
Revista nueva biologia c1
20
-
Upload
leticia-gibiaqui -
Category
Documents
-
view
228 -
download
0
description
Férnandez Lianeth Gómez Gabriel/Sánchez José G. Gónzalez Susan Bolivar Jetsimar Carreira Yuismary Gónzalez Keyri Gil Ángel
Transcript of Revista nueva biologia c1



La revista Curiosidades biológicas es una obra colectiva concebida, elaborada y diseñada por los alumnos de 5to año sección “C” del Colegio Parroquial “Jesús María Marrero” bajo la dirección pedagógica de la profesora Leticia Gibiaqui. Dirigida para estudiantes cursantes de 4to y 5to año de educación media y de ahí en adelante para personas con interés relacionados con el mismo.
Texto
Férnandez Lianeth – Artc. Heterotomía.
Gómez Gabriel/Sánchez José G. – Artc. Pectux excavatum.
Gónzalez Susan – Artc. Síndrome de Klippel Feil.
Bolivar J, Carreira Y, Gil A, Gónzalez K - Artc. Fibrodiplasia.
Edición
Bolivar Jetsimar
Carreira Yuismary
Gónzalez Keyri
Correción de estilo
Gil Ángel
2015 by Alumnos cursantes del 5to año sección “C”, C. P J. M. M.
Editado por Bolívar Jetsimar, Carreira Yuismary, Gónzalez Keyri.
PRIMERA EDICIÓN: 2015.
Venezuela, Miranda. Calle Arismendi, Colegio Parroquial “Jesús María Marrero”

Un mutación es un cambio en la secuencia o en la organización del ADN.
Las mutaciones cromosómicas son más comunes que las genómicas y
normalmente no son transmitidas a la descendencia porque sobrevivir con
una de estas mutaciones es casi imposible
Toda la información sobre los caracteres, estructura y
función del organismo de los seres vivos se encuentran
en los genes, dentro de los cromosomas, en forma de
ADN. En la fecundación, dos juegos simples de
cromosomas, uno de cada progenitor, se unen para
formar parte de la información genética de cada
individuo; es lo que se conoce como genoma. Si
durante este proceso se produce alguna mutación,
puede ocurrir lo que denominamos síndrome genético.
En muchos casos, el origen se debe a que uno o ambos
padres son portadores de una alteración genética que
es susceptible de ser transmitida a los hijos. En otros
muchos casos se desconocen las causas exactas por la
que un síndrome genético se da en un individuo
concreto sin haber antecedentes familiares.
durante la embriogénesis por una mutación heredada o
no, de otras patologías que pueden aparecer durante
el desarrollo del feto y que también conducen a
Los síndromes dentro de las Mutaciones Cromosómicas y nuestra vida

Es una anomalía de los ojos en la que el iris es de diferente color, también puede llegar a afectar a la piel o el cabello, pero el caso
más común es en los ojos, total o parcialmente. Los ojos pueden ser de
colores distintos o una sección del iris es distinta al resto en ambos ojos. La
heterocromía ocurre cuando una persona o un animal tienen demasiada o muy poca
Melanina en el cuerpo, este es un compuesto que crea pigmento y se encuentra en animales y plantas.
HETEROCHROMIA
IRIDUM

CAUSAS
La heterocromía puede ser causa de una
enfermedad, una lesión, un mosaicismo
genético, o un rasgo genético heredado.
Los ojos de diferente color también
pueden ocurrir debido a una hemorragia
o un objeto extraño en el ojo, un
glaucoma o algunos medicamentos para
tratarla. Incluso una leve inflamación
del ojo puede causar esta condición.

Tipos de Heterocromía
Heterocromía Completa y Heterocromía
Parcial.
La heterocromía completa es más común en los
gatos, aunque puede afectar a los seres
humanos. Con mayor frecuencia, el gato tiene
un ojo azul y un ojo normal. También ocurre
entre los perros, sobre todo en las razas Husky
Siberiano y de Dalmacia, algunos caballos,
vacas, búfalos y algunos hurones. La
heterocromía parcial es más común en los
perros de razas.

