
Santiago de Compostela, 27 Mayo 2010E9tic… · factor de pron óstico precario independiente de:...
29
Sandra Suárez Ordóñez Santiago de Compostela, 27 Mayo 2010
Transcript of Santiago de Compostela, 27 Mayo 2010E9tic… · factor de pron óstico precario independiente de:...

Sandra Suárez Ordóñez Santiago de Compostela, 27 Mayo 2010
Introducción §LMA: entidad heterogénea. Curso clínico, pronóstico diferentes.
§Conocimientos leucemogénesis:
Øadquisición secuencial lesiones moleculares: proliferación inadecuada y capacidad indefinida de autorrenovación
ØMayor conocimiento etiopatogénesis
ØIdentificación diferentes entidades diagnósticas características
ØEstrategias terapéuticas
ØNuevas dianas terapéuticas
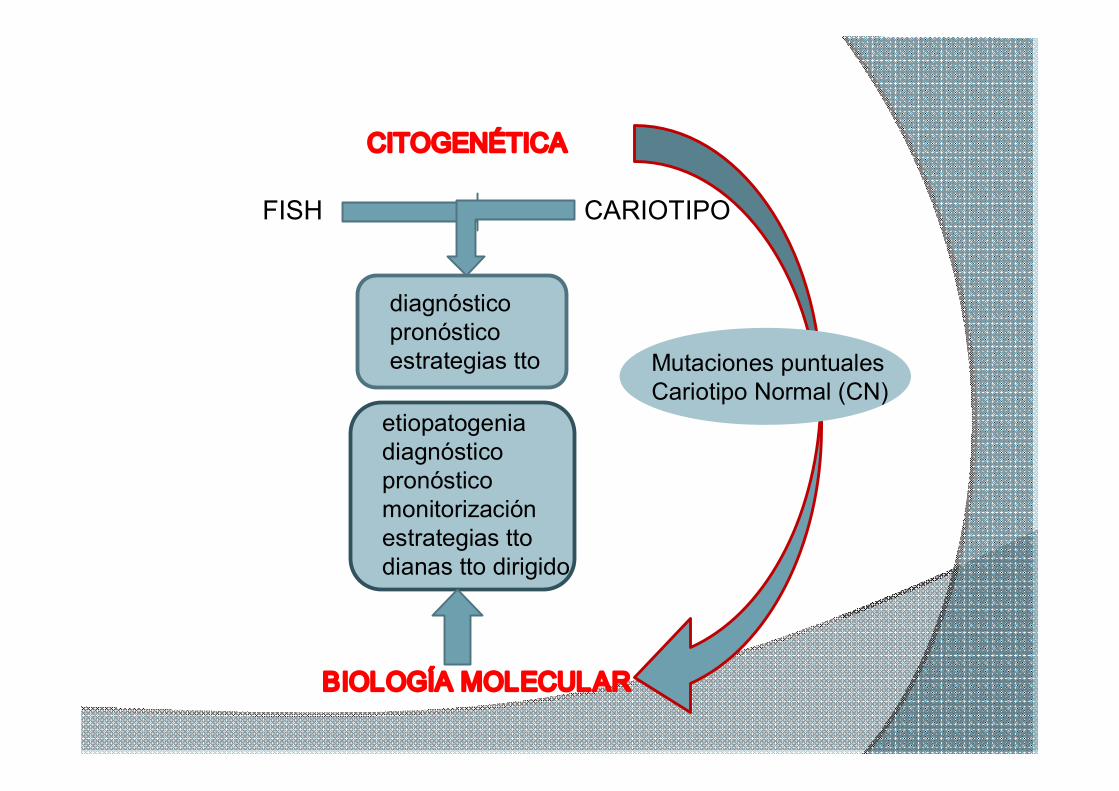
CITOGENÉTICA
CARIOTIPO FISH
BIOLOGÍA MOLECULAR
diagnóstico pronóstico estrategias tto
etiopatogenia diagnóstico pronóstico monitorización estrategias tto dianas tto dirigido
Mutaciones puntuales Cariotipo Normal (CN)

RESUMEN DE LA CLASIFICACIÓN DE LA OMS 2008
LMA con anormalidades genéticas recurrentes. LMA con t(8;21)(q22;q22); RUNX1RUNX1T1. LMA con inv(16)(p13q22) o t(16;16)(p13;q22); (CBFβ/ MYH11). Leucemia promielocítica aguda (LMA con t(15;17)(q22;q12)); (LMP/RARα) y variantes. LMA con t(9;11) (p22;q23); MLLT3MLL LMA con t(6;9) (p23;q34); DEKNUP214 LMA con inv(3)(q21q26.2) o t(3;3)(q21;q26.2); RPN1EVI1 LMA(megacarioblástica) con t(1;22)(p13;q13); RBM15MKL1 LMA con mutación de NPMM1 LMA con mutación de CEBPA
LMA relacionada con cambios mielodisplásicos.
LMA y SMD, relacionado con la terapia.
LMA no clasificada en otro grupo.
Sarcoma mieloide
