
Metodolog ía Sint ética Aplicada a la S íntesis de F … · 6.1. Modo de acción del nilotinib...
96
Metodolog Metodologí a Sint a Sinté tica Aplicada tica Aplicada a la S a la Sí ntesis de F ntesis de Fá rmacos rmacos Miguel Carda Miguel Carda Má ster en Qu ster en Quí mica Aplicada y Farmacol mica Aplicada y Farmacológica gica Universidad Jaume I Universidad Jaume I NH 2 HOOC MeO HN N N N N Ph3C (PriO)2B Br Br Síntesis de losartan
Transcript of Metodolog ía Sint ética Aplicada a la S íntesis de F … · 6.1. Modo de acción del nilotinib...

MetodologMetodolog íía Sinta Sint éética Aplicada tica Aplicada a la Sa la Sííntesis de Fntesis de F áármacosrmacos
Miguel CardaMiguel CardaMMááster en Quster en Qu íímica Aplicada y Farmacolmica Aplicada y Farmacol óógicagica
Universidad Jaume IUniversidad Jaume I
NH2
HOOCMeO
HN
N
NN
NPh3C
(PriO)2B
Br
Br
Síntesis de losartan

TEMA 8
Inhibidores de quinasas como agentes anticáncer
Máster en Química Aplicada y Farmacológica
Universidad Jaume I


Índice
1. Introducción
2. Quinasas como enzimas clave en el proceso de señalización celular
2.1. Estructura de las quinasas
2.2. Colocación del ATP en el bolsillo de unión de la quinasa
2.3. Mecanismo de fosforilación
2.4. Unión de la quinasa al sustrato diana
2.5. Estados activado e inactivado
3. Tipos de quinasas
3.1. Quinasas RTKs
3.2. Transducción de señal por activación de RTKs: vía RAS-RAF
3.3. Transducción de señal por activación de RTKs: vía JAK-STAT
4. Quinasas como dianas moleculares en el tratamiento del cáncer
4.1. Desregulación de la actividad quinasa
4.2. Inhibidores de quinasas
4.2.1. Fármacos inhibidores de quinasas hiperactivas
4.2.2. Fármacos inhibidores de Receptores Tirosina-Quinasa
4.3. Tipos de fármacos inhibidores de quinasas
5. Imatinib
5.1. Translocación cromosómica y formación de la quinasa híbrida bcr-abl
5.2. Quinasa abl
5.3. Modo de acción del imatinib
5.4. Unión del imatinib al centro activo de la quinasa abl
5.5. Síntesis de imatinib
5.5.a.1. Análisis retrosintético de imatinib
5.5.b.1. Síntesis de imatinib
5.5.c.1. Cuestiones
5.5.b.2. Síntesis alternativas de imatinib
5.5.c.2. Cuestiones
5.6. Resistencia a imatinib
6. Nilotinib
6.1. Modo de acción del nilotinib
6.2. Síntesis de nilotinib
6.2.a.1. Análisis retrosintético de nilotinib
6.2.b.1. Síntesis de nilotinib 6.2.c.1. Cuestiones 6.2.a.2. Análisis retrosintético alternativo de nilotinib
6.2.b.2. Síntesis
7. Dasatinib
7.1. Unión del dasatinib a la quinasa abl
7.2. Síntesis de dasatinib
7.2.a. Análisis retrosintético de dasatinib
7.2.b. Síntesis de dasatinib

7.2.c. Cuestiones
8. Sorafenib
8.1. Síntesis de sorafenib
8.1.a. Análisis retrosintético de sorafenib
8.1.b. Síntesis
8.1.c. Cuestiones
9. Gefitinib
9.1. Síntesis de gefitinib
9.1.a. Análisis retrosintético de gefitinib
9.1.b. Síntesis
9.1.c. Cuestiones
10. Erlotinib
10.1. Síntesis de erlotinib
10.1.c. Análisis
10.1.b. Síntesis
10.1.c. Cuestiones
11. Sunitinib
11.1. Síntesis de sunitinib
11.1.a. Análisis retrosintético de sunitinib
11.1.b. Síntesis
11.1.c. Cuestiones
12. Lapatinib
12.1. Síntesis de lapatinib
12.1.a. Análisis retrosintético de lapatinib
12.1.b. Síntesis
13. Pazopanib
13.1. Angiogénesis
13.2. Estructura del VEGF
13.3 Tipos de VEGF
13.4. Receptores del VEGF 13.4.1. Receptor VEGF-1
13.4.2. Receptor VEGF-2
13.4.3. Receptor VEGF-3
13.5. Uniones VEGF-VEGFR
13.6. Desarrollo del pazopanib
13.7. Síntesis del pazopanib
13.7.a. Análisis retrosintético del pazopanib
13.7.b. Síntesis
13.8. Otros compuestos antiangiogénicos

Inhibidores de quinasas 1
Inhibidores de quinasas como compuestos anticáncer
1. Introducción
La gran mayoría de fármacos antitumorales centran su acción en todas las células de crecimiento rápido, incluyendo las normales, de ahí los graves efectos secundarios de la quimioterapia. Desde hace algunos años se han empezado a aplicar nuevas terapias anticáncer denominadas terapias dirigidas. En estas terapias se emplean fármacos que bloquean el crecimiento y la diseminación del cáncer interfiriendo con biomoléculas implicadas en los procesos de señalización celular, consiguiendo de esta manera que las células cancerosas
detengan su crecimiento y su división incontrolada.1
2. Quinasas como enzimas clave en el proceso de señalización celular
Las quinasas son enzimas clave en el proceso de señalización celular encargándose de la transferencia de grupos fosfatos desde ATP a un sustrato diana. Este proceso es, desde el punto de vista químico, una fosforilación, por lo que a las quinasas también se las denomina como ATP(x)fosfotransferasas, donde (x)= molécula a la que se transfiere el grupo fosfato (véase el esquema 1). El proyecto del genoma humano ha permitido la identificación de 520 quinasas que constituyen lo que se denomina como el «quinoma» humano.2
Esquema 1
La fosforilación de las proteínas implica la adición de un grupo fosfato a un aminoácido de la cadena peptídica y es un proceso clave en las vías de señalización celular, ya que cada grupo fosfato lleva consigo dos cargas negativas, y la fosforilación de un aminoácido de la cadena peptídica provoca un cambio conformacional en la proteína que es fosforilada. El cambio conformacional en la proteína fosforilada permite un control regulatorio sobre la actividad de ésta.
La fosforilación por las quinasas es un proceso reversible. Las proteínas pueden ser desfosforiladas por enzimas denominadas proteína-fosfatasas. Estos dos grupos de enzimas, las quinasas y las fosfatasas, a menudo trabajan juntas para apagar y encender las señales celulares.
Las quinasas juegan un papel muy importante en varios procesos de señalización intracelular, incluyendo aquellos que controlan el crecimiento y la división celular.
2.1. Estructura de las quinasas
Las quinasas unen covalentemente un grupo fosfato, a partir de un ATP donador, a residuos de serina, treonina y/o tirosina de otras proteínas o sustratos. La reacción de fosfotransferencia requiere la presencia de tres sitios específicos:
a) Un sitio de unión de ATP.
1 http://www.cancer.gov/espanol/recursos/hojas-informativas/tratamiento/terapias-dirigidas. 2 Manning, G.; Whyte, D. B.; Martinez, R.; Hunter, T.; Sudarsanam, S. Science. 2002, 298, 1912-1934.

Inhibidores de quinasas 2
b) Un dominio que cataliza la transferencia de un grupo fosfato del ATP.
c) Un sitio de unión del sustrato.
Las quinasas constan de una estructura bilobular con un lóbulo N-terminal y uno C-terminal.
El lóbulo N-terminal, denominado dominio regulatorio, está constituido principalmente de láminas β y de una hélice α (hélice αC) que es parte del sistema de activación. El lóbulo N-terminal también contiene un bucle altamente flexible rico en glicina que cubre y ancla los fosfatos del ATP no transferibles (véase la figura 1).
El lóbulo C-terminal está constituido fundamentalmente de hélices α y se une al lóbulo N-terminal mediante un bucle denominado bisagra. En el lóbulo C-terminal se encuentra el bucle de activación. El sitio de unión del ATP está situado en el interfaz entre los dos lóbulos.
Figura 1. Estructura de una quinasa unida a ATP
En la figura 2 se muestra el complejo formado por una tirosina-quinasa receptora de insulina unida a ATP.3 En la parte a de la figura 2 se aprecia que el bolsillo de unión de ATP a la quinasa reside en un profundo surco colocado entre el lóbulo N-terminal y el lóbulo C-terminal.
En la parte b de la figura 2 se muestra con más detalle el centro activo del enzima. Los enlaces de hidrógeno que mantienen unido al ATP al bolsillo están indicados con líneas de trazos en color amarillo y son los que se establecen entre N1 y N6 de la parte de adenina con aminoácidos del dominio bisagra. El fosfato γ del ATP está colocado hacia el exterior y muy cerca del hidroxilo fenólico de un residuo de tirosina. Obsérvese también la presencia de dos cationes Mg2+ en el centro activo de la enzima. Estos cationes juegan un papel clave en el proceso de fosforilación.
3 Dar, A. C.; Shoken, K. M. Annu. Rev. Biochem. 2011, 80, 769-795.

Inhibidores de quinasas 3
Bisagra
Figura 2. ATP unido a una tirosina-quinasa
Todas las quinasas contienen 12 motivos o sub-dominios altamente conservados, y 12 residuos invariantes. En la figura 3 se representa esquemáticamente la estructura de una quinasa con indicación de los diferentes subdominios.
Especificidad
del sustrato
D5
Bucle Gly
Segm
entode
activación
Figura 3. Sub-dominios de una quinasa
D1 representa el sub-dominio de glicina que actúa como tapadera flexible coordinándose con los grupos fosfato no hidrolizables del ATP.
D2 y D3 indican los sub-dominios que contienen lisina y glutámico y forman un puente salino Glu-Lys orientado hacia los fosfatos α y β del ATP.

Inhibidores de quinasas 4
D5 indica el bucle bisagra, que une la región N-terminal con la C-terminal. El bucle bisagra se coordina con la parte de adenina del ATP.
D6 indica una región invariante de aspártico. Esta parte es la zona que aporta la funcionalidad básica encargada de la abstracción de un protón del grupo hidroxilo que va a ser fosforilado.
D7 marca una región invariante de aspártico denominada DFG, que contiene la secuencia Asp-Phe-Gly. En esta zona se coordina el catión Mg2+ que se unirá y orientará a los fosfatos β y γ del ATP.
D7.5 indica el lóbulo de activación, que abarca las regiones DFG y APE.
D8 marca la hélice αEF (región APE), que determina la especificidad de la quinasa hacia la fosforilación de serina/treonina o hacia la fosforilación de tirosina.
Los elementos estructurales que son clave en la actividad catalítica de la quinasa se indican en la figura 4 y son:
a) El bucle rico en glicina, que se ubica encima del ATP (en azul en la figura 4).
b) La hélice αC (en magenta en la figura 4).
c) La bisagra, que se une a la parte de adenosina del ATP formando un puente de hidrógeno bidentado.
d) El bucle de activación (en azul oscuro), que contiene una secuencia muy conservada de DFG y termina con APE.
…GxGxxG…
Helix-C
DFG……APE loopHinge ATP
…GxGxxG…
Helix-C
DFG……APE loopHinge ATP
Figura 4. Elementos clave en la actividad catalítica de la quinasa
En la figura 5 se esquematizan las regiones del bolsillo de unión de ATP y la colocación de esta molécula en dicho bolsillo. Como se verá más adelante, el bolsillo de unión de ATP es la diana farmacológica de la gran mayoría de los fármacos antitumorales inhibidores de quinasas. En este bolsillo se distinguen la siguientes zonas:
1) La zona hidrofóbica de unión de la parte de adenina. En esta zona los N-1 y N-6 del anillo de adenina de la molécula de ATP se coordinan mediante enlaces de hidrógeno con aminoácidos del dominio bisagra.
2) La zona hidrofóbica de unión de la parte de ribosa.

Inhibidores de quinasas 5
3) La zona hidrofílica de unión de la parte de fosfato. Esta región parece ser la menos importante en términos de unión de inhibidores debido a su elevada exposición al disolvente. El grupo trifosfato se encuentra constreñido por el bucle de glicina y está colocado en una región de aminoácidos básicos, los cuales, junto con el aspartato del dominio D6, que desprotona al OH fosfoaceptor, están implicados en el proceso catalítico.4
4) El bolsillo hidrofóbico o bolsillo de selectividad, denominado bolsillo adyacente en la figura 5. Este bolsillo no está ocupado por ATP, pero juega un papel clave en el proceso de fosforilación. Su tamaño está controlado por dos residuos de aminoácido en las posiciones 129 y 183. En esta zona se coloca el dominio DFG, cuyo residuo de aspartato es el encargado de orientar a los fosfatos β y γ del ATP mediante coordinación con un catión Mg2+.
Figura 5. Bolsillo de unión de ATP en una quinasa
En la figura 6 se representa la superficie molecular del bolsillo de unión de ATP. Se indican las regiones de unión de adenina, la zona de unión de la de ribosa, la zona de unión de fosfato y los bolsillos hidrofóbicos.
Figura 6. Representación de la superficie molecular del bolsillo de unión de ATP
4 Fabbro, D.; Ruetz, S.; Buchdunger, E.; Cowan-Jacob, S. W.; et al. Pharm. Ther. 2002, 93, 79-98.

Inhibidores de quinasas 6
2.2. Colocación del ATP en el bolsillo de unión de la quinasa
En la figura 7 se representan tres visiones consecutivas de una proteína quinasa tipo A (PKA), generadas por rotación de 90º alrededor del eje vertical. El lóbulo N-terminal es la región coloreada en gris, la región C-terminal está coloreada en ocre, la hélice αC en rojo, el bucle de activación en azul claro, la extensión N-terminal en púrpura, la extensión C-terminal en rosa, las proteínas asociadas en verde y la bisagra en azul oscuro.5
Figura 7. Representaciones de una proteína quinasa A
En la figura 8 se representa la estructura del complejo ATP-proteína quinasa A. La parte de adenina del ATP se coloca dentro del bolsillo de unión formando enlaces de hidrógeno entre los nitrógenos N1 y N6 con los aminoácidos de la zona bisagra. Los otros grupos no polares de la parte de purina establecen interacciones van der Waals con el bolsillo.
Figura 8. Complejo ATP-proteína quinasa A
5 Endicott, J. A.; Noble, M. E. M.; Johnson, L. N. Annu. Rev. Biochem. 2012, 81, 587-613.

Inhibidores de quinasas 7
Los grupos hidroxilo de la ribosa se unen mediante enlaces de hidrógeno a un residuo de glutamato de la cadena lateral y a un grupo carbonilo de E170 (glutamato-170, numeración de los aminoácidos de la quinasa A).6
El grupo trifosfato apunta hacia afuera del bolsillo a fin de poder transferir el grupo fosfato γ en la reacción de fosforilación. En el lóbulo N-terminal, y desde la hélice αC, un residuo de glutamato (E91) y una lisina (K72), colocada en la lámina β3, se coordinan con los fosfatos α y β (véase la figura 8). Los fosfatos α y γ se coordinan con un catión Mg2+ que, a su vez, se coordina con el aspartato D184 del dominio DFG y con una asparagina N171. Los fosfatos β y γ se coordinan con un segundo catión Mg2+ que, a su vez, se coordina con D184. Una tercera serie de interacciones adicionales con el bucle de glicina estabilizan aun más la conformación del ATP mediante unión a los fosfatos β y γ.
En la figura 9 se representa con más detalle la colocación del ATP en al bolsillo de unión de la quinasa. Los enlaces de hidrógeno que unen a la adenina con la zona bisagra se indican con líneas de trazos de color azul. Los enlaces de hidrógeno que une a los grupos hidroxilo de la ribosa con el bolsillo de unión se indican con líneas de trazos de color amarillo. Los cationes Mg2+ se representan mediante esferas de color púrpura. En el recuadro inferior de la figura 9 se indican las estructuras de los aminoácidos serina, treonina y tirosina. El grupo hidroxilo que contienen estos aminoácidos, que es el que experimentará el proceso de fosforilación, se remarca mediante una estrella de color purpura.
Figura 9. Colocación del ATP en el bolsillo de unión de la quinasa
En la figura 10 se representa de nuevo el ATP en el bolsillo de unión de la quinasa. En la conformación activa de la quinasa un residuo de lisina del dominio D2 forma un puente salino con el glutámico del dominio D3. Esta interacción asegura la coordinación del aminoácido aspartato (parte DFG del dominio D7) con el fosfato en γ y con el catión Mg2+.
6 Knighton, D. R.; Zheng, J. H.; Ten Eyck, L. F.; Ashford, V. A.; et al. Science 1991, 253, 407-417.

Inhibidores de quinasas 8
Figura 10. Coordinación de los fosfatos ββββ y γγγγ de ATP en el bolsillo de unión
2.3. Mecanismo de fosforilación
En la parte superior de la figura 11 se indican las interacciones de la parte de trifosfato de ATP con los aminoácidos del bolsillo de unión de una serina-treonina quinasa.
Parte de Ser o Thrque se va a fosforilar
Transferencia de fosfato
Estado detransición
ATP
ATP
ADP
Lys-72 (D2-D3)
Lys-168
Asp-184 (D7-DFG)
Asp-166 (D6)
Mg2+
Mg2+
Mg2+
Mg2+
Figura 11. Mecanismo de fosforilación de una serina/treonina quinasa
Los grupos fosfato en β y γ del ATP se coordinan con un catión Mg2+, coordinado a su vez a la parte de carboxilato de un aspartato (parte DFG del dominio D7).
Un segundo catión Mg2+ se coordina con los grupos fosfato α y γ del ATP.

Inhibidores de quinasas 9
Una lisina, Lys-72, protonada en el grupo amino, se coordina con los grupos fosfato en α y β de ATP (dominio D2 y D3).
Otra lisina, Lys-168, se coordina con el fosfato en γ, aumentando la reactividad electrofílica de esta parte del ATP. En tirosina-quinasas el residuo estabilizante de los fosfatos α y γ es arginina (véase más adelante la figura 12).
El hidroxilo nucleofílico del sustrato que se va a fosforilar se ioniza por cesión del protón al carboxilato de Asp-166 (dominio D6).
El ataque nucleofílico del anión alcóxido, que tiene lugar mediante la intervención del estado de transición indicado en la figura 11, provoca la transferencia de fosfato.
En la figura 12 se indica el mecanismo de fosforilación en la proteína kinasa A. El grupo hidroxilo del sustrato que se va a fosforilar se alinea de forma que el par solitario de electrones del oxígeno se dirige al átomo de fósforo del fosfato γ. El estado de transición es de tipo disociativo e implica un intermedio metafosfato en el cual la ruptura del enlace de P-O entre los fosfatos β y γ está muy avanzada, mientras que el enlace con el nucleófilo entrante está empezando a formarse.5
Figura 12. Mecanismo disociativo de fosforilación en la proteína quinasa A (PKA)
La carga negativa que va creciendo sobre el fosfato γ se compensa con los iones Mg2+ y sobre el residuo de lisina. A medida que la reacción progresa, la acidez del hidroxilo del sustrato se incrementa y su pKa va disminuyendo en relación con el pKa de aspartato, permitiendo de esta forma la transferencia del protón desde el hidroxilo del sustrato (pKa = 12) al aspartato (pKa =
4.5). Este protón es, probablemente, transferido al fosfato dianiónico del producto fosforilado, restableciéndose de esta forma la función carboxilato en el sitio catalítico de aspartato.5
Algunas quinasas también pueden llevar a cabo la fosforilación mediante un mecanismo asociativo, generándose en el estado de transición un fosforano pentavalente. En este mecanismo la formación del enlace entre el oxigeno del sustrato y el fosfato γ se produce simultáneamente con la ruptura del enlace entre los fosfatos β y γ, o incluso puede que la formación del enlace entre el sustato y el fosfato γ esté mas avanzada que la ruptura del enlace entre los fosfatos β y γ.7
En el esquema 2 se representa la quinasa con la letra E, el sustrato que se va a fosforilar con la letra S y el producto de la reacción con la letra P. K3 es la constante de velocidad para la etapa de fosforilación y k4 la constante de velocidad para la etapa de disociación de los productos. Se ha demostrado que la etapa catalítica es rápida (k3 ~300-500 s−1) y la etapa de disociación es lenta (k4 ~20-30 s−1), de lo que se deduce que, una vez que los sustratos se han orientado
7 Madhusudan, Akamine, P.; Xuong, N. H.; Taylor, S. S. Nat. Struct. Biol. 2002, 9, 273-77.

