
Comparación de Los Métodos Tradicionales y Constructivistas en El Aprendizaje de Idiomas Modernos
Upload
ruben-kapa-ticonaCategory
view
243download
1description

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
MÉTODOS MODERNOS DE ANÁLISIS QUÍMICO
Química Analítica
Es la ciencia que estudia el conjunto de principios, leyes y técnicas cuya finalidad es la determinación de la composición química de una muestra.
Al conjunto de técnicas operativas puestas al servicio de dicha finalidad se la denomina análisis químico. La química analítica es la creadora de dichas técnicas, por lo tanto se estudia los métodos de: Separación, Identificación y Determinación.
El análisis cuantitativo es indispensable para una variedad de operaciones técnicas y comerciales; aplicables en la industria, agricultura, medicina, geología y criminología.
Por lo tanto el químico debe contar con bases sólidas en otras ramas de la química y tener suficiente conocimiento en física y matemática.
El analista químico es un operador con poco conocimiento científico; en cambio el químico analítico es el profesional que interpreta los resultados, que puede modificar métodos y desarrollar métodos originales.
Métodos de Análisis
Los métodos de análisis se basan en determinar un compuesto o una especie que tiene que ser cuantificado o separado al que denominaremos ANALITO este puede constituir una pequeña o gran parte de la muestra analizada.
Si el analito es ¿1% se considera un componente principal. Si el analito se encuentra entre 0,01−1% se considera un componente menor. Si el analito es ¿0,01% se considera un componente vestigial.
Pasos para un Análisis
Son los siguientes:
Muestreo: Obtener una muestra representativa tanto de un sólido, líquido o gas.
Disolución de la muestra: Cualquier muestra que tengamos se debe llevar al estado líquido con disolventes indicados, de los cuales los más empleados son H 2O, HNO3 , HCl ,HClO4o AguaReg ia .
Medición: Puede basarse en métodos tales como: Gravimetría, Volumetría, Cromatografía, Espectrofotometría, Físicos, Químicos y Biológicos.
Determinación de la cantidad: Se puede saber mediante estequiometria y absorbancia.
1

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Clasificación General de los Métodos
Se puede clasificar de la siguiente forma:
Desde el punto de vista de los materiales analizados: los cuales pueden ser orgánicos e inorgánicos y se pueden estudiar con un análisis parcial o completo según se requiera o con análisis inmediato o último.
Análisis Inmediato Consiste en la determinación de sustancias que reaccionan en forma análoga ante un cierto tratamiento.
Análisis Último Llamado también elemental, determina el contenido de cada elemento presente.
Desde el punto de vista de la escala de análisis: se puede clasificar de acuerdo al tamaño de la muestra tomada o la cantidad de material que se determina.
Macro es 0,1−1,0o2,0g de muestra. Semimicro es 0,01−0,05g de muestra. Micro de 1,0 g a unos pocos miligramos. Ultra micro o microgramo del orden de es 1,0 μg.
Desde el punto de vista del método de medida que utilicen:
Precipitación Métodos Gravimétricos Electrodeposición
Volatilización
Neutralización Métodos Volumétricos Precipitación
Redox Valoración
Métodos Gasométricos medida del volumen de gas en condiciones normales.
Métodos de Análisis en General
Métodos Clásicos: Entre ellos tenemos.
Separación precipitación, extracción, destilación. Cualitativos punto de ebullición, punto de fusión, índice de refracción,
color, olor.
2

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Cuantitativos volumétricos y análisis gravimétricos.
Métodos Modernos o Instrumentales: podemos citar.
Separación cromatografía y electroforesis. Cualitativos y Cuantitativos espectroscopia, método electroquímico,
métodos radio químicos, RNM.
3

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
TRATAMIENTO ESTADISTICO DE LOS ERRORES
Introducción
Cuando se realiza cualquier medición científica es necesario considerar que se puede cometer un error e importante tener la capacidad de evaluar los datos y aprender a sacar conclusiones.
En un análisis químico no hay forma de medir el valor verdadero, lo mejor que puede aplicarse es una técnica la cual por experiencia se sepa que es de confianza.
Error
Tiene dos significados diferentes entre sí:
Se refiere a la diferencia entre un valor medido y el valor verdadero o conocido.
Puede denotar la incertidumbre estimada en una medición o experimento.
Cifras Significativas
Es el número mínimo de dígitos que se necesitan para expresar científicamente un valor sin que se pierda la exactitud.
Son cifras significativas todos aquellos valores que pueden leerse directamente del aparato de medición utilizado.
Cualquier digito diferente de cero es significativo (2,3; 4,6). El cero entre dígitos distintos de cero son significativos (5,008; 3,01). Cero a la izquierda del primer digito distinto de cero no son significativos
(0,001). Si en un valor numérico el primer número es mayor a uno todos los ceros a la
derecha del punto decimal son significativos (40,000).
4

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Si en el valor numérico el primer número es menor a uno únicamente los ceros que están al final del número y entre los dígitos distintos de cero son significativos (0,82301000).
Redondeo
6,72≅ 6,7
6,77≅ 6,8
6,65≅ 6,6
6,55≅ 6,6
Suma y Resta
18,998403+18,998403+83,80
19,00+19,00+83,80≅ 121,8
Multiplicación y División
1,632×105∗4,107×103∗0,984×106≅ 6,60×1014
Tipos de errores
Los más importantes son:
Errores crasos: Son errores tan graves que no queda otra alternativa más que abandonar el experimento y empezar de nuevo, las causas más comunes son:
Avería total del instrumento. Derramamiento accidental de una muestra. Descubrir que durante el desarrollo de un experimento el reactivo que
se suponía puro está contaminado.
Errores Sistemáticos: Son errores que afectan al resultado de una medida en la misma proporción y signo, es decir que los resultados son erróneos en el mismo sentido. Estos errores afectan a la exactitud y se pueden deber a: los cálculos que realizamos; los instrumentos de medida o las características del observador. También se los conoce como errores determinados, que se clasifican en constantes y proporcionales.
5

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Errores Constantes: este tipo de error es independiente del tamaño de la muestra analizada, se caracteriza porque el error absoluto es invariable mientras que el error relativo cambia al modificar dicho tamaño.
Errores Proporcionales: este tipo de error disminuye o aumenta en relación proporcional al tamaño de la muestra y se caracterizan porque el error absoluto varía con el tamaño de la muestra mientras que el error relativo permanece constante; una causa general de este error es la presencia de contaminantes en el proceso experimental.
Errores Aleatorios: Son conocidos como errores indeterminados que ocurren de modo casual, por lo tanto no pueden controlarse ni conocerse con anticipación, provocan que los resultados individuales caigan a ambos lados del valor medio. Entonces afectan a la precisión o reproducibilidad de un experimento, estos errores no pueden eliminarse totalmente y con frecuencia son la principal fuente de incertidumbre; se deben al cansancio del observador o a cambios de temperatura.
Conceptos Importantes
Debemos tomar en cuenta los siguientes conceptos:
Magnitud: todo aquello susceptible de ser medido. Medir: comparar una magnitud con otra de su misma especie, la cual se asuma
como unidad patrón. Exactitud: es el grado en que un valor experimental se acerca al valor
verdadero o aceptado como verdadero. Precisión: es el grado de concordancia entre dos valores. Media: es la media aritmética de todos los valores medidos.
x=∑i=1
n x i
n
Mediana: es el valor medio de un conjunto de datos dispuestos en orden ascendente o descendente.
19,4 ;19,5 ;19,6 ;19,8 ;20,1;20,3
19,6+19,82
=19,7
Moda: es el valor que se presenta con más frecuencia en la serie. En una distribución estadística verdadera en el que el número de datos es muy grande la mediana y la moda son idénticos.
6

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Desviación: es la diferencia entre un valor y la media que se expresa en valor absoluto.
|x i−x| Desviación media de una medida individual: es la media de las desviaciones.
d=1n∑i=1
n
(x i−x )
Desviación estándar de la población (): es una medida de la precisión de una población de datos.
σ=√∑ ( xi−μ )2
n; μ=
∑ x i
n
Desviación estándar de la muestra:
S=√∑ (x i−x )2
n−1N-1=grados de libertad
Desviación estándar de la media:
Sm=S
√N
Desviación estándar relativa (R.S.D): denominado también coeficiente de variación porcentual y se emplea con fines comparativos de la dispersión de los datos.
RSD=Sx×100
Población: conjunto de todas las medidas que interesan al experimentador. Muestra: subconjunto de medidas seleccionadas de la población. Error absoluto del conjunto:
E=x−μo
Error absoluto individual:Ei=xi−μo
Error relativo:
Er=Ex
Error relativo porcentual:%E r=E r×100
Dispersión (): es la diferencia entre el valor más alto y el más bajo del conjunto.
7

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Desviación estándar o incertidumbre de los resultados calculados
Contamos con dos tipos de incertidumbre
Incertidumbre absoluta: Es la expresión del margen de incerteza asociado a una medición.
BURETA :±0,02cm3
Incertidumbre Relativa: Es una expresión que compara la magnitud de la incertidumbre con la magnitud de la medición que le corresponde.
Incertidumbre Relativa= Incertidumbre AbsolutaVolumende muestra
OPERACION ECUACION FORMULA
Suma y Resta y=a+b+c Sy=√(Sa)2+(Sb)
2+(Sc)2
Multiplicación y División
y=a×bc
S y
y=√( Sa
a )2
+( Sb
b )2
+( Sc
c )2
Potenciación y=ax S y
y=x
Sa
a
Logaritmo y=log10a Sy=0,434Sa
a
Antilogaritmos y=antilog10aS y
y=2,303Sa
ojo :( Sa
a )=Valor Absoluto
Ejemplos:
1) Resuelva la siguiente suma:
+0,7±0,02+4,30±0,03−2,97±0,05
Sy=√(0,02 )2+(0,03 )2+ (0,05 )2
8

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Sy=0,06R=+2,00±0,06
2) Resuelva la siguiente división:
3,20 (±0,02 )×0,0060 (±0,0001 )2,97 (±0,04 )
y=3,20×0,00602,97
y=6,46×10−3
S y
6,46×10−3=√( 0,023,20 )
2
+( 0,00010,0060 )2
+( 0,042,97 )2
Sy=1,44×10−4
R=(6,46±0,14 )×10−3
3) La desviación estándar en la medición de una esfera es ±0,03cm; si el diámetro es de 3,15cm
V esfera=π6D3
V esfera=π6
(3,15cm )3=16,4 cm3
S y
y=x
Sa
a
Sy=16,4×3×0,033,15
=0,5cm3
V= (16,4 ±0,5 ) cm3
4) El producto de solubilidad (K ps ¿ para una sal de plata es de 4,0 (±0,4 )×10−8. La solubilidad molar de la sal de plata en agua es:
9

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
AgX→Ag+¿+X−¿¿ ¿
K ps=S2
S=√4,0×10−8
S=2,0×10−4
S y
y=x
Sa
a
Sy=(2,0×10−4)× 12×0,44,0
Sy=0,1×10−4
S= (2,0±0,1 )×10−4
Expresión de Resultados de Cálculos Químicos
Si se conocen las desviaciones estándar delos valores que conforman el cálculo final podemos utilizar las ecuaciones analizadas anteriormente sin embargo se pide con mucha frecuencia que se realicen cálculos con datos cuya precisión se indica solo por la utilización de cifras significativas, para estos casos se deben hacer suposiciones de sentido común en lo referente a la incertidumbre de cada alumno.
Para terminar el proceso con la utilización de las ecuaciones anteriores se debe aclarar que no se debe efectuar el redondeo hasta que se haya terminado el cálculo.
Ejemplo:
Una muestra de 3,4842[ g] de una mezcla sólida que contiene
C6H 5COOH (PM=122,123[ gmol ]) se disolvió y valoro con una base hasta el punto final
de fenolftaleína.
El ácido consumió 41,36 [ml] de NaOH 0,2328M ¿calcular el porcentaje de ácido benzoico en la muestra?
Datos:
PM=122,123[ gmol ]
10

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
m x=3,4842 [ g ]
V NaOH=41,36 [ml ] [ NaOH ]=0,2328M
Eq−gNaOH=Eq−gAcido
41,36mlsol×0,2328molNaOH
1000mlsol×1eq−gNaOH
1molNaOH
×1eq−g Ac
1eq−gNaOH
×122,123g Ac
1eq−gAc
mAcido=1,176[ g]
%Acido=m parcial
mtotal
×100%
%Acido=1,176[ g]3,4842[ g]
×100%
%Acido=33,8%
S y
y=√( Sa
a )2
+( Sb
b )2
+( Sc
c )2
Pero: Sc=√(Sc1 )2+(Sc 2 )2
Sc=√(0,02)2+(0,02)2=2,8×10−2
Sy=33,8×√( 0,00013,4842 )2
+( 0,00010,2328 )2
+( 2,8×10−2
41,36 )2
=0,03
%Acido=(33,8±0,03)%
Rechazo de Valores Anómalos
Cuando en una serie de cuatro o más valores aparece un valor que difiere mucho de los demás, se debe prescindir provisionalmente de él y; calcular con los otros valores la media y la desviación media, si esta desviación del valor en observación respecto a la media de los otros valores es mayor de cuatro veces la desviación media, el valor se rechaza completamente. Caso contario se conserva y se realiza nuevamente los cálculos.
El valor rechazable puede ser el que tenga un valor numérico más alto o más bajo en la serie.
490 ; 496 ;500 ;501;503
11

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
MÉTODOS GRAVIMÉTRICOS
Introducción
Son métodos cuantitativos que se basan en la determinación de la masa de un compuesto puro con el que el analito está relacionado químicamente y se fundamenta en relaciones estequeometricas y la constancia de las composiciones.
Requisitos para la determinación gravimétrica:
Se deberá precipitar totalmente la sustancia deseada. El precipitado deberá ser un compuesto estequeometrico de composición
desconocida. El precipitado deberá ser puro y poderse filtrar con facilidad.
Los métodos gravimétricos se pueden clasificar en:
Gravimetría de Precipitación Gravimetría de Volatilización Electro Gravimetría Valoraciones Gravimétricas Espectrometría de masas
Gravimetría de Precipitación
Este proceso consiste en que el analito se separa de los componentes de una solución en forma de precipitado que se trata y se convierte en un compuesto de composición conocida.
12
Se rechaza

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Agente Precipitante
Se clasifica en dos tipos de acuerdo a la muestra.
Agentes Específicos: Un reactivo es específico si reacciona con un solo analito, los reactivos específicos son una rareza. Dimetilglioxina precipitaal ¿2+¿¿.
Agentes Selectivos: Un reactivo es selectivo cuando reacciona con un número limitado de analito.
AgNO3→Cl−¿ ,Br−¿ ,SCN−¿¿¿ ¿
Para un precipitado pueda ser útil es necesario que sea insoluble en el medio en que se produce, que se pueda filtrar con facilidad, que sea puro, que tenga una composición constante y conocida.
Clasificación de las partículas del precipitado
Por su tamaño las partículas del precipitado pueden ser coloidales y cristalinas.
13

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Coloidal: Las partículas coloidales no pueden sedimentar con facilidad por lo que no pueden filtrarse por medios comunes, el diámetro de las partículas oscila entre 10−7−10−4 cm .
Cristalino: Son partículas que sedimentan con facilidad, su diámetro es del orden de los milímetros.
Factores que determinan el tamaño de una partícula
Los factores que determinan el tamaño de una partícula y la rapidez con que se mezclan los reactivos. Pueden explicarse en forma cualitativa asumiendo que el tamaño de las partículas está en función de una propiedad llamada sobresaturación.
SR=Q−SS
Dónde:
Q=concentración del soluto
S=solubilidad en equilibrio
SR↑↔COLOIDAL↔PARTICULAS PEQUEÑAS
SR↓↔CRISTALINO↔PARTICULASGRANDES
“La sobresaturación afecta al tamaño de la partícula”
Nucleación: Es el proceso por el que un número de iones o moléculas que se unen para formar una partícula que de por si se distingue en la solución como una segunda fase, la precipitación ocurre por la formación de núcleos dando origen a un precipitado coloidal.
Crecimiento de las partículas: Es el proceso por el cual iones o moléculas se van uniendo a los núcleos previamente formados aumentando el tamaño de la partícula del precipitado.
Manejo delos precipitados coloidales
Son difíciles de filtrar pero se pueden aglomerar para facilitar la filtración, mediante la coagulación, calentamiento de la solución, centrifugación, añadiendo un electrolito fuerte a la solución.
Contaminación de los precipitados
Los precipitados se pueden contaminar por dos mecanismos:
14

