
Manual de Practicas Cinetica y Catalisis
88
1 MANUAL DE PRÁCTICAS DE CINÉTICA Y CATÁLISIS Miguel Ávila Jiménez Elpidio Corral López Lilia Fernández Sánchez Hugo E. Solís Correa UNIVERSIDAD AUTÓNOMA METROPOLITANA UNIDAD AZCAPOTZALCO División de Ciencias Básicas e Ingeniería Departamento de Ciencias Básicas Coordenada de reacción Energía Complejo Activado ‡ Productos Reactivos
-
Upload
al-harith-as-sama -
Category
Documents
-
view
552 -
download
16
Transcript of Manual de Practicas Cinetica y Catalisis

1
MANUAL DE PRÁCTICAS DE CINÉTICA Y CATÁLISIS
Miguel Ávila JiménezElpidio Corral López
Lilia Fernández SánchezHugo E. Solís Correa
UNIVERSIDAD AUTÓNOMA METROPOLITANAUNIDAD AZCAPOTZALCO
División de Ciencias Básicas e IngenieríaDepartamento de Ciencias Básicas
Coordenada de reacción
Energía Complejo Activado ‡
Productos
Reactivos

2


Manual de prácticas de Cinética y Catálisis
Miguel Ávila JiménezElpidio Corral López
Lilia Fernández SánchezHugo E. Solís Correa
1ª. edición, 2005
UNIVERSIDAD AUTÓNOMA METROPOLITANAUNIDAD AZCAPOTZALCO
División de Ciencias Básicas e IngenieríaDepartamento de Ciencias Básicas

La ciencia no es más que una exploración del intrincado, sutil e imponente universo que habitamos. Quienes la practican conocen, aunque sólo sea ocasionalmente, aquel raro tipo de felicidad que Sócrates definiera como el mayor de los placeres humanos y, además, es un placer transferible.
Carl Sagan

i
PRESENTACIÓN
El manual de prácticas de cinética y catálisis, consta de ocho prácticas y un seminario que corresponden al programa de la uea experimental Laboratorio de Cinética y Catálisis de la carrera de Ingeniería Química.
Seis de las prácticas fueron escogidas del Banco de prácticas seleccionadas de cinética-química y catálisis para la carrera de ingeniería química.1 Se consultaron las fuentes originales. Las prácticas fueron probadas, modificadas de acuerdo a los objetivos del curso y adaptadas a los recursos disponibles para su mejor realización.
El criterio de selección fue la experimentación acorde con el concepto de Química Verde, que consiste en trabajar con el menor riesgo posible a la salud de los experimentadores y procurando al mismo tiempo proteger al medio ambiente. Las ocho prácticas cumplen con los requisitos anteriores, por lo que el manual tiene un enfoque ecológico. En cada práctica se incluye el tratamiento o disposición de los residuos. El enfoque ecologista, no se contrapone con la calidad del experimento ni de sus fundamentos.
La práctica número ocho, cinética de una reacción catalizada, no corresponde al Banco de prácticas, se implementa con la técnica de la Microescala2, la cual es una pedagogía acorde con la protección del medio ambiente y la cultura del ahorro. Además es innovadora en el uso de nuevas tecnologías, utiliza en la experimentación un sensor de presión calibrado previamente, lo que proporciona una mejor confiabilidad en los datos.
Deseamos agradecer a Angélica María Valdés Espinosa, Luis Alberto Pacheco Vela, Cristina Vilchis Pacheco, Karina Uribe Flores y Efrén Marco Antonio Camargo Fernández, el apoyo que nos brindaron en la prueba de algunas de las prácticas.
Los Autores
1 Icela D. Barceló Q., Hugo E. Solís C. y Alicia Cid Reborido. Banco de prácticas seleccionadas de cinética-
química y catálisis para la carrera de ingeniería química. Universidad Autónoma Metropolitana, Unidad Azcapotzalco, División de Ciencias Básicas e Ingeniería, Departamento de Ciencias Básicas.
2 Para una revisión de la Microescala, consultar: Rosa Ma. Mainero M., ¿Por qué Microescala?, Educ. Quím.,8[3], pp 166-167, 1997.

ii

iii
NORMAS DE SEGURIDAD
Medidas preventivas Durante la experimentación usar bata, guantes y lentes de seguridad (goggles) no comer durante la experimentación no fumar leer las etiquetas de los frascos de reactivos antes de abrirlos y en caso necesario abrirlos
en la campana de extracción documentarse en la literatura recomendada acerca de las propiedades de las sustancias
empleadas y las precauciones que deben observarse durante su manipulación no pruebe, no olfatee, ni toque directamente con las manos los reactivos químicos no usar la mano como tapón para agitar las soluciones no usar pupilentes durante la experimentación ya que los vapores de las sustancias
químicas irritantes pueden introducirse entre el ojo y el pupilente tener a la mano franela o papel secante para mantener limpia la mesa de trabajo de
sustancias derramadas usar el material de vidrio perfectamente lavado, enjuagado con agua destilada y seco e
igualmente lavarlo después de usarlo usar zapatos cómodos, cerrados y con suela antiderrapante en caso de cabello largo, recogerlo mantener la mesa de trabajo libre de objetos como mochilas, suéteres, chamarras y libros
que no sean la bitácora y el manual de prácticas evitar que mochilas, portafolios, bancos o cualquier otro objeto obstruyan el paso evitar bromas y distracciones que puedan provocar un accidente
Medidas correctivas En caso de quemaduras con ácidos o bases, tanto en ojos como en piel lavar con
abundante agua la zona afectada. En los ojos utilizar alguna de las siguientes soluciones Lav-Often, solución salina, suero o solución glucosada. En caso de requerirlo acudir al médico (Edif. E, cubículo E – 010)
en el caso de ingerir ácidos o álcalis, no provocar el vómito ni practicar el lavado gástrico, neutralizar con un antiácido a base de alúmina (hidróxido de aluminio), como el Melox. Se recomienda tomar leche como medida urgente
en el caso de ingerir un veneno o barbitúrico, inmediatamente provocar el vómito y practicar el lavado gástrico. Acudir al médico
en el caso de una cortadura, lavar el área afectada con agua y jabón, contener la hemorragia con la mano o con una gasa limpia y acudir al servicio médico
en caso de quemadura con flama u objeto caliente, enfriar la zona afectada con abundante agua o de ser posible con hielo hasta que cese el dolor. Aplicar pomada para quemaduras. Si la quemadura lo amerita, dirigirse al servicio médico

iv
el laboratorio cuenta con un botiquín y un extinguidor de polvo químico, localizar su ubicación. El técnico responsable del Laboratorio deberá informar a los usuarios acerca de los dispositivos de higiene y seguridad disponibles, en la primera sesión de trabajo.
Bibliografía recomendada para medidas de seguridad, primeros auxilios y manejo dereactivos. Catálogo de reactivos de Merck (Index Merck), 2001 Gessner G. Hawley. Diccionario de Química, Ediciones Omega, S. A., 1975 Improving Safety in the Chemical Laboratory: A Practical Guide; Second Edition, Edited
by Jay A. Young Green, M. E. and Turk, A. Safety in Working with Chemicals, Macmillan Publishing Co.,
Inc. 1978.

v
Bibliografía general
Barceló, I.D; Solís, H.E y Cid, A. Guía y antología de fisicoquímica. 1ª ed. Universidad Autónoma Metropolitana Azcapotzalco. 1993.
Daniels, F; Alberty R. A. Curso de fisicoquímica experimental. Mc Graw Hill. 1972.
Fogler, H.S. Elementos de ingeniería de las reacciones químicas. 3ª ed. Pearson Educación, 2001
Harris, G.M. Cinética química. 1a ed. Ed Reverté. 1973.
Laidler, K.J; Meiser, J.H. Fisicoquímica. 1a. ed. CECSA, 1997.
Levenspiel, O. Ingeniería de las reacciones químicas. 2ª ed. Ed Reverté, 1998
Logan, S.R. Fundamentos de cinética química. 1ª ed. Addison Wesley, 2000
Urquiza, M. Experimentos de fisicoquímica. Ed. Limusa. 1969.
Casado, J; López Quintella, M.A; Lorenzo Barral, F.M. The initial rate method in chemical kinetics. J. Chem. Educ., 63(5), 450, 1986.
Corsaro, G. A colorimetric chemical kinetics experiment. J. Chem. Educ., 41(1), 48, 1964.
Elias, H; Zipp, A.P. The study of a simple redox reaction as an experimental approach to chemical kinetics. J. Chem. Educ., 65(8), 737, 1988.
Lambert, J.L; Fina, G.T. Iodine clock reaction mechanisms. J. Chem. Educ., 61(12), 1037, 1984.

vi

vii
MANUAL DE PRÁCTICAS DE CINÉTICA Y CATÁLISIS
INDICE
Presentación i
Normas de seguridad ii
Bibliografía general v
Índice vii
PRÁCTICA Nο. 1 Cinética de saponificación del acetato de etilo. Análisis por el Método Diferencial
1
PRÁCTICA Nο. 2 Determinación de la constante de velocidad y el orden, dela reacción entre el yoduro I – y el persulfato S 2O 8
2 –. Análisis por el Método Integral
9
PRÁCTICA Nο. 3 Efecto del cambio de la concentración en la velocidad de reacción. Método de las velocidades iniciales
15
PRÁCTICA Nο. 4 Obtención de los parámetros termodinámicos del estado de transición de la reacción del yoduro I– con el persulfato S2O8
2–
25
PRÁCTICA Nο. 5 Cinética de una reacción redox a través de un observable 31
PRÁCTICA Nο. 6 Cinética de una reacción química, seguida colorimétricamente para verificar un mecanismo de reacción
41
PRÁCTICA Nο. 7 Cinética de una reacción de pseudoprimer orden seguida por el método colorimétrico
49
PRÁCTICA Nο. 8 Cinética de una reacción catalizada (en microescala) 55
PRÁCTICA Nο. 9 Obtención de los parámetros de la isoterma de adsorción de Langmuir. Cinética en fase heterogénea
63

viii

1
PRÁCTICA Nο. 1
CINÉTICA DE LA SAPONIFICACIÓN DEL ACETATO DE ETILO. ANÁLISIS POR EL MÉTODO DIFERENCIAL
OBJETIVO GENERALDeterminar experimentalmente la constante de velocidad y el orden, de una reacción monodireccional, homogénea en fase líquida, aplicando el método diferencial
OBJETIVOS ESPECÍFICOS Medir el consumo de reactivos durante la reacción de saponificación del acetato de etilo
por el método volumétrico
obtener la gráfica de concentración de reactivo contra tiempo – obtener la velocidad media r determinar el orden de reacción y la constante de velocidad de la ecuación de velocidad
linearizada Concluir a partir del coeficiente de regresión R2, si los datos obtenidos son confiables
FUNDAMENTOS TEÓRICOSEl objetivo de la termodinámica en las reacciones químicas, es el predecir en que dirección se desplazarán éstas en forma espontánea ∆ G < 0, mientras que la cinética química decide en que tiempo transcurrirán dichas reacciones.
REACCIÓN ENERGÍA DE GIBBS TIEMPO
La descomposición del agua en hidrógeno y oxígeno es una reacción no espontánea
∆ G > 0 ____
La reacción entre el dióxido de carbono, agua y luz para producir almidón (fotosíntesis) es una reacción espontánea que se realiza en un tiempo muy corto
∆ G < 0 10 ¯12 seg
La reacción de saponificación del acetato de etilo, es una reacción espontánea
∆ G < 0 1 hora
La reacción entre el cemento, el agua y el dióxido de carbono para dar dureza temprana al cemento es una reacción de algunos días
∆ G < 0 7 días
La conversión de grafito en diamante a presión, necesita millones de años para completarse
∆ G < 0 Millones de años

2
CR
t
CP
t
La cinética química estudia la velocidad (r) de las reacciones químicas (como la variación de la concentración de los reactivos o los productos en el tiempo) y los factores que la afectan.Antes de que los reactivos se consuman o alcancen el equilibrio químico, su concentración disminuirá con el tiempo, Figura 1.1a, en tanto que los productos comienzan a formarse e incrementan su concentración, Figura 1.1b
r R p P
(a) (b)Figura 1.1
(a) Variación en la concentración de reactivo (b) Variación en la concentración de producto
La velocidad instantánea en una reacción es el cambio de concentración respecto al tiempo, en un tiempo en particular, un punto en la curva CR vs. t. Un signo menos en la ecuación diferencial de velocidad indica que se consume el reactivo.
1 dCR 1 dCPVelocidad instantánea (r) = − r dt
=p dt
1.1
En la curva C vs. t, la velocidad instantánea se obtiene de la recta tangente que toca la curva en el punto de interés Figura 1.2.
Figura 1.2
La velocidad media r es el cambio de concentración CR en un intervalo de tiempo t
CR CPVelocidad media r = − t
= t
1.2
·r = − dCR/dt
CR
t
CP
t
r = dCP /dt
·

3
r = CP / tr = −CR/ t
CR
t
CP
t
En la curva C vs. t, la velocidad media se obtiene de la recta secante que toca la curva en dos puntos de interés Figura 1.3.
Figura 1.3
Ecuación de velocidad. La Ley de Guldberg y Waage relaciona la velocidad r con la concentración solo de los reactivos, siendo el factor de proporcionalidad la constante de velocidad k o coeficiente cinético en caso de reacción no elemental, Ecuación 1.3. Para la ecuación química
aA + bB + …. → qQ + sS + ….
Las ecuaciones de velocidad son:
1 dCA 1 dCB 1 dCQ 1 dCSVelocidad (r) = –
a dt= –
b dt + ... = q dt
= s dt + ...
r = kC A C
B .. 1.3
Donde , ... son los ordenes parciales de reacción en A, B, ...
Una reacción es elemental si la transformación química ocurre en una sola etapa o encuentro de las moléculas de los reactivos. Las reacciones no elementales ocurren en mas de una etapa, en procesos elementales sucesivos.
Si la reacción es elemental o se comporta como tal, los ordenes coinciden con los coeficientes estequiométricos a y b de la reacción anterior y a la constante de proporcionalidad se le llama constante de velocidad.
= a y = b
r = k CaA C
bB ... 1.4
Para el caso particular de una reacción bimolecular donde las concentraciones de los reactivos son iguales CA = CB se tiene
r = k CAa+b
1.5

4
Si el orden global de la reacción n es la suma de los órdenes parciales entonces n = a + b y
r = k CAn
1.6
La linearización de la ecuación de velocidad, proporciona una ecuación en donde la pendiente es el orden de reacción global n y la ordenada en el origen es el logaritmo natural de laconstante de velocidad k. Para la velocidad media absoluta r
ln r = ln k + n ln CA 1.7
En donde CA es la concentración media o promedio entre el intervalo de tiempo considerado
t = t final – t inicial
CA final + CA inicialCA =2
La representación gráfica de la ecuación 1.7 se muestra en la siguiente Figura 1.4
Los puntos en la gráfica 1.4 representan los datos experimentales, la línea entre ellos es la recta resultante de ajustar los datos con cuadrados mínimos (regresión lineal). En la ecuación de esta línea, la pendiente es el orden de reacción global y la ordenada en el origen, el logaritmo natural de la constante de velocidad. El coeficiente de correlación lineal R indica que tan próximos se encuentran los puntos experimentales a la línea de tendencia, un valor entre 0.99 y 1 es aceptable para considerar que la ecuación ajustada concuerda con la tendencia de los datos y que los errores en el experimento son menores.
Pendiente = n
Figura 1.4
}ln k
ln r |
ln CA

