
Redalyc.SÍNTESIS VÍA “ELECTROQUÍMICA DIRECTA” DE ... · de los ftalonitrilos con distintos...
27
Revista Cubana de Química ISSN: 0258-5995 [email protected] Universidad de Oriente Cuba Kharisov, B. I.; Garnovskii, A. D.; Kharissova, O. V.; Ortiz Méndez, U. SÍNTESIS VÍA “ELECTROQUÍMICA DIRECTA” DE FTALOCIANINAS Y AZOMETINAS: VENTAJAS Y DESVENTAJAS DE ESTA TÉCNICA EN COMPARACIÓN CON LOS MÉTODOS TRADICIONALES Revista Cubana de Química, vol. XIX, núm. 3, 2007, pp. 65-90 Universidad de Oriente Santiago de Cuba, Cuba Disponible en: http://www.redalyc.org/articulo.oa?id=443543708018 Cómo citar el artículo Número completo Más información del artículo Página de la revista en redalyc.org Sistema de Información Científica Red de Revistas Científicas de América Latina, el Caribe, España y Portugal Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto
Transcript of Redalyc.SÍNTESIS VÍA “ELECTROQUÍMICA DIRECTA” DE ... · de los ftalonitrilos con distintos...

Revista Cubana de Química
ISSN: 0258-5995
Universidad de Oriente
Cuba
Kharisov, B. I.; Garnovskii, A. D.; Kharissova, O. V.; Ortiz Méndez, U.
SÍNTESIS VÍA “ELECTROQUÍMICA DIRECTA” DE FTALOCIANINAS Y AZOMETINAS:
VENTAJAS Y DESVENTAJAS DE ESTA TÉCNICA EN COMPARACIÓN CON LOS
MÉTODOS TRADICIONALES
Revista Cubana de Química, vol. XIX, núm. 3, 2007, pp. 65-90
Universidad de Oriente
Santiago de Cuba, Cuba
Disponible en: http://www.redalyc.org/articulo.oa?id=443543708018
Cómo citar el artículo
Número completo
Más información del artículo
Página de la revista en redalyc.org
Sistema de Información Científica
Red de Revistas Científicas de América Latina, el Caribe, España y Portugal
Proyecto académico sin fines de lucro, desarrollado bajo la iniciativa de acceso abierto

65Vol. XIX, Nº 3, 2007
SÍNTESIS VÍA �ELECTROQUÍMICA DIRECTA�DE FTALOCIANINAS Y AZOMETINAS:
VENTAJAS Y DESVENTAJAS DE ESTA TÉCNICAEN COMPARACIÓN CON LOS MÉTODOS
TRADICIONALES B. I. Kharisov*, A. D. Garnovskii**, O. V. Kharissova*, U. Ortiz Méndez*
*Universidad Autónoma de Nuevo León, Monterrey, Mexico, **Rostov State University, Rostov-on-Don, Russia
Resumen
En este trabajo se reporta un análisis crítico y comparativo de las técnicas sintéticas tradicionalesy electroquímicas para la obtención de ftalocianinas y azometinas. Se muestra que el método electroquímicopermite la obtención de los compuestos de coordinación azometínicos libres de aniones de sales-precursores con más altos rendimientos a temperatura ambiente. La electrólisis de soluciones no acuosasde los ftalonitrilos con distintos grupos funcionales lleva a la formación de los ftalocianinatos correspondientesa temperaturas relativamente bajas (en 100 oC menores que en las condiciones sintéticas clásicas).Típicos problemas de procedimientos electroquímicos se examinan en detalle.
Palabras clave: técnicas sintéticas tradicionales, obtención de ftalocianinas y azometinas.
Resumen
This paper reports a critical and comparative analysis of the traditional synthetic techniques andelectrochemistry�s for obtaining ftalocianinate and azometines. It is shown that the electrochemicalmethod allows to obtain free azometínilical compounds of coordination of anions of salt-precursors withmore high performances at room temperature. Electrolysis of nonwater solutions of the ftalonitrite withdifferent functional groups takes to the formation of the ftalocianinate corresponding to relatively lowtemperatures (in 100 oC smaller than in the classic synthetic conditions). Typical problems of electrochemicalprocedures are examined in detail.
Key words: traditional synthetic techniques, obtaining ftalocianinate and azometines.
Introducción
La electrosíntesis es un método de una sola etapapara obtener compuestos de coordinación /1-9/ comose describe en el esquema (1) /8/:
M ........L (1 )M 0 - e L
Esquema 1.

66 Vol. XIX, Nº 3, 2007
C: LH + ne
A: M - ne
nL- + n/2H2
Mn+; Mn+ + nL- [MLn] (3)
La electrosíntesis de metalocomplejos,generalmente se lleva a cabo con uso de ánodossacrificiales de una serie de metales (Grupo IB � Cu,Ag, Au; Grupo IIA � Mg, Ca, Zn, Cd, Hg; Grupo IIIA� Al, In, Ga, Tl; Grupo IVA � Sn, Pb; Grupo IVB �Ti, Zr, Hf; Grupo V � Sb, V, Nb, Ta; Grupo VIB � Cr,Mo, W; Grupo VIIB � Mn, Re; Grupo VIIIB � Co, Ni,Pd; actínidos (Th, U) y algunos lantánidos), y el platinocomo cátodo en solventes no acuosos, con la constantedieléctrica (ε) de 3,5 al 64 /8, 9/.
Los solventes más utilizados son CH3CN (ε = 38)y alcoholes (ε ~ 30); los electrolitos soportes usualesson LiCl, LiClO4, NaBF4, R4NX (R = Alk, X = Br-,ClO4
-, BF4-).
Los ligantes, utilizados en este trabajo son lossiguientes: a) los precursores de las ftalocianinas {R-C6H4(CN)2, ftalonitrilos, R = H, 3-NO2, 4-NO2, 3-NH2, etc.}, y b) los ligantes azometínicos (fórmulasestructurales: favor de ver la parte experimental).Ambos tipos de ligantes contienen el enlace dobleC=N, cuyo átomo de nitrógeno puede coordinarse conátomos metálicos entrantes.
Con el uso del primer grupo de ligantes, la síntesises del tipo �templete� (�template� en inglés), cuandoel macrociclo se forma in situ; el otro tipo de ligantesconserva su estructura, pero puede formar quelatos(perdiendo el átomo H) o aductos (o complejos
moleculares), conservando este átomo en la molécula-precursor.
Equipo
El equipo especial para la electrosíntesis se describeen detalle en /3, 10/, y se muestra en las figuras 1-4.Las electrosíntesis se llevan a cabo en un matraz de100 mL, a veces en los espacios catódicos y anódicosseparados en atmósfera inerte, a temperaturas 20-80oC. La duración de reacciones varía de unos minutoshasta horas.
Fig. 1 Celda sonoelectroquímica /10/.
La misma reacción en solventes coordinativos puede ser presentada como se muestra en el esquema(2) /9/:
Esquema 2.
Para los ligantes donadores del protón, estos procesos tienen lugar sobre el cátodo (C) y ánodo (A), comose muestra en el esquema 3:
Esquema 3.

67Vol. XIX, Nº 3, 2007
Parte experimental
Procedimiento en general. Los experimentoselectroquímicos se llevan a cabo en las celdas arribamostradas (50-100 mL), tanto divididas como noseparadas. El acetonitrilo, alcoholes de bajo pesomolecular, DMF o DMSO, se utiliza como solventes.En el caso, en que el ligante tiene baja solubilidad en
estos solventes, se aplica la mezcla de CH3CN-benceno (o tolueno, hasta 30-50 % de peso). Losligantes (1-10 g) y electrolito soporte previamentesecados (LiCl, LiClO4, (n-Bu4N)ClO4, (n-Bu4N)Br,0-0,05 g), se añaden en el solvente (50-100 mL). Elnitrógeno seco pasa a través de la celda durante todoel tiempo de electrólisis. El platino (alambre 5-10 cmo lámina 2 × 2 cm) se utiliza como cátodo, y el metal
Fig. 2 Celda dividida para la obtención deftalocianinas /11/.
Fig. 3 Celda dividida comercial(�Bioanalytical systems�).
Fig. 4 Equipo combinado �electrosíntesis-ultrasonido�.
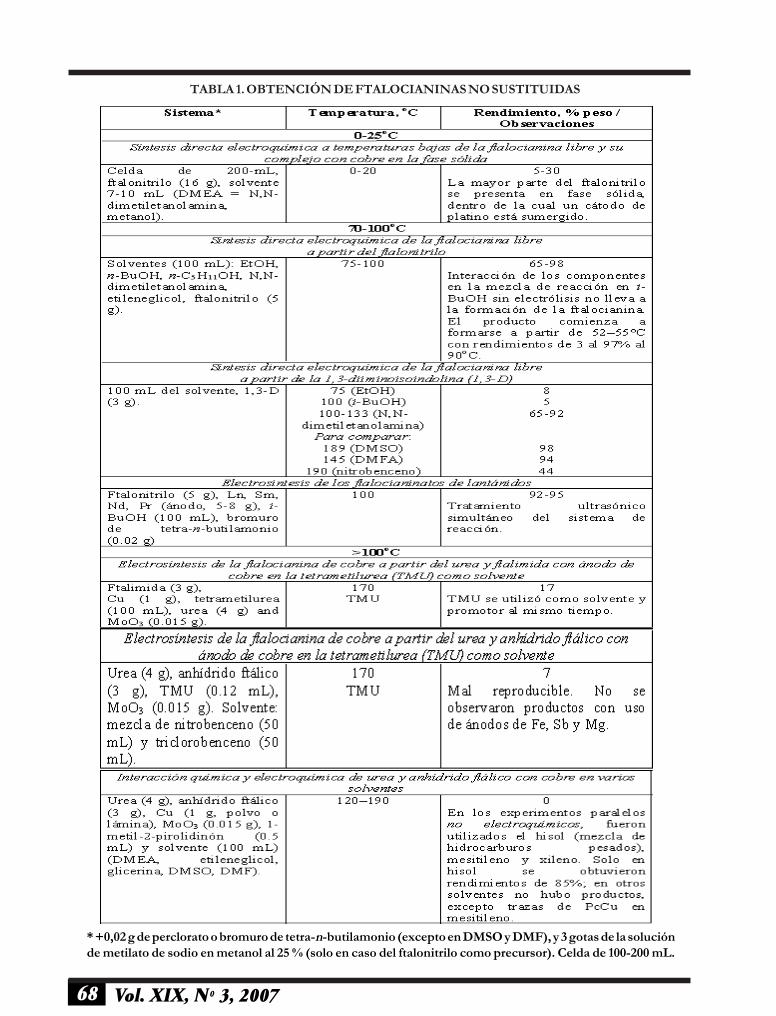
68 Vol. XIX, Nº 3, 2007
* +0,02 g de perclorato o bromuro de tetra-n-butilamonio (excepto en DMSO y DMF), y 3 gotas de la soluciónde metilato de sodio en metanol al 25 % (solo en caso del ftalonitrilo como precursor). Celda de 100-200 mL.
TABLA 1. OBTENCIÓN DE FTALOCIANINAS NO SUSTITUIDAS

