
SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA - Universidad de San Carlos de...
73
SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA
Transcript of SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA - Universidad de San Carlos de...

SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA
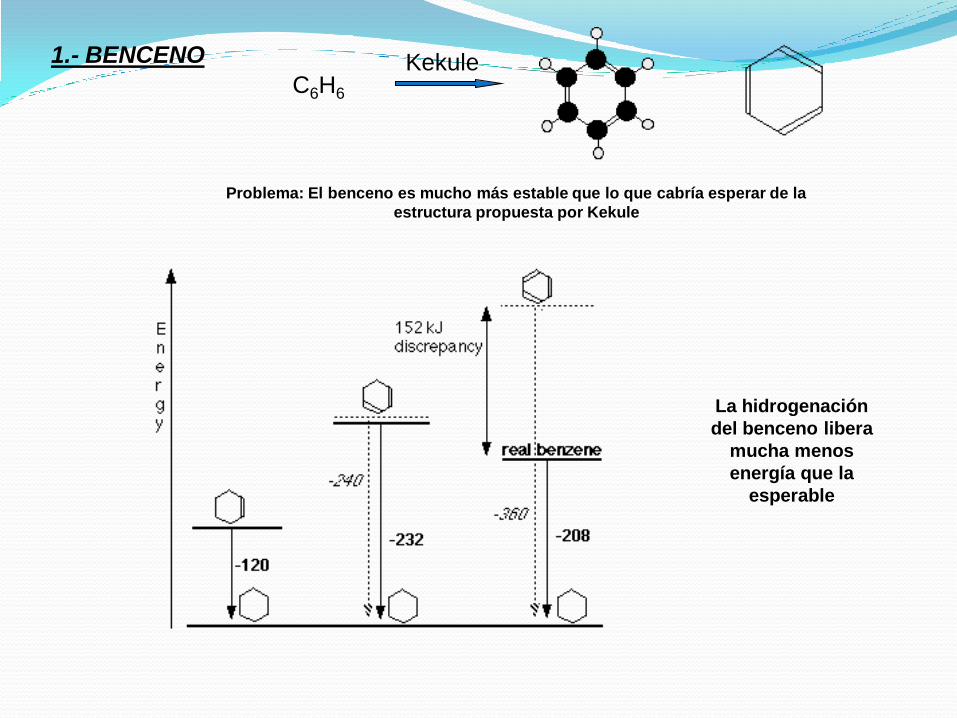
C6H6
Kekule
Problema: El benceno es mucho más estable que lo que cabría esperar de la
estructura propuesta por Kekule
La hidrogenación
del benceno libera
mucha menos
energía que la
esperable
1.- BENCENO
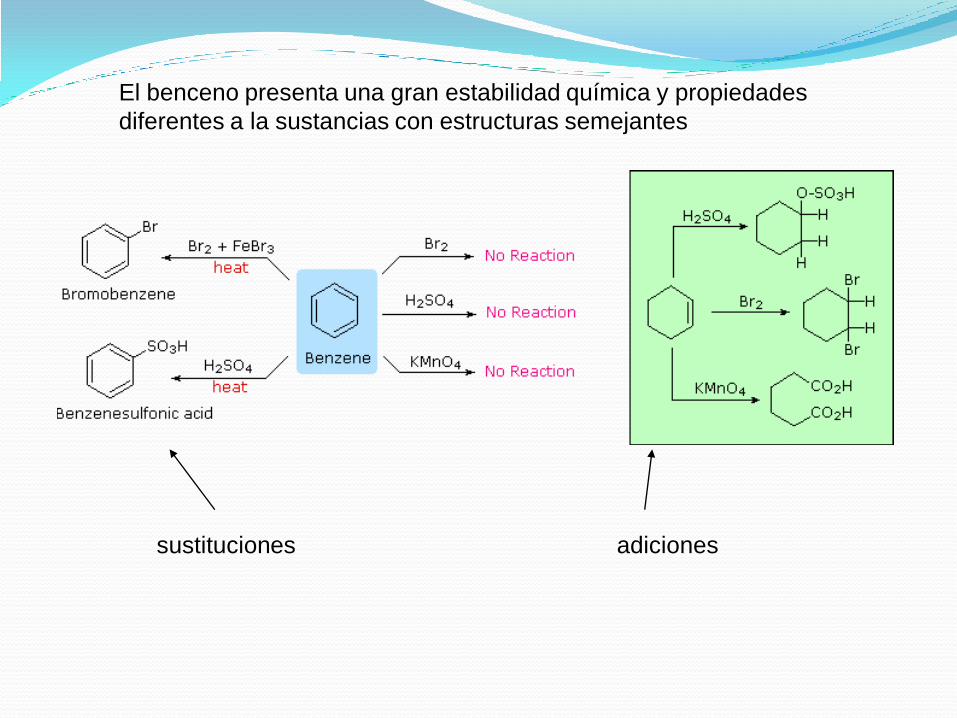
El benceno presenta una gran estabilidad química y propiedades
diferentes a la sustancias con estructuras semejantes
sustituciones adiciones

Sustitución electrofílica aromática.A pesar de que los electrones pi del benceno se encuentran en un sistema aromático estable, están disponibles para atacar a un electrófilo fuerte y dar lugar a un carbocatión. Este carbocatión estabilizado por resonancia se denomina complejo sigma debido a que el electrófilo se une al anillo del benceno mediante un nuevo enlace sigma.
La reacción es endotérmica porque el benceno pierde aromaticidad cuando ataca a un electrófilo. La aromaticidad se vuelve a recuperar mediante la pérdida de un protón. La reacción global le da el nombre de sustitución electrofílica aromática.
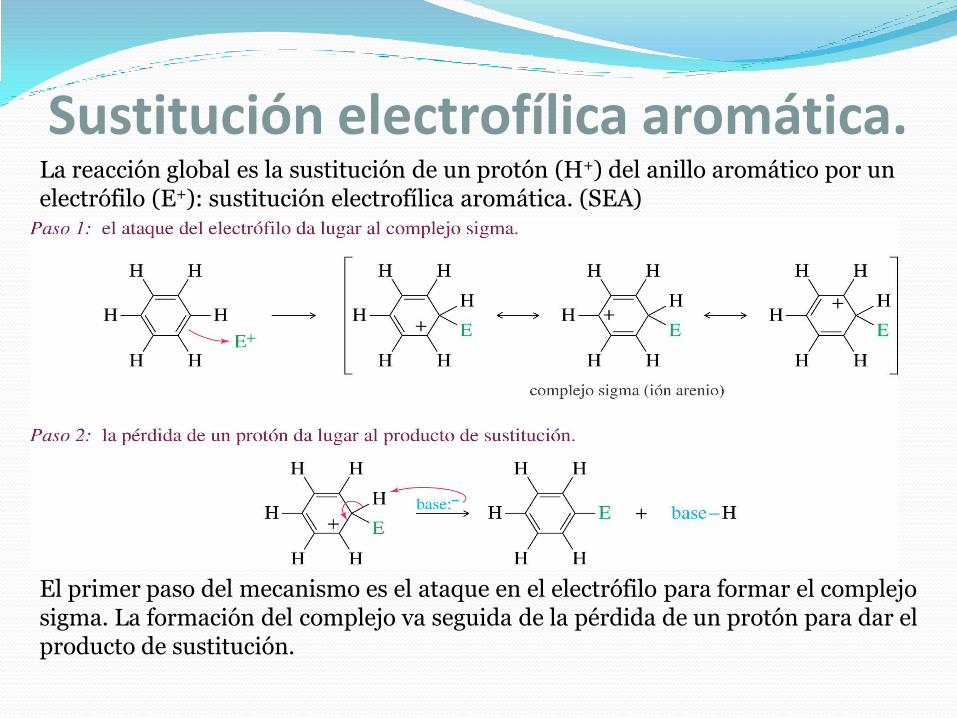
Sustitución electrofílica aromática.La reacción global es la sustitución de un protón (H+) del anillo aromático por un electrófilo (E+): sustitución electrofílica aromática. (SEA)
El primer paso del mecanismo es el ataque en el electrófilo para formar el complejo sigma. La formación del complejo va seguida de la pérdida de un protón para dar el producto de sustitución.

5.- SUSTITUCIÓN AROMÁTICA ELECTROFÍLICA
A pesar de su estabilidad
excepcional, el benceno y los
compuestos aromáticos están
lejos de ser inertes

Bromación del benceno. La bromación sigue el mecanismo general de la
sustitución aromática electrofílica. El bromo no es suficientemente electrofílico para reaccionar con el benceno, pero un ácido de Lewis fuerte, como el FeBr3, cataliza la reacción.
El primer paso del mecanismo es la formación de un electrófilo más fuerte. El catalizador reacciona con el Br2 para formar un electrófilo fuerte. El ataque del benceno en el electrófilo y la pérdida de protones da bromobenceno como producto mayoritario.

Bromación del benceno.

Diagrama de energía de la bromación del benceno
El diagrama de energía de la bromación del benceno muestra que el primer paso es endotérmico y el segundo paso es fuertemente exotérmico.
La reacción global es exotérmica, pero el ataque del electrófilo es el paso limitante de la velocidad porque el anillo pierde su aromaticidad.

En el caso del cloro el diagrama de energía de la reacción es el siguiente:
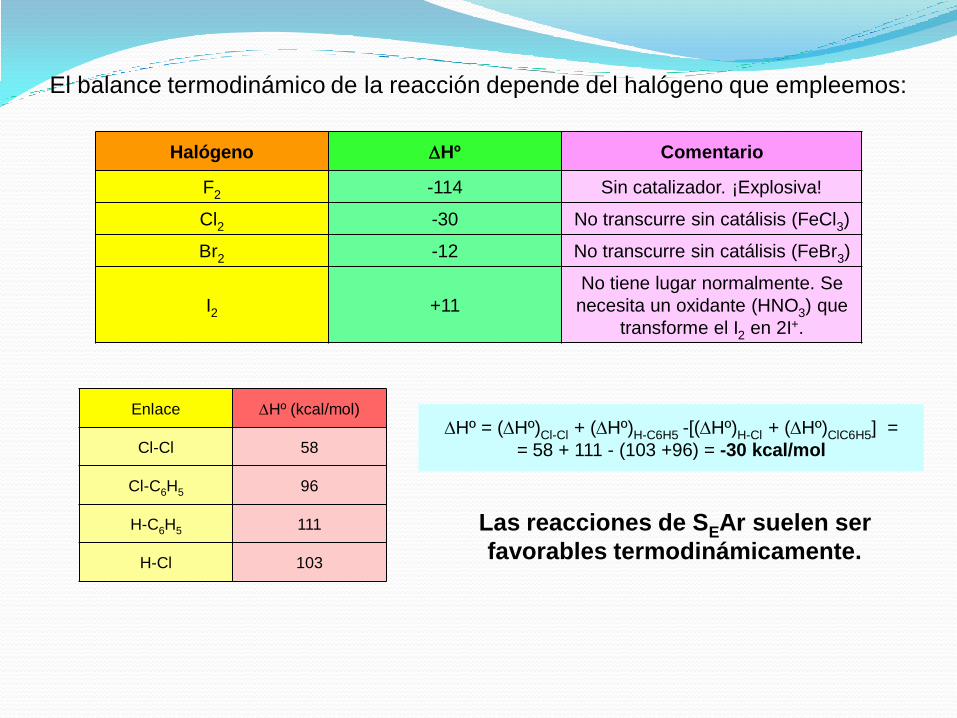
El balance termodinámico de la reacción depende del halógeno que empleemos:
Halógeno DHº Comentario
F2 -114 Sin catalizador. ¡Explosiva!
Cl2 -30 No transcurre sin catálisis (FeCl3)
Br2 -12 No transcurre sin catálisis (FeBr3)
I2 +11
No tiene lugar normalmente. Se
necesita un oxidante (HNO3) que
transforme el I2 en 2I+.
Enlace DHº (kcal/mol)
Cl-Cl 58
Cl-C6H5 96
H-C6H5 111
H-Cl 103
DHº = (DHº)Cl-Cl + (DHº)H-C6H5 -[(DHº)H-Cl + (DHº)ClC6H5] =
= 58 + 111 - (103 +96) = -30 kcal/mol
Las reacciones de SEAr suelen ser
favorables termodinámicamente.