5
DESARROLLO EXPERIMENTAL
Los residuos de la titulación y el sobrante de la mezcla acetato de etilo-solución de hidróxido de sodio, se colectan en un vaso de precipitados de 1 L, se calientan ligeramente durante 10 minutos para completar la reacción de saponificación y luego se desechan en la tarja.
PROCEDIMIENTOLos matraces y las pipetas deben estar perfectamente secos, para evitar la reacción del hidróxido de sodio con el dióxido de carbono disuelto en el agua destilada.
Llenar la bureta con la solución de HCl, no sin antes eliminar el aire de la punta y colocarla en el soporte universal. Registrar la concentración del ácido Tabla 1.1
medir con la pipeta volumétrica seca de 25 mL la solución de hidróxido de sodio y verter totalmente en un matraz Erlenmeyer de 250 mL, perfectamente seco, verificar si las pipetas son de vaciado total (con la leyenda PE)
Agregar 3 gotas de fenolftaleína, la solución se tornará rosa medir con la otra pipeta volumétrica PE, 25 mL de solución de acetato de etilo y verter
totalmente en otro matraz Erlenmeyer de 250 mL
MATERIAL REACTIVOS
2 matraces Erlenmeyer de 250 mL solución acuosa de acetato de Etilo
9 matraces Erlenmeyer de 125 mL 0.2 M (2mL/L)
2 pipetas volumétricas de 25 mL PE, solución de NaOH 0.2 M
vaciado total solución de HCl 0.1 M
1 pipeta volumétrica de 5 mL PE indicador fenolftaleína
1 vaso de precipitados de 1 L para residuos (uso común)
1 termómetro de 0-100ºC
1 bureta graduada de 50 mL
– agua destilada la cual ha sido hervida y dejada enfriar bien tapada, para que no se disuelva el CO2 atmosférico.
1 soporte universal con pinzas para bureta
propipetas
o jeringas con manguera
cronómetro
NOTA: Todas las soluciones deben ser preparadas con agua hervida y dejada
enfriar bien tapada, para que no se disuelva el CO2 atmosférico

6
− verter la solución de acetato en el matraz de la solución de hidróxido de sodio e inmediatamente accionar el cronómetro y registrar la temperatura
− tomar con pipeta volumétrica PE, una muestra (alícuota) de 5 mL de la mezcla reaccionante y colocarla en un matraz Erlenmeyer de 125 mL que contenga 20 mL de agua libre de CO2
− anotar el tiempo que ha transcurrido desde el inicio de la reacción sin detener el cronómetro
− titular rápidamente con la solución estandarizada de HCl hasta el vire de la titulación, de rosa a incoloro. No detener el cronómetro
− registrar en la Tabla 1.1 el volumen de titulante, la temperatura y el tiempo transcurrido− tomar sucesivamente 8 muestras espaciadas aproximadamente cada cinco minutos y repetir
el procedimiento anterior en cada muestra− los residuos se colectan en el vaso de 1 L. Al final se calientan levemente y se tiran
Tabla 1.1 Registro de datos experimentalesConcentración del HCl =
Núm. de muestra Temperatura[°C]
Tiempo[ min]
Volumen de titulante HCl[mL]
0123456789
Puesto que el hidróxido se consume conforme avanza la reacción:
CH3COO-Et + NaOH → CH3 COONa + Et OH Et = etilo
El método de seguimiento es la titulación ácido-base al vire con fenolftaleína. En estas condiciones, la concentración en el tiempo, de NaOH o de acetato de etilo en la mezcla está dada por la relación volumétrica siguiente
CHCl * VHClCNaOH =
Valícuota
La concentración inicial de NaOH o acetato de etilo será igual a la mitad de la concentración de la solución debido a que ésta se diluye en proporción 1:1 al momento de iniciar la reacción
CÁLCULOSa) Con los datos de la Tabla 1.1 calcular la concentración del NaOH. La concentración del
acetato de etilo es igual a la del NaOH en cada muestra

7
b) con los datos experimentales trazar las curvas de concentración de acetato de etilo contra el tiempo Figura 1.1a. Ajustar los puntos a la mejor curva suave posible
c) trazar la ordenada de cada 5 minutos en la gráfica anterior. Registrar en la Tabla 1.3 la concentración de acetato de etilo que se obtiene por la intersección de cada una de las ordenadas anteriores con la curva ajustada:
Tabla 1.3 Registro de datos según la instrucción c.No de
muestra N 0 1 2 3 4 5 6 7 8 9
Tiempo minutos 0 5 10 15 20 25 30 35 40 45
Cajustada mol / L
d) para la muestra número N, tomar los tiempos de N + 1 y de N – 1, tal que t siempre será de 10 minutos, por ejemplo, para el número de muestra 4, el tiempo de la muestra 5 es 25 minutos y el de la muestra 3 es de 15 minutos, por lo cual t es (25 – 15) = 10 minutos o 600 segundos. De la misma manera tomar las concentraciones ajustadas de las muestras N + 1 y N – 1 y con estos dos valores encontrar el valor de C. Con estos datos llenar la tabla1.4, excluyendo este cálculo para las muestras 0 y 9. Llenar los valores en la Tabla 1.4 para
__
el cálculo de la velocidad instantánea promedio rA __ __ __e) de los valores de rA obtener la curva Ln rA vs. Ln CA ( Figura 1.4 ), y /o la regresión
lineal de la Ecuación 1.7
f) de la curva anterior, y / o la regresión lineal calcular el valores de k y n. Donde k es el antilogaritmo de la ordenada al origen y n es la pendiente
Tabla 1.2 Concentración de acetato de etilo en el tiempo. Temperatura =_________Tiempo
[s]C acetato de etilo
[mol/L]Tiempo
[s]C acetato de etilo
[mol/L]0 (0.2*25 mL) / 50 mL = 0.1

8
RESULTADOS
Temperatura constante de velocidadk
Orden de reacción n
Coeficiente de correlación R
CUESTIONARIO1. El orden global n de la reacción de saponificación del acetato de etilo es ( ) A) uno B) dos C) tres
2. La reacción de saponificación del acetato de etilo es ( ) A) elemental B) no elemental
3. ¿Qué significa que una reacción bimolecular no sea elemental?
4. La constante de velocidad reportada en la literatura para la reacción entre el acetato de etilo y el hidróxido de sodio a 25°C, es de klit = 0.107 mol−1seg −1 (6.42 mol−1min −1). Calcular el porcentaje de error entre el valor experimental y el teórico, de la manera acostumbrada:
| k exp. − k lit | % de error =
k lit* 100
5. Escribir un posible mecanismo de reacción para la saponificación del acetato de etilo.
__ __
Tabla 1.4 Cálculo de la velocidad media absolutarA y la concentración promedio CA
Velocidad media absoluta Concentración promedio CAt[seg]
CA
[moles / L] rA = t= Cajustada de la Tabla 1.3
600 C2 – C0 = C1 =600 C3 – C1 = C2 =600 C4 – C2 = C3 =600 C5 – C3 = C4 =600 C6 – C4 = C5 =600 C7 – C5 = C6 =600 C8 – C6 = C7 =600 C9 – C7 = C8 =

9
PRÁCTICA Nο. 2
DETERMINACIÓN DE LA CONSTANTE DE VELOCIDAD Y EL ORDEN, DE LAREACCIÓN ENTRE EL YODURO I– Y EL PERSULFATO S2O8
2–. ANÁLISIS POR EL MÉTODO INTEGRAL
OBJETIVO GENERALObtener la constante de velocidad a dos temperaturas a partir de la ecuación integrada de segundo orden, utilizando el método integral para el análisis de datos cinéticos.
OBJETIVOS ESPECÍFICOS Determinar experimentalmente la concentración de yodo y persulfato en el tiempo
mediante el método volumétrico, a diferentes temperaturas obtener las gráficas concentración de persulfato contra tiempo obtener la ecuación cinética integrada para 2° orden aplicar la ecuación cinética integrada de 2° orden a lo datos, a diferentes temperaturas obtener el coeficiente cinético k, del ajuste de los datos a las cinéticas de 2° orden concluir si los datos cinéticos se ajustan a la cinética de 2° orden
obtener el tiempo de vida media t ½
FUNDAMENTOS TEÓRICOSLa mayoría de las reacciones químicas caen en las reacciones de orden 1 y de orden 2, pocas son de orden 0, 3 o fraccionario.
Para una reacción bimolecular elemental con CA y CB iguales se tiene
A + B qQ + sS + ...
r = k CA2 2.1
La ecuación diferencial es:
Para integrar la ecuación diferencial anterior, se separan variables y se integra desde la concentración en el tiempo cero hasta la concentración en el tiempo t
CA dCAt
– ∫CA0 CA
2 = k ∫0
dt 2.3
La solución de las integrales es:
dCAr = –dt
= k CA2 2.2

10
1 1– [ –CA
+ CA,0] = k t 2.4
El arreglo de la ecuación anterior a una recta, presenta una pendiente positiva igual a la constante de velocidad k
1 1
CA= CA,0
+ k t 2.5
Si los datos cinéticos de un experimento se ajustan al modelo anterior, el coeficiente de correlación lineal R tendrá un valor entre 0.99 y 1 y la cinética será de segundo orden.
La representación gráfica de la Ecuación 2.5 se muestra en la siguiente Figura 2.1
La reacción entre el yoduro y el persulfato de potasio presenta la siguiente ecuacióndiferencial de segundo orden Ecuación 2.6
2 KI + K2S2O8 → I2 + 2 K2SO4
dC S2O82 –
r = –dt
= k C I– C S2O8
2 – 2.6
Sustituyendo la concentración del yoduro en función de la concentración del persulfato:
C I ‾ = 2 C S2O82 –
se tiene:
d C S2O82 –
–dt
= k 2C S2O82 – ∙ C S2O8
2 – 2.7
t}1/CA,0
1 / CA
Pendiente = k
Figura 2.1

11
dC S2O82 ‾
–dt
= 2 k (C S2O82 ‾)2 2.8
La ecuación integrada es:
C S2O 8 2 – d C S2O8
2– t
– ∫ (C S2O 8 2 – ) 0 (C S2O8
2 – )2 = 2 k ∫0
dt 2.9
Cuya solución es:
1 1
C S2O82 – =
(C S2O82 –)0
+ 2 k t 2.10
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
1 bureta de 50 mL persulfato de potasio 0.1 M K2S2 O8
2 cronómetros yoduro de potasio 0.2 M KI
2 matraces Erlenmeyer de 250 mL tiosulfato de sodio 0.01 M Na2S2O3
6 matraces Erlenmeyer de 125 mL solución de almidón al 1%
1 pipeta volumétrica de 5 mL PE agua destilada
2 pipetas volumétricas de 25 mL PE hielo
1 pinza para bureta
vaso de precipitados de 500 mL
1 tapón de hule para el Erlenmeyer de 250 mL
PROCEDIMIENTO Tomar 25 mL de yoduro de potasio 0.2 M con una pipeta volumétrica PE y verter totalmente
en un matraz Erlenmeyer de 125 mL en otro matraz de 250 mL, verter totalmente 25 mL de persulfato de potasio 0.1 M, con una
pipeta volumétrica PE agregar el yoduro al persulfato y empezar a medir el tiempo (activar el cronómetro)
Colectar las soluciones residuales en un vaso de precipitados de 500 mL. Al final de la sesión neutralizar el yodo de las soluciones con la solución de tiosulfato o con unos cristales de tiosulfato, hasta eliminar el color oscuro, la solución incolora se desecha al drenaje.

12
colocar el tapón de hule al matraz y mezclar muy bien las soluciones registrar la temperatura en el momento de hacer la mezcla Tabla 2.1, en el caso de utilizar
un baño de agua a otra temperatura diferente de la ambiente mantener la isotermicidad del baño
tomar una muestra de 5 mL (alícuota) a los 5 minutos aproximadamente, tapar inmediatamente y verter en un matraz Erlenmeyer de 125 mL
el yodo liberado se titula rápidamente con tiosulfato 0.01M, agregando almidón cerca del punto final, cuando se aclare la coloración amarilla del yodo, la mezcla de reacción ahora adquirirá una coloración morada. El vire y final de la titulación es cuando la coloración morada del complejo yodo-almidón desaparezca momentáneamente. Cerrar la bureta, simultáneamente anotar el tiempo sin detener el cronómetro y el volumen gastado del tiosulfato, en la Tabla 2.1
la solución volverá de nuevo a tomar la coloración morada, colocarla en el vaso de 500 mL para su posterior tratamiento
tomar 5 muestras mas, en intervalos de aproximadamente 5 minutos, sin detener el cronómetro y titularlas siguiendo las mismas instrucciones
repetir el procedimiento con los reactivos en un baño de hielo (media hora antes) Las titulaciones se hacen a la temperatura del baño de hielo-agua (tener los matraces en el
baño de hielo y ahí hacer la titulación).
El seguimiento de la reacción se realiza mediante la determinación volumétrica del yodo producido con una solución estandarizada de tiosulfato de sodio. La reacción de la titulación yodo – tiosulfato es:
I2 + 2 Na2 S2 O3 → 2 Na I + Na2 S4O6
La concentración de yodo en el transcurso de la reacción será igual a:
Volumen de tiosulfato * C tiosulfatoCyodo = 2 * Volumen de muestra (alícuota)
Tabla 2.1 Registro de datos experimentales
Concentración del tiosulfato de sodio___________M
Temperatura ºC Tiempo (minutos:seg) Tiempo (minutos) Volumen de tiosulfato [mL]0:0 0 0

13
Para conocer la concentración de persulfato de sodio tenemos:
la concentración inicial será igual a la mitad de la concentración de la solución debido a que esta se diluye al momento de iniciar la reacción.
(C S 2 O 82 –)del frasco reactivo x V S 2 O 8
2 – 0.1 M x 25 mL(C S 2 O 8
2 –)0 = V S 2 O 8
2 – + V I– =
50 mL= 0.05 M
la concentración en el transcurso de la reacción (concentración en el tiempo) será igual a la concentración inicial de persulfato menos la concentración de yodo que se produce en ese mismo momento.
C S 2 O 82 – = (C S 2 O 8
2 –)0 – C I2 = (C S 2 O 82 –)t
CÁLCULOSa) Calcular la concentración de yodo y persulfato de sodio para completar la Tabla 2.2.
Tabla 2.2Tiempo[min]
C I2[ mol / L ]
C S 2 O 82 −
[ mol / L ]1 / C S2 O 8
2 −
[ L / mol ]0 0 (C S2 O 8
2 –)0 = 0.05
b) elaborar una gráfica de la concentración de persulfato C S2O82 – contra tiempo. Trazar la
curva que mas se aproxime a los datos obtenidos. Señalar el tiempo t ½ en que la concentración inicial del persulfato decae a la mitad, si es necesario extrapole la curva.
c) elaborar una gráfica 1/CS2O82 – contra tiempo y la regresión lineal. Determinar el
coeficiente cinético k, concentración inicial de persulfato y el coeficiente de correlación lineal.
d) proceder de igual manera para la cinética a la temperatura del baño de hielo-agua
RESULTADOS
TemperaturaºC
kmolar –1 min –1
C inicial de persulfato(C S2 O 8
2 –)0
Coeficiente de correlaciónR
T1 =
T hielo-agua =