69Vol. XIX, Nº 3, 2007
sacrificial (Cu, Co, Ni, Zn, etcétera, en forma dealambre o lámina, lavados previamente con HCl oH2SO4 diluidos) se usa como ánodo. Los procesos seefectúan con uso de la fuente de poder Sigma-Aldrich durante 1�3 h al 20�30 mA y 10�50 V. Enalgunos casos, la celda se mantiene a temperaturas0-130 oC; en casos necesarios, se pone en la cámaraseca para manipulaciones posteriores, tales comodecantación o limpieza de los productos. Si el sólido nose obtiene inmediatamente al terminar la electrólisis,la solución se evapora parcialmente directamente enla celda o en otro frasco a presión atmosférica oreducida; luego se filtra y lava con solventes.
Los rendimientos de complejos azometínicos varíande 50 hasta 98 %, de ftalocianinas /12/ � de 10 hasta95 %. El rendimiento electroquímico h se calculasegún la ecuación (4), donde Q es la cantidad deelectricidad aplicada, c es el equivalente electroquímico,c = M/nF (donde M es el peso atómico del metal, n elnúmero de electrones, y F = 96 500 C/mol).
∆mteor. = Q ⋅ χ = I (A) ⋅ t (seg.) ⋅ χ (g/C) (4)
Ftalocianinas no sustituidas. La electrosíntesis�templete� en solución se lleva a cabo en reflujo al 50-180 oC, con uso del ftalonitrilo no sustituido, 1.3-diiminoisoindolina, ftalimida o una mezcla del urea yanhídrido ftálico, durante 0,5-3 h, en una serie desolventes no acuosos: alcoholes, DMF, DMSO, N,N-dimetiletanolamina, nitrobenceno, etcétera. Los sólidosobtenidos se lavan con etanol en el extractor deSoxhlet, y se secan en aire a temperatura ambiente.
La electrosíntesis directa de la ftalocianinas librede metal y su complejo metálico en fase sólida atemperaturas bajas. En algunos experimentos, la celdaelectroquímica no contiene solución homogénea arribamencionada. Así, la fase sólida del precursor seelectroliza para obtener la ftalocianina (16 g) al 0�25oC en una celda de 200 mL. El ftalonitrilo se moja conuna pequeña cantidad del N,N-dimetiletanolamina o
metanol (7-10 mL) que contenía CH3ONa (3 gotas dela solución al 25 % en metanol) como agentenucleofílico, y (n-Bu)4NClO4 (0,02 g). El ftalonitrilosólido está en un contacto directo con el cátodo de altaárea superficial (malla de platino, ~ 30 cm2), sumergidocompletamente dentro del sólido.
El espacio anódico contenía la solución saturadadel ftalonitrilo y el ánodo de platino o cobre (2 ⋅ 2 cm).La temperatura se controla cuidadosamente durantetoda la reacción. Los rendimientos de ftalocianina(ánodo Pt) o ftalocianinato de cobre (ánodo Cu)fueron 5�30 %, dependiendo de temperatura y solvente(más altos en caso de N,N-dimetiletanolamina). Lascondiciones y observaciones para las ftalocianinas nosustituidas sintetizadas se muestran en la tabla 1.
La electrosíntesis directa de la ftalocianinassustituidas (aquí sería más mejor llamar a losprecursores como �dinitrilos�, a diferencia del nombreclásico �ftalonitrilo�, en el caso del ftalonitrilo singrupos funcionales). Los siguientes precursores fueronutilizados: 4-t-butilftalodinitrilo, 4-feniltioftalodinitrilo,3-feniltioftalodinitrilo, 3-fenoxiftalodinitrilo, 4-hidroxiftalodinitrilo, 4-a-naftoxiftalodinitrilo, 3-a-naftoxiftalodinitrilo, 3-nitroftalodinitrilo, 4-nitroftalodinitrilo, 3-b-naftoxiftalodinitrilo, 4-b-naftoxiftalodinitrilo, éter tetracianodifenílico (4,4´-oxodiftalodinitrilo), 4-aminoftalodinitrilo,tetracloroftalodinitrilo.
El procedimiento fue similar al descrito en la tabla1 (obtención de ftalocianinas a partir del ftalonitrilo).Los rangos de temperaturas fueron 25-78 oC (enEtOH) y 25-130 oC (en DMEA). Después de purificaciónen Soxhlet, los productos fueron recristalizados segúnlas técnicas, descritas en la tabla 2.
Complejos de β-aminovinilcetonas. Las β-aminovinilcetonas 1 (R = H, Me, Et), eportadosanteriormente en /13/, nos fueron prestadas en laUniversidad Estatal de Rostov (Rusia) y se utilizaronsin purificación adicional.

70 Vol. XIX, Nº 3, 2007
TA
BLA
2. D
AT
OS E
XPE
RIM
EN
TA
LES S
OB
RE
LA
ELE
CT
RO
SÍN
TE
SIS D
E L
AS F
TA
LOC
IAN
INA
S SU
STIT
UID
AS
Ftal
odin
itrilo
-pre
curs
or
Con
dici
ones
de
la sí
ntes
is
Ren
dim
ient
o
Peso
Prod
ucto
fin
al
Мet
al
Peso
m
olec
ular
Peso
m
ole-
cula
rg
mm
ol
Solv
ente
Т
о С
g m
mol
%
Puri
ficac
ión
aplic
ada/
Obs
erva
ción
es
4-Nitroftalodinitrilo
H
H
694,
4 17
3,1
0,99
2 5,
74
Etan
ol
78
(Rea
cció
n co
mie
nza
a te
mp.
am
b.)
0,01
3 0,
019
1,3
Rep
reci
pita
ción
des
de D
MF
con
18 %
HC
l, lim
pian
do c
on
DM
F+18
% H
Cl (
1:1)
, lue
go
18 %
HC
l (4
vece
s) y
agu
a. N
o ha
y pr
oduc
tos,
inso
lubl
es e
n D
MF
Cu
755,
9
1,59
8 9,
24
Etan
ol
78
(Rea
ctio
n be
gins
at r
.t.)
0,29
0,
384
16,6
Se
lava
con
DM
F+18
%
HC
l y a
gua
N
i 75
1,1
1,
161
6,71
Et
anol
78
(R
eacc
ión
com
ienz
a a
tem
p. a
mb.
)
0,10
4 0,
138
8,2
Rep
reci
pita
ción
des
de D
MF
agre
gand
o un
as g
otas
de
pirid
ina
y H
Cl c
once
ntra
do,
limpi
ando
con
DM
F+18
%
HC
l (1:
1), 1
8 %
HC
l y a
gua
Bi
(P
c lib
re
fue
obte
nida
)
1,7
305
10,0
0 Et
anol
78
(R
eacc
ión
com
ienz
a a
tem
p. a
mb.
)
0,13
1 0,
187
7,5
Tetr
akis(
4-ni
tro-Рс
)Н2
fue
obte
nido
Rep
reci
pita
ción
des
de D
MF
con
18%
HC
l (3
vece
s),
limpi
ando
con
18
% H
Cl y
ag
ua
4-(N
O2) 4
-PcМ
Zn
757,
8
1,1
340
6,55
Et
anol
78
(R
eacc
ión
com
ienz
a a
tem
p. a
mb.
)
0,16
2 0,
214
13,1
R
epre
cipi
taci
ón d
esde
DM
F co
n 18
% H
Cl (
3 ve
ces)
, lim
pian
do c
on 1
8 %
HC
l y
agua

71Vol. XIX, Nº 3, 2007
(contin
uación tabla 2
)

72 Vol. XIX, Nº 3, 2007
(con
tinua
ción
tabl
a 2)
Éter tetra
cianodifenílico (4,4´-o
xodiftalodinitrilo)
HH
1 08
2,8
270,
2
Et
anol
75
Tr
azas
So
lubl
e sol
o en
H2S
O 4НН
1 08
2,8
270,
2 0,
539
1,99
DM
EA
110
- 13
0 0,
130
0,
120
24,1
Pr
ecip
itació
n de
sde 1
8 %
HCl
, fil
tració
n y
limpi
eza c
on ag
ua
y eta
nol.
Rep
recip
itació
n de
sde D
MF
con
18%
HCl
, lim
piez
a con
DM
F+18
% H
Cl
y ag
ua
Cu
1 14
4,4
270,
2 0,
950
3,52
DM
EA
128
0,64
0,
130
14,8
Se
lava
con
HCl c
once
ntra
do,
agua
, etan
ol, a
ceto
na.
Solu
ble s
olo
en H
2SO 4
Ni
1 13
9,5
270,
2 0,
444
1,64
DM
EA
132
0,02
0,
010
2,5
(calc
u-la
do
para
Pc
H2)
Mez
cla d
e РсН
2 y РсN
i se
obtu
vo. R
epre
cipita
ción
desd
e HC
l con
c. co
n NH
3, lim
pian
do
con
agua
. Sol
uble
sólo
en
H 2SO
4
Bi
(Pc
libre
fu
e ob
teni
da)
27
0,2
0,51
4 1,
90
DMEA
13
2 0,
159
0,14
7 30
,9
(calc
u-la
do p
ara
РсН 2
)
РсН
2 se o
btuv
o Re
prec
ipita
ción
des
de H
Cl al
18
%, l
impi
ando
con
agua
y
aceto
na
4-[4
�,5�-
(CN)
2-PhO
] 4-Pc
M
Zn
1 14
6,2
0,
484
1,79
DM
EA
132
0,05
2 0,
045
10,1
Se
lava
con
etano
l, ac
etone
, DM
F +
18 %
HCl
, agu
a, ac
etona
. Rep
recip
itació
n de
sde
DMF
con
18 %
HCl
, lim
pian
do co
n DM
F +
18 %
HC
l y ag
ua
4-α-naftoxiftalodinitrilo
4-(α
-NfO
) 4-PcМ
HH
Etan
ol
75
Traz
as
3-β-naftoxiftalodinitrilo
3-(β
-NfO
) 4-PcМ
HH
Etan
ol +
to
luen
o 75
Tr
azas
en es
pect
ros (
en
benc
eno
y CH
Cl3).
Pr
ecip
itació
n de
sde H
Cl co
nc.,
filtra
ción,
lim
piez
a con
DM
F+18
% H
Cl y
agua

