
PROCEDIMIENTOS NORMALIZADOS DE TRABAJO - Inicio · • Orden SAS/3470/2009, de 16 de diciembre, por...
116
1 COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA DEL CONSORCIO HOSPITALARIO PROVINCIAL DE CASTELLÓN PROCEDIMIENTOS NORMALIZADOS DE TRABAJO Castellón, octubre de 2010
Transcript of PROCEDIMIENTOS NORMALIZADOS DE TRABAJO - Inicio · • Orden SAS/3470/2009, de 16 de diciembre, por...

1
COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA DEL CONSORCIO HOSPITALARIO PROVINCIAL
DE CASTELLÓN
PROCEDIMIENTOS NORMALIZADOS DE TRABAJO
Castellón, octubre de 2010

2
INDICE
1. INDICE Pág 2
2. DOCUMENTACION BASICA Pág. 4
• Declaración de Helsinki
• Normas de Buena Práctica Clínica (CPMP/ICH/135/95)
• Real Decreto 223/2004, por el que se regulan los ensayos clínicos con medicamentos
• ORDEN SCO/256/2007, de 5 de febrero, por la que se establecen los principios y las directrices detalladas de buena práctica clínica y los requisitos para autorizar la fabricación o importación de medicamentos en investigación de uso humano.
• Resolución de 16 de julio 2009, de la Consellería de Sanidad, por el que se aprueba el modelo de contrato que ha de suscribirse entre la gerencia de un centro sanitario, el promotor y los investigadores, para la realización de un ensayo clínico o estudios post-autorización observacionales de medicamentos y productos sanitarios en las organizaciones de servicios sanitarios en la Comunitat Valenciana.
• Decreto 73/2009, del Consell, por el que se regula la gestión de ensayos clínicos y estudios post-autorización observacionales con medicamentos y productos sanitarios.
• Resolución de 16 de julio de 2009, de la Consellería de Sanitat, de regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana.
• Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano.
3. COMPOSICIÓN Y ESTRUCTURA DEL CEIC, MIEMBROS QUE LO COMPONEN, MEDIOS CON QUE CUENTA Y
UBICACIÓN Pág.92
4. FUNCIONES DEL CEIC Pág.94
5. ELECCIÓN, SUSTITUCIÓN Y RENOVACIÓN DE LOS MIEMBROS DEL CEIC. PROCEDIMIENTO, CRITERIOS Y DURACIÓN DE LOS CARGOS Pág.98
6. ÁMBITO DE ACTUACIÓN Pág.99 7. PERIODICIDAD DE LAS REUNIONES Y TIEMPO MÁXIMO DE RESPUESTA DE LOS PROTOCOLOS. PROCEDIMIENTO
DE LAS CONVOCATORIAS DE LAS REUNIONES DEL CEIC. Pág.99 8. CALENDARIO DE EVALUACIÓN DE ENSAYOS MULTICENTRICOS Pág.100 9. DOCUMENTACIÓN QUE PRESENTARÁN LOS ENSAYOS A EVALUAR, TASAS DE EVALUACIÓN DEL CEIC Y EL
CONTRATO ECONÓMICO Pág.102
10. PROCEDIMIENTO DE EVALUACIÓN. Pág.107 11. MECANISMOS DE TOMA DE DECISIONES Y CASOS EN QUE SE PUEDE REALIZAR UNA REVISIÓN RAPIDA DE LA
DOCUMENTACIÓN Pág.108 12. PROCEDIMIENTO PARA IDENTIFICAR EL PROTOCOLO Y GARANTIZAR QUE ES EL APROBADO POR LA DIRECCIÓN
GENERAL DE FARMACIA Y MEDICAMENTOS Pág 109
13. MODELO DE ACTA DE LAS REUNIONES. Pág.109
14. PROCEDIMIENTOS PARA REALIZAR EL SEGUIMIENTO DEL ENSAYO, INFORMACIÓN SOBRE REACCIONES ADVERSAS Y SOLICITUD DE SUSPENSIÓN CAUTELAR Pág.109
15. ARCHIVO DEL CEIC: DOCUMENTACIÓN ARCHIVADA Y ACCESO A LA MISMA. TIEMPO MÍNIMO DE ARCHIVO Pág.111

3
16. PROCEDIMIENTO DE IDENTIFICACIÓN DEL INVESTIGADOR PRINCIPAL Y COLABORADORES DE CADA ENSAYO
CLÍNICO, ESPECIFICANDO QUIENES PUEDEN PARTICIPAR EN EL MISMO, SOLICITAR LA MEDICACIÓN A FARMACIA Y ADMINISTRAR LOS MEDICAMENTOS EN ESTUDIO. Pág.112
17. OBLIGACIONES DE LOS INVESTIGADORES Pág.112
18. RECEPCIÓN, IDENTIFICACIÓN, ALMACENAMIENTO, MANIPULACIÓN, DISTRIBUCIÓN Y DEVOLUCIÓN DE LAS
MUESTRAS PARA INVESTIGACIÓN CLÍNICA Y FINANCIACIÓN DE LAS MISMAS Pág 115
19. REVISIÓN/MODIFICACIONES DE LOS PROCEDIMIENTOS NORMALIZADOS DE TRABAJO Pág 116 20. ANEXOS DE LOS PROCEDIMIENTOS INDICADOS Pág 117

4
• Declaración de Helsinki
• Normas de Buena Práctica Clínica (CPMP/ICH/135/95)
• Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos
• Orden SCO/256/2007, de 5 de febrero, por la que se establecen los principios y las directrices detalladas de buena práctica clínica y los requisitos para autorizar la fabricación o importación de medicamentos en investigación uso humano.
• Resolución de 16 de julio 2009, de la Consellería de Sanidad, por el que se aprueba el modelo de contrato que ha de suscribirse entre la gerencia de un centro sanitario, el promotor y los investigadores, para la realización de un ensayo clínico o estudios post-autorización observacionales de medicamentos y productos sanitarios en las organizaciones de servicios sanitarios de la Comunitat Valenciana
• Decreto 73/2009, de 5 de junio del Consell, por el que se regula la gestión de ensayos clínicos y estudios post-autorización observacionales con medicamentos y productos sanitarios.
• Resolución de 16 de julio de 2009, de la Consellería de Sanitat, de regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana.
• Orden SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano.
Documentación Básica

5
DECLARACIÓN DE HELSINKI
A. INTRODUCCIÓN
1. La Asociación Médica Mundial (AMM) ha promulgado la Declaración de Helsinki como una propuesta de principios éticos para investigación médica en seres humanos, incluida la investigación del material humano y de información identificables
La Declaración debe ser considerada como un todo y un párrafo no debe ser aplicado sin considerar todos los otros párrafos pertinentes.
2. Aunque la Declaración está destinada principalmente a los médicos, la AMM insta a otros participantes en la investigación médica en seres humanos a adoptar estos principios.
3. El deber del médico es promover y velar por la salud de los pacientes, incluidos los que participan en investigación médica. Los conocimientos y la conciencia del médico han de subordinarse al cumplimiento de este deber.
4. La Declaración de Ginebra de la Asociación Médica Mundial vincula al médico con la fórmula "velar solícitamente y ante todo por la salud de mi paciente", y el Código Internacional de Ética Médica afirma que: "El médico debe considerar lo mejor para el paciente cuando preste atención médica.¨
5. El progreso de la medicina se basa en la investigación que, en último término, debe incluir estudios en seres humanos. Las poblaciones que están subrepresentadas en la investigación médica deben tener un acceso apropiado a la participación en la investigación.
6. En investigación médica en seres humanos, el bienestar de la persona que participa en la investigación debe tener siempre primacía sobre todos los otros intereses
7. El propósito principal de la investigación médica en seres humanos es comprender las causas, evolución y efectos de las enfermedades y mejorar las intervenciones preventivas, diagnósticas y terapéuticas (métodos, procedimientos y tratamientos). Incluso, las mejores intervenciones actuales deben ser evaluadas continuamente a través de la investigación para que sean seguras, eficaces, efectivas, accesibles y de calidad.
8. En la práctica de la medicina y de la investigación médica, la mayoría de las intervenciones implican algunos riesgos y costos.
9. La investigación médica está sujeta a normas éticas que sirven para promover el respeto a todos los seres humanos y para proteger su salud y sus derechos individuales. Algunas poblaciones sometidas a la investigación son vulnerables y necesitan protección especial. Estas incluyen a los que no pueden otorgar o rechazar el consentimiento por si mismos y a los que pueden ser vulnerables a coerción o influencia indebida.

6
10. Los médicos deben considerar las normas y estándares éticos, legales y jurídicos para la investigación en seres humanos en sus propios países, al igual que las normas y estándares internacionales vigentes. No se debe permitir que un requisito ético, legal o jurídico nacional o internacional disminuya o elimine cualquiera medida de protección para las personas que participan en a investigación establecida en esta Declaración
B. PRINCIPIOS PARA TODA INVESTIGACIÓN MÉDICA
11. En la investigación médica, es deber del médico proteger la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de la información personal de las personas que participan en investigación.
12. La investigación médica en seres humanos debe conformarse con los principios científicos generalmente aceptados, y debe apoyarse en un profundo conocimiento de la bibliografía científica, en otras fuentes de información pertinentes, así como en experimentos de laboratorio correctamente realizados y en animales, cuando sea oportuno. Se debe cuidar también del bienestar de los animales utilizados en los experimentos.
13. Al realizar una investigación médica, hay que prestar atención adecuada a los factores que pueden dañar el medio ambiente.
14. El proyecto y el método de todo estudio en seres humanos debe describirse claramente en un protocolo de investigación. Este debe hacer referencia siempre a las consideraciones éticas que fueran del caso y debe indicar cómo se han considerado los principios enunciados en esta Declaración. El protocolo debe incluir información sobre financiamiento, patrocinadores, afiliaciones institucionales, otros posibles conflictos de interés e incentivos para las personas del estudio y estipulaciones para tratar o compensar a las personas que han sufrido daños como consecuencia de su participación en la investigación. El protocolo debe describir los arreglos para el acceso después del ensayo a intervenciones identificadas como beneficiosas en el estudio o el acceso a otra atención o beneficios apropiados.
15. El protocolo de la investigación debe enviarse, para consideración, comentario, consejo y aprobación, a un comité de ética de investigación antes de comenzar el estudio. Este comité debe ser independiente del investigador, del patrocinador o de cualquier otro tipo de influencia indebida. El comité de considerar las leyes y reglamentos vigentes en el país donde se realiza la investigación, como también las normas internacionales vigentes, pero no se debe permitir que éstas disminuyan o eliminen ninguna de las protecciones para las personas para las personas que participan en la investigación establecidas en esta Declaración. El comité tiene el derecho de controlar los ensayos en curso. El investigador tiene la obligación de proporcionar información del control al comité, en especial sobre todo incidente adverso grave. No se debe hacer ningún cambio en el protocolo sin la consideración y aprobación del comité.
16. La investigación médica en seres humanos debe ser llevada a cabo sólo por personas con la formación y calificaciones científicas apropiadas. La investigación en pacientes o voluntarios sanos necesita la supervisión de un médico u otro profesional de la salud competente y calificado apropiadamente. La responsabilidad de la protección de las personas que toman parte en la investigación debe recaer siempre en un médico u otro profesional de la salud y nunca en los participantes en la investigación, aunque hayan otorgado su consentimiento.
17. La investigación médica en una población o comunidad con desventajas o vulnerable sólo se justifica si la investigación responde a las necesidades y prioridades de salud de

7
esta población o comunidad y si existen posibilidades razonables de que la población o comunidad, sobre la que la investigación se realiza, podrá beneficiarse de sus resultados.
18. Todo proyecto de investigación médica en seres humanos debe ser precedido de una cuidadosa comparación de los riesgos y los costos para las personas y las comunidades que participan en la investigación, en comparación con los beneficios previsibles para ellos y para otras personas o comunidades afectadas por la enfermedad que se investiga.
19. Todo ensayo clínico debe ser inscrito en una base de datos disponible al público antes de aceptar a la primera persona.
20. Los médicos no deben participar en estudios de investigación en seres humanos a menos de que estén seguros de que los riesgos inherentes han sido adecuadamente evaluados y de que es posible hacerles frente de manera satisfactoria. Deben suspender inmediatamente el experimento en marcha si observan que los riesgos que implican son más importantes que los beneficios esperados o si existen pruebas concluyentes de resultados positivos o beneficiosos.
21. La investigación médica en seres humanos sólo debe realizarse cuando la importancia de su objetivo es mayor que el riesgo inherente y los costos para la persona que participa en la investigación.
22. La participación de personas competentes en la investigación médica debe ser voluntaria. Aunque puede ser apropiado consultar a familiares o lideres de la comunidad, ninguna persona competente debe ser incluida en un estudio, a menos que ella acepte libremente.
23. Deben tomarse toda clase de precauciones para resguardar la intimidad de la persona que participa en la investigación y la confidencialidad de su información personal y para reducir al mínimo las consecuencias de la investigación sobre su integridad física, mental y social.
24. En la investigación médica en seres humanos competentes, cada individuo potencial debe recibir información adecuada acerca de los objetivos, métodos, fuentes de financiamiento, posibles conflictos de intereses, afiliaciones institucionales del investigador, beneficios calculados, riesgos previsibles e incomodidades derivadas del experimento y todo otro aspecto pertinente de la investigación. La persona potencial debe ser informada del derecho de participar o no en la investigación y de retirar su consentimiento en cualquier momento, sin exponerse a represalias. Se debe prestar especial atención a las necesidades específicas de información de cada individuo potencial, como también a los métodos utilizados para entregar la información. Después de asegurarse de que el individuo ha comprendido la información, el médico u otra persona calificada apropiadamente debe pedir entonces, preferiblemente por escrito, el consentimiento informado y voluntario de la persona. Si el consentimiento no se puede otorgar por escrito, el proceso para lograrlo debe ser documentado y atestiguado formalmente.
25. Para la investigación médica en que se utilice material o datos humanos identificables, el médico debe pedir normalmente el consentimiento para la recolección, análisis, almacenamiento y reutilización. Podrá haber situaciones en las que será imposible o impracticable obtener el consentimiento para dicha investigación o podría ser una amenaza para su validez. En esta situación, la investigación sólo puede ser realizada después de ser considerada y aprobada por un comité de ética de investigación.
26. Al pedir el consentimiento informado para la participación en la investigación, el médico debe poner especial cuidado cuando el individuo potencial está vinculado con el

8
por una relación de dependencia o si consiente bajo presión. En una situación así, el consentimiento informado debe ser pedido por una persona calificada adecuadamente y que nada tenga que ver con aquella relación.
27. Cuando el individuo potencial sea incapaz, el médico debe pedir el consentimiento informado del representante legal. Estas personas no debe ser incluidas en la investigación que no tenga posibilidades de beneficio para ellas, a menos que ésta tengo como objetivo promover la salud de la población representado por el individuo potencial y esta investigación no puede realizarse en personas competentes y la investigación implica sólo un riesgo y costo mínimos.
28. Si un individuo potencial que participa en la investigación considerado incompetente es capaz de dar su asentimiento a participar o no en la investigación, el medico debe pedirlo, además del consentimiento del representante legal. El desacuerdo del individuo potencial debe ser respetado.
29. La investigación en individuos que no son capaces física o mentalmente de otorgar consentimiento, por ejemplo los pacientes inconscientes, se puede realizar sólo si la condición física/mental que impide otorgar el consentimiento informado es una característica necesaria de la población investigada. En estas circunstancias, el médico debe pedir el consentimiento informado al representante legal. Si dicho representante no está disponible y si no se puede retrasar la investigación, el estudio puede llevarse a cabo sin consentimiento informado, siempre que las razones específicas para incluir a individuos con una enfermedad que no les permite otorgar consentimiento informado hayan sido estipuladas en el protocolo de la investigación y el estudio haya sido aprobado por un comité de ética de investigación. El consentimiento para mantenerse en la investigación debe obtenerse a brevedad posible del individuo o de un representante legal.
30. Los autores, directores y editores todos tienen obligaciones éticas con respecto a la publicación de los resultados de su investigación. Los autores tienen el deber de tener a la disposición del público los resultados de su investigación en seres humanos y son responsables de la integridad y exactitud de sus informes. Deben aceptar las normas éticas de entrega de información. Se deben publicar tanto los resultado negativos e inconclusos como los positivos o de lo contrario deben estar a la disposición del público. En la publicación se debe citar la fuente de financiamiento, afiliaciones institucionales y conflictos de intereses. Los informes sobre investigaciones que no ciñan a los principios descritos en esta declaración no deben ser aceptados para su publicación.

9
3. PRINCIPOS APLICABLES CUANDO LA INVESTIGACIÓN MÉ DICA SE COMBINA CON LA ATENCIÓN MEDICA.
31. El médico puede combinar la investigación médica con la atención médica, sólo en la medida en que tal investigación acredite un justificado valor preventivo, diagnóstico o terapéutico y si el médico tienes buenas razones para creer que la participación en el estudio no afectará de manera adversa la salud de los pacientes que toman parte en la investigación.
32. Los posibles beneficios, riesgos, costos y eficacia de toda intervención nueva debe ser evaluados mediante su comparación con la mejor intervención probado existen, excepto en las siguientes circunstancias:
* El uso de un placebo, o ningún tratamiento, es aceptable en estudios para los que no hay una intervención probado existente.
* Cuando por razones metodológicas, científicas y apremiantes, el uso de un placebo es necesario para determinar la eficacia y la seguridad de una intervención que no implique un riesgo, efectos adversos graves o daño irreversible para los pacientes que reciben el placebo o ningún tratamiento. Se debe tener muchísimo cuidado para evitar abusar de esta opción.
33. Al final de la investigación, todos los pacientes que participan en el estudio tienen derecho a ser informados sobre sus resultados y compartir cualquier beneficio, por ejemplo, acceso a intervenciones identificadas como beneficiosas en el estudio o a otra atención apropiada o beneficios.
34. El médico debe informar cabalmente al paciente los aspectos de la atención que tienen relación con la investigación. La negativa del paciente a participar en una investigación o su decisión de retirarse nunca debe perturbar la relación médico-paciente.
35. Cuando en la atención de un enfermo las intervenciones probadas han resultado ineficaces o no existen, el médico, después de pedir consejo de experto, con el consentimiento informado del paciente o de un representante legal autorizado, puede permitirse usar intervenciones no comprobadas si, a su juicio, ello da alguna esperanza de salvar la vida, restituir la salud o aliviar el sufrimiento. Siempre que sea posible, tales intervenciones deben ser investigadas a fin de evaluar se seguridad y eficacia. En todos los caso, esa información nueva debe ser registrada y, cuando sea oportuno, puesta a disposición del público.
Adoptada por la 18ª Asamblea Médica Mundial, Helsinki, Finlandia, Junio 1964, y enmendada por la:
29ª Asamblea Médica Mundial, Tokio, Japón, Octubre 1975
35ª Asamblea Médica Mundial, Venecia, Italia, Octubre 1983
41ª Asamblea Médica Mundial, Hong Kong, Septiembre 1989
48ª Asamblea General, Somerset West, Sudáfrica, Octubre 1996
52ª Asamblea General, Edimburgo, Escocia, Octubre 2000
Nota de Clarificación del Párrafo 29, agregada por la Asamblea General de la AMM, Washington, 2002

10
Nota de Clarificación del Párrafo 30, agregada por la Asamblea General de la AMM, Tokio, 2004 59ª Asamblea General, Seúl, Corea, octubre 2008

11
La guía de Buena Práctica Clínica (BPC) es una norma internacional de calidad ética y científica aplicable al diseño, realización, registro y comunicación de los ensayos clínicos en los que participen seres humanos. El cumplimiento de esta norma proporciona una garantía pública de la protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo de acuerdo con los principios de la Declaración de Helsinki, así como también garantiza la credibilidad de los datos del ensayo clínico. El objetivo de la guía de BPC de la Conferencia Internacional de Armonización (Internacional Conference on Harmonization, ICH) es proporcionar una norma única para la Unión Europea, Japón y los Estados Unidos, facilitando de este modo la aceptación mutua de datos clínicos por parte de las autoridades reguladoras de estas jurisdicciones.
Se deberá seguir esta guía cuando se generen datos de ensayos clínicos que se pretendan presentar a las autoridades reguladoras. Los principios que se establecen en esta guía se pueden aplicar también a otras investigaciones clínicas que puedan afectar a la seguridad y el bienestar de los seres humanos. El CEIC del Hospital Provincial de Castellón adopta las Normas de Buena Práctica Clínica y cumplirá sus funciones de acuerdo con las mismas, haciendo especial hincapié en los siguientes apartados:
Capitulo 2. LOS PRINCIPIOS DE LA BPC DE LA ICH 2.1. Los ensayos clínicos deben realizarse de acuerdo con los principios éticos que tienen su origen en la Declaración de Helsinki, y que sean coherentes con la guía de la BPC y la legislación vigente. 2.2. Antes de iniciar un ensayo, deberán considerarse los riesgos e inconvenientes previsibles en relación con el beneficio esperado, tanto para el sujeto individual del ensayo como para la sociedad. Un ensayo deberá iniciarse y continuarse únicamente en el caso de que los beneficios previstos justifiquen los riesgos. 2.3. Los derechos, la seguridad y el bienestar de los sujetos de un ensayo son las consideraciones más importantes y deberán prevalecer sobre los intereses de la ciencia y de la sociedad. 2.4. La información clínica y no clínica disponible sobre un medicamento en investigación deberá ser suficiente para avalar el ensayo clínico propuesto. 2.5. Los ensayos clínicos deberán estar científicamente justificados y estar descritos en un protocolo claro y detallado. 2.6. El ensayo se deberá realizar de acuerdo con el protocolo que previamente ha recibido un dictamen favorable de un CEIC. 2.7. El cuidado médico que reciben los sujetos y las decisiones médicas tomadas en su nombre serán siempre responsabilidad de un médico cualificado o, en su caso, un odontólogo cualificado. 2.8. Cada individuo implicado en la realización de un ensayo deberá estar cualificado, por su titulación, formación y experiencia, para realizar sus tareas y responsabilidades respectivas. 2.9. Se deberá obtener el consentimiento informado, otorgado de forma libre, de cada sujeto antes de su participación en el ensayo clínico.
Normas de la Buena Práctica Clínica (BPC ) (CPMP/ICH/135/95). Resumen de las Normas de Buena Práctica Clínica con referencia explicita a la traducción anotada realizada por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS)

12
2.10. Toda la información del ensayo clínico deberá ser registrada, manejada y archivada de forma que permita su comunicación, interpretación y verificación exactas. 2.11. Se deberá proteger la confidencialidad de los registros que pudieran identificar a los sujetos respetando la privacidad y las normas de confidencialidad de acuerdo con los requisitos legislativos pertinentes. 2.12. Los medicamentos en investigación deberán fabricarse, manejarse y almacenarse de acuerdo con las Normas de Correcta Fabricación (NCF) pertinentes y se deberán utilizar de acuerdo con el protocolo aprobado. 2.13. Se implantarán sistemas con procedimientos que aseguren la calidad de cada aspecto del ensayo. Capitulo 3. COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA ( CEIC) 3.1 Responsabilidades 3.1.1 Un CEIC deberá salvaguardar los derechos, la seguridad y el bienestar de todos los sujetos del ensayo. Deberá prestarse especial atención a los ensayos que puedan incluir sujetos vulnerables. 3.1.2 El CEIC deberá estar en posesión de los siguientes documentos: El protocolo del ensayo y sus modificaciones; el documento del consentimiento informado así como las actualizaciones del mismo propuestas por el investigador, los procedimientos de reclutamiento de sujetos (p.e. anuncios publicitarios); la hoja de información para el sujeto; el Manual del Investigador, la información disponible sobre seguridad, la información referente a la remuneración e indemnizaciones previstas para los sujetos, el currículum vitae actualizado del investigador y/u otra documentación que demuestre la cualificación del investigador así como cualquier otro documento que el CEIC pueda necesitar para cumplir con sus responsabilidades. El CEIC deberá evaluar la documentación que acompaña la solicitud de un ensayo clínico en un tiempo razonable y documentar por escrito sus criterios de evaluación, identificando claramente el ensayo, los documentos revisados así como las fechas de los siguientes documentos: • Dictamen favorable, • Modificaciones solicitadas antes del dictamen favorable, • Dictamen desfavorable y • Retirada o suspensión de cualquier dictamen favorable previo. 3.1.3 El CEIC deberá considerar la idoneidad del investigador para el ensayo propuesto, mediante su currículum vitae actualizado y/o cualquier información pertinente que el Comité solicite. 3.1.4 El CEIC deberá realizar el seguimiento de cada ensayo en marcha con una periodicidad proporcional al riesgo al que se exponen los sujetos del estudio. Este seguimiento será al menos anual. 3.1.5 El CEIC podrá solicitar que se proporcione a los sujetos más información de la señalada en el párrafo 4.8.10 cuando, a su juicio, la información adicional aumente significativamente la protección de los derechos, la seguridad y/o el bienestar de los sujetos. 3.1.6 Cuando se realice un ensayo no terapéutico con el consentimiento del representante legal del sujeto (véase 4.8.12, 4.8.14), el Comité deberá determinar si el protocolo propuesto y el resto de los documentos contemplan adecuadamente las cuestiones éticas y cumplen los requisitos legales para tales ensayos. 3.1.7 En aquellos casos en que el protocolo indique que no es posible obtener el consentimiento previo del sujeto de ensayo o de su representante legal (véase 4.8.15) el Comité deberá determinar si el protocolo propuesto y/u otros documentos contemplan adecuadamente las consideraciones éticas relevantes y cumplen la normativa vigente para tales ensayos (es decir, en situación de urgencia). 3.1.8 El CEIC deberá revisar tanto la cantidad como la forma de compensación a los sujetos del ensayo a fin de asegurar que no existe coacción o influencia indebida sobre los mismos. La contraprestación al sujeto se reducirá proporcionalmente a su participación y no dependerá completamente de que finalice el ensayo.

13
3.1.9 El Comité deberá asegurar que la información referente a la compensación a los sujetos, incluyendo la forma, las cantidades y el calendario de la misma, se refleje en la hoja de información para el sujeto y en alguna información escrita que se le entregue. Se deberá especificar así mismo la forma de prorrateo de la contraprestación. 3.2 Composición, Funciones y actividades 3.2.1 El CEIC debe estar constituido por un número razonable de miembros que conjuntamente posean la cualificación y la experiencia necesaria para revisar y evaluar los aspectos científicos, médicos y éticos del ensayo propuesto. Se recomienda que el CEIC esté integrado por1: a) Al menos cinco miembros. b) Al menos un miembro cuya área principal de interés no sea científica. c) Al menos un miembro que sea independiente del centro del ensayo. Solo aquellos miembros del Comité que sean independientes del investigador y del promotor del ensayo podrán votar u opinar sobre temas relacionados con el ensayo. Debe guardarse el listado de los miembros del Comité con sus respectivas cualificaciones. 3.2.2 El CEIC deberá realizar sus funciones con arreglo a unos procedimientos normalizados de trabajo (PNT), mantener registros por escrito de sus actividades y de las actas de sus reuniones así como seguir la guía de la BPC y la normativa vigente. 3.2.3 El Comité deberá tomar sus decisiones en reuniones convocadas en las que, al menos, debe haber el quórum estipulado en sus PNT. 3.2.4 Sólo los miembros del Comité que participen en la revisión y discusión del estudio deberán votar, opinar y/o aconsejar. 3.2.5 El investigador puede proporcionar información sobre cualquier aspecto del ensayo pero no debe participar en las deliberaciones ni en la votación/dictamen del Comité. 3.2.6 El Comité podrá invitar a expertos, que no sean miembros del mismo, en áreas especiales en las que necesite asesoramiento. 3.3 Procedimientos El CEIC deberá elaborar, documentar por escrito y seguir para su funcionamiento Procedimientos Normalizados de Trabajo (PNT) que incluyan: 3.3.1 La composición del Comité y los requisitos que deben cumplir sus miembros (nombres y cualificaciones), así como la autoridad competente que les acredita. 3.3.2 La forma de convocar a sus miembros, la periodicidad y la celebración de sus reuniones. 3.3.3 Los criterios para la evaluación inicial y el sistema de seguimiento de los ensayos. 3.3.4 La forma de determinar la frecuencia del seguimiento del ensayo clínico, si fuera necesario. 3.3.5 Los casos en que se contemple, conforme a la legislación vigente, la revisión urgente y el dictamen favorable de cualquier modificación menor en los ensayos iniciados que tengan un dictamen favorable del CEIC 3.3.6 Se especificará que no deberá ser admitido ningún sujeto en un ensayo, antes de que el CEIC emita un dictamen favorable por escrito del ensayo. 3.3.7 Se especificará que no deberá realizarse ninguna desviación o modificación del protocolo sin un dictamen previo favorable por escrito del CEIC, salvo cuando sea necesario para proteger a los sujetos de cualquier riesgo inmediato o cuando el cambio afecte solo a aspectos logísticos o administrativos del ensayo (p.e. el cambio de monitor, el número de teléfono) (véase 4.5.2).
1 Composición según el artículo 12 del RD 223/04: al menos 9 miembros para asegurar la independencia de las decisiones, competencia y experiencia. Compuesto por médicos (al menos un farmacólogo clínico), un farmacéutico de hospital, un diplomado universitario en enfermería, al menos un miembro independiente de los centros en que se lleve a cabo el proyecto. Al menos 2 de los miembros deben ser ajenos a las profesiones sanitarias (al menos un licenciado en derecho).

14
3.3.8 Se especificará que el investigador2 deberá informar sin demora al CEIC de: a) Las desviaciones o modificaciones del protocolo realizadas para proteger a los sujetos de riesgos inmediatos (véase 3.3.7, 4.5.2, 4.5.4) b) Las modificaciones que supongan un incremento del riesgo para los sujetos y/o afecten de forma significativa la realización del ensayo (véase 4.10.2) c) Todas las reacciones adversas al medicamento que sean graves e inesperadas. d) Cualquier información nueva que pueda afectar negativamente la seguridad de los sujetos o la realización del ensayo. 3.3.9 La garantía de que el Comité notificará rápidamente y por escrito al investigador3 y a la institución: a) Los dictámenes relacionados con el ensayo. b) La justificación de sus dictámenes. c) Los procedimientos para apelar sus dictámenes. 3.4 Documentos El Comité deberá archivar todos los documentos relevantes (p.e. los PNT escritos, la lista de miembros, la lista de las ocupaciones y/o afiliaciones de los mismos, los documentos presentados, las actas de las reuniones y la correspondencia) durante un periodo de al menos 3 años después de la finalización del ensayo y tenerlos a disposición de las autoridades competentes. Los investigadores, los promotores o las autoridades competentes. Los investigadores, los promotores o las autoridades competentes pueden solicitar al CEIC sus procedimientos de trabajo por escrito y la lista de sus miembros.
2 De acuerdo con el Real Decreto 223/2004, es el promotor el responsable de este tipo de notificaciones 3 De acuerdo con el Real Decreto 223/2004, el CEIC se lo notifica al promotor, a la AEMPS y si se trata del CEIC de referencia se lo notificará a los demás CEIC implicados.

15
Los ensayos clínicos con medicamentos han sido objeto de regulación en el título III de la Ley 25/1990, de 20 de diciembre, del Medicamento, y su desarrollo reglamentario en esta materia mediante el Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos.
La Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2001, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos de uso humano, ha venido a armonizar las legislaciones de los Estados miembros de la Unión Europea sobre ensayos clínicos con medicamentos en seres humanos, lo que ha hecho necesario la modificación de la legislación española vigente en esta materia. En este sentido, el artículo 125 de la Ley 53/2002, de 30 de diciembre, de medidas fiscales, administrativas y del orden social, ha introducido diversas modificaciones en el título III de la Ley 25/1990, de 20 de diciembre, del Medicamento, con la finalidad de eliminar las discrepancias de la citada norma con la Directiva 2001/20/CE, dotando así de la necesaria cobertura legal a este reglamento. Asimismo, se ha tenido en cuenta en la elaboración lo dispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica.
Este real decreto, por tanto, viene a incorporar en su totalidad al ordenamiento jurídico interno la Directiva 2001/20/CE, y sustituye al actual Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos, dotando de nuevo desarrollo reglamentario a la Ley 25/1990, de 20 de diciembre, del Medicamento, en cuanto a ensayos clínicos se refiere.
En este real decreto se han tenido en cuenta los principios básicos para la realización de ensayos clínicos con seres humanos fundamentados en la protección de los derechos humanos y la dignidad del ser humano respecto a la aplicación de la biología y la medicina, reflejados en la Declaración de Helsinki y en el Convenio de Oviedo sobre los derechos humanos y la biomedicina, así como las normas para la adecuada protección de los datos personales.
Por otra parte, se habilitan nuevos procedimientos administrativos para la autorización de los ensayos clínicos por parte de la Administración General del Estado, lo que supone agilizar y simplificar los trámites actualmente existentes, equiparando las distintas reglamentaciones en esta materia de los Estados miembros de la Unión Europea y permitiendo el mutuo reconocimiento entre las autoridades sanitarias de dichos Estados respecto a los resultados de los ensayos clínicos realizados.
En cuanto a la evaluación del ensayo clínico, en el ámbito que les concierne, por parte del Comité Ético de Investigación Clínica, además del establecimiento de plazos máximos para dicha evaluación se recoge la exigencia del dictamen único. En este sentido, en los ensayos clínicos multicéntricos, en los que participen dos o más centros ubicados en España, se designará un comité de referencia entre los distintos comités éticos implicados para la emisión del citado dictamen único, lo cual hace necesaria la creación de un organismo de coordinación denominado Centro Coordinador de Comités Éticos de Investigación Clínica.
Especial mención merece la obligación de aplicar las normas de buena práctica clínica a la planificación, realización, registro y comunicación de todos los ensayos clínicos que se realicen en España, como conjunto de requisitos éticos y científicos de calidad reconocidos a escala internacional y como garantía de la protección de los derechos, la seguridad y el bienestar de los sujetos del ensayo, así como la fiabilidad de sus resultados.
REAL DECRETO 223/2004 , de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Ministerio de Sanidad y Consumo

16
La verificación de la conformidad con las normas de buenas práctica clínica y la inspección de los datos, la información y los documentos para comprobar que han sido producidos, registrados y comunicados correctamente, así como el cumplimiento de las normas de correcta fabricación en la elaboración, importación y etiquetado de los medicamentos en investigación, la vigilancia de la seguridad de estos medicamentos y las comunicaciones entre las autoridades competentes en la materia, han sido también recogidos y tenidos en cuenta como requisitos indispensables para justificar la participación de seres humanos en los ensayos clínicos.
Por último, complementan este real decreto las normas de buena práctica clínica y las instrucciones para la realización de ensayos clínicos en España, o en su caso, las directrices de la Comisión Europea, que se publicarán por el Ministerio de Sanidad y Consumo.
Este real decreto mediante el que se incorpora al ordenamiento jurídico interno la Directiva 2001/20/CE desarrolla el título III de la Ley 25/1990, de 20 de diciembre, del Medicamento, y se dicta al amparo de lo dispuesto en el artículo 149.1.16.ª de la Constitución Española, en concordancia con el artículo 2.1 de la citada ley.
En la elaboración de este real decreto han sido oídas las comunidades autónomas y los sectores afectados.
En su virtud, a propuesta de la Ministra de Sanidad y Consumo, con la aprobación previa de la Ministra de Administraciones Públicas, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 6 de febrero de 2004,
D I S P O N G O :
CAPÍTULO I
Disposiciones generales
Artículo 1. Ámbito de aplicación.
1. Este real decreto se aplicará a los ensayos clínicos con medicamentos de uso humano que se realicen en España.
A estos efectos, no tendrá la consideración de ensayo clínico la administración de un medicamento en investigación a un solo paciente, en el ámbito de la práctica médica habitual y con el único propósito de conseguir un beneficio terapéutico para el paciente, que se regirá por lo dispuesto sobre uso compasivo en el artículo 28.
La práctica médica y la libertad profesional de prescripción del médico no ampararán, en ningún caso, la realización de ensayos clínicos no autorizados ni la utilización de remedios secretos o no declarados a la autoridad sanitaria.
2. Se excluyen del ámbito de aplicación de este real decreto los estudios observacionales, definidos en el artículo 2.c), que se regirán por su normativa específica.
3. Quedan prohibidos los ensayos clínicos con medicamentos de terapia génica que produzcan modificaciones en la identidad génica de la línea germinal del sujeto.
Artículo 2. Definiciones.
A los efectos de lo dispuesto en este real decreto, se aplicarán las siguientes definiciones:
a) Ensayo clínico: toda investigación efectuada en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de detectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia.
A estos efectos, se aplicará la definición de medicamento en investigación prevista en el párrafo d).

17
b) Ensayo clínico multicéntrico: ensayo clínico realizado de acuerdo con un protocolo único pero en más de un centro y, por tanto, realizado por más de un investigador.
c) Estudio observacional: estudio en el que los medicamentos se prescriben de la manera habitual, de acuerdo con las condiciones normales de la práctica clínica (aquellas establecidas en la autorización de comercialización). La asignación de un paciente a una estrategia terapéutica concreta no estará decidida de antemano por un protocolo de ensayo, sino que estará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica, y se utilizarán métodos epidemiológicos para el análisis de los datos recogidos.
d) Medicamento en investigación: forma farmacéutica de una sustancia activa o placebo que se investiga o se utiliza como referencia en un ensayo clínico, incluidos los productos con autorización de comercialización cuando se utilicen o combinen (en la formulación o en el envase) de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada, o para obtener más información sobre un uso autorizado.
e) Promotor: individuo, empresa, institución u organización responsable del inicio, gestión y/o financiación de un ensayo clínico.
f) Monitor: profesional capacitado con la necesaria competencia clínica, elegido por el promotor, que se encarga del seguimiento directo de la realización del ensayo. Sirve de vínculo entre el promotor y el investigador principal, cuando éstos no concurran en la misma persona.
g) Organización de investigación por contrato (CRO): persona física o jurídica contratada por el promotor para realizar funciones o deberes del promotor en relación con el ensayo clínico.
h) Investigador: médico o persona que ejerce una profesión reconocida para llevar a cabo investigaciones en razón de su formación científica y de su experiencia en la atención sanitaria requerida. El investigador es responsable de la realización del ensayo clínico en un centro. Si es un equipo el que realiza el ensayo en un centro, el investigador es el responsable del equipo y puede denominarse investigador principal.
i) Investigador coordinador: investigador responsable de la coordinación de los investigadores de todos los centros españoles que participan en un ensayo clínico multicéntrico.
j) Manual del investigador: conjunto de datos clínicos y no clínicos sobre el medicamento en investigación pertinente para el estudio de dicho medicamento en seres humanos.
k) Protocolo: documento donde se describen los objetivos, el diseño, la metodología, las consideraciones estadísticas y la organización de un ensayo. El término protocolo se refiere al protocolo original, a sus sucesivas versiones y a sus modificaciones.
l) Sujeto del ensayo: individuo que participa en un ensayo clínico, bien recibiendo el medicamento en investigación, bien como control.
m) Consentimiento informado: decisión, que debe figurar por escrito y estar fechada y firmada, de participar en un ensayo clínico adoptada voluntariamente por una persona capaz de dar su consentimiento tras haber sido debidamente informada y documentada acerca de su naturaleza, importancia, implicaciones y riesgos.
En el supuesto de que el sujeto tenga un impedimento para escribir, el consentimiento podrá otorgarse en casos excepcionales de forma oral en presencia de al menos un testigo.
Cuando el sujeto del ensayo no sea una persona capaz para dar su consentimiento, la decisión deberá adoptarse por su representante legal en los términos previstos en el artículo 7.
n) Comité Ético de Investigación Clínica (CEIC):

18
organismo independiente, constituido por profesionales sanitarios y miembros no sanitarios, encargado de velar por la protección de los derechos, seguridad y bienestar de los sujetos que participen en un ensayo y de ofrecer garantía pública al respecto, mediante un dictamen sobre el protocolo del ensayo, la idoneidad de los investigadores y la adecuación de las instalaciones, así como los métodos y los documentos que vayan a utilizarse para informar a los sujetos del ensayo con el fin de obtener su consentimiento informado. En el caso de ensayos clínicos multicéntricos, el Comité Ético de Investigación Clínica encargado de emitir el dictamen se denomina Comité Ético de Investigación Clínica de Referencia.
ñ) Inspección: revisión oficial por una autoridad competente de los documentos, las instalaciones, los archivos, los sistemas de garantía de calidad y cualesquiera otros elementos que la autoridad competente considere relacionados con el ensayo clínico y que puedan encontrarse en el lugar del ensayo, en las instalaciones del promotor y/o de la organización de investigación por contrato, o en cualquier otro establecimiento que la autoridad competente considere oportuno inspeccionar.
o) Acontecimiento adverso: cualquier incidencia perjudicial para la salud en un paciente o sujeto de ensayo clínico tratado con un medicamento, aunque no tenga necesariamente relación causal con dicho tratamiento.
p) Reacción adversa: toda reacción nociva y no intencionada a un medicamento en investigación, independientemente de la dosis administrada.
q) Acontecimiento adverso grave o reacción adversa grave: cualquier acontecimiento adverso o reacción adversa que, a cualquier dosis, produzca la muerte, amenace la vida del sujeto, haga necesaria la hospitalización o la prolongación de ésta, produzca invalidez o incapacidad permanente o importante, o dé lugar a una anomalía o malformación congénita. A efectos de su notificación, se tratarán también como graves aquellas sospechas de acontecimiento adverso o reacción adversa que se consideren importantes desde el punto de vista médico, aunque no cumplan los criterios anteriores.
r) Reacción adversa inesperada: reacción adversa cuya naturaleza o gravedad no se corresponde con la información referente al producto (por ejemplo, el manual del investigador en el caso de un medicamento en investigación no autorizado para su comercialización, o la ficha técnica del producto en el caso de un medicamento autorizado).
CAPÍTULO II
Protección de los sujetos del ensayo
Artículo 3. Postulados éticos.
1. Sólo se podrá iniciar un ensayo clínico cuando el Comité Ético de Investigación Clínica que corresponda y la Agencia Española de Medicamentos y Productos Sanitarios hayan considerado que los beneficios esperados para el sujeto del ensayo y para la sociedad justifican los riesgos; asimismo, sólo podrá proseguir si se supervisa permanentemente el cumplimiento de este criterio.
2. Los ensayos clínicos se realizarán en condiciones de respeto a los derechos del sujeto y a los postulados éticos que afectan a la investigación biomédica con seres humanos. En particular, se deberá salvaguardar la integridad física y mental del sujeto, así como su intimidad y la protección de sus datos, de acuerdo con la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal. Se obtendrá y documentará el consentimiento informado de cada uno de los sujetos del ensayo, libremente expresado, antes de su inclusión en el ensayo en los términos previstos en el artículo 7 de este real decreto.
3. Sólo se podrán realizar ensayos clínicos cuando se cumplan todos los requisitos siguientes:
a) Disponer de suficientes datos científicos y, en particular, ensayos farmacológicos y toxicológicos en animales, que garanticen que los riesgos que implica en la persona en que se realiza son admisibles.