Proliferaciones mieloides relacionadas con Sd Down
Neoplasia de células blásticas plasmocitoides dendríticas.
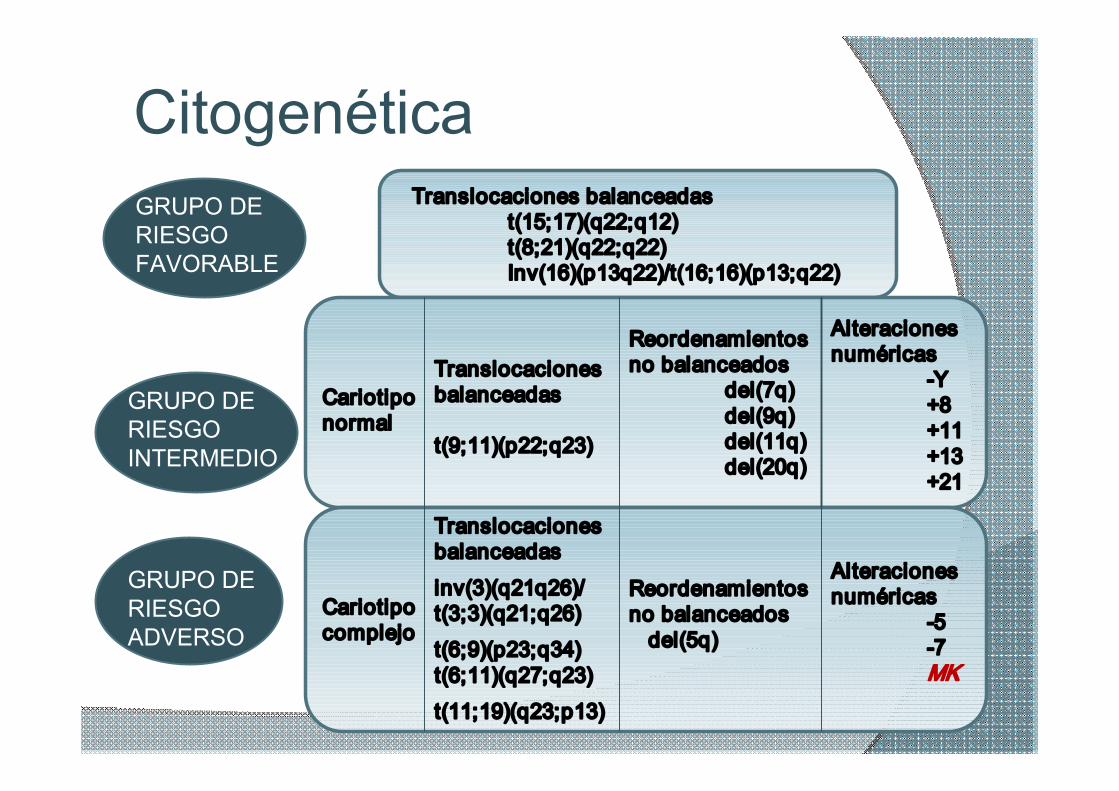
Citogenética GRUPO DE RIESGO FAVORABLE
GRUPO DE RIESGO INTERMEDIO
GRUPO DE RIESGO ADVERSO
Translocaciones balanceadas t(15;17)(q22;q12) t(8;21)(q22;q22) Inv(16)(p13q22)/t(16;16)(p13;q22)
Cariotipo normal
Translocaciones balanceadas
t(9;11)(p22;q23)
Reordenamientos no balanceados
del(7q) del(9q) del(11q) del(20q)
Alteraciones numéricas
Y+8 +11 +13 +21
Cariotipo complejo
Translocaciones balanceadas
inv(3)(q21q26)/ t(3;3)(q21;q26)
t(6;9)(p23;q34) t(6;11)(q27;q23)
t(11;19)(q23;p13)
Reordenamientos no balanceados del(5q)
Alteraciones numéricas
5 7 MK
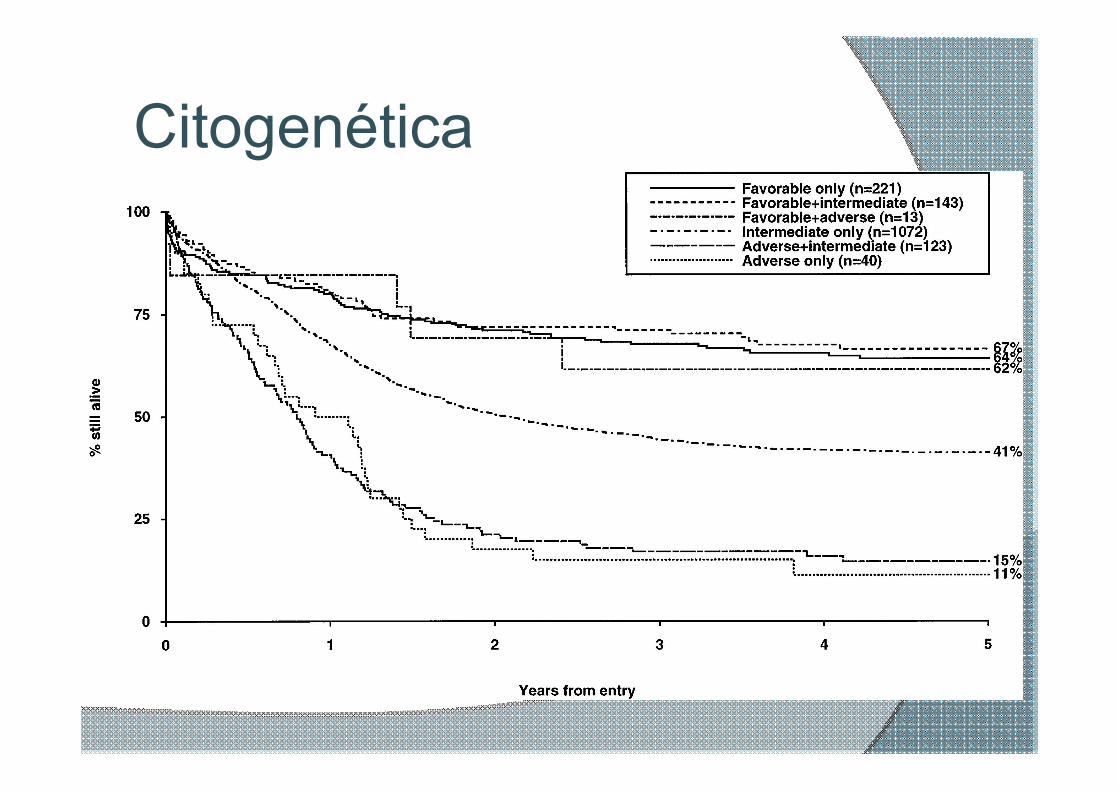
Citogenética

Citogenética: t(15;17) §t(15;17) (q22;q21) ; reordenamiento PML/RARα
§Determina un tratamiento y pronóstico específicos. §Tto con efectos diferenciantes del ácido transretinoico
§Resulta en la producción de una proteína de fusión que incluye RARA
§Otras traslocaciones del receptor pueden producir variantes LPA :
Øt(11;17)(q23;q21) PLZF/RARA §No resp ATRA. § Morfología hipogranular, núcleo regular, No bastones Auer. Pelgueroides
Øt(17;17)(q11;q21)STAT5B/RARA Resistencia ATRA Ø t(5;17)(q35;q21) NPM/RARA
§Sensible ATRA. §Morfología mixta: formas hipergranulares e hipogranulares. No bastones Auer.
Øt(11;17)(q13;q21) NUMA/RARA
§Morfología: citoplasma hipergranular, núcleo irregular. MPO+/ LMA M3 FAB
§Factor independiente de Pronóstico favorable
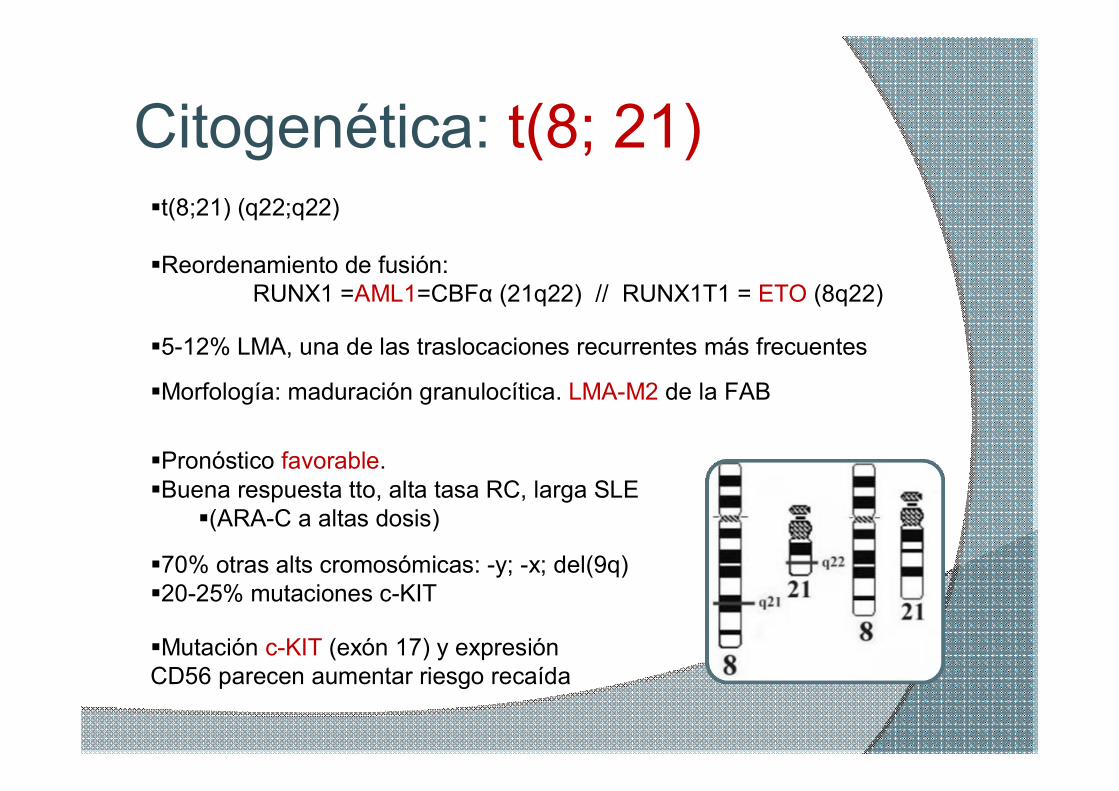
Citogenética: t(8; 21) §t(8;21) (q22;q22)
§Reordenamiento de fusión: RUNX1 =AML1=CBFα (21q22) // RUNX1T1 = ETO (8q22)