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Por precipitación simultánea: Es un inconveniente que rebela un enfoque incorrecto del análisis, no debe aplicarse métodos de análisis por precipitación sin conocer exactamente la composición general de la muestra.
Coprecipitación: Es un fenómeno en el que otros compuestos normalmente solubles en la disolución se separan de esta durante la formación del precipitado.
Proceso en equilibrio: se conoce la Adsorción en la superficie y la Formación de cristales mixtos.
Adsorción en la superficie: El efecto neto de la adsorción en la superficie es arrastrar un compuesto que otras condiciones seria soluble. Este fenómeno se puede disminuir a través de la digestión, durante este proceso se elimina el agua del sólido para obtener una más densa. También se puede lavar el coloide coagulado con una solución que contenga un electrolito volátil que reemplaza a cualquier electrolito no volátil que se haya agregado para lograr la coagulación.
Formación de cristales mixtos: Consiste en reemplazar uno de los iones de la red cristalina de un sólido por un ion de otro elemento con la característica que debe tener la misma carga y su tamaño no debe variar en más de un 5%.
Procesos cinéticos: Se conoce la Oclusión y el Entrampamiento mecánico.
Oclusión: Es el proceso en el cual un compuesto queda atrapado en los huecos formados durante el crecimiento rápido del cristal. Para evitar la oclusión se debe disminuir la velocidad de la precipitación.
Entrampamiento mecánico: Sucede cuando los cristales están muy juntos durante el crecimiento, una manera de evitar esta situación es mantener una sobresaturación baja y realizar una digestión.
Precipitación Homogénea: Consiste en producir el precipitado por medio de una reacción química que produce un precipitado cristalino y puro.
Gravimetría de volatilización o de desprendimiento
Se basa en la volatilización de la muestra y se lleva a cabo por lo general por calentamiento.
La cantidad de sustancia que se volatiliza se obtiene por diferencia de peso (antes y después de la volatilización).
15
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Los métodos más comunes son la determinación de agua y la de dióxido de carbono.
Ejemplos:
1) Calcular:
m=0,396 gBaCl22H 2O
V=50ml
0,328 gAgCl×35,5 gCl−¿
143,5 gAgCl×
244 gBaCl22H 2O
71 gCl−¿=0,279 gBaCl22H 2O¿¿
%BaCl22H 2O=0,279 g0,396 g
×100%
%BaCl22H 2O=70,5%
0,279gBaCl22H 2O
50ml×1mol
Ba2+¿
244 gBaCl22H 2O×1000ml1 l
¿
% ¿
0,279 gBaCl22H 2O×36 g H 2O
244 gBaCl22H 2O
m=4,1×10−2g H 2O
2) De una muestra de mineral que contiene Cu (65,4); Pb (209,9) y Zn (65,37) se separa 0,6214 g como muestra la cual produce 0,0126 g de PbSO4(303,25) y 0,5212 g de N H 4ZnPO4 (178,38). Determinar:a) La composición de la muestra original.b) Calcular el peso del Zn2P2O7 (304,68 ) que podrá obtenerse poniendo a
ignición el N H 4ZnPO4.
0,0126 gPbSO4×209,9 gPb2+¿
303,25 gPbSO4
=0,00872g Pb2+¿ ¿¿
0,5212 gN H 4ZnPO4×65,37 gZn2+¿
178,38gN H 4 ZnPO 4
=0,191g Zn2+¿ ¿¿
%Pb2+¿=0,00872
0,6214× 100%¿
16

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
% Zn2+ ¿= 0,191
0,6214×100% ¿
%Pb2+¿=1,40%¿ % Zn2+¿=30,74%¿ %Cu2+¿=67,86%¿
0,5212 gN H 4ZnPO4×65,37 gZn2+¿
178,38gN H 4 ZnPO 4
×304,68 gZn2P2O7
130,74 gZn2+¿¿¿
m=0,4451 gZn2 P2O7
3) Un mineral de dolomita es una mezcla de CaCO3 (100,07 ) y MgCO3 (84,32 ) . Una
muestra de 1,000 g de dolomita se calcina para volatilizar el CO2, el residuo obtenido consiste en una mezcla de CaO (56,08) y MgO(40,31) que pesa 0,5200 g. Determinar la composición de la muestra.
CaCO3→CaO+CO2
MgCO3→MgO+CO2
X+Y=1,000
XgCaC O3×
1molCaCO3
100,07 gCaCO3
×1molCaO1molCaCO3
×56,08gCaO1molCaO
=mCaO=0,5604 Xg
YgMgCO3×1molMgCO3
84,37MgCO3
×1molMgO1molMgCO3
×40,31gMgO1molMgO
=mMgO=0,4781Yg
0,5604 X+0,4781Y=0,5200
%CaCO3=50,91 %MgCO3=49,09
17

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
VOLUMETRIA
Introducción
El análisis volumétrico es un proceso basado en la medida de volúmenes de un reactivo necesario para que reaccione con el analito.
Términos volumétricos
Valoración: Es el proceso de adición de un volumen de una solución de concentración conocida para que reaccione con el constituyente buscado.
Disolución Patrón o Valorante Patrón: Es un reactivo de concentración conocida con exactitud, esta solución o este reactivo se usa para llevar a cabo una valoración. Debe cumplir las siguientes características: ser estable, reaccionar rápidamente con el analito y experimentar una selección selectiva con el analito.
Valoración por Retroceso: Es el proceso en el cual el exceso de una disolución patrón empleada para consumir un analito se determina por valoración con una segunda disolución patrón, estas valoraciones suelen ser necesarias cuando la velocidad de reacción entre el analito y el reactivo es lenta o cuando la disolución patrón carece de estabilidad.
Punto de equivalencia: Es un punto teórico que se alcanza cuando la cantidad de valorante añadido es químicamente equivalente a la cantidad del analito en la muestra.
NaCl+AgNO3↔AgCl+NaN O3
1 EqgCl−¿=1Eqg Ag+¿¿ ¿
18

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
El punto equivalente en una valoración es imposible de determinar experimentalmente.
Punto final: El punto final en una valoración es aquel en el que ocurre un cambio físico relacionado con la condición de equivalencia química. Se debe hacer lo posible para asegurar que cualquier diferencia de volumen o masa entre el punto de equivalencia y el punto final sea lo menor posible.
Error de valoración: Se expresa de la siguiente manera:
Er=V exp−V eq
V exp=Volumen final para alcanzar el punto final.V eq=Volumen necesario para alcanzar el punto equivalente.
Indicador: Es un reactivo auxiliar que exhibe un cambio como consecuencia de los cambios de concentración de las especies químicas, son sustancias que pueden existir en dos o más formas de equilibrio.
HInd+H 2O↔H 3+¿O+ Ind−¿¿¿
¿¿¿
Patrón Primario: Se utiliza como material de referencia en valoraciones gravimétricas y volumétricas, debe cumplir los siguientes requisitos:
Alto grado de pureza. Elevado peso molecular. Razonablemente soluble. Estable en el aire. Ausencia de agua de hidratación. Fácil de obtener y que sea barato.
Patrón Secundario: Es un compuesto cuya pureza ha sido determinada por un análisis químico y se usa como material de referencia en el análisis por valoración.
Alícuota: Fracción exacta de una solución cuyo volumen se conoce también con exactitud, en general se mide con una pipeta.
Titulación en Blanco: Es una titulación sin la muestra y se utiliza para detectar las impurezas que alteran el punto de equivalencia.
Métodos para establecer concentraciones
19

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
1. Método Directo: Se presenta cuando una cantidad cuidadosamente pesada del estándar primario se disuelve en un solvente apropiado hasta un volumen exacto.
2. Estandarización o normalización: Proceso mediante el cual se determina la concentración de la solución estándar que se utiliza para titular una muestra de patrón primario o secundario.
Unidades de Concentración
UNIDAD DE CONCENTRACION
FORMULA
Molaridad M=¿moles desoluto1 l de solución
Normalidad N=¿eq−gde soluto1 lde solución
Molalidad m=¿moles desoluto1Kgde solvente
Partes por millón ppm= masade solutomasadesolución
×106
Porcentaje en peso %pp=masa parcial
masa total×100
Partes por volúmen %pv= masa parcial
volúmen total×100
Porcentaje de volúmen
%vv= volúmen parcial
volúmen total×100
Clasificación de las volumetrías
Se conoce las siguientes:
Volumetría de precipitación y formación de complejos. Volumetría de neutralización. Volumetría de óxido - reducción. Volumetrías amperométricas. Valoraciones espectrofotométricas.
20

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
VOLUMETRÍA DE PRECIPITACIÓN
Introducción
Se basa en la aplicación de las reacciones que se acompañan por la sedimentación de precipitados o la formación de complejos, esta volumetría es escaza debido a:
Las reacciones en volumetría deben ser estequeometricas y para que esto ocurra no deben existir fenómenos de coprecipitación.
Las reacciones en volumetría deben ser rápidas y alcanzar el equilibrio en forma casi instantánea, pero las reacciones de precipitación suelen completarse lentamente.
NaCl+AgNO3↔AgCl↓+NaN O3
Ejemplos:
1) Una muestra de 27,73mg contiene FeCl2 y KCl. Disuelta en agua requirió 18,49ml de AgNO3 0,02237M para la titulación completa de sus cloruros.
Determine la masa de FeCl2 y el %pp
de Fe2+¿¿ en la muestra.
Fe2+¿=55,85 ¿; FeCl2=126,75; KCl=74,55; Cl−¿=35,4513¿; K+¿=39,0983¿
FeCl2+2 AgNO3↔2 AgCl+Fe (N O3 )2
KCl+AgNO3↔AgCl+KNO3
21

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
18,49mlsol×0,02237mmolAgN O3
1mlsol=0,4136mmolAgN O3
XmgFe Cl2×2mmol
Cl−¿
126,75mgFeCl2=0,01578 X ¿
YmgKCl×1mmolCl−¿
74,55mgKCl=0,01341Y ¿
X+Y=27,73
0,01578 X+0,01341Y=0,4136
X=17,61mgFeCl2 Y=10,12mgKCl%FeCl2=63,51 %KCl=36,49
17,61mgFeCl2×55,85mgFe2+¿
126,75mgFeCl2=7,760mgFe2+¿¿¿
%Fe2+¿=27,98 ¿
2) Se disuelve una muestra de 0,410 g de KBr (impuro) en 25,00ml de agua y se agregan a la disolución 50ml de AgNO3 0,0492N en exceso para precipitar todo el Br−¿¿ presente en la muestra. El exceso de AgNO3 se valoró utilizando 7,50ml de KSCN 0,0600N para valorar el exceso de Ag+¿¿. ¿Calcular el porcentaje de pureza de la muestra original?
AgNO3+KSCN↔ AgSCN+KN O3
7,50mlKSCN×0,0600eqgKSCN1000mlKSCN
×1molKSCN1eqgKSCN
×1molAgNO3
1molKSCN=4,5×10−4
¿moles=4,5×10−4molAgNO3
50mlsol×0,0492eqgAgNO3
1000mlsol×1molAgN O3
1eqgAgN O3
=2,46×10−3
¿moles1=2,46×10−3molAgNO3
¿molesAgN O3=¿moles1−¿moles
¿molesAgN O3=2,01×10−3mol
KBr+AgN O3↔ AgBr+KN O3
22

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
2,01×10−3molAgN O3×1molKBr1molAgN O3
×119,006 gKBr1molKBr
=mpura=0,239 gKBr
pureza= masa puramasa impura
×100%
pureza=0,239 g0,410 g
×100%
pureza=58,29% (KBr)
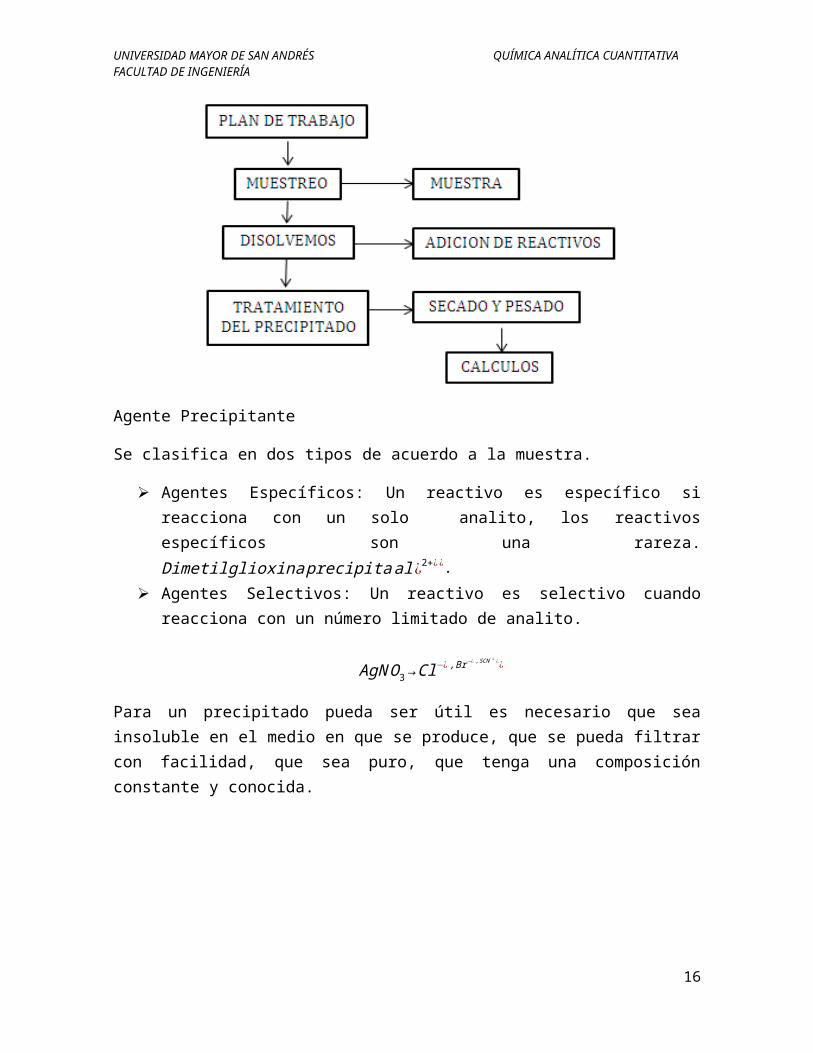
Curvas de Valoración por Precipitación
La curva de valoración es una representación gráfica de cómo varia la concentración de uno de los reactivos a medida que se añade el valorante. Se presentan dos tipos de curva:
Curva Signoidea: Se caracteriza porque las observaciones (cambios) se hallan en una pequeña región normalmente entre ±0,1 a ±0,5ml en torno al punto de equivalencia.
0 5 10 15 20 25 30 35 40 450
2
4
6
8
10
12
V (añadido)
pX
0,1-0,5
Curva de segmento lineal: Se caracteriza porque las medidas cerca del punto de equivalencia se evitan, realizando las medidas a ambos lados del punto de equivalencia y alejados de esta.
23

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
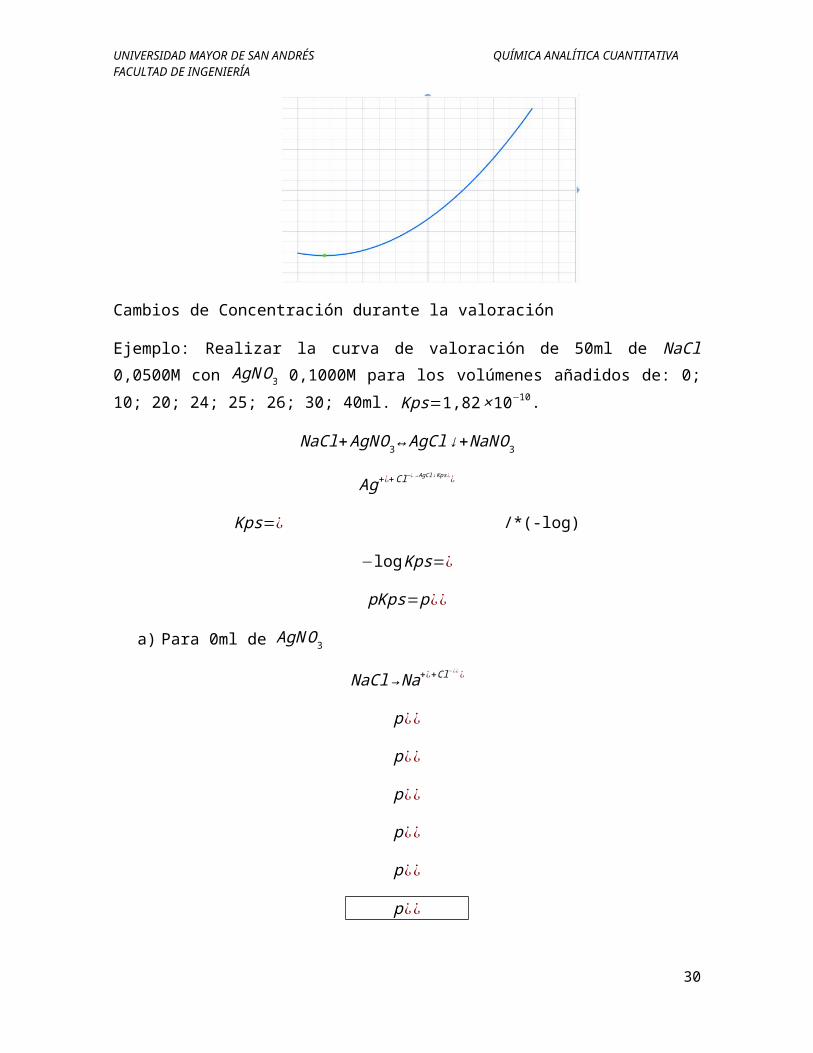
Cambios de Concentración durante la valoración
Ejemplo: Realizar la curva de valoración de 50ml de NaCl 0,0500M con AgNO3 0,1000M para los volúmenes añadidos de: 0; 10; 20; 24; 25; 26; 30; 40ml.
Kps=1,82×10−10.
NaCl+AgNO3↔AgCl↓+NaN O3
Ag+¿+Cl−¿ →AgCl↓ Kps¿ ¿
Kps=¿ /*(-log)
−logKps=¿
pKps=p¿¿
a) Para 0ml de AgNO3
NaCl→Na+¿+Cl−¿¿ ¿
p¿¿
p¿¿
p¿¿
p¿¿
p¿¿
p¿¿
b) Antes del punto equivalente
¿¿
24

