
Procesos de Extracion de Metales
148
CUSCO, 04 DE FEBRERO 2013 PROCESOS DE EXTRACCCION DE METALES UNIDAD DIDACTICA 1. Duración una semana 1.1. PLANTAS EXTRACTIVAS EN EL PERU. 1.2. OBJETIVOS ESPECÍFICOS Saber la Ubicación y Producción de los centros metalúrgicos del Perú y su importancia económica. Conocer el diagrama de flujo de cada planta 1.3 CONTENIDOS: 1.1. Introducción – La Metalurgia Extractiva METAL Según la química inorgánica, metal es un cuerpo simple que tiene tendencia a ceder electrones de la órbita periférica transformándose en un ion positivo, llamado catión y como tal suele encontrarse en sus compuestos. Los metales son dúctiles, maleables y buenos conductores de calor y electricidad, tienen un brillo característico denominado brillo metálico. Con excepción del mercurio a temperatura ambiente son sólidos y forman óxidos básicos, con poca o nula afinidad por el hidrógeno y mayor afinidad por el oxígeno y el azufre, con los que forman los minerales, fuente de la extracción de metales desde la naturaleza. Los metales se clasifican en: 1. Ligeros: a) Alcalinos: Sodio, potasio b) Alcalinotérreos: Calcio, magnesio c) Térreos: Aluminio 2. Pesados: a) Grupo del cobre: Cobre, plata, oro b) Grupo del Cinc: Cinc, mercurio c) Grupo del estaño: Estaño, plomo d) Grupo del Hierro e) Grupo del Platino Raramente se encuentran puros al estado natural, en general se hallan combinados con el oxígeno y otros no metálicos como el azufre, formando los óxidos metálicos y sulfuros
description
AREA DE PROCESOS DE COBRE
Transcript of Procesos de Extracion de Metales

CUSCO, 04 DE FEBRERO 2013
PROCESOS DE EXTRACCCION DE METALES
UNIDAD DIDACTICA 1. Duración una semana 1.1. PLANTAS EXTRACTIVAS EN EL PERU. 1.2. OBJETIVOS ESPECÍFICOS
Saber la Ubicación y Producción de los centros metalúrgicos del Perú y su importancia económica.
Conocer el diagrama de flujo de cada planta
1.3 CONTENIDOS:
1.1. Introducción – La Metalurgia Extractiva
METAL
Según la química inorgánica, metal es un cuerpo simple que tiene tendencia a ceder
electrones de la órbita periférica transformándose en un ion positivo, llamado catión y
como tal suele encontrarse en sus compuestos. Los metales son dúctiles, maleables y
buenos conductores de calor y electricidad, tienen un brillo característico denominado
brillo metálico. Con excepción del mercurio a temperatura ambiente son sólidos y forman
óxidos básicos, con poca o nula afinidad por el hidrógeno y mayor afinidad por el oxígeno y
el azufre, con los que forman los minerales, fuente de la extracción de metales desde la
naturaleza.
Los metales se clasifican en:
1. Ligeros:
a) Alcalinos: Sodio, potasio
b) Alcalinotérreos: Calcio, magnesio
c) Térreos: Aluminio
2. Pesados:
a) Grupo del cobre: Cobre, plata, oro
b) Grupo del Cinc: Cinc, mercurio
c) Grupo del estaño: Estaño, plomo
d) Grupo del Hierro
e) Grupo del Platino
Raramente se encuentran puros al estado natural, en general se hallan combinados con el
oxígeno y otros no metálicos como el azufre, formando los óxidos metálicos y sulfuros

respectivamente. El metal más difundido en la corteza terrestre es el aluminio, seguido por
el hierro, calcio, sodio, potasio y magnesio, los demás alcanzan proporciones mínimas.
MINERAL
Los MINERALES son sustancias naturales que se originan en la capa rocosa de la Tierra o
litósfera, y se caracterizan por tener una estructura homogénea y una composición química
bien definida.
El conjunto de los minerales es estudiado por la MINERALOGÍA, que es la ciencia encargada
de examinar las características físicas, químicas, morfológicas y estructurales de dichas
sustancias.
El Perú es uno de los principales países mineros del mundo y, como tal, alberga en sus
suelos numerosos minerales preciosos y semipreciosos, muchos de ellos aún desconocidos
pero de gran valor.
Los minerales que constituyen la corteza terrestre se han formado a partir de los elementos químicos que originaron el planeta, gracias a reacciones ocurridas en su interior. Por este motivo, la cantidad de combinaciones es inmensa. Para poner un poco de orden, se clasifican los minerales atendiendo a la forma en que se originan, a sus características cristalográficas, a su composición química, ... Mención aparte merecen los cristales y, entre ellos, los llamados "piedras preciosas" que siempre han cautivado a la humanidad.
Clasificación química
La clasificación química divide los minerales en grupos según sus compuestos químicos. Cualquier mineral conocido puede ser integrado dentro de estos grupos, pues la práctica totalidad de ellos incluyen alguno de estos compuestos.
1.- Elementos nativos: son los que se encuentran en la naturaleza en estado libre, puro o nativo, sin combinar o formar compuestos químicos. Ejemplos: oro, plata, azufre, diamante.
2.- Sulfuros: compuestos de diversos minerales combinados con el azufre. Ejemplos: pirita, galena, blenda, cinabrio.
3.- Sulfosales: minerales compuestos de plomo, plata y cobre combinados con azufre y algún otro mineral como el arsénico, bismuto o antimonio. Ejemplos: pirargirita, proustita.
4.- Óxidos: producto de la combinación del oxígeno con un elemento. Ejemplos: oligisto, corindón, casiterita, bauxita.

5.- Haluros: compuestos de un halógeno con otro elemento, como el cloro, flúor, yodo o bromo. Ejemplos: sal común, halita.
6.- Carbonatos: sales derivadas de la combinación del ácido carbónico y un metal. Ejemplos: calcita, azurita, mármol, malaquita.
7.- Nitratos: sales derivadas del ácido nítrico. Ejemplos: nitrato sódico (o de Chile), salitre o nitrato potásico.
8.- Boratos: constituidos por sales minerales o ésteres del ácido bórico. Ejemplos: borax, rasorita. 9.- Fosfatos, arseniatos y vanadatos: sales o ésteres del ácido fosfórico, arsénico y vanadio. Ejemplos: apatita, turquesa, piromorfita.
10.- Sulfatos: sales o ésteres del ácido sulfúrico. Ejemplos: yeso, anhidrita, barita.
11.- Cromatos, volframatos y molibdatos: compuestos de cromo, molibeno o wolframio. Ejemplos: wolframita, crocoita.
12.- Silicatos: sales de ácido silícico, los compuestos fundamentales de la litosfera, formando el 95% de la corteza terrestre. Ejemplos: sílice, feldespato, mica, cuarzo, piroxeno, talco, arcilla.
13.- Minerales radioactivos: compuestos de elementos emisores de radiación. Ejemplos: uraninita, torianita, torita.
METALURGIA EXTRACTIVA Los metales en la naturaleza se encuentran formando compuestos inorgánicos denominados minerales, por que las condiciones termodinámicas del planeta tierra no son favorables para la presencia de éstos al estado libre, sin embargo, los metales nobles como el oro, platino, plata y excepcionalmente el cobre, se pueden encontrar en estado metálico, por lo que para separar los metales desde sus minerales se recurre al uso de la metalurgia extractiva, entendida como aquellos procesos y operaciones aplicadas para la obtención del metal con distintos grados de pureza y la obtención de los concentrados de minerales, lo dejaremos al campo de la mineralurgia. La metalurgia extractiva es arte, ciencia y tecnología empleado para la obtención de los metales desde sus minerales y/o concentrados aplicando procesos pirometalúrgicos, hidrometalúrgicos y electrometalúrgicos o una combinación de éstos, según la naturaleza del mineral y/o concentrado Los procesos de metalurgia extractiva se han desarrollado para producir metales refinados de alta pureza, particularmente en los no ferrosos. En los últimos años se ha logrado un desarrollo rápido en el diseño, control y eficiencia en los procesos de extracción y

refinación de los metales, debido fundamentalmente, al conocimiento de la termodinámica y la cinética de los procesos extractivos experimentalmente obtenidos. Los siguientes diagramas de flujo, ilustran los procesos de extracción de metales desde sus minerales para algunos metales no ferrosos más importantes.
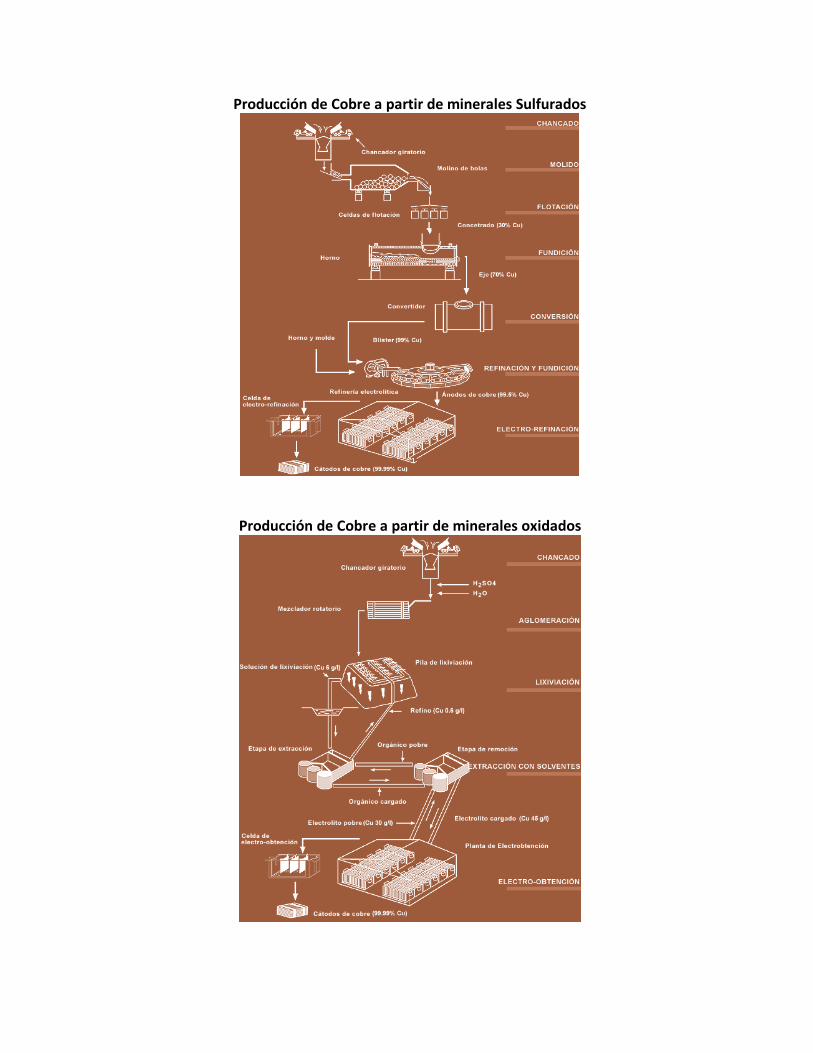
Producción de Cobre a partir de minerales Sulfurados
Producción de Cobre a partir de minerales oxidados

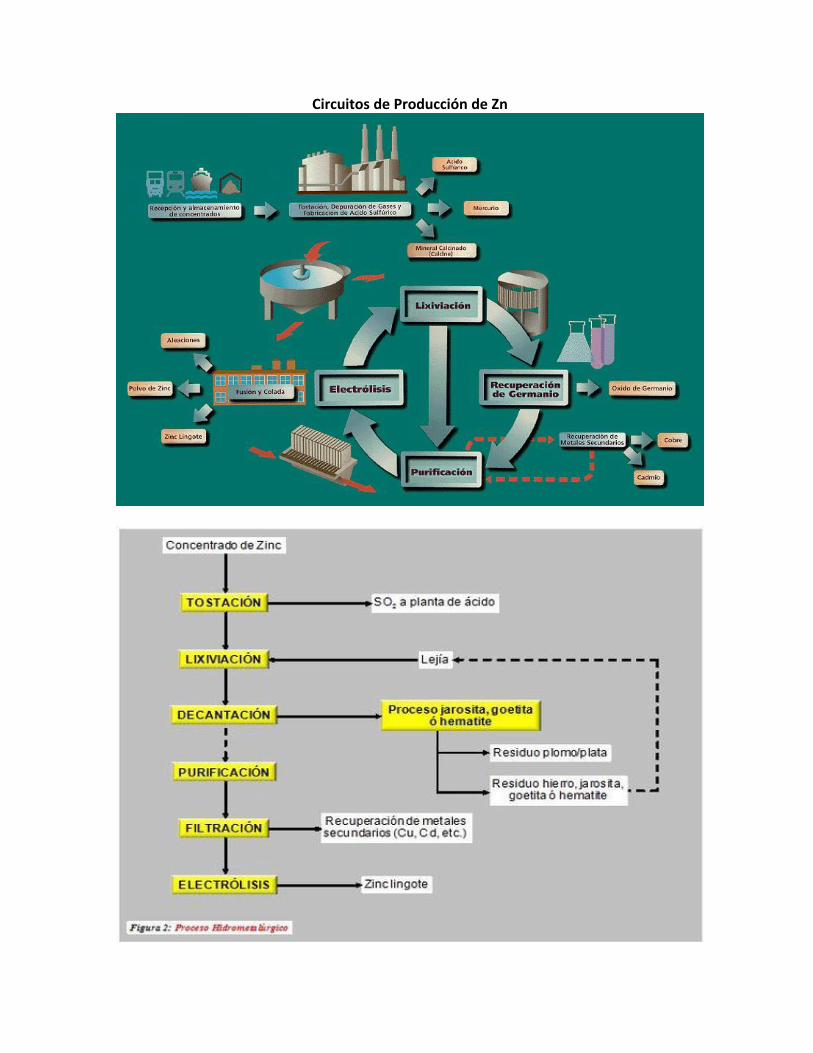
Circuito de Producción de Zn y Pb
1.2. Plantas de Extracción de Metales en el Perú. Las principales plantas de extracción de metales en el Perú son:
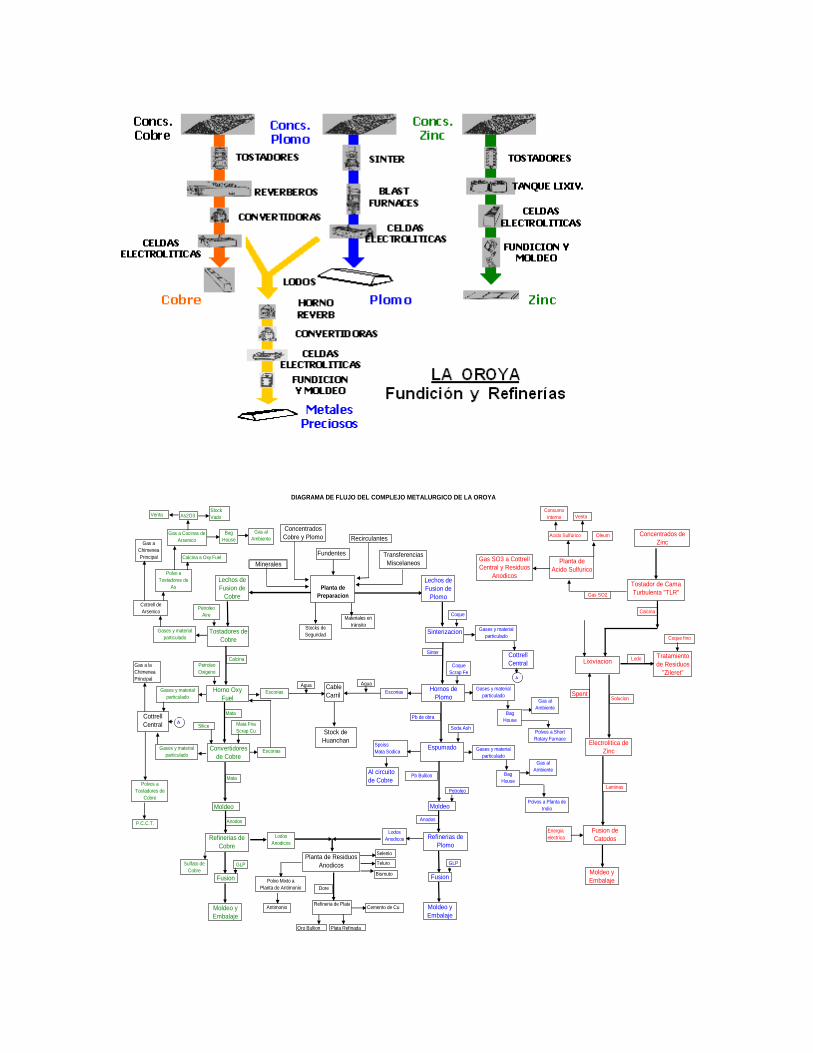
• El Complejo Metalúrgico de la Oroya, que produce: Zinc, Plomo, Cobre, Plata, Oro, Bismuto, Cadmio, Indio, Telurio, Antimonio, Selenio y como sub productos Polvo de Zinc, Acido Sulfúrico, Oleum, Trióxido de Arsénico, Sulfato de Cobre, Sulfato de Zinc, Concentrado de Zinc – Plata, Oxido de Zinc y Bisulfito de Sodio
• La Fundición y Refinería de Ilo, que produce cobre y del tratamiento de lodos anódicos obtiene Oro, Plata, Selenio, telurio, etc.
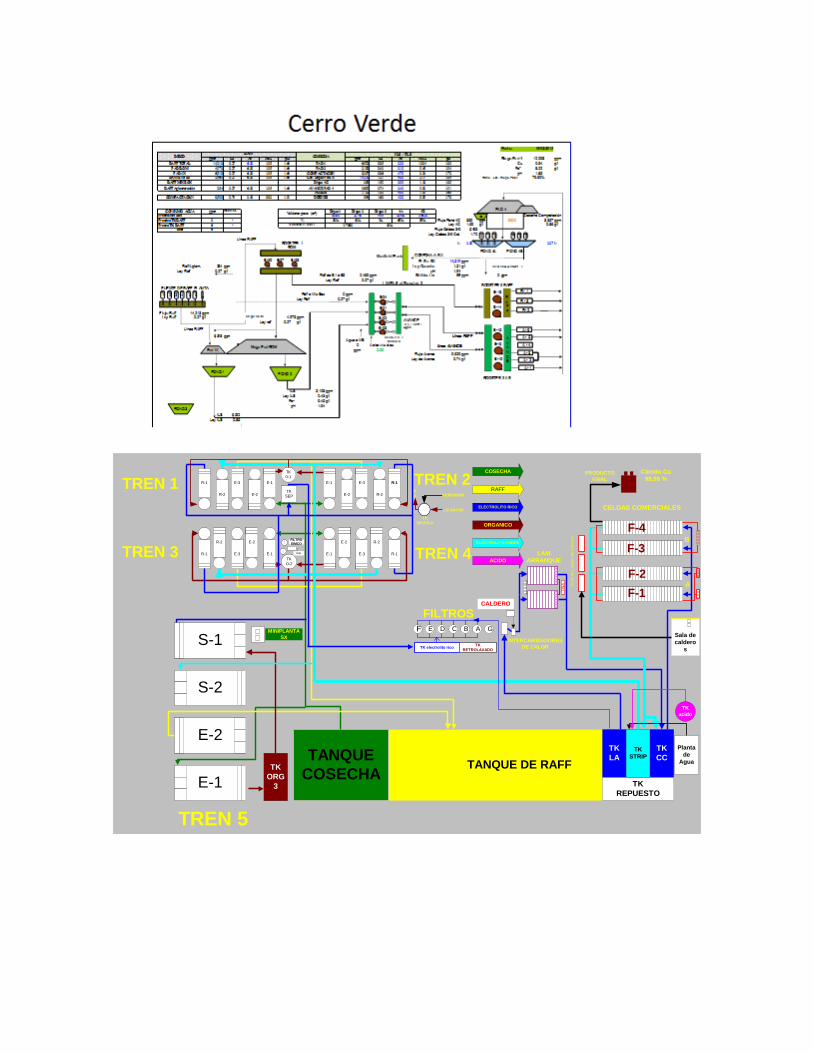
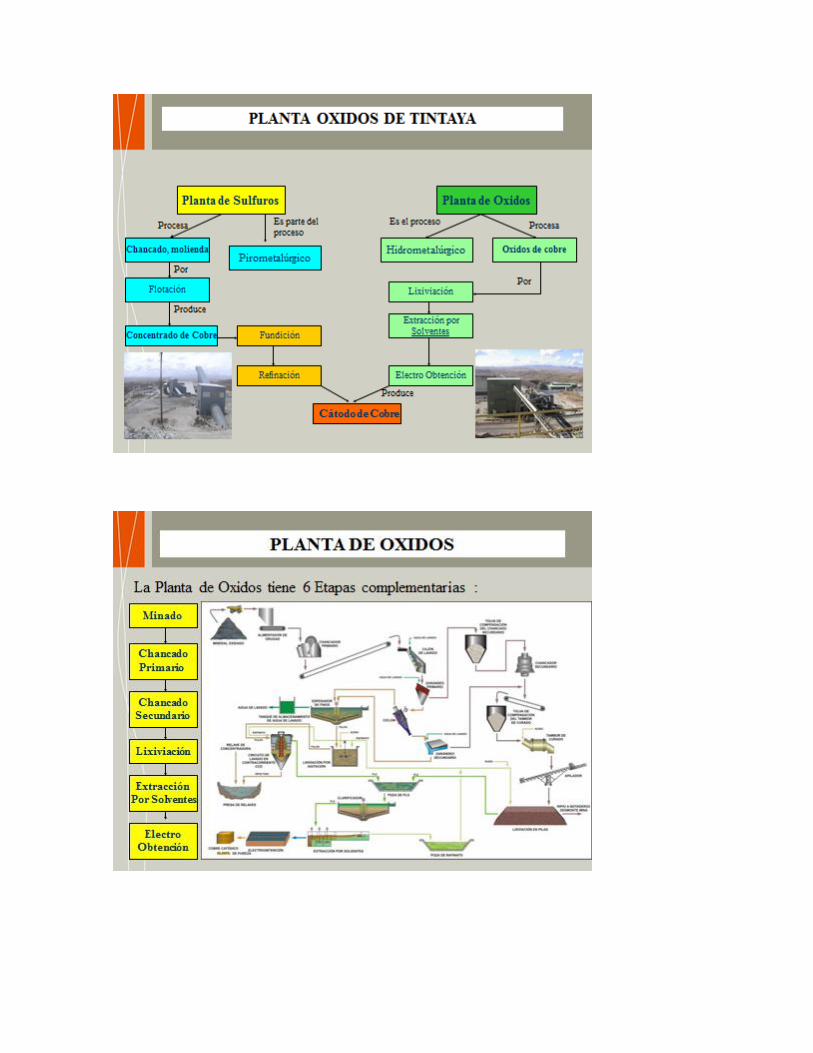
• Refinería de Cajamarquilla, produce Zinc y Cadmio principalmente • Cia. Minera Cerro Verde, produce cobre catódico a partir de óxidos de cobre • Xstrata Tintaya, produce cátodos de cobre a partir de óxidos de cobre • Southern Perú – Toquepala, produce cátodos de cobre a partir de los óxidos de
cobre de Toquepala y Cuajone • Yanacocha – Cajamarca, produce oro • Pierina - Ancash, produce oro • Existen muchas plantas de producción de oro y cobre a escala de mediana y
pequeña minería
1.3. El Complejo Metalúrgico de la Oroya – Circuito de Cobre, Plomo y Zinc.
Minerales
DIAGRAMA DE FLUJO DEL COMPLEJO METALURGICO DE LA OROYA
Gases y material
particuladoGases y material
particulado
Gases y material
particulado
Gases y material
particulado
Gases y material
particulado
Gases y material
particulado
EscoriasEscorias
Escorias
Lodos
AnodicosLodos
Anodicos
Calcina
Sinter
MataPb de obra
MataPb Bullion
Anodos Anodos
Cottrell
Central
Cottrell
Central
Cottrell de
Arsenico
Planta de Residuos
Anodicos
Stock de
HuanchanSpeiss
Mata Sodica
Bag
House
Bag
House
Gas al
Ambiente
Gas al
Ambiente
Polvos a Planta de
Indio
Polvos a Short
Rotary Furnace
Polvos a
Tostadores de
Cobre
P.C.C.T.
Gas a la
Chimenea
Principal
Polvo a
Tostadores de
As
Gas a
Chimenea
Principal
Concentrados
Cobre y Plomo Recirculantes
Planta de
Preparacion
Fundentes
Stocks de
Seguridad
Gas a Cocinas de
Arsenico
Calcina a Oxy Fuel
As2O3
Bag
House
VentaStock
Vado
Gas al
Ambiente
Cable
CarrilHorno Oxy
Fuel
Convertidores
de Cobre
Refinerias de
Cobre
Tostadores de
Cobre
Fusion Fusion
Moldeo y
EmbalajeMoldeo y
Embalaje
Refinerias de
Plomo
Espumado
Hornos de
Plomo
Lechos de
Fusion de
Cobre
Lechos de
Fusion de
Plomo
Sinterizacion
Silice
Petroleo
Oxigeno
Petroleo
Aire Coque
Coque
Scrap Fe
Soda Ash
Petroleo
Mata Fria
Scrap Cu
AguaAgua
A
A
Al circuito
de Cobre
Refineria de Plata
Dore
GLP GLP
MoldeoMoldeo
Transferencias
Miscelaneos
Concentrados de
Zinc
Tostador de Cama
Turbulenta "TLR"
Lixiviacion
Calcina
Electrolitica de
Zinc
Fusion de
Catodos
Planta de
Acido Sulfurico
Acido Sulfúrico
Venta
Consumo
interno
Gas SO3 a Cottrell
Central y Residuos
Anodicos
Spent
Moldeo y
Embalaje
Tratamiento
de Residuos
"Zileret"
Solucion
Gas SO2
Oro Bullion Plata Refinada
Selenio
Teluro
Bismuto
Oleum
Sulfato de
Cobre
Lodo
Coque fino
Laminas
Energia
electrica
Materiales en
tránsito
Cemento de Cu
Polvo Mixto a
Planta de Antimonio
Antimonio
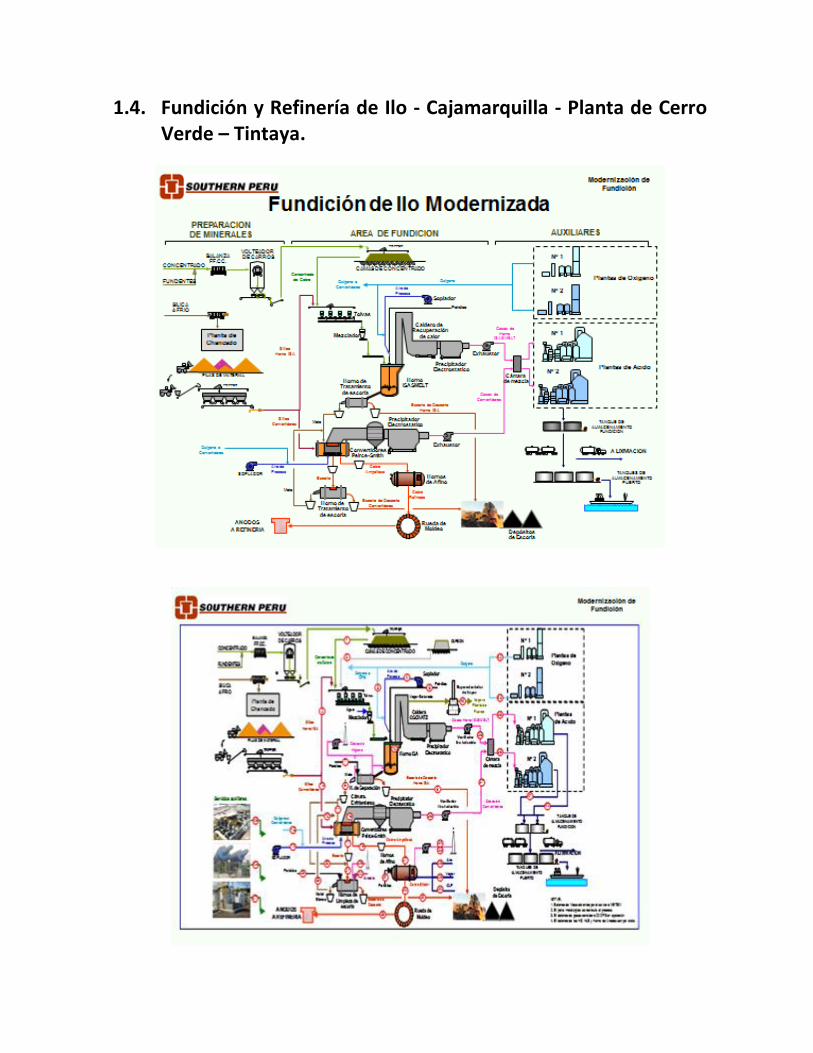
1.4. Fundición y Refinería de Ilo - Cajamarquilla - Planta de Cerro Verde – Tintaya.
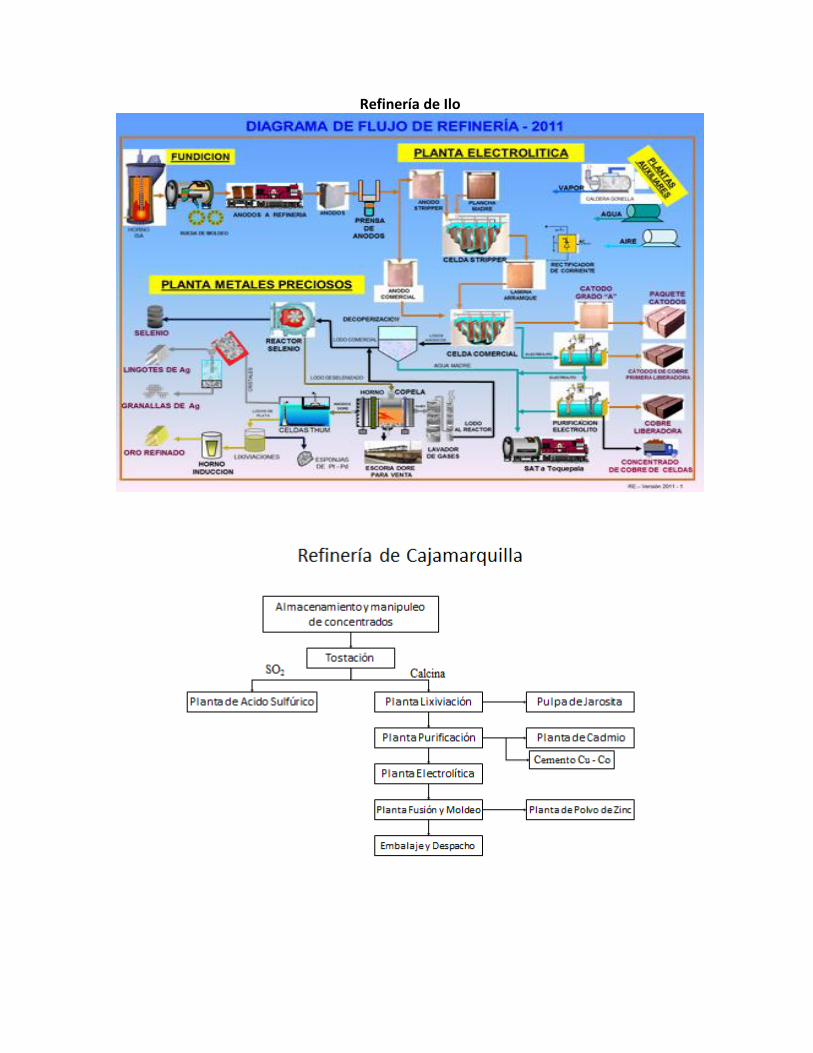
Refinería de Ilo
R-1
R-2
E-3
E-2
E-1 E-1
E-2
E-3
R-2
R-1
R-1
R-2
E-3
E-2
E-1 E-1
E-2
E-3
R-2
R-1
TANQUE DE RAFFTANQUE
COSECHATK
REPUESTO
TK
LATK
STRIP
TK
CC
S-1
S-2
E-2
E-1
TK
0-1
TK
SEP
TK
O-2
F E D C B A G
TK
ORG
3
CELDAS COMERCIALES
B
A
F-1
F-2
F-3
F-4
LAM.
ARRANQUE
TREN 5
TREN 1
FILTROS
INTERCAMBIADORES
DE CALOR
TREN 2
TREN 4
COSECHA
T
K
CALDERO
FILTRO
EIMCO
MINIPLANTA
SX
Planta
de
Agua
TK
acido
TK electrolito rico
Sala de
caldero
s
Tin
as d
e L
ava
do
de
Cá
tod
os
T
K
lodo
Cátodo Cu
99.99 %PRODUCTO
FINAL
TIN
A
RAFF
ELECTROLITO RICO
ORGANICO
ELECTROLITO POBRE
ACIDO
LIX 984 NC
KEROSENE
TK
MEZCLA
R
2
TREN 3
TK
RETROLAVADO

1.5. Plantas de cianuración de oro
UNIDAD DIDACTICA 2. Duración tres semanas
2.1. TERMODINAMICA DE LA EXTRACCIÓN DE METALES 2.2. OBJETIVOS ESPECÍFICOS
Saber la termodinámica aplicada a los procesos de extracción y purificación de metales.
Realizar cálculos Termodinámicos, Balance de materiales y Energía. 2.3. CONTENIDOS:
2.1. Introducción – estabilidad de los minerales. Los metales se encuentran en la corteza terrestre en una variedad de combinaciones químicas debido a que la atmosfera terrestre contiene oxígeno, bióxido de carbono y vapor de agua. Inicialmente la corteza estaba compuesto de sulfuros y silicatos metálicos, denominados, minerales primarios, pero debido a la interacción con la atmósfera, la mayoría de los metales se encuentran formando óxidos, sulfuros o silicatos. Existen también sulfatos y carbonatos como productos secundarios, así como los metales nativos, cuando la energía de enlace es muy pequeña en las condiciones imperantes.

El oro y los metales del grupo del platino se encuentran al estado nativo, junto a minerales de cobre, plata y mercurio El plomo se encuentra como sulfuros, sulfatos y carbonatos y el hierro como sulfuros y óxidos Los metales reactivos como el Al y el Si se encuentran formando depósitos grandes de óxidos o arcillas complejos de Al – Si Los metales alcalinos y muchos otros forman cloruros solubles que en parte son transportados al mar. Con algunas excepciones, como los depósitos de minerales de hierro, caliza y cuarzo, los minerales están diseminados en las rocas o gangas y están mesclados con metalíferos y otros compuestos que contienen valores o impurezas, por lo que un deposito de un mineral depende de muchos factores Así, un depósito comercial de mineral de hierro debe contener un 65% de Fe, con bajo contenido de S y P, los yacimientos de Cu y Ni una ley de 1% y los yacimientos de oro son trabajados con una ley de 8 g/ton o 8ppm. El proceso usado para la producción de un metal depende de la forma en que se encuentra como mineral y de las propiedades químicas y físicas del metal y sus compuestos, en tanto que éstos sean utilizados para lograr la máxima pureza del producto con el mínimo consumo de energía, trabajo y reactivos y la mínima generación de contaminantes sólidos, líquidos y gaseosos. Generalmente una primera etapa en el tratamiento de los minerales es la concentración por flotación, gravimetría o separación magnética, seguido por procesos de piro, hidro y//o electrometalúrgicos También se usan procesos directos como la cianuración para el oro, lixiviación ácida para la extracción del cobre, el uranio, etc, seguida de procesos de precipitación del metal o compuesto de valor por medios químicos o electroquímicos.
Estabilidad de los minerales Antiguamente se asumía que el calor de formación de un compuesto era el indicador de su estabilidad, de modo que a mayor valor del calor de formación se requería mayor energía para disociar dicho compuesto, este concepto no consideraba la influencia de la contribución de la entropía en el cambio de la energía disponible en la reacción de formación Tomando en cuenta la entropía, se ha demostrado que la afinidad o Energía Libre de Formación de Gibbs es un indicador seguro de la estabilidad de un compuesto y no se ha encontrado ninguna excepción. La Energía Libre de Gibbs (∆Go ) está expresada en función de la temperatura y la constante de equilibrio (ke) en condiciones estándar, según la ecuación: ∆Go = RT log ke . Para situaciones no estándar se aplican las correcciones. El estudio de la estabilidad de los compuestos hace posible la predicción del estado en el que es más probable que un metal pueda existir en la naturaleza, así como los principios a ser usados en el proceso más eficiente para la producción de un metal puro.

2.3. Afinidad de los metales por el oxígeno. La afinidad de los metales por el oxígeno es variado, los hay metales con poca afinidad y como el Au, Ag y Hg, con mediana afinidad como el Cu, Pb, Zn, otros con mayor afinidad como el Fe, Mn, P, y aquellos que son extremadamente afines como el Zr, Al, Ca, Mg, etc. Según la afinidad, los metales forman óxidos poco estables, estables y muy estables que es expresado a través de la variación de la energía libre de formación en función de la temperatura:
Para la reacción: M + O2 = MO
2
∆Go = RT log k
e ( condiciones estándar)
∆GT = ∆G
o – RT ln p
O2 ( Condición no estándar)
∆Go
T = ∆H
o
T - T∆S
o
T
La variación se representa gráficamente en el Diagrama de Ellingham-Richardson y Jeffes y es muy usado para la extracción de los metales desde sus óxidos. Según su afinidad, metales como el oro, tanto a condiciones estándar y atmosférica, se
encuentra al estado metálico, los óxidos de los metales como el Bi, Hg y Ag se
descomponen fácilmente por debajo de los 500oC, otros como los óxidos de Cu, Pb y Fe son
reducibles con C y los óxidos muy estables como los de Al, Ti y Ca requieren procesos
especiales para la producción del metal.
La afinidad de los metales por el oxígeno disminuye con el aumento de la temperatura, con
excepción del C que se mantiene constante.
Diagrama de Ellingham para Oxido

Diagrama de Ellingham para Oxidos

2.4. Afinidad de los metales por el azufre. El diagrama de Ellingham para los sulfuros muestra que la afinidad del azufre por los metales disminuye con el aumento de la temperatura, en la mayoría de los casos, debido a que hay una significativa disminución de la entropía cuando se forma el sulfuro. La alta afinidad del Mn, los metales alcalinos, alcalinotérreos y de metales como el Ce por el azufre es de gran importancia para eliminar el azufre del hierro y el acero, sin embargo la afinidad de los metales por el azufre es menor que por el oxígeno, con excepción de los metales alcalinos. Esta relativa diferencia de afinidad de los metales por el S y el O
2 es muy utilizado en los
proceso extractivos, principalmente pirometalúrgicos, como la tostación, sinterización,
fusión, conversión y afino de los metales, por reacciones como:
2MS + O2 = 2MO + S
2
2MS + 3O2 = 2MO + SO
2
Ejemplo:
Cu2O + FeS = Cu
2S + FeO
El Cu tiene mayor afinidad por el S y el Fe por el O2
Diagrama de Ellingham para Sulfuros
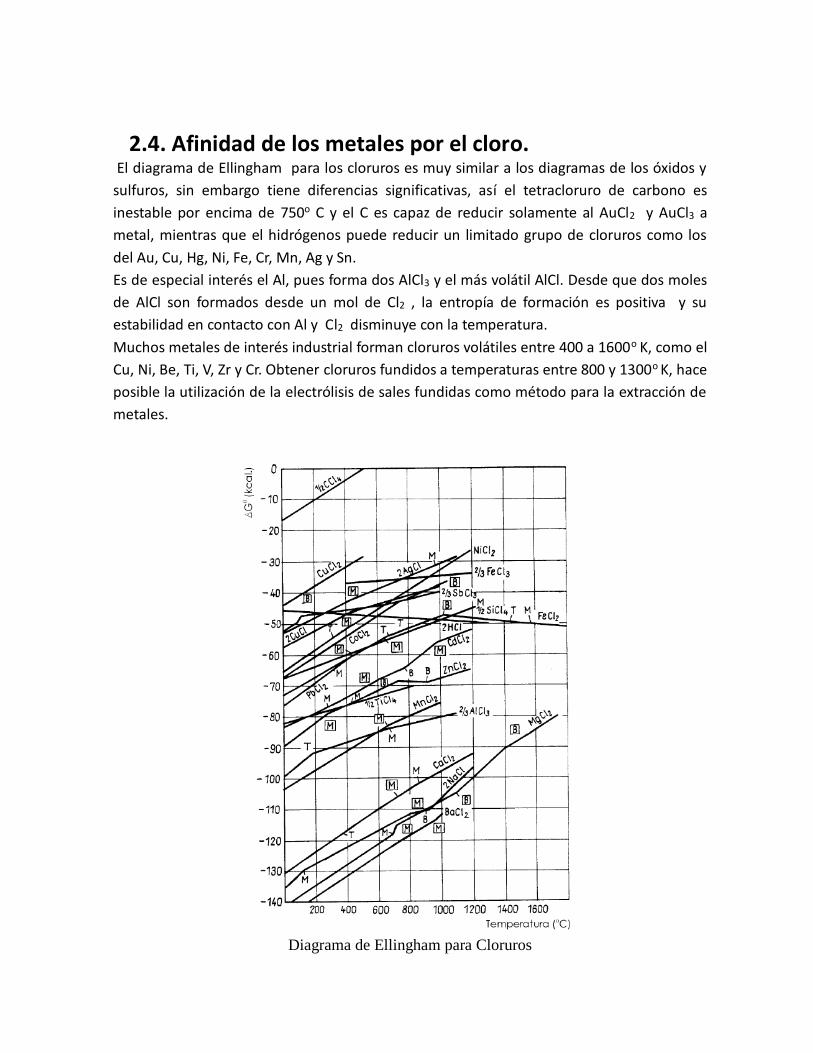
2.4. Afinidad de los metales por el cloro. El diagrama de Ellingham para los cloruros es muy similar a los diagramas de los óxidos y
sulfuros, sin embargo tiene diferencias significativas, así el tetracloruro de carbono es
inestable por encima de 750o C y el C es capaz de reducir solamente al AuCl2 y AuCl3 a
metal, mientras que el hidrógenos puede reducir un limitado grupo de cloruros como los
del Au, Cu, Hg, Ni, Fe, Cr, Mn, Ag y Sn.
Es de especial interés el Al, pues forma dos AlCl3 y el más volátil AlCl. Desde que dos moles
de AlCl son formados desde un mol de Cl2 , la entropía de formación es positiva y su
estabilidad en contacto con Al y Cl2 disminuye con la temperatura.
Muchos metales de interés industrial forman cloruros volátiles entre 400 a 1600o
K, como el
Cu, Ni, Be, Ti, V, Zr y Cr. Obtener cloruros fundidos a temperaturas entre 800 y 1300o K, hace
posible la utilización de la electrólisis de sales fundidas como método para la extracción de
metales.
Diagrama de Ellingham para Cloruros

