
Tesis Doctoral VALORIZACIÓN DE BIOMASA DE ORIGEN …
288
UNIVERSIDAD DE CASTILLA-LA MANCHA FACULTAD DE CIENCIAS Y TECNOLOGÍAS QUÍMICAS DEPARTAMENTO DE INGENIERÍA QUÍMICA Tesis Doctoral VALORIZACIÓN DE BIOMASA DE ORIGEN VEGETAL MEDIANTE PROCESOS TÉRMICOS Y TERMOQUÍMICOS DIEGO LÓPEZ GONZÁLEZ Ciudad Real, 2013
Transcript of Tesis Doctoral VALORIZACIÓN DE BIOMASA DE ORIGEN …

UNIVERSIDAD DE CASTILLA-LA MANCHA
FACULTAD DE CIENCIAS Y TECNOLOGÍAS QUÍMICAS
DEPARTAMENTO DE INGENIERÍA QUÍMICA
Tesis Doctoral
VALORIZACIÓN DE BIOMASA DE ORIGEN VEGETAL
MEDIANTE PROCESOS TÉRMICOS Y TERMOQUÍMICOS
DIEGO LÓPEZ GONZÁLEZ
Ciudad Real, 2013


UNIVERSIDAD DE CASTILLA-LA MANCHA
FACULTAD DE CIENCIAS Y TECNOLOGÍAS QUÍMICAS
DEPARTAMENTO DE INGENIERÍA QUÍMICA
VALORIZACIÓN DE BIOMASA DE ORIGEN VEGETAL
MEDIANTE PROCESOS TÉRMICOS Y TERMOQUÍMICOS
Memoria que para optar al grado de Doctor en Ingeniería Química
presenta
DIEGO LÓPEZ GONZÁLEZ
Directores: Dr. José Luis Valverde Palomino Dra. María Luz Sánchez Silva Composición del tribunal: Dra. Paula Sánchez Paredes Dra. Mª Pilar Coca Llanos Dr. Javier Dufour Andía Profesores que han emitido informes favorable de la tesis: Dr. Fernando Dorado Fernández Dra. Antonio Monzón Bescós
Ciudad Real, Julio de 2013


D. José Luis Valverde Palomino, Catedrático de Ingeniería Química de la
Universidad de Castilla-La Mancha, y Dª. María Luz Sánchez Silva, Profesor Titular
de Ingeniería Química de la Universidad de Castilla- La Mancha,
CERTIFICAN: Que el presente trabajo de investigación titulado: “Valorización e
biomasa de origen vegetal mediante procesos térmicos y termoquímicos”, constituye
la memoria que presenta D. Diego López González para aspirar al grado de Doctor en
Ingeniería Química y que ha sido realizada en los laboratorios del Departamento de
Ingeniería Química de la Universidad de Castilla-La Mancha bajo su supervisión.
Y para que conste a efectos oportunos, firman el presente certificado
En Ciudad Real a 2 de Julio de 2013
José Luis Valverde Palomino María Luz Sánchez Silva


vii
TABLE OF CONTENTS
Descripción del trabajo realizado 1
A. INTRODUCCIÓN 2
A.1. Cambio Climático y Sostenibilidad 2
A.2. Biomasa 3
A.3. Tipos de Biomasa 6
A.4. Aprovechamiento energético de la biomasa......................... 15
A.5. Energía Termosolar............................................................... 27
A.6. Objetivo del trabajo............................................................. 32
B. MATERIALES Y MÉTODOS.......................................................... 34
B.1. Reactivos empleados........................................................ 34
B.2. Instalación experimentales.................................................... 35
B.2.1. Instalación para el estudio termoquímico de biomasa 35
B.2.2. Instalación para el estudio de degradación de fluidos
de intercambio de calor.................................................... 35
B.3. Técnicas de caracterización............................................. 36
B.3.1. Específicas de biomasa......................................... 36
B.3.2. Caracterización de fluidos de intercambio de calor 38
C. DISCUSIÓN DE RESULTADOS...................................................... 41
D. CONCLUSIONES Y RECOMENDACIONES............................... 48
E. BIBLIOGRAFÍA......................................................................... 52
Abstract..................................................................................................... 58
CHAPTER 1: PYROLYSIS, COMBUSTION AND GASIFICATION CHARACTERISTICS OF NANNOCHLOROPSISGADITANAMICROALGAE
60

Table of contents
viii
1.1. Introduction 63
1.2. Experimental 66
1.3. Results and discussion 73
1.3.1. Pyrolysis of the NG microalgae 73
1.3.2. Combustion of the NG microalgae 78
1.3.3. Gasification of the NG microalgae 83
1.4. Conclusions 96
1.5. References 96
CHAPTER 2: THERMOGRAVIMETRIC-MASS SPECTOMETRIC ANALYSIS OF LIGNOCELLULOSIC AND MARINE BIOMASS PYROLYSIS
101
2.1. Introduction 102
2.2. Experimental 105
2.3. Results and discussion 112
2.3.1. Thermogravimetric study of pyrolysis of lignocellulosic and marine biomass
112
2.3.2. Effect of heating rate 117
2.3.3. Gas products analysis 122
2.4. Conclusions 134
2.5. References 134
CHAPTER 3: THERMOGRAVIMETRIC-MASS SPECTROMETRIC STUDY ON COMBISTION OF LIGNOCELLULOSIC AND MARINE BIOMAS
135
3.1. Introduction 136
3.2. Experimental 138
3.3. Results and discussion 144
3.3.1.Combustion of lignocellulosic biomass 144
3.3.2. Combustion of marine biomass 167
3.3.3. Combustion of Canadian biomass 184
3.4. Conclusions 199

Table of contents
ix
3.5. References 200
CHAPTER 4: 207
4.1. Introduction 209
4.2. Experimental 211
4.3. Results and discussion 215
4.3.1. Thermogravimetric analysis 215
4.3.2. Gasification kinetic analyses 223
4.3.3. Gas evolution analyses 229
4.4. Conclusions 233
4.5. References 234
CHAPTER 5: CHARACTERIZATION OF DIFFERENT HEAT TRANSFER FLUIDS AND DEGRADATION STUDY BY USING A PILOT PLANT DEVICE OPERATING AT REAL CONDITIONS
238
5.1. Introduction 239
5.2. Experimental 243
5.3. Results and discussion 252
5.3.1. Heat transfer fluids characterization for their use as thermal fluids in parabolic trough plants
252
5.3.2. Pilot plant assembly and tuning 261
5.3.3. Model validation 268
5.4. Conclusions 270
5.5. References 271
CHAPTER 6: General Conclusions and Recommendations 273
6.1. CONCLUSIONS 273
6.2. RECOMMENDATIONS 276
LIST OF PUBLICATIONS AND CONFERENCES 278

Descripción del trabajo realizado
1
DESCRIPCIÓN DEL TRABAJO
REALIZADO
Este trabajo da comienzo a una línea de investigación centrada en el desarrollo de
nuevas tecnologías para la valoración integral de biomasa en el Departamento de
Ingeniería Química de la Universidad de Castilla-La Mancha.
En particular, esta Tesis Doctoral tiene como objetivo la evaluación de los
principales procesos de conversión termoquímica de biomasa, principalmente pirólisis,
combustión y gasificación, mediante el sistema experimental de termobalanza
acoplada a un espectrómetro de masas. Adicionalmente, se estudió la degradación de
fluidos de intercambio de calor en su aplicación en plantas termosolares de
concentración. Este trabajo se encuadra dentro del proyecto CENIT VIDA basado en
un consorcio de colaboración de instituciones públicas (ministerio de economía y
competitividad) y privadas para el desarrollo de un nuevo concepto de BIO ciudad
basada en el aprovechamiento de biomasa. Concretamente, este proyecto ha recibido
la financiación de la empresa CT Ingenieros. Por otro lado, una parte de esta tesis ha
sido realizada en colaboración con el instituto de investigación canadiense

Descripción del trabajo realizado
2
IRDA(Institut de recherche et de developpement en agroenvironnement) y el centro de
investigación francés CNRS (Centre national de la recherchescientifique).
A. INTRODUCCIÓN
A.1. Cambio Climático y Sostenibilidad
La demanda energética se ha incrementado exponencialmente en los últimos años
debido al crecimiento de la población mundial. Este hecho, junto con el agotamiento
de recursos fósiles y el auge de la conciencia global sobre la degradación del medio
ambiente son las principales razones que se proponen para realizar un cambio hacia
una sostenibilidad global.
El desarrollo sostenible se define como el “desarrollo que satisface las necesidades
del presente sin comprometer la capacidad de las generaciones futuras para atender
sus propias necesidades”, este cambio debe producirseen base a tres pilares
fundamentales: eficiencia energética, dependencia energética y razones
medioambientales.
La eficiencia energética supone la mejora de los procesos energéticos actuales en
términos de ahorro y desarrollo de nuevas tecnologías. Respecto la dependencia
energética, la fuerte dependencia de nuestra sociedad de las fuentes de energía de
origen fósil no renovable (petóleo, carbón y gas natural, principalmente) derivan en un
continuo agotamiento de las mismas. Además, los yacimientos de origen fósil se
encuentran concentrados en pocas regiosnes, lo que facilita las presiones políticas por
parte de los países productores. En la Figura A.1 se representa el consumo de energía
primaria en España (a) y mundial (b), para el año 2011 observándose que para ambos
casos alrededor del 85% de los recursos energéticos que se utilizan son de origen fósil:
petróleo, carbón y gas natural [1]. Por último, el deterioro del medioambiente debido
el aumento rápido e importante de las concentraciones de gases de efecto invernadero
(GEIs) siendo consecuencia del uso masivo e incontrolado de combustibles fósiles
desde la época industrial hasta la actualidad.

Descripción del trabajo realizado
3
Las energías renovables, por su carácter autóctono, contribuyen a disminuir la
dependencia de los suministros externos, aminoran el riesgo de un abastecimiento
poco diversificado, favorecen el desarrollo tecnológico y la creación de empleo y
tienen un menor impacto medioambiental [1]. La utilización de tecnologías de
energías renovables como la eólica, la geotérmica, la hidráulica, la solar y la obtenida
a partir de la biomasa se presentan como alternativas para el reemplazo de los
combustibles fósiles. El presente trabajo está dedicado a dos de ellas: los procesos de
conversión de biomasa en energía y la mejora en la eficiencia de las plantas
termosolares.
Figura 2.1.Consumo de energía primaria (a) mundial y en (b) en España expresado en millones de toneladas equivalentes de petróleo [Mtep], año 2011 [1].
A.2. Biomasa
La biomasa ha sido desde siempre la mayor fuente de energía para el ser humano y
se ha estimado que actualmente contribuye un 14% al abastecimiento de la energía
mundial [2]. Una de las razones de que la biomasa haya tomado tanta importancia en
los últimos años es la elevada disponibilidad de la misma, estimándose en
aproximadamente 220 billones de toneladas secadas al año [3].
La biomasa se puede definir como “toda sustancia orgánica de origen vegetal o
animal que puede ser convertida en energía”[4]. Sin embargo, esta definición resulta
incompleta ya que estamos hablando de un vector energético que, a corto plazo, puede
ser básico en nuestra sociedad. Este término hace referencia a toda materia orgánica
Petróleo, 69,5
Gas natural, 28,9
Carbón, 14,9
Nuclear, 13
Hidráulica, 6,9Renovables,
12,7
CONSUMO DE ENERGÍA PRIMARIA EN ESPAÑA AÑO 2011, [Mtep]
Petróleo, 4059,1
Gas natural, 2905,6
Carbón, 3724,3
Nuclear, 599,3
Hidráulica, 791,5
Renovables, 194,8
CONSUMO DE ENERGÍA PRIMARIA MUNDIAL AÑO 2011, [Mtep]
a) b)

Descripción del trabajo realizado
4
originada de forma inmediata en un proceso biológico, espontáneo o provocado,
utilizable como fuente de energía[1]. La biomasa abarca un amplio rango de materias
orgánicas que se caracterizan por su heterogeneidad.
A.2.1. Aplicaciones de la biomasa.
La existencia de diferentes tipos de biomasa y métodos de transformación de la
misma, permite utilizarla como combustible para producción de energía térmica y
eléctrica, o como materia prima para la producción de biocombustibles líquidos y
gaseosos.
• Producción de Energía Térmica. Este tipo de energía se obtiene
principalmente de la combustión directa de residuos forestales, agrícolas, de
industrias transformadoras de la madera y algunos agroalimentarios (orujillo de
aceituna, orujo lavado de uva, cáscara de almendra, etc.). En el proceso se genera
calor, tanto para su uso doméstico como industrial.
• Producción de Energía Eléctrica. Este tipo de energía también se obtiene
por la combustión principalmente de diferentes tipos de residuos como los
utilizados para la producción de energía térmica, pero también de los cultivos
energéticos y del biogás procedente de la digestión anaerobia de algunos residuos.
El rendimiento de las plantas que emplean biomasa para producción de energía
eléctrica no es muy elevado debido al elevado porcentaje de humedad que presenta
la biomasa.
• Producción de Biocombustibles Líquidos. La producción de
biocombustibles líquidos que suplan a los derivados del petróleo (gasolina y diesel)
es una opción muy ventajosa en cuanto al empleo de energías renovables y
reducción de problemas medioambientales. Existen dos tipos de biocombustibles
líquidos: los bioalcoholes (bioetanol) que se obtienen a partir de la fermentación
mediante levaduras de materiales azucarados como caña de azúcar, remolacha,
maíz, etc., y los biogasóleos (biodiesel) que se obtienen del proceso de
transesterificación de materiales oleaginosos como girasol, colza, etc., o bien de
grasas animales.

Descripción del trabajo realizado
5
• Producción de Biocombustibles Gaseosos. La producción de
biocombustibles gaseosos a partir de procesos biológicos anaerobios es una opción
que presenta muchos beneficios. Este gas obtenido está formado principalmente
por metano, y aunque tiene bajo poder calorífico puede utilizarse en las propias
instalaciones donde es generado para producir tanto electricidad como calor. La
gasificación también conlleva la producción de un gas combustible rico en
hidrógeno y sobre todo en carbono.
A.2.2. Características de la biomasa para su aprovechamiento energético.
Son las propiedades inherentes de la biomasa las que van a determinar el proceso
de conversión y las consecuentes dificultades de proceso que puedan surgir[4]. Para
procesos de conversión de biomasa seca las propiedades más importantes son:
• Humedad:
Para la conversión térmica de biomasa son de interés aquellas biomasas que posean
baja humedad.Se pueden considerar dos formas de humedad en biomasa: humedad
intínseca, referida al contenido de humedad de la biomasa sin la influencia de los
efectos de la climatología, y humedad extrínseca, contenido de humedad de la biomasa
debido a los efectos de la climatología.Los procesos de conversión termoquímica
requieren materias primas con un contenido bajo de humedad (< 50%). Se podrían
usar materiales con mayor humedad pero el balance energético global para el proceso
de conversión se ve perjudicado por los procesos de secado.
• Poder calorífico (Energía/Masa) (MJ/kg):
El poder calorífico de un material es una expresión del contenido energético liberado
cuando el mismo se quema en aire.Se puede expresar de dos formas:
- HHV ( Higher heating value): Energía total liberada cuando el combustible es
quemado, incluyendo la del calor latente contenido en el vapor de agua y, por
tanto, representa la cantidad máxima de energía potencialmente recuperable dada
una fuente de biomasa determinada.

Descripción del trabajo realizado
6
- LHV ( Lower Heating Value): Contenido energético sin contar el calor latente
contenido en el vapor de agua.Se puede decir que el calor latente contenido en el
vapor de agua no puede ser usado efectivamente y, por lo tanto, el LHV será el
valor apropiado para considerar la potencialidad de uso de la biomasa como
combustible.
• Proporción de carbón fijo y volátiles:
Este parámetro da una medida de cómo de fácil una biomasa determinada puede
ser inflamada y, consecuentemente, gasificada u oxidada.
- Contenido en volátiles: Porción liberada de gas mediante calentamiento (950
ºC durante 7 min).
- Carbón fijo: Es la masa que queda después de la liberación de los volátiles,
excluyendo la ceniza y humedad.
• Contenido de Ceniza/Residuo:
La rotura de los enlaces de la biomasa por procesos termoquímicos o bioquímicos
produce un residuo sólido.El contenido de ceniza puede afectar al manejo y a los
costes de proceso. En los procesos termoquímicos, la magnitud del contenido en
ceniza afecta a la cantidad de energía disponible en el combustible.
• Contenido en metales alcalinos:
Los principales metales alcalinos contenidos en la biomasa son Na, K, Mg, P y Ca.
El contenido de estos metales en la biomasa es un parámetro importante ya que
estos metales pueden catalizar/inhibir los procesos dde conversión de biomasa en
energía. Además. pueden reaccionar con los componentes de la ceniza produciendo
compuestos que pueden producir bloqueos en los equipos.
A.3.Tipos de biomasa
A.3.1. Clasificación de biomasa
La clasificación de la biomasa más ampliamente aceptada responde a su origen:

Descripción del trabajo realizado
7
• Biomasa natural. Es la biomasa que se produce espontáneamente en la
naturaleza sin ningún tipo de intervención humana (recursos generados en las
podas naturales de un bosque).
• Biomasa residual. Es la biomasa que genera cualquier actividad humana. Se
distingue entre biomasa residual seca (aquella que procede de recursos generados
en las actividades agrícolas y forestales, en las industrias agroalimentarias y en las
industrias de transformación de la madera) y biomasa residual húmeda, como son
los vertidos biodegradables formados por aguas residuales urbanas e industriales,
los residuos ganaderos (generalmente purines) y también se incluyen los residuos
sólidos urbanos (materiales biodegradables sobrantes del ciclo de consumo
humano).
• Cultivos específicos (energéticos). Son cultivos realizados en terrenos
agrícolas y forestales dedicados exclusivamente a la producción de biomasa con
fines no alimentarios, sino únicamente energéticos (cardo, girasol, caña de azúcar,
maíz, remolacha, etc.). Éstos se dividen en leñosos y herbáceos.
• Biomasa marina. Como pueden ser algas, hierbajos marinos, juncos, etc.
El mayor punto de controversia encontrado en el uso de biomasa como fuente de
energía primaria reside principalmente en la competitividad con el abastecimiento de
humano comida. En este trabajo, se discutirá la conversión de biomasa lignocelulósica
y marina principalmente
A.3.2. Biomasa lignocelulósica
Una parte importante de la biomasa es lignocelulósica, siendo la celulosa, la
hemicelulosa y la lignina sus tres componentes principales. A diferencia de los
hidratos de carbono o el almidón, la lignocelulosa no es fácilmente digerible por los
seres humanos. Por ejemplo, se puede comer el arroz, que es un hidrato de carbono,
pero no podemos digerir la paja, que es lignocelulosa. La biomasa lignocelulósica no
forma parte de la cadena alimentaria humana y, por lo tanto, su uso para la obtención

Descripción del trabajo realizado
8
de biogás y de energía, no suponen una amenaza para el suministro mundial de
alimentos[5].
La celulosa no es un material fácilmente accesible como es el almidón o el azúcar,
al encontrarse íntimamente unida a otros materiales como son la lignina o las
sustancias pécticas.Las paredes lignocelulósicas son estructuras complejas y de difícil
accesibilidad para algunos componentes (Figura A.2). El material lignocelulósico está
constituido por tejidos vegetales que presentan una pared celular constituida por un
entramado de microfribillas de celulosa sobre las que se forman capas de
hemicelulosas y, posteriormente, se deposita la lignina.
En este sentido, el aprovechamiento global del material requiere métodos de
pretratamiento o fraccionamiento. Estos procesos son complejos y están alejados de
rendimientos elevados. Además, no son capaces de aislar completamente cada
componente sin modificarlo o degradarlo.
Para comprender qué es un material lignocelulósico y poder aprovecharlo
completamente, se deben conocer cuáles son los componentes principales de las
paredes celulares, cómo se distribuyen en la propia pared y qué tejidos las contienen.
Figura A.2..Matriz lignocelulósica[6].

Descripción del trabajo realizado
9
• Composición de la biomasa lignocelulósica.
Los componentes de los materiales lignocelulósicos se clasifican en estructurales y
secundarios.
Los componentes estructurales los forman tres polímeros, la celulosa, la lignina y
la hemicelulosa. Del total de compuestos que forman los materiales lignocelulósicos,
casi la mitad son celulosa y un 20% lignina.La unión entre celulosa y lignina puede
producirse directamente o generalmente a través de las hemicelulosas, como se
observa en la Figura A.2. En las paredes no lignocelulósicas aparece otro componente
formado por sustancias pécticas (pectina). En general, se puede establecer que entre un
60 y un 80% de los vegetales están constituidos por polisacáridos de elevado peso
molecular como son las holocelulosas. Entre las holocelulosas podemos distinguir
entre unos polímeros lineales de alto grado de polimerización, la celulosa y otros que
resultan fácilmente extraíbles en álcalis, las hemicelulosas.
- Celulosa: es un homopolímero lineal de elevado peso molecular y grado de
polimerización; entre 200 y hasta 10.000 unidades en estado nativo de β-D-
glucopiranosa unidas por enlace glicosídico o de tipo éter entre el carbono 1 y 4
(β,1�4).
En la Figura A.3 se muestra la estructura polimérica de la celulosa de forma
detallada.
Figura A.3. Estructura primaria de la celulosa[5].
- Hemicelulosas: forman cadenas ramificadas de menor grado de
polimerización que la celulosa y no tienen, por tanto, zonas cristalinas. Además, los

Descripción del trabajo realizado
10
puentes de hidrógeno son menos eficaces, haciendo de las hemicelulosas
polisacáridos más accesibles al ataque de reactivos químicos.
El xilano se usa como compuesto representativo de la hemicelulosa por ser uno de
los compuestos principales de la hemicelulosa y se ha demostrado que tiene un
comportamiento térmico parecido(Wang y col., 2008; Yang y col., 2006) En la
Figura 2.5 se muestra la estructura del xilano.
Figura A.4. Estructura molecular del xilano .
- Lignina : es un polímero aromático de estructura tridimensional bastante
compleja, muy remificada y amorfa, formada por la condensación de precursores
fenólicos unidos por diferentes enlaces. En la Figura A.5 se muestran las unidades
estructurales de la lignina.La variedad de enlaces y estructuras del polímero lignina
son debidas a la diversidad de reacciones de acoplamiento (al azar) entre las
distintas formas resonantes de los radicales fenóxido.
Figura A.5. Unidades estructurales de la lignina .
Los componentes secundarios se clasifican en solubles en agua, disolventes
orgánicos e insolubles.
- Solubles en agua y disolvente orgánicos: conocidos como terpenos, son
considerados polímeros del isopreno. Por otro lado, las resinas que contienen una

Descripción del trabajo realizado
11
alta variedad de compuestos no volátiles como son grasas, ácidos grasos, alcoholes,
resinas ácidas, fitoesteroides y otros compuestos neutros. Los fenoles, como los
taninos y también son solubles algunos hidratos de carbono de bajo peso
molecular, alcaloides y lignina soluble.
- Insolubles: dentro de este grupo se encuentran las cenizas, que son
principalmente carbonatos y oxalatos. Otros más raros y de poca proporción, pero
que también pueden ser insolubles, son pequeñas cantidades de almidón, pectinas o
proteínas.
A.3.3. Biomasa marina
Es la biomasa que producen los ecosistemas silvestres que se encuentran en los
océanos y corresponde al 40% de la biomasa que se produce en la Tierra (algas,
hierbajos marinos, juncos, etc.).
Las algas han atraído la atención desde hace mucho tiempo como posible materia
prima para la obtención de bioenergía[7-9], pero también la existencia de algunas
especies ricas en lípidos pueden ser explotadas como una alternativa interesante para
la producción de biodiesel[10; 11]. Son una biomasa muy prometedora por las
siguientes razones: alta velocidad de crecimiento, alto rendimiento por área, alta
eficiencia en la captura de CO2 y en la conversión de energía solar y no compiten con
la agricultura de alimentos. Además, pueden crecer en aguas abiertas (océanos o
estanques) y en bio-fotoreactores de tierra no cultivables [12]. La fijación de CO2 y las
principales etapas de transformación de biomasa marina se ilustran en la Figura A.6.
Las algas, que pertenecen al reino Protoctista y constituyen un grupo de
organismos muy variado y complejo, se encuentran en una amplia variedad de
ecosistemas acuáticos y terrestres gracias a su alta plasticidad y diversidad metabólica
y se pueden clasificar de acuerdo a su tamaño en los siguientes grupos:
• Microalgas: Incluyen todo tipo de microorganismos fotosintéticos,
procariotas o eucariotas, unicelulares o filamentosos, de tamaño inferior a 0,02
cm.

Descripción del trabajo realizado
12
• Mesoalgas: Se trata de microorganismos fotosintéticos, procariotas o
eucariotas, unicelulares o filamentosos, unialgal o plurialgal, cuyo tamaño se
encuentra entre 0,02 y 3 cm.
• Macroalgas: Engloba a algas pluricelulares de diversas formas y tamaños que
van desde los pocos centímetros a varios metros de largo.
Las microalgas han recibido más atención que las macroalgas para la producción
de biofuel, las cuales pueden ser cultivadas en estanques o fotobiorreactores con
suministro de nutrientes o aguas residuales [13; 14].
Figura A.6. Fijación de CO2 y principales etapas de transformación de biomasa marina [15].
Las microalgas contienen en diferentes proporciones proteínas (6-52%),
carbohidratos (5-23%) y lípidos (7-23%) [16]. De acuerdo con Ross y col. (2010)[17],
las microalgas con un alto contenido en lípidos pueden ser una futura fuente de
biocombustibles de tercera generación y productos químicos.
CO2 en atmósfera CO2 en atmósfera H2O
Conversión bioquímica
Sustancias iniciales biofijación
Crecimiento de microalgas
Procesamiento microalgas
Conversión termoquímica
Conversión directa Bioalcoholes
Biodiesel Biogás
Biohidrógeno
Aceite de algas Combustión
Alimentos origen animal Fertilizante
Biofuel Biocrudo Biodiesel
Gas
Luz solar
Organismos fotosintéticos

Descripción del trabajo realizado
13
A.3.4. Selección de la biomasa sometida a estudio
Como se ha comentado anteriormente, este trabajo se centra en el estudio de
biomasa lignocelulósica y marina, especialmente algas. La selección de los diferentes
tipos de biomasa sometidas a estudio se realizó en función de su composición.
• Selección de biomasa lignocelulósica
La elección de la biomasa depende, principalmente, de sus propiedades inherentes,
del proceso de conversión y de las dificultades de procesamiento posteriores que
puedan surgir. Las principales propiedades de interés para el tratamiento de biomasa
como fuente de energía son las siguientes como se comentó anteriormente:contenido
de humedad (MC), porcentaje de carbono fijo (FC) y proporción en volátiles (VM); la
relación cenizas / residuos (AC / AR), valor calorífico, contenido de metal alcalino y
la relación de celulosa / lignina[4].
En este sentido, se clasificaron diferentes especies de biomasa en un diagrama
ternario basándose en sus análisis inmediatos, realizados a partir de los datos
publicados por Yaman (2004) [18]. Se consideraron los siguientes parámetros:
cenizas, materia volátil y el contenido de carbono fijo (Figura A.7).
La biomasa se seleccionó de acuerdo con los siguientes criterios:
-Biomasa con alto contenido de VM y AC bajos.
-Biomasa de alto contenido FC y bajo AC.
De acuerdo con estos criterios, se identificaron dos zonas en el diagrama
claramente diferenciadas (señaladas con un círculo). La biomasa en estas dos zonas
correspondió a: madera de abeto y madera de eucalipto (ambos con elevada
proporción en volátiles) y corteza de pino (con el mayor contenido en carbono fijo).

Descripción del trabajo realizado
14
Figura A.7. Diagrama ternario con diferentes tipos de biomasa terrestre según
su análisis inmediato [19].
• Seleción de biomasa marina (microalgas)
Para la selección de la microalga a utilizar en esta investigación se ha llevado a
cabo un intenso estudio bibliográfico teniendo en cuenta su composición y las
cantidades recomendadas de sus componentes para lograr los productos deseados.
Para ello, se realizó un diagrama ternario con sus tres componentes principales:
proteínas, carbohidratos y lípidos (Figura A.8) en base a los datos publicados por
Brrown y col. (1991) [20] y Renaud y col. (1999) [21].
El criterio que se empleó, se basó en la selección de la biomasa con mayor
contenido en lípidos [10]. Atendiendo a esto, se determinó que las microalgas que
reunían mejores propiedades fueron la Nannochloropsis Gaditana, la Scenedesmus
Almeriensis y la Isochrysis sp. De éstas, se seleccionaron las
microalgasNannochloropsis Gaditana (microalga NG) y Scenedesmus Almeriensis por
0,0 0,2 0,4 0,6 0,8 1,0
0,00,00,00,0
0,20,20,20,2
0,40,40,40,4
0,60,60,60,6
0,80,80,80,8
1,01,01,01,00,0
0,2
0,4
0,6
0,8
1,0
Carbón fij o
Caña de azúcarUvaMaízOlivaColzaCáscara de arrozSerrínGirasolHierbajo marinoJacinto de aguaAbetoTabacoPinoDesechos de algodónEucaliptoPaja
Cen
iza
Volátiles

Descripción del trabajo realizado
15
su fácil disponibilidad y su comercialización en forma de polvo. Adicionalmente, se
seleccionó una especie de microalga con elevado contenido en proteínas a modo
comparativo. La especie de mciroalga con elevado contenido en proteínas elegida fue
la Chlorella Vulgaris.
Figura A.8. Diagrama ternario que representa la composición en proteínas, carbohidratos y lípidos de diferentes especies de microalgas.
A.4.- Aprovechamiento energético de la biomasa
Existen multitud de procesos para el aprovechamiento energético de la biomasa.
En la Figura A.9 se esquematizan los procesos más destacados. Este trabajo está
centrado en el aprovechamiento energético de biomasa mediante procesos de
conversión termoquímica, sin embargo se darán unas breves reseñas de otros procesos
de converión de biomasa.
0,0 0,2 0,4 0,6 0,8 1,0
0,0
0,2
0,4
0,6
0,8
1,00,0
0,2
0,4
0,6
0,8
1,0
Carbohidratos
Scenedesmus quadricauda Scenedesmus dinorphus Chlamydomonas rheinhardii Chlorella vulgaris Chlorella pyrenoidosa Spyroga sp. Dunaliella salina Tetraselmis maculata Porphyridium cruentum Spirulina maxima Synechoccus sp. A. coffeaformes Nitzschia sp. Cryptomonas sp. Rhodomonas sp. Nephroselmis sp. Tetraselmis sp. NT Isochrysis sp. Rhodosorus sp. Tetraselmis sp. TEQL Nannochloropsis gaditana
Prot
eína
s
Lípidos

Descripción del trabajo realizado
16
Figura A.9. Procesos para la conversión energética de biomasa[5].
A.4.1. Procesos de conversión bioquímica.
Consisten en la aplicación de diversos tipos de microorganismos que degradan las
moléculas de biomasa. Se utilizan para la transformación de biomasa húmeda en
compuestos simples de gran contenido energético. Dos de las técnicas más
importantes son:
• Digestión anaerobia:
Es un proceso de fermentación bacteriana en ausencia de oxígeno donde se genera
una mezcla de gases, principalmente metano y dióxido de carbono, conocida como
biogás, y también una suspensión acuosa o lodo que contiene los compuestos no
degradados y los minerales. Se utiliza principalmente para la fermentación de biomasa
húmeda del tipo de residuos ganaderos, aguas residuales urbanas o biomasa marina
húmeda. En este caso se deben controlar una serie de variables como temperatura
(aprox. 35 ºC), acidez (pH 6.6-7.6), contenido en sólidos (< 10%), nutrientes (carbono,
nitrógeno, fósforo, azufre y sales minerales para el crecimiento y la actividad
bacteriana) y compuestos tóxicos (bajas concentraciones de amoníaco, sales
Biomasa
�Bioquímicos
�Termoquímicos
oCombustión
oGasificación
o Pirólisis
oLicuefacción
oTratamientoHidrotérmico
Digestión anaerobiao
Fermentaciónalcohólicao

Descripción del trabajo realizado
17
minerales, detergentes y pesticidas que inhiben la actividad bacteriana). El biogás
puede utilizarse como combustible, mientras que el efluente (lodo) se puede utilizar
para la fertilización de suelos.
Este proceso ocurre en tres etapas consecutivas: hidrólisis, fermentación y
metanogénesis. En la hidrólisis, los compuestos complejos se dividen en azúcares
solubles. En ese momento, las bacterias fermentativas los convierten en alcoholes,
ácido acético, ácidos grasos volátiles y un gas que contiene H2 y CO2, el cual es
metabolizado principalmente en CH4 (60-70%) y CO2 (30-40%) por metanógenos
(Brennan y col., 2010).
• Fermentación alcohólica:
En el proceso de fotosíntesis las plantas almacenan la energía solar aportada en
forma de hidratos de carbono simples (azúcares) o complejos (almidón y celulosa). A
partir de estos hidratos de carbono se puede obtener por fermentación un bioalcohol,
denominado bioetanol, empleando diferentes etapas según el tipo de biomasa a
transformar. Estas etapas son las siguientes:
a) Pretratamiento: transformación de la materia prima para favorecer la
fermentación por medio de la trituración, molienda o pulverización.
b) Hidrólisis: transformación, en medio acuoso, de las moléculas complejas en
hidratos de carbono simples (azúcares) por medio de enzimas (hidrólisis
enzimática) o mediante reactivos químicos (hidrólisis química).
c) Fermentación alcohólica: conversión de los azúcares en bioetanol por la
acción de microorganismos (levaduras) durante dos o tres días bajo condiciones
controladas de temperatura (27-32 ºC), acidez (pH 4-5) y concentración de
azúcares (< 22%)
d) Separación y purificación del bioetanol: destilación de la masa fermentada
para obtener bioetanol comercial del 96% o destilación adicional con un disolvente
(benceno) para obtener un bioetanol absoluto del 99,5%.

Descripción del trabajo realizado
18
El bioetanol es utilizado como combustible alternativo a las gasolinas, o bien
mezclado con ellas, en el campo de la automoción.
A.4.2. Procesos de conversión termoquímica.
Se utilizan para la transformación de biomasa seca, es decir, residuos cuyo
contenido en humedad no es muy elevado (principalmente paja, madera, orujillo,
huesos, cáscaras). Son métodos basados en la utilización del calor como fuente de
transformación de la biomasa donde se distinguen tres tipos de procesos según la
cantidad de oxígeno aportada:
• Pirólisis:
Se puede definir como la degradación de la biomasa mediante calor en ausencia de
oxígeno, resultando la producción de un sólido carbonoso (carbonilla o char),
biocombustible (líquido) y fuel gas [22]. A través de la variación de los parámetros del
proceso de pirólisis es posible influir en la distribución y características de sus
productos.El proceso de pirólisis se puede representar como la siguiente reacción:
��������� � � → � ������ +������� +����í��� !
�"� + ��#ℎ %�
Desde un punto de vista térmico el proceso se puede dividir en cuatro etapas
principalmente:
- Secado (100ºC): Ocurre en la etapa inicial de calentamiento a baja
temperatura, perdiéndose la humedad y, por tanto, el agua que está débilmente
enlazada.
- Etapa inicial (100-300ºC): En esta etapa, se produce la deshidratación
exotérmica de la biomasa, liberándose agua retenida y gases de bajo peso
molecular como el CO y el CO2.
- Etapa Intermedia (>200ºC): Se produce una pirólisis inicial, entre 200 y
600ºC, produciéndose la mayor parte del vapor o precursor de bio-combustibles. Se

Descripción del trabajo realizado
19
comienza a romper las moléculas más grandes, descomponiéndose en el producto
sólido (Primary char), gases condensables (vapor y precursores del producto
líquido) y gases no condensables.
- Etapa Final (≈300-900ºC): La etapa final de pirólisis conlleva el craqueo
secundario de volátiles en producto sólido y gases no condensables. Si el tiempo de
residencia de la biomasa es suficientemente elevado, se puede producir el craqueo
de cadenas de elevado peso molecular en los gases condensables, incrementando el
rendimiento hacia el producto sólido y gases.Esta etapa ocurre principalmente por
encima de 300ºC. Si los gases condensables se retiran rápidamente del lugar de
reacción se produce la condensación hacia bio-combustibles o alquitrán.
• Combustión u oxidación:
La combustión es el proceso más directo para la conversión de biomasa en energía
útil, siendo usado en numerosas aplicaciones. Se basa en la oxidación completa de la
materia orgánica de la biomasa con exceso de oxígeno (cantidad de oxígeno superior a
la estequiométrica) convirtiendo la energía almacenada en calor, energía mecánica o
electricidad[23]. Además de calor, en el proceso se genera dióxido de carbono, agua y
cenizas.
El proceso de combustión se puede representar como la siguiente reacción:
��������� � � +�" → ��" +�"� + ��#ℎ %� + � &�% La ignición de la biomasa requiere elevadas temperaturas (≥ 550 ºC),
constituyendo la etapa más costosa del proceso el comienzo del mismo.
A pesar de su aparente simplicidad, la combustión es un proceso complejo desde
un punto de vista tecnológico, donde tienen lugar elevadas velocidades de reacción y
grandes cantidades de calor liberado. Además de obtenerse muchos productos y
caminos de reacción.
De forma general el proceso de combustión se divide en las siguientes etapas:
- Secado: Evaporación del agua contenida en el combustible.

Descripción del trabajo realizado
20
- Pirólisis y reducción: Descomposición térmica del combustible en volátiles y
un producto sólido (char).
- Combustión de los volátiles: Los productos obtenidos en la etapa anterior
son quemados en presencia de oxígeno.
- Combustión del char: Se produce la combustión del producto sólido.
• Gasificación:
La gasificación es un proceso termoquímico complejo que consiste en un número
de reacciones químicas elementales en presencia de un agente gasificante,
generalmente en atmósfera de aire, pobre de oxígeno (cantidad de oxígeno inferior a la
estequiométrica) o vapor de agua[24].
La importancia de este proceso se puede resumir en los siguientes puntos [5]:
- Incremento del valor calorífico de un combustible a través de la eliminación
de componentes como el nitrógeno y el agua.
- Eliminación de compuestos nocivos para el medioambiente, como pueden ser
los óxidos de nitrógeno y azufre.
- Reducción de la relación C/H del combustible.
- Obtención de productos químicos de gran interés comercial.
- Eliminación del oxígeno que constituye el combustible y, por lo tanto, se
produce un incremento de su densidad energética.
En general, cuanto mayor sea el contenido de hidrógeno de un combustible, menor
será la temperatura de vaporización y mayor la probabilidad de que el combustible
esté en estado gaseoso. La gasificación aumenta el contenido de hidrógeno en el
producto mediante una de las siguientes formas:
- Directa: Exposición directa al hidrógeno a alta presión.

Descripción del trabajo realizado
21
- Indirecta: Exposición al vapor de agua en unas condiciones de temperatura y
presión elevadas, donde el hidrógeno (producto intermedio) se añade al
producto. Este proceso también incluye el reformado con vapor.
Las principales reacciones que ocurren en el proceso de gasificación se describen a
continuación [4] :
C + O" → CO" (Combustión Completa)
C + 1 2+ O" → CO (Combustión Incompleta)
La presencia de agua como agente gasificante, permite aumentar la proporción de
hidrógeno generado de la siguiente forma:
C + H"O → CO + H" (Reacción Water gas)
En presencia de dióxido de carbono, el carbono de la materia orgánica reacciona
para producir monóxido de carbono, según la reacción de Boudouard:
C + CO" → 2CO (Reacción de Boudouard)
También son importantes en el proceso de gasificación las reacciones de
metanización:
� + 2�" → ��- (Reacción de metanización)
�� + 3�"↔��- +�"� (Reacción de metanización)
Al igual que la reacción de water gas, la reacción de water gas shift tiene lugar
cuando existe vapor de agua en el medio de gasificación.
�� + �"� ↔ ��" +�" (Water gas shift reaction)
Las flechas indican que las reacciones están en equilibrio y se pueden producir en
cualquier dirección, dependiendo de la temperatura, presión y concentración de las

Descripción del trabajo realizado
22
especies reaccionantes. De esto se deduce que el gas producto procedente de la
gasificación consiste en una mezcla de monóxido de carbono, dióxido de carbono,
metano, hidrógeno y vapor de agua.
• Otros Procesos:
Otros procesos para el aprovechamiento energético de la biomasa son el
tratamiento hidrotérmico y la licuefacción. El primero convierte la biomasa en una
atmósfera húmeda bajo presiones elevadas en hidrocarburos oxigenados parcialmente,
no obstante, este proceso está todavía en fase de planta piloto.
La licuefacción es la conversión de la biomasa en hidrocarburos líquidos estables
aplicando bajas temperaturas y elevadas presiones de hidrógeno. Este proceso está
atrayendo menos interés que la pirólisis ya que los reactores y los sistemas de
alimentación son más complejos y costosos [4].
A.4.3. Evaluación de procesos de conversión termoquímica mediante
tecnologías de análisis térmico (TA).
• Análisis termogravimétricos.
Durante los procesos de conversión termoquímica se presentan reacciones para las
cuales el estudio cinético resulta muy interesante. En esta tarea, el análisis
termogravimétrico (TGA) (realizado en un equipo denominado termobalanza) supone
una herramienta muy potente y es una de las técnicas más utilizadas a escala
laboratorio [25]. Consiste en medir la masa o el cambio de masa que experimenta una
sustancia en función de la temperatura mientras la muestra se calienta (o se enfría) con
un programa de temperaturas y bajo una atmósfera controlada (Montero, 2011).La
variación de masa puede ser una pérdida o una ganancia de la misma. El registro de
estos cambios nos dará información sobre si la muestra se descompone o reacciona
con otros componentes. La principal ventaja del análisis termogravimétrico es que
necesita un peso muy pequeño de muestra (escala de miligramos) para caracterizar un
proceso.

Descripción del trabajo realizado
23
La termogravimetría se está usando muy ampliamente acoplada a otras técnicas,
como por ejemplo el análisis térmico diferencial (DTA) o calorimetría diferencial de
barrido (DSC), y también técnicas de gases producidos (EGA) ya que permiten
obtener información complementaria sobre el comportamiento de la muestra.
• Calorimetría diferencial de barrido (DSC).
La calorimetría diferencial de barrido (DSC; diferential scaning calorimetry)
permite el estudio de aquellos procesos en los que se produce una variación entálpica
como puede ser la determinación de calores específicos, puntos de ebullición y
cristalización, pureza de compuestos cristalinos, entalpías de reacción y determinación
de otras transiciones de primer y segundo orden.
En general, el DSC puede trabajar en un intervalo de temperaturas que va desde la
temperatura del nitrógeno líquido hasta unos 600 ºC. Por esta razón, esta técnica de
análisis se emplea para caracterizar aquellos materiales que sufren transiciones
térmicas en dicho intervalo de temperaturas.
La finalidad de la calorimetría diferencial de barrido DSC es registrar la diferencia
en el cambio de entalpía que tiene lugar entre la muestra y un material inerte de
referencia en función de la temperatura o del tiempo, cuando ambos están sometidos a
un programa controlado de temperaturas. La muestra y la referencia se alojan en dos
crisoles idénticos que se calientan mediante resistencias independientes. Cuando en la
muestra se produce una transición térmica, se adiciona energía térmica bien sea a la
muestra o a la referencia, con objeto de mantener ambas a la misma temperatura. Por
tanto, la DSC permite medir la energía que es necesaria suministrar a la muestra para
mantenerla a idéntica temperatura que la referencia. La energía térmica es
exactamente equivalente en magnitud a la energía absorbida o liberada en la
transición. Por tanto, mediante el uso de la técnica DSC se puede evaluar la cantidad
de calor liberado durante los procesos de conversión termoquímica.
• Técnicas de análisis térmico acopladas al análisis de gases residuales.

Descripción del trabajo realizado
24
La principal limitación del análisis térmico en el estudio de procesos es que no te
proporciona informacción sobre los productos generados en los mismos. En este
sentido, se suelen utilizar técnicas complementarias acopladas al análisis térmico y
denominadas EGA, de sus siglas en inglés Evolved Gas Analysis. Existen diferentes
técnicas que se pueden utilizar con este fin, como la cromatografía de gases (GC),
espectroscopia de infrarrojo por transformada de Fourier (FTIR) ó la espetrometría de
masas (MS). Entre todas ellas destaca el acoplamiento de termogravimetría con la
especctrometría de masas (TGA-MS), siendo la única técnica experimental capaz de
monitorizar en tiempo real la distribución de productos generados en el proceso
partiendo de una muestra de bajo peso, enriqueciendo significativamente la
información del mecanismo de descomposición correspondiente [26].
A.4.4. Revisión de trabajos bibligraficos de los procesos de conversión
termoquímica de biomasa mediante TGA. DSC y TGA-MS.
• Referentes a biomasa lignocelulósica.
El proceso de pirólisis de biomasa lignocelulósica ha sido ampliamente estudiado
en biliografía mediante TGA y DSC [26]. Desde el estudio de descomposición de los
principales componentes de la biomasa lignocelulósica (celulosa, hemicelulosa y
lignina) [27-31] hasta diferentes ipos de madera [32; 33] u otros tipos de residuo [34].
Por otro lado, los estudios sobre el proceso de pirólisis de microalgas es
comparativamente mucho menor. Babich y col. (2011) [12] estudiaron la conversión
pirolítica de la microalga Chlorella mediante la técnica de TGA acoplada con MS. Las
muestras líquidas de biocombustible se recogen a partir de experimentos llevados a
cabo en un reactor de lecho fijo. Demirbas y col. (2011) [15]estudió la producción de
biocombustibles a partir de dos muestras de algas (Cladophora fracta y Chlorella
protothecoid). Para ello, investigó el efecto de la temperatura sobre la cantidad de
hidrógeno producido en los procesos de pirólisis y gasificación con vapor, estudiando
los gases producidos en dichos procesos. El proceso de pirólisis es muy importante ya
que es considerado como el primer paso de los procesos de combustión y gasificación.

Descripción del trabajo realizado
25
El estudio de combustión de biomasa lignocelulósica mediante TGA es
significativamente menor comparado con el de pirólisis. Zhang y col. (2011)[35]
investigaron sobre las características de combustión de biomasa como la “paja” de
arroz y la celulosa contenida en ella. Estudiaron las diferencias entres las curvas TG-
DTG-DSC y estimaron los parámetros de TG y los índices de ignición de las muestras
de biomasa, obteniendo información sobre sus características básicas de combustión.
Por otro lado, Zhang y col. (2012)[36], mediante sus estudios de los procesos de
combustión obtuvieron resultados que mostraban que el proceso de combustión se
puede describir como una reacción de primer orden. Joaquín Collazo y col. (2012)[37]
investigaron sobre un método para la determinación del máximo error de muestreo y
los intervalos de confianza de las propiedades térmicas medidas mediante TGA-DSC.
Mustafa Versan Kok y Emre Özgür (2013)[38] estudiaron las características de
combustión de muestras de biomasa como miscanto, madera de álamo, y cascarilla de
arroz.Amutio y col. (2012) [39]analizaron la pirólisis oxidativa de biomasa
lignocelulósica con diferentes concentraciones de oxígeno para establecer un modelo
cinético para dicho proceso. El estudio de combustión de microalgas en cambio, ha
sido poco estudiado. Chen y col. [40] evaluaron el fecto de la concentración de O2 en
la combustion de la microalga Chlorella Vulgaris.
El proceso de gasificación de biomasa es probablemente el menos estudiado. La
mayoría de los estudios han sido dirigidos a la evaluación del comportamiento de
diferentes tipos de carbón gasificándolos con vapor de agua o dióxido de carbono. En
este sentido, Shabbar et al. [41] analizó la termodinámica de carbones bituminosos.
Además, Tay et al. [42] evalúo el effecto de diferentes agentes gasificantes en
diferentes tipos de carbones. Sin embargo, el estudio del proceso de gasificación de
biomasa ha sido mucho menos estudiado. Mohammed et al. [43] evalúo las cinéticas y
las características térmicas de residuo de frutas. Otros estudios han ido dirigidos a la
evaluación del proceso de gasificación del char obtenido a partir de la pirólisis de la de
diferentes tipos de biomasa[44-46]. En cambio, estudios de gasificación de biomasa
marina mediante TGA no han sido encontrados hasta la fecha.

Descripción del trabajo realizado
26
Finalmente, el estudio EGA de los procesos de conversión termoquímica son
escasos en literatura. Huang y col. (2011)[47] investigaron la composición y las
propiedades térmicas de hemicelulosa, celulosa y lignina. Barneto y col. (2009)[48]
con el fin de optimizar el proceso térmico de pirólisis y tener un mayor conocimiento
de la evolución de los gases volátiles en el mismo para analizar dos muestras de
biomasa lignocelulósica.Li y col. (2003)[49]analizaron el comportamiento térmico y
caracterizaron los gases obtenidos en el proceso de combustión de trece especies
procedentes de China.Chul Yoon ycol. (2012) [50] estudiaron la pirólisis y la
gasificación de biomasa lignocelulósica y de sus principales componentes mediante
una combinación de termogravimetría y cromatografía de gases empleando aire o
vapor como agentes gasificantes en diferentes proporciones. Aghamohammadi y col.
(2011)[51] investigaron la emisión de los gases durante la combustión de madera
tropical, bambú, tronco de aceite de palma, acacia y madera de caucho utilizando la
técnica de análisis termogravimétrico acoplado a un espectrómetro de masas (TGA-
MS). Fang y col. (2006)[52] analizaron la pirólisis y la combustión de la madera bajo
diferentes concentraciones de oxígeno mediante la técnica TGA-FTIR, así como la
cinética de ambos procesos. Haykiri-Açma (2003)[53] estudió las características de
combustión de algunas muestras de biomasa terrestre tales como la cáscara de girasol,
las semillas de colza, el algodón y la piña mediante termogravimetría.
Babich y col. (2011) [12] estudiaron la conversión pirolítica de la microalga
Chlorella mediante la técnica de TGA acoplada con MS. Las muestras líquidas de
biocombustible se recogen a partir de experimentos llevados a cabo en un reactor de
lecho fijo. Demirbas y col. (2011) [15]estudió la producción de biocombustibles a
partir de dos muestras de algas (Cladophora fracta y Chlorella protothecoid). Para
ello, investigó el efecto de la temperatura sobre la cantidad de hidrógeno producido en
los procesos de pirólisis y gasificación con vapor, estudiando los gases producidos en
dichos procesos. Phukan y col. (2011)[14]caracterizaron el alga Chlorella sp mediante
espectroscopía FTIR y realizaron un estudio termogravimétrico de la misma a
diferentes velocidades de calentamiento para evaluar su viabilidad para la conversión
termoquímica. Miao y col. (2004)[54] utilizaron dos especies en sus experimentos,

Descripción del trabajo realizado
27
Chlorella protothecoides y Microcystis aeruginosa, para investigar la pirólisis rápida
de ambas especies en un reactor de lecho fluido en una atmósfera inerte de N2 para
proceder, posteriormente, a la comparación con resultados obtenidos de pirólisis lenta
en un autoclave. Minowa y col. [55] realizaron el proceso termoquímico de
licuefacción a la especie de microalga Botryococcus braunii para la obtención de
combustibles líquidos y la recuperación de hidrocarburos.
A.5. ENERGÍA SOLAR DE CONCENTRACIÓN: COLECTOR CILIN DRO
PARABÓLICO.
A.5.1. Generalidades.
El empleo de colectores cilindro-parabólicos se remonta a 1880, John Ericsson
construyó un sistema de espejos cilindro-parabólicos para alimentar un motor de aire
caliente. Frank Shuman y C.V. Boys, fueron los primeros en utilizar este tipo de
espejos para la generación de energía de forma significativa, construyendo en 1912
una planta para el bombeo de agua con vapor en Meadi (Egipto) utilizando espejos
con una superficie total de captación de 1200 m2. A pesar del éxito alcanzado, la
planta se cerró en 1915 debido al inicio de la Primera Guerra Mundial y a los bajos
precios del petróleo.
Debido a la crisis del petróleo renació el interés en este tipo de tecnología siendo
principalmente el Departamento de Energía de los Estados Unidos y el Ministerio de
Investigación y Tecnología alemán los que impulsaron diversos prototipos solares
cilindro-parabólicos para la producción de vapor y para el bombeo de agua.
Posteriormente y basándose en la tecnología de espejos cilindros-parabólicos se
consiguió producir electricidad solar para cubrir las necesidad de miles de habitantes
en California (900 GWh/año). Estas centrales podían funcionar en modo solar o en
combinación con gas natural, asegurando de esta forma su disponibilidad
independientemente de las condiciones climatológicas o del ciclo día-noche. Las
centrales se encuentran en el desierto de Mojave y hoy continúan su funcionamiento

Descripción del trabajo realizado
28
con 354 MW de potencia instalada, planificándose la construcción de más centrales en
sus alrededores.
En 1981, la Agencia Internacional de la Energía construyó y probó un sistema para
la producción de electricidad a base de captación solar mediante espejos cilindro
parabólicos de 500 Kw de potencia en la Plataforma Solar de Almería (Tabernas).En
esta plataforma se está investigando con todas las tecnologías termosolares. En 2008,
entró en funcionamiento Andasol I, en Granada, con 50 MW instalados, y también
cabe destacar la central de Iberdrola de 50 MW situada en Puertollano (Ciudad Real)
Aún así, este tipo de energía se considera que está en una fase de demostración de
viabilidad a gran escala, surgiendo cada día nuevos proyectos, con importantes retos
tecnológicos como el almacenamiento de calor o la hibridación con biomasa o gas
natural.
• Funcionamiento de una planta termosolar de colector cilindro-parabólico.
El esquema de funcionamiento de estas plantas es bastante simple. Se basan en un
campo de espejos con forma parabólica, que concentran la luz solar sobre un eje,
donde se encuentra una tubería por la que circula un fluido de intercambio de calor
(generalmente aceite). Este fluido caliente se introduce a la zona de generación, un
ciclo termodinámico convencional, donde se calienta agua para la producción de vapor
para el accionamiento de una turbina. Además, este tipo de centrales son combinadas
con otros tipos de combustibles, para los períodos de baja insolación.
En la Figura A.10 se muestra una imagen de un colector cilindro-parabólico en la
central termosolar de Almería y el diagrama de flujo de una planta termosolar (Flaberg
Solar International).Este tipo de plantas son capaces de calentar el fluido de
intercambio de calor hasta unas temperaturas entre 300 y 400 ºC (Razón de
concentración: 15-50), obteniendo rendimientos de hasta el 60 % y con capacidad de
320 MW.

Descripción del trabajo realizado
29
Figura A.10.- Planta termosolar de colector cilíndro-parabólico en España (Plataforma
Solar de Almería) y diagrama de flujo de una planta termosolar (Flaberg Solar International)
(Forristal, 2003).
A.5.2. Fluido de Intercambio de Calor (HTF).
Los fluidos utilizados comercialmente son principalmente aceites compuestos por
mezclas eutécticas de óxido de difenilo y óxido de bifenilo. Estos HTF presentan una
serie de inconvenientes que se describen a continuación:
• Riesgos para la salud de operarios de planta. La degradación del aceite
térmico puede tener como consecuencia la aparición de aromáticos, que son
nocivos.
• Son compuesto tóxicos e inflamables.
• Producen una disminución en su función de transmisor de energía y daños
provocados en los equipos y tuberías por los que circula el fluido.
• Poseen una presión de vapor elevada, generando elevadas sobrepresiones.
Esto incrementa el coste de los recipientes para el almacenamiento de energía.
• Tienen una temperatura de degradación baja, alrededor de los 300 ºC,
disminuyendo la eficiencia del ciclo termodinámico para la producción de energía.
Por tanto uno de los principales retos que presenta este tipo de tecnología es el
cambio del fluido de intercambio de calor (HTF). Diversos autores, se han
encaminado en la búsqueda de fluidos capaces de reemplazar a los utilizados

Descripción del trabajo realizado
30
comercialmente. Los principales esfuerzos, se han dirigido hacia las llamadas sales
fundidas. Estos estudios están encabezados por el Departamento de Energía de los
Estados Unidos[56; 57]. Otro tipo de fluidos, los líquidos iónicos, han abierto un
camino interesante para su sustitución [58; 59].
En la presente investigación, se pretenden estudiar diferentes HTF que puedan
mejorar los que actualmente se están empleando en la industria.
Con este fin, es importante un buen conocimiento de las propiedades específicas
requeridas para un buen intercambio de calor. Para el estudio preliminar de las
mismas, se consideró una lista de especificaciones propuesta por el Laboratorio
Nacional de Energías Renovables (NREL, 2000). En esta se especifica que la
capacidad de almacenamiento tiene que ser mayor de 1,9 MJ/m3, con un punto
decongelación inferior a 0 ºC y una estabilidad térmica por encima de los 430 ºC. La
presión de vapor debe ser inferior a la atmosférica para reducir el coste de recipientes
y debe tener una viscosidad adecuada para disminuir los costes de bombeo. Además,
como fluido de referencia se utilizarán las propiedades suministradas por el proveedor
del aceite térmico Therminol® VP-1 (Tabla A.1).
Tabla A.1.- Propiedades del HTF comercial Therminol®-VP1.
Propiedades Therminol-VP1
Punto de Cristalización 12 ºC
Humedad 300 ppm
Viscosidad Cinemática (40ºC) 2,48 cSt
Densidad 1060 kg/m3
Calor de fusión 97,3 Kj/kg
Temperatura de ebullición 257 ºC
Calor de vaporización 206 Kj/kg
Rango óptimo de uso, líquido 12-400 ºC
Rango óptimo de uso, vapor 260-400 ºC
Capacidad calorífica 100ºC 1,78 J/g ºC
Conductividad Térmica 100 ºC 0,1276 W/m K

Descripción del trabajo realizado
31
• Propiedades de un Fluido de Intercambio de Calor (HTF).
- Punto de congelación.
El punto de congelación de un líquido es la temperatura a la que dicho líquido se
solidifica debido a la reducción de temperatura.Este parámetro es muy importante, ya
que un punto de congelación elevado (>0ºC) limita el uso de la planta en climas fríos,
derivando en un elevado coste asociado a la protección a la congelación que
requerirían las tuberías.
- Estabilidad Térmica.
La estabilidad térmica de los fluidos proporciona el límite de temperatura en el cual
se puede operar.La necesidad de establecer estos parámetros debidamente, se traduce
en dos aspectos, cuanto mayor sea la temperatura de descomposición el fluido va a ser
capaz de almacenar más energía térmica, por lo que hace más eficiente el ciclo
termodinámico para la producción de energía.
- Viscosidad.
Al tratarse de sistemas de fluidos en movimiento la viscosidad aparece como una
propiedad importante a la hora de operar en la planta solar.
- Capacidad calorífica:
Mide la cantidad de energía térmica que un cuerpo puede almacenar. La
importancia de su cálculo, se debe a que es necesario su determinación para el cálculo
de la capacidad de almacenamiento energético.
- Densidad.
No es una propiedad térmica, pero su cálculo es importante, puesto que es
necesaria para el cálculo de la capacidad de almacenamiento energético como se
describirá posteriormente.

Descripción del trabajo realizado
32
- Capacidad de almacenamiento térmico: Calor sensible y Calor latente.
Esta variable define la capacidad de los mismos para almacenar calor.
La capacidad de almacenamiento térmico sensible, se puede calcular fácilmente
mediante la ecuación siguiente:
�0 = 2 ∙ �4 ∙ ∆6ª (A.1)
donde
HS = Capacidad de almacenamiento sensible (MJ/m3)
ρ = Densidad del fluido (kg/m3).
Cp= Capacidad calorífica del fluido (J/(kg K)).
∆T= Diferencia entre la temperatura de entrada y de salida del campo solar.
Para el cálculo de la capacidad de almacenamiento térmico latente se utilizará la
ecuación:
�8 = 2 ∙ ∆� (A.2)
donde
HL = Capacidad de almacenamiento latente (MJ/m3)
ρ = Densidad del fluido (kg/m3).
∆H= Entalpía de fusión/vaporización (J/kg).
A.6.- Objetivo del presente trabajo
En los apartados anteriores se ha puesto de manifiesto la importancia de las
energías renovables en el futuro desarrollo de nuestra sociedad. Entre estas tecnologías
cabe destacar el uso de la biomasa y la energía solar térmica como fuentes de energía
renovable. Sin embargo, el grado de desarrollo de las mismas no ha alcanzado una

Descripción del trabajo realizado
33
madurez tecnológica que permita un cambio en el modelo energético actual basado
principalmente en el consumo de combustibles fósiles.
Los procesos de conversión termoquímica de biomasa son los procesos más
interesantes para el aprovechamiento energético de biomasa puesto que permiten
transformar la energía química de la biomasa en diferentes formas, como la
trasformación directa en energía (combustión) ó en combustibles líquidos, sólidos y
gaseosos (pirólisis y gasificación) para su posterior procesamiento.
Por otro lado, el cambio de los fluidos de intercambio de calor (HTF) utilizados
comercialmente en plantas termosolares de concentración basados en hidrocarburos
(mezas de difenilo y bifenilo) por otros obtenidos desde fuentes de energía renovable
con la capacidad de incrementar el ciclo térmico para la obtención de energía es
necesario para la optimización de estos procesos.
Por todo lo anterior, se consideró de interés realizar una investigación enfocada al
estudio de los principales procesos de conversión termoquímica (pirólisis, combustión
y gasificación) de diferentes tipos de biomasa (lignocelulósica y marina).
Adicionalmente, se evaluaron las propiedades físico-químicas de diferentes HTF para
su uso en plantas termosolares de concentración de colector cilindro-parabólico y se
puso en marcha una planta piloto para la evaluación de los mismos a escala semi-
industrial.
A tal fin, se planteó el siguiente programa de investigación:
- Revisión bibliográfica y puesta a punto de las distintas instalaciones
experimentales (equipos de análisis, calibración de gases, equipos de reacción, etc.).
- Diseño y construcción de una planta piloto para el estudio de degradación de
HTF para su aplicación en plantas termosolares de concentración.
- Definición de las principales características de HTF.
- Selección de biomasalignocelulósica y marina en base a su composición
química.

Descripción del trabajo realizado
34
- Evaluación de las condiciones de operación óptimas en el sistema
experimental TGA-MS para el estudio de los principales procesos de conversión
termoquímicas (pirólisis, combustión y gasificación).
- Estudio de los procesos de pirólisis, combustión y gasificación de los
diferentes tipos de biomasa seleccionada.
- Modelización cinética de los procesos de pirólisis, combustión y gasificación.
- Caracterización de los HTF a estudio y selección del más apropiado para su
uso en plantas termosolares de concentración de colector cilindro-parabólico.
- Puesta a punto de la planta piloto para el estudio de degradación de HTF.
- Modelización de la degradación térmica del fluido comercial
MOBILTHERM® 605.
B. MATERIALES Y MÉTODOS
A continuación, se detallan tanto los reactivos como los gases utilizados, indicando
su concentración o pureza y la empresa suministradora.
B.1. Materiales.
Reactivos.
• Celulosa microcristalina con un tamaño de partícula medio de 50 µm. Fue
suministrada por la empresa Sigma-Aldrich.
• Lignina alcalina en forma de polvo marrón con un tamaño de partícula medio
de 50 µm. Fue suministrada por la empresa Sigma-Aldrich.
• Xilano elaborado a partir de madera de haya con un tamaño de partícula
medio de 100 µm. Se usó como referencia de la hemicelulosa y fue
suministrado por la empresa Sigma-Aldrich.
• Abeto, eucalipto y pino recogidos en la región de Castilla-La Mancha
(España). Estas muestras se secaron en un horno durante 5 horas y se
tamizaron para conseguir un tamaño de partícula medio entre 100 y 150 µm.