73Vol. XIX, Nº 3, 2007
Para evitar formación de una mezcla deproductos, no se añade electrolito soporte.Similarmente al trabajo /14/, la sal sódica delligante correspondiente es generada in situ, y seutiliza en calidad de electrolito soporte. Ésta seobtiene en la reacción de 2 mg de sodio metálico y0,001 mol del ligante 1 en 25 mL de metanol. Laelectrólisis se lleva a cabo a voltaje fijo durante más
de 2,5 h (M = Zn) ó 1,5 h (M = Cu). La corrienteaplicada es de 10 hasta 20 mA.
Después de electrólisis, los sólidos 2 y 3 se filtrany lavan con metanol caliente. Además, se realizan /15/las electrosíntesis sin generación in situ del electrolitosoporte, utilizando ligantes similares 4 (R = H, Me,reacción (5)), produciendo los complejos 5.
SN
O O
RO O
M/2
Ph
SN
O O
RO O
H
Ph
5
(5)
4
Los ligantes bi- y tridentados heteroazometínicosLH. Ligantes 6-9, reportados anteriormente en /16/,
se sintetizados en la Universidad Estatal de Rostov(Rusia) y se utilizaron sin purificación adicional.
N
N
CH3
C N
H NH
Ts 6
N-H
C=NN
N
R7
Ts
R = Me, Et
H
Las electrosíntesis se lleva a cabo con los ánodossacrificiales de Cu o Ni en metanol durante 1,2�3 h.
Los sólidos se filtran y lavan con metanol caliente, yse secan al vacío a 70 0C.
R = H, Et
O-H
C=NN
N
R8
N-H
C=N
9
Ts
HH

74 Vol. XIX, Nº 3, 2007
Los ligantes tridentados heteroazometínicosLH2. Los ligantes 10, reportados anteriormente en/17/, se sintetizaron en la Universidad Estatal deRostov (Rusia), y se utilizaron sin purificaciónadicional.
XH
N=C
H-Y
H
10X = NTs; Y = O (LH2), NTs (L1H2) Ts = -SO2C6H4-CH3-p
Las electrosíntesis se llevó a cabo con losánodos sacrificiales de Cu, Ni, Co, Cd ó Zn durante1,2�3 h. Los sólidos se filtraron y lavaron conmetanol caliente, y secados en vacío a 70 0C.
Los ligantes tridentados heteroazometínicosLH3. Los ligantes 11, reportados anteriormente en/18/, fuer sintetizados en la Universidad Estatal deRostov (Rusia), y se utilizaron sin purificaciónadicional. Las electrosíntesis se efectuaronsimilarmente al procedimiento anterior.
C
R1
X H
N
OR
12 13
X = NTs, O, S X = NTs, OR, R1 = H, Alk, Ar
C
R1
X
N
OR
M
N
X
RO
C
R1
C
R1
S M
N
RO
S
N C
OR R1
14
N
Ts
C=N N-C=O
H-OH
11H H
Las condiciones y observaciones en todas las síntesis de los complejos azometínicos se suman en latabla 3.
Oximas. Los ligantes 12 {2-(N-tosilamino)benzaldoxima}(L1H2) y {2-(N-tosilamino)benzal-O-metiloxima (L2H), reportadosanteriormente en /19/, fueron utilizados en laelectrosíntesis de complejos 13 y 14.

75Vol. XIX, Nº 3, 2007
TABLA 3. CONDICIONES Y OBSERVACIONES EN LAS SÍNTESIS DE LOSCOMPLEJOS AZOMETÍNICOS
Ligantes, condiciones Productos, rendimiento (%) observaciones β-Aminovinilcetonas 1, 4
(R=H, Me, Et) Cu, Ni, Co or Zn (ánodo);
metanol, sodio metálico (en caso de 1), 23 oC
2, 3, 5; 28-57 % Usando el ligante 1, su sal sódica se generó in situ para crear la electroconductividad en la celda. Las composiciones de complejos, sintetizados químicamente y electroquímicamente, corresponden a ML2
Ligantes heteroazometínicos bi- y tridentados LH 6-9
Cu, Ni o Zn (ánodo); metanol, 23 oC
N
N
CH 3
C =N
H
15
M /2 X
N
C H = NN
N
R
R = M e , E t R = H , E t
O
C H = NN
N
R
T s
C u /2
O H 2
O H 2 C u /2O H 2
1 6 1 7
85-95 % Una diferencia importante entre la síntesis química y electroquímica de estos complejos: en el primer caso se forman compuestos del tipo ML(OCOCH3), mientras electroquímicamente se forman complejos libres de aniones (es posible presencia de moléculas de agua coordinada)
Ligantes azometínicos tridentados LH2 10
Cu, Ni, Co, Cd or Zn (ánodo); metanol, 23 oC
X
N =C
Y
H
M
L
18L = S o lv , P y
87-96 % Complejos, obtenidos químicamente y electroquímicamente, son similares (la diferencia puede ser debido al número de moléculas de agua coordinada)
Ligantes azometínicos tridentados LH3 11
Cu, Zn (ánodo); metanol, 23 oC
n
N
Ts
N
M1O
O
OM 3
N
M 2N
N
TsN
O M = Co, Cu
19
H3C
SO
O
N
N
Zn
N
OH
O
Zn
O
N
SO
O
CH3
N
OH
N
20
90-95%
La única diferencia de las rutas química y electroquímica es el rendimiento más alto (90�95 %) de la segunda técnica al compararla con la primera (75-80 %)
Oximas 12 Cu, Ni, Zn (ánodo); metanol, 23
oC
13, 14; 83-92 % Complejos de cobre, obtenidos químicamente y electroquímicamente con uso del ligante L1H2, tienen composiciones Cu(L1H)2 y CuL2
2, respectivamente. Para el ligante L2H, en ambos casos se forman los complejos ML2
2 (M=Cu, Ni)

76 Vol. XIX, Nº 3, 2007
Resultados y discusión
Problemas observados durante laelectrosíntesis
Al llevar a cabo reacciones de electrosíntesis,nosotros frecuentemente observamos los siguientesproblemas:
1. Al utilizar metales como cobre y níquel (olantánidos en la obtención electroquímica deftalocianinas) como ánodos, el producto a veces sedeposita en la superficie metálica, resultando unaumento gradual del voltaje y un descenso de lavelocidad de disolución de metal. En este caso, esnecesario parar la electrólisis (ocasionalmente variasveces durante el proceso), y abrir la celda para limpiarla superficie anódica. En el caso del ánodo de cobre,a veces tiene lugar una contaminación del productocon el metal elemental al intentar quitar el productopegado desde la superficie metálica. Por lo tanto, aveces es imposible definir auténticamente la purezadel producto y su composición por análisis elemental.Para evitar este problema, se utiliza una combinaciónde la disolución electroquímica de metales contratamiento ultrasónico simultáneo de la celda. Elultrasonido aquí tiene las siguientes funciones:promueve aceleración de disolución de metal, aumentala transferencia de masa hacía o desde la superficiede electrodos, reduciendo así la duración de reacciones,y optimiza rendimiento químico y electroquímico deproductos.
2. En algunos sistemas con uso de cobre, seobserva una disolución anódica de cobre con susedimentación posterior en la superficie del cátodo deplatino. Esta transferencia de metal del ánodo hacia elcátodo, lleva al rendimiento cero o bajo del productofinal (complejo de cobre). Este fenómeno usualmentetiene lugar cuando la electrólisis se lleva a cabo apotenciales altos y conductividades bajas de solución.
3. En caso de alta solubilidad del producto ensolvente usado, a veces aparece el problema deseparación del producto y ligante-precursor. En estecaso, una evaporación parcial de solvente al vacío oaire (dependiendo de estabilidad de compuestos decoordinación) o �salting out� del complejo formadopuede resolver el problema.
4. Los metales tienen distinta habilidad de disolverseelectroquímicamente, y formar compuestos decoordinación con ligantes en solución. Por ejemplo,cuando el hierro y los solventes se secan muycuidadosamente, con frecuencia se forma una mezclade los hidróxidos de hierro en una cantidad apreciable,en lugar del producto esperado (complejo metálicocon ligante azometínico). En caso de los metalesrefractarios (W, Mo), se necesita HF /2/ o inclusooxidantes fuertes como peróxido de hidrógeno /2/, osales de nitrosonio /1/ para poder llevar a cabo laelectrólisis, de lo contrario, la disolución electroquímicade estos metales es prácticamente imposible o conrendimientos bajos (<3 %). En cualquier caso, eso noes un proceso puro electroquímico, sino una �mezcla�de la disolución química y electroquímica de metal, yel rendimiento electroquímico es difícil de calcularexactamente. Por otra parte, el cobre mismo sedisuelve rápidamente en la solución del disulfuro detetrametiltiuramo en acetonitrilo, aplicando un potenciala este proceso, la velocidad de disolución se aumentaconsiderablemente, en total llevando a la mismasituación: una �mezcla� de los procesos químico yelectroquímico. En general, teóricamente en cualquierproceso de disolución anódica de un metal ensoluciones no acuosas es imposible evitarcompletamente una disolución química del metal: éstasiempre tiene lugar, especialmente, con uso de metalescon baja energía de la celda cristalina, por ejemplocobre.
5. En caso de uso de aleaciones metálicas, cadacomponente de aleación se disuelveindependientemente. Como resultado, el cálculo delrendimiento electroquímico es imposible. De acuerdocon /20/, gracias a la presencia del �gradiente elevadode concentración de átomos metálicos en la superficiede una aleación en comparación con metales enforma individual�, las aleaciones, en especial aquellasque contienen metales químicamente activos (Al, Ca,Sn; sus aleaciones tienen propiedades de compuestosintermetálicos), se disuelven más rápidamente.
6. La electrosíntesis a temperaturas >100 oC (porejemplo, la obtención electroquímica de ftalocianinasa partir de urea y anhídrido ftálico) también tiene suspeculiaridades. En este caso, la celda debe de contenerun condensador para solventes orgánicos, sólidos a