2.5. Termodinámica de la tostación de minerales
El conocimiento de los datos termodinámicos permite construir diagramas que
muestran áreas de estabilidad o de predominio, llamados diagramas de Kellog,
mediante los cuales se puede predecir las fases sólidas que pueden estar presentes a
una temperatura y composición de gases dados y las posibles reacciones.
Las condiciones necesarias para la formación de los distintos productos de tostación
pueden ilustrarse mediante relaciones de equilibrio que existe en un sistema que
contiene metal, azufre y oxigeno. Se tiene tres componentes y de acuerdo con la regla
de fases, se puede tener un sistema de 5 fases, es decir 4 fases condensadas y una fase
gaseosa. Si la temperatura es constante se podría tener un sistema de 3 fases
condensadas y una sola fase gaseosa. La fase gaseosa contiene normalmente SO2 y O2.
Aunque SO3 y aun S2 pueden estar presentes.
Se debe recordar que la estabilidad de los compuestos está dado por la energía libre de
formación, mediante la ecuación de Gibbs:
∆Go = RT log k y k = aP / aR
aP = Actividad Productos y aR = Actividad reactantes
Durante la tostación, en un sistema Me-S-O, se producen cierto número de reacciones:
S2 + 2O2 = 2SO2 (1)
2SO2 + O2 = 2SO3 (2)
Me + SO2 = MeS + O2 (3)
2Me + O2 = 2MeO (4)
2MeS + 3O2 = 2MeO + 2SO2 (5)
2MeO + 2SO2 + O2 = 2MeSO4 (6)
MeS + 2O2 = MeSO4 (7)
Para una temperatura dada, la composición de la mezcla gaseosa esta definida por la
presión parcial de cualquiera de los 2 componentes gaseosos. También para una
composición constante de gas, la composición de las tres fases condensadas esta fija. A
las relaciones de fase en el sistema ternario a temperatura constante pueden
describirse por medio de los diagramas bidimensionales en donde las coordenadas son

las presiones parciales de dos de los componentes gaseosos. En la figuras se muestra
los diagramas de Kellogg. Las líneas que describen el equilibrio entre cualquiera de las
fases condensadas están dadas por ecuaciones de reacción.
En el caso de que el metal hubiera formado varios sulfuros y óxidos se deben
considerar mas ecuaciones para la formación de MeS2, Me2(SO4)3 etc. Pueden existir
también sulfatos básicos como MeO.MeSO4.
Para las reacciones dadas antes y para todas las fases condensadas en sus estados
estándar los equilibrios están dados por las expresiones siguientes:
logPo2 – log Pso2 = log K3
logPo2 = - logK4
2logPso2 – 3 log Po2 = log K5
2logPso2 + logPo2 = - log K6
2logPo2 = - log K7
Puede observarse que para una estequiometria de reacción dada la forma de la
expresión de equilibrio es la misma para todos los metales, es decir, la pendiente de las
curvas correspondientes en la fig. 1 es la misma. Solo los valores de las constantes de
equilibrio K3, K4 etc son distintos de metal a metal. Esto quiere decir que la posición de
las áreas entre las líneas. Estas áreas se denominan área de predominancia para la fase
particular. Puede observarse que en tanto exista solo una fase condensada. Las
presiones parciales de SO2 y O2 pueden modificarse independientemente una de la
otra. Es decir, el sistema a temperatura constante tiene dos grados de libertad. A lo
largo de las líneas de equilibrio entre dos fases condensadas. El sistema tiene solo un
grado de libertad. Finalmente cuando se encuentran presentes tres fases condensadas
el sistema no cambia a temperatura constante. En la fig. 1 se dan también líneas que
describen las reacciones (1) y (2) es decir la formación de S2 y de SO2. Estas están dadas
por las expresiones:
2logPso2 – 2logPo2 = logK1+ logPs2
logPso2 + 2logPo2 = -logK2+ 2logPso3
Se tiene entonces que para valores fijos de K1, K2 la relación entre logPso2 y logPo2
depende también de las presiones parciales de S2 o SO3 en la figura IV-1 las líneas son
para cuando Ps2 y Pso2 son iguales a una atmósfera. Para otras presiones, las líneas se
recorren hacia arriba o hacia debajo de acuerdo con las expresiones arriba dadas.
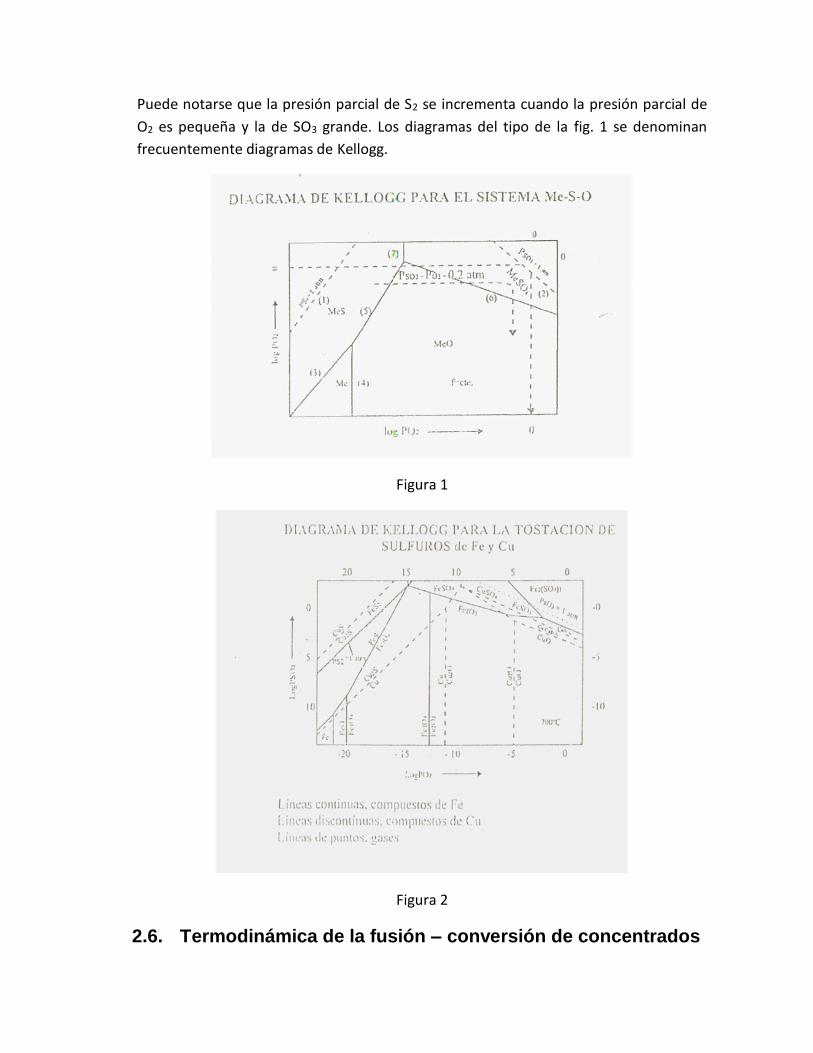
Puede notarse que la presión parcial de S2 se incrementa cuando la presión parcial de
O2 es pequeña y la de SO3 grande. Los diagramas del tipo de la fig. 1 se denominan
frecuentemente diagramas de Kellogg.
Figura 1
Figura 2
2.6. Termodinámica de la fusión – conversión de concentrados

Termodinámica de la Fusión La mayor aplicación industrial de fusión de mata es usado en la extracción de Cu y Ni de sus
concentrados sulfurados.
Las bases termodinámicas para la fundición de concentrados de cobre fue desarrollado por
Schuhmann, quién demostró la afinidad del Fe y el Cu por el O2 y el S, determinando por
que el Cu entra en la mata y no en la escoria y que el Fe tiene mucha mayor afinidad por el
oxígeno que el Cu.
El análisis de Schuhmann es el siguiente:
1. Las constantes de formación del FeS y el Cu2S, muestran que el Cu2S es algo más
estable:
2Cu(l) + ½ S2(g) = Cu2S (l) (1) k1300o
C = 760 ∆Go = - RT lnk = - 20 000 cal
Fe(ᴕ) + ½ S2(g) = FeS(l) (2) k1300o
C = 250 ∆Go = - RT lnk = - 17 370 cal
Sustraendo 1 – 2 se ve que el Cu tiene una afinidad ligeramente mayor por el S que el
Fe
2Cu(l) + FeS(l) = Fe(ᴕ) + Cu2S (l) (3) k1300o
C = 3.1 ∆Go = - RT lnk = - 3 530 cal
2. La posibilidad de la formación del Cu2O y el FeO fue también calculado por
Schuhmann:
2Cu(l + ½ O2(g) = Cu2O(l) (4) k1300o
C = 160 ∆Go = - RT lnk = - 16 000 cal
Fe(ᴕ) + ½ O2(g) = FeO(l) (5) k1300o
C = 2.1x 105 ∆Go = - RT lnk = - 38 500 cal
Combinando 3, 4 y 5 se tiene:
Cu2O(l) + FeS(l) = Cu2S (l) + FeO(l) (6) k1300o
C = 4000 ∆Go = - RT lnk = - 26 000 cal
El inmenso valor de la constante de equilibrio para la ecuación 5 muestra que el Fe
tiene una gran afinidad por el O2
3. Los valores mostrados de la ecuación 1 al 6 son para sulfuros y óxidos líquidos puros
1300OC. Para el sistema mata-escoria, la actividad del Cu2O, se puede estimar:
K = (aCu2Smata)(aFeOEscoria)/(aCu2OEscoria)(aFeSMata) = 4000 (7)
Schuhmann, asumió que la actividad del Cu2S y el FeS no se diferencian grandemente
para un grado de mata moderado dependiendo de las condiciones de operación (el

grado de mata es el porcentaje de Cu en la mata y varía entre 20 a 70%). Así por
ejemplo su radio puede ser aproximadamente 1 y que la actividad del FeO puede estar
ubicado dentro del rango de 0.3 a 0.9, sustituyendo estos valores en la ecuación 7, se
tiene: aCu2OEscoria = 10-4
Por consiguiente la presencia del Cu2O en sistema mata-escoria a una actividad mayor
a 10-4 hará que la ecuación 6 se desplace hacia la derecha y el FeS desplazará al cobre
de la escoria, por lo tanto la afinidad relativa del Cu y del Fe por el oxígeno es la razón
de que el Cu esté presente en la mata.
Termodinámica de la Conversión de matas En los procesos de fusión de concentrados de cobre y níquel se obtiene matas de Cu y
Ni o Cu-Ni, en este proceso de elimina la ganga y parte del Fe y S. La mata obtenida
principalmente está formada por Cu, Fe, S para las matas de Cu y por Cu, Ni, Fe y S
para las matas de Cu-Ni y otros elementos valiosos como el Au, Ag. El tratamiento de
conversión tiene por finalidad eliminar el Fe y el azufre insuflando aire, aire-oxígeno u
oxígeno a la masa fundida, aprovechando la mayor afinidad que tienen el Fe y el S por
el oxígeno frente al Cu y el Ni, lo que se puede apreciar claramente en el diagrama de
∆Go – T para los óxidos de Cu, Ni y Fe.
En el diagrama se observa que en presencia del O2 el Fe tiene mayor afinidad por el O2
seguido por el Ni y el Cu. De esta manera la ∆Go de formación es un indicador de la

estabilidad termodinámica de los compuestos oxidados, luego si la fase mata es
soplado con oxígeno, el Fe se oxidará preferentemente antes que el Ni y el Cu:
2FeS(Mata) + 3O2 = FeO(Escoria) + 2SO2 (Gas)
Esta reacción es exotérmica y proporciona la cantidad suficiente de calor para
mantener el baño al estado fundido, haciendo el proceso autógeno.
El tratamiento de las matas de cobre tiene dos etapas, una de formación de escoria, en
la que se elimina el Fe y parte de S y una segunda de formación de cobre blíster en la
que se elimina el S restante hasta alcanzar un contenido de cobre metálico entre 98 a
99%. Las posibles reacciones termodinámicas de la segunda etapa son:
2Cu2S(l) + 3O2 = 2Cu2O(l) + 2SO2 ∆Go1300
oC = - 107 000 cal
2Cu2O(l) + 2Cu2S(l) = 6Cu(l) + SO2(g) ∆Go1300
oC = - 3 500 cal
El Cu liquido es la fase estable, de modo que cuando el 2Cu2S(l), llamado metal blanco,
es expuesto al O2 y controlando cuidadosamente, se puede eliminar el S y producir el
blíster.
Las matas de Ni o Cu-Ni son tratados por otras técnicas, como el TBCR, por que
termodinámicamente, durante el soplado pueden ocurrir las siguientes reacciones de
oxidación:
2Ni3S2(l) + 7O2 = 6NiO(s) + 4SO2 ∆Go1300
oC = - 320 000 cal
4NiO(s) + Ni3S2(l) = 7Ni(l) + 2SO2 ∆Go1300
oC = +4 000 cal
2.7. Termodinámica de los procesos de reducción – fusión de óxidos
Los componentes metálicos de un concentrado pueden ser separados por reducción,
en pirometalurgia, para reducir un compuesto metálico MG se emplea un reductor R,
por que su afinidad por G es mayor que por M, de modo que la energía libre de
formación para la reacción de reducción es negativa.
MG + R = M + RG ∆Go = -
M = Metal G = Gas ( O , S , Cl, etc) R = Agente Reductor ( C, H2 , CO, etc)
Termodinámicamente la mayor o menor capacidad de reducción es una función de la
energía libre de formación. A altas temperaturas las barreras cinéticas son fácilmente
vencidas y las reacciones químicas ocurren rápidamente. Así, la extensión y secuencia
de las reacciones depende principalmente del equilibrio o la energía libre y la
velocidad a la que las reacciones pueden ser llevados.

Metales como el Fe, Mn, Cr y Ti se encuentran en la naturaleza como óxidos y otros
como el Zn y Pb que están presentes como sulfuros pueden ser transformados a óxidos
por tostación para luego ser reducidos. Los principales agentes de reducción son el C,
CO, H2 u otro metal con mayor afinidad por el oxígeno.
En resumen Reducción = f (-∆Go), cuanto más bajo es el valor de -∆Go, el compuesto es
más estable, entonces la reducción es más difícil.
La reducción de los óxidos metálicos es un proceso de transferencia del metal desde
sus óxidos a metal libre o a un estado de menor oxidación, que se puede dar por:
a) Por disociación en caliente
2MO → 2M + O2
MO2 → MO + ½ O2
b) Por acción de un agente reductor
MO + R → 2M + RO
MO + RO → M + RO2
MO2 + R → MO + RO
En las reacciones anteriores, la afinidad del agente reductor por el oxígeno es mayor que
por el metal, por lo que es desplazado del mineral oxidado y estas reacciones ocurrirán
espontáneamente siempre que ∆G de la reacción sea negativa. El cambio de la energía
libre en una reacción de oxidación:
2M + O2 = 2MO
Es llamado energía libre de formación del óxido a partir de sus elementos (∆GMO) y tiene un
valor generalmente negativo y cuanto más negativo más estable es el MO.
También para reducir un MO se puede emplear un metal u MO con mayor afinidad por el
oxígeno:
MO + Ml → M + MlO
MO + MlO → M + MlO2
Ejemplo: 4Fe3O4 + O2 → 6 Fe2O3 600oC ∆G = - 60 kcal
4/3 Al + O2 → 2/3 Al2O3 600oC ∆G = - 220 kcal
Luego el Al reduce al Fe2O3 por tener un ∆G menor.
PRESION DE EQUILIBRIO DEL OXIGENO
La reacción de formación de un oxido metálico MxOy por mol de oxígeno es :
2𝑥
𝑦M + 𝑂2 =
2
𝑦𝑀𝑥 O𝑦
La constante de equilibrio es KT = ( a2/yMxOy) / (aM
2x/y. pO2)
Si aMxOy = aM = 1 para el metal y el óxido puros, entonces kT = 1/pO2
y ∆GoT = - RT ln kT , luego ∆Go
T = RT ln pO2
pO2 = presión parcial del oxígeno en el equilibrio con el metal y óxido puros.
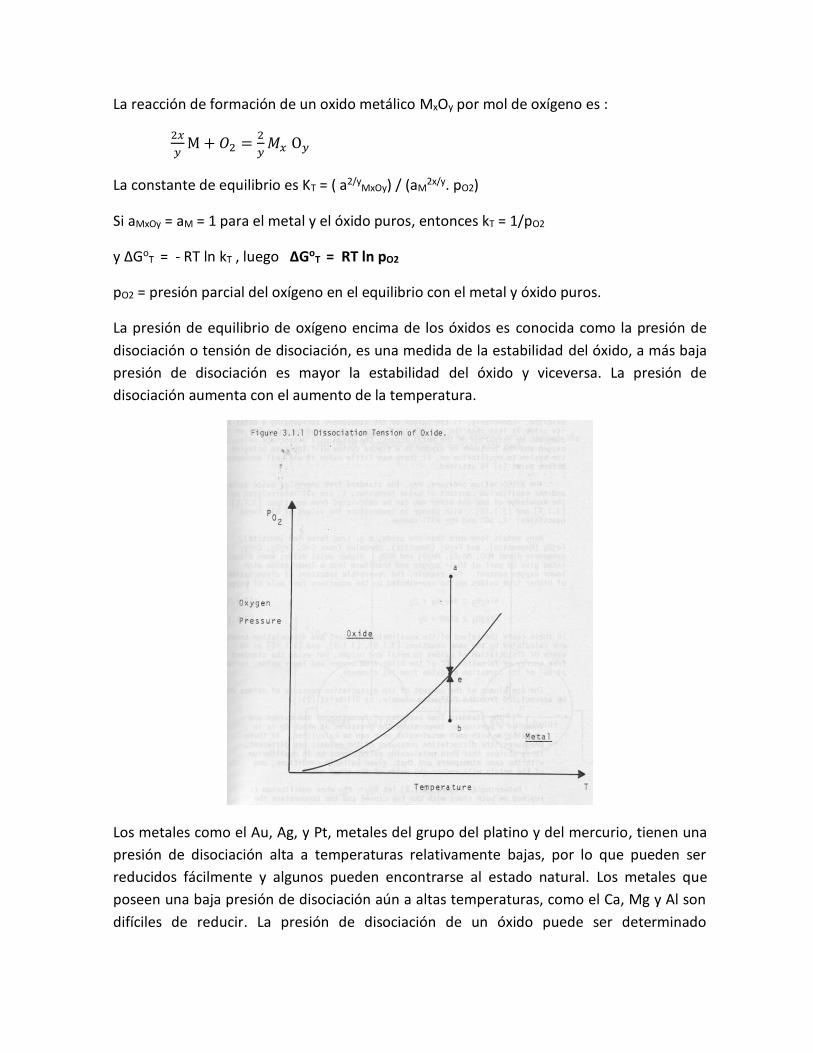
La presión de equilibrio de oxígeno encima de los óxidos es conocida como la presión de
disociación o tensión de disociación, es una medida de la estabilidad del óxido, a más baja
presión de disociación es mayor la estabilidad del óxido y viceversa. La presión de
disociación aumenta con el aumento de la temperatura.
Los metales como el Au, Ag, y Pt, metales del grupo del platino y del mercurio, tienen una
presión de disociación alta a temperaturas relativamente bajas, por lo que pueden ser
reducidos fácilmente y algunos pueden encontrarse al estado natural. Los metales que
poseen una baja presión de disociación aún a altas temperaturas, como el Ca, Mg y Al son
difíciles de reducir. La presión de disociación de un óxido puede ser determinado

experimentalmente, pero también puede ser calculado a partir de la energía libre estándar
o de la constante de equilibrio de la reacción de formación del óxido.
La oxidación de un metal es generalmente una reacción exotérmica reversible y la reacción
inversa, llamado de disociación es endotérmica. La dirección de la reacción puede ser
determinada comparando la presión de disociación pO2 con la presión parcial del oxígeno
en la atmósfera encima del metal y el óxido. La figura anterior muestra una curva típica de
la variación de la pO2 con la temperatura en el equilibrio para un oxido metálico, la curva
divide el campo en dos áreas de estabilidad definidas. Por encima de la presión de
equilibrio (punto e), el metal se oxida y es estable el MO (punto a), por el contrario por
debajo de la presión de equilibrio (e) el MO se disocia y es estable el metal (punto b).
La presión de disociación, pO2, la energía estándar libre de formación del óxido, ∆Go y la
constante de formación del óxido, k, están interrelacionados, de modo que conociendo
uno de ellos es posible calcular los otros dos, mediante la ecuación: ∆GoT = RT ln pO2
Muchos metales forman más de un óxido, ejemplo el Fe forma FeO, Fe3O4 y Fe2O3; el Cr
forma CrO, Cr2O3, CrO3; el Mn forma MnO, Mn3O4, Mn2O3 y MnO2. Los metales más
oxidados al disociarse ceden parte del oxígeno y se transforman metales menos oxidados
con bajo contenido de oxígeno, por ejemplo se tiene la reacción reversible de disociación
de del Fe de un grado de oxidación más alto a uno más bajo:
6Fe2O3 = 4Fe3O4 + O2
2Fe3O4 = 6FeO + O2
Para entender el concepto de presión de disociación de óxidos, J. D. Gilchrist, da el
siguiente ejemplo muy útil: “Si las energías libre de formación de dos óxidos son conocidas
a una temperatura dada, la presión a la cual el O2 está en equilibrio con el par M-MO,
puede ser calculado. Si estas presiones, la presión de disociación de los óxidos, son
diferentes, es obvio que ambos pares M-MO, no pueden estar en equilibrio en la misma
atmósfera y dado las condiciones adecuadas, uno de los metales reducirá al óxido del otro
metal.

En figura se aprecia que cuando la válvula está cerrada los metales y sus óxidos están en
equilibrio y que la p’O2 ˃ p”O2 a la misma temperatura. Cuando se abra la válvula el oxígeno
fluirá de la cámara 1 a la cámara 2, de modo que habrá una caída de presión en 1 y
aumentará en 2. En la cámara 1 se disociará el óxido para mantener la presión local en el
valor de equilibrio p’O2 y esta reacción continuará si la válvula sigue abierto y el metal en la
cámara 2 se oxidará mientras la presión de oxígeno sea mayor a p”O2. Si la válvula se
mantiene abierta, las reacciones continuarán hasta que todo el óxido de la cámara 1 se
disocie totalmente o todo el metal de la cámara 2 se oxide. El metal cuyo óxido tiene la
energía libre de formación estándar numéricamente más alto (-∆GoT) tiene la presión de
oxígeno en el equilibrio (pO2) menor, por consiguiente tiene mayor afinidad por el oxígeno y
tiene el óxido más estable y puede reducir a otros óxidos a metal bajo condiciones cinéticas
favorables.”
En la cámara 1 ocurre: M’O = M’ + ½ O2
En la cámara 2 ocurre: M” + ½ O2 = M”O
En la tabla siguiente, se muestra la presión de oxígeno en el equilibrio para algunos metales
y el carbón a 1000oC. El carbón puede ser usado como agente reductor para los óxidos
metálicos que están por encima de él en la tabla. Notar que la presión del sistema Ca - CaO
es menor que la del U - UO2, luego el Ca reduce al UO2 para producir el U. (Este proceso fue
usado para producir U metálico en 1942)

M - MO Presión de O2 (Atm)
Cu – Cu2O 8 x 10-8
Pb – PbO 3 x 10-8
Ni – NiO 5 x 10-11
Fe – FeO 1 x 10-15
C – CO2 7 x 10-17
Cr – Cr2O3 2 x 10-22
Mn – MnO 1 x 10-24
U – UO2 1 x 10-39
Ca – CaO 1 x 10-42
Fuente: L. C. Twidwell, Unit Processes In Extractive Metallurgy – Pirometallurgy – Montana College
DIAGRAMA ∆GoT – T ( Diagrama Ellingham)
La reacción de oxidación de un metal tiene la siguiente forma:
2𝑥
𝑦M + 𝑂2 =
2
𝑦𝑀𝑥 O𝑦
y el cambio de la energía libre ∆GT es :
∆GT = 2
𝑦 GMxOy -
2𝑥
𝑦GM – GO2
A cualquier temperatura T, las tres fases pueden coexistir solamente a la presión parcial de
equilibrio del oxígeno, a mayor presión de oxígeno el metal se oxida y a menor presión en
óxido es reducido a metal. Cuando se alcanza el equilibrio ∆GT = 0. Es importante conocer
el cambio de la energía libre ∆GT, por cuanto ésta determina la dirección de la reacción, las
condiciones del equilibrio y así la factibilidad termodinámica y el rendimiento del proceso.
Por definición: ∆GT = ∆HT - T∆ST
Por lo tanto ∆GT puede ser calculado solamente con los datos termodinámicos de ∆HT y
∆ST a una temperatura dada. Estos datos termodinámicos son determinados a partir de

ciertas condiciones estándar y éstos generalmente cambian para los reactantes y
productos, como líquidos, sólidos y gases a una atmósfera de presión parcial.
Por otra parte se sabe que: ∆GoT = - RT lnk y k = (a2/y
MxOy) / (aM2x/y. pO2) entonces : ∆Go
T
= RT ln pO2, esta ecuación muestra que ∆GoT está directamente relacionada con presión
de equilibrio o de disociación del oxígeno.
Las tres cantidades, ∆GoT, K y pO2 (e), son medidas de la afinidad química o estabilidad del
óxido y están interrelacionados unos con otros. Cuando ∆GoT es negativo, lnk es positivo y
k es mayor que la unidad y la pO2 es menor que una atmósfera. Cuanto más negativo es
∆GoT, mayor es el valor de k y más pequeño el valor de pO2, por consiguiente el óxido es
más estable y la reacción ocurre de izquierda a derecha. Por lado si ∆GoT es positivo, el lnk
es negativo y k es una fracción positiva menor que la unidad y pO2 es mayor que 1
atmósfera, entonces, cuando a una temperatura T, ∆GoT tiene un valor positivo, k es una
fracción muy pequeña y la reacción ocurrirá de derecha a izquierda y el óxido se disociará.
La representación gráfica del cambio de la energía libre estándar de formación de óxidos,
∆GoT, frente a la temperatura T fue desarrollado por Ellingham en 1944 y posteriormente
fue ampliado por Richardson y Jeffe. Estos diagramas son muy convenientes por que
contienen una gran cantidad de información y permiten ver la posibilidad termodinámica
de una reacción de oxidación o reducción, por lo que son de amplio uso en la
termodinámica metalúrgica y fisicoquímica.
Diagrama de Ellinghan – Richardson - Jeffe
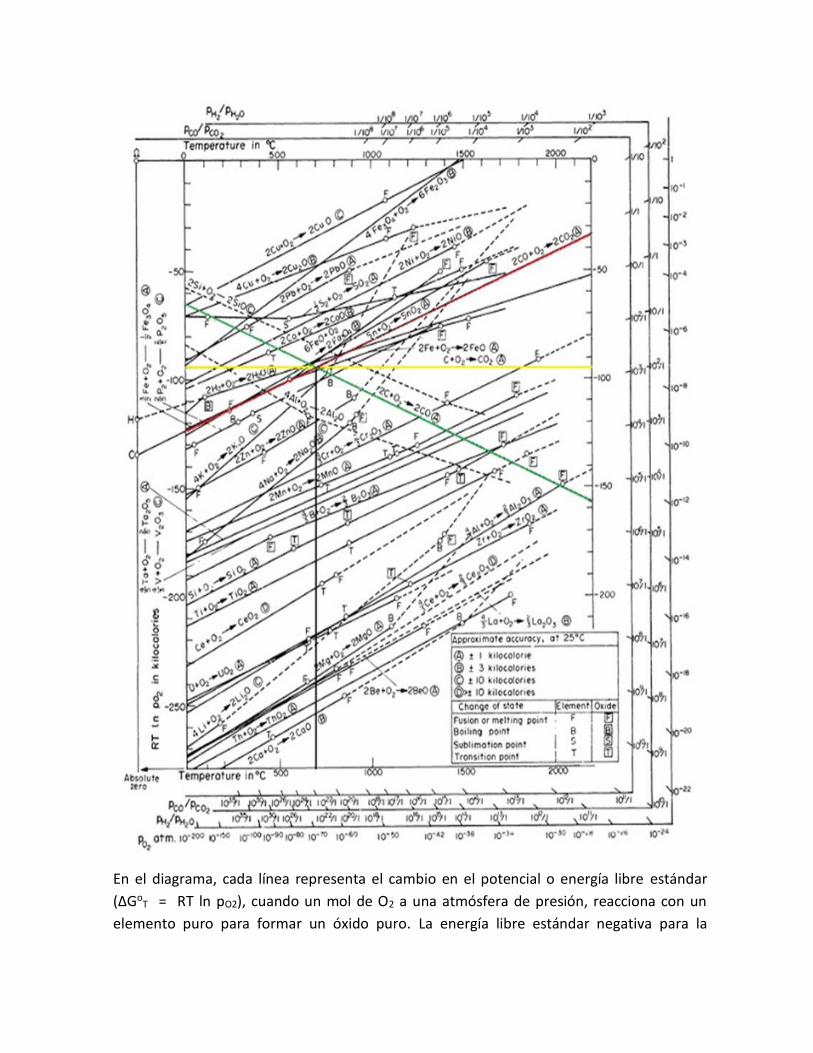
En el diagrama, cada línea representa el cambio en el potencial o energía libre estándar
(∆GoT = RT ln pO2), cuando un mol de O2 a una atmósfera de presión, reacciona con un
elemento puro para formar un óxido puro. La energía libre estándar negativa para la

formación de un óxido, se hace más negativo hacia abajo, por ejemplo la curva más baja
que aparece en el diagrama corresponde al oxido más estable y más difícil de reducir.
A cualquier temperatura superior al mínimo requerido por las consideraciones cinéticas, el
elemento combinado con el O2 y representado en una curva dada es capaz de reducir todo
los óxidos situados encima de él, así a 1200oC el C reducirá al FeO, H2O, Fe3O4, NiO, CoO,
Cu2O, Fe2O3.
Los aspectos más importantes del diagrama ∆Go – T son:
1) La energía libre por mol de O2 puede ser leído directamente en el gráfico a cualquier
temperatura. Los puntos de fusión y ebullición de los reactantes y productos también
están representados.
2) La gradiente de las líneas son iguales a la entropía negativa de la reacción, así:
∆GoR = ∆Ho
R – T∆SoR , derivando respecto a T
𝜕(∆𝐺𝑅
𝑜)
𝜕𝑇= 𝐺𝑟𝑎𝑑𝑖𝑒𝑛𝑡𝑒 = − ∆𝑆𝑅
𝑜
La gradiente llega a ser más positivo para un cambio de fase en el reactante y se hace
menos positivo para un cambio de fase en el producto, así:
Cuando hay cambio de fase en el reactante:
2M(S) + O2 = 2MO(S) ∆𝑆𝑅𝑜 = - (a)
2M(S) = 2M(L) ∆SFusión = + (b)
a - b: 2M(L) + O2 = 2MO(S) ∆𝑆𝑅𝑜 = ∆Sa - ∆Sb
= (-) - (+)
Por lo tanto la entropía de la reacción es más negativa y la gradiente es más positiva.
Cuando hay cambio de fase en el producto:
2M(S) + O2 = 2MO(S) ∆𝑆𝑅𝑜 = - (a)
2MO(S) = 2MO(L) ∆SFusión = + (b)
a + b: 2M(L) + O2 = 2MO(S) ∆𝑆𝑅𝑜 = ∆Sa + ∆Sb
= (-) + (+)
Por lo tanto la entropía de la reacción es menos negativa y la gradiente es menos
positiva.
Según la Regla de Knopp, la entropía de fusión y vaporización para todos los elementos
es aproximadamente el mismo:
∆SFusión ≈ 2.2 cal/mol oK, por lo que el cambio de gradiente en el punto de fusión es
pequeño
∆SVaporización ≈ 22 cal/mol oK, por lo que el cambio de gradiente en el punto de
vaporización es grande

3) La estabilidad de los óxidos está relacionado al valor negativo de la energía libre de
formación ∆Go, cuanto más negativo la energía libre más estable el óxido. Luego
cualquier metal reducirá al óxido encima de él.
Ejemplo: El Ca reduce al MgO a 1000oC
MgO + Ca = Mg + CaO
∆𝐺𝑅𝑜 = ∆𝐺𝑓
𝑜productos - ∆𝐺𝑓
𝑜reactantes
Del Diagrama: -(240 000/2) – (- 224 000/2) = - 8000 cal, reacción espontánea
4) El efecto del cambio d entropía en el valor de ∆G, es particularmente notorio en el caso
de la reacción entre el C y el O2. El dióxido de carbono se forma de acuerdo a la
reacción: C + O2 = CO2, el número de moles gaseosos no sufre cambio y hay un pequeño
cambio de volumen, luego ∆So ≈ 0 de modo que la representación gráfica de esta
reacción es casi una línea horizontal. Para la reacción C + O2 = 2CO, hay un aumento del
número de moles gaseosos que ocasiona un aumento considerable de ∆So, este cambio
positivo de la entropía origina una pendiente negativa y un valor cada vez más negativo
de ∆Go para la formación del CO con el aumento de la temperatura. Por tanto el CO es
mas estable con el aumento de la temperatura, lo que significa que el C puede reducir a
cualquier óxido metálico a una temperatura suficientemente alta.
Ejem. La reacción del ZnO con el C a 1100oC
2C + O2 = 2CO (1) ∆𝐺1𝑜 = - 110 kcal
2Zn + O2 = 2ZnO (2) ∆𝐺2𝑜 = - 90 kcal
(1 – 2): 2C + O2 = 2Zn + 2CO ∆𝐺𝑅𝑜 = ∆𝐺1
𝑜 - ∆𝐺 2 𝑜 = -20 kcal
La reducción el ZnO usando C se realiza a temperaturas mayores a 1100oC , sea en
retortas o altos hornos y se obtiene vapor de Zn, por que el punto de ebullición del Zn es
907oC.
5) Los óxidos metálicos pueden ser divididos en dos grupos:
a. Aquellos cuya línea de equilibrio es interceptado por la línea de equilibrio del CO a
temperaturas menores a 1600oC
b. Aquellos cuya línea de equilibrio es interceptado a temperaturas mayores a 1600oC,
es decir los más estables.
El primer grupo puede ser reducido por el C a metal a temperaturas fácilmente
alcanzables en hornos e manga ejm: Cu, Fe, Zn, etc , el segundo grupo puede ser
reducido por el C a temperaturas mayores a 1600oC, pero lo más probable es que se
formen carburos. Estos óxidos mas estables pueden ser recuperados por electrolisis,
como es el caso del Al. Este segundo grupo es importante para la formación de
aleaciones como: Fe – Ti, Cu – Al, aleaciones de cromo, etc.
6) Las presiones de equilibrio del O2 pueden ser determinados directamente a cualquier
temperatura observando la parte derecha e inferior del diagrama

k = 1
𝑝𝑂2
Ejm: a partir del cambio de energía libre estándar, calcular la presión de equilibrio para
la siguiente reacción:
Ti + O2 = TiO2 T = 1700oC ∆G = -130 000 cal
∆G = RT ln pO2 , entonces, ln pO2 =−130 000
1.98 (1973)
pO2 ≅ 5 x 10- 15
Este valor se puede leer directamente en el ∆G – T, haciendo pasar una recta desde el
punto O por la intersección de la temperatura a 1700oC con la línea de equilibrio hasta la
recta de pO2 ubicada a la derecha de gráfico.
7) En el mismo diagrama existen escalas que permiten la lectura directa de los radios de
presión H2/H2O y CO/CO2.Los puntos de referencia son H y C en la escala del cero
absoluto, estos son los puntos de energía libre en el cero absoluto para las reacciones:
2CO + O2 = 2CO2
2H2 + O2 = 2H2O
Ejem.: Ti(s) + 2CO2(g) = TiO2(s) + 2CO(g)
Ti(s) + 2H2O(g) = TiO2(s) + 2H2(g)
A 1600oC, cual es la relación pCO/pCO2 y pH2/pH2O
Del gráfico se tiene: pCO/pCO2 ≅ 4 x 104, entonces k = 𝑝𝐶𝑂
2
𝑝𝐶𝑂22 = (4x104)2
pH2/pH2O ≅ 4 x 104, entonces k = 𝑝𝐻2
2
𝑝𝐻2𝑂2 = (104)2
REACCION ESPONTANEA
Al exponer un metal y sus componentes a un radio de gases, cuya composición no es el
del equilibrio, se puede causar una reacción de oxidación o reducción. Esta posibilidad
puede ser calculado mediante la Isoterma de Van’t Hoff:
∆G = ∆G° + RT ln Q donde Q radio de gas al que es expuesto el metal
∆G = RT ln k + RT ln Q
∆G = RT ln 𝑸
𝒌
Si 𝑄
𝑘 < 1, entonces ∆G = - y la reacción es teóricamente espontánea
Si 𝑄
𝑘 > 1, entonces ∆G = + y la reacción es no espontánea.
Ejemplos:
1.- Puede el ZnO ser reducido en el vacío, donde pO2 = 10-5 atmósferas
2ZnO = 2Zn + O2
k = pO2(e) = 10-37 ( del gráfico de Ellingham) y Q = 10-5
k < Q , 𝑄
𝑘 > 1 entonces ∆G = +, por tanto no se reduce, por que
𝑄
𝑘 =
10−5
10−37 = 132
2.- Puede el Cu oxidarse en el aire a 1000°C?

4 Cu + O2 = 2Cu2O
k = 1/10-7 , Q = 1(𝑃𝑟𝑒𝑠𝑖𝑜𝑛 𝐴𝑡𝑚ó𝑠𝑓𝑒𝑟𝑐𝑎)
0.2 ≈ 5,
𝑄
𝑘 = 5 x 10-6
𝑄
𝑘 < 1 entonces ∆G = - y por tanto se oxida.
3.- Puede el Cu ser oxidado en presencia de CO y CO2, cuyo radio 𝑝𝐶𝑂
𝑝𝐶𝑂2 = 103, a 700°C?
Cu + CO2 = CuO + CO k = 10-5 del diagrama y Q = 103
𝑄
𝑘 =
103
10−5 = 1 x 108, 𝑄
𝑘 > 1 entonces ∆G = +, por tanto no se oxida
4.- Puede el Mg vapor a 0.001 atm. Ser oxidado por un gas cuyo radio CO/CO2 es de 105 a
1880°C?
Mg(v) + CO2 = MgO(s) + CO(g)
Del diagrama a 1880°C;
Mg(g) + ½ O2 = MgO(s) (1) ∆𝐺1° = - 73.5 kcal
CO(g) + ½ O2 = CO2(g) (2) ∆𝐺2° = - 24.5 kcal
(1 -2 ) Mg(g) + CO2(g) = MgO(s) + CO(g) ∆G° = - 49 kcal
Como el sistema no está en equilibrio usamos la Regla de Van’t Hoff: ∆G = ∆G° + Rt ln Q
∆G = - 49 000 + (1.98 x 2073) ln 𝑝𝐶𝑂
𝑝𝐶𝑂2 𝑥 𝑝𝑀𝑔
= -49000 + (4.575 x 2073) log 105
10−3 = +27000 cal
No se oxida.
2.8. Termodinámica de la lixiviación de minerales
Las reacciones de oxidación – reducción, particularmente, las reacciones electroquímicas
son muy importantes para el entendimiento de los procesos hidrometalúrgicos. El
intercambio de electrones para uno o más elementos resulta en un cambio de estado de
oxidación o valencia de ese elemento. En las reacciones redox, la pérdida de electrones de
un elemento debe estar acompañado por una ganancia de electrones de otro elemento, un
elemento pierde electrones al oxidarse y gana al reducirse. En las reacciones
electroquímicas la reacción de oxidación ocurre en el ánodo y la reacción de reducción en
el cátodo, en forma simultánea y separada a una distancia finita.
Las reacciones electroquímicas son reacciones redox heterogéneas en la que el
intercambio iónico ocurre en medias celdas y el concepto de media celda provee un

método conveniente para el balance de las reacciones redox, el que consiste de 4 etapas
básicas para cada media celda:
1) Establecer los productos de reacción de la media celda y balance de elementos
2) Balance del O2 con el H2O
3) Balance del hidrógeno con el H+
4) Balance de carga con electrones
Las media celdas balanceadas son ajustadas y sumadas de modo que el número de
electrones se cancela y obteniéndose la reacción redo total. La estabilidad de una reacción
de una media celda dada puede ser establecida de los datos de energía como es hecho
normalmente en los cálculos de equilibrio de soluciones. Alternativamente, la estabilidad
de la reacción de la media celda puede ser representada por el potencial de la media celda,
que está relacionada al cambio de la energía libre para las reacciones de reducción por la
siguiente relación:
∆G = -nFE
n= Número de electrones
E = Potencial de media celda
F = Constante de Faraday, 23.06 kcal/ Equivalente voltio
En este sistema al potencial de la media celda H+/H2 se le asigna un valor de cero por
convención y de forma arbitraria y todos los otros valores de media celda son medidos con
respecto a este valor de referencia designado como Eh, como resultado de esta convención
la energía libre estándar de formación de H+ es cero.
Usando el concepto de caracterización de las reacciones de media celda por un potencial,
es decir, su tendencia relativa a reaccionar, es posible construir diagramas de fase
electroquímicos para definir regiones de estabilidad para un sistema dado definido por un
conjunto de reacciones de media celda. Estos diagramas son conocidos como Diagramas de
Pourbaix o Diagramas Eh – pH. Estos diagramas se usan con mucha ventaja para entender
los fenómenos de la corrosión de los metales, la geoquímica, electroquímica, así como en
la hidrometalurgia.
Los diagramas de Pourbaix o diagramas electroquímicos están limitados por la estabilidad
del agua líquida, determinada por los potenciales a los cuales ocurre la oxidación y
reducción, así, la descomposición del agua ocurrirá, cuando la presión parcial del oxígeno
en equilibrio con agua líquida sea mayor a la presión total del sistema o cuando la presión
parcial del hidrógeno en equilibrio con el agua líquido excede la presión total de sistema,

siendo el punto crítico que define la región de estabilidad del agua cuando 𝑝𝑂2 o 𝑝𝐻2
iguala
a la presión total del sistema.
Las reacciones de media celda que describen la región de estabilidad electroquímica para el
agua líquida para un sistema a una presión total de 1 atmósfera son:
1
2 O2 (g) + 2H+ + 2e- → H2O (l)
Eo = - ∆G/nF = 56.7/2 x23.06 = 1.23 v
E = Eo - 𝑅𝑇
𝑛𝐹 ln
𝑎𝐻2𝑂
𝑎𝑂21/2
.𝑎𝐻+2
= Eo – 0.059
2 log
1
𝑎𝐻+2 0.0592 =
8.32 𝑥 298 𝑥 2.303
96 500
E = 1.23 – 0.059 pH
H+ + e- → 1
2 H2
Eo = - ∆G/nF = 0
E = Eo - 𝑅𝑇
𝑛𝐹 ln
𝑎𝐻2
1/2
𝑎𝐻+2 = Eo –
0.059
2 log
1
𝑎𝐻+
E = - 0.059 pH
La representación de estas ecuaciones de equilibrio para el agua, es:
La termodinámica de las reacciones de lixiviación están muy bien ilustradas en las
diagramas Eh – pH, en la que las especies disueltas deben de existir dentro de los dos
límites de estabilidad del agua, descrito por la ecuación de oxidación en la parte superior y
por la ecuación de reducción en la parte inferior.

Para estabilizar especies disueltas cerca de límite superior de la estabilidad del agua, se
requiere un ambiente oxidante y para estabilizar especies disueltas cerca del límite inferior
de la estabilidad del agua se requiere un ambiente reductor.
El diagrama Eh-pH para cada conjunto metálico es único, sin embargo se puede establecer
que la mayoría de los óxidos metálicos básicos cuando están en su estado de oxidación más
alto son fácilmente solubles en medios ácidos o medios alcalinos fuertes y la mayoría de los
metales, óxidos metálicos en sus estados de oxidación más bajos y sulfuros metálicos,
requieren, además de reactivos ácidos o reactivos alcalinos fuertes, agentes oxidantes para
su disolución. Es razonable asumir que los metales alcalinos o alcalinos térreos estén
presentes formando iones simples, sin embargo un número de metales básicos,
especialmente los metales de transición tienden a reaccionar con agentes complejantes
para formar iones complejos metálicos estables, un ejemplo es la cianuración del oro, es
importante destacar que los agentes complejantes juegan un rol determinante en los
procesos de lixiviación. Ejemplos de formación de iones complejos:
4 Au + 8 NaCN + O2 + 2 H2O = 4 Na[Au(CN)2] + 4 NaOH
CuO + 4NH3 + H2O = Cu(𝑁𝐻3)4+2 + 2OH-
La lixiviación de un mineral en una solución acuosa puede ser descrita por uno de los
siguientes tipos de reacción:
1. Lixiviación Acuosa
CuSO4 = Cu+2 + 𝑆𝑂4−2
CuCl2 = Cu+2 + 2 Cl-
Los minerales pueden ser de ocurrencia natural o ser producidos pirometalúrgicamente
por tostación sulfatante o clorurante
2. Lixiviación ácida
CuO + H+ → Cu+2 + H2O
ZnO + H+ → Zn+2 + H2O
Los minerales pueden ser de ocurrencia natural o ser fabricados
3. Lixiviación alcalina
Al2O3 + 2𝑂𝐻− = 2𝐴𝑙𝑂2− + H2O
Este ejemplo, proceso Bayer, demuestra el efecto de cambio de condiciones ácidas a
alcalinas para lograr selectividad en la lixiviación, en este caso frente al óxido de Fe,
insoluble en medio alcalino
4. Lixiviación por oxidación ácida
Cu2S + O2 + 4H+ = 2Cu+2 + 2H2O + S
El ion férrico puede servir solamente como un agente oxidante. En medio ácido el Fe
precipita como óxido de Fe en condiciones neutras y alcalinas
5. Lixiviación por oxidación alcalina
PbS + 2O2 + 3𝑂𝐻− = 𝐻𝑃𝑏𝑂2− + 𝑆𝑂4
−2 + H2O
6. Lixiviación por oxidación ácida con formación de iones complejantes
UO2 + 2Fe+3 + 2𝑆𝑂4−2 = UO2(𝑆𝑂4)2
−2 + 2Fe+2
Esta reacción complejante demuestra el cambio del catión uranilo al anión uranilo por
control del agente complejante. El complejo aniónico puede ser selectivamente
extraído por las operaciones unitarias de intercambio iónico o extracción por solventes.
7. Lixiviación por oxidación alcalina con formación de iones complejos
CuFeS2 + 4NH3 + 17
4 O2 + 2𝑂𝐻− = Cu(𝑁𝐻3)4
+2 + 1
2 Fe2O3 + 2𝑆𝑂4
−2 + H2O
8. Lixiviación reductora
MnO2 + SO2 = Mn+2 + 𝑆𝑂4−2
Existen solamente algunos ejemplos que se ajustan a esta categoría, como MnO2, Fe2O3
y SnO2, etc. Las especies de estado de oxidación más bajas son más solubles que las
especies de estado de oxidación más altas.
A continuación se presenta algunos ejemplos de diagramas de Eh – pH que muestran
las áreas de estabilidad de algunos metales y sus compuestos.

