
TEMA 8. ANEMIAS IIIs9af57bebc8a7f7c8.jimcontent.com/download/version/1294682394/module... · Cuando...
95
TEMA 8. ANEMIAS III ANEMIAS HEMOLÍTICAS (PERIFÉRICAS, REGENERATIVAS)
Transcript of TEMA 8. ANEMIAS IIIs9af57bebc8a7f7c8.jimcontent.com/download/version/1294682394/module... · Cuando...

TEMA 8. ANEMIAS III
ANEMIAS HEMOLÍTICAS(PERIFÉRICAS, REGENERATIVAS)

ANEMIAS HEMOLÍTICAS
1. CONCEPTO DE HEMÓLISIS
2. DATOS DE HEMÓLISIS2.1 DATOS CLÍNICOS2.1 DATOS CLÍNICOS2.2. DATOS DE LABORATORIO2.3 DATOS DE HEMÓLISIS INTRAVASCULAR.2.4 DATOS DE REGENERACIÓN.2.5 DATOS MORFOLÓGICOS

ANEMIAS HEMOLÍTICAS3. ESTUDIO SEGÚN CLASIFICACIÓN
3.1.- ANEMIAS HEMOLÍTICAS CORPUSCULARES
■ MEMBRANOPATÍAS# ESFEROCITOSIS HEREDITARIA O ENFERMEDAD DE MINKOWSKY–CHAUFFARD #
■ ENZIMOPATIAS# DEFICIT DE G6PD ## DÉFICIT DE PIRUVATO-# DÉFICIT DE PIRUVATO-
KINASA #
■ HEMOGLOBINOPATIAS# Hb S: AN. DREPANOCITICA# Β –TALASEMIA #
3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES:
■ INMUNESAHAI Anemia Hemolítica Autoinmunitaria■ NO INMUNES

1.- CONCEPTO DE HEMÓLISIS
� Las anemias hemolíticas son debidas a una destrucción de los hematíes por encima de los límites de la hemólisis fisiológica.
� Cuando la hemólisis esta aumentada, la vida media de los hematíes está acortada significativamentede los hematíes está acortada significativamente
� intravascular (dentro de las circulación) o extravascular (en los macrófagos del SMF de bazo, hígado y MO) Este segundo tipo es el mas frecuente.

2. DATOS DE HEMÓLISIS
� 2.1.- DATOS CLÍNICOS:tres síntomas indican la existencia de hemólisis:
� Anemia,� Ictericia� Esplenomegalia.� Esplenomegalia.
� Cuando la hemólisis es extravascular, el cuadro clínico es menos agudo ó crónico y se dan los tres síntomas de forma evidente
� Cuando la hemólisis es intravascular, el cuadro clínico es mas agudo (Aparece fiebre, malestar, escalofríos, nauseas, vómitos…) y predomina la anemia sobre la ictericia y hay menos esplenomegalia.

2. DATOS DE HEMÓLISIS2.2.- DATOS DE LABORATORIO
� 1. Aumento de la sideremia: se libera hierro del grupo hemo.
� 2. Aumento de la bilirrubina en sangre ó hiperbilirrubinemia: La cual produce un cuadro de ictericia prehepatica en el que predomina la BI con heces hipercólicas y orina sin coluria.predomina la BI con heces hipercólicas y orina sin coluria.
� 3. Aumento de urobilinogeno urinario: se produce mayor cantidad de urobilinógeno porque se metaboliza más proporción de Bilirrubina que accede más al intestino desde el que hay también mayor reabsorción.
� 4. Descenso de haptoglobina: se utiliza en retirar la Hbliberada por la hemólisis
� 5. Aumento de LDH ó Lactatodeshidrogenasa: Es una enzima que se libera desde los hematíes al suero cuando hay hemólisis.

2. DATOS DE HEMÓLISIS2.3.- OTROS DATOS DE LABORATORIO
HEMÓLISIS INTRAVASCULAR:
� Aumento de Hb libre en plasma �Hemoglobinemia
� Hemoglobinuria� Hemoglobinuria
� Hemosiderinuria
� Presencia de metahemalbumina

2. DATOS DE HEMÓLISIS 2.4.- DATOS DE REGENERACIÓN
� Debido a la regeneración medular con aumento de la eritropoyesis para compensar la hemólisis, aparecen
� Aumento de reticulocitos en sangre periférica (SP)(SP)
� Aumento de eritroblastos en MO� Eritroblastos en SP

2. DATOS DE HEMÓLISIS 2.5.- DATOS MORFOLÓGICOS
� 1.- La morfología general de las anemias hemolíticas es normocítica normocrómica
Anemia normocíticaReticulocitos ReticulocitosReticulocitos Reticulocitosbajos altos
Aplasia ó insuficiencia Hemólisis Hemorragia medular aguda

2. DATOS DE HEMÓLISIS 2.5.- DATOS MORFOLÓGICOS
� 2.- Si hay una reticulocitosis importante, morfológicamente la anemia puede ser macrocítica normocrómica
� 3.- Si hay muchos reticulocitos y � 3.- Si hay muchos reticulocitos y eritroblastos en SP, se observa Policromatofilia ó Policromasia:
� fenómeno que describe la aparición de numerosos RETICULOCITOS recién formados (gris-azulados) en la sangre periférica, en contraste con el tono rosadode los HEMATÍES maduros

2. DATOS DE HEMÓLISIS 2.5.- DATOS MORFOLÓGICOS
� 4.- Aparecen numerosas y variadas anomalías eritrocitarias, con poiquilocitosis: orientación del diagnóstico.
▪ Dianocitos: Característicos de las hemoglobinopatias: talasemias y otras
▪ Esferocitos: Característicos de la esferocitosis y otros procesos ▪ Esferocitos: Característicos de la esferocitosis y otros procesos como las anemias hemolíticas auto inmunes.
▪ Eliptocitos y dacriocitos: En las talasemias.▪ Estomatocitos: Característico de la estomatocitosis y
esferocitosis.▪ Hematíes Crenados: En las enzimopatias.▪ Excentrocitos: Se dan en el favismo▪ Drepanocitos: En la hemoglobinopatia S▪ Esquistocitos: Muy característicos de la hemólisis, sobre todo de
tipo mecánico.

ANEMIAS HEMOLÍTICAS3. ESTUDIO SEGÚN CLASIFICACIÓN
3.1.3.1.-- ANEMIAS HEMOLÍTICAS CORPUSCULARESANEMIAS HEMOLÍTICAS CORPUSCULARES
■■ MEMBRANOPATÍASMEMBRANOPATÍAS# ESFEROCITOSIS HEREDITARIA O ENFERMEDAD DE MINKOWSKY–CHAUFFARD #
■ ENZIMOPATIAS# DEFICIT DE G6PD ## DÉFICIT DE PIRUVATO-# DÉFICIT DE PIRUVATO-
KINASA #
■ HEMOGLOBINOPATIAS# Hb S: AN. DREPANOCITICA# Β –TALASEMIA #
3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES:
■ INMUNESAHAI Anemia Hemolítica Autoinmunitaria■ NO INMUNES

3.1.3.1.-- ANEMIAS HEMOLÍTICAS CORPUSCULARESANEMIAS HEMOLÍTICAS CORPUSCULARES
■■ MEMBRANOPATÍASMEMBRANOPATÍAS
� Una mayoría de estas alteraciones, es de carácter congénito y se incluyen procesos como:
_ Esferocitosis hereditaria o Enfermedad de Minkowsky – Chauffard **
_ Eliptocitosis hereditaria (mas de un 80% _ Eliptocitosis hereditaria (mas de un 80% son asintomáticas)
_ Estomatocitosis hereditaria_ Acantocitosis hereditaria (Acompañada
de una alteración de las β-lipoproteínas=Abeta lipoproteinemia)
� También existen defectos de la membrana de carácter adquirido como la denominada ‘ Hemoglobinuria paroxística nocturna (HPN) o Enfermedad de Marchafava – Micheli **

ESFEROCITOSIS HEREDITARIA O ENFERMEDAD
DE MINKOWSKY–CHAUFFARD
� Se caracteriza por la aparición de hematíes esféricos por una anormalidad intrínseca de su membrana, es de carácter hereditario.Etiología� Etiología
� Alteración genética, autosómica dominante, que determina un defecto a nivel de una ó más proteínas de la membrana eritrocitaria