14
CUESTIONARIO
1.- Con el valor de k obtenido en c) determinar el tiempo de vida media para la reacción de segundo orden yoduro-persulfato. a temperatura ambiente y baño de hielo
1t ½ =
k (C S 2 O 82 – )0
2.- ¿Se ajustan los datos experimentales a una cinética de segundo orden? Justifique su respuesta.
3. La reacción 2 KI + K2S2O8 → I2 + 2 K2SO4 es trimolecular, ya que en ella participan dos moléculas de yoduro de potasio y una de persulfato de potasio. Sugiera un mecanismo de reacción que justifique el orden dos obtenido. La reacción ocurre en dos pasos, en el primer paso (el determinante), reacciona un I– y un S2O8
2– para formar dos SO42– y un I +

15
PRÁCTICA No. 3
EFECTO DEL CAMBIO DE LA CONCENTRACIÓN EN LA VELOCIDAD DE REACCIÓN. MÉTODO DE LAS VELOCIDADES INICIALES
OBJETIVO GENERALMedir los efectos del cambio de concentración inicial en la velocidad de reacción inicial.
OBJETIVOS ESPECÍFICOS Efectuar las cinéticas de la reacción del ión yoduro con el ión persulfato en solución
acuosa, variando las concentraciones iniciales de los reactivos calcular las concentraciones del yodo I2 formado, yoduro de potasio y persulfato de amonio
(NH4)2S2O8 en el transcurso de la reacción construir las gráficas concentración del yoduro y de persulfato contra tiempo determinar la velocidad inicial en cada experimento, como la “tangente” a la curva
concentración vs. tiempo, en el tiempo cero comparar el efecto de modificar la concentración de los reactivos sobre las velocidades calcular los órdenes parciales y del yoduro I− y del persulfato S2O8
2−, comparando las velocidades iniciales a diferentes concentraciones iniciales, manteniendo la concentración del otro reactivo constante
determinar el valor de la constante de velocidad (coeficienten cinético) k, a partir de la ecuación de velocidad que resulta del análisis de la reacción entre el yoduro y el persulfato
FUNDAMENTOS TEÓRICOSSi una reacción es de orden cero respecto a un reactivo en particular, la modificación de su concentración no tendrá influencia sobre la velocidad
r [A] 0
Si la reacción es de primer orden con respecto a un reactivo, los cambios de concentración serán proporcionales a los cambios en la velocidad. Así al duplicar la concentración se duplicará la velocidad, si se triplica la concentración también lo hará la velocidad y así sucesivamente (relación lineal, ecuación de una recta)
r [B]
si [B1] = x, r1 [B1] y r1 [x]si [B2] = 2x, r2 [B2] y r2 [2x]si [B3] = 3x, r3 [B3] y r3 [3x]
entonces r3 3r1 , r2 2r1

16
Si la ecuación es de segundo orden respecto a un reactivo específico, al duplicar su concentración, la velocidad aumenta por un factor de 2 2 = 4; al triplicar la concentración, esto hace que la velocidad aumente por un factor de 3 2 = 9 y así sucesivamente
r [B] 2
si [B1] = x, r1 [B1]2 y r1 [x]2
si [B2] = 2x, r2 [B2]2 y r2 4[x]2
si [B3] = 3x, r3 [B3]2 y r3 9[x]2
entonces r3 = 9r1 y r2 = 4r1.
Para la reacción entre el persulfato y el yoduro
2 I −+ S2O8
2 − I2 + SO42 −
La ecuación de velocidad viene dada por
r = k CI –
∙ C S 2 O 8
2 ‾
donde: yson los ordenes parciales respecto al yoduro y al persulfato respectivamente
CI
– es la concentración del yoduro elevado a su orden parcial
C S 2 O 8
2 ‾ es la concentración del persulfato elevado a su orden parcial
k es la constante de velocidad específica o coeficiente cinético
El orden global n de una reacción es la suma de los órdenes parciales
n = +
La relación de velocidades cuando un reactivo cambia sus concentraciones iniciales manteniendo constante la concentración del otro, viene dada por las siguientes expresiones
r1 k (CI
– )1 (C S 2 O 8
2 ‾) 1
r 2=
k (CI
– )2 (C S 2 O 8
2 ‾) 2 3.1
Para la concentración de persulfato constante la expresión queda:
r 1 (CI
– ) 1 (C I
–) 1
r2=
(CI
–) 2=
(C I– ) 2
3.2

17
Donde el orden parcial del yoduro se obtiene a través de los logaritmos de la relación de velocidades y de concentraciones
r1 (C I– ) 1ln
r2= ln
(C I – ) 2
3.4
r1lnr2
(C I– ) 1
=
ln(C I
– ) 2
3.5
Por un procedimiento similar, el orden parcial con respecto al persulfato, cuando su concentración cambia manteniendo constante la concentración del yoduro es
r1lnr2
(C S 2 O 82 ‾ ) 1
=ln
(C S 2 O 82 ‾)2
3.6
Método de la velocidad inicial. Algunas reacciones tienen un comportamiento inicial que se ajusta a algún modelo de velocidad, pero la aparición de reacciones colaterales dificultan su seguimiento cinético, como es el caso de la reacción del yoduro con el persulfato en donde la reacción adicional del yodo producido con el yoduro inicial para formar el complejo yodo-yoduro I3‾, modifica la ecuación de velocidad por lo que es conveniente tratar la cinética en los primeros minutos, (cuando la concentración del complejo es mínima), con el método de la velocidad inicial
2 I ‾ + S2O8 2 ‾ I2 + 2SO4
2 – Reacción principal 3.7
I ‾ + I2 I3‾ Reacción colateral 3.8
2 I 3‾ + S2O8
2 ‾ 3I2 + 2SO42 – Reacción paralela 3.9
Por lo tanto la ecuación de velocidad viene dada por
r = k CI
_ C S 2 O 8
2 ‾ + k’ CI3 C
S 2 O 82 ‾ 3.10
Durante los primeros minutos de la reacción la concentración del triyoduro I3– es mínima y
r 1 (C I– ) 1ln
r2= Ln
(C I– ) 2
3.3

18
puede despreciarse de la ecuación de velocidad, la cual se reduce a
r inicial = k (CI
– ) 0 (C
S 2 O 82 ‾)0 3.11
donde (C I– ) 0 y (C S 2 O 8
2 ‾) 0 son las concentraciones iniciales del yoduro y del persulfato.
La velocidad inicial es la velocidad instantánea en el tiempo cero, como se muestra en la Figura 3.1 concentración vs. tiempo, cuando las concentraciones de los reactivos se conocen exactamente y son las concentraciones iniciales
Figura 3.1 Velocidad inicial
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
3 buretas de 50 mL solución de yoduro de potasio, KI 0.2 M
2 pipetas volumétrica de 1 mL PE solución de tiosulfato de sodio, Na2S2O3
1 vaso de precipitado de 400 mL 0.4 M (recién preparada)
1 vaso de precipitados de 100 mL solución de nitrato de potasio, KNO3 0.2 M
2 pinzas para bureta solución de persulfato de amonio
2 soportes universal (NH4)2S2O8 0.2 M
1 barra de agitación magnético solución sódica de etilendiaminatetracético
1 base de agitación magnética Na2EDTA 0.1 M (en frasco gotero)
1 termómetro solución de almidón al 1%. Hervida
1 cronómetro agua destilada
1 propipeta o jeringas con manguera
1 vaso de precipitados de 500 mL, para residuos (uso común)
* todas las soluciones se preparan con la solución de nitrato de potasio 0.2 M
t
r inicial = −CA/tCA
CA
t
Recolectar las soluciones residuales en un vaso de precipitados de 500 mL. Al final de la sesión experimental neutralizar el yodo de las soluciones con la misma solución de tiosulfato o con un poco de cristales de tiosulfato, hasta que el color oscuro desaparezca, la solución clara e incolora puede desecharse al drenaje.

19
PROCEDIMIENTO Colocar 3 buretas en un soporte universal. Utilizar estas buretas para medir los volúmenes
de las soluciones de KI, KNO3 y (NH4)2S2O8. Usar 2 pipetas volumétricas PE de 1 mL para medir volúmenes de las soluciones de
Na2S2O3 y de almidón. Cada solución deberá ser preparada en el momento de iniciar el estudio de la cinética de
velocidad, esto es preparar las soluciones 1, 2 y 3 a la vez que se vayan a utilizar. El EDTA se adiciona como secuestrante de metales pesados, que pudieran interferir con la
velocidad de reacción El yodo producido en la reacción principal entre el yoduro y el persulfato ( 3.7 ) reacciona
con el tiosulfato (1 mL), la mezcla de reacción permanece incolora hasta que el tiosulfato se agota y aparece la coloración azul-negro
I2 + 2 S2O32− 2 I − + S4 O6
2 – 3.12 Azul-negro incoloro
Con el cambio de color de incoloro a azul-negro, 2 x 10 − 4 mol de persulfato S2O82 − han
reaccionado en los 100 mL
Preparación de soluciones: Preparar 3 soluciones de reacción como sigue (una a la vez)
Solución 1 2 3Yoduro de potasio, mL 25 25 50Almidón, mL 1 1 1Tiosulfato de sodio, mL 1 1 1Nitrato de potasio, mL 48 23 23Solución de EDTA 1 gota 1 gota 1 gotaVolumen total, mL 75 50 75
Volumen de persulfato que deberá añadirse para iniciar la reacción
Persulfato de amonio, mL 25 50 25
Mediciones de la velocidad
Preparar la solución 1 en un vaso de precipitados de 250 mL, el cual deberá estar perfectamente limpio y seco ( sin el persulfato )
introducir al vaso la barra de agitación magnética y colocarlo en la base de agitación
agitar la solución. Medir y registrar la temperatura de la solución
en un vaso de precipitados de 100 mL, tomar con la bureta 25 mL de una solución de persulfato de amonio (NH4)2S2O8

20
rápidamente verter la solución de persulfato (NH4)2S2O en la solución 1, simultáneamente accionar el cronómetro. La reacción se inicia en el momento en que las soluciones son mezcladas (tiempo cero)
en el instante cuando aparece el color azul-negro registrar el tiempo en la Tabla 3.1 sin detener el cronómetro y simultáneamente (¡estar preparados!) añadir una alícuota de 1 mL de solución de tiosulfato de sodio Na2S2O3
registrar el tiempo cuando ocurra la reaparición del color azul-negro e inmediatamente después añadir otra alícuota de 1 mL de tiosulfato Na2S2O3 en la solución
repetir la instrucción anterior cada vez que reaparezca la coloración, registrar el tiempohasta haber agregado 5 alícuotas de tiosulfato a la solución 1
al terminar el trabajo con la solución 1, vuelva a medir y registrar la temperatura. Si la temperatura no se mantuvo constante, comuníquelo al profesor.
Para mejores resultados, las alícuotas de tiosulfato Na2S2O3 deben ser medidas en forma rápida, exacta y reproducible, tanto como sea posible.
Las soluciones 2 y 3 deben ser tratadas exactamente bajo el mismo procedimiento, excepto los 50 mL de la solución de persulfato de amonio (NH4)2S2O8 que deben ser agregados a la solución 2 mientras que a la solución 3 solo se le agregan 25 mL de solución (NH4)2S2O8.
Precaución: Estar alerta, las soluciones 2 y 3 reaccionan mas rápidamente que la solución 1.
Tabla 3.1. Tiempo acumulado cuando aparece el color azul-negro
Tiempo acumulativo[s]
Solución
Volumen agregado de Na2S2O3 0.2 M
[mL]1 2 3
0 0 0 0
1
2
3
4
5
6
21
CÁLCULOS
a) Calcular* las concentraciones molares iniciales de yoduro I− y persulfato S2O82 − para cada
una de las soluciones. Registrar en la Tabla 3.2
b) Calcular la cantidad de mol de yodo I2 producido, persulfato de amonio (NH4)2S2O8 y yoduro I − consumidos, considerando que por cada mililitro de tiosulfato Na2S2O3 0.4 Mañadido (4 x 10 −4 mol/mL), se producen 2 x 10 −4 mol de yodo (2x 10−3 mol/L) Ecuación 3.12, la misma cantidad se consume de persulfato y dos veces la de yoduro Ecuación 3.7. La cantidad remanente de persulfato y de yoduro, en cualquier tiempo, es la cantidad inicial menos la que se ha consumido. Registrados en la Tabla 3.3
c) Con los datos de la Tabla 3.3 calcular las concentraciones molares (mol/L) de yoduro de potasio KI y de persulfato de amonio (NH4)2S2O8, en el tiempo, para las mezclas 1, 2, y 3. Considerar, que el volumen de mezcla es de 100 mL (0.1 L). Registrar en la Tabla 3.4
d) Con los datos de tiempo acumulado y las respectivas concentraciones de persulfato, trazar en Excel las gráficas de concentración de persulfato contra tiempo en segundos, para cada una de las soluciones Figura 3.1 (las tres curvas pueden ser trazadas en un mismo gráfico)
Tabla 3.2. Concentraciones iniciales molares de yoduro y de persulfato *Solución 1 Solución 2 Solución 3
(C I– ) 0
(C S2O82 – ) 0
Volumen agregado x Molaridad*Concentración molar en la mezcla =
Volumen final de la mezcla
Tabla 3.3. Mol de yodo formado, persulfato y yoduro remanente para las mezclas 1, 2 y 3 Moles de
I2
Moles de S2O8
2 −
en 100 mLMoles de I −
en 100 mLSolución Solución
Volumen consumidode Na2S2O3
[mL] Solución1, 2, 3 1 2 3 1 2 3
0123456

22
e) Obtener la velocidad inicial (valor absoluto) para cada una de las soluciones. Esta se calcula al dividir la primera diferencia de concentraciones del persulfato (C= 0.0005) (Tabla 3.4), entre el tiempo cero y el siguiente (t) (Tabla 3.1). Anotar en la Tabla 3.5
f) Con los datos de velocidades iniciales de la Tabla 3.5 y las concentraciones iniciales de la Tabla 3.2, encontrar los coeficientes parciales y de reacción (Ecuaciones 3.5 y 3.6 ). Anotar en Tabla 3.6
Tabla 3.6. Cálculo de los coeficientes parciales ySolución Velocidad inicial Conc. inicial persulfato Conc. inicial yoduro
1 ―――2 ―――
1 ―――3 ―――
f) Con los valores calculados de y obtener el orden global de la reacción
Tabla 3.4. Concentración molar de persulfato y yoduro en el tiempo para las mezclas 1, 2 y 3 CS2O8
2 −
[Mol/L]C I
−
[Mol/L]Solución Solución
Volumen consumidode Na2S2O3
[mL]1 2 3 1 2 3
012
3
4
5
6
cantidad de molesConcentración molar =
Volumen en litros
Tabla 3.5 Velocidades iniciales para la cinética de las soluciones 1, 2 y 3
Solución C persulfato t r inicial =C/t123

23
g) Con los valores calculados de y calcular la constante de velocidad k (Ec. 3.11) para –
cada una de las soluciones y un valor de velocidad promedio k. Registrar en la siguiente Tabla 3.7
Tabla 3.7. Cálculo de la constante de velocidad de reacciónConstante de velocidad
[M –1 s–1]
r inicial
Solución
k=(C
I– )O
(CS2O8
2 – )O
Constante de velocidad promedio[M –1 s–1]
1 2
3
_k =
h) Comparar el valor promedio de k, con el valor reportado en la bibliografía a 25°C, que es de 4.6 x 10 − 3 M –1 s–1. Este valor menor al calculado experimentalmente, es debido al efecto iónico del nitrato de potasio en la reacción del “Reloj” como se conoce a esta práctica. El efecto iónico aumenta la velocidad de reacción y por tanto al valor de k.
CUESTIONARIO1. ¿Cuál es el orden parcial del yoduro, del persulfato y el orden global, de la reacción
anterior?
2. ¿Cuál es el valor de la constante de velocidad calculada en cada solución con el método de las velocidades iniciales?
3. ¿Cuál es el valor promedio de k y por qué es mayor al reportado en la literatura?

