Diapositivas del grupo formado por Rast, Juárez, Aguirre y Medina

Práctica MG-RAST Martha Lilia Colina Tenorio; Alexa Villavicencio Queijeiro
5 Marzo 2013 1. Acceda al servidor MG-RAST (http://metagenomics.anl.gov) 2. Describa brevemente cada una de las bases de datos y herramientas bioinformáticas de anotación del MG-RAST busque en el artículo del MG-RAST y en http://press.igsb.anl.gov/mg-rast/mg-rast-for-the-impatient-readme-1st/
Genbank Es una base de datos que agrupa todas las secuencias de nucleótidos disponibles a la comunidad científica de forma gratuita. Para obtener datos se pueden buscar identificadores de secuencia con Entez Nucleotide, que está dividida en tres grupos: CoreNucleotide, dbEST y dbGSS. Para alinear secuencias se puede usar BLAST, que obtiene información idependientemente de los tres grupos mencionados. Se pueden buscar, ligar y descargar secuencias usando NCBI e-utilities. IMG(Integrated Microbial Genomes) Es una base de datos para analizar y anotar tanto genomas como metagenomas en un contexto comparativo. Contiene genomas de bacterias, arqueas, eucariontes y fagos y una colección no redundante de secuencias de referencia muestras ambientales, de huéspedes y obtenidas por ingeniería. InterPro Es una base de datos que ofrece el análisis funcional de secuencias de proteínas y las clasifica en familias según sus dominios usando modelos predictivos conocidos como firmas que obtiene de diferentes bases de datos. Es útil porque combina datos de diversos tipos de bases de datos en una sola, disminuyendo así la redundancia y produciendo una herramienta integrativa. Se puede usar para el análisis a gran escala de proteomas, genomas y metagenomas y también si se busca caracterizar secuencias de proteínas individuales. InterPro se puede usar también para anotar secuencias de proteínas en UniProtKB. KEGG (Kyoto Encyclopedia of Genes and Genomes) Es una base de datos que incluye a 16 bases de datos clasificadas en información de sistemas, información genómica e información química, cada una con un código de color. Sirve para analizar las funciones y utilidades de una sistema biológico con base en información molecular generada por secuenciación de genomas. Esta base de datos contiene data objects para la representación en computadora de sistemas biológicos, por lo que la entrada para cada base de datos individual se llama KEGG object, y su identificador consiste de un prefijo que indica la base de datos y un número de cinco dígitos. KEGG ofrece redes moleculares, que son modelos que muestran interacción molecular, reacciones y relaciones entre elementos de un sistema biológico; estos modelos se construyen a partir de datos en la literatura y se organizan en tres grupos: KEGG PATHWAY (vías), KEGG BRITE (ontología) y KEGG MODULE y KEGG DISEASE.
M5NR (The M5 non-redundant protein database) M5NR integra muchas bases de datos de secuencias de forma unitaria y no redundante, por lo que al realizar una búsqueda en este servidor se hace de manera simultánea en diferentes bases de datos como BLAST o BLAT, KEGG y EGGnog, su mantenimiento permite eliminar la redundancia.

Patric (Pathosystems Resource Integration Center) Sistema de información diseñado para apoyar la investigación de enfermedades causadas por bacterias a través de la integración de información filogenómica disponible sobre diferentes patógenos y herramientas de análisis de datos. Este programa permite anotar un genoma bacteriano gratis en 24 horas usando RAST (igual que Patric) y hacer búsquedas basadas en taxonomía, nombre de genes, locus, función/familia de proteínas, números EC, interacciones huésped-patógeno, entre otros. Los datos y resultados de los análisis se pueden descargar gratis PhAnToMe (Phage annotation tools and methods) Plataforma para la anotación de genomas de fagos, desarrollada en base a SEED, busca establecer una nomenclatura consistente para los genes de fago y desarrollar herramientas que permitan identificar profagos. RefSeq Es una colección de secuencias integrada, no redundante y bien anotada, contiene información de DNA genómico, transcritos y proteínas. Es una referencia para anotación de genomas, identificación y caracterización de genes y análisis de mutaciones y polimorfismos. Contiene información de eucariontes, bacterias y virus y es accesible a través de BLAST, Entrez y NCBI FTP.