§512% LMA, una de las traslocaciones recurrentes más frecuentes
§Morfología: maduración granulocítica. LMAM2 de la FAB
§Pronóstico favorable. §Buena respuesta tto, alta tasa RC, larga SLE §(ARAC a altas dosis)
§Mutación cKIT (exón 17) y expresión CD56 parecen aumentar riesgo recaída
§70% otras alts cromosómicas: y; x; del(9q) §2025% mutaciones cKIT
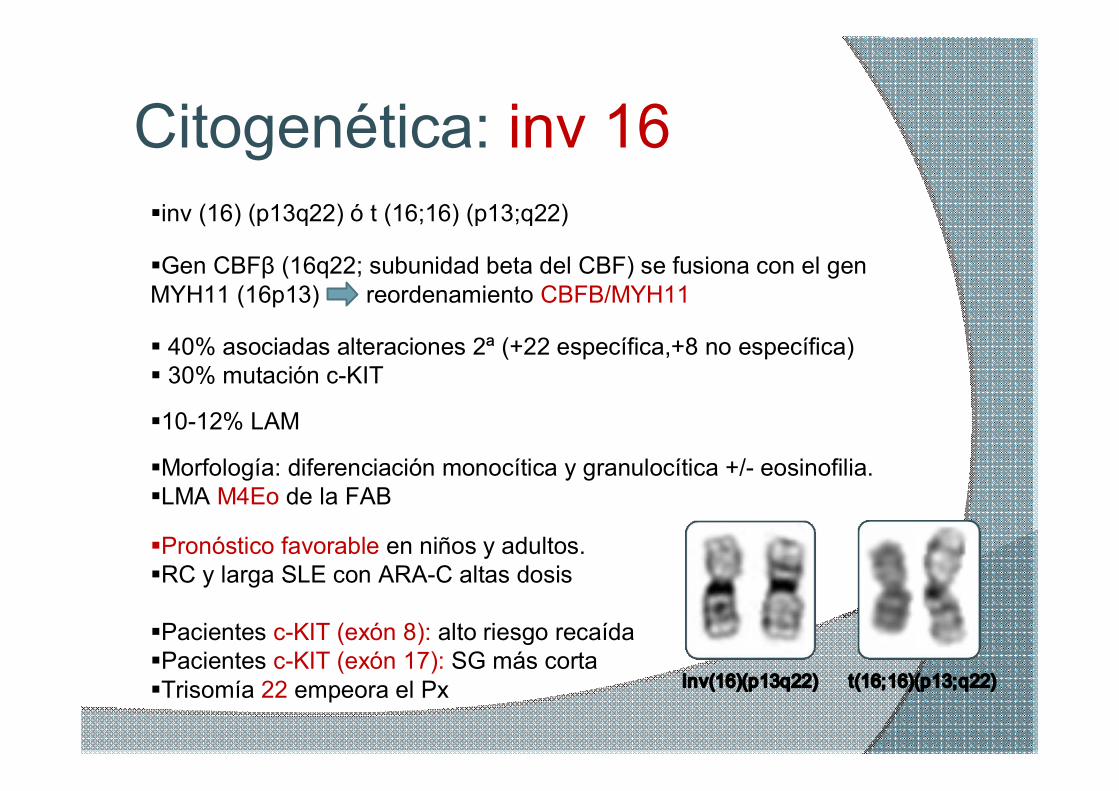
Citogenética: inv 16 §inv (16) (p13q22) ó t (16;16) (p13;q22)
§Gen CBFβ (16q22; subunidad beta del CBF) se fusiona con el gen MYH11 (16p13) reordenamiento CBFB/MYH11
§Morfología: diferenciación monocítica y granulocítica +/ eosinofilia. §LMA M4Eo de la FAB
§Pronóstico favorable en niños y adultos. §RC y larga SLE con ARAC altas dosis
§Pacientes cKIT (exón 8): alto riesgo recaída §Pacientes cKIT (exón 17): SG más corta §Trisomía 22 empeora el Px
§1012% LAM
inv(16)(p13q22) t(16;16)(p13;q22)
§ 40% asociadas alteraciones 2ª (+22 específica,+8 no específica) § 30% mutación cKIT
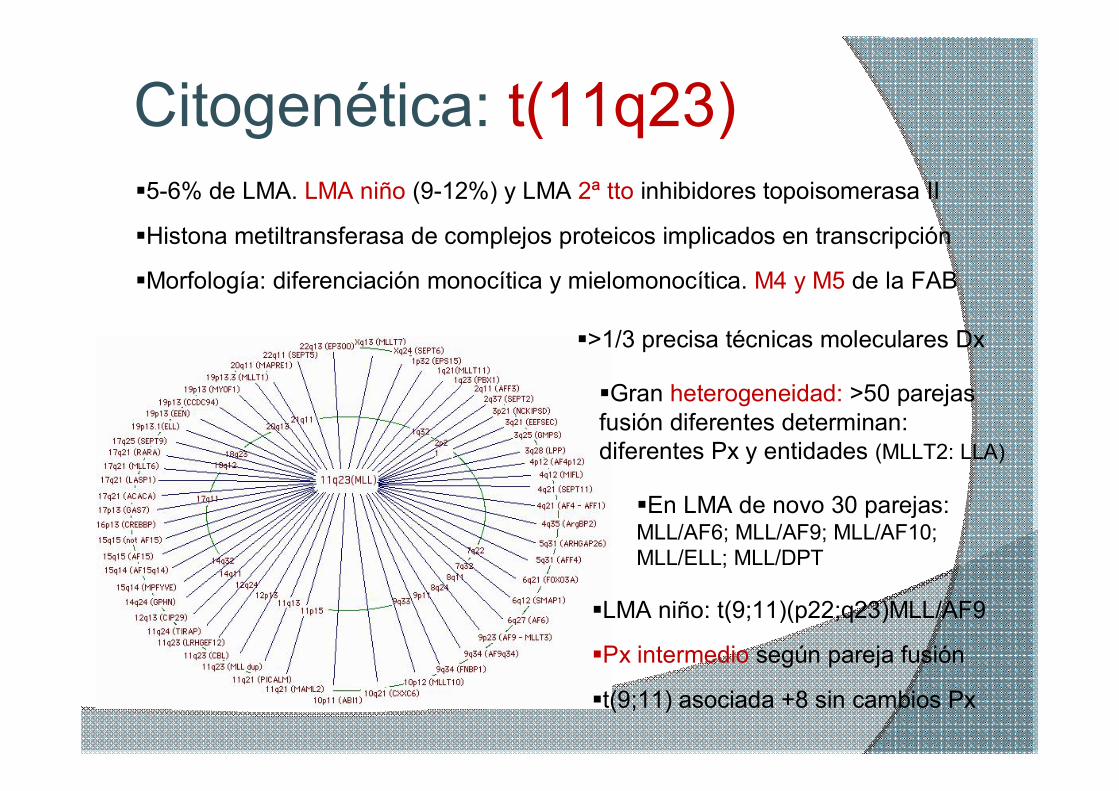
Citogenética: t(11q23) §56% de LMA. LMA niño (912%) y LMA 2ª tto inhibidores topoisomerasa II
§Morfología: diferenciación monocítica y mielomonocítica. M4 y M5 de la FAB
§Gran heterogeneidad: >50 parejas fusión diferentes determinan: diferentes Px y entidades (MLLT2: LLA)
§LMA niño: t(9;11)(p22;q23)MLL/AF9
§En LMA de novo 30 parejas: MLL/AF6; MLL/AF9; MLL/AF10; MLL/ELL; MLL/DPT
§Px intermedio según pareja fusión
§Histona metiltransferasa de complejos proteicos implicados en transcripción
§t(9;11) asociada +8 sin cambios Px
§>1/3 precisa técnicas moleculares Dx
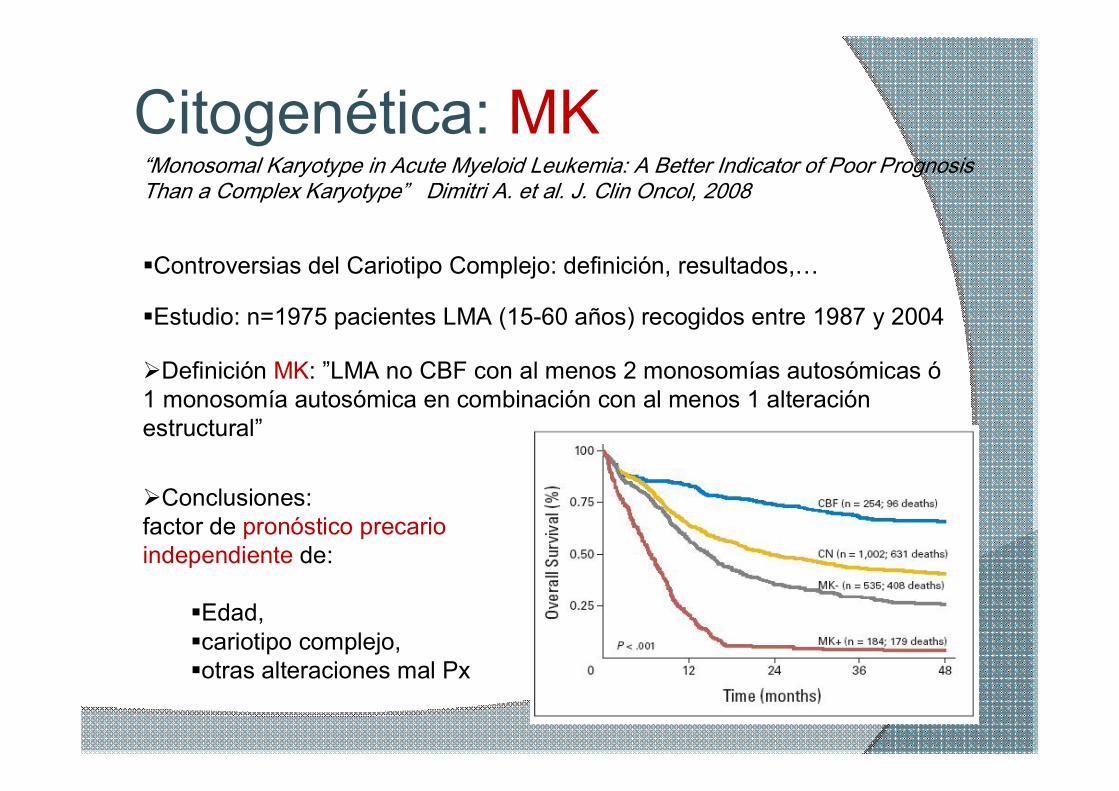