ESFEROCITOSIS HEREDITARIA Etiología
� PROTEINAS:alteración cuantitativa y cualitativa de la espectrina Según las alteraciones genéticas que se producen y que son variadas, también se pueden afectar otras proteinas como la ankirina, banda3 etcbanda3 etc
� La mayoría de pacientes presenta una deficiencia combinada de espectrina y ankirina.
� LÍPIDOS: Por la deficiencia de espectrina hay una disminución secundaria de los fosfolípidos y el colesterol de la membrana, la bicapa lipídica no tiene el sostén del esqueleto proteíco.
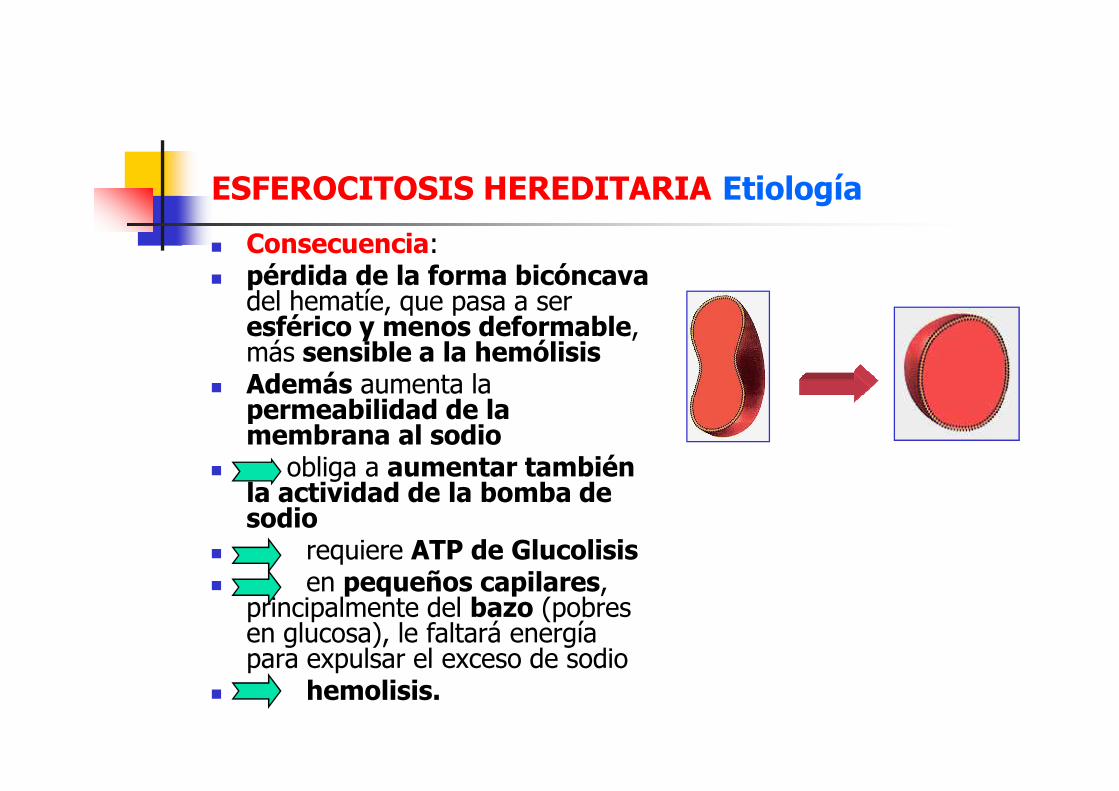
ESFEROCITOSIS HEREDITARIA Etiología
� Consecuencia: � pérdida de la forma bicóncava
del hematíe, que pasa a ser esférico y menos deformable, más sensible a la hemólisis
� Además aumenta la permeabilidad de la
� Además aumenta la permeabilidad de la membrana al sodio
� obliga a aumentar también la actividad de la bomba de sodio
� requiere ATP de Glucolisis� en pequeños capilares,
principalmente del bazo (pobres en glucosa), le faltará energía para expulsar el exceso de sodio
� hemolisis.

ESFEROCITOSIS HEREDITARIA Clínica
� La gravedad del proceso es variable� 50% de los casos, es bien tolerada� Casi siempre se diagnostica en la infancia pero hay
casos clínicamente asintomáticos que se diagnostican en adultos en adultos
� El cuadro clínico es el de una anemia crónica ( hemólisis extravascular crónica ) de intensidad variable. con anemia, ictericia y esplenomegalia
� Casos más graves: complicaciones� En niños: Retraso del crecimiento y alteraciones
esqueléticas por hiperplasia medular compensatoria� Litiasis biliar.

ESFEROCITOSIS HEREDITARIA Diagnostico
� ¬ Datos del catabolismo del hemo y otros de hemólisis▪ Aumento de la sideremia.▪ Aumento de la bilirrubina▪ Aumento del urobilinogeno. ▪ Aumento de LDH… ▪ Aumento de LDH…
� Datos de regeneración: Aumento de reticulocitos en SP.� Anemia normocítica normocrómica con esferocitos en sangre
periférica� Los esferocitos aparecen como hematíes con un menor diámetro e
hipercrómicos, con el área pálida central situada de forma excéntrica ó inexistente. También se presentan como microesferocitos.
� Hay poiquilocitosis, pueden aparecer otras formas anómalas como estomatocitos.
� Los índices eritrocitarios muestran:▪ VCM: disminuido▪ CHCM: esta alto. El aumento de CHCM es muy sugestivo de
esferocitosis.

ESFEROCITOSIS HEREDITARIA Diagnostico

ESFEROCITOSIS HEREDITARIA Diagnostico
SERIE ROJA: ANEMIAS
ESTOMATOCITOS
SP: ESTOMATOCITOS (ESFEROCITOSIS HEREDITARIA)

ESFEROCITOSIS HEREDITARIA Diagnostico
� Pruebas especiales� Se puede hacer un test de fragilidad
osmótica, que en estos hematíes, está aumentada. Significa que los hematíes son fácilmente hemolizados en contacto con aumentada. Significa que los hematíes son fácilmente hemolizados en contacto con soluciones hipotónicas.
� Existen otros test específicos.

ESFEROCITOSIS HEREDITARIA Tratamiento
� 1.- Tratamiento paliativo con trasfusiones de concentrado de hematíes y tratamiento intensivo en las crisis hemolíticas con vigilancia de la diuresis.
� 2.- Intentar un tratamiento curativo con esplenectomía o trasplante de MO.
la esplenectomía se realiza� la esplenectomía se realiza� en los casos mal tolerados, porque logra compensar a los
pacientes, aunque permanezcan algunos esferocitos � en mayores de 6 años. No se recomienda en niños menores de 6
años, habría tendencia a las infecciones y tampoco se recomienda en la adolescencia, porque puede haber alteraciones del desarrollo, como el hipogonadismo

HEMOGLOBINURIA PAROXÍSTICA NOCTURNA (HPN)
� Etiología
� Es un defecto adquirido de la membrana eritrocitaria, que consiste en la ausencia o déficit de factores que controlan determinadas proteínas del sistema de complementocontrolan determinadas proteínas del sistema de complemento
� La consecuencia es que se produce una especial sensibilidad de los hematíes al complemento, lo cual produce crisis hemolíticas

HPN
� Clínica
� mas frecuencia en adultos jóvenes (30-40 años),
� aparecen crisis hemolíticas, desencadenadas por diferentes circunstancias como infecciones, diferentes circunstancias como infecciones, menstruación, vacunas, etc.
� Las crisis con frecuencia son nocturnas y se manifiestan a veces con hematuria matutina por hemoglobinuria macroscópica.

HPN
� Diagnostico� ¬ En el hemograma:
La anemia se acompaña de algunas alteraciones de granulocitos y plaquetas, por
ejemplo:_ Disminución de granulocitos_ Disminución de granulocitos_ Disminución de plaquetas_ Alteraciones funcionales plaquetarias, a veces con
formación de trombos.Todo esto, añadido a los datos de hemólisis.
� ¬ Pruebas especiales:Hay una prueba, el test de Ham-Dacie, que consiste en la
determinación de la sensibilidad de los hematíes a los medios ácidos.
� Tratamiento: Es sustitutivo mediante transfusión
ANEMIAS HEMOLÍTICAS3. ESTUDIO SEGÚN CLASIFICACIÓN
3.1.- ANEMIAS HEMOLÍTICAS CORPUSCULARES
■ MEMBRANOPATÍAS# ESFEROCITOSIS HEREDITARIA O ENFERMEDAD DE MINKOWSKY–CHAUFFARD #
■ ENZIMOPATIASENZIMOPATIAS# DEFICIT DE G6PD ## DEFICIT DE G6PD ## DÉFICIT DE PIRUVATO# DÉFICIT DE PIRUVATO-- KINASA #KINASA ## DÉFICIT DE PIRUVATO# DÉFICIT DE PIRUVATO-- KINASA #KINASA #
■ HEMOGLOBINOPATIAS# Hb S: AN. DREPANOCITICA# Β –TALASEMIA #
3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES:
■ INMUNESAHAI Anemia Hemolítica Autoinmunitaria■ NO INMUNES

ENZIMOPATIASDEFICIT DE G6PD
� Etiología� Es una enfermedad congénita hereditaria que se
transmite igual que la hemofilia, por herencia recesiva ligada al cromosoma X. Se manifiesta clínicamente en hombres, que son X’ Y � Se manifiesta clínicamente en hombres, que son X’ Y y las mujeres, con un cromosoma alterado, son portadoras XXla enfermedad suele ser asintomática, aunque pueden presentar alteraciones. X’ X’ son embriones no viables, por lo tanto no suelen nacer
� La alteración genética produce un defecto de la vía glucolítica de las pentosas, por déficit de la enzima (G6PD).