SEED Es un proyecto que surge de la necesidad de tener una plataforma para llevar a cabo el análisis comparativo efectivo de los genomas disponibles. Su principal objetivo es la anotación de genomas, que se hace a través de subsistemas por un anotador experto a partir de varios genomas, no con una aproximación de gen por gen. Basados en estos subsistemas obtienen familias de proteínas que son el principal componente de su tecnología de anotación por RAST. Presentan el llamado SEED-Viewer como acceso de sólo lectura a los genomas anotados más recientes. Swissprot Swiss-Prot es la sección manualmente anotada y revisada de la base de datos UniProtKB. Es una base de datos de secuencias de proteínas de alta calidad y no redundante que integra resultados experimentales, datos computacionales y conclusiones obtenidas de la literatura, las proteínas que están aquí se encuentran bien caracterizadas, se conoce su estructura tridimensional, función, modificaciones post-traduccionales y variantes. Es accesible a través del sitio de UniProt.
TrEMBL UniProtKB/TrEMBL es una base de datos de secuencias de proteínas anotadas por computadora. Conjunta la información de traducción de secuencias codificantes de las siguientes bases: EMBL/GenBank/DDBJ y también aquellas reportadas en la literatura y guardadas en Swiss-Prot
COG (Clusters of Orthologous Groups of proteins) Los COGs son grupos de proteínas ortólogas que se obtienen al comparar las secuencias de proteínas en genomas completos. Cada COG está compuesto por proteínas individuales o grupos de paralogos de al menos 3 linajes y corresponde, por tanto, a un dominio conservado. GO (Gene Ontology) Iniciativa en bioinformática cuya meta es estandarizar la representación de un gen y de su producto en diferentes especies y bases de datos. Provee un vocabulario controlado de términos para describir las características del producto del gen así como herramientas para acceder y procesar a la información. KO (KEGG Ortology) En este se definen de forma manual grupos de ortólogos que corresponden a nodos de vías depositadas en KEGG y nodos jerárquicos de BRITE (Boston university Representative Internet Topology gEnerator)

Subsystems Un subsistema es un conjunto de funciones que un anotador decide que deben considerarse como relacionadas. Representan una colección de funciones que dan lugar a una vía metabólica, un complejo o una clase de proteínas BLAST (Basic Local Alignment Search Tool) Es un programa que encuentra una región de similitud entre secuencias, se pueden comparar secuencias de aminoácidos o de nucleótidos contra bases de datos y se calcula la significancia estadística del alineamiento. Se puede utilizar para inferir relaciones funcionales o evolutivas entre secuencias así como puede ayudar a identificar miembros de familias génicas. Glimmer (Gene Locator and Interpolated Markov ModelER) Es un sistema que permite encontrar genes en genomas de bacterias, arqueas y virus. Utiliza modelos interpolados de Markov para identificar las regiones codificantes y distinguirlas de las no codificantes. Greengenes Aplicación en línea que permite acceder a las secuencias de RNAr 16S con que se cuenta a la fecha. En ella se puede descargar, alinear, blastear y evaluar secuencias de 16S. Con las herramientas disponibles se pueden escoger sondas filogenéticamente específicas, interpretar resultados de micorarreglos y anotar/alinear secuencias nuevas. LSU
SSU El proyecto SILVA es un conjunto de datos de referencia de la subunidad pequeña ribosomal no redundante que se construye utilizando diferentes tipos de modelos (HSM/MWM/GNHM) y un criterio de identidad del 98%.
RDP (Ribosomal Database Project) Base de datos que conjunta secuencias anotadas de RNAr 16S de arqueas y bacterias, permite el análisis en línea de las secuencias entre ellos el alineador de estructura secundaria Infernal con el que se pueden evaluar secuencias parciales pequeñas con lo que se pueden detectar artefactos en la secuenciación.
