
Enfermedades de los leucocitos, los ganglios linfáticos, el bazo y el timo
Upload
aaron-rodriguezCategory
view
394download
0
Carrera : Medico Cirujano y Partero
Cátedra: PatologíaDocente: Guillermo Domínguez Ríos
BENEMÉRITA UNIVERSIDAD DE GUADALAJARACentro Universitario de los Altos
Facultad de ciencias biomédicas e ingenierías
Disertante: Aaron Rodríguez González

• Contenido• NEOPLASIAS MIELOIDES. • 1 Leucemia mieloide aguda; clasificación, morfología, inmunofenotipo, citogenética,
patogenia molecular, características clínicas, pronostico. • 2 Síndromes mielodisplásicos; morfología, patogenia, características clínicas,
pronostico. • 3 Trastornos mieloproliferativos; en cada uno de ellos investigar la patogenia,
morfología, etiología, características clínicas. • 1. Leucemia mieloide crónica. • 2. Policitemia vera. • 3. Trombocitosis esencial. • 4. Mielofibrosis primaria. • 4 Histiocitosis de células de Langerhans; características generales, clasificación
(entidad clínico patológica) y características generales de cada una de ellas. • 5 BAZO; arquitectura normal, características, funciones, definir esplenomegalia y
mencionar los trastornos asociados. • Esplenitis aguda inespecífica, morfología. • Esplenomegalia congestiva, morfología. • Infarto esplénico, morfología. • 6 TIMO, características generales, morfología, hiperplasia timica, timomas,
morfología, características clínicas.

Leucemias mieloides agudas LMA
SX Mielodisplásicos SMD
Trastornos mieloproliferativos TMP
Neoplasias mieloides
Cumulo de formas mieloides inmaduras en M.O
Hematopoyesis ineficaz
Producción de C. Mieloides totalmente diferenciadas
Leucemia mieloide Crónica
Policitemia Vera
Trombocitosis esencial
Mielofibrosis Primaria

• Grupo heterogéneo de neoplasias procedentes de células progenitoras hematopoyéticas de la serie mieloide
• Ocurren porque los mecanismos de la hematopoyesis sufren alteraciones neoplasias mieloides, que escapan de los controles homeostáticos
Neoplasias mieloides


Afecta personas de entre 15- 39 años Cumulo de formas mieloides inmaduras en M.O
Leucemias mieloides agudas LMA

• Línea predominante de proliferación.• Madurez de las células.• Menor predicción diagnóstica.
Morfológico. Inmunofenotípico. Según marcadores de superficie.Histoquímico Tinción o reacciones químicas. Ej. MieloperoxidasaCitogenético. Por medio de cariotipoMolecular. Detección de proteínas «quiméricas»
Franco-Americana-Británica (FAB):
Según la OMS:
Clasificación LMA.

Clasificación FAB.
Comienzo en precursores leucocíticos
Comienzo en formas tempranas de eritrocitos
Comienzo en formas tempranas de plaquetas

Clasificación OMS.

Inversiones (Ej.16) y Translocaciones (que conducen a formar genera quiméricos que codifica para una proteína de fusión)
Por ejeemplo: • LPMA t(15;17)
Produce gen de fusión codificador de Rc de ácido retinoico (RARα)
Se une a PMR
Represor que desactiva genes necesarios para diferenciación mieloide completa
Genera mutaciones puntuales en la tirosincinasa FLT3
seguirá proliferando o morirá precozmente
Neutropenia, Anemia, Trombopenia
Fisiopatología molecular

MieloblastosMP ( + )
Granulos azurofilos
LMA M1
MieloblastosMP ( - )
Cifra de celulas leucemicas en sangre P. de 10.000 o inferior (50% casos)
20% de blastos mieloides en M.O
Promielocitos neoplasicos MP( - )
Cuerpos de Auer LPMAMieloblastosMP ( - )
LMA M5b
Morfología

