NOTAS DE CLASES - raulbarrachina.com.ar · NOTAS DE CLASES CARRERA DE LICENCIATURA EN FISICA TEMAS...
240
Transcript of NOTAS DE CLASES - raulbarrachina.com.ar · NOTAS DE CLASES CARRERA DE LICENCIATURA EN FISICA TEMAS...


NOTAS DE CLASESCARRERA DE LICENCIATURA EN FISICA
TEMAS DE MECANICA CUANTICA
Raúl Oscar Barrachina
Con dos apéndices escritos por WilIy Pregliasco
Junio 1993

Indice
1 Operador densidad1.1 Estados puros de espín1.2 Vector polarización ..1.3 Estados de mezcla de espín .1.4 Operador densidad de espín1.5 Operador densidad .....1.6 Coherencia y oscilaciones cuánticas1.7 Ecuación de Liouville1.8 Bibliografía.............
2 Interpretación de la mecánica cuántica2.1 Introducción .2.2 Formalismo cuántico y realidad externa2.3 Interpretación de Copenhague .....2.4 Atributos de realidad de la función de onda.2.5 No localidad del formalismo cuántico2.6 Paradoja EPR . . . . . . . .2.7 Teorema de Bell .2.8 El problema de la medición.2.9 Observador consciente '"2.10 Interpretación de muchos mundos2.11 Teorías no locales de variables ocultas2.12 Interpretación ortodoxa.2.13 Decoherencia2.14 Bibliografía .
3 Momento cinético3.1 Momento cinético . . . . . . .3.2 Representación estándar Ijm)3.3 Espectro de j2 y Jo • • . . .
3.4 Suma de momentos cinéticos .3.5 Símbolos 3J de Wigner ....3.6 Suma de tres momentos cinéticos
111234577
99
10121213141517181920212223
25252627283031

3.73.8
Notación y convencionesBibliografía . . . . . . .
3434
4 Teorema de Wigner-Eckart 354.1 Operador de rotación . . . . . . . . 354.2 Operadores tensoriales irreducibles 364.3 Teorema de Wigner- Eckart . . . . . 384.4 Regla de suma . . . . . . . . . . . . 404.5 Cálculo del elemento de matriz reducido 414.6 Bibliografía............. 42
5 Ondas electromagnéticas en el vacío 435.1 Ondas electromagnéticas en el vacío 435.2 Polarización y helicidad . 455.3 Fotones . . . . . . . . . 475.4 Tensor de polarización 475.5 Parámetros de Stokes . 485.6 Grado de polarización. 495.7 Bibliografía... 50
6 Emisión de radiación 516.1 Interacción de un sistema cuántico con el campo de radiación. 516.2 Emisión de radiación . . . . 536.3 Transición dipolar eléctrica. . . . . . 546.4 Absorción de radiación . . . . . . . . 566.5 Validez del tratamiento perturbativo 576.6 Vida media y ancho de linea 586.7 Bibliografía......... 60
7 Reglas de selección 617.1 Transición entre estados desacoplados 617.2 Estados metaestables 617.3 Integral de transición 627.4 Series......... 637.5 Intensidad luminosa . 647.6 Polarización de la radiación emitida 657.7 Transición entre estados de estructura fina 67
8 Teoría de perturbaciones independiente del tiempo 698.1 Perturbaciones de estados estacionarios no degenerados 698.2 Perturbaciones de estados estacionarios degenerados 708.3 Efecto Stark en el átomo de Hidrógeno 718.4 Estados metaestables . . . . . . . . . . . . . . . . . 73

8.5 Apagado de líneas de emisión (Quenching) . . . . . 74
9 Teoría de perturbaciones dependientes del tiempo9.1 Perturbaciones dependientes del tiempo . . . . . . .
9.2 Desarrollo en autoestados instantáneos de la energía.
9.3 Aproximación adiabática .9.4 Cambio discontinuo del Hamiltoniano9.5 Aproximación súbita .
10 El átomo de Hidrógeno10.1 Introducción .....10.2 El átomo hidrogenoide10.3 Corrección isotópica ..10.4 Corrección relativista a la energía cinética10.5 Acoplamiento espín-órbita10.6 Término de Darwin10.7 Estructura fina .10.8 Efecto Lamb .10.9 Corrimiento isotópico por volumen nuclear10.10 Estructura hiperfina .10.11 Bibliografía .
11 El átomo de Helio11.1 Introducción.................11.2 Estados singletes y tripletes ..11.3 Aproximación de electrones independientes11.4 Apantallamiento .11.5 Tratamiento perturbativo .11.6 Cálculo variacional ....11.7 Estados excitados . . . . .11.8 Transiciones dipolares eléctricas11.9 Estados doblemente excitados y autoionización .11.10 Ion negativo de Hidrógeno11.11 Bibliografía .
12 Hartree-Fock y Thomas-Fermi12.1 Teoría de Harthee-Fock ..12.2 Modelo de Thomas-Fermi .12.3 Bibliografía .
777777787980
8383838788889090929394
95
9797989899
100101102103105105106
107107111114

13 La estructura de los átomos13.1 Atomos multielectrónicos13.2 Configuración electrónica .
13.3 Reglas de Madelung. . . .13.4 Interacción entre electrones. Reglas de Hund13.5 Términos espectrales .13.6 Estructura fina de los niveles atómicos13.7 Tabla periódica de los elementos .13.8 Electrones de valencia.
13.9 Berilio (Z=4) .13.10 Boro (Z=5) ...13.11 Carbono (Z=6) .13.12 Nitrógeno (Z=7) .13.13 Oxígeno (Z=8)13.14 Flúor (Z=9) ...13.15 Neón (Z=10)13.16 Segundo período corto13.17 Primer período largo .13.18 Metales de transición y tierras raras13.19 Acoplamiento J - J .13.20 Transiciones dipolares eléctricas13.21 Bibliografía .
14 Moléculas diatómicas14.1 Introducción .14.2 Molécula ion de Hidrógeno14.3 Orbitales moleculares ...14.4 Configuración electrónica de moléculas diatómicas14.5 Términos electrónicos . . . .14.6 Moléculas heteronucleares .14.7 Ligaduras covalente e iónica14.8 Bibliografía .
15 Moléculas poliatómicas15.1 El agua .15.2 Amoníaco .. . .15.3 Hibridización Sp3
1.5.4 Hibridización Sp2
15.5 Hibridización sp .15.6 Moléculas conjugadas15.7 Benzeno .15.8 Rotación molecular .
115115115116117118121124126128129129129130131131131132132133134135
137137137141143144145147148
149149150151152154155158159

15.915.1015.1115.1215.1315.1415.15
Oscilaciones molecularesModos normales . . . . .Espectro roto- vibracionalTransiciones electrónicasPrincipio de Franck-CondonAproximación de Born-OppenheimerBibliografía .
160161162163165166168
16 Teoría clásica de colisiones 16916.1 Sección eficaz diferencial . . . . . . . 16916.2 Definición clásica de la sección eficaz 17016.3 Dispersión de Rutherford . 17216.4 Efectos Gloria y Arco Iris. 17216.5 Bibliografía . . . . . . . . 175
17 Teoría cuántica de Colisiones 17717.1 Condición asintótica 17717.2 Operador de Moller . . . . 17817.3 Operador de Green . . . . 17917.4 Estados estacionarios de dispersión 18017.5 Sección eficaz cuántica . . . . . . . 18217.6 Límite asintótico de los estados estacionarios de dispersión 18317.7 Bibliografía . . . . . . . . . . . . . . . . . . . . . . . . . . 184
18 Aproximación de Born 18518.1 La serie de Born. . . . . . . . . 18518.2 Primera aproximación de Born . 18518.3 Potencial de Yukawa . . . . . . 18718.4 Dispersión elástica de partículas cargadas rápidas por átomos. 18818.5 Potencial coulombiano 19018.6 Bibliografía . . . . . . 192
19 Colisiones de baja energía 19319.1 Des!1rrollo de ondas parciales . . . . . . . . . 19319.2 Corrección a la aproximación de Born . . . . 19519.3 Propiedades de la amplitud de onda parcial. 19619.4 Bibliografía . . . . . . . . . . . . . . . . . . 198
20 Resonancias 19920.1 La función de Jost 19920.2 Ceros de la función de Jost y estados ligados 19920.3 Resonancias........ 20120.4 Resonancia de energía cero . . . . . . . . . . 203

20.5 Bibliografía . . . . . . . . . 204
21 Teoría semiclásica de colisiones 20521.1 Aproximación semiclásica. . . . . . . . . . 20521.2 Interferencia cuántica en el efecto Arco Iris 20721.3 Bibliografía . . . . . . . . . . . . . . . . . 209
22 Dispersión de partículas idénticas 21122.1 Descripción de un proceso de dispersión elástica en el sistema de
laboratorio. . . . . . . . . . . . . 21122.2 Dispersión de partículas idénticas 21222.3 Bibliografía . . . . . . . . . . . . 215
ApéndicesA Cuantificación del campo de radiación
por R. G. Pregliasco .B Cálculos de la teoría cuántica de colisiones . . .C Aproximación de Wentzel, Kramers y Brillouin
por R. G. Pregliasco .
217
217221
227

OPERADOR DENSIDAD
ESTADOS PUROS DE ESPIN
Consideremos un haz de partículas de espín 1/2 que pasaentre los polos de un imán. El haz se divide en dos partes,correspondientes a los dos posibles autovalores m =±h/2 de la
zproyección del operador espín en la dirección del gradiente delcampo magnético. Si eliminamos uno de los haces, las partículasemergentes tendrán todas el mismo autovalor, por ejemplom =+h/2.
zUn aparato del tipo descrito, denominado de Stern-Gerlach,
actúa como un filtro ya que, independientemente del estadoinicial del haz, a la salida éste estará formado exclusivamentepor partículas en un mismo estado de espín. Decimos que el hazha sido preparado en un estado puro del espín: Cualquiera de laspartículas del haz puede describirse con un único vector deestado I~> = a I+z>+ a I-z>, donde los coeficientes a y a
1 2 1 2
dependen de la orientación del filtro. Por otro lado es obvioque si el haz incidente se encuentra en un estado puro del espín,
siempre podrá encontrarse una orientación conveniente del filtrotal que el haz pase por él sin pérdida de intensidad.
VECTOR POLARIZACION
Definimos el vector polarización P de un estado puro I~>como el valor de expectación de las matrices de Pauli,
... ...P = <xla-Ix>
..• APor ejemplo, para el estado Ix>= I+z>, P z. Y como la dirección~ es arbitraria, vemos que en general el vector de polarizaciónapunta en la dirección en que el estado puro I~> tiene proyecciónde espín definida. Si parametrizamos los coeficientes del estadoIx>= a I+z>+ a I-z> con a = cos~/2 y a = ei~ sen~/2, el
1 2 1 2
1

correspondiente vector de polarización resulta
~ 1\ 1\ 1\P = sen~ cos~ x + sen~ sen~ y + cos~ z
Vemos que ~ y ~ son los ángulos polares del vector polarización.Si enviamos un haz en un estado puro Ix> a través de un filtroorientado en la dirección de su vector polarización, todo el hazpasará sin pérdida de intensidad.
ESTADOS DE MEZCLA DE ESPIN
Supongamos que preparamos de manera independiente dos hacesde partículas en los estados puros la> y lb> con flujos J y J
a b
respectivamente. Si ahora investigamos el estado de polarizacióndel haz combinado, pasándolo a través de un filtro Stern-Gerlach,no será posible encontrar una orientación que permita unatransmisión completa del haz. Decimos que el haz combinado noestá en un estado puro o, equivalentemente, que se encuentra enun estado de mezcla.
Nos enfrentamos ahora con el problema de como describir talestado de mezcla. Evidentemente no podemos caracterizar al hazcon un solo vector Ix>, ya que ello implicaría que todos losespines "apuntan" en la misma dirección: la dirección del vectorde polarización. Si pusieramos el filtro en esa direcciónpodríamos lograr una transmisión completa del haz. Y como ellono es posible, tampoco es posible describir una mezcla con unúnico vector de estado.
Desde otro punto de vista, conocemos las probabilidadesW=J/(J+J) y W=J/(J+J) de los estados la> y lb>, y con
a a a b b b a b
ellas, los módulos lA I = ~ Y lA I = ~ de losa a b b
coeficientes de un hipotético estado único Ix>= A la>+A lb>.a b
Pero, como ambos haces han sido preparados de maneraindependiente, no existe una relación de fase definida entreambos. Y sin una fase definida no es posible construir un estadoIx>= A la>+A lb> que describa a la mezcla.
a b
Aunque parezca obvio, una mezcla debe describirse
2

especificando la forma en que ha sido preparada. Por ejemplo ennuestro caso el haz combinado se ha construido con un flujo J de
apartículas en el estado I a> y J en el estado lb>. j y ésta es
b
toda la información que conocemos sobre el estado de mezcla!. Elvector polarización puede calcularse entonces, tornando elpromedio estadístico sobre ambos haces
~p = W
a
~<al<Jla> + Wb
~<bl<Jlb>
~ 222 22 ~ ~La magnitud de P está dada por P = W P + W P + 2 W W P .P,a a b b a b a b
~ W2 + W2 + 2W W = 1 ya que el producto escalar P . P de dosa b a b a b
vectores unidad no puede ser mayor que 1. La igualdad se da sólocuando P =P. En tal caso ambos haces tienen el mismo estado de
a b
espín y el haz compuesto está en un estado puro. En resumen
->donde IPI=l si y solo si el haz se encuentra en un estado puro.->
A un haz con IPI>O lo llamamos polarizado, y por contraste,->no polarizado Sl IP I=0. Los estados puros, correspondientes a
->IPI=l, forman un haz completamente polarizado donde todas laspartículas se encuentran en el mismo estado, con todos losespines apuntando en la misma dirección: la dirección del vectorpolarización P.
OPERADOR DENSIDAD DE ESPIN
Para describir al sistema anterior introducimos eloperador densidad
p = W la><al + W Ib><bla b
Este operador describe la preparación del sistema, y por lotanto incluye toda la información disponible sobre el mismo, yasea que se trate de una mezcla o de un caso puro. Puesto que W
a
es la probabilidad de encontrar una partícula de la mezcla en elestado la>; y l<xla>12 es la probabilidad de que Ix> esté en la>,
3

vemos que los elementos diagonales
<xlplx> = Wa
2l<xla>1 + wb
tienen una interpretación física directa, como la probabilidadtotal de encontrar una partícula en el estado Ix>. Además
tr (p) = W +W = 1a b
lo cual era de esperarse ya que representa la probabilidad deencontrar una partícula del sistema en "cualquier" estado.
OPERADOR DENSIDAD
Ahora generalizamos los resultados anteriores. Cuando no sedispone de la máxima información posible sobre un sistema dado,es necesario utilizar los métodos de la mecánica estadística paraestudiar su evolución. En mecánica clásica la máxima informaciónaccesible sobre un sistema de partículas está dada por laposición y el impulso de todas ellas. En Mecánica Cuántica, encambio, una medida simultánea y precisa de varias variablesfísicas es sólo posible si los operadores correspondientesconmutan. En otras palabras, la medición del mayor conjunto deobservables independientes que conmutan dará la caracterizaciónmás completa del sistema cuántico; y éste estará descrito por unautovector del conjunto de observables. Este estado de máximoconocimiento se denomina puro. Al realizar una medición completahemos preparado al sistema en un estado puro.
Sin embargo, en la práctica, tal preparación completa esraramente posible, es decir que las variables dinámicas medidasno forman un conjunto completo de observables. Corno resultado detal medida parcial el sistema no es puro y no puede representarsepor un único vector de onda. Decimos que se trata de un estadode mezcla estadística. A lo sumo podrá afirmarse que el sistematiene ciertas probabilidades W, W, de estar en los estados
1 2
puros I~ >, I~ >, ... , respectivamente. Y corno solo se dispone de1 2
tal información parcial es necesario utilizar una descripción
4

estadística. Por ejemplo, si queremos calcular el valor deexpectación de un obgervablQ 'A.. dQbQmog promQdiar log valorQgmedios de cada estado puro I~.> de probabilidad W .. O sea
1 1
<A> = L w. <~.IAI~.>1 1 1
Debe notarse que la estadística entra en esta fórmula de dosmaneras: Primero a través del valor de expectación <~.IAI~.> que
1 1
es un promedio inherente a la naturaleza cuántica del sistema.En segundo lugar, el promedio sobre el ensemble aparece en elcálculo debido a la falta de información respecto de en cual devarios estados puros puede estar el sistema.
Para describir este segundo promedio introducimos elconcepto de operador densidad
p = L w. l</>.><</>.I1 1 1
*que es un operador hermítico <</>lpIW> <Wlpl</», definidopositivo <</>Ipl</»~ O Y de traza unidad tr(p) = L w. = 1. Además
1
es fácil mostrar que el valor de expectación anterior puedeanotarse
<A> tr(Ap)
dice que el operador densidadde interés físico acerca del
Este importante resultado noscontiene toda la informaciónsistema.
En particular, si remplazamos A porobtenemos la probabilidad de encontrar alestado IW>
el proyector IW><WIsistema en un dado
W(W) = <WlpIW>
COHERENCIA Y OSCILACIONES CUANTICAS
La presencia de términos no diagonales en la matriz densidadse debe comúnmente a la coherencia existente entre algunos
5

estados de la base elegida. Este hecho se manifiesta en laaparición de fenómenos de interferencla. Un est.ado puro I porejemplo, representa el caso límite de un sistema completamentecoherente. En efecto, tal estado puede escribirse como unasuperposición lineal de estados de la base de autoestados delHamiltoniano
11/1>n
Es decir que existe una relación definida entre las amplitudes y
las fases (coherencia) de dichos estados. El operador densidades no diagonal en dicha representación
y esto da lugar a la aparición de oscilaciones cuánticas debidasa fenómenos de interferencia:
<A> = Ln,m
Definimos entonces:
Se dice que un sistema es una superposición
coherente de los estados de base 1<1>> si la matrizn
densidad <<1>Ip I<1>> es no diagonal. Si, además, eln m
sistema es·tá en un estado puro, se dice que lasuperposición es completamente coherente.
En el otro extremo, unapreparados independientemente,operador densidad p = L W
nn
mezcla de estados de base 1<1>>n
o sea de manera tal que el1<1>><<1>1 sea diagonal en dicha
n n
representación, no involucra una relación de fases bien definida,y por lo tanto decimos que se trata de una superposiciónincoherente de los estados de base. Más generalmente
6

Se dice que un sistema es una superposición
incoherente de estados I~ > (no necesariamenten
ortogonales) Sl su operador densidad puede anotarse
P = L W 1<1> ><<1> 1n n n
n
ECUACION DE LIOUVILLE
En el esquema dedel tiempo pues así loIljJ(t»= U(t)lljJ(O»,p(t) = U(t) p(O) U(t)tLiouville
Schrodinger el operador densidad dependehacen los estado que lo definen. Siendocon ih ~~ = HU (U(O)=l), resulta
y con ello obtenemos la ecuación de
BIBLIOGRAFIA
ih dpdt [H,p]
1. K.Blum: Density Matrix and Applications (Plenum Press, NewYork, 1981).
2. U.Fano: Rev. Mod. Phys. 29, 74 (1957).
7

INTERPRETACION DE LA MECANICA CUANTICA
INTRODUCCION
La mecánica cuántica fué creada a principios de este sigloen respuesta a ciertos resultados experimentales que resultabaninexplicables en el marco de la física clásica. Esta teoríaaportó tres grandes revoluciones. En primer lugar permitióinvestigar un nuevo rango de fenómenos que hasta entonces seconsideraban externos al dominio de la ciencia, tales corno laspropiedades de los átomos y las moléculas, el complejo mundo delas interacciones químicas, la física del núcleo atómico o lascaracterísticas de la materia sólida. La segunda revolución fuela eliminación del determinismo, que siempre había sido uningrediente incuestionable de la física clásica. Si ladescripción cuántica es correcta, el resultado de cualquierexperimento no es predecible. Todo lo que se puede hacer esestimar la probabilidad de que se dé un resultado particular.Este aspecto aleatorio de la física cuántica es de una naturalezadistinta del que ocurre, por ejemplo, al arrojar un dado. En esteúltimo caso la descripción probabilista se da por una falta deconocimento acerca del estado original del sistema, mientras queen la cuántica es de un carácter intrínseco. Sin embargo, estamosacostumbrados a observar un comportamiento aleatorio en el mundoque nos rodea, y por lo tanto podemos llegar a aceptarlo corno unacaracterística de la mecánica cuántica, sin violentar nuestrosentido común.
Este no es el caso con la tercera revolución, ya que pone aprueba una creencia básica, implícita en toda la ciencia y encasi todo el pensamiento humano, de que existe una realidadobjetiva, una realidad que no depende para su existencia de quesea observada o no. En general este punto es omitido por casitodos los libros de texto, a pesar de que expone claramente lasdificul tades conceptuales de la fundamentación de la mecánicacuántica, problemas que están muy lejos de ser resueltos, y quenos proponemos discutir a continuación.
9

FORMALISMO CUANTICO y REALIDAD EXTERNALa única realidad que uno conoce con seguridad absoluta es
la de la propia existencia corno ser consciente. Pero además unosupone que hay una realidad externa, que en parte es causa de lassensaciones que experimenta. Lógicamente no hay necesidad de estasuposición. Una postura que niegue la existencia de la realidadexterna es lógicamente defendible (G.Berkeley, 1685-1753), peroresulta mucho más fácil explicar las propias experiencias en basea un "modelo" que supone la existencia de un mundo real externo.Puede ser modificado, pero no se lo puede hacer desaparecer contan sólo dejar de observarlo.
Aparentemente este esquema consistente de la realidad esdestruido a nivel cuántico. Sabemos que una observación puedemodificar la realidad -esto no es sorprendente-, pero la mecánicacuántica parece indicar, además, que esta realidad es creada enel acto de observación.
Analizemos este problema con un experimento pensado.Consideremos una partícula, descripta por un paquete de onda, queincide sobre una barrera de potencial. Cuando el paquete alcanzala barrera se distorsiona y luego se separa en dos. En esta etapala función de onda es despreciable en casi todo el espacio salvoen dos zonas que describen la transmisión y la reflexión de lapartícula. El módulo cuadrado de esta función de onda da laprobabilidad de que, al realizar una medición, encontremos que lapartícula se ha reflejado o ha seguido de largo. Hasta estepunto, esta descripción del experimento pensado, dada por lamecánica cuántica, no está en contra de muestro sentido común. Dehecho podemos imaginar una explicación simple de lo ocurrido: lapartícula se mueve libremente hasta alcanzar la barrera. Allírealiza una elección y sigue de largo o rebota. Luego de uncierto tiempo la partícula se estará moviendo a uno u otro ladode la barrera con cierta probabilidad. Una observación sóloconfirmará esa realidad, indicando de que lado estaba lapartícula. En otras palabras el detector observa la realidad, nola crea.
Todavía podríamos discutir corno es que la partícula realizó
10

la elección de seguir o rebotar. Y en este punto hay dosescuelas. Las teorías ortodoxas indican que la probabilidad entraen esa elección de una manera intrínseca. Por otro lado, lasteorías de variables ocultas postulan la existencia de otrasvariables además de la velocidad, cuyos valores determinan que lapartícula pase o no. De esta manera se mantiene el determinismo,y el aspecto aleatorio sólo entra a través de nuestra ignoranciade esos valores, exactamente como en la física clásica. Dentro decualquiera de estos dos modelos, la explicación que dimos delexperimento no está en contra de nuestro "sentido común".Lamentablemente, y tal como veremos, esta explicación esincorrecta.
Supongamos que modificamos nuestro experimento colocando unpar de espejos, por ejemplo escalones de potencial, de manera talque, independientemente de que la partícula sea transmitida oreflejada por la barrera de potencial, se dirija hacia unapantalla de detección. Tenemos así un espectro de probabilidadesde observar la partícula en distintos lugares de la pantalla. Unoesperaría que dicho espectro fuese la suma de los queobtendríamos removiendo uno u otro espejo. Sin embargo sabemosque esto no es así. Tal como indica el formalismo cuántico, ladistribución observada cuando ambos espejos están presentes no esla suma de las distribuciones vistas con los dos espejosseparadamente, sino un esquema de interferencia.
Para poder interpretar este resultado debemos aceptar que lapartícula es influenciada por ambos espejos. Y esto no escompatible con nuestro modelo anterior donde la partícula erareflejada o transmitida por la primera barrera de potencial. Porel contrario, este experimento sugiere que cada partícula essimultáneamente reflejada y transmitida. Es como si se dividieraen dos partes, una de las cuales es reflejada por un espejo y laotra por el otro.
Este resultado no sólo está en contradicción con el primerexperimento, donde observamos que la partícula se reflejaba o setransmitía, pero nunca se dividía en dos partes, sino con nuestrosentido común. En las secciones siguientes analizaremos
11

diferentes interpretaciones del formalismo cuántico que intentanresolver esta contradicción.
INTERPRETACION DE COPENHAGUE
Para poder sobrellevar la contradicción anterior entre elmundo observado y nuestro esquema de la realidad se suele indicarque, mientra este último esquema se basa en nuestra experienciadel mundo macroscópico, las aparentes contradicciones ocurren aun nivel microscópico que nos es inaccesible en forma directa.Por lo tanto las palabras y metáforas que usamos para describirnuestra realidad no son adecuadas en el mundo de lo pequeño. Nohay contradicción. Solo debemos intentar construir un nuevoesquema de la realidad aplicable a ese mundo microscópicodisjunto del nuestro de las experiencias diarias. Por ejemplo, enese mundo microscópico existe una complementariedad entre lasnociones de partículas y ondas. Así la descripción ondulatoria esadecuada para explicar un fenómeno de interferencia, mientra quela noción de partícula es útil en todo proceso de medición.Puesto que esta dualidad ocurre en otro mundo que el de nuestraexperiencia diaria, no tiene sentido cuestionar su inserción ennuestra noción de la realidad.
Esta interpretación de la mecánica cuántica se debe en granmedida a Bohr, y es denominada interpretación de Copenhague.Indica que nuestra manera clásica de pensar debe abandonarse alanalizar el mundo microscópico. Todas aquellas contradiccionesque nacen al aplicar nuestros conceptos clásicos a lainterpretación de ese mundo pierden sentido y no debenformularse. Lo único que tiene sentido discutir es el resultadode los experimentos.
ATRIBUTOS DE REALIDAD DE LA FUNCION DE ONDA
Los detractores de la interpretación de Copenhague indicanque esa doctrina es anti-realista, pues implica que no debemosconstruir teorías que expliquen las observaciones, sino tan sólo
12

que estén de acuerdo con ellas, que sean 11 empíricamenteadecuadas". Al adoptar tal actitud positivista nos negamos aexplicar nuestras observaciones en base a un modelo de larealidad, aún cuando creemos en la existencia de tal realidadexterna. La cuántica deja de ser un modelo de la realidad y setransforma en una herramienta de cálculo. Quienes no están deacuerdo con esa actitud positivista, indican que, así como engeneral aceptamos que el campo electromagnético es real, tambiéndebemos considerar que la función de onda no es sólo un entematemático abstracto. Y en principio hay varias razones por las
cuales considerar esta idea. En primer lugar tenemos la evidenciade que las funciones de onda pueden interferir. Por otro lado, laindistinguibilidad entre electrones, plasmada en la simetría dela función de onda, da origen a importantes predicciones que hansido ampliamente verificadas, y que serían difíciles decomprender sin utilizar funciones de onda. De hecho todas lasformulaciones de la mecánica cuántica, incluyendo las teorías devariables ocultas, requieren el uso de funciones de onda.
Nuestra primitiva noción de existencia se refiere a objetoscon una localización espacial, es decir que están en algún lugar.No es difícil extender esta noción a la idea de "densidad", comohacemos, por ejemplo, con un gas. Esta densidad no está en unlugar particular, sino que en cada punto del espacio toma unvalor representativo.
La función de onda de una partícula es similar a unadensidad, con un valor complejo en cada punto de la región deespacio considerada. De esta manera podemos imaginar fácilmente ala función de onda como "algo" desparramado en el espacio con unacierta cantidad en cada punto. Posiblemente la mayoría de losfísicos tengamos esta imagen. Sin embargo hay dos dificultadesasociadas con ella. La principal se debe al hecho de que el mundono consiste en una sola partícula.
NO LOCALIDAD DEL FORMALISMO CUANTICO
La función de onda de dos partículas depende de dosposiciones. Si las partículas son distinguibles y no interactúan,
13

podemos escribirla como el producto de dos funciones de onda,
cada una dependiendo de una sola pogición. Cuando esto ocurre,
podemos mantener nuestra interpretación original, donde ahora haydos densidades independientes. Pero esto es verdad sólo si laspartículas son independientes, lo cual casi nunca ocurre. Dehecho t aún SI las partículas no interactúan, pero sonindistinguibles, los requerimientos de antisimetrización aseguranque la función de onda no es un simple producto de densidades. Yano podemos decir que en un lugar particular la función de ondatorna un valor específico, sino que asociado con cada dos puntosdel espacio hay un valor de la función de onda.
Por supuesto que no podemos detenernos en la consideraciónde dos partículas, y debemos incluir en general N partículas,describiéndolas con una función de onda que depende de N puntosdel espacio. En este nivel la función de onda comienza aparecerse más a un artefacto matemático. Además es ciertamentedifícil imaginarIa corno una densidad. Esta es local: en cadapunto del espacio torna un valor. La función de onda, en cambio,es no-local: Para establecer su valor necesitamos indicar variasposiciones en el espacio.
Esto es completamente distinto que cualquier noción quetuviesemos de la realidad. La función de onda no tiene una dadaposiclon y no tiene sentido preguntar cuanto de ella hay en undado lugar. La idea de que las cosas que son, lo son en lugaresindicados del espacio, parece ser contradicha por la teoríacuántica.
PARADOJA EPR
En 1935, Einstein Podolsky y Rasen (Phys . Rev. 47, 777 )propusieron un experimento pensado que -aunque no fuera ese suobj etiva original- hace explícito el conflicto entre lanaturaleza no-local de la teoría cuántica y nuestro esquema de larealidad. Esta paradoja ha tenido una enorme influencia en elanálisis de la interpretación de la mecánica cuántica, y sólorecientemente ha sido resuelta a favor del carácter no local dela teoría. Siguiendo la reformulación hecha por Bohm de esta
14

paradoja, consideremos una partícula inestable de espín nulo quedecae emitiendo do~ p~rti~ul~~ dé é~pi~ 1)~_ ~l é~tado dé é~píndel sistema está descripto por el espinor
11f1> = 1 (11,+>12,-> - 11,->12,+>)V2
de manera tal que [Sl+S2]1~> = O. O sea que, por conservación delespín, la medición de la proyección del espín del sistema da elvalor cero en cualquier dirección.
Ambas partículas se alejan una de otra, y al cabo de algúntiempo se encuentran totalmente separadas. La no-localidad delformalismo cuántico es aquí evidente, ya que, independientementede cuán separadas estén ambas partículas, sus estados de espínestán relacionados entre sí a través del espinor I~>. En efecto,si medimos la proyección del espín de una de las partículas enuna dada dirección, entonces sabemos que la otra tendrá espínopuesto en esa dirección. El espín de esta segunda partícula noestaba definido antes de la medición. Solo al medir el espín dela primer partícula, el estado de la segunda pasa de ser unamezcla coherente a ser un autoestado puro, no importa cuan lejosse encuentre. Las implicancias de esta observación sontrascendentes: Al efectuar la medición se produce unamodificación en la función de onda que es independiente de suextensión espacial. Esta predicción, debida a la no-localidad delformalismo cuántico, parece estar en contra de la hipótesis deque no puede haber influencia entre regiones espacio-temporalesno conectadas causalmente. En otras palabras, cabría esperar queambas partículas puedan estar lo suficientemente alejadas una deotra como para que la medición del espín de una de ellas noinfluya sobre la medición del espín de la otra.
TEOREMA DE BELL
El conflicto planteado por la paradoja EPR puede expresarseen forma cuantitativa mediante un sencillo teorema publicado porBell en 1964 (Physics 1, 195). Supongamos que en el experimentoEPR medimos simultáneamente el espín de una de las partículas en
15

la dirección ~ y el de la otra en la dirección ~. Sean A y B losresultados correspondientes, que pueden tomar lOQ valorQQ ±lJ1.Asumiendo una hipótesis de localidad, ambas mediciones sonindependientes y el producto AB puede adoptar los valores ±1/4.Si repetimos la medición sobre varios pares de partículas,productos de distintas des integraciones , obtendremos un valormedio para AB que verifica -1/4 ~ <AB> ~ 1/4.
Ahora elegimos dos direcciones g y g, para medir el espín deuna de las partículas y ~ y ~, para medir el espín de la otra.Efectuamos los cuatro pares de mediciones posibles aleatoriamentecon igual probabilidad sobre varios pares de partículas,obteniendo los resultados <AB>, <AB'>, <A'B> y <A'B'>. Con estosdatos calculamos la cantidad
<F> = <AB> + <A'B'> + <AB'> - <A'B>
que no es otra cosa que el valor medio de F = A(B+B')+A'(B-B').Ahora dos casos son posibles: B = B' o B = -B'. En el primer casoresulta F = 2 AB, Y en el segundo F = 2 A'B. Como A, A', B Y B'sólo pueden valer ±1/2, vemos que F = ±1/2, Y por lo tanto, enpromedio,
-1/2 ~ <F> ~ 1/2
En cambio, si usamos el formalismo cuántico obtenemos
1\-'> 11-'> 1\11<A.B> = <'lrl(a.Si)(0.S2)l'lr>= -a.0/4
con lo cual
En particular, si definimos ~, ~', Í) y Í)' por los ángulos 0°,90°, _45° Y 45° respectivamente, obtenemos <F> = -1/1Z. Así, elteorema de Bell establece un criterio para determinar cuálinterpretación es la correcta: Si al medir <F> obtenemos unresultado comprendido entre -1/2 y 1/2, la descripción local escorrecta. Si, en cambio, obtenemos un valor igual a -1/11 dentro
16

del error experimental, la naturaleza no-local de la formulacióncuántica es verificada.
El grupo de Alain Aspect de Francia realizó esteexperimento, empleando fotones en lugar de fermiones (Phys. Rev.Lett. 47, 460 (1981); 49, 1804 (1982)). Estos fotones nacen deldecaimiento de un estado excitado del átomo de calcio, y susimpulsos angulares deben estar correlacionados pues el impulsoangular total es nulo. El resultado experimental coincidetotalmente con la interpretación no-local de la mecánicacuántica, indicando que no existe separabilidad en la descripciónde dos porciones de un sistema cuántico, no importa cuán alejadasentre sí se encuentren esas partes. No es posible una descripciónlocal de la naturaleza.
EL PROBLEMA DE LA MEDICION
Dijimos que al considerar a la función de onda como atributode la realidad surgían dos dificultades. La primera era elcarácter no-local del formalismo cuántico, puesto en evidenciapor la paradoja EPR. La segunda dificultad tiene que ver con elproceso de medición. Cuando realizamos una medida la función deonda cambia de una manera súbita y discontinua. Además, y talcomo vimos, este cambio es fuertemente no-local, en el sentidode que las mediciones realizadas en un punto del espacio cambianla función de onda en otros puntos, instantáneamente, no importacuán alejados estén. El experimento de la barrera de potencialprovee un buen ejemplo de tal proceso de reducción de la funciónde onda. Si un detector ubicado a la derecha señala el paso de lapartícula, el trozo de función de onda a la izquierda se vuelvecero inmediatamente.
Suele decirse que la mecánica cuántica es una teoríauniversal que generaliza a la mecánica clásica. Por lo tantodebería poder aplicarse al aparato que usamos para "medir", y asíexplicar en que consiste la reducción de la función de onda. Sinembargo, es aquí donde se plantea el problema más serio. Dentrode este marco, el sistema "partícula + aparato" deberíaevolucionar de una manera continua y bien determinada, descrita
17

por la ecuación de Schrodinger, y no de una forma discontinua yaleatoria como en un proceso de medición. Vemos Que la mecánicacuántica parece contener una contradicción interna. Aparentementeno puede describir los instrumentos necesarios para realizarmediciones. Este es "el problema de la medición".
OBSERVADOR CONSCIENTE
Un amplio grupo de físicos, incluyendo verdaderas eminenciascomo Eugene Wigner (Am.J.Phys. 31, 6 (1963)), dieron la siguienteexplicación de como es que se produce la reducción de la funciónde onda. Un sistema aislado, no importa que tan complicado sea,evoluciona según las leyes de la mecánica cuántica. Sólo cuandoun observador consciente mide una propiedad del sistema, sóloentonces, el vector de estado se reduce al autoestadocorrespondiente.
Esta imagen de un proceso de medición plantea dificultadesimportantes. En efecto, supongamos que en el experimento de labarrera de potencial usamos placas fotográficas corno detectores.La explicación anterior nos dice que después de que la partículaalcanzó una de ambas placas y hasta que sea observada, ésta seencontrará en un estado indefinido donde tal vez fue velada y tal
vez no. Solo cuando un observador consciente mire la placa, enese mismo instante, la placa se velará por el impacto anterior dela partícula, aunque ese impacto haya ocurrido años antes.
Schrodinger fue uno de los que mas claramente advirtió laimportancia del problema de la medición. Le resultaba absurda lainterpretación "mentalista" de Wigner y para ilustrar estoinventó en 1935 la historia del "gato de Schrodinger"(Naurwissenschaften 23, 807, 823, 844), que consiste en otravuelta de tuerca a la paradoja de las placas fotográficas reciénmencionadas. Supongamos que en nuestro experimento de la barrerade potencial, el aparato de medición no es una placa fotográfica,sino un detector que, al ser alcanzado por el electrón, disparaun arma y mata un gato. Despues de realizado el experimento, ysegún la interpretación de Wigner, el gato se encontrará en unestado de mezcla coherente parte vivo - parte muerto hasta que un
18

observador consciente reduzca la función de onda hacia alguna deambas posibilidades. Este no parece ser un esquema razonable dela realidad.
INTERPRETACION DE MUCHOS MUNDOS
Otra solución posible al problema de la reducción de lafunción de onda fue propuesta por H. Everett III en 1957 (Rev.Mod. Phys. 29, 454) Y dice que tal reducción simplemente noocurre. Todo sistema aislado evoluciona según las prescripcionesde la ecuación de Schrodinger. Si el sistema es observado,debemos incorporar al observador en el sistema. El conjunto esnuevamente un sistema aislado y debe evolucionar según laecuación de Schrodinger. Aclaremos esto con nuestro experimentopensado de la barrera de potencial. Luego de la interacción conlos detectores, podemos incorporar a éstos en la función de onda
con la partícula moviendose a derecha (~ ) o izquierda (~ ) y losD 1detectores (D o r) disparados (+) o no (-) por ella. Según lainterpretación ortodoxa no tendré tal combinación lineal sino unareducción a los estados ~ D+r- o ~ D- r+ con probabilidades la ,2
D 1 D
Y la 12 respectivamente.
1
En la interpretación de Everett tal reducción no ocurre y laparadoja se resuelve incluyendo al observador en el sistema. Lafunción de onda del observador, o mejor dicho su matriz densidad,es muy complicada, pero la única parte relevante para nuestropropósito es aquella parte que "ve" a la partícula moviéndose aderecha o izquierda. Anotamos a estos dos estados como O y O , Y
D 1
los incluimos en la función de onda
Usualmente se dice que después de una medición la función deonda ha colapsado a una de ambas posibilidades. Everett en cambioindica que a través de la medición el observador ha separado al
19

mundo en dos. En uno él vivirá convencido de que la partícula sereflejó en la barrera de potencial. En el otro, su otro yo creeráque la partícula se transmitió. Cada medición separa al mundo enuna colección de mundos, uno por cada posible resultado de lamedición. En este esquema, el "error" de la interpretación usualreside en la suposición tácita de que nosotros podíamos observaral mundo desde afuera, y con ello concluir que un resultado de laobservación ocurría y los otros no. La realidad, en cambio, seríauna función de onda que contiene todos los posibles resultados.La conciencia demanda un resultado (es decir, realiza unamedición) y con ello elije una rama donde existir. Pero comotodas las ramas son equivalentes, la conciencia también seramifica, una por cada resultado.
TEORIAS NO LOCALES DE VARIABLES OCULTAS
Otra manera de evitar el problema de la medición está dadapor las teorías no locales de variables ocultas, en particularpor la teoría de onda piloto de de Broglie. Este nunca estuvo deacuerdo con la manera en que la teoría cuántica se habíadesarrollado, y mantuvo la idea de que las partículas siguentrayectorias definidas. El creía que el rol de la función de ondaera actuar como una onda piloto que guiaba estas trayectorias.Estas trayectorias no son observables, y por lo tanto constituyenuna variable oculta de la teoría. Estas ideas fuerondesarrolladas matemáticamente por David Bohm en 1952 (Phys. Rev.85, 166, 180): En este modelo la función de onda provee una"fuerza cuántica" que, sumada a las otras fuerzas clásicas delsistema, actúa sobre la partícula según las leyes de Newton. Losresultados que se obtienen de esta manera son completamenteequivalentes a los de la mecánica cuántica tradicional, salvo queahora no se necesita incluir la idea de la reducción de lafunción de onda para explicar el proceso de medición. La mediciónsólo consiste en una verificación del estado pre-existente de lapartícula.
A pesar de esto, esta teoría no fue aceptada por variasrazones. En primer lugar porque nació como una forma de mantener
20

el determinísmo en la teoría cuántica, y por lo tanto fue vistacomo una reacción conservadora en contra de la revoluciónconceptual que significaba la teoría cuántica en suinterpretación ortodoxa. Esta sería la razón sociológica delfracaso de los modelos de variables ocultas. Desde un punto devista más pragmático, hay que destacar que estas teorías sonbastante complicadas en contraste con la simplicidad, economía yelegancia de la teoría usual. Más aún, el modelo de onda pilotoes conceptualmente discutible. La 11 fuerza cuántica11 es de unanaturaleza muy extraña, y no verifica el principio de acción yreacción. La onda piloto afecta a la trayectoria de la partícula,pero no es afectada por ésta.
INTERPRETACION ORTODOXA
Hasta ahora hemos visto tres posibles respuestas al problemadel colapso de la función de onda. Dos de ellas, la teoría deonda piloto y la interpretación de muchos mundos, resuelven elproblema diciendo que el colapso nunca ocurre. El otro esquemapropone que la reducción de la función de onda se produce porintervención de un observador consciente.
Existe una cuarta postura, que premeditadamente hemos dejadopara el final, y que aparentemente goza del favoritismo de lamayoría de los físicos. Sin embargo, hay bastante confusión en loque respecta a sus lineamientos generales, hasta tal punto que esfrecuentemente redescubierta en una u otra de sus variantes conmarcada insistencia.
La idea es que los elementos no diagonales de la matrizdensidad del sistema completo (incluyendo al aparato demedición), en una representación correpondiente a estadosmacroscópicamente distinguibles, no pueden ser observados enningún experimento. Por lo tanto, la matriz densidad correcta,que es una superposición coherente de los posibles estadosmacroscópicos del detector, puede reemplazarse por lasuperposición incoherente (es decir, diagonal) que escaracterística de una descripción clásica. En otras palabras,esta interpretación dice que no es posible diseñar un
21

experimento que permita observar la interferencia entre esos
estados y, por lo tanto, pod~mog dQgcribir al sistema con unasuperposición incoherente clásica.
Para entender como es esto, supongamos que queremosverificar si el sistema formado por la partícula y los detectoresde nuestro experimento pensado está descrito, después de lamedición, por la superposición coherente de ambos posiblesresultados. Para ello tenemos que hacer interferir ambas partesde la función de onda. Sin embargo, aún cuando las partes de lafunción de onda correspondientes a la partícula reflejada y
transmitida sean reunidas por medio de espejos, no interferiránentre sí, ya que ahora contienen los estados macroscópicamentediferentes (de hecho, ortogonales) de los detectores. Para lograrinterferencia deberíamos llevar ambos detectores a exactamente elmismo estado, y esto está fuera de las posibilidades de cualquiertécnica experimental.
Hay que aclarar que esta propuesta no niega que lainterferencia entre estados macroscoplCOS pueda existir, sinosugiere que tal interferencia es prácticamente inobservable enlos experimentos usuales.
DECOHERENCIA
Recientemente la interpretación ortodoxa ha logrado unsustento formal a través de la teoría de la decoherencia (W. H.Zukek, Phys. Rev. D 26, 1862 (1982». Esta describe un efectodinámico por el cual los estados macroscópicos de un sistema sevuelven rápidamente ortogonales.
La idea de esta teoría es que la decoherencia tiene el mismoorigen físico que la disipación, ya que describe la interacciónentre las variables macroscópicas y microscópicas de un sistema.Esto significa que está relacionado con el coeficiente dedisipación A que entra en la ecuación clásica para la. evolucióntemporal de las variables macroscópicas. Por ejemplo, para unoscilador de masa m y frecuencia w, la ecuación clásica para laposición del centro de masa es
22

2d x t A dx t W2X = Odt2 dt
Para este sistema la teoría de la decoherencia muestra que loselementos no diagonales de la matriz densidad reducidacorrespondientes a dos estados macroscópicos caracterizados porposiciones x y x son proporcionales a (R. Omnes, Rev.Mod.Phys.
1 264,2(1992))
Con una masa de 1 gramo, un período de 1 segundo, y un tiempo derelajación de 10 minutos, se encuentra que para una diferencia enel estado macroscópico de apenas Ix1-X21 = 1 micrón, este factorse ha vuelto del orden de e-10000 después de transcurridoapénas un 1 nanosegundo desde la preparación del sistema. Vemosque este efecto de decoherencia está entre los más eficientesconocidos en la física, lo cual explica la imposibilidad prácticade observar ningún efecto de interferencia entre estadosmacroscópicamente distintos.
BIBLIOGRAFIA
1. R.Omnes: Rev.Mod.Phys. 64, 339 (1992).Una revisión completa y actualizada sobre este tema.
2. J. A. Wheeler & W. H. Zurek eds.: Quantum Theory and
Heasurement (Princeton University Press, 1983).Una recopilación de los más importantes artículos publicadossobre este tema entre 1926 y 1981.
3. B.J.Hiley & F.D.Peat eds.: Quantum Implications (Routledge &Kegan Paul, London, 1987)
23

Algunas otras referencias de carácter divulgativo son:
4. V.H.Ponce: El formalismo cuántico y el teorema de Bell(Informe CNEA-CAB 035 de 1987).
5. E.Squires: The Mystery of the Quantum World (Adam HilgerLtd., Bristol, 1986).
6. A.Rae: Quantum Physics: Illusion or reality (CambridgePress, 1988).
7. F.Laloe: La Recherche 17, 1360 (1986).8. B.d'Espagnat: Scientific American 241 (11), 128 (1979).
24

MOMENTO CINETICO
MOMENTO CINETICO
El operador vectorial ~J es un momento cinético si sus
componentes J, J Y J son observables que verifican lasx y z
relaciones de conmutación
[J ,J ] = ih Jx y z
[J ,J ] = ih Jy z x
[J ,J ] = iñ Jz x y
Estas reglas de conmutación pueden generalizarse en la forma
-7-7-7-7 • -7:-+-7[aoJ,boJ] = lh (axb)oJ
Son ejemplos de momentos cinéticos el operador momento angular-7 -7-7.'-7 ",-7L = rxp = -lhrx'i7y el espln S o
Introducimos los operadores adjuntos
1
v'2(J ±iJ )
x y
Resulta más cómodo -y así lo haremos de aquí en adelante-trabajar con las componentes {J, J ,J} (con J = J) que con
- o + o z
{J ,J ,J}o Sus relaciones de conmutación sonx y z
o
[J ,J ]+ -
- h Jo
2 2 2donde J se anota J =J -J J -J J oo + - - +
25

REPRESENTACION ESTANDAR /j,m>
2J conmuta con la componente J, por lo cual es posibleo
formar un sistema completo de vectores propios comunes. Como J2es hermítico y definido positivo, sus valores propios sonnecesariamente positivos o nulos. Convenimos en escribirlosh2j (j +1), con j~O. O sea que anotamos con I j ,m> los vectorespropios (que<jmlj'm'> = o .. o )
]J' rnrn'hm respectivamente
exigimos2de J y J,
o
que sean ortonormalizadosde valores propios h2j (j+l) Y
J Ij,m> = ñ m Ij,m>o
Ahora queremos ver cuál es el efecto de los operadores J± sobreestos vectores. Usando las relaciones de conmutación obtenemos
y
Jo J+ Ij ,m > .= (J+ J ± h J+ ) Ij ,m > = h (m± 1) J+ Ij ,m >_ _ o _ _
o sea que los vectores J±lj,m> son también vectores propios de J2y J de autovalores (j,m±l). Para normalizarlos utilizamos la
o
expresión J2 = J2 -J J -J J Y la relación de conmutacióno + - - +
[J ,J ] = -ñJ resultando+ - o
IIJ Ij,m>1I2= - <j,mIJ J Ij,m> =
+ - += - <j,ml(J J +J J )/2 + (J J -J J )/2Ij,m> =
-+ +- -+ +-
= <j ,m I(J2-J 2)/2 - ñJ /2 Ij ,m> =o o
= ñ2[j(j+l)_m2_m]/2 <j,mlj,m> =
= h2(j-m)(j+m+l)/2
y en forma similar
IIJ Ij,m>1I2= - <j,mIJ J Ij,m>+ -
ñ2(j+m)(j-m+l)/2
26

