranformar. Entonces se vierte la capa acuosa en 25 mi de ácido clorhídrico al 5%, frío, con agitación vigorosa. Esta mezcla deberá ser francamente ácida al indicador de rojo congo. Algunas veces el producto se separa como aceite; se induce la formación del sólido agitando o raspando. El derivado.se filtra y se «cristaliza de etanol- agua 1:1. Los puntos de fusión están anotados en la referencia Gt de la página 286.
Este procedimiento también puede usarse para Jas aminas y para las proteínas o péptidos que tengan grupos amino primarios libres en la cadena o en el extremo.
CARBOHIDRATOS Y GLUCOSIDOS
(Enlistados en la pág, 364),
Las osazonas [experimento 21 (b) , pág, 165] dan información ¿til ■. concerniente a los azúcares ordinarios. La forma cristalina de la oa^zona deberá compararse al microscopio con la de una muestra auténtica. De acuerdo con Midliken, las velocidades relativas de formación de laso sazonas son significativas. A menudo los puntos de fusión de las osazo- nas son demasiado cercanos para servir como medio de identificación.

L»
í t
r»r »
f *
Otros derivados valiosos para la identificación de los carbohidratos son les acetatos [procedimiento i l ( a ) u ÍI(6 )J , los benzoatos [proce« dimicnto 10 (c?) ]> los derivados de la acetona y los derivados benzalde- hídicos.
Se pueden obtener derivados característicos de las aldosas formando los dietilmercaptales aoctilados.
Hay una serie de pruebas sistemáticas desarrolladas por Dchn, Jacfc- son y Ballard, cuyo artículo deberá cónsul ta ra .
Referencias
Identificación de cart»lñdralo$. Dekfl, Jackson y Bailará, TnÁ. 8 n g . C fiera ., A n a iEd., 4, 413 (1932).
Acetatos, Hudson, Ind, Eng. Chem., 8, 360 (1916).G hicisdoc. Jackson y Deha, l n d . Eng. C him ., A n d . £d.> 6, 382 (193.4); Vogel
y Georg, TabeiUh Jer Zu¿<r u d ihref Derívate, J- Springer, Berlín, 1931. M crcaptalaccUit» ¿ e las aldosas. W oli: im v Karahinos, J. A m . Chem. Soc.j 67,
500 (1945).Fotomicrografías de Osazoflas. Ha«id y McCready> Ind . Eng. Ckettt., Anai. Ed..
14, 6&3 {1942).Tíoaccwk*. Wolfrom y Xsrabintw, / . Am . Chem. Soe-, 67, 5iX> (1945), Feííüwrctaoos, Heron, H iatt y Fordyce, ) . A m . Ch*m. Soc., 66, 995 (1944). Pígm&rt y Goepp, Chamiitry of Carbohychates, Acadcmic Press, Nueva York, 1940. Aduanéis in Carbohydfáte C htm iilry, volúmenes atxuaíes, 1945-1962, Acaderak
Press, Nueva York,
ESTERES
(Enlistados en las págs. 365-368)
Al preparar una lista de compuestos posibles, se encontrará que además del punto de ebullición, serán muy útiles los equivalentes de saponificación (procedimiento 30) y tes densidades específicas.
A causa de las dificultades involucradas en la separación y purificación de los productos de hidrólisis, es mejor preparar derivados por reacciones sobre el ésier original. Los esteres que contienen otros grupos funcionales a menudo pueden identificarse por referencia, a los derivados sólidos que se obtienen medíante reacciones tales como halogenaciónj nitradón y acilación.
Los derivados de la parte acilo del éster se pueden obtener por los métodos siguientes:
1. La reacción con bencilamina en presencia de una pequeña cantidad de cloruro de amonio proporciona W-bencilamidas, las cuales sirven como derivados (procedimiento 31),
R CO O R'-f CeHiCHjNHj—> RCONHCH 2C<H, + R'OH
290 P rep a ra c ió n d e d e r iv a d o s

£s!*t m 291
La reacción marcha bien cuando R ' es raedlo o etilo. Los ásteres de alcoholes superiores deberán previamente someterse a una metanóims,
R C O O R '+C H jO H — RCOOCHj-f R 'OHEl éster metílico así obtenido puede usarse para la aminólisis.
2, La hidrazina reacciona con loa ésteres metílicos y etílicos produciendo bidrazidas de ácido, las cuales sirven satisfactoriamente como derivados (procedimiento 32).
RCOOCH j+ H jNNHj - ^ RCO N H N H j-f CHjOlf.
Loa ésteres de alcoholes superiores deberán convertirse en los esteres metílicos correspondientes.
3, Algunos esteres sencillos reaccionan con amoníaco acuoso o alcohólico para producir amidas, las cuates sirven como derivados,
R C O O R '+ NH3 - * RCONHj + R 'O H
Sin embargo, la mayoría de los ¿Eteres deben calentarse a presión para que se efectúe esta reacción.
4, L a p-toluidida de la parte ácido de! éster puede obtenerse mediante las reacciones siguientes;
CjH6MgBr + CH30 N H 2 - C H ^ f ^ N H M g B r H Q H S
RCOORJ + 2CHa/ ^N H M gB r -
R Q N H ^ ^ C H a ) , + R'OMgBr
OMgBrI 2HCI
NHaCl
R C O N H / ^ C H a + ( f ^ ) - + MgBrQ
c h 2
Inversamente, se puede obtener un derivado sólido de la parte alcohólica de un éster sencillo efectuando una reacción de intercambio en tir el ácido 3>5-dinitrobeazoico y eí éster en presencia de ácido sulfúrico concentrado {procedimiento 33).
OgN OtN
+ R C O j R ' ( y * + R C O .K
0,Ñ~

2 9 2 P re p a ra c ió n d e d e r iv a d o s
Ei método puede aplicarse a un gran número de esteres sencillos, pero do puede usarse si el grupo R o el R ' del éster reacciona con el ácido sulfúrico concentrado, ni tampoco si los ésteres son de aleo peso molecular ( > 250).
Si no es posible obtener directamente derivados del éster, correspondientes a las partes ácida y alcohólica, se debe recurrir a la hidrólisis, la cual se realiza mejor por saponificación con álcali de acuerdo con el experimento 26, pág. 183. El procedimiento exacto que se ha de seguir al efectuar la saponificación depende de la naturaleza del componente ácido y del oxhidrilado. El ácido puede ser monobásico o polibásico y soluble o insoluble en agua. El compuesto oxhidrilado puede ser un alcohol monoxhidríjico, un alcohol polioxhidrüico o un fenol. De aquí que deba planearse cuidadosamente la separación y caracterización de los producios de saponificación de acuerdo con las sugestiones obtenidas de la lista de posibilidades y de los resultados de las reacciones de clasificación.
Referencia*
JV-bcimlamidas. D en rn r y Kin?, / . Org. Chem,, 8, 166 < 1?>43); Buehfer y M aclenzie, / . Am. Chem. Soc., 59, 421 {19*7).
H tán z itta i de écidot a p a rtir de é&teiei. $xh> Rec. Trav. Chim., 59, 1036 (194C). p-Toluldvdas % p a rtir de entere?, Koelích y Tenenbaü/a, / , A m . Chem. Soc., 55,
3049 (1933).A niüdas a p a rtir de a te ie s . H a jd y , / . Chem. Soc., 1936, 398. 3,S-D¡aitrQtauo&tos a partir de esteres, R e n f rw y Chaney, / . Am . Chem. Soe.,
68, 150 (194S).Etnaolammófiitt. R aurehcr y C lark, / . A m . Chem. Soc^ 70, 436 (194fl); Rauicher
y MaePeelt, A n d Chem., 22, 923 (1940). f/-{^-AoiiiiQetil}iiu>rfolina.Bo»t y Mullen, / . Am . Chem. Soc., 73, 1967 (1951),
P rocedim iento 3 0 . E quiva len tes de éaponificación de los ¿ulerea
a) M étodo del d iettíenglicot, Preparación de una solución de hidróxido de potasio en dietilengüeol. El reactivo se prepara disolviendo3 g de hidróxido de potasio Q.P. en 15 mi de dietílenglicol. Es necesario calentar suavemente la mezcla hasta que se efectúa la disolución. Deberá usarse un termómetro para la agitación y la mezcla no deberá, calentarse a más de 130°; las temperaturas más elevadas pueden hacer que el reactivo se coloree. Después que todo el sólido se ha disuelto, la solución tibia se vierte en 35 ral de dietilenglicol contenidos en un frasco con tapón de vidrio. La solución se mezcla perfectamente y se deja enfriar. Aproximadamente es LON y se Je valora pipeteando 10 mi a un matraz, añadiendo 10 mi de agua y titulando con solución de ácido clorhídrico 0.?5W, previamente valorada.
Saponificación del ¿íter. Se pipetean exactamente 10 mi del reactivo a un pequeño nwtraz Erlenmeyer con tapón esmerilado. El éster se

Coloca en un pesafiltros provisto de una pipeta pequeña. Se determina el peso del pesafiltros con el éster y la pipeta y mediante ésta se transfieren de 0.4 a 0.6 g del éstér al reactivo que está en el matraz Krlenmeycr, regresando después la pipeta al pcwíiltros. La pérdida en peso del pesa- filtros representa el peso de la muestra.
El ¿ster se mezcla con el reactivo mediante un movimiento circular del matraz. El tapón se conserva firmemente en su lugar y la mezcla se calienta en un baño de aceite procurando que en 2 ó 3 minutos se alcance una temperatura de 70-80°C. Durante el calentamiento el liquido se agita con movimiento circular. En este punto el matraz se saca del baño de calentamiento y se agita vigorosamente. El líquido se deja escurrir y el tapón se afloja con cuidado para dejar que escape el aire (¡precaución!). El tapón se vuelve a colocar en su lugar y la temperatura se eleva hasta 120-130°C. Con esteres de punto de ebullición muy elevado se puede quitar el tapón e insertar un termómetro.
Después de minutos a esta temperatura el matraz y su contenido se enfrían a 30-&0c/. ' v el tapón se quita y se lava con agua destilada en tal forma que los lavados escurran dentro del matraz. Se añaden unos 15 mi de agua destilada: el contenido se mezcla y entonces se titula con ácido clorhídrico 0.25/v, usando fenolftaleína como indicador. El equivalente de saponificación se calcula de acuerdo con la ecuación siguiente:
Equivalente de saponificación_ ________peso de la muestra X 10~[volum endei álcali (mi) XF«N} — [volumen del ácido (mi) XF-Ñ*]
V Comentarios, Con estos procedimientos se obtiene una saponificación 4 total de los ésteres que son insoluoles en agua. Los esteres como el ace
tato de bencilo, ftalato de butilo, sebazato de etilo, oieato de butilo y los ásteres del etilenglicol y glicol y glicerol se saponifican totalmente.7
b) M étodo del h idróxido de 9odio en alcohol. Preparación de una solución de hidróxido de sodio en alcohol. Ocho gramos de sodio se disuelven en 250 mi de etanol absoluto y después que se ha completado la disolución se añaden 25 mi de agua. Esta, solución se valora por titulación con una solución de ácido clorhídrico 0.25N, previamente valorada, o por titulación directa de una muestra pesada de ftaJato ácido de potasio puro.
Saponificación del éster. El cster se coloca en un pesafiltros que contenga una pipeta pequeña. Se determina el peso del pesafiltros y de !a pipeta y por medio de ésta se Transfiere una muestra de 0.2 a 0.4 g y del -
éster a un matraz Erlenmeyer de 150 mi; la pipeta se regresa al'pesa- filtros y los dos se pesan nuevamente. La pérdida de peso es el peso de la muestra. Mediante una bureta se añaden quince mililitros de la anterior
1 Artlemaon y Lucu, 1*4. £«f. Amtl EJ., é , 541 (1M7).
P ro ce d im ie n to 3 0 . E q u iv a le n te s d e sa p o n if ic a c ió n 2 9 3

9 7.94 P rep a ra c ió n d e d e r iv a d a s
v solución de hidróxido de sodio en etanol al matraz que contiene el éster.| B A! matraz se le coloca un eficiente refrigerante de reflujo por medio de
un tapón de hule limpiado escrupulosamente y la mezcla se calienta suavemente o reflujo durante 1 14 a I V¿ horas, después de este tiempo se le
r * * B deja enfriar un poco. Se afloja el tapón det refrigerante y el tapón de1 hule y el tubo del refrigerante se lavan con un chorra de agua proceden
te de una piseta: los lavados se dejan caer en 1? mezcla de saponificación. « ■ A Se añaden dos goeds de solución de fenolftale'ma y se titula el exceso| de álcali por roedio de ácido clorhídrico Q.25.W. El punto final debe1 quedar débilmente rosa. Es mejor titular la solución hasta que la fenol-
fialcína quede incolora y entonces se retitula con el álcali original. r 'Q P Comentarios, El ¿ster debe esiar puro y anhidro para dar un equiva
lente de saponificación exacto. Para obtener resultados exacto deben tomarse las siguientes precauciones:
p 9 p I. La solución de hidróx. ’o de sodio en etanol deberá valorarseinmediatamente antes de usarse v anotarse su factor de normalidad
1 (F.N.)>2. Las soluciones valoradas deberán medirse exactamente por medio
de una bureta, ya que el mínimo error en la cantidad de álcali producirá u r gran cnor en el equivalente de saponificación. Esto se observa espe- cialmentc con los esteres de peso molecular elevado.
3. La mayoría de los esteres se saponifican con un calentamiento [ de horas. Para algunos pueden necesitarse tiempos más prolongados i [2 a 24 horas).W B F 4. No deberán usara corchos para unir el matraz al refrigerante,
ya que ios vapores de alcohol extraen substancias que disminuyen laI concentración del álcali, Deberán usarse tapones de hule que se limpian| j B calentándolos con álcali diluido y luego lavándolos perfectamente con
agua destilada.
( 5, El punto final de la titulación deberá ser un rosa débil. Este es j B el color que tiene la fenolftaleína a pH 9.0, el cual representa la concen-
tración de los iones hidrógeno de las soluciones de las sales de sodio de la mayoría de los ácidos orgánicos.
rj j ^ 6. El peso molecular del éster es igual a A’ veces el equivalente deW p saponificación, en donde N es el número de grupos éster en ta molécula.
r »i
3 »
PrejruntM
1, ¿Q ué equivalente de eaponiíic&ctórt « obtendría para el acetoacelatO de eCÍto?, <p*ara «i /J-bromopropionai© de n-SjutiJo?, ¿para et io la io ¿c¡do de ítilo?, «para el *-cianoaceio<o de etilo?
2, ¿ Q ué sucedería ii el procedimiento an terio r se aplicara al benialdchído?3, Si un ésltr ve ha hidrolizado parcialm ente, ¿qué efecto tendría en el equiva
lente de íapomfitación?

Procedim iento 35, 3,S-Dinl1roben*ocrt©s do alquilo 29S
P roced im ien to 3 l . N-Benctiamida* a p a r tir de entere»
En un tubo de ensayo Pyrex, provisto de un refrigerante de dedo pequeño, se calienta durante 1 hora una mezcla de 1 g del éster, 3 mi de bencilamina y 0.1 g de cloruro de amonio pulverizado. La mezcla de reacción se deja enfriar* después se laya con agua para eliminar el exceso de bencilamina e inducir la cristalización. A menudo la adición de un poco de ácido clorhídrico diluido provoca La cristalización. Deberá evitarse un exceso de ácido ya que disuelve las A1 -benc il am idas. Ocasionalmente, Sa presencia de éster inalterado puede evitar la cristalización. Eti ese caso es mejor h e ñ ir la solución durante algunos minutos con agua, en una cápsula do porcelana, para volatilizar el éster, La amida sólida se filtra, se Java con un poco de ligroína y se recristaliza de etanol- agua o de acetona-agua.
Loa ésteres de alcoholes superiores al etanol deberán calentarse du rante 30 minutos con 5 mi de metanol absoluto en el que se ha disuelto un trozo pequeño de sodio (0.1 g ) , Al final del período de reflujo se evapora el metano! y el residuo se trata por el procedimiento anterior.
P ro ced im ien to 32 . H idr oxidas de ácido a partir d e ésteret
Se mezcla un grarno del ester metílico o etílico y 1 mi de hidrato de hidrazina al y la mezcla se calienta a reflujo durante 15 minutos. Entonces se añade, por la parte superior del refrigerante, suficiente etanol absoluto para obtener una solución transparente. Después que la mezcla se h a calentado a reflujo durante 2 horas, se evapora el alcohol y el residuo se enfria. Los cristales de la hidrazida se filtran y se recrístalizan de agua o de agua-etanol.
Los ésteres superiores deben someterse a una metanólisis, como se describió anteriormente, antes de tratarse con ia hidrazioa.
P roced im ien to 3 3 . 3 y5-Diniiroben%o<ttos de alquilo a p a r tirde ésteres
Unos 2 mi del éster se mezclan con 1.5 g de ácido 3,5-dinitroben- zoico y se añaden 2 gotas de ácido sulfúrico concentrado. Si cl éster hierve a menos de 150°, la mezcla se refluja calentándola suavemente. Si el éster hierve a más de 150°, la mezcla se calienta en un baño de aceite a 150°, agitando frecuentemente. El tiempo requerido varía de 30 minutos a 1 hora; un tiempo más prolongado se requiere en aquello* casos en los que el ácido 3,5-dimtrobeüzoÍco es incapaz de disolverse en unos 15 minutos. Desvié* que la mezcla se ha enfriado se añaden 25 m) de éter absoluto y la solución se extrae con dos porciones de 15 m i de

2 9 6 P re p a ra c ió n d e d « r ív o d « s
solución de carbonato de sodio al 5% para eliminar los áddca sulfúrico y 3,5-dmitrobenzoico. La capa, «terca se lava con 10 mi de agua y el disolvente se evapora. El residuo (ordinariamente un aceite) sé disuelve en 5 mi de etanol a ebullición. Después que la solución se ha filtrado se añade agua hasta turbiedad incipiente. La mezcla se enfría y se agita para inducir la cristalización del derivado.
E TER ES— ALIFATICOS
(Enlistados en las págs. 369-370)
El único método apropiado para la. preparación de derivados sólidos que se usan eu la identificación de cantidades pequeñas de étteres alifáticos involucra la escisión de los éteres y la formación de los correspondientes 3,5-dirútrobenioatoa.
£sto se logra tratando el éter con cloruro de 3,5-dinitrobenzoílo en presentía de dom ro de cinc (procedimiento 34).
n o 2 n o 2
COCI 4- ROR -* / V 0 0 R + RCJ
Este método no es apropiado para éteres mixtos en los que ambos radi* cales del éter son alifáticos. Los éteres alifáticos y los éteres de alquiladlo pueden escindirse con ácido yodhídrico (experimento pág. 146).
R >0-f 2H I —► 2RI 4-H jO AxOR-f H I A iO H + R I
XJsando una muestra grande (5 a 10 g) se puede aislar el yoduro de alquilo y convertirlo en un derivado (procedimientos 397 40, 41, 42). El fenol de,un éter de alquilarüo también puede aislarse y transformarse en un derivado (procedimientos 54, 55, 56),
Ya que la pirólisis a 500°C de los éteres produce principalmente hidrocarburos y compuestos carbonílícos, esta reacción puede usarse para Su caracterización.
(RCH^iO RCHi+RCHO (R3CH) ¡O -► RaCHj+ R,CO
Los vapores de la pirólisis se condensan y se tratan con reactivos apropiados, tales como o-tolilsemicarbazida, ¿-tolilsemicarbazida, m-nitro- benzhidrazida y p-clorobenzhidrazída, para producir semicarbazonas substituidas que se utilizan como derivados.
i^os éteres asimétricos producen una mezcla de aldehidos o cetonas y deben separarse por fraccionamiento para obtener derivados puros.

Efere* crom ático* 297
RCH jOGHjR' R C H j+ R 'CHj -f R C H O + R'CHO R 2CHOGHRrí -> R2CH2.f R '2C H ,+ R ?CO +R'*CO RCHjOCHR', -> R C H j-f-R 'jC H j+R G H O +R 'jC O
Refere a c lis
3,5 DinÍ!íobcra<«itc« de alquilo a p a rtir de éteres. Underw&od, Baríl y fo c n e , /.A m . C h tm . Soc., 52, 4067 (1930).
P iró & k de cu res . Sah, Hte. Trav. Chim., 58, 758 (1939).PícratOl de S-wtquiitíourOJlio a partir de grupos ídcoxilo. Kr&ul y OíW /bcrgcr,
M o n a u K 81, 998 (1950).P-Nítrobcnzoatos. Ward y JcnV.íns, J. Otg. Cktm,, 10, 371 (1945).
Procedimiento 34, 3,5*D¡nicrobenzoetv de alquilo a partird e é teret
U na me2cl¿ de 1 mi del éter, 0.15 g de cloruro de cinc anhidro y 0.5 g de cloruro de 3,5-dinitiobenzoílo se colocan en un tubo de ensayo unido a un refrigerante de reflujo. La mezcla se hierve durante \ hora y se enfría. Entonces se le añaden 10 mi de solución de carbonato de ¿odio. La mezcla se calienta a 90°C en un baila maría, se enfría y se filtra. El precipitado se lava con 5 mi de solución de carbonato de sodio al 5% y ÍÜ nal de agua destilada. El residuo se disuelve en 10 mi de tetra- cloruro de carbono caliente y la solución se filtra en caliente; entonces el filtrado se enfría en un baño con hielo. Si no precipita el éster, se evapora el tetraclorvro de carbono. El residuo se seca sobre una placa porosa, y se determina el punto de fusión.
ETERES — AROMATICOS
(Enlistados en Jas págs. 371-S72)
Los derivados de éteres aromáticos empleados con m is frecuencia son los que se obtienen por bromación (procedimiento 35) y miración [procedimientos 45(a) y 45 (6 )] . También se usan los picratos (procedimiento 3$).
Los éteres aromáticos que tienen una cadena lateral de alquilo pueden oxidarse hasta ácidos benzoicos substituidos [procedimientos ^ ( a ) ,y46(fr>].
Q H 4/
CH,CHa
\OCHj
COsH
OCHa

I *
t »
i».1
? •
Si *
br i
ón
óí>
*
Los éteres aromáticos reacciona fácilmente con ácido clorosulfónico a 0*C para producir cloruros de sulfoniio (procedimiento 36).
R f r f j ) + 2C1S03H — R O Q ^ C I + HCl + HjSO,
Estos últimos son aceites o sólidos de bajo punto d¿ fusión que se tratan con amoníaco o carbonato de amonio (procedimiento 37} . Las sultana-* midas obtenidas de esta m anera son derivados útiles.
2 9 8 P ra p a ra d ó n d e d e r iv a d o s
RoQ sO,C1 + (NKOCOa -
R Q ^ ^ S O ^ N H j + NH4Cl + COs 4- H20
Refere eteias
Picratoi. Baril f Mcjgrdkhíari, J. Am. Chem. Set., 58, 1415 (I936>.PicratOT de éteres de alquüaaftilo. E erroer y Dertuer. / . Org. Chem., 3, 289 (1938). Acidos ariolbcnzoicox, Reínheimer, Rresson* Holloway y Dat!eyf / . Am. C him .
Soe., 77* 1909 (1955).Acidos ariolpropitkúcos. Reinhcim cr y Sm ilh, / . Org. C hem ., 17, 1503 {1952), Sülfotíamidas <í« c t« r« de a rilo. Huntrcss y C artea, / . Am . Chem. Soc., 62, 603
(1940).
P roced im ien to 3 5 . B ro m o ción ,
Se disuelve un gramo del compuesto en 15 mi de ácido acético glacial y se añaden de 3 a 5 g de bromo líquido. La mezcla se deja reposar durante 15 a 30 minutos y entonces se vierte en 50 a 100 mi de agua, Eí compuesto bromado que precipita se filtra y se purifica por recrista’ lizarión de etanol diluido. En algunos casos puede usarse tetracloruro de carbono como disolvente en lugar del ácido acético: se destila él tetra- clorurc de carbono y el residuo se recristaliza.
P roced im ien to 3 6 . C loruros de tu lfo n iio
En un tubo de ensayo seco se pone una solución de 1 g del com- pueico en 5 mi de cloroformo seto y se enfría 0°C dentro de un vaso de precipitados con hieio. Unos 5 g de ácido clorosulfónico se añaden gola a gota y después que cl desprendimiento inicial de cloruro de hidrógeno se ha aminorado, se saca cl tubo del baño de hielo y se le deja alcanzar la temperatura ambiente (unos 20 m inutos). El contenido del tubo se vierte en un vaso de precipitados de 50 mi lleno de hielo molido. Se separa la capa clorofómnica y se lava con agua. Se evapora el cloro-

Húlvros — Da a tq im ilo 299
formo y el cloruro de sulfonilo residual se recristaliza de éter de petróleo de bajo punto de ebullición, de benceno o d* cloroformo.
El cloruro de sulfonilo (0,5 g) se mezcla con 2.0 g de carbonato de amonio seco y pulverizado y se calienta u 100CC durante 30 minutos. La mezcla se enfría y se Java con tres porciones de 10 ral de agua fria. La sulfonamida cruda se disuelve en 10 mi de solución de hidróxido <fe sodio al 5% t si es necesario se calienta suavemente y la solución se filtra para eliminar cualquier sulfbna o productos clorados* El filtrado se acidula con ácido clorhídrico diluido y la sulfonamida se filtra. Se purifica por recristalización de etanol diluido y se Je seca a 100°C,
P roced im ien to 38» Picraios d e éteres fenó lico t
Una solución de 1 a 2 g del éter en la mínima cantidad posible de cloroformo (2 a 10 mi) se aftade a una. solución, preparada por separado., de 2 g de ácido pícrico en 10 mi. de cloroformo a ebullición. La mezcla se agita perfectamente y se deja enfriar. El picrato cristaliza dejando la Jtnwla en reposo. Se 1c pui'de purificar por ivcristalización en la mínima cantidad posible de cloroformo a ebullición. EJ punto de fusión deberá determinarse tan pronto como sea postble ya que algunos picratos se descomponen,
HALUROS — DE ALQURNILO
(Enlistados en la pág. 373)
Muchos de Jos compuestos halogenados insaíurados pueden Identificarse mediante sus constantes físicas.
En determinados casos se pueden obtener reactivos de Grignard, usando procedimientos especiales* y convertirse en Jas anilxdas de ácidos insaturados. En ocasiones ocurren transposiciones. Por ejemplo, el bromuro de alilmagncsio y el isocianato de fer.ilo producen un aceite, el cual se transpone con ácidos formando Ja crotonaniüda.
C H i= CH CH 2M gB r+ C*HsN= C = O — > CéH jN = GCH2C H = GFI2
P rocedim iento 3 7 . SulfonamiéUts
OüvfgBr

3 0 0 P re p a ra c ió n d* d e r iv a d o s
Rrfcrftlicíl»
Aaüide». Sehwaxtx y Johttjoo, J . A m . C h e m , S o c ., 53, 1063 (1931). AUykaa£rv^ium Biomiát, Gilrtttn y JtfcClumphy, B tlii, S o c , C h itn . F r e n te , {4J,
43, 1322 (192A).
HALUROS— DE ALQUILO Y C ICLO ALQUILO
(Enlistados en las p%s. 373-374).
Les mejores métodos para, preparar derivados de haluros de alquilo y cídoalquiJo se basan en su conversión a los correspondiente reactivos de Grignard. Y los derivados usados con más frecuencia son las anilidas, toluididas y oc-naftalidas, preparadas a partir de los reactivos de Grignard (procedimiento 40} tratados con isocíanato de fenilo, de ¿-toluilo y et-oaftílo, respectivamente.
RR M g X + Q H jN ^ C ^ O ----->CJH5N = C O M g X ^ ( ^ s N H C O R
RRMgX-}-Ci9H ? N = 0 = 0 -----> C 1BH 7N=:CONf$X G10H?NHCOR
Si el haluro es primario el reactivo de Grignard puede convertirse en et correspondiente haluro de alquilmercurio por tratamiento con el haluro mercúrico apropiado (procedimiento 39).
R M gX + HgX, -+ R H gX -f MgX:
La sal de mercurio y el haluro de alquilo deben tener el mismo halógeno, ya que en otra íoroia se obtienen tneiclas de haluros de alquilmercurio, £stc m í todo es incapaz de formar derivados de los haluros terciarios de alquilo.
Entre los muchos otros derivados importantes de ios haluros de alquilo pueden mencionarse los 3,5-dimtroberaoatos, las N-alquilftali- midas, las Ay-alquil-3-nitroftalimidas, las tf-alquiltetradoroftalimidasj los éteres de alquil-p-naíülo {procedimiento 41), los picratos de 5-alquil- tioorea (procedimiento 42), las ,V-aIquiIsacarinas, los éteres de alquÜ- ttiyodofemlo, los ácidos ji-akoxibenzoicos, las /^-alquil-^bromobcncen- sulfon-p-atiisidas y las A'-alquil-/j-toluenuulfontoIuididas.
Los derivados de haluros de alquilo y cicloalquilo son particularmente útiles no sólo porque estos compuestos se encuentran con frecuencia, ano también porque se preparan rápidamente a partir de los alcholes y así pvopordonan una vía indirecta para identificar los alcoholes, Todos los métodos antes citados deben usarse con precaución en vista de que algunas veces ocurren transposiciones.

P ro ce d im ie n to 4 0 . A n i l in a s , t o lv id id a s y n a fc a lid o s 301
ívcfcrcoela*
Aíülidai, Schwfrrtz y Johnson, / . A m . Chem. Soc., 53, 1053 {1931).AdÜJdas, teluididaa oC'Daftaüdas. Undcrwood y C a lí, / . A m . Chem. Soc.. 56,
211? (1934).í^ 'D laJtrofcníItioéteres. Boj!, Stames y Wood, } . Am . Chem, Soc., 73, 1968
(1951).4<NitroffJJimida(. Biilroan y Cash, ‘'Derivativas Givin# a Sapoaiíication Equiva
len!” , J. A m . Chem. Joc., 75, 2499 (1953).Derivados del 2-ucrcaptobecaaúitxA. C u tte r y Goldcn, / . A m . Chem. Soc., 69,
831 (1947).Eteres de p.bidroiúdífcn¡latránA. Houston, / , Am. Chem. Sot., 71 395 (1949). jV-AJquil-S-nlfroftatímidas. Sah y M a, Be*., 65, 1530 (1932), A '-A lquíltttradoroflalimidas, Alien y NtcholU, / . Am . Chem. Soe., 56, 1409 (3534). F iera te* de S-alquUíHVioureJL Browu y Caropbetl, / . Chem . Soc., 1937, 1699. AlquiUacariaa.1, M crrilt, Levey y Cuttcr, / , A m . Chem. Soc., 6 Í , 15 (1939), Eterc* de alquiUríyodafcnilo. D i w y Sturtevant, / . Am. Chem. Soc., 61, 2666
(1939).Acidos j>-Alcoiib«nfc-'TcD4, tau e r, Sanders, leek ley y Ungrtadc, ] . Am . Chem. Soc.,
61, 3050 ( I9 3 s , ,Af-AIquil'p-'brtnoobejaccnsulfon>P'ftiitsid¡naj. Gillcspie, / . Am. Chem . So<^ 56,
2740 .(1934).rt-Alquil-)HfohKnBulfoataluÍ<iidas, Yeung, J. Am. C hem . Soc., 5C> 216?, 2?fl3
(1934); 57, 773 (¡935).
P roced im ien to 3 9 . H alures d e a lqu il m ercurio
E) reactivo de Grignard se prepara a partir del haluro de alquilo, tratando 0.3 g de magnesio en 13 mi de éter seco con 1 mi del lia)aro de alquilo, en un tubo de ensayo seco y limpio. Se añade un cristal de yodo para iniciar la reacción. Cuando se ha terminado la reacción, se filtra ia solución a través de un poco de lana de vidrio y se deja fluir el filtrado en un tubo de ensayo que contenga de 4 a 5 g de cloruro, bromuro o yoduro de mercurio, dependiendo del halógeno del haluro de alquilo original. La mezcla de reacción se agita vigorosamente, se calienta en el baño maría durante algunos minutos y se evapora a sequedad. Él residuo se hierve con 20 mi de etanol al 95% y la solución se filtra. El filtrado se diluye con 10 mi de agua y se enfría en un baño de hielo. El haluro de alquil mercurio que precipita se filtra y se rccristalúa de etanol al 60%.
P roced im ien to 40, A nüidas, lo lu id idas y nafudidas a partir de ha luros de a lquilo
El reactivo de Grignard se prepara como en el procedimiento 39 y se trata con 0.5 m3 de isocianato de fenilo, de ¿Moluílo o de a-naftílo, disueito en 10 mi de éter absoluto. La mezcla se agita y se deja reposar

3 0 2 P rep a ra c ió n d e d e r iv a d o s
10 minutos. Se aña Jen uno» 25 mí de árido clorhídrico al 2% con agí- tación vigorosa. Se separa !a capa etérea, se seca con sulfato de magnesio y se desrila el éter. El residuo SC recristaJiza de metano), éter o éter de petróleo. '
P roced í m iento 4 1 . E teres de úlquil-^-nafüliy
A una solución de 0.6 g de hidróxido de sodio en 25 rol de etanol se le añaden 2 g de JS-nafroI y 2 £ del haluro de alquilo. Si el haluro es un cloruro, también se añaden 0.5 g de yoduro de potasio.4 La mezcla ¿e calienta a reflujo durante 30 minutos y se vierte en 75 mi de agua helada. Esta mezcla se alcalimza hasta el viraje de la fenolítaleína y se agita vigorosamente. El éter de alquil-0-naitüo se filtra y se recrístalisa de etaool o de una mezcla de etanoUagua.
P rocedim iento 42, P icrctov de S-alquilis^tioitrea
En un tubo de ensayo se coloca una mezcla de 0.5 g de tiourca pul venada, 0.5 g de bromuro o yoduro de alquilo y 5 mi de etanol al 95% y se hierve durante 2 minutos. En otro tubo de ensayo se disuelven 0.4 g de áddo pícríco en la mínima cantidad de alcohol a ebullición. Las dos solucione* se mezclan y se dejan enfriar. El picrato de 5-alquil- tionfta se filtra y se «cristaliza de etanol.
En algunas ocasiones puede inducirse la reacción de Jos cloruros de alquilo mediante Ja adición de 1 g de yoduro de potasio a la mezcla de reacción original. Deberá añadirse suficiente etanol o agua para obtener una solución transparente en el punto de ebullición. El procedimiento subsecuente es cl mismo que se describió anteriormente.
HALUROS — DERIVADOS H ALO GE NA DO S DE LOS HIDROCARBUROS AROM ATICOS
(Enliitadvs en las págs. 375-377)
Se dispone de muchos métodos excelentes para preparar derivados apropiados de los derivados halogenados de los hidrocarburos aromáticos. El más general de éstos es la nitración [procedimiento 45 (6)3* Sin embargo, frecuentemente se oxidan los derivados bromados o clorados que tienen una cadena lateral hasta los ácidos correspondiente [procedimientos 46(a) y 4€(6)]< Para los yoduros y bromuros de arilo se recomienda la conversón a los correspondientes ácidos carboxflicos o anilidas (procedimiento 40).
* OcM knM bflfcftte c ite p ro e« C cn itd io Cs ¿ tü P*r» prtpW TW d c r ] v * d « d i tü h lfM «M tip® X ( C H 2)» T . S i * \ ©ojnpuwva CS u n d k h a lim j.l ,2 (« * 2 ) e o d e b e a á td ir ie j o d u n de p o tu io . ¿Par qn¿?

Proced ¡m ¡«n fo 4 3 . G o r d o s d e su lfo tt ifo 3 0 3
Los haluros de atilo reaccionan rápidamente con sí ácido cloros ul- fónico produciendo cloruros de sulfoniló {procedimiento 43), los cuales, cuando se tratan con amoníaco, producen sulfonamidas (procedimiento 44).
+ 2C[S03H - ) S 0 2CI + H Cl + H*SO,
x Q s O aCl + 2NHa — x /^ S s O ^ N H ^ + NH,CI
Durante la sulfonación pueden ocurrir dos reacciones anormales. El fluorobenceno, el yodobcnceno, el o-diclorobenceno y el o-dibromoben- ceno producen las correspondientes sulfonas cuando se tratan con ácido clorosulfónico a ñO^C en ausencia de disolvente. Estas sulfonas son sólidas y sirven como derivados. En otros casos se pueden producir cantidades pequeñas de ¿ulfonas, pero se pueden separar de las suifonamidas debido a que son insolubles en álcali. U na segunda reacción anormal es la doración nuclear. Esta reacción se efectúa en el ¿-diyodobenceno y el 1,2,4,5-tecracloiobenceno. En el caso de los derivados yodados de los hidrocarburos aromáticos no se obtienen resultados satisfactorios.
Los haluros de arilo en los que el átomo de halógeno está activado por grupos nitro, reaccionan rápidamente con la piperidína formando útiles derivados de piperidílo.
Las yoduros de arilo absorben cloro formando dicloruros de yodo* arilo, los cuales pueden servir como derivados,
A rl+ C lj —► ArICl2
Referencias
Arylstdfímsunides. H u n tre» y Carfeti, / ; Am. Chtm. Soe. 62, 511 (Í9 4 0 ), Derivados de pípcridilo de compuesto* aro milicos xtítrohalogetiados. Se¡k«l, / . A m .
Ch<m. Soc., 62, 750 (1940).D ic lo rarat de yodoaiüo. Nichol y Sandin, / . Am . Ch<m. Soc., 67, 130? (1945).
P roced im ien to 4 3 . Cloruros de su lfo n ilo
U ña solución de 1 g del compuesto en 5 mi de cloroformo seco, contenida en un tubo de ensayo, se enfría a 0o C dentro de un vaso de precipitados. con hielo. Gota, a gota se añaden aproximadamente 5 mi de ád d o clorosuífónico y después que lia disminuido el desprendimiento inicial de cloruro de hídrí¿enó, íc saca el tubo del baño de hielo y se le deja alcanzar la temperatura ambiente (unos 20 minutos). El contenido

3 0 4 P f6 p o ro o ¿ n c k d e r iv a d o s
de] tubo se vi arte en un vaso de precipitados de 50 mi lleno de hielo picado. Se separa la capa cloroformice se lava con agua y el cloroformo se evapora. El residuo de cloruro de suííonilo crudo se puede «cristalizar de é te í de petróleo de bajo punto de ebullición, de benceno o cloroformo.
N o t a . Las condiciones anteriores son satisfactorias para la mayoría de los haluros de arilo sencillos y para los alquiibencenos. Para los halo- toluenos es conveniente calentar la solución clorofórmica a 50 °C durante 10 minutos y entonces verter la mezcla sobre hielo picado. Los derivados polihaiogenados requieren un tratamiento más drástico. Para dichos compuesto* la. sulfonadón se efectúa sin disolvente y la mezcla en sul- fonación se caliente a 100°C durante 1 hora con un refrigerante a reflujo.
P rocedim iento 4 4 . S u ífo nam idas
Unos 0.5 g del cloruro de sulfonüo se hierven con 5 ral de hidróxido de amonio concentrado durante 10 minutos. La mezcla se diluye con 10 ral de agua, se enfria y se filtra.
L a sulfonamida cruda se disuelve en 10 mi de solución de hidróxido de sodio al 5%, si es necesario con calentamiento moderado, y la solución se filtra para eliminar cualquier sulfona o productos dorados. El filtrado se acidula con ácido clorhídrica diluido y la sulfonamida se filtra; Se purifica por recristalización de etanol diluido y se seca a 100*0.
HALUROS — DERIVADOS POLIHALOGENADOS DE HIDROCARBUROS N O BENCENOIDES
(Enlistados en las págs. 378-379)
No se han desarrollado métodos generales para preparar derivados sólidos de los derivados políhalogenados de los hidrocarburos no benceno ides y, por consecuencia, ordinariamente la identificación se basa en las propiedades físicas. Ya que entre los miembros de una determinada serie las variaciones en las constantes físicas son, en general, mayores que en los hidrocarburos fundamentales, constituyen una base más útil para su identificación.
Sin embargo, en varios casos se dispone de métodos especiales para preparar derivados de ciertas clases de derivados políhalogenados del tipo que se está considerando. Así, los haluros de alquilo que tienen substituyen tes baloarilo pueden tratarse como haluros de alquilo sencillos o corno monoaiquilbenotnos.

H id ro c a rb u ro s a ro m á t ic o s 305
HIDROCARBUROS — AROMATICOS
(Enlistados en las págs. 380-381)
Loa derivados más útiles de los hidrocarburos aromáticos son los que se obtienen por nítración (procedimiento 45), ¡Los bencenos muy alquilados han sido identificados por nitración, seguida de reducción de los compuestos nitrados hasta aminas, las cuales entonces se acetilan o benzoílan para dar derivados mono- o diacetoamino o benzamino.
ArH —> ArNOs -> ArNHj - * ArNHCOR
• Los hidrocarburos aromáticos que tien&n cadenas laterales pueden oxidarse hasta los ácidos correspondientes. Si sólo hay una cadena* ordinariamente éste e un método excelente. Para este propósito lo mejor es la oxidación con . s -^rmanganato [procedimiento 46(fc)J. Los ácidos aromáticos que tienen varios grupos carboxilo son a veces difíciles de manejar y por esta razón es limitada la utilidad del método de oxidación. Si hay dos cadenas laterales situadas en posiciones adyacentes del anillo* se recomienda la oxidación porque el ácádo resultante (ácido o-fcálico) es fácil de identificar. Los o-dialquilbencenos con el ácido crómico sufren una descomposición más profunda [procedimiento 46(a)}. Debido a que este agente oxidante puede conducir a resultados des- orientadores, siempre deberá usarse la oxidación con permanganato [procedimiento 4 í(¿ ) J . Los puntos de fusión de los ácidos obtenidos por oddación de los alquilbencenos pueden encontrare en la tabla de las págs. 335-338.
Los hidrocarburos aromáticos y sus derivados balogenados, mediante % una reacción de Friedel-Crafts con anhídrido ftáttco, producen ácidos
aroilbemoícos con buenos rendimientos (procedimiento 47),
CO
ArH + ' \ fiX N C 0A rk J coor
Análogamente, el cloruro de acetilo y el cloruro de aluminio dan meülarilcetonas (procedimiento 48), las, cuales pueden caracterizarse por medio de «¡micarbazonax o 2*4-dinitrúfenilhidrazonas,
El ácido pícrico se combina con los hidrocarburos aromáticos pro duciendo compuestos moleculares o picratos (procedimiento 38). La estabilidad de los picratos varía considerablemente; muchos de ellos se disocian fácilmente en sus componentes. De aquí que sea necesario consultar la bibliografía original antes de preparar dichos derivados.

i r
n
V*29
z
u¿ J
í »
: í
n
i »
= *
Las sulíonamidas también pueden usarse para identificar los hidrocarburos aromáticos. El hidrocarburo se convierte en cl cloruro de «ui- fonüo por acción del áddo clorosulfónico (procedimiento 43) y el cloruro de sulforíilo se trata con amoniaco (procedimiento 44) o carbonato de amonio para obtener la sulfonamida.
ArH ArSO»CI ->■ ArSOjNHj
Las sulfonamidas se recomiendan especialmente para los alquilhenccnos.
3 0 6 P rep a ra c ió n d e d e r iv a d o 3
Referencias
Deirmdo» aceUqúno y beozamino de los alquil bencenos, Ipatíeff y Schmerlins-, / , Am, Chem. Soc.f 59, 1056 (1937); 60, 1476 <J938>; 65, 2470 (1945).
Acidos aroflbemoitt*. Underwood y Wa)<h, r. Am . Chem. Sce., 57, 940 (1935);Lewenz y ScrijaD, ibld.t .75, 4087 (]9v-'t>
MetE* dichonas. Pincs y Shan-, }. Org. C h e m 20, 373 (1955).P ic ra t» . Baril y Haukwr, J. Am . Chem. Soe-, 53, 1037 (1931).Svlfoaanudas. Huntrcie y Aulcnrieth, / . A m . Chem. Soev 63, 3445 (1041).
P rocedim iento 4 3 . Wlirerclón
La nítración de un compuesto aromático, particularmente si es una substancia desconocida, deberá realizarse con precauciones especiales ya que muchos de estos compuestos reaccionan violentamente.
a) A 4 mi de ácido sulfúrico concentrado se ie añade aproxima-, damente 1 g del compuesto. Cuatro mililitros de ácidos nítrico concentrado íc añaden gota a gota, agitando después de cada adición. El matraz se une a un refrigerante de reflujo y se conserva en un vaso de precipitados con agua a 45° durante 5 minutos. La mezcla de reacción se vierte sobre 25 g de hielo picado y el precipitado se filtra. Se le puede recristalizar de etanol diluido.
b) Se sigue el procedimiento arriba descrito excepto que se usan4 mi de ácido nitrico fumante en lugar del ácido nítrico concentrado y la mezcla se calienta en un baño de vapor durante 10 minutos. Ocasionalmente, en los compuestos que son difíciles de nítrar se puede substituir el ácido sulfúrico concentrado por ácido sulfúrico fumante.
Comentarios. El procedimiento (a) produce m-dinitrobenceno a partir del benceno o nitrobenceno y el derivado />~njtro, del clorobenceno. bromobeneeno, cloruro de bcnci'o o tolueno. Él fenol, la acetanijída, el naftaleno y el bifcnilo producen derivados dínitrado?. Para los bencenos halogenados es preferible usar el procedimiento (&), que produce derivados dinitrados que son mucho más fáciles de purificar que los derivados mononitradea obtenidos por el procedimiento («0, El mesítileno, los xilenos y el pseudocumeno producen derivados tri nitrados.

P rocad im fen to 4 7 . A c id o s u ro U b e m o ito ? 3 0 7
P roced im ien to 46> O xidación de un a cadena lateral
a) O xidación con dicrotnaio . En un matraz pequeño se colocan 15 mí de agua, 7 g de dicromato de sodio y 2 a 3 g del compuesto que se va a oxidar. Se añaden diea mililitros de áddo sulfúrico concentrado, el matraz se une a un refrigerante de reflujo y la mezcla se agita perfectamente. El matraz se calienta cuidadosamente hasta que empiece la reaedón; entonces deberá suspenderse el calentamiento y á es necesario enfriar el matraz. Después que cesa la ebullición de la meacla a causa del calor de la reaedón, se calienta a reflujo durante 2 horas. El contenido del matraz se vierte en 25 mi de agua y el predpítado se filtra. El precipitado ss mezcla con 20 mi de ácido sulfúrico al 5% y Ja mezcla se calienta sobre un baño de vapor, agitando vigorosamente. Se le enfría y el predpitado se filtra y se lava con 20 mi de agua. El residuo se disuelve en 20 mi de soludón de hidróxido de sodio al 5% y la solución se filtra. El filtrado se vierte, con agitación vigorosa, en 25 mi de ácido sulfúrico al 10%. El precipitado se filtra, se lava, con agua y se purifica por recristaJizadón de etanol o benceno.
b) O xidación con pernum gútuU o. U n gramo del compuesto se añade a 80 mi de agua que contengan 4 g de permanganato de potasio. Se añade un mililitro de solución de hidróxido de sodio al 10% y la mezcla se calienta a reflujo, hasta que desaparezca el color púrpura del permanganato (% a 3 horas). Al finalizar este tiempo la mezcla se deja enfriar y se acidula cuidadosamente con ácido sulfúrico. La mezcla se calienta % hora y se enfría. Cualquier exesso de dióxido de manganeso se elimina por adidón de un poco de solución de bisulfito de sodio. El áddo precipitado se filtra y se recristaliza de benceno o etanol. Si el ácido es bastante soluble en agua, puede ser que no precipite de esta solución ácido diluida. En este caso el áddo se. puede extraer con cloro* formo, éter o benceno. En ocasiones, al addular puede aparecer un precipitado pequeño de áddo silícico, de aquí que sea importante rccris- talizar el ácido antes de determinarle el punto de fusión.
P roced im ien to 4 7 ♦ Acido* (troübcnxoicox
A unai soludón de L g del hidrocarburo aromático seco y 1.2 g de anhídrido itálico en 10 mi de disulfuro de carbono seco se le añaden 2.4 g de doruro de aluminio anhidro. La mezcla se calienta, con un refrigérame de reflujo, en un baño de agua hirvientc durante 30 m inutos y se enfría. Se decanta Ja capa de disulfuro de carbono y al residuo se le añaden 10 mi de áddo clorhídrico y 10 mi de agua. Al príndpio el áddo se añade lentamente, si es necesario enfriando con hielo, y ait final la me2cla deberá agitarse po Rectamente. Si el ácido aroilbenzoico pre- dpita, se le ñltra inmediatamente y se lava con agua helada. Si se

3 0 8 P re p a ra c ió n d e d e r iv a d o *
separa un acáte, la mezcla se enfria en im b.iño con hielo durante algún tiempo para inducir la cristalización. Sí el producto permanece aceiluso, se decanta el líquido sobrenadante y el aceite se lava con agua helada El producto crudo, ya sea sólido o líquido, se hierve durante 1 minuto con 30 m i de solución de hidróxido de amonio al 10% a la cual se le ha añadido aproximadamente 0.1 g de “Norite” La solución caliente se filtra y se enfria; entonces se añaden 25 g de hielo picado y la solución se acidula con ácido clorhídrico concentrado. El ácido aroílben- 20ico se filtra y se recristaliza de etanol diluido (30 a 80% ). Algunas veces es necesario que el producto se deje én reposo durante la noche para obtener cristales.
P roced im ien to 4 $ . M etüvrilceto tuu
En un matraz Erlenmeyer de 50 mi, provisto de un tubo con cloruro de caldo, se colocan 1.4 g de cloruro de aluminio anhidrOj pulverizado, 5 mi de disulfuro de carbono y 0.8 mi de cloruro de acetilo. Después de 5 minutos se añade una solución de 2 m i del hidrocarburo en 5 ral de disulfuro de carbono* imprimiéndole al contenido un movimiento de rotación. La mezcla se deja reposar a temperatura ambiente hasta que se disuelva todo cl cloruro de aluminio (de 10 a 15 minutos) y entonces se vierte sobre una mezcla de hielo y 5 mi de ácido clorhídrico concentrado. Se añaden otros 5 mi de disulfuro de carbono. La capa orgánica se lava sucesivamente con 3 mi de ácido clorhídrico al 10$>, agua, bicarbonato de sodio al 5% y agua. El lavado final se repite hasta que el agua de lavado sea neutra al tornasol. La capa orgánica se seca con cloruro de calcio y se evapora el disulfuro de carbono. El residuo se disuelve en etanol y se prepara por el método usual la 2/f-dímtrofencl- hídrazona o la semicarbazona.
HIDROCARBUROS — PARAFJNAS Y CICLOPARAFINAS
(Enlistadas en la pág. 382)
Existen pocos derivados apropiados para el trabajo de identificación de estos compuestos. Por lo tanto, es necesario fundar la identificación casi enteramente en constantes físicas. De éstas, las más útiles son el punto de ebullición, la densidad específica y el índice de refracción.

Hidrocarburos ¡nioturacfos 309
Rcíer«iciw
/tttcm ational Crüical TabUi,Dom, Physical Coüitants o f (h* Principal Hy¿rocarbon¡, The Texas Co. Nueva
York, 1939.Egloff, Phyiieel CamlaníS of. Hydrocarbont, VoJs. I y I I , R einhdd Publiihing
Corp. Nueva York, J939.Sólidos co* ficido dcssxícólico. H untre« y Phillips, / . Am. Chem. Soc., 71, 458
(1949).Separación de palatinas normales de Jai arborescentes con urea. Zimmerjchied,
D m enteín , Weitkamp y M arschoer, / , Am . Ch*m. Soc., 71, 2947 0 9 4 9 ) .
HIDROCARBUROS — INüATÜRADOS
(Enb'stados en las págs. S83-384)
Muchos hidrocArby os olefínicos sencillos se pueden identificar por medio de sus constantes físicas. En algunos casos los compuestos de adición con bromo (procedimiento 35), haluros de hidrógeno o cloruro de nitrosilo proporcionan derivados sólidos. El reactivo nombrado al último ha encontrado aplicación especial en los terpenos,9
La oxidación de una olefína o acetileno simétricamente substituidos, un atqucno-1 o un alquino-1 produce un ácido. Ordinariamente la oxidación se efectúa con perrnanganato [procedimiento 4 6 (6 )]
R C H ^ C H R -» 2RCOOH RC m CR 2RCOQH
RGH - GH2 - > R C O O H + COzR C w C H -» r c o o h +co2
Si el ácido cs sólido servirá como derivado. Si es líquido puede ser caracterizado por medio de un derivado apropiado (procedimientos 1, 2, 3)
Los compuestos acetilénicos que contienen el grupo — C**CH forman derivados de mercurio, los cuales son apropiados para propósitos de identificación.
2RG « C H + KjHgI4+ 2KOH ( RC ™ C ) ¿ ig 4* 4K I+ 2H^>
Los acetilenos disubstítuiejos pueden hidratarse hasta cetonas por la acción del ácido sulfúrico y el sulfato mercúrico en soluciones diluidas de etanol.
RC a GR+ HjO RCOCH^R
* SifW iK flj The Trrprnfi, Voli. t y II , 2* cé„ Cthibriiee U nívertity P r« tt, 1947, 194$.

4 m /
Mi
h *
f *
a»-9
4p
»
r»?».
T»s »
Entonces la cetona puede caracterizarse con un reactivo apropiado para carbonilo (procedimientos 16, 17* 18, 49).
Los mercaptanos y tiofenoles se condensan con Jas definas para producir fos sulfuros correspondientes.
CH3
R C H = C H 2+ R 'S H -> R ¿H S R '
Si hay ácidos presentes que actúen como catalizadores, la reacción pro* sigue de acuerdo con Ja regla de Markownikoff. En presencia de peróxidos puede invertirse el modo de adición.
RCH = C II2 + R'SH -+ R ~ CH2— CH jSR'
Aunque la mayoría de los sulfuras son líquidos, puede identificárseles con referencia a las sulfonas que dan cuando se les oxida. Las sulfon&S son sólidos y se purifican fácilmente.
Los hidrocarburos que contienen enla^ 's dobles conjugados se condensan fácilmente con el anhídrido maleico o la «-naftoquinona formando derivados sólidos.
3 1 0 P re p a ra c ió n d« d e r iv a d o s
Referencia*
Derivados mcrcúñcos de los hidrocarburos acctilciucos. Johnson y McExven, /.A m . Ch<m. Soc., 46, 469 (1926).
Compuestos de fldiri¿n con el anhídrido mtfeieo. D iek y *\lder, Bef., 62, 2081 (1929).
Compuestos de adiyión con Ja a-naítoquinoiia. Diels y Aldcr, Ser., 62, 2337 (1929).
Olcfinas y tiofenoL Ipaticff, Pines y Frícdm an, / . A m . Chem. Soc., GO, 2731 (1938); [patiefí y Fríedroan, ibid., 61 , 71, 664 (1939).
Olríioa» y mercaptanos. Jone» y Rcid, } . A m . Chem. Soc., 60, 2452 (1953), HEdr&tación de acetilenos. Johnson, Schwartz y Jacob*, / . A m . Chem. Sor., 60,
18S2 (1956); Thcnm s, Campbell y H enaion, ibid., 60, 716 (1935). DiiíocíaflatQs * partir de olefltun. Derm er y Dysinger, J . A m . Chem. Soe., 61,
750 (1959).Adición del cloruro de 2,4-dimtcosulfetillo. Kharasch y Buess, / . Am . C ktm . Soc.,
71, 2724 (1949).Adición de rúlrilos en presencia de ácido sulfúrico. R iit í r y M inierí, / . Airt.
Chem. Soc.. 70, 4045 (1948); R itter y K aliih , ibid., 70, 4048 (1948); Betuon y R itler, ibid , 71, 4123 (1949); K a ru e l y R ittcr, ibid., 71, 4130 (1949).

P ro ced im ien to 4 9 . O x im a s 3 í l
GETONAS
Los reactivos de carbonilo que se describieron bajo aldehidos (págs. 271-273} pueden ser usados para obtener derivados sólidos de las cetonas, Las cetonas de bajo peso molecular pueden caracterizarse por medio de las 2,4-dinitrofenÜhidrazOnas (procedimiento 18) > ¿-nítro- fenilhidrazonas (procedimiento 17) o semkarbazonas [procedimientos 16(a) 6 16(fc)]. Para las cetonas de peso molecular elevado son apropiadas las hidrazonas, las fenílbldrazonas (experimento 21, pág. 164) y las oximas (procedimiento 49).
Las me dice tonas pueden oxidarse selectivamente a ácidos con hipo- clorito de sodio. Este reactivo es particularmente útil para las metíl- cetonas insaturadas, ya que los agentes oxidantes ordinarios atacan el doble enlace.
R C H = CHOOCHj + 3NaOC) -> R C H = CHCOONa -j- OHClj+ 2NaOH
Las hidantoinas, obtenidas por calentamiento del compuesto car- bonílico con una mezcla de cianuro de potasio (o de sodio) y carbonato de amoniúj han sido propuestas como derivados, especialmente para los cetoéteres.
RCOR
(Enlistadas en las págd. 385-368)
Referencias
V er kn referencias ¿ik !loadas pa ra a ld e tid o j en laa págs. 273-274.Reacción de las xnctücetoaas con hI¡*xÜQjrit©4 VanArendonk y Cupcry, / . Am.
Chtm. Soc., 53, 3184 (1931); Hurd y Thomaj, ¡bi¿» 55, 1646 (1933). Oximas, Houbcn y Pfankuch, Bcr., 59, 2392 (1926); Buck y Idc, / . Am. Chtm.
Soc., 53, 1536 (1931 ) j Ryr&nt y Snuth, íbid.j 57, 57 (1935); Bachmann y Boatner, tbíd., 58, 209? (1936); Backcoín y Barton, / . Org. Ghenu, 3 , 300
.(1 9 3 5 ).H!dantoínas- Henzc y Speer, ]. Am. Chim. St*., 64, 522 (1942).
P roced im ien to 4 9 . O xim as
a) M étodo d e la p iríd ina . U na mezcla de I g del aldehido o cetona, 1 g de clorhidrato de hidroxílaminaj 3 m i de pirídina y 5 mi de etanol absoluto se calientan a reflujo durante 2 horas en un baño de vapor. Los disolventes se eliminan con corriente de aire y dentro de una campana. El residuo se tritura perfectamente con 5 mi de agua
R2C------- CO
NHCONH

3 ) 2 P repcu radón d e r iv a d o s
helada y la mezcla se filtra. La oxima se recristaliza de metano], etanol o de agua-etanol,
b) Aproximadamente 0.5 g de clorhidrato de hidrojri lamina se disuelven en 3 mi de agua; entonces se añaden 2 m i de soludón de hidróxido de sodio al 10$> y 0.2 g del aldehido o la cetona. Si el compuesto es insoluble en agua, ¿e añade a la mezcla el etanol justamente necesario para obtener una soludón transparente. La mezcla se calienta sobre el baño de vapor durante 10 minutos y se enfría en un baño con hielo. Para acelerar la cristalización se raspan las paredes del matraz con una varilla de vidrio. Ocasionalmente, la adición de unos cuantos militros de agua destilada ayuda a provocar la precipitación de la oxima. El producto puede recristalizarse de agua o etanol diluido*
Determinadas cetonas cíclicas, como el alcanfor» requieren un exceso de álcali y calentamiento par tiempos más prolongados. Si una cetona es incapaz de producir una oxima por cualquiera de I03 procedimientos (a) o ( ^ ] ,u n gramo de ella deberá tratarse con 1 g de clorhidrato de hidroxüamina, 4 g de hidróxido de potasio y 20 mi de etanol al 95%. L a mezcla se calienta a reflujo durante 2 horas y se vierte en 150 rol de agua. La suspensión se agita y se deja reposar para permitir que precipite la cetona que no reaccionó. La solución se filtra, se acidula con áddo clorhídrico y se deja reposar para que la oxima pueda cristalizar. El producto se «cristaliza de etanol o de agua*etanol.
MERCAPTANOS
(Enlistados en la pág. 389)
Los mercaptanos y tiofenoles pueden convertirse en t í t e r e s de 2,4>dinitrofenílo por reacción con el 2,4-dinitroclorobenceno.
RSNa + C l / ^ \ N 0 2 — RsA n O , + NaCl '
El cloruro de 3,5-dinitrobenzoilo puede usarse para preparar los tío-* ésteres.
COCI COSR
R S H + f ¡ + C>H6N - r j| + CcHaN-HClOjN ^ ^ N 0 2
Los mercaptanos también reaccionan con el anhídrido 3-nitroftá!ico.
'« i+ rfN » ^ C00HNO,

N it r i t o - 313
Los derivados de mercurio han sudo empleados para caracterizarlos* pero no son tan buenos como los otros derivados mencionados.
Referencias
Hoéterts de 2,4-dialtrofcnílo. Boat, T uroer y N orton, / . A m . Ckcm, $ot,} 54, 1995 (1932).
S-S-Dbutrobe&zoatoa y 3’nitrcftalataj. Wcrtliciro, J. Ar<t. Chem. Soc.t 51, 3661 (1929).
T íoétertá de autraquinoTia y SuKoa&í. Rcid, Maciail y MiJler, / . A*n* Chem. Soc., 43, 2104 (1921); Ho/fm an y Reíd, ib tí., 45, 1831 0 9 2 3 ) ; Bilis y Reíd, ibid., 54, 1C?4 (1932).
N ITRILO S
(Enlistados en la pág. 390)
Los nitrilos pueden huVolizaree a los ácidos correspondientes por medio de áddos (procedimiento 50) o álcalis (experimento 26, pág, 186),
R C N -f 2H20 + H G 1 -*■ RCOOH +NH*C1 R C N + H;0 + NaOH -*■ R G O O N a-fN H j
Sí el ácido resultante es sólido* sirve como im excelente derivado. Si el áddo es líquido o es soluble en agua, es difícil aislarlo en forma pura y Se caracteriza mejor por medio de Sü éster de ¿-bromofenaciío {procedimiento 1). Para datos sobre ácidos y sus derivados ver tabla 20.
La reduedón de tos nitrilos con sodio y un alcohol forma aminas primarias (procedimiento 51), las cuales pueden identificarse por conversión directa a feniltioureas substituidas,
R C N -f 2Hj &CH2NH2
El tratamiento de los nitrilos con el reactivo de Grignard y la hí- drólosis subsecuente produce cetonas, las cualea pueden caractcrizarse por sus sernicarba2onas.
R 'R C N + R 'M gX —* R C = NMgX
R ' R 'RC N M gX + H jO + 2HX - » RC = O + N I I*X+ MgX*
Sin embargo, determinados nitrilos experimentan una reacción al’ terna en la cual d grupo ciano es substituido por un grupo alquilo o arilo.50 Los compuestos de este tipo incluyen «t-ceto, a-hidroxi o atcoxi y a-amínonítrilos.
" Vef K|i»ruch y Rdnrouíi, GrífHsrii «f Uva-^ctaUie Suküajittr Prtntjcr.KnH,K ngkwooi Cüííi, N. J.t 1 * 4 , p4g, 776.

f *CL »
t *
i »
f »
r *
f »
f *i *»
i*
«J
f m
La reacción de Hocsch también puede servir para la identificación de nitritos. El floroglucinol se usa para obteuc: alquiltribidroxifenitce- tonas sólidas.
R C =N H H C 1 R C = 0H 0 Q 0 H + Tt C N + H a i ^ H 0 Q 0 H
OH S h
£3 ácido mercaptoaeético (ácido tioglicólico) se condensa con ios nitritos en presencia de cloruro de hidrógeno produciendo clorhidratos de ácidos a-iminoalquilmercaptoacéticos (procedimiento 52).
N H H C ls
RCN + HSCHjCOOH + HCJ -* RC\
. SCHjCOOHEstas sales pueden caracterizarse por sus puntos de descomposición y equivalentes de neutralización. Cuando se titulan con álcali, usando azul de timol como indicador, se comportan como ácidos dibásieos.
Todos estos métodos involucran adiciones al grupo ciano y fallan con los benzonitnlos orfo-íubsdtuidos tales como el o-toíuonítrilo o el o-clorobenzonitrilo. Los nitrilos con impedimento estérjeo pueden hidro- lizarse a ácidos calentándolos con ácido sulfúrico al 75% {procedimiento 50>,
3 1 4 P r& pd ra c tó ii do d e r iv a d o s
Referencia»
ALquüfenUcetóaas, Shríftcr y T tim er, / , A m . Chem. Soc., 52, 1267 (1930), A lquil 2,4,6-tnhidroxifcnílcetotuj. Howells y L ittle, / . A m . Chem. Soc., 54, 2451
(m '¿}.Clorhidratos de ácido? a-immcu 1 q táltnercap tw c&ícos. Condo, Hink«l, Faiscro y
Shrbicr, / . A m . Ckenj. íoc ., 59, 230 (1937).Rr.doccíóa de mtrilos y formación d* tenüurtas substituidas. G u tter y Taras, tnd .
Eng. Chem., Anal. E J., 13, 83D (1941).Hidrólisis en disolventes de punto d t ebullición elevado. Rovira y Palfray, Compt.
R tn d ., 211, 395 (1940).Bisa midas de xaetíkoo. M agat, C handler, Far¡s, R eith y Saliabury, / . A m . Chem.
Soe., 73, 1031 (1951).
Procedim iento 50. H idrólisis á c id a de nltrU ai
Aproximadamente 25 mi de ácido sulfúrico al 75% y 1 g de cloruro de sodio se colocan en un matraz de fondo redondo de 100 mi provisto de un refrigerante de reflujo. El matraz se calienta a l5ü-16ü°C en un baño de aceite y se aftaden 5 g del nitrilo, en porciones de 0.5 mi,

P ro ce d im ie n to 5 1 . R educc ión d e n ltrUo* 3T 5
a través de la parte superior del refrigerante., con agitación vigorosa ctopuís de cada adición. La mezcla se calienia, cor» agitación, a 160°C durante 30 minutos y a 190&C durante otra y2 hora. Entonces se enfria y se vierte en 10CT g de hielo picado contenidos en un vaso de precipitados y el precipitado se filtra. El precipitado se trata con un ligero exceso de solución de htdióxido de sodio al 10% y cualquier amida msoluble se elimina por filtración. Por acídulacióo del filtrado se obtiene el ácido orgánico, el cual puede purificarse por recmtalización de benceno o de agua-acetona.
Para muchos nitrilos11 el ácido clorhídrico es más efectivo que el sulfúrico. Sin embargo, para los nitrilos que son difíciles de hidrolizar cs aconsejable el ácido sulfúrico debido a su punto de ebullición más elevado. La adición de una cantidad pequeña de ácido clorhídrico (como cloruro de sodio) aumenta la velocidad de la reacción.
P ro ced im ien to 5 1 . R educción de n itrilos hasta aminas y su conversión a fen iltioureas substitu idas
En un matraz de fondo redondo de 200 mi, seco y limpio y provisto de un refrigerante de reflujo, se ponen 20 rnJ de etanol absoluto y 1 g de un nitrilo alifático (o 2 g de un nitrilo aromático). Por Ja parte superior del refrigerante se adicionan, tan rápido como sea posible y evitando que la reacción se vuelva demasiado violenta, 1.5 g de trozos muy delgados de sodio. Cuando se ha completado la reacción (10 a 13 minutos), la mezcla se enfría a 20*0 y por la pane superior del refrigerante se añaden 10 m i de ácido clorhídrico concentrado gota a gota y agitando vigorosamente el contenido del matraz. L a mezcla de reacción se prueba para asegurarse de que está ácida al tornasol y se pasa a un matraz de destilación de 200 mi unido a un refrigerante. Unos 20 m i de etanol y agua se eliminan por destilación. El matraz y su contenido se enfrian y en la parte superior del matraz de destilación se monta un pequeño embudo de goteo con 15 mi de solución de h idróxido de sodio al 40%. En cl extremo deí refrigerante se conecta uoa adaptación sumergida en 3 mi de agua contenidos en un matraz Erlen- meyer de 50 mJ. El álcali se añade gota a gota, con agitación. L a reacción es vigorosa y deberá tenerse cuidado para evitar que la adición del álcali sea demasiado rápida. Después que se ha añadido todo el álcali la mezcla se calienta hasta destilación total de la amina. L a destilación se susp< ?.de cuando el contenido del matraz se vuelve demasiado viscoso.
AI destilado *e le añaden Ü.5 mt de isotíocianaio de fenilo y la mezcla se agita vigorosamente durante 3 a 5 minutos. Si el derivado no precipita inmediatamente, se induce la cristalización por enfriamiento del
, » K ilp iificl, J . A*í. Cktrn. S í* . , t$ , 40 (1*7).