CD34 = C.Pluripotenciales CD64 = C. Mieloide madura CD33 = C. Mieloide inmadura CD15 = C. Mieloide madura
Citometría de flujo
1arias Mutaciones de novo asociadas con translocaciones equilibradas Ejeeemplo :
t(8;21) LMA con maduracion 35% t (15;27) LPMA 10 % inv (16) LMMonociticaA 15-20% 2rias Exposicion a sustancias dañinas para ADN Ejeemplo : Radio terapia ,quimioterapia Se asocian con monosomias del cromosoma 5 y 7
Inmunofenotipo CD34 y CD33
Etiología

Por desplazamiento de la MO: • Dolor óseo.Generalmente 3 meses antes del diagnóstico de LMA.
Manifestaciones clínicas.
Leucocitosis/leucopenia
Trombocitopenia:
Anemia
• Infecciones recurrentes y/o oportunistas. Ej: Hongos, seudomonas
Hemorragias (petequias en encias,GI)
• Debilidad, Fatiga ,Anorexia
PronósticoUn 60% consigue remisión completa con quimioterapia, pero solo 15 a 30% de ellos sigue sin tener signos o síntomas de la enfermedad 5 años despuésLas LMA asociadas a translocaciones poseen un mejor pronostico que las asociadas a deleciones o monosomias cromosómicas

• Defectos en la maduración, hematopoyesis ineficaz y riesgo de LMA, • M.O sustituida total o parcialmente por progerie clonal que conserva
capacidad de diferenciarse en pero de forma inefectiva
Los SMD aparecen en 2 contextos
• Idiopáticos o primarios personas superiores a 50 años
• Relacionado con tratamiento (SMD-t) es una complicacion por el tratamiento de Genotoxicos o Radiación aparece 2- 8 años tras terapia
SX Mielodisplásicos SMD
Etiología

• Desconocida• MO híper, normo e incluso hipocelular• Hematopoyesis ineficaz y alta tasa de
apoptosis
Monosomías 5 y 7 Deleciones de 5q, 7q y 20q Trisomía 8
Patogenia
Anomalías cromosómicas

Diferenciación alterada(displásica) en todos los linajes no linfoides en MOMorfología

Seudocelulas de Pelger-huet
Sideroblastos en anillo
Megacariocitos multinucleados
Eritrocitos nucleados bilobulados

Citopenias sanguíneas periféricas Infecciones Pancitopenia Anemia (Apnea Debilidad) Hemorragias LMA ocurre en 10 al 40%
SMD idiopatico Supervivencia media de 9 y 29 meses
SMD-t Supervivencia media de 4 a 8 meses
Pacientes jóvenes• Trasplante alogénico de
MO
Pacientes mayores• Antibióticos• Transfusiones
Evolución ClínicaTratamiento
Pronóstico

Diferenciación terminal relativamente conservada conduciendo a Hipercelulariedad medular , Hemopoyesis disminuida Hemopoyesis extramedular Transformación variable a leucemia aguda , LMC causa LMA invariablemente
Las C. Madre Neoplásicas tienen capacidad de circular y alojarse en órganos hematopoyéticos secundarios
Trastornos mieloproliferativos TMP
Leucemia mieloide Crónica
Policitemia Vera
Trombocitosis esencial
Mielofibrosis Primaria

• Algunos trastornos mieloproliferativos se asocian especialmente a mutaciones activadoras de tirosina cinasa especificas

Incidencia en adultos entre los 25 y 60 años
Presencia de un gen quimérico BCR-ABL derivado de las porciones del gen BCR en el cromosoma 22 y ABL en el cromosoma 9 se genera el Cromosoma
Leucemia mieloide Crónica
Anomalía cromosómica

1. La rotura y unión de BCR y ABL crea un gen BCR-ABL
2. Este gen contiene un dominio de dimerización que se autofosforila y activa la porcion ABL de la tirosina cinasa.
3. La cinasa ABL fosforila las proteínas que inducen la señalización a través de las vías que favorecen el crecimiento y supervivencia que activan los factores de crecimiento hematopoyéticos
Fisiopatología molecular

• La medula ósea es 100% celular (Normal 50% Cel y 50% grasa)• No. De megacariocitos,histiocitos diseminados,deposito de reticulina• Sangre periferica con leucocitosis (>a 100000c./mm) circulando neutrofilos,metamielocitos,• mielocitos, <10% de mieloblastos• Produce hematopoyesis extramedular ocasionando esplenomegalia
LMC
Morfología