Inhibidores de quinasas 10
correctamente, la etapa de fosforilación es fácil y rápida y que la etapa limitante de la velocidad es la liberación de los productos (ADP y la proteína fosforilada).8
Esquema 2
La eficiencia catalítica de las quinasas es enorme puesto que la reacción catalizada por la enzima es 1014 veces más rápida que la reacción no catalizada.
2.4. Unión de la quinasa al sustrato diana
La afinidad de las quinasas por los sustratos peptídicos que van a ser fosforilados es débil, del orden de 2 × 10−4 M, por lo que es necesario utilizar concentraciones milimolares del mismo para saturar la quinasa y conseguir la cristalización del complejo. Hasta el momento sólo se han conseguido cristalizar tres complejos de la quinasa con el sustrato proteínico. Uno de ellos es el formado por la quinasa PKR (proteína quinasa dependiente de RNA) con el factor de iniciación de la translación elF2α (véase la figura 13).9
Se observa en la figura 13 que la lámina β de elF2α se acopla con la hélice G de la quinasa PKR. El sitio de fosforilación sobre elF2α está colocado en una región desordenada entre los residuos 47-59.
Figura 13. Complejo PKR/ elF2αααα
8 (a) Skamnaki, V. T.; Owen, D. J.; Noble, M. E.; Lowe, E. D.; et al. Biochemistry 1999, 38, 14718-14730. (b). Adams, J. A.; Taylor, S. S. Biochemistry 1992, 31, 8516-8522. 9 Dar, A. C.; Dever, T. E.; Sicheri, F. Cell 2005. 122, 887-900.

Inhibidores de quinasas 11
2.5. Estados activado e inactivado
Las quinasas pueden adoptar dos estados conformacionales: el estado activado o el inactivado. El estado activado tiene una conformación abierta, con el bucle de activación colocado lejos del surco catalítico.
El estado inactivado se adquiere cuando el bucle de activación se desplaza hacia la zona de unión de ATP, bloqueando así la entrada de esta biomolécula.
En las quinasas el bucle de activación, que contiene el fragmento DFG, puede adquirir diferentes conformaciones denominadas DFG-in, DFG-out y DFG-like. En la conformación inactiva la posición de la fenilalanina del dominio DFG ocupa parcialmente el bolsillo de unión del ATP (véase la figura 14). La zona catalítica está desconectada debido a la ausencia de ATP en el bolsillo de unión. El aspartato del dominio D7 tampoco se dirige hacia el bolsillo de unión, lo que impide la coordinación del magnesio que se requiere para la catálisis. En la conformación inactiva DFG-out la hélice αC mantiene su orientación hacia dentro del bolsillo.
Figura 14. Conformación inactiva de la quinasa
El cambio conformacional desde DFG-out (quinasa inactiva) a DFG-in (quinasa activa) provoca que el residuo aspartato del dominio DFG ocupe la zona del bolsillo de unión ocupada en la conformación DGF-out por el anillo de fenilo de la fenilalanina, disponiendo a la quinasa para llevar a cabo su acción catalítica (véase la figura 15).10
Figura 15. Conformación activa de la quinasa
10 Jura, N.; Zhang, X.; Endres, N. F.; Seeliger, M. A.; Schindler, T.; Kuriyan, J. Mol. Cell 2011, 48, 9-22.

Inhibidores de quinasas 12
En la figura 16 se indican con más detalle las conformaciones que adopta el bolsillo de unión de la quinasa en sus estados activado e inactivado.
Figura 16. Conformaciones activa (parte A) e inactiva (parte B) de una quinasa
En el estado activado, parte A de la figura 16, la hélice αC (en rojo en la figura) se empaqueta contra el lóbulo N-terminal y el aspartato del dominio DGF se coordina con el catión Mg2+, orientando al fosfato γ hacia el sustrato que se va a fosforilar.
En la parte B de la figura 16 se representa el estado inactivado de la quinasa abl unida a Gleevec®, un fármaco inhibidor. En este estado inactivado, la interacción del aspartato con el catión Mg2+ está interrumpida y la fenilalanina del dominio DFG apunta hacia dentro del bolsillo de unión del ATP.
Se ha propuesto un mecanismo para explicar el cambio conformacional entre el estado activado (DFG-in) e inactivado (DFG-out) de las quinasas.11 Las estructuras marcadas como A y C en la figura 17 representan las conformaciones activa e inactiva, respectivamente. Estas dos conformaciones se diferencian en la conformación del dominio DFG.
En la conformación A (estado activado) la hélice αC se encuentra próxima al bolsillo de unión de ATP, de forma que la lisina del dominio D2 y el glutamato del dominio D3 pueden formar el puente salino.
En la parte B de la figura 17 se representa la conformación de transición entre el estado activado y el inactivado. La conformación B difiere de la A y de la C en dos aspectos significativos. El primero es que la hélice αC se coloca lejos del bolsillo de unión de ATP. El segundo es que el desplazamiento hacia fuera de la hélice αC crea un bolsillo en la base del lóbulo N-terminal que es ocupado por el grupo fenilo de la fenilalanina del dominio DFG.
El movimiento del dominio DFG promueve la liberación de ADP, que es la etapa limitante de la actividad catalítica de la quinasa.12 Cuando la quinasa está unida a ATP el aspartato del dominio DFG interacciona con un catión magnesio colocado a 2.3 Å que, a su vez, se coordina con los fosfatos β y γ del ATP (véase el apartado 2.3). Este catión magnesio puede ser liberado después de la transferencia de fosfato, reduciendo la carga positiva en las inmediaciones del aspartato del DFG y provocando la protonación de este residuo. La transferencia del protón
11 Shan, Y.; Seeligerb, M. A.; Eastwood, M. P.; Frank, F.; Xua, H.; Jensen, M.; Drora, R. O.; Kuriyanb, J.; Shawa, D. E. Proc. Natl. Acad. Sci. USA 2009, 106, 139.144. 12 Para un estudio computacional sobre el mecanismo de liberación de ADP del bolsillo de unión de la quinasa véase: Lu, B.; Wong, C. F.; J. McCammon, J. A. Protein Sci. 2005; 14, 159-168.

Inhibidores de quinasas 13
puede venir del disolvente o del aspartato que ha inducido la ionización del hidroxilo de la tirosina (o serina o treonina). Esta protonación es la que provoca el giro desde la conformación DFG-in a la DFG-out y facilita la liberación del ADP.
Phe dentro del
bolsillo
Bucle de
activación
Lóbulo C
Lóbulo N
Activa (DFG in) Inactiva (DFG out)Conformaciónintermedia
Figura 17. Cambio conformacional DFG-in/DFG-out
3. Tipos de quinasas
Existe un sistema de clasificación sistemático de las enzimas basado en los números EC (en inglés Enzyme Commission numbers). Cada número EC está asociado a las reacciones catalizadas por las enzimas de manera que enzimas diferentes, que proceden, por ejemplo, de organismos diferentes pero que catalizan la misma reacción, reciben el mismo número EC.
Cada código de enzimas consiste en las dos letras EC seguidas por 4 números separados por puntos. Estos números representan una clasificación progresivamente más específica. Por ejemplo, la enzima tripéptido aminopeptidasa tiene el código EC 3.4.11.4, construido así:
3 por hidrolasa (enzima que usa el agua como reactivo en el proceso químico).
4 por hidrolasa que actúa sobre los enlaces peptídicos.
11 porque actúa sobre el terminal amino del aminoácido del péptido.
4 por que actúa sobre el amino terminal de un tripéptido.
Las quinasas se clasifican según el código EC como EC 2, ya que son transferasas que trasfieren un grupo funcional desde una sustancia a otra.
Otra clasificación menos sistemática divide a las quinasas en dos superfamilias: la familia de las denominadas quinasas convencionales (ePKs) y la familia que engloba a las denominadas quinasas atípicas (aPKs). En la figura 18 se representa de forma gráfica el árbol taxonómico del quinoma humano (quinasas convencionales).
Como se aprecia en la figura 18, las quinasas convencionales se dividen en las familias:
1.- Familia AGC: quinasas enlazadas a proteínas G.
2.- Familia CAMK: quinasas reguladas por calmodulina.
3.- Familia CMGC: quinasas dependientes de ciclinas.
4.- Familia CK1: caseína-quinasas.
5.- Familia GYC: quinasas receptoras asociadas a guanilato-ciclasa.
6.- Familia STE: compuesta de tres subfamilias que se activan entre si y que luego activan a las MAP-quinasas (del inglés Mitogen Activaten Protein Kinases).
7.- Familia TK: tirosina-quinasas.

Inhibidores de quinasas 14
8.- Familia TKL (del inglés Tyrosine Kinase-Like). En este grupo se encuadran las quinasas similares a las TK pero que carecen del motivo específico TK.
Figura 18. Arbol taxonómico del quinoma humano
Las quinasas atípicas no tienen una secuencia muy similar a la de las quinasas convencionales, pero tienen actividad enzimática de quinasa. La familia está formada por cuatro subfamilias:
1.- Familia alfa
2.- Familia PIKK (del inglés Phosphatidyl Inositol 3'Kinase-related Kinases).
3.- Familia PDHK (del inglés Pyruvate Dehydrogenase Kinases)
4.- Familia RIO

Inhibidores de quinasas 15
De las 520 quinasas humanas identificadas, 90 son tirosina-quinasas, 43 son tirosina-quinasaslike y el resto, son serina/treonina quinasas.13
Las tirosina-quinasas se dividen en dos grupos:14
a) Tirosina quinasas que funcionan como receptoras (RTKs). Estas enzimas son proteínas transmembrana que contienen un dominio extracelular al que se unen diferentes ligandos, y un dominio intracelular, con actividad tirosina-quinasa.15 Se conocen 58 RTKs humanas (denominación EC 2.7.10.1) divididas en 20 subfamilias.
b) Tirosina quinasas citoplasmáticas no-receptoras (no-RTKs). Estas enzimas son proteínas intracelulares que funcionan por debajo de las RTKs en rutas de transducción de señal. Se conocen 32 no-RTKs humanas (denominación EC 2.7.10.2) divididas en 10 subfamilias.
3.1. Quinasas RTKs
Las tirosina-quinasas receptoras (RTKs) están unidas a receptores transmembrana. Existen seis grandes clases de receptores transmembrana y son los siguientes:
1- Canales Iónicos Dependientes de Ligando.
2- Receptores Tirosina-Quinasa (RTKs).
3- Receptores Serina/Treonina-Quinasa.
4- Receptores Guanilato Ciclasa.
5- Receptores Acoplados a Proteína G (GPCR, del inglés G Protein Coupled Receptor).
6- Receptores de Citoquinas.
Los receptores de la clases 1 a 4, entre los que se encuentran las tirosina-quinasas receptoras, son moléculas bifuncionales que unen hormona y sirven a su vez como efectores al actuar como canales iónicos o enzimas. En cambio, los receptores 5 y 6 ligan hormona pero deben reclutar otra molécula para catalizar su función
En la figura 19 se indican de forma esquemática las diferentes estructuras de los receptores con actividad tirosina-quinasa (RTKs).16
Como se explicará más adelante, muchos de los fármacos anticancerígenos inhibidores de quinasas dirigen su diana terapéutica a los receptores del factor de crecimiento epidérmico, denominados con el acrónimo EGFR del inglés Epidermal Growth Factor Receptor. La familia de receptores EGFR está compuesta por cuatro miembros: EGFR (ErbB1), HER2 (ErbB2, HER2/neu), HER3 (ErbB3) y HER4 (ErbB4), que pueden formar tanto homodímeros como heterodímeros.
13 Schlessinger, J. Cell. 2000, 103, 211-225. 14 Robinson, D. R.; Wu, Y. M.; Lin, S. F. Oncogene 2000, 19, 5548-5557. 15 Hubbard, S. R.; Till, J. H. Annu. Rev. Biochem. 2000, 69, 373-398. 16 Lemmon, M. A.; Schlessinger, J. Cell 2010, 141, 1117-1124.

Inhibidores de quinasas 16
Figura 19. Familia de receptores tirosina-quinasa
En la figura 20 se indica esquemáticamente la estructura de un receptor del factor de crecimiento epidérmico (EGFR).
Figura 20. Estructura de un EGFR
Los receptores EGFR están compuestos por:
1) Un dominio extracelular, que contiene la zona de unión al ligando.
2) De un solo dominio transmembrana.

Inhibidores de quinasas 17
3) De un dominio intracelular que contiene la región con actividad tirosina-quinasa (véase la figura 20).
La región extracelular contiene cuatro dominios (I-IV).
a) Los dominios I y III tienen una longitud de, aproximadamente, 160 aminoácidos y contienen unos dominios solenoide formados por hélices β de tipo LRR (del inglés Leucine Rich Repeat). Ambos dominios se unen a los ligandos.
b) Los dominios II y IV tienen una longitud de, aproximadamente, 150 aminoácidos y son dominios ricos en cisteina.
Cuando el ligando EGF se une al receptor pone en contacto dos sitios distintos en los dominios I y III de un único receptor EGFR (véase la parte 2 de la figura 21).
Esta unión bivalente promueve un cambio conformacional en la región extracelular que desenmascara un brazo de dimerización en el dominio II. Antes de la unión del ligando, este brazo de dimerización está completamente sepultado mediante interacciones intramoleculares con el dominio IV.
La unión del ligando permite que el brazo de dimerización del dominio II interaccione con un segundo complejo ligando-receptor povocándose la dimerización de las dos unidades receptoras (véase la parte 3 de la figura 21).
Figura 21. Proceso de dimerización de EGFR
En la figura 22 se indica una representación del dímero formado por la unión de EGF a la región extracelular de EGFR. El EGF está coloreado en verde en la parte izquierda del dímero y en magenta en la parte derecha. En la parte A de la figura se representa el dímero en diagrama de cintas y en la parte B en diagrama de superficie.17
Figura 22. Dímero EGF-EGFR
17 Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J-H.; Saito, K.; Sakamoto, S.; Inoue, M.; Shirouzu, M.; Yokoyama, S. Cell 2002, 110, 775-787.

Inhibidores de quinasas 18
El proceso de dimerización del receptor se puede llevar a cabo mediante otros mecanismos, que se indican a continuación.
1) Mediante ligando dimérico. Una subunidad del ligando se une a un receptor por un lado. La otra subunidad del ligando se une al otro receptor por el lado opuesto. En total se ligan dos subunidades receptoras (véase la figura 23).
Figura 23. Dimerización del RTK mediante unión a ligando dimérico
2) Mediante ligando que contiene dos sitios de unión. El ligando posee en su estructura dos sitios de unión al receptor, cada uno de los cuales se une a una subunidad receptora (figura 24).
Figura 24. Dimerización del RTK mediante unión dual
3) Mediante unión de ligando a dímero receptor preexistente. El receptor en este caso ya está dimerizado, aún en ausencia de ligando, pero dispuesto y orientado de tal forma que no puede activarse antes de su unión con el ligando, como ocurre con el receptor de la insulina (véase la figura 25).
Tyr Tyr Tyr Tyr
Insulina
PP
Figura 25. Dímero preexistente

Inhibidores de quinasas 19
La dimerización extracelular del EGFR provoca un proceso de dimerización asimétrica en los dominios tirosina-quinasa. Así, el lóbulo N-terminal de la parte intracelular quinasa del EGFR (véase la parte A de la figura 26-1) entra en contacto con el lóbulo C-terminal de la otra quinasa (véase la parte B de la figura 26-1). En la parte 2 de la figura 26 se aprecia cómo la dimerización asimétrica de los dominios quinasa se produce cuando la quinasa receptora, colocada a la izquierda, es activada mediante interacción de su hélice αC con la hélice B de la quinasa activadora.18 Este contacto entre los dominios quinasa los activa hacia el proceso de trans-fosforilación.
Figura 26. Dímerización asimétrica de los dominios quinasa en un EGFR
Como se acaba de explicar, la dimerización extracelular del EGFR provoca la dimerización asimétrica de los dominios intracelulares del receptor, que tienen actividad tirosina-quinasa. Esta dimerización intracelular provoca la autofosforilación de zonas proteínicas del dominio citosólico. Los dominios fosforilados interaccionan con proteínas plasmáticas iniciando el proceso de transducción de la señal (véase la figura 27).19
Figura 27. Efectos de la unión del EGF al receptor EGFR
18 Kuriyan, J.; Eisenberg, D. Nature 2007, 450, 983-990. 19 Para un vídeo sobre el proceso de dimerización y transducción de la señal véase: http://www.biooncology.com/research-education/hdis/index.html.

Inhibidores de quinasas 20
En la figura 28 se indica esquemáticamente una tirosina-quinasa receptora tipo EGFR. Se aprecia la región extracelular receptora de la molécula señalizadora, la región dentro de la membrana plasmática y la región intracelular que es la que tiene actividad tirosina-quinasa. Cuando la quinasa está inactiva existe en la membrana como monómero.
Figura 28. Proceso de dimerización y señalización mediado por una RTK
En la figura 28 se representa también la dimerización del receptor tirosonina-quinasa como consecuencia de la unión de una molécula señalizadora. Después de la unión del ligando, los monómeros se asocian en dímeros y los residuos de tirosina de la región intracelular se fosforilan. Este proceso promueve la fosforilación de otras proteínas que, a continuación, inician el proceso de señalización celular.
3.2. Transducción de señal por activación de RTKs: vía RAS-RAF
La cascada de acontecimientos que tiene lugar después de la dimerización del receptor es la siguiente:
1) La dimerización activa el dominio intracelular del receptor que se autofosforila en los residuos de tirosina.
2) Los residuos de tirosina fosforilados forman sitios de unión para proteínas que contienen dominios SH2, como la fosfolipasa C o la proteína adaptadora GRB2 (del inglés Growth factor Receptor-Bound Protein 2, véase la figura 29).
3) Por un lado, la unión de la fosfolipasa C provoca su activación y la subsiguiente ruptura de PIP2 (fosfatidilinositol 3,5-bifosfato) en InsP3 (inositol 1,4,5-trifosfato) y DAG (diacilglicerol).
4) Por otro lado, la unión del GRB2 al receptor fosforilado provoca la activación de la proteína Sos (del inglés Son Of Sevenless,20 véase la figura 29).
20 Con el nombre de Son of Sevenless (SOS) se agrupan una serie de genes que codifican factores de intercambio de nucleótidos de guanina y que actúan sobre la subfamilia Ras. El nombre de sevenless se da a una mutación de Drosophila melanogaster que produce la falta de la séptima célula en su órgano visual. Los genes se denominaron de esta forma porque la proteína SOS por ellos codificada operaba corriente abajo del gen sevenless en la vía RAS/MAP.

