
SINTESIS TOTALES -5 - pmcarda.files.wordpress.com · Síntesis de coraxeniólido A 3 En el esquema...
144
SINTESIS SINTESIS TOTALES TOTALES-5 Miguel Carda y Eva Falomir Miguel Carda y Eva Falomir Universidad Jaume I Universidad Jaume I O O H H Me Coraxeniólido A O O Me Me Me H H DEDAB
Transcript of SINTESIS TOTALES -5 - pmcarda.files.wordpress.com · Síntesis de coraxeniólido A 3 En el esquema...

SINTESIS SINTESIS TOTALESTOTALES--55
Miguel Carda y Eva FalomirMiguel Carda y Eva FalomirUniversidad Jaume IUniversidad Jaume I
O
OH
H
Me
Coraxeniólido A
O
OMe
Me
MeH
H
DEDAB


Tabla de contenidos
1. Síntesis de Coraxeniólido A (Corey, 2008) 1
2. Síntesis de Cortistatina A (Nicolaou, 2008) 13
3. Síntesis de (Z)-Desoxipukálido (Donohoe, 2008) 33
4. Síntesis de Didemniserinolípido B (Burke, 2007) 51
5. Síntesis de Dihidro-epi-desoxiarteannuina B (Dudley, 2007) 73
6. Síntesis de Elatol (Stoltz, 2008) 67
7. Síntesis de Espongidepsina (Negishi, 2007) 91
8. Síntesis de Exiguolido (Lee, 2008) 103
9. Síntesis de Fortucina (Zard, 2008) 119
10. Síntesis de Grandisina A (Danishefsky, 2007) 129


Síntesis de coraxeniólido A 1
SÍNTESIS DE CORAXENIÓLIDO A
Aislamiento: El coraxeniólido A, un diterpeno de la clase de los xenicanos, subclase xeniólidos, se aisló, en 1981, del coral rosa Corallium sp. Actividad biológica: Aunque no se ha publicado ninguna actividad biológica específica para el coraxeniólido hay que mencionar que muchos diterpenos xenicánicos poseen actividad antitumoral.
Retrosíntesis
El análisis retrosíntético del coraxeniólido A comienza con la desconexión de la cadena lateral en posición α al carbonilo lactónico (esquema 1).1 En el sentido sintético el coraxeniólido A se obtendrá mediante reacción de C-alquilación del enolato metálico derivado de 1 con el haluro de alquenilo 2 (X=halógeno).
La operación de interconversión de grupo funcional sobre la lactona bicíclica 1 conduce a la cetolactona 2, que se sintetizará a partir de la dienona monocíclica 4 mediante adición conjugada de un equivalente del sintón nucleofílico 5, seguida de reacción con formaldehído. La dienona 4 se preparará, mediante un proceso de fragmentación tipo Grob (X=grupo saliente), a partir del alcohol bicíclico 7, que se obtendrá del cetol 8 mediante reacciones de interconversión de grupo funcional.
1 C. V. Larionov, E. J. Corey, J. Am. Chem. Soc. 2008, 130, 2954.

Síntesis de coraxeniólido A
2
Esquema 1
Síntesis
1. Síntesis de la dienona (-)-4
En el recuadro anterior aparece dibujada la estructura de la cetona 4 en dos conformaciones diferentes. La denominada como 4 corresponde a la de un compuesto plano, sin estereocentros, y por tanto carente de actividad óptica. Sin embargo, la estructura numerada como (-)-4 se corresponde con la de un compuesto ópticamente activo, a pesar de que no contiene ningún centro estereogénico. Su actividad óptica se debe a la rotación restringida a lo largo de los enlaces C-C en el anillo de 9 eslabones. Esta restricción conformacional impide la racemización de (-)-4, siempre y cuando éste se conserve a temperaturas relativamente bajas.

Síntesis de coraxeniólido A 3
En el esquema 2 se indica la síntesis del compuesto (-)-4, que se inició con la preparación enantioselectiva del cetol 8. Este compuesto se obtuvo en una secuencia de dos pasos que comenzó con la adición conjugada Michael de la 2-metilciclopenta-1,3-diona a la metil vinil cetona.
Esquema 2
El producto de adición 11 se sometió a una reacción aldólica intramolecular
catalizada por el aminoácido (S)-prolina, lo que condujo a la obtención del cetol 8 con mas del 93% de exceso enantiomérico (véanse comentarios). La reducción estereoselectiva de 8 con triacetoxiborohidruro sódico llevó al diol 12, que se transformó regioselectivamente en el endiol 13 por reacción de eliminación de tipo Mitsunobu (véanse comentarios). La sililación del hidroxilo proporcionó el compuesto 14, el cual se sometió a continuación a un proceso de reducción estereoselectiva. La reducción con NaBH4 transcurrió con escaso estereocontrol, razón por la cual el compuesto 14 se trató con el complejo BH3·SMe2 en presencia de un 10 mol% de la oxazaborolidina quiral 15 (véanse comentarios). En estas condiciones se obtuvo estereoselectivamente el alcohol 16, el cual, por desililación, se transformó en el diol 17. La tosilación regioselectiva de este compuesto condujo al monotosilato 7 y dejó dispuesto el escenario sintético para intentar la obtención de (-)-4 mediante el proceso de fragmentación de Grob. Así, el tratamiento de 7

Síntesis de coraxeniólido A
4
con hidruro sódico en dimetilformamida proporcionó la dienona (-)-4 con un 93% de rendimiento. Este compuesto mantiene su actividad óptica durante mas de 1 mes si se guarda a -20ºC ([α]D=-89, c 5, CHCl3).
2. Pasos Finales
La transformación de la dienona quiral (-)-4 en el coraxeniólido A se inició con el proceso de adición conjugada Michael del silil enol éter derivado del acetato de metilo (esquema 3). Esta reacción se consiguió en presencia de cantidades catalíticas de perclorato de tritilo y condujo a la obtención del cetoéster 18. Los cationes tritilo actúan de ácidos de Lewis coordinándose con el oxigeno carbonílico, favoreciendo de esta forma la adición conjugada 1,4 sobre la adición 1,2. La desprotonación regioselectiva del cetoéster 18 se consiguió mediante reacción con t-pentóxido sódico en tetrahidrofurano, y el correspondiente enolato sódico se transformó en la cetolactona 3 por adición de formaldehído a la mezcla de enolización. La metilenación del carbonilo cetónico del compuesto 3 no funcionó cuando la reacción se intentó con Ph3PMeBr/n-BuLi en THF o CH2Br2/TiCl4/Zn. El compuesto 1 se consiguió cuando la reacción de metilenación se llevó a cabo con el, más reactivo, metilentrifenilfosforano cristalizado y libre de sales. La reacción del enolato lítico derivado de la lactona 1 con el yoduro 2 proporcionó una mezcla, en relación 1:6, formada por el coraxeniólido A y su epímero 19, respectivamente. El tratamiento de la mezcla anterior con la base de Schwesinger P2-Et (véanse comentarios), en tolueno, condujo a la formación de una mezcla, en relación 4:1, de coraxeniólido A y su epímero 19, de la cual se obtuvo puro el producto natural mediante separación en cromatografía de columna.

Síntesis de coraxeniólido A 5
Esquema 3
Comentarios 1. Síntesis enantioselectiva organocatalítica del cetol 9
La adición aldólica asimétrica intramolecular, catalizada por prolina, se conoce en química orgánica como la reacción de Hajos-Parrish-Eder-Sauer-Wiechert, en honor de sus descubridores, que a la sazón investigaban en las empresas Hoffmann-La Roche y Schering AG.2 Esta reacción, descubierta en 1970,
2 a) Z. G. Hajos, D. R. Parrish, J. Org. Chem 1974, 39, 1615; b) U. Eder, G.Sauer, R. Wiechert, Angew. Chem. Int. Ed. Engl. 1971, 10, 496.

Síntesis de coraxeniólido A
6
es, muy probablemente, la primera reacción organocatalítica de la química orgánica sintética.
El ciclo catalítico mediante el cual opera esta reacción se indica en el esquema 4. El proceso se inicia con la condensación regioselectiva de la prolina (de configuración S en el esquema 4) con la tricetona 12. Esta reacción forma la enamina I , que, a continuación, se transforma en la betaína II mediante adición intramolecular. La subsiguiente hidrólisis de la betaína II forma el cetol 9 y regenera la (S)-prolina.
Me
O
O
12
OMe
OH
O
O
9 (>93% ee)
N
H
COOH
Me
O
O
N
HOOC
Me
OH
O
N
OOC
H2OH2O
III
Esquema 4
El modelo estereoquímico que explica la formación estereoselectiva de la betaína II se basa en cálculos teóricos llevados a cabo por el grupo de Houk.3
Esquema 5
3 a) S. Bahmanyar, K. N. Houk J. Am. Chem. Soc. 2001, 123, 9922; b) S. Bahmanyar, K. N. Houk J. Am. Chem. Soc. 2001, 123, 11273; c) S. Bahmanyar, K. N. Houk, H. J Martin, B. List J. Am. Chem. Soc. 2003, 125, 2475.

Síntesis de coraxeniólido A 7
En el esquema 5 se indica el estado de transición del proceso de adición intramolecular, en el cual el sistema carbonado que va a generar el anillo ciclohexánico adopta una conformación de silla, colocándose en posición axial tanto la parte del anillo pirrolidínico de la prolina como el grupo carbonilo. Esta disposición favorece la activación del carbonilo cetónico mediante protonación intramolecular por parte de la agrupación carboxílica y, en consecuencia, facilita el subsiguiente ataque nucleofílico de la parte de la enamina.
2. Eliminación de Mitsunobu
El mecanismo del proceso de deshidratación Mitsunobu se describe en el esquema 6.
Me
OH
O
14
N N COOEtEtOOC + Ph3PN
PPh3
EtOOC NCOOEt1)
Me O
HO
13
N
PPh3
EtOOC NCOOEt+
2)Me
OH
O
O
N
PPh3
EtOOCHNCOOEt+
3) Me
OH
O
O
N
PPh3
EtOOCHNCOOEt+
Me
OH
O
OH
Ph3P+
NEtOOC
HNCOOEt
4) Me
OH
O
OPh3P
H
NCOOEtN
H
EtOOC
NH
EtOOCHNCOOEt
+ + Ph3P=O
I
III III
II IIIIV
V
IV
V
VI
OH
Esquema 6

Síntesis de coraxeniólido A
8
El mecanismo se inicia con la formación de la betaína I por adición tipo Michael de la trifenilfosfina al azodicarboxilato de dietilo. La reacción ácido-base de I con el diol 13 forma el alcóxido II y el intermedio III , electrofílico en el átomo de fósforo, que resulta atacado por el alcóxido para dar lugar al alcoxifosfonio IV y a la base nitrogenada V. Finalmente, el proceso de eliminación E2 sobre el alcoxifosfonio IV proporciona el alqueno 14. El ataque básico sobre IV es totalmente regioselectivo, puesto que solo se observa la formación del compuesto 14. Posiblemente, la presencia del hidroxilo libre vecinal al hidrógeno ácido sea el responsable de la elevada regioselectividad del proceso de eliminación. 3. Reducción estereoselectiva de la cetona 14
La reducción estereoselectiva de cetonas con borano, en presencia de
cantidades catalíticas de oxazaborolidinas quirales derivadas de prolina, se conoce como reacción de Corey-Bakshi-Shibata. El ciclo catalítico, y el modelo estereoquímico de este proceso, aplicado a la reducción de la cetona 14, se indica en el esquema 7.
El proceso reductivo se inicia con la formación del azaborohidruro quiral II , como consecuencia de la interacción ácido-base de Lewis entre el átomo de nitrógeno de la oxazaborolidina y el borano. A continuación, la coordinación de la cetona 14 se con el complejo II genera el estado de transición indicado en el esquema 7, que conduce al alcoxiborato III mediante reducción estereocontrolada del carbonilo cetónico. El ciclo catalítico se cierra con la regeneración del catalizador y la formación del alcoxiborano IV , que se transforma en el alcohol en el proceso de workup con MeOH y NaOH acuoso.
El principal factor de estereoinducción es la topología inherente al complejo quiral II , y en particular la presencia del grupo n-butilo enlazado al átomo de boro, que obliga a la cetona a coordinarse con el complejo en una disposición que expone la cara Re de aquélla al ataque del hidruro. La coordinación alternativa, con

Síntesis de coraxeniólido A 9
exposición de la cara Si del carbonilo cetónico, implicaría un fuerte impedimento estético con la cadena de butilo.
Esquema 7
4. Formación de la dienona (-)-4 mediante fragmentación de Grob
La reacción de fragmentación de Grob, denominada así en honor del químico británico Cyril A. Grob, es un proceso de eliminación provocado por la presencia de un grupo electrón-donante, negativamente cargado o con pares electrónicos

Síntesis de coraxeniólido A
10
libres, y ubicado en posición relativa 1,3 con respecto de un grupo saliente (esquema 8).
Esquema 8
Para que tenga lugar la fragmentación de Grob se requieren unos precisos efectos estereoelectrónicos en el sustrato que va a experimentar la reacción. En el esquema 9 se indican las estructuras de dos cloroaminas bicíclicas epiméricas.
En el primero de los sustratos se produce la reacción de fragmentación de Grob porque el orbital que contiene al par electrónico libre del átomo de nitrógeno y el orbital que contiene el enlace C-C que se va a romper están orientados en disposición anti-coplanar, como también están dispuestos en orientación anti-coplanar el enlace C-Cl y el enlace C-C que va a experimentar el proceso de fragmentación.
Sin embargo, en el segundo de los sustratos el enlace C-Cl no está orientado en disposición anti-coplanar con respecto al enlace C-C, motivo por el cual este sustrato experimenta un proceso de eliminación E2 en lugar de la reacción de fragmentación de Grob.
Esquema 9
El proceso de fragmentación de Grob que explica la formación enantioselectiva del compuesto (-)-4 se describe en el esquema 10. En este esquema también se representa el proceso Grob sobre el compuesto ent-7, que conduce a la formación de la dienona (+)-4. La configuración E del doble enlace

Síntesis de coraxeniólido A 11
generado en el proceso de fragmentación apunta a un proceso de eliminación concertado tipo E2.
Me
OH
OTs
(-)-4
OMe
NaH, DMFO
Me
OTs
OMe
7 I
Me
OH
OTs
NaH, DMF
O
H
Me
OMe
H
OMe
OTsH
IIent-7 (+)-4
H
Esquema 10 5. Bases de Schwesinger
Un fosfazeno es un compuesto químico que contiene un átomo de fósforo unido mediante un doble enlace a un átomo de nitrógeno, y a otros tres átomos o grupos mediante enlaces simples. Las bases de Schwesinger, que son alrededor de 18 órdenes de magnitud más básicas que la DBU o DIPEA, son fosfazenos que se caracterizan por poseer una fortaleza básica similar a la de las bases organolíticas.
El centro básico de los fosfazenos de Schwesinger es el átomo de nitrógeno, que genera un ácido conjugado estabilizado por deslocalización de la carga positiva. Las estructuras de algunas de las bases de Schwesinger se indican en el esquema 11.

Síntesis de coraxeniólido A
12
Esquema 11
La estructura de la base de Schwesinger P2-Et empleada en la isomerización del compuesto 19 se indica a continuación:

Síntesis de cortistatina A 13
SÍNTESIS DE CORTISTATINA A
Aislamiento: La cortistatina A se ha aislado de la esponja Corticium simplex. Actividad biológica: La angiogénesis es un proceso fisiológico que está implicado en la formación de nuevos vasos sanguíneos. Aunque la angiogénesis es un fenómeno normal durante el desarrollo embrionario, el crecimiento del organismo y la cicatrización de las heridas, su expresión aberrante favorece el crecimiento de los tumores cancerosos (angiogénesis tumoral).
Las paredes de los vasos sanguíneos están formadas por células vasculares endoteliales. Estas células raramente se dividen, haciéndolo solamente alrededor de una vez cada 3 años en promedio. Sin embargo, cuando la situación así lo requiere, la angiogénesis puede estimularlas para que se dividan. Se denomina angiogénesis tumoral a la proliferación de redes formadas por vasos sanguíneos que penetran dentro de los crecimientos cancerosos, proporcionando nutrientes y oxígeno, y eliminando al mismo tiempo los productos de desecho. La angiogénesis tumoral empieza cuando las células cancerosas del tumor liberan moléculas que envían señales al tejido huésped normal vecino. Este señalamiento activa a ciertos genes en el tejido huésped que, a su vez, producen proteínas para estimular el crecimiento de nuevos vasos sanguíneos. Se han identificado como angiogénicas más de una docena de proteínas diferentes, entre las que destacan dos: el factor de crecimiento vascular endotelial (FCVE) (vascular endothelial growth factor o VEGF, en inglés) y el factor básico de crecimiento de fibroblastos (FBCF) (basic fibroblast growth factor o bFGF, en inglés). Los factores de crecimiento FCVE y FBCF son sintetizados primero dentro de las células tumorales y después secretados en el tejido vecino. Cuando encuentran a células endoteliales se unen a proteínas específicas, conocidas como receptoras, localizadas en la superficie exterior de las células. La unión de alguno de los dos factores de crecimiento, FCVE o FBCF, provoca la activación de las células endoteliales y pone en marcha una serie de pasos encaminados a la creación de nuevos vasos sanguíneos. Primero, las células endoteliales activadas producen metaloproteinasas de la matriz o MPMs (matrix

Síntesis de cortistatina A 14
metalloproteinases o MMPs, en inglés), una clase especial de enzimas degradadoras. Estas enzimas se liberan de las células endoteliales al tejido vecino. Las MPMs degradan la matriz extracelular, que es el material de apoyo que llena los espacios entre las células, y que está compuesto de proteínas y polisacáridos. La degradación de esta matriz permite la migración de las células endoteliales. A medida que éstas migran a los tejidos vecinos, las células endoteliales activadas empiezan a dividirse, organizándose en tubos huecos que evolucionan gradualmente y se convierten en una red madura de vasos sanguíneos.
Aunque muchos tumores producen moléculas angiogénicas, como los factores FCVE y FBCF, su presencia no es suficiente para iniciar el crecimiento de vasos sanguíneos. Para que la angiogénesis se desarrolle las moléculas activadoras deben superar la acción de los inhibidores de la angiogénesis, que normalmente reprimen el crecimiento de los vasos sanguíneos. Entre los inhibidores de la angiogénesis destacan las proteínas denominadas angiostatina, endostatina y trombospondina. El balance entre las concentraciones de inhibidores y de activadores de la angiogénesis, tales como los factores de crecimiento FCVE y FBCF, determina si un tumor puede inducir el crecimiento de nuevos vasos sanguíneos. Para activar la angiogénesis, la producción de activadores se debe incrementar a medida que disminuye la producción de inhibidores.
Muchas terapias anti-cáncer se están centrando en la actualidad en el uso terapéutico de inhibidores de la angiogénesis. Estos compuestos se clasifican por su mecanismo de acción, como los que inhiben directamente a las células endoteliales, los que inhiben la cascada señalizadora de la angiogénesis, o los que bloquean la habilidad de las células endoteliales para degradar la matriz extracelular.
La cortistatina A es un potente anti-angiogénico: provoca la inhibición de las células endoteliales de la vena umbilical humana con un IC50=1.8 nM.
Retrosíntesis La cortistatina A es un alcaloide esteroideo que contiene un sistema
androstánico pentacíclico, cuyo anillo E se encuentra unido a un sistema isoquinolínico. El análisis retrosintético de la cortistatina A se inicia con una operación múltiple de interconversión de los grupos funcionales del anillo A, lo que conduce al derivado ciclohexanónico 1 (véase el esquema 1).4
4 K. C. Nicolaou, Y-P. Sun, X-S. Peng, D. Polet, D. Y.-K. Chen, Angew. Chem. Int. Ed. 2008, 47, 7310.