2.9. Diagramas potencial –pH Fue creado por el químico Ruso Marcel Pourbaix (1904-1998) que en el año 1963 publica
"Atlas of Electrochemical Equilibria", conteniendo los diagramas de Pourbaix de todos los
elementos conocidos en esa época. El diagrama de Pourbaix es conocido también como
diagrama Eh-pH.
Existe abundante información sobre las posibles reacciones que pueden ocurrir durante los procesos de corrosión, lixiviación, electrodeposición, electrorefinación, galvánicos, etc de los metales, por ejemplo, se sabe que un metal se disuelve con formación de iones metálicos a un potencial dado, a un potencial inferior no hay disolución y además dicho potencial es independiente del pH del medio. Se sabe también que si el metal se disuelve y da un óxido o hidróxido, la reacción ocurre a un potencial que depende del pH. Igualmente es conocido que si se neutraliza una solución ácida que contenga iones metálicos, al llegar a cierto pH comienza a formarse precipitados de hidróxidos, el pH al cual comienza esta precipitación se puede calcular en función de la concentración de los iones metálicos y también es sabido, en numerosos casos, que al aumentar el pH se llega a cierto umbral por encima del cual los hidróxidos precipitados se redisuelven y dan aniones o iones complejos. Por desgracia toda esa información se encuentra dispersa en la abundante literatura y resulta engorroso tener que revisar tablas y hacer cálculos cada vez que se requiera saber que ocurre con un metal a un potencial y pH dados y fue Pourbaix quien halló una forma ingeniosa de reunir todos estos datos en un solo diagrama, representando el potencial en función del pH. Sabiendo que la disolución o precipitación de un metal en un electrolito es básicamente una reacción electroquímica en la que se produce una reacción de oxidación y una de reducción junto a la circulación de iones a través del electrolito y debido a la relación directa que existe entre las actividades, el potencial de celda y las energías libres de Gibbs, es posible construir diagramas Potencial- pH que muestren los valores de estas variables en las que son estables las posibles especies metálicas, que forman el sistema M - H2O.
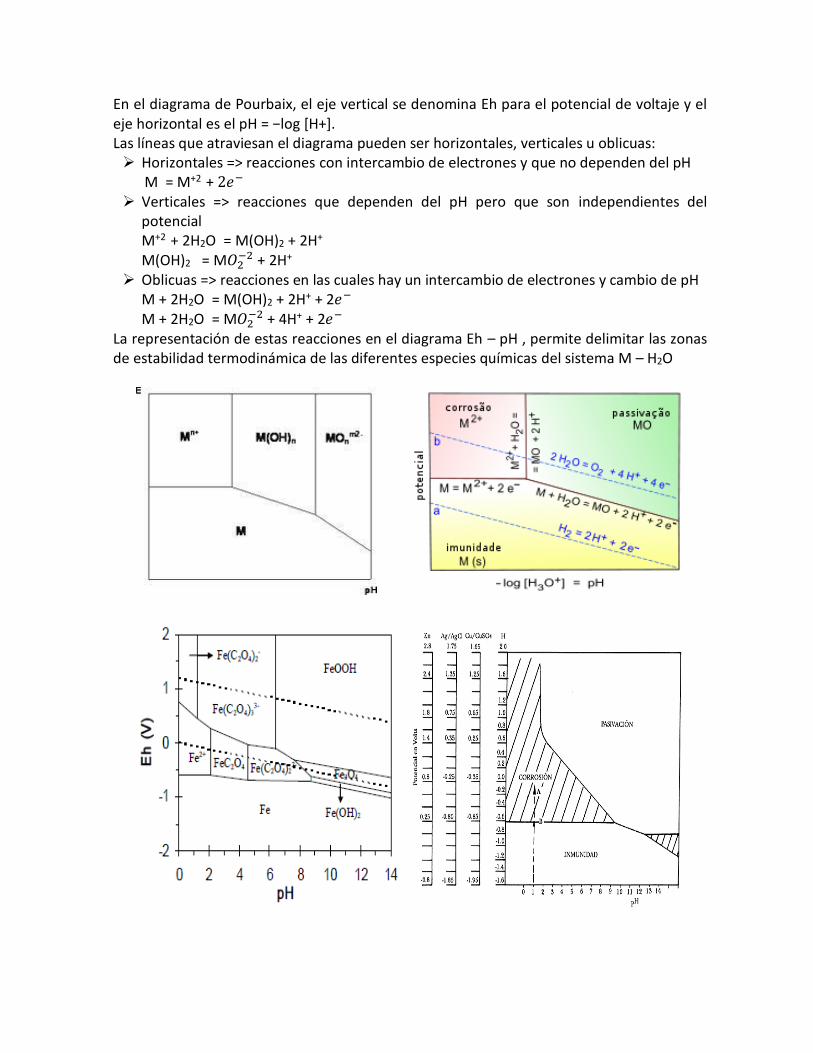
En el diagrama de Pourbaix, el eje vertical se denomina Eh para el potencial de voltaje y el eje horizontal es el pH = −log [H+]. Las líneas que atraviesan el diagrama pueden ser horizontales, verticales u oblicuas: Horizontales => reacciones con intercambio de electrones y que no dependen del pH M = M+2
+ 2𝑒− Verticales => reacciones que dependen del pH pero que son independientes del
potencial M+2
+ 2H2O = M(OH)2 + 2H+ M(OH)2 = M𝑂2
−2 + 2H+
Oblicuas => reacciones en las cuales hay un intercambio de electrones y cambio de pH M + 2H2O = M(OH)2 + 2H+ + 2𝑒− M + 2H2O = M𝑂2
−2 + 4H+ + 2𝑒−
La representación de estas reacciones en el diagrama Eh – pH , permite delimitar las zonas de estabilidad termodinámica de las diferentes especies químicas del sistema M – H2O
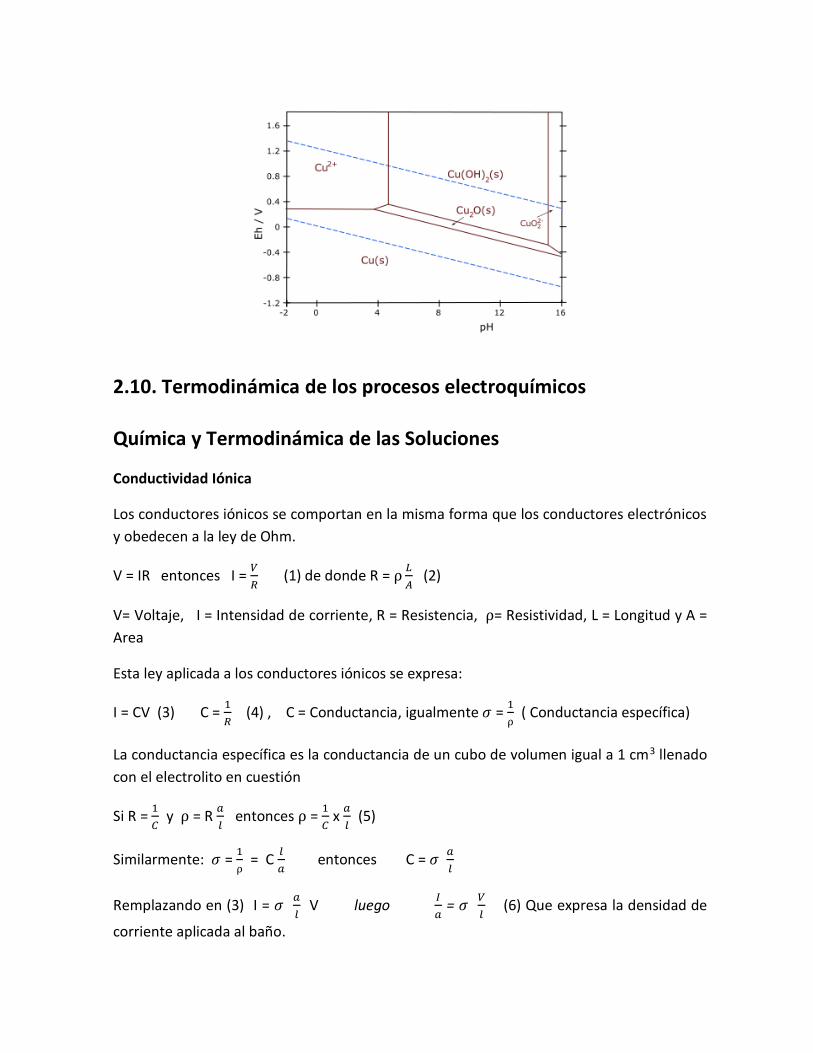
2.10. Termodinámica de los procesos electroquímicos Química y Termodinámica de las Soluciones
Conductividad Iónica
Los conductores iónicos se comportan en la misma forma que los conductores electrónicos
y obedecen a la ley de Ohm.
V = IR entonces I = 𝑉
𝑅 (1) de donde R =
𝐿
𝐴 (2)
V= Voltaje, I = Intensidad de corriente, R = Resistencia, = Resistividad, L = Longitud y A =
Area
Esta ley aplicada a los conductores iónicos se expresa:
I = CV (3) C = 1
𝑅 (4) , C = Conductancia, igualmente 𝜎 =
1
( Conductancia específica)
La conductancia específica es la conductancia de un cubo de volumen igual a 1 cm3 llenado
con el electrolito en cuestión
Si R = 1
𝐶 y = R
𝑎
𝑙 entonces =
1
𝐶 x
𝑎
𝑙 (5)
Similarmente: 𝜎 = 1
= C
𝑙
𝑎 entonces C = 𝜎
𝑎
𝑙
Remplazando en (3) I = 𝜎 𝑎
𝑙 V luego
𝐼
𝑎 = 𝜎
𝑉
𝑙 (6) Que expresa la densidad de
corriente aplicada al baño.

La conductancia específica de los electrolitos varía desde 10-6 Ω-1cm-1 para el agua
destilada, hasta pocos Ω-1cm-1 para soluciones acuosas muy concentradas y sales fundidas.
Esto es 10-12 a 10- 16 la conductividad del Cu o de cualquier otro metal.
La conductancia específica varía con la concentración y la temperatura de las soluciones,
en electrolitos fuertes es casi lineal para soluciones diluidas por la alta movilidad de sus
iones, hasta alcanzar un máximo y disminuir con el aumento de la concentración debido al
espesamiento de la nube iónica.
Conductividad Eléctrica Equivalente
Es la conductividad eléctrica de un volumen Q cm3 de solución que contiene 1 equivalente
– gramo de una sustancia disuelta, con los electrodos a 1 cm de distancia uno del otro
𝜎 = Ʌ
𝑉𝑒 Ʌ = Conductancia equivalente, 𝑉𝑒 = Volumen equivalente
Para un líquido puro 𝑉𝑒 = 𝑀𝑒
𝛿 donde 𝑀𝑒 = Masa Equivalente, 𝛿 = Densidad
Luego 𝜎 = Ʌ𝛿
𝑀𝑒 y como la concentración, usualmente se expresa en Equiv-g/litro,
entonces: Ʌ = 𝜎 1000
𝐶 cm2/ equiv-g –Ohmio
De esta relación se puede ver que la conductancia eléctrica equivalente disminuye al
aumentar la concentración y la conductancia específica aumenta con el aumento de la
concentración.
Migración y Número de Transporte
Para entender el mecanismo de la conducción iónica es necesario considerar la conducta
de las soluciones acuosas muy diluidas. En la tabla siguiente se dan los valores
experimentales de la conductancia equivalente de seis sales en soluciones acuosas con
dilución al infinito (Ʌo)
Tabla de Comparación de Conductancias Equivalentes a Dilución Infinita
KCl Diferencia KNO3 Diferencia ½ K2SO4
130.0 3.7 126.3 6.7 133.0
(21.1) (21.1) (21.1)

NaCl NaNO3 ½ Na2SO4
108.9 3.7 105.2 6.7 111.9
Comparando los valores de la conductancia equivalente de las tres sales de K con las tres
sales de Na (columnas) se ve que la diferencia es constante ( 130.0 – 108.9 = 126.3 – 105.2
= 133.0 – 111.9 = 21.1), muestran que no dependen del anión y puede ser atribuido a las
movilidades diferentes que poseen los iones Na y K. Una observación similar es hecha al
comparar las dos filas de números que corresponden a diferentes aniones con el mismo
catión.
Esta relación entre las conductancias equivalentes de electrolitos con dilución al infinito,
sugiere que los iones migran independientemente y que la conductancia equivalente de
una sal es igual a la suma de sus conductancias iónicas.
Ʌo = 𝜆+𝑜 + 𝜆−
𝑜 según la Ley de Kohlrausch, de migración independiente de los iones
Conductancia Iónica a Dilución Infinita
Recordando que:
I = 𝑉
𝑅 (Ley de Ohm) (1) Ʌ = 𝜎
1000
𝐶 (2) Ʌo = 𝜆+
𝑜 + 𝜆−𝑜 (3)
Y combinando estas tres ecuaciones, se tiene:
R = 𝑙
𝑎 y 𝜎 =
1
entonces =
1
𝜎 Luego R =
1
𝜎 x
𝑙
𝑎 remplazando en (1)
I = 𝑉𝜎𝑎
𝑙 de (2) 𝜎 =
Ʌ𝐶
1000 entonces I =
𝐶
1000 Ʌ (
𝑉
𝑙)a ; 𝑙 = longitud y a = área
Finalmente I = 𝐶
1000 (𝜆+
𝑜 + 𝜆−𝑜 )
𝑉
𝑙 a
La corriente total I, se puede dividir en I+ corriente catiónica e I- corriente aniónica y luego
se tiene: I+ = 𝐶
1000 𝜆+
𝑜 (𝑉
𝑙)a y I- =
𝐶
1000 𝜆−
𝑜 (𝑉
𝑙)a
Entonces I = I+ + I-
En número de transporte o transferencia es definido como la fracción de corriente que es
transportada por un ion dado, así:

t+ = 𝐼+
𝐼 =
𝜆+
Ʌ Numero de transporte del catión
t- = 𝐼−
𝐼 =
𝜆−
Ʌ Numero de transporte del anión
Cuando las soluciones son más complejas, se tiene:
Ii = Ziλi 𝐶
1000 𝑉
𝑙 a zi = Potencial químico de i
ti = Ii
I =
ZiλiCi
∑ ZiλiCi𝑛𝑖=1
y ∑ 𝑡𝑛𝑖=1 I = 1
n = número de especies iónicas con una carga o potencial zi y concentración Ci en
equivalentes por litro.
Movilidad Iónica y Coeficiente de Difusión
La conductancia del equivalente iónico es igual al producto de la movilidad iónica (μi) por
la carga transportada por un equivalente gramo llamado Faraday (F)
λi = μi F (1)
La movilidad es la velocidad de los iones cuando la fuerza que lo impulsa es la unidad.
Comparando la velocidad de transporte bajo una gradiente de potencial eléctrico y aquel
bajo una gradiente de concentración, se puede relacionar la movilidad iónica al coeficiente
de difusión Di , a través de la ecuación de Nernst – Einstein
μi = Zi F
𝑅𝑇 Di (2)
Aplicando la Ley de Stokes al movimiento difusional de los iones se tiene la ecuación de
Stokes – Einstein:
μi = Zi F
6𝜋𝑟𝑖𝜂 Di (3) 𝜂= Viscosidad del fluido y ri = Radio Iónico
Esta relación es una aproximación, puesto que la Ley de Stokes se aplica al movimiento de
esferas a través de un líquido incompresible, esto es bastante diferente al movimiento de
iones entre otras partículas en movimiento, sin embargo esta relación es muy útil para
predecir valores aproximados y para comparar cualitativamente diferentes electrolitos.
Combinando las ecuaciones 1, 2 y 3 se tiene:
λi 𝜂 = 𝑍𝑖 𝐹
2
6𝜋𝑟𝑖 (4)

Para un electrolito a diferentes temperaturas o en diferentes solventes, el producto de la
conductancia equivalente por la viscosidad es una constante y es conocido como la Regla
de Walden: λ𝜂 = Cte. (5)
Que también es una relación aproximada que es muy útil para estimar valores de
conductancia no conocidas.
Desde que la conductancia iónica está relacionada con la difusión, ésta puede ser tratada
en forma análoga a aquellos aplicados a la velocidad de otros procesos y ser expresado
como una función exponencial de la temperatura:
λ = 𝐴λ𝑒−𝐸λ𝑅𝑇 (6) 𝐷𝑜𝑛𝑑𝑒 𝐴λ = Constante y 𝐸λ = Energía de activación del proceso
La variación de la conductancia con la temperatura es a menudo expresado con bastante
exactitud por una ecuación parabólica:
λ (t) = λ (25) [1+ ∝ ( 𝑡 − 25) + 𝛽(𝑡 − 25)2 ] (7)
Donde ∝ y 𝛽 son constantes para un ion dado en un solvente dado y t es la temperatura en
grados centígrados. Derivando las ecuaciones 6 y 7 se tiene la relación:
𝐸λ = ∝ RT2
Para la mayoría de las soluciones acuosas, excepto para los iones H+ y OH- a 25oC, el
coeficiente de la temperatura es e 2%/oC y la energía de activación del proceso 𝐸λ para la
conductancia es de aproximadamente 3.6 kcal/ mol.
Teoría de la Disociación Electrolítica
Según Arrhenius, un soluto no está completamente dividido en sus iones, por lo que
introdujo el concepto de GRADO DE DISOCIACION ∝ , como la fracción de las moléculas que
están disociadas en iones. El grado de disociación se determina experimentalmente como
la relación de la conductancia equivalente Ʌ a la conductancia equivalente a dilución al
infinito λo
∝ = Ʌ
λo (I)
Si consideramos que un soluto univalente acuoso [ MX(acuoso)] se disocia en M+ + X-
MX(Aq) → M+ + X-
La constante de equilibrio k = [ 𝑀+].[𝑋−]
[𝑀𝑋(𝑎𝑞)]

Los coeficientes se presentan en concentración mol/l
La concentración total del soluto en equivalentes gramo/litro es C y ∝C es la concentración
de los iones
[ 𝑀+] = [𝑋−] = ∝C, entonces [𝑀𝑋(𝑎𝑞)] = ( 1 - ∝ )C
Luego k = ∝2𝐶2
(1−∝)𝐶 =
∝2𝐶
1−∝ (II)
Remplazando I en II:
k = (
Ʌ
𝜆𝑜 )2𝐶
1− Ʌ
𝜆𝑜
= (
Ʌ
𝜆𝑜 )2𝐶
𝜆𝑜− Ʌ
𝜆𝑜
= Ʌ2𝐶
𝜆𝑜(𝜆𝑜−Ʌ) Ley de Dilución de Ostwald
Esta ley es aplicada a valores experimentales de conductancia equivalente para el ácido
acético ( CH3 –COOH) y el KCl en agua. Un electrolito que se comporta como el ácido
acético con baja disociación se denomina electrolito débil, mientras que electrolitos con
mayor o total disociación, como el KCl se denominan electrolitos fuertes.
Termodinámica de las Soluciones
La relación entre la constante de equilibrio y la energía libre de una sustancia, está dada
por la relación isotérmica de Van’t Hoft:
∆G = -RTlnk + RTlnQ k = Constante de equilibrio y Q = Constante de actividad
Para la reacción:
aA + bB + ---------- = cC + dD + -------
Q = 𝑎𝐶
𝑐 . 𝑎𝐷𝑑
𝑎𝐴𝑎 . 𝑎𝐵
𝑏
La energía libre se puede expresar por la siguiente ecuación:
∆G = ∆Go + RTlnQ
Donde ∆Go es la energía libre en el equilibrio, es decir, cuando las actividades de los
constituyentes de la reacción es la unidad, por consiguiente en el equilibrio ∆G = 0 y
∆Go = -RTlnk
Para establecer la relación entre la energía libre y el potencial de electrodo, se debe
recordar que el primer y segundo principios de la termodinámica establecen:

dU = dQ – dW y dS = 𝑑𝑄
𝑇 o dQ = TdS
Luego dU = TdS - dW
En esta ecuación W representa a todo tipo de trabajo, como la compresión, expansión,
magnética, eléctrica, etc.
Bajo las condiciones de presión constante, que es el caso más general en los procesos
electrometalúrgicos, no se requiere compresión de volumen o trabajo de expansión, de
modo que todas las otras formas de trabajo no serán requeridas, excepto el eléctrico,
entonces tenemos:
dU = TdS – dW’
Por lado la energía libre G = H – TdS
También: G = U + PV – TS entonces dG = dU + PdV + VdP – TdS – SdT
A volumen y presión constantes:
dGV,P = dU – TdS – SdT
A volumen, presión y temperatura constantes:
dGV,P,T = dU – TdS , pero dU = TdS – dW’
Por consiguiente: dGV,P,T = TdS – TdS – dW’ entonces dGV,P,T = - dW’
Luego: ∆GV,P,T = W’
Por consiguiente se tiene trabajo realizado al transportar una carga eléctrica desde un
potencial a otro, pero este trabajo es igual al producto de la carga por la diferencia de
potencial:
W’ = Carga x Diferencia de potencial (E) W’ = Carga x E
La carga es igual al producto del número de electrones (n) transferidos al sistema
multiplicado por la constante de Faraday: Carga = nF
Luego W’ = nFE = - ∆G
∆G = - nFE = ∆Go + RT lnQ = - nFEo + RT lnQ
Para un proceso reversible a T y P constantes el trabajo hecho en el sistema, sin contar
trabajo de expansión, es igual al cambio de energía libre con signo negativo = - ∆G, n =

número de electrones transferidos o el numero de faradios, E = fuerza electromotriz (fem)
o diferencia de potencial aplicado al sistema y Eo = fem estándar
- nFE = - nFEo + RT lnQ dividiendo entre nF
E = Eo - 𝑅𝑇
𝑛𝐹 lnQ (Ecuación de Nernst)
Esta ecuación expresa la variación de la fem de una pila electroquímica en función de los
reactantes y productos del sistema y se conoce como la ecuación de Nernst.
Cuando se trabaja al estado estándar o al equilibrio se tiene que E = 0, siendo Eo el
potencial normal de la pila o celda;
Eo = 𝑅𝑇
𝑛𝐹 lnk
Al expresar Q en función de la concentración y actividad, vale decir la concentración de los
iones metálicos en g/l, podemos expresar los potenciales de electrodo para el catión y el
anión
E S1 = ESo + 0.0592
𝑛 log Cc (Cationes)
E S2 = ESo + 0.0592
𝑛 log Ca (Aniones)
Donde ESo es el potencial del electrodo normal en el equilibrio y la constante 0.0592 se
obtiene a 298oK, R = 8.32 joules y 96500 coulombs y convirtiendo el logaritmo natural a
logaritmo vulgar en base 10.
0.0592 = 8.32 𝑥 298 𝑥 2.303
96 500
Ecuación de Gibbs – Helmholtz
La relación entre la energía eléctrica de un sistema y el calor de reacción, es una función de
la temperatura y está dada por la ecuación de Gibbs – Helmholtz y se deriva de la
siguiente forma:
∆G = ∆H - T∆S y ∆S = - 𝑑𝐺
𝑑𝑇
Luego ∆G = ∆H + T 𝑑𝐺
𝑑𝑇 , como ∆G = - nFE
- nFE = ∆H – nFT 𝑑𝐸
𝑑𝑇

E = −∆H
𝑛𝐹 + T
𝑑𝐸
𝑑𝑇
ó E = −∆H+T ∆S
𝑛𝐹
Cuando varia 𝑑𝐸
𝑑𝑇 , la fuerza electromotriz de la pila voltaica reversible aumenta cuando
aumenta la temperatura y cuando es igual a cero, la energía eléctrica es igual a la energía
química. Los potenciales de los electrodos están referidos al potencial estándar del
electrodo de hidrógeno, que es igual a cero. Esta convención arbitraria se estableció en
adición a la convención de asumir que la energía libre de formación estándar del ion
acuoso hidrógeno es cero.
H+ + 1e- = ½ H2 (g) Eo = 0 y ∆Go = 0
UNIDAD DIDACTICA 3. Duración nueve semanas
EXTRACCION DE COBRE DE MINERALES SULFURADOS El cobre en la naturaleza se encuentra distribuido en la corteza terrestre principalmente formando parte de los minerales sulfurados, como la calcopirita, bornita y calcosina, y también está presente en forma de minerales oxidados, formado carbonatos, óxidos, silicatos y sulfatos. Para la extracción del cobre a partir de sus minerales se emplean procesos pirometalúrgicos, hidrometalúrgicos y electrometalúrgicos, en forma combinada de acuerdo a la naturaleza del mineral, así para los minerales sulfurados se combinan procesos piro y electrometalúrgicos y para los óxidos se usan principalmente los procesos hidro y electrometalúrgicos. El contenido de cobre en los yacimientos explotados es baja, varía de un 0.5% en minería a cielo abierto a 1 ó 2% en minería subterránea, por lo que se hace necesario un proceso de concentración previa para aplicar procesos de extracción. El proceso de extracción seleccionado depende de las características de mineral y es particular en cada caso. Los diagramas siguientes, ilustran las dos principales métodos de extracción del cobre a partir de sus minerales, sin que esto quiera decir que son los únicos, pues existen muchos métodos.


3.1. Concentrados de cobre – principales minerales. El 90% o más de cobre que se produce en el mundo proviene de los minerales sulfurados, que son difíciles de tratar hidrometalúrgicamente, por lo que la mayor parte se trata pirometalúrgicamente. Normalmente, el proceso de extracción comprende cinco etapas:
a. Concentración por flotación se obtiene como producto el concentrado b. Tostación, es optativo, depende de la composición del concentrado, así, si contiene
As, Sb, (es el caso peruano en la región Centro del Perú) será necesario la tostación. c. Fundición de matas, en procesos discontinuos o continuos d. Conversión de cobre blíster e. Piro o electrorefinación
Normalmente se hacen combinaciones según las características del concentrado y un paso siguiente de la electrorefinación del cobre es el tratamiento de los lodos anódicos para la recuperación del oro, la plata, el selenio, el teluro y otros metales preciosos. Los concentrados de cobre provienen de las celdas de flotación y son el resultado de la trituración, chancado y molienda de los minerales sulfurados de las minas. El producto de la flotación es el concentrado y un residuo que constituyen los relaves o colas. La composición química de los concentrados declarada, se reduce generalmente a tres elementos: cobre, oro, plata y se informa el contenido porcentual de cobre en el concentrado, del orden del 30% y en g/ton o Oz/ton los de oro y plata. Los concentrados de
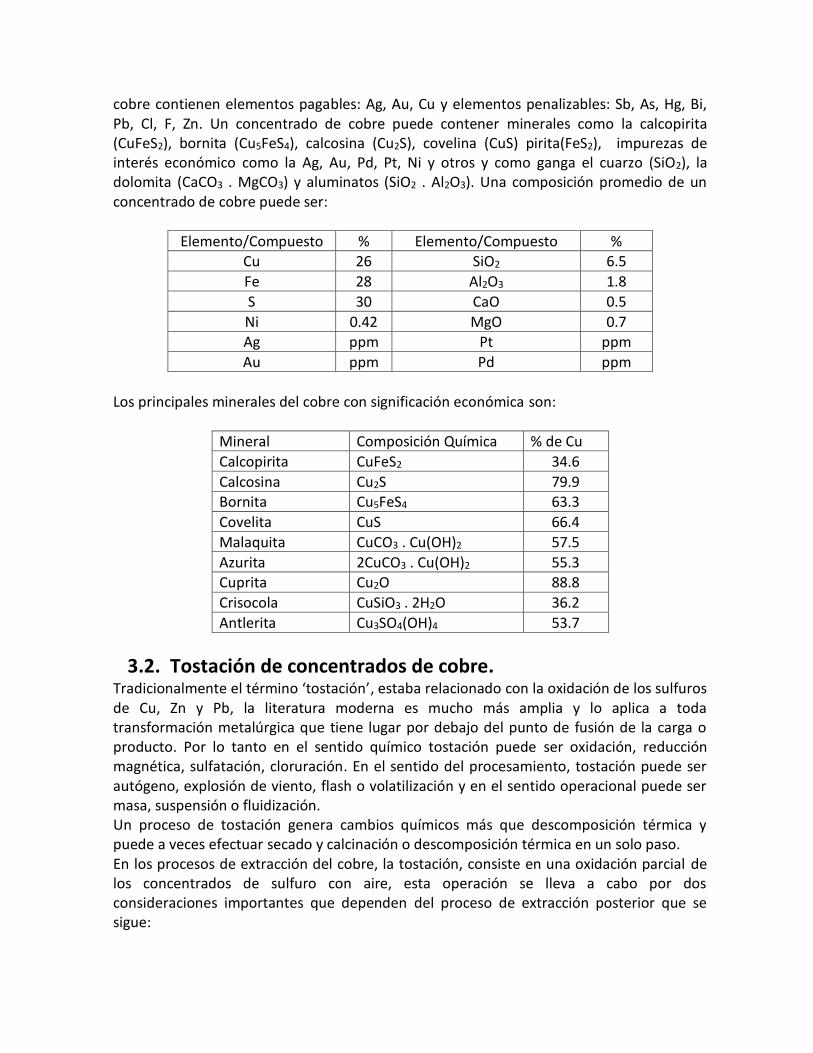
cobre contienen elementos pagables: Ag, Au, Cu y elementos penalizables: Sb, As, Hg, Bi, Pb, Cl, F, Zn. Un concentrado de cobre puede contener minerales como la calcopirita (CuFeS2), bornita (Cu5FeS4), calcosina (Cu2S), covelina (CuS) pirita(FeS2), impurezas de interés económico como la Ag, Au, Pd, Pt, Ni y otros y como ganga el cuarzo (SiO2), la dolomita (CaCO3 . MgCO3) y aluminatos (SiO2 . Al2O3). Una composición promedio de un concentrado de cobre puede ser:
Elemento/Compuesto % Elemento/Compuesto % Cu 26 SiO2 6.5
Fe 28 Al2O3 1.8
S 30 CaO 0.5
Ni 0.42 MgO 0.7
Ag ppm Pt ppm
Au ppm Pd ppm
Los principales minerales del cobre con significación económica son:
Mineral Composición Química % de Cu
Calcopirita CuFeS2 34.6
Calcosina Cu2S 79.9 Bornita Cu5FeS4 63.3
Covelita CuS 66.4
Malaquita CuCO3 . Cu(OH)2 57.5
Azurita 2CuCO3 . Cu(OH)2 55.3 Cuprita Cu2O 88.8
Crisocola CuSiO3 . 2H2O 36.2
Antlerita Cu3SO4(OH)4 53.7
3.2. Tostación de concentrados de cobre. Tradicionalmente el término ‘tostación’, estaba relacionado con la oxidación de los sulfuros de Cu, Zn y Pb, la literatura moderna es mucho más amplia y lo aplica a toda transformación metalúrgica que tiene lugar por debajo del punto de fusión de la carga o producto. Por lo tanto en el sentido químico tostación puede ser oxidación, reducción magnética, sulfatación, cloruración. En el sentido del procesamiento, tostación puede ser autógeno, explosión de viento, flash o volatilización y en el sentido operacional puede ser masa, suspensión o fluidización. Un proceso de tostación genera cambios químicos más que descomposición térmica y puede a veces efectuar secado y calcinación o descomposición térmica en un solo paso. En los procesos de extracción del cobre, la tostación, consiste en una oxidación parcial de los concentrados de sulfuro con aire, esta operación se lleva a cabo por dos consideraciones importantes que dependen del proceso de extracción posterior que se sigue:

a. Extracción hidrometalúrgica, los sulfuros de cobre no se lixivian fácilmente, pero sus sulfatos son solubles en agua y sus óxidos en ácido sulfúrico diluido, por lo tanto, un proceso de tostación controlado de sulfuros puede producir un producto calcinado rápidamente lixiviable. Este proceso se usa en varias partes del mundo como Zaire y Zambia.
b. Extracción pirometalúrigica, a menudo se practica la tostación como paso previo a la fundición en hornos reverbero y en horno eléctrico, con el propósito de utilizar el calor de tostación para secar y calentar la carga antes de introducir al horno de fundición y para aumentar la concentración del Cu en el producto fundido, así como disminuir el contenido de S en el la mata. En el caso del Perú, en la fundición de la Oroya, se hace la tostación para controlar el contenido de S y eliminar los contenidos de As y Sb de los concentrados sucios.
El proceso de tostación es la misma para ambos casos y se usan tostadores de hogares múltiples, flash o lecho fluidizado. La tostación para la extracción del cobre se realiza entre 500 a 700oC y normalmente es autógena, pero si los concentrados están muy húmedas se puede requerir combustible. El contenido de SO2 en los gases de tostación varía entre el 5 al 15%, lo que facilita su recuperación para la producción de ácido sulfúrico. 3.3. Tostación para fusión de concentrados – Tostación para lixiviación. Tostación para fusión de concentrados, La tostación controlada de los concentrados de cobre da por resultado la oxidación parcial del azufre a SO2 y la oxidación parcial de los sulfuros de Fe a sulfatos y óxidos de Fe. El gas SO2 se elimina durante la tostación y los óxidos y sulfatos de Fe se eliminan posteriormente en forma de escoria durante la fundición. Los óxidos y sulfatos de cobre también se forman durante la tostación parcial, pero se reducen de nuevo a sulfuro dentro del horno de fundición, por la afinidad diferenciada que tienen el Cu y el Fe por el oxígeno y el azufre, como se explicó en la termodinámica de fusión de concentrados. La tostación permite eliminar el S como SO2, acondicionar el Fe para su eliminación en las escoria de los hornos de fusión y lograr matas con alto grado de cobre. El grado de mata expresa el contenido de cobre en la mata. La etapa de tostación se usó principalmente en las fundiciones que empleaban hornos de reverbero, algunos todavía lo usan ejemplo la Fundición de la Oroya – Perú, que trataban concentrados de cobre con leyes menores al 20%. En la actualidad las técnicas mejoradas de beneficio de minerales han eliminado grandemente la necesidad de fundir concentrados de baja ley y por lo tanto la tendencia ha sido eliminar los tostadores del circuito de fundición de concentrados de cobre. Sin embargo, el empleo de hornos de lecho fluidizado de alta eficiencia, ha hecho que aún se usen los procesos de tostación en plantas que aún usan hornos reverbero y eléctrico, con el propósito de aumentar la capacidad de producción de estos hornos, por que las calcinas calientes se cargan directamente al horno y así se requiere menos energía para fundir concentrados frios o húmedos, teniendo por resultado un ahorro considerable de energía y mayor velocidad de fusión. Cargar una calcina caliente a un horno reverbero puede aumentar la producción de éste hasta en un 50% según datos bibliográficos.

Igualmente la velocidad de producción de cobre en los convertidores se incremente, debido a que en la tostación se elimina parte del S y la tostación/fundición se elimina parte del Fe, el resto se elimina en la etapa de conversión. Tostación para lixiviación, La lixiviación de los calcinados de tostación casi siempre se lleva a cabo junto con la lixiviación de las menas de óxido naturales. Las calcinas contribuyen al proceso total en dos formas:
a. Proporcionan sulfatos de cobre solubles y óxidos de cobre que agregan cobre al circuito de lixiviación
b. El sulfato de cobre en los calcinados adiciona iones 𝑆𝑂4−2 a la solución, los que
favorecen la formación del H2SO4 durante la electrodeposición del cobre, a través
de la reacción: Cu+2 + 𝑆𝑂4−2 + H2O
2𝑣→ Cuo + ½ O2 + H2SO4
Este ácido sulfúrico actúa como compensador por las pérdidas ocurridas durante la lixiviación de las menas de óxidos naturales. El producto del tostador debe contener suficiente sulfato de cobre para mantener el equilibrio de H2SO4 del proceso de lixiviación – electrodeposición, el resto del cobre está como óxido de cobre. Para lograr la suficiente cantidad de sulfato en la calcina la tostación debe ser oxido – sulfatante.
3.4. Hornos de tostación. Antiguamente la tostación se realizaba en montones y patios en áreas abiertas donde los gases de tostación escapaban al ambiente. Estos procesos fueron remplazados por procesos en hornos en columna a través del cual se forzaba el aire. El desarrollo de las técnicas se fueron renovando constantemente hasta llegar a los actuales procesos de tostación, como los hornos de hogares múltiples, tostación flash y lecho fluidizado. a.- Tostación en Hogares Múltiples, El tostador de hogar múltiple es un reactor cilíndrico revestido de material refractario de aproximadamente de 6 m de diámetro por 15 m de altura, acondicionado con 7 ó 14 hogares o pisos. Los concentrados se alimentan por la parte superior del tostador y se rastrillan en cada uno de los hogares antes de caer en el siguiente hogar inferior. A los rastrillos los mueve un eje central giratorio. El aire para la tostación ingresa por la parte inferior y sube en contracorriente con la carga que desciende y deja el horno a través de un ducto de humos instalado en la parte superior. La capacidad de estos hornos varía de 180 a 240 TM/día a más. El tostador se calienta hasta alcanzar una tempera tuta de ignición del concentrado, entonces se alimenta el concentrado y se inyecta el aire en sus direcciones a contracorriente normales provocando la combustión del concentrado. Los dos hogares superiores normalmente se utilizan para el secado y calentamiento de los concentrados, mientras que el encendido y las reacciones de oxidación ocurren en los hogares inferiores. Las reaccione principales de oxidación son exotérmicas, de modo que una vez iniciado el proceso la tostación se hace autógena, las reacciones que pueden ocurrir son: CuFeS2 + 4O2 → CuSO4 + FeSO4 ∆Ho 298 = - 360 000 kcal/kg mol CuFeS2

2CuFeS2 + 13
2 O2 → 2CuO + Fe2O3 + 4SO2↑ ∆Ho 298 = - 232 000 kcal/kg mol CuFeS2
Si los concentrados tienen mucha humedad, se utiliza combustible. Estos tostadores procesan de 0.5 a 1.0 toneladas de caga por m2 por día, lo que permite eliminar de 0.1 a 0.2 toneladas de azufre por m2 al día. En estos hornos la transferencia de masa y calor ocurre progresivamente a medida que el concentrado atraviesa los hogares, bajando por la columna a través de la serie de hogares, donde el concentrado es movido mediante rastrillos rotatorios, lo que causa que el concentrado avance a través de cada hogar y luego caiga en el siguiente hogar donde el movimiento de remoción continua. El contacto sólido gas es pobre a pesar del movimiento mecánico, por lo que una gran parte de la reacción de tostación se produce al caer el concentrado de un hogar a otro hogar inferior.
b.- Tostación Suspendida, Estos tostadores son una modificación del tostador de hogares múltiples, en éstos, el concentrado es secado y precalentado en los dos hogares superiores, luego el concentrado cae a través de la sección central del horno donde entra en contacto con los gases oxidantes, la recolección de la calcina se realiza en tres hogares situados en la parte inferior del horno. Un desarrollo de este tostador es la tostación flash, en el cual el mineral precalentado es inyectado a través de un ‘quemador’ simultáneamente con el aire precalentado, semejante a un flujo pulverizado, este proceso es más apropiado para la tostación de sulfuros que se oxidan exotérmicamente y no requieren combustible adicional. Para el control de la temperatura se dispone de una zona de expansión. Mediante este proceso se consigue una mayor capacidad de producción. El proceso de tostación suspendida es muy usado para la tostación sulfatante, la cual ocurre en una cámara especial situada debajo de la cámara de reacción principal, de modo que la sulfatación se da después de la oxidación.

Horno de Tostación Suspendida
c.- Tostación en lecho fluidizado, La tostación en lecho fluidizado consiste en la oxidación de las partículas de sulfuro mientras se suspenden en una corriente de aire uniformemente distribuida y se basa en el principio de que el aire inyectado a través de un lecho de sólidos finos tiende a elevar las partículas. A velocidades moderadas, las partículas estarán permanentemente suspendidas en un lecho expandido o fluidizado mientras que a velocidades altas, las partículas serán transportadas fuera del tostador junto con los gases. Son muy usados para la tostación de concentrados finos debido a que los coeficientes de calor y masa alcanzados entre el sólido y el gas son altos. El flujo gaseoso debe ser considerablemente alto, de modo que la caída de presión a través de la cama sea independiente de la velocidad del gas, la cámara bajo estas condiciones asemeja a un líquido hirviendo con burbujas reventando en su superficie. Este proceso llamado también ‘fluidización agresiva’, debe ser distinguido de la ‘fluidización particulativa’, donde la caída de presión a través de la cama, disminuye cuando aumenta el flujo de gas circulante. En la fluidización particulativa el contacto sólido-gas es parcial y se semeja a las condiciones encontradas en los equipos de clasificación hidráulicas, como el Jigs, Por contraste en la fluidización agresiva, el contacto sólido-gas es total, además el flujo gaseoso es altamente turbulenta, de modo que los sólidos son agitados en todas las direcciones con alta energía, pero la velocidad relativa entre el sólido y el gas no escapa de los límites de un flujo laminar. Las velocidades de transferencia de masa y calor en camas fluidizadas son altas. La distribución de la temperatura en el lecho es uniforme debido a la transferencia de calor por convección como consecuencia del movimiento rápido y al azar de las partículas. Se

supone que la transferencia de calor entre las partículas es por conducción y se da durante las colisiones entre éstas, y estas colisiones permiten romper las películas de gas formadas en la superficie. Las aplicaciones de la tostación en cama fluidizada en metalurgia extractiva, además de la tostación oxidante, es la tostación sulfatante, donde el control de la temperatura es muy importante, particularmente si se conduce una sulfatación diferencial, igualmente es importante el control de las presiones parciales del SO2 y el O2. En un proceso de sulfatación diferencial las condiciones de temperatura y presión deben ser tales que permitan la formación de un sulfato de uno de los metales que sea soluble en agua y los otros metales formen óxidos o silicatos insolubles, de modo que permita la separación por lixiviación en agua. La formación de sulfatos ocurre bajo condiciones oxidantes y a alta presión de SO2, ocurriendo simultáneamente las reacciones de oxidación y sulfatación. La temperatura de trabajo debe ser menor a la temperatura de descomposición del sulfato. Los gases generados en los hornos de lecho fluidizado son ricos en SO2 lo que facilita la producción del ácido sulfúrico.
TEMPERATURA DE DESCOMPOSICION DE ALGUNOS SULFATOS
Sulfato Temperatura oC Sulfato Temperatura oC
Al2(SO4)3 ± 700 NiSO4 900
CuSO4 850 CoSO4 ± 980
ZnSO4 850 MnSO4 ± 1000
Fe2(SO4)3 710 Cr2(SO4)3 ± 300
Horno de Tostación de Lecho Fluidizado

3.5. Química y cinética de la tostación. Las reacciones principales durante la tostación de concentrados de cobre, son la oxidación de los sulfuros de Fe y Cu para obtener sulfatos y óxidos, algunas e las reacciones más importantes son: CuFeS2 + 4O2 → CuSO4 + FeSO4
2CuS + 7
2O2 → CuO. CuSO4 + SO2↑
2CuFeS2 + 13
2O2 → 2CuO + Fe2O3 + 4SO2↑ (Oxidación completa)
Muchas reacciones se presentan durante un proceso de tostación parcial y es posible tener en el mismo producto tanto óxidos como sulfatos. La temperatura de operación del tostador es un factor importante debido a que determina el tipo de producto obtenido. Una buena estimación de la mejor temperatura de operación para un producto dado puede obtenerse de las composiciones de los gases junto con las relaciones de equilibrio conocidas en los sistemas gas-sólido de Cu-O-S y Fe-O-S. Diagramas de Equilibrio de Cu-O-S y Fe-O-S. En base a los datos termodinámicos se construyen los diagramas de equilibrio o Kellog, que muestran áreas de estabilidad o predominio de los compuestos, para de este modo predecir que fases sólidas pueden estar presentes a una temperatura y composición de gas dados y así predecir las posibles reacciones. Así los diagramas del sistema Cu-O-S indican, para una temperatura dada, los sólidos que estarán en equilibrio con un gas de concentraciones de O2 y SO2 dadas y estos diagramas pueden ser usados para predecir los sólidos que se producirán en un tostador si las reacciones sólido-gas llegan al equilibrio. La tostación parcial no es un proceso en equilibrio, no obstante los diagramas pueden ser usados para indicar los productos que más probablemente serán formados a medida que prosigan las reacciones hasta alcanzar el equilibrio.
Cualquier punto del diagrama de equilibrio está determinado por la intersección del log 𝑝𝑂2 vs log 𝑝𝑆𝑂2, lo que significa que a una temperatura dada es posible determinar las fases en equilibrio estable en el área, el límite y el punto de equilibrio, ejemplo para el sistema Cu-O-S a 627oC
Punto 𝑝𝑂2 𝑝𝑆𝑂2 Región Fases en Equilibrio Estable
A 10-4 1 Area CuSO4 CuSO4
B 10-5 10-1 Limite CuSO4/CuO.CuSO4 CuSO4 + CuO.CuSO4 C 10-5.3 101.5 Punto de intersección
CuO/Cu2O/ CuO.CuSO4 CuO + Cu2O + CuO.CuSO4
Los gases que salen de los tostadores industriales tienen composiciones que varían de 5 a 15% de SO2 y de 1 a 5% de O2, más de 10% de H2O y un 75% N2, los cuales son equivalentes a las presiones parciales de 𝑝𝑆𝑂2 = 10-1.5 a 10-0.5 y 𝑝𝑂2 = 10-2 a 10-1. Las fases sólidas en equilibrio potencial son fácilmente representadas al trazar estas presiones parciales del gas del tostador sobre los diagramas de equilibrio tanto para el Cu-O-S como para el Fe-O-S. También, estos diagramas permiten seleccionar la temperatura de tostación, así para evitar la formación del Fe3O4 y Fe2O3 o de las ferritas, que se producen por una sobreoxidación del Fe y son perjudiciales en el proceso de fusión, es mejor tostar a temperaturas relativamente bajas, así en la práctica industrial se trabaja entre 500 a 600oC. Cinética de la Tostación, La oxidación de los sulfuros con aire es de naturaleza heterogénea, las velocidades de tostación dependen del tamaño de la partícula 8área superficial) y la intensidad del contacto aire/partícula, así como de la concentración de oxígeno y la temperatura del gas. Un factor importante es la temperatura de ignición o encendido, que viene a ser la temperatura más baja la cual una partícula reacciona lo suficientemente rápido para conservar o aumentar la temperatura del reactor. En la siguiente tabla se presenta la temperatura de encendido de sulfuros normalmente presentes en la tostación de concentrado de cobre.
Temperatura de Ignición de Minerales Sulfurados
Mineral Temperatura de Ignición oC
CuS ≈ 400 Cu2S 450
CuFeS2 300
FeS2 400
FeS 400 La tabla indica que para concentrados secos se puede iniciar y mantener una temperatura de casi 400oC y cuando se trate concentrados húmedos se requerirá temperaturas mayores para vaporizar el agua. En general se obtienen velocidades mayores al operar a temperaturas altas y con una buena exposición de superficie de mineral a la atmósfera oxidante.