Inhibidores de quinasas 21
5) SOS causa la activación de la proteína RAS mediante liberación en ésta de GDP y adquisición de GTP (véase la figura 29). Las proteínas RAS forman una familia de unos 50 miembros.21 Son proteínas pequeñas que unen nucleótidos de guanina y son parte del sistema de transmisión de señales relacionado con la multiplicación y desarrollo celular. A la familia de enzimas RAS también pertenecen las proteínas ARF y RAB, vinculadas a la regulación del tránsito vesicular de proteínas, las RAN, relacionadas con la importación de proteínas en el núcleo y las RHO involucradas en la organización del citoesqueleto.
Figura 29. Transducción de señal por activación de RTKs: vía RAS-RAF
6) La RAS-GTP activa a la serina/treonina quinasa RAF22 que, a su vez, fosforila y estimula a MEK23 y ésta a ERK (del inglés Extracellular signal-Regulated Kinase, véase la figura 29).
7) La cascada de sucesos desemboca en la formación de AP-1, un factor de transcripción del núcleo celular que estimula la expresión de genes implicados en el crecimiento celular (véase la figura 29).24
3.3. Transducción de señal por activación de RTKs: vía JAK-STAT
Existen más vías de señalización en las que participan también receptores tirosina-quinasa. Una de las más importantes es la cascada de señalización que se inicia con un proceso de heterodimerización provocado por la unión de un monómero tirosina-quinasa con otro, también
21 Con el monbre de Ras (del inglés Retrovirus Associated Sequences) se denomina un oncogén aislado de sarcomas murinos inducidos por virus. Aunque inicialmente los genes Ras fueron aislados en virus de sarcomas murinos altamente oncogénicos, posteriormente fueron detectados también en tumores humanos y en células normales. 22 El acrónico Raf deriva del ingés Rapidly Accelerated Fibrosarcoma. 23 La quinasa MEK (del inglés MAPK kinase/ERK kinase) es una quinasa de doble especificidad (Ser/Thr y Tyr) que activa las MAPKs del tipo ERK fosforilándolas. 24 Para una animación de la vía de transduccción mediada por RAS-RAF (MAP quinasas) véase: http://www.bio.davidson.edu/courses/Immunology/Flash/MAPK.html

Inhibidores de quinasas 22
con actividad tirosina-quinasa, denominado JAK (del inglés Janus Kinase), de los cuales existen cuatro tipos: JAK-1, JAK-2, JAK-3 y JAK-4.
La cascada de acontecimientos que tiene lugar después de la heterodimerización del receptor es la siguiente.
1) La heterodimerización provoca la fosforilación cruzada de ambos monómeros receptores (véase la figura 30).
2) Las proteínas STAT (del inglés Signal Transducers and Activators of Transcription), que contienen un dominio SH2, se unen al receptor fosforilado y se activan por fosforilación.
3) Las STAT se disocian del receptor y se dimerizan mediante intervención de sus dominios SH2.
4) Las STAT diméricas migran al núcleo y se unen al ADN y a otras proteínas reguladoras de genes activando la transcripción génica (véase la figura 30).
P
STAT-1STAT-2
Sitios de unión del ligando Ligando
Dominio
SH2
Migración al núcleo
Dimerización
Otras proteínas
reguladoras de genes
P P P P
P
P P
P
P
P
P
PP
PP
Transcripción de genes Figura 30. Transducción de señal por activación de RTKs: vía JAK-STAT
4. Quinasas como dianas moleculares en el tratamiento del cáncer
Las quinasas son capaces de fosforilar una amplia variedad de sustratos, como proteínas estructurales, enzimas metabólicas, reguladores del ciclo celular, factores de transcripción, etc. Muchos de los sustratos fosforilados por las quinasas participan en las rutas de transducción de señal regulando procesos celulares fundamentales como el metabolismo, la proliferación celular, la diferenciación celular, la supervivencia, la migración o la angiogénesis.25
La perturbación de la actividad de las quinasas resulta en una transmisión de señal anormal que produce una desregulación de los procesos celulares que puede conducir al desarrollo de fenotipos malignos.26 De hecho, las quinasas son reguladores clave en las neoplasias, haciendo
25 Pawson, T. Cell. 2004, 116, 191-203. 26 Cohen, P. Nat. Rev. Drug. Discov. 2002, 1, 309-315.

Inhibidores de quinasas 23
que la biología del cáncer y de otras enfermedades se caracterice por una situación molecular de transmisión de señal y actividad proteína quinasa aberrante.27
4.1. Desregulación de la actividad quinasa
La desregulación de la actividad proteína-quinasa puede tener lugar por:
a) Una reordenación genética, que genera una proteína híbrida que contiene el dominio catalítico de la quinasa, como en el caso de la tirosina-quinasa c-abl híbrida responsable de la leucemia mieloide crónica.28 Esta quinasa se produce por la translocación del material genético entre los cromosomas 9 y 22. Así, el gen abl situado en el cromosoma 9, que codifica una tirosina-quinasa de la familia Src,29 se inserta en el gen bcr (del inglés Breakpoint Cluster Region) del cromosoma 22 (véase la figura 31).30 El gen resultante de la translocación es el encargado de codificar a la proteina quinasa aberrante.
Figura 31. Formación del gen bcr-abl por translocación
Esta translocación génica fue descubierta en 1960 por los científicos de Filadelfia Peter Nowell, de la Escuela de Medicina de la Universidad de Pensilvania, y David Hungerford del instituto Fox Chase Cancer Center. Desde entonces se conoce al cromosoma resultante de esta translocación como cromosoma Filadelfia, por el nombre de la ciudad donde se ubican ambos centros de investigación.
El gen de fusión, denominado gen bcr-abl, contiene secuencias de la c-Abl quinasa y se expresa a niveles muy superiores a los del gen que codifica la quinasa c-Abl normal. El resultado es la expresión de una vía que estimula el ciclo de división celular e inhibe la reparación del ADN, causando la inestabilidad del genoma y siendo una causa potencial de la temida «crisis en cadena» de la leucemia mieloide crónica, patología con una elevada tasa de mortalidad.
27 Bianco, R.; Melisi, D.; Ciardiello, F.; Tortora, G. Eur. J. Cancer 2006, 42, 290-294. 28 Hantschel, O.; Superti-Furga, G. Nat. Rev. Mol. Cell. Biol. 2004, 5, 33-44. 29 Las quinasas Src son tirosina-quinasas citoplasmáticas no receptoras que están codificadas por el proto-oncogen Src (abreviatura de sarcoma). 30 El nombre de gen abl deriva de Abelson, que es la denominación que se da a una proteína producida por un virus causante de leucemias.

Inhibidores de quinasas 24
b) Disrupción de la función normal, que se produce cuando una mutación hace a una quinasa constitutivamente activa, como es el caso del oncogen c-Kit en tumores gastrointestinales.31
c) Expresión aumentada o aberrante de la quinasa o del ligando para la quinasa receptora transmembrana (RTK), como ocurre en la amplificación de la tirosina-quinasa receptora HER-2 (una variante de EGFR) en el cáncer de mama (véase la figura 32), o la expresión aberrante del factor de crecimiento vascular (VEGF) y su receptor (VEGFR), que aumentan la angiogénesis y el crecimiento tumoral.
Figura 32. Amplificación del gen Her2 y aumento de la producción de la quinasa HER-2
d) Desregulación de la actividad quinasa por activación de oncogenes o pérdida de genes supresores, lo que puede contribuir también a la tumorogénesis, como ocurre en la mutación de la GTPasa Ras.32 La proteína Ras es clave en la proliferación celular y se encuentra mutada en el 30% de los tumores sólidos. Ras se activa mediante el reclutamiento de las proteínas señalizadoras Grb2 y Sos, como consecuencia de la activación del receptor con actividad tirosina-quinasa (RTK).
Figura 33. Señalización celular mediada por Ras
31 http://www.analesranf.com/index.php/mono/article/viewFile/808/778. 32 (a) Downward, J. Nat. Rev. Cancer. 2003, 3, 11-22. (b) Altomare, D. A.; Testa, J. R. Oncogene. 2005, 24, 7455-7464.

Inhibidores de quinasas 25
En la figura 33 se esquematiza el proceso de activación de Ras. Así, la activación del RTK activa a su vez a la proteína efectora Sos que activa a Ras abriendo el bolsillo de unión a GTP, lo cual favorece la salida de GDP y la entrada de GTP. La proteína Ras participa en una vía de señalización que gobierna el proceso de división celular. Ras se inactiva con el tiempo debido a la hidrólisis del GTP por la actividad GTPasa intrínseca de Ras. Las mutaciones que afectan a la actividad GTPasa de Ras producen proteína Ras constitutivamente activada y las células que la expresan proliferan sin control dando lugar a un cáncer.
4.2. Inhibidores de quinasas
Las estrategias convencionales en el tratamiento del cáncer se basan en una combinación de cirugía, radioterapia y quimioterapia. Sin embargo, dado que la base molecular que subyace en muchos tipos de cáncer es una disfunción de la actividad de las quinasas, estos enzimas constituyen dianas moleculares del mayor interés en el diseño de inhibidores específicos.
Para llevar a cabo su función, cualquier quinasa debe ser capaz de unir la proteína-sustrato y el ATP, antes de que la actividad catalítica pueda transferir el grupo fosfato. De acuerdo con ello, el diseño racional de inhibidores de quinasas se basa en impedir la unión de ATP, o del sustrato, o de ambos a la quinasa, de modo que quede bloqueada la función de la enzima.
Aunque la estructura 3D se conoce solamente para menos del 10% de las quinasas humanas, la información estructural de este reducido grupo de proteínas, facilita la predicción y comparación de los sitios de unión a ATP de moléculas relacionadas con los sustratos.33
Hay que señalar, que el desarrollo de inhibidores de quinasas como agentes terapéuticos estuvo durante algún tiempo ralentizado debido a dos factores:
1) La elevada concentración intracelular de ATP (2-10 mM), que se pensaba que competiría con el inhibidor, haciendo imposible la inhibición de la quinasa.
2) El origen común de todas las quinasas, que hace que todas tengan un mecanismo catalítico similar, por lo que se consideró improbable el desarrollo de inhibidores específicos, ya que el bloqueo de la actividad de una determinada quinasa inhibiría, simultáneamente, procesos fisiológicos no relacionados, lo que podría acarrear graves efectos secundarios.34
El procedimiento más usual en la búsqueda de inhibidores de quinasas ha sido el rastreo de grandes colecciones de sustancias químicas, compuestas por cientos de miles de productos naturales o sintéticos. La composición de una librería de estos compuestos suele estar basada en:
a) La estructura del bolsillo de unión del ATP de una quinasa (librería basada en la estructura).
b) La estructura de compuestos conocidos que se unen al bolsillo de ATP de la quinasa (librería basada en el ligando).
El grado de conservación de los sitios de unión de ATP en las diferentes quinasas no es absoluto y es posible desarrollar sustancias con elevada selectividad. Además de elevada selectividad, los inhibidores de quinasas deben ser capaces de unirse con una gran afinidad. Idealmente, ésta debe ser de varios órdenes de magnitud mayor que la afinidad de la quinasa por ATP, ya que el inhibidor solo estará presente en la célula en concentraciones de rango micromolar-nanomolar.
33 Noble, M. E.; Endicott, J. A.; Johnson, L. N. Science. 2004, 303, 1800-1805. 34 Dancey, J.; Sausville, E. A. Nat Rev Drug Discov. 2003, 2, 296-313.

Inhibidores de quinasas 26
4.2.1. Fármacos inhibidores de quinasas hiperactivas
Anteriormente se ha indicado que la desregulación de la actividad quinasa puede tener lugar por una reordenación cromosómica que origina un oncogen capaz de codificar una quinasa híbrida, como la quinasa bcr-abl. Las quinasas anómalas se caracterizan por su hiperactividad, lo que provoca procesos de fosforilación descontrolada que desembocan en la formación del cáncer. La inhibición de este tipo de quinasas aberrantes es una de las nuevas dianas farmacológicas en los tratamientos anticancerígenos.
En la parte A de la figura 34 se indica esquemáticamente el modo de acción de la quinasa bcr-abl. La unión del sustrato a la quinasa provoca la fosforilación de aquél y la subsiguiente activación del efector.
En la parte B de la figura 34 se indica la unión del inhibidor (imatinib) en el bolsillo de unión de ATP. Este hecho impide la actividad fosforilante del enzima. El sustrato no fosforilado no puede unirse al efector y la vía de señalización celular se interrumpe.
Figura 34. Bloqueo de la acción quinasa con un inhibidor (imatinib)
El primer fármaco inhibidor de quinasas empleado en terapias anticancerígenas fue el imatinib, que se comercializó por Novartis en 2001. Este producto es un inibidor de tirosina-quinasas y se emplea en el tratamiento de la leucemia mieloide crónica (LMC).
Figura 35. Estructura del imatinib
El nilotinib y el dasatinib son también inhibidores de quinasas, comercializados por Novartis y Bristol-Myers-Squibbs, respectivamente. Se emplean en el tratamiento de leucemia mieloide crónica resistente a imatinib.

Inhibidores de quinasas 27
Figura 36. Estructuras del nilotinib y del dasatinib
El sorafenib es un inhibidor multiquinasa ya que es capaz de inhibir a tirosina-quinasas y a serina/treonina quinasas. Se emplea en el tratamiento del cáncer renal primario avanzado y del cáncer hepático primario (hepatocarcinoma).
Figura 37. Estructura del sorafenib
4.2.2. Fármacos inhibidores de Receptores Tirosina-Quinasa (RTKs)
Los factores de crecimiento GF (del inglés Growth Factor) son proteínas que regulan la proliferación celular, se encargan de la supervivencia celular, estimulan la migración y la diferenciación celular y controlan la apoptosis.
El Factor de Crecimiento Epidérmico o EGF (del inglés Epidermal Growth Factor) es una pequeña proteína que contiene 53 aminoácidos y tres puentes disulfuro (véase la figura 38). Los puentes disulfuro del EGF se representan en amarillo en la figura 38 y son el Cys6-Cys20, Cys14-Cys31 y el Cys33-Cys42. Las cadenas laterales de los residuos 13 (Tyr), 41 (Arg) y 47 (Leu) juegan un importante papel en la funcionalidad del EGF.
Figura 38. Estructura del Factor de Crecimiento Epidérmico (EGF)
En el apartado 3.1 se ha explicado la estructura de un receptor del factor de crecimiento epidérmico (EGFR). En la figura 39 se da otra representación de este tipo de receptores.

Inhibidores de quinasas 28
Figura 39. Representación de un receptor del factor de crecimiento epidérmico (EGFR)
El proceso de transducción de la señal se lleva a cabo mediante vías como:
a) La cascada de las MAPK quinasas (del inglés Mitogen-Activated Protein Kinase), que es activada por la proteína Ras, estimulando la síntesis y activación de factores de transcripción como FOS y JUN. Estos factores inducen la producción de nuevos factores de crecimiento, de receptores para dichos factores y de proteínas que controlan la entrada de la célula en el ciclo celular.
b) La cascada de la PI3K (fosfoinositol 3-quinasa), que activa la quinasa Akt, implicada en la proliferación celular y la supervivencia celular por inhibición de la apoptosis.
Los receptores del factor de crecimiento epidérmico (EGFR) tienen actividad tirosina-quinasa intrínseca y están sobreexpresados en las células tumorales de origen epitelial. La desregulación de los EGFR activa la proliferación, invasión y supervivencia celular. También activa la metástasis y la angiogénesis y desactiva los mecanismos de apoptosis (véase la figura 40).
Figura 40. Consecuencias de la desregulación del EGFR

Inhibidores de quinasas 29
En la figura 41 se representa esquemáticamente las consecuencias positivas del bloqueo del receptor EGFR por parte de un inhibidor de tirosina-quinasa (TKI).
Figura 41. Consecuencias de la inhibición de la actividad EGFR
Los fármacos como gefitinib, erlotinib, sunitinib, lapatinib, vandetanib y pazopanib, basan su acción anticancerígena en el bloqueo del proceso de autofosforilación de receptores tirosina-quinasa, como los receptores EGFR, VEGFR, etc.
O
MeO
N
N
N
HN
F
ClO
Gefitinib
O
O
MeO
N
N
HN
Erlotinib
MeO
NH
NH
O
O
NH
N
SunitinibCl
O
F
HN
N
N
OHN
SO
OLapatinib
Br
HN
N
N
MeO
Vandetanib
N
N N
N
N
NH
SO O
NH2
PazopanibF
Figura 42. Estructuras de inhibidores de receptores tirosina-quinasa (RTKI)

Inhibidores de quinasas 30
4.3. Tipos de fármacos inhibidores de quinasas
La actividad inhibidora de quinasas se puede conseguir mediante tres tipos de inhibidores.
a) Inhibidores tipo I. Estos inhibidores ocupan la parte de adenosina del ATP en el bolsillo de unión induciendo una conformación del residuo DGF denominada DGF-in. El residuo DGF contiene la secuencia Asp-Phe-Gly, muy conservada en todas las quinasas. Esta secuencia se encuentra localizada muy cerca del bolsillo de unión de ATP. El residuo de aspartato de DGF es muy importante para la acción catalítica de la quinasa y apunta hacia el sitio de unión de ATP en la conformación DGF-in, donde se coordina con un ión magnesio unido a ATP y activa al fosfato γ para que tenga lugar la fosforilación del sustrato receptor.
Cuando la quinasa está activa, y dispuesta a su acción fosforilante, la conformación DFG es DFG-in.
En la figura 43 se indica la coordinación de un fármaco inhibidor tipo I en el bolsillo de unión de ATP.
Figura 43. Coordinación de un inhibidor tipo I al bolsillo de unión de ATP (HCK = del inglés Hemopoietic Cell Kinase, PP1= pirazolo pirimidina tipo I)
b) Inhibidores tipo II. Estos inhibidores, al igual que los de tipo I, también ocupan la parte de adenosina del bolsillo de unión pero, al contrario que éstos, inducen una conformación del residuo DFG denominado DGF-out (véase la figura 44). Cuando la conformación es DGF-out la quinasa está inactiva y no puede fosforilar porque el aspartato del dominio DFG se aleja del fosfato γ.
Figura 44. Coordinación de un inhibidor tipo II al bolsillo de unión de ATP

Inhibidores de quinasas 31
c) Inhibidores tipo III. Estos inhibidores bloquean la actividad quinasa sin desplazar el ATP de su bolsillo de unión a la enzima (véase la figura 45).
Hélice
Tipo III
Parte de
ATPInhibidor tipo III
Bisagra
Inhibición no
competitiva
de ATPHN
F
IBr
F
F
HN
OO
OH
HO
Figura 45. Coordinación de un inhibidor tipo III a la quinasa
5. Imatinib En los últimos años de la decada de 1980, investigadores de la entonces empresa
farmacéutica Ciba-Geigy (hoy Novartis) iniciaron, bajo la dirección de N. Lydon, A. Matter y Brian J. Druker, oncólogo del Oregon Health and Sciences University e investigador del Howard Hughes Medical Institute,35 un proyecto enfocado a la identificación de compuestos con actividad inhibitoria de quinasas. Uno de los compuestos que mostraban esta clase de actividad era la 2-fenilaminopirimidina (compuesto 7.88 del esquema 3). Este compuesto mostraba baja potencia y poca especificidad en la inhibición de quinasas pero fue el punto de partida en el desarrollo de la estructura de otros inhibidores más potentes.36
Un aumento de la actividad se consiguió con la adición de un grupo 3´-piridilo en la posición 3´ del anillo pirimidínico (estructura 7.89).
La introducción de un grupo benzamido en el anillo fenílico aumentó aun más la actividad inhibidora (estructura 7.90).
Un paso clave en el desarrollo del imatinib fue observar que las sustituciones en la posición 6 del anillo fenílico disminuían la actividad inhibidora (estructuras 7.91). Por el contrario, la introducción de un grupo metilo en esta posición 6 del anillo fenílico retenía o incluso aumentaba la actividad contra tirosina-quinasas (estructura 7.91, R=Me, véase la interacción de este inhibidor tipo II con el bolsillo de unión de ATP en la figura 42).
La primera serie de compuestos con estructura 7.91 tenían poca biodisponibilidad oral y poca solubilidad en agua. La introducción de una cadena lateral altamente polar, como la de N-metilpiperazina, aumentaba la solubilidad y la biodisponibilidad oral. Al nuevo compuesto se le nombró primeramente como STI571 y después imatinib.
El imatinib inhibía la actividad de la quinasa c-abl con un IC50 entre 0.1-0.35 µM. En pacientes con leucemía linfoblástica aguda el valor de IC50 estaba en el rango 0.1-0.5 µM, lo que demostraba que el fármaco penetraba la membrana celular. Novartis comercializó el imatinib,
35 Para una entrevista al Dr. Brian J. Druker, en la que explica el desarrollo del imatinib (Gleevec®) véase: http://www.nytimes.com/2009/11/03/science/03conv.html. Véase también Druker, B. J. Nat. Med. 2009, 15, 15-28. 36 (a) Zimmermann, J.; Buchdunger, E.; Mett, H.; et al. Bioorg. Med. Chem. Lett. 1996, 6, 1221-1226. (b) Zimmermann, J.; Buchdunger, E.; Mett, H.; Meyer, T.; Lydon, N. B. Bioorg. Med. Chem. Lett. 1997, 7, 187-192.

Inhibidores de quinasas 32
en forma de sal de mesilato, con el nombre de Gleevec® en Estados Unidos y como Glivec® en Europa y Australia.
Esquema 3. Desarrollo de la estructura del imatinib como inhibidor de quinasas
5.1. Translocación cromosómica y formación de la quinasa híbrida bcr-abl
El imatinib es un inhibidor específico de la quinasa bcr-abl y ha demostrado ser muy eficaz en pacientes con leucemia mieloide crónica, tumores del estroma gastrointestinal y otros cánceres.37
La leucemia mieloide crónica (LMC) es una enfermedad maligna de la sangre y la médula ósea de carácter progresivo y lento que se presenta en la edad madura, con mayor incidencia entre los 35 y 55 años de edad. Se caracteriza por una formación descontrolada de granulocitos inmaduros de forma que el tejido sanguíneo se vuelve tan espeso, por la acumulación de estas células sanguíneas anómalas, que las células sanas, como los glóbulos rojos y las plaquetas, no pueden llevar a cabo sus funciones. El resultado es anemia, disfunción del sistema inmune, infecciones y, en última instancia, leucemia.
La leucemia mieloide crónica resulta de una anomalía cromosómica adquirida, originada por el intercambio del gen abl del cromosoma 22 con el gen bcr del cromosoma 9 (véase la figura 46). El resultado de esta translocación cromosómica es la formación de cromosoma 22
37 Deininger, M. W.; Druker, B. J. Pharmacol. Rev. 2003, 55, 401-423.