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Para 10ml de AgNO3
¿¿
p¿
p¿
p¿¿
Para 20ml de AgNO3
¿¿
p¿
p¿¿
Para 24ml de AgNO3
¿¿
p¿
p¿¿
c) En el punto equivalente
¿¿
¿¿
p¿
p¿¿
d) Después del punto de equivalencia
¿¿
Para 26ml de AgNO3
¿¿
25

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
p¿¿
p¿¿
Para 30ml de AgNO3
¿¿
p¿¿
p¿¿
Para 40ml de AgNO3
¿¿
p¿¿
p¿¿
26

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 10 20 30 40 50 600
1
2
3
4
5
6
7
8
9
pAg
0,0500NaCl; 0,1000AgNO3 0,00500NaCl; 0,01000AgNO3
Volumen añadido[ml]
pAg
Efecto del producto de solubilidad en las curvas de valoración
27

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 10 20 30 40 50 600
2
4
6
8
10
12
14
16
PRODUCTO DE SOLUBILIDAD
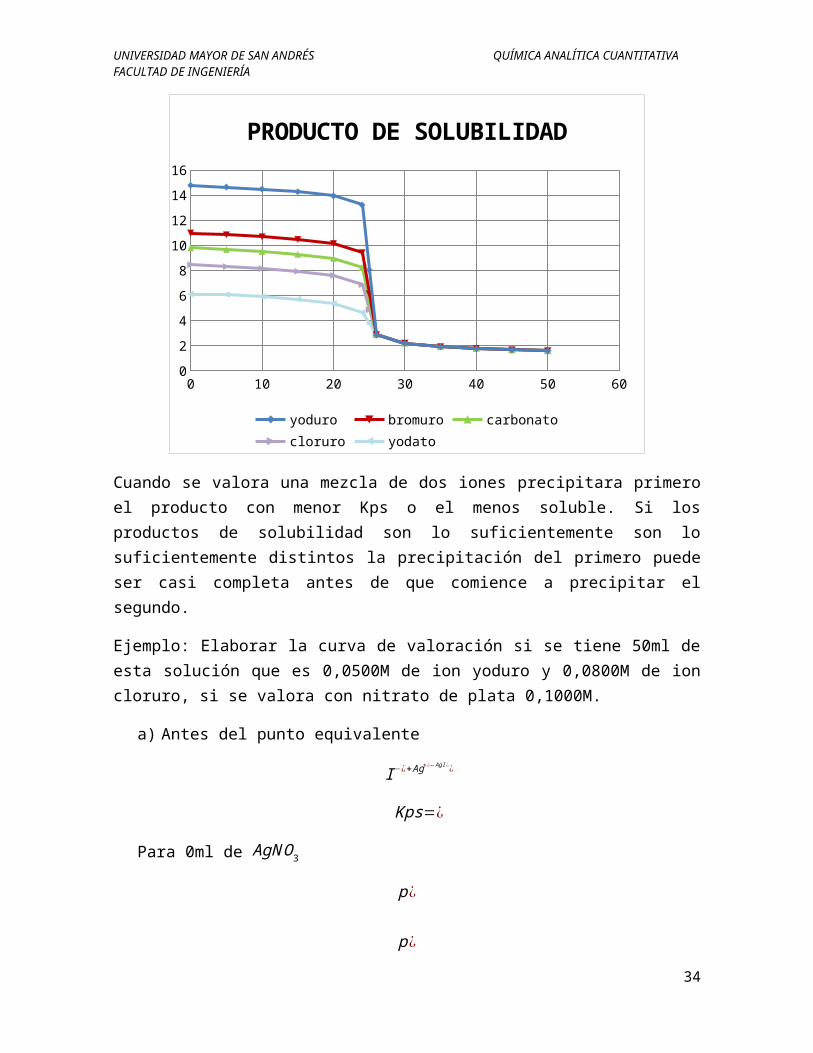
yoduro bromuro carbonatocloruro yodato
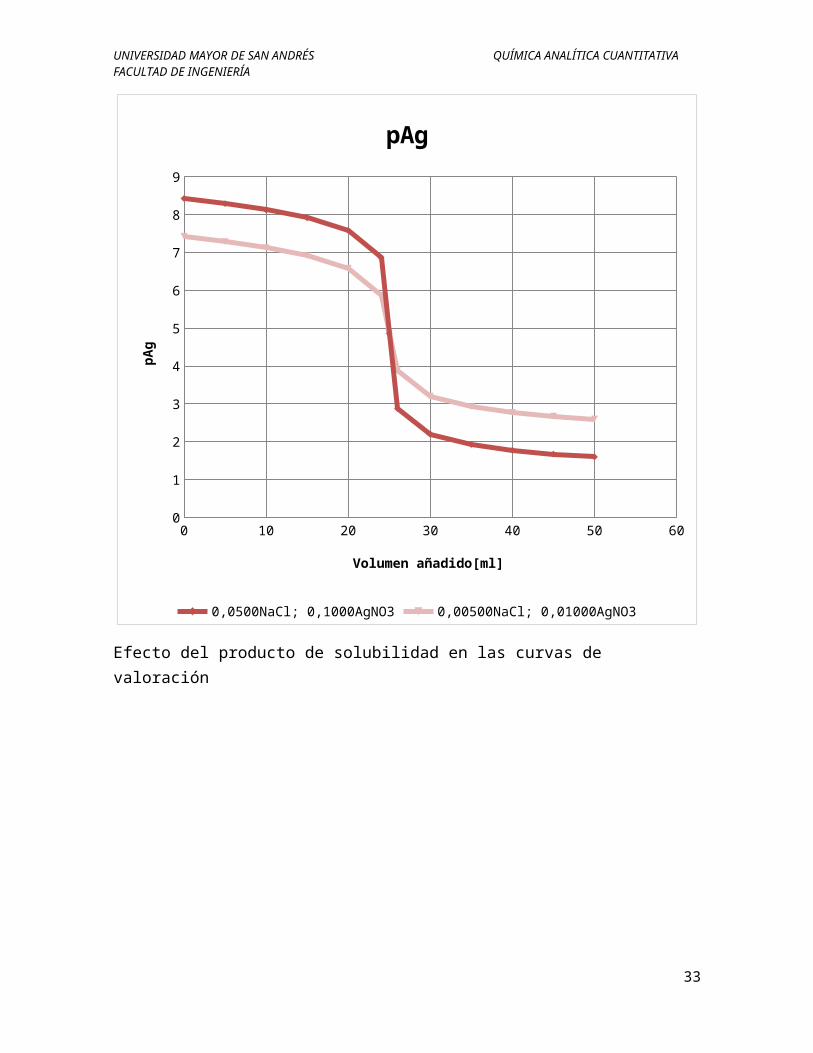
Cuando se valora una mezcla de dos iones precipitara primero el producto con menor Kps o el menos soluble. Si los productos de solubilidad son lo suficientemente son lo suficientemente distintos la precipitación del primero puede ser casi completa antes de que comience a precipitar el segundo.
Ejemplo: Elaborar la curva de valoración si se tiene 50ml de esta solución que es 0,0500M de ion yoduro y 0,0800M de ion cloruro, si se valora con nitrato de plata 0,1000M.
a) Antes del punto equivalente
I−¿+Ag+¿↔AgI ¿¿
Kps=¿
Para 0ml de AgNO3
p¿
p¿
p¿¿b) En el punto equivalente: ¿
I−¿+Ag+¿↔ AgI K ps1=¿¿ ¿
28
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Cl−¿+Ag+¿↔AgCl K ps2=¿¿ ¿
K ps1
K ps2
=¿¿
¿
V eq I−¿C1=V 2C2¿
Veq I−¿=50ml∗0,0500M
0,1000M=25ml¿
¿
¿
La cantidad de yoduro que no ha precipitado la hallamos en la ecuación (3)
¿
La concentración de plata será:
p¿
p¿¿
c) Después del puntos equivalente:
¿
¿
El potencial de plata será:
p¿
p¿¿
29
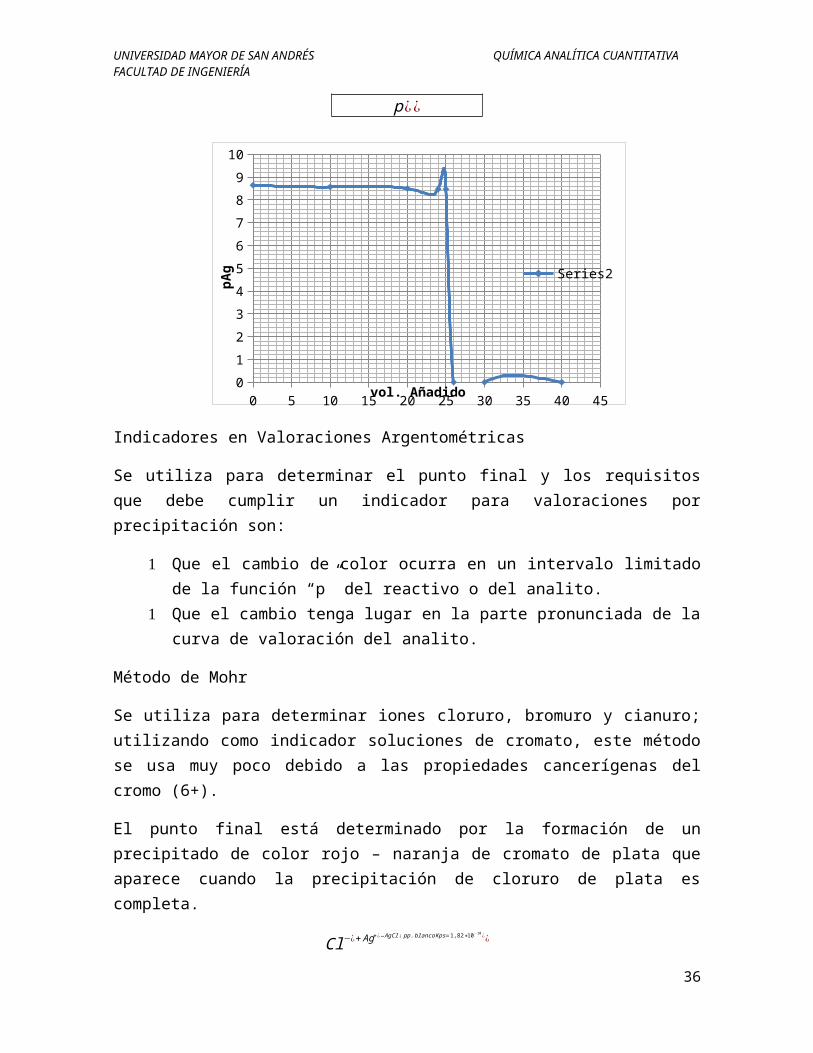
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 5 10 15 20 25 30 35 40 450
1
2
3
4
5
6
7
8
9
10
Series2
vol. Añadido
pAg
Indicadores en Valoraciones Argentométricas
Se utiliza para determinar el punto final y los requisitos que debe cumplir un indicador para valoraciones por precipitación son:
Que el cambio de color ocurra en un intervalo limitado de la función “p” del reactivo o del analito.
Que el cambio tenga lugar en la parte pronunciada de la curva de valoración del analito.
Método de Mohr
Se utiliza para determinar iones cloruro, bromuro y cianuro; utilizando como indicador soluciones de cromato, este método se usa muy poco debido a las propiedades cancerígenas del cromo (6+).
El punto final está determinado por la formación de un precipitado de color rojo – naranja de cromato de plata que aparece cuando la precipitación de cloruro de plata es completa.
Cl−¿+Ag+¿↔AgCl ↓ pp.blanco Kps=1,82∗10−10¿ ¿
CrO 42−¿+2Ag+¿↔Ag2 CrO 4↓ pp .rojo−naranjaKps=1,2∗10−12¿ ¿
Para las valoraciones por el método de Mohr el pH se debe encontrar entre los valores de 7 a 10. En el punto equivalente la concentración de la plata es igual a la raíz cuadrada de Kps.
La concentración de ion cromato necesario para la formación del cromato de plata es:
30

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿
A esta concentración (∝) la disolución tiene una coloración muy intensa que hace difícil detectar la formación de Ag2Cr O4 por lo que se utiliza soluciones un poco mas diluidas (aproximadamente 5∗10−3M).
Como consecuencia se requiere un exceso de AgNO3 para que empiece la precipitación.
Para eliminar el error sistemático en el método de Mohr, se puede utilizar una valoración en blanco con una solución de CaCO3 exenta de cloruros.
Método de Volhart
Es un método indirecto para la determinación de cloruros en el que se añade AgNO3 en exceso respecto a la cantidad de cloruros presente.
El exceso se valora por retroceso con disolución patrón de KSCN y alumbre de hierro
(FeK (SO 4 )2), (sulfato doble de hierro y aluminio) como indicador.
Ag+¿+SCN−¿↔AgSCN ↓ pp.blanco ¿¿
Fe3+¿+SCN−¿↔Fe( SCN )3↓ pp.rojo¿ ¿
Esta valoración se debe llevar a cabo en la disolución acida para evitar la precipitación del hierro como oxido hidratado.
Ag¿¿
Ag+¿+SCN−¿↔AgSCN ↓¿ ¿
Ojo: cuando se consume toda la plata recién reacciona con el hierro
VOLUMETRIA ACIDO BASE
Teoría de Arrhenius
Acido :HCl↔H+¿+Cl−¿¿ ¿
31

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Base :NaOH↔Na+¿+OH−¿¿ ¿
Bronsted Lowry
Acido :HCl+H 2O↔H 3+¿O+Cl−¿¿¿
acido base acidobase
Base :N H 3+H 2O↔OH−¿+NH 4+¿¿ ¿
base acido baseacido
AcidoBase
→ParConjugado
Solución patrón
Las disoluciones patrón usadas en las valoraciones Ácido Base son siempre ácidos o bases fuertes y generalmente son:NaOH , KOH ,HCl , H2SO 4 ,HClO4; los ácidos y bases débiles no se usan como sustancias porque reaccionan en forma incompleta con los analitos.
Indicadores ácido base
Son sustancias naturales, artificiales o sintéticas que presentan una coloración que dependen del pH en la solución en que se encuentran.
HInd+H 2O↔H 3+¿O+ Ind−¿¿¿
acido base
Ind−¿+H 2O↔HInd+OH−¿¿¿
base acido
Ka=¿¿ ¿
Acido :[HInd ]
¿¿
¿
p¿
p¿
Errores de valoración con Indicadores Ácido Base
32
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Un error cuando el pH con el cual se modifica el color del indicador difiere del pH correspondiente al punto de equivalencia. Este error se minimiza si se selecciona cuidadosamente el indicador o si se realiza una corrección mediante una titulación en blanco.
Un error indeterminado que se origina por la limitada capacidad del ojo humano para distinguir de manera reproducible el color indeterminado del indicador. Este error puede disminuirse si se compara el color de la disolución que se valora con el de un patrón de referencia que tiene una cantidad similar del indicador con el pH apropiado.
Valoraciones Ácido Fuerte Base Fuerte
La característica principal de estos ácidos y bases es la disociación total.
HCl↔H+¿+Cl−¿ NaOH↔Na+¿+OH−¿¿¿¿¿
pH=−log [Co ] pOH=−log [Co ]
H 2O↔H+¿+OH−¿¿ ¿
Kw=¿
14=pH+pOHEtapas de valoración
No se añade ningún reactivo. Antes del punto equivalente. En el punto de equivalencia. Después del punto equivalente.
Ejemplo: Realizar la curva de valoración para 50ml de HCl 0,0500M a la que se añade una solución de NaOH 0,1000M.
V eqCNaOH=V HClCHCl
V eq=50ml×0,0500M0,1000M
=25ml
HCl+NaOH↔NaCl+H 2O
a) No se añade ningún reactivo:
33
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
pH=−log [0,0500 ]
pH=1,30
b) Antes del punto equivalente
¿
Para 5ml de NaOH
¿
pH=−log [0,0364 ]
pH=1,44
Para 20ml de NaOH
¿
pH=−log [7,143×10−3 ]
pH=2,15
c) En el punto de equivalencia: la base y el ácido se neutralizan
pH=7,00
d) Después del punto de equivalencia
¿
Para 30ml de NaOH
¿
pH=14−pOH
pH=14−(−log [6,25×10−3 ])
34