3 1 6 P rep a ra c ió n d » d e r iv a d o *
matraz en un baño con hielo y raspando las paredes. El precipitado se filtra, se Uva con un poco de etanol al 50%, helado, y se re cristalina dos veces en una mezcla caliente de agva-etanol.
P rocedim iento 52 . C lorh idra to t d e ácido* &4niin<xdqutlnt6rcaptoacéiic<>8
En un tubo de ensayo limpio y seco se disuelven un gramo de nitrilo 7 2.0 g de ácido mercaptoacétíco (ácido tioglicólico) en 15 mi de ¿icr absoluto. La solución se enfría en un baño de hielo y se satura con cloruro de hidrógeno seco. El tubo se tapa perfectamente y se conserva en el baño de hielo o en el refrigerador hasta que precipiten Jos cristales del derivado. Los nitrilos aiifátícos forman el compuesto de adición en 15 a 30 minutos mientras que los nitritos, aromáticos ordinariamente tienen que dejarse una noche en el refrigerador.
Los cristales se filtuui, se lavan perfectamente con éter absoluto y se colocan en un desecador al vacío que contenga ácido sulfúrico concentrado en el fondo y vasos de precipitados pequeños, conteniendo perlas de hidróxido de potasio y para fina en la parte superior. El punto de descomposición se determina en el aparato usual para puntos de fusión y, si es necesario, se determina el equivalente de neutralización por titulación con un álcali valorado, usando como indicador azul de timo).
COM PUESTOS NITRADOS
(Enlistados en las págs. 391-395)
Por .reducción de los compuestos nitrados en medio ácido se obtienen aminas primarias (procedimiento 53).
R N O ,+ 6H -> . R N H j+ m jO
Estas pueden convertirse en derivados apropiados tales corno las berua- midas, acetamidas y arilsulfonamídas ^-substituidas.
Frecuentemente los compuestos aromáticos mononitrados pueden caracterizarse por su conversión a los correspondientes dinitro o tríni- troderivados [procedimiento 45{«) y 45 ( i ) ] . Muchos compuestos aromáticos pol'mitrados forman compuestos de adición característicos con el naftaleno.
En algunos casos la bromacíón del anillo (procedimiento 35) o la oxidación de una cadena lateral alqullica [procedimiento 46(c) y 46(fr)] ofrece el medio más apropiado para obtener un derivado.
Las nitropar Hnas pueden caracterizara: reduciéndolas coa cinc y ád d o clorhídrico. a las correspondientes aminas primarias, las cuales

Compuesto* nitroso, ozoxi, azo o hldrazo 317:>-■:
entonces se pueden convertir en derivados sólidos por reacción con is k tiocianato de fenilOj oxalato de etilo o 2 >4-d i nitroelorobenceno. Las sales dv. sodio de los compuestos nitrados alifáticos también se copulan con las sales de arildiazonio y en algunos casos los compuestos resultantes son sólidos que pueden servir como derivados,
Refectucúts
Conpuesto» moleculares de 3« cocopuestc* polínltrado* aromáticos coa el tufta- lcno. D erm cr y Sm jth, J. Am . Chem. Soc., 61, 7+3 (193$),
Derivados de nitroparafiflAs. Dérrn«r y Hutcheson, Okia, Aead. Sei., 23,60 (1943)j f e . A., 38, 2006 (194+)J.
COM PUESTOS N ITRO SO , AZOXI, AZO E HIDRAZO
(Enlistados en la pág. 39c
Los compuestos nitroso, azoxi, azo e hidrazo pueden reducirse con estaño y ácido clorhídrico (procedimiento 53} a las aminas correspondientes.
A r N O + 4 H - > A r N H j + H i O
A rN = N A r+ 6 H -> 2ArNH2+ H ,0 i
^ 1 O
A rN = N A r+ 4 H -> 2 A rN H 2 ArNHNHAr 2H -> 2ArNHz
Las aminas se identifican convirtiéndolas en apropiados derivadas de acilo. Las nitros aminas se reducen a hidrazinas substituidas.'
R2NNO + 4H ->■ R2NNH2+ HzO
Ocasionalmente pueden emplearse reacciones especiales. Por ejemplo, los. compuestos nitroso pueden condensarse con aminas primarias produciendo compuestos azo.
A rN O + A rN H j^ A rN ^ N A r+ H jO
El peróxido de hidrógeno puede oxidar los compuestos azo a compuestos azoxi,
ArN= N A r+ H20* —> A rN = N Ar+ HjO4»o
Los, compuestos azoxi pueden reducirse a compuestos azo mediante reactivos apropiado o transponerse a 4-hidroxiazob«ncenoS.

Los compuestos hidrato pueden acetilarse, ucnzoUarse o pueden sufrir transposición,
< ^ ) N H N n{ y ^ H5n Q - / V h 2
R eferencias
p-HWr«tíazí)beocenos a p a rtir de compuestos azox. Gaudry y Kíerstad, C<sn. J. fa tta rc h , 27, 997 (1949).
31 8 Prepcrraetón derivada
P roced im ien to 5 3 . R educción con estaña y ácido clorhídrico
U n gramo del compuesto que contiene nitrógeno (nitro, nitroso, azo., azoxi, o hidrazo) se añade a 2 g de es. -»ño granulado contenidos en un matraz pequeño. El matraz se conecta A refrigerante de reflujo y se añaden 20 mi de ácido clorhídrico al 10% , en porciones pequeñas, con agitación vigoiosa después de cada adición» Finalmente la mezcla se calienta en baño de vapor durante 10 minutos. La solución se decanta, aún caliente, sobre 10 mi de agua y se añade suficiente solución de hidróxido de sodio al 40% para disolver el hidróxido de estaño. La solución se extrae varias veces con porciones de 10 mi de cter. El extracto etéreo se seca y se destila eJ éter.
Con compuestos nitrados muy ínsolubJes se acelerará la reducción añadiendo 5 mi de etanol.
La amina primaria que queda después de la destilación del éter se convierte en uno de los derivados descritos en las págs. 27$'260.
FENOLES
. (Enlistados en las págs, 397-390)
Los fenoles, como los alcoholes, cuando se tratan con isoclanatos producen uretanos (procedimiento 56), Entre etlos, el a-naftiüsocianato quizá sea el reactivo generalmente más útil para la identificación de fenoles [procedimiento 9{u}]. Esta rcacción se cataliza por adición de unas cuantas gotas de pirídina seca.12
CJ0H7N = C = O + A iO H -> C JSH rNFICOOAr
O tro u re taño que se usa a menudo es el dífentturetano que deriva del cloruro de difenílcarbamilo, (C4H 5) jNC-OCt, de acuerdo con la ecuación siguiente [procedimiento 9(& )J:
a T a r b d t , k t« 3 l> tt 7 t f i f c o a , / . A n . S w , M , 2?29 < IW 2 ] .

F o n o U » 3 1 9
N íC O C l + ArOH
_C,h ,
Los u rétanos también pueden obtenerte tratando Jas azi das de ácido con el fenol. La azida se descompone en nitrógeno y el isocianaio, el cual entonces reacciona con el fenol.
RCON, - » R N = C = 0 + N ? R N = C ^ O + A rO H -> RNHCOOAr
En esta forma se lian preparado los fl*naftihiretanos así como los p-doix^, p-bromo-, /«-nitro-, 3,5-dirútro- y 3,5^mtro*4-metilfeniluretanos.
L a mayoría de Jos fenoles pueden identificarse fácilmente por referencia a sus productos de bromación (procedimiento 55), Asf} el fenol reacciona con bromo dando 2,4,6-tribroroofenol.
Br
H + 3Br* — R r f ^ y p H + 3HBr
BrEn presencia de álcalis los fenoles reaccionan fácilmente con áddo
doroacético dando ácidos ariloxiacéticos, Estos derivados cristalizan bien en agua y han demostrado ser extremadamente útiles en el trabajo de caracterización (procedimiento 54).
. A tO N a+ ClGHjCOOH ArOCH,COOH+ NaCT
Los ácidos ariloxiacéticos pueden compararse no tan sólo mediante sus puntos de fusión sino también por referencia a sus equivalentes de neutralización.
Otros tipos de derivados de los fenoles que se usan frecuentemente son los derivados nitrados [procedimiento 45(«)J, los picratos [procedimientos 26(<z) y 26(é) ] , los acetatos [procedimientos 21 (a) y 21(6)3, los 3,5-dinitrobenzoatos (procedimiento 12} y los ¿steres de áddos sulfónicos.
Pata identificar fenoles también pueden usarse los derivados de O^al- quiJsacarina que *e discutieron en relación con los alcoholes (pág. 266).
Referencias
U ittan as . V er alcoholes.Sales de p -tohjuüna de ád d o s arílsulfúricos. V er alcoholo.Ac&OKxacetazzndxs. Karaetkizt, Mel'nikov, B&stako y Bobarev", Zhvr, A na l K him .,
5, 7 (1950) [C. A ., 44, 4374- (1950)].Acidos A rilosúó lticos. Koelach, / . A ” , Chem. Soc., 53, 304 (I9 3 1 ) j Haya» y
B n n d i , «5, 1555 (1943).Esteres d e ¿ ¿ d o s wHónícco. Sclcora, / . A m , C him . Soc., 55, 421 (1033).
NCOOAr + HCI/
c ^h ;

rt d * d e r iv a d a ?
-.Mtimtefc BárS y íLtubcr, / . Am. Ch4tt*. Soe., 53, 1087 (3931).AceCstM. C iu ltuvay , }■ Chem. Soe., 1931, 2495,^^S-Dinicrobejyeoatde.Brown y K itm erf, / . A m . Yharm. Assoc., J l , 60? (1922);
Phiilipí f ScnoaQ, /• A m . Chem. Soc., 5 J , 1924 (1931 ),¡B(cret í c ^wtrobeiicilcx. Rckí, J. A m . Chein, Soc., 39, 30+ (1 9 1 ? ); Lym an y
R d d , ihtd., 42, 615 (1920),S-Niw>-4-2actiIfciifluíetiuios. Sah„ Li, Tsun* y W ang, J . C hínete Chem. Sor., 14,
«* (1946) { C J ., 43, 7+48 (1949)]. rtf-AriUWaoJrtot S a i, G ih, K an* y Fu, / . Chínese Chem. Soc:, 14, 65 (1946)
[C. A ., 45, 7447 (1949)].^^DxmtcOÍewHxreUUlOt. Sah y Ma, / . C him se Chem. Soc., 2y 229 (1934). Jcm iu ttU n cs. MeKoiiley, NkkcU y Stdhu, In d . £ n g . Chem., Anal. Ed., 16, 304
<l944>‘Ct'N'líliHircaik». French y W irtcl, / . A m . Chem. Soc:, 48, 1736 (1926),£ te res do 2j4*díaátwrfeiiJky. Boit y Nicholson, / . Am- Chem. í « . ; 57, 2368 (1935), p ^ a ro fe o S o n tu iM * Kao, Fxag y Sah, Sci. R tp ts . tfa tt. T fin g H m« Univ., (A )3,
109 (1935).£-N*ftünrel*iK»* Sah, Rec. Trav. Chim., 5S, 453 (1939), jkN itroítcU flrrüldoj.Sah y Cfiiao, Rec, Trav. C him ., 58, 595 (1939). jp»6m>ofeoihw<a»w>. Sah y Cheng, Rec. Trao. Chim., 58, 591 (1939). 3TW>iwtr<»^-Bttetilfen¡lureunoi.Sah, Rec. Trap. Chim ., 58, 582 (1939).
Procedim ientos 5 4 , A cidos arUoxiacélicos
A una mezcla de i g del fenol y 5 mi de una solución de hidróxido de sodio al S3#> se le añaden 1,5 g de ácido cloro acético. La mezcla se agita perfectamente y pueden adicionársele de 1 a 5 mi de agua, ai es necesario, para que se disuelva la sal de «odio del fenol- Entonces el tubo de ensayo que contiene la mezcla, se deja en un vaso de precipitados con agua a ebullición durante 1 hora. La solución se enfría, se diluye con 10 a 15 mi de agua, acidulada harta el vire del rojo congo con áddo clorhí- drico diluido, y se extrae con 50 mi de éter. La solución etérea se lava' con 10 mi de agua fría/después s i agita con 25 m i de solución de carbonato de sodio al 5% y la solución de carbonato de sodio se acidula con ád d o clorhídrico diluido; el ácido aríloxiacétíco se filtra y se recristal iza de agua caliente.
P rocedim iento 55* Brorrwción d e feno les
Se prepara una solución para la bromación disolviendo 15 g de bromuro de potasio en 100 mi de agua y añadiendo 10 g de bromo. Esta solución se aív .ie lentamente, con agitación, a una solución de 1 g del fenol disuelto en agua, etanol, acetona o dioxano. La solución para bromadón se adiciona en cantidad justamente suficiente para impartir

A c id o s « u l f ín k c i 321
un colcr amarillento a la mezcla. Entonce» se adicionan unos 50 mi de agua y la mezcla se agí La vigorosamente para romper los grumos. Él derivado bromado se filtra y se lava con solución diluida de bisulfito de sodio. Se le recristaliza de etanol o etanol-agua.
P roced im ien to $6 . FenUuretanoa
A una mezcla de 0,5 g del fenol seco y 0.5 mi de isocianato de fenito, contenidos en un matraz seco de 25 ml3 se le añade i gota de pin- dina seca. El matraz se tapa con un tapón de algodón flojo y se calienta sobre el baño de vapor durante 15 minutos. Si durante este tiempo no hay precipitación del derivado, se induce la cristalización enfriando el matraz y raspando las paredes. U na vez formados los cristales se añaden 10 mi de benceno seco ./> acetato de etilo seco), La mezcla se calienta sobre un baño de vapor, íj. filtra a través de un filtro de pliegues y se añade hexano hasta que se enturbie o se fórmen cristales. Se deja que Ja cristalización prosiga durante la noche; entonces el producto se filtra, se lava con hexano que contenga un poco de benceno y se seca al aire.
Sí en la muestra original o en los reactivos se encuentra presente una poca de agua, el producto estará contaminado con í-dlfenilurea (p.f. 298<?C ) . La purificación puede efectuarse por calentamiento del producto con 10 ral de tetracloruro de carbono y eliminación por filtración de la dxfenilurea Lnsoluble. El íeniluretano puede obtenerse por enfriamiento del filtrado. Ocasionalmente el filtrado deberá evaporarse hasta 2 ó 3 mi para provocar la cristalización.
ACIDOS SULFINICOS Y SALES DE ACIDOS SULFINICOS
Se han preparado varios ácidos sulfmicos aromáticos, pero los ácidos Jibres son inestables, transformándose en los correspondientes ácidos sultánicos y tiosulfonatos. Los tiosulíonatos experimentan hidrólisis produ- ciendo tiofenoles y ácidos sulfónicos.
S A rS O zH A rS O jH + Ar$ • SOiAr + H20* | hso
ArSH-hArSOjH
Los ácidos yulfinkos aiifátícos son menos estables que los aromáticos; de aquí que ordinariamente ambos tipos se encuentren como sales de «odio, potasio, o magnesio, que son más estables. Los mejores derivados son

;
I
N )is #
í »
* •
aquéllos que se obtienen poi alquiladón, la cua! produce sulfoaas en lugar de ¿teres. Por ejemplo, el yoduro de tnclilo reacciona con aribulíí- natos de sodio formando meiilsulfonas.
ArSOiNa -f- C H J -» ArSO,CH, 4* N al
El bromuro de etílcno se ha usado para preparar 1,2-diaIquilsuUo- niletanos sólidos a partir de las sales de los ácidos sulíínicos aiifátícos.
CH íSOjRRrOHjCHiBr-l-'JRSOjNa^)- | -f2N aBr
GHjSOtR
Las sales de sodio de los aril- y alquilsulfinatos reaccionan con cloruro mercúrico formando los correspondientes cloruros de api- y alquil- mercurio, los r.ualcs son sólidos cristalinos.
32 2 P re p a ra c ió n d e d e r iv a d o s
Referencias1,2'DíalquiWuIfonlÍMatios. Alien, ] . Org. Chem., 7, 23 (1942).Cloruros de arií- y alquilmerctirio a p a rtir de julfínatos» Pctecs, Ber., tó , 2567
(1905); K h a r t i th y Cbaikley, / . Am . Chem. Soc., 43, €07 (1921); Mflrit. morr, Hanulton y Thum iart, ibid., 45> 1066 (1 9 2 3 ); Coffey, / . Chem. $oe., 1926, $3?; M arvel, Adams y Johnson, / . A m . Chem. Soc., 68, 2735 (1946).
SULFONAMIDAS
(Enlistadas en las págs. 400*401)
Las sulfonamidas que tengan cuando menos un. átomo de hidrógeno en el átomo de nitrógeno pueden alquilarse tratando sus sales ¡de wdio con haluros reactivos o sulfates de alquilo,
R S 0 2NH2 + N'aOH -f- R 'X -»■ RSOjNH R '+ N áX 4 - H iO
RSO¿NHR,+ N a O H + R ''X R S 0 2N R 'R "+ NaX + H 20
Estas reacciones son útiles si el grupo alquilo se selecciona en ta! forma que se obtenga una sulfonamida conocida.
Las sulfonamidas primarias reaccionan con cloruro de ftalilo produciendo AT-sulfonilftalimidas.
r e c o c í^ ............... ,NS02R + 2HQ
v ^ J c O Q+ RSOjNHj
|Qy

Las amidas primarias de los áddos sultánicos, como las de 1er. ácidos carboxllicos (pág. 277), reaccionan con t í xaothidrol formando JV-xantil- sulfonamidas, Jas cuales son derivados satisfactorios (procedimiento 57).
\Qc o + h2o/ \ /
RSO.NH / = \
Las sulfonamidas se hidrolisan calentándolas con ácido clorhídrico al 25% (pág. 136).
RSOjNHR' + H20 -f HCl - » RSO¿H +R 'N TH3Cl
R S O * » '* * H rf> + H U - » RSOjH + R '2NH2C1i
La amina se puede aislar mediante adietan de álcali y cara< leráaiíc por un derivado apropiado (pág. 279), Si es necesario se puede recuperar el ácido sulfónico en forma de su sal de sodio (después de separar la amina) y convercirse en un derivado (procedimiento 59 ó 60).
Un método que puede ser satisfactorio consiste cu tratar la amida con ácido bromhjdrko al 48% y fenol (pág. Í36).
2ArSO,NHR -f 5 H B r+ SC^HjOI t ->ArSSAr+2RNHj + 5UrC*H|Ott + 4H ¿)
No sólo se puede caracterizar la amina sino también puede aislarse el disulfuro de díarilo y puede servir como derivado.
P ro ce d im ie n to 5 7 . /V -X cn flta u lfo n a m id a s 3 2 3
Referencia»
/V-xan<ilnilfonajra¡das. Phillij» y Frank, / . Org. C h w . 9, 9 {1944),
P rocedim iento 5 7 . N-Xantiltulfonatnidaf.
Aproximadamente 0.2 g de xanthidrol se disuelven a l ]0 mi <lo ácido acético glacial. Si la muestra no queda transparente, *c filtra o se centrifuga y a la solución transparente se !e añaden 0.2 g de la sultana* mida. La mezcla se agita y se deja reposar a temperatura ambiente hasta, que precipite.cl derivado; esto pw .ic requerir hasta 1.5 horas. La N - xantüsulfonamida se filtra y se recristaliza de dioxa«o-agua (3 :1 ) .

3 2 4 P re p a ra d é n d e d e r iv a d o s
ACIDOS 5ULFONICOS Y SALES DE ACIDOS SULFONICOS
(Enlistado* en las págs. 402-405 bajo Cloruros de sulfonilo)Los ácidos sul/ónicos y sus salea, en forma similar a los ácidos car-
boxOicos y sus sales, forma» fácilmente derivados característicos con cloruro de ¿ ‘bcndltíouronio. Esta reacción representa el método rpás rápido y directo para obtener derivados de estos compuestos (procedimiento 58).
Los ácidos sulíónicos y sus sales se convierten en cloruros de sul- fonüo por calentamiento con pentadoruro de fósforo (procedimiento 59).
Después, el doraro se trata con amoníaco o una am ina para obtener la amida (procedimientos 37 y 39)
Debido a que. los áddos sulfónícos son fuertes no es posible obtenerlos por íicidulación de sus sales, a menos que el ácido sultánico sea muy insohible en agua o en áddo clorhídrico. Los áddos sulíónicos se combinan con aminas para producir sales, las cuales tienen puntos de fusión o de descomposición definidos y, por lo tanto, pueden usarse como derivados;.
Tratando una soludón concentrada de la sal de sodio de un ácido sulfónico con áddo dorhidrico y p-toluidina es posible obtener las sales de /i-toluidína (procedimiento 60). Estas son derivados útiles.
Las sales de fenilhidrazina son útiles para identificar ácidos sultánicos alifáticos.
Cuando los sulfonatos de plata reaccionan con cloruro de ¿-nitro* bencüo y pirídina se forman las correspondientes sales de p-nítrobcndl-
RSOjNa+CySíCEiiSC (NH,) /G l“ ->CéHjCH2SC (NHj) i1- (RSOi) ’ 4- NaCI
RSOjNa -f PCI, RSOiCl+ POCi, -f- NaCl
RSO,Na -f HCl + CH;O n h 2 -+
:HZCI + CSH5N —

Estas sales cristalizan rápidamente, tienen puntos de fusión definidos y característicos y son apropiadas como derivados. Los ácidos aminusulfó- nicos de las series del benceno y del naftaleno pueden caracterizarse por substitución del grupo amino por cloro medíante una reacción de Sand- meyer, seguida por la conversión del ácido sulfónico a una sulfonamidao auffonanilida (procedimiento 61).
P ro ce d im ie n to 5 9 . C lo ru ro s d e s u lfe n ilo y s g lfo n a m íd a s 3 2 5
Referencias
Sultanatos de S 'benciltíourorJo, ChíUübe» y W att, / . 0 r£ . Chsm-, 6, 376 (1941). Sale* de p-toiuidina de Ácido» sulíóqícos. Fitsetj Org. Syn th ttes, Coü. Vol. 2 ,4 0 2 ;
f . A m . Chem. Soc.t 51, 2460, 2471 (1929).Sale? d e p-íoi nidios de monoaríbulfatos. Barton y Young, / . Am . Chtrn. Soc .,
65, 294 (1943).Sale» de fem lhidnui& a de ácidos sultánico*. Latícner y Bost, / . A m . Chetn. Soc.,
59, 2500 (1937).Sales de p-uirrobeMcUpiridoulo d i '.'cidos sultánico» aromáticos, H u n tren y Foote, .
/ . A m . C h tm . Soc., 64, 1017 \ 1942).Cloroarilsulfonauudai y cloroarilsulfoaamJJdaa. Alten y Fram e, / . Org- Chem.,
7, 15 (1942).Sultanato? d e arilamlna. Denrier y D enner, / . Org. C h tm ., 7, 581 (1942*.
P roced im ien to 5 8 . SulfonaUts de benciltiouronio
Aproximadamente 1 g de la sal de sodio o potasio d d ácido sulfónico se disudve en la mínima cantidad de agua y si es necesario se usa calor para efectuar la disolución. Si se parte del ácido sulfónico Ubre, se 1c disuelve en una solución de hidróxido de sodio 2N y cualquier exceso de álcali se neutraliza con ácido clorhídrico, empleando fenolftaleína. como indicador.
1 g de cloruro de benciltiouronio se disuelve en la mínima cantidad posible de agua. Esta solución y la del sulfonato se enfrían en .un baño de agua-hielüj se mezclan y se agitan perfectamente. En ocasiones es necesario raspar las paredes del tubo y enfriar en baño con hielo para inducir la cristalización. Los cristales del sulfonato de benciltiouronio se filtran, se lavan con un poco de agua fría y se recrístalizan de etanol caliente al 50%
P roced im ien to 5 9 . Cloruros de iu lfo n ilo y sulfonamxdas a p a rtir de sales de ácidos tu l fón icos
En un m a tm seco y limpio se mezclan dos gramos de la sal con 5 g de pentacloruro de fósforo. Se conecta a un refrigerante de reflujo y d matraz se calienta en un baño de aceite a 150°C durante 30 minutos.

V
La mezcla se enfría y se añaden 20 mt de benceno seco- Entonces se callenta la mezcla en el baño de vapor y la masa sólida se agita perfectamente. La solución se filtra a través de un papel filtro seco y el filtrado se lava con dos pociones de 15 mi de agua. Él benceno se elimina por destilación en baño de vapor, £1 cloruro de sulfonilo residual se puede recristali2ar de éter de petróleo o de cloroformo. Ya que los cloruros de sulfonilo usualmemc son compuestos de bajo punto de fusión, es mejor preparar la amida t>or adición de la solución benccnica a 20 mí de hidróxido di: amonio concentrado, con agitación vigorosa. Ocasional - mente la sulíonnmida precipita y se puede filtrar; de lo contrarío se 1c obtiene por evaporación de la capa bencénica. Las suífonarnidas se pue- den rccrístalizar de etanol.
326 Proporción do derivados
P ro íW ím ten ío 6 0 . Sales de ¡^to lu id ina d* ácidos suljónicos
a) A partir de ácidos aulf¿tucos libres. Un gramo del Ácido sulfónico se disuelve en la mínima cantidad de agua hirviente y se añade 1 g de ¿-toluidina. Para obtener una solución transparente se puc~ de añadir más agua o más ácido sulfónico. La solución se uniría y el matra/ se raspa para inducir la cristalización de la sal. La sal se filtra y se rccrístaliza en la mínima cantidad de agua hirviente.
b) A partir d e ¿ate* so lubles de ¡os ácidos sultánicos* Aproximadamente 2 g de b sal de sodio, potasio o amonio del ácido sulfónico se disuelven en la miníina cantidad de agua hirviente y se añaden 1 g de p-toluidina y de 2 a 4 mi de ácido clorhídrico concentrado, S¡ hay precipitación o si la p-tohudina no se disuelve totalmente, se añade más agua caliente y unas gotas de ácido clorhídrico concentrado hasta obtener una solución transparente en el punto de ebullición. La solución se enfría y las paredes del matraz se raspan para inducir la cristalización de la sal. El producto se filtra y se recristaliza de una pequeña cantidad de agua o etanol diluido.
P rocedim iento 61. C loraar'dsulfonam idas y cloraariU ulfo tuw üidas a partir ile ácidos am ínosu ifón icos
Aproximadamente i,5 g del ácido aminoarilsulfónico se disuelven en 10 mi de agua que contenga 0.5 g de carbonato de sodio. La dia- íxwción se efectúa añadiendo 2 mt de ácido clorhídrico concentrado y después, rápidamente, unos 5 mi de solución de r.itrito de sodio al 10%; la temperatura se mantiene entre 10°C y 15°C medíame adición de hielo.
:l : Mientras tanto, se prepara el cloruro cuproso mezclando una solución de 2,16 g d e sulfato cúprico y 0.56 g de cloruro de sodio en 10 mi

C lo ru ro s d e s u ífo n ilo 3 2 7
de agua con una soludón de 0.46 g de bisulfito de sodio y 0.33 g de hidróxido Je sodio en 10 i ni de agua. Después se disuelve eJ cloruro cuproso en 10 mi de ácido clorhídrico concentrado. L a soludón se enfria en hielo hasta 5°C y la soludón de diazonio se le añade rápidamente, con imitación. Se deja que la temperatura asdenda lentamente hasta llegar a la temperatura ambiente, la agitación se continúa durante I hora y entonces la solución se calienta a 60-70°C durante 30 minutos en el baño de vapor. El cobre se precipita con áddo sulfhídrico y el sulfuro de cobre formado se elimina por filtración, El ácido cloroarilsul- fónico se obtiene por evaporación de! filtrado a sequedad sobre el baño de vapor.
Después, en un vaso de precipitados pequeño se mezcla et ácido crudo con el doble de su peso de pentocloruro de fósforo. Cuando se ha terminado la reacción vigorosa, se calienta el vaso de precipitados, por uu tiempo corto, en un baño de aceite a 130-14G°C (dentro de la cam^ pana} para expulsar el oxicloruro de fósforo. Después que se lia enfriado el cloruro, se le lava por decantación con agua helada. El aceite resultante se añade a 45 mi dt; hidróxido de amonio concentrado y la solución se evapora a Sequedad sobre el baño de vapor. La sulfonamida cruda se recristaliza, con adidón de “¿Vorit” , de etanol o agua.
Para preparar la cloroaolsulfonanilida, una soludón de! doruro do sulfonilo crudo en 10 mi de benceno se mezcla con 2.5 g de anilina y Ja solución resultante se calienta, a reñu jo durante 1 hora. Entonces se concentra a la mitad de su volumen y se enfrja. Et sólido que precipita se filtra, se lava perfectamente con agua caliente y se recristaliza de cloro- benceno.
CLORUROS DE SULFONILO
(Enlistados en las págs, 402-405}
Los cloruros de sulfonilo se convierten rápidamente en amidas por tratamiento con hidróxido de amonio acuoso (procedimiento 59) o con carbonato de amonio (procedimiento 37). La reacción de Hinsberg (experimento 4, pág. 135) sirve pata obtener sulfonanilidas o Sulfonto- luid ¡das, que son buenos derivados.


i
CAPITULO
1 1
Tablas de derivados
Las t a b l a s de l a s p á g i n a s siguientes contienen compuestos c ián icos comunes distribuidos por clases. Los compuestos de cada tabla están enlistados en orden creciente de puntos de ebullición o puntos de fusión. Estas tablas tienen la ú ención de servir, principalmente, corno una lista de sugestiones de posi'.’es estructuras para el compuesto desconocí do. U na vez que una substancia se considera como posibilidad prometedora, deberá consultarse la. bibliografía original para obtener una lista completa de sus derivados y para una discusión de las características de esta substancia particular (ver págs. 39-21). Laa tablas de derivados no pretenden ser completas. U na razón para ello es la conveniencia de seguir las instrucciones específicas reportadas en la bibliografía para la conversión de un compuesto particular en su derivado. El uso de tales procedimientos aumentará las posibilidades de buen éxito del estudiante. Las instrucciones expuestas en cl capitulo 10 son instrucciones planeadas para que que se puedan aplicar a tantos compuestos de una clase particular corno sea posible; pero, obviamente bay muchos casos en Jos cuales no son satisfactorias y deberán usarse las instrucciones detalladas que se baya encontrado den resultados con el compuesto particular. O tra raión para dejar incompletas las tablas con respecto al contenido del curso es la creencia de que el curso debe proporcionar suficiente experiencia práctica sobre la búsqueda bibliográfica para que el estudiante pueda emplear diestramente la bibliografía.
Por supuesto que en las descripciones de los compuestos en la bibliografía es costumbre dar un intervalo de cierto número de grados para el punto de ebullición o el punto de fusión. Para evitar que las tablas siguientes fueran demasiado incómodas sólo se enlistó el punto de ebullición o punto de fusión más elevado; este valor se cita con el grado entero más próximo. Las densidades específicas se dan 4 2U°C, referidas al agua □ 4°C, a menos que se indique otra rosa. Loa índices de refracción se dan a 20°C para la línea D del sodio.
Los estudiantes deberán tener presente que el valor obtenido para un punto de fusión depende tanto del observador como del método que se haya empleado en la determinación. Asi, frecuentemente suce
3 2 9

3 3 0 T o b ía s d e d e r iv a d o s
de que la bibliografía cita vario* valores para Ja misma comeante. En tales casos se ha seleccionado el valor más elevado para incluirlo en las tablas siguientes. Sin embargo, es obvio que esto puede conducir a. serios errores. Consecuentemente, para información adicional el estudiante deberá consultar la bibliografía original.
T adla 18
Acetales (líquidos)
NombK del ccmpuctlp j». eL. Nomine d d C0ll>pUVt<0 p . til.ÍC .
Dimetoxúnctano (metilal) . . . . 45l,l*d¡m ctoxieiano....................... 64'^ tn e tíl - ljS -d io x o la n o ..................... 82Dictoximetano (ctilal) ............. 89]Fl-d¡írmxic<am> (¿ícmaI ......... 1021,3-dioxano ..................... i .......... 1052-mctiJ-l. 3-d¡t>Xanc................... 1301.1-diJWpropojtictano 1221.1-diotoxipropano... 124I ,] .d i» > t0 x i .2 - p T o p c r u > .......................... 1 2 6D U w .propw ciinc tano ........................ 140l,]* d > c l( tt tb u ta n o * ........................... 143
1.1-dw }'propo;'etano 14$I ,l-d¡etOxj*2*cL ic tano . . . . . . . 1571.1-d¡etoxi.2*l»n)i..jctano ....1701.1-di-ií<r-butax¡etan<*... ....1711.1-diisobutoxielano ....176Di-ft*butc«imft*no .............. ...........Í8J!,!'d¡etexl-2,2-didoro<tano . . . 184i,E^í>».bu(oi(ietai)o ................. ....186«,a-d»metc>xítol\i*nw ....................... 199Di.n-pentoxSmeiano ..................... 215a,o*djen>xtuflu«i)o ................... ... 222l,l*di*tt*pi:nt<!x;mno ............. ... 222

l a b io s d« d e r iv a d o s 331
T a bla 19
Haluros de adío y anhídridos (líquidos)
Dcrí'xJo*
*c AnikUi"C
Anlliáac*í:
Fluoruro de aceúlo ...................................... 21 Q'¿ 114A nhídrido trifluoroacélico .......................... -39Fluoruro de propiDoilo ................................ 45 8i 106
55 62 115Cloruro de oxalilo ......................................... 64 4l9d 245Fluoruro de b u tir jJ o ....................................... 67 115 96
76 85 105C loruro do proploniio ................................... 80 . 79 103
a i 82 11592 129 105
C loruro d e n -b u tir i lo ..................................... 1.00 115 90Brom uro de propionilo ................................ IOS
10581
119106 134 .
Cloruro de dicloroaceüfo .............. ............. 10/ 98 118103 B2 114
C loruro de m eto x iace tilo .............. ............... 113 97 58115 Í35 109
C loruro df; tridorOaCclito ............................ 115 141 94Cloruro de m etile tilaceu fo ................ ........... 116C loruro de crotonilo ..................................... 126 161 l i aYodura de propíonilo .................................. 127 81 306Cloruro de n-v&lerianilo ............................ .. 127 106 <53
127 119 134127 91 131
Bromuro de butirilo ..................................... 123 115 96isa 02 115
Bromuro de i&ovaJeri&rulo ....................... 140 135C loruro de iBOcaproito................................... 14? 321 112Yoduro de butirifo ............ , . , . .................. 148 115 96
149 91 131Bromuro de a-brom oprcpionilo ................ «53 123 99Clom ro de n -c a p ro ílo ......... . ...................... 15* 98 95
Fluoruro de b c n so lío .....................................154 , < 4
■ (59 12 fí 160160 267d. (di)163 79 103
C loruro de n -h e p ta n o ílo .............................. 176 94 71A nhídrido isobutlricri .................................... 162 12B 105C loruro de succinita ..................................... I90d. 242 226

Tapu* 19 ( continuación)
■ 3 3 2 ■ T o b la * <h d e r iv a d o s
Haluros de adío y anhídridos (líquidos)
Noo\i>« JrJ comp'W'dr
p. d>. *c
Derivado^
Awidu*C
Aailidai’C
Anhídrido n-butlrico .............................. 191 i Í5 90Cloruro da bereoílo ............................. 19? 126 160Cloruro d e fenilacetiJo ......................... 230 154 117A nhídrido dtraoórúeo ............................ 213 387d- 175A nhídrido tt'Vafcri¿T»Co ....................... 215 IW 63A nhídrido isovaleriánko ....................... 215 135 4 » -A nhídrido dicloro &cí tico ....................... 216d, 98 118Bromuro de bcnzoilo ........................... 218 128 160Cloruro de ¿-doroben2otfo „.............. 222 179 194C loruro de m-metoxib«iJ20í(o .............. 224 4 « - «..C loruro d e m-dorobcnzoílo ................ 225 134 J22C loruro de <>-cIaraben*íílo.................. 238 142 114Bromuro de «t-bromobcazoilo ............ 243 • ♦ k , , ,A nhídrido oro tónico ...................... 248 160 n eC loruro da tf-toeuodbeirzoíto .............. 254 128 62A nhídrido caproico ............................... 257 100 95A nhídrido c -h ep ian o íco ......... * .............C loruro de ftalilc ( tym ) .....................
96 7 i2?6 220 159
Haluros de acDo y anhídridos (sólidos)
iKmidc*K rail« Jel coapodio p.r.
•c Araíd**C
AoüidaCC
C loruro d e ¿-dcrobem oílo . . , 16 242 179 19416 188 242 226
C loruro de tf-ni?rcb«iZ3Ílo - . . 20 14Í 174 155'C loruro de cz tea rilo ................... 22 69 108 33C loruro de jlhíbüo......... ........... 26 184 162 168A nhídrido dei ácido c idohew n-
dicarboxílieo 'lí2 f d t ) .........C lo n u o d« m-m troten zoilo
32 192d. , ¥35 140 143 153
C loruro de dnam oílo .............. 36 133 142 . 15339 105 143

T o b ía s d e d e r iv a d a * 333
T a b la 19 (continuación)
Haluros de adío y anhídri<lo& (sólidos)
Nombre <M tomputsu»
_ .
p.T.‘C
Üeñvada»
Amida*c
AniR¿»-c
Bromuro de p -bromobenzoílo . . 42 189 197Anhídrido brom oacítico .......... 42 " 50 St 131A nhídrido benzoico ................ > +2 . 121 128 160Anhídrido láurico ..................... 42 44 n o 78Anhídrido doroaectico ............ 45 62 120 134Cloruro de 2,4-dmUrobcrtzaílo 46 s é é 203 . • •Anhídrido glutárico , ................. 56 97 176 224Anhídrido m a J e ic o ..................... 56 . isa 153 18?Bromuro de ¿«yodobenzotto . . 60 • • • 217 210Bromuro dé S^-dinitroberaoílo 60 103 234A nhídrido itacórüeo ----- ------- 68 Í65d. 192 190
70 70 109 95Anhídrido nj-ToIúico 71 113 94 126Anhídrido fcnilacéúco .............. ' 72 76 156 116Cloruro de 3,5-dinitrokenzoíJo 74 202 183 234Cloruro de p-Tlítrobenroílo . . ♦ 75 241 201 204A nhídrido o-dorobenzoíco 79 142 142 114Cloruro de ddenikarbarcúda . . 85 4 • • 189 % 4 ♦A nhídrido m -dorobciuoíeo . . . 95 158 134 122
95 179 160 145A nhídrido 4-nittXpítilico . , , , . • 119 165 200d. 192
120 188 242 / ( «Anhídrido a n ím ic o ................... 130 133 142 153A nhídrido ftáJico ............ . . . . . 131 184 149 .Anhídrido «‘üiwobenzoico . . . . 135 146 176 158
146 162 205 161Anhídrido m-mtrobenxoico . . . 160 140 143 154
162 218 2 0 id . (d i) 234 (d i)169 175d. 265d, 4 « .
Anhídrido p-aitrob*azoica Anhídrido ¿-dorobenzoico . . ,
IS9 241 201 . 211194 240 179 194 •221 187 192 210246 241d. .♦ . .« 4 ‘
A nhídrido tetradoroftálico . . . 256 2S0d.Anhídrido 1 ,8 -naftüko 274 . . . . . . • > 1Anhídrido trtrabroiooítóJico . . 275 266 ► * •A nhídrido W trayodoftilicc . , , 318 327 **♦

\m
V&
f *fe®
N i
ü »
I »
r*r*i *
l É
3 3 4 T a b la s d e d e r iv a d o s
T abla 20
Acidos (líquidos)
Derivados
Nombre díl icido p.«4i.Atiilid.’i
*C^•wtuididtt
-c
Ester
l^ntííico’C
Eslere-brorno»
TiiM^lko ........Fírfníco ....................Acético .....................AcrJIiw .....................1’ropiAnUo ...............Prop¡«J¡cO .................|*7Jsutír¡cci ..................
cucrtlico ..............«•butírico .................rivíJico ...........PWvifo ...........Cnm/ iVo (f\¡) ..,.VLnüneíikso ..........EtünwriUc/íko ......lioral«itnicA ..........Ontweéticn ..........«<l<)ríyn>p¡úni<rt . . . a-vaJenfraeo .............2,2-din>ctilbui»T.^«D-icferoac*tif« .......JsozipnjÍ£»> .........if/>2«mctílfKfl ejrtoko 3^i>MUpcnttn«ic« ...tífclü» _............«-lWmOÍJolwirricn ..Mcloxiacítico . . . . . .n-caproics a • be<i u>op r**(i á o icoKioxtttf'ko ..........Brtmcxicétieo ........¡-MÍIl»roUnoir*> . . . . 2-mrt¡Ihe»*íW«i . . . . a-WoiTitt-H-b'ítírlco . . ■♦•metiBK-Milwico«-ÍKptiroko ........«-rtl|.n<aprcüe« . . . . ft • brOrooixwmk ti inlco HjrakMroVníijica .. JDibroroae&kc .....«-cnvrilíco ...........*-broav>«-taj»rc*í« .LcVuJirico ..................Pt4.-uy4njco ........a-fcnilpropíóitlco . . . .Cijwico ......................UrWMMiOÍcj» ........
fcfcllitJ 7pil' níc« ... ndecdf ni<o ........
S3 ;i> 130i »101 47 55 '31
113 114 147 78 RJ140 104 141 ...540 m 124 ■fli 63144d. mX'j'j tos ■77(63 8716? 93 '72 '35 ‘¿3
76164. 129 120I&id 104 ice .. • ...
J«5d.lí.9
102 J32 82ift ... ’h176 (10 ’m _
1W 109 • 63m 1 « • ’D ... 1 »i-, 92 124m 70 75137 92 83\n DE 153 99193 124 11« ’ft; ...193 nt tó 79t96 95 81 . •.137 97 75w 7? 76 ¿i ’éfl200509 sá ...205205
■73I2 í
'72
200 •.. 32 jtó« a n i 91 ’áém M 120 ...210 98 85 •. • ...217¿. i 92 492IS 7 ? .. •224 71 ‘áó ... 72I ...232 i 42 ...234 •.. ... ,,,23G 57 70 ... 67240
ira...
25IW. if¿ 612 » r-7 64 _2(a _ ., _ •..2?ft (,? 78 6777i<{. 71 «0
‘3668
2 » 922 » ...

T a b la s dn d e r iv a d o s 33S
T a b l a . 20 (continuación)
Acidos (sólidos)
Dírivi.ulai
N w thre del leído p.í,’C Artilid*
JCAmífl*
’C
Esterí-ni&o-
wacílic©JC
E/iterí-b w o o -lrcv:(li«i
*cM cucrtllco ............. ...................., .dí-ltUito ...... ..............^broou>ítt>l>utírií»j .............T>ob«rtzo«<j ...... ............«-btOoioDfOptómeo ...........Utidc«*t¿nico .,...... .........Uadct&ndía» ..................H e<*hldroben*jia] ............. , ..........Ciprtc© .............. . . . . . . . . ........... .FJi»OTV*cfttco ..................Lcwltaíca .................... ................P iv ilíco .......................«-m«tc)h(dit)ciiiifnieo ....................2- f a i ! butírico ...................|-«loropropi«W ct> ..............THdta»»»lcc ....... ...............]«*br<Jrtoiiov<itaiáiiic6 ...................Aji*¿Kai ...... ...............D lS ^ ra o ic f tie o ................S-Ícnilprooi^nito (h¿dM*e¡cácúieo1aJ>r«ttOÍiob«tírícú ............R ro7»oakc< 11 ccs ..................TCWdieo ..........................................» “f«1 ifcotíroco ................................M i r l a d .....................................TrícJoreacítiw» ............................ "Mcuíárico ....,...............^bnamoproj>£óníe<} . . . , , .............
tttom»e¿ti«9 ........ ;........... i " iTígflco ........................«.y-^Jbren>óf4tip¿¿«lcrt ........CUaxtcítieo ..................................Qwlmoigñío .................Ertetrico .................. .Gn^tónleo ftrtn*) ..........................F«md»c*tko ,,,................GIWBíí. .........„............a-J\ltfro«iáofc«ttñco .........B-JexJopropiífiJtci ................ .V¿d0ac¿tic0 .................... . . . .«-beoMMlbocfnco ...........................DibencPfcCélíco ................«•fccfiiniJbeiuoico .......... , ........... ..Citrncijníca .........F w iox iic tóeo ........... , . ..................P talddebíifloo ..................................Otubiríco ............................ ...........Hotnovsrítáco (aphWro) ..........
1616192224242429SO503233
3742424344 44 <648 <8495051 5? 54 *? ms64646668ffí72K79»52 84 87899091 « 97 97 93
41S753
»
7¡14262
\ü2123
7680
Í2é’b i
Í3¡
8454
9033477
m«93
11*1179fi
136
ÍÍ3iis1951 »W
224
7610674
123« .
103186106108108164 1098610199
100133« 815682
14*919384
102141106
iós120
7619fr12310GiceITO154120198
1019S
14!»128165 187H. 101
174147
40
Ü2-- < ...
's é
‘¿7
'é i M-■r 76
*767S
'As ÍÓÍ... i , |,......¿i
'¿ñ4”? *8342 ’Mm
"04 68
4 • »'90
¿7 9665 89
107 14?80... ■■ .. ...
iñó " *71 ...... 148
‘Ü Í37... ...

TaWtJt Óo derivados
20 ( continuación)
Acidos (sólidos)
tkriv»io*
Nombre d íl itldo p l.’C AaiW»
"CAxoirU
*C •
EittrjUrtxv-
bncflícoCC
EstírMnoreo-tcp*cílko
*C
í ^ a i l í e o .......................................
C í t r ic o (h id ra ta d o ) .............o.nv^A üftir r . ^ l Q , , ...............O x iU o o ( h i d n w k ) ............»<dílcD ................Fi®élk*> ......................AieUcu . ........ .............mfVcAíica ...............gdhalAalcff ............MwftwtWw . . ..................
frhrn faifa» tO{B¿iucO ...........
b to d í r a ló a lc o ........... ...........A*H*UÜ€6 .................■ B -^ trv f tu lU c ^ tic O .......T r ic e fe c o t tc t ic o .....................
A o o a l t k c ...................................
Dietilj|ll26«vicd .............¿ « ñ trM a lfc fb o q i (b M tn ts d o ]o b d lO Í btruoico ............ ..H K n a o ü b m h ie » ...................Z,4-dbM rUa7C«M Í£o ......M»kfcn ..............4 í - r r t L - n ...................................? > < ü » > f t i» x :a x i Wn ..............Fwoka <pdr5CÉ-ÍQco) ....S tb tó c o ......................................4 - o te n M n á títoa .....................C Í I I< r t i r T ...............m íJutSco ...........MmtÓBtCO ...................................A c c to » d k « H M a ^ k o ......A s t i W l c f l k i > ............M e ü b u U d lk o ...........FcoUpro líUcB _..............Gíomíbím (<%i) .......C b ta c A i ik o ( i w u ) .....Z.S-dIclorailCMOÍoo ......*-CÍOrofx»W)4CO .......................«%-litxvb««uofaz> .....................
100
toaico102m105106 1)0 111ur»116116117H7d.lia120221124IZ>
125125126 12? 177130 230131 I» 1X3 133 133 153 133 23M.iaw.155133d.136136
1 »
1»140W
t97( 102'» >
124 179
199l l * (di»)
210 102 14?131 126 213257 4í9d. « 4 242d.125 142 91 3715$ . . . J3Í145 Í7Í 44 132123 07 Eti 1C8150 214 75 .. .200
( 165 >
150¿225 M i) )
m133169225
.. . .. .2¡7 Í19 »-.131 133 124 ...
110 .. . * ■isó m 89 119164 i«l?M, )
!nx>T>ol 1 200d, f s • - . . .(di) J
224<d¡) 91 (<ft)... 145 ...195 IC5 ióó• • • • r • • • • * > *
179Í¿7 isa ’é? Í68
123mi »1552241»196152 1251S6 (mono)
yocol > {d«>í
í¡4.-153
165 íd¡> 186 141 210 122 147 2»T l?0í»217102
202139142
133TÍiifi'tá■¿Ó7583
IOS241
W147
145
itt132

T a b re * d a d e r iv a d o * 3 3 7
T a 3 j la 2 0 ( coniinuación)
Acidos (sólidos)
D s iw ln
KorubN d íl icído P.f.*C Aaítda
• cAm>d*
*C
ÍM u r> e lc r f t .btncfliro
*C
E l t t f¿•broceo»iTDH-füCO
-.................................................140
S u b írv » ............................................ ...............140Faní»aítito ...................................H1ú-fllU 'ttíw lnoítico ......................... ............... 14](•M itU ae to M .................................. ...............142>(licrc4{tlicilic0 ................................ ...............M4d í«ülftc¿tk6 .................................. .............. ¡45«u¡crcl>enB ii« ................................ ...............144
r» 1 6 r ic o («flU dro) ..................... ...............14$
o-t^ -to iy iboaio ico .........................................146t -tiditnjícoU*£¿t»oi» ..................... ...............148-UíOn>obí*LM>»00 ................. I5ft
fcfntJKc© ................................ . .......... \ 150A dlpko ........................... .............. . . lMf-i: i trrrfejiUrétiro ...................................... 1322JM Íkk«W }\i9 Ía> ...................................... 153C ítrico ..................... .......................... .............. 155■B^MTJD&be^roiM ............................ ...............155^ .G -tr ix n c ti& tW tc o ................... ............... 155Stóiclüco ............................................ ............... 157«t-dtiiubéanM oo .............................. ...............15®2.4-dld«rei*c*ioíca ............... 160»-rocíe b to io leo ................................ ...............162«-Nnítoáca ....................................................... 1S2Tio**J»clfcc« ...................................... ............... 163?,5-<Lrl u ru b u tí» * » . ............... .....................1645.4-¿úní(¡i>c«¿«íoo .................... ................ 1644 ^ ¡ t i« ( tá ¡ iw .................................. ............... 165I ta c a te * .......................................................... 1654.
5 f c 2 & f f¿.'•d£>ro]«>CKJo;!rigp ....................... ............... 147d U a d lM w ín k ú ............................................. 1 CT¿~ q UiMTtierico ................................ ............... 1®S^dW tw M llcilko ............ ........... .............. 173>t¡»IÉxn ............................................ ............... 17?í,4 -e r« ¿ d e o ...................................................... I »A«ti!«wÍÍ£*rb<irflko .....................................1 »V j t e í S ^ k k L o ) ..................... ............... 1BIP flcnro txnnrico ............... ............. ............... i*24<Iar© »^»itro íx inak© ................................. IOT2/4-¿U trobcnxaía> ......................... ............... 183A»f>k/> ................................................ ............... 104p^uUtoJco .......................................... ............... 185Acctihíitr®aflicz> .............................. ............... 185¿-cw Jér^a ........................................ ............... 1Í7HWjiof ......................... ..........lgT^T*dot>c»*0«>» .............................................. 1*7Señale* ........................... ..........O*3*BÍtn0ubi«> .................................... ............... 130
w
m155
( l K J< (tnooo¡ > b » (A))
141175233
146
154122
142161
192190
Ü ?
222 Vd*) 1 » 1*1 140
1GS
ijíÍ6¿17316720J306
226Ifc
8
i *21616*161200145167 174
:«.m ?155*1761«7155154 220 139155 2l0d. 155168 159 IX
í*4205
15020M. 192 ida)
m 2m .
«Uf®197158
2«d.I «154156203162195171192IBSl«6242
35
112
1109}
106
J02145
%107
111
í «
m
¡U
i«7
207
Í4¿
140116
1101)5
117
Í3*
2 Í¿
155162M*í ; .
a-- i
: . *: .. ..íf-
136121
lií12*211

3 3 8 T a b la s d* d e r iv a d o s
T a b l a 2 0 ( continuación)
Acidos (sólidos)
•• D tr iv i io ;
N snO jrt á c l icbio p.f.*C AnUí&i AmkU f^-DÍtro-
-Q "C bAac(l¡ea ICrtMÍlko- I S . — ! £ —
Arenisco ..................J> l< ttp illm ii¿nfce • , . . . , . .
PriMocufíiiufrú .............Ki’fl'.InKJnlnilfio , ■ ...........2 < k * t -S , i • d t n l< ro t-s n i a I«íH*h ro!übeeio)oa .......3,a-d¡njtfOt>c7LZDÍe<> .......KTmtóoico ............................í-CUfcúíricO .................ít ilico ,...............................er BDliricO .................VairiiUfnko . . ....... .^ h íd ro x ib c iu o íc Qt- rC X 'r c ib a i ...............M úciío ............................./►•óánobeaioieo ..............Pip rUlo ....................J-ñHrüíd1ko ............2,4.^-lfljiítrofcíDioico . . . . á-Jrdrtfcí'S-nnTlaicí» . . . . . E -J ü il« > x l-2 - it tí« ¡c o ..........5-r>itros»litílien _................M')ilitnjoc*£íaafti«i ........DÍÍébkq .............................yipíyonílico ...............
...........C « ¡ 6 ......................Mqimuco _.................•C¡Lrcb rnro-<xv ...........¿"¿hroterzolco ...........Tctrocloref tilica ..........¿-hfODWUettrttco .........Quelld&nlíX'...............^y»Mxoi«c« «.........*-a itrtcÍ24 micoMriítíw ...................J ío í t tó c o ................................T c n í tá B » .........................
FuwAíko ..............................Trtaiiíco ..........................
(S iJS3íuW.r19920120220!20*2062fi6d.2072072J3213 213d.214 216 u ie 220d. 222 225 227 227d. m 229240 240d.241241242 2SM. 231 « 2 265a s 288d. X » 900 BihL ÍC2 ■a »
155234-115
1S9
2Ó2127
ÍW
234
245
224
230
207
■ w »194
Í97
2W
m
314IZOd.
250 76 195fcS> (<J¡) ai
212 ii»lilfi 174 ¡73
Í7Ó tófi ¡76183 1¿7176 134 é , ,2W 147 . • .m149 j 55 • i s i209d. 152
140162 isa iói222 3*9
310 223i 4 , 169 • s .ss % 145 ■ ■ •201 3B9 aa •264-d. • r .2ia r r • .2ia • 4 .225ie92121 «m245
m W i )2011 »
Í¿9
2172M
280
“ “ « S í '
1B3
í 32134160129ISO1 »
Í4Ím
2Ü265191
141
137
147
IBS224
197

Tobfere tí» «taivodft*
Tabla 21
Alcoholes (líquidos)
339
NDntf#* <jül compu'atoDerivad OT
p. «b. lC *níJt¡í«
urtUi»’C
FcrúJ-oret»»
*Cbenzoate
’CAlcohol metílico ......................... 66 124 47 107
7 a 79 52 93Alcohol ísopropílfco .................. 33 106 88 122
83 101 136 142B u tcn -3 -o t-2 ................................... 96
97 ¡09 ’?Ó 48Alcohol n -p ro p ílic o ............ 97 80 51 ■74
99 97 65 75Alcohol f¿r-amílico ..................... 102 7) 42 117-Alcohol isobutílico . ; ................ 108 104 86 86MetiUscprapílcaibinol ................ J 13 112 68 76Alcohol rt-butSlico....................... 116 71 57 64D ic tilca rh in o l................................ 116 71 48 9 7 ’
119 76 61Metil-2-p<ntaiiol-2 ..................... 121 239 72E ter monoinetílico <W ctilen*
.......................................... 125 113Clwo^l-propanol-2 ..................... 127 83MetjJ.*3-pcntanoI-3 ..................... 12a 50
123 97 62E tilendorhidrina ......................... 129 101 51 92Alcohol ísoaroilico ..................... 130 67 55 62M etil'4-pentanoU2 ..................... 131 86 143 65C loro-2 'propaoal-l ..................... 132 76E ter mojioetílíco det «ttlenglicol 135 67Hexanol-3 ....................... ............. 135 77Alcohol tt-amtlico ....................... 138 60 46 46C>clopcM3iK>|............................... 140 118 132 115TrietUcarbinoI ............................. 142
145H idroxiacíton* (acctol) ......... 146
148 76 51Etil-2-butanol-l ........................... 149 51Bromo*tano!-2 .............................. 150 86 76H cpunol-3 ................................ .. 156
156 80 64A lcohol-n*hexilíeo ......... , ........... 156 59 42 • 58
160 128 82 112H ep tan .o l-2 ............ ........................ 160 54 49Triraetilenclorhidrtr» ................ 1610. 76 38 772*nvstilcác3ohexanol ......... 165 155 103
170 129 45 80E ter roono'n-butilico del ctilcn-
glícol ........................... .. 171

; ' j T a b t e i d « r í v c t d o 4
T a b l a 21 (continuación)
Alcoholes (líquidos)
OtsmdMNo«aí<« del <xmip¿e»p P- cb.
*C a-c&ítü- 3,S-d¡ nitro--c °c *C
172 • • 9 215 ■ ■ ■4-metí3ddoh«anol ................... 174 160 125 130^rortikádohcícao&l ................... 175 122 96Düwbucílcariíod ..................... 175 « * • ». . ►..Ai cobd -ti -h tptüico ................... 176 62 68 47DIclcio-l,3-}iropaiioI-2 .............Tmoetiiftxibrorohidrína.............
176176d.
11573
73 129
Mcti|-2'propaiicKlioMJ2 .......... 176 • ► * Í4¡ • • 4AJocáiol tctrihkírttfurfurílico . . 178 - « - 61 84OcUnod-2 .................................... 179 63 114 32
181182 iió 96
Dicioro^S-propanat-í ............. 182 93 73 4 . .BuUmodíot-2,3 . - ....................... 183 ♦ J i 201 (di) ■ 4 4Etil-2-b<=oiooH ......................... 184 61 34Prop3«icIkol . , *..................... 188
190153
Alcohol n-octllico . ♦................. 192 66 74 6¡Eter mcaocnetllko del dwtckn-
gUcol ................................ 193Nonalol-5 ............................. -« . 194>íetD*2-j>cnuuiodiol-2,4 ........... 196
197 197
17653
15765
í 69 t
198 56 4 . . 43Eter monoctílUo d tí dietiler)'
202 • 14 4 4 * ► >, .203 106 94 95
A kotol bcndüco .................... 205 134 78 112ButenodioM ^ ........................... 208 184 . . . . 4 -
211 69 r • • 442l5d. •«► • • , , ,.215 65 62 52216 164 137 164
Bencildizúctilcarbjriol ............ 216 < i . - • i217 116 4 • 4 4 , .
MfctiJ'í’tolikajrbírtol , < Alcohol ^feoiletSKco . ■ D¡brcimo-2,S-propan£>14
EtíifeoUcarbinoI , . . . , EKbromo-1,3-propino]-2¿ta-texplud .............IX txonetol.....................
219219219
219219d.221222
119
102
152
9679 108 77 (A ddo a,$-
d í b r o r a o p r c -p¡faico64)
84113 79
IQCtila dí- pjco 89)

Tablas d e derivados 341
T a b l a 2 1 (continuación)
Alcoholes (líquidos)
DaividKNombre <£cl c a w p u a to fi. eb,
*C G-aifóT-orciasuj
*CFecif-
* » "?TWndlr&-b fflw »
*C
IdopropUícnilcarbinol ................G c ra n io l .................... ....................E ter mono-r^butUico del cü-
etilenglrcol .............................Alcohol n-dfctílico .....................S-íeniJpropflnol ...........................Peu u m etil e ngl ko l .....................Alcohol undccílico ................- •Dietüwialícol - . . ................E te r tn o n o fc n íiic o d e i c tilen -
gUcol .........................................Alcobcl-^^tojdbencílico A lc o h o l c in á m ic o ..................... ....Glioerol . ......................... .. ..........
Beraóhídrol ..................................
224 117 • • 4229 47 . . . 62
231231 7\ "¿Ó 57237 45 92238 147 (d i) 164 (di)243 ... 62 4 . .245 . . . . . . 149
245247 . ¡36 ...250 114 90 121290d. 191 180 (Ttüx
roato !297 136 140 141
Alcoholes (sólidos)
Nombre <3tl cotrpueflto p. f. VC iere«io
*CWÍUDO
°CDtvmoj
*C.
1516 í 23 82 3,5-diahrobcoxQ ito.
243d.112
Alcohol tricíoroetilícc» 19 120 87 , „ •Alcohol Jáuríco ----- 24 60 74 3,5-dimtrobenxoato . 60Alcohol p-metexiben
cílico . . . . . . . . . . . 25 94 Acido {m ilico ......... 184Alcohol Tn-aiirobcnci-
3íco .............. ..Hewuiwn-2,4-01‘1 , . ,
2731 . 4 , 79 3,5-dinícrobcnzoato . 35
Alcohol cinámico . . , 33 114 90 3,5-<ÍInítrcib<nrcíito . 12135 . . . 215 j-t< tram e tild id o ro - .
a-teipineol 35 147 112 3,5-<linitrc!xnrojUo .16078
o-tcüicarbmol . . . . . . 36 . . . 79Alcohol RÚríedoo , . . 39 . . . 71 4-yododifem lurrtano
í-n itrobcaioato . . , B e n zo a to ..................
146Alcohol rfWenquiüco 39 149 104 109(— )-m ental . . . . . . . 42 123 111 55 .

L»
ÍM
i
f imi
i »
? •a »
f e
I B
í *
i »jp§
N i
3 4 2 T ab la * <C« d e r iv e d o *
H idrato de p ir.sed . Alcohol cetilico . . ► Alcohol neopentílico Alcohol pjperoaílico p-tülílcirbmcl1,2-difeniletanol . . . B-Súíohidrot . E r ítrU o l.....................Alcohol o-nilrobcnc
! ic o ............. .. . . . ,Alcohol _ 2,2,2-tribro
mocrilico . . . . . Alcohol fenacUícv . Alcohol O'hidíoxiben
CÍlico . . . . . . . . . .Alcohol ¿-m tfotxnc
Jico .......................( 4 - ) ^ o r b i to l ............TerpinoJ ..........'HidrtWJ de tcrpinol.Btnzoína .................Sitotterol ..................
)-col«cerol -----Trifenitcarbinol . . .Ergottcrol ................(4-)-ttUüitol .........Bertzopirt*col .........D u ld to l ......... .. .í + )-bomco! .........Lnoeitol .....................Pemaeritritol .........
T a b l a 21 (continuación)
Alcoholes (sólidos)
N om bre <Jct tdtapo ts to p . i . a-nMcil- DIvtrsM• c “C ’ C
46 H rso 82 7$ 3,5-dinitrol;en2¡oato. 6653 100 144 • • .58 10260 ► i »67 . 4 s69 136 140 ík r tz o a lo ................... üi>72 . . .
74
SO66 ^-nitrobenioato . . . 129
87
93 s s98
102 Clorhidrato ............ 50117 Clorhidrato . . . . . . 51)133137
140 165 (Ver cetonas)127
148 160 169 B en sc* to ................... 150162 Tr¡ ferióme cano . . . . 92165 A c e ta to ..................... 180166 303 Hexaacctato ............ 120136 ► , ,189 ► * ,206 132 138 p-nitrebenzoaco . . . 137225 HexAaectato . . . . . . 216253 . . . . . . 64

T a b ( « dtt d* rívo< fo i $ 4 3
T a b l a 2 2
Aldehidos (líquidos)
D a 'w fa i
Nombre de] conpudto p. tfc.Oxúm
Scmi* íenil- 2,4-dW.Ctet*- tíácm. Crofteíl-
°C Z*o» toaa brffmto' '0• c *c ♦c ♦c
—21 Líq. 1 0 rM*. 166 18221 47 l « t l íá l m
PropwuiWtSCdo ................ 50 10 { .8 1 M «. 15* 124Glfcoul ................................ 50 178 270 180 32É 311AcroJoíni . .......................... 52 J7J 52*I»obutirtJdd)i<íci ................ 64 m >flfe 132a-mrti la «oleína .. .............. n 158 206«AnKiraldelildo ___'........... 74 IDt Líq. 122 92PívidaldeWdo ...................... 75 41 190 209 119I*OV*!«-»1cLtfcJ<ki ................ 52 43 m Llg. 125 101«-ITKtil^'bliLnikWlWo . . . 9 i 103 120C)c«il ................................. » 56 131*-v iten Web Sdn .................. 103 62 IOSCr4C4*aldehido . . ............ 103 119 l « ¿6 190 184
.. . . 116 , , 96 ■51124lía 51 IOS 104156 1» ios nl€l 202 97 2 »
Hotnh úíro fcx m 1 d e h kto 162 17513 ¡íc-e í 11 -a-c* j>r«*ld ................... 163 a v .J. 2 .3 ,$-tcbnüdraf>cnHdde~
h « o ................................. 15Ó • 76 154 163Cupnl&ldcKMo ................... 171 0) 101 106 *>Bptwal ................................. 174 115BeiualdeMdo ...................... 179 35 222 159 23?
185 , « 100 1005-mtlUfurfiuml .................... 187 <m ÍH l
CAÍi Sí f 211 148 212Feoúl¿eeu}d<bJdc> .............. 1 M 103 156 58 121SilkJhldyiWo .................... D96
1995760
231213
14284
25?d. ' 211
. . .200 45 209 101 156204 liíS I 221 114 233206 «2 77
*-c Joroba ¡ttiVkhMo .......... «08 75 225 K IC7^*«U3r<*«vaMcfeí<lo ........ TOS 70 z a 134 255
209 ÍS ICE J «SIScL 95 145 86
H idrocin* maldtiiMc ........ 224 IM 127 149CimJ ................................... 22M. U q . 1S4 116Bi-uveJOXibíBtaldeWdo . . 230 233d. 75 171«'browol»mzjirddiído ,, - 236 72 20S 141AniiíWcLído {p-ttMcaxjbcfV' < ®*45>
ttJdrh(4o> ...................... 247 •'-64}M3M
138
21(5 120 2Md, . . .
252 2 lS 168 255a.285 95 177 121 2» . . .
* Piraznlína.

3 4 4 Tab la* d e r iva d o *
T a b la 22 (continuación)
Aldehidos (sólidos)
Deríviáfl»
Nombre del poíajMKJtov‘c
p, ct>. ‘C Oaúb>
ÚC.
?ymLOLriui-tonai♦c
Fwiif-hür*-roct.J £ -
2,4-díril.CrafCflU-
a u m u t... V .
ÍWmituWítWo . . . ■•«uiíuldchklo * • . . FwihwüMAlÁi .
chído
<wnrtoililxtmli{th[<t> . - StfotnSfehU» ........ : , .¿MUOÍlMb*£lXfcíddbítifl . . .o-<Utrobea*al¿eUdQ ,...TjflTlVUtífc ...............BXftldeUda ....
d i ctorid .........__ .. . Jilfe■'ttlMUtUd* .............pJ*ixaobeuá}dehi<fck .• M ei»yg*> b«itsI< Idü< io ■J»-t¿ttvlKÍE*Í<Í«ÍÍCÍ[> . . .V«t*tndd«Wdo ............§-a*ítal¿rfcMo ..........Z.4 1 ig»TpbtTa:itd<ih((tft
' 2.4-dJclurpfct«»ldífc/«í<> P AIIKff .................F íd ^ m l
xíóo¿teflfceagjdthfda
WdéT«nJM*ldchíáa . dtfllttcddefcMo . Pr4^*l
SI BS m34 232 90 22334 135 99 35Í3? 2Q6 m 2<K37 263 310 2301S 2 « 92 215ja 89 139<0 1 »44 102 ü é45 ¿35 n U »47 214 110 230Sí 5S 9üd,54 205 94 231¿6 . . .SI . . . Íjth 15ft
loan* Ill{ 22657 4 • - 62 225X 120 $+658 385 95 17 ;€0 156 245© Tí» . . .71 136
22960 2£5d. 117tWdrtwl ♦ n í a t M l
(di 168C * 217a.-L lS —
106Tf) 1 3$ --
129 221115 . . . „ . .US ■ r . 72 224ll€ 245 200 . . .142 • • « 16M.154 157 230J.157 223
IOS6063 321T9
103 v m ... j 2»„ . tío221I5S 23CW.
IOS127 270
131138 K44-19J11315512* 293d,1 2 1 265206 i 770
305 27Í«t,<3! 152 ...
— H3i — -^ a c w .159 52ÓJ.
177 2804.í 154 )íd i 278Í ... .
¿i 132 107m 275d,
Tablas d e derivados 345
T a b l a 23
Amidas (liquidas)
Nombre del compuow p. eb. "C
ÍVctafcre del cocúfmo<« p. rt,'C
AryV-d imctil for tü&mid a .........A'^VKÜetiHorroiLniida Form anúda ............................. :
153176I95d.
222226
A m id as (só lid as}
Nwibrt) del c&mpntiia p. i. ‘C _ .
NciBibre <3et «oolpwsrta p. (. “C
O iean ilid* .................Fonnanilida ..............BenzoilpiperidilM . . . C-arbamato de etilo M akm am ida (mono) jV-rt-propilacccmJlid* DifluOroAcetaznidA . . C& rbanuta d e metilo FenUcarfcanuxo . , . < Aceto acetam idá . „ . . C arbajnato de «-butilo^-etilacetattilida ..........Cxrbamato d e iíobutifo jY“roetil-í>-acetoto!u'id ida M etoxiacetanilída . . . , . G irb an u to de n-propilc H ipurato de etilo . n-val?r¡anazul¡da . . C&rbamatc* de ¡«Osculom-acetotoluidida . . .Oxanilato d e etilo ,H c p ta n a rd lid a ..........jYjY-ditenilformamidaVímlacetanUidaGlcam ida ...................Propioxuvrúdíi . . . . . .
414648*9v?5Í.525233¿454-545556 58
.60 •6063646566 70 73 73 76 79
« ,8-d ie liIcarbaró lida .............. .......... 79Alilurea ................................ . , 60Jl-a 'doropropionam jda . . . . 80Acetamida .................. ............ .......... 82Etoxíac? «unida ................... ............ 82W -m eü^-acetotoluidída . . . 63AcatoaceUnilida 85A crikm ida . ......... , ............... .......... ¿5A K Iurea ............................................... 85o-cIoroacetAnilida .................. ...........83Oxamato de «-butilo ......... .......... 68i/-cJi3lmougranilida . . . ___ _____ 89Bromoacetemida ............................... 91Caíbanvato de inop*Opi!o . . 92£3-mCroacctaniH<3ft , .......................92(¿•íenílpropionam ida.........................92n-injpUnainida - ......... «• • ► 944-mctí(-2-tútraace(íinilída . . . 94Af- o%t tü-JV-a- r» ftilac k t ím id a . 95n-batiraiñlida ......................... ...........95Yodoicct¿8Úd& . . , . , .......................95ScmicM'baadda......................... ...........96MctojdaccUmida ............................. 97E&tearaniliáa ....................... * , 9?Tricloroieetanillda . . .......................97

Tablas de derivado!
T a b l a 2 3 (continuación.)
Amidas (sólidas)
Norafcrc del compuesto j p ,i.1 *C
Ncrabre «W «nnpuetto p, í.?G
p-acetox¡-A,'metil acctanüida 98 a,ft^Écloro*cetJUDÍdji ............ 98Hidrocinaxnaniüda - .............. 98A^YniífenüaoetajTiida ............ 101
«-ac«tíl-*P‘ícm lhídracina . . . . 128B em am ida .............................. 128Isobutiram lda ....................... 128o-m ctoxibepm nida . . . . . . . . 129
F ro p io n an itid a .............. .. 103Piruva&ílida , . ......................... 104Acrilanilida .............................. IOSHidrocínamaroida ............ 105
P;ferü]ptopk>namída .............. 105«M-dtOKtnunia ................ .. 106Palmttamtda ....................... 106
Huoroacetarnida ..................... Ifl8E stea ram íd a ............................. 109JsovaleranitW a......................... 109o -aceto tohúdida ....................... 112
3.4-Siraetitbenzamida 130Brorooacctanilida ................... 131«-metoxibeiuanHida .............. 131M alonan iH da.............. ............. 132
(mono)U rea (carbaoudíi) .............. 1322.4-díaietilacetanitíd a 1336-isopropilbenzan. ............ 133M esitilesam ida .............. 133doroacetard lida ............ .. 134Wi-clofobcnzatnida . . . . . . . . 134i/.^n aftilace tam id a .............. 134
*n*brcraobenzaidl¡da . . . . . . . 136(tiHítoxibcnEamida.................. 139
Oxamato de «tilo ................ 114 ftjaja-tricloroacetam ida - - - . 140
2-fcromo'4-tnetílacetatiilida •• 11?í»-wdobonzanílida ................... 141^Í>ronlobCrtzani|tda . . . . . . . H l
Dícloroatetaollida 118jY^tü-¿-nUro*cet»DÍ3ída ----- 118a-cloroacctamida ..................... J I9^-dim ctílcajcbanilída . . . . . . 121o-toluaniBda .................. .. 125S u cc io ira id a ......... . . . . . . . . . 125A ddo »'bvt»letilbarbiti5ricci . . 125m-Co lúa nítida ............................ 126Acida ctil*rt'hcxilbarb¡túríco, 126 j>-metoxiac<taniiSda ................ 127
Furw nida ................................ 142Cinam arnlda ............................ 142o-benrotoluidída . . . . . . . . . . 142rtt-n icobenzam ída ................... 143YodoaecUnilida ..................... 144p.toluamEida ............................ 1458-trííettilsuajúdina . . . . . . . . 145a-bromoUovalerÜurea . . . . . . 145Ckk>hexantarboxanil¡da . . . 146D ietilm alonajriidá .............. 146
ím ono)
T o b ías d « d sr ív o d a i 3 4 7
T a b l a 23 ( continuación)
Amidas (sólidas)
Nombre del owT>pHe*o P- (•<CH^hidrobenzamlida ..Penilurca ..........................Difenítguamdina .« ___4-meti|-3.futroacetani]ida Succinaniiidi ...................
Btrtdlurea ....................... . , ,M trourea ...........................«•acetotoluídida . ...................Af-rr»crit*-^-míroacctanilidi m-mtrohensanilida C inwnanitida . . . . . . .Acido ctüisoaizdlb&rbitúríco ¿•fluorobenzamida . . . . . . . .a-fenüacetaznida ...................Benci lamida ............................p-brom otienw niida .............m*brotttobcnzainida ..............2j5-dídorob«n24mida ........fc-naftilacetanilida . . . . . . . .m-nítroacítanilida . . . ........Dibromoacetaroída ..............jV-fenilíucciniraida................Sucán& m tdfi............................
p-benrotolukUda ..............¿V'tt.-itaftüacetamída . . . ,c-toluamida . . . . ..............Benxamlida .......................Ar-a-naftilbenzam 5da . . . .¿-anuncacetaníltóa .........ít-n a ftan ilid a ......................^brom o acetan3 i d a .........ti-bcnzoil-^-fenífhidraziraD i f enüaceu ni i d a ..............^údrcpdacetanilída.........Alófano .........................^•etoídbenaanSHda ............
14*147147148148
(mono)145I50d.153153153153154 154154155 155 155 155 155155156
. J56157
(morto)156159160 161 161 162 163167 163168169 170d.170
Nombre d<H cempocita. . 1
MaJeamida .......................Maionam ida .............. ..
t-/5AÍt»nih'da *4 4 , * * . . , . líHcnUbarbiMiríco, ácido DiaJUbarbitúrica, ácido .
¿>*íenetilurca .............. ..Mal canil id a . . . . . . .........d-nitrobenzamída . . . . . .¿•cloroacettm jída . . . . . .<X-aced]‘(i*mcCÍfurea . . . .DtíecdJac«ükmIida . . . . . .tt-paítilace (amida . . . . . .¿ ‘tolilurca .........................Diroetilurea f w m ) . . . .¿-yodo aceta ni I ida . . . . . .TíoureA .........................3,5-diyiitrobgngAmida . . . ó -y odobenzamida . . . . . .^ ^ ^ c e t i ta - f e x i t f e n d ia tm n Cidohexancarboxamida Hexahidrobenaamida . Tft*yt*dob<nzaimda . . . .Hipurico. icido Ditftilbarbitúriccj, ácido p -bra mobc nzamida . . . df-diííniturca N J F -diacetiJ’ fn
n t 44. .$-naítamida o-tclíhutaBiuret . . . .jEf-cJcn'óbcDzanllida . .......2-m díl-4-iúttoac«tanilida .(« a t in a ............*.............Mctilíuccinamída$-naítilaceUEQÍda ..............^s-nitrobenzamida . . . . . . .
f e n i lc n d ia m
p.í.
170 170 17t 172 172 "174175176 3 79 ISO 180 181 181 182 182
■ 182 183 183185186 186 186187188 189 189
191192 192 192d. 194 196 200
200 (di) 200 201

' 3 4 3 T a b ta t cíe d V ív a d o *
T a b l a 2 3 (continuación}
Amidas (sólidas)
No«itn dd,«mf«cit9 j>- . Nombro del c o i t a f K i e P - t.‘C
Acido ctiUsopwpilbsurtxitfrico 201 Tereft&hailida ...................... 237Cirb&nílida (r-diítnilujrea) . 238 ^4ddroxi-jY-iDCtlliLc<tíaxi!kla.. 240
<&f¡HúUucc4&aJ3iid& . . . . . . . . 211 (di)^4,6-(riúitrobemaii)ida . . ►, 2&4d.
^W'-dlicctíUjfr-íecttkndiam-
Cafeína ......... .. 234-
Sobl.