3. Describa los menús accesibles en el MG-RAST Browse Metagenomes: presenta el número de metagenomas disponbiles, el número de proyectos, número de secuencias, de cuántos ambientes, etc, y una tabla que organiza la información por proyecto, nombre, pares de bases, secuencia, bioma y tipo de secuenciación. About: es un cuadro que explica qué es el MG-RAST, cómo funciona, qué ofrece y cuánta información tiene disponible en ese momento. Register: es la página para crear una cuenta y registrarse en MG-RAST Contact: es un espacio para mandar preguntas o comentarios sobre el programa. Indica que la pregunta la lee todo el equipo y la responde el especialista en el tema. Help: incluye datos sobre las últimas modificaciones del programa. Upload: una vez creada una cuenta, esta sección permite subir la información a la página.

News: Despliega una página donde se indican modificaciones hechas al programa y nuevas utilidades adicionadas.
4. Acceda a la sección Browse 5. Seleccione un metagenoma por tipo de secuenciación de tipo Amplicones 6. ¿Qué datos le da la página de overview de cada muestra? Nombre del proyecto, visibilidad, link estático, descripción del proyecto (por qué surge, dónde, qué busca, cuántas muestras maneja, qué han encontrado y sus posibles implicaciones). También incluye qué instancia(s) apoya(n) ese proyecto, a quién contactar si se necesita y el número de metagenomas que se tienen ordenados en una tabla que los organiza por: MG-RAST ID, nombre, número de pares de bases, número de secuencias, bioma, localización, país, coordenadas, tipo de secuencia (en este caso amplicones), método de secuenciación y links de descarga. 7. Vaya al menu download del overview de la muestra elegida, ¿qué archivos le permite descargar? ¿En qué formato se encuentran? ¿Qué programa usaría para abrir los archivos? Tiene links de descarga para Metadata, Submitted, Analysis. La Metadata se descarga en formato .txt, es decir que se pueden abrir con cualquier procesador de texto. El link de Submitted descarga los datos comprimidos y se necesita usar un software de descompresión.
8. Ingrese al menú de análisis http://metagenomics.anl.gov/metagenomics.cgi?page=Analysis 9. En el menú 1 de la página de análisis seleccione: 1) Data type | Organism Abundance y seleccione las opciones (Representative Hit Classification, best hit classification, lowest comon ancestor) Describa que hace cada una de las opciones. Escoja una de las opciones y justifique porqué la va a usar para esta práctica. Representative Hit Classification: permite visualizar los datos en gráfica de barras, árbol, tabla, heatmap o PCoA. Best Hit Classification: permite las mismas formas de visualización que el anterior pero además tiene la opción de generar una gráfica de rarefacción y de usar información de la workbench. Lowest Common Ancestor: permite visualizar los datos igual que con la primera opción. Las tres clasificaciones tienen la opción de comparar los metagenomas individualmente o en grupos y de escoger la fuente de anotación; también permiten cambiar los valores de corte del e-value, de porcentaje de identidad y la longitud mínima de alineamiento. 10. Seleccione al menos 3 metagenomas de amplicones en la sección 2) Data Selection | Metagenomes | compare individually | public | amplicon. Use el menú de available metagenomes y paselos con las flechas de selección a selected metagenomes. Seleccione la fuente de anotación (Annotation Sources) y seleccione la(s) que considere adecuadas según el punto 2 de esta práctica.
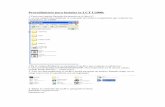
11. Genere los parámetros de búsqueda. Max e-Value cutoff, min % Identity Cutoff, Min. Alignment Length cutoff. Para darse idea de que parámetros usar puede guiarse de la descripción del punto 6. Justifique que parámetros de búsqueda utiliza.