Citogenética: MK “Monosomal Karyotype in Acute Myeloid Leukemia: A Better Indicator of Poor Prognosis Than a Complex Karyotype” Dimitri A. et al. J. Clin Oncol, 2008
ØDefinición MK: ”LMA no CBF con al menos 2 monosomías autosómicas ó 1 monosomía autosómica en combinación con al menos 1 alteración estructural”
ØConclusiones: factor de pronóstico precario independiente de:
§Edad, §cariotipo complejo, §otras alteraciones mal Px
§Estudio: n=1975 pacientes LMA (1560 años) recogidos entre 1987 y 2004
§Controversias del Cariotipo Complejo: definición, resultados,…
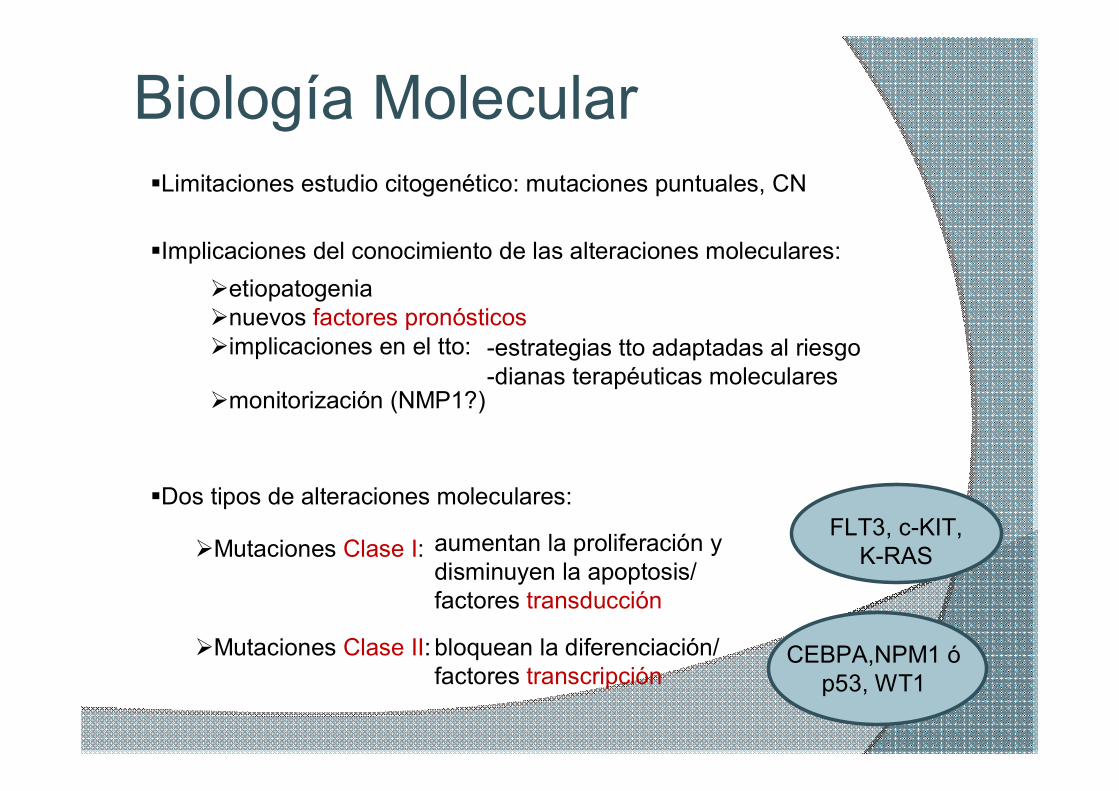
Biología Molecular §Limitaciones estudio citogenético: mutaciones puntuales, CN
§Implicaciones del conocimiento de las alteraciones moleculares: Øetiopatogenia Ønuevos factores pronósticos Øimplicaciones en el tto: estrategias tto adaptadas al riesgo
dianas terapéuticas moleculares Ømonitorización (NMP1?)
§Dos tipos de alteraciones moleculares:
ØMutaciones Clase I:
ØMutaciones Clase II: bloquean la diferenciación/ factores transcripción
aumentan la proliferación y disminuyen la apoptosis/ factores transducción
CEBPA,NPM1 ó p53, WT1
FLT3, cKIT, KRAS
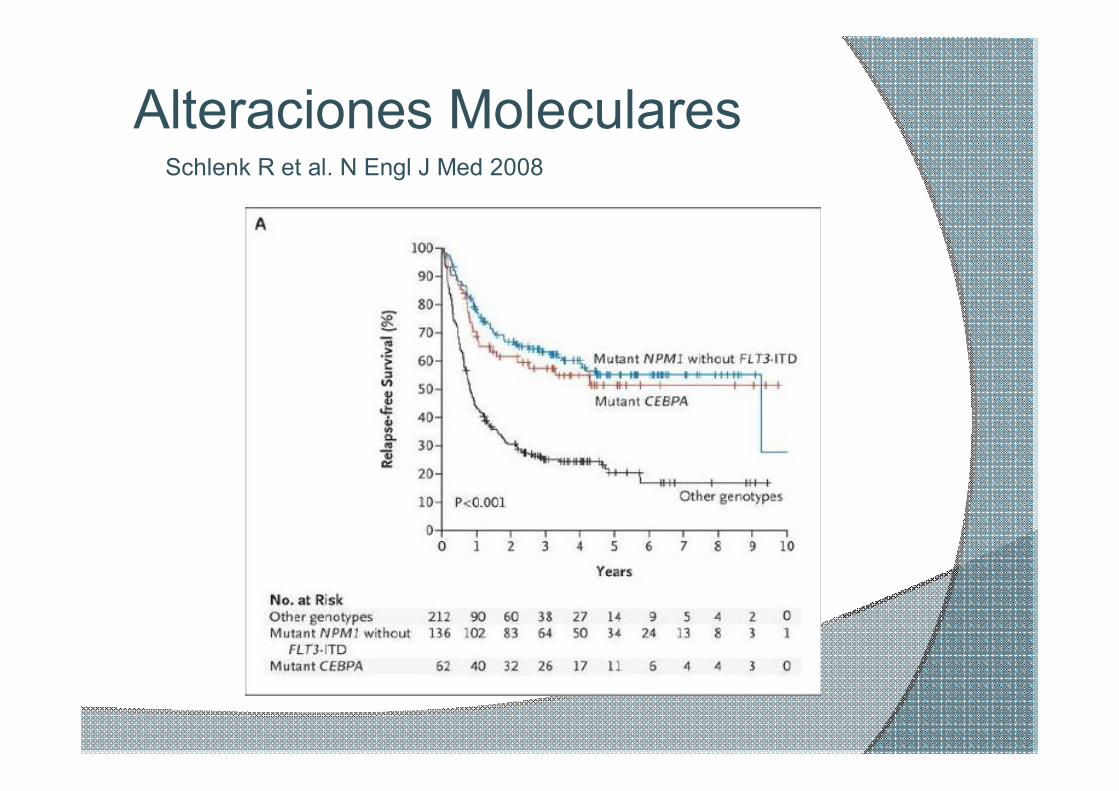
Alteraciones Moleculares Schlenk R et al. N Engl J Med 2008
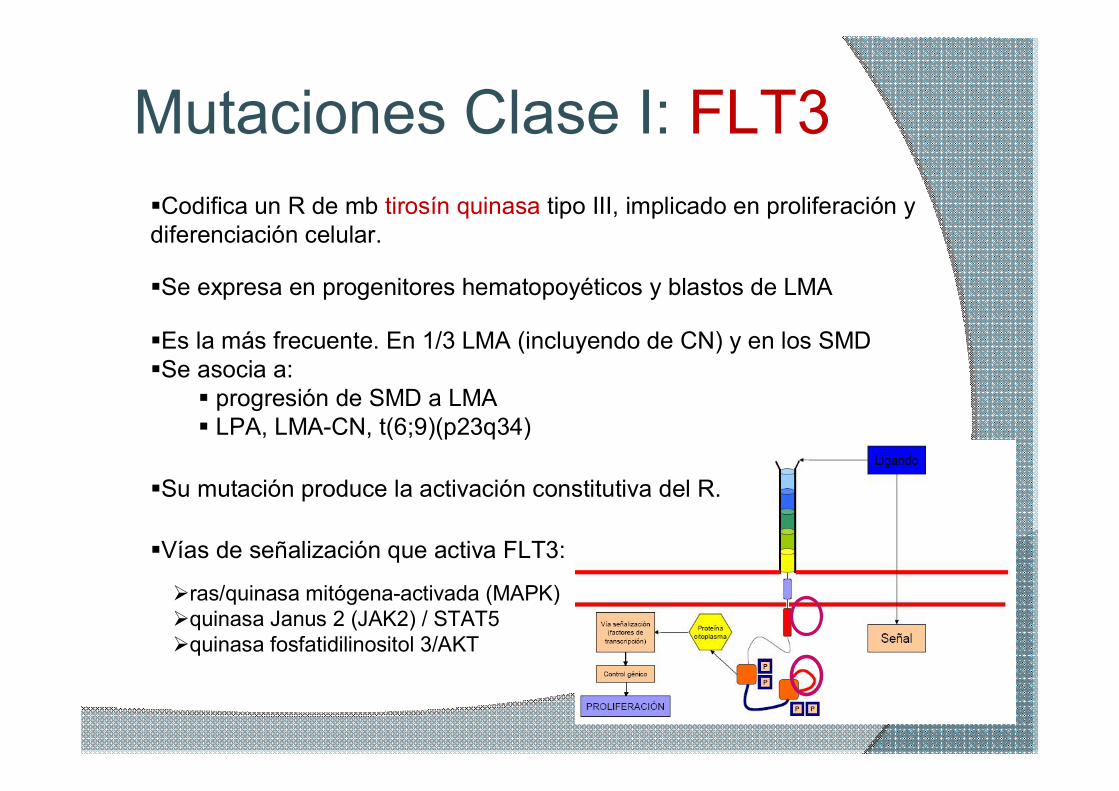
Mutaciones Clase I: FLT3 §Codifica un R de mb tirosín quinasa tipo III, implicado en proliferación y diferenciación celular.
§Se expresa en progenitores hematopoyéticos y blastos de LMA
§Es la más frecuente. En 1/3 LMA (incluyendo de CN) y en los SMD §Se asocia a: § progresión de SMD a LMA § LPA, LMACN, t(6;9)(p23q34)