Síntesis de cortistatina A 15
N
MeHO
OH
Me2N H
O
cortistatina A
AB
C
D
E
N
MeO
H
OAB
C
D
E
OPMe
O
H
OAB
C
D
Eretro-aldol
OPMe
O
H
OAC
D
E
O
adición 1,4
OPMe
O
H
HOAD
E
O
acoplamiento
O
AOP
Me
H
HOD
EX
+
O
H
OPMe
H
D
E
OOPMe
D
E
OMe
E
O
O
O+
1
2
34
5 6 7
8910
IGF
IGF
N
X+
MVC
Esquema 1 La siguiente operación desconecta el sistema quinolínico del sistema
pentacíclico. Esta operación origina la isoquinolina 2 y la dienona pentacíclica 3. Este derivado contiene un sistema carbonílico α,β-insaturado, que se podría construir mediante una reacción de condensación aldólica intramolecular en el cetoaldehído tetracíclico 4, que se desconecta en el anillo tetrahidrofuránico al derivado tricíclico 5. En el sentido sintético el anillo tetrahidrofuránico se instalará mediante una reacción de adición conjugada intramolecular del grupo hidroxilo sobre el sistema enónico del anillo A.
La siguiente operación retrosintética desconecta el anillo A del sistema bicíclico fusionado formado por los anillos D-E. La desconexión se basa en una reacción de acoplamiento carbono-carbono tipo Sonogashira, que se llevará a cabo entre el compuesto electrofílico 6 (X=halógeno, OTf) y el alquino terminal 7. La

Síntesis de cortistatina A 16
función de carbonilo aldehídico, que se encuentra libre en el compuesto 7, tendrá que estar protegida en este punto de la secuencia sintética. El compuesto 7 procederá de la cetona exo-metilénica 8, que, a su vez, se preparará a partir del compuesto bicíclico 9. Este derivado se obtendrá mediante una reacción de anelación Robinson entre la 2-metil-1,3-ciclopentanodiona 10 y la metil vinil cetona MVC. Este proceso se puede llevar a cabo en su versión enantioselectiva mediante la aplicación de la metodología desarrollada por Hajos y Parrish.
Síntesis
1. Síntesis del compuesto 8: sistema de anillos D-E
La preparación del compuesto bicíclico 8 se inició con la reacción de anelación Robinson enantioselectiva entre la 2-metil-1,3-ciclopentanodiona 10 y la metil vinil cetona (véase el esquema 2). Este proceso se inició con la adición conjugada Michael del enol derivado de la 2-metil-1,3-ciclopentanodiona 10 a la metil vinil cetona. Esta reacción proporcionó el compuesto 11, que se sometió a la reacción de adición aldólica intramolecular en presencia de (S)-prolina. La reacción catalizada por el aminoácido condujo enantioselectivamente al dicetol bicíclico 12 (véase en este volumen la síntesis del coraxeniólido A), el cual se convirtió en el compuesto 13 mediante deshidratación con ácido sulfúrico. La reducción quimio y estereoselectiva del carbonilo no conjugado, por reacción con tri-t-butoxihidruro de aluminio y litio, proporcionó el alcohol 14. Tras la sililación del grupo hidroxilo, el compuesto resultante 9 se sometió a calentamiento en DMF en presencia de carbonato de metilmagnesio (véanse comentarios). El resultado fue la formación del cetoácido insaturado 15, cuya hidrogenación estereoselectiva condujo al compuesto 16. La adición aldólica-descarboxilación de 16, por reacción con formaldehído y piperidina en DMSO, proporcionó la cetona exometilénica 8.
El estereocontrol que se consigue en la reducción de la dicetona 13 y en la hidrogenación del cetoácido 15 se debe a la presencia del metilo angular, que bloquea el acceso de los reactivos a la cara β del sistema bicíclico.

Síntesis de cortistatina A 17
Esquema 2
2. Síntesis del alquino 21
La conversión de la cetona exometilénica 8 en el alquino 21 comenzó con la dihidroxilación estereoselectiva del doble enlace de aquélla (esquema 3). Este proceso fue seguido de una reacción de cetalización que condujo al compuesto 17, el cual, mediante triflación de la forma enólica y reacción de carbometoxilación catalizada por paladio, fue convertido en el éster insaturado 18 (véanse comentarios). Este compuesto, mediante reducción con DIBAL y oxidación Dess-Martin, se transformó en el aldehído 19, que se sometió a reacción con 1,3-propanoditiol en presencia de BF3·Et2O. En estas condiciones se obtuvo el dihidroxiditiano 20 mediante conversión de la función aldehídica en ditioacetal, y eliminación concomitante del acetónido. El compuesto 20 se transformó en el alquino 21 mediante oxidación Parikh-Doering del hidroxilo primario, seguida de

Síntesis de cortistatina A 18
homologación con el reactivo de Ohira-Bestmann generado in situ (véanse comentarios).
Esquema 3
3. Síntesis del compuesto pentacíclico 3: sistema de anillos A-B-C-D-E
La construcción del compuesto pentacíclico 3 se inició con la reacción de acoplamiento Sonogashira entre el alquino 21 y el triflato de enol 22. La reacción se llevó a cabo en presencia de cantidades catalíticas de Pd(Ph3P)4 y CuI y de la base Et3N, y proporcionó el producto 23 con un 85% de rendimiento (véanse comentarios). Este compuesto se convirtió en el hidroxicetoaldehído 5 mediante destiocetalización inducida por ácido yodoxibenzoico (véanse comentarios), seguida de hidrogenación quimioselectiva del triple enlace. Cuando el compuesto 5

Síntesis de cortistatina A 19
se trató con carbonato potásico en dioxano, a 125ºC, se provocó un proceso de reacciones en cascada que se inició con una adición conjugada 1,4-intramolecular del alcóxido 24 sobre el sistema enónico. Esta reacción generó el enolato tetracíclico 25, que experimentó, a continuación, una reacción de adición aldólica intramolecular al grupo carbonilo aldehídico para dar lugar, después de la protonación, al cetol 27. La eliminación E1cB sobre este intermedio proporcionó el compuesto pentacíclico 3, con un rendimiento del 52% desde el compuesto 5.
Esquema 4

Síntesis de cortistatina A 20
4. Pasos finales
La transformación del derivado pentacíclico 3 en la cortistatina A exigía la instalación de la parte isoquinolínica. Con este objetivo el carbonilo cetónico del compuesto 3 se protegió en forma de etilidenacetal mediante aplicación del método de Noyori (esquema 5). La desililación del producto de cetalización condujo al alcohol 28, que se convirtió en el triflato de enol 29 mediante oxidación Parikh-Doering, seguida de reacción con N-feniltriflimida y hexametildisililamiduro sódico. El acoplamiento Suzuki del triflato de enol 29 con el éster borónico 2, promovido por Pd(Ph3P)4 y carbonato potásico, proporcionó el compuesto 30 con un rendimiento del 50% (véanse comentarios).
Esquema 5
El compuesto 30 transformó en el dienona 1 mediante eliminación de la
función acetálica, por reacción con ácido p-toluensulfónico en acetona acuosa, seguida de hidrogenación, quimio y estereoselectiva, del doble enlace aislado

Síntesis de cortistatina A 21
(esquema 6). Para la culminación de la síntesis de la citostatina A solo restaba la funcionalización del anillo A. Con este fin el anillo ciclohexanónico del compuesto 1 se deshidrogenó mediante su conversión en triflato de enol y reacción subsiguiente con el complejo IBX·MPO (véanse comentarios).
N
Me
H
O
OO
N
Me
H
O
O
N
Me
H
O
O
N
Me
H
O
OH
O
N
Me
H
O
OH
Me2N
HO
130
31
cortistatina A33
1. TsOH (88%)
2. H2, Pd/C (50%)
1. TMSOTf, Et3N
2. IBX·MPO, DMSO(46%, 2 etapas)
t-BuOOH, DBU
DMP
Me2NH, THF
Ti(iPrO)4 (45%)
N
Me
H
O
O
O
NaBH4, CeCl3 (80%)
(70%)32
Esquema 6 La epoxidación estereoselectiva del sistema de enona se consiguió por
adición conjugada 1,4 del anión t-butilhidroperóxido a la cara β de la ciclohexenona 31. A continuación, la reducción del cetoepóxido 32, con NaBH4 y CeCl3, condujo a la formación de una mezcla de epoxialcoholes diastereoisoméricos, en relación 1:1, de la que se separó el α-epoxialcohol 33 mediante cromatografía. La oxidación Dess-Martin del isómero β, seguida de reducción permitió aumentar el rendimiento en la formación del α-epoxialcohol. La reacción de este compuesto con dimetilamina, en presencia de Ti(iPrO)4,

Síntesis de cortistatina A 22
proporcionó una mezcla formada por la cortistatina A(46%) y el regioisómero de apertura del anillo oxiránico (36%). La separación cromatográfica de la mezcla anterior permitió la obtención de la cortistatina A pura.
Comentarios
1. Carboxilación de enonas con carbonato de metilmagnesio
La carboxilación de la enona bicíclica 9 en la carboxienona 15 se consiguió mediante calentamiento de aquélla en DMF, a 125ºC, en presencia de carbonato de metilmagnesio. En el esquema 9 se indica el mecanismo del proceso. El carbonato de metilmagnesio se descompone a 125ºC generando dióxido de carbono y metóxido de magnesio. El metóxido de magnesio provoca la enolización del sistema de enona. En las condiciones termodinámicas en las que tiene lugar la enolización se genera el enolato más estable, que es el que tiene el sistema π extendido (dienolato 34 del esquema 7). La carbonatación del dienolato de magnesio 35 conduce al cetocarboxilato de magnesio 35, estabilizado por quelación. La enolización de este compuesto forma el carboxidienolato 36, que se convierte en el cetoácido 15 en el procesado hidrolítico.
Esquema 7

Síntesis de cortistatina A 23
Esquema 7 (cont.)
2. Reacción de carbometoxilación
OTBSMe
OHO
O
OTBSMe
MeOOCHO
O
1. PhNTf2, NaHMDS
2. Pd(Ph3P)4, COEt3N, MeOH (72%)17 18
En el esquema 8 muestra el ciclo catalítico de la reacción de carbometoxilación del triflato de enol 37 derivado de la cetona 17. El proceso comienza con la etapa de inserción oxidante. El intermedio I que se genera en este proceso se transforma en el intermedio carbonilado II mediante el intercambio de ligandos del paladio con el monóxido de carbono. La etapa de inserción genera el nuevo enlace C-C y conduce al intermedio III , que se transforma en el complejo IV por intercambio de ligandos con el metanol. Finalmente, la etapa de eliminación reductora, que va precedida de la isomerización trans-cis del complejo IV (no dibujada en el esquema 8), proporciona el correspondiente metil éster insaturado 18 y regenera el catalizador. El ácido tríflico que se produce en el ciclo catalítico es neutralizado por la trietilamina.

Síntesis de cortistatina A 24
Esquema 8
3. Acoplamiento de Sonogashira
En el acoplamiento de Sonogashira se crea un nuevo enlace carbono-carbono mediante el acoplamiento de alquinos terminales con haluros o triflatos de arilo o

Síntesis de cortistatina A 25
vinilo. La reacción está catalizada por complejos de paladio y necesita un cocatalizador de Cu(I) y una base nitrogenada. La reacción puede llevarse a cabo en ausencia del catalizador de paladio, pero se ha demostrado que el paladio acelera el proceso y mejora los rendimientos. En este tipo de acoplamiento se genera in situ un reactivo de tipo organocuprato a partir del derivado acetilénico y la sal de cobre, de ahí que, a diferencia del ciclo catalítico que se establece en los acoplamientos de Stille y Suzuki, se establezcan en el acoplamiento de Sonogashira dos ciclos catalíticos independientes (véase el esquema 9).
Esquema 9

Síntesis de cortistatina A 26
4. Desprotección de ditioacetales con IBX
La eliminación de la función ditiocetálica en el sustrato 23 se consiguió mediante reacción con ácido yodoxibenzoico en dimetilsulfóxido. El mecanismo que explica esta reacción se indica en el esquema 10.5 El proceso de desprotección se inicia con la formación del intermedio de tipo sulfonio I , que se genera por adición nucleofílica de uno de los átomos de átomo de azufre del ditiano asistida por el azufre vecinal. A continuación, el ligando óxido de IBX se adiciona a la parte de sulfonio para formar el tioacetal mixto II . Éste experimenta una reacción de transposición que proporciona el aldehído, junto con ácido yodosobenzoico (IBA) y 1,2-ditiolano.
Esquema 10
5 K. C. Nicolaou, C. J. N. Mathison, T. Montagnon J. Am. Chem. Soc. 2004, 126, 5192.

Síntesis de cortistatina A 27
5. Acoplamiento de Suzuki
Lo primeros acoplamientos de tipo Suzuki se restringían a las reacciones,
catalizadas por complejos de paladio(0), entre haluros de arilo y ácidos arilborónicos. El gran desarrollo que han experimentado esta clase de procesos permite en la actualidad el empleo de sustratos electrofílicos tales como haluros de alquenilo, alquinilo e incluso alquilo. Los pseudohaluros, como los triflatos, también pueden intervenir como electrófilos en la reacción de acoplamiento de Suzuki. Por otro lado, el campo de los sustratos nucleofílicos también se han ampliado, y hoy en día se consiguen acoplamientos Suzuki no solo con ácidos borónicos, sino también con organoboranos, ésteres de boronato y trifluoroboratos de potasio.
A diferencia de los acoplamientos de Stille, las reacciones de Suzuki requieren de la activación del ácido, o éster borónico, en el proceso de transmetalación mediante su cuaternización con una base. Si el sustrato orgánico contiene grupos funcionales sensibles a bases se puede conseguir la activación del ácido, o éster borónico, con KF.
En la síntesis de la citostatina A se llevó a cabo el acoplamiento Suzuki entre el triflato de enol 29 y el éster borónico 2. A su vez., este compuesto se sintetizó mediante el acoplamiento Suzuki entre la 7-bromoisoquinolina 39 y el bispinacolato de diboro 40 (véase el esquema 11).
Esquema 11

Síntesis de cortistatina A 28
El ciclo catalítico que explica el acoplamiento Suzuki entre la bromoisoquinolina 39 y el boronato 40 se dibuja en el esquema 12. En esta reacción se emplea como generador del complejo de paladio(0) el complejo de paladio(II) 1,1´-[bis(difenilfosfino)ferroceno]dicloropaladio(II), que genera el 1,1´-[bis(difenilfosfino)ferroceno]paladio(0), mediante reducción in situ (véase estructura I del esquema 14). La adición oxidante del la bromoisoquinolina al complejo de paladio(0) forma el complejo II , con el paladio en estado de oxidación II. Por otro lado, la reacción entre el bispinacolato de diboro 40 y el acetato potásico genera el borato potásico III , que experimenta el proceso de transmetalación con el complejo II con la subsiguiente formación del complejo IV . La eliminación reductora en el complejo IV forma el producto de acoplamiento 2 y regenera el catalizador. La estructura rígida del catalizador de paladio facilita la etapa de eliminación reductora, puesto que en el complejo IV los grupos que se van a unir en el acoplamiento reductor ya se encuentran en posición cis en el complejo cuadrado-plano.
Esquema 12

Síntesis de cortistatina A 29
El mecanismo del acoplamiento Suzuki entre el triflato de enol 29 y el borato 2 se indica en el esquema 14. En esta reacción el complejo de paladio(0) que inicia el ciclo catalítico es el Pd(PPh3)2, y la base empleada en la cuaternización del pinacolboronato es el carbonato potásico. El ciclo catalítico del esquema 15 tiene un paso más que el ciclo catalítico en el que actúa el catalizador Pd(dppf)2, porque en aquél se tiene que producir la isomerización del complejo III , de geometría trans.
Esquema 13
8. Deshidrogenación de silil enol éteres con IBX

Síntesis de cortistatina A 30
El grupo de K. C. Nicolaou ha demostrado que los aldehídos y cetonas se pueden convertir en los correspondientes compuestos α,β-insaturados mediante deshidrogenación con ácido orto-yodoxibenzoico (IBX) en DMSO. Este grupo también ha descubierto que determinados ligandos, como los N-óxido de aminas, interaccionan con el IBX para formar complejos que son capaces de provocar la deshidrogenación de aldehídos y cetonas a temperatura ambiente (esquema 15).6
Además de los aldehídos y las cetonas, los silil enol éteres derivados de estos compuestos también se pueden deshidrogenar eficientemente con los complejos derivados de IBX, como el que se forma en la reacción de éste con el N-óxido de 4-metoxipiridina. Las reacciones ajustadas de estos procesos se indican a continuación:
Esquema 15
En el esquema 16 se indica el mecanismo de deshidrogenación del silil enol éter 41, que se inicia con el ataque de éste al complejo IBX·NMO. Esta reacción forma el intermedio I , el cual experimenta un proceso de transferencia monoelectrónica (SET) intramolecular y se convierte en el dirradical II . Este
6 K. C. Nicolaou, D. L. F. Gray, T. Montagnon, S. T. Harrison Angew. Chem. Int. Ed. 2002, 41, 996.

Síntesis de cortistatina A 31
intermedio, a través de la forma resonante III , sufre un proceso de eliminación que proporciona la enona 32, ácido yodosobenzoico (IBA) y el N-óxido de la p-metoxianilina.
NaHMDS, TMSCl
SET
+N
OMe
O
OI
O
HO
+
IBA
III
III
O
O
41
O
O
1
SiMe3
OI
O
HO OON
MeO
IBX·MPO
Me3SiOH
O
O
OI
O
O
ON
MeOO
O
OI
O
O
ON
MeO
O
O
OI
O
O
ON
MeO H31
O
O
Esquema 16


Síntesis de (Z)-desoxipukálido 33
SÍNTESIS DE (Z)-DESOXIPUKÁLIDO
Aislamiento: El (Z)-desoxipukálido se ha aislado de Leptogorgia sp., un coral blando (octocoral) que se encuentra en el océano Pacífico. Actividad biológica: El (Z)-desoxipukálido se encuadra dentro de la familia de los cembranólidos furánicos. Muchos miembros de esta familia de productos naturales son neurotóxicos y algunos exhiben actividad antiinflamatoria y antialimentaria.
Retrosíntesis
El (Z)-desoxipukálido contiene en su estructura un anillo furánico y uno de tipo furan-2(5H)-ona (anillo lactónico). Como se verá a continuación, ambos anillos se desconectarán a sistemas abiertos mediante la aplicación en el análisis retrosintético de la reacción de metátesis ciclante (RCM). De hecho la primera operación del análisis retrosintético se encarga de la desconexión, basada en una reacción RCM, del doble enlace C11-C12 contenido en el anillo lactónico (véase el esquema 1).7 Esta operación origina la furanolactona 1, la cual, por desconexión del enlace éster conduce al furanohidroxiácido 2. La desconexión del enlace C6-C7, basada en una reacción de acoplamiento catalizada por paladio, proporciona el intermedio electrofílico 3 (X=halógeno) y el componente nucleofílico 4 (M=metal).
Una operación de intercambio de grupo funcional en el intermedio 3 conduce al diol 5. Este compuesto se sintetizará mediante la reacción de carbo-halogenación del triple enlace que contiene el alquinodiol 6, el cual se obtendrá del epóxido 7 mediante apertura regioselectiva del anillo oxiránico con un acetiluro metálico.
El intermedio organometálico 4 se preparará por metalación regioselectiva del furano-ácido 8. Este intermedio se transforma en el cetofurano 9 mediante una operación retrosintética de intercambio de grupo funcional. En esta operación retrosintética también se ha convertido la parte de α-metilenácido (carbonos C12 y
7 T. J. Donohoe, A. Ironmonger, N. M. Kershaw, Angew. Chem. Int. Ed. 2008, 47, 7314.