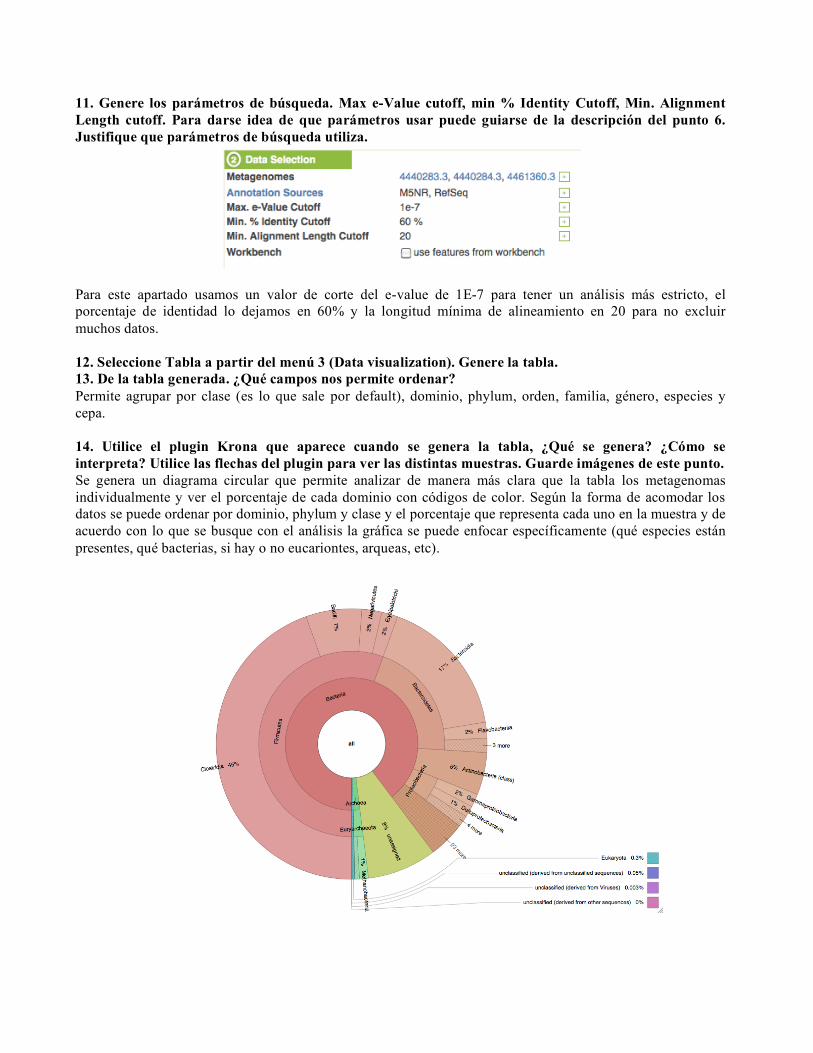
Para este apartado usamos un valor de corte del e-value de 1E-7 para tener un análisis más estricto, el porcentaje de identidad lo dejamos en 60% y la longitud mínima de alineamiento en 20 para no excluir muchos datos. 12. Seleccione Tabla a partir del menú 3 (Data visualization). Genere la tabla. 13. De la tabla generada. ¿Qué campos nos permite ordenar? Permite agrupar por clase (es lo que sale por default), dominio, phylum, orden, familia, género, especies y cepa. 14. Utilice el plugin Krona que aparece cuando se genera la tabla, ¿Qué se genera? ¿Cómo se interpreta? Utilice las flechas del plugin para ver las distintas muestras. Guarde imágenes de este punto. Se genera un diagrama circular que permite analizar de manera más clara que la tabla los metagenomas individualmente y ver el porcentaje de cada dominio con códigos de color. Según la forma de acomodar los datos se puede ordenar por dominio, phylum y clase y el porcentaje que representa cada uno en la muestra y de acuerdo con lo que se busque con el análisis la gráfica se puede enfocar específicamente (qué especies están presentes, qué bacterias, si hay o no eucariontes, arqueas, etc).