19
b) Que el estudio se base en los conocimientos disponibles, la información buscada suponga, presumiblemente, un avance en el conocimiento científico sobre el ser humano o para mejorar su estado de salud y su diseño minimice los riesgos para los sujetos participantes en él.
c) Que los riesgos e inconvenientes previsibles para los sujetos del ensayo se hayan ponderado con respecto a los beneficios previsibles para cada sujeto del ensayo y futuros pacientes.
4. Con el fin de garantizar una protección óptima de la salud y los derechos de los sujetos, no se podrán llevar a cabo investigaciones obsoletas o repetitivas.
5. El ensayo clínico debe estar diseñado para reducir al mínimo posible el dolor, la incomodidad, el miedo y cualquier otro riesgo previsible en relación con la enfermedad y edad o grado de desarrollo del sujeto ; tanto el umbral de riesgo como el grado de incomodidad deben ser definidos de forma específica y monitorizados durante el ensayo, especialmente cuando los sujetos del ensayo sean menores, adultos incapaces o constituyan una población especialmente vulnerable en razón de su situación económica, médica o social.
6. El tratamiento, comunicación y cesión de los datos de carácter personal de los sujetos participantes en el ensayo se ajustará a lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y constará expresamente en el consentimiento informado.
7. La atención sanitaria que se dispense y las decisiones médicas que se adopten sobre los sujetos serán responsabilidad de un médico o de un odontólogo debidamente cualificados.
8. Los sujetos participantes en ensayos clínicos sin beneficio potencial directo para el sujeto en investigación recibirán del promotor la compensación pactada por las molestias sufridas. La cuantía de la compensación económica estará en relación con las características del ensayo, pero en ningún caso será tan elevada como para inducir a un sujeto a participar por motivos distintos del interés por el avance científico.
La contraprestación que se hubiera pactado por la participación voluntaria en el ensayo se percibirá en todo caso, si bien se reducirá proporcionalmente según la participación del sujeto en la experimentación, en el supuesto de que decida revocar su consentimiento y abandonar el ensayo.
En los casos extraordinarios de investigaciones sin beneficio potencial directo para el sujeto en investigación en menores e incapaces, para evitar la posible explotación de estos sujetos, no se producirá ninguna compensación económica por parte del promotor, a excepción del reintegro de los gastos extraordinarios y pérdidas de productividad que se deriven de la participación del sujeto en el ensayo.
9. Los sujetos que participen en ensayos con un posible beneficio potencial directo para el sujeto de investigación o sus representantes legales únicamente podrán recibir del promotor el reintegro de los gastos extraordinarios y pérdidas de productividad que se deriven de su participación en el ensayo.
10. Los sujetos del ensayo dispondrán de un punto donde puedan obtener mayor información sobre el ensayo, que constará en la hoja de información para el sujeto.
Artículo 4. De los ensayos clínicos con menores.
Sin perjuicio de la aplicación de las disposiciones generales establecidas en el artículo anterior, solo se podrán realizar ensayos clínicos en menores de edad cuando se cumplan, además, las siguientes condiciones especiales:
a) Que los ensayos sean de interés específico para la población que se investiga, y solo cuando dicha investigación sea esencial para validar datos procedentes de ensayos clínicos efectuados en personas capaces de otorgar su consentimiento informado u obtenidos por otros medios de investigación. Además, la investigación deberá guardar relación directa con alguna

20
enfermedad que padezca el menor o bien ser de naturaleza tal que solo pueda ser realizada en menores.
b) Que el bienestar del sujeto prevalezca siempre sobre los intereses de la ciencia y de la sociedad, y existan datos que permitan prever que los beneficios esperados superan los riesgos o que el riesgo que conlleva el ensayo es mínimo.
c) Que la obtención del consentimiento informado se ajuste a lo especificado en el artículo. 7.3.
d) Que el protocolo sea aprobado por un Comité Ético de Investigación Clínica que cuente con expertos en pediatría o que haya recabado asesoramiento sobre las cuestiones clínicas, éticas y psicosociales en el ámbito de la pediatría.
e) Que se sigan las directrices científicas correspondientes de la Agencia Europea para la Evaluación de Medicamentos.
Artículo 5. De los ensayos clínicos con adultos incapacitados.
Sin perjuicio de la aplicación de las disposiciones generales establecidas en el artículo 3, solo se podrán realizar ensayos clínicos en adultos que no estén en condiciones de dar su consentimiento informado y que no lo hayan dado con anterioridad al comienzo de su incapacidad, cuando se cumplan, además, las siguientes condiciones especiales:
a) Que los ensayos sean de interés específico para la población que se investiga, y dicha investigación sea esencial para validar datos procedentes de ensayos clínicos efectuados en personas capaces de otorgar su consentimiento informado u obtenidos por otros medios de investigación. Además, la investigación deberá guardar relación directa con alguna enfermedad que padezca el adulto incapaz, y que ésta le debilite o ponga en peligro su vida.
b) Que el bienestar del sujeto prevalezca sobre los intereses de la ciencia y de la sociedad, y existan datos que permitan prever que reporta algún beneficio al paciente que prevalezca sobre los riesgos o no produzca ningún riesgo.
c) Que el consentimiento informado se ajuste a lo especificado en el artículo 7.3. En todo caso, los sujetos no deben haberse negado a dar su consentimiento informado con anterioridad al comienzo de su incapacidad.
d) Que el protocolo sea aprobado por un Comité Ético de Investigación Clínica que cuente con expertos en la enfermedad en cuestión o que haya recabado asesoramiento de este tipo de expertos sobre las cuestiones clínicas, éticas y psicosociales en el ámbito de la enfermedad y del grupo de pacientes afectado.
Artículo 6. De los ensayos clínicos sin beneficio directo para la salud de los sujetos.
1. En los ensayos clínicos sin beneficio potencial directo para la salud de los sujetos participantes, el riesgo que estos sujetos asuman estará justificado en razón del beneficio esperado para la colectividad.
2. En menores y en sujetos incapacitados podrán realizarse ensayos sin beneficio potencial directo para el sujeto únicamente si, además de tenerse en cuenta lo dispuesto en los artículos 4 y 5, el Comité Ético de Investigación Clínica considera que se cumplen los siguientes requisitos:
a) Que se adoptan las medidas necesarias para garantizar que el riesgo sea mínimo.
b) Que las intervenciones a que van a ser sometidos los sujetos del ensayo son equiparables a las que corresponden a la práctica médica habitual en función de su situación médica, psicológica o social.
c) Que del ensayo se pueden obtener conocimientos relevantes sobre la enfermedad o situación objeto de investigación, de vital importancia para entenderla, paliarla o curarla.

21
d) Que estos conocimientos no pueden ser obtenidos de otro modo.
e) Que existen garantías sobre la correcta obtención del consentimiento informado, de acuerdo con lo contemplado en el artículo 7.
3. En mujeres gestantes o en período de lactancia, sólo se podrán realizar ensayos clínicos sin beneficio potencial directo para ellas cuando el Comité Ético de Investigación Clínica concluya que no suponen ningún riesgo previsible para su salud ni para la del feto o niño, y que se obtendrán conocimientos útiles y relevantes sobre el embarazo o la lactancia.
Artículo 7. Del consentimiento informado.
1. La obtención del consentimiento informado debe tener en cuenta los aspectos indicados en las recomendaciones europeas al respecto y que se recogen en las instrucciones para la realización de ensayos clínicos en España o, en su caso, en las directrices de la Unión Europea.
2. El sujeto del ensayo deberá otorgar su consentimiento después de haber entendido, mediante una entrevista previa con el investigador o un miembro del equipo de investigación, los objetivos del ensayo, sus riesgos e inconvenientes, así como las condiciones en las que se llevará a cabo, y después de haber sido informado de su derecho a retirarse del ensayo en cualquier momento sin que ello le ocasione perjuicio alguno.
El consentimiento se documentará mediante una hoja de información para el sujeto y el documento de consentimiento. La hoja de información contendrá únicamente información relevante, expresada en términos claros y comprensibles para los sujetos, y estará redactada en la lengua propia del sujeto.
3. Cuando el sujeto del ensayo no sea una persona capaz para dar su consentimiento o no esté en condiciones de hacerlo, la decisión deberá adoptarse, teniendo en cuenta lo indicado en este artículo.
a) Si el sujeto del ensayo es menor de edad:
1.º Se obtendrá el consentimiento informado previo de los padres o del representante legal del menor ; el consentimiento deberá reflejar la presunta voluntad del menor y podrá retirarse en cualquier momento sin perjuicio alguno para él. Cuando el menor tenga 12 o más años, deberá prestar además su consentimiento para participar en el ensayo.
2.º El menor recibirá, de personal que cuente con experiencia en el trato con menores, una información sobre el ensayo, los riesgos y los beneficios adecuada a su capacidad de entendimiento.
3.º El investigador aceptará el deseo explícito del menor de negarse a participar en el ensayo o de retirarse en cualquier momento, cuando éste sea capaz de formarse una opinión en función de la información recibida.
4.º El promotor pondrá en conocimiento del Ministerio Fiscal las autorizaciones de los ensayos clínicos cuya población incluya a menores.
b) Si el sujeto es un adulto sin capacidad para otorgar su consentimiento informado:
1.º Deberá obtenerse el consentimiento informado de su representante legal, tras haber sido informado sobre los posibles riesgos, incomodidades y beneficios del ensayo. El consentimiento deberá reflejar la presunta voluntad del sujeto y podrá ser retirado en cualquier momento sin perjuicio para éste.
2.º Cuando las condiciones del sujeto lo permitan, este deberá prestar además su consentimiento para participar en el ensayo, después de haber recibido toda la información pertinente adaptada a su nivel de entendimiento. En este caso, el investigador deberá tener en cuenta la voluntad de la persona incapaz de retirarse del ensayo.

22
4. Cuando el ensayo clínico tenga un interés específico para la población en la que se realiza la investigación y lo justifiquen razones de necesidad en la administración del medicamento en investigación, podrá someterse a un sujeto a un ensayo clínico sin obtener el consentimiento previo en los siguientes casos:
a) Si existe un riesgo inmediato grave para la integridad física o psíquica del sujeto, se carece de una alternativa terapéutica apropiada en la práctica clínica y no es posible obtener su consentimiento o el de su representante legal. En este caso, siempre que las circunstancias lo permitan, se consultará previamente a las personas vinculadas a él por razones familiares o de hecho.
b) Si el sujeto no es capaz para tomar decisiones debido a su estado físico o psíquico y carece de representante legal. En este caso, el consentimiento lo prestarán las personas vinculadas a él por razones familiares o de hecho.
En ambos casos, esta eventualidad y la forma en que se procederá debe hallarse prevista en la documentación del ensayo aprobada por el Comité Ético de Investigación Clínica, y el sujeto o su representante legal será informado en cuanto sea posible y deberá otorgar su consentimiento para continuar en el ensayo si procediera.
5. El sujeto participante en un ensayo clínico, o su representante legal, podrán revocar su consentimiento en cualquier momento, sin expresión de causa y sin que por ello se derive para el sujeto participante responsabilidad ni perjuicio alguno.
Artículo 8. Del seguro u otra garantía financiera de los sujetos del ensayo.
1. Sólo podrá realizarse un ensayo clínico con medicamentos en investigación si, previamente, se ha concertado un seguro u otra garantía financiera que cubra los daños y perjuicios que como consecuencia del ensayo puedan resultar para la persona en que hubiera de realizarse, salvo que el ensayo se refiera únicamente a medicamentos autorizados en España, su utilización en el ensayo se ajuste a las condiciones de uso autorizadas y el Comité Ético de Investigación Clínica considere que las intervenciones a las que serán sometidos los sujetos por su participación en el ensayo suponen un riesgo equivalente o inferior al que correspondería a su atención en la práctica clínica habitual.
2. El promotor del ensayo es el responsable de la contratación de dicho seguro de responsabilidad o de la garantía financiera y éstos cubrirán las responsabilidades del promotor, del investigador principal y sus colaboradores, y del hospital o centro donde se lleve a cabo el ensayo clínico.
3. En el supuesto previsto al final del apartado 1 de este artículo, cuando no se concierte seguro u otra garantía financiera o, por cualquier circunstancia, el seguro o la garantía financiera concertados no cubran enteramente los daños, el promotor del ensayo clínico, el investigador principal y el hospital o centro donde se realice el ensayo serán responsables solidariamente, sin necesidad de que medie culpa, del daño que en su salud sufra el sujeto sometido al ensayo clínico, así como de los perjuicios económicos que se deriven, incumbiéndoles la carga de la prueba de que no son consecuencia del ensayo clínico o de las medidas terapéuticas o diagnósticas que se adopten durante su realización. Ni la autorización administrativa, ni el dictamen favorable del Comité Ético de Investigación Clínica eximirán de responsabilidad al promotor del ensayo clínico, al investigador principal y sus colaboradores o al hospital o centro donde se realice el ensayo clínico en estas circunstancias.
4. Se presume, salvo prueba en contrario, que los daños que afecten a la salud del sujeto del ensayo durante su realización y en el año siguiente a la terminación del tratamiento se han producido como consecuencia del ensayo. Sin embargo, una vez concluido el año, el sujeto del ensayo esta obligado a probar el nexo entre el ensayo y el daño producido.
5. A los efectos del régimen de responsabilidad previsto en este artículo, serán objeto de resarcimiento todos los gastos derivados del menoscabo en la salud o estado físico del sujeto sometido al ensayo clínico, así como los perjuicios económicos que se deriven directamente de

23
dicho menoscabo, siempre que éste no sea inherente a la patología objeto de estudio, o se incluya dentro de las reacciones adversas propias de la medicación prescrita para dicha patología, así como la evolución propia de su enfermedad como consecuencia de la ineficacia del tratamiento.
6. El importe mínimo que en concepto de responsabilidad estará garantizado será de 250.000 euros por sujeto sometido a ensayo clínico, como indemnización a tanto alzado. En caso de que la indemnización se fije como renta anual constante o creciente, el límite de la cobertura del seguro o de la garantía financiera será de al menos 25.000 euros anuales por cada sujeto sometido al ensayo clínico, pudiéndose establecer como capital asegurado máximo o como importe máximo de la garantía financiera un sublímite por ensayo clínico y año de 2.500.000 euros.
Se autoriza al Ministerio de Sanidad y Consumo para revisar los límites anteriormente establecidos.
7. Cuando el promotor e investigador principal sean la misma persona y el ensayo clínico se realice en un centro sanitario dependiente de una Administración pública, ésta podrá adoptar las medidas que considere oportunas para facilitar la garantía de los riesgos específicos derivados del ensayo en los términos señalados en los apartados anteriores, con el objeto de fomentar la investigación.
CAPÍTULO III
De los Comités Éticos de Investigación Clínica
Artículo 9. Del Centro Coordinador de los Comités Éticos de Investigación Clínica.
1. Con el objeto de facilitar el dictamen único se creará el Centro Coordinador de Comités Éticos de Investigación Clínica. Dicha organización se constituye como la unidad técnica operativa que tiene como objetivo facilitar que los Comité Ético de Investigación Clínica acreditados por las comunidades autónomas puedan compartir estándares de calidad y criterios de evaluación adecuados y homogéneos y favorecer la agilidad en el proceso de obtención del dictamen único.
2. El Centro coordinador de comités éticos de investigación clínica se adscribe al Ministerio de Sanidad y Consumo, a través de la Dirección General de Farmacia y Productos Sanitarios.
3. El Centro Coordinador de Comités Éticos de Investigación Clínica colaborará con las autoridades competentes de las comunidades autónomas y rendirá cuentas de su actividad en el Consejo Interterritorial del Sistema Nacional de Salud.
4. El Centro Coordinador de Comités Éticos de Investigación Clínica desarrollará las siguientes actividades:
a) Facilitar el dictamen único en los ensayos multicéntricos.
b) Coordinar con las comunidades autónomas el desarrollo de un sistema informático de comunicación entre Comités Éticos de Investigación Clínica.
c) Gestionar la base de datos de ensayos clínicos de la red nacional de Comités Éticos de Investigación Clínica.
d) Promover criterios de evaluación comunes en los Comités Éticos de Investigación Clínica.
e) Promover la formación de los miembros de los Comités Éticos de Investigación Clínica.
f) Promover foros de debate entre Comités Éticos de Investigación Clínica.
g) Actuar como punto de contacto para proporcionar información sobre el funcionamiento de la red nacional de Comités Éticos de Investigación Clínica.

24
h) Proporcionar asesoramiento a los Comités Éticos de Investigación Clínica en cuestiones de procedimiento.
i) Elaborar la memoria anual de actividades.
Ap. 2 modificado por disp. final 1 de Real Decreto núm. 590/2005, de 20 mayo.
Artículo 10. Funciones de los Comités Éticos de Investigación Clínica.
Los Comités Éticos de Investigación Clínica desempeñarán las siguientes funciones:
a) Evaluar los aspectos metodológicos, éticos y legales de los ensayos clínicos que les sean remitidos, de conformidad con lo establecido en la sección 2.ª del capítulo IV.
b) Evaluar las modificaciones relevantes de los ensayos clínicos autorizados.
c) Realizar un seguimiento del ensayo, desde su inicio hasta la recepción del informe final.
Artículo 11. Acreditación de los Comités Éticos de Investigación Clínica.
Los Comités Éticos de Investigación Clínica serán acreditados por la autoridad sanitaria competente en cada comunidad autónoma, quien determinará el ámbito geográfico e institucional de cada comité. Dicha acreditación deberá ser renovada periódicamente por dicha autoridad sanitaria según los procedimientos y plazos que ésta determine. Tanto la acreditación inicial como sus renovaciones deberán ser notificadas a la Agencia Española de Medicamentos y Productos Sanitarios y al Centro Coordinador de Comités Éticos de Investigación Clínica.
Artículo 12. Composición de los Comités Éticos de Investigación Clínica.
1. El Comité Ético de Investigación Clínica deberá estar constituido por al menos nueve miembros, de manera que se asegure la independencia de sus decisiones, así como su competencia y experiencia en relación con los aspectos metodológicos, éticos y legales de la investigación, la farmacología y la práctica clínica asistencial en medicina hospitalaria y extrahospitalaria.
2. Entre los miembros del citado comité figurarán médicos, uno de los cuales será farmacólogo clínico; un farmacéutico de hospital, y un Diplomado universitario en Enfermería.
Al menos un miembro deberá ser independiente de los centros en los que se lleven a cabo proyectos de investigación que requieran la evaluación ética por parte del comité.
Al menos dos miembros deben ser ajenos a las profesiones sanitarias, uno de los cuales deberá ser Licenciado en Derecho.
3. Se garantizará un sistema de renovación de miembros que permita nuevas incorporaciones de forma regular, a la vez que se mantiene la experiencia del comité.
4. Tal como establece el artículo 4 de la Ley 25/1990, de 20 de diciembre, del Medicamento, la pertenencia a un Comité Ético de Investigación Clínica será incompatible con cualquier clase de intereses derivados de la fabricación y venta de medicamentos y productos sanitarios.
Artículo 13. Requisitos mínimos respecto a los medios e infraestructura de los Comités Éticos de Investigación Clínica.
Las autoridades sanitarias de las comunidades autónomas correspondientes asegurarán que cada Comité
Ético de Investigación Clínica acreditado cuente al menos con los siguientes medios:
a) Instalaciones específicas que permitan la realización de su trabajo, en condiciones que garanticen la confidencialidad. Deberán disponer de un espacio apropiado para la secretaría del comité, para la realización de las reuniones y para el manejo y archivo de documentos confidenciales.

25
b) Equipamiento informático con capacidad suficiente para manejar toda la información generada por el comité y disponibilidad de un sistema rápido de transmisión de información.
c) Personal administrativo y técnico que permita al comité poder ejercer de manera apropiada sus funciones.
Artículo 14. Normas generales de funcionamiento de los Comités Éticos de Investigación Clínica.
1. Ni el Comité Ético de Investigación Clínica ni ninguno de sus miembros podrán percibir directa ni indirectamente remuneración alguna por parte del promotor del ensayo.
2. Los Comités Éticos de Investigación Clínica deberán elaborar y seguir para su funcionamiento unos procedimientos normalizados de trabajo que como mínimo se referirán a:
a) La composición y requisitos que deben cumplir sus miembros.
b) La periodicidad de las reuniones, que al menos deberá ser mensual.
c) El procedimiento para convocar a sus miembros.
d) Los aspectos administrativos, incluyendo la documentación que debe presentarse.
e) Los casos en que se pueda realizar una revisión rápida de la documentación correspondiente a un ensayo clínico y el procedimiento que debe seguirse en estos casos.
f) La evaluación inicial de los protocolos y sistema de seguimiento de los ensayos.
g) Los mecanismos de toma de decisiones.
h) La preparación y aprobación de las actas de las reuniones.
i) El archivo y conservación de la documentación del comité y de la relacionada con los ensayos clínicos evaluados.
3. En los casos que exista Comisión de Investigación o Comité de Ética Asistencial, deberá formar parte del comité un miembro de cada una de ellas.
4. Cuando el Comité Ético de Investigación Clínica no reúna los conocimientos y experiencia necesarios para evaluar un determinado ensayo clínico recabará el asesoramiento de alguna persona experta no perteneciente al comité, que respetará el principio de confidencialidad. De esta manera:
a) Cuando el comité evalúe protocolos de investigación clínica con procedimientos quirúrgicos, técnicas diagnósticas o productos sanitarios, contará con el asesoramiento de al menos una persona experta en el procedimiento o tecnología que se vaya a evaluar.
b) Cuando el comité evalúe ensayos clínicos que se refieran a menores o a sujetos incapacitados, contará con el asesoramiento de al menos una persona con experiencia en el tratamiento de la población que se incluya en el ensayo.
5. El investigador principal o los colaboradores de un ensayo clínico no podrán participar en la evaluación, ni en el dictamen de su propio protocolo, aun cuando sean miembros del comité.
6. Cada reunión del comité quedará recogida en el acta correspondiente, en la que se detallarán como mínimo los miembros asistentes, que para cada estudio evaluado se han ponderado los aspectos contemplados en este real decreto y la decisión adoptada sobre cada ensayo.
CAPÍTULO IV
De la intervención sobre los ensayos clínicos con medicamentos
SECCIÓN 1.ª DISPOSICIONES COMUNES

26
Artículo 15. Requisitos para la realización de ensayos clínicos.
Para la realización de ensayos clínicos con medicamentos se precisará del previo dictamen favorable del Comité Ético de Investigación Clínica correspondiente, de la conformidad de la dirección de cada uno de los centros donde el ensayo vaya a realizarse y de la autorización de la Agencia Española de Medicamentos y Productos Sanitarios.
El dictamen y la autorización citados en el párrafo anterior podrán solicitarse de forma simultánea o no, según las preferencias del promotor.
SECCIÓN 2ª DEL DICTAMEN DE LOS COMITÉS ÉTICOS DE INVESTIGACIÓN CLÍNICA
Artículo 16. Iniciación del procedimiento.
1. El promotor deberá solicitar por escrito el dictamen del Comité Ético de Investigación Clínica, de acuerdo con las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
2. La solicitud deberá acompañarse de la siguiente documentación:
a) El protocolo.
b) El manual del investigador.
c) Los documentos referentes al consentimiento informado, incluyendo la hoja de información para el sujeto de ensayo.
d) Los documentos sobre la idoneidad del investigador y sus colaboradores.
e) Los documentos sobre la idoneidad de las instalaciones.
f) Las cantidades y el modo en que los investigadores y sujetos puedan ser, en su caso, remunerados o indemnizados por su participación en el ensayo clínico, así como los elementos pertinentes de todo contrato previsto entre el promotor y el centro.
g) Una copia de la póliza del seguro o del justificante de la garantía financiera del ensayo clínico o un certificado de ésta, cuando proceda.
h) En los casos previstos en el artículo 8.3 de ausencia de seguro o de seguro con cobertura parcial, deberá acompañarse documento firmado de asunción de responsabilidad en caso de daños producidos como consecuencia del ensayo.
i) Los procedimientos y material utilizado para el reclutamiento de los sujetos del ensayo.
j) El compromiso de los investigadores que está previsto que participen en el ensayo.
Artículo 17. Criterios de evaluación para la emisión del dictamen.
1. El Comité Ético de Investigación Clínica correspondiente evaluará el protocolo, el manual del investigador y el resto de la documentación que acompañe a la solicitud y emitirá su dictamen tomando en consideración, en particular, las siguientes cuestiones:
a) La pertinencia del ensayo clínico, teniendo en cuenta el conocimiento disponible.
b) La pertinencia de su diseño para obtener conclusiones fundamentadas con el número adecuado de sujetos en relación con el objetivo del estudio.
c) Los criterios de selección y retirada de los sujetos del ensayo, así como la selección equitativa de la muestra.
d) La justificación de los riesgos e inconvenientes previsibles en relación con los beneficios esperables para los sujetos del ensayo, para otros pacientes y para la comunidad, teniendo en cuenta el principio de protección de los sujetos del ensayo desarrollado en el artículo 3.

27
e) La justificación del grupo control (ya sea placebo o un tratamiento activo).
f) Las previsiones para el seguimiento del ensayo.
g) La idoneidad del investigador y de sus colaboradores.
h) La idoneidad de las instalaciones.
i) La idoneidad de la información escrita para los sujetos del ensayo y el procedimiento de obtención del consentimiento informado, y la justificación de la investigación en personas incapaces de dar su consentimiento informado.
j) El seguro o la garantía financiera previstos para el ensayo.
k) Las cantidades y, en su caso, previsiones de remuneración o compensación para los investigadores y sujetos del ensayo y los aspectos relevantes de cualquier acuerdo entre el promotor y el centro, que han de constar en el contrato previsto en el artículo 30.
l) El plan previsto para el reclutamiento de los sujetos.
2. Las cuestiones indicadas en los párrafos g), h) y k) del apartado anterior deberán ser evaluadas para cada uno de los centros implicados en el ensayo clínico.
Artículo 18. Procedimiento para la emisión del dictamen en ensayos unicéntricos.
1. En el caso de los ensayos clínicos unicéntricos, la solicitud se presentará ante el Comité Ético de Investigación Clínica correspondiente. Éste, en el plazo de 10 días naturales, verificará que la solicitud reúne los requisitos previstos en el artículo 16 y, sin perjuicio de su subsanación cuando proceda, comunicará al promotor la admisión a trámite de la solicitud con indicación del calendario de evaluación o, en su caso, su inadmisión a trámite.
2. El Comité Ético de Investigación Clínica dispondrá de un plazo máximo de 60 días naturales, a contar desde la notificación de la admisión a trámite de la solicitud, para comunicar su dictamen motivado al promotor y a la Agencia Española de Medicamentos y Productos Sanitarios.
3. Durante el periodo establecido en el apartado anterior, el comité podrá solicitar una sola vez información complementaria al promotor ; en tal caso se suspenderá el cómputo del plazo de evaluación hasta que se reciba la información solicitada.
4. En el caso de ensayos clínicos que se refieran a medicamentos de terapia génica, de terapia celular somática o que contengan organismos modificados genéticamente, el plazo establecido en el apartado 2 será de 90 días naturales. Dicho plazo podrá prorrogarse por otros 90 días cuando se recabe dictamen de un comité de expertos.
5. En el caso de ensayos clínicos que se refieran a terapia celular xenogénica, no existirá ninguna limitación de plazo para la emisión del dictamen motivado.
Artículo 19. Dictamen único en ensayos clínicos multicéntricos.
1. En los ensayos clínicos en los que participen dos o más centros ubicados en España, se emitirá un único dictamen con independencia del número de Comités Éticos de Investigación Clínica implicados.
El dictamen único se adoptará de conformidad con el procedimiento previsto en los apartados siguientes.
2. El promotor presentará la solicitud de evaluación del ensayo ante el Comité Ético de Investigación Clínica que actuará como comité de referencia y que se responsabilizará de la emisión del dictamen único y al resto de los Comités Éticos de Investigación Clínica implicados.
3. El Comité Ético de Investigación Clínica de referencia, en el plazo máximo de 10 días naturales, verificará que la solicitud reúne los requisitos previstos en el artículo 16 y, sin

28
perjuicio de su subsanación cuando proceda, comunicará al promotor y a los Comité Ético de Investigación Clínica implicados en el ensayo la admisión a trámite de la solicitud con indicación del calendario de evaluación o, en su caso, su inadmisión a trámite.
4. El Comité Ético de Investigación Clínica de referencia dispondrá de un plazo máximo de 60 días naturales, a contar desde la notificación de la admisión a trámite al promotor para comunicar su dictamen motivado al promotor, a la Agencia Española de Medicamentos y Productos Sanitarios y a los demás comités implicados en el ensayo.
Cada comité implicado remitirá con tiempo suficiente al comité de referencia un informe sobre los aspectos locales del ensayo, así como sobre cualquier otro aspecto del ensayo que considere relevante.
5. Durante el periodo establecido en el apartado anterior, el Comité Ético de Investigación Clínica de referencia podrá solicitar una sola vez información complementaria al promotor ; en tal caso se suspenderá el cómputo del plazo de evaluación hasta que se reciba la información solicitada. Dicha información se presentará también a los demás comités implicados.
6. Los informes de los demás comités implicados deberán ser tenidos en cuenta por el Comité Ético de Investigación Clínica de referencia para la emisión del dictamen único, que habrá de ser motivado, especialmente, en caso de discrepar de la opinión de otro comité sobre cualquier aspecto del ensayo, pero sólo vincularán al comité de referencia respecto a los aspectos locales.
7. En el caso de ensayos clínicos que se refieran a medicamentos de terapia génica, de terapia celular somática o que contengan organismos modificados genéticamente, el plazo establecido en el apartado 4 será de 90 días naturales. Dicho plazo podrá prorrogarse por otros 90 días cuando se recabe dictamen de un comité de expertos.
8. En el caso de ensayos clínicos que se refieran a terapia celular xenogénica, no existirá ninguna limitación de plazo para la emisión del dictamen motivado.
SECCIÓN 3.ª DE LA AUTORIZACIÓN DE LA AGENCIA ESPAÑOLA DE MEDICAMENTOS Y PRODUCTOS SANITARIOS
Artículo 20. Iniciación del procedimiento.
1. La autorización del ensayo clínico deberá solicitarse mediante escrito del promotor dirigido al Director de la Agencia Española de Medicamentos y Productos Sanitarios, de acuerdo con las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
2. La solicitud deberá acompañarse de la siguiente documentación:
a) Protocolo del ensayo.
b) Manual del investigador.
c) Hoja de información para los sujetos del ensayo.
d) Expediente del medicamento en investigación, cuando proceda.
e) Acreditación del pago de la tasa prevista en el artículo 117.1, grupo V, epígrafe 5.2, de la Ley 25/1990, de 20 de diciembre, del Medicamento.
3. Además, será necesaria la calificación como producto en fase de investigación clínica para aquellos medicamentos en investigación que se definan en las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
Para la calificación del medicamento como producto en fase de investigación clínica, a los efectos previstos en el artículo 24, se deberá aportar la siguiente documentación:

29
a) Formulario de solicitud.
b) Expediente del medicamento en investigación.
c) Acreditación del pago de la tasa prevista en el artículo 117.1, grupo V, epígrafe 5.1, de la Ley 25/1990, de 20 de diciembre, del Medicamento.
Artículo 21. Validación de la solicitud.
1. La Agencia Española de Medicamentos y Productos Sanitarios, en el plazo de 10 días naturales, verificará que la solicitud reúne los requisitos previstos en el artículo anterior, y notificará al solicitante la admisión a trámite de la solicitud con indicación del procedimiento aplicable, así como del plazo para la notificación de la resolución.
2. En el caso de que la solicitud no reúna los requisitos establecidos en el apartado anterior, se requerirá al solicitante para que subsane las deficiencias en el plazo máximo de 10 días, con indicación de que, si así no lo hiciera, se archivará la solicitud previa resolución que se dictará en los términos establecidos en el artículo 42 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico para las Administraciones Públicas y del Procedimiento Administrativo Común.
3. En el plazo de 10 días naturales, a contar desde la presentación de la documentación requerida, se notificará al solicitante la admisión a trámite de su solicitud en los términos previstos en el apartado 1 de este artículo o, en su caso, su inadmisión a trámite.
Artículo 22. Procedimiento ordinario.
1. La autorización se entenderá otorgada si en el plazo de 60 días naturales, a contar desde la notificación de la admisión a trámite de la solicitud, la Agencia Española de Medicamentos y Productos Sanitarios no comunica objeciones motivadas al solicitante siempre y cuando se haya notificado de forma previa a la Agencia Española de Medicamentos y Productos Sanitarios el dictamen favorable del Comité Ético de Investigación Clínica y la conformidad de la dirección de los centros participantes en el ensayo.
2. En el caso de que se comuniquen objeciones motivadas, el solicitante dispondrá del plazo de 15 días naturales para modificar su solicitud de acuerdo con las objeciones planteadas o, en caso de discrepancia con dichas objeciones, efectuar las alegaciones y presentar los documentos que estime pertinentes en apoyo de su solicitud.
Transcurrido el plazo establecido en el párrafo anterior sin que el solicitante haya modificado la solicitud o presentado alegaciones, se le entenderá desistido de su solicitud.
3. A la vista de la modificación propuesta por el solicitante o, en su caso, de sus alegaciones, la Agencia Española de Medicamentos y Productos Sanitarios emitirá resolución expresa, autorizando o denegando el ensayo, que deberá ser notificada al solicitante en el plazo de 15 días a contar desde la entrada en su registro general del escrito de modificación o alegaciones.
4. La autorización del ensayo clínico se entenderá sin perjuicio de la aplicación, cuando proceda, de la legislación vigente sobre la utilización confinada de organismos modificados genéticamente, y la liberación intencional en el medio ambiente de organismos modificados genéticamente.
Artículo 23. Procedimientos especiales.
1. Sin perjuicio de lo previsto en al artículo anterior, no podrá iniciarse un ensayo clínico sin la previa autorización por escrito de la Agencia Española de Medicamentos y Productos Sanitarios en los siguientes casos:
a) Ensayos clínicos en los que la Agencia Española de Medicamentos y Productos Sanitarios haya comunicado objeciones al promotor dentro del plazo establecido en el apartado 1 del artículo anterior.

30
b) Ensayos clínicos con medicamentos que requieren la calificación de producto en fase de investigación clínica.
c) Ensayos clínicos con medicamentos de terapia génica, terapia celular somática (incluidos los de terapia celular xenogénica), así como todos los medicamentos que contengan organismos modificados genéticamente.
2. En los casos previstos en el apartado anterior, la Agencia Española de Medicamentos y Productos Sanitarios emitirá resolución expresa que autorice o deniegue el ensayo clínico, de conformidad con el procedimiento y los plazos previstos en el artículo 22, con las particularidades que se establecen en los apartados 3 y 4 siguientes.
Una vez transcurrido el plazo correspondiente sin que se notifique al interesado la resolución, se podrá entender desestimada la solicitud.
3. En los ensayos clínicos con medicamentos de terapia génica, terapia celular somática (excluidos los de terapia celular xenogénica), así como todos los medicamentos que contengan organismos modificados genéticamente, el plazo máximo para autorizar expresamente el ensayo clínico será de 90 días naturales. Dicho plazo se ampliará en 90 días naturales cuando resulte preceptivo recabar algún informe de acuerdo con la normativa vigente.
4. En los ensayos clínicos con medicamentos de terapia celular xenogénica, la Agencia Española de Medicamentos y Productos Sanitarios no tendrá límite temporal para la comunicación de objeciones ni para la autorización o denegación del ensayo.
Artículo 24. Productos en fase de investigación clínica.
1. La Agencia Española de Medicamentos y Productos Sanitarios, cuando autorice un ensayo clínico con un medicamento en investigación, hará constar en la autorización del ensayo la calificación de dicho medicamento como producto en fase de investigación clínica, en los casos que proceda.
2. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá un registro actualizado de los medicamentos en investigación calificados como productos en fase de investigación clínica, en el que se enumerarán las indicaciones concretas que pueden ser objeto de investigación clínica, así como las limitaciones, plazos, condiciones y garantías que, en su caso, se establezcan.
3. Para la autorización de posteriores ensayos clínicos con un medicamento en investigación previamente calificado como producto en fase de investigación clínica deberá actualizarse, cuando resulte necesario, la documentación citada en el artículo 20.3 conforme a las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
Las solicitudes que se acojan a lo dispuesto en el párrafo anterior harán referencia a dichas circunstancias.
Artículo 25. Modificación de las condiciones de autorización de ensayos clínicos.
1. Cualquier modificación en las condiciones autorizadas para un ensayo clínico que se considere relevante no podrá llevarse a efecto sin el previo dictamen favorable del Comité Ético de Investigación Clínica correspondiente y la autorización de la Agencia Española de Medicamentos y Productos Sanitarios.
Sin perjuicio de lo anterior, si la modificación se refiere exclusivamente a documentos específicos que deben ser evaluados por el Comité Ético de Investigación Clínica, únicamente se requerirá el dictamen favorable de dicho comité para su aplicación. Por el contrario, si la modificación se refiere a la documentación que deba ser evaluada únicamente por la Agencia Española de Medicamentos y Productos Sanitarios, solamente se requerirá la autorización de ésta.