Síntesis de (Z)-desoxipukálido 34
C20) en un sistema de α-hidroxicetona, protegido en el grupo hidroxilo. La operación clave que permite continuar el análisis del compuesto 9 es una interconversión de grupo funcional sobre el anillo furánico, que convierte a éste en un anillo dihidrofuránico como el que se indica en la estructura 10 (obsérvese la presencia de la agrupación acetálica en el anillo dihidrofuránico).
Esquema 1
La conversión del anillo furánico en uno dihidrofuránico permite la
desconexión del doble enlace de éste mediante una operación basada en una

Síntesis de (Z)-desoxipukálido 35
reacción de metátesis ciclante. El resultado de este proceso es la generación del éster triolefínico acíclico 11, que se desconecta en la función acetálica al α-metilen-β-hidroxiéster 12. La desconexión del enlace C3-C4 origina el sintón organometálico 13 (M=metal) y el aldehído 14, el cual, por reconexión 1,6 de las funciones carbonílicas conduce al alcohol ciclohexénico quiral 15, que es el alcohol (S)-perillílico, un alcohol monoterpénico de origen natural, comercialmente accesible.
Síntesis
1. Síntesis del furano-ácido 8
Como se ha explicado en el análisis retrosintético, el material quiral de partida para la preparación del furano-ácido 8 fue el alcohol (S)-perillílico 15. Todo el sistema carbonado de 15 se integrará en la estructura del (Z)-desoxipukálido, y el centro estereogénico de aquél constituirá el estereocentro en C1 del cembranólido. La síntesis del compuesto 8 se inició con la sililación del alcohol (S)-perillílico 15, que fue seguida de una reacción de ozonolisis. La regioselectividad en la ruptura oxidante del doble enlace trisustituido, más nucleofílico que el disustituido, se consiguió modulando la reactividad del ozono mediante adición de una cantidad estequiométrica de piridina (véase el esquema 2). Además, la reacción de ozonolisis se llevó a cabo en presencia de un exceso de isopreno, que actuó de protector externo del doble enlace disustituido, al ser oxidado con preferencia a éste. El resultado de la reacción de ozonolisis del alcohol 15 fue la generación del cetoaldehído 14. Este compuesto se transformó en el α-metilen-β-hidroxiácido 12 mediante reacción con el vinilalano generado in situ por adición, en presencia de HMPA, de hidruro de diisobutilaluminio (DIBAL) al propiolato de etilo (véanse comentarios). La transcetalización del hidroxiácido 12 con el dietil acetal de la acroleína, en presencia de p-toluensulfonato de piridinio PPTS, proporcionó el acetal mixto 11. Cuando este compuesto se sometió a la reacción de metátesis

Síntesis de (Z)-desoxipukálido 36
ciclante con el catalizador de Grubbs de 2ª generación, en diclorometano a reflujo, se generó, después de 16 horas de reacción, el acetal dihidrofuránico 10. A continuación, se añadió a la mezcla de reacción PPTS y el reflujo se continuó durante 2 horas más. El resultado de todo este proceso fue la obtención del compuesto furánico 9 con un rendimiento del 85% desde el β-hidroxiéster 12 (véanse comentarios). La metilenación de Wittig del carbonilo cetónico de 9, seguida de desililación, condujo al hidroxiéster diolefínico 18, el cual, mediante oxidación de Zhao, se transformó en el furano-ácido 8 (véanse comentarios).
OH
1. TIPSCl, imidazol2. O3, piridina, isopreno
O
OTIPS
CHO DIBAL, HMPA
COOMe
(48% 3 etapas)
O
TIPSO
HO
COOMe
EtO OEt
PPTS
O
TIPSO
O
COOMe
EtO
O
COOMe
EtO
10
TIPSO
O
O
COOMe
TIPSO
O
PPTS
(85% 3 etapas)
9
1. Ph3P=CH22. TBAF, THF
O
COOMe
HOTEMPO, NaClO2
NaOCl, CH3CN, H2OO
COOMe
O
HO(90% 3 etapas)
N N
RuPh
PCy3Cl
Cl
CH2Cl2, reflujo
15 14
1211
1617
188
Esquema 2

Síntesis de (Z)-desoxipukálido 37
2. Síntesis del yoduro de alquenilo 3
TBSO
I
3
O
COOMe
O
O
(Z)-desoxipukálido
1
3
12
6
11
20
15
7
101110
8 98 9
7
1919
El yoduro de alquenilo 3 contiene el segundo estereocentro del cembranólido, el situado en C10, y el sistema carbonado que va desde C7 hasta C11, además del grupo metilo C19. Para la construcción de este fragmento se eligió como material quiral de partida el (S)-glicidol 7. La sililación de este compuesto, seguida de reacción con el trimetilsililacetiluro de litio, en presencia de BF3·Et2O, proporcionó el alcohol homopropargílico 20. La eliminación del grupo TBS, por reacción con HCl en metanol, seguida de eliminación del grupo TMS con carbonato potásico, condujo al diol acetilénico 6.
Esquema 3

Síntesis de (Z)-desoxipukálido 38
El compuesto 6 se hizo reaccionar con trimetilaluminio en presencia de Cp2ZrCl2 (dicloruro de bis(ciclopentadienil)zirconio) en 1,2-dicloroetano, primero a 0ºC y luego a reflujo durante 3 días. El vinilalano generado en este proceso se adicionó, a -20ºC, a una disolución tetrahidrofuránica de yodo molecular. El resultado fue la obtención del (Z)-yoduro de alquenilo 21 con un rendimiento del 56% (véanse comentarios). La reacción de 21 con NaH y trisilimidazol 1-(2,4,6-triisopropilbencenosulfonil)imidazol, en THF, provocó la sulfonilación del hidroxilo primario y la subsiguiente formación del anillo oxiránico mediante reacción de desplazamiento intramolecular, tipo SN2, del grupo 1-(2,4,6-triisopropilbencenosulfonato. De esta forma se obtuvo, en un solo paso operativo, el epóxido 21, el cual, por reacción con el iluro generado mediante ionización del yoduro de trimetilsulfonio, se transformó en el alcohol alílico 23. La sililación de este compuesto llevó al yoduro de alquenilo 3.
3. Acoplamiento de los fragmentos y pasos finales
El acoplamiento de Negishi del yoduro de alquenilo 3 con el furano-ácido 8 se inició con la metalación regioselectiva del anillo furánico en este último compuesto. El proceso se llevó a cabo mediante litiación del carbono C6, por reacción con n-BuLi, seguida de transmetalación con ZnBr2. Esta secuencia de reacciones generó el derivado organozíncico 4, al que se le añadió una disolución tetrahidrofuránica compuesta por el yoduro de alquenilo 3, y el catalizador de paladio Pd(dppf)Cl2 (dicloro[1,1-bis(difenilfosfino)ferroceno]paladio (II)). Después de calentar a 50ºC durante 4 horas se obtuvo el producto de acoplamiento 24, con un rendimiento del 78% desde el compuesto 8 (véanse comentarios). La desililación de 24 proporcionó el hidroxiácido 2, que se sometió al proceso de lactonización en las condiciones de Shiina, mediante reacción con el anhidrido del ácido 2-metil-6-nitrobenzoico (MNBA) en presencia de Et3N y de 4-N,N-dimetilaminopiridina (DMAP) (véase pág. 65). Este proceso condujo a la lactona 1,

Síntesis de (Z)-desoxipukálido 39
que se convirtió en el (Z)-desoxipukálido mediante metátesis ciclante con el catalizador de Grubbs de 2ª generación.
TBSO
I
3
O
COOMe
O
O(Z)-desoxipukálido
8
O
COOMe
OHO
LDA, luego ZnBr2
O
COOMe
OLiO
BrZn
4
PdCl2(dppf),
luego workup
con NH4Cl ac.
O
COOMe
OTBS
HOOC
TBAF, THF
(78% desde 8)
O
COOMe
OH
HOOC
MNBA, Et3N, DMAP
(73%)
O
COOMe
O
O
N N
RuPh
PCy3Cl
Cl
tolueno, calor
(72%)
THF, -78ºC
6
6
7
6
7
242
1
Esquema 3
Comentarios
1. Síntesis del ββββ-hidroxi- αααα-metilenéster 12
Para la síntesis del compuesto 12 se utilizó el vinilalano generado in situ por
reacción del propiolato de metilo con DIBAL en presencia de HMPA. Así, una

Síntesis de (Z)-desoxipukálido 40
disolución toluénica de DIBAL se adicionó a una disolución de HMPA en THF. Luego
se añadió propiolato de metilo y después de 1.5 horas de agitación a 0ºC se añadió el
aldehído 14 disuelto en THF. La mezcla resultante se calentó a 45ºC durante 16 horas.
Operando en estas condiciones se obtuvo el β-hidroxi-α-metilenéster 12, con un 48% desde el alcohol perillílico 15.
En el esquema 4 se indica el mecanismo de este proceso, que se basa en la generación del vinilalano 13, por hidroaluminación regioselectiva del triple enlace del propiolato de metilo. Este intermedio organometálico se adiciona al aldehído con la subsiguiente formación del intermedio alcoxialumínico 25, el cual se transforma en el correspondiente β-hidroxi-α-metilenéster en el proceso de workup.
El HMPA disminuye la capacidad reductora del DIBAL, actuando como un ligando del aluminio, mas que como un cosolvente del proceso: la presencia de HMPA es vital para el éxito del proceso, puesto que este aditivo favorece la adición conjugada de hidruro al inoéster. En su ausencia se produce la reducción de la función éster.8
Esquema 4
8 T. Tsuda, T. Yoshida, T. Kawamoto, T. Saegusa, J. Org. Chem. 1987, 52, 1624.

Síntesis de (Z)-desoxipukálido 41
2. Síntesis del compuesto furánico 9
El anillo furánico que contiene el compuesto 9 se construyó en dos pasos a
partir del acetal mixto 11. En el primero de ellos el compuesto 11 se convirtió en el dihidrofurano 10 mediante reacción de metátesis ciclante con el catalizador de Grubbs de 2ª generación, en diclorometano a reflujo. En el segundo paso, el dihidrofurano 10 se transformó en el furano 9 por reacción con p-toluensulfonato de piridinio en diclorometano a reflujo. De hecho, el compuesto 10 no se llegó a aislar, puesto que se transformó directamente en el furano 9 por adición de PPTS a la mezcla de metátesis, seguida de calentamiento a reflujo de diclorometano durante 2 horas más. El mecanismo que explica la conversión de 10 en 9 se indica en el esquema 5, y se inicia con la reacción ácido-base entre la parte acetálica del dihidrofurano 10 y el PPTS. Esta interacción genera el acetal protonado I , piridina y el catión p-toluensulfonato (paso 1 del esquema 5). A continuación, el acetal protonado elimina etanol y se convierte en el cation oxonio II , el cual se transforma en el furano 9 mediante desprotonación provocada por el ataque básico de la piridina.
Esquema 5

Síntesis de (Z)-desoxipukálido 42
3. Oxidación de Zhao (modificación de la reacción de Anelli)
En 1987 Anelli y colaboradores demostraron que los alcoholes primarios se podían oxidar a aldehídos, o a ácidos carboxílicos, por reacción con hipoclorito sódico en una mezcla bifásica CH2Cl2/H2O, en presencia de bicarbonato sódico, bromuro potásico y una cantidad catalítica de 4-metoxi-2,2,6,6-tetrametilpiperidina-1-oxilo (4-MeO-TEMPO).9 La oxidación se podía parar en la etapa de aldehído si se acortaba el tiempo de la reacción. De manera alternativa, la oxidación hasta ácido carboxílico se podía conseguir de forma rápida por adición de un catalizador de transferencia de fase.
R CH2OH
N
O
NaClO,
NaHCO3, KBr, CH2Cl2/H2Ocat. transferencia de fase
R COOH
OMe
El mecanismo de la oxidación de Anelli se indica en el esquema 6 y se inicia
con la formación de la sal oxoamónica I por oxidación del 4-MeO-TEMPO con el hipoclorito. El ataque nucleofílico del alcohol a la sal oxoamónica genera la especie N-oxoamónica II , que origina el aldehído y la hidroxilamina III . La oxidación de la hidroxilamina por reacción con el ácido hipobromoso regenera la sal oxoamónica (el HOBr es mejor oxidante para la regeneración de la sal oxoamónica que el HOCl). El ácido hipobromoso se produce en el ciclo catalítico secundario por oxidación del bromuro con hipoclorito.
La reacción es bastante lenta al pH del hipoclorito sódico (pH=12.7) y mucho más rápida a pH=8.6, que es el que se genera cuando se adiciona NaHCO3.
9 P. L. Anelli, C. Biffi, F. Montanari, S. Quici, J. Org. Chem. 1987, 52, 2559.

Síntesis de (Z)-desoxipukálido 43
A pH muy básico la concentración de HOBr, el oxidante que regenera la sal oxoamónica, es muy baja en relación a la concentración del anión hipobromito.
N
OH
N
O O
H R
H
N
O Br
RCH2O
N
O
HOBr
BrNaOCl
oxidante
estequiométrico oxidante
catalítico
R
O
H
R
OH
H
OHR
O
OH
H2O
OMeOMe OMe
Cl
BrOel mismo mecanismo
que en la oxidación de
alcoholes a aldehídos
OMe
III
III
NaOCl
Esquema 6 Paradójicamente, la velocidad de la oxidación disminuye al incrementar la
temperatura. Este hecho se debe a la descomposición de la sal oxoamónica, que es muy estable a 0ºC, pero que descompone rápidamente en presencia de agua a 25ºC.
Por otro lado, la oxidación del aldehído al ácido carboxílico está mediada también por la sal oxoamónica, más que por el hipoclorito. De hecho, en ausencia de los radicales TEMPO, la velocidad de oxidación de aldehídos hasta ácidos carboxílicos es muy lenta.
Aunque el método original de Anelli utilizaba 4-MeO-TEMPO, otros derivados como 4-OH-TEMPO, 4-AcNH-TEMPO o el propio TEMPO, son igualmente eficaces en la reacción de oxidación. Usualmente un 1 mol% del radical TEMPO es suficiente para que la oxidación transcurra de forma eficiente.
Una limitación importante del método de oxidación de Anellli es la utilización de una cantidad estequiométrica de NaClO, un reactivo que puede provocar cloraciones en determinados sustratos. Esta limitación del método de

Síntesis de (Z)-desoxipukálido 44
Annelli puede ser evitada si se emplea la modificación de Zhao.10 En esta modificación, el oxidante secundario, el hipoclorito sódico, se emplea en cantidades catalíticas, y se regenera en el seno de la reacción por adición a ésta de clorito sódico, un compuesto que no provoca reacciones de cloración.
El mecanismo de la reacción de Zhao se inicia con la oxidación del radical TEMPO a la sal oxoamónica I por interacción con el hipoclorito sódico (véase el esquema 7.
N
OH
N
O O
H R
H
N
O Cl
RCH2O
N
O
NaOCl
NaCl
NaClO2
oxidanteestequiométrico
oxidantecatalítico
R
O
H
NaClO2
R
OH
H
O
Cl
O
R
O
OH+H O Cl
H2O
NaOCl
III
III
Esquema 7
La sal oxoamónica es atacada nucleofílicamente por el alcohol y convertida en la especie N-alcoxiamónica II , que genera el aldehído y la hidroxilamina III . El
10 M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski, P. J. Reider, J. Org. Chem. 1999, 64, 2564.

Síntesis de (Z)-desoxipukálido 45
clorito sódico oxida el aldehído a ácido carboxílico, mientras que la hidroxilamina III se convierte en la sal oxoamónica I por reacción con hipoclorito sódico.
En ausencia de hipoclorito sódico la reacción necesita un largo periodo de inducción antes de su iniciación. Esto es debido a lenta oxidación del radical TEMPO, o de la hidroxilamina, por parte del clorito sódico. Sin embargo, una vez iniciado el proceso éste se auto-sustenta debido a la continua regeneración del hipoclorito sódico. El problema de cloración que provoca este reactivo en el método de Anelli, queda muy disminuido en la modificación de Zhao, porque la concentración de NaOCl permanece en valores muy bajos a lo largo de todo el proceso de oxidación. El riesgo de epimerización cuando se generan aldehídos α-quirales también queda muy disminuido en el método de Zhao, debido a la rápida oxidación que éstos experimentan con el clorito sódico.
4. anti-Carboaluminación-yodonolisis de inoles homopropargílicos
La reacción entre alquinos y triaalquilalanos, como AlMe3, catalizada por
complejos de zirconio, proporciona los correspondientes productos de sin-carboaluminación, los cuales reaccionan con especies electrofílicas para dar lugar a los productos indicados en el esquema 8:
Esquema 8 El primer mecanismo propuesto para la reacción de carboaluminación,
catalizada por el complejo Cp2ZrCl2, se indica en el esquema 9. El mecanismo se basa en un intercambio Me-Cl reversible entre el Me3Al y el Cp2ZrCl2 (véanse intermedios I y II del esquema 9). La subsiguiente etapa de coordinación del alquino al átomo de zirconio genera el intermedio III , que es el que experimenta la reacción de carbozirconación del triple enlace, con formación del complejo IV .

Síntesis de (Z)-desoxipukálido 46
Una reacción de transmetalación intramolecular en este intermedio conduce al alquenilalano V, que forma el producto de carboaluminación y regenera el catalizador Cp2ZrCl2.
R
MeAl
Me
Me
Zr
Cl
Cl
MeAl
Me
Me
Zr
Cl
Cl
MeAl
Me
Cl
Zr
Cl
Me
MeAl
Me
ZrCl Me
Al
Me
Me
MeR
R
I
II
III
R
R RClIV
MeAl
Me
ZrCl Me
Cl MeR
R
Al
Me
Me
Zr
Cl
Cl
V
MeR
R
Esquema 9
Esta propuesta mecanística se modificó por el grupo de Negishi, el cual, basándose en experimentos de RMN, concluyó que la reacción tenía lugar mediante carboaluminación directa del alquino. En el esquema 10 se describe el mecanismo modificado, en el que se observa la coordinación del alquino al átomo de aluminio (intermedio VI del esquema 10), y la subsiguiente formación del alquenilalano VII .11
11 T. Yoshida, E. Negishi, J. Am. Chem. Soc. 1981, 103, 4985.

Síntesis de (Z)-desoxipukálido 47
Esquema 10 Si el sustrato sobre el que tiene lugar la carboaluminación es un alcohol
homopropargílico el producto de la reacción es un vinilalcoxialano, indicado con la estructura I en el esquema 11. Cuando este producto reacciona con electrófilos, como el yodo molecular, se forma el (E)-yoduro de alquenilo II , que en el workup hidrolítico conduce a la obtención del (E)-yodoalcohol homopropargílico III .
Esquema 11
Si el (E)-vinilalcoxialano I se calienta a reflujo de 1,2-dicloroetano (DCE) se provoca una reacción de isomerización que convierte a aquél en el isómero Z.12 El
12 S. Ma, E. Negishi, J. Org. Chem. 1997, 62, 784.

Síntesis de (Z)-desoxipukálido 48
mecanismo de esta transformación se indica en el esquema 12, y se inicia con la aluminación electrofílica intramolecular del doble enlace. Este proceso genera el intermedio IV , que se convierte en el alanaciclo V mediante transposición de metilo. La eliminación de trimetilaluminio forma el alanaciclohexeno VI , el cual, por reacción con especies electrofílicas como el yodo molecular, se convierte en el (Z)-yoduro de alquenilo VII . Finalmente, el proceso de workup hidrolítico proporciona el (Z)-yodoalcohol homopropargílico VIII .
O
H
Me AlMe2
AlMe
MeO
Al
Me AlH
Me
Me
Me
Me
O
Al
Me AlH Me
Me
Me
Me
O
Al
Me H
Me
AlMe3
O
I
Me H
Al
Me
IOH
I
Me H
workup
I IV V
VIVIIVIII
I2
Esquema 12 5. Síntesis del compuesto furánico 24 mediante acoplamiento de Negishi
Las reacciones de acoplamiento de Negishi permiten la unión entre reactivos organometálicos de tipo organozíncico y sustratos electrofílicos, usualmente haluros de alquilo, alquenilo, alquinilo o arilo.
El componente organometálico necesario para la síntesis del furano 24, mediante acoplamiento de tipo Negishi, se generó, in situ, por litiación del furano-ácido 8, seguida de transmetalación con bromuro de zinc (esquema 13).

Síntesis de (Z)-desoxipukálido 49
Esquema 13
La adición del yoduro de alquenilo 3, y del catalizador de paladio PdCl2(dppf), a la mezcla reactiva que contenía el organozíncico 27, desencadenó el proceso de acoplamiento y permitió la obtención del derivado furánico 24. En el esquema 14 se indica el mecanismo de la reacción.
Esquema 14 El catalizador que opera en el proceso (Pd(dppf), estructura I del esquema
13), se genera en el seno de la reacción por reducción del catalizador del PdCl2(dppf). En la primera etapa del ciclo catalítico se produce la inserción

Síntesis de (Z)-desoxipukálido 50
oxidante del yoduro de alquenilo 3 al catalizador I . Esta reacción genera el complejo II , el cual experimenta el proceso de trans-metalación con el compuesto organozíncico 27, con formación subsiguiente del complejo III . Finalmente, la eliminación reductora en el complejo III proporciona el producto de acoplamiento y regenera el catalizador.
La ventaja del catalizador Pd(dppf) reside en su estructura cíclica rígida, que origina complejos cis-cuadrado-planos de paladio (complejos II y III del esquema 14), que no pueden experimentar isomerización a complejos trans. Esta situación es particularmente favorable en el caso del complejo II , que experimenta directamente la reacción de acoplamiento reductor.