77Vol. XIX, Nº 3, 2007
temperatura ambiente y líquidos >100 oC, por ejemplo,el urea a veces se usa para obtener ftalocianinascomo solvente y precursor a la vez, su punto de fusiónes 135 oC; algunos hidrocarburos son sólidos atemperatura ambiente y líquidos a >100 oC, su medioneutro no afecta a la formación del macrociclo deftalocianina. E uso de plásticos en cables no esdeseable por una posible degradación bajo la acciónde vapores de solventes y compuestos disueltos, quefrecuentemente penetran para afuera en parte noherméticas de la celda. Los procesos de �sublimación� desublimación� de precursores también son posibles(por ejemplo, anhídrido ftálico) y llevan a unaredistribución de reactivos en varias partes del equipo,incluyendo superficie de los electrodos, y afectandoen la agitación. En cualquier caso, la electrosíntesisdirecta es más cómoda en condiciones más suaves, loideal a temperatura ambiente. Al contrario, atemperaturas altas (>100 oC), la preferencia deberáser dada a las técnicas sintéticas clásicas, �purasquímicas�.
7. A veces es necesario de efectuar la electrosíntesisde sustancias inestables (algunos compuestosorganometálicos) a temperaturas <0 oC. En este caso,se necesita uso de solventes deshidratados con bajospuntos de fusión, sin aplicación de corriente alta paraevitar calentamiento del sistema de reacción. Hastaahora, las técnicas de electrosíntesis no están bienadaptadas a temperaturas bajas, por esta razón estaárea es atractiva para investigaciones posteriores. Lamayor parte de los procesos electroquímicos se llevaa cabo a temperatura ambiente /3/.
Entre otras ventajas de la disolución electroquímicade metales en comparación con las técnicas clásicasa partir de sales o carbonilos, hay que mencionar unasola etapa de reacción, rendimientos frecuentementemás altos, así como la posibilidad de obtenercompuestos no accesibles por vías tradicionales. Alutilizar los ligantes azometínicos (con gruposfuncionales OH, SH y NH con el átomo H móvil)como precursores, estas ventajas se observanclaramente en la formación de quelatos. Además, losmetalocomplejos formados no contienen aniones desales.
Además, hay que mencionar la electrosíntesisdirecta con cátodo sacrificial. De acuerdo con /21/,
este método, que prácticamente no se utiliza ahora,fue aplicado sólo para obtener algunos compuestosorganometálicos de Pb, Sn y Hg. Desafortunadamente,los intentos para obtener complejos de ftalocianinas yazometinas de estos metales no han tenido éxitos.
La electrosíntesis de ftalocianinas no sustituidasy sus metalocomplejos
Según los datos de análisis elemental, los productoscorresponden a típicas ftalocianinas {la ftalocianinalibre de metal PcH2 o complejos MPc, M=Cu(II),Fe(II), etc.} /12/. En la síntesis de ftalocianinas libresa partir del ftalonitrilo como precursor, el uso desolventes apróticos (DMSO, Py, dioxano, THF, DMF,acetona, nitrobenceno) no produce PcH2 ni bajocondiciones de la electrosíntesis ni sin éste. La ausenciadel producto se puede explicar por una dificultad delataque nucleofólico por parte del metóxido de sodio algrupo CN del ftalonitrilo, en particular, debido a unainteracción entre el metóxido de sodio y solventeaprótico.
Los resultados más reproducibles se obtienen ensolventes próticos, tales como i-BuOH ydimetiletanolamina con rendimientos casi cualitativos.La síntesis exitosa en medios próticos está de acuerdocon la opinión de los autores /11/ que �un solventeprótico se requiere para la electrosíntesois de PcH2�.El metóxido desoído como fuente de iones alcóxidofavorece al ataque nucleofílico por el grupo ciano delftalonitrilo, para formar 1-alcoxi-3-iminoisoindolinacomo un producto intermedio, con una reducción yciclización posterior.
En contraste con N,N-dimetiletanolamina, lareacción en i-BuOH sin electrólisis no lleva a laformación de PcH2. Este hecho puede serexitosamente utilizado para electrosintetizar variosmetaloftalocianinatos, sincronizando la formación dela ftalocianina en el cátodo y disolución del ánodometálico. Esta ruta puede prevenir formación demezclas de la Pc libre y sus metalocomplejos. LaN,N-dimetiletanolamina también se usa como unsolvente-modelo para electrosintetizar Pc en la fasesólida del ftalonitrilo sin agitación a temperaturasbajas, cuando la formación del macrociclo tiene lugarcerca de la superficie catódica. En este procedimientotambién puede ser utilizado metanol, aunque con

78 Vol. XIX, Nº 3, 2007
rendimientos más bajos (2-8 %); así noo se formanftalocianinas en otros alcoholes.
La electrosíntesis (así como la síntesis química, sinelectrólisis) de la ftalocianina libre de metal a partir dela 1,3-diiminoisoindolina (1,3-D) se lleva a cabo tambiénen solventes apróticos como DMF o DMSO /12/. Essorprendente, que los rendimientos de la ftalocianinaen ROH son comparativamente menores (5-8 %).Entre otros solventes próticos con el grupo OH, en laN,N-dimetiletanolamina, los rendimientos son los másaltos (hasta 92 %).
Evidentemente, la síntesis exitosa química yelectroquímica de Pc a partir de 1,3-D en solventesapróticos, en comparación con el uso de ftalonitrilo,muestra que la influencia más alta de naturaleza delsolvente en la ruta de reacción tiene lugar en laprimera etapa de reacción (formación de 1,3-D).Para reacciones posteriores (ciclización y reducciónde 1,3-D), la naturaleza de solvente ya no tiene muchaimportancia. La formación de Pc a partir de 1,3-Dtiene lugar en todos los solventes utilizados; losrendimientos más altos se pueden lograr por laoptimización de procesos (variación deconcentraciones de 1,3-D, selección del mejor solvente,etcétera).
En caso de los metaloftalocianinatos, laelectrosíntesis se utilizó anteriormente para obtenerPcCu con ánodo de cobre /11, 22/ o CuSO4 /22/;además, algunas sales metálicas (de Ni, Co, Mg, Pb)fueron la parte del sistema de reacción electroquímicautilizando el ftalonitrilo como precursor /22/. Losprocesos electroquímicos en este caso se representancomo sigue (6, 7):
Disolución anódica en soluciones del ftalonitrilo(6):
Cátodo: 4PN + 2e- + 2ROH → PcH2 + 2RO-
Ánodo: M0 – 2e- → M2+
Luego: M2+ + PcH2 → PcM + 2H+
H+ + RO- → ROH
Formación electroquímica de ftalocianinatos apartir de sales metálicas (7):
Cátodo: 4PN + 2e- + 2ROH → PcH2 + 2RO-
Ánodo: 2Cl- - 2e- → Cl2*Luego: M2+ + PcH2 → PcM + 2H+
H+ + RO- → ROH*La coloración del macrociclo formado es posible.
Los intentos de uso de la ftalimida o mezcla de ureay anhídrido ftálico para la preparación electroquímicaen una etapa de Pc no tenían éxito /12/. Sin embargo,en caso del uso del ánodo de cobre, es posible obtenerPcCu con bajos rendimientos (7 %) en tetrametilurea(utilizada como solvente y promotor a la vez; �inerte�en relación al urea y anhídrido ftálico) a partir de losmismos precursores (tabla 1). No se producen PcMcon uso de otros metales tales como Fe, Mg y Sb,aunque los ftalocianinatos de cobre y hierro se formana partir de polvos de los metales mencionados y losprecursores de referencia sin electrólisis conrendimientos en el rango 69-77 %. Por lo tanto,utilizando los precursores dichos, la aplicación de laelectrosíntesis afecta en la ruta de reacción en lugarde aumentar rendimientos.
La conclusión principal acerca de la utilidad de laelectrosíntesis directa de ftalocianinas (tanto libres demetal como metálicos) es la siguiente: sí es posibleobtener PcH2 ó PcM a partir de distintos precursoresen un amplio rango de temperaturas desde 0 hasta190 oC con diferentes rendimientos. Los solventes depreferencia son los alcoholes de bajo peso molecularen caso del ftalonitrilo como precursor, los solventesapróticos en caso de la 1,3-diiminoisoindolina ytetrametilurea en caso de los precursores industriales(urea y anhídrido ftálico). Sin embargo, en el últimocaso la aplicación de electrosíntesis no es una accióndeseable, ya que afecta en la ruta de reacción, por lomenos de acuerdo con la comprensión actual de esteproblema.
Electrosíntesis de ftalocianinas con los gruposfuncionales (ftalocianinas sustituidas). Losresultados del estudio físico-químico de los productosobtenidos electroquímicamente muestran que enalgunos casos sí es posible obtener las ftalocianinas ysus complejos a temperaturas menores de 100 oC.Así, al utilizar el 4-nitroftalodinitrilo como precursor,se forma la tetrakis-(4-nitro)ftalocianina libre de metaly sus complejos con Cu, Ni y Zn. Al emplear el

79Vol. XIX, Nº 3, 2007
vanadio como ánodo sacrificial, no se observanftalocianinas ni en EtOH ni en DMEA. Fue observadovisualmente que comienza la formación de Pc en lasuperficie catódica, pero se termina rápidamente (en2-4 min.) cuando una concentración definida decationes de vanadilo se acumulaba en la solución. Porlo tanto, suponemos que en condiciones de la electrólisis,los iones vanadilo inhiben formación del macrociclo;al mismo tiempo, se sabe que los complejoscorrespondientes de vanadio se obtienen en las síntesisclásicas sin problemas.
Utilizando 4-nitroftalonitrilo, se puede fácilmenteefectuar la electrosíntesis a temperatura ambiente enuna celda no dividida. Comparándola con la ftalocianinano sustituida, cuya obtención es posible en solución atemperatura ambiente en la celda dividida /22/, en lafase sólida cerca de la superficie catódica a 0-25 oCy a >55 oC en solución en una celda no dividida /12/,se observa que la presencia del grupo NO2 en laposición 4 hace uno de los grupos CN del ligando másaccesible para un ataque nucleofílico por el activador(metilato de sodio o DBU = 1,8-diazabiciclo[5.4.0]undec-7-eno), con unaciclotetramerización posterior.
En realidad, 4-nitroftalodinitrilo es uno de losmejores precursores de ftalocianinas, cuyo uso permitela obtención de ftalocianinas con rendimientosrelativamente altos. Se ve, sin embargo, que losrendimientos electroquímicos son menores encomparación con la ftalocianina no sustituida /12/,probablemente debido a la formación simultánea desubproductos de reducción electroquímica del grupoNO2; pero, en cualquier caso, la posibilidad de obtener
electroquímicamente una ftalocianina no sustituida atemperaturas considerablemente menores (<100 oC)en comparación con las técnicas clásicas (>180 oC)parece muy atractiva para investigaciones posterioresen el área de las ftalocianinas.
Como se ve en la tabla 2, sólo tres ftalocianinassustituidas {4-nitroftalodinitrilo, 3-feniltioftalodinitriloy éter tetracianodifenílico (4,4´-oxodiftalodinitrilo)}producen electroquímicamente las ftalocianinascorrespondientes y sus complejos con rendimientossatisfactorios (8-31 %), típicos también para las síntesisclásicas de ftalocianinas sustituidas (10-55 %).
Existen casos, en los que usando ánodos metálicossacrificiales, se observa en los espectros UV (figuras5-8) la formación de ftalocianinas libres de metal o susmezclas con sus metalocomplejos {por ejemplo, en lossistemas Bi (o Ta) - 4-nitroftalodinitrilo, Bi (o Ni) - étertetracianodifenílico}. 4-α- y 3-β-naftoxiftalodinitrilosforman electroquímicamente sólo trazas de productos,como fue observado, por ejemplo, en los espectrosUV de 4-(α-NfO)4-PcHH (figura 8). Utilizando losdemás 9 ftalodinitrilos, no se obtienen ftalocianinascorrespondientes vía la electrosíntesis directa, aunquetodas ellas se forman con uso de técnicas tradicionalesa temperaturas altas.
Los espectros electrónicos de absorción de losmetaloftalocianinatos y ftalocianinas libres de metalcorrespondientes (tabla 4, figuras 5-8) y complejostetra-(4-nitro)Pc y tetra-(4-PhS)Pc, obtenidos por lastécnicas tradicionales, son casi idénticos. Los espectrosIR son típicos para las Pc sustituidas (tabla 4). Losdatos de análisis elemental se presentan en la tabla 5.
Fig. 5Espectro electrónico de absorción de latetrakis-(4-nitro)-ftalocianinato de cobre: línea continua� en ααααα-cloronaftaleno, línea discontinua � en H2SO4.

