
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09...
45
702 7 El paciente con problemas hematológicos ANEMIAS Y POLICITEMIAS Composición de la sangre La sangre está constituida por (Ver Imagen 1): • Un componente líquido (55% del volumen sanguíneo): el plasma san- guíneo. Es de color amarillo pálido y está compuesto en su mayor parte por agua y por solutos disueltos (proteínas, sustancias orgánicas e iones inorgánicos) (Ver Tabla 1). • Un componente sólido (40-45% del volumen sanguíneo): las células sanguíneas. – Hematíes, eritrocitos o células rojas (aproximadamente 5.000.000/mm 3 de sangre). Su función principal es el transporte de oxígeno. – Leucocitos o células blancas (5.000-10.000/mm 3 de sangre). Son las únicas células sanguíneas que poseen núcleo. Se clasifican en: • Granulocitos (neutrófilos, basófilos y eosinófilos): también de- nominados polimorfonucleares por presentar gránulos en el ci- toplasma y varios lóbulos. Su función principal es la fagocitosis. • Linfocitos: poseen un núcleo esférico de gran tamaño ro- deado por el citoplasma, formando un pequeño anillo, por lo que se les denomina leucocitos mononucleares, al igual que a los monocitos. Son los principales responsables de la res- puesta inmunitaria. • Monocitos: son los leucocitos de mayor tamaño, con un nú- cleo único y redondeado. Su función es la fagocitosis. – Plaquetas o trombocitos (150.000-450.000/mm 3 de sangre). Son el resultado de la segmentación del citoplasma de los grandes megacariocitos en la médula ósea, por lo que carecen de núcleo Imagen 1. Composición de la sangre Hematíes: 4,2-5,8 x 10 6 /mm 3 Leucocitos: 5.000-10.000/mm 3 Plaquetas: 250.000-400.000/mm 3 Albúmina: 55% Fibrinógeno: 6% Globulinas: 38% Nutrientes Iones Desechos Gases Hormonas 55% 7% 2% 91% 45% Plasma Células Proteínas Agua Otros solutos Componentes del plasma Agua Iones inorgánicos Na + K + Ca 2 + Cl - H 2 CO 3 /HCO 3 - HPO 42 - /H 2 PO 4 - Sustancias orgánicas Lípidos Carbohidratos Urea, ácido úrico Otras: hormonas, enzimas, vitaminas, fermentos Proteínas Albúminas Globulinas Fibrinógeno g/l de plasma del total 910 (91%) 3,25 0,2 0,1 3,65 1,65 0,4 6 1 0,30; 0,03 – 70 (6-8%) 44 (55%) 22 (38%) 3 (7%) Función Regulación de la temperatura corporal Medio de transporte de sustancias, etc. Mantenimiento de la presión osmótica Mantiene constante el pH Suministros celulares Suministro energético Productos de excreción del metabolismo celular – Reserva proteica y transporte de sustancias Inmunológica y transporte de sustancias Coagulación sanguínea Tabla 1. Componentes del plasma
Transcript of Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09...

702
7 El paciente con problemas hematológicos
ANEMIAS Y POLICITEMIAS
Composición de la sangre
La sangre está constituida por (Ver Imagen 1):
• Un componente líquido (55% del volumen sanguíneo): el plasma san-guíneo. Es de color amarillo pálido y está compuesto en su mayor partepor agua y por solutos disueltos (proteínas, sustancias orgánicas e ionesinorgánicos) (Ver Tabla 1).
• Un componente sólido (40-45% del volumen sanguíneo): las célulassanguíneas.– Hematíes, eritrocitos o células rojas (aproximadamente
5.000.000/mm3 de sangre). Su función principal es el transportede oxígeno.
– Leucocitos o células blancas (5.000-10.000/mm3 de sangre). Sonlas únicas células sanguíneas que poseen núcleo. Se clasifican en:• Granulocitos (neutrófilos, basófilos y eosinófilos): también de-
nominados polimorfonucleares por presentar gránulos en el ci-toplasma y varios lóbulos. Su función principal es la fagocitosis.
• Linfocitos: poseen un núcleo esférico de gran tamaño ro -deado por el citoplasma, formando un pequeño anillo, por loque se les denomina leucocitos mononucleares, al igual quea los monocitos. Son los principales responsables de la res-puesta inmunitaria.
• Monocitos: son los leucocitos de mayor tamaño, con un nú-cleo único y redondeado. Su función es la fagocitosis.
– Plaquetas o trombocitos (150.000-450.000/mm3 de sangre). Sonel resultado de la segmentación del citoplasma de los grandesmegacariocitos en la médula ósea, por lo que carecen de núcleo
Imagen 1. Composición de la sangre
Hematíes: 4,2-5,8 x 106/mm3
Leucocitos: 5.000-10.000/mm3
Plaquetas: 250.000-400.000/mm3
Albúmina: 55%Fibrinógeno: 6%Globulinas: 38%
NutrientesIonesDesechosGasesHormonas
55%
7%
2%
91%
45%
Plasma
Células
Proteínas
Agua
Otros solutos
Componentes del plasma
Agua
Iones inorgánicosNa+
K+
Ca2+
Cl-H2CO3/HCO3
-
HPO42-/H2PO4
-
Sustancias orgánicasLípidosCarbohidratosUrea, ácido úricoOtras: hormonas, enzimas, vitaminas, fermentos
ProteínasAlbúminasGlobulinasFibrinógeno
g/l de plasma del total
910 (91%)
3,250,20,1
3,651,650,4
61
0,30; 0,03–
70 (6-8%)44 (55%)22 (38%)
3 (7%)
Función
Regulación de la temperatura corporal Medio de transporte de sustancias, etc.
Mantenimiento de la presión osmóticaMantiene constante el pH
Suministros celularesSuministro energéticoProductos de excreción del metabolismo celular–
Reserva proteica y transporte de sustanciasInmunológica y transporte de sustanciasCoagulación sanguínea
Tabla 1. Componentes del plasma
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 702

703
enfermería médico-quirúrgica
y tienen forma de discos ovales o circulares, de 2 ó 3 μm de diá-metro. Desempeñan un papel fundamental en la coagulación dela sangre.
• Un componente gaseoso: fundamentalmente O2 y CO2. La mayor partede estos gases son transportados por los hematíes, mientras que unapequeña proporción se halla disuelta en el plasma.
Anemia
Se entiende por anemia o eritrocitopenia la disminución del número de he-matíes, reflejada por una disminución del nivel de hemoglobina (Hb) y del he-matocrito (Hto).
• Según su etiopatogenia, se pueden clasificar según se recoge en la Tabla 2.• Morfológicamente, se clasifican según el volumen corpuscular medio
(VCM), que es el Hto x 10/nº hematíes en millones (Ver Tabla 3).– Microcítica (menor de 80 micras cúbicas).– Normocítica.– Macrocítica (mayor de 100 micras cúbicas).
• Otra forma de clasificarlas es en función del nivel de saturación de Hb,medido mediante la Hb corpuscular media (HCM), que expresa la can-tidad de Hb media presente en el hematíe: Hb (g/l)/nº de hematíes (x1012/l) (picogramos):
– Hipocrómica (disminución del color, por disminución del contenidode Hb).
– Normocrómica (color normal).
Anemia ferropénicaEs una anemia microcítica e hipocrómica. La ferropenia es la causa más fre-cuente de anemia, producida por:
• Disminución del aporte procedente de una dieta inadecuada.• Excesivas pérdidas.• Malabsorción.• Aumento de las necesidades de hierro.
Técnicas y medios de diagnósticoEl diagnóstico se basará en las siguientes determinaciones de laboratorio:
• Determinación de la concentración de hierro y ferritina en sangre (dis-minuida).
• Determinación del VCM, HCM (microcítica e hipocrómica).• Valores de Hb proporcionalmente más bajos que los de hematocrito
(Ver Tabla 4).
Medidas terapéuticasSe debe buscar la causa primaria que provoca la anemia y su tratamiento. Laadministración del hierro puede llevarse a cabo de tres formas:
• Ferroterapia: sal ferrosa hasta complementar los depósitos de hierro,que ocurre normalmente a los 3-6 meses.
• Dieta rica en hierro.• Transfusión de eritrocitos.
Anemia por enfermedades crónicasGeneralmente es normocrómica y normocítica. Entre los mecanismos princi-pales de este tipo de anemia estaría una disminución de la función medularcomo consecuencia de sustancias como el interferón o el factor de necrosis tu-moral, producidos en el curso de enfermedades crónicas, disminución de lavida media del hematíe y un bloqueo en la utilización del hierro.
Medidas terapéuticasEl tratamiento irá encaminado a resolver la enfermedad de base. No está in-dicada la administración de hierro a pesar de una sideremia disminuida,puesto que la causa no es la falta de hierro, sino su mal uso.
Anemia megaloblásticaEs un tipo de anemia carencial causada por déficit de vitamina B12 o ácido fó-lico, lo que produce una disminución de la velocidad de síntesis del ADN.
Tabla 2. Clasificación etiopatogénica
Anemias regenerativas
Anemias arregenerativas
Sangrado agudo
Anemias hemolíticas• Corpusculares:
– Alt. membrana: esferocitosishereditaria, hemoglobinuriaparoxística nocturna, etc.
– Enzimopatía: glucosa 6 fosfatodeshidrogenasa
– Defectos hemoglobinas:talasemias, síndromes falciformes,etc.
• Extracorpusculares:– Inmunológicas: anticuerpos
calientes y fríos, enfermedadhemolítica del recién nacido,transfusionales, etc.
– Agentes tóxicos– Agentes infecciosos– Causas mecánicas:
microangiopatías, válvulas yprótesis vasculares
– Hiperesplenismo
Insuficiencias medularesAplasia medular
DesplazamientosLeucemias, linfomas y tumores sólidos
Trastornos metabólicos• A. ferropénica• A. megaloblásticas• A. por enfermedades crónicas• Déficit hormonal (EPO, hs. tiroideas, etc.)
Tabla 3. Clasificación morfológica de las anemias
VCMdisminuido
FerropeniaEnfermedad crónica,a vecesSideroblásticasUremiaTalasemias
VCMnormal
Enfermedad crónicaHemólisisMixdemaHepatopatía, a veces
VCMaumentado
MegaloblásticasHipotiroidismoAplasiaMielodisplasiaHepatopatía crónicaReticulocitosis
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 703

704
Manual DAE de enfermería. EIR. Oposiciones ••
La deficiencia de ácido fólico es la causa más frecuente de este tipo de ane-mia. La deficiencia de vitamina B12 es la causa más habitual de anemia per-niciosa.
Técnicas y medios de diagnóstico• Extensión de sangre periférica.• Por déficit de vitamina B12.• Por déficit de ácido fólico.
Medidas terapéuticasPor déficit de vitamina B12: administrar esta vitamina de forma parenteral; encaso de anemia perniciosa hay que administrarla de por vida. En estos casosse debe potenciar la adherencia al tratamiento a pesar de la mejoría clínica.En caso de déficit alimentario, los pacientes pueden responder al aporte oral.
Por déficit de ácido fólico: administrar ácido fólico o folínico si es por altera-ción de la folato-reductasa. Si la causa es farmacológica valorar su supresión.
Anemia hemolíticaSe entiende por anemias hemolíticas las que se deben a una mayor destruc-ción de hematíes (sobreviven menos de 15-20 días). Las anemias hemolíti-cas se clasifican en intracorpusculares, si el defecto está en el propio hematíe,o extracorpusculares, si se debe a una agresión externa al mismo. Con excep-ción de la hemoglobinuria paroxística nocturna, las anemias hemolíticas in-tracorpusculares son congénitas y las extracorpusculares adquiridas.
Anemias corpuscularesSe clasifican en tres grandes grupos:
• Alteraciones de la membrana.• Enzimopatías.• Defectos de la Hb:
– Talasemia: se puede clasificar en ß-talasemias y α-talasemias.• Dentro de las ß-talasemias, caracterizadas por tipocromía y
microcitosis, se encuentran la talasemia maior u homocigótica(anemia de Cooley) y la talasemia minor o heterocigótica.
• Dentro de las α-talasemias se distinguen cuatro grupos:• Rasgo α-2 talasémico (deleción de uno de los cuatro loci de
la α-globulina): es asintomático.
• Rasgo α-1 talasémico (deleción de 2 loci): similar a ß-talasemia.• Enfermedad por HbH (deleción de 3 loci).• Hidropesía fetal con Hb Bart (ausencia total de síntesis de α-
globina y formación de tetrámeros de gammaglobina, deno-minados Hb Bart).
– Hemoglobinopatías: la mutación de un aminoácido ocasiona laformación de cadenas de hemoglobinas anormales con funcióndefectuosa del transporte de oxígeno.
Anemias extracorpuscularesLa hemólisis en este tipo de anemias tiene lugar por una agresión externasobre el hematíe y se clasifican en autoinmune o no autoinmune. Los fárma-cos pueden inducir inmunidad de forma directa, produciendo anticuerposque atacan a los hematíes (metildopa), o de forma indirecta, el anticuerpoataca al fármaco que, a su vez, interactúa con el hematíe (penicilina).
El diagnóstico de las anemias hemolíticas se basa en:
• Recuento de reticulocitos (elevado).• Prueba del fenómeno drepanocítico.• Niveles séricos de hierro y bilirrubina (aumentados por la hemólisis).• Electroforesis de Hb para detectar deformaciones estructurales.• Prueba de Coombs (anemia hemolítica inmunitaria):
– Directa: determina la presencia de inmunoglobulina en la super-ficie de los eritrocitos.
– Indirecta: determina la presencia de inmunoglobulinas en elplasma.
Anemia por pérdida de sangreEstá causada por una pérdida sanguínea aguda o crónica. El tratamiento sebasa en el control de la hemorragia, la sustitución del volumen sanguíneo yla administración de suplementos de hierro si la pérdida es crónica.
Anemia aplásicaConsiste en un descenso en el número de células madre en la médula ósea,lo que provoca una disminución de todas las células sanguíneas (pancitope-nia). Este tipo de anemia puede ser congénita o adquirida. Es una anemianormocítica y normocrómica.
Hematíes
Hb
Fe sérico
Ferritina
Fe en médula ósea
Transferrina
Saturación de transferrina
Hb A2
CHCM
HCM
VCM
A. ferropénica
m
m
m
m
m
i
m
m
m
m
m
A. trastornos crónicos
m
m
m
i
i
m
m o ok
ok
ok o m
ok o m
ok o m
β-talasemia
ok o i
ok o i
ok
ok
i
ok
ok
i
ok o m
m
m
α-talasemia
ok o i
ok o m
ok
ok
i
ok
ok
m
ok o m
m
m
Tabla 4. Diagnóstico diferencial de las anemias
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 704

705
enfermería médico-quirúrgica
Hallazgos físicosDebido a la pancitopenia, además del síndrome anémico (fatiga, disnea, pa-lidez, etc.), el paciente presenta una mayor tendencia a hemorragias (sín-drome purpúrico) e infecciones.
Los individuos afectos de anemia de Fanconi presentan, además de lo ante-rior, diversas alteraciones, siendo las más frecuentes las cutáneas (manchascafé con leche) y las óseas (malformaciones del radio o hipoplasia del pulgar).
Técnicas y medios de diagnósticoEl diagnóstico se basa en la presencia de pancitopenia y recuento reticuloci-tario disminuido. El diagnóstico definitivo se consigue con la aspiración y biop-sia de la médula ósea, en la que se objetiva hipocelularidad. Se considerasevera cuando la celularidad en médula ósea es inferior al 25-30% del valornormal y además existen menos de 500 neutrófilos/mm3, menos de 20.000plaquetas/mm3 o menos del 1% de reticulocitos.
Medidas terapéuticasEl tratamiento de elección es el trasplante de médula ósea (si el paciente tieneun donante HLA-idéntico).
Policitemia
La policitemia, también denominada poliglobulia o eritrocitosis, es el au-mento de la masa eritrocitaria, por encima de 6.000.000 mm3, relacionado conel aumento del valor del hematocrito. Un Hto por encima del 55% en muje-res y el 60% en hombres implica casi siempre un aumento de la masa eritro-citaria.
La policitemia se clasifica en:
• Policitemia vera o primaria.• Policitemia secundaria a aumento de eritropoyetina.
– Por hipoxia.– Por tumor (hipernefroma).– Por andrógenos.– Por enfermedades renales.
• Policitemia relativa o pseudopoliglobulia.
Técnicas y medios de diagnóstico• Hemograma: aumento del número de eritrocitos, de la concentración de
Hb y del hematocrito.• Determinación de la velocidad de sedimentación (aumentada).• Determinación de vitamina B12 (aumentada).• Niveles séricos de bilirrubina (aumentados por el rápido recambio celular).• Niveles séricos de hierro (disminuidos).• Nivel sérico de ácido úrico (incrementado por hipermetabolismo de las
nucleoproteínas).• Histamina sérica aumentada en la policitemia vera.• Curva de afinidad de Hb por el oxígeno y saturación de oxígeno.• La diferencia principal entre policitemia primaria y secundaria es que en
la policitemia vera existe un aumento de la celularidad en las tres series,mientras que en las policitemias secundarias este aumento es exclu-sivo de la serie roja.
Medidas terapéuticas• De la policitemia primaria: flebotomía (extracción de sangre a interva-
los irregulares).• De la policitemia secundaria: solucionar el problema primario y, en caso
necesario, flebotomía para disminuir la hipervolemia e hiperviscosidad.
ALTERACIONES DE LA COAGULACIÓN
La hemostasia se define como un proceso para la prevención de la pérdidade sangre (Ver Imagen 2).
Clasificación
Defecto de los vasos sanguíneos• Púrpura simple y alérgica.• Telangiectasia hemorrágica hereditaria.
• Vasculopatía hipertensiva.
Alteración plaquetaria• Púrpura trombocitopénica idiopática (PTI).• Púrpura trombocitopénica trombótica (PTT).• Síndrome hemolítico urémico.
Alteración de factores de la coagulación
Congénitos• Hemofilia, A y B.
Te conviene recordar...
✔ El papel fundamental de los hematíes es el transporte de oxígeno a través de la Hb.✔ La prevalencia de los distintos tipos de anemia difiere según la zona geográfica en que se esté. Las causas principales de la ane-
mia son la pérdida excesiva de hematíes o la producción insuficiente.✔ La anemia ferropénica constituye el 85% del total de anemias.✔ Algunos tipos de anemia están condicionados fuertemente por factores socioculturales como hábitos nutricionales; por ello,
la educación para la salud se convierte en una herramienta básica.✔ El padecimiento de la enfermedad supone para el paciente grandes cambios en su rol.✔ La policitemia es el aumento de la población eritrocitaria. Puede deberse a una disminución del volumen plasmático (polici-
temia relativa) o a un verdadero aumento del número de hematíes, el cual se puede deber a una enfermedad primaria (poli-citemia vera) o a un proceso reactivo a diferentes alteraciones (policitemia secundaria).
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 705

706
Manual DAE de enfermería. EIR. Oposiciones ••
Adquiridos• Hepatopatías.• Intoxicación por cumarínicos.• Hipoprotombinemia.• Coagulación intravascular diseminada (CID).
Etiopatogenia. Factores de riesgo
Defecto de la pared de los vasos sanguíneosEs el proceso de hemostasia menos conocido. Esta alteración incluye incre-mento de la fragilidad capilar, innata o adquirida. Clínicamente está caracte-rizada por petequias cutáneas y púrpura con tendencia a formar hematomascon facilidad y sangrado.
Algunas de estas alteraciones son:
• Púrpura simple.• Telangiectasia hemorrágica hereditaria.• Púrpura alérgica.• Vasculopatía hipertensiva.
Alteración plaquetaria
La coagulación comienza con la adherencia plaquetaria y la formación de untapón de plaquetas. Cualquier trastorno que curse con disminución del númerode plaquetas o impida la capacidad de éstas para adherirse puede manifes-tarse como un trastorno hemorrágico (Ver Tabla 5).
Cuando el número real de plaquetas está por debajo de lo necesario parala coagulación normal se denomina trombocitopenia. Puede ser el resul-tado de:
• Defecto de producción (fármacos).• Aumento de la destrucción (radioterapia, quimioterapia, etc).
Los trastornos trombocitopénicos más frecuentes que afectan a los pacientesadultos son:
• Púrpura trombocitopénica autoimnune o idiopática (PTI): los pacientessintetizan un anticuerpo dirigido contra sus propias plaquetas, anti-cuerpo antiplaquetario.
• Púrpura trombocitopénica trombótica (PTT): se caracteriza por unaagregación plaquetaria anormal dentro de la microcirculación.
• Síndrome hemolítico urémico: trastorno caracterizado por la instaura-ción súbita de trombocitopenia y de hemólisis con hematíes fragmen-tados e insuficiencia renal aguda.
Alteración de factores de la coagulaciónSe produce como resultado de un defecto en alguno de los factores de la coa -gulación por:
• Incapacidad para sintetizar un factor específico.• Síntesis y cantidad insuficiente de un factor determinado.• Síntesis de una forma menos activa de factor.
CongénitosHemofilia: déficit de factor VIII o de factor IX. El 85% corresponde a la hemo-filia A, enfermedad genética ligada al sexo, transmitida por las mujeres y pa-decida por los hombres. La gravedad de la hemorragia guarda relación directacon el grado de déficit.
Adquiridos• Hepatopatías.• Intoxicación por cumarina.• Hipoprotrombinemia: la protrombina es una proteína que se sintetiza
en el hígado como parte de un proceso químico que depende de la vi-tamina K.
• Coagulación intravascular diseminada (CID): caracterizada por la for-mación anormal y difusa de muchos coágulos dentro de la microcircu-lación (Ver Imagen 3).
Imagen 2. Proceso de la hemostasia
Rotura del vaso
VasoconstricciónAdherencia plaquetariaLiberación de sustancias
(tromboplastina)
Tapón plaquetario
Trombina
Fibrinógeno
Fibrina
Coágulo estable
Protrombina
Vía intrínseca Vía extrínseca
Tabla 5. Fármacos que pueden contribuir a latrombocitopenia
Acohol
Quinina
Quinidina
Digoxina
Fenitoína
Ácido acetilsalicílico
Sulfonamidas
Compuesto de oroIbuprofeno
Furosemida
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 706