Síntesis de didemniserinolípido B 51
SÍNTESIS DE DIDEMNISERINOLÍPIDO B
Aislamiento: El didemniserinolípido B se ha aislado de un extracto del tunicado Didemnum sp. Actividad biológica: Aunque el didemniserinolípido B se ha obtenido de un extracto citotóxico del tunicado arriba mencionado, él mismo no posee actividad citotóxica. Un macrociclo relacionado, denominado ciclodidemniserinol trisulfato, ha demostrado ser inhibidor de la integrasa HIV-1.
Retrosíntesis
El análisis retrosintético del didemniserinolípido B se indica en el esquema 1, y se inicia con una operación de intercambio y adición de grupos funcionales (IGF/AGF) que conduce a la estructura 1.13 Este intermedio se ha generado a partir del producto natural mediante introducción de un doble enlace entre los carbonos C6-C7, y mediante la sustitución del doble enlace entre C2-C3 por un sistema saturado que posee en C2 una funcionalidad, indicada como X en la estructura 1. Esta funcionalidad X debe ser de tal naturaleza que permita, en su momento, la construcción del sistema de éster insaturado que contiene el didemniserinolípido B.
Por otro lado, la instalación del doble enlace entre C6-C7 en el compuesto 1
facilita la segunda operación del análisis retrosintético, que desconecta este enlace y genera los fragmentos 2 y 3. En el sentido sintético estos dos intermedios se conectarán mediante una reacción de metátesis cruzada.
El alqueno 2, mediante una operación de intercambio de grupo funcional, conduce al dieno 4. El fragmento de aminodiol que contiene el intermedio 4 se instalará mediante una reacción SN2 del aminodiol 6, adecuadamente protegido, sobre el dieno 5, en el cual Y simboliza a un grupo saliente. El aminodiol 6 se obtendrá de la D-serina.
13 C. C. Marvin, E. A. Voight, S. D. Burke, Org. Lett. 2007, 9, 5357.

Síntesis de didemniserinolípido B 52
Por otro lado, la desconexión del doble enlace del anillo dihidropiránico de 5 proporciona el trieno 7. En el sentido sintético el anillo dihidropiránico se construirá mediante una reacción de metátesis intramolecular. El compuesto 7 contiene una funcionalidad acetálica, cuya desconexión genera la enona funcionalizada 8 y el diol quiral 9. La enona 8 se sintetizará a partir del 16-hexadecanólido 10, mientras que el diol quiral 9 se obtendrá del D-manitol.
O
O
OO
NH2
S
OO
NaOH
OH
EtOOC
didemniserinolípido B
2930
31
1
8
9
10
13
28
COOH
HO
NH2
29
3031
D-serina
6
O
OO
P´O
NHP
H
EtOOC
2930
31
1
X
67
2
metátesiscruzada
EtOOC1
X
6
2
12
3
IGF/AGF
IGF
15O
OO
P´O
NHP
H
OH
2930
31
7
15
+
O
OY
H
7
15
1011
metátesisintramolecular
45
O
OY
H
7
15
1011
7
8
9
HO
HO
Y
O +
O
O
12
8 9
10
cetalización
OH
OHOH
OHOH
OH
89
7
10
7
10
D-manitol
OH
P´O
NHP
2930
31 +
OH
O
OO
P´O
NHP
H29
3031
7
15
11
8
9
15
SN2
Esquema 1

Síntesis de didemniserinolípido B 53
Síntesis 1. Síntesis de la enona funcionalizada 12
La enona 12 no se corresponde estrictamente con el fragmento 8 del análisis retrosintético, puesto que aquélla contiene un anillo aromático conjugado con el doble enlace. La presencia de la agrupación fenílica en la enona 12, como se verá a continuación, permite la colocación del doble enlace en la posición adecuada para llevar a cabo la proyectada reacción de metátesis intramolecular. Así, la preparación de 12 se inició con la adición del anión lítico derivado del metilfosfonato de dimetilo al 16-hexadecanólido 10 (esquema 2). Este proceso proporcionó el β−cetofosfonato 11, que se sometió a la reacción de olefinación Horner-Wadsworth-Emmonss con fenilacetaldehído y carbonato potásico, en metanol acuoso a reflujo. En estas condiciones, el anillo fenílico provocó la isomerización del doble enlace desde la posición original α,β a la posición β,γ obteniéndose, después de la mesilación del hidroxilo libre, la mesiloxienona 12.
Esquema 2

Síntesis de didemniserinolípido B 54
2. Síntesis del diol quiral 9
El diol quiral 9 se obtuvo a partir del D-manitol (esquema 3). Así, el tratamiento del D-manitol con bromuro de acetilo, seguida de acetilación del crudo de reacción, proporcionó el dibromoderivado 13 (véanse comentarios). El diol 9 se obtuvo por eliminación reductiva de 13 mediante reacción con zinc metálico, seguida de saponificación.
OH
OHOH
OHOH
OH
1. CH3COBr1,4-dioxano
2. Ac2O, piridina(62%)
Br
BrOAc
OAcOAc
OAc
1. Zn, NaOAcAcOH, reflujo
2. NaOMe, MeOH(81%)
OH
OH
9D-manitol 13
Esquema 3 3. Síntesis del intermedio bicíclico 4
La preparación del intermedio 4 se inició con la reacción de cetalización entre la enona 12 y el diol 9, que se consiguió por reacción a reflujo de benceno, en presencia de cantidades catalíticas de ácido canforsulfónico (esquema 4). Este proceso proporcionó el acetal 14, que se convirtió en el compuesto bicíclico 5

Síntesis de didemniserinolípido B 55
mediante reacción de metátesis intramolecular promovida por el catalizador de Grubbs de 1ª generación (véanse comentarios). La ionización del alcohol 6 (obtenido por reducción del aldehído de Garner) y la subsiguiente reacción SN2 sobre el mesilato 5 proporcionó el intermedio 4.
Esquema 4
4. Pasos finales
O
O
OO
NH2
S
OO
NaOH
OH
EtOOC
2930
31
1
8
9
10
15
7
O
OO
HO
NHFmoc
H
OH
EtOOC
2930
31
1
SePh
67
21
15
didemniserinolípido B
La conversión del intermedio 4 en el selenoéster 1 y la transformación de
éste en el didemniserinolípido B se indica en el esquema 5.

Síntesis de didemniserinolípido B 56
16
2
O
OOO
NBoc
H15
4
MCPBA, CH2Cl2
0-4ºC (60%)
O
OOO
NBoc
H15
10
11 O
15
LiAlH4THF (86%)
O
OOO
NBoc
H15
10
OH
2
EtOOC
SePh
RuCl
Cl
Cy3PPh
, CH2Cl2 (74%)
NNMes Mes
O
OOO
NBoc
H
OH
EtOOC1
SePh
67
2
15
NaOAc, H2OpCH3C6H4SO2NHNH2DME, reflujo (96%)
O
OOO
NBoc
H
OH
EtOOC1
SePh
67
2
15
1
16
MCBPA, CH2Cl2luego Et3N (89%)
O
OOO
NBoc
H
OH
EtOOC
15
17
HCl, EtOH(75%)
O
OOHO
NH2·HCl
H
OH
EtOOC1
15
18
FMocOSu, K2CO3H2O, THF (>99%)O
OOHO
NHFmoc
H
OH
EtOOC
15
19
SO3·pir, Na2SO4DMF, 110ºC, MW
O
OOO
NHFmoc
H
OH
EtOOC
31
15
20
S
NaO
O O
DMF, piperidina
O
OOO
NH2
H
OH
EtOOC
15
didemniserinolipido B
SNaO
O O
(27%, desde 19)
3
Esquema 5

Síntesis de didemniserinolípido B 57
La epoxidación regio y estereoselectiva del doble enlace endocíclico en el intermedio 4 se consiguió por reacción de éste con 1 equivalente de ácido m-cloroperoxibenzoico. Este proceso proporcionó el epóxido 15, que por reacción con aluminiohidruro de litio experimentó la apertura reductiva trans-diaxial del anillo oxiránico para dar el alcohol insaturado 2. El fragmento C1-C6 se instaló mediante calentamiento, a reflujo en diclorometano, de una mezcla del alcohol insaturado 2 (1 equivalente) y del selenoéster 3 (14 equivalentes) en presencia del catalizador de Grubbs de 2ª generación. En estas condiciones se obtuvo el compuesto de metátesis cruzada 1 con un rendimiento del 74% (véanse comentarios). La hidrogenación del doble enlace no funcionó cuando se empleó hidrógeno molecular en presencia de Pd/C, pero se pudo conseguir mediante la reacción de 1 con diimida. El producto de reducción 16, se convirtió en el éster α,β-insaturado 17 por oxidación con ácido m-cloroperoxibenzoico (véanse comentarios). La reacción de 17 con HCl 1N en etanol provocó la metanolisis ácida de la parte de serinol y condujo a la obtención del aminoalcohol 18, que se protegió quimioselectivamente en el grupo amino por reacción con carbonato de 9-fluorenilmetil succinimidilo. La introducción de la funcionalidad sulfato se consiguió mediante calentamiento en microondas, en presencia de sulfato sódico, de una disolución en dimetilfomamida del alcohol 19 y del complejo trióxido de azufre-piridina. Este proceso condujo a la obtención del correspondiente sulfato sódico que por Fmoc desprotección con piperidina proporcionó el didemniserinolípido B sintético (véanse comentarios).
Comentarios
1. Bromoacetilación de dioles
La reacción del D-manitol con bromuro de acetilo, seguida de esterificación
con anhidrido acético, proporciona el dibromo-tetraacetoxiderivado 13.14 El mecanismo de la primera de las reacciones se indica en el esquema 6. El proceso se inicia con la esterificación del hidroxilo primario, estéricamente más accesible.
14 C. Crombez-Robert, M. Benazza, C. Fréchou, G. Demailly, Carbohydr. Res. 1997, 303, 359.

Síntesis de didemniserinolípido B 58
Esta reacción forma el hidroxiacetato 21 y bromuro de hidrógeno. La presencia del ácido provoca la protonación de la función acetato, que es atacada intramolecularmente por el hidroxilo vecinal con formación del intermedio 23. El intercambio protónico y la subsiguiente deshidratación conducen al catión acetoxonio cíclico 25, que se transforma en el bromoacetato 26 por ataque regioselectivo del anión bromuro.
Esquema 6
El proceso de bromoacetilación del D-manitol forma el compuesto 27, que por acetilación de los hidroxilos remanentes se transforma en el dibromo-tetraacetoxiderivado 13 (esquema 7).
Esquema 7

Síntesis de didemniserinolípido B 59
2. Síntesis del compuesto bicíclico 5 mediante reacción de metátesis ciclante
La agitación de una disolución diclorometánica del compuesto 14, a
temperatura ambiente durante 40 horas, en presencia del catalizador de Grubbs de 1ª generación, proporcionó el compuesto 5 con un 53% de rendimiento (81% con respecto a producto de partida recuperado). Además del producto de metátesis intramolecular se obtuvo un 10% del producto 28, resultante de la metátesis cruzada con el estireno (véase el esquema 8), y 28% de producto de partida inalterado.
Esquema 8
En el esquema 9 se indica el ciclo catalítico de la reacción de metátesis
ciclante sobre el trieno 14. El intermedio I , que se genera como consecuencia de la cicloadición intermolecular entre el catalizador y el compuesto 14, forma, como consecuencia del proceso de ciclorreversión, el intermedio II y estireno. La subsiguiente cicloadición intramolecular, seguida de ciclorreversión, conduce al producto de metátesis y a la regeneración del catalizador.
El compuesto 28 se forma mediante la reacción de metátesis cruzada entre el producto 5 y el estireno, que se genera en el seno de la reacción en el proceso de metátesis intramolecular, según se indica en el esquema anterior. El ciclo catalítico que explica la formación de 28 se indica en el esquema 10.

Síntesis de didemniserinolípido B 60
Ru CHPh
cicloadiciónintermolecular
ciclorreversión
cicloadiciónintramolecular
ciclorreversión
I
II
III
O
O
MsO H
7
15
1011
Ph
14
O
OMsO
H
7
15
10
11
5
O
O
MsOH15
10
11
Ph
7
Ru
O
O
MsOH15
10
11
Ru
Ph
7
H2C CHPh
O
O
MsOH15
10
11Ru
7
PhPh
Esquema 9
Esquema 10

Síntesis de didemniserinolípido B 61
3. Síntesis del compuesto 1 mediante metátesis cruzada
El didemniserinolípido B se intentó obtener mediante la hidrogenación
selectiva del compuesto 30, sintetizado mediante metátesis ciclante entre el compuesto 2 y el dienoéster 29 (esquema 11). Sin embargo, esta estrategia sintética fracasó ante la imposibilidad de conseguir el compuesto 17 por hidrogenación selectiva del doble enlace C6-C7 en el compuesto 30. Este problema se solucionó mediante reacción de metátesis cruzada de 2 con el selenoéster 3, seguida de hidrogenación con diimida, oxidación con MCPBA y eliminación in situ del correspondiente selenóxido. Conviene señalar en este punto la compatibilidad del catalizador de metátesis de 2ª generación de Grubbs con la agrupación hidroxilo libre y, sobre todo, con la presencia de la función seleniuro. Como el selenoéster 3 es un compuesto racémico la reacción de metátesis cruzada proporcionó el compuesto 1 como mezcla de epímeros en C2.
6
O
OOO
NBoc
H
7
15
OH
2EtOOC
RuCl
Cl
Cy3PPh
, CH2Cl2
NNMes Mes
O
OOO
NBoc
H
OH
EtOOC
67
15
3029
O
OOO
NBoc
H
OH
EtOOC
67
15
17
Esquema 11

Síntesis de didemniserinolípido B 62
El mecanismo de la reacción de metátesis cruzada entre 2 y 3 se describe en el esquema 12.
Ru CHX
cicloadiciónintermolecular
ciclorreversióncicloadiciónintermolecular
ciclorreversión
I
II
III
H2C CHX
O
OMsO
H
7
15
Ru
(X=Ph,H)
6
O
OOO
NBoc
H
7
15
8
OH
2
EtOOC
SePh
O
OOO
NBoc
H
OH
EtOOCSePh
67
15
1
3
O
OOO
NBoc
H
7
15
OH
Ru
X
O
OOO
NBoc
H
7
15
OH
Ru
EtOOC
SePh
6
OH
Esquema 12 4. Síntesis del sistema de éster α,βα,βα,βα,β-insaturado (compuesto 17)
La introducción del doble enlace C2-C3 se consiguió mediante la reacción de 16 con ácido m-cloroperoxibenzoico en presencia de trietilamina. El mecanismo de

Síntesis de didemniserinolípido B 63
esta conversión se inicia con la oxidación de la mezcla epimérica de seleniuros y la subsiguiente formación de una mezcla epimérica de selenóxidos 31, que por sin-eliminación se convierte en el éster insaturado 17, con configuración E en el doble enlace (esquema 12). La formación exclusiva de la configuración E se debe a la participación de los dos estados de transición favorecidos, indicados en el esquema 13. Estos dos estados de transición se generan como consecuencia de la adopción de conformaciones de mínima interferencia estérica, que sitúan a la agrupación de éster etílico (COOEt) y R en posición trans.
Esquema 13 5. Fmoc protección/desprotección de grupos amino

Síntesis de didemniserinolípido B 64
La protección de grupos amino en forma de carbamatos de 9-fluorenilmetilo (Fmoc) permite la ortogonalidad con los usuales grupos Boc (t-butilcarbamato) y Cbz (bencilcarbamato), puesto que los grupos Fmoc son estables a ácidos, al contrario que los grupos Cbz, y además pueden ser eliminados en presencia de los grupos Boc y Cbz por reacción con bases nitrogenadas, como la piperidina.
En la síntesis del dideminiserinolípido B se empleó como reactivo para la introducción del grupo Fmoc el carbonato de 9-fluorenilmetil succinimidilo. El mecanismo de la reacción de protección se indica en el esquema 14, y se inicia con el ataque nucleofílico del grupo amino al grupo carbonilo del 9-fluorenilmetil succinimidilo 32. El intermedio tetraédrico 33, que se forma en la reacción anterior, regenera el grupo carbonilo con expulsión de N-hidroxisuccinimida 34 y formación del correspondiente carbamato de 9-fluorenilmetilo 35.
Esquema 14
El mecanismo de desprotección del grupo Fmoc por reacción con aminas, como la piperidina, se indica en el esquema 15. El proceso se inicia con la desprotonación de la parte de fluorenilmetilo provocada por el ataque básico de la piperidina y facilitada por la formación del anión aromático 36. El proceso de eliminación sobre el intermedio anterior forma el 9-metilen-fluoreno 37 y el carbamato 38, que experimenta un proceso de descarboxilación que conduce a la formación de la amina libre RNH2. El 9-metilen-fluoreno 37 se convierte en el aducto de piperidina 40 mediante adición nucleofílica de ésta al doble enlace, facilitada por la aromaticidad del intermedio 39.

Síntesis de didemniserinolípido B 65
Esquema 15


Síntesis de dihidro-epi-desoxiarteannuina B 67
SÍNTESIS DE DIHIDRO- EPI-DESOXIARTEANNUINA B
Aislamiento: De la planta medicinal china Artemisia annua L se extrae la droga antimalárica artemisinina, junto con el ácido artemisinico y la arteannuina B y otros metabolitos secundarios como la dihidro-epi-desoxiarteannuina B (DEDAB).
Actividad biológica: La malaria, también denominada fiebre palúdica o paludismo, y cuyo nombre deriva del italiano mal aire, es una enfermedad producida por parásitos del género Plasmodium. Es la primera en importancia de entre las enfermedades debilitantes, con más de 200 millones de casos cada año en todo el mundo. El vector de la malaria humana son las hembras de mosquitos del género Anopheles. Los machos no pican al ser humano, ya que únicamente se alimentan de jugos vegetales. Las especies reconocidas como causantes de la enfermedad son el Plasmodium falciparum, la especie más patógena y letal, puesto que es la responsable de cerca del 80 % de las infecciones y de, aproximadamente, el 90% de las muertes, y el Plasmodium vivax, una forma algo benigna que causa fiebres intermitentes con intervalos de tres días, razón por la que antiguamente se conocía al mal como tercianas, o fiebres tercianas. Otras especies son el P. ovale, que no es mortal, pero puede provocar recaídas a los cuatro o cinco años después de la primera infección, el P. malariae , que puede provocar recaídas en los veinte años siguientes, y P. knowlesi y P. semiovale, que pueden causar también malaria.

Síntesis dihidro-epi-desoxiarteannuina B 68
La única forma posible de contagio directo entre humanos es la transmisión, por vía trasplacentaria, de la mujer embarazada al feto. Los síntomas son muy variados, empezando con fiebre, ocho a treinta días después de la infección, acompañada, o no, de dolor de cabeza, dolores musculares, diarrea, decaimiento y tos. La primera vacuna contra la malaria ha sido desarrollada por el médico colombiano Manuel Elkin Patarroyo, aunque aún no cuenta con una efectividad suficiente. Una vez adquirido el contagio, la malaria se combate con una combinación de fármacos, uno de los cuales es la artemisinina, que ha demostrado ser mucho más eficaz que los medicamentos usados en la quimiprofilaxis o en los tratamientos curativos, tales como la cloroquina o la mefloquina. Recientemente las empresas farmacéuticas han acordado retirar paulatinamente los medicamentos para el tratamiento oral de la malaria o paludismo que sólo contienen artemisinina. Parece ser que el tratamiento basado exclusivamente en la artemisinina, utilizado sobre todo para tratar la malaria no complicada, acelera la aparición de resistencia a ese producto en los parásitos causantes de la enfermedad. En cambio, cuando este compuesto se utiliza correctamente en combinación con otros antimaláricos sintéticos (lo que se conoce como Tratamientos Combinados de Artemisinina, TCA), la eficacia como curación del paludismo no complicado alcanza casi el 95 por ciento y el parásito difícilmente se vuelve resistente al fármaco.
Retrosíntesis El esquema 1 muestra el análisis retrosintético de la estructura de la dihidro-
epi-desoxiarteannuina B (DEDAB).15 La desconexión del doble enlace C4-C5, basada en una reacción de metátesis ciclante, conduce a la lactona bicíclica 1, la cual, mediante una operación de intercambio de grupo funcional (IGF) se transforma en el intermedio 2. Este compuesto se obtendrá por adición de bromuro de vinil magnesio al carbonilo cetónico de la ciclohexanona 3, que a su vez se preparará mediante C-alquilación del enolato cinético derivado de la hidroxicetona 4 con el yoduro alílico 5. La cetona 4 se sintetizará del (-)-isopulegol.
15 G. B. Dudley, D. A. Engel, I. Ghiviriga, H. Lam, K. W. C. Poon, J. A. Singletary, Org. Lett. 2007, 9, 2839.