A diferencia de la calcinación o descomposición térmica, todas las reacciones en tostación son exotérmicas, por lo que no es probable controlar la velocidad de transferencia o difusión del calor, en la tostación la temperatura es lo suficientemente alta en todas partes, lo que permite la reacción química deseada del proceso o por lo menos en la isoterma de reacción delante del frente de reacción. Cada partícula es oxidada desde el exterior y el núcleo interno permanece inalterado, de modo que una cáscara de producto sólido está en contacto con el gas, en la atmósfera entre estas zonas ocurre las reacción de tostación a condición de que el radio 𝑝𝑂2 / 𝑝𝑆𝑂2 es localmente más alto que el radio de equilibrio para la reacción a la temperatura predominante, de modo que se cumpla la condición de que el flujo másico de oxígeno (J𝑜2) sea mayor o igual al flujo másico del SO2 (J𝑠𝑜2). El flujo depende del coeficiente de difusión D y la gradiente de la presión parcial, por debajo del cual tiene lugar la transferencia de masa. En la interface, el mecanismo de reacción probablemente ocurra en varios períodos: 1o.- El oxígeno es absorbido en la superficie del S 2o.- Esta produce electrones que van a incorporarse a la rejilla del mineral Los electrones neutralizan el ion del azufre cerca del sitio de la superficie como unidades con otro oxígeno molecular absorbido cerca de él. El SO2 formado se desabsorbe y migra hacia afuera, dejando una vacante en el sitio del ion azufre en la superficie del mineral, entonces otro ion azufre puede difundirse desde el interior a ocupar este sitio y así continuar la reacción. Cuando una capa de la superficie es agotada, la interface avanza hacia el interior del mineral hasta alcanzar más iones azufre. Una reacción secundaria importante en la tostación es la formación de sulfatos, probablemente en una zona de reacción más exterior y fría de la cáscara de óxido, por cuanto la mayoría de los sulfatos se descomponen por encima de 900oC. Con la provisión de SO2, la formación del sulfato debe permitir operar a un radio más bajo de 𝑝𝑂2 / 𝑝𝑆𝑂2, sin afectar la velocidad de la reacción principal. La sulfatación de un concentrado fino será restringido a una parte separada del horno, donde las temperaturas son más bajos y el contenido de SO2 de la atmósfera claramente más alta que en la zona de oxidación.
3.6. Desarsenización de Concentrados de Cobre Algunas empresas mineras del Perú, aún producen concentrados de cobre con mediano o alto contenido de As y Sb, elementos que afectan la calidad del cobre refinado final como también afectan la calidad ambiental. Una solución para ambos problemas es separar estos elementos por tostación parcial oxidante, llamado tostación desarsenizante. Los concentrados de cobre con contenidos de As se clasifican en:
a. Concentrados limpios, cuando el contenido de As < 0.1% caso Toquepala, Cuoajone, Tintaya, Cerro Verde, en general las minas ubicadas en el sur peruano
b. Concentrados medios, con contenidos de As > 0.1 < 0.3%, caso Condestable, Madrigal, Cobriza
c. Concentrado sucios, cuando el contenido de As es > al O.3%, como los de Minsur, Yauricocha, Casapalca, El Brocal, Huaron, etc, la mayoría ubicados en la región central peruana.
En el Perú, la tendencia de la producción de concentrados sucios es decreciente desde hace más de 30 años, debido a que las fundiciones no aceptan concentrados con alto contenido

de impurezas, sobre todo por las medidas de control ambiental que prohíben su tratamiento. La fundición de cobre del Complejo Metalúrgico de la Oraya, ubicado en la región central del Perú, es una de las pocas que aún trata este tipo de concentrados, por que en su circuito dispone de un sistema de obtención del As2O3 como un subproducto. Mecanismo de la Tostación Desarsenizante, Los investigadores peruanos José Vidalón y Henry Walqui, plantean que la tostación de desarsenización, viene a ser una tostación parcial en condiciones controladas de temperatura, entre 650 a 750oC, que impiden la fusión del material y de composición de la atmósfera del horno que permita sólo una oxidación parcial de los sulfuros, lo que se consigue limitando el acceso del aire al horno, de modo que se tenga una atmósfera neutro o ligeramente reductora. Al inicio del proceso de produce la descomposición térmica de algunos sulfuros y sulfosales, como se ve en las siguientes reacciones: Enargita: 2Cu3AsS4 = 4CuS + Cu2S + As2S3 Covelita: 4CuS = 2Cu2S + S2 – 8 400 cal Chalcopirita: 4CuFeS2 = 2Cu2S + 4FeS + S2 Pirita: 2FeS2 = 2FeS + S2 – 39 600 cal El sulfuro de arsénico y el azufre al estado gaseoso, pasan a la atmósfera del horno para luego ser oxidados con aire en una cámara de combustión o un nivel dado, según el tipo de horno empleado para el proceso. Las reacciones de oxidación del S2 y del As2S3 son: 2As2S3 + 9O2 = 2 As2O3 + 6SO2 S2 + 2O2 = 2SO2 + 141 920 cal. Algunos sulfuros, en presencia del oxígeno de la atmósfera del horno, reaccionan con dicho elemento aún a temperaturas inferiores a su punto de descomposición. Cuando hay deficiencia de oxígeno, el arsénico se volatiliza como As2O3, mientras que si la tostación se realiza en un gas inerte se logra As o As2S3. La tostación con exceso de oxígeno da lugar a la oxidación de los sulfuros de cobre y hierro, mediante reacciones exotérmicas siguientes: 2FeS + 3O2 = 2FeO + SO2 6FeO + O2 = 2Fe3O4 4FeO + O2 = 2Fe2O3 Cu2S + 2O2 = 2CuO + SO2 S + O2 = SO2 2As2S3 + 11O2 + 2As2O5 + 6SO2 En estas condiciones, y principalmente a mayores temperaturas, el Fe2O3 es estable y tiende a formar ferritas con los otros óxidos: Fe2O3 + CuO = CuO. Fe2O3 Fe2O3 + As2O5 = 2FeAsO = (As2O5. Fe2O3)

Los reactores que pueden ser usados para la desarsenización de concentrados de cobre son hornos de lecho fluidizado o de hogares múltiples, cuyos diagramas de flujo se presentan a continuación.
3.7. Fusión de concentrados de cobre. Los concentrados sulfurados, principalmente los concentrados que contienen sulfuros de Cu, sulfuros de Ni y sulfuros de Cu-Ni, son tratados por procesos de fusión y conversión. Son procesos que se llevan acabo a altas temperaturas con el fin de recuperar el metal valioso de sus concentrados. Básicamente el proceso de fusión consiste en calentar los concentrados por encima del punto de fusión, para separar los sulfuros metálicos en forma de mata y otros valores como el Au, la Ag de los componentes no deseables del concentrado y que generalmente está constituido por óxidos que al combinarse con los silicatos forman la escoria, que por ser menos densa e inmiscible con la mata, flota por encima de ésta. La mata obtenida por fusión, es tratada posteriormente en los convertidores para logar un producto metálico, eliminado por reacciones oxidación a los elementos no deseados, como son el Fe y el S, el Fe pasa a la fase escoria formando oxisilicatos y el azufre es eliminado del sistema como SO2. Los concentrados oxidados también pueden ser tratados por procesos de fusión, tenemos como ejemplos la reducción por el C del Fe, Pb, Pb-Zn en los altos hornos y el proceso de reducción electrotérmica para la obtención del Al. La producción de mata líquida y su posterior conversión a cobre blíster o ampolloso, es el método más importante de extracción de cobre a partir de minerales sulfurados y tiene como ventajas, frente a otros métodos, los siguientes:
a. Se obtiene cobre metálico a partir de sus sulfuros con un gasto de energía relativamente bajo
b. El cobre se produce a una alta velocidad
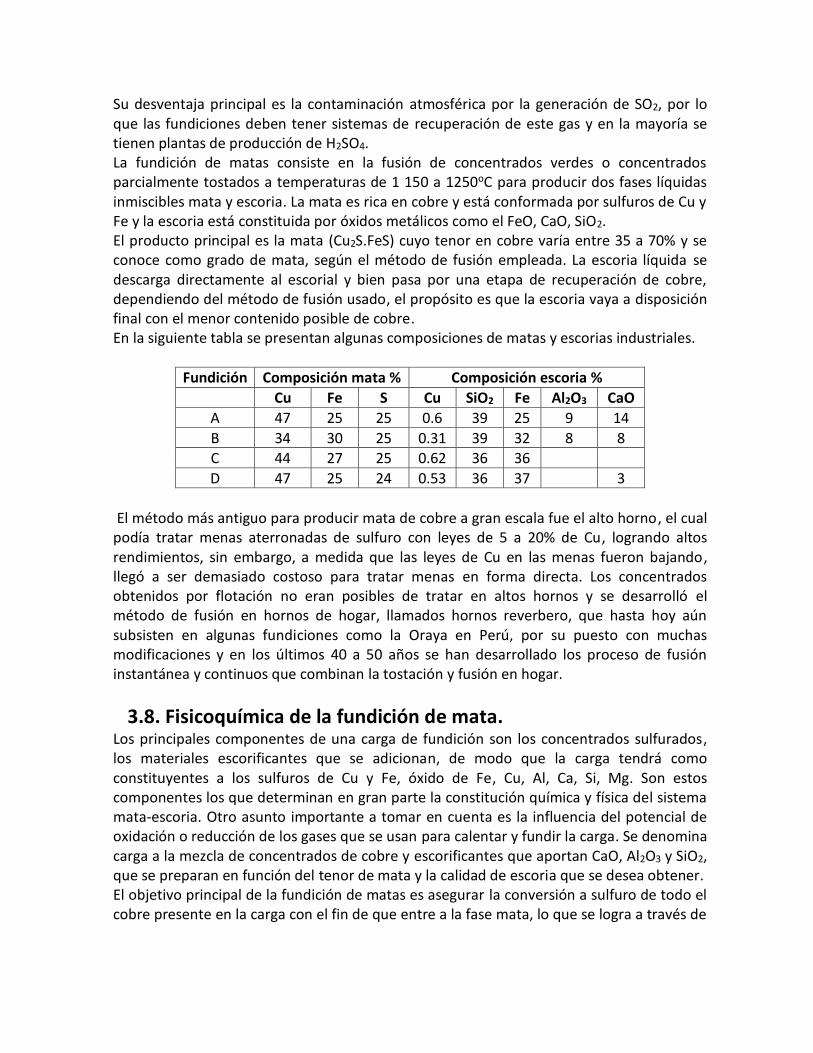
Su desventaja principal es la contaminación atmosférica por la generación de SO2, por lo que las fundiciones deben tener sistemas de recuperación de este gas y en la mayoría se tienen plantas de producción de H2SO4. La fundición de matas consiste en la fusión de concentrados verdes o concentrados parcialmente tostados a temperaturas de 1 150 a 1250oC para producir dos fases líquidas inmiscibles mata y escoria. La mata es rica en cobre y está conformada por sulfuros de Cu y Fe y la escoria está constituida por óxidos metálicos como el FeO, CaO, SiO2. El producto principal es la mata (Cu2S.FeS) cuyo tenor en cobre varía entre 35 a 70% y se conoce como grado de mata, según el método de fusión empleada. La escoria líquida se descarga directamente al escorial y bien pasa por una etapa de recuperación de cobre, dependiendo del método de fusión usado, el propósito es que la escoria vaya a disposición final con el menor contenido posible de cobre. En la siguiente tabla se presentan algunas composiciones de matas y escorias industriales.
Fundición Composición mata % Composición escoria %
Cu Fe S Cu SiO2 Fe Al2O3 CaO
A 47 25 25 0.6 39 25 9 14
B 34 30 25 0.31 39 32 8 8
C 44 27 25 0.62 36 36
D 47 25 24 0.53 36 37 3
El método más antiguo para producir mata de cobre a gran escala fue el alto horno, el cual podía tratar menas aterronadas de sulfuro con leyes de 5 a 20% de Cu, logrando altos rendimientos, sin embargo, a medida que las leyes de Cu en las menas fueron bajando, llegó a ser demasiado costoso para tratar menas en forma directa. Los concentrados obtenidos por flotación no eran posibles de tratar en altos hornos y se desarrolló el método de fusión en hornos de hogar, llamados hornos reverbero, que hasta hoy aún subsisten en algunas fundiciones como la Oraya en Perú, por su puesto con muchas modificaciones y en los últimos 40 a 50 años se han desarrollado los proceso de fusión instantánea y continuos que combinan la tostación y fusión en hogar.
3.8. Fisicoquímica de la fundición de mata. Los principales componentes de una carga de fundición son los concentrados sulfurados, los materiales escorificantes que se adicionan, de modo que la carga tendrá como constituyentes a los sulfuros de Cu y Fe, óxido de Fe, Cu, Al, Ca, Si, Mg. Son estos componentes los que determinan en gran parte la constitución química y física del sistema mata-escoria. Otro asunto importante a tomar en cuenta es la influencia del potencial de oxidación o reducción de los gases que se usan para calentar y fundir la carga. Se denomina carga a la mezcla de concentrados de cobre y escorificantes que aportan CaO, Al2O3 y SiO2, que se preparan en función del tenor de mata y la calidad de escoria que se desea obtener. El objetivo principal de la fundición de matas es asegurar la conversión a sulfuro de todo el cobre presente en la carga con el fin de que entre a la fase mata, lo que se logra a través de

la presencia del FeS en la mata, el cual tiende a convertir en sulfuro, virtualmente, todo el cobre que no está como sulfuro, por reacciones del tipo: FeS(l) + Cu2O(l, escoria) FeO(l,escoria) + Cu2S(l) ∆Go (Johansen, 1970) = - 35000 + 4.6ToK kcal(kg mol)-1
La constante de equilibrio para esta reacción es: KE = 𝑎𝐶𝑢2𝑆 .𝑎𝐹𝑒𝑂
𝑎𝐶𝑢2𝑂 .𝑎𝐹𝑒𝑆 y logKE =
−∆Go
4.576𝑇𝑜𝐾
KE para las temperaturas de fusión industrial, de aproximadamente 1200oC, es 10+4, valor que indica que el Cu2O, se transforma casi por completo en Cu2S por acción del FeS a las temperaturas de fusión, lo cual está en concordancia con la experiencia práctica. Esta reacción también se puede utilizar favorablemente para la recuperación del cobre oxidado presente en las escorias del convertidor. Normalmente las escorias de los convertidores son recirculados al horno de fusión, donde el Cu2O se transforma en Cu2S que pasa a la fase mata. El cobre oxidado puede estar presente en la carga en varias formas, ejemplo puede estar como CuO, CuSO4, CuO.CuSO4 o CuO.Fe2O3, estos compuestos también reaccionan para formar el Cu2S durante la fundición. A las temperaturas de fundición de mata el CuS y el FeS2, presentes en el proceso, son inestables debido a sus altas presiones de azufre y se descomponen para formar Cu2S y FeS. La presión de azufre para el CuS es aproximadamente de 100 atm a 600oC y para el FeS es de 5 atm a 700oC.
3.9. Formación Mata - Escoria. Como se ha visto en la sección 2.6 Termodinámica de la fusión – conversión de concentrados, Termodinámica de la Fusión, Schuhmann, demostró la afinidad del Fe y el Cu por el O2 y el S, determinando por que el Cu entra en la mata y no en la escoria y que el Fe tiene mucha mayor afinidad por el oxígeno que el Cu. La mayor afinidad del Cu por el S, frente al Fe está demostrada por el valor negativo ∆Go para la reacción:
2Cu(l) + FeS(l) = Fe(ᴕ) + Cu2S (l) (3) k1300o
C = 3.1 ∆Go = - RT lnk = - 3 530 cal
Igualmente la mayor afinidad del Fe por el O2 frente al cobre, está expresada por alto valor
negativo de ∆Go para la reacción:
Cu2O(l) + FeS(l) = Cu2S (l) + FeO(l) (6) k1300o
C = 4000 ∆Go = - RT lnk = - 26 000 cal
Estos valores han sido calculados para sulfuros y óxidos líquidos puros a 1300OC, para un
sistema mata-escoria industrial, Schuhmann, asumió que la actividad del Cu2S y el FeS no
se diferencian grandemente para un grado de mata moderado, entre 20 a 70%, y
dependiendo de las condiciones de operación, la actividad del Cu2O, se puede estimar:
K = (aCu2Smata)(aFeOEscoria)/(aCu2OEscoria)(aFeSMata) = 4000
Así por ejemplo su radio puede ser aproximadamente 1 y que la actividad del FeO
puede estar ubicado dentro del rango de 0.3 a 0.9, sustituyendo estos valores en la
ecuación anterior, se tiene que: aCu2OEscoria = 10-4
Por consiguiente la presencia del Cu2O en sistema mata-escoria a una actividad mayor
a 10-4 hará que el FeS desplace al cobre de la escoria, por lo tanto la afinidad relativa
del Cu y del Fe por el oxígeno es la razón de que el Cu esté presente en la mata.
Por lo tanto, en los procesos de fusión de concentrados de cobre, se busca la
formación de dos fases inmiscibles y con una marcada diferencia de densidades, que
facilita su separación , la fase mata constituida por sulfuros de Cu y Fe y la fase escoria
constituida por óxidos metálicos con escasa presencia de Cu.
La presencia del O2 en los hornos de fusión es una variable muy importante, por que
determina el contenido de cobre en la escoria, así a mayor presencia de oxígeno en la
atmósfera del horno mayor contenido de cobre en la escoria, una representación
gráfica del efecto de la presión parcial del O2 frente al contenido de Cu2O en la escoria
a 1300oC muestra que cuando la presión parcial del O2 aumenta la solubilidad del Cu2O
en la fase escoria también aumenta.
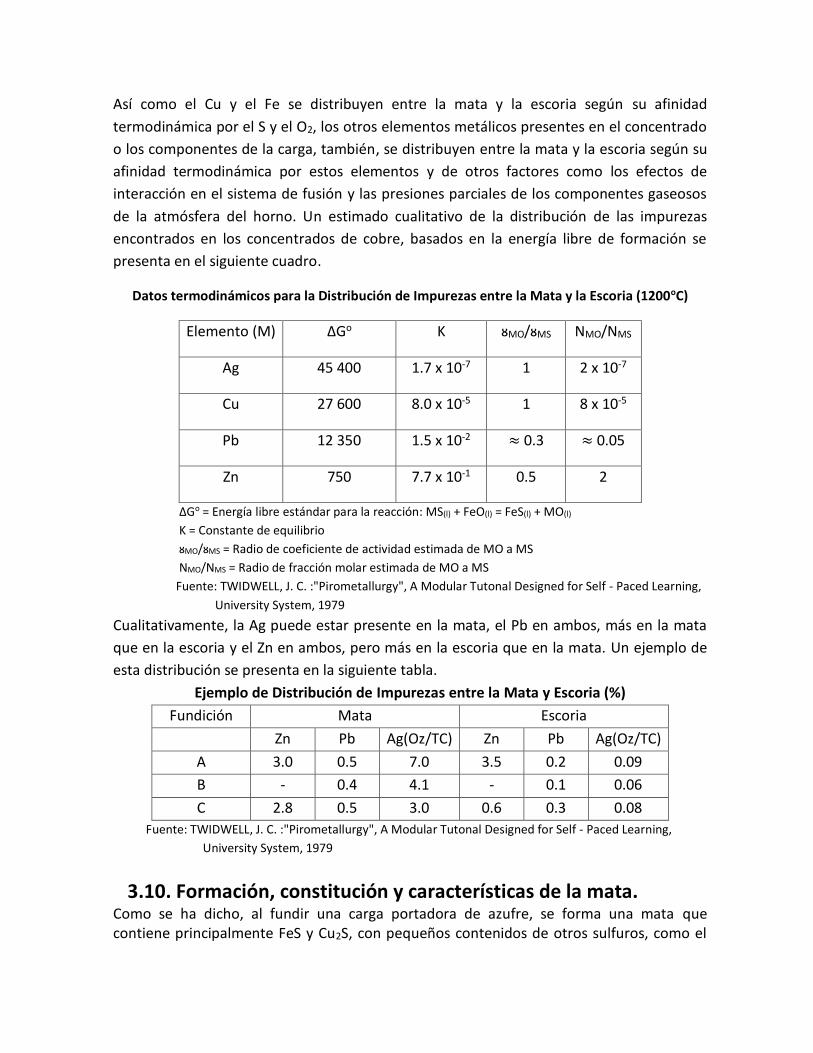
Así como el Cu y el Fe se distribuyen entre la mata y la escoria según su afinidad
termodinámica por el S y el O2, los otros elementos metálicos presentes en el concentrado
o los componentes de la carga, también, se distribuyen entre la mata y la escoria según su
afinidad termodinámica por estos elementos y de otros factores como los efectos de
interacción en el sistema de fusión y las presiones parciales de los componentes gaseosos
de la atmósfera del horno. Un estimado cualitativo de la distribución de las impurezas
encontrados en los concentrados de cobre, basados en la energía libre de formación se
presenta en el siguiente cuadro.
Datos termodinámicos para la Distribución de Impurezas entre la Mata y la Escoria (1200oC)
Elemento (M) ∆Go K ᴕMO/ᴕMS NMO/NMS
Ag 45 400 1.7 x 10-7 1 2 x 10-7
Cu 27 600 8.0 x 10-5 1 8 x 10-5
Pb 12 350 1.5 x 10-2 ≈ 0.3 ≈ 0.05
Zn 750 7.7 x 10-1 0.5 2
∆Go = Energía libre estándar para la reacción: MS(l) + FeO(l) = FeS(l) + MO(l)
K = Constante de equilibrio
ᴕMO/ᴕMS = Radio de coeficiente de actividad estimada de MO a MS
NMO/NMS = Radio de fracción molar estimada de MO a MS
Fuente: TWIDWELL, J. C. :"Pirometallurgy", A Modular Tutonal Designed for Self - Paced Learning,
University System, 1979
Cualitativamente, la Ag puede estar presente en la mata, el Pb en ambos, más en la mata
que en la escoria y el Zn en ambos, pero más en la escoria que en la mata. Un ejemplo de
esta distribución se presenta en la siguiente tabla.
Ejemplo de Distribución de Impurezas entre la Mata y Escoria (%)
Fundición Mata Escoria
Zn Pb Ag(Oz/TC) Zn Pb Ag(Oz/TC)
A 3.0 0.5 7.0 3.5 0.2 0.09
B - 0.4 4.1 - 0.1 0.06
C 2.8 0.5 3.0 0.6 0.3 0.08
Fuente: TWIDWELL, J. C. :"Pirometallurgy", A Modular Tutonal Designed for Self - Paced Learning,
University System, 1979
3.10. Formación, constitución y características de la mata. Como se ha dicho, al fundir una carga portadora de azufre, se forma una mata que contiene principalmente FeS y Cu2S, con pequeños contenidos de otros sulfuros, como el

Co3S, Ni3S, PbS y ZnS. La mata también es un gran disolvente de metales como el Au, Ag y los metales del grupo del platino. El 95 al 99% de estos metales entran a la mata junto con el As, Sb, Se y Te, la mata también contiene hasta un 3% de oxígeno disuelto. La obtención de la mata y su posterior conversión a cobre blisiter vienen a ser procesos de piroconcentración del cobre y los elementos valiosos para ser separados finalmente en etapa de electrorefinación, como metales de alta pureza o mezclas de ellos. El Sistema Cu-Fe-S La mata es una solución homogénea de FeS y Cu2S, como se puede apreciar en la figura
Existe algo de controversia en torno a la constitución exacta de la mata solidificada, pero se aceptan las líneas de liquidos, normalmente. En los trabajos de Reuleaux (1927) y de Krivsky y Schuhmann (1957) (BISWAS – DAVENPORT. “El Cobre – Metalurgia Extractiva”, Ed. Limusa,
España, 1993), se encuentran disponibles las descripciones más completas del sistema Cu-Fe-S, como se presenta en la figura siguiente:
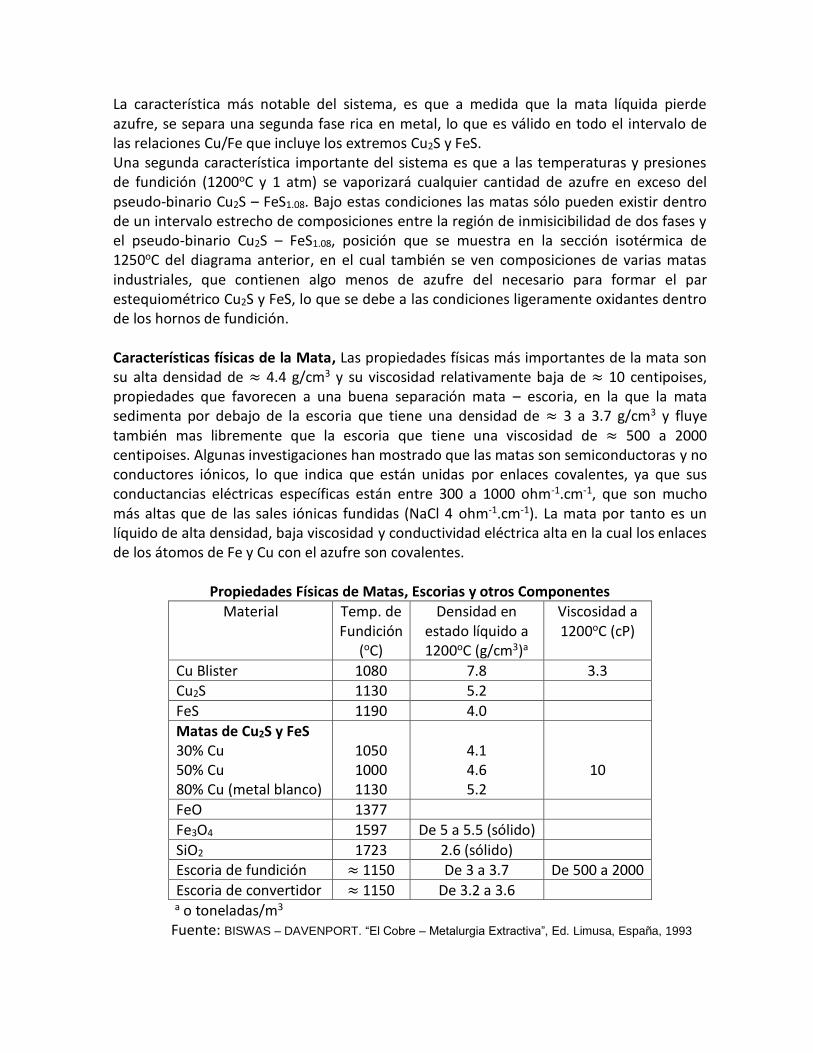
La característica más notable del sistema, es que a medida que la mata líquida pierde azufre, se separa una segunda fase rica en metal, lo que es válido en todo el intervalo de las relaciones Cu/Fe que incluye los extremos Cu2S y FeS. Una segunda característica importante del sistema es que a las temperaturas y presiones de fundición (1200oC y 1 atm) se vaporizará cualquier cantidad de azufre en exceso del pseudo-binario Cu2S – FeS1.08. Bajo estas condiciones las matas sólo pueden existir dentro de un intervalo estrecho de composiciones entre la región de inmisicibilidad de dos fases y el pseudo-binario Cu2S – FeS1.08, posición que se muestra en la sección isotérmica de 1250oC del diagrama anterior, en el cual también se ven composiciones de varias matas industriales, que contienen algo menos de azufre del necesario para formar el par estequiométrico Cu2S y FeS, lo que se debe a las condiciones ligeramente oxidantes dentro de los hornos de fundición. Características físicas de la Mata, Las propiedades físicas más importantes de la mata son su alta densidad de ≈ 4.4 g/cm3 y su viscosidad relativamente baja de ≈ 10 centipoises, propiedades que favorecen a una buena separación mata – escoria, en la que la mata sedimenta por debajo de la escoria que tiene una densidad de ≈ 3 a 3.7 g/cm3 y fluye también mas libremente que la escoria que tiene una viscosidad de ≈ 500 a 2000 centipoises. Algunas investigaciones han mostrado que las matas son semiconductoras y no conductores iónicos, lo que indica que están unidas por enlaces covalentes, ya que sus conductancias eléctricas específicas están entre 300 a 1000 ohm-1.cm-1, que son mucho más altas que de las sales iónicas fundidas (NaCl 4 ohm-1.cm-1). La mata por tanto es un líquido de alta densidad, baja viscosidad y conductividad eléctrica alta en la cual los enlaces de los átomos de Fe y Cu con el azufre son covalentes.
Propiedades Físicas de Matas, Escorias y otros Componentes Material Temp. de
Fundición (oC)
Densidad en estado líquido a 1200oC (g/cm3)a
Viscosidad a 1200oC (cP)
Cu Blister 1080 7.8 3.3 Cu2S 1130 5.2
FeS 1190 4.0
Matas de Cu2S y FeS 30% Cu 50% Cu 80% Cu (metal blanco)
1050 1000 1130
4.1 4.6 5.2
10
FeO 1377
Fe3O4 1597 De 5 a 5.5 (sólido)
SiO2 1723 2.6 (sólido) Escoria de fundición ≈ 1150 De 3 a 3.7 De 500 a 2000
Escoria de convertidor ≈ 1150 De 3.2 a 3.6
a o toneladas/m3
Fuente: BISWAS – DAVENPORT. “El Cobre – Metalurgia Extractiva”, Ed. Limusa, España, 1993

3.11. Formación, constitución y características de la escoria. Las escorias de fundición se forman partir de los óxidos presentes en la carga del horno y de los óxidos de Fe que se forman durante la fundición, siendo principalmente mezclas de silicatos fundidos y otros metales oxidados y son clasificados como escorias extractivas y escorias de refinación. Las escorias extractivas son mezclas de oxisilicatos fundidos formados de la ganga de los minerales asociados con el concentrado, más la adición de fundentes que permitan obtener una mezcla fluida a baja temperatura de fusión, es bueno aclarar que las escoria no tienen un punto de fusión sino un rango de temperatura de fusión. Las gangas silicosas requieren fundentes básicos como el CaO o el FeO, mientras que las gangas calcáreas pueden ser fluidizadas con sílica (arena). Las escorias de refinación son mezclas especialmente preparadas para desarrollar las operaciones de refinación y están ideadas para absorber impurezas, suministrar especies reactivas como el oxígeno al baño metálico, para promover las reacciones interfaciales, etc. Las escorias de interés en la fundición de sulfuros son las escorias extractivas, sin embargo, en algunos casos las escorias extractivas pueden actuar como escorias de refinación, por ejemplo las escorias formadas de las gangas oxidadas y silicatadas sirven como receptoras de impurezas si se usa un fundente apropiado. Las escorias producidas en la fundición de cobre están principalmente conformadas por FeO y SiO2 en una proporción nominal de 2:1 (como fayalita 2FeO.SiO2), los otros componentes, tales como el CaO, Al2O3, MgO, etc están presentes en cantidades menores. Las escoria de fundición, en promedio, contienen: Fe (como FeO, Fe2O3) de 30 a 40%, SiO2 (de los fundentes, escoria de convertidor reciclado o de ambos) de 35 a 40%, CaO hasta 10% y Al2O3 hasta 10%. Las escorias de la fundición de cobre pueden contener de 0.3 a 1.0% de Cu, dependiendo de parámetros como:
a. El sistema del horno de fundición b. Presión parcial del O2 ( ver 3.9. Formación Mata – Escoria) c. Grado de mata (ver figura siguiente) d. Composición de la escoria e. Temperatura del horno
La característica física más sobresaliente de las escorias es su alta viscosidad, que varia de 500 a 2000 cP, comparadas a las de las matas (10 cP) y el cobre líquido (3 cP). La presencia de magnetita sólida o sílice sólida en exceso aumenta aún más la viscosidad de la escoria. Las escorias líquidas son iónicas y se componen de cationes como el Ca+2, Fe+2, Mg+2, Fe+3, aniones como O-2, 𝑆𝑖𝑂4
−4 y las cadenas grandes y anillos de silicatos. Las escorias de fundición se clasifican en básicas, neutras y ácidas, en función a su contenido de sílice. La estructura de las escorias básicas es simple y son fluidas, mientras que las ácidas se componen por grandes iones complejos y su viscosidad es alta. Una buena escoria debe tener las siguientes propiedades:
a. Tener bajo costo b. Alta fluidez para asegurar una buena separación mata – escoria c. Bajo punto de fusión
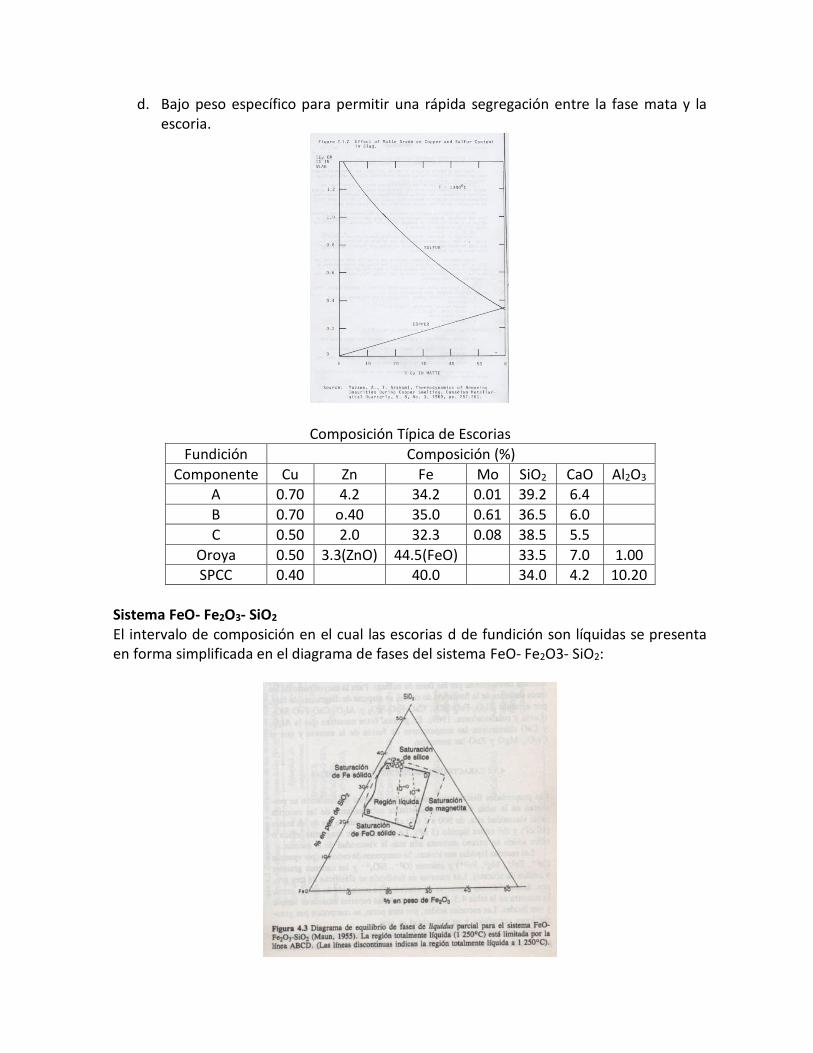
d. Bajo peso específico para permitir una rápida segregación entre la fase mata y la escoria.
Composición Típica de Escorias
Fundición Composición (%)
Componente Cu Zn Fe Mo SiO2 CaO Al2O3
A 0.70 4.2 34.2 0.01 39.2 6.4
B 0.70 o.40 35.0 0.61 36.5 6.0
C 0.50 2.0 32.3 0.08 38.5 5.5
Oroya 0.50 3.3(ZnO) 44.5(FeO) 33.5 7.0 1.00 SPCC 0.40 40.0 34.0 4.2 10.20
Sistema FeO- Fe2O3- SiO2 El intervalo de composición en el cual las escorias d de fundición son líquidas se presenta en forma simplificada en el diagrama de fases del sistema FeO- Fe2O3- SiO2:

El área determinada por ABCD señala el intervalo de composición dentro del cual las escorias se funden completamente a las temperaturas de fundición (1200oC). Este campo está rodeado por cuatro campos de saturación sólida de las cuales dos, saturación de Fe sólido (AB) y saturación de FeO sólido (BC), no se encuentran en los hornos de fundición debido a los potenciales altos de oxígeno y a los contenidos de sílice en las escorias de fundición. Pero, la línea AD es significativa, por que muestra la cantidad de sílice que se requiere para saturar las escorias de fundición, este factor es muy importante por que las separaciones óptimas mata-escoria se logran en condiciones cercanas a la saturación. Este diagrama muestra que se necesitan de 35 a 40% de SiO2 para la saturación de sílice en el sistema FeO- Fe2O3- SiO2, cuando existe apreciable cantidad de CaO en la escoria se requiere poco más de 40 a 42% de SiO2. Una segunda característica del diagrama es la línea de saturación de la magnetita sólida CD, este límite indica que la magnetita sólida será una fase en equilibrio cuando la presión parcial de oxígeno de los gases exceda en un10-9 (punto C) o 10-8 atm en la saturación con sílice (punto D). Los gases de combustión en la mayor parte de los hornos de fundición contienen en el orden el 1% de O2, o sea 𝑝𝑂2
≈ 10-2 atm, que indica que la magnetita sólida
se debe producir en la interfase gas-escoria durante la fusión. En la práctica, el FeS en el concentrado y la mata ejercen un potencial reductor fuerte sobre el sistema y la magnetita tiende a ser reducida por la reacción: 3Fe3O4(s) + FeS(l,mata) 10FeO(l, escoria) + SO2(g) En los sistemas de fundición siempre habrá formación de la magnetita por la oxidación atmosférica y su reducción por acción de las fases de sulfuro.
3.12. La magnetita en la fundición de la mata. La magnetita sólida, se forma en la fusión y conversión de cobre, por la reacción del oxígeno presente en los gases del horno con el óxido de hierro presente en la escoria, también puede estar presente en las calcinas que se cargan a los hornos de fundición. La presencia de la magnetita en los circuitos de fundición causa varios problemas operacionales importantes:
a. Los cristales de magnetita en las escorias de fundición las hacen viscosas, dificultando la separación mata-escoria y ocasionando pérdidas de cobre por arrastre de concentrado y mata en la escoria.
b. La magnetita sólida es más densa que la mata y la escoria, por lo que se asienta en la solera del horno, disminuyendo así el volumen y capacidad de producción del horno de fusión o conversión. Magnetita 5 a 5.5 g/cm3 ˃ Mata 4.5 g/cm3 ˃ Escoria 3.5 g/cm3
c. La magnetita se combina con otros óxidos, particularmente el Cr2O3, proveniente de los refractarios cromo-magnesita, para producir sólidos de densidades intermedias entre las de la mata y escoria, estos óxidos forman un “falso fondo” entre las capas de mata y escoria que obstruye la separación de la mata y escoria
Por las consideraciones expuestas la fundición se debe llevar a cabo en condiciones que mantengan en un mínimo la formación de magnetita sólida. Los factores de operación más importantes para lograr esta reducción son:
1. Temperatura, las altas aumentan la solubilidad de la magnetita en la escoria y mata líquida y disminuye la cantidad de magnetita sólida, igualmente favorecen la reducción de la magnetita por acción del FeS de la mata. Las constantes de equilibrio para la reacción de reducción: 3Fe3O4(s) + FeS(l,mata) 10FeO(l, escoria) + SO2(g)
Son 2x10-6 a 1100oC, 1x10-4 a 1200oC y 4x10-3 a 1300oC. 2. Material de carga, gran parte de la magnetita sólida está presente en la escoria del
convertidor recirculada a la fundición y en las calcinas de tostación de ser el caso. La carga de concentrados “verdes”, sin tostar y si no se agrega escoria de convertidor al horno de fundición disminuyen la cantidad de magnetita sólida en cualquier operación.
La cantidad de magnetita sólida también se reduce al evitar flamas excesivamente oxidantes en el horno de fundición, además un grado de mata bajo, que tiene un alto contenido de FeS, promueve la reducción de la magnetita, pues ésta actual como oxidante y pasa a la escoria a través de la reacción: 3Fe3O4(s, escoria) + FeS(l, mata) + 5SiO2(s, escoria) = 5Fe2SiO2(l, escoria) + SO2(gas)
∆𝐺1200𝑜𝐶𝑜 = - 34000 cal.
3.13. Comportamiento de otros metales en la fusión de concentrados de cobre.
Como se ha visto en el punto 3.9. Formación Mata – Escoria, los otros metales presentes en el concentrado de cobre, se distribuyen entre estas dos fases inmiscibles, según su afinidad termodinámica por el oxígeno o el azufre y su solubilidad, dependiendo además de las condiciones de fundición y el tipo del proceso. En el cuadro siguiente se presenta una distribución estimada de los componentes menores de un concentrado entre la mata y la escoria.
Distribución Estimada de Elementos en la Fusión de Matas (%)
Metal Mata Escoria Volatilizado
Alcalinos, alcalinotérreos, Al, Ti - 100 -
Au, Ag, grupo del Pt 99 1 - Sb 30 55 15
As 35 55 10
Bi 10 10 80
Cd 60 10 30 Co 95 5 -
Pb 30 10 60
Ni 98 2 -
Se 40 - 60 Te 40 - 60
Sn 10 50 40
Zn 40 50 10 Fuente: BISWAS – DAVENPORT. “El Cobre – Metalurgia Extractiva”, Ed. Limusa, España, 1993