707
enfermería médico-quirúrgica
Hallazgos físicos
Tegumentarios• Petequias.• Equimosis.• Hematomas.• Púrpura.• Palidez.• Piel fría.
Digestivos• Hemorragias gingivales.• Hematemesis.• Melenas.• Rectorragias.• Distensión y plenitud abdominal.• Dolor abdominal.
Renales• Hematuria.• Dificultad para orinar.• Oliguria.
Reproductores• Metrorragias.
Pulmonares• Epistaxis.• Hemoptisis.• Disnea.• Taquipnea.• Cianosis.
Cardiovasculares• Taquicardia.• Hipotensión.
Neurológicos• Vértigo.• Cefalea.• Alteraciones visuales.• Hemorragia conjuntival.
Músculo-esqueléticos• Dolor articular.• Hemartrosis.
• Acrocianosis.
Técnicas y medios de diagnóstico
Cuando se sospecha sangrado o existe evidencia de hemorragia, el procesodiagnóstico comienza con un recuento hematológico completo que incluye elnúmero de plaquetas.
Complicaciones potenciales
Estas complicaciones vienen determinadas por la severidad de la alteración ypueden variar desde ningún trastorno hasta crisis que ponen en grave riesgola vida:
• Anemia.• Insuficiencia cardiaca.• Insuficiencia respiratoria.• Insuficiencia renal.• Embolia pulmonar.• Trombosis.• Artralgia.• Shock hipovolémico.
Medidas terapéuticas
Dada la variedad de elementos que pueden ocasionar tanto alteraciones pla-quetarias como de coagulación, el objetivo del tratamiento será el control dela hemorragia y la coagulación, así como el reequilibrio ácido-base y el con-trol de la hemostasia.
• Púrpura trombocitopénica idiopática (PTI). Corticoesteroides, quimio-terapia a dosis bajas, transfusiones de plaquetas, administración de in-munoglobulinas y esplenectomía.
• Púrpura trombocitopénica trombótica (PTT). Corticoesteroides, recam-bio plasmático.
• Síndrome hemolítico urémico. Hemodiálisis y tratamiento de soporte.• Hemofilia. Reposición de factores.• Hepatopatías. Tratamiento de soporte, administración de plasma, de
crioprecipitados y de plaquetas.• Intoxicación por cumarínicos. Interrumpir el fármaco y administración
de plasma.• Hipoprotrombinemia. Administración de vitamina K, tratamiento de
soporte y administración de plasma.• Coagulación intravascular diseminada (CID). Administración de he-
parina, administración de factores y tratamiento de causa subya-cente.
Imagen 3. Proceso de la CID
Daño tisular Liberación de sustancias tisulares (tromboplastina)
Activación del proceso de coagulación
Hipoxia y necrosis tisular en zonas de microcirculación
HEMORRAGIA Consumo de factores y de PDF
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 707

708
Manual DAE de enfermería. EIR. Oposiciones ••
LEUCEMIAS Y LINFOMAS (EIR 99-00, 57)
Leucemias
ClasificaciónSegún los tipos celulares implicados, se dividen en linfoide (alteración de lin-focitos) y mieloide (alteración de monocitos, granulocitos-basófilos, neutró-filos y eosinófilos, eritrocitos y plaquetas). Según la historia natural de laenfermedad, en aguda y crónica (Ver Tabla 6).
EtiopatogeniaLa causa que provoca la leucemia es desconocida (Ver Tabla 7). Las célulasneoplásicas no pueden cumplir su función, ya que existe inmadurez de leu-cocitos o glóbulos blancos y de linfocitos (dificultad de combatir infecciones),de eritrocitos o glóbulos rojos (produciendo anemias) y de trombocitos o pla-quetas (alterando la coagulación).
Técnicas y medios de diagnóstico (Ver Tabla 8)• Evaluación hematológica: 50.000-300.000 leucocitos/mm3. Frecuente
aumento de eosinófilos y basófilos. Anemia en gran parte de los casos.• Biopsia medular ósea: gran porcentaje de las células nucleadas de la
médula ósea son formas de leucocitos inmaduros llamados blastos.• Biopsia de ganglios linfáticos, para valorar afectación ganglionar.• Radiografía de tórax, para objetivar ganglios mediastínicos e infiltra-
ción pulmonar.• Radiografía del esqueleto, con el fin de objetivar lesiones esqueléticas.
• Métodos inmunológicos empleados con el fin de detectar la línea ce-lular en la etapa de diferenciación.
• Demostración del pH en las células de la médula ósea en leucemia cró-nica mieloide.
Medidas terapéuticasLa utilización de fármacos antineoplásicos pretende impedir la reproducciónde las células tumorales. Existen distintos protocolos de tratamiento que per-miten combinar estos fármacos. El objetivo es llegar a la remisión de la enfer-medad (cuando la leucemia y sus síntomas pueden ser detenidos de formatemporal o permanente) para después mantenerla.
Los medicamentos más empleados en esta quimioterapia se reflejan en laTabla 9.
Asistencia de apoyoConsiste en la administración de hemoderivados (concentrados de hematíesy plaquetas), antibióticos, antimicóticos y antivíricos. Si se dispone de mé-dula ósea histocompatible con el paciente, se recurre al trasplante de médulaósea tras conseguir la remisión de la enfermedad.
Trasplante de médula óseaProcedimiento consistente en la extracción de una cantidad de médula óseadel donante. Después de filtrada se infunde por vía intravenosa a un pacientecompatible.
Al penetrar la médula en el torrente sanguíneo estas células se trasladan hacialas cavidades medulares donde se implantan, crecen y se dividen. De estaforma, el paciente puede producir células sanguíneas sanas. Se distinguenvarios tipos de trasplantes según la procedencia de la médula ósea:
• Sinérgico: la médula ósea procede de gemelos univitelinos, por lo tanto,es idéntica a la del receptor desde el punto de vista genético e inmuno-lógico. Está limitado por la escasa frecuencia de gemelos univitelinos.
• Autólogo: la médula procede del propio paciente.
Te conviene recordar...
✔ En condiciones normales, el cuerpo humano está protegido contra las pérdidas hemáticas abundantes y mortales como resul-tado de mecanismos numerosos, complejos e interrelacionados.
✔ Las anormalidades que predisponen a las enfermedades hemorrágicas pueden afectar a vasos, plaquetas o a cualquiera de losfactores de la coagulación. Algunos pacientes pueden tener varios defectos a la vez.
✔ La hemorragia puede ser una manifestación de un defecto primario de la coagulación (como en la hemofilia), ser secundariaa otra enfermedad, cirrosis, uremia o, incluso, deberse a fármacos como los cumarínicos.
Tabla 6. Clasificación de las leucemias
Agudas
Leucemia linfoblástica aguda (LLA)Leucemia mieloblástica aguda (LMA)
Crónicas
Leucemia linfoide crónica (LLC)Leucemia mieloide crónica (LMC)
Tabla 7. Posibles causas de las leucemias
Factores físicosRadiaciones (rayos X, tratamiento con quimioterapia)Factores químicosQuimioterapiaVapores de benzol y otros productos químicosFactores genéticosDeterminados virus
Tabla 8. Manifestaciones de laboratorio en leucemia
50.000-300.000 leucocitos/mm3
Aumento de eosinófilos y basófilosAnemia moderada normocítica normocromaPresencia de eritroblastos, en ocasionesTrombocitopeniaHiperuricemia
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 708

709
enfermería médico-quirúrgica
• Alogénico: la médula ósea procede habitualmente de un hermano ge-nético e inmunológicamente diferente al receptor, pero con compatibi-lidad del sistema HLA (antígenos leucocitarios humanos).
Linfomas
ClasificaciónSe clasifican en linfoma de Hodgkin (responsable del 14% de todos los lin-fomas malignos) y linfoma no Hodgkin, entre los que se encuentran linfosar-coma, sarcoma de células reticulares, linfoma de Burkitt y linfoma cutáneode células T.
Enfermedad de HodgkinSu causa es desconocida, aunque no se descartan:
• Factores víricos.• Factores inmunológicos.• Factores genéticos.
La célula característica de esta neoplasia es la de Reed-Sternberg (R-S) y la dela respuesta defensiva del huésped, el linfocito, por lo tanto, cuantos más lin-focitos se encuentren, mejor pronóstico tendrá el paciente, por el contrario,cuantas más células R-S haya, peor pronóstico. La clasificación de los linfomas
se realiza según la clasificación histológica de Rye (1965) y la clasificación clí-nica de Ann Arbor (1971) (Ver Tabla 10).
Linfomas no HodgkinNueva Working Formulation:
• Bajo grado: tipos favorables. Tumores de células B.– Linfocítico pequeño.– Folicular, células hendidas.– Folicular, mixto de células hendidas pequeñas y grandes.
• Grado intermedio: tipos desfavorables. Linfomas de células B y algu-nos de células T.– Folicular, células grandes.– Difuso, células hendidas pequeñas.– Difuso, mixto de células grandes y pequeñas.– Difuso, células grandes.
• Alto grado: tipos desfavorables. Linfomas inmunoblásticos sobre todode células B, tumores linfoblásticos de células T, tumores de células nohendidas pequeñas de Burkitt y no burkittiano de células B.– Inmunoblástico de células grandes.– Linfoblástico.
Vincristina
Prednisona
Daunorrubicina
Asparraginasa
Mercaptopurina
Metotrexato
Citarabina
Clorambucilo
Busulfán
Hidroxiurea
Ciclofosfamida
Tioguanida
Fludarabina
VP 16
Mitoxantrona
Idarrubicina
m-ANSA
VM 26
ATRA
Imatinib
Tabla 9. Quimioterapia de uso habitual en el tratamiento leucémico
Clasificación histológica (Rye)
Predominio linfocítico. 5-10%Proliferación de linfocitos e histiocitosCélulas de R-S escasas. Pronóstico excelente
Esclerosis nodular. 30-60%Aparecen bandas de colágenoCélulas R-S escasean. Buen pronóstico
Celularidad mixta. 20-40%Infiltración de cápsula ganglionarGran número de células de R-S y polimorfismo celularSupervivencia 5 años, 50-60%
Depleción linfocítica. 5-10%Histiocitos atípicos. Muchas células de R-S típicas Supervivencia menor al 50%
Clasificación histológica (Ann Habor)
Estadio IAfectación de una sola región ganglionar o de un solo órgano extralinfático
Estadio IIAfectación de dos o más grupos regionales linfáticos al mismo lado del diafragma o afectación de órgano extralinfático y/o más grupos ganglionares del mismo ladodel diafragma
Estadio IIIAfectación de grupos ganglionares a los lados del diafragma con afectación de órganos extralinfáticos o bazo o ambos
Estadio IVAfectación difusa o diseminada de uno o más órganos extralinfáticos (hígado,médula, pleura, pulmón, hueso, piel) o tejidos con o sin afectación de ganglios
Tabla 10. Clasificación de las enfermedades de Hodgkin
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 709

710
Manual DAE de enfermería. EIR. Oposiciones ••
– Células pequeñas no hendidas.
Etiopatogenia
Enfermedad de HodgkinLos ganglios linfáticos que se afectan tienen su arquitectura normal desor-ganizada con un infiltrado linfoide o pleomórfico compuesto por células lin-foides, macrófagos, eosinófilos y células plasmáticas. Habitualmente seobservan células de R-S que servirán para confirmar el diagnóstico, ya queno se encuentran en otros linfomas.
En principio es afectado un ganglio linfático y, posteriormente, llega al restodel cuerpo por los vasos linfáticos. También puede aparecer en el hígado, bazo,vértebras, uréteres y bronquios.
Linfomas no HodgkinForman un grupo heterogéneo de tumores malignos que pueden desarro-llarse a partir de cualquiera de los elementos celulares que forman los gan-glios linfáticos y/o restantes estructuras linfoides del organismo.
Hallazgos físicosLas manifestaciones clínicas de los linfomas se resumen en las Tablas 11 y 12.
Técnicas y medios de diagnóstico• Biopsia de ganglios linfáticos, para identificar las características histo-
lógicas comunes.• Hematología completa.• Radiología y tomografía del tórax para descubrir la afección mediastí-
nica, hiliar o intrapulmonar.• TAC para ver el lugar exacto de invasión ganglionar. Usado también
para planificar el tratamiento y la vigilancia.• Biopsia de médula ósea.• Pruebas de función hepática y gammagrama.• Linfangiograma, que muestra el tamaño de los ganglios linfáticos. Des-
cubre crecimientos de ganglios linfáticos abdominales, lo que probable-mente no se objetive en el TAC.
• Clasificación quirúrgica de la etapa (laparotomía con esplenectomía,biopsia hepática, múltiples biopsias de ganglios linfáticos, etc.) parapacientes seleccionados.
Medidas terapéuticas
Tratamiento del linfoma de HodgkinEl tratamiento óptimo del linfoma de Hodgkin no está plenamente estable-cido aún, aunque con el desarrollo de la radioterapia moderna, el índice de cu-ración se ha elevado hasta el 60-90%, dependiendo de la presencia o ausenciade factores pronósticos adversos (Ver Tabla 13).
Las bases del tratamiento son la radioterapia (en sus formas localizadas) y laquimioterapia combinada (en sus formas diseminadas) o unida con radiote-rapia.
Tratamiento de los linfomas no HodgkinEs necesario, antes de iniciar el tratamiento, establecer un diagnóstico histo-lógico exacto y determinar la extensión de la enfermedad. La mayoría de lospacientes reciben tratamiento combinado de citostáticos según el grado deafectación y la agresividad del tumor.
Linfomas de bajo grado de malignidad:
• Estadios localizados (I-II): la radioterapia es la indicación principal, contasas de control local mayores del 90%.
Tabla 11. Manifestaciónes clínicas de la enfermerdad deHodgkin
Adenopatías superficiales indolorasSobre todo cérvico-supraclaviculares
Síntomas constitucionales o síntomas B• Fiebre inexplicada• Sudoración nocturna• Pérdida de peso mayor del 10%
Síntomas inespecíficos• Anorexia• Astenia• Debilidad generalizada• Prurito
Según avanza la enfermedad• Infiltración• Esplénica• Hepática• Pleuropulmonar• Ósea• Neurológica• Renal
Tabla 12. Manifestaciónes clínicas del linfoma de no Hodgkin
Adenopatías indoloras y de consistencia variable: más frecuentes en cadenascervicales, axilares, paraaórticas e inguinales. En linfomas de alto grado, masasganglionares adheridas a piel con signos inflamatorios
Síntomas generales: anorexia, pérdida de peso, sudoración,etc.
Síntomas compresivos • Síndrome de vena cava superior, linfedema, compresión espina, etc. • Hepatomegalia, esplenomegalia, dolores óseos y complicaciones metabólicas
Afectación gastrointestinal: ictericia, calambres abdominales y diarreassanguinolentas
Datos de laboratorio • VSG acelerada o descenso de sideremia • Anemia hemolítica inexplicada
Tabla 13. Factores pronósticos adversos
Sexo masculino
Edad superior a 40 añosSíntomas B: fiebre elevada, sudoraciones nocturnas y pérdida de pesoEstadio avanzado
Infección extraganglionar múltiple
Invasión de la médula ósea, hígado o sistema nervioso central
Enfermedad voluminosa abdominal
Fracaso del tratamiento quimioterápico previo
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 710

711
enfermería médico-quirúrgica
• Estadios avanzados (III-IV): radioterapia linfoide, como indicación prin-cipal. La quimioterapia con cloramubucilo o ciclofosfamida con o sinesteroides o la poliquimioterapia dan una remisión más o menos rá-pida, aunque no consiguen curaciones, y con menos del 25% en remi-sión a los diez años.
• En jóvenes, con grupos seleccionados, la estrategia a seguir es intentar la re-misión de la enfermedad con poliquimioterapia agresiva e, incluso, con eltrasplante autólogo de médula ósea tras remisión con terapia alternativa.
Linfomas de grados intermedio o alto de malignidad:
• Estadios localizados (I-II): quimioterapia como primera elección, aun-que algunos estudios han demostrado buenos resultados en el uso deradioterapia en estadios localizados y no voluminosos.
• Estadios avanzados (III-IV): poliquimioterapia en todos.• Trasplante de médula ósea: es un procedimiento terapéutico empleado
en los linfomas, ya descrito anteriormente.
TRASPLANTE DE MÉDULA ÓSEA (TMO)
El TMO es una posibilidad terapéutica para algunos pacientes con trastornoshematológicos. Es un procedimiento que consiste en:
• Tratamiento con altas dosis de quimioterapia y radioterapia combina-das (tratamiento mieloablativo) para intentar eliminar la enfermedadhematológica de base.
• Infusión por vía intravenosa de células progenitoras hematopoyéticas re-cogidas de un donante con el fin de instaurar la hemopoyesis normal,sustituyendo la médula enferma por otra sana.
Tipos de TMO
Según el tipo de donante• Singénicos: cuando el donante es un gemelo con genotipo idéntico al
receptor.• Alogénicos: cuando el donante y el receptor son genotípicamente dife-
rentes. Se pueden dar los siguientes casos:– Emparentados: se recurre a un familiar.– No emparentados: se trata de donantes sin ningún nexo familiar
con el receptor. Merece un apartado especial el denominado tras-plante de sangre de cordón umbilical de donante no emparen-tado (TSC-DNE). La sangre del cordón umbilical es una fuentealternativa de progenitores hematopoyéticos. Ha quedado demos-trada su capacidad de regenerar la función medular tras un trata-miento mieloablativo.
• Autólogos: cuando la fuente medular es la propia médula del paciente,previamente obtenida y criopreservada.
Según la fuente de progenitores• Médula ósea: se obtiene mediante múltiples punciones y aspiraciones
en crestas ilíacas, esternón, mesetas tibiales, etc.• Sangre periférica: se induce la movilización de los precursores hemopo-
yéticos a la sangre periférica mediante factores de crecimiento, quimio-terapia o la combinación de ambos.
• Sangre de cordón umbilical: muy rica en precursores hemopoyéti-cos.
Según la intensidad del acondicionamiento
• Trasplante convencional.• Trasplante de intensidad reducida (minialotrasplante): se trata de un
trasplante en el que se hace un acondicionamiento (quimioterapia + ra-dioterapia) de intensidad reducida, no mieloablativa.
Procedimiento
Trasplante alogénico (emparentado y no emparentado) y singénicoUna vez seleccionado el donante se procede a la obtención de médula óseamediante uno de los siguientes procedimientos:
• Médula ósea: extracción en quirófano bajo anestesia general, mediante
Te conviene recordar...
✔ Dada su elevada incidencia en cuanto a morbimortalidad, las leucemias son enfermedades a tener en cuenta, si además se sabeque sus causas son desconocidas y, por lo tanto, no es posible realizar medidas preventivas. El tratamiento más habitual de estetipo de enfermedades es la poliquimioterapia, aunque también se puede combinar con la radioterapia y el trasplante de mé-dula ósea.
✔ El plan de cuidados de este tipo de neoplasias irá encaminado a llevar a cabo actividades para controlar los problemas que pue-den aparecer y que pueden ser letales para el paciente, como son la hemorragia y la infección y, en menor medida, la anemia.
✔ En cuanto a los linfomas, dada la importancia de su morbimortalidad, sobre todo en estadios avanzados, unido a la elevadaposibilidad de curación, hace que el diagnóstico y tratamiento precoz de los linfomas sea de vital importancia en este tipo depatologías.
✔ El tratamiento de elección va a depender del tipo de linfoma a tratar. La gama de tratamientos va desde la quimioterapia, ra-dioterapia o la unión de ambas y, en recaídas, el trasplante de médula ósea.
✔ El plan de cuidados de este tipo de pacientes irá encaminado a potenciar el conocimiento de la enfermedad, disminuir el miedoa una alteración de su imagen corporal, mantener una nutrición adecuada si hay metástasis gastrointestinal, controlar el dolorproducido por metástasis óseas y prevenir la aparición de infecciones.
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 711