80 Vol. XIX, Nº 3, 2007
Compuesto Solvente Bandas de absorción λλmax., nm
Bandas de IR νν , cm -1
(en inglés: s � fuerte, w � ancha, vs. � muy
fuerte) 4-(NO 2)4-PcНН α -Cl-naftaleno
H 2SO 4
720, 684, 652, 624, 350
783, 743, 705, 664, 307
740 (s), 1 025, 1 085, 1 345 (vs.), 1 530 (s, benzene rings C-C), 1 700 (s, w), 3 300- 3 000 (w, C-H)
4-(NO 2)4-PcCu α -Cl-naftaleno
H 2SO 4
703, 694, 627, 351
764, 740, 667, ∼410, 305
750 (s), 790 (s), 1 110 (s), 1 170 (s), 1 365 (vs.), 1 550 (s), 1 680
4-(NO 2)4-PcNi α -Cl-naftaleno
H 2SO 4
692, 622, 344
759, 676, ∼400, 308
750 (s), 780, 1 110, 1 180, 1340 (vs), 1 550 (s)
4-(NO 2)4-PcZn α -Cl-naphthalene
H 2SO 4
704, 698, 631, 344
764, 684, ∼410, 308
750 (s), 785 (s), 1 120 (s), 1 355 (vs.), 1 530 (s), 1 640
3-(PhS)4-PcHH CHCl3 738, 709, 676, 643, ∼450, 334
700, 740 (vs.), 800, 890, 1030 (vs.), 1 090, 1 120, 1 229, 1 322 (s), 1 490 (s, C-C de anillos pirólicos), 1 580 (anilos de benceno C-C)
3-(PhS)4-PcCu CHCl3
Triclorobenceno
720, ∼685, 647, 442, 333
724, ∼687, 489, 439, 340
690, 740 (vs.), 798, 902, 1 010, 1 110 (s), 1 140, 1 230 (s), 1 310 (s), 1 460 (s), 1 570 (s, anilos de benceno C-C)
4-[4�,5�-(CN)2-PhO]4PcНН
α -Cl-naftaleno
H 2SO 4
DM F+M eONa
704, 670, 644, 609, 345
870, 812, 770, ∼425, 306
675, 609, 340
740 (s), 840, 960, 1 020 (s, C-O), 1 110 (s), 1 220 (vs.), 1 300, 1 360, 1 480 (vs.), 1 600 (s), 1 710 (s), 2 250, 1 780
4-[4�,5�-(CN)2-PhO]4PcCu
H 2SO 4 818, 728, ∼504, 429, 305
760, 835, 960, 1 050 (C-O), 1 090 (s), 1 225 (vs.), 1 402, 1 459 (s, C-C de anillos de benceno), 1 590 (s), 1 660, 1 720, 1 801, 2 235 (de anillos de benceno C-C), 3 400- 3 300 (w, C-H)
4-[4�,5�-(CN)2-PhO]4PcZn
α -Cl-naftaleno
H 2SO 4
684, 619, 344
812, 734, 429, 309 4-(α-NfO)4-PcНН CH 2Cl2 702, 667, 640, 607,
∼400, 336 3-(β-NfO)4-PcНН CHCl3 722, 690, 660, 625,
∼410, 331 4 95, 755 (s), 1 040 (s), 1 250 (s), 1 340 (s), 1 450 (s), 1 655 (s)
TABLA 4. ESPECTROS ELECTRÓNICOS DE ABSORCIÓN E IR DE LASFTALOCIANINAS SUSTITUIDAS
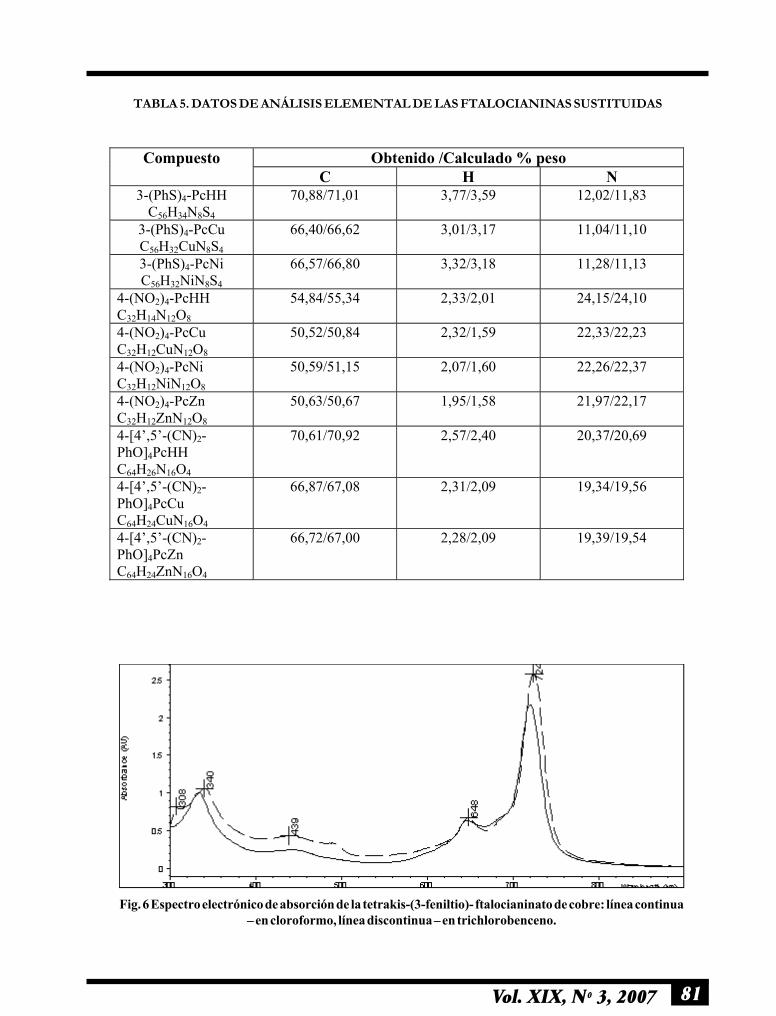
81Vol. XIX, Nº 3, 2007
TABLA 5. DATOS DE ANÁLISIS ELEMENTAL DE LAS FTALOCIANINAS SUSTITUIDAS
Obtenido /Calculado % peso Compuesto C H N
3-(PhS)4-PcHH C56H34N8S4
70,88/71,01 3,77/3,59 12,02/11,83
3-(PhS)4-PcCu C56H32CuN8S4
66,40/66,62 3,01/3,17 11,04/11,10
3-(PhS)4-PcNi C56H32NiN8S4
66,57/66,80 3,32/3,18 11,28/11,13
4-(NO2)4-PcННC32H14N12O8
54,84/55,34 2,33/2,01 24,15/24,10
4-(NO2)4-PcCu C32H12CuN12O8
50,52/50,84 2,32/1,59 22,33/22,23
4-(NO2)4-PcNi C32H12NiN12O8
50,59/51,15 2,07/1,60 22,26/22,37
4-(NO2)4-PcZn C32H12ZnN12O8
50,63/50,67 1,95/1,58 21,97/22,17
4-[4�,5�-(CN)2-PhO]4PcННC64H26N16O4
70,61/70,92 2,57/2,40 20,37/20,69
4-[4�,5�-(CN)2-PhO]4PcCu C64H24CuN16O4
66,87/67,08 2,31/2,09 19,34/19,56
4-[4�,5�-(CN)2-PhO]4PcZn C64H24ZnN16O4
66,72/67,00 2,28/2,09 19,39/19,54
Fig. 6 Espectro electrónico de absorción de la tetrakis-(3-feniltio)- ftalocianinato de cobre: línea continua� en cloroformo, línea discontinua � en trichlorobenceno.