Síntesis de dihidro-epi-desoxiarteannuina B 69
Esquema 1
Síntesis
1. Síntesis del yoduro de alilo 5
La síntesis del yoduro de alilo 5 comenzó con la hidromagnesiación del 2-
butin-1-ol catalizada por titano, seguida de quench con cloruro de trimetilsililo. Esta reacción proporcionó el alcohol alílico 6 con un 76% de rendimiento (véanse comentarios). El yoduro 5 se obtuvo por reacción de 6 con N-yodosuccinimida en presencia de trifenilfosfina.
Esquema 2

Síntesis dihidro-epi-desoxiarteannuina B 70
2. Síntesis del intermedio10
Para la síntesis del intermedio 10 se eligió como material de partida el (-)-isopulegol (esquema 2). Este producto natural se convirtió en la sililoxicetona 4 mediante hidroboración diastereoselectiva, seguida de sililación regioselectiva y oxidación. La enolización de 4 con LDA a baja temperatura generó el correspondiente enolato lítico, que se convirtió en el producto de C-alquilación 3 por reacción con el yoduro alílico 5, aunque la transformación tuvo lugar con muy bajo rendimiento. Por otro lado, la reacción de alquilación del enolato de litio con el hidroxilo protegido en forma de bencil éter, también proporcionó 3 con bajos rendimientos. El rendimiento del proceso se consiguió aumentar cuando la reacción de C-alquilación se llevó a cabo mediante transmetalación del enolato lítico a enolato de zinc. El rendimiento químico fue del 90% pero además la reacción fue altamente estereoselectiva, proporcionando, casi de forma exclusiva, el diastereoisómero 3 (véanse comentarios).
La adición del bromuro de vinil magnesio a la cetona 3 falló a la hora de preparar el alcohol terciario 7. Sin embargo, el tratamiento del bromuro de vinil magnesio con tricloruro de cerio activado, seguido de adición a la cetona 3 proporcionó el alcohol 7 con excelente rendimiento. La desililación de este compuesto, mediante hidrólisis ácida, generó un diol que se transformó quimioselectivamente en la lactona 8 por oxidación con perrutenato de tetra-n-propilamonio (TPAP) y N-óxido de N-metilmorfolina. La cetolactona 10 se consiguió mediante conversión de 8 en el epoxisilano 9, seguida de transposición de éste por reacción con ácido trifluoroacético.

Síntesis de dihidro-epi-desoxiarteannuina B 71
Esquema 3 3. Pasos finales
A fin de ensayar la construcción del anillo ciclohexénico de la dihidro-epi-
desoxiarteannuina, mediante la proyectada reacción de metátesis ciclante, la cetolactona 10 se transformó en el sustrato diolefínico 1 por reacción de

Síntesis dihidro-epi-desoxiarteannuina B 72
metilenación de Wittig. Sin embargo, todas las reacciones de metátesis sobre el sustrato 1 fallaron, debido, muy probablemente, a la elevada tensión del sistema tricíclico que se genera en esta reacción. El producto natural se obtuvo de manera indirecta, en una secuencia de tres pasos sintéticos. Así, la diolefina 1 se convirtió en el diol 11 por reducción con LiAlH4, y el producto de reducción se transformó, con un 37% de rendimiento, en el compuesto ciclohexénico 12 mediante reacción de metátesis ciclante con el catalizador de Grubbs de 2ª generación. La oxidación quimioselectiva del diol 12, con TPAP y NMO, proporcionó la dihidro-4-epi-desoxiarteannuina B.
Ph3P=CH2
(40%)
cat. de Grubbs
X
LiAlH4 (92%)
(37%)OH
OHMe
H
H
TPAP, NMO(62%)
1
12
Me
O
Me
H
Me
O
O 10
4
5
Me
O
Me
H
Me
O
4
5
11
Me
OH
Me
H
Me
OH
6
4
5 4
O
OMe
Me
MeH
H
DEDAB
4
5
N N
Ru
MesMes
Ph
Cl
ClPPCy3
Esquema 4
El bajo rendimiento químico obtenido en la reacción de metátesis ciclante llevó a ensayar la síntesis del producto natural mediante la aplicación de la estrategia de Hoye. Para ello, la cetolactona 10 se transformó en el compuesto triolefínico 14 mediante olefinación Wittig con el iluro de fósforo derivado de la sal de fosfonio 13. Cuando el compuesto 14 se calentó en tubo sellado, en diclorometano en presencia del catalizador de Grubbs de segunda generación, se obtuvo la dihidro-epi-desoxiarteannuina B sintética con un rendimiento del 90%.

Síntesis de dihidro-epi-desoxiarteannuina B 73
n-BuLi, éter (40%)O
Me
H
O
Ph3PI
Me
CH2Cl2, 40ºC,tubo sellado
(90%)
13
14
N N
Ru
MesMes
Ph
ClCl
PPCy3
Me
O
Me
H
Me
O
O 10
4
O
OMe
Me
MeH
H
DEDAB
4
5
5
4
5
Esquema 5
Comentarios 1. Hidromagnesiación de triples enlaces catalizada por titanio
La hidromagnesiación de triples enlaces, por reacción de alquinos con
reactivos de Grignard, en presencia de bis(ciclopentadienil)diclorotitanio(IV), permite la generación de reactivos alquenil-magnésicos, que pueden ser transformados en alquenos funcionalizados, con configuración definida en el doble enlace, mediante reacción con especies electrofílicas.
En el esquema 6 se muestra el mecanismo de la reacción de hidromagnesiación, que se inicia con la formación de la especie catalítica Cp2Ti(iBu)Cl (intermedio I ) por reacción del bis(ciclopentadienil)diclorotitanio(IV) con dos equivalentes de cloruro de isobutilmagnesio. El intermedio I se transforma en el hidruro de titanio II , que por adición cis al triple del alquino se transforma en el alqueniltitánico III . La transmetalación de III , por interacción con el reactivo de Grignard, conduce al compuesto alquenilmagnésico IV y a la regeneración de la especie catalíticamente activa.

Síntesis dihidro-epi-desoxiarteannuina B 74
Esquema 6
2. Reacción de alquilación de la cetona 4
LDA, THF, -78ºCluego Et2Zn, HMPA
luego yoduro 5(90%)
Me
O
Me
H
Z
SiMe3
Me
3
O
ZMe
Me
H
4
11
11
2
4
Los primeros intentos por conseguir el compuesto 3, mediante reacción del
yoduro 5 con el enolato lítico derivado de la cetona 4 (Z=OTIPS o Z=OBn), proporcionaron los productos de C-alquilación con bajos rendimientos (entradas 1 y 2 de la tabla 1). La dificultad en conseguir el proceso de C-alquilación se puso de manifiesto cuando las reacciones de alilación del enolato lítico derivado de la propia mentona (4, Z=H) proporcionaron el producto de C-alilación con bajos rendimientos (entradas 3 y 4 de la tabla 1). Un aumento sustancial del rendimiento se consiguió cuando el enolato lítico se convirtió en enolato de zinc por transmetalación con dietilzinc. Así, la alilación del enolato de zinc derivado de la mentona (4, Z=H), o de la sililoximentona (4, Z=OTIPS), proporcionó los

Síntesis de dihidro-epi-desoxiarteannuina B 75
correspondientes productos de C-alilación con rendimientos del 80% (véanse entradas 5 y 6 de la tabla 1). De hecho, el rendimiento en la C-alquilación de 4 (Z=OTIPS) con el yoduro 5 llegó a ser del 89% cuando el enolato lítico se convirtió en enolato de zinc (entrada 7 de la tabla 1).
Tabla 1 Entrada Z R-X Aditivo Rto.
1 OTIPS 5 HMPA 45 %
2 OBn 5 - 53%
3 H bromuro de alilo - 17%
4 H yoduro de alilo HMPA 53 %
5 H yoduro de alilo HMPA, Et2Zn 80 %
6 OTIPS yoduro de alilo HMPA, Et2Zn 80 %
7 OTIPS 5 HMPA, Et2Zn 89 %
En el esquema 7 se muestran las conformaciones que adopta el enolato metálico derivado de la cetona 4. La tensión alílica A1,2 que presenta la conformación I desplaza el equilibrio conformacional hacia el confórmero II , en el cual los dos sustituyentes alquílicos se colocan en posiciones pseudoecuatoriales, bloqueando de esta forma el acceso de los reactivos electrofílicos a las dos caras del enolato. La adición de dietilzinc al medio de reacción mejora drásticamente el rendimiento del proceso, debido, muy probablemente, a la mayor movilidad conformacional que poseen los enolatos de zinc en comparación con los enolatos de litio.
Esquema 7

Síntesis dihidro-epi-desoxiarteannuina B 76
3. Metátesis ciclante de trienos: activación de Hoye
Como se ha explicado en el apartado de síntesis la reacción de metátesis ciclante sobre el compuesto 1 no funcionó, y lo hizo con bajo rendimiento sobre el diol monocíclico 11. Recientemente, el grupo de Hoye ha demostrado que se puede conseguir la reacción de metátesis ciclante sobre sustratos con enlaces dobles electrónicamente desactivados o estéricamente impedidos.16 El concepto estratégico de Hoye consiste, básicamente, en la conversión del sustrato diénico en un sustrato triénico, que presenta un doble enlace fácilmente accesible al catalizador de metátesis, con lo que se favorece la primera reacción, de carácter intermolecular, entre el sustrato y el catalizador. El correspondiente carbeno de rutenio, generado en el proceso intermolecular, se transforma en el producto de metátesis ciclante mediante la intervención de reacciones intramoleculares cinética y entrópicamente favorecidas. La aplicación de esta estrategia a la síntesis de la dihidro-epi-desoxiarteannuina B se llevó a cabo del siguiente modo. En primer lugar el sustrato 10 se convirtió en la triolefina 14, cuyo enlace doble terminal debe permitir el proceso de metátesis debido a su nulo impedimento estérico (véase el esquema 5). En la práctica, la reacción de metátesis ciclante sobre 14 proporcionó la dihidro-epi-desoxiarteannuina B con un rendimiento del 90%. En el esquema 8 se indica el ciclo catalítico de esta conversión, que se inicia con la formación del intermedio I mediante cicloadición-ciclorreversión del catalizador al doble enlace terminal. La siguiente reacción, que ya es intramolecular y por tanto cinéticamente favorecida, forma ciclopenteno y el carbeno de rutenio II , que se transforma en la dihidro-epi-desoxiarteannuina B mediante un proceso de cicloadición intramolecular-ciclorreversión.
16 T. R. Hoye, C. S. Jeffrey, M. A. Tennakoon, J. Wang, H. Zhao, J. Am. Chem. Soc. 2004, 126, 10210.

Síntesis de dihidro-epi-desoxiarteannuina B 77
[Ru]=CHR
Me
O
MeH
O
Me Ru
CH2=CH2
I
II
14O
OMe
Me
MeH
HDEDAB
4
5
45
Me
O
MeH
O
Me
45
Me
O
MeH
O
Ru
Me
45
Esquema 8


Síntesis de elatol 79
SÍNTESIS DE ELATOL
Aislamiento: Los chamigrenos son una subclase de sesquiterpenos que se caracterizan por poseer en su estructura un núcleo de espiro[5.5]undecano. El elatol, un sesquiterpeno de tipo chamigreno, se ha aislado del alga marina Laurencia elata. Actividad biológica: El elatol exhibe un amplio rango de actividades biológicas, entre las que destacan su acción antibiótica contra bacterias patógenas de humanos, su actividad antifúngica y su citotoxicidad contra las líneas celulares tumorales humanas HeLa y Hep-2.
Retrosíntesis El análisis retrosintético del elatol se inicia con su conversión en la
ciclohexenona espirocíclica 1 (esquema 1).17 En el sentido sintético el producto natural se obtendrá a partir del compuesto 1 mediante operaciones de manipulación de grupos funcionales que comprenderán halogenación, isomerización de doble enlace y reducción estereoselectiva de grupo cetónico.
La interconversión de la función enónica de 1 en la agrupación de éster vinílogo conduce al intermedio 2, el cual, mediante una desconexión basada en una reacción de metátesis ciclante, origina el compuesto monocíclico 3. La siguiente operación del análisis retrosintético aborda la desconexión del estereocentro cuaternario cuya escisión conduce al sintón catiónico 4 y al enolato 5. Como se explicará en la parte sintética, la construcción enantioselectiva del estereocentro cuaternario se conseguirá mediante una reacción de alquilación alílica catalizada por un complejo de paladio quiral sobre el carbonato mixto 6. Este compuesto se preparará por carbonatación de la forma enólica derivada de 7, que a su vez se obtendrá mediante una reacción de C-alquilación regioselectiva de la enona 8. Este compuesto se sinterizará de la dimedona.
17 D. E. White, I. C. Stewart, R. H. Grubbs, B. M. Stoltz, J. Am. Chem. Soc. 2008, 130, 810.

Síntesis de elatol
80
Esquema 1
Síntesis 1. Síntesis de la ciclohexenona 3
La síntesis del elatol se inició con la preparación del compuesto 3. Para ello, la dimedona se convirtió en el éster vinílogo 8 por reacción con isobutanol en presencia de cantidades catalíticas de ácido p-toluensulfónico (esquema 2).
La enolización cinética de 8, con LDA a -78ºC, generó el enolato lítico 9, que se convirtió en el compuesto 10 por adición conjugada Michael a la metil vinil cetona. La subsiguiente reacción de metilenación Wittig proporcionó el intermedio 7, con un rendimiento combinado del 74% en las dos últimas etapas. Conviene indicar en este punto que la reacción de alquilación de 8 con 4-yodo-2-metil-1-buteno, que habría proporcionado directamente el compuesto 7, transcurrió con bajas velocidades y rendimientos. El carbonato de enol 6 se obtuvo por reacción

Síntesis de elatol 81
del enolato lítico derivado de 7 con el cloroformiato 11. Cuando 6 se hizo reaccionar con el complejo quiral generado por reacción de la fosfinooxazolina 12 con bis(3,5,3′,5′-dimetoxidibencilidenacetona)paladio(0) (Pd(dmdba)2), en benceno anhidro, se obtuvo el compuesto 3 con un 82% de rendimiento y un 87% de exceso enantiomérico (véanse comentarios).
Esquema 2
2. Pasos finales
La conversión del compuesto 3 en el elatol sintético se indica en el esquema
3

Síntesis de elatol
82
Esquema 3
El sistema espiránico del elatol se consiguió mediante calentamiento de una
disolución bencénica de 3, a 60ºC durante 18 horas, en presencia de 5 mol% del catalizador de rutenio 13. Estas condiciones experimentales proporcionaron el cloroalqueno espiránico 2 con un 97% de rendimiento. A continuación, la adición de metil-litio al carbonilo cetónico del compuesto 2 generó el alcohol alílico 14 el cual, mediante tratamiento con una disolución acuosa de ácido clorhídrico, se transformó en la clorociclohexenona espiránica 1 (véase el esquema 3 para los intermedios implicados en esta transformación). Finalmente, la halogenación de 1 con bromo molecular, en una mezcla de bromuro de hidrógeno/ácido acético, formó el derivado dibromado 17, que se convirtió en el elatol sintético por reducción-eliminación reductiva con DIBAL (véanse comentarios).

Síntesis de elatol 83
Comentarios 1. Alquilación asimétrica de carbonatos de enol catalizada por paladio
Se puede considerar la conversión enantioselectiva del carbonato 6 en la cloroenona 3 como una variante del método de alilación Tsuji-Trost.18 Este método permite la alilación, en presencia de cantidades catalíticas de complejos de paladio(0), de determinado tipo de nucleófilos, como enolatos, aminas, fenoles y metilenos activos, mediante reacción con acetatos y bromuros alílicos. El ciclo catalítico de la reacción de alilación Tsuji-Trost se indica en el esquema 4.
Pd0
adiciónoxidante
X
Pd
Nu
Nu
L LX
PdL
L
X
L
L
sustituciónnucleofílica
Pd
L L
descoordinación
X
I
coordinación
X
Pd
LL
complejo 2
0
IIII
complejo 3II
II
Nu
PdL
L
Nu
0
III
Esquema 4
18 a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 15044-; b) J. T. Mohr, D. C. Behenna, A. M. Harned, B. M. Stoltz, Angew. Chem. Int. Ed. 2005, 44, 6924.

Síntesis de elatol
84
En primer lugar se produce la coordinación del doble enlace con el catalizador de Pd(0). Este proceso conduce a la formación de un complejo η2 π−alílico (intermedio I del esquema 4). A continuación se produce la etapa de adición oxidante, llamada también de ionización, que provoca la expulsión del grupo saliente y la formación de un complejo η
3 π−alílico (intermedio II ). Dependiendo de la fuerza del nucleófilo la reacción puede, a continuación, tomar dos vías diferentes. Los nucleófilos blandos, como los derivados de ácidos conjugados con pKa<25, se adicionan directamente a la parte alílica y forman un complejo de paladio(0) (intermedio III del esquema 4), cuya descoordinación forma el producto de la reacción y regenera el catalizador.
Cuando el nucleófilo es duro se produce el ataque de éste al metal, con formación del complejo IV que se indica en el esquema 5. El producto de la reacción se forma mediante eliminación reductora en el complejo IV .
Esquema 5
El ciclo catalítico que explica la conversión del carbonato alílico 6 en el compuesto 3 se indica en el esquema 6. Así, la interacción de 6 con el catalizador de paladio(0) forma, después de las etapas de coordinación con el doble enlace y adición oxidante, el complejo I η3 π-alílico. La descarboxilación del complejo I forma un anión enolato, que es el nucleófilo del proceso. A continuación, el ataque

Síntesis de elatol 85
del enolato al metal conduce al complejo III , que por eliminación reductora se transforma en el producto de la reacción y regenera el catalizador.
Pd
descarboxilación
Pd0
adiciónoxidante
L
L
adiciónnucleofílica
eliminaciónreductora
CO2
L L**
Pd
L
L
*
* O O
O
R1
R2
Cl
Pd
L L**
O
R1
R2
Cl
Cl
O
R1
R2I
II
III
O
iBuO
Cl
O
iBuO
O
OCl
6
3
*
*
II
II
II
Esquema 6 En la reacción de alilación de enolatos se produce el ataque directo del
nucleófilo al metal, y el subsiguiente proceso de eliminación reductora forma el compuesto resultante de la C-alquilación del enolato, en detrimento del producto de O-alquilación.
El catalizador que se emplea en la reacción de formación de 3 es un complejo de paladio quiral que resulta del intercambio de ligandos entre el bis(3,5,3′,5′-dimetoxidibencilidenacetona)paladio(0) (Pd(dmdba)2) y la fosfinooxazolina 12 (véase el esquema 7).