Descripción del trabajo realizado
35
Gases.
• Argón, envasado en botellas de acero a 200 bares con pureza superior al
99,996% y suministrado por la empresa PRAXAIR.
• Nitrógeno, envasado en botellas de acero a 200 bares con pureza superior al
99,999% y suministrado por la empresa PRAXAIR.
• Oxígeno, envasado en botellas de acero a 200 bares con pureza superior al
99,99% y suministrado por la empresa PRAXAIR.
B.2. INSTALACIÓN EXPERIMENTAL
A continuación se detallan los diferentes equipos que se utilizaron para realizar los
diferentes desarrollos durante la presente investigación.
B.2.1. Calorimetría diferencial de barrido (DSC)
La diferente materia lignocelulósica fue analizada por calorimetría diferencial de
barrido (DSC) en un equipo TGA/DSC modelo 1 STAReSystem de METTLER
TOLEDO
B.2.2. Análisis termogravimétrico (TGA)
La pérdida de peso de los diferentes compuestos con la temperatura se analizó
usando un equipo TGA/DSC modelo 1 STAReSystem de METTLER TOLEDO. Este
equipo permite registrar con gran precisión la pérdida de masa de la muestra en
función de la temperatura/tiempo. Para ello, se debe establecer una secuencia de
calentamiento y configurar los gases circulantes por la cámara de reacción. La muestra
se coloca en unos crisoles de alúmina preparados para soportar las altas temperaturas
del ensayo.
B.2.3. Análisis termogravimétrico – Espectrometría de masas (TGA-MS).
Los productos liberados en el proceso de combustión se analizaron mediante el
acoplamiento de un espectrómetro de masas, ThermosStar-GSD320 con un analizador
de masa cuadrupolar y un potencial de ionización de 70 eV de PFEIFFER VACUUM
a un equipo TGA/DSC modelo 1 STAReSystem de METTLER TOLEDO.El principio
de funcionamiento de esta técnica se basa en la ionización de los componentes

Descripción del trabajo realizado
36
producidos por la degradación térmica de la muestra, separándolos por su relación
masa carga (m/z).
Los espectrogramas obtenidos en cada experimento son almacenados y
cuantificados por el propio software informático suministrado con el equipo.
B.2.4. Análisis elemental
El análisis elemental permite obtener el contenido de la muestra en los
principales elementos químicos, como son carbono (C), hidrógeno (H), nitrógeno (N),
oxígeno (O) y azufre (S). Para llevar a cabo este tipo de análisis se utiliza un
analizador elemental, que es un equipo capaz de detectar todos los elementos citados
mediante diversos mecanismos y dar el resultado en porcentaje en masa de cada uno
de ellos en base seca. En el analizador elemental la separación de elementos de la
muestra se produce por combustión a alta temperatura (950 ºC) mediante la inyección
de una dosis elevada de oxígeno puro. Antes de ser introducidas en el mismo, las
muestras deben ser secadas para eliminar el hidrógeno y el oxígeno procedente de su
humedad, y así poder obtener los resultados en base seca.
El porcentaje de C, H, N y S será una media de los valores obtenidos en los
diez ensayos realizados a la muestra. El analizador elemental calcula automáticamente
estos datos. El porcentaje de oxígeno (O) de la muestra se calcula según la ecuación
[4.1]:
� = 100 − �� + � + ; + < + #=>? �� [4.1]
siendo O, C, H, N, S y cenizas los porcentajes en masa de oxígeno, carbono,
hidrógeno, nitrógeno, azufre y cenizas en base seca, respectivamente. El porcentaje en
cenizas de la muestra se determina mediante el análisis inmediato.
B.2.4. Análisis inmediato
El análisis inmediato permite determinar cuatro de las características químicas más
importantes de cualquier tipo de combustible:
• Humedad. Es la proporción de masa de agua libre que contiene el
combustible. El agua en el combustible puede encontrarse de dos formas
diferentes: libre o combinada. El agua libre se denomina humedad y es la que
se puede separar del combustible por simple calentamiento a 105 ºC. El agua

Descripción del trabajo realizado
37
combinada forma parte de la estructura interna del combustible que, durante el
calentamiento, se combina con otros elementos para dar lugar principalmente
a hidrocarburos y para eliminarla es necesario calentar el combustible a
temperaturas comprendidas entre 150-185 ºC.
• Volátiles. Son las combinaciones de carbono, hidrógeno, oxígeno y otros
gases que contiene el combustible. El desprendimiento de volátiles es un
proceso exotérmico (desprende calor en el proceso de descomposición) que
ayuda al proceso de combustión de la biomasa.
• Cenizas. Son el residuo no orgánico de la combustión compuesto,
principalmente, por las materias minerales que acompañan al combustible. Se
trata de un residuo sólido no combustible, generalmente polvoriento, que
queda después de la combustión completa de la biomasa.
• Carbono fijo. El carbono fijo es la fracción residual de combustible,
descontadas las cenizas, que queda tras la desvolatilización del mismo. El
contenido en carbono fijo es un parámetro indicativo de la calidad del
combustible.
El equipo utilizado para realizar el análisis inmediato es el
analizadortermogravimétrico y permite medir la pérdida de peso de la muestra en
función de la temperatura en una atmósfera controlada. Para llevar a cabo los ensayos
se ha empleado el analizador termogravimétrico TGA/DSC modelo 1 STAReSystem
de METTLER TOLEDO.
El método utilizado para llevar a cabo este estudio consistió en calentar la muestra
de 25 a 950ºC a una velocidad de calentamiento de 10ºC/min en presencia de N2 con
un caudal de 70 ml/min. A continuación, se mantuvo la temperatura 950ºC durante 60
minutos en presencia de O2 con un caudal de 20 ml/min.
Las gráficas proporcionadas por el analizador termogravimétrico son pérdida de
peso vs temperatura y derivada peso vs temperatura, denominadas TGA y DTGA,
respectivamente. La curva TGA proporciona el contenido en volátiles, carbono fijo y
cenizas, mientras que la curva DTGA proporciona la velocidad de pérdida de masa en

Descripción del trabajo realizado
38
cada punto de calentamiento dando una idea de la estabilidad térmica de la
descomposición de la muestra. Mediante el análisis de las gráficas TGA y DTGA en
atmósfera inerte y oxidante se puede determinar los contenidos en volátiles, carbono
fijo y cenizas de una muestra.
Finalmente, el contenido en carbono fijo de la muestra en base seca se calcula
según la ecuación [4.2]:
%� %A�>�BC� = 100 − �%D�&áF&=� +%�=>? �� [4.2]
donde los contenidos en volátiles y cenizas están expresados también en base seca.
B.2.5.Espectroscopía de emisión atómica de plasma acoplado por inducción (ICP-
AES)
Mediante esta técnica espectroscópica se determinó la composición química de la
biomasa objeto de estudio. En concreto, se utilizó para calcular el porcentaje en peso
de los distintos elementos metálicos de la muestra. El equipo utilizado para realizar los
análisis es el modelo VARIAN LIBERTY RL sequential ICP-AES de análisis
multielemental. La espectroscopia de emisión atómica se fundamenta en la excitación
de los átomos metálicos mediante un plasma de Argón, capaz de alcanzar 10000 K,
asegurando la completa atomización de la muestra en estado líquido. Al cesar la
excitación, tiene lugar la emisión de radiación por parte del metal para volver al estado
enérgico fundamental. La intensidad de dicha emisión permite cuantificar la
concentración del elemento ya que depende de la cantidad de átomos del mismo.
B.2.6. Microscopía electrónica de barrido (SEM)
Para evaluar la morfología y el tamaño de la microalga NG se utilizó un microscopio
electrónico de barrido Quanta 250 SEM con filamento de wolframio.
El microscopio electrónico de barrido es un instrumento que permite la
observación y caracterización superficial de materiales orgánicos e inorgánicos,
proporcionando información morfológica del material analizado. La formación de la
imagen se produce por la dispersión de los electrones. Esta capacidad de dispersión va
a depender de las distintas estructuras atómicas de la muestra. El microscopio
electrónico funciona como un microscopio convencional cuando las muestras son

Descripción del trabajo realizado
39
conductoras. En cambio, cuando las muestras no son conductoras se pueden observar
utilizando el régimen de bajo vacío, y cuando las muestras son orgánicas se emplea el
régimen ambiental (ESEM). Para caracterizar la microalga NG se utilizó un detector
modelo GSED (Gaseous SED Detector).
B.2.7.Espectroscopía dispersiva de Rayos-X (EDAX)
Es una técnica analítica utilizada para el análisis elemental o caracterización
química de una muestra. Es una de las variantes de espectroscopia de fluorescencia de
Rayos X que se basa en la investigación de una muestra a través de interacciones entre
la radiación electromagnética y la materia, analizando los Rayos X emitidos por la
materia en respuesta al choque con partículas cargadas. Sus capacidades de
caracterización se deben en gran parte al principio fundamental de que cada elemento
tiene una única estructura atómica permitiendo que los Rayos X característicos de la
estructura atómica de un elemento sean identificados unos de otros.Para realizar este
análisis se utilizó el modelo APOLLO X acoplado a un microscopio electrónico de
barrido Quanta 250 SEM con filamento de wolframio.
B.2.8. Determinación de la cantidad de celulosa, hemicelulosa y lignina
El contenido de celulosa, lignina y xilano en las muestras de biomasa
lignocelulósica se calculó de acuerdo con el método descrito por Yang y col.
(2006)[27].
La determinación de la cantidad de extractos se llevó a cabo por extracción con
disolvente (100 ml de acetona para 1 gramo de muestra de biomasa seca) a 60 º C.
Después, la muestra de la biomasa se secó en un horno (110 º C) hasta que se obtuvo
un peso constante. Posteriormente, el residuo sólido se enfrió a temperatura ambiente
en un desecador y, finalmente, se pesó. La diferencia de peso antes y después de la
extracción es la cantidad de extractivos.
Para la determinación de la cantidad de hemicelulosa se añadieron 150 ml de
solución de NaOH (20 g / l) a 1 gramo de muestra de biomasa seca libre de extractos,
y la mezcla hirvió durante 3,5 h con agua destilada. El residuo se filtró y se lavó hasta
pH neutro y se secó en un horno. El residuo se enfrió posteriormente a temperatura

Descripción del trabajo realizado
40
ambiente en un desecador y posteriormente se pesó. La diferencia de peso antes y
después de este tratamiento es la cantidad de hemicelulosa (Li y col., 2004).
La determinación de la lignina se llevó a cabo por el método de Klason. Se
añadieron 30 ml de H2SO4 (72%) a una muestra de 1 gramo de biomasa seca libre de
extractos. La mezcla se calentó y se agitó durante 2 horas. Posteriormente, la mezcla
se diluyó al 4% de concentración de H2SO4. La mezcla resultante hirvió durante 4
horas con agua destilada. El residuo se filtró y se lavó. Por último, se secó y se enfrió
a temperatura ambiente en un desecador. La diferencia de peso antes y después del
tratamiento es la cantidad de lignina.
Finalmente, se calculó la cantidad de celulosa por diferencia de peso asumiendo
que las muestras de biomasa están compuestas principalmente por extractivos,
celulosa, hemicelulosa y lignina.
B.3. PROCEDIMIENTO EXPERIMENTAL
B.3.1. Análisis termogravimétrico del proceso de combustión
La combustión de la biomasa lignocelulósica así como sus principales
componentes se llevó a cabo en el equipo TGA (TGA-DSC 1, METTLER TOLEDO).
Las muestras se precalentaron a 105 ºC durante 10 minutos para eliminar la humedad.
Después, la biomasa se calentó desde 105 ºC hasta 1000 ºC empleando diferentes
velocidades de calentamiento (10, 20, 40 y 80 ºC/min) en una atmósfera compuesta
por un 21% de oxígeno y un 79% de argón. Estos estudios se realizaron de acuerdo
con los trabajos realizador por Sánchez-Silva y col. (2013) para evitar las limitaciones
de transferencia de materia y calor. En este sentido, la cantidad de muestra inicial fue
de 6 mg, el tamaño de partícula se mantuvo en un rango entre 100-150 µm y se utilizó
un caudal constante de 100 Nml/ min.
B.3.2. Análisis de los productos gaseosos desprendidos en el sistema TGA-MS.
El análisis de los productos gaseosos desprendidos durante el proceso de
combustión se llevó a cabo en una termobalanza (TGA-DSC 1, METTLER TOLEDO)
acoplada a un espectrómetro de masas (Thermostar-GSD320 con analizador de masa
cuadrupolar; PFEIFFER VACUUM) con un potencial de ionización de 70 eV que

Descripción del trabajo realizado
41
proporciona espectros hasta 300 a.m.u. La línea de conexión entre los equipos estaba
envuelta con hilo calefactor para evitar la condensación de los gases en esta zona.
Se realizó un análisis semicuantitativo usando un procedimiento de normalización.
Para ello, las intensidades de los iones se normalizaron con la intensidad del isótopo 38Ar para eliminar errores de instrumentación causados por la fluctuación del gas
portador, el peso de la muestra y cambios en la sensibilidad del espectrómetro de
masas [60]. Se calculó el área bajo la curva obtenida para cada uno de los gases
desprendidos, tomándose como criterio comparativo entre las diferentes muestras [61].
C. RESULTADOS Y DISCUSIÓN
El criterio empleado para la selección de la biomasa marina, se basó en la elección
de la microalga con mayor contenido en lípidos y una menor cantidad de proteínas y
carbohidratos. Por tanto, se realizó un diagrama ternario donde se determinó que la
microalga Nannochloropsis Gaditana (microalga NG) reunía mejores propiedades
para llevar a cabo este estudio.
En el Capítulo 1, el estudio de la pirólisis, combustión y gasificación de la
microalga NG se llevó a cabo mediante análisis termogravimétricos (TGA) y la
novedosa técnica de termobalanza acoplada a un espectrómetro de masa (TGA-MS),
siendo esta última la única herramienta capaz de detectar los compuestos que se
desprenden de una muestra de bajo peso a tiempo real.
Para la selección de las condiciones óptimas de operación en los procesos de
pirólisis y combustión se evaluaron las siguientes variables: masa inicial de muestra,
tamaño de partícula y caudal de gas reactivo.
En el estudio del proceso de pirólisis se observó que la microalga NG posee 3
etapas de degradación. Una primera etapa asociada a la eliminación de agua y
componentes más volátiles a temperaturas < 160ºC. La segunda etapa, donde se
produce la mayor pérdida de peso, asociada a la degradación de proteínas,

Descripción del trabajo realizado
42
polisacáridos y lípidos. Y una tercera etapa (> 450ºC) donde se produce la
degradación térmica de la carbonilla.
En el proceso de combustión de la microalga NG se dividió también en 3 etapas.
Una primera etapa de secado asociada a la pérdida de agua a temperaturas < 125ºC. La
segunda etapa, donde se produce la mayor pérdida de peso, asociada a la
descomposición de proteínas, hidratos de carbono y lípidos. Y una tercera etapa (>
450ºC) donde se produce la oxidación de la carbonilla resultante.
Del estudio de las diferentes variables se observó, de forma general, que al
aumentar la masa inicial de muestra se produce una aceleración en el proceso de
combustión así como un aumento en la velocidad de descomposición, a diferencia de
lo que ocurría en el proceso de pirólisis. En el proceso de pirólisis, el efecto del
tamaño de partícula tiene poca influencia en el mismo, mientras que en el proceso de
combustión la muestra de menor tamaño es la más reactiva y se volatiliza antes. El
caudal de gas no afecta a ninguno de los dos procesos.
Posteriormente, se llevó a cabo un análisis de las condiciones óptimas de operación
en el proceso de gasificación mediante TGA. Al aumentar la temperatura de
gasificación se produce un aumento en los valores de reactividad y conversión. La
reactividad aumenta y la conversión disminuye cuando la masa inicial de muestra
disminuye y la porosidad de la muestra aumenta. Al aumentar el caudal de Ar,
disminuye la conversión y aumenta la reactividad, al igual que ocurre al aumentar el
% de vapor de agua.
En la segunda parte de este trabajo se estudió la distribución de los productos
gaseosos generados en los procesos termoquímicos de pirólisis, combustión y
gasificación utilizando las condiciones óptimas para cada uno de ellos y empleando la
técnica TGA-MS.

Descripción del trabajo realizado
43
Mediante este análisis se concluyó que los productos generados durante el proceso
de pirólisis se liberan en tres etapas. En la primera etapa, se desprendió agua a
temperaturas < 160ºC. Posteriormente, en un rango de temperaturas entre 160 y 450ºC
se identificaron la mayor parte de los componentes, siendo el principal compuesto
detectado el CH3+ debido a la descomposición de los grupos carboxilos en las
proteínas y los polisacáridos, junto con HCN, CH4N, CO2, C3H8N, CO, C6H6 y otros
hidrocarburos volátiles como C2H5, C2H2 y CH4. Los compuestos nitrogenados son
liberados debido a la degradación térmica de las proteínas. Finalmente, a temperaturas
> 450ºC se produce la liberación de H2.
En cuanto a la segunda parte de la investigación, el estudio de la pirólisis de los
diferentes tipos de biomasa fue llevado a cabo mediante análisis termogravimétricos
(TGA) y mediante la novedosa técnica de termobalanza acoplada a un espectrómetro
de masas (TGA-MS). El uso de esta técnica se ha demostrado como la única capaz de
detectar los compuestos que se desprenden de una muestra de bajo peso a tiempo real.
Los tipos de biomasa sometidos a estudio fueron: la celulosa, la hemicelulosa y la
lignina (componentes mayoritarios de la biomasa terrestre), la madera de abeto y una
variedad de microalga (Nannochloropsis gaditana).
En el Capítulo 2, primero se llevó a cabo un análisis de las condiciones óptimas de
operación en el TGA-MS. Para el uso óptimo de esta técnica se tuvo en cuenta que a
elevadas cantidades de muestra inicial conllevan efectos de transferencia de materia y
de calor en el análisis termogravimétrico. Sin embargo, un bajo peso de muestra
inicial disminuye la detectabilidad en el espectrograma de masas. Por lo tanto, hay que
llegar a un compromiso en el que se utilice la mayor cantidad de masa inicial sin que
se produzcan limitaciones en el proceso por efectos de transferencia de materia y de
calor. Otras variables a estudio fueron la influencia del caudal de gas portador (He) y
de la velocidad de calentamiento. Las condiciones óptimas resultantes de este estudio
fue el empleo de un peso de muestra inicial de 10 mg, un flujo de gas portador He de
200 Nml/min y una velocidad de calentamiento de 40ºC/min

Descripción del trabajo realizado
44
Posteriormente se llevó a cabo la evaluación del proceso de pirólisis para los
diferentes tipos de biomasa seleccionados mediante un análisis termogravimétrico. En
primer lugar se observó que la madera de abeto en el proceso de pirólisis se
descomponía en cuatro etapas, una primera etapa asociada a la eliminación de agua a
temperaturas ≤ 120ºC y tres etapas posteriores atribuidas a la descomposición de la
hemicelulosa (≈ 220ºC), de la celulosa y la lignina (300-400 ºC) y la lignina (>400ºC)
respectivamente. También se comprobó que el proceso de pirólisis tuvo lugar en un
rango de temperaturas de 200 a 500 ºC, intervalo de temperaturas donde ocurre la
mayor parte de la descomposición de la biomasa. Por último, se observó que la
biomasa marina estudiada se degrada a mayores temperaturas (≈ 1000ºC) que la
biomasa terrestre (≈ 700ºC).
Una vez estudiado el proceso de pirólisis, se evaluó el efecto de la velocidad de
calentamiento en la descomposición térmica de la biomasa mediante
termogravimetría. Para llevar a cabo este estudio se utilizó 5, 15 y 40ºC/min. La
velocidad de calentamiento influyó significativamente sobre la temperatura a la que
comienza el proceso de pirólisis y en la que se produce la mayor pérdida de peso. En
cambio, la velocidad no mostró un efecto tan claro sobre la cantidad de residuo
generado.
Empleando la técnica TGA-MS se estudió la distribución de los productos
generados durante la pirólisis de la biomasa. Mediante este análisis se concluyó que
los productos en la pirólisis se liberan en tres etapas. En la primera etapa, se
desprendió principalmente agua a temperaturas ≤ 120ºC. Posteriormente, en un rango
de temperaturas comprendido entre 200 y 450 ºC se identificaron la mayor parte de los
componentes volátiles, siendo el principal compuesto detectado el CO2 junto con CH4,
C2H6 y pequeñas cantidades de CO. Finalmente, a temperaturas ≥500 ºC se produce la
liberación de H2.
Finalmente, se desarrolló un modelo cinético que permitió estudiar, mediante
termogravimetría, el comportamiento de la biomasa durante su pirólisis a diferentes

Descripción del trabajo realizado
45
velocidades de calentamiento. Con este fin, se empleó un modelo teórico de múltiples
saltos de descomposición basado en una expresión de velocidad tipo Arrhenius,
obteniéndose los parámetros cinéticos (energía de activación, factor pre-exponencial y
orden de descomposición) a cada velocidad considerada.
En el Capítulo 3, el estudio del proceso de combustión de la biomasa
lignocelulósica y de sus principales componentes se llevó a cabo mediante análisis
termogravimétricos (TGA), análisis de calorimetría diferencial de barrido (TGA/DSC)
y la novedosa técnica de termobalanza acoplada a un espectrómetro de masa (TGA-
MS), siendo esta última la única herramienta capaz de detectar los compuestos que se
desprenden de una muestra de bajo peso a tiempo real.
El proceso de combustión de la biomasa lignocelulóscia se divide en dos etapas
prinicipalmente. La primera etapa, donde se produce la mayor pérdida de peso,
llamada etapa de desvolatilización, en la cual se descomponen los principales
componentes de la biomasa (celulosa, hemicelulosa y lignina). Y una segunda etapa (>
441ºC) donde se produce la oxidación del residuo carbonoso (char) resultante.
La información obtenida mediante el análisis termogravimétrico se completó
mediante el estudio del proceso de combustión de las muestras de biomasa
lignocelulósica y de sus principales componentes por calorimetría diferencia de
barrido (DSC). En general, se observaron dos regiones exotérmicas, la primera se
atribuye a la etapa de desvolatilización y la segunda a la oxidación del char.
Una vez estudiado el proceso de combustión, se evaluó el efecto de la velocidad de
calentamiento en la descomposición térmica de la biomasa mediante
termogravimetría. Para llevar a cabo este estudio se utilizaron 10, 20, 40 y 80 ºC/min.
La velocidad de calentamiento influyó significativamente sobre la temperatura a la
que comienza el proceso de combustión y en la que se produce la mayor pérdida de
peso. En cambio, la velocidad de calentamiento no mostró un efecto tan claro sobre la
cantidad de residuo generado.

Descripción del trabajo realizado
46
Posteriormente, se estudió la distribución de los productos gaseosos generados en
el proceso termoquímico de combustión empleando la técnica TGA-MS. Mediante
este análisis se concluyó que los principales productos gaseosos generados fueron:
CO, CO2 y H2O. También se produjeron hidrocarburos ligeros, atribuidos a reacciones
secundarias, como son CH4 y C2H5. La mayoría de los productos detectados fueron
generados durante la etapa de desvolatilización, mientras que sólo el NO2, C2H5O+,
CO y CO2 se detectaron en la segunda etapa (etapa de oxidación). Se detectaron
compuestos de nitrógeno, en mayor proporción que los compuestos de azufre,
liberados en forma de aminas primarias y NOx.
Finalmente, se desarrolló un modelo cinético que permitió estudiar, mediante
termogravimetría, el comportamiento de la biomasa durante su combustión a
diferentes velocidades de calentamiento. Con este fin, se empleó un modelo teórico de
múltiples saltos de descomposición basado en una expresión de velocidad tipo
Arrhenius, obteniéndose los parámetros cinéticos (energía de activación y factor pre-
exponencial) a cada velocidad considerada. Se encontró el mejor ajuste de los datos
experimentales con los modelos basados en el orden de reacción (Oi), la nucleación
(Ni) y la difusión (Di). Mediante una aplicación Excel-VBA se evaluó el conjunto de
ecuaciones diferenciales ordinarias que definen el modelo cinético. Se obtuvo un
modelo teórico y se comparó con los datos obtenidos experimentalmente.
En el proceso de combustión también se pueden observar tres etapas en la
liberación de las emisiones gaseosas. En la primera etapa, se desprendió agua a
temperaturas < 125ºC. Posteriormente, se produce la liberación de los compuestos
CO2, SO2, NO2, C3H8N y NH3 en la segunda etapa del proceso. La liberación de SO2
se atribuye a los radicales sulfatos existentes en los polisacáridos y a la degradación de
los sulfuros en los residuos orgánicos. Finalmente, a temperaturas > 450ºC se produce
la liberación de H2, NH3, NO2, CO y CH3+. Los hidrocarburos volátiles como CH4,
C2H2 y C2H5 no se generaron durante el proceso.

Descripción del trabajo realizado
47
Los principales productos que se detectaron en el proceso de gasificación fueron
CO2, CO y H2, junto con CH4, C2H6, C2H5, C2H4 y C2H2 que también fueron
generados. Además, se estudió la influencia de la concentración de vapor en la
distribución de productos obtenida, utilizándose diferentes concentraciones de vapor
de agua. A medida que se incrementa la cantidad de vapor en el medio, también lo
hace la concentración de H2 y se produce la disminución de la producción de CH4.
Estos hechos indican que las reacciones water gas y water gas shift se ven favorecidas
al incrementar la concentración de vapor en el agente gasificante y la reacción de
reformado de metano podría estar teniendo lugar:
En el Capítulo 5 se concluyó que las propiedades más importantes que deben
reunir los fluidos térmicos para su aplicación en una planta termosolar son: un amplio
rango de temperaturas en estado líquido, una elevada temperatura de degradación, una
viscosidad baja, una temperatura de fusión baja, una densidad adecuada y una
capacidad calorífica elevada.
Posteriormente se realizó un estudio comparativo de cuatro tipos de HTF, dos sales
fundidas: Sal Solar y Hitec XL y dos líquidos iónicos: ([BMIM][BF4]) y
([EMIM][BF 4]). Del análisis comparativo se descartaron las sales fundidas ya que,
aunque poseían la mayor resistencia térmica (temperaturas de degradación ≥ 500ºC) su
temperatura de fusión es elevada, 230ºC para la Sal Solar y 120ºC para la Hitec XL,
estando limitada su aplicación a temperatura ambiente. Entre los dos líquidos iónicos
se determinó que el [BMIM][BF4] poseía las mejores propiedades para ser utilizado en
plantas solares, debido a que poseía una temperatura de degradación mayor y una
temperatura de fusión menor, y además contaba con propiedades térmicas similares al
[EMIM][BF 4].

Descripción del trabajo realizado
48
D. CONCLUSIONES Y RECOMENDACIONES
De los resultados obtenidos en esta investigación se pueden obtener las siguientes
conclusiones finales:
De los resultados obtenidos en esta investigación se pueden extraer las siguientes
conclusiones.
1. En el estudio termogravimétrico del proceso de pirólisis de la microalga NG
se identifican tres etapas. La primera pérdida de peso atribuida a la pérdida de
agua y componentes más volátiles de la microalga (< 160ºC). En la segunda
etapa se distinguen tres hombros, el primero a 180ºC asociado a la
degradación de proteínas y polisacáridos solubles y dos picos a altas
temperaturas (271 y 411ºC) atribuidos a la degradación de la celulosa de la
pared celular de la microalga y otros polisacáridos insolubles y lípidos,
respectivamente. En la última etapa (> 450ºC) se produce la degradación
térmica de la carbonilla.
2. En las curvas TGA-DTGA asociadas al proceso de pirólisis se puede observar
como, en el estudio del efecto de la masa inicial de muestra un aumento de la
misma desplaza el proceso térmico a temperaturas más elevadas y disminuye
la velocidad de descomposición. En el caso del estudio del efecto del tamaño
de partícula y del caudal de gas portador (Ar), se observa que no tienen
influencia en el proceso ya que todas las curvas se solapaban indicando que no
existen limitaciones por transferencia de materia y de calor.
3. Las condiciones de operación óptimas para llevar a cabo el proceso de
pirólisis de la microalga NG fueron: masa inicial de muestra de 9 mg con un
tamaño de partícula de 100-250 µm, caudal de Ar de 200 ml/min y velocidad
de calentamiento de 40ºC/min.

Descripción del trabajo realizado
49
4. Del estudio termogravimétrico del proceso de combustión se puede concluir
que el proceso está dividido en tres etapas. La primera etapa, que corresponde
a la etapa de secado, se atribuye a la pérdida de agua libre y débilmente ligada
a las biomoléculas. La segunda etapa (180-450ºC) se caracteriza por una
pérdida de peso importante debida a la descomposición de las proteínas, los
hidratos de carbono y los lípidos que constituyen la microalga. La última
etapa (> 450ºC) se corresponde con la oxidación de la carbonilla resultante.
5. En las curvas TGA-DTGA asociadas al proceso de combustión se puede
observar como la masa de muestra inicial tiene un efecto significativo en el
proceso. Al aumentar, desplaza el proceso de combustión a temperaturas más
bajas y aumenta la velocidad de descomposición. En el caso del efecto del
tamaño de partícula, la muestra con el menor tamaño es la más reactiva y se
volatiliza antes debido a que posee una mayor superficie que ofrece una
resistencia menor a la difusión. En cuanto al efecto del caudal de O2 no tiene
influencia sobre el proceso debido a que la concentración de O2 es constante.
6. Las condiciones de operación óptimas para llevar a cabo el proceso de
combustión de la microalga NG fueron: masa inicial de muestra de 10 mg con
un tamaño de partícula de 100-250 µm, caudal de O2 de 100 ml/min y
velocidad de calentamiento de 40ºC/min.
7. En las curvas TGA-DTGA del proceso de gasificación se pueden observar las
distintas variables estudiadas. Un aumento en la temperatura de gasificación
produce un aumento en los valores de reactividad y conversión. La reactividad
aumenta y la conversión disminuye cuando la masa inicial de muestra
disminuye y la porosidad de la muestra aumenta. Al aumentar el caudal de Ar,
disminuye la conversión y aumenta la reactividad, al igual que ocurre al
aumentar el % de vapor de agua.

Descripción del trabajo realizado
50
8. Las condiciones de operación óptimas para llevar a cabo el proceso de
gasificación de la microalga NG fueron: temperatura de gasificación de 850ºC,
20 mg de muestra inicial con un tamaño de partícula de 100-250 µm en
presencia de 200 ml/min de Ar y un 5% de vapor de agua.
9. El estudio TGA-MS del proceso de pirólisis permitió obtener la distribución
de los productos generados en el mismo. Se identificaron principalmente tres
etapas. En la primera, se desprendió agua a temperaturas < 160ºC.
Posteriormente, en un rango de temperaturas entre 160 y 450ºC se
identificaron la mayor parte de los componentes, siendo el principal
compuesto detectado el CH3+ debido a la descomposición de los grupos
carboxilos en las proteínas y los polisacáridos, junto con HCN, CH4N, CO2,
C3H8N, CO, C6H6 y otros hidrocarburos volátiles como C2H5, C2H2 y CH4.
Los compuestos nitrogenados son liberados debido a la degradación térmica
de las proteínas. Finalmente, a temperaturas > 450ºC se produce la liberación
de H2.
10. En el proceso de combustión también se pueden observar tres etapas en la
liberación de las emisiones gaseosas. En la primera etapa, se desprendió agua
a temperaturas < 125ºC. Posteriormente, se produce la liberación de los
compuestos CO2, SO2, NO2, C3H8N y NH3 en la segunda etapa del proceso.
Finalmente, a temperaturas > 450ºC se produce la liberación de H2, NH3, NO2,
CO y CH3+. Cabe destacar que en este proceso el compuesto con el pico de
mayor intensidad es el CO2 y la liberación de SO2 puede atribuirse a los
radicales sulfatos existentes en los polisacáridos y a la degradación de los
sulfuros en los residuos orgánicos.
11. Mediante la comparación de las curvas del MS de los procesos de pirólisis y
combustión es posible evaluar el efecto de la presencia de oxígeno en la
degradación térmica o liberación de productos. En la combustión de la
microalga se produjo una mayor cantidad de CO2, observándose además, a

Descripción del trabajo realizado
51
diferencia del proceso de pirólisis, emisiones de NO2 y SO2. Sin embargo, los
hidrocarburos volátiles como CH4, C2H2 y C2H5 no se generaron durante el
proceso.
12. Los principales productos que se detectaron en el proceso de gasificación
fueron CO2, CO y H2, junto con trazas de hidrocarburos ligeros como CH4,
C2H6, C2H5, C2H4 y C2H2. Además, se estudió la influencia de la
concentración de vapor en la distribución de productos emitida. A medida que
se incrementa la cantidad de vapor en el medio, también lo hace la
concentración de H2 y se produce la disminución de la producción de CH4.
Con objeto de ampliar y completar los resultados obtenidos en esta investigación se
recomienda:
- Investigar otros procesos menos comunes pero que pueden tener un gran interés
medioambiental como puede ser la oxy-combustión con CO2 y atmósfera deficiente de
O2.
- Escalar los experimentos de pirólisis, combustiión y gasificación en sistemas
experimentales para validar los resultados obtenidos mediante TGA-MS
- Uso de catalizadores para incrementar la generación de productos de interés,
como puede ser el H2 en el proceso de gasificación.
- Testeo de diferentes aceites de origen vegetal en la planta piloto diseñada y
optimizada para su uso en plantas termosolares de concentración.
- Evaluación de los aceites usados en el proceso de combustión, gasificación y
pirólisis en el sistema experimental TGA-DSC-MS.

Descripción del trabajo realizado
52
E. BIBLIOGRAFÍA
[1] I.D.A.E. Instituto para la Diversificación y el Ahorro de Energía. Biomasa.
(http://www.idae.es/index.php/idpag.233/relcategoria.1037/relmenu.48/mod.pags/me
m.detalle).
[2] Harun, N.Y., Afzal, M.T. 2010. Thermal decomposition kinetics of forest residue.
Journal of Applied Sciences, 10(12), 1122-7.
[3] WEC. World Energy Council 2010. Survey of Energy Resources.
[4] McKendry, P. 2002. Energy production from biomass (part 1): Overview of
biomass. Bioresource Technology, 83(1), 37-46.
[5] Basu, P. 2010. Biomass Gasification and Pyrolysis: Practical Design and Theory.
Elsevier Science.
[6] Bidlack, J., Malone, M., Benson, R. 1992. Molecular Structure and Component
Integration of Secondary Cell Walls in Plants.
[7] Catallo, W.J., Shupe, T.F., Eberhardt, T.L. 2008. Hydrothermal processing of
biomass from invasive aquatic plants. Biomass and Bioenergy, 32(2), 140-5.
[8] Patil, V., Tran, K.Q., Giselrød, H.R. 2008. Towards sustainable production of
biofuels from microalgae. International Journal of Molecular Sciences, 9(7), 1188-95.
[9] Miao, X., Wu, Q. 2006. Biodiesel production from heterotrophic microalgal oil.
Bioresource Technology, 97(6), 841-6.
[10] Chisti, Y. 2007. Biodiesel from microalgae. Biotechnology Advances, 25(3),
294-306.
[11] Xu, H., Miao, X., Wu, Q. 2006. High quality biodiesel production from a
microalga Chlorella protothecoides by heterotrophic growth in fermenters. Journal of
Biotechnology, 126(4), 499-507.
[12] Babich, I.V., van der Hulst, M., Lefferts, L., Moulijn, J.A., O'Connor, P., Seshan,
K. 2011. Catalytic pyrolysis of microalgae to high-quality liquid bio-fuels. Biomass
and Bioenergy, 35(7), 3199-207.

Descripción del trabajo realizado
53
[13] Bae, Y.J., Ryu, C., Jeon, J.K., Park, J., Suh, D.J., Suh, Y.W., Chang, D., Park,
Y.K. 2011. The characteristics of bio-oil produced from the pyrolysis of three marine
macroalgae. Bioresource Technology, 102(3), 3512-20.
[14] Phukan, M.M., Chutia, R.S., Konwar, B.K., Kataki, R. 2011. Microalgae
Chlorella as a potential bio-energy feedstock. Applied Energy, 88(10), 3307-12.
[15] Demirbas, A. 2011. Biodiesel from oilgae, biofixation of carbon dioxide by
microalgae: A solution to pollution problems. Applied Energy, 88(10), 3541-7.
[16] Brown, M.R., Jeffrey, S.W., Volkman, J.K., Dunstan, G.A. 1997. Nutritional
properties of microalgae for mariculture. Aquaculture, 151(1-4), 315-31.
[17] Ross, A.B., Biller, P., Kubacki, M.L., Li, H., Lea-Langton, A., Jones, J.M. 2010.
Hydrothermal processing of microalgae using alkali and organic acids. Fuel, 89(9),
2234-43.
[18] Yaman, S. 2004. Pyrolysis of biomass to produce fuels and chemical feedstocks.
Energy Conversion and Management, 45(5), 651-71.
[19] Sanchez-Silva, L., López-González, D., Villaseñor, J., Sánchez, P., Valverde,
J.L. 2012. Thermogravimetric-mass spectrometric analysis of lignocellulosic and
marine biomass pyrolysis. Bioresource Technology, 109, 163-72.
[20] Brown, M.R. 1991. The amino-acid and sugar composition of 16 species of
microalgae used in mariculture. Journal of Experimental Marine Biology and Ecology,
145(1), 79-99.
[21] Renaud, S.M., Thinh, L.V., Parry, D.L. 1999. The gross chemical composition
and fatty acid composition of 18 species of tropical Australian microalgae for possible
use in mariculture. Aquaculture, 170(2), 147-59.
[22] Demirbaş, A., Arin, G. 2002. An overview of biomass pyrolysis. Energy
Sources, 24(5), 471-82.
[23] Quaark, P., Knoef, H., Stassen, H.E. 1999. Energy from Biomass: A Review of
Combustion and Gasification Technologies. WORLD BANK PUBN.
[24] 2012. Review of the Literature on Catalytic Biomass Tar Destruction: Milestone
Completion Report. BiblioBazaar.

Descripción del trabajo realizado
54
[25] Brown, M.E., Gallagher, P.K. 2011. Handbook of Thermal Analysis and
Calorimetry: Recent Advances, Techniques and Applications. Elsevier Science.
[26] White, J.E., Catallo, W.J., Legendre, B.L. 2011. Biomass pyrolysis kinetics: A
comparative critical review with relevant agricultural residue case studies. Journal of
Analytical and Applied Pyrolysis, 91(1), 1-33.
[27] Yang, H., Yan, R., Chen, H., Zheng, C., Lee, D.H., Liang, D.T. 2006. In-depth
investigation of biomass pyrolysis based on three major components: Hemicellulose,
cellulose and lignin. Energy and Fuels, 20(1), 388-93.
[28] Grønli, M., Antal Jr, M.J., Várhegyi, G. 1999. A round-robin study of cellulose
pyrolysis kinetics by thermogravimetry. Industrial and Engineering Chemistry
Research, 38(6), 2238-44.
[29] Lin, Y.C., Cho, J., Tompsett, G.A., Westmoreland, P.R., Huber, G.W. 2009.
Kinetics and mechanism of cellulose pyrolysis. Journal of Physical Chemistry C,
113(46), 20097-107.
[30] Nakamura, T., Kawamoto, H., Saka, S. 2007. Condensation reactions of some
lignin related compounds at relatively low pyrolysis temperature. Journal of Wood
Chemistry and Technology, 27(2), 121-33.
[31] Liu, Q., Wang, S., Zheng, Y., Luo, Z., Cen, K. 2008. Mechanism study of wood
lignin pyrolysis by using TG-FTIR analysis. Journal of Analytical and Applied
Pyrolysis, 82(1), 170-7.
[32] Rao, T.R., Sharma, A. 1998. Pyrolysis rates of biomass materials. Energy,
23(11), 973-8.
[33] Stenseng, M., Jensen, A., Dam-Johansen, K. 2001. Investigation of biomass
pyrolysis by thermogravimetric analysis and differential scanning calorimetry. Journal
of Analytical and Applied Pyrolysis, 58-59, 765-80.
[34] Caballero, J.A., Conesa, J.A., Font, R., Marcilla, A. 1997. Pyrolysis kinetics of
almond shells and olive stones considering their organic fractions. Journal of
Analytical and Applied Pyrolysis, 42(2), 159-75.
[35] Zhang, C.C., Lu, H.J., He, Z. 2011. Combustion characteristics of biomass and
its cellulose, Vol. 306-307. Jinan, pp. 143-6.

Descripción del trabajo realizado
55
[36] Zhang, C., Lu, H., He, X. 2012. Combustion process's kinetics study of biomass
and its cellulose based on biomaterials in material engineering, Vol. 568. Wuhan, pp.
328-31.
[37] Collazo, J., Pazó, J.A., Granada, E., Saavedra, Á., Eguía, P. 2012. Determination
of the specific heat of biomass materials and the combustion energy of coke by DSC
analysis. Energy, 45(1), 746-52.
[38] Kok, M.V., Özgür, E. 2013. Thermal analysis and kinetics of biomass samples.
Fuel Processing Technology, 106(0), 739-43.
[39] Amutio, M., Lopez, G., Aguado, R., Artetxe, M., Bilbao, J., Olazar, M. 2012.
Kinetic study of lignocellulosic biomass oxidative pyrolysis. Fuel, 95, 305-11.
[40] Chen, C., Ma, X., Liu, K. 2011. Thermogravimetric analysis of microalgae
combustion under different oxygen supply concentrations. Applied Energy, 88(9),
3189-96.
[41] Shabbar, S., Janajreh, I. 2013. Thermodynamic equilibrium analysis of coal
gasification using Gibbs energy minimization method. Energy Conversion and
Management, 65, 755-63.
[42] Tay, H.L., Kajitani, S., Zhang, S., Li, C.Z. 2013. Effects of gasifying agent on
the evolution of char structure during the gasification of Victorian brown coal. Fuel,
103, 22-8.
[43] Mohammed, M.A.A., Salmiaton, A., Wan Azlina, W.A.K.G., Mohamad Amran,
M.S. 2012. Gasification of oil palm empty fruit bunches: A characterization and
kinetic study. Bioresource Technology, 110, 628-36.
[44] Khalil, R., Várhegyi, G., Jäschke, S., Grønli, M.G., Hustad, J. 2009. CO2
gasification of biomass chars: A kinetic study. Energy and Fuels, 23(1), 94-100.
[45] Mani, T., Mahinpey, N., Murugan, P. 2011. Reaction kinetics and mass transfer
studies of biomass char gasification with CO2. Chemical Engineering Science, 66(1),
36-41.
[46] Gómez-Barea, A., Ollero, P., Fernández-Baco, C. 2006. Diffusional effects in
CO2 gasification experiments with single biomass char particles. 1. Experimental
investigation. Energy and Fuels, 20(5), 2202-10.

Descripción del trabajo realizado
56
[47] Huang, Y.F., Kuan, W.H., Chiueh, P.T., Lo, S.L. 2011. Pyrolysis of biomass by
thermal analysis-mass spectrometry (TA-MS). Bioresource Technology, 102(3), 3527-
34.
[48] Barneto, A.G., Carmona, J.A., Alfonso, J.E.M., Blanco, J.D. 2009. Kinetic
models based in biomass components for the combustion and pyrolysis of sewage
sludge and its compost. Journal of Analytical and Applied Pyrolysis, 86(1), 108-14.
[49] Li, X., Matuschek, G., Herrera, M., Wang, H., Kettrup, A. 2003. A study on
combustion of Chinese coals by TA/MS. Journal of Analytical and Applied Pyrolysis,
67(2), 393-406.
[50] Yoon, H.C., Pozivil, P., Steinfeld, A. 2012. Thermogravimetric pyrolysis and
gasification of lignocellulosic biomass and kinetic summative law for parallel
reactions with cellulose, xylan, and lignin. Energy and Fuels, 26(1), 357-64.
[51] Aghamohammadi, N., Nik Sulaiman, N.M., Aroua, M.K. 2011. Combustion
characteristics of biomass in SouthEast Asia. Biomass and Bioenergy, 35(9), 3884-90.
[52] Fang, M.X., Shen, D.K., Li, Y.X., Yu, C.J., Luo, Z.Y., Cen, K.F. 2006. Kinetic
study on pyrolysis and combustion of wood under different oxygen concentrations by
using TG-FTIR analysis. Journal of Analytical and Applied Pyrolysis, 77(1), 22-7.
[53] Haykiri-Açma, H. 2003. Combustion characteristics of different biomass
materials. Energy Conversion and Management, 44(1), 155-62.
[54] Miao, X., Wu, Q., Yang, C. 2004. Fast pyrolysis of microalgae to produce
renewable fuels. Journal of Analytical and Applied Pyrolysis, 71(2), 855-63.
[55] Dote, Y., Sawayama, S., Inoue, S., Minowa, T., Yokoyama, S.y. 1994. Recovery
of liquid fuel from hydrocarbon-rich microalgae by thermochemical liquefaction.
Fuel, 73(12), 1855-7.
[56] Bradshaw, R.W., Cordaro, J.G., Siegel, N.P. 2009. Molten nitrate salt
development for thermal energy storage in parabolic trough solar power systems.
Conference Molten nitrate salt development for thermal energy storage in parabolic
trough solar power systems, 2, 615-24.
[57] Kearney, D., Herrmann, U., Nava, P., Kelly, B., Mahoney, R., Pacheco, J.,
Cable, R., Potrovitza, N., Blake, D., Price, H. 2003. Assessment of a molten salt heat

Descripción del trabajo realizado
57
transfer fluid in a parabolic trough solar field. Journal of Solar Energy Engineering,
Transactions of the ASME, 125(2), 170-6.
[58] Valkenburg, M.E.V., Vaughn, R.L., Williams, M., Wilkes, J.S. 2005.
Thermochemistry of ionic liquid heat-transfer fluids. Thermochimica Acta, 425(1-2),
181-8.
[59] Wu, B., Reddy, R.G., Rogers, R.D. 2001. Novel ionic liquid thermal storage for
solar thermal electric power systems. Proceedings of the Solar Forum 2001 Solar
Energy, 445-51.
[60] Jakab, E., Till, F., Várhegyi, G. 1991. Thermogravimetric-mass spectrometric
study on the low temperature oxidation of coals. Fuel Processing Technology, 28(3),
221-38.
[61] Jones, J.M., Harding, A.W., Brown, S.D., Thomas, K.M. 1995. Detection of
reactive intermediate nitrogen and sulfur species in the combustion of carbons that are
models for coal chars. Carbon, 33(6), 833-43.

Chapter 1:
PYROLYSIS,COMBUSTION AND
GASIFICATION CHARACTERISTICS OF
NANNOCHLOROPSISGADITANA
MICROALGAE
Pyrolysis, combustion and gasification characteristics
ofNannochloropsisGaditanamicroalgae (NG microalgae)were
investigatedby thermogravimetric analysis (TGA).NG microalgae
pyrolysis and combustion could be divided into three main stages:
dehydration, proteins and polysaccharides degradation and char
decomposition. The effects of the initial sample mass, particle size
and gas flow on the pyrolysis and combustion processes were
studied. In addition, gasification operation conditions such as
temperature, initial sample mass, particle size, sweep gas flow and
steam concentration, were experimentally evaluated.

Chapter 1
63
Theevolved gases were analyzed online using mass
spectroscopy (MS).In pyrolysis and combustion processes, most
of the gas products were generated at the second degradation
step. N-compounds evolution was associated with the
degradation of proteins. Furthermore, SO2releasefrom
combustion could be related tosulphated
polysaccharidesdecomposition.The main products detected
during gasification wereCO2, CO, H2, indicating that oxidation
reactions, water gas and water gas shift reactions,were
predominant.
1.1. INTRODUCTION.
Recently,the utilization of biomass for transport fuels, chemical commodities,
power generation and reduction of CO2 emissions is growing interest[1]. Thus,
biomass has the potential of being an important renewable energy source.
Algae are a very promising biomass for the following reasons: a high growth rate,
high yield per area, high efficiency in CO2 capture and solar energy conversion and no
competition with food agriculture. In addition, they can be grown in open water (sea
water or ponds) and in bio-photo reactors on non-arable lands[2].
The generic term microalgae refer to a large group of very diverse photosynthetic
micro-organisms of microscopic dimensions. They have received more attention than

Chapter 1
64
macroalgae for biofuels production, which can be cultured in ponds or
photobioreactors with supply of nutrients or wastewater . Moreover, the production of
microalgae does not require of high quality arable land and therefore it does not
compete with food crops.
Generally, microalgae varied in their proportions of protein (6-52 wt.%), carbohydrate
(5-23 wt.%) and lipid (7-23 wt.%). Eustigmatophytes are rich in one or both of the
20:5(n-3) and 22:6(n-3) polyunsaturated fatty acids. According to Ross et al. (2009),
microalgae with high lipid content could be a future source of third generation
biofuels and chemicals.The oil content itself can be estimated to be 64.4 % of the total
lipid component. Thus, Nannochloropsis Gaditana (NG) microalgae, belongs to
Eustigmatophytes microalgae specie, have been proposed as a candidate to carry out
this study.
Interest towards the quality and characteristics of bio-oil from microalgae is
revived nowadays, due to growing concerns over emissions, energy supply, oil prices
and availability [2]. The conversion technologies for utilizing microalgae biomass can
be divided into two basic categories of conversion: thermochemical and biochemical .
Thermal technologies to process algae include direct combustion, pyrolysis and
gasification. Combustion is the conversion of biomass fuels to several forms of useful
energy in the presence of air or oxygen. Pyrolysis is a process that can be employed to
convert algal biomass material into biofuel and gas in the absence of air or oxygen

Chapter 1
65
(350-700ºC). Gasification involves the partial oxidation of biomass into a combustible
gas at high temperatures (800-900ºC).
On the other hand, biological processes can allow the conversion of biomass into
other fuels by means of anaerobic digestion, alcoholic fermentation and
photobiological hydrogen production. Despite its inherent potential as a biofuel
resource, the commercial viability of algal biofuel technology has not been achieved
yet.
A large number of researches on microalgae pyrolysis have been carried out in
recent years[2-4].Very few studies have been focused on the combustion
ofmicroalgae. Chen et al., (2011) reported the combustion behavior of Chlorella
vulgaris microalgae under different oxygen concentrations by thermogravimetric
analysis (TGA). Furthermore, Tang et al. [5] investigated the combustion of Chlorella
protothecoides microalgae in N2/O2 and CO2/O2 atmospheres by means of same
technique. [6]studied the effects of temperature in the combustion of marine algae.
However, at the best of our knowledge, the gasification process of marine biomass has
not been explored yet.
The pyrolysis behavior of brown algae has been investigated using
thermogravimetry and pyrolysis-GC-MS (PY-GC/MS) [1; 7] and thermogravimetry-
differential scanning calorimetry (TG/DSC) [3].
During the process of thermochemical conversion of biomass, the composition of
the gas emissions should be determined before industrial application. In this sense, the

Chapter 1
66
evolution with time on stream of the volatile products evolved in the marine biomass
pyrolysis or combustion processes has been carried out using the on-line combination
of TGA and Fourier Transform Infrared Spectrometry (FTIR) [8]and thermal analysis-
mass spectrometry (TA-MS) [9; 10].
As aforementioned, thermogravimetric analysis coupled with mass spectrometry
(TGA-MS) could be a useful technique to obtain information at real-time of mass loss
and evolved gases for pyrolysis, oxidation and gasification processes.
The aim of the present study was to investigate the pyrolysis, combustion and
gasification characteristics of theNannochloropsisGaditanamicroalgae by means of
TGA.In addition, the effects of different operation conditions were studied. Moreover,
evolved gases for the thermochemical conversion of NGmicroalgae were also
evaluated using the MS technique.
1.2. EXPERIMENTAL
1.2.1. Materials
NannochloropsisGaditana(NG microalgae) from Alga EnergyCompany was used
in this work. It was collected in Cadiz Bay (Spain) and delivered in green powder with
100 µm average particle size.Its composition in dry basis is about 17.6 wt. % of lipids,
12.6 wt. % of fatty acids and 24.1 wt.% of proteins.
The proximate and ultimate analyses of the NG microalgae are shown in Table
1.The proximate analyses were carried outaccording to the technical specifications
UNE-EN UNE-EN 14775:2010, UNE-EN 15148:2010 and UNE-EN 1474-2 for ash,

Chapter 1
67
volatile matter and moisture determination, respectively. Metal salts contained in
biomass have a significant impact on the thermal conversion processes[4]. In this
research, the content of metals in the sample was determined by Inductively Coupled
Plasma Spectrometry (ICP) (Table 1).
Table 1.Proximate, ultimate analysis and mineral content determined by ICP of the
NannochloropsisGaditanamicroalgae.
Ultimate analysis (wt.%)
Biomass C H N S Oa
NannochloropsisGaditana
47.26 7.03 6.72 0.49 38.5
Proximate analysis (wt. %)
Moisture Ash Volatile matter
Fixed Carbon
5.12 10.68 75.91 8.29
Mineral content (ppm)
Ca Fe Na K P Mg Zn
8652
5
170
7 23817 1385 9042
189
2 127
a % of oxygen calculated from difference of C, H, N and S.
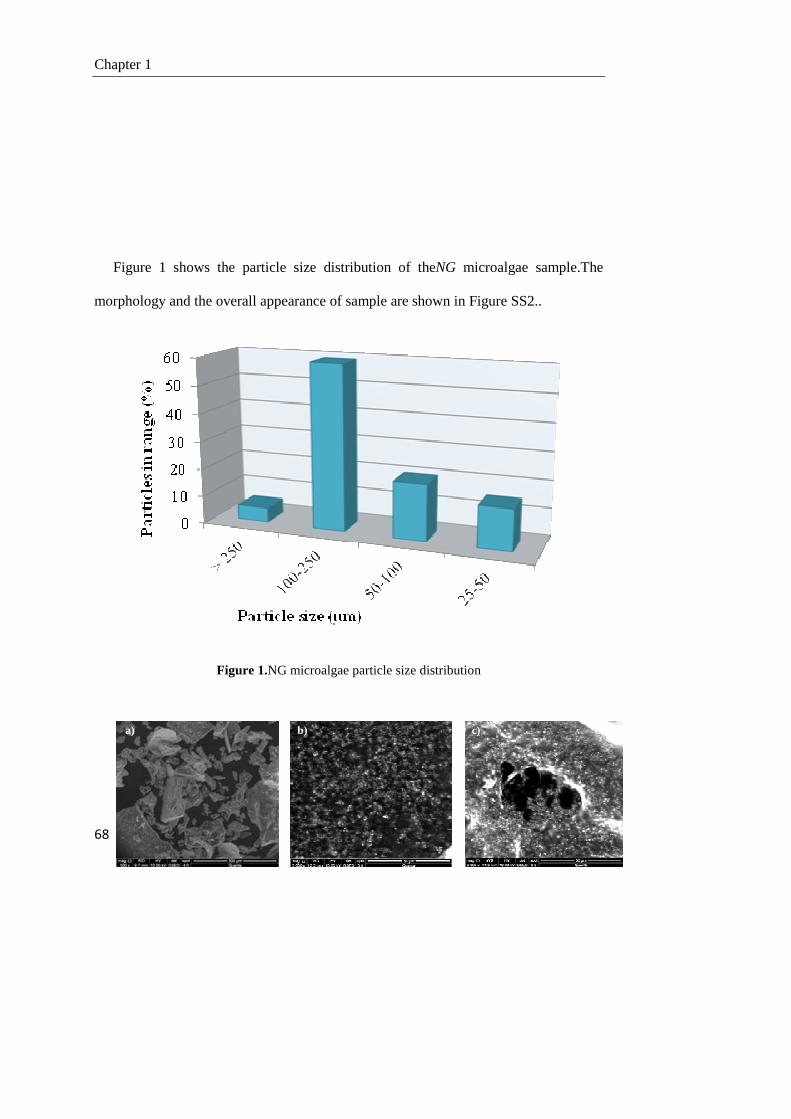
Chapter 1
68
a) b) c)
Figure 1 shows the particle size distribution of theNG microalgae sample.The
morphology and the overall appearance of sample are shown in Figure SS2..
Figure 1.NG microalgae particle size distribution

Chapter 1
69
Figure SS2. (a) SEM micrograph of NG microalgae sample. (b) SEM micrographs of the
resulting char after the devolatilization step for 25-50 µm and (c) SEM micrographs of the
resulting char after the devolatilization step for >250 µm.
1. 2.2. Equipment and Procedures
Pyrolysis, combustion and gasification experiments were carried out in a TGA
apparatus (TGA-DSC 1, METTLER TOLEDO). Each sample was analyzed at least
three times, being the average value recorded. The experimental error in theweight
loss and temperature measurements was ± 0.5% and ± 2 ºC, respectively.
1.2.2.1. Thermal Analysis for the Pyrolysis process
The sample was heated from 40 to 1200ºC at a heating rate of 40 ºC/min under
Argon (99.996 %) atmosphere. Initial sample weight, Argon flow rate and particle size
of the sample were varied in order to obtain the most suitable operating conditions to
avoid the effects of heat and mass transfer limitations.
1.2.2.2. Thermal Analysis for the Combustion process
The sample was preheated at 125 ºC for 10 min in order to remove the moisture
content.Subsequently, the sample was heated from 125 to 1000 ºC under a reactive
atmosphere of pure oxygen (99.99 %). Initial sample weight, oxygen flow rate and
particle size of the sample were evaluated in order to obtain the most suitable

Chapter 1
70
operating conditions to avoid the effects of heat and mass transfer limitations. Finally,
the oxygen concentration was evaluated. On this account, experiments were performed
under atmospheres containing 20 %, 40 %, 60% and 80 % of Oxygen in Argon.
1.2.2.3. Thermal Analysis for the Gasification process
Figure SS1 shows the experimental set-up used for the gasification process.
Gasification experiments were conducted in the presence of water vapor generated by
a bubbler system. Ar was bubbled through degassed water heated to different
temperatures. The effect of the gasification temperature, initial sample weight, Argon
flow rate, particle size of the sample and water vapour concentration were evaluated.
The gasification of the sample was performed in three steps:
• Drying: the sample was heated in an inert atmosphere of pure Ar from 30 to
125 ºC at a heating rate of 15 ºC/min.
• Pyrolysis: the sample was heated from 125 ºC to the operating temperature at
a heating rate of 40 ºC/min. Ar was used as the carrier gas (200 ml/min (25 ºC,
0.9 atm)).
• Gasification: the char obtained in the pyrolisis process was later gasified with
the reactive gas mixture (Ar + H2O) at the test temperature for one hour.
In this paper, X was the char conversion, which is defined as:
� =�� −�
�� (eq. 1)
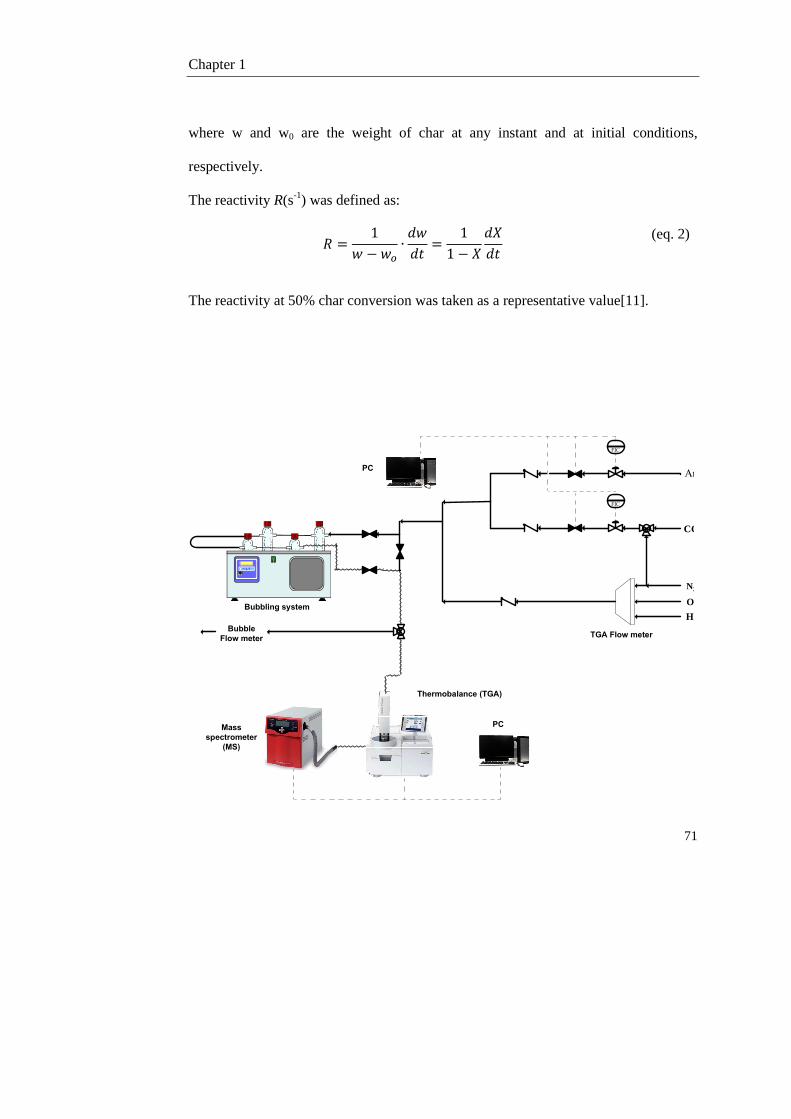
Chapter 1
71
FIC
FIC
Bubble
Flow meter
Thermobalance (TGA)
Mass
spectrometer
(MS)
N2
CO
O2
He
TGA Flow meter
Bubbling system
01
O 1
Ar
PC
PC
where w and w0 are the weight of char at any instant and at initial conditions,
respectively.
The reactivity R(s-1) was defined as:
� =1
� − ��∙�
=
1
1 − �
�
(eq. 2)
The reactivity at 50% char conversion was taken as a representative value[11].