82 Vol. XIX, Nº 3, 2007
Como conclusiones de la parte dedicada a lasftalocianinas sustituidas, se puede confirmar que lasPc sustituidas y sus complejos metálicos sí se obtienenelectroquímicamente en condiciones suaves a 25-130oC en medio de alcoholes vía electroquímica, a partirde 3-feniltioftalodinitrilo, 4-α-naftoxiftalodinitrilo, 4-nitroftalodinitrilo, 3-β-naftoxiftalodinitrilo y étertetracianodifenílico (4,4´-oxodiftalodinitrilo) comoprecursores.
Las composiciones y propiedades de los productosformados corresponden a aquellos sintetizados conuso de métodos clásicos a temperaturasconsiderablemente más elevadas (>160 oC). Algunos
metales utilizados (Bi, Ta) no producen ftalocianinasen condiciones electroquímicas. Se observa el efectode inhibición en la tetramerización de 4-nitroftalodinitrilopor los catones de vanadilo, cuando el vanadio seutiliza como ánodo sacrificial. Se muestra que esrealmente posible efectuar la electrosíntesis atemperaturas �bajas� (20-130 oC vs. técnicastradicionales a >170 oC), aunque con rendimientosbajos (8-31 %); la posibilidad de la electrosíntesisdepende de propiedades donador-aceptor del grupofuncional y su posición.
β-Aminovinilcetonas. En la electrosíntesis seutiliza una serie de ligantes /13, 15/ derivados de β-
Fig. 7 Espectro electrónico de absorción de 4-[4�,5�-(CN)2-PhO]4PcCu en H2SO4.
Fig. 8 Espectro electrónico de absorción de 4-(ααααα-NfO)4-PcHH: línea continua � en clorurode metileno, línea discontinua � en benceno.
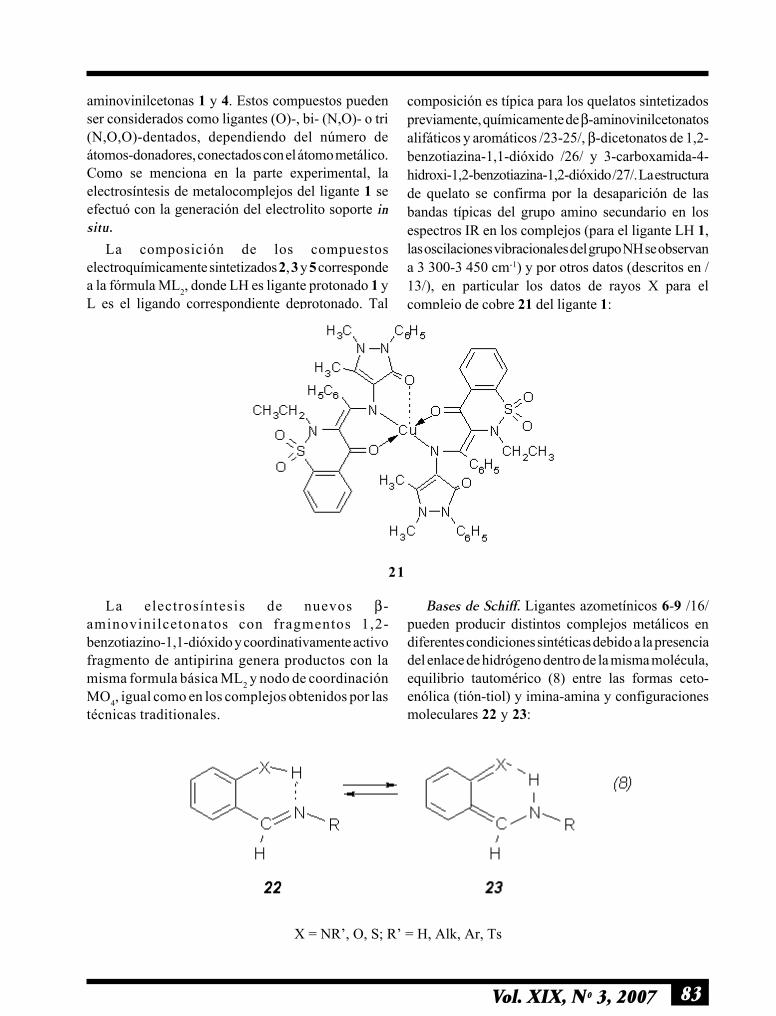
83Vol. XIX, Nº 3, 2007
aminovinilcetonas 1 y 4. Estos compuestos puedenser considerados como ligantes (O)-, bi- (N,O)- o tri(N,O,O)-dentados, dependiendo del número deátomos-donadores, conectados con el átomo metálico.Como se menciona en la parte experimental, laelectrosíntesis de metalocomplejos del ligante 1 seefectuó con la generación del electrolito soporte insitu.
La composición de los compuestoselectroquímicamente sintetizados 2, 3 y 5 correspondea la fórmula ML2, donde LH es ligante protonado 1 yL es el ligando correspondiente deprotonado. Tal
composición es típica para los quelatos sintetizadospreviamente, químicamente de β-aminovinilcetonatosalifáticos y aromáticos /23-25/, β-dicetonatos de 1,2-benzotiazina-1,1-dióxido /26/ y 3-carboxamida-4-hidroxi-1,2-benzotiazina-1,2-dióxido /27/. La estructurade quelato se confirma por la desaparición de lasbandas típicas del grupo amino secundario en losespectros IR en los complejos (para el ligante LH 1,las oscilaciones vibracionales del grupo NH se observana 3 300-3 450 cm-1) y por otros datos (descritos en /13/), en particular los datos de rayos X para elcomplejo de cobre 21 del ligante 1:
21
La electrosíntesis de nuevos β-aminovinilcetonatos con fragmentos 1,2-benzotiazino-1,1-dióxido y coordinativamente activofragmento de antipirina genera productos con lamisma formula básica ML2 y nodo de coordinaciónMO4, igual como en los complejos obtenidos por lastécnicas traditionales.
Bases de Schiff. Ligantes azometínicos 6-9 /16/pueden producir distintos complejos metálicos endiferentes condiciones sintéticas debido a la presenciadel enlace de hidrógeno dentro de la misma molécula,equilibrio tautomérico (8) entre las formas ceto-enólica (tión-tiol) y imina-amina y configuracionesmoleculares 22 y 23:
X = NR�, O, S; R� = H, Alk, Ar, Ts

84 Vol. XIX, Nº 3, 2007
Este equilibrio lleva a la formación de dos tipos de compuestos de coordinación � aductos 24 y quelatos25 con la molécula azometínica deprotonada:
Una comparación de complejos obtenidoselectroquímicamente de los ligantes 6-9 concomplejos-análogos, sintetizados en rutas �purasquímicas�, revela las siguientes diferenciasimportantes. Los complejos de Co, Zn y Cd con elligante 6 tienen composición ML2 ⋅ nH2O 15 (n= 0,1, 2, 5) formando los ciclos de 5 miembros, mientras
que los complejos electroquímicamente obtenidosde Cu, Ni y Pd con ligante similar 26-27 (existiendoen dos formas tautoméricas) correspondena la fórmula general MLAc ⋅ nH2O 28 (n =0, 1, 2).Éstos también contienen el anión acetato, queestá ausente en el producto sintetizadoelectroquímicamente.
Los ligantes 7 y 8 fueron utilizados comoformadores de quelatos con 6 miembros en el ciclo. Elligante 7 químicamente forma quelato de cobreCuLAc.H2O. La electrosíntesis con uso de 7 y 8permite obtener quelatos CuL2 ⋅ 2H2O 16, NiL2 yCuL2 ⋅ H2O 17. La esfera de coordinación del átomode cobre es saturada por la deprotonación de losgrupos o-tosilamino- ó o-hidroxo, y conservación deaniones acetato (síntesis química), o grupos OH(electrosíntesis). El ligante 9 con el grupo o-tosilamino,que no contiene un fragmento heterocíclico, formatanto químicamente como electroquímicamente losmismos productos ML2 /16/. Como se ve, la naturalezade metal y ligando, así como de la técnica sintética
aplicada, influye en la composición de productosfinales de los ligantes de referencia.
Ligantes azometínicos tridentados LH2. Enesta parte de trabajo, se utilizan los ligantestridentados 10 (H2L {2-N-tosilamino(2´-hidroxibenzal)anilina} y H2L1 {2-N-tosilamino(2´-tosylaminobenzal)anilina} /17/. Ya que H2L y H2L
1
disocian en dos etapas según el esquema H2L(H2L1)
« 2H+ + L2-(L1(2-)), las reacciones (9) en la celdapueden ser descritas como sigue /17a/:
Cátodo: H2L + 2e → H2 + L2- (9)Ánodo: M + L2- → ML + 2e

85Vol. XIX, Nº 3, 2007
En la base de este ligante, las síntesis tradicionalespueden llevar a los complejos monoméricos 18 [28,29]y diméricos 29 /30/:
Los complejos obtenidos electroquímicamentetienen composición general ML(L1) ⋅ nCH3OH oM2L2(L
12) ⋅ nCH3OH (n= 1, 2) y, en la base de
estudios realizados físico-químicos /17a/, la estructura18 (L=CH3OH).
Los complejos de los mismos metales, obtenidosquímicamente y electroquímicamente, son similares(las diferencias pueden ser relacionadas al número demoléculas solvatadas de metanol). Según /17b/, laformación de complejos ML ⋅ CH3OH se correlacionacon la inestabilidad de las moléculas diméricas 29,causada, en este caso, por la influencia estérica de losfragmentos NTs y moléculas-donadores de metanol,reacción (10):

86 Vol. XIX, Nº 3, 2007
Ligantes azometínicos tridentados LH3. Lasalicilhidrazona de 2-(N-tosilamino)benzaldehido11 /18/ existe en dos formas tautoméricas
3031; este equilibrio también es típico paraotros ligantes azometínicos, donde prevalece laforma izquierda 30:
El ligante 11 forma varios tipos de complejos,incluyendo estructuras bi- y trinucleares /31/. Ennuestros experimentos, se aislan los compuestosde coordinación [Cu3L2]n 19 (M = Cu) y [Zn(LH)]220 (LH3=11), tanto química comoelectroquímicamente. En caso del complejo decobre 19, tiene lugar deprotonación completa delsistema de ligante LH3 (todos los tres átomos H sedesplazan con el metal), mientras tanto, en elcomplejo de cinc 20 un átomo H se queda en lamolécula. En total, para este sistema de ligando, ladiferencia entre las técnicas química yelectroquímica es solo rendimiento más alto delproducto en caso de la electrosíntesis (90-95 %),comparándolo con el método clásico (75-80 %).
Oximas . Entre varias oximas, nosotrosseleccionamos los quelatos metálicos de 2-(N-tosilamino)benzaldoxima L1H2 (12, X=NTs,R=R1=H) y su derivado O-metílico L2H (12, X=NTs,R=Me, R1=H) /19/, los cuales no fueron obtenidosanteriormente por vía electroquímica.
Entre otros tipos de complejos /32, 33/, los mástípicos de ligantes 12 son complejos monoméricostrans-planares 13 (X = O) /34/ y cis-planares 14(X = S) /35/. En adicional a estructuras trans-planares s (M = Pd) /36/, la estructura octaédricadistorcionada se forma en los complejos 13 graciasa la coordinación intermolecular M-O (M= Cu) /37, 38/. Además, los aductos moleculares 32 puedenser aislados utilizando haluros metálicos MHaln enmedios no acuosos /39/:
Como resultado de nuestras investigaciones, loscomplejos ML1 (M=Cu) se forman químicamente apartir del ligante L1H2 y cloruro o acetato de cobre,mientras la electrosíntesis directa lleva a loscompuestos M(L1H)2. Sin embargo, utilizando el liganteL2H, se obtienen los complejos ML2
2 (M=Cu, Ni),tanto química como electroquímicamente. Loscomplejos de cobre Cu(L1H)2 y CuL2
2 (N=NTs)poseen la estructura tetraédrica.
La conclusión principal de esta parte del trabajo decomparación de la técnica química clásica yelectroquímica, es que la sustitución de la primera porla segunda, permite la obtención de complejosazometínicos teniendo distinta composición. De estamanera, en caso de oximas, los complejos de cobre yligantes 12 (X=NTs, R=R1=H; X=NTs, R=Me, R1=H),obtenidos química- y electroquímicamente, tienen