Nitración del benceno.El mecanismo es similar a las deshidrataciones catalizadas por ácido sulfúrico. El ácido sulfúrico protona el grupo hidroxilo del ácido nítrico, haciendo que se desprenda una molécula de agua y formando un ión nitronio.
El ácido nítrico no es el electrófilo, es un ión nitronio formado por la protonación y la deshidratación de HNO3 por H2SO4. La especie atacada por el benceno es el ión nitronio. La pérdida de un protón por parte del complejo sigma produce nitrobenceno.
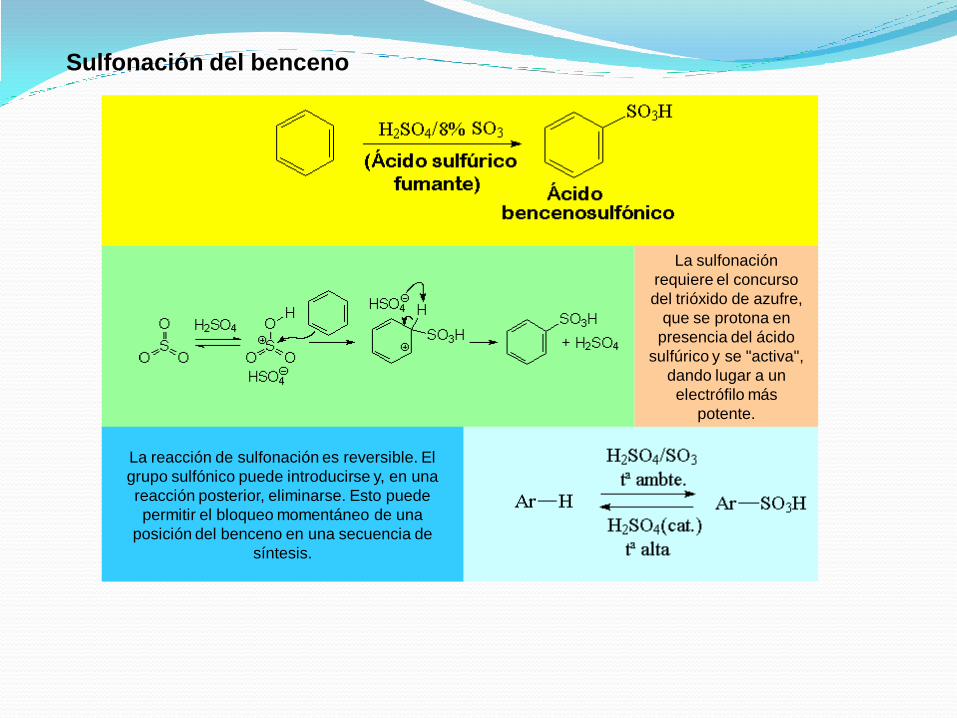
La sulfonación
requiere el concurso
del trióxido de azufre,
que se protona en
presencia del ácido
sulfúrico y se "activa",
dando lugar a un
electrófilo más
potente.
La reacción de sulfonación es reversible. El
grupo sulfónico puede introducirse y, en una
reacción posterior, eliminarse. Esto puede
permitir el bloqueo momentáneo de una
posición del benceno en una secuencia de
síntesis.
Sulfonación del benceno

Reacción de desulfonación.La sulfonación es reversible, el grupo del ácido sulfónico puede ser eliminado de un anillo aromático mediante el calentamiento en ácido sulfúrico diluido.
En la desulfonación, un protón se añade al anillo (el electrófilo) y la pérdida de trióxido de azufre vuelve a dar lugar al benceno.

Alquilación de Friedel-Crafts. Los carbocationes quizás son los más importantes para
las sustituciones en los anillos aromáticos, debido a que estas sustituciones forman un nuevo enlace carbono-carbono.
La adición de grupos alquilo a un anillo de benceno se llama alquilación Friedel-Craft. Un ácido de Lewis se utiliza como un catalizador para generar el carbocatión a partir del haluro de alquilo (secundario o terciario) o para activar el haluro de alquilo (primario o haluro de metilo) hacia el ataque nucleofílico.

Alquilación de Friedel-Crafts.Las alquilaciones de Friedel-Crafts se utilizan con una gran variedad de haluros de
alquilo primarios, secundarios y terciarios. Con los haluros secundarios y terciarios,
el electrófilo reaccionante es el carbocatión
El tricloruro de aluminio reacciona con el haluro de alquilo para formar un
carbocatión terciario. El catión reacciona con el benceno y, después de perder un
protón, dará t-butilbenceno a un rendimiento del 90 por ciento
Con los haluros de alquilo primarios, el carbocatión primario es demasiado
inestable. El electrófilo real es un complejo de cloruro de aluminio con haluro de
alquilo. En este complejo, el enlace carbono-halógeno es débil (como indican
las líneas de puntos) y hay una carga positiva considerable en el átomo de
carbono
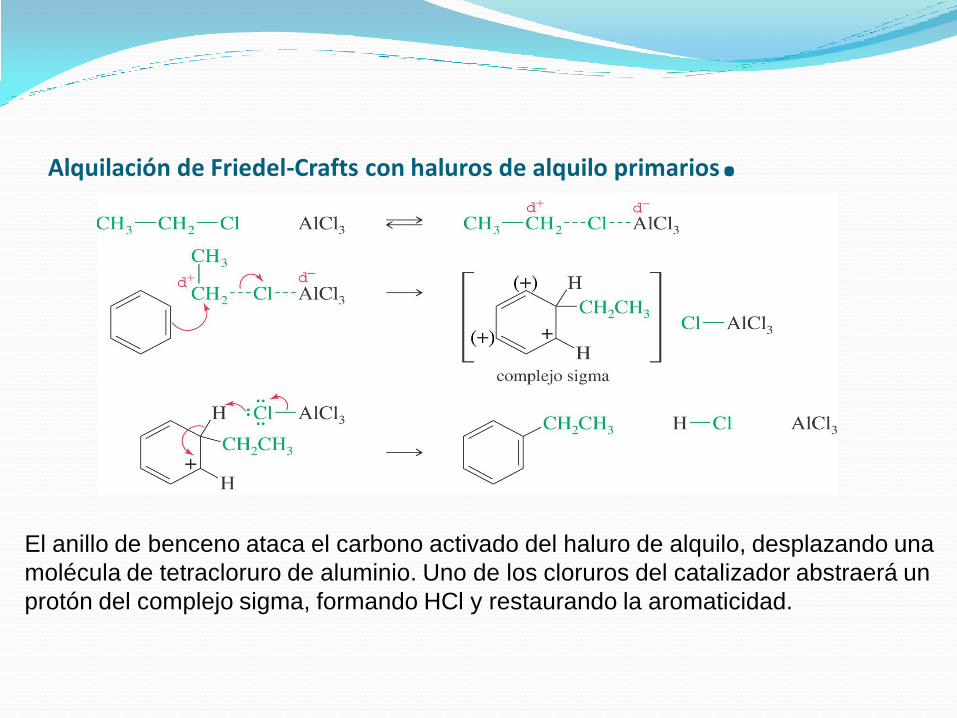
Alquilación de Friedel-Crafts con haluros de alquilo primarios.
El anillo de benceno ataca el carbono activado del haluro de alquilo, desplazando una
molécula de tetracloruro de aluminio. Uno de los cloruros del catalizador abstraerá un
protón del complejo sigma, formando HCl y restaurando la aromaticidad.

Reordenamientos del carbocatión durante la alquilación Friedel-Crafts.La alquilación Friedel-Crafts es susceptible a los reordenamientos del carbocatión.
Como resultado, sólo se pueden producir algunos alquibencenos usando la
alquilación Friedel-Crafts.
La reacción que tienen intermediarios de carbocatión sufre de posibles
reordenamientos del carbocatión para formar carbocationes más estables.

Alquilación de Friedel-Crafts.

A partir de haluros de alquilo:
A partir de alcoholes:
A partir de olefinas:
Los carbocationes
pueden generarse de
varias formas:
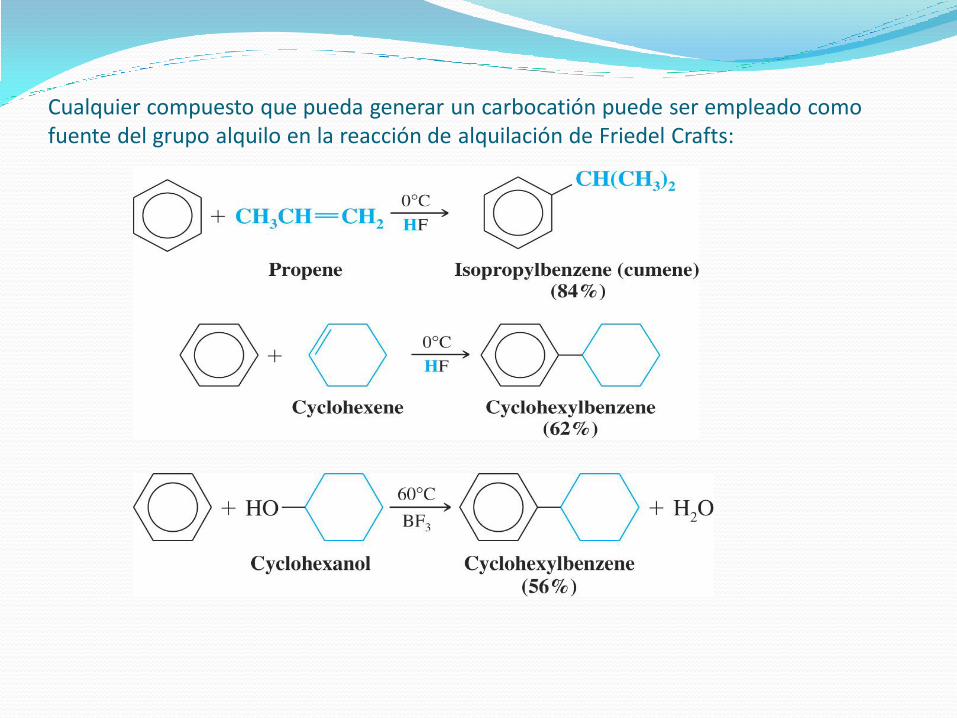
Cualquier compuesto que pueda generar un carbocatión puede ser empleado como fuente del grupo alquilo en la reacción de alquilación de Friedel Crafts:

Problemas de la alquilación de Friedel-Crafts
Transposición de carbocationes:
Polialquilación:
El 1-bromopropano reacciona con el tricloruro de
aluminio para dar un carbocatión 1º, muy
inestable, que sufre transposición hacia el 2º,
menos inestable, que es el que finalmente
reacciona, dando el 2-fenilpropano, en vez del 1-
fenilpropano
La introducción de un grupo alquilo en un anillo
aromático hace que la densidad electrónica de
éste aumente, ya que el resto alquilo es dador de
electrones. Esto hace que el derivado alquilado
sea más reactivo que el benceno, suponiendo una
seria competencia para él. Por tanto, a medida
que la reacción progresa y aumenta la cantidad de
alquilbenceno, la probabilidad de obtener
compuestos polialquilados aumenta, siendo éstos
un subproducto importante de la reacción.