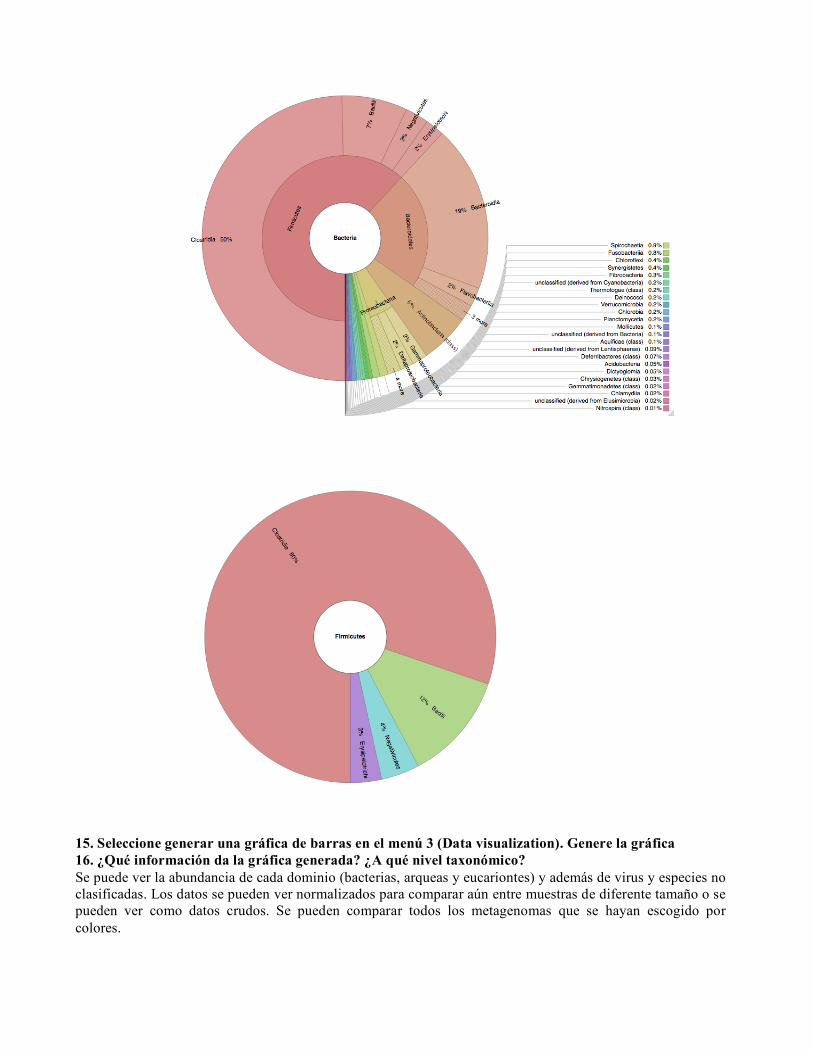
15. Seleccione generar una gráfica de barras en el menú 3 (Data visualization). Genere la gráfica 16. ¿Qué información da la gráfica generada? ¿A qué nivel taxonómico? Se puede ver la abundancia de cada dominio (bacterias, arqueas y eucariontes) y además de virus y especies no clasificadas. Los datos se pueden ver normalizados para comparar aún entre muestras de diferente tamaño o se pueden ver como datos crudos. Se pueden comparar todos los metagenomas que se hayan escogido por colores.

17. De click en la gráfica en cualquiera de las barras que pertenecen a Bacteria. ¿Qué sucede? ¿Si repite el proceso varias veces hasta donde se generan resultados? Se despliega desde dominio y de ahí se pueden seguir desplegando las siguientes distribuciones: phylum, clase, orden, familia, género, especie y hasta cepa; y se puede seleccionar el nivel de interés y guardar esos datos en el workbench. 18. Seleccionar generar un árbol en la barra del menu 3 (Data visualization). Genere el árbol (tree). 19. ¿Qué opciones adicionales aparecen al momento de desplegarse el árbol? Se puede modificar la forma de visualizar las leaf weights, ya sea en barras apiladas o en gráfica de barras, se puede escoger el nivel máximo de vista del mapa (phylum, clase, orden, familia, género, especie, cepa) y elegir por qué nivel colorearlo. También se puede restringir por dominio y así ver sólo bacterias o sólo arqueas o eucariontes o virus. 20. ¿Qué son las leaf weights? Son la representación gráfica de la abundancia de un clado específico en los metagenomas que se están analizando. 21. De click en un nodo ¿Qué pasa? ¿Que información extra se genera? Se marca con un cuadro rojo y se despliega una descripción detallada de las familias contenidas en esa rama del árbol en forma de gráficas de pastel. 22. Ubique un grupo único en una sola muestra y diga como llego a ubicarlo Lo que hicimos fue identificar visualmente un grupo único a partir de la imagen del árbol que obtuvimos y al darle clic y desplegar las gráficas de pastel pudimos comparar dentro de ese grupo seleccionado las diferencias a nivel metagenómico. 23. Guarde una imagen del árbol.