T ab ló n d e d e r iv a d o * 3 4 9
T*_s l a 2 4 — P
Aminas primarias (líquidas)
>Jpoftfc*« def ©omjn««*t<i p. «K ’C
iMctn-fuJtaa*.
mid*’C
I m u 'jtaida
'C
p-iDlutn-lol/ocj.mida°c
r«úi.TWirca
*c
'-~6 30 60 75 11319 58 71 63 10633 26 % - - ^ i « 101
ftfr-butí!wnina .............. 46 134 • . • 120n-propilamina ..................... 49 36 84 52 53
56 39 64 9863 70 76 55 101
IsQbuüJaran» ................................ 69 53 57 78 8277 42 6595 65 102
m < . ♦ 69
Aminas primarias (líquidas)
tfetñbr* det twojnfcito p. ib.Bctccn-•altpoa.
mida•c
Ac«u-jnkU‘C
&OUL'otid»
•c
^-tolu*a-tuífon*-
diá»*C
f <*b-w r a
*C
EtUcndiamina .............. 116 168 172 249 160 1021,2-diaminopropano . . . 120 . . . 139 192 103 . . .rt-higdlamina . . . . . . . . 128 96 . . . 40 • . . 77
154 89 104 149 1481,3-dwaÜQopropaiio . . 136 96 126 147 i 43
1,4-diaminobutano . . .155 ♦ • • • • * • 44 ♦ • • *75160 ♦ * • 168
2 -bíd ro n ttií&mina . , . . 171 «4a 135l^-d iarm nopcatano . . . 17B 119 ♦ * * • • • 148
180 4 • • , r . ♦ « «183 112 114 160 103 154
Bencilajniaa . . . . . . . . . 184 88 60 105 116 156a-fenüctiTamfaa » . ----- 185 57 120^C uoroaiuJiju . . . . . . . 188 4 . . 152 185 • m m.
198 69 114 116 . . . * 135199 124 112 143 108 236203 95 65 125 114 94
i-mcntílajni^a . . . . . . . 205 145 156 135207 129 87 99 i 05 156
4-am íno-l ,3-dimetílbcn-212 128 133 192 , . , 133
0!
Á : i
*
c
JS
Ül
ne
.vfr" - o»y<*'-

* 9r
L »
i »
r»r»
r *m
z
U
1
3 5 0 T obías <í« derivados
í »
! •
i *
T a» i ,a 24 — P (VeníinuaciánJ
Aminas primarias (líquidas)
Nocnhre dd «OinpiKrtO p.cb.»C
BclíCMl-nJfon¿-midj
Ací«Vo^ia’C
&CPZJ'ar¿¡da*C
yMoiu«i.vlfon*.
talida*C
Peall-tíout-ca
‘C
2-arom©4,4-dirnctí!b«n-ctno ........................... 215 ’ 138 139 140 119 140
j« -c tíla tu ltn a .................. 2>5 . . . a a * . i .2-arfüno*1 jS-dj rtxctilben-
ceno ............................ 216 176 168 204216 a a a 111 147
¿-ctüanilína 216 r r . 94 151 104c-w im o-A V M i mcálaru-
l i n a .............. *............. 2175-am ino-l ,3-dimetilben-
Otna . . ............. , 220 136 144 1592,3'd¡nKtilanilm^ . . . . 222 136 ‘ 189p -b ro n x ttm lín a .............. 225 *3 , 6 146
229 Í 02 79 11;- , . . £37rn-cloroanifiria , ............ 230 121 72 120 . . . [244-amj iio3-brojnolol u.enu 240 « a a 117 149 154S-cloro^-am inotolueno. 241 ► l . 140 . i .2 • amiao - 4 - isc^rttipilta-
lueno ................ . 242 71 102Fcnühidrarm a . . . . . . . 243 Í48 128 163 í 51 172«■amino-A'-mítÜaiü lina . 245 9 . . . . -« -íem tid in a 248 . . . 97 103 157 138m-aTiiúdina ..........- . >. 251 81 • • . 60 *4 ,ftt-bromea ni l in a . ............ 251 87 136 U3j^fenetidin* * - . - .......... 254 14 i 135 173 i 06 1363-broiYK>-2-metj]&níl¡na . 255 % s % 163 177 ■ i . . . >Jiodimdirt* ......... * . . , 255 > - . 211 4 n % • 4 . . . .3*bromo-4*metilanÍlina. .257 * ♦ * . . . . . .¿-aioino-tf-xnetílwulina. 258 - 4 4 í 65 . ► <Antro mixto d e m etilo .. 26úd. 107 101 100 , , ,p-amino-jY/i'-dietilanili*
na 262Antrcinilaia d e etilo . . 265d. 92 ’¿ i 93 •»►ar - tetrabidro - fc -naft il-
amina . . . . . ' ......... ; . 275 J 58¿r-to irah id ro -P -iia ftít- .
am ina ' . .....................: 275 107«-aniinolKtkzoato de eti
lo ......... *................. . . 294 . . . 110 143 . . . *. . . .
f

Tablas d e derivados 351
T a b l a 2 4 — P (continuación)
Aminas primarias (sólidas)
Nombre dd compuerta p. f. 4C
p. d>. *C
Bcíi*«ttnil.ion*.
Derivado
°c
Ptrtn-£B¿¿»*C
¿-le-¡neti-
aiIÍOna-n¿<i*aC
Fenil.Úo-tuxo.°c
A ntranilato de «tilo , . . 13 265 d . 92 61 982 'a ra in o - 1,4‘dcmetilben-
ceno . . . . . . . . . . . . . . 15 215 138 139 140 11918 251 . % 87 136 ‘ 97
PenÜ iiidm ina ................ 19 243 148 >26 163 Í5Í 172Antraijiiato de metilo .. 24 260d. 107 101 100 ♦ « sFsoduridim ..................... 24 255 * * * 211 ■ s * é , •4'3UiiírM>3-bromóto3ucnQ, 26 240 117 149 r a • J541 d iam in o b u tan o 27 159 [37 (di) 177 - fe r ♦ « am-yodoanilina ................. 2? 119
(di)157 128
5 -dora-í-am itiotojueno.. 29 24Í 140 . . . < . r é. 4 «30 % m m 148 ■ a t • a * . . .
«'brom oahiüna . . . . . . . . 31 250 99 116 r « a 146«zi-difenilhidrazina , . . . . 34 s s % • * 184- 192 % 4 1
¿-am íno’tf-m etílan ifina.. S6 258 • % % 63 165 - • fe«r*tetrahidro*3-nhítikj«í'
n a . ................................. 3$ 275 1075-ck>ro-4-3iiúnO“ 1,2-di-
metiíbencttio ........... . 40 148£-amino*Ar,7/-dimetilan¡-
lina . .. .................... 41 262 J30 2292*amÍno-6‘meti3piridina . 41 • • » + 4 * 90 90 . , s % * %1,6-diaminohexano 42 r t , 154 4 • • 155 a • fr a a a2J4'*diam ínoblíw iH o---- 45 363
(di)202 $ 8
A-toluídJna. . . . . . . . . . . .2-amínob¿fonil¿ . . . . . i .
45 200 . 120 153 158 1 ¡ 7 Í4Í‘ 45" 2 9 9 " 119 86 • • 4
4*areJno- l,2*dimetílb<n- ceno .............. 49 226 118 99 * ♦ • 154
50 251 132 120 % + é • ♦ ♦c t-n a f ti ia n ú m ............ 50 300 i 67 .159 160 157 1652 -amÍTto-3,5-dido3'oTolue-
n o ^ « . . . . ....................... 50 16653 302 171 230 4 • •56 • > * ♦ 4 • . . . 165 • . 4
oyodoanllina . . . . . . . . . 56 % m • • • * 109 139 , . . ./>*amÍaobena»ato d e n-bu-
56£>-MÚjidina .............. 57 243 95 ¿27 154 i 14 1542-araín6-5-brornoWfueno. 59 . . . . . . 156 . . . ... . . .

¿ fe .;
Tabica dw d w M o *
T a b la 2 4 — P ( continuación)
Aminas primarias (sólidas)
N<xabc< tW (xxuxk&q
4 * >pvtt>o-3, 5-dItJorotoI u g o ú ................. ................
SjS-^iamirtotoíwno r., ¿-yodaam iiiu . . . . . . . .ffw^n5coíli«nina . . . . ./^ im aeJIfanetíJaee tanll
d a . , . ► » ♦ ............. m •2.4-didoiw úliní. . . . . . S-ammopiñdijia2j S-diaxnin otohteno , , - 2,^-di»miaotojucno -.«¿-totílhidrerína ............#~bromo4mKn> .*♦».. ],8-dsm¡ü»otiJÍaJcaa . Pwudoaimírlina . . . . . .^cloroird llnft ................¿m itroíinillru . . . . . . . .2-doro.3í5-día,ínmatoUi<
no ............' 4-aitroniesidina . . . . ^«w nodííctuJainitva (an
h id ra ) ..........* «_4-4jDoÍDo-5-nitn>-l,S-<iÍTne
tílbcnceoo , . . ........4-ammo2-nitroColu«nQ ^•í^trídoroaniliiW L .« SiS-dibroaiQ - 4- aminoto»
I lleno2>4wübromoBLUiIi»u ..2.4-di«nÍTW Ícnol . . . , . 2*£Unl&odifcail»&ut& .3-wnLno-6<lortitducpo 2>fi*^ibr«rui4nilin* . .
2, í-dúuniooclorobcnceno
^-amiruTbcnzíiato de « ik» .................
3>4^1i«mnDtQlueno .2 -metü-6- nitro anilina
pA.*C
p. db. *C
« S s.íp«i-jXBÍa°C
Derivado««titulo
*c
4.1o-btn-
aúllan*-’C ••id»
°C
FcrikÜO'wc*■C
60 • * • • • • *..61 255 • • r .,.62 -r 4 , 18363 283 194 191
63 4 . . .63 245 í 28 14564 252 • • 4 13364 255 • • • « 4 •64 4 * ► • ► r 12065 9 9 ♦ 12166 245d, 134 16767 * 4 • , f , 9 ♦ 968 235 136 16170 232 121 17971 104 92
73 22«75 163 191
75 158
76 17377 i ¿Ó 14677 262 204
78 18379 ,, , • 4 1 14679d 4 m m80 • • ■ ► ♦ ♦ 4 • t83 241 , ,, 9183 4-4 210
17088 ..r (mono)
243(*)
89 ♦ 4 » 11089 265 ♦ » » - r r91 157
222240
117119
146204
16719294
169
203
185172174
134
178
148
167
72
26
01 148
95 152 142
153
64 145
34
í 215)K di)í
■£
iii
T a b la 2 4 — P (con tinuación)
T ebtat d» d#riv«do* ' 353
Aminas primarias (sólidas)
NomWc dd coínyuciio p. I. ♦c
p . cti. 'C
B » . ] cea»]- 1 !<»»- [ raid» 1 ’C 1
J)«r¡wfoMCtilado
*CBcitlírraid*‘C
fUuIIK».mida'C
Fcufl.tí©-v p
S-3 mi jKKjuiií oleína 94 172p .p ’ ► diamüiodifcnümeia-
no .................................. 94 . , t . , . 2282’n>etil*3-mtroanUina • - 95 w ■ ■ 4 . . 158 • ► 4 4 4- 4 » .6 ♦■cJorü-2>4<dibroinoan5]i-'
na. . . 4. . . . ,4 .............. 95 227 192'
1 ,2-diaroinoaüftaleno . . • 95 . .. 234 291 r ► t ,é .2,4-diyodoanjlLrta 96 , , . ♦ i • 181 r 4 4 4 1 rScmicarbazidíi ............ 96 4 4. 4 • h i 65 225 . • y • t .£-nitro»l-rtALftilaminü , 97 • ♦ > 194 191 rM2-atJi¡no4-mjctilpirídín3 , 98 . , , 103 114 .. 4 4 . .4-doro-í.n*ftil<Lioina . . , 98 4 • • 184 t t < 4 l .2^-diam inotoIucoo . , . . 99 280 \ 178 224 224 192 • 4 *o-feníleridttmíria ............ 102 256 v 185 185 301 201^O-diaznírKiiolucno . . . > 105 . , „ ... 203 (di) k • •/r-sunincacctcíctiuna---- 106 295 128 167 205 203 4 •p -tooraoícnHhidíaaina , . 106 4 • r , , , 9 * • 4 . . 4 , .2-aaiino-4*mtroto!ueno . 10? . . 172 150 4 %Ar-fenn.0-naftíl3unína . . 108 . . 93 ’ *J V. \P-rsfLilamina ............. 112 300 102 132 162 133 í 29m-nitroanilina ................ 114 284 136 155 155 138 1604 - biomo-8- pirro-1 -naftil-
am ína 116 202 . • 44-am¡no*3-mtrololuena 116 • • * 102 96 Í 48 i 66
2j4,6‘tribroTOoajiilin4i . ►, 1 ID StK)r 127
WO 232 190
5 'A Ítro l•naflilao jlna 4, , 119 183i (mono) ■
220l>4-diammon&ftzdenf> , 120 ■ « % 303 280 • • ♦ \ * '
122 . . *101
% 153 157 156.
4'amínQU>-mcn>. 1,3-din^-tilbi;fl.c«tK> ................
¿’-gnunobcuzofcnona . . ,123 149
(mono).
159 200124 , , 4 153 152
/H'anún&azobcnceriQ . . . . 125 360 144 ■l-n itro ^ n a ftila im n a . „ , 126 156 124 i 59
127 40Ó 232 317 ¿52 243 ► r

m
i *
r*m
Pm
35 4 T o b lo * d e d e r iv a d o s
T a b la 24- — P ('íon tírtttaeián j
Aminas primarías (sólidas)
Norata det compuesto p. f.’ C
0-to]idina 2-íunmo-S-nitrololijeno .3-aimno-S-nitrDtotü.cno -Bianííidjiia ......................./>-a«tofcnetidin3 ............2 ,6 -d in ítroan itína ............
«^Ícn ilín d iam in a ........ .2-amino-5"iiitro-l ,4*díme'
ttlteaccno ...................2 • amino-5 -nítronaftaJcna I ■ ar/iino- 2 -ni< ronaítafeno1-am ino íena rU r«no . . . .¿ -n ilro & n ilin a ...................4.arnJnoquinoicína , , . . ¿•nieroícmth¡dra*íiva . . p-amínoacetanilida - - . i-íiminO'3,5 “dim ito Co3ue
no .........................2>?-<iííraino^aítalcn(J . .
Acido pícrümico . . .4 ‘aromo-2,6.<3 initroiol üe
no ...........................
5 -amir.o-2'hídroxicoJueno
o-aroinofcnol , . .2/Wuútroanilíiu
íVair/mofeco!
(pj-2,4,6* trinitrijanilinacramida} .....................
1,5-djni{ro*2-naítiJ am ina, Dmitromesidína < < . . . . . 1 -■ajnjno-4'nitroiLafta]cno 2,4-diniirofftnílhicíra7jna. 4,4'-diaminocati]bcno . - . >tt-aininoa<*tam!ída . . . . 2 'arojnoantraqujnona ..
príl*.«C
Bto-| ÍÍOJlll-nuda°C
Í-IS-keen-
«Kttibdo mida suJFon*-°c °C mida
■’C
FmÚI.tío-unf*'C
129 314 265130 í 58 198 174 174135 • * 102 • s •335 • s % 242 236 . , r136 • . . . . 4 « .. t , ,136 ■ r • . . . 197 • • ■ ***
26? 247 304 300 266
142 162 J661+4 4 4 « 166 182 ' ...144 * m m 199 175 • <.14$ m • . 220 4 « • , , ,J47 139 210 • 199 191154 1 • • . . . 176 r • » 4 m •357d. . 4 . • • « 205 193 ■ • •162 ... . . . 304 . . .166 . . . ...166 2 6 i (di) 267
(d i)168 ... 201 220 . ..369 ... ...
103. . . . . .
173 ... (d i)179
(mono)194 ....
174 • * ■ 141 201 182 139 146*180 . . . 120
150202 219
I84d. ... 125 (d i)16$
.(m ono)234 . .. 150
190 211 230 196 4 ...191 s m » 203 • i % 182194 275 S é . ■ ■ ■ • 4 4195 158 190 224 16519fid. 197 206226 « s . 312 r . r « - • , . .279 • • » 191 . * . . . - r 4302 271 257 228 304
T a b l a 24 — S
Aminas secundaria* (líquidas)
Tabla» d« eU rivadot 355
[C<Mtibre dcJ CUrfipueMo p. tb, *c
Beicm*Juliana-
nóá*rc
B cp-irad*.DC
«otíoaa-raidí’C
ftflíl*i» w n
*CDimetílamma ....................... 7 47 43 79 135
3555 42 42 60 34
DíísopropUamina ................ 6669 123
205 93 48 96 101
A m in as se c u n d a ría s (liq u id a s)
Knrftlíí <Jel oompwíjw p. rl>.*c
Ikrwxft-flllíOD^
ifikU►c
Acít*-roidji•c
Bina-fit!d»aO b>14*
“C.
F«ni|-tiocn*
*c
Di-n^iropi3am3jijL......... n o131
51 . . . . . . ... 69
¿/-2-meti3píperidina . . . 316 . ►. 45 *55<ff-3-Triet53piperid5na . . . 126 . . . . . . . . , » 4 ,M oríoüna ....................... 3 30 118 75- 147 136DíjjobutjJamína ............ 139 55 "¿6 • é s . . . 133Di-»4>uiil¡Urana............ 160 s % » 66Ar»mctí{bencí3amina . . < 183 . . . . . . M , 95 . r ,Diisoamilamina ............ 187 . é m .. . 72MetilanilinA .................. 192 *79 102 63 94 87tf-etilbencilaim na , , , . 199 t . A . . . % r m 50 r . .Ar-etílani3ina . ................ 205 r • • 54 60 87 89Ar «tU 'ffl-tofuidirta . . 206 66 • • •
205 • • % , . . - - * . . . 172jV-mctílx)'toluidim . . . 207 55 66 120 ♦ ► 4W -mctn-^-toiuidiha . . * 208 64 83 53 60 89tf-íscpropilanílina ----- 213 * . t 39 , . . • 11iV-metit-íXloroanilina . 2(4 ». . • • •/^-Ctjl'p-toJvudínS........ 237 66 • • . 40 70 • • •AT-ettl-ff-toIuidína ►,«,. iV'etiUwi-toluidina . . . .
236 62 . . . 72 75 • • •221 . , . • • • % ♦ ►
4-W*met 33amino-1 > 3 *di- • nw ^lb tnclno . 4 # . 221 . . t • * * r, . • • * r r r
Af-isobutiJanilína ......... 231 . r , , , » % » kTetrahidroijoquinolina . 233 154 46 129 ♦ ♦ 4

T abla 24 — S (continuación)
Ajumas secundarias (liquidas)
Kfcaln dcJ oo«m»crto p. <fe. ’C
BcjCC»*jsMotU'
mida*C
Accu-HÜdi*c
BcM4-releíax
¿r-tohXft.alfojU'
«id»*C
FcnJt-tiourca
’G
236 * . 4 ►». . . .Ar-iacíi¡-éKjorainilina . T ctrahídroquinoldna . .
240 92 . •»250 75 , , . 4 . ,
^-óoiEoilam íini . « . , , 254 , , , i •, . . <W-metÜ-^-bromoaiiilina Di- (2*hj¿rcxictil) amina
260
(dieUQoljtRUns} 268Í2 Í
« - rJV-nx; tii-a-nafy¡amina , 293 95Ar*cndUimI£joa . - ......... 298 119 58 107 103°O ií^ndlam m a ----- , , , 300 68 , , , 312 « < rDífcraiamioa ................ 302 124 101 180 144'- 152Ar-o ic tíI^ ju fd l-am iiia . 309 107 • t .
, ►. 315 , 49 »..jV-ítil-o-rufúIamin» . . 325 63
Aminas secundarías (sólidas)
NmnbfC 4el copspiíttto p . f. *C
P--eb.• c
B evcecsukfcoa-Tni¿A*c
Denwi4omcetiUido
°c
P f ú a -Olida’C
P‘«y-3i*en-
Hjlíorin-Xnida*c
FeciJ.lia-
UtT»"C
T e tra h id ro q u in o lm a . . . 20 25 0 67 753Di- (2 -K id ro w c t il) a m in a
(dicULDObm ina) . . . . 28 2 6 8jV-fccnciLáíw 1 i na .............. 37 2 9 8 Ü 9 53* 107 4 . . 103»o * a it to - / í - in e ti la m lin a , . 37 70¿ í-ta c tíJ tr ib ro m o a n iK n a , 3 9 r * * 101 _ m m^ • ^ tD p s c u d b c u in k J ín a . 44 2 3 7 . . . ♦ » r4 - { N -m ttü m m n a )-2 - n i-
45 3 28f a d o l ........................... .. 5 2 25 4 ♦ .. 68
54 3 0 2 124 103 160 141 1524 - {AT*<tilaiíiLno) -3 - n i t ro -
59 ... . . . . . . 127
t a b t a t d e d e r iv a d o * 3 5 7
T a b l a 2 4 — $ (continuación)
Aminas secundarias (sólidas)/
Korabrt del am putil» P f- p. ib.B«a-
«and-ÍOfta-
Derivado[¿indi? miíi¿
F-iúUlio-
nG . rC rrtldi- c *C eC mUt
’ C eC
íY-femJc-naftílaciinA , , 62m<ittro»,Y--ciiU*mUna . . . 65Ar-met)J'í7J-mtrei>amtirta . . 66¿V-¿-toJÜ-<x-Daft¡Jamina . 79D¡-*‘-toli!amina .............. 794 - (Ar-m etilam tno}-3-nU
tro to iu e n o ..................... 84
335
330
p'hidrox¡-Ar-me(jlamtina, $5
o-iúdrcm-A'-inctikniLina. 87.V^til-^-nitroanilcna 96¿ - lo íií- f i’n a í t í la n i in a . . . 103Pjperazína ......... 104j,/^cü-2f4-dinítroaniJlñíL . 113¿-nítro-JV’-inctílanilínai 152W-mctü-2,4*dÍQÍtroajüUna 176Carbazol .......................... 24-3 351
140
8383
282
i 20
1158995
12485
64. 2 iO v {nnono)
97
$ 118 85
1$4
i 53
¿9
152
i 56 140 125
175
1509$
139191
n i
*98
10?
í 73

3 5 8 T a b la s d » deriveicfo*
T abla. 25
Am iau terciarías (liquidas)
► OraupUE«O» de *dld¿ri CeaAeÁdo (f-tohien*
Norobre del amcpctlto p. cb.*C
dWKputl-
nJ!omU de me Yogurt Addo
TÚCO tilo pteríeo*C “C °C °c
3 242 d. 230 d. 2168 9 -.. » . * 173
115 241 139 117 J67a-picolina ........................................ i as 195d. J 50 230 169Alcohol - 0 ‘díraecjlammoc tilico 133 . * * 962 j6 -d iin ed tp m d in a ............. .. 14-3 r % % . . . 233 168
143 150143 s • . . ►. 167149 < ■. J35
Tri-n-jywpilaTnána ..........,, -,T ria lü a rn m a .............. . .............
153155
116
2,4-lutídína . - ..........................„ 159 . . . 113 180275'dici>«tilpiHdiníi . . . . . . . . .Alcohol-p-diedlamiaocdlíco .
160161
169
2^-dím ctf Im ñdiaa .............. 164156 í 20
¡88
3-bramopiríditia . . ................... 170 156 1652,4,6-coliditva............. ............ 172 « . , 155jV^A'-rdÁmctil-o-toluidina........... 185 193d. -.. 210 122iV^/-dii>íctilb«ncUsimina , . . , 135 192 179 # s *D im cdiaiúlina ......................... 193 I73d. 161 22Sd. 1632-fcrom opirid ina....................... m 127 , , .3*ctLI-4-metílpiridina .............. 196 205d. • r r l » « 150tf-etí3-/i-metiianüina ............< 201 ♦ 1 r 125 . 134
206 224d. 180«wiloro-Ayí'dmietílaTviliní» . . 207 s m é
210 35 2Í9 127211 186 105
W,V*<Umettl'i7>-toluid¡na . , , . 212 177ÍTj^*d¡rnetüintiidür>a . . . . . . . 213
218 j % , • - 4 102 142A^Mdunetilpjvfrtidocumtdina 220 . . .
T a b k » d e d e r iv a d o s 3 5 9
T a b l a 2 5 ( continuación)
Aminas terciarias (liquidas)
CtoÉpoeMM de vE áfci co*
Norobrt d d w*ujn>tiln P- «*.«C
Acidoctoro-ptjlf-nloa°C
/r-toJixn-
de medio-c
Yodurode
mrtílo’C
Addo píctico
"C
22 9 184¿^loro-Z /jJV ’-d iin cU la iu ru ia , . V >iSr*dietil»m -to]ix id ína.........
23 0 • A 9 • A é é • •231 97
239 21 8 126
7 2 (h i~ 1d H itado ) 1 133 ( a n - | 2 0 3
IaoquÍTK.JdnE .................... 240 263d . 163h id ro ) J
159 222242 218
N ^ -J D í 'H ’p ro p iJ a n ilitw ....... 24 5 4 4* 4 r r 156 > ♦ *T r i i ^ m i l á n u n A .............. 245 * • s . 4 - 125
24 7 2 2 6 16 i 195 191257 s o d . . . . , , ,
6-ír>ctílqu:nolGÍníi. . 258 - k r 154 219 22 9S -b ro m o -JV ^ -d im e iíI ím ir tn a . 2 5 9 • • » . . . 259T v ^ ^ D i-a -b u ti la n il in a . . . . . . 261 . r « 180 . . . 125L e p i d in á ^ m e t i l q u m o le ln a ) . 26 3 . • « 174 2 1 22 ,4-d íitt« ti3qu ino l< iína ............. 26 4 2 2 9 ... 1942*fcn itp írid3na ............................. 26 9 ,.. ► .•-^ .A '-d jw e ti l - a -n a f t j la in in a .. 2 ? 2 . . . ... 14-5^bfom oqm nolelna 278 273 217Aí-avetildiíenilwtttníi.......... . 296 • » •214,6-tríbron>o-Ar>7/-dirnetilaní-
Iina .................. 301,Z/*dimedl-(l‘m ftiJam ína .. 305 .44 ... • • •
6-mcto>á<5uiuoleína ................ 305d. . . . . . . ► ♦ ♦7^bencíl-tf rmedlanilma. • * * < 30 6 164 103Ar‘bencil*tf-etüanilin¡i 3 l2 d . 161 117

3 6 0 T a b la s d « d e r iv a d o s
T a b l a 25 (continuación)
Aminas terciarias (sólidas)
OofnpUWW» «le adfcWti <v»l
N orotce dH cowpdeKo p . r,’C
j>. d>. -c
p.Loloc»- Sulícmalo
de me-V o iu w
dem etilo
•c
Acidopícrioo
*c
Ilo q a in o W n A ........................................ 246-njctoJcquJrvolcina 2?t-bnwio-W^/iMictiíaaiiifU . 34fr^ -c tilb e ñ c ila n ilín a ......... ............342 ^ o ro q u h > o M iu ......... . . . • 38?-metU<|uÍi>ol£íru ................ ........... 396-clorcH}UÍnalelni . ......... . . 41^^-dim«tíI-2-iufti]aiixin% . 472-broavoquLnolelna ....................... 498-aartoxMuútolttzta . . . . . . SO¿^ rcm o W ^d ím c tU tu ú lin a 552,6-d Lra^tilqui ooleirtf. . . . . 60JV^-dio>ctU->n-tD Ir vanilina 60A'-íetülpirxoI . . . . . . . . . . . . 61a Iff'-4ípiridili5 . . . . . . . . . . . 69W jtf-di&eDdUmlína.........................706«d»fl>etil»inipobcngald¿htdo 73th íd r o w iu m o le l i a ........................75p -n id n o á -N Jí-ó tmctilaní ti-
na ......... ....................................... 76W ,y ^ k tíl-2 i4-dÍn¡tTOanj3Lna 80A ^ -d I t t i^ l^ t ro « < * n i lm a . 84 mJjidrTO.AjAT-dinictilinili.
a» .................. ............................... 856-alttok>-MjV^iiitt£tilaLml¡-
» » ...................................................07¿^N-díioecU-2.4-<linitraaxtt-
fina ............ ................................... 87T ribendU znau . . . . . . . . . 91
240305d.270
267252262
283264265 285 234
SOCd.
266
268
380
163
43
?5
59
248
2106 085
23 7205
35
43d.
201
• 9 82
$4

Tabla* de derivados 361
T a b l a 25 (continuación)
Amina* terciarías (sólidas)
Cotnpuotoc dfl adJíiá'O Cotí
Nombre drí comtiueito r». i 'C
p. «b. *C
í-ÍOtuW»- luMoaato <£t Me
tilo «C
Ytxíuro de
án c<Uo*C
Acidoplcrweo
*C
JOB 224 2083^-díbrom oplridina......... 112 222 219 274Anttpirjn* . . « « ................... 113 , . , » , ,fl-d¡ríictila,minoaiobcnccno . n ? 174Trifcm laxnina..................... i 27Metílcnaminoaceconitrilo , 129g.nítroquinolcina ............... 149 . , , 245/«^•dinictü'-f-QÍtroAiiIlina . &-nItíOquma]dina
16316* t ,
¿,yrbU(d¿metilammo)-bcn' x o fe n o n a ,,.,. 174 » 105 156
193 . r , * 4 * 2362-hÍdroxíqutiiolcina ........... 199 , , , , , , , , %H«x*roedioHtttrnxnim$ , . 260 205 190 . . .

i »%
r b
P
3 6 2 Tablcr* d e d e r iv a d o *
T a b l a 26
Aminoácidos *
Nombra del CCoipuCtto
W*feni{jlic!na . . . ...........Acido antraníüco . . . . A ddo <it -hidrojdglutó-
mico ...................Acido ro-aitt'mobenzoic& Acido ¿>*am>riobcnj'A>ic00-aJanina ................ ..Acido j> o L-jjlutámíco
Acido p-aminofehilacé-t i c o ..............................
d í-p ro ljú a .....................-Sarco6ína _...................».l(—Jpraliná .............H> o l-liaina . . ...........j> o L-asparigin;Acido dl-%lulimi ¿í-serina
in a . 'ico
h .
Glicina ............................<ft*treom na . . . » ..............
i.( + )argioina . . . -----¿¿.arginma . . . . ; .........p *ki d rocáfeailg} leí na . . d?-ak>trttinuia ..............i> o L -tre o n in a ............t{—hostia* ........... .Glidígücina . . . . . . . . .Acido o- o L»aapirtko
L(— )htdroxíprolina . . c>* o L-alotrconina . . . . di'ioctíoniiia ...........
Puntodi
Dentado*<j£*CO«TVPW¿-«j6n*c
BctíoíI•c
FcaiUure«VC
#K«lUcn-PuKo OlVtr*»
*c
126 63 195144 182 181 Acctil , . , 185
150250174 248 27Ó . * w • Acctit . . .
186 278 300 • » » Acetil . - , 252196 174198 130 i i 7° a-naítn-
236170
137200203
198'Í 70 •
urea . , - Acetil . . ,
i -^rato . .210 102 r , , A^-ti! , . . 135222 170 139 Picrato . , 154224 184 Dibcnzoíl . 14-5226 164 ► « a Picrato , , 160d.227 157 • * ♦ - A 4 . . . . . . - a 4228 171 169 213 ^n a fta lcn -
220206232 187 163 147
sulíonil . Acetil . , .
235 148 }|(m ono);21 ?d.238 . > t A % m Plcrato . .
296 315 é % r M , Pierato . * 201248 117 • * * • s . Acetlt , . , 203252 177 ' • • •253 148 f 148 )
i(m ono))203d.260 181 117 , . .* » « ,« •
264 » * • 176270 185 162 í 40 CC-naftij-
115270 175 153
urca . ►.
272 128 4 - •......... . k »272 151 Acctil . . . 114
* l a roUdóa ¿ptka se indica con ( + ) 0 {—>. Lm Ucna » y L indican t u co*[[*üracÍ6n« «»q«$üvic*J relativas. Laj 41 eji bánltac<fífi* jndíciiri ia íof/na rftCéoucA.
T a b l a 2 6 ( ¿ o n i in u a c ió n )
Aminoácidos
T o b ía s d o d e r iv a d o » 3 6 3
¿/-feniíalamna . . . . .X .(~ -)h js tíd in a ..........d- o L-Uoleucina . A cido o-am inoísobutiri
oo .........................Acido á/-*»pártico. . . t (*^-) dihidroaífeniiila-
n in a ............... ..Jm etíouina . . .
Acido S-amino^aHcílico 283 Acido i- ( - f ) -o^anuno-
«-butírica . ► ............... 285
L{ + )norleucina . . . . . 285Jtriptoíano . . . . . 4 289
CrtatjQ ína .................... 292¿f-isoleudna ............ ... 292dí*alanina .................. 295xj- o L -aknína.............. 297
¿í-valm a 298Acido <fí-R*amíno-nJ)u-
tírico .......................... S07
D* o a>valina ............... 315W/'tifoíina . . . 4 . 4. . . . . 318eí- o t-feoilalaw na . . . 520«¿/-norleucina ................ 327¿M eucina ................ .. 332
o L-lcucina............. 33?l . ( — )t¡ttiiina . - r . . . 4 344
dí-orm tiaa •-.............. ..n- o L oro itin a ............................¿Miada ................... -¿/-tftOaerina ..................... . . .
— )¡Sos«fína ................................
165
135 Pkiolonato 182 204 Picrata . . 86132 Picrolonato 170
..4 a-naftH-urea . , 186
252 . . . .4 , Acetil . . . 218
121 . . . . . . a -naibl- yrca. . . 195
• • • . . . • » .« ..■ » «166 176 oc-nafli!'
urca 158• * ► . . . * • . Picrato , . 220118 « ■ t 140 . . . . . . . . .166 190 139 Picrolonato 214151 168 133 «-tuí til-
urea 4 4 T98132 163 UO Fotmil * . . 145
147 170 ,.4 ft-naíríl- Urca 4. 194
147 147 Fozmíl . . , 156197 « .. 226 Picfokmato 260146 181 161 Fonnil 4. . 167• r r 124 Formil . . . 114141 165 , Kcrolonaio 150107 115 1221G6 104- 114 a-naftü-
u iea . . 205267d. 192 A * • Picraio . . 19524fld. 190 • • s Picrato * . 204249 196 A % m Dibcnzoil . 146151 1A4 ....... , . . .109 • 1 •

■ 3 6 4 ’ T o b ía s <í« d o r iv a d o i
T a b ia 27
Carbohidratos y glucósidos *
KomVe del compítaloPúa lo de Petición
ejpedíie*
Tkm«trequeridof o l i ó n
Pcrlvaduc
tM JM
"Cde la
°C í«*¿Ona(mln.)
*COuzona
CGAce Uto
♦G
RAfinom (h k íra t^ f t) . , . fio + 104.5178
99P-meübiosQ íhMwLtada) , 65 + 129.5 -----
í í & t f ) Í H 2 <«>Glucosa (h idratada) . . . . 50 + 47.7 4 a 5 205
D-ribosa .................. ............. 95 - 21.5 . . . . 166Maltosa {hidratada) . . . . 100 + 129.0 . . . . 206 íoflLevukxsa (n-fructcna} , , . 104 - 92.0 2 205 J109 <$)
1 70 Ca)105 + 9.4 , , , , 182
oHxoca .............................. .. 105 + 13,5 4 • . . 163 . . . .Ra£no*x {anhidra} ......... 119 + 123 . 0 ,
20599
t H u n c u .............................. 132 + 14.1 0.3 . . . .142** . . . . . . . i . . 132 f(dim eridc)
¿ 154145 + 16.? 7 163 141
G ln co » (anhidra) ............ 14$ + 52.8 4 a 5 205M d i cito ¿a ....................... 147 + 88
160 + 104 10 166160 - 43.1164 ► * • r • • 169
CMaetüglucófidq . . . . . . . . 165 +157.6 101Arfcutma (h idratada) . . . 165 - 60.3 » » * < 136M altón . . . . . . . . . . . . . . . . 165 + 129 *% f ■ * 206 158<C-£ddo ascórtioo ............ 169c -ic ido asc<$rbi«> . . . . . . . - 43L ucido ztcbrbi c o .............. + 49 «44. . . . . • • • •
n o + 81-7 15 2 0 1 ÍI4 2 ($) i 95 (a )
f íe lk in a ................................ 175 - 60.4InuUua ......... ...................... 17B - 39.5 25Populina, ................ .. 160
2Ó5*Sacarosa ................................ 185 + 66.5 30 39185 - 66.9 125
a>metilmarv5srdo ................ 193 + 79.2201 - 62.4 ISOL a to s a ............ ................... 203 + 52.5 2 0 0 d. looCt,a-CrchaJosa ................ , , . 203 + 197.1
CdMÍOiA r . . , ............214225
- 41+ 35.0
-----198’
166
240 198d.
CduJoaa . . . . . . . . . . . . . . . á. ' % % • m
d.r . . « • • • • . . . .♦ r % • # * * * . . . .
* Ua* ietra BiayvKKuU Jiequeilk » ó i, *» Funde *itl docaRHXitkiáa,
Dolcc*d* anta det nombre del azúcw. índica con-
Tob ía s d e d e r iva d o * 3 6 5
T abla 28
Estires (líquidos)
Nombre do! oompuctto p. cb,•c D c o i. E d f .
Foftniito de metilo . . . .Fwinúito d« e t i lo ..........Aoeiuw ac me£U> . . . . . Fonnisto de ÍMpratnla - A«et*:o de vioilo Clorofúr0DÍjiU>> de mcUloACCMIO d« «tilo . .f............Prcplonto de metilo . . , . For/níflto de ir.proíiilo . . .Permuto da slilo . ■........Acfilaio de metilo ..........Cartón at» de metilo . . . . Aíttato de ijoprepilo . . . . líobutirtito do mirtilo . . . ClorafortTjUno de cilio . . Ac<ta>.0 de iiopropcoijo , Formato do ie*.butilo . . Fomúato de iiobuúlo . . . Propíonato de *tüo Acetato de /«--butilo . . . . MetocrjUto de raetito . . .P¡ va tato d i metilo ...........Acetato dr »-pt«pilo . . . .AcriUtO de etlto ............Or«jfor¡nbto de metilo , . n-lw in to de, metilo . . . .Ace u to de alilo; ................Cloroíonnjíto dé ¡*«>rop¡k> Formínlo de *.butifo . . . .l»batimK> <k etilo ........AccUtO de cfoioroetito . . AartHO de Jm>butilo . . . Glorofcroiiato de n-JWOpitoAcetato de iiobutikv........IwiJrriaowilO de BletiJo .«.butcnU» de «tifo ..........
- Propioaato de *.prppik» . Fono lato de itCdmiío . . .Acetato «¡.butilo ........Cübotutd de ctüo ..........X-butiratQ de ItopropBo . V s ltñ ia i» do metilo .... CWfpíermiato de j»obutilo Ace Cu (O de brOroanittito . Cío ro* Ce tato de rvjtib •• Formaio de a-amito . . . . hovfjeñanp» de cilio . . .Piruwito <k metilo ..........Propio nato de ttobuüla ..
32 D.SM*W tt.9to57 0.95968 0.635?? 0.9317í)475 1.336%77 0.924n 0.937»l 0.913
0.932'tíCI.9J7K
ÍM> l.055>»91 fl.917K a s m55 1.1441»% 0.931%i97 0.682S8 0.905 .9b 0.912 •93 0.857 ’•99 0.93ÍÍ
301 0.891M ■101 0.909 :1DI 032W] « D^74^102 0,919ICC 0.93S105JÍI7 Ó.Sl'ln o 0.B90m 1.195^n i 0J70i)3 1.094JJ5m o.w?l i s 0.901120 0.90012? 0.M2123 0.894125 0.902I2t> 0.975isa 0.879X130 0-910130 14)53'»ISO 1.65P»ISO 1.2381 » 0.902134 QM5136 1.154137 0 -Kfl '
Xo tabre <Jd compvewo p¿xb- _ c VKm. E*p.
Crotocate de etifa .......... 138°AecUto de Ucamib ........ 142a-butirato de a-propito . 143 PrcjHQtuLto de i-butilo . . 1 -H Í 4CU(o de metilo . . . . . . . 14+BrOnxnocUto de metilo . I44d. Acetato dd l u r moneroe-
tíbeo deí Mitengtícol , . 144 a-vaJeriwi*to de etilo . . . 1*5 Ctorofomúam de a^litUo US Ctorcmeeiato de etilo . . . . 145Qrttfonaiato de «tilo . . . 145Acetato de $<loroctita .. H5AeoUto d¿ n-aoiito . . . . . 1+5a-cJúrepropíenaUi de ctiío 146Cleroaéciato de boncilo . . i4?LobuÜTato de jp?but¡Jo . . 14? AecUto de mecilttobutil*
«rbújal ......................... ....... 145n^mprOito d« metilo . . . . 150LoitWú de oiíio ................... 152EtoxUcctatO de etílfl . . . . 1 S2CloroferroUto de Imwnlk» 154Formato da *-beidlo . . . lS íPüvrito de etOo .................. 155Acétalo del ít<r mOMctí-
Jico dd cúkngticol . . . . IMa-butúreto de is¿butlV> . . . 357Dkk<macctato de etilo . . I5SBrOoinaccUto d4 etilo . ■. 159JVopiotuítt de tojm ilo . . 1EDGllcoUto de etilo ............ ......160Fermúto de ckfobejtílo.. 163«-bruprwjropioiiato d« .etilo 1S2Aretito de fl-bromoetiJo , 153n-h*ilinto d* a.butilo . . . 16)n^Mrcoto da etilo . . . . . . ÍC6TrieJOi-Mce(»(o de rtiSo . 16?«•TiÜCíTJJiatO de n^ropito íí?Cwbaaato de a^rojpito . . Í6SLwtato de ¡topropilo . . . 16&P/oploMto de a^unilo . . 169^cecucobito de metilo . , 169h-]Ki>t*noato de Diftilo , , 1 ?3Aoeuto de olclohotílo . . . 175Clofoíkcetato de a^utito . 173Acetato de {iLcforJlo . . . . . 175B-butirato de isofcm¡k> . . I?Sfl-bromapropiooiito de etílo 17S
0.ÍI7»C.SMC¡89iaasf.I090«I.85JÜ1.306?;'*.1.0781.1580J971.1?*0.«96 t.0671.W» ÍJ875
OWWKO.WIJÜV%i.na^Q*m>ixao'HOL»««Sd0.8831.2821305osa*i.oiai j »1J240.B88o.sssI.Sffl'Ji0.6E2»0^9'K0.SSKo.e%, •D.972*
I.117S%
fS

¿ «ÍW
•
i f
irm
4
3 6 6 T a b la s «le de rivado»
T a b l a 2 8 ( continua ción)
Estere: (líquidos)
í ío m b re d d c o n p u fa to p . eb. D tru R¡^). 1 NoD tbty del eonopue*4ú p . cb . | D e iu . R jp .° £ "C í
F u re* (o de m etilo ............ 151M a to ítt t» d e m etilo ......... 131 I .I W 'SK 'A <Ca»Cetai □ ár, « tito . . . . MU l.Q31:£Am U q d e ^-bldnvLM lSn 182» 4 * ( in U ) d e i-taita ... 185« - a p n » < o {to n-^jrepíJn . l$6
O x i la to de «rilo ................. Ifll»M etil¿cet¿M e«»ato de «<ilu l¿7L u c b Jo d e b o ü ia .............. 180*^J|fl>l2a a a to d e e t i lo ......... 139DiarrtJitO del etileofflleol 190O m iIm o d e itofiTúpiV) . . . 190Gwfefttiato d e laobutílo . . ISOfo v iS e r iin a to d e ho»jtji)o I9rtE tiiiM io k c tU to de m etilo 190LevulinutiD d e m etilo . . . 191A cetato d e K -b tp tiJo . . . . 152C jq m la to do Jtieü la .......... 193Aeréalo d e Ic lrn h íd ro íu r-
tarífa .................................... 191S u a d o a lij d e lite tí lo ............ 1MM rtilm jfo n U a d i e tilo . . . 196A e tu io d e fmiw> . . . . . . . 1%M iV to t 'o d e « l i o ............ l$8f rf tu o a to de m etilo .......... IWC d lu tta it tU iW d e e t i lo . . 196Acetato do elil.2'Jie»il&-J.. 199C k r i> « * t» to de m ilito . . 200F e rm in a d« beneilo . . . . 2(0B « > » a tr d e vitt«lo ............ 203Val tr ia a a ta de «l-acúla . . 204>bü«nnlaetaa<v ...................... 2WL evulinato de e ti lo ......... ÍOJCapriUle* d e t l i la ............ 266CarfrwiatO «fe *-b\»tiJo . . 207y-Y*J '.TOÍaCl¿/U|............ .. 207A c e ta » d e « - c r a l lo . . . . V A D Eatetato d e l t r im e tiie n .
*6col ...................................... 2 t0
PrOpiaoaJ* do í c n i l o .......... 211FeroaJD de H-yTOplio , , , 211 D ipropio tw to dc4 e liltti-
gl¡e*t ...................................... 212A cetato de m <*ts¡k> . . . . 212
Aac«*t« d i í - c /e j l lo .......... 2 l2 1 -0 W SO x a lílo d e n-p rop ilo . . . 213 1.036Pe largo* a to d e m etilo . . . 21$ 0.692B o r a a to de e tilo ......... 213 1.066O>t«lualo d e m e tilo .......... 213 1.073'K
0.8ft«*0.8844»
1.062 l.024J* « W i o. « a 1.12» 1.0l0'« 0.919'!< D.97D 0 .!*» l.ifca 0.8891*0.ew
i.OG24«¡I.I20'«1.019'Xi.oat'K1.C77 1.103 0.972 0A71M» l.0P?í*üi.oai**1.065O.tól»
1.(1160.887a.s4i«tÍAttm1.(4$'*
1.070'%I.M4»*1.074i*5íIMS*
«•to hiato de metilo ........ .......2 1 1 1.066«»Fumarais de «ei ■»............ .......215 iM ? 2 *Acciito de hencálo .......... ...... 216 1.D6<¡1>¿Succinat» de «tito .................216 l-Dt^R¿>to*uau> Ae m etilo ........ .........217 .........Acetad det ,á(*r moroctí-
líco del dietilccplicoJ . . 259¿* i^rój>iJo . . 21$
Finili.eeia.to de metilo . . . 2 íflJ.«VüJ¡j|»t6 de it-propilo .. 221Salicilato de medio ........ .......224Maléalo de etilo .............. .......22?PelitKonato de etilo ........ .......22?n -be o rato de íerilfl ........ ...... 22?Acetato de í-oküIo .......... .......?27Carbonato de lvumlk> .. 228Oxataw de ifobutUo.................2MFeniUeeJíkto de otilo ...............229Bco2©*<o de n-propllo .. i '
H«ni£i»io de s l i lo ........ . 230 •m-clortil>coí<>j(i> <t« nwiilo 221Caprato de m etilo ............ .......232Chitaratü de eülo . . . . . . 234o*clorobení/wo di metilo 234SaJicibto i*, «tito . . . . . . . 22»Brotoon\fc!<><i»te de etilo . 2J¿Stdtclhw de ¡jopropilo . . . 237a-fcijlimto de beocilo . . . . 23SSaSiohto de a-propito . . . 239s-butilmalnoaio <ie ei¡io . . 240Be.n2£ib(4 de itobutilo . . . . 2410*a|»K> de 1t-i«itila . . . . 243K tttaco de tiroUo . . . . . . . 2*4&-L'i i»nioÍk-tvzgato de metilo 244Adipato de etilo ....................... 243Caprato dr e t i lo ................ ...... 245Succinafo de «-propilo . . . 2M
A retalo del ttWhulllipo de| dielitensliwt 246
reníta^rtaU» <fe liobutiio.. 247'Ufldeeitcn4IO du tnetiio . . 24R
Pea«ia(o «le «.butilo . . . . .......249 Acetondicjrbo*Hato de eti
lo ..................................... .......2J0 1.113F<nil««tíio de j iio t ilo .. 254 0.9^5'KPu-nefeto de eiílo .............. .......2J>5 0.M 4Wfiefitoilíuejniato de eiilo . . 2S7 . . . . .TiiaCetato de jliom lo . . . 258 I.IOl'JSOzalaio de Moamito ........ ...... 2 S2 0.9WK
1.0ll4*H«i.otiyto1.044'K0.98«¿í1 .18Wt.07441ÍI.656'h1.027'Wo.fiiÉA>.n0.912'tt l.OW'ft1.toi 1.025»»
J .06754<l'Á76<K1 .0Í 2
i'.wnI.4WÜ1l.OMW1.033^I.09B1SÜÓd2‘5iíxioJ 009
O.M7l2«j 0.999*
1.000

Tabtctj <íe d e r iv a d o * 3 6 7
T a b l a . 2 8 (continuación)
E s te r e s ( l íq u id o s )
ÍÍQirljrc rfel compueiti» f>. cfc- °C
Deox. E»p. Kocnljre de3 compuoto V «3v°e
Don. Ei0 .
Beezqftto de mombíIo . . . Í62 I.KH 291 0.97É61»SuMtJMtO de HOtiutiJo . . . . 265 0.974 2&4 1.137.Lfturüto de metilo .......... 26fl O.KíW 296 0.87»)!&4tk4ÜLtO d< K-butJÍo . . . 26fl 1.0741» 297 0.fl6 l>M
209 1.1 tStt n a 1.1172^9 0JB«IX 303 1.077**
Bcntcñlacrtiilo de etilo . . 2 Wd. I.I2JS44 Ft*]atO de woprtpilo . . . . 302 I.D&S L273 1 .0» 306 D^S^Í
SMIdbto de iy»m¡lo . . . m Í.OfiSl* SílMtíílo d< ciclo ---------- 307 D.9M>«OlaCeUto de JWrúnol .. -m 1.I80*S Bcnioato de jxtmíIo . . . . 307 3.114’«ht-tai traben acato de metilo 27? Tfibutiraío di jliccriln ., 33S•í-t2urtmt<> d i etilo . . . . . . 230 1.199 320Ftalita d i metilo ............ 282 i . m 'Z 323 I.II4H.Suberat© de etilo ............ 282 □.S622W « a I.OMMkTriyrrcpk>*ltO de güe^iilo 289 1.0831* FtaloJo de ¡Mamilo ........ « 9 1.024’k'
Esteres (sólidos)"
LMirtM» d« metilo ................ ......... S 2éflAuitit/» de rtilo .................. , .......... 7 269MiríiuitO de <*ik> ................ ......... It 3ÜI>"-Mfcrtto <fc elllti ............................17 2í*JMift'rtMo de üiítíla ............ ......... ffi 2Í*3SfCeinato i f , rtetDp , . . . . . . - 39 35JProoíroiito de íenilo ........ ......... 20 21XM-ctarobeníOato d4 metilo . . 2 1 231Acetato de cotilo .................. ......... 2?otirtr*to de nJwtila .......... ......... 22Fnliclt*<0 de etilo ......................... 24Acrt*<o de txjroa» ................ ......... 29 221Pp-lmltiito <k ittet'do ............ 30nt.bromóbenxAiUO.. de meato 52
5-tohttti» de ni c ti tu - .............- 33 217ai2*4to de tímilo .......................35
ErtcífílO de KÚlü .................. .........33Furoito de etilo ...................... .........33CSrtamalo d<! m«tílo .............. ......... 30 2S3Mjudcbto ¿e **5k > .......... 57SeljuMito de metilo ................ ..........3R 288d.Encanto de motilo ................ .........38
de bencU o .............. 39F ib ím o de beccílo ..................... 42SR.ltótato líe íwuio ................ 42Qxibto de cirtol.exilo . . . . . . 42//■<l0inbrn«ia<i> de meáli» . . . 44C in a m a w de cinanüta .......... 44Smaáanlo de bfflrilo ............ 45Aniwito de metilo .........., . . . 45¡Ujiilp^talito de efilú . . . . . . . 46»1>AÍtM>)e>iiMK> de «tilo. . . . . 4?o-t*ttrato de metilo .............. 4$<6t)Aetiato á( «-nalü3a .............. 49O l f a t o de m érito .............. 5ta-h«líioil b i n a n t e de metilo . . 32
d o f Cuita ................... 52b e tu o t to do m a n s i to . . . . . . . 54fr-n jtrobeo joato d e e tilo . . . . -‘>5Manecilla de roetifn ............ 38t i n t a n * (9 d e o -cre iilo . . . . . . 00Tñctopobitcuij df filio ^. •, •. 604 'P ¡ ( ro fa la ta (te itte tiio ........ 66Cubará» ............................... 67
2*5?*>ml£3352
If22»
* MudlOl íiftre» s6Udo* Cjtin íaUitadoi C<Múo derivado» de tó íd l , bJcoJioUí y fcnole* (T*-bt» 20, 21 y 45}.

3 ¿8 la b io s ¿9 d«r¡vadlo«
T a b l a . 2 8 (continuación)
Esteres (sólidos}*
Nombro dci ficmpuatc p. I. p. efe- _____ ! £ _
B tu t ito de (rallo . . . . 5-o¡tn)í«3ÜKtD de rottiloPtkktó de Jc«ib ..........M-¿idr0]ábeitta*(4 ¿o ttetJEoAcctaAo (Le p.pWflflo .........T r h f lM n to J » sJicrt-jkv ... Benio*K> de í-crc*Jk> ,..Ciaunató de fefiiU. .........<n*hkb%xíbenznito d« adío THlHozeaio de ptkerilo . D í^cnm io de ealcdglkolFtalida ........................^niirúdzuur^ta de rMCüo m-nitrobenzoato de atedio CutabJtto de fenRo .....C ítm o de (aetUo . . . . . . .H-nltn>CDUiiralo de etilo O x a b ta do bcftdlo . . . . . .Kbrccncfocitf&ato de c»e(ilo Ciraonilii de (WViniI . , it .b r tn ta de IBrtJo SJWwtr&be&BfcitD.dii ¿Mki—
'Sklic¡UtO de ¡J-naítilo . . .
H mW70 .. •70 • r»70 .. •7171 , , (Ti
¿Mnn t|Jn • ■ #n 2017371?n 305n 2a a .
- m*ii*h
7*Z• Sí . .fSj ...
Noxobrrdd <<n»pueitrtp-k
fr-GÍtrOÉKltiWA'tO de jretito >nltK4*UcilitO «fe etilo , ,Tríacowto de ílerogíudnol , Ad¡p*w de íedUo . . . . . . . . .Roiraxtó do3_,5 -d WtrobecKailfí de metiloUx u h U de t l i l í ................Orterwfp de ra-crcnlo . . .¿-/iidraa be mosto de tfifO ,DibíSttOa» de xa o n ta d . S-n¡U**ídÍ£Í5»ta de etilo 5*ti(trútaKdbto de roetilg , Suodcmto de (cada JOÍBcetata de hidwrjinera nMÚtzoüntiinato de xuetUoL*cdd» ,,.........fr-hidroxib«ii»ito de aiotjo ¿.nitroMdrdbto de metilo ¿.nUreittfcnuito de etilo Tcftíw U l© de metilo .<•- Tn»iieu(o dn ¡prcjfíilol . . . .^njtrtcíiMinkJtQ d* ««lila
p. «b. ♦C
« l*tt 10C, toe 107 10» 115 115 11 r, 117 llfi Uíl 12) 12S 124 123 I3T 132 137 14a ja 161
♦ Mech(J\ tttoxts tóUdo* d d n enE»Udot e*ooo derivado* (k icJdd, alcoholes y feiwk) (To- Ujl. 20. 2 \ y 43). .
Esteres de áddos inorgánicos
NotrtVe del canqpuCKO p-tfc. T>ciin. Ejp. Nombre de! eompuato p. eb. Detit. Etp.
Witritat Nitrt» de etilo . . . . NitHtCi de ^frajiilff Tíitritír de iaobutilo Kiftitó de it-kutit» , Nitrito de úoamito . Nitrito de mhÚJo •
S itr tla i N¡H*tQ de metilo ..
17 OUSOO'54+4 0.958fi7 0-í£8«
0.911*> 0.830*14
1 W
65 4.232?t
j V i í r f f / u f . contí'nuveiSm.Nitmto de «tilo .......... 87 1.130»Nitrjití» de «.propilo 1(0 1.075»Nitrato do icoDutilo , , , 123 1 ,CÍ)*iNitmto da i'W ü i) i - , , IS6 l.04flNitrato de ,b>amito 147 1,0005» S*JitiatS o íía ío dn w e t i te .......... l í í l.SSim
do etilo .......... 1,172*54
■' A \Tabla» dt ¿«rivadot:
T a b l a
Eteres alifáticos
Nnmfcre del e tn fac t!» p , efc.*c
Deai. Ef?.Pnelod* ioü¿o del 3¿*dkJtri».
bctwo4tode alquilo
E te r r e ti le tU x c o .............................. ............... 10 0 .7 2 5 2 *O x id o d e e d l f J i o ................ ............... 11 0 .8 8 2 %
32 0.9644X ,35 0.719 'X 93
O x id o d e p r o p S lc n o ..................................... 35 0 .8 3 0E t e r v in ileu J ico ........................................ 36 0 .7 6 0E te r c lo ro m c tílic o ........................................ 5 9 1.020'Sl^ -« p o x ib u t» n o ................ .. .......................... 62 ;. 0 .6 3 72 * m e tü fu n n o . i ............................... .. • 64 0 .9 1 3
65 0 .8 8 965
E te r M oprop llíco ................................ ...... 68 0 .7 2 4 122E te r a ie tíl-F J 'b u tílic c ........................ '■ 70 0 ,? 7 4T c trah id ifO iU vano ........................................... 79 0 8 5 5E t e r e tiíc lo ro m e ü lic o ................................... 80E te r d im c tllic o d e l e tü e t ig l ic o l ................ 85 0 .$ 6 ?
T c tra h id ro p ir - in o ............................. ..........8686
0 .9 2 30 .881
E te r **p ro p íJ ico * .................................... ..- 90 0 .7445¿ _E te r c t j t n * b u t n í ^ ................................ " S 2 .......... 0 .7 5 2 2 %
<54 0 .8 8 893
E te r m etil-n -aroD íco * ................................ 99 0.761102 1 .0353%105 0 .8 6 2 , s ,
E te r <r*ccr-dicJoroiD 4tílico . . . ................ 105 1.328%E te r ^ ó lc T o e tíiic o .............................. 107 1.057%
116117
1.138%1.204JÜ
. . .
118 0 .7 6 2121 0 ,8 4 2 4 %122 0.853
E te r i io b v tí í íc o ............................................. >22 0 .7 6 2 ^ 86E t e r ractil-TT.heJCÍlica ............................. ► ► 126 0 .7 7 2
127 1.370XE te r m e tü c íc lo ¿e x ílic o ........................... 134 0 .8 7 5E te r m o n o e tftiu o d e l eÓ IengliccJ » . , , 195 0 .9 3 1 1 %

- 370 Tobfos d# derivado*
T a b l a 2 9 (continuación)
Eteres alifátíco»
N«mV* «Jet « m p o tjto p. rf>. "C
Den*. £s¡>Punto de IuiÜddri 3 .diivltrt>-
fcwwoatode
E te r n-budlico ......................................... 140 0.769 64140 1 .1 7 4 *142 0.772
E te r etilcídohexílíco ............................. 1*9 0 .364E ter dibrornoroctílicc. ............................ 2.201E ter ditfictílico del dictilenglicof . . . . 162 .9 4 4 0 %Eter metjl bencílico ................................ 167 0 .98 IX
163E ter mono-n-butOico del etiien$[¿cDl.. 171 0 .9 0 2 %Eter isnamíluv» 172 0 .7 7 4 % 62Cmeol ......................................................... 176 0.927Eter O '-d ido roe tíJjca ..........................E ter aietllico del dietílcnglicol .........
m160
1 .2 3 % 0 .9 0 9 % '
. . .
E ter etilbencílico .................................... 189 o .9 4 y xE ter n-amílico . ....................................... a 90 « s • » • 46Eter manoroctÜico del dietílcnglicol. . m 1 .0354%E ter monoetílico d c] dietílenjlicol , . . 202 0 .9 9 0 2 %
208 5P,Eter isobutilbencDico ......................... .. 21 2
212E ter Y.'/'-dicloropropiUco .............. 215 1.040E ter monc-n-butílico del dieóJengücoI 231 0 .9 9 5 4 %Eter n-hexllico ......................................... 22 9E ter monoíenilieo del etilengltcol .„ . 245 1 .1 0 9 4 %E ter n-hrptílico .................. .................... 26 0 0-315/1Eter díroetílieo del tetraetUen^lico! . . 2 6 6 1.009
280 0 .6062 9 8 J .0 3 6 5£ 1X2

T o b ía s d e d e r iv a d o s 371
T a b l a . 30
Eteres aromáticos (líquidos)
N«nbre ¿el qompvmío D«\i. Eip. Picritft*C
OtiM dtrir*4.n*c _
Aniiol .............. ............................. 154 0988 81 Dínitro 87Eter metíU«<Tc.síUoo................ 171 0.96$?í 119 Bromo ......... 63
172 o w w 92 58Eter ra e til- í 'C re s íik o ................ 176 0.987% 89 Acido jinísíco 184Eter medí-rM-creif Jico ................ 177 0.9É5X 114 Trinitro . . . . 91E ter ft-proptlfcnitico ................ m 0.949 * - «Eter e td ‘0-cre*í!ico .................. 192 118 DinittO . . . . 51Eter e tü -m < ;e riU co ................... 192 115 Acido m-etoxi*
betuolca , 13?Eter eüJ^creJÍlico ................... 192 111 Acido ^etox i-
benzoico 195194195 9<i200 98205 89
Veratro! ....................................... 206 1.086‘Y- 57 D ibroiw . . . 92o«ck>rofenetol ................ ............ 208 r r • • • • • » Nitro . . . . . . 82
210 0 950 112g-cforofenetol .............................. 212 Dinitro . . . . 54Eter diroetítico de! rescrcinol 214 1.580% 58 Difcromo . . , 140
Trini i r o ---- 124Eter metUtlroilico ..................... 216 0.954ÍÍ Trímero . . . . 92«-bromoaniíol i ........................... 218 - r . - - • r r 106
223 0.944X - - ►223 1.494% 88224 98229 47
Sifrol ............................................ 232 u o m 105 Pcntabram© . 169Anetol ....................................... 235 0.989a# 70 T ríbromo .. 108Eter d ie tílko del rcBorcírto) ... 235 53a-yodoanisol ................................ 240 1.800 , , , 95Eter metílico del e u g c n o l---- 244 1.055'X 114 Tribromo ... 78
245 96Isosafrol ....................................... 246 1,125'Jí 75 Tribromo ,.» 109
252 1.073 110 Dibrotf» . . . 58E ter metü-cc-tiiftllíco .............. 265 u o m 113 Bromo ......... 46
267D'nitj-oanijol ................................ 272 1.254 ♦ « * O-ackrduLa 225Eter c til-a-naídüco ................... 278 1.074 100 4-brcmo . . . . 48
282 1.064 44 I-bromo . . . . 66

3 7 2 T a b la * d » d e r iv a d o ?
T a b l a 3 0 (continuación)
Eteres aromáticos (sólidos)
N o o b t r ¿ti COa3(X3j»rtO p . *■ Pícrntú*C
Otro» d«riva<(o("C
Veratro] , ....................................................6-ckxrofesetol . . . .. .............................Ac*«oI ........................ ..E ter ft-¿miL-2-aaítl!ieo ♦Eter isoarail-2-níiftílico .....................¿-yodofccrtol .......................................E ter J tn U íc o ................................ . , , -
E ter P-hromoctilfcnUico ...................A p ío i .......................................................
«►•meeojdbifenttoE ter itobutQ^2*QaftíIkco ...................m-níti'ofíaactol ..............................E ter írc-butíi-2-naf til i co ................E ter K-butü-2 - r¡ i f tí tico .....................E te r etU -^-w itílico ....................m^mtroiuuxol ................................
Ett i tm fü ic o del párogalol . . . . . .E te r bcpropil.2*i»ftjlico . . . . . . . .S te r ^*propíl“2»nsftílicQ ...............2.4.6-tridorcfenetoI E ter dietflico del e n te c o ! ................E ter T ri metílico del pirogaiol . . . . E te r monotoe tilico de la hidroqui-
nooa .......................... ....................... ...........
ájfflatllte# 4 í M hl4re(julnon«íMWKBjll ii i] 1111 > m i ■ i > i 11 ■ < •2 .4 .6 -ü id o ro én W .............................¿-nitrofc&etolE te r roetiJ'^Haaítflico^. . , ................Exet dactilico de l a h id roqu inona ..2.4.6-tribromofenetol 2,4,5-tríbfomofcix;tol . . . . . . . . . . . .E ter ‘taa c& a -u ítílíie**-i»eto¿bifew lo ........................2,4,(> ta b re moaniioJ . . . . . . . . . . . . .D íb e^o fa ran o . . . . . . . . . . . f ■2,4-dLcatroíumol .............................E ter beadl.2 'naftíJico . . ...................5¿Winitroarú*ol . . . . . . . . . . . . . . . .E ter dibcacilico dc-,Ut hidroqi” noi>a
21 57 Nitro .......... 9521 Dinitro . . , 5422 '70 TribrOmo , . 10625 6726 9127 • » » N i t r o .......... 9623 110 Dinitro 135
Dibromo , . 5432 « » a Bromo . • >. 5632 • • • Acido ajñó*
líoo, . . . . 175 .32 s . » Nitro . . . . . 9533 85 . . r . . . . . . .34 k r r Picrato de
lene ti dina 15834 86 • s » . «»»«»»36 67 . . ............... ..37 44 Bromo . . . . 6639 . . . Picrato de nt-
anisídina. 16&d.39 » * *40 9540 8143 • •» Dinitro . . , 10043 71 T rin itro . . . 12247 81
3354 l i nM 119 Dibremo ► ► 1426& 1 i i 1 • • 1 4 i 4 1 1 4«0 > , 4 Dinltro . . . 9560 F en acttin i . 13672 m Bromo . . . . 8672 Trinitro . . . 13072 • Nitro . . . . . 7973 ♦ 4 477 • • n85 ♦ 4 « 987 ♦ 4 •87 94 Nitro .......... 18295 * 1 4
102 123106 s • •127 « ■ • 33