DEFICIT DE G6PD Etiología
� La alteración genética produce un defecto de la vía glucolítica de las pentosas, por déficit de la enzima (G6PD).
� limitación en la producción de NADPH2
� la Hb y otras proteínas estructurales, se oxidan más fácilmente y desnaturalización de la Hb, hace que ésta precipite, formando inclusiones citoplasmáticas, llamadas ‘corpúsculos de Heinz
� hematíes se hacen más sensibles a la hemólisis

DEFICIT DE G6PD Clínica
� Esta anemia hemolítica tiene una incidencia característica en poblaciones, es mas frecuente en la raza negra, en la que existen variantes africanas y americanas y se da también mucho en la zona mediterránea. Existe alguna variante que afecta al 40 % de la población.
� El cuadro clínico es variable y puede presentar dos formas:� El cuadro clínico es variable y puede presentar dos formas:� 1.- Anemia hemolítica crónica: Extravascular, principalmente en el
bazo, y es bien tolerada
� 2.- Crisis hemolítica aguda: con hemoglobinemia y la hemoglobinuria.Se suele recuperar en 7 – 10 días y esta desencadenada por varios factores que actúan como factores oxidantes que provocan la hemólisis, los hematíes no resisten a la oxidación, porque tienen su poder reductor limitado. Entre estos factores están:
▪ Infecciones: Neumonías, infecciones víricas, salmonelosis, etc.▪ Medicamentos.▪ Favismo: Es una forma clínica mediterránea, en la que las crisis
hemolíticas están provocadas por la ingesta de habas en cualquier forma culinaria, o incluso por contacto directo con la planta o su ambiente, como por ejemplo el polen. En ocasiones puede cursar de manera asintomática
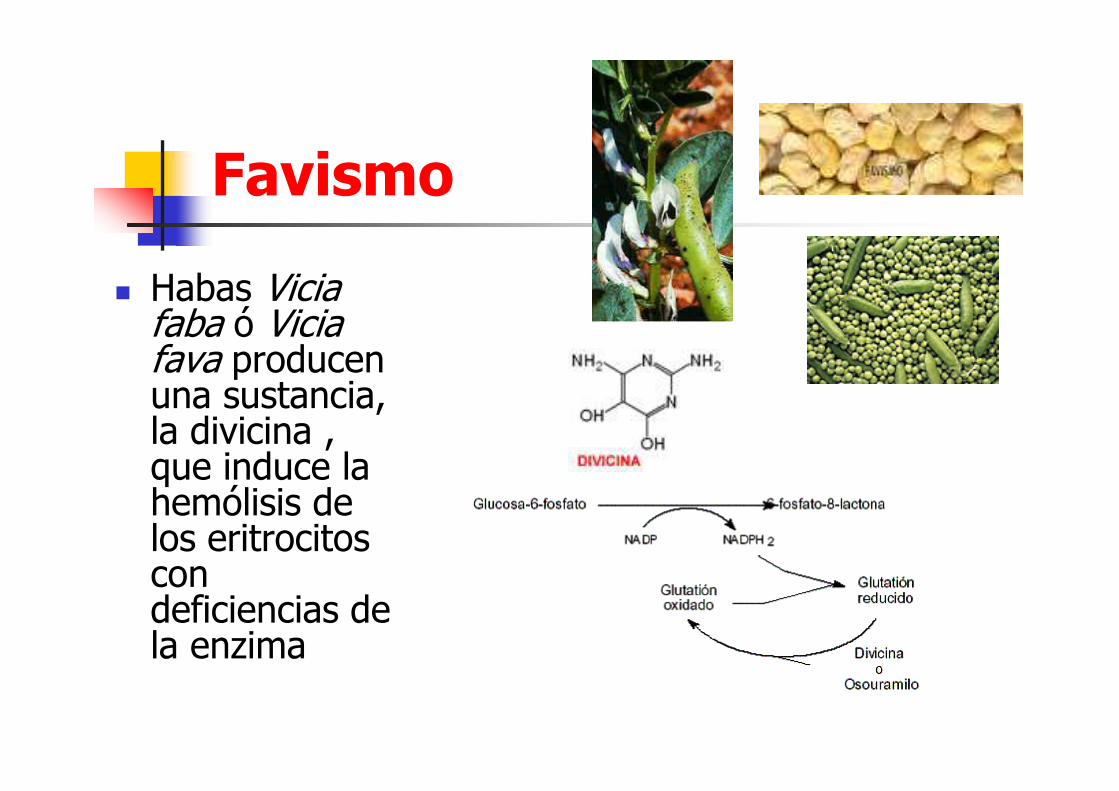
Favismo
� Habas Vicia faba ó Vicia fava producen una sustancia, una sustancia, la divicina , que induce la hemólisis de los eritrocitos con deficiencias de la enzima

DEFICIT DE G6PD Diagnóstico
� Además de los datos de hemólisis:
� Es característica la presencia de Corpúsculos de Heinz y de Excentrocitos, hematíes cuyo contenido en hemoglobina se hematíes cuyo contenido en hemoglobina se ha desplazado a un lado dejando una especie de bolsa vacía al lado contrario.
� Se pueden hacer pruebas para la determinación del déficit enzimático.

DEFICIT DE G6PD Diagnóstico
EXCENTROCITOS: DÉFICIT DE G6PD (FAVISMO)

DEFICIT DE G6PD
� Cuando están suficientemente repletos de -bolas» que son los cuerpos que son los cuerpos Heinz, los hematíes afectados o bien pierden fragmentos en forma de mordisco o bien son fagocitados por las células RE del bazo y de¡ hígado.

DEFICIT DE G6PD
La figura 22-1 muestra numerosas irregularidades morfológicas de los hematíes de un enfermo afecto de anemia hemolítica congénita con cuerpos de Heinz. Muchas células tienen contornos ¡rregulares y algunas parecen células algunas parecen células espiculadas. Una célula cerca de la base de la figura muestra la deformidad en mordisco que es casi diagnostica de la anemia con cuerpos de Heinz.
La aparición de hematíes repletos de cuerpos de Heinz tras la tinción supravital con metil (o cristal) violeta se muestra en la figura 22-2

ERITROBLASTO ORTOCROMÁTICO LINFOCITO
SERIE ROJA: ERITROBLASTOS EN SP
SP: CRISIS HEMOLÍTICA EN ENFERMO CON FAVISMO (DÉFICIT DE G6PD)
GRANULOCITOS
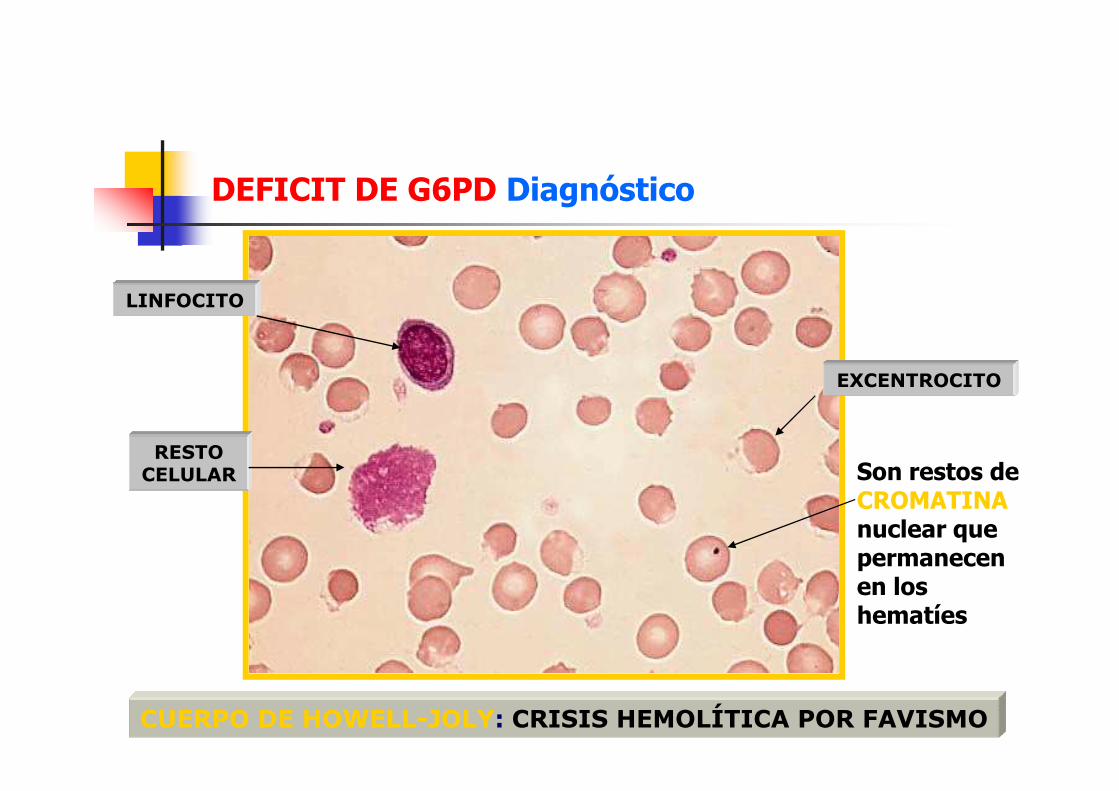
DEFICIT DE G6PD Diagnóstico
EXCENTROCITO
LINFOCITO
CUERPO DE HOWELL-JOLY: CRISIS HEMOLÍTICA POR FAVISMO
RESTO CELULAR Son restos de
CROMATINAnuclear que permanecen en los hematíes

DEFICIT DE G6PD
� Tratamiento
� 1. Profilaxis, evitando las sustancias o factores desencadenantes (medicamentos, habas, infecciones..).2. Tratamiento paliativo, con transfusión de infecciones..).2. Tratamiento paliativo, con transfusión de concentrado de hematíes y control de las crisis hemolíticas con vigilancia y garantizando una buena diuresis.Utilizar antioxidantes como la vitamina E.
� Esta enfermedad remite por sí sola, con una recuperación rápida, siempre y cuando no se repita la exposición.