Inhibidores de quinasas 33
alterado, denominado cromosoma Filadelfia, que contiene el gen bcr-abl. Este gen codifica una proteína quinasa alterada denominada quinasa bcr-abl que posee una actividad quinasa aumentada e incontrolada, lo que provoca en las células la desregulación del ciclo celular y la inhibición de la apoptosis. Por otra parte, la quinasa bcr-abl disminuye la adhesión de las células leucémicas a las células del estroma de la médula ósea, permitiendo la salida de aquéllas al torrente sanguíneo.
Figura 46. Formación del cromosoma Filadelfia
5.2. Quinasa abl
La quinasa abl es una tirosina quinasa no receptora con un peso molecular de 145 KDa. Existen dos variantes de esta quinasa: la abl-a y la abl-b. La diferencia entre estas dos quinasas es que la abl-b ha sufrido una modificación post-translacional mientra que la abl-a no tiene esta modificación. La quinasa abl-b está miristoilada en el lóbulo N-terminal. Por razones que se desconocen, la quinasa abl-b se encuentra en mayor cantidad en las células que la abl-a.
En la estructura de la quinasa abl se distinguen tres dominios denominados SH1, SH2 y SH3 (véase la figura 47). En el dominio SH1, en el que se encuentra la región catalítica, abarca desde el aminoácido 235 al 509.
Los dominios SH2 y SH3 están implicados en procesos de regulación. Ayudan a mediar las interacciones proteína-proteína y a regular el proceso de transducción de señales.
El dominio SH3 es el que controla y regula la actividad de la quinasa. Este dominio es el que se une a la proteína Grb238 y es el que pierde su función de control en la proteína híbrida bcr-abl, lo que resulta en una actividad quinasa incrementada.
El dominio SH2 tiene una elevada afinidad por los residuos de tirosina fosforilada y se sabe que identifica una secuencia de 3 a 6 aminoácidos en el motivo peptídico de la proteína diana.
La quinasa abl-a se localiza tanto en el citoplasma como en el núcleo celular. En el citoplasma participa remodelando el citoesqueleto durante el proceso de división, diferenciación y adhesión celular. Se cree que la quinasa abl-a utiliza su dominio de unión a la
38 Feller, S. M.; Knudsen, B.; Hanafusa, H. EMBO J. 1994, 13, 2341-2351.

Inhibidores de quinasas 34
actina para acoplarse a los filamentos de esta proteína y controlar sus movimientos durante las transformaciones citoesqueletales.
Figura 47. Estructura de la quinasa abl
La quinasa abl-a que se encuentra en el núcleo participa regulando el ciclo celular, en especial en el punto de control (checkpoint) de la fase G1/S. Así, si una célula es expuesta a una radiación dañina, como la de los rayos X o la de los rayos γ, la quinasa abl se activa y fosforila a la proteína p73 en el sitio Y99. La proteína p73 pertenece a la familia de proteínas p53, que son supresoras de tumores. La proteína p73 activada actúa sobre el gen p21 que codifica la proteína p21, que inhibe la actividad de la kinasa dependiente de ciclina. El resultado de esta vía de señalización es la parada del ciclo celular en el punto de control G1/S.
La quinasa abl-a se puede transferir del núcleo al citoplasma y viceversa, dependiendo de las funciones que la célula requiera en un momento dado.
5.3. Modo de acción del imatinib
El imatinib ocupa el bolsillo ATP de la quinasa bcr-abl impidiendo la fosforilación del sustrato, que ya no puede unirse al efector. De este modo, la vía específica de transducción de señales, activada anormalmente en el proceso de transformación leucémica, resulta inactivada por el imatinib interrumpiéndose la transducción de la señal (véase la figura 48).
La quinasa abl natural también se ve inhibida por imatinib, pero, debido a la redundancia en las vías de transducción de señales, no parece que este fenómeno altere los procesos normales desde el punto de vista clínico.

Inhibidores de quinasas 35
Figura 48. Modo de acción del imatinib
5.4. Unión del imatinib al centro activo de la quinasa abl
El imatinib se une a la quinasa-abl mediante interacciones del nitrógeno piridílico con el NH de metionina-318 (Met-318, CH3SCH2CH2CH(NH2)COOH), del NH anilínico con el OH de la treonina-315 (Thr-315, CH3CH(OH)CH(NH2)COOH), del NH del carbonilo amídico con el carboxilato de ácido glutámico-286 (Glu-286, (-)OOCCH2CH2CH(NH2)COOH), del oxígeno carbonílico con el NH del ácido aspártico-381 (Asp-381, HOOCCH2CH(NH2)COOH) y del nitrógeno piperazínico con los carboxilatos de isoleucina-369 (Ile-369, CH3CH2CH(CH3)CH(NH2)COOH) y de histidina-362 (His-362, Imid-CH2CH(NH2)COOH) Además de estas interacciones, otras de tipo van der Waals también son importantes en el reconocimiento del imatinib por la quinasa (véase la figura 49).
Figura 49. Interacciones del imatinib con el sitio de unión de ATP a la quinasa
En la figura 50 se indica la colocación del imatinib en el bolsillo de ATP de la quinasa abl. El resultado de la unión del imatinib a la quinasa es el solapamiento de los anillos de piridina y pirimidina con el sitio de unión del ATP, lo que impide la actividad fosforilante de la enzima.39
39 (a) Nagar, B.; Bornmann, W. G.; Pellicena,P.; Schindler, T.; R. Veach, D. R.; Miller, W. T.; Clarkson, B.; Kuriyan, J. Cancer Res. 2002, 62, 4236-4243. (b) Druker, B. J. et al. N. Engl. J. Med. 2001, 344, 1031-1037.

Inhibidores de quinasas 36
Bisagra
Figura 50. Colocación de imatinib en el bolsillo ATP de la quinasa-abl
El imatinib (Gleevec®) es un inhibidor tipo II y se une a la conformación inactiva de la quinasa abl, que es la que presenta el dominio DFG-out. En la figura 51 se representan las moléculas de imatinib (Gleevec®) y de ATP en el bolsillo de unión de la quinasa. Cuando la conformación del bucle activador es DFG-out (color verde en la figura) el ATP no puede ocupar el bolsillo de unión porque su grupo fosfato colisionaría con el resto fenilo del dominio DFG. En esta conformación DFG-out la hélice αC se coloca lejos del bolsillo (en color verde en la figura 48), dejando el suficiente hueco para que sea ocupado por el imatinib.
Figura 51. Conformaciones DFG out/DFG in en la unión de imatinib a la quinasa-abl
Cuando la conformación del bucle DFG es DFG-in el bucle activador se desplaza hacia fuera (véase la flecha en rojo y el bucle coloreado en azul). Al mismo tiempo, la hélice αC se mueve hacia el bolsillo de unión (véase la flecha en rojo y la hélice αC coloreada en azul). En esta situación el ATP encaja perfectamente en el bolsillo y la quinasa se encuentra activada para llevar a cabo la fosforilación.

Inhibidores de quinasas 37
5.5. Síntesis imatinib
5.5.a.1. Análisis retrosintético de imatinib
El análisis retrosintético del imatinib se inicia con la escisión del enlace amídico. Esta operación genera dos fragmentos. Uno de de los fragmentos es el cloruro de ácido 7.92 y el otro es la pirimidina 7.93 (véase el esquema 4).
Esquema 4
La desconexión del anillo pirimidínico conduce a la arilguanidina 7.95 y a la piridina 7.96. Estos compuestos se prepararán a partir de la orto-toluidina 7.97 y de la 2-acetilpiridina 7.98, respectivamente.
5.5.b.1. Síntesis de imatinib40
1) Síntesis de la arilguanidina 7.95
La síntesis de la 1-(2-metil-5-nitrofenil)guanidina se inicia con la nitración de la 2-metilanilina 7.97 que se lleva a cabo con ácido nítrico en etanol (esquema 5). Esta reacción proporciona la 2-metil-5-nitroanilina 7.99 que se transforma en la guanidina 7.95 mediante reacción con la cianamida.
Esquema 5
2) Síntesis de la piridina 7.93
Para la síntesis de la piridina 7.96 se elige como compuesto de partida la 3-acetilpiridina 7.98 (esquema 6). La enolización de este compuesto con metóxido sódico, seguida de reacción de formilación con formiato de etilo, conduce a la cetoaldehídopiridina 7.100. El tratamiento de
40 (a) Zimmermann, J. EP Patente 564,409, 1993. (b) Zimmermann, J. U.S. Patente 5,521,184, 1996. (c) Zimmerman, J.; Buchdunger, E.; Mett, H.; Meyer, T.; Lydon, N. B. Biorg. Med, Chem. Lett. 1996, 11, 1221.

Inhibidores de quinasas 38
este compuesto con dimetilamina y ácido acético, en tolueno a reflujo, proporciona la piridina 7.96.
Esquema 6
3) Síntesis del cloruro de ácido 7.92
El cloruro de ácido 7.92 se obtiene mediante aminación reductiva de la N-metilpiperidina con ácido p-formilbenzoico,41 seguida de conversión del correspondiente ácido en cloruro de ácido (esquema 7).
Esquema 7
4) Construcción del anillo de pirimidina y pasos finales
Los pasos finales en la síntesis del mesilato de imatinib se inician con la condensación entre la guanidina 7.95 y la piridina 7.96 (esquema 8).
O
N
N
7.96
O2N NH
NH
NH2
7.95
+NaOH, iPrOH
reflujo, 12 h O2N NH
N
N
N7.94
7.92, piridina
luegoCH3SO3H
N
N
NH
O
NH
N
N
N
Mesilato de Imatinib
· CH3SO3H
H2, Pd/CTHF, 21 h
H2N NH
N
N
N7.93
Esquema 8
La condensación se lleva a cabo en isopropanol a reflujo en presencia de NaOH y proporciona la piperazina 7.94. La reducción del grupo nitro mediante hidrogenación molecular proporciona 7.93, que por amidación con el cloruro de ácido 7.92 forma el imatinib. Este compuesto se convierte en el mesilato por adición de 1 equivalente de ácido metanosulfónico.
41 Koroleva, E. V.; Kadutskii, A. P.; Farina, A. V.; Ignatovich, J. V.; Ermolinskaya A. L.; Gusak, K. N.; Kalinichenko, E. N. Tetrahedron Lett. 2012, 53, 5056-5058.

Inhibidores de quinasas 39
5.5.c.1. Cuestiones
1) Explique la regioselectividad observada en la reacción de nitración de la 2-metilanilina 7.99.
2) Explique mecanísticamente la formación de la pirimidina 7.94.
5.5.b.2. Síntesis alternativas de imatinib
El principal inconveniente de la síntesis anterior estriba en el uso de la tóxica cianamida. En el año 2003 se patentó un proceso de síntesis de imatinib que tenía como paso clave un acoplamiento C-N catalizado por el complejo Pd2(dba)3CHCl3 en presencia de BINAP racémico. La síntesis alternativa de imatinib se describe en el esquema 7.51.42 En esta reacción se prepara el 4-[(4-metilpiperazin-1-il)metil]benzoato de metilo por aminación reductiva entre la N-metilpiperazina y el 4-formilbenzoato de metilo 7.104. El producto de la reacción, compuesto 7.105 se convierte en la bromoamida 7.107 por reacción con 3-bromo-4-metilanilona 7.106 en presencia de AlMe3. La reacció final es el acoplamiento entre la bromoamida 7.107 y la 4-(piridin-3-il)pirimidin-2-amina 7.108 en presencia del complejo Pd2(dba)3·CHCl3, BINAP racémico y t-butóxido sódico en xileno a reflujo. Esta reacción proporciona el imatinib base con un rendimiento del 72%.
Esquema 9
42 Loiseleur, O.; Kaufmann, D.; Abel, S.; Buerger, H. M.; Meisenbach, M.; Schmitz, B.; Sedelmeier, G. W.O. Patente 03/066,613, 2003.

Inhibidores de quinasas 40
El alto coste del catalizador de paladio empleado en la síntesis anterior hace desaconsejable el empleo de esta secuencia en una aplicación industrial a gran escala. Una síntesis de imatinib que evita los inconvenientes asociados a las síntesis anteriores es la diseñada por Wang y colaboradores (véase el esquema 10). La síntesis se inicia con la formación de la enaminona 7.96 mediante calentamiento, a reflujo de xileno, de la 3-acetilpiridina 7.98 en presencia del dimetil acetal de la N,N-dimetilformamida (esquema 10).43 La condensación de 7.96 con nitrato de guanidina, en n-butanol, proporciona la pirimidina 7.108. Este compuesto se somete a acoplamiento de Ullman con 3-bromo-4-metilnitrobenceno 7.109. El acoplamiento se consigue mediante calentamiento en dioxano a 100ºC en presencia de CuI, de N,N -́dimetiletilendiamina (DMEDA) como ligando, de KI como acelerante y de K2CO3 como base. La reacción proporciona el compuesto 7.94 con un 82% de rendimiento. La reducción de este compuesto con hidrato de hidracina, en presencia de FeCl3 en metanol a reflujo, conduce al compuesto 7.93, que se convierte en la amida 7.111 por N-acilación con el cloruro de 4-(clorometil)benzoilo 7.110, en THF en presencia de trietilamina. Finalmente, la reacción de la clorometilamida 7.111 con la N-metilpiperazina 7.101 proporciona el imatinib base.
Esquema 10
43 Liu, Y-F.; Wang, C-L.; Bai, Y-J.; Han, N.; Jiao, J-P.; Qi, X-L. Org. Process Res. Dev. 2008, 12, 490-495.

Inhibidores de quinasas 41
5.5.c.2. Cuestiones
1) Explique mecanísticamente la formación del compuesto 7.96 por reacción de la 3-acetilpiridina con el dimetilacetal de la N,N-dimetilformamida.
2) Proponga un mecanismo para la reacción de Ullmann que se emplea en la preparación del compuesto 7.94.
5.6. Resistencia a imatinib
Como se ha indicado anteriormente, el nitrógeno piridílico del imatinib interacciona con el NH de la Met-318. Esta interacción es similar a la que se produce entre el enzima y el ATP. Se ha demostrado que el imatinib no compite con el ATP por la unión a la quinasa-abl, sino que se une a ésta y estabiliza su conformación inactiva, que de esta forma es incapaz de unirse al ATP.
Si el dominio de unión del imatinib a la quinasa bcr-abl sufre mutaciones se pondría en peligro la unión del fármaco a la enzima, anulando la acción inhibidora de aquél. De hecho, uno de los inconvenientes de la terapia con imatinib es la resistencia al fármaco debida a mutaciones en el gen bcr-abl que provocan cambios en la secuencia de aminoácidos de la quinasa.
T315
E255
Figura 52. Mutaciones en la quinasa abl unida a imatinib

Inhibidores de quinasas 42
En la figura 52 se indican las 10 mutaciones diferentes que se han encontrado en la quinasa abl. Las mutaciones se han encontrado en las posiciones G250, Q252, Y253, E255, V289, T315, F317, M351, H396 y F486. Las dos mutaciones más frecuentes son las que afectan al aminoácido de la posición E255, en la cual la glutamina se ha cambiado por lisina (E255K) o valina (E255V) y la que afecta al aminoácido de la posición T315, en la cual la treonina es sustituida por isoleucina.
En la figura 53 se indican las aminoácidos que sufren mutaciones y que afectan a la unión del imatinib a la quinasa-abl.
Figura 53. Mutaciones que afectan a la unión del imatinib a la quinasa-abl
Los aminoácidos que pueden ser intercambiados en la mutación son los coloreados en verde (átomos de nitrógeno en azul, de oxígeno en rojo y de azufre en amarillo). Los aminoácidos que no sufren la mutación están coloreados en naranja. La superficie molecular de la quinasa, en azul, representa las interacciones de van der Waals entre el imatinib y la proteína. Los enlaces de hidrógeno están representados con líneas amarillas.44
En la figura 54 se muestra el imatinib encajado en el bolsillo de unión de ATP de la quinasa abl. En esta figura el aminoácido Thr-315 (CH3CH(OH)CH(NH2)COOH) se ha reemplazado por isoleucina (CH3CH2CH(CH3)CH(NH2)COOH, en amarillo en la figura). Se puede apreciar que esta mutación provoca el choque estérico del residuo de isoleucina con la molécula de imatinib (en color naranja en la figura) e impide el encaje de ésta en el bolsillo de unión del ATP.
44 Gambacorti-Passerini,C. B.; Gunby, R. H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. The Lancet Oncology, 2003, 4, 75-85.

Inhibidores de quinasas 43
Figura 54. Mutación deThr-315 por Ile-315 en la quinasa abl unida a imatinib
6. Nilotinib
Las mutaciones en el gen bcr-abl provocan resistencia a imanitib y explican el diseño de una segunda generación de inhibidores de la quinasa. Uno de estos fármacos de segunda generación es el nilotinib, cuya estructura está relacionada con la del imatinib.45 El nilotinib, que se comercializa con el nombre de Tasigna®, es un potente inhibidor de la quinasa bcr-abl y se utiliza en el tratamiento de pacientes adultos con leucemia mieloide crónica positivos a cromosoma Filadelfia y con intolerancia a tratamientos previos, incluidos tratamientos con imatinib.
Figura 55. Estructura del nilotinib
El nilotinib es entre 10-30 veces más potente que el imatinib e inhibe la autofosforilación de la quinasa bcr-abl sobre la Tyr-177.46
6.1. Modo de acción del nilotinib
El nilotinib se une a la forma inactiva de la quinasa abl mediante la interacción del enlace del nitrógeno piridílico con el N-H de la Met-318 (metionina, CH3SCH2CH2CH(NH2)COOH), del NH anilínico con el O-H lateral de la Thr-315 (treonina CH3CH(OH)CH(NH2)COOH), del NH amídico con el carboxilato del Glu-286 (glutámico, (-)OOCCH2CH2CH(NH2)COOH) y del oxígeno carbonílico con el NH de Asp-381 (aspártico HOOCCH2CH(NH2)COOH, véase la figura 56). El grupo 3-metilimidazol y el grupo triflurometilo son los responsables de
45 Manley, P.; Stiefl, N.; Cowan-Jacob, S.; Kaufman, S.; Mestan, J.; Wartmann, M.; Wiesmann, M.; Woodman, R. et al. Bioorg. Med. Chem. 2010, 18, 6977–6986. 46 (a) Weisberg, E.; Manley, P. W.; Breitenstein, W.;Brueggen, J.; et al. Cancer Cell 2005, 7, 117. (b) Abbour, E., Cortes, J., Kantarjian, H. Core Evid. 2009, 4, 207-213.

Inhibidores de quinasas 44
importantes interacciones de tipo van del Waals que se establecen entre el nilotinib y la quinasa abl.47
Figura 56. Interacciones del nilotinib con la quinasa abl
En la figura 57 se pueden observar dos representaciones del nilotinib unido la quinasa abl.
Figura 57. Nilotinib unido a la quinasa abl
La unión del nolitinib a la quinasa interrumpe la cascada de señalización de las células leucémicas, lo que disminuye la proliferación celular y promueve la apoptosis.48
En la figura 58 se compara la interacción del imatinib y del nilotinib con la quinasa bcr-abl. Se puede apreciar que el imatinib se ancla a la quinasa mediante intervención de seis enlaces de hidrógeno, mientras que el nilotinib solo emplea cuatro de estos enlaces. Los menores requerimientos en el nilotinib le permiten conseguir un encaje estérico más fuerte dentro del bolsillo de unión del ATP. De esta forma el nilotinib es menos susceptible a mutaciones porque su interacción lipofílica con la superficie de la proteína permite una mayor flexibilidad a ésta.49
47 Manley, P.; Stiefl, N.; Cowan-Jacob, S.; Kaufman, S.; Mestan, J.; Wartmann, M.; Wiesmann, M.; Woodman, R. et al. Bioorg. Med. Chem. 2010, 18, 6977-6986. 48 Aichberger, K. J.; Mayerhofer, M.; Krauth, M. T.; et al. Cancer Res. 2005, 65, 9436-9444, 49 Swords, R.; Mahalingam, D.; Padmanabhan, S.; Carew, J.; Giles, F. Drug. Des. Devel. Ther. 2009, 3, 89-101

Inhibidores de quinasas 45
Figura 58. Comparación de las interacciónes del imatinib y del nilotinib con la enzima
6.2. Síntesis de nilotinib
6.2.a.1. Análisis retrosintético de nilotinib
La desconexión del enlace amida del nilotinib genera el ácido 7.112, que se sintetizará a partir del 3-amino-4-metilbenzoato de metilo 7.113, y la anilina 7.114, que se obtendrá a partir del 3-fluoro-5-(triflurometil)benzonitrilo 7.115 (véase el esquema 11).
Esquema 11
6.2.b.1. Síntesis de nilotinib
1) Síntesis del ácido 7.112
La primera síntesis del nolitinib fue publicada por Novartis en forma de patente. No se dan rendimientos para ninguna de las etapas de la secuencia sintética.50 El fragmento de ácido carboxílico 7.112 se sintetiza a partir del 3-amino-4-metilbenzoato de metilo 7.113 (véase el esquema 12).
50 Breitenstein, W.; Furet, P.; Jacob, S.; Manley, P. W. WO 2004005281, 2004; Chem. Abstr. 2004, 140, 77161.