Como una norma no puede ser negativa, vemos que los estadosJ±lj,rn> sólo están definidos cuando
-J :::m :::]
en cuyo caso
J Ij,rn> = -h ¡ (j-rn) (jtrntl)/2' Ij,rntl>+
J Ij,rn> = th ¡(jtrn) (j-rntl)/2' Ij,rn-l>
donde hemos fijado arbitrariamente una convención de fases.En particular vemos que J± Ij,m> = O si y sólo si m = ±j,
respectivamente.
ESPECTRO DE J2 Y Jo
Si m < j podemos construir el estadoIj,m+l> = - + [(j-m) (j+m+l)/2r1
/2 J+lj,m>. A partir de este
estado podemos construir el estado Ij,m+2> ~ J Ij,m+1>. Siguiendo+
así, obtenemos una sucesión de estados I j ,m'> con m' > m. Sinembargo, como J Ij,m'> no está definido para m'~ j, esta sucesión
+
debe anularse en algún punto. Es decir que J Ij,m+p> = O para+
algún p que debe verificar m+p = j. En conclusión j-m es unentero no negativo. Podemos repetir el mismo argumento utilizandoel operador J_ para construir estados Ij,m-1 >, I j,m-2 >, encuyo caso concluiremos que j+m es también un entero no negativo.
De ambos resultados obtenemos las siguientes reglas
a. Los unlCOS autovalores posibles de J2 son de la formah2j(j+1) donde j es un entero o semientero no negativo:
j O, 1/2, 1, 3/2, ...
27

b. Los únicos autovalores posibles de J son de la forma hmo
donde m es un número entero o semientero
m = O, ±1/2, ±I, ±3/2, ...
c. Si h2j(j+l) Y hm son los autovalores respectivos de i y Jo
correspondientes a un autoestado común I j ,m> , los únicosvalores posibles de m son las 2j+l cantidades
m = -J, -j+l, ... , J
En conclusión, basándonos exclusivamente en las relacionesde conmutación de las componentes del operador vectorial momentocinético, hemos construído una base {Ij,m>} de autovectorescomunes de J2 y J, donde para cada valor propio h2j(j+l) de J2
o
(con j entero o semientero no negativo) le corresponde una seriede (2j+l) autovectores Ij,m> con m = -j, -j+l, ..., j, que sededucen unos de otros por la acción de los operadores J y J .
+
SUMA DE MOMENTOS CINETICOS
Sea un conjunto completo de autoestados Ij1,j2,m1,m2>== I j ,m > I j ,m > en el subespacio caracterizado por los momentos
1 1 2 2
cinéticos que conmutan J y J. Puesto que J2 y J2 conmutan con1 2 1 2
el momento cinético total J = J +J, podemos desarrollar el1 2
2subespacio en una base de autovectores Ij,m> de J y J. Pasamoso
de una representación a otra por medio de una transformaciónunitaria
Ij ,j ,j,m> = L Ij ,j ,m ,m ><j ,j ,m ,m Ij,m>12 12121212m ,m1 2
donde <j ,j ,m ,m I j ,m> son los denominados coeficientes de1 2 1 2
Clebsch-Gordan. Fijamos las fases relativas de los vectores Ij,m>(y con ellas, de los coeficientes de Clebsch-Gordan) para unmismo valor de j con la condición estándar de que los primerospuedan deducirse unos de otros con la aplicación de los
28

operadores J Y J. Los vectores I j,m> quedan así definidos a+
menos de una fase arbitraria que depende de j. Hacemosdesaparecer esta arbitrariedad con la condición de que elcoeficiente <j ,j ,j ,j-j Ij,j> sea real y positivo.
1 2 1 1Aplicando J y J a ambos miembros de la ecuación anterior
+ -se obtienen las relaciones de recurrencia
j (j-m)(j+m+1)' <j ,j ,m,m Ij,m+1> =1 2 1 2
= / (j +m )(j -m +1)' <j ,j ,m -l,m Ij,m> +1111 1212
+ / (j +m )(j -m +1), <j ,j ,m ,m -11j ,m>2222 1212
/ (j +m) (j -m+ 1) <j ,j ,m ,m 1j ,m-1 > =121 2
/ (j -m) (j +m +1), <j , j ,m +1,m 1j ,m> +1111 121 2
+ / (j -m )(j +m +1), <j ,j ,m ,m +11j,m>2222 1212
Cuando m = j el primer miembro de la primera ecuación se anula, yla aplicación reiterada de esta fórmula, junto con la condiciónde normalización de 1j ,j ,j,j>, L 1<j ,j ,m ,m Ij,j>12= 1, Y
1 2 1 2 1 2m ,m1 2
la convención de fases anterior, permite obtener cada coeficiente<j ,j ,m ,m Ij,j> a partir de <j ,j ,j ,j-j 1j , j >. Todos los
1212 121 1
demás coeficientes de Clebsch-Gordan se deducen aplicando lasegunda relación. Como estas relaciones de recurrencia son decoeficientes reales, con la convención de fase elegida resultaque todos los coeficientes de Clebsch-Gordan son reales. Luego,la relación inversa es
IJ",J",m ,m > = \' IJ",J",J",m><J",J",m,m IJ',m>1 2 1 2 .L-' 1 2 1 2 1 2
J ,m
Como siempre, a cada valor de j le corresponden 2j+1 valoresposibles de m desde -j hasta j. El mayor valor posible para m esm = j +j , y por lo tanto ése es también el mayor valor posible
1 2
de j, i.e.: j ~ j +j. El cambio de base debe mantener la1 2
dimensión del subespacio, por lo tanto el menor valor posible de
29

], que anotamos jmin' debe verificar
J1+J2
(2j1+1)(2j2+1) = L (2j+1) =J=J .rnln
(j +j -j. +1)(j +j +j. +1)1 2 rnln 1 2 rnln
de donde obtenemos J = IJ' -J' l. Resulta entonces el siguiente. 1 2rnln
teorema:
Teorema Fundamental de la suma:
En el subespacio de dimensión (2j +1) (2j +1) subtendido por los1 2vectores Ijl,j2,m1,m2> (j1 y j2 fijos)
a. Los valores posibles de j son
IJ' -J' 1, •.. , J' +J' -1, J' +J'1 2 1 2 1 2
b. A cada uno de ellos le corresponden los (2j+1) vectoresIj,m> con m = -j, -j+1, ..., j.
SIMBOLOS 33 DE WIGNER
En muchas aplicaciones, las propiedades de simetría de loscoeficientes de Clebsch-Gordan resultan muy confusas, conviniendousar los símbolos 3J de Wigner:
j -j +m(-1) 1 2
v2j+1<j ,j ,m ,m Ij,m>121 2
Estos coeficientes son reales y, en virtud del teoremafundamental de la suma, son nulos salvo que sus parámetrosverifiquen las condiciones dadas
[~: ~: ~:] = O
30

donde con 8(j1,j2,j3) indicamos que los números J1, J2 Y J3 debenverificar la desigualdad del triangulo
Los símbolos 3J toman el mismo valor ante una permutación par desus columnas
mientras que(-1) jl+j2+j3
una permutación impar introduce una fase
Además satisfacen la condición
[~: ~: ~:] o
y las reglas de ortogonalidad
SUMA DE TRES MOMENTOS CINETICOS
El problema de la adición de tres momentos cinéticos es. ..., ...,...,...,
formar los vectores proplos del momento total J = J +J +J en el1 2 3
espacio subtendido por los (2j +1)(2j +1)(2j +1) vectores123
31

I j ,j ,j ,m ,m ,m > = I j ,m > I j ,m > I j ,m >. Hay tres modos de123123 112233
acoplar estos vectores, acoplando dos de ellos entre sí, y luegocon el tercero. Por ejemplo, acoplando primero J y J
1 2
Ij ,j ,j,m> =12 3
o acoplando primero J y J , o J Y J. Para pasar de una de2 3 3 1
estas bases a la otra debemos realizar una transformaciónunitaria dada por los coeficientes W de Racah
=
En general es preferible reemplazar los coeficientes deRacah por los símbolos 6J de Wigner que presentan una mayorsimetría
de manera tal que
Con el fin de evitar algunas confusiones en el empleo de lossímbolos 6J resulta útil asociar a cada símbolo un tetraedro comomuestra la figura. Cada par de aristas opuestas está asociado alos dos momentos cinéticos de una misma columna. Además, los tresmomentos cinéticos de la primer fila corresponden a las aristasde una de las caras del tetraedro. Las simetrías del símbolo 6Json tales que para definirlo es suficiente con dar los seis
32

momentos cinéticos y sus posiciones relativas sobre el tetraedroasociado
Así el símbolo 6J es invariante cuando se permutan doscualesquiera de sus columnas, o cuando se intercambian doselementos cualesquiera de la primer fila por los que lescorresponden en la segunda. Por ejemplo
{j 1 j2 j3}Además, para que 1 1 1 sea distinto
123las ternas correspondientes a cada cara
de cero es necesario que
a) verifiquen las desigualdades del triángulob) Tengan como suma un número entero.
Finalmente indicamos las relaciones de ortogonalidad
~ (2j3+l) {~'J2 j3} {j, j2 j 3 } = 1 <51 l'1 1 1 1 l' 21 +1
J 1 2 3 1 2 3 3 3' 33
~ (_1)j3+13+1; F: J2
~3} e' 1j3 } {j,
1 ~:}(2j +1) 2 23 1 3 J2 jl l' J2 1
J3 2 3 1
y la regla de suma
33

L ( 1)m +m +m + 1 +1 +1 [~:1 ~:][~:1 ~:][~:1 ~:]2 3 1- 123123 =
m ,m ,m -m -m -m2 3 1
1 2 3
= [~:J2 ~:]F: J2 ~:}M 1
2 2
NOTACION y CONVENCIONES
No hay unanimidad en la notación o en las convenciones defase que se utilizan en el estudio del momento cinético y otras
cantidades relacionadas. Por ej emplo, en la definición de losoperadores adjuntos J+ + /I72 (J ±iJ) hemos usado unax y
estructura que los asemeja a los armónicos esféricos~r Y + (r) = + v'3/8rr (x±iy), en lugar de la definición estándar
1 _1
(usada por Messiah, por ejemplo) J+_ = J ±iJ . Esto no esx y
accidental, ya que la nuestra es la única elección coherente conla definición usual de las componentes estándar de los tensoresirreducibles, tal como veremos más adelante.
La convención de fases en la definición de los coeficientesde Clebsch-Gordan es la adoptada por la mayoría de los autores.
BIBLIOGRAFIA
1. A.Messiah: Mecánica Cuántica, tomo II (Ed. Tecnos, Madrid,1975).
2. M. Weissbluth: Atoms and Molecules (Academic Press, NewYork, 1978).
3. M. E. Rose: Elementary Theory of Angular Momentum (JohnWiley & Sons, New York, 1957).
4. A. R. Edmonds: Angular Momentum in Quantum Mechanics
(Princeton Univ. Press, 1957).
34

TEOREMA DE WIGNER - ECKART
OPERADOR DE ROTACION
Consideremos una partícula en un estado dado por la función-7de onda l/1 (r). Realizamos una rotación R del "sistema físico",
definiendo una nueva función de onda
A
Por ejemplo, ante una rotación de ángulo e alrededor del eje z->obtenemos R~(8) W(r) = W(x cos8 + y sen8, -x sene + y cos8, z).
En particular, si la rotación es infinitesimal, resulta
W(x + y 08, -x 08-> 8WW(r).+ 08 (y 8x -
l ->(1- h 08 Lz) W(r)
+ Y , z) '"x 8l/1 ) =
ay
Generalizamos este resultado a un sistema constituído por más deuna partícula: Si J es el momento cinético total, el operador derotación infinitesimal en ángulo 08 alrededor del eje ~ es:
i A->R~(08) = (1- h oe n.J)
Toda rotación finita puede considerarse como una sucesión derotaciones infinitesimales de manera tal que:
con lo cual
dRAn =d8 RA (O) = 1n
que integrando resulta:
35

1/\"h 8 (n.J)Rfi(8) = e
Vemos que el operador de rotación es un operador unitario RRt= 1que transforma los estados segGn la ley
IW') = R IW>
Por otra parte, un observable T representa una operación demedida, y la rotación en bloque del aparato de medición no debemodificar su resultado, siempre que efectuemos también unarotación del sistema físico. Es decir:
<I/1ITII/1> = <1/1' IT' 11/1' >
Por lo tanto
T' = R T Rt
o sea que los operadores se transforman siguiendo las mismasleyes básicas que los estados.
OPERADORES TENSORIALES IRREDUCIBLES
Por definición un operador T se transformasegún la ley RTRt. Ahora, si n operadores T q = 1,
qtransforman linealmente entre sí, es
por rotación2, ... , n sedecir si
t nRT R = La, T, decimos que esos operadores son lasq q ,= 1 qq q'componentes de un operador tensorial de dimensión n.
Para que la propiedad anterior se verifique cualquiera seala rotación, es suficiente con que se verifique para todarotación infinitesimal. Ahora bien
1 1\ ~= (1- h ~8 n.J) T (1+l !\ ~q
= Tq h 88 n.[J,TqJ
36
i 1\ ~h 08 n.J) =

Y, por lo tanto, para que se verifique la relación anterior esnecesario y suficiente que
~[J,T ] =
q
n
\' bL- qq'
q'=lT q'
Esta propiedad puede adaptarse como una definición alternativa deun operador tensorial de dimensión n.
Por último, si el espacio generado por estos operadores esirreducible, es decir si no existe ningún subespacio deoperadores cuyos elementos se transformen entre sí por rotación,éstos se dicen irreducibles. Definimos entonces las (2k+1)componentes estándar Tk de un operador tensorial irreducible Tk
q
de rango k, como aquellas que ante una rotación transforman comolos autoestados Ik,q> del momento cinético:
R Tk Rt = L <k,q'IRlk,q> Tk
q q'q'
Para una rotación infinitesimal
<k ,q' IRA (08) Ik ,q>ni A ~
= <k,q' 1,1-h 08 n.Jlk,q> =1 A ~
= oqq , h 08 n.<k ,q' IJ Ik,q>
con lo cual
RA(08) Tk RA(-08)n q' n = Tk_ 1 08 fL L <k,q' IJlk,q> Tk
q' h q'q'
y comparando con una expresión anterior resulta que
q'
= L <k,q' IJlk,q> Tk
q'
Puesto que
oq'q±l
h q
= :+:h j (k:+:q)(k±q+1)/2'o q'q
<k,q' IJ±lk,q><k,q' IJolk,q> =
37

podemos expresar la propiedadcomponentes irreducibles deldefinición alternativa:
anterior en términos de las mismas~vector J, obteniendo la siguiente
kLos operadores T son las componentes estándar deq
un operador tensorial irreducible Tk de rango k si ysolo si
componentes cartesianas de unestándar son
Con esta definicióntensoriales irreducibles(momento angular, espín,tensoriales irreducibles
vemos que los escalares son operadoresde rango O. Los operadores vectorialesvector posición, etc.) son operadoresde rango l. Si A, A, A son lasx y z
operador vectorial, sus componentes
A± :¡: 1 (A ±íA )= -v'2 x y
A = Ao z
Las (2i+1) funciones Cl = vi 4rr/(2l+1)Y (8,lj», cuando se lasm 1m
considera como operadores, son las componentes estándar de unoperador tensorial irreducible de rango l: el tensor de Racah Cl
•
TEOREMA DE WIGNER - ECKART
Consideremos ahora losConstruimos la siguientecoeficientes de Clebsch-Gordan
(2k+1)(2j'+1) vectores Tk/j'm'>.q
combinación lineal, usando los
Tklj',m'> <j'k,m'q/j",m">q
o, usando la propiedad de ortogonalidad,
38

Ahora bien
TkIJ",m" L 11/1 '/ = > <J'k m'q/]'" m")q j "m" , ,j "m"
(1)
= ím'q
([J ,Tk] + TkJ ) Ij' ,m'> <j'k,m'qlj",m"> =+ q q +
= L {-h ¡(k-q)(k+q+l)/2' Tk Ij' ,m'> -q+lm'q-h ¡-(-j-'---m-' -) {-j-'-+-m-'-+1-)-/-2'T:' j , ,m' +1 >} <j , k,m' ql j 11 ,mil> =
= - L {h ¡(k - q+1) (k +q) / 2' < j ,k , m ' q-11 j 11 ,m 11 > +m'q
+ h / (j'-m'+1)(j'+m')/2' <j'k,m'-l qlj",m">} T:lj' ,m'>
y, aplicando una relación de recurrencia de los coeficientes deClebsch-Gordan
m'qJ+ 11/1j "m" >
o sea
= - L h / (j" -m" ) ( j "+m"+1 ) / 2' <j , k, m' q 1 j " , m"+1> Tk 1 j , , m' >q
J 1,1, > = - h / (J'''-m'') (J'''+m''+1)/2' 1,1, >+ 'l'j"m" 'l'j"m"+l
De manera similar se demuestra que
J 11/1'''m''>- J= h / (J'''+m'') (J'''-m''+1)/2' 1,1, >
'1' j"m"-l
Entonces, los productos
quizás cuando j" = j y m"
el bra <j,ml
J I,h > = h m" 1,1, >'P . llmll 'P . 11m"o J J
De estas relaciones se deduce que, o bien I·h > = O para todo'P j "m 11
m", o bien, y salvo por una fase arbitraria, 1,1, > ()( 1 J'" m">.'P j 11m 11 ,
<J·,ml·l' > son todos nulos, excepto'P j"rn"
= m, con lo cual, multiplicando (1) por
39

<j,mITklj' ,m') O<. L <j,mlj",m") <j'k,m'qlj",m") =q ·11 11J m
= <j'k,m'qlj,m>
Obtenemos así el teorema de Wigner-Eckart:
En la representación estándar {J2,J} el elementoz
de matriz <a,j,mITkla' ,j' ,m') de la q-ésima componenteq
estándar de un operador tensorial irreducible de ordenk, es igual al producto del coeficiente deClebsch-Gordan <j'k,m'qlj,m> por una cantidadindependiente de m, m' o q.
<a,j,mITkla',j',m') =q
= 1 < j ,k , m ' q I j ,m > < a , j 11Tk 11a' , j ') =v 2j+l
. (' k J")(_l)J-m J-m q m' <a , j 11Tk 11a' , j , >
A esta cantidad <a,jIITklla',j' > se la denomina elemento de matrizreducido.
Corolario 1:
Para que el elemento de matriz <a,j,mITkla' ,j' ,m'>q
no sea nulo es necesario que 1j-j' 1 :S k :S j+j' yq = m-m' .
REGLA DE SUMA
Las propiedades de interés físico se relacionan con elmódulo cuadrado de los elementos de matriz 1<I/1ITII/1>12. Además
1 2
es muy común que no distingamos estados de diferentes númeroscuánticos magnéticos, por lo cual, sumando sobre ellos, obtenemos
40

Lm q
1 <a, j ,mITkla' , j' ,m' > 12
=m' q
1 I . Tk , ., I 2 \'= 2'+1 <a,JII lIa,J > L.J m q
<j' ,k,m' ,qlj,m>2m'
Sumando sobre q y m'
L l<a,j,mITkla' ,j' ,m'>12 =m q m' q
y la suma sobre m es inmediata
12j+l
jI < a , j 11T
k11a' ,j , > I 2 L 1
m=-j
\' I . I Tk I ' ., , > I 2L. <a,J,m a ,J ,mm q m' q
= l<a,jIlTklla' ,j'>12
Estos resultados permiten una clara interpretación física delmódulo cuadrado del elemento de matriz reducido como la sumasobre los números cuánticos magnéticos de las probabilidades detransición.
CALCULO DEL ELEMENTO DE MATRIZ REDUCIDO
En general el cálculo del elemento de matriz reducido de unoperador tensorial Tk depende de nuestra habilidad para calcularel elemento de matriz de al menos una componente de Tk
• Sea, por1 ->ejemplo, el operador T = J. Por un lado
<j,mlJ Ij'm'> = h m <5 <5o jj' rnm'
y por otro
-><j',l,m',Olj,m> <jIlJllj'> =<j,mIJ Ij',m'>
o
1= ---
v2j+1'1
=m «5 mm' -><jIlJllj'>
con lo cual
v2j+1' vj (j+1)'
-><jIlJllj'>= h vj(j+1)(2j+1)' <5j j'
41

BIBLIOGRAFIA
1. A.Messiah: Mecánica Cuántica, tomo II (Ed. Tecnos, Madrid,1975).
2. B. L. Silver: Irreducible Tensor Methods (Academic Press,New York, 1976).
3. U. Fano & G. Racah: Irreducíble Tensoríal Sets (AcademicPress, New York, 1959).
42

ONDAS ELECTROMAGNETICAS EN EL VACIO
ONDAS ELECTROMAGNE~ICAS EN EL VACIO
El campo electromagnético en el vacío y sin fuentes estádescrito por los vectores de campo eléctrico y magnético quesatisfacen las ecuaciones de Maxwell
Ley de Poisson divE = O
Conservación del flujo divB = O
Ley de Faraday rotE = _(, a-gat
Ley de Ampere rot-g (,aE
¡..L e ato o
donde e y ¡..L son la permitividad y la permeabilidad del vacíoo o
respectivamente, y (,es una constante de proporcionalidad
MKSA
(, 1¡..L 4rr l07Nw/Amp2
o
e 8.85 lO-12Cou12/N m2o
gaussiano
l/c
1
1
-7Como B es un campo de divergencia nula, podemos definir unpotencial vectorial tal que -g = rot A. Reemplazando en la ley de
~ aAFaraday obtenemos que rot (]:!; + (, at) O, por lo cual también
podemos definir un potencial escalar </> tal que E + (, ~~ = - V</>.Vemos entonces que ambos campos pueden generarse a partir desendos potenciales escalar </> y vectorial A como
rot A y E = - V</> _ (, aAat
Sin embargo los potenciales no están completamente definidos porestas ecuaciones. En particular E y B quedan inal terados anteuna sustitución A ~ A + Vx, </> ~ </> - (, ~~, con X cualquier campoescalar. Esta propiedad se denomina invariancia de gauge o de
43

medida y nos permite imponer una condición adicional sobre K, quedesignamos de gauge coulombi ano o transversal, div ~ = o.Cuando A satisface esta elección de Gauge, ~ = O~ y con ello
E - - ( aKat B = rot KLa ley de poisson se satisface automáticamente, mientras que laley de Ampere da origen a la ecuación de onda homogénea. Enefecto, reemplazando las expresiones anteriores para los camposeléctricos y magnéticos en la ley de poisson obtenemos
o sea
-7rot rot A = t; /J.o
con lo cual, aplicando la condición de gauge coulombiano,
= O
con c = 1/ (~ la velocidad de la luz en el vacío.o o
Una solución posible a esta ecuación está dada por una ondaplana monocromática de frecuencia w (donde se sobrentiende eltomar la parte real)
. Jt -7 tl(l\..r-w )eo
con A una amplitud compleja.o
La misma ecuación de ondas fija la magnitud del vector de onda:
k w/c
Además, el gauge coulombiano impone la condición
44

it.X oo
Es decir que la onda electromagnética es una onda transversal.Los campos eléctricos y magnéticos están dados, respectivamente,por:
E = i( w A
Vemos que ambos campos son perpendiculares entre sí, y respecto ala dirección de propagación. Además están en fase y con unarelación constante entre sus magnitudes.
El flujo de energía de la onda está dado por el vector dePoynting
s = (~11 Real(E) x Real(~)o
donde i\ es una constante de proporcionalidadsistema MKSA y 4rr en el sistema gaussiano.campos electromagnéticos E y ~ de la onda plana
igual a 1 en elReemplazando los
resulta
y, promediando sobre un período, obtenemos el flujo promedio deenergía o intensidad de la onda luminosa:
s = w lA 12 R2 i\11 oo
POLARIZACION y HELICIDAD
Dada la onda electromagnética plana
. ;t ~ tl(.l\..r-w )e k = w/c
podemos desarrollar la amplitud en una base de versores
45
A Ae y e1 2

ortogonales entre sí y respecto de la dirección de propagación
(~ x ~ = ~)1 2
A =A@+A@o 1 1 2 2
entonces
En un punto fijo del espacio el vector campo eléctrico gira en
una elipse con frecuencia w. Decimos que la onda está
elípticamente polarizada: Como caso límite, si ~~ es un múltiploentero de n, la elipse degenera en un segmento, y decimos que laonda está linealmente polarizada. En cambio si A y A son
1 2
iguales en magnitud, pero difieren en n/2 en la fase,
y por lo tanto
K = A 1°12
(Á +' Á) i (it ."f-wt )e _~e e
1 2
~Real(E) =-E°
1
12[ sen(it."f-wt)~ ± COS(it."f-wt)~ ]
1 2
Vemos que el campo eléctrico es constante en magnitud pero, en unpunto fijo del espacio, gira con frecuencia w. Decimos que laonda está polarizada circularmente. El sentido de giro define ladenominada helicidad de la onda. Con el signo superior decimosque la onda tiene una helicidad positiva. Por el contrario, parael signo inferior decimos que la helicidad es negativa. Ambosversores de helicidad definida
1
12(Á+'Á)e _~e
1 2
forman una base igualmente aceptable para la descripción delestado de polarización de la onda plana. Anotamos entonces:
[A ~ + A ~ ]+ +
46
. Tt -7 tl(r...r-w )e

FOTONES
La luz interactúa con la materia como si estuviera compuestade fotones. Un haz de luz es, de hecho, un haz de fotones. Unfotón es una partícula sin masa que podemos caracterizar con unvector de onda t y un versar de polarización ~ perpendicular a t.Su energía es hw y su impulso ht, con w = kc la frecuencia deoscilación. Por lo tanto, el flujo medio de energía de un fotón,
1 "hc2;7confinado a un volumen V es S = V hw c K = V K. Comparando con
... t' f .. ;7 A duna expreslon an erlor, vemos que un oton (h,e) se correspon econ una onda plana monocromática
~ ~ A i(K.I-wt)A(r,t) = A e eo
con
En lo que sigue diremos que un haz depolarizado si sus propiedades deespecificarse con un únicoEquivalentemente diremos que el haz espuede asignar una única frecuencia w.
las fuentes de luz consisten en un
1uz está completamentepolarización pueden
vector ~ = a ~ + a ~ .+ +
monocromático si se leSin embargo, en generalgran número de átomos
excitados que, independientemente uno de otro, emiten fotones dedistintas polarizaciones y frecuencias. Por lo tanto, no esposible encontrar un par de parámetros (K,~) que caractericen atodo el haz. Decimos que el haz luminoso se encuentra en unestado de mezcla.
TENSOR DE POLARIZACION
Consideremos una mezcla de varios haces de luz que han sido
47

preparados independientementeintensidades 1 . Definimos el
n
en estados de polarización ~ conn
ten~or de polari2ación en notacióndiádica
e = L ~n
n1
n
A *en
de manera tal que
A* A= e .C.e
es la intensidad del haz combinado cuando ~ste atraviesa unfiltro que solo admite luz de polarización ~.
Proyectando sobre estados de helicidad definida ~ podemos(J'
anotar en forma matricial
e = [~--~-.]+- ++
con
e 1\. 1\= e .C.e(J'(J" (J' (J"
intensidad total del•e e I por lo cual, y además de la
¡.LV V¡.Lhaz, sólo es necesario medir tres parámetrosdeterminar completamente al tensor e y con
polarización del haz. El conjunto másparámetros está dado por los denominados
Evidentemente
independientes paraél, al estado deconveniente de talesparámetros de Stokes
PARAMETROS DE STOKES
Definimos los siguientes parámetros
El grado de polarización lineal respecto de dos ejesortogonales
T}3
o o1(0 ) - 1(90 )
1
48

Igual medida con un desplazamiento de 45°o o
== 1(45 ) - 1(1].5 >1/ 1 I
El grado de polarización circular1+ - 1-
r¡2 = I
En términos de estos parárnetros, el tensar de polarización seanota
1e = -2- [
1-r¡ -r¡ -ir¡ ]231
- TI + i TI 1+TI3 1 2
Vemos que la normalización del tensor de polarización es tal que
traza C == 1
es la intensidad total del haz luminoso.En cambio traza (C2
) :s 12• En general la igualdad se da solo
cuando el haz es de polarización definida. En dicho caso elestado de polarización del haz puede representarse con un único
1\ 1\ 1\*vector e y por lo tanto C = e 1 e .
GRADO DE POLARIZACION
Definimos el grado de polarización del haz como
de manera tal que
2
traza C2== + (1+P2)
En vista de la discusión anterior diremos que un haz de luzestá completamente polarizado cuando P = 1. Cuando P < 1 decimos,simplemente, que el haz está polarizado. En el caso límite P == 0,en cambio, el haz está en un estado no polarizado. En este caso
49

todos los parámetros de Stokes se anulan y la matriz C está dadapor
1e = -2-
Una vez que los parámetros de Stokes de un haz de luz han sidodeterminados, es fácil calcular una expresión para la intensidadde luz transmitida por un filtro que sólo admita estados de
f
polarización @ = cos e/2 @1 + el~ sen e/2 @2. Esta es
/\- /\= e .C.e =1 (1-2- - + 1)1 sene cos~ + 1)2 sene sen~ + 1)3 cose)
BIBLIOGRAFIA
l. H. A. Bethe & R. W. Jackiw: Intermediate Quantum Mechanics
(Benjamin publ., Reading, 1968).2. M. Weissbluth: Atoms and Molecules (ACademic Press, New
York, 1978).3. H. Bethe and E. Salpeter: Quantum Mechanics of one and tvo
electron atoms (Springer-Verlag, Berlin, 1957).4. K. Blum: Density Matrix and Applications (Plenum Press, New
York, 1981).
50

EMISION DE RADIACION
INTERACCION DE UN SISTEMA CUANTICO CON EL CAMPO ELECTROMAGNETICO
Estudiaremos la interacción de un sistema cuántico con elcampo de radiación. En un tratamiento riguroso sería necesariocomenzar con una adecuada cuantización del campoelectromagnético. Sin embargo, aún con campos comparativamentedébiles, la densidad de fotones es suficientemente alta como paraque pueda tratarse como una variable continua. Por lo tantodesarrollaremos una teoría semiclásica de la radiación, tratandoal campo electromagnético clásicamente.
El Hamiltoniano de un electrón en un campo electromagnéticoestá dado por
donde hemos indicado explícitamente la carga negativa q = -e delelectrón. Teniendo en cuenta la condición de gauge coulombiano
-7-) .-7 -7-) -)-)resulta ~ = O Y p.A = -ih dlvA + A.p = A.p, con lo cual
Estudiaremos la ~ituación de campo débil, despreciando el términocuadrático y tratando al término lineal como una perturbaciónpequeña. O sea
H = H + ( !:: lt.po m
Desarrollamos ahora el vector de estado del sistema cuántico enuna base de autovectores del Hamiltoniano no perturbadoH In>
oE In>
n
11/1 >n
Reemplazando en la ecuación de Schrodinger
51

ih ~tIW> =resulta
y multiplicando por <ni vemos que los coeficientes c (t)n
satisfacen el siguiente sistema de ecuaciones acopladas
dcn
E En- mcon w = h la frecuencia de Bohr.nmSupongamos ahora que el electrón está inicialmente en un
dado autoestado I i> , y que se enciende un pulso de radiaciónmonocromática de polarización definida:
:A(t,t)
durante un tiempo t.para In> * li> que:
1 . ~ ~ tI\e e1(l\..r-w)+"2 [Aa c.c.]
A primer orden en la perturbación tenemos
c (t) = (e Jt <nllt~li> iw .t'dt'-ihm e ni -n
o i (w .-w) t 1(e [A <nlei"K."t 1\ 1" e nI -= - 2m e.V 1> i (w .-w)+
anI
* "l ~ 1\* i (w .+w)t 1+ A <nle-1 .r Vii> e nI -e. i (w .+w)a
nI
Vemos que la probabilidad I c 12 de encontrar al sistema en
n
el estado n es apreciable sólo cuando el denominador de uno deambos términos es prácticamente cero. Además, en ese caso, no hayinterferencia entre ambos términos. El primer término esimportante cuando E > E./ pudiéndose producir una interacción
n I
del electrón con un campo electromagnético de frecuencia w ~ w .ni
El segundo es importante sólo cuando E < E. Y la frecuencia deln I
campo es w ~ -w .ni
52

EMISION DE R1DI1CION
Estudiamos el decaimiento de un sistema cuántico de unestado excitado i a otro f de menor energía, como resultado de lainteracción con el campo de radiación. En este problema, sólo elsegundo término de la expresión anterior es importante
I ,2ICf
l-cos(w-wif)t2
(w-w i f )
y como lA (I{'~)12 está relacionado con la densidad N(}{,~)/v deo
fotones de parámetros (K,~) por
podemos anotar.
i\. h e2
e 2o m W
iK.t l-cos(w-wií)tI<il e ~.lJlf>12 -------
2(W-Wií)
Además, el factor temporal tiene un máximo muy agudo en W Wií'y por lo tanto podemos reemplazarlo por una delta de Dirac
l-cos (W-Wií)t2
(W-W i í) t
con lo cual
~ rr o (W-W i í ) •
1 2 rr i\.Ic I =-r- í eo
.;t ~I <l' I 1.1\.. r /\ ni f I 2 so ( )e e.v > u W-W
ií
Esta expreslon indica la probabilidad por unidad de tiempo de quese produzca una transición i ~ f en presencia de un haz luminosopuro con N(K,~) fotones de parámetros (K,~). Sin embargo, en lapráctica, el haz de luz cubre de manera incoherente por lo menos
-.7un pequeño espectro dI{ de números de onda. Luego debemos
53

multiplicar la expresión anterior por el número drt =
valores de K permitidos en dicho rango dK
v( 2Tr) 3
dK de
A28rr e
o
Ahora bien, en la electrodinámica cuántica se obtiene unresul tado similar salvo por el número de fotones, que apareceincrementado en uno: N ~ N+l (ver apéndice A). Evidentemente laaproximación semiclásica de despreciar el 1 frente a N esconsistente con la suposición inicial acerca de la presencia deun gran número de fotones en el campo electromagnético. Sinembargo, con esa aproximación no tenemos en cuenta la posibilidadde que se produzca una transición espontánea i ~ f en ausencia decampo electromagnético. Y es en este último proceso en el cualestamos particularmente interesados. Adoptamos entonces lacorrección provista por la electrodinámica cuántica y anotamos laprobabilidad por unidad de tiempo de que se produzca unatransición de un sistema cuántico desde un estado excitado i aotro f de menor energía, con la emisión de un fotón de impulso ñKy polarización @ como
dW =if
a =con
i\ =e
hm c
i\ e2~
4rrc ñ c ~o
= 0.0243 A1/137 la constante de estructura fina,
la longitud de onda de Compton del electrón.
y
TRANSICION DIPOLAR ELECTRICA
En muchos casos de interés práctico la longitud de ondaA = 2rrc/w
ifde la radiación es mucho mayor que las dimensiones de
la función de onda del electrón. Por ejemplo, si el sistema esun átomo tipo hidrogenoide de carga nuclear Z, sus dimensionesson del orden de a = a /Z, con a = ~ = 0.53 A el radio deo o mcaBohr, mientras que la energía de la transición debe ser menor que
54

la energía de
lOOO/Z.ionización ~E < Z2hcal2a •
oLuego Ala > 4rr/aZ ~
~ ~Esto signifi.ca que la cantidad K. r ~ 2rra/ A que aparece en el.;1 ~
1 t d t· .I 1!\. •r !\ I f - d 1e emen o e ma rlZ < 1 e e. 'íJ > es pequena compara a con aunidad y por lo tanto la exponencial puede eliminarse. Tenemosentonces:
.;1 -7<ile1!\..r ~.\7lf> ~ ~.<il\7lf>
donde hemos usado que [~,H ] = ihV H = ih p/m.o p o
Una transición para la cual la probabilidad puede calcularsesin error apreciable efectuando la simplificación anterior sedenomina transición dipolar eléctrica, dado que involucra alelemento de matriz dipolar -e~ = -e<iI1If>. Anotamos entonces
if
dWif
~ /\ /\ ~ 2 3"[N(K,e)+l] le.Kifl O(W-Wif) W dw dK dt
Si ~ <iI1If> es no nulo, decimos que la transición i ~ f estáif
Permitida. Por el contrario, si ~ = O la transición seif
denomina prohibida. En este último caso, sin embargo, puedeocurrir que alguno de los términos de orden superior en la seriei~.1 .~~ 1 ~ ~ 2e = 1 + lK.r - "2 (K.r) + ... (que corresponden a transiciones
dipolares magnéticas, cuadrupolares eléctricas, etc ...) no senanule. Pero la probabilidad de transición será (2rrajA) veces
más pequeña que para las transiciones permitidas. Si<ilei~.1 vlf> se anula, se dice que la transición estáestríctamente prohibida; aunque todavía puede ocurrir a través dela emisión simultánea de dos fotones. Esta posibilidad se debeal término cuadrático en A que despreciamos en un comienzo.
Supongamos ahora que la radiación de base es isotrópica y noestá polarizada, o sea que N(~,~) = N(w). En esta situación, oequi valentemente, si nos interesa analizar la probabilidad deemisión espontánea, podemos sumar sobre dos estados de
55

1\ 1\polarización opuesta (e ,e+ -
o ~,~ , etc.) obteniendo1 2
dWif
2 2 3 1\[N(w)+l] Rif
sen e o(W-Wif
) W dw dK dt
con e el ángulo entre Rif
y ~. Finalmente, integrando sobre todaslas direcciones posibles del vector de onda, encontramos laprobabilidad total de emisión en la aproximación dipolareléctrica:
dWif
dt = 4a w 3 [N(w1
•f)+1] Rl'f2
3c2 if
Para calcular el orden de magnitud de la probabilidad totalde emisión espontánea por unidad de tiempo consideramos que R
if
es del orden de un radio atómico típico, que para la transicióndesde un nivel de número cuántico principal n es
2 16 -1R ~ n a = a W /w, con W = exc/2a ~ 2.x 10 seg laif o o o o o
frecuencia de Bohr correspondiente a la ionización del átomo deHidrógeno. Entonces
dW 2
4ex W 3i f 2 o 3 ex 1.3 10-7= - ao -- W = 3 W ~ x W
dt 3c 2 2 i f i f i fW
i f
Obtenemos así los siguientes órdenes de magnitud
1010 -1 dW 103 -1 estructura hiperfinaW ~ seg dt ~ segbanda de rom.
1014 -1 dW 107 -1 de externaW ~ seg dt ~ seg e caparango visible
1018 -1 dW 1011 -1 de internaW ~ seg dt ~ seg e caparayos X blandos.
ABSORCION DE RADIACION
Hasta ahora estudiamos el segundo término de la expresión
56

c (t)f
= (e [A <fleiR.t ~.~Ii>- 2m o
i(w .-w)t 1e f 1 --~ ----- +i{wri-w)
*+ Ao
i(w .+w)t 1e f 1 -
i(wfi+w)
que era importante cuando Ef < Ei, Y representaba una transición
de un sistema cuántico desde un estado excitado i a otro f demenor energía como resultado de laelectromagnético. El primer término,transición de un estado 1 a otroresultado de la absorción de un fotónabsorción de radiación se estudia
interacción con el campoen cambio, representa unaf de mayor energía cornodel campo de radiación. Laexactamente igual que la
la
por
por el hecho de que ahora se requierecampo electromagnético no nulo que aporte el
;7 /\Por lo tanto debemos reemplazar N(K,e)+l
emisión, exceptopresencia de unfotón absorbido.
;7 /\N(K,e) •
Obtenemos así que la probabilidad por unidad de tiempo deque se produzca una transición de un sistema cuántico desde unestado i a otro f de mayor energía, con la absorción de un fotónde impulso ñ~ y polarización ~ es
dW =if
aA2
____ c_ N(~,~) I<flei~.t ~.Vli>12o(W+Wl.f)wdw d~ dt( 2rr ) 3
VALIDEZ DEL TRATAMIENTO PERTURBATIVO
Para obtener los resultados anteriores realizamos algunasaproximaciones que ahora debemos justificar. En primer lugar nosquedamos a primer orden en la expresión
dcn
dtiw te ron
reemplazando c (t) por la condición inicialm
Esto será válido mientras la probabilidad detransición se mantenga pequeña. O sea
c (t) ~ c (O) = o .'m m mlque se produzca una
57

dW tdtEsta condición impone un límite superior al tiempo.
Por otro lado, la separación que hicimos en cf (t) de lascontribuciones que son dominantes para w = w y w = -w es
fi fi
válida en tanto que ambas contribuciones no interfieran entre sí;o sea, en tanto que 271/t« 2/W
fil. Esto impone un límite inferior
al tiempo, por debajo del cual ambas posibilidades (emisión y
absorción) no pueden separarse.Vemos entonces que los resultados de este modelo semiclásico
son válidos en un intervalo de tiempo
3/cirr « t «w w
Puesto que rr « 3/a3~ 7.7 x 106, este intervalo es muy amplio.
VIDA MEDIA Y ANCHo DE LINEA
La probabilidad total de emisión espontánea por unidad detiempo
dWi f
dt =4a 33c2 W
if
sumada sobre todos los estados finales posibles
dWi f
li = L dtf < i
define la vida media del estado inicial
T = l/l.i 1
En este punto la teoría que hemos desarrollado se revelacomo no enteramente autoconsistente. Vemos que todos los niveles,excepto los más bajos, deben decaer eventualmente como resultadode un proceso de emisión espontánea, y por lo tanto no pueden ser
58

estacionarios como supusimos en un principio. En general todoproceso de decaimiento obedece una ley exponencial, es decir quela probabilidad de ocupación de un estado está dada porp (t) = P (O) e-t/r;, con -c la vida media. La vida media de lastransiciones estudiadas usualmente es mucho mayor que el períodocaracterístico del movimiento electrónico en el átomo,
-16r; ~ 10 seg, por lo cual estaba justificado despreciar elo
decaimiento en un primer orden de aproximación.La inclusión del proceso de decaimiento en la descripción de
los procesos de emisión y absorción de radiación fue planteadapor Weisskopf y Wigner en 1930 (Z. Physik 63, 54; 65, 18). En unsegundo orden de aproximación ellos suponen que la dependenciatemporal de todo estado debe tener· en cuenta su eventualdecaimiento por emisión espontánea en la forma
l/1 (t) ~ 'l1 (O)n n
-iE t/h -í t/2e n e n
lo cual, reemplazando en los cálculos realizados, lleva a laaparición de una frecuencia de Bohr compleja
W'i f
E -Ei f
=--h + i
í +1i f
2
Con esta modificación el factor temporaltiende, para tiempos grandes, a la deltauna Lorentziana
1 i (w~ -w) t 1e lf - 22rrtI w' -w I ya noif
de Dirac o(Wif-W) sino a
L(w) =(1
i+í
f)/2rr
(W-Wi
f) 2+ (íi +íf) 14
La radiación emitida no tiene una frecuencia bien definida,sino una cierta distribución centrada en la frecuencia de BohrW , y con un ancho í = í +í, denominado ancho de línea natural.
if i f
Adviértase que este resultado está de acuerdo con el principio deincerteza. La vida media ~ = l/í mide el tiempo durante el cualun estado está ocupado. Luego su energía no puede estardeterminada con una precisión mayor que ~E = hí. Si las energíasde los estados inicial y final son inciertas en esa cantidad, la
59

frecuencia de la radiación emitida estará indefinida en un rangot:.w ~ f +f .
i fEl ancho de línea descrito suele denominarse natural, para
diferenciarlo de un ensanchamiento adicional producido por otrosprocesos. Por ejemplo, un átomo excitado puede decaer no sólo poremisión espontánea, sino que puede entregar su energía deexcitación a otro átomo del gas en el curso de una colisión. Estoproduce una disminución de la vida media y un consecuenteensanchamiento de la línea de emisión. Como la frecuencia decolisión aumenta con la presión del gas, este proceso se conocecomo ensanchamiento de presión o colisión. En general esteensanchamiento de presión domina completamente sobre elensanchamiento natural.
El movimiento aleatorio de los átomos en un gas da origen aun ensanchamiento de las líneas de emisión por efecto Doppler. Adiferencia de los dos procesos anteriores, el perfilcorrespondiente no es lorentziano, sino de forma gaussiana, comoresultado de la distribución maxwelliana de velocidades.
Los ensanchamientos de Doppler y de presión sonindependientes. Cuando ambos efectos están presentes ladistribución en frecuencias resultante es una convolución deambas, que se denomina perfil de Voigt
00
V(x) = J L(y) G(x-y) dy-00
BIBLIOGRAFIA
1. B. H. Bransden & C. J. Joachain: Physics of Atoms and
Molecules (Longman, London, 1983).2. M. Weissbluth: Atoms and Molecules (Academic Press, New
York, 1978).3. H. A. Bethe & E. Salpeter: Quantum Mechanics of one and tvo
electron atoms (Springer-Verlag, Berlin, 1957).4. H. A. Bethe & R. W. Jackiw: Intermediate Quantum Mechanics
(Benjamin Publ., Reading, 1968).
60

REGLAS DE SELECCION
TRANSICION ENTRE ESTADOS DESACOPLADOS
Consideremos la emisión de radiación por un electrón en uncampo central al producirse una transición i m m 4 i 'm'm'.
s sEstamos despreciando la estructura fina de los nivelesenergéticos. Las componentes estándar del elemento de matrizdipolar son, en v~rtud del teorema de Wigner-Eckart,
g-m ( g 1 g,) ~< nernmIr In'e ' m 'm ,> = eS , (-1) , (ni11 r 11 n 'i' >s q s m m -m q m
s s
donde la delta en m aparece debido a que el espín no actúa sobres
las variables espaciales. El símbolo 3J nos provee además, y demanera explícita, otras reglas de selección
M = O ó ±1
He'?: 1
m = m'+ q
m = m's s
La regla de selección para m está asociada en el límiteclásico con el momento angular que se lleva la radiación. Si laluz es emitida en la dirección z y está polarizada circularmente,se lleva un cuanto de la proyección del momento angular en dichadirección, y por lo tanto ~m = ±1.
ESTADOS METAESTABLES
La condición e+t'?: 1 indica que la transición e O ~ e' = Oestá prohibida. Puede mostrarse explícitamente que
~ ->
<nOOleik.r Vln'OO> = O
por lo cual esta transición está estrictamente prohibida. Estonos dice que un nivel ns arbitrario no puede decaer directamente
61

al nivel fundamental 1s. Sin embargo, aún puede decaer a un niveln'p por medio de una trangici6n dipolar Ql~ctriC'a, y de allí.eventualmente, el sistema puede alcanzar el estado fundamental1s. Esta posibilidad no existe para el estado 2s que, por laregla anterior / no puede decaer al nivel 1s (que es su úniconivel inferior) por medio de una transición de un fotón. A pesarde esto / el sistema aún puede desexci tarse por medio de otrosmecanismos:
De hecho no es estrictamente cierto que el nivel 1s es elúnico ubicado debajo del 2s. Por efecto Lamb, el nivel 2p conj=1/2 está leve~ente por debajo del 2s. Sin embargo, ladiferencia de energia es tan pequefia que la probabilidad detransición espontánea es despreciable. Su vida media, definidacomo la inversa de la probabilidad de transición por unidad detiempo, es de 20 años aproximadamente.
Otro posible mecanismo de des excitación resulta del hecho deque el elemento de matriz de transición no es nulo cuando se usanfunciones de onda relativistas para describir los estados inicialy final. En tal caso puede ocurrir una transición dipolarmagnética, pero con una probabilidad que es menor, en un orden
6 6 1 -13 1 d' . . ~(Za) ~ Z x O , que a correspon lente a una tranSlClonpermitida. Su vida media es de 2 dias.
Finalmente, la mayor probabilidad para el decaimiento delnivel 2s proviene de la emisión simultánea de dos fotones, que esun orden Z2a3 menor que para transiciones permitidas. Así,mientras que la vida media del nivel 2p es 1.6 x 10-9 seg, parael nivel 2s es 0.14 seg. Debido a esta vida media tan alta, elnivel 2s se denomina estado metaestable.
INTEGRAL DE TRANSICION
Calculamos ahora la probabilidad total de transiciónespontánea entre dos niveles ni y n'i', utilizando una regla desuma deducida anteriormente
dWdt (ni-+n'i' )4a 3 2= 2 W L I< nimIr In'i 'm' > I =3c2 qm' q
62