ENZIMOPATIASDÉFICIT DE PIRUVATO- KINASA (PK)
� Etiología
� Es una enfermedad congénita hereditaria poco frecuentetransmisión es autosómica recesiva
� Alteración de la glucólisis por la vía principal, a causa del déficit de la enzima PKde la enzima PK
� se altera la síntesis normal de ATP
� los hematíes pierden el control osmótico
� se hemolizan mas fácilmente

ENZIMOPATIASDÉFICIT DE PIRUVATO- KINASA (PK)
� Clínica
� Es la de una anemia hemolítica crónica extravascular con anemia, ictericia y extravascular con anemia, ictericia y esplenomegalia

ENZIMOPATIAS DÉFICIT DE PIRUVATO- KINASA (PK)
� Diagnostico� Además de los
datos de hemólisis, es característica la característica la presencia de hematíes crenados.
� Pruebas especiales, se hace la demostración del déficit enzimático
EQUINOCITOS: H. Espiculados o “crenados”
EQUINOCITOS: Eritrocitos con prolongaciones en forma de “picos” cortos
Hepatopatía alcohólica Déficit de Piruvato KinasaInsuficiencia renal

ANEMIAS HEMOLÍTICAS3. ESTUDIO SEGÚN CLASIFICACIÓN
3.1.- ANEMIAS HEMOLÍTICAS CORPUSCULARES
■ MEMBRANOPATÍAS# ESFEROCITOSIS HEREDITARIA O ENFERMEDAD DE MINKOWSKY–CHAUFFARD #
■ ENZIMOPATIAS# DEFICIT DE G6PD ## DÉFICIT DE PIRUVATO- KINASA ## DÉFICIT DE PIRUVATO- KINASA #
■ HEMOGLOBINOPATIASHEMOGLOBINOPATIAS# Hb S: AN. DREPANOCITICA# Hb S: AN. DREPANOCITICA# # ΒΒ ––TALASEMIA #TALASEMIA #
3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES:
■ INMUNESAHAI Anemia Hemolítica Autoinmunitaria■ NO INMUNES

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� Son anormalidades moleculares de una de lascadenas de la globina, hay una alteración de la estructura molecular: hemoglobinopatías estructurales.estructurales.
� Generalmente porque hay una alteración en los genes que codifican para la cadena, sufren una mutación puntual que condiciona una sustitución de un aa de la cadena, por otro aa que es anómalo (aa’). consecuencias
patológicas

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� Las hemoglobinopatías estructurales se clasifican en cuatro grandes grupos:
� 1. Hemoglobinas con disminución de la solubilidad: HbS y HbC.cambios de aa superficiales dan anemia � cambios de aa superficiales dan anemia hemolítica
� La HbS con polimerización de la desoxi-hemoglobina y causante de la anemia falciforme
� La HbC aunque también cristaliza en el interior del eritrocito carece de las graves consecuencias de la HbS y es prácticamente asintomática
� Hay Hb D y Hb E: son poco frecuentes

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� 2. Hemoglobinas con disminución de la estabilidad molecular ó hemoglobinas inestables.
� Las mutaciones y cambios se sitúan en el interior � Las mutaciones y cambios se sitúan en el interior de la molécula y casi siempre en las zonas de contacto entre las subunidades por lo que al fallar esas uniones las hemoglobinas se hacen inestables.

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� 3. Hemoglobinas con alteración de la afinidad por el oxígeno
� Las mutaciones se sitúan en regiones próximas a la cavidad del hemoa la cavidad del hemo
� No dan anemia hemolítica.

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� 4. Metahemoglobinas (HbM).� También se forman por mutaciones y cambios
internos próximos a la cavidad del hemo. la mutación produce una oxidación permanente mutación produce una oxidación permanente del hierro y transformación de la hemoglobina en metahemoglobina incapaz de transportar O2
� No dan hemólisis, sí cianosis

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:
� Las Hb del apartado 1. con cambios de aa superficiales dan anemia hemolíticay se diagnostican por electroforesis(cambia la carga eléctrica electroforesis(cambia la carga eléctrica de la molécula de Hb), las restantes con cambios interiores no.

HEMOGLOBINOPATIASHEMOGLOBINOPATIASA) Hemoglobinopatías Cualitativas:A) Hemoglobinopatías Cualitativas:

HEMOGLOBINOPATIA S: AN. DREPANOCITICA,
DREPANOCITOSIS, Ó AN. DE CÉLULAS FALCIFORMES
� Etiología� Es una alteración
hereditaria del gen de la cadena β, que produce la β, que produce la sustitución del aa ácido glutámico por valina en la posición 6 de la cadena β (ß6 Glu → Val), con lo que se sustituye la Hb A por Hb S.
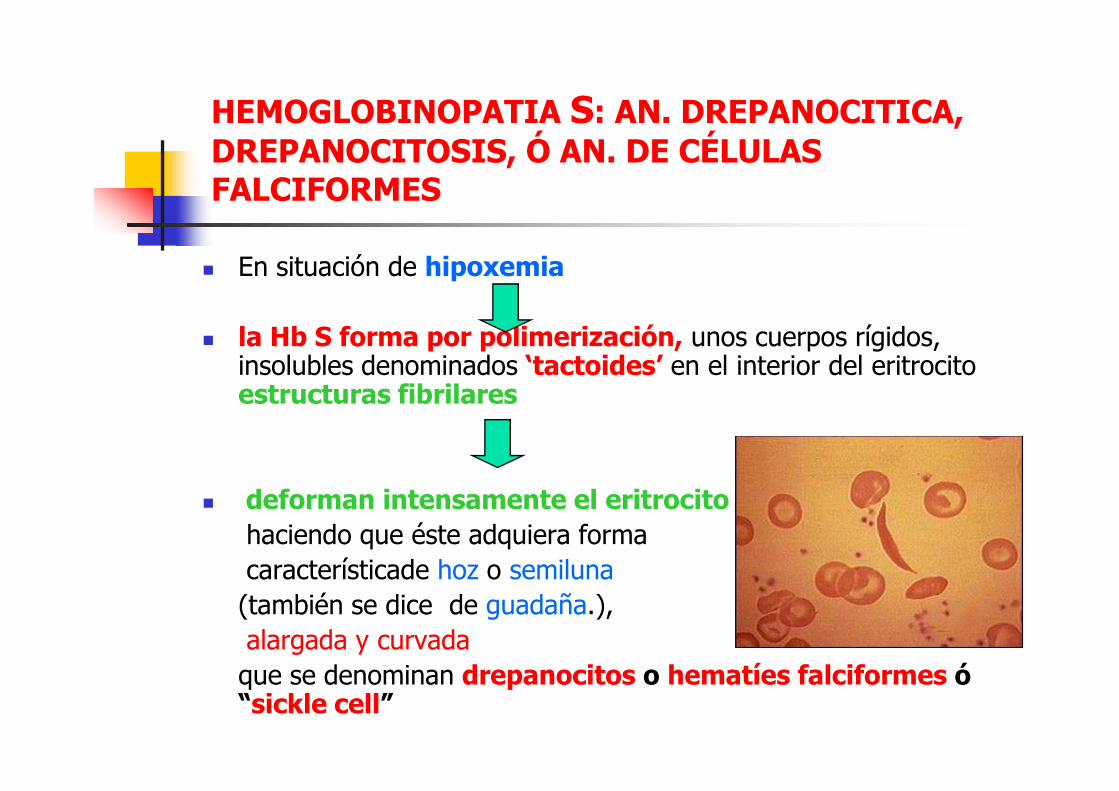
HEMOGLOBINOPATIA S: AN. DREPANOCITICA, DREPANOCITOSIS, Ó AN. DE CÉLULAS FALCIFORMES
� En situación de hipoxemia
� la Hb S forma por polimerización, unos cuerpos rígidos, insolubles denominados ‘tactoides’ en el interior del eritrocito estructuras fibrilaresestructuras fibrilares
� deforman intensamente el eritrocitohaciendo que éste adquiera forma
característicade hoz o semiluna
(también se dice de guadaña.),
alargada y curvada
que se denominan drepanocitos o hematíes falciformes ó “sickle cell”

HEMOGLOBINOPATIA S: AN. DREPANOCITICA, DREPANOCITOSIS, Ó AN. DE CÉLULAS FALCIFORMES
� Esta formación produce dos consecuencias� 1.- Los hematíes son más rígidos y mas
fácilmente hemolizables, por lo tanto hay hemólisis de predominio extravascular.
� 2.- Se favorecen las trombosis en órganos internos, porque los hematíes rígidos forman más fácilmente trombos, y obstruyen capilares de diferentes localizaciones con microinfartos.Estas localizaciones son: en el BAZO, en las zonas OSEAS, RENAL, y MUSCULAR en las cuales hay crisis dolorosas y un daño progresivo en esos órganos.


HEMOGLOBINOPATIA S Clínica
� 1.- HOMOCIGOTICA Hb S/S� Significa que de los dos genes de las cadenas β,
están alterados, en el cromosoma 11.� No se produce ninguna cadena β normal y en
consecuencia, ninguna Hb A. consecuencia, ninguna Hb A. � Estas personas manifiestan la enfermedad grave
con la anemia hemolítica, trombosis, crisis dolorosas (óseas, musculares, abdominales…) y complicaciones posibles como:
▪ Ulceras en piernas▪ Infartos orgánicos, los dos mas graves son el
de miocardio y el cerebral.