712
Manual DAE de enfermería. EIR. Oposiciones ••
múltiples punciones y aspiraciones en crestas ilíacas y esternón, si fueranecesario (Ver Imágenes 4 y 5).
• Sangre periférica: mediante la estimulación de la médula ósea del do-nante con un factor estimulante de la médula que se administra a tra-vés de inyecciones subcutáneas durante varios días.
• Sangre del cordón umbilical: se obtiene después del parto medianteuna punción en la vena umbilical. Se extraen entre 70-120 ml de san-gre rica en precursores hemopoyéticos que se congelan con un conser-vante llamado dimetilsulfoxido (DMSO).
Previamente a la administración de las células precursoras hematopoyéticas,el receptor es sometido a:
• Ingreso en una unidad de aislamiento con sistema de aire filtrado a pre-sión positiva, en habitación individual con medidas de aislamiento in-vertido simple.
• Colocación de una vía central.• El paciente es sometido a dieta cocinada, es decir, que todos los ali-
mentos que consuma tienen que estar perfectamente cocinados. Enningún caso puede consumir productos crudos: hortalizas, ciertas fru-tas, embutidos, etc.
• Profilaxis antiinfecciosa.
Posteriormente, el paciente es sometido a un tratamiento de acondiciona-miento, donde se administra quimioterapia o quimioterapia y radioterapiacombinadas, que aseguren la destrucción de la médula ósea del receptor yde su patología de base, además de una inmunosupresión eficaz para la acep-tación del injerto. En la Tabla 14 aparecen varios ejemplos de acondiciona-mientos y en qué casos se usa cada uno de ellos.
Trasplante de sangre de cordón umbilical (TSCU)Este tipo de trasplante presenta una serie de ventajas con respecto al resto detrasplantes alogénicos de DNE:
• No se requiere una compatibilidad HLA completa para su éxito.• La incidencia de la EICH es menor.• La búsqueda y localización de un cordón compatible con el receptor
suele ser más rápida.• Menor riesgo de transmisión de enfermedades infecciosas.• Ausencia de riesgos para el donante.
El proceso de TSCU es similar a los trasplantes alogénicos. El receptor se vesometido a las mismas medidas anteriormente citadas y también recibe untratamiento de acondicionamiento.
Trasplante autólogoEn este tipo de trasplante es el paciente el que dona sus propios precursoreshemopoyéticos antes de someterse al acondicionamiento y el posterior tras-plante.
El procedimiento de recolección de células se puede hacer a través de la ex-tracción de la médula ósea en quirófano o a través de la movilización y reco-gida de células germinales mediante aféresis.
Las principales desventajas de este tipo de trasplante son, por un lado, elriesgo de reintroducir en el paciente células tumorales que podrían provocaruna recidiva de la enfermedad de base y, por otro lado, la ausencia del efectoantitumoral que sí existe en el trasplante alogénico.
Previamente a la infusión de las células, el paciente será sometido a un acon-dicionamiento mieloablativo (Ver Tabla 15).
Complicaciones en los trasplantes
Las principales complicaciones que surgen durante un trasplante de progeni-tores hematopoyéticos (PH) suelen estar relacionadas con:
Imagen 4. Extracción de médula ósea en quirófano
© Ce
dida p
or las
autor
as
Imagen 5. Trócares y jeringas de extracción de médula ósea
© Ce
dida p
or las
autor
as
Tabla 14. Ejemplos de acondicionamientos
Acondicionamiento
Busulfán + ciclofosfamida (BuCy)
Ciclofosfamida + radioterapia corporaltotal (CY+TBI)
Ciclofosfamida + radioterapia linfoidetotal (CY+TLI)
VP16 + radioterapia corporal total
Citarabina + radioterapia corporal total
Busulfán + ciclofosfamida +radioterapia corporal total
Indicaciones más frecuentes
Leucemia agudaLeucemia mieloide crónicaSíndrome mielodisplásico
Leucemia agudaLeucemia mieloide crónicaSíndrome mielodisplásicoLinfoma linfoblástico
Anemia aplásica severaAnemia de Fanconi
Leucemia agudaLeucemia mieloide crónicaLinfoma linfoblástico
Leucemia aguda
Leucemia agudaLeucemia mieloide crónica
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 712

713
enfermería médico-quirúrgica
• Toxicidad del tratamiento de acondicionamiento.• Inmunosupresión severa provocada.• Enfermedad injerto contra huésped (EICH).
Las principales complicaciones que se pueden derivar de un trasplante son:
• Mielosupresión: durante esta fase, el riesgo de infección es muy impor-tante debido a la neutropenia tan severa que se genera.
• Mucositis (Ver Imagen 6).• Trastornos gastrointestinales.• Cistitis hemorrágica.• Trastornos hepáticos.• Trastornos renales.• Trastornos cardiacos.• Rechazo del injerto.• Enfermedad injerto contra huésped (EICH): esta complicación sólo apa-
rece en los trasplantes alogénicos, ya sean emparentados o no. En estecaso las células inmunocompetentes del injerto son las que reconocencomo extraño al receptor que en ese momento es inmunoincompe-tente debido al tratamiento de acondicionamiento recibido. La EICHpuede manifestarse en dos fases: aguda y crónica (Ver Tabla 16).
Medidas terapéuticas
Durante el TMO al paciente se le administran gran cantidad de medicamen-tos, así como diversos hemoderivados, para prevenir hemorragias, procesos in-
fecciosos y trastornos derivados de la alta toxicidad que genera el tratamientode acondicionamiento.
No hay que olvidar toda la medicación que debe recibir para evitar la EICH, fe-nómeno que puede provocar el fracaso del TMO:
• Concentrado de hematíes.• Plaquetas.• Plasma.• Profilaxis antibiótica, antifúngica y antiviral.• Medicamentos para prevenir efectos tóxicos del acondicionamiento y
otros tratamientos de soporte.• Medicamentos para prevenir o tratar la EICH.
Tabla 15. Tratamientos de acondicionamiento en el trasplante autólogo
BEAM: BCNU + etopósido + citarabina + melfalán
CBV escalado: BCNU + etopósidio/12 h + ciclofosfamida/12 h
BEAC: BCNU, etopósido + citarabina + ciclofosfamida
BuCy: busulfán/6 h + ciclofosfamida
CyICT: ciclofosfamida + ICT
BuMe: busulfán/6 h + melfalán
ICE: ifosfamida + carboplatino + etopósido
BuCyVP: busulfán + VP 16 + ciclofosfamida
Imagen 6. Mucositis oral
© Ce
dida p
or las
autor
as
Grado
1234
Piel
Rash < 25% scRash 25-50% scEritrodermia generalVesículas y descamación
HígadoBilirrubina (mg/dl)
2-33-6
6-15> 15
IntestinoDiarrea (24 h/ml)
500-1.0001.000-1.5001.500-2.500 Dolor, hemorragia y/o íleo
Tabla 16. Clasificación de la EICH aguda
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:31 Página 713

714
Manual DAE de enfermería. EIR. Oposiciones ••
Te conviene recordar...
✔ El TMO es un procedimiento que se utiliza como tratamiento definitivo de trastornos hematológicos.✔ El paciente es sometido a un tratamiento bastante agresivo de quimioterapia y/o radioterapia (“acondicionamiento”) que le
prepara para estar en las condiciones más adecuadas para recibir la nueva médula ósea.✔ En el paciente se provoca un estado de inmunosupresión durante un periodo importante de tiempo, lo que favorece el riesgo
de adquirir infecciones diversas y otros trastornos derivados del acondicionamiento, como son la toxicidad renal, hepática,mucositis y hemorragias, entre otras.
✔ Al mismo tiempo, el tratamiento inmunosupresor con ciclosporina evita la aparición de un trastorno igualmente importantey peligroso: la EICH.
✔ Entre los distintos TMO se encuentran los siguientes:– Autólogo: el donante y el receptor son la misma persona.– Singénico: el donante y el receptor son gemelos.– Alogénico: el donante puede ser un familiar (emparentado; el hermano nunca es gemelo) o una persona ajena al recep-
tor (no emparentado).– Cordón umbilical.
✔ Aunque la infusión de la médula ósea viene a durar sólo unos minutos, el proceso del TMO completo dura de cuatro a seis se-manas (dependiendo del tipo de trasplante realizado y si no surgen complicaciones importantes). Durante este tiempo, el pa-ciente es sometido a un completo y complejo tratamiento de mantenimiento y soporte del resto del organismo combinandoantibióticos, protectores gástricos y hepáticos, diuréticos, sueroterapia, etc.
• Acero Aguilar S, Guillén Cortijo V, Blanco Curví S, Herranz Márquez N, Carro Bravo MA. Sangre de cordón umbilical. Procedimientos de recogida, donación y aspectos le-gales. Metas Enferm 2007; 10(8):64-68.
• Beare PG, Myers JL. Enfermería médico-quirúrgica. 4ª ed. Madrid: Harcourt Brace; 2003.• Braunwald E, Kasper DL, Fauci A. Harrison. Principios de medicina interna. 17ª ed. Madrid: McGraw-Hill Interamericana; 2009.• Chocarro L, Venturini C. Procedimientos y cuidados en enfermería médico-quirúrgica. Madrid: Elsevier; 2006.• De la Fuente Ramos M (coord.). Enfermería Médico-quirúrgica II. 2ª ed. Madrid: DAE, 2009.• Farreras P, Rozman C. Medicina interna. 16ª ed. Barcelona: Masson; 2008.• Fernández-Rañada de la Gándara JM, Alegre Amor A. Manual de trasplante hemopoyético. 3ª ed. Barcelona: Elsevier; 2005.• Rodés Teixior J, Guardia Massó J. Medicina interna. 2ª ed. Barcelona: Masson; 2004.• Smeltzer SC, Bare BG. Enfermería médico-quirúrgica. Madrid: McGraw-Hill Interamericana; 2005.• Soler Gómez MD, Garcés Honrubia V, Zorrilla Ayllón I. Cáncer y cuidados enfermeros. Serie Cuidados Avanzados. Madrid: Difusión Avances de Enfermería (DAE); 2007.• Swearingen PL. Manual de enfermería médico-quirúrgica. 6ª ed. Madrid: Elsevier; 2008.
BIBLIOGRAFÍA
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 714

715
ALTERACIONES TRAUMÁTICAS
Esguince
Es una pérdida de la estabilidad con impotencia funcional de una articula-ción, producida por un desgarramiento o distensión de los ligamentos, debidoa un traumatismo directo sobre la articulación.
Clasificación• Grado I: leve rotura parcial del ligamento. Se conservan la estabilidad de
la articulación, la movilidad y la fuerza.• Grado II: rotura parcial con pérdida parcial de la estabilidad de la arti-
culación y de su funcionalidad.• Grado III: rotura completa del ligamento. Pérdida total de la estabilidad
y de la funcionalidad.
Esguince de tobilloGeneralmente se produce al realizar un giro excesivo del pie hacia el interior,lo que origina un esguince del ligamento lateral externo del tobillo. La mayorincidencia de este tipo de esguinces se debe a la morfología de la articulación(maléolo externo más largo) y a su mayor grado de movilidad hacia el exte-rior (Ver Imagen 1). Un diagnóstico y tratamiento incorrectos pueden causaruna morbilidad considerable (Ver Tabla 1).
Medidas terapéuticasEl tratamiento del esguince de tobillo se establece en función del grado:
• Grado I: conservador, inmovilización con vendaje.• Grado II: conservador, inmovilización mediante enyesado.• Grado III: quirúrgico, según edad y actividad.
Esguince de rodillaCuando se produce un movimiento de la articulación de la rodilla sin coordi-nación con el pie, sus ligamentos sufren distensiones de mayor o menor gradoen función de la posición de la rodilla con respecto a la posición del pie y elgrado de rotación que se haya originado. Los traumatismos directos son otromecanismo de producción de esguinces de rodilla.
La complejidad estructural de la articulación de la rodilla hace que haya quedistinguir entre varios tipos de esguinces en función del ligamento afectado(Ver Tabla 2).
Hallazgos físicos
Esguince de tobillo• Dolor: aumenta al repetir pasivamente el mecanismo de la lesión.• Equimosis localizada.• Edema, en las horas posteriores.• Disminución o impotencia funcional: indica lesión de grado II o III.• El explorador puede determinar el grado de lesión mediante la medi-
ción del grado de subluxación astragalina. Para ello se realiza la ma-niobra del “cajón anterior de Castaing”, consistente en hacer undesplazamiento anterior del pie en equino.
Al efectuar una imagen radiológica anteroposterior se observa el grado de “bos-tezo articular” secundario al esguince o rotura del ligamento lateral externo (elmás común), lo que permite valorar el grado de la lesión (Ver Imagen 2):
• Bostezo menor de 8°: esguince leve.• Bostezo entre 8 y 15°: esguince moderado.• Bostezo mayor de 20°: esguince grave.
8 El paciente con alteraciones del movimiento
Imagen 1. Esguince de tobillo: mecanismo de producción
© DA
E
Tabla 1. Recomendaciones en el esguince de tobillo
Durante tres o cuatro días el paciente hará reposo relativo con la extremidadlevantada
Evitará cuidadosamente apoyar el pie lesionado en el suelo hasta que hayatranscurrido ese tiempo
Durante tres o cuatro días hará reposo relativo con la extremidad levantada
Colocará sobre el tobillo afectado una bolsa de hielo durante media hora, tres veces al día, las primeras 48 h
Pasada ya la fase de reposo, iniciará apoyo progresivo según tolerancia.Paulatinamente irá incrementando la carga
Generalmente el vendaje puede ser retirado entre los 14 y 21 días tras la lesión,introduciendo previamente la pierna en agua caliente para facilitar la operación
Para cualquier duda o consulta relacionada con el tratamiento deberá dirigirse a sumédico de cabecera
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 715

716
Manual DAE de enfermería. EIR. Oposiciones ••
Esguince de rodillaSe encontrarán, como se ha dicho anteriormente, muchos hallazgos físicoscomunes con el esguince de tobillo, como pueden ser el dolor, hematoma cu-táneo e impotencia funcional en lesiones moderadas o severas.
La presencia de líquido intraarticular también resulta orientativa, puesto queun cúmulo de este líquido en las primeras dos horas indica una hemartrosis,mientras que si se produce en las 12 o 24 horas siguientes indica origen sino-vial (la presencia de líquido sinovial hace suponer una lesión lo suficiente-mente grave como para romper la cápsula articular).
En la Imagen 3 se puede observar la valoración de la lesión de ligamento la-teral interno, desde el análisis del mecanismo de producción hasta la explo-ración física.
Técnicas y medios de diagnóstico• Estudio radiológico.• Artroscopia.
Medidas terapéuticasLas medidas terapéuticas irán dirigidas principalmente a:
• Aliviar el dolor.• Impedir complicaciones y un agravamiento de la lesión.• Disminuir la inflamación.
Para ello se establecerán las siguientes acciones:
• Mantener reposo.• Disminuir la inflamación con frío local.• Comprimir mediante vendas elásticas y elevar el miembro afectado.
Estas medidas previenen y disminuyen el edema local.
Luxaciones
La luxación es la pérdida de contacto de las superficies de los huesos que for-man una articulación. Existen también las subluxaciones, en las que parte delas superficies articulares permanecen en contacto. La inmensa mayoría de lasluxaciones son consecuencia de un traumatismo, lo que implica que llevenasociadas lesiones de la estructura periarticular y de soporte como las que sedetallaban en el apartado anterior.
ClasificaciónLa clasificación de las luxaciones articulares se realiza en función de las arti-culaciones afectadas y de la importancia que éstas tienen en la capacidad delocomoción y movimientos de la persona.
Tipo de esguince
Interno
Externo
Posterior
Estructuras dañadas
Ligamento lateral interno y cruzado anterior. Se puedeasociar la rotura del menisco interno
Ligamento lateral externo y cruzado posterior. Escomún la asociación del menisco externo
Ligamento lateral interno y cruzado posterior
Mecanismo de producción
Aplicación de una fuerza en la zona externa de larodilla, con la pierna en extensión
Aplicación de fuerza sobre la parte interna de larodilla, con la pierna en extensión
Hiperextensión de la rodilla o golpe frontal sobre laparte superior de la tibia
Tabla 2. Clasificación del esguince de rodilla
Imagen 3. Lesión de ligamentos internos de la rodilla
© DA
E
2
2
1
3 45
6
7
8
9
10
EF
(1) La fuerza se produce sobre la zona externa de la rodilla, dondese aprecia una zona equimótica (9) (2) Esguince de rodilla (grado I): rodilla estable y leve desgarroligamentoso (3) La rotura de la zona más profunda del ligamento provoca que lamaniobra de flexión de la rodilla con el pie hacia dentro abra laarticulación más de lo normal (F)(4) La rotura total del ligamento provoca la rotura del meniscointerno (5 y 6). La maniobra de extensión forzada de la rodilla (E)permite notar que se abre levemente pudiéndose incluso palpar elborde del ligamento roto (7)(8) En los traumatismos graves se produce la rotura asociada delligamento cruzado anterior(9) Externamente se apreciará una equimosis en la zona externa dela rodilla y sensibilidad en el lado interno (10)
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 716

717
enfermería médico-quirúrgica
Luxación de hombroSe produce en la articulación glenohumeral, cuando la cabeza del húmero sesepara de la cavidad glenoidea, pudiendo quedar situada:
• Cabeza humeral delante de cavidad glenoidea: luxación anterior.• Cabeza humeral detrás de cavidad glenoidea: luxación posterior.• Cabeza humeral debajo de cavidad glenoidea: luxación erecta.
Técnicas y medios de diagnósticoEl estudio radiológico simple muestra claramente la luxación anterior de lacabeza del húmero. La enfermera establecerá las medidas habituales.
Medidas terapéuticasSe intentará la reducción de la luxación como primera medida, habitualmenteexitosa. Para ello se efectúa la maniobra de Kocher: suele ser dolorosa, por loque se recomienda la administración de anestesia local, analgésicos y ansio-líticos en función del estado y la colaboración del paciente. No es descartableel uso de anestesia general para llevar a cabo la reducción.
Luxación esterno-clavicularRaramente se produce la luxación completa, sino una subluxación moderadaoriginada por una caída con la mano extendida o golpe sobre la parte ante-rior del hombro.
Exploración físicaEs habitual la aparición de una asimetría de los extremos anteriores de lasclavículas, quedando la del lado afectado más baja y protuberante (Ver Ima-gen 4). Suele existir dolor localizado que se acentúa a la palpación.
Medidas terapéuticasPuede realizarse reducción bajo anestesia local en las luxaciones importantes.
Luxación acromioclavicularLa clavícula pierde toda conexión con la escápula, rompiendo los ligamentosy originando un desplazamiento que puede ser grave e ir acompañado deaparición de hematoma.
Exploración físicaCon el paciente de pie, frente al explorador, se comparan los hombros ob-servándose una prominencia en el extremo externo de la clavícula (VerImagen 5).
Técnicas y medios de diagnósticoLa radiografía puede permitir observar la luxación si se hace con el pacientede pie y los brazos en caída.
Medidas terapéuticasLas formas leves sin inestabilidad pueden tratarse de forma conservadoramediante la aplicación de un cabestrillo que cubra e inmovilice todo el miem-bro superior durante cuatro o seis semanas.
Luxación del codoRelativamente frecuente, tanto en niños como adultos, suele ser resultado deuna caída sobre la mano en extensión.
Exploración físicaEn principio irá dirigida a distinguir la luxación de la fractura supracondílea,para lo cual existe un signo clarificador que consiste en buscar un triánguloequilátero en la parte distal del antebrazo formado por el olécranon, el epi-cóndilo y la epitróclea que se mantiene en las fracturas y se modifica en lasluxaciones (Ver Imagen 6).
Medidas terapéuticasLa reducción se lleva a cabo bajo anestesia general, puesto que es precisotraccionar con mucha fuerza. Posteriormente se inmoviliza el brazo, en fle-xión de 90°, con una férula posterior durante dos o cuatro semanas.
Luxación de la caderaConsiste en la salida de la cabeza del fémur de su posición anatómica en elacetábulo. El compromiso vascular y neurológico que representa la convier-ten en una urgencia traumatológica.
El mecanismo de producción más frecuente puede ser:
Imagen 4. Luxación esternoclavicular
© DA
E
Imagen 5. Luxación acromioclavicular
© DA
E
En la exploración se aprecia la asimetría de los extremosinternos de la clavícula
Con el paciente de pie, frente al explorador, se comparan los hombrosobservándose una prominencia en el extremo externo de la clavícula
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 717