= 80: _1_ w3\ < ni \\r\\n Ii I > \ 2
3C2 U+l
donde el factor 2 resulta de considerar las dos posiblesproyecciones del espín en el estado final.
Vemos que el elemento de matriz reducido provee laprobabilidad total de transición entre niveles. Para un átomohidrogenoide este elemento puede calcularse fácilmente,resultando
<nlllrlln'l'> = v(2l+1)(2l'+1) [~~ ~'] I(nl,n'l')
donde hemos definido la integral de transición
JCXl • 31(ne, n ' e') = R (r) R (r) r dr
nI n' I'O
que, a través de la parte radial R de la función de ondanI
q, (r) = R (r) Y (~), contiene la única referencia al camponlm nI 1m
central donde se mueve el electrón. Por otra parte vemos queaparece una nueva regla de selección a través del símbolo 3J.Sabemos que
[e e e]123m m m
1 2 3
e e e [e e e]= (-1) 1+ 2+ 3 1 2 3-m -m -m
123
por lo cual, en muestro caso, debe ser e+1+e' = par. Esta es laregla de Laporte, e indica que, puesto que r es una función
impar, el elemento de matriz <nemlrln'e'm'> se debe anular entre
estados de igual paridad. Esta regla junto con una anterior nosdice que
M ±1
SERIES
El espectro de emisión del hidrógeno y de otros átomos se
63

espontáneamentecontribución a
clasifica en series, dependiendo de cual es el nivel final al quese produce el decaimiento. Estas son
Serie de Lyman n = 1f
Balmer 2
Paschen 3
Brackett 4pfund 5
Para el átomo de Hidrógeno, la serie de Balmer caemayormente en el rango visible, la serie Lyman en el ultravioletay el resto -de baja frecuencia- en el rango infrarrojo.
INTENSIDAD LUMINOSA
Consideremos una población p. de átomos hidrogenoides en el1
estado exci tado (n.,f!. , m.). Si estos átomos pueden decaer1 1 1
a un nivel (n, f! , m ), la correspondientef f f
la intensidad luminosa medida por un detector1\sensible a estados de polarización e, y que se encuentra a una
distancia R de la fuente, está dada por:
h Wif
= P a.i 2rrc2
que podemos anotar
con
dWi f =
1\* 1\e .C .eif
Cif
el tensor de polarización en notación diádica.En general la fuente luminosa no consiste en una mezcla
64

incoherente de átomos en estados (n.,i. ,m.), sino en un gas cuyo1 1 1
estado de mezcla está caracterizado por un operador densidad p.Usualmente es posible separar las distintas lineas de emisión defrecuencias w correspondientes a decaimientos desde distintos
ifniveles n . Por lo tanto suele aproximarse al operador densidad
ip corno una superposición incoherente de bloques de n. definido
1
p(t) = L Pn(t), Por otro lado, si no es posible distinguir entren ¡¡
decaimientos a estados finales con dietlutQ6 valores de t '1 IDf 1 f'
se debe promediar sobre ambos números cuánticos. Escribimosentonces al ten sor de polarización para el decaimiento del niveln al n de la siguiente manerai f
ex22rrc
~ ~L <n ,e ,m Ir p (t) rln ,e ,m >e m f f f ni f f ff f
POLARIZACION DE LA RADIACION EMITIDA
El estado de polarización dela luz emitida en un decaimiento
determinado por laentre los números
espontáneocompletamenterelación Lim
e'm' está z
kcuánticos magnéticos. En efecto,supongamos que observamos laradiación emitida en unadirección ~ que forma un ángulo econ el eje de cuantización ~. Laintensidad de la luz transmitidapor un filtro que sólo admitaestado de polarización~ = cos~/2 ~1 + eiosen~/2 ~2 esproporcional a
y
1/\ ~ 2 = /\ io /\ io /\ ~ 2e.R¡f I I(cos~/2 x + e sen~/2 cos~ y - e sen~/2 sen~ z).R
ifI
65

Consideremos primero el caso en que m = m' . Por las reglas deselección sólo la componente estándar R es no nula. Por lo
otanto A ~ 2 2 2 2le.R. I = R sen (3/2 sen'lJylr o
I(e,'lJ)=I
o2sen 'lJ
2(l-cos(3)
Comparando este resultado con la expresión general que dedujimosen un capítulo anterior
I(e) 1= -2- [ 1+ TI sen(3 coso + TI sen(3 seno + TI cos(3 ]123
vemos que la luz emitida está caracterizada por una intensidad2I = I sen 'lJy los siguientes parámetros de Stokes
o
TI = O TI = O TI = -11 '2 '3
que indican una polarización lineal en el plano (~,~).
En cambio, si ~m = q resulta
A 1oI(e,'lJ)=2 1 2122" (1+cos o(}-)+ 2" sen o(}-cos(3 + q coso(}-sen(3 seno ]
1con lo cual la intensidad total es I = ~ (1+cos2'lJ)y
= 2q coso(}-, Tl3=1+cos2
0(}-
sen20(}-
2l+cos o(}-
Nuevamente el haz está completamente polarizado, ya queestáhazelqueresulta'* OSiendo TI21.P = y1T12+TI 2+TI 2
123
elípticamente polarizado.Dos casos extremos son particularmente interesantes: Si
o(}-= O resulta TI = O, TI = q, TI = O, Y el haz está circularmente123
polarizado con helicidad q. En cambio, si 'lJ= rr/2 tenemos TI = O,1
TI = O, TI = 1, con lo cual la polarización es lineal en la2 3dirección perpendicular al plano (~,~).
66

s J) ( t 1 t') (t I S J I )m -M -m q m I m I m -M'8 8
Digamos, por último, que si la emisión ocurre en pr~s~ncia~ /\de un campo de radiación N(k,e), sólo se altera la intensidad
observada, pero no los parámetros de Stokes.~ /1. ~ /1. /1. -7 2I(k,e) <X [N(k,e) + 1] le.Rifl, y por lo tanto siguen siendo
válidos los resultados anteriores.
TRANSICION ENTRE ESTADOS DE ESTRUCTURA FINA
A veces es posible separar energéticamente las transicionesproducidas entr~ diferentes estados de estructura fina(l s J M) -> (t' s'J'M'). Las componentes estándar del elemento dematriz dipolar pueden calcularse a partir de los resultadosanteriores correspondientes a una base desacoplada (t s m m )
8
< ntsJM Ir In't I S I J I M'> =q
¿ <tsJMltsrnm><nEsrnm Ir Inltls'm'm'><tlslmlmlltlsIJIM'>m m'mm' s 8 q 8 8
8 8
Expresando los coeficientes de Clebsch-Gordan en términos de lossímbolos 3J, y reemplazando el elemento de matriz de r resulta:
q
-><ntsJM IrqIn I t ' S I J I M'> = °s 's v' ( 2J+1)(2J '+1) <nt11 r 11 n I t'> •
. ¿ (_l)~+s+M+t+m+t'+s+MI(~m m'm m
8 8
Esta suma puede realizarse explícitamente,expresión
<ntsJMlr In'tls'J'M'>q
resul tanda la
t+ M (J' 1 J) {J I 1 J } ->= 088' (-1) s- v'(2J+1)(2J'+1) M' q -M t s t' <ntllrlln'E'>
El símbolo 3J provee ciertas reglas de selección para que latransición tenga lugar. Vemos que debe ser
67

~J = -1, O Ó +1J=O ~ J'=O prohibido
Además selecciona la componente esférica del operador vectorial rresponsable de la radición emitida, siendo
q = M-M'
La delta de Kronecker o aparece en tanto que el operadorss'dipolar eléctrico no depende del espín:
~s = O
Además las propiedades del símbolo 6J introducen una regla de
selección adicional
M = -1, O ó +1
e =0 ~ e'=o prohibido
El cálculo explícito de estos elementos permite predeciralgunas relaciones entre las intensidades de las lineas deemisión espontánea. Por ejemplo, en una transición e ~ Hl, lalinea J ~ J+l es más intensa que la J ~ J Y ésta, a su vez, másque la J ~ J-l.
68

TEORIA DE PERTURBACIONES INDEPENDIENTES DEL TIEMPO
PERTURBACIONES DE ESTADOS ESTACIONARIOS NO DEGENERADOS
Supongamos que conocemos un autoestado no degenerado 11/1 (O) >
del Hamiltoniano Ho
Si ahora modificamos ese Hamiltoniano con un término H'
independiente del tiempo, el autoestado 11/1(0) > también se va amodificar. Para calcular este nuevo autoestado I~> y lacorrespondiente energía E, escribimos (H + H')I~> = E I~> en laoforma
(H - E) I~> = - H'I~>o
y multiplicamos por <~(O)I para encontrar
E E(Q)+ <~(O)IH'I~><~(O)I~>
A orden cero en la perturbación H', y tal corno era deesperarse, las dos ecuaciones anteriores se convierten en
E = E(O)
(H - E) I~> = Oo
que dan corno resultado I~> = I~(O» y E = E(Q).A primer orden, en cambio, resulta
<~(O) IH' I~(Q) ><~(O)I~(O»
Y, en general, construimos una técnica recursiva de resolución,
69

donde la aproximación enésima puede calcularse a partir de lan-l, como
< .1, ( O) I H' I .1, (n-l ) >E (n) = E (O) + __ 'P 'P _
<W(O) I W (n-l) >
Adviertase que para calcular la energía a cierto orden, sólonecesitamos conocer el autoestado IW> a un orden menor.
PERTURBACIONES DE ESTADOS ESTACIONARIOS DEGENERADOS
Si el nivel no perturbado E es degenerado, en general laoperturbación romperá esa degeneración, produciendo undesdoblamiento en varios autoestados. Si la perturbación H'tiende a cero, cada uno de esos autoestados tenderá a algunacombinación lineal de los autoestados no perturbados
Reemplazando en
IljJ> =H'->O
(H - E) IljJ> = - H' IljJ>o
y multiplicando por alguno de los autoestados no perturbados(O)
<ljJ 1, obtenemos al orden más bajo en la perturbaciónn
¿ a <ljJ(O) IH' IljJ(O) >m m n m
o sea
Este es un sistema homogéneo de ecuaciones lineales quedetermina a los coeficientes a del autoestado IljJ> a orden cero
m
en la perturbación. Para que este sistema tenga solución no
70

trivial el determinante debe anularse.
det [< 1/1 ( o ) I H I 11/1(O) > - (E - E (O)) Id] = O
A partir de esta ecuación secular podemos obtener las distintasenergías en que se desdobla el nivel a primer orden en laperturbación.
EFECTO STARK EN EL ATOMO DE HIDROGENO
Las modificaciones producidas en los niveles de energía deun átomo por la aplicación de un campo eléctrico uniforme E se
-7 -7denominan efecto Stark. La perturbación H'= e E.r altera el2Hamiltoniano de tal forma que ya no conmuta con L . Esto se debe
a que el campo eléctrico especifica una dirección preferencial, ypor lo tanto el sistema físico ya no es invariante anterotaciones arbitrarias. Sin embargo es aún invariante anterotaciones alrededor de la dirección preferencial definida por elcampo eléctrico, y por ende la proyección del momento angular Len esa dirección es una constante de movimiento.
Arbitrariamente aplicamos el campo eléctrico en la direcciónAz. Empleando el teorema de Wigner-Eckart vemos que los elementosde matriz
< nimI H I I n I i I m'> = eE < nimI z I n I i I m'>
se pueden escribir en la forma
(-1) e-m (-~ 1 el) -7< nlmI H I I n I e I m'> = eE < nillrllniI > =O m'
eE (_1)e-m ( e 1 i ') v' ( 2e+1) (2e I +1r (~1 ~') 1(ne,ne')= -m O m' O
con I(ni,ni') la integral de transición
I(ni,ne') = {XlR:i (r) Rni'(r) r3 drO
71

Los símbolos 3J nos indican que el elemento de matriz anterior seanula a menos que
M = ±1
Lim = O
o sea que los únicos elementos no nulos son
< nfm IH' In f-1 m> *= (n f-1 miH' Infm> =
t ¡; 2 2 2 '= (-1) eE (t -ID )/(4t -1) 1 (nf,n t-1)
La integral de transición puede calcularse fácilmente paraun átomo hidrogenoide de carga Z
3 a / 'a 2 21(nt, n t-1) = -2- -Z- n n-t
resultando finalmente
< nm IH' In t-1 m> = < n t-1 miH' Intm> =
= (_l)t /(n2_t2) (t2_m2)/(4t2_1)' _3_ ~ e E a2 Z a
Por ejemplo, para n 2 los únicos términos no nulos son
<210IH' 1200> = <200IH' 1210> = -3 e E
La ecuación secular
aa
Z
det [< 1/1 ( o ) IH' 11/1( o ) >- (E E (O» Id] = O
puede resolverse porsiguientes autovalores
simple inspección, resultando los
E = Ea, Ea , Ea - 3 e E
72
aa
Z
aa
Ea + 3 e E -Z-

con autovectores, a orden cero en el campo,
1
..¡z1
..¡z
Vemos que el nivel n = 2 se desdobla en tres nivelesequiespaciados. En general, cada nivel de número cuánticoprincipal n se desdobla en 2n-l niveles equiespaciados en energíacon ~E = ~ ~ e E a
o' y degenerados en el signo de ID.
Vemos que la separación en energía ~E aumenta con el númerocuántico principal n y disminuye con la carga Z. Esto es natural,ya que cuando más lejos del núcleo se encuentren los electrones,tanto mayor será el momento dipolar del átomo.
ESTADOS METAESTABLES
Aunque las predicciones de la teoría de perturbaciones parael efecto Stark son confirmadas por la experiencia, es naturalque surjan dudas sobre su validez, y por lo siguiente: La energíapotencial del electrón en el campo eléctrico E ~ es
A Ze2V (r) = - -4- - + eEz. Vemos que lo que antes era un pozo derre r
o
poten~ial, ahora es una barrera que, por más pequeño que sea elcampo eléctrico E, es penetrable. Existe entonces unaprobabilidad finita de que el electrón atraviese esa barrera depotencial y se aleje en dirección al ánodo, produciéndose así laionización del átomo.
Como ahora el movimiento del electrón es, estrictamentehablando, un movimiento infinito, el espectro energético pasa deser discreto a ser continuo. A pesar de ello, las solucionesobtenidas por medio de la teoría de perturbaciones tienen sentidofísico, como representativas de aquellos estados que, si bien noson del todo estables, son metaestables. Si el átomo se encuentraen uno de estos estados en un instante inicial, permanece en éldurante un intervalo muy largo de tiempo. Por ejemplo, la vidamedia del nivel fundamental de un átomo tipo hidrogenoide es (C.Lanczos, Z. Physik ~, 204 (1931»
73

1 1T - - -1!:l- 8 (.J
o
E [ 2 E ]E exp "3 EO
o
con Wo
= 2~a~Z ~ 2 Z2 x 1016 hertz la frecuencia correspondienteo
a la ionización de un átomo hidrogenoide de carga Z, y
E = _i\_ e 2 ~ 5 142 Z2 109 V' 1t Io 4Tl8 (a IZ) • x o cm
o o
el campo eléctrico producido por una carga unidad a una distanciaigual al radio de Bohr a ~ a IZ.
o
APAGADO DE LINEAS DE EMISION (QUENCHING)
La vida media es aún menor para niveles excitados,predominantemente para aquellos estados que tengan una mayordensidad de probabilidad en la zona del continuo en dirección alánodo. Para cada número cuántico principal n, esto ocurre conaquellos estados de menor energía en el desdoblamiento Stark. Porello, las componentes Stark de menor frecuencia (rojas)desaparecen al aumentar la intensidad del campo eléctrico. Si secontinúa aumentando el campo, líneas enteras comienzan adesaparecer, debido a la ionización por efecto Stark de losestados excitados, desapareciendo primero las líneascorespondientes a estados de mayor número cuántico principal.
Esta desaparición de líneas de emisión por efecto Stark sedenomina apagado (Quenching). De hecho, experimentalmente no seobserva la ionización de un estado excitado por efecto Starkdirectamente, sino el apagado de las correspondientes líneasespectrales. Para que ello ocurra, la vida media de ese estadopor efecto Stark debe ser menor que la vida media natural deemisión de radiación. Esta vida media es del orden de 10-8 segpara transiciones en el rango visible, lo cual, usando laexpresión anterior, conduce a la utilización de campos eléctricosmayores que E ~ 106 Volt/cm. Por ejemplo, para una transiciónBalmer n
i= 5 ~ n
f= 2 (Ha), la componente roja se apaga para
E = 0.69xl06 Volt/cm y la violeta para E = 1.01xl06 Volt/cm.
74

Calculando a segundo orden en el campo eléctrico, la teorlade perturbaciones predice una corrección que baja los niveles deenergía en una cantidad ~E ~ _E2n6/z4
• Como este corrimientoaumenta fuertemente con el número cuántico principal n, produceuna apreciable disminución de las frecuencias de emisión deradiación. Para las líneas Balmer hablamos de un corrimientohacia el rojo.
Comparando con el efecto Stark lineal, vemos que estacorrección es despreciable para campos eléctricos menores queE « E /n5• Para números cuánticos bajos, n ~ 6, en general se
oproduce el apagado de la línea antes de que su corrimiento haciael rojo sea muy fuerte.
BIBLIOGRAFIA
1. L. Landau & E. Lifchitz: Mécanique Cuantique (Editions MlR,Moscou, 1974).
2. L. l. Schiff: Quantum Mechanics (McGraw-Hill, New York,1968).
3. A. Messiah: Mecánica Cuántica (Ed. Tecnos, Madrid, 1975).4. H. Bethe & E. Salpeter: Quantum Mechanics of one and two
electron atoms (Springer-Verlag, Berlin, 1957).
75


TEORIA DE PERTURBACIONES DEPENDIENTES DEL TIEMPO
PERTURBACIONES DEPENDIENTES DEL TIEMPO
Si perturbamos al Hamiltoniano H con un término dependienteodel tiempo H'(t), es imposible hablar de correcciones a laenergía, ya que no existen estados estacionarios. El problema quese plantea es el de calcular aproximadamente la solución de laecuación de Schrodinger
ih dll/1> =crt (H +H') 11/1>o
como combinación lineal de los autoestados In> del Hamiltonianono perturbado
11/1 >n
Los coeficientes a (t) son solución den
danat i= - h L am(t) <nIH' 1m>
m
iw te nm Wnm
=E -En m
h
Estas ecuaciones pueden resolverse por iteraciones partiendo delorden cero atO)(t) = a (O). or ejemplo, a primer orden
n n
a~1)(t) = an(O) - ~ L am(O)«nIH' 1m> eiwnmt'dt'm O
Ya vimos una aplicación de esta técnica al estudiar lateoría semiclásica de emisión y absorción de radiación.
DESARROLLO EN AUTOESTADOS INSTANTANEOS DE LA ENERGIA
otra técnica para resolver una ecuación de Schrodinger conHamiltoniano dependiente del tiempo
77

ih dll/1> =dt H(t) 11/1>
consiste en realizar un desarrollo en autoestados instantáneos
del Hamiltoniano
con
11/1 (t». t
= L a (t) In, t> exp [ - ~ J E (t') dt']n h nn o
H(t) In,t> = En(t) In,t>
Suponemos que estos autoestados In,t> son ortonormales) nodegenerados y discretos.
Sustituyendo en la ecuación de Schrodinger y multiplicandopor <n,tl resulta
dan
=dtd JtL am ( t) <n ,t Idt 1m,t > exp [i Wnm ( t') dt' ]
m O
E -Econ wnm = n
ñm Este conjunto de ecuaciones es exactamente
equivalente a la ecuación de Schrodinger.
APROXIMACION ADIABATICA
Suponiendo que inicialmente el sistema se encuentra en unestado li,O>, y que la dependencia temporal del Hamiltoniano estal que la probabilidad de que se produzca una transición a otroestado In,t> distinto de li,t> es muy pequeña, podemos aproximara (t) ~ a (O) = o en el miembro de la derecha, resultando
m m mi
t t'
an (t) - - J <n,t' I~t' I i,t' > exp [iJ Wni (t") dt "] dt' n*iO O
El elemento de matriz <n,t I~t I i,t> puede calcularse derivandoH(t) li,t> = E.(t) li,t> respecto del tiempo, y multiplicando por
1
la izquierda por <n,tl, resultando
78

o sea
<n,tl~tli,t)<n,tl~li,t)
(E -E.)n 1
t'
< n, t' I ~~ ' I i, t') exp [i f Wni (t 11) dt 11] dt'O
varíe muy lentamente. Suponemos
son aproximadamente independientes
La aproximación adiabática será válida en tanto que todos
los coeficientes a (t), con n::¡:.l, se mantengan pequeños. Paran
ello es necesario que H(t)t ¡dH"en onees que <n dt l) Y W
nidel tiempo, resultando
a (t) ~n
1'h 21 W
ni
IdHl' iw t 1< n dt 1> (e ni - )
La condición la I « 1 requiere quen
Vemos que la variación del Hamiltoniano en un período de Bohr
debe ser menor que la diferencia de energía para la transiciónanalizada, en todo instante. En general es suficiente con probar
esta relación para el nivel de energía más cercano al inicial.
CAMBIO DISCONTINUO DEL HAMILTONIANO
Consideremos el caso de un Hamil toniano independiente deltiempo H. que se altera súbitamente, pasando a otro Hamiltoniano
1
(también independiente del tiempo) H en un instante dado t = t .f o
La solución general puede escribirse
11/1> L i Il/1i> -iEit/h si t<to= a e nn n
n
Lf Il/1f> -iEft/h si t>toa e nn n
n
donde son autoestados de los Hamil tonianos H y Hi f
79

respectivamente
Para t = t tenemoso
n n
Il> e-iE~t/hn
Multiplicando por <ll logramos despejar los coeficientes i enm n
términos de los coeficientes ai inicialesn
af L ai <~fl~i> exp[-hi(Ef-Ei) t ]n m nm nm o
m
obteniendo una solución completa y exacta del problema.
APROXIMACION SUBITA
La aproximación súbita consiste en usar la soluciónanterior, aún cuando el sal to del Hamil toniano ocupe un cierto
intervalo de tiempo -c pequeño pero no nulo. Para realizar una
estimación de las condiciones de validez de esta aproximaciónsupongamos que entre los instantes t = t Y t = t +-c el sistema
o o
se encuentra en una situación intermedia caracterizada por unHamil toniano H de autoestados I~o > y energías EO
• Pegando laso n n
soluciones en t = t Y t = t +-c obtenemoso o
fVemos inmediatamente que an se reduce a la expresión dada por la
aproximación súbita si el último término exponencial es próximo a
l. O sea que el error es pequeño si T« hl IEf_Eo l. En generaln q
aquellos estados intermedios con energías muy distintas de las
iniciales tendrán probabilidades de ocupación muy bajas, por lo
cual podemos aproximar la condición anterior por
80

f i-C « h/IE -E In m
Entre los ejercicios veremos varios ejemplos de aplicaciónde los métodos aproximados descritos en este capítulo.
BIBLIOGRAFIA
l. L. Landau & E. Lifchitz: Mécaníque Cuantíque (Editions MIR,Moscou, 1974).
2. L. l. Schiff: Quantum Mechanics (McGraw-Hill, New York,1968).
3. A. Messiah: Mecánica Cuántica (Ed. Tecnos, Madrid, 1975).
81


EL ATOMO DE HIDROGENO
INTRODUCCION
Comenzaremos nuestro estudio sobre átomos repasando algunasideas fundamentales: Un átomo está formado por un núcleorelativamente masivo, con dimensiones del orden de 10-12 cm, y uncierto número de electrones, de carga -e, que ocupan el resto delvolumen atómico de unos 10-8 cm de radio. El núcleo estácompuesto por A partículas llamadas nucleones, de las cuales Zson protones, de carga e, y el resto neutrones, eléctricamenteneutros. "A" se denomina número de masa y "Z" número atómico. Sihay tantos electrones cornoprotones, es decir Z electrones, elátomo es eléctricamente neutro. Si ello no ocurre, el átomoestará cargado positivamente o negativamente, dependiendo de queel número de electrones sea menor o mayor que el de protones enel núcleo. En tal caso el átomo se denomina ión.
La masa M de un nucleón es aproximadamente 1850 veces lamasa m de un electrón. Sin embargo, son los electrones quienesconfieren a un átomo la mayoría de sus propiedades, jugando unrol principal en procesos de emisión y absorción de luz,ligaduras moleculares, reacciones químicas o en las másimportantes características de la materia sólida.
Es esta estructura electrónica de los átomos, lo que nosinteresa estudiar en este curso. Comenzaremos discutiendo laspropiedades de átomos e iones que tienen tan sólo un electrón,para luego pasar al caso multi-electrónico.
EL ATOMO HIDROGENOIDE
El átomo más simple es el átomo de hidrógeno. Su núcleo estácompuesto por un protón al que está ligado un único electrón. Engeneral, un átomo o ión con números de masa A y átomico Zarbitrarios, pero con un único electrón ligado, tendrácaracterísticas similares a las del átomo de hidrógeno. Lollamaremos átomo hidrogenoide. Son ejemplos de átomos
83

hidrogenoides, los isótopos del Hidrógeno llamados Deuterio(A=2, Z=l) y Tritio (A=3, Z=l), el Helio ionizado He+ (A=4, Z=2)o el Litio doblemente ionizado Li2+ (A=7, Z=3).
En nuestro estudio del átomo hidrogenoide supondremos que elcampo eléctrico del núcleo es el de una carga puntual. Esto esrazonable, ya que el núcleo es muy pequeño (10-12 cm) encomparación con la distancia promedio del electrón al núcleo(10-8 cm). Eliminando el movimiento del centro de masa obtenemos,
7para la coordenada relativa electrón-núcleo r, la siguienteecuación de Schrodinger estacionaria
con
h2 2 7[- 2~ ~ + V(r)] w(r) =7
E w(r)
V(r) i\ Ze2
= - 4rrc ro
la interacción coulombiana correspondiente, y ~ la masa reducidadel sistema electrón-núcleo ~= rnM/(m+M).
Ahora reordenamos las constantes de la ecuación anteriorescribiendo
con
2 2[- (a /Z) ~ -o
ao
2Zr
4rrc f12o 5.3 -9ao = ~ x 10 cmi\
2~ eel radio de Bohr, y
1 i\ 2 4Ry ~ e 2.18 10-11 ergios2" (4rrc ) ~ x
o f12
una unidad de energía llamada Rydberg.Al escribir la ecuación de Schrodinger de esta manera
84

resulta evidente que los autovalores de energía serán2proporcionales a Z . En efecto, el espectro de energía está dado
por
Si
... , n-1 yla notación
... el denominado número cuántico principal. Enestados son múltiplemente degenerados.
2(L ,L) para formar, junto con elz
observables que conmutan, podemosortonormal de autofunciones
con n= 1/ 2/ 3/principio estosutilizamos el momento angularHamiltoniano, un conjunto deconstruir un conjunto
~ A~ (r) = R (Zr/ao) y (r). Vemos que, debido a la simetríanlm nI 1m
esférica del potencial coulombiano, hemos podido separarvariables, agrupando la dependencia angular de las autofunciones~ en los armónicos esféricos Y . Para cada valor de n, los
nlm 1m
números cuánticos l y m permitidos son l = O, 1, 2,m = -l, -l+l, ... , e. De aquí en adelante adoptaremosespectroscópica nl para designar los estados de n, l y m
mdefinidos, donde los momentos angulares e = O, 1, 2, 3, 4, 5,se representan por una letra minúscula s, p, d, f, g, h,respectivamente. Así el símbolo 2p designa al estado con n = 2,ol = 1 Y m = O. Sin embargo, debe recordarse que debido a ladegeneración de estos autoestados en e y m, cualquier combinaciónlineal de ellos para un dado valor de n es también autofuncióndel Hamil toniano de energía E. Aprovechamos esto para
n
representar la dependencia angular de las funciones reales(Y +Y )/2 y (Y -y )/2i, autofunciones de L2 y L 2, en un
1m l-m 1m l-m z
diagrama polar, donde el valor de cada función para una dadadirección angular es indicada por la distancia al origen. Estaestructura angular de la función de onda será de gran utilidad alestudiar las ligaduras moleculares.
Funciones angulares de onda para los estados p (l C~ 1).
85
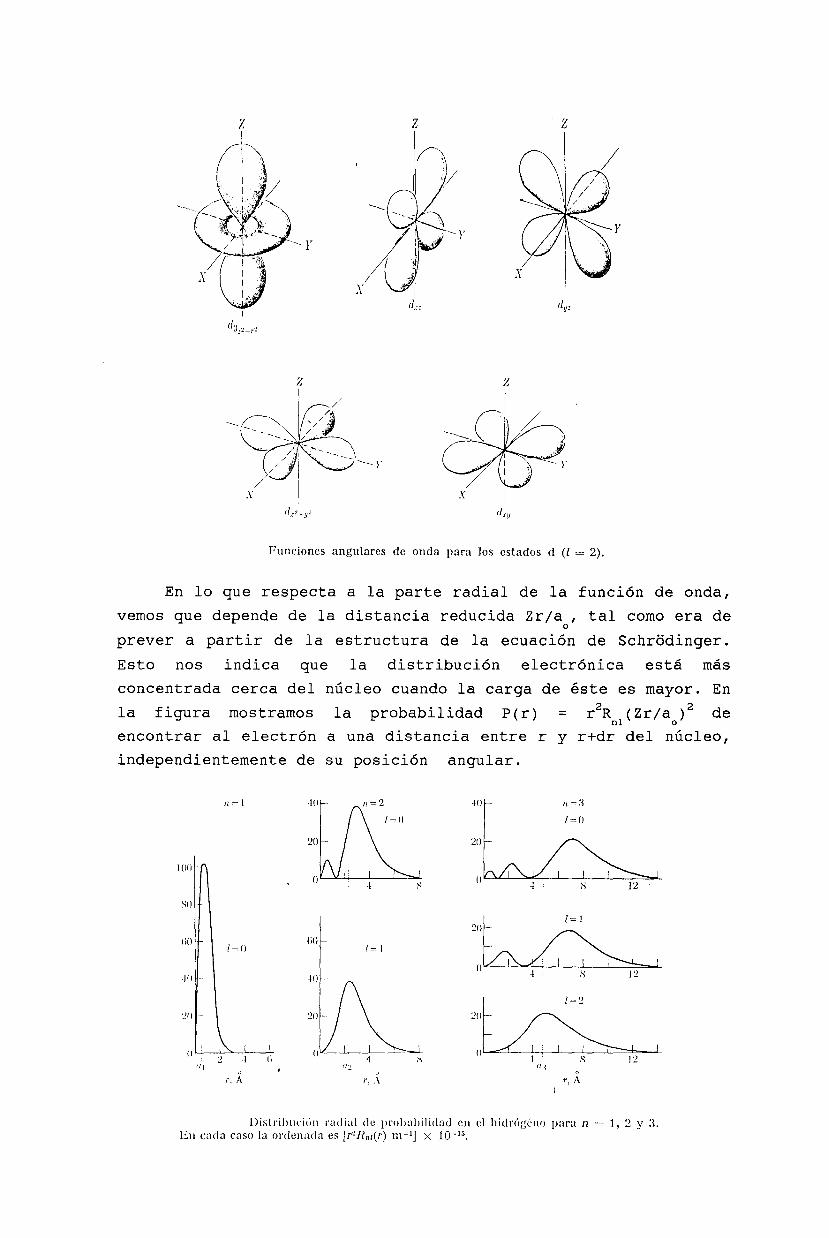
z
~.JJ~->lxVr
d"
z
z
Funciones angulares de onda para los estados d (l = 2).
En lo que respecta a la parte radial de la función de onda,vemos que depende de la distancia reducida Zr/a , tal corno era de
o
prever a partir de la estructura de la ecuación de Schrodinger.Esto nos indica que la distribución electrónica está másconcentrada cerca del núcleo cuando la carga de éste es mayor. Enla figura mostramos la probabilidad P(r) = r2R (Zr/a)2 de
ni o
encontrar al electrón a una distancia entre r y r+dr del núcleo,independientemente de su posición angular.
100
11=1
lA~j1=0
21l
(J ~ : s 12
so
lill -
lO
:!O -
11]
I=()
r, A
(jO
(('!.
1= 1
"r, ,\
~
20 •
01 : S 12
'J~1 . S 12
(/;~o
r, A
Dislribuciúll radial dc probabilidad en el hidrógl'llo para n = 1, 2 Y 3.En cada caso la ordcnada es [r2Rn¡(r) m-1J x 10-15•

Vemos que para un mismo valor de n, los autoestados conmenor valor de t tienen una probabilidad mayor de estar cerca del
núcleo. Decimos que son más penetrantes. Esto se debe a que, aldisminuir E, también disminuye la repulsión producida por el
2potencial centrífugo V (r) O< h g ( g +1) en la parte radial de la
e 2 2/lrecuación de Schrodinger. Tal como veremos, esta característicadel movimiento electrónico se refleja en algunas propiedadesimportantes de los átomos. Por ejemplo, los electrones s son mássensibles a la forma y estructura interna del núcleo, queaquellos en órbitas menos penetrantes.
CORRECCION ISOTOPICA
En general, podemos aprovechar que la masa del núcleo essuficientemente alta para aproximar la masa reducida del sistemaelectrón-núcleo por la del electrón exclusivamente,~ = rnM/(m+M) ~ m. De esta manera, la ecuación de Schrodinger, ycon ella la estructura electrónica del átomo, no depende delnúmero de masa A. Decimos que estamos despreciando posiblesefectos isotópicos, ya que de lo contrario, obtendríamos quedistintos isótopos de un mismo elemento tienen propiedadesdistintas. De hecho, como Ry O< ~, el pequeño corrimiento en losniveles de energía es del orden de
.6E lEn n
~-m~
m 1 -4= - M ~ - A 5.45 x 10
Esto hace que, por ejemplo, los niveles de energía E deln
Hidrógeno no coincidan exactamente con los correspondientesniveles E del Helio ionizado, como uno esperaría de no mediar
2n
efectos isotópicos. Fue esta pequenlslma diferencia la quecondujo en 1868 al descubrimiento del Helio por Frankland yLockyer al analizar el espectro de la luz solar. Igualmente elcorrimiento isotópico entre los niveles de energía del Hidrógenoy del Deuterio condujo a Urey y colaboradores al descubrimientode este último isótopo en 1932.
87

CORRECCION RELATIVISTA A LA ENERGIA CINETICA
En un tratamiento más preciso, basado en la ecuación deDirac, aparecen importantes correcciones relativistas a losniveles energéticos calculados en las secciones anteriores. Cornola velocidad media del electrón en un átomo hidrogenoide esaproximadamente Zac, con c la velocidad de la luz y
A 2 1e la constante de estructura fina, estasa=----~--41180 hc 137
correcciones serán de orden flEJE ()( ( Zex ) 2~ Z2 5 x 10-5 o superior.
En una formulación relativista la energía cinética es de laforma
T j 2 2+ 2 4' mc2= pc mc
donde hemos restado la energía en reposo para hacer coincidir elcero de energía con el caso no relativista. Ahora podemosaproximar
4P
8 3 2m c+ ...
en elVemos que aparece un término de perturbación H'
Hamiltoniano, que genera la siguiente correcciónenergía
4P
8 3 2m ca los niveles de
Vemos que, tal como esperábamos, esta corrección relativista esde segundo orden en Za. Además, al depender del momento angulart, rompe la degeneración accidental característica del potencialcoulombiano. Es siempre negativa y aumenta al disminuir l.
ACOPLAMIENTO ESPIN-ORBITA
Hasta ahora no tuvimos en cuenta la proyección m del espínsdel electrón. Esto se debe a que, en principio, el Hamiltoniano
88

no depende del espín y, por lo tanto, los estados son degeneradosen m . Tal degeneración se rompería de existir un campo magnético
s~B, ya que entonces deberíamos incorporar el término de ZeemanH' - - ~e s.a en el Hamiltoniano. Ahora bien, desde un sistema dereferencia fijo al electrón, el núcleo gira a su alrededor,produciendo un campo magnético paralelo al momento angular
-7orbital L. En efecto, usando la ley de Biot - Savart, lacorriente generada por el núcleo produce un campo magnético
~B =
-7 -7
(Zev)xr =3r
- Z471C
o
e2~mc
~ ~rxmv3r
= - Z i\
471Co
e2~mc
1 -7
- L3r
que interactúa con el momento magnético del espín del electrón,produciendo una contribución al HamiltonianoH' = Z 4~C (:C)2!3 s.L. Esta corrección, denominada
o racoplamiento espín-órbita, se obtiene inmediatamente en untratamiento relati vista del átomo hidrogenoide, excepto por unfactor 1/2, denominado factor de Thomas, y que incorporamos ahoraarbitrariamente. Tenemos entonces
Z i\H' = ---24m::
o
(~)2! -7 -7s.Lmc 3r
La incorporación de este término adicional hace que, ahora,ni L ni S conmuten con el Hamiltoniano. m y m ya no son
z z 1 S
buenos números cuánticos para caracterizar los autoestados deenergía definida. Sin embargo, puede verse fácilmente que lacorrección espin-órbita H' conmuta con el momento cinético total-7 -7 -7
J = L + S y, por lo tanto, es diagonal en la representaciónacoplada caracterizada por L2, J2 Y J. Vemos entonces que esta
z
corrección produce un desdoblamiento de cada nivel E enn
subniveles caracterizados, en princlplo, por autovalores n, t, jY M. Teniendo en cuenta que J2 = (1,+S)2=L2+ S2+ 2 L.s, elcorrimiento de energía resulta
LlE = < n, t, j ,M IH' In, t, j ,M> =
89

Z i\ (~)2 <n,l, j,M'l:.(l_L2_S2) In,l,j,M>= --- =4 4rrf:o
mc 3r
Z i\(~ ) 2 h2
[ j (j+1) -l (H 1) - 3/4] <n,l,j ,MI! In,l,j ,M>= --4 4rrco mc 3r
Vemos que esta corrección es nula para l = O, tal como era deesperarse. Calculando el elemento de matriz de 1/r3
, obtenemosfinalmente
!lE = (Z IX) 2 lE I ( j -1 )nlj n n(l+1/2) (j+1/2)
para l :t O Y con j = e ± 1/2. Esta corrección separa los nivelessegún los números cuánticos n, I!, Y J manteniendo unadegeneración en el número cuántico magnético total M.
TERMINO DE DARWIN
En un tratamiento relativista puede demostrarse que apareceuna perturbación adicional al Hamiltoniano, denominada término deDarwin, y que es proporcional a la divergencia del campoeléctrico. Aplicando la ley de Gauss tenemos entonces que estacorrección es proporcional a la densidad de carga nuclear,H ~ div(E) ~ Ze o(r). Para la corrección de la energía obtenemos
D
entonces que L\E ~ < nfm lo (r) I nfm> 11/1 (O) 12, que es distinta denlm
cero sólo cuando f=O. Resulta
ESTRUCTURA FINA
Las correcciones relativista, de espín-órbita y de Darwinson del mismo orden en Za. Sumándolas obtenemos
L\Enj
2= (Za)
Vemos que se produce un muy interesante resultado, por el cual,
90

aunque la corrección relativista a la energía cinética separa lostérminos con diferentes valores de l, el acoplamientoespín-órbita y el término de Darwin los vuelven a juntar, demanera tal que la degeneración en el número cuántico orbitalpersiste. Por ejemplo, para n = 2, se da la situación de lafigura. En ella hemos usado la notación espectroscópica de losestados acoplados ni., donde los valores l = O, 1, 2, 3, 4, 5,
Json representados por las letras mayúsculas S, P, D, F, G,
H, ••• , respectivamente. Cada nivel n, de degeneración 2n2, sesepara en n niveles caracterizados por el momento cinético totalj = 1/2, 3/2, 5/2, ... El conjunto de niveles para cada númerocuántico principal n forma así el denominado multiplete deestructura fina.
cor recc',ónr~ - ---.JA
,
relativista spin- órbita Oarwin Lamb
(zcG)2IEnln
\',\ ' ,\ ' ,2P1/2 2P3/2(" '\ ,\ ',
~--~.-+- "-\\\\\\\\\ 251/2 251/2
II
II
II
/I
II
I
2P:V2 2P3/2
251/22P1/2~ 251/7
2Pl/2
La degeneración en e y M se mantiene a cualquier orden enZa. En efecto, un cálculo relativista, en base a la ecuación deDirac, muestra que la energía de cada subnivel está dada por
Enj = mc2 {[[ 1 + (Za)2( n-j-l/2+!i+l/2-(Za)2' )-2J-1/2 -l}
que coincide con la expresión hallada por nosotros a orden (Za)2
y mantiene la degeneración en e y M a todo orden.
91

Si analizamos las transiciones dipolares eléctricas entreestos estados de estructura fina, usando las reglas de selecciónM = ±1 y llj = O, ±1, vemos que las líneas espectrales entreniveles s y p se han convertido en dobletes, y las líneas entreniveles p y d en tripletes. Sin embargo, en una transiciónt ~ t-l las líneas correspondientes a transiciones j ~ J son muydébiles en comparación con las correspondientes a transiciones j~ j-1, por lo cual la línea 3D ~ 2P / es muy débil frente a
3/2 3 2las líneas 3D ~ 2P Y 3D ~ 2P , Y por lo tanto el5/2 3/2 3/2 1/2triplete también se observa como un doblete. Un caso típico dedesdoblamiento de líneas de emisión está dado por la líneaamarilla del Sodio, que forma un doblete de longitudes de onda5890 Á Y 5896 Á. De hecho, la necesidad de explicar este tipo dedoblete fué lo que dió origen a la idea de un espín electrónicocon dos posibles orientaciones.
1=0 1=1 1=2
n=3
n=2
Desdof¡lamiento dI' los nive-les dt~ energía por la ilfl er:H'ción cspln-órbita y trallsiciones Jlosihles, La !lneatk Irazos indien una Il'allsiciúll eOIl (lI'O- 1l.~1habilidad lIJuy haJa.
EFECTO LAMB
Tal como dijimos, aún en la cuántica relativista de Dirac,la ruptura de degeneración dada por la estructura fina, no escompleta, manteniéndose la degeneración de los nivelesenergéticos respecto del momento angular orbital t. Sin embargo,un muy cuidadoso experimento de absorción de micro-ondasrealizado por Lamb y Retherford en 1947 mostró que estos nivelesdegenerados presentaban un muy pequeño desdoblamiento. Estacorrección adicional, denominada efecto Lamb, se debe a la
92

interacción del electrón con el campo electromagnético del vacío.El estado fundamental del campo de radiación no es nulo, sino queexiste una oscilación de punto cero que actúa sobre el electrónhaciéndole ejecutar un movimiento oscilatorio que desdibuja suposición. Por este motivo el potencial nuclear "neto" que sienteel electrón es levemente diferente del correspondiente a suposición media, lo que se manifiesta en un corrimiento de losniveles de energía, separando los niveles de diferente valor dee, o sea de diferente paridad. La diferencia de energía entre losniveles 28 y 2P es de apenas !lE~ 3.2 x 10-7 Ry para Z=l.
1/2 1/2
CORRIMIENTO ISOTOPICO POR VOLUMEN NUCLEAR
Las propiedades que distinguen a un núcleo de un centropuntual de campo coulombiano (masa finita, dimensiones, espín),influyen sobre los niveles de energía de un átomo. Uno de estosefectos es el corrimiento isotópico de los niveles que yaestudiamos anteriormente y que era debido a la masa finita delnúcleo. Este efecto disminuye en importancia al aumentar elnúmero de masa A, pero comienza a pesar una corrección debida altamaño finito del núcleo. En la práctica este efecto sólo seobserva en los niveles S que (en contraposición a las funcionesde onda de los estados con e * O) son penetrantes.
Si ~(r) es el potencial electrostático del campo del núcleo,la corrección a la energía correspondiente a un potencialcoulombiano puro es
f¡E <nsl<1>(r)-i\ Ze Ins>= -e 4m: - =ns r
o
-eS [~(r) i\ Ze ] 1/12 (r) dr= - 4rrc -r nso
La diferencia entre ambos potenciales es no nula tan sólo dentrodel volumen del núcleo, donde podemos aproximar a 1/1 (r) por sunsvalor en r=O,
f¡Ens
= - e 1/12 (O) J [~( r ) _ i\ Ze ] drns 4rrc r
o
93

Finalmente, considerando una distribución homogénea de la carganuclear en una esfera de radio R, resulta
!lEns
= - e 1/12 (O) A Ze 3 R2ns 4rre '5
o
y reemplazando 1/12 (O)ns
1 Z 3= ( ) obtenemos finalmenterr nao
!lE = ~ E 1 (ZR/a ) 2ns 5rr n n o
Corno el radio nuclear es del orden de R ~ A1I3 1.4 x 10-13cmpodemos calcular explícitamente la magnitud de este corrimientoisotópico, obteniendo
LiE lE = 1. 3 x 10-10ns n
Z2
n A2/3
que resulta ser tres órdenes de magnitud menor que el efectoLamb.
ESTRUCTURA HIPERFINA
Otro efecto debido a las propiedades del núcleo es eldesdoblamiento de los niveles de energía producido por lainteracción del electrón con el espín nuclear. Esta interacción
a la interacciónHamil toniano de la
el momento cinético
una perturbación delde manera similar
espín nuclear 1 con
está caracterizada por-7 -7forma H' ~ I.J que,
espín-órbita, acopla eltotal J del electrón.
Al igual que en el caso de la interacción espín-órbita, laforma del término de perturbación H' sugiere acoplar ambosmomentos cinéticos 1 y J definiendo F = I+J. H' conmuta con estemomento cinético total, y por lo tanto es diagonal en la
222representación acoplada caracterizada por J, 1, F Y F . Vemosz
entonces que esta corrección produce un desdoblamiento de cadanivel de estructura fina nL en subniveles caracterizados por el
J
94

autovalor F del momento cinético total. Las diferencias deenergía de este desdoblamiento, denominado de estructurahiperfina, son muy pequeñas en comparación con los intervalos deestructura fina. Por ejemplo, para el nivel 28 del átomo de
1/2
hidrógeno, como el espín nuclear es el correspondiente al protónI=1/2, los valores posibles del momento cinético total son F=O yF=l. La diferencia de energía entre ambos estados de estructurahiperfina es !lE ~ 4 x 10-7 Ry, que es del orden del efectoHFLamb. Una transición entre ambos niveles produce la emisión deluz de fecuencia w = 1420 MHz, es decir en la banda de lasmicroondas (A = 21.4 cm.). Esta frecuencia es una de lascantidades medidas con más precisión en la física, con docecifras significativas
w = 1420405751.800 ± 0.028 Hz
Desde el punto de vista teórico, el cálculo correspondienteinvolucra una descripción muy precisa de la estructura nuclear, yhasta el presente no hay ningún modelo que logre ajustar esteresultado experimental.
La radiación proveniente de esta transición juega un rolimportante en astrofísica. En un gas de átomos neutros, el estadoF = 1 tiene una alta probabilidad de excitarse por colisión. Asíel análisis de la intensidad de la linea de 21 cm producida porel decaimiento al nivel F = O da mucha información sobre ladensidad, temperatura y movimiento del gas de Hidrógenointerestelar.
BIBLIOGRAFIA
1. H. Bethe & E. Salpeter: Quantum Mechanics of one and twoelectron atoms (Springer-Verlag, Berlin, 1957).
2. M. Alonso & E. J. Finn: Física~ volumen III: Fundamentoscuánticos y estadísticos (Fondo Educativo Interamericano,Barcelona, 1971).
3. B. H. Bransden & C. J. Joachain: Physics of Atoms andMolecules (Longman, London, 1983).
95


EL ATOMO DE HELlO
INTRODUCCION
Estudiaremos ahoranuclear Z, como ser elionizado Li+ (A=7, Z=3)
un átomo de dos electrones y cargaátomo de Helio (A=4, Z=2), el Litioo el Berilio doblemente ionizado Be2
+
(A=9, Z=4).
En lo que sigue despreciaremos todo efecto isotópico,considerando que, siendo la masa del núcleo mucho mayor que la delos electrones, podemos 19norar su movimiento y reducir elproblema al de dos electrones en un potencial central. Debemosresolver, entonces, la siguiente ecuación de Schrodinger
2
[- ~ r¡i2m 1
~ iJ2 A Ze2
A Ze2 + _A_ e
2] . ~ ~)
2m 2 - 41lc r'- - 4m:' r 4rre r <P ( r 1 ' r 2o 1 o 2 o 12
~ ~= E <p(r ,r )1 2
~ -+ -+ .• •con r = Ir - r ! la dlstancla entre 108 dos electrones. Podemos12 1 2
escribir esta ecuación en la forma
conH
i
2 2~ iJ2 A Ze2m i - 4rrc ro i
el Hamiltoniano para un átomo tipo hidrogenoide de número átomicoZ, y
el potencial de interacción entre electrones.
97

ESTADOS SINGLETES y TRIPLETES
Puesto que estamos suponiendo que el Hamiltoniano delproblema es independiente de las variables de espín, podemosadoptar, para describir el estado de espín, la representación quequerramos. En particular, puesto que pone claramente demanifiesto las propiedades de simetría del sistema anteintercambio de partículas, adoptamos una representación acoplada
-7 -7 -7de vectores propios x(S,m ) del espín total S = S + S dados pors 1 2
X(O,O) + - - + /12= [Xl X2 - Xl X2]
x(l,l) + += Xl X2x(1,0) + - + / V2= [Xl X2 + X X2]1x(1,-1)= - -Xl X2
Por el principio de Pauli, el vector de estado total debeser antisimétrico ante intercambio de partículas. Esto implicaque la parte espacial <p (r ,r ) que acompaña al espinor X (O,O)
1 2
debe ser simétrica, y antisimétrica la que acompaña a x(1,m). Ossea que tenemos cuatro posibilidades
1/1(1,2)
- -- --= <p (r 1 ' r 2) X ( 1 , ms) ms 1,0,-1
de Schrodinger conestado se denomina
+ -- -- ,donde <p-(r ,r ) es la solucion de la ecuaClon,,12+__ __ +__ __ ,
la condlclón <p-(r ,r ) = ±<p-(r,r ). El prlmer1 2 2 1
singlete. Los últimos tres, correspondientesdenominan tripletes.
a s = 1, se
APROXIMACION DE ELECTRONES INDEPENDIENTES
Si ignoramos la repulsión entre electrones V(r ) en la12ecuación de Schrodinger podemos escribir los autoestadoscorrespondientes como combinaciones lineales, adecuadamentesimetrizadas, de productos de autoestados hidrogenoides. O sea
98