HEMOGLOBINOPATIA S Clínica
� 2.- HETEROCIGOTICA (Rasgo Drepanocitico)Hemoglobinopatia A/S
� solo esta presente un gen alterado. Los hematíes contiene mas de un 50% de Hb A, es decir, hay Hb A y Hb Sdecir, hay Hb A y Hb S
� en condiciones normales no hay clínica, es decir, las personas son asintomáticas
� en situaciones de hipoxia o acidosis, infección respiratoria, enfermedades metabólicas (como la diabetes) anestesia, estrés físico muy importante etc, cualquiera de estas causas pueden desencadenar crisis drepanocíticas con hemólisis y complicaciones vasculares.

HEMOGLOBINOPATIA S Clínica
� Tiene una alta incidencia en personas de raza negra, debido a que los individuos , especialmente los heterocigotos, son resistentes al paludismo (malaria), de forma que estos hematíes son menos afectados por el agente productor de esta afectados por el agente productor de esta enfermedad, el plasmodium, del cual existen diferentes especies (vivax, falciparum…). Por tanto no padecen anemia ni malaria, ésta es la razón que explicaría la persistencia de este rasgo genético en esta población


HEMOGLOBINOPATIA S Diagnostico
� 1.- Hemograma: Es una anemia normocítica normocrómica, con anisopoiquilocitosis y presencia de drepanocitos, esquistocitos, dianocitos
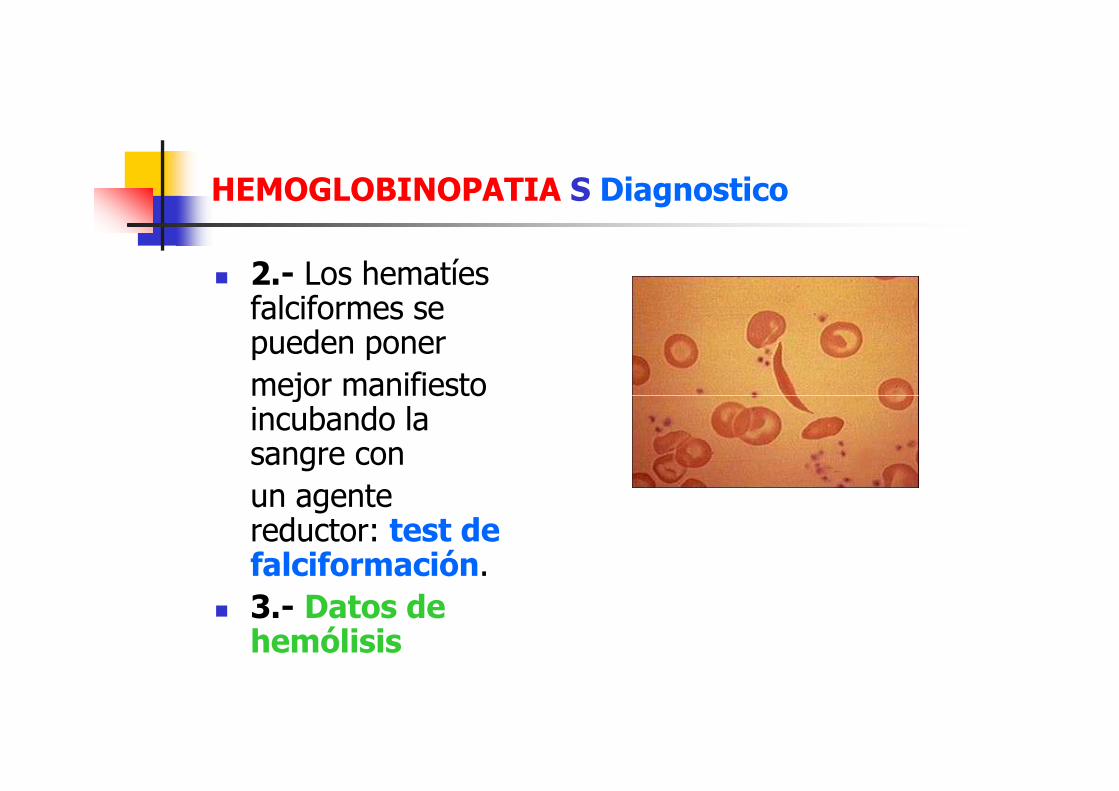
HEMOGLOBINOPATIA S Diagnostico
� 2.- Los hematíes falciformes se pueden poner
mejor manifiesto mejor manifiesto incubando la sangre con
un agente reductor: test de falciformación.
� 3.- Datos de hemólisis

HEMOGLOBINOPATIA S Diagnostico
� Electroforesis de la Hb:� Esta prueba permite detectar las anomalías hemoglobínicas. Se
puede ver la aparición de bandas de Hb anormal� Homocigotos:
1.- No hay Hb A, a no ser que haya sido transfundido recientemente.recientemente.
2.- Se encuentra Hb S, puede suponer un 80 – 90 % del total3.- Se produce un aumento de Hb F (1 – 20 %)4.- Se produce un aumento de Hb A2 (hasta un 4.5 %)
� Heterocigotos:1.- Sí hay Hb A, que puede suponer como mínimo un 50% hasta
un 70% 2.- Se encuentra Hb S, sobre un 30 %-40%3.- La Hb F es normal4.- La Hb A2 sufre un ligero aumento (hasta un 4.5 %)

HEMOGLOBINOPATIA S Tratamiento
� 1.- Profilaxis: tratando de evitar las causas de hipoxia2.- Régimen de transfusiones de concentrado de hematíes, el objetivo es evitar la anemia y reponer la Hb A al tiempo que se disminuye la síntesis de Hb S. Hb A al tiempo que se disminuye la síntesis de Hb S. Se debe mantener la Hb A en torno a un 70%3.- Tratamiento de las crisis hemolíticas y las complicaciones vasculares. Si hay trombosis, se tratan con analgésicos y anticoagulantes.4.- Se puede plantear el trasplante de MO (sobre todo en los homocigotos)

B) Hemoglobinopatias Cuantitativas
� Son enfermedades de carácter hereditario, en las que se forman cadenas de globina de estructura normal pero disminuye cuantitativamente la síntesis de algunas de las cadenas. Se denominan TALASEMIAS y según la cadenas. Se denominan TALASEMIAS y según la cadena afectada, hay diferentes variedades, entre ellas:
� • α-talasemias• β-talasemias• δ-talasemias• Combinadas: β-δ talasemias• Talasemias más otra Hbpatia:
• Hbpatia S -talasemias• Hbpatia C -talasemia
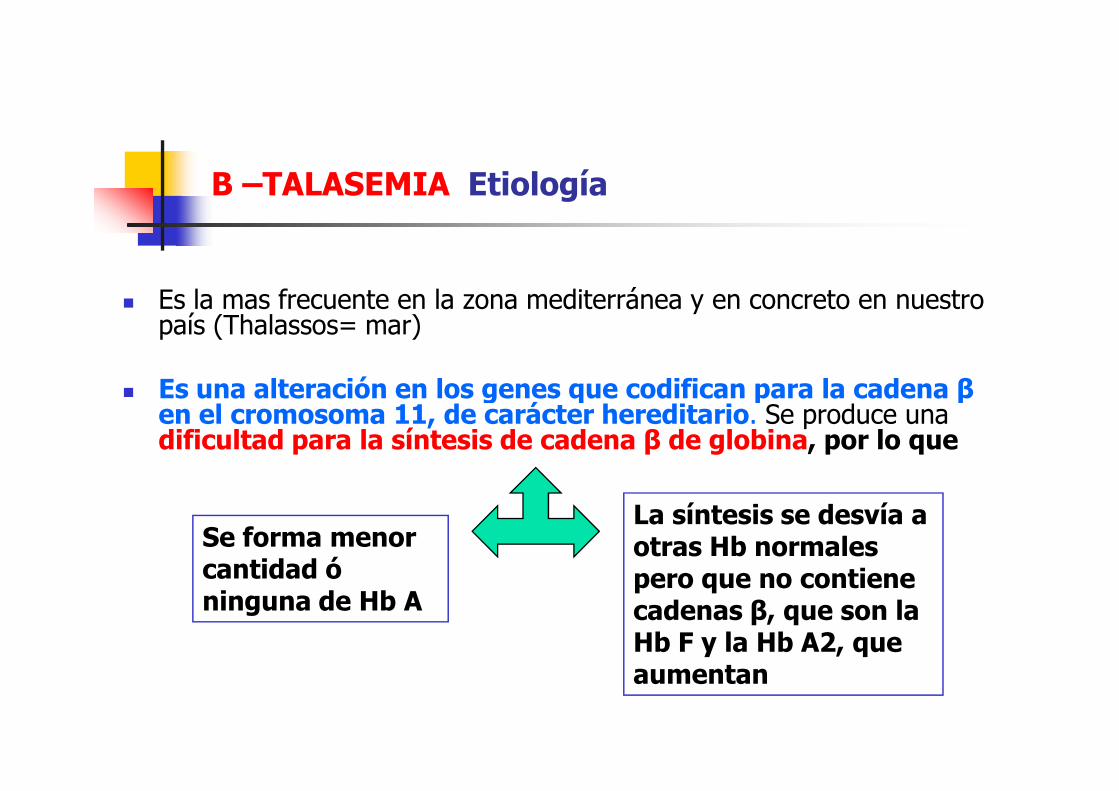
Β –TALASEMIA Etiología
� Es la mas frecuente en la zona mediterránea y en concreto en nuestro país (Thalassos= mar)
� Es una alteración en los genes que codifican para la cadena β � Es una alteración en los genes que codifican para la cadena β en el cromosoma 11, de carácter hereditario. Se produce una dificultad para la síntesis de cadena β de globina, por lo que
La síntesis se desvía a otras Hb normales pero que no contiene cadenas β, que son la Hb F y la Hb A2, que aumentan
Se forma menor cantidad ó ninguna de Hb A

Β –TALASEMIA Etiología
� Consecuencias de esta dificultad de síntesis, tenemos que:1.- Los hematíes poseen escasa Hb, que se manifiesta por una hipocromía y microcitosis a diferencia del resto de anemias hemolíticas y que diferencia del resto de anemias hemolíticas y que obliga a establecer un diagnostico diferencial con la anemia ferropénica.2.- Los hematíes presentan una carga hemoglobínica anormal, hay poca cadena β, exceso de alfa … Estos hematíes son destruidos mas fácilmente , tanto en la MO, con una eritropoyesis ineficaz, como a un nivel periférico, con unahemólisis extravascular.