87Vol. XIX, Nº 3, 2007
composiciones Cu(L1H)2 y CuL22, respectivamente,
y propiedades diferentes. Dependiendo de la naturalezadel metal, los sistemas de ligantes pueden portarsecomo sistemas ambidentados, formando, en caso deβ-aminovinilcetonas 1, los quelatos metálicos conestructuras tetraédricas (Cu+2 y Zn+2) o octaédricas(Co+2 y Ni+2). Al mismo tiempo, ambas técnicaspueden dar productos idénticos o similares, por ejemplocomplejos de los ligantes 1, 4 y 9. Además, lasventajas principales del método electroquímico sonaltos rendimientos (80-96 %) y ausencia de anionesde sales en los productos de reacciones (los anionesse presentan frecuentemente en quelatos de ligantestridentados de heteroarenos) /4/.
Optimización de la electrosíntesis con uso deltratamiento ultrasónico. Adicionalmente a lasventajas mencionadas arriba, la electrosíntesis directaposee desventajas tales como, formación de productossólidos insolubles en las superficies anódicas, enespecial las de cobre en soluciones de ligantesazometínicas, o las de lantánidos en soluciones noacuosas de ftalonitrilo /40/.
Observando el comportamiento de varios metales,se anota que la naturaleza de metal (particularmentedeterminando la solubilidad de sus complejos) tiene elpapel primordial en la adhesión de productos dereacción a la superficie anódica. Por ejemplo, cuandolas demás condiciones son iguales (los mismos ligandosazometínicos, solventes, temperatura, tiempo dereacción, voltaje y corriente, área de superficie de loselectrodos y distancia entre ellos, volumen de solución,etcétera), los complejos de níquel que son un pocomás solubles en comparación con los complejossimilares de cobre, no forman una capa estable en lasuperficie anódica, y se acumulan en el fondo de lacelda. Una pequeña cantidad del producto que todavíase queda en la superficie anódica se elimina fácilmentede manera mecánica. Además, las partículas metálicasde níquel no se eliminan desde el ánodo junto con elproducto, como en caso de cobre.
Para resolver tales problemas, se usa unacombinación de la disolución electroquímica de metalesy tratamiento ultrasónico simultáneo (figura 4). Sesabe muy bien, que el ultrasonido es un factor adicionalacelerando la disolución de metales en medios noacuosos /41-43/.
En caso del uso de ftalonitrilo (como precursor enla formación templete de la ftalocianina) y lantánidos(La, Nd, Pr y Sm) como ánodos, el uso de ultrasonidono solamente es deseable, pero realmente unprocedimiento necesario debido a serias dificultadesal intentar utilizar estos metales como ánodos /40/. Adiferencia del cobre, níquel y otros metales detransición, el uso de algunos metales de transicióninterna (lantánidos), incluso en forma bien pulida, llevaa la formación de una capa estable en la superficieanódica, que causa un aumento brusco del voltaje enla celda.
La electrosíntesis con lantánidos se lleva a cabo a100 0C en i-BuOH durante 2 h bajo, tratamientoultrasónico �débil� {reacciones electródicas (11)}.
Cátodo: 4C6H4(CN)2 + 2e → PcH2
Ánodo: Ln – 3e → Ln3+
Luego: 2Ln3+ + 3PcH2 → Ln2Pc3 + 6H+
Los rendimientos máximos (80-90 %) /44/ de los�super-complejos de tri-capa� Ln2Pc3 (encomparación con los compuestos XLnPc y LnPc2X)se logran (en la síntesis química) cuando la proporción�ftalonitril : sal metálica� (LnCl3, M=La, Sm, Gd, Tmy Lu) es 6:1. En nuestros experimentos, los rendimientosson de alrededor 95 % (con tratamiento ultrasónico)ó 35-50 % (sin éste), en el último caso, acompañándosecon las dificultades arriba mencionadas. Según /12/,no se forman ftalocianinas en los sistemas �i-BuOH-CH3ONa-(n-Bu)4NBr-ftalonitrilo� sin electrólisis a100 oC; aunque en otros alcoholes, tales procesospueden tener lugar formando (en caso del uso deánodos metálicos) una mezcla de la ftalocianina librey ftalocianinato metálico. Por lo tanto, precisamenteeste solvente es seleccionado para sincronizar ladisolución anódica de metal y formación de laftalocianina libre en la superficie catódica, evitandoasí la obtención de una mezcla �PcH2-Ln2Pc3�.Mostrando una diferencia clara de las técnicastradicionales para obtener ftalocianinatos de elementosde tierras raras a 170-290 oC /44, 45/, la electrosíntesis/46, 47/ permite reducir la temperatura hasta 100 oC.
Utilizando los ligantes heteroazometínicos 6-9 envarios solventes no acuosos, el problema de eliminacióndel precipitado insoluble desde el ánodo es, en general,
(11)

88 Vol. XIX, Nº 3, 2007
resuelto. Aplicando débiles fuentes de ultrasonido, lamayor parte de sólidos se quita del ánodo. Claro queel voltaje se aumenta gradualmente a lo largo deelectrólisis, pero no tan rápido como en caso deftalocianinas y lantánidos. Los rendimientos en casodel tratamiento ultrasónico son más altos (en 2-5 %)que sin éste. En los experimentos de control bajo, eltratamiento ultrasónico con el mismo metal, ligante yotras condiciones, pero sin electrólisis, para algunosligantes azometínicos se observa una disolución parcialde cobre (2-4 % del peso de metal) y menos la pérdidadel peso de níquel (0,5-0,8 %) formando los mismoscomplejos, debido a la disolución de metal �puraquímica�.
Observaciones finales
Sobre la base de estos y numerosos otrosexperimentos realizados con varios metales y ligantesque contienen nitrógeno y azufre como átomosdonadores, sin y con aplicación del tratamientoultrasónico simultáneo junto con la electrosíntesisdirecta, se ha concluido que la posibilidad de agravaciónde condiciones de electrólisis (inestabilidad de voltajeen la celda electroquímica) depende primero de lanaturaleza del metal y ligante y, respectivamente, depropiedades de los productos formados y menos denaturaleza del solvente (acetonitrilo, alcoholes o susmezclas con tolueno).
Conclusiones
Comparando los metales, generalmenteutilizados en la electrosíntesis (Cu y Ni), se puedeconcluir que la disolución electroquímica de níquelse lleva a cabo mucho más fácilmente,evidentemente debido a la solubilidad más alta desus metalocomplejos en solventes no acuosos, encomparación con los complejos similares de cobre.El uso del ultrasonido permite nivelar estadiferencia, efectuando la electrosíntesis sin lospoblemas mencionados. A veces (uso delantánidos), la aplicación del ultrasonido es parteobligatoria y necesaria del trabajo.
La electrosíntesis directa como una técnicanovedosa, en los últimos años se encuentra endesarrollo permanente /46 ,48-52/. En relación
con los ligandos y complejos estudiados en estetrabajo, el procedimiento electroquímico tiene tantoventajas como desventajas, en comparación conlos métodos clásicos y poco utilizados /47/. Desdenuestro punto de vista, para su mejorfuncionamiento, se requiere de una comprensiónmás profunda de los mecanismos con laparticipación de estos /53/ y otros metalocomplejos/54/, y sus precursores en condiciones de laelectrólisis.
Agradecimientos
Los autores agradecen mucho al CONACYT(proyecto 39,558-Q) y PAICyT-UANL, por elapoyo económico brindado a este trabajo.
Bibliografía1. Davies, J. A., Hockensmith C. M., Kukushkin V. Yu.,
Kukushkin, Yu. N., Synthetic Coordination Chemistry:Principles and Practice. Syngapore, London: World Scientific.,1992, 452 p.
2. Chakravorty, M. C., Subrahmanyam, G. V. P.Electrosynthesis of coordination compounds by thedissolution of sacrificial metal anodes. Coord. Chem. Rev.V.135-136, 1994, págs. 65-92.
3. Garnovskii, A.D., Kharisov B.I., Direct Synthesis ofCoordination and Organometallic Compounds, Eds.Amsterdam-Lausanne-New York-Oxford-Shannon-Singapore-Tokyo: Elsevier Science., 1999, 244 págs.
4. _______, Synthetic coordination & organometallic chemistry,Eds. New York, Basel: Marcel Dekker., 2003, 512 págs.
5. Garnovskii, A.D., Blanco L.M., Kharisov B.I., GarnovskiiD.A., Burlov A.S. Electrosynthesis of Metal Complexes:State of the Art. J. Coord. Chem. 48, 1999, págs. 219-263.
6. Grobe, I., Electrochemical Synthesis of Metal Complexes andHomogeneous Catalysts. Comments Inorg. Chem. 9(3), 1990,págs. 149-179.
7. Electroorganic synthesis (Little R.D., Weinberg N.L., Eds.).New York, Basel: Marcel Dekker., 1991, 472 p.
8. Lehmkuhl, H. Preparative Scope of OrganometallicElectrochemistry. Synthesis. 7, 1973, págs. 377-396.
9. Grobe, J., Keil M., Schneider B., Zimmermann, H.,Electrochemical Synthesis. II. Theoretical Aspects of theElectrochemical Synthesis of Complexes. Z. Naturforsch. B.35, 1980, págs. 428-432.
10.Compton, R. G., Eklund, J. C., Page, S. D., Rebbit, T. O.,Sono-Electrochemistry: the Oxidation ofbis(Cyclopentadienyl)Molybdenum Dichloride. J. Chem.Soc., Dalton Trans. (3), 1995, págs. 389-393.
11.Petit, M. A., Plichon, V., Belkacemi, H. Electrosynthesis ofPhthalocyanines, New J. Chem. 13(6), 1989, págs. 459-462.