Síntesis de elatol
86
Esquema 7 Los catalizadores que suelen emplearse en este tipo de alilaciones se
muestran en el esquema 8.
Ph2P N
O
t-Bu
(S)-t-Bu-PHOX
O
NH
PPh2
Ph Ph
HN
O
Ph2P
PPh2
PPh2
(R)-BINAP(S,S)-ligando de Trost
OMe
PPh2
(R)-MOP
N
PPh2
(R)-QUINAP
P P
(R,R)-Me-DUPHOS
O
O PPh2
PPh2
(R,R)-DIOP
Esquema 8

Síntesis de elatol 87
Con los ligandos anteriores se genera un complejo de paladio en forma de “bolsillo”, que permite la coordinación al sistema alílico sólo a través de una de sus dos caras (la parte abierta del bolsillo). El entorno asimétrico provocado por la quiralidad de los ligandos discrimina eficientemente una de las dos caras del sistema alílico, y la subsiguiente eliminación reductora proporciona el producto de la reacción enantioméricamente enriquecido (véase el esquema 9).
Esquema 9
Es interesante señalar que la reacción de alilación enantioselectiva sobre el
sustrato 6 en presencia del catalizador (S)-tBu-PHOX 18 (véase el esquema 8) proporcionó el producto de la reacción con un 81% de exceso enantiomérico, pero con tan sólo un 23% de rendimiento químico (esquema 10). Sin embargo, con el sustrato 19 la reacción de alilación transcurrió de forma casi cuantitativa (esquema 10).
Esquema 10
Los resultados del esquema anterior apuntan a falta de reactividad del
electrófilo alílico en el proceso de acoplamiento reductivo, debido, muy probablemente, a la disminución de la electrofílica del catión alílico clorado por el efecto resonante electrón-donante del átomo de halógeno. Nótese que la sustitución del átomo de cloro por un átomo de hidrógeno, como es el caso del sustrato 19,

Síntesis de elatol
88
aumenta la reactividad del catión alílico y en consecuencia explica el incremento espectacular del rendimiento en este sustrato. El catalizador 12, que contiene grupos electrón-atrayentes p-trifluorometilfenilo en lugar de fenilo, incrementa la reactividad de catión cloro-alílico al aumentar su electrofília. La consecuencia más importante desde el punto de vista experimental es el aumento del rendimiento en la formación del compuesto 3, que pasa del 23% al 82%.
2. Conversión del compuesto 1 en elatol
El mecanismo que explica la formación del compuesto di-halogenado 17 se indica en el esquema 11. La transformación se inicia con la enolización, catalizada por bromuro de hidrógeno, del anillo ciclohexenónico. El enol 21 que se forma en este proceso ataca nucleofílicamente al bromo molecular y forma, después de la etapa de desprotonación, el compuesto mono-halogenado 23. Un nuevo proceso de enolización, esta vez sobre el compuesto 23, genera el enol extendido 25, que por reacción con la molécula de bromo proporciona, después de la desprotonación, el compuesto di-halogenado 17.
Esquema 11

Síntesis de elatol 89
Esquema 11 (cont.) La reducción del compuesto 17 con DIBAL proporcionó una mezcla de
estereoisómeros en la cual la relación elatol(isómero sin)/isómero anti era 3.9:1 y la relación de productos SN2´/SN2 era de 11:1 (véase el esquema 12).
Esquema 12


Síntesis de espongidepsina 91
SÍNTESIS DE ESPONGIDEPSINA
Aislamiento: La espongidepsina se ha aislado de la esponja marina Spongia sp. Actividad biológica: La espongidepsina presenta actividad citotóxica y antiproliferativa contra las líneas celulares HEK-293 (relacionada con el cáncer de riñón) y WEHI-164 (relacionada con los fibrosarcomas).
Retrosíntesis La primera operación del análisis retrosintético convierte la espongidepsina
en el intermedio 1 (esquema 1). 19
Esquema 1
19 G. Zhu, E-i. Negishi, Org. Lett. 2007, 9, 2771.

Síntesis de espongidepsina
92
En el sentido sintético el producto natural se obtendrá por hidrogenación de 1 seguida de desprotección del hidroxilo primario, oxidación y homologación aldehído-alquino. La desconexión del doble enlace C5-C6, basada en una reacción de metátesis ciclante, origina el intermedio acíclico 2, que por escisión de los enlaces amida y éster conduce a la N-metilfenilalanina, al ácido insaturado 3 y al endiol 4.
El ácido 3 se obtendrá del diol monoprotegido 5, cuyo estereocentro en C4 se generará mediante reacción de carbometalación-oxidación en el compuesto 6. La desconexión del enlace C3-C4 de 6 conduce al bromuro de vinilo y al intermedio 7 (M=metal), cuyo estereocentro se instalará también mediante reacción de carbometalación en el alcohol alílico.
Esquema 2 El fragmento 4 se obtendrá por homologación del diol 8, cuyo estereocentro
en C7 se construirá mediante carbometalación-oxidación del alcohol insaturado 9. Este compuesto se obtendrá de forma enantioselectiva mediante reacción de alilación asimétrica del 5-hidroxipentanal protegido 10.

Síntesis de espongidepsina 93
Síntesis 1. Síntesis del ácido insaturado 3
La síntesis del fragmento 3 comenzó con la reacción de carboaluminación
asimétrica del alcohol alílico, seguida de quench con yodo y protección del hidroxilo libre. Para la reacción de carboaluminación se empleó trimetilaluminio, en presencia del complejo quiral de Zr(IV) bis[(+)-neomentilindenil]diclorozirconio(IV), y proporcionó el yoduro 11 con un 82% de exceso enantiomérico (véase el esquema 3 y comentarios). El intercambio halógeno-metal en el compuesto 11, por reacción con t-BuLi seguida de transmetalación con ZnBr2, generó el correspondiente reactivo organozíncico, que se transformó en la olefina 6 por reacción de acoplamiento de Negishi con bromuro de vinilo, en presencia de DIBAL y [bis(2-(difenilfosfino)fenil)eter]dicloropaladio(II) (véanse comentarios). La reacción de carboaluminación de la olefina 6 con trimetilaluminio y el complejo quiral bis[(+)-neomentilindenil]diclorozirconio(IV), seguida de quench de la reacción con oxígeno molecular, proporcionó el alcohol 5 con un rendimiento del 76%, y una relación diastereoisomérica de 5.5:1 (véanse comentarios). La proporción de diastereoisómeros pasó a ser >40:1 después de la separación en cromatografía de columna, aunque el rendimiento del proceso disminuyó del 76% al 45%.
La oxidación Swern del alcohol 5, seguida de metilenación Wittig, llevaron a la olefina 12 que, finalmente, se transformó en el ácido carboxílico 3 mediante desililación y oxidación con clorocromato de piridinio en dimetilformamida.

Síntesis de espongidepsina
94
3
HO
O
TBSO OH
TBSOTBSO IHO
5 (76%, d.r. 5.5:1)
611
1. AlMe3(+)-(NMI)2ZrCl2,luego I2
2. TBSCl(81%, e.e. 82%)
1. tBuLi, ZnBr2
2. Pd(DPEphos)Cl2,DIBAL, CH2=CHBr
(87%) AlMe3(+)-(NMI)2ZrCl2
luego O2
1. Swern
2. Ph3P=CH2(80%)12
TBSO1. TBAF
2. PDC(72%)
1
2 45 1
2 45
1
2 45
1
2
4
51
2
12
33 3
Esquema 3
2. Síntesis del alcohol 4
Para la síntesis del alcohol 4 se eligió como material de partida el 1,5-
pentanodiol, que se convirtió en el sililoxialdehído 10 por monosililación y oxidación de Swern (esquema 4). El tratamiento de 10 con el agente alilante generado in situ por reacción de bromuro de alilmagnesio con (-)-diisopinocanfeílmetoxiborano condujo al alcohol homoalílico 9. Este compuesto se transformó en el diol 8 por reacción de carboaluminación con trimetilaluminio y bis[(+)-neomentilindenil]diclorozirconio(IV) seguida de quench con oxígeno molecular. La relación diastereoisomérica obtenida en la reacción anterior fue de 3.5:1. Esta proporción de diastereoisómeros pasó a ser de >40:1 después de la purificación del producto mediante cromatografía de columna, aunque de nuevo a costa de una importante merma en el rendimiento químico que pasó del 73% al 42%. La oxidación selectiva del hidroxilo primario en presencia del hidroxilo secundario se consiguió mediante reacción con diacetato de fenilyodonio y

Síntesis de espongidepsina 95
TEMPO y proporcionó directamente la lactona 13. Este compuesto se convirtió en el alcohol 4 mediante reducción con DIBAL seguida de metilenación de Wittig.
Esquema 4
3. Pasos finales
La unión de los fragmentos constituyentes del esqueleto de la espongidepsina
se inició con la reacción de esterificación entre el alcohol 4 y la N-Boc-N-metilfenilalanina (esquema 5). El proceso se llevó a cabo en presencia de diciclohexilcarbodiimida y N,N-dimetilaminopiridina y condujo al aminoéster 14, el cual, mediante N-Boc desprotección con ácido trifluoroacético y amidación con el ácido 3, en presencia de 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDCI) y 1-hidroxibenzotriazol (HOBT), se transformó en el amidoéster 15. Este compuesto se sometió a la reacción de metátesis ciclante con el catalizador de Grubbs de segunda generación, lo que condujo a la formación del 16. La desililación de éste, seguida de hidrogenación, proporcionó 17, que se transformó en la espongidepsina mediante oxidación Dess-Martin seguida de homologación de Ohira-Bestmann.

Síntesis de espongidepsina
96
Esquema 5
Comentarios
1. Carboaluminación asimétrica de alquenos catalizada por zirconio (ZACA)
RER
1. AlMe3(+)-(NMI)2ZrCl2
2. E
La carboaluminación de olefinas promovida por trimetilaluminio en presencia de cantidades catalíticas del complejo quiral bis[neomentilindenil]diclorozirconio(IV) (ZACA) permite la metilación y metalación de alquenos monosustituidos de manera regio y enantioselectiva.
El ciclo catalítico de la reacción se inicia con el intercambio de ligandos entre las dos especies organometálicas. Esta operación forma el intermedio de Zr(IV) I , que se coordina con el alqueno para generar el intermedio II (esquema 6).

Síntesis de espongidepsina 97
A continuación, la reacción de sin-carbozirconación de la olefina forma el intermedio III , que transmetala al IV .
Esquema 6
La reacción de IV con electrófilos genera los correspondientes derivados (esquema 7).
Esquema 7
El catalizador del proceso en su versión asimétrica es el bis[neomentilindenil]diclorozirconio(IV) (ZACA), un complejo quiral de zirconio(IV). En el esquema 8 se muestra la estructura de los dos bis[neomentilindenil]diclorozirconio(IV) enantioméricos.

Síntesis de espongidepsina
98
(+)-(NMI)2ZrCl2
Zr
Cl
Cl ZrCl
Cl
(-)-(NMI)2ZrCl2
Esquema 8
La enantioselectividad del proceso se explica admitiendo que el paso de carbometalación (conversión de II en III , en el esquema 6) tiene lugar mediante la intervención de un estado de transición de cuatro eslabones, como se indica en los estados de transición ET-I (vía disociativa) y ET-II (vía asociativa) del esquema 9, para el complejo (+)-ZACA. En estos dos estados de transición el enlace σ Me-Zr y los orbitales d vacíos del zirconio se sitúan en la cara frontal del complejo, quedando, en función de la quiralidad del mentol, solo dos cuadrantes del catalizador accesibles a la trayectoria de aproximación de la olefina al metal. Si se emplea el (+)-ZACA se produce la interacción Zr-alqueno sobre la cara Re de la olefina (esquema 9).
Esquema 9
Después de mostrar el mecanismo general de este procesos, conviene
describir la estructura del complejo de Zr(IV). En el esquema 10 se indica la

Síntesis de espongidepsina 99
configuración electrónica del átomo de zirconio(IV) que posee todos los orbitales s, p y d vacíos.
Esquema 10
En el esquema 11 se indican los electrones que aportan los ligandos del
zirconio
Esquema 11
Cada anión indenilo cede 6 electrones al Zr llenándose seis orbitales del
metal cuya simetría es más parecida a la de los orbitales moleculares π enlanzantes del indenilo. Estos orbitales son: el orbital s, los tres orbitales p y dos orbitales dπ, concretamente el dxz y el dyz. Por otro lado, la hibridación de los tres orbitales d restantes (dx2-y2, dxy, dz2) genera tres nuevos orbitales híbridos (ds1, ds2 y dp), que se disponen en el espacio alejándose de los dos ligandos aromáticos, tal y como se muestra en el esquema 12. Cada anión cloruro cede 2 electrones al metal solapando su orbital híbrido sp3 lleno con un orbital híbrido d vacío del metal, quedando sin ocupar uno de los orbitales híbridos d del metal. Esta situación permite a este tipo de complejos actuar como ácidos de Lewis.
Esquema 12

Síntesis de espongidepsina
100
2. Obtención del alqueno 6 mediante acoplamiento de Negishi
TBSOTBSO I
67
1. tBuLi, ZnBr2
2.Pd(DPEphos)Cl2,DIBAL, CH2=CHBr (87%)
La transformación del yoduro 7 en la olefina 6 se consiguió mediante una
secuencia de transformaciones que se inició con el intercambio halógeno-metal, por reacción con t-BuLi, seguida de transmetalación con ZnBr2, y reacción subsiguiente con bromuro de vinilo mediada por un complejo de paladio(0), generado in situ por reducción de Pd(DPEphos)Cl2 con DIBAL (esquema 13).
TBSO
I
7TBSO Li TBSO ZnBr
ZnBr2
LiBr TBSO 6
Pd(DPEphos)Cl2,DIBAL, CH2=CHBr
(87%)
tBuLi
tBuI
Esquema 13
El ciclo catalítico que explica el acoplamiento C-C se indica en el esquema
14, y se inicia con la adición oxidante del bromuro de vinilo al complejo de paladio(0). El intermedio I , generado en el proceso anterior, experimenta la transmetalación al intermedio II , que por eliminación reductora proporciona el producto de acoplamiento y regenera el catalizador.

Síntesis de espongidepsina 101
Esquema 14


Síntesis de exiguolido 103
SÍNTESIS DE EXIGUOLIDO
O O
MeO2C
O
O
MeO2C
exiguolido
Aislamiento: El exiguolido, un macrólido de 16 miembros, se ha aislado de la esponja marina Geodia exigua Thiele. Actividad biológica: El exiguolido inhibe la fertilización de los gametos del erizo de mar pero no la embriogénesis de los huevos fertilizados.
Retrosíntesis
El análisis retrosintético del exiguolido se inicia con una doble etapa de interconversión de grupo funcional (IGF) que afecta al doble enlace C22-C23, de configuración Z, y al sistema de éster insaturado unido al anillo macrolactónico (esquema 1).20 La operación conduce al intermedio 1, que en el sentido sintético se transformará en el producto natural mediante cis-hidrogenación selectiva del triple enlace y reacción Wittig, o similar, sobre el grupo carbonilo cetónico que presenta el intermedio 1. La siguiente operación desconecta el sistema de enino (enlace C21-C22) para proporcionar el alquino 2 y el yoduro de alquenilo 3. A continuación, la desconexión del doble enlace C16-C17 origina el intermedio 4, que en el sentido sintético se obtendrá mediante reacción de metátesis ciclante sobre el intermedio 3. La desconexión de la agrupación éster en el intermedio 4 conduce al alcohol 5 (fragmento C17-C21) y al intermedio bis-tetrahidropiránico 6. El anillo tetrahidropiranónico que contiene 6 se construirá mediante una ciclación de tipo Prins sobre el β-alcoxiacrilato 7, que, a su vez, se preparará mediante adición conjugada tipo Michael del alcohol homoalílico 8 sobre el propionato de metilo.
20 M. S. Kwon, S. K. Woo, S. W. Na, E. Lee, Angew. Chem. Int. Ed. 2008, 47, 1733.

Síntesis de exiguolido
104
Esquema 1

Síntesis de exiguolido 105
El intermedio 8 se sintetizará mediante una reacción de alilación estereoselectiva sobre el aldehído 9, cuyo anillo tetrahidropiránico cis-2,6-disustituido se obtendrá mediante un proceso de adición radicalaria intramolecular provocado como consecuencia de la escisión homolítica del enlace C-X (X=halógeno) en el compuesto 10. Este compuesto se obtendrá a partir del alcohol homoalílico 11 mediante funcionalización del doble enlace seguida de adición conjugada Michael de la agrupación hidroxilo al propionato de metilo. El compuesto 11 se sintetizará mediante alilación estereoselectiva sobre el aldehído 12, que se obtendrá del éster de Roche 13 mediante un proceso de homologación.
Síntesis
1. Síntesis del intermedio 21 (fragmento C1-C16)
Para la síntesis del ácido 21 (fragmento C1-C16) se eligió como material de
partida el (R)-éster de Roche 13, cuyo centro estereogénico se convertirá en el estereocentro situado en el carbono C15 de la molécula de exiguolido. Así, el éster 13 mediante bencilación tipo Iversen y reducción con DIBAL se convirtió en el alcohol 14 (esquema 2). La mesilación y el desplazamiento nucleofílico del mesilato con cianuro sódico proporcionaron el nitrilo 15. El estereocentro en C13 se instaló mediante reducción a aldehído del nitrilo 15, seguido de un proceso de alilación tipo Brown con el reactivo alilante generado mezclando (-)-diisopinoclorocanfeilborano y bromuro de alilmagnesio. Esta secuencia de reacciones proporcionó el alcohol homoalílico 11 con una relación diastereoisomérica de 96:4. La funcionalización del carbono C10, necesaria para poder llevar a cabo la proyectada reacción de adición radicalaria intramolecular, se consiguió mediante hidratación anti-Markovnikov del doble enlace, seguida de sililación selectiva sobre el diol resultante del proceso anterior, lo cual condujo a la obtención del alcohol 16. La adición Michael del alcohol 16 sobre el propionato de metilo, en presencia de N-metilmorfolina (NMM), proporcionó el

Síntesis de exiguolido
106
β−alcoxiacrilato 17, que se convirtió en el yodoacrilato 10 mediante desililación y yodación.
El sustrato 10, que ya está adecuadamente funcionalizado para llevar a cabo la reacción de adición radicalaria intramolecular, se disolvió en etanol en presencia de hipofosfito de 1-etilpiperidinio y a la disolución resultante se le añadió, bajo aire, trietilborano. Después de 30 minutos a temperatura ambiente se obtuvo el compuesto tetrahidropiránico cis-2,6-disustituido 18 de forma casi cuantitativa. De esta forma, y en un solo paso sintético, el proceso radicalario no solo consigue la construcción del anillo tetrahidropiránico, sino que también permite la instalación estereocontrolada del centro estereogénico en C9 (véanse comentarios).
El compuesto 18 se convirtió en el alcohol homoalílico 8 mediante una nueva secuencia de reducción-alilación Brown. La adición conjugada de la agrupación hidroxílica de 8 al propionato de metilo llevó al β−alcoxiacrilato 7, que ya contiene todo el sistema carbonado C1-C16 necesario para la preparación del ácido 21.
El segundo anillo tetrahidropiránico se construyó mediante ciclación tipo Prins. Para ello se añadió ácido trifluoroacético a una disolución diclorometánica del β−alcoxiacrilato 7. Después de 1 hora de agitación a temperatura ambiente, la mezcla resultante del proceso anterior se sometió a saponificación por reacción con carbonato de potasio en metanol. Este proceso condujo a la obtención del compuesto 19 (mezcla epimérica en C5), junto con otros diastereoisómeros epiméricos en C3 y C7 (véanse comentarios).
La oxidación de la mezcla de epímeros, seguida de cetalización e hidrogenolisis condujo, después de la purificación cromatográfica, a una mezcla 7:1 formada por el compuesto 20 y el diastereoisómero epimérico en C3 y C7. La oxidación de la mezcla anterior proporcionó una mezcla de aldehídos, que se sometió a un proceso de metilenación catalizado por rodio (véanse comentarios). Esta reacción llevó a la obtención de una mezcla 7:1 de compuestos de metilenación, que se sometió a saponificación con hidróxido de litio. Después de la acidificación y separación cromatográfica se obtuvo el ácido 21 puro.

Síntesis de exiguolido 107
HOCO2Et 2. DIBAL
BnO
OH
1. MsCl, Et3N
2. NaCN, DMSO(95%)
BnO CN
1. DIBAL2. (-)-DIPClCH2CHCH2MgBr
(75%, d.r. 96:4)
BnO
OH1. (sia)2BH,H2O2, NaOH
2. TBSCl, imid.(77%)OBn
OH
TBSO
CO2Me
NMM (94%)
OBn
O
TBSO
CO2Me
1. HCl conc.
2. I2, PPh3(92%) OBn
O
I
CO2Me
H3PO2, 1-etilpiperidina
Et3B (99%)
BnO
O
CO2Me
O O
HO
MeO2C
BnO
O O
MeO2C
BnO
MeO2C
O
BnO1. DIBAL2. (-)-DIPClCH2CHCH2MgBrO
BnO
OH
CO2MeNMM (94%)
1. TFA, CH2Cl2
2. K2CO3, MeOH(81%)
1. Dess-Martin
2. HC(OMe)3, MeOH,H2SO4 cat, luego Et3N
O O
MeO2C
HO
MeO
MeO
1. Dess-Martin
2.TMSCHN2, [(Ph3P)3RhCl]
13 14 15
1116
1710
188
7 19
20
1.BnOC(NH)CCl3
(75%, d.r. > 99:1)
luego H2, Pd/C (74%)
3. LiOH, MeOH/H2Oluego HCl 2M (70%)
O O
O
HOOC
21
3
5
7
16
3
5
7
3
5
7
3
57
1
5
7
3
7
9
1316
7 9
13
16
7
13
10
7
9
13
13
9
7
16
15
14
9
Esquema 2

Síntesis de exiguolido
108
2. Síntesis de los fragmentos 2 (C22-C27) y 5 (C17-C21)
La síntesis del fragmento 2 (fragmento C22-C27) se inició con la reacción de
carboaluminación-yodonolisis del 3-butin-1-ol (esquema 3). Este proceso condujo al yodoalcohol 22, que se transformó en el enino 23 mediante acoplamiento de Kumada, catalizado por paladio, con el cloruro de etinilmagnesio. La oxidación Jones seguida de esterificación con diazometano dio el éster 2.
Esquema 3 La síntesis del fragmento 5 (C17-C21) se inició con la preparación del Z-3-
yodopropenoato de metilo 24 mediante adición Michael del anión yoduro al propinoato de metilo (esquema 4). La reducción del éster 24 con DIBAL, seguida de isomerización del doble enlace mediante tratamiento con el complejo trifluoruro de boro-eterato, proporcionó el E-3-yodopropenal 25. Este compuesto se sometió a un proceso de crotilación tipo Brown, que condujo a la formación de una mezcla de enantiómeros 5/26 con tan sólo un 39% de exceso enantiomérico en favor de 5. La proporción del enantiómero 5 se incrementó al someter la mezcla de enantiómeros a un proceso de resolución cinética tipo Sharpless, lo que permitió obtener 5 con un 89% de exceso enantiomérico.