Inhibidores de quinasas 46
Esquema 12
La reacción de 7.113 con cianamida genera la guanidina 7.116 que se convierte en la pirimidinoéster 7.117 mediante ciclación con 3-(dimetilamino)-1-(3-piridinil)prop-2-en-1-ona 7.96 (véase la síntesis del imatinib para la preparación de este compuesto). El ácido 7.112 se obtiene por saponificación del pirimidinoéster 7.117.
2) Síntesis de la anilina 7.114 y pasos finales
La síntesis de la anilina 7.113 se inicia con una reacción de sustitución nucleofílica aromática. Así, el 3-fluoro-5-(triflurometil)benzonitrilo 7.115 se trata con 4-metilimidazol en DMF a 145ºC (esquema 13). En estas condiciones se obtiene una mezcla formada por dos regioisómeros, el de N-arilación en el N-1 del 4-metilimidazol (compuesto 7.119) y el de N-arilación en N-3 del 4-metilimidazol.
Esquema 13
En la patente de Novartis no se indica la proporción de los regioisómeros formados en el proceso SNAr. Después de la separación de la mezcla de regioisoméros, el compuesto 7.119 se

Inhibidores de quinasas 47
somete a hidrólisis básica con NaOH acuosa 1 M en dioxano a 95ºC, lo que conduce al ácido 3-(4-metil-1H-imidazol-1-il)-5-(trifluoro-metil)benzoico 7.120. El calentamiento de este compuesto en t-butanol, en presencia de difenilfosforilazida, provoca una reacción de transposición de Curtius, en la cual el isocianato intermedio reacciona con el t-butanol y proporciona el carbamato 7.121. La hidrólisis acida de este compuesto conduce a la 3-(4-metol-1H-imidazol-1-il)-5-(trifluorometil)anilina 7.114.
El acoplamiento amidíco entre el ácido el ácido 7.112 y la anilina 7.114 se lleva a cabo mediante reacción de estos dos compuestos en presencia de cianofosfonato de dietilo y trietilamina, en DMF a 60ºC, lo que permite la obtención del nilotinib.
6.2.c.1. Cuestiones
1) Explique mecanísticamente la formación del compuesto 7.119 mediante reacción SNAr del 4-metilimidazol 7.118 con el 3-fluoro-5-(triflurometil)benzonitrilo 7.115. ¿Por qué no se lleva a cabo la reacción SNAr sobre la 3-fluoro-5-(triflurometil)anilina, lo que permitiría obtener directamente el compuesto 7.114?
2) Explique mecanísticamente la transposición de Curtius que se emplea en la conversión del ácido 7.120 en el carbamato 7.121.
3) Explique mecanísticamente el acoplamiento amídico entre 7.112 y 7.114.
6.2.a.2. Análisis retrosintético alternativo de nilotinib
En una síntesis reciente del nilotinib se ha llevado a cabo la síntesis de este fármaco mediante el empleo, como pasos clave, de reacciones de acoplamiento N-X (X=halógeno).51 El análisis retrosintético de esta nueva secuencia sintética se indica en el esquema 14. La retrosíntesis se inicia con la desconexión de la parte de 4-(piridin-3-il)pirimidinamino (véase el
51 Huang, W-S.; Shakespeare, W. C. Synthesis 2007, 2121-2124.x.x.207

Inhibidores de quinasas 48
esquema 14). Esta operación genera la 4-(piridin-3-il)pirimidin-2-amina 7.108 y el derivado halogenado 7.122 (X=halógeno). La escisión del enlace amídico en este último compuesto forma el ácido 3-yodo-4-metilbenzoic 7.122 y la 3-(4-metil-1H-imidazol-1-il)-5-(trifluorometil)anilina 7.123. Finalmente, la desconexión del fragmento imidazolilo conduce al 4-metilimidazol 7.118 y a la halogenoanilina 7.124.
Esquema 14
6.2.b.2. Síntesis
La síntesis alternativa del nilotinib se inicia con el acoplamiento de Ullmann entre la 3-bromo-5-(trifluorometil)anilina 7.124 y el 4-metilimidazol, que se consigue empleando CuI y 8-hidroxiquinolina en DMSO a 120ºC (esquema 15).52 En estas condiones se obtiene una mezcla formada por dos regioisómeros en relación 85:15 (el regioisómero de N-arilación en el N-1 del 4-metilimidazol (compuesto 7.123) y el de N-arilación en N-3 del 4-metilimidazol). La separación por cromatografía de columna de la mezcla de regioisómeros permite la obtención del compuesto 7.123 puro.
El compuesto 7.123 se convierte en la yodoamida 7.122 por reacción con el cloruro del ácido 3-yodo-4-metilbenzoico. El último paso en la síntesis del nilotinib es el acoplamiento entre la yodoamida 7.122 y la 4-(piridin-3-il)pirimidinamina 7.108 (para la síntesis de este compuesto véase el esquema 15). El acoplamiento se lleva a cabo en presencia de Pd2(dba)3 y de Xantphos [(9,9-dimethyl-9H-xanteno-4,5-diil)bis(difenilfosfina)] en una mezcla de dioxano/t-butanol, y proporciona el nilotinib con un 89% de rendimiento.
52 (a) Zhou, J. C.; Confalone, P. N.; Li, H.-Y.; Oh, L. M.; Rossano, L. T.; Clark, C. G.; Teleha, C. A. WO 200208199,2002; Chem. Abstr. 2002, 136, 134674. (b) Liu, L.; Frohn, M.; Xi, N.; Dominguez, C.; Hungate, R.; Reider, P. J. Org.Chem. 2005, 70, 10135.

Inhibidores de quinasas 49
Esquema 15
7. Dasatinib
El dasatinib es un inhibidor de tirosina-quinasas que se emplea como alternativa al imatinib en el tratamiento de la leucemia mieloide crónica y de la leucemia linfoblástica aguda positiva para cromosoma Filadelfia. El fármaco lo comercializa Bristol-Myers-Squibb con el nombre de Sprycel® (clorhidrato de dasatinib).
Figura 59. Estructura del dasatinib
El fármaco fue denominado dasatinib en honor al químico Jagabandhu Das, miembro del equipo de Bristol Myers Squibb que trabajó en el proceso de descubrimiento y desarrollo de este compuesto.
7.1. Unión del dasatinib a la quinasa abl
El dasatinib es 325 más potente que el imatinib es su capacidad inhibitoria de quinasas de la familia bcr-abl y src.53 La unión del dasatinib a la quinasa abl exige menos requerimientos conformacionales en el receptor que el imatinib, por lo que aquél es más potente pero menos selectivo que éste. El dasatinib es un inhibidor de quinasas tipo I ya que se une exclusivamente a la conformación activa de la quinasa abl, al contrario que la mayoría de los inhibidores de quinasas, que se unen a las conformaciones inactivas.54
La unión del dasatinib a la quinasa abl humana se indica en la figura 60.55 En la parte A de esta figura el lóbulo NH2-terminal está coloreado en azúl y el lóbulo COOH-terminal está coloreado en rosa. La cadena bisagra que conecta los dos lóbulos está coloreada en rojo en la parte A de la figura 60. El bucle coloreado en verde es el bucle catalítico y el coloreado en amarillo es el bucle de activación.
53 Manley, P.; Cowan-Jacob, S.; Mestan, J. Biochim. Biophys. Acta 2005, 754, 3-13. 54 Eck, M.; Manley, P. Curr. Opin. Cell. Biol. 2009, 21, 288-295. 55 Tokarski, J. S.; Newitt, J. A.; Chang, C. Y. J.; et al. Cancer Res. 2006, 66, 5790-5797.

Inhibidores de quinasas 50
La zona de unión a ATP está situada en una profunda hendidura entre los dos lóbulos y está situada por debajo del bucle-P, coloreado en naranja en la parte A de figura 60. El dasatinib se une al bolsillo de unión de ATP envuelto por los dos lóbulos, con la parte de amidotiazol ocupando el sitio donde usualmente se coloca el grupo adenina del ATP.
En la parte B de la figura 60 se indican dos conformaciones del dasatinib unidas a la quinasa abl. El anillo de 2-cloro-5-metilfenilo se encuentra colocado ortogonalmente a la parte de tiazolcarboxamida y dentro de un bolsillo hidrofóbico cerca de la treonina Thr-315 (CH3CH(OH)CH(NH2)COOH), zona que no ocupa el ATP.
El anillo de piperazina apunta hacia la zona bisagra de unión al ATP. Otros puntos de coordinación importantes son los puentes de hidrógeno que se establecen entre el dasatinib y el enzima. Así, el nitrógeno N-3 del anillo de aminotiazol se une mediante enlace de hidrógeno con el nitrógeno amídico de la metionina Met-318 (CH3SCH2CH2CH(NH2)COOH), el NH del dasatinib forma enlace de hidrógeno con el grupo carbonilo de Met-318 (CH3SCH2CH2CH(NH2)COOH) y el oxígeno de la cadena lateral del Thr315 (CH3CH(OH)CH(NH2)COOH) forma enlace de hidrógeno con el nitrógeno amídico del fármaco.
Figura 60. Unión del dasatinib a la quinasa abl humana
Otros contactos son de tipo van der Waals, como los que se establecen entre la parte de pirimidina, que ocupa un bolsillo hidrofóbico creado por la leucina Leu-248 ((CH3)2CHCH2CH(NH2)COOH) y la glicina Gly-321 (NH2CH2COOH), y la parte del anillo de 2-cloro-6-metilfenilo, que ocupa un bolsillo hidrofóbico formado por el grupo metilo de treonina Thr-315 (CH3CH(OH)CH(NH2)COOH), por la metionina Met-290 (CH3SCH2CH2CH(NH2)COOH), la valina Val-299 ((CH3)2CHCH(NH2)COOH), la isoleucina Ile-313 (CH3CH2CH(CH3)CH(NH2)COOH) y la alanina Ala-380 (CH3CH(NH2)COOH).
El grupo hidroxietilo terminal interacciona en una de las conformaciones con el grupo carbonilo de la tirosina Tyr-320 (4-HOC6H4CH2CH(NH2)COOH). En la otra conformación, el grupo hidroxietilo se coloca en la dirección opuesta exponiéndose al disolvente, lo que indica la alta movilidad conformacional de esta parte de la estructura.

Inhibidores de quinasas 51
En la figura 61 se indica otra visión del encaje del dasatinib en el bolsillo de unión de ATP en la quinasa abl. Los residuos de aminoácidos coloreados en verde (F317) y amarillo (T315) son posiciones que pueden experimentar mutación.
Figura 61. Colocación del dasatinib en el bolsillo de unión de ATP en la quinasa abl
El dasatinib se coloca en el bolsillo de unión de ATP lejos del bucle de activación. Por tanto, cualquier mutación que afecte a esta zona no afecta a la unión del dasatinib. En la figura 62 se representan solapadas las estructuras del imatinib (en verde) y del dasatinib (en rojo) en el bolsillo de unión de ATP. Se puede apreciar que el dasatinib se coloca más lejos del bucle de activación que el imatinib.
Figura 62. Colocación del imatinib y del dasatinib en el bolsillo de unión de ATP
En la figura 63 se comparan las uniones del imatinib, nilotinib y dasatinib a la quinasa abl. En la parte inferior de la figura 63 se observa que las posiciones del bucle P (en color rojo) y del

Inhibidores de quinasas 52
bucle A (en color magenta) varían dependiendo de si la quinasa está en su forma activa o inactiva.
Figura 63. Uniones del imatinib, nilotinib y dasatinib a la quinasa abl
El imatinib y el nilotinib son inhibidores de tipo II y bloquean a la quinasa uniéndose a la conformación inactiva (DFG-out), mientras que el dasatinib es un inhibidor de tipo I y bloquea la conformación activa de la quinasa (DFG-in).
En la conformación inactiva de la quinasa el bucle P se dobla sobre el inhibidor y el bucle de activación adopta una conformación denominada DGF-out, como se puede observar en las uniones del imatinib y del nilotinib a la quinasa representadas en la figura 63.
En la conformación activa el bucle P adopta una conformación extendida y el bucle de activación adopta la conformación DGF-in. En la unión del dasatinib a la quinasa representada en la figura 63, se puede observar que la hélice αC, coloreada en verde, cambia su conformación con respecto a la que adopta en el estado inactivo de la quinasa (DFG-out, interacciones con el imatinib y el nilotinib).56
La mutación del aminoácido treonina-315 es especialmente crítica ya que este aminoácido forma un puente de hidrógeno con los inhibidores permitiendo fijarlos en el bolsillo hidrofóbico. En la figura 64 se representan superpuestas las estructuras del imatinib (en verde) y del dasatinib (en rojo) en el bolsillo de unión de ATP. Se puede apreciar la formación de un enlace de hidrógeno de ambos compuestos con el resiudo de treonina-315.
56 Weisberg, E.; Manley, P. W.; Cowan-Jacob, S. W.; Hochhaus, A.; D. Griffin, J. D. Nature Rev. Cancer 2007, 7, 345-356.

Inhibidores de quinasas 53
Figura 64. Enlaces de hidrógeno de imatinib y dasatinib en el bolsillo de unión de ATP
La mutación de la treonina-315 por isoleucina (I-315) impide la fijación del imatinib o del dasatinib en el bolsillo de unión de ATP (véase la figura 65), comprometiéndose de manera muy acusada la eficacia de estos fármacos.
Figura 65. Colocación de imatinib y dasatinib en el bolsillo de ATP de una forma mutada (I-315 por T-315) de la quinasa abl
7.2. Síntesis de dasatinib
7.2.a. Análisis retrosintético de dasatinib
El análisis retrosintético del dasatinib se inicia con la desconexión, basada en una reacción SNAr, del fragmento de 2-(piperazin-1-il)etanol. Esta operación conduce al compuesto 7.126 y a la pirimidina 7.127 (X=halógeno, véase el esquema 16). La desconexión del fragmento de halometilpirimidinamina, basada también en una reacción SNAr, proporciona la compuesto 6-halo-2-metilpirimidin-4-amina (compuesto 7.128) y la tiazolocarboxamida 7.129 que se obtendrá mediante adición del compuesto halotiazolil-lítico 7.131 al isocianato 7.130. El compuesto organolítico se obtendrá a partir del 2-halotiazol 7.132.

Inhibidores de quinasas 54
Esquema 16
7.2.b. Síntesis de dasatinib
Para la síntesis del dasatinib se elige como material de partida el 2-clorotiazol 7.132 (véase el esquema 17).57 El tratamiento de este compuesto con metil-litio genera el compuesto organolítico 7.131 que se adiciona al isocianato 7.13058 y forma la tiazolilcarboxamida 7.129. Antes de proceder a la proyectada reacción SNAr, el compuesto 7.129 se protege en el nitrógeno amídico mediante reacción con cloruro de p-metoxibencilo (PMBCl) en presencia de NaH. El producto de la protección, compuesto 7.133, se somete a reacción SNAr con el anión resultante de la ionización de la 6-cloro-2-metilpirimidin-4-amina 7.128 con NaH. En este proceso se obtiene el compuesto 7.134 que se convierte en 7.127 mediante eliminación del grupo PMB con una mezcla de ácido tríflico y ácido trilfuoroacético en diclorometano. El calentamiento del compuesto 7.127 con 2-(piperazin-1-il)etanol 7.126, en dioxano, proporciona el dasatinib base, que por tratamiento con HCl se convierte en el correspondiente clorhidrato.
57 Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; et al. J. Med. Chem. 2006, 49, 6819-6832. 58 Este compuesto se puede preparar por reacción de 2-cloro-6-metilanilina con carbonato de bis(triclorometilo (Cl3CO)2CO en diclorometano a reflujo: Usharani, V.; Bhujanga Rao, A. K. S.; Reddy, M. P.; Dubey, P. K. Asian J. Chem. 2011, 23, 1802-1806.

Inhibidores de quinasas 55
Esquema 17
7.2.c. Cuestiones
1) ¿Por qué se protege el átomo de nitrógeno amídico en el compuesto 7.129?
2) Explique mecanísticamente la formación del compuesto 7.134 mediante reacción de 7.333 con 6-cloro-2-metilpirimidin-4-amina 7.128 en presencia de NaH. ¿Por qué se debe emplear NaH en esta reacción?
3) Explique mecanísticamente la formación del dasatinib por reacción de 7.127 con 2-(piperazin-1-il)etanol 7.126 ¿Por qué no se sustituye el cloro de la parte de clorofenilamida?

Inhibidores de quinasas 56
8. Sorafenib
El sorafenib es un inhibidor de tirosina-quinasas que se emplea en el tratamiento del cáncer renal primario avanzado (carcinoma de células renales) y del cáncer hepático primario (hepatocarcinoma).
Figura 66. Estructura del sorafenib
El sorafenib es codesarrollado y comercializado por Bayer y Onyx Pharmaceuticals con el nombre de Nexavar®. La Comisión Europea autorizó, en octubre de 2007, la comercialización del sorafenib para el tratamiento de pacientes con carcinoma hepatocelular, la forma más frecuente de cáncer de hígado. La FDA aprobó el empleo del sorafenib en noviembre de 2007, pero en Estados Unidos el fármaco solo se ha autorizado para el tratamiento del carcinoma hepatocelular no resecable.
El desarrollo del sorafenib se inició al descubrir que el compuesto 7.135 (véase el esquema 18) presentaba actividad inhibitoria de la quinasa Raf-1 (IC50 = 17 µM).59
La potencia inhibitoria se aumentó al añadir un grupo metilo en la posición para del anillo aromático (compuesto 7.136, IC50 = 1.7 µM). Una nueva mejora se consiguió al demostrar que el compuesto 7.137, con un grupo fenoxi en lugar del grupo metilo, tenía un IC50 de 1.1 µM. Cuando se cambió el grupo fenilo por un grupo piridilo la potencia inhibitoria llegó a 0.23 µM. Cuando se cambió el grupo isoxazol por un grupo 3-trifluorometil-4-clorofenilo y se introdujo un grupo carboxamida en al anillo piridínico se consiguió un compuesto con un IC50 de 0.006 µm al que se denominó sorafenib.
59 McDonald, G. B.; Chen, W.; Ellis, B.; Hoffman, C.; Overton, L.; Ronk, M.; Simth, A.; Marshalland, C. J.; Wood, E. R. Anl. Biochem. 1999, 268, 318-329.

Inhibidores de quinasas 57
Esquema 18. Desarrollo de la estructura el sorafenib como inhibidor de quinasas
Inicialmente se descubrió que el sorafenib era un inhibidor de la serina-treonina quinasa Raf-1 (IC50 = 6 nM) y B-Raf (IC50 = 22 nM).60 La quinasa Raf-1 es una MAP quinasa que interviene corriente abajo en la cascada de señalización. Una vez activada puede fosforilar otras quinasas como MEK1 y MEK2 que, a su vez, fosforilan las quinasas ERK1 y ERK2. Estas dos quinasas son efectores de la fisiología celular y juegan un importante papel en el control de la expresión génica implicada en la apoptosis, la división, la migración y la diferenciación celular.
En la figura 67 se representa esquemáticamente la cascada de señalización en la que participan las quinasas RAF.61
60 Chong, H. et al. Cell Signal 2003, 15, 463-468. 61 Para un vídeo con animación sobre la acción del sorafenib véase: http://wn.com/cancer_sorafenib_nexavar_fran%C3%A7ais

Inhibidores de quinasas 58
Figura 67. Cascada de señalización mediada por RAF
Posteriormente se demostró que el sorafenib bloqueaba muchas más quinasas, incluyendo a los receptores del factor de crecimiento vascular endotelial VEGFR-2 (IC50 = 90 nM) y VEGFR-3 (IC50 = 20 nM), al receptor del factor de crecimiento derivado de plaquetas PDGFR-B (IC50 = 57 nM), a c-Kit (IC50 = 68 nM), a FIt-3 (IC50 = 58 nM) y a RET.
El sorafenib también inhibe selectivamente otras quinasas como MEK-1, EPK-1, EGFR y al factor receptor de insulina humana. El bloqueo multi-quinasa del sorafenib disminuye el crecimiento tumoral mediante efectos antiangiogénicos, antiproliferativos y proapoptóticos (véase la figura 68).62
Figura 68. Efectos de la inhibición multi-quinasa provocada por sorafenib
62 Wilhelm, S.; Carter,C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R. A.; Schwartz, B.; Simantov, R.; Kelley, S. Nature Rev. Drug. Disc. 2006, 5, 835-844.