Chapter 1
72
Figure SS1. Experimental set-up for the gasification process.
1.2.2.4. TGA-MS Analysis of the Gaseous Products
The analysis of the gas products distribution coming from the thermal analysis was
carried out in a thermogravimetricanalyzer (TGA-DSC 1; METTLER TOLEDO)
coupled to a mass spectrometer (Thermostar-GSD 320/quadrupole mass analyzer;
PFEIFFER VACUUM) with an electron ionization voltage at 70 eV and provided
mass spectra up to 300 a.m.u. The interface was wrapped with heating wire to
circumvent condensation of exhausting gases. Pyrolysis, combustion and gasification
experiments were carried out under the selected operating conditions. In order to
identify ions with m/z in the range 0-300, a preliminary broad scan was performed at a
heating rate of 40 ºC/min.
Although a quantitative analysis was not performed in this work, a comparison of the
intensity peak areas between different samples (semiqualitative analysis) was carried
out by using a normalization procedure. The method used in this work was based on
the relative integrated peak linear intensity normalized to total integrated peak linear
intensities and to sample weight (eq. 3), that are reported elsewhere [12; 13].
�(� )� =� �
((∑ � �) ∙ �)
(eq. 3)
where R(Int)i is the relative integrated peak linear intensity, Inti is the integrated
intensity of a gas species, and m is the mass of the sample. Ion fragments with R(Int)
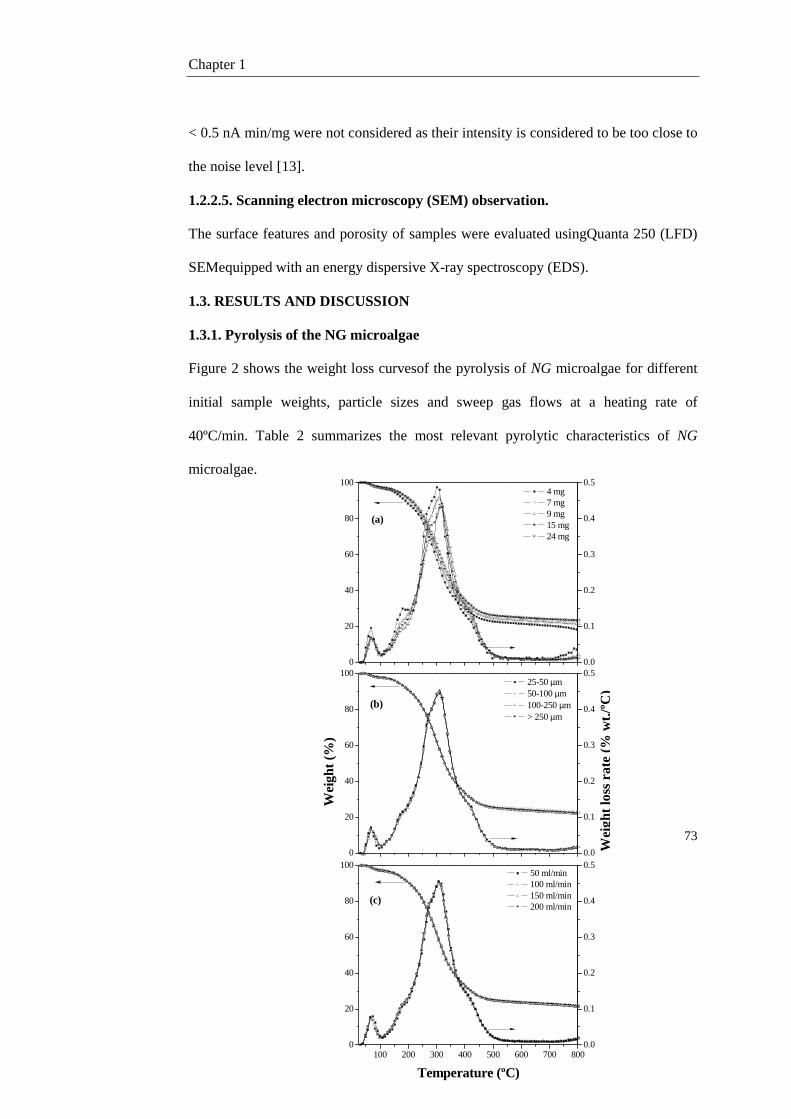
Chapter 1
73
0
20
40
60
80
100
0
20
40
60
80
100
100 200 300 400 500 600 700 8000
20
40
60
80
100
(c)
(b)
4 mg 7 mg 9 mg 15 mg 24 mg
(a)
0.0
0.1
0.2
0.3
0.4
0.5
Wei
ght (
%)
25-50 µm 50-100 µm 100-250 µm > 250 µm
0.0
0.1
0.2
0.3
0.4
0.5
Wei
ght l
oss
rate
(%
wt./
ºC)
Temperature (ºC)
50 ml/min 100 ml/min 150 ml/min 200 ml/min
0.0
0.1
0.2
0.3
0.4
0.5
< 0.5 nA min/mg were not considered as their intensity is considered to be too close to
the noise level [13].
1.2.2.5. Scanning electron microscopy (SEM) observation.
The surface features and porosity of samples were evaluated usingQuanta 250 (LFD)
SEMequipped with an energy dispersive X-ray spectroscopy (EDS).
1.3. RESULTS AND DISCUSSION
1.3.1. Pyrolysis of the NG microalgae
Figure 2 shows the weight loss curvesof the pyrolysis of NG microalgae for different
initial sample weights, particle sizes and sweep gas flows at a heating rate of
40ºC/min. Table 2 summarizes the most relevant pyrolytic characteristics of NG
microalgae.

Chapter 1
74
Figure 2.Thermogravimetric (TGA) and differential thermogravimetric (DTG) curves of the
NG microalgae pyrolysis process as a function of: (a) initial weight, (b) particle size and (c) gas
flow rate at a heating rate of 40 ºC/min.
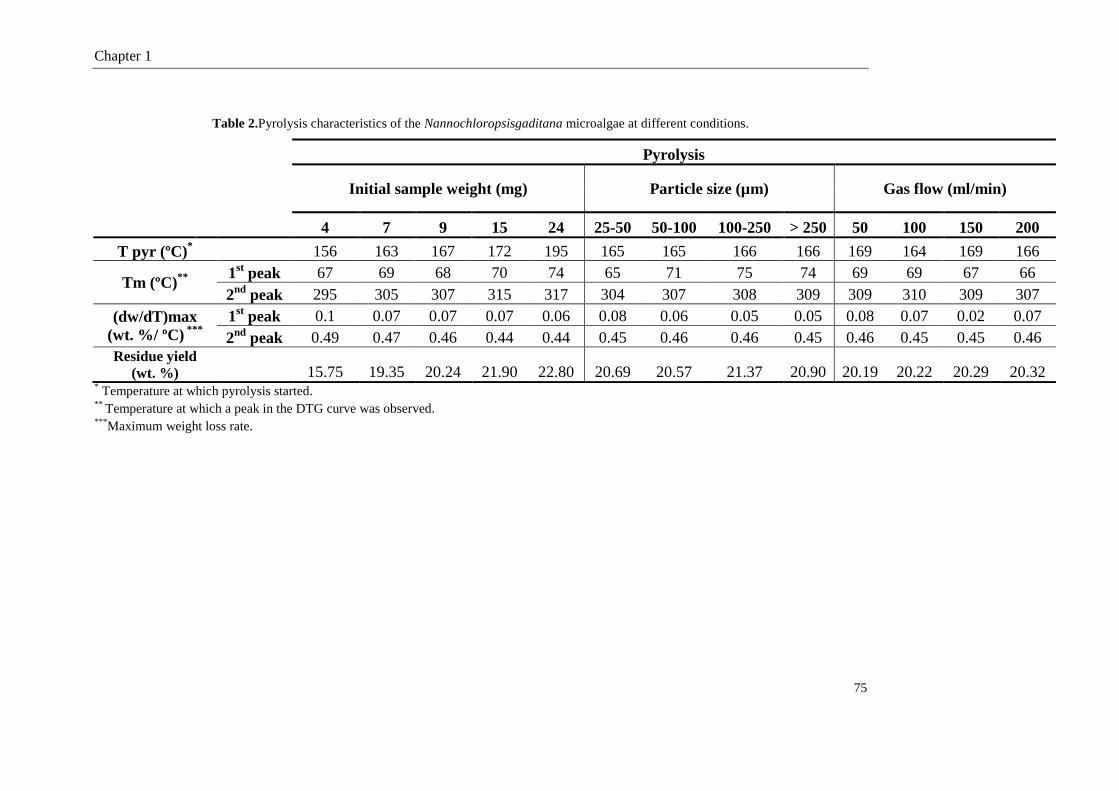
Chapter 1
75
Table 2.Pyrolysis characteristics of the Nannochloropsisgaditana microalgae at different conditions.
Pyrolysis
Initial sample weight (mg) Particle size (µm) Gas flow (ml/min)
4 7 9 15 24 25-50 50-100 100-250 > 250 50 100 150 200
T pyr (ºC)* 156 163 167 172 195 165 165 166 166 169 164 169 166
Tm (ºC)** 1st peak 67 69 68 70 74 65 71 75 74 69 69 67 66 2nd peak 295 305 307 315 317 304 307 308 309 309 310 309 307
(dw/dT)max (wt. %/ ºC) ***
1st peak 0.1 0.07 0.07 0.07 0.06 0.08 0.06 0.05 0.05 0.08 0.07 0.02 0.07 2nd peak 0.49 0.47 0.46 0.44 0.44 0.45 0.46 0.46 0.45 0.46 0.45 0.45 0.46
Residue yield (wt. %) 15.75 19.35 20.24 21.90 22.80 20.69 20.57 21.37 20.90 20.19 20.22 20.29 20.32
* Temperature at which pyrolysis started. ** Temperature at which a peak in the DTG curve was observed. *** Maximum weight loss rate.

Chapter 1
76
In good agreement with literature [3; 7-9], the thermogravimetric (TGA) and
differential thermogravimetric (DTG) curves revealed three degradation steps
common to all studied work conditions.The first stage (40-160ºC) was associated with
a small weight loss due to dehydration (cellular water and external water). The second
stage represented the main devolatilization reactions, where most of the sample weight
was lost as volatile matter (160-450 ºC). Three shoulders canbe distinguished in this
stage, being the low-temperature peak (180 ºC)mainly associated to the degradation of
protein and soluble polysaccharide whereas the higher temperature peaks (271 and
411 ºC) would correspond to the degradation of crude cellulose in the cell wall, other
insoluble polysaccharides and crude lipid[14]. Finally, the laststagetook place at
temperatures above 450 ºC leading to char formation[8].
The effect of the initial mass of the sample on the NG microalgae pyrolysis was
also examined (Figure 2a). Experiments were performed using different initial sample
weights (4-24 mg) with particle sizeof 100-250 µm. Argon flow rate was fixed at200
ml/min (25 ºC, 0.9 atm). In agreement with Antal (1998) and Stenseng et al. (2001),
increasing sample mass shifts the pyrolysis process to higher temperatures turning into
higher residue yields (from 16 to 23 wt.%). The height of the DTG peaks
decreasedwhereas the width increased withincreasing sample weights. However,
TGA/DTG curves for weights of 7 and 9 mg overlapped, indicating negligible
internal-thermal and external-mass transfer limitations. As described by Antal (1998),
the lower peak height would correspond with the increase in the peak width and
ahigher char yield.

Chapter 1
77
Therefore, high mass loadings caused heat-transfer and mass-transfer problems
delaying the pyrolytic process[15]. In addition, the shape of the peak was slightly
distorted at the high mass sample[15]. On the basis of the results described above,
initial mass sample of 9 mg was selected for the following experiments.
Figure 2b shows TGA/DTG plots versus temperature obtained from the pyrolysis
of the NG microalgae at different particle sizes (25-50, 50-100, 100-250 and >250 µm)
with initial mass of 9 mg and Ar flow rate of 200 ml/min (25 ºC, 0.9 atm). In all
cases,the second and third stagesof thepyrolyticTGA/DTG profiles were similar for
the second and third stages. However, the first stage was delayed for sample
sizesbigger than 50µm. According to Mani et al. (2010), this fact could be attributed to
the fact thatsmaller particles have larger surface area leading to less diffusion
resistance for the pyrolysis reaction.On the other hand, the residueproduced (≈21
wt.%)remained constant regardless of the particle sizeused(Table 2). Therefore, a
particle size of 100-250 µmwas selected to avoid the grinding or milling of the sample
due to the NG microalgae sample had a narrow particle size distribution centered on
this particle size range (Figure 1a).
TGA/DTG curves for different sweep gas flows (50, 100, 150 and 200 ml/min) (25
ºC, 0.9 atm) using initial mass of 9 mg and a particle size range of 100-250 µm are
shown in Figure 2c. As it canbe seen, the gas flow did not affect the pyrolysis
outcomes [15]. In all cases, the amount of residue obtainedwas practically constant (20
wt.%). However, higher sweep gas flows were required in order to avoid secondary
reactions due to long residence times inside the TGA[16].
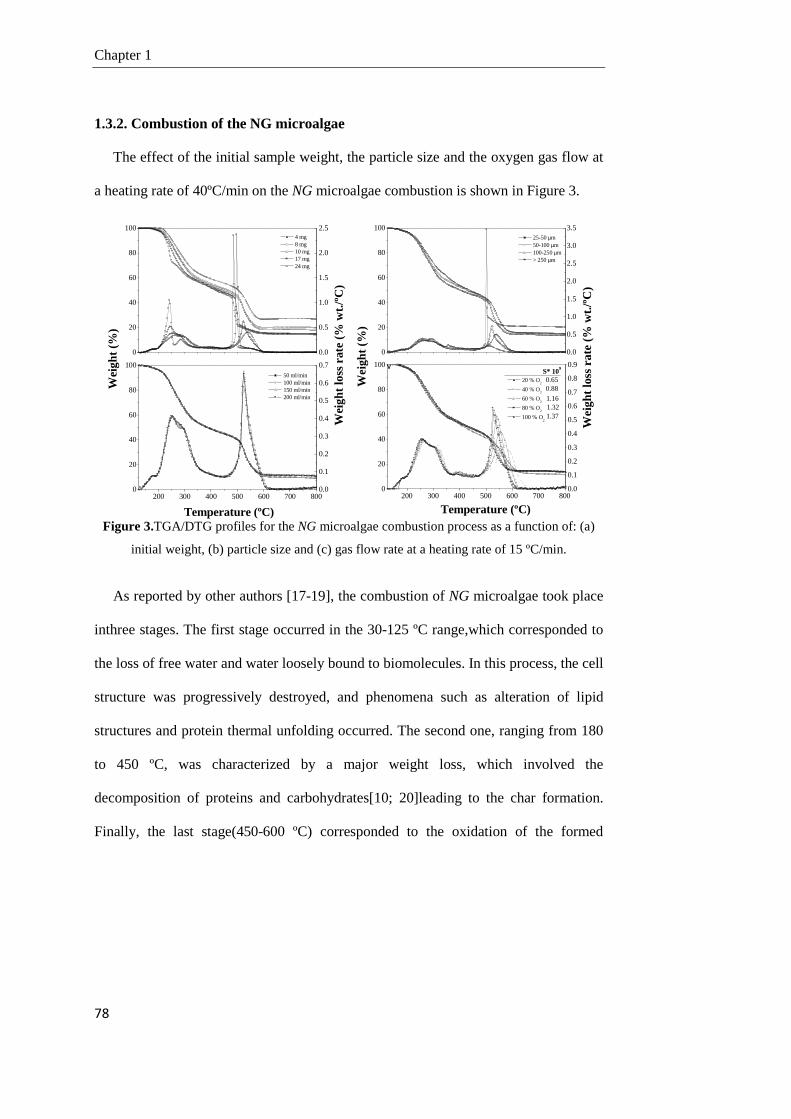
Chapter 1
78
0
20
40
60
80
100
0
20
40
60
80
100
200 300 400 500 600 700 8000
20
40
60
80
100
200 300 400 500 600 700 8000
20
40
60
80
100
4 mg 8 mg 10 mg 17 mg 24 mg
0.0
0.5
1.0
1.5
2.0
2.5
25-50 µm 50-100 µm 100-250 µm > 250 µm
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Temperature (ºC)
Wei
ght (
%)
50 ml/min 100 ml/min 150 ml/min 200 ml/min
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
W
eigh
t los
s ra
te (
% w
t./ºC
)
1.371.321.160.880.65
Wei
ght (
%)
Temperature (ºC)
20 % O2
40 % O2
60 % O2
80 % O2
100 % O2
S* 109
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
Wei
ght l
oss
rate
(%
wt./
ºC)
1.3.2. Combustion of the NG microalgae
The effect of the initial sample weight, the particle size and the oxygen gas flow at
a heating rate of 40ºC/min on the NG microalgae combustion is shown in Figure 3.
Figure 3.TGA/DTG profiles for the NG microalgae combustion process as a function of: (a)
initial weight, (b) particle size and (c) gas flow rate at a heating rate of 15 ºC/min.
As reported by other authors [17-19], the combustion of NG microalgae took place
inthree stages. The first stage occurred in the 30-125 ºC range,which corresponded to
the loss of free water and water loosely bound to biomolecules. In this process, the cell
structure was progressively destroyed, and phenomena such as alteration of lipid
structures and protein thermal unfolding occurred. The second one, ranging from 180
to 450 ºC, was characterized by a major weight loss, which involved the
decomposition of proteins and carbohydrates[10; 20]leading to the char formation.
Finally, the last stage(450-600 ºC) corresponded to the oxidation of the formed

Chapter 1
79
remaining char.At the end of this stage, it was observed that between 25 and 8 wt.% of
the char was not completely oxidized, depending on the conditions used.The main
thermogravimetric features in the combustionof NG microalgae aresummarizedin
Table 3.The influence of the initial weightwas investigated under oxygen atmosphere
for different initial masses (4, 8, 10, 17 and 24 mg)of the NG microalgae samplewith
particle sizeranging from100 to 250 µm and an oxygen flow rate of 100 ml/min (25
ºC, 0.9 atm)(Figure 3a). The initial weight had a significant effect on the thermal
degradation behavior. In agreement with some studies reported in the literature [21;
22], the higher the sample weight, the lower both the temperature needed for the
combustion process and the residue yields were (from 25 to 14 wt.%). Likewise, the
DTG peaks heightincreased and the width decreased with increasing sample weights.
As observed for the pyrolysis process, TGA/DTG curves for sample weights of 8 and
10 mg overlapped, indicating negligible internal-thermal and external-mass transfer
limitations[15]. On the basis of the results discussedabove, a sample weight of10 mg
was chosen for the experiments of the next section in order to ensure the detection of
the gas products distribution coming from the combustion of the NG microalgae by
means of a mass spectrometer.
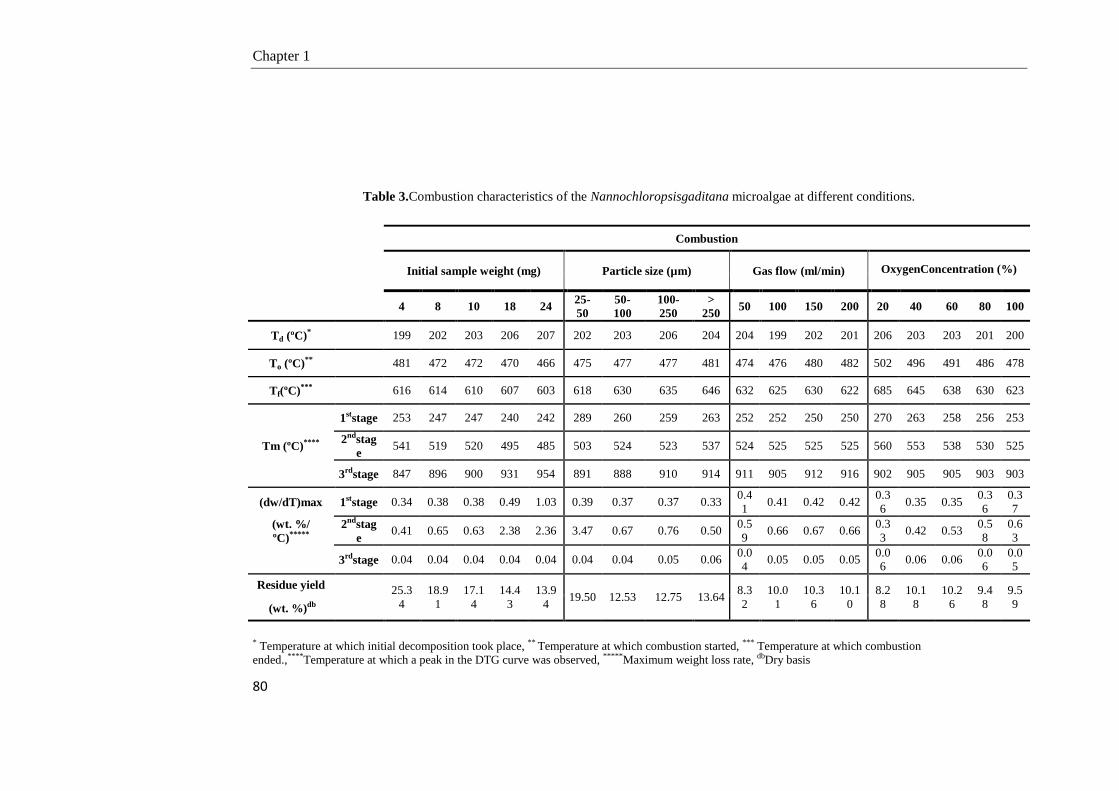
Chapter 1
80
Table 3.Combustion characteristics of the Nannochloropsisgaditana microalgae at different conditions.
Combustion
Initial sample weight (mg) Particle size (µm) Gas flow (ml/min) OxygenConcentration (%)
4 8 10 18 24 25-50
50-100
100-250
> 250 50 100 150 200 20 40 60 80 100
Td (ºC)* 199 202 203 206 207 202 203 206 204 204 199 202 201 206 203 203 201 200
To (ºC)** 481 472 472 470 466 475 477 477 481 474 476 480 482 502 496 491 486 478
Tf(ºC)*** 616 614 610 607 603 618 630 635 646 632 625 630 622 685 645 638 630 623
Tm (ºC)****
1ststage 253 247 247 240 242 289 260 259 263 252 252 250 250 270 263 258 256 253
2ndstage
541 519 520 495 485 503 524 523 537 524 525 525 525 560 553 538 530 525
3rdstage 847 896 900 931 954 891 888 910 914 911 905 912 916 902 905 905 903 903
(dw/dT)max 1ststage 0.34 0.38 0.38 0.49 1.03 0.39 0.37 0.37 0.33 0.41
0.41 0.42 0.42 0.36
0.35 0.35 0.36
0.37
(wt. %/ ºC)*****
2ndstage
0.41 0.65 0.63 2.38 2.36 3.47 0.67 0.76 0.50 0.59
0.66 0.67 0.66 0.33
0.42 0.53 0.58
0.63
3rdstage 0.04 0.04 0.04 0.04 0.04 0.04 0.04 0.05 0.06 0.04
0.05 0.05 0.05 0.06
0.06 0.06 0.06
0.05
Residue yield
25.34
18.91
17.14
14.43
13.94
19.50 12.53 12.75 13.64 8.32
10.01
10.36
10.10
8.28
10.18
10.26
9.48
9.59 (wt. %)db
* Temperature at which initial decomposition took place, ** Temperature at which combustion started, *** Temperature at which combustion ended.,**** Temperature at which a peak in the DTG curve was observed, ***** Maximum weight loss rate, dbDry basis

Chapter 1
81
The effect of the particle size on the combustion of the NG microalgae was studied
for four different particle sizes (25-50, 50-100, 100-250 and >250 µm) usingan initial
weightof 10 mg and an oxygen flow rate of 100 ml/min (Figure 3b). According to the
TGA/DTG curves, the most reactive sample was the NG microalgae with the smallest
particle size(25-50 µm). In addition, the residue yield was slightly reduced at higher
particles sizes (from 19 to 13 wt.%).Chouchene et al. (2010) noted that the char
oxidation of the finest particles samplestook place at lower temperaturesthan those
corresponding to higher sizes. These results are in agreement with those reported in
previous works focused on the study of the influence of the particle size on biomass
combustion[18; 23].Finally, TGA/DTG profiles for particle sizesranging from 50 to
100 andfrom 100 to 250 µm overlapped.
Figure 3c shows TGA/DTG curves of the NG microalgae for different pure oxygen
gas flows (50, 100, 150 and 200 ml/min) (25 ºC, 0.9 atm), an initial sample weight of
10 mg and particle sizesranging from 100 to 250 µm. Regardless the oxygen flow
rates used, the combustion TGA/DTG profiles remained practically the same. Thus,
the combustion behavior of the NG microalgae was not significantly affected by the
oxygen flow rate due to the oxygen concentration was kept constant anyway.
The effect of oxygen concentration on the combustion of the NG microalgae was
studied for five different oxygen concentration (20/80, 40/60, 60/40, 80/20 and 100/0
oxygen/Argon ratios) using an initial weight of 10 mg, a total flow rate of 100 ml/min
and a particle size range of 100-250 µm (Figure 3.d). TGA-DTG profiles show that
the first decomposition step (180-400 ºC) was not influenced by the oxygen

Chapter 1
82
concentration as the curves almost overlap. This phenomenon is attributed to the
thermal decomposition of the sample is in the kinetic control zone, being mainly
affected by the temperature, and the effect of oxygen concentration is almost
negligible [17]. However, in the temperature range between 380 and 475 ºC a small
peak appeared in the DTG curves for oxygen concentrations of 20, 40 and 60 % being
unappreciated for higher values of oxygen concentration. The effect of the oxygen
concentration is highly remarked between 480 and 600 ºC, where the oxidation of the
char was taking place. As the oxygen concentration is increased both, the initial
oxidation temperature and the peak temperature in the DTG curve shifted to lower
temperatures, whereas the final temperature of oxidation was decreased. On the other
hand, the maximum weight loss was higher for increasing values of oxygen (Table 3).
Thus, the oxygen concentration enhanced the combustion of the remaining char. These
results agreed well with literature [18; 24], being attributed to the fact that the
combustion reaction of the NG microalgae is in the diffusion control zone and the
oxygen concentration becomes the major influencing factor [24]. In these types of
tests, combustion characteristic index S is usually used to evaluate the combustion
behavior of biomass [24]. S is defined as follows:
� =(��
��)��� ∙ (
��
��)����
��� ∙ ��
(eq. 4)
where (dw/dt)max and (dw/dt)mean are maximum and average mass loss rates,
respectively. Ti and Tb are the ignition and burnout temperatures. It is established, than
the bigger the value S is, the higher the combustion activity [24]. Figure 3.d shows the

Chapter 1
83
value of S for the different oxygen concentrations studied. It can be observed that S
increased as the concentration of oxygen was increased. However, for oxygen
concentrations above 60 %, the increasing trend of S was stabilized. Thus, optimum
values of oxygen concentrations were found to be between 60 and 100 %.
1.3.3. Gasification of the NG microalgae
Biomass gasification depends mainly on the biomass type and the operating
conditions, such as particle size, char porosity, temperature and partial pressure of the
gasifying agents [11; 25; 26]. The gasification process generally includes a
devolatilization step (pyrolysis) and a char gasification step.The char obtained after
the devolatilization step was later gasified.In this study, a devolatilization step was
carried out by heating the samples from 125 to 850 ºC at a heating rate of 40ºC/min
under Aratmosphere.Figure 4 displays the char conversion (X) vs timeon stream
obtainedat different temperatures, initial sampleweights, particle sizes,Argongas flows
and steam concentrations. Furthermore, Table 4 lists the most relevant gasification
characteristics of the NG microalgae.
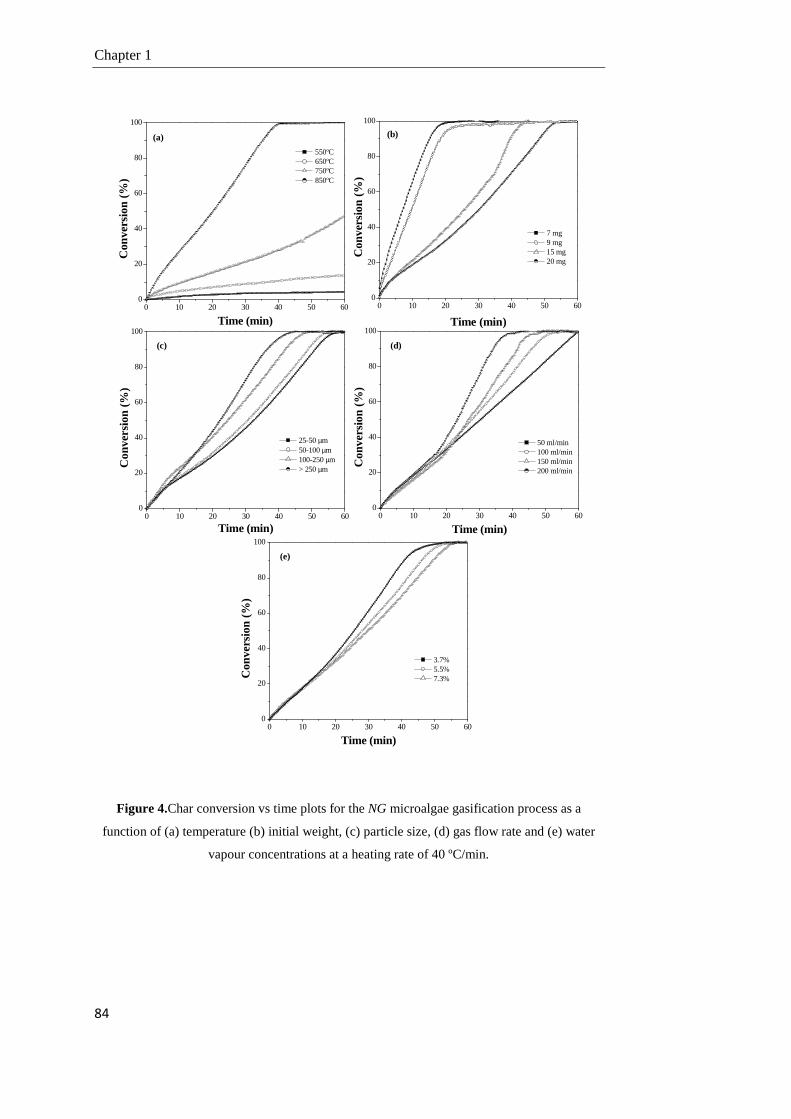
Chapter 1
84
Figure 4.Char conversion vs time plots for the NG microalgae gasification process as a
function of (a) temperature (b) initial weight, (c) particle size, (d) gas flow rate and (e) water
vapour concentrations at a heating rate of 40 ºC/min.
0 10 20 30 40 50 600
20
40
60
80
100
0 10 20 30 40 50 600
20
40
60
80
100
0 10 20 30 40 50 600
20
40
60
80
100
0 10 20 30 40 50 600
20
40
60
80
100
0 10 20 30 40 50 600
20
40
60
80
100
(d)
Con
vers
ion
(%)
Time (min)
50 ml/min 100 ml/min 150 ml/min 200 ml/min
(b)
7 mg 9 mg 15 mg 20 mg
Con
vers
ion
(%)
Time (min)
Con
vers
ion
(%)
Time (min)
550ºC 650ºC 750ºC 850ºC
(a)
(c)
Con
vers
ion
(%)
Time (min)
25-50 µm 50-100 µm 100-250 µm > 250 µm
(e)
Con
vers
ion
(%)
Time (min)
3.7% 5.5% 7.3%

Chapter 1
85
Table 4.Gasification characteristics of the Nannochloropsisgaditana microalgae at different
conditions.
Nannocholoropsisgaditana (NG) Temperature(ºC)
550 650 750 850
Time X50 (min) >60 >60 >60 19.9
Reactivities ((s-1)*104) - - - 9.1
Initial sample weight (mg)
7 9 15 20
Time X50 (min) 7.1 9.8 25.8 30.2
Reactivities ((s-1)*104) 18.3 16.4 9.2 8.5
Particle size (µm)
25-50 50-100 100-250 > 250
Time X50 (min) 32 30.2 24.2 23.6
Reactivities ((s-1)*104) 6.3 7.4 7.6 8.2
Gas flow (ml/min)
50 100 150 200
Time X50 (min) 33.0 30.1 27.9 24.4
Reactivities ((s-1)*104) 5.3 7.5 9.9 48.2
Water vapour concentration (%)
3.7 5.5 7.3
Time X50 (min) 33 30.1 27.9
Reactivities ((s-1)*104) 6.2 7.5 8.3

Chapter 1
86
The effect of the temperature(550, 650, 750 and 850 ºC) on the NG microalgae
chargasification was studied considering an initial weight of 9 mg, a particle size
ranging from 100 to 250 µm,an Argon flow rate of 200 ml/min, a steam concentration
of 5 vol.% in Argon and a heating rate of 40ºC/min. As expected, the higher the
gasification temperature, the higher both, the char conversion and the reactivitywere.
The same behavior was reported elsewhere[11; 25; 26].50% of char conversion at
850ºC was achieved after 20 min, whereas at lower temperatures this level of
conversion was achieved after 60 min. In fact, for the period of 60 min, only 48% of
the char was converted at 750 ºC. According to Mani et al. (2011), the char
gasification at temperatures lower than 1000 ºC, the chemical reaction was the rate-
determining step.This way, 850 ºC was chosenas the gasification temperature for the
following experiments due to the fact that a high amount of char is converted in less
time.
Figure 4b shows the conversionof the NG microalgae char in the gasification
process versus time on stream for different initial weights (7, 10, 15 and 20 mg) at
850ºC for particle sizes ranging from 100 to 250 µm, an Ar flow rate of 200 ml/min
(25 ºC, 0.9 atm), a steam concentration of 5 vol.%in Arand a heating rate of 40ºC/min.
The reactivity increasedwith decreasing initial sample weights.It is clear that the
sample weight had a significant influence on the time to reach a plateau in all
conversion curves. In fact, a constant conversion (50%) for 7 mg was obtained after 18

Chapter 1
87
min, whereas for 20 mg was achieved after 30 min. As expected, the reactivity
decreased with the sample weight. On the basis of the results described above, an
initial weightof the sample of 20 mg was selected. This value allowed to achieve
reproducible experimental TGA data and to improve the sensitivity of the mass
spectrometer.
The effect of the particle size on the gasification of the NG microalgae char was
studied for four different particle sizes (25-50, 50-100, 100-250 and >250 µm)at
850ºC foran initial sample weight of 20 mg, an Ar flow rate of 200 ml/min (25 ºC, 0.9
atm), a steam concentration of 5 vol.% in Argon and a heating rate of 40ºC/min
(Figure 4c).Among the particle size ranges investigated, the higher the particle size,
the shorter the time to reach a constant conversion was.However, according to the
reported literature [26; 27], the reactivity should decrease with increasing particle
sizes as a consequence of an increase in the diffusion resistance in the gasification
process.In order to explain this finding, the surface feature and the porosity of the
resulting char after the devolatilization step for each particle size was evaluated
bySEM analyses (Figure SS2). SEM micrographs clearly showed that the porosity
increasedwith the particle size justifying this unexpected behavior.In order to confirm
SEM results, Nitrogen adsorption-desorption isotherms were carried out to the
samples according to J.A. Díaz procedure [28]. This way, bigger samples of the NG
microalgae were pyrolyzed in a flux bed reactor keeping the same operating
conditions (Ar flow rate of 200 Nml/min, heating rate of 40 ºC/min and a final
temperature of 850 ºC) for obtaining a bigger amount of char. The results obtained

Chapter 1
88
backed up the SEM analyses showing than the char produced from the pyrolysis of the
NG microalgae had a non-very porous structure. Isotherms obtained can be assigned
to a type II according to the IUPAC classification with a small hysteresis loop due to
capillary condensation [28]. Type II corresponds to non-porous materials. The char
left from the minimum particle range sample was the least porous (0.006 g/cm3), and
the sample within the maximum particle range shown the highest porosity (0.01
g/cm3).The increase in the sample porosity affected the profile of total conversion-
time on stream relationship (Figure 4c). The reactivity increased with porosity due to
the reduction in gasifying agent diffusion resistance, which consequently decreased
the time to reach a plateau in conversion[29].According to these results, particle sizes
ranging from100 to 250 µm were selected for the following experiments.
The sweep gas flow determines the gas residence time during the biomass gasification.
Figure 4d shows the conversion versus time on stream curves of the NG microalgae
charat 850ºC for different argon gas flows (50, 100, 150 and 200 ml/min) (25 ºC, 0.9
atm), an initial weight of 20 mg, a particle size ranging from 100 to 250 µm, a steam
concentration of 5 vol. % in Argon and a heating rate of 40ºC/min.As the argon gas
flow increased, the conversion vs time curves shifted to a higher conversion
rate.According to Zhang et al. (2010), low sweep gas flow (long residence time)results
in the formation of carbon deposits, consequently decreasing the gas yield. Thus, a
value of argon flow of 200 ml/min (25 ºC, 0.9 atm)was chosen in order to minimize
secondary reactions such as thermal cracking, repolymerization and
recondensation[30] and increase the char conversion [29; 31].

Chapter 1
89
The effect of the steam concentration on the gasification of the NG microalgae char
was studied (3.7, 5.5 and 7.3 % in Argon) at 850ºC, for an initial weight of 20 mg, a
particle size ranging from 100 to 250 µm, an argon flow rate of 200 ml/min (25 ºC, 0.9
atm)and a heating rate of 40ºC/min (Figure 4e). As expected, the reactivity increased
with increasing steam concentrations. According to Florin and Harris (2008), water
vapour has been identified as a catalyst for the char formation mechanism, enhancing
the concentration of H2during the char gasification due to the occurrence of the water-
gas shift reaction. This is in agreement with previous reported studies [26]. However,
a steam concentration of 5.5 % was selected for the following experiments since at
higher concentrationsoverpressure problems in the bubbler system were observed.
1.3.4. Gas product analysis
The main productsderived from the pyrolysis, oxidation and gasification of theNG
microalgae were evaluatedby TGA-MS analysis. On the basis of a preliminary scan, a
list of key molecular ions was compiled by rejecting signals when the maximum
intensity was close to the noise level[32].Thedatabase of National Institute of
Standards and Technology (NIST) were usedfor the atomic mass units (a.m.u.)
selection.Furthermore, elemental analyses by Energy Dispersive X-ray Spectroscopy
(EDS) were performed on the NG microalgae and the resulting solid residue obtained
after the pyrolysis and oxidation process (Table 5).Characteristic peaks of C, N, O,
Na, Mg, P, S, Cl, K and Ca were presented in the analysis of the NG microalgae.
However, peaks correspondingto the N element were not detected in the EDS analysis
of the solid residues obtained after the pyrolysis and combustion of the microalgae.
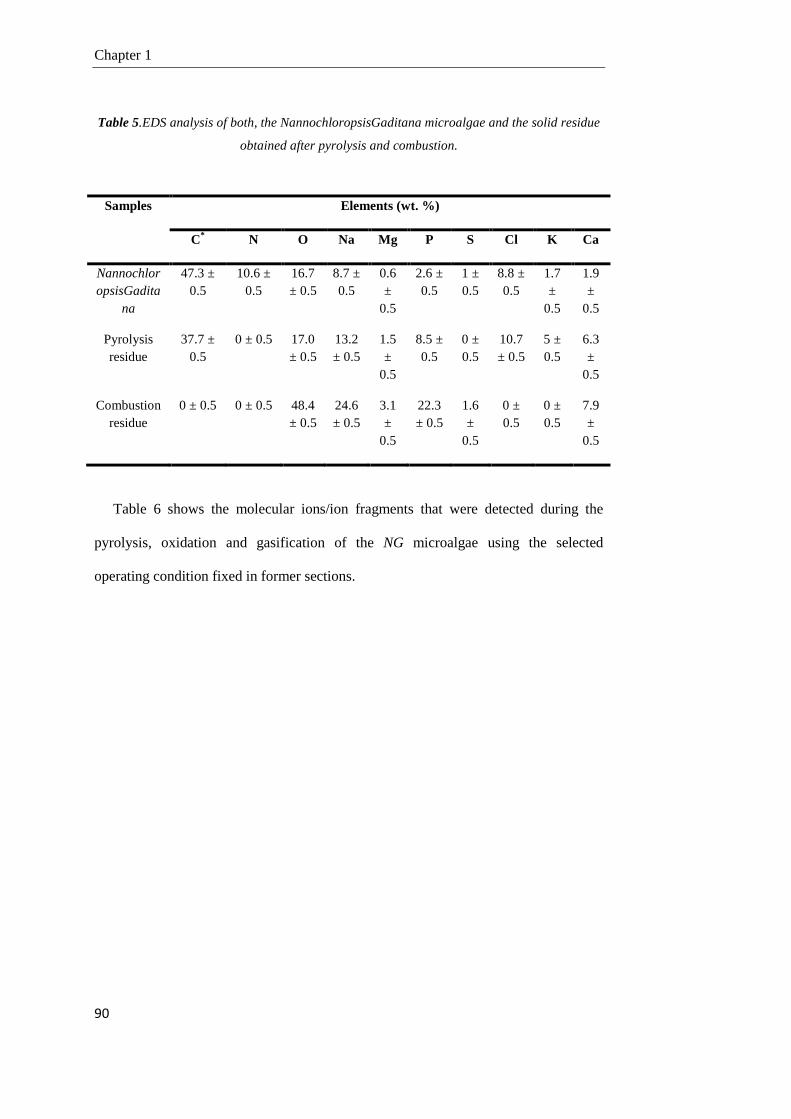
Chapter 1
90
Table 5.EDS analysis of both, the NannochloropsisGaditana microalgae and the solid residue
obtained after pyrolysis and combustion.
Samples Elements (wt. %)
C* N O Na Mg P S Cl K Ca
NannochloropsisGadita
na
47.3 ± 0.5
10.6 ± 0.5
16.7 ± 0.5
8.7 ± 0.5
0.6 ±
0.5
2.6 ± 0.5
1 ± 0.5
8.8 ± 0.5
1.7 ±
0.5
1.9 ±
0.5
Pyrolysis residue
37.7 ± 0.5
0 ± 0.5 17.0 ± 0.5
13.2 ± 0.5
1.5 ±
0.5
8.5 ± 0.5
0 ± 0.5
10.7 ± 0.5
5 ± 0.5
6.3 ±
0.5
Combustion residue
0 ± 0.5 0 ± 0.5 48.4 ± 0.5
24.6 ± 0.5
3.1 ±
0.5
22.3 ± 0.5
1.6 ±
0.5
0 ± 0.5
0 ± 0.5
7.9 ±
0.5
Table 6 shows the molecular ions/ion fragments that were detected during the
pyrolysis, oxidation and gasification of the NG microalgae using the selected
operating condition fixed in former sections.

Chapter 1
91
Table 6. Molecular ions and probable parent molecules detected in the pyrolysis, combustion
and gasification processes for the NannochloropsisGaditanamicroalgae.
m/z
NannochloropsisGaditanamicroalgae
Key molecular ions/Ion fragment
Probable parent molecule
Pyrolysis Combustion Gasification
2 H2+ H2 X X X
15 CH3+ CH4 X X -
16 O+; CH4+ CH4 X - X
17 NH3+ NH3 - X -
18 H2O+ H2O X X -
26 CN+; C2H2+ C2H2(acetylene) X - X
27 HCN+; C2H3+ HCN (nitriles) X - X
28 C2H4+; CO+ CO X X X
29 C2H5+
C2H5 (ethyl derivates)
X - X
30 C2H6+;
CH2NH2+
CH4N (primary amines)
X - X
44 CO2+ CO2 X X X
46 NO2+;
C2H4O+
NO2 - X -
56 C3H6N+;
C4H8+
C4H8 (alquenes) - X -
58 C3H8N+ C3H8N (amines) X X -
64 SO2+ SO2 (sulfones) - X -
78 C6H6+ C6H6 (benzene) X - -
Mass spectra of the pyrolysis and oxidation processes for NG microalgae are
shown in Figure 5.MS curves could be divided into three stages that could be related
to the three degradation steps described in the TGA/DTG curves.

Chapter 1
92
200 300 400 500 600 700 800
H2OCO
H2
CH+
3
CH4N
C2H
2
CH4
CO2
C6H
6
C3H
8N
C2H
5
Inte
nsity
(a.
u.)
Temperature (ºC)
HCN
200 300 400 500 600 700
CO2
H2
NO2
SO2
CH+
3
H2O
C4H
8
NH3
C3H
8N
Inte
nsity
(a.
u.)
Temperature (ºC)
CO
(a) (b)
Figure 5.Mass spectra of the (a) pyrolysis and (b) combustion of the
NannochloropsisGaditana microalgae.
Figure 5a shows the MS curves of the gaseous products released during the
pyrolysis process. H2O, CO and CO2 were detected in the MS spectrum at <160 ºC
due to the moisture content in the NG microalgae. In this case, most of the gas
products (H2O,CO, C6H6, C3H8N, CO2, CH4N, HCN)and volatile hydrocarbons such
as CH3+, CH4, C2H2, C2H5were generated at the second degradation step (160-450 ºC).
As reported elsewhere [2; 8; 33], the pyrolysis of the carbohydrates and proteins of
algaewould mainly take place in this stage.The N-containing compounds in the NG
microalgae could be released in form of amines (C3H8N andCH4N) andnitrile
(HCN)due to the thermal degradation of proteins[33]. According to other authors [8],
CO2 was mainly produced by the cracking and reforming of carboxyl groups in protein
and saccharides.HCN, C2H5, C2H2, CH4Nand CH3+were also producedin the last
decomposition step (>450ºC), corresponding to the slow decomposition of the solid
residue [8].A slow H2 release was observed at around 450-650 ºC, which could be
caused by the further dehydrogenation of remaining carbonaceous species[2].Among

Chapter 1
93
various gaseous hydrocarbons released, the content of CH3+ was the highestone [4].
Similar decomposition profiles have been reported for different algae species by many
authors [8; 14]. These results agree with the low nitrogen and carbon contentsin the
pyrolysis solid residuemeasured by EDSanalysis (Table 5).
The evolution profiles of the gaseous species, CO2, NO2, CO, SO2, C3H8N, H2O,
NH3, CH3+, H2and C4H8, from the combustion of the NG microalgae are shown in
Figure 5b. Two peaks at 265 and 515 ºCfor the H2O intensity associated with moisture
and combustion of volatiles and char were obtained [10].The release of CO2and CO
were observed at around 250-350 and 475-600 ºC due to the combustion of fixed
carbon [10].Furthermore, the SO2release took place at225-300 and 350-425 ºC, which
could be relatedto the decomposition of sulphated polysaccharideexisting in the NG
microalgae [10; 14].The N-containing compounds evolution (NO2, C3H8N and NH3)at
225-350 ºCwas associated with the degradation of protein in the NG
microalgae.Finally, H2, NH3,NO2and CH3+were detected in the last step (>500 ºC)
corresponding to decomposition of the solid residue.
By comparing pyrolysis and combustion MS curves, it is possible to evaluate the
effect of the oxygen presence on the gas emission.As expected, the content of CO2in
the combustion processwas the highestone whereas no generation of volatile
hydrocarbons such as CH4, C2H2, and C2H5was observed. In addition, NO2 and SO2
were also released.
Finally, as it can be seen in Table 5, some of the inorganic materials that were
present in the EDS analysis of the NG microalgae were not found in the combustion or

Chapter 1
94
pyrolysis results, as chloride and potassium for combustion and sulfur in the pyrolysis
residue, pointing out that the evolution of these compound took place during the
combustion and pyrolysis of the sample. This fact is mainly due to the signals of these
ion fragments were rejected as they were too close to the noise level. Further studies
are required for the evaluation of the release of inorganics during combustion and
pyrolysis as they are a potential source of contaminants.
TGA/DTG-MS curves of the gasification process of the NG microalgaeare shown
in Figure 6a.As can be seen in TGA/DTG curves, the time to reach the total
conversion was 55 min. The main products detected during the gasification at 850ºC
were CO2, CO, H2, indicating that oxidation reactions, water gas and water gas shift
reactions were the predominant ones.In addition, traces of CH4, C2H6, C2H5, C2H4and
C2H2were also generated. Similar evolution profiles have been reported elsewhere[31;
34; 35].
Figure 6b shows the yield of the main gases during the gasification process (CO2,
CO, CH4 and H2) at different steam concentrations. It can be observed that as the
proportion of water in the reactive gas was increased, the product distribution varied,
enhancing the production of H2 and decreasing the CH4yield. This fact would indicate
that water gas (C + H2O � CO + H2), water gas shift (CO + H2O � CO2 + H2)and
methane reforming (CH4+ H2O � CO + 3H2)reactions werebeing promoted[32;
35].Anyway, CO and CO2 emissions were kept constant.
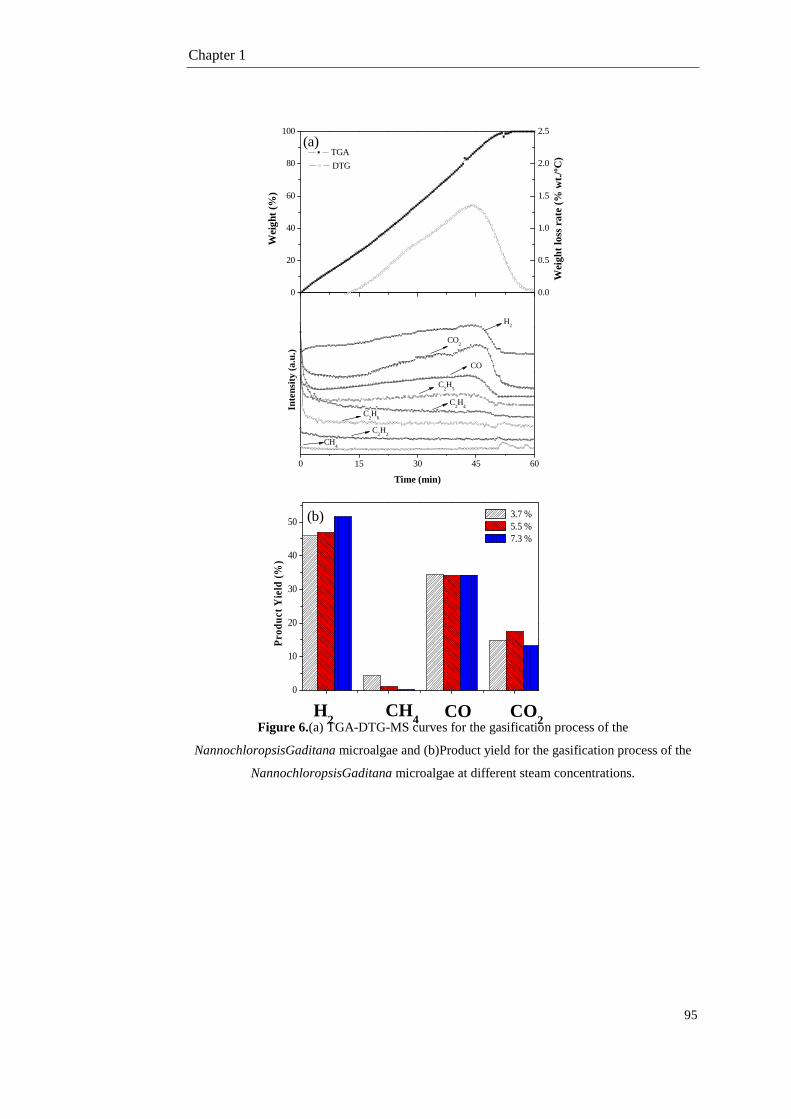
Chapter 1
95
0 15 30 45 60
0
20
40
60
80
100
In
tens
ity (
a.u.
)
Time (min)
CH4
C2H
2
C2H
5
C2H
4
C2H
6
CO2
H2
CO
Wei
ght (
%)
TGA
0.0
0.5
1.0
1.5
2.0
2.5
DTG
Wei
ght l
oss
rate
(%
wt./
ºC)
(a)
H2 CH4 CO CO2
0
10
20
30
40
50
Pro
duct
Yie
ld (
%)
Main Products
3.7 % 5.5 % 7.3 %
H2 CH
4 CO CO
2
(b)
Figure 6.(a) TGA-DTG-MS curves for the gasification process of the
NannochloropsisGaditana microalgae and (b)Product yield for the gasification process of the
NannochloropsisGaditana microalgae at different steam concentrations.

Chapter 1
96
1.4. CONCLUSIONS
Pyrolysis, combustion and gasification of NG microalgae were analyzed by means
of TGA-MS. Pyrolysis and combustion processes were divided into three stages.
During pyrolysis, the main devolatilization step took place between 160 and 450 ºC,
associated to the degradation of protein and soluble polysaccharide. In combustion the
oxidation of the sample took place between 450 and 600 ºC. As oxygen concentration
increased, the oxidation of the char shifted to lower temperatures.
N-compounds evolution was associated with the microalgae proteins degradation.
SO2release during combustioncould be related to sulphated polysaccharides
decomposition.H2production was enhanced by steam concentration.
1.5. REFERENCES
[1] Ross, A.B., Anastasakis, K., Kubacki, M., Jones, J.M. 2009. Investigation of the
pyrolysis behaviour of brown algae before and after pre-treatment using PY-GC/MS
and TGA. Journal of Analytical and Applied Pyrolysis, 85(1-2), 3-10.
[2] Babich, I.V., van der Hulst, M., Lefferts, L., Moulijn, J.A., O'Connor, P., Seshan,
K. 2011. Catalytic pyrolysis of microalgae to high-quality liquid bio-fuels. Biomass
and Bioenergy, 35(7), 3199-207.
[3] Li, D., Chen, L., Zhang, X., Ye, N., Xing, F. 2011. Pyrolytic characteristics and
kinetic studies of three kinds of red algae. Biomass and Bioenergy, 35(5), 1765-72.
[4] Pan, P., Hu, C., Yang, W., Li, Y., Dong, L., Zhu, L., Tong, D., Qing, R., Fan, Y.
2010. The direct pyrolysis and catalytic pyrolysis of Nannochloropsis sp. residue for
renewable bio-oils. Bioresource Technology, 101(12), 4593-9.
[5] Tang, Y., Ma, X., Lai, Z. 2011. Thermogravimetric analysis of the combustion of
microalgae and microalgae blended with waste in N2/O2 and CO2/O2 atmospheres.
Bioresource Technology, 102(2), 1879-85.

Chapter 1
97
[6] Pane, L., Franceschi, E., De Nuccio, L., Carli, A. 2001. Applications of thermal
analysis on the marine phytoplankton, Tetraselmis suecica. Journal of Thermal
Analysis and Calorimetry, 66(1), 145-54.
[7] Anastasakis, K., Ross, A.B. 2011. Hydrothermal liquefaction of the brown macro-
alga Laminaria Saccharina: Effect of reaction conditions on product distribution and
composition. Bioresource Technology, 102(7), 4876-83.
[8] Marcilla, A., Gómez-Siurana, A., Gomis, C., Chápuli, E., Catalá, M.C., Valdés,
F.J. 2009. Characterization of microalgal species through TGA/FTIR analysis:
Application to nannochloropsis sp. Thermochimica Acta, 484(1–2), 41-7.
[9] Sanchez-Silva, L., López-González, D., Villaseñor, J., Sánchez, P., Valverde, J.L.
2012. Thermogravimetric-mass spectrometric analysis of lignocellulosic and marine
biomass pyrolysis. Bioresource Technology, 109, 163-72.
[10] Wang, S., Jiang, X.M., Han, X.X., Liu, J.G. 2009. Combustion Characteristics of
Seaweed Biomass. 1. Combustion Characteristics of Enteromorpha clathrata and
Sargassum natans. Energy & Fuels, 23(10), 5173-8.
[11] Ollero, P., Serrera, A., Arjona, R., Alcantarilla, S. 2002. Diffusional effects in
TGA gasification experiments for kinetic determination. Fuel, 81(15), 1989-2000.
[12] Arenillas, A., Rubiera, F., Pis, J.J. 1999. Simultaneous thermogravimetric-mass
spectrometric study on the pyrolysis behaviour of different rank coals. Journal of
Analytical and Applied Pyrolysis, 50(1), 31-46.
[13] Widyawati, M., Church, T.L., Florin, N.H., Harris, A.T. 2011. Hydrogen
synthesis from biomass pyrolysis with in situ carbon dioxide capture using calcium
oxide. International Journal of Hydrogen Energy, 36(8), 4800-13.
[14] Wang, S., Jiang, X.M., Wang, N., Yu, L.J., Li, Z., He, P.M. 2007. Research on
pyrolysis characteristics of seaweed. Energy and Fuels, 21(6), 3723-9.
[15] Lin, Y.C., Cho, J., Tompsett, G.A., Westmoreland, P.R., Huber, G.W. 2009.
Kinetics and mechanism of cellulose pyrolysis. Journal of Physical Chemistry C,
113(46), 20097-107.

Chapter 1
98
[16] Shuping, Z., Yulong, W., Mingde, Y., Chun, L., Junmao, T. 2010. Pyrolysis
characteristics and kinetics of the marine microalgae Dunaliella tertiolecta using
thermogravimetric analyzer. Bioresource Technology, 101(1), 359-65.
[17] Chen, C., Ma, X., Liu, K. 2011. Thermogravimetric analysis of microalgae
combustion under different oxygen supply concentrations. Applied Energy, 88(9),
3189-96.
[18] Chouchene, A., Jeguirim, M., Khiari, B., Zagrouba, F., Trouvé, G. 2010.
Thermal degradation of olive solid waste: Influence of particle size and oxygen
concentration. Resources, Conservation and Recycling, 54(5), 271-7.
[19] Ross, A.B., Jones, J.M., Kubacki, M.L., Bridgeman, T. 2008. Classification of
macroalgae as fuel and its thermochemical behaviour. Bioresource Technology,
99(14), 6494-504.
[20] Yu, L.J., Wang, S., Jiang, X.M., Wang, N., Zhang, C.Q. 2008. Thermal analysis
studies on combustion characteristics of seaweed. Journal of Thermal Analysis and
Calorimetry, 93(2), 611-7.
[21] Stenseng, M., Jensen, A., Dam-Johansen, K. 2001. Investigation of biomass
pyrolysis by thermogravimetric analysis and differential scanning calorimetry. Journal
of Analytical and Applied Pyrolysis, 58-59, 765-80.
[22] Antal Jr, M.J., Várhegyi, G., Jakab, E. 1998. Cellulose Pyrolysis Kinetics:
Revisited. Industrial and Engineering Chemistry Research, 37(4), 1267-75.
[23] Bridgeman, T.G., Darvell, L.I., Jones, J.M., Williams, P.T., Fahmi, R.,
Bridgwater, A.V., Barraclough, T., Shield, I., Yates, N., Thain, S.C., Donnison, I.S.
2007. Influence of particle size on the analytical and chemical properties of two
energy crops. Fuel, 86(1-2), 60-72.
[24] Luo, S.Y., Xiao, B., Hu, Z.Q., Liu, S.M., Guan, Y.W. 2009. Experimental study
on oxygen-enriched combustion of biomass micro fuel. Energy, 34(11), 1880-4.
[25] Mani, T., Mahinpey, N., Murugan, P. 2011. Reaction kinetics and mass transfer
studies of biomass char gasification with CO2. Chemical Engineering Science, 66(1),
36-41.

Chapter 1
99
[26] Gómez-Barea, A., Ollero, P., Fernández-Baco, C. 2006. Diffusional effects in
CO2 gasification experiments with single biomass char particles. 1. Experimental
investigation. Energy and Fuels, 20(5), 2202-10.
[27] Mani, T., Murugan, P., Abedi, J., Mahinpey, N. 2010. Pyrolysis of wheat straw
in a thermogravimetric analyzer: Effect of particle size and heating rate on
devolatilization and estimation of global kinetics. Chemical Engineering Research and
Design, 88(8), 952-8.
[28] Díaz, J.A., Díaz-Moreno, R., Silva, L.S., Dorado, F., Romero, A., Valverde, J.L.
2012. Nickel supported carbon nanofibers as an active and selective catalyst for the
gas-phase hydrogenation of 2-tert-butylphenol. Journal of Colloid and Interface
Science, 380(1), 173-81.
[29] Kirubakaran, V., Sivaramakrishnan, V., Nalini, R., Sekar, T., Premalatha, M.,
Subramanian, P. 2009. A review on gasification of biomass. Renewable and
Sustainable Energy Reviews, 13(1), 179-86.
[30] Pütün, A.E., Özean, A., Pütün, E. 1999. Pyrolysis of hazelnut shells in a fixed-
bed tubular reactor: Yields and structural analysis of bio-oil. Journal of Analytical and
Applied Pyrolysis, 52(1), 33-49.
[31] Zhang, M., Chen, H.P., Gao, Y., He, R.X., Yang, H.P., Wang, X.H., Zhang, S.H.
2010. Experimental study on bio-oil pyrolysis/gasification. BioResources, 5(1), 135-
46.
[32] Florin, N.H., Harris, A.T. 2008. Enhanced hydrogen production from biomass
with in situ carbon dioxide capture using calcium oxide sorbents. Chemical
Engineering Science, 63(2), 287-316.
[33] Maddi, B., Viamajala, S., Varanasi, S. 2011. Comparative study of pyrolysis of
algal biomass from natural lake blooms with lignocellulosic biomass. Bioresource
Technology, 102(23), 11018-26.
[34] De Lasa, H., Salaices, E., Mazumder, J., Lucky, R. 2011. Catalytic steam
gasification of biomass: Catalysts, thermodynamics and kinetics. Chemical Reviews,
111(9), 5404-33.

Chapter 1
100
[35] Kojima, T., Assavadakorn, P., Furusawa, T. 1993. Measurement and evaluation
of gasification kinetics of sawdust char with steam in an experimental fluidized bed.
Fuel Processing Technology, 36(1-3), 201-7.
(1)

Chapter 2:
THERMOGRAVIMETRIC -MASS SPECTROMETRIC ANALYSIS OF
LIGNOCELLULOSIC AND MARINE BIOMASS
PYROLYSIS
The pyrolysis characteristics of three lignocellulosic biomasses
(Fir Wood, Eucalyptus and Pine Bark) and a marine biomass
(NannochloropsisGaditana microalgae) were investigated by
thermogravimetric analysis coupled with mass spectrometry (TGA-
MS). Thermal degradation of lignocellulosic biomass was divided
into four zones, corresponding to the decomposition of their main
components (cellulose, hemicellulose and lignin) and a first step
associated to water removal. Differences in volatile matter and
cellulose content of lignocellulosic species resulted in different
degradation rates. Microalgae pyrolysis occurred in three stages
due to the main components of them (proteins), which are greatly
different from lignocellulosic biomass. Heating rate effect was also
studied. The main gaseous products formed were CO2, light

Chapter 2
102
hydrocarbons and H2O. H2 was detected at high temperatures,
being associated to secondary reactions (char self-gasification).
Pyrolysis kinetics were studied using a multiple-step model. The
proposed model successfully predicted the pyrolitic behaviour of
these samples resulting to be statistically meaningful.
2.1. INTRODUCTION
Depletion of world fossil fuel reserves and the external dependence that these types
of fuels produce together with the environmental risks derived from its use are the
main reasons of the increasing attention that renewable energies sources are receiving.
In this context, biomass conversion for transportation fuels, chemical commodities and
power generation is getting growing interest.
Biomass is a term for all organic material that stems from plants including algae,
trees and crops that are susceptible to be converted into energy (McKendry, 2002).
One of the most controversial points in the use of biomass as an energy source is its
possible competition with human food supply. Nevertheless, there are different types
of biomass that fit into this definition without compromising world food supply. The
main ones refer to lignocellulosic biomass (Basu, 2010) and marine biomass
(especially algae). Algae are a good candidate because they present the following
advantages: large amount available, fast growth, low priced, environment protection
and suitable for pyrolysis (Wang, 2006).
There are many conversion technologies for utilizing biomass, such as direct
combustion, thermochemical, biochemical and agrochemical processes.