§Vías de señalización que activa FLT3:
Øras/quinasa mitógenaactivada (MAPK) Øquinasa Janus 2 (JAK2) / STAT5 Øquinasa fosfatidilinositol 3/AKT
§Su mutación produce la activación constitutiva del R.

Mutaciones Clase I: FLT3 Dos tipos de mutación:
Ø 7580%: mutación en el dominio yuxtamembrana: duplicaciones interiores en tandem (FLT3ITD) con variaciones en su longitud:
Ø 2035%: mutación del dominio TK (FLT3TDK):
§2027% LMA adultos // 5% LMA niños <5 años §Factor de mal Px independiente. Alto RR y baja OS §Ratio alelo mutado/alelo normal §Tamaño del segmento DTI §Morfología: variante microgranular (M3v) del LPA e hiperleucocitosis
§D835 y D836 (2º dominio TK) §57% LMA §No claro valor Px
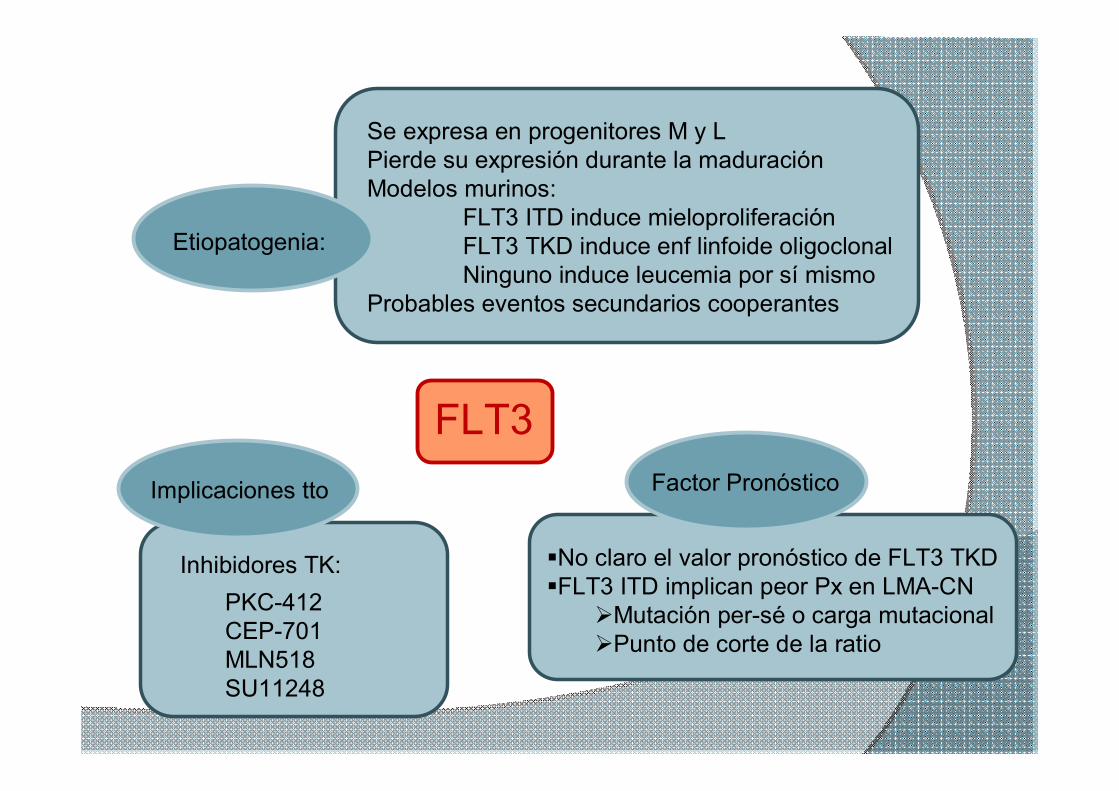
Etiopatogenia:
Se expresa en progenitores M y L Pierde su expresión durante la maduración Modelos murinos:
FLT3 ITD induce mieloproliferación FLT3 TKD induce enf linfoide oligoclonal Ninguno induce leucemia por sí mismo
Probables eventos secundarios cooperantes
Factor Pronóstico
§No claro el valor pronóstico de FLT3 TKD §FLT3 ITD implican peor Px en LMACN ØMutación persé o carga mutacional ØPunto de corte de la ratio
Implicaciones tto:
Inhibidores TK:
FLT3
PKC412 CEP701 MLN518 SU11248
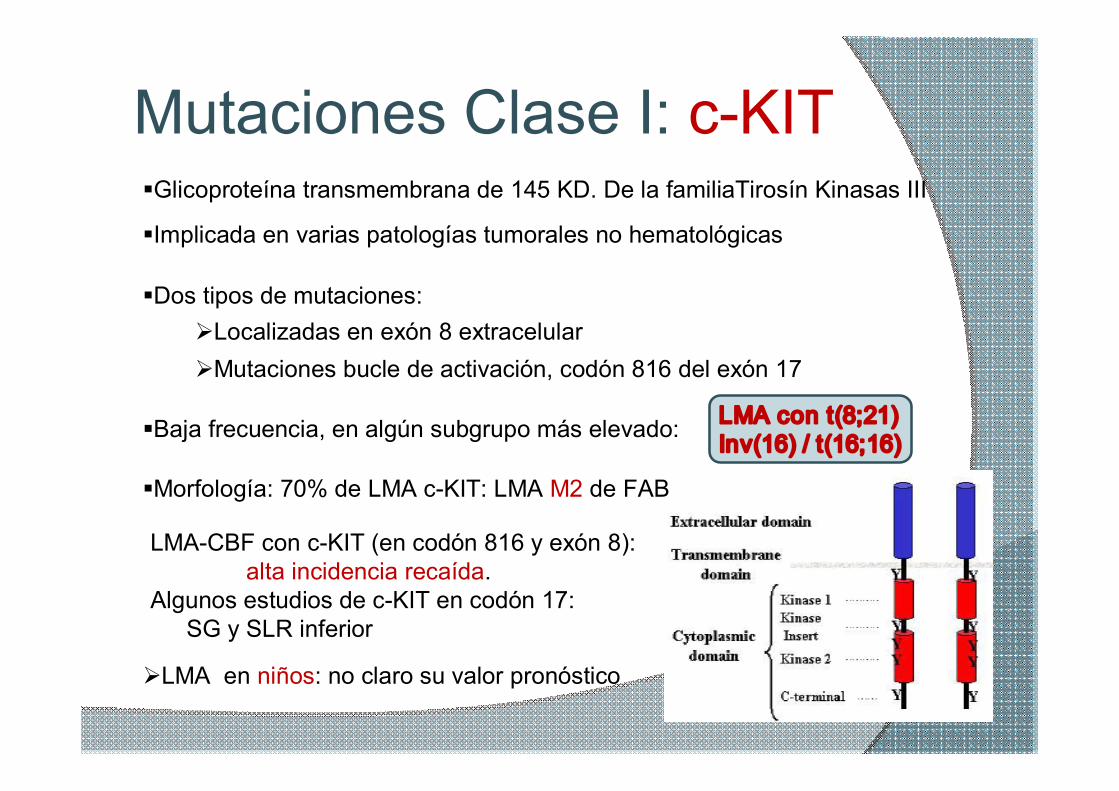
Mutaciones Clase I: cKIT
§Dos tipos de mutaciones: ØLocalizadas en exón 8 extracelular ØMutaciones bucle de activación, codón 816 del exón 17
§Baja frecuencia, en algún subgrupo más elevado: LMA con t(8;21) Inv(16) / t(16;16)
§Morfología: 70% de LMA cKIT: LMA M2 de FAB
ØLMA en niños: no claro su valor pronóstico
LMACBF con cKIT (en codón 816 y exón 8): alta incidencia recaída.