Inhibidores de quinasas 59
8.1. Síntesis de sorafenib
8.1.a. Análisis retrosintético de sorafenib
El análisis retrosintético del sorafenib se inicia con la escisión de la parte de urea tal y como se indica en el esquema 19. Esta desconexión genera el sintón electrofílico no natural 7.139, cuyo equivalente sintético será el isocianato 4.140, y la anilina 7.141. La desconexión del puente diaril éter, basada en una reacción SNAr, origina el 4-aminofenol 7.142 y la 4-halo-N-metilpicolinamida 7.143. Este compuesto se convierte, mediante una operación de interca,bio de grupo funcional, en el ácido 4-halopicolínico 7.143 que se sintetizará a partir del ácido picolínico 7.145.
N
OO
NH
NH
NH
O
CF3
Cl
Sorafenib
NC
O
CF3
Cl
N
O
NH2
NH
O+
7.139 7.140
SNAr
HO
NH2
+N
NH
O
X
N
HO
O
X
N
HO
O
IGF
7.141
7.1427.1437.144
O
NH
CF3
Cl
7.145
Esquema 19
8.1.b. Síntesis
La síntesis del sorafenib se inicia con el tratamiento del ácido picolínico 7.145 con el reactivo de Vilsmeier, generado in situ por reacción del cloruro de tionilo con N,N-dimetilformamida (véase el esquema 20).63 En esta reacción se obtiene directamente el cloruro del ácido 2-cloropicolínico 7.146. Este compuesto se convierte en la la 4-cloro-N-metilpicolinamida 7.143 por reacción con metilamina. La unión del 4-aminofenol 7.142 al compuesto 7.143 se lleva a cabo mediante reacción SNAr en presencia de t-butóxido de potasio en DMF. En esta reacción también se añade carbonato potásico, no indicado en el esquema 18, que permite acortar el tiempo de reacción de 16 a 6 horas y proporciona el compuesto 7.141 con un rendimiento del 87%. Finalmente, la reacción de 7.141 con el isocianato 7.140, sintetizado mediante reacción de la 4-cloro-3-(trifluorometil)anilina con carbonildiimidazol, permite la obtención del sorafenib.
63 Bankston, D.; Dumas, J.; Natero, R.; Riedl, B.; Monaham, M-K.; Sibley, R. Org. Pro.Res. Deve. 2002, 6, 777-781.

Inhibidores de quinasas 60
Esquema 20
8.1.c. Cuestiones
1) Explique mecanísticamente la formación del cloruro del ácido 2-cloropicolínico 7.146 por reacción del ácido picolínico con el reactivo de Vilsmeier.
2) Explique mecanísticamente la formación del isocianato 7.140 por reacción de la 4-cloro-3-(trifluorometil)anilina con carbonildiimidazol.
9. Gefitinib
El receptor del factor de crecimiento epidérmico (EGFR) es una tirosina quinasa de la familia erb-B implicada en la activación de la cascada de señales que controlan la proliferación celular. El gefitinib inhibe la autofosforilación del EGFR e interrumpe la vía de señalización celular mediada por este receptor. El gefitinib se comercializa por la farmacéutica AstraZeneca con el nombre de Iressa®.
O
MeO
N
N
N
HN
F
ClO
Gefitinib Figura 69. Estructura del gefitinib

Inhibidores de quinasas 61
El gefitinib es un inhibidor del receptor EGFR, que está sobreexpresado en células de determinado tipo de tumores como los de pulmón y mama. La sobreexpresión de EGFR conduce a una activación inapropiada de la cascada de señalización mediada por Ras que finalmente desemboca en proliferación celular descontrolada, inhibición de la apoptosis y activación de la metástasis y de la angiogénesis (véase la figura 70).
Figura 70. Acción del gefitinib
Se ha demostrado que la mutación en el dominio tirosina-quinasa de EGFR es responsable de la activación de las vías anti-apoptóticas.64 Estas mutaciones tienden a conferir mayor sensibilidad a los inhibidores de tirosina-quinasas como el gefitinib y el erlotinib.
En la figura 71 se representa la unión del gefitinib al domino tirosina-quinasa de un mutante de EGFR.65
Figura 71. Unión del gefitinib al receptor EGFR mutado
64 (a) Pao, W.; Miller, V.; Zakowski, M.; et al. Pro. Nat. Acad. Sci. USA 2004, 101, 13306-13311. (b) Sordella, R.; Bell, D. W.; Haber, D. A.; Settleman, J. Science 2004, 305, 1163-1167. 65 Yun, C.-H.; Boggon, T. J.; Li, Y.; Woo, S.; Greulich, H.; Meyerson, M.; Eck, M. J. Cancer Cell 2007, 11, 217-227.

Inhibidores de quinasas 62
En la figura 72 se indican los efectos que provoca la acción del gefitinib. Así, la inhibición de la actividad fosforilante del receptor EGFR bloquea las vías de señalización RAS/RAF y STAT, lo que provoca en última instancia la activación de la apoptosis y la desactivación de los procesos de metátesis, angiogénesis, invasión y proliferación celular.
Figura 72. Efectos provocados por la inhibición del EGFR mediante gefitinib
9.1. Síntesis de gefitinib 9.1.a. Análisis retrosintético de gefitinib
El análisis retrosintético se inicia con la desconexión de la cadena lateral de 4-propilmorfolina que se instalará mediante una reacción O-alquilación SN2 del compuesto 7.148 sobre un compuesto de tipo 4-(3-halopropil)morfolina 7.147 (véase el esquema 19). La desconexión de la 3-cloro-4-fluoroanilina 7.149, basada en una reacción SNAr, genera la quinazolina 7.150. Este compuesto se obtendrá de la quinazolinona 7.151 que se preparará de la 6,7-dimetoxiquinazolin-4(3H)-ona 7.152.
O
MeO
N
N
N
HN
F
ClO
Gefitinib
SN2
Cl
N
O
HO
MeO N
N
HN
F
Cl
+
H2N
F
Cl
SNAr
PO
MeO N
N
X
+IGFPO
MeO N
NH
OMeO
MeO N
NH
O
7.147 7.148
7.1497.1507.1517.152
Esquema 19

Inhibidores de quinasas 63
9.1.b. Síntesis
La síntesis del gefitinib se inicia con la O-desmetilación regioselectiva de la 6,7-dimetoxiquinazolin-4(3H)-ona 7.152 (véase el esquema 20).66
O
MeO
N
N
N
HN
F
ClO
Gefitinib
ClN
O
AcO
MeO N
N
HN
F
Cl
H2N
F
Cl
MeO
MeO N
NH
O
7.148 7.150
7.1517.152
HO
MeO N
NH
O
AcO
MeO N
NH
O
SOCl2, DMF
AcO
MeO N
N
Cl
i-PrOH, 90ºC, 5h(56% 2 pasos)
MeS
NH2
COOH
MeSO3H, reflujo 5h
Ac2O, pir.
(75% 2 pasos)
7.153
90ºC, 4h
NH4OH, H2O
K2CO3, DMF, 90ºC (55%)
HO
MeO N
N
HN
F
Cl
MeOH (95%)
7.149
Esquema 20
La desmetilación se lleva a cabo mediante calentamiento a reflujo de 7.152 en presencia de L-metionina y ácido metanosulfónico y proporciona la 6-hidroxi-7-metoxiquinazolin-4(3H)-ona 7.153. Este compuesto se somete a acetilación del hidroxilo fenólico y el producto resultante, 7.151, se trata a 90ºC con cloruro de tionilo en presencia de cantidades catalíticas de DMF. En estas condiciones se obtiene la cloroquinazolina 7.150, la cual, por calentamiento con 3-cloro-4-fluoroanilina en isopropanol, se convierte en el compuesto 7.148. La desacetilación de este compuesto seguida de reacción de O-alquilación con la 4-(3-cloropropil)morfolina proporciona el gefitinib.
9.1.c. Cuestiones 1) ¿Por qué la desmetilación del compuesto 7.152 es regioselectiva?
1) Explique mecanísticamente la formación de la cloroquinazolina 7.150
66 (a) Gibson, K. H. 1996, WO, 96/13980. (b) Barker, A. J.; Gibson, K. H.; Grundy, W.; Andrew, A.; Godfrey, A. A.; et al. Biorg. Med. Chem. Lett. 2001, 11, 1911-1914.

Inhibidores de quinasas 64
10. Erlotinib
El erlotinib (Tarceva®) inhibe la autofosforilación intracelular del EGFR tipo 1, también conocido como Her1. Se emplea en el tratamiento del cáncer de pulmón no microcítico y en el cáncer de páncreas.
O
O
MeO
N
N
HN
Erlotinib
MeO
Figura 73. Estructura del erlotinib
En la figura 74 se indica la colocación del erlotinib en el bolsillo de unión de ATP en el dominio tirosina-quinasa de EGFR-Her1. El bucle catalítico P está coloreado en azul oscuro, el bucle de activación está coloreado en verde y la hélice αC en azul claro. Las cadenas laterales G695, L834 y L837 están marcadas con esferas de color amarillo.
Figura 74. Unión del erlotinib al dominio tirosina-quinasa de EGFR
La principal interacción que mantiene unido al erlotinib con el EGFR-Her1 es de tipo hidrofóbico, mientras que las interacciones electrostáticas contribuyen solo en pequeña medida. Los enlaces de hidrógeno que se generan en la unión del erlotinib a la quinasa son los del N1 de

Inhibidores de quinasas 65
la quinazolina con el NH de Met-769 y los del Gln-767 con el HC2 de la quinazolina (véase la la figura 75).67
Figura 75. Interacciones del erlotinib con la quinasa EGFR
10.1. Síntesis de erlotinib 10.1.a. Análisis retrosintético de erlotinib
La retrosíntesis del erlotinib se inicia con la desconexión, basada en una reacción SNAr, del fragmento de 3-etinilanilina. Esta operación conduce a la quinazolina 7.154 y a la propia 3-etinilanilina 7.155 (esquema 21). La operación de intercambio de grupo funcional en la quinazolina 7.154 proporciona la quinazolinona 7.156 que se obtendrá del aminobenzoato 7.157 que a su vez se preparará del nitrobenzoato 7.158. Este compuesto derivará del benzoato 7.159 que se sintetizará del ácido 3,4-dihidroxibenzoico 7.160.
O
O
MeO
N
N
HN
Erlotinib
MeO
SNArO
O
MeO
N
N
X
MeO H2N+
IGF
O
O
MeO
N
NH
O
MeO
O
O
MeO
NH2
OR
O
MeO
IGF
O
O
MeO
NO2
OR
O
MeO
SEArO
O
MeO OR
O
MeO
HO
HO
OH
O
7.154 7.155
7.1567.157
7.158 7.159 7.160
Esquema 21
67 Choowongkomon, K.; Sawatdichaikul, O.; Songtawee, N.; Limtrakul, J. Molecules 2010, 15, 4041-4054.

Inhibidores de quinasas 66
10.1.b. Síntesis de erlotinib
La síntesis del erlotinib se inicia con el calentamienteo en DMF del ácido 3,4-dihidroxibenzoico con 1-cloro-2-metoxietano en presencia de carbonato potásico y de yoduro de tetra-n-butilamonio (esquema 22).68 La reacción provoca la O-alquilación de los hidroxilos fenólicos y la esterificación del grupo carboxilo y proporciona el éster 7.161. La saponificación de este compuesto seguida de esterificación de Fischer conduce al benzoato de etilo 7.159 que se convierte en el nitrobenzoato 7.158 mediante reacción con ácido nítrico en ácido acético. La reducción del grupo nitro a amina se consigue con formiato amónico en isopropanol y permite la obtención del aminobenzoato 7.157. Cuando este compuesto se calienta a 160ºC en formamida en presencia de formiato amónico se obtiene la quinazolinona 7.156, que se transforma en la cloroquinazolina 7.154 mediante cloración con N,N-dimetilformamida en cloroformo en presencia de dicloruro de oxalilo. Finalmente, la reacción SNAr de 7.154 con la 3-etinilanilina en agua en presencia de HCl proporciona el erlotinib en forma de clorhidrato.
Esquema 22
10.1.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 7.157.
68 Barghi, L.; Aghanejad, A.; Valizadeh,H.; Barar1,J.; Asgari1, D. Adv. Pharm. Bull. 2012, 2, 119-122.

Inhibidores de quinasas 67
2) Explique mecanísticamente la reacción de cloración de 7.156.
11. Sunitinib
El sunitinib, comercializado por Pfizer con el nombre de Sutent®, fue aprobado por la FDA en enero de 2006 para el tratamiento del carcinoma de células renales y tumores del estroma gastrointestinal (GIST) resistentes al imatinib.
NH
NH
O
O
NH
N
Sunitinib
Figura 76. Estructura del sunitinib
El sunitinib inhibe la señalización celular bloqueando diferentes receptores de tipo tirosina-quinasa entre los que se incluyen los receptores de los factores de crecimientos derivados de plaquetas (PDGFR) y los receptores del factor de crecimiento vascular endotelial (VEGFR). Estos receptores juegan un papel crucial en la proliferación celular y la angiogenesis.
El sunitinib se une al bolsillo de unión del ATP de PDGFRs y de VEGFRs, impidiendo la fosforilación del receptor. La inhibición de PDGFRs y de VEGFRs reduce la vascularización del tumor (efecto antiangiogénico) y provoca la muerte celular.
El sunitinib también inhibe KIT, un receptor de citoquinas. Las mutaciones del gen KIT provocan sobreexpresiones del receptor KIT que están asociadas con muchos tumores gastrointestinales. El sunitinib inhibe al receptor KIT mediante estabilización de su conformación inactiva (IC50 = 40nM para la conformación inactiva y 21 nM para la conformación activa). En la parte A de la figura 77 se indica la colocación del sunitinib en la forma inactiva de la quinasa KIT. En esta imagen se superponen las dos conformaciones de la quinasa, la no enlazada a sunitinib (en color amarillo) y la unida a sunitinib (en color gris). Se puede observar que las dos conformaciones son muy similares.
En la parte B de la figura 77 se indica el encaje del sunitinib en el bolsillo de unión de ATP en la quinasa. En la parte C de la figura 77 se indica el sunitinib unido a la forma mutada de la

Inhibidores de quinasas 68
quinasa (coloreada en azul oscuro) y superpuesta a la forma salvaje (no mutada) de la quinasa (en color amarillo). Se puede observar que las dos conformaciones son muy similares. 69
Figura 77. Unión del sunitinib a la forma salvaje y a la mutada de la quinasa KIT
11.1. Síntesis de sunitinib 11.1.a. Análisis retrosintético de sunitinib
La retrosíntesis del sunitinib se inicia con la desconexión del sistema carbonílico α,β-insaturado que se obtendrá mediante reacción de condensación de tipo aldólico entre la indolin-2-ona 7.172 y el pirrolcarbaldehído 7.163 (esquema 23). La desconexión de la función amídica conduce a la diamina 7.164 y al pirrol 7.165. Una operación de intercambio de grupo funcional proporciona el pirroldicarboxilato 7.166 que se sintetizará mediante ciclocondensación entre 3-oxobutanoato de alquilo 7.167 y el aminocetoéster 7.168.
NH
NH
O
O
NH
N
Sunitinib
Aldólica
NH
ONH
O
NH
N
O
H+
Amidación
NH
O
OH
O
HH2N
N+
NH
O
OR IGF
O
R´O
7.172 7.163
7.164
7.1657.1667.1677.168
O
O
OR
NH2O
R´O
O
+
Esquema 23
11.1.b. Síntesis
La síntesis del sunitinib se inicia con la condensación del 3-oxobutirato de t-butilo 7.169 con ácido nitroso. La reacción forma la hidroxi-imina 7.170 que, a continuación, se trata con 3-oxobutirato de etilo en presencia de Zn en ácido acético a 65ºC (esquema 24).70 En estas condiciones la hidroxi-imina 7.170 se reduce in situ a la amina 7.168 y se condensa con el cetoéster para formar el pirrol 7.166. Cuando el compuesto 7.166 se trata con ácido
69 Gajiwala K. S.; Wu, J. C.; Christensen, J.; Deshmukh, G. D,et al. Proc. Natl. Acad. Sci. U S A. 2009, 106,1542-1547. 70 Tang, P. C.; Miller, T.; Li, X.; Wei, C . C.; Shirazian, S.; Liang, S.; Vojkovsky, T.; Nemetalla, A. S. WO2001060814, 2001.
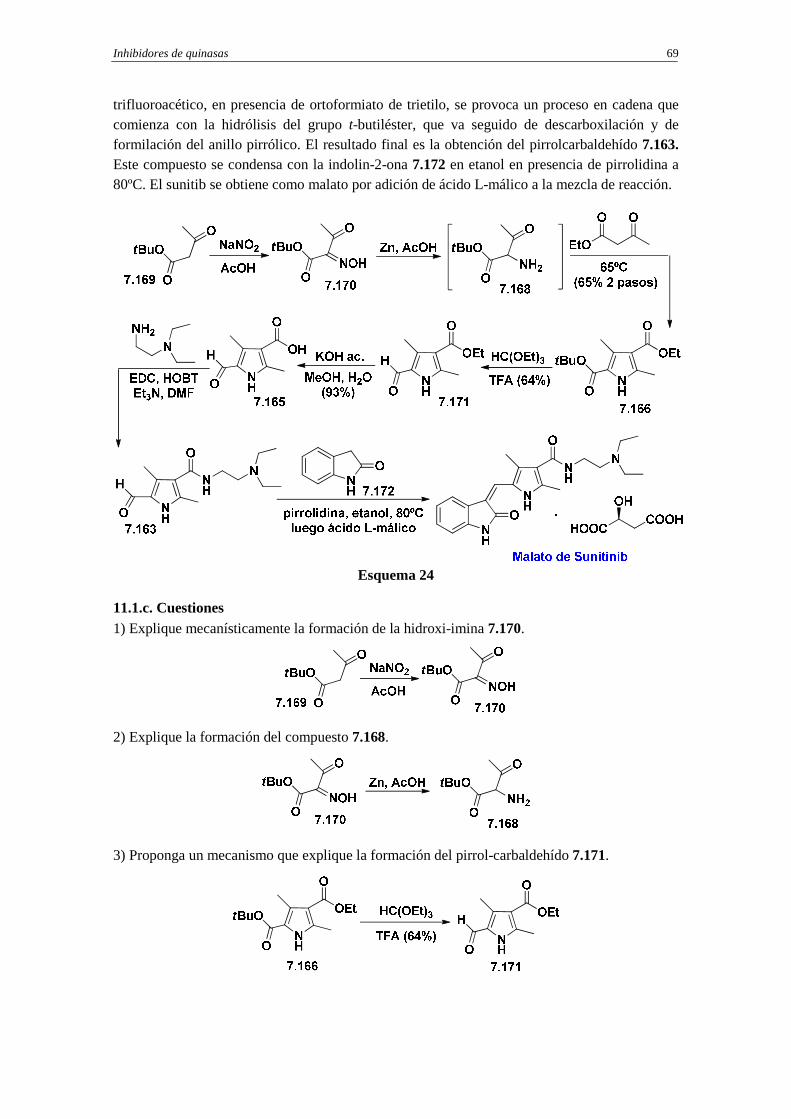
Inhibidores de quinasas 69
trifluoroacético, en presencia de ortoformiato de trietilo, se provoca un proceso en cadena que comienza con la hidrólisis del grupo t-butiléster, que va seguido de descarboxilación y de formilación del anillo pirrólico. El resultado final es la obtención del pirrolcarbaldehído 7.163. Este compuesto se condensa con la indolin-2-ona 7.172 en etanol en presencia de pirrolidina a 80ºC. El sunitib se obtiene como malato por adición de ácido L-málico a la mezcla de reacción.
Esquema 24
11.1.c. Cuestiones 1) Explique mecanísticamente la formación de la hidroxi-imina 7.170.
2) Explique la formación del compuesto 7.168.
3) Proponga un mecanismo que explique la formación del pirrol-carbaldehído 7.171.