24

25
PRÁCTICA No. 4
OBTENCIÓN DE LOS PARÁMETROS TERMODINÁMICOS DEL ESTADO DE TRANSICIÓN DE LA REACCIÓN DEL YODURO I– CON EL PERSULFATO S2O8
2–
OBJETIVO GENERALObtener los parámetros termodinámicos de activación del estado de transición de la reacción de la práctica número 2
OBJETIVOS ESPECÍFICOS Describir la Ecuación de Arrhenius, que relaciona la dependencia de la constante de
velocidad con la temperatura definir el factor de frecuencia de colisiones de Arrhenius describir la Ecuación de Eyring, como una alternativa de la ecuación de Arrhenius definir el estado de Transición obtener:
a. de la ecuación de Arrhenius, la energía de activación Ea y el factor de frecuencia A de la reacción entre el yoduro y el persulfato (práctica número 2)
b. la entalpía de activación H ‡ a partir de la energía de activaciónc. la Energía de Gibbs de activación G‡ de la ecuación de Eyringd. la constante de equilibrio de activación K ‡ de la relación entre ésta y la energía de Gibbse. la entropía de activación S ‡ de la relación G ‡ = H‡ – TS ‡
FUNDAMENTOS TEÓRICOSLa constante de velocidad k de una reacción química varía en forma exponencial con el recíproco de la temperatura
Svante Arrhenius y vant´Hoff relacionaron a la constante de velocidad con la temperatura de acuerdo a la siguiente expresión
k = A e – Ea / RT 4.1
k e – 1 / T
k
Temperatura

26
Donde:
k = constante de velocidadA = factor de frecuencia o de colisiones o factor preexponencial
Ea = energía de activación, J∙mol–1
R = constante molar del estado gaseoso, 8.314 J∙mol–1∙K
–1
T = temperatura termodinámica o kelvin
La energía de activación Ea es la mínima energía requerida por las moléculas de reactivo para interaccionar y formar los productos
El factor de frecuencia A se refiere a la “frecuencia” con la que las moléculas de los reactivos colisionan y si además se necesita una determinada orientación entre ellas para que reaccionen(factor de orientación), también queda incluido en esta constante A.La fracción de moléculas con mínima energía Ea para reaccionar esta dada por: e–Ea/RT
R1 + R2 → P + Q
Eyring también relacionó a la constante de velocidad con la temperatura de acuerdo a la siguiente ecuación:
T1 T2
Energía cinética
Fracción de moléculas con suficiente energía Ea para colisionar y dar productos
Fracción de moléculas con determinada energía
T1
T2
Ea
R1
R1R1
E
QPR2R1
Ea
R1R2
E
R2
Ea
Dos moléculas de R1, no reaccionan
R1 y R2 no tienen la energía ni la orientación para reaccionar
R1 y R2 si tienen la energía para reaccionar, pero no la orientación
R1 y R2 si tienen la energía y la orientación para reaccionar,

27
Donde:
k = constante de velocidad
k B = constante de Boltzman, 1.38 x 10– 29 J∙K
–1
T = temperatura termodinámica, K
h = constante de Planck, 6.63 x 10– 34 J∙s
G ‡ = Energía de Gibbs de activación, J∙mol –1
R = constante molar de los gases, 8.314 J∙mol –1∙K –1
Eyring basó su teoría en la formación de una especie en un estado de transición entre los reactivos (en el estado base) y los productos, al que llamó Complejo Activado (en el estado de transición), el cual es una especie transitoria de máxima energía
Con las ecuaciones de Arrhenius (Ecuación 4.1) y de Eyring (Ecuación 4.2) pueden calcularse, todos los parámetros termodinámicos del estado de transición. La linearización de las anteriores ecuaciones da por resultado la ecuación de una recta, cuyas pendientes son proporcionales a la Energía de Activación Ea (Ecuación 4.3) y a la Energía de Gibbs de
k B Tk = h
e – G‡/ RT
4.2
R1
R1 + R2 → R2
= ‡
→ P + Q
Complejo activado
Estado base
EnergíaComplejo Activado ‡
Reactivos
Productos
Coordenada de reacción
Estado de transición

28
Activación G ‡ (Ecuación 4.4). La ordenada en el origen de la ecuación de Arrhenius proporciona el valor del Factor de Frecuencia A
La Entalpía de Activación H‡, se calcula a partir de la Energía de Activación como sigue:
Ea = E + RT 4.5Donde E es la Energía Interna
E = H – PV) 4.6En un proceso a presión constante, como normalmente se trabaja en un laboratorio, se tiene que:
E = H – PV 4.7Para las fases condensadas sólidos y líquidos, el cambio de volumen V es despreciable, por tanto
E = H 4.8De aquí que la energía de activación se relaciona con la entalpía de acuerdo con la siguiente ecuación:
Ea = H ‡ + RT 4.9Finalmente
H ‡ = Ea – RT 4.10
La entropía de activación se calcula a partir de
G ‡ = H ‡ – TS ‡ 4.11
Finalmente la Constante de Equilibrio en el estado activado K ‡, se calcula con la Energía de Activación de Gibbs como sigue:
G ‡ = – RT ln K ‡ 4.13
K ‡ = e – G‡/ RT 4.14
Ea 1ln k = ln A –
R∙
T 4.3
k kB G ‡ 1ln
T= ln
h–
R∙
T 4.4
H ‡ – G ‡
S ‡ =T
4.12

29
DESARROLLO EXPERIMENTAL.Realizar nuevamente la práctica No. 2, a temperaturas diferentes de las que ya se hayan realizado.
Registrar en la siguiente Tabla 4.1 los datos de la reacción entre el yoduro y el persulfato a temperatura ambiente, a temperatura de un baño de hielo - agua y alguna otra temperatura, de la cinética de la práctica número 2
2 KI + K2S2O8 → I2 + 2 K2SO4
CÁLCULOS
a) Obtener la Energía de Activación Ea, de la ecuación de Arrhenius ( Ecuación 4.3 ) por cualquiera de los siguientes métodos:
a.1) despeje directo de la ecuación 4.3 a dos temperaturas T1 (k 1) y T2 (k 2) (T2>T1):
k2 Ea T2 – T1lnk1
= –R
(T2 ∙ T1
) 4.17
a.2) a varias temperaturas, a partir de la pendiente de la regresión lineal ln k contra 1/ T
EaPendiente = –
R 4.18
b) Calcular el Factor de Frecuencia A de la ecuación de Arrhenius por cualquiera de los siguientes métodos
b.1) por sustitución de la Energía de Activación en la Ecuación de Arrhenius 4.1 o 4.3
b.2) a varias temperaturas, a partir de la ordenada en el origen de la regresión lineal ln k
contra 1/T
c) Calcular la Energía de Gibbs de Activación G ‡, por cualquiera de los siguientes métodos a partir de la ecuación de Eyring (Ecuación 4.4 ):
Tabla 4.1Temperatura
[°C]Constante de velocidad k
[L mol–1s–1 ]

30
c.1) por despeje directo sustituyendo valores de k, T, kB y h en la Ecuación 4.4
c.2) por despeje directo de la ecuación a dos temperaturas T1 (k1) y T2 (k2)
k2 / T2 G ‡ 1 1ln k1 / T1
= –R ( T2
–T1
) 4.15
c.3) a varias temperaturas a partir de la pendiente de la regresión lineal ln (k/T) contra 1/T
G ‡
Pendiente = –R
4.16
c) Calcular la Entalpía H ‡, Entropía S ‡ y Constante de Equilibrio K ‡ a partir de las Ecuaciones 4.10, 4.12 y 4.14
CUESTIONARIO1.- ¿Es la reacción entre el yoduro y el persulfato más rápida a mayor temperatura? Justificar
la respuesta a través de la comparación de las constantes de velocidad a diferentes temperaturas.
2.- ¿El valor de G‡ es variable con la temperatura? Justificar la respuesta.

31
PRÁCTICA Nο. 5
CINÉTICA DE UNA REACCIÓN REDOX A TRAVÉS DE UN OBSERVABLE
OBJETIVO GENERALSeguir la cinética de una reacción química a través de un observable.
OBJETIVOS ESPECÍFICOS Seguir la cinética de una reacción química a través de la absorbancia como observable de la
reacción de formación del complejo yodo-yoduro I3¯ variando la concentración de los reactivos, por el procedimiento de las velocidades iniciales
obtener la pendiente inicial de la absorbancia en el tiempo determinar el orden parcial de los reactivos a través del método de concentración constante
de uno de los reactivos y la velocidad inicial determinar el orden global de la reacción a partir de los ordenes parciales calcular el valor de la constante de velocidad aplicar la Ley de Lambert y Beer para relacionar la concentración del complejo yodo-
yoduro con la absorbancia comparar el valor de la constante experimental de velocidad con el valor reportado en la
literatura
FUNDAMENTOS TEÓRICOSLa velocidad de una reacción química puede monitorearse a través de un método indirecto de medir el cambio de la concentración en función del tiempo. Ese método consiste en medir el valor de una propiedad física (variable) que sea proporcional a la concentración de alguno de los reactantes. A esta propiedad física se le llama observable de la reacción.
Por ejemplo la reacción en fase gaseosa del acetileno con el hidrógeno, puede monitorearse por el cambio de presión en el sistema, cambio que es proporcional a la concentración.
C2H4 (g) + H2 (g) → C2H6 (g)
PV = nRT 5.1n
P V
= Concentración
La presión total del sistema P disminuye conforme se consume el etileno y el hidrógeno. Como se observa en la ecuación de gas ideal, la concentración molar (M) corresponde a las moles (n) por litro (V), R y T son la constante molar del estado gaseoso y la temperatura termodinámica, respectivamente.La reacción de descomposición del agua oxigenada puede monitorearse a través del aumento del volumen del oxígeno desprendido

32
H2O2 (l) → H2O (l) + O2 (g)↑
El aumento de volumen en la jeringa es el volumen generado por el oxígeno, el cual se mide en el tiempo
La reacción de acomplejamiento del Cr3+ (amarillo) a Cr3+•EDTA (azul) puede seguirse espectrofotométricamente por el cambio de color de amarillo a azul, en las etapas intermedias, el color verde se debe a la combinación del azul del cromo coordinado con el amarillo del Cr3+. La reacción se sigue por la disminución del color amarillo, el observable, es la transmitancia o la absorbancia de la luz amarilla
Cr3+ (amarillo) → Cr3+•EDTA (azul)
Medir la conductividad de una reacción también es un método indirecto para evaluar la concentración de los reactantes, siendo la conductividad el observable
C4H9Cl + H2O → C4H9OH + H + + Cl ‾
Los sistemas observables, son sistemas cerrados con un sensor que detecta la propiedad física observable, por ejemplo: manómetro, jeringa, espectrofotómetro, celda de conductividad, etc.
Para un seguimiento de observables se necesita que:
a) el sensor no interfiera con la reacción b) la medida de la propiedad sea directamente proporcional (lineal) a la concentración de
al menos una especie química. Por ejemplo que la conductividad sea proporcional al número de iones
c) se conozca el valor de la propiedad física observable en el tiempo cero , a diferentes tiempos t y al final de la reacción ∞. Siendo el valor de la propiedad física observable:
tiempo Volumen de O2
0 01 V1
2 V2
3 V3
SoluciónNo conductora
SoluciónConductora

33
La Ley de Lambert y Beer relaciona la absorbancia A con la concentración C de la solución y la longitud de la celda , la constante de proporcionalidad es el coeficiente de extinción o de absorción molar ε, que es característico de cada sustancia
A = ε C 5.2
Cuando un haz de luz I 0 incide sobre una solución, parte de esta luz es absorbida y la que sale, I, es detectada por el espectrofotometro
La relación entre la luz transmitida (I) y la incidente (I 0) se le denomina transmitancia T,
de tal manera que la absorbancia es el logaritmo (exponente) negativo de base diez, del valor de la transmitancia: T = 10 –A 5.4
A = – log 10 T 5.5
La reacción del yoduro con el persulfato (peroxodisulfato), puede ser cinéticamente monitoreada en un fotocolorímetro, midiendo la absorbancia A del complejo yodo-yoduro que se forma colateralmente durante la reacción
2 I ¯(ac) + S2O8 2 –
(ac) → I 2 (ac) + 2SO42 –
(ac)
I ¯(ac) + I2(ac) → I3¯(ac)
IT =I 0
5.3
∞
t
o
tiempo
C A0
Figura 5.1
II 0
Figura 5.2

34
La variación de la absorbancia con el tiempo es proporcional a la velocidad media inicial r ide la reacción, donde m es la pendiente de esta relación
Como la pendiente m es proporcional ( a la velocidad de reacción media inicial, esta aumenta linealmente con la concentración inicial de yoduro C I ¯, 0 de diferentes corridas cuando se mantiene constante la concentración inicial del persulfato, lo que indica un orden parcial de 1 en el ión yoduro. Alternativamente la pendiente correlaciona linealmente con la
concentración inicial de persulfato C S2O82–, 0 de diferentes corridas, manteniendo constante la
concentración inicial del yoduro, indicando también un orden parcial de 1, ahora en el persulfato.
La ecuación diferencial de velocidad de la reacción con respecto a cada uno de los reactivos debe ser:
CI3 ¯ A Ar i = t
=ε t
α t
m
Figura 5.3
m r i
t = 1 minuto
A
Figura 5.4
Concentración inicialde yoduro o persulfato de diferentes corridas
m
m CS2O8 2–, 0
m C I ¯, 0

35
1 dC I ¯ d CS2O8 2–
r = –2 dt
= – dt
5.7
Entonces:
dC I ¯ d CS2O8 2–
dt= 2
dt5.8
Lo cual significa que la pendiente de la velocidad inicial con respecto a la concentración del yoduro debe ser el doble de la pendiente de la velocidad inicial con respecto a la concentración del persulfato (Figura 5.4)
Una vez que el orden de la reacción con respecto a cada uno de los reactivos se ha determinado, se puede calcular la constante de velocidad, correlacionando la pendiente m con el producto de las concentraciones iniciales de yoduro y persulfato, para las diferentes corridas (Ecuación 5.9). El valor del coeficiente de extinción molar ε reportado en la literatura para esta reacción es de 26 000 cm¯
1 M ¯1
El valor de la constante de velocidad reportada en la literatura2 a diferentes temperaturas y utilizando diferentes equipos se muestran en la siguiente Tabla 5.1:
2 Horst, E.; Arden, P. Z. J. Chem. Educ.1988, 65, No.8, 737
m / ε k =(C I ¯,0) (C S2O8
2 –,0)
*5.9
Figura 5.5
(C I ¯, 0) (C S2O82 –
, 0)
de las diferentes corridas
m / ε
k

36
Efecto de la Fuerza Iónica del medio en la velocidad de la reacción: La adición de sales al disolvente (KNO3) puede modificar la velocidad de la reacción, si este efecto salino la aumenta, entonces el medio iónico interactúa con el estado de transición (complejo activado) estabilizándolo, la barrera energética disminuye, aumentando el valor de la constante de velocidad y por consiguiente la velocidad. Si ocurre lo contrario, que el medio iónico desestabilice el estado de transición y/o estabiliza el estado base ( reactivos ), aumentando la distancia energética entre ellos ( barrera ), disminuye el valor de la constante de velocidad y por consiguiente la reacción se hace mas lenta.
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
Espectrofotómetro Genesys 10 (= 353 nm)
celdas del espectrofotómetro
solución de persulfato de potasio K2S2O8 0.02 M
1 jeringa de 5 mL
1 jeringa de 3 mL
solución de yoduro de potasio KI 0.04 M
1 jeringa de insulina de 1 mL
1 termómetro
un vaso de precipitados de 100 mL
pinzas para tubo de ensayo
1 vaso de precipitados de 500 mL para residuos
– Horno de microondas
solución de nitrato de potasioKNO3 0.1 M
Tabla 5.1 Valor de la constante de velocidad k en diferentes condiciones de la reacción entre el yoduro y el persulfato
CondicionesTemperatura
° CEquipo de detección
103 kM –1 s–1
Agua destilada y desionizada 25 Beckman 4.77 – 4.96Agua desionizada 25 Beckman 4.49 – 4.14Agua destilada y desionizada 20 – 21 Spectronic 3.08Agua desionizada 23 Spectronic 4.01
Utilizar guantes al usar las celdas. No lavarlas con jabón, solo con agua destilada. Usarlas y guardarlas siempre secas. Usar el horno de microondas para secarlas, precaución están calientes usar pinzas para tubo. Para enfriarlas colocar las celdas en un vaso de precipitados de 100 mL.Recolectar las soluciones residuales en un vaso de precipitados de 500 mL. Al final de la sesión, neutralizar el yodo de las soluciones con un poco de cristales de tiosulfato hasta que el color oscuro desaparezca, la solución puede desecharse al drenaje.