Chapter 2
103
Thermochemical conversion of biomass is considered as one of the most promising
processes for biomass utilization (Shen et al., 2010). There are four thermochemical
technologies: pyrolysis, gasification, combustion and liquefaction. Pyrolysis is of
special interest since it is a prior step in combustion and gasification processes.
Therefore, it seems essential to obtain a deep knowledge of biomass pyrolysis in order
to gain further understanding of the combustion and gasification processes.
Pyrolysis can be described as the biomass conversion by heat in the absence of
oxygen in a relatively low range of temperatures (300-600 ºC), which results in the
production of charcoal (solid), bio-oil (liquid) and fuel gas products. Thermal analysis
has shown to be a powerful tool for investigating the pyrolysis of biomass. Numerous
studies based on thermogravimetic analysis (TGA) and derivative thermogravimetry
(DTG) have been carried out. Many of them focused on the main components of
lignocellulosic biomass, mainly constituted by cellulose, hemicellulose and lignin
(Wang et al., 2008; Yang et al., 2006), different types of lignocellulosic biomass
(Barneto et al., 2011; Stenseng et al., 2001), and algae (Li et al., 2011; Peng et al.,
2001). The effects of heating rate and amount of sample have been also reported in the
literature (Lin et al. 2009).
Pyrolysis kinetics is other of the aspects that has been widely studied by thermal
analysis. Most studies have been focused on cellulose pyrolysis (Grønli et al., 1999,
Lin et al., 2009), lignin and xylan (Rao and Sharma, 1997), lignocellulosic biomass
(Órfão et al., 1999) and marine biomass (Wang et al., 2006). The determination of the
kinetics corresponding to biomass thermal decomposition involves the knowledge of
the reaction mechanisms. However, pyrolysis is an extremely complex process, where

Chapter 2
104
numerous reactions take place, practically making impossible to develop a kinetic
model that takes into account all these reactions. Thus, the pyrolysis is usually studied
in terms of pseudo-mechanistic models (Caballero et al., 1997). White et al. (2011)
reported that kinetics of biomass decomposition can be divided into three principal
types of models: single-step global reaction models, multiple-step models and semi-
global models.
Thermal analysis itself might not seem sufficient for a thorough study based on
kinetics. Therefore, other techniques must be used to obtain valuable results (White et
al., 2011). The combination of thermogravimetric analysis coupled with mass
spectrometry (TGA-MS) appears to give a deeper insight of the process. Some studies
concerning TGA-MS of the biomass pyrolysis have been carried out (Grønli et al.,
1999; Widyawati et al., 2011). One of the most attractive advantages of TGA-MS is
that it is able to afford real-time and sensitive detection of evolved gases, which is an
important and often a difficult task in many thermal applications (Huang et al., 2011).
The aim of this study was to investigate the pyrolysis characteristics and gas
products distribution of lignocellulosic (Fir Wood, Eucalyptus Wood and Pine Bark)
and marine (Nannochloropsisgaditana microalgae) biomass by means of the TGA-MS
technique. This work pretends to establish and gain further understanding of the
possible relationships among these components, from those present in lignocellulosic
materials to those that constitute terrestrial and marine biomass. Moreover, the effect
of heating rate on the pyrolysis behaviour of these samples was also studied. Finally,
experimental data obtained using thermogravimetric analysis were interpreted using a
multi-step kinetic model.

Chapter 2
105
2.2. EXPERIMENTAL
2.1.1 Materials
Cellulose, Xylan and Lignin were purchased from Sigma Aldrich. Xylan was used
as a representative of the hemicellulose component in the pyrolysis (Wang et al.,
2008; Yang et al., 2006). These chemicals are as follow: Cellulose (microcrystalline
cellulose with 50 µm average particle size), Lignin (alkali lignin in brown powder
form with 50 µm average particle size) and Xylan (xylan processed from beechwood
with 100 µm average particle size, was used as hemicellulose). The selected terrestrial
biomass (FirWood, EucalyptusWood and PineBark) were taken from the region of
Castilla-La Mancha (Spain). These samples were dried in an oven for 5 hours, milled
and sieved to less than 240 µm. The microalgae NannochloropsisGaditana(NG
microalgae) were purchased from AlgaeEnergy Company. This compound is delivered
in green powder with 100 µm average particle size.
2.2.2. Biomass selection
The choice of biomass mainly depended on its inherent properties, determining the
conversion process and any subsequent processing difficulties that may arise. The
main properties of interest for the biomass processing as an energy source are the
following (McKendry, 2002): moisture content (MC); proportion of fixed carbon (FC)
and volatiles (VM); ash/residue content (AC/AR); calorific value; alkali metal content;
cellulose/lignin ratio.
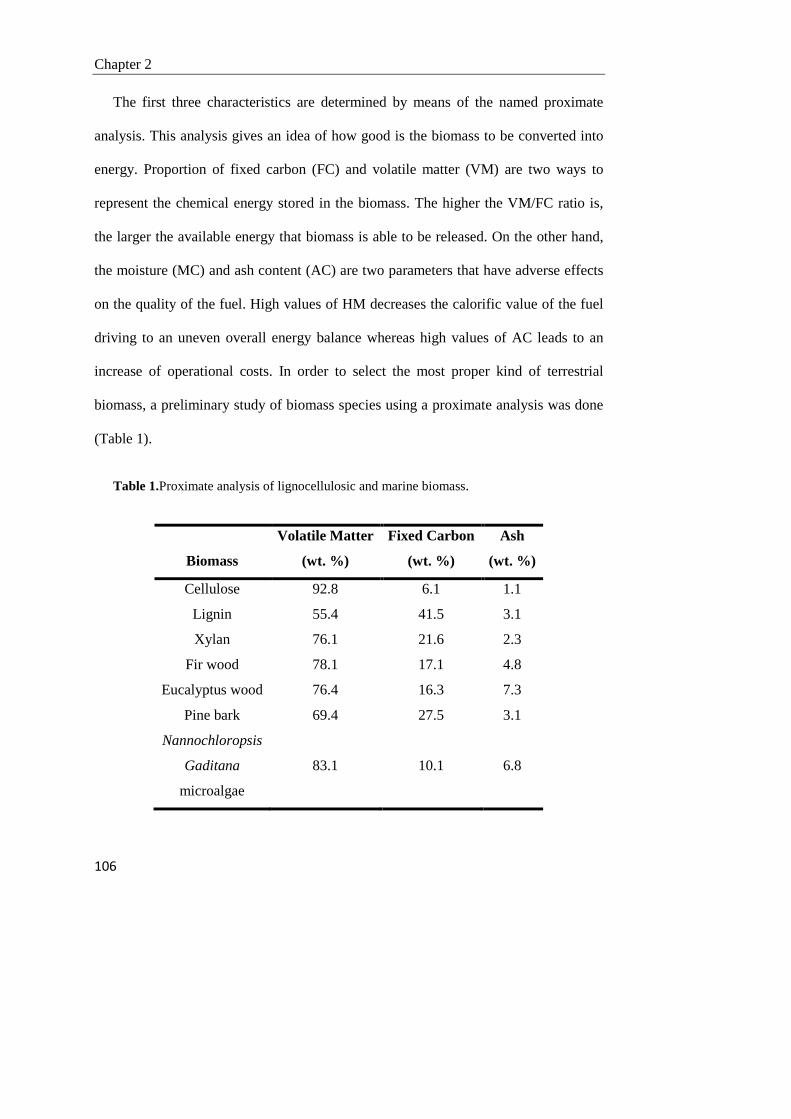
Chapter 2
106
The first three characteristics are determined by means of the named proximate
analysis. This analysis gives an idea of how good is the biomass to be converted into
energy. Proportion of fixed carbon (FC) and volatile matter (VM) are two ways to
represent the chemical energy stored in the biomass. The higher the VM/FC ratio is,
the larger the available energy that biomass is able to be released. On the other hand,
the moisture (MC) and ash content (AC) are two parameters that have adverse effects
on the quality of the fuel. High values of HM decreases the calorific value of the fuel
driving to an uneven overall energy balance whereas high values of AC leads to an
increase of operational costs. In order to select the most proper kind of terrestrial
biomass, a preliminary study of biomass species using a proximate analysis was done
(Table 1).
Table 1.Proximate analysis of lignocellulosic and marine biomass.
Biomass
Volatile Matter
(wt. %)
Fixed Carbon
(wt. %)
Ash
(wt. %)
Cellulose 92.8 6.1 1.1
Lignin 55.4 41.5 3.1
Xylan 76.1 21.6 2.3
Fir wood 78.1 17.1 4.8
Eucalyptus wood 76.4 16.3 7.3
Pine bark 69.4 27.5 3.1
Nannochloropsis
Gaditana
microalgae
83.1
10.1
6.8

Chapter 2
107
Taking into account the analysis listed in this Table and those reported by Yaman
(2004) for different biomass, a ternary diagram was plotting by considering the
following parameters: ash, volatile matter and fixed carbon contents (Figure 1).
Biomass to be used in this study was selected according to the following criteria:
- Biomass with high VM content and low AC.
- Biomass with high FC content and a low AC.
According to these criteria, two areas in the diagram were clearly identified
(they have been outlined with a circle). Biomass within these two zones corresponded
to: Fir Wood and Eucalyptus Wood (both with pretty higher VM) and Pine Bark (with
the highest FC).
Figure 1.Ternary diagram with different kinds of terrestrial biomass according to their
proximate analysis (biomass data was obtained from Yaman et al., 2004).
0.0 0.2 0.4 0.6 0.8 1.0
0.00.00.00.0
0.20.20.20.2
0.40.40.40.4
0.60.60.60.6
0.80.80.80.8
1.01.01.01.00.0
0.2
0.4
0.6
0.8
1.0
Fixed Carbon
Sugarcane bagasseGrapeMaizeOliveRapeseedRice huskSawdustSunflowerBrown Kelp GiantWater hyacinthFir woodTobaccoPine barkCotton wastesEucalyptus woodStraw
Ash
Volatile Matter

Chapter 2
108
2.2.3. Equipment and Procedures
2.2.3.1. Pyrolysis
The pyrolysis of biomass components was firstly carried out in a TGA apparatus
(TGA-DSC 1, METTLER TOLEDO). The sample was heated from 40 ºC to 900ºC at
different heating rates (5, 15 and 40 ºC/min). From previous studies in TGA analysis
the following operating conditions were chosen in order to avoid the effects of heat
and mass transfer limitations: sample weight was kept at 10 mg, helium (99.99 %) at a
flow rate of 200 ml/min was used as the carrier gas to provide an inert atmosphere and
the particle size was kept lower than 300 µm. Each sample was analyzed at least three
times, and the average value was recorded. The experimental error of these
measurements was calculated, obtaining an error for all studied samples of ± 0.5% in
weight loss measurement and ± 2 ºC in temperature measurement.
2.2.3.2. TGA-MS
The analysis of the gas products distribution coming from the pyrolysis was carried
out in a thermogravimetricanalyzer (TGA-DSC 1; METTLER TOLEDO) coupled to a
mass spectrometer (Thermostar-GSD 320/quadrupole mass analyzer; PFEIFFER
VACUUM) with an electron ionization voltage at 70 eV and provided mass spectra up
to 300 a.m.u. The interface was wrapped with heating wire to circumvent
condensation of exhausting gases. Approximately 10 mg of sample was loaded into an
alumina crucible pan and heated from room temperature to 900 ºC at a heating rate of
40 ºC/min. In all experiments, He was used as the purge gas (99.99 %) with a
constant flow rate of 200 ml/min. In order to identify ions with m/z in the range 0-300,

Chapter 2
109
a preliminary broad scan was performed at a heating rate of 40 ºC/min. The signals
identified corresponded to the mass spectra of 2, 15, 18, 27, 28, 30, 32, 44 a.m.u,
which corresponds to the main components of the pyrolysis gas (H2, CH4, H2O, C2H4,
CO, C2H6 and CO2, respectively).
2.2.4. Kinetics
Kinetic data from solid state pyrolysis was obtained using thermogravimetric
analysis. The model proposed in this work is similar as that reported by Sun et al.
(2011) for the determination of the kinetic parameters of decomposition of bagasse
fibers, and is based on works previously reported (Órfão et al., 1999). Considering
npyrolizable compounds, the kinetic rates of thermal decomposition of a material
assuming independent parallel nth-order reactions and an Arrhenius dependence of the
rate constants are:
( )inn
ii
s
iaioi RT
Ekc
dt
d∑=
−
−=
11exp αα
(1)
( ) ini
s
iaio
iRT
Ek
dt
d αα −
−= 1exp
(2)
where α is the degree of conversion of the material, kioand Eiaare the pre-exponential
factor and the activation energy for the individual components; R is the gas constant;
ni is the reaction order; and αi is the degree of conversion for the individual
component defined by

Chapter 2
110
io
ipioi m
mm −=α
(3)
wheremio and mip represent the mass at t=0 and t=t for each component, respectively.
The constant ci is related to the initial composition of the different components.
Finally, Ts is the actual sample temperature that may differ from the external
temperature by a thermal lag. Lin et al. (2009) proposed the following equation as a
function of a fitting experimental factor C to correct the thermal delay of the
apparatus:
( )CtbTT os −+=
(4)
In this equation, To and b represent the initial temperature and the heating rate,
respectively.
2.2.4.1. Parameter estimation
A VBA-Excel application was developed to solve this model (de la Osa et al.,
2011; Valverde et al., 2004). The Bader-Deufhard method was used in the evaluation
of the set of ordinary differential equations (Press, 1992), whereas the Marquardt-
Levenberg algorithm was used in the nonlinear regression procedure (de Lucas et al.,
2006; Froment et al., 1990; Marquardt, 1963). The ordinary differential equation, Eq.
(1) was solved by considering the following initial conditions:
α(t=0) = 0 and αi(t=0) = 0 (5)
The weighted sum of the squared differences between the observed (Exp) and the
calculated (Pr) degree of conversion was minimized according to the following
equation described elsewhere (de Lucas et al., 2006):

Chapter 2
111
( )∑ ∑= =
−=n
i
m
jjiExpiSSQ
1 1
2Pr αα
(6)
wherei represents the number of equations to be fitted, j the specific experimental data
and n and m the total number of equations and experiments (more than 4500 if all the
data generated in a TGA analysis are considered), respectively.
AF-test is a statistical test in which the test statistic has a F-distribution under the
null hypothesis. The procedure was based on the comparison between the tabulated F-
value (F-test) and Fc, which was defined elsewhere (Froment, et al., 1990):
( )
( ) ( )∑ ∑
∑ ∑
= =
= =
−∗−=
n
i
m
jjiExpi
n
i
m
ji
c
pmn
k
F
1 1
2Pr
1 1
2Pr
/
/
αα
α
(7)
wherep represents the total number of parameters.
If Fc is larger than F(p, n-p, 1-α), assuming a value of α = 0.05, 95% confidence
level, the regression was considered to be meaningful, although there is no guarantee
that the model is statistically suitable since the meaningfulness of each parameter in
the model must be also evaluated.
Hence, a complementary test, named t-test, was used. The t-test considers that the
statistical hypothesis test follows a Student’s t distribution and allows to verify if the
estimate of the parameter bfi differs from a reference value (generally zero). Thus, a
parameter is meaningful (at α = 0.05) each time that the following inequality occurs:

Chapter 2
112
( )
−−>=2
1,α
pntbV
bt
iif
fici
(8)
where[V(bf)]ii represents the diagonal ith term of the covariance matrix used in the last
step of n-linear regression procedure.
2.3. RESULTS AND DISCUSSION
2.3.1. Thermogravimetric study of pyrolysis of lignocellulosic and marine
biomass
The TGA/DTG profiles of the main components of the biomass here considered
(hemicellulose (Xylan), lignin and cellulose) as a function of temperature at a heating
rate of 15ºC/min during the pyrolysis process are shown in Figure 2a. There are
substantial differences between the pyrolysis behaviour of these components. The
decomposition of Xylan showed two peaks, starting at about 200 ºC and reaching its
maximum weight loss rate at 250ºC, being the residue yield equal to 28 wt.%. These
two peaks can be associated to the decomposition of the xylan side units and the
cracking of the main xylan chains (Severian, 2008). The pyrolysis of Cellulose
occurred between 290 ºC and 390 ºC reaching its maximum value at 340 ºC. It can be
noticed the pronounced DTG profile of this sample. The residue yield of cellulose was
the lowest one (9 wt.%). Lignin showed the highest thermal stability, decomposing in
the whole range of temperatures studied (200-700 ºC). Furthermore, the DTG profile
of Lignin was the flattest being the residue yield obtained the highest one (>40 wt.%).
This fact could be due to the slow carbonization of lignin, being the main responsible

Chapter 2
113
for the biomass char formation (Yang et al., 2006). According to Wang et al. (2008),
the differences in the thermal behaviour of these components can be attributed to their
different chemical structures. Hemicellulose has a random and amorphous structure
with little strength whereas cellulose has a crystalline and strong structure and it is
resistant to hydrolysis. On the other hand, Lignin is the most different one due to the
fact that it is a complex, heavily cross-linked and highly branched polymer (Basu,
2010).
Figure 2b shows the TGA/DTG plots versus temperature for Fir Wood,
Eucalyptus Wood and Pine Bark at a heating rate of 15 ºC/min. The criterion for
biomass selection was explained in Section 2.2. Generally, the thermal degradation
profiles of lignocellulosic biomass are interpreted as the addition of the independent
degradations of their main components (Caballero et al., 1997). According to this fact,
the pyrolysis process can be divided into four stages: moisture evolution (<120ºC);
hemicellulose decomposition (150-310 ºC); lignin and cellulose degradation (310-400
ºC) and lignin decomposition (> 450ºC).
In spite of these four well identified stages, there are some differences in the
behaviour of these materials. The second zone, corresponding to the hemicellulose
decomposition, took place at different temperatures as a function of the raw materials.
This area is represented by a shoulder in the DTG curve (Grønli et al., 2002). In the
case of the Fir Wood sample, hemicellulose decomposition took place at lower
temperatures than for Eucalyptus Wood and Pine Bark. Nevertheless, regardless the
sample, the third zone occurred in a similar range of temperatures (340-350 ºC). This

Chapter 2
114
temperature range agrees with that of the volatilization of cellulose shown in Figure
2a. In the last zone ascribed to lignin decomposition (zone IV), Pine Bark and Fir
Wood were almost overlapped whereas that of Eucalyptus occurred with different
weight loss rates, showing two peaks in the DTG curve at 505 ºC and 650ºC,
respectively. Finally, the residue yield was 34, 29 and 26 wt.% for Pine Bark,
Eucalyptus Wood and Fir Wood, respectively.
The differences in the thermal behaviour of all lignocellulosic samples can be
attributed to content variations of hemicellulose, lignin and cellulose. The higher
content of hemicellulose in Fir Wood could explain the peak in the DTG curve
obtained at low temperatures. In addition, the different contents of lignin could justify
the differences among the residue yield observed, which is related to the fixed carbon
content present in the sample. Pine Bark had the highest fixed carbon content, being
the one leading to the largest residue yield. Finally, the differences in the maximum
weight loss rate (in the order: Eucalyptus Wood> Fir Wood > Pine Bark) can be
explained attending to the volatile matter and cellulose content in these samples
(Damartzis et al., 2011).
Figure 2c shows the TGA/DTG curves for the thermal decomposition of the
variety of microalgae NannochloropsisGaditana(NG microalgae). Its thermal
degradation behaviour can be divided into three zones. The first zone was attributed to
a dehydration process at temperatures below 130ºC. The second zone, between 140 ºC
and 540 ºC, corresponded to a devolatilization process. Three shoulders can be
distinguished in this zone, being mainly associated to the decomposition of different

Chapter 2
115
kind of triglycerides and other hexane-soluble compounds (Marcilla et al., 2009).
Finally, the last zone took place at temperatures above 540 ºC, being mainly related to
residue decomposition.
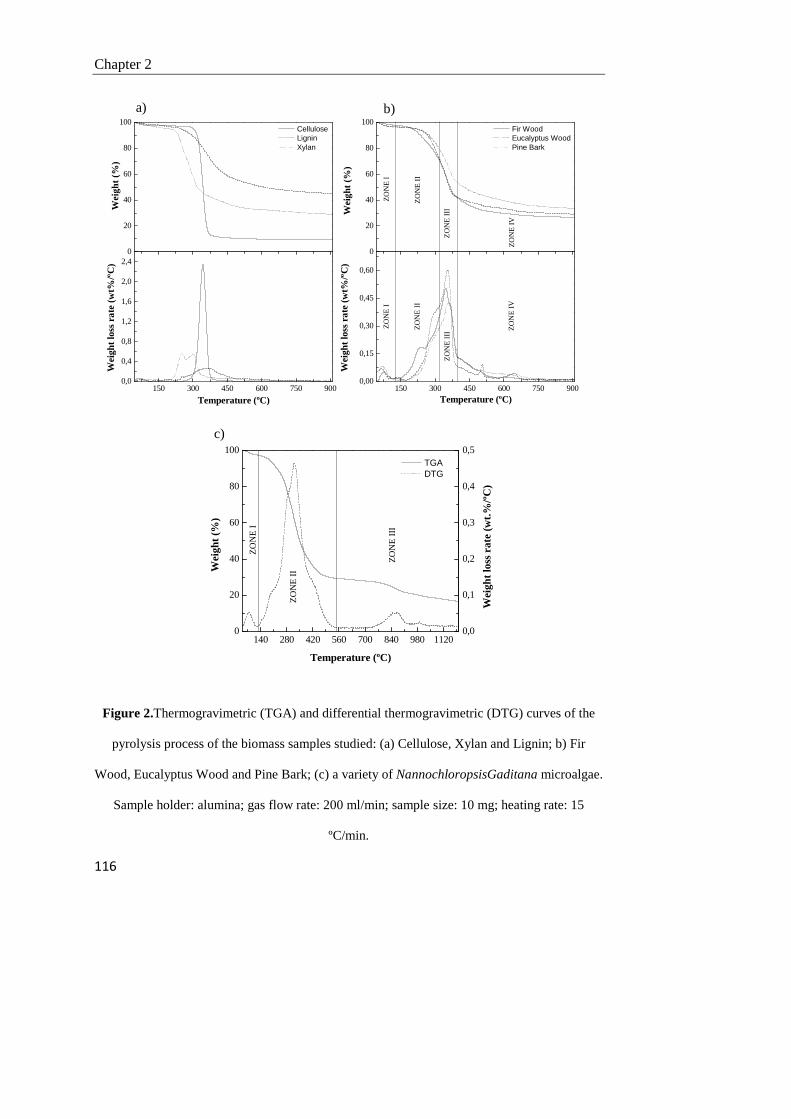
Chapter 2
116
Figure 2.Thermogravimetric (TGA) and differential thermogravimetric (DTG) curves of the
pyrolysis process of the biomass samples studied: (a) Cellulose, Xylan and Lignin; b) Fir
Wood, Eucalyptus Wood and Pine Bark; (c) a variety of NannochloropsisGaditana microalgae.
Sample holder: alumina; gas flow rate: 200 ml/min; sample size: 10 mg; heating rate: 15
ºC/min.
0
20
40
60
80
100
150 300 450 600 750 9000,00
0,15
0,30
0,45
0,60
Wei
ght (
%)
Fir Wood Eucalyptus Wood Pine Bark
ZO
NE
IV
ZO
NE
III
ZO
NE
II
ZO
NE
I
ZO
NE
IV
ZO
NE
IIIZ
ON
E I
I
Wei
ght l
oss
rate
(w
t%/º
C)
Temperature (ºC)
ZO
NE
I
0
20
40
60
80
100
150 300 450 600 750 9000,0
0,4
0,8
1,2
1,6
2,0
2,4
W
eigh
t (%
)
Cellulose Lignin Xylan
Wei
ght l
oss
rate
(w
t%/º
C)
Temperature (ºC)
140 280 420 560 700 840 980 11200
20
40
60
80
100
ZO
NE
III
ZO
NE
II
ZO
NE
I
Wei
ght l
oss
rate
(w
t.%/º
C)
Wei
ght (
%)
Temperature (ºC)
0,0
0,1
0,2
0,3
0,4
0,5 TGA DTG
a) b)
c)

Chapter 2
117
To sum up, NG microalgae showed higher thermal stability than
lignocellulosicbiomass, decomposing in a broader temperature range (the residue does
not remain constant until temperatures above 900 ºC). Furthermore, the residue yield
obtained for the microalgae was lower (about 15 wt. %) than those for the other types
of biomass considered in this work. Nevertheless, the main loss weight, corresponding
to the pyrolytic process, occurred at the same temperature range as that of the
terrestrial biomass (200-500 ºC). On the other hand, the shape of the DTG curves for
the analyzed samples showed evident differences. For the lignocellulosic biomass, a
well-defined shoulder was observed in the DTG curves whereas three little humps
were detected in the marine one. These differences were attributed to the different
compositions of these materials. NG microalgae are mainly composed of proteins (>60
%) whereas lignocellulose biomass is constituted by cellulose, hemicellulose, lignin
(>90%) and a little amount of extractives (Shuping et al., 2010).
2.3.2. Effect of heating rate
Figure 3 shows TGA/DTG plots versus temperature obtained from the
pyrolysis of Cellulose at different heating rates (5, 15 and 40 ºC/min). This figure
represents the general trend of biomass samples studied during the pyrolysis process.

Chapter 2
118
Figure 3.Effect of the heating rate in the pyrolysis process of Cellulose at 5, 15 and 40 ºC/min.
Sample holder: alumina; gas flow rate: 200 ml/min; sample size: 10 mg.
Table 2 shows the most relevant experimental results for all raw materials. As
it can be seen in Table 2, the behaviour of all of them is quite similar. Generally, as
the heating rate increased, the pyrolysis temperature (Tpyr) and all characteristic
temperatures shifted to higher values (Table 2). It can also be observed that the
maximum weight loss rate decreased as the heating rate was increased. These results
are similar as those reported by other authors (Li et al., 2010; Peng et al, 2001).
0
20
40
60
80
100
250 300 350 400 450 5000,0
0,5
1,0
1,5
2,0
2,5
Wei
ght (
%)
C/min؛ 5 C/min؛ 15 C/min؛ 40
Wei
ght l
oss
rate
(w
t%/º
C)
Temperature (ºC)

Chapter 2
119
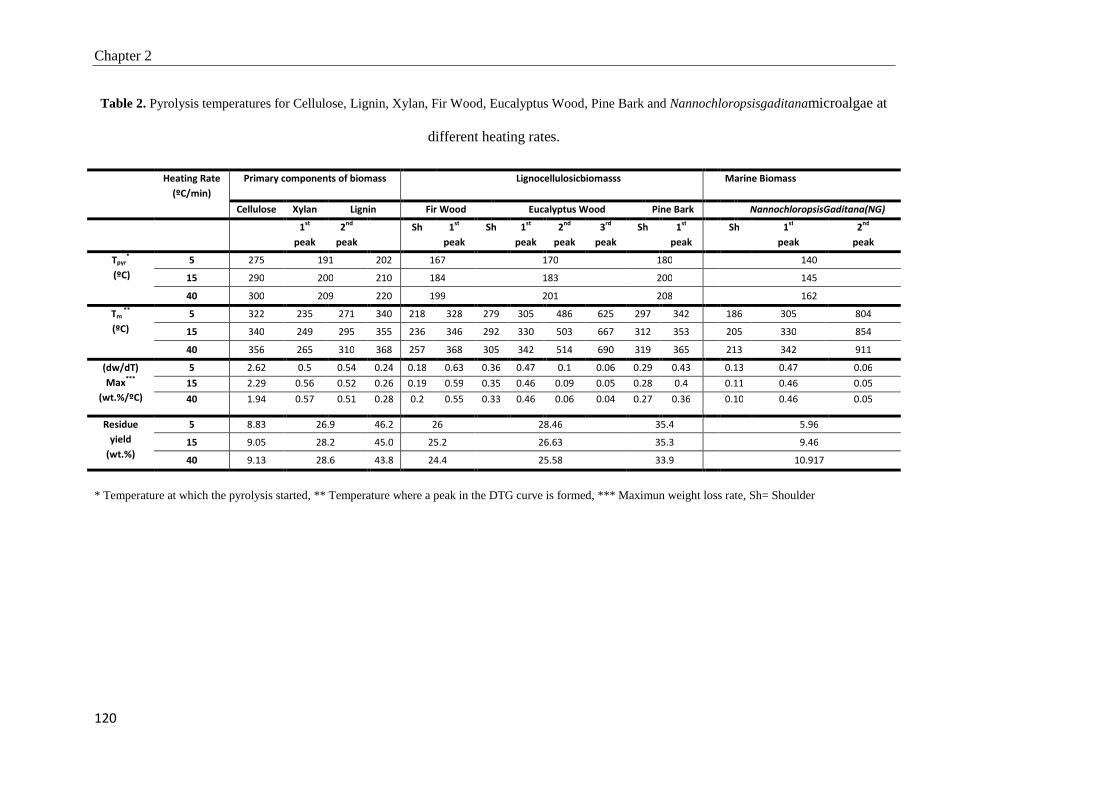
Chapter 2
120
Table 2. Pyrolysis temperatures for Cellulose, Lignin, Xylan, Fir Wood, Eucalyptus Wood, Pine Bark and Nannochloropsisgaditanamicroalgae at
different heating rates.
* Temperature at which the pyrolysis started, ** Temperature where a peak in the DTG curve is formed, *** Maximun weight loss rate, Sh= Shoulder
Heating Rate
(ºC/min)
Primary components of biomass Lignocellulosicbiomasss Marine Biomass
Cellulose Xylan Lignin Fir Wood Eucalyptus Wood Pine Bark NannochloropsisGaditana(NG)
1st
peak
2nd
peak
Sh 1st
peak
Sh 1st
peak
2nd
peak
3rd
peak
Sh 1st
peak
Sh 1st
peak
2nd
peak
Tpyr*
(ºC)
5 275 191 202 167 170 180 140
15 290 200 210 184 183 200 145
40 300 209 220 199 201 208 162
Tm **
(ºC)
5 322 235 271 340 218 328 279 305 486 625 297 342 186 305 804
15 340 249 295 355 236 346 292 330 503 667 312 353 205 330 854
40 356 265 310 368 257 368 305 342 514 690 319 365 213 342 911
(dw/dT)
Max***
(wt.%/ºC)
5 2.62 0.5 0.54 0.24 0.18 0.63 0.36 0.47 0.1 0.06 0.29 0.43 0.13 0.47 0.06
15 2.29 0.56 0.52 0.26 0.19 0.59 0.35 0.46 0.09 0.05 0.28 0.4 0.11 0.46 0.05
40 1.94 0.57 0.51 0.28 0.2 0.55 0.33 0.46 0.06 0.04 0.27 0.36 0.10 0.46 0.05
Residue
yield
(wt.%)
5 8.83 26.9 46.2 26 28.46 35.4 5.96
15 9.05 28.2 45.0 25.2 26.63 35.3 9.46
40 9.13 28.6 43.8 24.4 25.58 33.9 10.917

Chapter 2
121
These changes could be mainly attributed to changes in the decomposition
kinetics (Peng et al., 2001) and the fact that an increase of the heating rates provided
higher thermal energy, ensuring a better heat transfer between the surrounding
environment and the sample inside (Li et al., 2010). On the other hand, the residue
yield is one of the parameters that did not remain constant. Several differences can be
observed between the primary components of biomass and lignocellulosic biomass.
First of all, Cellulose and Lignin followed opposite trends than Xylan. The residue
yield for Cellulose and Lignin increased at increasing heating rates, whereas Xylan
residue decreased. Shen et al. (2010) ascribed this fact to structure differences. The
structure of cellulose is chemically and physically rearranged after the “preheating
process”, enhancing the final production of char residue (Maschio et al. 1992). Thus,
the char residue from Cellulose pyrolysis would increase with the longer pre-heating
process at the low heating rate. In Xylan, the structure of the char residue formed at
the low heating rate is less stable than that formed at the high heating rate, leading to
secondary cracking reactions. The char formation in Lignin is enhanced at low heating
rates. According to Nakamura et al. (2007), the formation mechanism of char in
Lignin is attributed to condensation reactions.
The effect of the heating rate in the residue yield for lignocellulosic biomass
followed the same trend in all cases. The higher the heating rate, the lower the residue
yield was. According to White et al. (2011), this fact can be attributed to the
completion of thermal degradation reactions at high heating rates. Finally, NG
microalgae showed a decrease in the residue yield with increasing heating rates. This
is due to the fact that lower heating rates resulted in longer residence times inside the

Chapter 2
122
reactor favouring secondary reactions such as cracking, re-polymerization and re-
condensation, thus leading to char formation (Shuping et al., 2010).
2.3.3. Gas products Analysis
The pyrolysis behavior of biomass by means of TGA-MS has been studied by
different authors (Lin et al., 2009; Widyawati et al., 2011). TGA-MS measurements
reproduce the evolution of the main gas products during the pyrolysis of biomass. This
technique is the only one to simultaneously measure in real time the thermal
decomposition and the gas product distribution of a very small sample. The present
study was focused on the main volatile products of biomass pyrolysis on the basis of
both their relative intensities across the temperature range 40-900ºC and on their
relevancy. H2, -CH3, CH4, C2H4, C2H6, CO, CO2 assigned to the ion/mass intensities
(m/z) 2, 15, 16, 27, 28, 30 and 44, respectively (according to the database of National
Institute of Standards and Technology (NIST)). Mass spectra curves for all samples
are shown in Figure 4.
Mass spectrometry analysis for Xylan, Cellulose and Lignin are shown in
Figure 4a. As aforementioned, the pyrolysis process for Cellulose and Xylan occurred
in a relatively narrow range of temperature (200-500 ºC) coincidental with most of the
gas product detection whereas thermal decomposition of Lignin took place in a wide
temperature range. The main gas detected was in all cases CO2. Compared to Xylan
and Lignin, Cellulose pyrolysis released most of gaseous products in a narrow
temperature range (300-400 ºC). On the other hand, Xylan and Lignin released CH4
and -CH3 groups at 500 ºC.

Chapter 2
123
Firstly, CH4 was generated at 450ºC in Xylan and at 500 ºC in Lignin.
Secondly, H2 was produced as CH4 and –CH3 groups are consumed. This fact can be
attributed to CH4 steam reforming reactions (Eq. (9)) (Widyawati et al., 2011). Finally,
Lignin showed the highest reactivity in the whole range of temperatures. These results
are in good agreement with previous works using TGA-MS techniques (Widyawati et
al., 2011). On the other hand, most of the H2 formation was observed at high
temperatures (>500ºC). H2 production is attributed to secondary reactions as steam
reforming of methane and/or tar cracking (Widyawati et al., 2011; Huang et al. 2011):
CH4 + H2O ↔ CO + 3H2 CH4 steam reforming (9)
CnHmOp + (2n-p)H2O ↔ nCO2 + (1/2m + 2n-p)H2 Tar steam reforming (10)
CnHm ↔Cn-xHm-y+H2+CH4+C Thermal craking (11)
Figure 4b shows the MS spectra for lignocellulosic biomass as a function of
temperature. The process could be divided into 4 stages. Firstly, peaks at low
temperatures (<150 ºC) represented the drying process of the samples. Furthermore,
methyl groups were also detected in a similar way than in Lignin pyrolysis mass
spectra. In the second stage (150-250 ºC), the main pyrolysis products detected were
CO2, CO, H2O and light hydrocarbons (CH4 and C2H6). CH4 and CO2 productions
were also detected at temperatures ranging from 400 to 500ºC. Additionally, at
temperatures above 500 ºC two peaks were detected. The first peak occurred in all
samples at a similar temperature (about 530 ºC); the second one shifted depending on
the sample (650, 675 and 698 ºC for Fir Wood, Pine Bark, and Eucalyptus Wood,
respectively). In spite of these differences, the CO2 and CH4 evolution was similar. On

Chapter 2
124
the other hand, two peaks related to H2 evolution was observed when the rate of CO2
and CH4 formation was decreasing, reaching its maximum values at about 750 ºC.
The product distribution observed in the last stage (470-800 ºC) suggested that
secondary reactions took place. These reactions could be attributed to tar cracking,
(Eqs. 10 and 11), being CO2, CH4 and H2 mainly formed, Eqs. (10-11), self
gasification of samples (Eq. 12) (Huang et al., 2011), and CH4 consumption by steam
reforming (Eq.(9)) (Widyawati et al., 2011).
C+H2O ↔ CO2 + H2 Self Gasification (12)
This way, it can be concluded that most of the H2 produced from
lignocellulosic biomass pyrolysis came from secondary reactions (Widyawati et al.,
2011).
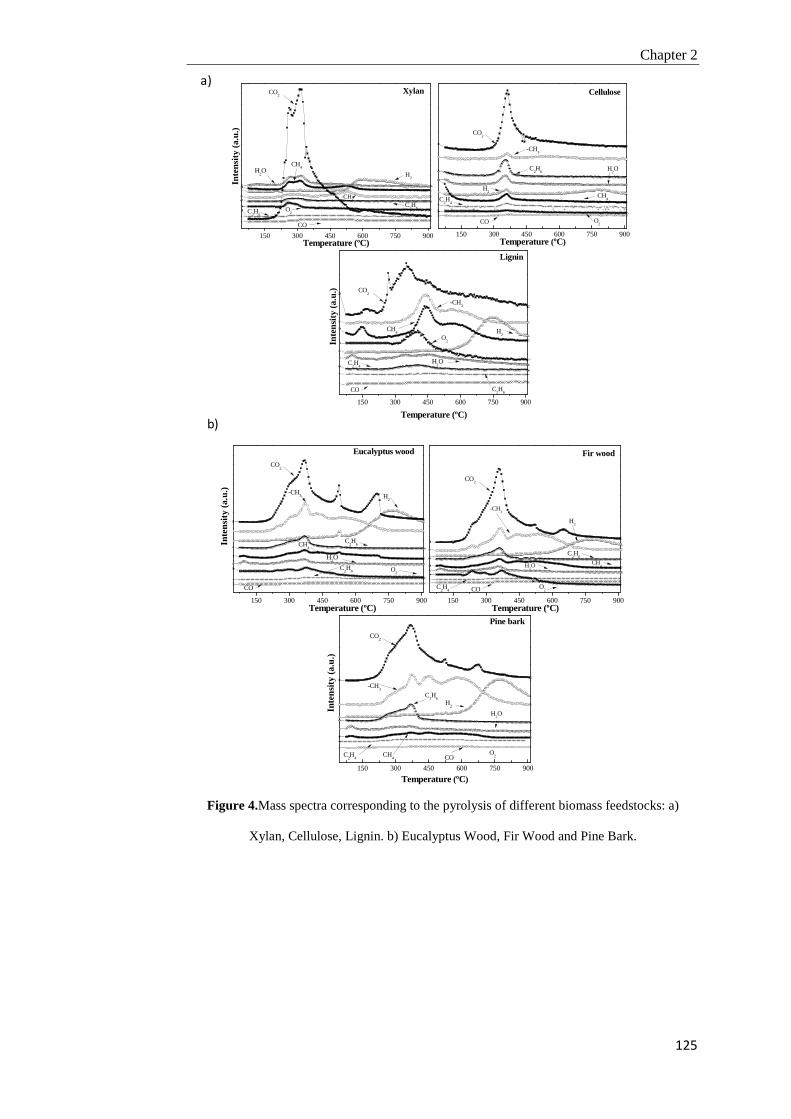
Chapter 2
125
150 300 450 600 750 900 150 300 450 600 750 900
150 300 450 600 750 900
Inte
nsity
(a.
u.)
Temperature (ºC)
O2
CO
C2H
4
CH4
H2
H2O
C2H
6
-CH3
CO2
Xylan Cellulose
O2CO
C2H
4
CH4
H2
H2OC
2H
6
-CH3
Temperature (ºC)
CO2
Lignin
Inte
nsity
(a.
u.)
Temperature (ºC)
O2
CO
C2H
4
CH4 H
2
H2O
C2H
6
-CH3
CO2
150 300 450 600 750 900 150 300 450 600 750 900
150 300 450 600 750 900
Eucalyptus wood
Inte
nsity
(a.
u.)
Temperature (ºC)
O2
CO
C2H
4
CH4
H2
H2O
C2H
6
-CH3
CO2
Fir wood
Temperature (ºC)
O2COC
2H
4
CH4
H2
H2O
C2H
6
-CH3
CO2
Pine bark
O2COC
2H
4CH
4
H2
H2O
C2H
6
-CH3
Inte
nsity
(a.
u.)
Temperature (ºC)
CO2
a)
b)
Figure 4.Mass spectra corresponding to the pyrolysis of different biomass feedstocks: a)
Xylan, Cellulose, Lignin. b) Eucalyptus Wood, Fir Wood and Pine Bark.

Chapter 2
126
Figure 5 shows the mass spectra of NG microalgae. No previous studies have
been found in the literature about the pyrolysis process of microalgae by means of
TGA-MS technique. Nevertheless, the pyrolytic characteristics of microalgae have
been studied by using TG-FTIR (Marcilla et al., 2009) and GC-MS (Ross et al., 2008).
As mentioned above, the pyrolysis process was divided into three stages (Figure 2c).
The first zone (<140 ºC) corresponded to the loss of moisture and very light volatiles
compounds. H2O and CH4 were released in a similar process as that described for
lignocellulosic biomass. In the second zone, associated to the major weight loss, three
well-identified products were detected. A first peak for CO2 and CH4 were detected at
190 ºC. Then, the main pyrolysis products (CO2, CO, CH4 and H2O) were detected
between 240 and 440 ºC. The third stage corresponded to a similar process than that
described for lignocellulosic biomass, where CH4, CO, CO2 and H2 were evolved. This
product distribution agrees well with that reported by Ross et al. (2008). Four stages
were identified: decomposition of carbohydrates (180-270 ºC) and proteins (320-450
ºC), loss of volatile metal and carbonate decomposition (<500 ºC), and char
decomposition (>750ºC) present in NG microalgae leading to H2 and CO2 evolution,
together with a significant proportion of inorganic material decomposed, probably
metal carbonates (Ross et al., 2008).
From the viewpoint of the pyrolysis of lignocellulosic biomass and marine
biomass, two main differences can be observed. Firstly, pyrolysis products from NG
microalgaewere detected at temperatures below 200 ºC. This behaviour is mainly due
to the fact that microalgae are composed by different kind of extractives, triglycerides
and hexane soluble components. These components are less thermal resistant than

Chapter 2
127
150 300 450 600 750 900 1050 1200
Inte
nsity
(a.
u.)
Temperature (ºC)
Algae
O2
CO
C2H
4
CH4
H2
H2O
C2H
6
-CH3
CO2
hemicellulose, cellulose and lignin present in about 90 % of the lignocellulosic
biomass composition (McKendry, 2002). Secondly, H2 production from microalgae at
high temperatures was lower than that from lignocellulosic biomass samples. This fact
could be due to the fact that the char, formed during the pyrolysis of NG microalgae
was less reactive than that occurred for Pine Bark, Fir Wood and Eucalyptus Wood.
Figure 5.Mass spectra of the pyrolysis of NannochloropsisGaditana microalgae.
2.3.4. Kinetic model
Figure 6 shows the experimental (solid line) compared to the predicted curve
(dotted line) obtained by non-linear regression of the kinetic model described in
Section 2.1 for Cellulose, Eucalyptus Wood and NG microalgae pyrolysis at a heating
rate of 40 ºC/min. It can be observed that the proposed model adequately reproduces
the experimental values.

Chapter 2
128
200 400 600 8000
20
40
60
80
100
200 400 600 800 10000
20
40
60
80
100200 400 600 800
0
20
40
60
80
100
Wei
ght (
%)
Experimental Theoretical
Experimental Theoretical
Wei
ght (
%)
Temperature (ºC)
Wei
ght (
%)
Experimental Theoretical
Figure 6.Comparison between experimental and theoretical results for the pyrolysis of a)
Xylan; b) Eucalyptus Wood; c) NannochloropsisGaditana microalgae.
The Marquardt-Levenberg algorithm was used to obtain kinetic parameters
(Marquardt, 1963). Table 3 shows the weight loss steps, the activation energy, the pre-

Chapter 2
129
exponential factor and the reaction order (n) for each weight loss step during the
pyrolysis of biomass samples.
Table 3.Estimated kinetic parameters for the pyrolysis of different types of lignocellulosic and
marine biomass.
Sample
Step
EA (KJ/mol)
log Ko
(log mol1-nln-1/s)
n
Cellulose 1 191.25 14.54 1
Xylan
1 94.09 14.22 2
2 125.26 12.72 3
3 181.35 13.29 6
Lignin
1 88.93 13.84 1
2 94.13 12.07 2
3 99.14 8.61 4
Fir Wood
1 95.58 14.83 1
2 128.28 13.91 2
3 154.22 14.01 5
Eucalyptus
Wood
1 57.14 8.43 1
2 129.68 12.98 2
3 159.97 13.94 2
4 176.75 12.30 2
5 202.82 11.29 3
Pine Bark
1 91.35 14.05 1
2 142.56 12.90 2
3 166.44 13.20 8
Nannochloropsis
Gaditana microalgae
1 93.64 14.17 2
2 83.46 8.60 2
3 122.71 12.55 4
4 132.38 6.03 3

Chapter 2
130
Cellulose pyrolysis kinetics has been broadly studied (Grønli et al., 1999; Lin et al.,
2009) due to two facts: cellulose is the main component in biomass structure and, its
structure is more homogenous than hemicellulose and lignin (Basu, 2010). Cellulose
thermal decomposition kinetics can be well fit by a single step first order reaction with
an activation energy in the range of 180-240 KJ/mol and a pre-exponencial factor
(log(ko)) of 14-19 log (1/s). (Grønli et al., 1999; Lin et al., 2009; Órfão et al., 1999).
These values agreed well with the experimental results obtained (Table 3).
Xylan and Lignin have also been studied in literature (Rao and Sharma, 1998). The
decomposition of Xylan and Lignin was divided into three steps. Both of them showed
lower activation energies than cellulose in the pyrolysis temperature range (step 2).
The values of activation energies for the three steps in the pyrolytic decomposition of
Lignin were the lowest ones, showing that is the most active material in the whole
range of temperatures.
Lignocellulosic biomass pyrolysis was divided into three steps: moisture evolution
(0-150 ºC), main devolatilization process (150-400 ºC) and char decomposition (400-
900 ºC). In the case of Eucalyptus Wood, the last step was divided into two more
substeps since the char showed higher reactivity. The kinetics parameters for each
substep were in the same range of values, although no similarities were found in
literature (Caballero et al., 1997; Órfão et al., 1999; Shen et al., 2010).
For the kinetic evaluation of NG microalgae pyrolysis and that of for
lignocellulosic biomass four steps were considered (Table 3). The value of the kinetics
parameters obtained in the pyrolysis of lignocellulosic biomass samples and

Chapter 2
131
microalgewere in a similar range, though a lower activation energy for the microalgae
pyrolysis were observed. These results agreed well with those reported by Li et al.
(2010) under similar operating conditions. However, the values of activation energies,
pre-exponential factors and reaction orders did not show a general trend. This fact
suggested than the pyrolysis kinetics was greatly influenced by the type and
composition of the biomass feedstock (Li et al., 2010; Shuping et al., 2010). Table 4
shows the comparison between the pyrolysis kinetic parameters of different types of
biomass sources and the results obtained in this work.
As aforementioned, the discrimination of kinetic parameters was done applying the
F-test and the t-test at the 95% confidence level.
The resulting parameters obtained from the computational non-linear regression are
summarized in Table 5. In terms of statistical results, F-test considered the regression
to be suitable in all cases since the corresponding values to the Fc/Ftest ratio was
larger than one. The t-test was also used for evaluating each parameter in the model.
As shown in Table 5, the values of tc/t-test ratio were also larger than one, showing the
statistical significance of the proposed models and their corresponding parameters.
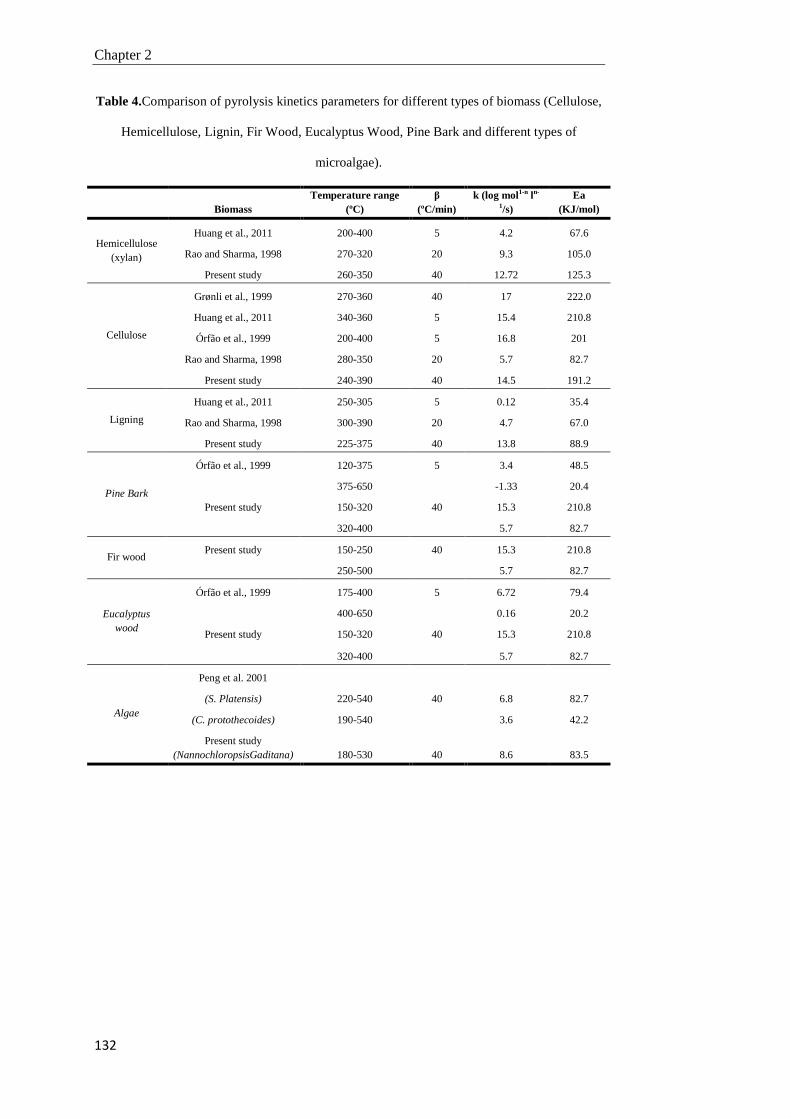
Chapter 2
132
Table 4.Comparison of pyrolysis kinetics parameters for different types of biomass (Cellulose,
Hemicellulose, Lignin, Fir Wood, Eucalyptus Wood, Pine Bark and different types of
microalgae).
Biomass
Temperature range (ºC)
β (ºC/min)
k (log mol1-n ln-
1/s) Ea
(KJ/mol)
Hemicellulose (xylan)
Huang et al., 2011 200-400 5 4.2 67.6
Rao and Sharma, 1998 270-320 20 9.3 105.0
Present study 260-350 40 12.72 125.3
Cellulose
Grønli et al., 1999 270-360 40 17 222.0
Huang et al., 2011 340-360 5 15.4 210.8
Órfão et al., 1999 200-400 5 16.8 201
Rao and Sharma, 1998 280-350 20 5.7 82.7
Present study 240-390 40 14.5 191.2
Ligning
Huang et al., 2011 250-305 5 0.12 35.4
Rao and Sharma, 1998 300-390 20 4.7 67.0
Present study 225-375 40 13.8 88.9
Pine Bark
Órfão et al., 1999 120-375 5 3.4 48.5
375-650 -1.33 20.4
Present study 150-320 40 15.3 210.8
320-400 5.7 82.7
Fir wood Present study 150-250 40 15.3 210.8
250-500 5.7 82.7
Eucalyptus wood
Órfão et al., 1999 175-400 5 6.72 79.4
400-650 0.16 20.2
Present study 150-320 40 15.3 210.8
320-400 5.7 82.7
Algae
Peng et al. 2001
(S. Platensis) 220-540 40 6.8 82.7
(C. protothecoides) 190-540 3.6 42.2
Present study (NannochloropsisGaditana) 180-530 40 8.6 83.5
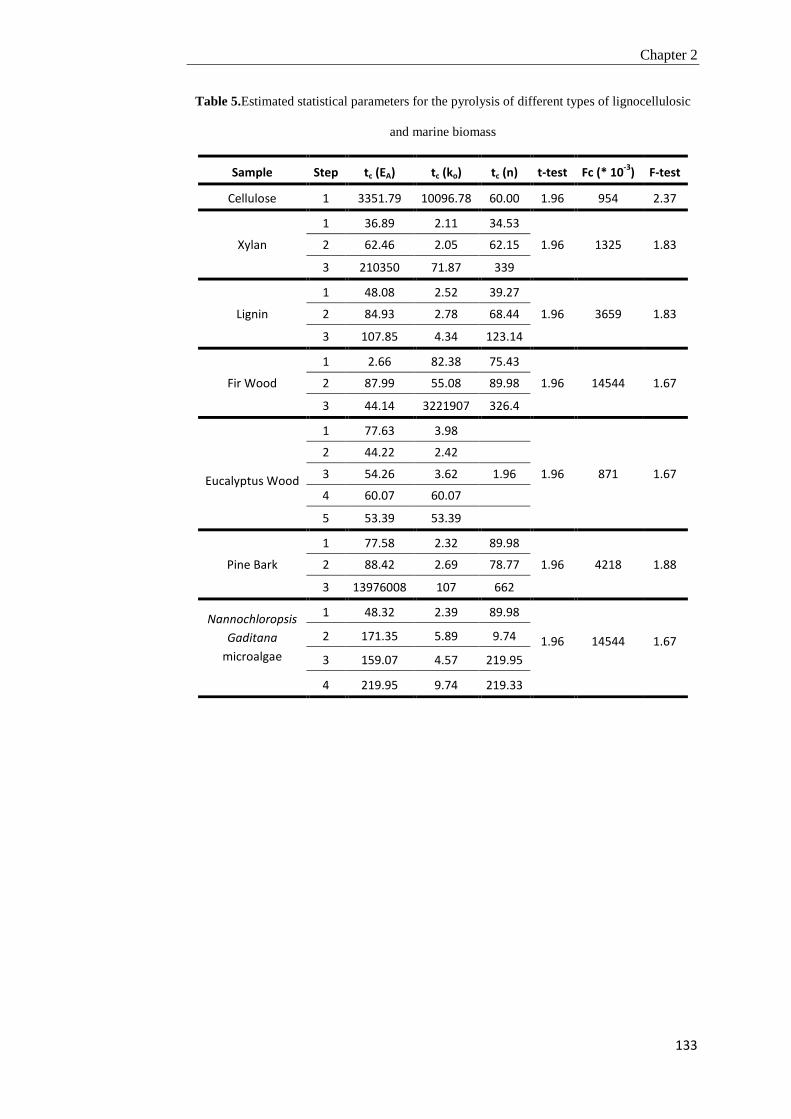
Chapter 2
133
Table 5.Estimated statistical parameters for the pyrolysis of different types of lignocellulosic
and marine biomass
Sample Step tc (EA) tc (ko) tc (n) t-test Fc (* 10-3
) F-test
Cellulose 1 3351.79 10096.78 60.00 1.96 954 2.37
1 36.89 2.11 34.53
Xylan 2 62.46 2.05 62.15 1.96 1325 1.83
3 210350 71.87 339
1 48.08 2.52 39.27
Lignin 2 84.93 2.78 68.44 1.96 3659 1.83
3 107.85 4.34 123.14
1 2.66 82.38 75.43
Fir Wood 2 87.99 55.08 89.98 1.96 14544 1.67
3 44.14 3221907 326.4
Eucalyptus Wood
1 77.63 3.98
2 44.22 2.42
3 54.26 3.62 1.96 1.96 871 1.67
4 60.07 60.07
5 53.39 53.39
1 77.58 2.32 89.98
Pine Bark 2 88.42 2.69 78.77 1.96 4218 1.88
3 13976008 107 662
Nannochloropsis
Gaditana
microalgae
1 48.32 2.39 89.98
2 171.35 5.89 9.74 1.96
14544
1.67
3 159.07 4.57 219.95
4 219.95 9.74 219.33

Chapter 2
134
2. 4. CONCLUSIONS
Thermal characteristics and gas formation during pyrolysis of Fir Wood,
Eucalyptus Wood, Pine Bark, NG microalgae and three individual components of
lignocellulosic biomass (hemicellulose, lignin and cellulose) were analyzed by TGA-
MS. Pyrolysis of lignocellulosic biomass was divided into four zones: moisture
evolution, hemicellulose decomposition, lignin and cellulose degradation and lignin
decomposition. NG microalgae showed the highest thermal stability. The main
products (CO2, light hydrocarbons and H2O) were generated between 200 and 450 ºC.
H2 was produced at high temperatures (>700 ºC). Kinetic model satisfactorily
predicted the pyrolysis of biomass. Furthermore, the statistical significance of the
model was proved.
2.5. REFERENCES
(1) Barneto, A.G., Vila, C., Ariza, J., 2011. Eucalyptus kraft pulp production:
Thermogravimetry monitoring. Thermochim. Acta. 520, 110-120.
(2) Basu, P., 2010. Biomass Gasification and Pyrolysis: Practical design and
theory, first ed. Elsevier, Oxford.
(3) Caballero, J.A., Conesa, J.A., Front, R., Marcilla, A., 1997. Pyrolysis kinetics
of almond shells and olive stones considering their organic fractions. J. Anal. Appl.
Pyrol. 42, 159-175.

Chapter 2
135
(4) Damartzis, Th., Vamvuka, D., Sfakiotakis, S., Zabaniotou, A. 2011. Thermal
degradation studies and kinetic modelling of cardoon (Cynaracardunculus) pyrolysis
using thermogravimetric analysis (TGA). Bioresour. Technol. 102, 6230-6238.
(5) De la Osa, A.R., De Lucas, A. Romero, A., Valverde, J.L., Sánchez, P., 2011.
Kinetic models discrimination for the high pressure WGS reaction over a commercial
CoMo catalyst, Int. J. Hydrogen. Energ. 36, 9673-9684.
(6) De Lucas, A., Sánchez, P., Dorado, F., Ramos, M.J., Valverde, J.L., 2006.
Kinetic model of the n-Octane hydroisomerization on PtBeta agglomerated catalyst:
Influence of the reaction conditions, Ind. Eng. Chem. Res. 45, 978-985.
(7) Froment, G.F., Bischoff, K.B., 1990 Chemical reactor analysis and design,
Wiley, New York.
(8) Grønli, M., Antal, M.J., Varhegyi, G., 1999. A round robin study of cellulose
pyrolysis kinetics by thermogravimetry. Ind. Eng. Chem. Res. 38, 2238-2244.
(9) Grønli, M.G., Várhegyi, G., Di Blasi, C., 2002. Thermogravimetric analysis
and devolatilization kinetics of wood. Ind. Eng. Chem. Res. 41, 4201-4208.
(10) Huang, Y.F., Kuan, W.H., Chiueh, P.T., Lo, S.L., 2011. Pyrolysis of biomass
by termal analysis-mass spectrometry (TA-MS). Bioresour. Technol. 101, 1968-1973.
(11) Li, D., Chen, L., Yi, X., Zhang, X., Ye, N., 2010. Pyrolytic characteristics and
kinetics of two brown algae and sodium alginate. Bioresour. Technol. 101, 7131-7136.
(12) Li, D., Chen, L., Zhang, X., Ye, N., Xing, F., 2011. Pyrolytic characteristics
and kinetic studies of three kinds of red algae. Biomass bioenergy. 35, 1765-1772.
(13) Lin, Y-C., Cho, J., Tompsett, G.A., Westmoreland, P.R., Huber, G.W., 2009.
Kinetics and Mechanism of Cellulose Pyrolysis. J. Phys. Chem. C. 113, 20097-20107.

Chapter 2
136
(14) Marcilla, A., Gómez-Siurana, A., Gomis, C., Chápuli, E., Catalá, M.C.,
Valdés, F.J., 2009. Characterization of microalgal species through TGTA/FTIR
analysis: Application to nannochoropsis sp. Thermochim. Acta. 484, 41-47.
(15) Marquardt, D.W., 1963. An algorithm for least-squares estimation of
nonlinear parameters, J. Soc. Ind. Appl. Math. 11, 431-441.
(16) Maschio, G., Koufopanos, G., Lucchesi, A., 1992. Pyrolysis, a promising
route for biomass utilization. Bioresour. Technol. 42, 219-231.
(17) McKendry, P. 2002. Energy production from biomass (part 1): overview of
biomass. Bioresour. Technol. 83, 37-46.
(18) Nakamura, T., Kawamoto, H., Saka, S., 2007. Condensation reactions of some
lignin related compounds at relatively low pyrolysis temperature. Journal of wood
chemistry and technology. 27, 121-133.
(19) Órfão, J.J.M., Antunes, F.J.A., Figueiredo, J.L., 1999. Pyrolysis kinetics of
lignocellulosic materials-three independent reactions model. Fuel. 78, 349-358.
(20) Peng, W., Wu, Q., Tu, P., Zhao, N., 2001. Pyrolytic characteristics of
microalgae as renewable energy source determined by thermogravimetric analysis.
Bioresour. Technol. 80, 1-7.
(21) Press, W.H., Teukolsky, S.A., Vetterling, W.T., Flannery, B.O., 1992.
Numerical recipes in Fortran, second ed. Cambridge University Press, Cambridge.
(22) Rao, T.R., Sharma, A., 1998. Pyrolysis rates of biomass materials. Energy. 23,
973-978.

Chapter 2
137
(23) Ross, A.B., Jones J. M., Kubacki, M.L., Bridgeman, T. 2008. Classification of
macroalgae as fuel and its thermochemical behavior. Bioresour. Technol. 99, 6494-
6504.
(24) Severian, D., 2008. Polysacharides: Structural diversity and functional
versatility, Second ed. Marcel Dekker, New York.
(25) Shen, D.K., Gu, S., Bridgwater, A.V., 2010. The thermal performance of the
polysaccharides extracted from hardwood: Cellulose and hemicellulose. Carbohydr.
Polym. 82, 39-45.
(26) Shuping, Z., Yulong, W., Mingde, Y., Chun, L., Junmao, T., 2010. Pyrolysis
characteristics and kinetics of the marine microalgae Dunaliellatertiolecta using
thermogravimetric analyzer. Bioresour. Technol. 101, 359-365.
(27) Stenseng, M., Jensen, A., Dam-Johansen, D., 2001. Investigation of biomass
pyrolysis by thermogravimetric analysis and differential scanning calorimetry. J. Anal.
Appl. Pyrolysis. 58-59, 765-780.
(28) Sun, L., Chen, J.Y., Negulescu, I.I., Moore M.A, Collier B.J., 2011. Kinetics
modeling of dynamic pyrolysis of bagasse fibers, Bioresour. Technol. 102, 1951-1958.
(29) Valverde, J.L., De Lucas, A., Carmona, M.S., González, M., Rodríguez, J.F.,
2004. A generalized model for the measurement of effective diffusion coefficients of
heterovalent ions in ion exchangers by the zero-length column method, Chem. Eng.
Sci. 59, 71-79.
(30) Wang, G., Li, W., Li, B., Chen, H., 2008. TG study on pyrolysis of biomass
and its three components under syngas. Fuel. 87, 552-558.