24. Seleccione generar un heatmap en la barra del menu 3 (Data visualization). Genere el heatmap. 25. ¿De qué formas se puede agrupar el heatmap? Phylum, clase, orden, familia, género, especie y cepa 26. ¿Es lo mismo usar datos crudos (raw) que normalizar? ¿Que usaría y porqué? No, usaría normalizados para poder comparar varios metagenomas de manera simultánea ya que se establece la misma escala para todos. 27. ¿Qué es clustering? ¿Que es ward, single, complete, mcquity, median, centroid? Clustering es la tarea de aglomerar un conjunto de objetos, en este caso genes o proteínas en el mismo grupo en base a su similitud entre sí respecto a otro conjunto o cluster. Para esto se pueden utilizar diferentes métodos: unión entre grupos, intra grupos, vecino más cercano, más lejano, por centroide, por mediana y por el método de Ward. Los métodos de distancia mínima, máxima y entre centroides denotan similitud y distancia entre grupos en tanto que los de signgle, complete, mediana y Ward sirven para analizar el cluster.
El método de Ward es un criterio que se aplica en el análisis jerárquico de clusters. El criterio para elegir el par de grupos o clusters que se unen en cada paso está basado en el valor óptimo de una función objetiva, que podría ser cualquiera que refleje las necesidades del investigador. En este método se usa la varianza mínima. Para el vecino más cercano o single se representan las diferencias entre el cluster A y el cluster B por el mínimo de todas las distancias posibles entre los casos del cluster A y del cluster B. El método de vecino más lejano o complete representa las diferencias entre el cluster A y el cluster B por el máximo de todas las distancias posibles entre los casos del cluster A y del cluster B. En el método de agrupamiento por centroide las diferencias entre el cluster A y el cluster B se representan por la distancia entre el centroide de los casos en el cluster A con el centroide de los casos en el cluster B. La distancia entre 2 clusters se define como la distancia Euclidiana cuadrada entre los centroides. Este método es más robusto que otros métodos jerárquicos pero no tan eficiente como el de Ward. Si al utilizar el método del centroide se fusionan dos grupos de tamaño muy diferente, el centroide del nuevo grupo queda más cerca del grupo de mayor tamaño y más alejado del de menor tamaño, con lo que al ir fusionando se pueden perder propiedades de los grupos pequeños. Para prevenir esto se puede asumir que los grupos son de igual tamaño con lo que la distancia entre individuos de diferentes grupos sería la misma por lo que este método se denomina de la mediana. El método de McQuitty calcula la distancia promedio entre el cluster A y el cluster B basándose en el centro de gravedad del cluster. 28. ¿Qué distancia será la adecuada? ¿Bray-curtis, euclidiana, maxima? Cuando se usan los métodos centroide, mediana y Ward se debe usar la distancia Euclidiana cuadrada, la distancia euclidiana es la distancia entre dos puntos, puede merdirse aritméticamente y está dada por el teorema de pitágoras. La disimilitud de Bray-Curtis es una medida estadística que sirve para cuantificar la diferencia en composición entre dos sitios con base en cuentas en ambos lugares. Se representa con valores entre 0 y 1 donde 0 significa que son iguales y 1 que son totalmente distintos. Este valor no es una distancia sino un índice de disimilitud. Un cluster puede describirse por la distancia máxima que se necesita para conectar a dos partes del cluster. A diferentes distancias se formarán diferentes clusters, que pueden representarse usando un dendograma en el

que los datos se agrupan de forma jerárquica, en el eje Y se ilustra la distancia en que los clusters se agrupan en tanto que los objetos en el eje de las X no se mezclan. 29. Pruebe distintos parámetros y re-dibuje el heatmap. 30. ¿Qué le dicen los árboles verticales del heatmap? Los árboles verticales muestran la similitud que existe entre los metagenomas en análisis.
31. ¿Qué le dicen los árboles horizontales del heatmap? Los árboles horizontales muestran la similitud entre las categorías. 32. ¿Qué dicen las gráficas de caja y bigotes abajo del heatmap? Estas gráficas permiten visualizar de manera sencilla cinco parámetros: mínimo, primer cuartil, mediana, tercer cuartil y máximo de cada muestra. La primera se hace utilizando las cuentas de abundancia crudas y la segunda (la de los bigotes) se hace con los datos normalizados. Estas gráficas permiten saber de ver el tamaño de las cajas si las muestras son lo suficientemente similares para hacer comparaciones significativas.