37
PROCEDIMIENTO
Calibración del espectrofotómetro El espectrofotómetro es un instrumento delicado. Antes del experimento, consultar el manual de operación para su correcto uso (ver páginas 40 y 41)
encender el espectrofotómetro y estabilizar durante 30 minutos
durante el tiempo de estabilización del equipo, marcar una celda como 1 y la otra como 2
preparar en ambas celdas la misma solución A (usar respectivas jeringas), sin agregar el KI de acuerdo a la siguiente Tabla 5.1a
Tabla 5.1a Corridas mL de H2O mL de K2S2O8 0.02 M mL de KI 0.04 M
A 3.45 0.75 0.30B 3.30 0.75 0.45C 3.15 0.75 0.60D 3.00 0.75 0.75E 3.60 0.15 0.75F 2.70 1.05 0.75G 2.40 1.35 0.75
en Test seleccionar la opción de cinética, absorbancia y la longitud de onda (353 nm), con los parámetros de intervalo de tiempo 10 segundos y el tiempo total de 1 minuto.
tiempo de retardo 00:00 y con el modo pendiente por factor encendido
Corrida AEsta reacción es muy rápida, hay que coordinar cada paso antes de iniciar el experimento.
La cinética se efectuará a temperatura ambiente. Registrarla en la Tabla 5.2a
seleccionar correr análisis, en el modo tabular (quedará marcado gráfico)
introducir la celda 1, cerrar la tapa y correrla como blanco, al terminar suena la alarma
con la misma celda correr “medir muestra”, en el momento de apretar la tecla, se añade rápidamente con jeringa limpia, la solución de KI al tubo 2, marcada en la Tabla 5.1a.
agitar el tubo 2 e intercambiarlo por el tubo 1, antes de los 10 segundos.
anotar la absorbancia que aparece a los 10 segundos, en la Tabla de datos 5.2a (Corrida A). La absorbancia se deberá medir cada 10 segundos hasta completar 60 segundos. Anotar los datos y el valor de la pendiente m que aparece al final del experimento(absorbancia por minuto).
repetir el procedimiento anterior (preparar tubo 1 y tubo 2 con la misma solución) para todas corridas de la Tabla 5.1a (B a G)
Para secar las celdas usar el horno de microondas, precaución al sacarlas utilizar unas pinzas para tubo, están calientes colocarlas en un vaso de precipitados para evitar choque térmico con la mesa

38
se propone otra serie de corridas como se describe en la Tabla 5.1b. En estas corridas se pondrá de manifiesto el efecto de la ionicidad del medio sobre la velocidad de la reacción. El volumen total de la reacción es constante (4.5 mL) y los volúmenes de los reactivos persulfato y yoduro en las corridas H, I, J y K son los mismos que los de la corrida A. El volumen del KNO3 se va incrementando con el objeto de lograr un aumento en la fuerza iónica del medio con respecto al de la corrida A que no contiene KNO3. El agua se adiciona para igualar el volumen final de los sistemas reaccionantes.
Anotar la absorbancia cada 10 segundos hasta completar 60 segundos en la Tabla de datos 5.2b. Registrar el valor de la absorbancia por minuto, pendiente m.
Tabla 5.1bCorridas mL de H2O mL de KNO3 0.1 M mL de K2S2O8 0.02 M mL de KI 0.04 M
H 3.0 0.45 0.75 0.3I 2.0 1.45 0.75 0.3J 1.0 2.45 0.75 0.3K 0.0 3.45 0.75 0.3
Tabla 5.2b AbsorbanciaTiempo
[s] H I J K0
102030405060
Pendientem
Tabla 5.2a Temperatura AbsorbanciaTiempo
[s] A B C D E F G0
102030405060
Pendientem

39
CÁLCULOSa) calcular las concentraciones iniciales de yoduro y persulfato, en cada una de las corridas
de acuerdo con la siguiente expresión. Tabular en la Tabla 5.3
b) Anotar el valor de la pendiente (absorbancia por minuto) para cada corrida. Esta pendiente es proporcional a la velocidad (Figura 5.3), anotar sus valores en la Tabla 5.3.
c) graficar las pendientes de las corridas A, B, C y D contra la concentración inicial del yoduro (el persulfato permanece constante), ajustar con cuadrados mínimos e indicar el valor del coeficiente de regresión lineal R, si el valor esta dentro del intervalo de 0.98 y 1, el orden de reacción parcial con respecto al yoduro es uno. Figura 5.4
d) graficar las pendientes de las corridas D; E; F y G contra la concentración inicial del persulfato (el yoduro permanece constante), ajustar con cuadrados mínimos e indicar el valor del coeficiente de regresión lineal, si el valor esta dentro del intervalo de 0.98 y 1 el orden de reacción parcial con respecto al persulfato es uno. Figura 5.4
e) con el valor de los ordenes parciales de la reacción obtenidos de e) y f), las concentraciones iniciales de yoduro y persulfato de cada corrida así como del valor de las pendientes para cada corrida, el valor del coeficiente de extinción molar y el valor de la longitud de la celda (l=1 cm) calcular la constante de velocidad k para la reacción del
yoduro y el persulfato (Figura 5.5) a través de la regresión lineal m/(l ε) contra (CI ¯, 0)
(CS2O82 –, 0)
f) comparar el valor de k ¨[M −1min−1] del punto anterior con el reportado en la literatura que es de 0.26 M–1 min–1, obtener el porcentaje de error si la temperatura de trabajo es de 25ºC.
g) comparar la pendiente de la corrida A con las pendientes de las corridas H, I, J y K. Correlacionarlas con la fuerza iónica (concentración de KNO3).
Fuerza iónica y velocidad de reacciónCorrida A H I J KPendiente mmL de KNO3 0 0.45 1.45 2.45 3.45Conc. de KNO3 0 0.010 0.032 0.054 0.076
Molaridad del yoduro o persulfato x Volumen empleadoC,0 = Volumen total de la solución (yoduro, persulfato y nitrato)
5.10
Tabla 5.3
Corrida A B C D E F G
CI ¯, 0
CS2O82 –, 0
Pendiente

40
CUESTIONARIO1.- Mencionar tres propiedades físicas (observables) que se pueden usar para seguir la cinética
de una reacción2.- ¿Cuál es el orden global de la reacción anterior?3.- ¿Cuál es el porcentaje de error entre la constante de velocidad k experimental de la
reacción y el reportado en la literatura si la temperatura de trabajo es de 25ºC?4.- Definir velocidad inicial de una reacción química.5.- Del inciso g de los cálculos, observar si hay variación en las pendientes y concluir a cual
estado (base o de transición o a ambos) estabiliza el medio iónico.
* la ecuación 5.9 se deduce de las ecuaciones siguientes:la ecuación de Lambert y Beer es: A = lC, por lo cual A = l C. La pendiente se obtiene
como A/t y eso es A Ct
= lt
= lri = lk (C I ¯,0) (C S2O82 –
,0)
Y el valor de A/t es el valor de m. El despeje de k es la Ecuación 5.9
GUÍA RÁPIDA DE OPERACIÓNESPECTROFOTÓMETRO THERMOSPECTRONIC GENESYS 10 VISIBLE
Guía rápida Espectrofotómetro Genesys 10 visible
GRETHEL MENDOZA GUTIÉRREZ LAURA MELINA LÓPEZ RAMÍREZLILIA FERNANDEZ SÀNCHEZ
TestUtility
Cursor o flechas
Esc DeletEnter

41
1. Conectar el cable a la corriente eléctrica2. encender el interruptor (ON) que está atrás del equipo. Aparece la palabra Genesys3. esperar a que se calibre el equipo, 2 minutos aproximadamente. Se oye un ruido, esperar
hasta que aparezca en la pantalla la longitud de onda (4. oprimir la tecla UTILITY, se despliega un menú
5. del menú UTILITY y con ayuda del cursor , señalar las opciones a cambiar, dar ENTER,
seleccionar y volver a dar ENTER para fijar la selección.
a) cambiar el modo de espera (stand by), a modo apagado ( u off) b) expiración de línea base, también apagc) Idioma, Español o Inglés
6. oprimir la tecla Test7. seleccionar cinética. ENTER
8. seleccionar en modo de medición, absorbancia y teclear ENTER
9. si la longitud de onda no es la deseada, oprimir ENTER y escribir el valor deseado ENTER
10. seleccionar tiempo retardado. ENTER, seleccionar 00:00 (min:seg) ENTER
11. seleccionar con las flechas, absorbancia. ENTER
12. seleccionar tiempo de intervalo, ENTER. Escribir el tiempo min:seg, que se necesite para reportar la absorbancia, dar ENTER. Ejemplo 30 segundos 00:30
13. seleccionar tiempo total, ENTER. Escribir el tiempo total que se necesite que a la reacción se le determine la absorbancia. Ejemplo un minuto 01:00
14. seleccionar modo pendiente por factor encendido, la pendiente es proporcional a la velocidad media (entre lecturas)
15. seleccionar correr análisis.16. seleccionar modo tabular.(quedará el nombre de gráfico). Registrará la absorbancia en el
tiempo seleccionado en forma tabular tiempo (seg) Absorbancia
0 010 0.05120 0.052
30 0.053 17. insertar la celda blanco al espectrofotómetro con las marcas blancas diagonal a la
izquierda del usuario, (hay un volumen de 5 mL hasta la parte inferior del rectángulo blanco, pero pude usarse menos volumen de muestra, mínimo 2.5 mL, después de la línea blanca. Cerrar el compartimiento
18. oprimir la tecla: medir blanco, solo si han transcurrido 30 minutos de encendido el equipo. Se oye una alarma
19. retirar la celda con el blanco, oprimir la tecla medir muestra e introducir la celda con la reacción lo más rápido posible
20. al terminar el tiempo total de la cinética la toma de lecturas se detendrá, suena alarma.Aparecerá la ultima medida de Abs/tiempo
21. se puede regresar al modo gráfico

42
22. para otra corrida, medir blanco y después la muestra23. lavar las celdas solo con agua destilada, si hay alcohol, también. Dejar secar y guardar24. con la tecla ESC, se va saliendo de las pantallas, hasta llegar a longitud de onda25. Con el botón posterior OFF-ON apagar el equipo, desconectar y guardar.

43

44
PRÁCTICA Nο. 6
CINÉTICA DE UNA REACCIÓN QUÍMICA SEGUIDA COLORIMETRICAMENTE PARA VERIFICAR UN MECANISMO DE REACCIÓN
OBJETIVO GENERALEstablecer un mecanismo de reacción usando una técnica fotocolorimétrica y el método de las velocidades iniciales.
OBJETIVOS ESPECÍFICOS Seguir la cinética a temperatura ambiente de la reacción entre el cristal violeta y el
hidróxido de sodio a través de un observable, la absorbancia del cristal violeta Obtener la pendiente m de la variación de la absorbancia con el tiempo de diferentes
corridas de la reacción anterior, como un método indirecto de medir la velocidad de reacción
Relacionar la pendiente de dos corridas, con el reactivo que cambia su concentración inicial en cada corrida para determinar el orden parcial con respecto a éste
Determinar el orden global y la constante de velocidad, para confirmar si el mecanismo de reacción es SN1 o SN2
FUNDAMENTOS TEÓRICOSUn mecanismo de reacción explica cómo se forman los productos a partir de los reactivos, describe, paso por paso, el proceso de ruptura y formación de enlaces
La reacción entre el cristal violeta y el hidróxido de sodio puede ocurrir a través de un mecanismo SN1 o un SN2 (tetraedral).
El mecanismo SN1 que corresponde a una sustitución nucleofílica unimolecular. Ocurre a través de un intermediario catiónico, por lo que solamente se involucra un reactivo en el paso determinante y la reacción es de orden uno. La cinética de tal reacción solo dependerá de la concentración del sustrato que forma al carbocatión y no de la concentración del nucleófilo OH ¯, la velocidad de reacción con respecto al nucleófilo sería cero. Figura 6.1.
El estado de transición es iónico y se ve estabilizado en medios polares, aumentando la velocidad de reacción, esto se logra agregando sales que aumenten la polaridad del medio.
El mecanismo SN2 tetraedral ocurre en un solo paso concertado, por lo que requiere de la participación de los dos reactivos involucrados en la sustitución nucleofílica y la reacción es de orden 2. La cinética de tal reacción dependerá de la concentración tanto del sustrato como del nucleófilo OH ¯. Un medio iónico eleva la energía del estado de transición y hay una disminución en la velocidad de reacción. Figura 6.2

45
OH + Cl ¯
Producto incoloroCloruro de p- rosanilinao Cristal violeta
N(CH3)2
N(CH3)2
N(CH3)2Cl ¯
COH ¯
(CH3)2 N
N(CH3)2
N(CH3)2
C
Cuando a una reacción de segundo orden se le agrega uno de los reactivos en exceso, este prácticamente permanecerá constante con respecto al otro y la cinética de la reacción será ahora de pseudoprimer orden.
Figura 6.2 Mecanismo SN2, tetraedral
La reacción entre el cristal violeta y el hidróxido de sodio3 se seguirá a través de la disminución de la absorbancia del cristal violeta CV en la región del espectro visible con longitud de onda de 590 nm, en tres corridas manteniéndose constante en dos de ellas, la
3 Corsaro, G. A colorimetric chemical kinetics experiment. J. Chem. Educ., 41(1), 48, 1964.
Figura 6.1 Mecanismo SN1
OH
N(CH3)2
SustratoCloruro de p-rosanilina
o cristal violeta
IntermediarioCarbocatión
C
N(CH3)2
OH ¯
N(CH3)2
+ (CH3)2 N
N(CH3)2
C
N(CH3)2Cl ¯N(CH3)2
Producto incoloro
N(CH3)2
C (CH3)2 N