Tablas d » d trivados ¡ 373 C
T a b l a S I
Haiuros de aiquenilo
J&emzdoi
Norwbr» del cmupueari» p , «V- c
D e n , Eip.Auüid»
“c
Haloro-dcJiiqc íd l.nMrcurlo
*C
Bromuro de vinito ......... . 16 1.517% 1042-doropropeno ............. , . . . 30 0.918X1-clorcipropcno .................... , 36 ........... J HCloruro de a U lo ..................... 46 0,938 *2-bromoprOpeno .. 46 1,362Yoduro de v in í lo ................ . 56 2-080ÍÍC lo ro p reu o ................................ 59 0.956%UbrxráwpropcLnc. ..................... 60 1.428 114Broznuro do a l i l o .................. 71 1.396 +Y oduro do a l i l o ..................... 103 1.648%g 'c la ro s itli 'cu o ..................... . ► 199 'N.l 12’ 115P'bromocjtírttna ..................... 221 1 .'2 7 1(5 91
• CretoaaiMH4A ll+*C.
T a b l a S2
Haluros de alquilo y cicloalquilo
Derivado*
Nombre <W «rapucJlo p. «b. DC AnilM*
*G«•ofcíUlLAi
H*hirc de tDuit"
toercurio 'C
Cloruroj d*Etilo ........................................... 12 104 126 192
36 103 , M 9746 92 121 14751 128 14767 108 12966 109 12577 63 112 12785 131 • » , 118
7 > r- jin u lo ................................... 86 92 138100 108 111 86 •107 96 112 110134 69 106 125142 146 188 164160 57 95 119179 117 166184 57 , 91190 97 * * * i195 133 - 1,202 . . . . . .

i »
í •
n»
r »
f
3 7 4 T o b ld t d é d e r iv a d a s
f
T ab ta 32 f¿^níin««cíók)
Halurüs de alqaüo y cíclosjquílo
NofTibM dd eompuesio y, íb. *C Aailld»
’C
Dirivadai
■c
Halará de alquil-
roe rcvró *C
BotojIo ....................................... 207 . . .¿-cforobencilo .......................... 214 166
241 . « .223 , . .
Cetilo .......................................... 299d. . . . 102
Bromuro* deM e d io .......................................... 5 114 160 160Etilo ............, .............................. 3e 104 126 193
60 103 •Í2 Í
93re»propí]o ................................... 71 92 .138¿«•-butilo ................................... 72 126 147 .Síc-butllo ................................... 90 106 129 39Iiobutólo ................................ 91 109 125 55« -b u tilo ........................... ........... 100 63 112 129
108 92 J38Neopentiío ................................ JOS 126
¿ÓIsoamilo . . . . , .......................... l i e 108 111113 93 104
3-pcndlo .................................... 119 121 r . .129 96 112 122
C idopcntilo .............................. 137 4 * ► , , .>57 69 106 116
Oíclohexiío ................................. 165 146 188 ■ 153174 57 95 118196 117 166 119
n - o c ü lo ....................................... 20* 57 91 109a-íerúletílo ............ - ................. 205 1332-etücícloiisídlo . . * , .............. 212 -»< • < t
218 97 • • < 169220 *«■ 109
Y o d u ra ¿4145‘M e ti lo ......................... * ............. 43 114 160
72 104 126 182IiopropUo ................................... 89 103 125ffr-butilo ----- . . . . - ........... 98 126 147
102 92 121 112119 108 129
" 72Iw butilo . . „ .............................. 120 *09 125ter- a m i to ..................................... 126 92 138 > ■ r
130 63 112 U 7142
Sos i i i 122148156 96 112 110180 69 106 110
ít-híptílo ..................................... 204- . 57 95 103Octilo ......................................... 226 1' • 1 • •
T o b ía s d e d e r iv a d o s 375
T abla 33Haluros —'Derivados ¡lalogeuado!» de liidiocarljuros aromáticos (líquidos)
Nombre d<i Cúmpuélto P- A."G
Deriv¿<tos
PiutJ&Cta de nitfláín Diversa
Pcxíción P.Í.*C Coronen* o p. f.°GFluorobenccno . . . . . . . . 85o'fluorotoJueno .............. 114
115117132 2 ,4 52*
Bromobeyceno ................ 157 2 ,4 75 Ct-naí Calida ......... 161O'cbrQtoliMnú . . . . . . . . 159 3 ,5 63 Acide» o-doroben*
140162 4 ,6 91 Acido ra-d&robcn-
158¿•dorotolueno ................ 162 2 38 Acido ^-ctoíoben-
24?w-dicJorobenceno ......... 172 4 ,6 103l-cloro*2-ettlbenccuo . . . 178 • % % Acido o-cJorobcn-
140o-diderobencano . . . . . . 179 4 ,5 110
181 3 ,5 82 Acido o-tífomoben*MICO ................ 147
pH jtoroetilbenc^no......... 182 . . - . » . • »»4 « • • M 4 •J.83 4 ,6 103 Acido tfi-brcwobeji-
2OIC0 1551 •dorú'3'etilb<»nc«tto . . . 184 Sé* Acido m-doroben-
zoico ................ 158¿•bromotolueno .............. 165 2 47 A ddo ¿>bromobeñ'
TOICO ................ 251Yodobenceno , <......... .. 188 4 171 ír.ljroniDyodobcnc«*
no ...................... 911 -cloro‘2 'iiop ropilbenco
191 * * l • • % A ddo o-doroben-ZQtCQ.............. 140
2j4<t¡c]orotolu<;no ......... 195 3 ,5 104 Acido 2,4-didoro-160
o-bromodorobenceno . . . 195 * 1 4 %wm1,3-bromodorobertcrno < 196 . • * * • • ............................. .. ♦ 4 4£*dorocumcno ................ 198 A ddo p-clorobCíV-
zotCO . . . . . . . . . 2422,5-diclorototueno ......... 199 • • » Acido 2,6-dickn'o*
b«n2oico............ 130l - b ro m ^ ^ t i lb e a u t io . 1.99 , » » Acido ©-bromoben
zoico ................. 147*rt-yodoto!ueno 204 * 4 4 Acido m-yodober.-
186l*bromo-4-ctilb< ncet»o 205 A ddo jCr-bromobtU'
2Oigo 2511 - bremo- 2 ■ ieopropilbcn-
210 * - . Acido o-bronwbtn*147
o-yodotoluíno . . . . . . . . 211 6 103 A ddo a-yodoben-162
¿-yodotolucno . . . . . . . . 211 A ddo p-yodoben-265
l s2,4*tridorob«ic«rt<> - - 213 5 56rn^ibrom obcnceno . 219 4 61 ........................... .
219 Acido ¿-bromoben-2 0 1 C O 251
9 TimWín ie rep4 ti»n formii uvcf*»ldhl4 <|4í fondeo o 2?*C y 43'C-

3 7 6 . T o W a i d « derrvopdoj
T a b l a 33 (con tinuación)
Haluros
Derivados halogenados de hidrocarburos aromáticos (líquidos)
EMrivjriol
Nombr* dd eo&if>»K=rto p. e!>. ♦C
4Ífc»CÍ6l Dívfnwf
Pcaci6aP‘í
Cato pu«Mto*<?•
o-dibromabenCono . . . . . 224225
4 ,5 114
234 ?, ? 97 1432 ^ -dibrocac tolueno — . 236 Acido 2,_5^Jii>romO'
b e n z o ic o ............ 1573,4^ÜbroznotnlDQao . . . . 241 . . . Acido S,4-dibroiDo-
233o-brosaoTodobcnccno . , 257 ► < * 4 4 4 »< •
............ 263 4 ,5 160 P íe n lo ................... 137aixrewtKaiaítakao . . . . . 279 4 05 , , > ■ , 4 4 4 * 4 4 4 . * * ,
297 A ddo o-broma benzoico ................... 147
1-yodooAÍtaleao . . . . . . 305 Picrato 123
Haluros
Derivados halogenados de hidrocarburos aromáticos (sólidos)
Dflfívxtoi
N—nbci <td confKfXn p. e.’C
t i t a d i t PÍWKt
?c*ie»6a p .í.r e
GotapvM«>
/♦broarotohieao < 28
2-^Iázpbiferulo ♦ ,, * ♦,< , 34
¿-yodotohit&c ............. ........ 35
tt,^d»do?oiuiítalcno . . . 36m-díyodobe&ceao ■........ ........ 401.2.4-?ribrcra»b<;Dceíx> . . 441.2.3.4-tctra -’Tfot>enc« c í* 46 l^^tetrtóorobeiKXiKí 51
S-yadon^S!^^ . . . . . . 55
47 Acido í-brom obtiv2o i c o .............. .... 251
Acido ^dojrobcn*zoíoo ................... -140
Acido >-yodoben- x o íc o ................... 265
5654
Anilida .................. 170

T a b l a 33 (c o n tin u a c ió n )
Halaros
Derivados halogenados de hidrocarburos aromáticos (sólidos)
Tobías d*
Wortbre del CODWK*© p.í.’C
ProduCU-eícwtclín
Ptaúáón p-
cloronaruleno . . .K LronionaftaJcsno , . 2,2 ''diclorobLfemlo .
?-fluoronaítal«Jio . . ,3,5-lridorobenccna
1,2-dibronKinatbilerio .
ú-bromoclorob«ncciK] . 5,5-dicJorO'^'XÍIcno ♦ « ,
H-cltirobifcnUo ...........2,2 'rd ibí'ons obif« nii o . , J(4-dibix>manAÍUle*o .
nt-dífluorobccccno . , , pr.ntadorobenccno * , ,1 ¿,3-tnbrom obfinccna p ’ü ’iifuorobfinccxw . . . . p -d i bromobe n ceno . , 4,í.h ro n io b ifc m k ) ..........¿-broawyodobepc^no . a-diíluorobcriccno , . . .
ibifeaik» -tríbromobcnccno
4-yod o 1 fl£*¡/xdiyodobeuceno
1,2 .4 3 -tc tra c la r o b c n c e n o 4 ,4 -a ic iorcb ífn n ito ► ►.«
4>4'-<líbrttUG b ifen ilo . . .Í ,2 ,4 ,5 'te trab rw n ob cn cfl-
no ..........................-Tctwdarooaftaleno . , .
Hexjkdorobenceno
565960 60 63 67
6768
778182
8286.8788 89 89 92 92
114120129
136149
169
180182
229
1 ,S 175
68
a
3 166
D«riv#di
DítoíOj
Picraw............... 01A n ilid a .................. 170
Picrato .................. 101
Acido 3,4*díbror&o- fOUco .............. 196
Acido 2.5-diclaro- tcrcítáUoo 306
Acido 3>6-dibrciP£>- f tilico . . . . . . . . 135
Dicloruro ........... 102d.
í-yodoni trobenee- no I7t
Acido /-clorobea- aoico . . . , , 4 4 . . 242
1,3-dicloroELaitíJo-po .....................* 61
Clorando 290

»
m
r *
i *m
3 7 8 T a b la s d e d e r iv a d o s
T abla 34
Haluros
Derivados polihalogcnados de hidrocarburos no bencenoi.dcs {líquidos)
y »r
L»
[ •
i »r »i
f »
m
Nombre <je[«Mtpual» p. «b. *c
| JDea*, Esp.
36 1.693% . . . . . .42 i . m % 1.4237
Dácloroctileno (cis) ................................ 48 1.265 'X 1.4490Perfl\4oro-n-hfc*ano .................................. 57 1.699% 1.2515%
60 1.291» 1.451860 i . ie o ’Jí 1.416661 1.504% 1.4467
Cloruro de rt-nitrolienial ..................... 65 ......... . . . . . .2,2-dtcloropropano .................................. 70 1.093 1.40931,1,1-tricJoroeiano ...................«............... 74 1.325^ 1,4349Tetracloruro de carbono ....................... n 1.591% 1.4607
82 ........ .........«3 1.657'X
Cloruro de eLifeftO .................................. 83 1.256 *' * .44+3Tfícloroetileno ........................................... 90 1.440'% 1.47S2Bromuro de m e ti le n o .............................. 96 2.498% .........Cloruro de propileno ............................TriflüOruro ae íxn ia l , „ .......................
93 1.166% 1.4338102 * - - * r r .........105 • • « • % %
l*bronio2<lorDet*nc> ............... ............. 107 1.689'% . . . . . .Bromuro de «tílideno ......... «............... 112 2.100% 1.5-12 31,1.2-mcloroelano ..................................... 114 1.457% 1.47112,3-diclorobutauo ..................................... 116
121 5.63IX i .50551,2-dÍdorobutano .................................... )24 • - * - r rCloruro d e trimetílcno ................. 125 1.130%
130 2.178 1.53791.1.1.2-tetracloroetano ♦.........................1.2-dtbronKjpropeno
131 M M f |132 2.008%140 » • . . i .
Bromuro d e p ro p ile n o ............................ 142 1.933 1.5203143 1.593^ 1.4708
■í'tctractoroelano ............'. ►«........... 147 1.6! 4% 1.4942Bromuro de isobutijeno .......................... 149 1.759 1.509D ibrom o'I,2-butcno-l .............................. 150 1,88?>í .........
15! 2.904% 1.5891,2,3-cridoropropano . . ; ......................... 155 1.4171,3-dibromapropeno ................................ 356 2.097/» 1.5*8*2,3-dibro8K>butano , .............. 353 1.830

T o b ía s d« d e r iv a d o s 3 7 9
T a b l a 3 4 (continuación)
Haluros
Derivados polihalogenados de hidrocarburos no bencenoides (líquidos)
Nombre del «ompcwio P- r t ,“C
Dea, E#p. fco
Pentacloroctuno ................ . . .................. 161 1.693’X 1.5041,2-dihfoiaobLttario ................................... 165 1.820^
165 1,973^ 1.523169 1.877*' 1.54B169
1,3-dibromobutano ................................... 174 1.807 1.507180d. 3.285‘>; 1.7422
1 ,1 ,2 'tribrom oetarto .................... . ........... 189 2.6211*55 1.5933\ ,4-díbrornobüCano ................................... 198 l.&47% . • » «
200d. 2.971% 1.638212 1.5515
Cloruro de o-clorobenctlo ..................... 214214
Cloruro de m-dorobcjicílo ................... 216 1.2695?*'1,2,3'trihronx.propaiio ............................ 2 i 9 2.4369Í 1.584Benzotridoruro ................................... 220 1.3S0% 1.5573Bromuro de pentametíieno ................... 221 1.706'S1,8-dibromooetatio ..................................... 272 1.468’X 1-501 <15’C)
Haluros
Derivados polihalogenados de hidrocarburos no be/iccnoides (sólidos)
Nombre de) compuesto
G [oruro de 0-dorobencilo . . . .Hepttcloropropajio . . . . . . . . .Bromuro de o-bromabencilo • . Brompjv de w-bromyljcnciJo . . Cloruro de p-bromobenciJo , , Bromuro de ^-bromobcncilo . . Yoduro de ctjletK ».....................
Tetrabrom uro de carbono . . . 1,1.1 -tricloro '2 »2-bis ( p -tlo r o í c
n il) eiano ( D D T ) ' ................Y odoformo ...................................
a^bccictorobenceno . . . ¿•tetrAmetildidoroctAcio í'tetram ctildibrom oetano H exadoroetóno . . . . . .(S-hexadorobcnceno . . ,
P.F.‘C
292930 41 50 63 32
92
103119
1575601691673i0
I)cri\ulc4
Acido ^-dorobcnaoico .
Acido o-bcMHOberuoico Acido m-bromobeníoico Acido /i-bromobenzoico Acido ít-bromobecuoico E ter d*-$-rt&fttlíco del «til
enslicoJ ....................
Compuesto de adición co q u ln o lc ín a ................
p. í.

3 8 0 ' T a b la » d o d e r iv a d o *
T a b la 3 5
Hidrocarburos aromáticos (líquidos)
D tsw £ 9 .
P todaetodQD¿l/4£u>* A ck k
Pkrntó
JC Poóciánlo ica *C
Ben ceno .................................. 30Tolueno . ....................... . . . . 111EtübcDoeno ............................ 135¿-XÍIcno .................. 13?m-yálnao ............ .. ►.............. 139O-XÜCLUO - > . • - ........ 4 4 4.. 142»ItopropUbenceuo (eumeno} 153«-propUbeoceno ► ♦ < . . . . . . 15$M eánleno ....................... .. 164¿HfftUtolueoo .......................... 162o-eái tolueno .......................... 165Pteudociu&cno ..................... 163/«/-butUbenceno .................. 169Itotm tübcocciio ................... 173«f-butólbenceno ........... 173#»-<£n»rno .............. 175^c im en o ............ 1751,2,3-trinietílbenceno 176H idriijdeao .................. . . . . 177xútiCDO .............................. - 178Indetto . . . . , ... ....................- 180fR-propiltoiueno ................... 182TO-&etilbettc*:oo , , , . .......... 182w-buñl benceno ......... .. 182^ o ro p i] tolueno .................. J84o-¿ja o l benceno ............ 184¿süetilbcuC cno . , *»............ 184no d u reao .............................. 195n-amilbcnceno ....................... 202m -diboprcpílbeoceno . . . . . 203o-diUopiTjpílbc nccao . . . . . . 204P ir ib n ite n o .............................. 204l»2j3r+“tetrsm etilbcnc«io . . 205T e trá lira ................... ........... 206¿MÜiiópropUbexKcno . . . . . . 2101 ,3 ,5 'U ietilbcB ceno......... .. 218nroexilbeacecLO ..................... 226
0.874 1 ,30.881* 2 ,40 876’íí 2 ,4 , 60.866% 2 ,3,50.8? 1% 2 ,4Ú.890X 4 ,50.875# 2 , 4 , 60.861 .........0.869% 2 ,4 0.8610.881 ..........0.895 3, 5, 60-867 2 ,40 .853 ...........0.862 ..........0.861 ..........0,857 2 ,60.894 ..........0.965 ..........0 .877 ...........1X100 ........0.8623 ..........0.860 2 , 4 , 60.862 ..........0.859 ..........0.881 ..........0.862 ................... 4 ,60.866% ........0.856 . . . . .0.877 ..........0 .901% 5 j 6 0 .905 5 , 6 0 .971 5 , 70.857 ..........0 .8 6 3 2, 4. 6 0 .878
89 127 8470 137 8837 122 96
137 132 9083 125 9171 178 88
109 133 , , ,# 4 125 10386 211 97
185. . .
62-.4
54 i 23
*62 í 1497
157 213
i 76. . .
176 « • .95 153
ióé i¿9• • • ♦ V V

T o b íc i d e c f o r r /o d c s 3 8 1
T a b l a 3 5 (continuación)
Hidrocarburos aromáticos (líquidos)
fíorebw p. cb. fC
Deoi, tai.Producto do
niinciin A<ldouail-beo»B?'lCO’C
PicruU.■cPorieián
t !Cidohexilbenceno 237 0.955 4 58 é 4 .l-metUnafuJcnc ............* * • 240 1.001'X 4 7! í 69 141I- ítiln a fu ltn o ....................... 250 1.008 r r ■ • ♦» 78I,4-dimetíJ«aftateno ............ 263 1.008 « • V • é 140I jl-d ifc n ilíta r io ............ 270 1.003* . .. . . . . . .
Hidrocarburos aromáticos (íóiidos)
, Producto flr »¡tr*dón Acido«rail-
GC PotiaÍDIx»*releo *c
DjfeniimcUtno ....................... .........26 2 ,4, V , 4 ' 172B>mecilnaítaleno ..................... .........32 1 81Pentaraetltbcncefto ................ ........ 51 6 154Dibcnc.no .................................. .........52 4 ,4 ' 100B íte n ilo ....................................... .........*7(i 4 ,4 ' 233D u r e n o ................................................79 3 ,6 205Nft£<aler»0 ............................. 8 0 1 51m -difenilbencezio..............................85 ................... .Trifenilmetano , ............ .................92 4, 4, 4" 206ACénaíteno . . . * ....................... ........ 95 5 101Retamo .............................., , . . 98 ...................................F«m ntreno .............. ............... .........100 .......... ..2,S-dimetilnaftal«u> . . ......... .........104 .................................P lu o rc u o ....................... „ .......... .........115 2 ,7 199Pireno ........... . . . . 148 .................................CLa’J>jnaft¡]o ........................... .........160 .............................. ..H exam etilbcnceno ................ ...........162 ................ 176p.fj'-binaftílo .....................................188j-totrafeniJetarto . 211 '- 4 , 4 , 4 , 4 143p-terfenilo ♦.........., ...........................213 ................................Aatraccoo ................................ .........216 .................................Cris«no .............. ................... ............. 251 ................................ 214Piceno . '..............................................364 ................................
224263172
98
227
116131
149
16112414412484
220145 170 184
138273

382 Toblft* d a derivados
1 *
£
T i
m
T a b l a ” 6
.Hidrocarburo*, parafinas y cicloparfcfinas
Neopeniano ................................Ijopen tino ..................................P entano .........................................CiclopcnLano ..............................2* tn e t i^ e n tu » .................. . • •3*m eúlp«ntano ......................««hexano .....................................MctilcJclopfntano .....................Ciclohexano . , ...........................! ,1 -d iro e tiltid o p cn tan o ............1-mctilhexan o .3.2-dimetilcidopcntano (trens)3-m ctílh««kno.............................3*Ctilpcntai>o ..............................it-hcptano .....................................2,2,4-trimeulpeniano ................M ctilddohew uia ..................... ..1.2-djtfie tí3eick>p«rtano ( c h ) .fc tílc td o p ttítan o .........................1,1,3-trúnetÍlcidapentano . . . . 4'íUetühcpUmo . ............ . ..........2-meliIhepUno 3-ctilhcxano... Ijl-dimctilcíclohcxauo ..............M -dím etildcIohcxana (trans) 1¿-dírnetSkicIohexano ( c it) .n*ocl*no ....................................Rtilcidohtxano ..........................n - N o n a n o .......................................... ...A.£ropílcklohcxanD . . . . . . . . .DiisoainJlo ..................................../f-m cntano ......................Cictononatio ......................... %. *«'dec&no .......................................D e c a lin a f ( r a t i s j ........................D c ca lio a ( < ¡ t ) ..............................n -u n d cean o ................ ..CtclocUjcano . ............' ............n -d o d ecan o ..................................o-trfdccano ................................TctraJecanc*........... ...............P c n tad c ca n o ......................... . - •KfxsdctJnci ............... .........
p. cb. ’C
Día). Efjj.
9 0.6135)31 0.633’#36 0.63150 0.75060 0.65363 0.66463 0.66072 0.74980 0.79088 0.75590 0.67992 0.75192 0.68794 0.69898 0.68499 0.692%
100 0.769100 0.773104 0.766105 0.7.4R118 0.705118 0.693119 0.7E4320 0.791119 0.763124 0.783125 0.703132 0.783149 0.717157 0.795158 0.735%169 0.796%172 0:8534‘?í173 0.730185 0.870194 0.896394 0.745rX201 0.8577?;215 0.755'JÍ236 0.7563254 0.764271 0.7G9207 0.773
1.3513(0)1.3551.35701.40931.37161.37641.37541.41001.42631.41361.38551.41201,30871.39341.3851.39161.42351.42221.41981.41121.39$51.39501.44401.42901.42001.42971.33901.43321.4051.43701.4081.4371.4328£1.4151.4&9?1.40111.41841.46921.42091.42561.42893.43101.4352
T a b la 37
Hidrocarburos insaturados
TcrbTc» de derivado* 383
N om brí p, tfc. P - ! - C
C ejo!, Eíp. no D ttrvaJo p . i .
M etil-3'butcno*l , ......... 21P tn tad ltaa-1-4 . . . . . . . . 28Butíno.2 ....................... .... 27P ín ten o s .......................... 30Metil*2-buteno-l . . . . . . 31Isopreno . . . « . ................ 34Ptnteno-2 ................... , 3 6T nm ctik tilcno .............. .. 38P c n tln o l .......................... 40Cktepentadicno ............... 42Pentidíeno-1,3 . . . . . . . . 42P entadJenol,2 . . ............ 45C id o p en te n o .............. ...... 46Pefltadieno-2,3 48Pcntíno-2 . . . . . . . . . . . . ’ 55
................................. 59Hex«no-l .......................... 66Hcxcno-3 67Hcxeno-2 ....................... 68H «drto-I ............................ 70Tetram cdletiletio . . . . . . 72H c»uri eno-1, 3 , 5 .............. 78.Dihidrobenccno-1,2 . . . . 80Hexadseno-2,4 ................ 81HcúnO‘3 ............................ 81Cidohexsno . . ................. 34Heptcno-1 ................... .. 94Hcptino-1 .......................... 100Diisobutíleno < . ............ .. ♦ 101¿ ’-ttfrahidrocoluenc» . . . 103
A^tctrahidrotolucno . . . 105
A’-tctrahjdrotoIueno . . . >11C itfo h rp te jw ..................... 115Octeno-1 ............................ 121Octíno-1 ............................. 132F en ilacetiíe tio ..............* ► 140Estira no ............................ 146
0.660% 0.661 1.3887 ....................... . .0.688 1.3893 Tetrabrom ura 2430.641 i.3710 ............................0 650 1.3778 ...........................0.681 1.4219 ...........................0.651 1.3789 ...........................0 .668^ 1,3855 .. ........................0.688 1.4079 Mercúrico . . 1180.805*% 1.4470 ............................0.6803 1.4309 Tctrabrotnuro 1140.693 1.4209 ...........................0.7 7 4 ^ 1.4218 ...........................0.695 1.4284 ...........................0.710 1.4045 ...........................0.690 1.4010 ...........................0.673 1.3858 ...........................0.682 1.3942 ...........................0.681 1.3928 ...........................0.712 1.3930 Mercúrico 990.728* 0.710 1.4330 ...........................0 841 1.4740 ...........................0.715 1.4493 T etrabrcw uro 1850.724 1.4115 ...........................0.809 1,4492 Acido adípíco 1520.697 1.3998 ...........................0.750% 1.418 Mercúrico . . 610.715% 1.4082 ...........................0.799 1.4430 Acido fl-metíl.
adípico . . . 930.800 1,4426 Acido cc-metil-
adipico . . , 640.809 1.4496 Nitrojocloruro 970.823 1.4580 ...........................0.716 1.4088 ...........................0.747 1.4172 ...........................0.930 1.5524 Mercúrico . . 1250.925 1.5483 Díbromuro . , 73

3 8 4 • YaWcw d« óeflvodo*
T a p l a 37 (con tinuación)
Hidrocarburos insaturados
M Vztt-Up. W9 J}rtrfr*<fc> *4av :ü-
AlübtóiCííio . . . . . .í-cjtoíína ...........IiopíopemTbfrncttio
D tcfcíio-1 ► ♦ , o-T7>etJ«íireD.o
Silvestre DO
Ind«ru>
Uadcct&o-LDodocamo-l . . .
D iÍBnilbutadieno - 1 j 4 (tráttf)
Diicnjlbutadiffno-1,4 ( c is) 1,1,2-trifeniletÜeAo
T4fi 113T 4? 0.7291.V5 0.858 1.4653 Djbromuro < •1S7 o . m160 42 0-822 1.4621 Dibrom gro . .165 0.910 1.5386167 0.798 1.4706
0.900 1.542169 0.S97 1.541171 0.741 1.4220171 0.916 1.550 ............176 0.846JX 1.4727 T etrabrom uro176 0.85 l’K 1.4774 T e trabrc nw ro177 0.914% 1.5143 ..........., m . • •180 1.000 1.5710 Picrato181 0.854’X 1,4730 TetrabromufO133 0.750 1.4261 ............ .71? 15 0.998 1.5740213 0.758 1.4300277 a 1.033'K 1.5967?Sfi .......306 125 0.970'% Dibromuro . .
350 143 % % % • % ...................70 -.«. s - - « -
, 4 , 73 . . . . ■............................
164
89
10+135
9E124
2 3 7
Tobfai do derivados 365
T abla 38
O e to iu s (Jíqu ídas)
Dtj tyjiívi
N ombí« ¿el cofiipucUo p . d»,>c Oxiroji
°G
S m í-
OOflfl, ----- l í — ,
Fenit*bJ4ra<tonaCC
Z(4^3W.troíejUJ-
t i d n z o u♦ c
f r m t-hWrtn-ona
* rAcetam .......................... 56Met'úetilcxtflPa .................... 80Met¡Jvini]<c<onj ................ 80
»ÍW*ti|o ........ ............ 8fcfe^CuopropJlctKrfia .......... $4
........ 102Dktüwtcnfi ........................ 102PiQftC&tCM ............................ 106M^cilbatiDtilcetcM ............ 119CtúftttteCon* ........................ 119«^ttMíxloTowstona .......... ■ i ¿0DíiiznxKrpllcetOQa .............. 125M táí-H-tiUtliCetona ........ , , 129Oxida d« cnailD o.......... . . lMCicJapcBtujloffÉ .............. .. 131Srosxwetioji» ................ .. 13$
AcctÜaccton» ...........ÍS9
2-joecflaclop«i«iuiúB» 159Dl-*‘J*rop[leero'na 345A«iofoa ............................. 145Hldfoxiawttma {bííwI) . , 14&Mclil-re-ñW¡ketMÍ* . , ........ J5!CfclcllíwmoQn .................... J55Dl-rtr-fcüüteetoot .............. |Í2ü-wdileklofcjaBon* . . . . . I6íAlrahol dU m onj .............. IDiUohviilcoiona .................. 168J-Wsúleictí'hcajiofia ........ 161)4-nteltkicUibCTGmaiiR 1§9Mctil.n-fa«dlcotof]« , .......... 172Mutilciclobcjiiltttcin» ........ lfclCjctobeptarvoeii . . . . . . . . . . 181
..................... 187
59
{ (mofla) l
&8es74.w
344?4»56X
349
7i'w
‘435 7
2lú" ñ‘«óS3
167 146 141
m
1131101 »
ISSi« ¿ .163160m154205
18*133 1 » 1% 127 1 É€ 84
105
iáil&üisaJ22177163*5
42
24S
50
2 4 3 d .IftJ2 0777
1 »H7
S1511714415612595
85106mJ42
¡ V )l(dl>209j
75315129 «9162
13715992
155130 55
140m41
152129
117 14l t »
BS1M154
l_

386 Tobías d a derivado*
T a b l a 3 8 (continuación)
C lo n as (líquidas) t
1H O utb e c <3cl C0TOpH<«O p .e l> .
, O x Jo u iJ C
S e m i-emtfcfl-UUU♦c
F m i íUl ú d t i -z e o aIS _
A entorilo < «t6o t . . . . ....................Butiroío* ..................................... 19»Fo/ijrta ........................................... iMA«WÍ«ivMia ................................. 200 0.túyi>n* ....................................... ¿02 ftníntwia , ............................... 2P7fKifoKMia ...................................... 214MetH-U-ticnifcettiiu .................... 2J4 Mc<5f -<DpfMton<L ..................... 21ftMctiltKBciIcctan. ....................... 2162-hjdro>cja«[óíeflimi ............... . . 216PropárfínorMi ............................... - - ?MeiQ-ni'tó))ice(ocia ............ 220I«©buf¡rtfei*Win ..... ............. "'t
CarwMa ...................................... 22JCii-n*«noílMtoíji ......................... ''-'tEtiJbcncitcefcid*. .................. 7KMptil*p>tolilceuina .................... 226m-d<ifCí«tofefion» ............... . i ¿j>l>«3p!lfcfúfcttOfi» ...................... 250í-doro*«tofo*»* ..................... 232Mctif-P-leni[¿ÚlcetOt\a ................ 235 w-uirtwi»c<[«ícnjjru .................. 240«.VatwoíeiWDa ............................. 242o>nk«t0jein<:ct0ri:j«>ra .................. 243 Aceto fó£S(tl£jBO ........................... i4 J
........................ 250Dipnoft* ....................... . 5fS
(di) 137 (di) 220 (di) 320
■4S C¿6199 ió í
35 174y¡ 181 " úK <*B1 w S5
117 210 11070 J98 W61 2065* 174 - . »*7 200 lM
¡ 8 } ‘ ■81 73174 • *♦
74 210i 0672 142
135¿S 21»t t 232 1 »50 1W 20095 201 1X465 142 *• i
V.%’íá lóG • if.2S3 lflj 114
fxín í 301 ida/i 7SJ 151
-3L.

TobJos d e d e r iv a d o » 3&7
T a b l a 3 3 (continuación)
Cetonas (sólidas)
Derivada
Nesrtifc dil corafitcno p. í.‘C
V. «*»->C 0*Lm»
'CSecni-CJLTbj.2DJL»
°c.
Ftoil»hürt-tíKJjl‘C
2,+*)iní-tratoii'UrfrxwuiaC
itífoenJIc^ona ..............JfúWrt* -“ i..................l+meÚUQtuAt**** .........l5-W¿rtX“*“ wítJ>ona ......Vp¡bcxit«<royi ...............*—-•» ¿.naltdeefoBa .........
dtce<orU ..............„_jprcpjokaona .......pEM)li»ceti»ííIV)*ia -......
:»n& .........- . . . .-I ..................ClurMMloiv» .......OM'udMV> i . . . . . .......
Jtoav» ........... .,^ ._ j!» a a £ < le í« o i> J V » (« ■ > -
Kimt^oeclaíeOOfti .......|jílílfl-í-trt((¡lceto*'i .......
l.p.toUlct«>«ia ........_____ JcíKwia (ctCruhO
jT-d¡b'í*»»tíJo) ..............[Dc*ttiT*enn>5»i* ............edlbcazatenona ........ll««Uüixi ....................
*l»otto<íft©na .......... . .
BCiaa&eBiOÍetKiri* ......alcípínoOa .............. .. ..
____ _ (orv* ..... . . .h).< t-fcrem w Jc.» n fo r . . . .4—-ílmet»»!!» ...........
ifeiwa ........etoIíBoii* ........u ..................üiktfótt» .............
Dicctofefiocui ..... v ¿* /Kbcoraoítaaclto
2728 26 263334 M 36384142 45 45 tó50515334
39 M W SI &
62 « 69 TI ?5 73 73 SI W 92
95
96 109
216ISOm218
330
2¿82G22*21732323 »
&300326
2+1320
m
3A5
27*
202S41334
347
163132isa163
f 137 , ■í ÍOIOW>>1*37 (dl> iié
193IBé205210229146176isa18723a120202364
145m237122
156UBJ22
f ¡® í i m
179179
VA
2J7231
87
*110
146120
Í4213?133
'93*137
126I7S109
llfi109
I »
í » !130»
10612fi131100
tóis}} ® i »
i »
258
w
22Ó223258
iáó*239
2302622CÍI2122CH202
J89128*
27Í
I&5228283229
389

3 8 8 T o b ía s <f* derivado#
T a b la . 33 {continuación)
Ce tonas (sólidas)
N© totee <W coapUCTii) p .L«C
I*, «b.°C
D lbrtuatacetoa* .................. 112£c&»*F¿a<>Cu» ...................... 1 »JI-a*ílO<jn«Wmi ................... 130A cantono»». ....................- 121(•MÍlequilMlIl .................. 12>Bromuro <fc p-ícnclfcsfrdto 525V*iaQUcGn*«toní ............ 330Bonoín* ........ , ........... 1333P « t9& a.......... .............33?frÜdrti«3>«i»arE|Wp|»ji . . . . . 15Í-Z,i^\Mroxñcctnúíum* ». 147>-&¿r<}x5jcT>í<toía*<xv» 148 4,V-iid¿rcvb*iizaiax>ntM etan* (5>4l2DCtÍWiUdr6-
m w á D c J ) .......................... M í
BcoEtatTcnu ....................... 170OclalúdttitL* ...................., 171XtUBRU. .............................. 173G*iic*tQÍtoocí .................. 173« ■ alcafa r .......................... 178f+)-*fc*»far ..........4...' J79^ccrtQjuKnK)OÍxMa& .......... 379
C ttfoq tfalO » . . . i ........ .. 1*8
P c ttr tro q u ln o a i ................. 20?
FkpccicoSty'cBoa» ................ 219Antraquliui** .................... 285A tow ir» .............................. 289C kraD a ............................. 230
Oxtat»C
144S40d.16337*207
343
150
2Q&2 »
m
£>«nv»ác*
enrb«>»»
_ °C
2431*4¿47
Foníl-Wdr*.XOHll__1C
152»13S»50
206*
131161lf-2189
¿ té d .
IS4234
12830681
144>53
J ll5 (á i) l
H W...
* . •
i«i163338119
225235237
Í52
iüí H r nU « M il Í Í ITT
22í¿. 365*
224:::
iáá*220
2|4-4iai.
':'C180231»
27Í*
m245P 7244
2 »
177
í 36?»
3I3J,
<k U* quítuMus y U> h3dtidMi
T a b la s d e d e r iv a d o s 3 8 9
T a bla 39
Mercaptanos
P *•Tioítrr
¿fertoptano p. «b- 2,4>d;nEitt'«C -c fc-íitüco
NiCtil v ♦ 4 < < . * • 4 4 < • • • i « « • • • 6 4 *' 129Etll 4 a 4. * 4 • • ■ • • • i * « • • r r r r 1 4 • • » 36 a 1 a 115IsOprOpil « « 4 • • »4 «4 < » *4 t 1 4 * * • • 1 - - 4 * 56 » r » 94n-propi{ «• i • • •» j • • 4 • r j • • ••• a a 1 4 4 a a 67 ‘ r ■ 81IflDbutll • SS i • 4 ?6Allí a * a r « i t t • r4 r 4 • • r 1 . . * * * 90 72f t rremj l . « j a a t ^ i a a i • a • • s a a 97fl-hut^l a r rt « 4 r » r i i « r • . . • a r 97 662-pemÍI » r r • 4 4 • « n i • r 1 r * 4 * > 112 a a al$03HUl a a a • i i a a a * * a a r a a a « a 117 59tt"2UHll a a a a • i « a a a i 4 4 a r r r r 4 a 126 SOl^ t* fl< ÍÍÚ O l - , > . ------ > , , 4 - 147 a a 1 243
« a p 4 * « (l a 1 • a a 1 a k »a a 151 * - 4 74G k lo h c x ll f r « a a . i 4 . r a r a a a 153 a % • 143TiCn^I a a a a a r « a a a i • \ » « r r * 4 * » 1 • • r 4 • 4 166 a a 1 119Penll (tíofenol) ......... > , . . 4 4 * - 4 - r 4 4 • 169 » » 4 121l^ p r o p a n d i t i c d ....... , ............. ,I! C P t l a a k * < a e a . e a a r . t i a a
4 4 a a • 4 4 a a a i 1 a a
169176
a i i 19432
BCdCll a a 4 a » r i 4 « a i r«arr aaaa 4 W • 1 4 4 194 130C tJOCTCVOl • » 1 4 4 « » »4 < a » .4 • 4 4 - r t ' ' 194 101R lWGXBOl a a . 4 a a r ............. a • a a a 1 4 a 195 91j)atÍOCrt!!OÍ • r • 4 • 4 ‘ M a • • • » 1 • 195 103
* f ^ a a « i A a a a i a j a a < k a 199 1 a a ?aCt-ÍüLílctÍl a a a .« a a a a i ..........a a 199 ♦ 4 r 89H TLOlUl « 4 i i a r » 4 4 4 r » 4 « 4 a 4 4 4 220 4 a * 86 .
^ a f f a a Aj a a a a ^ a a a a k i a r 170 (3 mm} 50 91f/ncloro tiofc n ol »♦ 4 4 «»¥««««»« *4 • 53 » ► ♦^-bn>nx>tÍGfenol *....................... 75 • ► ♦m-nicrotiorena! ......................... a 4> • • » 4 4 4 75 , ,,p -tion a íto l ♦ 4 4 > ♦ ♦ 4 4 > ♦ ♦ ♦ 4 a ♦ 4 ► 4 4* ► ♦ 4 4 SI 1454-x«ú tpercapU iw , . - *......... .............. a " ■ 111 146

T '
3 9 0 T ab fe s d e d«rlv<xJos
r *r »
s»W
MI
L»• • f i
r»H
*i»
T abla 40
N itr i to (líquidos)
Nwribre del cOflipuwto p . d>. *C
Seifljcnrbufcoa <)r b «fciuil- (cotkítoM
*c
Alquil ?,4.é-trí' biáríOTtoiit-
Cirtonn"C
K f.t ilon itx íte ................................................ 78A c íto n itr i l» ................................................. Í1P n jp to« ii» íJo . , ............................................. 97l» b ii l¡n > j]k r ilo ................ ......................... ICfl* - l* i ( f o a i t r i lo ............................................. 118É¡.balíivotiiu>k»_ (c ia n u ro di; a l i J n J . . USa-K jd m ^ iu o b u ú ro R rtttlo , . . . .............. 120M e w ti^ c r to n ít r ilo ................... 120 ;CJw nB4€iD nlm Jo ....................................... I i7í io y a J íto n itr ík v .......................................... 13S0íl-VfllCTOlÚllile ................................... 141
FKir^oi»rilft ................ . ............................. .. 146lK « a p r tc i( r i!o ............................................ 335
«(•eipm oStrito . . . , ................ .... ................ !6<P.fcjc'W ocitriW j .......................................... 3 'M-iK lo K ip ro f tio n itiilo ................................. l í t íÍA c la n a tri te .................................................E f lu u o n itrJh i .............................................. 1 S íGlicolümirijo . . . . . _______ ___ . . . ia3>i.B f.fizanjiril* ................................................ 191Y -ctef o b u tiro n ílr ilo ................ ................ 19?C ap rilo iijlñ i& .............................................. 2000-M Junitrílo .................................- ............. 205C L io o in ew ü J de /ü lo ......................... 207
i-jUrJlo .............................................. 212c-iatum «r¡J© ................................................. 217M atc in o n lin io .............................. . . . . . 219
f.bidroülproiitenirrilo ........................ 221ea tlacM O nilrllo ....................................... 234
Y ^ i d i^ j l í u b r w ú í i i l o .............................. 240l ic té n o j i l t r í te ............................................ 245C ioM nofú trilo ............................................ 254GJqUíWlltrito ..................................... 28GA d ip o o i t r íb ................................................ 295
Nitrilos (sólidos)
a -n aJ(O o ítft|o . . . . . nj-i>rcfi_iobí^n>i\llríla^-toJunitrite ...........«1-c I a rúfct n ion i 5 ti Joo-daKibe-TttocjUrílo■> b ro tn oh t nzoo i Cri V> S iK dcion ic riio . . . . . .«■rodobí^u-onitríte . P 'TCiUoxlb<n>«nitríte
35 38 38 41 43 53 5 i 5S G2
29$225217
¿32
¿éw.257
fcnaí >J1icrí o .............jd o c ia iio sc ííic a . . . .
■clot^bcBíOnict'ik»fnilfObfriEuoifriJo ......í-bioiod^azonilciV . . . . p o> ¡tri> íf* iíliiC fJ& n ¡l*üoítt*nUro4>ír)Zíiriitrilo . . . .M.:tíleriumioi>.Kr»¿>u¡irilri /"i>ilrob<«lrori«r¡lo ........
ClArHídma ddicááoakjvitroercjp-
«MKititt»*c
199174
? is175
XÍ4124
3 ¿4 i s i i?5
157 149 {hidrato 89 i
Í53137
ÍÍ6
m
>22(hidrato 104)
. 121 (Kjdi-.iio 96j
i?ó
156
. I ! ‘‘ i »
. . . ¡24
" • Í34
Í69■91
i+ i
Nombre dd Cotnpüejlo í>. fl>, ! S'&jnlws del cdniputsto p. i. ■ }>. d>.JC | ’C -c.
«66921Í0
112116ne129J47
mrew.
.'1

T ab ta s d a d e r iva d o * 391
T a b l a 41
Compuestos n itro (líquidos)
D < m u t« (id lk ' tía» ¿c U » m ü i
No«nbr« del u m p u tilo p. <&. ‘C Btcccti-
■uifoii»,-OiwU
’C
Bttam-w¡4a
°C
O ttt«denvwioi ’ C
101 30 mCloropicrina . ...................... 133Nítroctano .......................... 1142-Jiitropropano ..................... 120Tetramcroinetaiw ............. J.261-rütropfojwtao 1302>nitro&Utena ......... 1402-mcdl-l -nitroprcpano . . . 1412-nxcül'2-nitrobu¿no . ►.»< 150I -níüobuta no . > . . ......... •., 1522‘jútropentano *1543-W«tíI'l'lÚlTQbut«lK > l$41-nitrchexano ....................... 1931-nitropcntano .. l?3Nitrobencttto . . . . > , .......... 209<5‘hitrotolueno ....................... 224o-wtroetilbenceno .............. 2242,$KUmetíInhrobenceao . . . .225
FenilmtrometAno ................ 2?.$d.m-nitrotolucno .................... 2312,5-djm«ilii»ttob<5rtCcno . . . , 2342,4-dimetilnitrobettccno . . . 238
£-nítroctill>eiic£no .............. 241ZjíMimetilmtrobeaceno . . 250^-allroeitircdo. - - ................. 260d.2-idtrodtncno ....................... 254o-mtroanüol .......................... 265o-nítrofenetol ................ ...... 268
5a26
367053
96
112124
6895
133128
89102
71
547655
40
Í6Ó143147
105125140192
151
102
H Cl de la jmúna ,
HCl de la amina .
Trt-dmitrobenceno .2,4-dhvtrotolucno
A ddo 2-nitTDÍsoícá l í c o .............. ..
Trimlro-.£-xtte:l0 ljB-dimetiWjfi-djrií
irobencíüo . . . Trinitríxtilbettcen©
2.6-diiiitrodmef)0 .2.4.6-trirjtroaiiísol 2,4-<lÍnítrofeneto! .
140
157
9070
30 0
137
9337
5 46386

T a b la s d o d e r iv a d o s
T a b l a 41 (c c x t in v a c ió n )
Compuestos nitro (sójidos)
d d «aipwctto p-í. P- «b.♦c UC
Daitodotuili' <k4 de U Un i na
BctJcwv(UÜOfl*.r a id *, °c .
2eflta- in id*’C
93
115103
Tjibroa*>nittonicttcto , . 10a . . . .Tclranitrotnctano ......... .... 13 126° T iinltrarneU no ...................15 4» n j-íiitio to lu eu o .............. ......15 231 95° 125*AJcotol m-nitrobencílíco 27 ►.2-rootü-2 -ni tcop r«p ¿no , 26 12? <3,4-dlmítilnjtrobeÍKciK) , 29 258 » . . < ..o-doroaitrobenceno . . . . 32 246 129
2-cItrofclícnÜo ..................... 3344mm>o3-iiitrotoluem> , 332,4*dickrronitrobeaíeno . 33 258 129jn-nitro/cnetol *...................346 - d a r > 2 - n i tro to ! u e n o . . 3 7 2 3 3 « . .
4*<Joro*2'iiitroto3ueno . , 33 240
m-yodomtrofeenccno . , . 33ra-rütroanÍK>I ......... .. ............38 256abrom onitrobenceno . . 43 261
C lorare d e ^-n ítrobcjzal 43 . 4.
wx-cloromtfobenceao . 4 4 235 121
Nitrojaciiieikoo . - 4« . . . 44 245
C lororo de O'iUtrobíücüo 48 ♦
116
120
204
Derivada»n fC tttla*c
S 4 d ¿ N Í¿‘!
Benzoato . . .
2 ,4* d i ai troció- ro b e n Ce n o ,
Acido _ 6-cIoro- 2 «ni Croben- íoico ..........
Acido 4-cIoco- 2 - n i t roben- zaico .........
m-yodoanüüia.
2.4-dínitrobro. roobenceno .
Acido ¿-n itro benioico . .
3.4-dxnítroclcK robenteao .
D uiitrom witik- «o ...........
Tablas d« derivados . 393
T a b l a 4 1 ( c o n t in u a c ió n )
Compuestos nitro (sólidos)
Derivados orilA- dos de U «min»
Nombre tfcl c*K*>UttK> p. í. °C
p. «v. *c S4CKC1I-
»df<KWl-llUdi’ O
Beou.'raída■’C.
’ C
o-nitroyodobenccno . . . . 492.4-dinitroctorobenccno . 52* 315 d. 2,5*diclc<roDÍiroiienceno . 54 . . .
■^-¿•nitroanisoj................... 54 256rt-brx>manitrobciKCrjo , . 54 257
■^-nitroiolueno ................ 54 238
4-yodo-3.nitroioluc. ■> . . 55 ►-,
Bromuro de m-iútroben*CttO ....................................... . .
3- n i a r a ti reno , , . ; ------ 58 260d.n-nicofcfíetol ................... 60 233
- 3-iütrobifenilo ................ 61 .. R-rnUonaícaleño 61 304
Cloruro de m-nitroben-zal . , ....................... 65 1 . . .
Sjfi-dinitrotoluerto ......... 662,4,6‘b initroaaííol . . . . . .-^62-. ►♦,2.4-dimtiotolueno... 70
• Cloruro de ¿-nitiribencilo 71 . . . 2-nitr£>-/>-dorobromab«:tt-
«eno ........................... 722,4 'düthrobrom obepceno 72
Alcohol o-rutrobencílíco. 74 270d,3.5-dimelilnitrobenceno . 75 2733.5-dinUro*o-xileno . . . . 76 2,4,6-trimtrofeiKjtol . . . . 78
. . . Í7& 2,4-difiitroícnol . 114 [20 2 ,5 'd ic lo ro -1,3-
dinilrobcncccio 104 95 154 2,4-dinitroan¡sol. 89
136 3 ,4 -d in itro l> ro-m o b c v e n o t<
120 J58 . 2>4-dinitrotoIuc*• na ............ ♦ ►. 70
. . . , ,» 4-ycdo-3-ainmo-tcluerto . . ► ♦« 38
í 43 i 73 2,4^dini troíérvétol 86 ..................... 3-arainobifenilo. 30167 160 .............. ...............
Acido pícrico , .2,4,6-trin itro<0 '
lueno ............p-toh iid itv* . . . .
2,4-dinitrofcnol A c e ta to ............
ÑitAjjálidina A cido p íc ric o
122
8245
11435
75122
♦ Tumblén »« jnqwrtM foríiMi inclutables qi*o funde* » 27'C y W'C-

3 9 4 T o b ía s cto d e r iv a d o »
T a b l a 4 1 (continuación)
Compuestos nitro (sólidos)
- »
»
i »
r »
P
F*
* Ouiyiilodicik. rtOJ <l« 1* Urtio»
Dulvadofetp«cUto*c
del compuwto P-í.PC
p, tb. °c Bcocco-
» mU*c
Beño*.ni id* *C
J^nitroniítiltno . . . . . . 78 136 162 ■3,4-dímiro-o-xikno 82 . . . 2-rútro*3,4-díme-
liíaróíina . . . . 66Bromuro á/i ¿-nitrobenza! 82 . . . . . . . . . Acido ¿>*mtro-
benzoico . . . . 2412,4;6-ti5nim>to]u#no . . . 82 . . . . . . Acido 2,4,6‘tríní-
irobeoioicc . . 220¿■-doronitrobencena 4. . . 83 242® 121 192 í-mtroíeaol ».. 114Cloruro de píenlo . , , , 83 * ► • 211 • • « Acido pícrica ., 1222,4-ditiItrCHin-xiIcno 84 4 . . • 3-njtrO'2,4-dime*
tiliaiHaa . . . . 844-bromo * 1 -nttronaftaJ«no 85 . . . 4-brctT>o-l-ttAÍ til-
amina . . . . . . 1022,5 ‘dibromenitrobenceno 55 M , Acetamíd* . . . . 1712,4-djnitrociciítílcno ..« S5 . . . 2,4,6-trimirorrte-
utiíeno ........ 2322,4-dinitroani$o[............ 89 4 . . • • % - « # 2,4-dmitroíenol . 114
90 302 194 240 «i-cútro*nilina . 1143,6-<lií3Ítr0'O»xtJtxKi . . . . 90 . . . 4-nitro.2,3‘dimc-
titanilin» . . . . 1142,4,5-trinilra-modJeno . . 90 t4 .3,5-dimtrotoIu«no , , . . . 2,4-diroctil-1 ,3-diniteo-
92 . . . . . .
93 ►.. . . . 2,4-dímciil-l,3,5- crinittobence- no ............... 125
2,4'*díii?bit>bifm»lo........ 93 .442>3'din¡tro^-rilBTio . . . . 93 .4. Acido 2,3-dini-
tro-^-toluico . 249Alcohol /^nltrobencílico. 93 . . . 4 . . . . . Acetato . . . . . . . 78í'Cloro^-nitmanUol . . . 98 * ’ 4 . . . 4-cloro-o-ahÍ8Ídí.
na 4. . . . . . . . 84

T cb fa» d e d e r iv a d o s 395
T a b l a 41 (continuación)
Compuestos nitro (sólidos)
D<riv»ó«i *cüb. <fc* dí biArtiaa
Nombre del p. /. JC
p. ib. X £«BCMl>
aitíjyjt»’n id i*C
Bcim.mitU,*c
Bromuro de ¿-nittoben*cilo .............. ..
2(5-d¡nitrc>‘m-¿Uct>o , , , ,99
1Ü1. . . . . . . . .
2J3 '^icútrobiíc« 'ü lo .........
<MjÍRÍUX>btoeeno . . . . . .4J5*dinc(ro-o-xíleriO . . . .
110119ml i e
319 106230301
1,3,5-trinitrofcencena . . . 122 . . . . . .
í^ -d in itro ^ -w len o . . . . 125 , , , . . .
2,4,6-tribromcniirobcnec- no ................................... 125 « • • 198
£.bnSmonUrobeiK«no . . 2,2'-diniLrobifcníto ►,. , ,
126123
259 134 204
4,5-dimcro.rtt-xÜeno . . . . 132
Eter /^-d irtitrofenO ico. 2,5-dinitro-^-xiteno . . . .
14314?
. . .. . .
1,8-dinitronaftaleno . . . . 170
^¿itroyodobenceiw . . . . 171172 299 247
210300
SjS^im trobiícnílo . . . . . 200
I^-dínitronaftaleno . . . . 214
4,4í-dimtrobifcrulo > . , , , 237
Duindatc^xiila’C
4 -n itro -2 ,6 -d ¡m c- tit&niHna. . . . .
■j-nitroaniliiiA . .2-n¡tro-4,5-d¡mc»
d ía ni lina . . . .NafuJeno, corap.
de adn. . . . . .3-miTO-2,5-dime*
(«[anilina . . . .
158
71
m
153
9B
2 ,4 ,6 - tr ib rom o.anilina ......... 122
c-ottrofenol : . . 114 2,2'-diaininol>iíe-
nílo ................ 812-nítfO;4.Mim(¡‘
cjlaniliria. . . . , 76
4-n¡lro‘2.5-dime- tilacutina . . . . 145
IjS.S-trijiitronaf-taleno ........... 21fí
p-nitroícnol . . . 114 J^d iam itio b ííe*
nito 94 1 S -triru tronaf-
tatc t» ......... - IMBcnddina . . . . . 128

T a b l a 42
Compuestos nitroso, azoxi, azo e hidrazo
3 9 6 Tobías d e derivados
CoroptMtla p.t._________________________ _ 2 L _Compueítos nífroso:
I .n iiro so -2 ,4 -d im « c i]b en ec n o . 41¿ -rá tftn o tó lu en o ............. . . . 48N-aicrosoac«tanUída 5 Id .íK-nitrajotolueno .................. 53A r.iJu ro so d ifc rJlan iif ta . . . . . 6 6nitT O sobcnceno , , . ........... . - , Gí¡0-nitrosotolucno ................... 72^■nitrcio-JV-ctil&ntliiV» . . . . . ?S¿ -m tro w -N ^ iV -d ie iiJan jljjia . 34/•n ítio ao -.V ^ V -d m ic títa riitín a . 871-nittosan&ítakno 985'nícriXi>-t>-ftnÍBÍ<i)na ............. 1071 -n ltr a so ^ n a íto í ................ . 109
^-mtroso>A'-metilanílíaa . . . 116V niü-osofcQ ol . . . . . . ............. 125d.S-nitrosQ-tf-creíoí ................. 335¿.nítrow -difcnilaim i-ia ......... 144 2-mtro5o.l-na/iol... . . . . . . . . 152d, Nitmsotiraol ....................................162/¡-nitraw atúlina *............- . . 1744 .a h r© s o - |-n a f to l - . . . 4 . . - , 394d.
Compurstos azoxi:A¿>xit*ocjeQo......................... 36m-awcdtolueno . . , ............. 39p-jLzoxiiolvicno ....................... 59¿-aa&xiloJuena ....................... 75l^ r-a iox jnaíuJeno . . . . . . . 127 ¿jí'-«3¡cTon?ji20X¡bence/Jí> . . . 154o-azoxihi/enilo ....................... 1582 ,2 , -4 z o x m a f ta le n o .................. 16B4,4'-di nitroaicxibcnccik> . . r 200í-ajcoxibiíenilo .............. .. 2J.2
Ccmpwio
C om puaios Azo:» > - a z o t o l u c n o ............................nt-azotolwno . . . . . . . . .Azobcnceno .......................1-bcncenazonaícaleno . . . O'liidroxiaiwbenccno . , , . DenccnazodiíenUamioa . . /r-dimei ilaro inoaw benceno p-am m oazobcnceno Benecnazo-ocresol . . . . . .I -bencawizcwí-naítol , . . .o-azofenetol <«. .2-bcnccnauonaftaleno . . .l^-azonaf caleño ...........2*beneeaazc>~] -naftol . . . . /v-azotoluerto . . . . . . . . . . .o-«obiíetú3ü............ * h ,p-hidroxiaj# benceno . . . . ,^-aswfcnctol ................ , , . , Bcncenazorworcínol . . . . . .l ,r-a ro n aító lcn o . . . . . . . .4-bencenazo‘l-naftoJ .........2,2'-azonaíta)eno . . . . . . . .
Contbi¿e;t&s hidfózo:rrc-bidrazotojueno . . Htdrazobencem» . , . .
Í-bidrazotolncno . . . ,2’-hidrazonafia]eno
1,1'-hidra zonaftaleno0-hidrazotolueno . . . í 'h idrazobífcnilo . , . p-hidrazobitenilo . . .
55 55 68 70 83 96
117 125 128 131 131 131 136isa145145152160l?0190206d.20$
36130134140153161170182
T ab la i de derivado* 397
T a b l a 4 3
Fenoles (líquidos)
Nombre «fel «itnjn»«to p. «b.°c
p.<.•r.
o-dorofcaol .............................Fino! .........................................o-ctodI ........................ ¿ j . l i r o i c o f e n o l ........................................3*üoro-ji-<retól ........................gC-crtul ......................................
i«l ...................... .
S»!SSi :;í , 4 - d i c í o r o ( e M p | .............................2.4- )mrt\H«i>ol m.cl0rc¿«aol ................ .é-clcrcfcnol .......... ...................zv<tjtf«aol . . . . . . . . . . ..........«<ttxif<fiot .......... •’ ..........C'P(Vq>11í«rtOl ....... '. .......fl.«pbixlK«ool * n - t) ro m o t« & o l Curwrol ........................2.4-dlbríihK>tef]ol Xm o k ím I, í*cr morvoc^tifir» MlUtilÍÉBol Eugmol .........................C~ajnilí'ntt>l ..........................Isocgjieivol ............ .........
1 » 7leo 42190 31195197202 3é202 3205 2e20?2>M 43212 272H 33217 43237 <. -217 Jfl232 222 » ...236 32295 0m 30243245 222¿0253 23267 . ...
Drñwictf
tihire-taoo*C120133ltó12&146128w
13$Itó
1(6129
¡22
isó
TñUfltno Ribmmo . TribnJtrui
Dibxorr-QTribrornoTnWomo¿xónio . .
Biflttto .........Tíihnín*o . . . . Trlbrtrao . . . .
TítnÍiroiw> . . .
D ib r c iR iO ...............
OcAcido
ttiioü.MÍtítO
1 «r . «56 1 Y¿ -95 14S
ioa4S i »
103\ 1 6 121
141€8 134:
lid156... 77
125W
■W 151-V, 153IIH lt8
SIm so
9094 94
Fenoles (sólidos)
Derivad®*
Niírabr« dd ccKJip««4e> p- í. • c iWiaíOt* 1
ttrrümo .» c ¡
Dw)»*k«brotowlot
°CEiícríi
'C
i4-dúaetíHovfll Cunrdtcl .....................<-crcw! ........... .f.-brCBiotc»uJ' . . . . . 2.lKrt>TiV)-4-clor<’lf»10[ S-nJftvJi-cmol ....t>-crcn>i ..................2,4-4jl>r«niofcnol m-y C/JerJo) .........F«0<tl .................¡0-cl0/T>fíool ............
1252(t mn31 m
n>4
:i4:w m
4042 1334 t i w
3&.. 315 BunZOaCo . . . . 5?
Eoft«v»to .... ai56 3,i-dnlitrobí>-
133
BíMMIu ---- 100----- 1U¿
.. m S » m t 4 ... - VI
.. 95 ScOK>ito « /
'.’. '95 BeoMilito .... 68Bírttojto --- ai

3 9 8 T o b ía s d e d e r iv a d o s
T ab la 43 (continuación)
Fenoles (sólidos}
Norftbre «Ííl cowpueswn</ivados
v.t."C a-níJtCl. | Dtrivados «lf6Ur>o • tirtmodOJ
*C 1 *C.F4(írti
'C
iV-yíKfcjfírnjl .................. 434.3 Bromo ........ 4 ñ8 Bcr.»ato .... í>7ú-íiltioí nol .................. 45 Ü3 4/é-Íibroino ... tí? 3.S-d'mi«ri:lxn.í-cJoio-3-rüelilíefiol ..................í»-Ct:líenol ..................................... 475>«l0TO*»-<Tí»al ..................... . . . 45S,S-di bn?m<f-p-«fTTZi[ .................. 432^-djrotijirciwJ .......................... <9
2-dtmj-3-nvc«íf«no"t .................... 5Úí-cicloKíxiireoo! .......................... 53HídPO«omOfia, éttr iDWOmetllkl 53i»4>*ti<jlftnel ............................... M3^-¿brcma-r;-<reío] .................. 5?Í.S-didortJÍeool . . . . .................... 372-l\i'iro*»-3.5-<íibr<m»<olue<liJ . . 57Oroool (hidratado) ................ W<i-f«uir«»l , . . . .......................... 594-claro4i»not ......................... .. W24WieÍ£wT>íírtOl ............... ........... 55f-brninofcnol ............................... 633.4-díwietitíeoDl SSi-clow-i-hMroxttolucno ............ 662.4.6-trí e &3.5-<ilclirriícnol . ........... S92,4,^-tridó»íenoJ . ........ . ........... 633^^Jicnetilfcool ................ . ......... 65
2.4.6-triracíilí«floJ 09■t'ic-Jiextli'escrctm») .............. ; . . . í9Mestol .......... . ............................. 70Studacutntnol ............................. 71¿^•^iyúdciítfioJ ........................ 72J,5-<fi[iwXUrEOQ[ ............................ 742,3-diEnetlire&ol ............3-hidroxIbiIt*i3l6...i , ...................... 7ÍI4-«lor»-2vyo4flí<oi>3 .--■ 7SIiódimtiriot . . . ............................ 81M^ncllfefiot . , ........................... 84'¿-hirfríxi-Í.S-djmlrtitC'lBCúo . . . . Wa*mftol ................ - ...................... 94p-yo¿°im A .................................. 5*fr-Afr-b«tdtc»oJ ....................... ■• 9j5»4,5*tribrocno!Mipl ...................... 95^-«7-cmníirefiol ............................ 96m-íiilroícnot ................................. 97
......................................... 104Clo«4údN5«aci>(HUi .................... I0fiOrcúwl T.......................... 107IjS-dihiárftcirotláieea ............... 108
ba»<0 ...... 1421» BenzoatoBenzoatoBenioíitoBenzoato
31 so7195
ICO Bronw ......... .... 54 Benzoato . . . . V‘K
. . . Benzoato . . . . «7
. . JPI?>fo«no .......... 90A-ll¡Uob<Ji«iato 137
160 T ribwmo ........ 101 Di)j«izi>a(o , . «t. . . . . . Dinitro . . . . . . 2V7
T/iltfiwivo ........<•.0
: « 95 f0?142 TriJKwuui ........ 171 Benzoato . . . . ífi15* ............................. Benzoato . . . . MIM . , , ............ .. 10
Tfibromo ........ J89 íl«lZO¿Cfi . . . . 53Tlibrónio ........ Í6Á
TBriiroJíi> . . . . Sj^-dmitrobeif
W
Üibrojiio .......... líift• 201W . . . . . )l?3
• tDíWojúc .......... 159 '¿¿
63. * . BtCrúa .............. 'J5 BcníOalo . . . .- . -
Tribromo ........UH
173 1Í4¡ Rísuoitf» . . . . 01
Brnzonw . . . . 61. . . Bcnw>*t& . . . . «9. . . Benzijilo . . . . n
Bcrzonía . . . . H72,4-<Jibromft . . . 132
152 m ASm1M
50TctwbfímKi , , . m ft?Dibromo . . . . . .
611S7 91 ai153 Trt/4b«x>oio . . . ir> DUcetnW . . . . «i. , . . , , tJ ........... is . 7?lUf Dihnofíií» 104 m
Acetato .......... 106
T a b la * d e d e r iv a d o s 3 9 9
T a 6 la . 43 (con tinuación)
Fenoles (sólidos)
Nombre dd compittllo p . I.•c
EV.rivjuío»
a-nnítll-ivet»!»CDcrlvjxlúibffimidn“C
tw w-c2,2’-4iW4rti!ot>«títilo ...................... i 10KefúrciboJ .............. .........................1103>h¡drox|»2,4l&tnnltroi|?luen<> . . 110Brútoofcidroqucnoii* ....................... 1IQp.ailcnrcoM ........ , ...........................1142.4-d¡critTO<ínGl ...114¿4lJdr©*jl»fllizñJ<3ebJ«U> . . . . . . . . . 1154-tkjfn-3J^íbro«uiftt»l ............ ... 121Eter moretxBcJJiGo de Ib bi-
<Uuqaíno«* ................................ ...122K M o y ferieo .............................. ....122
......................................... ....122TotuMrMgbmvt ...................... ... 124fcckWtólítfv»! .................. ..mF nvrtM ........................................ ... 1932,43baiutMifi4l ........ ................ ....13(Bl,U M 4 i« » u ii4 l(n 4 ................ ...1401.2.4-trihMmíbcDCCTU] . . . . . . . . 1402,4>Í*kJOfrwiütV6] .................... ... I4R2,4,&-4riv«cWcnol ........................ ... 159Mn^fobcl .................................... ... 165HI¿ÍOqO)i>W» ..................................16SGfclhfittofenoiu ............................ ....173l/xübktwilMÍti^ivrt ................ ....176Aúdo trtlfnUo ............................... 1A0217'diKidto*líuJokJ«io ................ ... lí-SPaottíJoitifenol .......... ................ ....1WCvWMznle ................................... 203■J>triibrorr*>-<S-CíT'Kif .................. ... 2104^1oío-;i315.6-«etr»brofí*oíc3>ol . 215FlorpgfudBol ................ .............. ... 21SOl-0-naftol .................... ............... ....2ISS^J^-Tetntctorohidr^unoBa . 237l^ihidrotinufLiJeno ................ ... 256F«»al(t»klni ............................... ... 241
f i ¡ ¡S íS r í ! “ „ ::::::::: §8
J5t
157
4,W¡t>rooio .Dibroot* . . . 2,fc-DJbr*iTx> fy>BrnnO . . ■ 3 d¡b»orti» .
BrciM
Di broma
Dilirofi»
Tríbrooqo
112!RS142lidIfll
Sí
158
(«6
151
AcelMo . . . DibeBttiu AceOÍ.) . . DíaietatO AOMst» . . ,V>üu . . lextMto Beewato
A « u t o . .Auum ..D iia á toBtswtaT rU ú e b toB e n m u >OiiMtil»Trincento
ítatMlO A c * U » . . DitautoHkxclíinD&MMlO A c e t» » . .
A cetato . .Br.nxoata7>l(Kelalo
D Ü ie tJ laD íacet* ioDuie/Ü«t»Oi»ceUtt>
93í”13572SI729093
767052
1181»174l«97
i 37 W 123
123
¡30150
Í54203105
245160146ífia