Acilación de Friedel Crafts Es poco probable que los halogenuros primarios formen
carbocationes discretos una vez formado el complejo con el AlCl3, pero desarrollarán una polarización permanente los suficientemente elevada como para reaccionar como
electrófilos:
NO reaccionan
Halogenuro de viniloHalogenuro de arilo
Sin embargo:

Acilación de Friedel-Crafts.En presencia de cloruro de aluminio, un cloruro de acilo reacciona con benceno (o
con un derivado activado del benceno) para dar lugar a una fenilcetona o
acilbenceno
Esta reacción es análoga a la alquilación, pero el producto es la fenilcetona

Ion acilio, estabilizado
por resonancia
El catalítico ácido de Lewis reaccionará con el halogenuro de
ácido o con el anhídrido de ácido para formar un complejo,
que se disocia para generar el verdadero electrófilo, un ion
acilio, que es un híbrido de las dos estructuras de resonancia
formadas. Este ion NO sufre ningún tipo de reordenamiento,
a diferencia de los carbocationes formados para la
alquilación.
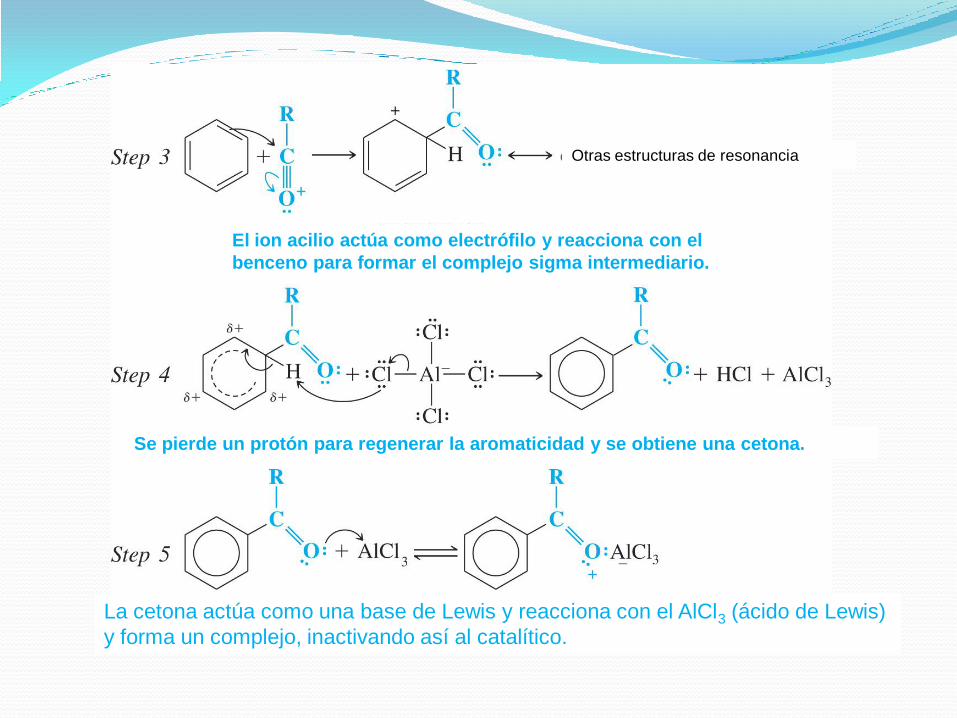
Otras estructuras de resonancia
El ion acilio actúa como electrófilo y reacciona con el
benceno para formar el complejo sigma intermediario.
Se pierde un protón para regenerar la aromaticidad y se obtiene una cetona.
La cetona actúa como una base de Lewis y reacciona con el AlCl3 (ácido de Lewis)
y forma un complejo, inactivando así al catalítico.
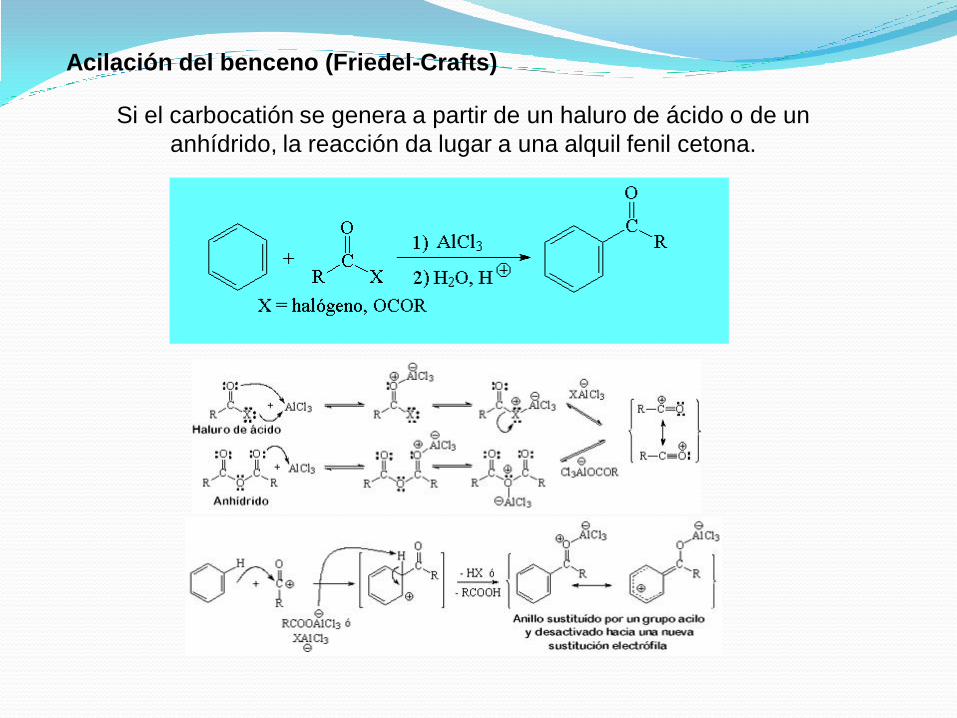
Acilación del benceno (Friedel-Crafts)
Si el carbocatión se genera a partir de un haluro de ácido o de un
anhídrido, la reacción da lugar a una alquil fenil cetona.

Ejemplos
Cloruro de
acetilo
Acetofenona (metil fenil
cetona)
Anhídrido
acético
Acetofenona

Ejemplos:
Una acilación de Friedel
Crafts intramolecular
Nuevo enlace C-C
La posibilidad de formar un nuevo anillo en la estructura es una
de las ventajas de las acilaciones intramoleculares.

Tanto la alquilación como la acilación de Friedel-Crafts no ocurre en anillos aromáticos desactivados.

Limitaciones de las reacciones de Friedel Crafts No se pueden dar si en el anillo hay grupos como –
C=O, -NO2, - CF3, SO3H, - (desactivadores frente a la SEA), -NH- , -OH (reaccionan con el catalítico)
Esta limitación es común tanto para alquilaciones como para
acilaciones de Friedel y Crafts

Limitaciones de las reacciones de Friedel CraftsAlquilación
No es posible usar halogenuros de vinilo o bencilo.
Se dan reordenamientos.
Se dan polialquilaciones.
Acilación
Debe añadirse AlCl3 en cantidad equimolar, no catalítica.