31
Si se dieran circunstancias que pudieran poner en peligro la seguridad de los sujetos, el promotor y el investigador adoptarán las medidas urgentes oportunas para proteger a los sujetos de cualquier riesgo inmediato. El promotor informará lo antes posible tanto a la Agencia Española de Medicamentos y Productos Sanitarios como a los Comités Éticos de Investigación Clínica implicados en el ensayo de dichas circunstancias y de las medidas adoptadas.
2. Se consideran modificaciones relevantes aquellas que se detallen en las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
3. La solicitud deberá presentarse por escrito, fechada y firmada por el promotor e investigador, ante la Agencia Española de Medicamentos y Productos Sanitarios y ante los Comités Éticos de Investigación Clínica correspondientes. La solicitud se adecuará a lo establecido en las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
4. El dictamen previo se adoptará conforme al procedimiento establecido en los artículos 18 y 19. No obstante:
a) El Comité Ético de Investigación Clínica correspondiente dispondrá de un plazo máximo de 35 días naturales, a contar desde la notificación de la admisión a trámite de la solicitud, para comunicar su dictamen motivado al promotor, al Centro Coordinador de Comités Éticos de Investigación Clínica, a la Agencia Española de Medicamentos y Productos Sanitarios y, en el caso de ensayos multicéntricos, a los Comités Éticos de Investigación Clínica implicados.
b) En los ensayos multicéntricos, el informe de los Comités Éticos de Investigación Clínica implicados distintos del comité de referencia sólo resultará preceptivo cuando la modificación suponga la incorporación de nuevos centros o investigadores principales al ensayo, y sólo será vinculante en los aspectos locales del ensayo. Sólo será necesario el informe del comité ético correspondiente al centro o investigador que se incorpore.
5. La autorización de la Agencia Española de Medicamentos y Productos Sanitarios se adoptará conforme al procedimiento establecido en los artículos 21, 22 y 23. No obstante, la autorización se entenderá otorgada si en el plazo de 35 días naturales, a contar desde la notificación de la admisión a trámite de la solicitud, la Agencia Española de Medicamentos y Productos Sanitarios no comunica objeciones motivadas al solicitante.
6. En caso de modificaciones en ensayos clínicos con medicamentos de terapia génica, de terapia celular somática (incluidos los de terapia celular xenogénica), así como todos los medicamentos que contengan organismos modificados genéticamente, los plazos para su autorización podrán ser ampliados, notificando al promotor el nuevo plazo.
Artículo 26. Suspensión y revocación de la autorización del ensayo clínico.
1. La autorización del ensayo clínico se suspenderá o revocará, de oficio o a petición justificada del promotor, mediante resolución de la Agencia Española de Medicamentos y Productos Sanitarios en los siguientes supuestos:
a) Si se viola la ley.
b) Si se alteran las condiciones de su autorización.
c) Si no se cumplen los principios éticos recogidos en el artículo 60 de la Ley 25/1990, de 20 de diciembre, del Medicamento.
d) Para proteger a los sujetos del ensayo.
e) En defensa de la salud pública.
2. La resolución por la que se suspenda o revoque la autorización del ensayo se adoptará previa instrucción del oportuno procedimiento, con audiencia al interesado que deberá

32
pronunciarse en el plazo de siete días a contar desde la notificación del inicio del procedimiento.
Una vez adoptada la resolución citada en el párrafo anterior, la Agencia Española de Medicamentos y Productos Sanitarios notificará la decisión adoptada, con expresa indicación de los motivos, a los Comités Éticos de Investigación Clínica participantes, a la Comisión Europea, a la Agencia Europea para la Evaluación de Medicamentos, a las autoridades sanitarias de las comunidades autónomas y a las autoridades sanitarias de los demás Estados miembros.
3. Las autoridades sanitarias de las comunidades autónomas, por propia iniciativa o a propuesta del Comité Ético de Investigación Clínica correspondiente, podrán resolver la suspensión cautelar del ensayo clínico en los casos previstos en el apartado 1, y lo notificarán de inmediato a la Agencia Española de Medicamentos y Productos Sanitarios, la cual, conforme a lo establecido en el apartado 2, resolverá la suspensión o revocación de la autorización del ensayo o, en su caso, el levantamiento de la medida cautelar.
Artículo 27. Informe final del ensayo clínico.
1. Una vez finalizada la realización del ensayo clínico, en el plazo de 90 días, el promotor notificará a
la Agencia Española de Medicamentos y Productos Sanitarios y a los comités éticos implicados el final del ensayo.
2. En caso de terminación anticipada, en el plazo de 15 días el promotor remitirá a la Agencia Española de Medicamentos y Productos Sanitarios y a los Comités Éticos de Investigación Clínica implicados un informe que incluya los datos obtenidos hasta el momento de su conclusión anticipada, así como los motivos de ésta, y en su caso las medidas adoptadas en relación con los sujetos participantes en el ensayo.
3. En el plazo de un año desde el final del ensayo, el promotor remitirá a la Agencia Española de Medicamentos y Productos Sanitarios y a los Comités Éticos de Investigación Clínica implicados un resumen del informe final sobre los resultados del ensayo.
4. Cuando la duración del ensayo sea superior a un año, será necesario además que el promotor remita un informe anual sobre la marcha del ensayo.
5. En todos los casos se seguirán las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
CAPÍTULO V
Del uso compasivo
Artículo 28. Uso compasivo de medicamentos.
1. Se entiende por uso compasivo de medicamentos la utilización en pacientes aislados y al margen de un ensayo clínico de medicamentos en investigación, incluidas especialidades farmacéuticas para indicaciones o condiciones de uso distintas de las autorizadas, cuando el médico bajo su exclusiva responsabilidad considere indispensable su utilización.
2. Para utilizar un medicamento bajo las condiciones de uso compasivo se requerirá el consentimiento informado del paciente o de su representante legal, un informe clínico en el que el médico justifique la necesidad de dicho tratamiento, la conformidad del director del centro donde se vaya a aplicar el tratamiento y la autorización de la Agencia Española de Medicamentos y Productos Sanitarios.
3. El médico responsable comunicará a la Agencia Española de Medicamentos y Productos Sanitarios los resultados del tratamiento, así como las sospechas de reacciones adversas que puedan ser debidas a éste.

33
Artículo 29. Continuación del tratamiento tras la finalización del ensayo.
Una vez finalizado el ensayo, toda continuación en la administración del medicamento en investigación, en tanto no se autorice el medicamento para esas condiciones de uso, se regirá por las normas establecidas para el uso compasivo en el artículo anterior.
CAPÍTULO VI
Aspectos económicos
Artículo 30. Aspectos económicos del ensayo clínico.
1. Todos los aspectos económicos relacionados con el ensayo clínico quedarán reflejados en un contrato entre el promotor y cada uno de los centros donde se vaya a realizar el ensayo. Esta documentación se pondrá a disposición del Comité Ético de Investigación Clínica correspondiente.
2. Las Administraciones sanitarias competentes para cada servicio de salud establecerán los requisitos comunes y condiciones de financiación, así como el modelo de contrato de conformidad con los principios generales de coordinación que acuerde el Consejo Interterritorial del Sistema Nacional de Salud.
3. En el contrato constará el presupuesto inicial del ensayo, que especificará los costes indirectos que aplicará el centro, así como los costes directos extraordinarios, considerando como tales aquellos gastos ajenos a los que hubiera habido si el sujeto no hubiera participado en el ensayo, como análisis y exploraciones complementarias añadidas, cambios en la duración de la atención a los enfermos, reembolso por gastos a los pacientes, compra de aparatos y compensación para los sujetos del ensayo e investigadores. También constarán los términos y plazos de los pagos, así como cualquier otra responsabilidad subsidiaria que contraigan las partes.
CAPÍTULO VII
Medicamentos en investigación
Artículo 31. Fabricación.
1. La fabricación de medicamentos no autorizados en España para su utilización en el ámbito de un ensayo clínico únicamente podrá realizarse previa autorización de la Agencia Española de Medicamentos y Productos Sanitarios. Esta autorización estará vigente durante el tiempo de realización del ensayo clínico en el que se utilicen.
2. El fabricante de un medicamento en investigación ha de estar autorizado para el ejercicio de su actividad de acuerdo con lo establecido en el Real Decreto 1564/1992, de 18 de diciembre, por el que se desarrolla y regula el régimen de autorización de los laboratorios farmacéuticos e importadores de medicamentos y la garantía de calidad de su fabricación industrial.
3. En el caso de que alguna de las fases de la fabricación, como el acondicionamiento final, se realicen en un servicio de farmacia hospitalario, dicho servicio quedará excluido de la autorización contemplada en el apartado 2.
4. En todas las fases de la fabricación de un medicamento en investigación se han de seguir las normas de correcta fabricación de medicamentos en la Unión Europea, incluido su anexo 13, publicadas por el Ministerio de Sanidad y Consumo.
Artículo 32. Importación.
1. La importación de medicamentos en investigación, para su utilización en el ámbito de un ensayo clínico, únicamente podrá realizarse previa autorización de la Agencia Española de Medicamentos y Productos Sanitarios.

34
2. El laboratorio farmacéutico importador garantizará que el medicamento ha sido elaborado por un fabricante debidamente autorizado en el país de origen y que cumple normas de correcta fabricación, al menos equivalentes a las establecidas por la Unión Europea, sin perjuicio de la responsabilidad del promotor establecida en el artículo 35.
3. La solicitud de fabricación o importación de medicamentos en investigación podrá solicitarse en unidad de acto con la solicitud de realización del ensayo clínico al que estén destinados.
La fabricación o importación de medicamentos hemoderivados, estupefacientes o psicótropos se regirán por su normativa específica en la materia.
Artículo 33. Etiquetado.
El etiquetado de los medicamentos en investigación deberá figurar al menos en lengua española oficial del Estado y adecuarse a lo establecido en el anexo 13 de las de normas de correcta fabricación de medicamentos en la Unión Europea.
CAPÍTULO VIII
Normas de buena práctica clínica
Artículo 34. Normas de buena práctica clínica.
Todos los ensayos clínicos con medicamentos que se realicen en España deberán llevarse a cabo de acuerdo con las normas de buena práctica clínica publicadas por el Ministerio de Sanidad y Consumo, siempre que no se opongan a lo dispuesto en este real decreto.
Artículo 35. Promotor.
1. El promotor o su representante legal habrá de estar establecido en uno de los Estados miembros de la Unión Europea.
2. Corresponde al promotor firmar las solicitudes de dictamen y autorización dirigidas al Comité Ético de Investigación Clínica y a la Agencia Española de Medicamentos y Productos Sanitarios.
3. Son responsabilidades del promotor:
a) Establecer y mantener un sistema de garantías y control de calidad, con procedimientos normalizados de trabajo escritos, de forma que los ensayos sean realizados y los datos generados, documentados y comunicados de acuerdo con el protocolo, las normas de buena práctica clínica y lo dispuesto en este real decreto.
b) Firmar, junto con el investigador que corresponda, el protocolo y cualquiera de sus modificaciones.
c) Seleccionar al investigador más adecuado según su cualificación y medios disponibles, y asegurarse de que éste llevará a cabo el estudio tal como está especificado en el protocolo.
d) Proporcionar la información básica y clínica disponible del producto en investigación y actualizarla a lo largo del ensayo.
e) Solicitar el dictamen del Comité Ético de Investigación Clínica y la autorización de la Agencia Española de Medicamentos y Productos Sanitarios, así como suministrarles la información y recabar las autorizaciones que procedan, sin perjuicio de la comunicación a las comunidades autónomas, en caso de modificación o violación del protocolo o interrupción del ensayo, y las razones para ello.
f) Suministrar de forma gratuita los medicamentos en investigación, garantizar que se han cumplido las normas de correcta fabricación y que las muestras están adecuadamente envasadas y etiquetadas. También es responsable de la conservación de muestras y sus protocolos de fabricación y control, del registro de las muestras entregadas y de asegurarse

35
que en el centro donde se realiza el ensayo existirá un procedimiento correcto de manejo, conservación y uso de dichas muestras.
Excepcionalmente, se podrán acordar con el centro otras vías de suministro.
g) Designar el monitor que vigilará la marcha del ensayo.
h) Comunicar a las autoridades sanitarias, a los investigadores y a los Comités Éticos de Investigación Clínica involucrados en el ensayo las sospechas de reacciones adversas graves e inesperadas de conformidad con lo establecido en los artículos 43 a 46.
i) Proporcionar al investigador y al Comité Ético de Investigación Clínica, de forma inmediata, cualquier información de importancia a la que tenga acceso durante el ensayo.
j) Proporcionar compensación económica a los sujetos en caso de lesión o muerte relacionadas con el ensayo. Proporcionar al investigador cobertura legal y económica en estos casos excepto cuando la lesión sea consecuencia de negligencia o mala práctica del investigador.
k) Acordar con el investigador las obligaciones en cuanto al tratamiento de datos, elaboración de informes y publicación de resultados. En cualquier caso, el promotor es responsable de elaborar los informes finales o parciales del ensayo y comunicarlos a quien corresponda.
l) El promotor dispondrá de un punto de contacto, donde los sujetos del ensayo puedan obtener mayor información sobre éste, que podrá delegar en el investigador.
Artículo 36. Monitor.
Son responsabilidades del monitor:
a) Trabajar de acuerdo con los procedimientos normalizados de trabajo del promotor, visitar al investigador antes, durante y después del ensayo para comprobar el cumplimiento del protocolo, garantizar que los datos son registrados de forma correcta y completa, así como asegurarse de que se ha obtenido el consentimiento informado de todos los sujetos antes de su inclusión en el ensayo.
b) Cerciorarse de que los investigadores y el centro donde se realizará la investigación son adecuados para este propósito durante el periodo de realización del ensayo.
c) Asegurarse de que tanto el investigador principal como sus colaboradores han sido informados adecuadamente y garantizar en todo momento una comunicación rápida entre investigador y promotor.
d) Verificar que el investigador cumple el protocolo y todas sus modificaciones aprobadas.
e) Comprobar que el almacenamiento, distribución, devolución y documentación de los medicamentos en investigación es seguro y adecuado.
f) Remitir al promotor informes de las visitas de monitorización y de todos los contactos relevantes con el investigador.
Artículo 37. Investigador.
1. El investigador dirige y se responsabiliza de la realización práctica del ensayo clínico en un centro, y firma junto con el promotor la solicitud, corresponsabilizándose con él.
2. Solamente podrá actuar como investigador un médico o persona que ejerza una profesión reconocida en España para llevar a cabo las investigaciones en razón de su formación científica y de su experiencia en la atención sanitaria requerida.
3. Son responsabilidades del investigador:
a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.
b) Conocer a fondo las propiedades de los medicamentos en investigación.

36
c) Garantizar que el consentimiento informado se recoge de conformidad a lo establecido en este real decreto.
d) Recoger, registrar y notificar los datos de forma correcta y garantizar su veracidad.
e) Notificar inmediatamente los acontecimientos adversos graves o inesperados al promotor.
f) Garantizar que todas las personas implicadas respetarán la confidencialidad de cualquier información acerca de los sujetos del ensayo, así como la protección de sus datos de carácter personal.
g) Informar regularmente al Comité Ético de Investigación Clínica de la marcha del ensayo.
h) Corresponsabilizarse con el promotor de la elaboración del informe final del ensayo, dando su acuerdo con su firma.
Artículo 38. Publicaciones.
1. El promotor está obligado a publicar los resultados, tanto positivos como negativos, de los ensayos clínicos autorizados en revistas científicas y con mención al Comité Ético de Investigación Clínica que aprobó el estudio.
2. Cuando se hagan públicos estudios y trabajos de investigación sobre medicamentos, dirigidos a la comunidad científica, se harán constar los fondos obtenidos por el autor por o para su realización y la fuente de financiación.
3. Se mantendrá en todo momento el anonimato de los sujetos participantes en el ensayo.
4. Los resultados o conclusiones de los ensayos clínicos se comunicarán preferentemente en publicaciones científicas antes de ser divulgados al público no sanitario.
No se darán a conocer de modo prematuro o sensacionalista tratamientos de eficacia todavía no determinada, ni se exagerará ésta.
5. La publicidad de medicamentos en investigación queda terminantemente prohibida, tal como se establece en la Ley 25/1990, de 20 de diciembre, del Medicamento, el Real Decreto 1416/1994, de 25 de junio, por el que se regula la publicidad de los medicamentos, el Real Decreto 1907/1996, de 2 de agosto, sobre publicidad y promoción comercial de productos, actividades o servicios con pretendida finalidad sanitaria, y la Ley 34/1988, de 11 de noviembre, General de Publicidad.
Artículo 39. Archivo de la documentación del ensayo clínico.
Los documentos que constituyen el archivo maestro de un ensayo clínico deberán conservarse durante el tiempo y conforme a las especificaciones establecidas en las Instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea que publicará el Ministerio de sanidad y Consumo.
CAPÍTULO IX
Verificación del cumplimiento de las normas de buena práctica clínica
Artículo 40. Inspecciones.
1. La Agencia Española de Medicamentos y Productos Sanitarios y las autoridades sanitarias competentes de las comunidades autónomas, en el ámbito de sus respectivas competencias, verificarán la aplicación de este real decreto, de las normas de buena práctica clínica y de las normas de correcta fabricación en los ensayos clínicos que se realicen en España, a través de las correspondientes inspecciones.
2. La Agencia Española de Medicamentos y Productos Sanitarios informará a la Agencia Europea de Evaluación de Medicamentos de las inspecciones efectuadas y de sus resultados.

37
Asimismo, será responsable de la introducción de los datos relativos a inspecciones en la base de datos europea de ensayos clínicos EUDRACT, de acuerdo con lo especificado en las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
3. Las inspecciones serán llevadas a cabo por inspectores debidamente cualificados y designados para tal efecto en los lugares relacionados con la realización de los ensayos clínicos y, entre otros, en el centro o centros en los que se lleve a cabo el ensayo, el lugar de fabricación del medicamento en investigación, cualquier laboratorio de análisis utilizado en el ensayo clínico y/o en las instalaciones del promotor.
4. Tras la inspección se elaborará un informe que se pondrá a disposición de los inspeccionados. También se podrá poner a disposición del Comité Ético de Investigación Clínica implicado, de las autoridades competentes en España y demás Estados miembros de la Unión Europea y de la Agencia Europea de Evaluación de Medicamentos, 5. Corresponde a la Agencia Española de Medicamentos y Productos Sanitarios recabar la colaboración de las autoridades competentes de otros Estados miembros de la Unión Europea y terceros Estados para la inspección del centro del ensayo, instalaciones del promotor o instalaciones del fabricante del medicamento en investigación establecido fuera del territorio nacional.
CAPÍTULO X
Comunicaciones
Artículo 41. Bases de datos.
1. La Agencia Española de Medicamentos y Productos Sanitarios se responsabilizará de la inclusión en la base de datos europea de ensayos clínicos EUDRACT de los datos relativos a los ensayos clínicos que se lleven a cabo en el territorio nacional, de acuerdo con lo establecido en las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
2. La Agencia Española de Medicamentos y Productos Sanitarios se responsabilizará de mantener actualizada su base de datos de los ensayos clínicos autorizados que se lleven a cabo en el territorio nacional.
Las autoridades competentes de las comunidades autónomas tendrán acceso a los datos relativos a los ensayos que se realicen en su ámbito territorial.
3. La Agencia Española de Medicamentos y Productos Sanitarios pondrá a disposición de los ciudadanos a través de su página web información referente al título del ensayo, promotor, centros implicados, patología y población en estudio de los ensayos clínicos autorizados.
Se considerará que no existe oposición, por parte del promotor del ensayo, a la publicación de los datos antes indicados de los ensayos promovidos por él, siempre que no se haga indicación expresa en su contra en la solicitud de autorización del ensayo clínico dirigida a la Agencia Española de Medicamentos y Productos Sanitarios.
CAPÍTULO XI
De la vigilancia de la seguridad de los medicamentos en investigación
Artículo 42. Obligaciones de los investigadores en el registro y comunicación de acontecimientos adversos.
1. El investigador comunicará inmediatamente al promotor todos los acontecimientos adversos graves, salvo cuando se trate de los señalados en el protocolo o en el manual del investigador como acontecimientos que no requieran comunicación inmediata. La comunicación inicial irá seguida de comunicaciones escritas pormenorizadas. En las comunicaciones iniciales y en las

38
de seguimiento se identificará a los sujetos del ensayo mediante un número de código específico para cada uno de ellos.
2. Los acontecimientos adversos y/o los resultados de laboratorio anómalos calificados en el protocolo como determinantes para las evaluaciones de seguridad se comunicarán al promotor con arreglo a los requisitos de comunicación y dentro de los periodos especificados en el protocolo.
3. En caso de que se haya comunicado un fallecimiento de un sujeto participante en un ensayo clínico, el investigador proporcionará al promotor y a los Comités Éticos de Investigación Clínica implicados toda la información complementaria que le soliciten.
Artículo 43. Obligaciones del promotor en el registro, evaluación y comunicación de acontecimientos adversos.
1. El promotor mantendrá unos registros detallados de todos los acontecimientos adversos que le sean comunicados por los investigadores. Estos registros se presentarán a la Agencia Española de Medicamentos y Productos Sanitarios cuando ésta así lo solicite.
2. El promotor tiene la obligación de evaluar de forma continua la seguridad de los medicamentos en investigación utilizando toda la información a su alcance.
Asimismo, debe comunicar sin tardanza a la Agencia Española de Medicamentos y Productos Sanitarios, a los órganos competentes de las comunidades autónomas donde se realice el ensayo clínico y a los Comités Éticos de Investigación Clínica implicados cualquier información importante que afecte a la seguridad del medicamento en investigación. Dicha comunicación se realizará según los criterios que se especifican en los artículos siguientes y de acuerdo con los procedimientos establecidos en las instrucciones para la realización de ensayos clínicos en España o, en su caso, directrices de la Comisión Europea, que publicará el Ministerio de Sanidad y Consumo.
3. La comunicación de información de seguridad del promotor a los investigadores seguirá lo especificado en las normas de buena práctica clínica.
Artículo 44. Notificación expeditiva de casos individuales de sospecha de reacción adversa a la Agencia Española de Medicamentos y Productos Sanitarios.
1. El promotor notificará a la Agencia Española de Medicamentos y Productos Sanitarios todas las sospechas de reacciones adversas graves y a la vez inesperadas asociadas a los medicamentos en investigación, tanto si ocurren en España como en otros Estados, y tanto si han ocurrido en el ensayo clínico autorizado como en otros ensayos clínicos o en un contexto de uso diferente, siempre que dichos medicamentos no se encuentren comercializados en España. Para los productos comercializados, incluyendo el medicamento utilizado como control o los medicamentos utilizados como concomitantes, la Agencia Española de Medicamentos y Productos Sanitarios emitirá unas directrices específicas que tendrán en cuenta los requerimientos de farmacovigilancia al objeto de evitar posibles duplicaciones en la notificación.
2. El plazo máximo de notificación será de 15 días naturales a partir del momento en que el promotor haya tenido conocimiento de la sospecha de reacción adversa.
Cuando la sospecha de reacción adversa grave e inesperada haya ocasionado la muerte del sujeto, o puesto en peligro su vida, el promotor informará a la Agencia Española de Medicamentos y Productos Sanitarios en el plazo máximo de siete días naturales a partir del momento en que el promotor tenga conocimiento del caso. Dicha información deberá ser completada, en lo posible, en los ocho días siguientes.
3. Cuando las sospechas de reacciones adversas graves e inesperadas ocurran en un ensayo clínico doble ciego, se deberá desvelar el código de tratamiento de ese paciente concreto a efectos de notificación. Siempre que sea posible, se mantendrá el carácter ciego para el investigador, y para las personas encargadas del análisis e interpretación de los resultados, así como de la elaboración de las conclusiones del estudio. En aquellos casos en que se considere

39
que este sistema de notificación pueda interferir con la validez del estudio, podrá acordarse con la Agencia Española de Medicamentos y Productos Sanitarios un sistema de notificación específico.
4. Las sospechas de reacciones adversas atribuibles a placebo no estarán sujetas a este sistema de notificación individualizada.
5. Las notificaciones se realizarán preferiblemente utilizando el formato electrónico estándar europeo.
Cuando esto no sea posible, debido a un motivo justificado, se utilizará el formulario de notificación en papel para las notificaciones de sospechas de reacción adversa que ocurran en España. Para las notificaciones de sospechas de reacción adversa que ocurran fuera de España, podrá utilizarse un formulario estándar internacional. Las notificaciones que ocurran en España, con independencia del formato utilizado, tendrán que ser comunicadas en la lengua española oficial del Estado.
6. La Agencia Española de Medicamentos y Productos Sanitarios mantendrá una red de proceso de datos para registrar todas las sospechas de reacciones adversas graves inesperadas de un medicamento en investigación de las cuales tenga conocimiento. Dicha red permitirá, además de la recepción de comunicaciones electrónicas, el acceso en tiempo real de las comunidades autónomas y la comunicación electrónica a la Agencia Europea de Evaluación de Medicamentos de las sospechas de reacciones adversas graves inesperadas que hayan ocurrido en España.
Artículo 45. Notificación expeditiva de casos individuales de sospecha de reacción adversa a los órganos competentes de las comunidades autónomas.
1. El promotor notificará a los órganos competentes de las comunidades autónomas en cuyo territorio se esté realizando el ensayo, de forma individual y en el plazo máximo de 15 días, todas las sospechas de reacciones adversas que sean a la vez graves e inesperadas asociadas al medicamento en investigación y que hayan ocurrido en pacientes seleccionados en sus respectivos ámbitos territoriales. Este plazo máximo será de siete días cuando se trate de sospechas de reacciones adversas que produzcan la muerte o amenacen la vida. Para los medicamentos comercializados, incluyendo el utilizado como control o los utilizados como concomitantes, la Agencia Española de Medicamentos y Productos Sanitarios emitirá unas directrices específicas que tendrán en cuenta los requerimientos de farmacovigilancia al objeto de evitar posibles duplicaciones en la notificación.
2. El promotor notificará cualquier otra información sobre reacciones adversas graves e inesperadas asociadas al medicamento en investigación cuando así lo dispongan las normativas específicas de las comunidades autónomas y, en cualquier caso, si la información supone un cambio importante en el perfil de seguridad del producto investigado.
3. Cuando el promotor realice la comunicación en formato electrónico no será precisa la notificación a las comunidades autónomas, dado que dicha información les será accesible en tiempo real a través de la red de proceso de datos.
Artículo 46. Notificación expeditiva de casos individuales de sospecha de reacción adversa a los Comités Éticos de Investigación Clínica.
1. El promotor notificará a los comités éticos implicados, de forma individual y en el plazo máximo de 15 días, todas las sospechas de reacciones adversas que sean a la vez graves e inesperadas asociadas al medicamento en investigación y que hayan ocurrido en pacientes seleccionados en sus respectivos ámbitos. El plazo máximo será de siete días cuando se trate de sospechas de reacciones adversas que produzcan la muerte o amenacen la vida.
2. El promotor notificará cualquier otra información sobre reacciones adversas graves e inesperadas asociadas al medicamento de investigación cuando así lo dispongan los comités éticos implicados en el momento del dictamen favorable del estudio y, en cualquier caso, si la

40
información supone un cambio importante en el perfil de seguridad del producto investigado. Los comités éticos implicados podrán establecer que esta información adicional le sea suministrada periódicamente de forma resumida. La Agencia Española de Medicamentos y Productos Sanitarios, en consonancia con las disposiciones europeas, publicará las correspondientes directrices para orientar a los promotores.
Artículo 47. Informes periódicos de seguridad.
1. Adicionalmente a la notificación expeditiva, los promotores de ensayos clínicos prepararán un informe periódico en el que se evalúe la seguridad del medicamento en investigación teniendo en cuenta toda la información disponible.
2. El informe periódico de seguridad se presentará a la Agencia Española de Medicamentos y Productos Sanitarios, a los órganos competentes de las comunidades autónomas correspondientes y a los Comités Éticos de Investigación Clínica implicados, anualmente hasta el final del ensayo y siempre que lo soliciten las autoridades sanitarias o los comités éticos implicados.
3. La Agencia Española de Medicamentos y Productos Sanitarios determinará el formato del informe periódico de seguridad teniendo en cuenta la normativa europea al respecto. El informe periódico de seguridad no sustituirá a la solicitud de modificaciones a los documentos del ensayo, que seguirá su procedimiento específico.
4. Sin perjuicio de la periodicidad señalada para los informes de seguridad, el promotor preparará un informe de evaluación ad hoc siempre que exista un problema de seguridad relevante. Dicho informe se presentará sin tardanza a la Agencia Española de Medicamentos y Productos Sanitarios, a los órganos competentes de las comunidades autónomas y a los Comités Éticos de Investigación Clínica concernidos.
5. El informe periódico de seguridad podrá ser una parte del informe anual y final correspondientes o bien ser preparado de forma independiente.
CAPÍTULO XII
Infracciones
Artículo 48. Infracciones administrativas en materia de ensayos clínicos.
Constituirán infracciones administrativas las previstas en el artículo 108 de la Ley 25/1990, de 20 de diciembre, del Medicamento, y serán sancionadas de acuerdo con el artículo 109 de la misma ley.
Disposición adicional única. Ensayos clínicos con productos sanitarios.
Los ensayos clínicos con productos sanitarios se regirán por los principios recogidos en este real decreto en lo que les resulte de aplicación.
Disposición transitoria primera. Régimen transitorio.
Los procedimientos iniciados con anterioridad a la entrada en vigor de este real decreto se regirán por lo establecido en el Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos.
Disposición transitoria segunda. Aplicación de este real decreto al ámbito de los servicios sanitarios de las Fuerzas Armadas.
En tanto no se desarrollen las previsiones contenidas en la disposición adicional segunda de la Ley 25/1990, de 20 de diciembre, del Medicamento, en la aplicación de este real decreto al ámbito de los servicios sanitarios de las Fuerzas Armadas se observarán las siguientes normas:

41
a) Corresponderá al Ministerio de Sanidad y Consumo la acreditación de los Comités Éticos de Investigación Clínica.
b) El Ministerio de Defensa, a través de la Inspección General de Sanidad de la Defensa, ejercerá las competencias en materia de inspección, recepción de comunicaciones y notificaciones y las demás que este real decreto atribuye a las comunidades autónomas.
Disposición derogatoria única. Derogación normativa.
Queda derogado el Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización de ensayos clínicos con medicamentos, sin perjuicio de lo dispuesto en la disposición transitoria primera, así como cualquier otra disposición de igual o inferior rango que se oponga a lo dispuesto en este real decreto.
Disposición final primera. Título habilitante.
Este real decreto tiene carácter de legislación sobre productos farmacéuticos a los efectos previstos en el artículo 149.1.16..ª de la Constitución Española, y se adopta en desarrollo del título III de la Ley 25/1990, de 20 de diciembre, del Medicamento.
Disposición final segunda. Facultad de desarrollo.
Se faculta al Ministro de Sanidad y Consumo para dictar las disposiciones necesarias para el desarrollo de este real decreto, conforme al avance de los conocimientos científicos y técnicos y de acuerdo con las orientaciones de la Unión Europea.
Asimismo, se faculta al Ministro de Sanidad y Consumo para la adopción de las normas de buena práctica clínica y las instrucciones para la realización de ensayos clínicos en España, de acuerdo con las directrices que adopte la Comisión Europea.
Disposición final tercera. Entrada en vigor.
El presente real decreto entrará en vigor el día 1 de mayo de 2004.
Dado en Madrid, a 6 de febrero de 2004.
JUAN CARLOS R.
La Ministra de Sanidad y Consumo,
ANA MARÍA PASTOR JULIÁN
REFERENCIAS ANTERIORES • DEROGA el REAL DECRETO 561/1993, de 16 de abril . • TRASPONE parcialmente la DIRECTIVA 2001/20/CE, de 4 de abril • DE CONFORMIDAD con el título III de la LEY 25/1990, de 20 de diciembre REFERENCIAS POSTERIORES
Afectado – por • art. 9 - ap. 2 modificado por disp. final 1 de Real Decreto núm. 590/2005, de 20 mayo • art. 28 - Derogado por disp. derog. única j) de Real Decreto núm. 1345/2007, de 11 octubre • art. 29 - Derogado por disp. derog. única j) de Real Decreto núm. 1345/2007, de 11 octubre • art. 30 -Aplicado por Resolución de 9 diciembre 2005

42
Documentación Básica
Los ensayos clínicos con medicamentos de uso humano están regulados en el título III de la Ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, que recoge en su articulado parte muy importante de los principios garantistas que ya contaban con previsiones normativas precedentes. En particular, el vigente Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, que incorporó al ordenamiento jurídico español la Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2001, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos de uso humano. Esta Directiva estableció la obligación de adoptar los principios y directrices de buena práctica clínica y de las normas de correcta fabricación de medicamentos en investigación, así como las directrices detalladas para la autorización de la fabricación o importación de medicamentos en investigación. Posteriormente, el Real Decreto 2183/2004, de 12 de noviembre, por el que se modifica el Real Decreto 1564/1992, de 18 de diciembre, por el que se desarrolla y regula el régimen de autorización de los laboratorios farmacéuticos e importadores de medicamentos y la garantía de calidad en su fabricación industrial, transpuso la Directiva 2003/94/CE de la Comisión, de 8 de octubre de 2003, por la que se establecen los principios y directrices de las prácticas correctas de fabricación de los medicamentos de uso humano y de los medicamentos en investigación de uso humano. Más recientemente, la Directiva 2005/28/CE de la Comisión, de 8 de abril de 2005, por la que se establecen los principios y las directrices detalladas de las buenas prácticas clínicas respecto a los medicamentos en investigación de uso humano, así como los requisitos para autorizar la fabricación o importación de dichos productos, incluye asimismo las directrices detalladas sobre la documentación relativa a los ensayos clínicos, el archivo, la cualificación de los inspectores y los procedimientos de inspección. Mediante esta disposición se incorpora al ordenamiento jurídico interno la Directiva 2005/28/CE de la Comisión, de 8 de abril de 2005. En la aplicación de los requisitos y las directrices contempladas en esta Orden se deberán tener en cuenta las directrices comunitarias publicadas por la Comisión Europea en los distintos volúmenes de las normas sobre medicamentos de la Unión Europea. En la tramitación de esta Orden han sido oídos los sectores afectados. Esta Orden se dicta al amparo de lo establecido en la disposición final segunda del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. En su virtud, con la aprobación previa del Ministro de Administraciones Públicas y de acuerdo con el Consejo de Estado, dispongo: Artículo 1. Objeto. Esta Orden regula los principios y las directrices de buena práctica clínica en los ensayos clínicos con medicamentos en investigación de uso humano, los requisitos para autorizar la
ORDEN SCO/256/2007, de 5 de febrero, por la que use establecen los principios y las directrices detalladas de la buena práctica clínica y los requisitos para autorizar la fabricación o importación de medicamentos en investigación de uso humano (BOE num. 38, de 13 de febrero)

43
fabricación o importación de estos medicamentos, las directrices detalladas sobre la documentación relativa a dichos ensayos, así como su archivo, y la cualificación de los inspectores y los procedimientos de inspección. Artículo 2. Principios y directrices de buena práctica clínica. En la realización de ensayos clínicos con medicamentos en investigación de uso humano se deberá observar lo siguiente: a) Los derechos, la seguridad y el bienestar de los sujetos del ensayo prevalecerán por encima de los intereses de la ciencia y de la sociedad. b) Cada persona que participe en la realización de un ensayo estará capacitada por su titulación, formación y experiencia para ejecutar sus tareas. c) Los ensayos clínicos, en todos sus aspectos, deberán tener una sólida base científica y deberán regirse por principios éticos. d) Deberán adoptarse los procedimientos necesarios que aseguren la calidad de cada uno de los aspectos del ensayo clínico. e) La información disponible sobre un medicamento en investigación, tanto clínica como no clínica, deberá ser adecuada para avalar el ensayo clínico propuesto. f) Los ensayos clínicos deberán realizarse de acuerdo con la Declaración de Helsinki sobre los principios éticos para las investigaciones médicas en seres humanos, aprobada por la Asamblea General de la Asociación Médica Mundial. g) El protocolo deberá establecer los criterios de inclusión, exclusión y retirada de los sujetos que participen en un ensayo, el plan de monitorización y el plan de publicación. h) El investigador y el promotor tendrán en cuenta todas las instrucciones para la realización de ensayos clínicos en España o, en su caso, las directrices de la Comisión Europea relativas al inicio, realización y finalización del ensayo clínico publicadas por la Agencia Española de Medicamentos y Productos Sanitarios. i) Toda la información sobre el ensayo clínico deberá registrarse, tratarse y conservarse de forma que pueda ser comunicada, evaluada y verificada de manera precisa, protegiendo al mismo tiempo el carácter confidencial de los registros de los sujetos del ensayo. j) Los Comités Éticos de Investigación Clínica adoptarán los procedimientos normalizados de trabajo necesarios para la realización de sus funciones, de acuerdo con lo establecido en el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Los Comités conservarán todos los documentos esenciales, relacionados con cada ensayo clínico evaluado, durante al menos tres años tras la finalización del mismo, o durante un período más largo si así lo establece la Agencia Española de Medicamentos y Productos Sanitarios o la autoridad competente de la Comunidad Autónoma correspondiente. Esta documentación debe archivarse preferentemente agrupada por protocolos, en un lugar que permita garantizar la confidencialidad de la información durante el tiempo de archivo requerido. En caso de cese de la actividad del Comité, la institución en la que esté constituido el Comité debe mantener el archivo de la documentación durante el plazo establecido. Para cada ensayo clínico debe incluir, como mínimo, lo siguiente: 1º El protocolo, las modificaciones y toda la documentación presentada por el promotor o su representante legal. 2º Los dictámenes o informes emitidos por el Comité (Comité Ético de Investigación Clínica de Referencia si el ensayo clínico es multicéntrico), especificando la versión de los documentos revisados. 3º Copia de la correspondencia con el investigador y/o el promotor o su representante. 4º La documentación relativa a las actividades de seguimiento del ensayo clínico. 5º El informe anual sobre la marcha del ensayo clínico. 6º Copia de cualquier correspondencia con la Agencia Española de Medicamentos y Productos Sanitarios y/o con las autoridades competentes de la Comunidad Autónoma donde este localizado el comité.