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
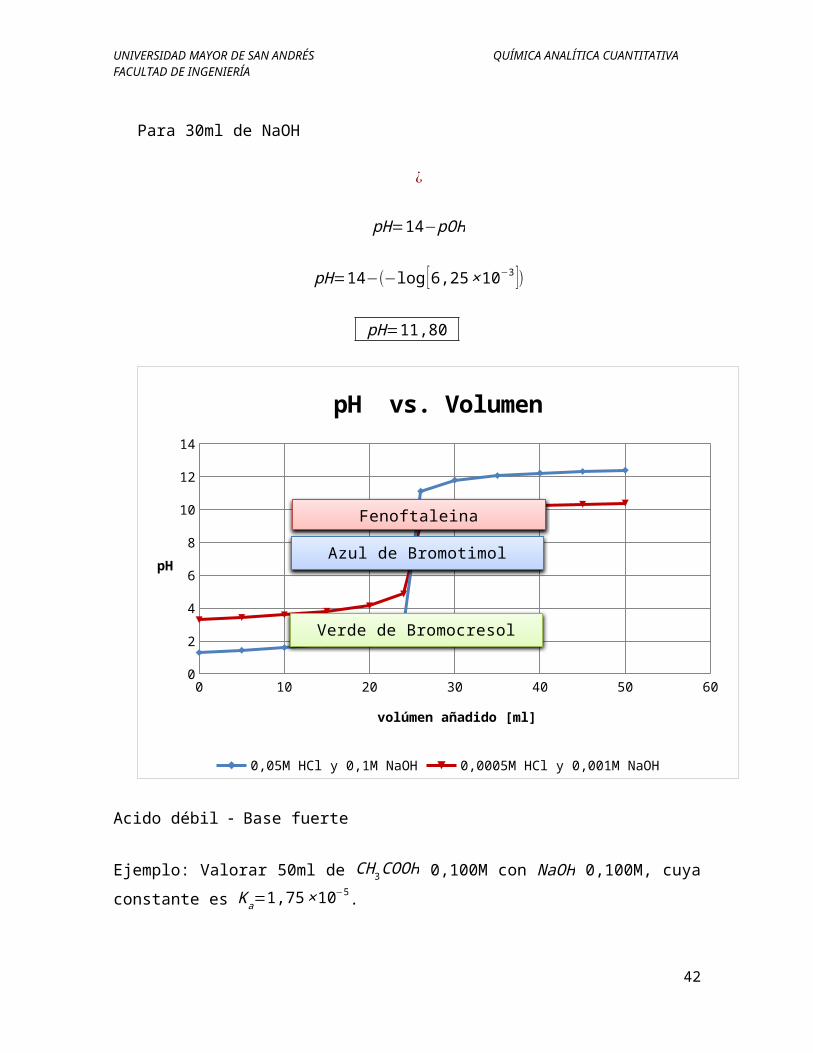
pH=11,80
0 10 20 30 40 50 600
2
4
6
8
10
12
14
pH vs. Volumen
0,05M HCl y 0,1M NaOH 0,0005M HCl y 0,001M NaOH
volúmen añadido [ml]
pH
Verde de Bromocresol
Azul de Bromotimol
Fenoftaleina
Acido débil Base fuerte
Ejemplo: Valorar 50ml de CH 3COOH 0,100M con NaOH 0,100M, cuya constante es
Ka=1,75×10−5.
a) Sin añadir reactivo
Para 0ml de NaOH
H 2O↔H+¿+OH−¿¿ ¿
CH 3COOH+H 2O↔H 3+¿O+CH 3COO−¿ ¿¿
c 00−x x xc−x x x
35
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ka=¿¿
Ka=x2
Ca−x
x=√Ka×Ca
x=√ (1,75×10−5 ) (0,100 )=1,323×10−3M
pH=−log [1,323×10−3 ]
pH=2,88
b) Antes del punto de equivalencia
Para 25ml de NaOH
CHAc=¿moles iniciales HAc−¿molesañadido NaOH
Volúmen total
CHAc=(50×0,100)−(25×0,100)
50+25=0,0333M
CAc−¿=¿moles añadido NaOH
Volúmen total¿
CAc−¿=
(25× 0,100)50+25
=0,0333M ¿
¿
¿
pH=−log [1,75×10−5 ]
pH=4,76
c) En el punto de equivalencia
CCH3COONa=¿moles de NaOH añadidos
V T
=¿moles de HAcinicialesV T
36

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
CCH3COONa=50×0,10050+50
=0,050M
CH 3COO−¿+H 2O↔CH3COOH+OH−¿ ¿¿
cb00
−x x xcb−x x x
Kb=x2
Cb−x
x=√ K w
K a
×Cb
x=√ 1×10−14
1,75×10−5×0,050=5,345×10−6M
pH=14−(−log [5,345×10−6 ])
pH=8,73
d) Después del punto de equivalencia
CH 3COO−¿+H 2O↔CH3COOH+OH−¿ ¿¿
¿¿
CNaOH=¿moles debase−¿moles de ácido
Volumen total
CNaOH=(51×0,100)−(50×0,100)
50+51=9,901×10−4M
pH=14−(−log [9,901×10−4 ])
pH=11,00
37

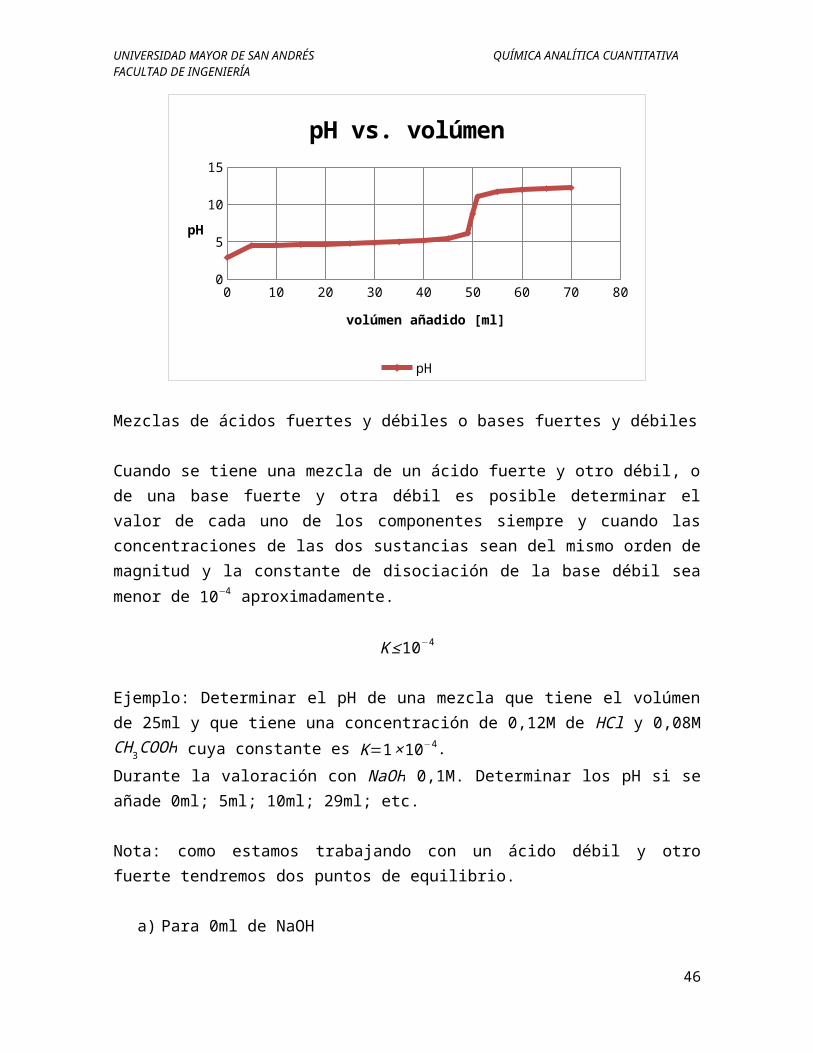
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 10 20 30 40 50 60 70 8002468
101214
pH vs. volúmen
pH
volúmen añadido [ml]
pH
Mezclas de ácidos fuertes y débiles o bases fuertes y débiles
Cuando se tiene una mezcla de un ácido fuerte y otro débil, o de una base fuerte y otra débil es posible determinar el valor de cada uno de los componentes siempre y cuando las concentraciones de las dos sustancias sean del mismo orden de magnitud y la constante de disociación de la base débil sea menor de 10−4 aproximadamente.
K ≤10−4
Ejemplo: Determinar el pH de una mezcla que tiene el volúmen de 25ml y que tiene una concentración de 0,12M de HCl y 0,08M CH 3COOH cuya constante es K=1×10−4
.Durante la valoración con NaOH 0,1M. Determinar los pH si se añade 0ml; 5ml; 10ml; 29ml; etc.
Nota: como estamos trabajando con un ácido débil y otro fuerte tendremos dos puntos de equilibrio.
a) Para 0ml de NaOH
CH 3COOH+H 2O↔H 3+¿O+CH 3COO−¿ ¿¿
c 00−x x xc−x x x
38

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ka=x2
Ca−x
x=√Ka×Ca
x=√ (1×10−4 ) (0,08 )=2,828×10−3M
HCl↔H+¿+Cl−¿¿ ¿
¿¿
pH=−log [ (2,828×10−3 )+(0,12)]
pH=0,91
Ka=¿¿
¿¿
[CH 3COOH ]T=¿0,08=¿
¿
b) Antes del primer punto equivalente
Para 5ml de NaOH
[ HCl ]=¿moles HCl inicial−¿moles NaOH añadidoV T
[ HCl ]= (25×0,12 )−(5×0,1)25+5
=0,0833M
[CH 3COOH ]=¿moles de [CH 3COOH ]
V T
[CH 3COOH ]=25×0,0825+5
=0,0667M
39

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿¿
pH=−log [(0,0833)]
pH=1,08
Hallando la concentración de acetato para confirmar si es despreciable
0,0667=¿
¿
Para 10ml de NaOH
[ HCl ]= (25×0,12 )−(10×0,1)25+10
=0,0571M
[CH 3COOH ]=25×0,0825+10
=0,0571M
¿¿
pH=−log [(0,0571)]
pH=1,24
Hallando la concentración de acetato para confirmar si es despreciable
0,0571=¿
¿
Para 29ml de NaOH
[ HCl ]= (25×0,12 )−(29×0,1)25+29
=1,852×10−3M
[CH 3COOH ]=25×0,0825+29
=0,0370M
40

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿¿
pH=−log [(1,852×10−3)]
pH=2,73
Hallando la concentración de acetato para confirmar si es despreciable
0,0370=¿
¿
Comparando podemos verificar que el aporte del ácido acético no es despreciable por lo que se tiene la siguiente relación.
¿¿
Reemplazando datos:
¿¿
¿
pH=−log [(1,945×10−3)]
pH=2,71
c) En el primer punto equivalente
El ácido clorhídrico se neutraliza y solo queda ácido acético.
HCl+NaOH↔NaCl+H 2O
CH 3COOH+H 2O↔H 3+¿O+CH 3COO−¿ ¿¿
c 00−x x xc−x x x
Ka=x2
Ca−x
41
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
[CH 3COOH ]=25×0,0825+30
=0,0364M
x2+Ka x−KaC HAc=0
x=¿
pH=−log [(1,858×10−3)]
pH=2,73
d) Después del punto equivalente del HCl y Antes del punto equivalente del CH 3COOH .
CHAc=¿molesdel HAc−(¿moles deañadido−¿moles de HCl)
V inicial+V añadido
Para 35ml de NaOH
CHAc=(25×0,08 )− ( (35×0,1 )− (25×0,12 ) )
25+35=0,025M
x2+Ka x−KaC HAc=0
x=¿
pH=−log [(1,532×10−3)]
pH=2,81
Para 40ml de NaOH
CHAc=(25×0,08 )− ( (40×0,1 )−(25×0,12 ) )
25+40=0,0154M
x2+Ka x−KaC HAc=0
x=¿
42

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
pH=−log [(1,192×10−3)]
pH=2,92
Para 49ml de NaOH
CHAc=(25×0,08 )− ( (49×0,1 )−(25×0,12 ) )
25+49=1,351×10−3M
x2+Ka x−KaC HAc=0
x=¿pH=−log [(3,209×10−4)]
pH=3,49
e) Después del punto de equivalencia del CH 3COOH .
CNaOH=¿moles deañadido−(¿moles delHAc+¿molesde HCl)
V inicial+V añadido
Para 51ml de NaOH
CNaOH=(51×0,1)−((25×0,08)+(25×0,12))
25+51=1,316×10−3M
pH=14−(−log [1,316×10−3 ])
pH=11,12
43

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 10 20 30 40 50 60 70 800
2
4
6
8
10
12
14
Volúmen añadido [ml]
pH
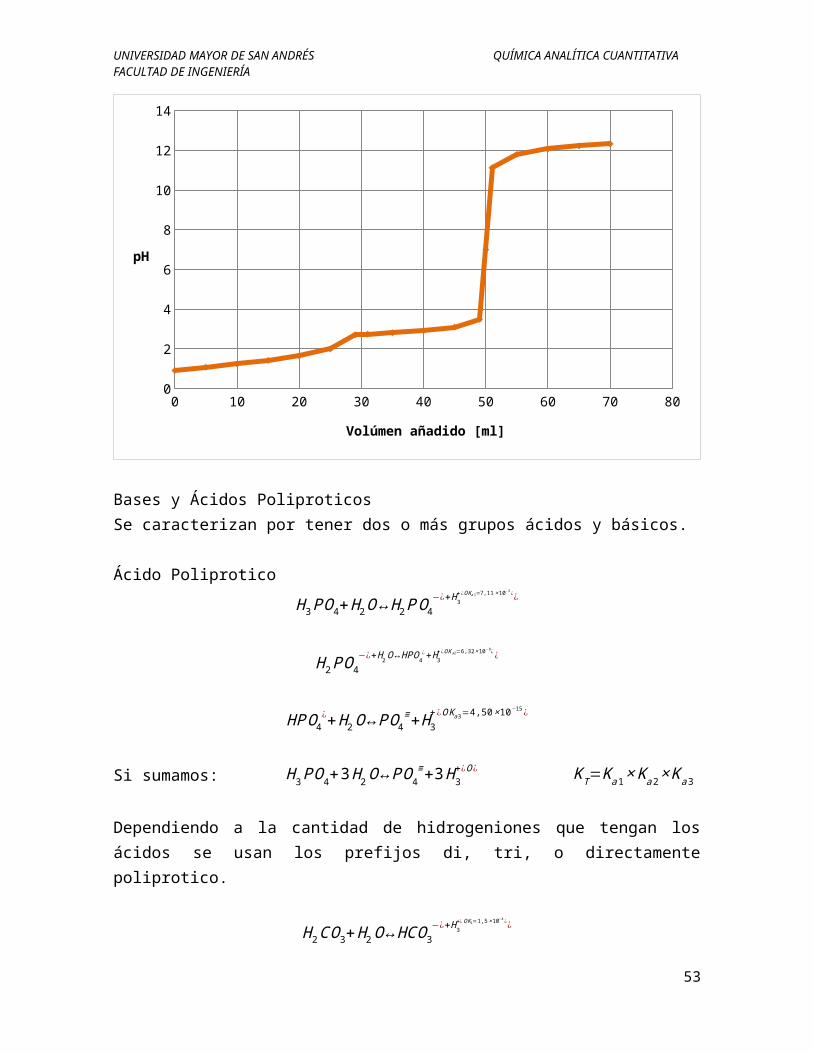
Bases y Ácidos PoliproticosSe caracterizan por tener dos o más grupos ácidos y básicos.
Ácido Poliprotico
H 3PO 4+H 2O↔H 2 PO4−¿+H 3
+¿O Ka1=7,11×10−3¿ ¿
H 2PO4−¿+H 2O↔HPO 4
¿+H 3+¿OK a2=6,32×10
−8¿ ¿
HPO 4¿+H 2O↔PO 4
≡+H 3+¿OK a3= 4,50× 10
−15 ¿
Si sumamos: H 3PO 4+3H 2O↔PO4≡+3H 3
+¿O¿ KT=Ka1×Ka2×Ka3
Dependiendo a la cantidad de hidrogeniones que tengan los ácidos se usan los prefijos di, tri, o directamente poliprotico.
H 2CO3+H 2O↔HCO3−¿+H 3
+¿O K1=1,5× 10−4 ¿¿
HCO3−¿+H 2O↔CO3
¿+H3+¿OK 2=4,69×10
−11 ¿¿
Sumando: H 2CO3+2H 2O↔CO3¿+2H 3
+¿O¿ KT=K1×K2
44

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ejemplo: Determinar el pH de una solución 0,3M CO2, siendo KT=7,035×10−15 y
Kh=2,8×10−3.
CO2+H 2O↔H 2C O3
H 2CO3+H 2O↔HCO3−¿+H 3
+¿O ¿ ¿
CO2ac+2H 2O↔HCO3−¿+H 3
+¿O ¿ ¿
KT=¿¿
Balance de masa CO2
CO2= [C O2ac ]+¿
Balance de carga
¿
De (1): De (2):
CO2= [C O2ac ] ¿Entonces:
¿
¿
pH=−log [(3,550×10−4)]
pH=3,45
Estas suposiciones son válidas cuando las constantes sucesivas del ácido o la base difieren en una factor de 103.
Disolución Tampón
45
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Se denomina solución tampón a una disolución a la cual se puede añadir o eliminar iones hidronio por adición de un ácido o una base sin que se modifique notablemente el pH.
Ejemplos:
1) Calcular la concentración de iones hidronio en una disolución tampón de H 3PO 4 2M y K H 2PO4 1,5M.
H 3PO 4+H 2O↔H 2 PO4−¿+H 3
+¿O ¿ ¿
¿
¿
pH=−log [(9,48×10−3) ]
pH=2,02
H 2PO4−¿+H 2O↔HPO 4
¿+H 3+¿O ¿¿
K2=[HPO4¿ ]¿¿
[HPO 4¿]=K2¿¿
[HPO 4¿]=(6,32×10−8)(1,50)
(9,48×10−3)=1×10−5M
El valor de [HPO 4¿] es despreciable por lo tanto el primer análisis es el correcto.
2) Calcular la concentración de iones hidronio de una solución tampón de ftalato
ácido de potasio 0,05M (KHP) y ftalato de potasio 0,15M. K2=3,92×10−6,
K1=1,12×10−3.
HP−¿+H 2O↔P¿+H 3+¿O ¿¿
K2= [P¿ ]¿¿
46