Inhibidores de quinasas 70
12. Lapatinib
El lapatinib es comercializado por GlaxoSmithKline en forma de ditosilato bajo el nombre deTykerb/Tyverb®. El lapatinib es un inhibidor dual de tirosina-quinasas puesto que inhibe la acción de los receptores ErbB1 y ErbB2.
El lapatinib se emplea, en combinación con capecitabina, como terapia de primera línea en el tratamiento de pacientes con cáncer de seno triple-positivo (positivo para los receptores ER/EGFR/HER2).
Cl
O
F
HN
N
N
OHN
SO
O
Lapatinib
Figura 78. Estructura del lapatinib
El oncogen HER-2/neu (también conocido como erbB-2) es un gen que codifica el factor de crecimiento epidermal humano tipo 2 (HER2). Este receptor se encuentra en niveles moderados en algunas células normales y está involucrado en las respuestas celulares a los factores de crecimiento. El HER-2/neu se encuentra amplificado en hasta un 30% en muchos de los cánceres de mama. El incremento del número de copias del gen HER-2/neu lleva al aumento de la expresión de los receptores Erb2 en la superficie de la célula, lo que conlleva un aumento en la proliferación celular.
El lapatinib actúa intracelularmente bloqueando el sitio de unión de ATP, impidiendo la fosforilación subsiguiente y, en consecuencia, interrumpiendo la vía de señalización celular. La habilidad del lapatinib en la inhibición de ErbB1 y ErbB2 es única entre los inhibidores de tirosina-quinasas y contrasta con los efectos del gefitinib y del erlotinib, que únicamente bloquean el receptor ErbB1.71
La inhibición dual presenta una serie de ventajas ya que el bloqueo de dos receptores implicados en la proliferación tumoral puede tener efectos sinergísticos en la inhibición del crecimiento del tumor. Además, la inhibición simultánea de ErbB1 y ErbB2 por parte del lapatinib es más efectiva a la hora de impedir la formación de heterodímeros de ErbB1 y ErbB2.72
En la figura 79 se indican las principales interacciones del lapatinib en el bosillo de unión de ATP de la quinasa EGFR.
71 Johnston, S.; Leary, A. Drugs Today 2006, 42, 441-453. 72 Wood, E. R.; Truesdale A. T.; McDonald, O. B.; Yuan, D.; et al. Cancer Res. 2004, 64, 6652-6659.

Inhibidores de quinasas 71
Figura 79. Principales interacciones del lapatinib en el bolsillo de unión de ATP
El N1 del anillo de quinazolina se une mediante puente de hidrógeno al NH de la Met-769, mientras que el N3 del anillo de quinazolina se une mediante puente de hidrógeno mediado por agua al OH de la cadena lateral de NH de Thr-830. El anillo de quinazolina se encuentra emparedado entre las cadenas de Ala-719 y Leu-820.
El grupo 3-cloro-4-[(3-fluorobencil)oxi]anilina se orienta hacia dentro del bolsillo de ATP mediante interacciones hidrofóbicas. El grupo 3-cloro-anilina se coloca en un bolsillo formado por las cadenas laterales de Val-702, Lys-721, Leu-764, Thr-766, Thr-830 y Asp-831.
El grupo 3-fluorobenciloxi ocupa un bolsillo formado por las cadenas laterales de Met-742, Leu-753, Thr-766, Thr-830, Phe-832 y Leu-834.
El nitrógeno anilínico y el átomo de oxígeno del puente éter no están implicados en ninguna interacción directa de tipo puente de hidrógeno.
El grupo metilsulfoniletilaminometilfurilo se coloca en la superficie del interfaz con el solvente y no tiene ninguna interacción significativa con la proteína.
12.1. Síntesis de lapatinib 12.1.a. Análisis retrosintético de lapatinib
La retrosíntesis del lapatinib se inicia con la desconexión de la parte de metilsulfonil-etanamina 7.172 que se instalará mediante aminación reductiva con el aldehído 7.173 (esquema 25). La desconexión de la parte de furanocarbaldehído se basa en una reacción de acoplamiento sp2-sp2 catalizada por paladio y origina el metal-furanocarbaldehído 7.174 (Z=metal) y la quinazolina 7.175. La desconexión de la parte de quinazolina en el compuesto 7.175 se basa en una reacción SNAr y proporciona la dihaloquinazolina 7.176 (X y X´= halógeno) y la 3-cloro-4-[(3-fluorobencil)oxi]anilina 7.177. Una operación de intercambio del grupo amino por nitro proporciona el 2-cloro-1[(3-fluorobencil)oxi]-4-nitrobenceno 7.178. Este compuesto se sintetizará mediante reacción SN2 del 2-cloro-4-nitrofenol 7.179 sobre el haluro de 3-fluorobencilo 7.180.

Inhibidores de quinasas 72
Esquema 25
12.1.b. Síntesis
La síntesis del lapatinib se inicia con la reacción SN2 del fenóxido derivado de 2-cloro-4-nitrofenol 7.179 sobre el bromuro de 3-fluorobencilo 7.180. La reacción proporciona el 2-cloro-1[(3-fluorobencil)oxi]-4-nitrobenceno que se transforma en la 3-cloro-4-[(3-fluorobencil)oxi]-anilina 7.177 mediante hidrogenación en presencia de Pd/C (esquema 26).73 La reacción SNAr del compuesto 7.177 con 4-cloro-6-iodoquinazolina se lleva a cabo en isopropanol a reflujo y proporciona el compuesto 7.175. La unión del fragmento de furanocarbaldehído se confía a una reacción de acoplamiento de Stille y se lleva a cabo por reacción de 7.175 con el furanilestannano 7.181 en presencia de PdCl2(PPh3)2 y DIPEA en DMF. La reacción permite la obtención del compuesto 7.182 que se transforma en lapatinib mediante hidrólisis de la función acetálica seguida de aminación reductiva con metilsulfonil-etanamina 7.172.
73 Whitehead, B. F.; Ho, P. T. C.; Sutle, A. B.; Pandite, A. WO143483, 2007, p. 49.

Inhibidores de quinasas 73
Esquema 26
1) Explique mecanísticamente la formación del compuesto 7.175 ¿Por qué se sustituye el átomo de cloro y no el átomo de yodo?
2) Explique mecanísticamente la formación de 7.182 ¿Por qué se sustituye el átomo de yodo y no el átomo de cloro o el de flúor?

Inhibidores de quinasas 74
13. Pazopanib
El pazopanib es comercializado por GlaxoSmithKline con el nombre de Votrient®. Este fármaco se receta en el tratamiento de pacientes que sufren sarcoma avanzado de tejidos blandos y cáncer de riñón. El pazopanib es un inhibidor multiquinasa y sus dianas terapéuticas son los receptores del factor de crecimiento endotelial vascular VEGFR (del inglés Vascular Endotelial Growth Factor Receptor).
El pazopanib es un inhibidor de la angiogénesis. Ejerce su acción anticancerígena impidiendo el crecimiento de nuevos vasos sanguíneos tumorales.
Figura 80. Estructura del pazopanib
13.1. Angiogénesis
Se define la angiogénesis como el proceso que conduce a la formación de nuevos vasos sanguíneos a partir de la vasculatura preexistente. El proceso de la angiogénesis incluye:
1) La migración y proliferación de células endoteliales (ECs).
2) La formación y organización de grupos celulares en estructuras tubulares.
3) La formación de vasos sanguíneos estables.
Los principales reguladores del proceso de angiogénesis son los factores de crecimiento vascular endotelial VEGF (del inglés Vascular Endotelial Growth Factor) y sus receptores VEGFR.
Se pueden distinguir dos tipos de angiogénesis:74
a) La angiogénesis fisiológica/normal. Este proceso tiene lugar durante la embriogénesis y continúa después del nacimiento en el desarrollo temprano post-natal, aportando el oxígeno y los nutrientes requeridos por los órganos en crecimiento, proporcionando señales promotoras morfológicas y siendo fundamental para la formación del hueso endocondral75 y en el crecimiento del esqueleto.
b) La angiogénesis patológica/anormal. Este proceso se produce cuando se alcanza y sobrepasa el umbral entre los estimuladores e inhibidores angiogénicos.
Se pueden distinguir dos tipos de angiogénesis patológica:
b1) La angiogénesis activada, en la que dominan los niveles de moléculas pro-angiogénicas. Este tipo de angiogénesis está asociada, muy frecuentemente a enfermedades malignas, además de estar implicada en la obesidad, el asma, la diabetes, la cirrosis, la esclerosis múltiple, la endometriosis, el SIDA, las infecciones bacterianas y las enfermedades auto-inmunes.
74 Martínez-Ezquerro, J. D.; Herrera1, L. A. Cancerología 2006, 1, 83-96. 75 El hueso endocondral es el que se forma en los fetos a partir de tejido cartilaginoso. El término endocondral significa que la osificación se realiza desde el interior del cartílago hacia el exterior.

Inhibidores de quinasas 75
b2) La angiogénesis inhibida, en la que dominan los niveles de moléculas anti-angiogénicas. Este proceso está asociado al mal funcionamiento de células endoteliales (ECs) e impide la revascularización, la cicatrización y la regeneración de los vasos sanguíneos.
Además del VEGF, existen distintas moléculas implicadas en la regulación positiva de la angiogénesis, incluyendo al factor ácido de crecimiento de fibroblastos (aFGF), el factor básico de crecimiento de fibroblastos (bFGF), el factor de crecimiento transformante alfa (TGF-α), el factor de crecimiento transformante beta (TGF-β), el factor de crecimiento de hepatocitos (HGF), el factor necrosistumoral alfa (TNF-α), la angiogenina, la interleucina 8(IL-8)m, las angiopoietinas (Ang-1 y Ang-2), el factor de crecimiento derivado de plaquetas (PDGF), VEGF derivado de la glándula endócrina (EG-VEGF), la leptina, las prostaglandinas y los lípidos, entre los que cabe incluir al TGF-α y al bFGF, que actúan regulando la expresión del VEGF.
La expresión del VEGF se aumenta mediante estímulos tales como hipoxia/isquemia76 principalmente mediante el factor inducible de hipoxia 1 (HIF-1), mediante factores de crecimiento como EGF (factor de crecimiento epidérmico), TGF-α y TGF-β (Factor transformante alfa y beta), KGF (factor de crecimiento derivado de queratinocitos), IGF-1 (factor de crecimiento insulínico tipo 1), FGF (factor de crecimiento de fibroblastos) y PDGF (factor de crecimiento derivado de plaquetas), mediante oncogenes activados (por ejemplo Ras), así como mediante distintas citoquinas (IL-1-α y IL-6), mediante p53 mutado77 y mediante estrógenos y óxido nítrico (NO).
13.2. Estructura del VEGF
El factor de crecimiento vascular endotelial (VEGF) es un homodímero unido mediante puentes disulfuro. El VEGF es expresado en diferentes isoformas que contienen desde 121 a 206 aminoácidos (VEGF121, VEGF165, VEGF189 and VEGF206). Todas las variantes comparten la misma parte N-terminal y seis residuos C-terminales. El dominio de unión al receptor del VEGF comparte mucha homología con el factor de crecimiento derivado de plaquetas (PDGF) uniéndose a la parte extracelular de su receptor VEGFR por los dominios segundo y tercero (véase más adelante la estructura del VEGFR).
En la figura 81 se indica una representación del homodímero de VEGF humano.78 Un monómero del dímero se representa en rojo y el otro en azul. Cada monómero contiene siete láminas β (de la β1 a la β7) y dos segmentos helicoidales α (α1 y α2). La lamina β3 está conectada con la β7 mediante dos puentes disulfuro (coloreados en amarillo en la figura 81). Un tercer puente disulfuro conecta la lámina β1 con la β4.
La dimerización en el VEGF se produce de forma antiparalela. Los monómeros se unen covalentemente mediante dos enlaces disulfuro entre Cys51 y Cys60.
76 La hipoxia se define como el estado de deficiencia de oxígeno en sangre, células y tejidos del organismo con compromiso de la función de éstos. 77 El gen p53 resulta esencial para inducir la respuesta de la célula ante el daño al ADN. Se encarga de detener el ciclo celular en caso de mutación. El gen p53 es un gen supresor de tumores y desempeña un papel importante en la apoptosis y en el control del ciclo celular. Un gen p53 defectuoso podría permitir que las células anormales proliferen dando por resultado un cáncer. Alrededor de un 50 % de todos los tumores humanos contienen mutaciones en el gen p53. 78 Muller, Y. A.; Christinger1, H. W.; Keyt, B. A.; de Vos, A. M. Structure 1997, 5, 1325-1338.

Inhibidores de quinasas 76
Figura 81. Representación del homodímero de VEGF humano
En la figura 82 se indican dos vistas del homodímero VEGF (en colores azul y rojo) formando un complejo mediante unión al dominio D2 del receptor VEGFR-1 (en color verde).79
Figura 82. Complejo VEGF-dominio 2 de VEGFR-1
En la figura 83 se representan los cuatro primeros dominios del receptor VEGFR-1 y el dímero VEGF (en colores azul y rojo) unido al dominio 2 del receptor. 79 Wiesmann, C.; Fuh, G.; Christinger, H. W.; Eigenbrot, C.; Wells, J. A.; de Vos, A. M. Cell 1997, 91, 695-704.

Inhibidores de quinasas 77
Figura 83. Complejo VEGF/dominios 1-4 VEGFR-1
La actividad del VEGF está modulada por su unión a heparina, lo que resulta en una mayor
eficiencia de la actividad de la quinasa receptora.
13.3 Tipos de VEGF
1) VEGF-A. Este factor es una glicoproteína homodimérica de 45 KDa que está codificada por un solo gen organizado en ocho exones separados por siete intrones.80
Figura 84. Unión de VEGF-A a VEGFR-1 y VEGFR-2
El VEGF-A emplea sitios de unión simétricos en cada polo del dímero para unirse tanto al VEGFR-1 como al VEGFR-2 y puede inducir heterodímeros entre estos dos receptores.81
80 Ferrara, N.; Gerber, H. P.; LeCouter, J. Nat. Med. 2003, 9, 669-676. 81 Veikkola, T.; Karkkainen, M.; Cleasson-Welsh, L.; Alitalo, K. Cancer Res. 2000, 60, 203-212•

Inhibidores de quinasas 78
Hasta el momento se han descrito seis isoformas del VEGF en seres humanos las cuales contienen 121, 145, 165, 183, 189 y 206 residuos de aminoácidos, generados como resultado del procesamiento alternativo del RNAm.
El VEGF-A165, la isoforma predominante del VEGF-A, es una molécula cargada positivamente que se une a la heparina. El VEGF-A145 y el VEGF-A183 son las variantes menos frecuentes.
Las isoformas del VEGF-A difieren tanto en su masa molecular como en sus propiedades biológicas. Las distintas isoformas del VEGF-A incrementan la permeabilidad vascular, estimulan la proliferación y migración de células endoteliales (ECs), proveen a dichas células con señales de supervivencia y anti-senescencia y además promueven la neuroprotección en desórdenes isquémicos.
2) VEGF-B. De VEGF-B se conocen dos isoformas, el VEGF-B167 y el VEGF-B186, cada una con 167 y 186 residuos de aminoácidos, respectivamente. La expresión del VEGF-B no está regulada por hipoxia. El VEGF-B estimula ligeramente la proliferación celular y es un ligando tanto para el VEGFR-1 como para la NRP-1 (Neuropilin Receptor-1) que actúa como co-receptor de los VEGFR.
El VEGF-B se expresa en tejidos humanos normales, principalmente en el miocardio en desarrollo, pero también se expresa en tumores humanos benignos y malignos.82
Figura 85. Unión de VEGF-B a VEGFR-1 y NRP-1
3) VEGF-C. El VEGF-C es un potente factor de permeabilidad, aunque es 4-5 veces menos potente que el VEGF-A. Se sintetiza como dímero pre-péptido (proteínas que requieren procesamiento post-traduccional) con un tamaño de 61 KDa por subunidad. La subsiguiente maduración proteolítica forma un homodímero de 21 KDa. Tanto las formas maduras del VEGF-C como las parcialmente procesadas se unen al VEGFR-3 con gran afinidad, mientras que solo las formas completamente procesadas se unen al VEGFR-2.
82 Partanen, T. A.; Paavonen, K. Microsc. Res. Tech. 2001, 55, 108-121.•

Inhibidores de quinasas 79
VEGF-C
VEGFR-2
S S SS
VEGFR-3
Figura 86. Unión de VEGF-C a VEGFR-2 y VEGFR-3
El VEGF-C estimula la migración y proliferación de ECs. Su unión al VEGFR-3 regula la señalización del VEGFR-2 actuando de manera sinérgica con el VEGF-A. Además, está asociado a las células neuroendócrinas (NE), aunque sin participación aparente en el desarrollo de la vasculatura del sistema neuroendocrino.
4) VEGF-D. El VEGF-D es un mitógeno para las ECs y un ligando tanto para el VEGFR-2 como para el VEGFR-3. Este factor es un compuesto linfangiogénico de menor potencia que el VEGF-C, que parece estar sub-expresado durante el desarrollo, así como después de la organogénesis.
VEGF-D
VEGFR-2
S S SS
VEGFR-3 Figura 87. Unión de VEGF-D a VEGFR-2 y VEGFR-3
5) VEGF-E. El VEGF-E fue descubierto en el genoma del parapoxivirus Orf (cepa NZ-7), un virus de cadena lineal doble de ADN que causa dermatitis pustular contagiosa en ovejas, cabras y ocasionalmente en seres humanos. El VEGF-E, también nombrado como VEGForf,

Inhibidores de quinasas 80
carece del dominio de unión a heparina que si presenta el VEGF-A. EL VEGF-E se une y activa específicamente al VEGFR-2, resultando en un efecto mitogénico y en actividad de permeabilidad vascular similar al producido por el VEGF-A165.
VEGF-E
VEGFR-2 Figura 88. Unión de VEGF-E a VEGFR-2
13.4. Receptores del VEGF
Los VEGF se unen con alta afinidad a receptores específicos denominados VEGFR, que tienen actividad tirosina-quinasa (RTKs). Estos receptores se localizan en la superficie de las ECs vasculares y en la superficie de células derivadas de la médula ósea.
Como se ha indicado en el apartado anterior, todas las isoformas del VEGF son capaces de unirse a alguno de estos tres receptores: VEGFR-1 (también conocido como Flt-1), VEGFR-2 (denominado también como KDR o Flk-1) y/o a VEGFR-3 (denominado Flt-4). La estructura de estos tres receptores está formada por:83
1) Siete dominios homólogos a inmunoglobulina (dominios Ig-like), colocados en la parte extracelular.
2) Una región transmembranal (TM).
3) Un dominio yuxtamembranal (JM).
4) Un dominio intracelular de señalización con actividad tirosina quinasa, que está dividido por una inserción de quinasa (KI) y acaba en la parte carboxi-terminal.
En la figura 89 se comparan las estructuras de los receptores VEGFR-1 y VEGFR-2. El dominio quinasa de estos dos receptores es la región más conservada, con un 70% de homología. La parte carboxi-terminal representa la región más divergente, con sólo el 28.1% de homología.
83 Holmes, K.; Roberts, O. L.; Thomas, A. M.; Cross, M. J. Cell. Signal. 2007, 19, 2003-2012.