44
7º Copia de las notificaciones y correspondencia relevantes con los Comités implicados y/o el Comité de referencia, de un ensayo multicéntrico. 8º Las sospechas de reacciones adversas recibidas y los informes de seguridad presentados por el promotor. 9º Notificación de la finalización del ensayo clínico, ya sea prematura o programada. 10º Resumen del informe final del ensayo clínico presentado por el promotor. 11º Cualquier otra documentación relevante. Los Comités deben mantener archivada la documentación relacionada con su funcionamiento y actividad. En caso de cese de la misma, esta documentación debe conservarse en la institución durante al menos tres años, transcurridos desde la finalización del último estudio evaluado. Debe incluir como mínimo, lo siguiente: 1º Resoluciones de acreditación y de posteriores modificaciones. 2º Currículum vitae de los miembros actuales o que hayan pertenecido al Comité. 3º Convocatoria y actas de las reuniones del Comité. 4º Procedimientos normalizados de trabajo del Comité, versión actual y archivo histórico. k) La Agencia Española de Medicamentos y Productos Sanitarios establecerá los procedimientos que garanticen la transmisión de información entre la Agencia y los Comités Éticos de Investigación Clínica. l) El promotor de un ensayo clínico podrá delegar la totalidad o una parte de sus funciones en un particular, empresa, institución u organismo, que deberá poner en marcha un sistema de garantía y control de calidad. Las obligaciones del promotor establecidas en las normas de buena práctica clínica que se hayan delegado serán de aplicación al particular, empresa, institución u organismo contratado. No obstante, en estos casos, el promotor seguirá siendo el responsable de garantizar que la realización del ensayo clínico y los datos finales generados en dicho estudio se ajustan a lo dispuesto en el Real Decreto 223/2004, de 6 de febrero, y en esta Orden. Cualquier transferencia de funciones del promotor en relación con un ensayo clínico debe quedar específicamente documentada. m) El investigador y el promotor podrán ser la misma persona física. n) La información del manual del investigador y sus actualizaciones se presentarán de forma concisa, sencilla, objetiva, equilibrada y no promocional, comprensible para los posibles investigadores y que les permita realizar una evaluación no sesgada de los riesgos y beneficios y de la pertinencia del ensayo clínico propuesto. Este documento servirá como referencia para la evaluación del carácter esperado, o no, de las reacciones adversas graves que pudieran ocurrir durante la realización del ensayo. ñ) En caso de medicamentos autorizados en algún Estado miembro de la Unión Europea, cuando el medicamento se utilice en las condiciones de uso autorizadas, el manual del investigador podrá reemplazarse por la ficha técnica autorizada. o) El manual del investigador debe validarse y actualizarse de manera regular por el promotor, al menos una vez al año. Artículo 3. Requisitos de la solicitud para la autorización de fabricación o importación. 1. Será necesaria la autorización previa otorgada por la Agencia Española de Medicamentos y Productos Sanitarios para la fabricación total o parcial, incluyendo las diversas operaciones de división, acondicionamiento o presentación, de medicamentos en investigación, incluso en los casos en que los medicamentos fabricados estén destinados a la exportación. La autorización será necesaria también para las importaciones procedentes de terceros países. 2. No se exigirá la citada autorización para el acondicionamiento final, en caso de que se realice en un Servicio de Farmacia autorizado, siempre que los medicamentos en investigación estén destinados a ser utilizados únicamente en un centro sanitario dependiente de dicho servicio. Cuando en el contexto de un ensayo clínico específico cuyo promotor sea un investigador o un grupo de investigadores, un Servicio de Farmacia autorizado desee realizar una operación de fabricación distinta de las contempladas anteriormente, deberá solicitar una autorización previa a la Agencia Española de Medicamentos y Productos Sanitarios y únicamente se podrá utilizar

45
el medicamento en el ensayo clínico concreto. La Agencia Española de Medicamentos y Productos Sanitarios acordará con las Comunidades Autónomas los procedimientos de verificación de las normas de correcta fabricación en estos casos. 3. El solicitante deberá disponer de manera permanente y continua de una persona cualificada o de un director técnico, conforme a lo establecido en el Real Decreto 1564/1992, de 18 de diciembre, por el que se desarrolla y regula el régimen de autorización de los laboratorios farmacéuticos e importadores de medicamentos y la garantía en su fabricación industrial. 4. La solicitud para la autorización de fabricación o importación deberá incorporar los datos y documentos siguientes: a) Los tipos de medicamentos y formas farmacéuticas que se pretenden fabricar o importar. b) Las operaciones de fabricación o importación de que se trate. c) El proceso de fabricación, cuando proceda, como en el caso de inactivación de agentes virales o no convencionales. d) La identificación del lugar en el que se fabricarán los productos, que deberá contar, para la fabricación o importación de los medicamentos y formas farmacéuticas, de los locales, el equipo técnico y los medios de control adecuados y suficientes que se ajusten a los requisitos que establece el Real Decreto 1564/1992, de 18 de diciembre, en lo que respecta a la fabricación, el control y el almacenamiento de los productos. 5. Los tipos de medicamentos referidos en el apartado 4.a) de este artículo incluyen medicamentos hemoderivados, inmunológicos, de terapia celular, de terapia génica, biotecnológicos, de origen humano o animal, plantas medicinales, homeopáticos, radiofármacos y de origen químico. 6. La autorización establecida en el apartado 1 de este artículo se notificará en el plazo de 90 días desde la presentación de una solicitud válida, por la Agencia Española de Medicamentos y Productos Sanitarios, una vez comprobada la exactitud de los datos facilitados por el solicitante. En caso de que la Agencia precise datos complementarios, el plazo para resolver quedará en suspenso hasta que se faciliten los mismos. La citada autorización podrá estar condicionada al cumplimiento de determinadas obligaciones impuestas en el momento de la concesión de la autorización o en una fecha posterior, con el fin de garantizar el cumplimiento de los requisitos establecidos en la misma. La autorización únicamente será válida para las instalaciones, los tipos de medicamentos y las formas farmacéuticas solicitadas. Artículo 4. Obligaciones del titular de la autorización de fabricación o importación. 1. El titular de la autorización deberá cumplir, como mínimo, los requisitos siguientes: a) Disponer de los medios personales, materiales y técnicos necesarios, que respondan a las exigencias legales para la fabricación y control establecidos en el Real Decreto 1564/1992, de 18 de diciembre. b) Disponer de los medicamentos en investigación o autorizados, conforme a lo dispuesto en el Real Decreto 223/2004, de 6 de febrero. c) Permitir en todo momento el acceso a sus instalaciones a los inspectores. d) Permitir que la persona cualificada o el director técnico pueda cumplir las funciones establecidas en función de su cargo. e) Respetar los principios y directrices de las normas de correcta fabricación de medicamentos previstos en la normativa europea. 2. Para modificar los requisitos establecidos en los apartados 3 y 4 del artículo 3, el titular de una autorización de fabricación deberá solicitarlo a la Agencia Española de Medicamentos y Productos Sanitarios. En el caso de que la modificación se refiera a los tipos y formas farmacéuticas autorizadas o afecte al director técnico, la duración del procedimiento relativo a esta solicitud no será superior a 30 días. En casos excepcionales, este plazo podrá ser prorrogado hasta 90 días. 3. El incumplimiento de los requisitos establecidos será causa de suspensión o revocación de la autorización de fabricación de forma parcial o total.

46
4. Sólo podrán importar medicamentos en investigación los importadores que estén en posesión de la autorización de importación prevista en el artículo 3, otorgada por la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 5. Adquisición de medicamentos para ensayos clínicos. 1. Medicamentos comercializados en España y en el resto de la Unión Europea: Cuando los medicamentos que se vayan a utilizar en un ensayo clínico estén comercializados enEspaña o en otro Estado miembro de la Unión Europea, el promotor de un ensayo clínico podrá obtenerlos bien a través de un distribuidor autorizado, tal como un almacén mayorista, o directamente del titular de la autorización de comercialización, previa presentación de la autorización de la Agencia Española de Medicamentos y Productos Sanitarios para ese ensayo clínico. 2. Medicamentos procedentes de un tercer país: Podrá importar medicamentos en investigación un importador autorizado por la Agencia Española de Medicamentos y Productos Sanitarios, previa solicitud de la importación del medicamento en investigación de un ensayo clínico a la Agencia. Para obtener la autorización de importación de medicamentos procedentes de un tercer país sin un acuerdo de mutuo reconocimiento vigente, es necesario que aporte un certificado del director técnico o persona cualificada en el que se acredite que cada lote importado se ha fabricado en unas condiciones equivalentes a las normas de correcta fabricación de la Unión Europea o, si no se puede emitir ese certificado, un certificado de liberación de estos lotes firmado también por el director técnico, en el que se acredite que se han llevado a cabo, en la Unión Europea, los análisis, pruebas o controles necesarios para evaluar su calidad conforme a la información presentada para la autorización del ensayo. Artículo 6. Archivo maestro del ensayo y métodos de archivo. 1. La documentación relativa al ensayo clínico constituye el archivo maestro del mismo y constará de los documentos esenciales que permitan la evaluación de la realización de un ensayo clínico y de la calidad de los datos obtenidos. Estos documentos deberán demostrar el cumplimiento por parte del investigador y el promotor de los principios y directrices de buena práctica clínica y de todos los requisitos aplicables y, en particular, del anexo II sobre normas y protocolos analíticos, farmacotoxicológicos y clínicos relativos a la realización de pruebas de medicamentos del Real Decreto 767/1993, de 21 de mayo, por el que se regula la evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso humano fabricados industrialmente. 2. El archivo maestro del ensayo proporcionará la base para las auditorías que pueda realizar el auditor independiente del promotor y las inspecciones de las autoridades competentes. 3. El contenido de los documentos esenciales deberá ajustarse al carácter específico de cada fase del ensayo clínico. Se deberán tener en cuenta las orientaciones suplementarias sobre el contenido de dichos documentos publicados por la Comisión Europea. 4. El promotor y el investigador conservarán los documentos esenciales de cada ensayo clínico durante al menos cinco años tras la finalización del ensayo, o durante un período más largo si así lo disponen otros requisitos aplicables, como en el caso de que el estudio se presente como base para el registro de un medicamento en que se deberá cumplir el anexo II del Real Decreto 767/1993, de 21 de mayo, o un acuerdo entre el promotor, el investigador y el centro. 5. Los documentos esenciales deberán archivarse de forma que se puedan poner fácilmente a disposición de las autoridades competentes, en caso de que los soliciten. 6. La historia clínica del sujeto del ensayo deberá ser custodiada con arreglo a lo dispuesto en la Ley 41/2002, de 14 de noviembre, Básica Reguladora de la Autonomía del Paciente y de Derechos y Obligaciones en materia de Información y Documentación Clínica, y conforme al período máximo permitido por el hospital, la institución o la consulta privada. 7. Todos los cambios en la titularidad de los datos y documentos deberán documentarse. El nuevo titular asumirá las responsabilidades de las tareas de archivo y conservación de los datos.

47
8. El promotor nombrará a la persona de su organización responsable de los archivos y el acceso a los mismos deberá limitarse a las personas designadas. 9. Los soportes utilizados para conservar los documentos esenciales deberán garantizar que los documentos permanecen completos y legibles durante el período previsto de conservación y que estén a disposición de las autoridades competentes en caso de que los soliciten. Cualquier modificación de los registros habrá de ser trazable, permitiendo conocer el dato inicial y el corregido, así como la fecha y la firma del autor. Artículo 7. Titulación, formación y experiencia de los inspectores de buena práctica clínica. 1. Los inspectores deberán tener la debida cualificación y formación universitaria en medicina, farmacia, farmacología, toxicología u otras disciplinas pertinentes. 2. Las administraciones sanitarias garantizarán la formación de los inspectores, asegurándose de que: a) Reciban la formación adecuada, se evalúen periódicamente sus necesidades de formación y se adopten medidas adecuadas para mantener y mejorar su cualificación. b) Tengan información referente a: 1º Los principios y procedimientos referentes al desarrollo de medicamentos y de la investigación clínica. 2º La legislación europea y nacional aplicable a ensayos clínicos. 3º Las directrices aplicables a la realización de ensayos clínicos y a la concesión de autorizaciones de comercialización. 4º Los procedimientos y sistemas de registro de datos clínicos, así como sobre la organización y regulación del sistema de asistencia sanitaria de su competencia y, cuando proceda, de terceros países. 3. Las administraciones sanitarias garantizarán que los inspectores tienen experiencia previa suficiente para llevar a cabo una inspección. 4. Las administraciones sanitarias competentes deberán designar los inspectores. A tal efecto mantendrán un registro actualizado que incluya la titulación, formación y experiencia del inspector en temas de inspección de buena práctica clínica. Artículo 8. Normas comunes para la inspección de buena práctica clínica. 1. Para armonizar la realización de inspecciones por parte de las distintas administraciones sanitarias competentes, los inspectores deberán seguir para su actuación procedimientos normalizados de trabajo, que se aprobarán en el seno del órgano competente de coordinación técnica de inspecciones de la Agencia Española de Medicamentos y Productos Sanitarios creado a tal efecto, se actualizarán periódicamente de acuerdo con el avance científico y técnico y se harán públicos. 2. Las inspecciones se realizarán de conformidad con los documentos sobre inspecciones elaborados por la Agencia Europea de Medicamentos, para apoyar el reconocimiento mutuo de las conclusiones de las inspecciones dentro de la Unión Europea. Estos documentos serán utilizados a nivel nacional como orientadores de los procedimientos normalizados de trabajo de inspección, aprobados por el órgano competente de coordinación técnica de inspecciones de la Agencia Española de Medicamentos y Productos Sanitarios creado a tal efecto. 3. Se facilitará a cada inspector un documento con dichos procedimientos normalizados de trabajo, así como sus responsabilidades y funciones. 4. Los inspectores de buena práctica clínica deberán firmar un documento de confidencialidad antes de iniciar las actividades como inspector. 5. La actividad de inspección de buena práctica clínica tendrá carácter confidencial. Deberán cumplir los requisitos en cuanto a la protección de los datos personales, establecidos en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal. 6. Los inspectores contarán con los medios adecuados de identificación facilitados por la administración sanitaria correspondiente.

48
7. El inspector firmará una declaración de intereses, para comunicar cualquier vínculo económico o de otro tipo con las partes que serán objeto de inspección. Esta declaración se tendrá en cuenta a la hora de designar a los inspectores para una inspección determinada. 8. Sin perjuicio de las incompatibilidades establecidas para los inspectores en el ejercicio de sus actividades, asimismo serán incompatibles con cualquier tipo de intereses económicos directos derivados de la investigación clínica, así como de la fabricación, elaboración, distribución y comercialización de los medicamentos. 9. Se podrán nombrar equipos de inspectores y expertos con cualificaciones y experiencia adecuadas para cumplir de forma conjunta la cualificación necesaria para realizar la inspección. Artículo 9. Inspecciones de buena práctica clínica. 1. Las inspecciones de buena práctica clínica podrán realizarse antes, durante o después de la realización de los ensayos clínicos y podrán iniciarse como parte de la verificación de las solicitudes de autorización de comercialización, como seguimiento de éstas o como consecuencia de una denuncia. 2. Se podrán hacer inspecciones, entre otras, al centro de investigación, al lugar de fabricación del medicamento en investigación, a cualquier laboratorio de análisis utilizado en el ensayo clínico, a las instalaciones del promotor y/o de las organizaciones o empresas de investigación implicadas por contrato en la realización del ensayo y al Comité Ético de Investigación Clínica. 3. Los objetivos principales de las inspecciones de buena práctica clínica serán: a) Verificar que se han protegido los derechos, el bienestar y la seguridad de los sujetos que participan en los ensayos clínicos. b) Garantizar la validez de los datos procedentes de ensayos clínicos que se presentan como base para la autorización de comercialización de los medicamentos o como seguimiento de ésta. c) Garantizar la calidad de los ensayos clínicos que se realicen en centros sanitarios públicos y privados. 4. Realizada la inspección de buena práctica clínica, se elaborará un informe de inspección que deberá enviarse al inspeccionado y poner a disposición del promotor salvaguardando los aspectos confidenciales. 5. El informe de la inspección, previa solicitud motivada, se podrá poner a disposición de: a) El Comité Ético de Investigación Clínica. b) La Agencia Española de Medicamentos y Productos Sanitarios y las autoridades sanitarias de las Comunidades Autónomas. c) Las autoridades sanitarias de la Unión Europea. d) La Agencia Europea de Medicamentos. 6. En caso de que las desviaciones encontradas durante la inspección fueran calificadas de muy graves, de acuerdo con el artículo 101 de la Ley 29/2006, de 26 de julio de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, se enviará el informe al Comité Ético de Investigación Clínica, así como a la Agencia Española de Medicamentos y Productos Sanitarios o a la Comunidad Autónoma correspondiente según quien haya realizado la inspección. Artículo 10. Competencias en materia de inspección de buena práctica clínica. 1. La Agencia Española de Medicamentos y Productos Sanitarios llevará a cabo actuaciones de inspección en los siguientes casos: a) Para verificar los resultados de los ensayos clínicos presentados en las solicitudes de autorización de medicamentos mediante los procedimientos de reconocimiento mutuo, descentralizado y nacional. b) Para verificar los resultados de los ensayos clínicos presentados en las solicitudes de autorización de un medicamento mediante el procedimiento centralizado y coordinadas por la Agencia Europea de Medicamentos, dentro del ámbito de aplicación del Reglamento (CE) núm. 726/2004 del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos.

49
c) Cuando se trate de inspecciones a realizar en el territorio de las Comunidades Autónomas que no hubieran recibido los correspondientes traspasos de la competencia de ejecución de la legislación de productos farmacéuticos. 2. Podrán realizar inspecciones de buena práctica clínica, en todo el territorio español, los inspectores españoles acreditados, previa conformidad por la autoridad competente de la inspección del centro a inspeccionar. La Agencia Española de Medicamentos y Productos Sanitarios y las Comunidades Autónomas con competencia en materia de inspección de buena práctica clínica podrán solicitar la colaboración y/o participación de inspectores no adscritos a su ámbito de competencia cuando lo estimen necesario. Si alguna Comunidad Autónoma considera necesaria la realización de una inspección de buena práctica clínica fuera del ámbito territorial de su competencia, lo solicitará a la Agencia Española de Medicamentos y Productos Sanitarios, que dará traslado de la solicitud a la Comunidad Autónoma competente de la inspección. Corresponde a la administración sanitaria de esta última, autorizar la inspección y determinar si se lleva a cabo por inspectores propios o de la comunidad que la solicitó. 3. La relación con las autoridades competentes de los Estados miembros de la Unión Europea en materia de inspección de buena práctica clínica, se llevará a cabo a través de la Agencia Española de Medicamentos y Productos Sanitarios. 4. Las autoridades competentes de terceros países que vayan a realizar inspecciones de buena práctica clínica en España, deberán notificarlo a la Subdirección General de Inspección y Control de Medicamentos de la Agencia Española de Medicamentos y Productos Sanitarios con tiempo suficiente para que pueda acudir a la citada inspección, si fuera pertinente. La Agencia informará de la inspección a la Comunidad Autónoma donde se encuentre el centro a inspeccionar. El promotor será responsable de informar a la autoridad competente del tercer país de esta obligación de notificación. 5. La Agencia Española de Medicamentos y Productos Sanitarios elaborará los procedimientos pertinentes para las siguientes actividades: a) Solicitar inspecciones o ayuda de otros Estados miembros de la Unión Europea y cooperar en las inspecciones realizadas en centros de otro país de la Unión Europea. b) Realizar inspecciones en terceros países. Modificado por art. único de Orden núm. SCO/362/2008, de 4 febrero 2008 . Artículo 11. Recursos y bases de datos de las inspecciones de buena práctica clínica. 1. Las administraciones sanitarias competentes en materia de inspección de buena práctica clínica proporcionarán recursos personales y materiales suficientes para garantizar la comprobación efectiva del cumplimiento de las normas de buena práctica clínica. 2. La Agencia Española de Medicamentos y Productos Sanitarios creará una base de datos con las inspecciones nacionales y, en su caso, internacionales, incluyendo la situación de cumplimiento de buena práctica clínica, así como de su seguimiento. Para ello, las administraciones sanitarias competentes en materia de inspección remitirán a la Agencia los siguientes datos de todas las inspecciones de buena práctica clínica que se realicen en su territorio: código EudraCT del ensayo inspeccionado, datos del centro donde se realiza la inspección (nombre y dirección completa), tipo de centro inspeccionado, fecha de inspección y situación de cumplimiento de buena práctica clínica. Disposición Adicional primera. Normas de buena práctica clínica y directrices de la Unión Europea. La Agencia Española de Medicamentos y Productos Sanitarios publicará las normas de buena práctica clínica aplicables en los ensayos clínicos realizados en España. También se tendrán en cuenta las directrices publicadas por la Comisión Europea. Disposición Adicional segunda. Exención de la autorización de fabricación para medicamentos de terapia celular. En el caso de medicamentos de terapias avanzadas procesados en entidades de naturaleza pública integradas en el Sistema Nacional de Salud, dichas entidades no precisarán autorización de fabricación hasta la publicación de una norma específica.

50
Disposición Final primera. Título competencial. Esta Orden se dicta al amparo del artículo 149.1.16ª de la Constitución Española, que atribuye al Estado competencia exclusiva en materia de legislación sobre productos farmacéuticos. Disposición Final segunda. Incorporación de derecho de la Unión Europea. Mediante esta Orden se incorpora al derecho español la Directiva 2005/28/CE de la Comisión, de 8 abril de 2005, por la que se establecen los principios y las directrices detalladas de las buenas prácticas clínicas respecto a los medicamentos en investigación de uso humano, así como los requisitos para autorizar la fabricación o importación de dichos productos. Disposición Final tercera. Entrada en vigor. La presente Orden entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado». Afectado-por: • art. 10 - Modificado por art. único de Orden núm. SCO/362/2008, de 4 febrero 2008

51
La Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, dedica el Título III a las garantías de la investigación de los medicamentos de uso humano, estableciendo en el artículo 60.6 que ningún ensayo clínico podrá ser realizado sin informe previo favorable de un Comité Ético de Investigación Clínica debidamente acreditado. Todos los aspectos económicos relacionados con el ensayo clínico quedarán reflejados en un contrato entre el promotor y cada centro donde se vaya a realizar el ensayo, de acuerdo con lo establecido en el artículo 30 del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, con el fin de conseguir la mayor transparencia económica de los pactos existentes. Para ello, la Conselleria de Sanidad especificará los requisitos comunes, condiciones de financiación y contratación de investigadores, así como los sistemas adecuados que se han de utilizar para la percepción de la remuneración acordada en el contrato por parte del centro u hospital y por el investigador principal u otros colaboradores del ensayo. La entrada en vigor del contrato propuesto en esta resolución, unifica los diferentes modelos existentes en los centros sanitarios de la Comunitat Valenciana, normaliza el contenido a acordar entre las partes y debe conseguir una mayor agilización administrativa así como una disminución en los tiempos de tramitación que se traduce en una más rápida iniciación del ensayo clínico y estudios post-autorización observacionales. La existencia de diferentes modelos de contratos que difieren en función del centro y del tipo de estudio a realizar es uno de los principales motivos que demoran el comienzo efectivo de los mismos. La existencia de un modelo de contrato único y la aplicación de criterios económicos comunes a todos los actores constituye una herramienta para mejorar y agilizar el procedimiento. Por todo ello y de acuerdo con las competencias y funciones asignadas a la Agencia Valenciana de Salud de la Conselleria de Sanidad por el Decreto 120/2007, de 27 de julio, del Consell, por el que se aprueba el Reglamento Orgánico y Funcional de la Conselleria de Sanidad, y por el Decreto 25/2005, de 4 de febrero, del Consell de la Generalitat, por el que se aprueban los Estatutos reguladores de la Agencia Valenciana de Salud, dispongo: Artículo 1. Modelo de contrato que ha de suscribirse entre la Gerencia de un centro sanitario, el promotor y los investigadores y en su caso la Fundación, para la realización de un Ensayo Clínico o estudios post-autorización observacionales con medicamentos y productos sanitarios en las organizaciones de servicios sanitarios de la Comunitat Valenciana. 1.1. De conformidad con lo dispuesto en el artículo 30 del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, los aspectos económicos del ensayo clínico con medicamentos quedarán reflejados en un contrato entre el promotor y cada uno de los centros donde se vaya a realizar el ensayo. En esta resolución se establece un modelo único de contrato a suscribir entre el promotor, la gerencia y los investigadores, para la realización de un ensayo clínico con medicamentos y productos sanitarios en cualquier organización de servicios sanitarios de la Comunitat Valenciana. En el caso de los centros que gestionan sus actividades mediante fundación de derecho privado, constituida al amparo de lo previsto en la Ley 50/2002, de 26 de diciembre, de Fundaciones, el contrato será firmado además por el representante legal de la misma. La
Resolución de 16 de julio 2009, de la Consellería d e Sanidad, por el que se aprueba el modelo de contrato que ha de suscribirse entre la gerencia de un centro sanitario, el promotor y los investigadores, para la realización de un ensayo clínico o estudios post-autorización observacionales de medicamentos y productos sanitarios en las organizaciones de servicios sanitarios de la Comunitat Valenciana (2009/8925)

52
gestión de los cobros y los pagos derivados de la realización del ensayo clínico, deberá tramitarse a través de la fundación. 1.2 De igual manera, todos los aspectos económicos relacionados con un estudio post-autorización de tipo observacional con medicamentos y productos sanitarios quedarán reflejados en un contrato que se suscribirá, en todo caso, por las gerencias de los centros donde se vaya a realizar el estudio, los investigadores y el promotor y en su caso la Fundación. En este contrato se especificarán los siguientes aspectos: confidencialidad de los datos, compensación por costes indirectos derivados de la realización del estudio, contribución a la investigación biomédica y condiciones de publicación de los resultados del estudio. En el contrato podrán preverse compensaciones económicas por el promotor del estudio a los investigadores. Estas compensaciones serán explícitas, transparentes y proporcionales al tiempo y responsabilidades adicionales asumidas. 1.3 El anexo I establece el modelo de contrato único en las organizaciones de servicios sanitarios para ensayos clínicos y estudios observacionales en los centros sanitarios de la Comunitat Valenciana. 1.4 En el plazo máximo de un mes desde la comunicación del dictamen favorable del CEIC en el ámbito de la Comunitat Valenciana, los gerentes de los centros sanitarios firmarán el contrato con el promotor y los investigadores. El investigador suscribe el contrato en prueba de aceptación y conformidad de las obligaciones asumidas en calidad de investigador principal del estudio. 1.5. Los Gerentes suscribirán el documento de conformidad con lo dispuesto en la presente resolución. Artículo 2. Requisitos comunes del contrato económico 2.1 Para la compensación de la gestión administrativa, el promotor abonará la cantidad de 500 euros + IVA, cantidad efectiva a la firma del contrato y que no es susceptible de devolución 2.2 El contrato económico observará las siguientes especificaciones básicas: 2.2.1 Costes directos extraordinarios: Gastos que no se hubieran producido en el centro sanitario de no participar el sujeto en el ensayo. La fijación de los precios será establecida por la norma que fija los precios públicos de los servicios sanitarios prestados por centros dependientes de la Agencia Valenciana de Salud. El reembolso por gastos extraordinarios ocasionados a pacientes, y que no se hubieran producido de no haber participado el sujeto en el ensayo, así como, en su caso, las compensaciones a pacientes (voluntarios sanos o pacientes en ensayos clínicos que no supongan un potencial beneficio para ellos) por su participación en el ensayo, serán abonadas por el promotor al paciente a través del Centro/Fundación. 2.2.2 Costes indirectos: El Centro establecerá el porcentaje asignado a costes indirectos, que será al menos el 20% del presupuesto establecido por cada paciente reclutado. 2.2.3 Compensación al equipo investigador. Como compensación al equipo investigador por su participación en el ensayo clínico, y al considerarse una actividad extraordinaria, el equipo investigador percibirá una compensación económica máxima del 70% del presupuesto calculado por cada paciente reclutado evaluable, según protocolo, o por las cantidades correspondientes a pacientes que no completen el ensayo. Dicha cantidad se entrega al investigador principal, investigadores colaboradores y al personal que participa de forma efectiva en la realización y/o ejecución del ensayo clínico. Cada Centro establecerá las normas para cobrar por los trabajos realizados fuera del horario laboral. Del referenciado 70%, y siempre que sea posible, como mínimo un 20% del mismo será reinvertido en el Servicio o será aplicado por la dirección del centro al fomento de la I+D+I de las unidades servicios en los

53
que el investigador desarrolle el proyecto, así como las unidades de apoyo que puedan intervenir en el mismo. 2.3. Compensación por la participación de los servicios centrales del centro sanitario. La retribución por la participación del servicio de farmacia figurará en un anexo específico del contrato en función de la actividad generada por el estudio. En este anexo se podría incluir el resto de servicios clínicos centrales implicados en el estudio. La retribución por la participación del Servicio de Farmacia u otros servicios centrales será hasta un 10% del presupuesto establecido por cada paciente reclutado. Artículo 3. Comisión técnica de evaluación de los contratos de ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios. 3.1 A los efectos de profundizar en la repercusión de los efectos de los contratos de ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en los ámbitos correspondientes a régimen del personal estatutario y/o laboral, incompatibilidades, sistema retributivo, régimen vigente en materia presupuestaria, patrimonial y tributaria, pronunciamientos del Tribunal de Cuentas del Estado y fiscalidad de este tipo de contratos, se constituye una Comisión Técnica para el análisis de todos los aspectos referenciados. 3.2 La Comisión Técnica propondrá en su caso, en el plazo máximo de 12 meses desde la publicación de esta resolución, una norma con rango suficiente que diera respuesta a todos los aspectos referenciados en este apartado.
Instrucciones finales La presente resolución entrará en vigor al día siguiente de su publicación oficial en el DOCV. El modelo de contrato a que se refiere la presente resolución, será objeto de publicación en el portal de salud de la Consellería de Sanitat. Valencia, 16 de julio 2009.– El conseller de Sanidad: Manuel Cervera Taulet.

54
La Ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, ha intensificado el impacto de dos ideas-fuerza: la ampliación y reforzamiento de un sistema de garantías que gire en relación a la autorización del medicamento y la promoción del uso racional del mismo. El desarrollo tecnológico, la globalización, el acceso a la información así como la pluralidad de agentes que progresivamente intervienen en el ámbito de la producción, distribución, dispensación y administración de medicamentos aconsejan ampliar la transparencia y el control de sus resultados en el uso de medicamentos y productos sanitarios. El legislador, tanto europeo, estatal y autonómico, ha diseñado un marco normativo donde a los medicamentos y productos sanitarios se les dota de entidad propia a los efectos de la observación, análisis y control de resultados para la aplicación en la práctica clínica asistencial de los conocimientos derivados de la investigación básica en medicamentos y productos sanitarios. Fruto de ello es la inclusión de los ensayos clínicos dentro del marco competencial y regulatorio de los productos farmacéuticos. La promulgación de la ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, vuelve a reafirmar el entorno propio, excluyéndose esta materia del ámbito de aplicación de la Ley 14/2007, de 3 de julio, de Investigación Biomédica. El marco de actuación y tramitación en ensayos clínicos de medicamentos y productos sanitarios se complementa con las directrices estatales en farmacovigilancia y estudio post-autorización observacionales de interés para la salud pública, ostentando competencias de ejecución tanto la Agencia Española de Medicamentos y Productos Sanitarios como el Centro Coordinado de Comités Éticos de Investigación Clínica, a través de la Dirección General de Farmacia y Productos Sanitarios, del Ministerio de Sanidad y Política Social. La presente norma moderniza la estructura de gestión de los ensayos clínicos y estudios postautorización observacionales en medicamentos y productos sanitarios, facilitando la elección de la Comunitat Valenciana como lugar para el inicio de los estudios referenciados, desde el punto de vista de la agilidad en la tramitación y control de le ejecución de los estudios clínicos en medicamentos y productos sanitarios. La estructura de gestión mantiene una separación organizativa de las entidades que coordinan, impulsan, seleccionan o ejecutan las materias terapéuticas objeto de los protocolos de estudio, reforzando la independencia y transparencia en el control y la toma de decisiones sobre la idoneidad ético-clínica de los referenciados estudios. A los efectos de obtener el mayor grado de consenso, se ha recabado el apoyo y asesoramiento de las entidades de la Generalitat que impulsan materias de investigación sanitaria, así como de las fundaciones para la promoción de la investigación, estructuras que impulsan, promueven, favorecen y desarrollan la investigación científico-técnica en los centros sanitarios de la Comunitat Valenciana. La Consellería de Sanidad viene realizando actividades reguladoras e inspectoras en materia de ensayos clínicos de medicamentos y productos sanitarios desde la promulgación de la primera Ley del Medicamento del periodo constitucional. La Ley 25/1990, de 20 de Diciembre, del Medicamento, dedicó su título II a los ensayos clínicos con medicamentos, combinando la necesidad de su existencia como mecanismo necesario para los avances científicos y el obligado respeto a los derechos fundamentales de quienes sean cometidos a los mismos. El Real Decreto 561/1993, de 16 de abril, desarrolló la citada Ley atribuyendo a las Comunidades Autónomas la competencia en la especificación de los aspectos económicos de la realización de los ensayos clínicos en el procedimiento de acreditación de los Comités Éticos de Investigación Clínica (en adelante, CEIC) y en las facultades inspectoras en materia de ensayos clínicos respectivamente.
DECRETO 73/2009, de 5 de junio, del Consell , por el que se regula la gestión de ensayos clínicos y estudios post-autorización con medicamentos y productos sanitarios (2009/6667)

55
Mediante Orden de 6 de julio de 1994, de la Consellería de Sanidad y Consumo, se regulan estas competencias, unificando los criterios en la acreditación de CEIC y en la inspección sanitaria en materia de ensayos clínicos, estableciendo las medidas que se hablan de adoptar en lo que se refiere a contratos económicos y velando por el cumplimiento de las Normas de Buena Práctica para ensayos clínicos con medicamentos. En la misma línea, la ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, y el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, reafirman las competencias de las Comunidades Autónomas en esta materia y la necesidad imperativa de que todos los ensayos clínicos tengan un informe previo favorable de un CEIC debidamente acreditado. Aunque los medicamentos han contribuido decisivamente a la mejora de la esperanza y al aumento de la calidad de vida, en ocasiones plantean problemas de efectividad y de seguridad, necesitando las Administraciones Públicas adoptar políticas innovadoras que incorporen el concepto de farmacoepidemiología y gestión de los riesgos, así como la garantía de seguimiento continuando del balance beneficio/riesgo de los medicamentos autorizados. El Real Decreto 1344/2007 de 11 de octubre, que regula la farmacovigilancia de medicamentos de uso humano, persigue agilizar la tramitación de los estudios post-autorización observacionales de interés para la salud pública con medicamentos y productos sanitarios. Actualmente, se necesita potenciar y facilitar su desarrollo para generar un conocimiento que permita evaluar de un modo más efectivo la relación beneficio-riesgo de los medicamentos. Estos estudios se llevarán a cabo de acuerdo con las condiciones que establezcan las Administraciones Sanitarias en el ámbito de su competencia. La Consellería de Sanidad reguló esta potestad mediante la Orden de 8 de marzo de 2004, por la que se crea la Comisión Asesora de la Consellería de Sanidad para la Evaluación de los Estudios Post-Autorización Observacionales Prospectivos. La publicación en octubre de 2008 de la nueva versión de la declaración de Helsinki de la Asociación Médica Mundial (Seúl, 2008), presenta en su texto algunas novedades, en sus artículos 1, 14, 15 y 25, que afectan tanto a ensayos clínicos y estudios observacionales y que se incorporan en la presente regulación. En el marco expuesto se hace necesaria una ordenación y actualización del marco normativo valenciano, clarificando la participación de la Agéncia Valenciana de Salut, a través de su Dirección General de Farmacia y Productos Sanitarios, como órgano competente de gestión y control de la información referente a ensayos clínicos autorizados y estudios post-autorización observacionales de medicamentos y productos sanitarios. La presente disposición desarrolla el modelo valenciano mediante el establecimiento del Programa de Estudios Clínicos en Medicamentos y Productos Sanitarios de la Comunitat Valenciana (en adelante, PECME), modernizando el sistema de información autonómico sobre esta materia y regulando las actividades de formación y coordinación de los CEIC locales. Se constituye el Comité Ético Autonómico de Estudios Clínicos de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEC), que, adicionalmente a las funciones propias de evaluación clínica, se configura como un órgano consultivo encargado de asistir y asesorar en todo aquello relacionado con el desarrollo de los procesos de evaluación clínica en medicamentos y productos sanitarios. En virtud de todo lo anterior, de conformidad con lo dispuesto en el artículo 28 de la Ley del Consell, previa audiencia de los sectores implicados, a propuesta del conseller de Sanidad, conforme con el Consell, Jurídic Consultiu de la Comunitat Valenciana y previa deliberación el Consell, en la reunión del día 5 de junio de 2009,

56
DECRETO Artículo 1. Ámbito y objeto de aplicación La presente norma tiene por objeto establecer la estructura organizativa para la realización de ensayos clínicos y estudios post-autorización de tipo observacional con medicamentos y productos sanitarios de uso humano en la Comunitat Valenciana, de acuerdo con la normativa estatal en la materia. Articulo 2. Definiciones 1. Ensayo clínico: estudio efectuado en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos, y/o demás efectos farmacodinámicos, y/o detectar las reacciones adversas, y/o estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia. 2. Ensayo clínico multicéntrico: ensayo clínico realizado de acuerdo con un protocolo único pero en más de un centro y, por tanto, realizado por más de un investigador. 3. Estudio observacional: estudio en el que los medicamentos se prescriben de manera habitual, de acuerdo con las condiciones normales de la práctica clínica (aquellas establecidas en la autorización de comercialización). La asignación de un paciente a una estrategia terapéutica concreta no estará decidida de antemano por un protocolo de ensayo, sino que estará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica, y se utilizarán método epidemiológicos para el análisis de los datos recogidos. 3.a) Estudio observacional prospectivo: aquel estudio en el que los pacientes son seleccionados por su exposición a un determinado medicamento y son después seguidos durante un tiempo suficiente, siendo el periodo de estudio posterior al inicio de la investigación. 3.b) Estudio observacional retrospectivo: aquel estudio en el que los pacientes son seleccionados por su exposición a un determinado medicamento y son después seguidos durante un tiempo suficiente, siendo todo el periodo de estudio anterior al inicio de la investigación. 3.c) Estudio observacional transversal: aquel estudio en el que se registran observaciones sobre numerosos factores en el mismo momento y luego se comparan entre ellas. 4. Estudio post-autorización: cualquier estudio clínico o epidemiológico realizado durante la comercialización de un medicamento según las condiciones autorizadas en su ficha técnica, o bien en condiciones normales de uso, en el que el medicamento o los medicamentos de interés son el factor de exposición fundamental investigado. Este estudio podrá adoptar la forma de un ensayo clínico o un estudio observacional. Los estudios post-autorización deberán tener como finalidad el complementar la información obtenida durante el desarrollo clínico de los medicamentos, previo a su autorización, y no podrán condicionar directa o indirectamente la prescripción o dispensación. No se planificarán, realizarán o financiarán estudios post-autorización con finalidad de promover la prescripción o dispensación de los medicamentos. 4.a) Estudio post-autorización observacional: estudio epidemiológico que cumple las condiciones de ser post-autorización y observacional. 4.b) Estudio post-autorización observacional de seguimiento: todo aquel estudio post-autorización de tipo observacional en el que los pacientes son seleccionados por su exposición a un determinado medicamento y son después seguidos durante un periodo de tiempo

57
suficiente, en relación con el acontecimiento de interés. Se consideran prospectivos cuando el periodo de estudio posterior al inicio de la investigación y retrospectivos cuando el periodo de estudio es todo él anterior al inicio de la investigación. Cuando se realice un estudio post-autorización observacional de seguimiento prospectivo, el o los promotores e investigadores deberán expresar específicamente en el protocolo los procedimientos que se emplearán para garantizar que la realización del estudio no modificará los hábitos de prescripción del médico o de dispensación del farmacéutico (en caso de medicamentos que no requieran prescripción) 5. Reacción adversa: cualquier respuesta a un medicamento que sea nociva y no intencionada, y que tenga lugar a dosis que se apliquen normalmente en el ser humano para la profilaxis, el diagnóstico o el tratamiento de enfermedades, o para la restauración, corrección o modificación de funciones fisiológicas. Este término incluye también todas las consecuencias clínicas perjudiciales derivadas de la dependencia, abuso y uso incorrecto de medicamentos, incluyendo las causadas por el uso fuera de las condiciones autorizadas y las causadas por errores de medicación. 5.a) Reacción adversa grave: cualquier reacción adversa que ocasione la muerte pueda poner en peligro la vida, exija la hospitalización del paciente o la prolongación de la hospitalización ya existente, ocasione una discapacidad o invalidez significativa o persistente o constituya una anomalía congénita o defecto de nacimiento. A efectos de su notificación, se tratarán también como graves aquellas sospechas de reacción adversa que se consideren importantes desde el punto de vista médico, aunque no cumplan los criterios anteriores, como las que ponen en riesgo al paciente o requieren una intervención para prevenir alguno de los desenlaces anteriores. Así mismo, a efectos de su notificación, se tratarán como graves todas las sospechas de transmisión de un agente infeccioso a través de un medicamento. 5.b) Reacción adversa inesperada: cualquier reacción adversa suya naturaleza, gravedad o consecuencias no sean coherentes con la información descrita en ficha técnica. 6. Medicamento en investigación: forma farmacéutica de un principio activo o placebo, que se investiga o utiliza como referencia en un ensayo clínico, incluido los productos con autorización cuando se utilizan o combinen (en la formulación o en el envase) de forma diferente a la autorizada, o cuando se utilicen para tratar una indicación no autorizada, o para obtener más información sobre un uso autorizado. Artículo 3. Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana (PECME) 1. El Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana (PECME), dependiente de la Agéncia Valenciana de Salut, gestiona las funciones citadas en esta disposición propias de ensayos clínicos y estudios post-autorización observacionales, así como las establecidas en esta materia en el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, el Real Decreto 1344/2007, de 11 de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano, y el Real Decreto 1143/2007, de 31 de agosto, por el que se modifican los Reales Decretos 634/1993, de 3 de mayo, sobre productos sanitarios implantables activos; 414/1996, de 1 de marzo, por el que se regulan los productos sanitarios; y 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnóstico ¨in vitro¨. 2. El Programa tendrá los objetivos siguientes: a) Promover la correcta realización de ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios tanto en la aplicación de la normativa vigente como desde el punto de vista científico, así como el adecuado conocimiento de los aspectos éticos y metodológicos por parte de todos los agentes implicados en la realización y evaluación de dichos ensayos y estudios.