4 0 0 T a b la s d» derivado*
T a b l a 44
Sultonamidus (sólidas)
SuKoo amida p . í .’C
Suironwviidíi J». í.*c
11. 1 »hllTí r>« 3 1502-;tv rti¡-1 -p ro p ai* . ............................. 16 4 J6 -d ic lo r< r2 J5 •d im etílb en ccn * , , 1501 -b u ta n - ............................................. .. 45 1531 «k kWr*r*inm 52
53153153
2 -p ío p ím - ............................................. 60 m -brom obe»cert- ............................. 1544-iícipropi3-2'W 'iírtilbcncen- ........... 73 2 -y o d o - l-n a íc a lc n ' ........................... 1541 -h ffp tin - ............................................2 , 4 ^ ‘tr im c to x ;b e n c c n - ...................
75 2 ,6 -d ick iro -4*m etilbec tcen - . . . . 15576 2 ,4 -d icucrobencen - .......................... 157
3 < tU b e iK c n - ........................................ 86 213 -d ic lo ro » 5 » m e tilb c n c e n - '........... 158M e ta n - .................................................. 90 T rt'n itrobencil- < ................................ 1592 ,6 'd im c tílb rn c e n - ........................... 96 5 -c lo ro -2 -n ic ro b cn ceJv ................... 159C e d í ............................................................. 97 3-c[oro-2*m eti3-5-nitr<>benc<ín' , . 1612 -« tí lb cn c e n .............................................. 100 ¿ -a rn in o b en ccQ - ... .................. .... 4 < 163S y m e tíM -ílu o ro b e n c c n - ................ 105 4 -d o ro -2 'n it ro b c n c c n - 164
105 3 .6 -d ¡c lo rr> -2 ,5 -d irneü Ibcnccn - . , 165in -to lu e n - .............................................. 108 5 - c lo r o .3 r m tr o b c n c e n - .................. 1654 -c tílb e n c en - ........................................ 309 W-ni Crubcnccn- ........................ .. 166i-ró e til* 6 -n a í ta le n - ..................... 116 2 -^ c tH -5 -b » m o b € n c e n - .............. 166Im íd a d e l ¿Lado o iu lío b c n z o ic o
( s a c a r in a ) ................... .................... 121 p -b ra rd o b en c e n ..................................... 1662 - íe n il-1 -e ran - . . » ..................... .... - 122 2 ,S -d u n e t¡lb e n c ¿ n ................ ............... 167p -fluoM benccD - ....... ......................... 125 2 ,4^Jim eto2dbenceri- ..................... 1674-rrtctíl-3-<JorobetK :cn- 126 5 -c lo ro -2 -m e á [.3 -m tro b c n c c n - • . 1674-don>-3-Tnetílbc»>ceil- ................... 128 2 -m etil-4 -brom ob«ricen* ........... .. 1663 < fo ío 4 -n j« to w b < tn cc n - . . . . . . . 131 2 t4-d¡c3or& .6-m etilbcncen» . . . . 168(4 - ) -c a n fo ir -1 0 - ................ .. ............. 132 2J5 ^ ix n e ti l '3 -n 3 tro b e n c e n - . . . . 173S ^ -d á m e titb e n c e u - ........................... 135 2-m ctít-4-fluor<ibenc>;n- . . . . . . . 1733 ,4 -d íd o ro b c n c e n - ........................... 135 l - n ^ í t i l ^ n a f t a l e n ' ......................... 174< 4 - ) -c * n fo r .S - .................................. 137 3 ,4 ’d íbT O m obeijC tífi-................ ...... 1752,4-dim ctilbenCci>- ............. 137 2 >4-d io lo rO '5 -rnetU bcnccn- 176p 'tO lU ttl- ..................................... .. 137 4-< ]oro*3-n itrobcnccn» . . 7 ........... 176o - r n t r o b e n d l ' . . . . ....., ................. 137 4 - b r o n » 3 - m tr o b e D c e n ' ............. 1772 -b m m o -1 -n a ftsü en - ........................ 140 Fcno l-4 - .............. 1772 -n c til-5 'ÍIu o ro b cn cfc tt- ................ 141 2-clopO '5-m ctil-6.m U fO *benceri- ,• 177
142 ¿ 'r t íe rcb e n c en - ........................... .. i s o '2 -m ?t¡l-$-clí> robcnc€n- ................ .. 342 180 ■2 ,4 ,6 * trim e tílb eac* íi............................ 142 2 ,4 ,5 -tr in je ti!b e n c en - ...................... 181C 4 -) 'C « í ío r -3 - ♦ . ♦ „ ................... .. 143 3.<Tor<>-2-n>ein-6-mtroh<íiK*rt- . . 1815<lw o*2-m et31b<:iicav 143
344181
3,4~d1metÜbencctK. ........................... 5 -c lo ro -4 -m c tíl-2 - tiitro b eo o e n - . . 181144 5 -c lo ro -4 -w e til-3 -n itN > h cn ccn - 4 4 182
1 -m e tC -3 -n jiíu lc n - * . ..................., 144 5-cforo-2-m eti3-6 'iri¡tíob«iic£tt-' . . 1834 - r¡>e t¡i -3 -b rom obc rvee n- 146 185
148 4 -c to /0 -2 >5 -d tin c tiIb e o « :n - . . . . 185143 185
T a b i a 44 (continuación)
S u U on am ida s (só lida s)
SvJfoparaUl»
& n:3oro-3-nhrobe:nccn- ..............a-bromc*benceri‘ ................. ; •4-dOro-l-naílalcn........................4r5-dkli>ro-3<rt«i¡fl'JcíJCcrt' . -.2.3-dicloro-6-meti]l>cnccr:- . . , 3j5'd¡cioro-2'm<ít¡llrt:nccr.- . -, 2.cloro-4-melllb<:nccr.- . . . . . .2,+-di anii nobencejv1,5 -di - . . .3-ycdo- \ -naftaEcn............ . . . . ,2-mclü-5-nUrobcnccn- ........0-clorobencen- ......................2.6-dicloro-3-metiJbcric«rti- . . 2-c3orO-5> inct i l-4-n ¡ trofcc nccn- p -b ro m o b cn c il 1-metil-7-nafta 4*et>3bcnc£rt-l,3-->i - ................2.4-d3bromobcnce.................... n o-n tlrobcnC en- . . ........................■í'itteliloeíKeTi-Ij^-di- ...........2,5^imet^6*r»Urc>b<2íicen- . .2 .7 -a n tra q u m o rid í- .... S-brotno-'T-naíialcn- ..............2.5-dibrorrt<iben«^rt- . . . . . . .2*doro-5-mctil-3-nítrobencen' ¡J^-dimelil-^nitrObencen- ..8-tlorí>-1 -naítalcn-. . ..............p-nítroboncií- ............. ..2,+J5-tridí>robencenr '..............3,4-4ic]crO'í,5'dimelilbcncen-4-clor©-3-rtttt¡l-5-mcrob«ncen- 3>yodo-4-nafta]«ri' ........
: ’ 2,4-dicloro-3-metítbence»- .. p-yodobencil- .....................2-n>«dl*6-naí.talen...................
3B6186186]B6186186196IC7[«7187 l«8188 188 188 139190 )9<1191191192192193194 196 198 !99 200
> 2 0 32012012022 0 32 0 6206
■ 2 ,6 ^ ím c lít-3 -n a fu lo n - ................ 207£ 4'hidrcxN2>6rdmiétilb«iiic«n' 1,3-
d¡- ................................ i ............. 208
*ic2,4>&-trtcJorobeiKcn. .........1-uitro-2-naftalctv ..................3-Tnctübcnccn-l,S-di- . . . . . . .2-naftalc...................n ................................2,4,6-tribrt'inObi: nccn................3-d.orobcncC»-1,5 <1 ¡...............2 -m cti]bcm ,c n * l ,4 * d í - ...........2,3r4~ertclorí>bence«............S ^ -d ic lo rv - í-m e ü lb e n c c ti- .1.3-beneend i 4-bifenii- 1-yodo*5-naítakT T ...............4 'm e t j lb e n c c f l '1 ,2 -d [ - ...........2.3-d¡cloro '4 'm c(ilb< nc«fn*4*t‘to ró -2 'fi ¡ trobc nccn ..............l,2 'd ím clíll)C nccn*3 ,5-d i* .4 - rn e to x ib e n c c n -l,3 .d í. . . .2.7-naftalcnHi- ..................1.5-antraffwinondi - 2.4-dirncúH >ciiC «ri'l,5-d]- ,2 .3 -d im ct» ll‘c n r c n - l J4-di« ,l,2-benccn<li............. ' . ...........3 .b ro m o b ^ c cn » { ,5 -d ¡- . . .2-aLminO'5-mctilbcnccn-tj3*<! 2'r>«tilb€rvCcn-l»S-di- Á ntraG ucnon-JJ...........................1.4-riaftal«;ndi - . . . .2 ,4*d iclo robC ücen-l,5 -d ¡- . .1.4-bervccndi- .¿ ¿ - d i r / ie t i lb c n c c i i 'l^ - d i - ,lj6-jiftfUl«nd¡- .....................Biíeml-4,4'-dí..........................f^ .n a ftak n d i- ............; . . .2J5-dítntííJber>cen-l,4.di- .1.3.5-bencentri-... l - c ia n o J M ia f t t le n -
1 .8 - a n W íü in o n d i -
2 1 2 d 21* 216 217 2 2 0d . 224 224 22 6 d . 228229230236237 237 237239240 242 246 249 251 254256 .257 2 6 0 261 273 2 7 6 2 8 8 2 9 5 2 9 8 3 0 0 3 1 0 3 1 0 31 2 336
340

4 0 2 T o b ía s do d e r iv a d o s
T a bla 45
Cloruros de sulfonilo (líquidos)
CJo*\loú <lc niHoqUo p. cb. "O
Derivado*
60/21 iam 1.449070/20 mm 1.450661/0 mm 1.452267/3 Mm 1,45187 3 /n mtn 1.452075/10 a m 1.451,788/9 mm 1.4530
125/9 mm
Addo p. <£>.
VC
AinicUM -*c
Ah¡lid* U-1.*n
167/10 mm 90 9953 5860 04
336/1 mm 52 - 1 0....... 16 38147/Oü mm 45
3 4275
M e te n .....................EUn- ................2-propaci-.........1-J>n>j>a n 2-mctü'J'propain'1- b u te n - ............3'jnct>3»l-hutí*n-i-heptu)* .........
Cloruros de sulfonilo (sólidos)
Cloruro «fe ¡nJIonílo
o - to lu tn - ............................M-tcduen* .......................2 * « ijlb c n c tn ..............................+-clilbcnccr\ ...................Bety*n-..........................H e p ta n - . . . * ........................3.4-diclorobencín- 2,6-didoro-3-i75ctilbcncen* 2*rattí]-5'Clorob«icftn- . * - 2.5-dinttülbcncen .........o-clorobencen- ...............2*fenil'l-etan......................2.4.5-triclorob«nccr i 2.4-dim<itilb«noeri ~ S^-dibrooiobencen*........2“rnc61-5rbrr>mobenctf\' . .fr-fliKirobeTjcen- ......... . . .3*cloio-4-metílbcncen« - .2 .5 -d íc lo rc b en c e n - 2.5-din>eíi!benccn 2,4,6«triclc>rob«icen- . . . . 2,3 •dícloro-4-metilbcncen-4-cloro-3-nitrobencen- . . ¿ 14-¿idt>rO'_6*metilbenccti' 2-jneti!-5'mtrohenceri'2 -doro*4-m¿tiJbencen- . . * 2,3-dimetilbcncen...........
i».í.C
Dcrivadoi
Addp’C
1 Amidai -y :
Aníltdft’C
10 57 153 13612 • • s 108 9612 • % • 1Ü012 » , . 11014 66 153 i ió16 - - r 7519 13519 , r , 13821 - • r 14225 48 14328 • » • m33 91 122 7734 i r .34 62 Í37 i ió34 • s . 17535 ■ • i 16636 , , , 125 ♦ 4 •38 • a » 134 9633 161 16039 ’óá 96 ...40 2l2d.41 • 4 - 23741 • % . 176 •.,43 - r . 16844 S m . 187 14846 * • • 1864? ... 167 ...
T ablas de d e r iv a d a s 4 0 3
T a b l a . 4 5 (continuación)
Clonuros de sulfonilo (sólidos)
Cloruro de rultohiJo j». í.DuradM
ÚC Arcdda ( ArúW*’C ’C 1 *C
2 .5 -d ic to rO '6*m etilbenc«n* . . ................2 > m e til '4 'b ro m o b c n c ín .................................^ c lo r o - S ^ - d im e t i l b e n c e n - ........... ..........4-b*>mo-3-n je ti I bcnccn» ................... o-broroobcncen- ................. ..................3,4*dim ctilbM \ccn- . . . . .............................3r4-diflon>-2-inetilbrnccn>................. .2,4*d>cí ° r<v n,:cri............................................./r-clor«>bcnccn- ................... ..................2-rnct¡M -cli>rt>bc.ncen - ........3.5-dictora'2-meljlbcnccn- .4* < d o co -2 -m ctilb en c eü -................... ........Cctil ................. ...................................2 .6 -d » cl« o * 4 -m e tttb en cen - 4-rnetilbeaceo-l,3-di- , ...........................2 ,4>ft'lr in jc ti]b en c * n .....................................2 .4-dictoro*3*m cLübcncen* 5-cloro-2-metí)-3*niírobcncen- 4 ‘m cü l'3 -b rc in o b cn c e n * ........... ..3-doro-2*metil-6-nitrobonf:ejv . . . . .2 .4 .6 -ír ib r^ m c b c u cc ft.... . . . .2.4.5-irio]ctilb<inoe n 2 J5 < lin ,>«tLK¿*n¡crob€iv:cn- ...................3 ,4 * d íc fo ro .2 .5 -d im c tilb en c cn ‘ ...........« t-n U ro b en c en - ................................... .. ►..4*mctil'3-<l<*nkl>cnc4ín* ...........................1 .3 -benccnd i- ....................... .....................2 -< Jo ro .5*m ctit-3 -riU rob ínce n 2.3-<íicli5ro-5“iT K tilbencen“ .............. .3 -c lo n > 2 * m etíl* 5 -m tro b en ce iv 2 .3 .4 -tr ic lo ro b en cen * .... ...............................( - 4 - ) -canfor* 1 0 ..............................................1-naft& len- ^ •n ítro b c n c e n - .............................................6 - to lu en .......................................................- ...................... * -3 ,5^ i¡c lo ro -4 .rt^e t¡!benccn . 5*< !o rú -4 -fnct¡)-3 -n itrobenccn - . . . . . 2,4*d im c io x ib e n c cn .......................................2 .5 -d ib ro m o b c n c en - .. .....................3 ^ -d id o rO '2 ,5 -d im c tilb « n c e n ..................2.4-didon>-5*metilbcncc n 2J5 -d » n x til-4 -n itro b c rK « n ‘ ....................£ -b ro in o b e n í£ n ........................................ ..2 -n a fta Jcn - . .................
. í,i-d i2 jro n * sli¿ n c en - ...................................I,2 -d > m etilb cn o cn -3 ,5 ‘d i - ........................0 -n 'u ro b c n c tn - .............................................
49 .. ► 18550 168 4 • •50 100 185 15550 , 146 • • •51 186 , . r52 '¿4 144 • ■ •52 22353 180 . » •53 68 !44 10453 18554 t .. 186 «•.54 185 »-«54 97 . • •56 % * • 135 - r •56 191 18956 77 142 10959 203 . « •60 % m • 167 « • •60 • * 146 , . .60 is i • . •G0 720d.61 112 181 , , ,61 200 173 14462 201 15763 166 >2663 12863 229 A 4 é63 % 196 • « •64 158 4 - •64 16165 226d. . . t67 193 132 12168 90 150 11263 70 191 11569 92 137 10369 191 k , .70 162 % 4 .71 192 167 % • •71 19471 165 17172 176 >..75 140 190 13175 103 166 11976 91 217 13279 190 ...79 239 20080 95 180 171

4 0 4 YebJos d* derivados
T a b la 45 (continuación)
Cloruros de sulfonilo (sólidos)
DoiwfctClonáis de toitíoeilo p. i.
'C Addo Anuid Anllid*«C ’G 'C.
2,5-dímclilbc neen - 3 ,S-di- .............................. 81 295 1744í6-4ícloro.2J5-<Iin>«tilbencen- ..................... t i , . . 150 175i-m ctiM -naítakn- 81 • ► ♦ 174 158
82 . . . 133 , . r83 . . . 193 , 4 ,
4HnwtDxibeacen'],3K3i- ............... ............8586
183240
143209
( + )-caníor-3- ............... ..................... 83as
143116
124
88 • • 9 1862-tactU bencen-l^^i- ...«.«.......................... 88 • . * 260 162
90 186 , . .91 104 . , « • r fc92 103 102
2<lort>*5-n\ctil'4-mfTobencen- . . . ►............ 92 188S,3*dínjc:tjlb«howi' .............. 94 4 • • 135 119
95 104 69S-metUbcü£<n-l15-<dt- ..................................... 95 216 153
95 • • 1 198 14398 224 17899 123 18199 235
«t-lúlrobíncít- . . * . ........... . ................... ♦ , . 100 I59d.34)rojaBob«ittn-l^*<ií- .......................... 100 f , . 2568-clor^I-naTulcii- . , ............ 101 • 1992/4-<liwtrcbcnain- *....................... . ............... 102 157
108 lOOd. 224110 145 192 182n o 154
4 ’roctilb«nccn-l,2-<3i ................................ .. 111 237 190113114115 113
236
i 87 230 •
237140125 ■ *
n * •
4*hidwixi-2,6*dÍTOetilbenceri-!,$-<ii- ........... 119 208 207121 202 133 •121 105 214 202122 177122123 276!23 189
DUciülmetan-4'4Mi....................... . 124 59 178125 144128 125 298129 249 196 • v130 76 170138 . . . i
Tabla* d* d»ríva<k* 405
T a b la 45 (continuación)
Cloruros de sulfonilo (sólidos)
C tw uro de «lllfonilo
Derivado*
d-canfo'r.S- .............. ......... .....................l ,4 ‘benc«ndi- . , ................................................l r2-benccn<Jí- ....................................................l»S^-na£tal«itri- . ................ ..........................5-doro*2«r>ietil-$>mciahcftten.....................1.4.5-íiaftalentr í .......................2-anüni>5-rfteri!beitcen-l,3-<ii- ...................27?-naffaú«MJi- . . . , .........................................SjS-^lmílifbencen'l^^i* ........................1,4-naítafendi» ........................... ...............2-doro>7-ancraKiuinQn' ..................................Iy5-na/talcj»d¡'. . ................................................2 ,7 -a n tra q u ín o i. ......................, . . . ►...............1.3.5-boncentri- . , . ■ ,............................. . .ljSjG-naítssdcncri- . . . - ............................ ..Antwjulüon-P- , , : ........................ .........1.6-autraquinandí- ¿^jC-naftálenúi- ......................................2'dori>5'antraqwinon- .......................... ,Bifcnü'í^-di- ................................ . . . . . AntrAquínon*^......................... , ..............^HWidoroLantraíen-S-............................3 ,8-antraquSaondi- .................... ....................2.6-rjAftnlencü-... Antracen-l,&-diU ..................... - ......................1.7-AntrnquinaJwJi - . .......... - ...........2,^antrBquinor»d.i- *............... ............I j5-ancniquJnoiidt- ............... - ................2/Wi»niínob<MCen4,5-d¡- . . . . . . .............
p - f- ‘C Aod„ Arofrfa
4CAnilló»
*C
138 137 4 4%
139 28 8143 25 4 241146 9 9 < . . . . 4 4154 r • • 183156 • 4 - ... * ♦ V156 « • • 257 192162 242 . ..164 310 22 3166 273 179176 • * ♦ , .,183 245 310 24 9186 . . , 192187 ió ó d . 312 237193 , , , ... . . .197 261 193198 2 Í7 ♦ » • 227<L200 ♦ 4 « - - « . . .202 , , , . r ,203 72 360 • • ♦21 4 218 ... 21 6221 279 2 48223 2 9 4 3 4 0 2 3 8225 ... *..22 5 4 • * 333 224232 120 • ♦ 4 23 82 50 ... 3212 6 5 d . 3 l0 d . 246 2 7 M .27 5 .»♦ 187 2363 10 .. • . . .

i *
í »
r»r »I
B»
U»
u
á>
r *r i
i
i »a

CAPITULO
12Introducción a la solución de problemas estructuralesE l e x a m e j í d e v n c o m p u e s t o en el laboratorio consiste en la acumulación de datos concernientes a las propiedades físicas, particularmente solubilidad y datos espectroscopia», comportamiento frente a determinadas clases de reactivos y varias pruebas especiales. Todos estos hechos observados deben ser correlacionados e interpretados para poder llegar a las fórmulas estructurales posibles para el compuesto en estudio. Es necesario demostrar qué grupos funcionales tiene, determinar la naturaleza del núcleo al cual están unidos y encontrar las posiciones ocupadas por estas uniones*
El proposito de esta discusión es señalar, por medio de varios ejemplos específicos, el modo de ataque y el razonamiento involucrado en la deducción de 1a estructura a partir de la información proporcionada por los datos experimentales.
INTERPRETACION D E LAS FORMULAS MOLECULARES
En investigación, los análisis cuantitativos, junto con las determinaciones de peso molecular, proporcionan los datas necesarios para, el cálculo de la fórmula molecular de cualquier substancia desconocida. A menudo se puede deducir mucha información acerca de los grupos funcionales posibles tan sólo con dicha fórmula y, por esta razón, es pertinente considerar la transcendencia de la fórmula molecular.
U n hidrocarburo saturado, sin aníUos, tiene la fórmula general OriHn‘2- La introducción de oxígeno para dar un alcohol, éter, aceta! o cualquier otro compuestos acíclico saturado, nc cambia la relación car- bono-hidrógeno y la fórmula molecular es La formación deun enlace doule 0 un anillo en la molécula ¿aturada requiere la eliminación de dos átomos de hidrógeno y la formación de un enlace triple involucra la eliminación de cuatro átomos de hidrógeno. Así, medíante un examen de la relación carbono-hidrógeno se pueden deiducir conclusiones concerniente* al posible número de enlaces múltiples o de anillos que pueda tener una molécula.

4 0 $ P ro b lw t ia * M tru c tv ra l* $
Por ejemplo, la substancia CjHoO debe tener un enlace doble (ya sea oleflruco o carbonílico) o un anillo* pero no puede tener un enlace triple ya que sólo le faltan dos átomos de hidrógeno para ser saturada. El compuesto C*Hi¿0 debe tener tres enlaces dobles o tres anillos o alguna combinación de enlaces dobles, enlaces triples y anillos, que corresponda a los tres pares de átomos de hidrógeno fallantes. Sin embargo, no puede tener un anillo bencénico debido a que se requeriría la falta de cuatro pares de átomos de hidrógeno para la saturación. (Así, inmediatamente se excluye la posibilidad de que fa función oxigenada sea un grupo oxhidrilo íenólico).
Además, la introducción de un átomo de halógeno en una molécula saturada requiere Ja eliminación de un átorno de hidrógeno y, consecuentemente, la fórmula general de un monohaluro odclicQ saturado es CtHaniX. Por otra parte, la, introducción de un átomo de nitrógeno para dar una amina acíclica. saturada también requiere: la. adición de un átonio de hidrógeno, por lo que la fórmula es C JK ^ jN . Una consecuencia de estas generalizaciones, que es valiosa para la derivación de una fórmula molecular a partir de La. determinación de carbono c hidrógeno por el analista, es, que una molécula que sólo contenga los elementos carbono, hidrógeno y oxígeno debe tener un número par de átomos de hidrógeno; un número impar de átomos de halógeno o nitrógeno requiere ún número impar de átomos de hidrógeno y un número par de átomos de halógeno o nitrógeno requiere un número par de átomos de hidrógeno. Así, una fórmula molecular C jH jxO j, calculada a partir de datos analíticos, obviamente es incorrecta; la fórmula correcta debe ser QsH ijOj ó CsHgOj.
PROBLEMAS DE EJEM PLO
En el capítulo 9 se describieron dos constantes físicas muy útiles, los equivalentes de neutralización de los ácidos y las bases y los equivalentes de saponificación de lo* ésteres. Estos datos numéricos, junto con la dase de solubilidad 'y comportamiento frente a los reactivos, frecuen- teme ate proporcionan valiosos indicios concernientes a la estructura molecular del compuesto. Su empleo se explica mejor mediante el estudio de ejemplos.
e j e m p l o L U n ácido orgánico tiene u n equiva len te de neutra#nación de 45 ± 1, .
Gomo se indicó en la pág, 2é0, el equivalente de neutralización de un leído depende del número de grupos carboxilo que haya en la molécula. Si sólo hay un grupo carboxilo, el equivalente de neutral!-

Problem as d* ejem plo 409
zadón es igual al peso molecular. Si «1 compuesto desconocido es monobásico, su peso molecular1 debe ser 44, 45 ó 46, U n grupo cadjoxilo pesa 45; por lo tanto, si el peso molecular fuera 45 nada podría estar unido al radical carboxilo. U n peso molecular de 44 obviamente es imposible, peno ur* peso molecular de 46 deja uri residuo de I después de substraer el peso del radical carboxilo. Sólo un elemento tiene un peso atómico de 1; por lo tanto, el ácido fórmico (HCOOH) es una posibilidad.
Sin embargo, el compuesto podría ser dibásico, en cuyo caso el peso molecular sería 90 ± 2. ¿tos grupos carboxilo son igual a 2 X 45 ~ 90. Por lo tanto, queda un posible residuo de 0, 1 ó 2 unidades. No hay átomos bivalentes con este peso atómico; de aquí que el único áddo dibásico posible sea el ácido oxálico, en el cual los dos grupos carboxilo están unidos; CO2K. Asi, suponiendo primero un ácido monobásico y
ICO*H
después un ácidi dibásico, se han deducido dos estructuras posibles partiendo tan sólo de; equivalente de neutralización. Para decidir entre las dos serviría el estado físico del compuesto o su clase de solubilidad. Si este compuesto, con un equivalente de neutralizadón de 45 ± i , es un líquido soluble en agua, debe ser áddo fórmico; si es un sólido soluble en agua, es ácido oxálico anhidro.
La consideración de los pesos moleculares indica en forma análoga que el compuesto no puede ser tribásico (peso mol. 135 ± 3 } o tetra- básico (peso mol, 160 ± 4),
e jem plo 2. Un compuesto tiene una fórmula molecular Q H jO y muestra ' tina fuerte absorción de carbonilo a 1740 cm"1.
La fórmula molecular tiene un átomo de oxígeno el cual puede encontrarse como alcohol, éter, aldehido o cetona. La rcladón carbono- hidrógeno muestra que el número de enlaces dobles o anillos es de dos. La absorción de carbonilo indica que un enlace doble Se encuentra como enlace carbomüco. Aunque no se podría excluir un enlace doble carbono- carbono por la ausencia de absorción en la región correspondiente del espectro en el infrarrojo, la posición de la absorción del carbonilo se puede explicar mejor colocando el grupo carbonilo en un anillo de anco miembros. Por lo tanto, no puede haber otro enlace doble y la substancia debe ser la cidopentanona.
e j e m p l o 3. Un ácido (A) posee un equivalente de neutralización de 136 ± i. Dio pruebas negativas para halógenos, nitrógeno y azufre, No decoloró en frió una solución de permanganato de potasio, pero cuando
’ Confto )m prODvíltoi de a n enchufo ion Uutfrtu- hL <Je tooh er probfenMi j«ítciuar Joi necmric*, h«m«a íuad» núrrxrot ía lttm por* ta pooc •'¿mico) de Coi«tcmQfilo* carbono, fatdpígcfiO, ■iicójrno y bromo. Lot peío» «ióuÚüm VrrA»dtnn
) (quti deheo CfnpfeATK tlt Codos Icú anJUi*rt Ctsa(iUtivox e'4 « « ) difitft» 4a y Jarra Ttdon-, en UT># Cánlidjd meao* /4UC el mrnr^ «prríjretjHl imvlvcJído en la d<trrmínio6a ¿c tai- * Q b r í i l i T l « j d e a c a f r a f i i i c j i e j ■ a p o n i i í a ’C Iv fl.
fí:r. •
>■■ .

una solución alcalina del compuesto se calentó con este reactivo duran-* te una hora y se aciduló, se pteápitó un nuevo compuesto (B ), Este compuesto tuvo un equivalente de neutralización de 83 + í.
Primero considere el compuesto B. Suponga que es monobásico.Peso molecuJar = $3 ± I
Menos un — C O O H = 45sa* i
Este residuo al ctial está unido el grupo carboxíio debe estar formado por alguna Combinación do carbono, hidrógeno y quizá oxigeno, que es estable a la solución caliente de permangaoato de potado* El siguiente examen muestra que esta suposición no es posible.
Residuo c= 36 i 1T ris átomos de carbono = 3 X 12 ~ 36___
Remanente = 2 ± IEl residuo podría ser CjH CjH* ó C3Hí, ninguno de los cuales
corresponde a un compuesto e¿able al permanganato. La paraíina requeriría CjHj. como compuesto iundamental y cl radical alquilo tendría que ser C»H7—; análogamente, la cídoparafina tendría que ser C¿H¿ y el radical ciclopropilo C iH j—.
También se excluye la presencia de oxígeno. Si se Supone estar presente, se obtienen loa valores siguientes:
Residuo = 38 ±. 1 o Residuo = 38 ± lUn átomo d e oxigene» = J6 Dos átomos de oxígeno = 32
± 1 Remanente =ü 6 ± 1U n átomo de carbono — 12____
Remanente = 10 ± I
¡Ninguno de estos remanentes corresponde a un átomo o grupo de átomos. Asi, ahora se concluye con seguridad que el compuestos E no puede ser monobásico.
Suponga que el compuesto B es díbástco.Fcgo molecular ^ 2 X 83 ± 1 s= 166 =b 2
Des grupo» carboxilo ~ 90Residuo =* 70 ± 2
Si este residuo cs parafínico debo estar formado por unidades — (CHj) —.Cinco — C H j — = 5 X 14 = 70
Sci, - C H , - = 6 X 14 = S i
Ninguno de estos corresponde al peso del radical residual, 76 cb 2.Otro gTupo que es estable al permanganato caliente es ei núcleo
bencánieo. Este está formado por seis grupos CK 6 6 X 13 = 73 = peso molecular del benceno mismo. Si están presentes dos grupos carboxilo se habrán desplazada dos átomos do hidrógeno y el residuo resultante es
— 2 — 76. Este valor corresponde al que se calculó arriba para el residuo y de aquí que la posible estructura para B sea C¿H+(COOHh» esto cs> que puede ser uno de los ácidos itálicos.
4 1 0 P ro b le m a s estructural© *

P r o b le m a * d e e j i m p t o 4 1 1
Ahora se plantea la pregunta de » el compuesto B podría ser tribásico. Asi) tenemos los valores siguientes:
P e ro m o le c u la r = 3 X 3 3 ± J = 2 4 9 ± 3 T r « í g ru p o s c a tb o x ilo = 3 x 4 5 = 135
Residuo — 11 * ± 3Este residuo no puede ser aromático ya que no corresponde a uno o
más anillos bencénicos. U n anillo bcncénico más una cadena lateral queda excluido porque la cadena lateral oxidaría con el permanganato. Sin embargo, el valor 114 ¡fc 3 corresponde a ocho grupos — CH2— > dentro del error experimental (8 X H = 112). De aquí CigH^COOH ij, con un peso molecular de 246, queda dentro del limite 249 ± 3. Aun» que este ácido tricaiboxíÜco representa una estructura posible para un compuesto con un peso molecular de 249 ± 3, sería imposible obtenerlo a partir de un compuesto A, que tiene un equivalente de neutralización de 136 ±: I,
suponiendo que A es monobásico.Pew molecular d e A = 136 ± 1
U n —C O O H *= 45ft**iduo = 91 ± 1
Ya que B tiene un grupo C6H 4í estable al permanganato, este mismo grupo también debe estar presente en A.
Residuo = 91 ± JC,H, - 76____
Remanente = 15 ± 1 Este remanente de 15 -± 1 corresponde a un grupo metilo, cl cual
debe estar unido al anillo.COOH COOH
-+ G J I ,
Hj \ s O O HCtmÉpUAtfn A C««iavKit9 ¡BE.N . t= 1 » B.N. = «3
El compuesto original debe ser d ácido o , w o ¿-toliiico; los tres darían las reacciones citadas. Se necesitarían datos adicionales, tales como punto de fusión o un derivado, para diferenciarlos.2
1 Nota Pora rf j>r*í*;or, Oxr*o todM lea pfofocxt* <*í CWW »3e ¡de»l¡íit*cíóii fctltín,uit» d t bis ra&yort* dificxúLad«a q«c te *1 oeritór jttotA-.ma* de « le t''f*a c* Jtaiicíjuirsc* *o4oi lo» rim ín rp tju t Je(sulr r l « h íd iá íi te par» ftw l-.trio*. Et advenim iento IK U "f.ru«ím ic»" ¿<jb tu ^ tnuilluciv i) cb d ¡olrrji par 1ot ¡tólopai b i tnuonealida rít» iü(iculli<I. Puf q —íplp, en « f pfoblettU te pueden «ocoutror <*T»J rU juctunu ¿ ubi C” , M . Cí, ,H X ? lO O ] [ tfeaie Un I>tin roofccütar tk M y to* re<jwbít<>* <¿e*¡a»f «írS corajn»e*lo H. U n a tn d b iU eU « a puede jurgrumtfUarr <tu« rttr, <ddl> y. jKXÍfil obtener, u n bwí'i* K tttfiralw 'lo, ■ Jkirtlr de *«a
• ‘ * ife r t» a c V 1*5 « c * iw :;i r . t o
: • • <C»Eü)iO»! 'C"IQC*'KiC"OOH« ttí t í tn r o n p a o moleeatirr J 37 y m í poóth. te t u n a « tím C u r j poúWe p ar* «I rnm puctin A. En© tipo da a iyurteftto k enfa» tu n «I m tuítik* J í que rn b » m n u tH i i íi í í w *a tab len** *• utilícen» »W<j W topcü wrfHí*le*.
CeH4

4 1 2 P roblem as Mfrvctv roles
Este ejemplo también ilustra el hecho de que la oxidación casi invariablemente convierte un compuesto con un determinado equivalente de neutralización en un producto que dene un equivalente de neutralización menor. Esta generalización es consecuencia natural del incremento en el número de grupos carboxilo o escisión de la molécula en fragmentos más pequeños.
bjempjjO 4 . -í/ftc substancia (A) tiene la fórmula. C^H». For hidroge- nación con platino en condiciones moderadas se convierte en B, Cuando se ozoniza el compuesto A, forma dos producios: acetaldehldo y gtioxal.
La fórmula molecular muestra que el número máximo de enlaces dobles y anillos es de dos, o bien, un enlace triple. L a absorción de dos moles de hidrógeno indica, oue hay, de hedió, dos enlaces dobles o un enlace triple. La oaonólisis al dar fragmentos de dos carbonos, sugiere que t i esqueleto de seis carbonos que originalmente se tenía debe haberse escindido en dos puntos en lugar de en uno para dar tres fragmentos de dos carbonos. Esto significa que debe haber dos enlaces dobles y no un enlace triple. Finalmente, la distribución probable de los tres fragmentos de dos carbonos es la que se muestra en seguida:
CH3C H = rC H C H =C H C H |
ejem plo 5, Un compuesto cristalino, incoloro (A) dio prueba positiva para nitrógeno pero no para halógenos o azufre. Fue insoluble en agua, ácidos diluidos y álcalis. Reaccionó con cloruro de acetilo pero no con fentlhidraztna. El compuesto A se disolvió en solución caliente de hidróxido de sodio con desprendimiento de amoníaco y formación de t*rw solución transparente. Por acidulación de esta solución se produjo el compuesto B, el cual no tuvo nitrógeno y dio un equivalente de neutra* litación de 182 ± 1. Por oxidación de B con solución caliente de permanganato Je potasio se produjo C, el cual tuvo un equivalente de neutralización de 98 ± 1. Cuando A o TI se calentó con ácido bromhl- drico durante algún tiempo, precipitó un compuesto D. Este compuesto tuvo bromo> pero no nitrógeno; dio un precipitado con agua de Bromo, color violeta con cloruro férrico y rápidamente redujo e l permanganato de potasio diluido, D fue soluble en solución de bicarbonato de sodio.
Estas reacciones pueden resumirse en el esquema siguiente:
_ , n«oh _Compuesto A -------► NH3 +. Compuesto B
jm tr j KMBo<
Compuesto D Compuesto C

P roW w no* d * « Jom p lo -413
L a eliminación de nitrógeno por hidrólisis alcalina del compuesto A sugiere la presencia de un gtupo nitrilo o amida, ya que estos grupos funcionales liberan amoníaco cuando sufren hidrólisis.
RCN 4- NaOH + H 20 - > R C 0 0 N a + NHi RCONH* + NaOH RCOONa + NH3
El grupo ¡mida, — CON H CO —, que también desprende amoníaco, queda excluido por que el compuesto A no cs soluble en solución de hidróxido de sodio. Ya que el compuesto B no tuvo nitrógeno, se excluyeron aminas con substituyentes negativos, tales corno la 2,4-dinítroanilina. La ausencia de nitrógeno en B y el hecho de que fue ácido y no básico, igualmente eliminan ureas substituidas, que también desprenden amoníaco cuando se hidrolízan,
RNHCONH* + H*0 - » RNH, + C 0 2 + NH5
La prueba posa. 'a con cloruro de acetilo sugiere un alcohol; se excluye un grupo amino o fenójíco por que el compuesto A es neutro. El hecho de que ¿1 compuesto D, producido a partir de A por acción del ácido bromhídrico, tenga bromo, proporciona una evidencia adicional de la presencia de un grupo oxhidrilo,
R O H + HBr RBr + H30
Las propiedades del compuesto D sugieren fuertemente Ja presencia de un grupo oxhidrilo fenólico. La facilidad de bromación, la sensibilidad al permanganato y el color con cloruro férrico son propiedades características de los fenoles substituidos. Este grupo oxhidrilo fenólico se produjo por acción del ácido bromhídrico sobre algún grupo funcional presente en A y en B, ya que ninguno de éstos tenía originalmente el grupo fenólico. U n tipo de compuesto que produce un fenol cuando se tra ta con ácido bromhídrico es un éter de alquilarilo.
. ArOR 4" HBr —> AiOH 4 RBr
Sí el grupo alquilo es pequeño, se pierde como bromuro de alquQo durante el tratamiento con ácido bromhídrico. Asi, kw compuestos A, B y C probablemente tienen dicho grupo éter mixto y también un núcleo aromático, ya que el fenol substituido D tiene uno. La solubilidad de D en solución de bicarbonato de sodio se debe probablemente a la presencia de un grupo carboxilo, ya que tanto los grupos nitrilo como los amida se hidrolizan a grupos carboxilo con ácidos o con álcalis. Por lo tanto, el compuesto D es un áddo hidroxibeazoico con cl bromo unido a una cadena lateral. Esta cadena lateral también debe encontrarse en el compuesto A, con un grupo alcohólico en lug3r del átomo de bromo.
Ahora se pueden considerar los equivalentes de neutralización de los compuestos B y G. Se observará que el equivalente de neutraliza-

f *I
92»u
i B
í i f *
f * *r
á#í •
ción del compuesto <J, producido por oxidación con permangaaato, es menor que e! del compuesto B, que -ojo adquirió sus propiedades ¿sidas después de una reacción de hidrólisis. Obviamente esta oxidación afecta la cadena latera), y el compuesto C debe tener más grupos carboxilo que B.
Suponiendo que el compuesto C sea dibásico;Peso m olecular = 2 X &a i I = 196 ± 2
Dos gnipoj carboxilo = 2 X 45 = 3 0106 ± 2
Un átomo de oxígeno en cl enlace ti¿rco = 3690 ± 2
Benceno menos lc«6 átomos de hidrogeno (C<H») ~ 75Residuo = 15 ± 2
Este residuo de 15 corresponde a un grupo CHS— y de aquí que el grupo etéreo debe ser C H jO —.
Ahora es necesario encontrar la •'ngitud de la cadena lateral a la Cual está unido cl grupo alcohólico en A y en B.
Si C es dibáxico, B es monobásico.
E.N. - Peto Mol. = 1*2 ± I G rupo carboxilo = 45
'll7 ± 1 Grupo inctoxilo = 3 1 _
1 0 6 ± 1 Grupo oxhidrilo = i?
“ 89 ± INúcleo bencénico (C*ih) — _?5
R esiduo = 14 ± l
Este residuo representa el peso de la cadena lateral y obviamente corresponde a —CH2 —, por lo que las estructuras posibles para A, B, C y D son:
414 Problem as estructurólos
CONH2 (orCN )
CflH ^C H *O H
X o c h 3CompijWÍO A
COOH
Q H 3-C H *B r
ComcM*B<o D
CÓOH
Q H ^ C H 5OH
so c h 5Compuesto B
1KMnO
COOH
Q H 3—COOH
OCHaCooijWHtoC

Problem as «fe ejem plo 415
Este ejemplo ilustra el hecho de que un determinado reactivo puede afectar más de un grupo funcional. Así, el ácido bromhídrico a ebullición afectó tres grupos funcionales en el compuesto A y dos en el compuesto B.
Eji la identificación de compuestos nuevos es costumbre hacer análisis cuantitativos de los elementos y determinar el peso molecular. A partir de estos datos se puede calcular la fórmula molecular. Considerando las fórmulas moleculares de varios compuestos y las reacciones que los produjeron, frecuentemente se pueden deducir las fórmulas posibles. El problema siguente implica la deducción de una gran cantidad de información a partir de sólo unos cuantos datos.
e j e m p l o 6 . Un compuesto neutro (A ), C jjH mO , da prueba de Baeyer -negativa y no lo ataca el ácido bromhídrico; A se oxido a un ácido(B), CkHjoOj, con. deido crómico.
Primero observe que la oxidación ha causado la pérdida de un átomo de carbono y .cuatro átomos de hidrógeno, pero ha habido una ganancia de dos átomos de- oxígeno. Es necesario encontrar un grupo funcional o varios grupos en que esto pueda ocurrir. Para este propósito es útil tabular el comportamiento en la oxidación de los grupos funcionales comunes, tomando nota de la ganancia y perdida en la composición.
En la tabla de la pág. 416 se ve que la oxidación del grupo etilo de una cadena lateral corresponde exactamente a la oxidación de A a en ambos casos hay ganancias de dos átomos de oxígeno y pérdida de
: cuatro átomos de hidrógeno y un átomo de carbono.
Substrayendo un grupo etilo de A (C^H ^O ) o un grupo carboxilo de B queda un radical Este radical se deriva de algún compuestofundamental (O ), C^H ^O , el cual es estable a la oxidación.
El pioblema siguiente concierne al carácter del grupo funcionat que _ contiene el átomo de oxigeno; ¿Qué grupos funcionales que sólo contie-
nen un átomo de oxígeno so» estables a la oxidación? La consideración >. de varios grupos funcionales conduce a la conclusión de que una podbi- ' lidad es el enlace etéreo y una segunda, una cetona substituida conve
nientemente. Las cetonas que no tienen átomos de hidrógeno en los ; átomos de carbono a, ordinariamente son estables a la oadación. Los .ejemplos más comunes son las diarilcetonas.

4 l6 Problem a! estructuróles
Grupo / k « M Producto d< oxidación GatonctA . Pérdida
RCHO -► R C O O H 1 -0RCH iO H -*• R C O O H 1 -0 2-Hk ,g h o h -> R iC O 2-H
O HyR jC -> R ,C O 4-H 1-C
\ h ,R C H = CHX —> R C O O H 2-0 2-H 1-CRG m C H -*■ RCOO H 2 -0 l-CA iC H jC H iO H -*■ ArC O O H 1*0 4-H 1-CAtCO CH j -♦ A tC O O H 1 -0 2-H 1-CArCH> • -+ ArCOOH 2 -0 2-HArCHaCHs -> ArCOOH 2 -0 4-H 1CAriGHCH, AriCO 1-0 4-H 1-C(A ÍG cH mO - * (B )C hH iiO) 2*0 4-H 1-C
El siguiente paso consiste en considerar la relación carbono-hidrógeno en los compuestos A y B y en el compuesto fundamental hipotético(C ), CtfHicO. Éstos átomos de carbono e hidrógeno deben estar combinados en tal forma que el compuesto resultante sea estable a la oxidación. Un compuesto saturado, C «H ^27 requeriría una fórmula CuHy para un compuesto {C) con trece ¿tornos de carbono. Aun considerando dos átomos de hidrógeno com a .equivalentes a un ¿tomo de oxígeno, es obvio que el compuesto no tiene tal relación de átomos de carbono e hidrógeno. U n compuesto alirídico requeriría oC jjH». Los compuestos olefínicos y los compuestos acetílénicos con suficientes enlaces dobles o triples para disminuir la relación de hidrógeno a carbono quedan excluidos por la estabilidad de (G) a Ja oxidación.
La única dase de compuestos con dicha relación baja de átomos de hidrógeno a carbono es de naturaleza aromática. Como el benceno tiene seis átomos de carbono y el compuesto fundamental (C ) tiene trece, se sugiere la posibilidad de que haya dos anillos bencílicos. Esto deja sin explicación la naturaleza de un átomo de carbono.
Restando los dos radicales fenilo (Cu FIw) del compuesto C (G |SHwO) queda un resto CO. Se recordará que las diarilcetonas son estables a la oxidación. Evidentemente, el compuesto fundamental (C ) es benzofe- nona, CíH jC O C J^ , y los compuestos A y B son, probablemente, una etilbenzofenona (CíH jCOC íH íCH^CHj) y un ácido benzoilbenzoico (Q H jCOC íH+OOOK), respectivamente,
e j e m p l o 7. Un ésier, que sólo contiene carbono, hidrógeno y oxígeno, potee un equivalente de saponificación de 74 ± 1.
El primer paso es considerar todas las posibilidades originadas por la sup*. ádón de que la molécula sólo tiene un grupo éster. En este caso el peso molecular es igual al equivalente de saponificación. La

P ro b le m a s d« « [«m plo 4 1 7
fórmula tipo para un éster es R — C O O — R ' y, por lo tanto, <1 primer paso es restar el peso de —CO O — del peso molecular,
Peio molecular = 74 ± 1 - C O O - - 44
rwkíuo s= 30 d; ]
Este residuo representa el pe«o combinado de R y R', En los «teres saturados* que sólo contienen carbono, hidrógeno y oxígeno, este residuo siempre debe ser igual a CJFr^.j y, además, siempre debe ser un número par.4 Así que los pesca residuales de 31 y 29 son imposibles y el valor 30 representa el peso molecular de En este caso, a simplevist# se deduce que el resto de hidrocarburo es CjH j, pero la forma lógica es despejar el valor de n multiplicando por los pesos atómicos de los elementos que haya en la fórmula e igualando con el peso residual.
I 2 n + 1(2» 4- 2) = 3014a = 28 y r = 2
Este resto CjH¿ representa Ja suma de R y R ' y ahora es necesario escribir Las posibilidades.
R R' = CjH*H + CjH»
C H ,+ C H ,
Por lo tanto, para la suposición de que el compuesto sea un mono- éster hay dos posibilidades.
H C O O C iH j y CHiCOO CH,/onmalO efe tlilq acr|;iC6 dr liH'lilo
Si en la molécula hay dos gruj»s éster, entonces el peso molecular es el doble del equivalente de saponificación. Dos grupos éslcr tendrán dos —C O O — por lo que:
. Pcbo molecular = 2 X 74 ± 1 t= 148 ± 2 Dos - C O O - = 2 x 44 = fiS
Residuo = 60 ± 2Bl ^ator 60 ± 2 representa la suma de aquellas partes de la molécula que no sean los dos grupos —CO O —, Nuevamente, CnHí,* = 60 ±. 2 y
12* + 1(2* + 2) = 60 ± 2 34» * 58 ± 2
Ya que n debe ser un entero, el único valor del lado derecho de la ecuación que satisface este requisito es 56 y, en consecuencia, n = 4. El resto debe ser C^Hjo y tiene que dividirse entre los diferentes radicales
5 E n u n ¿ * 4 * t « f c l í n k o e l tM W n C - H j . ; t u u n I r t t T i c r tQ íc W s o ,4 Véa p«d de ptgtafc: Jr j»ig. <09 y 2, p ig . 411.

41 8 P ro b le m a s e s tru c tu ró le s
cooA COOR|
(G H O , (R C H ),
CO O R' ¿ O O R
hidrocarbonados que se encuentren en las fórmulas tipo para un compuesto con dos grupos éster. Algunos de los posibles tipos de fórmulas son los siguientes:
RCOO RCOO
(¿ H ,) , (¿ H R ),
RCOO RCOO
Usando estas fórmulas tipo para los éateres de un ácido dicarboxU lico o un alcohol dioxhidrílico, se pueden escribir posibles estructuras en las que los cuatro átomos de carbono y diez átomos de hidrógeno estén distribuidos apropiadamente en cada una de las fórmulas anteriores.
Este ejemplo ilustra el uso de los equivalentes de saponificación en la deducción de posibles estructuras. También muestra que el equivalente de saponificación no es tan útil t 'rao el equivalente de neutralización de un áddo. Siempre es convenid. e tener algunos datos concernientes ya sea al áddo, al alcohol o a ambos productos de la saponificación del éster, para disminuir el número de ésteres isómeros que posean el requerido equivalente de saponificación.

CAPITULO
13ProblemasLos p r o b l e m a s s i o u i e u t e s e s t á n concebidos para proporcionar a l estudiante una experiencia adicional en k» tipos de razonamientos ilustrados en los ejemplos del capítulo 12. Es de importancia primordial buscar Jas respuestas mediante procedimientos sistemáticos y se apremia a los estudiantes para que eviten atacar los problemas en forma desorganizada.
Grupo 1
En la investigación de compuestos desconocidos frecuentemente se observan los tipos de comportamiento siguientes. En cada ejemplo indi- ■ que las deducciones quo puedan hacerse sobre la naturaleza dei compuesto.
1. U n compuesto neutro, amarillo, que «Slo contiene carbono, hidrógeno y oxígeno, se convirtió en un Ácido pt>r acción del peróxido de hídrógeao.
2 U n compuesto neutro, reaccionó con feoilhidrazina dando un producto que diferia de la esperada fenilhidrazona en los elementos del etanol (C iH ,O H );
decir, la condensación involucró no tan sólo la eliminación de los elem&ntoa del agua «n o también de los del etanol.
3. U n compre*te que <63o contiene carbono, hidrógeno y oxigeno reaccionó con cloruro de acótilo, pero no con femlhidraxina. El tratam iento coa íd d o per- yódico lo convirtió en u n compuesto que reaccionó con fenilhidraána, pero no con cloruro de acetílo. .
4. U n com puesto n eu tro , am arillo , d io u n d erivado con ¿>-fe&)1endiamina.5. U u alcohol dio positiva la prueba del yodoíormo y negativa la prueba
de Lucas,S. U n compuesto neu tro que sólo contiene carbono, hidrógeno y oxígeno
reaccionó con cloruro de acctilo, pero no con fenilhidrazir.;., El calentamiento con ácidos minerales lo convirtió en u n compuesto irca p a i d e reaccionar con cloruro de .acetilo, pc“0 que dio prueba* positivas con fcnilhidrazina y bromo en tetracloruro de carbono,
7. U n compuesto que contiene nitrógeno dio positiva la prueba del ácido nitroso para aminas secundarias, pero su derivado con cloruro d e bencensutíodlo fue soluble en álcali*.
8. Guando un compuesto *í trató con ortoformiato de etilo y huellas de ácido, se encontró que su fórmula m olecular se había increm entado en los elementos del éter etílico (CjHjOCfeH,)»
4 1 9

4 2 0 P ro b le m a s
9. U n compuesto M u tro <jne contiene carbono, hidrógeno y oxígeno itiírió u n í tümerywciún en solución trl&nólica de cianuro fíe sodio.
10. Un Compuesto básico fue incapaz d e reaccionar con cloruro de b<i>- ccnEulfonilo, pero dio un derivado con ácido nitroso.
II» U n é*trr tuvo un equivalente de saponificación d e 59 ± 1.12. U n ácido sólido, soluble en agua, tuvo un equivalente de neutralización
de 54 ± I , A temperatura* mayores que su punto d e fusión, el com pueito perdió anhídrido carbónico y re obtuvo u n nuevo ácido lólido con un equivalente de nzutraJUación de 59 ± Ir
13, U n compuesto ácido, soluble ert agua, que contiene nitrógeno y azufre, dio un equivalente d e neutralización d e 142 ± 1. L a adición de cloruro d e bario a una solución acuosa produjo un precipitado ¡«soluble en ácidos. El álcali cauió la precipitación de un corapuesco básico cuyo clorhidrato tuvo un equivalente de neutralización d e *30 ± 3.
14. U n compuesto que condene nitrógeno y e i soluble en agua, dio un equivalente dé neutralización d e 73 ± 1 cuando ie tituló con ád d o clorhídrico valorado y con rojo d e metilo como indicador. Guando se trató con cloruro de bencensulfonilo e hidróxido de sodio dio un precipitado.
15, Urea m uestra de 12 g de un compuesto, CgHiO, se trató con una solución de 60 g de bromo en tctracloruro de carbono. Se desprendió ácido bromhídrico y u n a determinación cuantitativa demostró que se liberaron 16.2 g. Después que se term inó la reacción, la solución todavía permaneció ra ja y se encontró que quedaban 12 g de brom o sin consumir. Calcule el número de átomos d e bromo que k introdujeron p o r substitución j , si también hubo adición, el número de átomo» de bromo que se adicionaron. Calcule con Br = 50, H ^ 1,C = 12, O = 16.
Grupo 2
AI resolver los problemas de este grupo y los de los grupos 3 y 4, siga los pasos ex-poestos en el capítulo 2, usando los datos citados en los problemas. Considere las concesiones usuales a los errores experimentales en los puntos de ebullición y puntos de fusión.
I . 1) Cristales blancos; p.f. 117-118“C.2 ) A ñil ¡ai» elemental para X, N, S— negativo.5) Solubilidad— agua (4-)J4 ) P/vebas de clasificación;
CJKjNHNH j— negativa CH>COCl— positivaKM nO,— positiva H !0<—positiva Br, en CCU— negativa E J f e 151 ± I
5 ) D eriv ad o :E íte r de ^-nitrobencilo, pX 123ÚC.
11. I ) L íqu id o inco lo ro ; p.eb, 259-261 "C ,2 ) A nilb i. elemental para Br—positivo; para C!, I , N , S— negativo.3} S o lu b ilid ad -ag u a ( —) , N aO H ( - ) , HCI { - ) , HjSO, < + ) .

4 ) Pruebas de d e if ic a r a n :H íN O H • HGl— negativa KMnCV-~<'-eg»tiv3,CHjCOCl—negativa Br? en GC1<—-ncgaüvjiAgNOj— negativa N a l— negativaHidróxido de Bcdio caliente—solución tram parent*, la quo acidulada dio cristales blanco», p.f, 250C'C . que contienen bromo, Equiva- valente de eaponifieación = 229-
5) Derivados:a) Por tratarm enio con hidraarva dio edítales incoloros, p.f, 1S4°C.b) Por tratam iento con ácido 3*S-dinitrobcnzoico y ácido sulfúrico
dio c ris ta l» amarillo pálido, p.f. 92*0.
I I I . I) L iquido café ; p.eb. 198-200*0.2 ) Análisis elemental para X, N, S— negativo.3) Solubilidad—agua ( - ) , N a O H (4 -) . N a H C O » (-)p H C l ( - ) ,4) Pruebas do clasificación;
CHjCOCl— positiva Brj, en H jO— precipitadoC*HjNHNH j —negativa F eQ ,—violetaCeíNO»)*- positiva
5 ) Derivado: Por tratam iento con ácido cloroacético dio cristales blancos, p.f. I0 2 -í0 3 eC ,
IV . 1) Líquido incoloro; p.eb. 194-195".2) Análisis elemental para X, N, S— negativa.3) Solubilidad—en todo ( — ),4 ) Pruebas de clasificación:
H¿SO« • SO;— negativa A1CI» + CHC11-—amarillo claro
5) Derivados; ninguno,Dens. ejp. 2 0 /4 = 0.8963 n “ =^J.4B Jl
V . 1) Cristales blancos; p .f. 18 7-1089 C.2) Análisis elemental pa ra X , N, S— negativo.8) Solubilidad— agua ( + ), L a «oluejón acuosa cs ádcLa al tornasol.4) Pruebas d e dasiiicación:
KM nO í—negativa G íHjNH NHr—negativaBr* en CClf—negativa GHjGOC!— negativaEquivalente de neutralización — 59. Coeficiente de partición = 7.7
5) Derivado: Por calentamiento con fcnilhidrazina dio cristal*» blancos, p.f. 209-210*C.
v r , J ) Sólido café-rojizo; p.í. 55-70cC.2) Análisis elemental pa ra N — potitivo; para X , S—negativo.3) Solubilidad en agua ( — ) . N aO H ( — )¡ H C l (4* ). Una solución d d
compuesto en ácido clorhídrico diluido se decoloró con Norit y se añadió álcali. El compuesto, purificado en esta forma, fundió a 7 3 * 0 .1
4) Pruebas de clasificación:K M n O r—positiva C*H»SOxCi + NaOH— residuo
to lu b k en ácido clorhídrico Brj eo CCI«—prcdpítado Fe ( OH )r—negativaBrj en agua— precipitado AgtO— positivaC*H;NKNH}— pexitiva
Grupo 2 421

ma»
m
r
3&
[ * La
El hidróxido de sodio caliente ¿«com puso la substancia. Después de reparar por filtración un sólido cate obscuro, ei filtrado se r.«u* tral¡>¿ y s« obtuvo un precipitado de color canela claro. Despulí, de recm talizado d e una n » 2d a agua-ct3nol, fundió cori dezcompo*
' »ición a 236-24úcC. Fue soluble <*n HCJ y NaOH c iwiolubic en bicarbonato de sodio* En acetona e hidrópico de sodio dio un precipitado amarillo, p.f. ISI-ISS^C.
5) Derivadas:Oxima. p.f, l*H°C.Fcnilhidrazona, p.f. 143*0.Semicarbazona. p.f. 224°C.
4 2 2 P ro b le m a *
G ru p o 3
1. Un liquido caf¿ ( ! ) hirvió a 193-193'C . Tuvo nitrógenn, pero dio pruebas negativa? para azufre y halógenos. Fue insoluble en agua, pero soluble en ácido diluido. No reaccionó con cloruro de acetílo o cloruro de bencensulfonito. El tratam iento de una solución en ácido dorHtdric- . d d compuesto desconocido con nitrito de sodio, seguido de neutralización, dio ■ n compuesto ( I I ) , <1 cual fundió a 83*04 "C. Él compuesto I I fue ínsotuble en álcali*. pero ae disolvió en solución concentrada y caliente de hidróxido de «xlío. con desprendimiento de un gas ( IH ) , Éste gas f l í l ) absorbió en agua y la solución acuosa s* tra t¿ con isotiocianato dé fenilo. formándose un compuesto (IV ) con un punco de fusión de J34*135*C. Por acidulación cuidadosa de la solución alcalina anterior, seguida de extracción, se obtuvo un compuesto (V ) que fundió a 125-326'’C.
2. Un ISqukl» incoloro fue soluble en agua y en éter. Hirvió a S4-96°C y dio negativas laj pruebas pa ra halógenos, nitrógeno y azufre. R edujo una Soludón diluida de permanganato d e potasio, decoloró el bromo en tetraeleruro de carbono, «accionó con cloruro de acetilo y desprendió hidrógeno cuando se trató con sodio. No dio yodoformo cuando se trató con hipoyodito de sodio y no reaccionó con fenilhid ratina. El tratam iento con clorure» de 3,5-d:nitmbcnzoilolo convirtió en un compuesto que fundió a -t7-4&°C.
3. Un sólido amarillo ( í ) t que fundió u l t. 'M l4 °C , tuvo nitrógeno, peni no halógeno», adufre o metales. Fue inwluble en agua y en álcalis, pero soluble en ácidos diluidos, L a solución ácida de I se trató con nitrito de jodio en frío y después se hirvió. El producto ( I I ) de esta reacción precipitó al enfriar la solución. Contuvo nitrógeno y fundió a 95-$S6C : fue iníoluble «n ácidos y en solución de bicarbonato de sodio, pero soluble en solución de hidtóxtdo de sodio. Los productos obtenidos a( tra ta r los compuestos 1 y I I con cinc y solución de cloruro de amonto a ebullición redujeron fácilmente el reactivo de Toltens. El compuesto original ( I ) je trató con clonjra de bencm tulíoniJo y álcali. La acidulación de h solución residíante dio un compuesto ( I I I ) que fundió a i 35-136"C.
4. Un compuesto ( I ) cristalino, incoloro, fundió ¿* JñC-lb?*: tuvo nitrógeno, pero no halógenos ni azufre. Fue insolublc en agua > ácidos diluidos, pero soluble en solución diluida de bicarbonato de sodio. Dio un equivalente de neutra* Ezadón de ISO ± 2; no reaccionó con bromo en tetracloruro de carbono, ni con solución diluida de perm anganato de potasio, ni con d o ru ro de acetílo o con fenJl- h id raána. El compuesto I se trató- por algún tiempo con ácido clorhídrico a ebullición- Cuando se enfrió esta m eid a de reacción, precipitó un compuesto ( I I ) que fundió a 120-12l* C y tuvo un equivalente de neutralización de 121 S 3. El filtrado remanente después de la separación de II se evaporó a sequedad y el residuo (1H ) se purificó p o r recristal ¡ración. El compuesto I f l tuvo nitrógeno

G tvpo 4 42 3
y clora, fue bastante higroscópico y se descompuso cuando i* Viítq u c intento pa ra determ inarle su punto de fusión, Fue insohible en éter y « solución acuosa dio u n precipitado con nitrato de plata. U na solución de III je trató con ácido nitroso en frío. Se observó desprendimiento vigoroso de un gas. El compuesto I I I se tra tó con cloruro de bencensulfonilo y solución de hidróxido de sodio. La acidutación de la solución rem ítante dio »n nuevo producto ( tV ) que fundió a IC4-I65CC.
5 U n compuesto ( I ) cristalino, incoloro, fundió a 162-165*C con descom- pojicióo. D io prueba* negativas para nitrógeno, halógeno!, azufre y metales. Fue soluble en agria, pero insoluble en éter. Reaccionó con cloruro de acetito, decoloró Ib solución de permanganato de potasio, redujo la solución de Fehlinj: y el «¡activo de Tcllens, pero no dio color con el reactivo de Schtíf. El compuesto I reaccionó con fenilhidrazina dando un producto ( I I ) , d cual fundió con descomposición a 199-20¡“C. Cuando 1 te calentó con ácido nítrico concentrado ocurrió una reacción vigoróla y cuando se enfrió la mezcla de reacción precipitó un compuesto ( I I I ) . Ejte compuesto ( I I I ) fue insoluble en agua, pero fácilmente soluble en ¿ leal» ; dio un equivalente de neutralización de 104 ± 1. El compuesto I I I reaccionó con cloruro de acetílo, pero no con fenilhidrarina y fundió alrededor d e 212*213#C con descomposición. M anteniéndolo durante algún tiempo a tem peratura superior a su punto de fusión se convirtió en un nuevo compuerta (IV ) cl cual, después de ser rccrblaliM do, fundió a J32-!33*C. El compuesto IV fue insoluble en agua, pero soluble en solución d e bícatbonato de «odio y dio un equivalente d e neutralización de 111 =t 1. El tratam iento de la sal de sodio d e IV con brom uro de ¿-bromofenadlo dio un compuesto V que fundió a 137-138*0. El compuesto original ( I ) fue ópdcam ente activo, teniendo una rotación especifica d e + 8 1 6C, pero los productos de degradación I Í I , IV y V fueron ópticamente inactivos.
Grupo 4
1. U n compuesto iruolubte en agua, hidróxido de sodio y ácido clorhídrico, pero soluble en Scido sulfúrico y conteniendo nitrógeno, fundió a 68*C. Cuando $e trató con estaño y ácido clorhídrico se obtuvo u n a substancia, la cual reaccionó con clorar© de beneensuffonflo p a ra dar un derivado soluble en álcali G uando el compuesto original se tra tó con cinc y solución caliente de hidróxido de tedio, se convirtió en una cueva substancia que fundió a I30*C,
2. U n compuesto hirvió a 166-169*0 y tuvo azufre, pero no nitrógeno o halógenos. Fue insoluble en agua y áddos dütftdo^ pero se dwoMÓ en solución de hidróxido de sodio. Su derivado con lodio reaccionó coa 2,4-dinítrocíoroben- ccno dando un compuesto que fundió a 116-119#C¡. Cuando *e dejó a l aire, el conrpuesto original se oxidó lentamente a un derivado que fundió a 60-6 P C .
3. U n compuesto fundió a 141*142* y tuvo nitrógeno, pero no halógenos c azufre, Fue inaoluble en agua, ácidos diluidos y álczKs dtlatdos. E l compuesto no se afectó por tratam iento con estafio y ácido clorhídrico. C uando se trató duran te largó tiem po con solución caliente d e hidróxido de «odio, reaccionó form ando u n &\ ñte insoluole ( I ) . El aceite fue soluble en ád d o clorhídrico diluido y ««accionó con cloruro de acetDo para d a r u n sólido que fundió a 111-112*C. L a addulación de la solución alcalina, de la cual se había eliminado I, dio un sólido que fundió a 120-121 *C y cuyo equivalente d e neutralización íu« 122 ± 1 . •
4. Un compuesto hirvió a l S ^ v l 4C y tuvo cloro, pero no nitrógeno ni adufre. Fue insoluble en agua» en ácidos y álcalis diluidos y en ácido sulfúrico

424 Problema*
concentrado y frío. Se dinolvió en ácido sulfúrico fum ante. Con solución caüente de nitrato de p la u en c w o í na dio precipitado. Ei tratam iento con solución caliente de pcrpxaxi^nato de potasio hixo el compuesto se disolviera lentamente. L a solución resultante, a l se r acidulada con id d o sulfúrico, dio un precipitada que fundió a 1311-139*0 y tuvo un equivalente de neutralización de 157 a: 1 .
5. Un líquido incoloro hirvió x 188-192*C, Sólo tuvo carbono, hidrógeno y oxígeno. Fue iniolublc en aguo, ácidos diluidos y álcali!, pero te di «oí vi ó fácil, mente en ácido iwilfúrico concentrado j frío. No reaccionó co a fenilhidrazina o cloruro de acctilo y no decoloró una solución de brom o en tetracloruro de carbono, Los álcalis a ebullición lo disolvieron lentamente. L a mezcla resultante k destiló cou arrastre de vapor. £1 destilado contuvo un compuesto que, cuando se purificó, hirvió k J29«I30*C y reaccionó con a-naftilisncianato dando un derivado que fundió a 65-60^0.
El residuo alcalino que quedó después de la destilación per arrastre con vapor, te aciduló con ácido foifóríco y se destiló por arrastre con vapor. £1 des- til id o contuvo un ácido, el cual dió los siguientes números de D u d au x : 28.6, 23.0, 16.9. El ácido dio una atttlida que fundió a 108-109°C¡.
6. U n compuesto desconocido fue un sólido rosa que fundió a 109-112’C. E l tra tan ten te con carbón activado y rccrijtalízadón eliminó el color y elevó el punto de fusión a 112-1 H ^C . E l compuesto ardió con llanta fuliginosa y no dejó residuo» El análisis elemental mostró q u e había nitrógeno presente, pero que no había azufre ni halógenos.
El compuesta fue insoluble ea agua y álcali; diluidos, pera se disolvió en óter y ácidos diluidos. Reaccionó con cloruro de bencensulfonilo dando un deriva- de soluble en álcali que fundió a lOl-lOZ^C. El derivado acetilado fundió a J132°C.
Grupo 5
Para cada uno de los problemas ele éste y los grupos siguientes, dé la fórmula estructura] de un compuesto orgánico que satisfaga Jas condiciones enunciadas y demuestre medíante ecuaciones los cambios que experimente.
1. U n ád d o (A ) que sólo contiene carbono, hidrógeno y oxigeno, dló un equivalente d e neutralización de 103 ± I . Dio prueba negativa con fcíúlhidra- zma. El tratam iento con-ácido sulfúrico lo convirtió en un nuevo ácido (B ), cl cual decoloró las soluciones de brom o y permanganato y tuvo un equivalente d e neutralización de 87 ± I. El ád d o original (A ) dio, con hipoyodito, yodo- formo y un nuevo ácido (C ) cuyo equivalente de neutralización fue 52 ± 1.
2. U n ácido dio un equivalente de neutralización de 9?. No se pudo lograr con íad lid ad la substitución de hidrógeno por bromo, aun en presencia de tribro- m uro de fósforo. Por oxidación vigorosa xo transformó en u n n ’tevú ácido cuyo equivalente de neutralización fue 83.
*» U n hidrocarburo, ópticamente activo, se disolvió en ácido mlfúrico concentrado y frío, decoloró la solución d e peroanganato d e po tado y rápidam ente absorbió bromo. Por oxidación se convirtió en un ácido con el n i t r ^ número de átomos d e carbono que la substancia fundam ental y con un equivalente de neutralización de 66.
4 ; U n compuesto tuvo un equivalente de neutralización de 66. L a substancia no se afectó con bromo en tetracloruro de carbono, pero el calor ía transformó en nq ád d o cuyo equivalente do neutralización fue 86,

G rupa 6 425
5« U n a ba*a dio un equivalente d e neutralización de 121 ± 1 . Por oxidación vigorara te convirtió t n un ácido coa un equivalente de nrutraJizaciórt de 121 ± I.
6. U n ácido, cuyo equivalente de neutralización fue 166, no reaccionó con bromo en tetracloruro de carbono, pero dio positiva. Ia prueba de yodofomio.
7• U n compuesto (A ) dio negativas las pruebas para nitrógeno, azufre y halógenos. Fue insolutos en agua, pero »e disolvió en solución diluida de hidróxido de sodio. El compuesta (A) no dio coloración con cloruro fénico y no decoloró la solución de permanganato de potasio.
El tratamiento con ácido bromhídrico concentrado convirtió A en dos compuestos. Uno fue un compuesto (inaoluble en todai las pruebas) que contenía bromo y dio un precipitado con yoduro d e so lio en acetona. Eí otro fue un nuevo ácido (B ), el cu li decoloró las solucione* d e bromo y dio coloración con cloruro férrico. El compuesto B no tuvo halógeno. Los equivalente* de neutralización de los compuestos A y B fueron, respectivamente* ISO ± 2 y J 3 7 ± l .
0. Un ácido ópticamente activo tuvo un peso molecular de 9£-9, Un compuesto (T^ dio coloc rojo con cloruro iónico y precipitado con
2,4-dirdtrofenilHidrazina. Ei compuesto I se calentó con hidróxido de sodio acuoso al 25% y la mezcla se dest. '► parcialmente. El destilado contuvo un compuesto que reaccionó con sodio y dio prueba de yodoíormo positiva. La prueba de Lucas fue negativa.
Et residuo de Ia destilación se aciduló con ád d o fosfórico y la mezcla destiló por arrastre con vapor. Se aisló u n ácido volátil. Sw éster de p-brorno fenacilo dio un número de saponificación de 257 ± l.
Grupo 6
U n ácido {I}* conteniendo nitrógeno, dio Un equivalente de neutralización de 197 ± 2. Et tratam iento con cloruro de tionilo, seguido por hidróxido de amonio, convirtió I en un compuesto neutro ( H ) , el cual reaccionó can solución alcalina de hipobromito de sodio formándose un compuesto básico ( I I I ) . L a hidrólisis del compuesto I I I produjo un nuevo compuesto ( IV ) , soluble tanto en ic ido j como en bases y que dio u n equivalente de neutralización de 186 ± 2. El tratamiento de este compuesto con ácido nitro3o en presencia de ácido sulfúrico dio una solución transparente. Cuando Se añadió cianuro cuproso a cata solución so regeneró el compuesto I.
' La hidrólisis de I d io un ácido (V ) con un equivalente d e neutralización d e 107 ± L El calor convirtió V en una substancia neutra ( V I ) , la cual por hidrólisis se volvió a convertir en V.
L a oxidación del compuesto IV dio un nuevo ácido (VJI) con un equivalente de neutralización de 71 ± 1.
2. U n compuesto desconocido (I) fue insoluble en agua, pero soluble tanto en ácido diluido como en álcali diluido. El compuesto I tuvo nitrógeno y bromo. No Se pudo obtener un equivalente de neutralización satisfactorio- El tratam iento con anhídrido acético lo convirtió en ácido ( I I ) , et cual dio un cqu¡váleme de neutralización de 270 ± 3. C uando I se trató en frío Con ácido n itro » , se desprendió un gas y le p rodujo un compuesto { I I I ) , el cual tu v o « n equivalente de neutraliíactó/i de 230 ±. 2. Los compuestos I, I [ y I I I , cuando *e oxidaron vigorosamente, dieron el mismo producto-— un Acido que contuvo bromo y cuyo equivalente de neutralización fue 199 dfc '¿~
3. Un éiter sólido ( I ) se saponificó (equivalente de saponificación, 173 ± 2) y la solución acuosa alcalina se evaporó cari a sequedad. El destilado fue e*íhi-

4 2 6 P rob lem a*4
1jiram ente *gua- El residuo se aciduló y se destiló; se aiiló u n aceite incoloro ( U ) . ':! L a substancia n reaccionó con «■loruro de acetílo y dio odoración con solución de doruro férrico. El residuo de la destilación co n iu v j un íójido ( I I I ) soluble en álcaji. Cuando I I [ «e calentó a tem peratura superior » su pun to de íución, ' se convirtió en IV ; IV íc disolvió al agitado con álcali acuoso caliente y Ja xolución *e aciduló; el sólido que precipitó fue idéntico a I I I ,
4. Un sólido ( I ) , que dio positiva la p rueba para nitrógeno, fue «dublé en agua e insoluWe en éter. L a adición d e álcali frío liberó un compuesto ( I I ) soluble en agua, wlublc en éter, con olor amoniacal y que reaedonó con cloruro de bencensulfonilo dando un derivado insoluble en álcali. L a adición de ácido dorhídríco a una solución acuosa dilu ida de I dio un ácido sólido ( I l í ) con un equivalente de neutralización de 167 ± 2. Después que se hirvió con cinc en polvoy solución de cloruro de amonio, ia substancia en que se convirtió Í I I redujo el. 4 reactivo de Toltcns.
5. Un compuesto (CiH.uO) ópticamente activo, ligeramente soluble en agua.^ Su solubilidad no aum entó apreciablem eate con hidróxido de so<iio o ácido cíorhídríeo. Dio pruebas negativas con fenilhidrazina, reactivo de Lucas c Kipoyodr- u>; decoloró las soludones de bromo y de permanganato. El compuesto reaccionó con cloruro de acetilo. L a oxidación con permangar: 'o lo convirtió en un ácido que dio un equivalente de neutralización de 59 ¡fc 1. V -ando el ácido se calentó perdió anhídrido carbónico y se convirtió en un nuevo áddo con un equivalente de neutralización de 73 ± !.
6. Un compuesto ( I ) conteniendo carbono, hidrógeno, oxígeno y nitrógeno, fue soluble en soludón diluida de hidrójddo de sodio y en ácido clorhídrico diluido, pero jnsoluble en solución de bicarbonaío de sodiu. El compuesto I reaccionó con u n cxceio de anhídrido acético dando un producto (I I) insolublc en agua, en ácidos diluidos y en álcalis diluidos. El compuesto I decoloró el agua d e brom o; cuando se disolvió en un exceso d e ácido dorhidríco diluido y la soludón irla se trató con nitrito de sodio, precipitó uti nuevo producto ( H t) sin desprendimiento de nitrógeno.
1. Cuando un sólido, CiH,Oa ( I ) , so calentó con wlucióo diluida de carbonato de potasio, se convirtió en la sal potásica d e un áddo, C*HiOi ( I I ) , Cuando la sal se trató con ácido* se regeneró I . E l compuesto I reaedonó con bromo en te erad orare de carbono obteniéndose CyH¿0>Br2 ( I I I ) . Cuando el compuesto I I I se calentó con soludón de hidróxido de. sodio, «e disolvió lenta* mente. Al acidular la solución precipitó un compuesto, CÍ,H*Oj (IV ).
2. Un compuesto, CtKiONBr, cuando se trató con Bolución caliente de hidróxtdo d e potasio dio la sal de potasio d e un ácido, C|H»Oj, la cual se pudo resolver en sus formas i y i.
3. Un compuesto, C icH hO« ( I ) , reaccionó con soludón caliente de hidróxido de «odio obteniéndose una sal, C jiHuOjNa ( I I ) , E l tratam iento de la tal con ácido sulfúrico diluido y caliento la convirtió en ixq áddo , C*H*Ot ( I I I ) . El calor convirtió el compuesto 1ÍT en un compuesto de punto de iusíón elevado, C,,H«Ol (IV ).
4. U n compuesto, CsH«0¿ (3 ), se convirtió en CjHiQjCI ( I I ) por trata~ miento con ácido dorhidrico concentrado" hirviente. El compuesta I reaccionó lentamente con soludón de hidróxido d e potasio dando C5H,03K < 111). Este compuesto se convirtió en CsH«Oi (IV ) por tratam iento con solución alcalina de
Grupo 7

G ru p o 8 4 2 7
permanganato. Cuando d compuesto IV k uutó con solución de Wjw'dorito d e jodio se transformó en 0*HtO, (V ) , un aculo cuyo equivalente de neutralización fue 58 ± 1. Cuando este ácido « calentó d io C»H«Oj (V I) . C uando la solución acuosa de I I k Uevó a neutralidad con solución de hidróxido de «odio, Jen lam ente m regeneré d compuesto I .
5. U n compuesto líquido, neutro, tuvo la fórmula C jH íNO*6. U n compuesta, C t tH ^ , formó sales con ácidos minerales fnert« . Las
sale* se hidrolizaron con agu3.7. U n compuesto, C|»HitO, dio C jjH hO v cuando »e oxidó con pernvangx-
nato alcalino. El compuesto original reaccionó con sodio dando C tJifO N a.8. Un ácido, con equivalente de neu tra liación de 57, no ee afeetó con
bromo en tetracloruro do carbono.
Grupo 8
1. U n líquido, QHtO* ( I ) , decoloró la solución d e pcnrtanganato y tam bién redujo Ja solución de Bencdict. C uando el compuesto I «e calentó con un cianuro alcalino, se dtm erúó. El tratam iento del compuesto I con ortoformiato de etilo lo convirtió en una nueva substancia, C jHhO j.
2. U n compuesto, C iILO . ( I ) , íuc soluble en solución diluida de hidróxido de sodio y cuando bc trató con bromo se convirtió en C<H*0«Brj ( I I ] . El compuesto I «e obtuvo a p a rtir de I I tratando este últim o con polvo de cinc. El compuesto I I se convirtió en O H ) por calentamiento con ácido yodhí- dríco. El compuesto I I I tuvo un equivalente de neutralización de 59 y cuando »e calentó perdió una molécula de agua. El anhídrido así obtenido reaccionó cx>n benceno, en presencia de cloruro de aluminio, para d a r C»H mO>, el cual íuc soluble en álcali, reaedonó con fenilhidrazina y poi oxidación vigorosa se convirtió en ácido benzoico.
3. U n compuesto, CuKuNi, i e convirtió por oxidación vigorosa e n ChUiNjO.4. U n compuesto, CjHuC), decoloró una solución atcalina de perrnanganato
de potasio, pero no se afectó con bromo en tetracforura d e carbom».5. U n compuesto, Q tH uO , por oxidación con ácido crómico se convirtió
en un ácido cuyo equivalente de neutralización fue 226.6. U n compuesto neutro, C»HiO« ( I ) , cuando se calentó con solución do
hidróxido de sodiii su convirtió en Ja sal de un ácido ( I I ) que tuvo un equiva* lente de neutralización de 209 — 2. El compuesto I reaccionó con peróxido de hidrógeno produciéndose un nuevo ácido { III} que tuvo u n equivalente de neutralización de 111 ± 1.
1. U n compuesto, CkILOzN ( I ) , cuando se trató con hierro y ácido clorhídrico se convirtió er.'CuHrN ( H ) , compuesto que fue soluble en ácido clorhídrico d ilu ido . Por oxidación vigoras» de I y I I ie produjeron, respectivamente*, CiUfO^N y CtH<0<. Ambas productos de Oxidación fueron solubles en álcali.
Grupo 9
I . Un producto natural,' CnHi^O* ( I ) , fue inerte al cloruro d e acetilo y la fenilhidrazina. Sin embargo» calentándolo con hidróxido d e potasio ®e isomoritó. El isómero (IÍJ también fue incapaz d e reaccionar t«n cloruro de acetilo y fenilhidrazina. Ambos isómeros decoloraron las soluciones d e bromo y pennan- ganato. El ozono convirtió el isómero I I en el compuesto I I I , el cual dió una fenilhídrazona. L a oxidación del compuesto I I o del I I I con perm anjanato de potasio alcalino produjo un ácido, CqfLCX (IV ) .