Síntesis de exiguolido 109
Esquema 4
3. Unión de los fragmentos y pasos finales
El asalto final a la molécula de exiguolido se inició con la reacción de
esterificación del ácido 21 con el alcohol 5. Esta reacción, que se llevó a cabo en presencia de diciclohexilcarbodiimida (DCC) y N,N-dimetil-4-aminopiridina (DMAP), proporcionó el éster 4, el cual, mediante reacción de metátesis ciclante inducida por el catalizador de Grubbs-Hoveyda, proporcionó la cetomacrolactona 3 (esquema 5). El compuesto 29 se obtuvo de forma selectiva (5.8:1) cuando la cetomacrolactona 3 se hizo reaccionar con el anión derivado del fosfonato quiral 28. A continuación, se procedió a instalar el fragmento C22-C27, lo cual se consiguió mediante el acoplamiento Sonogashira entre el enino éster 2 y el yoduro de alquenilo 29. Ester proceso proporcionó el compuesto 30, que se convirtió en el exiguolido sintético mediante hidrogenación Lindlar del triple enlace.

Síntesis de exiguolido
110
Esquema 5
Comentarios 1. Ciclación radicalaria de yodo−β−−β−−β−−β−alcoxiacrilatos

Síntesis de exiguolido 111
La ciclación radicalaria intramolecular sobre sustratos que contengan sistemas de yodo−β−alcoxiacrilatos, tales como el que presenta el compuesto 10, permite la obtención de anillos tetrahidropiránicos cis-2,6-disustituidos. Experimentalmente la conversión del compuesto 10 en el tetrahidropirano 18 se consiguió mediante reacción con hipofosfito de 1-etilpiridinio y trietilborano en atmósfera de aire. El hipofosfito de 1-etilpiridinio se genera in situ mediante reacción del ácido hipofosforoso con la 1-etilpiperidina. En el esquema 6 se muestra el mecanismo de la reacción, que comienza con el proceso de generación radicalaria (etapa de iniciación).
Esquema 6
El hipofosfito de 1-etilpiridinio se convierte en la especie radicalaria I centrada en el fósforo (segunda etapa del proceso de iniciación).

Síntesis de exiguolido
112
El radical I provoca la escisión homolítica del enlace carbono-yodo en el yodoacrilato 10, y la subsiguiente formación del radical II centrado en el carbono (primera etapa del proceso de propagación). A continuación, el radical II experimenta el proceso de ciclación radicalaria intramolecular, que es estereoselectivo y que conduce a la formación del radical tetrahidropiránico IV , cuya reducción, por reacción con hipofosfito de N-etilpiridinio, conduce al compuesto tetrahidropiránico neutro 18.
Aunque el hipofosfito de 1-etilpiridinio es un precursor eficiente de radicales se requiere, muy a menudo, del empleo de un exceso de este reactivo. De hecho, si se suman las tres etapas del proceso de propagación se accede a la reacción ajustada del proceso, en la que se observa que se necesita una cantidad estequiométrica de hipofosfito de 1-etilpiridinio. Aún así, este precursor radicalario, y dador de átomos de hidrógeno, constituye una alternativa económica y benigna, desde el punto de vista medioambiental, a otros dadores de hidrógeno empleados en los procesos radicalarios, como es el caso de los hidruros de estaño, ya que los residuos que contienen fósforo no son tóxicos, y se eliminan fácilmente de la mezcla de reacción mediante un work-up acuoso.
La estereoselectividad del proceso se explica en el esquema 7, en el cual se observa la presencia de una interacción estérica desestabilizante en el estado de transición I , que conduciría a la formación del anillo tetrahidropiránico 2,6-trans-disustituido. Esta interacción desfavorable no existe en el estado de transición II , que es el que explica la formación del anillo tetrahidropiránico 2,6-cis-disustituido.
Esquema 7

Síntesis de exiguolido 113
2. Ciclación de Prins de β−β−β−β−alcoxipropionatos
O O
HO
MeO2C
BnO
O O
MeO2C
BnO
1. TFA, CH2Cl2
2. K2CO3, MeOH(81%)
7 19
1
3
5
7
1
3
57
La reacción de Prins sobre β−alcoxipropionatos homoalílicos permite la
obtención de anillos tetrahidropiránicos cis-2,6-disustituidos. El mecanismo que explica la conversión del compuesto 7 en el bis-tetrahidropirano 19 se describe en el esquema 8. El proceso se inicia con la protonación quimio y regioselectiva del sistema de β-alcoxipropionato, lo que conduce a la formación de los cationes oxonio isoméricos I y II . El ataque nucleofílico intramolecular del doble enlace forma los carbocationes oxaciclohexánicos II y IV , que son estabilizados por el ataque nucleofílico del ácido trifluoroacético, o del anión trilfuoroacetato. El proceso forma una mezcla de tetrahidropiranos epiméricos en C3 y C5, que por saponificación se convierten en los compuestos 19 y 31.
O
R
MeO2C
7
1
3
57
TFA
TFA
O
MeOOCH
RH
O
H H
R
MeOOC
O
MeOOCH
RH TFAO
R
MeO2C
3
5
7OF3C
O
O
H H
RMeOOC
TFAO
R
MeO2C
3
5
7OF3C
O
LiOH O
R
MeO2C
3
5
7HO
LiOHO
R
MeO2C
3
5
7HO
33
33
77
77
19
31
I III
II IV
V
VI
Esquema 8

Síntesis de exiguolido
114
La reacción Prins sobre el sustrato 7 provoca la epimerización parcial del estereocentro en C7. En el esquema 9 se explica este proceso, que se inicia con la reacción de transposición [3,3] sigmatrópica sobre el catión oxonio I . Este proceso forma el catión oxonio isomérico VII , en el cual el carbono C7 exhibe hibridación sp2. La isomerización de VII forma el catión oxonio VIII , que conduce al carbocatión oxaciclohexánico IX por ataque nucleofílico intramolecular del doble enlace. La estabilización de este carbocatión, por reacción con el ácido trifluoroacético, y la subsiguiente saponificación, proporcionan el compuesto tetrahidropiránico 32 epimérico en C7.
Esquema 9 3. Metilenación de aldehídos catalizada por rodio
O
HR
N2
HTMS (1.4 eq)
ClRh(PPh3)3 (2.5 mol%)i-PrOH (1.1 eq), PPh3(1.1 eq)
CH2
HR
La metilenación de aldehídos, por reacción con trimetilsilildiazometano,
trifenilfosfina e isopropanol en presencia de cantidades catalíticas de ClRh(PPh3)3, constituye una alternativa a los conocidos procesos de metilenación de Wittig y Tebbe.

Síntesis de exiguolido 115
Las condiciones de reacción son tan suaves y neutras que permiten la conversión de sustratos enolizables, como los aldehídos α-quirales, sin que se produzca pérdida de pureza óptica.
El mecanismo de la reacción implica la formación, in situ, de metilentrifenilfosforano, que va seguida de una reacción de Wittig convencional entre dicho fosforano y el aldehído.
El ciclo catalítico que explica la formación del metilentrifenilfosforano se muestra en el esquema 10. En primer lugar se produce la activación del trimetilsilidiazometano mediante coordinación con el rodio. Este proceso forma el intermedio I , que resulta protonado por reacción con el isopropanol. A continuación, el aducto protonado II es atacado por la trifenilfosfina, lo que provoca la regeneración del catalizador y la formación de la sal de fosfonio sililada III . La subsiguiente descomposición de III forma isopropóxido de trimetilsililo y metilentrifenilfosforano.
Esquema 10

Síntesis de exiguolido
116
4. Reacción de olefinación Horner-Wadsworth-Emmons con el fosfonato quiral 28
El fosfonato quiral (S)-28 es capaz de diferenciar las dos caras enantiotópicas
de los grupos carbonilo para proporcionar, preferentemente, la olefina de configuración Z.
La ionización del fosfonato 28 forma un quelato cíclico 32 cuya estructura se muestra en el esquema 11. La formación de este quelato impide la libertad conformacional y provoca el bloqueo estérico de la cara Re.
32
OP
O
OH
OOMe
M H
H
cara Re
estéricamente
bloqueada
cara Si
accesible
Esquema 11
El proceso de olefinación con el quelato 32 implica, en primer lugar, la adición al grupo carbonilo, y a continuación el proceso de sin-eliminación, que forma el alqueno y un éster fosfórico (véase el esquema 12). La etapa que determina la velocidad global del proceso es la de adición al grupo carbonilo, y puede transcurrir a través de los dos estados de transición mostrados en el esquema 12, en los cuales el quelato 32 siempre expone su cara Si en la aproximación al compuesto. Sin embargo, el quelato puede atacar la cara Re o la cara Si del compuesto carbonílico, como se muestra en el esquema 12. Cuando se produce el ataque a la cara Re del compuesto carbonílico se genera el estado de transición ET-
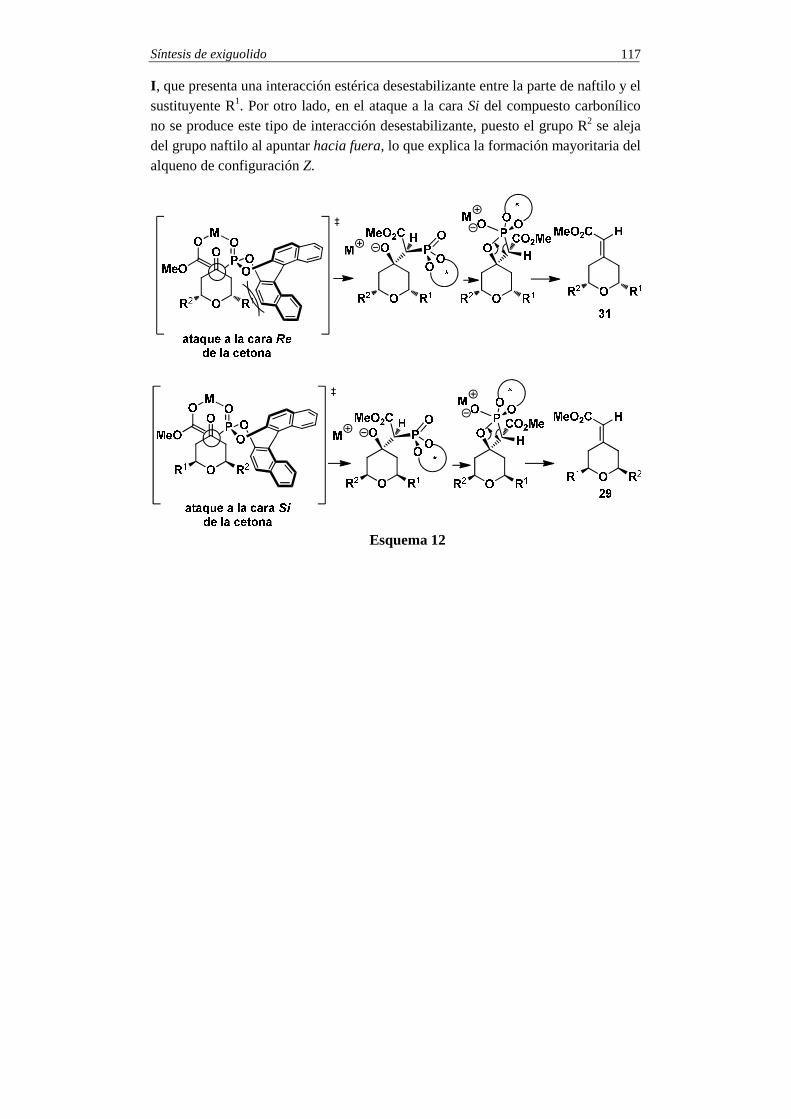
Síntesis de exiguolido 117
I , que presenta una interacción estérica desestabilizante entre la parte de naftilo y el sustituyente R1. Por otro lado, en el ataque a la cara Si del compuesto carbonílico no se produce este tipo de interacción desestabilizante, puesto el grupo R2 se aleja del grupo naftilo al apuntar hacia fuera, lo que explica la formación mayoritaria del alqueno de configuración Z.
Esquema 12


Síntesis de fortucina 119
SÍNTESIS DE FORTUCINA
Aislamiento: El alcaloide fortucina se ha aislado de las hojas de una variedad de narcisos denominada Fortuna. Actividad biológica: Los alcaloides de tipo licorina, como la fortucina, la licorina y el γ-licorano, se caracterizan por poseer en su estructura el sistema de anillos de galantano (perpirrolo[d,e]fenantridina) y constituyen una subclase dentro de la familia de los alcaloides de Amaryllidaceae. La gran mayoría de los alcaloides de tipo licorina presentan fusión trans entre los anillos B/C, como en el caso de la propia licorina (véase figura 1). Sin embargo, se han aislado unos pocos alcaloides de esta subclase que presentan fusión cis entre los anillos B/C, como es el caso del γ-licorano y de la fortucina.
Figura 1
Muchos compuestos de la familia de alcaloides de Amaryllidaceae poseen propiedades antivirales, antineoplásicas, antialimentarias, de regulación del crecimiento vegetal o de disrupción de la síntesis de proteínas.
Retrosíntesis La retrosíntesis de la estructura de la fortucina se inicia con una operación de
intercambio de grupo funcional que conduce a la enona tetracíclica 1.21 La
21 A. Biechy, S. Hachisu, B. Quiclet-Sire, L. Ricard, S. Z. Zard, Angew. Chem. Int. Ed. 2008, 46, 1436.

Síntesis de fortucina
120
presencia de la agrupación alcoxi (RO) en el carbono angular permitirá dirigir el sentido de la reacción de eliminación mediante la cual se instalará el doble enlace angular del anillo C de la fortucina. La agrupación amino del intermedio 1 se preparará por reducción de la función amida que contiene el compuesto 2. En este punto se aborda el paso clave del análisis retrosintético que consiste en la desconexión homolítica de los ciclos B y D, con la consiguiente generación de la especie radicalaria centrada en el nitrógeno 3. En el sentido sintético, la generación del radical imidilo 3 deberá provocar la formación del sistema de anillos de galantano mediante un proceso de adición radicalaria intramolecular en cascada. El precursor del radical nitrogenado 3 será la amida 4, en el cual la estructura funcional del grupo R´ deberá ser de naturaleza tal que permita la generación del radical imidilo mediante escisión homolítica del enlace N-R´. La escisión del enlace amida en 4 origina el compuesto 5 y el derivado de ácido carboxílico 6. La desconexión del compuesto 5 conduce a la p-quinona 7 y al sintón organometálico 8 (M=Metal), cuyo equivalente sintético será la hidrazona 9.
Esquema 1

Síntesis de fortucina 121
Síntesis 1. Síntesis del precursor radicalario 18
El compuesto 18 es el precursor de la especie radicalaria centrada en el nitrógeno que debe desencadenar el proceso de formación en cascada de los anillos B y D de la fortucina (véase retrosíntesis).
Esquema 2

Síntesis de fortucina
122
La preparación de 18 se inició con la obtención de la hidrazona 9 por condensación del acetaldehído con la 1-(metiltio)tiocarbonil-1-fenil-hidracina 11 (esquema 2). Por otro lado, el p-metoxifenol 13 se transformó en el monocetal de p-quinona 14 por reacción con etilenglicol y PIFA (trifluroroacetato de fenilyodonio, véanse comentarios). La metalación de la hidrazona 9 con LDA generó el azaenolato lítico 12, que se adicionó al grupo carbonilo de 14 para dar el alcohol 15 con rendimientos cuantitativos. La protección del hidroxilo, seguida de reducción del doble enlace C=N con borohidruro de sodio, condujo a la hidracina 17, que se convirtió en el compuesto 18 por amidación con el cloruro de ácido 6. 2. Obtención del intermedio 2 mediante ciclación radicalaria intramolecular y su conversión en fortucina
Conviene señalar en este punto que la síntesis de la fortucina de Zard y
colaboradores no es enantioselectiva. Aunque en el esquema 3, y en todos los que siguen, únicamente se dibuja la estructura de un enantiómero, todos los compuestos quirales se obtuvieron en su forma racémica, incluida la fortucina sintética.
Los últimos pasos en la síntesis de la fortucina se iniciaron con el proceso de ciclación radicalaria intramolecular. Así, la amida 18 se calentó durante siete horas, a reflujo de 1,2-dicloroetano, en presencia del iniciador radicalario peróxido de lauroilo. En estas condiciones se obtuvo, con un 60% de rendimiento, el pentaciclo todo-cis 19 (véanse comentarios). En esta reacción no se formó ningún diastereoisómero con fusión trans de los anillos B/C, aunque se obtuvo una mezcla 14:1 de isómeros para/orto, con relación al grupo benciloxi. La compresión estérica entre la agrupación etilencetal y el grupo benciloxi explica la formación muy minoritaria del isómero orto.