58
b) Garantizar que los miembros de los CEIC acreditados en la Comunitat Valenciana están capacitados para evaluar los protocolos de los ensayos clínicos y estudios post-autorización observacionales en sus aspectos metodológicos, ético y legal. c) Establecer y organizar sistemas de información adecuados y fluidos entre la administración Sanitaria autonómica, la Agencia Española de Medicamentos y Productos Sanitarios, el Centro Coordinador de los CEIC, los CEIC y los promotores e investigadores de los ensayos clínicos y estudios post-autorización observacionales. 3. El Programa se articula con la intervención de las siguientes entidades: a) El Consejo de Dirección y la comisión delegada del PECME. b) El Coordinador del PECME. c) El Comité Autonómico de Estudios Clínicos de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEC) d) El Comité Autonómico de Estudios Post-autorización Observacionales de Medicamentos y Productos sanitarios de la Comunitat Valenciana (CAEPO). e) Los CEIC locales y corporativos acreditados en el ámbito de la Comunitat Valenciana (CEIC) Articulo 4. El Consejo de Dirección y la Comisión delegada del Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana (PECME) 1. El Consejo de Dirección del PECME tutela las actividades y acciones para el desarrollo de los objetivos de Programa en la Comunitat Valenciana. El Consejo conocerá y aprobará el plan anual y la memoria de actividades del PECME. 2. El Consejo de Dirección de PECME estará constituido por los miembros siguientes: a) Presidente: el director Gerente de la Agéncia Valenciana de Salut. b) Vicepresidente: el director general de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut. c) Secretario: el Coordinador del PECME. d) El director general de Ordenación, Evaluación e Investigación Sanitaria, de la Consellería de Sanidad. e) El director general de Asistencia Sanitaria, de la Agéncia Valenciana de Salut. f) El director general de Calidad y Atención al Paciente, de la Agencia Valenciana de Salut. g) El director general d e Salud Pública, de la Conselleria de Sanidad h)El Jefe de Área de Farmacia y Productos Sanitarios. i) El Jefe de Área de Inspección, Evaluación e Investigación Sanitaria. j) El Jefe de la Oficina de Investigación Sanitaria. k) Los Presidentes de cada uno de los CEIC acreditados en la Comunitat Valenciana. Artículo 4.2 corregido según la corrección de errores del decreto 73/2009, publicada en el Diari Oficial de la Comunitat
Valenciana el 30 de julio de 2009
El Consejo de Dirección se reunirá con carácter al menos semestral, o cuando se precise con carácter de urgencia. 3. La Comisión Delegada del PECME mantendrá el nivel de decisiones operativas entre periodos de reuniones del Consejo de Dirección de PECME, realizando jornadas de trabajo preceptivamente con carácter bimestral. Estará presidida por el Jefe de Área de Farmacia y Productos Sanitarios, actuando como Secretario el Coordinador del PECME. La Comisión Delegada estará integrada por aquellos miembros del Consejo designados por el Presidente del Consejo de Dirección del PECME.

59
Artículo 5. Coordinador del Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana. 1. El Coordinador del PECME será nombrado por el director Gerente de la Agéncia Valenciana de Salud, a propuesta del director general de Farmacia y Productos Sanitarios, de dicha Agéncia. 2. Serán funciones del Coordinador de PECME: a) Recibir, informar y tramitar en la Dirección General de Farmacia y Productos Sanitarios, de la Agencia Valenciana de Salut, las solicitudes de acreditación y reacreditación de los CEIC, el ámbito geográfico de actuación y comunicar el resultado de dicha acreditación al centro solicitante. Comunicar la Agencia Española de Medicamentos y Productos Sanitarios las acreditaciones y reacreditaciones de los CEIC. b) Comunicar a los centros la necesidad de reacreditación de cada CEIC en los plazos pertinentes o cuando así se disponga, recibir las memorias para reacreditación de los Comités, tramitar la solicitud de la reacreditación y comunicar el resultado de la misma. c) Archivar copia de los informes de aprobación o desestimación de los CEIC acerca de los protocolos recibidos. También se recogerán y archivarán las modificaciones relevantes de los protocolos aprobados y los certificados emitidos de adecuación deontológica de protocolos de investigación en humanos. d) Regular los trámites para el traslado de un ensayo clínico de un centro de la Comunitat Valenciana a otro y recibir y archivar copia de los documentos tramitados, una vez autorizado por la Agencia Española de Medicamentos y Productos Sanitarios. e) Recibir y evaluar las comunicaciones de reacciones adversas graves o inesperadas que acontezcan durante el desarrollo del ensayo clínico, conocidas por el CEIC, y comunicarlas al resto de centros implicado en el mismo ensayo o en otros con el mismo producto de estudio. f) Recibir, informar y tramitar a la Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, la propuesta de los CEIC de suspensión cautelar de un ensayo clínico que se esté llevando a cabo, o realizar propuestas de suspensión. g) Proponer la realización de inspecciones en materia de ensayos clínicos, colaborar en la elaboración y comunicación de los protocolos de dichas inspecciones, recibir y archivar una copia de los resultados de los resultados de las mismas e informar de ellas a las autoridades de la Consellería de Sanidad. h) Solicitar, recoger y archivar la notificación por el promotor de la finalización del ensayo clínico, o de la justificación de su suspensión. i) Realizar revisiones bibliográficas de las publicaciones de ensayos clínicos realizados en centros sanitarios de la Comunitat Valenciana, comprobando que han sido llevados a cabo en los términos que establece la normativa vigente. Asimismo, aconsejar la publicación o notificación de resultados tanto positivos como negativos de los ensayos clínicos realizados. j) Ser vehículo de coordinación con la Agencia Española de Medicamentos y Productos Sanitarios y otros Comunidades Autónomas en materia de desarrollo y realización de ensayos clínicos. En este sentido, recibirá la comunicación sobre autorización, denegación, suspensión o modificación de ensayos clínicos de la Agencia Española de Medicamentos y Productos Sanitarios.

60
k) Recepción de los CEIC acreditados en la Comunitat Valenciana de todos los estudios por ellos autorizados. Esta función será sustituida si la información residente en la Agencia Española de Medicamentos y Productos Sanitarios se encuentra disponible con medios electrónicos. l) Gestión de una base de datos y un archivo documental donde se recoja toda la información relativa a su actividad. m) Elaboración de una memoria anual de sus actividades. 3. La Agéncia Valenciana de Salut, a través de su Dirección General de Farmacia y Productos Sanitarios, impulsará los medios telemáticos y electrónicos necesarios para agilizar todos los trámites descritos en el apartado anterior. Artículo 6. Procedimiento de acreditación de los CEIC 1. El responsable o responsables de los centros, servicios y establecimientos sanitarios de la Comunitat Valenciana autorizados por la Conselleria de Sanidad podrán solicitar la acreditación de los CEIC de dichos centros ante la Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut. 2. La acreditación del CEIC tendrá una validez de 3 años. Según los términos indicados en la legislación vigente, los miembros de CEIC serán provistos de la correspondiente acreditación de pertenencia al CEIC expedida por la Dirección General de Farmacia y Productos Sanitarios. 3. La renovación de la acreditación de los CEIC deberá realizarse cada tres años. La Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, podrá revocar la acreditación concedida a cualquier CEIC que no cumpla los requisitos mínimos exigidos en la presente disposición y en el resto de la norma aplicable. Articulo 7. Requisitos de la acreditación de los CEIC 1. El ámbito geográfico e institucional de actuación de cada CEIC será determinado por la Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, y se establecerá en función del número de CEIC acreditados, abarcando, necesariamente, el departamento de salud correspondiente y, en su caso, otros departamentos de salud, oído el centro o centros interesados, y teniendo en cuenta la distribución geográfica de éstos. 2. Sus miembros serán nombrados por el director general de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de salud, a propuesta de la dirección de la institución de que se trate, tras informe de la coordinación de PECME, de acuerdo con los criterios establecidos en la presente disposición en su artículo 5.2 a). La renovación de la composición de los CEIC se realizará por el mismo procedimiento, tras consulta con el propio CEIC. 3. Los requisitos mínimos para la acreditación de un CEIC serán los siguientes: a) Estar formado, como mínimo, por once miembros, y en todo caso formar parte de él:
1º. El director del centro 2º. Médicos: Un farmacólogo clínico Dos médicos especialistas con labor asistencial activa en un centro hospitalario Un pediatra 3º. Farmacéuticos: Un farmacéutico de hospital Un farmacéutico de Atención Primaria 4º. Un diplomado en Enfermería

61
5º. Dos miembros ajenos a las profesiones sanitarias, debiendo ser uno de ellos licenciado en Derecho. Uno de estos miembros será una persona independiente de la organización asistencial, entendiendo por independiente la desvinculación laboral en relación a los centros en los que se lleven a cabo los proyectos de investigación que requieran la evaluación ética por parte del Comité, a ser posible propuesto por el Consejo de Salud de la Comunitat Valenciana. b) Tener garantía explicita, por parte del titular del centro, de que el CEIC cuenta con los medios necesarios para poder realizar su cometido. Los medios básicos a disponer son: 1º. Instalaciones específicas que permitan la realización de su trabajo, en condiciones que garanticen la confidencialidad. Deberán disponer de un espacio apropiado para la Secretaria del Comité, para la realización de las reuniones y para el manejo y archivo de documentos confidenciales. 2º. Equipamiento informático con capacidad suficiente para manejar toda la información generada por el Comité y disponibilidad de un sistema rápido de transmisión de información. 3º. Personal administrativo y técnico permita al Comité poder ejercer de manera apropiada sus funciones. c) Disponer de los correspondientes procedimientos normalizados de trabajo. 4. La renovación de la acreditación de los CEIC deberá cumplir los requisitos mínimos siguientes: a) Los de su previa acreditación. b) Realizar con carácter mensual, al meno, 11 reuniones anuales, con evaluación de
ensayos clínicos y seguimiento de los aprobados. c) Superar favorablemente las inspecciones de la Consellería de Sanidad. Artículo 8. Funcionamiento de los CEIC 1. Además de las funciones señaladas en el Real Decreto 223/2004, de 6 de febrero, los CEIC en la Comunitat Valenciana llevarán a cabo las funciones siguientes: a) Evaluar la información escrita sobre las características del estudio que se dará a los posibles sujetos de la investigación, así como la forma en que dicha información sea proporcionada y el tipo de consentimiento que fuera a obtenerse. b) Conocer y evaluar el alcance de las compensaciones ofrecidas a los investigadores y a los sujetos de la investigación, así como la forma en que dicha información sea proporcionada. c) Recabar el asesoramiento de personas expertas no pertenecientes al Comité en los supuestos a que se refiere el artículo 14.4 del Real Decreto 223/2004, de 6 de febrero, que respetarán el principio de confidencialidad. d) Realizar un informe acerca de las modificaciones de los protocolos de los ensayos, velando por la corrección de los protocolos propuestos desde el punto de vista metodológico, ético y legal. e) Informar al Coordinador del PECME de todos aquellos protocolos de investigación que aprueben y que se eleven al órgano competente para su autorización, y también de los que finalmente se desestimen. f) Remitir al Coordinador del PECME una copia de los certificados que se emitan sobre la adecuación deontológica de los protocolos que impliquen la investigación en humanos. g) Comunicar al Coordinador del PECME las reacciones adversas graves o inesperadas que acontezcan durante el desarrollo del ensayo, mediante copia de la notificación del investigador o promotor. h) Proponer a la Conselleria de Sanidad, a través del Coordinador del PECME, la suspensión cautelar de un ensayo clínico en los supuestos previstos en la legislación vigente. i) El CEIC hará explícitos los criterios de idoneidad de los investigadores y del Centro para ser objetivos en la evaluación.

62
2. El CEIC desarrollará sus funciones con independencia y según sus procedimientos normalizados de trabajo, de conformidad con lo previsto en el artículo 6 de la Ley 29/2006, de 26 de julio, de Garantías y Uso Racional de Medicamentos y Productos Sanitarios, y en el artículo 14.2 del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Los miembros del Comité deberán respetar el principio de confidencialidad en lo que se respeta a la documentación recibida para la evaluación del protocolo y la identidad de los pacientes. 3. La pertenencia a un CEIC será incompatible con cualquier clase de intereses derivados de la fabricación y venta de medicamentos y productos sanitarios. Los miembros harán constar ante la Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, antes de su toma de posesión la no concurrencia de dicha circunstancia. El investigador principal o los colaboradores de un estudio no podrán participar ni en la evaluación ni en el dictamen de su propio protocolo, aun cuando sean miembros del CEIC. Esta norma es extensiva a los promotores y monitores de los ensayos clínicos cuyo proyecto sea sometido al estudio del comité. Ni el CEIC ni ninguno de sus miembros podrán percibir, directa o indirectamente, remuneración alguna del promotor del ensayo o estudio observacional. Artículo 9. Organización de los CEIC 1. Los CEIC estarán compuestos de un Presidente, un Vicepresidente, un Secretario y los Vocales. 2. Corresponde al Presidente del CEIC: a) Presidir las reuniones del Comité.
b) Elaborar, junto con el Secretario, las actas y el orden del día de las reuniones correspondientes.
c) Elaborar, junto con el Secretario, la memoria anual del Comité. d) Velar por la consecución de los objetivos asignados al Comité. e) Realizar cuantas funciones sean inherentes a su condición de Presidente. 3. El Vicepresidente del CEIC asistirá en sus funciones al Presidente del Comité y le sustituirá
en caso de vacante, ausencia o enfermedad. 4. Son funciones del Secretario del CEIC:
a) Efectuar la convocatoria de las reuniones por orden del Presidente y las citaciones correspondientes.
b) Redactar las actas de las reuniones. c) Expedir las certificaciones de los dictámenes y acuerdos adoptados.
d)Redactar y firmar, junto con el Presidente, la memoria anual del Comité, así como las funciones generales de Secretario contempladas en la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
e) Realizar cuantas otras funciones sean inherentes a su condición de Secretario. 5. Son funciones de los vocales del CEIC: a) Asistir a las reuniones a las que hayan sido convocados.
b) Evaluar la documentación que reciban correspondiente a la posterior valoración por el Comité.
c) Realizar aquellas tareas que les sean asignadas por el presidente. 6. Para la válida constitución del órgano, a efectos de celebración de sesiones, deliberaciones o toma de acuerdos, se requerirá la presencia del Presidente, o en su caso del Vicepresidente, y del Secretario, o, en su caso, de quien le sustituya, y de la mitad más uno de sus miembros. En el caso de no concurrir suficiente número de miembros para considerarse válidamente constituida la sesión, transcurrida media hora, o el tiempo establecido en la convocatoria, se tendrá por realizada una segunda convocatoria, en cuyo caso para que se constituya

63
válidamente bastará con la concurrencia del Presidente, o, en su caso, del Vicepresidente, del Secretario o persona que le sustituya, y, al menos, de tres miembros más. 7. En lo no previsto expresamente en este artículo se aplicará lo dispuesto en el capítulo II del título II de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común. Artículo 10. Funciones de la dirección del centro o establecimiento sanitario con CEIC acreditado en la Comunitat Valenciana. La dirección del centro llevará a cabo las funciones siguientes: 1. Solicitar la acreditación del CEIC. 2. Firmar el documento correspondiente de conformidad con la realización del ensayo clínico en el centro y con el contrato firmado con el promotor en el que se especifican todos los aspectos económicos de dicho ensayo clínico. 3. Garantizar los medios necesarios para la correcta realización de las funciones del CEIC, descritos en el artículo 7.3.b) de esta disposición. 4. Coordinar las actuaciones del CEIC con las distintas Comisiones existentes en el centro. 5. Solicitar, en los casos que así se determine, la autorización del centro para la realización de ensayos clínicos sin finalidad terapéutica. Artículo 11. El Comité Ético Autonómico de Estudios Clínicos de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEC) y los CEIC de ámbito corporativo. 1. El Comité Ético Autonómico de Estudios Clínicos de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEC) es un órgano colegiado de carácter técnico sobre ensayos clínicos y estudios post-autorización observacionales, con ámbito de actuación en la Comunitat Valenciana. 2. El CAEC de la Comunitat Valenciana tendrá las siguientes atribuciones y actividades: a) Evaluación de las solicitudes de ensayos clínicos con medicamentos/productos sanitarios o estudios post-autorización observacionales, que, de forma ordinaria o extraordinaria, requieran su consideración remitidos por los directores de los centros sanitarios de la Agencia Valenciana de Salut o por los CEIC acreditados en la Comunitat Valenciana o por fundaciones dependientes de los centros sanitarios de la Agéncia Valenciana de Salut. b) Establecer los requisitos mínimos curriculares de los miembros de un CEIC, el contenido de la memoria de acreditación y el sistema de evaluación de las actuaciones de los CEIC acreditados en la Comunitat Valenciana. c) Realizar la evaluación de la actividad de los CEIC locales acreditados, garantizando la unidad de criterio y dictamen y prestando el apoyo y asesoramiento técnico que requieran en el ejercicio de sus funciones. d) Velar por la correcta aplicación de los principios éticos, metodológicos y legales de los CEIC acreditados en la Comunitat Valenciana, realizando propuestas para la normalización de los procedimientos de trabajo y criterios de evaluación. e) Proponer medidas para agilizar los trámites burocráticos y reducir los tiempos de evaluación y respuestas en los ensayos clínicos y proyectos de investigación. f) Promover reuniones periódicas para abordar temas de interés común que apoyen y aumenten la calidad de los ensayos clínicos y de los proyectos de investigación. g) Promover las relaciones entre las instituciones y personas relacionadas con los ensayos clínicos y proyectos de investigación. 3. El CAEC estará constituido por el Presidente, el Vicepresidente, el Secretario y los Vocales. Podrán asistir a las reuniones del Comité expertos que asesoran cuando sena requeridos para ello. El CAEC adoptará los procedimientos de trabajo que a tal efecto se establezcan y actuará de acuerdo con lo que se determine en sus normas internas de funcionamiento. El

64
Comité se reunirá con carácter mensual al menos once veces al año. De cada reunión se levantará acta en al que se consignarán los acuerdos adoptados y los miembros asistentes. 4. La designación de los miembros será realizada por el director general de Farmacia y productos Sanitarios, de la Agéncia Valenciana de Salut, a propuesta del coordinador del PECME, que actúa como Secretario del CAEC. El director general de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, nombrará, entre ellos, al Presidente y Vicepresidente. El CAEC estará formado por un mínimo de trece miembros, entre los que figurarán: a) Médicos, entre los cuales habrá al menos un farmacólogo clínico, dos médicos especialistas con labor asistencial en un centro hospitalario, un médico con labor asistencial en un centro de Atención Primaria, un pediatra y un experto en epidemiología clínica. b) Farmacéuticos, entre los cuales, al menos, habrá un farmacéutico de hospital, un farmacéutico de atención primaria y un farmacéutico del Centro Valenciano de información de Medicamentos. c) Un diplomado en Enfermería. d) Al menos dos miembros ajenos a la profesión sanitaria, debiendo ser uno de ellos Licenciado en Derecho. e) Una persona independiente de la organización asistencial. f) Un representante del Centro de Farmacovigilancia de la Comunitat Valenciana. g) El Coordinador del PECME. 5. A los efectos de agilizar la evaluación de los ensayos clínicos y estudios post-autorización observacionales en determinado ámbitos asistenciales, se conforman los CEIC corporativos de Atención Primaria de la Comunitat Valenciana y de Salud Pública (Centro Superior de Investigaciones en Salud Pública), de ámbito multicéntrico, facilitando el fomento de la investigación en sus respectivos ámbitos sanitarios. 6. El CEIC corporativo de Salud Pública del Centro Superior de Investigación en Salud Pública se regirá, en cuanto a su régimen de creación, composición y funcionamiento por lo dispuesto en los artículos 6, 7, 8, 9, y 10 de la presente norma. 7. El CEIC corporativo de Atención Primaria de la Comunitat Valenciana se regirá, en cuanto a su régimen de creación, composición y funcionamiento por lo dispuesto en los artículos 6, 7, 8, 9 y 10 de la presente norma. La designación de los miembros será realizada por el director general de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, a propuesta del Coordinador del PECME. El director general de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, nombrará entre los miembros al Presidente y Vicepresidente.
Artículo 11.7 corregido según la corrección de errores del decreto 73/2009, publicada en el Diari Oficial de la Comunitat
Valenciana el 30 de julio de 2009
Artículo 12. El Comité Autonómico de Estudios Post-autorización Observacionales de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEPO) 1. El Comité Autonómico de Estudios Post-autorización Observacionales Prospectivos de Medicamentos y Productos Sanitarios de la Comunitat Valenciana (CAEPO) es un órgano consultivo para la autorización de dichos estudios en su ámbito de competencias. 2. El CAEPO estará formado por: a) Un Presidente, que será el Jefe del Área de Farmacia y Productos Sanitarios. b) Un Vicepresidente, que será el Jefe de Servicio de Provisión y Asistencia Farmacéutica.
c) Los Jefes de Servicio de Prestación Ortoprotésica y Ordenación y Control de Medicamentos.
d. Un profesional del Centro Autonómico Valenciano de Información de Medicamentos y Productos Sanitarios, que actuará como Secretario.

65
e) Dos profesionales, preferentemente médicos, que pertenezcan o hayan pertenecido a los CEIC de Atención Primaria acreditados por la Consellería de Sanidad.
f) Dos profesionales, preferentemente médicos, que pertenezcan o hayan pertenecido a los Comités Éticos de Investigación Clínica de Hospital acreditados por la Consellería de Sanidad.
g) Un farmacéutico de atención primaria. h) Un farmacéutico de atención especializada. i) Un especialista en oncología. j) Un especialista de medicina interna. k) Dos médicos de Atención Primaria. l) Un farmacólogo clínico. m) Un experto en epidemiología y estadística. 3. Son funciones del CAEPO: a) Evaluar la pertinencia de los estudios post-autorización observacionales prospectivos
presentados en la Dirección General de Farmacia y Productos Sanitarios. b) Establecer los criterios de valoración por los que se regirá el CAEPO para la evaluación
de la pertinencia de los estudios post-autorización observacionales prospectivos. c) Emitir informe motivado al director general de Farmacia y Productos Sanitarios sobre la
pertinencia de los estudio post-autorización observacionales prospectivos. 4. Los miembros del CAEPO serán nombrados por el director general de Farmacia y Productos Sanitarios. El nombramiento tendrá una duración inicial de dos años, pudiendo renovarse, sucesivamente y por una sola vez, por otro periodo adicional de dos años. El comité establecerá sus normas de funcionamiento. Todo lo que no esté dispuesto en esta norma de creación se ajustará a las disposiciones que regulan los órganos colegiados. Artículo 12 corregido según la corrección de errores del decreto 73/2009, publicada en el Diari Oficial de la Comunitat
Valenciana el 30 de julio de 2009 Artículo 13. Aspectos legales y económicos de los ensayos y estudios post-autorización observacionales con medicamentos y productos sanitarios. 1. Todos los estudios en humanos con medicamentos y productos sanitarios, incluyendo los supuestos donde sólo se recoge información de la historia clínica, deben ser presentados en forma de protocolo, y evaluados por un CEIC acreditado por la Consellería de Sanidad. Se incluyen tanto los ensayos clínicos como los estudios observacionales, tanto prospectivos como retrospectivos. El protocolo debe incluir información sobre financiación patrocinadores, afiliaciones institucionales, otros posibles conflictos de interés e incentivos para las personas del estudio. 2. De conformidad con lo previsto en la Ley 41/2002, de 14 de noviembre, Básica Reguladora de la Autonomía del Paciente y de Derechos y Obligaciones en Materia de Información y Documentación Clínica, en los ensayos clínicos y estudios post-autorización de tipo observacional prospectivos es imprescindible que el sujeto otorga libre y voluntariamente su consentimiento informado antes de ser incluido en el estudio, debiendo ser informado sobre la naturaleza, importancia, implicaciones y riesgo del ensayo clínico. En el caso de menores de edad o personas incapaces, este consentimiento lo debe otorgar su representante legal. 3. El consentimiento debe obtenerse por escrito. Si el sujeto del ensayo o estudio no está en condiciones para escribir, podrá dar, en caso excepcionales, su consentimiento verbal en presencia de, al menos un testigo mayo de edad y con capacidad de obrar. El sujeto participante en un ensayo clínico o estudio, o su representante, podrá revocar, en todo momento, su consentimiento sin expresión de causa. 4. En los estudios post-autorización de tipo observacional retrospectivos y transversales que requieran entrevistar al sujeto, o en aquellos en los que, utilizando otras fuentes de

66
información, no sea posible adoptar un procedimiento de disociación segura, se solicitará el consentimiento informado de los sujetos. Podrá haber situaciones en las que será imposible o impracticable obtener el consentimiento para dicha investigación o podría ser una amenaza para su validez. En esta situación, la investigación sólo puede ser realizada después de ser considerada y aprobado por un CEIC. Artículo 14. Sistema de información autonómico en materia de ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios. 1. Para facilitar la comunicación con los CEIC y disponer de un repositorio de información sobre ensayos clínicos y estudios post-autorización con medicamentos y productos sanitarios, la Dirección General de Farmacia y Productos Sanitarios, de la Agéncia Valenciana de Salut, dispondrá de un portal de trabajo exclusivo del PECME, garantizando la confidencialidad en todo momento. 2. La Agéncia Valenciana de Salut facilitará al PECME un sistema de gestión operativo y bases de datos para realizar la gestión de la información y trámites administrativos de ensayos clínicos y estudios post-autorización observacionales en medicamentos y productos sanitarios en la Comunitat Valenciana. El sistema facilitará la integración o la comunicación con los sistemas de información de la Agencia Española de Medicamentos y Productos Sanitarios, en especial SIC-CEIC. 3. Las historias clínicas de los pacientes dispondrán de un sistema permanente, ágil y rápido para identificar que un paciente participa o ha participado en un ensayo clínico o estudio post-autorización observacional. En la historia clínica del paciente se archivará una copia del consentimiento informado. Por parte de la Agéncia Valenciana de Salut se potenciará la identificación el consentimiento informado electrónico por parte del paciente en el sistema de información sanitario de la Agéncia Valenciana de Salut. En cualquier caso, y en ausencia de firma electrónica del paciente, en aquellos centros sanitarios que dispongan de historia clínica electrónica, deberá constar su firma de consentimiento en formato de papel. 4. Para facilitar la comunicación y el manejo de datos en los proyectos de estudios de medicamentos y productos sanitarios, la documentación deberá ser presentada por el promotor en formato digital. Artículo 15. Formación y normalización en la toma de decisiones de los miembros de los CEIC 1. El PECME impulsará las actividades periódicas de formación y jornadas de normalización de criterios entre los profesionales que integran los CEIC acreditados en la Comunitat Valenciana, para minimizar las variaciones en la toma de decisiones, intentando fomentar, en el ámbito de la Comunitat Valenciana, unos criterios de evaluación homogéneos y adecuados. 2. Para agilizar el proceso de evaluación, el PECME promoverá jornadas de coordinación con todos los agentes implicados, evaluadores, promotores e investigadores con la colaboración del Centro Coordinador de Comités Éticos de Investigación Clínica, de la Dirección General de Farmacia y Productos Sanitarios, del Ministerio de Sanidad y Política Social, y el Comité de Coordinación de Estudios Post-autorización, de la Agencia Española de Medicamentos y Productos Sanitarios. Artículo 16. Inspección del PECME por la Consellería de Sanidad 1. Se entiende por inspección la revisión oficial por una autoridad competente de los documentos, as instalaciones, los archivos, los sistemas de garantía de calidad y cualesquiera otros elementos que la autoridad competente considere relacionadas con el ensayo clínico/proyecto de investigación y que puedan encontrarse en el lugar del ensayo, en las

67
instalaciones del promotor y/o de la organización de investigación por contrato, o en cualquier otro establecimiento que la autoridad competente considere oportuno inspeccionar. Tras la inspección se elaborará un informe que estará a disposición de los inspeccionados. 2. En un proceso de inspección de un ensayo clínico/proyecto de estudio, se auditará al menos: a) Hoja de información al paciente. b) Aprobación del estudio y/o modificaciones del protocolo por parte del CEIC y las
autoridades sanitarias. c) Grado de adhesión al protocolo: cumplimiento de los criterios de selección, asignación al
tratamiento, administración y dosificación de fármacos, preservación del ciego y pruebas complementarias.
d) Control de la medicación suministrada: almacenamiento, dispensación, recogida y recuento medicación sobrante.
e) Verificación de los datos de las fuentes originales. f) Validación de las variables eficacia y seguridad. g) Registro de las notificaciones de efectos adversos. 3. La Inspección de Servicios Sanitarios de la Consellería de Sanidad llevará a cabo las actuaciones necesarias para verificar la observancia de las Normas de Buena Práctica Clínica en los ensayos clínicos. Los datos relativos a las inspecciones serán remitidos al PECME y la Agencia Española de Medicamentos y Productos Sanitarios para su introducción en la base de datos europea de ensayos clínicos EUDRACT, de acuerdo con las directrices de la Comisión Europea. Artículo 17. Infracciones El incumplimiento de los dispuesto en la presente disposición será sancionado de conformidad con el régimen previsto en el capítulo II del título VIII de la Ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios. DISPOSICIÓN ADICIONAL Única. Indemnizaciones y gratificaciones por servicios extraordinarios Los profesionales que formen parte de los Comités referenciados en esta disposición participarán en ellos mientras el PECME se encuentre en desarrollo. Su actuación será exclusivamente de colaboración asesora, sin que origine creación de puesto de trabajo, si bien la Consellería de Sanidad abonará, en su caso, las indemnizaciones o gastos que su asistencia a la Comisión pueda ocasionar, de acuerdo con lo establecido en el Decreto 24/1997, de 11 de febrero, del Consell, sobre indemnizaciones por razón del servicio y gratificaciones por servicios extraordinarios, modificado por el Decreto 88/2008, de 20 de junio. DISPOSICIONES TRANSITORIAS Primera. Plazo de adaptación de los CEIC. Los CEIC acreditados en la Comunitat Valenciana deberán acomodar sus actuaciones a la presente disposición en el plazo de 90 días. Segunda. Comités de Ética de la Investigación En tanto se desarrolle la normativa regulada por la Ley 14/2007, de 3 de julio, de Investigación Biomédica, los CEIC de las fundaciones gestoras y promotoras de investigación biomédica que

68
habitualmente no evalúen protocolos de ensayos clínicos de medicamentos y productos sanitarios, sino estudios comprendidos dentro del campo de aplicación de la citada Ley, podrán cambiar su denominación a Comité de Ética de la Investigación sin cambiar su composición ni su ámbito geográfico de influencia. Esta acreditación tendrá validez hasta que se publiquen nuevas normas en este sentido por las autoridades competentes. DISPOCIÓN DEROGATORIA Única. Derogación normativa 1. Quedan derogadas la Orden de 6 de julio de 1994, de la Consellería de Sanidad y Consumo, por la que se regulan las competencias de la Comunitat Valenciana en materia de ensayos clínicos con medicamentos, y la Orden de 8 de marzo de 2004, de la Consellería de Sanidad, por la que se creó la Comisión Asesora de las Consellería de Sanidad para la Evaluación de los Estudios Post-autorización Observacionales Prospectivos. 2. Asimismo, quedan derogadas cuantas disposiciones de igual o inferior rango que se opongan a lo dispuesto en este decreto. DISPOSICIONES FINALES Primera. Habilitación normativa Se faculta el conseller de Sanidad para dictar las disposiciones precisas para el desarrollo y ejecución del presente decreto. Segunda. Entrada en vigor El presente decreto entrará en vigor al día siguiente de su publicación en el Diari Oficial de la Comunitat Valenciana. Afectado – por • art. 4 - ap. 2 corregido por la Corrección de errores del Decreto 73/2009, de 5 de junio, publicada en el Diari Oficial de la Comunitat Valencia num. 6068 el 30 de julio de 2009. • art. 11 - ap 7 corregido por la Corrección de errores del Decreto 73/2009, de 5 de junio, publicada en el Diari Oficial de la Comunitat Valencia num. 6068 el 30 de julio de 2009. • art. 12 – ap 1 y 3 corregido por la Corrección de errores del Decreto 73/2009, de 5 de junio, publicada en el Diari Oficial de la Comunitat Valencia num. 6068 el 30 de julio de 2009.

69
Preámbulo
La Orden de 6 de julio de 1994, de la Conselleria de Sanidad y Consumo, por la que se regulan las competencias en la Comunitat Valenciana en materia de ensayos clínicos con medicamentos, estableció, entre otros, los criterios en la acreditación y funcionamiento de los Comités Éticos de Investigación Clínica (en adelante CEIC) dictándose las Circulares 2/1998 y 1/1999 que regulaba los procedimientos de funcionamiento de los CEIC de la Comunitat Valenciana, así como contemplar la especial composición de los CEIC de Atención Primaria que estaban funcionando en la Comunitat Valenciana. El Decreto del Consell por el que se regula la gestión de ensayos clínicos y estudios post-autorización observacionales con medicamentos y productos sanitarios ha actualizado estas disposiciones normativas.
Por otra parte la Circular 15/2002 de la Agencia Española del Medicamento, de fecha 30 de septiembre, en el anexo VI que lleva por título directrices sobre estudios post-autorización de uso humano, se recogen una serie de requisitos comunes exigibles en la realización de este tipo de estudios, sin perjuicio de que las diferentes comunidades autónomas puedan establecer unas exigencias adicionales. Estas competencias son reguladas por las instrucciones de 27 de septiembre de 2004 de la Dirección General para la Prestación Farmacéutica, sobre la realización de estudios post-autorización en la Comunitat Valenciana.
Los estudios post-autorización con medicamentos se consideran necesarios para la obtención de un conocimiento que no aportan los ensayos clínicos realizados durante el desarrollo clínico de los medicamentos. La realización de estudios post-autorización de tipo observacional, permite la obtención de datos de seguridad y efectividad del medicamento, incrementando la información disponible. En definitiva, el estudio post-autorización de tipo observacional se contempla como una continuación necesaria del proceso de evaluación y análisis que determinó su autorización y registro, y constituye una herramienta importante para avanzar en el conocimiento científico. La realización de estos estudios deberá contar con el dictamen favorable de un Comité Ético de Investigación Clínica (en adelante CEIC) acreditado y de la autorización administrativa de la Dirección de Farmacia y Productos Sanitarios cuando se trate de estudios post-autorización de tipo observacional de carácter prospectivo.
La presente Resolución regula los procedimientos relacionados con los acreditación curricular y sustitución de los miembros del CEIC, acreditación y reacreditación de los CEIC, procedimientos en ensayos clínicos y procedimientos en estudios post-autorización observacionales.
Por todo ello y de acuerdo con las competencias y funciones asignadas a la Agencia Valenciana de Salud de la Conselleria de Sanidad por el Decreto 120/2007, de 27 de julio, del Consell, por el que se aprueba el Reglamento Orgánico y Funcional de la Conselleria de Sanidad, y por el Decreto 25/2005, de 4 de febrero, del Consell de la Generalitat, por el que se aprueban los Estatutos reguladores de la Agencia Valenciana de Salud,
Resolución de 16 de julio 2009, de la Consellería d e Sanidad, de regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana (2009/8823)

70
Dispongo la siguiente regulación:
Articulo 1. Procedimiento de cambio de vocales de los CEIC.
Cuando en un CEIC acreditado se decida el cambio de un vocal por otro que cubra su puesto, se tramitará la solicitud con el siguiente procedimiento:
1.1 Nombramiento del nuevo miembro: el director del centro solicitará el nombramiento al coordinador del Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios (PECME), y acompañará a dicha solicitud el currículum vitae actualizado (anexo I), con fotocopia de las titulaciones académicas y las publicaciones científicas pertinentes, y la declaración, por parte del miembro propuesto, de carecer de intereses derivados de la fabricación y venta de medicamentos y productos sanitarios. En dicha solicitud se indicará el miembro actual del CEIC al que se propone sustituir, en su caso, y que cesará como miembro del CEIC. La Comisión Delegada de PECME informará positiva o negativamente sobre dicha propuesta, para su resolución por el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud.
1.2 Si la resolución es favorable, se tramitará a continuación el cese propuesto por el director del centro, que debe resolver, como en el caso anterior, el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud.
1.3 En el caso de que la decisión sobre el nombramiento del nuevo miembro sea desfavorable, no se tramitará el cese del miembro antiguo si dicho cese supusiera la inhabilitación del CEIC por quedar en situación de incumplimiento de la normativa vigente, hasta poner este hecho en conocimiento del director del centro.
1.4 Se tramitarán sin dilación las propuestas de cese de un miembro de un CEIC solicitadas por el director del centro al que pertenece el CEIC, previo acuerdo de éste, siempre que no estén asociadas al nombramiento de un nuevo miembro. También se tramitarán aquellas propuestas de renuncia solicitadas a título personal por un miembro de un CEIC, tras ponerlo en conocimiento del presidente del CEIC y del director del centro.
1.5 El director del centro no podrá realizar nombramientos provisionales ni definitivos de miembros del CEIC.
1.6 Los nombramientos y ceses no condicionados se evaluarán en un plazo máximo de tres meses.
Articulo 2. Criterios de acreditación curricular de un miembro de CEIC.
2.1 Los criterios de acreditación curricular serán los siguientes:
2.1.1 Por razón de cargo (director del centro) que se tramitarán automáticamente, y cuyo nombramiento tendrá carácter provisional, hasta que se remita un informe favorable por parte de la Comisión Delegada del PECME.
2.1.2 Por razón de titulación o condición: en ellas se tendrá en cuenta fundamentalmente la posesión del correspondiente título o condición: diplomado en Enfermería, miembros ajenos, titulación en Derecho, farmacéutico de hospital, farmacéutico de atención primaria y farmacólogo clínico. No obstante la Comisión Delegada del PECME puede considerar, a la vista del currículum vitae del solicitante, la no idoneidad del candidato.
2.1.3 Por razón curricular: en el resto de casos y en los anteriores cuando así lo estime la Comisión Delegada del PECME, sólo se aceptarán como miembros a aquellos que presenten un currículum suficiente que garantice su capacitación para evaluar los protocolos de los ensayos clínicos en sus aspectos metodológico, ético y legal será considerado en principio