+ ~ ~q>-(r ,r ) =
1 2
~ ~~ (r) ~ (r) ] /~
nlm 2 nlm 111122 2
si los estados hidrogenoides son distintos, y
+ ~ ~q> (r Ir ) =
1 2
~ ~~ nlm ( r1 ) ~ nlm ( r2 )
en caso contrario.Los niveles de energía en este modelo están dados entonces
por la suma de las energías de dos átomos hidrogenoides.
En1
+ En
2
= _(_1_ + _1_) Z2 Ry2 2n n
1 2
Así, la energía del estado fundamental es el doble de la de unátomo hidrogenoide de igual número atómico. Para el Helio, porejemplo, E 2 = -8 Ry que es un 38 % superior que el valor
ls
experimental E 2 = -5.808 Ry. Esta discrepancia proviene de quels
hemos despreciado la repulsión entre electrones. Para númerosatómicos Z mayores, el potencial interelectrónico es menosimportante en comparación con la atracción electrón-núcleo y esde esperar que el modelo de electrones independientes déresultados más precisos. Por ejemplo, el valor teórico
+4E 2 = -72 Ry para el estado fundamental del ión de carbono els
es sólo un 12 % mayor que el valor experimental E 2 = -64.4 Ry.ls
APANTALLAMIENTO
Los niveles anteriores se miden desde un cero de energíacorrespondiente a ambos electrones infinitamente separados entresí y del núcleo. También podríamos usar como cero la energía delátomo ionizado, es decir cuando un electrón se encuentra en elinfinito y el otro en el estado fundamental del ión hidrogenoideresidual
Ecero
= E1
+ E00
2= -Z Ry
Si observamos los valores experimentales de los niveles de
99

energía, medidos desde ese cero de ionización, vemos que, dejandode lado el estado fundamental, estos se apartan poco respecto delos niveles de un átomo hidrogenoide de carga nuclear Z-l. Estose debe a que, si consideramos un estado excitado 1s ni, elefecto de la repulsión entre electrones se comporta sólo como unapantallamiento de la carga nuclear. El electrón en el órbital niexterior ve una carga nuclear Z-l reducida por la presencia delelectrón 1s más penetrante. De hecho, la carga del electrónexcitado está distribuida casi completamente fuera de la delnivel 1s. Los niveles de energía, medidos desde el cero deionización, son entonces
Elsnl
2~ _Z2Ry _ (Z-l) Ry - E2 ceron
(Z_1)2 R2 Y
n
Este argumento justifica la definición de una carga efectiva*Z = Z-l para describir los niveles de los átomos de dos
electrones. Inclusive podemos definir una carga efectiva para elnivel fundamental, de manera tal que, medido desde el cero deionización
* 2E1s
2 = - (Z ) Ry
Para el Helio y demás iones livianos hasta Z ~ 9 (F+7) severifica experimentalmente que
•Z 2 ~ Z - 0.63ls
TRATAMIENTO PERTURBATIVO
Vamos a mejorar nuestra descripción del estado fundamentaldel átomo de dos electrones por medio de un tratamientoperturbativo del potencial V(r ). Calculamos el valor medio
12 + ~ ~ ~ ~del Hamiltoniano entre estados cp (r ,r ) = 4> (r) 4> (r), con1 2 ls 1 ls 2
100

~~ (r)15 = !Z3jlla3' exp[-Zrja ], obteniendoo o
E 2ls
2 + +~ -2Z Ry + <~ IVI~ > =
2 5= -2Z Ry + 4" Z Ry
En particular, para el Helio, tenemos E 2 = -5.5 Ry que ahorals
difiere en sólo un 5% del valor experimental. Aquí hemostrabajado con una función de onda donde los electrones actúanindependientemente uno de otro. Sabemos, sin embargo, que lapresencia de uno de ellos apantalla la carga nuclear que ve elotro. En la siguiente sección tendremos en cuenta este hecho pormedio de un cálculo variacional.
CALCULO VARIACIONAL
Un método más exitoso para obtener el valor de la energíadel estado fundamental del Helio es el método variacional.utilizamos corno función de onda de prueba
+ -> ->rp(r,r)
1 2
3= (Z-<T)
3rcao
r +r ]1 2
ao
donde el factor de apantallamiento <T es el parámetro variacional.Resulta entonces
Minimizando corno función de <T, resulta <T = 5/16. O sea que lafunción de onda hidrogenoide da el mejor valor de la energíacuando la carga nuclear se reduce en <T = 5/16, lo cual indica queel electrón apantalla la carga nuclear que ve el otro.Encontramos que el nuevo valor de la energía del estadofundamental es E 2 = -2(Z-5/16)2 Ry, que es un 1.9% mayor que el
ls
valor experimental para el átomo de Helio.*La carga efectiva Z de un átomo hidrogenoide de igual
101

energía fundamental (medida desde el cero de ionización) resultaser
= Z - 0.625 en el límite de Z
que laPara el
los valores experimentales meJor~ Z - 0.63 descrita anteriormente.
*Z - 0.698 Y Z
Esta función ajusta*dependencia simple Z
*Helio tenemos Z =
grande.
ESTADOS EXCITADOS
El primer estado excitado corresponde a una configuración 1s2f • O sea
m
la corrección a este
electronesde5 2- 4" Z Ry. A primer orden
nivel
aproximación
en la repulsiónenergético es
La energía correspondiente, en la2 Z2independientes, es E = -Z Ry - - Ry =
22
entre electrones,
!lE±
= f± f
dr1
dr1
El primer término tiene la forma familiar de una interacciónelectrostática entre dos nubes electrónicas distribuidas con unadensidad 11> 12• Es una simple generalización de la corrección dela energía para el estado fundamental. El segundo término notiene una interpretación clásica. Tiene su origen en el principiode pauli y su signo depende del espín total s = O (signopositivo) o s = 1 (signo negativo). Debido a este término de
102

intercambio los estados singlete y triplete no tienen la mismaenergía. Ya no son más degenerados en S.
La primera integral es manifiestamente positiva. Y lo mismopuede demostrarse para la segunda. Luego los estados tripletesestarán más ligados que el correspondiente estado singlete. Estose puede entender, también, desde un punto de vista cualitativo.Para el estado triplete la parte espacial es antisimétrica, y loselectrones están restringidos a mantenerse alejados uno de otro.De esta manera el apantallamiento de la carga nuclear es menor y,además, la repulsión es menos efectiva que para un estadosinglete. Estas conclusiones son válidas para cualquier nivelexcitado, y suelen expresarse en la denominada primera regla deHund, que dice que si otras cosas son iguales los estados de
mayor espín tienen menor energía. Un aspecto interesante de este
resul tado es que, aunque la repulsión entre los electrones no
depende del espín, la simetría de la función de onda hace que el
potencial actúe como si dependiera del espín.
En general las fuerzas que dependen del espín son muydébiles. Un ejemplo es el acoplamiento espín-órbita que es de
orden (Za) 2 respecto de las interacciones electrostáticas. Ladependencia con el espín debida al intercambio es mucho másfuerte que esto. De hecho es del mismo orden que las fuerzas
electrostáticas.
TRANSICIONES DIPOLARES ELECTRICAS
En la siguiente figura mostramos los primeros niveles delHelio. Las reglas de selección para transiciones dipolares
eléctricas entre estos niveles son
f1S Of1L = O, ±I
f1J O, ±l
L=O ~ L'=O prohibida
J=O ~ J'=O prohibida
a las cuales agregamos la regla de inversión de paridad que, paratransiciones entre configuraciones (n e ,n e ) ~ (n' e' ,n' e' ) ,
11 22 11 22
indica que
103
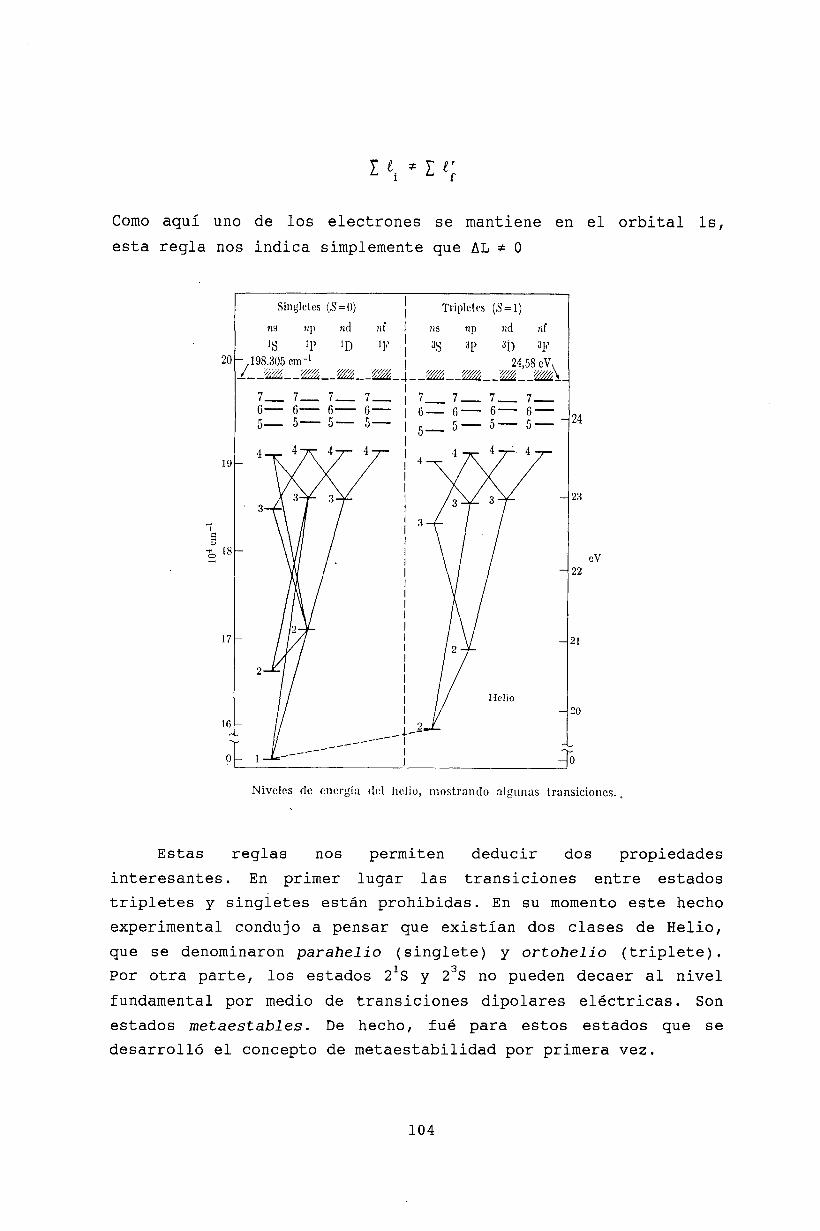
Como aquí uno de los electrones se mantiene en el orbi tal 1s,esta regla nos indica simplemente que ~L * O
Singlctcs (8 = O) 1 Triplctcs (8 = 1)
n8 np nd nf I ns np nd nfIS lp 11) 11<': 3S 3p 31) 31<'
20 198.305 cm-I I 2458 eVj--~--~--~--~-4--~--~--~~-~\
19
'18<.Jb 18-~
17
16
o~
7_(j-
5-
4
3
2
I7_ 7_ 7_ I 7_ 7_ 7_ 7_(j- (j- (j- I u- 6- (j- 6-r. r. r. • •i)- i)- i)- I 5- 5- a- a-
II 4IIIIII 3IIIIIIIIIIIIIIIII HclioII 2J.--------1
------ II
24
23
22
21
20
eV
Niveles de encrgia del heJio, mostrando algunas transiciones ..
Estas reglas nos permiten deducir dos propiedadesinteresantes. En primer lugar las transiciones entre estadostripletes y singletes están prohibidas. En su momento este hechoexperimental condujo a pensar que existían dos clases de Helio,que se denominaron parahelio (singlete) y ortohelio (triplete).Por otra parte, los estados 21S y 23S no pueden decaer al nivelfundamental por medio de transiciones dipolares eléctricas. Sonestados metaestables. De hecho, fué para estos estados que sedesarrolló el concepto de metaestabilidad por primera vez.
104

ESTADOS DOBLEMENTE EXCITADOS Y AUTOIONIZACION
Hasta ahora estudiamos estados excitados donde uno de loselectrones permanece en el nivel fundamental 1s. Consideremosahora un estado doblemente exci tado ni ~ n' i'. La energíacorrespondiente en el modelo de electrones independientes es,respecto del nivel de ionización
~ Z2 Ry - Z2 Ry - EE ~ - -- =nln' l' 2 ,2 ceron n
Z2 Ry [1 l/n 2 1/n,2] Z2 Ry /2= - ~
Vemos que estos estados se encuentran bien por encima delnivel de ionización. El sentido físico de estos niveles discretossumergidos en un contínuo es similar al de los estados de unátomo perturbado por efecto Stark. Ya sea por colisión o porabsorción de radiación, el átomo tiene una alta probabilidad deexcitarse a uno de estos seudoestados discretos, en lugar deionizarse directamente. Esto se manifiesta cornoun salto bruscode la sección eficaz o el espectro de absorción correspondientescuando la energía entregada al átomo es la de uno de estosniveles. Este efecto se denomina resonancia. Una vez poblados,estos niveles pueden decaer a un estado ligado emitiendoradiación, o bien emitiendo un electrón y decayendo a un niveldel átomo ionizado. Este último proceso se denominaautoionización. Para el nivel 2s2 del Helio, por ejemplo, la
-15autoionización ocurre con una vida media ~ = 5 x 10 seg.
ION NEGATIVO DE HIDROGENO
El ion H es un caso muy interesante de átomo tipo Helio. Esapenas estable, con una energía de ionización de 0.0554 Ry.Además de ese estado fundamental no posee ningún otro estadoligado. El estado fundamental se mantiene ligado como resultadode una penetración mutua de ambos orbitales, de manera queninguno de ellos apantalla completamente la carga nuclear que ve
105

el otro. Calcular su energía significa una prueba de fuego paralos métodos de cálculo aproximado. Por el método variacional, porejemplo, obtenemos E == -2 (1-5/16)2 Ry - E == 0.055 Ry, queceroestá por encima del nivel de ionización. Sólo métodosvariacionales más sofisticados, con funciones de onda de variosparámetros ajustables, permiten obtener valores realistas para laenergía del estado fundamental.
El ión negativo de Hidrógeno juega un papel muy importanteen conexión con la teoría de hídridos alcalinos. Talesestructuras pueden pensarse como la combinación de un ión álcalipositivo y un ión de Hidrógeno negativo.
Tambien es de gran importancia en el estudio de la opacidadde la atmósfera del Sol y otras estrellas. EL potencial deionización de 0.0554 Ry equivale a una temperatura T = 8700° K,que es levemente superior a la de la atmósfera solar. Estaatmósfera está constituida principalmente por Hidrógeno neutro yuna proporción de iones negativos H-. Debido a la temperaturarelativamente baja de la atmósfera, los iones de H- no se ionizanpor colisión sino, principalmente, por absorción de radiación.Los electrones emitidos son recapturados por el hidrógeno neutro,repitiendo el proceso. Este mecanismo es el principal responsablede la opacidad de la atmósfera solar.
En un acelerador TANDEM, la creación de un haz de ionespositivos requiere la creación de iones negativos que sonacelerados la mitad del camino, ionizados en una cámara deintercambio, y vueltos a acelerar en la segunda mitad. Vemos asíque la fabricación de iones H- en el laboratorio es un problemade interés práctico para la obtención de haces de protones.
BIBLIOGRAFIA
1. H. Bethe & E. Salpeter: Quantum Mechanics of one and two
electron atoms (Springer-Verlag, Berlin, 1957).2. M. Alonso & E. J. Finn: Física~ volumen III: Fundamentos
cuánticos y estadísticos (Fondo Educativo Interamericano,Barcelona, 1971).
3. B. H. Bransden & C. J. Joachain: Physics of Atoms and
Molecules (Longrnan, London, 1983).
106

HARTREE -FOCK y THOMAS-FERMI
TEORIA DE HARTREE Y FOCK
Ahora generalizaremos el método de cálculo empleado parahallar el nivel fundamental de un átomo de dos electrones, alcaso de un átomo mul tielectrónico cualquiera. Usando la mismanotación del capítulo anterior, tenemos que resolver la siguienteecuación de Schrodinger para un átomo de n electrones y de númeroatómico Z
dondef12 "\ Ze2
H (-7) = íJ2 _ 11.Z r - 2m 4m:: r
o
es el Hamil toniano para un átomo tipo Hidrogenoide de númeroatómico Z, y
V(r)
es el potencial de interacción entre electrones.Nuevamente escribimos a la función de onda como un producto
antisimetrizado de estados de cada electrón. Para elloaprovechamos la propiedad de cambio dedeterminantes ante intercambio de dos de sus
signo de losfilas o columnas,
para escribir la función de onda de la siguiente manera
I/J = 1
vnT [
q; (r)x (1) •••••• q; (r)x (n)]1 1 1 1 n 1. .
det: :-7 -7
q; (r)x (1) •••••• q; (r)x (n)n 1 n n n n
El conjunto de todos los determinantes de Slater distintos que sepueden formar con una base completa de autoestados q; es una basecompleta para las funciones de onda antisimetrizadas del espacio
107

de n electrones. La solución general de la ecuación deSchrodinger se puede escribir como una combinación lineal de loselementos de esta base. Sin embargo, y al igual que en el casodel átomo de dos electrones, nos restringiremos a trabajar con unúnico determinante de Slater. De esta manera persiste en lafunción de onda una correlación dada por el principio deexclusión, pero no la dada por las interaccionesinterelectrónicas.
Repitiendo los cálculos que realizamos en el caso del átomode dos electrones obtenemos para el valor de expectación de laenergía
<l/1IHIl/1> ~ I1
+ Li < j
Reconocemos los mismos términos obtenidos en el caso delátomo de Helio. El primero tiene en cuenta las interaccioneselectrón-nucleo. El segundo, denominado término directo,incorpora la repulsión entre electrones como una interacciónelectrostática entre dos nubes electrónicas distribuídas con unadensidad 14>(h 12
• El último es el término de intercambio que,como ya sabemos, se debe al principio de exclusión de pauli.Advertimos que se anula a menos que el estado de espín sea elmismo para ambos orbitales.
Ahora podemos considerar una dada configuración electrónicaen un modelo de electrones independientes y calcular lacorrespondiente corrección a la energía debida a la repulsiónentre electrones, en un primer orden de aproximación. Para ellousamos la expresión anterior para el valor de expectación de laenergía, reemplazando los estados 4>. por adecuados orbitales
1
hidrogenoides. Sin embargo, este método no da muy buenosresultados en el caso de átomos multielectrónicos y, por lotanto, conviene ir un paso más allá, ajustando variacionalmentela forma funcional de los estados monoelectrónicos 4>, de manera
1
de minimizar la energía. Debemos recordar que ahora tenemos
108

ciertas condiciones de ligadura dadas por la ortonormalización delos estados ~. Al estudiar el estado fundamental del átomo deHelio esto lo tuvimos en cuenta al considerar como función deprueba un orbital 18 normalizado de carga variable. Ahora, encambio, vamos a realizar una variación arbitraria ~. -7 ~.+o~., y
1 1 1
la normalización la vamos a mantener utilizando el método de losmul tiplicadores de Lagrange. Esa técnica nos indica que lasfunciones ~ que minimizan el valor de expectación de la energíaverifican el siguiente problema de autovalores de ecuacionesacopladas
-7E.4>.(r)1 1
donde la densidad de carga efectiva que interactúa con esei-ésimo electrón es
-7 •.•
p.(r,r)=1
..• 2 * ..• ..•L el4>.(r)1 + L e o(m ,m ) </>.(r)</>.(r)J s S J 1
j j j
...4>.(r)J ...</>.(r)1
De esta forma 4>es la solución de una ecuación de Schrodinger conun Hamiltoniano que es suma de la energía cinética, una energíapotencial debida al núcleo y una energía de interacción con losrestantes electrones. En la densidad p(r,r') que define a estainteracción distinguimos nuevamente un término directo y otro deintercambio. Al respecto notemos que un electrón no puede ejercerninguna acción sobre sí mismo y, en efecto, el término i-ésimodel término de intercambio cancela un término similar en eltérmino directo. Si integramos sobre el espacio, aplicando laortonormalidad de las funciones </>,resulta Jp(f,f')d31'=-(n-1) e.Esto nos dice que el término de intercambio es interpretable comola acción de una carga eléctrica positiva e sobre el electrón deforma que la carga electrónica total que actúa sobre el electrónen estudio es - (n-1)e. En el punto donde está el electrón la
. -7-7 -7 2densldad es Pi
(r,r) = - L [l-o(IDs,ms ») el</>.(r) I , que tiene el. .. JJ 1 J
valor de la densidad de carga en el punto r debida a todos loselectrones con espín opuesto al del electrón en estudio. En
109

el radio mediolo tanto,
del término de
Podemos concluir que el término dela distribución de carga negativa del
unitaria situada alrededor de la
difiere de r en más que* 4 ~<1>. (r')<1>.(r) ::O y, porJ J
O sea que la contribución
~r'cuandocambio,
de los orbitales,P. (t,l') =-¿ el<l>.(1,)¡2.
1 . JJ
intercambio tiende a cero.
sistema atómico una carga~coordenada r del electrón en estudio, y debida a la distribución
de carga de los electrones del mismo espín que el estudiado.Esto es una confirmación de la presencia del principio deexclusión de pauli en la ecuación de Hartree-Fock. El resultado
intercambio remueve de
es como si el electrón cuya función de onda estamos buscandoarrastrase consigo un agujera que elimina una carga -e de susinmediaciones. Este agujero es el resultado del principo deexclusión de Pauli que mantiene a los electrones de igual espínlejos del electrón en estudio.
Para completar esta sección debemos dar una interpretaciónfísica del autovalor E. Si calculamos el valor medio de la
i
energía para un átomoelectrones, digamos el<~IHI~> correspondiente
en el que hemos eliminado</>, y lo comparamos con el
k
al átomo original, obtenemos
uno de losvalor medio
<H> - <H> = Jn n-l3~ • ~ ~ ~d r </> (r) H (r) </> (r) +
k Z k
+ L
= J ~·(t) [H (~) A~k Z r - 4rre
oe J
o sea
E representa entonces la energía que hay que entregarle al átomok
de n electrones para arrancar uno situado en el orbital k. ¡Peroojo! : Esto es sólo una aproximación, puesto que los orbitalesrestantes van a variar debido a la ausencia de un electrón. Sinembargo la aproximación es muy buena, especialmente cuando N es
110

grande (Teorema de Koopmans).
es la energía total del átomo.energía puede escribirse
Vemos además que la suma L E nok
kEn efecto, el valor medio de la
-7 -7
L J <P~(t)~ 1 i\ J P; (r,r')
dt,)] <P. (t) d3t=<H>= [H (r) - 2" 41lC e1 z It-t' I 1
o
1 i\
JP. (t,t·)
d3t d3t,= L E + - --e L 1<P.(t)12 1i 2 4rrc
1 Il-l'lo
Esto se debe a que, si consideramos sólamente todas las energíasE necesarias para arrancar cada uno de los electrones pori
separado del conjunto de N electrones, estaríamos contando dosveces la repulsión entre ellos. Ese es justamente el términorestante en la expresión anterior.
MODELO DE THOMAS-FERMI
Los cálculos numéricos de la distribución de carga y delcampo en un átomo siguiendo el método de Hartree-Fock sonextraordinariamente engorrosos, en particular para átomoscomplejos. Pero precisamente para estos átomos existe otrométodo aproximado cuyo valor consiste justamente en susimplicidad. En todo caso sus resultados pueden utilizarse cornopunto de partida para el método de Hartree-Fock.
Consideraremos que los electrones atómicos están sometidos auna energía potencial V(r) que se anula en el infinito y es unafunción suave de la distancia r al núcleo. Además suponemos quehay una buena cantidad de electrones de modo tal que la carga deuno de ellos es una pequeña porción de la carga total, por lo quela energía potencial V(r) que actúa sobre un electrón esprácticamente igual al potencial generado por la carga total.Bajo estas suposiciones el número de estados electrónicos en un
3--7volumen d r donde V (r) es sensiblemente constante resulta ser,d3i d3paplicando condiciones periódicas de contorno, ---- A este
h3
número debemos multiplicarlo por 2 para tener en cuenta los dos
111

d31d3pposibles estados de espín dn = 2 ---o Entonces ( si en unh3
~dado punto r todos los estados están ocupados hasta cierto nivelde energía cinética p2(~) 12m, la densidad de carga es
o
~ J d3-7 dnp(r) = -e p 3-7 3 =p:sp d r d p
o
2rr 8rrp3
2 t' 2 r de J d</J o= -e - p dp sine = -eh3 O O O 3h3
(De no
máximo2
~m + V(r). El valorindependiente de r
La energía total de cada electrón es2de esta energía, E = P 12m + V(r), eso
serIo, los electrones pasarían a los puntos con valor menor de E)y negativa (Pues si no, el electrón escaparía al infinito).Obtenemos así
p(r) = 8n e [2m(E-V(r»]Y23 h3
Por otro lado, el potencial electrostático V(r)/e estádeterminado por la ecuación de poisson
n2V(r) = A p(r)v e 4nc
o
Definiendo
V(r)
con a el radio deo 2/3b = 2(4/3rr) ~ 1.13,
Bohr y b una constante numérica igual aesta ecuación diferencial puede escribirse
en forma adimensional corno
= <1>3/21/2
X
En el límite r ~ O el potencial nuclear debe ser dominante, porlo cual,
V(r) para r -7 O
112

Esto nos provee una condición de contorno en el origen
<P(O) = 1
En este punto nos topamos con dos inconvenientes. Por unlado, con esta única condición de contorno, la ecuacióndiferencial anterior nos provee toda una familia de soluciones.Para decidir cuál de ellas tiene sentido físico precisamos otracondición de contorno. Por otra parte, aún no conocemos laenergía de Fermi E. Para resolver estos dos puntos advertimos quep(r) se anula cuando V(r) = E. Es evidente que p(r) debe sercero también para V(r) > E, dominio en donde la relación anteriorconduciría a una energía cinética máxima negativa. De esta forma,en el límite r = R del átomo debe ser V(r) = E. Por ejemplo, para
ión de número atómico •un Z, carga Z e y radio R tenemos que
E = V(R)i\= - 4rrc
o
• 2Z eR
En tanto que no hay singularidades en r = R el potencialdebe ser continuo allí y, por lo tanto,
dV! =drR
i\
4rrco
En términos de la función universal ~(x) estas dos ecuaciones nosindican que en el punto x = x donde ~(x ) = O debe sero o
•d~1 = _ Z /Zdx xx oo
Esta es la condición de contorno que nos faltaba para fijar a lafunción ~(x). Esta función ha sido tabulada para distintosvalores del cociente z·/Z. Vale 1 en x=O y decae monótonamente
•anulándose en cierto x=x cuando Z /Z * O. En el caso de un átomoo
neutro el mismo carece de fronteras y se extiende formalmentehasta el infinito.
113
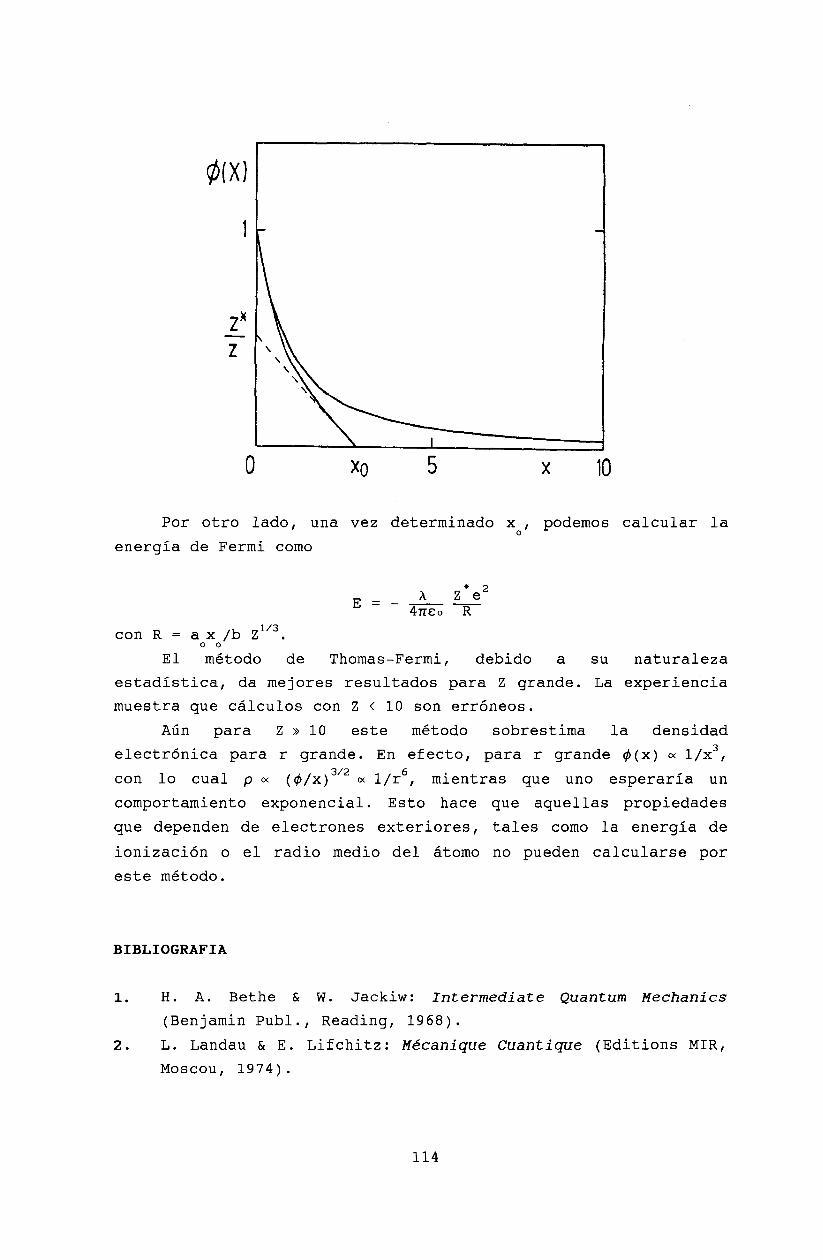
~(X)
z)l
z
o Xo 5 x 10
Por otro lado, una vez determinado x, podemos calcular lao
energía de Fermi como
A z·e2
E=-----4rrco R1/3con R = a x lb Z .o o
El método de Thomas-Fermi, debido a su naturalezaestadística, da mejores resultados para Z grande. La experienciamuestra que cálculos con Z < 10 son erróneos.
Aún para Z» 10 este método sobrestima la densidadelectrónica para r grande. En efecto, para r grande </>(x)O< 1/x3
,
con lo cual p O< (</>Ix) 3/2 O< 1/r6, mientras que uno esperaría un
comportamiento exponencial. Esto hace que aquellas propiedades
que dependen de electrones exteriores, tales como la energía de
ionización o el radio medio del átomo no pueden calcularse poreste método.
BIBLIOGRAFIA
1. H. A. Bethe & W. Jackiw: Intermediate Quantum Mechanics(Benjamin Publ., Reading, 1968).
2. L. Landau & E. Lifchitz: Mécanique Cuantique (Editions MIR,Moscou, 1974).
114

LA ESTRUCTURA DE LOS ATOMOS
ATOMOS MULTIELECTRONICOS
La discusión sobre átomos de dos electrones ilustra la clasede lógica que se debe aplicar al estudiar átomosmultielectrónicos . Como un punto de partida razonable vamos ausar nuevamente el modelo de electrones independientes. A cadaelectrón le vamos a asignar una órbita identificada por losnúmeros cuánticos ni. Ahora, cada estado ni puede acomodar varioselectrones con distintos valores de número cuántico magnético mgy proyección de espín m. Tales electrones se denominansequivalentes.
La especificación completa del estado del átomo en estemodelo de electrones independientes requiere indicar cuántoselectrones equivalentes hay en cada estado neo Esto constituye loque se llama una configuración. Si hay x electrones en el estadone, se indica con un supraíndice como n~. Así, la configuracióndel estado fundamental del Helio es 1s2, y la del primer estadoexcitado ls 2s.
CONFIGURACION ELECTRONICA
Ahora vamos a determinar la configuración electrónica de losátomos. Cada nivel ne puede acomodar hasta 2(2l+l) electrones,correspondientes a distintos valores de me = -e, -l+l, ..., e ym = ±lj2. Entonces, para cada átomo de número atómico Z, vamos asir ocupando cada nivel, empezando por los de menor energía, hastallenarlos. Cuando un nivel ha recibido el mayor número posible deelectrones que es permitido por el principio de exclusión,seguimos con el nivel siguiente. Y así continuamos hasta terminarcon los Z electrones del átomo neutro.
En general, dentro de nuestra aproximación de electronesindependientes, uno esperaría que los niveles ne sean degeneradosen el momento angular orbital e. Sin embargo ello no es así. Dehecho, estados con distinto valor de e no son degenerados. Los
115

estados con menor g , para un n dado, tienen menor energía. Estose debe a que las órbitas con menor g son más penetrantes y, porlo tanto, la interacción con el núcleo está menos apantallada porel resto de los electrones que para un e grande.
Esta misma diferencia entre órbitas penetrantes y extendidashace que, en varias oportunidades, un nivel ne dado tenga menorenergía que otro n'e' de menor número cuántico principal n' < n,siempre que el momento angular orbital del primero sea menor queel del segundo e < e'. Así, por ejemplo, el nivel 4s puede teneruna energía levemente menor que el 3d.
REGLAS DE MADELUNG
En la figura indicamos el ordensucesivos niveles ne. Puede haberciertos átomos particulares, pero enindica.
en que se van llenando lospequeñas diferencias para
general el orden es como se
i\úmero total deelectrones
Designación Electrones al completarsedel nivel en la capa cada capa
6d7P----6-,~. ------1l8(?)---!O 325í-======= J.!
í( "2--'
6p---.-5i ------86 (Rn)~_' i~~326s'~----2-.-1
5p----6' ------54 (Xc)4d---1O ~18 .5~/---·_;) i
4p---·-61 ------36 (Rr)30---10 :'-1845---·-2 ¡
.J
3p-----6--: ------18 (Ar)&3----2]-8
2p 6' 8 ------10 (Ne)213-----2J1:;--- 2 2 ------2 (He)
Estructura en capas de losniveles de energía atómicos_
En primer lugar vemos que se producen ciertas "brechas deenergía" entre los niveles np y (n+1)s. Aquellos niveles
116

agrupados entre dos brechas sucesivas constituyen una capa.Consecuentemente el conjunto de orbitales de cada nivel ni sellama subcapa.
En este punto debemos aclarar que los espectros copistas usanel término capa con otro sentido, como designando a todos losestados con igual número cuántico principal n. Así, los estadoscon n = 1, 2, 3, 4, constituyen diferentes capas que seespecifican con una letra mayúscula K, L, M, N, ...respectivamente.
Volviendo a nuestra nomenclatura, vemos que cada capa secompleta al llenarse una subcapa np. Y ello ocurre para númerosatómicos Z = 2, 10, 18, 36, 54 Y 86.
Madelung formuló las siguientes reglas empíricas sobre elllenado de niveles en los átomos neutros:
Los estados se llenan en orden de n+t creciente, y a partirdel t máximo compatible con ese valor de n.
Una capa queda cerrada cuando los orbitales p han sidoocupados completamente, excepto por la primer capa que se cierracon 1s2
•
INTERACCION ENTRE ELECTRONES. REGLAS DE HUND
Tal como vimos al estudiar el átomo de Helio, laconfiguración electrónica resultante de una aproximaclon deelectrones independientes no basta para especificar unívocamenteel estado de un átomo. De hecho, la interacción entre electronesproduce un desdoblamiento de cada uno de estos niveles, según losvalores del momento angular orbital L y de Espín S totales. Laseparación entre estos niveles caracterizados por valores de L y
S distintos suele ser del orden de 0.01 Ry, o sea entre uno y dosórdenes de magnitud menor que entre niveles correspondientes aconfiguraciones distintas.
Acerca de la distribución relativa de niveles de una mismaconfiguración, pero con valores de L y S distintos, existen lassiguientes reglas empíricas, denominadas reglas de Hund
117

1) La mínima energía la posee el término con el maXlmo valorposible de S para la configuración electrónica dada,
2) ... y el mayor valor de L posible para ese valor de S.
No es posible dar una demostración de las reglas de Hund.Ellas son ciertas experimentalmente y se verifican al realizarlos cálculos sobre átomos individuales. Sin embargo podemosentenderlas con algunos argumentos simples.
La primer regla dice que el término del multiplete con mayorvalor de S tendrá la menor energía. Ahora, tal como vimos alestudiar el átomo de Helio, cuando mayor es S, más simétrica esla parte de espín de la función de onda y más antisimétrica laparte espacial. Esto hace que los electrones se mantenganapartados entre sí, reduciendo la interacción entre electrones yaumentando la energía de ligadura. De la misma manera, y en unaanalogía clásica, cuando mayor es L más abiertas son las órbitasde los electrones y estos pueden separarse más, reduciendonuevamente la repulsión entre ellos.
Ahora, una descripción completa del estado del átomo nos vaa exigir, junto con la enumeración de los estados de todos loselectrones, la indicación de los valores del momento angular L yel espín S totales, en la notación usual 2S+1L. Así el símbolo1s2p 3p representa el estado de un átomo de Helio donde L=l, S=ly los dos electrones se encuentran en los estado 1s y 2p.
TERMINaS ESPECTRALES
Veamos cómo se pueden hallar los términos atómicos oespectral es posibles para una dada configuración electrónica. Sise da la situación de electrones no equivalentes la determinaciónde los valores de L y S posibles se efectúa directamenteaplicando la regla de composición de momentos. Así, por ejemplo,para la configuración (np, n'p) con n y n' distintos, el momentocinético orbital L puede tomar los valores 2,1,0 y el espín totalS los valores 1 ó O. Combinándolos entre sí obtenemos los
118

M =0 M =1/2.L ' s
tabla.
términos 3D, 3p, 3S, lD, lp Y lS. Sin embargo, en los casos máscorrientes, se tiene cierto número de capas llenas (que nocontribuyen a los L y S totales) y una capa incompleta con ciertonúmero de electrones equivalentes. Nos encontramos en tal casocon limitaciones impuestas por el principio de Pauli.Consideremos, por ejemplo, una configuración de 3 electrones pequivalentes. Siendo l=l, m
lpuede tomar los valores 1, O ó -1,
de forma tal que son posibles seis estados electrónicos, con lossiguientes pares de índices mi y ms:
(1, 1/2), (O, 1/2), (-1, 1/2)(1,-1/2), (0.-1/2), (-1,-1/2)
Los tres electrones se pueden distribuir en trescualesquiera de estos seis posibles estados ...
m m M Ms 1 S L
+ + + 1 O -1 3/2 O
+ + - 1 O 1 1/2 2
O 1
-1 O
-1 1 1
O O
O -1 1 O
Podemos dejar de escribir los estados con valores negativos de ML
Y M porque sólo dan una información redundante.sLa existencia de un estado con M =2 Y M =1/2 demuestra que
L sdebe tenerse un término L=2, 8=1/2 o sea un término 2D, a estetérmino deben corresponder también los estados M =1, M =1/2 YL S
Retiramos estos tres estados ya analizados de la
119

Ms ML
3/2 O
1/2 1
O
O
Queda aún un estado con M ~1, M ~1/2 de forma que debe tenerse unL stérmino L~l 8=1/2, o sea un término 2p, al cual correspondeademás un estado M =0, M ~1/2. Retirando también estos estados
L squeda
Ms ML
3/2 o
1/2 O
Ambos estados corresponden a un término L=O S=3/2, o sea 4S. Así,para la configuración de tres electrones p equivalentes esposible tan sólo un término da cada uno de los tipos 2D, 2p Y 4S.El término de espín máximo es único e igual a 4S, constituyendoel término fundamental. Este término fundamental se puede hallardirectamente de la siguiente manera. Por ejemplo, si analizamosuna capa incompleta con cuatro electrones tipo d, el máximo espínposible es S=2. Atribuimos luego a los electrones los máximosvalores posibles de m (-2~m ~2) de forma de obtener el mayor
1 1
valor de M =¿ m, o sea m = 2, 1, O Y -1. Luego M = 2. EstoL 1 1 L
significa que también es igual a 2 el mayor valor de L posiblepara S = 2. El término fundamental es 5D.
Es interesante destacar la coincidencia del carácter de lostérminos espectrales que corresponden a dos configuraciones, cadauna de la cuales posee tantos electrones como faltan en la otrapara llenar una capa. Por ejemplo, para cuatro electrones pequivalentes obtenemos los mismos términos posibles que para doselectrones p equivalentes: lS,lD,3p. Esto se debe a que la falta
120

de un electrón en una capa se puede considerar como un hueco cuyoestado se determina por los mismos números cuánticos quecaracterizan al estado del electrón que falta.
ESTRUCTURA FINA DE LOS NIVELES ATOMICOS
Los términos relativistas en el hamiltoniano de un átomo sedistribuyen en dos categorías: unos son lineales respecto de losoperadores de espín de los electrones, los otros dependencuadráticamente de dichos operadores. Los primeros se deben a lainteracción del movimiento orbi tal de los electrones con susespines. Es la llamada interacción espín-órbita. Los segundos,en cambio, corresponden a la interacción entre los espines de loselectrones (interacción espín-espín). Ambas formas de interacciónson de segundo orden respecto de v/c (el cociente de la velocidadelectrónica respecto de la velocidad de la luz). Sin embargo, enlos átomos pesados, la interacción espín-órbita esconsiderablemente mayor que la interacción espín-espín. Ello sedebe a que la interacción espín-órbita crece rápidamente alaumentar el número atómico, mientras que la interacciónespín-espín es independiente de Z. La interacción espín-órbitaH no es diagonal en los estados IL,M ,S,M > pues no conmuta con
so L S
L Y S. En cambio sabemos que debe conmutar con el impulsoz zangular total j. Entonces los estados IL, S,J, M > son
J
aproximaciones en orden cero de las autofunciones del átomo conacoplamiento espín-órbita incluído, y la corrección al primerorden de la energía está dada por elementos diagonales de laforma
!lE == <L,S,J,M IR IL,S,J,M> == A [J(J+l)-L(L+l)-S(S+l)]W J W J
donde A es positivo si la capa incompleta esta llena como máximohasta la mitad, y negativo en caso contrario.
Vemos así que cada nivel con valores dados de L y S Y dedegeneración (2L+1)(2S+1) se desdobla, por acción de los efectosrelativistas, en una serie de diferentes niveles próximos entresí que difieren en los valores del momento cinético total J.
121