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿
¿
pH=−log [(1,307×10−6) ]
pH=5,88
Curvas de valoración (cálculo de pH)
H 2 A+NaOH↔NaHA (1)
NaHA↔Na+¿+HA−¿(2)¿ ¿
HA−¿+H 2O→A ¿+H 3+¿OHA
−¿+ H2O→H
2A+OH
−¿¿¿¿ ¿
K2= [ A¿ ]¿¿[ A¿ ]=K2¿¿
Balance de masa y balance de carga
CNaHA=¿
¿
H 2O↔H+¿+OH−¿¿ ¿
Kw=¿
Reemplazando (5) en (6):
¿
¿
Reemplazando (3), (4) y (7) en (8):
¿
47

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Realizando operaciones y despejando los hidrogeniones tenemos:
¿
Que es una fórmula para hallar el pH en el primer punto de equivalencia, aunque debe tenerse en cuenta ciertas consideraciones.
Si CNaHA≅ ¿. Si CNaHA /K1>>1 y KaCNaHA>K w ¿.
Ejemplo: elaborar la curva de valoración de 25ml de ácido málico 0,1M con NaOH 0,1M.
H 2M+H 2O→HM−¿+H 3+¿OK1=1,3×10
−2 ¿¿
HM−¿+H 2O→M ¿+H 3+¿O K2=5,9× 10
−7¿ ¿
a) 1˚ punto para 0ml de base añadida
La 1˚ disociación es la más importante
CH 2M=¿
[H 2M ]=CH 2M−¿
[H 2M ]=CH 2M−¿
K1=¿¿ Haciendo los cálculos correspondientes:
¿¿
¿¿
¿
pH=−log [(0,0301)]
pH=1,52
48

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
b) Antes del 1˚ punto equivalente
2˚ punto: para 5ml de NaOH
CH 2M=¿moles iniciales−¿molesde NaOH
V inicial+V añadido
CH 2M=
(25∗0,1)−(5∗0,1)25+5
=0,0667M
CNaHM=¿moles de NaOHV inicial+V añadido
CNaHM=(5∗0,1)25+5
=0,0167M
H 2M+H 2O→HM−¿+H 3+¿O ¿¿
HM−¿+H 2O→H 2M +OH−¿¿ ¿
CNaHM=¿
CH 2M=[H 2M ]−¿
De ( ): α ¿ y de ( ): β [H 2M ]=CH 2M−¿
K1=¿¿
Ordenando:
¿¿
¿¿
¿
pH=−log [(0,0181)]
49

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
pH=1,74
3˚ punto: para 15ml de NaOH
CH 2M=
(25∗0,1)−(15∗0,1)25+15
=0,025M
CNaHM=(15∗0,1)25+15
=0,0375M
¿¿
¿
pH=−log [(5,775×10−3)]
pH=2,24
4˚ punto: para 20ml de NaOH
CH 2M=
(25∗0,1)−(20∗0,1)25+20
=0,0222M
CNaHM=(20∗0,1)25+20
=0,0444M
¿¿
¿
pH=−log [(4,651×10−3) ]
pH=2,33
4˚ punto: para 24,9ml de NaOH
H 2M+H 2O→HM−¿+H 3+¿O ¿¿
HM−¿+H 2O→M ¿+H 3+¿O ¿ ¿
50

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
K1=¿¿
K2= [M ¿ ]¿¿
Balance de masa y de carga:
CH 2M+C HM−¿=¿ ¿
¿
CH 2M=
(25∗0,1)−(24,9∗0,1)25+24,9
=2,004×10−4M
CNaHM=(24,9∗0,1)25+24,9
=0,0499M
Reemplazando (*) y (**) en (2):
¿
Ordenando:
¿¿
¿¿
¿
pH=−log [(1,014×10−4)]
pH=3,99
c) En el 1˚ punto equivalente
CNaHM=¿moles de NaOHV inicial+V añadido
CNaHM=(25∗0,1)25+25
=0,05M
¿
51

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿
pH=−log [(7,80×10−5)]
pH=4,11
d) Para 25,01ml añadidos
NaHM+NaOH↔Na2M+H 2O
Balance de masa:
CNaHM+CNa2M=¿
Balance de carga:
¿
Luego tendremos:
CNa2M=¿molesde NaOH añadidos−¿moles de NaHM formados
V o+V añadido
CNa2M=
(25,01×0,1)−(25×0,1)25+25,01
=1,9996×10−5M
CNaHM=¿moles de NaHM−¿moles de NaOH añadidos−¿molesde NaHM
V o+V añadido
V o+V añadido
CNaHM=(25×0,1 )−1,9996×10−5
25+25,01=0,04999M
¿
¿
Restando B.M. de B.C.
52
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
CNaHM+CNa2M−¿
¿¿
Ordenando:
¿¿
Reemplazando valores:
¿¿
¿
pH=−log [(7,434×10−5)]
pH=4,13
e) Después del 1˚ punto equivalente
¿
|M−2|≡C Na2M=9,9×10−4M
pH=4,54
Para 49,9ml
M−2+H 2O↔HM−¿+OH−¿ ¿¿
Kb1=Kw
Ka2
=¿¿
¿
|M−2|=CNa2M−OH−¿¿
Reemplazando y ordenando tenemos la siguiente ecuación:
53

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
¿¿
CNa2M=¿molesde NaOH añadidos−¿moles de NaHM formados
V o+V añadido
CNa2M=
(49,9×0,1)−(25×0,1)25+49,9
=0,0332M
CNaHM=¿moles de NaHM−¿moles de NaOH añadidos−¿molesde NaHM
V o+V añadido
V o+V añadido
CNaHM=(25×0,1 )−0,0332
25+49,9=1,33×10−4M
¿¿
¿
pH=14−(−log [4,1×10−6 ])
pH=8,61
f) 2˚ punto equivalente (50ml)
NaHM+NaOH↔Na2M+H 2O
|M−2|=¿molesañadidos−¿moles inicialesV T
|M−2|= (50×0,1 )−(25×0,1)25+50
=0,0333M
¿
¿pH=14−(−log [2,377×10−5 ])
pH=9,38
54

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
g) Después del 2˚ punto equivalente
Para 50,01ml de NaOH
NaHM+NaOH↔Na2M+H 2O
M−2+H 2O↔HM−¿+OH−¿ ¿¿
C|M−2|=¿moles formados
V T
C|M−2|=(50×0,1 )25+50,01
=0,0667M
¿¿
¿¿
¿¿
Kb=¿¿
Balance de masa:
|M−2|=CM−2−¿
¿¿
Reemplazando en la anterior ecuación y ordenando porque además ¿.
¿¿
¿¿
¿
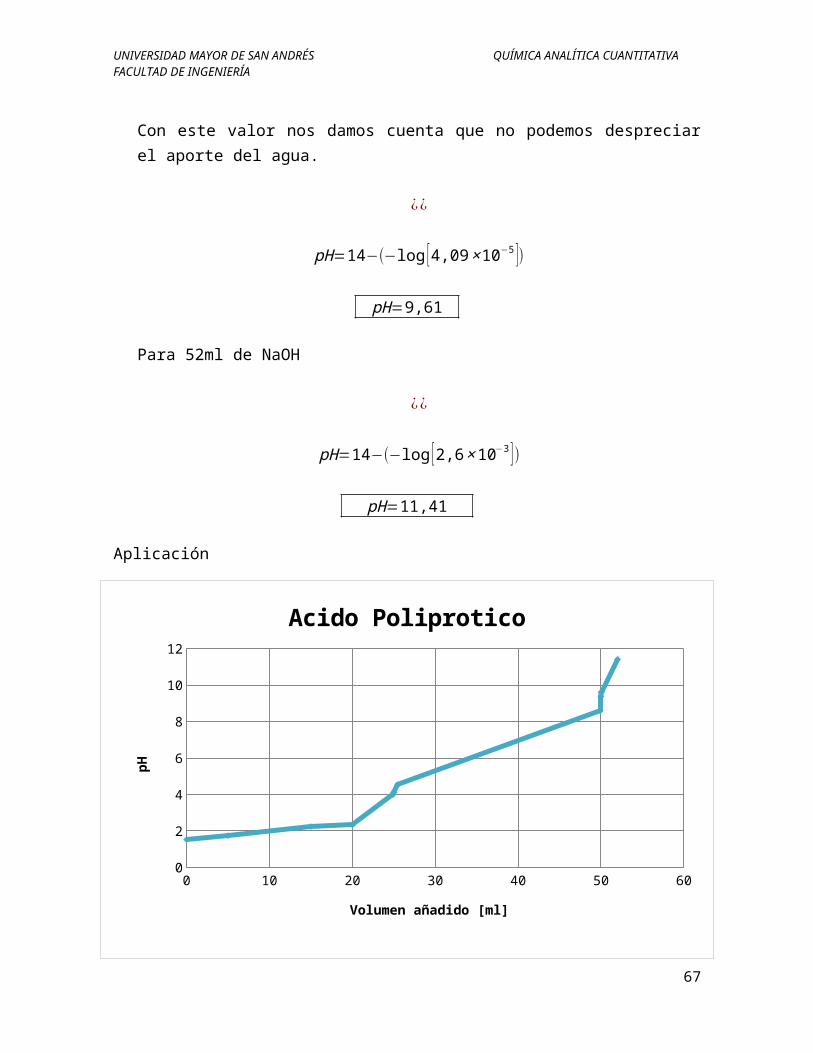
Con este valor nos damos cuenta que no podemos despreciar el aporte del agua.
¿¿
55

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
pH=14−(−log [4,09×10−5 ])
pH=9,61
Para 52ml de NaOH
¿¿
pH=14−(−log [2,6×10−3 ])
pH=11,41
Aplicación
NaOH }básico→fenoftaleina
HClNa2CO3 , NaHCO3 }Naranjade metiloo verdedebromocresol
Ejemplo:
56
0 10 20 30 40 50 600
2
4
6
8
10
12
Acido Poliprotico
Volumen añadido [ml]
pH

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Una muestra de 1,2g de una mezcla de hidróxido de sodio con carbonato de sodio que contiene impurezas inertes, se disuelve y se titula en frio con ácido clorhídrico 0,5N. Con fenolftaleína como indicador la solución se vuelve incolora después de la adición de 30ml de ácido. Se agrega naranja de metilo como indicador y se requiere 5ml más del ácido antes que este indicador cambie de color. ¿Determinar el porcentaje de hidróxido de sodio y de carbonato de sodio en la muestra?
NaOH+HCl→NaCl+H 2O
30mlHCl+Na2C O3→NaHCO3+H 2O
NaHCO3+HCl→Na2C O3→CO2↑+NaCl
CO3−2+H 2O→HCO3
−¿+OH−¿¿ ¿
HCO3−¿+H 2O→H 2CO3+OH−¿¿ ¿
5ml HCl−→NaHCO3
10ml HCl5ml HCl−→Na2C O3
V T (HCl )=35ml
V T (HCl )=V NaOH+V HCO 3−¿+V
CO3−2 ¿
V NaOH=V T (HCl )−V HCO3−¿−V
CO3−2=25ml ¿
Eq−gHCl=Eq−gNaOH
V∗C=25ml∗0,5mEq−gml
=12,5mEq−gNaOH
Eq−gHCl=Eq−gNa2CO3
V∗C=10ml∗0,5mEq−gml
=5mEq−gNa2CO 3
12,5mEq−gNaOH ×40mgNaOH1mEq−gNaOH
×1gNaOH
1000mgNaOH=0,5 gNaOH
5mEq−g Na2CO3×106mgNa2CO3
1mEq−gNa2CO3
×1gNaOH
1000mgNaOH=0,53 gNa2CO3
57

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ahora calculamos los porcentajes en peso de Na2CO3 y NaOH :
%NaOH=mNaOH
mT
×100%
%NaOH=0,5g1,2
×100%=41,7%
%Na2CO3=mNa2CO3
mT
×100%
%Na2CO3=0,53g1,2g
×100%=44,2%
58

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
REACCIONES Y VALORACIÓN DE FORMACIÓN DE COMPLEJOS
Formación de Complejos
M+L→ML metal ligando
acepta e−¿¿ dona e−¿¿
Equilibrio de formación de complejos
M+L→MLM+ L→MLML+L→ML2M+2 L→ML2ML2+L→ML3M+3 L→ML3ML3+L→ML4M+4 L→ML4
Las constantes se obtienen con ayuda de estas ecuaciones:
K1=|ML||M||L|
K2=|ML2|
|ML||L|K3=
|ML3||ML2||L|
K4=|ML4|
|ML3||L|
Poniendo en función de M y L:
K1∗K 2=|ML2|
|M||L|2K1∗K2∗K 3=
|ML3||M||L|3
K1∗K2∗K3∗K 4=|ML4||M||L|4
Valoraciones EDTA
HOOC−H 2CC H 2−COOH
N−C H 2−C H 2−N
HOOC−H 2CC H 2−COOH
Ácido etilendiaminotetraacético (H 4Y )
Las disoluciones de EDTA son valiosas como valorantes ya que el reactivo se combina con los iones metálicos en proporción de uno a uno independiente de la carga del protón.
Las valoraciones del EDTA (H 4Y ) se llevan a cabo en soluciones tamponadas que facultan la solución de los ejercicios.
Ag+¿+Y−4→AgY−3¿
59

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Al+3+Y−4→AlY−¿¿
En general:
M +n+Y−4→MY n−4
KMYn−4=|MY n−4|
|M+n||Y−4|
Ahora expresamos el valor de EDTATO |Y−4|, en función del grado de disociación:
α 4=|Y−4|CT
→|Y−4|=α 4CT
Donde CT= concentración de EDTA no acomplejado.
CT=|HY−3|+|H 2Y−2|+¿
KMYn−4=|MY n−4|
|M+n|α 4CT
KMY n−4'=
|MY n−4||M +n|CT
α n=K1 ¿¿¿
D=¿¿
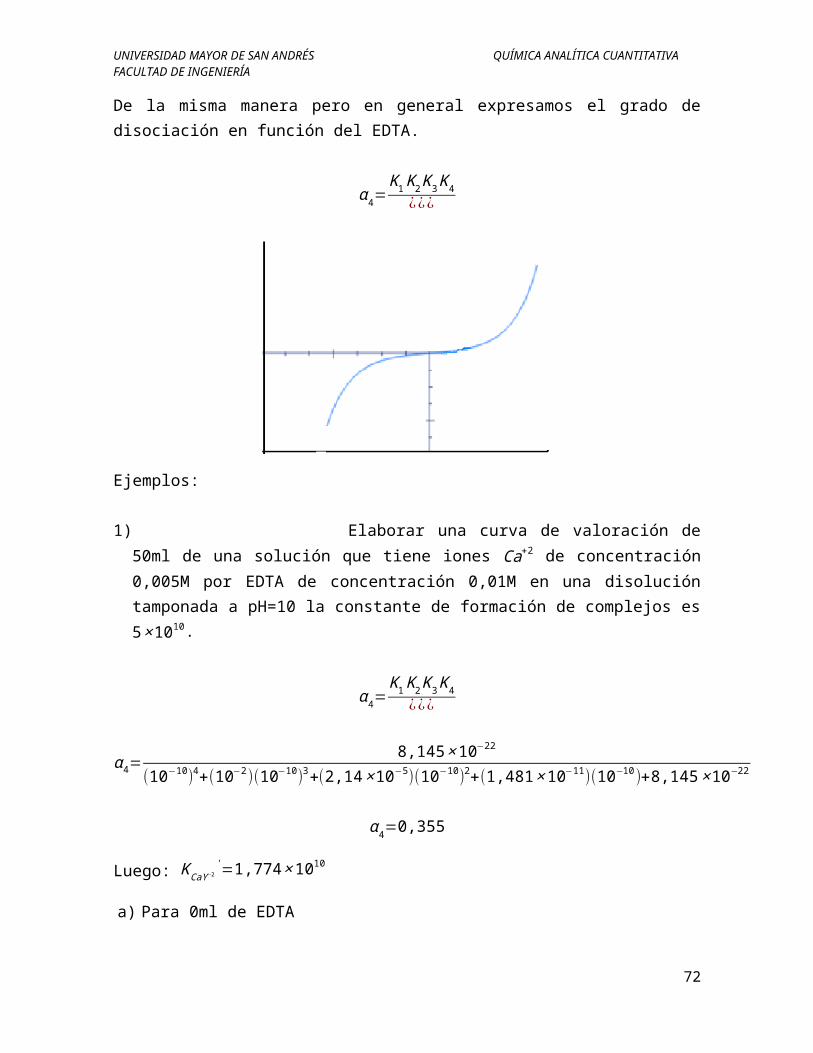
De la misma manera pero en general expresamos el grado de disociación en función del EDTA.
α 4=K1K2K3K4
¿¿¿
60