Chapter 2
138
(31) Wang, J., Wang, G., Zhang, M., Chen, M., Li, D., Min, F., Chen, M., Zhang,
S., Ren, Z., Yan, Y., 2006. A comparative study of thermolysis characteristics and
kinetics of seaweeds and fir wood. Process Biochem. 41, 1883-1886.
(32) White, J.E., Catallo, W.J., Legendre, B.L., 2011. Biomass pyrolysis kinetics:
A comparative critical review with relevant agricultural residue case studies. J. Anal.
Appl. Pyrolysis. 91, 1-33.
(33) Widyawati, M., Church, T.L., Florin, N.H., Harris, A.T., 2011. Hydrogen
synthesis from biomass pyrolysis with in situ carbon dioxide capture using calcium
oxide. Hydrogen Energy. 30, 1-14.
(34) Yaman, S. 2004. Pyrolysis of biomass to produce fuels and chemical
feedstocks. Energy Convers. Manage. 45, 651-671.
(35) Yang, H., Yan, R., Chen, H., Zheng, C., Lee, D.H., Liang, D.T., 2006. In-
Depth Investigation of Biomass Pyrolysis Based on Three Major Components:
Hemicellulose, Cellulose and Lignin. Energy Fuels. 20, 388-393.

Chapter 3:
THERMOGRAVIMETRIC -MASS SPECTROMETRIC ANALYSIS ON COMBUSTION
OF LIGNOCELLULOSIC AND MARINE BIOMASS
The combustion characteristics of lignocellulosic biomass and
marine biomass were investigated by means of TGA-DSC-MS.
Additionally, Canadian crops were investigated. The combustion
process can be mainly divided into two main steps: devolatilization
(Dev. stage) and char oxidation stage (Oxid. stage). The thermal
behavior of both types of biomass could be related to the
decomposition of their main components. During the Dev. stage of
lignocellulosic biomass the hemicellulose and cellulose decompose
whereas the oxidation stage was mainly ascribed to lignin. On the
other hand, in the Dev. stage for microalgae all of their main
components decompose in the following order: carbohydrates,
lipids and proteins. Combustion kinetics were studied by Pseudo
multi-components separate-stage models (PMSM). Models based
on reaction order (Oi), nucleation (Ni) and diffusion (Di) achieved
the best fitting to the experimental data. Additionally, the process
was successfully modeled obtaining errors below ± 3.35 %. CO,
CO2 and H2O were the main components evolved from

Chapter 3
136
combustion. Additionally, light hydrocarbons (CH4 and C2H5) were
also present. Finally, nitrogen compounds were in a higher
proportion than sulfur compounds being released as primary
amines and NOx. The NOx release was higher for the combustion
of microalgae than for lignocellulosic biomass due to their high
initial nitrogen content. The high ash content of microalgae and
Canadian biomass samples catalyzed the volatile compounds
release and shifted the process to lower temperatures. Furthermore,
this fact implied that sample pre-treatment is required before being
used in thermal applications Finally, the main pollutants released
during the combustion process of Canadian biomass were
analyzed. Nitrogen (NO, NO2 and HCN) and sulfur (SO and SO2)
compounds were found in high proportions. Nitrogen compounds
were released in both combustion stages, whereas sulfur
compounds evolved mainly in the a lower temperature range. Other
pollutants were found in lower concentrations (CH3Cl and C6H6).
3.1. INTRODUCTION
As already mentioned in Chapter 2, thermochemical conversion of biomass is
considered as one of the most promising processes for biomass utilization. These
processes are employed for power generation, production of liquid biofuels, chemicals
and charcoal.Thus, a good understanding of the decomposition of biomass during
thermochemical conversion is important for developing efficient processing
technology [1].
Combustion can be defined as the conversion of biomass fuels to several forms of
useful energy in the presence of air or oxygen. Thermogravimetric techniques have
commonly been used to investigate the thermochemical conversion of solid raw
materials as coal and woods [2-4].Unlike pyrolysis, the combustion of biomass using

Chapter 3
137
thermogravimetric analysis has not been studied intensively yet [5].Recently, the
combustion behavior of different types of wood has been performed[1; 5;6].
Furthermore, Kay et al., (2011) studied the effect of biomass components on the co-
combustion of biomass and coal.
Biomass characteristics and kinetics of biomass combustion are essential for
modelingthe combustion in industrial processes [7]. Furthermore, a knowledge of the
kinetics of the process has great importance for a correct design and product yield
control [8]. Although there are significant differences in operating conditions,
thermogravimetric analysis provides a powerful tool to accomplish preliminary kinetic
studies on the thermal decomposition of solids.Algae are a very promising biomass for
the following reasons: rapid growth rate, high yield per area, high efficiency in CO2
capture and solar energy conversion and no competition with food agriculture. Among
the different types of algae, microalgae have received more attention than others
because they can be cultured in ponds or photobioreactors with supply of nutrients or
wastewater [9].
In Canada, the average annual wood cut has been estimated at 167.5 million m3
creating over 60 million tons of residues. The annual harvests are taken from
approximately 1 million ha, constituting of only about 0.25 % of the total forestland in
Canada[10]. Thus, due to the great potential of Canada soil and variety, it seems
mandatory to invest on crops for energy production.
Dedicated energy crops can come from multiple sources. However, it is recognized
the high potential of woody crops (hardwoods and pines) and non-woody, high-
yielding annual and perennial crops (Miscanthus, switchgrass and sweet sorghum) due
to multiples advantages. Some of the benefits of energy crops include: less capital-
intensive conversion technologies, attractive opportunity for local and regional self-
sufficiency, reduction in greenhouse gas emissions and viable alternative to fossil fuel
use[11]. In spite of the environmental advantages, some aspects concerning the release
of contaminants during biomass combustion must be taken into account. In this regard,
NOx and SOx emissions depend on raw biomass composition, which usually is

Chapter 3
138
variable[12]. The control of NOx and SOxfrom biomass combustion can contribute to
decrease the emissions of these pollutants in Canada which are around 0.7 and 1.2
million tons per year for NOx (as NO2) and SO2, respectively[13]. Furthermore, the
chloride amount in biomass might turn into operational problems such as corrosion.
Other organic compounds such as benzene and toluene are considered to be part of the
most dangerous emissions from biomass combustion causing diseases as lung
infection and leukemia[14]. Therefore, the knowledge of pollutant release during
biomass conversion is truly important in order to reassure the use of biomass from the
environmental point of view.
As reported by Naik et al. (2010), suitable mathematical models can be derived for
a better comprehension of the thermal behavior of these complex feedstock that allow
to perform economic analysis and develop technology for a more efficient biomass
conversion[10].
The aim of this work was to perform a comprehensive study of the combustion
behavior of lignocellulosicand marine biomassby means of the TGA-DSC. Firstly,
three different types of lignocellulosic biomass(fir wood, eucalyptus wood and pine
bark) and their main components (cellulose, hemicellulose and lignin)
technique.Secondly, three different microalgae species were selected:Chlorella
vulgaris, NannochloropsisGaditana and ScenedesmusAlmeriensis. Finally, two
different types of Canadian biomass were considered: woody crops (black spruce and
Pinusbanksiana mixtures and willow) and different non-woody and perennial crops
(common reed, reed phalaris and switchgrass). In addition, the effect of the heating
rate on the combustion behavior of lignocellulosicsamples was also studied.
Furthermore, the kinetics of the oxidation process were evaluated. Finally,the gases
released during the combustion process were analyzed by MS. The main pollutant
gases released during the combustion process of Canadian biomass were analyzed.
3.2. EXPERIMENTAL
3.2.1. Materials
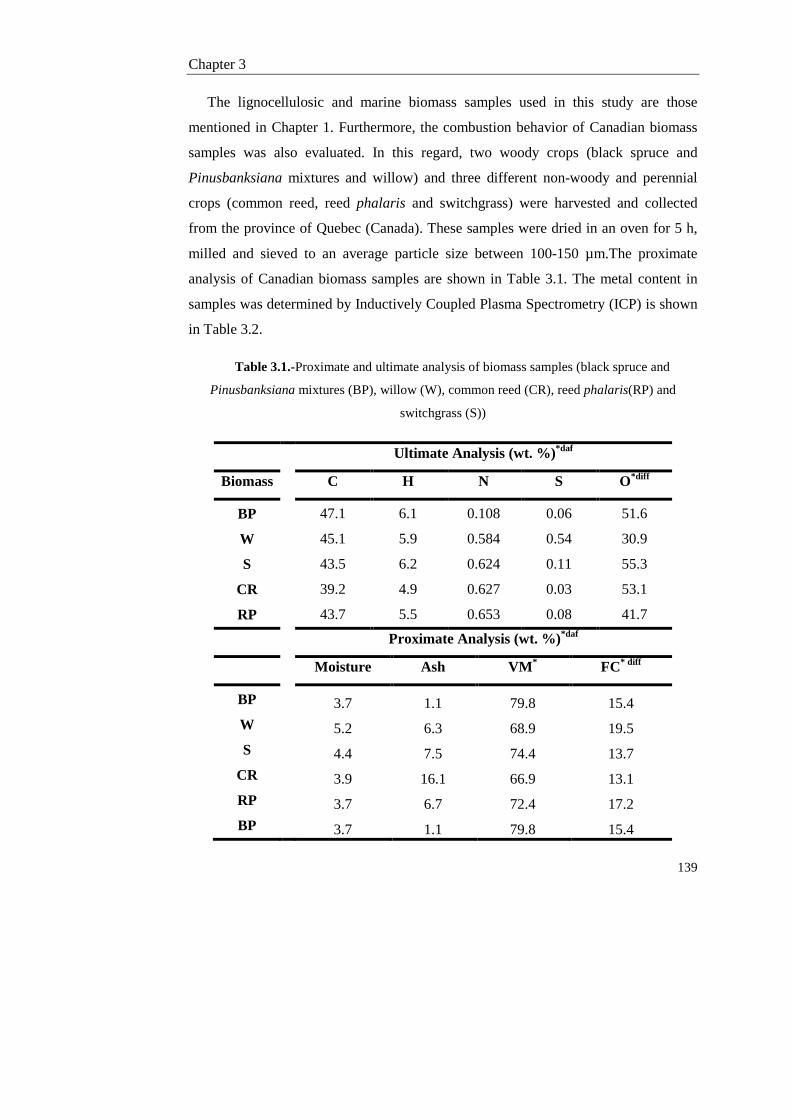
Chapter 3
139
The lignocellulosic and marine biomass samples used in this study are those
mentioned in Chapter 1. Furthermore, the combustion behavior of Canadian biomass
samples was also evaluated. In this regard, two woody crops (black spruce and
Pinusbanksiana mixtures and willow) and three different non-woody and perennial
crops (common reed, reed phalaris and switchgrass) were harvested and collected
from the province of Quebec (Canada). These samples were dried in an oven for 5 h,
milled and sieved to an average particle size between 100-150 µm.The proximate
analysis of Canadian biomass samples are shown in Table 3.1. The metal content in
samples was determined by Inductively Coupled Plasma Spectrometry (ICP) is shown
in Table 3.2.
Table 3.1.-Proximate and ultimate analysis of biomass samples (black spruce and
Pinusbanksiana mixtures (BP), willow (W), common reed (CR), reed phalaris(RP) and
switchgrass (S))
Ultimate Analysis (wt. %)*daf
Biomass C H N S O*diff
BP 47.1 6.1 0.108 0.06 51.6
W 45.1 5.9 0.584 0.54 30.9
S 43.5 6.2 0.624 0.11 55.3
CR 39.2 4.9 0.627 0.03 53.1
RP 43.7 5.5 0.653 0.08 41.7
Proximate Analysis (wt. %)*daf
Moisture Ash VM* FC* diff
BP 3.7 1.1 79.8 15.4
W 5.2 6.3 68.9 19.5
S 4.4 7.5 74.4 13.7
CR 3.9 16.1 66.9 13.1
RP 3.7 6.7 72.4 17.2
BP 3.7 1.1 79.8 15.4
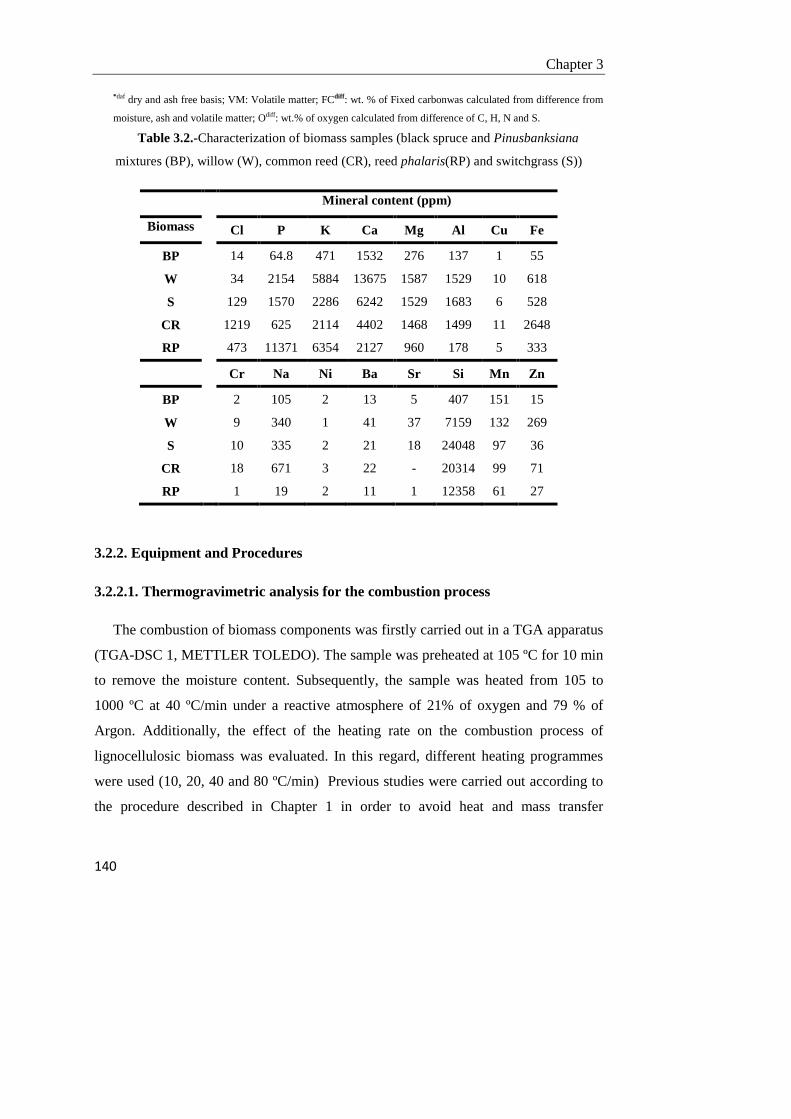
Chapter 3
140
*daf dry and ash free basis; VM: Volatile matter; FCdiff : wt. % of Fixed carbonwas calculated from difference from
moisture, ash and volatile matter; Odiff: wt.% of oxygen calculated from difference of C, H, N and S.
Table 3.2.-Characterization of biomass samples (black spruce and Pinusbanksiana
mixtures (BP), willow (W), common reed (CR), reed phalaris(RP) and switchgrass (S))
Mineral content (ppm)
Biomass Cl P K Ca Mg Al Cu Fe
BP 14 64.8 471 1532 276 137 1 55
W 34 2154 5884 13675 1587 1529 10 618
S 129 1570 2286 6242 1529 1683 6 528
CR 1219 625 2114 4402 1468 1499 11 2648
RP 473 11371 6354 2127 960 178 5 333
Cr Na Ni Ba Sr Si Mn Zn
BP 2 105 2 13 5 407 151 15
W 9 340 1 41 37 7159 132 269
S 10 335 2 21 18 24048 97 36
CR 18 671 3 22 - 20314 99 71
RP 1 19 2 11 1 12358 61 27
3.2.2. Equipment and Procedures
3.2.2.1. Thermogravimetric analysis for the combustion process
The combustion of biomass components was firstly carried out in a TGA apparatus
(TGA-DSC 1, METTLER TOLEDO). The sample was preheated at 105 ºC for 10 min
to remove the moisture content. Subsequently, the sample was heated from 105 to
1000 ºC at 40 ºC/min under a reactive atmosphere of 21% of oxygen and 79 % of
Argon. Additionally, the effect of the heating rate on the combustion process of
lignocellulosic biomass was evaluated. In this regard, different heating programmes
were used (10, 20, 40 and 80 ºC/min) Previous studies were carried out according to
the procedure described in Chapter 1 in order to avoid heat and mass transfer

Chapter 3
141
limitations. In this sense, sample weight was kept at 6 mg, the particle size was kept in
the 100-150 µm range and a constant flow rate of 100 Nml/min was used.
3.2.2.2. TGA-MS analysis of the Gaseous Products
The analysis of the gas products distribution coming from the thermal analysis was
carried out in a thermogravimetricanalyzer (TGA-DSC 1; METTLER TOLEDO)
coupled to a mass spectrometer (Thermostar-GSD 320/quadrupole mass analyzer;
PFEIFFER VACUUM) with an electron ionization voltage at 70 eV and provided
mass spectra up to 300 a.m.u. The interface was wrapped with heating wire to
circumvent condensation of exhausting gases. A semiquantitative analysis was
performed by using a normalization procedure. The ion intensities were normalized to
the intensity of 38Ar isotope to eliminate systematic instrumental errors [15].
3.2.2.3. Kinetic analysis
Kinetic data from solid-state combustion was obtained using thermogravimetric
analysis. The devolatilization curve is usually obtained as a sum of the corresponding
individual components contributions [2; 16]. However, solid-state reactions are a more
complex process, involving processes such as nucleation, adsorption, desorption and
surface/bulk diffusion [17].
The model proposed in this work is similar to that reported by Gil et al.[2] for the
determination of the combustion kinetic parameters of coal/biomass blends.The
kinetic rates were based on the Arrhenius equation[2; 6].
�� ��� = � ∙ �(�) (1)
� = � ∙ � �/�∙� (2)
wheref(α) represents the hypothetical model of the reaction mechanism;k is the
reaction rate; Arepresents the pre-exponential factor (min-1); Eis the activation energy
(kJ mol-1);Tis the absolute temperature (K); trepresents the time (min), and αis the
degree of conversion defined by:

Chapter 3
142
� = (�� −��) (�� −��)� (3)
wheremoand mtrepresent the mass at t=0 and t=t, respectively, and mfis the final mass
of the sample.
For a constant heating rate β(K min-1), β= dT/dt, Eq. (1) can be transformed into:
�� �(�)� = � �� �� (4)
By integrating Eq. (4) gives:
�(�) = � �� �(�)��� = � �� � � � ����
� �� (5)
whereg(α) is the integral function of conversion.
Eq. (5) is integrated by using the Coats-Redfern method [18]:
ln[�(�) �$� ] = ln[� ∙ & � ∙ ' ∙ (1 − 2&� '� +� ] − ' & ∙ �� (6)
Generally, the term 2RT/Ecan be neglected as it is much less than unity[19]. It has
been demonstrated that for both, the temperaturesof combustion range and most values
of E, the expression ln[AR/βE] in Eq. (6) is essentially constant [20]. Thus, if the
correct expression of g(α) is used, the plot of ln[g(α)/T2] against 1/T should give a
straight line with a high correlation coefficient of linear regression analysis, from
which the values of E and A can be respectively calculated from the slope of the line;
and by the intercept term in Eq. (6).The functions f(α) and g(α)referredto the different
models for reaction are presented in Table 3.3(White et al., 2011).The Marquardt
algorithm was used in the linear regression procedure to obtain the kinetic parameters
(E and A). Furthermore, the statistical significance of the estimated parameters based
on the F-test and t-test was performed according to the procedure described
elsewhere[21].

Chapter 3
143
Table 3.3.- Expressions for the most common reaction mechanisms in solid state reactions [17]
Reaction model f(α) g(α)
Reaction order
O0 (1-α)n α
O1 -ln(1-α)
O2 -(1-α)-1
O3 1/2 (1-α)-2
Phase boundary controlled
reaction
R2 (1-α)(1-1/n) 1-(1-α)(1/2)
R3 1-(1-α)(1/3)
Power Law
P1 n(α)(1-1/n) α1/4
P2 α1/3
P3 α1/2
P4 α3/2
Nucleation and growth (Avrami-Erofeev equation)
N1 n(1-α)[-ln(1-α)](1-1/n) [-ln(1-α)](1/1.5)
N2 [-ln(1-α)](1/2)
N3 [-ln(1-α)](1/3)
N4 [-ln(1-α)](1/4)
Diffusion
D1 1/2α α2
D2 [-ln(1-α)]-1 (1-α)ln(1-α)+α
D3 3/2(1-α)2/3[1-(1-α)1/3]-1 [1-(1-α)1/3]2
D4 3/2[(1-α)1/3-1]-1 1-2/3α-(1-α)2/3

Chapter 3
144
3.3. RESULTS AND DISCUSSION.
3.3.1. Combustion of lignocellulosic biomass
Thermogravimetricstudy on combustion of lignocellulosic biomass
Figure 3.1 shows the TGA-DTG profiles ofcombustion between 105 and 1000 ºC
for the main components of lignocellulosic biomass (cellulose, xylan and lignin) and
different types of lignocellulosic biomass (eucalyptus wood, fir wood and pine bark)
at a heating rate of 40 ºC/min. Table 3.4 summarizes the most relevant combustion
characteristics of lignocellulosic biomass at heating rates of 10, 20, 40 and 80 ºC/min.
Figure 1.-Thermogravimetric curves for the combustion process of: a) main components of
lignocellulosic biomass (cellulose, lignin and xylan) and b) lignocellulosic biomass (eucalyptus
wood, fir wood and pine bark).
0
20
40
60
80
100
125 250 375 500 625 750 875 10000.0
0.5
1.0
1.5
2.0
2.5
3.0
0
20
40
60
80
100
125 250 375 500 625 750 875 10000.0
0.2
0.4
0.6
0.8
Wei
ght (
%)
Lignin Xylan Cellulose
Wei
ght l
oss
rate
(%
wt./
ºC)
Temperature (ºC)
Wei
ght (
%)
Eucalyptus Wood Fir Wood Pine Bark
Wei
ght l
oss
rate
(%
wt./
ºC)
Temperature (ºC)

Chapter 3
145
Table 3.4.-Combustion characteristics for cellulose, lignin, xylan, fir wood, eucalyptus wood and pine bark at different heating rates
Primarycomponents of biomass Lignocellulosicbiomass
Cellulose Xylan Lignin Firwood Eucalyptuswood Pine bark
1st
peak 2ndpeak 1st
peak 2ndpeak 3rd
peak 4rd
peak 1st
peak 2ndpeak 3rd
peak 4rd
peak 1st
peak 2ndpeak 3rd
peak 1st
peak 2ndpeak 3rd
peak 1st
peak 2ndpeak 3rd
peak 4th
peak
Tdo
*(ºC) 244 196 138 184 185 186
262 205 142 203 196 196
270 217 147 212 206 199
283 226 166 228 214 212
Tpo* (ºC) 244 438 196 262 385 578 138 348 455 - 184 372 575 185 397 566 186 357 443 -
262 455 205 277 396 561 142 302 434 - 203 395 587 196 407 588 196 370 446 607
270 479 217 288 429 - 146 309 446 - 212 413 607 206 413 608 199 380 452 616
283 518 226 304 443 - 166 313 459 832 228 434 640 214 436 624 212 388 481 653
Tpf* (ºC) 413 556 262 368 530 693 348 455 555 - 360 505 652 355 484 653 342 443 531 -
429 584 277 380 556 703 302 434 619 - 383 529 658 378 505 675 356 446 561 645
443 616 288 383 618 - 309 442 757 - 393 553 679 390 525 699 368 452 589 667
459 649 304 396 680 - 313 459 832 911 405 622 711 418 606 735 388 481 653 715
Tp* (ºC) 325 493 243 285 485 617 285 444 481 - 318 424 608 290 431 620 314 422 469 -
339 517 253 289 491 605 297 395 499 - 330 438 630 303 438 636 322 427 473 627
354 532 264 298 550 - 309 397 518 - 340 455 652 314 424 667 333 431 491 645
373 544 274 304 557 - 312 404 537 880 350 449 682 328 451 694 344 465 528 658
(dw/dT)max*
(dwt.%/ºC) 3.247 0.142 0.675 0.616 0.387 0.256 0.144 0.590 0.905 - 0.719 0.532 0.042 0.701 0.458 0.057 0.542 0.440 0.450 -
3.129 0.106 0.778 0.619 0.335 0.302 0.141 0.277 0.603 - 0.692 0.506 0.024 0.697 0.435 0.029 0.521 0.431 0.435 0.016
2.739 0.102 0.841 0.631 0.254 - 0.132 0.271 0.336 - 0.678 0.329 0.021 0.657 0.405 0.027 0.509 0.363 0.37 0.012
2.264 0.081 0.978 0.699 0.152 - 0.123 0.213 0.301 0.02 0.652 0.222 0.020 0.634 0.222 0.027 0.451 0.248 0.080 0.08
Residue yield (%)
0.13 5.81 5.48 4.82 5.43 5.98
0 3.46 5.51 2.74 3.48 2.69
0 4.44 2.61 3.49 2.68 1.76
2.24 4.05 2.93 3.21 3.47 2.89

Chapter 3
146
There are substantial differences in the thermal behavior of the main components
of lignocellulosic biomass (Figure 3.1a), which are mainly attributed to their different
chemical structures [3]. Lignin was the first component to decompose (146 ºC)
whereas xylan and cellulose started decomposing at 187 and 266 ºC, respectively. This
behavior is attributed to the fact that lignin and xylan have methoxy functional groups,
which tend to break easily[3].
Lignin was the most thermal stable component decomposing in two steps, starting
at 146 and 550 ºC, respectively. DTG curve for lignin oxidation presented the smallest
and widest peaks. This fact is due to lignin is polymeric in nature with a three-
dimensional structure consisting of phenylpropane coupled with C-C or C-O-C bonds
whose activity covers a wide range of temperatures [3]. The DTG curve for lignin
combustion shows two main peaks at 397 and 518 ºC (decomposition rate of 0.295
and 0.326 %/ºC, respectively). The burnout temperature was established at 757 ºC.
Xylanwas the least thermally stable component of biomass. Two strong
decomposition peaks overlapped in the temperature range of 147-371 ºC, having the
maximum DTG peak at 264 ºC (0.841 %/ºC). This degradation step is mainly
attributed to C-O-C and some pyranose C-C bonds breakdown[5]. A second step was
observed between 429 and 615 ºC.
The primary decomposition of cellulose, in which 87 % by weight was lost, took
place between 266 and 423 ºC. The DTG peak for cellulose oxidation was found at
354 ºC and presented the highest weight loss rate of all samples (2.74 %/ºC). During
this stage, a complex set of reactions as denitration and deacetylation, scission of O-N,
C-O, CC and C-H bonds might take place [22]. A smaller peak was found between
441 ºC and 637 ºC (0.10 %/ºC) that can be attributed to char oxidation.
Figure 1b shows the TGA-DTG profiles for the combustion process of
lignocellulosic biomass (eucalyptus wood, fir wood and pine bark). The thermal
degradation of lignocellulosic biomass is often reported as the addition of the primary
decomposition of their main components [3; 5;23]. As can be seen from Figure

Chapter 3
147
3.1.b,the lignocellulosic combustion biomass presented similar TGA-DTG profiles,
exhibiting three decomposition peaks. In the temperature range between 180 and 388
ºC, a marked peak with a shoulder corresponding to the decomposition of cellulose
and hemicellulose, was detected. According to different authors [3; 24;25], this stage
represents the release of volatiles and their ignition leading to char formation. Then, a
broad peak between 368 and 600 ºC,related to char oxidation,was observed. Lignin is
the main contributor in this stage as it is the main responsible for biomass char
formation [23]. Finally, a smaller peak was observed for all samples at temperatures
above 625 ºC. This step is mainly related to inorganic matter decomposition as
carbonates [26].
In spite of their similarity, several differences can be observed in the combustion
behavior of the lignocellulosic samples here considered. Eucalyptus wood is the
biomass sample with high cellulose content and low hemicellulosecontent. According
to Kai et al.[3], a high content in cellulose shifts the devolatilization stage to lower
temperatures, increasing the decomposition rate. Furthermore, the shoulder in the
DTG curve, indicating hemicellulose decomposition, was less marked. On the other
hand, pine bark had the minor cellulose content, presenting the lowest weight loss rate,
whereas the shoulder related to the hemicellulose decomposition shoulder was the
most pronounced. The second stage, corresponding to the oxidation of the char formed
during the devolatilization stage, was characterized by the presence of lignin in the
corresponding tested sample [3]. In this case, the DTG peak for the pine bark
combustion was the widest one, from 368 to 589 ºC compared to the fir and
eucalyptus woods. Finally, the last stage was similar in all samples studied, pointing
out that cellulose, hemicellulose and lignin content had no influence on the residue
generated at the end of the thermal process[3].
Effect of the heating rate.
Figure 3.2 shows the DTG profiles for the combustion of biomass main
components (cellulose, xylan and lignin) and lignocellulosic biomass(eucalyptus

Chapter 3
148
wood, fir wood and pine bark) at heating rates of 10, 20, 40 and 80 ºC/min. Table
3.4shows the most relevant characteristics of the combustion process.
Figure 3.2.- Effect of the heating rate in the combustion process of: a) main components of
lignocellulosic biomass (cellulose, lignin and xylan) and b) lignocellulosic biomass (eucalyptus
wood, fir wood and pine bark) at 10, 20, 40 and 80 ºC/min.
Figure 3.2a shows the DTG plots for the combustion of the main components of
biomass at different heating rates. Generally, the higher the heating rate, the higher the
temperature at which the peak appeared. This fact is attributed to the poor thermal
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
125 250 375 500 625 750 875 10000.0
0.2
0.4
0.6
0.8
0.0
0.2
0.4
0.6
0.8
1.0
0.0
0.2
0.4
0.6
0.8
0.0
0.2
0.4
0.6
0.8
125 250 375 500 625 750 875 10000.0
0.1
0.2
0.3
0.4
0.5
Wei
ght l
oss
rate
(%
wt./
min
)
10 ºC/min 20 ºC/min 40 ºC/min 80 ºC/min
Cellulose
Lignin
Temperature (ºC)
10 ºC/min 20 ºC/min 40 ºC/min 80 ºC/min
Xylan
Fir wood
Wei
ght l
oss
rate
(%
wt./
min
)
Eucalyptus wood
Pine bark
Temperature (ºC)

Chapter 3
149
conductivity of lignocellulosic biomass, resulting in a particle gradient temperature
[17]. Furthermore, the weight loss rate decreased with increasing values of theheating
rate. Cellulose main peak shifted from 325 ºC at 10 ºC/min to 373 ºC at 80 ºC/min,
whereas char oxidation peak turned from 493 ºC to 544 ºC. The maximum weight loss
rate shiftedfrom 3.247 %/ºC to 2.264 %/ºC whereas the peak for the char oxidation
varied from 0.142 to 0.081. Xylan and lignin combustion behavior followed a similar
trend, although several differences can be observed. Maximum weight loss rate
increased in the devolatilization stage with increasing heating rates for Xylan (from
0.675 to 0.978 %wt./ºC).These findings agree well with those obtained by William
and Besler[27]. Furthermore, at low heating rates,10 and 20 ºC/min, an additional
peak was found at temperatures above 500 ºC. This peak is mainly attributed to the
presence of impurities that could not be extracted from the raw material [5]. These
small impurity peaks vanished at 40 and 80 ºC/min by the overlapping effect caused
by the use of high heating rates.Finally, lignin experimented an irregular behavior. In
this case, three peaks were identified at 10, 20, and 40 ºC/min, whereas an additional
peak was found at temperatures above 832 ºC. At 10 ºC/min the combustion of lignin
was mainly performed between 300 and 525 ºC, occurring two peaks at 444 and 481
ºC in a similar way as that reported by Cheng et al. [5]. However, when the heating
rate was increased, the first peak shifted to lower temperatures whereas the second one
did to higher temperatures. As aforementioned, both peaks became broader by the
overlapping effect caused by using high heating rates.
Figure 3.2b shows the DTG combustion profiles for lignocellulosic biomass.
Concerning the heating rate effect on lignocellulosic biomass combustion behavior, all
the samples followed the general trend. The higher the heating rate, the higher the
peak temperature was whereasthe maximum weight loss rate, for all DTG peaks,
decreased. In all cases, the oxidation peak was broader when heating rate was
increased.

Chapter 3
150
Kinetic analysis
The kinetic model used in this work was derived fromthe PMSM (Pseudo multi-
component separate-stage models) approach. In this type of models, the biomass
sample is composed of multiple pseudo components [19]. In this regard, the kinetic
parameters can be determined assuming single separate reactions for the different
stages of thermal conversion. As aforementioned, the DTG plots represented different
decomposition peaks dividing the combustion process of biomass samples in different
stages. Each stage represents a separate reaction. Biomass combustion was clearly
defined by three main stages: devolatilization stage (Dev. stage), char oxidation stage
(Oxid. stage) and remaining char burning stage (Rem. stage). However, some samples
experimented additional decompositions. For example, the Dev. Stage for xylan was
represented by two peaks. In order to differentiatethem, the Dev. stagefor xylan was
divided into two stages:Dev. stage A and Dev. stage B. Furthermore, xylancombustion
showed a peak athigh temperatures named as Imp. stage.Additionally, the Oxid.stage
for lignin and pine bark combustion was characterized by two peaks. In a similar way,
these stages were named as Oxid. stage A and Oxid stage B, respectively.This way,
eq. (6) was used separately to each of the stages commented above.In order to obtain
reliable kinetic data, operating parameterswere established according to the procedure
described in Chapter 1.
The model representing the form of g(α)(Table 3.3), which delivered the highest
correlation coefficient,was considered to be the function representing the mass loss
kinetics for the samples under study. The function g(α) depends on the mechanism
controlling the reaction and the size and shape of the reacting particles [28]. Figure 3.3
and 3.4 shows the plots of ln[g(α)/T2] versus 1/T that provided the best linearity at 10,
20, 40 and 80 ºC/min for biomass main components and lignocellulosic biomass
samples, respectively. Table 3.5 and 3.6 summarizes themain kinetic parameters of
biomass samples.It can be seen from Figure 3.3,Table 3.5 and Table 3.6 that all the
stages fitted well into a straight line. All cases showed an acceptable linear fit
regression (r2> 0.9).However, only models based on reaction order (Oi), nucleation

Chapter 3
151
Figure 3.3.- Plot of ln(g(α)/T) vs 1/T for the combustion process of: a) main components of
lignocellulosic biomass (cellulose, lignin and xylan) and b) lignocellulosic biomass (eucalyptus
wood, fir wood and pine bark)at 10, 20, 40 and 80 ºC/min.
0.0012 0.0016 0.0020 0.0024
-24
-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
ln (g
( αα αα)/T
2 )
Dev. Stage - D3
Oxid. Stage A - O3
Oxid. Stage B - O1
Dev. Stage - D3
Oxid. Stage A - O1
Oxid. Stage B - O1
10 ºC/min 80 ºC/min40 ºC/min
20 ºC/min Dev. Stage - D3
Oxid. Stage A - O1
Oxid. Stage B - O1
1/T (K -1)
Dev. Stage - D3
Dev. Stage - O1
Dev. Stage - O1
Dev. Stage - O1
0.0012 0.0016 0.0020 0.0024
-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024
-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024
-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024
-22
-20
-18
-16
-14
-12
-10
ln (g
( αα αα)/T
2 )
Dev. Stage - O1
Oxid. Stage - O1
10 ºC/min 80 ºC/min40 ºC/min
Dev. Stage - O1
Oxid. Stage - O1
20 ºC/min
1/T (K -1)
Dev. Stage - O1
Oxid. Stage - O1
Dev. Stage - O1
Oxid. Stage - O1
Cellulose
Lignin
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
-8
-6
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
-8
-6
-4
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
-8
-6
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
-8
-6 Dev. Stage A-N1
Dev. Stage B-O2
Oxid. Stage -O1
Imp. Stage -O2
ln
(g( αα αα
)/T2 )
Dev. Stage A-N1
Dev. Stage B-O2
Oxid. Stage -O1
Imp. Stage -O1
10 ºC/min 80 ºC/min40 ºC/min
20 ºC/min Dev. Stage A-N1
Dev. Stage B-O2
Oxid. Stage -O1
1/T (K-1)
Dev. Stage A-N1
Dev. Stage B-O2
Oxid. Stage -O1
Xylan
0.0012 0.0016 0.0020 0.0024-20
-18
-16
-14
-12
-10
-8
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
ln (g
( αα αα)/T
2 )
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
10 ºC/min 80 ºC/min40 ºC/min
20 ºC/min Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
1/T (K-1)
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
Fir wood
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
ln (g
( αα αα)/T
2 )
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
10 ºC/min 80 ºC/min40 ºC/min
20 ºC/min Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
1/T (K-1)
Dev. Stage - O1
Oxid. Stage - O1
Rem. Stage - O1
Eucalyptus wood
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10
0.0008 0.0012 0.0016 0.0020 0.0024-22
-20
-18
-16
-14
-12
-10 Dev. Stage - O
1
Oxid. Stage A - O1
Oxid. Stage B - O1
ln (g
( αα αα)/T
2 )
Dev. Stage - O1
Oxid. Stage A - O1
Oxid. Stage B - O1
Rem. Stage - O1
10 ºC/min 80 ºC/min40 ºC/min
20 ºC/min Dev. Stage - O1
Oxid. Stage A - O1
Oxid. Stage B - O1
Rem. Stage - O1
1/T (K-1)
Dev. Stage - O1
Oxid. Stage A - O1
Oxid. Stage B - O1
Rem. Stage - O1
Pine bark

Chapter 3
152
Table 3.5.-Estimated kinetic parameters for the combustion of the main components of lignocellulosic biomass (cellulose, xylan and lignin)
BiomassSample HeatingRate (ºC/min)
E (kJ/mol)
A (1/min)
r 2 E (kJ/mol)
A (1/min)
r 2 E (kJ/mol)
A (1/min)
r 2 E (KJ/mol)
A (1/min)
r 2
Stages Dev. Stage Oxid. Stage - - Mechanism O1 O1 - -
Cellulose 10 164 3.5·1013 0.9978 187 1.21012 0.9932 - - - - - -
20 166 1.3·1014 0.9937 193 2.1·1012 0.9930 - - - - - -
40 171 1.3·1014 0.9910 159 3.4·109 0.9905 - - - - - -
80 173 8.3·1013 0.9908 181 8.6·1013 0.9906 - - - - - -
Stages Dev. Stage A Dev. Stage B Oxid. Stage Imp.Stage Mechanism N1 O2 O1 O1/ O2
*
Xylan 10 128 4.8·1012 0.9975 107 7.6·109 0.9954 146 4.3·109 0.9943 188 1.7·1010 0.9911
20 125 1.9·1012 0.9947 106 3.6·109 0.9945 142 4.3·109 0.9946 252 1.1·1015 0.9761
40 131 8.8·1012 0.9931 105 2.3·109 0.9935 129 4.3·107 0.9951 - - -
80 131 3.7·1012 0.9925 104 1.0·109 0.9954 80 1.1·104 0.9900 - - -
Stages Dev. Stage Oxid. Stage A Oxid. Stage B Rem. Stage Mechanism D3 O1 O1 O1
Lignin 10 96 6.6·109 0.9941 55 1.8·105 0.9931 60 8.2·105 0.9918 - - -
20 84 5.6·106 0.9930 89 2.3·106 0.9965 119 1.2·107 0.9946 - - -
40 83 5.4·106 0.9901 96 8.6·106 0.9976 74 3.6·103 0.9906 - - -
80 70 2.4·105 0.9925 88 1.4·106 0.9958 52 6.1·101 0.9913 599 6.9·1026 0.9932
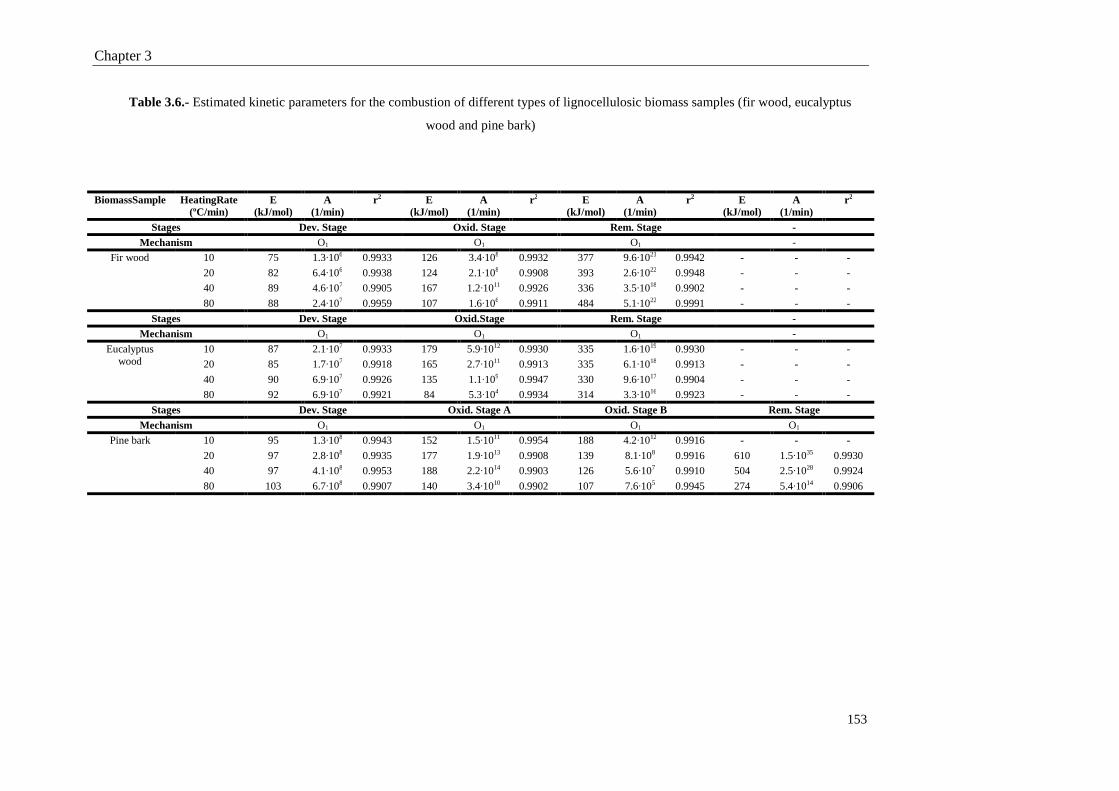
Chapter 3
153
Table 3.6.- Estimated kinetic parameters for the combustion of different types of lignocellulosic biomass samples (fir wood, eucalyptus
wood and pine bark)
BiomassSample HeatingRate (ºC/min)
E (kJ/mol)
A (1/min)
r 2 E (kJ/mol)
A (1/min)
r 2 E (kJ/mol)
A (1/min)
r 2 E (kJ/mol)
A (1/min)
r 2
Stages Dev. Stage Oxid. Stage Rem. Stage - Mechanism O1 O1 O1 -
Fir wood 10 75 1.3·106 0.9933 126 3.4·108 0.9932 377 9.6·1021 0.9942 - - -
20 82 6.4·106 0.9938 124 2.1·108 0.9908 393 2.6·1022 0.9948 - - -
40 89 4.6·107 0.9905 167 1.2·1011 0.9926 336 3.5·1018 0.9902 - - -
80 88 2.4·107 0.9959 107 1.6·106 0.9911 484 5.1·1022 0.9991 - - -
Stages Dev. Stage Oxid.Stage Rem. Stage - Mechanism O1 O1 O1 -
Eucalyptus wood
10 87 2.1·107 0.9933 179 5.9·1012 0.9930 335 1.6·1019 0.9930 - - -
20 85 1.7·107 0.9918 165 2.7·1011 0.9913 335 6.1·1018 0.9913 - - -
40 90 6.9·107 0.9926 135 1.1·109 0.9947 330 9.6·1017 0.9904 - - -
80 92 6.9·107 0.9921 84 5.3·104 0.9934 314 3.3·1016 0.9923 - - -
Stages Dev. Stage Oxid. Stage A Oxid. Stage B Rem. Stage Mechanism O1 O1 O1 O1
Pine bark 10 95 1.3·108 0.9943 152 1.5·1011 0.9954 188 4.2·1012 0.9916 - - -
20 97 2.8·108 0.9935 177 1.9·1013 0.9908 139 8.1·108 0.9916 610 1.5·1035 0.9930
40 97 4.1·108 0.9953 188 2.2·1014 0.9903 126 5.6·107 0.9910 504 2.5·1028 0.9924
80 103 6.7·108 0.9907 140 3.4·1010 0.9902 107 7.6·105 0.9945 274 5.4·1014 0.9906

Chapter 3
154
(Ni) and diffusion (Di) achieved a regression coefficient above 0.98. These results
agreed well with those reported in literature [2; 7;19]. The general disadvantage of
dynamic thermal analysis is that in many cases more than one function g(α) fits the
experimental results. Consequently, selection of the responsible mechanism and
estimation of the real kinetic parameters can be difficult as previously reported [7].
Furthermore, separate stage models fail to predict the transition region between two
mass losses processes and do not consider chemical processes.
Regarding the Dev. stage, the cellulose combustion followed a first reaction order
mechanism. That of lignin followed a D3 mechanism whereasxylanfollowed a N1and
O2onesforDev. stage Aand Dev. stage B, respectively. The oxidation stage followed
aO1 functionfor cellulose and xylanwhereas the lignin combustion fitted betterinto a
O3 and O1for Oxid. stage Aand Oxid. stage B, correspondingly. The Imp.stage for the
xylanoxidation was only found at 10 and 20 ºC/min. Different mechanism were found
to be meaningful for each heating rate. O1model showed the best r2at 10
ºC/minwhereas O2 model was the one that better described the Imp stage at 20 ºC/min.
Finally, the Rem. stage forlignin oxidation at 80 ºC/min followed an O1mechanism.
Anyway,model function of first order (O1) yielded the best correlation coefficient
for lignocellulosic biomass combustion process (Table 3.6). These results agreed well
withthose reported elsewhere[19] and indicate that the composition of biomass do not
influence the overall reaction mechanism of lignocellulosic biomass oxidation.
Up to now, the effect of the heating rate on biomass thermal decomposition
kinetics is still unresolved [17]. Different authors have proposed that the heating rate
has minimal impact on the frequency factor, which is mainly related to the structure of
the material [7; 17]. Anyway, the activation energy is the main characteristic attributed
to the reactivity of a biomass sample [2; 7]. Figure 3.5 shows the estimated activation
energies at different heating rates for the different stages of the combustion process
ofbiomass samples.Concerning the kinetic parameters obtained for the main
components oflignocellulosic biomass (Figure 3.5a and Table 3.5), several differences
can be observed. Activation energies of biomass main components can beranked as

Chapter 3
155
follows: E(cellulose)>E(xylan)>E(lignin). This order determined that the
decomposition of cellulose is the rate-determining step of the biomass combustion
process[17]. On the other hand, the activation energy valueswere hardly affected by
the heating rate.
Figure 3.4.-Comparison of activation energy for the combustion process of: a) main
components of lignocellulosic biomass (cellulose, lignin and xylan) and b) lignocellulosic
biomass (eucalyptus wood, fir wood and pine bark) at 10, 20, 40 and 80 ºC/min.
The study of the Oxid.stage showed a similar trend as that commented for the
previous stage. Activation energies for cellulose and xylan were higher than that for
lignin. However, the higher the heating rate, the slightly lower the activation energies
were.Finally, the activation energies for the Rem. stage werehigher than those
obtained for the previous ones. This fact can be attributed to the high energy required
to decompose inorganic matter [26]. The effect of the heating rate on the kinetic
parameters for lignocellulosic biomass samples was similar to that commented for
0 20 40 60 80 10050
100
150
200
250
0 20 40 60 80 1000
50
100
150
200
250
300
0 20 40 60 80 10050
75
100
125
0 20 40 60 80 10050
100
150
200
250
0 20 40 60 80 100200
300
400
500
600
700
Dev. Stage
EA
(Kj/m
ol)
Heating Rate (ºC/min)
Xylan A Xylan B Cellulose Lignin A
Lignin B
Oxid. Stage
Heating Rate (ºC/min)
EA
(Kj/m
ol)
Heating Rate (ºC/min)
Dev. Stage Fir wood Eucalyptus Wood Pine bark A Pine bark B
Heating Rate (ºC/min)
Oxid. Stage
Heating Rate (ºC/min)
Rem. Stage

Chapter 3
156
their main components (Figure 3.4.b and Table 3.6). Activation energies for the Dev.
stage did not differ for different heating rates [1]. However, for the Oxid.stagethe
higher the heating rate, the lower the activation energy was observed. Shen et
al.[1]related this finding with the occurrence of gradient temperatures within the
particle, which increased at higher heating rates. As above commented and expected,
the value of activation energies for the Rem. stage were the highest one.
In order to corroborate the kinetic analysis, the reconstruction of the weight loss
curves was performed. Considering n separate reactions, the kinetic rates of thermal
decomposition of a material can be easily derived from Eq. (4) as follows:
dα. dT� = A. β� e 34 56� f.(α.) (7)
whereαi, Ai, Ei and fi(αi) are the degree of conversion, the pre-exponential factor,
activation energy and model functions obtained for each stage of the combustion
process, respectively.
A VBA-Excel application was developed to solve this model based on the Runge-
Kutta-Fehlberg method for the evaluation of the set of ordinary differential equations.
Figure SS1 shows the experimental data (solid line) compared to the predicted one
(dotted line) for the combustion process of biomass samples at a heating rate of 10
ºC/min, obtained by substituting the calculated activation energy and pre-exponential
factor for each stage into Eq. (7). It can be observed that the proposed model
adequately reproduces the experimental values, obtaining a low error for all cases.
Finally, in order to ensure the reliability of the proposed models, the discrimination of
kinetic parameters was done applying the F-test and the t-test at the 95% confidence
level [23]. The resulting parameters obtained from the linear regression are
summarized in Table 3.7 and 3.8. In terms of statistical results, F-test considered the
regression to be suitable in all cases since the corresponding values to the Fc/Ftest ratio
were larger than one. The t-test was also used for evaluating each parameter in the
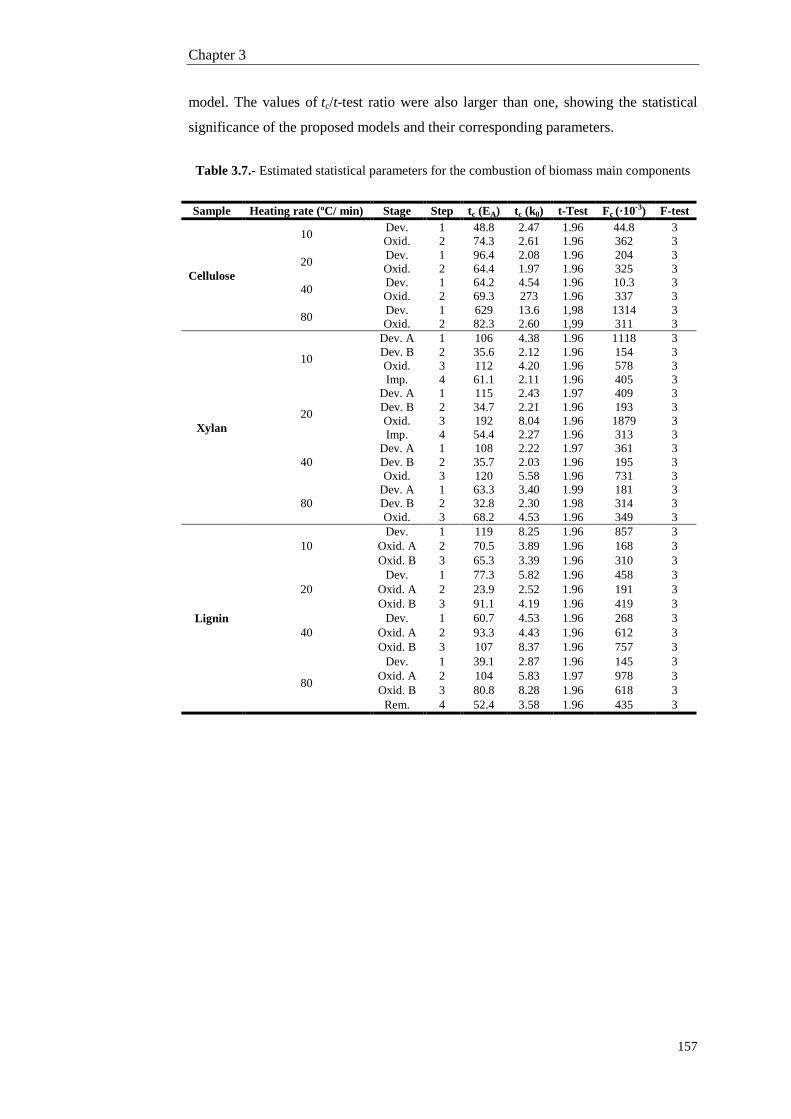
Chapter 3
157
model. The values of tc/t-test ratio were also larger than one, showing the statistical
significance of the proposed models and their corresponding parameters.
Table 3.7.- Estimated statistical parameters for the combustion of biomass main components
Sample Heating rate (ºC/ min) Stage Step tc (EA) tc (k0) t-Test Fc (·10-3) F-test
Cellulose
10 Dev. 1 48.8 2.47 1.96 44.8 3 Oxid. 2 74.3 2.61 1.96 362 3
20 Dev. 1 96.4 2.08 1.96 204 3 Oxid. 2 64.4 1.97 1.96 325 3
40 Dev. 1 64.2 4.54 1.96 10.3 3 Oxid. 2 69.3 273 1.96 337 3
80 Dev. 1 629 13.6 1,98 1314 3 Oxid. 2 82.3 2.60 1,99 311 3
Xylan
10
Dev. A 1 106 4.38 1.96 1118 3 Dev. B 2 35.6 2.12 1.96 154 3 Oxid. 3 112 4.20 1.96 578 3 Imp. 4 61.1 2.11 1.96 405 3
20
Dev. A 1 115 2.43 1.97 409 3 Dev. B 2 34.7 2.21 1.96 193 3 Oxid. 3 192 8.04 1.96 1879 3 Imp. 4 54.4 2.27 1.96 313 3
40 Dev. A 1 108 2.22 1.97 361 3 Dev. B 2 35.7 2.03 1.96 195 3 Oxid. 3 120 5.58 1.96 731 3
80 Dev. A 1 63.3 3.40 1.99 181 3 Dev. B 2 32.8 2.30 1.98 314 3 Oxid. 3 68.2 4.53 1.96 349 3
Lignin
10 Dev. 1 119 8.25 1.96 857 3
Oxid. A 2 70.5 3.89 1.96 168 3 Oxid. B 3 65.3 3.39 1.96 310 3
20 Dev. 1 77.3 5.82 1.96 458 3
Oxid. A 2 23.9 2.52 1.96 191 3 Oxid. B 3 91.1 4.19 1.96 419 3
40 Dev. 1 60.7 4.53 1.96 268 3
Oxid. A 2 93.3 4.43 1.96 612 3 Oxid. B 3 107 8.37 1.96 757 3
80
Dev. 1 39.1 2.87 1.96 145 3 Oxid. A 2 104 5.83 1.97 978 3 Oxid. B 3 80.8 8.28 1.96 618 3 Rem. 4 52.4 3.58 1.96 435 3

Chapter 3
158
Table 3.8.- Estimated statistical parameters for the combustion of lignocellulosic biomass
Gas evolved analysis
The main products derived from the combustion of lignocellulosic biomass and its
main components were evaluated by TGA-MS analysis. On the basis of a preliminary
scan, a list of key molecular ions was compiled and gathered inTable 3.9.
Sample Heating rate (ºC/ min) Stage Step tc (EA) tc (k0) t-Test Fc (·10-3) F-test
Eucalyptus wood
10 Dev. 1 131 6.16 1.96 537 3 Oxid. 2 53.1 3.78 1.96 24.5 3 Rem. 3 105 2.19 1.96 673 3
20 Dev. 1 108 5.27 1.96 354 3 Oxid. 2 98.8 2.13 1.96 669 3 Rem. 3 65.6 2.02 1.96 261 3
40 Dev. 1 112 5.33 1.96 370 3 Oxid. 2 108 2.45 1.96 892 3 Rem. 3 75.2 2.96 1.96 474 3
80 Dev. 1 79.2 4.01 1.96 185 3 Oxid. 2 70.1 4.42 1.96 605 3 Rem. 3 99.1 2.44 1.97 659 3
Firwood
Dev. 1 56.7 1.12 1.96 79.1 3 10 Oxid. 2 96.7 4.08 1.96 567 3 Rem. 3 124 2.42 1.98 523 3 Dev. 1 146 3.29 1.96 1826 3
20 Oxid. 2 82.2 3.43 1.96 435 3 Rem. 3 125 2.26 1.97 1147 3 Dev. 1 146 3.29 1.96 1825 3
40 Oxid. 2 69.6 2.74 1.96 425 3 Rem. 3 91.9 2.37 1.96 543 3 Dev. 1 99.8 4.96 1.96 396 3
80 Oxid. 2 74.5 5.15 1.96 685 3 Rem. 3 57.3 2.04 1.96 433 3
Pine bark
10 Dev. 1 150 6.54 1.96 716 3
Oxid. A 2 71.8 2.46 1.96 483 3 Oxid. B 3 79.5 2.44 1.96 542 3
20
Dev. 1 160 7.05 1.96 825 3 Oxid. A 2 72.5 2.17 1.96 486 3 Oxid. B 3 66.9 2.62 1.96 366 3 Rem. 4 78.6 3.18 1.96 567 3
40
Dev. 1 183 8.14 1.96 1030 3 Oxid. A 2 235 10.7 1.96 1832 3 Oxid. B 3 72.1 3.18 1.96 412 3 Rem. 4 89.6 3.38 1.98 1195 3
80
Dev. 1 126 5.58 1.96 471 3 Oxid. A 2 63.5 2.44 1.98 467 3 Oxid. B 3 147 6.70 1.96 716 3 Rem. 4 13.2 3.49 2.02 110 3

Chapter 3
159
Table 3.9.-Molecular ions and probable parent molecules detected in the combustion of lignocellulosic biomass and its main components.
(m/z)
Key molecular ions/Ion fragment Probable parent molecule Cellulose Lignin Xylan Fir wood Eucalyptus wood Pine bark
2 H2+ H2 X X X X X X
15 CH3+ CH4 X X X X X X
16 O+, CH4+ CH4 - X X X X X
18 H2O+ H2O X X X X X X
26 CN+, C2H2+ C2H2 X X X X X X
27 HCN+, C2H3+ HCN (nitriles) X X X X X X
28 C2H4+, CO+ CO X X X X X X
29 C2H5+ C2H5 (Ethylderivates) X X X X X X
30 C2H6+, CH2NH2
+ CH4N (Primary amines) X X X X X X
44 CO2+ CO2 X X X X X X
45 C2H5O+, C2H7N
+, CHS+ C2H5O (hydroxyderivates) X X X X X X
46 NO2+, C2H5OH+ NO2 X X X X X X
47 CH3S+, CCl+, C2H5OH+ CH3Cl X X - X X X
48 CH3SH+, CHCl+, SO+ SO - X - X X X
50 C4H2+, CH3Cl+, CF2
+ CH3S X X - X X X
51 C4H3+, CHF2
+ C4H3(aromatics) X - - X X X
52 C4H4+ C4H4 (aromatics) X - - X X X
53 C4H5+ C4H5 (aromatics) X - - X X X
54 C4H6+, C2H4CN+ C4H6 (aromatics) X - - X X X
55 C4H7+, C3H3O
+ C4H7 (aromatics) X - X X X X
56 C3H6N+, C4H8
+ C4H8 (alquenes) X - X X X X
57 C4H9+, C3H5O
+, C3H2F+ C3H5O (cyclopentanol) X - X X X X
58 C3H8N+ C3H8N (amines) X - X X X X
60 COS+ COS X X X X X X
64 SO2+ SO2 - X X - - -
68 C5H8+, C4H4O
+, C3H6CN+ C4H6O(cyclohexenones) X - - X X X
70 C5H10+, C4H6O
+, C4H8N+ C4H6O (cycloalkanones) X - - - - -
72 C4H8O+, C4H10N
+, C6+ C4H8O(alkanones) X - - - - -
84 C5H10N+ C5H10N (pyrolidines) X - - - - -
95 C5H3O2+ C5H3O2(furycarbonil-derivates) X - - - - -
96 C7H12+ C7H12 (alicyclics) X - - - - -

Chapter 3
160
Mass spectrometry analysis for the oxidation of xylan, cellulose and lignin are
shown in Figure 3.5. MS curves were move up and down in order to obtain a
clearevolved gas profile. Magnifying picture of the curves was placed in the upper
right corner. MS spectra of biomass main components could be divided into different
stages related to their degradations steps studied in the TGA/DTG curves and
described in previous sections.Table 3.10 summarizes the most representative MS ions
detected, their integrated peak intensities in the whole temperature range and the
temperature where the maximum emission peak was found. Cellulose sample
combustion showed the major emission peak for all productsat the devolatilization
stage at temperatures around 350 ºC which is in good agreement with its maximum
DTG peak.In this stage, the degradation of glycosyl units in cellulose produced H2O,
CO and CO2 leading to the formation of char residue.Xie et al.[29]stated that during
the devolatilization of cellulose the complete decomposition of
glycosidicstructuresproceeded. Furthermore, rapid depolymerization of cellulose
turned into the breakdown of the molecule,producing a variety of low molecular
weight products[5]. Inthe char combustion stage only H2O, CO, CO2, NO2,
C2H5O+(hydroxyderivates) and C2H5were detected at 510 ºC. CO and CO2 evolution
from cellulose sample oxidation is believed to be a consequence of the loss of
carbonyl and carboxyl groups previously formed by the oxidation of hydroxyl groups
[5]. This fact can also be corroborated by the presence of C2H5O+ in this temperature
range.In a similar way, xylanevolution was resolved in two stages. This way, volatiles
were released between 200 and 450 ºC whereas only CO2, CO, CH3+,C2H5O
+, HCN
and NO2 were found at higher temperatures. In this case, the maximum evolution rate
for most compounds was found at temperatures between 260 and 270 ºC. The
formation of two peaks for CO, CO2 and H2O in this stage showed the good
correlation with its DTG profile. However, a second group of compounds (H2, CH3,
C2H2, and HCN) had their maximum emission peak at 466 ºC, coincidental to the
shoulder in the CO2 curve. This second peak could be due to char elimination and
rearrangement reactions. Finally, CO, CO2, C2H5O+and NO2decomposed at higher
temperatures. Lignin sampleMS spectra showed a more diversified profile, volatile

Chapter 3
161
compounds evolved within the whole temperature range as expected from the TG-
DTG analysis, which could beattributed to the presence of aromatic compoundsin the
raw material thatremained after their oxidation in air at 240 ºC [29], widening the
temperature range for volatiles evolution.As abovementioned, lignin sample was the
first biomass component to start decomposing. Most volatiles started evolving in the
130-200 ºC temperature range, finding the main emission peak for H2O, CH3Cl,
CH3S+,SO and SO2at 340 ºC.The low temperatures at which oxygenated compounds
evolved pointed out that aliphatic HOgroups were easily removed from lignin[30]. In
this stage, CO, CO2 and H2O were released by C-C bond scission [31]. Furthermore,
the decomposition ofsulphoxide and sulphone groups facilitated lignin
depolymerization[30]. C2H2, CH4N and CH3+ emission peaks occurred at 392, 397 and
427 ºC, respectively.The evolution of these compounds was associated to dehydration
and demethylation reactions [30].H2O, C2H2, HCN, COS, CO and CH4maximum
peaks took place between 483 and 501 ºC. H2 was later released at 529 ºC being
attributed to the hydrogen splitting from aromatic rings [30]. Finally, CO2, NO2 and
C2H5O+peak was found between 553 and 575ºC where the devolatilization of char
took place. A last CO2 peak was found at high temperatures related to carbonates
decomposition. This peak was not observed in cellulose and xylansamples MS profiles
which is due to their low inorganic content. This fact confirmed that the peak observed
in the DTG profiles at high temperatures can be attributed to the inorganic matter
present in the formed ash. Finally, it can be observed that cellulose sample MS spectra
showed the highest intensity peak for all the observed products in the devolatilization
stage being in agreement with its high DTG peak. Furthermore, the low CO2 peak in
the oxidation stage compared to that of lignin and xylan samples was due to the low
amount of fixed carbon. On the contrary, lignin and xylan samples showed their
highest CO and CO2 peaks in the oxidation stage.