Este desdoblamiento se llama estructura fina. J toma todos losvalores entre 1+8 y 11-81, por ello un nivel con valores dados de1 y 8 se desdobla en 28+1 (si 1>8) o 21+1 (si 1<8) nivelesdiferentes. Cada uno de estos niveles es degenerado respecto dela dirección del vector J. Debemos completar la simbología delos términos espectrales de los átomos agregando el valor delmomento cinético total J, como un número a la derecha y en laparte inferior. Así, el símbolo 2p representa a un nivel con3/21=1, 8=1/2 Y J=3/2.
Ahora introducimos una regla de Hund adicional que determinael valor de J en el estado fundamental del átomo con una capaincompleta: 11 8i en esta última se encuentra no más de la mitaddel número máximo de electrones posibles en ella será J= IL-S I •Si, en cambio, la capa está ocupada en más de la mitad se tieneJ=L+S" .
122
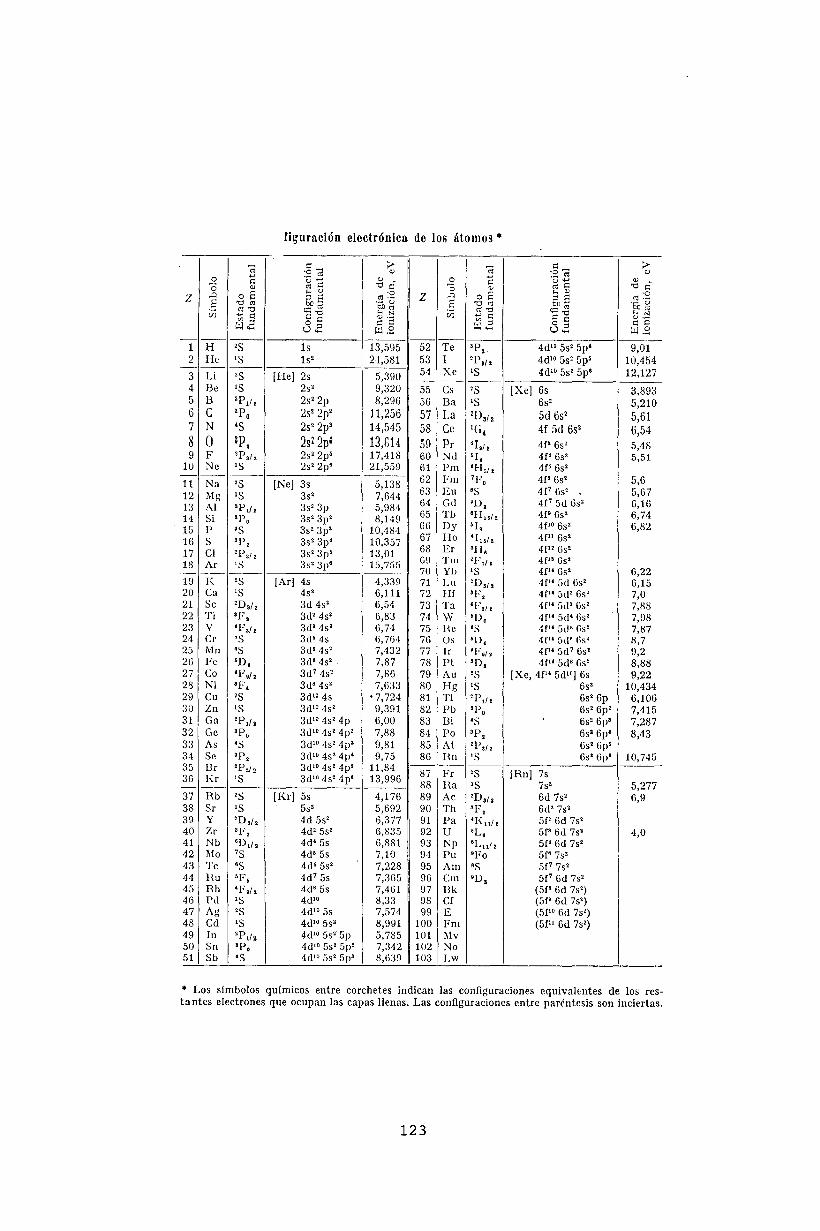
figuración electrónica de los átomos·
(;1 .§ ó1 ;> (;1 :§ (;1;>-
'" '"o .•.. ·u +..l '" o e u .•.• '"¡::::'O '" Ol ¡:::: 'O ¿ 'O '" Ol ¡:::: 'O ¿.Q o S •... '" '0 Z .Q o S •... '" '0z É?ú s Ol .- ª,S Ol .-.5 'OOl .- u S 'O '"
._ u
,"'O c.::: Ol tú '" ,"'O c.:::'" tú",VJ .•.• ¡:::: ¡::::'O •..•N éii .•.• ¡:::: ¡::::'O •..•N'" .- "'.-en ;:l O ¡:::: ¡:::: ¡:::: w2 O ¡:::: ¡:::: ¡::::w ..... u2 r.:¡ .2 u2 W.8
- -- --1 H 'S 1s 13,595 52 Te 'P, 4d'0 5s' 5p' 9,012 He 'S 1s' 24,581 53 1 'p./, 4d'0 5s' 5p' 10,454- -- -- 54 Xe 'S 4d1O 5s' 5p' 12,1273 Li 'S [He] 2s 5,390 -- --4 Be 'S 25' 9,320 55 Cs 'S [Xc] 6s 3,8935 B 'p,/, 25' 2p 8,296 56 Ba 'S 65' 5,2106 C apo 2s' 2p' 11,256 57 La 'Da/, 5d 652 5,617 N 'S 2s' 2pa 14,545 58 Ce 'G, 4f 5d 6s' 6,548 O iP, 2s22pl 13,614 M Pr '1,/, 4f' 65' 5,489 F 'p./, 25' 2p' 17,418 60 Nd '1, 4f' 65' 5,51
10 Ne 'S 25' 2p' 21,559 61 Pm 'H,/, 4f' 65'- -- G2 Fm 71'0 4f' 65' 5,611 Na 'S [Ne] 35 5,138
63 Eu 'S 4[1 65' 5,G712 Mg 'S 35' 7,64464 Gd 'D, 4f7 5d 65' 6,1613 Al 'p,/. 35' 3p 5,984
14 Si 'Po 35' 3p' 8,149 65 Tb 'H"t, 4f' 65' 6,7415 p 'S 35' 3p' 10,484 66 Dy '1, 4f1° 65' 6,8216 S 'P, 35' 3p' 10,357 67 Ho '1,,/, 4f11 65'
17 CI 'P,t, 35' 3p' 13,01 68 Er 'Il, 4f" 65'18 Ar lS 3s' 3p' 15,755 Gil Tlll 'F,/. 4f" Gs''- -- 70 Yb IS 4f" 65' 6,2219 1<. 'S [Ar] 45 4,339 71 Lll 'D,t. 4f" 5d 6s' 6,1520 Ca 'S 4s' 6,111 72 lIf '1", 4f" 5d' 6s' 7,021 Sc 'D,/, 3d 45' 6,54 73 Ta 'F,{, <lf" 5d' 6s' 7,8822 Ti '1', 3d' 45' 6,83 74 \V 'Do 4f" 5d' ¡¡s' 7,9823 V 'F,/, 3d' 4s' 6,74 75 He 'S 4f" 5<1' ¡'s' 7,8724 Cr 'S 3d' 4s G,7IH 7G Os 'D, 4f" [,d' Gs' 8,725 Mn oS 3d' 4s' 7,432 77 Ir "'Fg/2: 4[1' 5d 7 6s' 9,22G Fe 'D, 3d' 45' 7,87 78 pt 'D, 4ft< 5d' Gs' 8,8827 Co 'F,/, 3d74s' 7,86 79 Au 'S [Xc, 4[1' 5d1O] 6s 9,2228 Ni 'F, 3d' 4s' 7,633 80 Hg 'S 65' 10,43429 Cl! 'S 3d1O 4s '7,724 81 TI 'p,/, 65' 6p 6,10630 Zn 'S 3d1O 4s' 9,391 82 Pb 'Po 6s' 6p' 7,41531 Ga '1\/, 3d'o 4s' 4p 6,00 83 I3i 'S 6s' 6p' 7,28732 Ge 'Po 3d1O 45' 4p' 7,88 8·1 Po 'P, 6s' 6p' 8,4333 As 'S 3d1O 4s' 4p' 9,81 85 Al '1>,/, 65' Gp'34 Se 'lO, 3d'o 45' 4p' 9,75 86 Hn IS Gs' Gp' 10,74535 I3r 'p,/, 3d'° 45' 4p' 11,84 -- ---
87 Fr 'S [Hn] 7536 I<.r 'S 3d10 45' 4p' 13,99688 Ha 'S 75' 5,277- --
37 Rb 'S [Kr] 55 4,176 89 Ac 'Da/, 6d 75' 6,938 Sr 's ,
55' 5,692 90 1'11 'F, 6d' 7s'39 y 'D,/, 4d 55' 6,377 91 Pa '1<.,,/, 5f' 6d 75'40 Zr 3F2 4d' 5s' 6,835 92 U 'L, 5í' 6d 75' 4,041 Nb 'D,/, 4d' 55 6,881 93 Np 'L,,/, 5r' 6d 75'42 Mo 7S 4d' 55 7,10 94 Pu 'l"o 5r' 75'43 Tc 'S 4d' 5s' 7,228 95 Am 'S 5[1 75'44 Bu '1', 4d75s 7,3G5 9G Cm 'D, 5r7 6d 75'45 Hh '1',/, 4d' 55 7,461 97 I3k (5f' 6d 75')4G Pd IS 4d10 8,33 98 Cf (5f' Gd 75')47 Ag 'S 4d1O 55 7,574 99 E (5pO 6d 75')48 Cd 'S 4d'O 5s' 8,991 100 Fm (5f11 6d 75')49 In 'P1/2 4d10 55' 5p 5,785 101 J\lv50 Sn 'Po 4dlO 55' 5p' 7,:1<12 102 No51 Sb 'S 4d10 55' 5p' 8,G39 103 Lw
• Los símbolos químicos entre corchetes indican las configuraciones equivalentes de los res-tantes electrones que ocupan las capas llenas. Las configuraciones entre paréntesis son inciertas.
123
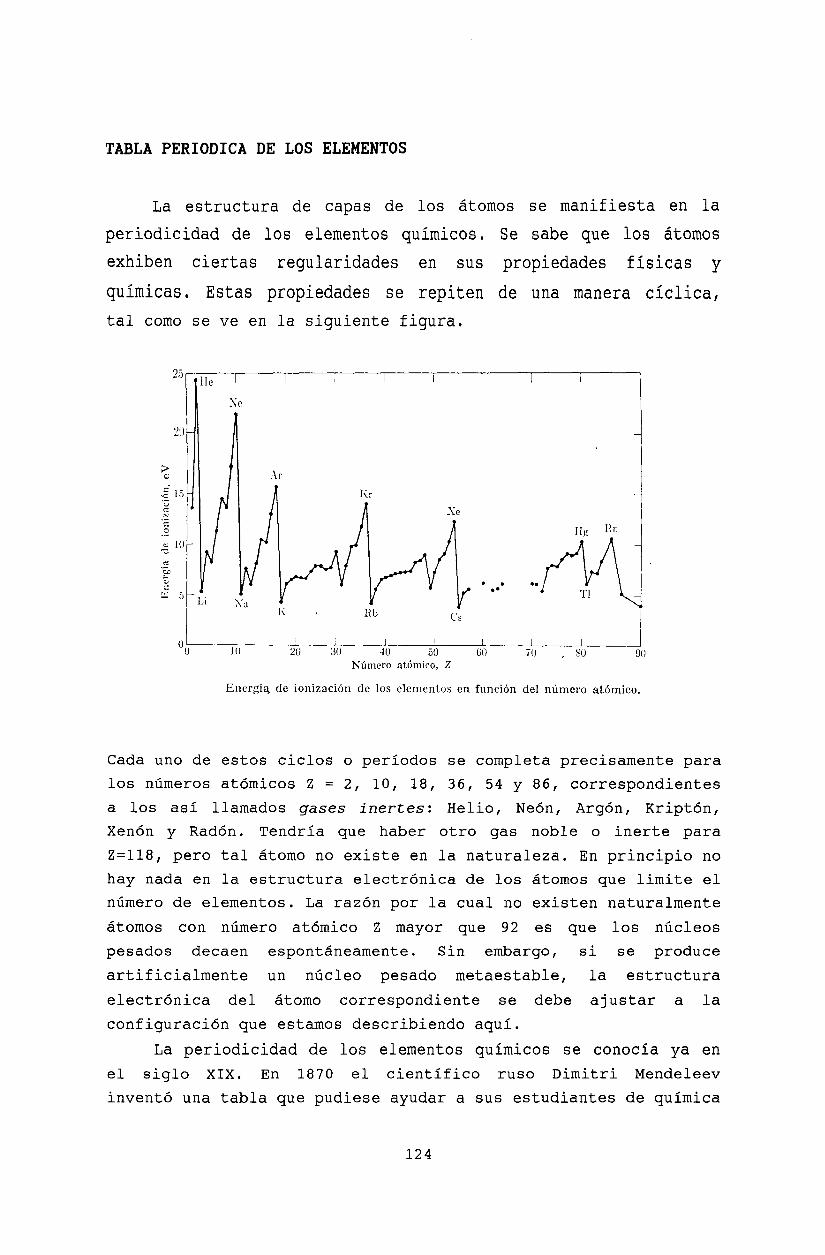
TABLA PERIODICA DE LOS ELEMENTOS
La estructura de capas de los átomos se manifiesta en laperiodicidad de los elementos químicos. Se sabe que los átomosexhiben ciertas regularidades en sus propiedades físicas y
químicas. Estas propiedades se repiten de una manera cíclica,tal como se ve en la siguiente figura.
DO
1I¡; !ln
.. ...'es!lb
I
;)0 40 50Númcro atómico, Z
20
2;'jHe
;\e
211
;.>ArQ
r:;' 15-o'ü""(3
~ lO
"'EC•...'",.. 5
K
OIIIO
Energía, de ionÍzaCÍón de los elementos en funCÍón del número atómÍco.
Cada uno de estos ciclos o períodos se completa precisamente paralos números atómicos Z = 2, 10, 18, 36, 54 Y 86, correspondientesa los así llamados gases inertes: Helio, Neón, Argón, Kriptón,Xenón y Radón. Tendría que haber otro gas noble o inerte paraZ=118, pero tal átomo no existe en la naturaleza. En principio nohay nada en la estructura electrónica de los átomos que limite elnúmero de elementos. La razón por la cual no existen naturalmenteátomos con número atómico Z mayor que 92 es que los núcleospesados decaen espontáneamente. Sin embargo, si se produceartificialmente un núcleo pesado metaestable, la estructuraelectrónica del átomo correspondiente se debe ajustar a laconfiguración que estamos describiendo aquí.
La periodicidad de los elementos químicos se conocía ya enel siglo XIX. En 1870 el científico ruso Dimitri Mendeleevinventó una tabla que pudiese ayudar a sus estudiantes de química
124

inorgánica a recordar las propiedades de los 63 elementosconocidos en aquel entonces.
o n bl T'h e H e T E M bl 3 JI E hf E H T o B'b •
OCH08AHHO~ HA HXb ATOl1HO,li1> OtCt H Xl\MH~ECKOII1> CXOj¡CTn.
TI;;;:; 50V =51ern 52
M n == 55tc=56
Nj:=Co""S9H •• I Cu==63,f
Be= 9,4 Mg"" 24 Zn = 65.1B=II AI==27,. ?=68C==12 51=28 ?=70N=14 P=31 As=7:)0=16 5=32 Se=79 .•F= 19 CI==35,lBr=80
Li=7 Na=23 K==39 flb=85 .•Ca".40 Sr=87.e
1""45 Ce=92?Er •• 56 La~94?YI=60 DI=9:)?In •• 75.6 Th -118?
Zr= 90 ?=180.Nb ••• 94 T8.= 182.Mo= 96 W = 186.Rh=-I04,4 Pl= 197, ••Ru= 104 •• Ir= 198PJ=IOG.o 05==199.
Ag: 108 Hg:..::200
Có= 112Ur=116 Au:c197?Sn = 118Sb= 122 BI=210?Te= 128?
1=127es= 133 TI =204l3a= 137 Pb=207.
Tabla periódica de los elementos de Mendeleev
El descubrió que esas propiedades se repetían periódicamentecorno función del peso atómico. Así pudo agrupar los elementos enfamilias, construyendo un esquema que, desde entonces, se conocecon el nombre de tabla periódica. Esta tabla, tal corno aparece ensu publicación "Sistema Experimental de los elementos", difierede la tabla moderna en el desconocimiento de algunos pesosatómicos (Por ejemplo, los elementos Osmio (Os), Iridio (Ir) y
Platino (Pt) aparecen en orden invertido), y en la ausencia dealgunos elementos, corno por ejemplo todos los gases nobles. Sinembargo las similitudes con la tabla moderna son notables.Aparecen bien ordenados todos los alkali, con excepción delFrancio (Z=87) que no se conocía en aquella época. Los dosperíodos cortos, y la porción derecha de la tabla moderna tambiénestán correctas; lo mismo que los metales de transición, conexcepción del Rutenio (Ru) y el Rodio (Rh) que aparecen malordenados y mal apareados por ausencia del Tecnesio (Tc) que noexiste naturalmente.
Debido a las regularidades que se observaban, Mendeleev sedió cuenta de que la lista de elementos conocidos estaba
125

incompleta. Por lo tanto dejó huecos en la tabla correspondientesa elementos por descubrir.
En la tabla periódica moderna vemos que el primer grupo ocapa se llena con el Hidrógeno y el Helio. La ocupación del 2do.y 3er. grupo corresponden a los dos primeros períodos, llamadoscortos, que contienen ocho elementos cada uno. Siguen luego dosperíodos largos de 18 elementos y uno de 32 que contiene lastierras raras. El séptimo grupo no llega a ocuparse por completocon los elementos que existen en la naturaleza, y ni aún con lostransuránidos artificiales.
2p2,
3,
4,
5,
6,
7,
r-¡--H
3 4l. Be
11 12Na Mg
19 20K ea
37 38Rb Sr
55 5óCs 5a
87 88fr Ra
3d
4d
5d
6d
21 22 23 24 25 26127CQ
28 29 30Se TI V Cr Mn fe NI Cu In
4,13d·' 4,13dlO
39 40 41 42 43 44 145 46 ' 47 48Y Ir Nb 1.10 Te Ru Rh Pd Ag Cd
5,14d' 5s14di . 5;14d8 5s04dlO 5,14d1O
57 72 73 74 75 76
1
77 78 179 80La Hf Ta W Re Os Ir Pt Au Hgla.1t.'la-
r.!~ 6s15d9¡6.!lSdlO
89 -, 1AeAc:J~ldes
3p
4p
5p
6p
7p
~He
5 6 7 8 9 10S e N O f No
13 14 15 16 17 18Al SI P S o A
31 32 33 34 35 36Ga Ge As Se Sr Kr
49 50 51 52 53 54In Sn Sb Te I XO
81 82 83 84 85 86Ti Pb Si Po Al Rn
5rActinidos
FIGURA
f6 f7
tiP
Tabla periódica de los elementos mostrando la configuración electrónica para cada elemento.
ELECTRONES DE VALENCIA
En los capítulos anteriores estudiamos detalladamente a los
126

de la tabla periódica: el Hidrógeno y el2 2 1fundamentales son 1s 8 y 1s 8,
1/2 o
dos primeros elementosLitio, cuyos estadosrespectivamente.
El principio de exclusión impide que el siguiente elemento,el Litio, tenga una configuración (ls)3. Así que la configuraciónaccesible de menor energía es la 1s22s. Puesto que estamosagregando un solo electrón a una capa cerrada 18 , la descripciónoespectroscópica es la misma que para el Hidrógeno: 28
1/2Los electrones que ocupan la capa cerrada constituyen el
carozo o kernel del átomo, el electrón restante se denominaelectron de valencia. La energía de ligadura de los electronesdel carozo es mucho mayor que la de los electrones de valencia, ycrece muy rápidamente con el número atómico. Esto hace que loselectrones del carozo estén fuertemente ligados y no seanperturbados en la mayoría de los procesos en los que participe elátomo. Luego el o los electrones de valencia son los responsablesde las propiedades qUlmlcas de los átomos, participando dereacciones y ligaduras. Esto hace que el Litio tenga propiedadesquímicas similares a las del hidrógeno, explicando su lugar en latabla.
Si el apantallamiento de los electrones del carozo fueraperfecto, el electrón de valencia vería una carga Z=l y siendon=2, tendríamos una energía de ionización de 1/4 Ry. Sin embargo,como el orbital 2s es penetrante, el apantallamiento no esperfecto, y la energía de ionización es algo mayor
•0.4 Ry = 5.4 eV, correspondiente a una carga efectiva Z = 1.3.Sin embargo, sus estados excitados 1s2nl consisten en orbi talesde valencia menos penetrantes, y los correspondientes niveles deenergía se asemej an a los del átomo de hidrógeno, tal corno semuestra en la siguiente figura
127

Sodio
2s
Hidrógeno LitioI I
Or- iW&--i~~~~+-~~WJi~6 I 6s 6p 6d 6f I 6d 6f---¡-----r------- ----r-----r- - -~ L5S 5p 5d 5f 1_ 6s 6p 5d 5f
-1 - 4 : _ 4P id if I _ 5p 4cf 4f4s I 5s 4])
11_---t--------t----- ----3 I 3P 3d 1 3d
-2 - 1 I1- II 3s I 4sI II II II II I 3p
- --t--------t------ ----2 -I 2p I
I IIIIIIIII 3sIIIIII
-4-
-5'-
-6-
del hidrógeno, el ¡¡tio y el sodio.
Niveles de energía
BERILIO (Z=4)
El cuarto electrón ocupa el lugar vacante en el orbital 2s,completando una subcapa con la configuración 1s22s2
• Ladescripción espectroscópica es similar a la del Helio, con elmismo término espectral 1S . Energéticamente la situación es muy
oparecida a la del Helio. Si el apantallamiento de los electrones1s del carozo fuese perfecto, la única diferencia con un átomo deHelio sería que los dos electrones restantes están en estados conn=2. Así, uno esperaría tener una energía de ionización del ordende un cuarto de la del Helio: 1.808 Ry/4 = 0.45 Ry. Sin embargo,como el apantallamiento no es perfecto, el valor experimental esalgo mayor 0.68 Ry = 9.3 eVo
En el Helio, se necesita una energía muy alta para excitarun electron: 1.47 Ry = 2O eV. Esta alta energía de excitación,junto con el hecho de que los electrones forman una capa cerrada,es lo que hace del Helio un elemento químicamente inerte. En elBerilio, en cambio, y aunque se ha llenado una subcapa, serequiere poca energía para excitar un electrón del orbital 2s al2p. Por ello, en presencia de otro átomo se puede producirfácilmente una reordenación de la nube electrónica que rompa lasubcapa. Así el berilio no es tan inerte como el helio.
128

BORO (2=5)
Según las reglas de Madelung el quinto electrón ocupa elorbi tal 2p. La configuración es, entonces, 1s22 S22p. Al aplicarlas reglas de Hund para determinar la descripciónespectroscópica, hay que considerar únicamente la subcapaincompleta, ya que los momentos cinéticos y de espín de loselectrones en las subcapas llenas son nulos. Aquí tenemosentonces S=1/2 y L=l. Como la subcapa está menos que semillena,el momento cinético total J es el mlnlmo posible, o sea
2J = IL-SI = 1/2. El estado fundamental es, entonces, P .1/2
La energía de ionización debe ser menor que para el Berilio,ya que el orbital 2p está menos ligado que el 2s. El valorexperimental es 0.61 Ry = 8.3 eVo
CARBONO (Z=6)
La configuración electrónica es 1s22s22p2. El espín totalpuede ser S = O o 1, y el momento angular L = O, 1 o 2. Corno lafunción de onda debe ser antisimétrica para los electrones, unestado singlete (S=O) debe corresponderse con un L par y unotriplete (S=l) con un L impar. Luego las únicas posibilidadespara el término fundamental son lS, iD o 3p Ahora
o 2 0,1,2
invocamos las reglas de Hund, tornando el término con mayor S,3mayor L y menor J. O sea P.
oEl segundo electrón 2p puede mantenerse lejos del primero,
ubicándose en un orbital con m distinto. Así, la repulsión entreelectrones es menos importante que la atracción nuclear, queahora tiene carga mayor que para el boro. Por todo esto es deesperar una mayor ligadura, y la energía de ionización aumenta a0.83 Ry = 11.3 eVo
NITROGENO (Z=7)
La configuración electrónica es ls22s22p3. Debido a lasreglas de Hund el espín total del estado fundamental es el máximoposible, o sea S=3/2. Si no tenemos en cuenta la interacción
129

espín-órbita, este estado fundamental, caracterizado por losmomentos angulares L y de espín 8, es degenerado en M
Ly Ms'
Luego el nivel fundamental incluye un estado con M=3/2,sdegenerado con el resto. Las tres proyecciones del espín apuntanen la misma dirección, y por lo tanto los electrones debenencontrarse en orbitales 3p con valores distintos de m. O sea
m
que M solo puede valer M =0, SI quiero ser consistente con elL L
principio de exclusión. Luego, el estado fundamental tiene L=O.Por último el momento cinético total solo puede valer J=3/2.Resulta entonces que el nivel fundamental es 48
3/2Nuevamente esperamos que la energía de ionización sea algo
mayor que para el elemento anterior, ya que Z aumenta en 1 y eltercer electrón puede ubicarse en un orbital 3p que no sesuperpone significativamente con los otros dos. El valorexperimental es 1.07 Ry = 14.5 eVo
OXIGENO (Z=8)
más queLuego el
Hund, como la subcapa estáel mayor valor posible de J.
f . . ~ 1 22 22 4 C t 1 t 1La con 19uraclon es s s p. on cua ro e ec rones en asubcapa incompleta, uno esperaría que la determinación del estadofundamental fuese muy difícil, pero no es así. Sabemos que cuandola subcapa está llena el estado tiene L=O, S=O. Luego podemospensar al Oxígeno como formado por una subcapa 2p llena con dos"agujeros" en ella. Los posibles estados consistentes con laantisimetrización de los dos agujeros serán los del Carbono, osea lS, lD Y 3p La única diferencia está en que, al
o 2 0,1,2aplicar las reglas desemillena, debemos tomart ~ . 1 3ermlno espectra es P.
2
Cuando agregamos el cuarto electrón 2p, debemos ubicarlo enun orbital con un valor de m ya ocupado. Así que el solapamientoentre electrones es grande, aumentando la correspondienterepulsión electrostática. La energía de ionización cae entonces a1 Ry = 13.6 eVo
130

FLUOR (Z=9)
L f . .~ 1 22 22 5 ~l' 1a con l.guracl.ones s s p. Tenemos un so o agujero en eorbital 2p. Como en el boro el estado fundamental tiene 5=1/2 y1=1, pero ahora debemos tomar el mayor de los dos posibles
2valores de J = 1/2 o 3/2. Luego resulta P3/2
NEON (Z=10)
La subcapa 2p queda cerrada, y con ella se cierra tambien lacapa L. La energía de ionización toma un valor máximo de 1.59 Ry= 21.6 eVo El estado fundamental es 1S . Nuevamente se requiere
omucha energía para excitar a este átomo, y por ello compartejunto con el helio las propiedades de un gas inerte, cerrando unperíodo de la tabla de elementos.
SEGUNDO PERIODO CORTO
El siguiente período también tiene ocho elementos. Primerose llena la subcapa 3s con Sodio (Z=11) y Magnesio (Z=12), yluego la 3p que incluye al Aluminio (Z=13), Silicio (Z=14),Fósforo (Z=15), Azufre (Z=16), Cloro (Z=17) y Argón (Z=18). Estoselementos son químicamente muy parecidos a los del períodoanterior Litio-Neón, y la descripción espectroscópica de losestados fundamentales es la misma. La única diferencia está enque al llenar subcapas menos ligadas, las energías de ionizaciónson menores que para el período anterior.
Los elementos de la primer columna, con excepción delHidrógeno, se llaman metales alcalinos. Nunca aparecennaturalmente en estado elemental, pues reaccionan rápida ycompletamente con virtualmente todos los metales alcalino-terreos
de la segunda columna.Así como llama la atención la similitud entre los miembros
de la misma familia en los dos grupos anteriores, los dossiguientes muestran algunas propiedades en abierto contraste. Porejemplo el Boro es semimetálico, mientras que el Aluminio es un
131

metal.
Las siguientes tres columnas incluyen la mayoría de 106
elementos no metálicos, entre ellos los halógenos de la séptimacolumna. Los gases noble ocupan la octava columna.
PRIMER PERIODO LARGO
En el comienzo del siguiente período se llena la subcapa 4scon el Potasio (Z=19) y el Calcio (Z=20), repitiendo laspropiedades químicas de los dos primeros elementos de losperíodos anteriores.
A continuación se llena la capa 3d, ocupando en ello diezelementos, desde el Escandio (Z=21) hasta el Zinc (Z=30). Apartir de allí se comienza a ocupar la subcapa 4p con elementoscuyas propiedades son similares a las de aquellos con subcapas 2py 3p semillenas en los dos períodos cortos anteriores.
METALES DE TRANSICION y TIERRAS RARAS
La serie de elementos en que la subcapa 3d se va llenandomerece un comentario especial. El nivel 4s está debajo del 3d,pero no mucho. Así se desarrolla una competencia entre ambos, yal pasar del Vanadio al Cromo es energéticamente favorable sacarun electrón del nivel 4s para llenar la subcapa 3d hasta lamitad. Algo similar ocurre al pasar del Niquel al Cobre, donde unelectrón se saca del nivel 4s para terminar de llenar el 3d.Todos estos elementos, denominados metales de transición tienenpropiedades químicas similares. Esto se debe a que las órbitas 3dtienen radios menores que las 4s, y por lo tanto, estas últimastienden a apantallar las primeras de influencias externas,independientemente de cuantos electrones estén ocupando esasórbitas 3d. Esto se manifiesta en una energía de ionización quevaría de manera relativamente lenta con respecto a Z.
Una competencia similar ocurre entre los niveles 4d y Ss delperíodo largo siguiente, y entre los niveles 5d y 6s de la serieque comienza después de las tierras raras. Todos los elementos en
132

estas series de transición comparten propiedades químicamentesimilares.
Los lantánidos o tierras raras en el sexto período y losactínidos en el séptimo también muestran una competencia entreniveles casi degenerados. Estos son el 4f y el 5d en la primerserie y el 5f con el 6d en la segunda. En el Gadolinio (Z=64) y
en el Curio (Z=96), es energéticamente más favorable ocupar elnivel d manteniendo la subcapa f semillena.
Podemos agrupar la tabla periódica de los elementos enbloques, dependiendo de que orbitales s, p, d o f son llenados encada uno de ellos. Los bloques s y p se suelen denominar deelementos representativos, el bloque d de elementos de transicióny el f, que se suele representar separado por convención, sellama de elementos de transición internos.
Aunque la tabla fue diseñada para enfatizar las relacionesentre elementos ubicados verticalmente, hay ciertas propiedadesque muestran regularidades en la dirección horizontal. Esto esparticularmente cierto, tal corno vimos, en los elementos detransición.
ACOPLAMIENTO J-J
Para Z pequeño es una muy buena aproximación tratar lainteracción espín-órbita corno una pequeña perturbación a losmultipletes L-8. Esta descripción del átomo se llama acoplamientoL-8 o de Russell-8aunders.
Por otro lado, para números atómicos muy grandes, lainteracción espín-órbita ya no se puede considerar corno unaperturbación y debe tenerse en cuenta desde el principio. Estosignifica que ya no es posible caracterizar a cada orbitalelectrónico con números cuánticos n, 1, m y m, sino que se debe
1 s
usar una descripción acoplada n,l,j,m. Después se construyen losmultipletes acoplando los correspondientes autoestados. Esteprocedimento se llama acoplamiento j-j.
133

TRANSICIONES DIPOLARES ELECTRICAS
Hemos dedicado un gran esfuerzo para calcular los posiblestérminos espectrales para una dada configuración y sin embargotodavía no los hemos usado. La utilidad de calcular los términos
t 1 25+1 d' t" t' despec ra es LJ ra lca en que es os numeros cuan ICOS son egran interés en el estudio de la emisión de radiación por átomosmultielectrónicos.
El gran número de niveles de energía de los átomos de muchoselectrones muestra claramente que el espectro de emisión debe sermuy complicado. Sin embargo sabemos que las transiciones posiblesestán severamente limitadas por ciertas reglas de selección: Enprimer lugar tenemos la regla de Laporte que nos indica que estánprohibidas transiciones entre estados de igual paridad. Cuando esposible hacer una descripción en términos de orbitales, laparidad de un estado está dada por la suma de los momentosangulares L e.. Entonces la regla anterior nos conduce a la
1
condición
!J. Le. = ±1.1
Debemos agregar las reglas de selección para el momentocinético total
!J.J = O, ±1 J=O ~ J'=O prohibido
Estas tres reglas son válidas independientemente del tipo deacoplamiento que se aplique (L-S o J-J).
Si es posible aplicar un acoplamiento L-S tenemos dos reglasde selección adicionales. Por un lado, como r conmuta con S, nopueden ocurrir transiciones entre niveles de diferentemultiplicidad
!J.S = O
Además sigue valiendo la regla de selección para el momentocinético orbital
134

f1L = O, ±l L=O ~ L'=O prohibido
Es importante destacar que ahora están permitidastransiciones con llL = O. La correspondiente prohibición en elcaso de un átomo monoelectrónico era una consecuencia de la reglade Laporte ya que, en dicho caso, llL = M = O significaría uncambio de paridad. En el caso multielectrónico, en cambio, L notiene una relación directa con la paridad, como la tiene la sumade los momentos angulares t. de los orbitales individuales.
1
llL = O es compatible con un cambio de paridad.
BIBLIOGRAFIA
1.
2.
M. Alonso & E. J. Finn:cuánticos y estadísticos
Barcelona, 1971).B. H. Bransden & c. J.
Física~ volumen III: Fundamentos(Fondo Educativo Interamericano,
Joachain: Physics of Atoms and
Molecules (Longman, London, 1983).
135


MOLECULAS OlATOMICAS
INTRODUCCION
Básicamente una molécula es un grupo de núcleos rodeados porelectrones de tal manera que el conjunto es estable. Esta es unaextensión natural del concepto de átomo, que es un núcleo rodeadopor electrones. En esta concepción los átomos que forman unamolécula pierden su individualidad, pero no es enteramente así.Cuando varios átomos se combinan para formar una molécula, loselectrones del carozo, más fuertemente ligados, prácticamente noson perturbados, manteniéndose unidos a sus núcleos originales.Sólo los electrones de valencia en las subcapas incompletas sonafectados, y se mueven bajo el efecto resultante de lainteracción con todos los núcleos y carozos. Estos electrones devalencia son los responsables de las ligaduras químicas y de lamayoría de las propiedades químicas de las moléculas.
MOLECULA ION DE HIDROGENO
H+, con dos protones y un electrón, es la molécula más2
simple. Debido a que las masas de los protones son mucho mayoresque la del electrón, las velocidades del movimiento nuclear sonpequeñas respecto de la velocidad del electrón, por lo cual, ycon muy buena aproximación, podemos estudiar el movimientoelectrónico suponiendo que los núcleos permanecen en reposo. Laenergía potencial del problema es, entonces,
i\ e2V=--- 4m: r
o 1
2 2_i\_ ~ + _i\_ ~4rre r 4rre R
o 2 o
donde la distancia R entre los protones es, en principio, unparámetro del problema.
Si R fuese muy grande, la situación de mínima energíaestaría dada por el electrón orbitando alrededor de uno de losprotones en un estado 1s del átomo de Hidrógeno, y el otro protón
137

suficientemente alejado como para no alterar de manera apreciablea ese estado. Si esta configuración fuese la resultante de haberdesintegrado la molécula, el electrón tendría que tener igualprobabilidad de quedar ligado a uno u otro de los protones. O seaque una descripción adecuada de esa situación estaría dada poruna combinación lineal, ya sea simétrica o antisimétrica, deestados 1s ligados a ambos centros.
Ahora supondremos que esta función de onda constituye una+buena aproximación del estado molecular del ión H. Esto puede2
considerarse como un primer orden de aproximación para el orbitalmolecular, así como la aproximación de electrones independienteslo era para la descripción de átomos multielectrónicos. Estateoría que estamos desarrollando se denomina combinacion lineal
de orbitales atómicos, abreviado CLOA (LCAO en inglés).En un primer orden de aproximación calculamos la energía
+correspondiente a los orbitales moleculares 1/1- como el valormedio del Hamiltoniano
Un cálculo elemental nos permite obtener
C 15A ± B1 ± S
donde A, B Y S son funciones de R dadas por
i\e
2J(j>2 (r)
i\2 R -2R/a ]15 dr e
A 4rrc 4rrc R [1-(1+-) e o~ ~ ao Ir - RI o o
~ ~ ~i\
e2J
(j> (r) (j> (r-R) i\2
(1+~) -R/a15 15 ~ eB = 4rrc dr = 4rrc - e or a a
o o o o
J~ ~ ~ ~ R 1 (~ ) 2 ] e-R/aoS = (j> (r) (j> (r-R) dr = [l + - + 3"1 5 1 5 a a
o o
138

+El primer término en E-(R) es la energía e del nivel ls de15un átomo de Hidrógeno. El segundo da la energía de repulsiónentre los protones. El término A también tiene una interpretaciónclásica como la energía de interacción entre los orbitales 1s y
el otro protón. Las integrales de intercambio B y solapamiento S,en cambio, son de naturaleza puramente cuántica, y no tienen unaanalogía clásica. Estos términos son apreciables sólo si ambosorbitales atómicos ~ (~) y ~ (~-R) se solapan en alguna región
1 8 1 8
del espacio. En particular, si R es suficientemente grande, o seasi R » a, entonces tanto B como S tienden a cero, y nos queda la
o
expresión clásica
Eel
Ahora debemos evaluar la distancia de equilibrio R,variacionalmente, corno aquella que minimiza la energía. Vemos queclásicamente la interacción atractiva entre el electrón y losprotones no alcanza a compensar la repulsión entre estos últimos.La energía clásica crece monótonamente al disminuir R
2E e + _i\_ ~ (1+~) e -2R/ao
el 18 4rre R ao o
En otras palabras, la existencia de moléculas estables es unefecto puramente cuántico debido al solapamiento, combinado conla simetría de la función de onda. En efecto, si graficamos E±(R)como función de R, vemos que la energía correspondiente a lafunción de onda molecular simétrica 1// presenta un mínimo paraR = 1.2 a. El que solo el orbital par de lugar a una molécula
o
estable tiene sentido. En ese caso hay una alta probabilidad deque el electrón se encuentre entre ambos protones. El electrónactúa como un cemento que tiende a juntar a ambos protones,compensando la repulsión entre ellos. En el orbital impar, encambio, la función de onda se anula en el punto medio entre ambosprotones y, por lo tanto, no logra formar una configuraciónestable. Decimos que 1// es una función de onda ligante y 1/1- esantiligante.
139
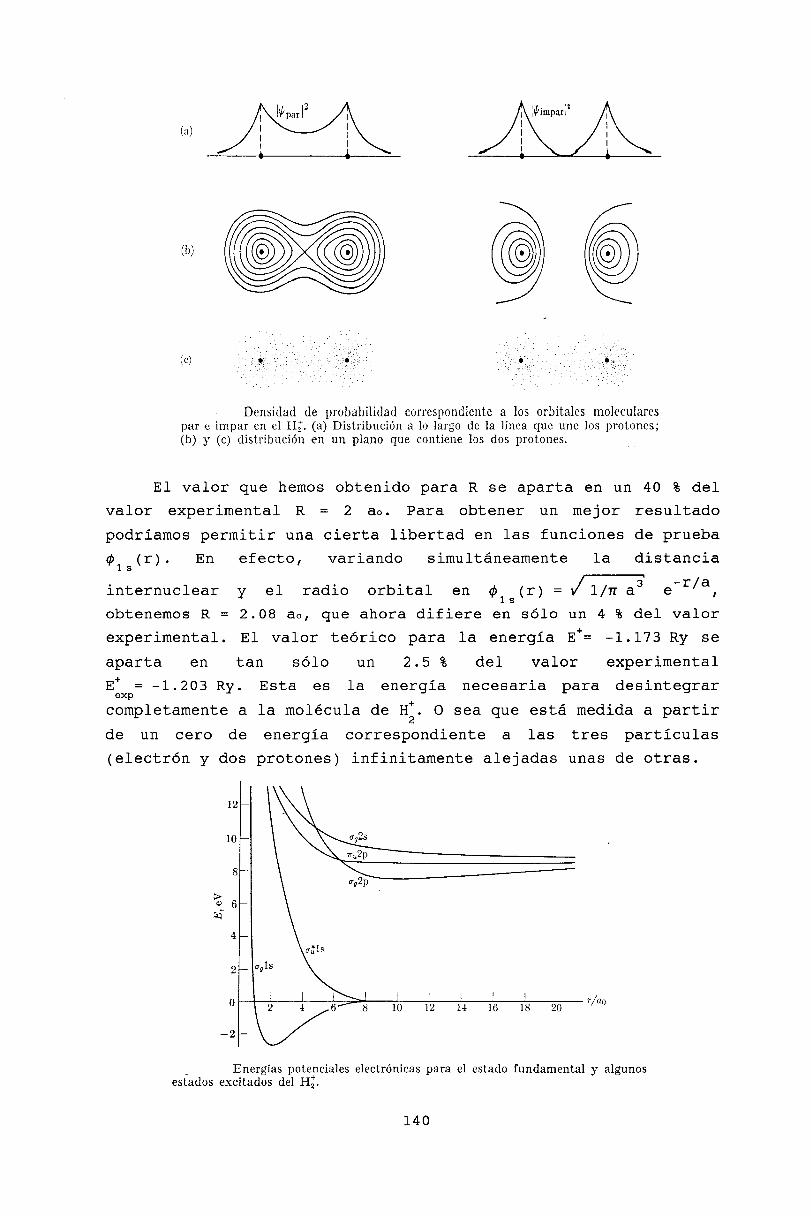
(a)
(b)
(e)
Densidad de probabilidad correspondiente a los orbitales molecularespar e impar en el H;. (a) Distribución a lo largo de la línea que une los protones;(b) Y (e) distribución en un plano que contiene los dos protones.
El valor que hemos obtenido para R se aparta en un 40 % delvalor experimental R = 2 ao. Para obtener un mejor resultadopodríamos permitir una cierta libertad en las funciones de prueba<p (r). En efecto, variando simultáneamente la distancia
1 s
internuclear y el radio orbital en <p (r) = 11/rr ai e-r/a,1s
obtenemos R = 2.08 ao, que ahora difiere en sólo un 4 % del valorexperimental. El valor teórico para la energía E+= -1.173 Ry seaparta en tan sólo un 2.5 % del valor experimental
+E = -1.203 Ry. Esta es la energía necesaria para desintegrarexp
completamente a la molécula de H+. O sea que está medida a partir2
de un cero de energía correspondiente a las tres partículas(electrón y dos protones) infinitamente alejadas unas de otras.
12
10
8
:>6el)
~-4
2
o r/ao
-2
_ Energías potenciales electrónícas para el estado fundamental y algunosestados excitados del H;.
140

ORBITAS MOLECULARES
Ahora vamos a generalizar los resultados anteriores al casode moléculas diatómicas mul tielectrónicas. Tal corno hicimos alestudiar átomos, vamos a comenzar por un modelo de electronesindependientes, analizando el movimiento orbital de cada electrónpor separado en un campo medio producido por los núcleos y elresto de los electrones.
Los electrones no se mueven en un potencial central y por lotanto en una molécula no se conserva el momento angular 1. Sinembargo, hay simetría axial respecto de un eje que pasa por ambosnúcleos, conservándose la proyección del momento cinético orbitalsegún dicho eje. Sus autovalores son m = O, ±1, ±2, ..., donde
1
el signo determina el sentido de rotación alrededor del eje. Sinembargo, la energía del electrón es independiente del sentido derotación. Por lo tanto, para especificar su estado, sólonecesitamos indicar el valor absoluto de m. Entonces vamos a
1
clasificar los orbitales electrónicos según los valores de 1m 1,1
designándolos con letras griegas
A = 1m I = O1 '
(J'
1, 2, 3,rr eS ~
Salvo los estados (J',todos los demás van a tener, además de ladegeneración de espín, una doble degeneración según el signo dem. O sea que cada orbital molecular (J'puede acomodar dos
1
electrones, mientras que los restantes orbi tales rr, eS, ~,
pueden ser ocupados hasta por cuatro electrones.En nuestro modelo de electrones independientes y combinación
lineal de orbitales atómicos, vamos a completar la notaciónanterior indicando los orbitales atómicos que fueron combinadospara formar cada orbi tal molecular en la forma Ant. Así (J'2pindica un orbital (J'formado por la combinación lineal de estadoshidrogenoides 2p. Cada uno de estos orbitales moleculares secorresponde con una energía distinta.
En las moléculas homonucleares, formadas por dos núcleosidénticos (H+,H, ...), tenemos una importante simetría adicional.
2 2Estas moléculas tienen un centro de simetría y la distribución de
141

probabilidad de un electrón debe ser simétrica respecto de él.Esta es, por ejemplo, la situación que encontramos al analizar lamolécula de H;, con funciones de onda simétricas 1// oantisimétricas w-. Entonces, en estos casos, podemos clasificarlos orbitales moleculares teniendo en cuenta también su
(a) OI'IJita/"s 1I",lec"larcs r,·",llarilcs ,1,· orlJilales alólllieos s
antiligante. Enestados excitados.
indicamosasterisco
paridad, que indicamos con los subíndices g (gerade = par) y u(ungerade = impar). Con esta notación resulta que los estados ~+
y I/J que estudiamos para la molécula de H+, correspondían a• 2orbitales moleculares u 1s y u 1s, respectivamente. Con el
g ti
que el orbital correspondiente esla figura mostramos, también, algunos otros
(b) Ol'bitales moleeulares resultantes ,le orbitales atómicos p,
z
(e) Ol'bitales moleculares resultantes de ol'bilalcs atúmicos Jlx Ó !Jy
z
z
Orbitalcs muleculares cn lllo!(>culas dialómicas ]¡omolludearcs.

una configuración
a poner uno de losPero el efecto
CONFIGURACION ELECTRONICA DE MOLECULAS DIATOMICAS
Ahora vamos a analizar algunas moléculas diatómicas simplesusando un método similar al que utilizamos al estudiar laconfiguración electrónica de los átomos multielectrónicos.
En la molécula de Hidrógeno H, ambos electrones se ubican2
en los orbitales ligantes (J 1s, dando la configuración (J 1s2 queg ges estable. La separación entre los núcleos en equilibrio es1.4 ao, y su energía de disociación en dos átomos de Hidrógeno es0.33 Ry.
El siguiente nivel de energía corresponde•electrones en el siguiente orbital (J 1s.u
antiligante de este domina, y no se alcanzaestable.
En la molécula de He+, dos electrónes están en el estado2 •ligante (J 1s, y el otro en el antiligante (J 1s. El resultado es
9 uuna molécula estable con una energía de disociación de 0.18 Ry.La molécula de He requeriría poner dos electrones en el nivel
2•antiligante (J 1s, lo cual conduce a una configuración no estable.u
Esto explica porque el helio es un gas monoatómico.Energía
Longi- Estado1\10- de (liso.llTlIlu Conllgllracíóll ciaeilÍn, tlltl dt· flllidll-
cV enlacl', A Illl'nlal-----_.-- -----~-._- _ ..-----"---------_._- ----- ----
n~ls n:1s ou'2s o:'2s rr,,'2po~'2p rr:'2p a:2p
I-H ITJ 2,\)5 1,06 2~g
Il2 01] 4,48 0,74 IV••..•g
He~ [TI] [] 3,1 1,08 2Lul.
111'. [TI] [TI] Inestable lLg
Li2 [TI] [TI] [ill 1,03 2,67 lLg
lk. [TI] OIJ [ill OIJ J lIl'stahle 'l:g
B2 [ill WJ WJ WJ [[] 3,6 1,S\! 3l:g
e2 [ill [0J WJ [uJ [DJ] :3,(j 1,:¡1 IV••..•g
N [ill [ill WJ WJ fmT] WJ 7,37 1,OH l:Eg2
O2 lill [j]J lill lliJ [Uill WJ [TI ¡ G,OH 1,'21 3\'••..•g
F [ill [ill WJ lliJ [iiliJ lliJ Uilll 2,8 1,44 lLg2
Nc. [TI] [ill UD WJ ITillJ WJ r TI lil WJ Incstable 1'".....g
---- ._--._-_._-------_. __ .~-~-------_._-_._-------.---
eonriguración electrónica tic moléculas tllatómicas bomollucleares

TERMINOS ELECTRONICOS
Tal como ocurría con los átomos multielectrónicos, laconfiguración electrónica resultante de una aproximación deelectrones independientes no basta para especificar unívocamenteel estado de una molécula. De hecho, la interacción entreelectrones produce un desdoblamiento de cada uno de estosniveles según los autovalores de operadores totales. En el casode los átomos, ellos correspondían al momento angular orbital L y
de espín S totales. Ahora, debido a las simetrías del problema,los términos electrónicos se clasifican según el módulo de laproyección del momento angular según el eje que une ambos núcleosde la molécula diatómica. Estos valores se designan nuevamentecon letras griegas, pero ahora mayúsculas.
A = O, 1, 2,¿ TI .b.
Cada estado electrónico se caracteriza, además, por el espíntotal S. Cuando S es distinto de cero se produce la degeneraciónasociada con las direcciones posibles de S, cuya multiplicidad es2S+1, que anotamos como un supraíndice. Así, 3rr representa untérmino con A=l y S=l.
Como en el caso atómico, la repulsión entre electronesfavorece energéticamente a la función de onda que es másantisimétrica ante intercambio . Esto requiere nuevamente, y deacuerdo con el principio de exclusión, que la parte de espín sealo más simétrica posible. En otras palabras, para moléculastambién vale la primera regla de Hund: "La mínima energía laposee el término con el máximo valor posible de S para laconfiguración electrónica dada".
Cuando un orbital molecular Ane está completo, tiene M=O yS=O, y por lo tanto no da ninguna contribución al momento angulartotal o al espín. Es así como en las moléculas H, He, Li, C,
2 2 2 2
N Y F, por ejemplo, todos los orbitales ocupados están2 2
completos, y por lo tanto el término espectral es 1¿,
Si sólo hay un electrón en la capa incompleta, es obvio que
144

A="A Y 8=1/2. Tal es la situación para el H; y He;, cuyo nivel2fundamental el E.
Cuando el orbi tal incompleto tiene más de un electrón, encambio, pueden darse varios términos posibles. Tal es el caso deB y O, ambos con la configuración II2p2. El posible arreglo de2 2números cuánticos para ambos electrones, compatible con elprincipio de exclusión, es
ID ID M Ms 1 S L
+ + 1 -1 1 O
+ - 1 1 O 2
-1 o
-1 1 o
Como en el caso atómico no hemos escrito los estados con valoresnegativosadicional.
de M YsEn esta
M porque no aportanL
tabla descubrimos losninguna información
siguientes estadosmoleculares
M = Os0,1O
M = 2L
O
O
De ellos el que corresponde al estado fundamental es el de mayormultiplicidad, o sea 3¿. Por esto la molécula de oxígeno tiene unmomento dipolar magnético permanente, lo que explica porque elOxígeno es un gas paramagnético, mientras que la mayoría de losgases diatómicos homonucleares son diamagnéticos.
MOLECULAS HETERONUCLEARES
Cuando los dos núcleos que componen una molécula diatómicason distintos, ésta se llama heteronuclear. Ahora ya no existe un
145

centro de simetría y, por lo tanto, pierde sentido laclasificación gerade-ungerade que veníamos usando. Consideremoscomo ejemplo la molécula de sal NaCl. El problema consiste endescribir el movimiento de 28 electrones (11 del sodio y 17 delcloro) en el campo eléctrico producido por los núcleos de Na y elseparados una cierta distancia R. Tal como dijimos en uncomienzo, este problema puede simplificarse enormemente si seconsidera que los electrones en capas atómicas cerradas estánfuertemente ligados al núcleo, y no son afectados. Así, puedeconsiderarse que el Sodio aporta tan sólo un electrón de valenciaa la ligadura molecular, mientras que el resto de los electronespermanece ligado al núcleo. Al cloro, en cambio, le falta unelectrón para cerrar la capa 3s23p6. Lo que ocurre, entonces, esque el electrón de valencia del Na se desplaza en dirección al Clpara formar dos iones de carga opuesta Na+ y Cl-con configuraciónde capa cerrada que se atraen entre sí. El resultado es unamolécula que tiene una distribución de carga no simétrica, y quepor lo tanto está polarizada. El momento dipolar eléctrico es3.54 ea. Si el electrón 3s del Na se transfiriera completamente
o
al Cl tendríamos un dipolo de carga e y longitud igual a laseparación internuclear en equilibrio, R = 4.74 ao, con lo cualel momento dipolar sería 4.74 ea. Vemos así que el 75 % de la
odistribución de carga del electrón de valencia está desplazadahacia el Cloro, y por ello nuestro modelo de la molécula de salcomo compuesta por dos iones que se atraen por interaccióncoulombiana es adecuado. Podemos expresar esta situaciónescribiendo Na+Cl-. Este tipo de ligadura molecular se denominaiónica, mientras que la situación descrita anteriormente para lasmoléculas homonucleares, con una distribución electrónicasimétrica, se denomina covalente.
La figura muestra la energía del Cloruro de Sodio en su fasegaseosa corno función de la separación internuclear. A grandesdistancias la interacción entre los átomos del Na y Cl es muypequeña, y la energía es constante. Para una separación de 20 a
ola transferencia de carga entre ambos átomos comienza a tenerefecto, y la curva se asemeja a un potencial coulombianocorrespondiente a la interacción entre los iones Na+ y Cl-. Porúltimo, a distancias muy pequeñas, la repulsión entre los núcleos
146
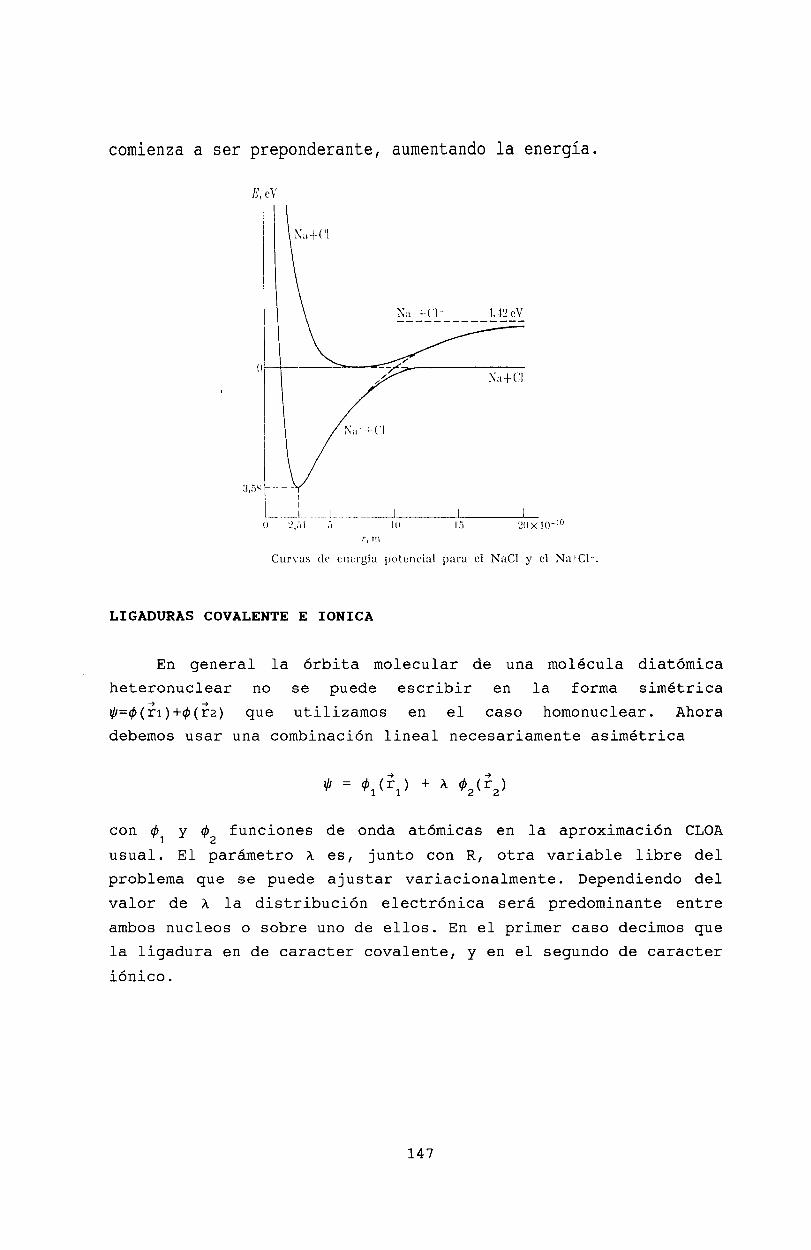
comienza a ser preponderante, aumentando la energía.
E, eV
:\a' t('l-
()
- 3,:.~ - --II
__l I L L-o ~/,¡;. lO 15
r, In
1.42 eV
I20X 10-10
Curvas de energía potencial para el NaCl y el Na+Cl-.
LIGADURAS COVALENTE E IONICA
En general la órbita molecular de una molécula diatómicaheteronuclear no se puede escribir en la forma simétrica
~ -7 • •w=~(rl)+~(r2) que utlllzamos en el caso homonuclear. Ahoradebemos usar una combinación lineal necesariamente asimétrica
~ ~~ (r ) + A ~ (r )
1 1 2 2
con ~ y ~ funciones de onda atómicas en la aproximación CLOA1 2usual. El parámetro A es, junto con R, otra variable libre delproblema que se puede ajustar variacionalmente. Dependiendo delvalor de A la distribución electrónica será predominante entreambos nucleos o sobre uno de ellos. En el primer caso decimos quela ligadura en de caracter covalente, y en el segundo de caracteriónico.
147