Algunos estudios de cKIT en codón 17: SG y SLR inferior
§Glicoproteína transmembrana de 145 KD. De la familiaTirosín Kinasas III
§Implicada en varias patologías tumorales no hematológicas
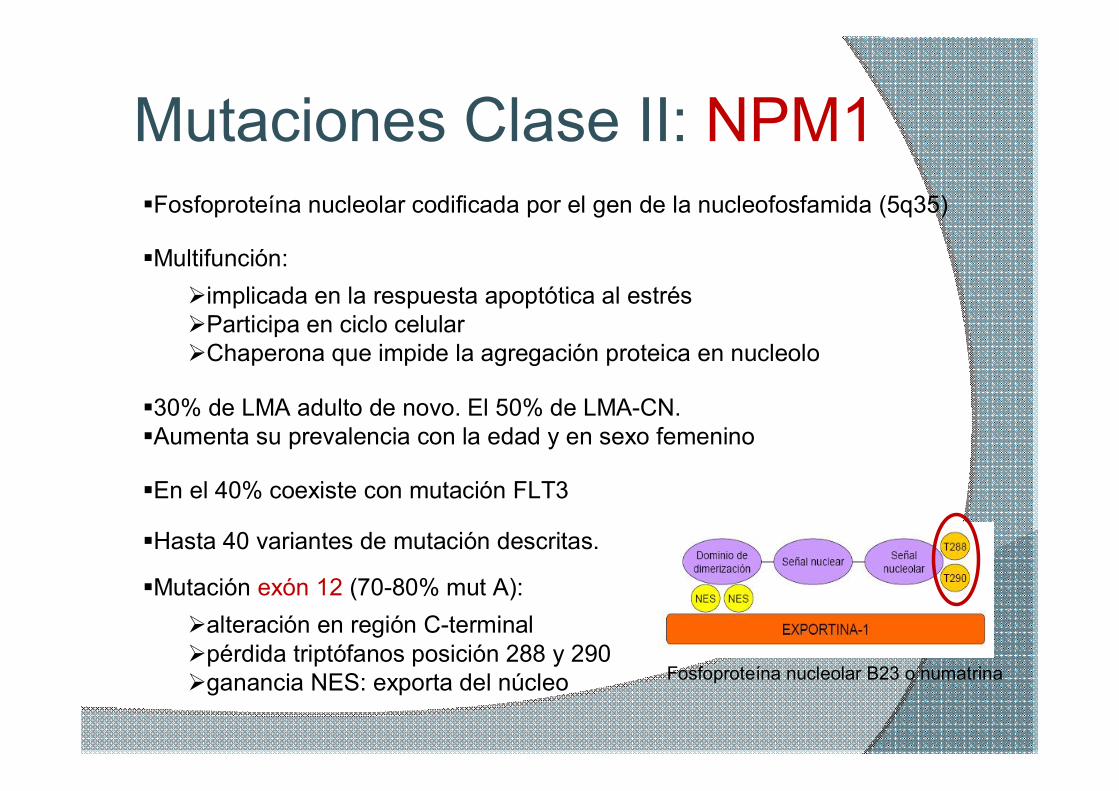
Mutaciones Clase II: NPM1
Øimplicada en la respuesta apoptótica al estrés ØParticipa en ciclo celular ØChaperona que impide la agregación proteica en nucleolo
§Fosfoproteína nucleolar codificada por el gen de la nucleofosfamida (5q35)
§Multifunción:
§30% de LMA adulto de novo. El 50% de LMACN. §Aumenta su prevalencia con la edad y en sexo femenino
§Mutación exón 12 (7080% mut A): Øalteración en región Cterminal Øpérdida triptófanos posición 288 y 290 Øganancia NES: exporta del núcleo Fosfoproteína nucleolar B23 o numatrina
§Hasta 40 variantes de mutación descritas.
§En el 40% coexiste con mutación FLT3

Mutaciones Clase II: NPM1
§Pronóstico favorable en función de FLT3: ØNPM1 con FLT3 negativo: Buen pronóstico y respuesta tto ØNPM1 con baja carga mutacional de FLT3: dudoso ØLMA niños: menos frecuente, menos estudios sobre Px
§Implicaciones tto: ØDianas terapéuticas: ATRA ØSu mutación confiere buen pronóstico (si FLT3 no mutado) y condiciona estrategia terapéutica en grupo de riesgo intermedio
§Morfología: Blastos de núcleo hendido. LMA M4, M5 y M5b de FAB. §Fenotipo: presenta baja expresión de CD34
§Etiopatogenia: localización aberrante en citoplasma células leucémicas. Alteración ciclo celular y apoptosis (alteración actividad y estabilidad p53)
§Inmunohistoquímica: detecta la proteína aberrante en citoplasma blastos
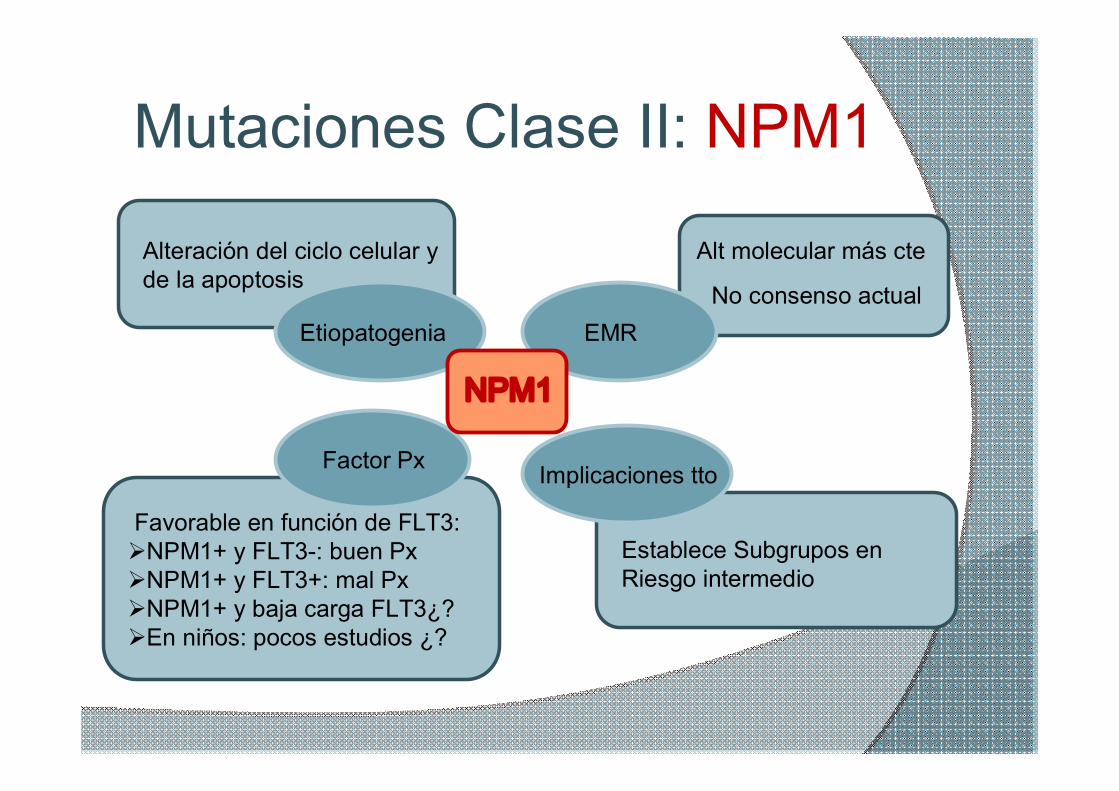
Mutaciones Clase II: NPM1
NPM1
Etiopatogenia
Alteración del ciclo celular y de la apoptosis
Factor Px
Favorable en función de FLT3: ØNPM1+ y FLT3: buen Px ØNPM1+ y FLT3+: mal Px ØNPM1+ y baja carga FLT3¿? ØEn niños: pocos estudios ¿?