Chapter 3
162
Figure 3.5.-Mass spectra of the combustion of the main components of lignocellulosic biomass (cellulose, xylan and lignin) at 40 ºC/min.
200 400 600 800 1000
CH3
+
NO2
H2O
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
HCN
C2H
2
CO
C4H
7
200 300 400 500 600
Inte
nsity
(a.
u)
Temperature (ºC)
H2
C3H
8N
CH4
COS
C4H
8
C3H
5O
200 400 600 800 1000
SO2
CH3
+
NO2
H2O
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
HCN
CO
200 300 400 500 600
Inte
nsity
(a.
u)
Temperature (ºC)
H2
C2H
2
CH4
COS
CH3S+
C4H
8O+
200 400 600 800 1000
C3H
5OC
4H
6O+COS
CH3Cl
CH3
+C
4H
8
C4H
6
C3H
8NNO
2
H2O
Inte
nsity
(a.
u.)
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
HCN C2H
2
COC4H
7
C4H
5
+ C4H
4O
200 300 400 500 600
Inte
nsity
(a.
u)
Temperature (ºC)
H2
C5H
12
+
C5H
3O
2
+
C5H
10N+
CH3S+
C4H
8O+
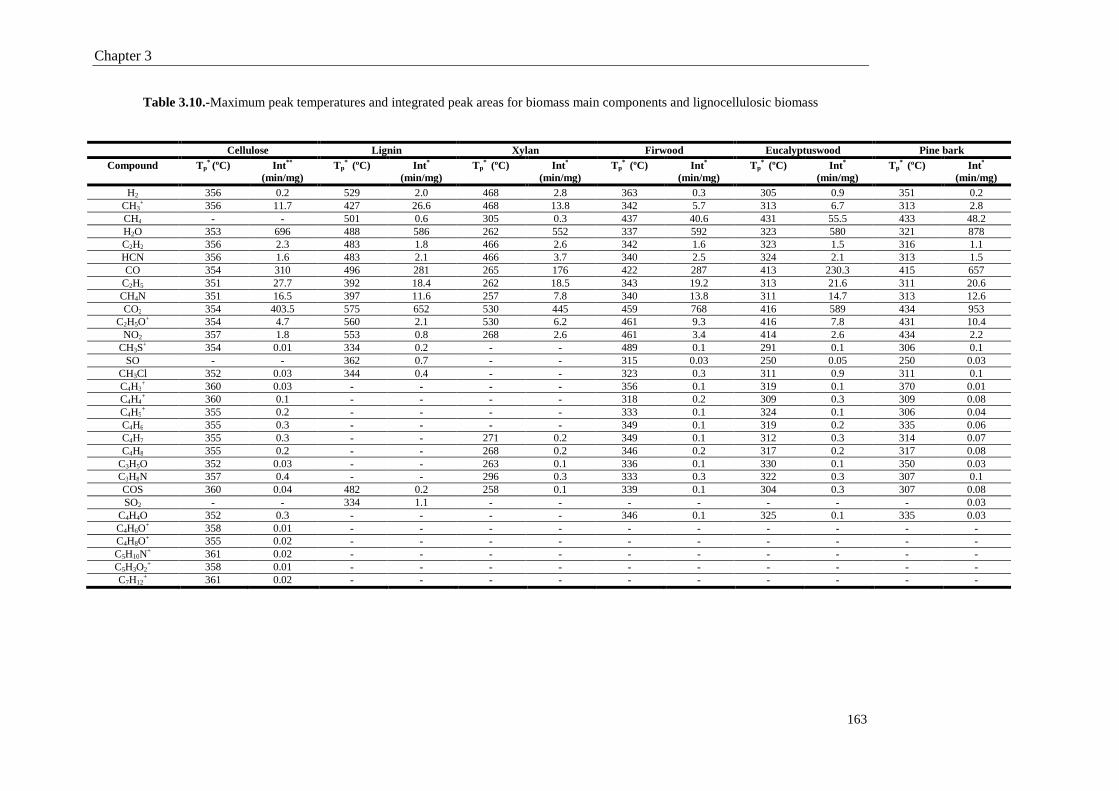
Chapter 3
163
Table 3.10.-Maximum peak temperatures and integrated peak areas for biomass main components and lignocellulosic biomass
Cellulose Lignin Xylan Firwood Eucalyptuswood Pine bark Compound Tp
* (ºC) Int ** (min/mg)
Tp* (ºC) Int*
(min/mg) Tp
* (ºC) Int* (min/mg)
Tp* (ºC) Int*
(min/mg) Tp
* (ºC) Int* (min/mg)
Tp* (ºC) Int*
(min/mg) H2 356 0.2 529 2.0 468 2.8 363 0.3 305 0.9 351 0.2
CH3+ 356 11.7 427 26.6 468 13.8 342 5.7 313 6.7 313 2.8
CH4 - - 501 0.6 305 0.3 437 40.6 431 55.5 433 48.2 H2O 353 696 488 586 262 552 337 592 323 580 321 878 C2H2 356 2.3 483 1.8 466 2.6 342 1.6 323 1.5 316 1.1 HCN 356 1.6 483 2.1 466 3.7 340 2.5 324 2.1 313 1.5 CO 354 310 496 281 265 176 422 287 413 230.3 415 657
C2H5 351 27.7 392 18.4 262 18.5 343 19.2 313 21.6 311 20.6 CH4N 351 16.5 397 11.6 257 7.8 340 13.8 311 14.7 313 12.6 CO2 354 403.5 575 652 530 445 459 768 416 589 434 953
C2H5O+ 354 4.7 560 2.1 530 6.2 461 9.3 416 7.8 431 10.4 NO2 357 1.8 553 0.8 268 2.6 461 3.4 414 2.6 434 2.2
CH3S+ 354 0.01 334 0.2 - - 489 0.1 291 0.1 306 0.1 SO - - 362 0.7 - - 315 0.03 250 0.05 250 0.03
CH3Cl 352 0.03 344 0.4 - - 323 0.3 311 0.9 311 0.1 C4H3
+ 360 0.03 - - - - 356 0.1 319 0.1 370 0.01 C4H4
+ 360 0.1 - - - - 318 0.2 309 0.3 309 0.08 C4H5
+ 355 0.2 - - - - 333 0.1 324 0.1 306 0.04 C4H6 355 0.3 - - - - 349 0.1 319 0.2 335 0.06 C4H7 355 0.3 - - 271 0.2 349 0.1 312 0.3 314 0.07 C4H8 355 0.2 - - 268 0.2 346 0.2 317 0.2 317 0.08
C3H5O 352 0.03 - - 263 0.1 336 0.1 330 0.1 350 0.03 C3H8N 357 0.4 - - 296 0.3 333 0.3 322 0.3 307 0.1 COS 360 0.04 482 0.2 258 0.1 339 0.1 304 0.3 307 0.08 SO2 - - 334 1.1 - - - - - - - 0.03
C4H4O 352 0.3 - - - - 346 0.1 325 0.1 335 0.03 C4H6O+ 358 0.01 - - - - - - - - - - C4H8O+ 355 0.02 - - - - - - - - - - C5H10N+ 361 0.02 - - - - - - - - - - C5H3O2
+ 358 0.01 - - - - - - - - - - C7H12
+ 361 0.02 - - - - - - - - - -

Chapter 3
164

Chapter 3
165
Figure 6.-Mass spectra of the combustion of lignocellulosic biomass (eucalyptus wood, fir wood and pine bark) at 40 ºC/min.
200 400 600 800 1000
COS
CH3+
C3H
8N
NO2
H2O
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
C2H
2
CO
C4H
7
HCN
200 300 400 500 600
C4H
8
C4H
4
+
Inte
nsity
(a.
u)
Temperature (ºC)
CH4
H2
CH3Cl
C4H
6
SO2
C4H
4O
C4H
5
+
SO C4H
5
+
200 400 600 800 1000
COS
CH3
+CH3Cl
C4H
8
C3H
8N
NO2
H2O
Inte
nsity
(a.
u.)
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
C2H
2
CO
C4H
7
C4H
4
+
HCN
200 300 400 500 600
Inte
nsity
(a.
u)Temperature (ºC)
H2
CH3S+CH
4
C4H
6
C3H
5O
C4H
5
+
C4H
3
+
SO
C4H
4O
200 400 600 800 1000
COS
CH3
+
CH3Cl
C4H
8
C4H
4
C3H
8N
NO2
H2O
Temperature (ºC)
CO2
C2H
5
CH4N
C2H
5O+
C2H
2
CO
C4H
7
C4H
4
+
HCN
200 300 400 500 600
Inte
nsity
(a.
u)
Temperature (ºC)
H2
CH3S+
SO2
C3H
5O
C4H
5
+
C4H
3
+
SO
C4H
4O
CH4

Chapter 3
166
Figure 3.6 shows the MS spectra for firwood, eucalyptus wood and pine bark.The
gas products distributionwas pretty similar andmay be divided into three stages. As
commented above, most compounds evolved during the devolatilization stage. Sulfur
compounds were the first to be detected as SO and CH3S+. An intermediate emission
peak was found, where CO and CH4 evolved. H2 was also emitted between these two
stages. Finally, CO, CO2, C2H5O+, and NO2 were the main peaks detected during the
char oxidation stage. The same pattern was followed by all the samples. However, the
emission peaks temperatures changed. As expected from biomass samples DTG
profiles, peaks for eucalyptus wood and pine bark took place at lower temperatures
than those for fir wood. This fact could be due to compositional differences among
them (volatile matter, cellulose and hemicellulose content).
H2O, CO and CO2 were the main products obtained during biomass combustion
(Table 3.10). CO and CO2 evolved over the whole temperature range with a higher
proportion of CO2.CO is assumed to be formed by the creation of molecular oxide
complexes that further rearrange turning into the evolution of CO [32].However,
CO2presented a more complex formation process [32], as its production can be
catalyzed or inhibited by the formation of carbon intermediates[33]. Pine bark released
the highest amount of CO2 which is attributed to its high lignin content. However, the
CO evolution from combustion of lignocellulosic biomass cannot be directly
correlated to that for biomass components since CO signal has a great contribution of
the CO2 one. Furthermore, there is also a minor contribution of ethylene to the ion
corresponding to m/z=28[34].H2O was released in three steps. Firstly, the water
released in the low temperature range was associated to the dehydration of the sample.
Then, the higher peak for water was formed in the main devolatilization stage, being
associated to the evolved aliphatic OH groups [15; 33]. Finally, a shoulder appeared in
the MS spectra, which was related to the water formed by the oxidation of H2 and
calcium carbonate decomposition [33]. The higher amount of H2O evolved from pine
bark sample combustion was mainly due to its higher initial moisture content.Light
hydrocarbons and especially CH4 and C2H5 were also predominant. CH4had two main
origins related to devolatilizationand charring processes [6].It was also interesting to

Chapter 3
167
note the release of nitrogen compounds (N compounds) in form of HCN, NO2 and
CH4N. However, it has to be careful with relative amount of amines as the associated
ion (m/z=30) can be related to NO and C2H6compounds. Thus, N compounds emission
should be related to NO2 rather than NO and CH4N. Two emission zones were found
for N compounds. One stage related to the release of amines and NO2, which is
attributed to the decomposition of proteins [12].In the second one,NO2 was only
detected and attributed to the oxidation of the retained N in the
char[35].Furthermore,Darvel et al.[12]reported a possible explanation for HCN
formation (C(H)1 + C(N)1 → HCN + Cfas, where C(H)1 and C(N)1 arelocalized surface
species and Cfas is a "free active site"). Finally, the rest of the products were found in
low content. Among them, special attention to the release of sulfur and chloride
compounds should be paid since they behave as hazard pollutants. Sulfur compounds
were released as SO, CH3S and COS (SO2 signal was hardly detected during biomass
combustion, which can be explained by the easy fragmentation of this ion within the
mass spectrometer) whereas thechloride fraction was detectedin form of CH3Cl.
3.3.2. Combustion of marine biomass
Thermogravimetric Analysis (TGA)
Figure 3.7 shows TGA-DTG profiles for the microalgae Scenedesmusalmeriensis
(SC), Nannochloropsisgaditana(NG) and Chlorella vulgaris (CV) at a heating rate of
40 ºC/min. Table 3.11 shows the most relevant combustion characteristics. Ignition,
peak and burnout temperatures are the most characteristic parameters when evaluating
the combustion performance of a material [36; 37]. Peak temperature (Tp) refers to the
temperature where maximum loss weight rate (dw/dT)maxis reached. Peak temperature
and its corresponding rate is a measure of combustibility and reactivity, respectively.
Thus, the lower the Tp, the easier the ignition of a material is. The ignition temperature
(Ti) is the temperature at which a sudden decrease in weight loss on the DTG curve is
observed. Ti was calculated as the intersection between the tangent line to the point
which decomposition started and the tangent line to the maximum weightloss rate. The
burnout temperature (Tb) is the temperature where the process is finished.
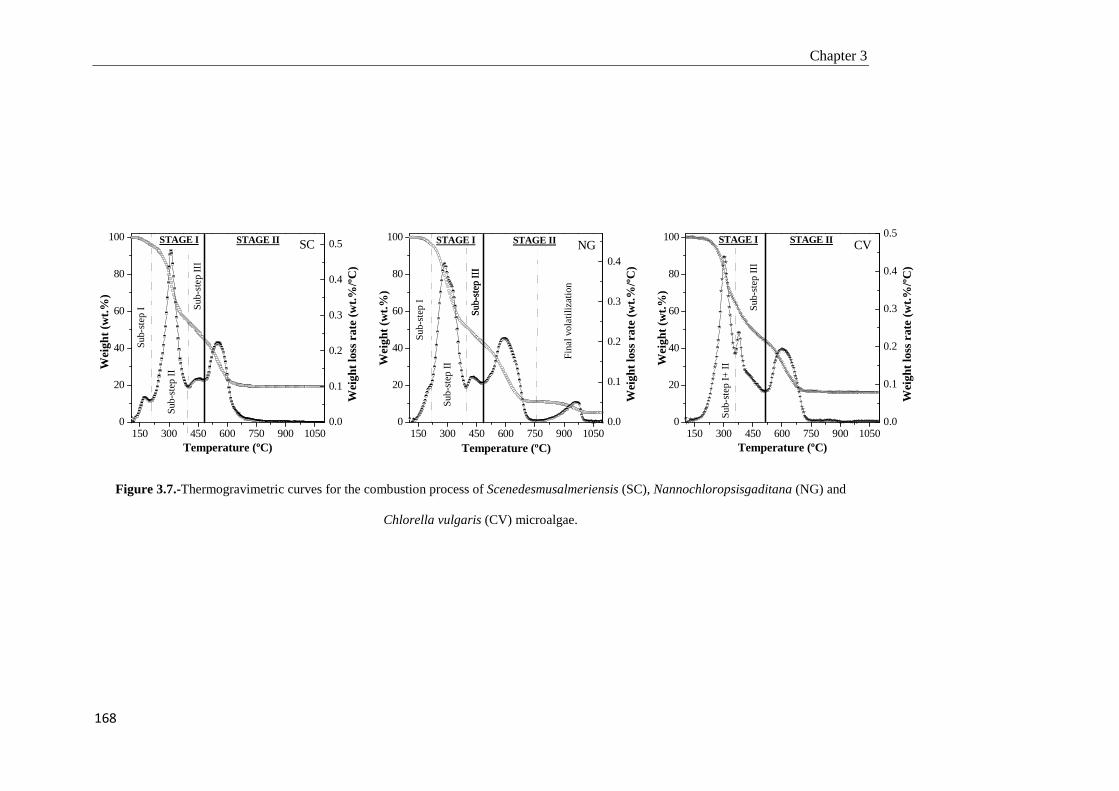
Chapter 3
168
Figure 3.7.-Thermogravimetric curves for the combustion process of Scenedesmusalmeriensis (SC), Nannochloropsisgaditana (NG) and
Chlorella vulgaris (CV) microalgae.
150 300 450 600 750 900 10500
20
40
60
80
100
150 300 450 600 750 900 10500
20
40
60
80
100
150 300 450 600 750 900 10500
20
40
60
80
100
Sub
-ste
p I+
II
Fin
al v
olat
iliza
tion
Sub
-ste
p III
Sub
-ste
p III
Sub
-ste
p II
Sub
-ste
p I
Sub
-ste
p III
Sub
-ste
p II
Sub
-ste
p I
STAGE ISTAGE II
Wei
ght (
wt.%
)
Temperature (ºC)
SC STAGE I
0.0
0.1
0.2
0.3
0.4
0.5
Wei
ght l
oss
rate
(w
t.%/º
C)
STAGE I STAGE II
Wei
ght (
wt.%
)
Temperature (ºC)
0.0
0.1
0.2
0.3
0.4
NG
Wei
ght l
oss
rate
(w
t.%/º
C)
Sub
-ste
p III
STAGE II
Wei
ght (
wt.%
)
Temperature (ºC)
CV
0.0
0.1
0.2
0.3
0.4
0.5
Wei
ght l
oss
rate
(w
t.%/º
C)
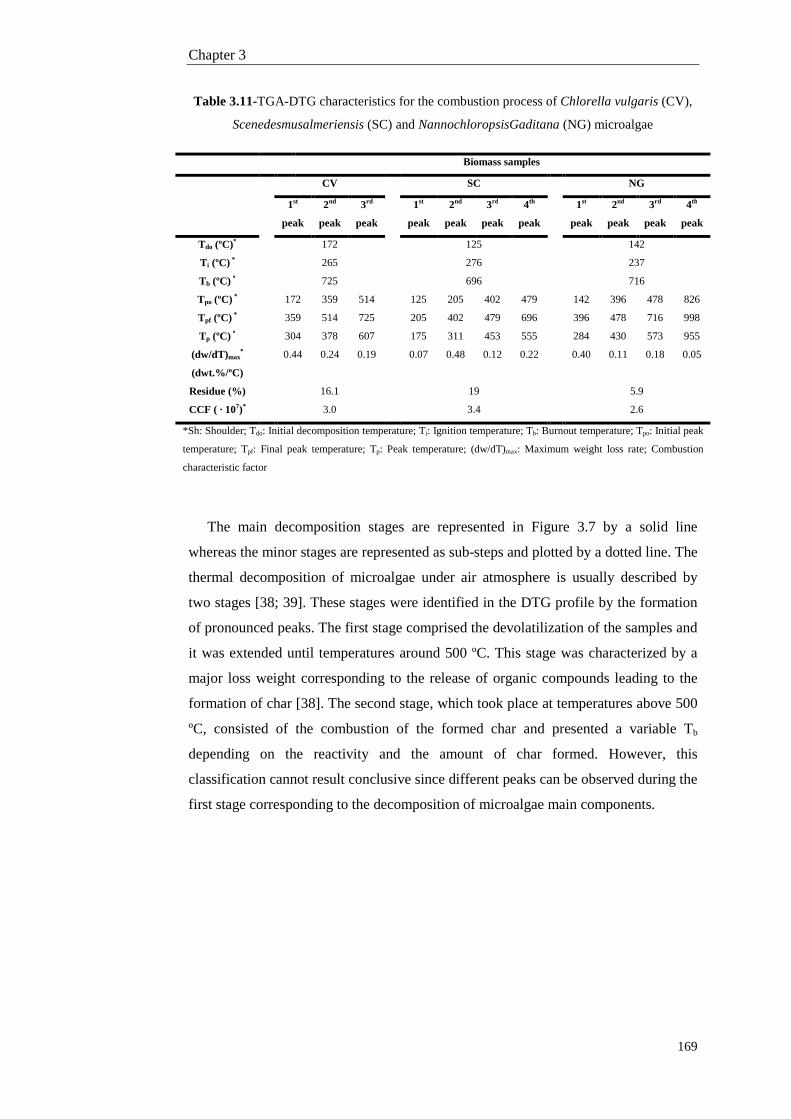
Chapter 3
169
Table 3.11-TGA-DTG characteristics for the combustion process of Chlorella vulgaris (CV),
Scenedesmusalmeriensis (SC) and NannochloropsisGaditana (NG) microalgae
Biomass samples
CV SC NG
1st
peak
2nd
peak
3rd
peak
1st
peak
2nd
peak
3rd
peak
4th
peak
1st
peak
2nd
peak
3rd
peak
4th
peak
Tdo (ºC)* 172 125 142
T i (ºC) * 265 276 237
Tb (ºC) * 725 696 716
Tpo (ºC) * 172 359 514 125 205 402 479 142 396 478 826
Tpf (ºC) * 359 514 725 205 402 479 696 396 478 716 998
Tp (ºC) * 304 378 607 175 311 453 555 284 430 573 955
(dw/dT)max*
(dwt.%/ºC)
0.44 0.24 0.19 0.07 0.48 0.12 0.22 0.40 0.11 0.18 0.05
Residue (%) 16.1 19 5.9
CCF ( · 107)* 3.0 3.4 2.6
*Sh: Shoulder; Tdo: Initial decomposition temperature; Ti: Ignition temperature; Tb: Burnout temperature; Tpo: Initial peak
temperature; Tpf: Final peak temperature; Tp: Peak temperature; (dw/dT)max: Maximum weight loss rate; Combustion
characteristic factor
The main decomposition stages are represented in Figure 3.7 by a solid line
whereas the minor stages are represented as sub-steps and plotted by a dotted line. The
thermal decomposition of microalgae under air atmosphere is usually described by
two stages [38; 39]. These stages were identified in the DTG profile by the formation
of pronounced peaks. The first stage comprised the devolatilization of the samples and
it was extended until temperatures around 500 ºC. This stage was characterized by a
major loss weight corresponding to the release of organic compounds leading to the
formation of char [38]. The second stage, which took place at temperatures above 500
ºC, consisted of the combustion of the formed char and presented a variable Tb
depending on the reactivity and the amount of char formed. However, this
classification cannot result conclusive since different peaks can be observed during the
first stage corresponding to the decomposition of microalgae main components.

Chapter 3
170
The first decomposition stage can be subdivided in three more sub-steps for
samples SC and NG and in two more sub-steps for sample CV. The first sub-step was
represented by a peak for sample SC and as a shoulder for sample NG, which is
related to intrinsic lipid decomposition, such as aldehydes and ketones [40]. In
addition, the decomposition of carbohydrates started in this temperature range (170-
180 ºC) [41]. This sub-step was not found for sample CV and might be due to its
lower lipid and carbohydrate content. The second sub-step was detected by a peak at
284, 304 and 311 ºC for samples NG, CV and SC, respectively, and was associated to
carbohydrates and proteins decomposition [42]. The maximum weight loss rate was
observed in this sub-step for all samples. Sample SC had the highest (dw/dT)max (0.48
wt.%) followed by samples CV (0.44wt.%) and NG (0.40wt.%). Carbohydrates,
proteins and ash content could influence this order. In this sense, the higher their
content in the raw material, the higher the (dw/dT)maxwas. Regarding the ash content,
this was much higher for samples SC and CV than for sample NG. In this sense, alkali
metals present in the ash could catalyze the combustion process increasing the
volatiles yield [43]. Finally, a third sub-step, common to all samples, was observed
close to the char oxidation stage. This peak appeared at lower temperatures for sample
CV (378 ºC) than for samples NG (430 ºC) and SC (453 ºC). In this stage, the final
decomposition of lipids took place and it was mainly associated to the break-down of
hydrocarbon chains of fatty acids [38; 40; 41]. In sample CV, this peak appeared at
lower temperature and with a higher weight loss rate. This fact can be related to the
higher protein content in this sample. In this regard, Kebelmann et al. [40] found a
shoulder for the thermal decomposition of microalgae proteins close to the main
decomposition stage.
The second decomposition stage took place between 478 and 725 ºC. For sample
SC, this stage took place at lower temperatures and with a slightly higher weight loss
rate, if compared to that of samples CV and NG. This fact pointed out that SC
devolatilization led to the formation of a bigger amount of char. This is in agreement
with Ross et al. [41] who reported that high levels of K in the sample promoted the
formation of char. Additionally, a last decomposition step was observed for sample

Chapter 3
171
NG between 826 and 998 ºC in a similar way as reported by Wang et al. for the
combustion of seaweeds [26]. This step is mainly related to volatile metal loss and
carbonate decomposition.
Concerning the general burning profile of the microalgae samples, it can be
observed that sample SC was the first to decompose (125 ºC) whereas samples NG
and CV decomposed at 142 and 172 ºC, respectively. On the other hand, sample SC
was the most difficult sample to ignite. Finally, the sample CV showed the highest Tb
(725 ºC) compared with samples NG (716 ºC) and SC (696 ºC).
Samples SC and CV left a high amount of ash (19 and 16 wt.%, respectively)
compared to sample NG (5.9 wt.%). This fact restricts the use of sample SC and CV
for direct combustion and gasification due to the catalyst/inhibiting effect of the ash.
Thus, a pre-treatment based on water, acid or alkali washing to reduce the influence of
minerals may be needed [44].
The combustion characteristic factor (CCF) can be used to preliminary assess the
microalgae combustion performance [45]. This factor is based on the energy required
to burn a material in terms of low Ti and Tb values and high (dw/dt)max and is
expressed as follows:
889 = (:;:� )<=> ∙ (:;:� )<?=@ �A$ ∙ �B
� (7)
where (dw/dt)max is the maximum burning velocity (%/min); (dw/dt)mean is the average
burning velocity (%/min); Ti is the ignition temperature (K) and Tb is the burnout
temperature (K).
CCF values for all samples are shown in Table 3.11. In all cases, these values were
bigger than 2 indicating the good general burning performance [37]. Sample SC
required less energy than the other samples to perform the combustion. However,
these data must be used only as a reference since they do not give any information
about the heat released during the combustion process.

Chapter 3
172
Differential scanning calorimetry (DSC)
In order to complete the information obtained by TG analyses, the marine biomass
(samples CV, SC and NG) was also investigated by the DSC technique. Experimental
DSC curves are presented in Figure 3.8. DSC main temperatures and heat of
combustion (Hcomb) are included in Table 3.12 DSC analysis of lignocellulosic
biomass has been studied by different authors [24; 37;46]. However, at the best of our
knowledge, the DSC analysis of microalgae combustion has not been explored yet.
Figure 3.8.-DSC curves for the combustion process of Nannochloropsisgaditana (NG),
Scenedesmusalmeriensis (SC) and Chlorella vulgaris (CV) microalgae.
Table 3.11.-DSCcharacteristics for the combustion process of Chlorella vulgaris (CV),
Scenedesmusalmeriensis (SC) and NannochloropsisGaditana (NG)microalgae.
Biomass samples
CV SC NG
1stPeak 2ndPeak 1stPeak 2ndPeak 1stPeak 2ndPeak
T (ºC)* 212-516 516-798 152-428 428-802 156-426 426-774 Tp (ºC)* 491 617 366 570 336 601
Hcomb (kJ/g)* 7.9 7.8 8.8 * T: temperature interval where a thermal event takes place; Tp: peak temperature; Hcomb: Heat released during combustion
120 240 360 480 600 720 840 9600
5
10
15
20
25
30 NG SC CV
Hea
t Flo
w (
W/g
)
Temperature (ºC)

Chapter 3
173
When studying the biomass combustion by the DSC technique, two different
exothermic regions are generally observed [37; 46]. The first region is associated to
the combustion of light volatile matters, which provides reactivity of biomass fuels.
This peak is short and lower, so less heat was released. The second one represents the
combustion of fixed carbon [46]. As can be seen in Figure 3.8, the first exothermic
region for sample CV was represented as a wide shoulder rather than a peak.
Concerning the heat released during the combustion of the different microalgae
samples, it can be observed that samples with wider combustion peaks in the stage of
fixed carbon oxidation showed a higher heat release than samples whose combustion
was mainly developed in the stage of volatiles release. The first exothermic region
appeared in all samples at a similar temperature interval (300 ºC). However, the
second exothermic region covered a higher temperature range for sample NG (500 ºC)
than for samples SC and CV (370 and 280 ºC, respectively). Biomass samples used in
this work were ranked according to the combustion heat as follows: NG > SC > CV.
This trend did not agree well with that obtained if the combustion characteristic factor
(CCF) is considered. In the latter case, sample NG presented the lowest CCF value. As
aforementioned, the CCF measures how easy a combustible is burnt in terms of low
energy requirement to carry out the combustion process (low Ti and Tb). However, it
does not give information about the exothermic reactions taking place during the
combustion. DSC analysis helps to obtain a more realistic approach to the combustion
process of biomass and determine the amount of energy contained in the char.
Therefore, the devolatilization of sample NG led to the formation of the most
energetic char. Furthermore, the high ash content in samples CV and SC might affect
the char oxidation process [43].
Kinetic analysis
The kinetic model used in this work was derived from the pseudo multi-component
separate-stage models (PMSM) approach. In this type of models, the biomass sample
is composed of multiple pseudo components [19]. In this regard, the kinetic
parameters can be determined assuming single separate reactions for the different

Chapter 3
174
stages of thermal conversion. Microalgae combustion is usually described by two
main stages: devolatilization stage (Dev. stage) and char oxidation stage (Oxid. stage).
However, as aforementioned this classification may result unclear due to the fact that
it does not consider different thermal events that take place during these stages. Thus,
an additional sub-classification was carried out as in latter sections. In this regard, the
different event occurring during the Devolatilization stages are named to as sub-step 1,
2 and 3. Furthermore, the last stage for sample NG combustion, related to volatile
metal loss and carbonate decomposition, was named to as rem. step. Therefore, Eq. (6)
was separately used to each of the stages above commented.
The model representing the form of g(α) (Table 3.3), which delivered the highest
correlation coefficient, was considered to be the function representing the mass loss
kinetics for the samples under study. Figure 3.9 shows the plots of ln[g(α)/T2] versus
1/T that provided the best linearity at 40 ºC/min. Table 3.15 summarizes the main
kinetic parameters for the biomass samples here studied. It can be seen that all the
stages fitted well to a straight line. All samples showed the best regression coefficient
for the model O1, which is the most used mechanism for the kinetic calculation of
biomass thermal decomposition [17].
All microalgae samples showed a similar kinetic behavior. The main differences
can be attributed to the different stages considered. In this regard, sample CV did not
show the sub-step 1. Sample NG showed a slightly higher activation energy (116.8
kJ/mol) than sample SC (93.6 8 kJ/mol), which can be explained by the low
temperature required for sample SC to decompose. In a similar way, little difference in
the Ea values was observed for the sub-step 2. Sample NG showed the lowest Ea value
(62.9 kJ/mol), pointing out that it was the most reactive sample and the easiest to
ignite. This way, the combustion reaction is more continuous than that observed for
the other microalgae [47]. Regarding sub-step 3, the low amount of lipid in sample CV
and the closure of sub-step 2 and 3 would explain the lower Ea value obtained for this
sample. Sample SC showed the highest Ea value, which may be due to its lipid
composition. In this regard, Kebelmann et al. [40] reported different thermal behaviors

Chapter 3
175
between the lipids extracted from different types of microalgae being attributed to
different fatty acids compositions. Concerning the second stage of combustion, Ea
values were almost the same for all samples. This is in agreement with the results
obtained by Yu et al. [47] for the combustion of different seaweeds. Finally, sample
NG was the only one that showed a last sub-step at high temperatures. The Ea in this
stage was markedly higher than that for the previous stage, pointing out to the high
energy required for metals and carbonates volatilization.
Figure 3.9.-Plot of ln(g(α)/T) vs 1/T for the combustion process of Scenedesmusalmeriensis
(SC), Nannochloropsisgaditana (NG) and Chlorella vulgaris (CV) microalgae.
In order to corroborate the kinetic analysis, the reconstruction of the weight loss
curves was performed. Considering n separate reactions, the kinetic rates of thermal
decomposition of a material can be easily derived from Eq. (4) as follows:
:�C:� =
�A �� � �C ��� �A(�A) (8)
whereαi, Ai, Ei and fi(αi) are the degree of conversion, the pre-exponential factor,
activation energy and model functions obtained for each stage of the combustion
process, respectively.
A VBA-Excel application was developed to solve this model based on the Runge-
Kutta-Fehlberg method for the evaluation of the set of ordinary differential equations.
Figure 3.10 shows the experimental data (solid line) compared to the predicted one
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-20
-18
-16
-14
-12
-10
-8
0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-20
-18
-16
-14
-12
-10
-8
-6
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-20
-18
-16
-14
-12
-10
-8
-6CV
ln (g
( αα αα)/T
2 )
1/T (1/K) * 103
Sub-step 2 Sub-step 3 Oxid-step
NG
1/T (1/K) * 103
Sub-step 1 Sub-step 2 Sub-step 3 Oxid-step Rem-step
SC
1/T (1/K) * 103
Sub-step 1 Sub-step 2 Sub-step 3 Oxid-step

Chapter 3
176
(dotted line) obtained by substituting the calculated activation energy and pre-
exponential factor for each stage into Eq. (8). It can be observed that the proposed
model adequately reproduces the experimental values. The mean error between the
experimental and theoretical curves was calculated and shown in Figure 3.10. The
obtained error was lower in all cases than 3.1 %.
Table 3.14.-Estimated kinetic parameters for the combustion of Nannochloropsisgaditana
(NG), Chlorella vulgaris (CV) and Scenedesmusalmeriensis(SC) microalgae
Figure 3.10.-Comparison between experimental and theoretical results for the combustion
process of Scenedesmusalmeriensis (SC), Nannochloropsisgaditana (NG) and Chlorella
vulgaris (CV)microalgae.
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
Mean error (%): 2.7
Sc
(1- αα αα
)
Mean error (%): 3.1
NG
Temperature (K)
Mean error (%): 1.9
CV
Theoretical Experimental
Stage 1 Sub-step 1 Sub-step 2 Sub-step 3
Biomass Ea
(kJ/mol) A
(1/min) r 2 Ea
(kJ/mol) A
(1/min) r 2 Ea
(kJ/mol) A
(1/min) r 2
NG 116.8 4.7·1013 0.9901 62.9 3.1·105 0.9911 157.8 4.1·1011 0.9933 CV - - - 80.9 1.9·104 0.9915 135.27 5.2·107 0.9901 SC 93.6 1.2·1011 0.9915 71.3 1.4·106 0.9907 178.9 1.3·1013 0.9915
Stage 2
Oxid. step Rem. step
NG 113.2 3.4·106 0.9912 326.5 1.0·1013 0.9907 CV 124.9 5.2·107 0.9921 - - - SC 126.1 3.9·107 0.9908 - - -

Chapter 3
177
Evolved gas analysis
The main products derived from the combustion of the marine biomass (samples
CV, SC and NG) were evaluated by TGA-MS analysis. In this regard, a preliminary
scan was performed in order to identify the main gaseous products released during the
combustion of microalgae samples. The most prominent ions were detected at (m/z)=
2, 15, 18, 27, 28, 29, 30, 41, 44, 45, 46, 48, 50, 58, 60 and 64 corresponding to the
following compounds: H2, CH4, H2O, HCN + C2H4, CO, C2H6, NO + CH4N (primary
amines), C3H5+ (alkenes), C2H5O + CHO2 (esters and ethers + carboxylic groups),
CO2, NO2, SO, CH3Cl, C3H6O (ketones), COS and SO2, respectively. Special attention
must be taken into account when reporting some ions due to they could belong to
multiple compounds. Thus, ions with m/z 27, 30 and 45 are related to the evolution of
different compounds.
Mass spectrometry analyses for the different types of microalgae here considered:
Chlorella vulgaris (CV), Scenedesmusalmeriensis (SC) and
Nannochloropsisgaditana(NG) are shown in Figure 3.15 and 3.16. MS spectra of
microalgae could be divided into different stages related to their degradations steps
studied in the TGA/DTG curves and described in previous sections. Table 3.11
summarizes the most representative MS ions detected, their integrated peak intensities
in the whole temperature range and the temperature where an emission peak was
found.
Microalgae combustion is a chemical process where the organic matter contained
in them is oxidized to release heat [26]. However, the oxidation of microalgae
involves many complex reactions, both in parallel and series, such as thermal
cracking, condensation and depolymerization due to the complex composition of
microalgae. The thermal decomposition of microalgae can be considered step-wise
were carbohydrates, proteins and lipids are decomposed. Thus, the study of gases
evolving during combustion is of high importance from a fundamental point of view
in order to gain further understanding of the complex reactions occurring during
combustion.

Chapter 3
178
Figure 3.11.-Gas evolution profile of CO, CO2, H2O, CH4, C3H5+ (alkenes), C2H5, H2 and C3H6O for the combustion process of
Scenedesmusalmeriensis (SC), Nannochloropsisgaditana (NG) and Chlorella vulgaris (CV) microalgae.
200 400 600 800 1000
200 400 600 800200 400 600 800 1000 200 400 600 800 1000
200 400 600 800
200 400 600 800
200 400 600200 400 600 800
Temperature (ºC)
C2H
6
Temperature (ºC)
NG SC CV
CH4
Inte
nsity
(a.
u.)
Temperature (ºC)
CO
Temperature (ºC)
CO2
Temperature (ºC)
H2
Temperature (ºC)
H2O
Temperature (ºC)
C3H
6O (ketones)
Inte
nsity
(a.
u.)
Temperature (ºC)
C3H
5
+ (alkenes)

Chapter 3
179
Figure 3.12.-Gas evolution profile of NO, NO2 SO, SO2, COS, CH3Cl, HCN and CH4N for the combustion process of Scenedesmusalmeriensis
(SC), Nannochloropsisgaditana (NG) and Chlorella vulgaris (CV) microalgae.
200 400 600 800 1000200 400 600 800200 300 400 500 600 700 200 400 600 800 1000
200 400 600200 400 600 800 1000 200 400 600 800 1000200 400 600 800 1000
Temperature (ºC)
C2H
5O + CHO
2
Temperature (ºC)
COS
Inte
nsity
(a.
u.)
Temperature (ºC)
SO
Temperature (ºC)
SO2
Temperature (ºC)
NG SC CV
CH3Cl
Temperature (ºC)
CH4N + NO
Temperature (ºC)
HCN + C2H
4
Inte
nsity
(a.
u.)
Temperature (ºC)
NO2
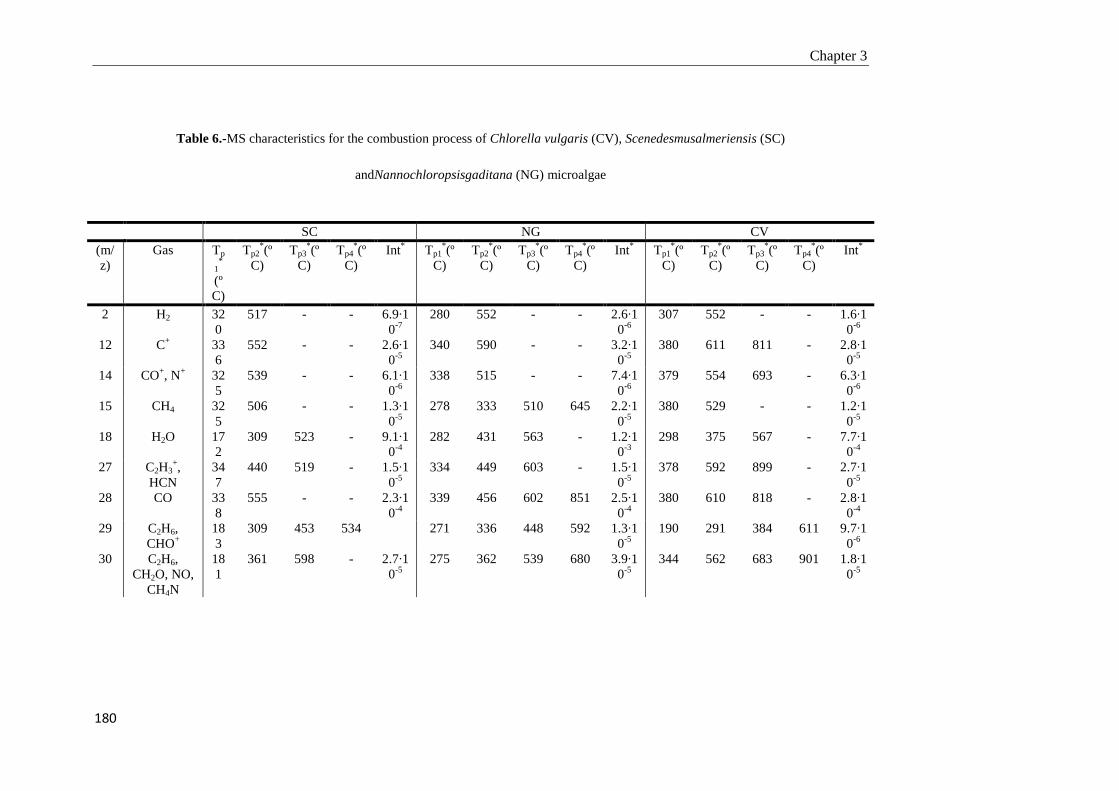
Chapter 3
180
Table 6.-MS characteristics for the combustion process of Chlorella vulgaris (CV), Scenedesmusalmeriensis (SC)
andNannochloropsisgaditana (NG) microalgae
SC NG CV (m/z)
Gas Tp
1*
(ºC)
Tp2*(º
C) Tp3
*(ºC)
Tp4*(º
C) Int* Tp1
*(ºC)
Tp2*(º
C) Tp3
*(ºC)
Tp4*(º
C) Int* Tp1
*(ºC)
Tp2*(º
C) Tp3
*(ºC)
Tp4*(º
C) Int*
2 H2 320
517 - - 6.9·10-7
280 552 - - 2.6·10-6
307 552 - - 1.6·10-6
12 C+ 336
552 - - 2.6·10-5
340 590 - - 3.2·10-5
380 611 811 - 2.8·10-5
14 CO+, N+ 325
539 - - 6.1·10-6
338 515 - - 7.4·10-6
379 554 693 - 6.3·10-6
15 CH4 325
506 - - 1.3·10-5
278 333 510 645 2.2·10-5
380 529 - - 1.2·10-5
18 H2O 172
309 523 - 9.1·10-4
282 431 563 - 1.2·10-3
298 375 567 - 7.7·10-4
27 C2H3+,
HCN 347
440 519 - 1.5·10-5
334 449 603 - 1.5·10-5
378 592 899 - 2.7·10-5
28 CO 338
555 - - 2.3·10-4
339 456 602 851 2.5·10-4
380 610 818 - 2.8·10-4
29 C2H6, CHO+
183
309 453 534 271 336 448 592 1.3·10-5
190 291 384 611 9.7·10-6
30 C2H6, CH2O, NO,
CH4N
181
361 598 - 2.7·10-5
275 362 539 680 3.9·10-5
344 562 683 901 1.8·10-5

Chapter 3
181
41 C3H5+
(alkenes) 329
450 - - 2.5·10-6
331 437 - - 4.5·10-6
379 425 501 - 2.6·10-6
44 CO2 329
555 - - 5.3·10-4
193 339 592 870 6.9·10-4
380 610 818 - 4.3·10-4
45 C2H5O+;
CHO2 340
554 - - 6.1·10-6
203 344 587 895 7.6·10-6
372 609 899 - 4.4·10-6
46 NO2; C2H5OH
337
573 - - 1.9·10-6
193 339 591 - 2.5·10-6
392 608 830 - 1.5·10-6
48 SO 279
443 - - 4.1·10-6
275 386 - - 4.2·10-7
322 429 - - 2.8·10-7
50 CH3Cl 306
445 - - 1.2·10-7
279 421 - - 5.3·10-7
303 382 - - 1.5·10-7
58 C3H6O(ketones)
326
- - - 1.2·10-7
326 424 - - 1.9·10-7
395 573 - - 5.5·10-8
60 COS 388
- - - 8.6·10-8
413 - - - 1.6·10-7
262 - - - 1.4·10-7
64 SO2 28
1 375 582 - 4.4·1
0-7 275 383 597 - 4.1·1
0-7 312 390 641 - 2.8·1
0-7

Chapter 3
182
The gaseous emissions followed a similar pattern in all microalgae samples. CO,
CO2 and H2O were the main components produced and evolved over the whole
temperature range. CO and CO2 main peaks took place at temperatures between 555
and 610 ºC being coincident with their DTG peak. The CO and CO2 emissions
detected in this temperature range were due to the fixed carbon oxidation [26].
Emission peaks at lower temperatures were associated to the decomposition of
carboxyl groups in protein and saccharides [48]. Furthermore, an additional peak was
found for samples CV and NG at temperatures above 800 ºC, which was attributed to
the decomposition of mineral matter, as carbonates, in the ash [26; 49]. On the other
hand, H2O emissions reached their maximum evolution rate at 282, 298 and 309 for
samples NG, CV and SC, respectively. The H2O produced at this step was mainly
associated to the oxidation of oxygen containing functional groups (especially
hydroxyl groups). H2O peaks at lower temperatures were attributed to the loss of
cellular water and external water bounded by surface tension [44]. Finally, the water
produced at higher temperatures was associated to the evolution of H2. In this regard,
H2 was produced by the dehydrogenation of the char [50] and reached their maximum
yield at 517 and 552 ºC for samples SC and NG and CV. In addition, H2O emissions
peaks were found at slightly higher temperatures. This fact pointed out that H2O at this
stage was produced by the oxidation of produced H2. H2O peaks were found at lower
temperatures than CO2 ones, indicating that microalgae samples decomposition started
via dehydration of the algae components followed by combustion [50]
Light hydrocarbons (HC), especially CH4, were the main secondary products
detected. The origin of HC is attributed to the decomposition of carbohydrates and
lipids. In this sense, Marcilla et al. [51] reported that the main source of methyl groups
was the decomposition of lipids. Their results agreed well with those reported in this
study as maximum peaks for CH4 were obtained at 506, 510 and 529 ºC.
Carbohydrates decomposition also led to the formation of HC, as it can be observed
from the C2H5 and C3H5 (alkenes) emissions between 180 and 450 ºC. Emission peaks
for HC were also observed at higher temperatures as reported by Bae et al. [52]. The
evolution of oxygen containing hydrocarbons such as ketones (C3H6O) and carboxylic

Chapter 3
183
acids, esters and ethers (C2H5O + CHO2) was attributed to the breaking up of carbonyl
groups from fatty acids[53]. Ketones were mainly detected between 326 and 396 ºC
whereas carboxylic acids, ester and ethers evolved in two steps. The C-C scission of
these compounds may turn out in direct CO emissions, or they can be later combined
with oxygen to form CO2.
The evolution of N-compounds took place forming different emission peaks.
Nitrogen compounds (N-compounds) evolved as CH4N (primary amines), NOx and
HCN. The first peak between 200 and 400 ºC was mainly associated to the
decomposition of proteins. The second peak was related to the ignition of N-
containing compounds between 400 and 500 ºC. In this temperature range, the
presence of primary amines and HCN was less likely and their signal may have an
important contribution of other compounds related to their ions such as HC and NO.
The presence of NO at temperatures above 400 ºC has been reported by different
authors [26; 48]. The last peak at temperatures above 500 ºC corresponded to the
oxidation of the remaining nitrogen in the char. The maximum yield of NO2 was
reached in this stage. The release of NOx is of high importance as they are the primary
components of photochemical smog [26].
Chloride and sulfur compounds were released in lower proportions than nitrogen
compounds. Chloride compounds were mainly detected as CH3Cl and emitted between
200 and 400 ºC, the evolution of this compound was much higher for the sample NG
than for samples SC and CV which is in good agreement with their compositional
analysis (Table 1.2). Concerning the release of sulfur compounds, SO and SO2 were
the main products detected. Their release is mainly associated to the decomposition
and oxidation of sulphated polysaccharides[48]. SO and SO2 maximum peaks were
found at similar temperatures for all samples (270-300 ºC). Furthermore, a second
emission peak was found in the lipid decomposition temperature range (350-480 ºC)
for both compounds due to the degradation of organic sulfides in organic residues
[48].These peaks were characterized by the apparition of SO2 at lower temperatures
than SO. Finally, only SO2 was produced at higher temperatures being quite consistent

Chapter 3
184
with the combustion of fixed carbon temperature range. Furthermore, COS was
produced, which can be originated by the partial oxidation of organic sulfur or the
reaction of SO2 with carbon complexes [54].
Samples SC and NG showed peaks at temperatures above 700 ºC for different
compounds (HCN + C2H4, C2H5, CH4N + NO, NO2, CO and CO2). The evolution of
compounds in this temperature range is mainly associated to the catalytic effect of
some compounds in the ash. Furthermore, the volatilization of some mineral matter
and carbonates may take place as above commented. However, this fact was unusual
as no appreciable weight loss was detected at this temperature for these samples
(<0.05 wt.% for both samples). The interactions of the ash in the combustion of
microalgae were out of the scope of this work. However, the problematic associated to
their presence in combustion processes can be considered of high importance and
further studies will be carried out in order to achieve a deeper insight of their behavior.
3.3.3. Combustion of Canadian biomass
Thermogravimetric analysis
Figure 3.13 shows the thermogravimetric (TGA) and derivative thermogravimetric
(DTG) profiles for different types of biomass here considered: two woody crops
(black spruce and Pinusbanksiana mixtures (BP) and willow (W)), and three
herbaceous non-perennial energy crops (common reed (CR), reed phalaris (RP) and
switchgrass (S)). Table 3.17 shows the main relevant combustion characteristics. The
thermal decomposition of biomass under oxidative environment is usually described
by two stages[19; 37;43]. Firstly, the devolatilization of the sample takes place at low
temperatures (160-400 ºC), leading to char formation. Then, the oxidation of the
sample occurs at temperatures higher than 400 ºC. Generally, each stage is attributed
to the decomposition of the biomass main components (hemicellulose, cellulose and
lignin)[3; 5].Hemicellulose and cellulose are assumed to decompose during the
devolatilization stage[55]. Hemicellulose usually appears as a shoulder in the DTG
curve in the devolatilization stage at low temperatures, whereas cellulose oxidation
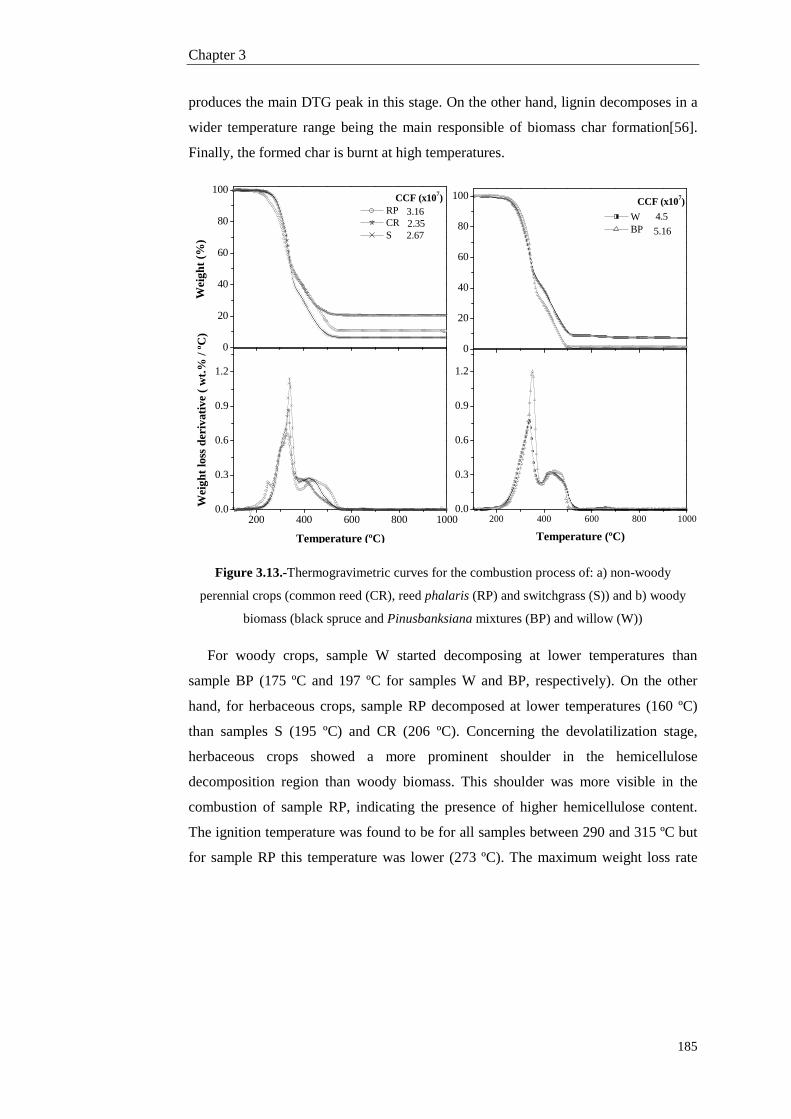
Chapter 3
185
produces the main DTG peak in this stage. On the other hand, lignin decomposes in a
wider temperature range being the main responsible of biomass char formation[56].
Finally, the formed char is burnt at high temperatures.
Figure 3.13.-Thermogravimetric curves for the combustion process of: a) non-woody
perennial crops (common reed (CR), reed phalaris (RP) and switchgrass (S)) and b) woody
biomass (black spruce and Pinusbanksiana mixtures (BP) and willow (W))
For woody crops, sample W started decomposing at lower temperatures than
sample BP (175 ºC and 197 ºC for samples W and BP, respectively). On the other
hand, for herbaceous crops, sample RP decomposed at lower temperatures (160 ºC)
than samples S (195 ºC) and CR (206 ºC). Concerning the devolatilization stage,
herbaceous crops showed a more prominent shoulder in the hemicellulose
decomposition region than woody biomass. This shoulder was more visible in the
combustion of sample RP, indicating the presence of higher hemicellulose content.
The ignition temperature was found to be for all samples between 290 and 315 ºC but
for sample RP this temperature was lower (273 ºC). The maximum weight loss rate
0
20
40
60
80
100
200 400 600 800 10000.0
0.3
0.6
0.9
1.2
0
20
40
60
80
100
200 400 600 800 10000.0
0.3
0.6
0.9
1.2
CCF (x107)
2.672.353.16
Wei
ght (
%)
RP CR S
CCF (x107)
Wei
ght l
oss
deriv
ativ
e (
wt.%
/ ºC
)
Temperature (ºC)
4.5
5.16
W BP
Temperature (ºC)

Chapter 3
186
was found for samples BP and S, which pointed out to a higher content in cellulose.
The char oxidation stage started at similar temperatures for all samples (375-391 ºC).
However, it ended at a slightly higher temperature for herbaceous crops, 540 ºC,
compared to 507-534 ºC temperature range for the woody crops. An additional mass
loss weight was found in the combustion of sample W. This fact was attributed to the
burning of the remaining semi-coke. The highest value of Tb was found for sample W,
which is related to the presence of high lignin content. Finally, the amount of residue
(ash) that remained after the combustion process is of great interest. Sample CR left
the highest amount of residue (18.2wt.%) whereas BP yielded a very low one
(1.6wt.%). The ash composition is dominated by metal oxides, especially silica,
calcium oxide and potassium oxide. An high ash content can contribute to the
development of the combustion process due to its catalytic effect[43]. In the opposite,
a high ash content contribute to operational problems due to the occurrence of fouling
and slagging phenomena.
The relative amount of biomass main components is important when determining
the quality of a biomass fuel. In this regard, a high content in hemicellulose and
cellulose turns out in a low Ti and a high (dw/dT)max. On the other hand, a higher
content in lignin produces a high amount of residue to be burnt. Wang et al.
(2009)[37] described the combustion characteristic factor (CCF), that can be used as a
criterion for fuel combustion performance according to the mentioned parameters (the
higher the CCF value is, the easier to ignite a sample is), as follows:
889 = (:;:� )<=> ∙ (:;:� )<?=@ �A$ ∙ �B
� (8)
where (dw/dt)max is the maximum burning velocity (%/min); (dw/dt)mean is the average
burning velocity (%/min); Ti is the ignition temperature (K) and Tb is the burnout
temperature (K).
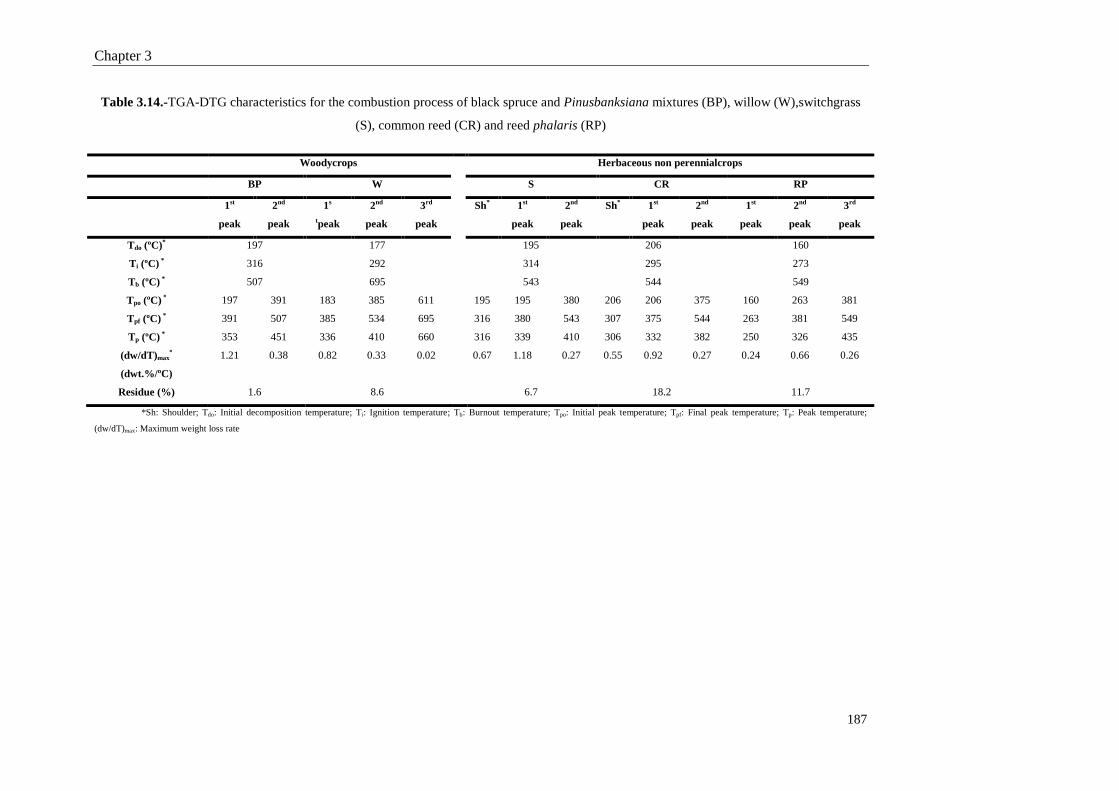
Chapter 3
187
Table 3.14.-TGA-DTG characteristics for the combustion process of black spruce and Pinusbanksiana mixtures (BP), willow (W),switchgrass
(S), common reed (CR) and reed phalaris (RP)
Woodycrops Herbaceous non perennialcrops
BP W S CR RP
1st
peak
2nd
peak
1s
tpeak
2nd
peak
3rd
peak
Sh* 1st
peak
2nd
peak
Sh* 1st
peak
2nd
peak
1st
peak
2nd
peak
3rd
peak
Tdo (ºC)* 197 177 195 206 160
T i (ºC) * 316 292 314 295 273
Tb (ºC) * 507 695 543 544 549
Tpo (ºC) * 197 391 183 385 611 195 195 380 206 206 375 160 263 381
Tpf (ºC) * 391 507 385 534 695 316 380 543 307 375 544 263 381 549
Tp (ºC) * 353 451 336 410 660 316 339 410 306 332 382 250 326 435
(dw/dT)max*
(dwt.%/ºC)
1.21 0.38 0.82 0.33 0.02 0.67 1.18 0.27 0.55 0.92 0.27 0.24 0.66 0.26
Residue (%) 1.6 8.6 6.7 18.2 11.7
*Sh: Shoulder; Tdo: Initial decomposition temperature; Ti: Ignition temperature; Tb: Burnout temperature; Tpo: Initial peak temperature; Tpf: Final peak temperature; Tp: Peak temperature;
(dw/dT)max: Maximum weight loss rate

Chapter 3
188
CCF values for all samples are plotted in Figure 3.13. In all cases, these values
were bigger than 2 indicating the good general burning performance[37]. However,
these data must be used only as a reference since they do not give any information
about the heat released during the combustion process.
Differential scanning calorimetric analysis
Woody crops (samples W and BP) and herbaceous non-perennial energy crops
(samples S, CR and RP) were also investigated by the DSC technique. Experimental
DSC curves are presented in Figure 3.14. This way, it is possible to identify the kind
of mass loss event explained in the TG analyses[57]. DSC main temperatures and heat
of combustion (Hcomb) are included in Table 3.18.
Figure 3.14.-DSC curves for the combustion process of: a) non-woody perennial crops
(common reed (CR), reed phalaris (RP) and switchgrass (S)) and b) woody biomass (black
spruce and Pinusbanksiana mixtures (BP) and willow (W))
When studying the biomass combustion by the DSC technique, two different
exothermic regions are generally observed[37; 46]. The first region is associated to the
combustion of light volatile matters, which provides reactivity of biomass fuels. The
second one represents the combustion of fixed carbon[46]. As can be seen from Figure
3.14, most biomass samples followed this trend. However, both exothermic regions in
120 240 360 480 600 720 840 9600
5
10
15
20
25
30
35
120 240 360 480 600 720 840 9600
5
10
15
20
25
30
35
40 RP CR S
Hea
t flo
w (
W/g
)
Temperature (ºC)
W BP
Temperature (ºC)