718
Manual DAE de enfermería. EIR. Oposiciones ••
• Fuerza transmitida hacia arriba a través del fémur (accidentes frontalesde tráfico).
• Golpe a nivel lumbar.
Exploración física• Es característica la posición en aducción y rotación interna. También se
encuentra acortamiento de la pierna.• Es significativo el intenso dolor producido por la luxación posterior de
cadera.
Técnicas y medios de diagnósticoLa radiografía en proyección anteroposterior es claramente definitoria.
Medidas terapéuticasLa reducción precoz es esencial para la posterior evolución de la lesión, de-biendo hacerse ésta, al menos, en las seis horas siguientes.
Fracturas (EIR 97-98, 37)
Cuando se aplica una tensión sobre un hueso, sobrepasando la capacidad decarga de éste, aparece una fractura (pérdida de la solución de continuidad deun hueso). En la mayoría de los casos esta fuerza proviene de un trauma-tismo, que puede ser:
• Directo: el impacto se produce directamente sobre el hueso, fracturán-dolo en ese punto.
• Indirecto: el hueso se fractura en un punto diferente al que sufrió eltraumatismo, por un efecto de torsión, cizallamiento o angulación.
En ocasiones se originan fracturas patológicas en las que la capacidad de cargadel hueso disminuye de tal forma que una tensión normal puede fracturarlo.
Clasificación
Según el grado de compromiso óseo (Ver Imagen 7)• Fracturas incompletas:
– Fracturas “en tallo verde”, propias del niño.– Fracturas “por cansancio o fatiga”, constituidas por fisuras óseas,
corticales, propias de huesos sometidos a exigencias de flexoex-tensión, compresión o rotación de pequeña intensidad, pero repe-tidas una y otra vez. Se observan en deportistas, atletas, etc.
• Fracturas de rasgo único.• Fracturas de doble rasgo segmentarias con formación de tres fragmen-
tos óseos.• Multifragmentaria, esquirlosa, conminuta, por estallido.
Según la dirección del rasgo• Fractura transversal.• Fracturas de rasgo oblicuo.• Fracturas de rasgo helicoidal. Muy frecuentes en tibia y húmero. Son de
muy difícil reducción, notoriamente inestables, de rasgos agresivos: cor-tantes y punzantes, y de ellas es factible esperar compromiso de vasos(arteria femoral en fractura de la diáfisis del fémur), de nervios (nervioradial en la fractura de la diáfisis humeral) o de la piel (fractura de la diá-fisis tibial).
Según la desviación de los fragmentos• Sin desviaciones, fractura de rasgo único y horizontal.• Con desviaciones. Éstas pueden ser:
– Laterales.– Con angulación de los ejes (en varo o valgo) en rotación.– Con impactación de los fragmentos.– Con cabalgamiento de los fragmentos, determinado por la con-
tractura muscular; frecuentes en fracturas de diáfisis humeral y fe-moral.
Problemas interdependientes
Embolia grasa asociada con fractura de huesos largos• Prioridad. La enfermera detectará precozmente los signos de embo-
lismo graso.• Intervenciones:
– Vigilar signos de dificultad respiratoria.– Vigilar el aumento de la frecuencia cardiaca.– Detectar disminución del nivel de consciencia.– Controlar el estado de confusión y agitación.
Imagen 6. Luxación del codo
© DA
E
La luxación de codo esfrecuentemente confundidacon la fractura supracondílea.Para evitar esta confusión seutiliza la maniobra de lafigura, identificando eltriángulo (1) que semantiene en las fracturas (2),pero se modifica en lasluxaciones (3)
Imagen 7. Algunas de las fracturas más frecuentes
© DA
E
1
2
3
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 718

719
enfermería médico-quirúrgica
– La determinación de gases arteriales suele ser clarificadora en eldiagnóstico del embolismo graso.
Otras alteraciones articulares
TendinitisAfectación inflamatoria y dolorosa de los tendones implicados en una articu-lación. Se presenta en forma aguda generalmente tras un uso prolongado ode forma crónica por degeneración del tendón (tendinosis), calcificaciones ypor causas orgánicas (diabetes) y artritis.
Hallazgos físicos• Dolor y sensibilidad articular a lo largo de un tendón, generalmente
cerca de una articulación.• El dolor empeora con el movimiento o la actividad.• Se presenta dolor en la noche.• La zona puede aparecer enrojecida y caliente y dolorosa a la palpación
y en los movimientos forzados del músculo de inserción.
Medidas terapéuticasEn general se intentará recuperar la funcionalidad del tendón aliviando los sín-tomas mediante el reposo y/o inmovilización y la reducción de la inflamacióncon AINE.
Tendinitis del hombroLa afectación es en el tendón del bíceps y en el manguito de los rotadores(Ver Imagen 8). La lesión puede manifestarse como una leve inflamación opuede llegar a afectar a la mayor parte del manguito rotatorio.
Hallazgos físicos• Imposibilidad de sostener el brazo en ciertas posiciones.• Dolor o aumento de la sensibilidad en el hombro.
• La exploración y la visión radiológica suelen ser suficientes para el diag-nóstico.
Medidas terapéuticasEn etapas iniciales, el reposo durante dos semanas y los antiinflamatorios sue-len remitir el proceso. Las formas más agudas suelen requerir de la infiltracióncorticoidea.
Tendinitis del codoTanto la tendinitis como la bursitis en el codo se identifican en el proceso máshabitual de esta articulación: la epicondilitis (epicondilalgia o codo de te-nista).
Hallazgos físicosLos síntomas tienen un comienzo generalmente insidioso, más raramenteagudo. Presentan dolor localizado en la inserción de los músculos epicondí-leos, sobre todo extensores, que aumenta con la presión local sobre el epi-cóndilo, por la extensión activa de la muñeca y por su flexión pasiva queimpide hacer ciertos movimientos cotidianos (dar la mano, levantar un peso,usar una herramienta, etc.). Puede haber una ligera tumefacción y aumentodel calor local. Suele ser unilateral y tiene un curso clínico autolimitado conevolución cíclica.
Medidas terapéuticas• Reposo; en los casos agudos se puede colocar una férula.• Masaje con hielo.• Los AINE orales no son eficaces.• Fisioterapia.• Ortesis de antebrazo.
El tratamiento más útil a corto plazo es la infiltración local en el punto de má-ximo dolor, con una mezcla de corticoides + anestésico local (1 cm3 de Scan-dicaín® al 2% + 1 cm3 de celestone cronodose). No se deben poner más de2 cm3 de líquido, pues aumenta el riesgo de efectos secundarios locales (atro-fia subcutánea y cambios en coloración de la piel). Se produce una mejoría im-portante en el plazo de dos a seis semanas.
Tendinitis de la muñecaEl uso excesivo y los movimientos repetidos pueden tener repercusiones sobrela mano y la muñeca y causar diversas afecciones como tendinitis y síndromedel túnel carpiano.
Hallazgos físicos• Dolor, hipersensibilidad, hinchazón menor y limitación del movimiento.• El dolor por tendinitis en la muñeca tal vez baje hasta los dedos de la
mano o ascienda hasta el codo.• La tendinitis en los dedos de la mano puede afectar a uno o más dedos
de la mano a la vez. Puede sentir dolor constante o sólo con ciertos mo-vimientos.
• El área alrededor del tendón puede estar muy sensible.• Puede referir un crujido cuando dobla o flexiona el dedo o la muñeca.
Tendinitis de De QuervainIrritación de los tendones que se encuentran en el borde del pulgar de la mu-ñeca (Ver Imagen 9). La irritación causa que el revestimiento del comparti-miento alrededor de los tendones se inflame, cambiando así su forma; estohace difícil a los tendones moverse como debieran.
Hallazgos físicos• Dolor y sensibilidad en el borde del pulgar de la muñeca, el que usual-Imagen 8. Anatomía del hombro
© DA
E
Ligamentoacromiocoraideo
Acromion
Cavidad glenoideade la escápula
Ligamentoacromiocoraideo
Ligamentoglenohumeral
Acromion
Ligamentoacromioclavicular
Húmero
Ligamentoglenohumeral
Apófisiscoracoides
Escápula
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 719

720
Manual DAE de enfermería. EIR. Oposiciones ••
mente se manifiesta al tratar de hacer un puño, agarrando o tomandocosas o rotando la muñeca.
• El dolor puede aparecer de forma brusca o gradual.• Se siente en la muñeca pero puede irradiarse hacia el antebrazo.• Es posible evidenciar inflamación sobre el lado del pulgar de la mu-
ñeca, también puede haber un quiste en esta zona.
• La irritación del nervio que está encima de la vaina tendínea puede cau-sar adormecimiento en la parte de atrás de los dedos pulgar e índice.
Medidas terapéuticasComo en otros casos, inmovilización, evitación de movimientos dolorosos yantiinflamatorios. Cuando los síntomas son severos o no mejoran, la cirugíapuede estar recomendada.
Síndrome del túnel carpianoEs una afección causada por presión sobre un nervio grande en la muñecacuando pasa por un “túnel” formado por tendones (Ver Imagen 10).
Hallazgos físicos• Dolor que puede diseminarse hacia la mano y el antebrazo.• Entumecimiento y hormigueo en los dedos, en especial el pulgar, el ín-
dice y el dedo medio, y pérdida de la fuerza en la mano que puede pro-vocar la caída de objetos a menudo y que despierte por la noche por elhormigueo y el entumecimiento.
BursitisTérmino que indica la inflamación de la bolsa lubricante o bursa (sinovial)ubicada en las proximidades de las articulaciones ya referidas (Ver Imagen11):
• Las bolsas son cavidades llenas de líquido ubicadas cerca de las articu-laciones donde los tendones o los músculos pasan por encima de lasprotuberancias óseas. Su función es ayudar con el movimiento y redu-cir la fricción entre las partes movibles.
• La sintomatología y tratamientos iniciales no difieren excesivamentede los descritos en las tendinitis. Sólo en las formas en las que la infla-mación no responde al tratamiento inicial es posible que sea necesarioextraer líquido de la bolsa e inyectar corticosteroides.
• Rara vez se requiere cirugía.• Puede tener etiología infecciosa, en cuyo caso precisa terapia antibió-
tica y drenaje quirúrgico.
Imagen 9. Tendinitis de De Quervain
© DA
E
Imagen 10. Síndrome del túnel carpiano
© DA
E
Revestimientohinchado
Vaina tendínea
Tendón inflamado
Imagen 11.Bursitis©
DAE
Túnel carpiano sano Nervio mediano
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 720

721
enfermería médico-quirúrgica
AMPUTACIONES: SÍNDROME DEL MIEMBROFANTASMA
Se desconoce la causa exacta del síndrome del miembro fantasma. Se pre-sume que las sensaciones que el paciente experimenta se deben al intento delcerebro de reorganizar la información sensorial que sigue a la amputación.
Factores periféricos
• Las sensaciones dolorosas de espasmo y compresión en un miembrofantasma reflejan la tensión muscular en el miembro residual. Así, amayor contracción muscular, mayor será el dolor espasmódico.
• En los miembros amputados:– Las terminaciones nerviosas del muñón siguen siendo sensibles a
los estímulos.– La disminución del flujo sanguíneo en la extremidad causa un des-
censo de su temperatura, lo que aumenta la intensidad del dolor.
• Otro mecanismo periférico importante es la descarga ectópica a partirde un neuroma formado en el muñón. Esta descarga puede ser provo-cada por la estimulación del muñón (p. ej.: frío o calor) u ocurrir espon-táneamente.
• La formación de un neuroma y la aparición de dolor fantasma dependendel tipo de amputación, de la estimulación del muñón y de la predispo-sición genética de la persona al dolor neuropático. Los neuromas apare-cen al seccionar el tejido nervioso, son la forma en la que cicatrizan losnervios y se presentan como nódulos muy sensibles y dolorosos.
Factores centrales
Tras una amputación se produce una reorganización a nivel cortical (Ver Ima-gen 12).
Hallazgos físicos
El paciente que ha sufrido una amputación refiere formas diversas de dolor;calambres, ardor, hormigueo, dolor pulsátil y dolor ardiente. Cuando estossíntomas son experimentados por él en el miembro fantasma y no en elmuñón, se dice que se trata de un dolor fantasma.
Medidas terapéuticas
Tratamiento farmacológicoLa elección de un tratamiento farmacológico u otro se realiza según un as-pecto fundamental: diferenciar el dolor del muñón del dolor del miembrofantasma.
El dolor del muñón se combate con analgésicos, antiinflamatorios y opiáceos.El del miembro fantasma, descrito por el paciente como una sensación dequemazón, hormigueo y parestesia, es más difícil de controlar. Los utilizadosprincipalmente son:
• Antidepresivos.• Anticonvulsivantes.• Clorpromazina.• Opiáceos.• Clonidina.• ß-bloqueantes.
Tratamiento rehabilitadorResulta fundamental para fortalecer el miembro residual. Tiene como objeti-vos generales:
• Restaurar y/o mantener la movilidad articular.• Fortalecer la musculatura.
Te conviene recordar...
✔ Las alteraciones traumáticas más frecuentes son el esguince, la luxación y la fractura.✔ Los esguinces de tobillo y de rodilla son los más frecuentes y su tratamiento depende del grado de gravedad.✔ Entre las luxaciones destacan las de hombro, esternoclavicular, acromioclavicular, de codo y de cadera.✔ El tratamiento común a todas ellas es la reducción, con anestesia local o general.✔ Las fracturas se clasifican según el compromiso óseo, la dirección del rasgo y la derivación de fragmentos.✔ Su tratamiento es la reducción o cirugía.
Imagen 12. Reorganización cortical tras una amputación
Impulso dolorosopersistente desde el
miembro
Aparición de unamemoria de dolor
corticalAMPUTACIÓN
Reorganización de lazona de la amputación
en la cortezasomatosensorial
• Impulsos aleatoriosdesde el neuroma delmuñón
• Activación simpática
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 721

722
Manual DAE de enfermería. EIR. Oposiciones ••
Otros tratamientos• Vasodilatación del miembro residual.• Disminución de la tensión muscular.
• Estimulación nerviosa eléctrica transcutánea (TENS).• Simpatectomía regional.• Bloqueo simpático lumbar.
ALTERACIONES ARTICULARES DEGENERATIVAS
Las artrosis son procesos articulares crónicos caracterizados por cambios de-generativos en el cartílago articular y crecimiento marginal óseo, así comoproliferación del hueso subcondrial.
Clasificación
Clínicamente es útil clasificar la artrosis en dos grandes grupos:
• Primaria o idiopática: cuando su causa es desconocida o hereditaria.• Secundaria: cuando se conoce la causa directamente implicada en el
proceso artrósico.
Ahora bien, es importante diferenciar además entre lesión artrósica y artro-sis con manifestaciones clínicas:
• La artrosis anatómica es exclusivamente histológica y a partir de los 50años (edades medias de la vida) es prácticamente constante en algu-nas articulaciones.
• La artrosis radiológica es la artrosis anatómica que en ocasiones, de-bido a su intensidad, se detecta radiológicamente.
• Artrosis clínica o enfermedad artrósica: es cuando la artrosis radioló-gica, en una pequeña proporción de casos, se acompaña de manifes-taciones clínicas imputables a ella.
Etiología
Artrosis primariaSe desconoce la causa de la artrosis primaria, pero parece estar en relacióncon factores predisponentes, tales como:
• Factor genético o herencia.• Edad.• Sexo.• Obesidad.
Artrosis secundariaLa artrosis secundaria aparece en una articulación que, por definición, estápreviamente sana y el cartílago se altera bajo diversas condiciones.
Una anormalidad morfológica como resultado de una fractura articular deuna luxación, de una displasia acetabular, de un deslizamiento epifisario y deuna enfermedad de Perthes será causa de que se incrementen las presionesde contacto debido a la reducción de las áreas de carga (Ver Imagen 13).
Fisiopatología
La integridad del cartílago depende de dos elementos antagonistas: por unlado, la carga mecánica y, por otro, la calidad de la matriz cartilaginosa.
Te conviene recordar...
✔ La amputación de un miembro supone un gran impacto en la vida de cualquier persona, sea cual sea su edad, y requiere el tra-bajo de un equipo multidisciplinar para conseguir la evolución favorable del proceso, tanto a medio como a largo plazo.
✔ El paciente tiene que participar de forma activa en el plan de cuidados para conseguir que el nivel de independencia sea má-ximo.
✔ El plan de cuidados de cada paciente debe ser individual y mucho más en el caso de los niños, cuyas inquietudes, temores ysentimientos resulta fundamental conocer, contando siempre con el apoyo de la familia.
✔ Los cuidados del muñón y la realización de un vendaje adecuado resultan cruciales para la buena evolución del mismo y la fu-tura implantación de una prótesis, que facilitará la movilidad y la independencia del paciente.
Imagen 13.La anormalidad morfológica aumenta las presiones de contacto
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 722

723
enfermería médico-quirúrgica
La artrosis es la consecuencia de la pérdida de la matriz cartilaginosa de su ca-pacidad para soportar la carga mecánica que corresponde a cada articulaciónen particular, siendo muchos los factores que pueden incidir en la pérdida deesta matriz: síntesis escasa, matriz inadecuada, degradación de proteoglica-nos y colágeno, etc. Estas alteraciones de los caracteres físico-químicos delcartílago disminuyen su resistencia a las fuerzas compresivas y de tensión. Lasuperficie cartilaginosa se vuelve más blanda e irregular, desarrollándose fi-brilaciones, hendiduras profundas, fragmentación y, finalmente, erosión com-pleta que pone al descubierto el hueso subcondral.
Técnicas y medios de diagnóstico
Pruebas de laboratorio (VSG, proteína C reactiva, FR, ANA ylíquido sinovial)No hay pruebas diagnósticas de laboratorio específicas para la artrosis. Tam-poco existen anormalidades características de la artrosis en las pruebas de la-boratorio. A pesar de que estas pruebas son negativas, se deben realizar paraestablecer el diagnóstico diferencial.
RadiologíaConstituye el método diagnóstico fundamental. Según avanza el proceso, laartrosis anatómica se detecta radiológicamente, la artrosis radiológica, apa-reciendo los signos radiológicos característicos, entre ellos:
• Pinzamiento o estrechamiento del espacio articular.• Esclerosis, eburnación o condensación del hueso subcondral.• Presencia de osteofitos marginales.• Quistes subcondrales o geodas.• Deformidad articular acompañada, muchas veces, de subluxación.
Medidas terapéuticas
Pueden ser farmacológicas, no farmacológicas e incluso quirúrgicas. Un re-sumen de todas ellas se muestra en la Tabla 3.
OSTEOPOROSIS
Etiopatogenia
La osteoporosis es una enfermedad que se incluye dentro de otro grupo de en-fermedades denominado osteopatías metabólicas, ya que todas tienen encomún que la afectación esquelética está condicionada por alteraciones en losmecanismos reguladores del remodelado óseo. Es un proceso que se carac-
teriza por una disminución de la masa ósea y modificaciones en la arquitec-tura y resistencia mecánica del hueso.
Existe un equilibrio entre la actividad de los osteoblastos y de los osteoclas-tos, lo que permite una remodelación ósea constante (especialmente duranteel crecimiento) que hace que la masa ósea sea la adecuada (Ver Imagen 14).Existen numerosos factores que pueden alterar este equilibrio, generando unaumento o una disminución de la masa ósea.
Tabla 3. Opciones terapéuticas en la enfermedad articulardegenerativa
Intervenciones no farmacológicas
Ejercicio físico moderadoAplicación de calorReducción de peso (en obesos)
Intervenciones farmacológicas
AnalgésicosAntiinflamatorios no esteroideos
Medidas quirúrgicas
OsteotomíasDesbridamientosArtroplastiaArtrodesis
Te conviene recordar...
✔ La artrosis es un proceso articular crónico degenerativo. Puede deberse a causa desconocida o ser secundaria a procesos infla-matorios, metabólicos, biomecánicos, hormonales, etc.
✔ Parece empezar en la tercera década de la vida, pero las alteraciones no se manifiestan hasta los 55-65 años.✔ Afecta por igual a ambos sexos, pero a partir de los 55 años la incidencia es mayor en mujeres.✔ No hay pruebas diagnósticas de laboratorio específicas para la artrosis. La radiología constituye el método diagnóstico funda-
mental.✔ La presentación clínica de la artrosis es variable y depende de las articulaciones afectas, la duración, la intensidad del proceso
y de la respuesta del paciente al estímulo doloroso.✔ La terapia debe ir destinada a aliviar el dolor y a mejorar la función articular, reduciendo la progresión de la lesión de las es-
tructuras articulares y la rigidez articular.✔ El cuidado del paciente artrítico ha de obedecer a un auténtico programa individualizado en el que es preciso contemplar la
localización de las lesiones artríticas sintomáticas, la severidad del dolor y la incapacidad funcional, así como las característi-cas del entorno social del paciente.
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 723