Hipermetabolismo Pérdida de peso AnorexiaDebilidad
Anemia Pérdida de peso
Peso en hipocondrio derecho
Características clínicas
Pronostico50% Crisis blastica intermedia
50% entra en fase acelerada
Superivencia de 3 años
Inmunofenotipo CD 10 CD 19
Marcadores de linaje Lin.B precoces

Policitemia vera PCV Neoplasia en C. Madre multipotente 1-3 casos por 100,000 habitantes y año Producción aumentada de productos Eritroide, Granulocitos, y trombociticos Niveles séricos de eritropoyetina bajos Mayor viscosidad y aglutinación de la sangre
Fisiopatología Molecular>97% Mutaciones puntuales activadoras de tirosina cinasa JAK2
Dos copias mutadas de
• Líneas celulares hematopoyéticas independientes de • Factor de crecimiento
• Panmielosis

M. O hipercelular
Aumento de reticulina
Basófilos y Plaquetas Grandes
Fibrosis medular
Hematopoyesis extramedular
Etapa agotada
Etapa temprana
Tomografía C.Morfología

Aumento de volumen sanguineoAumenta riesgo de hemorragias y trombosis
“La mayoría de los síntomas están relacionados con el incremento de la masa de los eritrocitos y del hematócrito”
En adultos > 60 años
Las hemorragias leves (epistaxis, encías sangrantes) son frecuentes.
La concentración de hemoglobina varía entre 14 y 28 g/dl
El hematócrito es del 60% o mayor
Recuento de plaquetas es a menudo mayor de 500.000 plaquetas/mm 3El recuento de leucocitos varía de 12.000 a 50.000 células/mm 3.
En ocasiones, las hemorragias deficiencia de hierro
Curso clínico
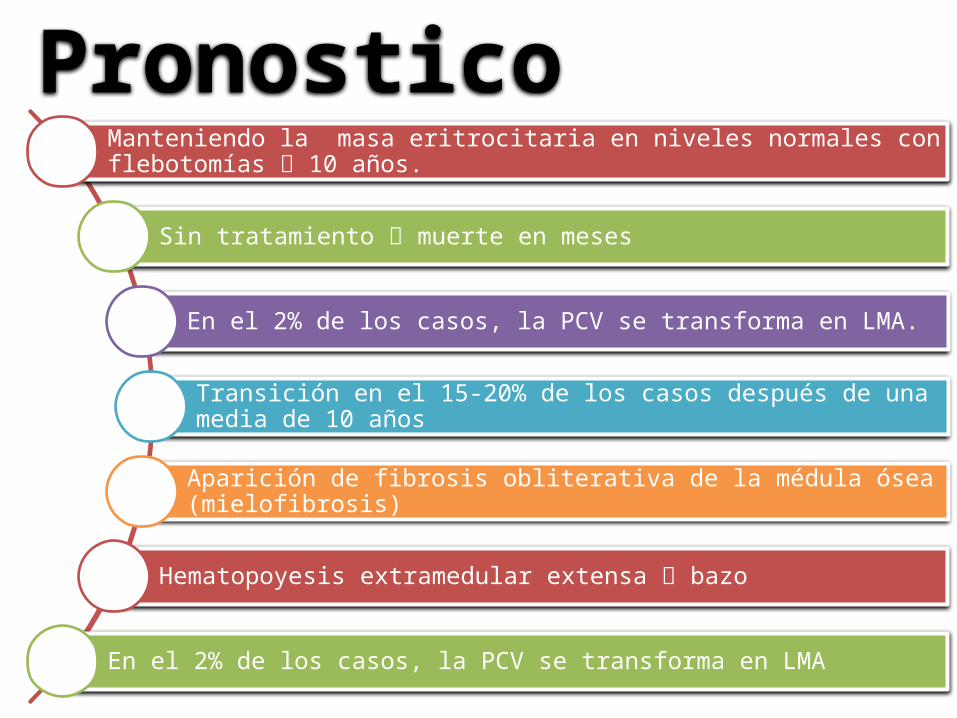
Manteniendo la masa eritrocitaria en niveles normales con flebotomías 10 años.
Sin tratamiento muerte en meses
En el 2% de los casos, la PCV se transforma en LMA.
Transición en el 15-20% de los casos después de una media de 10 años
Aparición de fibrosis obliterativa de la médula ósea (mielofibrosis)
Hematopoyesis extramedular extensa bazo
En el 2% de los casos, la PCV se transforma en LMA
Pronostico