Chapter 3
189
sample CR overlapped turning into only one peak at an intermediate temperature
between those corresponding to volatile matters release and fixed carbon combustion,
respectively. This fact can be explained attending to the low fixed carbon content in
sample CR (Table 3.1). Furthermore, the high ash content might reduce the char
combustion temperatures due to its catalytic metal content[43].
Concerning the heat released during the combustion of the different biomass
samples, it can be observed that samples with more prominent DSC peak in the stage
of combustion of fixed carbon showed a higher heat release than samples whose
combustion was mainly developed in the stage of volatiles release. Biomass samples
used in this work were ranked according to the combustion heat as follows: W> RP
>CR>S >BP. This trenddid not agree well with that obtained if thecombustion
characteristic factor (CCF) is considered. In the latter case, sample BP presented the
highest CCF. As aforementioned, the CCF measures how easy a combustible is burnt
in terms of low energy requirement to carry out the combustion process (low Ti and
Tb). However, it does not give information about the exothermic reactions taking place
during combustion. DSC analysis helps to obtain a more realistic approach to the
combustion process of biomass and determine the amount of containing energy in the
char. Therefore, the devolatilization of samples W and RP led to the formation of the
more energetic char. The combination of this fact and a high CCF value might
determine the combustion quality of a biomass[37; 46].
Kinetic analysis
The kinetic model used in this work was derived from the pseudo multi-component
separate-stage models (PMSM) approach. In this type of models, the biomass sample
is composed of multiple pseudo components[19]. In this regard, the kinetic parameters
can be determined assuming single separate reactions for the different stages of
thermal conversion. As abovementioned, biomass combustion was clearly defined by
two main stages: devolatilization stage (Dev. stage) and char oxidation stage (Oxid.
stage). However, additional decompositions occurred for some samples. For example,
the Dev. Stage for herbaceous crops was represented by two peaks. In order to

Chapter 3
190
differentiate them, the Dev. stage for them was divided into two stages: Dev. stage A
and Dev. stage B. This way, an additional weight loss step took place for sample W
combustion due to the remaining char burning. This stage was named as Rem. stage.
Therefore, eq. (6) was used separately to each of the stages above commented.
The model representing the form of g(α)(Table 3.3), which delivered the highest
correlation coefficient, was considered to be the function representing the mass loss
kinetics for the samples under study.Figure 3.15 shows the plots of ln[g(α)/T2] versus
1/T that provided the best linearity at 40 ºC/min. Table 3.19 summarizes the main
kinetic parameters for the biomass samples here studied. It can be seen that all the
stages fitted well to a straight line (r2> 0.99).All samples showed the best regression
coefficient for the model O1, which is the most used mechanism for the kinetic
calculation of biomass thermal decomposition[17; 19]. However, a good correlation
was also obtained by the model D3 for the Dev. stage 1 in herbaceous crops. This stage
corresponded to the hemicellulose decomposition temperature range. High correlation
coefficients for diffusion mechanisms during the devolatilization stage of
coal/biomass blends were also observed by Gil et al. (2010)[2]. Yorulmaz and Atimtay
et al. (2009)[7] reported that different models can be suitable to describe the biomass
combustion process by thermal analysis. Further research combining dynamic and
isothermal studies should be carried out in order to elucidate the exact mechanisms of
the oxidation process.
Calculated activation energies for the multiple-step model for different types of
woody and herbaceous biomass are listed in Table 4. Activation energies obtained for
the first stage of woody biomass during the oxidation process were quite similar (91
kJ/mol and 101 kJ/mol for samples BP and W, respectively). These values are in good
agreement with those obtained by different authors[1; 2]. Higher values were obtained
for the oxidation stage. Thus, sample BP showed the highest one (143 kJ/mol),
pointing out that the char obtained from this sample is less reactive. Finally, the Rem.
stage in the oxidation of sample W showed a high value of the activation energy (372
kJ/mol). This fact could be attributed to the little amount of remaining char-semi-coke,
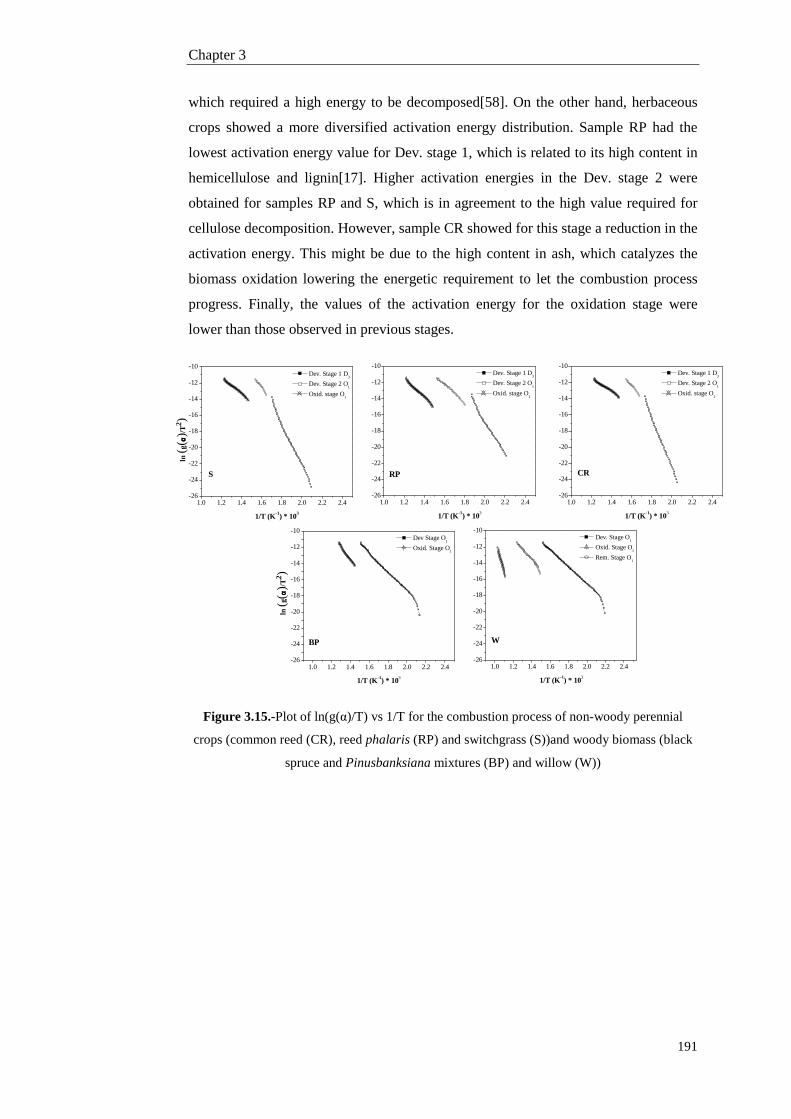
Chapter 3
191
which required a high energy to be decomposed[58]. On the other hand, herbaceous
crops showed a more diversified activation energy distribution. Sample RP had the
lowest activation energy value for Dev. stage 1, which is related to its high content in
hemicellulose and lignin[17]. Higher activation energies in the Dev. stage 2 were
obtained for samples RP and S, which is in agreement to the high value required for
cellulose decomposition. However, sample CR showed for this stage a reduction in the
activation energy. This might be due to the high content in ash, which catalyzes the
biomass oxidation lowering the energetic requirement to let the combustion process
progress. Finally, the values of the activation energy for the oxidation stage were
lower than those observed in previous stages.
Figure 3.15.-Plot of ln(g(α)/T) vs 1/T for the combustion process of non-woody perennial
crops (common reed (CR), reed phalaris (RP) and switchgrass (S))and woody biomass (black
spruce and Pinusbanksiana mixtures (BP) and willow (W))
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-26
-24
-22
-20
-18
-16
-14
-12
-10
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-26
-24
-22
-20
-18
-16
-14
-12
-10
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-26
-24
-22
-20
-18
-16
-14
-12
-10
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-26
-24
-22
-20
-18
-16
-14
-12
-10
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4-26
-24
-22
-20
-18
-16
-14
-12
-10
W
1/T (K -1) * 103
RP
1/T (K -1) * 103
CR
1/T (K -1) * 103
Dev. Stage 1 D3
Dev. Stage 2 O1
Oxid. stage O1
Dev. Stage 1 D3
Dev. Stage 2 O1
Oxid. stage O1
ln (g
( αα αα)/T
2 )
1/T (K -1) * 103
Dev. Stage 1 D3
Dev. Stage 2 O1
Oxid. stage O1
S
Dev. Stage O1
Oxid. Stage O1
Rem. Stage O1
Dev Stage O1
Oxid. Stage O1
BP
ln (g
( αα αα)/T
2 )
1/T (K -1) * 103

Chapter 3
192
In order to corroborate the kinetic analysis, the reconstruction of the weight loss
curves was performed. Considering n separate reactions, the kinetic rates of thermal
decomposition of a material can be easily derived from Eq. (4) as follows:
:�C:� =
�A �� � �=C ��� �A(�A) (8)
whereαi, Ai, Eai and fi(αi) are the degree of conversion, the pre-exponential factor,
activation energy and model functions obtained for each stage of the combustion
process, respectively.
A VBA-Excel application was developed to solve this model based on the Runge-
Kutta-Fehlberg method for the evaluation of the set of ordinary differential equations.
Figure 3.16 shows the experimental data (solid line) compared to the predicted one
(dotted line) obtained by substituting the calculated activation energy and pre-
exponential factor for each stage into Eq. (8). It can be observed that the proposed
model adequately reproduces the experimental values. The mean error between the
experimental and theoretical curves was calculated and shown in Figure 3.16. The
obtained error was small in value for all samples (lower than 3.4%).

Chapter 3
193
Figure 4.-Comparison between experimental and theoretical results for the combustion process
of non-woody perennial crops (common reed (CR), reed phalaris (RP) and switchgrass (S))
and woody biomass (black spruce and Pinusbanksiana mixtures (BP) and willow (W))
Evolved gas analysis
Although contaminant emissions associated to biomass are lower than those in
fossil fuels, they must be taken into account due to the high development of biomass
conversion technologies[59]. In this regard, a preliminary scan was performed in order
to identify the main contaminants released during the combustion of woody and
herbaceous biomasses. The most prominent ions related to contaminants were detected
at (m/z) = 18, 27, 28, 30, 36, 44, 46, 48, 50, 64 and 78 corresponding to the following
compounds: H2O, HCN, CO, NO, CO2, NO2, SO, CH3Cl, SO2 and C6H6, respectively.
Figure 3.17 and 3.18 shows the mass spectra obtained for the combustion process
of woody and herbaceous crops. MS curves in Figure 3.18 were moved up and down
in order to clarify the results due to the fact that they mostly overlapped. Table 3.20
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
400 600 800 1000 12000.0
0.2
0.4
0.6
0.8
1.0
Mean error (%): 1.86Mean error (%): 0.95
RP
Temperature (K)
S
Temperature (K)
Mean error (%): 3.35
Temperature (K)
W
CR
Temperature (K)
Theoretical Experimental
Mean error (%): 1.82Mean error (%): 2.98
(1- αα αα
)
BP
(1- αα αα
)
Temperature (K)

Chapter 3
194
summarizes the most representative MS ions detected, their integrated peak intensities
in the whole temperature range and the most relevant temperatures.
The mass spectra showed two emission peaks for most detected products. The first
one that took place at lower temperatures, corresponded to the so-called oxidative
pyrolysis or devolatilization of the sample. The second one corresponded to the
oxidation of the char. CO, CO2 and H2O were the main compounds formed in the
combustion process of woody and herbaceous biomass. CO and CO2 evolved for all
samples in the whole oxidation temperature range. Both compounds showed emission
peaks at similar temperatures in the first emission temperature range, whereas in the
char oxidation stage the CO2 peak was detected at higher temperatures. The maximum
CO and CO2 yield was observed in the Oxid. stage. CO formation during the first
stage was associated to decarbonylation reactions, secondary reactions between
volatiles and rearrangement of the char skeleton[33]. Furthermore, the CO evolution
during the char oxidation stage was mainly due to the formation of active carbon sites
in the char which were later oxidized, releasing CO and leaving oxygen atoms
attached to carbon surface[32]. The CO2 evolution during the first decomposition
stage followed a similar path than that for CO. However, the later appearance of CO2
compared to CO during the second stage, pointed out that a fraction of the produced
CO reacted with oxygen increasing the CO2 yield. Additionally, Li and Brown et al.
(2001)[32] established different pathways for CO2 evolution during char combustion
involving the formation of different carbon-oxygen complexes. On the other hand,
H2O spectra was released into one step between 334 and 356 ºC. In this stage, the
water formed is known as pyrolityc water[34]and is produced due to hydroxyl groups
bond scission. A small shoulder could also be observed at slightly high temperatures.
The H2O shoulder is mainly attributed to the oxidation of hydrogen produced at
temperatures higher than 400 ºC due to char cracking reactions and the occurrence of
the reverse water-gas shift reaction[33; 34].

Chapter 3
195
Figure 3.17.-Gas evolution profile of CO, CO2, H2O, NO and NO2 for the combustion process of non-woody perennial crops (common reed
(CR), reed phalaris (RP) and switchgrass (S)) and woody biomass (black spruce and Pinusbanksiana mixtures (BP) and willow (W))
200 400 600 800 1000 200 400 600 800 1000 200 400 600
Inte
nsity
(a.
u.)
Temperature (ºC)
CO
Temperature (ºC)
CO2
Temperature (ºC)
BP W S CR RP
H2O
200 400 600 800 1000 200 400 600 800 1000200 400 600
Inte
nsity
(a.
u.)
Temperature (ºC)
NO
Temperature (ºC)
HCN
Temperature (ºC)
NO2
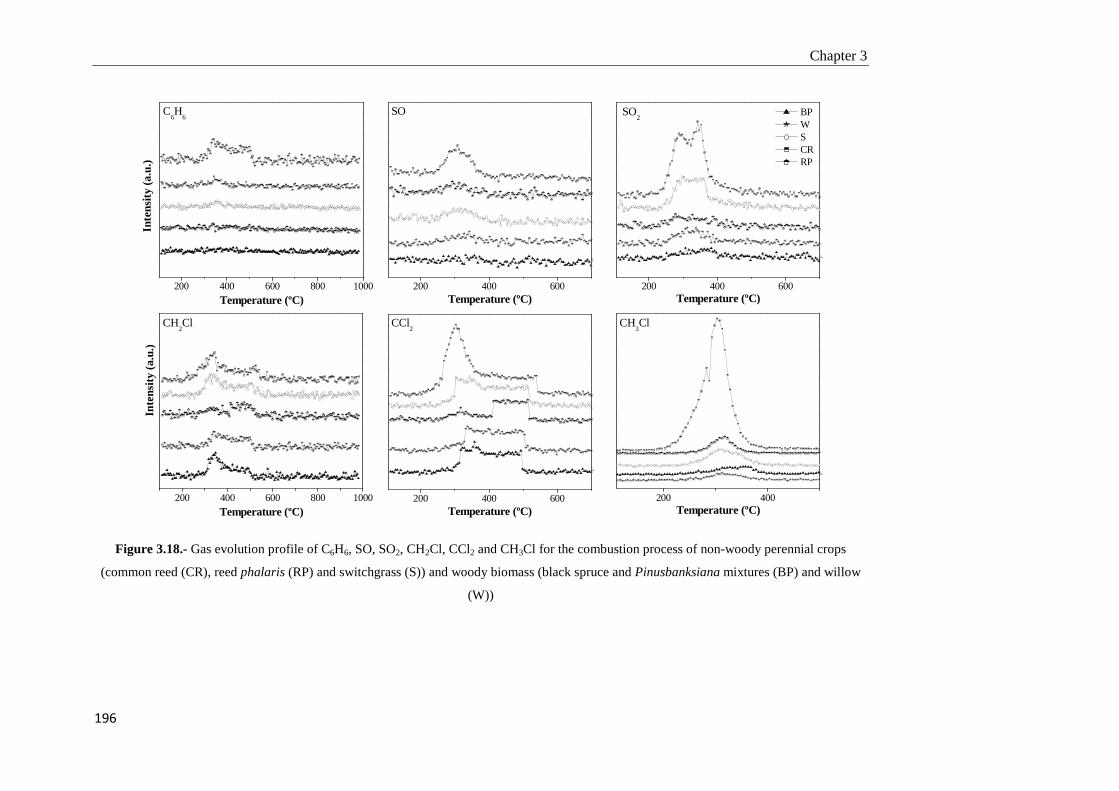
Chapter 3
196
Figure 3.18.- Gas evolution profile of C6H6, SO, SO2, CH2Cl, CCl2 and CH3Cl for the combustion process of non-woody perennial crops
(common reed (CR), reed phalaris (RP) and switchgrass (S)) and woody biomass (black spruce and Pinusbanksiana mixtures (BP) and willow
(W))
200 400 600
200 400 600 200 400 600
200 400 600 800 1000
200 400 600 800 1000
200 400
Temperature (ºC)
CCl2
Temperature (ºC)
SO
Temperature (ºC)
BP W S CR RP
SO2
Inte
nsity
(a.
u.)
Temperature (ºC)
CH2Cl
Inte
nsity
(a.
u.)
Temperature (ºC)
C6H
6
Temperature (ºC)
CH3Cl
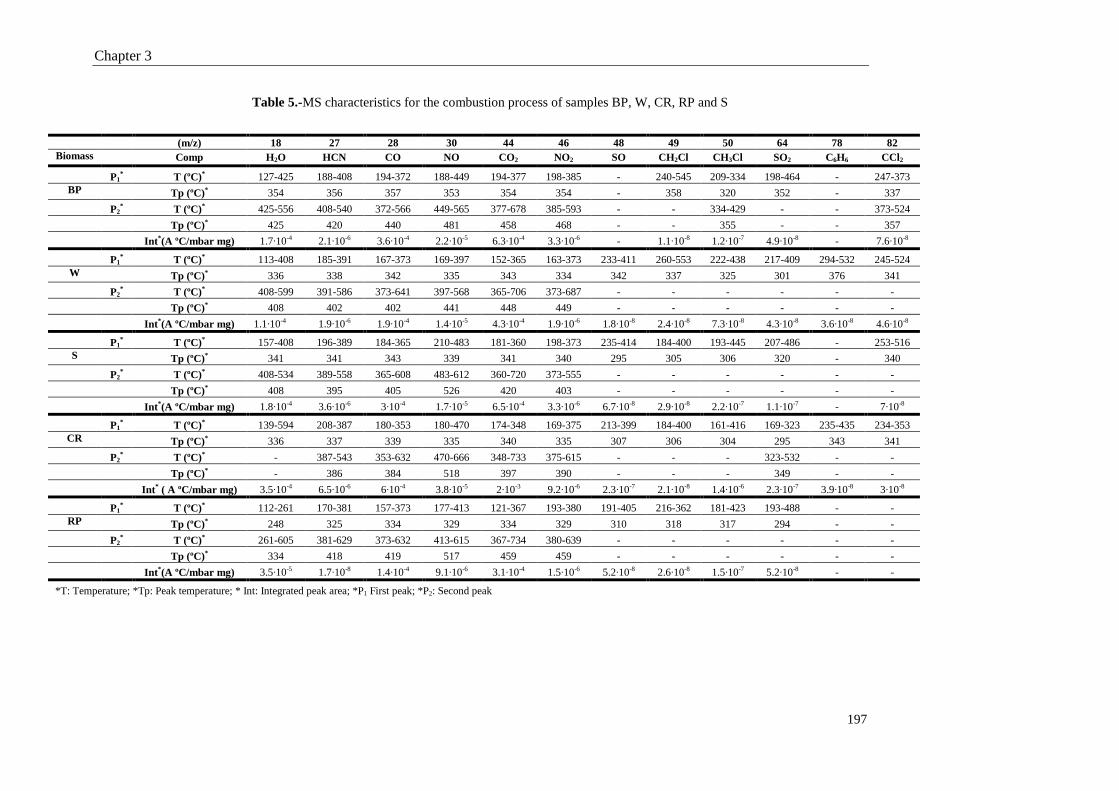
Chapter 3
197
Table 5.-MS characteristics for the combustion process of samples BP, W, CR, RP and S
(m/z) 18 27 28 30 44 46 48 49 50 64 78 82 Biomass Comp H2O HCN CO NO CO2 NO2 SO CH2Cl CH3Cl SO2 C6H6 CCl2
P1* T (ºC)* 127-425 188-408 194-372 188-449 194-377 198-385 - 240-545 209-334 198-464 - 247-373
BP Tp (ºC)* 354 356 357 353 354 354 - 358 320 352 - 337 P2
* T (ºC)* 425-556 408-540 372-566 449-565 377-678 385-593 - - 334-429 - - 373-524 Tp (ºC)* 425 420 440 481 458 468 - - 355 - - 357 Int *(A ºC/mbar mg) 1.7·10-4 2.1·10-6 3.6·10-4 2.2·10-5 6.3·10-4 3.3·10-6 - 1.1·10-8 1.2·10-7 4.9·10-8 - 7.6·10-8
P1* T (ºC)* 113-408 185-391 167-373 169-397 152-365 163-373 233-411 260-553 222-438 217-409 294-532 245-524
W Tp (ºC)* 336 338 342 335 343 334 342 337 325 301 376 341 P2
* T (ºC)* 408-599 391-586 373-641 397-568 365-706 373-687 - - - - - - Tp (ºC)* 408 402 402 441 448 449 - - - - - - Int *(A ºC/mbar mg) 1.1·10-4 1.9·10-6 1.9·10-4 1.4·10-5 4.3·10-4 1.9·10-6 1.8·10-8 2.4·10-8 7.3·10-8 4.3·10-8 3.6·10-8 4.6·10-8
P1* T (ºC)* 157-408 196-389 184-365 210-483 181-360 198-373 235-414 184-400 193-445 207-486 - 253-516
S Tp (ºC)* 341 341 343 339 341 340 295 305 306 320 - 340 P2
* T (ºC)* 408-534 389-558 365-608 483-612 360-720 373-555 - - - - - - Tp (ºC)* 408 395 405 526 420 403 - - - - - - Int *(A ºC/mbar mg) 1.8·10-4 3.6·10-6 3·10-4 1.7·10-5 6.5·10-4 3.3·10-6 6.7·10-8 2.9·10-8 2.2·10-7 1.1·10-7 - 7·10-8
P1* T (ºC)* 139-594 208-387 180-353 180-470 174-348 169-375 213-399 184-400 161-416 169-323 235-435 234-353
CR Tp (ºC)* 336 337 339 335 340 335 307 306 304 295 343 341 P2
* T (ºC)* - 387-543 353-632 470-666 348-733 375-615 - - - 323-532 - - Tp (ºC)* - 386 384 518 397 390 - - - 349 - - Int * ( A ºC/mbar mg) 3.5·10-4 6.5·10-6 6·10-4 3.8·10-5 2·10-3 9.2·10-6 2.3·10-7 2.1·10-8 1.4·10-6 2.3·10-7 3.9·10-8 3·10-8
P1* T (ºC)* 112-261 170-381 157-373 177-413 121-367 193-380 191-405 216-362 181-423 193-488 - -
RP Tp (ºC)* 248 325 334 329 334 329 310 318 317 294 - - P2
* T (ºC)* 261-605 381-629 373-632 413-615 367-734 380-639 - - - - - - Tp (ºC)* 334 418 419 517 459 459 - - - - - - Int *(A ºC/mbar mg) 3.5·10-5 1.7·10-8 1.4·10-4 9.1·10-6 3.1·10-4 1.5·10-6 5.2·10-8 2.6·10-8 1.5·10-7 5.2·10-8 - -
*T: Temperature; *Tp: Peak temperature; * Int: Integrated peak area; *P1 First peak; *P2: Second peak

Chapter 3
198
Two emission regions were also observed for nitrogen compounds. NO and NO2
spectra showed two well developed peaks whereas HCN showed a clear peak in the
Dev. stage and kept evolving as a shoulder up to temperatures around 400 ºC. The first
emission peak for HCN, NO and NO2 took place at similar temperatures (330-356 ºC).
In this stage, the nitrogen containing volatiles are mainly attributed to the
decomposition of proteins[60]. Protein decomposition leads to the formation of
volatile cyclic amides which, due to cracking reactions, produces HCN among other
components[60]. The second peak stood out for the early apparition of NO2 followed
by the NO emission at higher temperatures for all samples except for W. These results
agree well with those reported by Darvell et al. (2012)[12] who found two stages for
the combustion of different biomass char model samples. These stages were
characterized by the release of NO at higher temperatures and the presence of sharper
peaks. Peak areas for NO and HCN showed a higher amount of these compounds
compared to NO2. This fact is due to ions selected as NO and HCN compounds, (m/z)
= 30 and 27, respectively belong to other compounds such as light hydrocarbons,
which are also common products from biomass combustion.
Sulfur compounds (SO and SO2) were found in a lower proportion than nitrogen
ones, which is in agreement with biomass samples ultimate analysis (Table 3.1). Both
compounds were detected in all samples, but in sample BP, which had the lowest
sulfur content in the original material and only SO2 was observed. SO and SO2 were
released during the first stage at temperatures between 300 and 340 ºC. As observed
by Otero et al. (2002), the SO2 peaks occurred at lower temperatures than CO2 peaks
in a narrow temperature range and with a shallow shape. Chloride compounds were
mainly detected as CH3Cl for all studied samples, which showed the clearest spectra,
whereas Cl- ions were more diffused due to the fact that they were found close to the
sensitivity level of the mass spectrometer. In the same way, C6H6 was only observed
in samples W and CR, being an indicative of lignin decomposition.
Sample CR showed the highest yield for volatiles, which were released at lower
temperatures than observed for other biomasses. As above mentioned, the high content

Chapter 3
199
in ashes of sample CR, catalyzed the volatile release, increasing products yields and
shifting them to lower temperatures[61].
3.4. conclusions
Combustion behavior and gas formation from the oxidation process of fir wood,
eucalyptus wood, pine bark and three individual components of lignocellulosic
biomass (cellulose, hemicellulose and lignin) were analyzed by TGA-MS.Biomass
combustion took place into two main stages:devolatilization stage (Dev. stage) and
oxidation stage (Oxid. stage). Most products detected in the combustion of
lignocellulosic biomass were released during the Dev. stage whereas only NO2,
C2H5O+, CO and CO2 were detected at the Oxid.stage. Nitrogen compounds were
released as CH4N, HCN and NOx. Lignocellulosic biomass combustion was fitted to a
first order reaction model (O1). The combustion behavior of marine biomass was
carried out by TGA-DSC-MS. Three different types of microalgae
(Nannochloropsisgaditana(NG), Scenedesmusalmeriensis(SC) and Chlorella vulgaris)
were selected due to their chemical composition. Combustion of microalgae took
place into two main stages: devolatilization stage and oxidation stage. However, up to
three sub-steps could be identified during the microalgae combustion attributed to the
decomposition of carbohydrates, proteins and lipids. The ignition characteristic
showed that samples CV and SC required less amount of energy to develop the
combustion process. However, NG sample released a higher amount of heat during the
combustion. The kinetic analysis of microalgae combustion showed that the most
representative mechanism for representing the process was a first order reaction model
(O1). The excellent fitting between the experimental and theoretical curves (maximum
mean error was 3.1%, for NG sample) confirmed the selection of model O1. CO, CO2
and H2O were the main products released during combustion. Other compounds
detected during the combustion of microalgae were light hydrocarbons (especially
CH4); nitrogen compounds (mainly released as NO, NO2 and HCN); sulfur
compounds (SO, SO2 and COS); hydrogen and other oxygen containing hydrocarbons

Chapter 3
200
(ketones, esters, ethers and carboxylic acids). Nitrogen compounds were found in
higher proportions than sulfur ones.
Combustion behavior and gas formation from the oxidation process of two woody
crops (black spruce and Pinusbanksiana mixtures (BP) and willow (W)), and three
herbaceous non-perennial energy crops (common reed (CR), reed phalaris (RP) and
switchgrass (S)) were studied by TGA-DSC-MS. Samples W and RP showed the best
burning profile by combining a high combustion characteristic factor (CCF) and a
high release of combustion heat (Hcomb). The kinetic analyses of the oxidation process
was performed using pseudo mulit-component separate-stage models (PMSM). The
combustion process was divided into three stages: Devolatilization stage (correlated
with the hemicellulose and cellulose content in the samples), Oxidation stage
(influenced by the initial amount of lignin in the samples) and Remaining burning
(associated to the final char burning and devolatilization of inorganic matter). The
high ash content of CR sample enhanced the amount of volatiles released during the
combustion process lowering its activation energy. The good fitting of experimental
curves with theoretical ones validated the proposed model (mean error below 3.4 %).
H2, CO and CO2were the main product obtained from energy crops combustion
process. Furthermore, NOx were detected in a higher proportion than other pollutants
such as SOx, chloride compounds (CH3Cl) or aromatic ones (C6H6).
3.5. References
[1] Shen, D.K., Gu, S., Luo, K.H., Bridgwater, A.V., Fang, M.X. 2009. Kinetic study
on thermal decomposition of woods in oxidative environment. Fuel, 88(6), 1024-30.
[2] Gil, M.V., Casal, D., Pevida, C., Pis, J.J., Rubiera, F. 2010. Thermal behaviour
and kinetics of coal/biomass blends during co-combustion. Bioresource Technology,
101(14), 5601-8.
[3] Kai, X., Yang, T., Huang, Y., Sun, Y., He, Y., Li, R. 2011. The effect of biomass
components on the co-combustion characteristics of biomass with coal. Zhangjiajie,
Hunan. pp. 1274-8.

Chapter 3
201
[4] Stenseng, M., Zolin, A., Cenni, R., Frandsen, F., Jensen, A., Dam-Johansen, K.
2001. Thermal analysis in combustion research. Journal of Thermal Analysis and
Calorimetry, 64(3), 1325-34.
[5] Cheng, K., Winter, W.T., Stipanovic, A.J. 2012. A modulated-TGA approach to
the kinetics of lignocellulosic biomass pyrolysis/combustion. Polymer Degradation
and Stability, 97(9), 1606-15.
[6] Barneto, A.G., Carmona, J.A., Alfonso, J.E.M., Blanco, J.D. 2009. Kinetic models
based in biomass components for the combustion and pyrolysis of sewage sludge and
its compost. Journal of Analytical and Applied Pyrolysis, 86(1), 108-14.
[7] Yorulmaz, S.Y., Atimtay, A.T. 2009. Investigation of combustion kinetics of
treated and untreated waste wood samples with thermogravimetric analysis. Fuel
Processing Technology, 90(7-8), 939-46.
[8] Cordero, T., Rodríguez-Maroto, J.M., Rodríguez-Mirasol, J., Rodríguez, J.J. 1990.
On the kinetics of thermal decomposition of wood and wood components.
Thermochimica Acta, 164(C), 135-44.
[9] Phukan, M.M., Chutia, R.S., Konwar, B.K., Kataki, R. 2011. Microalgae
Chlorella as a potential bio-energy feedstock. Applied Energy, 88(10), 3307-12.
[10] Naik, S., Goud, V.V., Rout, P.K., Jacobson, K., Dalai, A.K. 2010.
Characterization of Canadian biomass for alternative renewable biofuel. Renewable
Energy, 35(8), 1624-31.
[11] Hood, E.E., Nelson, P., Powell, R. 2011. Plant Biomass Conversion. Wiley.
[12] Darvell, L.I., Brindley, C., Baxter, X.C., Jones, J.M., Williams, A. 2012.
Nitrogen in biomass char and its fate during combustion: A model compound
approach. Energy and Fuels, 26(11), 6482-91.
[13] NPRI. Environment Canada. National Pollutant Release Inventory-2010 NPRI.
[14] Miranda, T., Román, S., Montero, I., Nogales-Delgado, S., Arranz, J.I., Rojas,
C.V., González, J.F. 2012. Study of the emissions and kinetic parameters during
combustion of grape pomace: Dilution as an effective way to reduce pollution. Fuel
Processing Technology, 103, 160-5.

Chapter 3
202
[15] Jakab, E., Till, F., Várhegyi, G. 1991. Thermogravimetric-mass spectrometric
study on the low temperature oxidation of coals. Fuel Processing Technology, 28(3),
221-38.
[16] Heikkinen, J.M., Hordijk, J.C., De Jong, W., Spliethoff, H. 2004.
Thermogravimetry as a tool to classify waste components to be used for energy
generation. Journal of Analytical and Applied Pyrolysis, 71(2), 883-900.
[17] White, J.E., Catallo, W.J., Legendre, B.L. 2011. Biomass pyrolysis kinetics: A
comparative critical review with relevant agricultural residue case studies. Journal of
Analytical and Applied Pyrolysis, 91(1), 1-33.
[18] Coats, A.W., Redfern, J.P. 1964. Kinetic parameters from thermogravimetric
data Nature, 201(4914), 68-9.
[19] Liu, N.A., Fan, W., Dobashi, R., Huang, L. 2002. Kinetic modeling of thermal
decomposition of natural cellulosic materials in air atmosphere. Journal of Analytical
and Applied Pyrolysis, 63(2), 303-25.
[20] Zhou, L., Wang, Y., Huang, Q., Cai, J. 2006. Thermogravimetric characteristics
and kinetic of plastic and biomass blends co-pyrolysis. Fuel Processing Technology,
87(11), 963-9.
[21] Sánchez-Silva, L., Gutiérrez, N., Romero, A., Sánchez, P., Valverde, J.L. 2012.
Pyrolysis and combustion kinetics of microcapsules containing carbon nanofibers by
thermal analysis-mass spectrometry. Journal of Analytical and Applied Pyrolysis, 94,
246-52.
[22] Li, X.G. 1999. High-Resolution Thermogravimetry of Cellulose Esters. Journal
of Applied Polymer Science, 71(4), 573-8.
[23] Sanchez-Silva, L., López-González, D., Villaseñor, J., Sánchez, P., Valverde,
J.L. 2012. Thermogravimetric-mass spectrometric analysis of lignocellulosic and
marine biomass pyrolysis. Bioresource Technology, 109, 163-72.
[24] Haykiri-Açma, H. 2003. Combustion characteristics of different biomass
materials. Energy Conversion and Management, 44(1), 155-62.

Chapter 3
203
[25] Gani, A., Naruse, I. 2007. Effect of cellulose and lignin content on pyrolysis and
combustion characteristics for several types of biomass. Renewable Energy, 32(4),
649-61.
[26] Wang, S., Jiang, X.M., Han, X.X., Liu, J.G. 2009. Combustion characteristics of
seaweed biomass. 1. combustion characteristics of enteromorpha clathrata and
sargassum natans. Energy and Fuels, 23(10), 5173-8.
[27] Williams, P.T., Besler, S. 1996. The Influence of Temperature and Heating Rate
on the Slow Pyrolysis of Biomass. Renewable Energy, 7(3), 233-50.
[28] Gadalla, A.M.M. 1984. Kinetics of the decomposition of hydrated oxalates of
calcium and magnesium in air. Thermochimica Acta, 74(1-3), 255-72.
[29] Xie, X., Goodell, B., Zhang, D., Nagle, D.C., Qian, Y., Peterson, M.L., Jellison,
J. 2009. Characterization of carbons derived from cellulose and lignin and their
oxidative behavior. Bioresource Technology, 100(5), 1797-802.
[30] Amen-Chen, C., Pakdel, H., Roy, C. 2001. Production of monomeric phenols by
thermochemical conversion of biomass: a review. Bioresource Technology, 79(3),
277-99.
[31] Liu, Q., Wang, S., Zheng, Y., Luo, Z., Cen, K. 2008. Mechanism study of wood
lignin pyrolysis by using TG-FTIR analysis. Journal of Analytical and Applied
Pyrolysis, 82(1), 170-7.
[32] Li, C., Brown, T.C. 2001. Carbon oxidation kinetics from evolved carbon oxide
analysis during temperature-programmed oxidation. Carbon, 39(5), 725-32.
[33] Shen, D., Hu, J., Xiao, R., Zhang, H., Li, S., Gu, S. 2013. Online evolved gas
analysis by Thermogravimetric-Mass Spectroscopy for thermal decomposition of
biomass and its components under different atmospheres: Part I. Lignin. Bioresource
Technology, 130, 449-56.
[34] Widyawati, M., Church, T.L., Florin, N.H., Harris, A.T. 2011. Hydrogen
synthesis from biomass pyrolysis with in situ carbon dioxide capture using calcium
oxide. International Journal of Hydrogen Energy, 36(8), 4800-13.

Chapter 3
204
[35] Darvell, L.I., Jones, J.M., Gudka, B., Baxter, X.C., Saddawi, A., Williams, A.,
Malmgren, A. 2010. Combustion properties of some power station biomass fuels.
Fuel, 89(10), 2881-90.
[36] Idris, S.S., Rahman, N.A., Ismail, K. 2012. Combustion characteristics of
Malaysian oil palm biomass, sub-bituminous coal and their respective blends via
thermogravimetric analysis (TGA). Bioresource Technology, 123, 581-91.
[37] Wang, C., Wang, F., Yang, Q., Liang, R. 2009. Thermogravimetric studies of the
behavior of wheat straw with added coal during combustion. Biomass and Bioenergy,
33(1), 50-6.
[38] Tang, Y., Ma, X., Lai, Z. 2011. Thermogravimetric analysis of the combustion of
microalgae and microalgae blended with waste in N2/O2 and CO2/O2 atmospheres.
Bioresource Technology, 102(2), 1879-85.
[39] Chen, C., Ma, X., Liu, K. 2011. Thermogravimetric analysis of microalgae
combustion under different oxygen supply concentrations. Applied Energy, 88(9),
3189-96.
[40] Kebelmann, K., Hornung, A., Karsten, U., Griffiths, G. 2013. Intermediate
pyrolysis and product identification by TGA and Py-GC/MS of green microalgae and
their extracted protein and lipid components. Biomass and Bioenergy, 49, 38-48.
[41] Ross, A.B., Jones, J.M., Kubacki, M.L., Bridgeman, T. 2008. Classification of
macroalgae as fuel and its thermochemical behaviour. Bioresource Technology,
99(14), 6494-504.
[42] Shuping, Z., Yulong, W., Mingde, Y., Chun, L., Junmao, T. 2010. Pyrolysis
characteristics and kinetics of the marine microalgae Dunaliella tertiolecta using
thermogravimetric analyzer. Bioresource Technology, 101(1), 359-65.
[43] Bridgeman, T.G., Darvell, L.I., Jones, J.M., Williams, P.T., Fahmi, R.,
Bridgwater, A.V., Barraclough, T., Shield, I., Yates, N., Thain, S.C., Donnison, I.S.
2007. Influence of particle size on the analytical and chemical properties of two
energy crops. Fuel, 86(1-2), 60-72.

Chapter 3
205
[44] Li, D., Chen, L., Chen, S., Zhang, X., Chen, F., Ye, N. 2012. Comparative
evaluation of the pyrolytic and kinetic characteristics of a macroalga (Sargassum
thunbergii) and a freshwater plant (Potamogeton crispus). Fuel, 96, 185-91.
[45] Wang, S., Jiang, X.M., Han, X.X., Liu, J.G. 2009. Combustion Characteristics of
Seaweed Biomass. 1. Combustion Characteristics of Enteromorpha clathrata and
Sargassum natans. Energy & Fuels, 23(10), 5173-8.
[46] Kok, M.V., Özgür, E. 2013. Thermal analysis and kinetics of biomass samples.
Fuel Processing Technology, 106(0), 739-43.
[47] Yu, L.J., Wang, S., Jiang, X.M., Wang, N., Zhang, C.Q. 2008. Thermal analysis
studies on combustion characteristics of seaweed. Journal of Thermal Analysis and
Calorimetry, 93(2), 611-7.
[48] Wang, S., Jiang, X.M., Wang, N., Yu, L.J., Li, Z., He, P.M. 2007. Research on
pyrolysis characteristics of seaweed. Energy and Fuels, 21(6), 3723-9.
[49] Marcilla, A., Gómez-Siurana, A., Gomis, C., Chápuli, E., Catalá, M.C., Valdés,
F.J. 2009. Characterization of microalgal species through TGA/FTIR analysis:
Application to nannochloropsis sp. Thermochimica Acta, 484(1-2), 41-7.
[50] Babich, I.V., van der Hulst, M., Lefferts, L., Moulijn, J.A., O'Connor, P., Seshan,
K. 2011. Catalytic pyrolysis of microalgae to high-quality liquid bio-fuels. Biomass
and Bioenergy, 35(7), 3199-207.
[51] Marcilla, A., Gómez-Siurana, A., Gomis, C., Chápuli, E., Catalá, M.C., Valdés,
F.J. 2009. Characterization of microalgal species through TGA/FTIR analysis:
Application to nannochloropsis sp. Thermochimica Acta, 484(1–2), 41-7.
[52] Bae, Y.J., Ryu, C., Jeon, J.K., Park, J., Suh, D.J., Suh, Y.W., Chang, D., Park,
Y.K. 2011. The characteristics of bio-oil produced from the pyrolysis of three marine
macroalgae. Bioresource Technology, 102(3), 3512-20.
[53] Li, D., Chen, L., Zhang, X., Ye, N., Xing, F. 2011. Pyrolytic characteristics and
kinetic studies of three kinds of red algae. Biomass and Bioenergy, 35(5), 1765-72.
[54] Jones, J.M., Harding, A.W., Brown, S.D., Thomas, K.M. 1995. Detection of
reactive intermediate nitrogen and sulfur species in the combustion of carbons that are
models for coal chars. Carbon, 33(6), 833-43.

Chapter 3
206
[55] Tapasvi, D., Khalil, R., Várhegyi, G., Skreiberg, O., Tran, K.Q., Grønli, M.
2013. Kinetic behavior of torrefied biomass in an oxidative environment. Energy and
Fuels, 27(2), 1050-60.
[56] Yang, H., Yan, R., Chen, H., Zheng, C., Lee, D.H., Liang, D.T. 2006. In-depth
investigation of biomass pyrolysis based on three major components: Hemicellulose,
cellulose and lignin. Energy and Fuels, 20(1), 388-93.
[57] Leite, B.S., Andreuccetti, M.T., Leite, S.A.F., d'Angelo, J.V.H. 2012. TG and
DSC analyses of eucalyptus black liquor as alternative methods to estimate solids
content. Journal of Thermal Analysis and Calorimetry, 1-6.
[58] Fang, M.X., Shen, D.K., Li, Y.X., Yu, C.J., Luo, Z.Y., Cen, K.F. 2006. Kinetic
study on pyrolysis and combustion of wood under different oxygen concentrations by
using TG-FTIR analysis. Journal of Analytical and Applied Pyrolysis, 77(1), 22-7.
[59] Miranda, T., Román, S., Montero, I., Nogales-Delgado, S., Arranz, J.I., Rojas,
C.V., González, J.F. 2012. Study of the emissions and kinetic parameters during
combustion of grape pomace: Dilution as an effective way to reduce pollution. Fuel
Processing Technology, 103(0), 160-5.
[60] Hansson, K.-M., Samuelsson, J., Tullin, C., Åmand, L.-E. 2004. Formation of
HNCO, HCN, and NH3 from the pyrolysis of bark and nitrogen-containing model
compounds. Combustion and Flame, 137(3), 265-77.
[61] Fahmi, R., Bridgwater, A.V., Darvell, L.I., Jones, J.M., Yates, N., Thain, S.,
Donnison, I.S. 2007. The effect of alkali metals on combustion and pyrolysis of
Lolium and Festuca grasses, switchgrass and willow. Fuel, 86(10-11), 1560-9.

Chapter 4:
GASIFICATION OF LIGNOCELLULOSIC BIOMASS CHAR OBTAINED FROM
PYROLYSIS: KINETIC AND EVOLVED
GAS ANALYSES
The pyrolysis and gasification process of three types of
lignocellulosic biomass (Eucalyptus wood, fir wood and pine bark)
and biomass main components (cellulose, xylan and lignin) were
studied by thermogravimetric-mass spectrometric analysis (TGA-
MS). Pyrolysis was used to obtain a solid fuel (char) that was later
gasified with steam. The reactivity profiles of the studied biomass
samples showed a clear catalytic effect at high conversion values
which was directly correlated with their ash composition.
Gasification apparent rates were obtained by a preliminary
kinetic analysis. Three standard models were used to reproduce the
gasification process. Only cellulose and pine bark samples
obtained an accurate fitting being attributed to their low ash

Chapter 4
209
content and subsequently low catalytic activity. A semi-empirical
model, based on the catalytic effect observed, was proposed which
highly improved the obtained fitting. H2, CO and CO2 were the
main products obtained. Furthermore, secondary products such as
CH4 pointed out the existence of methanation reactions. NOX were
also observed indicating that part of the initial nitrogen in the
sample was retained in the char after the pyrolysis process.
4.1. INTRODUCTION
The production of clean and sustainable fuels arethe main challenges to tackle
upcoming energy crises and global warming [1]. Among all renewable energy sources,
biomass fuels are gaining particular attention as a potential alternative to increase
energy independence on fossil fuels and reduce environmental pollution [2, 3].
Thermochemical conversion of biomass is the most promising route for biomass
utilization. These processes mainly include the direct combustion, to generate heat and
electricity, pyrolysis and gasification, to produce liquid and gaseous fuels which are
suitable for feeding efficient gas engines and gas turbines [4, 5].Pyrolysis plays an
important role in these processes,being the first chemical step in both gasification and
combustion processes. Generally, pyrolysis can be considered as a two-stage process
involving the devolatilization of biomass and the slow heterogeneous conversion of
char [5]. The char generated during pyrolysis is a high energy-density solid fuel
suitable for combustion and gasification processes.
Gasification is of special interest due to the fact that it is compatible with new
applications in the area of biomass conversion coal to liquid and superior
environmental performance especially with regard to CO2 capture and sulfur removal.
Furthermore, it is economical in a wide range of capacities (5 kWe onwards) [1, 6].
Gasificationcan be defined as the conversion of biomass to a gaseous fuel by heating
in a gasification medium such as air, oxygen or steam involving a complex set of
reactions [7, 8]. Char gasification is an important step during thermochemical

Chapter 4
210
conversion of lignocellulosic biomass because it often represents the rate-controlling
phenomenon in the gasifier[9]. The progress of char gasification is a function of
several factors such as particle size, porosity, gasifying agent chemical composition,
gasifying agent partial pressure, reactor temperature, pore structure, number of active
sites and ash content, among others[10].
Thermogravimetric analysis (TGA) has been commonly used for the study of the
thermochemical conversion of biomass [11]. Compared to pyrolysis and combustion,
there are few works reported in literature concerning the gasification of biomass. Most
works have been focused on the study of coals. In this regard,Shabbar et al. [1]
performed a thermodynamic analysis of bituminous coals. Furthermore, Tay et al. [7]
studied the effect of different gasifying agents on the char structure of Victorian
brown coal during the gasification. On the other hand, few studies have been reported
on the gasification of biomass. Mohammed et al. [12]evaluated the thermal
characteristics and kinetics of empty fruit bunches. Additionally, different authors
have investigated the carbon dioxide gasification of biomass chars [13, 14].
During the process of thermochemical conversion of biomass, the composition of
the gas emissions should be determined before industrial application. Complementary
techniques to TGA must be used in order to obtain qualitative information of biomass
transformation during the analyses. Very few studies have been found in literature
concerning gas evolution from biomass gasification. Yang et al. [15] studied the steam
gasification of tobacco by TGA coupled with gas chromatography (GC). Furthermore,
Yoon et al. [16] used TGA-GC to perform an kinetic analysis of thewoody biomass
gasification. In this sense, the use of TGA coupled with mass spectrometry (TGA-MS)
can give a deeper insight of the gasification process being able to afford real-time and
sensitive detection of evolved gases during the thermal analysis [17] .
The aim of this work was to study the pyrolysis and gasification of different types
of lignocellulosic biomass (fir wood, eucalyptus wood and pine bark) and their main
components (cellulose, hemicellulose and lignin) by means of TGA. The pyrolysis of
biomass samples was carried out to obtain a solid fuel (char) which was later gasified

Chapter 4
211
using steam. In addition, a preliminary kinetic analysis of the gasification process was
performed in order to obtain the apparent gasification rates. Finally, the gases released
during the gasification process were analyzed by MS.
4.2. EXPERIMENTAL
4.2.1. Materials
Cellulose, xylan and lignin were purchased from Sigma-Aldrich. Xylanwas used as
representative of that of the hemicellulose. These chemicals are as follow: cellulose
(microcrystaline cellulose with 50 µm average particle size), lignin (alkali lignin in
brown powder form with 50 µm average particle size) and xylan (xylan processed
from beechwood with 100 µm average particle size). The selected terrestrial
biomasses (fir wood, eucalyptus wood and pine bark) were taken from the region of
Castilla-La Mancha (Spain) on the basis of a preliminary analysis [18]. These samples
were dried in an oven for 5 h, milled and sieved to an average particle size between
100-150 µm.
The proximate analysis, ultimate analysis and composition of biomass samples are
shown in Table 1. The metal content in samples was determined by Inductively
Coupled Plasma Spectrometry (ICP) (Table 1). The content of hemicellulose, lignin
and xylan inlignocellulosic biomass samples was calculated according to the method
reported elsewhere [19] (Table 1).
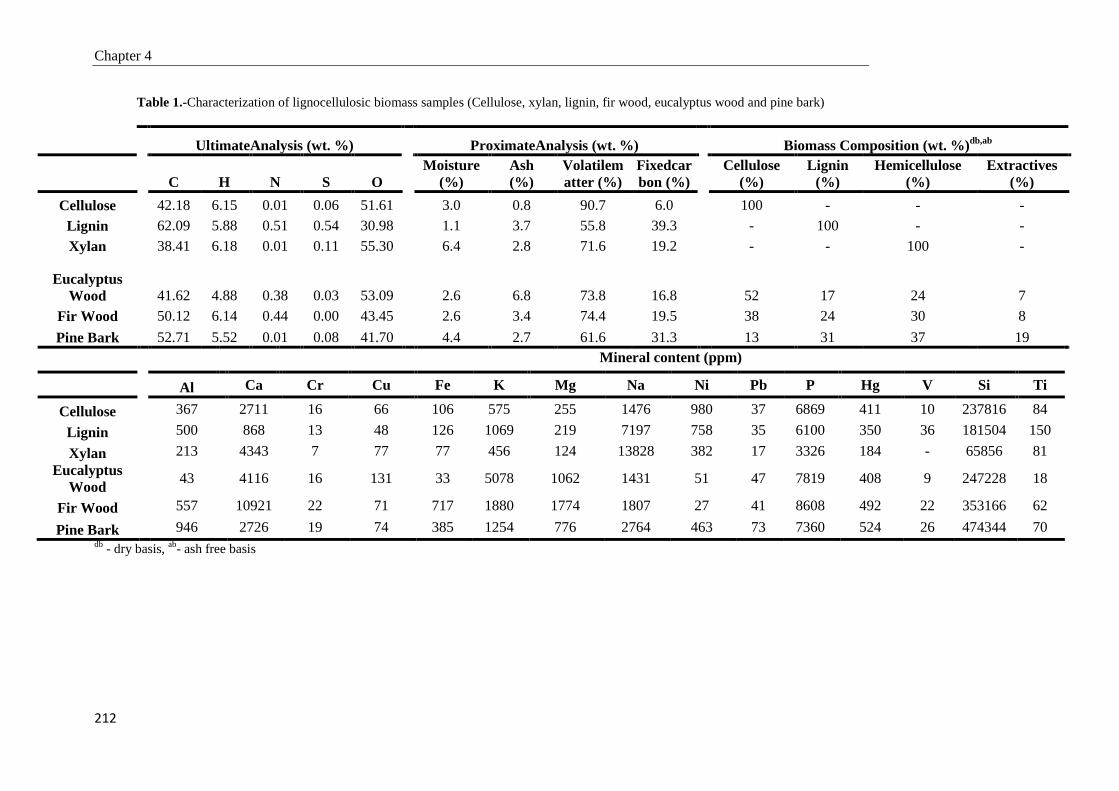
Chapter 4
212
Table 1.-Characterization of lignocellulosic biomass samples (Cellulose, xylan, lignin, fir wood, eucalyptus wood and pine bark)
UltimateAnalysis (wt. %) ProximateAnalysis (wt. %) Biomass Composition (wt. %)db,ab
C H N S O Moisture
(%) Ash (%)
Volatilematter (%)
Fixedcarbon (%)
Cellulose (%)
Lignin (%)
Hemicellulose (%)
Extractives (%)
Cellulose 42.18 6.15 0.01 0.06 51.61 3.0 0.8 90.7 6.0 100 - - -
Lignin 62.09 5.88 0.51 0.54 30.98 1.1 3.7 55.8 39.3 - 100 - -
Xylan 38.41 6.18 0.01 0.11 55.30 6.4 2.8 71.6 19.2 - - 100 -
Eucalyptus Wood
41.62 4.88 0.38 0.03 53.09
2.6 6.8 73.8 16.8
52 17 24 7
Fir Wood 50.12 6.14 0.44 0.00 43.45 2.6 3.4 74.4 19.5 38 24 30 8
Pine Bark 52.71 5.52 0.01 0.08 41.70 4.4 2.7 61.6 31.3 13 31 37 19
Mineral content (ppm)
Al Ca Cr Cu Fe K Mg Na Ni Pb P Hg V Si Ti
Cellulose 367 2711 16 66 106 575 255 1476 980 37 6869 411 10 237816 84
Lignin 500 868 13 48 126 1069 219 7197 758 35 6100 350 36 181504 150
Xylan 213 4343 7 77 77 456 124 13828 382 17 3326 184 - 65856 81
Eucalyptus Wood
43 4116 16 131 33 5078 1062 1431 51 47 7819 408 9 247228 18
Fir Wood 557 10921 22 71 717 1880 1774 1807 27 41 8608 492 22 353166 62
Pine Bark 946 2726 19 74 385 1254 776 2764 463 73 7360 524 26 474344 70
db - dry basis, ab- ash free basis

Chapter 4
213
4.2.2. Equipment and Procedures
4.2.2.1. Thermogravimetric analysis for the combustion process
The pyrolysis and gasification of biomass components was carried out in a TGA
apparatus (TGA-DSC 1, METTLER TOLEDO). The experimental setup used for the
gasification experiments was described in a previous study[20]. Steam was generated
in a bubbler system.Ar was bubbled through degassed water heated to 33 ºC.
Assuming the Ar-H2O mixture was saturated, a current with approximately 5% Vol. of
H2O in Ar was obtained. The pyrolysis of the sample was carried out by preheating the
sample at 105 ºC and then kept at 105 ºC for 10 min to remove the moisture content.
Subsequently, the sample was heated from 105 to 1000 ºC at 40 ºC/min under a 200
Nml/min of Ar. The temperature was kept at 1000 ºC for 10 min to ensure the
completion of the pyrolysis reaction. The sample was then cooled down to the
gasification temperature (900 ºC). The gasification step was carried outunder
isothermal conditions until the entire char was consumed. Previous studies were
carried out according to Sanchez-Silva et al. in order to avoid the effects of heat and
mass transfer limitations[20]. In this sense, initial sample weight was kept at 20 mg,
the particle size was kept in the 100-150 µm range and a constant flow rate of 200 and
50Nml/min were used for pyrolysis and gasification experiments, respectively.
4.2.2.2. TGA-MS analysis of the Gaseous Products
The analysis of the gas products distribution coming from the thermal analysis was
carried out in a thermogravimetric analyzer (TGA-DSC 1; METTLER TOLEDO)
coupled to a mass spectrometer (Thermostar-GSD 320/quadrupole mass analyzer;
PFEIFFER VACUUM) with an electron ionization voltage at 70 eV and provided
mass spectra up to 300 a.m.u. The interface was wrapped with heating wire to
circumvent condensation of exhausting gases. In order to identify ions with m/z in the
range 0-300, a preliminary broad scan was performed at a heating rate of 40 ºC/min.
Although a quantitative analysis was not performed in this work, a comparison of
the intensity peak areas between different samples (semiqualitative analysis) was

Chapter 4
214
carried out by using a normalization procedure. The ion intensities were normalized to
the total ion current to eliminate systematic instrumental errors caused by the
fluctuation of carrier gas flow, the shift in the sensitivity of the mass spectrometer
method and the sample weight. The evolved gas peak areas are useful for comparing
relative amounts of products from different samples [21].
4.2.2.3. Kinetic analysis
In this work, the char gasification was considered as an overall reaction, and a
general kinetic expression can be written as follows [22]:
����� = �(�, �) ∙ �(�) (1)
where k is the apparent gasification reaction rate, which includes the effect of
temperature, T, and the effect of the gasifying agent concentration, Pw, and f(α)
described the changes in the physical or chemical properties of the sample as the
gasification proceeds[22].
Three different modelswere used in this work, the volumetric model (VM) (Eq. 2), the
shrinking core model (SCM) (Eq. 3) and the random pore model (RPM) (Eq. 4):
�(�) = (1 − �) (2)
�(�) = (1 − �)�� (3)
�(�) = (1 − �) ∙ �1 − � ∙ ln(1 − �) (4)
whereα is the degree of conversion and Ψ is a parameter related with the initial pore
structure of the sample (α=0).
For the parameter estimation a VBA-Excel application was developed to solve this
model [23-25]. The Runge-Kutta-Fehlberg methodwas used in the evaluation of the
set of ordinary differential equations. Furthermore, the statistical significance of the
estimated parameters based on the F-test and t-test was performed according to the
procedure described elsewhere [18].