Implicaciones tto
Establece Subgrupos en Riesgo intermedio
EMR
Alt molecular más cte
No consenso actual

Mutaciones Clase II: CEBPα §Factor de transcripción localizado en crom 19q13. §Diferenciación progenitores a neutrófilos maduros (inhibe expresión cMYC). § Presente en las células mielomonocíticas.
§Su mutación bloquea maduración progenitores a neutrófilos sin afectar otras líneas celulares
§Morfología: LMA M1 y M2 de la FAB
§Dos tipos de mutaciones: ØNterminal: impide expresión proteina ØCterminal: dificulta su unión al ADN o inhibe su dimerización
§Pronóstico: ØEn ausencia FLT3: Buen Px ØEn LMACN: Px similar a grupo de Bajo Riesgo (SG y SLE favorable) ØCoexistencia otras mutaciones: no claro valor Px
§15% LMA adultos con CN (<60 a) / 5% LMA niños
ADN
cremallera
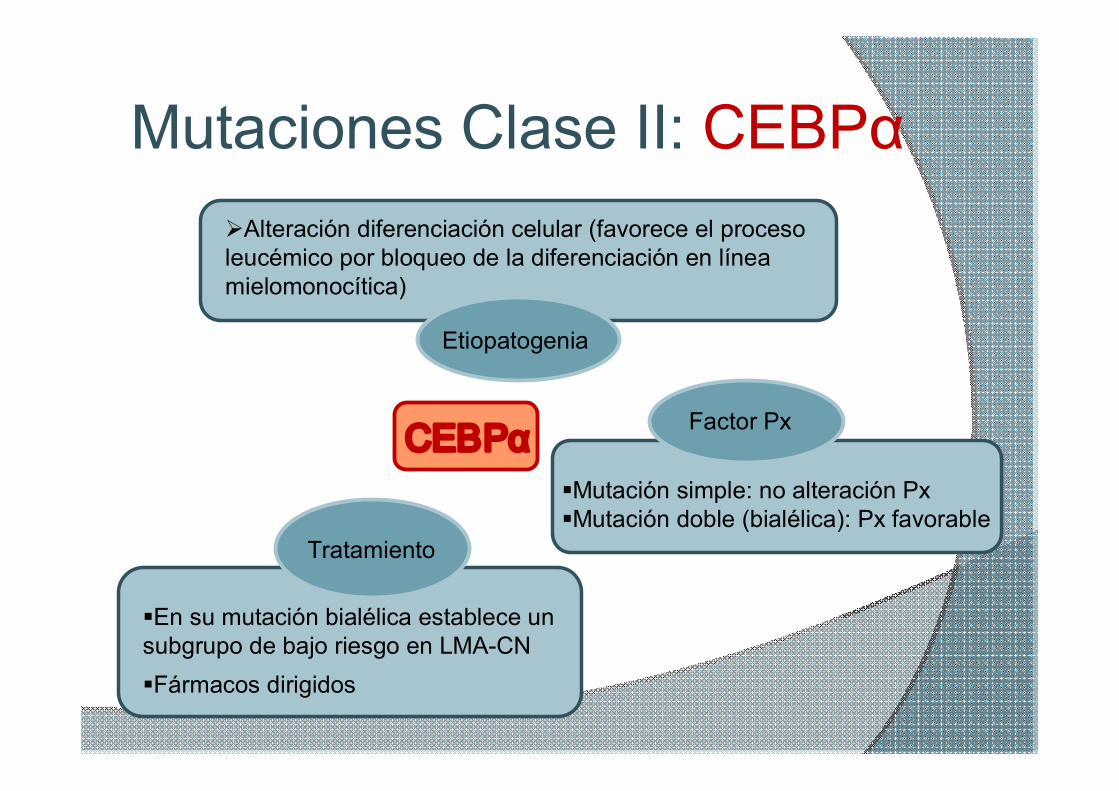
Mutaciones Clase II: CEBPα
Factor Px
§Mutación simple: no alteración Px §Mutación doble (bialélica): Px favorable
§Fármacos dirigidos
CEBPα
Etiopatogenia
ØAlteración diferenciación celular (favorece el proceso leucémico por bloqueo de la diferenciación en línea mielomonocítica)
Tratamiento
§En su mutación bialélica establece un subgrupo de bajo riesgo en LMACN

Mutaciones Clase II: WT1
§El papel de gen WT1 o tumor de Wilms (11p13) aun no ha sido clarificado en la hematopoyesis pero su sobreexpresión se detecta en varios tipos de leucemia, por lo que se podría utilizar como marcador de enfermedad residual.
§Aporta peor Pronóstico a LMA con cariotipo normal:
ØPeor tasa de RC tras tto inducción en LMACN y FLT3ITD ØSLE y OS disminuyen
¿ ¿ ? ?

Sobreexpresión génica ØMutaciones de los genes implicados en el ciclo celular y apoptosis
1 la sobreexpresión de : BAALC, ERG, MN1 o EVI1, en ausencia de translocaciones, parecen tener importancia pronóstica.
2 El gen BAALC (8q22) se expresa en progenitores hematopoyéticos. En LAM CN se asoció a inferior SG, SLE en pacientes con FLT3WT.
3EL gen ERG pertenece a una familia de factores de transcripción implicados en la regulación de la diferenciación celular y apoptosis. En LAM CN: su sobreexpresión se asoció a pronóstico adverso.