Reacciones de disustitución Al diseñar una síntesis de un producto aromático con
dos sustituyentes en el anillo, la secuencia en que vamos a llevar la reacción es de extrema importancia.
Recuerde que se debe tomar en cuenta el efecto orientador del grupo que se introduce primero al anillo, así como su efecto sobre la reactividad del anillo.
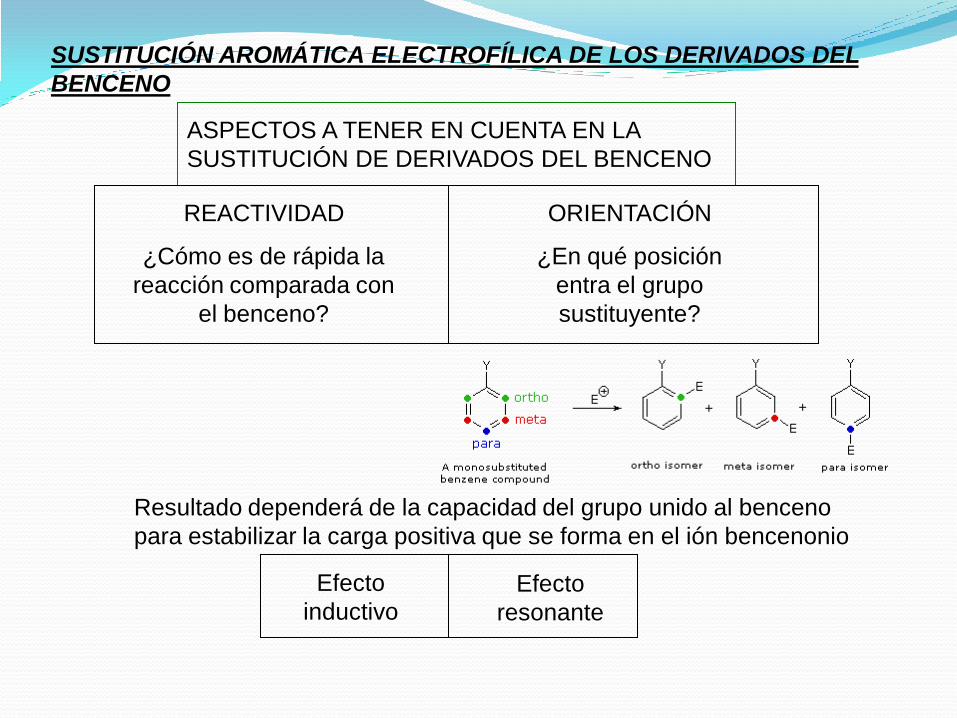
SUSTITUCIÓN AROMÁTICA ELECTROFÍLICA DE LOS DERIVADOS DEL
BENCENO
ASPECTOS A TENER EN CUENTA EN LA
SUSTITUCIÓN DE DERIVADOS DEL BENCENO
REACTIVIDAD
¿Cómo es de rápida la
reacción comparada con
el benceno?
ORIENTACIÓN
¿En qué posición
entra el grupo
sustituyente?
Efecto
inductivoEfecto
resonante
Resultado dependerá de la capacidad del grupo unido al benceno
para estabilizar la carga positiva que se forma en el ión bencenonio
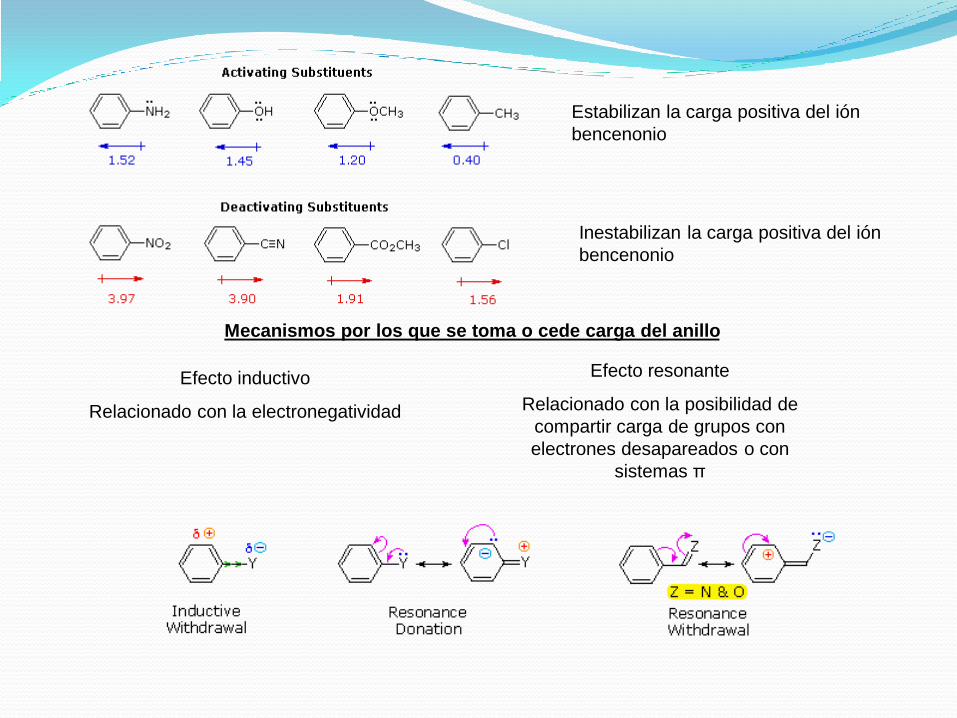
Estabilizan la carga positiva del ión
bencenonio
Inestabilizan la carga positiva del ión
bencenonio
Efecto inductivo
Relacionado con la electronegatividad
Efecto resonante
Relacionado con la posibilidad de
compartir carga de grupos con
electrones desapareados o con
sistemas π
Mecanismos por los que se toma o cede carga del anillo
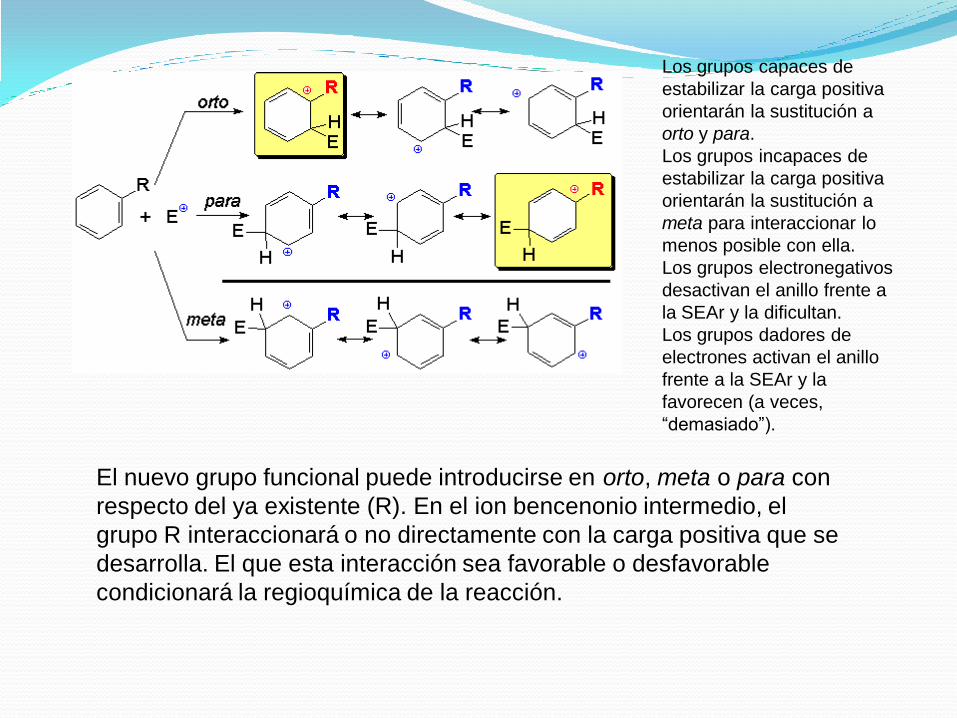
El nuevo grupo funcional puede introducirse en orto, meta o para con
respecto del ya existente (R). En el ion bencenonio intermedio, el
grupo R interaccionará o no directamente con la carga positiva que se
desarrolla. El que esta interacción sea favorable o desfavorable
condicionará la regioquímica de la reacción.
Los grupos capaces de
estabilizar la carga positiva
orientarán la sustitución a
orto y para.
Los grupos incapaces de
estabilizar la carga positiva
orientarán la sustitución a
meta para interaccionar lo
menos posible con ella.
Los grupos electronegativos
desactivan el anillo frente a
la SEAr y la dificultan.
Los grupos dadores de
electrones activan el anillo
frente a la SEAr y la
favorecen (a veces,
“demasiado”).
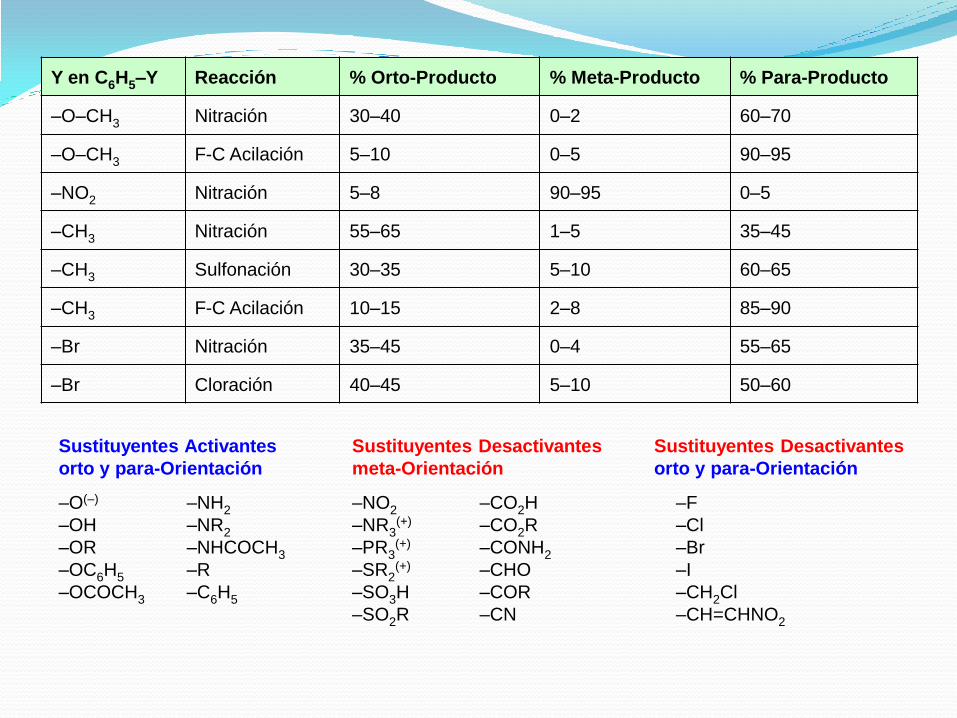
Y en C6H5–Y Reacción % Orto-Producto % Meta-Producto % Para-Producto
–O–CH3 Nitración 30–40 0–2 60–70
–O–CH3 F-C Acilación 5–10 0–5 90–95
–NO2 Nitración 5–8 90–95 0–5
–CH3 Nitración 55–65 1–5 35–45
–CH3 Sulfonación 30–35 5–10 60–65
–CH3 F-C Acilación 10–15 2–8 85–90
–Br Nitración 35–45 0–4 55–65
–Br Cloración 40–45 5–10 50–60
Sustituyentes Activantes
orto y para-Orientación
Sustituyentes Desactivantes
meta-Orientación
Sustituyentes Desactivantes
orto y para-Orientación
–O(–)
–OH
–OR
–OC6H5
–OCOCH3
–NH2
–NR2
–NHCOCH3
–R
–C6H5
–NO2
–NR3(+)
–PR3(+)
–SR2(+)
–SO3H
–SO2R
–CO2H
–CO2R
–CONH2
–CHO
–COR
–CN
–F
–Cl
–Br
–I
–CH2Cl
–CH=CHNO2

Nitración del tolueno.El tolueno reacciona unas 25 veces más deprisa que el benceno en las mismas condiciones. Se dice que el tolueno está activado para la sustitución electrofílica aromática y que el grupo metilo es un grupo activante.
La nitración del tolueno da tres productos: o-nitrotolueno (60 por ciento), m-nitrotolueno (4 por ciento) y p-nitrotolueno (36 por ciento).
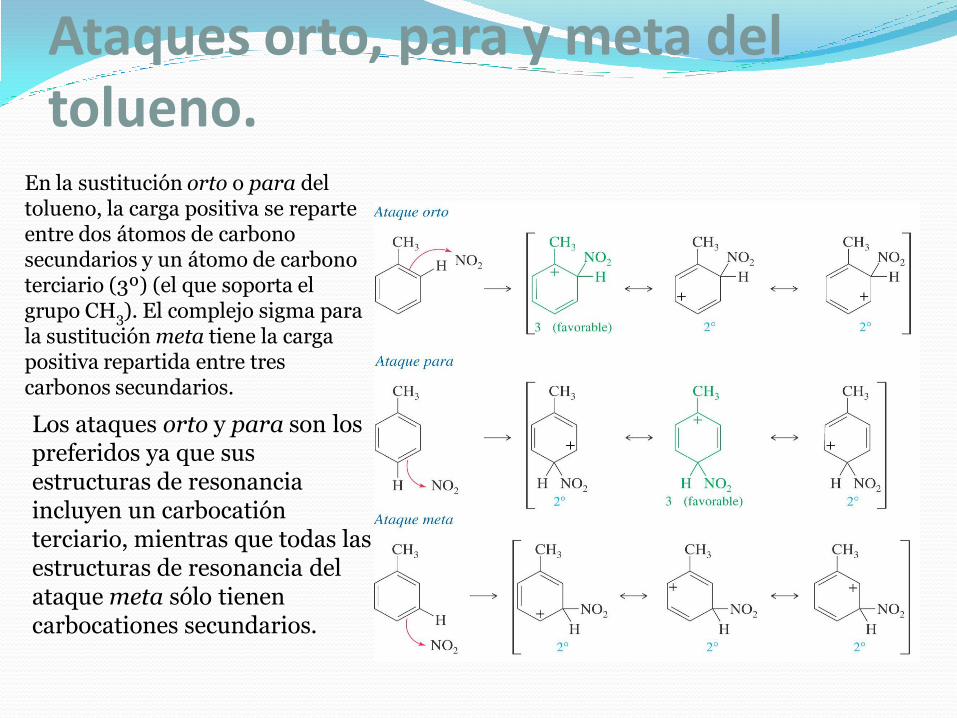
Ataques orto, para y meta del tolueno.
En la sustitución orto o para del tolueno, la carga positiva se reparte entre dos átomos de carbono secundarios y un átomo de carbono terciario (3º) (el que soporta el grupo CH3). El complejo sigma para la sustitución meta tiene la carga positiva repartida entre tres carbonos secundarios.
Los ataques orto y para son los preferidos ya que sus estructuras de resonancia incluyen un carbocatión terciario, mientras que todas las estructuras de resonancia del ataque meta sólo tienen carbocationes secundarios.
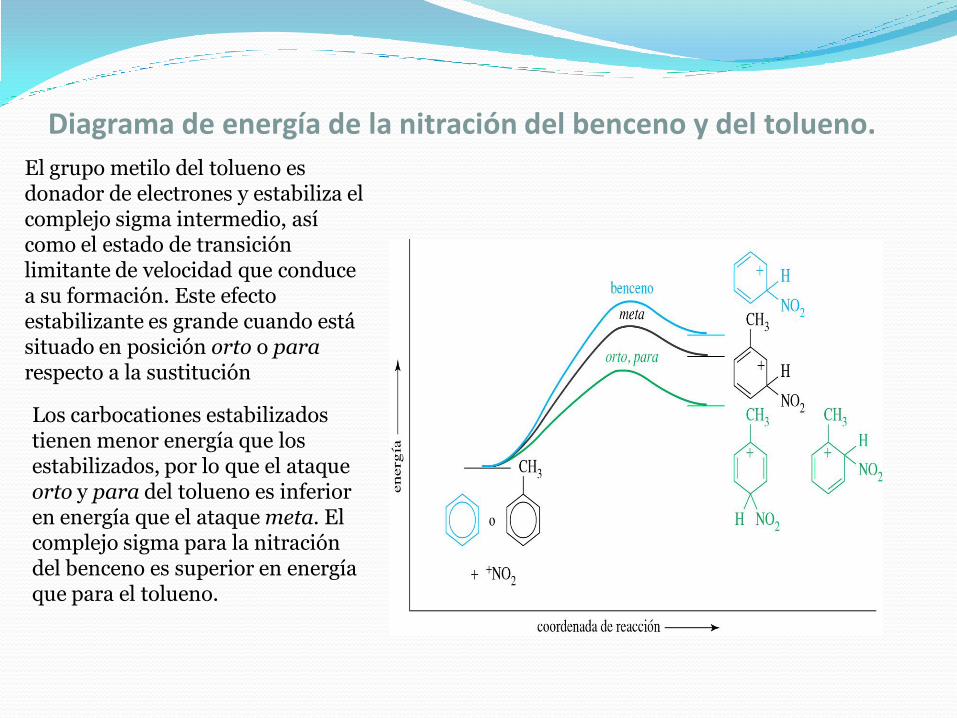
Diagrama de energía de la nitración del benceno y del tolueno.
El grupo metilo del tolueno es donador de electrones y estabiliza el complejo sigma intermedio, así como el estado de transición limitante de velocidad que conduce a su formación. Este efecto estabilizante es grande cuando está situado en posición orto o pararespecto a la sustitución
Los carbocationes estabilizados tienen menor energía que los estabilizados, por lo que el ataque orto y para del tolueno es inferior en energía que el ataque meta. El complejo sigma para la nitración del benceno es superior en energía que para el tolueno.