Síntesis de fortucina 123
NBnO
MeOOTBS
O N
Ph
S
SMe
18
peróxido de lauroilo
1,2-dicloroetano (60%)
NH
H
O
BnO
MeO
O
OTBS
2
TFA
H2O(88%)
O
O
NH
H
BnO
MeO
O
OTBS
19
O
O
HS OMe
THF, Et3N, RT(80%)
NH
H
O
BnO
MeO
O
OTBS
20
SAr
LiAlH4, THF
NH
H
HO
BnO
MeO OTBS
21
SAr
dimetildioxiranoN
H
H
HO
BnO
MeO OTBS
22
SO2Ar
H2, Pd/C
THF (94%)
NH
H
HO
HO
MeO OTBS
23
SO2Ar
NH
H
HO
HO
MeO
fortucina
Na/Hg, MeOH
reflujo (50%)
RT (66%)
TFA, acetona (92%)
A
B
D
A
B
D
C
A
B
D
C
A
B
D
C
A
B
D
C
A
B
D
C
A
B
D
C
A
C C
Esquema 3
El compuesto 19, mediante hidrólisis ácida de la función acetálica, se
convirtió en la enona tetracíclica 2. La conversión de este compuesto en la fortucina implicaba la reducción estereoselectiva del grupo carbonilo cetónico y la isomerización formal del doble enlace disustituido a la posición angular trisustituida. Con este fin el compuesto 2 se transformó en el tioéter 20 por adición conjugada Michael de 2-metoxitiofenol. La reducción de la función amida y cetónica de 20, por reacción con LiAlH4, proporcionó la amina terciaria 21. La reducción estereoselectiva del grupo carbonilo cetónico se explica por el ataque del reductor a la cara convexa de la molécula. La conversión de 21 en la fortucina se consiguió mediante una secuencia de tres pasos, que se inició con la oxidación del sulfuro a sulfona, por reacción de 21 con dimetildioxirano. La desbencilación

Síntesis de fortucina
124
hidrogenolítica proporcionó el fenol 23, cuyo tratamiento reductivo con la amalgama Na/Hg condujo a la fortucina sintética (véanse comentarios).
Comentarios
1. Síntesis de monocetales de p-quinona
La reacción ajustada para la conversión del fenol 13 en el cetal 14 se indica en el esquema 4.22 En esta transformación se emplean 1.3 equivalentes de bis(trifluoroacetato de fenilyodonio, PIFA) y 1.5 equivalentes de etilenglicol.
Esquema 4
El mecanismo que explica la reacción anterior se describe en el esquema 5. El proceso comienza con el ataque nucleofílico del fenol a la molécula de PIFA. Esta reacción forma el intermedio 24 y ácido trifluoroacético. A continuación, el compuesto 24 experimenta un proceso de eliminación que conduce a la formación de yodobenceno y del trifluoroacetato de oxonio 25. Este compuesto, altamente electrofílico, es atacado por la molécula de etilenglicol con formación del intermedio 26 y ácido trifluoroacético. Los últimos pasos, indicados en la etapa 4 del esquema 5, son los propios de un proceso de cetalización intramolecular, que en este caso está catalizado por el ácido trifluoroacético.
22 M.-E. Traân-Huu-Daâu, R. Wartchow, E. Winterfeldt, Y.-S. Wong , Chem. Eur. J. 2001, 7, 2349.
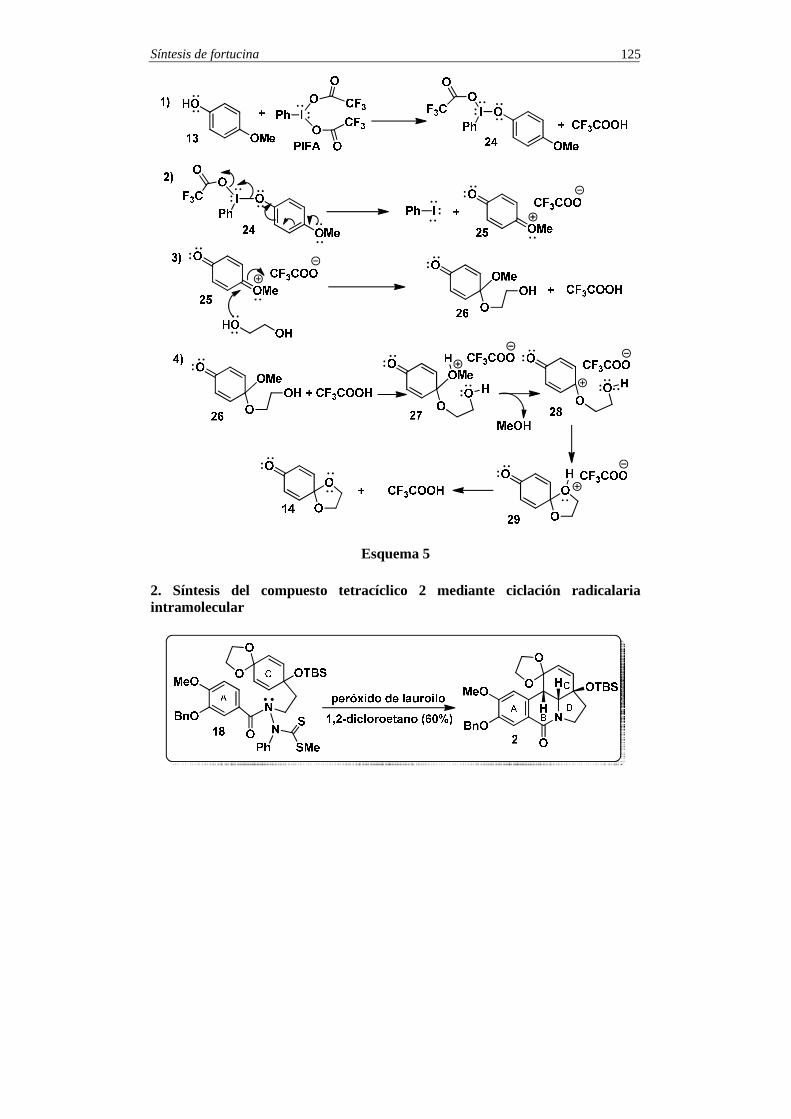
Síntesis de fortucina 125
Esquema 5
2. Síntesis del compuesto tetracíclico 2 mediante ciclación radicalaria intramolecular

Síntesis de fortucina
126
La síntesis del sistema de anillos de galantano se consiguió por reacción del compuesto 18 con peróxido de lauroilo a reflujo de 1,2-dicloroetano. El mecanismo general de este proceso radicalario, que se describe en el esquema 6, incluye, como etapas principales, la de iniciación y la de propagación.
NBnO
MeOOTBS
O N
Ph
S
SMe
18
O
O
NH
H
BnO
MeO
O
OTBS
2
O
O
2 CO2 +
1)
a) Iniciación
b) Propagación
C11H23N
BnO
MeOOTBS
O N
Ph
S
SMe
O
O
C11H23
calor
NBnO
MeOOTBS
O N
Ph
S
SMe
O
O
C11H23
2)
NBnO
MeOOTBS
O
N
Ph
S
SMe
O
OC11H23
+
3)
NBnO
MeOOTBS
O
O
O
NH
H
BnO
MeO
O
OTBS
O
OH
NH
H
BnO
MeO
O
OTBS
O
OH
4)
N
Ph
S
SMe
C11H23
++
PhNH SMe
S
+
O
C11H23 OO
O
C11H23 2 C11H23
30
3031
31 33
33
32
32
34
C11H23
Esquema 6

Síntesis de fortucina 127
En la etapa de iniciación se generaran radicales centrados en el carbono por la descomposición térmica del peróxido de lauroilo. El proceso de propagación se inicia con la adición del radical centrado en el carbono al doble enlace C-S y con la formación de la nueva especie radicalaria 30, cuya escisión homolítica genera el radical centrado en el nitrógeno 31 y la especie neutra 32. En la tercera etapa del proceso de propagación tiene lugar la reacción de adición radicalaria intramolecular en cascada que convierte al radical 31 en el radical 32, que ya contiene el sistema de anillos A/B/C/D de galantano. En la última etapa del proceso de propagación se produce la conversión del radical 32 en el compuesto neutro 2 y la generación de radicales centrados en el carbono, que inician un nuevo ciclo de propagación. 3. Obtención de fortucina mediante olefinación tipo Julia
En las reacciones de olefinación de Julia un aldehído, o cetona, por reacción con un anión de sulfona, se convierte en una β−alcoxisulfona, la cual se transforma en una olefina por esterificación, seguida de tratamiento reductivo con un dador monoelectrónico (esquema 7).
Esquema 7

Síntesis de fortucina
128
La etapa reductiva del mecanismo anterior, y la transformación de 2 en fortucina, son mecanísticamente similares, razón por la cual se clasifica a esta conversión como de tipo Julia. El mecanismo del proceso, que se dibuja en el esquema 8, se inicia con una etapa de cesión monoelectrónica desde el metal a la sulfona 2. Esta reacción genera el radical 34, que es reducido al anión 35 mediante un nuevo proceso de cesión monoelectrónica. Finalmente, el anión experimenta un proceso de eliminación intramolecular, con formación del doble enlace y expulsión del grupo t-butildimetilsililóxido.
El modesto rendimiento en la formación de fortucina se debe a la generación del producto de desulfonilación reductiva sin eliminación del grupo t-butildimetilsililóxido.
NH
H
HO
HO
MeO OTBS
2
S
NH
H
HO
HO
MeO
O O
Ar
ArSO2+ 1e NH
H
HO
HO
MeO OTBS
+
NH
H
HO
HO
MeO OTBS
1)
2)
+ 1e
OTBS
NH
H
HO
HO
MeO
3)
NH
H
HO
HO
MeO OTBS
+ TBSO
fortucina
34
34 35
35
Esquema 8

Síntesis de grandisina 129
SÍNTESIS DE GRANDISINA A
Aislamiento: Las grandisinas, una familia de alcaloides de indolizidina, han sido aisladas del arbol Elaeocarpus grandis. Actividad biológica: Se denominan opiodes aquellas sustancias, endógenas o exógenas, que tienen un efecto análogo al de la morfina. Los compuestos opiodes se unen selectivamente en el organismo a los denominados receptores opiodes, de los cuales hay tres tipos: mu (µ), delta (δ) y kappa (κ).
Las grandisinas provocan un efecto opiode mediante su unión al receptor delta (δ). Estos alcaloides tienen un efecto agonista puesto que su unión al receptor provoca algún tipo de respuesta en la célula. Por otro lado, los compuestos que se unen al receptor, pero no desarrollan efecto alguno, y carecen de actividad intrínseca, se denominan antagonistas opioides.
La actividad intrínseca se define como la capacidad que tiene un compuesto de provocar la inducción de determinados efectos en un receptor. Por ejemplo, la morfina y la metadona tienen la misma especificidad por el receptor opioide y como agonistas puros provocan la máxima actividad intrínseca. En principio producen el mismo efecto, aunque se diferencian en su comportamiento farmacocinético, es decir en su capacidad para atravesar la barrera hematoencefálica, en su distribución por los diferentes compartimentos (sangre, tejido cerebral, órganos internos), en su metabolismo y en su excreción.
Los receptores opioides se localizan frecuentemente en la porción final del axón presináptico de la célula nerviosa y modulan la liberación de los neurotransmisores al inhibir la entrada en funcionamiento del potencial de acción, por lo que provocan una disminución de la cantidad de sustancia transmisora liberada. El efecto del receptor opioide es muy marcado en las células nerviosas que transmiten el dolor, en las cuales provocan la inhibición de la liberación de la sustancia transmisora de aquél, lo que explica su efecto analgésico. Los opioides con mayor efecto analgésico, que son los preferidos por los adictos, son los que

Síntesis de grandisina 130
actúan en los receptores µ. Las grandisinas disminuyen el dolor sin los efectos laterales adversos, como hipertensión y náuseas, que provocan los opiodes clásicos.
Retrosíntesis
Lo más llamativo de la estructura de la grandisina A es la presencia de la fusión cis entre los anillos B-C-D, puesta de manifiesto en la configuración de los estereocentros en C7, C8 y C9. Desde el punto de vista termodinámico, la fusión cis entre los anillos B-C es menos estable que la fusión trans. Esta disposición estructural obligará a llevar a cabo la fusión de estos dos anillos en condiciones de control cinético, a fin de evitar la formación de la más estable fusión trans.
El análisis retrosintético de la grandisina A se indica en el esquema 1.23 La primera desconexión de la grandisina A, que tiene lugar sobre el anillo D, conduce al intermedio tricíclico 1, que a su vez se podría obtener mediante homologación del compuesto 2.
Esquema 1
23 D. J. Maloney, S. J. Danishefsky, Angew. Chem. Int. Ed. 2007, 46, 7789.

Síntesis de grandisina 131
La escisión del anillo dihidropiranónico A conduce a la estructura 3, que por desconexión del sistema 1,3-dicarbonílico genera el β−alcoxibutanal 5 y el compuesto bicíclico 4. La interconversión del grupo funcional cetónico en el compuesto 4 conduce al enol éter 6. El análisis del anillo B de este último compuesto, mediante aplicación de una operación retro-Diels-Alder, genera acetaldehído (heterodienófilo) y el compuesto 7 (dieno). Una etapa de interconversión de grupo funcional convierte 7 en la azaenona 8, que se podría obtener a partir de la dihidropiridona 9.
Síntesis
1. Síntesis del compuesto 8: anillo C funcionalizado
O
N
O
Me
OH
H
H
Me
grandisina A
A
B C78
9
8
D
Me NCbz
O
8
7
910
C
La dihidropiridinona 9, que constituirá el anillo C de la grandisina A, se obtuvo a partir de la 4-metoxipiridina 10 mediante tratamiento con borohidruro de sodio en metanol a -78ºC, adición de cloroformiato de bencilo a la mezcla de reacción, agitación durante 1 hora a -78ºC y luego adición de agua y calentamiento hasta temperatura ambiente (véanse comentarios) (esquema 2).
Esquema 2

Síntesis de grandisina 132
La adición conjugada de bromuro de vinilmagnesio a la dihidropiridinona 9, en presencia de yoduro cuproso, generó el correspondiente enolato metálico que se adicionó al acetaldehído para dar el compuesto 11. La sililación del hidroxilo, seguida de reducción del carbonilo cetónico, proporcionó el alcohol 12, que por mesilación y desililación se convirtió en 13. La enona 8 se obtuvo, en forma racémica, por oxidación Swern del alcohol 13 seguida de β−eliminación del grupo mesiloxi con DBU (1,8-diazabiciclo[5.4.0]undec-7-eno). 2. Síntesis del intermedio bicíclico 4
Se puede considerar la conversión de la azaenona 8 en el compuesto bicíclico 4 como la secuencia clave en la síntesis de la grandisina A, puesto que en ella se deberán construir, de forma estereocontrolada, los centros estereogénicos en C7, C8, C9 y C12. La transformación comenzó con la obtención del silil dienol éter 7 por enolización-sililación de la azaenona 8 (esquema 3). La reacción de este compuesto con acetaldehído, en presencia de BF3·Et2O, proporcionó el compuesto bicíclico 6. El estado de transición 14 del esquema 3 explica la estereoselectividad observada en la reacción de cicloadición hetero-Diels-Alder entre el acetaldehído y el silil dienol éter 7. La aproximación del heterodienófilo se produce desde la cara opuesta a la que ocupa el grupo benciloxicarbonilo (Cbz). Por otro lado, la parte de ácido de Lewis, coordinada con el oxígeno carbonílico del acetaldehído, se sitúa en disposición exo a fin de disminuir las interacciones estéricas con el grupo vinilo axial. Esta disposición obliga a la colocación endo del grupo metilo, y permite la construcción de los centros estereogénicos de C7 y C12 con la configuración requerida para la síntesis de la grandisina A (véanse comentarios). Finalmente, el compuesto bicíclico 4 se obtuvo por desililación de 6 con fluoruro de tetra-n-butilamonio en ácido acético. En este proceso se construyó el estereocentro en C8 por protonación axial del enolato 15, tal y como se indica en el esquema 3.

Síntesis de grandisina 133
Esquema 3
2. Síntesis del intermedio tricíclico 2
El compuesto bicíclico 4, preparado según se indica en el esquema 3, se obtuvo en forma racémica. A fin de conseguir una síntesis enantioselectiva de la grandisina A se procedió a la separación de la mezcla racémica mediante HPLC preparativa en columna quiral (esquema 4).

Síntesis de grandisina 134
Esquema 4
El compuesto (+)-4, obtenido en el proceso de separación enantioselectiva, se enolizó, en condiciones de control cinético, con hexametildisililamiduro de litio (LiHMDS), y el correspondiente enolato de litio se convirtió en enolato de zinc por transmetalación con cloruro de zinc. La adición del β−sililoxialdehído 17 a la mezcla de reacción proporcionó el producto de adición aldólica 18, que se oxidó con el reactivo de Dess-Martin a la dicetona 19. La adición de ácido trifluoroacético a la mezcla de oxidación, seguida de agitación a temperatura ambiente durante 1 hora, proporcionó el compuesto tricíclico 2, con un 73% de rendimiento desde (+)-4.

Síntesis de grandisina 135
4. Pasos finales
En el esquema 4 se indica la secuencia sintética que permitió la conversión del compuesto 2 en la grandisina A.
O
N
O
Me
OH
H
H
Me
grandisina A
O
NCbz
O
Me
OH
H
H
Me
2
O3, MeOH, Sudan III
-78ºC, luego SMe2O
NCbz
O O
Me
OH
H
H
Me
20
H
Ph3P=CHCOOMe
benceno, 60ºC
(80% desde 2)
O
NCbz
O
Me
OH
H
H
Me
21
H COOMe
H2, Pd/C
MeOH
O
NH
O
Me
OH
H
H
Me
1
COOMe
O
N
O
Me
OH
H
H
Me
O tolueno
reflujo
O
N
O
Me
OH
H
H
Me
S Ni-Raney, THF
25ºC (94%)
reactivo de Lawessontolueno, 65ºC (98%)
22
23
Esquema 4
El proceso se inició con la ozonolisis de 2, lo que provocó la ruptura
oxidante del doble enlace vinílico y la formación del aldehído 20, que se transformó en el éster insaturado 21 mediante reacción de olefinación Wittig. La hidrogenación selectiva de 21, en presencia de Pd/C, proporcionó el éster 1, que se

Síntesis de grandisina 136
convirtió en la lactama 22 por calentamiento en tolueno. El tratamiento de la lactama 22 con el reactivo de Lawesson (2,4-bis(4-metoxifenil)-1,3,2,4-ditiadifosfetan-2,4-disulfuro) generó la tiolactama 23 (véanse comentarios), que se convirtió en el grandisina A sintética por desulfuración con Ni-Raney.
Comentarios
1. Síntesis de la dihidropiridinona 9
La tetrahidropiridinona 9 se obtuvo del siguiente modo. A una disolución
metanólica de la 4-metoxipiridina, se la añadió, a -78ºC, NaBH4. Después de 15 minutos de agitación se añadió una disolución etérea de cloroformiato de bencilo y se continuó la agitación durante 1 hora a -78ºC. La reacción se detuvo mediante adición de agua y calentamiento hasta temperatura ambiente.
El mecanismo de la conversión se indica en el esquema 5, y se inicia con la adición de hidruro al anillo piridínico. Esta reacción forma el intermedio 24 que conduce al carbamato 25 por adición de cloroformiato de bencilo a la mezcla de reducción.
Esquema 5

Síntesis de grandisina 137
La adición de agua debe provocar la hidrólisis de la parte de metil enol éter. Este tipo de funcionalidad se hidroliza en medio ácido, sin embargo las condiciones de reacción no son tales, a tenor de la parte experimental descrita más arriba. Un posible mecanismo de hidrólisis se iniciaría con la protonación del intermedio 25 por ataque básico de la parte de metil enol éter a la molécula de agua. Esta reacción conduciría a la formación del catión oxonio 26, que resultaría atacado nucleofílicamente por el anión hidróxido con formación de metanol y de la tetrahidropiridinona 9.
2. Ensayos alternativos para la obtención del compuesto bicíclico 4
Como se ha comentado anteriormente, uno de los desafíos que planteaba la síntesis de la grandisina A era la construcción del sistema de estereocentros de los carbonos C7, C8, C9 y C12. En una de las primeras aproximaciones a la síntesis del producto natural se llevó a cabo una reacción de cicloadición entre el acetaldehído y el compuesto 27 (esquema 6).
Esquema 6

Síntesis de grandisina 138
Sin embargo, el producto obtenido en esta secuencia sintética fue el compuesto 30, con disposición anti de los anillos B-C con respecto a D. Muy probablemente, la conformación doblada del compuesto 27 provoca la cicloadición del acetaldehído desde la cara exo, menos impedida, como se pone de manifiesto en el estado de transición 28 del esquema 5.
Otra de las aproximaciones iniciales al sistema de anillos B-C-D de la grandisina A se indica en el esquema 7. En este caso la reacción del compuesto 31 con hidrógeno molecular, en presencia de Pd/C, proporcionó el producto de hidrogenación y de reducción concomitante del grupo carbonilo. Cuando el producto de hidrogenación se oxidó se obtuvo únicamente el producto 32, con fusión trans entre los anillos B-C.
Esquema 7 Los ejemplos presentados en los esquemas 6 y 7 ponen de manifiesto la
dificultad en la preparación, en presencia del anillo D, del sistema de anillos B-C con fusión cis. A fin de evitar el sesgo conformacional que impone el anillo D, la síntesis del sistema de anillos B-C se llevó a cabo sobre el sustrato 7. Este compuesto sitúa al grupo vinilo en posición pseudoaxial a fin de evitar las interacciones de tipo alílico con el grupo Cbz y con el grupo sililoxi. Aun con el grupo vinilo en posición pseudoaxial, la aproximación del complejo acetaldehído-BF3 tiene lugar desde la cara α, con formación del enlace C7-O en sentido axial con respecto al anillo B (véase el esquema 8).
NCbz
TIPSO
8
7
910CH3CHO
BF3·Et2O, -78ºC
NO
OBn
H
TIPSO
OH3C
BF3
H
9
10
7
12
11
NO
OBn
H
TIPSO
OH3C
H
9
10
7
12
11
H
8
8
614
7
B
Esquema 8

Síntesis de grandisina 139
3. Empleo del reactivo de Lawesson: síntesis de la tiolactama 23
El reactivo de Lawesson (2,4-bis(4-metoxifenil)-1,3,2,4-ditiadifosfetan-2,4-
disulfuro) se emplea en la tionación de compuestos carbonílicos. La tionación de cetonas, amidas, lactamas y lactonas es usualmente más rápida que la de los ésteres, que suelen ser inertes al reactivo de Lawesson.
El reactivo de Lawesson es comercialmente accesible, pero se puede preparar en el laboratorio por reacción del anisol con pentasulfuro de fósforo. La estructura y el mecanismo de tionación del reactivo de Lawesson se indican en el esquema 9.
Esquema 9

Síntesis de grandisina 140
Al calentar el reactivo se produce la escisión de éste y la formación del iluro de ditiofosfina 33. El ataque nucleofílico del grupo carbonilo forma un tiaoxafosfetano 34, que experimenta una reacción de ciclorreversión para formar el derivado azufrado 36. La fuerza impulsora del proceso es la formación del muy estable enlace P=O.


