33. ¿Si agrupa el heatmap a nivel de género que observa? ¿Es lo mismo que hacerlo a otros niveles taxonómicos? Que se hace mucho más grande porque se desglosan todos los componentes, es decir, todos los géneros agrupados en una familia se despliegan. 34. Genere un PCoA en la barra del menu 3 (Data Visualization). 35. ¿Qué es un PCoA? Análisis de coordenadas principales 36. ¿Tiene alguna similitud con el heatmap? No, sólo da una idea global de la relación entre metagenomas pero no da información sobre la relación de sus componentes. 37. ¿Qué se trata de responder con un PCoA? ¿Qué puede decir de la relación entre sus muestras analizadas? Es un gráfico que muestra qué tan diferentes son los metagenomas entre sí. Este tipo de gráfico es más útil para analizar un número grande de metagenomas y se pueden apreciar diferencias individuales y entre grupos. 38. Genere una gráfica de rarefacción en la barra del menú 3 (Data Visualization). 39. ¿Qué puede decir de las muestras que compara en base a lo que ve en la gráfica? ¿Qué es la diversidad alfa? ¿Cómo lo calcula en este caso? Las tres curvas siguen un patrón similar (hiperbólico) pero la pendiente de una de ellas empieza a aumentar antes de la de las otras dos, lo que sugiere que con menos reads se tiene un mayor número de especies. La diversidad alpha es un número simple que ilustra la diversidad de especies a escala local en cada metagenoma y se relaciona con cuántas especies diferentes son detectables en una muestra. El valor se obtiene de evaluar la riqueza de especies diferentes en cada metagenoma.
ANÁLISIS DE FUNCIONES 40. Vuelva al menú de selección de Datos (2) y seleccione ahora metagenomas WGS solamente, al menos seleccione 3. (2 Data Selection | Metagenomes | compare individually | public | WGS | available metagenomes -> ) 41. Seleccione el tipo de fuente de Anotación en Subsistemas. ¿Porqué le llaman sistemas jerárquicos de clasificación? Porque agrupan funciones que se consideran relacionadas a distintos niveles. 42. Seleccione un valor de corte de e-value (CutOff) %ID y longitud de alineamientos y justifique su elección, en esta vez utiliza amino ácidos. Para este apartado dejamos el valor de corte del e-value en 1E-5, el porcentaje de identidad en 70% y la longitud mínima de alineamiento en 20 porque el tamaño de proteínas es muy variable. 43. Genere una gráfica de barras. Menú 3, Data visualization, barchart. 44. ¿Qué obtiene? ¿Qué pasa si da click en una de las barras? ¿Se puede hacer recursivamente hasta que punto? Se despliegan tres niveles de distribución y el último de función

45. De la gráfica anterior seleccione el nivel máximo al que pueda acceder y en la parte inferior de la página seleccione el botón TO WORKBENCH. 46. En el worbench (pestaña verde entre el menu 3 y las gráficas). De click, ¿Qué puede hacer en el workbench? Se pueden descargar esos datos en formato FASTA y cambiar a otras formas de visualización. 47. Genere una tabla funcional. (2 Data Visualization -> Table -> generate) 48. Ordene la tabla y encuentre la función más representada por abundancia. (ponga un valor númerico y de Intro). Aquí encontramos que la función más representada es el agrupamiento basado en subsistemas. 49. Seleccione la función más representada con un click en la última columna y luego de click en el boton TO WORKBENCH. ¿Que obtiene de esto en el workbench? Esos datos se pasan separados y se pueden descargar en formato FASTA 50. Genere un Krona Graph en el menú que aparece por encima de la tabla. Identifique la función más representada. Se obtiene el mismo resultado que en la tabla funcional. 51. Genere un heatmap funcional. (2. Data Visualization -> Heatmap -> generate) 52. Compare la agrupación del Heatmap por el nivel 1 hasta el nivel 3. ¿Que nivel de comparación sugiere usar? En este caso, para los pocos metagenomas con los que estamos trabajando, escogimos el nivel 2 porque es el que permite apreciar diferencias en funciones sin ser muy reductivo pero tampoco tan extenso que vuelva muy difícil su interpretación. Además, en las gráficas de caja y de bigotes se puede ver que tanto los datos crudos como normalizados son comparables. 53. Del Menú 3 seleccione el botón open KEGG Mapper 54. Seleccione los mismos metagenomas en el DATA Selection con los que ha estado trabajando (los números de acceso ayudan a identificar esto rápidamente e.j. 4447970.3, 4447971.3) 55. Tiene que seleccionar primero el Target A, definir el metagenoma(s) a usar dar load data. Repetir lo mismo para el Target B y dar load data. después puesde seleccionar el menú para desplegar los datos únicos, los compartidos de A y B. 56. ¿Qué utilidad tiene dicha gráfica? ¿En qué formatos podemos guardar los datos de salida? Guarde una imagen de la gráfica para el reporte. Es una buena forma de comparar metabólicamente los metagenomas, lo que comparten y lo que es único para cada uno y en este caso puede ser más útil cuando se comparan pocos metagenomas. Los datos se exportan como archivos de texto.