46
concentración del cristal violeta, y en otras dos la concentración de la base. Por último se repetirán las tres corridas pero ahora con un exceso de la base.La variación de 2 absorbancias A iniciales con el tiempo t (pendiente m), es directamente proporcional ()a la velocidad inicial de la reacción r. Ecuación 6.1
At
= m r 6.1
Figura 6.3
Las velocidades iniciales son proporcionales() a las concentraciones iniciales, medidas con la absorbancia, siendo la constante de proporcionalidad la constante de velocidad. Así el logaritmo natural de la relación de pendientes iniciales m 0 con respecto al logaritmo natural de la relación de concentraciones iniciales C0 del reactivo que cambia en dos corridas, proporciona el valor del orden parcial de tal reactivo.
CV+Cl¯ + Na+OH¯ CV−OH + Na+Cl ¯
m0,1 k CCV, 0,1
���CNaOH,
0
m0,2
=k CCV, 0, 2
CNaOH, 0
6.3
m 0 kCCV, 0 CNaOH, 0
6.2
m0,1 k CCV,0
���CNaOH,0,1
m0,3
=k CCV,0
CNaOH,0,3
6.5
ln ( m 0,1/ m0,2)ln [ CCV,0,1/ CCV,0,2]
6.4
ln ( m0,1/ m0,3)ln [ CNaOH,0, 1/ CNaOH, 0, 3]
6.6
t seg
A

47
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
2 pipetas graduadas de 5 mL PE solución de tinte cristal violeta (0.02 g
2 pipetas beral de 1 mL en un litro de agua) 5 x 10 –5 M
1 termómetro solución de hidróxido de sodio NaOH
1 propipeta o jeringas con manguera 2 a 0.02 M
1 espectrofotómetro Genesys con celdas solución de nitrato de potasio KNO3 1 M
Papel tisú (Kleenex) Agua destilada hervida, libre de CO2
1 vaso de precipitados de 500 mL para residuos (uso común)
PROCEDIMIENTOAntes de preparar las mezclas para las corridas 1-3 revisar el resumen en la Tabla 6.1
Calibración del espectrofotómetro (ver página 41)
longitud de onda de 590 nm
Programar el tiempo de lectura de la absorbancia 4 minutos y el total de lecturas 4
El equipo dará las pendientes medias y al final de la corrida (16 minutos) la pendiente media final, la cual es proporcional a la velocidad.
Utilizar guantes
−Se preparan cinco tubos del espectrofotómetro Genesys , el tubo 1 es el blanco, el tubo 2 el testigo de violeta en las tres corridas. Los tubo 3, 4 y 5 serán los reactores para cada corrida como se muestra en la siguiente Tabla 6.1
Tabla 6.1Blanco Testigo Reactores
CORRIDA TUBO 1, 2.5 mL de agua
TUBO 2, 2.5 mL de CV
TUBO 3, 2.5 mL de CV
TUBO 4, 2.5 mL de CV
TUBO 5, 2.5 mL de CV
1+ 1 gota de
NaOH Tubo 2+ 1 gota de
NaOH
2+ 1 gota de
NaOH Tubo 2+ 2 gotas de
NaOH
3+ 1 gota de
KNO3
+ 1 gota deKNO3
+ 2 gotas de NaOH1 gota de KNO3
Recolectar las soluciones residuales en un vaso de precipitados de 500 mL. Al final de la sesión experimental el técnico del laboratorio se hará cargo de desechar la solución.
Utilizar guantes durante la experimentación

48
−Se realizan las tres corridas en el Espectrofotómetro, utilizando el mismo tubo 1, como blanco, el tubo 2 proporcionará la absorbancia inicial en las tres corridas, intercambiándolo, antes de los 4 minutos de la primera lectura, por el tubo reactor correspondiente
Corrida 1 seleccionar correr análisis. seleccionar modo tabular.(quedará el nombre de gráfico). Registrará la absorbancia en el
tiempo seleccionado en forma tabular insertar la celda blanco que consiste en 2.5 mL de agua con una gota de NaOH, al
espectrofotómetro con las marcas blancas diagonal a la izquierda del usuario, cerrar el compartimiento
oprimir la tecla: medir blanco, (solo si han transcurrido 30 minutos de encendido el equipo). Se oye una alarma cuando el equipo termina de leer el blanco
retirar la celda con el blanco, introducir la celda testigo (2.5 mL de Cristal violeta) oprimir la tecla medir muestra y simultáneamente por fuera introducir 1 gota de NaOH al tubo reactor agitar e intercambiarla por el testigo (tiene un poco menos de 4 minutos para intercambiar el testigo por el reactor)
al terminar el tiempo total de la cinética la toma de lecturas se detendrá, suena alarma.Aparecerá la ultima medida de Abs/tiempo
aparecerán los datos en forma de tabla se puede regresar al modo gráfico y observar la curva de pendiente negativa
Repetir el procedimiento para la Corrida 2
El blanco es el mismo tubo de la corrida 1, solo hay que agregar otra gota de NaOH. El testigo es el mismo tubo con 2.5 mL de cristal violea. El tubo reactor se intercambia por el testigo antes de los 4 minutos de la primera lectura, no sin antes haber agregado dos gotas de NaOH por fuera y simultáneamente al correr la muestra con el testigo
Corrida 3 (efecto salino primario de Brönsted)
Repetir la corridas 2, pero a la misa solución del blanco agregar 1 gota de una solución de KNO3 1 M (sal de Brönsted), al testigo y reactor también, al disparar correr análisis simultáneamente por fuera al reactor agregar 2 gotas de NaOH
Estos datos probarán el efecto de sal primaria de Brönsted (aumenta el carácter iónico del medio y si hay formación de carbocatión este se estabilizará y favorecerá la reacción SN1aumentando la “velocidad” con respecto a la de la corrida 1). o disminuyendo la velocidad (pendiente ) si el proceso es SN2 con intermediario concertado.
Registrar los datos en la Tabla 6.2

49
Tabla 6.2 Registro de datos de las corridas.
Tiempo[min] 0 4 8 12 16 Pendiente media inicial (entre 0 y 4
minutos)Corrida 1
Absorbancia
Tiempo[min] 0 4 8 12 16Corrida 2
Absorbancia
Tiempo[min] 0 4 8 12 16Corrida 3
Absorbancia
CÁLCULOSa) Con los datos obtenidos elaborar gráficas de absorbancia vs. tiempo, para cada corrida.
Efectuar las gráficas en Excel. En el mismo gráfico trazar las curvas de la corrida 1 y 3, si estas no coinciden, la reacción es de primer orden en la NaOH
d) la corrida 3 corresponde a un experimento realizado en medio iónico. Si el complejo activado tuviera carga eléctrica (carbocatión, mecanismo SN1), la ionicidad del medioestabilizaría tanto al estado base iónico (cristal violeta), como al estado de transición, no se afectaría o sufriría un aumento en el valor de la pendiente m en la corrida 3 con respecto a la corrida 2, por otro lado si el complejo activado es neutro (mecanismo SN2), el medio iónico estabilizaría solo el estado base y no al complejo activado y la pendiente m de la corrida 3 sería menor que la de las corridas 2 (reacción mas lenta)
CUESTIONARIO1.-De acuerdo a los valores de las pendientes cuales seríam los ordenes parciales y , de la
reacción entre el cristal violeta y el NaOH. ¿Cuál es el orden global de la reacción? ¿es ésta una reacción S N1 o S N2 ?. Justificar

50
O O
CH3–C–O–C–CH 3 (ac) + H 2 O (l) → 2 CH3COOH (ac)
anhídrido acético agua ácido acético
PRÁCTICA Nο. 7
CINÉTICA DE UNA REACCIÓN DE PSEUDOPRIMER ORDEN SEGUIDA POR EL MÉTODO COLORIMÉTRICO
OBJETIVO GENERALModificar la cinética de segundo orden a pseudoprimer orden, por el exceso de uno de los reactivos, el agua, que es también el disolvente en la reacción de hidrólisis del anhídrido acético
OBJETIVOS ESPECÍFICOS Seguir la cinética a temperatura ambiente de la reacción de hidrólisis entre el anhídrido
acético y el agua a través del observable absorbancia. medir la absorbancia del complejo yodo-yoduro I 3¯ obtenido mediante una reacción
acoplada a la reacción de hidrólisis determinar el orden y la constante de velocidad de la reacción de hidrólisis confirmar el mecanismo de seudoprimer orden con el valor del coeficiente de correlación R
FUNDAMENTOS TEÓRICOSLa reacción de hidrólisis entre el anhídrido acético y el agua, como se vio en la práctica de saponificación del acetato de etilo, es una reacción de segundo orden, donde el nucleófilo es el agua, la cual por ser el disolvente también, se encuentra en exceso y esto transforma a la ecuación de velocidad en una ecuación de pseudoprimer orden
r = k Canh CH2O
r = (k CH2O) Canh
r = k´ Canh ecuación de velocidad de pseudoprimer orden
La reacción anterior puede seguirse a través de la absorbancia ( 353 nm ) del complejo yodo-yoduro que se forma cuando se acopla la reacción anterior a otra donde el ácido acético formado en la reacción principal es empleado para reaccionar con yoduro y yodato como se muestra a continuación
5 KI (ac) + KIO3 (ac) + 6 CH3COOH (ac) → 3 I2 (ac) + 6 CH3COOK (ac) + 3 H2O(l)
I ¯(ac) + I2 (ac) → I 3¯ (ac)La suma de las reacciones anteriores es:

51
CH3–CO–O–CO–CH 3 + H 2 O → 2 CH3COOH
5 13
I ¯ +3
IO3¯ + 2CH3COOH →I2 + 2 CH3COO¯ + H2O
I ¯ + I2→ I 3−
8 13
I ¯ +3
IO3¯ + CH3–CO–O–CO–CH3 →I 3¯ + 2 CH3COO¯
Entonces la velocidad de reacción se puede definir como:
dCanh dC I3−
r = –dt
=dt
La ecuación anterior justifica que la reacción puede ser seguida mediante la concentración de anhídrido acético o del ion triyoduro. La ventaja del seguimiento mediante el ion triyoduro es que este es colorido y absorbe la luz UV que tiene la longitud de onda de 353 nm y la reacción puede seguirse por el aumento de la absorbancia del I 3
− , Figura 7.1
En la Figura 7.1 a la izquierda se muestra una cinética seguida midiendo concentraciones respecto al tiempo t. A la derecha, la misma cinética seguida mediante medidas de Absorbancia del producto triyoduro respecto al tiempo. Se observa un periodo de invariabilidad de la absorbancia del triyoduro en el tiempo cero (A 0), llamado período de inducción.
Si la reacción fuera de primer orden, la ecuación integrada, monitoreando la CI3ˉ, es:
CI3ˉ, f
k t = lnCI3
ˉ, f – CI3ˉ, t
Figura 7.1
A f
A t
A 0tiempo
Canh, 0
Canh, t
Canh, f
Período de inducción
tiempo

52
Si la concentración CI3ˉ, es seguida mediante la absorbancia del ión triyoduro esta ecuación se
debe transformar en:
A final – A 0k t = lnA final – A t
En el tiempo cero no hay triyoduro, por lo tanto A0 = 0
A finalk t = lnA final – A t
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
1 pipeta graduada de 5 mL anhídrido acético, QP
1 jeringa de insulina o pipeta beral yoduro de potasio KI, cristales
1 espátula yodato de potasio KIO3, cristales
1 termómetro agua destilada hervida y fría, libre de
1 espectrofotómetro Genesys con celdas CO2
1 gradilla
PROCEDIMIENTO
Calibración del espectrofotómetro
Encender el equipo y estabilizar por 30 minutos en una longitud de onda de 353 nm
oprimir la tecla Test
seleccionar cinética. ENTER
seleccionar en modo de medición, absorbancia y teclear ENTER
si la longitud de onda no es 353 nm, oprimir ENTER y escribir el valor
seleccionar tiempo retardado. ENTER, seleccionar 00:00 (min:seg) ENTER
La solución residual se neutraliza con un cristal de tiosulfato de sodio o hasta que la solución quede incolora. Desechar en la tarja.
Utilizar guantes durante la experimentación

53
seleccionar tiempo de intervalo, ENTER. Escribir el tiempo min:seg, que se necesite para reportar la absorbancia, dar ENTER. Ejemplo 2 minutos 02:00
seleccionar tiempo total, ENTER. Escribir el tiempo total que se necesite que a la reacción se le determine la absorbancia. Ejemplo 36 minutos 36:00
seleccionar modo pendiente por factor encendido, la pendiente es proporcional a la velocidad media (entre lecturas)
seleccionar correr análisis. Aparecerá una pantalla con un plano cartesiano, que registraráabsorbancia VS. Tiempo
seleccionar modo tabular.(quedará el nombre de gráfico). Registrará la absorbancia en el tiempo seleccionado en forma tabular.
Preparación del blanco:
Las celdas del espectrofotómetro al momento de usarse deben de estar limpias y secas. No lavar con jabón, solamente con agua destilada y alcohol tanto en la parte interna como externa, secar suavemente la parte externa con papel kleenex. Siempre usar guantes, para evitar depositar grasa.
Llenar una celda, con 2.5 mL de agua destilada, libre de CO2 colocar un cristalito de KI y otro de KIO3, agitar para disolver
elegir correr análisis
introducir el blanco al equipo, cerrar la tapa
oprimir la tecla correr el blanco, al terminar se oye una alarma
Para iniciar la cinética se prepara la mezcla de reacción como sigue:
retirar el blanco y añadir una micro gota de anhídrido acético, con la pipeta beral o jeringa de insulina, agitar perfectamente, introducir la celda al equipo e inmediatamente teclear correr la muestra
La cantidad de anhídrido acético que se debe poner es muy pequeña, de lo contrario la reacción será muy rápida y no podrá leerse la absorbancia, el equipo titilará
registrar en la Tabla 7.1, los datos de absorbancia contra tiempo, durante 36 minutos(opcional registrar todos) y el tiempo y la absorbancia que permanezca constante (absorbancia final). la invariabilidad de la absorbancia en las últimas mediciones comprueba que la reacción ha terminado

54
al terminar el tiempo total de la cinética la toma de lecturas se detendrá, suena una alarma
regresar al modo gráfico, oprimiendo esta opción, aparecerá la curva absorbancia del triyoduro contra tiempo.
lavar las celdas solo con agua destilada, si hay alcohol, también. Dejar secar y guardar
con la tecla ESC, se va saliendo de las pantallas (no guardar análisis) hasta llegar a longitud de onda. Con el botón posterior OFF-ON apagar el equipo
desconectar y guardar.Tabla 7.1
Tiempo[min]
Absorbancia
02468
1012141618202224262830323436
Abs. I3–
tiempo seg