4 2 8 P ro b le m a s
2. U n compuesto, C jH iO íd ( I ) , cuando « calentó con etanol absoluto dio . Cj«H»Ü4 ( I I ) , d cual p o r ex idadón to n pcm iar.ganato dcalino ae convirtió en GtH*Qi, T ratando I con anilina te obtuvo CnHj»ON>
3. U n compuesto, CiHkOj ( I ) , fue soluble en solución diluida de hidróxido d e sodio y dio coloradón roja con soludón d e d o ru ro férrico. L a ícnilhidracina lo convirtió co Cj>Hi*ON* ( I ) ) y calentando I con ácido dorhídrico a! 20% pe transformó en C sH j»0 ( I I I ) , Ei compuesto I I I dio un precipitado cristalino cuando ie trató con semicarbazida, pero cuando bis trató con solivión de hipo* yodito de «odio no dio yodoformo.
4. U n compuesto, C*HTN ( I ) , M convirtió por teduedón catalítica en C*HuN ( I I ) . C uando c l compuesto I I «e trató con un exceso de yoduro de metilo, seguido por óxido de plata, y el producto d e la reacción se calentó y se produjo el compuesto C»H » ( I U ) . E íte compuesto se convirtió (a ) por osidadón vigorosa en C>K«04 ( I V ) ; (¿ ) por tratam iento con ozono e hidrólisis del producto de reacción, en C ^H uO N (V ). El producto de ozonijadón, cuando se calentó con solución diluid? de cianure- de potasio, dio C^H nOjN, ( V i ) . Por oxidación vigoro» i kl el compuesto V I be convirtió en IV.
5. U n com puejto, C ^H ^O ( I ) , reaedonó con sodio dando un derivado CnHiONa, d cuaj je hídrolizó totalmente con agua. E l tratam iento d d compuesto I coa ácido sulfúrico al 80% y frío, en presencia de sulfato mercúrico, y después con agua, d io CttHuQj ( í l ) . Este compuesto se disolvió lentamente en solución d e hipocloríto de sodio y por aciduladón de la solución re obtuvo C»H»Q> (1X1). El compuesto I I I , hervido ooq ácido bromhídrico al 4Z% } se convirtió en C V H jO iB r ( I V ) , compuesto que por o rid ad ó n vigorosa ae transformó en C 1B4O4.
6. Un compue-lCo que tiene la fórmula molecular ChHjj, por oxidación con perm anganato se convirtió en u n derivado cuya fórm ula molecular es C uH ftO y que no te afecta p o r un tratam iento posterior con permangiuiato.
7. U n compuesto ópticamente sctivOj C»HmN, p o r o rid ad ó n vigorosa ac convirtió en un ád d o cuyo equivalente de neutraliaadón «s 03,
6. Un compuesto no decolora d agua d e bromo, pero reacciona con sodio dando CíHíOiNaj. Ei compuesto tiene u n equivalente d e neutralización d e 152.
Grupo 10
1. U n sólido (I) cristalino, incoloro, tuvo nitrógeno, pero no halógenos ni azufre. Fue soluble en agua,, pero insoluble en ¿ter. Su solución acuosa fue Adda y d io un equivalente de neutralizadón de 123 ds 2. Se le trató con nitrito do sodio y 'ácido dorh íd rico y se desprendió u n gas, un te td o del cual fue soluble en soludón de hidróxido de potasio. L a soludón d d tratam iento con ád d o nitroso se evaporó a sequedad] sólo quedó d oru ro de sodio, nitrato de sodio y nitrito de sodio. El compuesto I se calentó con soludón diluida d e hidróxido de sodio; se desprendió amoníaco y por a d d u la d ó a d e la sd u d ó n se desprendió un gas. AI evaporar cata soludón sólo quedaron talca inorgánicas.
2. U n .ompuesto destonoddo que contiene carbono, hidrógeno, otfigeno y nitrógeno, fue iusoluble en agua, en ácidos diluidos y en álcalis düuidoa. No reaccionó con cloruro de acctüo tú con fcnilh idraána y no se redujo con facilidad. Guando se trató con álcali acuoso caliente, Ja substancia se disolvió lentamente. L a destilación d e esta solución alcalina dio un destilado que contuvo u n compuesto que p red p h ó , por adición d e carbonato d e potasio. Este compuesto dio la p rueba del yodofonoo, pero no reaedonó con soludón de cloruro de d n c en ¿ d d o clorhídrico. L a solución alcalina original (residuo de l a destilación) se ad d u ló

con ácido sulfúrico y *e destiló d e nuevo; en el destilado se obtuvo un ácido volátil, Este dciülado redujo el nerrnangariato. £1 liquido residual de esta segando destilación se neutralizó c>3ctamentc y te obtuvo un *ÓKdo. hzxt sólido tuVO nitrógeno y un equivalente d e n ru tralizadón do 137 ± I.
3. U n ácido, C JitG , ( I ) , se convirtió en C*H*OxCl> <H ) coa pentadowro de lósforo, C uando el derivado clorado « trató con benceno en presencia de cloruro de aluminio anhidro, d io C^H uO; ( I I I ) . Eate compuesto tk> decoloró el permanganato, pero fácilm ente d io un dibrom uro y una diorama. Bajo la influencia d d penttcloniro d e fósforo I» dicwima s t transpuso a CjiHuOsN* (IV )> c o m p ac to que cuando le hídrotízó dio el árido original ( I ) .
4, U n compuerto ( I ) conteniendo carbono, hidrógeno, oxígeno, nitrógeno y d o ro , fue soluble en agua, pero insoluble en éter. L a solución a ^ o sa dio inmediatam ente un precipitado con nitrato de p lata. Cuando la solución acuosa do I ce neutralizó exactamente, precipitó u n nuevo compuesto ( I I ) Ubre de cloro, que reaccionó con anhídrido acético dando un compuesto ( I I I ) soluble en álcalis, insoluble en ácidos y que tuvo, un equivalente de neutralización de 207 ± 2. El compuesto I I reaccionó con cloruro d e bencensulfonilo, dando un producto soluble en álcali y tratado con ácido nitroso no ie desprendió ningún gas aun cuando se calentó. El p> lucto de « t e último tratam iento aún contenía nitrógeno y íue soluble en álcalis insoluble en ácido». L a oxidación vigorosa de I , de I I o de I I I di») un ácido ¿ n nitrógeno, insoluble en agua y con un equivalente de ncutralitación de 82 ± Z.
3. U n compuesto, CuH*Oi ( í ) , decoloró el permawgatiato alcalino y reaccionó con hidroxH amina. Se descompuso cuándo se destiló a presión ordinaria obteniéndote C*H,Oj ( H ) , compuesto que dio un derivado mono-sódico y *e oxidó fácilmente a CjH«0« ( I I I ) , El compuesto 111 fue un ácido que Cuando w: calentó con cal-sodada ie convirtió en C?H»Oj (IV ) . El compuesto }V je deícotnpuio por calentamiento a p reáón con ácido clorhídrico diluido, dando un compuesto débilmente ¿cido de íórmuja CcH»Oj,
G rupo H 4 2 9
Grupo II
1. U n compuesto (A ) que sólo contiene carbono, hidrógeno y oxígeno, fue insoluble en agua, ácidos diluidos y álcalis diluidos, pero se disolvió en ácido sulfúrico concentrado y frío. D io negativas las pruebas con fenilhídraxina y cloruro de acerilo, Cuando se calentó con hidróxido de sodio acuoso' se obtuvo
. u n aceite (B) y un ád d o volátil con ur. equivalente d e neutralización de 59 * 1. El compuesto B reaccionó con cloruro d e acetilo, pero no con fenilhidiazína. El tratam iento de B con solución concentrada de ácido bromhídrico lo tranrformó en un compuesto (C ) que tuvo bromo y que dio positivas laa pruebas con ni* trato de p lata , cloruro férrico y agua de bromo.
U na oxidación m oderada convirtió B en u n ácido (D ) que tuvo un equivalente d e neutralización de 164 í : 2-
2. TJn compuesto ( I ) dio pruebas positivas p a ia nitrógeno y cloro, fue insoluble en agua y ácido clorhídrico, pero soluble en soludón de bicarbonato de sodio. El compuesto I d io un equivalente de neutralízadón de 210 ± - í . T am bién reaccionó con cloruro d e acetilo, pero no con solución alcohólica caliente de n itra to de plata. El derivado acctil&do ( I I ) dio un equivalente de neutral?* «ación de 253 2. Por ebullidón de I con álcali se desprendió amoniaco y por acidulacíón do la solución resultante precipitó un nuevo ád d o ( I I I)* t i cual dio un equivalente de neutralización de 115:fc 1, El compuesto I I I tuvo cloro, p^ro no nitrógeno. C uando el compuesto I I I se hirvió con solución de pennar*-
. ganato de potasio, je produjo un nuevo compuesto que tuvo las mumav c a ra o

m - tI '¡ -S
* <■
■ »
»
»
r»r»f»8»m
terísti” : d e solubilidad que I, todavía tuvo cloro y d io un equivalente de neutra- lizadón de 81 ± 1.
Z. "Un compuesto neu tro (A ) , sólido, incoloro, dio pruebas negativa» para nitrógeno,. azufre y halógeno*. D io pruebas positivas con fenilhidraána y cloruro d e acttflo. U na oxidación m oderada Jo convirtió en un nuevo compuesto, un sólido amarillo (B). El nuevo compuesto reaccionó con fenchidrajara dando el mismo derivado que se obtuvo a partir de A, Por oxidación vigorosa B se convirtió en un ád d o (C )j «1 cuat dio un equivalente de neutralización de 121 ± i.
4. U n liquido ( I ) que contenía d o ro fue insoluble en agua> ácido clorhídrico c hidróxido de sodio diluido. Se disolvió en ácido sulfúrico concentrado y Trío, pero no en ád d o fosfórico. No dio p redp itado en caliente con nitrato de plata alcohólico y no reaedonó con cloruro de acetilo. D io un precipitado con fenilbidraana, pero no dio color con el reactivo de Schiff. El compuesto T « disolvió cuando ie hirvió con soludón concentrada d e hidróxido de sodio. El destilado de esta solución alcalina dio positiva la prueba del yodoformo. Por aciduladón de la solución alcalina precipitó un compuesto { II) , el cual tuvo cloro y un equivalente de neutralizarán de 156 ± 1. El compuesto I I no se afectó con soludón de permanganaco. Después de eliminar el compuesto I I , se destiló una parte del "loado ácido y *e encontró que d destilado era ácido al tornasol. El compuesto d 'rinal (T) dio un equivalente de saponificación de 113 ± 1.
5. U n líquido tuvo un equivalente de saponificación de 163 ± J, La saponificación produjo un aceite, el cual dio positiva la prueba deí cloruro íórrico, y un áddo con u a equivalente de neutralización de 60 ± 1.
4 3 0 P ro b le m a s .
Grupo 12
3. U n compuesto ( I ) Con nitrógeno c insoluble en éter, se disolvió en agua dando un w!uctó« alcalina. L a titulación d e esta solución con un ácido valorado dio un equivalente de neutralización d e $7 ± l . El tratamiento de una solución fría de I con nitrito de sodio y ácido clorhídrico liberó un gas. L a solución resultante se alcalinízó y Se le añadió cloruro d e bcnzoilo. P redpitó un compuesto neutro (11). Et compuesto I I dio un equivalente de saponificación de 142 ± J.
2. Un liquido incoloro ( I ) no dio pruebas pa ra halógenos, nitrógeno, azufreo metales. Fue insotuble en agua, ád d o clorhídrico diluido, hidróxido de sodio diluido y áddo fosfórico, pero soluble en ád d o s-utffirico concentrado y frió. No. reaccionó con cloruro de acetilo o feoilhédrazina y no se afectó por calentamiento con hidróxido de sodio. Se le hirvió con ád d o fosfórico diluido y al enfriar la
. «elución-se separó un aceite { II) . El compuesto I I dio un precipitado con feoit-
. h id m in a y con tolución de bisulfito de sodio, pero no reaccionó con cloruro de ..•M etilo. Cuando I I se agitó vigorosamente con un álcali fuerte, de la solución
alcalina predpitó un compuesto ( I I Í ) . Este producto ( W ) reaccionó con do- •: .;'..n iro de acetilo, pero no con fenühidradna. L a aciduiación de la solución alcalina : dio IV» el cual tuvo un equivalente d e neutralizadón de l3 ó á : l« La oxidación
• vigorosa de ÍV dio un ád d o {V) COD un equivalente d e neutralización de1.* ,
L a soludón de áddo fosfórico, d e la cual se aisló H , se destiló. El destilado '■««•"¿¿(•jttuió oetn carbonato d e potasio y se obtuvo un compuesto (V I). Este com-
puesto.-reaedonó con sodio, con d o ru ro de acetilo, y con hipoyodito de eodio dio* precíph*áo amarillo.-No reaccionó con el reactivo de Lucas.
:^U n líquido mcoktro {I) d io negativas las pruebas para nitrógeno, azufre Fue iduM e en agua y en óter. N o reaccionó con sodio., cloruro de
ijj fenJQiidrtn¿na o soludón diluida de petm anganato. No decoloró el bromoS # i> ' •

en te tradoru ro de carbono y no w afectó p o r ebullición coa álcalis. C ian d o ei c o m p lic ó I *¡ calentó von un w w »a de ávido Woxnhídricv, x> separó u n aceite( I I ) . Eate acetite ( I i ) tuvo bromo j rápidam ente dio un precipitado con nitrato de plata alcohólico. Fue insoluble en agua, á d d o j y álcalis. Después de que I I se ttcó y purificó, se Je trató con magnesio en é te r puro- Hubo reacción to a desprendim iento de un gas ( I I I ) , No pe pudo poner en evidencia la presencia de un reactivo d e GrignArtL Por tratam iento de I I con hidróxido de potado alcohólico Se desprendió un gas ( IV ) , el cual d io n n precipitado cuando *e burbujeó en nitrato d e plata amoniacal. £1 gas ( I I I ) no dio precipitado con cloruro cuproso amoniaca?. T anto I I I como IV decoloraron el agua d e bromo y redujeron lai soluciones de permanganato. U n examen cuidadoso de la acción d d ád d o bromhí- drico sobre et compuesto I mostró que I I fue el único compuesto orgánico producido y que durante la reacción no te desprendieron gases,
4 , U n compuesto (I) que contiene carbono, hidrógeno, nitrógeno y oxígeno, fue insoluble en álcalis y Acido» diluido*. C uando se calentó du ran te algún tiempo con ¿cido clorhídrico, el compuesto I produjo un ácido sólido ( I I ) que tuvo un equivalente d e ceutxalizaeión de IfiO ± 1. Por oxidación del conrpruerto I con dicromato d e potasio y ícido sulfúrico, *c obtuvo u n ácido sóHdo ( I I I ) con nitrógeno y u n equivalente de neutralización de 166 ± 1. E l compuesto I reaccionó con betwaldehído en presencia de álcalis forreando un derivado de bcnjaL
T ratando eJ compuesto I con cloruro estañóse j cloruro d e hidrógeno en éter «eco y después, ia mezcla resultante con agua, se produjo un nuevo compuerto (IV ) cuya fórmuta molecular fue C«HjN. U na determinación de Z erw i- tinoff costró que IV tiene un ¿tomo de hidrógeno activo. El compuesto IV fu« débilmente básico y j* resinificó con ácidos.
Grupo 13
1. U n líquido (1) aceitoso, inooloro, de olor agradable y conteniendo sólo carbono, hidrógeno y oxigeno, reaccionó con ícnilhidrazioa, pero no coa cloruro de acetilo y decoloró rápidam ente la solución de permanganato d e potasio. Cuando se trató con solución concentrada de hidróxido de sodio y la mezcla de reacción se aciduló, se obtuvieron dos productos: un compuesto ( I I ) que tuvo oxígeno y que reaccionó con cJoruro de acetilo y u n ácido ( I I I ) que reaedonó con ád d o wlfú- rico fumante' y que a 200 "C perdió anhídrido carbónico para dar un compuesto con oxígeno (CUHtO). Este últim o decoloró !a solución d e permanganato, pero no reaccionó con sodio o íen ílh ldn tina. Guando el compuesto I je calentó con cianuro de potasio, se produjo un compuesto (IV ) q tó dio una osazona y redujo la solución alcalina de cobre y que, por ox idadón con ád d o peryódico, se convirtió en el compuesto original ( I ) y en e l á^ido ( I I I ) .
2. U n compuesto ( i ) débilmente ád do , tuvo nitrógeno, pero no azufre, ni halógenos. Reaccionó con cloruro de ACetilo, pero no con fenilhidrazina. Cuando el compuesto I se calentó con áddos dituídos, se obtuvo un nuevo compuesto (II ) que se a¡B?ó p o r dettiladón de la solución ¿ d d a y sa tu rad ó a del destilado con carbonato d e potasio. El compuesto I I no reaedoaó con cloruro de acetilo o reactivo d e Schífí, pero dio pruebas positivas con fcntlhidratina y bisulfito de sodio.
C uando el compuesto I se calentó con peatacloruro de fósforo y íe Vertió en agua, se obtuvo nn nuevo compuesto ( I I I ) . £1 compuesto I I I todavía tuvo nitrógeno, pero no halógeno, fue neutro y los álcali* lo descompusieron. Añadiendo cloruro d e benzoílo a esta solución alcalina se obtuvo u n ácido (IV ) que dio ún equivalente de neturaJlzadón de 220 ± 2. C uando el compuesto IV se calentó
G ru p o 13 431

duran te algún tiempo con Addos diluidos y se destiló, se obiuvkron dos productos: U n o (V ) tic contuvo nitrógeno, fue ¿cid» y dio un equivaliente de neutral& tdón d e 120 ± 2 y e l otro re<ult6 ser id ín tico a XII. G uindo I I I ie « a tó en irlo con ¿ d d o clorhídrica concentrado, n obtuvo un compuesto (V I) con nitrógeno y cloro, «(dublé en agua. Guando V I je tra tó con solución ir ía de nitrito de sodio, te desprendió no gas.
3- U n compuesto (I) soluble en agua, pero no en étér, ae descompuso por calentamiento t n u n compuesto ( H ) insolublc. cn todas las pruebes y un compuesto básico ( I I I ) . Cuando I I y I I I se calentaron jiin to i, te regeneró I, TantoI como I I , cuándo te trataron con nitrato d e plata, dieron inmediatamente tin predpitado. El compuesto I l t n o dio benccnsulfonamída, pero sí ua derivado nitroso.
4 . U n compuesto neutro ( I ) d ió positivas las pruebas para cloro, bromo y yodo. C on nitrato de p la ta alcohólico dio u n precipitado blanco, el cual fue fácilmente soluble en hidróxido de amonio. Con íenílhidrazána se obtuvo un pre- espitado, pero con d o ru ro de aoetílo no reaccionó. El permanganato se decoloró lentam ente lo ratsmo que el bromo en tetracloruro de owbono. Cuando el compuesto I te agitó con álcali diluido y frío du ran te algún tiempo, se disolvió. La acidulacíón de la solución alcalina produjo un compuesto <111 que dio pruebas positivas para bromo y yodo y tuvo un equivalente de neutralización de 369 ± Z. Cuando ae hirvió el compuesto I con álcali diluido y después ie aciduló, precipitó un compuesto ( I I I ) , el cual dio positiva la prueba para yodo. £1 compuesto I I I dio u n equivalente de neutralización de 306 i 3 y reaedonó tan to con fenilhidra- riñ a como con donnro de acetilo. Por tratam iento de I o I I con hipodorito de sodio y acíduladón se produjo' un ácido que tuvo yodo y dio un equivalente de neutralización de 146 ± I.
5. U n compuesto neutro (A) tuvo bromo, pero no nitrógeno u otros halógenos. No reaodonó con soludón caliente d!c n itrato d e p la ta en alcohol, ni con cloruro d e acetilo, n í con íenílhidraaina, ni con bromo en tetracloruro de carbono. Se disolvió en solución de hidróxido de s»jdio a ebullición, pero r i destilado de e*ta soludón alcalina no contuvo compuestos orgánicos, por addulación de la soludón alcalina con á d d o fosfórico precipitó un compuesto (B ), el cual tuvo brom o y un equivalente de neutralización de 200 ±. 2. L a destilación por arrastre con vapor d e la solución i d d a dio u n destilado que se extrajo varias veces con cloroformo. A l evaporar d cloroformo se obtuvo un líquido incoloro ( C ) , d cual se purificó p o r destilación. E l compuesto C no tuvo bromo y fue soluble en solución d e bicarbonato de «odio. D io u n equivalente de neutralización de JG2±;I. Después d e obtener C, la so ludón rem anente en el m atraz de la destilación por arrastre ooo vapor *e alcalificó, se añadió cloruro de benzoflo y la mezcla. « agitó vigorosamente. De la solución alcalina p red p itó u n nuevo compuestos (D ). E l compuesto D no tuvo bromo y fue neutro. T uvo un número de saponifica* d ó n de 135 ± 1.
4 3 2 Problema*
Grupo 14
1. U n compuesto (I) conteniendo sólo carbono, hidrógeno y oxigeno, tuvo u n equivalente de neutralización de 179 ± 1. C uando se le csJeptó con hidróxido de sodio acuoso la mezcla d e reacción se, ad d u ló con á d d o sulfúrico, precipitó un sólido ( I I ) toluble en álcalis y con un equivalente de neutralización de 136 ± 1, El compuesto I [ decoloró el agua d e brom o y d io coloración con doruro férrico. Por destilación del filtrado I I se obtuvo u n ád d o ( I I I ) con u n equivalente de -neutralizadón de 60 ± 1.

2. U n Compucato (1) con nitrógeno fue ú to lu b le en agua y álcalis diluidos, pero soluble en ácido clorhídrica düuido. Ej tretaroientA con d o ru ro de bencensulfonilo y álciH dio una solución transpajoct*. Por aciduladóu de e»ta solución se obtuvo un precipitado que se disolvió en ex ce » de ácido. El compuCTto original no reaccionó con fenilhidrazina, pero te deacoropuso por ebullición con solución <'¿liente de hidróxido d e sodio. De la solución alcalina { III) se separó un aceice ( I I ) . El aceite todavía tuvo nitrógeno y fue soluble en ácido clorhídrico diluido. El «impuesto I I con a su a de bromo dio un precipitado y reaccionó con cloruro do acetilo y con sodio. C uando el compuesto II se trató con cloruro de beacensulfonilo y álcali, se obtux'o un aceite soluble en ácido clorhídrico. El compuesto í l «e disolvió en ¿ter y la solución se «aturó con cloruro de hidrógeno; precipitó un compuesto sólido (IV ). £3 compuesto IV fue soluble en agua y dio un equivalente de neutralización de 187 ± 1,
Por a d d u la d ó a de la solución alcalina, an terior ( I I I ) se obtuvo un precipitado (V ), <1 cual íe disolvió en exceso d e ‘ácido. L a adición de nitrito de jodio a una solución ácida y helada de V dio un solución transparente sin desprendí* m iento do nitrógeno. L a adición de esta solución a una solución de P-naftówdo de sodio dio una solución roja. El compuesto V dio un equivalente de neutra-I i ¡Lición d e 137 ± I,
3. U n sólido ( I ) dio prueba» p ^ itiv a s pa ra nitrógeno, azufre y bromo, fue insoluble en éter, pero se disolvió en agua dando u n a lolución ácida. Dio un equivalente d e neutralización de 221 ± 1, L a adición d e álcali irlo hizo que se separará un aceite ( I I ) , el cual tuvo bromo y nitrógeno, pero no azufre. El compuesto Tí reaccionó con cloruro de bcncensulíonilo y álcali dando una solución transparente de la cual precipitó el ácido I I I que tuyo bromo, nitrógeno y azuf-'e. El compuesto I I no dio precipitado con n itra to de p lata, pero decoloró tan to el agua de bromo como una solución diluida de permanganato d e potasio. El tratam iento de I con ácido nitroso en frió dio una solución sin que se desprendiera ningún gas. Esta solución se vertió sobre una solución de cianuro cuprosó y precipitó un compuesto ( IV ) . E l compuesto IV todavía contuvo bromo y nitrógeno, pero fue insoluble en álcalis y ácidos diluidos. Cuando IV se hirvió coa ád d o sulfúrico diluido duran te algón tiempo, se convirtió en V. Este compuesto ya no contuvo nitrógeno, pero li bromo. El compuesto V fue insoluble en agua y ád d o clorhídrico diluido, pero soluble en bicarbonato de sodio. Dio un equivalente de neutralización de 200 ± 2. El compuesto V no reaccionó con nitrato de plata ni con permanganato. , .
4. U n compuesto ( I ) cristalino, amarillo clavo, dio positivas las pruebas pa ra nitrógeno y bromo, fue insoluble en agua, ácidos diluidos y álcalis. No reaccionó con fenilhidrazina, 'doruro d e . acetilo o perm anganato diluido y frío. Con nitrato de plata alcohólico y irla no reaedonó, pero cuando la soludón se hirvió duran te elgún tiempo, se formó un predpitado de bromuro d e p iala. Cuando el- compuesto I re calentó con cinc y soludón d e cloruro de amonio y la mezcla se filtró, el filtrado redujo el reactivo d e Tollen». L a oxidadón vigorosa de I dio un nuevo compuesto ( I I ) que todavía tuvo bromo y nitrógeno y fue insoluble en ád d o dorhídrico, pero soluble en solución d e bicarbonato de sodio. El compuestoI I dio un equivalente de neutralización de 145 ±. 1.
El tratam iento del compuesto I con estaño y ácido clorhídrico, seguido de álcali, dio un nuevo compuesto ( I I I ) el cual tuvo nitrógeno y bromo y í u e soluble en ád d o clorhídrico diluido. El compuesto ( I I I ) dio un precipitado con «gua de bromo, reaccionó con cloruro d e acetilo y cuando se trató con d o m o de bencensulfonilo y álcali dio una so ludón transparente. Por oxidadón vigorosa d e i l l se obtuvo un áddo' cristalino, blanco ( IV ) . El compuesto IV dio negativas las pruebas para bromo y nitrógeno y tuvo un equivalente de neutralización de82 ± I.
G uipo 14 433

5. U n lA íd* neutro ( I ) , amarillo claro, tuvo doro, pero ~c nitrógeno. No reaccionó con nitrato de piala alcohólico caliente, clorure* acétale, o con'bm- mo en tetracloruro de carbono. Con íenilhídi*zin& dio un precipitad^. £] compuesto I £ue inerte a loj álcalis en frío, pero cuando se calentó durante algún tiempo con hidnfcódo de «jdto concentrado, se obtuvo una solución transparente. EJ deslúado de esra solución alcalina no contuvo compuestos orgAnicos. La acidulación de la solución alcalina con ácido fosfórico dio un precipitado ( I I ) que se filtró. No je pudieron obtener compuestos orgánicos de filtrado, ni 'p « r rvaporarión a sequedad.
El compuesto I I tuvo cloro, un equivalente de neutralización de 29? ± 2 y reaccionó con anhídrido acético dando un compuesto ( I I I ) , e! cual tuvo un equivalente de neutralización de 340 ± 3. El compuesto II I no reaccionó con Agua de bromo, ni con bromo en tetracloruro de carbono, ni con Íenithidrazin3.
L a oxidación vigorosa d e I con perm anganato alcalino dio muy buen rendimiento d e -un producto, IV, que tuvo cloro y un equivalente de neutralización de 156 ± J. No se pudo encontrar otro producto de oxidación. La oxidación vigorosa de I I o I I I con dicromato de potasio y ácido sulfúrico tatribiín produjoIV , pero con muy bajo rendimiento.
Grupo 151. Un ácido ( I ) tuvo un equivalente de neutralización de 151 ± 2 . El Ira-
tsm iento de I en frío con d o ru ro d e acetilo lo convirtió en u n nuevo ád d o ( I I ) que dio un equivalente de neutralización de 193 ± 2. L a oxidación moderada de I con soludón irla de peim anganato de potasio Ja transformó en u n ácido ( I I I ) que dio un equivalente de neutralización de 149 ± 2 . El compuesto I I I d io un derivado con lenilhádrazina. Por oxidación vigorosa de I , I I o 1H « obtuvo un ácido (IV ) con un equivalente de neutralización de 122 ± 1,
2. U n compuesto ( I ) dio un equivalente de neutralización de 223. Por oxidadón vigorosa se convirtió en un nuevo á d d o ( I I ) con un. equivalente de neutralización de 167. T ratando I con d n c y á d d o clorhídrico se obtuvo ur. áddo( I I I ) con un equivalente de neutralización de 179. Leu compuestos I , I I y I I I tuvieron nitrógeno.
3. U n compuesto con azufre iue aoJublo en álcalis fuertes, pero no en soludón de bicarbonato de sodio. Por oxidación vigorosa te obtuvo un áddo que tuvo azufre y un equivalente de neucralizadón de 102 ± 1. Cuando e*tc compuesto se trató con vapor sobrecalentado, se obtuvo un á d d o libre de anafre.
4. U n compuesto ( I ) que contenía cloro dio pruebsa positivas con nitrato d e plata alcohólico, solución de hipoyodito de sodio y d oru ro de acetilo. Por hidróloíii con bicarbonato de sodio se obtuvo un compuesto ( I I ) libre de cloro, d cual dió positiva Ja prueba d e l yodoformo y se oxidó con á d d o pexyódico a un compuesto 6n k o ( I I I ) . El compuesto I I I t-imbífin dio positiva la prueba del yodoíoemo.
5. Un compuesto (A) conteniendo sólo carbono, hidrógeno y oxígeno, reaedonó con doruro de acetilo, pero no con fenilhidrañaia. L a oxidación con ácido peryódtCD lo convirtió en u n nuevo compuesto (B ) , .e l cual redujo oí reactivo de Tollens pero no el de Fehling. T ratando B con cianuro de potasio en etanol acuoso te convirtió en un nuevo c o m p u s o (C ), cí cual dio pruebas positivas con cloruro de acetilo y íenilhidradna, Por oxádadón de C con idución d e Fehüng o ád d o nítrico se obtuvo u n 1 compuesto amarillo (D ) , el cual dio un derivado con o-fenilendiaauina. C uando D se trató con peróxido de hidrógeno, »e obtuvo un á d d o (E ) que dáo un equivalente de neutralización de 135 ± 1- Por hidrogenación catalítica de C o D se obtuvo el compuesto original (A ).
434 Problemas

6. XJna substancia (A) sólida, neutra, tu ro nitrógeno. Se le recuperó después de in ten tar hidroJixaHa coa áddo* y baj*^ diluido». Dio negativas las pruebas con cloruro de acetilo, bromo en tctracloruro de carbono, solución de hipoyodito de sodio y reactivo de Taliens. Reaccioné lentamente coa dimtro- fenilhidrazina y, después de tratarla con cinc y cloruro d e amonio, redujo el reactivo de Tolíens.
Guando A se trató con clorhidrato de hidroxtlaxnina en piridína, se convirtió lentam ente en un nuevo compuesto B. La subí tan d a B se trató con pentadoruro de fósforo, obteniéndose C. Hirviendo C con ácido se convirtió cu D, un ácido que tuvo n iíróg íno y un equivalente de neutralización de 168 ± 2, y la «1 de una base E. Et clorhidrato d e E tuvo un equivalente de neutralización {por titulación con álcali) de 195 ± 2.
El ácido D se trató con cloruro de tíonilo y el producto se añadió a hidróxido de axaonio. L a substancia neutra que se obtuvo se tra tó con bromo y solución de hidróxido de sodio. Él producto (F ) fue una substancia soluble en ácidos, la cual, despuót de tratares con nítrico de sodio y ácido clorhídrico, dio coloración con fl-naícol. T ratando F con estaño y ácido clorhídrico se produjo G, substancia fácilmente atacable p o r agente» oxidantes. El compuesto G na día derivado cristalino cuando se trató con bemílo.
L a base E, obtenida en la reacción-con pentacloruro d© fó*foro, reaccionó con nitrito de aodk> y ácido sulfúrico diluido. La. solución resultante dio coloración con jl*naftoJ, AI hervir la sofudón acuosa, se p rodujo tina substancia H , libre d e nitrógeno, 3a cual se disolvió en álcali acuoso^ pero no en bicarbonato d e jod io acuoso. Por oxidadón d e E o de H íe p rodujo un ád d o (c<juívalente de neutralización, 83 ) j el cual íádlment© se convirtió en u n anhídrido. L a subitan-' cía H no se copuló con solución de bencendiaronio.
G ru p o 15 4 3 5


Apéndice
L a s i g u i e n t e l i s t a r z p r s s e n t a ' í i v a de los reate ríales y reactivos del laboratorio apropiados para un curso de identificación de compuestos orgám^ eos ha sido incluida corno resultada de numerosas solicitudes al respecto. Se verá que las variaciones en el equipo disponible en uno u otro laboratorio pueden requerir que se modifique u omita mucho del equipo aquí sugerido. Dichos cambios a menudo se pueden realizar sin que se afecte seriamente la naturaleza del cursa.
APARATOS
E q u ip o p a r» c a d a g a v e ta in d iv id u a l
Se ha encontrado conveniente asignar a cada estudiante una gaveta en el laboratorio de química orgánica, equipada con los aparatos ordinariamente requeridos para el primer año de trabajo en preparaciones orgánicas, Estos se complementan con un equipo especial que contiene los aparatos necesarios para efectuar ias pruebas de clasificación y preparación de derivados en escala pequeña.
E q u ip o com plem entario que se sugiere
12 Tubos d« ensayo, P jtoc, 16 x 125 mw 12 Tubo* de ensayo, Pj7W<, i 3 X 100 wm12 Tubos de ensayo, vidrio lu&ve> JO X 75 o r a
i Pipeta graduada de 1 nú1 Pipeta graduada de 10 mJ 5 G otcroi medicinales2 Vasos de precipitados, Pyrex, 50 nrJ I Embudo Hirsch, N’ 000i Embudo Hirsch, N e 0000 I Embudo Büchrter, 50 mmI M ana* K itajato p a i a filtración, 50 miI Espátula pequeña de acero inoxidable [ C rúol de porcelana con tapa, N» 00

r *M¡ 438 Apéndice
9jh tN iN ii
S»
W
ui »
1»r»r *f»
:
t Balanza con do» pesas de 0.1 g, una de 0.2 g y una de 0.0 g1 Desecador pequeño3 M atraz de dejtíf&tíón, 25 mi1 M atraa de destilación, 10 ral2 Tubo» de ensayo con tubu’adura lateral, Pyrex, 15 x 125 ritm 1 Tubo» de ensayo con tubuladura lateral, Pyrcx, 20 x 150 jntn
Equipo general de laboratorio
Hornos de sccado, eléctricos o de vaporBalanza), tipo triple escalaBloque de Maquenne, calentado con gas
Equipo especial de laboratorio
Loa aparatos siguientes deberán conservarse en u r cuarto especial, alejados de todos los vapores corrosivos.
B alanzas a n a lític as , d e las e m p le a d a s e n a n á lis is c u a n t i ta t iv o , y eu? n ia ix e í d e pesas
Rcíraciómeiro, tipo de Abbe, como el Valentina (Ir,duít«-o-$cier<tfíic Co., Philadelplüa, Pa. o BauscJi & JLcmb, Rochester, N- YO
Densitómotro Fiscbcr-Davidson Polarímelr»Aparatoi para punto» de fusión Fisbec-Johns y Natge-Axctrod
Artículos obtenidos por préstamo temporal del instructoro del almacén
Alambre de platino, aproxim adam ente de 5 ero de longitud, N* 20, soldado a una varilla de vidrio
Lámina de platino, aproxim adam ente de 2.5 era1, soldada a una varilla de vidrio
Tubo» para polarímetro, uno de 1 dm de largo, tino d e 2 dm
SUBSTANCIAS PARA EL ESTARTE DEL LABORATORIO .
Compuestos orgánicos
Las substancias siguientes son útiles para eíectuar las pruebas de solubilidad y clasificación y para preparar algunos derivados {pero no todos), Se ha encontrado conveniente proporcionar frascos de unos 100 mi de capacidad, de boca ancha, con tapón esmerilado, para los sólidos y boca angosta, con tapón esmerilado, para los líquidos. Deberán usarse frascos más grandes (de unos 500 mi) para los disolventes comU' nes, tales como acetona, benceno, cloroformo, tctracloruro de carbono,
.:S£r•>í

C o m p u e rta * o rg án ic o s 4 3 9
dioxano, éter, ügroína (70-90^) y tolueno* Para una clase de veinte estudian les, de 20 a 50 g de los siguientes compuestos orgánicos deberán colocarse en. los frascos de los estantes, aunque, por economía en ia compra de reactivos, pueden comprarse en unidades de 50, 100 6 500 g. Las cantidades realmente necesarias por estudiante variarán, como es de esperarse, con ia naturaleza de las substancias desconocidas, b atingencia con que se seleccionen Jas pruebas de clasificación y la habilidad de manipulación del estudiante.
Acetaniíida A nhídrido acético Acetona Acctoíenona Alcohol atllicop AroinofenotAlcohol í«-bul(l¡coAlcohol fer-hutiliooAlcanforAmilenoAnilinaAnisolA20xibenccno Acido benzoico Alcohol bencílico Alcohol fc-buttlíco Acido cloroacétko Acido cinámico Acido 3,5-dinttrobenzoico A cetato de etilo A cetatoaceuco de etilo Acido í¿r¿nico Alcohol n-hcptüico Acido ^-hidroxiben2oíco Alcohol isoamilíco Alcohol isopropllico Acido láctico (85% )Alcohol láuríco A nhídrido malefco Acido mercaptoac¿t¡co (áddo ti'
célico)A cido p-mcrebenzoicr A nhídrido 3-júircfcálico Acido oxálico A nhídrido ítilíco Acido picrico Alcohol metílico Alcohol >i-propSlico Acido salicOico
Acido sulfenilico Acido tartárico A nhídrido tetradorvítálico Acido />'to!ucn3UIf6nico Sene i lamina Benzoato de amonio Bromuro de n-butilo Bromuro de ící-butito Bromuro de ¿-butilo n-Budraldchido Benzaldehído Bcnzamida Benceno Bejiddina Benzonítrilo Benzoícnona ¿-brom oanilina Bromobenceno Bromuro de 0-bromobencito Bromuro de p-bromo fena rilo ^-hrom ofenílhidraana Bisutíuro d e carbono Bromuro do ¿-dorofcnacilo Benzoato de etilo Bromuro d e etilo Bromuro de otüeno Bromuro de ¿-yodofenadto Bromuro de p-fenllfcnadb Benzoato de metilo Cloruro d e alilo C lorhidrato d e anilina C loruro de bencilo C loruro de iS-bcnciltiouronío C loruro d e f-bromobencenaulfonllo C loruro d e ¿>-bronaobervdlo C loruro de 5-(¿*bron>0bencíl)ti0'
uronio a*C(oroacetofcnona Clorobenceno p-Clorobenzhidrazida

v -4 4 0 A p Á n d k *
C ia n u ro d e •S -(/>-<Joi'(ib«ncil)tiouro- n ío
C lo ro fo rm o ¿ •C lo ru n itro b e n ce n o C ic lo hexanOCloruro de S^-dinitnsbcnzoíIo Cloruro de difenikarbam ílo Clorhidrato d e hldrcoálamina Cloruro de w aitrobencensulíonílo Cloruro de /i-rútrotwnzoüo Cloruro de £-nitrobencilo Clorhidrato d e íenilhidrazina Cloruro de metánsulfonilo
, C bru ro de oc-naftalenrulfoxnloCloruro de pieudoiacarina C lorhidrato d e ienúcatbaúda Cloruro de entilo Difcnilop , j f -Diamiiwdiíeniinintano p -T h biOmoixrnCcnoD«etilan£n&Dietitenglicol/■'DimetiiammobeiualdehídoI Mtngálanilina2.4-Diwlroanilina «•Dinítrobencena2 .4 -D ú ú tro c Io ro b e n c ea o 2,4~D¿QÍtroferúlib»dra3Ína .DioxanoEter b+ u tilico EtilectgficolE ter d e petróleo (40-60°}Form alizaFructosaFenolo-FemJcndiamina
, se-Fc pile ti laminaFrnilhi|ji^pn^FcnXI lexoicarbazid a
. Florogtudnol Ftalimida GalactosaG arduol (sulfato de lauriitodio^GlucosaOlicerinftn-HeptaldehídoHexa no
*
Hidroquinonar*ocianato d e ¿-bromofenílo Isocianato d e 3,5-dinitrofcmlo -* Isodanato de ¿«nítrofenilo Isocianato d e f3*naít¡lo Jíojí acia nato d i « n a ítito LactosaL íg r o l t t a (70*90°)Maltosa,MeaitücnoM etonaMetitarülinaTO-N¡troaí¿lina/ ¡ - N it ro a n i l in a
^-NüijobenauidaNitrobencenom-Nitrobeiwhidraxida¿•Nitrofenal^-Nicrofcrúlhidrazana N aítdcno f^Naítol tz-Naítoquinona(J-Naftilhidra-ziaaPentanoPentanol-2Penteno-2PiperaaoaPiperidinaPiridina (robre liidróxido d e po ta
s a )Reactivo d e icido yódico ReiorduolSdiciJaldchídoSacarosaTetraclom ro de carf;or>o /-Toluenjvlfonato de metilo ^-Toluküna j^Tclib«imcarbazída Trietanolaüiina Trietlíam ina1,3,5-Trijiítrobeneeno TlcureaTolueno (anh.)U rea .XanthidrolXilenoYoduro do metilo

Reactivos y to lw ín i i t t J ^ S E
Las cantidades sugeridas son apropiadas para una clasq do veinte estudiantes, aunque algunos de lo» frascos con los reactivos usados con más frecuencia tendrán que llenarse varias veces en un semestre.
H ierro en polvo Limadura* de ma$ne»io N itrato mercúrico N itrato de jodio N itrito de sodio N itropruíiato de jodio Oxido mercúrico Perm anganato de potasio Persulfato de potado Peróxido de plomo Sodio metálico (en tolue
no)Sulfato cúprico (anhidro)Sulfato feiToto amónico Sulfato d e hidraaina Sulfato de magnesio {Anhi
dro)Sulfato de eodio (anhidro)Sulfuro de sodio (cm tal)Sulfato mercúrico T íocianato de potasio Tiosulfato de sodio
(cristal.)Trióxido de cromoY o d o (rw u b lim a d o )Yoduro de potasio Yoduro mercúrico
Compuestos inorgánicos
Acetato de plomo 150 sAcetato de sodio (anhidro) 200 aAcetato de jodio (cm t.) 100 sAcido yodhidrico (5?$5) 100 %A ddo fosfórico (£5% ) 1Bicarbonato de sodio 1Bisulfito de sodio (anhidro) 100 sBisulfito de Godio I kgBromuro de potasio 100 SBromuro mercúrico 10 sCarbonato d e amonio 100 sCarbonato de potasio
(anhidro) 1*C arbonato de *odio {anhi
dro ) ' 1 k sC ianuro de potasio 100 8C inc (en polvo) 200 SCloruro de amonio 100 gCloruro de calcio (anhidro) l fc*Cloruro de sodio 1 ksCloruro cslanoso 100 £Cloruro de cinc (anhidro) 100 gDicromato de sodio 1 fc*Estaño (granular) 200 gH idrato de hidraiina 25 SHidróxido de potasio (Jen*
tejas) 1Hidróxido de sodio (kn«
teja») 1 **
100 s100 *100 8100 s200 g
5 ¿50 8
200 s25 8
100 s
50 s50 g
100 *50 s
1 t gI
50 G50 s
100 s
100 s100 B100 g100 s10 s
Compuestos diversos, reactivos y soluciones
Ee conveniente preparar de 500 a 1000 mi de las soluciones y conservarlas en un estante o mesa lateral del laboratorio. Las instrucciones para su preparación se dan en el capitulo 8 y en los libros de texto de análisis cualitativo y cuantitativo inorgánicos.
Aceite mine ral (Starvolind)Aceite de semilla de algodónAcido clorhídrico, solución valorada (0.25W) (garrafón de IS litrw) Acido nítrico diluido 5%Acido sulfúrico diluido (lO 'ft)Agua de bromo (saturada)A gua de d o ro (reciente)AlgodónBisulfito de sodio, acuoso-alcohólico

DxtcoEtóxkío de sodio alcohólico (1 % )Hidrólogo de potajloj alcohólico (10% )Hidróxído de ¿odio, solución valorada (0 .1 //) (garrafón de 18 litro»)Kicsd^uirLana de vidrioMezcla magneaio*c&rbona$o de potasio N itrato d« p lata, acuoso (5 % )N itrato de plata, alcohólico (2 % )N itrato mercúrico xN’itroprusiato de zodto NoritPapel de rojo congo Papel reactivo de ahnídón-yoduro Papel reactivo de zírconio-alisarina Peróxido de hidrógeno ( 2 %)Plúmbico de sodioReactivo de ácido peryódicoReactivo de BenedictReactivo de 2,4-dirtitfoíenHbídrazmaReactivo de clorhidrato de hidroxíJaminaReactivo de cloruro de niquel-bisulFuro de carbonoReactivo de cloruro de nSquel-S-nitrosalicilaidehidoReactivu de fucsina para aldehidosReactivo de nitrato céricoSolución de acetato de plcm o (1 % )Solución de ácido elorhidrico-cloruro de cinc (reactivo de Lucas) Solución de ácido fosfórico (20% )Solución de bicarbonato de sodio (5 % )Solución de bromo en tetradoruro de carbono (59o)Sotución de carbonato de sedio (1 0 % )Solución de cloruro d e calcio (saturada)Solución de cloruro férrico (5 % )Solución de Fehling N 0 i Solución de Fehling N9 2 Solución de fluoruro de potasio (20% )Solución de hidróxido de amonio (2 %)Solución de hidró>ádo de sodio (2 0 % )Solución de pciroanganato de potasio ( 2 %)Sotuccón de sulfato fcrrcKo anjónico (5(1 g/1)Solución de yodo'yoduro de potasioSolución indicadora de azul de limol (0 .2% en ctanol al 95% ) Soludón indicadora de EogeoSolución indicadora de fenolctaldna (1% en elanol al 95% )Solución indicadora de Grarnmetcy Yoduro de sodio en acetona
¿ 4 2 A p é n d ic e
Substancias conservadas dentro de la campana
Acido clorojÜJfónico BromoAcido nítrico fumante Bromuro d e feriadloAcido sulfúrico fumante Cloruro de acetiloAgua saturada con ácido sulfhídrico Cloruro de aluminio (anhidro)

S u b ito n e ra s e spec ía lo s 4 4 3
Cloruro de henceraulíonifo Cloruro de bemoíto Cloruro de ítalilo Cloruro de tionilo C loruro de ^-toluensulfonilo Isadañato de a-n&ftíb Isocíanato de íeuífo
Poliíulfu/o de amonio Tricloruro de íósfoco
FsOÜOCÍanato d t fcnitci Oxicloruro de fóiforoPenlacloruro de íórforo
Isocianato de ¿-tolilo fMjüociajLdtc <¡e a-naliilo
Reactivos para las gavetas de los estudiantes
SuhjUociíLE, grado reactivo, en frascos de 250 m i con tapón esmerilado Acido acético glacialHidróxido de amonio, dens. cap. 0,90, 28% NHi Acido clorhídrico concentrado, dens. «;sp, 1,19, 37% HCI Solución de ácido clorhídrico, 10%Acido nítrico concentrado, dena. csp. 1-42, ?0% HNOj Solución d e hidróxido do &odio, 10%Acido sulfúrico concentrado, dens. t ip . 1.84, 95% HjSO»
Substancias especíales obtenidas del afniacén
Etanol 95% Eter «tilico absolutoEtanol ahsotulo Acido doroplaiínico
Substancias desconocidas
Estos compuestos deberán seleccionarse cuidadosamente por su pu- reza y «cogiéndose ejemplos típicos de los mencionados en las tablas del capítulo I I , pero no necesariamente limitándose a los compuestos en ellas enlistados. Es suficiente de 5 a 10 % de un sólido y de 10 a 15 mi de un liquido. Las mezclas también deberán contener de 5 a 10 g de un sólido y de 10 a 15 mi de un líquido además de suficiente cantidad del disol* vente apropiado.
A medida que se aproxima el fin del semestre mejora la experiencia y técnica del estudiante, pudiéndosele d ar cantidades mucho menores de las substancias desconocidas y las pruebas de solubilidad, clasifica* ción y preparación de derivados deberán efectuarse en una escala mucho más pequeña (aproximadamente una décima de las cantidades especificadas en los experimentos y procedimientos). Ver sugestiones en la pág. 10.


Indice alfabético
Nota; Los números dentro de paréntesis redondos son puntos de ebullición. Los números dentro de paréntesis rectangulares {corchetes) son puntos de fusión. Todos los demás números que están Ubres se refieren a las páginas correspondientes.
La nomenclatura es bastante v'; nada. Se usan tanto los nombres comunes como nombres sistemáticos. Hay que advertir que existen muchas variaciones en el uso diario y por lo tanto el estudiante debe consultar libros de texto o Jos índices del Chemical Abstr<ul$t para localizar las estructuras correspondientes a los nombres.
Los principales compuestos de las tablas del capítulo 11 están incluidos en este índice (con puntos de fusión, puntos de ebullición y números de página), pero los derivados no figuran en él. Los procedí miento* para preparar derivados del capítulo 10 aparecen en el índice, pero no se han incluido en él lo? numerosos derivados especiales y las reacciones. Estos pueden encontrarse mediante la consulta de las referencias citadas bajo cada clase de los compuestos discutidos en el rnismo capítulo.
A
Abbe, rcfractóroetno de, 64 Absorción, m&xinxu de, 229 Absorción, áreas de las señalca, 220 Ace na í ceno {95}, 381 Aoenaflenona [121), SBC Acctal (102), 330 Acctaldehído {21), 3+3 Aeetaíes, derivados de, 252
tablas de, 330 A cettunidi Í82J, 345 A ctU nilida (114], 3+6 Acetatos, 269 Acético, ácido ( l i d ) , 334 Acético, anhídrido ( i 3 8 ), 331 AcctopropWnico, anhídrido (15 4 ), 331
Acetil-tírí-buiífico, alcohol (164), 385 Acítoacetameda [54], 345 A c e t o a c e t a n i l í d a (85], 345 2'Aceto-¿-cimeno (250), 386 Acctoína (1 4 5 ), [15), 339. 341 Acetol (146), 339, 385 .Acetomesitífeno (245), 386 Acetona (56 ), 3B5 Acetondicarbcxílicv, ácido J135 d.j,
336Acetoniirilo (8 1 ), 390 Acctonitaeetona (188), 386 ¿•Acetofenetídina {138]T 354 Acctofenon& (200), 366 m-Acetotoluidída (651, 345o A ottotoluídida [112], 346 ¿'Acetotoluididá [153], 347
4 4 5

In d ic e a l f a b é t i c o
/-acetOxwV-Tnedlacetaryílida [98J, 346 Ace ¿üac« tona (13 9 ), 385 Acctilantranílieo, ácido [185], 337 Acótilo, bromuro d e (81 ), 331 Acetilo, cloruro de (5 5 ) , 331 Acetilendicarboxflico, ácido [179], 337 Acetilo, fluoruro de (21), 331 Acetilo, yoduro dn (108), 331 a-acetil-fj-raetilurca [180], 34? a-acctil-jj-fenilhidra2Ína [128], 34$ Acetílpiperidina (226), 345 Acetiisalicííieo, ácido [135J, 336 Acetilurea 1218], 343 Acido, derivados de anhídridos de, 253
tab la de, 333 Acido, cloruros de , 128 9-AdlamÍdoxanc©nos, 278 Acílo, derivados de bal unos de, 253
tabla de , 333 Acides, efecto de estructura sobre 3a.
91efectos cleccrónicos sobre, 9(
' efectos esférico* sobre, 93 Acido?, derivado* de, 254
tabla de, 334 ■Aconítico, ácido [19 í], 338 Aconítico, h idrato de ácido [125], 335
< Acopiamiento sptn-ipin, 220 Acridina [108J, 361 Acrilamida [8J»}, 345 Aerilanilida [105], 346 Acriüco, ácido {140}, 334 AcrilonitriJo {78), 390 Acrililo, cloruro de (7 $ ) , 931 Acrolcína, acc&J de (126), 330 A croldna Í5 2 ), 343 Adiuvidad, principio de, 233 Adipamida {mono) [130J, 346 Adiparoida (d¡) [220), 348 Adípico, á d d o [152], 337
solubilidad de, 88Adiponitrilo (295), 390 n-Alanina (29? d.J 363 L-AI*n¡na [297 d i 363 dí-A kaina. [295 d j , 363 0-Alanina [196), 362 { + ) -Alcanfor (205) (179], 388 ¿AAlcanfor (205), [178], 388 Alcohole», derivados de, 263
tab la de , 339 Aldehidos, derivado* de, 271
metona, derivado» de, 276 oxidación de, 274 tab la de, 343
Alilo, acetato de (103), 3G5
Alílico, alcohol {97), 339 AlUamína (56), 349 Aiilbenccno (157), 384 Alilo, benzoato de (23 0 ), 366 Alilo, brom uro de <71), 373 Alilo, clorura d e -(4 6 ) , 373 Alilo, c i a n u r o . d e 118), 390 Ali id ílico , ¿ter (6 5 ) , 369 Alilo, íorroi&to de {$3), 365 Alilo, yoduro de (1 0 3 ), 373 Alilmercaptano (9 0 ), 389 Al tlurca [80], 345 Alizarina [289], 388 Almidón [d], 364 o-Alotrcoaina [2 72 d.], 362 k-Alotreonina [272 d.], 362 ¿/-Alotreonina [252 d.J, 362 Alootano [170 d.], 347 Alquenilo, derivados l haluros do,
299 tabla de, 373
Alquilo, derivados de haluros de, 300 tabla de, 373
¿•Alquilisotímirca, picratos de, 302 AlquiJmercurio, haluros de, 301 Alquil-^ naftil*/», iteres, 302 O-Alquilsacarína, derivados de, 265 Aluminio, cloruro de , anhidro, I3Q Amidas, derivados de, 277
preparación de, 257, 277 tabla de, 345
Aminas, primarias y secundarlas, de- rivadof de , 278
sale» cuaternarias de amonio de, 284 tabla» de , 349, 355 terciaria»,’ derivados, d e 283 tabla de, 358
ro-Aminoacetanilida [279], 354 ^-Aminoacetanilida [162], 354 p-AmínoaCetofenona (29 5 ), [Í06J, 353 Aminoácidos, derivado? de, 285
derivados d e ¿-toluensulfonilo de,- 288
prueba de la mullid riña para, 206 rotación óptica de, 287 tah la de, 362
2-aTn*noaniraquínOna [302], 354 /-A m inoajobenceno (3601, [125], 353,
396o-Arfúnobeiualdchído [401, 344
. m-ArninobenCensulfonamida [142], 400o-Aminobenccnsulfonanúda [i53¿ 400 /j-AminobencejJBulíonaiuida [1633, 400 >n-Aminobcnjoico, á d d o [174 d.J, 362 Í-Am ínobenzoko, ácido [186 d j , 362
In d ice a lfa b é t ic o 4 4 7
¿•Aminobcnzofenocia [124J, 353 2'Aminobifcnilo {299;, {45J, 351 'wAminobifenilo (302) [53], .351 2*Amino.5'br0mototueno [59], 351 4-Amino-3-brc>tnololucno (240), [26],
350, S5lva-A nuno-n .bu tírico , ácido ¡285 d.3,
363 _ ■' ’dAct-Amino-n-butirico, ácido [307 d.?,
3633*An)inO'6-clorotolueno (241), [83],
?522-An>ino-3.5-dicloro tolueno Í50], 3514-Aroino-3 ¿ ‘didorotolueno [60J, 352 í-Aminov.VrlV-dietjlaniJina (262), 350 o-Arm no-Ar,iV-dím cti!atu|tna (217),
350-p- Ami no-Ar, A7 - dimctitanilina {262),
[4 U, 3512 • Amino -1 ,3 - d ímetií benceno (216),
3502 - Amino -1 ,4 • dimetilbenceno (215), % [15], 350, 351
4'-A m ino-1 , 2 -dimetilbenceno {226), [49], 351
5-ArmnO'C-cforotolucr.O (2 4 1 ), [83],352
2-Ainino^3,5'díclorotolueno [50], 3 5 14-Am ino.3r5-dídoroe©luetio [60J, 352 ^-Amino-A'^Y-dictilanilina (262), 350 C ~ Amino - Ñ t N - d ímctílanilina (217),
3304 - Amino *1 .3-dim etilbenceno {212),
3495 -A m in o -1 ,3 -dimetilbenceno .(220),
3504-Amino-2,6-dinitrotoIaeno [169J, 354 4-Amino-3,5-dínitrctolueno (166J, 354 2-Ajninodiíenilaminft [80], 352 4-Am inodifenÍtaiaina [75], 352 5*Amino-2-hídr,0)dtaluen0 [173J, 354 a-Amínoisobutírico, ácido [280 d.], 363 2-Am5nó-4-uoprópiltolueoo (242), 350 '^•Amino-iV-ifletilacetanilida [63], 352o-Am ino-tf-iuetilanilina (245), 350 p-Amíno-W-metilanilina (258), [36L
350, 3512-Araíno-5 -metilbenceno-1,3 -disi11 *on-
amida [257J, 401 2'Atníno-5-metilbenceno-lJ3-su3fonil-
cloruro de [156], 405 2'Aniíno:6-metilpiridina 141], 351 2-An3Íno-4-metilpirídÍDa [98J, 353 2-Ainíno-S-niuck- 1,4-dímetil benceno
: [142], 354 4“- Ainino-5-nítro-l ,3-dimctilbcnceno
[76], 352
4-Amino-'6*n¡tro-l,3-din«:ti]benceno Í123J. 353
UArtiíno-2-nítronafta!etM> [144] 3541-Amino-4-nitronaflaleno {195], 3542-Amino.5'ni<ronaílaleno [144], 354 2‘Amino-4-nicrotoluí!no ÍIG7], 353 2-Amino-5-ni Cro tolueno [130], 354 3»Amíno-6-ni tro tolueno [135], 354 4-Armno-2-nitroto!ueno [77], 352 4-Attúno-3-nitrotoTueno TI 16], 3531-Artúnofcruntreno TI46], 354 W-Aminoícnol [122] 353o-Aroinofcnol [174], 354 ¿-AminOÍcno) [18+ d,], 334 p-Aminoícnilacítico, ácido [200 d.],
362
2-Aminop>ridina [56) 3513-Amtnopíridina [64], 3523-AmcnoquinOleína [94], 3534-Amínoqu‘cnolcína [154], 3545-Aminosalicílico, ácido [283 d.], 363 Amígdalina [214 d.], 364 n-Amilo, acetato de (146), 365i*-Amílico, alcohol (138), 339 ícc-Amílico, alcohol' (119), 339 ¿ír-Amílieo, alcohol {102), 339 rj-Amilamina (104), 349 Fi-Aii}ilmcíilcetor.a (151), 385 fí-AiniHjenceno (202), 380 «-Amilo, bromuro de (129), 374 ¿«r-AmiJo, bromuro de (108), 374 n^Amilo, n>butirato d e (185), 366 n-Amilo, clorura de {107), 373 íír-Amilo, d o ru ro de (8 6 ), 373 «-Amílico, í le r (19C), 370 n-Ami le tilico, éter (118), 369 ' n-Amilo, ío rn ia to de (130), 365 n-Amílo, yoduro d e (156), 374 í<r-Ajnilo, yoduro de (128), 374 n-Amilmercaptano (126), 389 jec-Am ilm rrcaptano (112), 389 ffr-¿Vmilmerc3ptano (97 ), 389 n-Amitmetilico, éter (99 ), 369 n 'j\m il-2-naítílíco, éter (25), 372 n-Amito, nitrito de (104), 368 ^A m ilfenol (2 5 3 ), [231, 337 ^•isr-Amilfenol [96], 398 r 7!-Amilo, propionato d e (169), 365 u-AmiJo, valcrianato d e (204), 366 Amonio, sales de, cuaternarias, 284 Anetol (23 2 ), P 2 L 371, 372Atvíóteros. .-wlulnlídad de compuesto»,
98
AngéGco, ácido [46], 335 Anilida», 128, 255, 258