BIBLIOGRAFIA
1. M. Alonso & E. J. Finn: Física} volumen III: Fundamentos
cuánticos y estadísticos (Fondo Educativo Interamericano,
Barcelona, 1971).2. B. H. Bransden & C. J. Joachain: Physics oE Atoms and
Molecules (Longman, London, 1983).
148

MOLECULAS POllA TOMICAS
EL AGUA
En el caso de una molécula diatómica vimos que ladistribución espacial de los electrones ligantes era máxima en laregión entre ambos nucleos. Para ello las funciones de ondaatómicas que se combinaban para formar el orbital moleculardebían superponerse lo más posible a lo largo de la linea queunía ambos nucleos. Podemos formalizar este requerimiento desuperposición máxima diciendo que
"La ligadura entre dos átomos se produce en unadirección en la cual las funciones de onda atómicasrespectivas que constituyen el orbital molecular sesuperponen. La fuerza de la ligadura depende del gradode superposición de ambas funciones de onda."
El principio de máxima superposición es muy útil paradeterminar la forma geométrica de una molécula. Consideremos comoejemplo la molécula de agua H O que contiene tres nucleos y diez
2
electrones. En el oxígeno podemos olvidarnos, en primeraaproximación, de todos los electrones en subcapas cerradas. Delos cuatro electrones equivalentes restantes, ubicados en lasubcapa 2p, dos se encuentran en el mismo nivel, digamos el p ,
zcon espines opuestos. Los dos restantes, con espines paralelos,se ubican sobre orbitales lo más alejados posible entre sí y delorbital p para minimizar la repulsión electrostática. Podemos
zasumir, entonces, que estos electrones desapareados se ubican enorbitales p y p. Los dos átomos de Hidrógeno, cada uno con unx y
electrón 1s, se van a ubicar de manera de acoplar sus electronescon los dos desapareados del Oxígeno, logrando la mayorsuperposiclon posible. Concluimos entonces que ambos átomos seubican en los ejes x e y, ortogonales entre sí, y a igualdistancia del átomo de O. El resultado es una molécula en formade triángulo rectángulo, como se indica en la figura.
149

rDistribuciún cIedrúnil':l en
la molécula de H20.
Vemos que los orbitales p aparecen deformados o polarizadospor la presencia de los átomos de H. Además el ángulo entre ambasligaduras O-H es ligeramente mayor que 90° (es 104.5°) debido ala repulsión entre ambos átomos de Hidrógeno.
Un cálculo detallado muestra que la distribución electrónicaque tiende a concentrarse en las ligaduras deja un desequilibriode cargas que produce, a su vez, un momento dipolar eléctrico de0.73 ea a lo largo de la bisectriz entre ambas ligaduras.
o
AMONIACO
Ahora consideraremos la molécula de amoníaco NH. Los3
tres electrones 2p desapareados del Nitrógeno se concentran enorbitales p , p y p en las direcciones x, y y z. De esta manera
x y zla molécula de NH tiene una estructura de tetraedro, con el
3
átomo de N en un vértice y los átomos de H en los otros tres. Losángulos en el vértice formado por el N son de 107.3° en lugar de90°, debido a la repulsión entre los átomos de Hidrógeno.Nuevamente se produce un desequilibrio de carga que genera unmomento dipolar eléctrico de 0.59 ea .
o
- - -'.\r¡¡/¡A' II, '~-
107.:lo __ ---1.,.,..--c" .. , {.~~l~\~t.'~
}'
Dist rihución clectrónica cnla molécula de NHa.
150

HIBRIDIZACION Sp3
La manera como se liga el átomo de Carbono es muyinteresante. En su estado fundamental tiene sólo dos electrones2p desapareados, que no alcanzan para explicar la formación de lamayoría de los compuestos del Carbono. El primer estado excitadoconsiste en una configuración lS22s12p3, es decir, en un electrón2s y tres electrones 2p desapareados. Los electrones 2p actuaríancomo los del Amoníaco, pero el electrón 2s, debido a su falta dedireccionalidad, no podría producir una ligadura tan fuerte comoaquellas. De esta forma no se podría explicar una molécula concuatro ligaduras equivalentes, como ocurre en el metano CH. Tal
4
tipo de ligadura se produce por medio de un proceso denominadohibridización de funciones de onda. La diferencia de energíaentre los niveles 2s y 2p es tan pequeña en comparación con lasenergías de disociación típicas, que se pueden suponerdegenerados. Entonces, podemos construir funciones de ondahíbridas, todas de igual energía, como combinaciones lineales delos orbitales s, p , p y p . Las combinaciones más simples
x y z
1/111 (s + p + p + P )= "2 x y z
1/121 (s + p p )= "2 Pyx z
1/131 (s + p pz)"2 Px y
1/141 (s + P )"2 Px Py z
constituyen funciones de onda híbridas cuyos máximoshacia los vértices de un tetraedro, formando ángulos deEste tipo de hibridización se denomina Sp3.
z
apuntan109.5°.
(a) Funcioncs de onda s, Px. py Y P.
y
151
(ll) Funciones dc onda híbridas sp'

Volviendo a la molécula de metano CH, las ligaduras más4
fuertes se obtienen al superponer las órbitas ls de los átomos de
H con las cuatro híbridas sp3 del e, formando una estructura de
tetraedro.
De forma similar, la molécula de etano HC-CH tiene la3 3
estructura que se muestra en la figura. Los átomos de carbono se
mantienen unidos por medio de una superposición de dos órbitas
híbridas Sp3. Esto se denomina ligadura (j por su similitud con
los orbitales (j de las moléculas diatómicas.
la) (1.)
Elllaces de orhilales IllOleclIlarcs sp" localizados en (a) metano, (11) ctano.
La hibridización Sp3 ocurre también en otros átomos conestructura electrónica similar a la del carbono. Por ej emplo:Silicio Si, Germanio Ge, ión Nitrógeno N+, etc.
HIBRIDIZACION Sp2
Además de la hibridización Sp3 hay otras posibilidades deformar funciones de onda híbridas, incluso con orbitales atómicosde momento angular mayor que uno. Sin embargo, aquí noslimitaremos a discutir sólamente las hibridizaciones spn, conn = 1, 2 y 3. En la hibridización Sp2, por ejemplo, los orbitaless, Px y Py se combinan para producir tres funciones de ondahíbridas, cuyos máximos apuntan en direcciones coplanares queforman ángulos de 1200 entre sí. Estas funciones de onda son
152

1/111= -13'
1/12
1= -13'
1/131= -13'
l
(s + v'Z p )y
(S _ L +13' p )px yyz yz
1 13'(8 - - p --p)
yzxyzy
(a) Funciones dc onda híoridas sp' (o) Función de onda pz,FlllH:iuncs de onda <IUt~rcsllllan lk la hilJridizaCÍún sp".
Este es el tipo de hibridización que se necesita paraexplicar la estructura del etileno H C=CH. Las órbitas s, px y
2 2
P del Carbono se hibridizan. Los átomos de Hidrógeno se ligan ay
dos de estas órbitas híbridas, mientras que la tercera genera unaligadura u entre los carbonos. Los orbitales p, por otro lado,
zse superponen entre si, formando una ligadura n, asi llamada porsu similitud con los orbitales n de las moléculas diatómicas.Como resultado de todo esto, la molécula de etileno es plana. Ladoble ligadura un hace que la molécula tenga una cierta rigidezrespecto a torsiones alrededor del eje C-C. Esta rigidez, que noestá presente cuando sólo hay una ligadura u como en el etano,tiene algunos efectos sobre las propiedades moleculares.
El grado de solapamiento en una ligadura n es mucho menorque en una ligadura u, y por lo tanto contribuye menos a laenergía de ligadura. Por ejemplo, se requieren 0.46 Ry pararomper una ligadura u, y sólo 0.29 Ry para romper una dobleligadura un y transformarla en una u única.
153
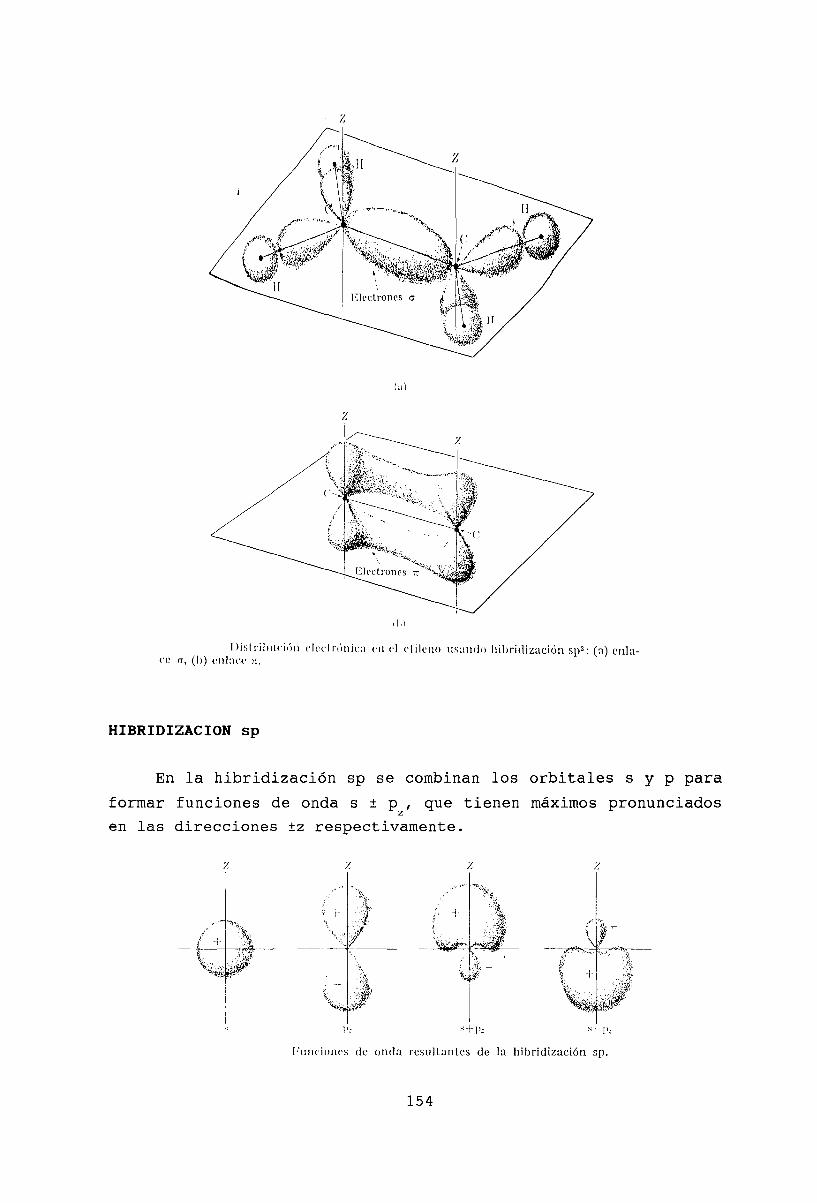
la)
IlislriiJlIciÚIl dl'rtr('lllica l'n e1elileno usando hibridización Sp2: (a) enla-ce a, (iJ) enlace 1t,
HIBRIDIZACION Sp
En la hibridización sp se combinan los orbitales s y p paraformar funciones de onda s ± p, que tienen máximos pronunciados
z
en las direcciones ±z respectivamente.
Funciones de olHla resullantes de la hibridización sp.
154

Esto permite explicar la estructura del acetileno HC=CH. Laligadura triple entre los dos átomos de carbono resulta delsolapamiento de dos órbitas híbridas sp (en una ligadura ~) y dedos solapamientos entre órbitas p y p (en ligaduras rr). Los
x y
átomos de hidrógeno se unen a los restantes orbitales sp. Comoresultado de esto, el acetileno es una molécula lineal.
lb)
MOLECULAS CONJUGADAS
z
Distribución electró-nica en el acetileno usando hibri-dización sp. (a) Funciones deonda de los átomos separados.(b) Enlaces cr y 7t.
En la siguiente figura mostramos la estructura del butadienoCH. Los átomos de carbono forman una cadena unida por ligaduras
4 6
~ usando una hibridización Sp2. Además hay cuatro electrones pz
que forman ligaduras n. Estos electrones n, uno por cada átomo,actúan de una manera muy especial; y en lugar de estarlocalizados en regiones particulares de la molécula, como ocurreen las ligaduras ~, pueden moverse más o menos libremente a lolargo de la molécula.
155

Distribución elec.trónica en el butadieno. (a) Enlaces localizados cr;(b) enlaces no localizados 7r ••
Tal como se indica en la figura, hay básicamente cuatromaneras posibles de acoplar orbitales atómicos p para formar las
zfunciones de onda moleculares de estos electrones IT. Estosorbitales moleculares tienen energías diferentes, que aumentan alaumentar el número de nadas. Luego, en el estado fundamental, loscuatro electrones se agrupan en los dos orbitales de menorenergía. Uno de ellos es ligante para todos los átomos, mientrasque el otro liga los pares extremos pero no el central. estosignifica que la fuerza de la ligadura central es menor que enlos extremos. Experimentalmente se observa que la ligaduracentral tiene una longitud de 2.76 a, mientras que en los
o
extremos es 2.55 a .o
156
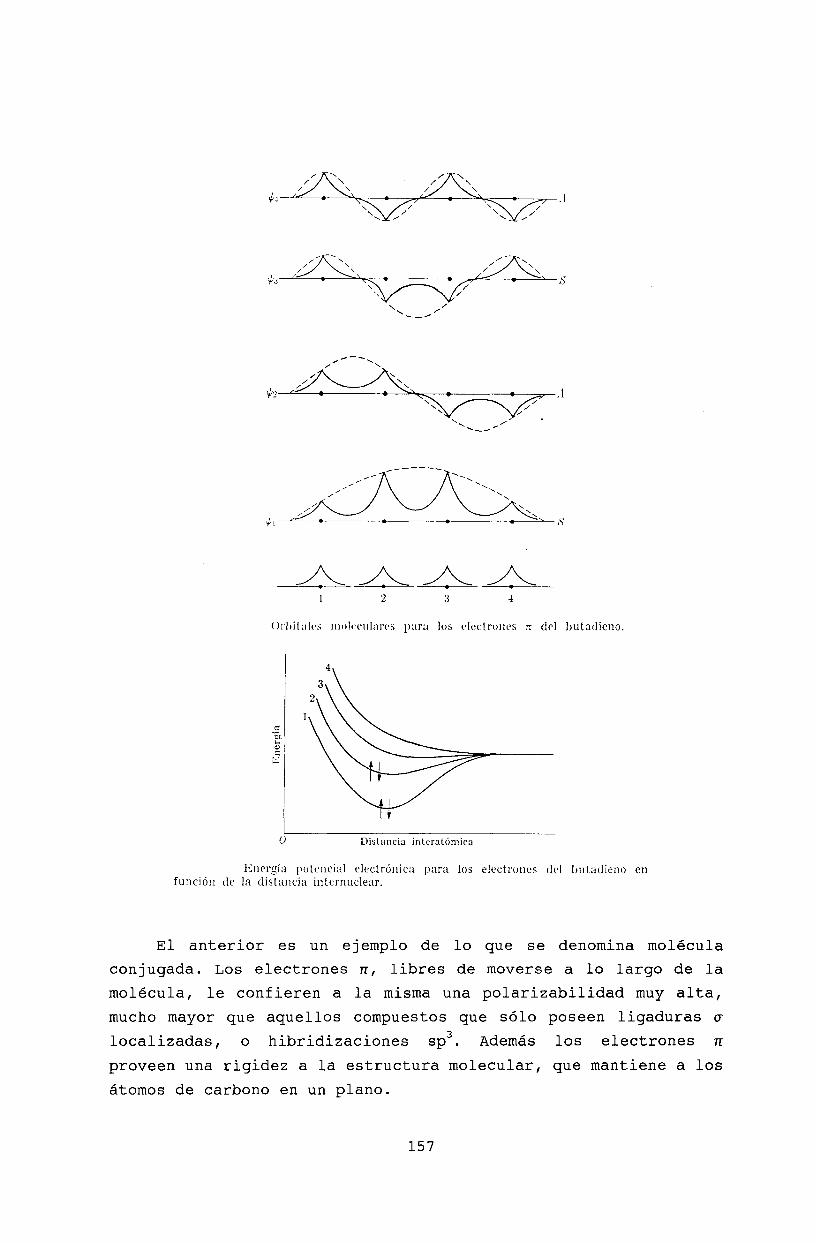
Orhitaks 11lOkclItarcs para los electrunes •. del butadienoo
~05't...
~I~-O Distancia interatómica
Energía potcneial electrónica para los electruncs del blltadicno enfunción de la distancia internuclear.
El anterior es un ejemplo de lo que se denomina moléculaconjugada. Los electrones n, libres de moverse a lo largo de lamolécula, le confieren a la misma una polarizabilidad muy alta,mucho mayor que aquellos compuestos que sólo poseen ligaduras ~localizadas, o hibridizaciones Sp3. Además los electrones nproveen una rigidez a la estructura molecular, que mantiene a losátomos de carbono en un plano.
157
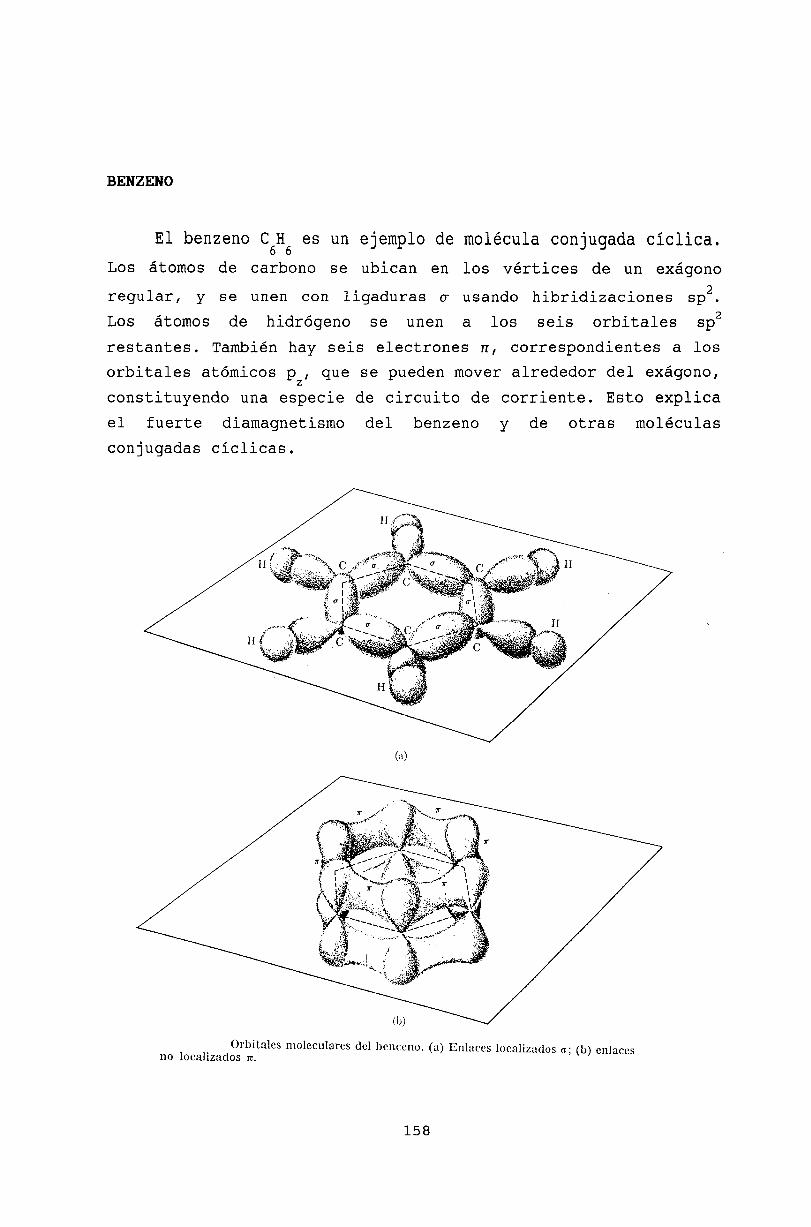
BENZENO
El benzeno e H es un ejemplo de molécula conjugada cíclica.6 6Los átomos de carbono se ubican en los vértices de un exágonoregular / y se unen con ligaduras (j usando hibridizaciones sp2.Los átomos de hidrógeno se unen a los seis orbitales Sp2restantes. También hay seis electrones rr, correspondientes a losorbitales atómicos p / que se pueden mover alrededor del exágono,
zconstituyendo una especie de circuito de corriente. Esto explicael fuerte diamagnetismo del benzeno y de otras moléculasconjugadas cíclicas.
(a)
. Orbitales molcculares del bcnceno. (a) Enlaces localizados cr; (b) enlacesno loca!¡zados 1t.
158

ROTACION MOLECULAR
Hasta ahora habiamos considerado que los nucleos en lamolécula se mantenían fijos en sus posiciones de equilibrio. Estaes una aproximación que vamos a eliminar, comenzando por estudiarel movimiento nuclear más simple, el de rotación de la moléculacomo un cuerpo rígido alrededor de su centro de masa.
Puesto que las masas electrónicas son muy pequeñas, podemosdespreciar el momento de inercia de una molécula diatómicaalrededor del eje de simetría que pasa por ambos nucleos. Porello el momento angular L de la molécula es perpendicular a dichoeje. Si R es la separación internuclear en equilibrio y
o/.l f:f M M / (M +M) la masa reducida de la molécula, el momento de
1 2 1 2
inercia es 1 = /.lR 2. Con ello la energía cinética de rotación eso
E = L2/21 = L2/2/.lR2. Por último, y en virtud de la cuantizaciónr odel momento angular, obtenemos
h2
E = - e(t+1)r 21 e = 0, 1, 2, ...
La energía característica ñ2/21 toma valores entre-6 -42.3x10 Ry para Cl y 5.9x10 Ry para H. Estos son valores tan
2 2pequeño en comparación con las energías de traslación en un gasque, inclusive a temperatura ambiente, donde kT f:f 2x10-3 Ry, unamuy alta proporción de las moléculas se encuentra en estadosexcitados de rotación que se alcanzan por colisión.
Cuando la molécula posee un momento dipolar eléctricopermanente, el movimiento de rotación puede interactuar con elcampo de radiación, dando lugar a procesos de absorción defotones. Las moléculas diatómicas homonucleares, que no tienen unmomento dipolar eléctrico permanente, no pueden interactuar conel campo de radición. En las moléculas heteronucleares, encambio, esta interacción será más fuerte cuando mayor sea elcarácter iónico de la ligadura. Nuestra discusión también esaplicable a moléculas poliatómicas lineales, tales como ca, que
2
en general tienen mayores momentos de inercia y,correspondientemente, menores frecuencias de radiación.
Habiendo estados caracterizados por momentos angulares e que
159

emiten y absorben radiación por medio de transiciones dipolareseléctricas, se debe verificar la regla de selección
M = ±1
Luego sólo son posibles transiciones entre estadosrotacionales adyacentes. Las frecuencias características son
w = b.E/h = ht/I
con f el momento angular del estado más excitado que intervieneen la transición.
Vemos que el espectro de rotación consiste en una serie delineas igualmente espaciadas en una cantidad ~w = h/I. Midiendo~w podemos calcular el momento de inercia y estimar la separaciónnuclear R. Las frecuencias características son del orden de
o
~w f::f 1012 Hz, que están en el rango de las microondas oinfrarrojo lejano, con longitudes de onda típicas de 1 rnm. En lafigura mostramos un espectro de absorción para el Hel.
/(#),
d::l NI....~Q.;<J.. GOoo.:: 40'-e:§E"'o::..E- I
I
40 60 80 100 120 140 ltiO lbO 2iHl 220 :.?~'l 2i;(1 2.':11 3/10v, cnl-l
Espectro rotacional de absorción del Hel en fase gaseosa.
OSCILACIONES MOLECULARES
Hasta ahora consideramos a los nucleos como manteniéndose auna distancia fija uno respecto del otro. Sin embargo, esto no esasí, y los nucleos pueden oscilar alrededor de sus posicionesrelativas de equilibrio.
160

Podemos aproxlmar a la energía potencial de una molécula
diatómica, cerca de su separación de equilibrio, por un potencialarmónico V(R) == i k (R-Ro)2, con frecuencias características de
oscilación w = vk/~ que van de lx1014 Hz para el Cl hastao 2
148.lx10 Hz para el H .2
Cuantizando el movimiento de oscilación nuclear, obtenemos
niveles de energía
En = (n + i) hWo n = O, 1, 2, ••.
La existencia de una energía de punto cero E = hw /2, noso o
indica que las energías de disociación D no deben medirse desdeel mínimo de la energía potencial, sino desde esa energía mínima,o sea D - hw /2.
o
Para que pueda ocurrir una transición entre niveles devibración con emisión o absorción de radiación, debe existir unmomento dipolar eléctrico, ya sea permanente o inducido por elmismo movimiento oscilatorio, que se acople con el campoelectromagnético. Ninguna de las dos situaciones puede darse enel caso de moléculas diatómicas homonucleares, tales como H o
2
N, Y por lo tanto estas no pueden realizar transiciones2
radiativas entre niveles de vibración.Las moléculas heteronucleares, en cambio, poseen momentos
dipolares eléctricos permanentes, y por lo tanto pueden efectuartal tipo de transiciones. Estas están reguladas por la regla deselección
Lin = ±1
Vemos entonces que las transiciones vibraciona1es se efectúansólo entre niveles adyacentes, y todas con la misma frecuenciacaracterística w que cae en el rango infrarrojo.
o
MODOS NORMALES
El espectro de oscilación de las moléculas poliatómicas debeanalizarse en términos de los modos normales de vibración
161

correspondientes. A cada modo normal le corresponde, en general,una frecuencia característica distinta. En la figura mostramoslos modos normales del dióxido de carbono eo y del agua H O.2 2
Modos normales de vibraciónde las moléculas poliatómicas. (a) CO2: 001 == 1337 cm-l, 002 = 667 cm-l, 003 = 2349¡;m-l; (b) H20: 001 = 3657 cm-l, 002 == 1595 cm-l, 003 = 3756 cm-l,
(a)
w¡ W2
(b)w3
Para que una frecuencia sea activa en el espectroinfrarrojo, el correspondiente modo normal debe inducir un dipoloeléctrico oscilante en la molécula. Por ejemplo, la molécula ea
2
es simétrica y no tiene, por lo tanto, un momento dipolareléctrico permanente. La primer oscilación indicada en la figurapreserva esa simetría, no induce un momento dipolar, y por lotanto ese modo vibracional es espectroscópicamente inactivo.
ESPECTRO ROTO-VIBRACIONAL
La cantidad h2/21 _ 10-5 Ry es mucho menor quehw
o~ 10-2 Ry. Vemos entonces que cada nivel vibracional n está
desdoblado en varios niveles rotacionales t. Entonces, cuando seproduce una transición entre niveles vibracionales adyacentes,esta debe ocurrir entre subniveles rotacionales cuyos momentosangulares verifiquen la regla de selección ~t = ±1. Esto da lugara un espectro de frecuencias roto-vibracional dada por
162
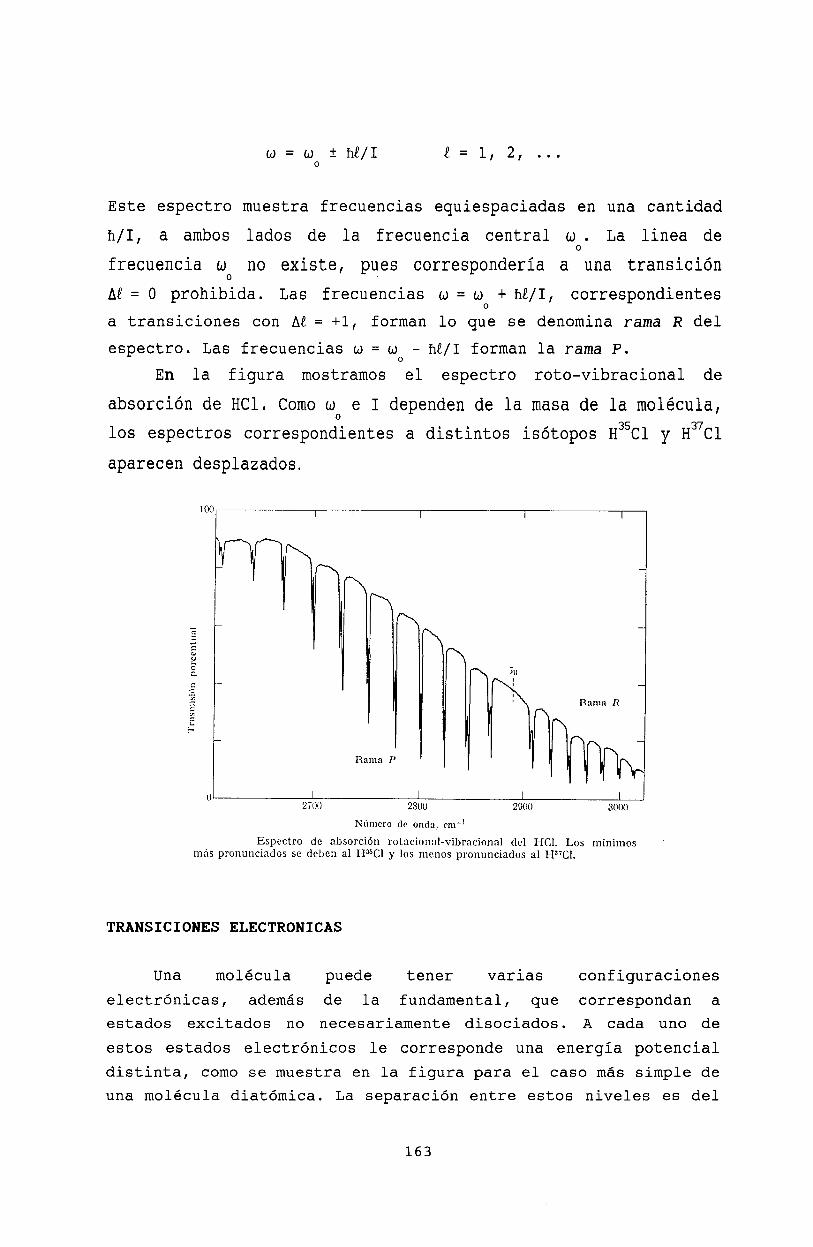
w = w ± hilIo
i = 1,2, ...
Este espectro muestra frecuencias equiespaciadas en una cantidadh/I, a ambos lados de la frecuencia central w. La linea de
ofrecuencia w no existe, pues correspondería a una transición
o 'M = O prohibida. Las frecuencias w = w + hi/I, correspondientes
o
a transiciones con Al = +1, forman lo que se denomina rama R delespectro. Las frecuencias w = w - MI! forman la rama P.
o
En la figura mostramos el espectro roto-vibracional deabsorción de HC1. Como w e I dependen de la masa de la molécula,
olos espectros correspondientes a distintos isótopos H35Cl y HTIClaparecen desplazados.
100
o 2700 2800Número de onda, rm-I
2900 30()0
Espectro de absorción rotacional-vibracional del HC\. Los mínimosmás pronunciados se deben al H35Cl y los menos pronunciados al H37C\.
TRANSICIONES ELECTRONICAS
Una moléculaelectrónicas, ademásestados excitados no
puede tener variasde la fundamental, que
necesariamente disociados.
configuracionescorrespondan aA cada uno de
estos estados electrónicos le corresponde una energía potencialdistinta, como se muestra en la figura para el caso más simple deuna molécula diatómica. La separación entre estos niveles es del
163

orden de 0.1 a 10 Ry, por lo cual, cuando una moléculaexperimenta una transición entre estados electrónicos distintos,la radiación involucrada está en el rango visible o ultravioletaw ~ 1015 Hz.
A cada nivel electrónico E le corresponde varios nivelese
vibracionales (n+1/2)hw, y a cada uno de estos, a su vez, leo
2corresponde varios niveles rotacionales ht(t+1)/2I. Juntando lastres contribuciones tenemos, para la energía molecular
E = E + (n+1/2) hw + h2t (t+1)/21e o
En una transición electrónica, las frecuenciascaracterísticas de la radiación emitida y absorbida son suma detres términos
w=w +w(n.,n) +w((,J!)e v 1 f rlf
donde w (E .-E )/h. Las frecuencias de transición entree el ef
niveles de vibración w y rotación w, en cambio, no tienenv rexpresionesanteriores.momento de
tan simples corno las halladas en seccionesEllo se debe a que tanto la frecuencia w, corno el
o
inercia 1, cambian al cambiar de nivel electrónico.Luego, en general,
w = (n.+1/2) w. -(n +1/2) wv 1 01 f of
y
Para determinar los posibles valores de w y w necesitamosv rreglas de selección. Cuando consideramos transiciones dipolareseléctricas, tenemos para los niveles rotacionales
M = O, ±1 J!i = O ~ J!f = O prohibido
La transición M = O está permitida ahora puesto que, alcambiar la configuración, no hay que preocuparse porconsideraciones de paridad. Sin embargo la transición J! =0 ~ J! =0
i f
sigue estando prohibida por conservación del momento angular.
164

PRINCIPIO DE FRANCK-CONDON
Las reglas de selección para los niveles vibracionales sonalgo más complicadas. Las transiciones electrónicas ocurren entiempos del orden de 10-16 seg, mientras que el movimiento deoscilación nuclear tiene un período de 10-13 seg, o sea mil vecesmayor. Luego, puede suponerse que durante la transiciónelectrónica el movimiento oscilatorio de los nucleos permanececongelado.
Clásicamente los nucleos se encuentran preferentemente enlos extremos de la oscilación, donde la velocidad se anula, y porlo tanto aumenta el tiempo de permanencia. La forma de lasfunciones de onda de un oscilador armónico muestra que lo mismovale en el caso cuántico, con excepción del nivel fundamental.
Ahora bien, las transiciones entre niveles vibracionales sevan a producir preferentemente entre aquellos estados para loscuales sea más fácil ajustar, con la menor distorsión posible,las posiciones nucleares (que se mantienen congeladas) a la nuevaconfiguración electrónica. O sea que las transiciones másprobables ocurren entre niveles cuyos extremos de oscilacióncoinciden. Por ejemplo, en el caso de la siguiente figura, si elestado n.=5 en la configuración electrónica fundamental sufre una
1
transición electrónica por absorción de radiación, va a pasar conmayor probabilidad al nivel n =1, para el cual coincide uno de
f
los extremos de la oscilación. O bien se va a disociar con unaenergía E .
D
Cuando la transición involucra un estado fundamental n =0 oi
n =0, los nucleos se encuentran preferentemente en el punto def
equilibrio. De esta manera, las transiciones de absorción másprobables que involucran a estos dos estados son, en el caso dela figura, n = O ~ n = 4 Y n = 2 ~ n = O.
i f i f
Hasta ahora hemos considerado transiciones por absorción deradiación, pero la misma lógica se aplica al caso de emisión.
Las reglas que hemos explicado se denominan principio deFranck-Condon. Fueron formuladas primero en término clásicos porJames Franck y reformuladas para el caso cuántico por EdwardCondon en 1926.
165
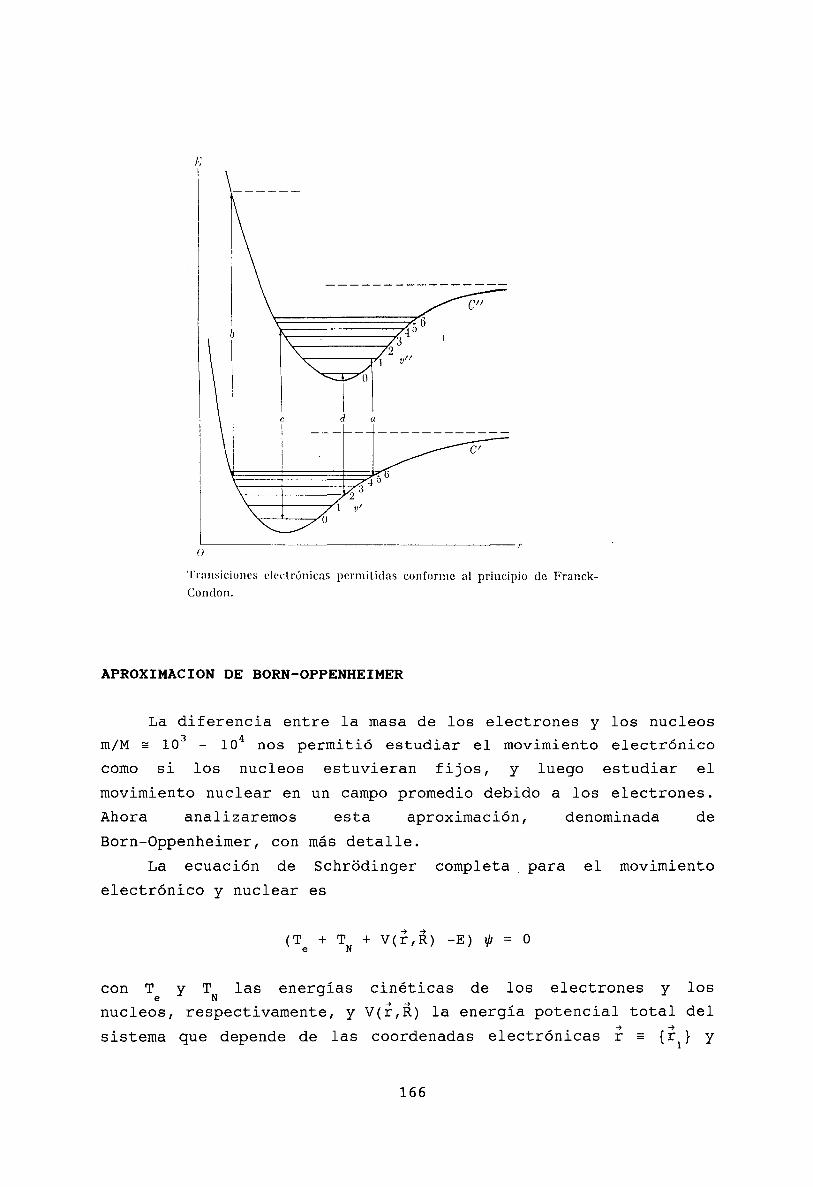
l,;
()
e d (l
---~- ------------I e'
• ti3-!u
21 v'
Transiciones electrónicas permitidas conformc al principio dc Franck-Con don.
APROXIMACION DE BORN-OPPENHEIMER
La diferencia entre la masa de los electrones y los nucleosm/M - 103
- 104 nos permitió estudiar el movimiento electrónicocomo si los nucleos estuvieran fijos, y luego estudiar elmovimiento nuclear en un campo promedio debido a los electrones.Ahora analizaremos esta aproximación, denominada deBorn-Oppenheimer, con más detalle.
La ecuación de Schrodinger completa para el movimientoelectrónico y nuclear es
-> ->(T + T + V(r,R) -E) l/J = O
e N
con T Y T las energíase N
nucleos, respectivamente, ysistema que depende de las
cinéticas de los electrones y losV(r,R) la energía potencial total del
;. ~ ~coordenadas electronlcas r == {r.} y1
166

~ ~nucleares R - {R.}.1
Ahora expandimos W en autofunciones del Hamiltonianocorrespondiente al movimiento electrónico considerando a losnucleos fijos. O sea
~ ~w(r,R) ~ ~ ~= L F (R) u (r,R)
n nn
con~ ~[T + V(r,R)
e
~ ~ ~E; (R)] u (r,R) = O
n n
Reemplazando en la ecuación de Schrodinger original resulta
~ ~ ~ ~L [T + e (R) - E] F (R) u (r,R) = ON n n nn
Hasta ahora no hemos hecho ninguna aproximación. Ahora sí,vamos a despreciar en el término de energía cinética nuclear alos términos de la forma F ~2U y ~ F .~ u frente a u ~2F .nRn Rn Rn nRnPara ello advertimos que, mientras u es una función de onda que
n
se extiende en una longitud del orden de las ligadurasmoleculares R, la función de onda nuclear F se extiende sobreo nuna distancia característica de las oscilaciones x ~ vh/Mw. Ahorabien, mientras que por el principio de incerteza las energíaselectrónicas e son del orden de e ~ (h/R)2 /2m, la frecuencia
n n ode oscilación debe verificar e ~ kR2/2= Mw2R2/2, con lo cualn o ox/R ~ (m/M)1/4 y las energías de oscilación son un orden v'rri/M
o
menores que las electrónicas hW/c ~ v'rri/M. Luego, tenemos quen
h2 ~2F h2 1 hw2M u ~ ~n R n 2M 2X
h2F .~ h2 1 1
2M ~R u ~2M Rn R n Xo
h2 ~2U h2 12M Fn ¡::: -R n 2M R2
o
¡::: (g) 3/4 en
Vemos que los últimos dos términos son órdenes de magnitudmenores que el primero y, por lo tanto, podemos despreciarlos.Obtenemos entonces
167

~ ~ ~ -tL u (r,R) [TN + e (R) - E] F (R) = On n n
n
y usando la ortonormalidad de las funciones de onda electrónicasu, logramos separar el movimiento nuclear en un potencial dado
n
por la energía electrostática de cada nivel electrónico
~ ~[T + e (R) - E] F (R) = O
N n n
De esta manera queda confirmada la validez de la aproximaclon queutilizamos para estudiar la estructura molecular.
BIBLIOGRAFIA
1. M. Alonso & E. J. Finn: Física, volumen III: Fundamentoscuánticos y estadísticos (Fondo Educativo Interamericano,Barcelona, 1971).
2. B. H. Bransden & C. J. Joachain: Physics of Atoms and
Molecules (Longman, London, 1983).
168

TEORIA CLASICA DE COLISIONES
SECCION EFICAZ DIFERENCIAL
Consideremos un flujo J de partículas que inciden conimpulso p sobre un blanco formado por N centros de fuerza. Elnúmero 1 de partículas detectadas por unidad de tiempo en undiferencial dO de ángulo sólido no es adecuada para describir elproceso de colisión, ya que siendo proporcional al flujo deproyectiles y al número de blanco8, depende de lascaracterísticas particulares del experimento.
I.dQ ex J.N
El factor de proporcionalidad debe ser un diferencial deigual orden, y lo llamaremos sección eficaz diferencial d~
De esta ecuación vemos que la sección eficaz ~ tiene unidades deárea. Es claramente independiente de la intensidad del hazincidente, del número de partículas en el blanco o de laresolución del detector. Está definida exclusivamente por lascaracterísticas de la interacción entre cada proyectil y cadapartícula del blanco, y es función únicamente del impulso p ydel ángulo de dispersión B.
169

DEFINICION CLASICA DE LA SECCION EFICAZ
Consideremos un proyectil que se acerca a un centro defuerza con cierto apartamiento p respecto de la trayectoria decolisión frontal, para luego ser desviado en un cierto ángulo dedispersión e por la acción del potencial central actuante.Supondremos que la relación entre el ángulo de dispersión e y elparámetro de impacto p es biunívoca. Esto es claramente ciertopara el potencial coulombiano en el cual el ángulo de dispersiónes una función monótona del parámetro de impacto. En este casosólo aquellas partículas con parámetros de impacto entre p y p+dpserán dispersadas en el cono de ángulo sólido entre e y e+de.
dA= 2rcpdp
dP
d Q = 2 TI sen e d e
Luego I dO = J dA, o si consideramos N centros dispersoresI dO = N J dA. O sea
I 2rr sene de = N J 2rr p dp
170

Y, por definición de sección eficaz diferencial, tenemosfinalmente
dcr _ p dpdO - sene Idel
El cálculo de la sección eficaz diferencial se ha reducido aconsiderar el problema de la dispersión de una partícula por uncentro de fuerzas. Sabemos que la trayectoria de la partícula enun campo de fuerzas central es simétrica respecto de una líneapor el punto más cercano al centro de fuerzas. Por lo tanto, sillamamos ~ al ángulo de dicho perihelio, tenemos una relaciónbastante simple con el ángulo de dispersión e
2cp+e = rr
El impulso angular t = pp y la energía Edurante la colisión. O sea
2 •pp = mr 'lJ
~; = ~ m (r2+r2~2)+V(r)
2P 12m se conservan
Eliminando dt de ambas ecuaciones resulta la ecuación de lasórbitas
d'lJ2(plr )dr
j 1-(plr)2_(2mV/p2)'
Finalmente, integrando entre el punto en el infinito y elperihelio obtenemos
Esta ecuación nos permite calcular eltérminos del parámetro de impactocompleta al problema.
171
ángulo de dispersión e enp, dando una resolución