La heterocromía central aparece cuando la parte central de la pupila del iris es de un
color diferente al de la parte media-periférica o ciliar, formando un anillo central
alrededor de la pupila del ojo. Este tipo es más común en los diafragmas que contienen
bajos niveles de melanina. El verdadero
color es en realidad el anillo exterior, mientras que el anillo central muestra el
color afectado por la heterocromía

La forma congénita es la más rara, y está
presente desde el nacimiento. Puede estar
asociada con algunas enfermedades raras,
como la neurofibromatosis, el Síndrome de
Waardenburg o el Síndrome de Claude-
Bernard-Horner. Se presenta de forma
habitual en los gatos y en los perros de
raza Husky siberiano, Collie de la frontera
o Bobtail, dálmata, Gran Danés y Pastor
Australiano.

La forma adquirida puede deberse a un
traumatismo, al depósito de pigmentos, a la
administración de fármacos a nivel ocular,
sangrado en el ojo producido por un golpe;
inflamación ocular consecuencia de otras
condiciones médicas o problemas inmunes, o
una inflamación de la capa media de ojo,
conocida como uveítis. En casos de
heterocromía adquirida, se debe realizar un
completo análisis del ojo y el resto del cuerpo,
para dar con la causa y así comenzar el
tratamiento adecuado, ya que también puede
ser una señal de tumores en el ojo.

Personas Famosas con Heterocromía
Mila Kunis
Kiefer Sutherland
Christopher Walken
Demi Moore
Tim McIlrath
OdedKattash. Dominic Sherwood.
Kate Bosworth.
Jane Seymour
Madeleine McCann
Alice EveAilyn

Es producto de un defecto en el
desarrollo embrionario, entre las 3 y 8
semanas de gestación. Los cuerpos
vertebrales de la columna cervical no se
separan, permanecen fusionados. La
causa de este defecto no se conoce
SINDROME DE KLIPPEL FEIL
Es una enfermedad que se caracteriza por la fusión de los cuerpos
vertebrales de la región cervical. Fue descrita por primera vez en 1912 por
dos médicos: Klippel y Feil, de ahí su nombre.
El síndrome de Klippel-Feil ocurre en un grupo heterogéneo de pacientes
unificado sólo por la presencia de un defecto congénito en la formación o
la segmentación de la columna cervical.
¿CÓMO SE EXPRESA?
Es producto de un defecto en el
desarrollo embrionario, entre las 3 y 8
semanas de gestación. Los cuerpos
vertebrales de la columna cervical no se
separan, permanecen fusionados. La
causa de este defecto no se conoce.

¿CÓMO SE CONTRAE?
Ocurre en un grupo heterogéneo de
pacientes unificado sólo por la
presencia de un defecto congénito
en la formación o la segmentación
de la columna cervical.
CUÁL ES SU TRATAMIENTO?
Si el médico lo estima oportuno y necesario, se corrige con una sencilla operación.
Analgésicos en los casos leves, artrodesisprofiláctica de vértebras hipomóviles, o en los casos en que hay compromiso neurológico. El método de Bolona se utiliza para aumentar la longitud aparente del cuello extirpando la primera costilla.
Según la técnica de Bonola se hace una resección de los primeros cuatro arcos costales, esto da la impresión estética de un cuello más corto. La intervención se realiza en dos tiempos, primero un grupo de costillas y luego el otro.
1. Cuello corto.
2. Implantación baja del pelo en la nuca.
3. Rigidez del cuello.
Determinados factores incrementan el riesgo de presentar cervicalgia:
género (en pacientes jóvenes el 70% de las cervicalgias afectan a varones,
invirtiéndose el porcentaje según avanza la edad), edad mayor de 40
años, influencia genética, tabaquismo, escasa actividad física, sobrecarga
laboral, posiciones laborales sedentarias,