724
Manual DAE de enfermería. EIR. Oposiciones ••
Factores de riesgo
Los principales factores de riesgo que favorecen el proceso osteoporótico se re-sumen en la Tabla 4. Determinados fármacos, que se recogen en la Tabla 5, in-crementan el riesgo de padecer osteoporosis.
Hallazgos físicos
La osteoporosis no siempre presenta síntomas, siendo con frecuencia la frac-tura el primer aviso. Sus principales manifestaciones clínicas son las fracturasde Colles (fractura de la parte distal del radio), las fracturas de cadera (que seproducen, habitualmente, por caídas accidentales) y las vertebrales (Ver Ima-gen 15). Las fracturas vertebrales se producen en su mayoría en los cuerposdorsales y lumbares (generalmente D8 a L2). Este tipo de fracturas aparece porcompresión o aplastamiento vertebral y se desarrollan gradualmente. Sue-len ser asintomáticas y el diagnóstico se realiza de forma casual en exámenesradiológicos rutinarios.
Imagen 14. Mecanismo regulador del calcio
Aumenta laabsorciónintestinal de calcio
Disminuye la absorciónintestinal de calcio
Aumenta laexcreción renal de calcio
Calcio plasmático
Concentración plasmáticadisminuida
Concentración plasmáticaaumentada
Aumenta lasecreción de PTH
Vitamina D
Disminuye la secreción dePTH
Aumenta lasecreción de calcitonina
Tabla 4. Principales factores de riesgo de la osteoporosis
Ingesta inadecuada de calcio y vitamina D
Déficit de estrógenos
Sedentarismo
Enfermedades crónicas
Medicación
Tabaquismo
Alcoholismo
Antecedentes familiares de osteoporosis
Bajo peso corporal
Caídas recurrentes
Nuliparidad
Ingesta elevada de cafeína
Ingesta elevada de proteínas
Tabla 5. Fármacos que aumentan el riesgo de osteoporosis
Glucocorticoides
Inmunosupresores (ciclosporina)
Heparina
Anticonvulsivantes (fenitoína)
Antiácidos que se unen a fosfatos
Fármacos citotóxicos
Exceso de tiroxina
Litio
Imagen 15. Fractura conminuta intraarticular de radio distal derecho y fractura estiloides cubital en mujer de 68 años
© Ce
dida p
or las
autor
as
A B
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 724

725
enfermería médico-quirúrgica
Cuando las fracturas son múltiples se produce una pérdida de altura de varioscentímetros, cifosis, dolor y molestias en la espalda (Ver Imagen 16).
Técnicas y medios de diagnóstico
Actualmente existen varias técnicas diagnósticas no invasivas que permitendeterminar la densidad ósea del esqueleto:
• Radiografías simples anteroposterior y lateral (Ver Imagen 17): propor-cionan un diagnóstico de las fracturas osteoporóticas. No son útiles paradeterminar la pérdida de densidad ósea.
• Densitometría ósea.• Tomografía computarizada cuantitativa (TCC).• Ecografía.
Entre las pruebas de laboratorio, se suelen estudiar los siguientes marcadores:
• Calcio sérico.• Calcio en orina.• Nivel sérico de PTH.• Nivel sérico de TSH y/o cortisol en orina.• Albúmina sérica.• Colesterol.
Medidas terapéuticas
Tratamiento farmacológicoExisten varios grupos de fármacos en el tratamiento de la osteoporosis:
• Agentes antirresortivos (estrógenos, bifosfonatos, calcio y calcitonina,entre otros).
• Agentes estimuladores de la formación ósea (flúor, PTH).• Diuréticos tiacídicos.• Suplementos de calcio y vitamina D.
Medidas dietéticas• Aconsejar la ingesta de alimentos que contengan calcio y vitamina D.• Otros elementos de la dieta:
– Es necesario limitar el consumo de alcohol y cafeína, ya que estaúltima en dosis excesivas incrementa la calciuria.
– La dieta ha de ser equilibrada: baja en colesterol y fosfatos y sin ex-ceso de proteínas (Ver Tabla 6).
• Ejercicio físico. Ya se ha mencionado que la falta de ejercicio físico favo-rece el riesgo de osteoporosis, al disminuir la masa ósea del esqueleto,lo que constituye un factor de riesgo esencial de fracturas patológicas(Ver Tabla 7).
• La soja en la prevención de la osteoporosis. Los fitoestrógenos, o estró-genos vegetales, están presentes en todos los alimentos de origen ve-getal y se clasifican en:– Isoflavonas (genisteína, daidzeína, biochanina A, etc.): poseen la
mayor capacidad estrogénica. Se encuentran en las legumbres,fundamentalmente en la soja.
– Lignanos (enterodiol y enterolactona): en cereales, frutas y vege-tales.
– Coumestanos (coumestrol): en las semillas de girasol.Imagen 16. Radiografía de aplastamiento vertebral en mujer de 68 años
© Ce
dida p
or las
autor
as
Imagen 17. Radiografía de fractura bifocal de rama iliopubiana derecha en mujer de 68 años
© Ce
dida p
or las
autor
as
A B
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 725

726
Manual DAE de enfermería. EIR. Oposiciones ••
ALTERACIONES DE LA COLUMNA VERTEBRAL
Escoliosis
La desviación lateral de la columna vertebral asociada a una rotación de loscuerpos vertebrales, originando una curva, se conoce como escoliosis, que esuna anomalía frecuente en la infancia y la adolescencia. Los tipos de curva-tura dan origen a diferentes aspectos de la escoliosis (Ver Tabla 8).
ClasificaciónLa escoliosis puede clasificarse en dos tipos:
• Actitud escoliótica o no estructural. Se presenta cuando la inclinaciónen el plano frontal no se acompaña de rotación y nunca se produce unadeformidad permanente (Ver Imagen 18).
• Estructural. Se presenta cuando se ha completado la osificación de lasvértebras afectadas y se fijan las deformidades ligamentosas y discales,produciéndose rotación de los cuerpos vertebrales con curvas que sevan haciendo rígidas. Éstas se clasifican en:– Idiopáticas. La escoliosis evoluciona durante el crecimiento y tiene
riesgo de progresión durante toda la vida (Ver Imagen 19).– Congénitas. Son menos frecuentes que las idiopáticas.– Neuromusculares. La forma más frecuente hasta hace unos años
ha sido la escoliosis paralítica postpoliomielítica, que ha ido des-apareciendo debido a la vacunación, ocupando el primer lugar la
parálisis espástica. En este tipo de escoliosis la incurvación es de-bida a un desequilibrio muscular.
Tipos de alimentos
Alimentos energéticosGrupo 3º: patatas, legumbres y frutos secosGrupo 6º: pan, pastas, cereales y azúcarGrupo 7º: grasas, aceite y mantequilla
Alimentos plásticosGrupo 1º: leche y derivadosGrupo 2º: carne, pescados y huevosGrupo 3º: patatas, legumbres y frutos secos
Alimentos reguladoresGrupo 4º: verduras y hortalizasGrupo 5º: frutas
Sustancias nutritivas
Hidratos de carbonoGrasas
Proteínas (aminoácidos)Calcio
Minerales y vitaminas
Tabla 6. Grupos de alimentos
Te conviene recordar...
✔ La osteoporosis es el resultado, principalmente, de dos procesos: en la mujer, la disminución de los valores de estrógenos des-pués de la menopausia produce una alteración en la remodelación ósea; en ambos sexos, a partir de los 65 años, la disminu-ción fisiológica de la absorción digestiva de calcio provoca un hiperparatiroidismo secundario.
✔ Las medidas preventivas para mantener la masa ósea deben iniciarse lo antes posible, desde la infancia, mediante una dietaequilibrada y rica en calcio y vitamina D y mediante la práctica de una actividad física regular para optimizar el pico de masaósea hacia los 20 años.
✔ Para disminuir la pérdida ósea acelerada que aparece durante los diez años que siguen a la menopausia, el tratamiento hor-monal sustitutivo con estrógenos y progesterona es una medida esencial.
✔ Para la pérdida ósea tardía, propia de ambos sexos después de los 60 años, el objetivo irá encaminado a que el paciente man-tenga el consumo adecuado de calcio y vitamina D e informarle de las medidas necesarias para disminuir el riesgo de caídas.
Tabla 8. Tipos de curvatura en la escoliosis
Curva torácica derecha
Clara asimetría de la caja torácica. Cuando la curva es severa puede llegar a un deterioro cardiorrespiratorio.Se da en el adulto
Curva toracolumbar
Puede crear un desequilibrio en el tronco y ocasionar una alteracióncardiorrespiratoria.Más severa que las curvas torácicas derechas
Doble curva primaria dorsal derecha y lumbar izquierda
Estas dos curvas se equilibran pero conducen a un tronco corto (poco perjuicioestético)
Curva lumbar
La más frecuente es la curva izquierda.Provocan una falsa dismetría de las extremidades, son curvas evolutivas y puedendesencadenar una artrosis lumbar en el adulto
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 726

727
enfermería médico-quirúrgica
– Postraumáticas.• Óseas: fracturas y cirugía.• Extraóseas: toracoplastias, quemaduras, etc.
– Secundarias a fenómenos irritativos. Tumores intra y extrarraquídeos.– Conectivopatías.
• Congénitas (enfermedad de Marfan).• Adquiridas (artritis reumatoide).
Hallazgos físicos
Músculo-esqueléticos• Dolor de espalda, limitación de movimientos y dificultad progresiva en
la deambulación.• Desigual altura de hombros, un brazo está más cerca del cuerpo que el
otro. Asimetría pélvica.• El paciente cojea de manera notoria.
• Curvatura visible en la columna vertebral o una joroba pronunciada enlas costillas, con dificultad postural.
Neurológicos• Presencia de parálisis en la mano y dedos en garra.• Atrofia de las eminencias tenar e hipotenar.
Técnicas y medios de diagnóstico
RadiológicosLas exploraciones radiológicas aportarán datos de las curvas estructuradas yde las de compensación:
• Radiografía anteroposterior en bipedestación: para medir la angulacióny la rotación.
• Radiografía lateral en bipedestación: para medir lesiones asociadas (es-pondilolistesis).
• Radiografía anteroposterior en decúbito: muestra la reductibilidad. Es lamejor manera de diferenciar la escoliosis de la actitud escoliótica.
• Tomografía axial computarizada (TAC).• Radiografía simple de mano izquierda: para valorar la edad ósea.
Forma de medir una escoliosisPara comparar los resultados y valorar la evolución hay que utilizar un sis-tema para medir las curvas. El método más empleado es el de Coob, que cal-cula la inclinación lateral por medio del ángulo que forman las perpendicularesa las placas limitantes superior e inferior, de las respectivas vértebras limitan-tes. Además, se debe medir la rotación (ésta se valora teniendo en cuenta laposición de los pedículos) (Ver Imagen 20).
Medidas terapéuticasDependen de la curvatura y de la edad del paciente:
• Curva de 0 a 20º y edad inmadura: observación y examen radiológicocada dos meses.
• Curva de 0 a 20º y edad madura: habitualmente no requieren evalua-ción adicional.
• Curva de 20 a 30º y edad inmadura: se evalúa cada tres o cuatro meses
Imagen 18. Actitud escoliótica y escoliosis
© DA
E
Imagen 19. Evolución de la escoliosis durante el crecimiento
© DA
E
5 años 10 años 15 años
72o34o
20o
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 727

728
Manual DAE de enfermería. EIR. Oposiciones ••
con Rx, AP, etc., en bipedestación. Se emplea ortesis cuando la curvaprogresa más de 25° o para las pequeñas curvas (menores de 20°) queproducen deformidad que afecta a la estética.
• Curva de 30 a 40º y edad inmadura: utilización de corsé ortopédico.• Curva más de 40°, inmadura o madura: cuando el tratamiento con el
corsé no ha sido exitoso o la curva excede a los 40°. Se realiza cirugíacorrectiva con injertos óseos e instrumentación.
Complicaciones potenciales• Derivadas de la insuficiencia cardiaca derecha.• Derivadas de la insuficiencia respiratoria.• Alteraciones neurológicas.• Derivadas del tratamiento ortopédico y quirúrgico:
– Alteración de la integridad cutánea, relacionada con los puntos depresión, derivados del tratamiento ortopédico.
– Complicaciones pulmonares como resultado de la inmovilizaciónpostoperatoria.
– Íleo paralítico.
HERNIA DISCAL
Etiopatogenia
La hernia de disco es una patología frecuente que afecta a una gran parte dela población y por igual en ambos sexos. Es la causa más frecuente de inca-pacidad laboral en personas por debajo de los 45 años. La hernia discal lum-bar o la patología degenerativa discal asociada son la causa más frecuente dedolor lumbar (lumbago o lumbalgia) y ciático (lumbociatalgia). La mayoríade las hernias de disco aparecen en la columna lumbar baja, especialmenteen los niveles L4-L5 y L5-S1 (Ver Imagen 21).
Hay muchos factores de riesgo para la hernia discal, aparte del desgaste pro-pio de la edad. Algunos de ellos son:
• Malas posturas.
Imagen 20. Medición de la escoliosis
© DA
E
D = vértebra dorsal
1º. Medir el ángulo2º. Valorar la rotación
D5
D6
D7
D8
D9
70o70o
70o
Te conviene recordar...
✔ La escoliosis es una desviación lateral de la columna vertebral, siendo un trastorno que se desarrolla con lentitud durante elcrecimiento de los niños. Se suele descubrir en los primeros años de la adolescencia y suele presentarse como un desequilibrioo falta de simetría del tronco o del talle de la cintura.
✔ De un 2 a un 3% de la población presenta un cierto grado de desviación; de este porcentaje sólo requiere tratamiento del 0,3al 0,5 %. Un 85% de las veces el trastorno corresponde a la categoría de escoliosis idiopática y en el 15% depende de otras pa-tologías, por lo que es importante que se diagnostique el origen de la escoliosis.
✔ Es mucho más frecuente en las niñas que en los niños, desconociéndose la causa. No es hereditaria, pero tiende a presentarsemás en algunas familias.
✔ La escoliosis no se cura, pero es posible controlar la desviación y evitar que aumente durante el crecimiento.✔ Cuando la persona alcanza su estatura total, la desviación suele estabilizarse y no se requiere más tratamiento.✔ Aproximadamente una de cada 20 ó 30 escoliosis precisarán cirugía.
Imagen 21. Hernia discal lumbar
© DA
E
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 728

729
enfermería médico-quirúrgica
• Mecánica corporal incorrecta.• Falta de ejercicio físico regular.• Levantar pesos de forma incorrecta; movimientos repetitivos de flexión-
extensión, cargando peso.• Sobrepeso.• Atrofia de la musculatura dorsolumbar.
Las fases que se siguen hasta la producción de una hernia discal son:
• Degeneración: el disco se debilita.• Protrusión o prolapso: el disco ocasiona una deformación de la parte
más debilitada del anillo fibroso.• Fisura: se produce un desgarro en el anillo fibroso del disco.• Extrusión: el núcleo pulposo sale fuera del anillo fibroso y se ubica en
el canal medular.
Hallazgos físicos
Neurológicos• Dolor sordo o agudo, intermitente o continuo, que se irradia hacia los
miembros y se agudiza con ciertas posiciones o movimientos, al tosero estornudar y al estar sentado.
• Ciática: es un dolor que se inicia en la zona lumbar o las nalgas y seirradia hacia el pie. Se produce al ser presionado el nervio ciático.
• Hipoestesia.• Claudicación intermitente para la marcha de tipo neurógeno.• En casos graves, cambios en el funcionamiento de la vejiga o intestinos.
Músculo-esqueléticos• Dificultad de marcha (lumbar) o movimientos finos de las manos (cervical).• Parestesias, debilidad y adormecimiento de las piernas o los pies.• Espasmos en los músculos de la espalda.
Técnicas y medios de diagnóstico• Rx de columna.• Resonancia magnética (RNM).• Tomografía axial computarizada (TAC).• Electromiografía.• Exploración:
– Signos de Ramond: contractura muscular paralumbar uni o bilateral.– La flexión del tronco produce dolor en la pierna.– Hacerle caminar de puntillas y de talones (para explorar la fuerza).– Maniobras de Lasségue (elevación de la pierna extendida, apare-
ciendo dolor a menos de 45º) y Lasségue cruzado, induciendodolor ciático al elevar el miembro no doloroso.
– Reflejos osteotendinosos (rotuliano, aquíleo, etc.): suelen estarabolidos.
Medidas terapéuticas
Tratamiento no quirúrgico• Inicialmente, reposo relativo de dos días a dos semanas, como máximo,
acompañado de analgésicos, antiinflamatorios no esteroideos (AINE) yrelajantes musculares.
• Tratamiento conservador mediante fisioterapia o rehabilitación y rela-jantes musculares, para hacer desaparecer los síntomas.
Tratamiento quirúrgicoLa cirugía se considera cuando el tratamiento no quirúrgico no alivia los sín-tomas o cuando se sospecha compresión de la médula espinal. También está
indicada cuando hay recurrencia de periodos incapacitantes. Si hay síndromede compresión de cola de caballo, con paresias y pérdida de control de esfín-teres, se requiere cirugía urgente.
• Discectomía: consiste en extirpar exclusivamente el material discal her-niado, sin alterar el hueso vertebral.
• Microdiscectomía: es una discectomía que se hace usando un micros-copio, con una incisión y manipulación quirúrgica muy pequeña y, porlo tanto, un plazo de recuperación muy breve.
• Artrodesis: consiste en fijar dos vértebras.
Modificación del estilo de vida• El sobrepeso empeora cualquier dolor de espalda. Por ello, son necesa-
rios el ejercicio y la dieta para remediarlo.• Es importante la fisioterapia para todos los pacientes con problemas
discales.
Estenosis de canal
EtiopatogeniaEs el estrechamiento del canal de la columna cervical o lumbar que producecompresión en la médula o en las raíces del nervio. Ocurre en personas deedad mediana o avanzada y puede ser causada por cambios degenerativospropios de la edad, osteoartritis, enfermedad de Paget o como resultado deuna lesión que causa compresión en las raíces del nervio y/o en la mismamédula espinal. También se presenta la espondilolistesis (deslizamiento deuna vértebra sobre otra), que conduce a la compresión (Ver Imagen 22).
Hallazgos físicos
Neurológicos• Pérdida de los reflejos en las extremidades.• Con frecuencia, el dolor en la pierna mejora cuando se flexiona hacia de-
lante.• El canal cervical estrecho puede ocasionar síntomas similares en los
hombros, brazos y piernas.• En algunos pacientes el dolor empieza en las piernas y avanza hacia
arriba en dirección a los glúteos.
Imagen 22. Estenosis de canal lumbar
© Ce
dida p
or la
autor
a
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 729