Sustitución aromática electrofílica del metoxibenceno.
Las formas de resonancia indican que el grupo metoxilo estabiliza de forma efectiva el complejo sigma si la posición de sustitución es orto o para, pero no en el caso de que la posición sea meta. La estabilización por resonancia se debe al enlace pi entre el sustituyente -OCH3 y el anillo.
El par no enlazante de electrones en el heteroátomo puede deslocalizar más adelante la carga positiva del carbocatión, haciendo que las sustituciones orto y parasean especialmente estables.
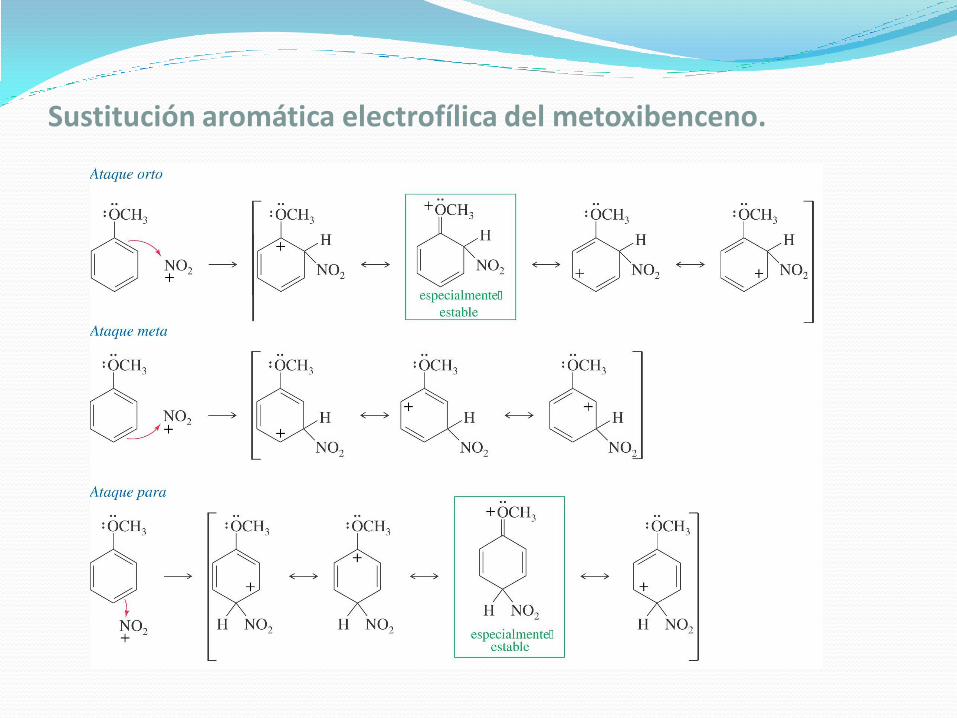
Sustitución aromática electrofílica del metoxibenceno.
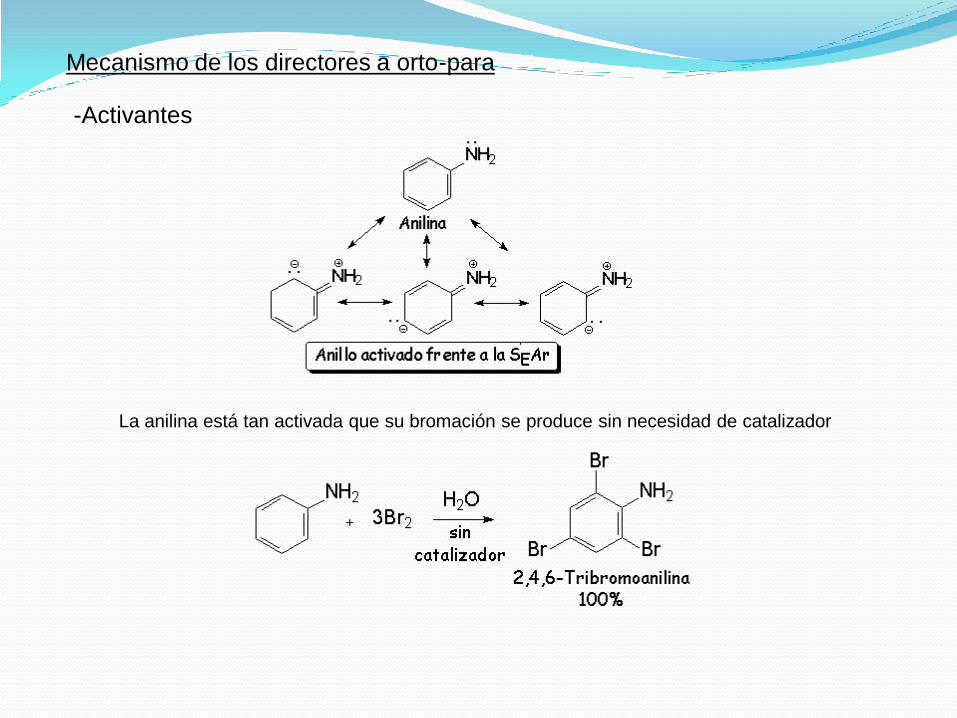
Mecanismo de los directores a orto-para
-Activantes
La anilina está tan activada que su bromación se produce sin necesidad de catalizador
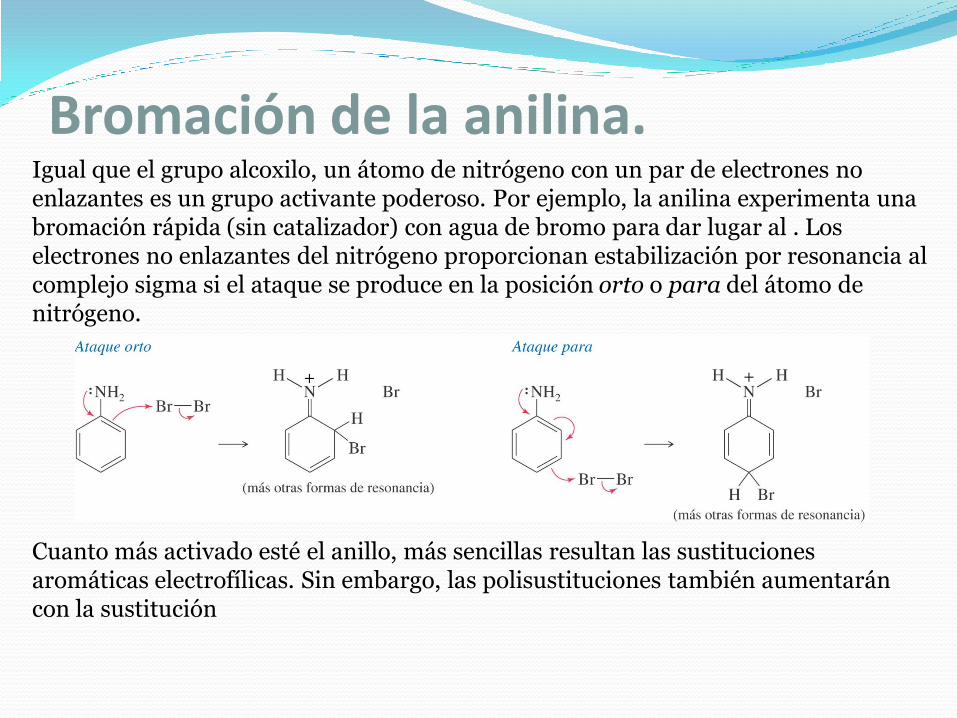
Bromación de la anilina.Igual que el grupo alcoxilo, un átomo de nitrógeno con un par de electrones no enlazantes es un grupo activante poderoso. Por ejemplo, la anilina experimenta una bromación rápida (sin catalizador) con agua de bromo para dar lugar al . Los electrones no enlazantes del nitrógeno proporcionan estabilización por resonancia al complejo sigma si el ataque se produce en la posición orto o para del átomo de nitrógeno.
Cuanto más activado esté el anillo, más sencillas resultan las sustituciones aromáticas electrofílicas. Sin embargo, las polisustituciones también aumentarán con la sustitución
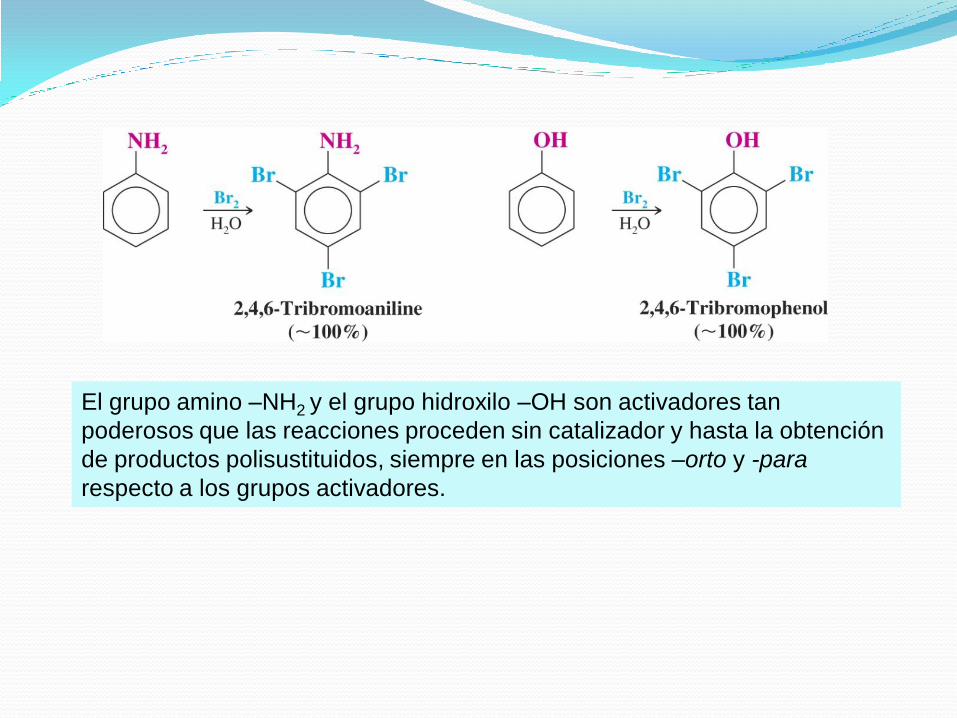
El grupo amino –NH2 y el grupo hidroxilo –OH son activadores tan
poderosos que las reacciones proceden sin catalizador y hasta la obtención
de productos polisustituidos, siempre en las posiciones –orto y -para
respecto a los grupos activadores.
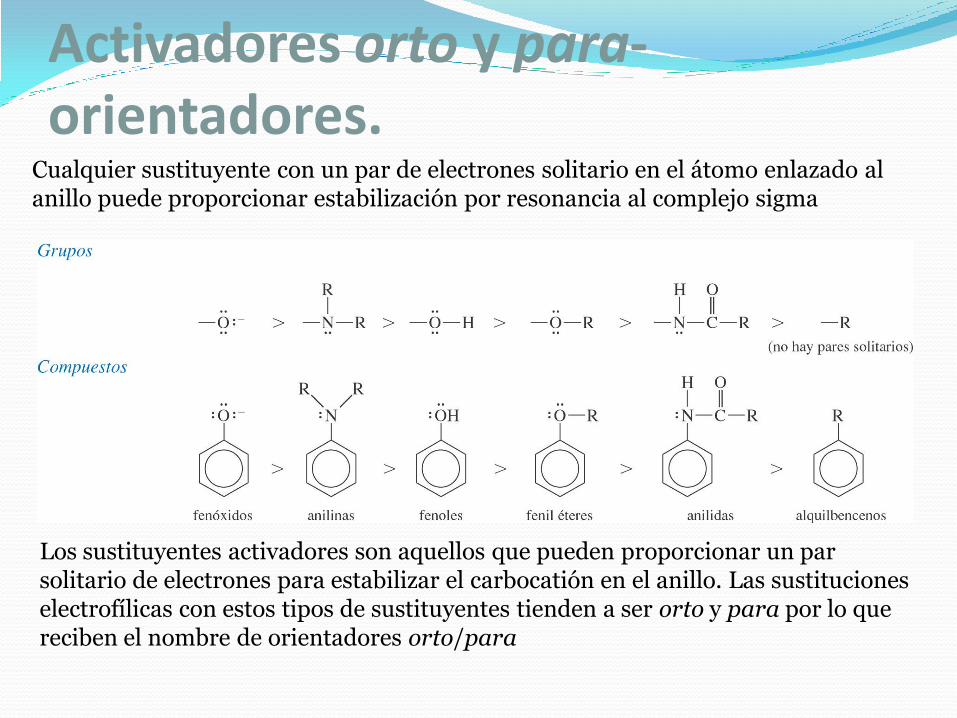
Activadores orto y para-orientadores.
Cualquier sustituyente con un par de electrones solitario en el átomo enlazado al anillo puede proporcionar estabilización por resonancia al complejo sigma
Los sustituyentes activadores son aquellos que pueden proporcionar un par solitario de electrones para estabilizar el carbocatión en el anillo. Las sustituciones electrofílicas con estos tipos de sustituyentes tienden a ser orto y para por lo que reciben el nombre de orientadores orto/para