4El gen MN1 y EVI1 se han asociado a pronóstico adverso §EVI1 (3q26) asociado o no a MDS1 implica pronóstico precario con muy mala respuesta al tratamiento
5No punto de corte de la normalidad/sobreexpresión ni consenso actual
EMR
§Sobreexpresión génica: WT1 sobreexpresado en blastos Øno claro cutpoint Øno consenso actual
§Alteraciones moleculares: NMP1 ??? Øvariabilidad inter e intraindividual Øno consenso actual
§Alteraciones citogenéticas: Øseguimiento mediante cariotipoFISHPCR: t(15;17); t(8;21)…
§Progresivamente + objetividad y sensibilidad:
§Detección de : 1. targets específicos de leucemia: Øgenes de fusión: PMLRARA; RUNX1RUNXT1;CBPBMYH11 Øo mutaciones: NPM1
2. sobreexpresión génica: WT1
morfología inmunofenotipo citogenética molecular
Tratamiento: dianas moleculares
FLT3 inhibidores
PKC412 CEP701 MLN518 SU11248
ØFase I y II: monoterapia en recaída o refractariedad §Respuesta en LMA con FLT3 activa y no activa mediante vías alternativas
ØEstudios in vitro: sinergia con QT §Fase III: CEP70 + MEC o HiDAC en recaída PKC412 + AraC y Daunorubicin / HiDAC en LMA de novo §Fase I/II MLN518 + AraC y daunorubicin en LMA de novo
Inhibidores farnesyl transferasa
R115777 (Zarnestra) Tipifarnib
ØIn vitro: Zarnestra inhibe proliferación, induce apoptosis
ØFase II: Tipifarnib monoterapia; mantenimiento en >2ª RC; tipifarnib + AraC e idarubicina
ØInhiben farnesilación Ras, inhibiendo su paso a mb
Tratamiento: dianas moleculares ØReexpresión genes supresores y proapoptóticos
Demetilantes ADN HDAC inhibidores
Moduladores transcripción ØDemetilantes:5azacitidina y 5aza2’deoxicitidina
ØHDAC i: MS275; MG0103; vorinostat; Ác valproico; depsipeptido(FK228); fenilbutirato sódico
ØFase I/II: demetilantes+iHDAC
Moduladores multirresistencia1
Oligonucleótido anti Bcl2
Oblimersen ØDegrada ARNm de la proteina bcl2 ØCombinado con Qt
Ø3ªG: especificidad Pgp, menos interacción CYP3A4 XR9576(Tariquidar);
LY335979(Zosuquidar); R101933(Laniquidar) ONT 093:
1ªgeneración 2ªgeneración 3ªgeneración
Ø1ªG: Ciclosporina Ø2ªG: PSC833 (Valspodar): poca eficacia y toxicidad
Øbcl2: estabilizador mb mitocondria: antiapoptosis
§Gen resistencia multidrogas1: 170 kD pgp: “pump”
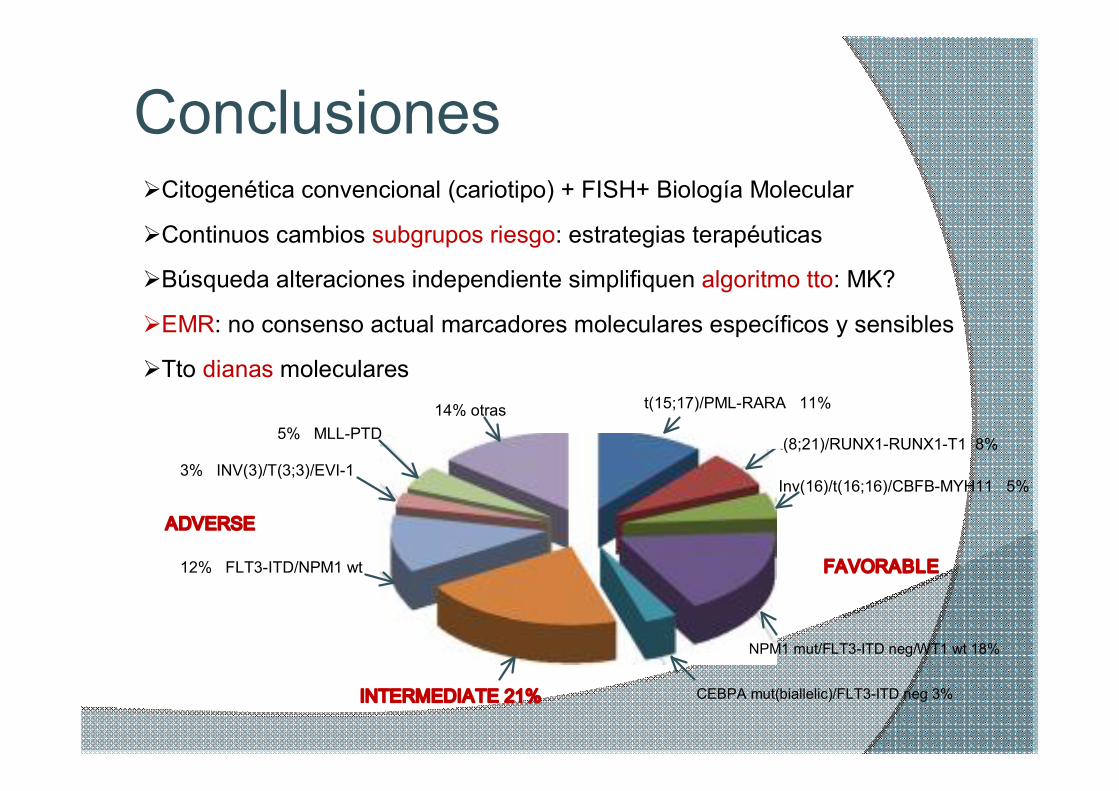
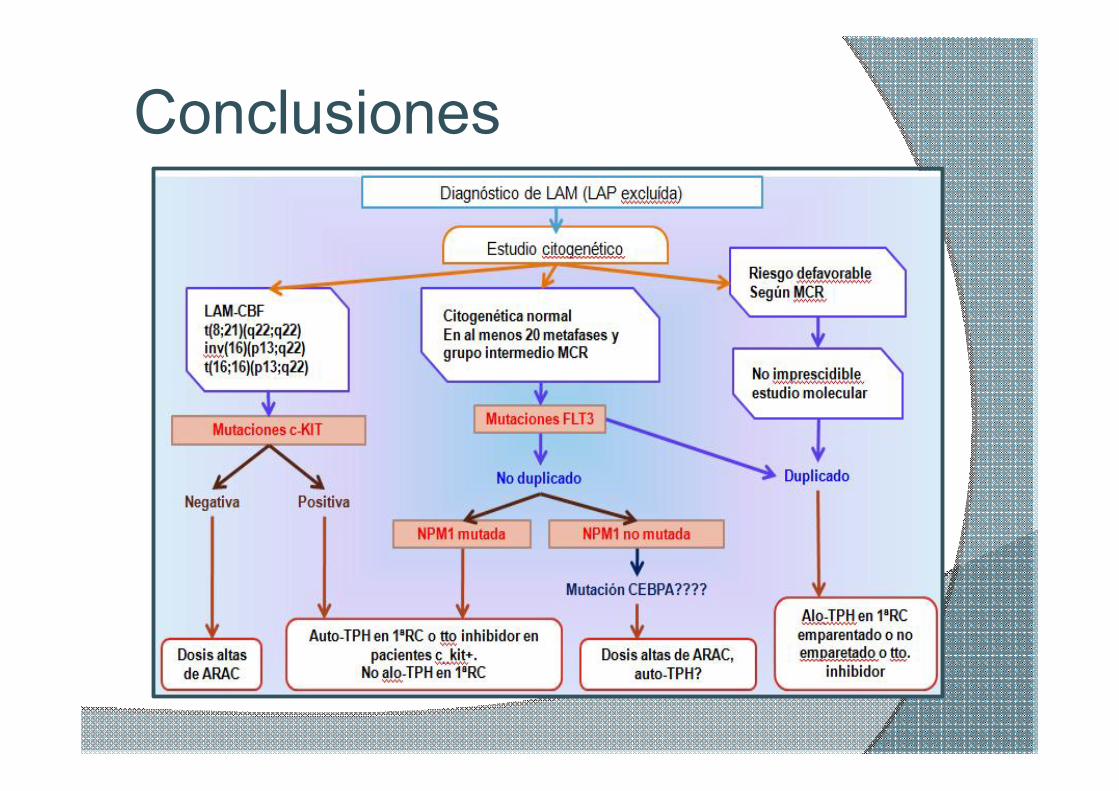
Conclusiones
5% MLLPTD
t(15;17)/PMLRARA 11%
t(8;21)/RUNX1RUNX1T1 8% 3% INV(3)/T(3;3)/EVI1
ADVERSE
12% FLT3ITD/NPM1 wt
INTERMEDIATE 21% CEBPA mut(biallelic)/FLT3ITD neg 3%
NPM1 mut/FLT3ITD neg/WT1 wt 18%
FAVORABLE
14% otras
Inv(16)/t(16;16)/CBFBMYH11 5%
ØCitogenética convencional (cariotipo) + FISH+ Biología Molecular
ØContinuos cambios subgrupos riesgo: estrategias terapéuticas
ØEMR: no consenso actual marcadores moleculares específicos y sensibles
ØBúsqueda alteraciones independiente simplifiquen algoritmo tto: MK?
ØTto dianas moleculares
Conclusiones


