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ejemplos:
1) Elaborar una curva de valoración de 50ml de una solución que tiene iones Ca+2 de concentración 0,005M por EDTA de concentración 0,01M en una disolución tamponada a pH=10 la constante de formación de complejos es 5×1010.
α 4=K1K2K3K4
¿¿¿
α 4=8,145×10−22
(10−10)4+(10−2)(10−10)3+(2,14×10−5)(10−10)2+(1,481×10−11)(10−10)+8,145×10−22
α 4=0,355
Luego: KCaY −2'=1,774×1010
a) Para 0ml de EDTA
pCa+2=−log|Ca+2|
pCa+2=−log (0,005 )
pCa+2=2,30
b) Antes del punto equivalente
Para 5ml de EDTA
61

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
|CaY−2|=V añadido∗C
V T
|CaY−2|=5∗0,0155
=9,1×10−4M
|Ca+2|=¿moles iniciales−¿molesañadidosV T
|Ca+2|= (50∗0,005 )−(5∗0,01)50+5
=3,63×10−3M
pCa+2=−log (3,63×10−3 )pCa+2=2,44
c) En el punto equivalente
|CaY−2|=¿moles inicialesV T
=¿moles añadidosV T
|CaY−2|=(25∗0,01)50+25
=3,33×10−3M
|Ca+2|=√|CaY−2|K '
CaY −2
|Ca+2|=√ 0,003331,774×1010
=4,332×10−7M
pCa+2=−log (4,332×10−7 )
pCa+2=6,36
d) Después del punto equivalente
Para 26ml de EDTA
C|CaY−2|=¿moles formados
V T
62

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
C|CaY−2|=(50∗0,005)50+26
=3,289×10−3M
|EDTA|=¿moles añadidos−¿moles inicialesV T
|EDTA|= (26∗0,01 )−(50∗0,005)50+26
=1,316×10−4M
|Ca+2|= |CaY−2|CT (K ¿¿ ' ¿¿CaY−2)¿¿
|Ca+2|= (3,289×10−3)(1,316×10−4)(1,774×1010)
=1,409×10−9M
pCa+2=−log (1,409×10−9 )
pCa+2=8,85
0 10 20 30 40 50 600
2
4
6
8
10
12
pCa
Volumen Añadido[ml]
pCa
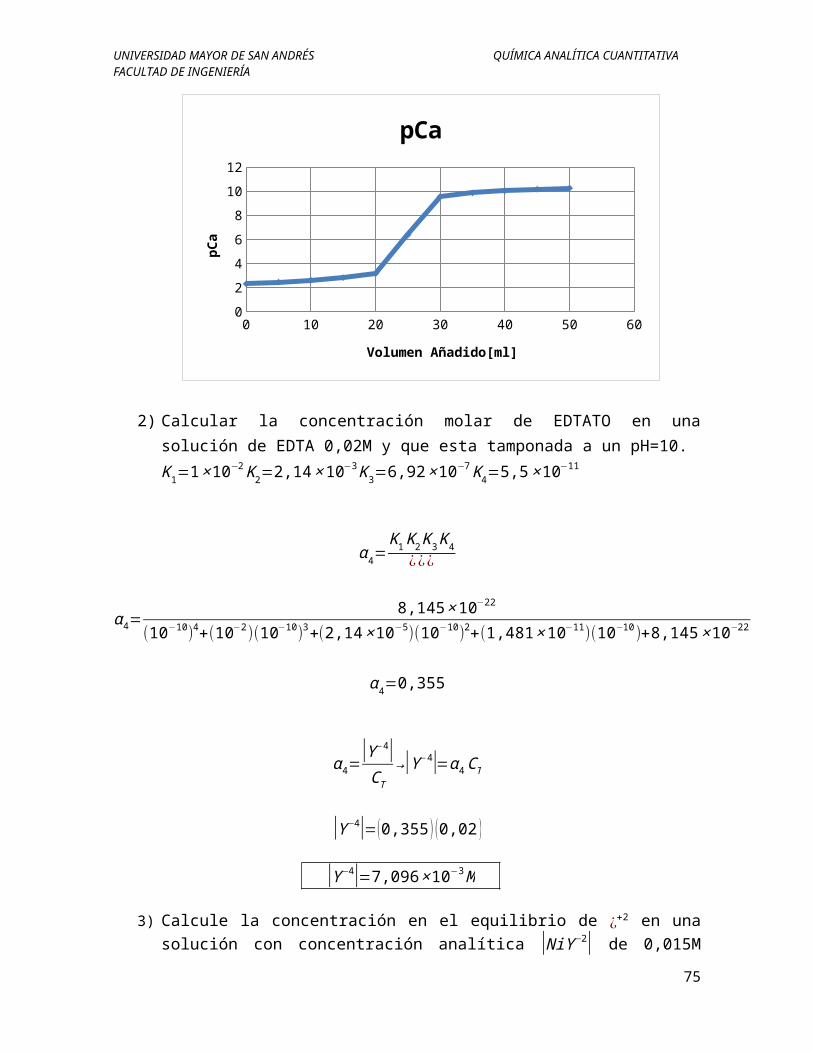
2) Calcular la concentración molar de EDTATO en una solución de EDTA 0,02M y que esta tamponada a un pH=10.
K1=1×10−2K2=2,14×10
−3K3=6,92×10−7 K4=5,5×10
−11
α 4=K1K2K3K4
¿¿¿
63

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
α 4=8,145×10−22
(10−10)4+(10−2)(10−10)3+(2,14×10−5)(10−10)2+(1,481×10−11)(10−10)+8,145×10−22
α 4=0,355
α 4=|Y−4|CT
→|Y−4|=α 4CT
|Y−4|=(0,355 ) (0,02 )
|Y−4|=7,096×10−3M
3) Calcule la concentración en el equilibrio de ¿+2 en una solución con concentración analítica |NiY−2| de 0,015M donde la constante para la reacción tiene el valor de 4,2×1018. Para un pH de 3,8 y 8 respectivamente.
a) A pH=3,8
¿+2+Y−4↔NiY−2
K Niy−2=|NiY−2||¿+2||Y−4|
K Niy−2'=
|NiY−2||¿+2|CT
=|NiY−2||¿+2|2
|¿+2|=√|NiY−2|K Niy−2
'
α 4=K1K2K3K4
¿¿¿
α 4=8,145×10−22
(10−3,8)4+(10−2)(10−3,8)3+(2,14×10−5)(10−3,8)2+(1,481×10−11)(10−3,8)+8,145×10−22
α 4=1,404×10−9
Siendo: K Niy−2'=5,895×109
|¿+2|=√ 0,0155,895×109
=1,595×10−6M
64

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
b) A pH=8
α 4=K1K2K3K4
¿¿¿
α 4=8,145×10−22
(10−8)4+(10−2)(10−8)3+(2,14×10−5)(10−8)2+(1,481×10−11)(10−8)+8,145×10−22
α 4=1,291×10−6
Siendo: K Niy−2'=5,420×1012
|¿+2|=√ 0,0155,420×1012
=5,261×10−8M
4) Hallar la concentración de Ni (II) en 50ml de solución de concentración 0,03M, con 50ml EDTA 0,05M. la mezcla esta tamponada a pH=3.
¿+2+Y−4↔NiY−2
K Niy−2'=
|NiY−2||¿+2|CT
α 4=K1K2K3K4
¿¿¿
α 4=8,145×10−22
(10−3)4+(10−2)(10−3)3+(2,14×10−5)(10−3)2+(1,481×10−11)(10−3)+8,145×10−22
α 4=2,513×10−11
Siendo: K Niy−2'=1,055×108
|¿+2|=|NiY−2|KNiy−2
' CT
|NiY−2|=¿moles inicialesV T
|NiY−2|=50∗0,03100
=0,015M
65

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
|EDTA|=¿moles añadidos−¿moles inicialesV T
|EDTA|= (50∗0,05 )−(50∗0,03)100
=0,01M
|¿+2|= (0,015)(1,055×108)(0,01)
=1,421×10−9M
66
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
MÉTODOS ELECTROQUIMICOS
Estos métodos se basan fundamentalmente en reacciones de óxido reducción también llamados REDOX.
5e−¿+8H +¿+MnO4↔ Mn+ 2+4H 2OReducción ¿ ¿
Fe+2↔Fe+3+1e−¿Oxidación¿
Estas reacciones se llevan a cabo en celdas electroquímicas que están separadas por un puente salino que aísla los reactivos pero mantiene el contacto eléctrico entre las dos soluciones.
Tipos de celdas
Celda Galvánica: Se caracteriza porque almacena energía eléctrica el flujo de electrones va del cátodo hacia el ánodo por un flujo de corriente externa. El cátodo es el electrodo donde se produce la reducción y el ánodo es el electrodo donde se produce la oxidación.
Celda electrolítica: Es la producción de una reacción química a través del suministro de energía eléctrica.
Representación de una celda
67

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ánodo↗↗CátodoZn0(s )↗ Zn+2
( Acu, 0.1M )↗↗Cu+2(Acu, 0.1M )↗Cu0(s)
Ánodo: Zn0(s )→Zn+2+e−2
Cátodo: Cu+2+e−2→Cu0(s )
Cuando una celda alcanza el equilibrio el voltaje es cero; el flujo de corriente es inversa u opuesta al flujo de electrones.
V=0(condiciónde equilibrio)
∆G=0∆G=−nFE(E=potencial del electrodo)
Potencial del electrodo
La diferencia de potencial que se genera entre los electrodos es una medida de la tendencia para que una reacción proceda desde un estado de no equilibrio hasta la condición de equilibrio.
∆G≥0 EQUILIBRIOEcelda=0
∆G<0PROCESO ESPONTÁNEO Ecelda>0
∆G>0PROCESONO ESPONTÁNEOEcelda<0
Ecelda=Ecátodo−Eánodo
En el equilibrio: Ecátodo=Eánodo
Efecto de las concentraciones y la ecuación de Nerst
aA+bB+cC→dD+ fF
a ,b , c , d ,e→coeficientes esqueoometricos
A ,B ,C ,D , F→compuestos
E=E°− RTnFln
[D ]d [ F ] f
[ A ]a [B ]b [C ]c
Dónde:
R= Constante universal de los gases (8,314 [J /mol° K ]).
68

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
E°= Potencial estándar [V ].F= Constante de Faraday (96500 [C ]).T= Temperatura de trabajo [° K ].n= # de moles de electrones.
En la constante de equilibrio no ingresa la concentración de los sólidos porque su concentración es igual a 1.
Si algún componente es gaseoso se utiliza la presión parcial en vez de su concentración.
Ejemplos:
1) Calculé el potencial de una solución de plata (I) sumergido en una solución de cloruro de sodio 0,05M. E°
¿¿, KpsAgCl=1,82×10−10.
1e−¿+Ag+¿→Ag 0¿ ¿
Ag+¿+Cl−¿ →AgCl¿ ¿
Kps=¿
¿
E=E°− RTnFln1¿¿
E=0,799−(8,314)(298)1(96500)
ln1
3,64×10−9
E=0,300[V ]
Potencial formal
El potencial formal (E° ¿ de un sistema es el potencial de la semi celda con respecto al electrodo estándar de hidrogeno medido de manera que las concentraciones analíticas de reactivos y productos tal como aparecen en la ecuación de Nerst sea exactamente la unidad; y las concentraciones de todas las otras especies del sistema estén cuidadosamente especificadas.
2) Calcular el potencial termodinámico de la celda siguiente y el cambio de la energía libre relacionado con la reacción de la celda.
Cu0/Cu+2 (0,02M )↗↗ Ag+¿ (0,02M ) /Ag0¿
1e−¿+Ag+¿→Ag 0E0=0,799[V ]¿¿
69

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
2e−¿+Cu+2→Cu0 E0=0,337 [V ]¿
Cu0→Cu+2+2e−¿E0=0,337[V ] ¿
Hallando el potencial para cada uno
E¿ ¿
E(Cu0 /Cu+2 (0,02M ) )=0,337 [V ]− (8,314 ) (298 )(2 ) (96500 )
ln [ 10,02 ]=0,287 [V ]
ES=E¿¿
ES=0,698 [V ]−0,287 [V ]=0,411[V ]
∆G=−nFE
∆G=−(2 ) (96500 ) (0,411)=−79323 [J ]
Curvas de valoración REDOX
Se considera que los potenciales de electrodo de todas las semi reacciones son idénticas cuando un sistema redox está en equilibrio, por lo tanto el sistema está en equilibrio durante la valoración.
Fe+2+Ce+4↔Fe+3+Ce+3
Para determinar los valores de la curva de valoración se debe considerar lo siguiente:
Antes del punto de equivalencia se utiliza la ecuación de Nerst para el analito. En el equilibrio se saca un promedio con los potenciales estándar de las
semiceldas. Para después del punto de equivalencia se utiliza la ecuación de Nerst para el
valorante.
Ejemplo:
Realizar la curva de valoración de 50ml de una solución que es 0,05M de hierro (II) con una solución que tiene una concentración de 0,1M de cerio (IV) en una solución que es en todo momento 1M en H 2SO 4.
Ce+4+1e−¿→Ce+3E°=1,440[V ] ¿
Fe+3+1e−¿→Fe+2 E°=0,680 [V ]¿
70

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
a) Antes del punto de equivalencia
Para 5ml añadidos de Cerio
|Fe+2|=¿moles iniciales−¿moles añadidosV T
|Fe+2|= (50∗0,05 )−(5∗0,1 )50+5
=0,0364M
|Fe+3|=¿molesañadidosV T
|Fe+3|=(5∗0,1)50+5
=9,09×10−3M
Eeq=E0(Fe+3 /Fe+2)−0,059 log(|Fe+2|
|Fe+3|)Eeq=0,680 [V ]−0,059 log( 0,0364M
9,09×10−3M )Eeq=0,644 [V ]
Para 10ml añadidos de Cerio
|Fe+2|= (50∗0,05 )−(10∗0,1 )50+10
=0,025M
|Fe+3|=(10∗0,1)50+10
=0,017M
Eeq=0,680 [V ]−0,059 log( 0,025M0,017M )Eeq=0,670 [V ]
Para 15ml añadidos de Cerio
|Fe+2|= (50∗0,05 )−(15∗0,1 )50+15
=0,0154M
71
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
|Fe+3|=(15∗0,1)50+15
=0,0231M
Eeq=0,680 [V ]−0,059 log( 0,0154 M0,0231M )
Eeq=0,690 [V ]
b) En el punto de equilibrio
Eeq=E0(Ce+4 /Ce+3)+E0(Fe+3 /Fe+2 )
2
Eeq=1,440 [V ]+0,680 [V ]
2
Eeq=1,060 [V ]
c) Después del punto de equivalencia
Para 26ml añadidos de Cerio
|Ce+3|=¿molesañadidosV T
|Ce+3|=(26∗0,1)50+26
=0,0329M
|Ce+4|=¿moles añadidos−¿moles inicialesV T
|Ce+4|= (26∗0,1 )− (50∗0,05 )50+26
=1,316×10−3M
Eeq=E0(Ce+4/Ce+3 )−0,059 log(|Ce+3||Ce+4|)
Eeq=1,440 [V ]−0,059 log( 0,0329M
1,316×10−3M )Eeq=1,360 [V ]
72

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 5 10 15 20 25 30 35 40 450
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6 E[V]
V[ml]
E[V]
Efecto de las variables en las curvas de valoración REDOX
Concentración de reactivos: Las curvas de valoración redox tienden a la independencia respecto a las concentraciones del analito y de los reactivos.
Grado de extensión en la reacción: El cambio del potencial en la región del punto de equivalencia de una valoración redox es mayor a medida que avanza la reacción.
73

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Indicadores de reacciones REDOX
Los indicadores redox se dividen en generales y específicos.
Indicadores redox generales
Son sustancias que cambian de color cuando se oxidan o reducen, en contraste con los indicadores específicos los cambios de color de los indicadores redox verdaderos dependen de los cambios del potencial del electrodo del sistema con forme avanza la titulación sin importar la naturaleza química del analito valorante.
¿ox+ne−¿⇄ ¿red ¿
[¿red ][¿ox ]
= 110
[¿ox ][¿red ]
=10
E=E°−RTnFln
[¿red ][¿ox ]
E=E°±0,0592
n
INDICADOR COLOR EN LA REDUCCION
COLOR EN LA OXIDACION
E˚[V]
Índigo monosulafato Incoloro Azul 0,26Azul de metileno Azul Incoloro 0,86
Di fenilamina Incoloro Violeta 0,76Di fenilamina-Sulfato de
BarioIncoloro Purpura 0,84
Ferroina Rojo Azul pálido 1,06
Aplicación de la valoración REDOX
Ejemplo: Una muestra de 5ml de Brandy se diluyo a un litro en un matraz volumétrico, de esta solución se toma una alícuota de 25ml de etanol diluyéndose, posteriormente se destila en 50ml de bicromato de potasio 0,02M que es utilizado como solución patrón. Al calentar el alcohol se oxida a ácido acético y el bicromato a cromo (III), según:
C2H 5OH+Cr2O7−2→Cr+3+CH 3COOH
Después se enfría y se añade un volumen de 20ml de hierro (II) que tiene una concentración de 0,1253M. El exceso de hierro se valora con 7,46ml de bicromato de potasio patrón hasta el punto final con ácido di fenilamina sulfonico. Determinar el porcentaje p/v de etanol presente en el brandy.
74
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Solución: igualamos la reacción por el método ión electrón
H 2O+C2H 5OH→CH 3COO−¿+5H +¿+4 e−¿∗(3)¿ ¿¿
(sumando)6e−¿+14 H¿→2Cr+3+7H 2O∗(2)¿¿
13H+¿+3C2H 5OH+2Cr 2O 7
−2→3CH3COO−¿+4Cr +3+11 H 2O ¿¿
16H+¿+3C2H 5OH+2Cr 2O 7−2→4Cr+3+3CH3COOH +11H 2O ¿
Para la segunda reacción:
6e−¿+14 H¿→2Cr+3+7H 2O¿¿(sumando)
Fe+2→Fe+3+1e−¿∗(6)¿
14H+¿+Cr 2O7−2+6Fe+2→2Cr+3+6Fe+3+7H 2O¿
(50+7,46 )mlsol×0,02mmol K2Cr2O7
1mlsol=1,1492mmol K2Cr2O7
20mlFe+2×0,1253mmol Fe+2
1mlFe+2
×1mmolCr2O7
−2
6mmolFe+2 =0,4177mmolCr2O7−2
(1,1492−0,4177 )mmolCr2O7−2×
3mmolC2H 5OH
2mmolCr2O7−2 ×
46mgC2H 5OH
1mmolC2H 5OH=50,4735mg
Ahora calculamos el volumen:
5mlbrandy×25mlC2H 5OH
1000mlbrandy=0,125mlC2H 5OH
El porcentaje (p/v) será:
% (p /v )= masavolúmen
×100%
% (p /v )=50,4735mg0,125ml
×100%
% (p /v )=40,38%
75