Es una deformidad congénita de la caja torácica caracterizada por pecho
hundido en la región del esternón.
Algunas veces se le llama "pecho en embudo". La mayoría de los casos
corresponde a hallazgos aislados, es decir, que no se asocian con ninguna
otra condición. Sin embargo, el pectusexcavatum forma parte de algunos
síndromes.
Es causado por un crecimiento anómalo excesivo del cartílago costal
durante el desarrollo de la pared torácica. Estos cartílagos son demasiado
largos y empujan el esternón hacia atrás.
Por lo general, se hace aparente a los 2 ó 3 años de edad. Puede volverse
severo más adelante durante la infancia y seguir progresando con el
crecimiento de la pubertad. Si bien, aunque menos frecuentemente, puede
ocurrir que el hundimiento comience tardíamente durante los picos de
mayor crecimiento. Estos son casos de aparición tardía. Es 4 veces más
común en los varones que en las niñas y ocurre con más frecuencia en las
familias en las que un miembro tiene esta malformación. La misma se
asocia con otras malformaciones de los músculos y huesos,
particularmente la escoliosis.
Los pacientes con PectusExcavatum suelen adoptar una postura anormal
que se caracteriza por el encorvamiento de la columna dorsal y
rotaciónde los hombros hacia adelante. Esta postura suele empeorar el
aspecto del hundimiento y resulta imprescindible corregirla antes de
indicar la cirugía.

TERAPIA CON LA CAMPANA DE SOLUCIÓN
Este tratamiento consiste en la aplicación de una campana sobre el
defecto depresivo que crea un vacío y pone a plano la pared torácica,
como se puede observar en la imagen del TAC.

REMODELACIÓN TORÁCICA MEDIANTE DISPOSITIVOS MAGNÉTICOS
Jamshidi y Harrison han evaluado los resultados de un sistema en el cual
aplican campos de fuerzas magnéticas para elevar el esternón. Para ello
implantan un dispositivo magnético, en principio inocuo para el organismo,
en el esternón. Por fuera se coloca un aparato que abraza las costilla y
sube el esternón. Mediante fuerzas magnéticas que se aplican durante un
período largo se consigue un moldeamiento. Estos métodos todavía
requieren una mejor evaluación.
La intervención quirúrgica está indicada cuando la solicita el paciente o
cuando compromete de forma severa el funcionamiento cardiopulmonar.
Habitualmente estos pacientes se encuentran en un rango de edad entre
los 4 o 5 años y entre los 18 y 20 años. Salvo casos excepcionales, no se
recomienda la intervención en edades tempranas ya que el sistema óseo
aún no está consolidado. Existen dos técnicas quirúrgicas para corregir la
malformación, la de Ravitch y la de Nuss

Tiene distintas complicaciones respiratorias, el sujeto tiene cierta
predisposición a sufrir procesos congestivos e infecciones
broncopulmonares. Además de las respiratorias, también
presentará habitualmente una disminución de talla y peso,
aparición notable de ojeras y fatiga constante en el cuerpo
junto con un menor gasto cardíaco y desarrollo muscular. A
todo esto hay que añadir las consecuencias estéticas obvias del
cuadro que pueden llegar a originar daños psicológicos.
También algunas de las personas con pectusexcavatum tienen
un 5% menos de esperanza de vida, a diferencia de las
personas que no sufren esta anomalía.