Sustituyentes meta-orientadores.El nitrobenceno es unas 100.000 veces menos reactivo que el benceno respecto a la sustitución electrofílica aromática. Por ejemplo, la nitración del nitrobenceno requiere ácidos nítrico y sulfúrico concentrados, y una temperatura superior a 100ºC; la nitración es lenta, dando lugar al isómero meta como producto mayoritario.
Los grupos sustractores de electrones desactivan el anillo y orientan cualquier grupo entrante hacia la posición meta.
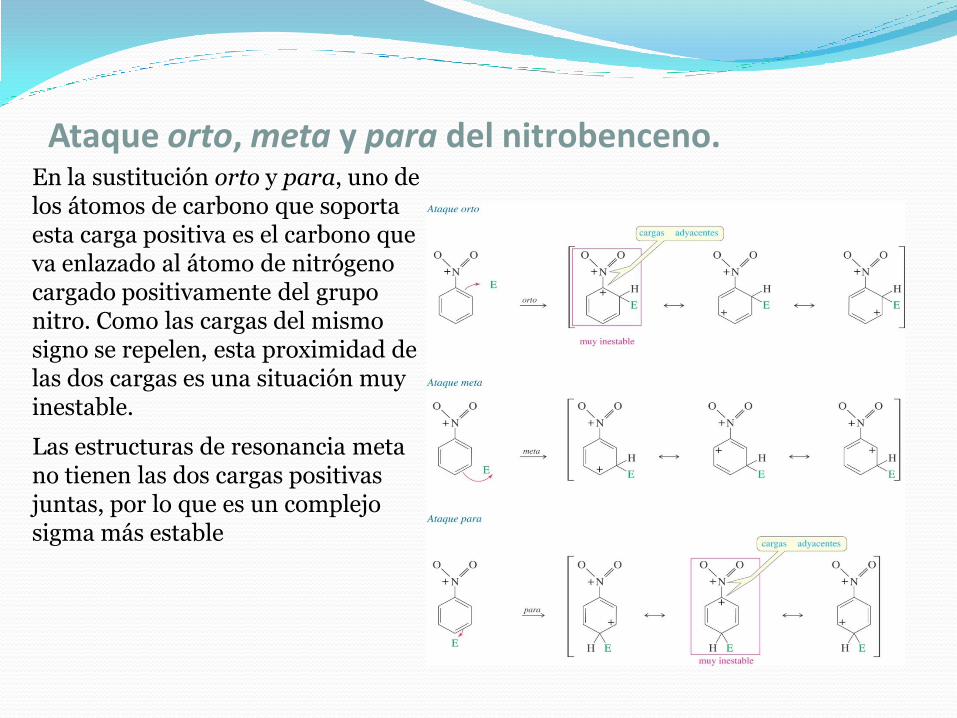
Ataque orto, meta y para del nitrobenceno.En la sustitución orto y para, uno de los átomos de carbono que soporta esta carga positiva es el carbono que va enlazado al átomo de nitrógeno cargado positivamente del grupo nitro. Como las cargas del mismo signo se repelen, esta proximidad de las dos cargas es una situación muy inestable.
Las estructuras de resonancia meta no tienen las dos cargas positivas juntas, por lo que es un complejo sigma más estable

Diagrama de energía para la sustitución del nitrobenceno.
El nitrobenceno está desactivado respecto a la sustitución electrofílica aromática en cualquier posición, pero la desactivación es más fuerte en las posiciones orto y para. La reacción se produce en posición meta, pero es más lenta que la reacción del benceno.
El grupo nitro desactiva el anillo hacia la sustitución, de manera que las reacciones tardan más incluso bajo condiciones fuertes

Diagrama de energía para la sustitución del nitrobenceno.
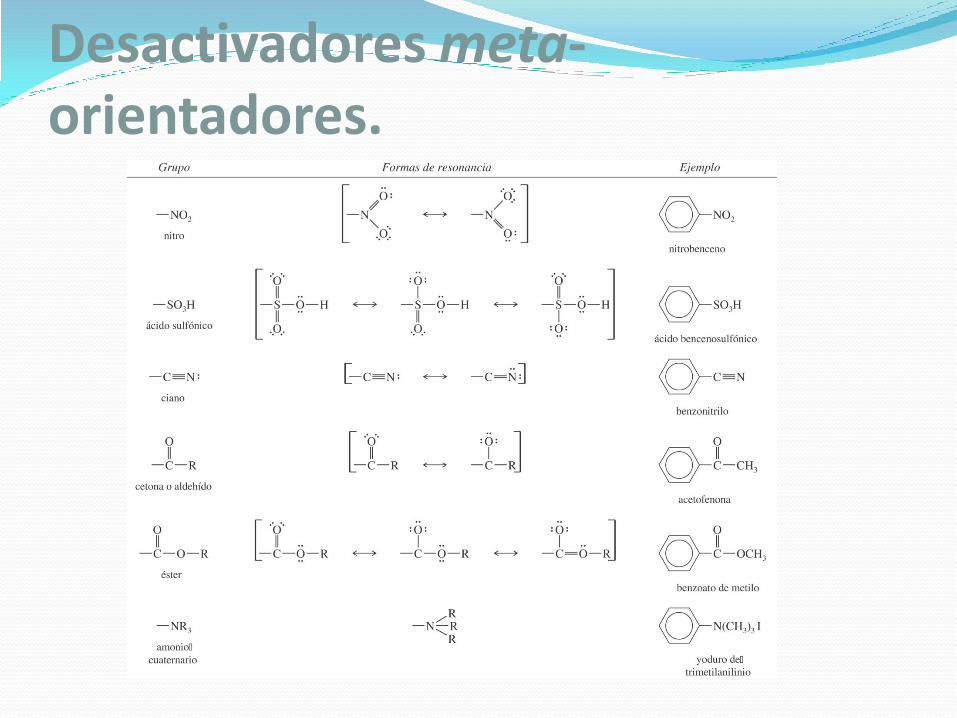
Desactivadores meta-orientadores.
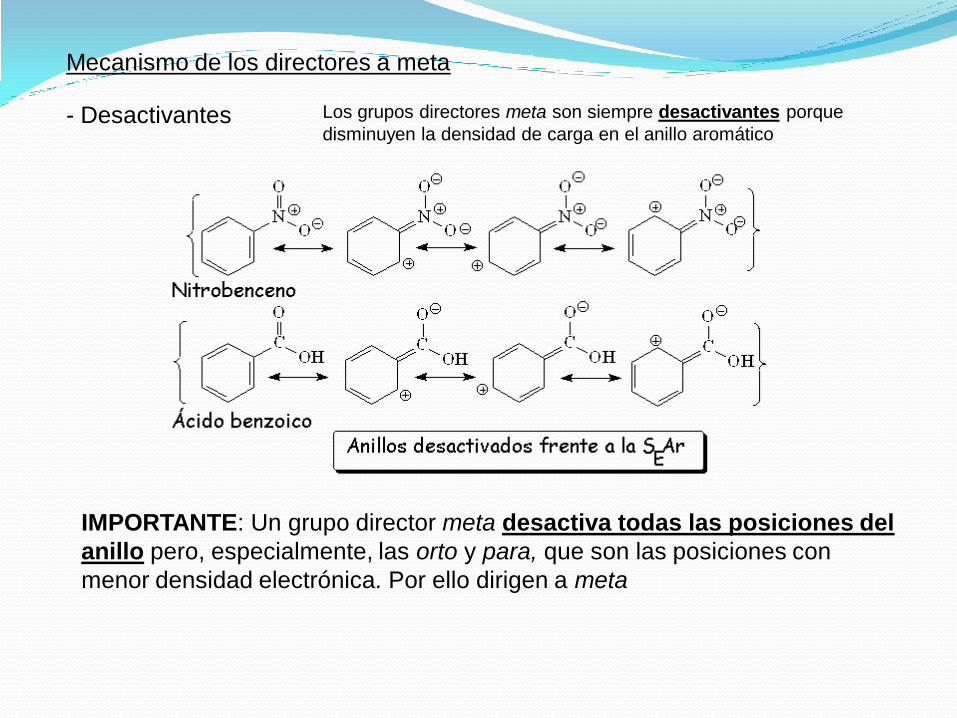
Mecanismo de los directores a meta
- Desactivantes Los grupos directores meta son siempre desactivantes porque
disminuyen la densidad de carga en el anillo aromático
IMPORTANTE: Un grupo director meta desactiva todas las posiciones del
anillo pero, especialmente, las orto y para, que son las posiciones con
menor densidad electrónica. Por ello dirigen a meta

La bromación del nitrobenceno, mucho más difícil que la del benceno,
se da en la posición menos desfavorable, la meta, como lo muestran
las formas resonantes del ion bencenonio intermedio.