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
76

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
EQUILIBRIOS HETEROGÉNEOS
EXTRACCIÓN LÍQUIDO - LÍQUIDO
Introducción
Un proceso de separación consiste en obtener un componente determinado de una mezcla de componentes. Por lo tanto el proceso de separación implica el transporte de material y la redistribución de sus componentes, algunos de estos procesos de separación son la precipitación, destilación, extracción, cromatografía e intercambio iónico.
Por lo tanto en este capítulo se realizara el estudio de las separaciones analíticas que tiene como objetivo eliminar o reducir las interferencias para que se pueda obtener información cuantitativa acerca de una mezcla de complejos.
Separación por extracción líquido – líquido
Es la separación de un constituyente de una mezcla liquida por contacto con otro líquido que disuelve preferentemente a uno de los constituyentes de la disolución original.
Principios
Cuando un soluto se distribuye (reparte) entre dos fases no miscibles se trata de un fenómeno de equilibrio que se rige por la ley de distribución que expresa lo siguiente a “a temperatura constante la relación de las concentraciones de equilibrio de las especies distribuidas es constante”.
A(acuoso)⇄ A(orgánico)
[A (orgánico)][ A(acuoso)]
=K REPARTO
Para determinar la concentración del analito después de n-extracciones con un disolvente inorgánico la ecuación a utilizar es:
[ A ]i=[ A ]0( V acuoso
V organicoK+V acuoso)n
Dónde: K= Constante de reparto del soluto.
77

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
[ A ]i= Concentración de “A” que queda en la disolución acuosa después de extraer el volúmen acuoso de concentración inicial con “n” porciones de disolvente inorgánico.
[ A ]0= Concentración inicial.
V organico= Volumen del disolvente orgánico.
V acuoso= Volumen de la disolución acuosa.
N= Número de extracciones.
78

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
MÉTODOS CROMATOGRÁFICOS
Introducción
La cromatografía es un método muy utilizado que permite la separación, identificación y determinación de los componentes químicos en mezclas complejas.
Se basa en que la mezcla de componentes se separa a partir de la diferencia de velocidades a la que son transportadas.
Fase Estacionaria
La fase estacionaria esta fija en un lugar, ya sea en una columna o en una superficie plana que puede ser sólida o líquida.
Fase Móvil
La fase móvil se mueve sobre la fase estacionaria o a través de ella arrastrando consigo la mezcla de analitos. La fase móvil puede ser un gas o un líquido.
Este método fue inventado por el ruso Mikhail Tswett (1900), lo utilizó inicialmente para separar los pigmentos de la clorofila, pasándola por una columna de CaCO3. Proviene de la voz griega: Croma= Color, Gaphein = Escrito.
Clasificación
Se puede dividir en dos grandes grupos:
79
FASE ESTACIONARIA
FASE MÓVIL

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Cromatografía en columna: La fase estacionaria está contenida en un tubo estrecho y se fuerza el paso de la fase móvil a través del tubo ya sea a presión o por gravedad.
Cromatografía plana: La fase estacionaria esta sostenida sobre una placa plana o en los poros de un papel, la fase móvil se desplaza a través de la fase estacionaria por el fenómeno de capilaridad o por efecto de la gravedad.
Tipos Fase móvil Fase estacionariaCromatografía en papel Líquido Líquido ( moléculas de agua contenidas
en la celulosa del papel )Cromatografía en capa
finaLíquido Sólido
Cromatografía de gases Gas Sólido o líquidoCromatografía líquida
en fase inversaLíquido (polar) Sólido o líquido
(menos polar)Cromatografía líquida
en fase normalLíquido
(menos polar)Sólido o líquido
(polar)Cromatografía líquidade intercambio iónico
Líquido (polar) Sólido
Cromatografía líquidade exclusión
Líquido Sólido
Cromatografía líquidade adsorción
Líquido Sólido
Cromatografía defluidos supercríticos
Líquido Sólido
Cromatografía en columna
CROMATOGRAFIA DE GASESMÉTODO
ESPECÍFICOFASE ESTACIONARIA TIPO DE EQUILIBRIO
Gas – líquido Líquido absorbido o unido a una superficie sólida
Reparto entre gas y líquido
Gas – sólido Sólido Adsorción
CROMATOGRAFIA LIQUIDO – LIQUIDO
MÉTODO ESPECÍFICO
FASE ESTACIONARIA TIPO DE EQUILIBRIO
Líquido - líquido o reparto
Líquido absorbido o unido a una superficie sólida
Reparto entre líquidos inmiscibles
Líquido - sólido Sólido AdsorciónIntercambio
iónicoResina de intercambio iónico Intercambio iónico
El proceso
80

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Elución: Es un proceso en el cual los solutos son arrastrados de una fase estacionaria por una fase móvil. La fase móvil que sale de la columna se denomina ELUATO.
Eluyente: Es el disolvente que se usa para transportar los componentes de una mezcla a través de una fase estacionaria.
Cromatograma
El cromatograma es una gráfica de alguna función de la concentración versus el tiempo de la elución o el volumen de la solución.
81
Analito (A+B) Eluyente
EluatoElución

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
0 10 20 30 40 50 60 700
2
4
6
8
10
12
14
16
CROMATOGRAMA
tiempo [s]
Seña
l del
det
ecto
rAαC
Existen algunas características cuando la fase móvil atraviesa la fase estacionaria, como ser:
El movimiento del soluto solo puede ocurrir en la fase móvil. La velocidad promedio a la cual la banda del soluto migra en la columna
depende de la fracción de tiempo que pase en la fase estacionaria. El tamaño de la banda del soluto aumenta con el tiempo debido al proceso de
dilución.
Velocidad de migración
La velocidad de migración se determina por la magnitud de la constante de distribución.
A(móvil)⇄ A (estacionario )
K=C(est acionaria)
C (móvil)
Tiempo de retención (tR)
Es el tiempo transcurrido entre la inyección de una muestra y la aparición de un pico de un soluto en el detector de la columna cromatográfica.
Tiempo muerto (tM)
82

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Es el tiempo que una especie no retenida tarda en pasar a través de una columna cromatográfica. Todos los componentes pasan esta cantidad de tiempo en la fase móvil.
V M= LtR
=longitud de lacolumnatiempode retención
=[ cms ]La velocidad promedio lineal “u” del movimiento de las moléculas de la fase móvil es:
u= LtM
Relación entre la velocidad de flujo volumétrico y la velocidad de flujo lineal
83
Caudal o flujo volumétrico [Q]
Espacios vacíos [Є]

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Q=uo [cm /s ]∗A [cm2 ]∗∈
V tubo=V vacio+V ocupado
Dónde:
uo= velocidad lineal a la salida de la columna [cm /s ].A= Área transversal [cm2 ]=π r2.
Relación entre velocidad de migración y la constante de distribución
V=u∗fraccióndel soluto
fracciondel soluto=molesde solutoen la fasemóvilmoles totales
X s=V MCM
VM CM+V sC s
V=( 1
1+( V s
V M)K )u
Velocidad de migración del soluto
El factor de retención “K”
Es un parámetro experimental que se usa para comparar velocidades de migración de solutos en las columnas y podemos definirla como la cantidad de tiempo que pasa un soluto en la fase estacionaria en relación con el tiempo que pasa en la fase móvil.
K 'A=K A
V S
VM
V=u ( 11+K ' A )
Pero: u=L/ tM ;V =L/ tR
LtR
=
LtM
∗1
1+K ' A
84

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
K 'A=tR−tMt M
Características
Cuando el factor de retención para una especie es mucho menor que la unidad la elución tiene lugar tan rápidamente que es difícil determinar con exactitud los tiempos de retención.
Cuando el factor de retención es del orden de 30 a 20 o mayor los tiempos de elución son excesivamente largos. En este sentido idealmente las separaciones se realizan en condiciones en las que los factores de retención se encuentran en el rango de 2 a 10.
Velocidades de migración relativas
Factor de selectividad
Es la relación entre las constantes de distribución del soluto (factores de retención más fuertemente retenidos con respecto del soluto retenido con menos fuerza).
α=K 'BK ' A
=( tR )B−tM
(tR ) A−t M
Ensanchamiento de banda y eficiencia de la columna
La eficiencia de una columna cromatográfica se ve afectada por el ensanchamiento de la banda que se produce cuando los compuestos pasan a través de la columna, por lo tanto para entender esta influencia debemos analizar las razones de este ensanchamiento.
Teoría de la velocidad cromatográfica
Al tener miles de transferencias entre la fase móvil y la fase estacionaria, el tiempo que el soluto pasa en cada fase después de una transferencia es muy irregular y depende de la ganancia de energía térmica, por lo tanto su migración también es irregular, unos pueden moverse más rápidamente porque se encuentran en la fase móvil y otros más lentamente porque están en la fase estacionaria.
Como consecuencia se tiene una dispersión simétrica de las velocidades alrededor de su valor medio. Por lo tanto la altura de banda aumenta a través de la columna.
Anchodebanda∝tR
Anchodebanda∝ 1V
85

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Eficiencia de la columna
Se utilizara dos conceptos para medir la eficiencia de la columna:a) En función de la altura de plato que denominaremos “H”.
b) En función de la cantidad de platos o número de platos teóricos que denominaremos “N”.
Dónde las variables se relacionan mediante la siguiente ecuación:
N= LH
La eficiencia de la columna cromatográfica aumenta si “N” aumenta y “H” disminuye.
Definición de la altura de plato (H)
Para este análisis asumiremos que las bandas cromatográficas tienen forma Gaussiana y se relaciona con la desviación estándar de una medida. Por lo tanto la eficiencia de una columna se refleja en la amplitud de los picos cromatográficos; de esta forma los cromatógrafos utilizan a la varianza por unidad de longitud de la columna como una medida de la eficiencia de estas.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 260
1
2
3
4
5
6
7
8
9
-1σ 1σ
Determinación experimental del número de platos de una columna
86

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
N i=16 ( tRiw i)2
N=∑i=0
n
N i
n
Variables que afectan la eficiencia de la columna
El ensanchamiento de banda afecta la eficiencia de la columna. Cuanto más lento es el proceso de transferencia de masa, más ancha será la banda a la salida de la columna; por lo tanto existen diferentes variables que influyen en este proceso:
VARIABLES SÍMBOLO UNIDADESVelocidad lineal de la fase móvil u [ cms ]Coeficiente de difusión en fase móvil DM [ cm2s ]Coeficiente de difusión en fase estacionaria DS [ cm2s ]Factor de retención K [ Adimencional ]Diámetro de partículas del empaquetamiento d P [cm ]Espesor de recubrimiento líquido de la fase estacionaria
d f [cm ]
Efecto de la velocidad de ensanchamiento de la fase móvil
87
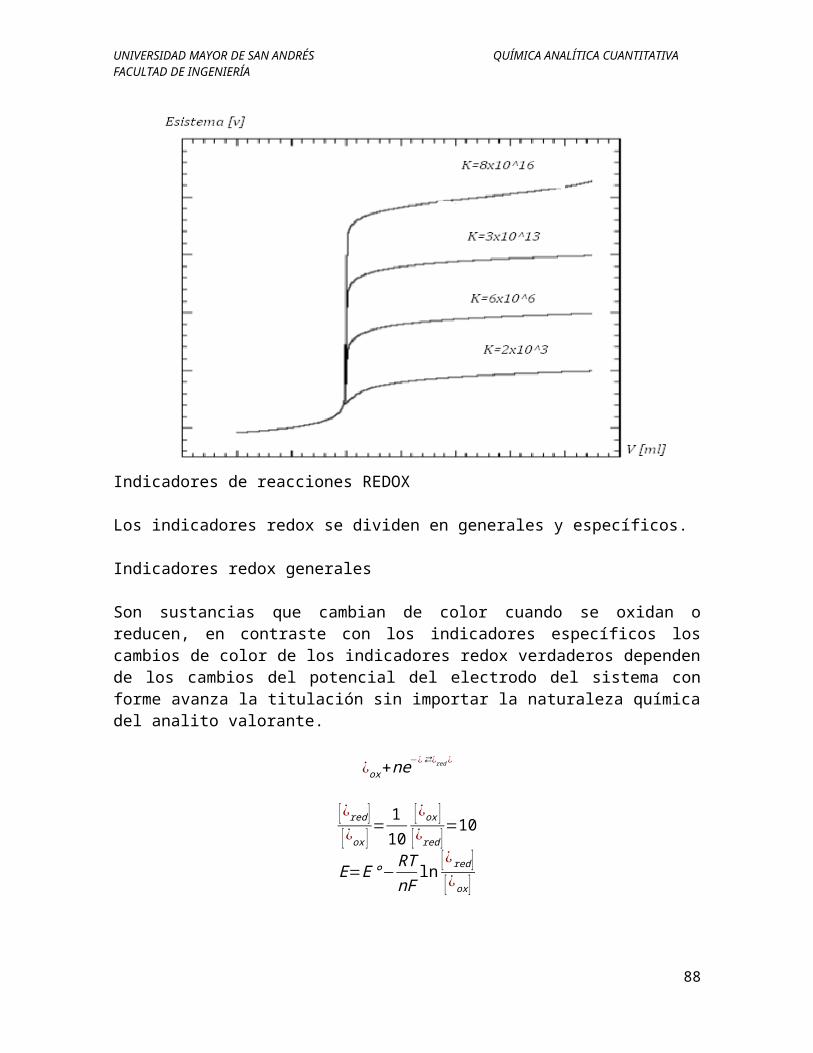
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
El grado de ensanchamiento de banda depende de la cantidad de tiempo que la fase móvil este en contacto con la fase estacionaria, la cual depende a su vez de la velocidad de flujo de la fase móvil.
De acuerdo a las gráficas no es práctico utilizar columnas cromatográficas de líquidos que tengan una longitud mayor entre 25 a 50 [cm], por el contrario las columnas cromatográficas de gases pueden tener una longitud mayor de 50 [m].
Si comparamos la cromatografía de gases y la de líquidos, la cromatografía de gases es capaz de realizar la cromatografía con rapidez y eficiencia.
Teoría del ensanchamiento de banda
Se han propuesto una gran variedad de expresiones para calcular la altura de plato, pero ninguna de ellas es del todo adecuada para explicar las complejas interacciones físicas y los efectos que dan lugar al ensanchamiento de la zona y reducir así la eficiencia de la columna.
Sin embargo alguna de estas ecuaciones se usa con frecuencia a pesar de no ser perfectas con la finalidad de mejorar el rendimiento de la columna.
Baja Velocidad: Para columnas cromatográficas capilares y empaquetadas la eficiencia se puede calcular con la siguiente ecuación.
H= Bu
+CSu+CMu
H = Altura de plato [cm].u= Velocidad lineal en la fase móvil [cm/s].
88
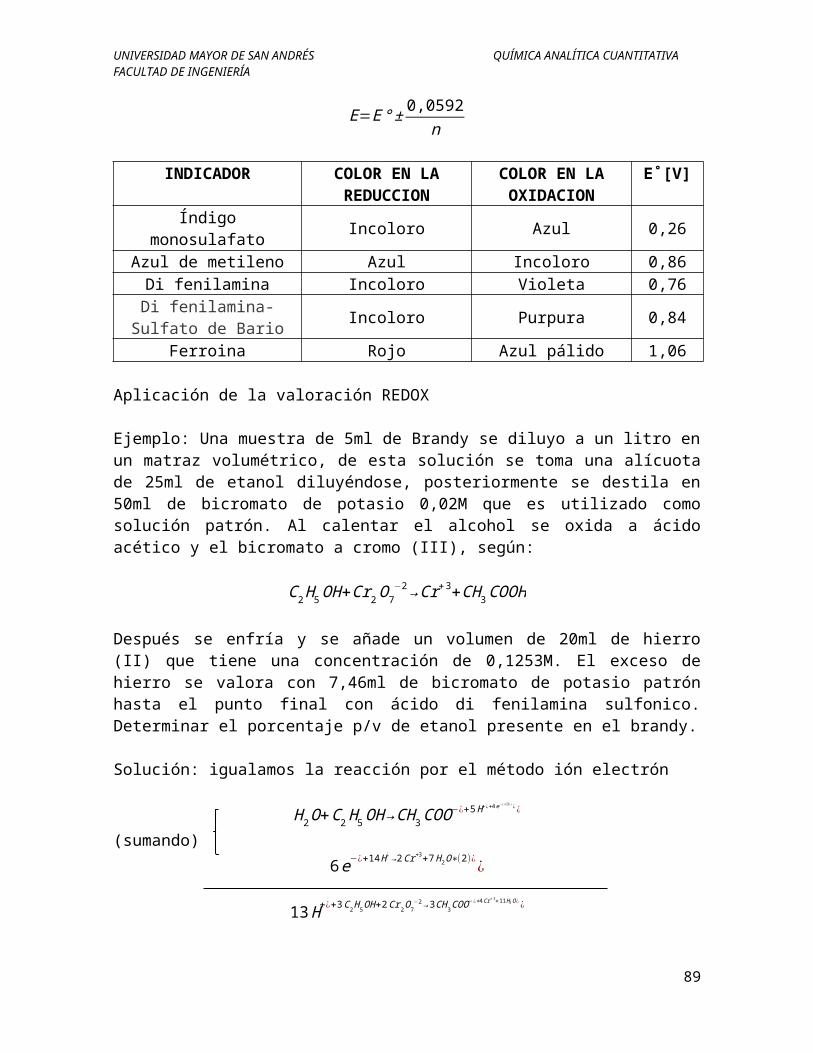
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
B= Coeficiente de difusión longitudinal.CS ;CM= Coeficientes de transferencia de masa.
Altas Velocidades: En columnas empaquetadas donde los flujos que dominan el proceso son la difusión.
H=A+Bu
+CSu
A= Coeficiente que describe los efectos de la trayectoria múltiple (Difusión aparente).B/u= Difusión longitudinal (fuente común de ensanchamiento de banda en cromatografía de gases, despreciable en cromatografía líquida).CSu= Transferencia de masa en fase estacionaria.
DESCRIPCIÓN FÓRMULA SIGNIFICADOSi la fase estacionaria es un líquido inmovilizado.
CS∝d t2
CS∝ (1/DM )
Espesor de la película.
Coeficiente de difusión.
Si la superficie estacionaria es una superficie sólida
CS∝t
CM∝ (1/DM )
Tiempo que tarda la especie en ser absorbido o de sorbido.Difusión del analito en la fase móvil.
Para columnas empacadas CM∝d P2 Diámetro del material de
empaquetamiento.Para columnas capilares CM∝dC
2 Diámetro de la columna y es función de la velocidad de flujo.
89