Figura 1. KEGG con los datos únicos resaltados en el metagenoma A.
Figura 2. KEGG con los datos únicos resaltados en el metagenoma A.
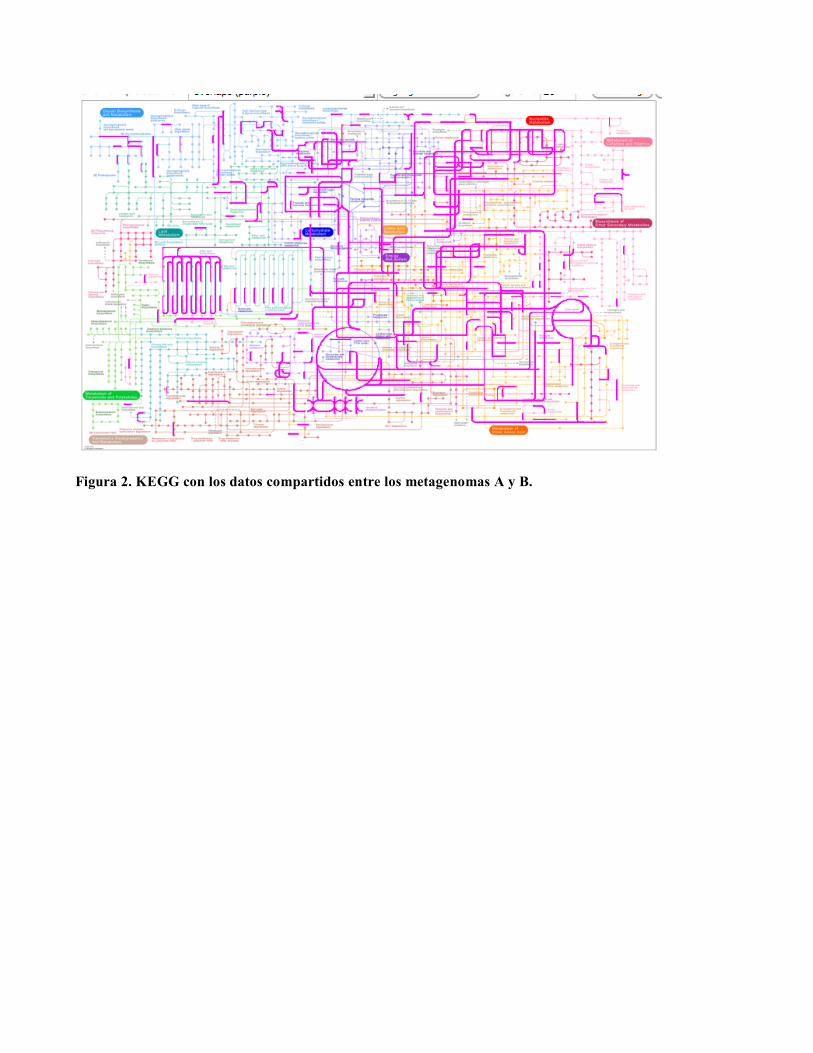
Figura 2. KEGG con los datos compartidos entre los metagenomas A y B.