DISPERSION DE RUTHERFORD
Reemplazando V(r) = Z/r en la expresiónefectuando una integración elemental obtenemos elimpacto p en función del ángulo de dispersión 8 que,en la sección eficaz, nos dá corno resultado laRutherford dedujo en 1911
anterior yparámetro dereemplazandofórmula que
14sen (8/2)
D~b~ nct~r~~ qu~ la fórmula d~ Ruth~rford eg independientedel signo de la carga Z, con lo cual el resultado es igualmenteválido, ya sea para campos coulombianos repulsivos o atractivos.
EFECTOS GLORIA Y ARCO IRIS
Consideremos un potencial interatómico típico, repulsivo acortas distancias y atractivo a grandes distancias.
v (r)
r
Así, si el parámetro de impacto p es grande, el ángulo dedispersión e es negativo y decrece con p. Para parámetros deimpacto pequeños, en cambio, e crece al disminuir p hastaalcanzar el valor 8 = Tl correspondiente a la dispersión haciaatrás.
172
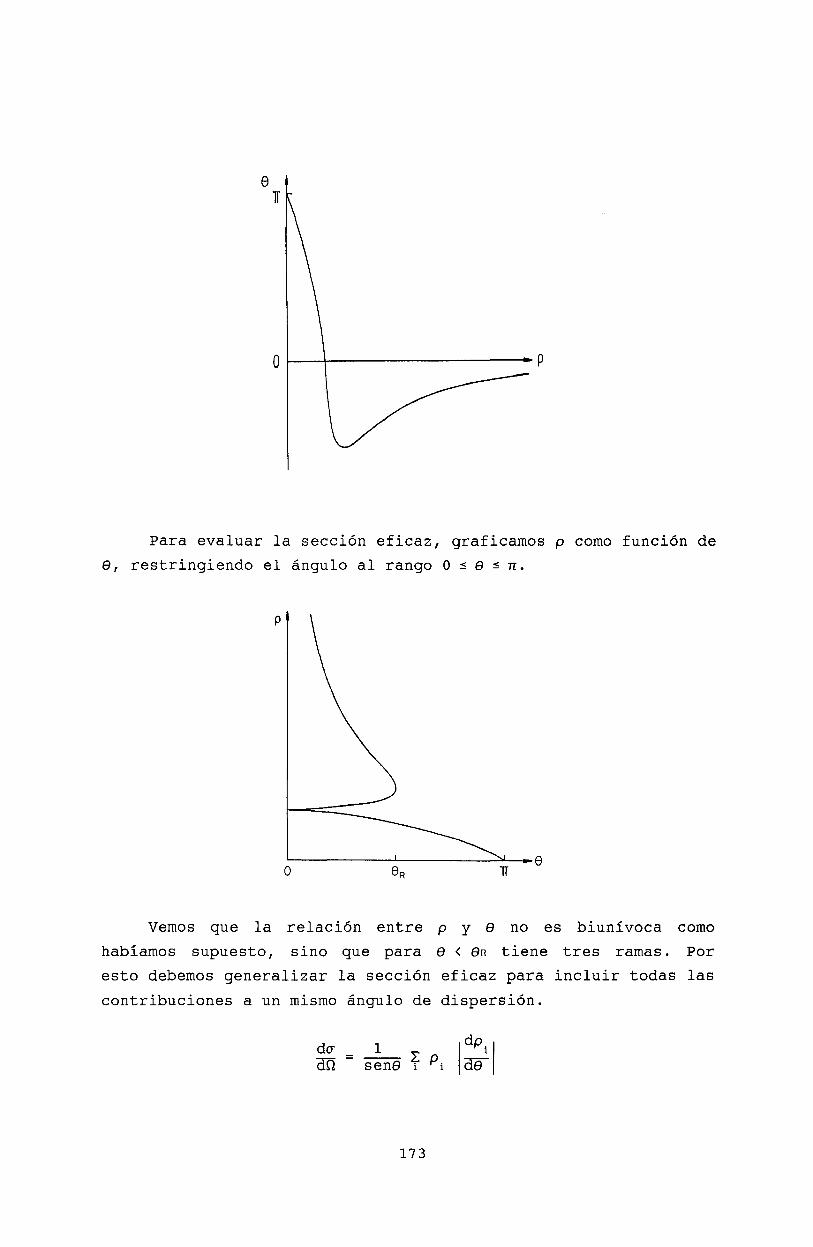
elf
o p
Para evaluar la sección eficaz, graficamos p como función dee, restringiendo el ángulo al rango O ~ e ~ IT.
p
e
Vemos que la relación entre p y e no es biunívoca comohabíamos supuesto, sino que para e < eR tiene tres ramas. Poresto debemos generalizar la sección eficaz para incluir todas lascontribuciones a un mismo ángulo de dispersión.
173
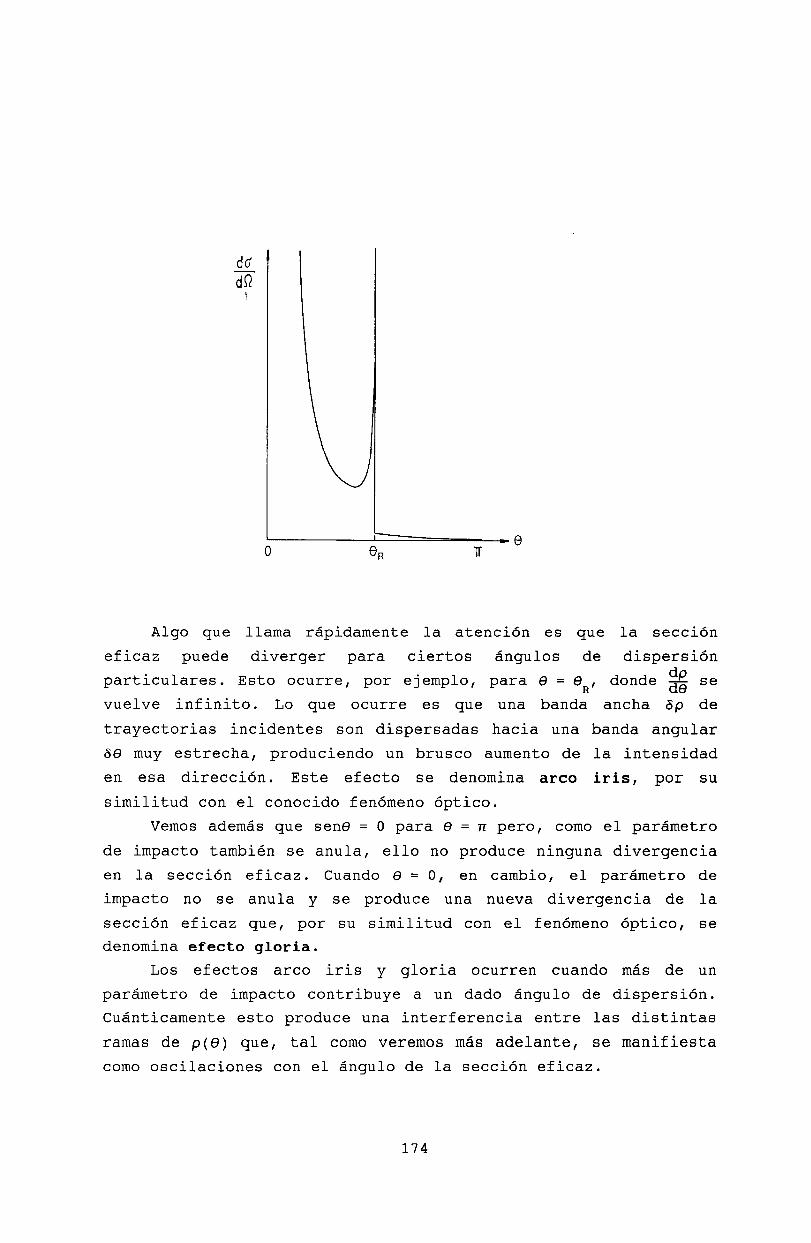
dedñ,
e
Algo que llama rápidamente la atención es que la seccióneficaz puede diverger para ciertos ángulos de dispersiónparticulares. Esto ocurre, por ejemplo, para 8 = 8R, donde ~~ sevuelve infinito. Lo que ocurre es que una banda ancha op detrayectorias incidentes son dispersadas hacia una banda angular08 muy estrecha, produciendo un brusco aumento de la intensidaden esa dirección. Este efecto se denomina arco iris, por susimilitud con el conocido fenómeno óptico.
Vemos además que sen8 = O para 8 = rrpero, como el parámetrode impacto también se anula, ello no produce ninguna divergenciaen la sección eficaz. Cuando 8 = O, en cambio, el parámetro deimpacto no se anula y se produce una nueva divergencia de lasección eficaz que, por su similitud con el fenómeno óptico, sedenomina efecto gloria.
Los efectos arco iris y gloria ocurren cuando más de unparámetro de impacto contribuye a un dado ángulo de dispersión.Cuánticamente esto produce una interferencia entre las distintasramas de p(e) que, tal corno veremos más adelante, se manifiestacomo oscilaciones con el ángulo de la sección eficaz.
174

BIBLIOGRAFIA
1. M. R. C. McDowell and J. P. Coleman, Introduction to the
Theory of Ion - Atom Collisions (North Holland, Amsterdarn,1970).
2. L. D. Landau and E. M. Lifshitz, Mechanics (pergamon Press,Oxford, 1976).
175


TEORIA CUANTICA DE COLISIONES
CONDICION ASINTOTICA
Comenzaremos discutiendo el proceso de colisión más simple:la dispersión elástica de una partícula sin espín por un centrode fuerzas fijo de potencial V(r). Mucho antes de la colisióndescribiremos al proyectil por un paquete de onda cp desplazadouna distancia p perpendicularmente a la trayectoria de colisiónfrontal. (En lo que sigue usaremos unidades atómicas)
con <ql~> fuertemente centrado en el impulso inicial q = p. Lejosdel centro de fuerzas este estado evoluciona libremente según laley
con H el Hamiltoniano de partícula libre.o
Si esta ausencia de interacción se mantuviera inclusivehasta el instante mismo de la colisión, que tomamosarbitrariamente como t=O, el vector de estado en ese instantesería I~. >. Sin embargo, la presencia del centro de fuerzas hace
inque el proyectil se mueva en una "órbita" distinta
u(t) I ~ > = e-iHt I ~ > , H H +Vo
que identificamos unívocamente por el correspondiente vector deestado I~> en el instante t=O.
Estamos suponiendo que, si el potencial es de corto alcance,antes y después de la colisión el movimiento es indistinguibledel de un paquete de onda libre, y está caracterizado por unoperador de evolución libre U (t). O sea
o
177

antes de la colisión
después de la colisión
U(t) 11/1>
U(t) 11/1>--) U (t) 11/1 >t~ 00 o out
A estos dos estados 11/1. > y 11/1> los llamaremos asíntotasIn out
entrante y saliente de la órbita U(t)II/1>,respectivamente.
Uo~\ ~\~,i~
~>
OPERADORES DE MOLLER
Definimos los operadores de Moller
que proyectan los estados asintóticos sobre la órbita a t=O.
11/1> = 0+ 11/1. > = O 11/1 >In - out
Debe destacarse que no toda órbita 11/1> representa a unestado de dispersión con asíntotas entrantes y salientes, ya quesi 11/1> es un estado ligado, nunca actuará libremente. En otraspalabras, los operadores de Moller mapean el espacio de Hilbert ~en el subespacio ~ de estados de dispersión conservando la norma
178

(oto = 1). Decimos que los operadores de Moller son isométricos,pero no -en general- unitarios. Sólo si el Hamiltoniano no tieneestado ligados, entonces ~ = ~ y O es unitario.
Los operadores de Moller verifican la relación deentremezclado
hacerasíntota
sinla
ser -en general-tienen el mismo
expresarentrante,
permitenasíntota
Mollerde la
operadores deen términos
Esta relación muestra claramente que O no puedeunitario, pues ello significaría que H y H
oespectro de energía. Llegamos nuevamente a la conclusión de queel operador de Moller es unitario sólo cuando H no tiene estadosligados.
Lossalientereferencia a la órbita a t=O
Il/J > = º tº+ Il/J . >out - ln
Este producto de operadores, que se suele denominar operadorde scattering S, contiene toda la información de interés físico.Si sabemos como calcularlo, el problema de colisión estaráresuel to. Ya veremos como hacer efectivo este cálculo y comoobtener, a partir de él, la sección eficaz de dispersión.
OPERADOR DE GREEN
La herramienta más importante en la teoría de colisiones esel operador estacionario de Green
-1G(z) = (z-H)
Es evidente que no está bien definido cuando z es igual a unautovalor del Hamiltoniano, es decir, sobre el eje real positivoy sobre algunos puntos del eje real negativo. Fuera de esa zonaG(z) es analítico. Además, siendo H hermítico, verifica
• tG(z ) = G(z) .
179

Los operadores de Moller y de Green están relacionados por(ver apéndice B.l)
con Ep
2 += p 12m y e ~ O •
Por ser de futura utilidad anotamos el elemento de matrizdel operador de Green libre G (z)=(z-H )-1 (ver apéndice B.2)
o o
±iplr-r' I~ ~ -m e<rlG (E ±ie) Ir'>=-2 -----o p Tl 11-1' I
Vemos que, debido al corte que presenta a lo largo del eje realpositivo, G (z) toma valores distintos al aproximarse según E +ie
o p
o E -ie.p
Conocer G(z) es equivalente a tener solucionado el problemade autovalores de H. Este es un problema muy difícil, por lo cualse buscan soluciones aproximadas. Tal como veremos más adelante,la clave de estas técnicas de aproximación es la ecuación deLippmann-Schwinger
G = G + G V Go o
G + G V Go o
que resulta de
G G G-1G = G (z-H ) G G (G-1+V) Go o o o o
ESTADOS ESTACIONARIOS DE DISPERSION
Como siguiente paso hacia la obtencióncalculamos la amplitud de probabilidad
~partícula dispersada tenga impulso k despues
180
de la sección eficaz~<kl~ > de que laout
de la colisión.

= I
donde hemos introducido los estados estacionarios de dispersióncomo las órbitas a t=O correspondientes a estados asintóticos deonda plana
I~> + G(E ±ie) V I~>p
Estos estados son autovectores del Hamiltoniano total
~ ~Hlp±> = E Ip±>p
y satisfacen la ecuación de Lippmann-Schwinger
Ip±> = I~>+G (E ±ie)VI~±>o p
Con estos resultados podemos completar el cálculo del elemento de~ t ~ ~ ~matriz del operador de Scattering <klº_ º+Iq> = <k-Iq+>
resultando (ver apéndice B.3)
~ ~ ;7 ~ .;7 ~<k-Iq+> = <l{lq>-21U<l{IVlq+>o(E -E )k q
o, definiendo el operador de transición T(z) = V+VG(z)V,
El significado de ambos términos en la expresión anterior esfácil de comprender. El primer término expresa la ausencia dedispersión. El segundo término representa al fenómeno de colisiónpropiamente dicho: el impulso de la partícula cambia, pero nocambia su energía cinética.
181

~Reemplazando en <kl~ > obtenemosout
Suponemos que la dirección de observación k es exterior a la~pequeña zona alrededor de la dirección de incidencia donde <kl~>
es no nulo.
J.-? -?
i7 . i7 ...• e-1q.p <-?qlA.> d'2<l\.11/1 > =-21U <l\.IT(Eq+H~) Ip> o(E -E ) 'f' Yout q k
SECCION EFICAZ CUANTICA
Ahora, por fin, estamos en condiciones de calcular laseCClon eficaz. Consideremos un flujo J de partículas de masa m eimpulso p que inciden sobre un centro de fuerzas. Cada proyectilque se acerca a dicho centro de fuerzas con un apartamiento prespecto de la trayectoria de colisión frontal tendrá una ciertaprobabilidad 1<1~11/1> 12 de ser dispersado con impulso ~. De estaoutmanera, el número de partículas dispersadas con impulso ~ porunidad de tiempo es J J(p) I <~11/1 > 1
2 d2p; o si consideramos unout
flujo J uniforme y N centros de fuerza
y, por la definición de la sección eficaz
...Reemplazando <kll/1 > en esta expresión obtenemos (apéndice B.4)out
182

Con q la componente del vector q en la dirección del impulso11 4inicial p. Suponemos ahora que la variación de la matriz de
transición es insignificante en la región donde l<ql~>12 esapreciablemente distinto de cero. Entonces podemos reemplazar
-t -t 21<kiT Iq> I /q 11 por su valor en el impulso inicial para obtener laexpresión final de la sección eficaz elástica
dO'dk
4m 4 42= (2n) - l<kIT(E +ü:)lp>1p p8(E -E )
k P
o, integrando sobre la variable energía,
dO"dO
donde E = E •k P
LIMITE ASINTOTICO DE LOS ESTADOS ESTACIONARIOS DE DISPERSION
En la expreslon anterior para la sección eficaz vemos quetodo el problema radica, ahora, en calcular el elemento de matrizde transición
<k IT( E +ic) Ip> = (~IVlp+>p
Un método usual para realizar ésto es estudiar el límiteasintótico de los estados estacionarios de dispersión ya que, talcomo mostramos en el apéndice B. 5, el estado estacionario dedispersión <rIP±> es, a grandes distancias, la superposición deuna onda plana y una onda esférica de amplitud proporcional a lamatriz de transición
-t -t<r Ip±> '" 1 ip-t r 2 I\-t------ [e . - (2rr) m <±prIVlp±>( 2rr ) 3/2
±iprer
Esta ecuación nos da una técnica paratransición, despejándola del desarrollosolución del problema de autovalores para
183
calcular la matriz de-t -tasintótico de <rlp±>,
el Hamiltoniano total.

BIBLIOGRAFIA
1. J. R. Taylor, Seattering Theory (John Wiley and Sons, NewYork, 1972).
2. A. Galindo y P. Pascual, Mecáni ea Cuánti ea (EudernaUniversidad, Madrid, 1989).
184

APROXIMACION DE BORN
LA SERIE DE BORN
Hemos visto que el cálculo de la sección eficaz elástica sereduce a la evaluación del elemento de matriz de transición
~ . ~ ~ ~< kiT (E +H~) Ip> = < k IVIp+ >
p
~ ~ ~Si iteramos en la ecuación de Lippmann-Schwinger Ip+>=lp>+GVlp+>o
obtenemos la serie de Born
~ . ~ ~ ~ ~ ~<kIT(E +lc)lp> = <klvlp> + <kIVG (E +ic)Vlp> +p o p
+ <kIVG (E +ic)VG (E +ic)Vlp> + ...o p o p
En muchos casos de interés práctico esta serie converge, y lohace con suficiente rapidez como para que sólo unos pocostérminos sean importantes. Existen teoremas de convergencia que,en general, resultan en condiciones de validez innecesariamenterestrictivas. Sin embargo, teniendo en cuenta que cada términoincluye potencias sucesivas del potencial y de la función deGreen -que en cierto sentido es inversamente proporcional a laenergía- podemos anticipar que la convergencia será buena parapotenciales débiles y energías altas.
PRIMERA APROXIMACION DE BORN
El primer término de la serie es
J. ~ k ~
<kiT Ip> = <klvlp> = (2rr) -3 e1(P-)·r V(r) dr =lB
= dr
Es posible obtener algunas propiedades importantes de esteprimer orden de aproximación sin necesidad de referirse a ningún
185
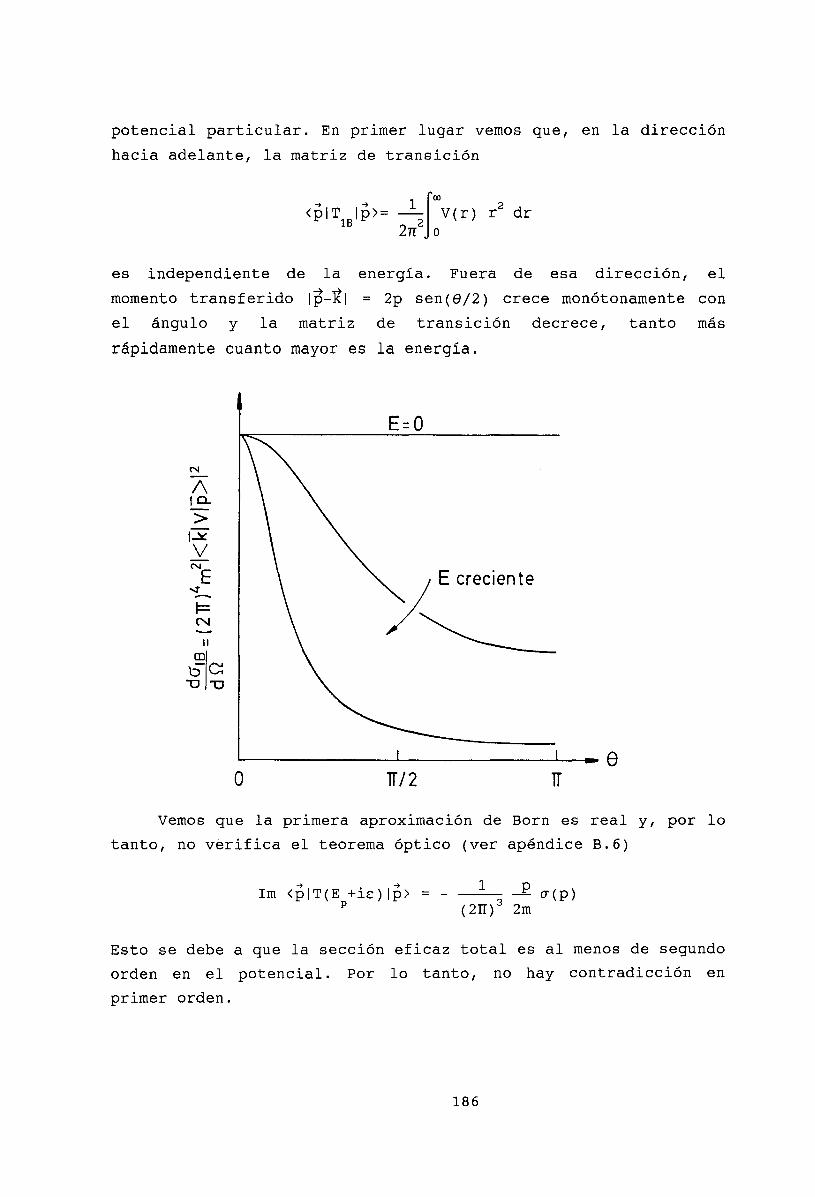
potencial particular. En primer lugar vemos que, en la direcciónhacia adelante, la matriz de transición
<plT Ip>= -l-IOOv(r) r2 drlB 2 2rr o
es independiente de la energía. Fuera de esa dirección, elmomento transferido IP-RI = 2p sen(8j2) crece monótonamente conel ángulo y la matriz de transición decrece, tanto másrápidamente cuanto mayor es la energía.
E=O
FN
11~Ic:-0-0
eo Tr/2 TT
Vemos que la primera aproximación de Born es real y, por lotanto, no verifica el teorema óptico (ver apéndice B.6)
1m <pIT(E +ic) Ip> = - 1 .....E <T(p)P (2II)32m
Esto se debe a que la sección eficaz total es al menos de segundoorden en el potencial. Por lo tanto, no hay contradicción enprimer orden.
186

POTENCIAL DE YUKAWA
Como primera aplicación de la serie de Born consideramos eld Z -r/apotencial e Yukawa V (r) = r e para el cual
~ ~<kiT Ip> =lB
Za2 12 ~ ~ 2 2
2rr 1+ Ip-k I a
La sección eficaz
dO'=dQ
4m2Z2a4
(1+IP_RI2a2)2
=4m2Z2a4
2 2 2 2(1+4p asene/2)
alturay
anteriormente,118'"lipa
indicamosancho
quede
propiedadesángulo ceroa
lastodasmuestraformando un picodO' I = 4 2Z2 4dQ m a.
oPara analizar la validez de esta aproximaclon deberíamos
~ -+compararla con el término de segundo orden <kIVGVlp>. Esteo
término puede calcularse exactamente sin mucha dificultad, peroen nuestro caso basta con conocer su expresión a ángulo cero
-+ -+<pIVG Vlp>o
Za2 mZa=-----2rr2l-2ipa
Vemos que, siendo
-+ -+<p!VG Vlp> mZao
I ~ ~ I =<p IVIp > / 1+ ( 2pa )2'
debería verificarse que mZa « 1 para que la aproximación de Bornfuese válida independientemente de la energía. Pero, como encolisiones atómicas típicas m, Z y a son del orden de la unidad omayores, esta condición no puede satisfacerse. En cambio, a altasenergías tenemos la condición
p/m » Z/2
que se satisface con un proyectil suficientemente rápido.
187

DISPERSION ELASTICA DE PARTICULAS CARGADAS RAPIDAS POR ATOMOS
Consideremos ahora el choque de un proyectil de carga Zp
contra un átomo de número atómico Z . Este es, en realidad, unB
problema de muchas partículas. Sin embargo es posible obtener unaaproximación razonablemente buena para la dispersión elástica sise considera a los electrones del átomo blanco como formando unadistribución fija de carga p(r), sin tener en cuenta efectos depolarización o intercambio. En esta aproximación estática elpotencial de interacción es
-7V(r)-7
p(r') )-dr'
-7 -7
I r-r' I
Reemplazando en la primera aproximación de Born, obtenemos
dO' (2mz p ) 2 -7 -7 2-dO = -IP-_-~-12 [ZB - F(p-k)]
donde-7F(q) = J
,-7 -7-7 lq.r -7p(r) e dr
es el llamado factor de forma atómico.Para átomos simples podemos describir la densidad de carga
por
3ex -2exrp(r) = Z - eB rr
con ex una carga efectiva obtenida variacionalmente (ex = 1 para elHidrógeno atómico, 1.69 para el Helio). El factor de formaresulta
F(q)
y la sección eficaz
188
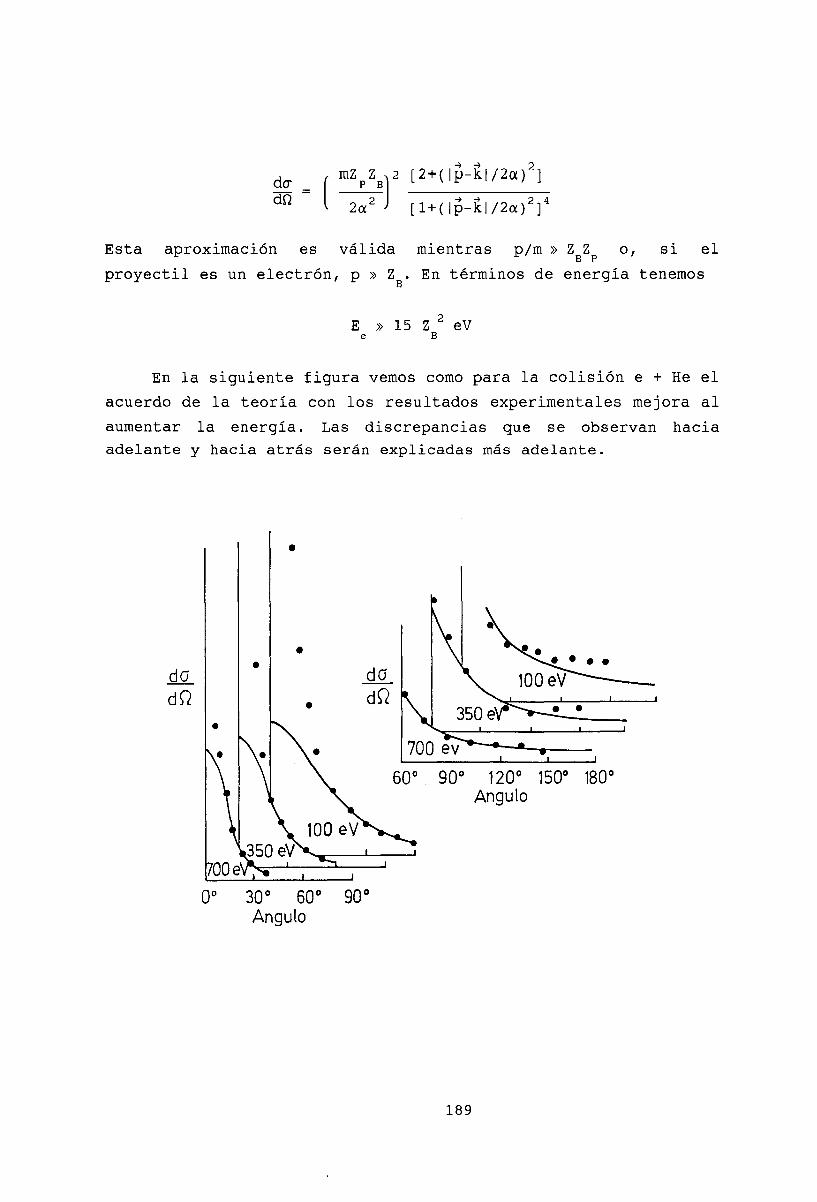
~ ~ 2[2+( Ip-kl/2a) ]
~ ~ 2 4[1+( Ip-kl /2cx) ]
Esta aproximación es válida mientras p/m» Z Z o, si elB Pproyectil es un electrón, p » Z . En términos de energía tenemos
B
E » 15 Z 2 eVe B
En la siguiente figura vemos como para la colisión e + He elacuerdo de la teoría con los resultados experimentales mejora alaumentar la energía. Las discrepancias que se observan haciaadelante y hacia atrás serán explicadas más adelante .
•
• •
•
900 1200 1500 1800
Angula
189

POTENCIAL COULOMBIANO
En la base de la teoría de colisiones desarrollada hastaaquí está la suposición de que mucho antes y mucho después de lacolisión el movimiento del proyectil es indistinguible del de unpaquete de onda libre o, equivalentemente, que los operadores deMoller están bien definidos. Puede demostrarse (M.N.Haek, NuovoCimento .2., 731 (1958)) que esta condición asintótica es válidasiempre que el potencial sea localmente integrable y verifiquerV(r) ~ O. Vemos que el potencial coulombiano V(r)=Zjr no está
r-7OOincluido en esta categoría y, por lo tanto, no se le puedeaplicar la teoría de colisiones. Para entender la naturaleza deesta dificultad conviene analizar el comportamiento asintótico deuna trayectoria coulombiana clásica.
En el límite r ~ 00 la órbita hiperbólica tiendeasintóticamente a una trayectoria rectilínea y su velocidad a unlímite bien definido -t9 = v2E/m. Sin embargo, separando los
00
diferenciales espacial y temporal en la ecuación de conservación~ 1.2 e2 Zde energla E = -2mr + -- + - obtenemos en el límite asintótico
2 rZ 1 2mr-t9 dt ~ (1+--- -)dr, que conduce al siguiente comportamiento
00 2m-t9 r
00
Zr ~ -t9 Itl - - lnltl + 0(1)00 2
m-t900
Vemos que se mantiene una diferencia logarítmica entre la órbitay su aproximación de movimiento libre. En otras palabras laórbita nunca converge a una trayectoria asintóticamente libre.
Teniendo en cuenta el resultado clásico anterior, es deesperar que las órbitas coulombianas cuánticas tampoco converjana estados asintóticamente libres, sino que mantengan unadiferencia logarítmica ya que, aproximadamente,
Jt Z
U (t) ~_ exp [-i (H o + -t9 I t I 1) dt'] =t~+oo o 00
-iH t .Zo 1
= e exp [±-- enltl]-t9
00
190

Vemos que el estudio de la dispersión por un potencialcoulombiano requiere modificar la teoría de colisiones desde sumisma base, es decir a partir de la condición asintótica. Enprincipio dicha condición es un requerimiento muy restrictivocuando se la compara con la situación experimental real. En lapráctica nunca se mide el vector de estado U(t)I~> para verificarsi está evolucionando libremente para t ~ +00. A lo sumo se mideun pequeño conjunto de observables como ser el impulso, laenergía cinética o el espín. Es cierto que si vale la condiciónasintótica usual entonces esos observables tienden a unaconstante para t ~ +00, pero la inversa es falsa. Esto sugiereconstruir una teoría de colisiones basada en una forma más débilde la condición asintótica, con el único requisito de que losobservables medidos sean asintóticamente estables. Esta teoríageneral, desarrollada por J.D.Dollard (J. Math. Phys. ~, 729(1964» logra absorber a la teoría usual, incorporando ademáspotenciales de largo alcance como el Coulombiano. El estudio deesta teoría supera el marco del presente curso. Nos limitaremos aindicar que la matriz de transición para un potencial coulombianopuede calcularse en forma exacta, resultando
t k ~ = _Z_ ['(1+in) 1 [ 4p 2 ] ine ( , p) 2 [' ( 1-in) ~ ~ 2 ~ ~ 22rr Ik-pl Ik-pl
con n = mZ/p. Si ahora reemplazamos en la sección eficaz,dO' 4 2 ~ ~ 2dQ = (2rr)m I te(k,p) I , obtenemos
Vemos que esta expresión coincide con la fórmula clásica deRutherford que obtuvimos en un capítulo anterior. Más aún, estaexpresión también coincide con la primera aproximación de Bornque resulta de tomar el límite a ~ 00 en el resultado obtenido enla sección anterior para el potencial de Yukawa. Esto último sedebe a que la matriz de transición se puede escribir como
191

~ ~ Z 1donde t1B
(k,p) = -- es la primera aproximación de Born y2rr2 Ik-p¡2
rjJ (n) es una fase que se anula para n = O. De esta manera se dauna muy extraña situación: Todos los órdenes de aproximación deBorn, que corresponden a desarrollar ei~(n) en potencias den = mZ/p, difieren del resultado exacto, a excepción del primerorden, que coincide exactamente con él.
BIBLIOGRAFIA
1. J. R. Taylor, Scattering Theory (Jahn Wiley and Sans, NewYork, 1972).
2. H. A. Bethe & W. Jackiw: Intermediate Quantum Hechanics(Benjamin publ., Reading, 1968).
192

COLISIONES DE BAJA ENERGIA
DESARROLLO EN ONDAS PARCIALES
Podemos desarrollar el estado estacionario de dispersión enpolinomios de Legendre
., .,<rlp+>
donde las funciones de onda ~fp(r) verifican la ecuación deSchrodinger radial
[d 2 _ f ( f +1) _ 2mV(r) + p2J ifJfp (r) = Odr2 r2
con la condición ifJn (O) = O Y la normalización{,p
rr"2 8(p'-p)
Reemplazando este desarrollo en ondas parciales dela matriz de transición, obtenemos
.,Ip±> en
con
~ . ~ ~ ~<kl T(Ep+u:) Ip> = <k IVIp+>00
-1 ¿ (2H 1) f f (p) Pt (1~.~)(2Tc)2m f=o
donde
2~ IOO ~~(pr) V(r) ~f (r) drp o p
~~(Z) = vrrz/2' Jt+~(Z)2
son las funciones de Ricatti-Bessel, solución de la ecuación deSchrodinger radial en ausencia de interacción. Es decir que~tp(r) = ~~(pr) para V(r) = O.
193

Finalmente, Sl reemplazamos este desarrollo en la seccióneficaz e integramos sobre el ángulo resulta
~ ~A partir del teorema óptico 1m <p\Tlp>
<r(p)
2qu e 1m f t (P ) = p \ f t (p) I ,debe ser de la forma
-1 P= -- 2m <r(p) vemos( 2rr) 3
por lo cual la amplitud de onda parcial
con 8t(P) una cantidad real llamada "desfasaje".
La importancia del desarrollo en ondas parciales radica enque, tal como veremos, a bajas energías sólo algunas fases 8t sonsignificativamente distintas de cero. Esto permite truncar eldesarrollo, obteniendo una solución aproximada de la matriz detransición que, a diferencia del desarrollo de Born, esautomáticamente consistente con el teorema óptico.
La forma más simple de calcular la matriz de transic~ón (osea, de hallar los desfasajes 8t) es en base a la formaasintótica de los estados estacionarios. Reemplazando la ondaplana y la matriz de transición en la forma asintótica
-> -><r Ip+> '"iprer
por sus correspondientes desarrollos en ondas parciales,obtenemos
-> -><rlp+> '" 1 1 00 • t'/lO i8t ipr A A)--- ¿ (2l+1) [l 'f'o(pr)+ e sen8t e ] pt(r.p(2rr)3/2pr t=o (,
o i(pr-trr/2+8t)por lo cual l/1t (r) ~ l/1t(pr)+ sen8t e o,
p r~ooreemplazamos l/1~(Z)por su forma asintótica l/1~(Z)'"sen(z-trr/2),
194
si

iOt---7 e sen(pr-trr/2+o
Q)
r~
Esta expresión da una manera de calcular los desfasajesresolviendo la ecuación de Schrodinger radial (con la condiciónde que la solución se anule en r = O) Y tomando el límiteasintótico para despejar OtO
CORRECCION DE LA APROXIMACION DE BORN
Si el potencial es muy débil la función de onda radial Wtpse aproxima a la solución de onda libre W~ y ello se refleja enque el desfasaje 0g lo hace a algún múltiplo entero de n. Pero si
1 iOgesto es así, la parte imaginaria de fi(p) = P e senoi esdespreciable frente a su parte real, y por lo tanto laaproximación de Born que reemplaza fi por un valor real
no ha de violar significativamente la condición de unitariedaddada por el teorema óptico. Entonces, y a partir de la ecuaciónde Schrodinger para Wgp' vemos que -tal como ya sabíamos- laaproximación de Born Wgp(r) '"W~(pr) es válida cuando elpotencial es débil comparado con la energía. Pero ahora vemos,además, que esto también es cierto para momentos angulares ialtos. Este último resultado es fácil de entender, pues cuandomayor es g más repulsiva es la barrera centrífuga y menor es laprobabilidad de que la partícula penetre en la zona donde elpotencial V(r) es apreciable. O sea que si a es el alcance del
i2potencial, el desfasaj e será próximo a cero para E «-- es
p 2ma2
decir para i» pa. Esto nos indica que una manera de mejorar laaproximación de Born consiste en corregir las ondas parciales debajo momento angular g < g '"pa
o
195

Para ángulos grandes el primer orden de Born decrece fuertementey esta corrección (que, en particular para l = O, no depende delángulo) es dominante.
PROPIEDADES DE LA AMPLITUD DE ONDA PARCIAL
Para p pequeño podemos reemplazar en
2~ JOO W~(pr) V(r) Wt (r) drp o p
tanto I/J~(pr)como I/Jep(r)por sus límites de baja energía, ambos. 1 H1 1 11proporclona es a p ,con o cua
o, equivalentemente,
0e(P) -----7 -ae p2H1 mod(rr)p-7ü
donde ae es una constante, necesariamente real, llamada "longitudde scattering". Vemos entonces que a bajas energías todas lasamplitudes de ondas parciales, excepto la correspondiente a laonda s, tienden a cero, y tanto más rápido cuando mayor es e.Esto justifica el interés práctico del método de ondas parciales,pues indica que a bajas energías sólo es necesario calcularalgunos pocos términos del desarrollo.
En general la indeterminación en módulo rr de los desfasajessuele eliminarse exigiendo arbitrariamente que 0e(oo)=O. Con estacondición puede demostrarse (Teorema de Levinson) que0e(O) = nerr,con ne el número de estados ligados de momento e.
V-3fe(P) = O(p ).
acotado.
resultado1Este
en
caso
el
Si
texto
es
Ver)
sigue
válidoV
O(1/r )
siendo
si
para
vál ida,
el potencial está exponencialmente
r~, entonces la expresión dada
e V-3excepto cuando > - , en cuyo
2
196
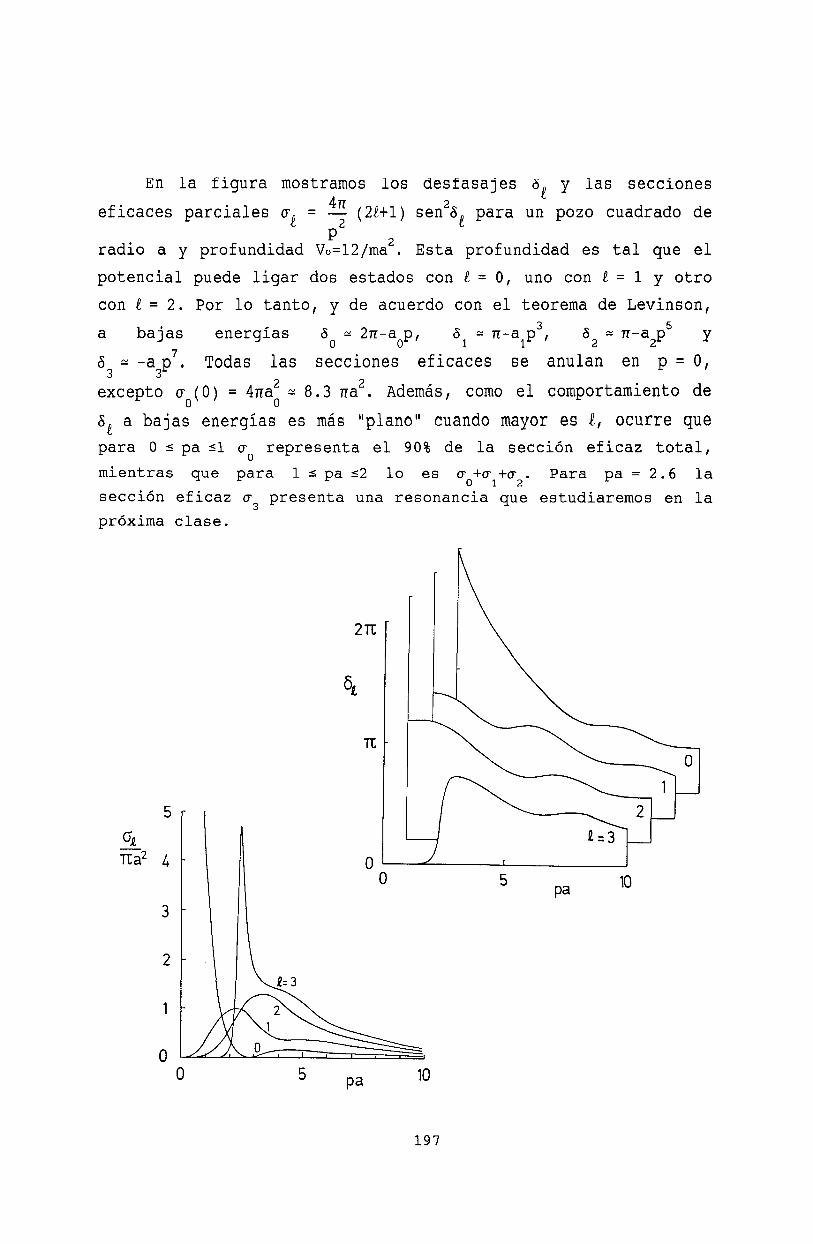
y
En la figura
eficaces parciales
mostramos los desfasaj es ot y las secciones41l 2(Jt = 2 (2H 1) sen o t para un pozo cuadrado dep
radio a y profundidad Vo=12/ma2• Esta
potencial puede ligar dos estados concon t = 2. Por lo tanto, y de acuerdoa
profundidad es tal que elt = O, uno con t = 1 Y otro
con el teorema de Levinson,3 5bajas energías o '" 2n-a p, o '" n-a p , o '" n-a po o 1 1 2 2
7Ó '" -a p. Todas las secciones eficaces se anulan en p = O,
3 3excepto (J (O) = 4ni ~8.3 ni. Además, como el comportamiento deo oSi a bajas energias es más "plano" cuando mayor es t, ocurre quepara O ~ pa ~l (J' representa el 90% de la sección eficaz total,omientras que para 1 ~ pa ~2 lo es (J' +(J' +(J'. Para pa = 2.6 la
012
sección eficaz (J' presenta una resonancia que estudiaremos en la3
próxima clase.
10pa5oo
n:O
5 2u.e 2=3rta2 4 O
O 5 10pa3
2
2rt
197
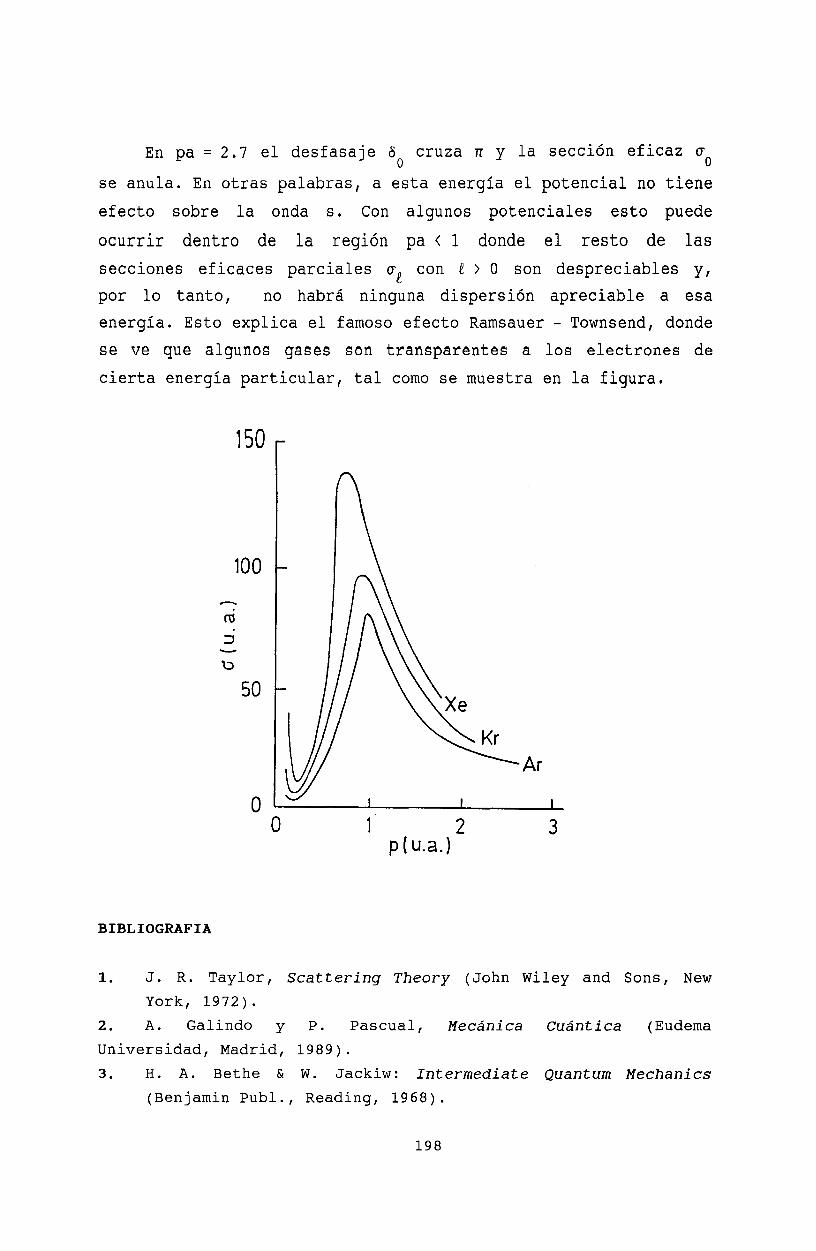
En pa = 2.7 el desfasaj e <5 cruza rr y la sección eficaz (jo ose anula. En otras palabras, a esta energía el potencial no tieneefecto sobre la onda s. Con algunos potenciales esto puedeocurrir dentro de la región pa < 1 donde el resto de lassecciones eficaces parciales (jl con l > O son despreciables y,por lo tanto, no habrá ninguna dispersión apreciable a esaenergía. Esto explica el famoso efecto Ramsauer - Townsend, dondese ve que algunos gases son transparentes a los electrones decierta energía particular, tal como se muestra en la figura.
150
100
50
o
BIBLIOGRAFIA
o 2p (u.a.)
Ar
3
1. J. R. Taylor, Scattering Theory (John Wiley and Sons, NewYork, 1972).
2. A. Galindo y P. Pascual, Mecánica Cuántica (EudemaUniversidad, Madrid, 1989).3. H. A. Bethe & W. Jackiw: Intermediate Quantum Mechanics
(Benjamin publ., Reading, 1968).
198