89Vol. XIX, Nº 3, 2007
12.Kharisov, B. I., Blanco, L. M., Torres-Martínez, L. M.,García-Luna, A., Electrosynthesis of Metal Phthalocyanines:Influence of Solvent. Ind. Eng. Chem. Res. 38(8), 1999, págs.2880-2887.
13.Garnovskii, A. D., Kharisov, et al., New β-aminovinylketonates with annealated 1,2-benzothiazine-1,1-dioxide fragment. Polyhedron. 23(11), 2004, págs. 1909-1914.
14.Méndez-Rojas, M. A., Cordova-Lozano, F., Gójon-Zorrilla,G., González-Vergara, E., Quiroz, M. A. DirectElectrosynthesis of Cu, Cd, Zn Complexes of Piroxicam (4-Hydroxy-2-Methyl-N-(2-Pyridyl)-2H-1,2-Benzothiazine-3-Carboxamide-1,1-Dioxide) and Isoxicam (4-Hydroxy-2-Methyl-N-(5-Methyl-3-Isoxazolyl)-2H-1,2-Benzothiazine-3-Carboxamide-1,1-Dioxide) in Nonaqueous Media by in-situ Generation of Supporting Electrolyte. Polyhedron.18(20), 2651-2658, 1999.
15.Bicherov, A.V., Kharisov, et al. Metal chelates of NewLigands: 1,2-Benzothiazine-1,1-Dioxide Derivatives. J.Coord. Chem. 54, 337-342, 2001.
16.Kharisov, B. I., et al., Synthesis of Transition MetalComplexes with Heteroazomethinic Ligands: A Comparisonof Traditional and Electrochemical Methods. Polyhedron.17(2-3), 1998, págs. 381-389.
17.a) Kharisov, B. I., et al., Direct Electrochemical Synthesis ofNovel Transition Metal Complexes of Tridentate AzomethinicLigands. Polyhedron. 18(7), 985-988 (1999); b) Burlov, A. S.,et al., Synthesis and Spectral-Luminescent Properties ofTridentate o-Tosylaminoazomethines and Their Zinc andCadmium Complexes. Koord. Khim. 24(12), 1998, págs. 915-918.
18.Blanco, L. M., et al., Direct Electrochemical Synthesis of theChelates of a Novel Ligand: Salicylhydrazone of 2-(N-tosylamino)Benzaldehyde. Current Separations. 18(2), 1999,págs. 41-46.
19.Burlov, A. S., et al., Conventional Chemical and DirectElectrochemical Synthesis of the Chelates of 2-(N-Tosylamino)Benzaldoxime and 2-(N-Tosylamino)Benzal-O-Methyloxime. J. Coord. Chem. 47, 1999, págs. 467-478.
20.Kubiak, R., Janszak, J., A Simple, Novel Method for thePreparation of Metallophthalocyanines, J. Alloys &Compounds, 200, 1993, L7-L8.
21.Tuck, D. G., Direct Electrochemical Synthesis of Inorganicand Organometallic Compounds. Pure & Appl. Chem. 51,1979, págs. 2005-2018.
22.Yang, C. H., Lin, S. F., Chen, H. L., Chang, C. T.,Electrosynthesis of the Metal Phthalocyanine Complexes,Inorg. Chem. 19, 1980, pág. 3541.
23.Holm, R. H., O�Connor, M. J., Stereochemistry of Bis-chelate Metal(II) Complexes, Progr. Inorg. Chem. 14, 1971,págs. 214-401.
24.Tung, Y.-L., Tseng, W.-C., Lee, C.-Y., Hsu P.-F., Chi Y., PengS.-M., Lee G.H. Synthesis and Characterization of Allyl(?-Ketoiminato)Palladium(II) Complexes: New Precursors forChemical Vapor Deposition of Palladium thin Films.Organometallics. 18(5), 1999, págs. 864-869.
25.Garnovskii, A. D., Nivorozhkin, A. L., Minkin, V. I., LigandEnvironment and the Structure of Schiff Base Adducts andTetracoordinated Metal-Chelates. Coord. Chem. Rev. 126(1-2), 1993, págs. 1-69.
26.Korshunov, O. Yu., Bicherov, A. V., Garnovskii, A. D., NewAmbidentate Ligands Derivatives of 1,2-Benzothiazine-1,1-Dioxide. Koord. Khim. 26(12), 2000, págs. 949-950.
27.Cini, R., Giorgi, G., Guantini, A., Rossi, C., Sabat, M., MetalComplexes of the Antiinflammatory Drug Piroxicam., Inorg.Chem. 29(26), 1990, págs. 5197-5200.
28.Holm, R. H., Everett, G. W., Chakravorty, A., MetalComplexes of Schiff Bases and ß-Ketoamines, Progr. Inorg.Chem. 7, 1966, págs. 83-214.
29.Maggio, F., Pizzino, T., Romano, V., Complexes of SomeFirst Series Transition Metals Formally Tricoordinated,Inorg. Nucl. Chem. Lett. 10(11), 1974, págs. 1005-1008.
30.Garnovskii, A. D., Vasilchenko, I. S., Russ. Chem. Rev. 71,2002, pág. 943.
31.Biradar N.S., Mahale V.B., Havinale B.R. Four and sixcoordinate complexes of zinc(II) with aryl hydrazones. Rev.Roum. Chim. 25(1), 55-61 (1978).
32.Chakravorty, A., Structural Chemistry of Transition metalComplexes of Oximes, Coord. Chem. Rev. 13(1), 1974, págs.1-46.
33.Kukushkin, N. Y., Tudela, D., Pombeiro, A.J. L., Metal-IonAssisted Reactions of Oximes and Reactivity of Oxime-Containing Metal Complexes, Coord. Chem. Rev. 156, 1996,págs. 333-362.
34.Keeney, M. E., Osseo-Assere, K., Woode K. A., TransitionMetal hydroxyoxime Complexes, Coord. Chem. Rev. 59,1984, págs. 141-201.
35.Vasilchenko, I. S., First Representative ofThiosalicylidenoximates: Synthesis and Molecular Structureof Bis[Thiosalicylidene(O-Methyl)Oximato]-Nickel(II).Zhurn. Neorg. Khim. 37(5), 1992, págs. 1047-1051.
36.Pfeuger, C. E., Harlow, R. L., The Crystal and MolecularStructure of Bis(Salicyl-Aldoximato)Palladium(II), ActaCryst. 26B(10), 1970, págs. 1631-1633.
37.Jarski, M. A., Lingafelter, E. S., The Crystal Structure ofBis(Salicylaldoximato)-Copper(II), Acta Cryst. 17(9), 1964,págs. 1109-1112.
38.Orioli, P. L., Lingafelter, E. S., Brown, B. W., The CrystalStructure of Bis(5-Chlorosalicylaldoximato)Copper(II), ActaCryst. 17(9), 1964, págs. 1113-1118.
39.Garnovskii, A. D., Type-Controlled Synthesis of CoordinationCompounds, Koord. Khim. 18(7), 1992, págs. 675-698.
40.Kharisov, B. I., Blanco, L. M., García-Luna, A., DirectElectrochemical Synthesis of Metal Complexes. LanthanidePhthalocyanines: Optimization of the Synthesis. Rev. Soc.Quím. Méx. 43(2), 1999, págs. 50-53.
41.Luche, J.-L., Cintas, P., Ultrasound-Induced Activation ofMetals: Principles and Applications in Organic Synthesis. In:Active Metals (Fuerstner A., Ed.). Weinheim: VCH. 1996,págs. 133-190.

90 Vol. XIX, Nº 3, 2007
42. Ultrasound: its Chemical, Physical, and BiologicalEffects (Suslick K.S., Ed.), New York: VCH. 1988, pág.336.
43. Kharisov, B. I., Blanco, L. M., Salinas, M. V.,Garnovskii, A. D., Direct Electrochemical Synthesis ofCopper Dimethyldithiocarbamate: an Optimization UsingUltrasonic Treatment. J. Coord. Chem. 47, 1999, págs.135-143.
44. Sokolova, T. N., Lomota, T. N., Morozov, V. V.,Berezin, B. D., Complex Compounds of Lanthanides withPhthalocyanine: Double Sandwich., Koord. Khim. 20(8),1994, págs. 637-640.
45. Kirin, I. S., Moskalev, P. N., Ivannikova, I. V., AboutNew complex compounds of phthalocyanine with rare-earth elements. Zhurn. Neorg. Khim. 12(3), 1967, págs.707-712.
46. Kharisov B.I., Garza-Rodríguez L.A., Méndez-RojasM.A., Blanco L.M. Técnicas para la preparación deftalocianinas. Ingenierías. VII(22), 2004, págs. 71-84.
47. Kharisov B.I., Ortiz Méndez U., Rivera de la Rosa, J.Low-temperature synthesis of metal phthalocyaninates.Russ. J. Coord. Chem. 32(9), 2006, págs. 643-658.
48. Sánchez Vergara, M. E., Islas Bernal, I. F., Rivera, M.,Ortíz Rebollo, A., Alvarez Bada, J. R., Formation andCharacterization of thin Films from PhthalocyanineComplexes: An Electrosynthesis Study Using the Atomic-
Force Microscope. Thin Solid Films. 515(13), 2007, págs.5374-5380.
49. Skompska, M., et al. Electrosynthesis and Propertiesof Poly(3,4-Ethylenedioxythiophene) FilmsFunctionalized with Titanocene Dichloride Complex,Electrochimica Acta, 51(11), 2006, págs. 2108-2119.
50. Antiñolo, A., et al. New Reactivity of Cp�2NbHs, Cp�= h5-C5H4SiMe3. Synthesis, Electrosynthesis andReactivity of New Carboxylato Niobocene Complexes, J.Organomet. Chem., 690(13), 2005, págs. 3134-3141.
51. Budnikova Yu.H., Tazeev D.I., Trofimov B.A.,Sinyashin O.G. Electrosynthesis of nickel phosphides onthe basis of white phosphorus. Electrochem. Comm. 6(7),2004, págs. 700-702.
52. E. Duñach, A. P. Esteves, M. J. Medeiros, D. Pletcher,S. Olivero, The Study of Nickel(II) and Cobalt(II)Complexes with a Chiral Salen Derivative as Catalysts forthe Electrochemical Cyclisation of Unsaturated 2-Bromophenyl Ethers. J. Electroanal. Chem., 566(1), 2004,págs. 39-45.
53. Aravindakshan, A., Copper Phthalocyanines. Website http://www.specialchem4coatings.com/resources/articles/article.aspx?id=2479, 2007.
54. Berezin, B. D., Lomova, T. N., Reactions ofDissociation of Complex Compounds. Moscow: Nauka,2007.


