730
Manual DAE de enfermería. EIR. Oposiciones ••
Músculo-esqueléticos• Limitaciones del movimiento en la columna y problemas de equilibrio.
De eliminación• Los casos severos de canal estrecho también pueden ocasionar proble-
mas de vejiga e intestino, pero no es común.
Técnicas y medios de diagnóstico• Rx de la columna vertebral.• RNM.• TAC.• Electromiografía.• Gammagrafía ósea.
Medidas terapéuticas
Tratamiento no quirúrgicoPuede aliviar el dolor en muchos casos. Se trata básicamente con AINE, aso-ciados a un programa adecuado de terapia física y rehabilitación.
Tratamiento quirúrgico• Laminectomía descompresiva.• Laminotomía y foraminotomía.• Facetectomía medial.• Discectomía y fusión cervical anterior.• Corpectomía cervical.• Laminoplastia.
Espondilolistesis
Consiste en un deslizamiento de una vértebra sobre otra. Existen dos tipossegún se deslice hacia delante (anterolistesis) o hacia atrás (retrolistesis) (VerImagen 23). El deslizamiento puede ser sólo del cuerpo o de toda la vértebra.Frecuentemente se localiza a la altura de la columna lumbosacra. Se clasificaen cuatro grados en función del desplazamiento:
• Grado I: al menos un desplazamiento equivalente, igual o menor al25%.
• Grado II: 25-50%.• Grado III: 50-75%.• Grado IV: 75-100%.
Se habla de espondiloptosis cuando el desplazamiento es mayor del 100% yuna vértebra “se cae” por delante de la vértebra inferior.
La espondilolistesis puede variar de leve a severa. Puede producir un incre-mento de la lordosis (curvatura anormal en cualquier parte de la espalda),pero en las etapas finales puede ocasionar cifosis (espalda redonda) a me-dida que la columna superior desciende sobre la columna inferior.
Clasificación• Displásica o congénita: producida por defecto en las articulaciones de
la columna, se da a nivel L5-S1.• Ístmica: resulta de la fractura de la porción interarticular pars articula-
ris. Esta fractura se denomina espondilolisis y se produce a nivel L5-S1.• Degenerativa: principalmente a nivel L4-L5, debida a enfermedad dis-
cal degenerativa y artritis. Se suele acompañar de estenosis de canal.• Traumática: debida a fractura de una parte de la vértebra distinta a la
pars articularis. Afecta principalmente al arco neural de L4-L5 y se vemás frecuentemente en adultos jóvenes (accidentes deportivos y detrabajo).
• Iatrogénica: en pacientes a los que se les ha realizado una laminecto-mía o descompresión que les ha ocasionado inestabilidad.
• Patológica: se agrega a afecciones generalizadas del esqueleto: enfer-medad de Paget, mal de Pott, metástasis ósea, sífilis, etc.
Hallazgos físicosLa persona con espondilolistesis puede estar asintomática.
Neurológicos• Ciatalgia o lumbociatalgia.• Sensibilidad localizada sobre la columna justo por encima de la pelvis.• Alteraciones de los reflejos (aquíleos, rotulianos, etc.).• Puede haber daños neurológicos (debilidad en la pierna o cambios en
la sensibilidad como consecuencia de la presión ejercida sobre las raí-ces nerviosas y puede causar dolor que se irradia hacia abajo).
• En espondilolistesis severas se produce dolor irradiado.• Claudicación intermitente de origen neurológico.
Músculo-esqueléticos• Postura hiperlordótica con acortamiento de los músculos isquiosura-
les.• En ocasiones, escoliosis en pacientes con espondilolistesis displásica.• Dolor al levantar la pierna en posición recta.• Puede haber dolor a la palpación profunda de las últimas vértebras
lumbares.• En ocasiones contractura muscular refleja de la región paralumbar que
hace persistir el dolor, incluso en reposo.• El dolor se pone especialmente de manifiesto al realizar una hiperexten-
sión lumbar o extensión y rotación espinal.• Marcha sui generis: la contractura de los músculos isquiotibiales hace
que camine con las caderas y las rodillas flexionadas y el tronco haciaadelante.
Técnicas y medios de diagnóstico• Radiografía simple: en bipedestación, anteroposterior y lateral. Imagen 23. Retrolistesis
© DA
E
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 730

731
enfermería médico-quirúrgica
• Electromiograma: si se sospecha que se está iniciando una compresiónnerviosa.
• Resonancia magnética.
Medidas terapéuticasControl de los síntomas:
• Reposo según la magnitud del dolor.• Tratamiento farmacológico: analgésicos, antiinflamatorios, relajantes
musculares, etc.
• Calor superficial y profundo. El tratamiento depende en gran medidadel grado de desplazamiento vertebral.
La mayoría de las espondilolistesis que causan dolor responden a los trata-mientos conservadores sin que sea necesario operarlas.
La cirugía sólo es necesaria en las espondilolistesis de grado III y IV en lasque se demuestra que el deslizamiento provoca compresión nerviosa cau-sando pérdida de fuerza importante o progresiva, o dolor irradiado a laspiernas.
TUMORES ÓSEOS
Clasificación
En general, los tumores óseos se clasifican con criterio histológico, es decir,atendiendo al tejido del que proceden (hueso, cartílago, médula ósea, vasoso tejido conectivo) y a su carácter benigno o maligno (Ver Imagen 24).
Según el tejido de origen• Tejido óseo:
– Osteoma osteoide.– Osteosarcoma.– Tumor de células gigantes.
• Tejido cartilaginoso:– Condroblastoma epifisiario.– Condrosarcoma.
• Tejido conectivo: fibrosarcoma.• Tejido hematopoyético: sarcoma de Ewing.
Según su ubicación• Epífisis:
– Tumor de células gigantes.– Condroblastoma.
• Metáfisis: bajo el cartílago de crecimiento, que es una barrera para queel tumor avance hacia la articulación. La existencia en esta región delcartílago de crecimiento implica que sea la zona de mayor producciónde tumores óseos:– Osteosarcoma.– Osteocondroma.– Fibrosarcoma.– Quiste óseo simple.– Quiste óseo aneurismático.
• Diafiso-metafisiarios:– Sarcoma de Ewing.– Fibrosarcoma.
Te conviene recordar...
✔ Hay muchos factores de riesgo para la hernia discal, aparte del desgaste propio de la edad. Algunos de ellos son: malas pos-turas, mecánica corporal incorrecta, falta de ejercicio físico regular, levantar pesos de forma incorrecta, movimientos repetiti-vos de flexión-extensión, cargar peso, sobrepeso y atrofia de la musculatura dorsolumbar.
✔ Tras un episodio de dolor, y una vez realizado el reposo, es conveniente plantearse la modificación de pautas anómalas en elestilo de vida o en la forma de moverse que afecten a la columna vertebral.
✔ La espondilolistesis consiste en un deslizamiento de una vértebra sobre otra. Frecuentemente se localiza en la columna lum-bosacra.
✔ La estenosis de canal es el estrechamiento del canal de la columna cervical o lumbar que produce compresión en el saco duraly/o en las raíces del nervio.
✔ Las lesiones relacionadas con mala mecánica corporal se evitan mediante una higiene postural adecuada.
Imagen 24. Signos radiológicos en partes blandas de tumores benignos y malignos
© DA
E
Malignos (con destrucción
de periostio)Benignos
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 731

732
Manual DAE de enfermería. EIR. Oposiciones ••
• Diáfisis:– Mieloma.– Metástasis.
Según su malignidad• Tumores benignos (Ver Tabla 9):
– Osteocondroma.– Condroma.– Defecto fibroso cortical.– Osteoma osteoide.– Condroblastoma.– Tumor de células gigantes.– Fibroma.– Displasia fibrosa.– Quiste óseo esencial.– Quiste óseo aneurismático.– Tumores latentes o inactivos.– Tumores activos.– Tumores localmente agresivos.
• Tumores malignos:– Osteosarcoma.– Sarcoma de Ewing.– Condrosarcoma.– Mieloma.– Neuroblastoma.– Fibrosarcoma.– Condroma.
• Tumores metastáticos. Más frecuentes en los huesos o zonas de huesoque son más ricos en médula ósea:– Columna vertebral.– Pelvis.– Extremos superiores del fémur y húmero.– Cráneo.
– Costillas.
Hallazgos físicos
En general, los tumores óseos provocan dolor en la zona donde se asientancomo síntoma principal; pueden provocar tumefacción e impotencia funcio-nal.
Cuando se trata de tumores malignos, incluidos los metastáticos, se altera elestado general y aparece fatiga, fiebre, cansancio, pérdida de peso y pérdidade apetito. El dolor es más intenso en estos tumores y pueden causarse frac-turas patológicas.
Técnicas y medios de diagnóstico
• Radiografía simple.• Resonancia nuclear magnética (RNM).• Tomografía axial computarizada (TAC).• Gammagrafía.• Biopsia.• Laboratorio.
Medidas terapéuticas
Las opciones de tratamiento dependen del tipo, tamaño, sitio y estadio de laenfermedad, así como de la edad del paciente. La cirugía, la quimioterapia yla radioterapia son los pilares del tratamiento de los tumores óseos.
CirugíaLa extirpación estaría indicada cuando producen dolores, deformidades o enlos casos en que exista riesgo de malignización.
En los tumores malignos, el tratamiento ha de individualizarse en función deltipo de tumor, grado histológico y extensión, pero la cirugía sigue siendo eltratamiento de elección inicial. La cirugía puede ser:
Osteocondroma
Tumor de células gigantes
Condroblastoma
Condromas benignos
Osteoma osteoide
Osteoblastoma
Edad
10-20 años
20-30 años
10-20 años
10-30 años
10-30 años
10-30 años
Localización
• Huesos largos de lapierna
• Pelvis• Escápula
• Rodilla• Pelvis• Esternón
• Extremos de huesoslargos
• Cadera• Hombro• Rodilla
• Parte central del hueso
• Brazos y piernas
• Columna vertebral
Síntomas
• Masa dura e inmóvil• No dolor
• Masa visible• Dolor• Limitación movilidad
• Dolor• Limitación movilidad• Aspecto reseco del
músculo adyacente
• Algunos, dolor; otros, no
• Atrofia muscular• Dolor que empeora
durante la noche
• Dolor
Tejido origen
• Metáfisis• Tejido cartilaginoso
• Epífisis• T. óseo
• Epífisis• T. cartilaginoso
• Metáfisis• T. cartilaginoso
• T. óseo
• T. óseo
Tratamiento
Observación
Quirúrgico
Quirúrgico
Observación
Quirúrgico
Quirúrgico
Tabla 9. Tumores benignos
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 732

733
enfermería médico-quirúrgica
• Conservadora.• Radical.
QuimioterapiaSe emplea en algunos tumores como complemento de la cirugía, antes (neo -adyuvante) y/o después (adyuvante) de la intervención, y es el tratamientode elección para las metástasis óseas de tumores quimiosensibles.
RadioterapiaLa radioterapia es también un complemento de la cirugía en aquellos tumo-res sensibles a ella, como el sarcoma de Ewing y los linfomas óseos. El osteo -sarcoma es menos sensible.
Tumores benignos
La clasificación de los tumores benignos se puede observar en la Tabla 10.
Hallazgos físicos• Masa detectable dura e inmóvil que no produce dolor.• Estatura más baja de lo normal para la edad.
Medidas terapéuticasSi no hay señal de debilitamiento del hueso o crecimiento adicional del tumorse recomienda el control periódico y la observación.
Tumor óseo de células gigantesLos tumores de células gigantes aparecen con mayor frecuencia una vez com-pletado el crecimiento óseo del esqueleto. Este tumor está compuesto poruna gran cantidad de células benignas y se origina cerca del extremo delhueso, en las inmediaciones de una articulación. La ubicación más habituales la rodilla, pero también puede aparecer en los huesos de los brazos y pier-nas o en los huesos planos como el esternón o la pelvis (Ver Imagen 25). Enciertos casos, su causa se ha asociado con la enfermedad de Paget.
Hallazgos físicos• Dolor en la articulación adyacente.• Masa visible.• Hinchazón.• Fractura del hueso.• Limitación de la movilidad en la articulación adyacente.
Medidas terapéuticas• Cirugía para extraer el tumor y el hueso afectado.
• Injerto óseo para rellenar el vacío de la zona afectada.
CondroblastomaEl condroblastoma es un tumor benigno poco frecuente que puede afectar apersonas de cualquier edad.
Este tumor, también llamado tumor de Codman, se origina a partir del cartí-lago. Suele afectar a los extremos de los huesos largos de los brazos y las pier-nas, la cadera, el hombro y la rodilla.
Se cree que los tumores se originan en las células denominadas condroblas-tos.
Tumores malignos
La clasificación de los tumores malignos se refleja en la Tabla 10.
Imagen 25. Posibles localizaciones del tumor de células gigantes
© DA
E
Localización frecuenteLocalización menos frecuente
Osteosarcoma
Mieloma múltiple
Sarcoma de Ewing
Condrosarcoma
Edad
10-20 años
> 60 años
5-20 años
50-70 años
Localización
• Huesos largos querodean la rodilla
• Pelvis, hombro, etc
• Capa externa del hueso
• Brazos y piernas• Pelvis
• Pelvis, muslos, escápula,fémur, etc.
Síntomas
• Dolor• Hinchazón• Fractura patológica
• Dolor• Astenia• Infecciones repetidas
• Dolor• Hinchazón
• Dolor
Tejdo origen
Tejido conjuntivo y óseo
Células de la médula ósea
Tejido hematopoyético
Tejido cartilaginoso
Tratamiento
• Quimioterapia• Cirugía
• Quimioterapia• Radioterapia• Cirugía
• Quimioterapia• Radioterapia• Cirugía
• Cirugía
Tabla 10. Tumores malignos
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 733

734
Manual DAE de enfermería. EIR. Oposiciones ••
OsteosarcomaLos osteosarcomas (sarcoma osteogénico) constituyen alrededor del 20% detodos los tumores óseos malignos primarios. Aunque su incidencia es mayoren individuos de entre 10 y 20 años, pueden aparecer a cualquier edad. Lagran mayoría no tienen una etiología conocida y se consideran, por tanto,idiopáticos o primarios.
Entre los osteosarcomas primarios, los medulares son el tipo más frecuente;constituyen aproximadamente el 85% de todos los tipos de osteosarcoma.
Hallazgos físicos• Dolor en el hueso afectado, moderado e intermitente al principio, que
se hace progresivamente más intenso y continuo.• Tumefacción alrededor de la zona afectada.• Aumento del dolor con la actividad y al levantar peso.• Cojera.• Reducción del movimiento del miembro afectado.
Medidas terapéuticasCombinación de quimioterapia y cirugía.
Mieloma múltiple
EtiopatogeniaEs el tumor maligno primario más habitual. Se presenta con mayor frecuen-cia en personas de edad avanzada y su incidencia es del doble en las muje-res que en los hombres.
El mieloma afecta a determinados glóbulos blancos, denominados célulasplasmáticas, que empiezan a crecer en exceso y se convierten en células anor-males y similares entre sí. A estas células plasmáticas anormales se las deno-mina células de mieloma, las cuales se instalan en la médula ósea y en lacapa externa del hueso.
Hallazgos físicos• Dolor en el hueso.• Fracturas.• Debilidad.• Fatiga.• Pérdida de peso.• Infecciones repetidas.
Medidas terapéuticas• Quimioterapia.
• Radioterapia.• Cirugía.
Sarcoma de EwingEl sarcoma de Ewing es el tumor óseo responsable de alrededor del 30% delos tumores óseos pediátricos.
El sarcoma de Ewing se puede originar en cualquier hueso, pero aparece conmayor frecuencia en las extremidades y puede comprometer a los músculosy al tejido blando que rodea el tumor. Sus células también pueden producirmetástasis en otras zonas del cuerpo, como la médula ósea, los pulmones,los riñones, el corazón, la glándula suprarrenal y los tejidos blandos.
Hallazgos físicos• Dolor alrededor de la zona del tumor.• Hinchazón y/o enrojecimiento alrededor de la zona del tumor.• Fiebre.• Síntomas relacionados con la compresión de los nervios causada por el
tumor: adormecimiento, sensación de hormigueo, parálisis, etc.
Medidas terapéuticas• Quimioterapia.• Radioterapia.• Cirugía: se efectúa biopsia del tejido afectado para confirmar el diagnós-
tico.
CondrosarcomaEl condrosarcoma es un tumor óseo que se desarrolla en las células del cartí-lago. Es el tercer tumor primario maligno del hueso en frecuencia, después delmieloma y del osteosarcoma. Constituye aproximadamente el 10% de todoslos sarcomas óseos primarios.
Afecta preferentemente a huesos planos de la cintura escapular y pelviana ya porciones proximales de los huesos tubulares largos. La mayoría de las vecesse produce a partir de células del cartílago normales, aunque puede origi-narse a partir de un tumor benigno.
Hallazgos físicosDolor, que puede estar presente durante años e intensificarse paulatinamente,que no se alivia con el descanso y empeora por la noche.
Medidas terapéuticas• Efectuar biopsia para confirmar diagnóstico.• Cirugía.
Te conviene recordar...
✔ La edad del paciente y la determinación del número de lesiones (única o múltiple) son los puntos de partida en la aproxima-ción diagnóstica de los tumores óseos.
✔ La edad del paciente es el factor aislado más importante de toda la historia clínica, pues puede utilizarse junto con los hallaz-gos radiológicos para establecer un diagnóstico.
✔ Con frecuencia, la cirugía es el tratamiento de elección. Aunque algunas veces es necesaria la amputación de una extremidad,la quimioterapia pre y postoperatoria ha hecho posible que, en muchos casos, se lleve a cabo la cirugía y se salve la extremi-dad afectada.
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 734

735
enfermería médico-quirúrgica
ARTRITIS REUMATOIDE (EIR 94-95, 59)
La artritis reumatoide es una patología que afecta al tejido conjuntivo, deforma inflamatoria, crónica, progresiva, sistémica y poliarticular. También cursacon manifestaciones extraarticulares, siendo una enfermedad autoinmune,no infecciosa (Ver Imagen 26).
En la AR las articulaciones más afectadas por la inflamación y la degeneraciónson las diartrodiales o sinoviales. Estas articulaciones están dotadas de granamplitud de movimiento. El cartílago articular es una superficie lisa y resis-tente que facilita los movimientos, además de avascular, por lo que no puederegenerarse. La membrana sinovial reviste la superficie interna de la cápsulafibrosa y secreta un líquido en el espacio articular (líquido sinovial) que tienefunciones de absorción de impactos y de lubricación, con lo que posibilita lalibertad de movimientos de la articulación.
El inicio de la enfermedad se caracteriza por inflamación del tejido sinovial delas articulaciones (sinovitis). El sinovio se engrosa, se acumula líquido en elespacio articular y se forma pannus (tejido de granulación vascular compuestode células inflamatorias que erosionan el cartílago articular y destruyen elhueso). El resultado es la formación de calcificaciones y adherencias fibrosascon pérdida de la densidad del hueso. Asimismo, los músculos se ven afecta-dos por pérdida de elasticidad y contractilidad.
Etiopatogenia
Aunque se ha avanzado mucho en el conocimiento de los mecanismos queproducen el daño articular, la causa inicial se desconoce. Se habla de ciertosfactores que predisponen a la aparición de AR, reflejados en la Tabla 11.
Imagen 26 . Frecuencia de afectación de diversas articulaciones en la AR
© DA
E
Temporomandibular 30%
Columnacervical 40%
Cricoaritenoidea 10% Acromioclavicular 50%
Externoclavicular30%
Codo 50%
Muñeca50%
Metacarpofalángicas,interfalángicas 90%
Tobillo, subtalar 80%
Rodilla 50%
Metatarsofalángicas 90%
Cadera50%
Hombro 60%
Tabla 11. Factores implicados en la producción de AR
Factores genéticosSe les atribuye un riesgo de un 20-30%. Los más estudiados
son los antígenos de histocompatibilidad HLA de
clase II y el receptor de linfocitos T
Factores iniciadoresAgentes infecciosos: virus, bacterias
Factores localizadores• Integrinas: permiten la transmisión de señales
entre el citoplasma y el exterior celular• Selectinas: implicadas en la fase de contacto
entre leucocitos y endotelio vascular• Inmunoglobulinas
Factores reguladores• Citoquinas: mediador polipeptídico
responsable del estímulo y diferenciación dedistintos tipos celulares
• Quimioquinas: – A: atraen a neutrófilos– B: atraen a linfocitos T y monocitos
Inicio de mecanismos efectores
Factores perpetuadores• Colágeno tipo II que actúa como antígeno
oculto• Factor reumatoide
Mantenimiento de mecanismos efectores
Cambios estructurales en articulaciones
Lesiones extraarticulares
Inflamaciónarticular
aguda
Inflamaciónarticularcrónica
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 735