55
CALCULOS
a) trazar la gráfica absorbancia vs. tiempo (para los32 minutos)
b) calcular la constante de velocidad de reacción observada (pseudo) con el procedimiento tabular, a partir de los datos de absorbancia A y tiempo, solo en una región recta de la gráfica anterior. Se debe tomar en cuenta que la reacción de hidratación del anhídrido acético es seudomolecular,
e) ajustar los datos con cuadrados mínimos
AfinallnAfinal ¯ A t
vs. t
f) obtener nuevamente el valor de kobservada (pendiente), el coeficiente de correlación lineal R, de regresión lineal R2 y confirmar si la cinética es de pseudoprimer orden
CUESTIONARIO
1.- Calcular el tiempo de vida media de la reacción mediante la ecuación t1/2 = ln 2/ kobservada, y comparar con el tiempo de vida media que corresponde en la gráfica del inciso a)absorbancia vs. t cuando, A t = A final / 2. Escriba sus conclusiones acerca de estos datos.
2.- La reacción desarrollada en la práctica es de pseudoprimer orden debido a que un reactivo se agregó en exceso. Si quisiera determinar el tiempo de vida media de la reacción ¿cuál ecuación debe usar para calcularla, la de orden uno o la de orden dos?
Afinal = _____________
AfinaltSeg
AbsorbanciaA t
Afinal ¯ A tAfinal ¯ A t
= A ln A kobservada = ln A / t

56
3.- En esta práctica, el seguimiento de la reacción ocurre por un procedimiento indirecto, ya que se determina la concentración del I 3¯ y no la concentración de un reactivo (anhídrido acético) o de un producto. Escribe una justificación del porqué no es necesario hacer una determinación cuantitativa de la concentración del ión triyoduro para desarrollar la ecuación de velocidad.
4.- La reacción entre el anhídrido acético y el agua se puede monitorear por absorbancia, gracias a la reacción acoplada del yoduro para producir yodo. ¿puede proponer otro observable para monitorear la reacción?
5.- En la siguiente gráfica se observa el consumo de aceético, la formación del yodo simultáneamente en el tiempo cero, pero hay un intervalo en el tiempo en que el triyoduro no se forma, ¿cómo se le llama a este intervalo común en las reacciones consecutivasABC?
I2Acético
I3¯
t00

57
PRÁCTICA No. 8
CINÉTICA DE UNA REACCIÓN CATALIZADA.(en microescala)
OBJETIVO GENERALObservar el efecto de un catalizador sobre la velocidad de reacción
OBJETIVOS ESPECÍFICOS Seguir la cinética de la reacción
2H2O2 (l)Yoduro
→ 2H2O ( l ) + O2 ( g )
catalizada mediante el ion yoduro midiendo la producción del oxígeno a través del aumento de presión del sistema de reacción utilizando un sensor de presión.
comprobar el modelo cinético de primer orden para la reacción catalizada. tratar los datos obtenidos de presión de acuerdo con el modelo propuesto. obtener el coeficiente de regresión y correlación para confirmar el modelo. desarrollar la reacción en microescala para utilizar menores cantidades de reactivos con
respecto a las técnicas experimentales tradicionales
FUNDAMENTOS TEÓRICOSLa velocidad de una reacción química puede aumentarse con el uso de un catalizador.
La reacción entre el dióxido de azufre y el oxígeno para formar el trióxido de azufre en la obtención de ácido sulfúrico es un paso muy lento pero cuando se le agrega pentóxido de vanadio la reacción se acelera:
V2 O5SO2(g) + O2(g) → SO3(g)
La reacción de descomposición del agua oxigenada de acuerdo a la siguiente reacción es muy lenta
2 H2O2 (l) → 2 H2O(l) + O2 (g)
Cuando al agua oxigenada se le agrega una solución de yoduro de potasio la velocidad de descomposición aumenta
2H2O2 ( l)Yoduro
→ 2H2O ( l) + O2 ( g )
En los ejemplos anteriores el pentóxido de vanadio V2O5 y el yoduro I − son catalizadores.
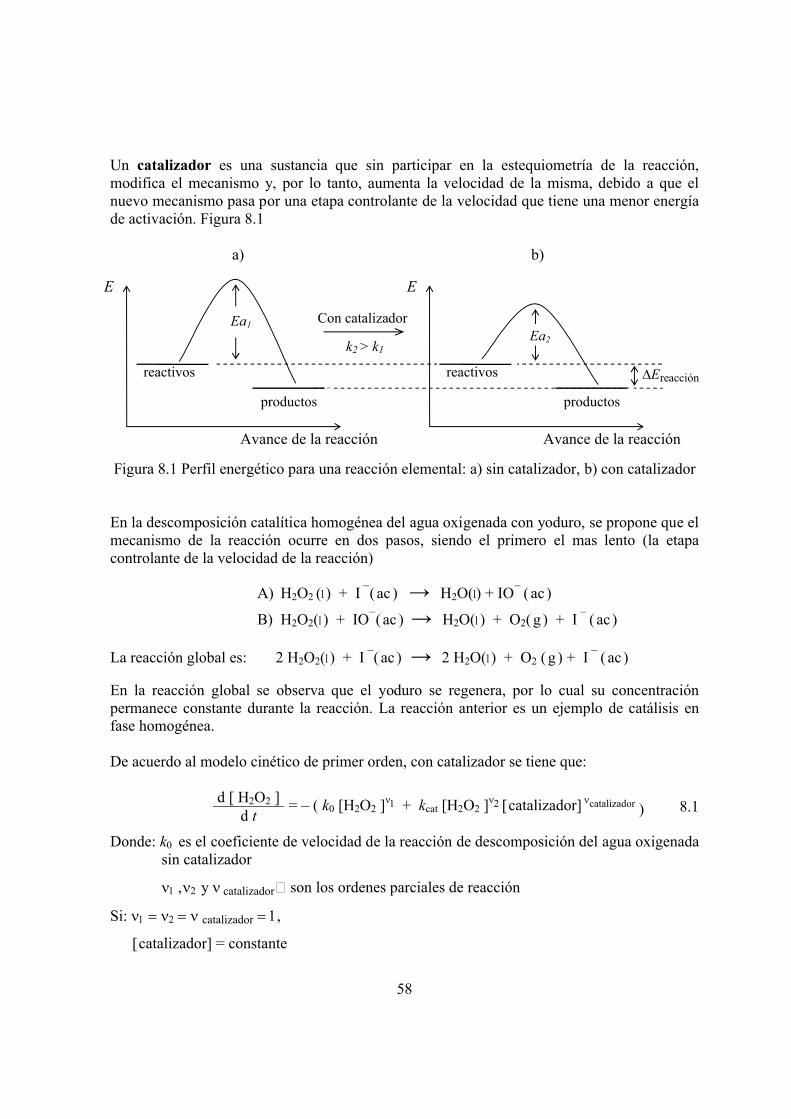
58
Un catalizador es una sustancia que sin participar en la estequiometría de la reacción, modifica el mecanismo y, por lo tanto, aumenta la velocidad de la misma, debido a que el nuevo mecanismo pasa por una etapa controlante de la velocidad que tiene una menor energía de activación. Figura 8.1
Figura 8.1 Perfil energético para una reacción elemental: a) sin catalizador, b) con catalizador
En la descomposición catalítica homogénea del agua oxigenada con yoduro, se propone que el mecanismo de la reacción ocurre en dos pasos, siendo el primero el mas lento (la etapa controlante de la velocidad de la reacción)
A) H2O2 (l ) + I −( ac ) → H2O(l) + IO− ( ac )
B) H2O2(l ) + IO−( ac ) → H2O(l ) + O2( g ) + I − ( ac )
La reacción global es: 2 H2O2(l ) + I −( ac ) → 2 H2O(l ) + O2 ( g ) + I − ( ac )
En la reacción global se observa que el yoduro se regenera, por lo cual su concentración permanece constante durante la reacción. La reacción anterior es un ejemplo de catálisis en fase homogénea.
De acuerdo al modelo cinético de primer orden, con catalizador se tiene que:
d [ H2O2 ]d t
= – ( k0 [H2O2 ] + kcat [H2O2 ]
catalizador]catalizador ) 8.1
Donde: k0 es el coeficiente de velocidad de la reacción de descomposición del agua oxigenada sin catalizador
y catalizador� son los ordenes parciales de reacción
Si: catalizador
catalizador] = constante
Ea1
Ea2
Avance de la reacción
k2 > k1
E E
Ereacción
b)
Avance de la reacción
reactivos
Con catalizador
a)
productos productos
reactivos

59
y, si el valor de la constante de velocidad k0 es muy pequeño, la ecuación cinética es:
d [ H2O2 ]d t
= – kobs [ H2O2 ] 8.2
Donde la constante de velocidad observada es kobs = kcat catalizador]
La integración de la ecuación anterior es:
[H2O2]0ln[H2O2]t
= kobs t 8.3
El seguimiento cinético de la reacción puede ser realizado mediante la medición de la evolución del oxígeno, que es la única sustancia en fase gaseosa. En un reactor a volumen constante, la generación del oxígeno se manifiesta con un aumento de presión en el sistema, y la reacción puede ser monitoreada con un sensor de presión. El desarrollo de la reacción se puede seguir mediante la curva de presión vs. tiempo, en lugar de la curva de concentración vs. tiempo, como se muestra en la Figura 8.2, donde P0 es la presión inicial del sistema, Pt es la presión en el tiempo t y Pf la presión final, cuando la reacción no avanza más y ésta permanece constante.
Figura 8.2 Comparación del monitoreo de la reacción midiendoConcentraciones o presiones
Se puede observar que la concentración inicial [H2O2]0 es proporcional a la cantidad total del oxígeno producido durante toda la reacción [O2]f que se puede medir por la diferencia total de presiones en el experimento cinético Pf – P0, por lo tanto
[H2O2]0 = 2 [O2]f, 8.4o sea
[H2O2]0 = 2κ (Pf – P0) 8.5
donde κ es la constante de conversión de la presión producida por el oxígeno generado a las unidades de concentración. κ es igual 1/RT. En la reacción global se observa que la relación estequiométrica entre el H2O2 y el O2 es 2:1.
CH2O2,0
C H2O2,t
Pf
Pt
P0tiempo tiempo

60
Una vez iniciada la reacción, la concentración de H2O2 en el tiempo es [H2O2] t, menor que [H2O2]0, ya que el agua oxigenada se degrada en la producción del oxígeno gaseoso:
[H2O2]0 – [H2O2] t = 2 [O2] t 8.6
El oxígeno producido es proporcional a Pt – P0:
[O2] t = κ (Pt – P0) 8.7
entonces [H2O2] t = [H2O2]0 – 2 [O2] t = 2κ (Pf – P0) – 2κ (Pt – P0) = 2κ (Pf – Pt) 8.8
y la ecuación integrada en término de presiones es
o sea
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
1 matraz de tres bocas agua oxigenada comercial al 3% p/v
1 sensor electrónico de presión solución de yoduro de potasio KI 0.1 M
1 sensor electrónico de temperatura
1 transductor CBL
1 calculadora Texas Instruments
2 jeringas de 3 mL
1 tapón de plástico Ultra-Ión
1 cronómetro
1 tapón de hule o septum
1 agitador magnético con barra
1 soporte universal con pinzas
PROCEDIMIENTO
Conectar el transductor CBL a la corriente, a la calculadora, al sensor de presión en el canal 1 y al sensor de temperatura en el canal 2. Figura 8.3
calibrar el sensor de presión tomando como referencia la presión atmosférica y otra presión utilizando el manómetro de mercurio
2κ (Pf – P0)ln2κ (Pf – Pt)
= kobs t 8.9
(Pf – P0)ln(Pf – Pt)
= kobs t 8.10

61
Figura 8.3 Calibración de la presión
armar el equipo como se muestra en la Figura 8.4 apoyado apropiadamente en un soporte universal con base de agitación magnética
Sensor detemperatura
Eliminador de corriente
Sensor de presión
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
1234567890 =
Eliminador decorriente
Sensor de presión
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
1234567890 =
Eliminador de corriente
Sensor de presión
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
1234567890 =
Eliminador de corriente
Sensor de presión
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
Sensor de presión
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
calculadora
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
Q W E R T Y U I O P ` + 7 8 9 $ % + *
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Z X C V B N M , . - 1 2 3 0 & = .
1234567890 =
-
1234567890 =1234567890 =
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Q W E R T Y U I O P ` + 7 8 9 $ % + *
Z X C V B N M , . 1 2 3 0 & = .
calculadora
1234567890 =
Eliminador de corriente
Eliminador de corriente
Sensor de presión
Sensor de temperatura
Figura 8.4 Instrumentación para la práctica
Manómetro de mercurio
A S D F G H J K L Ñ ´ 4 5 6 ( ) _ /
Q W E R T Y U I O P ` + 7 8 9 $ % + *
Z X C V B N M , . 1 2 3 0 & = .
calculadora
1234567890 =
Sensor de presión
Eliminador de corriente
Válvula de tres vías
Jeringa

62
insertar el sensor de temperatura a una boca del reactor y el sensor de presión a otra de las bocas, utilizando como boquilla una porción de manguera como se muestra en la Figura 8.5
Figura 8.5
revisar que las conecciones no presenten fuga
antes de cerrar el reactor, añadir con la jeringa 2 mL de agua oxigenada
cerrar el reactor con el tapón de hule o septum.
programar la calculadora para registrar la temperatura y la presión del sistema, cada 30 segundos durante 15 minutos (30 lecturas)
sin la jeringa, accionar el sistema de adquisición de datos. Durante 2 minutos observar que la reacción es muy lenta
después de ese tiempo, agregar al reactor 1 mL de solución de yoduro de potasio, con la jeringa, cuidando no introducir aire. Retirar la jeringa después de la adición. Continuar la adquisición de datos
observar que la reacción se acelera
la reacción habrá terminado cuando la presión del sistema permanece constante(aproximadamente 15 minutos)
obtener de la calculadora la información registrada de tiempo, presión y temperatura. Corroborar que durante el experimento la variación de la temperatura sea menor a 2°C
registrar en la Tabla 8.1 los datos de tiempo y presión
─
Sensor de presión
Solución de agua oxigenada
Reactor
Tapón de hule o séptum
Sensor de temperatura
Solución de yoduro de potasio
Agitador magnético

63
Tabla 8.1 Datos de presión y tiempo de la cinética catalizada del agua oxigenada con yoduro
Tiempo 0 30 60 90 120 150 180 210 240 270 300
Presión
Tiempo 330 360 390 420 450 480 510 540 570 600 630
PresiónTiempo 660 690 720 750 780 810 840 870 900 930 960PresiónTiempo 990 1020 1050 1080 1110 1140 1170 1200 1230 1260 1290PresiónTiempo 1320 1350 1380 1410 1440 1470 1500 1530 1560 1590 1620PresiónTiempo 1650 1680 1710 1740 1770 1800Presión
CÁLCULOSa) Trazar la gráfica de presión vs. tiempo
b) Trazar la gráfica de ln{(Pf – Po)/(Pf - Pt)} vs. tiempo, solo en la parte recta de la curva.
c) Ajustar con cuadrados mínimos los valores de ln{(Pf – Po)/(Pf - Pt)} vs. tiempo, obtener la pendiente (k observada), el coeficiente de correlación R y el de regresión R2.
CUESTIONARIO1.- ¿Qué modelo cinético se propone para la reacción de descomposición del agua oxigenada
catalizada con yoduro?2.- ¿Los valores de R y R2 confirman el modelo propuesto?3.- Escribir los comentarios al respecto de la gráfica de presión vs. tiempo, en particular
respecto al punto de “inicio” de la reacción (cuando t = 300 segundos)4.- Justificar el hecho de que la ecuación de velocidad de descomposición del H2O2 contiene 2
términos:
5.- En una reacción, cuál es el efecto de un catalizador sobre:a) La velocidad de la reacciónb) la forma como se lleva a cabo la reacción (mecanismo)c) la constante de equilibriod) el rendimiento
d [ H2O2 ]d t
= – ( k0 [H2O2 ] + kcat [H2O2 ]
catalizador]catalizador )

64

65
PRÁCTICA No. 9
OBTENCIÓN DE LOS PARÁMETROS DE LA ISOTERMA DE ADSORCIÓN DE LANGMUIR
(CINÉTICA EN FASE HETEROGENEA)
OBJETIVO GENERALDeterminar los parámetros de la ecuación de adsorción de Langmuir y la superficie específica de un adsorbente
OBJETIVOS ESPECÍFICOS Definir los conceptos de: adsorción, desorción, sorbato y sorbente explicar las características de la adsorción química y de la física definir isotermas de adsorción. describir la isoterma de adsorción química y la isoterma de adsorción física aplicar el modelo de adsorción-desorción de Langmuir calcular con el modelo matemático de Langmuir ajustado a una línea recta, la constante de
equilibrio de adsorción-desorción K y la cantidad de moles de sorbato N necesarias para cubrir la superficie del sorbente
determinar con el valor de N, la constante de Avogadro L y el área transversal de la molécula de sorbato , el área superficial A en m2/g, del carbón activado empleado.
FUNDAMENTOS TEÓRICOSLa adsorción es un fenómeno superficial que ocurre cuando dos fases están en contacto, hay una región llamada interfase donde la composición es diferente a la del resto de las fases. El aumento en la concentración de una sustancia en la interfase, comparada con la concentración en la mayor parte, es conocida como adsorción. En concreto, adsorción es cuando un soluto se adhiere sobre una superficie.
Sobre la superficie de un sólido pueden ser adsorbidas o fijadas sustancias de un gas o un líquido (fluido). Al sólido se le llama adsorbente o sorbente y a las partículas adsorbidas sorbato o adsorbato.
Aún las superficies mas cuidadosamente pulidas vistas microscópicamente, presentan irregularidades en toda el área. Estas superficies son particularmente susceptibles a la presencia de campos de fuerzas residuales. En estos lugares (también llamados “sitios activos”), los átomos de la superficie del sólido pueden atraer a otros átomos o moléculas de la fase fluida circundante (ver Figura 9.1). Es así que la actividad superficial de los sólidos es responsable del mecanismo de la adsorción.