Anilina (183), 349 Anísaldehído (247), 343 Ankíco, íd d o [184J, 337 m-Aniíidina. (251), 350 ¿-Amsidina (243), [57], 351 AflSúco» .alcohol (¿5J, 341 Aniailo, dc ru ro d e [26], 332 Aniso! (154), 371 Antraceno (216], 381 AntranHico, ícido (144 d.), 362 Antraquinona [286], 3881.5-Antraqumondisulíonamida (246], 401 l,8'Antrcquinond¿Bulfonamida [304], 401I.S.Antraquinondisulfonüo, cloruro de
[265 d.], 4051.6-Antraquinoridísulfomto, cloruro de
[192], 405 % .1.7-AntraquinondUulfonilc), cloruio de
[232], 405l^A ntraqu ísondisu lfon ilo , cloniro de
[223], 405 2,6*AntraquinondisuIíonila, clorura de
1250J, 4052.7-Antraquínonditulfonjlo, cloruro de
(186], 405 .Antraquinotvp-sulfonamida [261], 401 Antraquinon ~a. -juíCodíÍo, cloruro de
(214J, 405 Antraquinon-0-su1fonilo, cloruro de
(197L 405 A m ipirina [113L 361 Apiol [32], 372 t-Arabinosa [160 d.], 364 dí*Arabinosa [164 tí.], 364 Arbutina {165 tí.], 364 jl( + )-Ar$ínina [233 d j 362 dl-Arginin» [238 d.], 362 Arijo, halaros de, 375 Aritmetílcetonas, 308 Arilcwdacéticos, ácidos, 320 Arikulfonamidaj, 283, 325 AromitíoosL, derivados d e hidrocarbu
ros, 305 labia de, 380
Aroilbetuoico*, ácidos, 307 Arrastre con vapur, 106, 261 r;-Asc¿rbi''o, ácido, 364 L'Ascórbico, ácido, 364 ¿/-Aícórhíco, ácido, [ 169], 364 Asoeiadón, 89 i>Aspara$íaa [226 d.], 362 L-Aspangin* [226 d.], 362 D-Aipárrico, ácido [270 d.], 362 L^Aipártico, ácido (270 d,}, 362
j44£ Indica alfabético
I ¿J-Aspártko, ácido [280 <L], 363 ! Azelaico, ácido [1061, 336 1 iolubiüdad de, 88
Azobcnceno [68}, 396 O'Azabifenilo [145], 396 Aso, compuesto* de, 31?, 396
derivados de, 317 tabla de, 396
I,r-Azoíiafta!«no [190], 396l-2'-A3otwítaleno [136], 396 2,2*A7onaftaler»o (208), 356
I o*Azofenctol [131], 396 | ¿-Azofenetot [160], 396■ m-Azololucno (55), 396 | o-Aro tolueno [55], 396
^-Axotalueno [145], 336 Azoxibenecno [36], 396 o-Azoxibifenilo [1581, 396 ¿-Azoxibifenilo [212J^ 396 Aro>á, compuestos derivado* de, 317
tabla de, 396 Í.l'-Auoxinfiftaleno [127], 396 2.2‘-A*o*¡naf taheño [168], 396 m-Azoxítolueno [39], 3960-Azoxitolueito [59], 396 ¿•Axoxitolueno [75], 396
B
Baeycr, prueba de, 166 B a/bitárico, ácido [245J, 348 Basicidüd, efecto de la estructura ta
bre, 91, 96 efectos electrónico! B o b re , 91 efectos estéricoj sobre, 92
BeiHtein, prueba de, 72 Benedict, solución 132 B encei» (8 0 ) , 380 Bencenazo-o-cresol [128], 396 Bencenazodifenilamida. [96], 3961-Beneenazonaftaleno [70], 3962-BencenazonaftaIeno [131], 3962-Bcncenazo-l-iiaftol [138], 398 4-Bencenazo-l-naftol [206 d-], 396 t-Bencenazo-2-r>aíto! (131], 396 Bencenazortsorstnol £170]s 396 I^-B encendisulionam ida [254], 4011.3-Benccndisulfoiiamida [229], 4011.4-BcnceadifluJfonaniida [288], 4011.2-Bcnceudúulfonilo, cloruro de [143j,
4051.3-Boncendísulfonilo, cloruro de. [63],
4031.4-Benccndisulionilo, cloruro de [139?,
405
Benceno «-hexacloro [137], 379 Benceno fi-hexacioro [310], 379 Bencemolfínico, ácido, solubilidad de,
96BenccnMdfotiajnida [153], 400 BencenEulío^amidas, 283 Benccnsulfónico, id d o , solubilidad de,
96Bencensulíonilo, cloruro de '14], T 35»
. 4021.3.5-Rencentiisulfonamida [312], 4011.3.5-BcnccntrísulfoniIo, cloruro de
. [187], 405Benciditva (400), [127], 353 Bencilo (347), [95], 387 Bcncilamida [155], 347 Bencílico,'ácido (150), 337 Bencilanm a (184), 349 AT-BencilantUa» (298), '37J, 356 BendJo, acetato de (216), 366 Bencílico, alcohol (2 0 5 ), 340 .V-Bendlamidas, 295 Benciio, benzoato de (323), 367 Bencilo, bromuro de (198), 374 Bcncil-a-buiftioo, éter (212), 370 B endlo, n-butirato de (2 3 8 ), 366 Benciio, cloruro de (179), 375 Bentito, doroiicctato de (14-7), 365 Bencilo, tinam ato de [39], 367 Bencildimetitcarbínol (2 1 6 ), 340 Bencílico, ét*r (2 9 8 ), 370 jV-Bendl-A^olanSlina (312 d .) , 359 B endletílko, i t cr (1 8 9 ), 370 Benciletíleetcna (226), 386 Bendlo, íorm iato de (203), 366 BencHísobutíríco, ¿ter, (212), 370 Bencilmalónico, ácido [117 d.J, 336 Bencilmercaptano (194), 389 W'Bcnc3‘W-metilanilina (306), 359 Beneilmeiilico, ¿ te r ( Í 6 7 ) . 370 Bcudlmetilcetona (216), [27], 385, 387 Bcncíl-Ct-naftilico, éter [77J, 372 Bencíl-2-ftflf«Ü¡co, éter [102], 372 Bencilo, o b la to d e (80], 368 o-Bencilfcnol [54], 398 /i-BendJfenol [84], 396 Bencilo, í calato d e [42], 367 Bettciltiouronío, sales de, 259 Bencilo, salicilato d e (320), 367 Bencilo, suednato de [45J, 367 Bencilsulíonamida [105], 400 Bcricilsulíooamidas, 283 Benciliulfoniloj cloruro de (92], 404 Bencütiouronio, w líatos ácidos de al
quil. 270
Bcnciltjoi.ror.io, . 325Benc Jurea [149]. 357 Bentalacetona (262), (4 l i 387 - Benzalacelofenona (348), (62L 387 Bcnzaf, cloruro de (212), 379 . Benzaldebldo (179), 343 \ Bemamida [128], 3 4 6 . ^ ¿1.¿ ..- , Benzamidaí, 232 Bcníanilida [161), 347 .- . B enan tro^a [170], 388 !Benzoatos, 129, 268 Bciutohtdrol (297), [69], 341, 342 Benzo-.co. ácido Ü2 JÍ. 336 Benzoico, anhídrido {42], 333 Benwína (343), [133], 383 Benzonitrilo (191), 390 Bcnrofenona (305), (48], 387Bcnzopinacol [185], 342Benjoquinooa [115], 388
, o-Bensotoluidida (142), 346 ^•Benzotoluidida [158], 34-7 Benzotricloruro (220), 379 BcnzoUacetona *61], 387o-benjLoilbenzoico, ácido (126], 336o-Bcnsoilbenzoico, ácido, hidrato [90],
335Bcnxoilo, brom uro de (218), 332 ct'Beníoilbuiiríco, ácido [87], 335 Y-benzoilbiUirico, ácido [127], 336 Eenzoílo. cloruro de (197), 332 BenzoUo, fluoruro de (159), 33! a*BenzoSl-(J-feniihidra«na [168], 347 BcjjiOilptpcridina [48}, 3453-Benzoilpropiónico, ácido (116], 336 Biacerilo (83), 335 Bianisídina [135], 354 .K-ft'-Binaftilo (160), 381 . V -H '-B i ia f tn o [188], 381 Bifenilo (70], 381 , : . .Bifenil-4,4#-dis\ilíonaEaida [300J, 401 B¡fcnil-4,4-‘-disulfoniío, cloruró de [203],
405bilemlrnercapiano [ i J l j , 389 p 'f f 'B ii (d imetiiamino) benzofenona
(174], 361 Biuret [192 d.], 347 ( + > Borr.col (208), 342 Barnito, acetato de [29], 367 BomiJo, cforuro de (2 0 7 ), 374 i^Borndeno (1 4 6 ), [113], 384 Bromal (174), 343 Brontación, 293, 320 Bromo en tetradoruro de carbono, 137 Brotno, a su a d e . 159 Bromoaceiamida [91], 345 et-Eromoacctanilida [T31], 346

p -Bromoíc t taniLkL» (16 7j, 347 Bromoacético, ácido *20B), [30], 334,
335Bromoacético, anhidrído (42], 333
Bromo&cetona (136), 385 ¿•Bromoacetofertona (255), [51], 387 to-BronJoacctofenoaa {50), 30? Brotnoaecuío, bromuro de (149), 3 3 1 ’ Bromoacetilo, cloruro d e <127), 331 ¿>-Bromoanitidas, 258 m-BromoantlSna (251), [183, 35í1j 351 o-BjcrooaniJina (250), [31], 351 p-uromoanilina (245 d ,) , [66], 352 o-BromoanisoI (213), 3 ; l ¿-Biomoanisol (223), 371 jrt*Bromobe>iz*3dch5do {236), 343 /•Bramobeazaldehído [57], 344 m-Bron»ob«ti?an¿iia [155J, 347 ú-Brojnobem»mid& [155J, 347 yvBroma b«n wunída [189J, 3$7 ttJ&romobensanílida. [136), 346 o-Bromob«n2anílída [ l4 lj , 346 Bromobenceno (157), 3753-BroflaObencen-J,5-dísulfonzmida,
[255J, 4013-£romobencett-I,5-disulfoni(o, cloruro
de [100], 404rn-Brcmobenccasulíoitainida {(541, 400o-Bromobenccnsvtlfonaimda [18éi}f 403 Íf-Broraobencensulfonaímda [166], 400 ¿'Bromobencenauiibtiamída*, 283 d-Bromobcncensulfonik», cloruro de
[51], 403p - BromobenceittulíOruJo., cloruro de
[75], 403 m-Biomofcenwicíi, ácido [155], 337o-BromobenZ0¡C0> ácido [150], 337 ¿■Eromobenzoico, ácido [251], 338 m-BrOmObenzonitrílo J[33¿ 390o-Bromobcnzanitrilo [53], 390 J-Bromobenzorntrilo [112], 390 rtt-Bromobeiwot!o, bromuro de (245),
332p-Bromobcmoílo, bromuro de [42], 333 nt-Bronoobencilo, bromuro de [41], 370o-BronJobeacálo, bromuro de [30], 379 ¿-Broroobe&ctlo, bromuro de [63J, 379 /►-BromobencHo, cloruro de [50], 379 ^B rom obeodltíouw nio, sales de, 259 • )>-Brom«>bcücil5ulfonajmda [I88J, 4012-Bromobifenílo (297), 3764-BrofHobifcnilo [89], 377
V
4 5 0 ' Indico d ia b é tico
«-Bromo-4'butírico, ácido (217 <L),334
( + )^B rw uO íJcaa ío r {274), [76}, 387 cc-Broírto-n-caprotco, ácido (240), 334 J,3-Bromoclorübenceno (196), 375 ísBromockwobeticeno (195), 375 />-Bromodorobence«o [67], 377 I -Bromo- I-tíoroetano (83), 3781-Bromo-2-cioroetario, (1 0 J), 3782-Brorao-4-<JorofeiK>l [34}, 397 ¿-Bromocumeno (219), 3752-Bromodmcrw (234), 376 í-Bromo-jV^-diecjlatiiltna (270), [34],
3603-Bromo-¿V>^-dimeTÍfanilina (25?) 359 ¿ -B ro n x i-tf jA’-dimetitaoilína (2&4),
[55], 3602-Bromoetanol (150), 339 jS-Bromoetílo, acetato de (163), 365l-Bromo-Z-etilbínceüo ( l . 'J), 375 (-Bromo-4iitílbertCeno (20; , 375 (i-Bromoetílíco, éter (127), 369 [J-BromoetilfeníUco, éter [32], 3721,2-Bromofluorocü3<ao (3 6 ) , 378 BrOmofóilnD (151), 378 Bromohidroquinona [110], 3990-Bromoyodobenceno (257), 376 p-Eromoyodobenceno [92], 377 a-Brorootjobu lírico, ácido (200), [49],
334, 335 p-Broraoisobutírico, ácido [22j, 3351-Bromo-2-iEopropiíbencena <210}, 375 K'Bromoisovateriánieo, ácido (230),
[44], 334f 335 a-Bromoisovaleriaruturea (145), 346 Bromom«átílenD (225), 3762-Bromo*4-metílacfttamlida [117], 346 Bromornetilo, acetato d e (13 0 ), 365 3»BromO-2-metilatulina (2 5 5 ), 350 3‘Bromo-4*mcti!arii1ma (2 5 7 ), 350 a-Bromom&ftaleno (2 7 9 ), 376 (J-Bromonaftaleno [59), 3771-B ronx^-nafalensulíonam ida [193],
4002-Bromo-l-naftaletisülfonarmda [140},
401l-Brom a4'ítafU lem ulÍ0m ]0, cloruro de
[83], 404ra-Bramon¡trobenceno (2 5 7 ), [54], 393o-Broroonitrabenceno (26 1 ), [43], 392 ¿•Bromonitrobenceno (2 5 9 ), [126], 3954-Brom o-l-nkronaítaIeno [05], 3944- Bromo - 8-nítro -1 - naítilam ina [116],
3534»Bromo-3-nitroto!uc'no [33], 392
t n d í t e o l f o b é r i c o 4 S 1
/►-Bromofenacilo, bromuro de fl09l, 387
f-Bromóle nacito, ¿atere* de, 257 O-Brorrtoícneto] Í2 2 4 ), 371 í'B rom oícnctol (2 2 9 ), 37! wi-bromc-fenol (23 6 ), [32], 397 o-Bromoíenol (595), 397 ¿-Bromofeno!. [63], 3980-Bromoíenilhidraatna [106], 3531-Bromopropeno (60 ), 3732-Bromopí,opcno (48 ), 373 a-Bromopropiónico, ácido (205V [241
3S4, 335(Í-Brotnopropiówco, ácido [62], 335 a-Bromopropíonilo, bromuro de (353)
3312-Bromopiridioa (194), 3533-Bromopiridina f 170), 3583-BromcqmnoleÍHa [49], 360 6-BromoquírwTelna (2 78), 359 ^-Bromocatireno (22 1 ), 373 ¿-Brorooliofeno] [75), 375 m-bromotoiueno (18 3 ), 3780-Bromotolucr*o (181), 375 ¿-Bromotolucno (185), [28], 3?5, 376 Bromot r:cloror/ietano (105)* 378 B utanodioM ,3 (208), 340 Bttanodiol-2,3 {183), 3401-Butansuifonamida [45], 4001-Bufcansutfoniío, cloruro de (75/10
' rnm ), 402 Bütea-3-ol-2 (9 6 ) , 339 (3-Butenonitrilo (118), 390 »B utilo , acetato de ( (2 5 ) , 365 ¿«r-Butílo, acetato d e (311), 365 n-butítico, alcohol (1 1 5 ), 339 jíff-Butilico, alcohol (9 9 ) , 339 fer-ButiJíco, alcohol (8 3 ) , 339 n-Buti Camina (7 7 ), 349 «c-B uiilam ina (6 3 ), 349 «r-B utitam ina (4 6 ) , 349 n-Butí?o, ¿-aminobenzoato de [56],
351A^rt-Budlanilirta (236), 356 K-B»ti3benceno ( Í8 2 ) , 380 w -B utilbcnceno (1 7 3 ), 380 íer^Butílbcnccno (1 6 9 ;, 380 n-Butílo, benzoato de (249), 3S6 >t-Butilo, bromuro de (100)» ,374 ííí-B ulilo , brom uro de (90 ), 374 ¿ef-Butito, Bromuro de (12 ), 374 n-Butílo, fl-b'.itírato d e [165], 365 n'Bucüo,carb?m ata de [54], 345 jrí-Butílcarbirval (12 8 ), $39 «-Butifo, carbonato de (207), 3$6 n-Butilo, cloruro de (77 ), 373
i^f-Butilo, clortíro de (67), 373 irfr-Buiilu, d o ia to ¿c [ 5 1 ), 373 n-Butílo, c loroaceuto de (1 7 5 ), 365 n-Buti3o, cloroformialo de (145), 365 a-Buül-a-crcsílico, éter (223), 37 | «'Butilico, éter (140), 370 rt’Butilctitbarbilárico, ácido [125] 346 n-Bulilo, íorm iato do (107), 365 ¿« 'B u tilo , íorm iato de (97) ,365 n*B«ti3y, yoduro de (130), 374 iíí-B utifo , yoduro de (1 (9 ), 374 ftr-fiucilo, yoduro de (9 8 ), 374 *-Butilo, actato <Je (188). 366 73-ButiljiiercaptanO (97), 389 rt*Bu til metílico, í k r (70 ), 369 fj-Bu tjJmctíJcetona (129), 385 ?i‘Bu<iJ-2-naítífico, éter [36], S72 s«-B u til-2-oaítilico, í te r [34], 372 n-B\jtí]o, n itrato d e (136), 36S n-Bvtjlo, nitrito de (75), 368 n-Bufilo, o x ah to de (243), 366 n-Bulílo, o^am alo <tc [88], 345 ¿•ButiJfcr,ol (248), {22], 39? í-íc r-B u tilíen d [95], 398 rt-Butilo, ícntlacclato de (254), 366 n-ButilfonHico, éter (2 Í0 ), 37J «■Butilo, íla lato de (333), 367 «-Butilo, propionato de (144), 365 7?-Bui¡lo, saücilato de (268), 367¿-n-ButUo, tartrato de [22], 367 Butino-2 (2 7 ), 383 rt-Butira?dehído (7 4 ), 343 n-Butiram ida [Í15J, 346 n-Buttranilida [95J, 345 rt-Butirico, ácido (Í6 3 ), 334 n-Butírico, anhídrido (191), 332 Butiroína (190), 340, 386 y-Eutirolactona (2 0 4 ), 366 n-Butironitrílo (118), 390 n-Butírílo, bromuro de (128), 331 n-Butirifo, cloruro de (300), 331 n-Butfrílo, fluoruro de {$?), 331 n-Bulirclo, yoduro de (148), 331
CCafeína [234], 348 Ir-Cánfcna (1 6 0 ), [42], 384 n^Canfórico, ácido (187], 337
anhídrido [221], 333 CanfoquinOna ((98}, 388 ( + )-Canf<ir-3-iulícnamida |(4 3 j, 400 í + }-Canfor-B-au3fonamida 1(37], 400 ( + )-Canfoxvj0-su3fcmamidji (32], 400 í + )'C*;nfor-3-5u3íonílo. cloruro de
[8B], 404

(+)-C aafoiv8*igIfon¡lo , d o ru ro de[138J, 405
í - f ) -Canfor*! O-buIíodüú. cloruro de . [671 403 CapraWeWdo (209), 343 Ciprino, árido (270), £30L 334, 335 n-CaproaMehldo (128), 343 *-Caproamída [101], 346 Ji-Caproico, ¿cído (205), 334 c-Capcoico, anhídrido (2 5 ? ), 332 n-Caprotutrilo (164), 390 ji-CaproSEo, cloruro de (153), 331 CaprilaJdehído (171), 343 C aprilko , ácido (236), 334 Caprilam trilo (200), 390 Carbam ida (132], 346 Caibarülída [238], 348 C a it* io I (3 5 1 ), [243J, 357 Carbohidratos, derivados de , 299
rotación óptica, 364 tabla de , 364
Carbono, tetrabrom uro de [92}, 379 Carbono, tetracloruro de (78 ), 378 Carbowcirifo [200], 399 Carvacrol (237), (0J, 397 C arvons (225), 386 C atecol [104], 398 Catecol, dietUico, éter [43], 372 Celobiosa [225 d.], 364 Celulosa [d-]. 364 Celulosa, acetato de [d.], 364 Cérico, reactivo de nitrato, 140 O d io , acetato de [22J, 367 Ce til o, alcohol [50], 342 CetUo, d o ru ro d e (289 d ,) , 374 CetUmeícapcino (170/3 min)> [50],• 369Ceta 034, derivado* de, 311
tabla de, 383 CSanoacétíco, ácido [66], 335 f^C ianobcnzoíco, id d o (214], 338l-CÍ3J>o-8-nafta3«Jiíulíonamídfi, [336J,
401Cicjo alquilo, derivados de haluros de,
300 tabla de, 373
Ciclodecano <201 >, 382 Cídohept&nona (181), 365 C klohepteno (¡1 5 ) , 333 Ctclobcxano (80 ), 382 CKkthexanboxamida [186], 347 Cídobexancarboxamida [ 186], 347Cidohexan-1 ,2-diCarboxí ¿ico, anhídrido
del ácido (c u ) [32], 332 Ctclohownol (160), [16], 339, 341.
:• 452 V;, Indice o ffab á tto
CícJohexanona (155), 385 Ciciohtfxtuiu (6 4 ) , 383 C iclohoálo, acetato de (175), 365 CidohexiU raina (134], 349 C idoe*ü benceno (237), 381 Cidohcadlo, bromuro de (165), 374 CicTohexilcarbinol (18 2 ), 340 Cidohejrilo, cloruro de (142), 373 C idoexiletíljco, é te r (149), 370 CÜdoh'exilo, forraiato de (162), 365 Ciclohexilmetlüeo, é te r (134), 369 Cidobesdlm ercaptano (159), 389 Ciclohexilo, oxaiatc de [42], 367 o-Cidobexilfenoi [53], 398 ¿*C idob«ilfenoI [132], 399 C idononano (172), 382 Cicloparafinas, derivados de, 308
tab la d e , 382 Cictopetttadicno (4 2 ) , 383 C idopentano (5 0 ), 382 CicJopeotanol (14 0 ), 339 C tdopcntanona (131), 385 C idopenteno (4 6 ) , 383 CíctopentUa, Bromuro de (137), 374 Cidopentiletílico, éter (122), 369 Cidopeütílm etílico, éter (105), 369 w-Ciroeno <175), 380 p-Címeno (1 7 8 ), 380 ^•C im ero (1 7 5 ), 380 C inc y d o ru ro de amonio, 192 L-Cistina [260 d.J, 362 Cineol (17 6 ), 370 CinamaldehM o (252), 343 C inam am ída [142J, 346 Cínam anílida [153], 347 Cinámico, á d d o fl33L 336
anhídrido [130], 333 Cinamonitrilo (25 4 ), 390 Cinamoílo, d o ru ro de [35], 332 CinamOico, alcohol (250), [33], 341 Ciaantilo, cinam ato de [44], 367 C itjacón ito , ácido [91], 335 Citraeónico, anhídrido (213), 332 C itral (223 d .)» 343 Cítrico, ácido [153J, 337 Cítrico, h id rato d e l . á^ido [100], 336 Citroa<la| (2 0 6 ), 343 C itrondol (22 2 ), 340 Claiacn, álcali de , 93 Clasificación, reac tiv o de, 123- Benedict, fo ludón de, 132• benccjjiulfortila d o ru ro de, 135
bromo en tetradoruro de carbono, 137
bromo, agua de, 139
In d ic » a l f a b é t i c o 4 5 3
cérico, nitrato, 140 d n c y doruro de amonio, 192 clorhídrico, ácido-cloruro de cinc,
146cloruros de ad ío , 123 d o ru ro de aluminio, 130 crómico, anhídrido, 1412,4-<ünítroíeníIhid ruana, 142 Fehlin, prueba de; 134 fenilhidrazina, 164 férrico, cloruro de, 143 ferroso, hidróxido, 144 fucsina, reactivo para aldehidos,
.145H insbírg, prueba de, 135 hidroxüam ina clorhidrato de, 150 n fqud , cloruro de, y disulíuro de
carbono, 156 ñique!, do ru ro de, y 5-nitrotalídl-
aldehído, 157 >-nítrofcná]hidrarin&, 164 nitroso, ácido, 158 n itra to de p lata , 169 periódico, ácido, (62 permanganato de potasio, soludón,
166 sodio, 180sodio, bisulfito de, 181 jodio, hidróxido d«, 182 sodio, yoduro de, en acetona, 188 sulfúrico, ácido fum ante, 190 ToUens, reactivo de, 192 yodhídrico, ácido, 146 yodo-hidrósido de sodio, 154 yodofonno, prueba de, 154
Cloral (98 ), 343 Cloial, h idrato de [53], 344 Cloranilo [290], 388 Clorhídrico, ácido-doruro d e Cinc, re
activo, (48 cc-Cloxoaceiamida [119], 346 O-Cloroacctanilida [134], 346o-Cloroacetanilida [88], 345 ^•Cloroacetanilída [179], 34? Cloroacético, ácido (185), [63], 334,
335Cloroacético, anhídrido [45], 3S3 Cloroacetona (119), 385 Cloxoacetonitrilo (127), 390 p-Cloroacetofenona (232), 386 wCloroacetofenona (244), (59T, 387 Cloroacetilo, bromuro de (127), 331 Cloros ce tilo, d o ru ro de (105), 331 5*ClorO'4-arnino-i,2'dicocál benceno,, 140], 351
5’Cloro-2-a*ninot9lueno (241), {29], S50, 351
m-Cloroanilina (230), 3500-Cloroanilina (20 7 ), 349 Í-C íoroanilina (232), [70], 352 fft-Cloroatüial (1 9 t) , 3716-Cloroantsol (19 5 ), 3711-Cloroanixo] {200), 3712-C]oro-6-antraquinonsu|foniio, doruro
d e [202], 4052-Cioro-7'antraquinonsulfonilo, cloruro
de [176?, 405Ooroarilsutfonamidas, 326 CloroariUulfonaniüdas, 326 m-Clorobenzaldebido (2 0 8 ), 34S o-Clorobenzaldehído (20S), 343 ^•Clorobeozaldehklo (2 1 4 ), [47]/ 344 m-CIorobenzamtda [134], 346 c-CIorobennnilída [114], 346
\ í-Ck>robenanilida (1943, 347 Clorobcnceno (1S2), 3753-Clorobenceri'1,5-dÍsulfonamída,
[224], 4013-Clorobencen-l,5<distilfonilci, doruro
de [1061, 404 w*Clorobenccnjulfonamida [H 8], 400 o-Clorobenceusulíonamida [188], 401 ¿■Clorobenccnjulfonamida [144], 400 o-Clorobenccnsulfonilo, doruro de [28],
402¿•Clorohencwwulfomlo, cloruro de {53],
403ni'Clorobcjxwico, á d d o [I58J, 337 o-Clorobcnroico, ácido fl40], 336 ¿•Cloroberreoicc, ácido [242], S3S m-Clorobenzoico, anhídrido [95J, 333o-Ciorobenzoko, anhídrido [79], 333 ^-Ctorobenzoico, anhídrido [194], 333 m-Clorobentonitrilo [41], 390 lí-Clorobcnzonítriio [43], 390 ¿-Clorobensonítrílo [92], 390 p-Clorobenzcfcnona [78], 387 m-CloroberiaoUo, cloruro de • (225),
332o-CIorobenzodo, d o ru ro de (238),
332¿ ‘Clorobenzoíío, d o ru ro de (222),
116], 332m-CIorobencilo, cloruro de (216), 379 o*Clorobcncilo, d o m o de (214), 379 Í-C lorobendlo, cloruro de (214), [29],
374, 379Í-Clorobencildouronio, sales de, 2592-dorobifenilo (34L 3764-Clorobifenilo [77], 377

454 b d k c a f f r f i f t J m
Y-Clorobutíronítrilo (197), 390 ¿■Cloro-c-crwol {43], 3983-C1oro-¿-cre»ol (197), 397 ¿-Clorocumeno (198), 375 ^'Cloro-S^-díaminotolucno [73], 352 6'C]oro-2,4'dibromcanilina [95], 3534-C1oro-3,S-diüromoíenol [[21], 399 ¿■Cloro-tf,//-dipieiilariiJina (230), 355 4-ClOK>*2,5-<iinKti1benccnsulf[>namida
[185], 4004*C(oro*2,5-d)mctilbcncm5ulfomlo,
cloruro do {50], 4032-Cloro-3,5*dinilrobtnj:oicc>J ácido
[199], 338 (J-Cloroctilo, acetato de (145), 365l-Cloro-2-clilbcnccno (176), 375l-Cturo-3-eÜtbcnccDO (184), 375 ^CloroetilbcncenD (162), 375 «t-Ctoroetílico, éter (93), 369 JJ*Cloroetiüco, Éter (107), 369 Ctoroformo (61 ), 373 Clorohidroíjuinorta {106], 390 2 ‘Clon5*5-hcdroxitolucno [66], 398 4»Cior^2'yodofer>ol [73], 3961-Cloro-2-i$oprop'tlbeneeno (191), 375 3^i1 o ro-4-me t oxi J>c ac e n w 1 f a mid a
[131], 4003-Cloro*4-me<owbeni:en5u]foTÚlc> cío*
ruro de [82]). 404Clorometclo, acetato de í 111) j 3652-Cloro-4-mcljlhertcen ju [foruenida
[166], 4014-Ck>ro-2-nvct3lbcnccnsulíor»iaida
[165], 4002-01oro-4-raetifb«ncensu]foii3loj cloruro .1 de [46], 402
' 4-Qoro*2'mctilbcneenSulfonílo, cloruro de [34], 403
Goromeiflico, éter (59) 369 Clorametiletíiico, é te r-(80), 369 2*Cloro-5-metil-3-nitrobeíiceíVUJlfona-
mida [196], 4012*Cloro*5-mctiM-nitrobe»cen2u]fona.-
• ‘ mida [188], 4012-C]orO'&-metÍ1-l ,6-nitrob«ncensulíona-
. mida [177], 4003*Clon>-2-melil'5-nilrobcnc<nsulíona-
m id a [ l6 J l4 0 0 3'Cloro>2-inctÍ1'6*nilTobcn«n9ulfona-
mida [181], 400 iu5-Ckiro-2-melil-3-nitrobcnc<i«ulíona-
m ída [167], 400 . 5«CloíO<2*metil‘(>-nilrobenceniutfona-
mida [183], 400
5-Cíor?-4*metLl-2-nitrobence wu fíbna> mída [161], 400
5*Ctoro-4-metil-3-jútrobencensulfcrc*- mida [162], 400
2*Cloro-5‘met itrobencemulfon í* ít>, cloruro de [63], 403
2-Cloro-S -metil-4-nü robencensulf on i - lo, cloruro de (92), 404
2'C]oro-5-rt*etiJ-6-nit-obeticcr.sulfanik>. cloruro de [122], 404
3-C loro*2-met¿t-5*n i trobcncensulfoTi i*lo, cloruro de [64], 403
3-C]oro-2-mettN6-n3irol.>cnceoM.i1for’.t-lo, cloruro d« (60], 4C'J
5 -Cloro- 2-mo<rt« 3-ni trobcncensuJfcv nilo, cl&turo de [60], 403
5-Cfntu-2»m«úl*6-nurobcnccmulf«>n5l<>% cloruro de (154), 405
5-CJoca-4-meti]-2*n¡trobcr.?cnBulíoniJo, cloruro de {99], 404 .
5-Cloro-4-met»l'3 -miH)b*ncc' «uEíonilo, cloruro de [70], 403
2-Cloro-3'met5)fenol [50], 398 $.CJoro.3<netilfenol [4G], 398 a-Cloronaftaleno (263), 376 P-Ctoroiiaftaleno {56], 377 ] -C l0r0 '4-naital<nsuíl’onam t¿a {165],
4004-C loro-1 -naítal«nsu(ío»£tnida [106],
401fi-Cloro - 1 - naítalensutíonacr.ida {199],
4014*C]oro* l*naíialensuJfont!o, cloruro de
{95], 404B-Cloro-l-jiaftalensulfonilo, cloruro de
[101], 404 4-Cloro-l-naftUaznína [98], 353 4-Cloro-2-nitroaniíol t98], 394 ■ w-Cloronitrobenceno {235), [44], 392 O'Cloronílrobenceno (24 6 ), [32], 392 p-Cloronitrobenceno (242), [83], 394 4-Cloro-2-nitn>beocen$ulíona>™da
[164]; 400 4-Cloro-3-nitrobenzoiro, ácido [182],
3376-Cloro-2'n«trotolueno (2 3 8 ), [37], 392 4'Gloro-2-núrotolueno (240), [38].
392¿-Clorof-nacito, Atures de, 358o-Clorofenctol (2 0 8 ), 371 .¿ -C W c n c to l (212), [21], 371, 372 1 ro-Cloroíenol (2 1 4 ), [33], 397o-Cloroíenol (17 5 ), [?], 397 ¿C loroíenol (217), [43], 397 Cloropicrína (113), 391
CJoroptotm itas, 2$5Cloropreno (5 9 ) , 373 3‘Cloro-1,2-propanodicd (215 <L), 340I-Gloro-2-propanol (127), 339 2'ClOro-l -propanol (132), 3391-Cloro^ropeno (3 6 ) , 373 2 'C loropropeno (3 0 ), 373 di-a-Gloropropíonam ída [60], 345 « 'C loropropiónico, ácido (186), 334 P-CIoroproplótlico, ácido [42], 335 fl-Cloropropionierilo (178), 390 Í-C loropropíoíenona [36], 3872-Cloropiridina (1 6 6 ), 3583-dorop irid ina (14 9 ), 358 2'Cloroquinofcína (2 6 7 ), [38], 3606-Cloroquínoleína (262), [41], 360 pt-ClorocEtireao (19 9 ), 3734-Cloro*2,3,5,6*tctrabromofenol [215],
399p-Clorotiofeno! [53J, 389 4-Clorotimol [59J, S9B m^Clorotolueno (16 2 ), 3?5 ¿«Clorotolucno (1 5 9 ), 375 p-Clorotolueno (162), 3?5 < ~)-C olestcro t [148], 3422,4,6-Colidma (1 7 2 ), 358 Cotor, 37Congelación, puntos de, 46 Coniíerína {165 d.J, 364 Copulaci6n, 158 C reatina [315 d.], 348 C reatin ina [292 d.J, 348, 363 m-Crcsot (20 2 ), [3], 397 «•Cresol (1 9 0 ), [31], 397 /■Cresol (2 0 2 ), [36L 397 2,4‘C reaótko, ácido [178], 337 m -C r« ito , acetato de (212), 366 ó'CresUo, acetato d e (208), 366
Cresílo, ace ta to de (212, 366 m-Crcsilo, benzoato. de [54], 367 «•Creailo, benzoato d e (30 7 ), 367 ¿-Cresito, benzoato de [71], 366 m -Creúlo, carbonato de [115], 368 ^-Creólo, carbonato de [60], 367 «5-Crcsi)eií(ko, é te r (192), 371 o-Crtsileiílíco, é te r (192), 371 /'C r& siletilko, éter (192), 371 m-Cresiimctílico, ¿ccr (177) 371o-Crcsilmi» tífico, é te r (171), 3/1 ¿-Cresílmetílico, éter (1 7 6 ), 371 Criseno [251], 381 C rotonaldehído (1 0 3 ), 343 • Crotónico, ácido (cis) (165 d.), 334 C íoténico, ácido (irans) [72], 335
. Crotóníco, anhídrido (2 4 8 ), 332
Crotonüo, cloruro de (126). 331 Curoeno (15 3 ), 380o-Cumárieo, ácido [207], 33$ ¿-C um irico , ácido [206], 338 Cum artna [67], 367
CHChalm ougram ída {106J, 346 ¿Chalm ougramlida [89], 345 Chalmo(«jrico, ácido [63], 335
D
DecaJina ( l is ) , (194), 382 Decalina ( tra ía ) , (165), 382 n-Decano (1 7 3 ), 332 Decanol-2 (2 1 1), 340 Dccanonitrilo (245)j 390 Dcce«v>I (1 7 1 ), 384 n-Decílico, alcohol (231), 34 J. a-Decilo, cloruro de (223), 374 Densidad especifica 55 Densitómetro, 55Derivados, preparación de, 249 pro
piedades de, 21 Dcíoxibcnzoina (320), [60], 387 Despt&iaxnientoj químico», 218
tabla de, 219A'/Z^Diacetil-rM-íenilendiamína [191],
347A">A"-Di»cet¡l'<>-íenílendiamina [185],
347 ^V '-D iacetil./'fen ilend iam ina. [300],
348D iacetona alcohol (164)t 365 Dialilo (5 9 ) , 383 Dialílam ina (11 i ) , 355 DiaiilbaLrbkúrico, ácido [172], 347 2,4‘DLuuinobenceno-1,5-diaulfotiaaiida
{137], 401 _ •2.4-Diaininobonceno-l,5-dUuJíoni{o,
cloruro d e {275L 4052,4 '-D iam bobifenilo (363), [45L 351 1,4'D iam inobutano (160), [27], 349,
3512.4-Diaroinociorobenc<no [88J, 352 p^D iam inodííenÜ naetano [94], 353 l^D iam in o h ex an o [42], 351 l^D iam in o n a fta ten o [95J, 3531.4-D iam ínonaíuleno [120], 3531,8-D iaminoaaftaleno [67], 352 2,7~Diam)nonaftaleno (166), 354l,5 .D ip^iinopentano (178), 349 2,4‘Diaminofenol [79 d.], 3521,2-Dianúi'Jopropano (120), 349
M t t «ftUM tfc» 45S

4 5 6 ■ IndU o a ífa b S lic o
Ly3 DLurinopropiíyi ( 136), 349 4,4'<-D¡axmiiocítilbeno [226), 354 2*3*Di&imnotdueDO (225), [61], 352 2,4~Diumnotolueao (2 8 0 ), [99J, 353 2^D ianUnotolueno [64], 3522,6-Diaminotdueno [105], 3533.4-Dianúnotolueno (2 6 5 ), [89]» 352 P i-E -a m f la m in a (2 0 5 ) , 355Di-n-Amilcetona. (22 6 ), 326 DxazO&ción, 158 Dibcoza lace ion» ¿112], 363 Dibenzoíurtno [8?], 372 Diben^oíh’CieuiK» [78], 38?D ibendlo [52], 381 DibíTWÍlacétko. ácido [89], 335 D ibeiidlannna <300), 356 N ^ -D ílx a á la n ilm (300 d .) , [70],
360D ibenálcetom {330), [34], 33?D ib romo ace am id a [156)), 347 Dibromo&céúco, ¿cido (234), [46],
334, 3353.5-Dibrom o^-amino tolue no [78], 352 2,4'Dibromoanilina [79], 352 2,6'Díbrornoanílina [83], 352 re-Dibromobenccno (2 1 9 ), 375 fr-Díbromobenceno (22 4 ), 376p-Dibroraobcnceno [89J, 3772.4-DibromobenccnsuJfonamida [ f $0],
• 4012,5‘Díbromobcncensuííonamida [194],
4013,4JDibromobenCcrwuUonaiTjida [175],
4002l4'Dibromob«r»ceri3,nlíotiüoJ cloniro de
[79], 4032.5-Dibromot>enc«n*ulíomlo, cloruro de
(71). 4033.4-Dibron»obcnceri*ulíonjlo, cloruro de
[34], 4022,2'.Difaroroob¡íen¡lo [61], S77 4,4’*Dibroc]ob5fcni!o [169], 3771.2-Dibromobutano (1 6 5 ), 379 1,3'Dibromobutano (174), 379 1,4'lKbroraobuUno (1 9 8 ), 3792.3-D¡bromobu<ímo ( I5 « ) , 378 |,3-D$bro!Ho.2'buUmo (1 6 9 ), 3793.5-DíbroiaO'O-cresd [57], 3983.5-DibromQ-/)-c.'e$cl [49], J96 2,2'Dibroroíxtatwl (1 8 1 ), 340 Dibromometdíeo, éter (150), 3701,2-DibroinoiiaíCaleño [67}, 3771.4-Dibromonaftaleno £82], 3772.5-Dfbrc3inonicrQb<nceao [65], 3941.6-Dibrooctano (2 7 2 ), 3792,4-Díbjrcniofenct (2 3 8 ), [36], 397
1,3-DiLromo-2-proj^anol (219 d .) , 340 2 ,3 'D ibconw i-p ropand (219), 3401.2-Dibroinopropeno (132), 3731.3-Dibromopiopcno (156), 378 2,$-Dibremoprop«no (140), 378 a-P'JDibromopLropWnico, ácido [64], 335 3 ,5-D ibron»piridina (2 2 2 ), [112], 361 ¿¿-Dibromosucdmco, ád d o [167], 3373.4-DibromótolúeiK> (2 4 1 ), 376 Di-n-butoxietano (18 6 ), 330 Di-n-butoximetano (16 1 ), 330 DÍ'í <n;-bu toxieumo (1 7 1 ), 330 Di-n-butilaraina (1 6 0 ), 355 ^ ^ ’-Di'fl'butOanílixLa. (261), 359 Di-n-butilcetona (16 7 ), 385 D í-w -butilcetona (16 2 ), 365 a>a-Dicl0f0cetanúda [96], 346 a,a-D iclorocettnilida [116*, 346 Dicloroac Ético, ácido (189), 334 Didoroacético, anhídrido (216 d .),
332d,tt-D ídóroacctona (1 2 0 ), 385 aV -D iclcroacetottt (1 7 3 ), [45], 387 DidoroacetÜo, cloruro de (107), 3332.4-dicloroani(ina (2 4 5 ), [63], 3522.5-Dicloiwinílina (2 5 ! ) , [50?, 351 ¿¿/-D ídoroaaoxibericeno [154], 3962.4-Dídoroben2alaJdchIdo [737, 34-12.5-DidorobcnKimida [155], 3472.6-Didoiobenzamida [202], 343 m-Dide>robenccno (172), 375 o-Dídorobenceno (179), 375$.-E>idoro¡t*nceno p j ] , 3762.4-Didorobencen-1¿j-diíutfonamida
[276], 4012.4-Didorobenccn-l,5-disulfonilo, do-
n iro de [123], 4042,5J5iciorob«ncen-I,3-dísulfonjlo, do-
. Juro de [114], 404 2 J4 ‘ D3clorobcncensulfonam3da [130],
4002.5-Diclorcbcncen¿uUonara¡da [181],
4003.4-Dic|orobexlccnsulfonamída. [1351,
4002.4-Didorobenccrtiutíorúio, doruro de
[53], 4032.5-DicJorobcncenwlfonilo, doruro de
[33], 4023.4-Díclorobencen5ulfonüo, doruro de
; [I9J, 4022,3'’Dtcforober)zoico, ácido [164J, 3372.4-Dklorobenroico, io d o [160], 3372.5-Didorobcnzoico, ácido [153), 3372.6-Dícbrobenzoico, ácido [139], 336
4,4'-Dic!on>benzoTerion* la48]» 386 ' 2,2-D idorobifenilo [60], 377
4,4'-Did<ji\ibtfertiIo [149], 3773.2-Dickirobutano (124), 3782.3-Diclorobutano (U6), 378 3,4~Didoro*2,5-dinteá]bencensulfona-
m ida [201], 40 1 •3.6-Dicloro-2,5-diinctnbéncéíUulíona-
m ida [165], 4004.6-D¡doro-2,5-Dinjt: tilbíDcenaulfona-
irjida [150], 4003.4-Didofo-2,5-dimcülbcnccn3ulfonílo,
d o ru ro de [62], 403SJ6-DiclorO'2,5-djmctilbcttcensulfonila,
cloruro d e [71], 4034.6-Didoro-2j5-diractilbcncen5u)font]o,
d o ru ro d e [3 !] , 404D td o ro c t i le n o ( c ú ) , (4 0 ) .. 370 D idO rO C tileno ( t rans ) , ( 6 0 ) , 3 ? 6 a t^ -D id o ro e .t í t jc o , é te r (1 1 6 ) , 36 9 a ,6 'D id o « * t i l i c o ( 1 4 0 ) , 370 kfJ '-D icIoroetSU co , ¿ te r ( 1 7 9 ) , 3 7 02.3-Didoro-4-roeti3benccnsulíonamida
[237], 4012.3-DidQro-5*metilbenceniu)fonaraida.
[150], 4002.3-Díc1orO'6-mctílbertcetisu{íonarriida
[106], 4012.4-Didoro-S-metilbcnccnau] fonaroid a
[203], 4012.4-D¡doro-5-metilbenccniulforiaLraida
[176], 4002.4-D¡dorO'6-íneti3bcnGcn5.ulfonaimda
[168J, 4002,6-P¡dwo*3-nictílb*«censu3ronamida
[163], 4012,tí-DidoJ,o-4-njetílbenccnruífonaimda
[155], 4003.4-DidorO'2-meÚlbínCcnsu]fonatnida
[226], 4013.5-Dídoro-2-m£;tilbeiJCeai!:ulíonara{da
[186], 40 14,5“DidorO'3-medJ b*rjcensu]forui»¡da
[166], 401 2J3-D¡cloco*4-mctilt'ertCensu]fonHo, do-
íu ro de [41], 4Ú2 2,3*Didoro-5*nn:tilbc:ncensulíon)lo, clo
ru ro ¿ e [64], 403 2*3-Didoro.6-mctilbct\cen¿d foniío, d u
na ro de [49], 4032,4-JDidoto-S-meCÍ]l>encen3uJforiilo, clo
ru ro de [59], 403 2»4-D¡doro-5-melilbcncensu!fon5]o, cfo-
ru ro de [72], 403
Indica alfabético 457■ •
2.4-üídorO‘6-inetilbcncensulíoníIc.J do- ruTO da [43], 402
2t6-D id0r0‘-3-metilbcncenxulÍ0nilo, do- ruro de [19], 402
2,6-Dicloro-4-nveti¡b«ncen3ulfoni3i>, cloruro de [56], 403
3.4-DidorO'2-me61bcncenju!íonilo, cloruro de [52), 403
3,5 -D id oro-2'meúlljencensulíoniki, d o ruro de [54], 403
4J5-Didoro-3-ractitbencensu3fonüo, cloruro de [83], 404
tt.a'-Didoronvstiüco, «ter (105), 369 a.p 'D idoronaítaJeno [36], 3762.4-Djdoronitrobenceno (258), [33K
3922.5-Djclororiilrofc<:nccno [54], 3932.3-DicJorofenol [57], 3952.4-Didoroíeiio! (20 9 ), [43], 397, 398
••2,5-Didorofend [59], 398 ■^,5-Didoroíeiid [683, 3932.2-Dicloropropano (70 ), 37$2.3-DidOrO-l-propanol (182)^ 340 l,$ 'D idoro-2-propand (176), 340 Y,Y’-Dic?oropropWico, éter (215), 3702.4-Didorotolueno (19 5 ), 3752.6-Didorotoliitfio (199), 3752.5-Djclorg-^-xiJeno f66j, 377 D iríanodiam ida [207], 348 C ietalonam ina (266), [23], 3561,1 -Die lo>d'2-bromostaño (! 70), 3301.1-Dietoxibutano (14 3 ), 330 l,l*Dietoxi«2-doroetano (157), 3301.1-DíetDjri*2,2-didoroetajio (184), 3301.1-D ktoáetano (102), 330 DietOXÍmeÉano (3 9 ), 330 l,l^DietOxjpropano H 2 4 ), 3301.1-Díctoxi*2'Pr0pena (126), 330 a,a-Díetoxitolueno (222), 330 Dieülacético, ácido (19 3 ), 334 D iedlam ina (55 ), 355 (í-Dieülamjnoc{£Líco, alcohol ( 1 6 1 ) ,
356jVJV-DjeütarijJitia (2 Í8 ) , 356 Dietílbarbilúrico, ácido [188), 347 rrt-D3eli (benceno (162), 360 o-Dietilbenccfto (184), 380 ¿ ‘Dictilbcneerio (184), 360 a.p-Díetilcarfcanílida [79], 345 D ietilcarb índ (116), 339 A, -I> ieti]-2J4-dinitroaniUna [00], 360 Dietilengticol (245), 341 Dietcleriglieol, cter diettlico (183), 370 Diílílenglicol, éter dtmetílico (162),
370

Indice alfabético
Dktfteúglicol, é ter m or.o-n-butilico, (2 3 1 ), 341, 370
DietilengUcoJj acetato del ¿ te r mano- «•butílico {246), 366
Dícütcoflicol, éter monoetiKco (202), 340, 370
Dietilenglicol, soitato del éter mono- etílico d d (218), 366
Dietilenglicol, é ter monanietllico {153), 340, 370
ALV'DictíHorrqanúdA (176), 345 Dicttlcctona (102), 305 Dx£XÍlinatorLaniid3 (m ono), [146), 346 Dietilmalcnamida {di), [2241, 348 DielUmatómco, ácido [125], 336 AV^-Díetil-iw-toluidina {231), $59 W ^JD jetil^'toluidtiiA (206), 356 Nftf-D ietil-í'toluidjúa (229), 359 DiKnicp, ácido [228], 338 tfpY'DifemlceUfiaida [101], 346 DifctúlacítanudA [166], 347 DiftníJcctamlida [180J, 347 Difeniiw,ético, ácido {145], 337 Diíeailacetileno (298), 384 D ifenilsuaka (302), [54], 356 m-Díícnilbcnceno (65], 3611.4-D'jfcnUbutadieno (cis), [70], 334 l,4*Diícj)31butadieno (Ir&m), (350),
(1481, 364 Difenilcarbamida, cloruro de [05], 333 M 'D ifcniIctanu {270), 381 1,2'Difenilctanol [67], 3421.1-Difewietüetw {277), {8j, 384 tf^ 'D ifcm tíerrnan 'üda ¿73], 34-5 DKenilgu&mdina [147], 347 <n-Dií«v3ihidraann ¿34], 351 Difetulrnetano [26}, 381 Difenilmetan -4 ,4 '- dí sulfonílo, ciar uro
de (1241, 404iv-Difenflufea [189], 347 Difem lart* [238], 348 Dífeniluíctanos, 268Difluoroacelsmida [52], 345 m-Ditfuorobenceno [62], 377&-Difluorobcn,'£no {92], 3?? />-Dj£luo*ot>enceiio [68], 3?7 Dihexilcetona [33], 3871.2-Díhidrobenccno (80), 383 Dihidronaítaleno (212), [J.o], 3tt4 Dihidropirano (86), 369 Dihidroxiacetona {72], 3672.4-Dihidrüixiacctoíenona [147], 38Í53.5-DihÍiiroxtbeníaldchído [157], 23 4-,
344
2J2’'DiH3drojáfeifcnüo [110], 399 ^f'-D ü tid row liiíenyo [272], 399 Dí-(24udcox3i;ij])-amiiia (268), [28],.
3561.2-Dihjdfoxiñahaler.a [108J, 398 Í^D ihidrcoónaftaleno {176J, 399 1,5-Dihidroxtnaftateno [256], 399 1,8-Díhidroxkiaítaleno ¿140},. 399 2,7-Dlhidroxinaftalcno [186], 399 L-DihidroxiíemlaJanJna (280 d.J, 363 Diísoamílo (158). 382 D iiroaraitanutu 187>, 355 Diisobutoxietñrto (176), 330 Diieobtitilaroína (139), 355 Diisobutilcaíbiaol (175), 340 Diisobutileno (101), 383 O»3obuliíceton3 (1 6 8 ), 365 l,l*DiÍ30propoxie»no (122), 3SO Düsopropilaaüna (86 )> 355 fn-Diisopropilbcnceno (203), J30 ■o-Düaoprcpilbcnceno (204), 3 •P-DSiaopropilWnceno (210), 330 Diisopropiicetona (125), 3852.3-Dimetoxibenzaldehído [54], 3442.4-D5mctoxibenialdehído [69], 3442.4-DÍ»rietoxibcnci:niulfoiiaínjda [16?J,
4002.4-DjrtietoxJbenccnsulfonilo, cloruro de
[?1J, 4031.1-Dimetoxtetano (64 ), 330 DimctoxJinetano (4 5 ) , 3302,tt-D5metoxitclueno (199V, 330 Diraetilacctal (6 4 ) , 3302.4-Dímetilacetarviiída {133}, 346 DimetiJamina (7 ) , 355 ¿-Djitw:ütanúr.oa20bciiceno (117), 361,
396i?-E}jmcriUimnobcri«ild«hído [73], 360 (l-Djmctítaminoctílico, alcohol (133),
358DimctíJanilina (1 9 3 ), 35B2.3-Ditnetilalaaina (2 2 2 ), 350 3,4<Din]etíIbcnzaaitda (1 3 0 ), 3461.2-DimetJtf>cncen*3,5Klts«]£onamida
[239], 4012.4-DÍmeiilbcncisjv 1 jS-disuIíonamtda
{249], 4012,5 •Diroetilbenceci' 1,3-d jaulfonamida
[295J, 4012.5-Dímctílbenc«ii-l,4-disulfonfcjnida
. [310], 401I ;2-DimetUbencfiji-3,5-<íisu]fon¡lo, cío*
rtico d e [79], 403 2,4 'D íntctilbenccn'l ,5 'diw tíonílo , clo
ruro d e [129], 404
Indica alfetbélfeo 459
2.5-D¡metilbenceii*l,3-duu]fonílo> etc. míe* de [81], 404
2,5'DiJBctiIbcncen’l,4-<Jixul/oriilo, cloru ro de [164], 405
2.3-Di.metilbcricenjulfonamida {I67L 400
2.4-D jm ctilbcncem utfonwnida {{37],400
2,5'I>íin,ct[ibenceniuIfoaamída [148],400
2.6-Dimeblbtncensulíonamida[961,4003.4-DirrjitilbencenBulfonamida [144],
4003 . 5-DimcUlbencenjulfonamida [135],
400 _2.3-Dimctilbenawiaulíonilo, cloruro de
[47], 4022.4-‘DLmetílbenceaíuIíani]o, clom to de
[34], 4022.5-DmKtilbenceittulfan:k>, cloruro de
[25], 4022.6-Dimetilbenccnj:ulfowíloJ cloruro de
[39], 4023,4‘DimeíilbenceruuIfonJÍci, cloruro de
[52], 4033 ^ ‘Ditnctilbtncenasjlíonílo, cloruro ce
[94], 4042,4'Dirflct33benzoíco, ácido [127], 3362,5-Djmfttílbeftza'jco, ácido [132], 3363.4-Dimetilbertzoico, ácido [164], 337 .VpV-Dimetiíbcncííarrina (185), 358fr , AT-Bíroet i l - p - bromoanilina ( 26 4),
3602.2-D imetiibuianoico, ácido (187), 344 cc,^-DimetilcarbanÜída [121], 346 ttjA'-fcjmctif-G-cloroaoíIina {207), 353 WA’-Diroetil-¿‘cloroan5lina (230), 359 IjI'D iiriétileiclohexano (120), 382 l,4*D5jnc(j7ciclohexano ( ira n i) J (119),
382l,4.D¡mcti!cicloh*xano (c u ) , (124),
382l , l ‘-Dimeíilciclop«itano (88 ), 3821.2-DiinctiteicIopentano (tran t), (92),
332I,2'Din)etiIcicIopentano (cis), (100),
3825,5'DimeliIdihidrorejnretnol [149J ,388 jV^/-Diinetií-2,4'dinífroaniJina[87Í,‘3602 .4 - Dtm e til—1 ,9 - d irntrobenceno [93],
394A ^-D úifctllfom 'iam ida [153], 345 2,5'DÍm etilfurana (9 4 ) , 369 Dimetiimalóníco, ácido {193], 338 AV^-DimetÜmesidína (213), 3561-,4'DinictjIftaftalcno (263), 381
2^-D laK íitrjifu Icno {104], 3812.6-]Din»e til-6-na í talo as iil fottíuxJda
[207], 401/^ 'I> ím ctilH 2 'r» íü lan u ria (27 2 ), 35$ A 'JV -D W tü-fkiaftílam iíja (3 0 5 ), [47],
359, 3602.3-D>mt:tilrtitrobectceno (2 5 0 ), 3912.4-DxmetiliutrobencenO (2 3 8 ), 391 2^-Dim etilnitrobenceno (2 3 4 ), 3912.6-Dimetílnilirobenceno (225), 391 3,4'Dimctílnitrobenccuo (256), [29],
3923.5-Dimotifnitrobenceno (2 7 3 ), [75],
3932,5 •Dímctil-3-ni trobencetuulfonamida
(173], 400 2 ¿ ‘Dime'.ttl-4'njtrobcncensul£onaniida
{198], 401 2,5“Dir/y:til-^'ry trabe occatulfortanúda
[192], 401 2^-D¡meül-3-nitrobericervíulfoniJo, clo
ru ro de (61], 4032.5-D ¡m ei)M 'nitto1b<TJ«rt5uIíbmfo,
cloruro de [751, 4032,5‘Dimet¡l-6-níLrobencen$ulfonilo, cío-
ry ro de fllO ], 404 2J3-D:meliIíeQ3l [75], 396 •2.4-Di»YJctil(cnol (212), {27], 397. 2^-Dirneiitfeool [74], 3962.6-Diroetilícnol [49], 396 S^D im etH fenoI [63], 3983.5-Dii«otífíenol [68], 398 A^-V-Dimctífpaeudocumidina ( 2 2 0 ) ,
3582,3*D j^t]Ipíridijxa (164), 358 2,5'DiiTietílpirídína (160), 356 2,5‘DimetUptñdina (143), 3582.4-Dimetilquírtoleína (264), 359 2,6>D¡metilquinolclna (265), [60], 360 i^ -D im edl-M '-to lu id ina (21 2 ), 358 /^^-Dim etil-«>toluidína (1 8 5 ), 358 ¿V,?/-DimetU-/)-toluidina ( 210 ) , 350 ArA >-DliT]Btíl-2>4,6- fc-íbromoamlína
(30 1 ), 359 jim-Dimelílurea [106), 346 iMi'm'Dicnetííurra [182], 347O i-a-iufto l [300], 399 líi-p-naftol [2 [8], 3992.4-Diriitroanilina [180], 354 2,6*Diri5troartilina [136], 3542.4-DinitroaniioJ {95], 372 3^-DÍnitrí>anijol [106], 3724,4'-D¡nitcot*_^'ábenceno [200], 396 2,4‘Diaitrobcn?am ida {203], 3483.5-D im trolxnzaiiúda [183], 347

-D initrolxjraniiida [234], 348 'ra-DJnitrobeaccno (302 }, [9GJ, 39* ¿-Dinítrobcaceno (31$), [118], 395 ¿-Dinitrobenceno (299). [172], 395 S^Dinitrobenzoatos, 264, 269, 2972.4-Dmítrobcozoícu, ácido Í183J, 337 S^DinitTobenKHCO, ácido [202], 3385.5-Dirtitrobenioilo, bromuro de £50],
3332.4-Dinitiobeinoílo, cloruro d e (46],
3333.5-Dinítrobetuoílo, cloruro de [74J,
3332,2''DÍB3trobifeflilo [128], 395 2,3'Dinhrobííenüo [110], 393 2>4'-DmitrtWfeBÍ(o [93], 394 S^'-Dimirobífcnílo [200], 3954,4,-Dinitrobifenilo [237], 3952.4-DÍQÍtfobronvoí;cticct»o [72], 3932.4-Dinityoclorobenteno (315 d .) , [52],
S93Dinitrom csidini [194], 3542.4-DinitrorDesitilemí [85], 3941.5-DinUrorut/talcno [214], 395 t,8*Dimtroriafta]eno 1170], 3952.4-Dinitronaítol (138], 399 {.S-Dinitro^-naftilamina [191], 354 2,4‘Diniuofenol [114], 399 p.p'-Dini c r o f eüííko, é t e r [143], 3952.4-DinitrofeniJhcdrazina (198 d.], 354
derivado* con, 275¿tactivo de clasificación, 142
2,4'Dinitrofcmlhiiárazona, 2752.4-DmitroresortinoI [148), 399 SjS-DimtrottKcíIico, ácido [173], 337 2,4*Dinilroto1umo [70], 3932.6-Dimtrotolueno [66], 393 '3.5-Dimtrocolueno (92], 394 2^'D ínitro-te-xileno [10IJ 3952.4-Dimtró-m‘XÜeuo [84], 3944.5-Dinitro-m'Xilcno (132], 395 • 3,4<&mitro-o-iá)eiia (82L 394 SjSJKnítro-o-jrileno [76J, 393 . 3,&'Din¡tro-o--KÜeno [90], 394 4i5-DIttitro-^‘>3]eno [118], 395 2^'D¡nitro-¿->áIeno [93], 394 2,5‘Dinítro'P-xileno [i 47], 395 2,6*Dioitro*p-xyeno [123], 395 Dioxano (102) ,'3 6 91,3-Dioxano (105), 330 Dipenteno <181), 384 }rUDl-^-pcntozietazio (22 2 ), 330 lyl-Di-rt-pentaiiinet&fio (219), 330 Dipjiona (345), 385 Dl-a-jwopcxiítano (148), 330
Ind ice a lfa b é t ic o
Dí-fl-propoxinietaiw (140), 330 Di-R-propilamina ( i 10), 355 jV^-Di-n-propilanilina (245), 359 Di-n-propilcarbiiiol (156), 339 D j'ft-propücctona (1 4 5 ), 385 «.a'-D ipIridilo [69], 360 Dispersión, 66Dispersión óp tica .ro tato ria , 71 D e^taíaaúento,'' teoría de reaccione!
de, 170Di'^-toíílam jna (330), [79], 357 D i-M olilcetona (334), (92], 387 j-Di-m-tolilurea [218], 348 í'D í-o-tolilurea [250], 348 /'Di-p-to5üurea [268], 348 tt-Dodecano (2 1 5 ), 382 l.D odcceno (2 1 3 ), 384 Ductaux, constantes de, 262 Duldcol [189], 342 Durcno (79 ), 3812.4-Dryodoanilina [96], 353 írt-Diyodobenceno (40], 376 ¿-Diyodobenceno [129], 3772.4-Diyodoíeno) [72?, 398
E
Ebullición, puntos de, 46 efectos de la presión sobre, 49
Elaíqioo, ácido [51], 335 Elementos, p ru e b a p a ra , 76 Enantronitrilo (1 8 3 ), 390 EpicJorbidrína (117), 3691,2-Epo.xibutano (62), 369 Ergosterol [165J, 342 Ericritol (72], 342 Ejpectrotcópkoj, método». 197
Infrarrojo, 192 resonancia m agnética de proco»».
' 214ultravioleta, 227
Esteres, derivados de, 230 inorgánicos, tabla de, 363 Labia de, 365
Estcaraldehido [38], 344 Estearam ida [109], 345 Estcaranilida (97], 345 Esteárico, ácido £69}, 335 Esteárico, anhídrido [70], 333 Eltearilo, cloruro de (22], 332
• Estiibeno (3 0 6 ), [125], 384■ ' Estireno (1 4 6 ), 383
Eídfnico, ácido (180), 399 Eteres aiifátícos, derivadas de, 296 •
tabla de, 369 & '
b i t f i c t o f f ü h é t k o 461
aromáticos, brominación de, 298 derivados de, 297 tabla d -, 371
1.2-Etanditio (147), 3891.2-Etandisulíonjlo, cloruro de (91],
404Etansulfonilo, cloruro de 70/20 m m ),
402Etoxiacctaixiida (32], 345 Etoxiacétaco, ácido (206), 334 rn-etoxibenzanúda [139], 346 p-Etoxiberusaraida (202], 343 ¿-Etoxibenzanilida (170], 347o-Etoxifenol (217), (28], 397 AMEtilacctariilida (54], 345 Etilo» acetato de (77 ), 365 Etilo, acetaacetauj de (131), 365
solubilidad de, 96 Etilo, acetotvíicarborilato de (250),
366Etilo, acril&io de (101), 365 Etilo, adipato de (245), 366 E lilal (39 ), 330 Etílico, alcohol (73), 339 EtiJam ma (19), 345 EtiJo-ro-ainiaobcnzoato de (294 5, 350 Etilo, p-ajniaobetuoato de [89], 352 4-(Ar‘Etiíaraino) -3 -n itro tdueno (59J.
356ra-EtilaoJlina (215), 350 //-E tífanilina (205), 355 O'Etilanilína (216}, 350 ¿E tila n iiin a (216), 350 Etilo, amanto de (269), (7], 367 Etilo, antranilato de (265 d ,), [13],
350, 351 Etilo, azclato de (291), 367 Etílbenceno (135), 380 4'Etübcncen-l,3-disuHonam ída [190],
4012-Etilbcncenawjfonamida [100], 400 3'Etilbencensuiforjamida (86], 400 4-Etilbcncensulfonamída [109J, 400 Etilo, benzoato de (213), 366 Etilo, ben2cúlac*tato de (270 d .)j 367 Etilo, bcnaoilforrntato de (257), $66 AT-Etiibencílaniina (199), 355 Ar,#-E ttlbencilanilína [34], 360 EUlbencilico, éter (139), 370 Etilo, bencílmaíonato do (300), 367 Etilo, brom uro de (38), 374 Etilo, bromoacetato de (159), 365 Etifo, brom onialoiuto de (235), 366 E tilo, ct-bronjopropionato do (162),
365
Etilo, 0-hromopropÍDuato do (179), 565
2-Etil-l -butanol (149), 339 Etil-a-butílico, éter (9 2 ), 369 Etilo, n^bucilmalonato de (240), 366 a-Etíl>n-bütiraldehído ( 116 ) , 343 E tib , n*butirato de (120), 365 a-Etíl-n-bucíríco, ácido (197), 334 Etilo, captaio de (245), 366 <t-Etil-n-caproaldehído (163), 343 Etilo, n-caproato de (166), 365 a-Etilo-n-caproico, ácido (224), 334 Etilo, caprilato de (206), 366 Etilo, carbam ato de (49J, 345 Etilo, carbonato de (126), 365 Etilo, cloruro de (12 ), 373 Etilo, eloroacetato de (145), 365 Etilo, clotoformiato de (93 ), $65
t Etilo, íwJoropropionato de (146), 365 '%v-ti(o, cinamato de (271), $67 Etilo, citrato de (294), 367 Etilo, crotonato de (138)j 365 Etilo, cianoacetato de (207), 390 Etilciclohexano (132), 382 2-Etílcictohexilo, bromuro de (2 t2 ) ,
. 374 . ¿Etilciclopentano (104), 332 Etilo, dicloroacetato de (158), 365 .V*Eti(-2,4HdÍ75ÍerG3úilina [113], 357 Etilo, SjS'dínitiobenzoato de [94], 368 Etilcno, bromuro de (ISO ), 378 Etilenbromhidrina (150), 339Etíleno, d o m o de (83), 378 Etilcndorhídrina (129), 339 Etilendiamina (116), 349 EtilengUcol (197), 340 Etilen^liccl, diacetato deí (190), $66 Etilenglicol, dibenzoato d e l [7$], 368 Etilcji^licol, éter diclOico del (121),
369Etilenglicol, éter dimetílico de! (85),
369Etilenglicol, dipropionato del ' (212),
366Etilenglicol, í te r moúo-«-butílico del
(171), 339, 370 Etilenglicol, éter monoetílico del (133),
339. $69Etilenglicol, acetato del éte? monOetí'
lico del (156), 365 Etilenglicol, éter monomeitlico (125),
339Etilenglicol, acetato d d éter monome-
tilico dcJ (144). 365 Etilenglicol, éier maooíenüico ■ del
(2 4 5 ), 341, 370