Inhibidores de quinasas 81
Figura 89. Estructura de VEGFR-1 y VEGFR-2 y uniones a VEGF
Al igual que otros RTKs, los VEGFRs se dimerizan y experimentan trans-autofosforilación después de la unión al ligando, desencadenando una cascada de señalización mediante fosforilación de distintas proteínas, como la proteína-quinasa C (PKC), la fosfolipasa C-gamma (PLC-γ), el fosfatidilinositol 3-quinasa (PI3K), la sintasa de óxido nítrico endotelial (eNOS) y el blanco de rapamicina en mamíferos (mTOR).
La fosforilación del domino quinasa de los VEGFRs regula mecanismos involucrados en los procesos de angiogénesis.84
13.4.1. Receptor VEGF-1 (VEGFR-1)
El VEGFR-1 fue el primer receptor identificado de alta afinidad para el VEGF. El VEGFR-1 es una glicoproteína transmembranal de 180 KDa cuyo RNAm puede ser cortado y empalmado produciendo una proteína más corta (sFlt), con únicamente seis dominios extracelulares de tipo Ig-like. Este receptor más corto mantiene la actividad de unión al VEGF-A y a PlGF, (del inglés Placental Growth Factor), funcionando así como un regulador negativo de estas proteínas al secuestrar y suprimir sus niveles fisiológicos.
El VEGFR-1 de la superficie celular se une al VEGF-A, VEGF-B, PlGF-1 y PlGF-2, provocando la activación de la PI3K y otras proteínas de transducción de señales.85
El VEGFR-1 se encuentra expresado principalmente en las células endoteliales (ECs), aunque también se encuentra en células trofoblásticas, monocitos, células renales mesangiales, células del músculo liso uterino y en distintos tipos celulares tumorales.
La transcripción del VEGFR-1 es aumentada por la hipoxia y, aunque probablemente no induce proliferación celular, debido a que las proteínas quinasas activadas por mitógeno (MAPKs) no son activadas por este receptor, sí promueve la generación de proteasas, como la
84 Dvorak, H. F. J. Thromb. Haemost. 2005, 3, 1835-1842. 85 Maru, Y.; Yamaguchi, S.; Shibuya, M. Oncogene 1998, 16, 2585-2595.

Inhibidores de quinasas 82
metaloproteasa de matriz extracelular 9 (MMP-9), las cuales se emplean en la degradación de la membrana basal de los vasos sanguíneos en las etapas iniciales de la angiogénesis.
11.4.2. Receptor VEGF-2 (VEGFR-2)
El VEGFR-2 fue el segundo receptor del VEGF en ser identificado. Es una proteína de 230 KDa de la cual se conocen dos variantes funcionales producto del procesamiento del RNAm. El VEGF-2 se une a VEGF-A y a formas maduras del VEGF-C y VEGF-D.
Únicamente la forma glicosilada final del VEGFR-2 es capaz de autofosforilarse en respuesta al VEGF y participa de manera crucial en la angiogénesis, en el desarrollo y en la hematopoyesis,86 siendo el mayor mediador de los efectos mitogénicos, angiogénicos, y de aumento de la permeabilidad del VEGF. Además, cuando es activado el VEGFR-2 también provoca la respuesta migratoria celular.
El VEGFR-2 se encuentra expresado principalmente en las ECs, además de en células madre hematopoyéticas, en megacariocitos y en células progenitoras retinales. Aunque la producción de este receptor aumenta bajo condiciones de hipoxia, esta no es su principal inductora.
Se ha propuesto que un mecanismo post-transcripcional pudiera ser el responsable de la sobre-expresión de VEGFR-2. Su activación por fosforilación, vía unión a VEGF, resulta en la activación de las MAPKs por fosforilación de las tirosinas y de la vía del PI3K-PLC-γ-PKC, sin emplear la activación de la vía de Ras.
La activación del VEGFR-2 aumenta y activa la sintetasa endotelial de óxido nítrico (eNOS). El NO juega un papel crucial en la proliferación de ECs, en la migración celular, en la formación de tubo, en el aumento de la permeabilidad vascular, en la hipotensión y en la angiogénesis inducidas por VEGF.
La activación de VEGFR-2 activa la transducción de señales involucradas en la litogénesis y migración celular, la angiogénesis y la permeabilidad vascular.
Cuando VEGF se une a los dominios 2 y 3 de un nomómero de VEGFR-2 se incrementa la probabilidad de que otro monómero VEGFR-2 se una al ligando y forme un dímero. Una vez se ha producido la dimerización del receptor, las interacciones entre sus dominios 7s lo estabilizan significativamente. La dimerización permite la orientación precisa de los dominios intracelulares con actividad tirosina-quinas, produciéndose la autofosforilación y la activación de la vía se señalización mediada por quinasas.
El dominio tirosina-quinasa de VEGFR-2 está separado por dos segmentos con una longitud de 70 aminoácidos denominada dominio de inserción de quinasa (véase la figura 90).
86 Se denomina hematopoyesis al proceso de formación, desarrollo y maduración de los elementos formes de la sangre (eritrocitos, leucocitos y plaquetas) a partir de un precursor celular común e indiferenciado conocido como célula madre.

Inhibidores de quinasas 83
SecuenciaN-terminal
Zona de unióndel ligando
Dominios Ig
Dominio extracelular
Membrana plasmática
Dominio intracelular
Dominiotransmembrana
Yuxtamembrana
Dominioquinasa 1
Dominioquinasa 2
Dominio de inserciónde quinasa
Cola C-terminal
TyrY951
Y1054
Y1059
Y1175
Y1214
Figura 90. Dominios del receptor VEGFR-2
El VEGFR-2 se autofosforila en el dominio de inserción de quinasa, en el dominio quinasa-2 y en el dominio carboxi-terminal. Los residuos de tirosina que resultan fosforilados son 951 (domino de inserción de quinasa), 1054, 1059 (dominio quinasa-2) y 1175 y 1214 (cola carboxi-terminal). De estos sitios, el 1175-Tyr-P es la región de unión al dominio SH2 de la fosfolipasa-Cy (PLCy). Después de la fosforilación de la PLCy se activa la corriente de transducción de la señal de las MAP-quinasas, desencadenando en última instancia el proceso de angiogénesis.
13.4.3. Receptor VEGF-3 (VEGFR-3)
El VEGFR-3 es un receptor tirosina quinasa de superficie celular altamente glicosilado y relativamente estable de 180 KDa aproximadamente. Actualmente se conocen dos formas transcritas que difieren entre sí por sus extremos carboxilo. La forma predominante en los tejidos corresponde a un transcrito de 5.8 Kb, mientras que la variante alternativa consta de 4.5 Kb. Después de la biosíntesis, la forma glicosilada del VEGFR-3 de 195 KDa, es cortada proteolíticamente en el quinto dominio de Ig, aunque las cadenas resultantes de 120 KDa y 75 KDa, permanecen unidas por un enlace disulfuro.

Inhibidores de quinasas 84
El VEGFR-3 y sus ligandos (las formas no procesadas del VEGF-C y VEGF-D), están involucrados en la linfangiogénesis, y también en la angiogénesis y en la migración celular, a pesar de que el VEGF-A no se une a este receptor.
El VEGFR-3 se expresa en las células endoteliales (ECs) vasculares en las etapas tempranas del desarrollo, mientras que después de la organogénesis permanece restringido a las ECs linfáticas. Se puede sobre-expresar tanto en tumores vasculares benignos como malignos de niños y adultos. Este receptor se encuentra activado en los capilares sanguíneos de reciente formación que rodean a los tumores sólidos, siendo importante tanto para la génesis de la neo-vascularización inducida por el tumor, como para el mantenimiento del recubrimiento de las ECs durante la angiogénesis tumoral.
13.5. Uniones VEGF-VEGFR
El VEGFR-1 se une a VEGF-A, VEGF-B y PlGF. Este receptor se expresa en las células madre hematopoyéticas, macrógafos y monocitos y también en el endotelio vascular. Provoca migración celular y hematopoyesis (véase la figura 90).
Figura 91. Efectos de la activación de los receptores VEGFR
El VEGFR-2 se expresa en los endotelios vascular y linfático. El endotelio vascular está compuesto de una monocapa de células endoteliales, con firmes uniones inter e intra-endoteliales. En este endotelio las células se encuentran rodeadas por la membrana basal y están estabilizadas por células musculares denominadas pericitos. El endotelio linfático carece de uniones inter-endoteliales, de membrana basal y de pericitos de soporte, lo que permite la permeabilidad de los vasos linfáticos.

Inhibidores de quinasas 85
El VEGFR-2 se une a VEGF-A, VEGF-C, VEGF-D y VEGF-E. Los efectos corriente abajo del VEGFR-2 son la activación del endotelio vascular, la proliferación celular, la migración, la permeabilidad y la supervivencia celular, lo cual provoca vasculogénesis y angiogénesis (véase la figura 90).
El VEGFR-3 sólo se expresa en el endotelio linfático. Los efectos corriente abajo del VEGFR-3 son la proliferación celular y la migración celular, lo cual provoca vasculogénesis y linfangiogénesis (véase la figura 91).
13.6. Desarrollo del pazopanib
El desarrollo del pazopanib se inició con un cribado de una colección de compuestos de la empresa GlaxoSmithKline. En este estudio se identificó el compuesto 7.183 como inhibidor de VEGFR-2, con un IC50 de 400 nM (véase el esquema 27).87
Un avance significativo se consiguió al descubrir que la pirimidina 7.184 inhibía el VEGFR-2 con un IC50 de 6.3 nM. El aumento de la potencia inhibitoria de este compuesto se achacó a un enlace de hidrógeno entre el hidroxilo fenólico y el Asp-1046 en el bolsillo de unión de ATP de la quinasa. El perfil farmacocinético de 7.184 era bajo ya que el clearance (depuración)88 era de 28-83 mL/min/Kg, debido a la glucorodinación del hidroxilo fenólico. La biodisponibilidad en ratas era de menos del 11%.
El siguiente avance se consiguió al cambiar el grupo 4-metil-3-hidroxianilina por un grupo 3-metilindazol. Este cambio condujo al compuesto 7.185 con buena potencia contra VEGFR-2 (IC50 = 6.3 nM). Además, la acción inhibitoria contra v-HUVECs (células endoteliales de la vena de cordón umbilical humano) era de IC50 = 0.18 µM. Este es un parámetro que mide la selectividad del compuesto ya que la estimulación de la proliferación de v-HUVECs por el b-FGF (Factor de crecimiento básico de fibroblastos), no debería verse afectada por un inhibidor de VEGFR-2, siendo la situación ideal la de elevada inhibición de VEGFR-2 y baja inhibición de v-HUVECs. La farmacocinética de 7.185 se incrementó notablemente, en comparación con 7.184, con una clearance (depuración) de 16 mL/min/kg y una biodisponibilidad oral del 85% con dosis de 10 mg/kg en ratas.
La estructura cristalina del complejo formado por 7.185 y VEGFR-2 puso de manifiesto la presencia de enlaces de hidrógeno entre el nitrógeno pirimidínico N1 y el NH del nitrógeno anilínico en C2 con la cisteina Cys919 del bolsillo de unión de ATP (véase la parte A de la figura 92). El mapa de densidad electrónica del anillo indazólico mostraba la colocación del compuesto en dos conformaciones alternativas, una en forma de S, con el anillo indazólico colocado dentro del bolsillo lipofílico y otra en forma de U, que se muestra en la figura 92, con el anillo indazólico colocado hacia fuera del bolsillo de unión de ATP.
Para evitar la presencia de las dos conformaciones en el centro activo de la quinasa se preparó el compuesto N-metilado 7.186. La estructura cristalina del complejo fomado por 7.186 y VEGFR-2 mostraba solamente la presencia de la conformación S (véase la parte B de la figura 92). La conformación U no se puede adquirir en el compuesto 7.186 por el impedimento estérico que se produciría entre el hidrógeno H5 del anillo imidazólico y el grupo N-Me.
87 Harris, P. A.; Boloor, A.; Cheung, M.; Kumar, R. et al. J. Med. Chem. 2008, 51, 4632-4640. 88 El clearance o depuración es un parámetro farmacocinético descriptivo y consiste en el análisis de la capacidad que tiene el organismo para eliminar un fármaco.

Inhibidores de quinasas 86
Esquema 27. Desarrollo del pazopanib
La sulfona 7.187 tenía una potencia inhibitoria similar a 7.186. Su perfil farmacocinético en ratas era mejor con clearance más bajas (10 mL/min/kg) y biodisponibilidades orales del 65%.
El compuesto 7.187 presentaba un buen perfil farmacocinético pero inhibía algunas isozimas del citocromo P450, muy probablemente debido a la unión del nitrógeno indazólico con el átomo de hierro del grupo hemo del citocromo P450. Una forma de disminuir la inhibición del citocromo 7.187 es mediante el aumento del impedimento estérico en el anillo indazólico. Este razonamiento llevó a la estructura del pazopanib que contiene un grupo metilo en la posición 2 del sistema indazólico. Este compuesto poseía una buena combinación de potencia celular en VEGF y bFGF dirigidas a HUVEC, con IC50 de 0.021 y 0.72 µM, respectivamente. Además, el pazopanib mostró muy buena actividad inhibitoria contra todos los VEGFR con IC50 de 10, 30 y 47 nM para VEGFR-1, VEGFR-2 y VEGFR-3, respectivamente.

Inhibidores de quinasas 87
Figura 92. Parte A: unión de 7.185 (conformación U) a VEGFR-2. Parte B: unión de
7.186 (conformacíón S) a VEGFR-2.
El pazopanib presenta buenos parámetros farmacocinético en ratas, perros y monos, con clearance entre 1.4-1.7 mL/min/kg. Las disponibilidades orales en ratas, perros y monos eran del 72, 47 y 65% en dosis de 10, 1 y 5 mg/kg, respectivamente. La inhibición del citocromo P450 también se mejoró, con inhibiciones de >10 µM contra las isozimas ensayadas, con excepción de la 2C9 (7.9 µM).
En la figura 93 se presenta la producción de factores de crecimiento vascular endotelial (VEGF) provocada por las condiciones de hipoxia que están actuando sobre la célula tumoral. En presencia de oxígeno, la proteína HIF-1α (Factor Inducible por Hipoxia) sufre modificaciones post-traduccionales por parte de hidroxilasas las cuales hidroxilan residuos de prolina y de asparagina. Estas modificaciones permiten la unión de la proteína VHL (von Hippel Lindau) a los sitios hidroxilados de HIF-1α provocando su ubiquitinación y degradación.
El crecimiento rápido de los tumores determina que su incremento de tamaño no se acompañe de un desarrollo vascular suficiente, lo que genera zonas de hipoxia en el interior de la masa tumoral. En ausencia de oxígeno (condiciones de hipoxia) se activa HIF-189 que, además, se relaciona con la inactivación funcional del gen supresor VHL, lo que disminuye la degradación y aumenta la síntesis de HIF-1.90
El HIF se une a secuencias específicas localizadas en el gen promotor de VEGF y provoca un aumento en la expresión de este factor. La sobre-expresión de VEGF activa la vía RAS/RAF que induce el proceso de angiogénesis.
Los inhibidores de VEGFR, como el pazopanib y el sunitinib, inhiben la acción fosforilante de los VEGFR inhibiendo la vía de señalización celular y bloqueando el proceso de angiogénesis.91
89 Quintero, M.; Mackenzie, N.; Brennan, P. A. Eur. J. Surg. Onco. 2004, 30: 465-468. 90 (a) Kanno, H.; Kondo, K.; Ito, S.; et al. Cancer Res.1994; 54:, 4845-4847. (b) Gnarra, J. R.; Tory. K.; Weng, Y.; et al. Nat. Genet. 1994, 7, 85-90. 91 Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; et al. J. Clin. Oncol. 2009, 27.3126.

Inhibidores de quinasas 88
Figura 93. Efectos antiangiogénicos de pazopanib y sunitinib
13.7. Síntesis del pazopanib
13.7.a. Análisis retrosintético del pazopanib
La retrosíntesis del pazopanib se inicia con la desconexión de la 5-amino-2-metilbencenosulfonamida 7.187 (esquema 28). Este fragmento se instalará mediante reacción SNAr sobre el pirimidinoindazol 7.188 (X=halógeno). La siguiente desconexión se centra en el anillo de pirimidina y se basa también en una reacción SNAr. La operación conduce a la 2,4-dihalopirimidina 7.190 y al aminoindazol 7.191. Una operación de intercambio de grupo funcional proporciona la nitroindazolina 7.192 que se sintetizará de la 2-etil-5-nitroanilina 7.193.
N
N NHN NN
SO2NH2
Me
MePazopanib
O2NNN
CH3
NH2O2N
SNAr
NH2
SO2NH2
Me
N
N N NN
Me
X+
N-Met
N
N NH
NN
X
SNArN
NX X H2NNN+
IGF
7.190 7.191 7.189
7.1887.187
7.192 7.193 Esquema 28

Inhibidores de quinasas 89
13.7.b. Síntesis
Para la síntesis del pazopanib se elige como compuesto de partida la 2-etilanilina 7.194 (esquema 29).92 La nitración de este compuesto proporciona la 2-etil-5-nitroanilina 7.193 que se convierte en el 3-metil-6-nitro-2H-indazol 7.195 mediante reacción con nitrito de isoamilo en ácido acético. El tratamiento de este compuesto con ortoformiato de trimetilo en presencia de trifluoruro de boro-eterato permite la obtención del 2,3-dimetil-6-nitro-2H-indazol 7.192. La reducción del grupo nitro se consigue mediante tratamiento de 7.192 con formiato amónico en presencia de paladio sobre carbón. En esta reacción se obtiene el 2,3-dimetil-6-amino-2H-indazol 7.191 el cual, por reacción SNAr con 2,4-dicloropirimidina 7.190, se convierte en el pirimidinoindazol 7.189. La metilación del grupo NH con yoduro de metilo proporciona el compuesto 7.188, que se convierte en pazopanib por reacción SNAr con la 5-amino-2-metilbencenosulfonamida 7.187.
N
N NHN NN
SO2NH2
Me
Me
Me
Clorhidrato dePazopanib
O2NNNH
O2NNN
HC(OMe)3, BF3·Et2O
CH2Cl2, 0ºC (65%)
NH4HCO2, Pd/C
MeOH ac. (97%)H2NNN
N
NCl Cl
NaHCO3, EtOH, THF85ºC (90%)
N
N NH
NN Me
Cl
MeI, Cs2CO3
DMF, t.a. (83%)
N
N N NN Me
Cl
CH3
H2N
SO2NH2
Me
HCl, iPrOHreflujo (81%)
CH3
NH2
HNO3 fumante
CH3
NH2O2N
Nitrito de isoamilo
AcOH (98%)H2SO4 (60%)
7.194
7.188
7.189
7.191 7.192
7.1957.193
7.187
7.190
· HCl
Esquema 29
1) Explique mecanísticamente la formación de 7.193 ¿Por qué la reacción es regioselectiva?
2) Explique mecanísticamente la formación de 7.195.
92 Sorbera, L. A.; Bolos, J.; Serradell, N. Drugs Future 2006, 31, 585.

Inhibidores de quinasas 90
3) Explique mecanísticamente la formación de 7.192.
13.8. Otros compuestos antiangiogénicos
Como se ha explicado anteriormente, el estado de hipoxia (baja tension de O2) en los tumores que están en expansión provoca la estabilización del factor inducible de hipoxia (HIF-1α) que se une al elemento de respuesta a la hipoxia HRE (del inglés Hypoxia Responsive Element) en el gen promotor de VEGF-A, lo que incrementa la expresión del gen y su transcripción al VEGF-A. Este factor de crecimiento se une al receptor VEGFR-2, expresado en la superficie de las células endoteliales, estimulando la angiogénesis (véase la figura 94). La señalización del VEGFR-2 puede ser inhibida mediante secuestro del VEGF-A, por ejemplo con anticuerpos como el bevacizumab, o mediante la inhibición del VEGFR-2 con pequeñas moléculas como el el sorafenib, el sunitinib, el vatalanib, el vandetanib o el recentin.
Figura 94. Inhibidores anti-angiogénicos
Las estructuras, el modo de acción y la síntesis del sorafenib y del sunitinib ya se han explicado en apartados anteriores. El vatalanib (PTK787/ZK 222584) es un inhibidor de VEGFR-2 (IC50=39 nM), así como de VEGFR-1, FlK, PDGFR y c-Kit, con IC50 de 54 nM, 270

Inhibidores de quinasas 91
nM, 730 nM y 580 nM, respectivamente. Está siendo investigado en fase III para el tratamiento del cáncer colorrectal metastático.
Figura 95. Estructura del vatalanib
El vandetanib ha sido desarrollado por AstraZeneca, con el nombre de Caprelsa®. Se emplea en el tratamiento de ciertos tumores de la glándula tiroides. Es un inhibidor de VEGFR-2 (IC50=40 nM), así como de VEGFR-3 y de EGFR, con IC50 de 110 nM y 500 nM, respectivamente.
Figura 96. Estructura del vandetanib
El recentin es un inhibidor de VEGFR-2 (IC50 <1 nM), así como de c-Kit, VEGFR-3, VEGFR-1 y PDGFR con IC50 de 2 nM, 3 nM, 5 nM y 50 nM, respectivamente. Está siendo estudiado en fase III para el tratamiento de cánceres como el colorrectal, de pulmón o riñón.
Figura 97. Estructura del recentin


