Todos los caminos de la SEAr del nitrobenceno son más altos en
energía que los del benceno porque el grupo nitro desactiva todo el
anillo. El camino que conduce al isómero meta, aún siendo más
costoso que el del benceno, es el menos desfavorable de los tres.
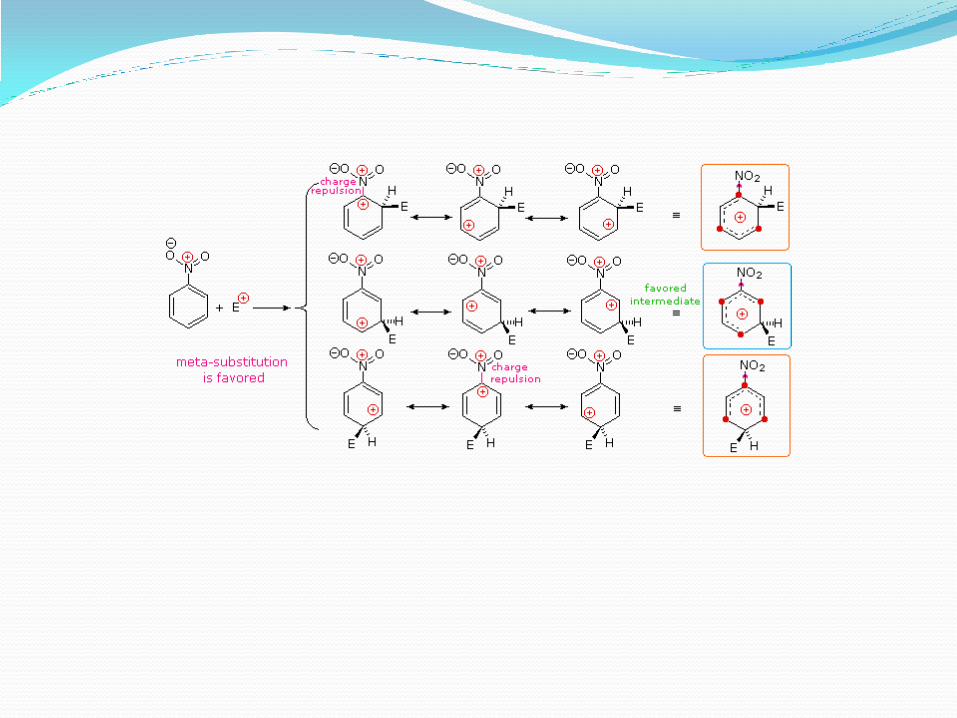



Diagrama de energía de las sustituciones electrofílicas en el clorobenceno.
Las reacciones del clorobenceno requieren las energías más altas, sobre todo para el ataque en la posición meta.
A pesar de que los halógenos son grupos desactivantes, el diagrama de energía del clorobenceno muestra que son orto/paraorientadores. La electronegatividad de los halógenos desactivará el anillo, pero los pares no enlazantes de electrones estabilizarán los carbocationes producidos durante los ataques orto y para.

- Desactivantes débiles
Mecanismo de los directores a orto-para
La mayor electronegatividad de los halógenos retira algo de carga del anillo aromático y esto dificulta
ligeramente la SEAr. Sin embargo, una vez producida la reacción, los pares de electrones no
compartidos que poseen los halógenos son capaces de deslocalizar la carga positiva del ión
bencenonio estabilizándolo. Por ello dirigen la SEAr a orto-para.
El Cl estabiliza la carga +
una vez producida
El Cl estabiliza la carga +
una vez producida
El Cl no puede
interaccionar con la carga +
y no la estabilza
adicionalmente

Efectos orientadores de los sustituyentes.
Los activadores son orto, para-directores y los desactivadores son meta- directores, excepto en el caso de los halógenos.
Los grupos más activantes son aquellos con electrones no enlazantes como -OH, -NH2, y -OR. Los desactivadores son grupos sustractores de electrones que tienen un átomo cargado positivamente directamente adherido al anillo. Los halógenos son la excepción porque a pesar de que desactivan el anillo, son orto, para-directores.

Efecto de múltiples sustituyentes sobre la sustitución electrofílicaaromática.
Cuando hay dos o más sustituyentes, éstos ejercen un efecto combinado en la reactividad del anillo aromático. Si los grupos se refuerzan entre sí, el resultado es fácil de predecir
En el caso del m-xileno, ambos grupos metilo son orto, para-directores, por lo que ambos dirigirán los grupos entrantes a los mismos carbonos. Las posiciones estéricamente impedidas no reaccionarán. En el caso del p-nitrotolueno, el metilo dirigirá el orto mientras que el nitro dirigirá el meta. En estos compuestos ambos grupos se dirigen a las mismas posiciones, por lo que sólo se obtendrá un producto.

Efectos contrarios de los sustituyentes.Cuando los efectos directores de dos o más sustituyentes son contrarios, es más
difícil predecir dónde reaccionará el electrófilo. En muchos casos, se obtienen
mezclas de productos; por ejemplo, el o-xileno está activado en todas las
posiciones, por lo que da lugar a mezclas de productos de sustitución
En la nitración del o-xileno, los grupos metilo dirigen a distintas posiciones orto y
para. Hay dos posiciones diferentes en el compuesto con las que puede reaccionar
el electrófilo. Se obtendrá una mezcla de sustituciones en ambos lados.
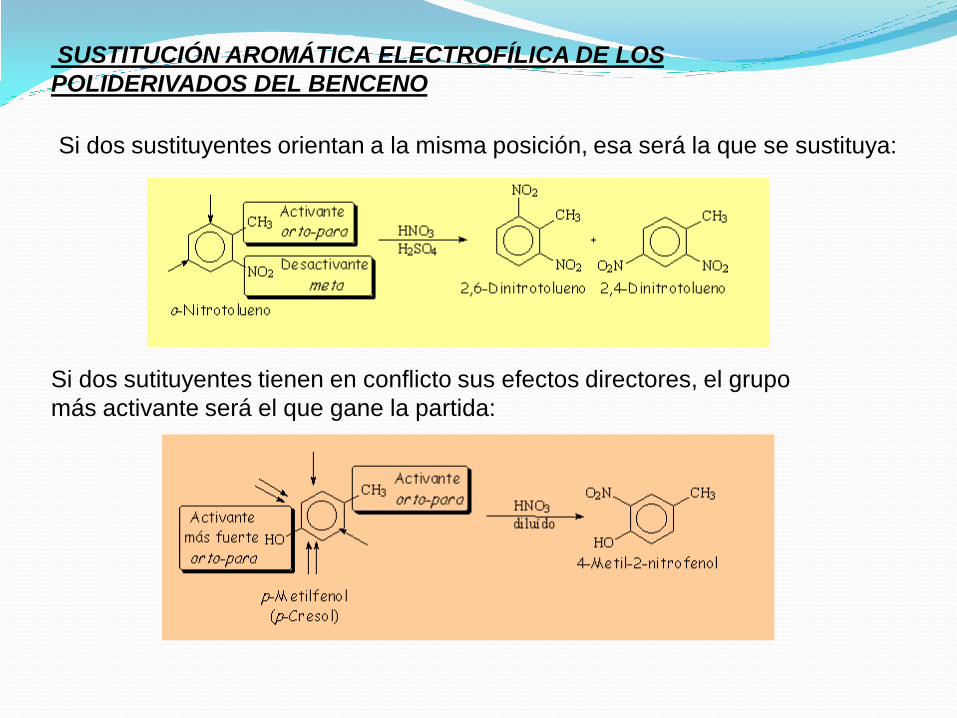
SUSTITUCIÓN AROMÁTICA ELECTROFÍLICA DE LOS
POLIDERIVADOS DEL BENCENO
Si dos sustituyentes orientan a la misma posición, esa será la que se sustituya:
Si dos sutituyentes tienen en conflicto sus efectos directores, el grupo
más activante será el que gane la partida:
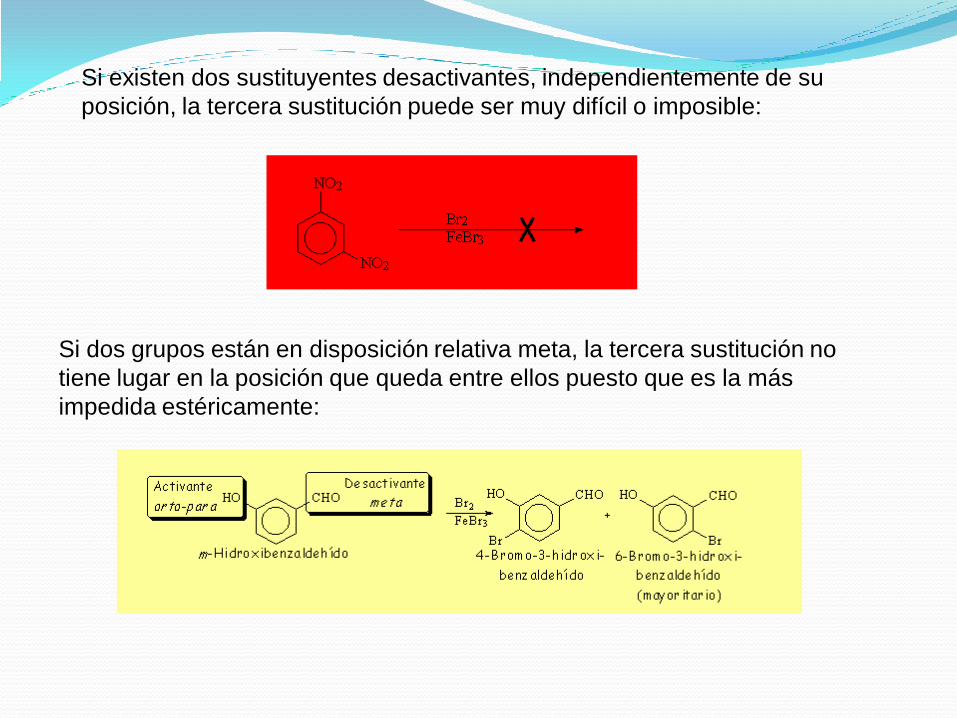
Si existen dos sustituyentes desactivantes, independientemente de su
posición, la tercera sustitución puede ser muy difícil o imposible:
Si dos grupos están en disposición relativa meta, la tercera sustitución no
tiene lugar en la posición que queda entre ellos puesto que es la más
impedida estéricamente:
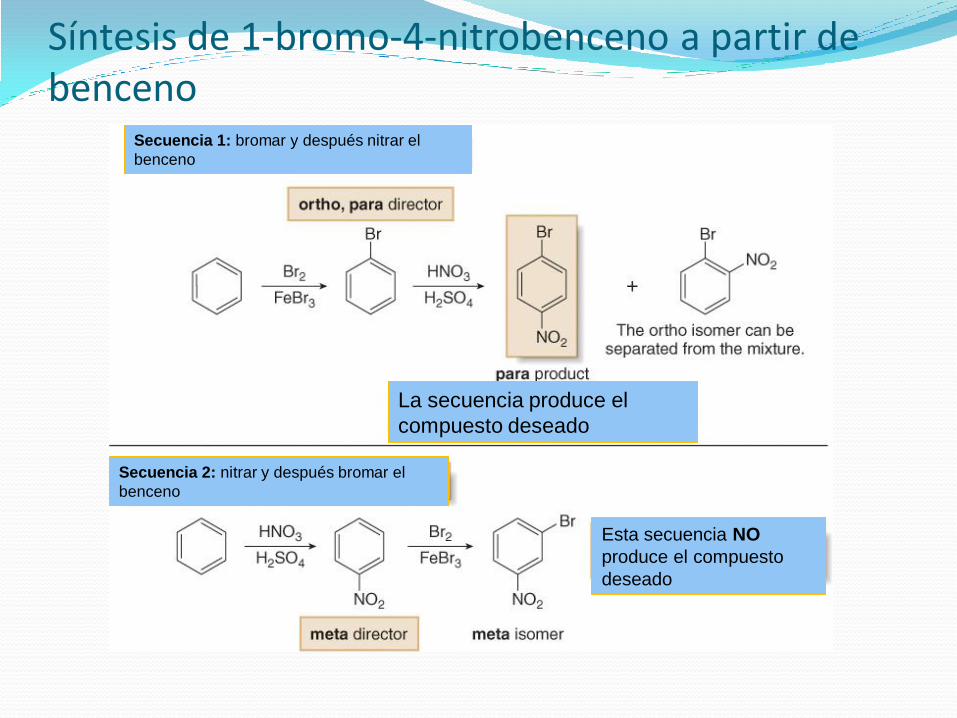
Síntesis de 1-bromo-4-nitrobenceno a partir de benceno
Secuencia 1: bromar y después nitrar el
benceno
La secuencia produce el
compuesto deseado
Secuencia 2: nitrar y después bromar el
benceno
Esta secuencia NO
produce el compuesto
deseado