En el cuadro se destaca: a) El Au, Ag, metales del grupo del Pt, Co y Ni entran casi completamente a la mata y
son transportados a la etapa de conversión, donde son disueltos en la fase de cobre blíster o ampolloso para finalmente ser recuperados como subproductos durante la electrorefinación de los ánodos de cobre.
b) Cantidades importantes de impurezas perjudiciales a la calidad del cobre también entran a la mata, estos son el Sb, As, Bi, Pb, Se y Te. Algunos de estos metales se recuperan como subproductos durante las operaciones posteriores de conversión y electrorefinación.
c) El Zn presente en la escoria se puede recuperar por el proceso de “escoria humeante”, si la cantidad presente lo justifica.
3.14. Fusión de concentrados en hornos reverbero. Con el desarrollo de la concentración de minerales por flotación, los hornos reverbero resultaron ser las unidades de fusión más idóneos para la fundición de concentrados de cobre, pero, con el avance de la tecnología, debido a la crisis energética de los años 70 del siglo pasado y la implementación de medidas de control ambiental a nivel mundial, después de la cumbre de Río (1992), estos hornos han sido progresivamente remplazados por tecnologías más eficientes y menos contaminantes. En la actualidad ya quedan pocos hornos en funcionamiento. En el Centro del Perú, en el circuito de cobre del Complejo Metalúrgico de la Oroya, se ha utilizado el horno reverbero hasta mediados de 2011 y se ha paralizado por el incumplimiento de Doe Run Perú con sus programas del PAMA (Plan de Adecuación de Manejo Ambiental) y es probable que superado los problemas que afronta el Complejo Metalúrgico ce la Oroya aún se opere estos hornos por un tiempo más. La fusión de concentrados sulfurados de Cu y Ni en hornos reverbero es similar y existen dos formas de fundición:
a) Hornos que usan calcina como alimentación, caso La Oroya b) Hornos funden directamente concentrados secos o húmedos
Una tostación previa elimina del 20 al 50% del azufre como SO2, que al ser concentrado en forma suficiente sirve para producir H2SO4, por cuanto en los tostadores de hogares múltiples la corriente gaseosa contiene del 5 al 10% de SO2 y en los tostadores de lecho fluidizado la concentración del SO2 puede ser de 12 a 14%. En el proceso de tostación también se forma FeO, que posteriormente en la etapa de fusión en horno reverbero pasa a la escoria, por lo que la mata producida será de mayor grado, pero este mayor grado de mata también ocasionará una mayor pérdida de cobre en la escoria. En el caso de emplear concentrados verdes (sin tostar) secos o húmedos como carga en el horno reverbero se logra matas de menor grado y también se pierde menos cobre en las escorias, lo que significa mayor tiempo de soplado en la etapa de conversión de matas, por contener mayor concentración de Fe y S. Para el funcionamiento del horno reverbero se pueden emplear como combustibles petróleo, gas natural o carbón pulverizado, los que se queman encima del baño fundido inyectados mediante quemadores instalados en el extremo opuesto a las toberas de descarga de mata y escoria. Una modificación para mejorar la capacidad de fusión de estos

hornos fue instalar quemadores verticales en la bóveda del horno e utilizara aire enriquecido con oxígeno. Concentrado Concentrado
20-50% S 20-40%S Calcina Mata 10-30% S Mata 60-80%S 40-50% S Cu Blister Cu Blister Para el proceso de combustión se requieren grandes volúmenes de aire, para mantener la temperatura del horno entre 1200 a 1300oC, así por ejemplo para quemar un volumen de gas natural se requiere de 10 a 12 volúmenes de aire, lo que significa que los gases que se generan en el horno son de gran volumen y principalmente contienen nitrógeno, por lo que el contenido de SO2 en los gases de salida es relativamente baja, entre 0.5 a 2%, que lo hace prohibitivo su tratamiento para producir H2SO4 y por tanto el SO2 se descarga a la atmósfera.
Tostación
Fundición
Conversión
Conversión
Fundición

El contenido de SO2 en los gases del horno depende de muchos factores:
Del concentrado a ser fundido Del proceso de fusión (tostado o no tostado) Del combustible a ser quemado Del exceso de aire empleado De la cantidad de escoria de convertidor recirculado De la infiltración de aire en el horno y ductos
Una característica importante del proceso de fusión en hornos reverbero es el tratamiento de escoria recirculada de los convertidores, que contienen mucho cobre, entre 3 a 6%. Este cobre es pasado a la fase mata, durante el proceso de fusión a través de la reacción: Cu2O(escoria) + FeS(mata) = Cu2S(mata) + FeO(escoria) ∆G1300
oC = -26 000 cal
Las escoria recirculadas de los convertidores, también, contribuyen al aumento del contenido de SO2 en los gases, por cuanto contienen de25 a 45% de Fe3O4 que actúan como oxidantes frente al FeS 3Fe3O4(escoria) + FeS(mata) + 5 SiO2(escoria) = 5Fe2SiO4(escoria) + SO2(gas) ∆G1300
oC = -34 000 cal
La magnetita que no reacciona se acumula en la solera del horno, originando la disminución de la capacidad del horno. La recuperación del cobre de las escorias de los convertidores se puede realizar por otros métodos, como el enfriamiento, molienda y flotación, reducción con pirita en hornos eléctricos u otros hornos, pero, el método más eficiente es la recirculación a los hornos reverbero, si es parte del circuito. Las escorias de los hornos reverbero tienen bajo contenido de Cu y se debe a tres causas:
1) Por la disolución del cobre en la matriz Fe-silicato 2) Partículas de mata atrapadas en la escoria o mata no sedimentada 3) Partículas de concentrado no reaccionados y que han sido atrapadas en la fase
escoria El mayor o menor efecto de estos factores depende a su vez de:
Operación del horno Temperatura alcanzada Presión parcial de oxígeno

Grado de mata Tipo de concentrado Cantidad de escoria recirculada de los convertidores Composición de la escoria del horno Experiencia del personal
Descripción del Proceso Los concentrados de cobre al ser fundidos en los hornos reverbero forman dos capas líquidas inmiscibles: mata y escoria, la mata por ser más densa se acumula en la solera del horno y la escoria flota sobre ella por ser menos densa. El concentrado o calcina alimentada al horno se funde sobre la escoria. El horno tiene dos secciones principales:
a) Sección de fundición, que se ubica en la parte superior del horno b) Sección de sedimentación
Se debe tener presente que la carga se funde cerca a la zona de los quemadores y fluye hacia el área de sedimentación del horno y por l parte final del horno, en lado opuesto a los quemadores, se extrae la escoria a recipientes grandes denominados cuchas u ollas para ser transportados hacia el área de descarga o escorial, en otros caso la escoria es enfriada con agua para obtener un producto granulado de fácil transporte en fajas transportadoras. Los hornos más grandes producen de 1200 a 2000 toneladas de escoria al día. La mata se extrae por un costado el horno, siendo el orificio de colada ubicada generalmente en la zona de sedimentación del horno, esta mata es descargada a cucharas u ollas de gran capacidad, la producción de mata varia entre 1200 a 1800 toneladas por día. La temperatura del líquido oscila entre 1150 a 1250oC y el calor requerido para la fusión es suministrado por la quema de combustibles como el petróleo, gas natural o carbón pulverizado. Los gases salen del horno a una temperatura de 1200 a 1300oC. La velocidad de fusión en los hornos reverbero es de 4 a 9 toneladas por metro cuadrado de solera u hogar por día, las velocidades bajas son para hornos que tratan concentrados húmedos y las velocidades altas cuando se tratan concentrados secos y/o calcinados, se requiere mayor energía para alimentadores de calcinas. La alimentación al horno de los materiales a fundir se realiza dejando caer una capa fina a lo largo de las paredes laterales, de esta manera se logra una gran superficie de fundición y al mismo tiempo de protege los ladrillos refractarios. Otra forma de alimentar es por el techo del horno en forma de spray (material atomizado o pulverizado) hacia la cama fundida o lecho, en modificaciones recientes se usó un sistema de alimentación con fajas corta a gran velocidad, denominados “slinger” (lanzador), con lo que se logra también atomizar la carga encima del lecho fundido, consiguiendo, así, una forma de fusión instantánea. Cerca de 80% de la carga se deposita en la primera mitad del horno, donde la temperatura es alta y el resto a lo largo de la parte final. Una representación gráfica de la variación de la temperatura del gas y la distribución de la carga a lo largo del horno se representa en las gráficas siguientes:
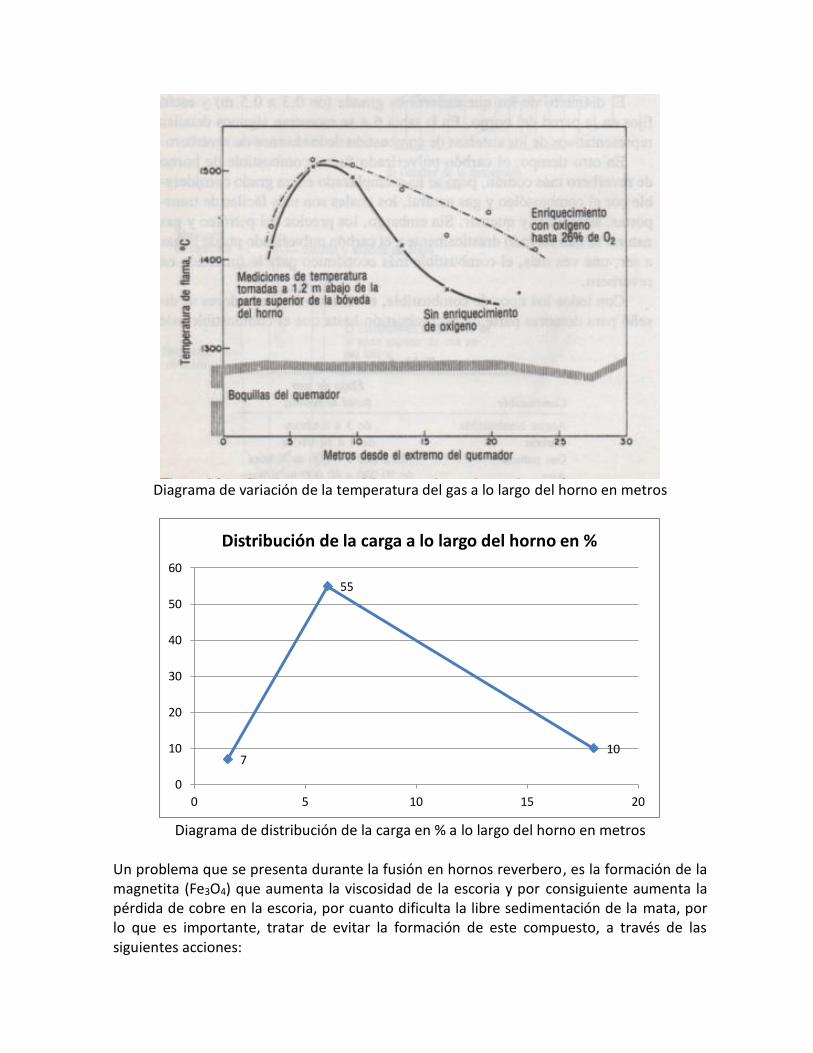
Diagrama de variación de la temperatura del gas a lo largo del horno en metros
Diagrama de distribución de la carga en % a lo largo del horno en metros
Un problema que se presenta durante la fusión en hornos reverbero, es la formación de la magnetita (Fe3O4) que aumenta la viscosidad de la escoria y por consiguiente aumenta la pérdida de cobre en la escoria, por cuanto dificulta la libre sedimentación de la mata, por lo que es importante, tratar de evitar la formación de este compuesto, a través de las siguientes acciones:
7
55
10
0
10
20
30
40
50
60
0 5 10 15 20
Distribución de la carga a lo largo del horno en %

Manteniendo la temperatura de operación alta Manteniendo alto el contenido de sílice en la escoria, por ejemplo un radio de
SiO2/FeO en % en peso mayor a 0.8 Operar a una velocidad alta de producción para así lograra levantar la magnetita
sólida a través del horno Es necesario hacer un comentario de las ESCORIAS DE REVERBERO, cuyas relaciones de fase para las escorias de reverbero se pueden aproximar en el diagrama de fases CaO- FeO – SiO2 (diagrama siguiente), el uso de este diagrama se facilita al considerar todo el hierro en la escoria como “FeO “y al combinar AL2O3, CaO, y MgO como “CaO” total. Un examen de las escorias industriales de reverbero sobre esta base indica que todas estas se sitúan en el valle de baja temperatura del sistema CaO - FeO – SiO2. Estas escorias también tienden a la saturación con SiO2, es decir, contienen 35% o más de SiO2. Existe un caso excepcional, la escoria de reverbero de El teniente en la cual el contenido de SiO2 es solamente de 27%, pero la concentración de Al2O3 en esta escoria es alta (11.9%) y la Al2O3 actúa un tanto como un sustituto para la SiO2. En general, los contenidos de SiO2 son de 35 a 42% los cuales están de acuerdo con el requerimiento teórico de saturación- próxima para separaciones buenas mata – escoria.
Diagrama de equilibrio de fases CaO-FeO-SiO2
Además, para tener una buena separación de Cobre en equilibrio entre las fases mata escoria, esta debe ser razonablemente fluida para permitir el asentamiento de la mata o concentrados arrastrados a través de la capa de escoria. Para esto la escoria debe tener una viscosidad lo mas baja posible. Las viscosidades de las escorias de reverbero se grafican en función del módulo de composición Kv, donde:
Kv = %𝑝𝑒𝑠𝑜 𝐶𝑎𝑂+%𝑝𝑒𝑠𝑜𝐹𝑒𝑂+%𝑝𝑒𝑠𝑜𝐹𝑒3𝑂4+%pesoMgO
%𝑝𝑒𝑠𝑜𝐴𝑙2𝑂2 +%𝑝𝑒𝑠𝑜𝑆𝑖𝑂2

A grandes contenidos de SiO2 y Al2O3 en la escoria el valor del modulo Kv, es bajo, lo que significa que la viscosidad es alta, como se aprecia en la figura siguiente. La SiO2 y Al2O3 actúan como constructores de la estructura de la escoria, mientras que el CaO, FeO y MgO, actúan como destructores de la estructura y tienen un efecto de disminuir la viscosidad y aumentar el valor del módulo Kv. Datos adicionales proporcionados por algunos investigadores muestran que el Cr2O3 aumentan la viscosidad de las escorias de reverbero y el MnO lo reduce. En la figura también se aprecia que la viscosidad aumenta al disminuir la temperatura, debido a que los iones se vuelven menos móviles. La presencia de sólidos en la escoria, particularmente la magnetita, también aumenta la viscosidad, por lo que debe Evitarse lo más posible su presencia. Los sólidos tienden a disolverse en un mayor grado a mayores temperaturas, por lo que es recomendable mantener temperaturas altas en todas las partes del horno y garantizar una escoria fluida.
Diagrama Viscosidad de Escorias
Para lograr menores costos de operación y mayor eficiencia en las hornos reverbero, se han desarrollado muchos avances, entre ellos podemos mencionar:
a. La reintroducción de la tostación como etapa previa a la fundición b. El uso de aire precalentado para la combustión c. El enriquecimiento con oxígeno del aire de combustión d. Mejoramiento de la captura de SO2 de los gases del horno e. Instalación de quemadores oxi-fuel en los techos de los hornos

3.15. Fusión de concentrados en hornos eléctricos Es un horno de hogar calentado con energía eléctrica y desempeña las mismas funciones que el horno reverbero. Se usa en varias localidades donde la energía eléctrica es barata y procede de hidroeléctricas. Estos hornos trabajan con electrodos de carbón que están sumergidas en la capa de escoria, que ofrece suficiente resistencia para el paso de la corriente eléctrica y así alcanzar la temperatura de fusión necesaria. La carga es muy similar a los usados para la fusión en hornos reverbero, pero tiene que ser seca o parcialmente tostado. La fusión en estos hornos puede generar gases con bajo contenido de SO2, si es que no existen altas infiltraciones de aire. Operados apropiadamente son eficientes en consumo de energía, en el orden del 40 al 50% de la energía proporcionada por combustibles con carga seca. Se usan en casos especiales, como para fundir concentrados complejos o especiales y cuando la energía procede de una hidroeléctrica, además permite un buen control de las emisiones de SO2. El horno eléctrico tiene dos ventajas sobre el horno reverbero:
1. Produce cantidades más pequeñas de gases efluentes, debido a la ausencia de productos gaseosos de combustión
2. La concentración del SO2 en los gases efluentes son fácilmente controladas por la regulación de la cantidad de infiltraciones de aire hacia el interior del horno. Una mínima cantidad de infiltraciones de aire, permite lograr concentraciones de SO2 del orden de 0.4%, que es un valor muy bajo y se puede descargar directamente a la atmósfera, mientras que una gran infiltración de aire, ocasiona mayor oxidación del azufre y por consiguiente mayor contenido de SO2 en los gases de descarga, que pueden llegar al orden de hasta 5%,en este caso, estos gases se mezclan con los gases del convertidor y el SO2 se extrae de ambos como H2SO4.
Las dimensiones de los hornos eléctricos son del orden de 35 m de largo por 10 m de ancho y 5 m de alto del hogar a la bóveda. Un horno de este tamaño empela seis electrodos tipo Soderberg de auto cocción. El flujo de corriente y tensión entre los electrodos son del orden de 30 000 A y 500 V respectivamente, lo cual da una potencia total nominal de 40 000 kw. Esta potencia permite fundir entre 1500 a 2300 toneladas de carga seca por día. Las ventajas más importantes de un horno eléctrico de fundición de matas son:
a) Es completamente versátil y se puede usar para fundir cualquier material b) Produce pequeños volúmenes de gas efluente, que contiene N2, proveniente de la
infiltración del aire, Co y CO2, originados por las reacciones escoria-electrodo y SO2 de la oxidación del azufre.
c) La concentración del SO2 en los gases efluentes se controla fácilmente al controlar la infiltración del aire en el horno
d) Hace uso eficiente de la energía eléctrica Sus desventajas son:
a) No usa toda la energía que está potencialmente disponible, debido a la oxidación de los minerales sulfurados
b) Los costos de operación tienden a ser altos debido al precio elevado de la energía eléctrica

3.16. Fusión de concentrados en altos hornos Ya como reseña histórica, diremos que los altos hornos se usaron extensamente en el pasado para la producción de grandes cantidades de mata a partir de menas sulfuradas en trozo. También se uso al mismo tiempo para producir “cobre negro” no refinado contaminado con hierro, partiendo de menas de oxidadas. La disminución de las menas ricas en trozo y el predominio de los concentrados por flotación han eliminado poco a poco el alto horna para la fusión de matas. Este horno no es el adecuado para fundir material finamente molido, que es el caso de los concentrados por flotación, sin embargo dicho horno se usaba en Africa, para fundir mezcla de sulfuros y óxidos sinterizados. Para 1979 sólo quedaban 10 altos hornos en el mundo. La carga para el horno consistía en concentrados sinterizados, mena en trozo, fundente de sílice, escoria sólida del convertidor y coque metalúrgico. En general los sólidos son mayores a 1 cm de diámetro para evitar que sean arrastrados del horno por la corriente gaseosa ascendente y asegurar un flujo de gas uniforme a través de la columna de la carga. Se inyecta aire, normalmente a temperatura ambiente, a través de las toberas ubicadas cerca al fondo del horno para combustionar el coque y parte de los sulfuros de hierro de la carga, con el propósito de:
a) Proporcionar calor para el proceso de fusión b) Tostar parcialmente los sulfuros, para producir, así, una mata de grado mejorado,
cuando la mata se funde próximo al fondo del horno. Los productos obtenidos son:
1. Una mata líquida rica en cobre ( ≈ 50% Cu) 2. Una escoria líquida que permite el asentamiento de la mata 3. Gases con cera de 5% de SO2. Los polvos arrastrados por el gas son recuperados en
ciclones y precipitadores electrostáticos

3.17. Fusión de concentrados en hornos flash La necesidad de ahorrar combustible en los procesos de fusión de concentrados sulfurados de cobre y níquel ha permitido desarrollar tecnologías que permitan el aprovechamiento del calor generado por las reacciones de oxidación del azufre y el hierro presentes en los concentrados sulfurados, es así que se desarrollan los procesos de fusión flash (fusión instantánea). En estos hornos se utiliza el calor de oxidación de una parte de la carga de sulfuro para proporcionar gran parte o la totalidad de la energía necesaria para la fundición. La fusión flash es un proceso pirometalúrgico usado para fundir concentrados de sulfuros metálicos, inicialmente se uso para fundir concentrados de cobre y posteriormente se extendió para concentrados de níquel, también se ensayó para tratar concentrados de sulfuros de plomo. La fusión flash de concentrados de cobre consiste en inyectar dentro del horno caliente el concentrado de cobre seco de granulometría fina y fundente de sílice con aire, aire enriquecido con oxígeno u oxígeno. El ingreso de estos materiales en el horno caliente ocasiona la reacción instantánea del mineral sulfurado con el oxígeno y como resultado se tiene: i.- Una oxidación controlada del Fe y el S del concentrado ii.- Generación de gran cantidad de calor iii.- La fusión de los sólidos Los productos que se obtienen son:

a) Una mata fundida Cu-Fe-S, rica en cobre, 45 a 65% de Cu, que contienen casi todo el cobre del concentrado, más el Fe y S no oxidado.
b) Una escoria fundida que contiene óxido de hierro procedente de la oxidación del hierro del concentrado y el hierro presente en la ganga y óxidos de los fundentes.
c) Una corriente gaseosa que contiene SO2, originado por la oxidación del azufre y nitrógeno por el aire inyectado al horno, además CO2 y vapor de H2O, si se ha usado combustible en forma complementaria.
La mata es enviada a los hornos convertidores, donde se elimina el Fe y S remanentes y se obtiene el cobre ampolloso (blíster) que finalmente se envía a la refinería o al mercado. La escoria que puede contener entre 0.5 a 2% de Cu es enviada a un proceso de tratamiento para la recuperación del Cu y la escoria final es llevado a un escorial o utilizado en construcción de vías. Los gases del horno que pueden contener entre 10 a 80% de SO2, son enfriados, usualmente en calderas que aprovechan el calor residual, y limpiados para recuperar las partículas en suspensión, son enviadas a una planta de recuperación de SO2, sea para la producción de ácido sulfúrico o SO2 líquido. Las reacciones químicas que ocurren en los hornos de fusión instantánea son:
CuFeS2 + 5
4 O2 →
1
2 (Cu2S . FeS) +
1
2 FeO + SO2 (mata) ∆H298
𝑜 = -78 000 cal
FeS + 1
2 O2 → FeO + SO2 (escoria) ∆H298
𝑜 = -115 000 cal
2FeO + SiO2 → 2FeO.SiO2 (escoria) ∆H298
𝑜 = -10 000 cal Estas reacciones generan suficiente energía térmica para calentar, fundir y sobrecalentar la carga del horno. En la práctica industrial, cuando se usa oxígeno industrial o aire enriquecido con oxígeno con más del 40%, la fundición instantánea es autógena. La combustión (oxidación) de las partículas de sulfuro en el horno es extremadamente rápida, el calor producido por las reacciones de oxidación es suficiente para fundir rápidamente los minerales que se oxidan parcialmente. Las gotas fundidas que tienen una apariencia de una neblina luminosa, caen sobre la capa de escoria, donde se terminan las reacciones de formación de mata y escoria y donde cualquier cobre oxidado se reduce de nuevo a Cu2S, por la reacción: Cu2O + FeS → Cu2S(mata) + FeO(escoria) Las gotas de mata formadas se sedimentan a través de la capa de escoria para formar la capa de mata. Durante el proceso de combustión la FeS2, CuS y CuFeS2 liberan el azufre al estado gaseoso, este azufre al combustionar en forma rápida genera altas velocidades de oxidación y fusión. La productividad de los hornos de fusión flash, es del orden de 8 a 12 toneladas de carga por día/ m2 de área de hogar, que es de 2 a 4 veces la productividad de un horno reverbero y eléctrico. La cantidad de Fe y S que se oxidan mediante las reacciones que ocurren en el horno, determinan el grado de mata (% de Cu en la mata) y se controla ajustando el radio:
𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒 𝑂2 𝑖𝑛𝑦𝑒𝑐𝑡𝑎𝑑𝑜 (𝑒𝑛 𝑎𝑖𝑟𝑒, 𝑎𝑖𝑟𝑒 + 𝑜𝑥í𝑔𝑒𝑛𝑜 𝑢 𝑜𝑥í𝑔𝑒𝑛𝑜)
𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒 𝑎𝑙𝑖𝑚𝑒𝑛𝑡𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑐𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎𝑑𝑜
A mayor valor del radio, mayor cantidad de Fe y S oxidados y viceversa, igualmente a mayor cantidad de Fe y S oxidados se tendrá un mayor grado de mata y viceversa, pero hay que tener presente que se debe conservar suficiente cantidad de Fe y S en la mata para garantizar el calor necesario para la etapa de conversión. Se pueden lograr grado de mata de 45 a 65% de Cu. El grado de mata para una operación de fusión específica se escoge para:
a) maximizar el uso del calor generado por las reacciones de oxidación del Fe y S que ocurren en el horno
b) maximizar la captura del SO2 generado en el proceso c) dejar suficiente cantidad de Fe y S en la mata, para que sirva como combustible en
la etapa de conversión d) evitar la excesiva formación de Cu2O y Fe3O4 en la escoria.
En el concentrado están presentes todas las impurezas que acompañan al cuerpo mineralizado y durante la fusión se distribuyen, según su afinidad termodinámica, entre la mata y la escoria, así, metales como el Au, Ag y metales del grupo del platino estarán mayoritariamente en la mata, los metales alcalinos y alcalino térreos pasarán a la escoria en un 100% y algunos metales como el As, Bi se volatilizarán, por su puesto que el Cu en un 99% se fijará en la mata. En el siguiente cuadro se presenta una distribución estimada.
Tabla de Distribución Estimada Impurezas para una Mata de Grado 55
Metal Distribución en %
Mata Escoria Volatilizado Cobre 99 1
Alcalino y alcalino térreos, Al, Ti 100
Ag, Au, metales del grupo del Pt 90 5 5
Sb 30 30 40 As 10 10 80
Bi 15 5 80
Co 40 55 5
Pb 20 10 70 Ni 50 45 5
Se 75 5 20
Zn 15 45 40 Fuente: Davenport, W.G, Partelpoeg E.H, “Flash Smelting” , Pergamon Press, USA, 1987
Existen dos tipos básicos de fundición instantánea:
a) Proceso Outokumpu, que usa aire precalentado o aire enriquecido con oxígeno precalentado o a temperatura ambiente. No es autógeno, usa combustible, hidrocarburos, energía eléctrica más combustible.
b) Proceso INCO que usa oxígeno comercial y es completamente autógeno.
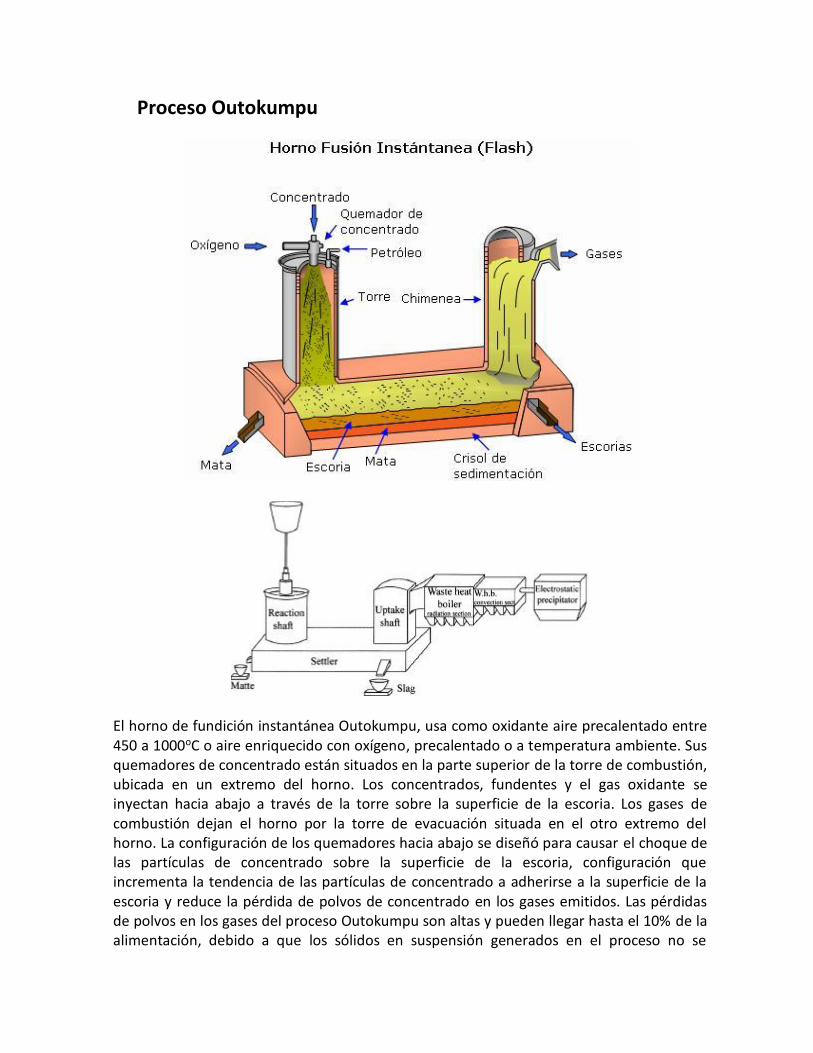
Proceso Outokumpu
El horno de fundición instantánea Outokumpu, usa como oxidante aire precalentado entre 450 a 1000oC o aire enriquecido con oxígeno, precalentado o a temperatura ambiente. Sus quemadores de concentrado están situados en la parte superior de la torre de combustión, ubicada en un extremo del horno. Los concentrados, fundentes y el gas oxidante se inyectan hacia abajo a través de la torre sobre la superficie de la escoria. Los gases de combustión dejan el horno por la torre de evacuación situada en el otro extremo del horno. La configuración de los quemadores hacia abajo se diseñó para causar el choque de las partículas de concentrado sobre la superficie de la escoria, configuración que incrementa la tendencia de las partículas de concentrado a adherirse a la superficie de la escoria y reduce la pérdida de polvos de concentrado en los gases emitidos. Las pérdidas de polvos en los gases del proceso Outokumpu son altas y pueden llegar hasta el 10% de la alimentación, debido a que los sólidos en suspensión generados en el proceso no se

sedimentan por la alta velocidad del gas dentro del horno. Para la recuperación de estos polvos se dispone de un sistema de limpieza de gases. Las dimensiones internas de un horno Outokumpu varían entre 19 a 23 m de largo, 6 a 9 m de ancho y 2 a 4 m de alto (de hogar a bóveda). Las torres de combustión o reacción miden entre 4 a 8 m de diámetro y entre 6 a 10 m de alto (sobre la bóveda), mientras que las torres de evacuación de gases, tienen dimensiones de largo x ancho entre 2.5 x 5 a 6 x 8 m o diámetros entre 2.5 a 7 m y un alto entre 6 a 18 m; hornos de estas dimensiones pueden tratar entre 850 a 2200 toneladas de concentrado por día. El sistema de quemador de concentrado consiste en dos tubos concéntricos, en la que el concentrado y fundentes se alimentan por gravedad a través del tubo central, de 0.4 m de diámetro y los gases oxidantes se inyectan a través del tubo anular de 0.8 m de diámetro. Los hornos grandes se equipan hasta con 4 quemadores, que tratan de 10 a 20 toneladas secas de carga cada uno y de 8000 a 1200 m3N de oxidante por hora. El proceso no es autógeno menos que el aire de entrada contenga 40% de oxígeno o más, caso contrario se disponen quemadores de combustible en la parte superior de la torre de reacción. Como producto del proceso, dentro del horno de obtienen dos capas líquidas inmiscibles, una fase mata y una fase escoria, que se mantienen a una profundidad de 0.75 m y son extraídas intermitentemente a través de toberas de colada sumergidos, el de la escoria debajo de la torre de evacuación de gas y el de la mata situado en una de las paredes laterales.
Proceso INCO

El proceso INCO (Merla y colaboradores, 1972) se restringe a dos instalaciones en operación (Sudbury, Canadá y Almalyk, URSS). Es algo difícil de entender ya que este proceso parece ser un método de fundición excelente y versátil. La unidad Sudbury es un horno tipo hogar pequeño, de 23 m de longitud (interior), 6 m de ancho y 5 m de alto (de hogar a bóveda). Dicho horno funde 1 600 toneladas de carga seca por día. Los concentrados, fundentes y oxígeno se inyectan horizontalmente hacia el interior del horno desde ambos extremos y los gases generados se extraen a través de un gran extractor ubicado en la parte central del horno. Este diseño produce una llama de alta temperatura sobre el área total del hogar, provocando la fusión instantánea de la carga, para formar la mata y la escoria. La mata se sangra a través de una tobera ubicada en la parte central de horno (debajo del sistema de evacuación de gas) y la escoria se sangra debajo de los quemadores por un extremo del horno. Las profundidades de la mata y escoria se mantienen a 0.6 m y 0.6 m, respectivamente; el agujero de colada de la escoria siempre se sumerge para reducir la extracción de mata sin asentar. Los quemadores son de un diseño simple en la que los concentrados y fundentes se alimentan por gravedad en la corriente de oxigeno para transportarlos por aspiración al interior del horno. Los concentrados secos se comportan bien en estas condiciones pero es improbable que los concentrados húmedos puedan usarse con esta configuración simple. El horno tiene cuatro quemadores, cada uno de los cuales trata de 15 a 20 toneladas de carga y de 2 000 a 2 500 m3N de oxígeno por hora. El horno INCO se cubre completamente por una coraza de acero dulce soldada de 1 cm de espesor la cual se apoya sobre pilares de ladrillo. El hogar es un arco invertido de ladrillo de magnesita, en tanto que la bóveda es un arco cóncavo de cromo-magnesita con un espacio de aire de 15 cm entre el refractario y la coraza de acero de la bóveda. Unas camisas de acero enfriadas con agua (revestidas de refractario de cromo-magnesita) cubren aproximadamente el 20 % de las paredes laterales, principalmente en la región inferior de la toma de gas. Esta área central es la zona de combustión de sulfuros de intensidad más alta y las paredes se protegen contra las temperaturas altas del gas de esta zona. El horno INCO normalmente funciona en forma continua durante dos años, después de los cuales se inspecciona y reviste con refractario donde sea necesario. El proceso INCO usa oxígeno comercial y es completamente autógeno y tiene varias ventajas sobre el proceso Outokumpu de aire precalentado, como son:
a) tiene un requerimiento de energía total mucho más bajo b) el volumen de gas generado (por tonelada de carga) es pequeño debido a que no
hay nitrógeno y productos de combustión de hidrocarburos, lo cual significa que su requerimiento para el equipo de colección de gas es pequeño
c) la concentración de SO2 en el gas generado es muy alta (80%) lo cual hace posible que el SO2 se recupere como ácido sulfúrico o SO2 líquido
d) las pérdidas de polvo son bajas debido a que la velocidad de flujo volumétrico de gas relativamente pequeña
e) su productividad (toneladas de carga/día/m2 de área del hogar) es casi 30% más alta que la del proceso Outokumpu.
Además, las escorias del proceso INCO son relativamente bajas en cobre (0.7%) y se desechan sin tratamiento adicional. Sin embargo, aún esta pérdida de cobre es excesiva y

es probable que en una nueva instalación las escorias INCO deban volverse a tratar para recuperar cobre. El método más probable puede ser la flotación del cobre a partir de la escoria lentamente enfriada, solidificada y finamente molida, este método permite lograr relaves (escoria final) con 0.35% de Cu. El producto gaseoso de las reacciones de oxidación de los sulfuros es el S02 y se producen cantidades considerables de éste por las reacciones de la fundición instantánea. Los gases que se producen por el proceso INCO contienen alrededor de 80% de S02, que se recupera eficientemente como S02 líquido mientras que el SO2 del proceso Outokumpu preferentemente se fija como ácido sulfúrico.
HORNOS ISASMELT
Southern Peru Copper Corporation (SPCC) ha modernizando su fundición de cobre de Ilo, instalando un horno ISASMELT para sustituir a sus dos hornos reverberos existentes y un convertidor modificado el Teniente (CMT), cumplimiento el compromiso del PAMA (Plan de Adecuación de Manejo Ambiental) para la Fundición de Ilo, acordado con el gobierno peruano. El horno ISASMELT tiene una capacidad nominal de 1.200.000 toneladas por año de concentrados nuevos y producir una mata conteniendo 62% de cobre y escoria de descarte. En la fundición se trata los concentrados de cobre de Cuajone y Toquepala, los cuales contienen aproximadamente 27% de Cu. Es probable que el Complejo Metalúrgico de la Oroya, también instale un horno ISASMELT en su circuito de cobre, por lo tanto, el futuro de la fundición de concentrados de cobre en el Perú estará ligado al proceso Isasmelt. El proceso ISASMELTTM emergió en la industria mundial de los metales durante la década de los 90 y actualmente está procesando alrededor de cuatro millones de toneladas de concentrados y materiales secundarios cada año. La tecnología de fusión por lanza

sumergida produce tanto como plomo metálico, matas de cobre o cobre metálico en las plantas ubicadas en Australia, EE.UU., Bélgica, India, Alemania, Malasia, China, Perú y Zambia. El proceso se basa en la lanza sumergida con entrada superior refrigerada por aire, denominado Sirosmelt, desarrollado por el Dr. John Floyd en la División CSIRO de Ingeniería de Minerales y Proceso en los años 70. El horno ISASMELT es un reactor cilíndrico vertical soportado sobre una base de concreto y forrado con ladrillos refractarios de cromo magnesita. La base es enladrillada en forma de plato. Un elemento clave del proceso es la lanza ISASMELT, la cual se baja desde la parte mas alta del horno dentro del baño fundido para inyectar aire enriquecido con oxigeno industrial de 95% de O2. La alimentación cae dentro del horno desde la puerta (compuerta) de alimentación ubicada en la parte alta del horno, el concentrado reacciona con el aire y el oxigeno inyectado por la lanza. La inyección de aire y oxígeno dentro del baño crea una alta turbulencia en el baño el cual constituye un método muy intenso de fusión de mata y una operación extremadamente eficiente requiriendo un horno relativamente pequeño para tratar altos radios de alimentación de concentrado. El horno ISASMELT incluye un caldero de tubos de pared, la carcasa de acero recubierto con refractario, blocks antisalpicaduras enfriado con agua, blocks para sacar material fundido y canales de transferencia de la mezcla mata-escoria a los hornos de retención rotatorios con enfriamiento por agua y todo el equipo auxiliar necesario. El techo del horno contiene un ducto de salida de gases y está construido con paneles de tubos del caldero. El vapor producido en el techo del horno es conectado al sistema del caldero recuperador de calor. Los gases salen por la parte más alta del horno e ingresan dentro de la sección de radiación del caldero recuperador de calor. Bajo condiciones normales la transición entre el horno y el caldero es ligeramente sellado. Una compuerta enfriada con agua es ubicada cerca de la transición entre el horno y el caldero y es insertada de manera que sirve para aislar el horno del caldero y de esta manera permitir el mantenimiento del caldero mientras el horno se mantiene caliente. Los ductos de ventilación secundaria son incorporados dentro del diseño del chute de alimentación para colectar emisiones fugitivas. Puertas en los chutes de alimentación son instalados para permitir el ingreso de la barra de medir y quemadores que son insertados dentro del horno a través de los chutes de alimentación. Una cámara del circuito cerrado de TV (CCTV) es también instalado sobre el chute de alimentación para detectar atoros en la carga. La barra de medir es usada para medir la profundidad del baño cuando no está fundiendo. La barra de medir está suspendida con un winche eléctrico. Una caja de escoria es instalada encima de la lanza en el techo del horno y cumple diversas funciones. La caja de escoria, junto con los sellos de la lanza, provee un efectivo sellado alrededor de la lanza para prevenir emisiones fugitivas. La caja de escoria es conectada hacia el sistema de ventilación secundaria para prevenir emisiones fugitivas. La caja de escoria también actúa como un embudo para colectar escoria fría la cual cae desde la lanza cuando es removida desde el horno para ser limpiada o remplazada.