736
Manual DAE de enfermería. EIR. Oposiciones ••
Hallazgos físicos
Dermatológicos• Nódulos reumatoides (Ver Imagen 27), abultamientos duros que pue-
den aparecer en zonas de roce (codos, dorso de manos y pies, etc.).• Sequedad de piel y mucosas.• Manchas en la piel.• Quistes de Baker en casos de enfermedad avanzada (Ver Imagen 28).
Músculo-esqueléticos• Deformidad articular que afecta sobre todo a las articulaciones proxi-
males de las manos (Ver Imagen 29).• Ensanchamiento de los dedos y desviación cubital de la mano (Ver Ima-
gen 30). Aumento de temperatura en la articulación afectada (calorlocal).
• Atrofia muscular.• Inflamación de las articulaciones afectadas, como rodillas en flexo (Ver
Imagen 31).
Respiratorios• Ronquera mantenida, sin que anteriormente haya notado catarro.
Cardiovasculares• Fiebre inexplicable.• Cansancio fácil.
Imagen 27. Nódulos reumatoides subcutáneos
© Ce
dida p
or las
autor
as
Imagen 29. Lesiones iniciales
© Ce
dida p
or las
autor
as
Imagen 30. Desviación cubital inicial
© Ce
dida p
or las
autor
as
Imagen 31. Desviación cubital inicial
© Ce
dida p
or las
autor
as
Imagen 28. Gran quiste de Baker que ha ido abriéndose por la parte posterior de lapantorrilla
© Ce
dida p
or las
autor
as
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 736

737
enfermería médico-quirúrgica
Oculares• Enrojecimiento o sensación de arenilla en los ojos.• Sequedad ocular.
Ginecológicos• Picor vaginal.• Sequedad vaginal.• Dolor coital (dispareunia).
Técnicas y medios de diagnóstico
Dado el carácter no migratorio de la AR, en la que la afección de las articu-laciones es simultánea o sucesiva, es fundamental conocer los criterios diag-nósticos de esta enfermedad para valorar su persistencia (Ver Tabla 12).
Datos de laboratorioAumento de la velocidad de sedimentación globular (VSG); cuantificaciónde la proteína C reactiva (PCR); anemia normocítica, normocrómica o hipo-crómica; FR IgM en el suero (seropositivos); anticuerpos antinucleares (ANA).En caso de enfermedad avanzada, en el proteinograma suele detectarse unaumento de la alfa-2-globulina y de la gammaglobulina con carácter poli-clonal.
Radiología y otras técnicas de imagen• Observación de la osteopenia epifisiaria: la pérdida de cartílago arti-
cular es un signo temprano de lesión irreversible. La radiografía de lasmanos es definitiva con hallazgos bilaterales y simétricos (Ver Ima-gen 32).
• Resonancia magnética: puede detectar precozmente las erosiones.• Gammagrafía: puede registrar el incremento de la actividad inflamato-
ria articular.
• Ecografía: puede delimitar los quistes sinoviales y otras alteraciones detejidos blandos.
• Artrocentesis: aumento del líquido sinovial de las articulaciones afec-tadas, de aspecto opaco y con niveles de glucemia inferiores a los nive-les séricos.
Medidas terapéuticas
Tratamiento farmacológicoEs un objetivo primordial el inicio precoz del tratamiento para retardar o de-tener la lesión articular y mejorar así la calidad de vida del paciente.
• Primer grupo: los destinados a aliviar los síntomas pero que no tienenningún efecto sobre la progresión de la enfermedad, es decir, que no lafrenan y que, por tanto, no van a evitar el daño articular permanente.Se habla de los fármacos conocidos como antiinflamatorios que puedenser esteroideos o no esteroideos (Ver Tabla 13).
• Segundo grupo: aquellos fármacos que evitan el daño articular perma-nente y, por tanto, frenan la progresión de la enfermedad. Estos conjun-tamente reciben el nombre de FARME (fármacos antirreumáticosmodificadores de la enfermedad), también llamados FARAL (fármacosantirreumáticos de acción lenta) o, en inglés, DMARD (disease modif-ying antirheumatic drug) (Ver Tabla 14).
Tratamiento quirúrgicoSon varias las técnicas quirúrgicas, por lo que la elección de una u otra va a de-pender especialmente de la zona afectada:
• Sinovectomía.• Artrodesis.• Osteotomía.• Artroplastia.
Tratamiento rehabilitadorEl tratamiento rehabilitador precoz es fundamental en la recuperación y prác-ticamente el 100% de los pacientes experimentan alivio del dolor y mejoransu capacidad de movimiento respecto a su situación previa.
Tabla 12. Criterios diagnósticos de la AR
Criterios
1. Rigidez matutina
2. Artritis de tres o más articulaciones
3. Artritis de articulaciones de las manos
4. Artritis simétrica
5. Nódulos reumatoides
6. Factor reumatoide sérico
7. Cambios radiográficos
Especificaciones
Una hora de duración como mínimo
Observada por un médico comoaumento de partes blandas o derrames.Articulaciones IFP, MCF, muñeca, codo,rodilla, tobillo, MTF
IFP, MCF, muñeca: una al menos
Artritis simultánea de dos articulacionessimétricas: la afección de IFP, MCF o MTFpuede ser bilateral y no simétrica
Observados por un médico enprominencias óseas,superficiesextensoras o para articulaciones
Determinado por un método que seapositivo en menos del 5% de los sujetossanos
Cambios típicos de AR en la radiografíaposteroanterior de mano y muñeca quedeben incluir erosiones o decalcificaciónepifisaria evidente
• Los criterios 1 a 4 son valorables si persisten más de seis semanas• El diagnóstico de artritis reumatoide requiere cuatro de los siete criterios• Se suprime la designación de artritis reumatoide clásica definida o probable
Imagen 32. Erosiones en el carpo, metacarpofalángicas e interfalángicas conestrechamiento de los espacios articulares y osteopatía
© Ce
dida p
or las
autor
as
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 737

738
Manual DAE de enfermería. EIR. Oposiciones ••
NombreAntiinflamatorios no esteroideos
AINE TRADICIONALESIbuprofenoNaproxenoDiclofenacoAceclofenacoIndometacinaMetamizolPiroxicamNabumetonaKetorolacoSulindaco
Paracetamol
Ácido acetilsalicílico
INHIBIDORES DE LA COX-2CelecoxibRofecoxibEtoricoxib
Antiinflamatorios esteroideos
CORTICOIDESPrednisonaMetilprednisolonaDeflazacort
Usos
Reducen el dolor y la inflamación en horas o díasdespués de su ingesta. Existe variabilidad de respuesta
Analgésico
A dosis de más de 3 g al día es antiinflamatorio
Son capaces de reducir el dolor y la inflamación. Al noinhibir la COX-1, reducen el riesgo de úlcera o sangradogástrico
Usados a dosis mínimas eficaces
Efectos secundarios
Dolor abdominal, náuseas, vómitos, sangrado digestivo,perforación digestiva, ardor esofágico, cefaleas, mareos,fallo renal, incremento de la presión arterial, tinnitus
A dosis altas puede producir hepatotoxicidad
Trastornos digestivos, úlcera y sangrado de estómago oduodeno, fallo renal
Pueden incrementar la presión arterial y las molestias,ulceración o sangrado digestivo
Osteoporosis, edemas, aumento de peso, debilidadmuscular, hiperglucemias, cataratas, hipertensión arteriale hiperlipemia
Tabla 13. Clasificación del primer grupo de fármacos utilizados en el tratamiento de la AR
Nombre
FARME CLÁSICOSMetotrexato
Sulfasalacina(Salazopirina®)
Antipalúdicos
Leflunomida(Arava®)
OTROSSales de oro, AzatriopinaD-Penicilamina
ANTI-TNFInfliximab (Remicade®)
Adalidumad (Humira®)
Etanercept (Enbrel®)
ANTI-INTERLEUQUINA-1Anakinra (Kineret®)
Usos
Es el más utilizado. Se emplea en dosis únicas semanalespor vía oral el mismo día de la semana (o como muchoen dos dosis el mismo día) o intramuscular
2-3 g día durante tres meses por vía oral
Cloroquina disfosfato (Resochin®250 mg) una al día ohicrocloroquina (Dolquine® 200 mg) dos al día
20 mg/día por vía oral
A veces se usan cuando fallan otros medicamentos
Vía endovenosa cada 4-8 semanas asociado ametotrexato
Vía subcutánea cada dos semanas, solo o asociado ametrotexato
Vía subcutánea, dos veces a la semana, solo o asociado aMTX
Vía subcutánea en inyecciones diarias
Efectos secundarios
Miolosupresión: leucopenia, anemia Trombocitopenia Vómitos, diarrea, gingivitis, mucositis, hemorragiadigestiva. Toxicidad hepática, toxicidad pulmonar aguda(neumonitis, fibrosis), fotodermatitis Cefaleas, mareos, afasia. Vasculitis, prurito,hiperpigmentación
Náuseas, diarrea, cefalea, úlceras bucales, alopecia,oligospermia transitoriaRara vez leucopenia
Náuseas, cefalea, retinopatíaRara vez dolor abdominal, miopatía
Náuseas, diarrea, rash, alopecia. Alta teratogenicidad. Trombocitopenia Rara vez leucopenia, hepatitis
Su toxicidad limita su uso
Aumento del riesgo de infección incluyendo reactivacióndel TBC Rara vez desmielinizante
Reacción local en zona de punción
Infección, neutropenia, cefaleaVértigo, náuseas
Tabla 14. Clasificación del segundo grupo de fármacos utilizados en el tratamiento de la AR
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 738

739
enfermería médico-quirúrgica
ENFERMEDAD ÓSEA DE PAGET
El tejido óseo nunca está en reposo metabólico, constantemente remodela yredistribuye sus reservas minerales de calcio y fósforo. La formación y la des-trucción (resorción) de hueso en el esqueleto adulto están equilibradas por unproceso denominado acoplamiento. En la enfermedad ósea de Paget (EOP)hay una alteración de los mecanismos de formación (mediados por los os -teoblastos) y destrucción (dirigidos por los osteoclastos) del hueso, de maneraque operan a un ritmo que no es el adecuado (Ver Imagen 33).
La EOP es un trastorno metabólico crónico exclusivo del tejido óseo, al queafecta de forma focal. Se caracteriza por un anómalo proceso de remodela-ción del hueso en el que se produce un marcado aumento de la resorción, se-guido de una formación ósea anormal. El hueso que se genera es de estructuradesorganizada, de mayor tamaño, menor resistencia y más susceptible a de-formidades y fracturas (Ver Imagen 34). La elevación del recambio óseo, quepuede llegar a ser 20 veces mayor de lo normal, depende de la extensión deesta patología.
Estas lesiones pueden afectar a un solo hueso, 17% de los casos (monostó-tico), o a varios (poliostótico), y en cada una puede haber más de un foco
Te conviene recordar...
✔ La artritis reumatoide (AR) es la segunda enfermedad del tejido conjuntivo más destructiva. Es una enfermedad inflamatoriacrónica, poliarticular y sistémica que puede afectar a cualquier articulación sinovial. Dicha afección de las articulaciones es bi-lateral y simétrica, pudiendo cursar con manifestaciones extraarticulares.
✔ El comienzo, curso clínico y pronóstico es muy lento y variable, por lo que el enfoque terapéutico debe individualizarse.✔ Es una enfermedad sin tratamiento curativo en la actualidad. Sin embargo, el inicio precoz del tratamiento puede retardar o
detener la lesión articular y mejorar así la calidad de vida del paciente.✔ Entre los fármacos que alivian los síntomas están los antiinflamatorios, que pueden ser esteroideos o no esteroideos.✔ Aquellos fármacos que evitan el daño articular permanente y, por tanto, frenan la progresión de la enfermedad, reciben el
nombre de FARME (fármacos antirreumáticos modificadores de la enfermedad).✔ Puede ser necesaria la cirugía reparadora.✔ Existen programas de ejercicios para mejorar la fuerza y la resistencia musculares.
Imagen 33. Remodelado óseo
© DA
E Destrucción ósea
Crecimiento óseo normal
Imagen 34. Diferencias entre el tejido óseo normal y el pagético
© DA
E
Hueso normal
Cavidad medular Incremento de la vascularización
Hueso pagético
Resorción ósea conneoformación irregular
Arqueamientode hueso largo
Aumento dela densidad
cortical
Fractura
Hueso esponjosoHuesocortical
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 739

740
Manual DAE de enfermería. EIR. Oposiciones ••
(Ver Imagen 35). La EOP puede ocasionar complicaciones neurológicas, or-topédicas y cardiovasculares:
• Las complicaciones neurológicas están provocadas por el crecimiento delhueso pagético, que puede provocar la compresión de estructuras neu-rológicas (nervios, masa encefálica).
• Las complicaciones ortopédicas pueden ser artrosis (principalmente enla cadera y la rodilla), fisuras y fracturas patológicas.
• Las complicaciones cardiovasculares vienen dadas por el aumento delflujo sanguíneo hacia el hueso pagético.
Etiopatogenia
Su etiología se desconoce hasta el momento, aunque se especula que deter-minados factores ambientales y alimentarios podrían iniciar o propagar la en-fermedad en personas con una cierta predisposición genética.
Hallazgos físicos
Músculo-esqueléticos• Dolor óseo.• Deformidad y arqueamiento óseo: es típica la incurvación de la tibia en
“sable” (Ver Imagen 36) y del fémur “en cayado de pastor”.• Cojera y alteraciones en la marcha.• Deformidad e inflamación articular en relación con artropatías de vecin-
dad.• Fracturas patológicas completas e incompletas.• Osteoartritis.
• Cifosis dorsal y disminución en la estatura.• Osteosarcoma.
Neurológicos• Paresia.• Hidrocefalia.• Platibasia.• Cefalea.• Mareos.• Vértigo.
Imagen 35. Porcentaje de los huesos más afectados por la EOP en el adulto joven
© DA
E
Cráneo 43%
Húmero 14%
Columna dorsal40%
Columna lumbar 40%
Pelvis 67%
Fémur 50%
Tibia 19%
Imagen 36. Tibia en “sable”
© DA
E
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 740

741
enfermería médico-quirúrgica
• Parálisis facial.• Paraplejia.• Epilepsia.• Disartria.
Metabólicos• Hipercalcemia.• Hipercalciuria.
Sensoriales• Pérdida de audición o sordera.• Alteraciones visuales.• Exoftalmos.• Diplopía.• Acúfenos.• Parestesias.• Epífora (lagrimeo copioso y persistente).
Cardiovasculares• Bloqueos cardiacos.• Insuficiencia cardiaca.• Edemas.• Varices.• Calor local en el área afecta.• Elevación del gasto cardiaco (en enfermedad generalizada y que afecta
a 1/3 o más del esqueleto).
Renales• Disuria.• Tenesmo vesical.• Hematuria.• Infecciones urinarias recurrentes.• Expulsión de arenilla al orinar, en relación con la mayor incidencia de
cálculos renales en estos pacientes.
Digestivos• Maloclusión• Hipercementosis.• Aumento del sangrado de las encías.• Estreñimiento.
• Disfagia.• Dificultad en la masticación.• Diastema.
Conductuales• Demencia.• Alteración en el comportamiento.
Técnicas y medios de diagnóstico
El diagnóstico se sospecha por el hallazgo casual de la elevación de la fosfa-tasa alcalina total (FAT) o por los hallazgos radiológicos.
Pruebas de laboratorioEl incremento de la resorción y la formación óseas se evalúa con los marca-dores de recambio óseo (MRO).
Radiología y otras técnicas de imagenLa radiología simple es la base del diagnóstico en la mayoría de los casos. Lasimágenes van a variar según la fase evolutiva y el hueso afecto.
Medidas terapéuticas
Actualmente no existe tratamiento curativo, aunque los nuevos fármacos, in-troducidos muy recientemente, son capaces de modificar el curso de la enfer-medad. Los objetivos son controlar la progresión de la enfermedad, suscomplicaciones y sus manifestaciones clínicas.
Tratamiento farmacológico (Ver Tabla 15)
Tratamiento ortopédico: cirugíaSe recomienda sólo cuando surgen complicaciones propias de la EOP, como:
• Fracturas.• Artrosis degenerativa severa.• Deformidad ósea.
Tratamiento no farmacológicoCentrado principalmente en fisioterapia rehabilitadota para mejorar la calidadde vida.
Fármacos
Bifosfonatos orales
Leve a grave
Risedronato (Actonel®)
Alendronato (Fosamax®)
Leve
Tiludronato (Skelid®)
Dosis
30 mg/día (2 meses) VO
40 mg/día (6 meses) VO
400 mg/día (3 meses) VO
Evaluación% referidos a pacientes
SAP(fosfatasa alcalina sérica)
e SAP 54%Normalización en un 54%
e SAP 88%Normalización en un 63%
e SAP 50-60%Normalización en un 25-30%
Precauciones
1/2 h antes de la primera in-gesta de la mañana y con unvaso lleno de agua del grifo (nomineral). Se aconseja tomar unsuplemento de calcio y vita-mina D, si la ingesta dietéticaes inadecuada
2 h antes o después de comer(sobre todo productos lácteos oricos en calcio) y con un vasolleno de agua del grifo (nomineral).
Efectos secundarios
Administración oral
• Gastrointestinales: náuseas,vómitos, pirosis, úlceraesofágica, dolor abdominal,esofagitis, dispepsia y úlcerapéptica
• Cefaleas y dolor óseo,muscular o articular
• Efectos secundarios pocofrecuentes: uveítis, escleritis, conjuntivitis,
Tabla 15. Fármacos antirresortivos utilizados en la EOP
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 741
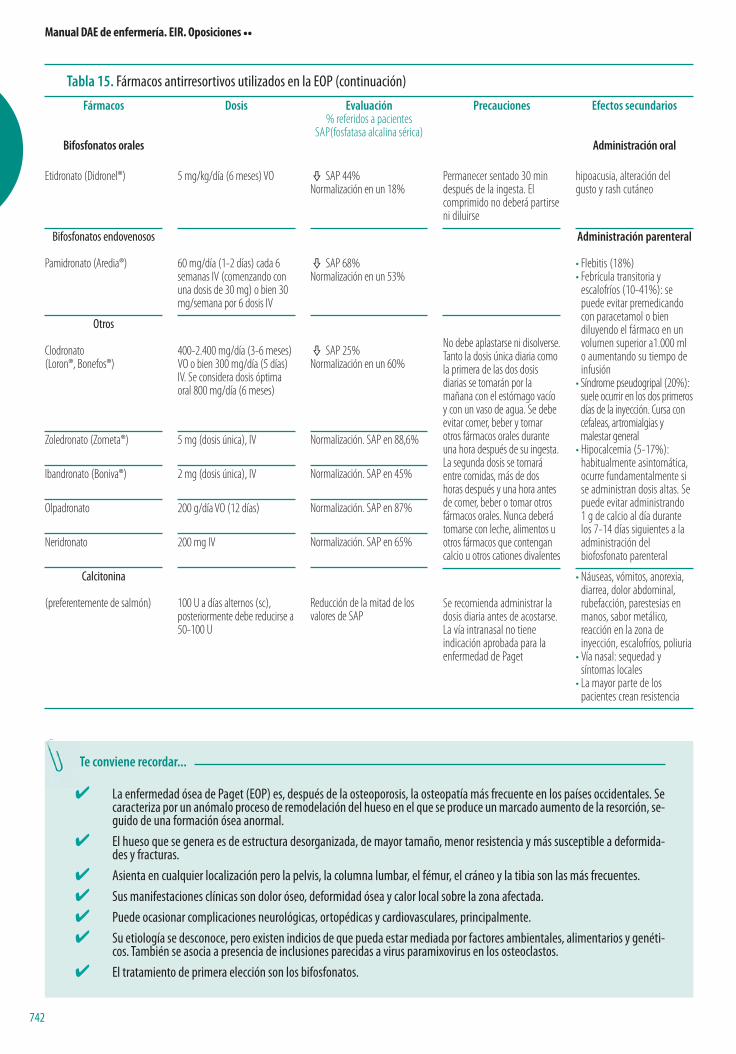
742
Manual DAE de enfermería. EIR. Oposiciones ••
Fármacos
Bifosfonatos orales
Etidronato (Didronel®)
Bifosfonatos endovenosos
Pamidronato (Aredia®)
Otros
Clodronato(Loron®, Bonefos®)
Zoledronato (Zometa®)
Ibandronato (Boniva®)
Olpadronato
Neridronato
Calcitonina
(preferentemente de salmón)
Dosis
5 mg/kg/día (6 meses) VO
60 mg/día (1-2 días) cada 6semanas IV (comenzando conuna dosis de 30 mg) o bien 30mg/semana por 6 dosis IV
400-2.400 mg/día (3-6 meses)VO o bien 300 mg/día (5 días)IV. Se considera dosis óptimaoral 800 mg/día (6 meses)
5 mg (dosis única), IV
2 mg (dosis única), IV
200 g/día VO (12 días)
200 mg IV
100 U a días alternos (sc),posteriormente debe reducirse a50-100 U
Evaluación% referidos a pacientes
SAP(fosfatasa alcalina sérica)
e SAP 44%Normalización en un 18%
e SAP 68%Normalización en un 53%
e SAP 25%Normalización en un 60%
Normalización. SAP en 88,6%
Normalización. SAP en 45%
Normalización. SAP en 87%
Normalización. SAP en 65%
Reducción de la mitad de losvalores de SAP
Precauciones
Permanecer sentado 30 mindespués de la ingesta. Elcomprimido no deberá partirseni diluirse
No debe aplastarse ni disolverse.Tanto la dosis única diaria comola primera de las dos dosisdiarias se tomarán por lamañana con el estómago vacíoy con un vaso de agua. Se debeevitar comer, beber y tomarotros fármacos orales duranteuna hora después de su ingesta.La segunda dosis se tomaráentre comidas, más de doshoras después y una hora antesde comer, beber o tomar otrosfármacos orales. Nunca deberátomarse con leche, alimentos uotros fármacos que contengancalcio u otros cationes divalentes
Se recomienda administrar ladosis diaria antes de acostarse.La vía intranasal no tieneindicación aprobada para laenfermedad de Paget
Efectos secundarios
Administración oral
hipoacusia, alteración delgusto y rash cutáneo
Administración parenteral
• Flebitis (18%)• Febrícula transitoria y
escalofríos (10-41%): sepuede evitar premedicandocon paracetamol o biendiluyendo el fármaco en unvolumen superior a1.000 mlo aumentando su tiempo deinfusión
• Síndrome pseudogripal (20%):suele ocurrir en los dos primerosdías de la inyección. Cursa concefaleas, artromialgias ymalestar general
• Hipocalcemia (5-17%):habitualmente asintomática,ocurre fundamentalmente sise administran dosis altas. Sepuede evitar administrando 1 g de calcio al día durantelos 7-14 días siguientes a laadministración delbiofosfonato parenteral
• Náuseas, vómitos, anorexia,diarrea, dolor abdominal,rubefacción, parestesias enmanos, sabor metálico,reacción en la zona deinyección, escalofríos, poliuria
• Vía nasal: sequedad ysíntomas locales
• La mayor parte de lospacientes crean resistencia
Tabla 15. Fármacos antirresortivos utilizados en la EOP (continuación)
Te conviene recordar...
✔ La enfermedad ósea de Paget (EOP) es, después de la osteoporosis, la osteopatía más frecuente en los países occidentales. Secaracteriza por un anómalo proceso de remodelación del hueso en el que se produce un marcado aumento de la resorción, se-guido de una formación ósea anormal.
✔ El hueso que se genera es de estructura desorganizada, de mayor tamaño, menor resistencia y más susceptible a deformida-des y fracturas.
✔ Asienta en cualquier localización pero la pelvis, la columna lumbar, el fémur, el cráneo y la tibia son las más frecuentes.✔ Sus manifestaciones clínicas son dolor óseo, deformidad ósea y calor local sobre la zona afectada.✔ Puede ocasionar complicaciones neurológicas, ortopédicas y cardiovasculares, principalmente.✔ Su etiología se desconoce, pero existen indicios de que pueda estar mediada por factores ambientales, alimentarios y genéti-
cos. También se asocia a presencia de inclusiones parecidas a virus paramixovirus en los osteoclastos.✔ El tratamiento de primera elección son los bifosfonatos.
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 742