5.4- SÍNTESIS DE DERIVADOS DEL BENCENO
Los bencenos polisustituídos han de ser creados mediante una secuencia de
sustituciones electrófilas cuidadosamente planeadas.
Hay que tener en cuenta las propiedades de orientación y activación de los grupos que se van
introduciendo.
En una secuencia de síntesis el orden de factores SÍ puede afectar el producto.
TRUCO 1: Unos grupos funcionales pueden transformarse en otros, con lo que sus
propiedades de orientación y activación pueden cambiarse:
Conversión de un grupo NO2,
orientador meta, en NH2 orientador
orto-para y viceversa
Conversión de un grupo cetona, orientador
meta, en grupo alquilo orientador orto-
para.
Orientador meta Orientador orto-para

•TRUCO 2: La sulfonación reversible permite el bloqueo de la posición para y, por tanto, la
síntesis eficiente de bencenos orto-sustituídos:
•TRUCO 3: Transformación del grupo NH2 en sal de diazonio:
¿Cómo se puede
sintetizar el 1,4-
dinitrobenceno?
Dos nitraciones consecutivas dan lugar al
isómero 1,3 porque el grupo NO2 orienta
meta.
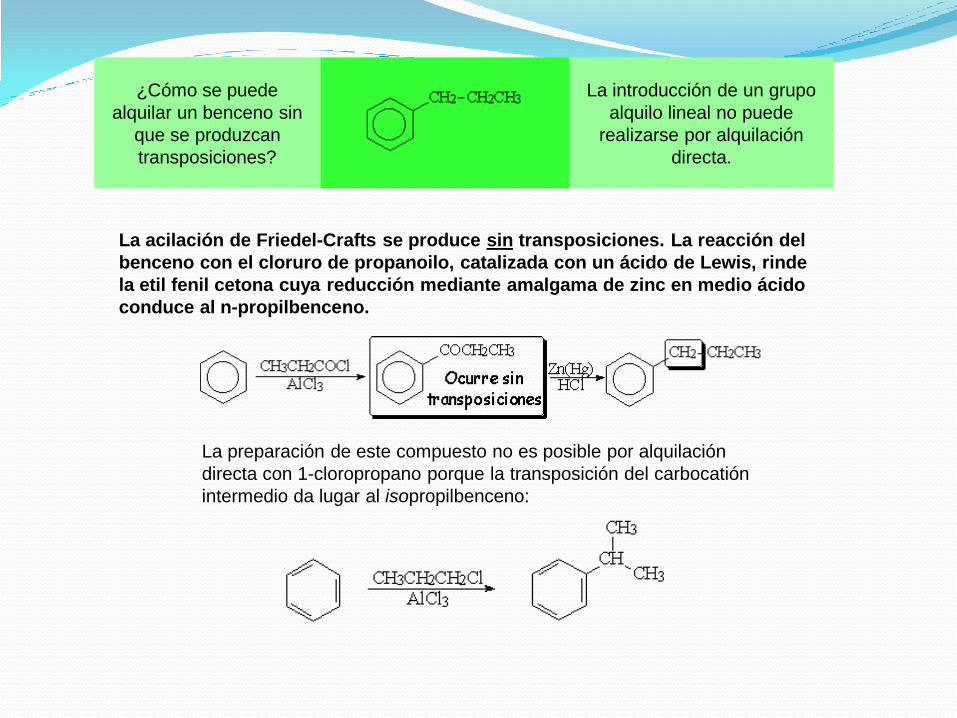
¿Cómo se puede
alquilar un benceno sin
que se produzcan
transposiciones?
La introducción de un grupo
alquilo lineal no puede
realizarse por alquilación
directa.
La acilación de Friedel-Crafts se produce sin transposiciones. La reacción del
benceno con el cloruro de propanoilo, catalizada con un ácido de Lewis, rinde
la etil fenil cetona cuya reducción mediante amalgama de zinc en medio ácido
conduce al n-propilbenceno.
La preparación de este compuesto no es posible por alquilación
directa con 1-cloropropano porque la transposición del carbocatión
intermedio da lugar al isopropilbenceno:
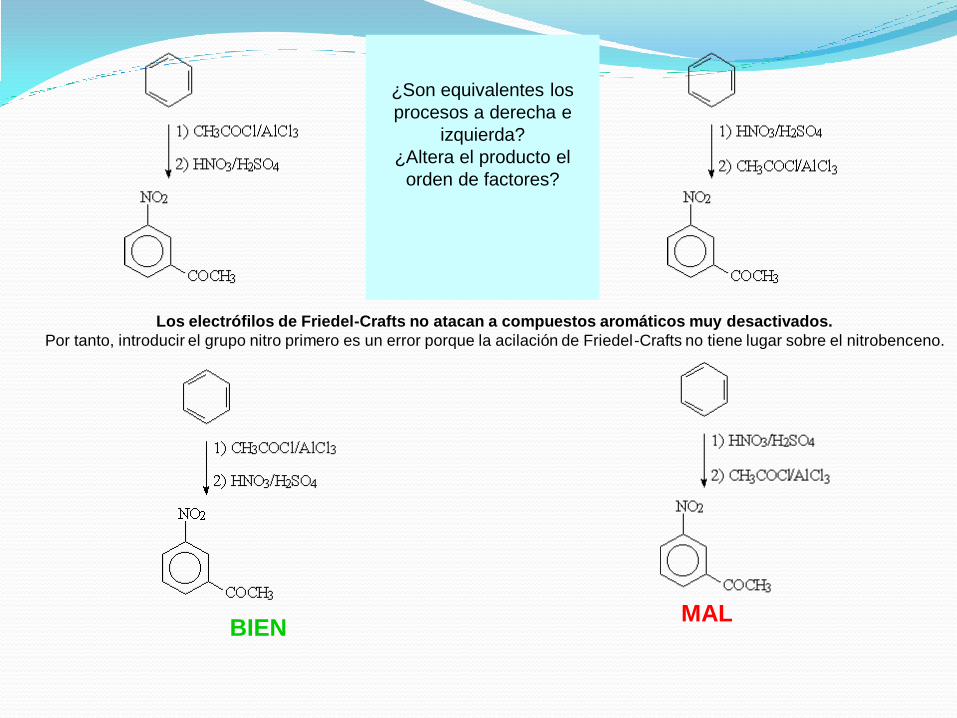
¿Son equivalentes los
procesos a derecha e
izquierda?
¿Altera el producto el
orden de factores?
Los electrófilos de Friedel-Crafts no atacan a compuestos aromáticos muy desactivados.
Por tanto, introducir el grupo nitro primero es un error porque la acilación de Friedel-Crafts no tiene lugar sobre el nitrobenceno.
BIENMAL

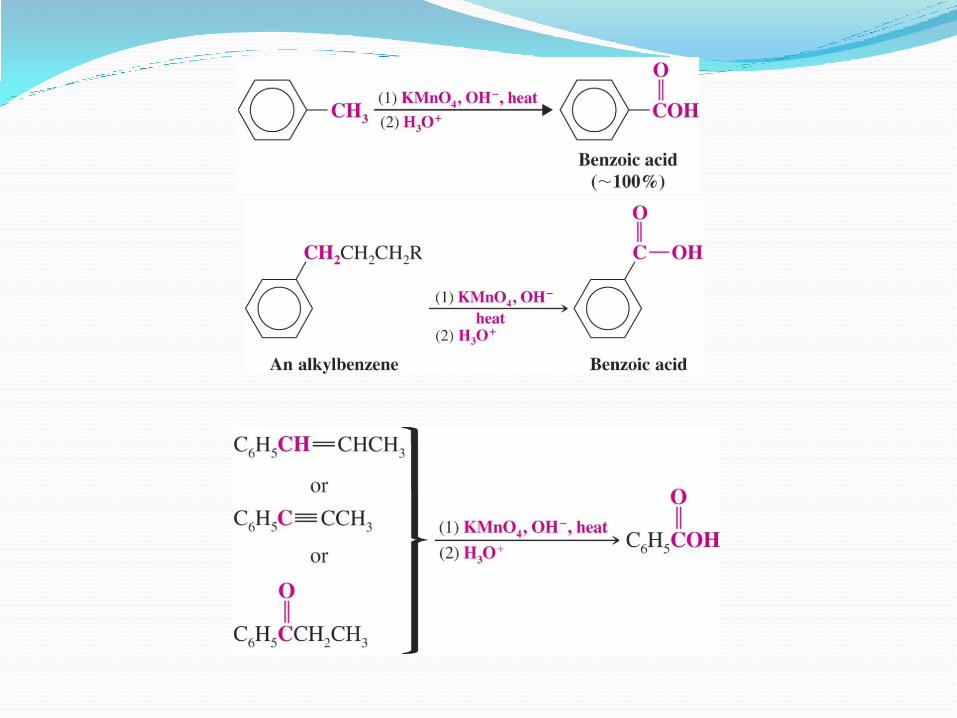
Reacción de arenos con KMnO4 El benceno es inerte frente a soluciones concentradas,
alcalinas y calientes de permanganato de potasio.
Sin embargo, anillos aromáticos con una parte alifática, sufrirán una reacción de oxidación en la cadena alifática (llamada cadena lateral.)
El producto de la oxidación de la cadena lateral será un grupo –COO- (porque se trabaja en medio alcalino), así que es un método para obtener ácidos carboxílicos aromáticos.

Referencias Carey. Química Orgánica. 6a. Edición. McGraw-Hill.
2005
McMurry, J. Química Orgánica. 7a. Edición. Cengage. 2008
Wade, L. Química Orgánica. 5a. Edición. Pearson Education. 2004
Solomons and Frhyle. Organic Chemistry. 9th. Edition. Wiley. 2007
Dr. Carlos Antonio Rius Alonso
Depto. de Química Orgánica
Facultad de Química. UNAM. septiembre 2007


