71
adecuado el currículum en alguna de las siguientes circunstancias: cuando alguna universidad haya emitido un informe positivo de suficiencia investigadora, cuando el solicitante tenga el grado de doctor, cuando haya publicado artículos de investigación en revistas de reconocido prestigio, o cuando haya participado como investigador principal en proyectos de investigación financiados por organismos públicos en convocatorias oficiales.
2.2 Cuando la Comisión Delegada del PECME no acepte la propuesta de algún miembro deberá comunicar por escrito las razones del rechazo.
Artículo 3. Procedimiento para la acreditación de los Comités Éticos de Investigación Clínica.
3.1 Los responsables de los centros, servicios y establecimientos sanitarios de la Comunitat Valenciana autorizados por la Conselleria de Sanidad, podrán solicitar la acreditación de un CEIC en el centro que dirigen, mediante instancia según el modelo (Anexo II) adjunto, enviada al coordinador del PECME.
3.2 La Comisión Delegada del PECME evaluará la propuesta de acreditación.
3.3 El informe final, favorable o desfavorable, sobre la propuesta de acreditación, será tramitado para su resolución por el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud.
3.4 Documentación y requisitos:
3.4.1 Por parte del centro:
a. Centro autorizado por la Conselleria de Sanidad.
b. Garantía explícita por parte del titular del centro de que el CEIC cuenta con medios para realizar su cometido. Para acreditar y garantizar el cumplimiento de sus funciones, cada CEIC de la Comunitat Valenciana contará, al menos, con los siguientes medios:
1. Espacio reservado para Secretaría del CEIC en el que exista suficiente mobiliario en función de su actividad, así como para garantizar la confidencialidad de los documentos.
2. Soporte informático con suficiente capacidad para almacenar y procesar toda la información generada por el CEIC.
3. Los recursos humanos suficientes para garantizar el mantenimiento y gestión de la base de datos del CEIC, la elaboración de las actas de reuniones, la comunicación entre éste y las autoridades sanitarias y el resto de funciones administrativas correspondientes al CEIC. Para ello contará como mínimo con una persona cuya dedicación, total o parcial, estará en función de la actividad que el CEIC desarrolle.
4. Disponer de los correspondientes Procedimientos Normalizados de Trabajo.
5. Para garantizar el cumplimiento de los apartados anteriores, se incorporarán los gastos del CEIC al presupuesto general del centro, que se realizarán con la justificación correspondiente o se llegará a los acuerdos pertinentes con la fundación para la investigación que dispongan dichos centros. Deberán cubrirse los gastos extraordinarios de funcionamiento del Comité, incluyendo los servicios externos que pudiera necesitar.
c. Centro con acreditación o solicitud de acreditación docente.
d. Compromiso de remitir la memoria anual de actividades al coordinador de PECME.
e. Compromiso de notificar las modificaciones de la composición o características del CEIC en un plazo de 15 días.
3.4.2 Composición del CEIC.
Estará formado como mínimo por once miembros, de suficiencia investigadora probada deberán y en todo caso figurar en él:
• director del centro Médicos:
• Un farmacólogo clínico

72
• Dos médicos especialistas con labor asistencial activa en un centro hospitalario
• Un médico con labor asistencial activa en un Centro de Atención Primaria
• Un pediatra
• Farmacéuticos:
• Un farmacéutico de Hospital
• Un farmacéutico de Atención Primaria
• Un diplomado en Enfermería.
• Dos miembros ajenos a las profesiones sanitarias, debiendo ser uno de ellos licenciado en Derecho.
Uno de estos miembros será una persona independiente de la organización asistencial, entendiendo por independiente la desvinculación laboral en relación a los centros en los que se lleven a cabo los proyectos de investigación que requieran la evaluación ética por parte del Comité y se recomienda que sea propuesto por el Consejo de Salud.
3.4.3 Los miembros:
a. No pueden delegar su participación en otros profesionales..
b. Serán nombrados por el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud a propuesta de la dirección del centro, después que la Comisión delegada del PECME haya emitido el informe preceptivo.
c. Serán elegidos entre profesionales que se presenten y/o acepten voluntariamente.
d. El presidente y secretario serán elegidos por el CEIC de entre sus miembros, por mayoría simple, habiendo un quórum de dos tercios del CEIC, y serán nombrados por la dirección del centro.
e. Los miembros del CEIC harán constar su compromiso de cumplir las funciones especificadas concretando el sistema de evaluación en sus procedimientos normalizados de trabajo.
3.4.4 Procedimientos normalizados de trabajo: ya elaborados, o bien aceptada su estructura y el envío al PECME antes de que transcurran seis meses desde su acreditación. En caso contrario será revocada esta acreditación. Estructura:
a. Respetará los criterios de las Normas de Buena Práctica Clínica de la Unión Europea vigentes.
b. Serán aprobados y firmados por todos y cada uno de los miembros del Comité.
c. Se harán públicos, enviando al menos una copia de los mismos a cada unidad funcional con capacidad investigadora del centro, o estarán expuestos en la página de la Web interna del Hospital, o en la intranet del centro.
d. Constará, al menos, de los apartados establecidos en el Anexo III.
Artículo 4. Procedimiento para la reacreditación de los Comités Éticos de Investigación Clínica.
4.1 Cuando hayan transcurrido tres años desde la fecha de acreditación o reacreditación de un CEIC, el responsable del centro deberá solicitar la reacreditación del mismo, según del modelo del Anexo IV.
Dicha acreditación es condición indispensable para que la actuación del CEIC continúe siendo válida.
4.2 La Comisión Delegada del PECME evaluará la propuesta de reacreditación, en base a los siguientes criterios:

73
4.2.1 Que la composición actual del CEIC cumpla las normas de la Conselleria de Sanidad para acreditación de los CEIC de la Comunitat Valenciana.
4.2.2 Que tenga publicados sus procedimientos normalizados de trabajo Se entenderá como cumplido dicho requisito cuando, al menos, estén a libre disposición en la Dirección Médica del centro.
4.2.3 Que realice el seguimiento de los ensayos desde su inicio hasta la recepción del informe final
4.2.4 Que haya superado favorablemente las inspecciones de la Conselleria de Sanidad.
4.2.5 Que haya realizado con carácter mensual al menos once reuniones anuales con evaluación de ensayos clínicos y seguimiento de los aprobados.
4.2.6 Que haya remitido al coordinador del PECME un informe sobre los protocolos de investigación aprobados y desestimados, al menos con periodicidad mínima anual, y una copia de los certificados de adecuación deontológica de protocolos que impliquen investigación en humanos).
4.3 El informe final, favorable o desfavorable, sobre la propuesta de reacreditación, será tramitado para su resolución por el director general de Farmacia y Productos Sanitarios.
Artículo 5. Procedimientos en el curso de un ensayo clínico. Estimación, adecuación deontológico, modificaciones y reacciones adversas.
Los comités éticos de investigación clínica, en el ámbito de la Comunitat Valenciana, vendrán obligados a:
a. Informar al coordinador del PECME de todos aquellos protocolos de investigación que aprueben y que se eleven al órgano competente para su autorización, y también de los que finalmente se desestimen, según el modelo del anexo V de esta Orden. En dichos informes los protocolos se identificarán de acuerdo con lo establecido por la Dirección General de Farmacia y Productos Sanitarios.
b. Remitir al coordinador del PECME una copia de los certificados que se emitan sobre la adecuación deontológica de los protocolos que impliquen la investigación en humanos.
c. Realizar un informe acerca de las modificaciones de los protocolos de los ensayos, según el modelo del anexo VI de esta norma, remitiendo al coordinador del PECME.
d. Comunicar al coordinador del PECME las reacciones adversas graves o inesperadas que acontezcan durante el desarrollo del ensayo, mediante copia de la notificación del investigador o promotor.
Artículo 6. Procedimientos en el curso de un ensayo clínico. Tutela.
6.1 Los CEIC evaluarán los protocolos cuya propuesta de realización implique a centros sanitarios pertenecientes al ámbito geográfico e institucional determinado por la Conselleria de Sanidad en el documento de acreditación del Comité, en las mismas condiciones en que evalúen los protocolos del propio centro.
6.2 Los investigadores principales pertenecientes a centros que no estén situados en el ámbito geográfico de alguno de los CEIC actualmente acreditados, podrán remitir a cualquiera de los CEIC acreditados una propuesta de evaluación de un protocolo de ensayo clínico. El equipo investigador y el promotor contactarán previamente con la Secretaría del Comité con el fin de iniciar los procedimientos establecidos en los procedimientos normalizados de trabajo del CEIC. Ningún CEIC podrá rechazar la tutela de un ensayo clínico de fuera de su ámbito geográfico sin una justificación razonada y pertinente.
6.3 La responsabilidad que asume el centro tutor es aquella que le encarga el Real Decreto 223/2004 de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos: establecer un sistema de comunicación que le permita conocer cuándo se ha producido un

74
efecto adverso grave y seguir el ensayo clínico desde su inicio hasta la recepción del informe final. De cualquier modo es conveniente que conste, en el comunicado de informe del CEIC sobre la evaluación del protocolo, que la responsabilidad del centro tutor se restringe a estos puntos, además de asumir las funciones reflejadas en el Real Decreto y que el resto de apartados obligatorios señalados en el citado real decreto debe asumirlos el centro que realice el ensayo.
Artículo 7. Procedimientos en el curso de un ensayo clínico. Suspensión.
7.1 Sólo procederá la suspensión de un ensayo clínico:
a. Por petición justificada del promotor.
b. Por decisión de la Dirección General de Farmacia y Productos Sanitarios.
c. Cautelarmente, por iniciativa de las administraciones sanitarias.
d. A propuesta del CEIC correspondiente.
7.2 Para que pueda estimarse favorablemente la solicitud de suspensión deberán concurrir cualquiera de los supuestos previstos en la Ley 29/2006, de 26 de julio, de garantías y uso racional de medicamentos y productos sanitarios
7.3 Si es el CEIC quien propone la retirada, debe hacerlo argumentando su solicitud y, basándose en ella, solicitar a la Conselleria de Sanidad, a través del coordinador del PECME la suspensión cautelar del ensayo.
7.4 Los CEIC no poseen la facultad de suspender un ensayo clínico en curso.
Artículo 8. Procedimientos para la realización de estudios post-autorización de tipo observacional en la Comunitat valenciana.
8.1 El objetivo de los estudios post-autorización de tipo observacional con medicamentos es generar información adicional sobre los efectos de los medicamentos y las características relacionadas con su utilización, en las condiciones habituales de la práctica clínica, en aquellas indicaciones para las que fueron autorizados, con el fin de completar la información obtenida durante las fases I, II y III y contribuir a su mejor utilización.
8.2. Los estudios post-autorización de tipo observacional con medicamentos pueden realizarse con alguno de los siguientes fines:
a. Determinar la efectividad de los fármacos en las condiciones de práctica habitual, así como los factores modificadores de la misma.
b. Identificar y cuantificar las reacciones adversas del medicamento, en especial las no conocidas antes de la autorización, e identificar los posibles factores de riesgo.
c. Obtener nueva información sobre los patrones de utilización de medicamentos (dosis, duración del tratamiento, cumplimiento, utilización apropiada) y sobre su eficiencia, valorando la relación entre los resultados sanitarios y los recursos utilizados, mediante el empleo de análisis farmacoeconómicos.
d. Conocer los efectos de los medicamentos desde la perspectiva de los pacientes, medidos en términos de calidad de vida y satisfacción.
8.3 En la tramitación y autorización de estudios post-autorización de tipo observacional:
a. Los estudios post-autorización de tipo observacional prospectivos deberán contar con una autorización de la Dirección General de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud.
b. Los estudios post-autorización de tipo observacional retrospectivos y transvesales deberán contar para su realización con la aprobación de un CEIC acreditado, a excepción de aquellos estudios que se realicen mediante la utilización de registros ya existentes que no contengan

75
datos de carácter personal. En cualquier caso los promotores de estos estudios deberán remitir, con carácter previo al inicio del estudio, la copia del informe favorable del CEIC correspondiente, la conformidad de la Gerencia del centro y la fecha prevista de inicio del mismo a la Dirección General de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud.
8.4. Procedimientos en estudios post-autorización observacionales prospectivos.
8.4.1 Solicitud. El promotor deberá solicitar, mediante escrito dirigido al director general de farmacia y productos sanitarios de la Agencia Valenciana de Salud, la autorización para realizar el estudio postautorización prospectivo de medicamentos en la Comunitat Valenciana previo pago de las tasas establecidas. La solicitud (Anexo VIII) deberá acompañarse, entre otros de:
a. Protocolo del estudio, que debe ajustarse a los puntos recogidos en el Anexo VII. Se presentarán en formato electrónico y en papel. Se deberá especificar en el mismo los procedimientos que se emplearán para garantizar que la realización del estudio no modificará los hábitos de prescripción del médico.
b. Dictamen favorable de un CEIC acreditado.
c. Relación de centros sanitarios de la Comunitat Valenciana e investigadores propuestos para realizar el proyecto.
d. Cuaderno de recogida de datos.
e. Ficha técnica del medicamento investigado.
f. Compromiso del investigador coordinador.
g. Compromisos de los investigadores de los centros sanitarios de la Comunitat Valenciana.
h. Hoja de información a los sujetos.
i. Formulario de consentimiento informado.
j. Propuesta de Contrato y Memoria económica.
k. En aquellos casos en que la solicitud sea presentada por una organización de investigación por contrato (CRO), deberá adjuntarse un documento emitido por el promotor del estudio en el que se haga constar la delegación de responsabilidades en la gestión administrativa del estudio, así como el alcance de esta delegación.
l. Ejemplar para la administración del impreso modelos 046 de tasas por servicios sanitarios, sellado por la entidad bancaria colaboradora, que justifique el abono de la tasa establecida.
8.4.2 Evaluación y autorización El Comité Autonómico de Estudios Post-autorización Observacionales de medicamentos y productos sanitarios de la Comunitat Valenciana (CAEPO), será el que evaluará la pertinencia de estos estudios y emitirá informe motivado al respecto al director general, teniendo en cuenta, entre otros, los siguientes aspectos:
a. Utilización de los medicamentos y productos sanitarios de acuerdo con la ficha técnica autorizada o a las condiciones habituales de uso cuando ésta no exista.
b. Justificación científica contrastable del estudio.
c. Inducción a la prescripción o dispensación.
d. Finalidad promocional del estudio.
e. Adecuación con las recomendaciones en materia de farmacoterapia recogidas en las guías, protocolos o líneas de trabajo que se adopten desde la administración sanitaria o en los centros sanitarios donde esté prevista su realización.
f. Interferencia con los cometidos asistenciales y/o utilización de procedimientos diagnósticos o de seguimiento no empleados en la práctica habitual.
8.4.3 Resolución del procedimiento y desarrollo del estudio La Dirección General de Farmacia y Productos Sanitarios dictará resolución y la notificará en el plazo máximo de noventa días

76
contados desde la fecha en que toda la documentación haya tenido entrada en el Registro de la Conselleria de Sanidad. En el caso en que la solicitud no reúna los requisitos establecidos se requerirá al solicitante para que subsane las deficiencias en el plazo máximo de diez días, con indicación de que, si así no lo hiciera, se archivará la solicitud. El plazo máximo establecido se suspenderá cuando se requiera al interesado para la subsanación de deficiencias, y la aportación de documentos o aclaraciones sobre su solicitud, por el tiempo que media entre la notificación del requerimiento y su efectivo cumplimiento:
a. Si la resolución de autorización es desfavorable, el promotor del estudio no podrá realizarlo en ningún centro sanitario de la Comunitat Valenciana.
b. Si la resolución es favorable, el promotor, antes de iniciar el estudio, deberá contar con el Visto Bueno de la Dirección del centro y formalizar por escrito el contrato económico. La Dirección General de Farmacia y Productos Sanitarios dará traslado de la Resolución: al promotor, a los CEIC de los centros sanitarios donde se quiera llevar a cabo el estudio y a las Gerencias de los Departamentos a que pertenezcan los centros sanitarios.
8.4.4 Seguimiento del estudio postautorización de tipo observacional prospectivo tras su aprobación. Una vez aprobado el estudio y previamente a su inicio, el promotor deberá notificar a los CEIC locales, como responsables del seguimiento, la autorización del mismo y remitir a la Dirección General de Farmacia y Productos Sanitarios la conformidad de las Gerencias implicadas y la fecha prevista de comienzo del estudio una vez formalizado el contrato con las Gerencias.
a. Las Gerencias respectivas contarán con un plazo de treinta días desde la autorización de la Dirección General de Farmacia y Productos Sanitarios para formalizar el contrato. Transcurrido dicho plazo sin que éste se haya formalizado, se entenderá que la conformidad de la Gerencia no ha sido ratificada y por tanto no se llevará a cabo el estudio en dicho centro.
b. El promotor del estudio deberá comunicar al CEIC la fecha de inicio del estudio, que será siempre posterior a la fecha de la firma del contrato con la Dirección del centro donde se realice, adjuntando la conformidad de la misma.
c. Asimismo, deberá comunicar cualquier enmienda relevante o cualquier incidencia relevante (interrupción del estudio, problema grave de seguridad, etcétera) al protocolo inicial. En el caso de que la Dirección General de Farmacia y Productos Sanitarios emita nuevo dictamen sobre un protocolo previamente autorizado con motivo de enmiendas a este protocolo, el promotor deberá informar de ello a las Gerencias y CEIC implicados.
d. La Dirección General de Farmacia y Productos Sanitarios podrá interrumpir y, en su caso, suspender definitivamente el desarrollo del estudio, si se detecta cualquier incumplimiento.
e. Una vez finalizado el estudio, y como máximo quince días después de su interrupción o finalización, el promotor deberá comunicarlo a la Dirección General de Farmacia y Productos Sanitarios y a los CEIC locales implicados, facilitándoles una copia del informe final debidamente aprobado en el plazo máximo de seis meses desde su interrupción o finalización.
8.5. Procedimientos en estudios post-autorización observacionales retrospectivos y transversales. Para la realización de este tipo de estudios en la Comunitat Valenciana se requiere:
• Que el promotor notifique a la Dirección General de Farmacia y Productos Sanitarios que va a realizar el estudio, especificando los centros sanitarios de la Comunitat Valenciana donde se quiere llevar a cabo y la conformidad de las direcciones de los centros.
• El promotor deberá remitir el informe final del estudio a la Dirección General de Farmacia y Productos Sanitarios.
Este tipo de estudios no precisa de la autorización previa por parte de la Dirección General de Farmacia y Productos Sanitarios para su realización.
8.6 De conformidad con lo previsto en la Ley 41/2002, de 14 de noviembre, reguladora de la autonomía de los pacientes, en los estudios post-autorización de tipo observacional prospectivos es imprescindible que el sujeto otorgue libre y voluntariamente su consentimiento

77
informado antes de ser incluido en el estudio. Si el sujeto del estudio no está en condiciones de escribir, podrá dar, en casos excepcionales, su consentimiento verbal en presencia de, al menos, un testigo mayor de edad y con capacidad de obrar. El sujeto participante en un ensayo clínico o estudio o su representante podrá revocar, en todo momento, su consentimiento sin expresión de causa.
8.7 En los estudios post-autorización de tipo observacional retrospectivos y transversales que requieran entrevistar al sujeto o en aquellos en los que, utilizando otras fuentes de información, no sea posible adoptar un procedimiento de disociación seguro, se solicitará el consentimiento informado de los sujetos. Podrá haber situaciones en las que será imposible o impracticable obtener el consentimiento para dicha investigación o podría ser una amenaza para su validez. En esta situación, la investigación sólo puede ser realizada después de ser considerada y aprobada por un CEIC.
La presente Resolución entrará en vigor el día siguiente de su publicación en el Diari Oficial de la Comunitat Valenciana.
Valencia, 16 de julio de 2009.
El Conseller de Sanidad, Manuel Cervera Taulet.

78
El Real Decreto 1344/2007, de 11 de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano, en su artículo 19 apartado 2 dispone que la Agencia Española de Medicamentos y Productos Sanitarios establecerá un Comité de Coordinación de Estudios Posautorización. Dicho Comité contará con la participación de los representantes de todas las comunidades autónomas y de la Agencia Española de Medicamentos y Productos Sanitarios. Así mismo, dicho artículo prevé que las administraciones sanitarias establecerán de común acuerdo las condiciones en las que se llevarán a cabo los estudios posautorización de tipo observacional, con la finalidad de favorecer aquellos que puedan contribuir al conocimiento del medicamento o a mejorar la práctica clínica, así como que las directrices de los procedimientos comunes que cada comunidad autónoma ejercerá en su ámbito competencial, serán objeto de debate en el citado comité.
En cumplimiento de lo anteriormente expuesto, el Comité de Coordinación de Estudios Posautorización, fue constituido con fecha 24 de septiembre de 2008, procediendo en ejercicio de sus funciones a adoptar unas directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano. Mediante esta orden se publican las «Directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano», que establecen los procedimientos comunes que cada comunidad autónoma ejecutará en su ámbito competencial, con el fin de facilitar su difusión y conocimiento. En su tramitación se ha obtenido el informe preceptivo de la Agencia Española de Protección de Datos, y han sido oídos los sectores afectados y consultadas las comunidades autónomas. En su virtud y previa aprobación de la Vicepresidenta Primera del Gobierno y Ministra de la Presidencia, dispongo: Artículo único. Publicación de las Directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano. Esta orden tiene por objeto publicar las Directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano, que figuran como anexo a la misma, y que han sido adoptadas por el Comité de Coordinación de Estudios Posautorización, en el ejercicio de sus funciones. Disposición final primera. Título competencial. Esta orden tiene carácter de legislación de productos farmacéuticos a los efectos previstos en el artículo 149.1.16ª de la Constitución. Disposición final segunda. Entrada en vigor. La presente orden entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado». Madrid, 16 de diciembre de 2009. – La Ministra de Sanidad y Política Social, Trinidad Jiménez García-Herrera.
ORDEN SAS/3470/2009, de 16 de diciembre, por la que se publican las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano.

79
ANEXO Directrices de estudios posautorización de tipo obs ervacional para medicamentos de uso humano 1. Antecedentes y base legal Los estudios posautorización se consideran necesarios para la obtención de un conocimiento que los ensayos clínicos controlados realizados durante el desarrollo clínico de los medicamentos no aportan. Dicho conocimiento es fundamental para orientar la práctica clínica y favorecer un uso racional de los medicamentos. El Real Decreto 711/2002, de 19 de julio, por el que se regulaba la farmacovigilancia de medicamentos de uso humano y la Circular 15/2002 de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) constituyeron un primer paso en el control de estos estudios y en la garantía de los fines de los mismos. En este real decreto se prohibía expresamente la utilización de estudios posautorización como este tipo de prácticas promocionales encubiertas y se establecía que las administraciones sanitarias, en el ámbito de sus competencias, debían regular las condiciones en las que se realizarían dichos estudios, al objeto de favorecer los que tuvieran verdadero interés científico e impedir aquellos con fines promocionales. Este mandato se desarrolló a través de normativas específicas por cada una de las comunidades autónomas (CC.AA.) con competencias en ejecución de la legislación sobre productos farmacéuticos, que tomaron como referencia el anexo VI de la citada Circular 15/2002. A la AEMPS le asignaba dicho real decreto la responsabilidad de mantener un registro común para todo el Estado de todas las propuestas de estudio que se solicitaran. Dicho registro (base de datos GESTO) está operativo desde 2003 y es accesible para todas las CC.AA. La citada regulación, y en particular, la autorización previa de los estudios posautorización de seguimiento prospectivo por parte de las CC.AA., ha permitido mejorar el control y la calidad de estos estudios. Superada esta etapa, por tanto, procede actualizar la normativa de referencia con el objeto de diferenciar con claridad los distintos tipos de estudios posautorización, e introducir mecanismos que permitan reducir la carga burocrática de aquellos que sean de gran interés para la salud pública, como son, entre otros, los requeridos por las autoridades sanitarias por motivos de seguridad o los financiados con fondos públicos. Las bases legales para esta diferenciación fueron introducidas por el nuevo el Real Decreto 1344/2007, de 11 de octubre, por el que se regula la farmacovigilancia de medicamentos de uso humano. Un elemento clave que ha introducido el Real Decreto 1344/2007 de 11 de octubre ha sido el mandato de crear un Comité de Coordinación de Estudios Posautorización, formado por representantes de todas las CC.AA. y de la AEMPS, como foro de debate en el que se acuerden las directrices de los procedimientos comunes que tendrán que aplicar las CC.AA. en su ámbito de competencia. Dicho Comité fue constituido el 24 de septiembre de 2008. Las presentes directrices son el fruto de las deliberaciones de dicho Comité y reflejan por tanto la participación de todas las CC.AA. y la AEMPS en el proceso de toma de decisiones y el común acuerdo sobre su redactado. Para la elaboración de estas directrices se ha tenido en cuenta, además, lo estipulado en el Capítulo I.7 sobre Estudios Posautorización promovidos por compañías farmacéuticas del Volumen 9A de las Normas sobre Medicamentos de la Unión Europea. 2. Ámbito de aplicación de las directrices Las directrices se aplican a todos los estudios posautorización de tipo observacional que se realicen con medicamentos de uso humano. No obstante, se establecen cuatro procedimientos diferentes en función del tipo de estudio posautorización observacional de que se trate. Cuando un estudio posautorización tenga carácter de ensayo clínico, de conformidad con la definición que figura en el artículo 58 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de medicamentos y productos sanitarios, no se regirá por lo dispuesto en las

80
presentes directrices sino que le resultará de aplicación la normativa específica sobre ensayos clínicos, en particular el Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. 3. Definiciones Datos de carácter personal : (Real Decreto 1720/2006, de 21 de diciembre, por el que se aprueba el Reglamento de desarrollo de la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal): Cualquier información numérica, alfabética, gráfica, fotográfica, acústica o de cualquier otro tipo concerniente a personas físicas identificadas o identificables. Procedimiento de disociación: (Real Decreto 1720/2006, de 21 de diciembre): Todo tratamiento de datos personales que permita la obtención de datos disociados. Estudio posautorización : (Real Decreto 1344/2007, de 11 de octubre): Cualquier estudio clínico o epidemiológico realizado durante la comercialización de un medicamento según las condiciones autorizadas en su ficha técnica, o bien en condiciones normales de uso, en el que el medicamento o los medicamentos de interés son el factor de exposición fundamental investigado. Este estudio podrá adoptar la forma de un ensayo clínico o un estudio observacional. Estudio posautorización de seguridad : (Real Decreto 1344/2007, de 11 de octubre): Estudio farmacoepidemiológico o ensayo clínico efectuado de conformidad con las disposiciones de la autorización de comercialización y realizado con el propósito de identificar, caracterizar o cuantificar los riesgos asociados a los medicamentos autorizados. Estudio observacional : (Ley 29/2006, de 26 de julio): Estudio en el que los medicamentos se prescriben de la manera habitual, de acuerdo con las condiciones establecidas en la autorización. La asignación de un paciente a una estrategia terapéutica concreta no estará decidida de antemano por el protocolo de un ensayo, sino que estará determinada por la práctica habitual de la medicina, y la decisión de prescribir un medicamento determinado estará claramente disociada de la decisión de incluir al paciente en el estudio. No se aplicará a los pacientes ninguna intervención, ya sea diagnóstica o de seguimiento, que no sea la habitual de la práctica clínica, y se utilizarán métodos epidemiológicos para el análisis de los datos recogidos. En el Capítulo I.7 del Volumen 9A, se aclara que «las entrevistas, los cuestionarios y las muestras de sangre se pueden considerar como práctica clínica habitual». En todo caso, para el manejo de muestras biológicas se seguirá lo dispuesto en la Ley 14/2007, de 3 de julio, de Investigación biomédica. Estudio posautorización de tipo observacional : Estudio epidemiológico que cumple las condiciones de ser posautorización y observacional. Estudio posautorización observacional de seguimient o: Todo aquel estudio posautorización de tipo observacional en el que los pacientes son seleccionados por su exposición a un determinado medicamento y son después seguidos durante un período de tiempo suficiente en relación con el acontecimiento de interés. Se consideran prospectivos cuando el periodo de estudio es posterior al inicio de la investigación y retrospectivos cuando el periodo de estudio es todo él anterior al inicio de la investigación. Fuente de información : Origen de los datos que se utilizan para la realización del estudio. 4. Objetivos de los estudios posautorización El objetivo de los estudios posautorización es generar información adicional sobre los efectos de los medicamentos, así como las características relacionadas con su utilización, en las condiciones autorizadas en su ficha técnica o bien en condiciones normales de uso, con el fin de completar la información obtenida durante las fases I, II y III y contribuir a su mejor utilización. Más concretamente, los estudios posautorización pueden realizarse con alguno de los siguientes fines: - Determinar la efectividad de los fármacos, es decir sus efectos beneficiosos en las condiciones de la práctica clínica habitual, así como los factores modificadores de la misma,

81
tales como el incumplimiento terapéutico, la polimedicación, la gravedad de la enfermedad, presencia de enfermedades concomitantes, grupos especiales (ancianos, niños, etc.), factores genéticos o factores relacionados con el estilo de vida. - Identificar y cuantificar los efectos adversos del medicamento, en especial los no conocidos antes de la autorización, e identificar los posibles factores de riesgo o modificadores de efecto (características demográficas, medicación concomitante, factores genéticos, etc.). Con frecuencia, esto solo podrá estudiarse con precisión en grupos amplios de población y durante tiempos de observación prolongados. - Obtener nueva información sobre los patrones de utilización de medicamentos (dosis, duración del tratamiento, utilización apropiada). - Evaluar la eficiencia de los medicamentos, es decir la relación entre los resultados sanitarios y los recursos utilizados, utilizando para ello análisis farmacoeconómicos, tales como los de coste-efectividad, coste-utilidad, coste-beneficio o comparación de costes. - Conocer los efectos de los medicamentos desde la perspectiva de los pacientes (calidad de vida, satisfacción con los tratamientos recibidos, etc.). 5. Consideraciones éticas Para que un estudio sea éticamente justificable debe estar bien diseñado y cumplir con los principios éticos básicos contenidos en la Declaración de Helsinki de la Asociación Médica Mundial sobre principios éticos para las investigaciones médicas en seres humanos, y en sus posteriores revisiones. Todos los estudios posautorización de tipo observacional deben ser sometidos a la consideración de un Comité Ético de Investigación Clínica (CEIC) (o Comité de Ética de la Investigación, en su caso) acreditado, con la excepción de aquellos estudios que se realicen mediante la utilización de información ya existente que no contengan datos de carácter personal. En los estudios que requieran entrevistar al sujeto o en aquellos en los que, utilizando otras fuentes de información, no sea posible adoptar un procedimiento de disociación seguro que garantice que la información que se maneja no contenga datos de carácter personal, se solicitará el consentimiento informado de los sujetos, el cual deberá ser otorgado por escrito, de acuerdo con la normativa vigente. Cuando los sujetos sean menores o incapaces se solicitará el consentimiento informado de los tutores legales. El promotor y los investigadores del estudio deben garantizar la confidencialidad de los datos de los sujetos y velar porque se cumpla en todo momento con lo establecido por la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal y Real Decreto 1720/2007, de 21 de diciembre. Los estudios posautorización de tipo observacional están exentos de la obligatoriedad de suscripción de un seguro. Cuando se trate de un estudio posautorización observacional de seguimiento prospectivo, el promotor y el investigador principal deberán expresar específicamente en el protocolo los procedimientos que se emplearán para garantizar que la realización del estudio no modificará los hábitos de prescripción del médico, o de dispensación del farmacéutico (en caso de medicamentos que no requieran prescripción). La prescripción del medicamento habrá de seguir los cauces habituales. Si se considera necesario un suministro del medicamento diferente al habitual, deberá justificarse apropiadamente en el protocolo. Los investigadores deberán asegurarse de que su participación en el estudio no interfiere con sus cometidos asistenciales.

82
Los investigadores podrán recibir una compensación proporcional al tiempo y responsabilidades adicionales dedicadas al estudio, sin perjuicio de las normas internas de las entidades empleadoras de los investigadores relativas a esta cuestión. La percepción económica habrá de ser, en todo caso, explícita y transparente y deberá ser puesto en conocimiento del CEIC que evalúe el estudio. La participación en el estudio de los investigadores habrá de ser libre, voluntaria e independiente.
6. Identificación de los responsables y elementos que debe contener el protocolo del estudio 6.1 Identificación de los responsables del estudio. En todos los estudios deben identificarse las figuras del promotor y el investigador principal o coordinador como responsables últimos de la investigación. Es promotor de un estudio posautorización toda aquella persona física o jurídica que tiene interés en su realización y asume las siguientes obligaciones: a) Firmar con el investigador coordinador el protocolo y cualquier modificación del mismo. b) Suministrar a los investigadores el protocolo y la ficha técnica de los medicamentos a estudiar. c) Remitir el protocolo al CEIC (o Comité de Ética de la Investigación, en su caso). d) Solicitar la autorización de la Administración, cuando proceda, y presentar la documentación correspondiente. e) Presentar los informes de seguimiento y final, en los plazos establecidos y comunicar, en su caso, la interrupción y las razones de la misma. f) Entregar copia del protocolo y de los documentos que acrediten el seguimiento de los procedimientos establecidos en las presentes directrices a los responsables de las entidades proveedoras de servicios de atención a la salud donde se vaya a realizar el estudio. g) Aplicar un control de calidad en la obtención y el manejo de datos para asegurar que los datos son fiables. h) Comunicar las sospechas de reacciones adversas graves que surjan a lo largo del estudio al punto de contacto designado por el órgano competente en materia de farmacovigilancia de la comunidad autónoma donde ejerza su actividad el profesional sanitario que comunicó el caso. i) Identificar las fuentes de financiación del estudio j) Firmar, en su caso, el contrato con la entidad competente k) Hacer públicos los resultados del estudio, a ser posible, a través de una revista científica. El promotor podrá designar a un monitor como vínculo entre él y los investigadores. Su principal obligación es asegurarse de que el estudio se está realizando conforme a lo exigido en el protocolo. Para ello, podrá realizar cuantas comprobaciones considere necesarias. Los profesionales sanitarios que contribuyan al estudio recogiendo información serán considerados como investigadores. Son obligaciones de éstos: a) Firmar un compromiso en el que se reconocen como investigadores del estudio y afirman que conocen el protocolo y cualquier modificación del mismo, y están de acuerdo con él en todos sus términos. b) Informar a los sujetos de investigación y obtener su consentimiento. c) Recoger, registrar y notificar los datos de forma correcta respondiendo de su actualización y calidad ante las auditorías oportunas. d) Notificar al promotor los acontecimientos adversos según se establezca en el protocolo. e) Respetar la confidencialidad de los datos del sujeto. f) Facilitar las visitas de monitorización del monitor, las auditorías del promotor y las inspecciones de las autoridades sanitarias. g) Saber responder sobre los objetivos, metodología básica y significado de los resultados del estudio ante la comunidad científica y profesional.

83
Es investigador coordinador el profesional sanitario que dirige científicamente el estudio. Son obligaciones específicas del investigador coordinador las enumeradas anteriormente más las siguientes: a) Firmar el protocolo y cualquier modificación del mismo junto con el promotor. b) Co-responsabilizarse con el promotor de la elaboración de los informes de seguimiento y finales. c) Contribuir a difundir los resultados del estudio, en colaboración con el promotor. 6.2 Elementos del Protocolo. Siempre que sea posible, los protocolos deberán ajustarse, de forma general, a la siguiente estructura (basada en Capítulo I.7. Volumen 9A, Tabla I.7.B): A. Título descriptivo y versión del protocolo. B. Responsable del estudio (nombres, títulos, grados, especialidad, lugar de trabajo y direcciones de todos los responsables, incluyendo investigador coordinador, otros investigadores y monitor cuando proceda). C. Promotor (nombre y dirección. En su caso, titular de la autorización de comercialización). D. Resumen:
1. Identificación del promotor y dirección. 2. Título del estudio. 3. Código del protocolo (según normas oficiales de codificación). 4. Investigador principal y dirección. 5. Tipo de centros donde se prevé realizar el estudio. 6. CEIC que lo evalúa. 7. Objetivo principal. 8. Diseño. 9. Enfermedad o trastorno en estudio. 10. Datos de los medicamentos objeto de estudio. 11. Población en estudio y número total de sujetos. 12. Calendario. 13. Fuente de financiación.
E. Plan de trabajo (tareas, hitos y cronología del estudio). F. Objetivos generales y específicos. Fundamentos. G. Revisión crítica de la literatura. H. Métodos:
1. Diseño y justificación. 2. Población de estudio. 3. Fuente de información. 4. Definición operativa de variables de resultado, exposición y otras. 5. Tamaño de la muestra previsto y bases para su determinación. 6. Métodos para la obtención de los datos. 7. Manejo de los datos. 8. Análisis de los datos. 9. Control de calidad. 10. Limitaciones del diseño, de la fuente de información y de los métodos de análisis.
I. Aspectos éticos/protección de los sujetos participantes: 1. Evaluación beneficio-riesgo para los sujetos de investigación. 2. Consideraciones sobre información a los sujetos y consentimiento informado.. 3. Confidencialidad de los datos. 4. Interferencia con los hábitos de prescripción del médico.
J. Manejo y comunicación de reacciones adversas. K. Planes para la difusión de los resultados. L. Recursos para la realización del estudio y asignación de tareas. Forma de suministro del medicamento. Financiación. M. Bibliografía. N. Modificaciones del protocolo. Ñ. Consideraciones prácticas:

84
1. Informes de seguimiento y final. 2. Difusión de los resultados O. Anexos (al menos los siguientes):
1. Anexo 1: Cuaderno de recogida de datos. 2. Anexo 2: Compromiso del investigador coordinador. 3. Anexo 3: Conformidad del CEIC. 4. Anexo 4: Ficha Técnica del medicamento investigado. 5. Anexo 5: Hoja de información a los sujetos. 6. Anexo 6: Formulario de consentimiento informado. 7. Anexo 7: Memoria económica.
6.3 Identificación de estudio. Sin perjuicio del código interno del protocolo de cada promotor, todos los protocolos de estudios posautorización deben identificarse con un código de 12 dígitos siguiendo las siguientes normas de codificación: Posiciones 1-3: tres primeras letras del promotor Posiciones 4-6: tres primeras letras del principio activo de interés (uno de ellos si hay más de uno) Posiciones 7-10: año en curso Posiciones 11-12: Número secuencial de 2 dígitos Ejemplo: El promotor PANDORA desea hacer un estudio con el principio activo ACTIVINA, en el año 2002 siendo el cuarto que hace este año con dicho principio activo. Código: PAN-ACT-2002-04 Solo se consignará un código por protocolo. Por ejemplo, si un mismo estudio se va a realizar en 4 CC.AA., en todas ellas se identificará por el mismo código. 7. Procedimientos administrativos para los estudios posautorización de tipo observacional En el Real Decreto 1344/2007, de 11 de octubre, en su artículo 19.3 establece que «la Agencia mantendrá un registro de las propuestas de estudio posautorización de tipo observacional, al que tendrán acceso los órganos competentes de las comunidades autónomas, e informará a cada promotor sobre los procedimientos a seguir en cada caso. A tal efecto, el promotor del estudio deberá remitir a la Agencia Española de Medicamentos y Productos Sanitarios el protocolo del estudio». De acuerdo con esto, la AEMPS realizará una clasificación previa de todos los estudios clínicos o epidemiológicos no aleatorizados, que se realicen con seres humanos o con registros médicos y que tengan uno o varios medicamentos como exposición de interés en el plazo máximo de 30 días naturales desde su recepción. Para ello el promotor deberá remitir a la AEMPS el protocolo completo o un resumen amplio del mismo, donde queden perfectamente explícitos los objetivos y el diseño del estudio, indicando que el destinatario es la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS. Es relevante que en esta comunicación se indique claramente que se remite para su CLASIFICACIÓN, así como la dirección postal y electrónica y el teléfono de contacto del promotor. Se podrá presentar el protocolo en paralelo a la AEMPS para clasificación y al CEIC para evaluación. Una vez recibido el protocolo o su resumen amplio, la AEMPS remitirá al solicitante la resolución correspondiente indicando la clasificación del estudio y la ruta administrativa que el mismo deberá seguir (ver cuadro I). Para los casos de dudosa clasificación la AEMPS solicitará formalmente un informe al Comité de Coordinación de Estudios Posautorización. En dicho caso se producirá una suspensión del plazo por el tiempo que medie entre la petición de dicho informe, que será comunicada al interesado, y la recepción del mismo. Este plazo de suspensión no podrá exceder en ningún caso de tres meses. Toda aquella documentación que se remita a la AEMPS para la clasificación de un estudio, no tendrá que ser reenviada en el momento de la solicitud de autorización y/o registro del mismo.