Β –TALASEMIA Clínica
� 1.- HOMOCIGOTOS β-Talasemia Mayor o “Major” ó Anemia de Cooley
� Gran incidencia en países mediterráneos como Italia y Grecia� Se diagnostica desde la infancia con un síndrome de hemólisis
extravascular, de anemia, ictericia y esplenomegaliaA esto se añade� A esto se añade� Predisposición a infecciones� Retraso en el crecimiento y desarrollo, con posibles
alteraciones óseas (dolor, deformación) debido a el aumento de actividad eritropoyetica de la MO.
� Con el tiempo, puede desarrollar una sobrecarga de hierro en el organismo debido a la elevación de la sideremia por la hemólisis y al régimen de transfusiones sanguíneas. se desarrolla una enfermedad denominada “Hemosiderosis”que puede causar alteración de órganos internos, por ejemplo en el hígado, en el corazón, etc.

Β –TALASEMIA Clínica
� 2.- HETEROCIGOTOS β-Talasemia menor o “minor” ó Rasgo talasémico ó Portador.
� Es la forma clínica más frecuente de ß talasemia en el área mediterránea Es frecuente en EspañaClínicamente es una forma bien tolerada, los � Clínicamente es una forma bien tolerada, los pacientes son asintomáticos o presentan una anemia discreta con esplenomegalia leve, a veces en la infancia, hay cierto retraso del crecimiento o cierta tendencia a la infecciones
� Con un régimen de vida adecuado, no suelen presentar problemas médicos o quirúrgicos, y suelen mostrar una supervivencia normal.

Β –TALASEMIA Clínica
� En realidad existen otras dos formas clínicas, en total cuatro de β –talasemia
� Talasemia mínima. Carece de expresividad clínica o biológica y su hallazgo es siempre resultado de un estudio familiar. No existen, por tanto, clínica o biológica y su hallazgo es siempre resultado de un estudio familiar. No existen, por tanto, alteraciones del VCM ni de la concentración de HbA2 y el patrón electroforético de hemoglobinas es normal.
� Talasemia intermedia. Cursa con un síndrome hemolítico crónico con anemia moderada o intensa, palidez, ictericia intermitente, esplenomegalia y ocasionalmente alteraciones óseas no asociadas a retraso del crecimiento corporal o gonadal. Prácticamente nunca requiere transfusiones.

Β –TALASEMIA
Diagnostico
� A. Por hemograma� Anemia microcítica e hipocrómica� En la β-talasemia menor, los datos habituales son� 1 Anemia discreta, Hb ligeramente baja, el recuento de
hematíes poco disminuido, incluso puede estar normal ó ligeramente elevado lo que representa una situación particular en hematíes poco disminuido, incluso puede estar normal ó ligeramente elevado lo que representa una situación particular en relación a la microcitosis
� 2 La microcitosis es más uniforme que en la anemia ferropénica, con menor anisocitosis. Esto se refleja en una menor alteración del RDW
� 3 Hay poiquilocitosis, con presencia caracteristica de dianocitos y eliptocitos, también puede haber esquistocitos
� 4 Anemia de carácter regenerativo (con reticulocitos altos)
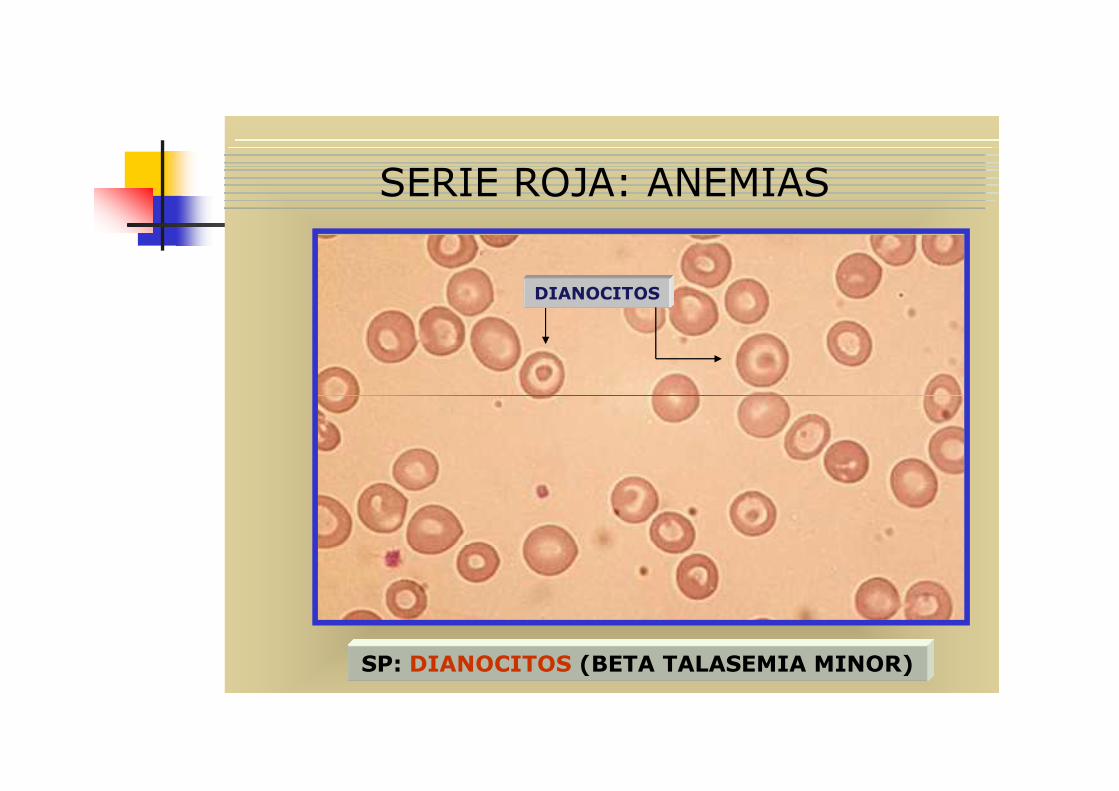
DIANOCITOS
SERIE ROJA: ANEMIAS
SP: DIANOCITOS (BETA TALASEMIA MINOR)

SERIE ROJA: ANEMIAS
SP: ELIPTOCITOS (BETA TALASEMIA MINOR)

SERIE ROJA: ANEMIAS
SP: ESQUISTOCITO (BETA TALASEMIA MINOR)
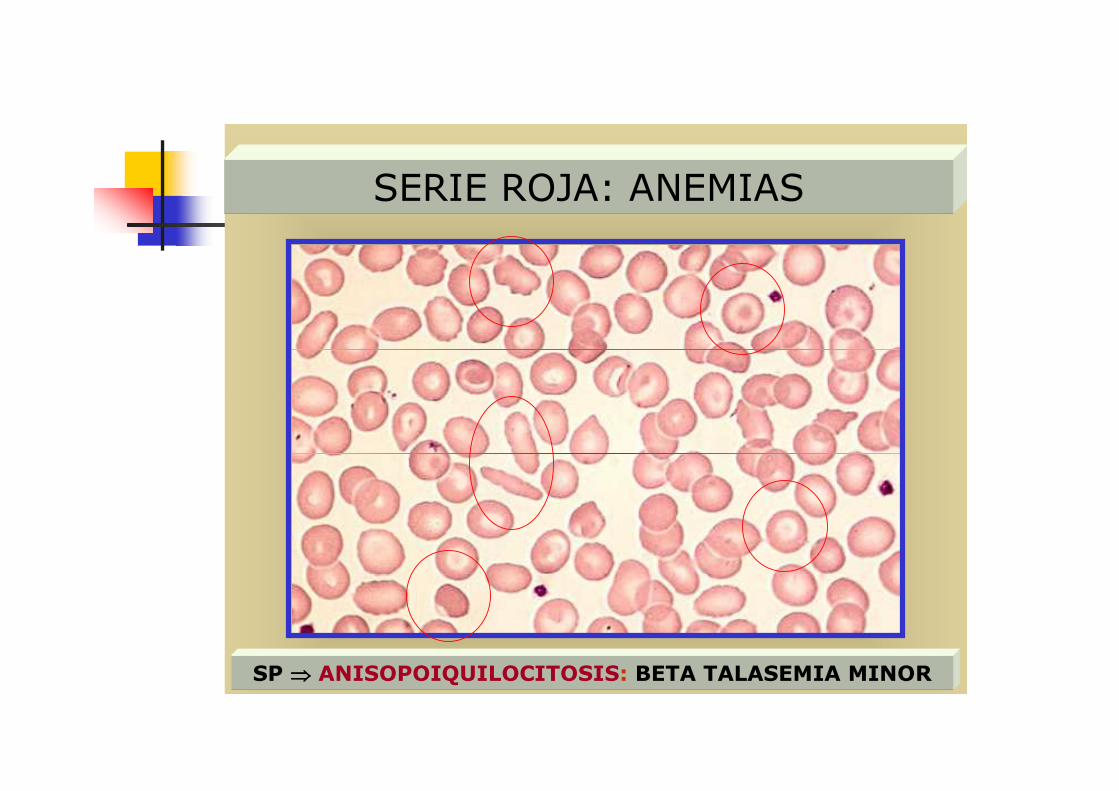
SERIE ROJA: ANEMIAS
SP ⇒⇒⇒⇒ ANISOPOIQUILOCITOSIS: BETA TALASEMIA MINOR