66
Figura 9.1
La adsorción de un soluto sobre una superficie puede ser física o química.
En la adsorción física el adsorbato se enlaza al adsorbente con fuerzas relativamente débiles, del tipo van der Waals, es reversible, la energía de activación del proceso es muy pequeña o no se aprecia y, además, se puede formar mas de una capa de soluto adsorbidos.
En la adsorción química hay compartición de electrones entre el adsorbato y la superficie del adsorbente, así que ocurre una reacción química. El enlace formado entre el adsorbente y el adsorbato es esencialmente químico y por tanto mas fuerte que en la adsorción física. La quimiadsorción algunas veces requiere de energía por lo que es también llamada adsorción activada y como en toda reacción química, se manifiesta una energía de activación en el proceso, el calor de adsorción es usualmente mayor que en la adsorción física, da lugar solamente a la formación de una capa de grosor monomolecular y es irreversible.
La relación entre la cantidad de sorbato sobre una determinada cantidad de masa de adsorbente y la concentración de la fase fluida en un experimento realizado a temperatura constante, cuando se representan gráficamente, se llama isoterma de adsorción, debido a que es necesario que el experimento de adsorción se realice a temperatura constante. La forma del trazo que aparece en estas gráficas varía cuando cambian cualesquiera de los dos componentes del experimento de adsorción, el sólido o el soluto. Sin embargo, las formas mas frecuentemente encontradas se clasificas en 5 tipos, de las que vamos analizar dos.
La isoterma de adsorción de tipo I se caracteriza porque el tramo inicial es de pendiente alta, que gradualmente se va aproximando a una recta paralela al eje de las abcisas. La parte inicial se debe a que la superficie no cubierta es fuertemente adsortiva y es mas probable el fenómeno de adsorción que el de desorción y, en la medida de que los sitios activos se van ocupando, la probabilidad de que ocurra la desorción aumenta, hasta que ambos procesos se hacen igualmente probables. La cantidad de soluto adsorbido (medido en número de moles de moléculas adsorbidas o en forma del volumen que estas moléculas ocuparían si estuvieran en fase gaseosa en condiciones normales de presión y temperatura) depende de dos características del sistema: la fuerza de atracción entre las moléculas adsorbibles y la superficie y de la cantidad disponible de superficie. Cuando la superficie queda cubierta con
Soluto adsorbido(sorbato)
Soluto en soluciónCentro activo
Sorbente

67
todas las moléculas que puede adsorber se dice que queda “saturada” y esto explica la parte final de la isoterma.
Figura 9.2 Isoterma de tipo I
La isoterma de tipo II inicia de la misma manera que la isoterma de tipo I, pero no presenta el fenómeno de saturación, sino que al final la curva es monótonamente creciente. Esto se debe a que las moléculas adsorbidas atraen a otras moléculas de la fase libre formando adsorbatos de 2, 3 o más moléculas apiladas.
Figura 9.3 Isoterma de tipo II
La adsorción química o quimisorción produce casi exclusivamente isotermas de tipo I, a diferencia de la adsorción física o fisisorción que generalmente da isotermas de tipo I o de tipo II.
Concentración o presión del fluido (sorbato)
Volumen adsorbidode sorbato o soluto
por unidad de masa de sorbente
Volumen adsorbido de adsorbato por
unidad de masa de sorbente
Presión o concentración relativa del adsorbato

68
En 1916 Irving Langmuir propuso un modelo sencillo para el comportamiento del fenómeno de adsorción, que en particular es muy útil para la quimiadsorción, limitada en su forma original, a una capa monomolecular.
Langmuir consideró que en el sistema de una superficie sólida y un soluto, las moléculas chocarían continuamente con la superficie, y una fracción de ellas quedarían adheridas al sólido. Sin embargo, debido a las energías, rotacional y vibracional de las moléculas adsorbidas las de mayor energía se desprenderían continuamente de la superficie. Se establecerá un equilibrio en condiciones de estado estacionario (equilibrio dinámico), de tal manera que la velocidad a la cual las moléculas golpean la superficie ra y permanecen en ella por una cantidad apreciable de tiempo, se equilibraran exactamente con la velocidad a la cual las moléculas se desprenden de la superficie rd. Figura 9.4, Ecuación 9.1.
Figura 9.4
Sólido + molécula [sólido – molécula]
donde: velocidad de adsorción ra = ka C(1–) 9.2 velocidad de desorción rd = k d 9.3
las ecuaciones de velocidad anteriores corresponden a una cinética de primer orden.donde:
1– = fracción de superficie no ocupada = fracción de superficie ocupada o cubiertaka = constante de velocidad de adsorciónkd = constante de velocidad de desorciónC = concentración del soluto en la solución en equilibrio con el
adsorbente
En el equilibrio adsorción-desorción:
ra = rd 9.4 ka C(1–) = kd 9.5
ka
kd
9.1
rard

69
k a =k d
C(1–) 9.6
La constante de equilibrio del proceso adsorción-desorción de acuerdo a la Ecuación 9.1 es:
Concentración de productos K= Concentración de reactivos =
C(1–) 9.7
de las Ecuaciones 9.2 y 9.3 se tiene:rd =kd
raC (1–)=ka
rd /kdK=ra /ka
En el estado estacionario, las velocidades de adsorción-desorción son iguales por tanto la relación ka/kd ha sido sustituida por una constante K que cumple la definición de la constante de equilibrio para las reacciones bidireccionales.
kaK=kd
9.8
Por lo que sustituyendo en la Ecuación 9.6 se tiene
= K C(1–) 9.9
= KC– KC
KC + = KC
[1 +KC] = KC
= KC / (1 +KC)
La fracción de superficie cubierta por el sorbato es:
= n/N 9.10
donde:n = cantidad de moles de soluto adsorbido por gramo de adsorbenteN = cantidad de moles de soluto adsorbido por gramo para cubrir totalmente la superficie

70
Sustituyendo el valor de se tiene:
n /N = KC / (1 +KC) 9.11
N/n = (1 +KC) / KC 9.12
N/n = 1/ (KC) + 1 9.13
1/n = 1/ (NKC) + 1/N 9.14
C/n = 1/ (NK) + C/N 9.15
La Ecuación 9.15 corresponde a una línea recta donde la pendiente es 1/N y la ordenada en el origen contiene a la constante de equilibrio de adsorción–desorción Figura 9.5
El área especifica del adsorbente (A) se puede calcular si se conoce el área transversal del soluto adsorbido (σ), es decir el área ocupada por una molécula de adsorbato en el adsorbente y si se supone que sus moléculas están muy cerca una de la otra formando una monocapa:
A =NxLxσx10– 20 9.16
Donde: A = área superficial del adsorbente en m2/g σ = área transversal del adsorbato en Å2/ molécula L = constante de Avogadro, 6.022 x 1023 moléculas/mol
El último factor convierte los Å2 en m2
Figura 9.5C
C/n

71
DESARROLLO EXPERIMENTAL
MATERIAL REACTIVOS
2 buretas de 50 mL carbón activado comercial
6 embudos Bückner de filtración ácido acético 0.4 M, preparado con agua
6 Kitazatos con manguera de vacío hervida (fría, libre de CO2)
12 matraces Erlenmeyer de 125 mL hidróxido de sodio 0.1 M, preparado con
6 matraz volumétrico de 50 mL agua hervida (fría, libre de CO2)
1 pipeta volumétricas de 10 mL* fenolftaleina
1 soporte universal piseta con alcohol para secar el material
1 pinzas con nuez de vidrio
1 pinzas para bureta agua destilada hervida (fría, libre de CO2)
1 base para agitación magnética recipiente con hielo
6 agitadores magnéticos
papel filtro Wathman No. 5 para elembudo Bückner (6 por equipo)
6 tapones de hule del No 4 para el kitazato, monohoradados
6 tapones de hule para matraz Erlenmeyer de 125 mL
1 pistola de aire
bomba de vacío
trampa de cloruro de calcio anhidro
*La pipeta volumétrica puede sustituirse por jeringa de 10 mL, con la ventaja de que no introducen CO2 al sistema. La titulación puede hacerse con jeringa con la misma ventaja.
PROCEDIMIENTO Pesar 1 g de carbón activado (con una tolerancia de 1 mg) en cada uno de 6 matraces
Erlenmeyer de 125 mL enumerar cada uno de los matraces anteriores y agregar a cada uno, una de las siguientes
soluciones colocar en una bureta de 50 mL la solución de ácido acético 0.4 M. preparar en un matraz volumétrico de 50 mL, las siguientes soluciones con ácido acético
0.4 M. Enjuagar el matraz con agua destilada cada vez que se prepare una nueva solución (solo para las soluciones 2 hasta la 4).

72
Solución 1: tomar con la bureta 1 mL de ácido acético ( Vacético ) y aforar a 50 mL con agua destilada. Mezclar la solución y agregar al matraz número 1 que contiene el carbón.
Solución 2: tomar con la bureta 5 mL de ácido acético, aforar, mezclar y adicionar al matraz 2.
Solución 3: tomar con la bureta 10 mL de ácido acético, aforar, mezclar y adicionar al matraz 3.
Solución 4: tomar con la bureta 15 mL de ácido acético, aforar, mezclar y adicionar al matraz 4.
Solución 5: tomar con la bureta 20 mL de ácido acético, aforar, mezclar y adicionar al matraz 5.
Solución 6: tomar con la bureta 25 mL de ácido acético directamente de la bureta al matraz 6
Solución 7: tomar con la bureta tomar con la bureta 30 mL de ácido acético, aforar,mezclar y adicionar al matraz 7.
Solución 8: tomar con la bureta 35 mL de ácido acético, aforar, mezclar y adicionar al matraz 8.
Solución 9: tomar con la bureta 40 mL de ácido acético, aforar, mezclar y adicionar al matraz 9.
Solución10: tomar con la bureta 45 mL de ácido acético directamente de la bureta al matraz 10
Solución 3: verter con la bureta 50 mL de ácido acético, aforar y adicionar al matraz 11.
agitar manualmente los matraces Erlenmeyer con el carbón activado y las respectivas soluciones, durante 15 minutos.
filtrar al vacío las soluciones Figura 9.6
Figura 9.6 de cada uno de los filtrados: tomar 10 mL y vaciarlos en un matraz Erlenmeyer limpio.
Adicionar dos gotas de fenolftaleína y titular con hidróxido de sodio 0.1 M. Registrar los gastos obtenidos en la Tabla 9.1. (incluir diluciones de 1, 15, 25, 35 y 45 mL)
Filtrado
Matraz Kitazato con embudo
Baño de hielo
Trampa de CaCl2 anh.
Bomba de vacío

73
CÁLCULOSa) Calcular la concentración del ácido acético inicial C0 con la fórmula:
C0 = V acético x 0.4M /50 mL
y la del equilibrio C después de la adsorción:
C = (MNaOH x VNaOH) / 10 mL
Registrar en la Tabla 9.2
b) calcular la cantidad de mol de acético adsorbido n por gramo de adsorbente:
n = ( C0 - C ) 0.05L
Registrar en la Tabla 9.3.
c) Realizar el gráfico n contra C equilibrio (verificar si es de tipo 2)
Tabla 9.1
Temperatura _________ Ácido acético 0.4 M, Hidróxido de sodio _________M
Matraz V acético mL V NaOH mL1 12 53 104 155 206 257 308 359 4010 4511 50

74
c) calcular la relación de la concentración C ( Tabla 9.2 ) de soluto en equilibrio con la cantidad n de mol adsorbidos solo en la zona de monocapa, C/n y registrar los cálculos en la Tabla 9.3
Tabla 9.3 datos de la monocapaMatraz 1 2 3 4 5 6
Cn
C / n
d) hacer una gráfica de C/n contra C. Comparar la gráfica obtenida con la Figura 9.5
e) ajustar los datos experimentales C/n contra C a una línea recta con el método de cuadrados mínimos. Escribir en la Tabla 9.4 los valores de pendiente, ordenada en el origen, el coeficiente de correlación R y el de regresión lineal R2.
f) determinar con la pendiente y la ordenada en el origen del ajuste, la cantidad N y el coeficiente de adsorción K según la Ecuación 9.16.
Tabla 9.4 Regresión lineal
Coeficiente lineal dePendiente
Ordenada en el
origenN K correlación R regresión R2
g) calcular el área especifica del adsorbente, considerando que el área (σ) del ácido acético es igual a 21 Å2. Ecuación 9.16.
CUESTIONARIO
Tabla 9.2
MatrazC0
[mol/ L]C
[mol /L]1234567891011

75
1. Definir adsorción, soluto, adsorbato y adsorbente.
2. Mencionar dos diferencias entre la adsorción física y la química
3. ¿Qué es una monocapa en el contexto del fenómeno de la adsorción?
4. En la presente práctica ¿qué sustancia es el soluto? _______________ y ¿cuál el adsorbente?______________
5. Dibuje la gráfica n contra C.
6. Dibuje la gráfica de ln n contra ln C y ajuste los datos por el método de mínimos cuadrados. Compare el valor del coeficiente de correlación R de este ajuste con el que aparece en la Tabla 9.4 y comente al respecto.









![CATALISIS 1-[14]](https://static.fdocuments.es/doc/165x107/563db7b8550346aa9a8d5130/catalisis-1-14.jpg)