La lanza ISASMELT es manufacturada de acero bajo en carbono y acero inoxidable. Remolinos internos en la lanza aseguran que el aire de proceso enfríe la lanza tal que una capa fría de escoria se forma sobre la lanza y la protege de la erosión y corrosión del baño fundido. El diseño de la lanza fue desarrollado para alcanzar la mínima caída de presión consistente con un aceptable tiempo de vida de la lanza (típicamente cerca de 10 días). La lanza de aire es suministrada para un determinado flujo de aire de proceso. Los cambios de lanza son normalmente llevados a cabo durante la rutina o en otras plantas durante las paradas por mantenimiento y toma alrededor de 45 minutos. Válvulas independientes controlan el aire de proceso proveniente del blower del ISASMELT y el oxigeno proveniente de las plantas de oxigeno. Mediciones exactas y control del flujo másico de cada uno es importante para el control del proceso en el horno. La lanza de aire enriquecido (mezcla de aire de proceso y oxígeno) es conducido a la lanza del ISASMELT por una tubería articulada. La tubería articulada permite que la lanza sea completamente removida del horno. Líneas de servicio adicional son también conectadas a la lanza (petróleo y líneas de retorno). La lanza esta diseñada para usar petróleo diesel N°2. La lanza es soportada en un porta lanzas el cual es subido y bajado por medio de un grúa eléctrica. La lanza es controlada por el PCS para permitir un posicionamiento automático y control de flujo. Cuando la fusión se detiene, la lanza es completamente removida del horno. Cuando es removido, la lanza esta cubierta de escoria. La escoria fría debe ser removida de la lanza en una forma controlada. Escoria en bloques cierran alrededor de la lanza en la posición alzada y van a caer directamente dentro de la caja de escoria. La lanza es golpeada con un martillo soportado por una grúa pescante. Cuando una lanza requiere cambio, es removida desde el porta lanzas usando la grúa, almacenada en el edificio ISASMELT y remplazada por una lanza reparada. Lanzas de repuesto están almacenadas dentro del edificio del ISASMELT. Cuando es necesario, lanzas usadas son removidas del edificio usando la grúa. Los productos de la fusión del horno ISASMELT son extraídos como una mezcla física de mata y escoria a través de dos placas hacia 2 hornos de retención rotatorios (RHF). Cada RHF es alimentado desde el horno ISASMELT vía una placa y un canal de cobre refrigerado con agua. Cada placa consiste de dos blocks (uno interior y otro exterior) El block exterior es remplazado periódicamente cuando el desgaste es excesivo. El block interior normalmente permanece en servicio mientras dure la campaña del ladrillo refractario. Cada canal ISASMELT consiste de múltiples secciones de cobre enfriado con agua. La sección de la cabecera del canal (muy cerca del agujero de colada) es protegida del flujo inicial de mata por un refractario especialmente diseñado. Cada agujero de colada es mecánicamente abierto y cerrado por medio de una máquina tapadora. Ambos agujeros de colada son servidos por la misma máquina viajando entre agujeros de coladas vía rieles adecuados para el caso. Si las máquinas tapadoras no se pueden utilizar, los agujeros de colada pueden ser abiertos usando lanzas de oxígeno. Los agujeros pueden ser tapados manualmente usando barro con un dolle (cono de arcilla
unido al extremo de una barra larga).
Quemadores de petróleo diesel (N°2) y aire comprimido son usados en las cabezas de sección (muy cercano al agujeros de colada) de cada canal para minimizar las acreciones durante la colada. La sección del canal después de la cabecera de sección tiene una pendiente pronunciada para evitar la formación de acreciones. Los gases de combustión y los humos procedentes de la operación de sangrado son colectados por una campana ubicada en la parte superior. La campana se mueve en el mismo nivel mediante rieles al igual que las máquinas tapadoras. Cuando la máquina tapadora esta sobre el canal la campana de ventilación esta en posición de parqueo, fuera del canal. Un quemador petróleo-aire (combustible N°6) es usado para mantener la temperatura del horno cuando no se alimenta concentrado. Para atomizar el petróleo se requiere de vapor de mediana presión. Un soplador de aire de combustión suministra el aire de combustión al quemador. El soplador de aire de combustión también suministra el aire de combustión secundario al horno a través de la lanza durante la fusión. El quemador es insertado dentro del horno a través del chute de alimentación. Termocuplas dentro del refractario del horno son usados para asegurar que la temperatura del horno es suficiente para asegurar la combustión del petróleo.
Tabla 1 - Parámetros de operación típicos - ISASMELT de cobre
Tasa Unidades
Tasa fusión concentrado 160 tph
Contenido cobre (base seca) 23,8 %Cu
Flujo de sílice 3.4 tph
Material Circulante 1.6 tph
Coque menudo 07 tph
Gas natural 706 Nm3/h
Aire en la lanza 20.210 Nm3/h
Oxígeno de la lanza (95%) 23.580 Nm3/h
Enriquecimiento con oxígeno de la lanza 60,8 %
Temperatura del baño 1172 Grados C
Ley de la mata 57,0 %
3.18. Conversión de matas de cobre En la conversión de matas de cobre, el proceso comercial es una adaptación del proceso Bessemer usado para la industria del acero, para convertir el arrabio obtenido en el alto horno a acero. Cualquiera sea la procedencia de la mata (hornos reverbero, hornos flash, eléctricos) ésta contiene básicamente Cu, Fe y S y hasta 3% de oxígeno disuelto, además contiene As, Sb, Bi, Pb, Ni, Zn y metales preciosos (ver cuadro siguiente), elementos que han estado presentes desde el concentrado y no se han podido separar en la etapa de fusión.
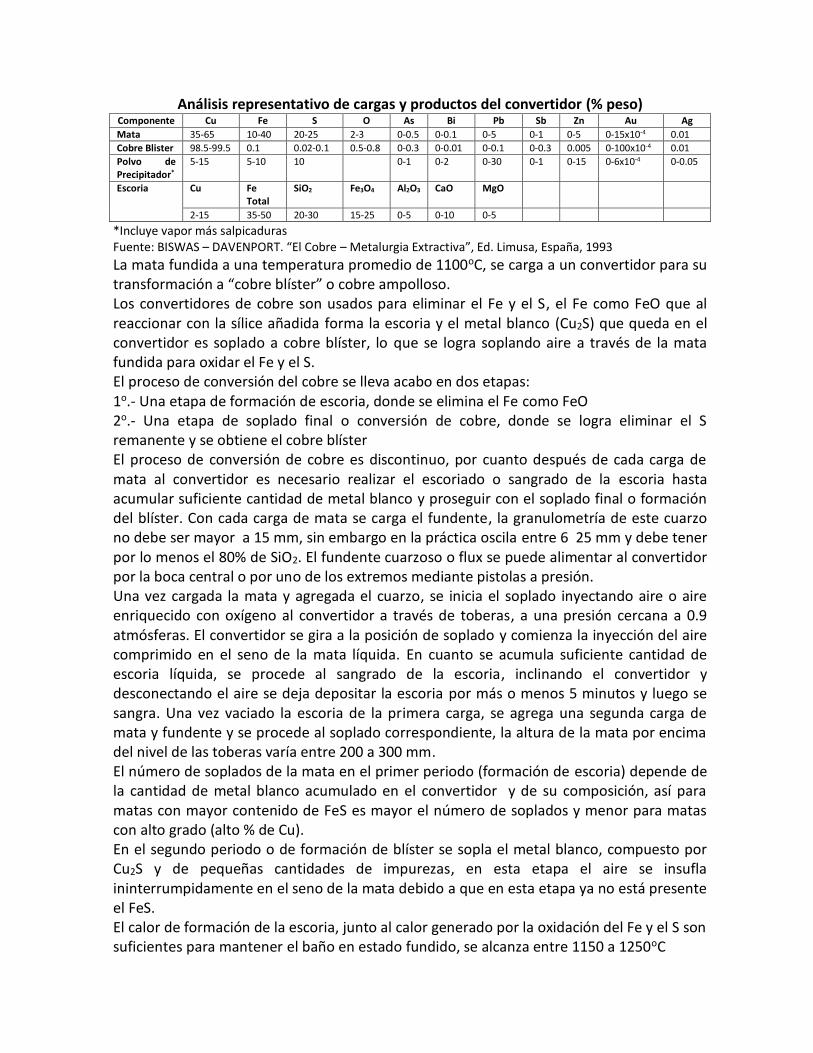
Análisis representativo de cargas y productos del convertidor (% peso) Componente Cu Fe S O As Bi Pb Sb Zn Au Ag
Mata 35-65 10-40 20-25 2-3 0-0.5 0-0.1 0-5 0-1 0-5 0-15x10-4 0.01
Cobre Blister 98.5-99.5 0.1 0.02-0.1 0.5-0.8 0-0.3 0-0.01 0-0.1 0-0.3 0.005 0-100x10-4 0.01
Polvo de Precipitador*
5-15 5-10 10 0-1 0-2 0-30 0-1 0-15 0-6x10-4 0-0.05
Escoria Cu Fe Total
SiO2 Fe3O4 Al2O3 CaO MgO
2-15 35-50 20-30 15-25 0-5 0-10 0-5
*Incluye vapor más salpicaduras Fuente: BISWAS – DAVENPORT. “El Cobre – Metalurgia Extractiva”, Ed. Limusa, España, 1993
La mata fundida a una temperatura promedio de 1100oC, se carga a un convertidor para su transformación a “cobre blíster” o cobre ampolloso. Los convertidores de cobre son usados para eliminar el Fe y el S, el Fe como FeO que al reaccionar con la sílice añadida forma la escoria y el metal blanco (Cu2S) que queda en el convertidor es soplado a cobre blíster, lo que se logra soplando aire a través de la mata fundida para oxidar el Fe y el S. El proceso de conversión del cobre se lleva acabo en dos etapas: 1o.- Una etapa de formación de escoria, donde se elimina el Fe como FeO 2o.- Una etapa de soplado final o conversión de cobre, donde se logra eliminar el S remanente y se obtiene el cobre blíster El proceso de conversión de cobre es discontinuo, por cuanto después de cada carga de mata al convertidor es necesario realizar el escoriado o sangrado de la escoria hasta acumular suficiente cantidad de metal blanco y proseguir con el soplado final o formación del blíster. Con cada carga de mata se carga el fundente, la granulometría de este cuarzo no debe ser mayor a 15 mm, sin embargo en la práctica oscila entre 6 25 mm y debe tener por lo menos el 80% de SiO2. El fundente cuarzoso o flux se puede alimentar al convertidor por la boca central o por uno de los extremos mediante pistolas a presión. Una vez cargada la mata y agregada el cuarzo, se inicia el soplado inyectando aire o aire enriquecido con oxígeno al convertidor a través de toberas, a una presión cercana a 0.9 atmósferas. El convertidor se gira a la posición de soplado y comienza la inyección del aire comprimido en el seno de la mata líquida. En cuanto se acumula suficiente cantidad de escoria líquida, se procede al sangrado de la escoria, inclinando el convertidor y desconectando el aire se deja depositar la escoria por más o menos 5 minutos y luego se sangra. Una vez vaciado la escoria de la primera carga, se agrega una segunda carga de mata y fundente y se procede al soplado correspondiente, la altura de la mata por encima del nivel de las toberas varía entre 200 a 300 mm. El número de soplados de la mata en el primer periodo (formación de escoria) depende de la cantidad de metal blanco acumulado en el convertidor y de su composición, así para matas con mayor contenido de FeS es mayor el número de soplados y menor para matas con alto grado (alto % de Cu). En el segundo periodo o de formación de blíster se sopla el metal blanco, compuesto por Cu2S y de pequeñas cantidades de impurezas, en esta etapa el aire se insufla ininterrumpidamente en el seno de la mata debido a que en esta etapa ya no está presente el FeS. El calor de formación de la escoria, junto al calor generado por la oxidación del Fe y el S son suficientes para mantener el baño en estado fundido, se alcanza entre 1150 a 1250oC

Las reacciones de la primera etapa son: 2FeS(l) + 3O2 → 2FeO(l) + 2SO2 ∆H = - 224 500 cal FeO(l) + SiO2 → FeO.SiO2(l) ∆H = - 5900 cal En esta etapa de formación de escoria, cualquier Cu2O que se forme, se vuelve a sulfurar hasta transformarse a Cu2S por la reacción: FeS(l) + Cu2O(l, escoria) → FeO(l, escoria) + Cu2S(l) k 1200oC = 104 Este valor de la constante de equilibrio muestra que el FeS forma sulfuros con el Cu2O, casi completamente. También es posible que se produzca cobre metálico líquido durante la formación de escoria, pero este cobre se vuelve a sulfurar con el FeS, según la reacción:
FeS(l) + Cu(l) + 1
2 O2 → Cu2S(l) + FeO(l)
En esta etapa parte del Fe se transforma en Fe3O4, según la reacción:
3FeO(l) + 1
2 O2 → Fe3O4(s)
Cuando se ha oxidado todo el S asociado con el Fe, el Cu2S comienza a oxidarse, generando Cu2O y tan pronto alcanza una concentración suficiente este óxido de cobre reacciona con el Cu2S para formar el cobre blíster, prosiguiendo estas reacciones hasta eliminar todo el S. También una parte del As, Sb, Pb y Zn presentes se volatilizan como óxidos, mientras que el Au y la Ag se disuelven en el blíster. Las reacciones de esta segunda etapa son: 2Cu2S(l) + 3O2 → 2Cu2O(l) + 2SO2 ∆H = - 118 900 cal Cu2S(l) + 2Cu2O(l) → 6Cu + SO2 2Cu2S(l) + O2 → 2Cu(l) + 2SO2 Si la cantidad de As y Sb presentes son suficientes, mayor al 0.8%, una parte de estos elementos se oxidan. 2As2S3 + 9O2 → 2As2O3 + 6SO2 2Sb2S3 + 9O2 → 2Sb2O3 + 6SO2 El encargado de la operación del horno controla el progreso del soplado observando los cambios registrados en la llama que sale del convertidor, debiendo tener mucho cuidado de no proseguir demasiado el soplado ya que ello podría provocar la solidificación de la carga del convertidor o la saturación del cobre fundido con Cu2O. El color rojizo original de la llama se transforma en verde al final del periodo de escorificación y por último al azul al final de la segunda etapa o formación de blíster. La conversión de matas de cobre se lleva a cabo casi universalmente en el convertidor Peirse-Smith.

3.19. Operaciones de conversión industriales
El convertidor de eje horizontal, llamado también convertidor Peirce – Smith, es el convertidor más comercial y consta de un cilindro horizontal recubierto con refractario, con una abertura en un costado que funciona como boca de carga y descarga del convertidor. El convertidor puede rotar sobre su mismo eje unos 120O para descargar y más o menos 60o para cargar. El aire comprimido o aire enriquecido con oxígeno es suministrado al convertidor a través de una caja de viento situado a lo largo de la parte posterior del convertidor, del cual una serie de tuberías horizontales (toberas delgadas) proporcionan pasajes a través del forro al interior. La mata de cobre se carga por la boca del convertidor y también se adiciona los fundentes por la boca o por un dispositivo ubicado en uno de los extremos del horno, cuando el convertidor está en la posición de carga y las toberas están ubicadas por encima del baño fundido; después de cargar la mata y los fundentes se comienza a suministrar el aire a través de las toberas bajo una presión adecuada, comenzando el soplado y finalmente se rota el horno hasta que la boca alcance su posición vertical. Las toberas se sumergen 2 a 4 pulgadas en el seno de la mata, tan pronto el aire entra, la mata que rodea la tobera se solidifica formando incrustaciones, los que se eliminan limpiando mecánicamente las toberas mediante la introducción de barras de acero. Las posiciones que adopta el convertidor durante la primera etapa de formación de escoria se presenta en el diagrama siguiente. La primera etapa del soplado o formación de escoria y colada de escoria se realiza tantas veces sea necesario, hasta que la cantidad de Cu2S (metal blanco) acumulado en el convertidor alcance su nivel de trabajo para iniciar el soplado final o de formación de cobre blíster, es decir la oxidación del metal blanco para producir Cu y SO2. Por consiguiente el

convertidor sólo contiene cobre metálico o cobre blíster del 98 al99%, que es colado en cucharas para ser llevado a instalaciones de moldeo o hacia las refinerías o al mercado.
CARGA SOPLADO ESCORIADO Generalmente en las fundiciones existen 3 a 6 convertidores asociados a cada horno de fundición. Dependiendo del grado de mata un convertidor puede hacer 1 a 3 ciclos por 24 horas.
Grado de mata Interrupciones de soplado en %
Tiempo del ciclo en horas
Utilización del convertidor en %
30 4.3 17.6 75
40 3.2 12.1 73
50 2.5 8.8 71
En la etapa de formación de escoria se desprende más calor que en la etapa de soplado de blíster, esto se debe a que la oxidación de 1 libra de FeS desprende 200 BTU, según la reacción: FeS + 3O2 + SiO2 → 2 FeO.SiO2 + 2SO2 Mientras que una libra de Cu2S, al ser oxidado desprende solamente 600 BTU, según la reacción: Cu2S + O2 → 2Cu + SO2 El siguiente diagrama ilustra las posibles reacciones que ocurren dentro del convertidor en la etapa del soplado final.

Con el propósito de aprovechar el exceso de calor generado en las reacciones y proteger el revestimiento de los ladrillos, se añaden al convertidor residuos de fundición, chatarras o fragmentos de cobre, en algunos casos concentrados de cobre, después de la primera extracción de escoria. Teóricamente, durante el soplado de la escoria, los gases deberían contener 15% de SO2 y durante el soplado del blíster deberían contener 21% de SO2, cosa que no se cumple en la práctica, debido a las sulfataciones que se producen dentro del horno y las infiltraciones de aire en el sistema. El diámetro del convertidor horizontal varía entre 3 a 4 metros, generalmente es de 4 metros y la longitud puede estar entre 6 a 11 metros. Un convertidor de 3.9 m de diámetro interno por 9 m de longitud puede tener hasta 40 toberas de 3.75 cm de diámetro, distribuidos a una distancia de 16.5 cm de centro a centro. La construcción de un convertidor comprende una coraza de acero de 4 a 5 cm de espesor, revestidos con ladrillos de magnesita o cromo magnesita de 25 a75 cm. Los convertidores de estas dimensiones tratan de 300 a 500 toneladas de mata por día para producir 100 a 200 toneladas de cobre. Existen dos modificaciones de los convertidores horizontales:
1. Convertidor Hood (chimenea) 2. Convertidor Hoboken
En ambos casos se logra reducir las infiltraciones de aire en los gases de salida y por consiguiente se aumenta la concentración de SO2. El convertidor Hood dispone de una chimenea con compuertas herméticas a la salida de los gases, logrando así evitar las infiltraciones de aire y el convertidor Hoboken, dispone en el extremo del convertidor una campana de extracción en forma de “U” invertido que rota con el convertidor, este convertidor usa aire enriquecido con oxígeno.
CONVERTIDOR HOOD CONVERTIDOR HOBOKEN
3.20. Formación de la magnetita en la conversión La fase de óxido estable proveniente de la oxidación del FeS es la magnetita sólida. Aunque se desea algo de magnetita como depósito sobre las paredes del convertidor para proteger los refractarios, una cantidad excesiva conduce a escorias viscosas y al arrastre de cantidades grandes de mata. La tendencia hacia la formación o reducción de magnetita dentro de los convertidores se da según la reacción:

3Fe3O4(s) + FeS(l, mata) 10FeO(l, escoria) + SO2 Para esta reacción ∆Go = 162 000 – 92.1 T oK (Johansen, 1970) y la constante de equilibrio
K = (𝑎𝐹𝑒𝑂)10 𝑝𝑆𝑂2
(𝑎𝐹𝑒3𝑂4)3𝑎𝐹𝑒𝑆, es 10-4ª 1200oC. La formación de la magnetita se reduce al disolverse el
FeO en una escoria de silicato, lo cual disminuye la aFeO, causando así que la reacción de formación de FeO anterior proceda a la derecha. Los cálculos muestran que la aFeO, se debe reducir hasta casi 0.6 para evitar la formación de la magnetita, cuando aFeS ≅ 0.5 y pSO2 ≅ 0.1. Esto se logra con una escoria que contenga de 20 a 30% de SiO2 y de 70 a 80% de FeO. La formación de magnetita se vuelve particularmente aguda al final de la etapa de formación de escoria por que existe poca cantidad de FeS y su actividad es baja y por tanto no puede reducir la magnetita, por lo que estas escorias finales contienen entre 10 a 20 % de magnetita sólida. La mejor práctica del convertidor para reducir la formación de magnetita es mantener la mata tan concentrada como sea posible en FeS hasta el final de la etapa formadora de escoria al oxidar sólo parcialmente la mata después de cada adición de mata con lo que se asegura una cantidad suficiente de FeS para reducir la magnetita. La reducción de formación de magnetita sólida también se favorece con una temperatura alta del proceso de conversión por que:
a) Causa la combinación rápida del FeO y el SiO2 para formar la escoria b) Aumenta la solubilidad de la magnetita sólida en la escoria c) Tiende a promover la reducción del Fe3O4 por el FeS, por que la constante de
equilibrio k aumenta con la temperatura.
3.21. Procesos de fusión – conversión continúas Los procesos pirometalúrgicos tradicionales de obtención de cobre ampolloso o blíster a partir de concentrados sulfurados de cobre comprende dos o tres etapas de tratamiento, según la naturaleza del concentrado, así puede ser: fusión y conversión o tostación, fusión y conversión. Estos procesos exigen instalaciones grandes, equipos diversos para trasladar los materiales de una etapa a otra, alto consumo de energía, sobre todos combustibles, que significa altos costos, por lo que los ingenieros metalúrgicos han buscado alternativas de tratamiento más rápidos, eficientes y económicos, llegando a desarrollar procesos continuos y autógenos o semiautógenos. Con ese propósito se realizaron experimentos en el decenio de 1950, en las que se demostró que el cobre blíster podía ser directamente y continuamente producido a partir de una mata de Cu2S-FeS soplando aire y oxígeno al interior de una mata líquida, trabajos posteriores de los años 1960 mostraron que los concentrados pueden ser fundidos simultáneamente con la producción continua de cobre ampolloso, con lo que se logró la verdadera fundición de una etapa. Las primeras patentes fueron publicadas en 1967, siendo la patente 3326671 de Estado Unidos de Worcra Process y la patente 758020 de Canadá de Noranda Process. El desarrollo de estos procesos continuos, que se ha dado en los últimos 40 años, ha sido una respuesta al alto costo de los combustibles y a las cada vez más estrictas leyes de protección ambiental y minimizar los costos de producción. Una representación esquemática de un proceso ideal de una sola etapa se presenta en el diagrama siguiente:

Los materiales de entrada al proceso deben ser concentrados, fundentes y aire, aire más oxígenos u oxígeno (el uso de aire más oxígeno u oxígeno disminuye las perdidas de calor sensible en el nitrógeno si se usa aire) y los productos pueden ser cobre blíster, un gas con alta concentración de SO2 y una escoria de bajo contenido de cobre que se deseche directamente, el proceso puede ser continuo en un solo reactor o en varios. Las ventajas de un proceso de una sola etapa en la producción del cobre blíster pueden ser:
a) Reducida manipulación de materiales debido a la ausencia de etapas intermedias b) Baja o nula demanda de energía debido al uso eficiente de la energía utilizada de las
reacciones de oxidación de los sulfuros en un solo reactor c) Producción de una sola corriente de gas con alta concentración de SO2 apta para la
producción de ácido sulfúrico o SO2 liquido d) La posibilidad de la aplicación del control automatizado a todo el proceso
Además está el hecho de que el costo de capital es menor comparado con los procesos de más de una etapa, la baja demanda de energía y la eficiente capacidad de recuperar el SO2 de la corriente de gas que se genera en el proceso. Se han probado varios procesos continuos de una sola etapa como el Proceso Noranda, Worcra, Convertidor Teniente Modificado, CONTOP, algunos han tenido uso industrial. Una característica de los procesos continuos de una sola etapa es la presencia permanente de tres fases liquidas dentro del reactor: escoria, mata y cobre blíster. En esencia, el sistema de una etapa siempre está en estado equivalente con respecto al momento de la conversión normal cuando:
Se oxida a su nivel mínimo el último FeS Se produce el primer cobre blíster
En los procesos de conversión común se puede remover la escoria final y comenzar la etapa de formación del blíster, sin embargo, en los procesos de una etapa la escoria no se elimina completamente, de manera continua la mata es reabastecida con sulfuro de cobre y hierro por la carga continua de concentrados. Este procedimiento permite que siempre haya escoria, mata y cobre blíster dentro del horno todo el tiempo, tales elementos conducen a la oxidación simultánea de los sulfuros de hierro a óxidos de hierro y los sulfuros de cobre a cobre blíster, siendo la reacción total del proceso:
4CuFeS2(concentrado) + 21
2O2 4Cu(l, blíster) + FeO(l, escoria) + Fe3O4(s) + 8SO2
También se han probado y usado industrialmente un proceso industrial de etapas múltiples, el Proceso Mitsubishi, que tiene como ventaja principal la producción continua y uniforme de gas que permite recuperar el SO2 en forma eficiente.

3.22. Proceso Noranda Es propiedad de la compañía Noranda Mines – Quebec – Canadá y empezó a operar a nivel de planta piloto en 1968, originalmente se concibió como un proceso de obtención de cobre blíster en una sola etapa. El proceso Noranda ha sido probado a escala de planta piloto (90 toneladas de concentrado por día) y a escala industrial (1100 toneladas de concentrado por día). Están en operación 4 hornos a escala industrial (1969) uno en Noranda, Quebec y tres en Garfield, Utah (Kennecott). Aunque la intención original del proceso fue producir cobre blister directamente en una sola etapa, la totalidad de estas unidades producen comúnmente mata de grado muy alto (70-75% de Cu). DESCRIPCION DEL REACTOR Los reactores industriales son de 21 metros de longitud con un diámetro de carga interior de 5 metros. El reactor Noranda es un recipiente cilíndrico revestido de cromo-magnesita que se asemeja a un convertidor Peirce Smith, el aire o aire enriquecido con oxígeno es introducido en la capa de mata mediante 50 o 60 toberas situadas a lo largo de uno de los lados del recipiente. Las toberas permanecen sumergidas durante todo el proceso y solamente se les hace girar hacia fuera de los líquidos en el caso de una para, falla del soplador u otra situación de urgencia El reactor Noranda utiliza un horno cilíndrico con revestimiento refractario para la fundición. El concentrado peletizado y los aditivos se cargan en el baño de escoria fundida por el extremo superior del horno. Los quemadores alimentados a gas natural o aceite, situados en ambos extremos, producen el calor necesario para el proceso. Se inyecta aire enriquecido con oxigeno en el baño fundido mediante toberas, lo que produce la oxidación del azufre y el hierro.

OPERACIÓN DEL PROCESO
Carga de concentrado verde aglomerado (10% de agua), carbón y fundente de sílice sobre la superficie de la escoria por medio de un banda de slinger.
Inyección de aire o aire enriquecido con oxigeno por medio de las toberas sumergidas.
Sangrado de escoria en el lado opuesto al extremo de carga.
Sangrado de cobre blíster (1 a 2% S) intermitentemente desde el fondo del reactor por un agujero de colada caliente.
Quemado de gas natural o aceite en quemadores en ambos extremos del reactor.
En el reactor se produce en un gas que contiene de 8 a 15% de SO2, que depende del nivel de enriquecimiento con oxigeno del aire de entrada. El gas sale por la boca hacia un sistema de colección de polvos. La boca se usa solamente para sacar el gas desde el reactor, por consiguiente, puede estar perfectamente cubierto para evitar fuga de gas a los alrededores o infiltraciones de aire. La cubierta hermética también impide la dilución del SO2 por la infiltración del aire y de ese modo los gases provenientes del reactor tienen la concentración suficiente de SO2 como para que se pueda recuperar de manera eficaz en forma de H2SO4 o SO2 líquido. El cobre blíster producido se asienta en un “pozo” en el fondo del reactor desde el cual se vacía (1200ºC) en ollas de colada que son enviadas a los hornos de ánodos para eliminar el S remanente. El contenido de S en el cobre blíster del proceso Noranda es del 1 a 2% y es considerablemente más alto comparado con el convertidor común que es de 0.02 a 0.1% y por lo tanto requiere más aire y un periodo de oxidación mucho más prolongado en el horno de ánodos, lo cual es una desventaja de proceso. La escoria es sangrado (1200ºC) en ollas y son coladas en piezas en forma de grandes ladrillos y se les deja enfriar lentamente. Dichos ladrillos se quiebran y muelen, el cobre se recupera de la escoria por flotación. La escoria inicialmente contiene de 8 a 12% de cobre y las colas finales de flotación contienen 0.5%. El concentrado obtenido del tratamiento la escoria con un contenido de 55% de Cu se combina con el concentrado fresco y refundido en el reactor.
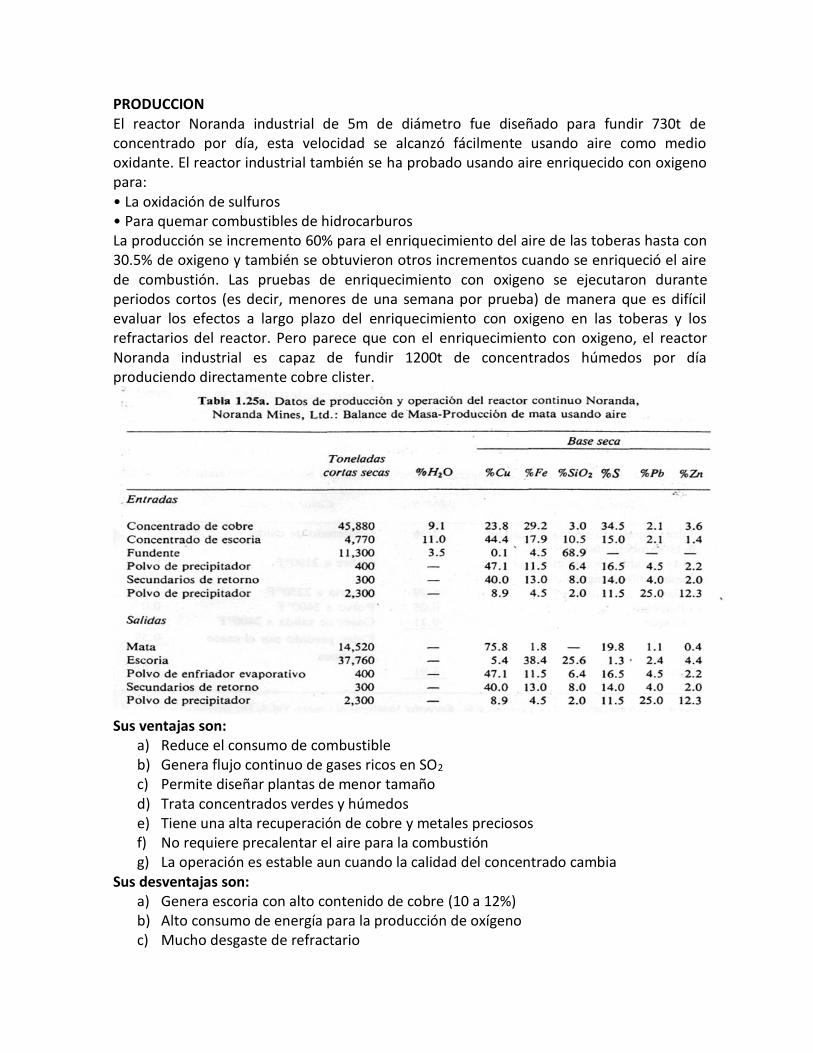
PRODUCCION El reactor Noranda industrial de 5m de diámetro fue diseñado para fundir 730t de concentrado por día, esta velocidad se alcanzó fácilmente usando aire como medio oxidante. El reactor industrial también se ha probado usando aire enriquecido con oxigeno para: • La oxidación de sulfuros • Para quemar combustibles de hidrocarburos La producción se incremento 60% para el enriquecimiento del aire de las toberas hasta con 30.5% de oxigeno y también se obtuvieron otros incrementos cuando se enriqueció el aire de combustión. Las pruebas de enriquecimiento con oxigeno se ejecutaron durante periodos cortos (es decir, menores de una semana por prueba) de manera que es difícil evaluar los efectos a largo plazo del enriquecimiento con oxigeno en las toberas y los refractarios del reactor. Pero parece que con el enriquecimiento con oxigeno, el reactor Noranda industrial es capaz de fundir 1200t de concentrados húmedos por día produciendo directamente cobre clister.
Sus ventajas son:
a) Reduce el consumo de combustible b) Genera flujo continuo de gases ricos en SO2 c) Permite diseñar plantas de menor tamaño d) Trata concentrados verdes y húmedos e) Tiene una alta recuperación de cobre y metales preciosos f) No requiere precalentar el aire para la combustión g) La operación es estable aun cuando la calidad del concentrado cambia
Sus desventajas son: a) Genera escoria con alto contenido de cobre (10 a 12%) b) Alto consumo de energía para la producción de oxígeno c) Mucho desgaste de refractario

CONVERTIDOR MODIFICADO EL TENIENTE
En 1977 nació el Convertidor Modificado el Teniente, en la Fundición de Caletones de CODELCO, Chile y por el éxito industrial alcanzado se expandió a fundiciones de Zambia, Canadá y Perú. En Perú estuvo operando en la Fundición de Ilo en remplazo de dos hornos reverberos y debido a la modernización por las exigencias ambientales ha sido también remplazado por un horno Isasmelt. El convertidor Teniente, desarrollado y patentado por la División El Teniente de Codelco, es un horno basculante, formado por un cilindro metálico de 5 m de diámetro por 22 m de largo, dispuesto en posición horizontal y revestido por ladrillos refractarios en su interior. Este horno está montado sobre un sistema de cremalleras que le permiten oscilar. El convertidor Teniente es cargado en forma continua con concentrado de cobre y sílice (cuarzo) por una abertura ubicada en su parte superior. La sílice tiene por objeto captar el hierro contenido en los minerales sulfurados fundidos y concentrarlo en la parte más liviana de la mezcla fundida. El convertidor Teniente tiene un sistema de cañerías en el interior, las cuales insuflan aire enriquecido con oxígeno, el cual permite la oxidación del hierro y del azufre presentes en los minerales que constituyen el concentrado. El hierro
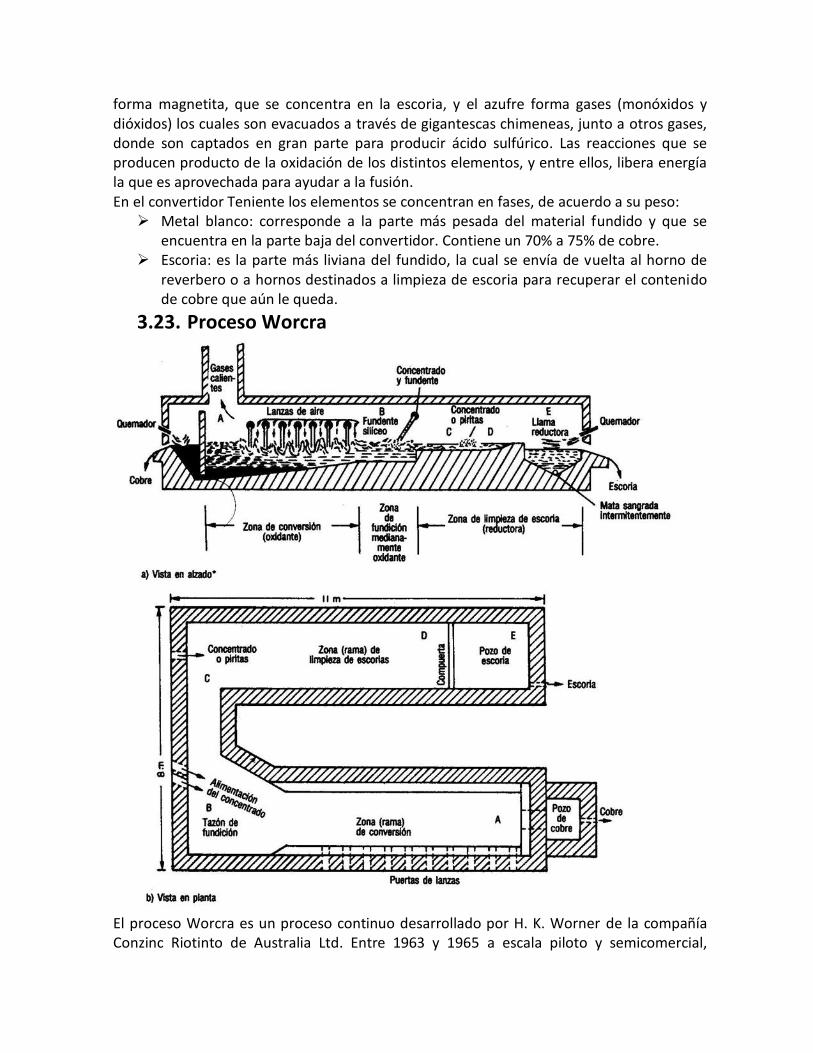
forma magnetita, que se concentra en la escoria, y el azufre forma gases (monóxidos y dióxidos) los cuales son evacuados a través de gigantescas chimeneas, junto a otros gases, donde son captados en gran parte para producir ácido sulfúrico. Las reacciones que se producen producto de la oxidación de los distintos elementos, y entre ellos, libera energía la que es aprovechada para ayudar a la fusión. En el convertidor Teniente los elementos se concentran en fases, de acuerdo a su peso:
Metal blanco: corresponde a la parte más pesada del material fundido y que se encuentra en la parte baja del convertidor. Contiene un 70% a 75% de cobre.
Escoria: es la parte más liviana del fundido, la cual se envía de vuelta al horno de reverbero o a hornos destinados a limpieza de escoria para recuperar el contenido de cobre que aún le queda.
3.23. Proceso Worcra
El proceso Worcra es un proceso continuo desarrollado por H. K. Worner de la compañía Conzinc Riotinto de Australia Ltd. Entre 1963 y 1965 a escala piloto y semicomercial,

habiendo estado en operación una planta de 80 toneladas por día de concentrado y demostrando que es posible producir cobre blíster con una ley de 98.2 a 99.5%, sin embargo no se alcanzó a instalar planta alguna a nivel comercial. Este proceso se caracteriza por que produce metal en lugar de mata y que la mayoría de las reacciones exotérmicas se generan y continúan dentro del horno. Dentro del horno se genera una turbulencia consistente en la zona de fusión y conversión por la inyección de gas con oxígeno en la masa fundida que fluye continuamente; en la zona de conversión la escoria se mueve por gravedad en contracorriente a la mata y al metal y la escoria se limpia en la zona de respectiva. Los gases generados en el proceso contienen de 9 a 12% de SO2, concentración que facilita el proceso de producción de H2SO4. El reactor consiste de un cilindro forrado con refractarios, dispone de y toberas para la descarga de la escoria por un extremo y de cobre ampolloso por el otro. En el interior del horno se producen una serie de reacciones y flujos de materiales. Los concentrados y fundentes se inyectan por medios mecánicos o neumáticos al centro e interior del horno en atmósfera oxidante y a un ángulo apropiado para asegurar la penetración de las partículas en la masa fundida y ayudar a la circulación continua de la mata y la escoria en la taza central. En cierta forma se consigue un efecto de extracción por solventes en caliente al forzar a la escoria a moverse en contracorriente a la mata, de modo que los componentes no volátiles que se desea eliminar, como el Fe, son transferidos continuamente de la mata a la escoria después de ser oxidados. Por otro lado en las zonas de fusión y limpieza de escoria en especial, se retorna el cobre de la escoria a la mata mediante la acción del FeS presente en la mata, por esto se requiere agregar concentrados o pirita en la zona de limpieza de la escoria a fin de separar y sedimentar la mata atrapada que regresa por gravedad a la zona de fusión, debido a la pendiente del fondo del horno. A medida que la mata avanza hacia las zonas de fusión y conversión, es soplado con aire o aire enriquecido con oxígeno, oxidando primero el FeS de la mata hasta metal blanco y luego a cobre blíster que pasa a moldeo. La escoria limpia contiene de 0.3 a 0.5% de cobre, según se use pirita o concentrado para la limpieza. Los gases ricos en SO2 pasan por un equipo convencional de recuperación de calor sensible y de polvos para ser tratados en la planta de +acido sulfúrico. El principio de este proceso es similar al proceso Noranda, es decir tratar concentrados de cobre hasta obtener cobre blíster, sin embargo difiere en tres puntos fundamentales:
1. El aire es introducido a la fase mata por medio de lanzas que se extienden hacia el interior de la capa de mata desde la bóveda o la parte superior de las parees laterales
2. El reactor es un horno de tipo hogar 3. En el horno hay una zona de asentamiento de manera que las escorias pueden ser
descargadas directamente. El uso de lanzas significa que el reactor puede ser estático, no siendo necesaria la rotación como es el caso del reactor Noranda. El mayor inconveniente es la manufactura de lanzas durables de gran resistencia que se puedan sumergir en la fase mata. La forma en “U” del reactor, visto en planta, es útil por que proporciona dos ramas aisladas, una se usa para alimentar el concentrado, zona de fusión, y para la oxidación

continua de la mata y la segunda proporciona una zona relativamente quieta en la cual el cobre se asienta desde la escoria bajo condiciones reductoras debido a la adición de FeS o FeS2. Aunque la forma en “U” del proceso Worcra es útil para la sedimentación del cobre a través de la escoria, tiene dos desventajas:
1. El calor generado en las reacciones de oxidación no se aprovecha en la rama de sedimentación, por lo que es necesario quemar combustible de hidrocarburos, lo que produce grandes volúmenes de gas que diluye el contenido de SO2 a niveles de 1 a 2%
2. Solamente una pequeña parte del horno se aprovecha para la reacciones de formación de cobre, por lo tanto, la productividad del horno es baja, 2 a 3 toneladas de carga por m2 de área de hogar.
3.24. Proceso Mitsubishi Un proceso de producción continua de cobre es el proceso Mitsubishi (Suzuki, 1974; Suzuki y colaboradores, 1974, EMJ, 1972; PATENTE CANADIENSE 952319,19749). El proceso Mitsubishi emplea tres hornos interconectados por un flujo continuo por gravedad de mata y escoria y tiene similitud con los procesos Noranda y Worcra por producir continuamente cobre blíster por la oxidación continua de la mata en un “horno de conversión “final. El proceso Mitsubishi ha sido probado a escala de plata piloto con 65t de carga por día y a escala de prototipo con 150t de carga por día. La primera planta comercial que ha entrado en producción tenía una capacidad de 650t por día. En las figuras siguientes se observan los tres hornos Mitsubishi conectados en forma de cascada de manera que la mata y la escoria fluyan por gravedad en tres de los hornos.
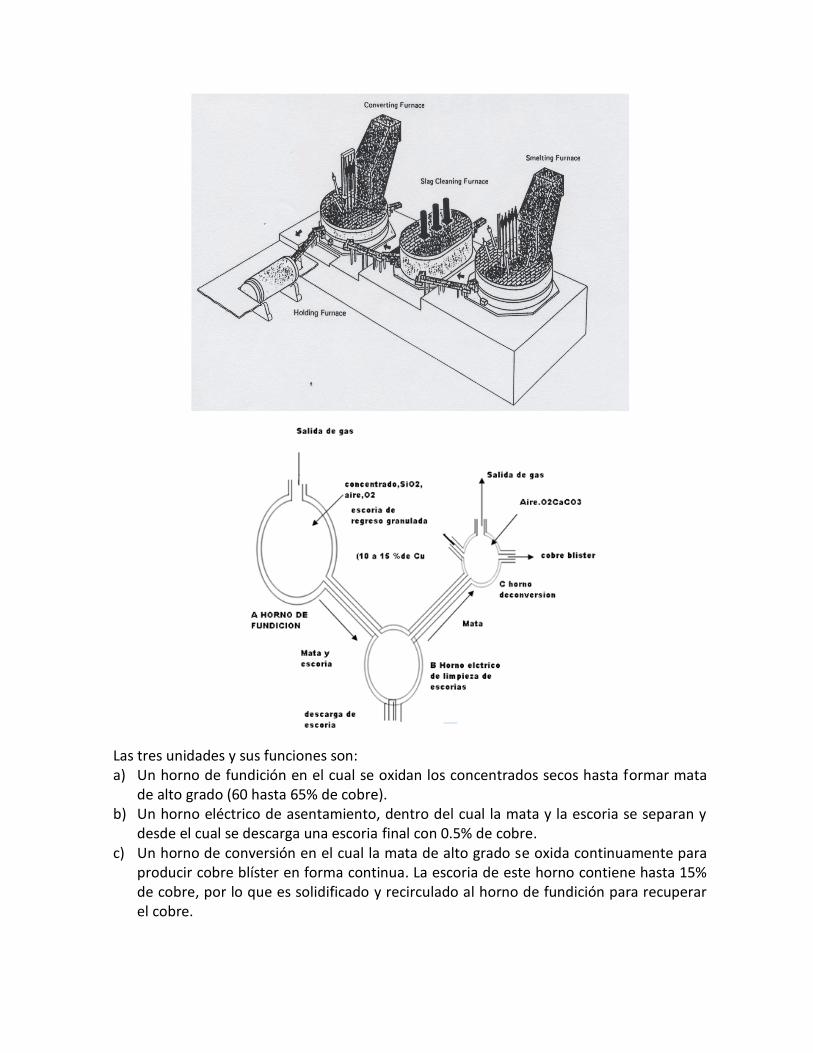
Las tres unidades y sus funciones son: a) Un horno de fundición en el cual se oxidan los concentrados secos hasta formar mata
de alto grado (60 hasta 65% de cobre). b) Un horno eléctrico de asentamiento, dentro del cual la mata y la escoria se separan y
desde el cual se descarga una escoria final con 0.5% de cobre. c) Un horno de conversión en el cual la mata de alto grado se oxida continuamente para
producir cobre blíster en forma continua. La escoria de este horno contiene hasta 15% de cobre, por lo que es solidificado y recirculado al horno de fundición para recuperar el cobre.