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Resolución de la columna
RS=∆ Z
wA
2+wB
2
=2 {(tR )B− (tR )A }
wA+wB
RS=√N4 (α−1
α )( K 'B1+K 'B )
(tR )B=16 RS
2Hu ( α
α−1 )2( (1+K '
B )3
K 'B2 )
Dónde: K 'B= factor de retención de la especie que se mueve más lentamente.
90

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Ejemplo:
Las sustancias A y B tienen tiempos de retención de 16,40 y 17,63 [min] respectivamente, en una columna que tiene una longitud de 30[cm]. Una especie no retenida pasa a través de la columna en 1,3[min], las anchuras máximas de los picos en su base son 1,11 y 1,21 [min] para A y B respectivamente. Calcular:
a) La resolución de la columna.b) El número medio de platos en la columna.c) La altura del plato.d) La longitud necesaria para lograr la resolución de 1,5.e) El tiempo necesario para eluir la sustancia B en la columna que da un valor de
resolución de 1,5.
91

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Solución:
RS=2 {(tR )B−(t R )A}
w A+wB
=2 {17,63−16,40 }
(1,11+1,21)
RS=1,06
N i=16 ( tRiw i)2
N A=16( 16,401,11 )2
=3493
N B=16( 17,631,21 )2
=3397
Sacando el promedio tenemos:
N=N A+NB
2=3493+3397
2
N=3445
H= LN
=30[cm]3445
H=8,7×10−3 [cm ]
RS1=√N1ctte
RS2=√N2 ctte
Dividiendo (2) entre (1):
N2=N 1( RS2
RS1)2
92

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
N2=3445 ( 1,51,06 )2
N2=6899
L=H N2
L=0,0087∗6899
L=60,02 [cm ]
(tR )1=RS12 ctte (t R )2=RS2
2ctte
Luego de dividir (4) entre (3):
(tR )2=(tR )1( RS2
RS1)2
=17,63 [min ]( 1,51,06 )2
(tR )2=35,30[min]
Factores externos que influyen en el ensanchamiento de las bandas cromatográficas
Introducción de la muestra
Cuando se introduce un volumen finito a un cromatógrafo (cromatógrafo de gases), este volumen al ser calentado o vaporizado sufre un incremento en su volumen; este volumen puede llegar a ocupar una gran longitud de la columna que ocasiona un ensanchamiento de banda del cromatograma, además debemos considerar que el calentamiento no es instantáneo sino se produce lentamente.
Entonces tenemos otro factor que ocasiona un ensanchamiento de banda del cromatograma.
Otro de los factores que ocasiona ensanchamiento de banda son los volúmenes muertos que existen en el sistema de inyección, en el sistema detector y entre los tubos de conexión; por volumen muerto se debe considerar el existente entre el punto de inyección de la muestra y el punto de detección, excepto el ocupado por la fase estacionaria.
(FALTA TEORIA)
Principios Básicos
Las principales diferencias entre gases y líquidos pueden sintetizarse en lo siguiente:
93

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Las moléculas de los gases a diferencia de los líquidos suelen ser pequeñas, por otra parte los gases suelen tener poca capacidad para disolver las moléculas de soluto o de solutos no volátiles que se fijan en la fase estacionaria, por este motivo en la cromatografía gaseosa la fase móvil no interacciona con las moléculas del analito siendo su única función el transporte del analito a través de la curva.
Los gases se difunden entre sí mucho mejor que los líquidos, lo cual influye en la resolución y la velocidad de separación por lo tanto a velocidades de fase móvil próximo a su valor óptimo. La cromatografía líquida proporciona mejor resolución, aunque en la cromatografía gaseosa la velocidad óptima de flujo es del orden de 104 a 105 veces superior que la cromatografía líquida, con lo que pueden obtenerse separaciones en muy poco tiempo.
Las propiedades superficiales de los líquidos (tensión superficial) es una característica muy importante que se utiliza en la cromatografía sobre papel y de capa fina que permite realizar separaciones sin columna. Los gases carecen de esta propiedad por lo tanto este tipo de separaciones no se pueden realizar en gases.
La mayor densidad de los líquidos respecto a los gases permite usar la gravedad o la fuerza centrífuga como fuerza motriz de la fase móvil, mientras que la pequeña viscosidad de los gases hace necesario el empleo de columnas largas.
Debido a que los gases se comprimen fácilmente en cromatografía de gases es más aconsejable utilizar volúmenes de retención en lugar de tiempos de retención.
Los volúmenes de retención y muerto dependen de la fracción media en el interior de la columna por lo cual también es necesario corregir estos volúmenes a través del factor de compresibilidad.
J °=32 ( (
Pi
Po)2
−1
( P i
Po)n
−1 )En cromatografía de gases para estandarizar los volúmenes de retención suele usarse la siguiente relación:
V neto=V R−V M
94
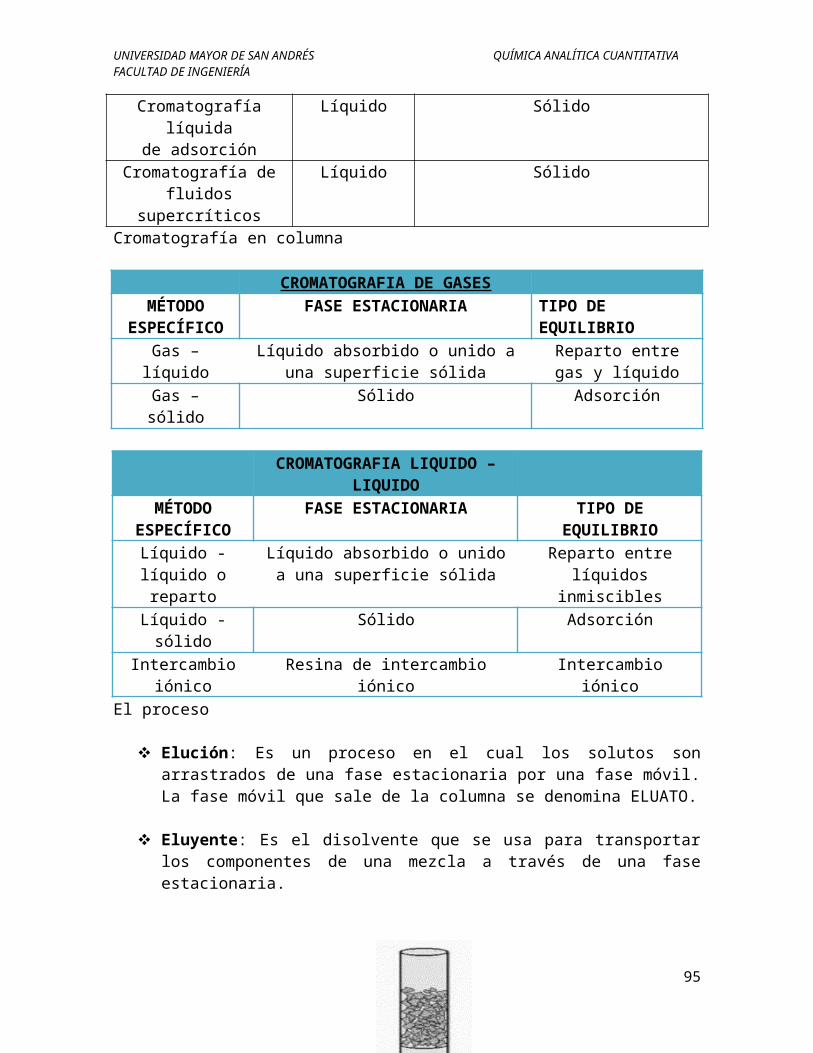
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Además del volumen de retención específico que está definido como el volumen neto del analito por gramo de fase estacionaria a 0˚C.
V g=
V neto
m∗273
T
El volumen de retención específico está relacionado con la constante de retención “K”.
V g=
Kρ∗273
T
Por lo tanto a una determinada temperatura el volumen de retención específico depende únicamente de la constante “K“, y de la densidad del líquido que constituyeρ la fase estacionaria
Por lo que en principio podría ser un parámetro característico con fines de identificación, en la cromatografía gaseosa la constante de distribución depende marcadamente de las presiones de vapor de los componentes y este a su vez de la temperatura, por lo tanto este parámetro deberá mantenerse de la forma más precisa o simple.
Las muestras en general deben cumplir las siguientes condiciones:
Los compuestos en estado gaseoso, líquido o sólido con presiones de vapor por lo menos de 0,3mmHg a la temperatura máxima de la fase estacionaria empleada.
No se descomponen por el calor a la temperatura de separación, no absorbibles por el soporte solido de la columna.
Que sean detectables a la salida.
Instrumentación
95
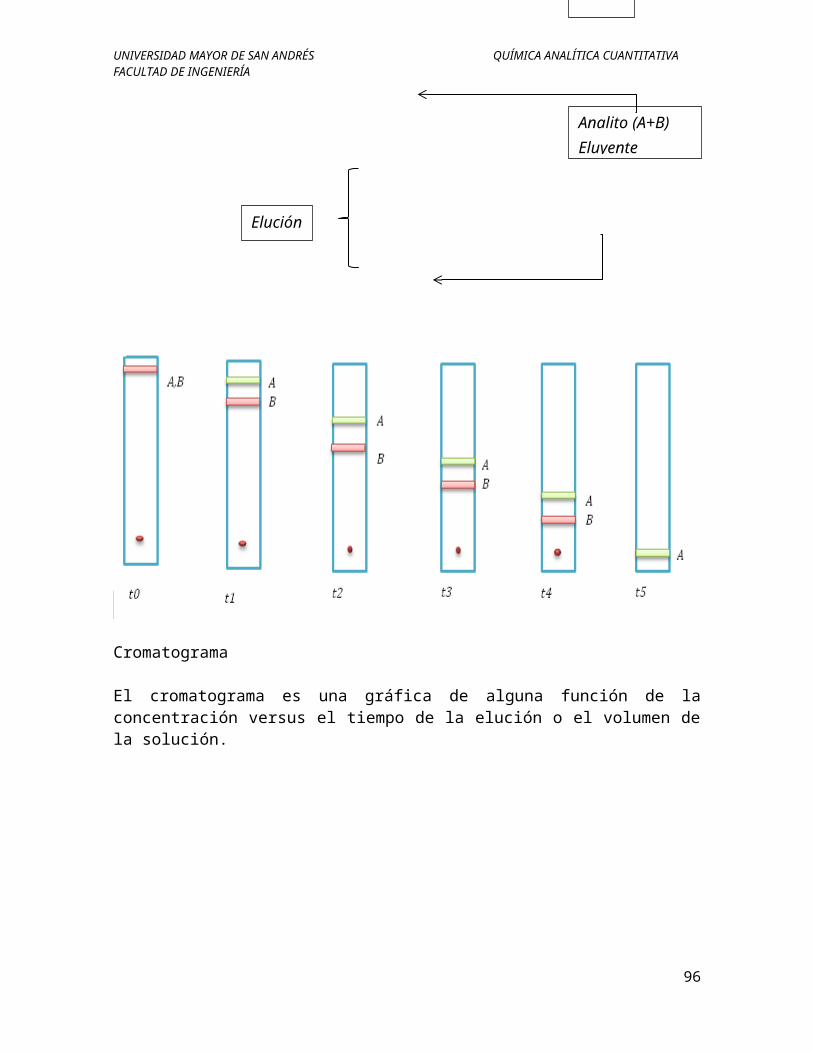
UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
Gas portador
Actúa como fase móvil y transporta los componentes de la muestra a través de la columna hasta el detector y una de las características más importantes que debe cumplir es que debe ser una especie químicamente inerte respecto al analito y relativamente barato.
Los más utilizados son: He, N2, H2, Ar, CO2; por lo tanto la elección de la fase móvil se debe realizar en función de costo disponibilidad, en función del detector utilizado y ocasionalmente de acuerdo a la eficacia de la separación.
Un factor muy importante en las separaciones por cromatografía de gases es el ajuste del caudal, debido a que esta condiciona la velocidad de separación, estos caudales se controlan con los reguladores de presión que se colocan en el botellón del gas portador o que ocasionalmente pueden estar instalados en el cromatógrafo.
El caudal se puede determinar mediante un rotámetro que puede estar situado en la cabeza de la columna o lo más simple utilizar un medidor de pompas de jabón que puede estar situado a la salida de la columna.
Sistema de introducción de muestras
Las muestras a separar suelen ser de diferente naturaleza y estado físico pero necesariamente tienen que pasar al estado vapor y este procedimiento tiene gran importancia desde el punto de vista de la eficacia de la separación.
El tamaño de la muestra viene condicionado por el detector utilizado en consecuencia “deberá inyectarse” la cantidad adecuada de muestra en el menor tiempo posible y conseguir la vaporización total de las muestras no gaseosas, en este sentido las
96

UNIVERSIDAD MAYOR DE SAN ANDRÉS QUÍMICA ANALÍTICA CUANTITATIVAFACULTAD DE INGENIERÍA
muestras gaseosas se introducen en la columna usando válvulas rotatorias y bucles de volumen conocido.
Alternativamente la inyección puede realizarse directamente en la columna, en estos casos la aguja hipodérmica debe llegar hasta el relleno de la columna donde se depositara directamente la muestra líquida.
El tamaño de la muestra para columnas analíticas ordinarias varía entre 1 L y 20 Lμ μ además se pueden utilizar repartidores de caudal que en ocasiones se pueden colocar a la salida del detector.
Columna y la fase estacionaria
Es la parte más importante del cromatógrafo dependiendo de esta el éxito o el fracaso de los análisis, antiguamente se utilizaban básicamente columnas empaquetadas o de relleno, en la actualidad las más utilizadas son columnas tubulares abiertas o capilares. Las primeras tienes una longitud entre 1 y 6[m] con un diámetro interno entre 2 y 6[mm] mientras que las columnas capilares tienen una longitud de 10 a 100[m] y diámetros ente 0,1 y 0,6[mm], están fabricadas de acero inoxidable o vidrio para las columnas empaquetadas y sílice para las capilares.
Temperatura
La temperatura es una de las variables más importantes en cromatografía y es necesario tener un control preciso para obtener datos reproducibles, por este motivo las columnas cromatográficas suelen disponerse en rollos de 10 a 30[cm] de diámetro.
En el análisis práctico la temperatura debe ser superior al punto de ebullición medio de la muestra, de esta forma obtener tiempos de elución razonable. A veces se puede tener muestras en las que el punto de ebullición pueden ser cercanos (H2O, C2H5OH).
Para evitar estos inconvenientes se suele emplear un aumento de temperaturas programado aumentando esta ya sea en forma lineal o por etapas.
97


