Β –TALASEMIA Diagnostico
� B. Estudio del Hierro� _ Sideremia = Normal o elevada
_ Transferrina= Normal_ IST = Normal o elevado_ Ferritina = Normal o elevada._ IST = Normal o elevado_ Ferritina = Normal o elevada.
Es importante realizar éste estudio y el diagnostico diferencial con la Anemia Ferropénica con el objetivo de evitar un tratamiento inadecuado de administración de hierro a estos pacientes que además de no necesitarlo, pueden padecer una sobrecarga.

Β –TALASEMIA Diagnostico
� C. Electroforesis de la Hb y Cuantificación de las fracciones HbA2 y HbF.:
� En la β-talasemia mayor� ▪ La Hb A está muy disminuida o incluso ausente
▪ La Hb F está aumentada (podría suponer mas de un 50 %)▪ La Hb F está aumentada (podría suponer mas de un 50 %)▪ La Hb A2 esta aumentada, más de un 3.5 %
� En la β-talasemia menor� ▪ La Hb A está algo disminuída
▪ La Hb F esta normal.▪ La Hb A2 está aumentada más de un 3.5 % (3,8%-7%)� Cuando existe también un aumento de HbF (> 2%), los
criterios clínicos permiten orientar el diagnóstico hacia otras formas de ß -talasemia, especialmente la intermedia

Β –TALASEMIA Tratamiento
� En la β-talasemia mayor� 1.- Régimen de transfusiones de concentrados de hematíes periódicamente� 2.- Esplenectomia� 3.- Tratamiento de la sobrecarga de hierro “desferoxamina”, � 4.- Eritropoyetina: el efecto de concentraciones aun más elevadas de rHu EPO
produce una mejora de la eritropoyesis y disminuye la intensidad de la anemiaproduce una mejora de la eritropoyesis y disminuye la intensidad de la anemia� 5.- Trasplante de MO, el cual permite prescindir de la transfusión periódica� 6.- Medicina preventiva
� Diagnostico sistemático de los Portadores heterocigotos para prevenir la transmisión de la enfermedad. Se hace con pruebas de laboratorio, con estudios familiares y con diagnostico genético mediante análisis del ADN
� Diagnostico prenatal, mediante el análisis del ADN en micro muestras fetales
� Diagnóstico pre-implantacional y selección de embriones libres de la enfermedad
� Se ha realizado recientemente Diagnóstico pre-implantacional con una selección de embrión libre de la enfermedad y además Histocompatible para realizar un TMO a un hermano con beta talasemia,

Diagnostico sistemático de los Portadoresheterocigotos
� ¿Por qué es importante que Ud. sepa que es portador de Beta-Talasemia?
� La Talasemia menor está presente desde el nacimiento, permanece igual toda la vida, y puede nacimiento, permanece igual toda la vida, y puede transmitirse de padres a hijos. Esto significa que es hereditaria.Por lo tanto, es muy importante saber si somos o no portadores porque 2 personas con talasemia menor pueden tener hijos con Talasemia Mayor, una enfermedad grave
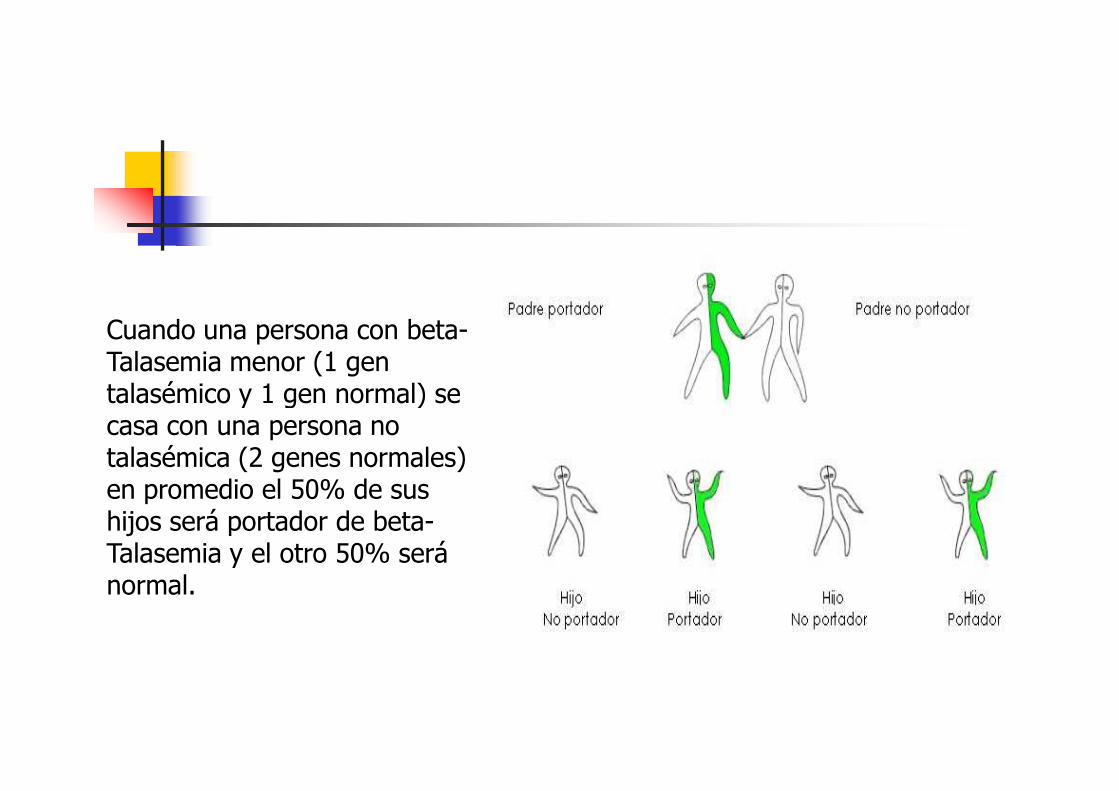
Cuando una persona con beta-Talasemia menor (1 gen talasémico y 1 gen normal) se talasémico y 1 gen normal) se casa con una persona no talasémica (2 genes normales) en promedio el 50% de sus hijos será portador de beta-Talasemia y el otro 50% será normal.

En cambio, si ambos son portadores de beta-Talasemia, las posibilidades de herencia en cada de herencia en cada embarazo, son las siguientes:1.- hijos absolutamente normales (los 2 genes sanos) el 25%2. hijos portadores de beta-Talasemia (1 gen sano y 1 gen talasémico) el 50%3. hijos con beta-Talasemia Mayor (2 genes talasémicos) el 25%

Β –TALASEMIA Tratamiento
� En la β-talasemia menor� 1.- Con un régimen de vida adecuado no suelen
presentar problemas médicos o quirúrgicos
2.- En situación de descompensación debe haber una � 2.- En situación de descompensación debe haber una vigilancia de la anemia y de las crisis hemolíticas
� La supervivencia es prácticamente normal.