743
enfermería médico-quirúrgica
FIBROMIALGIA
La fibromialgia es una enfermedad reumática crónica y compleja que cursa condolores generalizados y en puntos selectivos o diana, fatiga, sueño no repa-rador y otros síntomas como, por ejemplo, cefalea y colon irritable.
No es una forma de artritis, pues no produce inflamación de las articulacio-nes. Es una forma de reumatismo de los tejidos blandos que no puede ser ex-plicada por la presencia de trastornos inflamatorios o degenerativos de origenmúsculo-esquelético.
Su origen es idiopático, no conociéndose con certeza las alteraciones fisiopa-tológicas que la desencadenan. A menudo se asocia con trastornos de tipopsiquiátrico.
Evolución y pronóstico
Los estudios a largo plazo de pacientes con fibromialgia han demostrado quela evolución natural del proceso es la cronificación. El impacto que tiene sobrelas actividades habituales y la calidad de vida del paciente es muy variable (VerImagen 37). La mayoría de los pacientes no pueden trabajar, por lo que elcoste social por baja laboral es muy importante.
Clasificación
Se puede distinguir entre:
• Fibromialgia primaria: no hay ningún proceso concomitante o subya-cente que pueda explicar los signos y síntomas del paciente.
• Fibromialgia secundaria: se presenta acompañando a una enfermedadde base y mejora cuando lo hace ésta.
• Fibromialgia concomitante: se presenta asociada a otros procesos detipo sistémico como el lupus, el síndrome de Sjögren, artritis reuma-toide y neoplasias sin que la evolución muestre un curso paralelo.
• Fibromialgia localizada o regional: síndrome del dolor miofascial.• Fibromialgia de la senectud: suele presentarse como forma secundaria
o asociada.• Fibromialgia juvenil: aparece en personas menores de quince años.
Etiopatogenia
Actualmente no se conoce la causa de la fibromialgia. Las investigaciones yahan revelado mucho acerca de esta enfermedad, pero sigue siendo un capí-tulo abierto en medicina.
Se han descrito casos que comienzan después de procesos concretos, estre-santes desde el punto de vista físico o psicológico (infección bacteriana o ví-rica, traumatismo, separación matrimonial, etc.). En otros casos aparecedespués de otra enfermedad conocida como la artritis reumatoide, el lupus oel hipotiroidismo.
Estos agentes desencadenantes no parecen causar la enfermedad, sino que loque probablemente hacen es “despertarla” en una persona que tiene una al-teración latente en la regulación de la capacidad de respuesta a determina-dos estímulos estresantes.
Hallazgos físicos
Dolor• El dolor muscular generalizado es el síntoma más característico de la fi-
bromialgia. Es descrito como dolor muscular profundo, quemazón oardor o como si algo se clavase.
• Suele ser generalizado (frecuentemente se describe como un dolor depies a cabeza), aunque puede comenzar en una región del cuerpo, comocuello y hombros, y extenderse a otras áreas al cabo de cierto tiempo.
• Las localizaciones más frecuentes son la región del músculo trapecio yla región lumbar de la espalda.
• Su severidad varía según el día y la hora y es peor por la mañana.• Se exacerba con las situaciones que causan estrés físico (ejercicio mayor
del habitual) o psicológico (ansiedad, depresión, etc.), con los cambiosclimáticos (frío y cambios en la humedad relativa, empeorando con losambientes muy secos), fluctuaciones hormonales (periodo premens-trual, menopausia) y con el empeoramiento de la calidad del sueño.No cede con el reposo.
• Además del dolor generalizado, los pacientes presentan puntos locali-zados de dolor intenso a la palpación o a la presión conocidos comopuntos gatillo.
Fatiga• Alrededor del 90% de los pacientes experimenta fatiga y menor resis-
tencia al esfuerzo, resultando a veces más problemática que el dolor.
Trastornos del sueño• El sueño es poco reparador.
Otros síntomas y síndromes que frecuentemente presentan los pacientes confibromialgia y que se exacerban con los factores ya mencionados son:
• Rigidez y anquilosamiento.• Dolores de cabeza y de cara.• Síndrome de dolor miofascial (MPS o SDM).
Condición neuromuscular caracterizada por la presencia de puntos muy do-lorosos o hipersensibles en áreas específicas (típicamente cuello, hombros ycintura).
Se ha descrito sintomatología referida a otros aspectos como son los gas-trointestinales, genitourinarios, torácicos, sensaciones en los brazos y enlas piernas, hipersensibilidad a la luz, sonidos, toques y olores, dermo-
Imagen 37. La fibromialgia afecta fundamentalmente a las mujeres y a su vidafamiliar y laboral
© L.
Rojo
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 743

744
Manual DAE de enfermería. EIR. Oposiciones ••
grafismo positivo, dificultad de concentración, problemas de desequili-brio, prurito cutáneo, reacciones similares a las alérgicas, ansiedad o de-presión.
Como se puede ver en la Imagen 38, se establecen relaciones en ambos sen-tidos o “círculos viciosos” que hacen que los problemas se mantengan en eltiempo.
Técnicas y medios de diagnóstico
El diagnóstico es eminentemente clínico. Debe realizarse una historia clínicadetallada, así como una exploración física con examen general, neurológicobásico y osteoarticular.
Desde el año 1990, la fibromialgia se diagnostica según los criterios del Ame-rican College of Rheumatology, que son:
• Presencia de dolor crónico generalizado músculo-esquelético durantemás de tres meses en cada uno de los cuatro cuadrantes del cuerpo(dolor generalizado significa dolores arriba y debajo de la cintura y enambos lados del cuerpo, además de en el esqueleto axial).
• Existencia de puntos de dolor exagerado a la palpación o a la presiónde unos 4 kg, denominados puntos gatillo (trigger points o tenderpoints en la literatura anglosajona). Se requiere la presencia de doloren 11 o más de los 18 puntos hipersensibles (Ver Imagen 39). Paraque un punto doloroso sea considerado como positivo, el paciente ma-nifestará que la palpación es dolorosa. La simple molestia no se con-sidera dolor.
Diagnóstico diferencial
La fibromialgia tiene que diferenciarse de cuadros generalmente sistémicosque cursan con afectación inflamatoria articular o muscular, enfermedadesendocrinas, neoplásicas y puramente psiquiátricas (Ver Tabla 16).
Imagen 38. Círculos viciosos
Síntomas de la fibromialgia:
• Dolor
• Cansancio
• Rigidez
• Otros: cefalea, colon irritable, etc.
• Dificultades con las actividades de la vida diaria:
– Familia
– Trabajo
– Ocio
• Pensamientos negativos y preocupaciones
• Problemas de:
– Sueño
– Memoria
– Relaciones sexuales
• Otros problemas
TENSIÓN
ANSIEDAD
DEPRESIÓN
Tabla 16. Cuadros con los que se puede confundir lafibromialgia
Enfermedades dolorosas locales
Enfermedades reumáticas
Enfermedades endocrino-metabólicas
Enfermedades infecciosas
Enfermedades psiquiátricas
Otras enfermedades
Síndrome de dolor miofascialSíndrome de disfunción de laarticulación temporomandibular
Otros reumatismos de tejido blandoLupus eritematoso sistémico (LES)Síndrome de fatiga crónica (SFC)(1)
Artropatías inflamatorias Artritis reumatoidePolimiositis y dermatomiositis Polimialgia reumáticaOsteoporosis OsteomalaciaSíndrome de Sjögren
Hipo e hipertiroidismoHipo e hiperparatiroidismoInsuficiencia suprarrenalMiopatías metabólicas
Infección por virus de Epstein-Barr VIHOtras viriasis
NeurosisDepresión mayor Reumatismo psicógeno(2)
Neoplasias Enfermedad de Parkinson en fasediscinética
(1) Síndrome de fatiga crónica (SFC): cuadro caracterizado por debilidad severa y que es losuficientemente grave como para obligar al paciente a reducir su actividad diaria en un 50%o más. Se ha investigado su relación con infecciones virales sin llegar a conclusionesdefinitivas. Con relativa frecuencia aparece como proceso entremezclado con la fibromialgia
(2) Reumatismo psicógeno: cuadro doloroso sin causa orgánica. A veces se confunde con lafibromialgia
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 744
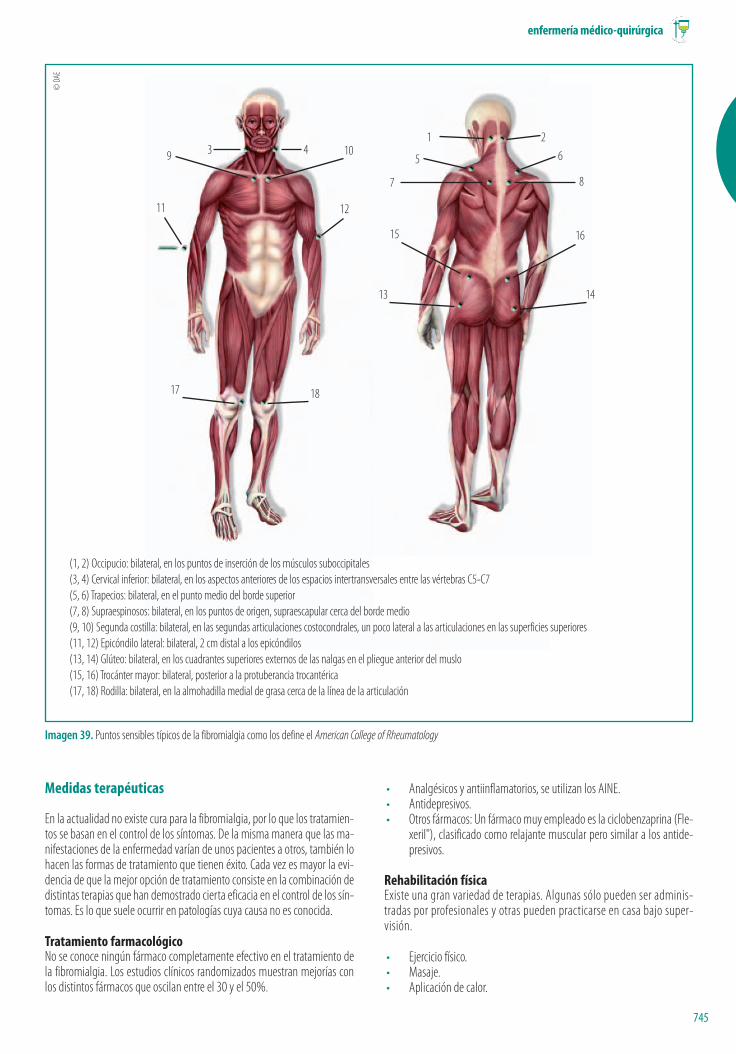
745
enfermería médico-quirúrgica
Medidas terapéuticas
En la actualidad no existe cura para la fibromialgia, por lo que los tratamien-tos se basan en el control de los síntomas. De la misma manera que las ma-nifestaciones de la enfermedad varían de unos pacientes a otros, también lohacen las formas de tratamiento que tienen éxito. Cada vez es mayor la evi-dencia de que la mejor opción de tratamiento consiste en la combinación dedistintas terapias que han demostrado cierta eficacia en el control de los sín-tomas. Es lo que suele ocurrir en patologías cuya causa no es conocida.
Tratamiento farmacológicoNo se conoce ningún fármaco completamente efectivo en el tratamiento dela fibromialgia. Los estudios clínicos randomizados muestran mejorías conlos distintos fármacos que oscilan entre el 30 y el 50%.
• Analgésicos y antiinflamatorios, se utilizan los AINE.• Antidepresivos.• Otros fármacos: Un fármaco muy empleado es la ciclobenzaprina (Fle-
xeril®), clasificado como relajante muscular pero similar a los antide-presivos.
Rehabilitación físicaExiste una gran variedad de terapias. Algunas sólo pueden ser adminis-tradas por profesionales y otras pueden practicarse en casa bajo super-visión.
• Ejercicio físico.• Masaje.• Aplicación de calor.
Imagen 39. Puntos sensibles típicos de la fibromialgia como los define el American College of Rheumatology
© DA
E
(1, 2) Occipucio: bilateral, en los puntos de inserción de los músculos suboccipitales (3, 4) Cervical inferior: bilateral, en los aspectos anteriores de los espacios intertransversales entre las vértebras C5-C7(5, 6) Trapecios: bilateral, en el punto medio del borde superior(7, 8) Supraespinosos: bilateral, en los puntos de origen, supraescapular cerca del borde medio (9, 10) Segunda costilla: bilateral, en las segundas articulaciones costocondrales, un poco lateral a las articulaciones en las superficies superiores(11, 12) Epicóndilo lateral: bilateral, 2 cm distal a los epicóndilos(13, 14) Glúteo: bilateral, en los cuadrantes superiores externos de las nalgas en el pliegue anterior del muslo (15, 16) Trocánter mayor: bilateral, posterior a la protuberancia trocantérica (17, 18) Rodilla: bilateral, en la almohadilla medial de grasa cerca de la línea de la articulación
3 49 10
7 8
65
21
12
15 16
13
17 18
14
11
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 745

746
Manual DAE de enfermería. EIR. Oposiciones ••
Te conviene recordar...
✔ La fibromialgia es una enfermedad reumática caracterizada por dolor generalizado y por la presión en unos puntos específi-cos (puntos gatillo), fatiga y mala tolerancia al esfuerzo físico y sueño de mala calidad. Otros síntomas frecuentes son las ce-faleas, el colon irritable, dolores en la menstruación, trastornos de la circulación en manos y pies, ansiedad y depresión.
✔ Los síntomas empeoran con el cambio de tiempo, el frío, las fluctuaciones hormonales (periodo premenstrual y menopausia),ejercicio físico mayor del habitual y afectividad negativa. Es una enfermedad crónica. No es degenerativa ni supone una ame-naza para la vida. Su causa es desconocida. Se han identificado agentes desencadenantes como traumatismos, infección viralo bacteriana, estrés psicológico u otra enfermedad (artritis reumatoide, lupus, etc.).
✔ Los estudios de investigación implican la alteración del sistema nervioso central, detectándose niveles bajos de sustancias im-portantes en la regulación del dolor (serotonina) y niveles elevados de sustancias productoras de dolor (sustancia P). Otras po-sibles causas hacen referencia a alteraciones genéticas, endocrinas, del sistema inmune, de la fisiología del sueño y trastornospsicológicos.
✔ El diagnóstico de la enfermedad se lleva a cabo utilizando los criterios determinados por el American College of Rheumatology. ✔ El tratamiento va dirigido al control de los síntomas, ya que no existe cura. Últimamente se están llevando a cabo tratamien-
tos de tipo interdisciplinar, que engloban el tratamiento médico-farmacológico, la terapia física, psicológica y ocupacional. Lacombinación de ejercicio físico no fatigante, el masaje y la aplicación de calor, la psicoterapia menor y la administración de anal-gésicos y de pequeñas dosis de antidepresivos para mejorar la calidad del sueño y disminuir la percepción del dolor resumenel tratamiento actual.
• Arboleya Rodríguez L (coord.). Guía práctica de enfermedades del metabolismo óseo y mineral. Madrid: Adalia Farma; 2006. • Barrio Guirado MA, Feijoo Cid M, Finestres Capdevila A, Gómez Felices E. Diseño de un programa de educación sanitaria para la prevención de lumbalgias y cervicalgias.
Metas Enferm 2000; 3(29):16-20.• De la Fuente Ramos, M (coord.). Enfermería Médico-quirúrgica. 2ª ed. Madrid: DAE; 2009.• De la Torre Aboki J, Hill J. Desarrollo y momento actual de la enfermería reumatológica. Metas Enferm 2008; 11(5):20-24.• Farreras P, Rozman C. Medicina interna. 16ª ed. Barcelona: Masson; 2008.• González Viejo MA, Cohí O, Salinas Castro F. Escoliosis: realidad tridimensional. Barcelona: Masson; 2001.• Greenspan A, Remagen W. Tumores de huesos y articulaciones. Madrid: Marbán; 2001.• Guañabens N. Enfermedad ósea de Paget. Madrid: Novartis; 2006.• Hakim A. Manual Oxford de reumatología. 2ª ed. Madrid: Aula Médica; 2008.• León Cabezas MJ. La consulta de enfermería en reumatología. Educare21 2008; 47.• Long BC, Phipps WJ, Cassmeyer VL. Enfermería médico-quirúrgica. 3ª ed. Madrid: Harcourt Brace; 2004.• Martínez Abril C. Enfermería en cirugía ortopédica y traumatológica. Madrid: Elsevier; 2003.• Paulino Tevar J, Mulero Mendoza J, Peña Arrebola A. Guía de información de fibromialgia. Asociación de Fibromialgia de la Comunidad de Madrid (AFIBROM). [En línea]
[fecha de acceso: 22 de junio de 2009] URL disponible en: http://www.medicinainformacion.com/fibromialgia_libros.htm• Sterling GW, Gene VB. Tratamientos en reumatología. Madrid: Marbán; 2003.• Swearingen PL. Manual de enfermería médico-quirúrgica. 6ª ed. Madrid: Harcourt Brace; 2008.• Swiontkowski MF. Manual de ortopedia y traumatología. Barcelona: Masson; 2004.
BIBLIOGRAFÍA
Médico-quirúrgica II-caps7-9:maquetaEIR.qxp 22/07/09 12:32 Página 746


