Ocurre en adultos jóvenes y mas común en adultos >60 años
↑ del recuento de plaquetas, mas grandes y anormales
Se diferencia de PCV por ausencia de policitemia
Se diferencia de MF1aria por ausencia de fibrosis medular
Hematopoyesis extramedular, 50% de los casos.
En el 2% de los casos, la PCV se transforma en LMA
Eritromelalgia (Sensacion de ltidos y ardor de manos y pies
Trombocitosis esencial TE

Supervivencia media de 12 a 15 años
Morfología
Pronostico

Sustitución de medula por tejido fibroso, anulando hematopoyesis medular promoviendo hematopoyesis extramedular neoplásica del bazo, hígado y ganglios linfáticos
Deposito extenso de colágeno por fibroblastos no neoplásicos de la medula
PDFG
TGF-β
Liberacion inapropiada de factores fibrogenicos
Mitogenos
Mutaciones en JAK2, 50% de los casos, MPL, 1-5% afecta megacariocitos
Mielofibrosis Primaria
Fisiopatología
Etiología

M.O Hipercelular
Megacariocitos grandes
Esplenomegalia de hasta 4 kg
Hígado ligeramente agrandado
Presencia de leucoeritroblastosis en sangre periférica
Dacriocitos( Hematies en lagrima) Dacriocitos
Morfología

Personas > de 60 años
Anemia progresiva
Esplenomegalia notoria
Cansancio, pérdida de peso y sudores nocturnos.
5 a 20% se transforma a LMA
Supervivencia de entre 3 a 5 años
Curso Clínico
Pronostico

Trastornos proliferativos monoclonales neoplásicos de las células dendríticas o macrófagos en ganglios linfáticos
CCR6
CCR6 CCR7CCL20 Piel hueso li g de CCR6CCL19 y 21 linfoides lig CCR7AG CD1a HLA-DR, S100
Cel.Langerhans Piel
Granulos de Birbeck
Histiocitosis de Cel. De Langerhans HCL

HCL Multisistemica multifocal (Enfermedad de Letter-Siwe)
HLC Unisistemica, unifocal, (Granuloma Eosinofilo)
HCL Unisitemica ,multifocal (ENF. De HAND SCHULLER CHRISTIAN
HCL Pulmonar
Subtipos HCL

HCL Multisistemica multifocal (Enfermedad de Letter-Siwe)
Frecuente en niños menores de 2 años. Lesiones cutáneas que simulan erupción seborreica, por infiltrado de CL sobre la cara post. y ant. del tronco y cuero cabelludo Hepatoesplenomegalia Anemia, Trombocitopenia--------Enf .infecciosas Ej .Otitis , Mastoiditis
Pronostico Si no se trata es mortal Con quimioterapia intensiva 50% sobreviven 5 años
Curso clinico

HLC Unisistemica, unifocal, (Granuloma Eosinofilo)
Se caracteriza por proliferación de las CL mezcladas con eosinófilos, linfocitos, células plasmáticas y neutrófilos.Surge dentro de las cavidades medulares del hueso. Ej. Boveda craneal ,femur,costillas

HCL Unisitemica ,multifocal ENF. De HAND SCHULLER CHRISTIANAfecta niños jóvenes en forma de múltiples masas erosivas que se extienden a tejidos blandos En 50% de pacientes se afecta el tallo hipotalámico del lóbulo post. de hipófisis que conduce a diabetes insipida
Triada de Hand Schuller Christian • Defectos oseos de boveda craneal• Diabetes insipida• Exoftalmos

TC
Se encuentra frecuentemente en fumadores adultos. Indicando que es una hiperplasia reactiva más que una verdadera neoplasia
HCL Pulmonar

BIBLIOGRAFÍA

Gracias!!!


