RESONANCIAS
LA FUNCION DE JOST
Introducimos otra solución cP lp (r) de la .... deecuaClonSchrodinger radial que difiere de Wlp (r) en su normalización.Mientras que para Wlp(r) pedíamos que ~lp(O) = O Y
Joo *Wl ,(r) Wl (r) dr = ; ~(p'-p)o p p
a esta nueva función le exigimos que
Estas funciones ~lp(r) deben ser reales, ya que lo son laecuación y la condición de contorno, y proporcionales a lasfunciones de onda radiales normalizadas
donde el coeficiente de proporcionalidad Fl(P) se denomina*función de Jost. Puede demostrarse qu~ Fl(P) = F1(-p). Además, en
lOlel límite asintótico ~lp(r) <:::: Fl(P) e sen(pr-l71/2+o1) y por lotanto, como ~lp(r) es real, la función de Jost debe verificar que
en módulo 7l
CEROS DE LA FUNCION DE JOST y ESTADOS LIGADOS
Reescribimos el límite asintótico de ~lp(r) como
-i (pr-l71/2) i (pr-l71/2)e - F1( -p) e ]
y continuamos analíticamente Fl(P) al plano complejo. Supongamos
199

ahora que Fg(P) tiene un cero en algún punto Po = ia del semieje
imaginario positivo. Entonces
-ar-ilrr/21 .
~lp (r) ~ - '2 Fl (-la) eo
tiende a cero para r ~ 00 y, por lo tanto, representa una soluciónnormalizable de la ecuación de Schrodinger con momento angular ey energía negativa -ci 12m. Es decir que es un estado ligado.Vemos entonces que existe una relación entre ceros de la funciónde Jost en el semieje imaginario positivo y estados ligados.
Supongamos ahora que multiplicamos al potencial por unparámetro de acoplamiento A. Si, a partir de A = 1, disminuimoseste parámetro, la capacidad del potencial para mantener estadosligados disminuye y los ceros de Ft<P) se mueven hacia el origen.Puede demostrarse que, cuando t = O, los ceros cruzan el origen yse meten en el semiplano inferior siguiendo el eje imaginario.Estos ceros ya no representan estados ligados y se los llama11 estados virtual es ". Cuando t > O, en cambio, por cada cero en eleje imaginario positivo hay otro en el eje imaginario negativo yambos alcanzan el origen para el mismo valor de A. Si se siguedisminuyendo A, estos ceros se separan del origen moviéndosetangencialmente al eje real, dentro del semiplano inferior. Pormotivos que veremos enseguida, al cero de la derecha se lodenomina resonante.
plano p
.Q=o
estado ligado
estado virtu al
200
plano p
•
estado ligado
• •resonancia
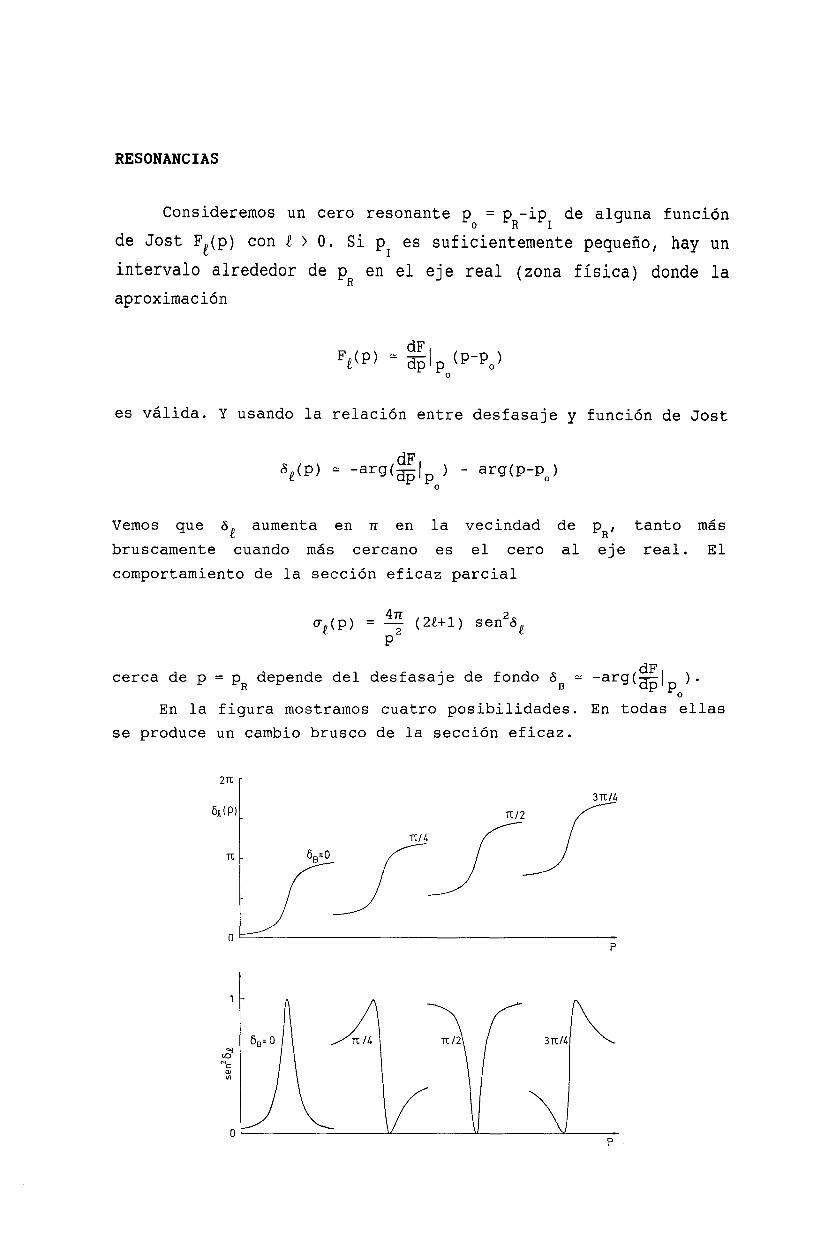
RESONANCIAS
Consideremos un cero resonante p = p -ip de alguna funcióno R 1
de Jost Ft(p) con t > O. Si PI es suficientemente pequeño, hay unintervalo alrededor de p en el eje real (zona física) donde la
Raproximación
es válida. y usando la relación entre desfasaje y función de Jost
Vemos que °t aumenta en rr enbruscamente cuando más cercano
la vecindad dees el cero al
tantoreal.
másEl
comportamiento de la sección eficaz parcial
4rr 2<Tt(P) = 2 (2t+l) sen 0t
p
dFcerca de p = PR depende del desfasaje de fondo 0B ~ -arg(dplp)'o
En la figura mostramos cuatro posibilidades. En todas ellasse produce un cambio brusco de la sección eficaz.
2n:3n:/4
n:/4
n:
op
p
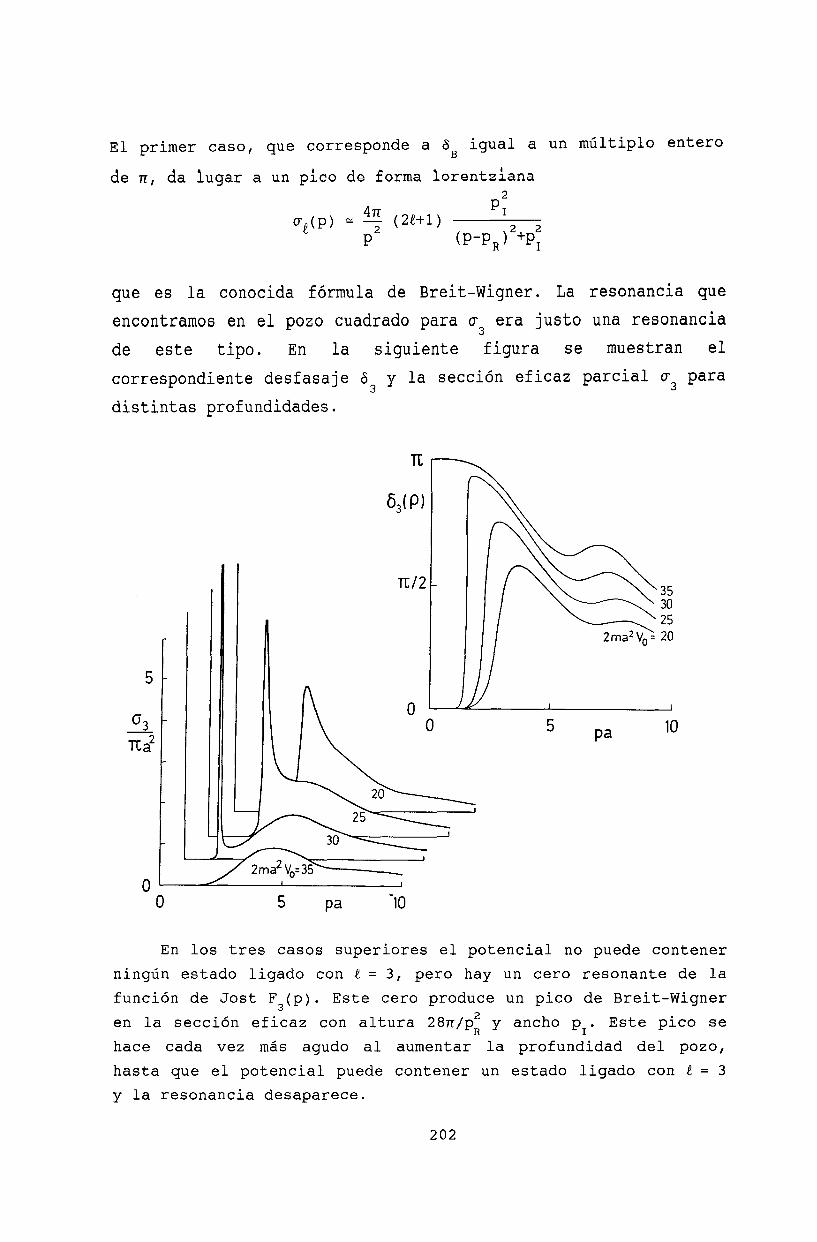
~ 4rr (2e+ 1)2
P
El primer caso, que corresponde a 0B igual ade rr, da lugar a un pico de forma lorentziana
2PI
(p_p )2+p2R 1
un múltiplo entero
que es la conocida fórmula de Breit-Wigner. La resonancia queencontramos en el pozo cuadrado para ~ era justo una resonancia
3
de este tipo. En la siguiente figura se muestran elcorrespondiente desfasaje o y la sección eficaz parcial ~ para
3 3
distintas profundidades.
10
353025
2ma2yo= 20
pa5
TI:
rr/2
pa5
5
OO
En los tres casos superiores el potencial no puede contenerningún estado ligado con e = 3, pero hay un cero resonante de lafunción de Jost F (p). Este cero produce un pico de Breit-Wigner
3
en la sección eficaz con altura 28rr/p2 y ancho p. Este pico seR 1hace cada vez más agudo al aumentar la profundidad del pozo,hasta que el potencial puede contener un estado ligado con e = 3Y la resonancia desaparece.
202

Físicamente las resonancias ocurren como una capturatransitoria del proyectil en un egtado metaegtable de energía
2p 12m del cual es posteriormente emitido, con un decaimientoR
exponencial de vida media T ~ Lii~ PmLip~ PmPR R 1
RESONANCIA DE ENERGIA CERO
En la sección anterior Vlmos como los ceros de la función deJost de momento angular l > O podían producir una variaciónbrusca de la sección eficaz parcial correspondiente. Veamos ahoraque ocurre cuando l = O. En este caso sabemos que los ceros estánubicados sobre el eje imaginario. Sea entonces p = i/a el ceroo omás cercano al origen. Tenemos que, para p pequeño,
F (p) '"i(3(p-i/a )o o
donde puede demostrarse que la constante (3 es real. De estamanera la amplitud de dispersión es
f (p)o
io1 e °senop o
i'" -p--~i-/a-
o
de donde deducimos que a = -f (O) es la longitud de scatteringo o
de onda s. Finalmente, reemplazando en la sección eficaz resulta
(J' (p) 2= 4rrIf (p) I '"o o
4rra2o
'" 1+p2a2o
Vemos que la sección eficaz presenta un máximo en el origen quees tanto más agudo cuando mayor es la longitud de scattering a ,
o
es decir, cuando más cerca se encuentra el cero p = i/a delo oorigen. Este tipo de resonancia se denomina de "energía cero". Esimportante señalar que estos resultados son independientes de que
203

el cero corresponda a un estado ligado ( Imp > O ) o un estadoo
virtual ( Imp < O ). Una resonancia de este tipo se observa en elo
caso ya analizado del pozo cuadrado de radio a y profundidadV = 12/ma2
, donde la sección eficaz (J' (O) es varias veces mayoro oque la sección geométrica. El ejemplo más conocido se da en elsistema n-p singlete donde la sección eficaz es 200 veces mayorque cualquier estimación razonable de su tamaño geométrico.
BIBLIOGRAFIA
1. J. R. Taylor, Scattering Theory (John Wiley and Sons, NewYork, 1972).
204

TEORIA SEMICLASICA DE COLISIONES
APROXIMACION SEMICLASICA
Volvamos al desarrollo en ondas parciales
dO' =dO1 00 2io t 2
I ¿ (2H 1) (e -1) Pn (cose) I4p2 t=O <-
Sabemos que sólo aquellos términos con t < pa, siendo a elalcance del potencial, dan una contribución significativa. Sinembargo, usualmente, pa es un número muy grande, y se necesitancien o más términos para lograr la convergencia del desarrollo.Por esto se buscan aproximaciones que lo simplifiquen. Porejemplo, para ángulos de dispersión no muy pequeños podemosreemplazar los polinomios de Legendre por sus expresionesasintóticas
t sene » 1
Además, y siempre que el desfasaje 0t sea una función suave de t,podemos reemplazar la suma por una integral, con lo cual
1 I {Xl 2t+1
2rrp2sene o Vl2iot
(e -1) cos
Debemos tener presente que esta aproximación no es válida paraángulos pequeños, y no puede representar correctamente lacontribución de las ondas parciales de bajo orden. Vemos que elintegrando está compuesto por términos fuertemente oscilantes
1 I {):) 2t+1
2rrp2sene o Vl
excepto en las regiones de valores de t donde el exponentepresenta una variación suave con t. En los dos últimos términos
205

esto ocurre para ángulos pequeños e ~ o, es decir fuera de la
zona de validez de la expresión anterior. En los dos prim~rostérminos, en cambio, la condición de "fase estacionaria" es(extendemos el ángulo e a valores negativos para eliminar laindeterminación en el signo)
o sea que sólo ciertas ondas parciales l. (dadas por esta1
ecuación) contribuyen significati vamente a la dispersión en un
dado ángulo e. Esto es análogo a lo que ocurre en la teoría
clásica, donde sólo ciertas trayectorias (representadas porparámetros de impacto Pi=li/p) contribuyen a la dispersión en unadirección dada.
De hecho si para cada punto estacionario i. aproximamos1
aOl I 1o ""-" + - (l-l) + -l li al I l . 2i
obtenemos
L [-2_l -de-] 1/2
p sene dllli
i(20n - i.e)<- 1 12e i
o, en términos del parámetro de impacto P.=i./P,1 1
dO' ~dn -
P . dp. 1/2 i (2 o n - l. e ) 21 1 <- 1L [sene de] e i I
Esta expresión es idéntica a la sección eficaz clásica,excepto que ahora puede producirse un efecto de interferenciaentre las distintas trayectorias que contribuyen a la dispersiónen un cierto ángulo. Este es un fenómeno que ya habíamosmencionado al discutir los efectos gloria y arco iris.
Tampoco tienen porque ser similares la relación entre elángulo de dispersión y el parámetro de impacto dada por la
aocondición de fase estacionaria 2 al l = e, y la obtenida
206
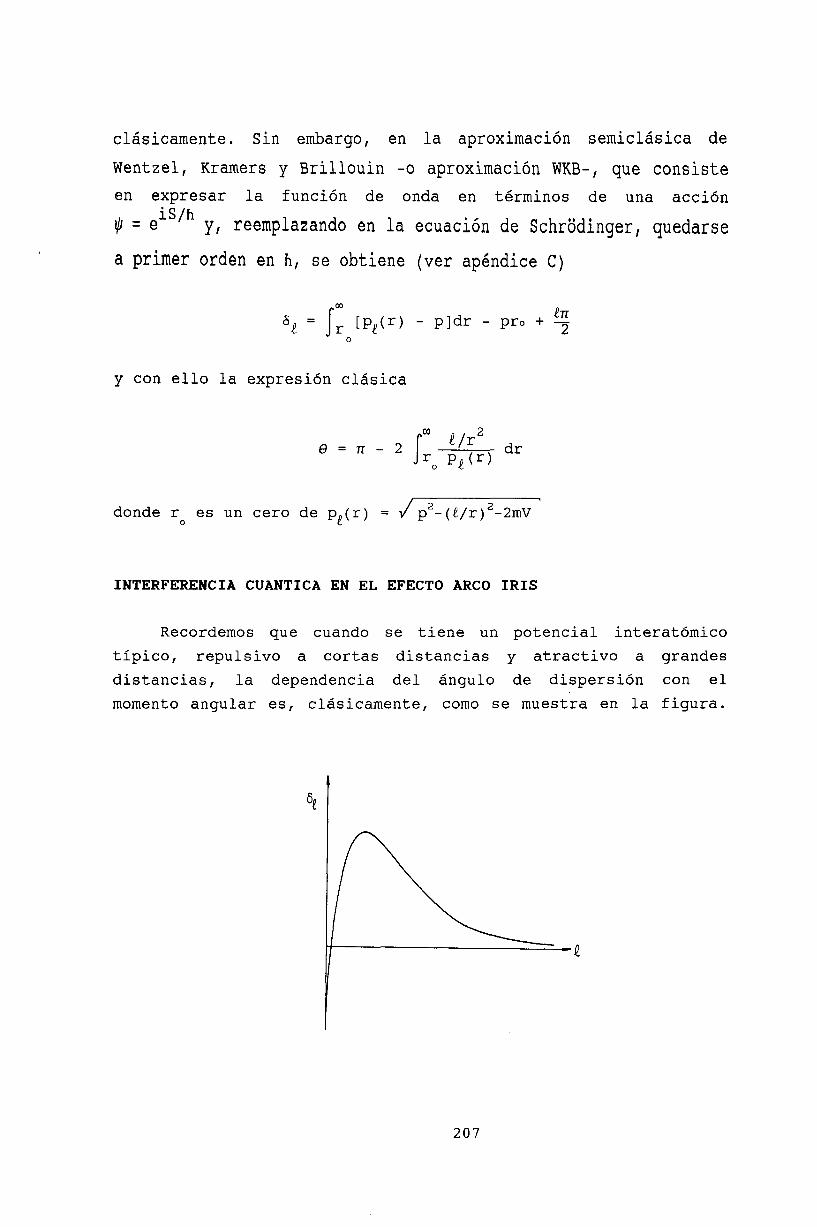
quedarseecuación de Schrodinger,(ver apéndice C)
clásicamente. Sin embargo, en la aproximación semiclásica deWentzel, Kramers y Brillouin -o aproximación WKB-, que consisteen expresar la función de onda en términos de una acción
iS/hW = e y, reemplazando en laa primer orden en n, se obtiene
fOO ln0l = r [Pl(r) - p]dr - pro + 2
o
y con ello la expresión clásica
e = n - 2 JOOr_R _/_r_2_ dro p l (r)
INTERFERENCIA CUANTICA EN EL EFECTO ARCO IRIS
Recordemos que cuando se tiene un potencial interatómicotípico, repulsivo a cortas distancias y atractivo a grandesdistancias, la dependencia del ángulo de dispersión con elmomento angular es, clásicamente, corno se muestra en la figura.
Q
207
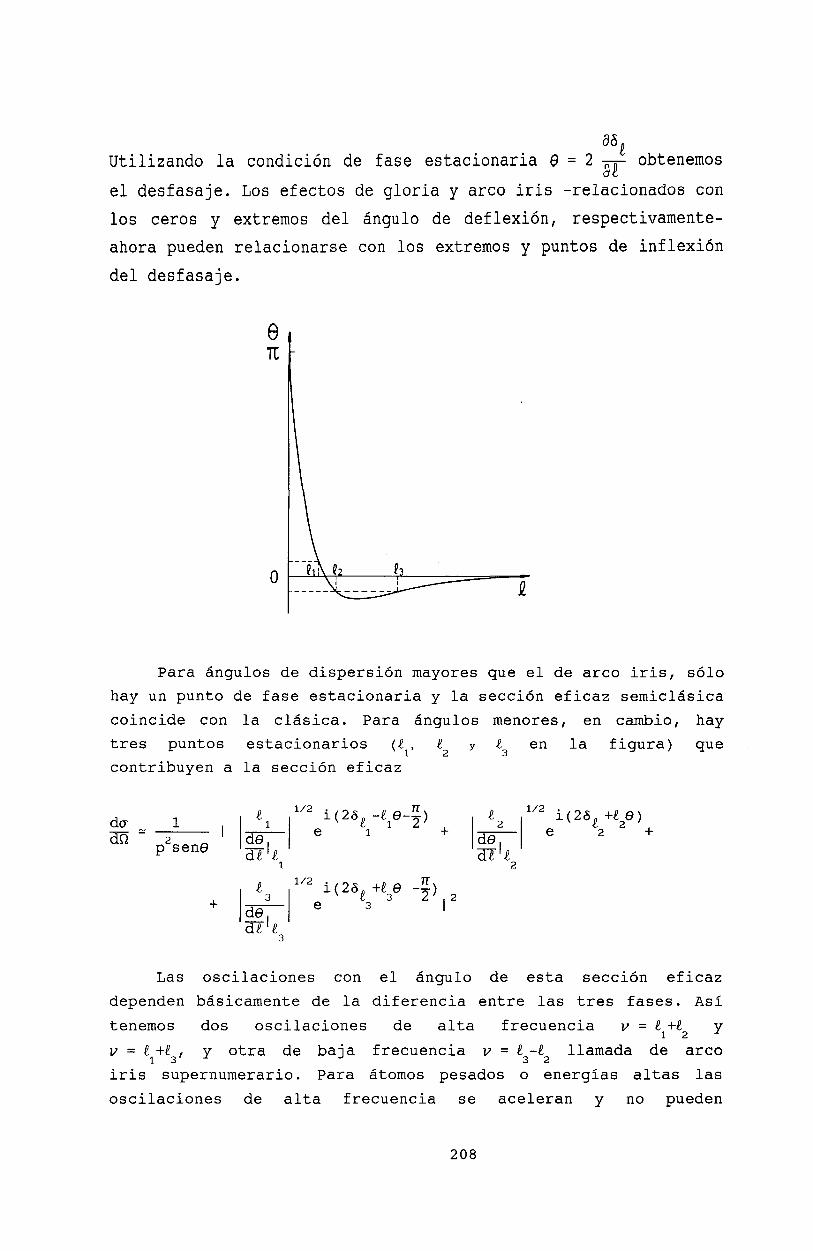
BOlUtilizando la condición de fase estacionaria e = 2 8r obtenemosel desfasaje. Los efectos de gloria y arco iris -relacionados conlos ceros y extremos del ángulo de deflexión, respectivamente-ahora pueden relacionarse con los extremos y puntos de inflexióndel desfasaje.
ere
oQ
Para ángulos de dispersión mayores que el de arco iris, sólohay un punto de fase estacionaria y la sección eficaz semiclásicacoincide con la clásica. Para ángulos menores, en cambio, haytres puntos estacionarios (e,
1
contribuyen a la sección eficaze
2y e
3en la figura) que
rr--)2 I 2
12P sene
+
e 1/2 i(2o -e e-!!.)I de 1 I e el 1 2 +de le
1
i 1/2 i (2 o +i eIde3 I e i3 3
de le3
Las oscilaciones con el ángulo de esta sección eficazdependen básicamente de la diferencia entre las tres fases. Así
y
laspueden
altasno
11 = i +i1 2
llamada de arcofrecuenciaaltadeoscilacionesdostenemos
v = i +i, y otra de baj a frecuencia v = i -i1 3 3 2
iris supernumerario. Para átomos pesados o energíasoscilaciones de alta frecuencia se aceleran y
208
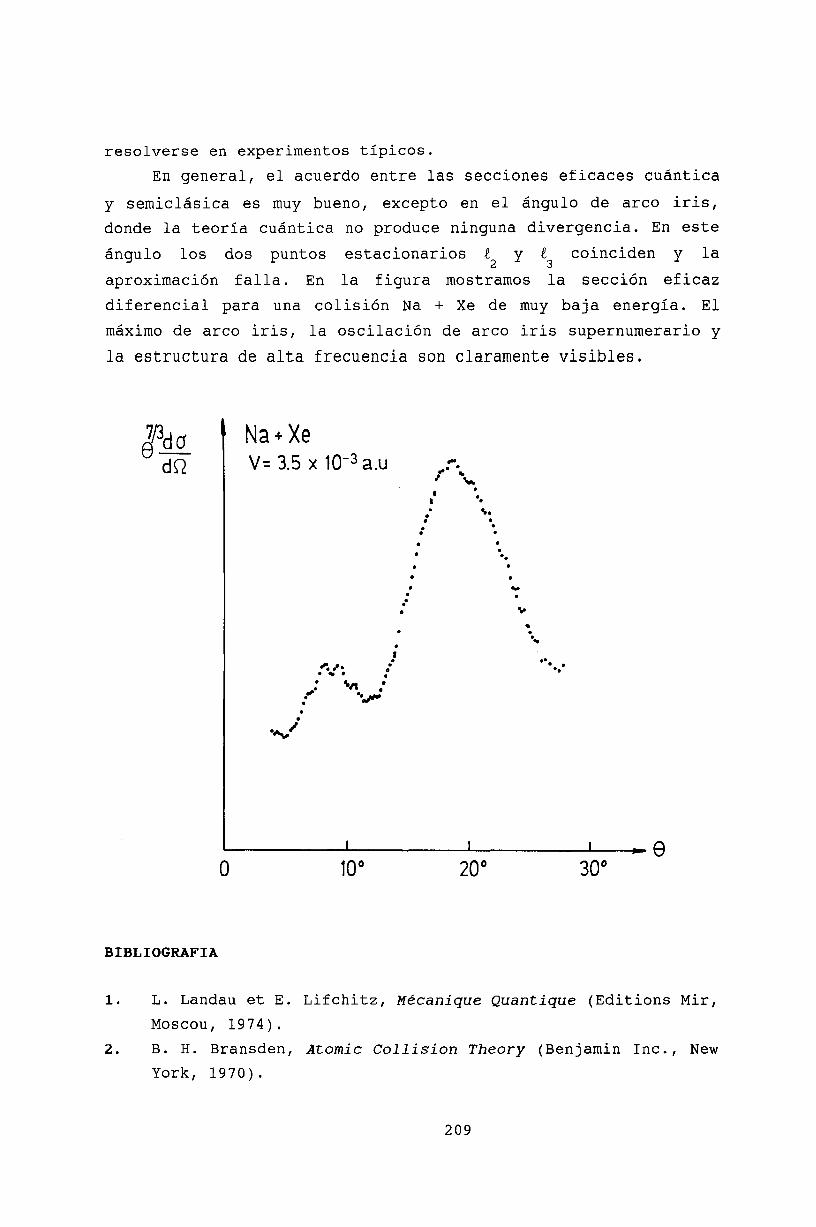
resolverse en experimentos típicos.En general, el acuerdo entre las secciones eficaces cuántica
y semiclásica es muy bueno, excepto en el ángulo de arco iris,donde la teoría cuántica no produce ninguna divergencia. En esteángulo los dos puntos estacionarios t y t coinciden y la2 3aproximación falla. En la figura mostramos la sección eficazdiferencial para una colisión Na + Xe de muy baja energía. Elmáximo de arco iris, la oscilación de arco iris supernumerario yla estructura de alta frecuencia son claramente visibles.
Na + XeV= 3.5 X 10-3 a.u •••~..,
I "'".• •, .
~....•.'..
....•
·•·~... :.... .. .." ..",. .• "JP
.••••
...........
... ...
l..--------IL-- __ ---L --L..__ 8o
BIBLIOGRAFIA
1. L. Landau et E. Lifchitz, Mécanique Quantique (Editions Mir,Moscou, 1974).
2. B. H. Bransden, Atomic Collision Theory (Benjamin lnc., NewYork, 1970).
209


DISPERSION DE PARTICULAS IDENTICAS
DESCRIPCION DE UN PROCESO DE DISPERSION ELASTICA EN EL SISTEMA DELABORATORIO
Hasta ahora hemos estado describiendo procesos de colisiónde un proyectil contra un centro de fuerzas que permanece fijo.Aunque esta es una buena aproximación cuando el blanco tienemucha más masa que el proyectil, en general ha de retroceder porefecto de la colisión. En tal caso la colisión de una partículade masa m contra otra de masa m inicialmente en reposo puede
P B
describirse muy simplemente en el sistema centro de masa como ladispersión por un centro de fuerzas de una partícula de masareducida m =m m / (m +m) y velocidad igual a la velocidad
P B P B
relativa. De esta manera el problema de colisión se reduce alcaso que hemos estado describiendo hasta ahora de la colisión deuna partícula por un centro de fuerzas fijo. Por conservación deimpulso y energía es fácil ver que si e es el ángulo dedispersión en el sistema centro de masa, el ángulo de dispersióndel proyectil en el sistema de referencia del laboratorio, dondela partícula blanco se encuentra inicialmente en reposo es
m senee arctg __ B _P m +m coseP B
Vemos que si m < m el proyectil puede ser dispersado, enP Bprincipio, en cualquier dirección. En cambio si m > m, sólo
P B
podrá ser dispersado en un ángulo que no exceda un cierto valorm
máximo, e ~ arcsen B • Además puede demostrarse que el átomoP mP
blanco retrocede en una dirección eB = ~(rr-e)
Calculada la sección eficaz en el sistema centro de masadO'dO ' el pase al sistema de laboratorio sólo requiere cambiar e
CM
por e , o seaP
211

resultando
dO'
dOSL
dO dO'CM= =dO dOSL CM
2n sene de dO'
2n sene de dQP P CM
dO' m- = [28(m-m) P cose + [2-8(m-m)]dO BPm P BP
SL B
2l+(m 1m ) cos2e ]
P B P
/ l-(m 1m )2sen2e 'P B P
dO'
dOCM
con 8(x) = 1 si x ~ O Y 8(x) = O si x < O.En particular, si ambas partículas tienen igual masa resulta
eP
1= - e2 y 1e = - (n-e)B 2
o sea que después de la colisión las partículas se mueven endirecciones que forman un ángulo recto entre ellas.
Además la sección eficaz en el sistema de laboratorioresulta
con e :S rr/2.p
DISPERSION DE PARTICULAS IDENTICAS
Hasta ahora hemos estudiado colisiones donde las partículasactivas pueden considerarse distintas. Sin embargo, hay muchosexperimentos que no satisfacen esta condición (colisiónelectrón-electrón o electrón-átomo). Hay varias maneras deconstruir una teoría de colisiones de partículas idénticas. Porejemplo, a través de un formalismo de segunda cuantización. Sinembargo aquí desarrollaremos un método más directo basado en laadecuada simetrización de los resultados obtenidos hasta ahora.Veamos como afecta este requerimiento ados partículas idénticas de espínadecuadamente simetrizado es
212
la colisión elástica des. El estado inicial

li> = 1
VI:'( IP ID ID > + (-1) 2S I - P ID ID »
1 2 2 1
donde ID Y ID son las proyecciones de espín de ambas1 2
partículas. Similarmente el estado final correspondiente a la
dispersión indicada en la figura
debe simetrizarse con el estado obtenido al intercambiar ambaspartículas
o sea que el estado asintótico adecuado es
I f > = _1_ (1 ~ m'm' > + (-1) 2S I- ~ m'm' > )V2"' 12 21
Ahora la sección eficaz resulta
d<T = (2rr) 4m2, < f IVI i> ,2 =dO4 2 ~ ~ 2S ~ ~ 2= (2rr) m I<k m'm' ITlp mm > + (-1) <-k m'm' ITlp mm > I
12 12 21 12
213

~ ~ ~ ~donde hemos usado que <k m'm' ITlp m m > = <-k m'm' ITI-p m m >. Si12 12 21 21
la interacción no depende del espín, entonces
dO'=dQ
Finalmente, si los haces iniciales no están polarizados y nomedimos la polarización final obtenemos
dO'=dQ
42 1 ~ ~ 2S ~ ~ 2(271) m --2 L / <kITlp>om m,om m,+(-l) <-k/Tlp>om m,om m' I
(2s+1) m mil 2 2 1 2 2 11 2
m'm'1 2
o sea
25= (21T)4m2[I<kITlp>I+I<_kITlp>12+ 2(2~ll Real«kITlp><-kITlp>'")]
Los dos primeros términos representan el resultado que hubiesemosobtenido clásicamente sumando las probabilidades para ambosprocesos indistinguibles. El término de interferencia es deorigen puramente cuántico.
Si la interacción es coulombiana obtenemos la llamadafórmula de Mott
14sen 8/2
+1 (_1)254 + 2 2s+1cos 8/2
Z 2COS(p¡m ln(cot 8/2» ]2 2sen 8/2 cos 8/2
Vemos que el término de interferencia se superpone en formaoscilatoria al resultado clásico. En particular la sección eficazcuántica para la dispersión en ángulo recto es el doble de laclásica para partículas de espín O. Este efecto fue verificado en1930 por Chadwick para la colisión entre partículas alfa (nucleosde Helio). Cuando las partículas tienen espín 1/2, en cambio, lasección eficaz para la dispersión en ángulo recto es nula. Estosresultados ilustran el hecho de que aún interacciones que sonindependientes del espín conducen a resultados que si dependen deél.
214

La sección eficaz anterior Duede convertirse fácilmente alsistema de laboratorio, resultando
2 Z 2
dO'(m
2:l [ 1 1 cos( - ln(cot B ))]±
v p cosedO = +4 4 2 2 PSL sen e cos e sen e cos e
p p p p p
con e ::::rr/2 y v la velocidad inicial del proyectil de masa m.p p
BIBLIOGRAFIA
1. J. R. Taylor, scattering Theory (John Wiley and Sons, NewYork, 1972).
2. L. D. Landau et E. M. Lifshitz, Mécanique Quantique
(Editions MIR, Moscou, 1974).3. A. Galindo y P. Pascual, Mecánica Cuántica (Eudema
Universidad, Madrid, 1989).
215


APENDICE A
CUANTIFICACION DEL CAMPO DE RADIACION
R. G. Pregliasco
Vamos a cuantificar el campo de radiación. El procedimientoserá el de escribir el hamiltoniano de forma tal que lacuantificación sea inmediata. Desarrollamos el potencial vector
"como una suma de ondas planas monocromáticas de polarización e(donde se sobrentiende el tomar la parte real)
~ A i(k.r-wt)A=A ee
o
y reemplazando los campos eléctrico y magnético
E - - ( BA/Bt = i(WA
"8= rot A= ikxA
en la siguiente expreslon para la energía del campo de radiaciónen un dado volumen V,
resulta
Finalmente, definiendo las "coordenadas normales"
1+- ( -iwt * iwtq = "2 A e + A eo o
1+- (w -iwt * iwtP TI A e A e
o o
217

obtenemos ... ¡magial
H l( 2+ 22)= '2 p w q
Esta es la ecuación de un oscilador armónico unidimensional demasa unitaria. Las coordenadas q y p son cantidades canónicamenteconjugadas. En efecto, tenemos que p = dq/dt y por lo tanto dq/dt= aH/ap. En forma similar se demuestra que dp/dt = - 8H/aq. Lacuantificación se logra 11 imponiendo 11 a estas coordenadascanónicamente conjugadas las relaciones de conmutación cuánticas
[q ,q = [p ,p ] = o
[q ,p = ih
Aunque esta cuantificación es simple y plausible, nohacerse a priori un juicio sobre su validez. Sininternamente consistente y, tal como veremos,adecuadamente los fenómenos electromagnéticos.
es posibleembargo es
describe
-iwtVemos que las relaciones entre q, p y A e presentan unao
gran semejanza formal con las definiciones de los operadores decreación y destrucción del oscilador cuántico,
t -1 (p + i w q)a =v'2hw
1 (p i w q)a = -v'2hw
que verifican las siguientes relaciones de conmutación
[a ,a ] t t O[a ,a ] =
[a t 1,a ]
•Esto nos lleva a definir los operadores A y A que no conmutano o
y están dados por
218

A = /2 h, ao C (2 V W
o
lI</ 2 hA t
A = ao C (2 V W
o
De esta manera resulta:
lI< 2;\ hA .A = -- No o (2C W
o
tcon N = a ·a el operador de número cuyos autoestados In>, talesque N In> = n In>, verifican
atln> = v-n+I In+l>
a In> = v-n- In-l>
donde en la última igualdad aln> = O si n = O.
lI<La expresión anterior para A.A es la misma con lao o
introdujimos el número de fotones N en la teoría semiclásicapágina 53). Sustituyendo en la expresión del Hamiltonianocampo electromagnético resulta H = hw (N + 1/2).
que(verdel
Vamos a hacer de nuevo parte del cálculo de emisión yabsorción de radiación con el campo electromagnéticocuantificado. El hamil toniano de partida era, en la medida deCoulomb (ver página 51):
p2 -7 eH = 2 m + V(r) + ( m A-7 -7 + r2 e A-7 A-7l1<
.p '" 2 m .
campos
Corno elatat, atafotones.
-7con la única novedad de que ahora A es un operador.último término contiene productos proporcionales a aa,y aat se dice que involucra transiciones de dosNuevamente podemos despreciarlo en una situación de
219

débiles. La emisión y la absorción pueden tratarse de la mismaforma que antes, resultando
dWif ~ ~ 2ilLr A ~
I i ni> I ()(dt ()( I <f n I A e e.pf o
I<nflalni> <fl;7 ~ 2ilLr A ~
li>\()( e e.p
ydWifdt ()(
respectivamente. El primer factor de cada término es exactamenteel elemento de matriz de los operadores de destrucción y
creación, que son diferentes de cero, sólo en los siguientescasos
Emisión n = n + 1f i
l<n+lla In>12 = n+l
Absorción n = n - 1f i
De esta manera queda justificada la irrupción del n+l en lugar den para el caso de la emisión.
220

=
APENDICE B
CALCULOS DE LA TEORIA CUANTICA DE COLISIONES
1 RELACION ENTRE LOS OPERADORES DE MOLLER y DE GREEN
Para relacionar los operadores de Moller y de Green anotamos
o± = Ur:: U(t)tuo(t) = J Ur:: ei(H-Ep)tIP><PI dpt~+ 00 t~+oo
Reemplazamos ahora el límite t ~ +00por una integral de Bochner
tL~ f(t) = +e I+OOe±etf(t) dt (J. Howard: J. Math. Anal. Appl.t~+oo o
+20,22 (1967)), con e-+O.
+eI ¡I:OOei(H-Ep.ie)t dt] IP><PI dp =
= +eIi(H-~~+ie) IP><PI dp =
que también podemos escribir
2. MATRIZ DE GREEN LIBRE
El elemento de matriz del operador de Green libreG (z)=(z-H )-1 está dado poro o
221

<tlG (z) /t' > = J <tlG (z) I~> d~ <~It'> =o o
J'K (-7 -7,)
1 1. • r-re d~= =(2rr)3
2z -k 12m
2im 1 r ei:1t-t. I k dk=(2rr)2 It-t' I -00 k -2mz
Considerando Im(z) '* O, esta integral puede evaluarse porresiduos,
iv2mz It-t' I-m e2rr --11---1-'-1--
Como caso particular, en el límite Im(z) -7 0, obtenemos
±ipI1-1' I-m e= -----2rr 1-7 -7, 1r-r
3. ELEMENTO DE MATRIZ DEL OPERADOR DE SCATTERING
Calculamos
~ ->< k-I P+>
->Ip+> es
ladonde hemos aprovechado el hecho de queHamiltoniano total. Aplicamos ahoraLippmann-Schwinger
autoestado delecuación de
= <K Ip> + [1 + 1 ] <K IVI p+ >E -E +ic E -E +icp k k P
222

El término entre corchetes es una representación de la delta denirac -2ni$(R -R ) con lo cual resulta
p k
~ ~<k-Ip+> ;7~ .;7 ~= <Klp>-2rrl<KIVlp+> 8(E -E )
p k
4. CALCULO DE LA SECCION EFICAZ CUANTICA
Reemplazando I<RIW > en la sección eficazout
obtenemos
da-
dK
El término entre corchetes es
[ ... ]2 ~ ~
= (2rr) o(E -E ) o(E -E ) o(p -p )k P1 k P2 2~ 1~
= 2m (2rr)2 o(E -E ) o(p2_p2) o(P -P )=k P1 2 1 2~ 1~
= m~2rr) o(E -E ) [o(P2-P1) + o(P -P )o(p +p )]
k P1 2~ 1~ 211 1111 11
Como <ql~> está fuertemente centrado en el impulso inicial q = p,~ ~ *el producto <p I~ ><p I~ > es despreciable si p = -p , con lo1 2 211 111
cual, obtenemos finalmente
da-
dk = (2rr)4 m J dq l<kIT(E +ic)lq>12 l<ql~>12 o(E -E )~ q k P
223

5. LIMITE ASINTOTICO DE LOS ESTADOS ESTACIONARIOS DE DISPERSION
De la ecuación de Lippmann-Schwinger resulta:
~ ~ ~ ~ J (r IG (E ±ic ) Ir' > V ( r' )~ ~ ~<rlp±> = <rlp> + <r' Ip± > dr'=
o p
. ~ ~~ ~ J ±lplr-r'l ~ ~ ~m e= <rlp>
- 2rr I r.-rol IV(r') <r' Ip±> dr'
Cuando r es mucho mayor que el alcance a de V(r') (que es la zonade integración) podemos desarrollar
. ~ ~±lplr-r'le~ ~Ir-r' I
. A ~1 ±ipr :¡:lpr.r''" - e er
2r » a , r » p.a
de tal forma que
-> -><r Ip±> '" -> -> m<rlp> - 2rr
-> -> m<rlp> 2rr
e±riprJ . A ->e+1pr.r' V(r') <r' Ip±> dr'=
e±ipr 3/2J A -> -> ->r (2rr) <±prlr'> V(r') <r' Ip±>
=-> ->
1 [ ip. r---- e -(2rr)3/2
2 A->(2rr)m <±prIVlp±>±iprer
6. TEOREMA OPTICO
Debido a que el operador de Moller es isométrico, eloperador de scattering S = n tn es unitario. Por lo tanto
- +
y, recordando que
<i~lntn Ip> = <klp> - 2rrio(E -E ) <kIT(E +ic) Ip>- + k p P
224

obtenemos
~ -+<klp>
~ -+. -+.-+= <klp> + 2rrl 8(E -E ) <kIT(E -lC) Ip> +k p k
~ . ~- 2rri o(E -E ) <kIT(E +lc)lp> +
k p p
ó(E -E ) J<iCIT(E -ic)lq> dq ó(E -E ) <qIT(E +ic)lp>k p k k P P
-+ -+Despejando la delta de Dirac y tomando k = P resulta
-+ • -+2 -+l<qIT(E +lc)lp>1 o(E -E ) dq =
p q p
= 1 --E J d~ dq(2II) 3 2m dq
o sea,
1m <pIT(E +ic)lp> = _ 1 --E ~(p)p ( 2II) 3 2m
Este teorema indica que la matriz de transición no puedeser puramente real.
225


APENDICE C
APROXIMACION DE WENTZEL, KRAMERS y BRILLOUIN
R. G. Pregliasco
Vamos a realizar una interpretación clásica de la función deonda. Para ello la escribimos como
.-7 -7 l-7w(r,t) = A(r,t) exp( h S(r,t) )
Reemplazando en la ecuación de Schrodinger:
h22 -> • d) ->(- 2m íI + V (r) - lh dt l/1 (r,t)=0
y separando las partes real e imaginaria obtenemos:
as (ílS)2at + 2m + V
aA2
1 2+ íI ( A ílS ) = Oat . m
(1)
(2 )
y a esta ecuación la conozco de alguna parte... j Es la ecuaciónde continuidad!. En forma consistente con las definiciones de ladensidad y el flujo de probabilidad, uno podría interpretar quela función de onda representa a un fluído de partículas nointeractuantes de densidad
y corriente
-7 ( •J = Re l/1h
Im
Ahora bien, si la expresión (2) es la ecuación de continuidad, laprimera debería representar la ecuación de movimiento del fluído.
227

Tornando el límite clásico, y reemplazando ~ -tV = ]/p = 'í/S/m,
esta ecuación pu~d~ ~gcribirg~ como
2as + mv - _ vat 2-
Finalmente, calculando el gradiente de ambos miembros resulta:
~mv = d ~dt mv = -'i1V
que es la ecuación de Newton para el movimiento del fluido. Laanalogía es completa.
Estos resultados nos dan una pista para buscar una soluciónsemiclásica de la ecuación de Schrodinger. A partir de este puntovamos a limitarnos a estudiar un problema estacionariounidimensional. En este caso podemos escribir
A(r,t) - A(x) S(r,t) - S(x) - E t
y las expresiones (1) y (2) que representan exactamente laecuación de Schrodinger quedan:
S,2 + 2m [ V(x)-E ] A' ,= h2-A-
d (S' A2) = Odx
Esta segunda ecuación puede ser integrada fácilmente, resultandoA= cte (S,)-1/2. Reemplazando en la primera obtenemos finalmente
[_34 (_SS',') 2S,2 = 2m [E-V] + h2
1 ~]2 S'
Esta ecuación diferencial es "exactamente" equivalente a laecuación de Schrodinger original. El único inconveniente es queparece muy difícil de resolver. La aproximación de Wentzel,
228

Kramers y Brillouin -o aproximación WKB- consiste en expandir la
solución en una serie de potencias de h2
s = S + h2 S + h
4 S + ...o 1 2
Y reemplazando en la ecuación anterior, resolverlarecursivamente. Por ejemplo, a orden O y 1 resulta
orden o: s (x)o Jx= ±
xop(x') dx'
orden 1: s (x)1
:¡: ¡h A' (x) 1 JX± 8h
xo
A'(X')--- dx'A (x')
donde hemos definido el impulso clásico
p(x) = (2m [E-V(x)] )1/2
y la longitud de onda local A(X) = h/p(x).
A orden cero, la función de onda resulta ser combinaciónlineal del siguiente par de soluciones independientes
1u+_(x) O< ---
P (x) 1/2exp (± ~ J x p (x ') dx' )
xo
Obviamente en los puntos de retorno p(x) se anula y esta solucióndeja de ser válida. De hecho, para que la solución de primerorden S de una contribución despreciable a la fase de la función
1
de onda, debe satisfacerse la condición lA' I « 1, que estableceuna cota para la variación espacial del potencial.
Vemos entonces que tendremos problemas con la solución de orden Ocuando el potencial varíe muy bruscamente o en aquellos casos enque p sea muy pequeño. Es decir que la aproximación WKB vale en
229

casi todo el espacio menos en los alrededores de los puntos deretorno. Pero es justamente allí donde tenemos que empalmar lassoluciones de la zona clásicamente permitida con las de la regióninaccesible clásicamente. Aparentemente la única alternativaconsiste en empalmar las soluciones WKB resolviendo en formaexacta la ecuación de Schr6dinger cerca de los puntos de retorno.Este asunto es muy trabajoso y no lo vamos desarrollar en detalleaquí. Solo expondremos los resultados obtenidos por Langer (PhysRev 51 669 (1937)). Las fórmulas de enlace que vamos a mostrarserán válidas cuando:
1) La energía E-V sea aproximadamente lineal con laposición x en un rango de varias longitudes de onda aambos lados del punto de retorno.
2) Cada una de estas regiones empalme con otras dondela aproximación WKB es válida.
Consideremos que E > V para x > a (barrera por la izquierda).Puede demostrarse que las soluciones clásicamente permitidas
1/11
p-1/2cos ( ~ fp(x' )dx'- : + e )a
y
-1/2 (1 JX Tl )1/12 = P cos ti p(x' )dx'- 4"a
se acoplan en la región clásicamente prohibida con
sen(e) Ip 1-1/2 exp ( 1 r lp(x') I dX')1f)1 = ñx
y
I -1/2 ( 1 r Ip(x')1 dx')1f)2 = pl expñ
x
respectivamente. El sentido de la conexión es t/J ., If) y If) ., t/J ,1 1 2 2
Y no al revés. Si la barrera está a la derecha, bastaráintercambiar ~ y 'ª en las desigualdades y en los límites deintegración.
230

Como prim~ra aplicación d~ ~~t~ r~~ultado, con~id~r~mo~ unpozo de potencial lo más sencillito posible, que para la energíaE tenga sólo dos puntos de retorno a y b (a < b). En las regionesinaccesibles clásicamente proponemos las soluciones:
rpa = A IP 1-112exp [-k r Ip (x' ) I dX']
x
<¡lb = B IPI-l12 exp [-j¡ ( Ip(x')1 dX']
x < a
X > b
donde A Y B son constantes arbitrarias. De acuerdo con lasfórmulas de enlace aplicadas a los puntos de retorno a y bobtenemos las correspondientes soluciones en la regiónclásicamente accesible a < x < b
cos (x
)I/Ja
2 A -1/2 1 J p(x' ) dx' Tl= P - -h 4
a
cos(b
)I/Jb 2 B -1/2 1 J p(X') dx' Tl= P - -h 4
x
Para que la vida sea más linda estas soluciones deben ser lamisma función: I/J = I/J • Pueden darse dos casos: Si A = B entonces
a bbk J p(x' )dx'= Tl (2n+l/2). Por otro lado, si A = -B, entonces debea
b
ser k J p(x' )dx'= Tl (2n+l+l/2). Obtenemos así la regla dea
cuantificaciónbJ P (x' )dx '= hTl ( N+1/2 )a
donde N es un número entero. Como el método exige que en laszonas límites alrededor de los puntos de retorno, V(x) debe seraproximadamente lineal sobre varias longitudes de onda, estaregla de cuantificación se justifica sólo para números cuánticosmuy grandes, es decir N » 1.
Una ecuación de Schrodinger radial representa otro problema"unidimensional" al cual podemos aplicarle la aproximación WKB.
231

Por ejemplo, en la página 193 definimos las funciones de ondaradiales Wtp(r), soluciones de una ecuación de Sch5dino@r
2unidimensional de energía E = P 12m y potencial efectivo
V (r) = V(r) +efl(l+l)
22mr
Intentemos encontrar la solución WKB para este problema. Para rpequeño existe un punto de retorno r dado por la condición
oV (r) = E Para r < r proponemos una solución
ef o o
exponencialmente decreciente. Si la barrera centrífuga es losuficientemente grande (l » 1), la función de onda va a llegar ar = O con un valor despreciablemente pequeño y la condiciónWip(O) = O se satisfará casi exactamente. La fórmula de conexiónnos indica que para r > r la solución es de la forma:o
Wip(r)= 2 A pi(r)-1/2 sen( r Pi(r') dr' + ~ )r
o
Cuando r ~ 00 el potencial tiende a cero y pi(r) tiende a p, demanera que la solución WKB tiene el límite asintótico:
-r-~-00-)2 A p-1/2 sen({ Pi(r') dr' + : )r
o
Comparando esta expresión con la forma asintótica
r ~ 00
obtenemos la siguiente expresión para el desfasaje
00
0i = J [pi(r)-p]dr - pro + (i+~) ;r
o
Esta es la misma expresión que utilizamos para desarrollar lateoría de colisiones semiclásica.
232