85
La AEMPS mantendrá una base de datos con la información relativa a todos los estudios posautorización que se remitan para su registro, a la que tendrán acceso para consulta y, en su caso, carga de la información correspondiente, los órganos competentes de las CC.AA. En dicha base de datos constarán los aspectos administrativos y metodológicos fundamentales de los estudios. Todos aquellos estudios que se consideren ensayos clínicos deberán seguir la normativa específica. En el Cuadro I se resumen de forma esquemática los procedimientos administrativos que se detallan a continuación para cada tipo de estudio. 7.1 Procedimiento para la autorización de estudios posautorización de tipo observacional que sean una condición establecida en el momento de la autorización de un medicamento, o bien constituya una exigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad del medicamento, o forme parte del plan de gestión de riesgos. Este tipo de estudios se abreviarán como EPA-LA. La normativa aplicable es el Real Decreto 1344/2007, de 11 de octubre, donde se recoge en el artículo 19.5 lo siguiente: «Cuando la realización de un estudio posautorización de tipo observacional sea una condición establecida en el momento de la autorización de un medicamento, o bien constituya una exigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad del medicamento, o forme parte del plan de gestión de riesgos que debe llevar a cabo el titular requerirá únicamente la autorización de la Agencia Española de Medicamentos y Productos Sanitarios, según los procedimientos que se establezcan al efecto. La Agencia Española de Medicamentos y Productos Sanitarios informará de estos estudios a las comunidades autónomas donde se vayan a realizar y los incluirá en el registro referido en el apartado 3». Para todos los estudios clasificados como EPA-LA, el promotor deberá presentar la documentación siguiente, indicando como destinatario la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS: - Carta de presentación dirigida a la División de Farmacoepidemiología y Farmacovigilancia de la AEMPS donde se solicite la autorización del estudio e indique la dirección y contacto del solicitante y la relación de documentos que se incluyen. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre. - Formulario resumen del protocolo (disponible en: www.aemps.es). - Protocolo completo, incluidos los anexos y cuaderno de recogida de datos, y donde conste el número de pacientes que se pretenden incluir en España. - Dictamen favorable del estudio por un CEIC acreditado en España. - Acreditación documental de que el estudio ha sido requerido por la autoridad competente, o que forme parte del plan de gestión de riesgos del producto. - Acreditación documental de la revisión por parte de la autoridad sanitaria competente que instó la realización del estudio. - Listado de CC.AA y centros sanitarios en los que se pretende realizar el estudio, así como los investigadores participantes. - Si el estudio se pretende realizar en otros países, situación del mismo en éstos.
La AEMPS evaluará la pertinencia del estudio y resolverá favorable o desfavorablemente el mismo en el plazo máximo de 60 días naturales desde su recepción. Si pasado este plazo, la AEMPS no ha emitido informe desfavorable se dará por aprobado el estudio

86
En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento. Una vez aprobado el estudio por la AEMPS, deberá contratarse la realización con los centros sanitarios donde se vaya a realizar, siguiendo los requisitos que cada comunidad autónoma establezca, lo que incluirá necesariamente la resolución de la AEMPS por la que se autoriza el estudio. Para los estudios que formen parte del plan de gestión de riesgos, es necesario que se solicite su autorización de manera independiente a la presentación en la AEMPS del plan de gestión de riesgos. La AEMPS mantendrá informados a los órganos competentes de las CCAA. sobre la autorización de estos estudios, a través de la aplicación informática de registro de estudios posautorización u otro procedimiento alternativo que se acuerde. No se podrán realizar estudios EPA-LA sin la preceptiva autorización de la AEMPS. El incumplimiento de lo anteriormente expuesto dará lugar a la incoación del correspondiente expediente sancionador. 7.2 Procedimiento simplificado para la autorización de estudios posautorización de tipo observacional de seguimiento prospectivo promovidos por las Administraciones sanitarias o financiados con fondos públicos. Este tipo de estudios se abreviarán como EPA-AS De acuerdo con el artículo 19.6 del Real Decreto 1344/2007, de 11 de octubre, «cuando se trate de estudios promovidos por las Administraciones sanitarias o financiados con fondos públicos, se establecerán procedimientos simplificados al objeto de facilitar su realización y que se acordarán en el Comité de Coordinación de Estudios Posautorización». El promotor de un estudio posautorización de seguimiento prospectivo que responda a los criterios mencionados, enviará el protocolo del estudio, junto con el formulario resumen del mismo (disponible en www.aemps.es) y el dictamen favorable del CEIC, a la Secretaría del Comité de Coordinación de Estudios Posautorización (División de Farmacoepidemiología y Farmacovigilancia de la AEMPS). La Secretaría del Comité distribuirá el protocolo del estudio a sus miembros, junto con un informe en el que consten las razones de su inclusión en la categoría de estudios promovidos por las Administraciones sanitarias o financiados con fondos públicos. El Comité emitirá informe sobre la solicitud de autorización del estudio, correspondiendo a la AEMPS, de acuerdo con el pronunciamiento previo del Comité, emitir la resolución correspondiente. Dicha resolución se emitirá en un plazo máximo de 30 días naturales a contar desde la solicitud de autorización. Si pasado este plazo, la AEMPS no ha emitido informe favorable se dará por autorizado el estudio. En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento.

87
El promotor informará a los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio, o a los organismos que la comunidad autónoma haya designado para este fin, y les entregará copia del protocolo y de los documentos que acrediten su aprobación. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma. 7.3 Procedimiento para la autorización de estudios posautorización de tipo observacional de seguimiento prospectivo con medicamentos, no incluidos en los epígrafes anteriores (7.1 y 7.2). Este tipo de estudios se abreviarán como EPA-SP. Una vez clasificado el estudio como EPA-SP por la AEMPS, el promotor presentará el protocolo del estudio, junto con el material informativo que se prevea enviar a los profesionales sanitarios, a los órganos competentes de las CC.AA. donde se vaya a realizar el estudio y simultáneamente a la AEMPS, una vez obtenida la conformidad de un CEIC acreditado, junto con el formulario resumen del estudio (disponible en www.aemps.es). El promotor hará constar en el protocolo si el estudio se ha presentado con anterioridad a algún otro CEIC e informará del resultado de su evaluación. El promotor deberá presentar en las CC.AA. donde se pretenda realizar el estudio la siguiente documentación: - Carta de presentación dirigida a los responsables de esta materia en la comunidad autónoma en la que se solicite la autorización del estudio e indique la dirección y contacto del solicitante y la relación de documentos que se incluyen. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre. - Formulario resumen del protocolo (disponible en: www.aemps.es). - Resolución de la AEMPS sobre la clasificación del estudio - Protocolo completo incluidos los anexos, y donde conste el número de pacientes que se pretenden incluir en España, desglosado por comunidad autónoma. - Dictamen favorable del estudio por un CEIC acreditado en España. - Listado de centros sanitarios donde se pretende realizar el estudio, desglosado por comunidad autónoma - Listado de investigadores participantes en la comunidad autónoma. - Si el estudio se pretende realizar en otros países, situación del mismo en éstos - Documento acreditativo de haber satisfecho las tasas correspondientes, en aquellas CC.AA. donde se exijan. Los órganos competentes de las CC.AA. evaluarán la pertinencia del estudio y resolverán favorable o desfavorablemente el mismo en el plazo máximo de 90 días naturales desde su recepción. Si pasado este plazo la comunidad autónoma correspondiente no ha emitido informe desfavorable se dará por aprobado el estudio en el ámbito de dicha comunidad autónoma. En el caso de que fuera precisa la aportación de cualquier documentación o información adicional a efectos de la autorización del estudio, y una vez requerida la misma, se podrá suspender el plazo para la resolución del procedimiento de acuerdo con lo establecido en el artículo 42.5 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, siendo en este caso notificada dicha suspensión al interesado en la comunicación del requerimiento. La comunidad autónoma informará de la resolución a la AEMPS (normalmente a través de la aplicación informática), y al promotor. El promotor informará a los responsables de las entidades proveedoras de servicios sanitarios donde prevea llevarse a cabo el estudio, o a los organismos que la comunidad autónoma haya designado para este fin, y les entregará copia

88
del protocolo y de los documentos que acrediten su aprobación. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma. Para la evaluación de los protocolos de los estudios y la revisión de los informes de seguimiento y finales, las CC.AA. podrán solicitar la colaboración de expertos, entre los que se encontrarán los técnicos correspondientes del Sistema Español de Farmacovigilancia de medicamentos de uso humano. No se podrán realizar estudios EPA-SP sin la preceptiva autorización de los órganos competentes de las comunidades autónomas involucradas. El incumplimiento de lo anteriormente expuesto dará lugar a la incoación del correspondiente expediente sancionador. 7.4 Procedimiento a seguir para otros estudios posautorización de tipo observacional. Este tipo de estudios se abreviarán como EPA-OD. En este grupo se encuentran los estudios posautorización que no hayan sido categorizados como EPA-LA y respondan a diseños diferentes al de «seguimiento prospectivo», por ejemplo, estudios de casos y controles, estudios transversales o estudios de cohorte retrospectivos. Una vez clasificado el estudio como EPA-OD por la AEMPS, el promotor deberá remitir a la AEMPS el protocolo junto con la aprobación del mismo por un CEIC acreditado en España y el formulario resumen del protocolo (disponible en www.aemps.es). Si para la clasificación del estudio se presenta esta documentación, no requerirá ningún trámite adicional. La AEMPS procederá a registrar oportunamente el estudio e informará a los órganos competentes de las CC.AA. En el caso de que el promotor no sea quien presente la documentación, se deberá incluir en la misma un documento que indique las responsabilidades delegadas por el promotor a la persona o empresa que actúa en su nombre. Los estudios EPA-OD no requieren autorización por parte de la AEMPS ni de los órganos competentes de las CC.AA. donde se vayan a realizar, si bien el promotor tendrá que informar a los responsables de las entidades proveedoras de servicios sanitarios donde se lleve a cabo el estudio y les entregará copia del protocolo y de los documentos que acrediten la aprobación por parte del CEIC y la clasificación de la AEMPS. Asimismo estos documentos se entregarán a los órganos competentes de las CC.AA., cuando sea requerido. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma. 7.5 Procedimiento para la realización de estudios observacionales que no sean posautorización. Estos estudios se abreviarán como No-EPA. Se trata de aquellos estudios en los que el factor de exposición fundamental investigado no son medicamentos. En caso de que se recoja información sobre medicamentos, el protocolo tendrá que ser presentado a la AEMPS para su clasificación. La AEMPS procederá a registrar oportunamente el estudio e informará a los órganos competentes de las CC.AA. Los estudios No-EPA no requieren autorización por parte de la AEMPS ni de los órganos competentes de las CC.AA. donde se vayan a realizar, pero sí es necesario presentarlo a un CEIC acreditado y obtener su dictamen favorable. El promotor tendrá que informar a los responsables de las entidades proveedoras de servicios sanitarios donde se lleve a cabo el estudio y les entregará copia del protocolo y de los documentos que acrediten la aprobación por parte del CEIC y, en su caso, la clasificación de la AEMPS. Asimismo estos documentos se entregarán a los órganos competentes de las CC.AA., cuando sea requerido. La gestión y formalización del contrato estará sujeta a los requisitos específicos de cada comunidad autónoma. 8. Seguimiento de los estudios posautorización Todos los estudios posautorización requerirán para su inicio el oportuno contrato del promotor con los responsables de las entidades proveedoras de servicios sanitarios donde prevea

89
llevarse a cabo el estudio o el visto bueno del director del centro conforme a los procedimientos específicos que se manejen en cada centro. El promotor comunicará la fecha efectiva de comienzo del estudio a los órganos competentes de las CC.AA. donde se prevea realizar y a la AEMPS, a menos que ya venga indicado en el propio protocolo. 8.1 Enmiendas al protocolo Cuando la enmienda afecte a aspectos fundamentales del protocolo del estudio (en particular a los apartados de objetivos, métodos, aspectos éticos – ver 6.2), se someterá de nuevo a la evaluación del CEIC que informó favorablemente sobre el mismo y se solicitará autorización administrativa para dicha enmienda a quien corresponda en función del tipo de estudio. Para el resto de enmiendas bastará con que se informe de las modificaciones efectuadas, justificando por qué dicha enmienda no afecta a aspectos fundamentales del protocolo del estudio. En caso de duda sobre la consideración de la enmienda, se puede remitir consulta a la AEMPS a través de la dirección de correo electrónico [email protected]. 8.2 Informes de seguimiento y finales. Para los estudios EPA-LA, EPA-SP y EPA-AS que sean considerados como de seguimiento prospectivo el promotor deberá enviar un informe de seguimiento anual, o antes si así se solicita. Asimismo, el promotor informará de forma inmediata sobre cualquier incidencia relevante (interrupción, problema grave de seguridad, etc.) que pueda producirse en el transcurso del estudio. Todas estas comunicaciones se presentarán a los órganos competentes de las CC.AA. involucradas y a la AEMPS. Para todos los estudios posautorización, el promotor deberá remitir un informe final entre los tres y seis meses después de la finalización del estudio, a la AEMPS y a los órganos competentes de las CC.AA. donde se realizó. 8.3 Comunicación de sospechas de reacciones adversas Para los estudios catalogados como EPA-SP, EPA-AS y para los EPA-LA que sean considerados como de seguimiento prospectivo, las sospechas de reacciones adversas graves que se detecten durante el transcurso del estudio se notificarán al punto de contacto designado por el órgano competente en materia de farmacovigilancia de la comunidad autónoma donde ejerza su actividad el profesional sanitario que notifique el caso, en el plazo máximo de 15 días naturales desde que se tuvo conocimiento de la sospecha de reacción adversa. Esta notificación se realizará obligatoriamente de forma electrónica si el promotor es una compañía farmacéutica, por parte del responsable de farmacovigilancia siguiendo las instrucciones publicadas la AEMPS. La AEMPS pondrá a disposición del titular de la autorización de comercialización dentro de los 15 días naturales siguientes a su recepción, las notificaciones sobre sospechas de reacciones adversas graves que se detecten en el transcurso de un EPA, haciendo referencia al código del estudio. En el caso de que el promotor sea un grupo de profesionales (vgr. sociedades científicas) podrá optar por una de la dos siguientes opciones: 1) tarjeta amarilla al centro autonómico de farmacovigilancia correspondiente, indicando en observaciones el nombre y el código del estudio del cual proviene; o 2) utilizar la carga on-line a través del portal SINAEM de la AEMPS, siguiendo las instrucciones publicadas por la AEMPS (esta segunda opción es la recomendada). Si el promotor es un profesional sanitario notificará las sospechas de reacciones adversas a través de tarjeta amarilla al centro autonómico de farmacovigilancia correspondiente, indicando igualmente el nombre y el código del estudio del cual proviene. De acuerdo con el Capítulo I.7 del Volumen 9A, aquellos estudios en los que no sea posible o no sea apropiado hacer una evaluación individual de la relación de causalidad entre los

90
acontecimientos clínicos y los medicamentos de interés, la notificación individual de sospechas de reacciones adversas no será necesaria. Los estudios etiquetados como EPA-OD, o EPA-LA que no sean de seguimiento prospectivo, entran dentro de esta categoría y para ellos no se precisará la notificación expeditiva de sospechas de reacciones adversas, a menos que se indique otra cosa por la AEMPS en el momento del registro del estudio. Además de lo especificado en el párrafo anterior, cualquier problema de seguridad relevante que se detecte durante el transcurso del estudio será puesto en conocimiento de la AEMPS y de los órganos competentes de las CC.AA. involucradas, con independencia del diseño y catalogación del estudio. 8.4 Archivo de los documentos del estudio: La documentación relativa al estudio posautorización constituye el archivo maestro del mismo y constará de los documentos esenciales que permitan la evaluación de la realización de un estudio posautorización y de la calidad de los datos obtenidos. Estos documentos deberán demostrar el cumplimiento por parte del investigador y del promotor de los requisitos establecidos para los estudios posautorización. El archivo maestro del estudio posautorización proporcionará la base para las auditorías que pueda realizar el promotor a través de auditores independientes y para las inspecciones de las autoridades competentes. El promotor y el investigador conservarán los documentos y materiales esenciales de cada estudio durante al menos cinco años tras la finalización del mismo, o durante un período más largo si así lo disponen otros requisitos aplicables. Los documentos y materiales esenciales deberán archivarse de forma que se puedan poner fácilmente a disposición de las autoridades competentes, en caso de que los soliciten. La historia clínica de cada paciente del estudio deberá ser custodiada con arreglo a los dispuesto en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica, y la legislación autonómica aplicable, y conforme al período máximo establecido por el hospital, la institución o la consulta privada. Asimismo, se deberá contemplar lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de protección de datos de carácter personal y el Real Decreto 1720/2007, de 21 de diciembre, por el que se aprueba el reglamento de desarrollo de dicha ley Todos los cambios en la titularidad de los datos, documentos y materiales deberán documentarse. El nuevo titular asumirá las responsabilidades de las tareas de archivo y conservación de los datos. El promotor nombrará la persona de su organización responsable de los archivos y el acceso a los mismos deberá limitarse a las personas designadas. Los soportes utilizados para la conservación de los documentos esenciales deberán garantizar que los documentos y materiales permanezcan completos y legibles durante el período de conservación previsto y que estén a disposición de las autoridades competentes en caso de que los soliciten. Cualquier modificación de los registros habrá de ser trazable, permitiendo conocer el dato inicial y el corregido, así como la fecha y firma del autor del cambio. 8.5 Inspecciones: Las autoridades sanitarias competentes de las CC.AA., en el ámbito de sus competencias, verificarán el cumplimiento de las prescripciones legales relativas a los estudios posautorización que se realicen en España, a través de las correspondientes inspecciones y de acuerdo con los procedimientos que se establezcan.

91
Las inspecciones serán llevadas a cabo antes, durante o después de la realización del estudio por inspectores debidamente cualificados. Se podrán hacer inspecciones en los lugares relacionados con la realización del estudio y, entre otros, en el centro o centros sanitarios en los que se lleve a cabo el estudio, en cualquier laboratorio de análisis o centro de diagnóstico utilizado, en las instalaciones del promotor y/o de las organizaciones o empresas de investigación implicadas por contrato en la realización del estudio y al CEIC que lo evaluó.
Ver imagen en documento original
Abreviaturas y leyendas en el Cuadro I: * En alguna comunidad autónoma este paso es previo a la autorización de la Administración Sanitaria. EPA: Estudio posautorización. EPA-LA: Estudios posautorización cuya realización tiene lugar a instancia de las autoridades reguladoras y ligada a la autorización de comercialización (AEMPS o Agencia Europea de Medicamentos). Se incluyen en esta vía administrativa tanto los estudios ligados a la autorización como los estudios de seguridad a requerimiento de las autoridades sanitarias y los incluidos en los planes de gestión de riesgos. EPA-AS: Estudio posautorización promovido por las Administraciones Sanitarias (AS) o financiado con fondos públicos. EPA-SP: Estudio posautorización de seguimiento prospectivo que no corresponde a ninguna de las dos categorías anteriores. EPA-OD: Estudios posautorización con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos. No-EPA: Estudios en los que el factor de exposición fundamental investigado no es un medicamento; por ejemplo estudios de incidencia o de prevalencia de enfermedades, etc.

92
3. COMPOSICIÓN Y ESTRUCTURA DEL C.E.I.C, MIEMBROS QUE LO COMPONEN, MEDIOS CON QUE CUENTA, UBICACIÓN 3.1 Composicón del C.E.I.C
Estructura Miembro, Carácter con que actúa, Titulación
Adscripción Actual
Presidente Dr. Nicolas Martinez Tornero Facultativo especialista en cirugía
Director Gerente Consorcio Hospitalario Provincial de Castellón
Vicepresidente Dr Manuel Bruscas Maicas Facultativo especialista en Medicina
Interna
Director Médico Consorcio Hospitalario Provinical de Castellón
Secretaria Dña. Eva Felip Viciano Miembro ajeno a la profesión
sanitaria. Diplomada
Secretaria Administrativa Consorcio Hospitalario Provincial de Castellón
Vocales D. Miguel Llorens Izquierdo Miembro ajeno a la profesión
Sanitaria
Director Económico Consorcio Hospitalario Provincial de Castellón
Dr. Francisco Morales Olivas Farmacólogo Clínico Doctor en Medicina
Profesor Titular de Farmacología. Dpto. de Farmacología Univ. Valencia
Dña. Belen Bertolin Olmos Facultativo Especialista en Farmacia
Hospitalaria
Farmaceutica Hospitalaria Consorcio Hospitalario Provincial de Castellón
Dr. Carlos Ferrer Albiach Facultativo especialista en Oncología
Radioterápica
Director Instituto Oncológico Consorcio Hospitalario Provincial de Castellón
Dr. Eduardo Martinez de Dueñas Facultativo Especialista Oncológica
Médica
Oncólogo Médico Consorcio Hospitalario Provincial de Castellón
Dr. Enrique Boldo Roda Facultativo Especialista Cirugía
Cirujano Consorcio Hospitalario Provincial de Castellón
D. Pablo Sebastiá Tirado Miembro ajeno a la profesión
Sanitaria
Director de Comunicación y Marketing de la Fundación Lubasa.
D. Enrique Ochoa De Aranda Biólogo
Jefe Sección Biopatología Molecular Consorcio Hospitalario Provincial de Castellón
Dña. Elena Ferrada Daudi Miembro ajeno a la Profesión
Sanitaria. Licenciada en Derecho
Jefa Servicio Jurídico Consorcio Hospitalario Provincial de Castellón
Maria Jose Beltran Lavernia Enfermera
Directora de enfermeria
Marian Bonet Dean Farmacéutica Atención Primaria
Farmacéutica Atención Primaria Hospital General
Dra. Ana Esther Felip Gimeno Pediatra
Pediatra Complejo Socio Educativo Penyeta Roja
Dr. José Mª Sabater Melchor Médico Atención Primaria
Médico Atención Primaria Centro Salud Alcora (Castellón)
Dr. Juan Pablo Aracil Kessler Facultativo Especialista Cirugía Plástica
Jefe Área Quirúrgica Consorcio Hospitalario Provincial de Castellón
Dr. Diego Castell Campesino
Director Fundación C.V. Hospital Provincial de Castellón
TOTAL : 18 miembros

93
El Comité Ético de Investigación Clínica del Hospital Provincial de Castellón estará formado al menos por:
- El Director Médico del centro o Director Gerente del Consorcio Hospitalario Provincial de Castellón. - Farmacólogo clínico. - Farmacéutico de hospital. - Farmacéutico de Atención Primaria - Diplomado en Enfermería. - Dos miembros ajenos a las profesiones sanitarias, uno de ellos licenciado en Derecho. Uno de estos
miembros no estará vinculado laboralmente al centro, y se recomienda que sea propuesto por el Consejo de Salud.
- Al menos dos médicos más: - Con labor asistencial activa. - Un médico de Atención Primaria. - Un pediatra - De suficiencia investigadora probada, valorándose especialmente que sea en el campo de
ensayos clínicos. En los casos en que exista Comisión de Investigación o Comité de Ética Asistencial, deberá formar parte del comité un miembro de cada una de ellas. Los miembros serán nombrados por el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud, después que la Comisión Delegada del Programa de Estudios Clínicos de medicamentos y Productos Sanitarios (PECME) haya emitido el informe preceptivo a propuesta de la dirección del centro, y serán elegidos entre profesionales que se presenten y/o acepten voluntariamente y hagan constar lo especificado en el artículo 8, punto 3, del Decreto 73/2009 del Consell. 3.2. Medios con que cuenta y ubicación : Sala de reuniones : Sala de reuniones Dirección Clínica ( Planta primera) Area de trabajo y archivo : Sala de Reuniones Dirección Clínica y despachos de Dirección y Servicios Jurídicos Recursos materiales: 2 PCs, 2 impresoras, teléfono, fax y mueble archivador en una habitación bajo de cierre con llave para garantizar la confidencialidad de la documentación. Recursos personales : Secretaria de Dirección y Jefa Servicios Jurídicos Dirección : Consorcio Hospitalario Provincial de Castellón. Eva Felip Viciano. Secretario CEIC. Avda. Doctor Clara 19 12002 Castellón. Teléfono : 964 35 97 00 Fax : 964 35 43 01 e-mail: [email protected] o [email protected]

94
4. FUNCIONES DE COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA, INCLUYENDO LAS ESPECIFICADAS EN EL R.D. 223/2004, Y EL DECRETO 73/2009, DE 5 DE JULIO DEL CONSELL. FUNCIONES DE LOS MIEMBROS, PRESIDENTE Y SECRETARIO, PROCEDIMIENTOS DE COMUNICACIÓN CON EL PROGRAMA DE ENSAYOS CLÍNICOS DE LA COMUNIDAD VALENCIANA Y CON EL MINISTERIO DE SANIDAD Y CONSUMO
A. FUNCIONES DEL CEIC
ENSAYOS CLINICOS El CEIC evaluará los aspectos METODOLÓGICOS, ÉTICOS y LEGALES de los ensayos clínicos que les sean remitidos, para ello estudiará los siguientes aspectos:
1. La pertinencia del ensayo clínico, teniendo en cuenta el conocimiento disponible.
2. La pertinencia de su diseño para obtener conclusiones fundamentadas con el número adecuado de sujetos en relación con el objetivo del estudio.
3. Los criterios de selección y retirada de los sujetos del ensayo, así como la selección equitativa de la muestra.
4. La justificación de los riesgos e inconvenientes previsibles en relación con los beneficios esperables para los sujetos del ensayo, para otros pacientes y para la comunidad, teniendo en cuenta el principio de protección de los sujetos del ensayo desarrollado en el artículo 3.
5. La justificación del grupo control (ya sea placebo o un tratamiento activo).
6. Las previsiones para el seguimiento del ensayo.
7. La idoneidad del investigador y de sus colaboradores.
8. La idoneidad de las instalaciones.
9. La idoneidad de la información escrita para los sujetos del ensayo y el procedimiento de obtención del consentimiento informado, y la justificación de la investigación en personas incapaces de dar su consentimiento informado.
10. El seguro o la garantía financiera previstos para el ensayo.
11. Las cantidades y, en su caso, previsiones de remuneración o compensación para los investigadores y sujetos del ensayo y los aspectos relevantes de cualquier acuerdo entre el promotor y el centro, que han de constar en el contrato previsto en el artículo 30.
12. El plan previsto para el reclutamiento de los sujetos.
13. En el caso de los ensayos clínicos multicéntricos en que se actúe como CEIC implicado, únicamente se evaluarán las cuestiones indicadas en los apartados 7, 8, y 11.
14. Aprobará sus normas de funcionamiento interno
15. Velará por la corrección de los protocolos propuestos, desde el punto de vista metodológico, ético y legal.
16. Informará al Coordinador del Programa de Estudios Clínicos de Medicamentos y Productos Sanitarios en la Comunitat Valenciana (PECME) de todos aquellos protocolos de investigación que aprueben y que se eleven al órgano competente para su autorización, y también de los que finalmente se desestimen.
17. Remitirá al Coordinador del PECME una copia de los certificados que se emitan sobre la adecuación deontológica de los protocolos que impliquen la investigación en humanos.

95
18. Realizará un informe acerca de las modificaciones de los protocolos de los ensayos, velando por la corrección de los protocolos propuestos desde el punto de vista metodológico, ético y legal (según el modelo del anexo VI de la Resolución de 16 de julio de 2009, de la Consellería de Sanitat, de regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana).
19. Comunicará al Coordinador del PECME las reacciones adversas, graves o inesperadas que acontezcan durante el desarrollo del ensayo, mediante copia de la notificación del investigador o promotor.
20. Propondrá a la Conselleria de Sanidad, a través Coordinador del PECME, la suspensión cautelar de un ensayo clínicos en los supuestos previstos en legislación vigente.
ESTUDIOS POST-AUTORIZACION OBSERVACIONALES El CEIC evaluará los aspectos METODOLÓGICOS, ÉTICOS y LEGALES de los estudios post-autorización observacionales valorando: 1. El diseño del estudio, si es correcto y cumple con los principios éticos básicos de la declaración
de Helsinki. 2. El interés científico que se pretende conseguir con el estudio y la relación beneficio–riesgo
para los sujetos de investigación. 3. Los procedimientos para garantizar que la realización del estudio no modificará los hábitos de
prescripción del médico, debiéndose prescribir los medicamentos de la manera habitual, de acuerdo con las condiciones normales de la práctica clínica y para aquellas indicaciones para las que fueron autorizados.
4. La hoja de información y el formulario de consentimiento informado cuando se requiera entrevistar al sujeto, o en aquellos en los que, utilizando otras fuentes de información, no sea posible adoptar un procedimiento de disociación seguro.
5. Que la participación en el estudio no interfiere los cometidos asistenciales de los investigadores.
6. La idoneidad de los investigadores y de las instalaciones. 7. Que la compensación económica es explicita, transparente y proporcionada con la dedicación y
responsabilidad.
B. FUNCIONES DEL PRESIDENTE, SECRETARIO Y VOCALES
Corresponde al Presidente: 1. Representación del órgano. 2. Convocatoria de las Sesiones ordinarias y extraordinarias, así como la fijación del orden del
día, según las peticiones realizadas por los demás miembros, formuladas con la suficiente antelación.
3. Asistir a las sesiones con voz y voto. 4. Presidir, moderar y suspender las sesiones con causa justificada. 5. Visar las actas y certificaciones de los acuerdos del órgano.

96
Corresponde al Secretario 1. Efectuar la convocatoria de las sesiones del órgano por orden de su Presidente, así como las
citaciones a los miembros del mismo por escrito o correo electrónico. 2. Asistir a las sesiones con voz y voto. 3. Levantar acta de las sesiones conforme a lo dispuesto en el art. 14.6 del R.D. 223/2004 4. Recibir las comunicaciones que se efectúen al CEIC. 5. Expedir certificación de las actas, consultas, dictámenes y cualesquiera otros acuerdos
adoptados. 6. Archivar y custodiar la documentación del CEIC. Corresponde a los vocales 1. Asistir a las sesiones con voz y voto. 2. Efectuar las propuestas que estimen oportunas, por escrito y dirigidas al Presidente, con una
antelación mínima de diez días naturales previas a la convocatoria, para que sean incluidas en el orden del día.
3. Recibir con una antelación mínima de una semana las convocatorias conteniendo el orden del día.
4. Ejercer su derecho al voto y formular su voto particular, así como expresar el sentido de su voto y los motivos que lo justifican.
5. Formular ruegos y preguntas 6. Obtener la información precisa para cumplir las funciones asignadas. LOS MIEMBROS NO PUEDEN DELEGAR EN OTRO Los miembros harán constar su compromiso de cumplir las funciones especificadas en los arts. 10 y 17 del R.D. 223/2004 y art. 8 del Decreto 73/2009, de 5 de junio, del Consell, y están obligados a guardar la máxima confidencialidad en lo que respecta a la documentación recibida para la evaluación del protocolo y la identidad de los pacientes.
C. PROCEDIMIENTOS DE COMUNICACIÓN CON EL PROGRAMA DE ENSAYOS CLÍNICOS DE LA COMUNIDAD VALENCIANA Y CON EL MINISTERIO DE SANIDAD Y CONSUMO.
Conforme a la Resolución de 16 de julio de 2009, de la Consellería de Sanitat, de regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana, y el Decreto 73/2009, del, Consell, por el que se regula la gestión de ensayos clínicos y estudios post-autorización observacionales con medicamentos y productos sanitarios, se remite al Coordinador del Programa de Estudios Clínicos de medicamentos y Productos Sanitarios en la Comunitat Valenciana (PECME) comunicación de los ensayos/estudios post-autorización evaluados, conforme al siguiente esquema.
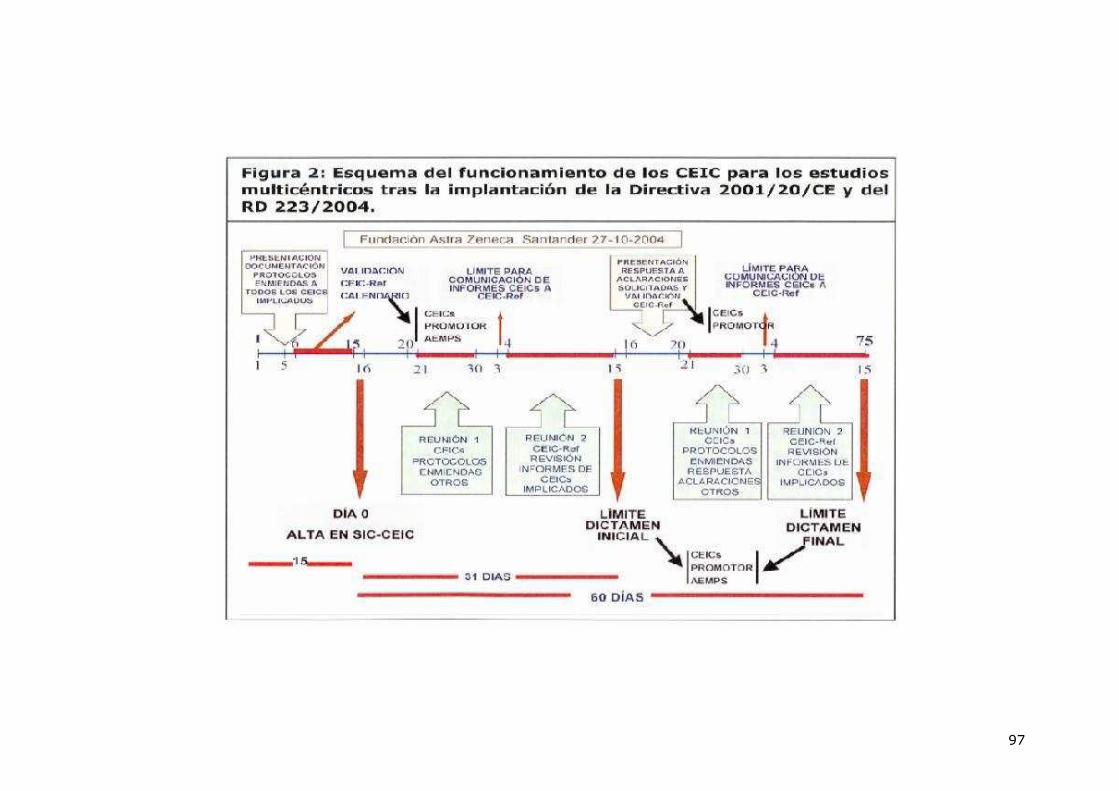
97

98
5. ELECCIÓN, SUSTITUCIÓN Y RENOVACIÓN DE LOS MIEMBROS DEL C.E.I.C: PROCEDIMIENTOS, CRITERIOS Y DURACIÓN DE LOS CARGOS.
5.1. PROCEDIMIENTO DE CAMBIO DE VOCALES
Según art. 1 de la Resolución de 16 de julio de 2009, de la Conselleria de Sanitat sobre regulación de los procedimientos, documentación y plazos a observar en la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana.
5.2. CRITERIOS DE ACREDITACIÓN CURRICULAR DE UN MIEMBRO DE CEIC
Art. 2 de la Resolución de 16 de julio de 2009, de la Conselleria de Sanitat.
5.3. NOMBRAMIENTO Y CESE DE LOS MIEMBROS
Conforme con el artículo 1 de la Resolución de 16 de julio de 2009, de la Consellería de Sanitat, el director del centro solicitará el nombramiento al Coordinador del PECME, y acompañará a dicha solicitud el curriculum vitae actualizado (anexo 8 de este documento), con fotocopia de las titulaciones académicas y las publicaciones científicas pertinentes, y la declaración, por parte del miembro propuesto, de carecer de intereses derivados de la fabricación y venta de medicamentos y productos sanitarios. En dicha solicitud se indicará el miembro actual que se propone sustituir, en su caso, y que cesará como miembro del CEIC. La Comisión delegada del PECME informará positiva o negativamente sobre dicha propuesta, para su resolución por el director general de Farmacia y Productos Sanitarios de la Agencia Valenciana de Salud. Será causa de cese como miembro del CEIC la ausencia sin justificar a tres reuniones consecutivas en periodo de actividad laboral. La justificación deberá ser documentada y aportar por escrito opinión de los estudios que aparezcan en el orden del día.
5.4. ELECCIÓN DE LOS MIEMBROS
Serán elegidos entre profesionales que se presenten y/o acepten voluntariamente y hagan constar que carecen de cualquier clase de intereses derivados de la fabricación y venta de medicamentos y productos sanitarios.
5.5. ELECCIÓN DE LOS CARGOS
EL Presidente será el Director Gerente o en su defecto el Director Médico del Consorcio. El secretario de dicho comité será designado de entre el personal administrativo del centro.
5.6. DURACIÓN DE LOS CARGOS
Los cargos de Presidente y Secretario se renovarán en cada reacreditación del CEIC (cada 3 años), sin perjuicio de su reelección o de su renuncia.

99
6. ÁMBITO DE ACTUACIÓN El ámbito de actuación será en el Consorcio Hospitalario Provincial de Castellón. 7. PERIOCIDAD DE LAS REUNIONES Y TIEMPO MÁXIMO DE RESPUESTA A LOS PROTOCOLOS. PROCEDIMIENTO DE LAS CONVOCATORIAS DE LAS REUNIONES DEL CEIC Y QUORUM. CALENDARIO DE EVALUACIÓN DE LOS ENSAYOS MULTICÉNTRICOS.
7.1. PERIODICIDAD DE LAS REUNIONES
Mensuales, con carácter ordinario. Preferiblemente el último miércoles/jueves de cada mes a las 13 h. Con el fin de cumplir los plazos para la obtención del dictamen único, de conformidad con el R.D. 223/2004.
7.2. TIEMPO MÁXIMO DE RESPUESTA A LOS PROTOCOLOS
En los ensayos multicéntricos el último día para que el CEIC notifique su informe al CEIC de referencia (mediante la aplicación informática SIC-CEIC II) será el día 3 del mes siguiente al de la presentación de la documentación valida por parte del solicitante. En los ensayos unicéntrico o estudios post-autorización de tipo observaciónal la respuesta máxima seria de tres meses.
7.3. PROCEDIMIENTO DE LAS CONVOCATORIAS
De conformidad con el art. 9 del Decreto 73/2009, del Consell, por el que se regula la gestión de ensayos clínicos y estudios post-autorización observacionales con medicamentos y productos sanitarios, y, en lo no previsto expresamente en este articulo, se aplicará lo dispuesto en el capitulo II del título II de la Ley 30/92, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
7.4. QUORUM
Para la valida constitución del órgano, a efectos de la celebración de sesiones, deliberaciones o toma de acuerdos, se requerirá la presencia del Presidente, o en su caso el Vicepresidente, y del Secretario, o, en su caso, de quien le sustituya, y de la mitad más uno de sus miembros. En el caso de no concurrir suficiente número de miembros para considerarse válidamente constituida la sesión, transcurrida media hora se tendrá por realizada una segunda convocatoria, en cuyo caso para que se constituya válidamente bastará con la concurrencia del Presidente, o en su caso el Vicepresidente, del Secretario o personal que le sustituya, y, al menos, 3 miembros más. Respecto a los votos delegados, estos no contabilizarán para establecer el quórum, solo se contabilizan los miembros presentes. Ningún protocolo en el que participe un miembro del comité podrá ser evaluado en presencia de éste. Del mismo modo ningún protocolo será evaluado en presencia del promotor, investigador o monitor, sin perjuicio de que puedan ser invitados, a los solos efectos de exponer una defensa de su ensayo. A las sesiones del Comité podrán asistir, con voz, pero sin voto, profesionales expertos en las materias sometidas a su consideración, a criterio del Comité.