El oxidante en los hornos de fundición y de conversión es aire enriquecida por oxigeno (33% de O2 y 26% de O2, respectivamente). El aire se inyecta en la superficie de escoria por 18 lanzas de acero inoxidable en posesión vertical. Los que diseñaron el proceso Mitsubishi (Suzuki, 1974), informan que la inyección de aire u oxigeno en la superficie en la parte superior del baño de mata y escoria no puede usarse para producir continuamente cobre blíster en una sola unidad de fundición. Esto se debe probablemente, a que se forme demasiada magnetita sólida sobre la superficie de escoria debido al choque del aire. La presencia de una capa de magnetita sólida puede obstaculizar la alimentación de concentrados en lo líquidos lo que inhibe grandemente el proceso de fundición. El uso de tres hornos, donde los dos primeros están en condiciones relativamente reductores por la presencia del FeS, impide la formación excesiva de magnetita.
REACCIONES QUÍMICAS DEL PROCESO MITSUBISHI
Las reacciones en el horno de fundición del sistema Mitsubishi son químicamente, pero no físicamente, similares a las de un horno de fundición instantánea Outokumpu, es decir, que los concentrados son oxidados y fundidos para formar mata de alto grado. Existen, sin embargo dos diferencias significativas: a) El horno de fundición Mitsubishi `produce mata de un grado más alto (65% de cobre)
que la mayoría de los hornos de flash Outokumpu (55% de cobre). b) El horno de fundición Mitsubishi trata escorias de horno de
Conversión solidas y recicladas (15% de Cu) para fundirlas y lograr asentamiento parcial de cobre.
Una característica más significativa del sistema Mitsubishi es que la mata y escoria se extraen juntas del horno de fundición. Se separan en un pequeño horno eléctrico de sedimentación el cual también se usa, con adiciones de coque, FeS o FeS2, para recuperar el cobre contenido en la escoria del convertidor. Las reacciones en el horno de sedimentación Mitsubishi son similares a las de los hornos de sedimentación de escoria comunes. Las reacciones en el horno de conversión del sistema Mitsubishi son químicamente similares a aquellas de los hornos de fabricación de cobre de una sola etapa. La mata se oxida continuamente para producir cobre blíster y escoria (más magnetita) y continuamente se suministra sulfuro de hierro y cobre. En el caso Mitsubishi, la fuente de sulfuros nuevos es la mata liquida proveniente de los hornos de fundición y sedimentación y no los concentrados recién cargados. Una diferencia mayor entre las condiciones en el horno de conversión Mitsubishi y las de los reactores de una etapa es la composición de la escoria. La del horno de conversión Mitsubishi es básica (es decir, poco o nada de SiO2 se agrega) mientras que la escorias de los reactores de una sola etapa contienen 20 a 35% de SiO2. Las principales consecuencias de usar una escoria sin sílice (es patente canadiense 954700, 1974) son: a) El contenido de azufre en cobre blíster es bajo (0.1 a 0.9% de S) comparado con el
proceso de una etapa (1 a 2%).

b) Virtualmente, todo el producto de oxido de hierro es magnetita, pero la escoria es moderadamente fluidas debido a las temperaturas de fusión relativamente bajas en sistema CaO-Cu20-Fe3O4.
c) La producción de escoria es pequeña (0.33 t por tonelada de Cu producido).
A pesar de sus composiciones completamente diferentes, las concentraciones de Cu en las escorias del horno de conversión Mitsubishi (10 a 15% de Cu) son bastantes similares a las composiciones de las escorias del proceso de una etapa (8 a 12% de Cu en las zonas de reacción). El uso de una escoria básica en el horno de conversión Mitsubishi tiene dos ventajas principales: a) el cobre blíster requiere relativamente poca refinación térmica debido a su contenido
de azufre (0.1 A 0.9 % DE S). b) la recirculación de escoria al horno de fundición se mantiene un mínimo por la pequeña
producción de escoria. La escoria del horno de conversión Mitsubishi solidifica en forma granulada con chorros de agua y se recircula al horno de fundición por medio de un sistema trasportador de cangilones para recuperar el cobre. La práctica de cargar escoria solida fría al horno de fundición representa un gasto excesivo de energía y, por esta razón, es importante mantener a un mínimo las recirculaciones escoria.
IMPUREZAS EN EL COBRE BLISTER DEL PROCESO MITSUBISHI (SUZUKI Y SHIBASAKI, 1975)
Se ha publicado los niveles de antimonio, arsénico y bismuto en el cobre blíster Mitsubishi (EMJ.19729), pero no se a analizado en términos de las proporciones retenidas desde la alimentación original hasta el lugar de fundición. Existe la posibilidad de la retención de As, Sb y Bi en el cobre del proceso Mitsubishi pueda ser alta debido al contacto continuo entre la mata y el metal dentro del horno de conversión, por esta razón se debe poner especial cuidado con la electrorefinación del cobre Mitsubishi y como el proceso Noranda podría

ser que el proceso Mitsubishi no fuera el apropiado para el tratamiento de concentrados impuros.
PRODUCCION Y DEMANDA DE COMBUSTIBLE
La producción del proceso Mitsubishi es de 5 toneladas de carga por metro cuadrado de área en planta total de los tres hornos, que es un poco más baja que la unidad industrial Noranda, esto es debido que el proceso Mitsubishi emplea uno de sus hornos para par la recuperación de cobre a partir de la escoria (y no de molienda de escoria) lo que explica en cierto grado, su baja productividad total. La demanda de combustible para el proceso Mitsubishi es alrededor de 5x105 Kcal que es del mismo orden que los hornos de flash Outokumpu. Esta demanda sería más baja si el enriquecimiento con oxigeno en los hornos de fundición y conversión se incrementara por arriba de los niveles comunes de 36 y 26 %. El proceso Mitsubishi a escala industrial es operado bajo el control automático.
3.25. Proceso Kivcet - Proceso CONTOP
PROCESO KIVCET Desarrollado en Rusia, se basa esencialmente en el uso de un ciclón especialmente diseñado para fundir concentrados hasta matas, que se iniciaron a escala piloto , combinado el ciclón con hornos reverbero, para lo cual se colocó primero el ciclón en un extremo, sin ningún éxito y luego se le puso encima del reverbero a la vez que se introdujo el uso de oxígeno puro en el ciclón en lugar de aire o aire enriquecido con oxígeno, esta innovación tecnológica mejoró la eficiencia de la combustión, redujo la cantidad de elementos desechables en los gases y aumentó la concentración de SO2 recuperable. En un diseño más avanzado, la instalación de una pared divisoria y refrigerada con agua, entre la sección de fusión y sedimentación ha permitido separar el gas en esos compartimientos, haciendo posible el tratamiento de concentrados complejos que contienen metales como Zn y Pb, que son recuperados además del cobre. Este diseño del horno KIVCET permite tener condiciones altamente oxidantes en el ciclón, mientras se lleva acabo una reducción en la zona de sedimentación calentada por resistencias eléctricas. De esta manera se obtienen tres productos que se descargan independientemente: la mata de cobre de alta ley, hasta 50% de cobre, conteniendo metales valiosos como Ag, Au, Ni, que sigue su marcha normal en convertidores; el plomo crudo de calidad convencional que se trata por refinado térmico y la escoria que puede desecharse por contener metales no ferrosos en menor porcentaje que los valores mostrados por escorias producidas por procesos convencionales. Los gases producidos con elevado contenido de SO2, hasta un 85%, pasan por instalaciones de enfriamiento y separación de polvos finos antes de ingresar a la planta de ácido sulfúrico.

PROCESO CONTOP Es propiedad de la KHD Humboldt Wedag AG de Alemania y es el resultado de una combinación de la tecnología de fusión en un horno ciclón usando oxígeno con el proceso de soplado por el techo usado para el tratamiento secundario de las escorias producidas en la fusión y es similar al proceso LD en la conversión de aceros. En el proceso CONTOP (Continuous Top Blowing Process ), el horno ciclón produce el metal blanco y una escoria alto contenido de FeO, el proceso de fusión es autógeno y usa el Fe y S de concentrado como combustible, usando oxígeno puro para el soplado en la etapa de conversión. La masa fundida en el horno ciclón es descargada a una cámara de sedimentación, donde la escoria es soplada por el techo con un reductor para limpiarla, desde esta cámara la mata es continuamente descargada a un convertidor estacionario o reactor de refinación que está dividida en tres cámaras: de conversión, de oxidación y de reducción, obteniéndose como producto final un ánodo de cobre de 98.8% y poca cantidad de escoria. La temperatura alcanzada en el horno de ciclón es del orden de 2000oC, por lo que requiere refrigeración y esta temperatura permite mantener la viscosidad de la escoria en niveles bajos. En 1978 la KHD Humboldt instaló una planta piloto de 20 a 24 toneladas de concentrado por día y luego ofreció instalar plantas con capacidad de 25 000 TM por año de ánodos de cobre. Las ventajas de este proceso son:
a) La fusión de los concentrados es totalmente autógena, pues usa como combustible el Fe y el S del concentrado
b) El uso del oxígeno permite emplear equipos de tratamiento de gases pequeños y menos complejos
c) No hay formación de óxidos de nitrógeno que son tóxicos d) Se obtiene gases con alto contenido SO2

3.26. Pre afino ígneo de cobre blíster La refinación térmica del cobre tiene por finalidad producir dentro de un marco económico, ánodos que sean química y físicamente aptos para su refinación en las celdas electrolíticas, mediante este proceso se logra producir ánodos con una superficie más regular y al mismo tiempo se elimina parte de las impurezas. Este proceso se lleva acabo en dos etapas:
1. Etapa de oxidación, que se realiza insuflando aire al lecho fundido, donde las impurezas como el Zn, Pb, S, Sb, son volatilizados y impurezas como el Fe, Zn, Mg, Pb, Al, Mn, Ni, Co, Bi son oxidados y pasan a la escoria y el Cu queda saturado con Cu2O, pasando parte a la escoria. Las reacciones de oxidación son: 4Cu + O2 → 2Cu2O Cu2O + SiO2 → Cu2O.SiO2 5Cu2O + 2As → As2O5 + 10Cu As2O5 + Cu2O → Cu2O. As2O5 5Cu2O + 2Sb → Sb2O5 + 10Cu Sb2O5 + Cu2O → Cu2O. Sb2O5 2Cu2S + 3O2 → 2Cu2O + 2SO2
2. Etapa de reducción, por cuanto el baño fundido queda saturado con Cu2O, es necesario una etapa de reducción, para lo que se pueden emplear troncos verdes, que es un método tradicional también conocido como berlingado, petróleo vapor, amoniaco gaseoso, gas natural, gas propano, mezcla de carbón pulverizado con aire.
TECNOLGIAS DE REDUCCIÓN EN LA PIROREFINACION DEL COBRE

a) Reducción con troncos verdes, es un método tradicional para la reducción del cobre y consiste en sumergir troncos verdes en el cobre líquido, generando un proceso de destilación de la madera desprendiendo H2, CO e hidrocarburos que aceleran la reducción del cobre oxidado Cu2O + C → 2Cu + CO Cu2O + H2 → 2Cu + H2O Cu2O + CO → 2Cu + CO2 La desoxidación de cobre con troncos verdes es un proceso relativamente costoso, de difícil abastecimiento y manipuleo que ocasiona un deterioro prematuro del revestimiento refractario de los hornos.
b) Reducción con petróleo vapor, en este caso se emplea una mezcla de petróleo – vapor de agua, la que es constantemente inyectada debajo de la superficie del baño metálico a través de una lanza y se genera la reacción entre el vapor de agua y el petróleo para generar H2 y CO, que luego reaccionan con el Cu2O, mediante las reacciones: Cu2O + CO → 2Cu + CO2 Cu2O + H2 → 2Cu + H2O Cu2O + C → 2Cu + CO La eficiencia de la desoxidación dependerá considerablemente del área de contacto
c) Reducción con amoniaco, la ventaja de este proceso es que el gas no contiene impurezas como el S u otros productos de combustión que si contienen el petróleo y la madera. 3Cu2O + 2NH3 → 6Cu + N2 + 3H2O La velocidad de la reacción del amoniaco con el cobre fundido es muy rápida y completamente turbulenta, siendo la eficiencia de reducción H2 muy alta debido a su fuerte actividad termodinámica.
3.27. Tratamiento de gases y polvos El control de los contaminantes procedentes de las plantas metalúrgicas ha sido desde hace mucho tiempo un problema difícil y costoso y su importancia ha aumentado en los años recientes. Se ha dado mayor importancia a la reducción de la contaminación al mínimo y se están estableciendo en muchos países normas de control más rígidas sobre los contaminantes permitidos en el aire y en el agua mediante estrictas medidas de reglamentación. La advertencia de este problema en la industria metalúrgica no es nueva, ya desde 1821, a principios de la Revolución Industrial, había preocupación acerca de las emisiones de S02 que lanzaban los hornos de las fundiciones de cobre de Swansea, Gales, en donde, dichas emisiones dañaban las cosechas de las áreas circunvecinas y tenían que controlarse. Para 1860 las fundiciones ya tenían cierto control de su S02 gaseoso, del que trataban una parte para producir ácido Sulfúrico (con aproximadamente 40% del azufre total que iba a las fundiciones, recuperado como tal) hasta el grado que permitía el consumo comercial del ácido producido, y el S02 restante lo expulsaban por chimeneas elevadas por disiparlo en las capas altas de la atmósfera.

En general, el gas de proceso por tratarse contiene partículas sólidas en suspensión, las cuales pueden separarse por distintos tipos de colectores de polvo, y compuestos químicos gaseosos de los que el anhídrido sulfuroso es, con ventaja, el más común, aunque hay otros gases como el cloro, el ácido clorhídrico gaseoso y los fluoruros que son también subproductos de algunas operaciones metalúrgicas. Estos productos gaseosos se eliminan por métodos químicos de combinación. El agua que ha de purificarse contiene también cantidades considerables de materia sólida la cual puede separarse por varios pasos de clarificación, y también puede contener compuestos contaminantes de metales pesados en disolución, los cuales pueden removerse por precipitación. Además, puede tener un alto contenido de ácido que puede neutralizarse agregando un material básico para bajar el pH a niveles aceptables. Existen varias razones importantes para depurar los gases y el agua procedentes de los procesos metalúrgicos; las más importantes son las siguientes:
1. Recuperar partículas en suspensión portadoras de valores para regresarlas a la planta y reprocesarlas. Una fundición de cobre en la que se procesen concentrados de flotación de menos de 200 mallas puede perder hasta 8% de la materia prima en forma de polvo de chimenea, y si no se recupera y recicla este tonelaje, la economía del proceso en conjunto se verá afectada seriamente.
2. La contaminación ambiental, tanto desde el punto de vista de los trabajadores expuestos a la emisión de gases tóxicos en concentraciones peligrosas (en la norma estadounidense EPA, 5.0 ppm para (os trabajadores expuestos a S02) como por la expulsión de gases tales como SO2 que ocasionan daños a la agricultura combinándose con el vapor de agua de la atmósfera para formar anhídrido sulfúrico y ácido sulfúrico que queman las cosechas. La contaminación de las fuentes de abastecimiento de agua por compuestos de metales pesados y alto contenido de ácido se encuentra también bajo estricta reglamentación gubernamental por razones de salud. (El Ministerio Canadiense del Ambiente establece 1 ppm como concentración máxima segura para estos metales.)
3. Separar subproductos gaseosos portadores de metales, tales como el S02, que se pueden usar como materia prima para producir ácido sulfúrico o de azufre elemental, que son productos comerciales.
4. Depurar productos gaseosos de alto poder calorífico que contienen grandes porcentajes de CO combustible, que habiendo sido liberados de (os sólidos suspendi-dos, pueden llevarse a quemadores, para usarse como gas combustible en cualquier parte de la planta.
5. Muchos gases metalúrgicos son producto de las operaciones de hornos a alta temperatura, y salen del proceso con un alto contenido de calor sensible. Este calor puede recuperarse en dispositivos tales como calderas recuperadoras y sistemas de recuperación de calor, en general, antes de dejar escapar a la atmósfera los gases ya enfriados.
6. Clarificar o depurar el agua de la planta, separando la materia sólida, para que se pueda reciclar como agua limpia para proceso o para descargarla a corrientes cercanas sin peligro de contaminación.

COLECTORES DE POLVO - SEPARACIÓN DE SOLIDOS SUSPENDIDOS EN GASES
La recolección de polvo se efectúa por diversos métodos, y se debe tomar en cuenta en la selección del método lo siguiente: 1) el tamaño de partículas por remover, 2) la temperatura del gas, 3) el volumen de gas, 4) la velocidad del gas, 5) si el colector ha de usarse solo o formando parte de una serie, y 6). si ha de recuperarse o no el calor sensible de los gases. Los tamaños y tipos de partículas se clasifican en cuatro categorías (1 miera = 1/1000,000 metro = 1/25,400 pulgada = 3.937 x 10"5 pulgada):
Vapores 0.05 a 1.0 micras; partículas sólidas finas formadas por la condensación de vapores metálicos.
Polvo 1.Oa 50 micras; partículas sólidas, pequeñas, formadas por la fractura de partículas de mayor tamaño.
Niebla 0.5 a 10 micras; pequeñísimas gotas de líquido generadas por condensación.
Humo 0.05 a 1.0 micra; partículas sólidas finas resultantes de la combustión incompleta de materiales orgánicos.
Los contenidos de polvo se dan en granos por pie cúbico o en gramos por metro cúbico (1.0 grano por pie cúbico = 2.3 gramos por m3; 1 grano = 0,065 gramo). Hay cuatro designaciones del contenido de polvo:
a) Ligera 1/2 a 2 granos por píe cúbico (1.15 a 4.6 gramos por m3) b) Mediana 2 a 3 granos por pie cúbico (4.6 a 6.9 gramos por m3) c) Moderada 3 a 5 granos por pie cúbico (6.9 a 11.5 gramos por m3) d) Pesada 5 o más granos por pie cúbico (11.5 o más gramos por m3)
Existen ocho tipos de colectores de polvo en uso común, cada uno de los cuales tiene su eficiencia de colección óptima dentro de una cierta gama de lámanos de partícula. Estos tipos de colectores y sus intervalos de eficiencia son los siguientes:
Nro Tipo Colector Eficiencia
1 Cámara de sedimentación 90% mayor a 50 micras
2 Ciclón 70 micras, 20%; 100 micras, 92% 3 Multiciclón o ciclón múltiple 3 micras, 20%; 70 micras 99%
4 Filtro de bolsas 0.5 a 100 micras, 99%
5 Torre de rocío 10 micras, 88%; 90 micras, 98%
6 Lavador de Venturi 0.2 micras, 30%; 5 micras, 99% 7 Precipitador electrostático 0.1 micras, 82%; 2 micras 99%
8 Lavador húmedo 0.3 micras, 20%; 9 micras, 99% Fuente: GILL, G.D.; "Nom Ferrous Extractive Metallurgy"; A Wily Interscience Publication; Toronto, Canadá -1980

Los dos siguientes diagramas muestran el equipo y la eficiencia según el tamaño de partícula
CÁMARA DE SEDIMENTACIÓN (cámara de sedimentación por gravedad, cámara de ex-pansión, ducto de balón). Este es uno de los métodos más antiguos y simples para recolectar polvo y se emplea para separar partículas gruesas mayores de 50 micras. La cámara de sedimentación funciona con base en el principio de un pequeño ducto que conduce gas a alta velocidad y que descarga a una cámara de mucho mayores dimen-siones, cuya área de sección transversal aumenta considerablemente. La velocidad del gas disminuye, debido al mayor volumen de la cámara, y por esta razón ya no es capaz de arrastrar las partículas de polvo más grandes que lleva la corriente de gas por lo que caen. El gas limpiado sale por un pequeño ducto de descarga. La temperatura del gas no es importante, ya que la cámara es de acero y puede revestirse con ladrillo refractario resistente al calor. Es de construcción simple sin partes mecánicas que se desgasten rápidamente y puede hacerse de dimensiones suficientes para pasar por ella un volumen grande de gases. La cámara de sedimentación puede ser de diseño vertical u horizontal, y como el gas se hace pasar seco por la unidad, no hay enfriamiento ni pérdida de calor sensible. La cámara de sedimentación es una unidad primaria que sólo puede usarse si las partículas de polvo son todas grandes, o como la primera unidad de una serie, si se tiene una variedad de tamaños de partícula para ser recuperado por medio de varios colectores. La eficiencia de una cámara de sedimentación horizontal puede obtenerse por la ecuación
Eficiencia por peso = (𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑡𝑒𝑟𝑚𝑖𝑛𝑎𝑙 𝑑𝑒 𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎) (𝑙𝑜𝑛𝑔𝑖𝑡𝑢𝑑 𝑑𝑒 𝑙𝑎 𝑐á𝑚𝑎𝑟𝑎)
(𝑎𝑙𝑡𝑢𝑟𝑎 𝑑𝑒 𝑙𝑎 𝑐á𝑚𝑎𝑟𝑎)(𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒𝑙 𝑔𝑎𝑠)
O sea Nw = (𝑈𝑡)(𝐿)
(𝐻)(𝑣𝑔) ; 𝑈𝑡 en pies/s, H en pies, L en pies y 𝑣𝑔 en pies/s.
La ecuación para calcular el tamaño mínimo teórico de partícula que puede recolectarse con eficiencia al 100% en una cámara horizontal es:
Diámetro mínimo de partícula= [18(𝑣𝑖𝑠𝑐𝑜𝑠𝑖𝑑𝑎𝑑 𝑑𝑒𝑙 𝑔𝑎𝑠)(𝑎𝑙𝑡𝑢𝑟𝑎 𝑑𝑒 𝑐á𝑚𝑎𝑟𝑎)(𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒 𝑔𝑎𝑠 𝑒𝑛 𝑐á𝑚𝑎𝑟𝑎)
(𝑙𝑜𝑛𝑔𝑖𝑡𝑢𝑑 𝑑𝑒 𝑐á𝑚𝑎𝑟𝑎)(𝑝𝑒𝑠𝑜 𝑒𝑠𝑝𝑒𝑐í𝑓𝑖𝑐𝑜 𝑑𝑒 𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎−𝑝𝑒𝑠𝑜 𝑒𝑠𝑝𝑒𝑐í𝑓𝑖𝑐𝑜 𝑑𝑒 𝑔𝑎𝑠)]1/2

O sea Dp mínimo = [18(𝜇𝑔)(𝐻)(𝑣𝑔)
(𝐿)(ϒ𝑝−ϒ𝑔)]1/2 ; Dp en pies, 𝑣𝑔en pies/s, ϒ𝑝en lb/pie3, ϒ𝑔 en lb/pie3
y H en pies.
Cámara de sedimentación
LOS CICLONES son también colectores sin partes movibles que se utilizan en seco y conservan el calor sensible de los gases calientes que pasan por ellos. El ciclón trabaja en forma óptima en el tratamiento de partículas de tamaño mediano, y tiene una eficiencia de sólo 20% de recuperación en partículas pequeñas de 7 mieras, aumentando ésta al 92% con partículas más grandes, del orden de 100 micras. El ciclón es un colector de bajo costo inicial y bajo costo de operación, y es de tamaño relativamente reducido; en la variedad de formas en que se fabrica es el tipo de colector de uso más frecuente, siendo los más eficientes los de cuerpo de diámetro pequeño (menor de 9 pulgadas, 22.5 cm). Estos aparatos funcionan bajo el principio de que al entrar una corriente de gas a alta velocidad cerca de la sección cilíndrica superior del ciclón e incidir tangencialmente sobre la superficie curva, la fuerza centrifuga que resulta de la velocidad tangencial del gas lleva las a partículas de polvo hasta la pared del ciclón que al chocar con ésta se deslizan hacia abajo por su propio peso y salen por el fondo cónico del ciclón. El gas depurado escapa por una abertura de descarga que hay en la parte superior del colector. Sí se produce un desgaste excesivo del casco de acero del ciclón por el efecto de choque de las partículas finas de polvo que entran en la unidad a gran velocidad, tal desgaste puede controlarse mediante un revestimiento formado por insertos de porcelana aplicado en la sección cilíndrica de alimentación del ciclón. El ciclón puede usarse únicamente como unidad primaria, o como colector intermedio si se utiliza formando parte de una serie. La fuerza centrífuga que separa a las partículas de una masa en particular, se expresa mediante la ecuación:
Fuerza Centrífuga = (𝑚𝑎𝑠𝑎 𝑑𝑒 𝑙𝑎 𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎)(𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑡𝑎𝑛𝑔𝑒𝑛𝑐𝑖𝑎𝑙 𝑑𝑒 𝑙𝑎 𝑝𝑎𝑟𝑡í𝑐𝑢𝑙𝑎)2
(𝑟𝑎𝑑𝑖𝑜 𝑑𝑒𝑙 𝑐𝑖𝑐𝑙ó𝑛)(𝑎𝑐𝑒𝑙𝑒𝑟𝑎𝑐𝑖ó𝑛 𝑔𝑟𝑎𝑣𝑖𝑡𝑎𝑐𝑖𝑜𝑛𝑎𝑙)
O sea, Fc = (𝑚𝑝)(𝑉𝑝𝑡)2
𝑔𝑟𝑐 con Fc en lb, 𝑉𝑝𝑡 en pies/s, g en pies/s2 y rc en pies.
De acuerdo con la ecuación anterior, la magnitud de la fuerza centrífuga que puede ser ejercida sobre una partícula dada en un cierto tamaño de ciclón es función de la masa de la partícula y de la velocidad tangencial, dependiendo la velocidad tangencial de la velocidad del gas a la entrada del ciclón. Teóricamente, la eficiencia de separación de partículas para un ciclón dado aumenta al aumentar

el radio de la fuerza centrífuga a la fuerza de arrastre:
Radio = (𝑝)(𝑣𝑝𝑡)(𝐷𝑝)2
(𝜇𝑔)(𝐷𝑐)
Dp = diámetro de la particular en pies 𝑝 = densidad de la partícula en lb/pie3
𝑣𝑝𝑡 = velocidad tangencial de la partícula en pies/s
𝜇𝑔 = viscosidad del gas en lb-s/pie2
𝐷𝑐 = diámetro del ciclón en pies
LOS CICLONES MÚLTIPLES son una forma de colectores ciclónicos que también se usan en seco, en los que se combinan varios ciclones de diámetro pequeño dentro de un solo casco de contención que trabajan en conjunto como una sola unidad. Estos aparatos aprovechan la mayor eficiencia de operación de los ciclones de diámetro pequeño, y como los ciclones pequeños toman sólo su parte de la carga de polvo y trabajan en paralelo, estos grupos de ciclones que forman los multiciclones pueden procesar volúmenes grandes de gases con carga de polvo importante con bastante buena eficiencia. Este equipo también opera mejor en la recolección y separación de partículas de tamaño mediano y tiene una eficiencia de sólo 20% con partículas de 3 mieras, pero de 99% con partículas de 70 micras.

LOS FILTROS DE BOLSA (Bag House) son colectores de tipo filtro y de los más antiguos y confiables, su uso todavía está muy generalizado. El gas que contiene los sólidos suspendidos se inyecta a cierta presión al interior de bolsas de tela, por las que pasa el gas hacia afuera del colector, dejando atrás los sólidos, el polvo o los polvillos que son retenidos por la tela. Después del primer instante de contacto, la acción filtrante la realiza en realidad la torta de polvo que se ha depositado sobre el filtro de tela. Las bolsas se usan en grandes grupos y se sacuden periódicamente, o bien se invierte momentáneamente la corriente de gases para desprender la capa de partículas que se ha acumulado en el interior de las bolsas. El polvo cae en una tolva situada en el fondo del filtro, y de allí se le extrae. La vida de las bolsas varia de seis meses a dos años, y el tamaño de las mismas de 5 a 20 pulgadas de diámetro (12.5 a 50 cm), y alcanza una longitud hasta de 44 pies (13.2 m). Cuando se rasga una bolsa hay que cambiarla inmediatamente, porque ocasiona fugas y deja de filtrar. El filtro de bolsas puede usarse en una amplia gama de tamaño de partículas, 0.05 a 100 micras, con eficiencia hasta del 99%, pero generalmente se le considera mejor como colector de partículas de los tamaños más pequeños y para gases con carga de polvo ligera a mediana, de otra manera, la instalación de este tipo de colector para procesar grandes volúmenes de gas con cargas de polvo grandes seria enormemente grande y poco práctico. La tela de la bolsa y lo cerrado del tejido se hacen corresponder a las condiciones de trabajo, las cuales son: 1.- tamaño de partícula, 2.- temperatura del gas, 3.- resistencia a la abrasión y 4.- resistencia al ataque de ácidos o álcalis. A medida que aumenta el espesor de la torta de polvo que se acumula sobre la tela, aumenta también la presión necesaria para forzar el gas a través de las bolsas. Esta caída de presión se expresa como la suma de dos resistencias: la resistencia debida a lo cerrado del tejido de la tela misma y la resistencia debida a la capa de polvo que se forma sobre la superficie de la tela. La caída de presión continúa aumentando y varía según:
1. la velocidad del gas a su paso por la tela 2. la carga de polvo, 3. el tamaño del polvo y 4. la tela que se emplee
Y se controla mediante la separación periódica de la torta de polvo que se acumula en las bolsas. ∆P = (vs)[Kf + (Kc)(Wc)] ∆P = caída de presión en lb/pie2 Kf = coeficiente de resistencia de la tela sin dimensión Kc = coeficiente de resistencia de la torta sin dimensión Wc = peso de la torta por áreas en lb/pie2 El filtro de bolsas puede usarse solamente como una unidad para condiciones en las que las partículas sean pequeñas y las cargas de polvo o los volúmenes de gas no sean excesivos, o bien puede usarse como colector final de una serie para separar las últimas partículas finas
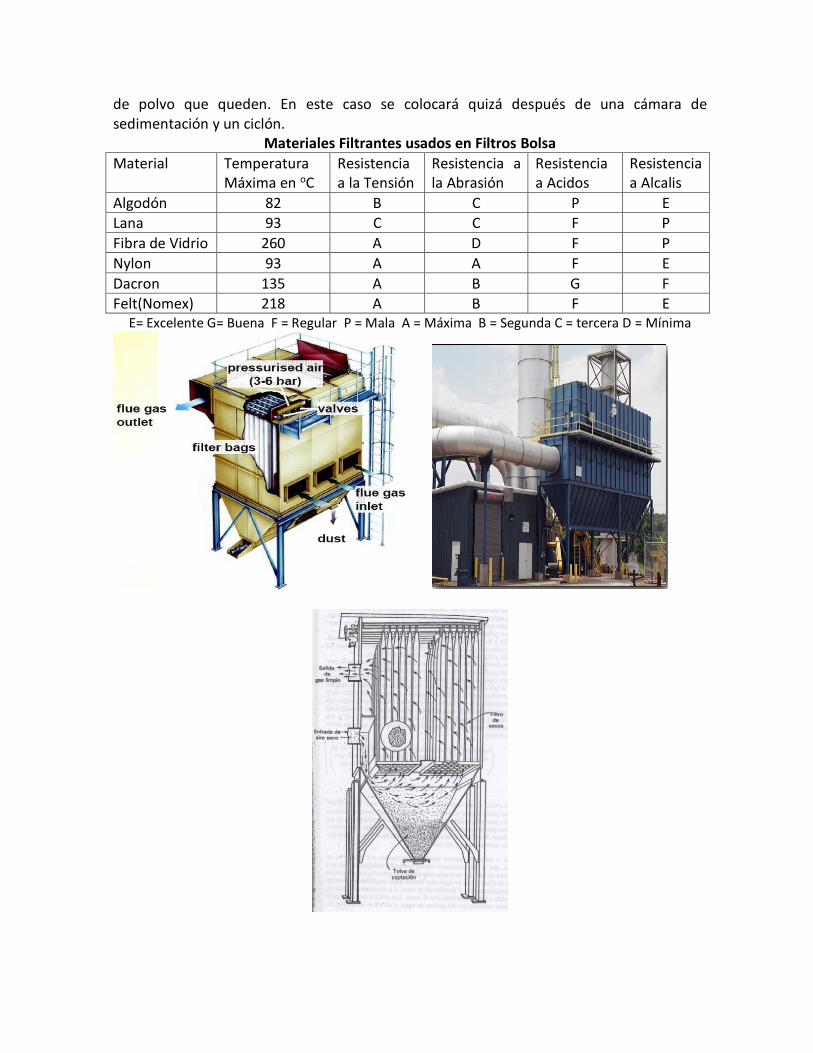
de polvo que queden. En este caso se colocará quizá después de una cámara de sedimentación y un ciclón.
Materiales Filtrantes usados en Filtros Bolsa
Material Temperatura Máxima en oC
Resistencia a la Tensión
Resistencia a la Abrasión
Resistencia a Acidos
Resistencia a Alcalis
Algodón 82 B C P E Lana 93 C C F P
Fibra de Vidrio 260 A D F P
Nylon 93 A A F E
Dacron 135 A B G F Felt(Nomex) 218 A B F E
E= Excelente G= Buena F = Regular P = Mala A = Máxima B = Segunda C = tercera D = Mínima

LAS TORRES DE ROCÍO, recolectan partículas por medio de pequeñas gotitas de agua, que al hacer contacto con las partículas sólidas que van en el gas, las moja y las separa de éste. En su forma más simple, la torre de rocío tiene una corriente descendente de finas gotas de agua que lanzan unas boquillas de atomización; el rocío cubre completamente el interior de la torre. La corriente de gases sucios por depurar asciende a contracorriente con el rocío. Los sólidos son acarreados hasta el fondo de la torre de rocío por las gotitas de agua, y de allí se les extrae, decanta y recupera. Existen boquillas atomizadoras para una variedad de formas, para llenar el in tenor de la torre y dar la finura requerida de las gotitas de agua, y aportar el volumen de agua requerido. Estas boquillas se hacen corresponder al tamaño de partícula del polvo, al volumen de gas y a la velocidad de éste en su paso por la torre para lograr la máxima eficiencia de depuración. Pueden separarse en partículas de todos los tamaños, aunque las partículas menores del miera son más difíciles de separar, por lo que se considera a la torre de rocío como una unidad primaria 0 secundaria, teniendo eficiencias del 88% en material de 10 micras y hasta del 98% en partículas de 90 micras. La torre de rocío es un colector relativamente simple y de bajo costo con el que se pueden manejar grandes volúmenes de gas con todos los grados de carga de polvo, pero se usa con mayor ventaja cuando las condiciones de depuración de gases no son extremas. Por ser un colector de tipo húmedo, el gas se enfría a su paso por la torre y se pierde su calor sensible.
EL LAVADOR DE VENTURI es un dispositivo depurador de gas de tipo húmedo en el que se atomizan el gas sucio y el agua para lavado en una corriente de gases en movimiento. La velocidad relativa entre el gas y las gotitas de agua es muy alta, y las velocidades del gas son del orden de 200 a 400 pies por segundo (3660 a 7320 m por minuto), y esta alta velocidad permite una gran capacidad de tratamiento. El lavador tiene una garganta de Venturi convergente — divergente en su diseño, y es en este lugar en donde se agregan las gotitas de agua para dar el máximo contacto de líquido-gas. La eficiencia de recolección es

proporcional a la caída de presión y aumenta con ésta. Por ello, los lavadores de Venturi de alta caída de presión, mediante ajustes de la caída, el gasto del rocío de agua y la velocidad de entrada del gas, son de eficiencia lo suficientemente alta para recolectar partículas hasta de tamaño inferior a 1 micra. Las gotitas de liquido que han hecho contacto con las partículas de polvo se acumulan sobre las paredes de la sección divergente inferior del lavador, abajo de la estricción de la garganta, y fluyen hacia abajo en flujo laminar hasta llegar al fondo del colector. De allí se les extrae, y las partículas sólidas se separan del líquido. El gas depurado sale por un conducto de descarga situado en la sección inferior de la unidad. El lavador de Venturi es un colector barato, de tipo simple que es muy eficiente para separar partículas pequeñas, + 99% para 5 micras, y que puede procesar grandes volúmenes de gases en unidades bastante pequeñas, debido a su rápido régimen de tratamiento. Se utiliza como unidad primaria única cuando todas las partículas por separar son pequeñas, o como unidad para tratamiento final para las partículas más pequeñas si forma parte de una serie de colectores. Como éste es también un colector húmedo, se enfría el gas caliente al hacer contacto con las gotitas de agua y se pierde su contenido de calor sensible.

LOS PRECIPITADORES ELECTROSTÁTICOS utilizan electricidad para separar las partículas mojadas o secas que lleva una corriente de gas. Si bien es un colector costoso, en general se le considera de lo mejor para la separación de partículas muy pequeñas, con una eficiencia de 82% para tamaños de 0.1 micra y de +99% para sólidos de 2 micras. El precipitador utiliza dos electrodos, uno de descarga de potencial negativo y un electrodo colector de potencial positivo. Un rectificador convierte la energía que se alimenta a estos electrodos a corriente directa y eleva la tensión a 80,000 volts. Existen precipitadores de varios diseños diferentes. Un tipo común tiene los dos electrodos como placas situadas a cierta proximidad unas de otras (tipo de placas), mientras que en otro tipo el electrodo de descarga es un alambre suspendido dentro de un tubo que es el electrodo colector (tipo de tubo). Debido al elevado potencial de operación del precipitador, se forma una corona de moléculas de gas ionizado en torno al electrodo de descarga negativo, y los iones de gas negativo y positivo son atraídos hacia la superficie del electrodo de polaridad contraria. Los iones de gas cargados negativamente tienen que recorrer una distancia mayor hasta el electrodo colector, y en su camino hacen contacto y pasan su carga negativa a la mayoría de las partículas de polvo que lleva la corriente de gas que está pasando por el precipitador. Estas partículas cargadas negativamente son también atraídas hacia el electrodo colector que tiene carga positiva, y forman sobre éste una capa de partículas de polvo descargadas. De manera semejante, las partículas de polvo que adoptan una carga positiva, recorren la distancia corta que hay al electrodo de descarga cargado negativamente y se juntan sobre él para formar una capa de partículas descargadas. En un precipitador de tipo seco, los sacudidores o vibradores desprenden periódicamente las capas acumuladas de partículas de polvo para que caigan a una tolva para su extracción. En un precipitador de tipo húmedo, los sólidos húmedos pueden desprenderse por lavado y descender con una película de agua por los electrodos. Por su alta eficiencia de recolección de partículas pequeñas y su capacidad para manejar grandes volúmenes de gas, el precipitador electrostático puede usarse como unidad única

cuando sólo tienen que separarse partículas, o bien puede usarse en la etapa final de una serie de varios colectores instalados para separar una variedad de tamaños de partícula. La eficiencia teórica de un precipitador electrostático la da la expresión
𝜂 = 1 – e-(A/Q)(w) 𝜂 = Eficiencia del precitador A = Area colectora en pie2 Q = Gasto de gases que pasa por el precipitador en pie3/s w = velocidad de migración de las partículas hacia el electrodo pie/s Las eficiencias más altas ocurren para un gasto bajo de gases (Q), alta velocidad de migración (w) y área de recolección grande (A). Se pueden obtener eficiencias más altas si se aumenta el área de recolección, aunque para aumentar la eficiencia del 90 al 99% se requiere duplicar dicha área, y del 90 al 99.9%, se necesita triplicar el área. Al aumentar el gasto de gases se reduce la eficiencia, y aun un pequeño incremento del gasto conduce a un gran incremento del número de partículas que escapan del colector. Un descenso del 99% de eficiencia al 97% triplica las emisiones, y son estas emisiones las que constituyen la variable importante en todas las operaciones de purificación de gases.

LAVADORES HÚMEDOS, hay muchos tipos de lavadores húmedos que trabajan bajo el principio de sumergir el gas sucio directamente en un baño de agua, en el cual, por el contacto que tiene lugar entre el agua y las partículas en suspensión en el gas, se se-paran éstas de la corriente de gas y sale de la unidad del gas depurado. Las capacidades son algo limitadas por el tamaño del baño de agua que se requeriría para el tratamiento de cualquier volumen grande de gas. Este tipo de lavador es muy útil como unidad única para aplicaciones de lavado de gases en escala pequeña a mediana. La eficiencia baja hasta 30% tratándose de tamaños inferiores a una micra (0.2 miera) pero aumenta hasta 99% con tamaños de partículas mayores de 9 micras.
3.28. Producción de ácido sulfúrico El anhídrido sulfuroso es el compuesto que más comúnmente se encuentra en los gases producto de los tratamientos metalúrgicos, debido al hecho de que muchos de los metales no ferrosos (cobre, plomo, zinc, níquel, cobalto) se presentan en forma de sulfuros, y su procesamiento consiste en tostarlos, fundirlos y convertirlos; todas estas operaciones son oxidantes y producen grandes cantidades de gas S02. Como consecuencia, el SO2 se ha considerado desde hace mucho tiempo como un contaminante indeseable en la atmósfera, por el daño que causa al aparato respiratorio humano y a la agricultura, y como se está legislando en muchos países los reglamentos son cada día más estrictos para la limpieza del aire para controlar las emisiones de las fundiciones, tiene gran importancia la eliminación del SO2 de los gases de los procesos metalúrgicos. Por ejemplo, los controles legislativos sobre el S02, elevados recientemente al carácter de Actas por la Agencia de Protección del Ambiente de Estados Unidos (U.S. Environmental Protection Agency) han establecido los límites siguientes: No más del 10% del azufre que entra en una fundición puede ser emitido a la atmósfera. Los trabajadores no se deben exponer a una atmósfera que contenga más de 5.0 ppm de S02.