1
4 é l Ind ice o í f a W t lr o
Etíleno, óxido de (11)» 369 Etíleno, yoduro de [62], 37?Etílico, écer *(35), 369 Etilo, etoxiacettco d e (152), 365 Etilo, etílacetoacetato de (198), 366 Etilo, íormiato de {54), 365 Etilo, fum arato d e (215), 3S6 Etilo, (u n rfo de [33]. 367 Etilo,jgfutarato de (234), 366 E tilpfslicolato de (ISO ), 365 Efño¡ n-heptarvoato de (189), 366
'Etilhexano (119), 382 2‘Etil-l-hcxanol (18 4 ), 3402-EtiM-hexilo, acetato de (199), 366 Etíl»«-hexilb*ibitúríco, ácido [ i26],
346Etilo, hípucato de (60], 345 Etilo, m-h*e(ro»beiiíoato de (282),
[72], 368Etilo, ¿-hidroxibenroato d e [116], 368 Etilídeno, bromuro de (112), 3?8 EtiÜdeivo, cloruró d e (60), 378 Etil isoamil bwbitúriro, Ácido [*54],
347Etilo, iiobutii&io de (110), 365 Eli) úopropi] harbhiríco . ácido 120*3»
348Etilo, isovalerato de (134), 365 Etilo, l&ctato de (15 2 ), 365 Etilo, U uraco de (269). 36 “Etilo, levulinato de (20 5 ), 366 Etilo, maleato de (225), 366 'Etilo, maJoDato d e (198), 366 Etil xnalónico, ácido [ l i l i , 336 Etilo, mandelato de £37), 367 Etil mercaptano (36 ), 389 Etil metílacétioo, ácido (17 6 ). 334 Etilo, metilacetctacetaco de <187)>
366í/.EtU.//-iTjetílw5Ílitta (201), 35B Etilmc tilico, éter (10), 369 Etil tnetíJ tetona (8 0 ), 385 Etilo, metil m^lonato de (196), 366 3'£tif-4-met3pirid¡na (196), 358 Etilo, m iristato de (3 0 6 ), [II], 367l-Etitnaftaleno (258), 381 //-Elil-<i-naftitarotna (325), 356 If^EíU’fi xiaftiLamLoa (31 5 ), 356 Et¡l*a-naft1|i«>, í te r (27B), 371 Rtil-fl-naf tilico, éter (282), [37], 371 E tík , nitrato de (67J, 368 Etilo, nitrito da (17), 368 tf-Etil-/>-nitroacetanil]da [118], 346 /V-Etil^-nitroatuLina (96], 357 Etilo, m-nítrobenzoalo de (296), (4-7J,
367
Etilo, /»-mtrobenzoato de [55], 367 Etilo, ro-nitrorínam ato de [79), 368
.Etilo, p-nitrocinam ato de [137], 368
.Etilo, 3-nitroftatato d e [46], 367 ‘Etilo, 3‘nílro«dicilato de (118], 36H Etilo, 5-nitrosalídJato (102], 368 Etilo, ortoíonrtiato de (145), 365 Etilo, oxatato d e (186), 366 Etilo, oxamato de (114], 346, 368 Etilo, oxanilato d e [66], 345 Etilo, palm ¡tato d e [24], 367 Etilo, pelatr^onato de (227), 366 3‘Etilpeatano (9 4 ), 382 2'Etitpentanoico, ácido (209), 334 m-EtilfcnoE (217), 397 o-Etilfenol (2 0 7 ), 397 p-Etilfenoí [47J, 398 Etilo, íenilacetato de (229), 366 Etilíeniíbarbitúríco, ác ]o (172), 347 EtÜ fdiiluriiinú! (21 9 ), *40 Etilo, ítafato de (29 8 ), 367 Etilo, psm rlaio de (25 5 ), 366 Etilo, propionato de (98), 365 £ tt|o , p intvato de (15 5 ), 365 Etilo, salicilato de (234), 366 Etilo, jebazata de (307), 367 Etilo, estcaralo de [33], 367 Etilo, suberato de (23 2 ), 367 Etilo, suednato d e (21 6 ), 366 Etilo, sulfato de (208), 36b Etilo, ivt&r trato de (280), (17], 367 ti-EtiltoIuejio (165), 380 í»*Etílt0lüen0 (162), 380 W-Etil-rrt-tolmdína (2 2 1 ), 355 A '-Eiil-o-tcluidína (2 1 8 ), 355 ^ -E til^ -to lu jd tn a (2 1 7 ), 35S Etilo, trícloroaceiato de (167), 365 Etilo, trkloroJactato de (162), [66].
367Etilo, n*vaIeríana«o de (145), 365 Etilviotlico, éter (3 6 ), 369 Etilo, yoduro d e (7 2 ) , 374 Eogenol (250), 397 Eugenof, ítr¡r metílico dei (244), 371
F
Feblitip, solución de, 134 Fenacetina [135], 346 Fenacdico, alcohol (86], 342 Fenacilo, brom uro de [50], 387 < Feoacálo, cloruro de (244), (59], 387 Fenacilo, ésterea de, 257 Fem antroquinona (360), [207], 3S8 Fenantreno [Í00], 381
m-Fenetidina (248), 350 o-Fenetidioa (229), 350 p-FenetkTma (254), 350 Fenetol (1 7 2 ), 371 ¿-Fcnetiíurca [174], 347 Fenol (1 8 0 ), [42], 397
solubilidad de, 96 Fenolftaleíoa [261], 399 Fenoles, bromación de, 139, 320
derivado* de, 318 tabla de . 397
F cnoxdiu [59], 372 FenoxSacetaldcfiído (215 d .) , 343 Fenoxtacítko, ácido (96}, 335 Ferúlacetafdehido (194), [34], 343,
344<t-FctulacetamÍda [154J, 347 «•Fenilacetanilida [117], 346 Fenílo, acetato de (196), 366 Fenilicético, ácido [76J, 335 FenHacétlco, a&hfctñáo [72], 333 Fenilacetonitrílo (234), 390 Fenílacetilo, cloruro de (2 1 0 ), 332 Feniiacetífeno (14 0 ), 383 Fenilo, adipato d e [106], 365&-Fenilalanína [320 cLJ, 363 3>Fcnílalanina (320 tL), 363 ¿/-Fem ialanína [273 d.], 363 Fenilo, benzoato de (299), [60], 368 Fenílo, «-butirato de [22 7J, 366 2-FenilbutSrico, ácido (42], 335 r~Ferúlbutíríco, áeido (52], 335 Fenilo, carbamato de (53], 343 Femto, carbonato d e (306), [78], 368 Fenílo, cínam ato de (72], 368 •n-Fenilendiamina ‘(235) (63], 352 ^FenU eodiarnína (256) > [102], 353 ¿-Femlfcndí am ina (267), [140], 354 2 'FeniM -etansulíonam ida [1221, 4002-Fenil-l-ctanaulfonUo, cloruro de [33],
402Ferúlico, éter (2 5 2 ), [28], 371, 372 «-FenDctílícD, alcohol (203), 340
■ P-Fenilctílico,. alcohol (219), 340 a-Fentlctilarrina (I8 5 )j 3493-FeflÍletílamina (198), 349 a-Feniletílo, bromuro de (205), 3740-Fenitetilo, .bromuro de (218), 374 et-Feiuletílo, cloruro de (t**»), 373 p-Fertiletilo, cloruro de (190), 373 «'Feaífetilm ercaptano (199), 389 W-Fenílglicina [126 d ) , 362 Penílgliox&l [91], 34Í Fenilhidratidai, 258 Fenilhídrszm a (243), [19], 350, 351
reactivo de clasificación, 164
Fenilhidrazonío, sslei de, 258 Feaulm^capt&no (369), 389 iV-Fcnil-a-iiaftiUmina (335), [62], 357 Fenitnitrometano (2-26 d .) , 391 Í-Fenillenacílo, bromuro de [J25], 388 ¿-Fenilfenacilo, éjtercs de, 25?o-Fenilfenol [58], 398 j»»Fenilfenol [165], 399 FeniTo, fiatato de [70], 368 .V-Fenilftalímída (¿05], 3483-FenilpropanoI (237), .341 FemtpropióHco, ácido (1363, 536 «-Fcnilpropíon3m¡da (92], 345 P-Fenilpropionamida [105], 346 Fenilo, propionato de (21 1 ), [20J, 366,
367« ‘FcnilpropiómCo, ácido (2 6 5 ), 334 p-Fenilpropiómco, ácido (2 8 0 ), Í4S1,
334, 335Y-Fenilpropllico, alcohol (23 7 ), 341 Fenil-n-propilico, éter (18 8 ), 371 Fcnil-if-propilcetona (230) t 3S6 2-Fem lpiridina (269), 359 AT-Fenifpirrol (234), [6J], 360 Fenilo, «alicilato de [42], 367 Fenilo, e j t« r a to de [52], 367 ’• rfí-Fenilsuccínamida (d i) , [211], 348 ^f-Feiul&uce¡nani!ida (d i) [222], .348 Fenilo, succinato de [121], 368 ^-F enikuodnim ida [156], 347 FeuiltiourcaB, 283 Fenil-í-tolilcetona (326) v [54), 387 Fenilurea [147], 347 Fcniluretwws, 268, 321 FJoroacetofenona [219], 388 Florogluciaol [218L 395 Floroslucino!, tríacetato de [106], 368 Forona (19 8 ), [28], 386, 387 Ftalaldcbido (56], 344 Ftalaldehidico, ácido [97], 335 Ftalam id* [219 d.], 343 Ftálico, Acido [206 d \ 338 Ptálico, anhídrido (131), 333 Ftalida (290), [73], 368 Ftalim ida [233], 348 Ftalónico, ácido T14í], 337 FtalÜc», cloruro de (27(¡)t 332 dí-FenquíÜCO, alcobol [39], 34Í Férrico, solución de cloruro, 143 Ferroso, hidróxido, 144 FÍBher-E>avidion, deniítétnetro de, Í55 Fiaher-Johní, aparato de, 44 ■ Fluoreno [115], 381 Fluozcnona (3 4 1 ), [83], 387 Fhioroacetam ída [108], 346
Indice d ia b é tic o 463

4 6 4 '• ín d ic a a lfa b é t ic o
Fluoroac¿tico, ácido [S2J, 333 ¿•Fluorobervr*rdda [154], 347 ^-F lu o rw iñ m a (183), 349 Fluorobenceno (8 5 ) , 375 f-FluorobenoemuItoníunida fl25J, 400 f-Fluorobcnceniu|foniJo, cloruro de
{36], 402 ^•Fluarobetuoico, ácido [1B2), 337 p Fh ioronafukoo [60], 377 Pi-Fluoro1olucno (115), 3750-FIuorotoiueqo {114), 375 /J-Fluorotolueno (117), 375 FornwJdchído { - 2 1 ) , 343 Formamiida (135 d .) , 345 Formanílida (46], 345 Fórmico, ácido ( ÍQ l) , 334 Fortrúlpipcridina (22¿^-, §45 xvFructau [104 ¿ $ r$ 6 4 F yctina^ ld eh jíi^ rcac tív o de, 145 Fum árico, áidüo [200 »ubl], [302 tubo
ccn^Qo], 338 soiujtffidad de, $6
Fuaííram ida (m ono), [142], 346 R fW a m k l» (d i) , [266 d.], 348
’T uroanm ilída [314], 348 Furoarilo, cloruro de (160), 331 Furano (3 2 ) , 369 Furfural (161), 343 Furfurilo, acetato de (176), 365 Furfurflíco, alcohol (1 7 0 ), 333 Furoico, ácido [133], 336 Furoína [135], 388 Fnronitrilo (1 4 6 ), 390 Fuiilacrflico, ácido [141], 337
‘F iuión, puntos de, 39
- G
u-Calictosa [170 ó \ 364 G alvetofeoon* [173J, 383, 399 Gálico, ácido [240 d.J, 338 Gerank»! (229), 341 Gluco&a [90 d., 146 d.J, 364 Ghilacónico, ácido ( cit)> [136], 336 Glutacóntoa, ácido (tr« n i), [138], 336 p>Clutáxni< á, ácido [1^8 d.3, 362 w-Glutámico, ácido [193 d.], 362 ■ «U-Gluiámico ácido [227 d.], 362 G lutirico, ácido [97), 335
solubilidad de, 88 Glutáríco, anhídrido [56], 333 Glutaronitrílo (286), 390 ¿i-Gliccraldehldo [142], 344, 364 Glícerol (290 d . ) 341 .
GUcerilo, triaoetato d e (258)y 366 C licedlo, tribenzoau> d e [72], 368 Gliccrilo, tributiruto de (3 1 8 ), 367 Glicerilo, trípropionato de (209), 367 Glicerilo, trícstcArato de [71}, 360 Glicina [232], 362 G lie to n o (240 d.), 364 GUcótico, ácido [79), 335 GticoTonítrilo {183 d .) , 390 Glucósido*, derivado» de , 289
rotación óptica de, 364 tabla de, 364
GUcilglicina [264 d.], 362 'Glioxal (5 0 ), 343 Guayacol (2 0 5 ), [28J, 397 Guayacol, carbonato de [86], 363
H '
Helicina [175 d .j, 364 K eptacloroprapano [29 j, 379 «.Hcptaldehido (1 5 6 ), 343 n-Kept&mida [94], 345 K eplananilida [70J, 345 n-H cptano (98 ), 382l-Heptansulfonam ida [73], 400l-HeptansuIfonUo, cloruro de (125/9
m m ), 402 Heptaool-2 (16 0 ), 339 Heptanol-3 (15 6 ), 339 H eptenoM (9 4 ), 383 n-Hept»noico, ácido (224), 334 «-H eptanoko, anhídrido (258), 332 n-Hcptanoüo, cloruro de (176), 331 , n-Heptilo, aceta to d e (192), 366 a-Hcptílíco, alcohol (176), 340 n-Heptilam lna (1 5 5 ), 349 n-Hcptilo, brom uro de (174), 374 n-Hcptila, cloruro de {160), 373 tt-Heptilico, éter (260), 370 n-Heptilo, yoduro de (204), 374 n-Heptüm ercaptario (17 6 ), 389 HeptinO-1 (1 0 0 ). '*83 Hexaclorobenceno [229], 377 ' Hexacloroetano (187], 379 Hexadecano (2 8 7 ), 382 Hexadien.2,4-o].| [31], 341 H oatbídrobcnzaldehldo (362), 343
* Hexahídrobenxamida [186], 347 Hexahidrobenmnilída [146J, 347 Hexahídrobcnzoico, ácido (232), [30),
334, 335 • i;.Hexametilbenccno. [162], 301 . ¿Vi1-
Fndk* a lfa b é t ic a 465
Hexametílentetramir* ¿280] 361 n-Kexano (68 ), 382 Hexar.c!*3 (135), 339 Hcxadieno‘2,4 (8 1 ), 383 Hexatrieno-1,3,5 (78), 383 Hexeno-1 (66 ), 383 Hexeno-2 (6 8 ) , 383 Hexeno-3 (6 7 ), 383 n-Hcxltico. alcohol .-(156), 339 n-Hexilam¡na (1 2 8 ) ,r349 8-Hexilbenceno (226), 380 n-Hexilo, bromuro de (157), 374 n-Hexiio, cloruro de (134), 373 n-Hcxítcco, éter (229), 370 ít'H «rilet3íco, é te r (142),-370 n^Hcxilo, formíaco de (154), 365 n-Heaáto, yoduro de (180), 374 n-Rexür&trcaptana (151), 389 n-Hexilmetilico, éter (126), 369 n-Hexilmetilcetona (172), 3854-rj-Hexi]reíorcinol [69], 398 Hexino-1 (7 0 ), $83 HexÍQo-3 (81 ), $83 Hinsberg, método de, 135 H ipúrico, ácido [I87J, 337, 347 L-Hiitidiua [277 d.], 363 H idan to ína [218), 348 H idantoínai, 272, 311 Hidrazo, 295Hídrazobenceno [130], 396 Í»-Hidraiobífenilo [182], 396 ¿.Ridnm>bifenil<> [170], 396 Hidrazo, derivados de compuestos, 317
tabla de, 896 I,l'~HidrázonaFta3cno [153J, 396 2,2'-HidrazDnaftakno [140), 296 TO'Hidrazotolueno [38], 396 o-Hidrazotoluena (161], 396 p 'H idrazotolueno [134], 396 Hidriodeno (177), 380 Hidrocarburos insa turada, derivados
de, 309 tabla de, 383
Hidracánamaldchído (224), 343 Hidrocinanmimda [105], 346 H idrociium ariüida [98], 346 Hídrocínámico, ácido [483, 335 H idroquinona [169], 399 Hidroquinona, d iaec ta to d e (3 23), 360 H idroquinona, éter dibenoílico [127],
372Hidroquinona, éter dietílino [72], 372 H idroquinona, éter dimetdico [55],¡ 372
H id ro q u in o n a , é te r monobencÜico [122), 399
Hidroquinona, éter rooncroetílico [53], 398, 372
p-Hidroxiacetanilida [169}, 347 Hidroxíacctona (146), 339, 385 2'Hidroxiacctofenona (28], (218), 386,
3873*Hidroxiacetofenona [96], 387 O-Hidroxiazobenceno [83], 396 ^'Hídroxiazobcnceno [352], 396 m-Hídrcttibcnzfddehido [105J, 344o-Hidroxibcnzaldehtdo (196), 343 p-HidrOxibenzaldehido [115], 344, 399 rn-HidrozenüOko, ácido {201], 338 j^Hidroxibenzoico, ácido [213], 338 p-H¡droxibenzoícnona [135], 388o-Hidroxibencílico, alcohol [87], 342
V. 3-Hidro«biíínU& [78L 398Y-HidroxibutironuriJo (240), 390 2-Hidroxi'3^-dibromotolueno (5 7],-398 rn-Hidroxi»Ar -d im ctü am lin a (263),
[85], 360
^HidroW 'A'^v'-dimctilaniliaa P6), 3604-Hidrci5d-2>6-di mctiibenceno- 3,3-di-
sulfor.amída [208], 4014-H ídroxi- 2,6-dímed] heneen-1,3-disul •
lonilo, cloruro d e [119], 404 2-Hidro»-3,5-dúufcroto3uena {86], 398 fV-Hidroxxetilo, acetato de (182), 366 2-Hídroxictilamina (171), 349 ^H ldrox íg ju tá ttiico , ácido [150 d.],
362«-HidTOxjisotJUÜrico, ácido [79J, 335a-HidroxiisobudronitrUo (120), $90Hidraxilam ina, clorhidrato de, 150 ^'H idroxi-N -iiK tílacetarü lida [240],
348o-Hidrojd'iV-metilamüna [87], 357 p-Hkfroxi-iV-roetilanjUna [85], 3572-Hidrori-3-naítoíco, ácido [222], 3383 *Hi<íroxi.2-naflojco, ácido Í226], 338 ^•Hidroxiíenilacéxíco, ácido [148], $37 p-Hidroxifenilglidna [248 d.j, 362 t^HidroxiproIína [270 d.], 3620-Hidnnópropiotútriki (221), 390 ¿-Hidroxipropiofenona [148], 3882-Hidrroáquinoleína [199), 3^16-HidroxiquinoIeína [193], 361S-Hidrojiiquinoleína (2 6 6 ), [75J, $603.H ídroxi-2,4,6-trinítrotolueno [1103,
399Horooverárico,. i e d o [99], 335

4 6 6 - - In d ice c lfo b é tic s
I
ignición, pni»Ka de, 36 I mida i, derivados de, 277
tabla de, 345a-Imin&iiftjuilirtrcapfoacéticoj, cíorht*
drato* de ácido», 316 Indanona.J (2*2), [4-2], 367 fndeno (180), 384, 380 TimIcoc de refracción, 62 Indol (254), 152], 356 infrarroja, espectroscopia. 15?
acetilenos, 20? áddos, 20? alcoholes, 20? aldehidos, 202 amidas, 208 aminaj, 208anhídridos de ácido, 207 cartwáticos, ácido*, 207 cetonai, 211 esteres, 209 éteres, 210 Kenepfce&tionee, 200 bidíros de acílo, 207
/'Hidrocarburos, 210 nitril&s, 213 nitrocompueítOí, 213 nitrógenos, grupos funcionales, 21Sokfinas, 213
Inositol [225], 342 Intermolcculares, fuer2a?, 08 Ioulina [3 78 d.}, 364 Isatina 1200), 347 Isoamil©, acetato de (14 2 ), 365 Isoamüico, alcohol (130), 339 Iso&mil&nuna {95), 349 ¿V-isoamilanüma (254), 356 Iwam ílo, benzoato de (2 6 2 ), 36? Isoamlfo, bromuro de {118), 374 Isoamilo, c -bu ti rato de (17 8 ). 365 IioasúlO; earb&mato de {$4], 345 Isoamilo, carbonato de (2 2 8 ), 366 Zsxunilo, cloruro de (10 0 ), 373 ísoaimlo, elorofomúato d« (154), 365 Isoamil ico, éter (172), 370 isoamilo, íorm iato de {123), 365 IsoatüOo, yoduro de (14 8 ), 374 Isoamilo, úovderanialo de (19 0 ), 366 Isoam ilm treapano (1 1 7 ), 3B9 Ijoamil-p-nAftilico, éter [26], 372 Isoamilo, nitrato de {147), 368 Tsoajxülo, nitrito de (9 9 ), 368 Isoatoilo, o ya lato de (2 6 2 ), 360 IstornUo, ftaJato de (349) 367
Isoamilo, propionato de ( l6 ü ) , 365 Lo&milo, salicilain d e (277), 367 boam ilo, í^ c ú n a to de (297), 3b7 Isotutflo, acetato d e (116), 365 isobutllico, alcohol (108), 339 Isobütílamina (6 9 ), 349 AMaobutiTauílina (231), 355 ísobutilbenceno (173), 380 ísobutilo, benzoato de (241), 366 Ijobutilo, bromuro de (91 ), 374 líobutilo, q-butíralo de (15?), 365 ísobutifo* carbam ato de [55], 345 Ijobutilo, carbonato de (190), 366 Isobutilo, cloruro de (68 ), 373 ísobutilo, cloroíotmiato de (ISO), 365 Tsobtlttkno, brom uro de (H 9 ) , 3?8 laobulítico, é te r (122), <369 Isobutilo, íorm iato d e (98), 365 Isobutilo, yoduro d e (120), 374 Isobutilo, «obutir^to de (147), 365 líobutüm eieaptam . ..(88), 389 Isohutilm etíltctona ; '.1 9 ), 395 Isobutil-2-nafilljco, éter [33], 372 ísobutilo, nitrato de (123), 358 Isobutilo, nitrito (67 ), 368 r.Bobytilo, oxalato de (225), 356 ^IsobuUlfenol (236), 397 Isobutilo, fenilacetato de (247), 366 Ijobutiioj p/opionato de (137), 365 Isobutilo, succinato de (265), 36? Iv)bütíraldeh(do (6 4 ), 343 ísobutim m ida [128), 346 Isobutiranilida £105], 346 Isobutírico, ácido (155), 334 Isobutírico, anhídrido (182), 3 3 1 Isobutironitrilo (1 0 8 ), 390 líDbutiroíenona (22 2 ), 386 Iaobutirlo , cloruro de (92 ), 331 Isocaproico, ácido (1 9 5 ), 334 rsaeapronítcito (15 5 ), 390 IsacaprciSlo, cloruro de (147), 331 rsodyreno (1 9 5 ), 380 Isoduresol [81], 398 líodurid ína (25 5 ), [24],' 350, 351 Ijoeugenol (26 7 ), 397 D-I&oleudiu {279 d.]» 363 L-Isoleucína [279 d.], 363 ¿M ioteuciaa [292 d,J, 363 leopentano {31), 382 Jsoforona (214), 386 ísoftálico, ácido (300], 338 Isopreno (3 4 ), 383 Isopropenílo, acetato de )9 $ ), 365 Isopropemlbenceno (165), 334 Iiopropílo, aceta to de (91 ), 365

In d ice a lfa b é t ic o 467
Jjopropílico, alcohol (83), 339 Iscproprl&núsa (33), 343 jV-Isopropilanilina (213), 355 /r-IsopropübenzamídA [133], 346 JsopíTjpUbcnceno (153), 380 I:opropiJo, benzoato de (218), 366 í-IjoprDpilbcruoiCOj ácido [1163, 336 I*opropilo, brom uro de (60 ), 374 Isopropifo, n-butirato de (128), 363 ísopropiio, carb amato de [92), 345 Jsopropib, cloruro de [361, 373 Isopropilo, do rofortdato de (10 5 ),
S65Isopropílíoo, éter (68 ), 369 leopropilo, formiato de (69 ), 365 tsopropilo, yoduro de (89) t 374 Jiopropilo, lactato do (1 6 8 )/ 365 Isopropil mercaptano (56 ), 3894-rjopropH*2-mctilbencenst:Jfonamida
[731, 400 TíopropiJciettlcetoüa (94), 335 Ijoprop>]-2-naftilico, ¿ter [401, 372 ÍWpropüo, o b la to de (190), 366 IsopropilfcnjJcarbmol (224), 341 Isopropilo íta la to de (302), 367/ Isopropilo. sa lid Jato de (237), 366 Iaoquinoleina (240), [24], 359, 360 Isosafrol (246), 37 1 u*I*oserina id.], 363 dM joserina [d,], 363 IecvalcraldchUo (92 ), 343 Lsovaleranilida [109], 346I so valeriánico, áddo (176), 334
solubilidad de, 90 Isovalerlaaitrilo (130), 390 • Jsovsileroffrnona (225), 336 IsovsJerianílo, bromuro de (140), 331 íiovaleriantlo, cloruro de (115), 331 [lacónico, ácido [165 d.J, 337 Itacónko , anhídrido [68], 333
L
¿/-Láctico, id d o fie ), 335 L ie tid a (255), [128], 368 Lactonitxílo (182 d ,) , 390 Lactosa [203 d.], 364 ' ' LsuraídehSdo £45], 344 L iurico, ácido (431 335 L áurko , anhídrido [42}, 333 L aurona [69] 387 L áu rk o , alcohol (24J, 341 Lepídina (263), 359 •
0-Leudn» [337 d.), 363 t-L cucína [337 d.J, 3C3 ¿ /-L eudna (332 d ] , 363 Lcvulínico, áddo (250 d .) , [33], 334,
335L evulott [104 d j , 364 Licbemiann, reaedón de nitroso de,
159Limoncno (176), 384 Línalol (197), 340 j>L5iina [224 ¿ I 362 L-Liiina [224 d.J, 362 ¿J*L»ina [d), 3631-L íxou» (105 d.), 364 Lucas, prueba de, 1482,4-Lutidina (159), 358
M
M aíeamida [170), 347 M aleanilida [175), 347 Maleíco, ácido [130], 336
solubilidad de, 88 M aleteo, anhídrido [56), 333 d.’-Málico, ácido (131], 336 L-Málico, ácido [100], 336 M alonaraída [170], 347,MalonamicLa (m ono), [50J, 345 M alonanilida (mor*©), [132], 346 M aJonanilida {di), [225], 348 Mal&nico, ácido [133 d-), 336
solubilidad de, 88 Makwonitril© (219), 390 MaJtoia [100 d ., 165 d .) , 364 ivMandéliGO, áddo [133], 336 L-Mandólioo, áddo [133], 336 dA M andako , áddo [118], 336 Mandelonitrily (170 d ,) , 390 o-M anitoI'[166], 342 i>M arx«a [132 d.], 364 Xlaquennc, bloque de, 43 M argárico, ád d o [60], 335 Mcüciloea [147 ¿ l 364 /í-Me]ibíosa (h id ratada}, [85], 364 M díeico, ád d o [288 d ], 338 Í-M entano (169) 382 (— )-Mcntol [42). 341 i>M entoaa (207), 386 (— )*Mentilo, acetato d e (2 2 7 ), 366 ( - ) -M e n ¿ la m in a (2 0 5 ), .349 Ti'Metilcetona (151), 385 M ercaptalacctatte d e aldosas, 290 . W ^rcaptanoa, d e r i v o s d e , 3X2 •
tab la de, 389

4 6 S . Ind io» a lfa b é t ic o
ercuri b u a á l ara Idas, 277 M m cónioa, áddo [202 subí,}, [24Í],
338Mtü'tilaDO (164), $80
solubilidad de, 99 M eútüenaaúda [135], 546 Madtíléoico, Acido (166}, 33?Meaiálo, óxido de (13 0 ), 385 MetaIdehído, Ti 3], 344 iíctoerflam id* [116), 346 M etaoflíco, ácido (1 6 3 ), [16], 334,
335Metasulionamida [90], 400 M eusulfonaaudía, 233 Metasulfonflo, cloruro de (60/21 w m ),
402l^M etíonina [283 d.J, 363 dZ’-líetionitta [272J, 362 M eton* (149), 388
derivado* c d o , 276 Metoxiacetaroida (97), 345 Mctcociacctanílida [58], 345 ¿-M etoxiacetanílida [1271, 346 M etoóac^dco, ád d o (203) j 334 MetoxiacetotútriJo (1 2 0 ), 390 jn-MetoxLicetofencma (2 4 5 ), 386 ¿-MetojáacetofenoBa (2 5 8 ), [33J, 387 Metoxiacetilo, cloruro d e (113)» 331 «n-MetoxibeoBaldehído (230), 343 o-MetoxjlxnzaJdeiiído [38], 3+4 ^M eto tíx a& ld eW d o (247), 343 o-M^M&benzamida [129], 346 ^Mcvcmbcrücuülida f lS l j , 346
(^Mctwábcocen-ljS-disulfonamída [240], 401
4-Metoxibenccn-I,3<lisuifoníJo, cloruro de (86), 40+
o-MeloxibentoicD, ácido [100], 336 ¿-Metoxibcnzonitrilo [62), 390 ¿•M etoribenzcfenona (355), [62;, 387o-Metaxibersoílo, cloruro de (254),
332e-Metoxibemoílo, cloruro de (224),
332o-Metojdbiíemlo [32), 372 f-M etaxibífeailo [85j, 3726-Metoxic|mnoIeína (305 d .), [28],
359, t-‘08-Melojtiquiúoleína (2 8 3 ), [50], 360 N.M etilaceUftüida [102], 340 Metilo, acetato de (5 7 ) , 365 M etilo, aetto&cetato d e (169), 365 ^M etílaceto ícao iu (2 2 6 ), [28), .38? ?/-n>eti 1 -o-acctotduidida [56], 345 ¿/-'M e&'i-acetotoluidida [63], 345
a-M cdlacroJclna (7 3 ), 343 Metilo, acrilato de (85), 365 M ctilal (45], 330 M etílica, alcohol (66 ), 339 M etüam hia ( —6 ) , 3494 - (Ar-M etüam ino ) - l ,3-dimetilbenceno
(2 2 1 ), 3554-(2V-Métilamm©)-2-níU‘Otolueno [45J,
35é4-(tf-M etiIandno)'3-nítrotolucno [84],
357MedlanHina (192), 355 M etilo, anisato de (255), [45], 367 M etilo, antraniiato de (260 d .), [24),
350, 351 fl-M etilantraquínona [V79], 3882-Metil6rnceu-l,3KÍüu]íoiwnúda [2503,
4012-M ctilbenccn-l,4-diíulfanamida [224],
4013-Mctilbencen-l,5-disuífonaniid* [216],
4014-Metühen.ccn'J ,2-disulíonajvnda [237T,
<014-Metil heneen-I,3-dúuIforiArtLÍd& [191],
4012-M«tiJbencen-l,3-diíulfoiijloJ cloruro
de [88], 4042-Medlbcnceü-í,4-dí$u!íotüío, cloruro
de [98J, 4043-M otilbcnc«n']J5-<fou1lfanifo, cloruro
d e [95J, 4044-M etilbencen4>2-disulfoniJo, cloruro
de [111), 4044-Metilbencc»'l,S'<iijulíonílo, cloruro
de [56), 403 M etilo, benzoato d e (193), 366 Í-M etilbenzofeuona [60], 387 Metilo, o-benzoílberttoato de [52], 367 W -M etilbeodlamina (185)T 355 Metilo, brom uro de (5 ) , 374 Metilo, bromoacetato de (144 d .), 365 //•M etil-^-brom oanjlm » (260), 3562-Mcti|-4-bromobencem«lfonaifjjda
Í168], 4002-Metil-5-bromobertCenJuJfonamida
[166), 4004-Mcti I - 3 -b r ©moben cc o*u) f onarn id a,
[146], 4004 2-MctiJ-4^«t>mobencen$ulfonilo, cloru
ro de [50], 4032-Meti]-5-brom©bencensulf©n>lo, cloru
ro d e (35), 402
4-Metü-3-bromobc^ccnaulfoniloJ cloruro de [60], 403
Metilo, fTj-bromobenzoato de [32], 367 Metilo, o-bromobenzoaui <lc (244),
366Metilo, ¿-broroobenzoato de [81], 3683-MetiM-buttwsuLfonainida [3), 4003-M etil-l -butatwulÍQnilo, .• cloruro de
86/9 n un), 4022-Metíl-jl-butcno (31 ), 3833-Metil-l-buseno (21 ), 383 a-M etil-n-butiraldehído (93 ), 343 Metilo, n-butirato de (102), 365 Metilo, caprato de (232), 366 Metilo, rt-caproato de (150), 365 Metilo, caprilaio de (193), 366 M etilo, carbamato de [52], 345 M etilo, carbonato de (90 ), 365 Metilo, cloroHcetato de (130), 365 \ tf-M etilo-o-doroanilída (214), 355 A'-Metil-^-cloroanilina (240), 356 2-MetIl-4*dorobencenjulf©namída
[185], 400 2-Metil-5-claróbenceiisulforiíiinida
Í165J, 4002-Metil*5-clorob€iiccns«) foria:ri>da
[1421 4004-Mctil-3-c]orobcncensulíonarnÍda
[128], 4002*Mctil-4-d0r0bericeimilÍ0í’.il©, cloruro
de [53], 4032 - M e ti t -5 -cío roben ce nsulfoni t o, cloruro
de (21], 4024-MctiI-3^1orobencei»ulfonilo, cloruro
de [63], 403 Metilo, m-dorobenzoato de (231),
[21], 366, 367 Metilo, «p-dorobenzoato de (2 3 4 ), 366 Metilo, ^clorobenzoato de [44], 36? Metilo, cjorofomüat© de (75), 365 Metilo, dnam ato de (263), [36), 367 Medio, d tra to de (285 d .} , [79], 366 M edio, danoacetato de (200), 366 M etilddocxa»o (10 0 ), 382 2’M ctüddohexariol (16 5 ), 3393-M etilddohexanol (175), 3404-Mctilddohey.attol (174), 3402-M*tílcídohex2nona (169), 3353-Metil)dc]ohcxanon3 (16 9 ), 3054-MctiWclohexanona (169), 385 Metilcicloexilcetona ’ (180), 3 85 M etilcidopentano (7 2 ) , 3822-M etilddopenianona < 139), 385 tt-M etil-2,4-dím troanilina 1176], 357
In d ice a lfa b é t ic o 469
Metí©, 3^-dilútrobonzoato de [108],368
2-Met¡l-l,3-dioxan© (11 0 ), 3302-MetíM,3-dioxo<#rto (82 ), 330 //-Métildiíenilaixdna (296), 359 Xíetilenactúnoacetoiútrilo (129], 361
390Metileno, bromuro de (98 ), 378 Metileno, cloruro de (4 2 ), 373 MetiJeno, yoduro de (180 d .) , 379 Metilo, etilacetoaceta(o de (190), ,
366MetíletilaCetilo, cloruro de (116),
331JY-Metitetilamina (35), 355 Metiletílíco, éter (10), 3692-Metil-4-fluorobcnceruulfon amida
[173], 4002-Metil-5-Fluorobencensulfonam»da
[141], 4003-Meti3-4-fluorobcncensu]fonamida
[105], 400Metilo, íormiato de (32), 3652-M*ti3furano (64), 3695-Metilfurforal (1 3 7 ), 353 MetilOjíuroato de (161), 366 a-Mecilslucósúio [165 d . l 3642-Metilheptano (11 8 ), 3624-Metilheptano (118), 382 Metilo, B'hepíanoato de (173), 365 2'Metithexano (90), 3823-Metilhexzno (9 2 ), 3822-MetiHiexanoico, ácido (210), 3344-Metiihexanoíco, ácido (216), 334o-Mctilhidrocináimieo, ácido [37], 335 Metilo, PTi-hidroxibeozoato de (70], 368 Metilo, £-h¡drOxibenzoato de [1313,
368Metiliminodiacético, ácido [227 d.], 338 Metilo, yoduro de (43), 374 Metilisobutilcarbínol (131), -339 M e t i l i jo b u t í l c a r b i n o l , acetato de
(148), 365 Metilo, isobutirato de (92), 365 Metilisopropilcarbiool (1 )3 ), 339 Metilo, Uova! cria ru to de (116), 365 Metilo, lactato d e (1 4 4 ), 365 Metilo, lagrato de (268), [5], 367 Metíto, levufinato de (19 1 ), 366 Metilo, malonato de (181), 366 MetilmalónicD, ácido [135 d.J, 336 Metilo, m andelato de [58], 367 a-Met([manóe¡do [193 d,j, 364 Metilmercaptano (6 ), 389 Metilo, m iríitato de (296), [18], 367

l-M edliuftaheño (240), M I P-M ctilnaftskno [32], 381 t-Mctil - 3 - ruLÍtalenjulfonamida
4001-M «dí'4» nahl]cn$ui forjamída
400I -Metíl - 6 - naítalensuJfonamida
.4001-Metil -7 ‘ nafta]ensulfortaraid3
4012‘M<ítíl-6* naíwüensulfonanuda
401I-Meti3-3-natia!eniutfonilo, doruro de
(I25J, 404I-Meiíl-i-naítaterisulfortjlo, doruro de
(31), 404J-Meul-ó-naftalensulíonilo, doruro de
ÍSgj, 4041-MetiU7»naftA]ensulfonilo, cloruro de
[123], 404jV-Mctil-A^-naftilaccLirnida [95], 345 W-Mctil-a-natVilareuna (293), 356 /í*M«üI-fl-naLftí1araina (309), 35G Metíl-ct-is’aítílico, éter (265), 37 í Metil-fl-naítíiíco, éter [72], 372 Metil-0-naítilcetona (300), [53], 387 Metilo, nitrato de (65), 3í>B .Y-Metil-p-nitroacetariÜida [353], 347 2‘MetiM-rvitroacctanilida [I96J, 347 4‘Metil-2»nicroact:antljda (94J. 345 4‘Metil-3-mü-o»cctani!ida [148], 347 A’-Metil-wi-nitroajiilina [66], 357 2'Mc<j]-3»attroan>]¡na [95], 3532-Metil-4j-mtroanitÍtwi [91], 352 Metilo, «Mtitrobensoato de (279),
(76], 3S7, 360 Metilo, ¿Miátxobemoato de [96], 363 2*Metil*2-nítxobutano (150), 391 3*MetiH*nitrobütano (164), 391 Metilo, m-nitrocinamato de [124], 368 Mcdto, o*nitroeinamato de (73), 368 Metilo,fl*fll?todiiamAto de [161J, 363 Metálíf’ j-mtconaftalato de [69J, 368 Mtfmo, 4-nitronaftalato de [66], 367 "ÍMetiH-nitropropano (141), 3912-M etil‘2-nUropropano (126), (26],
392Metilo, 3-wtroialiciUto de [132], 353 Metilo, 5-rotmalidlato de [119], 363 Me tito, orrofcrniiaio de (101), 365 Metilo, oxmlato de (163), [51], 367 Metilo, palmitato de [30], 367 Metilo, pelai^onato de (213), 3662-Melilpentano (60), 3823-Metilpcnuno (63), 382
4 7 0 Indtco a lfa b é t ic o
2-M í iil-2,4-pcatanodIol (196), 340 ¿¿■2-Metiípentanoico, ácido (I9 6 ); 3343-Mcti3pentauoioo, ácido (197), 334 2*Metíl-Up«nUnol (148), 3392-Mctií-2-pe n*anol (121), 3393-Metil-3-pentanol (128), 3394-Metí3-2'pentftno3 (131), 339 M etilo, feni lace tato d e (21 8 ), 36$ Metilfenilcarbinol (203), 340 Met¡l-^-feníletilcetona (235), 386 Metilo, ítalato de (282), 3$7 ¿¿•2-Metílpipcridina (116), 355 dW -M elilpiperidina (J2 8 ), 3552-M «tjl-l,2-propanodioI (178), 3402-M etíM -propansulfonaintda [16], 400 2'Metii-l-propamulíon:10j doruro de
(73/11 m m ), 402 Metilo, propionato de (79), 365 M etíl-ti'proptlCirbinol '119), 339 Metil-tt-prcipilcetona ( l i ? ) , 385
solubilidad, 90 A'-Metilpseudocumidin* Í237), (44].
356M etilo, piruvato de ( Id 6 ), 3654-Metilquinoíefrta (263), 359 8-Met;lquino]eína (253). 3597-Melilquinole>JU (252), [39], 360 M etilo, íalicilato de (224), 366 MetiJo, s e b a s to de (263 d .) , '38],
367Me til. estes rato de [38J, '3670i-M etitestireno (163), 304 <>Mctilestireno (1 7 J) , 384 ¿•M etilestireno (16 9 ), 384 M etíluicdnarzúda (d i) [200], 347 Metilo, succíjiato de (195), [J9j, 366,
367M etiljucdnico, ¿d d o (115], 336 M ed io , sulfato de (388), 3C3 Metilo, D*tartratO de (280), [48], 367 Metilo, d l-tartra to de (90], 368 Metilo, tercftalato de [140?, S68 Metil-2-tiemlcctona (214), 386 Metiltimílico, éU í (216), 371 Medio, m -toluato de (235), 366 Metilo, o-tolualo d e (213), 366 Metilo, p-toluato de (217), [331, 366,
367ft'*Metit-m'toluidi»a (206), 355 /V'Metil'tf-lcluidnia, (207), 355 íí-M etí3-/i'toIuidína (208), 355 M etil-p-tolilcarbinol (219), 340 Meüf*7Ti-to)ilcetona (220), 38$ Mctü-o-toKJcetona (21 6 ), 38$
[ 1 4 4 ] ,
[116],
(389},
[206),
1 ridk« . a lfa b é t ic o 471
Metil-p-toliic«torui (226), 38$ ¿/•MetUcriLrcanoluailiiw [39], 356 Metilo, undetilcn&to de (248), 366 M edí urea [101], 346 M etilo, a-vaJerianato d e (130), 3$5 Metílvinilcetona (80 ), 385 Meiitol [70], 398 Mezclas, puntos de fusión, 39 Mezclas, 103
binarias, 104examen prelim inar de, 111 dastíladón con vapor de, 95 insoluble» en a$ua, U 6 , 117 sintéticas, 118
Mezclas soluble» en agua, 113, 114 con tres componentes, 109
Molecular, refracción, 6$Molecular, peso, 72 M aiío tina (1 3 0 ), 355 Móeico, á d d o (213 d.j, 333 M irceno (16 7 ), 384 M irííiieo, ácido [54], 335 M-irtítico, alcohol (39), 341
N
Nalje-Axelrod, aparato para puntos de fusión, 45
«•N aítaldehído [34], 344 P-KaítaJdehklo [60], 344 NaítaJeno (80], 3811.4-NaítaJündísulfonamida [273], 4011.5-Naftalendisulfonamida [310], 401 1,6*Naftatendisulfon&mída [298], 401 2,7*Naítalcodisul£onamida [242], 4011.3-NaftalendÍEulfonilo, doruro de
[138], 4041 .4-Nafta3cndísulfomlo, c lo ru ro de
[166], 4051.5-N aítalendisulfom io, c lo ru ro de
[183), 405l ,6 'N aítalendisulfoniIo, c lo ru ro de
[1281, 404 l , 7 -NaCtalendiiulfomlo, c lo ru ro de
[ 122], 4042 .6-Naftalendisulfonilo, cioraro de
[225J, 4052.7-N aítálendisulfoniIo, c lo ru ro de
(162], 4051*Naftalciuiulfonajmda [150], 4002-NaítaIcosulfonftmída [217], 401 1 -Naítalcnsulíonilo, doruro de [68],
403
2-Nafulen*ulfoni lo. doruro de [76j,403
Naftaíeno, tetractaruxo de [182], 377 1,3,5,7-N afttlentetm ulfom lo, cloruro
de [310], 4051.3.5-Naf talentm ulfonilo, d o ru ro de
[14$3, 4051.3.6-Naíudentrisulfonilo, d o ru ro de
[193], 4051 ,4-5 -NaftaIentrisulíoiub, d o ru ro d e
(156), 405 2,3,6‘Naftaüentriiulfonita, d o ru ro de
[200}. 4051,2-Naftilico, anhídrido [169], 3331,8-N aftálko, anhídrido [274J, 333 2,3'Naftil»co, anhídrido [246], 333 N&ftalidas, 301 p-N aitaraida [192], 347 a N afU nilida [163J, 347 p-N aftanüida [171J, 347 a*Naftoico, ád d o [152], 337 p-Xaítoico, á d d o (185], 337 a-Naftoico, anhídrido ( i46], $33 a-Naftol [94], 398 £-Naftol [122], 399 A-Naftonitnlo (2 9 9 ), [351. 3900-Naftonitrilo (3 0 $ ), [$$}, 390 • a -N aftoquinotu (125], 308 ^-N aftoquiuona [120], 3Sfa a-N aí;ilácetajnida [181], 347 ^N dftil3cetam ida [200], 347 Ív<c-Nafc¡la«tam ida [159], 347 N -p .N afQ W tam id* (134], 346 Ot-Naftilacetanilida [155], 347 a-N aftílo , aceta to de (49], 367 fi-Naftilo, acetato de (70], 36S út-í/af til acético, áddo [133], 336 fl-Nafrilacético; áddo [142}, 337 a-N aftilam ina (300), [50], 351, * p-NaftHauzuna (300), (112L 353 ^-a-N aftilbenzaiiúda [181], 3470-NafriIo, benzoato de [107], 363 (J-Naítilo, é te r« , 302 ci>Nafti|cneti]cetona [34], 387 P-NaftiEo, salícilato de [95], 368 «"Naftilsulfoníunidas, 283 «-Naftiluretauos, 268 Weopencuio (9 ) , 332 Neopentllico, alcohol (53], 342 Neopcnúlo, brom uro d e (10 9 ), 374 Neopentüo, d o ru ro de (85)," 373 Neufr -Uxadón, equivalente d e , 259 Níquel, d o ru ro d e , y 5*cútXOSa]¡álal>
deídoj 157

Me
v: i»- ^
tn -t. 'O ~o>- « S s s g -<¿> S f á '- ’Cl „cv °?o <7> «o OI^M i n “ „ <■> .. ai ~’
~ t n < o __o * u o * "O ~
¡ Í K
' *»o »í I s I e
I
_. .- ce<* /0 o r t
'A 03<o «r. •rt qíj
.r£
2 3 - *• t t i
XJ —O c-1
■o- os <© •* •WbOS - ° .'S 5 «5 «'Cfs!0£«>to vrv ,*: - r* o _ ~ 9 « ^ —s
^ s; 5 S g 5 s
I ¡ ? ¿ l l r f r f i r f i ¿ s£ 2 c c ? - = ,3 ,a ' a ' ; ,a S _3 $ J $ W W $ g iü r Q ü í e r i e e r < JU
se 2
* 3
00
<£>r^.o o „ S 00 J i »s i 3 tí 0 0
~?~=f
l í í M i a i.2 .5 •? 3 ® -o o3> -o £e e l ¡ g - l i I -i ' i oog 3 15 5 jf 3* £ i i ^ o o o o 6
5 . "
o £ iC ,f'^ r o •O cxd <£>
s a s | ¿ o - ■1 s i l 3 S” 'l i l i l í I'5 'c ’c ’c ’c '£ *= : vü ^ Já síá ^ 5 . 2 S 2 S S S S5 i; £ i; i> ü ü9. z % y. £ % iQ, -Cl. O. O. -Cl, O.v'j <
¡•§ -g” g : rt “ » * 3 !■§" 8 l i ’a; 5 | 8 2 '3
o “3 g* g- 8* ¿ ¿ t! t C£ 2 2 y. y. <A tJ- h fi ¿
- *r '-^'^'7^— °y~4 « rt
j J l § 3l l §*3 *5% g - 3 3 3Y í c c e^ 3 ^ 7 Ss
C*Mrten to eo.“
* “ * •*_ j s l á i - gt ~ en ü « .r
u ~ CO ^ ^ ^ :£
K¿ M M U ¿J
E
.9 5 -8 5 - í S 'sSU S« •£ “ «c c í; «S í X ¿ o2 § - S-5 .2 -5 s
2 Z “ * -¿ -Cl
►O£*
"OK
(SEK.*■
<p.OT
<r> -4> r-;
_ V5 S 2 „ «
|
: i | - S »
«•» <v^|„ r—>0 *—> fvj21■" n « pÍr t f l C CM —r s l § ^ í i
rfl gc a
„ 2 = Si I l a l l i g l X z g
í f l - E E o
40 ^ 51
-C 'S B3 «J »T»■§ «3^75 *3-0■* *o
i f i s s i a i f f i l s j í s s s i s á j g g g | g g g g g « U 8 U E U A E

n
m*
r
474 lodJe# alfabético
Oxidación, dicromato, 307 perroa/Jíwiato, 35?
O am as, 313
Palm itaktchídoJSí), 344 Palmitamida [106], 346 Palmítico, ácido [62], 335 Pamfmas, derivados de, 308
ta t ía de, 3S2 Paraldehído (124), 343 Partición, coefírientes de, 262 Pelarjonaldehído (1£ 5 ), 345 Pelargónico, ácido (253), 334 Pentaclorobénceno [86], 377 Pentacloroetano (161), 379 Pcntaclorofenol (150), 399 PeDtadecano (271), 382 P en(ad íeos 1,2 (45), 383 Pen(adienc*l,3 (42), 383 PentadiertO*l,4 (26 ), 383 Pentteritñtol [253], 342 Peniarnetilbenceno (51), 3&{ Pentamculeno, bromuro de (221), 379 Pentamctilenglicoí (238), 341 PíDiano (36 ), 382 P«ateno-I (30 ), 383 P«nterio*2 (36 ), 3832-Pentílo, bromuro de (U S ), 3743-remífo, bromuro da (119), 3741-Pcnttno (40 ), 3832-Pcntino (55), 383 Percolador, aparato para puntos de
ebullición, tipo, 48 PcrfluoTo-n-hexano (57), 378
I Péryódtco, icido, 162 L ^ ^ ^ P ic e n o [364], 381 V ^ B r - P ic o l in a (129), 358 * ^ ^ p -P w o U n a (143), 358
Y-PicoÜna (143), 358 '{enómetro, 56,'Ireno tI48}, 381
___ 'indina (116), 558 "" Píngalo! [I3SJ, 399 . Piroftalol, tríacciato de £161}, 3C8 « ^ ■ c o g a lo ! , óter irietílieo {39] 372 jS B P ro g a lo I , óter trúneiílico [47], 372 r ~ Piromúcíco, árido (133], 336 ! Pirrodilina [89], 355 ip É ^ u v a n i l Id a [104), 346 B ó r i c o , ácido (165 d .), 334 f ^ P k rá m ic o , ácido (168], 354 ' Picramida {190), 354. m tos, 285. 299, 302
t»mmY~-
m
T*<
3 b lw ¡
Picrieo, á n d o {1221, 399 Píenlo, cloruro de (83), 394 Pimelico, ácido t i 05], 336 ' ■
solubilidad de, 88 Pinato! (1 7 2 ), [35], 340, 341 Pínaeol, h idrato d e {46}, 342 Pinacolona ( l0 6 ) , 385 ¿-Pinena (15 6 ), 384 J^Pineno (156), 384 Piperazma (14 0 ), [104), 357 Pipírico, ácido (216], 338 Pipcridina (10 5 ), 355 Pipcrina [129], 346 Pipcroaal (2 6 3 ), [37], 344 PiperoníJico, alcohol {58), 342 PiperonUico, ácido [229], 338 Pjvalaldehldo (7 5 ) , 343 Píválico, ácido (164), (35], 334,
335Polaritnetro, 69 Polaridad, 86Polialogenadoí, derivados de hidrocar
buro* no bencenoides, derivados de, 304
tabla de, 378 Polioxímctileno, solubilidad de, 89 Populina [180 d ] , 364 Potasio, solución de permanganato de,
166Prehiútetio (20 4 ), 380 Problemas* 419
solución de, 407 t-P ro lina [222 d.j, 362 iU Prolm a (203 d.\, 362 Propandiriol-i,3 (1 6 9 ), 3891-PropansuUonamida [521, ^002-PropanBulíonatnida [60), 400 1‘PropansulfomIo, cloruro de (67 /9
.mm ), 4022-PropansulíoniIo, cloruro «le (61/9
m m ), 402 Proporúlbenccno (1 ? ? ) , 384 Propiólíco, á d d o (144 d .} , 334 Propionaldebldo (5 0 ) , 343 Ptopíonam ida [79], 345 Propionanilida [103], 346 Propiónico, ácido (1 4 0 ), 334 Propiónico, anhídrido (163), 331. Propíonitrilo (9 ? ), 390 Propionilo, brom uro de (103), 331. Propionilo, cloruro de (80 ), 331 Propionilo, fluoruto de (45 ), 331 Propionilo, yoduro d e (1 2 7 ), 331 Propiofenona (218), 386
m

ín d ic e a lfa b é t ic o 4 7 5
W-n-PropUacetaiiiUda [50], 345 «-Propitak acetato d r (101), 365
solubilidad de, 90 n-Propllice, akokol (9 7 ), 359 n-Propilaniina (49), 349 r. Propilbenceno (158), 380 n-Proptfo,* benzoato de (230), 366 n-Prcpilo, bromuro de (71), 374 rt-Prbpilo, n-feutirato (145), 345 n-Propilo, n-caproato de (186), 366 n*Prop¡lo, carbamato de [60], 345 n -Propilo, carbonato de (168), 365 n-Prop»lot cloruro de (46 ), 373 n-Prapilo, dorofcrm iato de (113), 355
' rt-Prop¡Icidohexano (157), 382 Propileoo, bromuro d e (142), 375 Propileno, cloruro d e (98), 373 Propilenerlicol (188), 340 Propileno, óxido de (35 ), 369 n-PropIlico, ¿ter (90 ), 369 n-Propilo, fcrmiaio de (81), 365 «'Propíl-a-furfuríiíoo, éter (168), 370 u-Proptlo, furoato de (211), 366 n-Propilo, yoduro d e (102), 374 Tj-Pjvpilo, iewdinato d e (221), 366 n*Propllmcrcaptano (67), 389 ft-Propíl-2-cvaítllíco, í tc r [40], 372 n-Propilo, nitrato de (110), 368 fi-Propilo, nitrito de (4 4 ) , 368 «•Propilo, oxalato de (213), 366 Í-P ropüícnol (232), [22], 397 n-PropUo, propiaruto de (122), 365 n-Propilo, tóJicOato de (239), 356
Propilo, succinato d e (246), 366 m -Propiltolueno (182), 380 ¿-Propil tolueno (184), 380 r-Propilo , n-valeríanato de (167), 365 Protocateealdehido (154], 344 Protocatéquico, ácido fl9 4 d.], S38, Protones, resonancia nwigaéuca de,
214Pseudocumeno ■ (168), 380 Pscudocumcnol [71], 398 Pseudocuwidina (235), [68}, 352 Pseudosacarina, cloruro de, 365 Pulcgona (224), :186
Q
Quelidónico, Ácido [262], 338 Q u ina lde ti» (247), 359 Q uinhidrona [171J, 388 Q uinolcína (2 3 9 ), 359^
R
R a íin o » (80 d., 119 d.], 364 Rast, peso» moleculares con alcanfor
je#ún, 72 Reíractómctro, 63-66 Reacciones de nutitucíón, teoría de,
170Resolución de problemas estructúra
le., 407Resonancia roasnóüca nuclear, 214 RcBorcinol (110], 399 Rcrorrinol, diacetato de (2 7 8 ), 367 Rcsorcinol, díbensoato d e [117), 368 RciotcÍDol,_éter dietílico del (235),37J Rcsordnol, éter dírrjetllico del (214),
371Reaordnol, í t e r monoroccilico d d
(2 4 3 ), 397 ft-RtíorrlIico, ic id o [213], 336 Rcteno {98}, 381 L.Rhamnosa [105 d.j, 364 n-Ribosa [95 d.}, 364 Rotación óptica, 68
m agnética, 71
S
Sacarina [220], 348 Safrol (232), 371 Sacarosa [185 d.j, 364 Saücina [201 d j , 364 SaHalaldehído (196), 343 Salicüamida [139J, 346 SalicQjeo, t ó á o [157], 337 •Saponificación, equivalentes de, 292 Sarcosina [210 dL), 362 Schotten-Baumann, reacción de, 129 Sehácico, Acido [133], 336
solubilidad de, 83 Sem ícarbadda [96], 345, 353 SemicarbaJiotias, 275
obtención de, 149 Wí-Serina [22B], 3G?Silvcstreno (176), 384 $»tosten*l (137], 342 Sodio, 18»Sodio, solución de disulGto d e , 181 Sodio, solución de hidróxido de, 182 Sodio, hipoyodito de, 154 Sodio, yoduro, solución en acetona,
188Solubilidad, 81
compútalos aníótexoj, 98 gráfica, 82

4 7 6 btdte# a lfa b é t ic o
Solubilidad, definición de, 61 dicnrbox2ícoi, ácidos, 88
tabla dcj $8 efecto d e cadenas arborescentes so
bre, 90 efecto» dectrónicos, 91 efecto» estáñeos, 93 ejercido® de laboratorio en, 83, 66 e n ácido sulfúríoo concentrado, 98 en ácido clorhídrica diluido, 95 en solución d ilu id a de bicarbonato
de sodio, 96 eo solución diluida de hidróxido de
sodio, 96 e n agua, 94f u t r a s intermotecutaxcs en, 88 generalizado n « , 66 polaridad, 86relación del peso molecular, con, 89
ScJ vóUí Íi , 170-179< + ) Sorbttol [98], 342 Subérico, ácido (140), 337
solubilidad de, 88 Sustancias desconocida», cantidades de,
U , 443 pureza, I I reportes de, 24
Sucdoam ida (250], 348 Sucdnaniiida [226J, 343 Sucdnam tda (mono), (157], 347 Succuutnilida (m ono), (148], 347 Succliúco, á d d o [188), 337
solubilidad de, 88 Succíoico, anhídrido (120?, 333 S ucdoim ida [125), 346 SucdaonitrU o (267 d .) , [54], 390 Su c d nito, cloruro de (190 d .) , (16),
331, 332 SuIJlnicos, ácidos, 321
tales de, 321 Sullonarnidu, derivados de, 322
preparación de, 283, 299, 304, 325 tabla de, 400
Sultánicos, derivados de áddos, 324 «alca de Jstoluidina, 326
Sulíoniío, derivado» de cloruro de, 327 p rep arad ó n de, 298, 303, 325 tab la de, 402 -s
Sulfúrico fumante, ácido, 190 SÜveitrcno (176), 384
T •
¿-Tartárico, ácido [169J, 337 {♦Tartárico, áddo [169], 337
¿/-Tartárico, ácido [204], 338 j/w o-Tartárico, ácido [140), 337 Tereftalaldefcido (245), {116], 344 T erd tan iü d a [237], 343 T ereftilico, ácido (300 subí.], 338 f-Terfenilo [213), 381 d¿-*-Terpiaeol (221), 340 a-Terpíneol [35], 341 Tcrpinol [102], 342 Terpinol, hidrato de [117], S4-21 .íjijS-Tetrabrom obenceno [180], 377 Tetrabiotno-o-crwol [210], 399 r-Tetrabrom oetano (200 d . ) , 379 Tetrabrvmoitálico, anhídrido [275],
333\,2,4,5-Tctraclorobenccno (l?8], 234,
3771,2,3,4-Tetracloiobenceno [46], 376 1,2.3,5’Tetradorobenceno [51], 376 l , l ;'l,2 -T ctm lo rae tan o [131], 378 s-Tetríicloroeiano (1 4 7 ), 378 Tetracloroetileno (121), 378 2,3^,6-TetradorobÍdroquinona [237],
399TetradoroftáKco, á d d o (250 d.]> 333 Tetracloroí tífico, anhídrido (256], 335 Tetradecano (254), 382Tctractifercjjlicol, é te r dj metílico del
(266), 3701,2,3,6-TetrahidrobeniaIdehído (165),
343Tetrahidroíurano (6 5 ), 369 Tetrahidroíurfurilo, acetato de 0 9 4 } ,
366Tétrahidroíurfurílico, alcohol (178),
340 'TetTabidroijóquínoIdnft (233), 355 «r -T etrahidro - a - naft¡lamina ( 2 7 5 ) ,
.350er- Tetrafiklro - 0 - naí litamina ( 2 7 5 ) ,
{38], 350, 351 T ttrahidropirano (8 8 ), 369 Tetrahidnxjuinolcína (2 5 0 ), (20), 356 A'-Tetrahidrotolueno ( I I I ) , 383 A’-TetrabtdrotoJucno (105), 383 A '-Tetrahidm olueno (103), 383 Tetrahidrosilvano (7 9 ), 369 Tetrayodofcálico, anhídrido [318], 333 Tetra! ina (206), 3801,2,3,4-Tetrametflbcncena (205), 380 /-Tetram etildibrom oetano [169], 379 í-Tetram ctildicloroetano [160], 379 Tetram etiletilehó (72)* 383 Tetxanitrometano (126), [13], 391,
Jn d íc o .o J fc b é lic o 4 7 7
i-Tetrai?níletano (211], 381 Teobromina [337 subí.], 348 Teofilina [264], 348 T ienilm ercaptano (166), 389 Tjoacédco, ád d o (93), 334 Tiobenzoico, ácido [24], 335 w-TíocrcioI (195), 389 O-Tiocrcsol (194), 389
-Tioeresol (1 9 5 ), [43], 389 -Tionaftol (SIL 309
Tiofenol (169), 389 •Tiosalícllico, ácido (163], 337 Tiourca [182], 347v-Tirosina [344 d.], 363 ¿¿-Tirowna (318), 363 D-Treonína (253 d.), 362 L-Treonina [253 d.j, 362 rfJ-Troonina (235 d.], 362 P-Tuyona (2 0 2 ), 386 Tiraol (50], 398 Túno^uinona (232), (45], 387 Tiroilo, acetato de (244), 366 Timilo, benzoato de [33], 367 T il ic o , ácido (198), [64], 334, 335 o-Tolidtna [J29], 354 Tollens, reactivo de, 192’ m-Toluafdchido (199)', 343. o-Tolualdchido (200), 343 í-ToIualdcb(do (204), 343 o-Toluamida [140], 346 ¿-Toluamida (160], 347 ' ©‘Toluanilida 1125], 346 m -Toluanilida (126], 346; ^T aíuanJlida (145), 346 Tolueno (111), 380 rn-Toluetuulfonanvda [108], 400 0-ToIucnsulfonarnida [153J, 400 /i-Tolueníulfonamjda [137), 400 /)'To!uen9ulíonamid«, 283 m-Tolucnsulfonilo, cloruro de [12], 402 O-Toluensuffonilo, cloruro de [10], 402 /j-Toluensulíotúlo, d o ry ro de (69], 403 Toluhidroquinona (124), 399 m -Tolúko, á d d o [110], 336 a-Tolúico, ád d o (102], 336 p-To!úico, ád d o (177], 337 m-Tolúico, anhídrido [71), 333 o-Tolúico, anhídrido [39), 332 Í-T olúico, anhídrido [95], 333 p-Toluididaí, 258, 301 fn-Toluidina (203), 349 o*Tolu¡d¡na (199), 349 p-Toluidioa (200),.(45], 351 m-Tolupitrilo (212), 390 o-ToIunitrilo (20 5 ), 390
Í-T olunitrilo (217), [38L 390 • p-ToJuquinon* [68J, 367 o-(p*ToJuií)-benzoico, ácido [130 hi
drato]. [146), 337 m-Tolilairbm ol (217), 340 o-ToUlcarbínol [36], 341 ¿-ToUlcarbind [60], 342 /5*ToliIhidra¿na [65), 352 ¿-ToJiimercapUno (195), [43], 389 ti-fi-Tolll-a-naftilanuna [79], 357 Af- í 'T o lil“3-fiaídlamina [103], 357 m-Tolilurca [142], 3460-Tolfturc* [192], 347 Í-T oltlurea (181), 347 Trialilam ína (155), 358 Tri-fi-amilaroina (257), 359 Tribencilamina (380), [91], 3602,4,6-Tribroraoanilina (300), (119),
353.\ 2,4,6-Tribromoaaisol (87), 372
1,2,3-Tribromobenccno (87), 377 1,2,4‘Tribromobcnceno [44], 3761.3.5-Trtbromobcnceno [120], 3772.4.6-Tribromobencenuilfotumida [220
d.}, 4012,4,6*Tribrümobeacensulfoailo dom ro
de [60], 4031.1.2-Tribromoetano (189), 379 2,2,2*Tribromoeti)ico, alcohol (60], 342 Tribrom on’MfometsnO [10], 3922.4.6-Tríbrofúonitrobcnceno [125], 3952.4.5-Tríbromofenetoí [73], 3722.4.6-Tribromoíenetol (72J, 3722.4.6-Tribromofenol (95), 398 1,2^-Tribrom opropano (219),, 379 Tri-n-butílantina ( 2 U ) , 358 Tricarbalflico, ácido [166], 337 a.ct.a-Trídgroacctaroidi [140], 346 Tridorcacetaailida [97], 345 Tridoroacédco, ácido [57], 335 Tridoroacetilo, donuro de (115), 3312.4.6-Tricloroanilina (262), [77], 3522.4.6-TridoroanUol [60], 3721.2.3-Triclorobenceno [52], 3761.2.4-Triclorobcnceno (213), 3751.3.5-Triclorobenceno [63], 377 2,S>4'Triclorcbcnccnsuliíonam3da [226
d.J, 4012,4^-Triclorobcnceniulforainjda •
[ > 200], 4012.4.6-Tricloróbcnceníulfonarrúda [212
d.J, 4012,3,4-TridoroberKünsuIíontlo, : cloruro
de [65], 403

4 7 6 I n d i a a l f a b é t i c o
2,4,5‘TridorobencenEuIíonilo, c lo ruro de [54], 402
2,4,6'Tridorcibencemulfonilo, cloruro de ^OJ, 4 *
1.2.3-Tridorobatano (169), 379 1,1,1 -Tricloro-2,2-bis (¿•elorofenil) -
etano fl>DTJ, [108], 3791,1,1-TrícIoroetono (74 ), 378 [,1,2-Tridoroetano (114), 378 Tridoroetílico, alcohot [19}, 341 TricloroetUtivo (90), 378 Tridorotictico, áddo [124], 3362,4,6-Triclorofenetol [43], 3722.4.5-Tricloroí«no) [68], 3932.4.6-Triclorofcnol [67], 3931.2.3-Tridoroprapano (155), 373 rj'Trkkcano (236), 392 Tridccauoieo, ácido {44}, 335 TríetíJamina (B9), 358I*3,5-Trietilbcaccno (21 8 ), 380 Trietilcarbínol (142), 339 Triflucroacérieo, anhídrido (39 ), 331 I,?,4-Trihidro:dbcnC«W [140], 399 TriiioarcdUjmna (245), 359 Trimfsico, áddo [350], 333 Trimrtilaimnn (3 ), 3581.2.3-TriroetilbeDceiia {176), 3802.4.5-Tnraetübencensu] fonarmda,
[181], 4002,14,6‘TrimeÜ bcncentulíonamtda,
[142], 4002.4.5-TñmetiJbencerisulfonilo, cloruro
dg [611 4032,4^-TricneiIlbencensulfonilo, cloruro • de [56], 4032.4.6-Trimetílben20ic0, á d d o [155],
3371.1.3-TrtnjetilcWopcntano (105), 382 Ttttoétíl«io, bl^muro de (165), 379 •TrimítSenbronjhidrina (176 <!.), 340 •Trimetílcno, doruro de (125), 378 Triraefiléno, dorobromuro de {143), v - . 378TrinietilcrtClortiídrim {161 d ) , 339 Triractíleno, cianuro de (28 6 ), 390 Triratrifengliccl (216), 340 TnmetilengNcol, diacetato (2 f0 ) . 366 Trimetiletikna <38), 3832.2.4-TrittwtilpentaíK) (9 9 ), 382 2j4,6*TfimetiIfenol [65], 3982.4.6-Tiinitwanilina [190], 3542.4.6-TxÍQÍtroanisol [68], 393 2>4,6'Trinitirob«izanüda [264 d.], 348 l^J-Trioitxobenceno [122], 396
2.4.6-Trin'trobenz0it0, á d d o [220 d«],338
Trinítrom etano [15], 3922.4.6-Trinitrofenctoí [78]. 394 2,4>&*Trinitrotoluerio [82], 3992.4.5-Trinilro-m'XÍIfttio [90], 394 TrifenUamítu [127], 361 Trifenilcarbínol [162], 3421,1,2-Trifeni letileno [73?, 354 a-Trifenilgu&nidina [145], 346 Trifenihnctano {92], 381 Tri-a^propílamina (153), 358 Tritilo, éteres de, 265 dí-Trópico, ácido (117], 336i.-Tript¿fano [289 d.]> 3633 jS-Triyodobenroato*. 2652.4.6-Tri>-odofeuol [159], 399
OUltravioleta, espectroscopia, 22'/
Aminas, 240compuestos carbonilicos, 239 compuestos «Morvcfurícior.s'eJ, 230,
239catructuTSi, compuesto* modelo en, seotnelrfa molecular, 235 definas no corrugadas, 239 principia de adilividad, 233 sistemas conjugados, 237
n -U n d « an o (194). 382 •' Undícanoico, ácido (275 d ) , [29],
334, 335 Uhdeceiw-1 (19 3 ), 384 UndecilénicOj ácido (29 5 ), [24], 334,
335Undecüíco, alcohol (2 4 3 ), 341 Undccilo, cloruro de [241], 374 U rea [132], 346 Urctanos, 268, 321 Urico, ácido (d.j, 348
V
Ts-Vaterialdehído (103), 343 n-V&leriananílida [63], 345 n-Valeriánxco, ácido (1 8 6 ), 334
solubilidad de, 90 n-Valeriánico, anhídrido (215), 332 y-Valerilanona (14 1 ). (2 0 7 ), 366,
* rt-Valeronitrilo (1 4 1 ), 390 n-Valeriañilo, cloruro de (127), 33). o*Val)iu. [315 <L]), 383 L 'Valina [315 d ], 363

Ind ico q lFahático 4 7 9 '
¿I-VaJina [296 d.}, 363 VaimUieo, ¿d d o [207], 338 VainílJídinacetcna {130), 388 V ainillina (285 d .) , [80], 344 VeCKKH, participación de grupos, 176 VeriíricO; ácido [181], 33?Verátrico, aldehido (285), [58], 344 Veratrol (206), (21], 371, 372 Vínilacetanilídá [73J, 345 Virnlo, acetato de [72), 365 VimlíacétJcn, ád d o (169), 334 V inila, benzoato de (203), 366 Vinilo, bromuro de (16), 373 Vinilo, yeduro de (56), 373
X«
•Xantítia (360), 343 X an to tu (350), [173], 3^8 ¿\’-Xantitsu]fonamída£, 3234-Xenilmcrcaptano [111], 389 m.Xil<no (139), 380o-Xileno (14 2 ), 380 ¿ X ile n o (137), 380 oOtilosa [545 d ] , 364
Y
TK-Yodonítrabencero J38J, 392 4*Yodo-3-n\trotolueno (55], 3932-Yodopcntano (142), 374 ij-Yodoienetol (245), 371 *-Y«dofet»toI [27], 372 m-Yodofenol [40j, 397o-Yodofenol [43], 398 Í-Yodofenol ([94], 396 P'Yod&propiánico, Acido [82], 335 rrt-Yodotoluer»o (204) > 375o-Yodotolueno (211), 375 p-Yodotolueno (211), [35), 375, 376 YodMdricO, ácido, 146 Yodoacetatmda [95), 345 ct-Yod oacetanilid a (L44], 346 p-Yodoacetanilída (182), 347
Yodoacítióo, ácido (84), 335 m-Yodoanilina [27], 351 o-Yodoanilina, (56], 351 p-Yodoanilina [62¿ 352 o-Yodoaniaol (240), 371 YodoariJo, dicloruros de, 303 o-Yodobcuialdchído [37], 344 «i-Yodob*nza]dchída [57], 344 }n~Yodob<&zaxmck [186], 347 O-Yodobúnzamida [183], 347 ¿-Yodobenzunída [217], 348 O-Yodobeaianilida [141 ¿ 346 ¿ ‘Yodobemanilida [210], 348 . Yodofcenceno (188), 375 m-Yodúhenzoico, ácido [187], 337 «•Yodobenroico, áddo [162], 337 /»-Yodobezuoico, ád d o [265], 3380-Yodobenzoñítrüo (55], 390 ^-Yodobcntóilo, bromuro de [60], 333 ^-Yodobcnccnsulíonamida [2CHS], 401 Yodoíormo [1191 379
prueba, 1541-Yodonafu1«no (305), 376 fNYodonaiíaleno [55], 376l-Yodo-4-naíUtetisulfonamida [202],
4011-Yodo*5*Naft3lenwUonamida [236],
4012-Yodo-l*naítalensul/onanúda 1154J,
4008-Yodc*l-nafta!cntulíouamida (Í87),
40)UYodo-4-naftoleniulfcniIo, do m ro de
[121], 404l-Yodo-5-naftaleníulfonilo, cloruro de
[113), 404 2'Yodo-l-naftalenwlíoiúIo, cloruro de
[110], 404¿uYodo-l-naftalensuTíonilo, cloruro de
[I15J, 404 -
Z
Zdsef, método de, para alcoxilos, 146

















![Teologia sistematica [smalling]](https://static.fdocuments.es/doc/165x107/549cd2e7b479599b318b4857/teologia-sistematica-smalling.jpg)