Es una enfermedad hereditaria, autosómica
dominante, que provoca una osificación
progresiva de los músculos esqueléticos,
fascias, tendones y ligamentos. Es decir, algunos
tejidos del organismo, como músculos o tendones,
se osifican y se convierten en hueso. Afecta,
según algunos estudios, a unas 3.300 personas
aproximadamente en todo el mundo.
Historia
Desde el siglo XIX ha habido referencias
esporádicas en la literatura médica describiendo
a pacientes que, aparentemente, "se volvieron
como piedra". Es posible que algunos de estos
casos pueda haber sido atribuible a la FOP.Quizá
el caso más conocido de FOP en tiempos
modernos sea el de Harry Raymond Eastlack Jr.,
nacido en Filadelfia, PA en noviembre de 1933. Su
condición comenzó a desarrollarse a la edad de
Fibrodisplasia
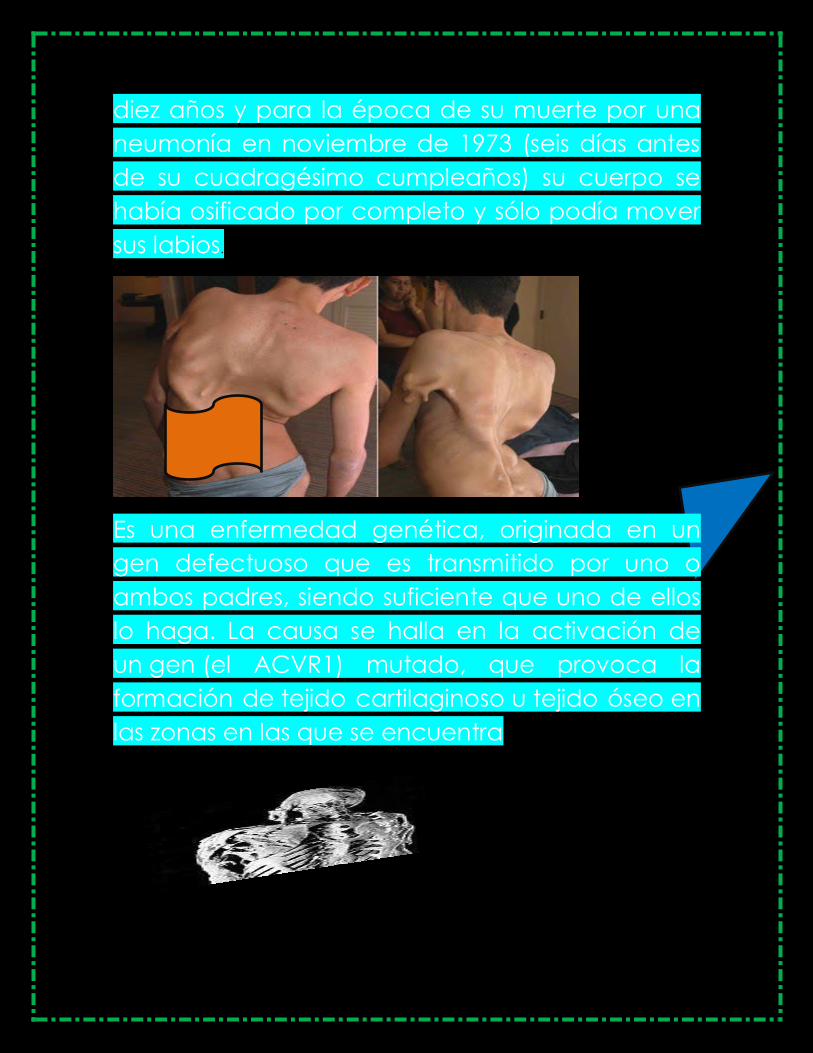
diez años y para la época de su muerte por una
neumonía en noviembre de 1973 (seis días antes
de su cuadragésimo cumpleaños) su cuerpo se
había osificado por completo y sólo podía mover
sus labios.
Es una enfermedad genética, originada en un
gen defectuoso que es transmitido por uno o
ambos padres, siendo suficiente que uno de ellos
lo haga. La causa se halla en la activación de
un gen (el ACVR1) mutado, que provoca la
formación de tejido cartilaginoso u tejido óseo en
las zonas en las que se encuentra

Patologia Estas formaciones óseas se producen por brotes desde
la infancia. Tras la inflamación de los tejidos blandos en
cuestión (músculos, tendones o ligamentos), éstos se
convierten en hueso, lo que poco a poco supone la
pérdida de movilidad. Los primeros signos de la
enfermedad se suelen dar en las zonas cercanas a
la columna vertebral y posteriormente se van
propagando hacia otras articulaciones y grupos
musculares como los codos o las rodillas. El avance de
la patología es progresivo, según se desarrolla aparecen
deformidades, discapacidades funcionales y
perturbaciones conductuales. Por otro lado, tanto los
músculos faciales como los músculos indispensables
para las funciones vitales del organismo como la
respiración, el diafragma, o el corazón para la
circulación sanguínea, están característicamente fuera
de peligro.


