100
8. CALENDARIO DE EVALUACIÓN ENSAYOS MULTICENTRICOS CON REFERENCIA EXPLICITA A LOS CRITERIOS Y PROCEDIMIENTOS PARA LA OBTENCIÓN DE DICTAMEN ÚNICO DE LA AGENCIA ESPAÑOLA DEL MEDICAMENTO Anexo II (versión 30 de abril de 2004). La aplicación del Real Decreto 223/2004 requiere la adopción de una serie de medidas de coordinación y de sincronización por parte de los Comités Éticos de Investigación Clínica (CEICs) con el fin de hacer posible la obtención del dictamen único en los plazos legalmente establecidos. A tal efecto, se considera necesario que los promotores y los CEICs acreditados en nuestro país sigan las siguientes normas:
Consideraciones generales 1. Para facilitar el intercambio de informes y dictámenes y cumplir los trámites de notificación a los CEICs implicados, al promotor, al Centro Coordinador de CEICs y a la AEMPS, que establece el Real decreto 223/2004 para la obtención del dictamen único, los CEICs estarán conectados mediante una aplicación informática denominada "Sistema Informático de Conexión de los CEICs¨ (SIC-CEIC II). 2. Los CEICs deberán tener un registro de entrada a fin de que quede constancia de la fecha de recepción de cada solicitud de evaluación, tanto para nuevos ensayos clínicos, como para modificaciones y respuestas a las aclaraciones solicitadas. 3. El promotor elegirá el CEIC de referencia de común acuerdo con éste. 4. El solicitante deberá presentar las solicitudes de ensayo clínico o modificación relevante acompañada de la documentación correspondiente de manera simultánea a todos los CEIC implicados. La documentación debe ser completa e idéntica para todos los CEIC, excepto la correspondiente a los aspectos específicos de centro. 5. El solicitante deberá identificar los centros en que se prevea realizar el ensayo y los CEICs implicados, indicando cual de ellos será el CEIC de referencia. Presentación de la solicitud y calendario de evaluación 1. El CEIC de referencia tendrá un plazo máximo de 60 días naturales para emitir un dictamen definitivo motivado sobre el ensayo, y podrá pedir información adicional al solicitante una sola vez. Con el fin de hacer posible el cumplimiento de este requisito, es necesario que el plazo máximo para emitir el dictamen inicial no supere los 31 días. De esta manera, en el caso de que se hayan pedido aclaraciones, quedarán 29 días para evaluar la respuesta que presente el promotor a las mismas. 2. El plazo máximo para emitir el dictamen definitivo en el caso de modificaciones relevantes es de 35 días naturales. 3. La presentación de documentación a los CEICs por parte del solicitante debe realizarse únicamente dentro de los periodos siguientes:
• Protocolos y modificaciones relevantes: días laborables del 1 al 5 de cada mes.
• Reclamación de documentación y validación: del día 6 al día 15 de cada mes.
• Respuesta a las aclaraciones mayores solicitadas: días laborables del 16 al 20 de cada mes. El promotor deberá asegurarse de que todos los CEICs han recibido correctamente toda la documentación en los periodos citados e informará de ello al CEIC de referencia.

101
4. Cada CEIC verificará la validez de la documentación específica de centro recibida y contactará con el solicitante para subsanar posibles deficiencias. El CEIC de referencia verificará, además, la validez de la documentación común para todos los comités. Cuando un CEIC implicado no comunique al CEIC de referencia la existencia de deficiencias en la documentación específica de centro, el CEIC de referencia dará por válida la solicitud. 5. El CEIC de referencia deberá agotar el plazo de 10 días naturales de validación antes de considerar la documentación válida, dar de alta el documento (protocolo o modificación) en la aplicación SIC-CEICS y notificar el calendario de evaluación a los CEICs implicados, al solicitante y a la AEMPS. 6. El día 16 de cada mes se considerará día 1 del calendario de evaluación para todas las solicitudes de nuevos ensayos o modificaciones consideradas válidas en ese mes. 7. Todos los CEICs deberán reunirse entre el día 21 y fin de mes con el objeto de evaluar los protocolos, las modificaciones y las respuestas a aclaraciones que se hayan recibido en dicho mes. 8. El último día para que los CEICs implicados notifiquen su informe al CEIC de referencia (mediante la aplicación informática SIC-CEICS) será el día 3 del mes siguiente al de la presentación de la documentación válida por parte del solicitante. A partir del día 4, el CEIC de referencia podrá consultar los informes de los CEIC implicados. 9. El día 15 del mes siguiente al de la presentación de una solicitud válida, el CEIC de referencia notificará el dictamen correspondiente (favorable, petición de aclaraciones o desfavorable) al solicitante, los CEICs implicados y a la AEMPS. El cómputo del plazo de evaluación se detendrá cuando el CEIC de referencia solicite aclaraciones. Si en el dictamen del CEIC de referencia se solicitan aclaraciones, el solicitante deberá presentar la respuesta a las mismas a todos los CEIC implicados en el ensayo entre los días 16 y 20 del mes en curso o, a lo sumo, del mes siguiente. La ausencia de respuesta en dicho plazo motivará un dictamen desfavorable por parte del CEIC de referencia. 10. Los CEIC que actúen como CEIC de referencia tendrán que hacer necesariamente dos reuniones al mes: - Una para la evaluación de los nuevos ensayos, modificaciones relevantes y respuestas a aclaraciones previamente solicitadas (entre el día 21 y fin de mes). - Otra para la revisión de los informes de los CEICs implicados y elaboración del dictamen correspondiente (entre el día 4 y 15 de cada mes).

102
Resumen secuencial del calendario propuesto
Fecha
1. Del 1 al 5 Presentación de la documentación a los CEICs de nuevos ensayos y modificaciones relevantes.
2. Del 6 al 15 Validación de la documentación presentada y notificación por el CEIC de referencia de acuse de recibo de una solicitud válida y calendario de evaluación a los CEICs implicados, al solicitante y a la AEMPS.
3. Día 16 Se considerará el día 1 del procedimiento
4. Del 16 al 20: Presentación de las respuestas a las aclaraciones solicitadas por el CEIC en el mes actual o en el anterior. El CEIC de referencia debe comprobar que la respuesta del promotor a las aclaraciones es válida y comunicar el calendario de evaluación a los CEICs implicados y al promotor.
5. Del 21 a fin de mes
Reunión de los CEICs
Cambio de mes
6. Día 3 del mes siguiente
Fecha límite para que los CEIC implicados comuniquen su informe (favorable, objeciones, desfavorable) al CEIC de referencia.
7. Entre el 4 y el 15 Reunión del CEIC de referencia para revisar los informes de los CEIC implicados y preparar el dictamen (favorable, petición de aclaraciones o desfavorable).
8. Día 15 Fecha límite para que el CEIC de referencia notifique el dictamen a los CEICs implicados, solicitante y a la AEMPS.
9. Día 16 al 20 Presentación, en su caso, de las respuestas a las aclaraciones solicitadas. El proceso continúa en el apartado 4.
Dictamen único: evaluación de nuevos ensayos Si un CEIC implicado no ha remitido su informe sobre los aspectos específicos de centro del ensayo en el plazo fijado, se considerará que su opinión es desfavorable. Dictamen único: evaluación de modificaciones relevantes Los CEICs implicados siempre podrán emitir un informe respecto a las modificaciones relevantes de los ensayos clínicos multicéntricos. Cuando la modificación suponga la incorporación de un nuevo centro o investigador principal la falta de informe del CEIC implicado que corresponda, se considerará indicativa de informe desfavorable. Para otras modificaciones relevantes, la falta de informe por parte de un CEIC implicado se considerará indicativa de opinión favorable. 9. DOCUMENTACIÓN QUE PRESENTARÁN LOS PROTOCOLOS A EVALUAR, CON REFERENCIA EXPLÍCITA AL ART 16 DEL REAL DECRETO 223/2004 Y A LAS ACLARACIONES DE LA AGENCIA ESPAÑOLA DEL MEDICAMENTO SOBRE APLICACIÓN DE LA NORMATIVA DE ENSAYOS CLÍNICOS CON MEDICAMENTOS DE USO HUMANO A PARTIR DE 1 DE MAYO DE 2004 (VERSIÓN NUM. 6, MAYO DE 2008). Constará de la siguiente documentación:

103
DOCUMENTACIÓN A PRESENTAR: ENSAYOS CLÍNICOS CON MEDICAMENTOS
Carta de acompañamiento (solicitud de nuevo ensayo clínico), según modelo, junto con la notificación de código Eudract (1 copia) Identificará el ensayo clínico mediante el código de protocolo del promotor, el título y el nº EudraCT e irá firmada por el solicitante. Cuando el diseño del ensayo haya tenido en cuenta un asesoramiento científico emitido por la AEMPS, por la EMEA o la agencia responsable de la autorización de los medicamentos en cualquier otro país, se hará constar este hecho, identificando el lugar en el que se incluye dicho documento en la documentación aportada. Se especificará si el ensayo es uni o multicéntrico. Cuando el protocolo prevea un procedimiento de notificación de reacciones adversas distinto del habitual se hará constar aquí. En las reiteraciones de una solicitud de ensayo clínico se hará referencia al desistimiento de la solicitud, dictamen desfavorable del CEIC o no autorización de la solicitud previa y se indicarán los cambios introducidos en la solicitud actual. Se incluirá cualquier otra información que se considere relevante.
Solicitud de autorización de un ensayo clínico con medicamentos de uso humano a las autoridades competentes y de dictamen por los comités éticos en España (anexo 1A) junto con el apartado k (1 copia)
Copia de la autorización que le habilita para actuar en nombre del promotor (1copia) Cuando el solicitante no sea el promotor ni el representante legal.
Copia del poder notarial o documento equivalente que acredite el nombramiento del representante legal (1 copia) Cuando el promotor no esté ubicado en un Estado Miembro de la Unión Europea.
Relación de investigadores y centros de CEIC (1 copia). En el caso de ensayos clínicos multicéntricos se especificarán todos los centros españoles donde se propone la realización del ensayo, especificando los CEICs implicados y cual será el CEIC de referencia.
Protocolo (2 copias). Deberá tener en cuenta lo especificado en las guías de desarrollo de la Directiva 2001/20/CE sobre el formato de solicitud y documentación referente a las solicitudes de autorización de ensayos clínicos por las autoridades competentes y de dictamen sobre ensayos clínicos con medicamentos dirigidas a un comité ético y la guía ICH sobre normas de buena práctica clínica (CPMP/ICH/135/95 Topic E6 step 5 Note of guidance on Good Clinical Practice)”. Estos documentos se encuentran en el volumen 10 – Ensayos clínicos de la normativa europea sobre medicamentos http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev10.htm . Firma de protocolo por el investigador principal (1 copia).
Manual de investigador/Ficha técnica o ficha técnica más resumen de datos no clínicos / clínicos
(2 copias) que avale la utilización de cada medicamento en el EC. Se ajustará en su estructura y contenido a la guía ICH de la Comunidad CPMP/ICH/135/95. En caso de medicamentos autorizados en algún Estado Miembro de la Comunidad, cuando el medicamento se utilice en las condiciones de uso autorizadas el Manual del Investigador podrá reemplazarse por la ficha técnica autorizada.

104
Cuando los medicamentos en investigación estén autorizados en España y el protocolo admita la posibilidad de utilizar cualquier marca comercial, el manual del investigador podría consistir en una información general equivalente a la de ficha técnica aplicable a todas las especialidades farmacéuticas que tengan igual composición.
Cuaderno de recogida de datos (2 copias). Debe tenerse en cuenta que con el fin de respetar la Ley de Protección de Datos, la identificación del sujeto en este documento debe ser mediante un código que no permita su identificación directa.
Los documentos referentes al consentimiento informado, (2 copias) incluyendo la hoja de información al paciente (HIP). Los documentos referentes al consentimiento informado deberán tener en cuenta lo especificado en la “Guía detallada sobre el formato de solicitud y documentación referente a las solicitudes de dictamen sobre ensayos clínicos con medicamentos que se debe remitir a los Comités Éticos” que puede consultarse en www.agemed.es vínculo “Investigación clínica” “Ensayos clínicos con medicamentos de uso humano” y “Normativa europea” La hoja de información para el sujeto deberá estar identificada con una fecha o número de versión.
Los documentos sobre la idoneidad del investigador y sus colaboradores (1 copia) a) El compromiso de los investigadores que participen en el ensayo b) Currículum Vitae, resumido (2 folios máximo) c) Certificado de idoneidad de cada colaborador d) Certificado de idoneidad del equipo colaborador
Los documentos sobre la idoneidad de las instalaciones (1 copia) a) Recursos estructurales para llevar a cabo el ensayo b) Compromiso de aceptación de los servicios implicados – directa o indirectamente – en la
realización del ensayo.
Memoria económica: las cantidades y el modo en que los investigadores y sujetos puedan ser, en su caso, remunerados o indemnizados por su participación en el ensayo clínico, así como los elementos pertinentes de todo contrato previsto entre el promotor y el centro (1 copia)
10 carpetillas individuales, en papel, conteniendo: a) Resumen del protocolo b) Apartado k c) Hoja de información para los posibles participantes, así como el anuncio para el reclutamiento de los sujetos del ensayo, si procede. d) Una copia de la póliza del seguro y del justificante de la garantía financiera del ensayo clínico o un certificado de esta, cuando proceda. e) Propuesta compensación económica.
Una copia de la póliza del seguro (1 copia) o del justificante de la garantía financiera del ensayo clínico o un certificado de ésta, cuando proceda.
Documento firmado de asunción de responsabilidad de daños producidos como consecuencia del ensayo (1 copia) en los casos previstos en el articulo 8.3 del Real Decreto 223/2004 de ausencia de seguro o de seguro con cobertura parcial.
Los procedimientos y material utilizado para el reclutamiento (1 copia) de los sujetos del ensayo
Copia de los asesoramientos científicos (1 copia) emitidos por alguna Agencia de Medicamentos (cuando existan)
Una copia de toda esta documentación en formato electrónico (pdf) en un CD.
Justificante de pago de tasas de evaluación/solicitud de exención de tasas (1 copia)

105
DOCUMENTACIÓN A PRESENTAR: ESTUDIOS POST-AUTORIZACIÓN OBSERVACIONALES
Protocolo (2 copias). Se deberá especificar en el mismo los procedimientos que se emplearán para garantizar que la realización del estudio no modificará los hábitos de prescripción del médico.
Ficha técnica o ficha técnica más resumen de datos no clínicos / clínicos del medicamento
investigado. (2 copias ) Clasificación por parte de la AEMPS del tipo de EPA (EPA-LA, EPA-AS, EPA-SP o EPA-OD) (1 copia)
según Orden SAS/3470/2009, de 16 de diciembre, de las directrices sobre estudios posautorización de tipo observacional para medicamentos de uso humano
Los documentos referentes al consentimiento informado, (2 copias) incluyendo la hoja de información al paciente (HIP).
Cuaderno de recogida de datos (2 copias)
Relación de investigadores, centros de CEIC implicados en la realización del ensayo clínicos multicéntrico. (1 copia)
Memoria Económica: las cantidades y el modo en que los investigadores y sujetos puedan ser, en su caso, remunerados o indemnizados por su participación en el ensayo clínico, así como los elementos pertinentes de todo contrato previsto entre el promotor y el centro (1 copia)
Los documentos sobre la idoneidad del investigador y sus colaboradores (1 copia) a) El compromiso de los investigadores que participen en el ensayo. En el caso de ensayos multicéntricos la documentación completa del ensayo se presentará a todos los CEICs implicados en los plazos establecidos, pero cada uno de ellos sólo recibirá la documentación específica de su centro. b) Currículum Vitae, resumido (2 folios máximo) c) Certificado de idoneidad de cada colaborador d) Certificado de idoneidad del equipo investigador.
Los documentos sobre la idoneidad de las instalaciones (1 copia) a) Recursos estructurales para llevar a cabo el ensayo
b) Compromiso de aceptación de los servicios implicados – directa o indirectamente – en la realización del estudio.
Los procedimientos y material utilizado para el reclutamiento (1 copia) de los sujetos del estudio.
Copia de la autorización que le habilita para actuar en nombre del promotor (1copia) Cuando el solicitante no sea el promotor ni el representante legal.
Copia del poder notarial o documento equivalente que acredite el nombramiento del representante legal (1copia) Cuando el promotor no esté ubicado en un Estado Miembro de la Unión Europea.
Una copia de toda esta documentación en formato electrónico en un CD.
Justificante de pago de tasas de evaluación/solicitud de exención de tasas (1 copia)

106
INDICE DE DOCUMENTACIÓN PRESENTADA – ENMIENDAS
Formulario correspondiente a la solicitud de evaluación de enmienda (anexo 1C) (1 copia)
Copia del protocolo actualizado con la enmienda a evaluar, indicando claramente cuáles son los cambios específicos (1 copia)
Copia de la firma de aceptación del investigador principal de la enmienda (1 copia)
En el caso de que la enmienda implique nuevas pruebas, documento de aceptación de los diferentes servicios implicados (1 copia)
Contenido y justificación de la enmienda (2 copias)
Nueva hoja de información al paciente y copia de todos los documentos que hayan sido modificados por la enmienda (2 copias)
Una copia de toda la documentación en formato electrónico (PDF) en un CD. (1 copia)
Justificante del pago/solicitud de exención de tasas (1 copia) TRAMITACIÓN La documentación completa deberá entregarse a: Secretaria CEIC Departamento de Calidad Consorcio Hospitalario Provincial de Castellón Avda. Dr Clará, 19 12002 Castellón Telf: 964 359 700 extn. 54379 El CEIC emitirá el correspondiente dictamen en los plazos establecidos en el RD 223/2004 9.1 TASAS DE EVALUACIÓN COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA (CEIC) CONSORCIO HOSPITALARIO PROVINCIAL DE CASTELLÓN El promotor abonará a la Fundación de la CV Hospital Provincial de Castellón la cantidad correspondiente al protocolo a evaluar (ensayo clínico, estudio post-autorización o enmienda) según las tasas establecidas por la Fundación (anexo 3), previa solicitud de factura (anexo 4). El justificante del pago se entregará con la documentación a presentar en el CEIC. Cuando el promotor es una entidad sin ánimo de lucro, o una persona física, se mantienen las tasas anteriores, considerando, previo escrito motivado del promotor, la solicitud de exención de tasas (anexo 5). 9.2 CONTRATO ECONÓMICO El promotor abonará la cantidad de 500 euros + IVA en concepto de compensación de la gestión administrativa a la Fundación de la CV Hospital Provincial de Castellón, cantidad efectiva a la firma del contrato y que no es susceptible de devolución. Al menos 20% del presupuesto establecido en el contrato por paciente reclutado se destinará a la Fundación de la CV Hospital Provincial de Castellón para cubrir los costes indirectos.

107
10. PROCEDIMIENTO DE LA EVALUACIÓN, CON ACLARACIÓN EXPLICITA DE LOS CRITERIOS REFERIDOS EN EL R.D 223/2004 ARTS. 3 Y 17.
El Comité Ético de Investigación Clínica correspondiente evaluará el protocolo, el manual del investigador y el resto de la documentación que acompañe a la solicitud y emitirá su dictamen tomando en consideración, en particular, las siguientes cuestiones:
a) La pertinencia del ensayo clínico, teniendo en cuenta el conocimiento disponible.
b) La pertinencia de su diseño para obtener conclusiones fundamentadas con el número adecuado de sujetos en relación con el objetivo del estudio.
c) Los criterios de selección y retirada de los sujetos del ensayo, así como la selección equitativa de la muestra.
d) La justificación de los riesgos e inconvenientes previsibles en relación con los beneficios esperables para los sujetos del ensayo, para otros pacientes y para la comunidad, teniendo en cuenta el principio de protección de los sujetos del ensayo desarrollado en el artículo 3.
e) La justificación del grupo control (ya sea placebo o un tratamiento activo).
f) Las previsiones para el seguimiento del ensayo.
g) La idoneidad del investigador y de sus colaboradores.
Experiencia y capacidad investigadora para llevar adelante el estudio, en función de: Sus obligaciones asistenciales. Los compromisos previamente adquiridos con otros protocolos de investigación
h) La idoneidad de las instalaciones.
i) La idoneidad de la información escrita para los sujetos del ensayo y el procedimiento de obtención del consentimiento informado, y la justificación de la investigación en personas incapaces de dar su Consentimiento informado.
Respecto al consentimiento de las personas no capaces o que no estén en condiciones de prestarlo y menores de edad debe cumplir lo indicado en el Artículo 7 de R.D. 223/2004. También se regulan en este artículo los casos excepcionales en que se puede someter a un sujeto a un ensayo clínico sin obtener el consentimiento previo
El sujeto participante en un ensayo clínico, o su representante legal, podrán revocar su consentimiento en cualquier momento, sin expresión de causa y sin que por ello se derive para el sujeto participante responsabilidad ni perjuicio alguno.
El consentimiento se documentará mediante una hoja de información para el sujeto y el documento de consentimiento. La hoja de información contendrá únicamente información relevante, expresada en términos claros y comprensibles para los sujetos, y estará redactada en la lengua propia del sujeto.
Es un documento escrito, específico para cada ensayo clínico, que se entregará al posible participante antes de que éste otorgue su consentimiento para ser incluido en el mismo. Contendrá información referente a los siguientes aspectos del ensayo clínico: 1) Objetivo. 2) Metodología empleada 3) Tratamiento que puede serle administrado 4) Beneficios esperados para él y la sociedad 5) Incomodidades y riesgos derivados del estudio, número de visitas, pruebas complementarias a
que se someterá. 6) Posibles acontecimientos adversos. 7) Tratamientos alternativos disponibles. 8) Carácter voluntario de su participación, así como posibilidad de retirarse del estudio en
cualquier momento, sin que por ello se altere la relación médico-enfermo ni que produzca perjuicio para su tratamiento.

108
9) Personas que tendrán acceso a los datos del voluntario y forma en que se mantendrá la confidencialidad.
10) Modelo de compensación económica y tratamiento en caso de daño o lesión por su participación en el ensayo, tal como consta en la ley del medicamento.
11) Investigador responsable del ensayo y de informar al sujeto y contestar sus dudas y preguntas y de contactar con él en caso de urgencia.
j) El seguro o la garantía financiera previstos para el ensayo.
k) Las cantidades y, en su caso, previsiones de remuneración o compensación para los investigadores y sujetos del ensayo y los aspectos relevantes de cualquier acuerdo entre el promotor y el centro, que han de constar en el contrato previsto en el artículo 30.
l) El plan previsto para el reclutamiento de los sujetos.
Las cuestiones indicadas en los párrafos g), h) y k) del apartado anterior deberán ser evaluadas para cada uno de los centros implicados en el ensayo clínico. Y serán las únicas que evaluadas por los CEICs locales, resultarán vinculantes para el CEIC de referencia.
Además, y respecto a los postulados éticos, el CEIC comprobará los siguientes aspectos:
1. Que los beneficios esperados para el sujeto del ensayo y para la sociedad justifican los riesgos.
2. Que el ensayo se realice en condiciones de respeto a los derechos del sujeto y a los postulados éticos que afectan a la investigación biomédica con seres humanos
3. Que se disponga de suficientes datos científicos y, en particular, ensayos farmacológicos y toxicológicos en animales, que garanticen que los riesgos que implica en la persona en que se realiza son admisibles.
4. Que el estudio se base en los conocimientos disponibles, la información buscada suponga, presumiblemente, un avance en el conocimiento científico sobre el ser humano o para mejorar su estado de salud y su diseño minimice los riesgos para los sujetos participantes en él.
5. Que los riesgos e inconvenientes previsibles para los sujetos del ensayo se hayan ponderado con respecto a los beneficios previsibles para cada sujeto del ensayo y futuros pacientes.
6. Con el fin de garantizar una protección óptima de la salud y los derechos de los sujetos, no se podrán llevar a cabo investigaciones obsoletas o repetitivas.
7. Que el ensayo clínico esta diseñado para reducir al mínimo posible el dolor, la incomodidad, el miedo y cualquier otro riesgo previsible en relación con la enfermedad y edad o grado de desarrollo del sujeto ; tanto el umbral de riesgo como el grado de incomodidad deben ser definidos de forma específica y monitorizados durante el ensayo, especialmente cuando los sujetos del ensayo sean menores, adultos incapaces o constituyan una población especialmente vulnerable en razón de su situación económica, médica o social.
8. Que el tratamiento, comunicación y cesión de los datos de carácter personal de los sujetos participantes en el ensayo se ajustará a lo dispuesto en la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, y constará expresamente en el consentimiento informado.
9. Que la atención sanitaria que se dispense y las decisiones médicas que se adopten sobre los sujetos serán responsabilidad de un médico o de un odontólogo debidamente cualificados.
11. MECANISMOS DE TOMA DE DECISIONES Y CASOS EN QUE SE PUEDE HACER UNA REVISIÓN RAPIDA DE LA DOCUMENTACIÓN.
De conformidad con la Ley 30/92, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común, los acuerdos serán adoptados por mayoría simple.

109
Cuando un ensayo/estudio haya sido evaluado en reunión del CEIC y se acuerde su aprobación condicionada a la aclaración de cuestiones menores, se permitirá que la revisión de dichas aclaraciones, y consiguiente aprobación del ensayo, se realice en reunión de Presidente y Secretario. Si se dispone de las aclaraciones en formato electrónico, podrán enviarse por correo electrónico a todos los miembros del CEIC para que expresen su opinión en el plazo que se indique.
12. PROCEDIMIENTO PARA IDENTIFICAR EL PROTOCOLO Y GARANTIZAR QUE ES EL APROBADO POR LA DIRECCIÓN GENERAL DE FARMACIA Y MEDICAMENTOS. Identificación del ensayo: Cada ensayo clínico se deberá identificar mediante dos códigos únicos e invariables que figurarán en toda la documentación del ensayo: Código de protocolo del promotor: Código alfanumérico con una extensión mínima de 6 y máxima de 35, en el que no se tendrán en cuenta mayúsculas. Numero EudraCT: Se otorga de forma centralizada y tiene formato AAA-NNNNNN-CC, donde:
• AAAA es el año en el que se emite dicho número.
• NNNNNN es un número secuencial de seis dígitos
• CC es un número de control
Una vez aceptada la realización del ensayo, el Promotor y al Investigador enviarán una copia del certificado de aprobación de la AEMPS (en el caso de ensayos clínicos o las enmiendas) al CEIC. En el caso de estudios postautorización de tipo observacional prospectivo, una vez aprobado el estudio y previamente a su inicio, el promotor deberá notificar a los CEIC locales, como responsables del seguimiento, la autorización del mismo y remitir a la Dirección General de Farmacia y Productos Sanitarios la conformidad de las Gerencias implicadas y la fecha prevista de comienzo del estudio una vez formalizado el contrato con las Gerencias.
13. MODELO DE ACTA DE LAS DECISIONES DEL CEIC, CONFORME A LA NORMATIVA VIGENTE (ART 14.6 DEL R.D 223/2004). Conforme con el art. 27 de la Ley 30/1992, de 26 de noviembre, de Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común. Ver modelo de acta (anexo 9) Una vez aprobado por el Comité, se firmará el Modelo de Conformidad de la Dirección (anexo 1B ) y el contrato económico con la Dirección del Centro (anexo 18 ).
14. PROCEDIMIENTOS PARA REALIZAR EL SEGUIMIENTO DEL ENSAYO/ESTUDIO, DE INFORMACIÓN SOBRE REACCIONES ADVERSAS, Y SOLICITUD DE SUSPENSIÓN CAUTELAR.
Se solicitará del Investigador principal que presente informe sobre la evolución del ensayo/estudio post-autorización como mínimo anualmente o con mayor frecuencia si así se le requiere (ver anexos 14-16). Sin perjuicio de estos informes del investigador, el promotor deberá presentar informe final al terminar el estudio. 14.1 Solicitud de Suspensión Cautelar
El CEIC no posee la facultad de suspender un ensayo clínico en curso. Sólo puede proponer su suspensión cautelar, según lo previsto en el art 7.4 del la Resolución de 16 de julio de 2009, de la Consellería de Sanitat, de regulación de los procedimientos, documentación y plazos a observar en

110
la presentación y modificaciones en procesos relacionados con los ensayos clínicos y estudios post-autorización observacionales de medicamentos y productos sanitarios en la Comunitat Valenciana. Procedimiento de suspensión: según lo previsto en el art. 8 apartado h) del Decreto 73/2009, de 5 de junio, del Consell, y el articulo 7. de la Resolución de 16 de julio de 2009, de la Consellería de Sanitat. Contenido de la solicitud de suspensión : Deberá concurrir cualquiera de los supuestos previstos en el art. 26 del R.D. 223/2004:
a) Si se viola la Ley
b) Si se alteran las condiciones de su autorización
c) No se cumplan los principios éticos recogidos en el art. 60 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de medicamentos.
d) Para proteger a los sujetos del ensayo o,
e) En defensa de la salud pública Si el CEIC propone la suspensión o retirada del ensayo clínico, deberá argumentarlo en su solicitud y, basándose en ella, solicitar a la Consellería de Sanidad, a través del Coordinador del PECME, la suspensión cautelar del ensayo (Resolución de 16 de julio, de la Consellería de Sanitat).

111
15. ARCHIVO DEL CEIC: DOCUMENTACIÓN ARCHIVADA Y ACCESO A LA MISMA Y TIEMPO MÍNIMO DE ARCHIVO CON REFERENCIA EXPLICITA AL ORDEN SCO/256/2007
El archivo del CEIC tiene CARÁCTER CONFIDENCIAL y un ACCESO RESTRINGIDO En él se archivará la documentación detallada a continuación:
- El protocolo, las modificaciones y toda la documentación presentada por el promotor o su
representante legal. - Los dictámenes o informes emitidos por el Comité (Comité Ético de Investigación Clínica de
Referencia si el ensayo clínico es multicéntrico), especificando la versión de los documentos revisados.
- Copia de la correspondencia con el investigador y/o el promotor o su representante. - La documentación relativa a las actividades de seguimiento del ensayo clínico. - El informe anual sobre la marcha del ensayo clínico. - Copia de cualquier correspondencia con la Agencia Española de Medicamentos y Productos
Sanitarios y/o con las autoridades competentes de la Comunidad Autónoma donde este localizado el comité.
- Copia de las notificaciones y correspondencia relevantes con los Comités implicados y/o el Comité de referencia, de un ensayo multicéntrico.
- Las sospechas de reacciones adversas recibidas y los informes de seguridad presentados por el promotor.
- Notificación de la finalización del ensayo clínico, ya sea prematura o programada. - Resumen del informe final del ensayo clínico presentado por el promotor. - Resoluciones de acreditación y de posteriores modificaciones. - Currículum vitae de los miembros actuales o que hayan pertenecido al Comité. - Convocatoria y actas de las reuniones del Comité, con nombre de los participantes en las mismas,
conclusiones a las que se han llegado y resultados de las votaciones efectuadas. El acta reflejará explícitamente, que para cada estudio evaluado se han ponderado los aspectos del art. 17 del R.D. 223/2004
- Procedimientos normalizados de trabajo del Comité, versión actual y archivo histórico. - Cualquier otra documentación relevante. Los documentos se conservarán durante un mínimo de 3 AÑOS, después de la comunicación de la finalización del Ensayo, o durante un periodo más largo si así lo establece la Agencia Española de Medicamentos y Productos Sanitarios o la autoridad competente de la comunidad autónoma correspondiente. Se destruirán previa notificación al Comité

112
16. PROCEDIMIENTO DE IDENTIFICACIÓN DEL INVESTIGADOR PRINCIPAL Y COLABORADORES DE CADA ENSAYO CLÍNICO, ESPECIFICANDO QUIENES PUEDEN PARTICIPAR EN EL MISMO, SOLICITAR LA MEDICACIÓN A FARMACIA Y ADMINISTRAR LOS MEDICAMENTOS EN ESTUDIO.
1. Los datos relativos de los intervinientes serán los siguientes :
1. Nombre, dirección, teléfono y fax del promotor. En el caso de que el promotor esté ubicado fuera de España, los mismos datos del responsable autorizado en España.
2. Nombre, dirección, teléfono y fax del investigador principal y colaboradores, incluyendo su lugar de trabajo en cada Centro.
3. Director Técnico responsable de la elaboración / control de las muestras Nombre : Tfno. : Firma :
4. Identificación del monitor
5. Código de laboratorio (Nº D.G. Atención Primaria y Farmacia)
2. Sobre la idoneidad del equipo investigador para el ensayo propuesto
Ver apartado 10.g) de los Procedimientos Normalizados de Trabajo
3. Sobre quienes pueden solicitar la medicación a farmacia y administrar los medicamentos en estudio.
Peticiones de Medicamentos / Productos Sanitarios al Servicio de Farmacia. Con el fin de que los medicamentos utilizados en los protocolos de los ensayos Clínicos aprobados por el CEIC del Consorcio Hospitalario Provincial de Castellón, lleguen de manera adecuada a manos del investigador y con el control exigido por la normativa vigente: El Servicio de Farmacia entregará a cada grupo de trabajo y en el momento de la recepción de la medicación, el modelo de la “Hoja de Dispensación de Medicamentos / Productos Sanitarios sometidos a Ensayos Clínicos”, que deberán de rellenar y firmar, en todos y cada uno de los casos en que se vaya a hacer uso de los mismos. Por tanto, es necesario que los Investigadores Principales de los Ensayos Clínicos comuniquen al Servicio de Farmacia, el nombre y la firma de los colaboradores autorizados para la solicitud y dispensación de la medicación correspondiente. 17. OBLIGACIONES DE LOS INVESTIGADORES, HACIENDO ESPECIAL REFERENCIA A LOS ARTICULOS 37 Y 42 DEL REAL DECRETO 223/2004
16.1 OBLIGACIONES DEL INVESTIGADOR CONFORME R.D.223/2004 Y NORMAS DE B.P.C. DE LA U.E.
a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.

113
b) Conocer a fondo las propiedades de los medicamentos en investigación.
c) Garantizar que el consentimiento informado se recoge de conformidad a lo establecido en este real decreto.
d) Recoger, registrar y notificar los datos de forma correcta y garantizar su veracidad.
e) Notificar inmediatamente los acontecimientos adversos graves o inesperados al promotor.
f) Garantizar que todas las personas implicadas respetarán la confidencialidad de cualquier información acerca de los sujetos del ensayo, así como la protección de sus datos de carácter personal.
g) Informar regularmente al Comité Ético de Investigación Clínica de la marcha del ensayo.
h) Corresponsabilizarse con el promotor de la elaboración del informe final del ensayo, dando su acuerdo con su firma.
El investigador comunicará inmediatamente al promotor todos los acontecimientos adversos graves, salvo cuando se trate de los señalados en el protocolo o en el manual del investigador como acontecimientos que no requieran comunicación inmediata. La comunicación inicial irá seguida de comunicaciones escritas pormenorizadas. En las comunicaciones iniciales y en las de seguimiento se identificará a los sujetos del ensayo mediante un número de código específico para cada uno de ellos.
En caso de que se haya comunicado un fallecimiento de un sujeto participante en un ensayo clínico, el investigador proporcionará al promotor y a los Comités Éticos de Investigación Clínica implicados toda la información complementaria que le soliciten.

114
17.2 OBLIGACIONES EN CUANTO AL ARCHIVO DEL INVESTIGADOR
El investigador mantendrá la CONFIDENCIALIDAD de este archivo 1. Póliza de seguro 2. Acuerdos firmados entre las partes implicadas 3. Instrucciones para la apertura del ciego 4. Autorización AEM 5. Aprobación CEIC – referencia/composición 6. Currículum Vitae del Investigador Principal 7. Manual Investigado/ficha técnica 8. Protocolos/enmiendas firmados y CRD 9. Hoja de Información al Paciente 10. Valores normales/certificados calidad 11. Medicamento en investigación: manejo. Registro, envios. 12. Hoja de firmas 13. Comunicaciones promotor/monitor/investigador 14. Consentimiento informados firmados 15. Notificación SAEs: Investigador – promotor – AEM y CEIC 16. Informes intermedios/anual 17. Registro selección sujetos 18. Lista códigos de identificación de sujetos 19. Registro de inclusión 20. Registro de muestras biológicas 21. Registro contabilización del medicamento en investigación 22. Registro de destrucción del medicamento en investigación 23. Informe final del investigador al CEIC 24. Informe final ensayo

115
18. RECEPCIÓN, IDENTIFICACIÓN, ALMACENAMIENTO, MANIPULACIÓN, DISTRIBUCIÓN Y DEVOLUCIÓN DE LAS MUESTRAS PARA INVESTIGACIÓN CLÍNICA Y FINANCIACIÓN DE LAS MISMAS.
1. Las muestras de medicamentos o productos en fase de investigación clínica para utilización de ensayos clínicos serán proporcionadas gratuitamente por el promotor. Todas las muestras sobrantes serán devueltas al promotor una vez finalizado el período de tratamiento del ensayo clínico. En situaciones especiales podrán autorizarse ensayos en los que se contemplen otras vías de suministro, constando en el acta en que forma se procederá a su financiación. 2. El Director Técnico responsable de las muestras de un ensayo clínico garantizará la fabricación y adecuada calidad de las mismas según las normas de correcta fabricación. En caso de que las muestras sean productos de importación avalará la calidad de las mismas, debiendo para ello adoptar las comprobaciones y controles adecuados. Asimismo remitirá a las autoridades competentes muestras de los productos que serán utilizados en el ensayo clínico cuando le sean solicitadas. 3. IDENTIFICACIÓN. Las muestras para un ensayo clínico irán envasadas y acondicionadas convenientemente. Su etiquetado o rotulación permitirá, en cualquier momento, su perfecta identificación. En la etiqueta constarán, como mínimo, los siguientes datos :
a) Código del protocolo.
b) Número de unidades y forma galénica.
c) Vía de administración
d) Nombre y dirección de la entidad farmacéutica responsable.
e) Nombre del Director Técnico responsable
f) Número de lote.
g) Fecha de caducidad, si la hubiera
h) Condiciones especiales de conservación, si las hubiera.
i) La inscripción “Muestra para investigación clínica”. En los ensayos de carácter doble ciego, el número de lote, el nombre y dirección de la entidad farmacéutica elaboradora y el nombre del técnico responsable de las muestras no se incluirán en la etiqueta, sino en el documento que contenga la identificación del tratamiento, con el fin de no romper la igualdad entre las muestras. Con este mismo fin, cuando difieran las fechas de caducidad o las condiciones de conservación de los productos en comparación, figurará en las etiquetas la más restrictiva de ellas.

116
4. DISTRIBUCIÓN. La distribución al investigador de las muestras para el ensayo se realizará a través del servicio de farmacia del Centro donde se realice la investigación. Dichos servicios acusarán recibo por escrito de la entrega de los productos y se responsabilizarán de su correcta conservación y dispensación; asimismo controlarán la medicación sobrante al final del ensayo. S el ensayo se realiza en el medio extrahospitalario, las obligaciones fijadas en este punto serán asumidas por los servicios farmacéuticos de las estructuras de atención primaria o, en caso de no existir, por los servicios de farmacia de los hospitales de referencia y, de forma extraordinaria, por el investigador principal del ensayo. 5. El promotor conservará en el archivo principal del ensayo los protocolos de fabricación y control de los lotes de productos fabricados para el ensayo clínico. Asimismo, las muestras de cada lote se conservarán hasta doce meses después de la fecha de caducidad. 6. Peticiones de Medicamentos / Productos Sanitarios al Servicio de Farmacia. Con el fin de que los medicamentos utilizados en los protocolos de los ensayos Clínicos aprobados por el CEIC del Consorcio Hospitalario Provincial de Castellón, lleguen de manera adecuada a manos del investigador y con el control exigido por la normativa vigente, el Servicio de Farmacia entregará a cada grupo de trabajo y en el momento de la recepción de la medicación, el modelo de la “Hoja de Dispensación de Medicamentos / Productos Sanitarios sometidos a Ensayos Clínicos”, que deberán de rellenar y firmar, en todos y cada uno de los casos en que se vaya a hacer uso de los mismos. Por tanto es necesario que los Investigadores Principales de los Ensayos Clínicos comuniquen al Servicio de Farmacia, el nombre y la firma de los colaboradores autorizados para la solicitud y dispensación de la medicación correspondiente. 19. REVISIÓN/MODIFICACIÓN DE LOS PROCEDIMIENTOS NORMALIZADOS DE TRABAJO (PNT) Los Procedimientos Normalizados de Trabajo (PNTs) incorporarán automáticamente todas las exigencias legales que les afecten. De forma rutinaria se revisarán los Procedimientos Normalizados de Trabajo: - cada 3 años - cuando cambie de forma significativa la composición del CEIC y/o la normativa. - Siempre que se solicite por parte de alguno de los miembros, presentando propuestas concretas a los apartados a revisar.


