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES
� Son anemias hemolíticas adquiridas, producidas por algún factor del plasma o del sistema vascular que provoca la o del sistema vascular que provoca la hemólisis
� Se dividen en dos grupos
■ INMUNES■ NO INMUNES
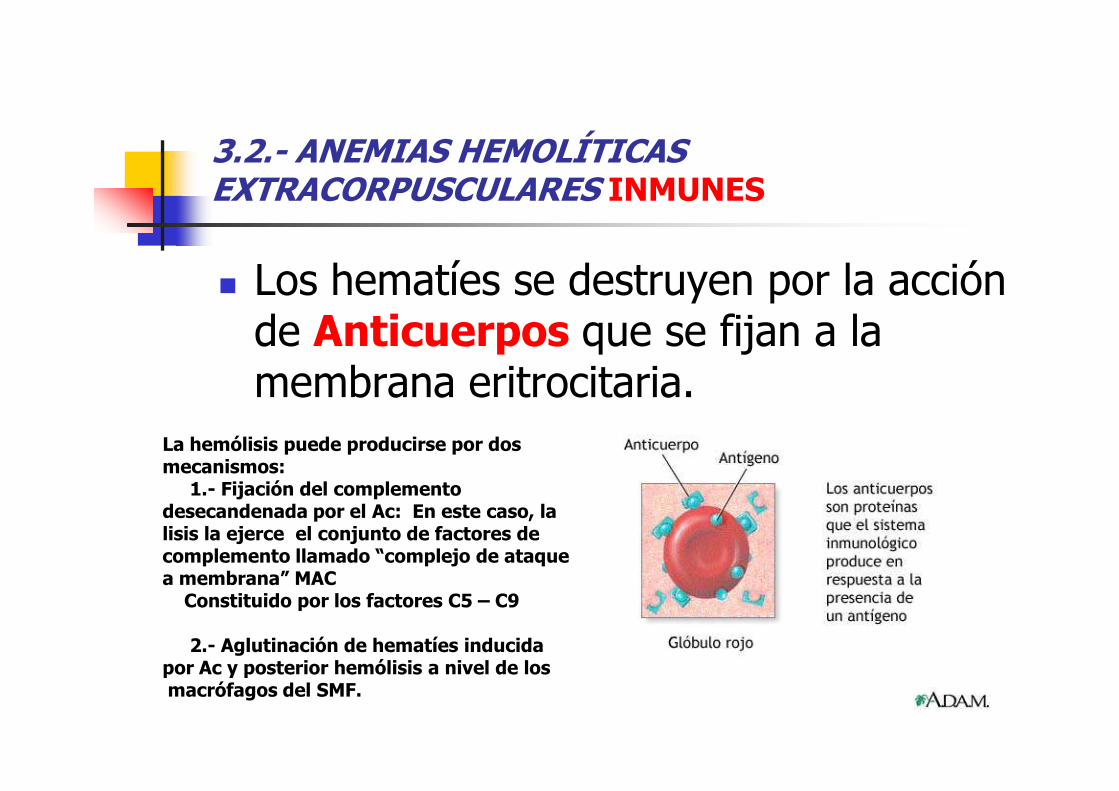
3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES INMUNES
� Los hematíes se destruyen por la acción de Anticuerpos que se fijan a la membrana eritrocitaria. membrana eritrocitaria.
La hemólisis puede producirse por dos mecanismos:
1.- Fijación del complemento desecandenada por el Ac: En este caso, la lisis la ejerce el conjunto de factores de complemento llamado “complejo de ataque a membrana” MAC
Constituido por los factores C5 – C9
2.- Aglutinación de hematíes inducida por Ac y posterior hemólisis a nivel de los macrófagos del SMF.

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES INMUNES
� ▪ Isoanticuerpos Los hematíes son destruidos por Ac procedentes de otra persona de la misma especie. � se incluyen dos patologías:
� La reacción hemolítica postransfusional: La crisis hemolítica es predominantemente intravascular crisis hemolítica es predominantemente intravascular por incompatibilidad de grupo, de manera que los hematíes del donante son destruidos por Ac presentes en el receptor.
� Enfermedad hemolítica del recién nacido: En este caso hay hemólisis de los hematíes fetales por Ac maternos que atraviesan la placenta hasta la circulaciónfetal. ( Se estudia en la Patología Inmunológica EHRN)

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES INMUNES
� ▪ Autoanticuerpos: AHAI Anemia Hemolítica Autoinmunitaria
� Los hematíes son hemolizados porque existe una respuesta inmune anómala existe una respuesta inmune anómala que provoca la síntesis de Anticuerpos contra los propios hematíes
� Se pueden clasificar atendiendo a dos criterios

AHAI Anemia Hemolítica Autoinmunitaria
� 1.- Respecto a la etiología� Anemia Hemolítica Secundaria a una enfermedad
subyacente o a medicamentos� ▪ A infecciones: Determinadas enfermedades infecciosas
cursan con esta complicación hemolítica, destacan, la mononucleosis infecciosa, producida por el virus de mononucleosis infecciosa, producida por el virus de Epstein Barr y la neumonía por mycoplasma pneumoniae.▪ A Neoplasias: (Cáncer)▪ A hemopatías malignas (leucemias, linfomas…)▪ A enfermedades del colágeno, como el lupus ( LED o LES:
Lupus eritomatoso sistémico o diseminado).▪ A Otros factores: medicamentos. Algunos medicamentos
desencadenan la producción de anticuerpos contra antígenos de los hematíes Ej alfa metildopa (anti- hipertensivo), y otros se fijan a la MB eritrocitaria y se comportan como antígenos (haptenos) contra los que actúan autoanticuerpos produciendo hemólisis.Ej. antibióticos (cefalosporina, tetraciclina..), anti-inflamatorios(diclofenac.) etc

AHAI Anemia Hemolítica Autoinmunitaria
� Anemia Hemolítica Primarias ó Idiopaticas: No se puede identificar la causa, se desconoce la causa.. La Anemia Hemolítica Autoinmunitaria Idiopática es responsable de la mitad de todos Idiopática es responsable de la mitad de todos los casos de anemias hemolíticas inmunitarias. El comienzo de la enfermedad puede ser muy rápido y muy serio

AHAI Anemia Hemolítica Autoinmunitaria
� 2.- Respecto al tipo de Ac: Se distinguen dos clases
� Por Crioaglutininas (Ac Fríos): Son de clase IgM y producen hemólisis en temperaturas inferiores a 32 º C. En este caso, se puede hacer una determinación del titulo de aglutininasPor Ac calientes: Son de clase IgG y producen hemólisis a la � Por Ac calientes: Son de clase IgG y producen hemólisis a la temperatura corporal (37 º C). Son mas frecuentes. � Se utiliza como prueba diagnóstica un test de Coombs
directo ó Prueba de antiglobulina directa, en la cual se añade el suero de Coombs, para lograr que se forme el aglutinado mediante los anticuerpos anti-inmunoglobulina (Ac anti-anticuerpo) del suero de Coombs que se unen a los anticuerpos patológicos fijados a los hematíes.

AHAI Anemia Hemolítica Autoinmunitaria Diagnóstico AHAI
� Diagnóstico AHAI Específicamente:� La prueba de Coombs directa es
positiva. Se utiliza para detectar autoanticuerpos en la superficie de los autoanticuerpos en la superficie de los glóbulos rojos. Una prueba de Coombs directa anormal (positiva) significa que la persona tiene anticuerpos que actúan contra sus glóbulos rojos
� En todas las AHAI, es característica la presencia de esferocitos


AHAI Anemia Hemolítica Autoinmunitaria
MECANISMO DE FORMACIÓN DE ESFEROCITOS EN LA AHAI

AHAI Anemia Hemolítica Autoinmunitaria Diagnóstico AHAI
� Además:
� El conteo de glóbulos rojos y de hemoglobina sérica es baja= Anemia
� Los niveles de bilirrubina están elevados
La haptoglobina sérica es baja A. Hemolítica� La haptoglobina sérica es baja A. Hemolítica� Se presenta hemoglobina en orina
� El conteo de reticulocitos es elevado= A. Regenerativa

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES NO INMUNES
� En todas ellas hay una hemólisis traumática por algún factor bien sea, físico, químico, biológico, etc… presente en el plasma o en el sistema vascular
En todas es frecuente la presencia de � En todas es frecuente la presencia de esquistocitos.
ESQUISTOCITO: ERITROCITO FRAGMENTADO

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES NO INMUNES
ESQUISTOCITOS
SERIE ROJA: ANEMIAS
SP: ERITROCITOS FRAGMENTADOS (ANEMIA HEMOLÍTICA MICROANGIOPÁTICA)

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES NO INMUNES
� Mecánicas: Se produce una alteración traumática de los hematíes al circular por un sistema cardiovascular adverso, ya que el corazón o los vasos presentan alteraciones
� 1.- Macroangiopatias: Que son lesiones a nivel del corazón y los grandes vasos, por ejemplo en prótesis de válvulas cardiacas, válvulas alteradas, injertos vascularescardiacas, válvulas alteradas, injertos vasculares
� 2.- Microangiopatias: Que son lesiones a nivel de pequeños vasos. Los hematíes se fragmentan al circular a través de pequeños vasos cuyo endotelio está alterado. Por ejemplo en la Hipertensión arterial, Diabetes Mellitus, Coagulación intravascular diseminada CID, Síndrome hemolítico- uremico.

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES NO INMUNES
� Infecciosas: Se deben a la acción de microorganismos o sus toxinas, como por ejemplo:_ El plasmodium (agente productor de la malaria), es un protozoo_Bacterias que producen toxinas que se denominan hemolisinaspor su acción hemolítica: Clotridium, estafilococos, estreptococos, E. Coli etc...E. Coli etc...
� Tóxicas: Se debe a hemólisis producidas por sustancias químicas como el plomo, el benzol, o algunos medicamentos.
� Secuestro en SRE (Sistema Reticulo Endotelial) : Equivale al SMF, y se refiere a la situación de hiperesplenismo que se produce cuando hay esplenomegalia, de tal manera que el bazo cuando aumenta de tamaño capta una mayor cantidad de hematíes y causa hemólisis de hematíes normales en los macrófagos, por ejemplo en una enfermedad hepática crónica. La hemólisis desaparece al tratar el proceso de base.

3.2.- ANEMIAS HEMOLÍTICAS EXTRACORPUSCULARES NO INMUNES
TROFOZOITOS
PALUSDISMO GRAVE POR PLASMODIUM FALCIPARUM



