El aire emitido al ambiente a nivel de tierra no debe exceder de 0.03 ppm de S02 en promedio anual. El contenido medio de S02 para un día cualquiera no debe exceder de 0.14 ppm. En Bélgica, la legislación establece que la descarga de S02 no debe exceder del 0.1% por volumen del gas descargado, mientras que en Suecia se han dispuesto lineamientos provisionales a los siguientes límites de S02: promedio de una hora, 0.25 ppm; promedio de una semana, 0.10 ppm; promedio de un mes, 0.02 ppm. Existen tres métodos de tratamiento para los gases que contienen S02, que sirven para remover el azufre. El primero, y con ventaja el de mayor aplicación, consiste en convertir el S02 en ácido sulfúrico por el proceso de contacto. En el segundo, puede extraerse el azufre en forma de azufre elemental, y en el tercero, se combina el SO2 con óxido de calcio para formar sulfito de calcio y sulfato de calcio. El proceso de contacto para la fabricación de ácido sulfúrico, trabaja mejor y más económicamente con gases que contienen por lo menos 3.5% de S02, y de preferencia de 10 a 14% de S02. Sin embargo, se ha hecho trabajar, con la inclusión de ciertos adicionales y a un costo considerablemente mayor, con gases que contienen hasta 2.2% de S02. La concentración del ácido producido varía un poco de una planta a otra, y por lo general está en el intervalo de 98 a 99.6% de H2SO4, el cual se diluye luego al 93% de H2SO4, que es la concentración usual del producto comercial. El gas caliente de la fundición tiene que enfriarse y purificarse para evitar la contaminación durante el procesamiento, y es particularmente importante eliminar toda impureza, como el trióxido de arsénico, el cual se forma durante la tostación de los materiales areníferos. Los gases calientes del proceso metalúrgico se hacen pasar primero por una caldera recuperadora de calor para recuperar el calor sensible, y luego a un precipitador electrostático seco para separar la materia en suspensión; Si el contenido de polvo es muy alto, puede instalarse un colector ciclónico antes del precipitador electrostático para dividir la separación de los sólidos en dos etapas. El gas depurado se hace pasar luego por una torre lavadora con agua para lavarlo, seguido por un enfriador de refrigeración para bajar la temperatura por debajo del punto de rocio. En un recipitador electrostático húmedo se separa la neblina formada en el ciclo de enfriamiento y el polvo que haya quedado después del tratamiento inicial de separación de polvo. Al enfriar los gases de tostación se forma una neblina de ácido sulfúrico que contiene al As203 que esté presente, y la eliminación de está neblina en el precipitador húmedo evita también cualquier problema de pudiera causar el As2O3 en los pasos siguientes. En particular, elimina el problema de envenenamiento del catalizador de pentóxido de vanadio durante la operación de conversión catalítica. El gas limpio y frío pasa en seguida a una torre en la que se seca rodándolo con ácido sulfúrico al 93% y luego entra al sistema del proceso de contacto. Este sistema está formado por un convertidor y un intercambiador de calor, seguidos por un enfriador de gas y una torre de absorción. El convertidor es un casco de acero cilíndrico revestido de ladrillo, que por lo general contiene cuatro camas de catalizador de pentóxido de vanadio. El anhídrido sulfuroso se oxida catalíticamente a trióxido por oxígeno atmosférico,

2S02 + 02 →2S02 y la oxidación ocurre sobre la superficie del catalizador a la presión atmosférica. Se logra una conversión de 95 a 98% en un tiempo de contacto es de 2 a 4 segundos. La reacción es exotérmica y se desarrolla entre 790 a 1100°F (420 a 600°C) para obtener un máximo régimen de conversión. El contenido de calor de los gases que salen de la cámara de contacto puede aprovecharse por medio de un intercambiador para calentar el gas S02 frío a la temperatura inicial necesaria; si no se emplea un intercambiador de calor, dicho calentamiento inicial puede hacerse por medio de un precalentador con quemadores de combustible. El gas S03, enfriado a casi 400°F (200°C), se envía a una torre de absorción en la que el S03 es absorbido circulando H2S04 al 98.5%. El S03 se combina con el agua del ácido para producir H2S04 del 99.6%: S03+H20 → H2S04 Esta concentración del 98.5% del H2S04 da la óptima eficiencia de absorción. El hierro fundido y los aceros aleados no son atacados por estas altas concentraciones de ácido, y esto simplifica notablemente la elección de materiales para el equipo que se necesita para el bombeo y la recirculación. Una parte del ácido circulante se remueve en forma continua del sistema de absorción y diluida con agua al 93% de H2S04, que es la concentración usual del mercado. Los gases de cola que salen de la torre de absorción contienen neblinas de ácido sulfúrico e incluso pueden contener hasta 0.1 a 0.2% de S02. El lavado de estos gases con solución de cal precipita el azufre como sulfito de calcio y sulfato de calcio, y en un precipitador electrostático se remueven ambos productos y cualquier neblina de H2S04 antes de descargar finalmente el gas a la atmósfera por una chimenea. Este gas de liberación final contiene menos de 0.01% de SO2, y se le ha removido más del 99% del S02 que contenía el gas alimentado a la planta de ácido. Los siguientes diagramas muestran la fabricación de ácido sulfúrico por contacto.
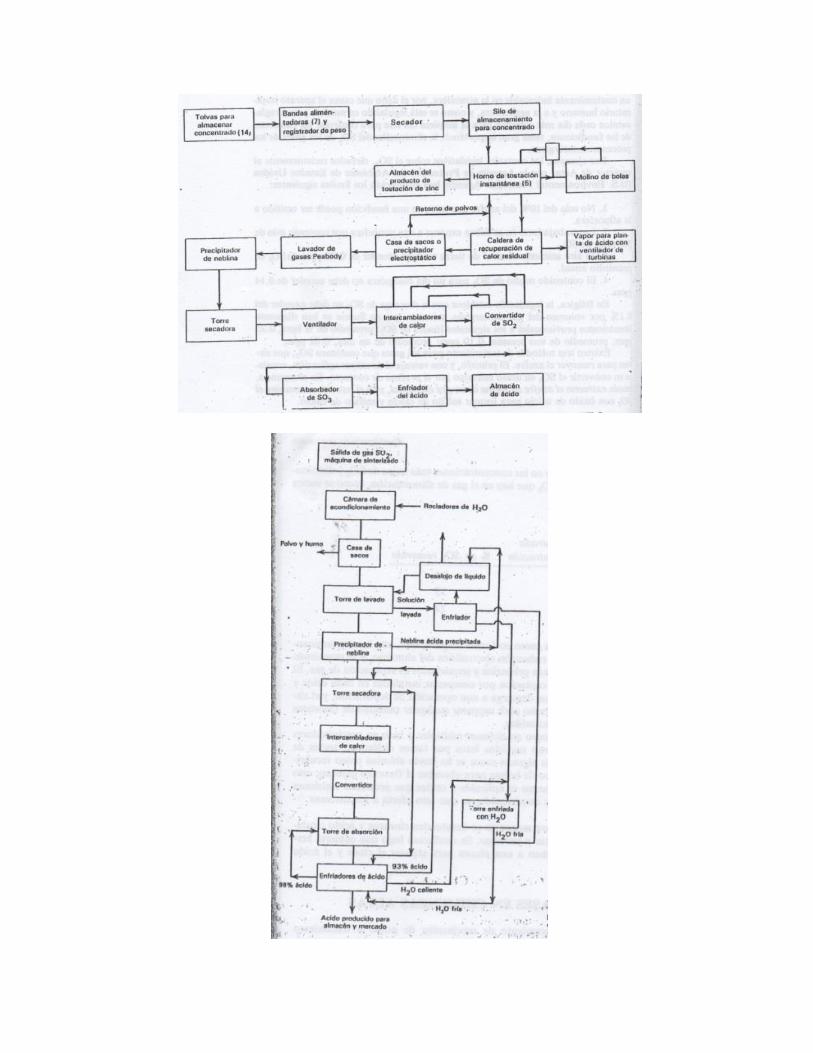

La manufactura de azufre elemental es un segundo proceso que puede usarse para tratar gases de fundición de alto contenido de S02, particularmente los que contienen más del 10% de S02. El azufre elemental tiene buenas características para el almacenaje prolongado y es adaptable para otros usos diferentes al del ácido sulfúrico. En este proceso se enfría primero el gas, se depura y se seca en forma similar a la que se aplica para el proceso de contacto en la fabricación del ácido. Luego se le reduce con carbón (gas natural o carbón mineral) o con hidrógeno a temperaturas de 9320F (500°C): S02 + C → C02 + S(g) S02+2H2 → 2H2O + S(g) Continuamente se hace pasar una mezcla de S02 y aire a través de un horno, y los gases resultantes se condensan a 340°F (170°C) para dar azufre elemental líquido, el cual se saca mediante piquera del condensador y se vacía en bloques. El gas que sale de este primer condensador contiene todavía algo de SO2, por lo que se recalienta a 465°F (240°C) y se le hace reaccionar con ácido sulfhídrico en una reducción de una o dos etapas para dar gas de azufre, 2H2S + S02 → 2H20 + 3/2S2(g) Gas que se condensa de nuevo al estado líquido, se extrae y se moldea. El gas de cola contiene alrededor del 1 % de S02 y se libera entonces por una chimenea a la atmósfera. El lavado húmedo por medio de óxido de calcio fino o caliza fina, de gases que contienen pequeñas cantidades de S02, se lleva acabo cuando la concentración de S02 es demasiado baja para efectuar un tratamiento para producción de H2S04 o de azufre elemental, y también para limpiar aún más el gas de cola limpio procedente de las plantas de contacto para ácido antes de su descarga final a la atmósfera. Se emplean torres de rocío y lavadores de Venturi con pulpas de sólidos y agua de CaO o de CaC03 finamente molidos (—200 a —325 mallas). Las reacciones que ocurren con el S02 en el gas son: CaO + S02 → CaS03
y en cierto grado ocurre la oxidación del sulfito: CaS03 +1/202 → CaS04 Los sólidos se remueven en tanques de sedimentación, lavadores de Venturi y centrífugas, para ser descargados a un sitio de desechos, mientras que el gas se descarga por una chimenea. El proceso es más eficiente en las concentraciones más bajas de S02 y disminuye al aumentar la cantidad de S02 que hay en el gas de alimentación, como se indica a continuación: % de S02 en gas de entrada % de SO2 removido 0.3 a 0.6 85 1.0 75 1.5 72
3.29. Electrorefinación del cobre – Celdas La metalurgia extractiva, tiene tres campos básicos: Pirometalurgia, hidrometalurgia y electrometalurgia. La electrometalurgia se ocupa de la obtención de los metales a partir de

sus iones mediante la aplicación de le energía eléctrica. Ejemplos se tiene la producción de Al a partir de sales fundidas, la producción de Cu por electrorefinación y electrodeposición, la producción de Zn por electrodeposición, etc. Las reacciones electroquímicas que ocurren dentro de las celdas son de oxidación y reducción y ocurren en sitios distintos y a una distancia definida a diferencia de las reacciones químicas en las que la oxidación y reducción ocurren en un mismo sitio, un ejemplo es la disolución del Zn en HCl. Zn + HCl → ZnCl + H2↑ Zno → Zn+2 + 2e- Oxidación (ánodo) 2H+ + 2e- → H2 Reducción (cátodo) Zn + 2H+ → Zn+2 + H2 La reacción total de la celda es la suma de la reacción anódica más la reacción catódica, donde e- representa el movimiento de los electrones a través del metal al sitio de reducción. Para un mejor entendimiento de las reacciones electroquímicas, es importante revisar algunos factores y nomenclaturas usadas en la electrometalurgia.
Las reacciones involucran especies cargadas o iones que se dividen en: a) Cationes, con carga positiva, ejemplo: Cu+2, H3O+, H+ b) Aniones, con carga negativa, ejemplo: Cl-, OH-, CN-
Los iones se forman en un medio apropiado o solvente para la disolución de una sal, siendo el agua el solvente más usado para los procesos electrometalúrgicos. Se realizan las modificaciones más convenientes para ajustar el pH y/o el tipo de anión para lograr la solubilidad conveniente.
Ciertos sistemas reaccionan con el agua y por tanto requieren un electrolito no acuoso, en este caso se tienen dos posibilidades: usar soluciones acuosas o sales fundidas, en ambos casos es posible llevar acabo procesos electrometalúrgicos bajo condiciones apropiadas.
Oxidación, es del término usado para describir una reacción donde ocurre pérdida de electrones, ejemplos 2Cl- → Cl2 + 2e- Zn → Zn+2 + 2e- Fe+2 →Fe+3 + e-
Reducción, es el término usado para describir una reacción en el cual ocurre ganancia de electrones, ejemplos Cl2 + 2e- → 2Cl- Cu+2 +2e- → Cu [Cd(CN)4]-2 + 2e-2 → Cd + 4CN-
Reacción de media celda, cuando se escriben las reacciones de oxidación y reducción por separado se denominan reacciones de media celda. Para escribir la reacción total de la celda, es necesario combinar las dos reacciones de media celda. Ejemplo: Zn → Zn+2 + 2e- reacción de oxidación de media celda Cu+2 + 2e- → Cu reacción de reducción de media celda Zn + Cu+2 → Zn+2 + Cu reacción total de la celda

Se debe recalcar que el número de electrones involucrados en la reacción de oxidación es siempre igual al número de la reacción de reducción y que los cationes migran hacia el cátodo y los aniones al ánodo. Condiciones esenciales para una reacción electroquímica, Entendido la naturaleza de la reacción electroquímica, es fácil visualizar las condiciones esenciales. Como existe un flujo de electrones o carga durante la ocurrencia de la reacciones, es obvio, que se debe mantener un circuito eléctrico completo, si el circuito fuera interrumpido, pararía el flujo de corriente, lo que significa que las reacciones de oxidación y reducción cesarían. Este circuito está compuesto por cuatro partes principales y deben estar presentes si se quiere mantener la reacción.
1. Sitio anódico, sitio de oxidación o pérdida de electrones 2. Sitio catódico, sitio de reducción o ganancia de electrones 3. Conductor electrolítico, medio de contacto entre los electrodos (ánodo y cátodo) a
través del cual fluyen los iones, se denomina conductor iónico 4. Conductor electrónico, conecta el ánodo con el cátodo y permite el flujo de
electrones con algún grado de facilidad, la mayoría de los metales como el cobre y el aluminio, son ejemplos de conductor electrónico. El gráfico siguiente muestra una representación esquemática de un circuito electroquímico
Conductores eléctricos, La naturaleza y tipo de materiales usados en las reacciones electrometalúrgicas, pueden tener un gran efecto sobre el resultado obtenido, por lo tanto es necesario tener familiaridad con los muchos factores que pueden influir en la conducción de la electricidad a través de una celda electroquímica.

La conductividad eléctrica es el movimiento de una carga eléctrica y la habilidad de los diferentes materiales para permitir la transferencia de la carga eléctrica está determinada por la movilidad de la carga transportando electrones o iones dentro del material. Se tiene cuatro tipos de conductores.
1. Conductores de primera clase (metálicos), son algunas sustancias o sólidos que tienen la habilidad de transportar cargas eléctricas y presentan conductancias específicas altas, en cada tipo se tiene que la transferencia de carga o electrones se realiza sin transferencia sustancial de masa, ejemplos son los metales, grafito, óxidos (PbO2, MnO2), metales en transición, algunos carburos y nitruros. En esos materiales, al aumentar la temperatura, disminuye su capacidad de transmisión de corriente por que se produce una excitación de los electrones en el átomo.
2. Conductores de segunda clase, tienen una conductancia electrolítica o conductancia iónica que transporta iones, por consiguiente existe una transferencia de masa asociada con la conductividad, en estos materiales, al aumentar la temperatura aumenta la conductancia por una mayor movilidad de los iones, ejemplos son las soluciones acuosas de sales, sales iónicas, etc.
3. Conductores de tercera clase o conductores mixtos (semiconductores), presentan una conductancia electrónica e iónica, generalmente es la iónica la que prevalece, esta conductancia es baja, pero aumenta al aumentar la temperatura. Ejemplos: La mayoría de los óxidos metálicos, ZnO, NiO, etc, algunos metales como el Si, Ge, etc.
4. Aislantes, la conductancia es sumamente difícil, ejemplos: mica, cera, SiO2, etc. UNIDADES – DEFINICIONES Y TERMINOS
Ley de Ohm : E = IR E, es la diferencia de potencial que indica la fuerza de empuje o manejo para la reacción electroquímica, se expresa en voltios I, es la intensidad de corriente expresada en amperios e indica el flujo de electrones R, es la resistencia total del sistema al flujo de la corriente, se expresa en ohmios (Ω)
Potencia, es el producto de E x R x T (en segundos) y representa el trabajo requerido para una reacción, se expresa en watt
Energía, se expresa en voltios por segundo, voltios por coulomb o kw-h. En términos económicos es el parámetro más usado para comparar los costos relativos del proceso electrolítico para la producción del metal, se expresa en costo de energía/lb o kg de metal
Cantidad de electricidad, se expresasen coulombios igual a amperios por segundo Densidad de corriente, expresa la cantidad de corriente que pasa total que pasa por
una unidad de área, se expresa en A/m2, A/ft2 Eficiencia de corriente, se define como el % obtenido, cuando la cantidad real de
metal depositado es dividido por la cantidad de metal teórico predecido por la ley de Faraday
Eficiencia (𝜂) = 𝑃𝑒𝑠𝑜 𝑟𝑒𝑎𝑙 𝑑𝑒𝑝𝑜𝑠𝑖𝑡𝑎𝑑𝑜
𝑃𝑒𝑠𝑜 𝑡𝑒ó𝑟𝑖𝑐𝑜 (𝐿𝑒𝑦 𝑑𝑒 𝐹𝑎𝑟𝑎𝑑𝑎𝑦) x 100

Esta relación indica la cantidad de corriente que realmente es usado para la deposición del metal
Corriente directa o continua, la corriente continua es usado para todo proceso de electrólisis, siendo las fuentes baterías o rectificadores de corriente alterna a corriente continua
Culombímetros, son celdas electrolíticas especiales usados para medir el número de coulombios que fluyen a través de un sistema, se usa mucho para cálculos de eficiencia de corriente.
LIMITACIONES DE LOS PROCESOS ELECTROMETALÚRGICOS 1. No permiten variaciones amplias como en el caso de las reacciones químicas 2. El proceso debe ser lo más simple posible 3. Operan satisfactoriamente bajo condiciones constantes 4. Las materias primas deben ser tan puras como las limitaciones económicas lo
permitan 5. La alimentación y composición de las materias primas es constante 6. El suministro de la energía eléctrica debe ser barato y permanente
DIVISION DE LA ELECTROMETALURGIA
1. Electrólisis acuosa a) Electrodeposición (Electrowining), abarca la extracción de un metal a partir de
una solución, la que generalmente es obtenida por lixiviación de un mineral que contiene el metal de interés. El metal es reducido y depositado en el cátodo y sobre el ánodo se produce oxígeno. Este proceso se conoce como el proceso del “ánodo insoluble”
b) Electrorefinación (Electrorefining), es similar a la electrodeposición el la mayoría de los detalles, con la diferencia de que el suministro de los iones metálicos proviene de un ánodo soluble
c) Electroplateado, es la electrodeposición de metales en capas finas sobre un metal denominado base, generalmente el espesor del metal depositado es de 0.2 mm o algo más. Ejem. Cobreado, niquelado, cromado, dorado.
2. Electrólisis de sales fundidas, cuando loe metales no pueden ser electrolizados a partir de soluciones acuosas debido a su alta reactividad con el agua y su posición relativa en la serie electromotriz, entonces, estos metales son reducidos a partir de otros medios, como las sales fundidas que pueden ser floruros, cloruros, que sirven como electrolitos para producir metales como el Al, Mg, etc.
3. Producción de energía, es inverso a los procesos de extracción de los metales, en éstos se aprovecha algunas reacciones químicas para producir energía eléctrica, ejemplos son las baterías, que pueden ser recargadas con sólo invertir el sentido de las reacciones.
4. Misceláneos a) Electroformado, es la reproducción de un artículo por electrodeposición de un
metal sobre una matriz y el metal es posteriormente retirado. La

electrodeposición puede alcanzar espesores de hasta un cuarto de pulgada, las matrices pueden ser metálicas o no metálicos.
b) Anodización, consiste en provocar la formación de una capa de óxido metálico sobre un metal, con el fin de mejorar su resistencia a la corrosión. El metal a ser anodizado constituye el ánodo en un electrolito adecuado con desprendimiento de oxígeno y siendo la reacción principal la formación de la capa de óxido metálico. Es un proceso muy usado en el acabado de metales como el aluminio
c) Electroplateado sin corriente, se consigue un recubrimiento metálico sobre un metal por reacción química sin aplicar energía eléctrica, la fuente es un agente reductor químico en la solución en la el metal está en forma iónica
d) Depósito por inmersión, es un proceso en el cual un metal de mayor potencial (más noble) que está al estado iónico en la solución se deposita sobre un metal de menor potencial (menos noble). Este último se oxida y pasa a la solución. Ejm. Fe → Fe++ + 2e- Cu++ + 2e- → Cu
CELDAS ELECTROQUIMICAS, existen dos tipos de celdas electroquímicas: Celdas galvánicas y celdas electrolíticas que tienen cuatro componentes esenciales un ánodo, un cátodo, un conductor electrónico y un conductor electrolítico. Celdas Galvánicas, o voltaicas, son usadas para generar energía eléctrica o flujo de electrones por reacción química espontánea. Estas celdas convierten la energía química en energía eléctrica, ejemplos son las baterías secas y las baterías de plomo. En estas celdas el ánodo es el polo negativo y es donde ocurre la reacción de oxidación, un ejemplo típico es la disolución del Zn Zn → Zn++ + 2e- El cátodo es el polo positivo y es donde ocurre la reacción de reducción, un ejemplo típico es la reducción del hidrógeno 2H+ + 2e- → H2 El material del electrodo debe ser un sólido o conductor de primera clase, así el platino es usado para la generación del hidrógeno. Para el flujo de la corriente o los electrones es necesario un conductor metálico que una el ánodo y el cátodo. El conductor electrolítico es la fuente de los iones. Los electrolitos pueden estar separados por un diafragma poroso para evitar la mezcla física o contacto entre los productos de las reacciones anódicas y catódicas, pero deben permitir la transferencia de iones o el movimiento libre de la corriente eléctrica. La solución que rodea el ánodo se denomina anolito y catolito a la que rodea al cátodo.

Celdas electrolíticas, son celdas a las que es necesario aplicar energía desde una fuente externa, normalmente un rectificador de corriente, para forzar una reacción química en el sentido deseado. En estas celdas se usa la energía eléctrica para causar la reacción electroquímica deseada, ejemplo la recarga de una batería agotada, la descomposición del agua en hidrógeno y oxígeno. Estas celdas son muy usadas en la metalurgia extractiva para la refinación de metales impuros como el cobre, oro, plata, plomo que se disuelven en el ánodo y se depositan en el cátodo a un estado más puro y también se usan para la electro obtención o electrodeposición de metales desde soluciones procedentes de la etapa de lixiviación, por este método se producen metales de alta pureza como el cobre y el zinc. Para el funcionamiento de estas celdas se requiere una fuente de fuerza eléctrica y así forzar las reacciones que de por sí no son espontáneas. El ánodo en estas celdas por convención es el polo positivo y dependiendo del proceso las reacciones de oxidación pueden ser de disolución del metal o descomposición del agua. Cu° → Cu++ + 2e- en electrorefinación del cobre 2H2O → O2 + 4H+ + 4e- en electrodeposición de Cu o Zn El cátodo es el polo negativo y es donde ocurre la reacción de reducción del metal deseado, sea en electrorefinación o en electrodeposición. Cu++ + 2e- → Cu Zn++ + 2e- → Zn

LEYES DE FARADAY A mediados de los años 1800 Farady estableció entre la cantidad de corriente que fluye a través de una solución y la cantidad de material liberado o depositado en una celda electrolítica, sus investigaciones le permitieron formular dos leyes de la electrólisis.
1. La masa de cualquier sustancia liberada o depositada en un electrodo es proporcional a la cantidad de electricidad (coulombs) que pasa a través del electrolito
2. La cantidad de masa de diferentes sustancias producidas por la misma cantidad de electricidad son proporcionales a su peso equivalente
Un equivalente de cualquier sustancia será liberado (oxidado o reducido) por un Faraday, F, que es igual aproximadamente a 96 500 coulombs.

w = 𝑒𝐼𝑡
96 500
w = peso en gramos
e = peso equivalente; 𝑝𝑒𝑠𝑜 𝑎𝑡ó𝑚𝑖𝑐𝑜
𝑣𝑎𝑙𝑒𝑛𝑐𝑖𝑎
I = intensidad de corriente en A T = tiempo en s
3.30. Cinética de la electrorefinación Cuando se emplea una celda electroquímica para convertir energía química en energía eléctrica, se obtiene esta energía como consecuencia de la disminución de la energía libre asociada a la reacción química espontánea dentro de la celda. Bajo condiciones reversibles el trabajo eléctrico es igual al aumento de la energía libre, sin embargo si la celda opera irreversiblemente la energía eléctrica disponible para realizar trabajo es menor que la disminución de la energía libre y la diferencia se disipa en forma de calor, de esto se deduce forzosamente que las reacciones que van acompañadas de un aumento de energía libre no pueden utilizarse para realizar trabajo y para hacer posibles tales reacciones debe de proporcionarse energía a fin de elevar el contenido de la energía libre de los productos sobre los reactantes, electroquímicamente esta adición de energía va acompañada por el paso de electricidad bajo un potencial aplicado a la celda en la que ocurre la reacción particular. Por medio de la electrólisis se provoca cambios electroquímicos en los electrodos y una acción química a expensas de la energía eléctrica. De las consideraciones termodinámicas podemos anticipar que la energía eléctrica mínima requerida para forzar una reacción no espontánea deberá ser igual al incremento de la energía libre que acompaña al cambio y este ∆G es igual a su vez, pero de signo contrario, a la variación de la energía libre que acompañe al proceso espontáneo inverso. Esto será cierto cuando la electrólisis se efectúa reversiblemente, es decir cuando los electrodos son totalmente reversibles y cuando sólo pasa a través de la celda una corriente muy pequeña, cuando no se satisfacen estas condiciones, la energía eléctrica requerida será la mínima teórica más la necesaria para sobrepasar o vencer la irreversibilidad, el potencial aplicado para la electrólisis tiene que ser mayor que la fuerza electromotriz (fem) de la celda. Una celda que exige un sobrevoltaje respecto al teórico se encuentra POLARIZADA y el exceso de polarización se llama VOLTAJE DE POLARIZACIÓN, mientras que el fenómeno en general se llama polarización. La polarización es la diferencia de potencial cuando una corriente apreciable pasa a través del límite de un electrodo y la solución. La polarización del electrodo puede ser vista como una forma de resistencia del electro al paso de la corriente eléctrica. Cuando una corriente eléctrica es pasada a través de un electrodo, el potencial se traslada en una dirección opuesta a la corriente aplicada por una diferencia mínima de potencial, este traslado en el potencial es llamado “sobrevoltaje” y decimos que tal electrodo es polarizado.
3.31. Curvas de polarización

La polarización de un electrodo es bien descrita ploteando el potencial del electrodo frente a la intensidad de corriente o densidad de corriente. Un circuito simple (circuito galvanostático) para la medición de las curvas de polarización consiste de una fuente de corriente eléctrica variable aplicado sobre el electrodo a medir, un electrodo de referencia con una unión capilar a la superficie del electrodo a medir y un milivoltímetro que medirá la diferencia de potencial entre el electrodo a medir y el electrodo de referencia. También se usan circuitos potenciostáticos que permiten mantener constante el potencial de la probeta. Ambos sistemas se ven en la figura siguiente.
Una muestra de una curva de polarización es la figura que se adjunta y es un ejemplo de las muchas formas posibles como curvas pueden haber, la razón por la que las curvas de polarización pueden mostrar una multitud de formas, es que ellas pueden resultar de procesos de polarización completamente distintas no relacionadas, las cuales tienen solamente un factor común, todos son de naturaleza irreversible y originan la disipación de la energía eléctrica en calor. La forma compleja de la curva podría resultar por ejemplo de un predominio de transferencia de masa por polarización de la región más grande de corriente anódica. El grado de polarización también varía sustancialmente de un sistema a otro. Algunos electrodos muestran muy bajos sobrevoltajes aún a densidades de corriente altas, mientras

que otros sistemas de electrodos muestran sobrevoltajes muy grandes aún para densidades de corriente bajas.
CARÁCTER IRREVERSIBLE DE LA POLARIZACIÓN Termodinámicamente el potencial de un electrodo reversible está relacionado con el valor de ∆G, en una celda compuesta por el electrodo en cuestión y un electrodo de hidrógeno. El potencial de un electrodo polarizado se puede expresar: Ԑ(I) = Ԑ(o) + 𝜂 𝜂 = Ԑ(I) - Ԑ(o)
Donde Ԑ(o) = Ԑo - 𝑅𝑇
𝑛𝐹 ln Q Q =
𝐴𝑐𝑡𝑖𝑣𝑖𝑑𝑎𝑑 𝑃𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠
𝐴𝑐𝑡𝑖𝑣𝑖𝑑𝑎𝑑 𝑅𝑒𝑎𝑐𝑡𝑎𝑛𝑡𝑒𝑠
El sobrevoltaje 𝜂 puede ser de signo positivo o negativo, depende de la dirección de la corriente. Una forma de determinar el signo correcto, es recordar que el sobrevoltaje corresponde a la energía eléctrica (o ∆G) irreversible e inútilmente convertida en calor. Por lo tanto en una batería electroquímica que produce energía eléctrica los valores absolutos del ánodo y del cátodo se restan del potencial de equilibrio de la celda Ԑ(o), reduciéndose el voltaje obtenido de la batería a una cantidad menor que el voltaje teórico de Ԑ(o). Desde que por el efecto de Joule se pierde un potencial en el electrolito, también representa disipación de energía, es conveniente incluir este término en el sobrevoltaje. El voltaje obtenido de la batería es por lo tanto (Usando valores absolutos de sobrevoltaje): V = Ԑ(o) – RI – η𝐴 − η𝐶 (∆G ˂ 0) En el caso de un dispositivo electroquímico que consume energía eléctrica y lo convierte en energía libre del sistema , tal como una celda de electrodeposición o electrorefinación, el valor absoluto de los sobrevoltajes y el término IR es añadido al potencial de la celda, tal que el potencial es más grande que el potencial de equilibrio Ԑ(o), es realmente necesario lograr la asociación de la reacción dada con un ∆G positivo.

V = Ԑ(o) + RI + η𝐴 + η𝐶 (∆G ˃ 0) En el caso de la electrorefinación de metales, ∆G y por consiguiente el potencial de la celda están próximos a cero y el voltaje de la celda es la suma de los tres componentes correspondientes al proceso irreversible en la celda. V = RI + η𝐴 + η𝐶 (∆G ˃ 0) GENERACION DE CALOR Aunque no se puede usar la termodinámica electroquímica para predecir los valores del sobrepotencial o los voltajes de la celda de las celdas de baja corriente, se puede hacer uso de los cálculos termodinámicos si los componentes del potencial irreversible o por lo menos el voltaje de las celdas de baja corriente son conocidos. Una tarea muy usual en ingeniería electroquímica es calcular el calor desprendido en una celda electroquímica operando a una corriente. Desde que el calor total desprendido Q debe incluir tanto el calor reversible asociado con la reacción Qrev = - T∆S = ∆G - ∆H = nF Ԑ(o) - ∆H ------- (1) Como la energía irreversible disipada es: Qirr = nF(IR + η𝐴 + η𝐶 ) = nF(V - Ԑ), Ԑ → 0 -------(2) El calor neto generado será la suma de 1 y 2 Q = nFV - ∆H ----------- (3) Debe destacarse que mientras el calor irreversible Qirr, es siempre positivo (el calor es disipado), el calor reversible Qrev, en la mayoría de los procesos electrolíticos de interés, es negativo, de modo que el calor neto generado (o disipado) es a menudo menor que el Q irr. En las operaciones electrometalúrgicas, parte de la corriente eléctrica es a menudo consumido debido a reacciones como: Fe++ → Fe+++ + 1e- , reacción que se presenta en la electrodeposición del cobre. O simplemente debido a los cortos circuitos entre los electrodos. La eficiencia de corriente en la electrodeposición del cobre es siempre menor al 100%. Tales procesos deben estar considerados, cuando se realiza el cálculo del calor disipado (generado, liberado). En los ejemplos citados no tiene lugar un proceso electroquímico neto y la porción de corriente eléctrica correspondiente es completamente disipada como calor. El término adicional:
Qirr = 100− 𝜂𝐼
𝜂𝐼 nFV --------- (4), donde 𝜂𝐼 es la eficiencia de corriente,
debe ser añadido a la ecuación de generación de calor neto (3) que combinado con la ecuación (4), se tiene:
Q = 100
𝜂𝐼 nFV - ∆H ----------- (5)

Un ejemplo del cálculo de calor neto generado es una celda de electrodeposición del cobre operando a un V = 2 voltios y eficiencia de 85%.
Cu++ + H2O → Cu + 1
2 O2 + 2H+
Sustituyendo los valores en la ecuación (5)
Q = 100
85 (2
𝑒𝑞
𝑚𝑜𝑙 )96500
𝑐𝑜𝑢𝑙𝑏
𝑒𝑞 x 2 volt - ∆H
Si ∆H = 56000 cal/mol
Q = 454100 𝑐𝑜𝑢𝑙𝑏− 𝑣𝑜𝑙𝑡
𝑚𝑜𝑙 – 56000
𝑐𝑎𝑙
𝑚𝑜𝑙
Q = 45100 𝑐𝑜𝑢𝑙𝑏− 𝑣𝑜𝑙𝑡
𝑚𝑜𝑙 (0.239
𝑐𝑎𝑙
𝑐𝑜𝑢𝑙𝑏−𝑣𝑜𝑙𝑡) – 56000
𝑐𝑎𝑙
𝑚𝑜𝑙
Q = 53 000 cal/mol. TIPOS DE POLARIZACIÓN La polarización puede ser causada por fenómenos irreversibles que tienen lugar cerca de los electrodos y la polarización resultante es clasificada según el fenómeno que lo causa en:
1. Polarización por transferencia de carga o polarización de activación o transición, que es causada por la energía de activación, por lo que la carga eléctrica que lleva una partícula debe vencer la interface solución – electrodo
2. Polarización por transferencia de masa o polarización por concentración o difusión, es causada por una baja difusión de iones hacia o lejos de los electrodos, el proceso irreversible aquí es la difusión.
3. Polarización por reacción, es similar a la polarización por transferencia de masa, con la diferencia de que el bajo suministro de los iones reactantes al electrodo es originado por una baja velocidad de reacción química que generan las especies reactantes en el electrodo.
4. Polarización por cristalización, la causa de esta polarización es la baja velocidad con que se incorporan los átomos dentro de una red cristalina del electrodo metálico o para un proceso inverso, ejemplo en el desprendimiento de iones metálicos hacia la solución
5. Polarización por resistencia o polarización óhmica, es causado por una caída de potencial a través de una capa porosa o la misma película sobre la superficie del electrodo.
Muchos tipos de polarización están a menudo presentes en el electrodo al mismo tiempo, aunque uno puede predominar sobre los otros, la suma de los sobrepotenciales asumidos es:
𝜂 = 𝜂𝑇𝐶 + 𝜂𝑇𝑀 + 𝜂𝑅𝑥 + 𝜂𝐶𝑟 + 𝜂𝛺 En los procesos electrometalúrgicos extractivos, tienen mucha importancia la polarización causada por transferencia de masa y de carga.

POLARIZACIÓN POR RANSFERENCIA DE MASA Los iones pueden ser transportados hacia o desde los electrodos por tres mecanismos: migración, difusión y convección. En los procesos electrometalúrgicos casi siempre están presentes los tres mecanismos. 1.- Transporte de iones por migración La causa de la migración iónica es la fuerza electrostática generada por la interacción entre el campo eléctrico y los iones eléctricamente cargados. También se debe tener presente que los diferentes iones sujetos a un mismo campo migran a diferentes velocidades debido a sus diferentes tamaños y sus velocidades se relacionan a sus respectivos números de transporte o movilidad. En un electrolito de un solo componente, tal como el CuSO4 diluido, la fuerza del campo eléctrico puede ser alta, especialmente en los electrolitos agotados en la capa de difusión catódica, bajo esas circunstancias, la contribución de la migración iónica a la velocidad del proceso de difusión puede ser sustancial y desafortunadamente algo difícil de calcular. En polarografía, la cual está basada en la medición cuantitativa de las velocidades de difusión, la incertidumbre o dificultad de la medición se elimina añadiendo un electrolito soporte, esto es aumentando la conductividad del electrolito que reduce la fuerza del campo eléctrico y reduciendo el efecto de la migración a valores despreciables. En electrometalurgia se usa una aproximación similar, aunque por una razón diferente. La mayoría de los electrolitos industriales contienen entre 50 a 250 g/l de ácido sulfúrico libre o alguna sal conductora para incrementar la conductividad y reducir el consumo, ejemplo, en la electrorefinación y electrodeposición del cobre, electrodeposición del Zn. Los electrolitos usados para la refinación y electrodeposición de metales menos, ejemplo el Ni, usualmente contienen sales de Na, por lo tanto, el efecto de la migración sobre la transferencia de masa en procesos electrometalúrgicos es relativamente pequeña y pueden ser, afortunadamente, ignorados por el electrometalurgista. 2.- Transporte de iones por difusión El paso de una corriente eléctrica a través de una celda electrolítica origina cambios de concentración local de las especies reaccionantes cerca de los electrodos, dando por resultado un proceso de difusión controlado. Un buen ejemplo para ilustrar esta forma de transferencia de masa es una celda de refinación de cobre, que se muestra en la figura siguiente:

Antes de aplicar la corriente eléctrica, la concentración de los iones cobre es uniforme en todo el electrolito, típicamente ≅ 0.7 M de CuSO4, 0.3 M NiSO4 y 2 M H2SO4. Después de permitir el flujo de corriente, la concentración de iones cobre (Cu++) cerca del ánodo aumenta debido a la disolución del ánodo y la concentración de iones cobre cerca del cátodo disminuye debido a la deposición. La electroneutralidad del electrolito no es alterada por que lo iones negativamente cargados de 𝐻𝑆𝑂4
− migran hacia el ánodo. Si solamente tiene lugar la difusión, el proceso eventualmente puede alcanzar el establecimiento de un estado lineal estable de gradiente de concentración desde el ánodo hasta la superficie del cátodo y esta gradiente debe impulsar el transporte de masa por difusión de iones Cu++ desde la superficie anódica hasta la superficie catódica. La máxima velocidad de un proceso de refinación hipotético, solamente por difusión, puede ser estimada asumiendo que el máximo estado constante (estable) de la gradiente puede ser obtenido cuando la concentración de Cu++ en el cátodo es cero y consecuentemente 1.4 x 10-3 mol/cm3 en el ánodo. El flujo de difusión para una celda dada será:
JCu++ = DCu
++ (𝑑𝐶
𝑑𝑙)
= (1.0 x 10-5 𝑐𝑚2
𝑠)
1.4 𝑋 10−3 𝑚𝑜𝑙
𝑐𝑚3
2 𝑐𝑚
= 0.7 x 10-8 𝑚𝑜𝑙
𝑐𝑚2−𝑠
JCu++ = Flujo por difusión de iones cobre
DCu++ = Coeficiente de difusión del cobre
Y la densidad de corriente J será: j = nF JCu
++ = 2 x 96500 x 0.7 x10-8 = 1.35 x 10-3 A/cm2 j = 13.5 A/m2 La máxima densidad de corriente que debe alcanzar en una celda de determinada concentración de cobre en ausencia de cualquier mecanismo de transporte de masa debe entonces ser de 13.5 A/m2, pero en las refinerías de cobre se puede operar fácilmente con densidades de 15 a 20 veces mayor que este valor debido a la presencia de la convección natural. 3.- Transporte de iones por convección Las capas finas de electrolito adyacentes a los electrodos y a las paredes de la celda, están virtualmente estancadas o estacionarias, mientras que el resto del electrolito está siempre en movimiento. Esto es cierto, aún, cuando el electrolito no sufre agitación artificial. El electrolito está caliente entre los electrodos y frio sobre las paredes de la celda, hecho que genera una gradiente de temperatura, que a su vez genera una gradiente de densidades, estas gradientes provocan una lenta pero efectiva circulación del electrolito. Igualmente

una causa más efectiva del movimiento del electrolito por convección son las gradientes de concentración que se dan cerca de los electrodos, estas diferencias producen densidades diferentes y por lo tanto provocan el movimiento del electrolito. En una celda de refinación de cobre, el electrolito se agota en Cu++ cerca del cátodo y tiene una menor densidad que en el resto del electrolito y entonces tiende a fluir hacia arriba a lo largo del electrodo, mientras que la capa de electrolito cerca del ánodo se enriquece en su contenido de Cu++ y por lo tanto es más denso que el resto del electrolito y produce un movimiento hacia abajo a lo largo del ánodo. Estas fuerzas que producen una suave circulación del electrolito entre los electrodos, se conocen como fuerzas convectivas naturales, y su tendencia es igualar las diferencias de concentración entre los dos electrodos y así efectivamente causan un transporte neto de Cu++ desde la vecindad anódica a la catódica. Sin embargo una capa de electrolito adyacente a los electrodos es estacionario debido a las fuerzas moleculares, en esta capa estacionaria el movimiento del electrolito es muy limitado o amortiguado debido a las fuerzas de la viscosidad. La existencia de tal capa fue determinada por Nernst y se denomina “MODELO DE LA CAPA DE DIFUSION DE NERNST”. De acuerdo a este modelo, el electrolito está completamente estacionario a través de toda la CAPA DE DIFUSION adyacente al electrodo, pero, el electrolito se mueve y es agitado por convección en el resto de la masa del electrolito. Según este modelo el flujo de difusión de los Cu++ hacia el cátodo es:
JCu++ = (DCu
++) ∆𝐶
𝛿𝑁
Y la densidad de corriente respectiva será:
j = nFD∆𝐶
𝛿𝑁 , donde ∆C = CB - CS
∆C = gradiente de concentración CB = concentración de Cu++ en el seno del electrolito CS = concentración de Cu++ sobre la superficie del electrodo 𝛿𝑁 = Espesor de la capa de difusión de Nernst, aproximadamente igual a 0.03 cm Usando los valores del ejemplo del transporte de iones por difusión para una celda de electrorefinación de cobre, pero asumiendo el modelo de la Capa de Difusión de Nernst = 0.3 mm, valor experimentalmente determinado, es posible obtener el flujo máximo de iones, cuando la concentración de Cu++ cerca de la superficie del cátodo tiende a cero.
JCu++ = (1.0 x 10-5
𝑐𝑚2
𝑠)0.7 𝑋 10−3
𝑚𝑜𝑙
𝑐𝑚3
0.03 𝑐𝑚
= 23 x 10-8 𝑚𝑜𝑙
𝑐𝑚2−𝑠
Y la densidad de corriente J será: j = nF JCu
++ = 2 x 96500 x 23 x10-8 = 0.04439 A/cm2 j = 443.9 A/m2

La densidad de corriente máxima permitido según el modelo de la Capa de Difusión de Nernst, usualmente se expresa:
j = nFD 𝐶𝐵
𝛿𝑁
y se le denomina densidad de corriente límite (jlim). Es importante hacer notar que la mayoría de las refinerías de cobre operan a densidades de corriente que oscilan entre 200 a 250 A/m2, es decir con cerca de la mitad de la densidad de corriente límite. Esto se debe a que la calidad de los depósitos se deteriora rápidamente cuando se opera a densidades de corriente cercano al límite.
Capa de Difusión Real, Es obvio que el modelo de Nernst de la Capa de Difusión es sólo un artificio. La transición del electrolito estacionario cerca de la interfase, donde el transporte de masa solamente tiene lugar por difusión (y migración) y la masa de electrolito, donde prevalece el transporte de iones por convección, es indudablemente gradual, entonces el perfil de la concentración real cerca del electrodo es la que muestra la figura anterior. Luego el flujo total de las especies catódicamente reducibles será la suma de los flujos por difusión y convección, excepto en las primeras capas muy cerca de la superficie catódica, donde el flujo total está determinado solamente por difusión. Puesto que en un estado constante el flujo total debe ser la misma en cualquier punto de la capa de difusión, éste puede ser simplemente definido por:
JCu++ = D (
𝑑𝐶
𝑑𝑙) y cuando l→ 0 JCu
++ = D (∆𝐶
𝛿𝑁)

3.32. Selectividad anódica – selectividad catódica La electrorefinación consiste en la disolución electroquímica del cobre de los ánodos impuros y el depósito selectivo de este cobre disuelto en forma más pura sobre el cátodo y tiene dos objetivos:
a) Eliminar las impurezas que dañan las propiedades eléctricas y mecánicas del cobre b) Separar las impurezas valiosas para luego recuperarlas como subproductos
metálicos El principio de la refinación electrolítica del cobre consiste en aplicar un potencial eléctrico entre un ánodo de cobre (electrodo positivo) y un cátodo de cobre (electrodo negativo), ambos sumergidos en una celda que contenga una solución de sulfato de cobre acidificada, para ocasionar las siguientes reacciones y procesos:
a) El cobre del ánodo se disuelve electroquímicamente dentro de la solución Cuo
ánodo → Cu++ + 2e- E = - 0.34 v b) Los electrones producidos por la reacción de disolución del cobre son conducidos
hacia el cátodo a través del circuito y suministro de energía externa c) Los cationes Cu++ en la solución migran por difusión y convección hacia el cátodo d) Los electrones y los iones Cu++ se recombinan sobre la superficie del cátodo para
producir cobre metálico que se deposita sobre el cátodo Cu++ + 2e- → Cuo E = + 0.34
Los efectos finales son la disolución electroquímica del cobre del ánodo, la migración de los electrones y iones de cobre hacia el cátodo y el depósito de cobre sobre la superficie del cátodo. La reacción electroquímica total es la suma de la reacción de oxidación en el ánodo y la reacción de reducción en el cátodo: Cuo
Impuro - ánodo → Cuopuro – cátodo, para la cual el
potencial teórico reversible, o sea, la diferencia entre los potenciales de electrodo es cero. En la práctica real, la resistencia al flujo de corriente en el electrolito (por transporte de iones) y la resistencia al flujo de electrones en las barras de distribución y conexiones eléctricas deben ser vencidas aplicando una tensión entre los ánodos y cátodos. También existe una sobretensión de aproximadamente 0.06 v necesaria para el depósito de cobre sobre el cátodo y uno más pequeño en el ánodo. La tensión en las celdas industriales, entre ánodos y cátodos es de 0.25 a 0.3 v. El propósito de la electrorefinación es obtener un metal de alta pureza a partir e un metal impuro mediante un método electrolítico y una ventaja derivada del proceso es que los metales que están presentes como elementos impurificantes en el metal original son separados y pueden ser recuperados posteriormente en una forma de alta pureza, por esto, la electrorefinación es la única manera de separar los elementos menores, lo que se consigue a través de un proceso anódico selectivo entre solubles e insolubles y un proceso catódico de deposición selectiva.
a) Anodo Soluble, El bullón metálico procedente de la fundición puede contener de 90 a 99% del metal principal, tal como Cu (ánodos de cobre), Pb o Ni y pequeñas cantidades de metales impurificantes. Las impurezas pueden incluir metales nobles como el Au, Ag, Pt, Se, etc y algunos elementos menos nobles que el metal principal como el Fe, Bi, As, Sb, etc. El grado de nobleza se define según su posición en la serie electromotriz.

Cuando el metal impuro es colocado en un electrolito y conectado como ánodo a una fuente de poder, algunos de los elementos son selectivamente disueltos y pasan a la solución. Los elementos que son menos nobles que el metal principal tendrán una fuerte tendencia a disolverse, mientras que los metales que son más nobles permanecerán en el estado metálico. La secuencia de la selectividad que indica el grado de nobleza de cada elemento se indica en la figura siguiente. Pt Au Se Ag Cu As Sb Pb Bi H2 Fe Ni Zn Mg Al Aumenta Nobleza Disminuye Nobleza
Las impurezas nobles forman un lodo, llamado lodo anódico, término usado para describir el residuo metálico algo gelatinoso que queda después de que el ciclo anódico es completado. En las refinerías se distinguen dos tipos distintos de lodos. En las refinerías de cobre las partículas de metal no disueltas en el ánodo se depositan en el fondo de la celda, desde son periódicamente cosechadas, estos lodos por ser acumuladas en el fondo de las celdas se les conoce como lodos de caída. El otro tipo de lodo son los generados en al electrorefinación del plomo que están presentes como una red de cintas, semejante a un esqueleto y quedan colgados en el ánodo, por lo que se les conoce como lodos colgantes. Los lodos pasan a la planta de tratamiento de Lodos Anódicos, donde se recupera los metales valiosos, como el oro y la plata. Los elementos menos nobles pasan a la solución junto con el metal principal y son separados desde la solución en un proceso de tratamiento del electrolito purgado en forma periódica desde el circuito del electrolito de la planta industrial.
b) Selectividad catódica, En el cátodo, los iones de Cu++, metal principal, son electrolíticamente reducidos a metal y desde que la reducción es un proceso inverso a la oxidación, la selectividad trabajará de un modo opuesto al del ánodo, es decir que los iones de metales más nobles y los iones del metal principal se reducirán sobre el cátodo, mientras que los iones de los metales menos nobles no serán afectados por la electrólisis.
Por tanto la purificación o refinación electrolítica del cobre tiene lugar por que solamente las impurezas menos nobles que el metal principal pueden ser disueltos desde el ánodo y solamente los metales más nobles y el metal principal pueden ser reducidos en el cátodo.
3.33. Equipos de la casa tanque 3.34. Procedimientos de la casa tanque - control 3.35. Electrolito – Purificación del electrolito 3.36. Densidad de corriente y producción 3.37. Aditivos – control



