Chapter 4
215
4.3. RESULTS AND DISCUSSION
4.3.1. Thermogravimetric analysis
In this work, the pyrolysis of biomass samples was carried out to obtain a solid fuel
(char) which was later gasified using steam. The pyrolysis of biomass samples was
studied previously and described elsewhere [18]. Thus, a comprehensive evaluation of
the pyrolysis process of lignocellulosic biomass was not the objective of the present
study. However, some remarks of this process are described in order to get a better
understanding of the char formation mechanism.
4.3.1.1. Pyrolysis and gasification of lignocellulosic biomass main components
Figures 1 shows the TGA/DTG profiles for the pyrolysis and gasification of
biomass main components (cellulose, xylan and lignin). Table 2 summarizes the most
relevant pyrolysis characteristics for all biomass samples (biomass main components
and lignocellulosic biomass).The pyrolysis of biomass main components took place
between 200 and 700 ºC as it can be seen from their DTG profile (Figure 1.b).Xylan
sample was the first one to decompose showing two peaks at 262 and 306 ºC. On the
other hand, cellulose sample decomposition took place in one stage between 220 and
500 ºC. The cellulose showed the highest weight loss rate (88 wt.%/min) beingthe
sample which released thehighest amount of volatiles. Finally, lignin decomposed
over the whole temperature range (215-700 ºC). Lignin thermal decomposition is a
slow carbonization process producing the highest amount of char (43 wt.%) compared
to that formed in xylan (28 wt.%) and cellulose (8 wt.%) pyrolysis.
Figure 1.c shows the DTG profiles for the steam gasification of the char produced
from biomass main components pyrolysis. It can be seen that the gasification of
biomass chars started as soon as the gasifying agent reached the surface of the char
particle. Lignin and xylan samples produced the most reactive char and it took around
26 and 29 min to be totally gasified whereas the char produced from cellulose sample
was decomposed at a lower rate (~120 min). These facts agreed well with those
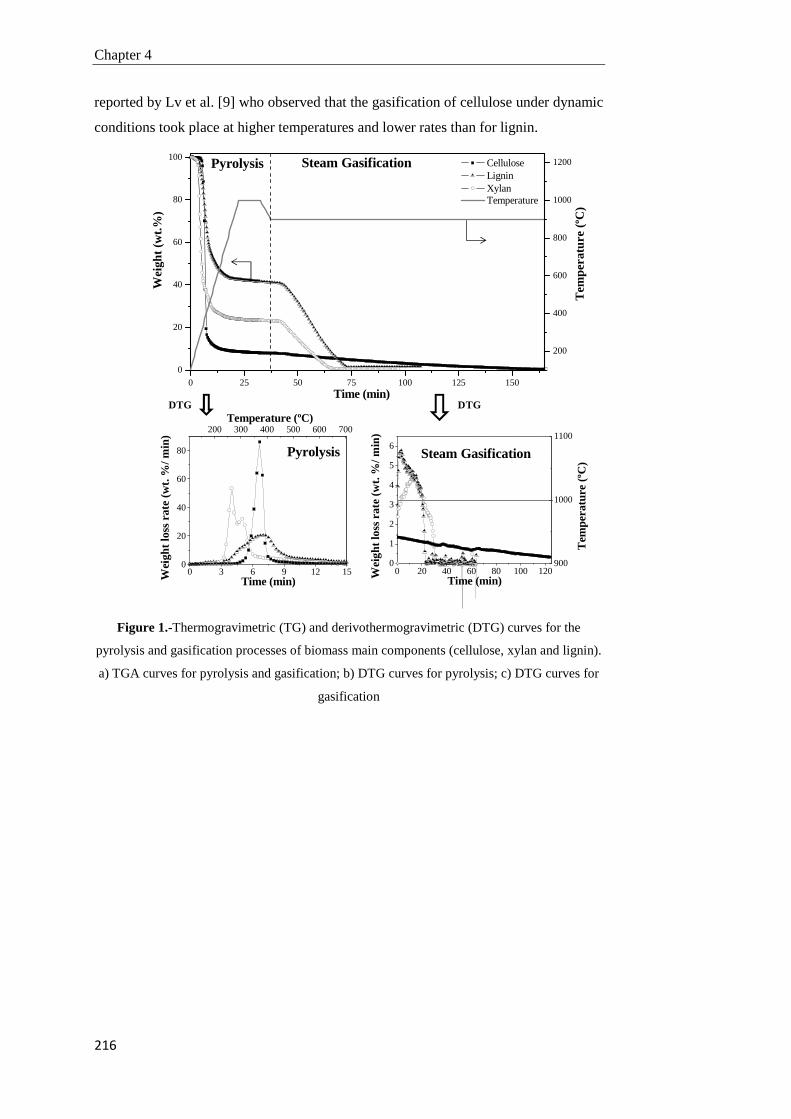
Chapter 4
216
0 25 50 75 100 125 1500
20
40
60
80
100
0 3 6 9 12 150
20
40
60
80
0 20 40 60 80 100 1200
1
2
3
4
5
6Pyrolysis Steam Gasification
Pyrolysis
Wei
ght (
wt.%
)
Time (min)
Cellulose Lignin Xylan
Steam Gasification
200
400
600
800
1000
1200
Temperature
Tem
pera
ture
(ºC
)
Wei
ght l
oss
rate
(w
t. %
/ min
)
Time (min) Wei
ght l
oss
rate
(w
t. %
/ min
)
Time (min)
900
1000
1100
Tem
pera
ture
(ºC
)
200 300 400 500 600 700 Temperature (ºC)
DTG DTG
reported by Lv et al. [9] who observed that the gasification of cellulose under dynamic
conditions took place at higher temperatures and lower rates than for lignin.
Figure 1.-Thermogravimetric (TG) and derivothermogravimetric (DTG) curves for the
pyrolysis and gasification processes of biomass main components (cellulose, xylan and lignin).
a) TGA curves for pyrolysis and gasification; b) DTG curves for pyrolysis; c) DTG curves for
gasification
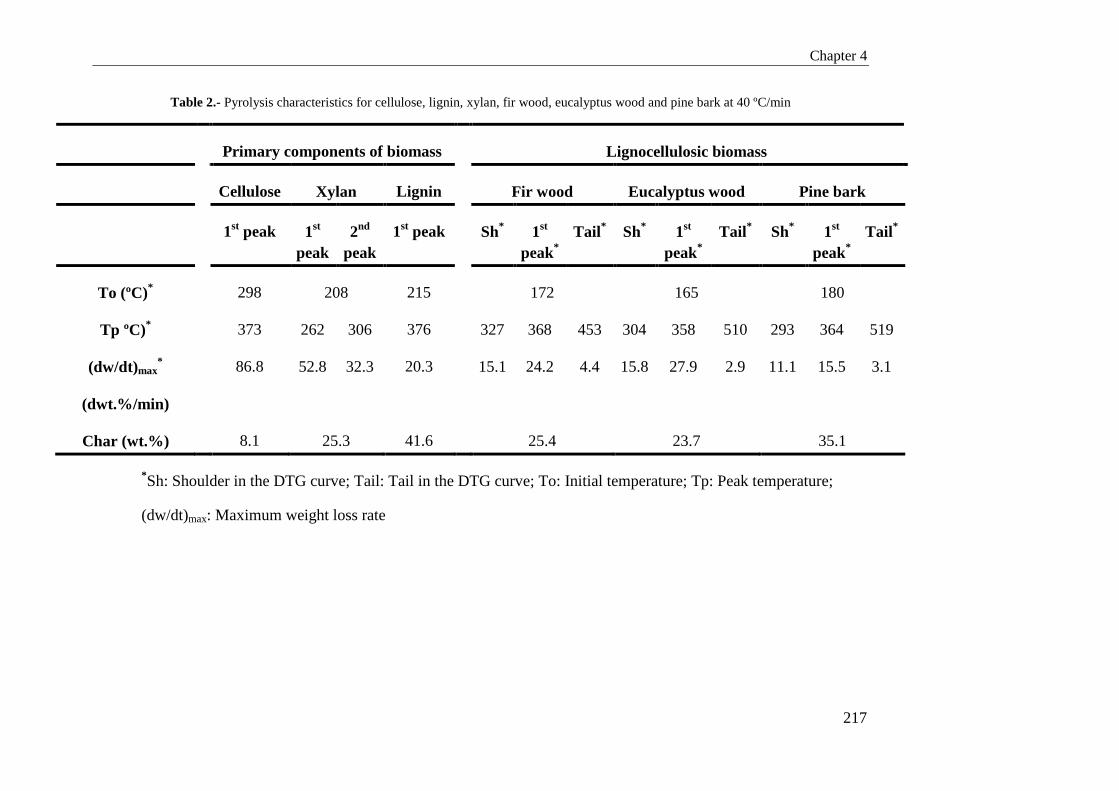
Chapter 4
217
Table 2.- Pyrolysis characteristics for cellulose, lignin, xylan, fir wood, eucalyptus wood and pine bark at 40 ºC/min
Primary components of biomass Lignocellulosic biomass
Cellulose Xylan Lignin Fir wood Eucalyptus wood Pine bark
1st peak 1st peak
2nd peak
1st peak Sh* 1st peak*
Tail * Sh* 1st peak*
Tail * Sh* 1st peak*
Tail *
To (ºC)* 298 208 215 172 165 180
Tp ºC)* 373 262 306 376 327 368 453 304 358 510 293 364 519
(dw/dt)max*
(dwt.%/min)
86.8 52.8 32.3
20.3 15.1 24.2 4.4 15.8 27.9 2.9 11.1 15.5 3.1
Char (wt.%) 8.1 25.3 41.6 25.4 23.7 35.1
*Sh: Shoulder in the DTG curve; Tail: Tail in the DTG curve; To: Initial temperature; Tp: Peak temperature;
(dw/dt)max: Maximum weight loss rate

Chapter 4
218
0 25 50 75 100 125 1500
20
40
60
80
100
0 3 6 9 12 150
5
10
15
20
25
30
0 20 40 60 80 100 1200
1
2
3
4
5
6
Pyrolysis Steam Gasification
Pyrolysis
Wei
ght (
wt.%
)
Time (min)
Pine bark Fir wood Eucalyptus wood
Steam Gasification
200
400
600
800
1000
1200
Temperature
Tem
pera
ture
(ºC
)
Wei
ght l
oss
rate
(w
t. %
/ min
)
Time (min) Wei
ght l
oss
rate
(w
t. %
/ min
)
Time (min)
900
1000
1100
Tem
pera
ture
(ºC
)
200 300 400 500 600 700 Temperature (ºC)
DTG DTG
4. 3.1.2. Pyrolysis and gasification of lignocellulosic biomass
Figures 2 shows the TGA/DTG profiles for the pyrolysis and gasification of
lignocellulosic biomass (fir wood, eucalyptus wood and pine bark). The shape of the
DTG curve was similar for all samples.
Figure 2.-Thermogravimetric (TG) and derivothermogravimetric (DTG) curves for the pyrolys
and gasification processes of lignocellulosic biomass (eucalyptus wood, fir wood and pine
bark). a) TGA curves for pyrolysis and gasification; b) DTG curves for pyrolysis; c) DTG
curves for gasification

Chapter 4
219
Firstly, a shoulder can be observed at temperatures around 300 ºC which is
attributed to hemicellulose decomposition. This shoulder was more sharped for pine
bark sample which is in agreement with its high hemicellulose content (Table 1).
Secondly, the maximum weight loss rate was observed at358, 364 and 368 ºC for
eucalyptus wood, pine bark and fir wood samples, respectively. This stage is ascribed
to cellulose decomposition. Eucalyptus wood sample showed the highest weight loss
rate (27 wt.%/min) compared to that for fir wood (24 wt.%/min) and pine bark (16
wt.%/min) samples due to its high cellulose content. Finally, the maximum peak was
followed by a wide tail which is essentially related to the lignin decomposition leading
to char formation [5]. As expected, the pine bark sample, that was the sample with the
highest lignin content, produced a higher amount of char (35 wt.%). On the other
hand, eucalyptus and fir wood samples, with a similar lignin content, generated a
similar char yield (25 and 24 wt.%, respectively). According to these evidences, the
mechanism of lignocellulosic biomass pyrolysis can be divided into two main stages:
devolatilization of raw biomass, where hemicellulose and cellulose mainly
decompose, and the slow carbonization of the remaining biomass, associated to lignin
decomposition turning into the production of the final char.
Figure 2.c shows the DTG profiles for the steam gasification of the char produced
from lignocellulosic biomass samples pyrolysis. Unlike their pyrolysis behavior, the
gasification of lignocellulosic biomass chars could not be described according to their
initial chemical composition. The eucalyptus wood sample, with the highest cellulose
content, was the one that needed the least time to be gasified. Additionally, the char
produced from pine bark sample pyrolysis, which was expected to decompose at lower
times than eucalyptus and fir wood samplesdue to its high hemicellulose and lignin
content, was the one thattook longer to be gasified. These results suggested that the
char formation from lignocellulosic biomass may be affected by the presence of other
components in the complex matrix of the wood. Therefore, the process cannot be
explained by considering the proportional interactionsbetween their main components.

Chapter 4
220
In this regard, the morphology of the formed char is a factor usually employed to
compare the reactivity of different chars [15].
Char conversion is more complicated than solid devolatilization as it is a
heterogeneous process where the surface is the location of the chemical reactions [5].
It is recognized that the heterogeneous rates of char conversion are determined by the
fundamental components, represented by surface area, surface accesibility, carbon
active sites and catalytic active sites created by indigenous or added inorganic matter,
and the local gaseous reactant concentration. Consequently, the reactive depends on
three chief characteristics of the sample: chemical structure, porosity and inorganic
constituents [5].Furthermore, the concentration of the gasifying agent also plays an
important role in the process. The two first factors might be influenced by the initial
cellulose, hemicellulose and lignin content in lignocellulosic samples. According to
Lv et al.[26], biomass rich in lignin component produced a high surface area and
porous charwhich makes easier the diffusion of the reactive agent turning intohigh
gasification rates. On the contrary, biomass with a high cellulose content produced a
fibrous structure char, lowering the char reactivity. However, this trend was not found
in the experimental results obtained in this study. Therefore, assuming that the
reactive gas concentration was kept constant in all experiments, these results pointed
out that the catalytic activity of the indigenous inorganic matter in the biomass played
a significant role in the gasification of the studied biomass samples.These results agree
well with those reported byXie et al. [27]who observed that specific surface areas and
porosities of lignin and cellulose char prepared at temperatures higher than 700ºC did
not have a meaningful role in the oxidative mass loss process. These results are
discussed in more detailed in the next sub-section.
4.3.1.3. Char Reactivity
The reactivity of char is an important parameter when evaluating the gasification
process. Several definitions were used to evaluate the char reactivity, however the
more extended one refers to the intrinsic reactivity (Ri) and it can be described as
follows [5, 22, 28, 29]:

Chapter 4
221
�� = −1 �� ∙ �� ��� = 11 − ��� ∙ ��� ��� (10)
wherexiand wiare the conversion and weight of charat any time, respectively. The
reactivity is dependent on the temperature and gas composition and varies with the
conversion degree [5, 30]. Thus, a representative value of reactivity must be presented
in order to make reliable comparisons. In this work, the reactivity at 50 % char
conversion is taken to be representative (R50) 28, 29][30, 31]. R50 values and the time
to achieve 100 % char conversion are summarized in Table 3. As aforementioned the
reactivity of biomass main components was ranked as: Xylan> Lignin>Cellulose. On
the other hand, lignocellulosic biomass samples was: Eucalyptus wood > Fir wood >
Pine bark.Thus, this order is not correlated with the biomass samples chemical
composition.
The gasification rate (ri) is also used to describe the gasification reaction and was
calculated by Eq. (11) [32]:
�� =�����
(11)
Figure 3 shows the typical reactivity and gasification rates versus conversion plots on
a comparative basis for biomass main components (cellulose, xylan and lignin) and
lignocellulosic biomass (fir wood, eucalyptus wood and pine bark) samples. It can be
observed that reactivity increase slowly up to conversion values of 0.8. However, for
xylan, lignin, eucalyptus and fir wood samples a sudden rise of reactivity took place
beyond 0.8 conversion, whereas pine bark and cellulose samples showed a lower
rise.This behavior can be explained by a high activity of the inorganic matter
contained in biomass samples[33]. As the gasification proceeds the carbon material is
consumed and the metal to carbon ratios increase which strengthen the catalytic effect
[33, 34].Furthermore, gasification rates versus conversion plots also corroborated this
fact. Pine bark and cellulose samples showed a decreasing trend and no maximum was
obtained whereas xylan, lignin, eucalyptus and fir wood samples showed a maximum.
This fact points out that the catalytic activity of indigenous inorganic matter

Chapter 4
222
increasethe gasification rate of xylan, lignin, eucalyptus and fir wood samples. This
reactivity profiles are similar to those reported by Blasi et al.[35] for the air
gasification of wheat straw, olive husks and grape residues.
Figure 3.-Reactivity versus conversion profiles for a) lignocellulosic biomass main
components (cellulose, xylan and lignin); b) lignocellulosic biomass (eucalyptus wood, fir
wood and pine bark).
Numerical indices such as the alkali index (A.I.) have been defined to describe the
catalytic efficiency of the overall influence of catalytically active species within the
ash [36]. This index is calculated as the ratio of the sum of the fraction of the basic
compounds (catalytic nature) in the ash (CaO, MgO, K2O, Na2O and Fe2O3) to the
fraction of the acidic compounds (non-catalytic nature) (Al2O3 and SiO2):
. " = #ℎ(��.%) ∙ ('()*+�)*,-)*.(�)*/0�)�)(12�)�*3�)�)
(12)
0.0
1.5
3.0
4.5
6.0
7.5
0.0 0.2 0.4 0.6 0.8 1.00.0
0.5
1.0
1.5
0.0
1.5
3.0
4.5
6.0
7.5
0.0 0.2 0.4 0.6 0.8 1.00.0
0.5
1.0
1.5
2.0
2.5
Gas
ifica
tion
rate
(1/
min
)
Rea
ctiv
ity (
1/m
in)
Conversion (X)
Cellulose Xylan Lignin
Gas
ifica
tion
rate
(1/
min
)
Rea
ctiv
ity (
1/m
in)
Conversion (X)
Eucalyptus wood Fir wood Pine bark
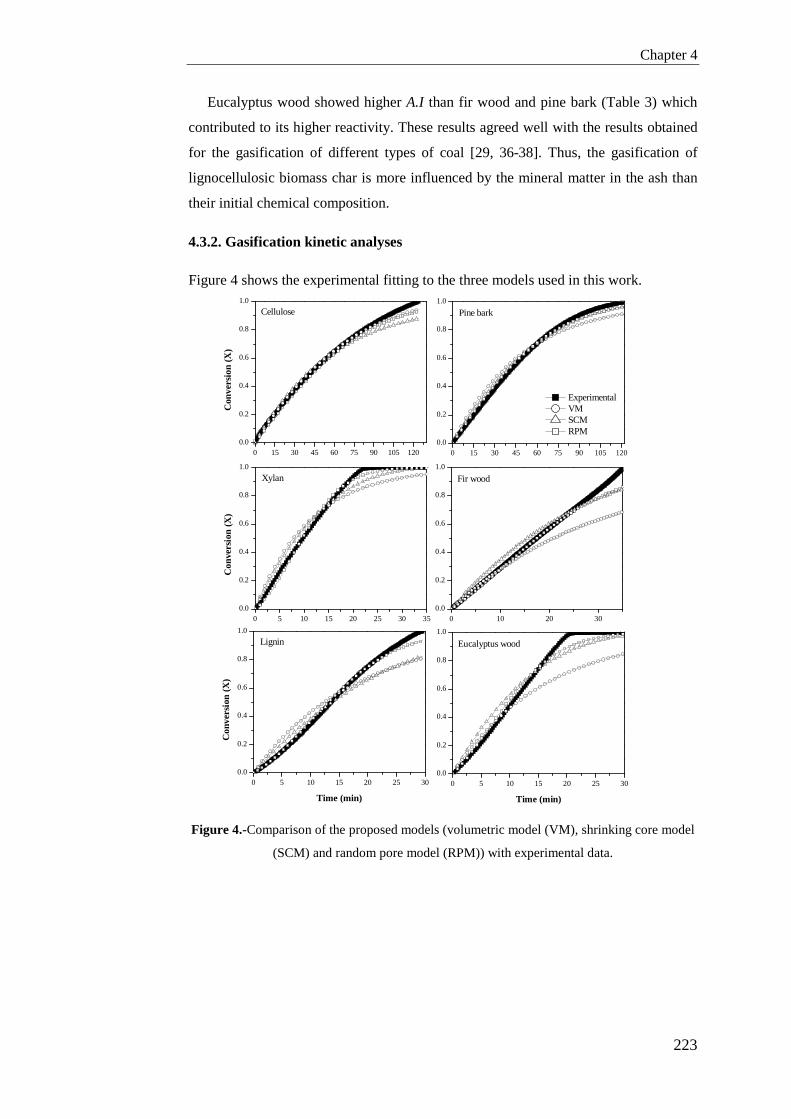
Chapter 4
223
0 15 30 45 60 75 90 105 1200.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 20 25 300.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 20 25 30 350.0
0.2
0.4
0.6
0.8
1.0
0 10 20 300.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 20 25 300.0
0.2
0.4
0.6
0.8
1.0
0 15 30 45 60 75 90 105 1200.0
0.2
0.4
0.6
0.8
1.0
Con
vers
ion
(X)
Cellulose
Con
vers
ion
(X)
Time (min)
Lignin
Con
vers
ion
(X)
Xylan
Fir wood
Time (min)
Eucalyptus wood
Experimental VM SCM RPM
Pine bark
Eucalyptus wood showed higher A.I than fir wood and pine bark (Table 3) which
contributed to its higher reactivity. These results agreed well with the results obtained
for the gasification of different types of coal [29, 36-38]. Thus, the gasification of
lignocellulosic biomass char is more influenced by the mineral matter in the ash than
their initial chemical composition.
4.3.2. Gasification kinetic analyses
Figure 4 shows the experimental fitting to the three models used in this work.
Figure 4.-Comparison of the proposed models (volumetric model (VM), shrinking core model
(SCM) and random pore model (RPM)) with experimental data.

Chapter 4
224
Table 4 summarized the obtained gasification constants and the statistical
significance of the models. In general, it can be observed that volumetric model (VM)
obtained the worst fitting to the experimental curves, whereas shrinking core model
(SCM) and random pore model (RPM) achieved a better fit. VM assumes a
homogeneous reaction throughout the particle and a linearly decreasing reaction
surface area with conversion [22, 28]. On the other hand, SCM and RPM describe the
evolution of the solid structure with conversion [34]. SCM assumes that a porous
particle consists of an assembly of uniform nonporous grains and the reaction takes
place on the surface of the grains assuming spherical shape of the porous. This model
predicts a monotonically decreasing reaction rate and surface area because the surface
area of each grain is receding during the gasification which agrees with profiles
described for cellulose and pine bark (Figure 3). Finally, the RPM considers the
overlapping of pore surfaces which reduces the area available for reaction[28]. This
model is able to predict a maximum for the reactivity as the reaction proceeds, as it
considers the competing effects of pore growth during the initial stages of gasification,
and the destruction of the pores due to the coalescence of neighboring pores.As it can
be seen from Figure 4, none of the models accurately predicted the biomass samples
behavior for conversion values greater than 0.8 except for cellulose and lignin
samples. Furthermore, it is clearly observed how these models clearly underpredicted
the conversion values. These results agree well with literature [22, 28, 34]. This
behavior is mainly due to the fact that these models fail to predict the catalytic activity
of the ash and are only valid under the chemical controlled regime [22]. The good
fitting of cellulose and pine bark sample to the proposed models, especially SCM,
corroborated the low activity behavior of their indigenous inorganic matter.Anyway,
curves predicted with SCM models obtained a slightly lower error than SCM for the
gasification of the biomass samples used in this work. In order to ensure the reliability
of the proposed models, the discrimination of kinetic parameters was done applying
the F-test and the t-test at the 95% confidence level [18] (Table 4). In terms of
statistical results, F-test considered the regression to be suitable in all cases since the
corresponding values to the Fc/Ftest ratio were larger than one. The t-test was also used

Chapter 4
225
for evaluating each parameter in the model. The values of tc/t-test ratio were also
larger than one, showing the statistical significance of the proposed models and their
corresponding parameters.
A semi-empirical model was proposed in order to obtain a model that accurately
reproduce the gasification process of biomass samples (xylan, lignin, eucalyptus
wood and pine bark). An expression representing the activity of biomass ashes at high
conversion values was added to the SCM. SCM was chosen due to two facts. Firstly,
the RPM includes an additional adjustable parameter (Ψ) which is difficult to be
measured and secondly, the error obtained was not too different than that obtained for
RPM.The proposed model include two parameters, an activation constant (ka) and
activation order (na) and is described as follows:
����� = �(�, �) ∙ (1 − �)
45� + �( ∙ �78 (13)

Chapter 4
226
Table 4.- Estimated parameters for the proposed models (VM, SCM and RPM) and their statistical analysis
Biomass samples Model Parameters tc ttest Fc Ftest Error (%)
Cellulose
VM 1.71 76 24723 3.84 6.8
SCM K (min -1)(·102) 1.48 55059 1.96 175522 3.84 1.1
RPM 1.21 69809 32677 3.00 5.1
Ψ 1.9 13991
Lignin
VM 5.58 2251 1602 3.84 31.7
SCM K (min -1)(·102) 4.48 98519 1.96 7025 3.84 22.5
RPM 2.14 38039 20194 3.00 8.9
Ψ 20.7 30254
Xylan
VM 8.81 2265 36796 3.84 19.0
SCM K (min -1)(·102) 7.32 89 1.96 110675 3.84 10.5
RPM 2.49 204 217283 3.00 8.8
Ψ 33.5 25350
Fir wood VM 3.31 60942 1102 3.84 17.7
SCM K (min -1)(·102) 3.98 10221 1.96 9177 3.84 11.8
RPM 2.29 20953 32749 3.00 8.8
Ψ 7.3 15236
Eucalyptus wood VM 6.26 4715 6744 3.84 18.9
SCM K (min -1)(·102) 7.23 64 1.96 44861 3.84 12.9
RPM 3.79 4458 73100 3.00 9.3
Ψ 10.2 27593
Pine bark
VM 2.08 1343 11357 3.84 13.7
SCM K (min -1)(·102) 1.67 10011 1.96 132563 3.84 2.6
RPM 1.46 301 32128 3.00 1.2
Ψ 1.8 294
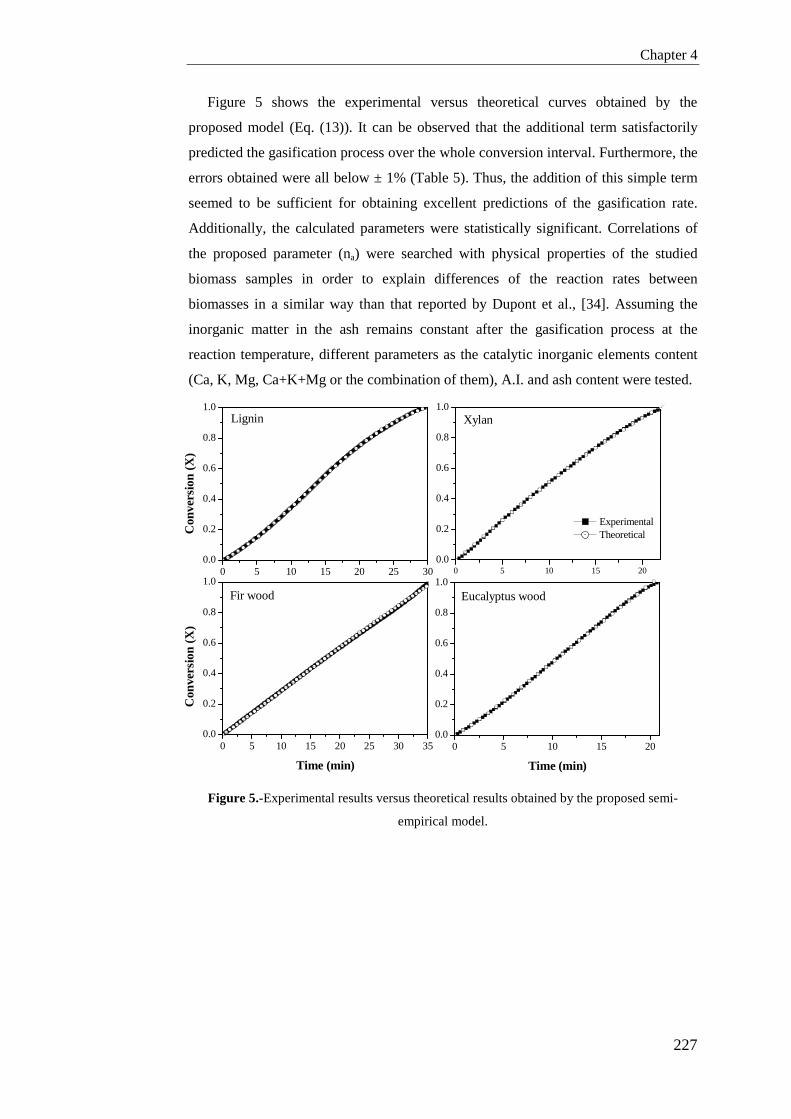
Chapter 4
227
0 5 10 15 20 25 300.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 200.0
0.2
0.4
0.6
0.8
1.0
0 5 10 15 20 25 30 350.0
0.2
0.4
0.6
0.8
1.00 5 10 15 20
0.0
0.2
0.4
0.6
0.8
1.0
Con
vers
ion
(X)
Lignin
Time (min)
Eucalyptus wood
Con
vers
ion
(X)
Time (min)
Fir wood
Experimental Theoretical
Xylan
Figure 5 shows the experimental versus theoretical curves obtained by the
proposed model (Eq. (13)). It can be observed that the additional term satisfactorily
predicted the gasification process over the whole conversion interval. Furthermore, the
errors obtained were all below ± 1% (Table 5). Thus, the addition of this simple term
seemed to be sufficient for obtaining excellent predictions of the gasification rate.
Additionally, the calculated parameters were statistically significant. Correlations of
the proposed parameter (na) were searched with physical properties of the studied
biomass samples in order to explain differences of the reaction rates between
biomasses in a similar way than that reported by Dupont et al., [34]. Assuming the
inorganic matter in the ash remains constant after the gasification process at the
reaction temperature, different parameters as the catalytic inorganic elements content
(Ca, K, Mg, Ca+K+Mg or the combination of them), A.I. and ash content were tested.
Figure 5.-Experimental results versus theoretical results obtained by the proposed semi-
empirical model.

Chapter 4
228
Table 5.- Estimated parameters for the proposed semi-empirical model and their statistical
analysis.
The parameter nashowed a really good linearity with K content with a correlation
coefficient higher than 0.97 (Figure 6). Thus, the proposed model can be generally
described as:
����� = �(�, �) ∙ (1 − �)
45� + �( ∙ �(9.4:;<'(=*5.;∙>9
?�) (14)
This correlationship of the proposed parameters with active catalytic species might
help to understand the role of the ashes in the gasification process. However, future
work must be carried out in order to understand the process from a phenomenological
point of view.
Biomass
samples
Parameter tc ttest Fc Ftest Error
(%)
Lignin k a 1.54·10-2 141 1.96 5282310 2.37 0.1
na 0.94 221
Xylan ka 3.55·10-2 100 1.96 37838 2.45 0.1
na 0.67 36
Fir wood ka 2.56·10-2 3702315 1.96 1431060 2.37 0.3
na 0.76 80
Eucalyptus
wood
ka 4.92·10-2 152 1.98 199429 2.45 0.7
na 0.43 13
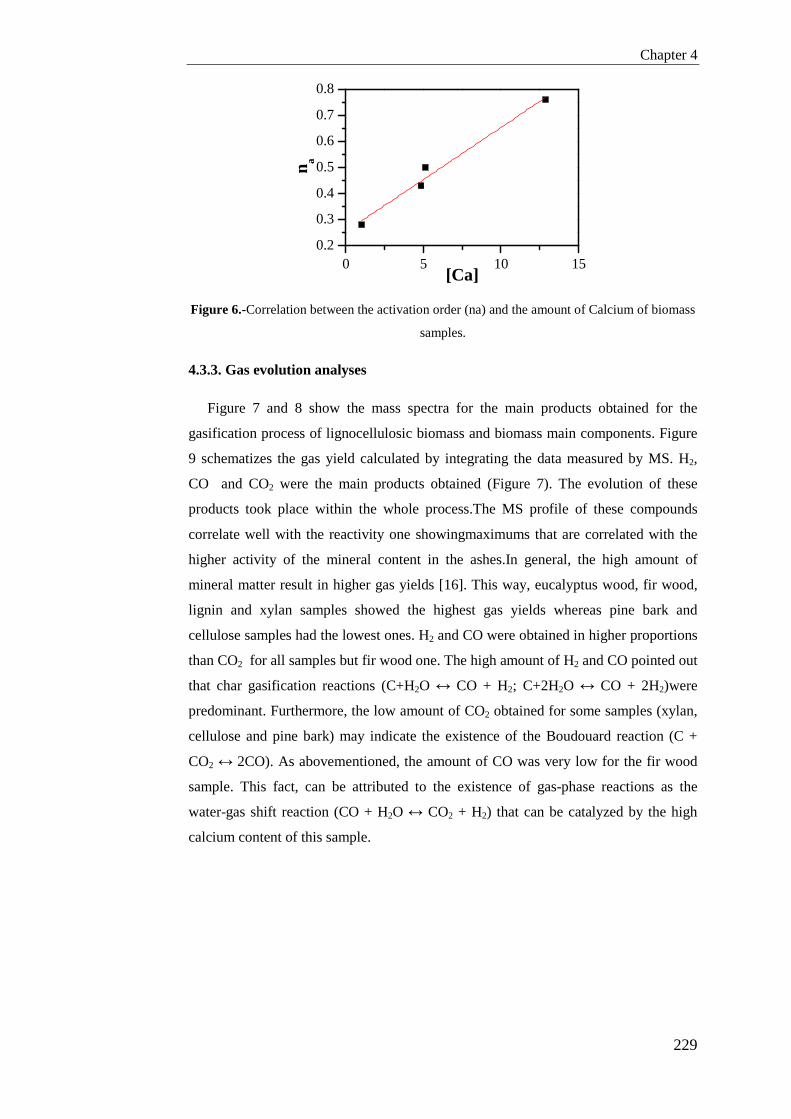
Chapter 4
229
0 5 10 150.2
0.3
0.4
0.5
0.6
0.7
0.8
n a
[Ca]
Figure 6.-Correlation between the activation order (na) and the amount of Calcium of biomass
samples.
4.3.3. Gas evolution analyses
Figure 7 and 8 show the mass spectra for the main products obtained for the
gasification process of lignocellulosic biomass and biomass main components. Figure
9 schematizes the gas yield calculated by integrating the data measured by MS. H2,
CO and CO2 were the main products obtained (Figure 7). The evolution of these
products took place within the whole process.The MS profile of these compounds
correlate well with the reactivity one showingmaximums that are correlated with the
higher activity of the mineral content in the ashes.In general, the high amount of
mineral matter result in higher gas yields [16]. This way, eucalyptus wood, fir wood,
lignin and xylan samples showed the highest gas yields whereas pine bark and
cellulose samples had the lowest ones. H2 and CO were obtained in higher proportions
than CO2 for all samples but fir wood one. The high amount of H2 and CO pointed out
that char gasification reactions (C+H2O ↔ CO + H2; C+2H2O ↔ CO + 2H2)were
predominant. Furthermore, the low amount of CO2 obtained for some samples (xylan,
cellulose and pine bark) may indicate the existence of the Boudouard reaction (C +
CO2 ↔ 2CO). As abovementioned, the amount of CO was very low for the fir wood
sample. This fact, can be attributed to the existence of gas-phase reactions as the
water-gas shift reaction (CO + H2O ↔ CO2 + H2) that can be catalyzed by the high
calcium content of this sample.
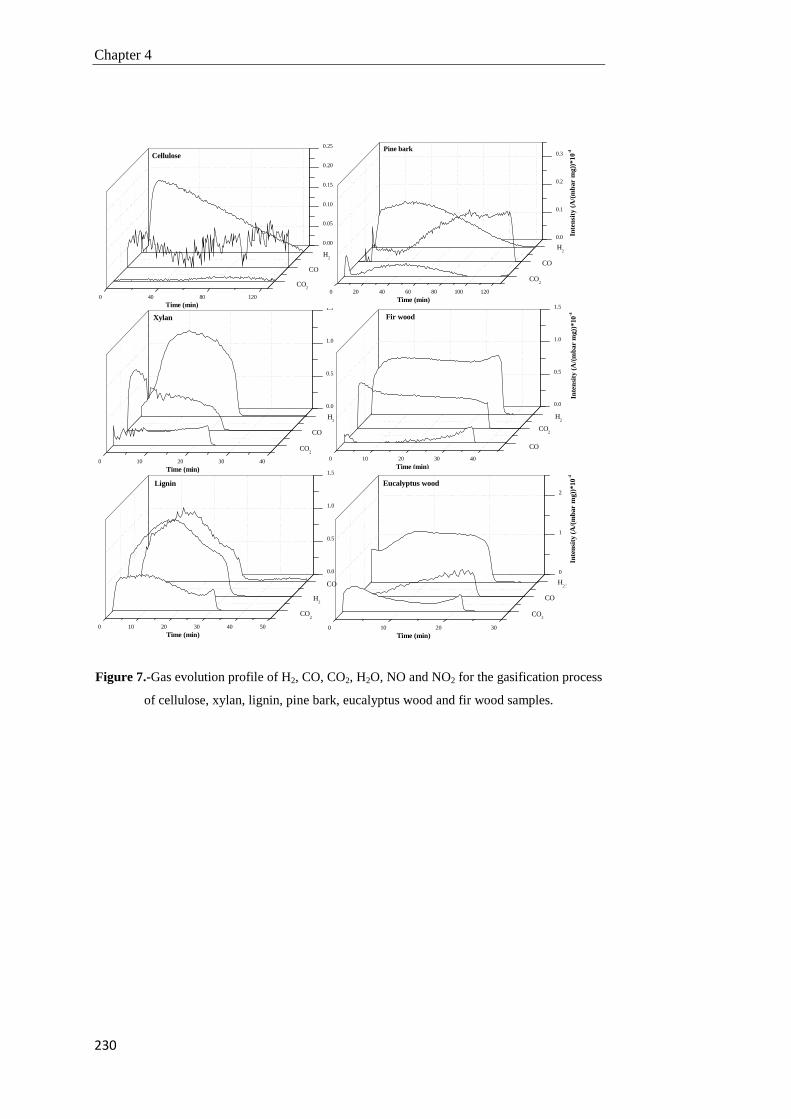
Chapter 4
230
0 40 80 120
0.00
0.05
0.10
0.15
0.20
0.25
H2
CO
CO2CO2
H2
Time (min)
CO
Cellulose
0 20 40 60 80 100 120
0.0
0.1
0.2
0.3
H2
CO
CO2CO2
H2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-4
CO
Pine bark
0 10 20 30 40
0.0
0.5
1.0
1.5
CO2
CO2
H2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-4
CO
Fir wood
0 10 20 30 40
0.0
0.5
1.0
1.5
H2
CO
CO2CO2
H2
Time (min)
CO
Xylan
0 10 20 30 40 50
0.0
0.5
1.0
1.5
CO
H2
CO2
Lignin
CO2
H2
Time (min)
CO
0 10 20 30
0
1
2
H2
CO
CO2CO2
H2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-4
CO
Eucalyptus wood
Figure 7.-Gas evolution profile of H2, CO, CO2, H2O, NO and NO2 for the gasification process
of cellulose, xylan, lignin, pine bark, eucalyptus wood and fir wood samples.
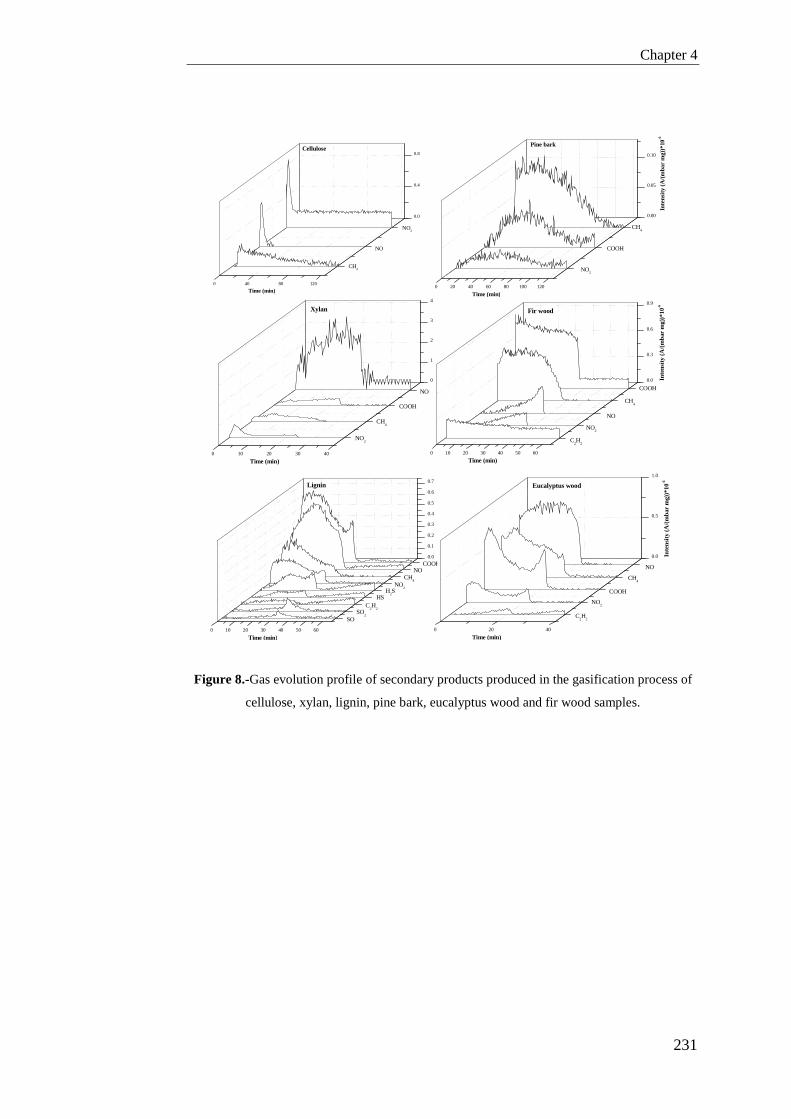
Chapter 4
231
0 40 80 120
0.0
0.4
0.8Cellulose
NO
CH4
NO2
Time (min)0 20 40 60 80 100 120
0.00
0.05
0.10
Pine bark
COOH
CH4
NO2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-6
0 10 20 30 40 50 60
0.0
0.3
0.6
0.9
Fir wood
COOH
NO
CH4
NO2
C2H
2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-6
0 10 20 30 40
0
1
2
3
4
Xylan
COOH
NO
CH4
NO2
Time (min)
0 10 20 30 40 50 60
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
COOHNO
CH4
NO2
H2S
HSC
2H
2SO
2SO
Time (min)
Lignin
0 20 40
0.0
0.5
1.0
Eucalyptus wood
COOH
NO
CH4
NO2
C2H
2
Time (min)
Inte
nsity
(A
/(m
bar
mg)
)*10
-6
Figure 8.-Gas evolution profile of secondary products produced in the gasification process of
cellulose, xylan, lignin, pine bark, eucalyptus wood and fir wood samples.
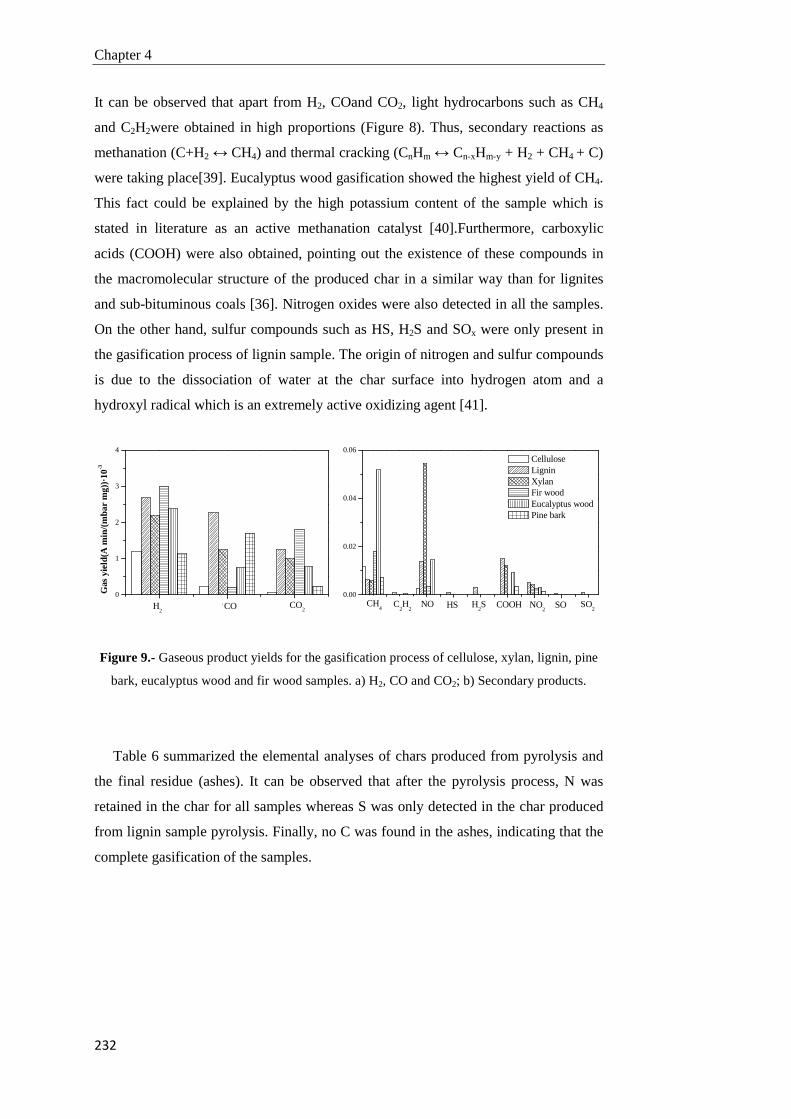
Chapter 4
232
It can be observed that apart from H2, COand CO2, light hydrocarbons such as CH4
and C2H2were obtained in high proportions (Figure 8). Thus, secondary reactions as
methanation (C+H2 ↔ CH4) and thermal cracking (CnHm ↔ Cn-xHm-y + H2 + CH4 + C)
were taking place[39]. Eucalyptus wood gasification showed the highest yield of CH4.
This fact could be explained by the high potassium content of the sample which is
stated in literature as an active methanation catalyst [40].Furthermore, carboxylic
acids (COOH) were also obtained, pointing out the existence of these compounds in
the macromolecular structure of the produced char in a similar way than for lignites
and sub-bituminous coals [36]. Nitrogen oxides were also detected in all the samples.
On the other hand, sulfur compounds such as HS, H2S and SOx were only present in
the gasification process of lignin sample. The origin of nitrogen and sulfur compounds
is due to the dissociation of water at the char surface into hydrogen atom and a
hydroxyl radical which is an extremely active oxidizing agent [41].
Figure 9.- Gaseous product yields for the gasification process of cellulose, xylan, lignin, pine
bark, eucalyptus wood and fir wood samples. a) H2, CO and CO2; b) Secondary products.
Table 6 summarized the elemental analyses of chars produced from pyrolysis and
the final residue (ashes). It can be observed that after the pyrolysis process, N was
retained in the char for all samples whereas S was only detected in the char produced
from lignin sample pyrolysis. Finally, no C was found in the ashes, indicating that the
complete gasification of the samples.
CH4 C2H2 NO SH H2S C2H5O NO2 SO SO20.00
0.02
0.04
0.06 Cellulose Lignin Xylan Fir wood Eucalyptus wood Pine bark
SO2NO
2SOCOOHH
2SHSC
2H
2NO
CH4H2 CO CO2
0
1
2
3
4
CO2COH
2
Gas
yie
ld(A
min
/(m
bar
mg)
)·10
-3

Chapter 4
233
Table 6.-Elemental analysis of biomass char and ash
daf: dry ash
basis, *Oxygen
was calculated
by difference,
N/A: Not
available
4.4. CONCLUSIONS
Thermal characteristics and gas formation during the pyrolysis and gasfication of
eucalyptus wood, fir wood, pine bark and biomass main components (cellulose, xylan
and lignin) were analyzed by TGA-MS. The presence of indigenous inorganic matter
in the gasification process of biomass samples played an important role compared
withtheir initial chemical composition. The reactivity of biomass samples was
Ultimate analysis (wt.%)daf Char
C H N S O*
Cellulose 91.28 0.44 0.07 - 8.25
Lignin 68.02 0.49 0.88 0.16 30.44
Xylan 82.32 0.55 0.58 - 16.57
Fir wood 79.68 0.63 1.22 - 18.51
Eucalyptus wood 69.69 0.61 0.90 - 28.91
Pine bark 84.58 0.46 0.35 - 14.76 Ash
C H N S O*
Cellulose - - - - N/A
Lignin - - - - N/A
Xylan - - - - N/A
Fir wood - - - - N/A
Eucalyptus wood - - - - N/A
Pine bark - - - - N/A

Chapter 4
234
correlated with their alkali index and was ranked as follows: Xylan> lignin > cellulose
and Eucalyptus wood > fir wood > pine bark. The high relevance of inorganic matter
was proved by the inaccuracy of the results obtained by three standards models (VM,
SCM and RPM) which fail to predict the effect of catalytic active species. A semi-
empirical model was proposed in order to accurately model the gasification process.
The proposed model showed errors below 1 %. Furthermore, the models used in this
work were statistically validated. The high production of H2 and CO showed the
predominance of solid-gas reactions. On the other hand, gas phase reactions as water-
gas shift had a higher relevance in the gasification of fir wood due to its high calcium
content. Methanation reactions also took place especially for eucalyptus wood sample
and was correlated to the catalytic effect of potassium.
4.5. REFERENCES
[1] Shabbar S, Janajreh I. 2013. Thermodynamic equilibrium analysis of coal
gasification using Gibbs energy minimization method. Energy Conversion and
Management; 65: 755.
[2] Meng X, de Jong W, Badri FS, Benito P, Basile F, Verkooijen AHM. 2012.
Combustion study of partially gasified willow and DDGS chars using TG analysis and
COMSOL modeling. Biomass and Bioenergy; 39: 356.
[3] Seo DK, Lee SK, Kang MW, Hwang J, Yu T-U. 2010. Gasification reactivity
of biomass chars with CO2. Biomass and Bioenergy; 34(12): 1946.
[4] Ollero P, Serrera A, Arjona R, Alcantarilla S. 2003. The CO2 gasification
kinetics of olive residue. Biomass and Bioenergy; 24(2): 151.
[5] Di Blasi C. 2009. Combustion and gasification rates of lignocellulosic chars.
Progress in Energy and Combustion Science; 35(2): 121.
[6] Kirubakaran V, Sivaramakrishnan V, Nalini R, Sekar T, Premalatha M,
Subramanian P. 2009. A review on gasification of biomass. Renewable and
Sustainable Energy Reviews; 13(1): 179.

Chapter 4
235
[7] Tay HL, Kajitani S, Zhang S, Li CZ. 2013. Effects of gasifying agent on the
evolution of char structure during the gasification of Victorian brown coal. Fuel; 103:
22.
[8] McKendry P. 2002. Energy production from biomass (part 2): Conversion
technologies. Bioresource Technology; 83(1): 47.
[9] Umeki K, Moilanen A, Gómez-Barea A, Konttinen J. 2012. A model of
biomass char gasification describing the change in catalytic activity of ash. Chemical
Engineering Journal; 207-208: 616.
[10] Ahmed II, Gupta AK. 2011. Particle size, porosity and temperature effects on
char conversion. Applied Energy; 88(12): 4667.
[11] White JE, Catallo WJ, Legendre BL. 2011. Biomass pyrolysis kinetics: A
comparative critical review with relevant agricultural residue case studies. Journal of
Analytical and Applied Pyrolysis; 91(1): 1.
[12] Mohammed MAA, Salmiaton A, Wan Azlina WAKG, Mohamad Amran MS.
2012. Gasification of oil palm empty fruit bunches: A characterization and kinetic
study. Bioresource Technology; 110: 628.
[13] Khalil R, Várhegyi G, Jäschke S, Grønli MG, Hustad J. 2009. CO2
gasification of biomass chars: A kinetic study. Energy and Fuels; 23(1): 94.
[14] Mani T, Mahinpey N, Murugan P. 2011. Reaction kinetics and mass transfer
studies of biomass char gasification with CO2. Chemical Engineering Science; 66(1):
36.
[15] Yang Y, Jin S, Lin Y, Huang S, Yang H. 2012. Catalytic gasification of
tobacco rob in steam-nitrogen mixture: Kinetic study and fuel gas analysis. Energy;
44(1): 509.
[16] Yoon HC, Pozivil P, Steinfeld A. 2012. Thermogravimetric pyrolysis and
gasification of lignocellulosic biomass and kinetic summative law for parallel
reactions with cellulose, xylan, and lignin. Energy and Fuels; 26(1): 357.
[17] Huang YF, Kuan WH, Chiueh PT, Lo SL. 2011. Pyrolysis of biomass by
thermal analysis-mass spectrometry (TA-MS). Bioresource Technology; 102(3): 3527.

Chapter 4
236
[18] Sanchez-Silva L, López-González D, Villaseñor J, Sánchez P, Valverde JL.
2012. Thermogravimetric-mass spectrometric analysis of lignocellulosic and marine
biomass pyrolysis. Bioresource Technology; 109: 163.
[19] Yang H, Yan R, Chen H, Zheng C, Lee DH, Liang DT. 2006. In-depth
investigation of biomass pyrolysis based on three major components: Hemicellulose,
cellulose and lignin. Energy and Fuels; 20(1): 388.
[20] Sanchez-Silva L, López-González D, Garcia-Minguillan AM, Valverde JL.
2013. Pyrolysis, combustion and gasification characteristics of Nannochloropsis
gaditana microalgae. Bioresource Technology; 130: 321.
[21] Jones JM, Harding AW, Brown SD, Thomas KM. 1995. Detection of reactive
intermediate nitrogen and sulfur species in the combustion of carbons that are models
for coal chars. Carbon; 33(6): 833.
[22] Fermoso J, Arias B, Pevida C, Plaza MG, Rubiera F, Pis JJ. 2008. Kinetic
models comparison for steam gasification of different nature fuel chars. Journal of
Thermal Analysis and Calorimetry; 91(3): 779.
[23] Sánchez-Silva L, Gutiérrez N, Romero A, Sánchez P, Valverde JL. 2012.
Pyrolysis and combustion kinetics of microcapsules containing carbon nanofibers by
thermal analysis-mass spectrometry. Journal of Analytical and Applied Pyrolysis; 94:
246.
[24] Jiménez-Borja C, Delgado B, Dorado F, Valverde JL. 2013. Experimental
data and kinetic modeling of the catalytic and electrochemically promoted CH4
oxidation over Pd catalyst-electrodes. Chemical Engineering Journal; 225: 315.
[25] Valverde JL, De Lucas A, Carmona M, González M, Rodríguez JF. 2004. A
generalized model for the measurement of effective diffusion coefficients of
heterovalent ions in ion exchangers by the zero-length column method. Chemical
Engineering Science; 59(1): 71.
[26] Lv D, Xu M, Liu X, Zhan Z, Li Z, Yao H. 2010. Effect of cellulose, lignin,
alkali and alkaline earth metallic species on biomass pyrolysis and gasification. Fuel
Processing Technology; 91(8): 903.

Chapter 4
237
[27] Xie X, Goodell B, Zhang D, Nagle DC, Qian Y, Peterson ML, et al. 2009.
Characterization of carbons derived from cellulose and lignin and their oxidative
behavior. Bioresource Technology; 100(5): 1797.
[28] Mandapati RN, Daggupati S, Mahajani SM, Aghalayam P, Sapru RK, Sharma
RK, et al. 2012. Experiments and kinetic modeling for CO2 gasification of indian coal
chars in the context of underground coal gasification. Industrial and Engineering
Chemistry Research; 51(46): 15041.
[29] Zhang L, Huang J, Fang Y, Wang Y. 2006. Gasification reactivity and kinetics
of typical Chinese anthracite chars with steam and CO2. Energy and Fuels; 20(3):
1201.
[30] Gómez-Barea A, Ollero P, Fernández-Baco C. 2006. Diffusional effects in
CO2 gasification experiments with single biomass char particles. 1. Experimental
investigation. Energy and Fuels; 20(5): 2202.
[31] Mitsuoka K, Hayashi S, Amano H, Kayahara K, Sasaoaka E, Uddin MA.
2011. Gasification of woody biomass char with CO2: The catalytic effects of K and
Ca species on char gasification reactivity. Fuel Processing Technology; 92(1): 26.
[32] Huang Y, Yin X, Wu C, Wang C, Xie J, Zhou Z, et al. 2009. Effects of metal
catalysts on CO2 gasification reactivity of biomass char. Biotechnology Advances;
27(5): 568.
[33] Marquez-Montesinos F, Cordero T, Rodríguez-Mirasol J, Rodríguez JJ. 2002.
CO2 and steam gasification of a grapefruit skin char. Fuel; 81(4): 423.
[34] Dupont C, Nocquet T, Da Costa JA, Verne-Tournon C. 2011. Kinetic
modelling of steam gasification of various woody biomass chars: Influence of
inorganic elements. Bioresource Technology; 102(20): 9743.
[35] Di Blasi C, Buonanno F, Branca C. 1999. Reactivities of some biomass chars
in air. Carbon; 37(8): 1227.
[36] Hattingh BB, Everson RC, Neomagus HWJP, Bunt JR. 2011. Assessing the
catalytic effect of coal ash constituents on the CO 2 gasification rate of high ash,
South African coal. Fuel Processing Technology; 92(10): 2048.

Chapter 4
238
[37] Skodras G, Sakellaropoulos GP. 2002. Mineral matter effects in lignite
gasification. Fuel Processing Technology; 77-78: 151.
[38] Sakawa M, Sakurai Y, Hara Y. 1982. Influence of coal characteristics on CO2
gasification. Fuel; 61(8): 717.
[39] Widyawati M, Church TL, Florin NH, Harris AT. 2011. Hydrogen synthesis
from biomass pyrolysis with in situ carbon dioxide capture using calcium oxide.
International Journal of Hydrogen Energy; 36(8): 4800.
[40] Nanou P, Van Rossum G, Van Swaaij WPM, Kersten SRA. 2011. Evaluation
of catalytic effects in gasification of biomass at intermediate temperature and pressure.
Energy and Fuels; 25(3): 1242.
[41] Chaudhari ST, Dalai AK, Bakhshi NN. 2003. Production of hydrogen and/or
syngas (H2 + CO) via steam gasification of biomass-derived chars. Energy and Fuels;
17(4): 1062.

Chapter 5:
CHARACTERIZATION OF DIFFERENT HEAT
TRANSFER FLUIDS AND DEGRADATION
STUDY BY USING A PILOT PLANT DEVICE OPERATING AT REAL CONDITIONS
A pilot plant was designed to evaluate the degradation of heat
transfer fluids (HTF) for their application in concentrating solar
power plants (CSP). Firstly, the characterization of sixHTFs was
carried out: two ionic liquids ([BMIM][BF4] and [EMIM][BF4]),
two molten salts (Hitec XL and solar salt), a commercial HTF
(Mobiltherm 605) and an oil extracted from
NannochloropsisGaditanamicroalgae(NG oil). Mobiltherm 605
was selected for tuning the pilot plant due to its similarity to HTFs
used in CSP, low cost and easy acquisition. The operating
conditions were set according to thermogravimetric analysis. Thus,
three isothermal experiments were carried out at 140, 160 ant 180
ºC for 15 days. Mobiltherm 605 viscosityincreased with time
indicating that polymerization of hydrocarbon chainstook place.
Two mathematical models were developed to assess the HTF
behaviour in the pilot plant.
A mathematical model for the estimation of the most
representative parameters (viscosity, heat capacity and overall heat

Chapter 5
240
transfer coefficient) of HTF performance was proposed.
Furthermore, an activation/deactivation model was proposed to
predict the variation of the estimated parameters with time. This
model was validated with experimental viscosity measurements
(average error of about 3 %). Finally, the statistical significance of
the model wasproved.
5.1. INTRODUCTION
Nowadays, the electricity power is mainly generated bycentral power plants or
distributed generation systems. Renewable energy resources is one of the solutions to
the dependence on the petroleum import, the energy efficiency and the
conflictsusually arisen from the building of new large power plants based on fossil
fuels [1].
Among all renewable energy sources, the solar energy is by far the most abundant
one. There are two methods to extract electricity from solar radiation: photovoltaic
(PV) and concentrating solar power (CSP) [2]. According to a study of the
International Energy Agency,CSP is three times cheaper than PV[3].
Solar energy is not very dense. It is necessary to concentrate it to produce usable
exploitable temperatures for the production of energy[4].There are two viable
technologies to concentrate solar energy, those that concentrate iton a line and those
that concentrate it on a point. Within the first group, the most developed techniques
are the parabolic trough and linear Fresnel reflector technologies. In the second group,
two technologies stand out: parabolic dishes and solar tower concentrated power
plants[5].Among all of them, parabolic trough is the most mature technology, having a
great promotion worldwide, which has turned out into great cost and performance
improvements.

Chapter 5
241
Parabolic trough plants consist of large fields of parabolic trough collectors, a heat
transfer fluid/steam generation system, aRankine steam turbine/generator cycle and
optional storage and/or fossil-fired backup systems[6]. However, in spite of the great
experience accumulated into this technology, there are still several technique gaps to
make it competitive with traditional fossil-fuel based technologies. The main efforts
have led to the improvement ofthe collector heat transfer [7, 8], the efficiency of the
mirror [9], the integration of a thermal storage system [10]and the substitution of the
heat transfer fluid [11, 12].
The substitution of the current heat transfer fluid (HTF) appears as one of the most
prominent pathways to reduce costs due to the fact that very large quantities of HTF
are needed, entailing high capital investment costs[12]. The present generation of
commercial fluids used are organic synthetic oils composed by eutectic mixtures of
diphenyl oxide and biphenyl[13]. This synthetic oil currently offers the best
combination of low freezing point (12ºC) and upper temperature limit (393ºC) [12].
However, these oils are toxic and high flammable products, resulting into a direct
danger to the plant operators. Furthermore, their use is limited by their degradation
temperature (<400ºC), limiting the efficiency of the thermodynamic cycle for power
generation. Additionally, they have high vapour pressures, exceeding atmospheric
pressure, making difficult its use as thermal storage media as it would require
impractically large pressure vessels[14].
In the recent years, different heat transfer fluids have been proposed. The fluids
that have brought more attention are based on molten salts, mainly inorganic nitrate
salts [14]and ionic liquids[13, 15]due to their excellent thermal properties. Van
Valkenburg et al. [13] established a list of the properties that the new generation of
HTF must satisfyto be considered adequate candidates.Wide liquid temperature range,
high heat capacity, high density, high thermal and chemical stability,low vapour
pressure and non-harmfulness are required[13, 15].
One of the problems when a HTF is evaluated is the difficulty of predicting its
durability as an effective medium as energy carrier in a solar plant. However, no

Chapter 5
242
previous studies in literature about predicting the life cycle of the HTF at large scale
have been reported. Thus, it seems necessary to design an experimental setup in order
to evaluate the thermal performance of HTF. Furthermore, it could be helpful to
establish mathematical models that reproduce the behaviour of the thermal fluids
under different operating conditions.
The aim of this work was to carry out the thermal and physical characterization of
the different thermal fluids that have come up lately as feasible alternatives to be used
in parabolic trough plants, such as molten salts, ionic liquids and microalgae oil.
Furthermore, a comprehensive comparison among the studied HTF was done.
Moreover, the design, assembly and tuning of a pilot plant based on CSP working
principle have been carried out in order to evaluate the degradation of the most
suitable HTF. Finally, mathematical models to predict the thermal behaviour of HTF
under selected operating conditions have been proposed. The suggested models were
validated and their statistical significance was proved.
5.2. MATERIALS AND METHODS
5.2.1 Materials
1-Butyl-3methylimidazolium tetrafluoroborate ([BMIM][BF4]) and 1-ethyl-3-
methylimidazolium tetrafluoroborate ([EMIM][BF4]) were purchased from Sigma
Aldrich.
The molten salts mixtures were prepared using NaNO3, KNO3 and Ca(NO3)2-
tetrahydrate. Reagent grade salts were provided by Sigma Aldrich. Hitec (60%
NaNO3, 40% KNO3, Ca(NO3)2-tetrahydrate), Solar Salt (60% NaNO3, 40% KNO3,
Ca(NO3)2-tetrahydrate) and Hitec XL (60% NaNO3, 40% KNO3, Ca(NO3)2-
tetrahydrate) were synthesized according to Bradshaw et al. [12]procedure.
A commercial heat transfer fluid, MOBILTHERM 605, was purchased from
EXXON MOBIL.

Chapter 5
243
Oil extracted from the microalgae NannochloropsisGaditana (NG microalgae) was
provided by the University of Almeria (Spain).NG microalgae raw material was
purchased from AlgaEnergy Company (Spain). NG microalgae belong
toEustigmatophytes microalgae species with an average composition of 17.5 % lipids,
12.6% fatty acids and 24.1 % of proteins.The extraction process was carried out over
lyophilized biomass, which was previously grounded and sieved to obtain a particle
size between 100 and 300 µm. The extraction was done continuously in a distillation
column joined with a stirring tank, using hexane as the extractor agent.
5.2. Equipment and procedures
5.2.1. Thermogravimetric Analysis (TGA)
Thermal gravimetric analyses were carried out on a Mettler Toledo–TGA-DSC 1.
The initial mass sample was kept between 4 and 15 mg for all tests. Each sample was
analyzed at least twice in order to ensure the reproducibility of the measurements.
Fast scans were performed in the temperature range 40-700 ºC in order to
determinate the degradation temperature (Td) at a heating rate of 10ºC/min under a N2
flow of 60 ml/min. Isothermal TGA experiments were also used for long-term stability
scans. The weight loss of the HTF was evaluated as a function of time under
isothermal conditions at temperatures close to the calculated Tdby fast scans
measurements.
5.2.2 Differential Scanning Calorimetry (DSC) and Modulated DSC (MDSC).
The melting point and heat of fusion of studied samples were measured by using a
differential scanning calorimeter (DSC), TA Instruments model Q100.
Thesemeasurementswerecarried out byvaryingthetemperature in the range from -50 to
300 ºCwith a heating rate of 10 ºC/min under 60 ml/min N2flow.

Chapter 5
244
For themeasurementsofspecificheatcapacity (Cp), thesamplesweresubjectedto a
heatingrampfrom 30 ºCto 300 ºCusing a modulation amplitude of ± 0.5 ºC, with 100
secondsperiodandanunderlyingheating rate of 0.5 ºC/min.
5.2.3. Viscosity
Kinematic viscosity was measured using a Canon–Fenske capillary viscometer.
The viscosity measurements were carried out in the temperature range of 25-100 ºC.
The solution temperature was controlled by a thermostat in a circulating bath
(TAMSON TV2000) monitored by a thermometer. The stopwatch with a resolution of
0.1 s was used to measure the flow times.
5.2.4. Density
The density of different heat transfer fluids was measured by means of a Coriolis
mass flow measuring system, Promass 80. This device allows the measurement of the
density in a large temperature range (-50 – 350 ºC). The calibration was carried out
with air and water at 23.3 ºC and 23.6 ºC in a laboratory certified by ISO/IEC 17025.
5.2.5. Heat Storage
Sensible heat storage is easily calculated from the heat capacity, density and the
temperature change chosen (Eq. 1).
�� = � ∙ �� ∙ (�� − ��) (1)
whereEsis the sensible heat storage (J/m3), ρ is the density of the fluid (kg/m3), Cp is
the heat capacity (J/(kg ºC)), Tout and Tin are the inlet and outlet temperatures of the
solar field, respectively. For this work a rise in temperature of 100 ºC was selected due
to the fact that it is often used in solar applications[13].
5.3. Design of the HTF degradation pilot plant
A pilot plant was designed and constructed for the thermal performance study of
different HTF. The plant with an outer size of 1.15 m x 0.8 m x 2 m is composed of

Chapter 5
245
N2
FI
H-1
V-1
P-1
O-1
Sampling
DI
TI
PI
TIC
TIC
TI
TI
TIFIMITI
TICPI
PI PI
P-67
six functional units: a feedstock vessel, a pumping system, a tubular oven, a heat
exchanger unit, a coriolis mass flow meter and an automatic control system. The
installation is shown schematically in Fig. 1.
Figure 1.Schematic diagram of the HTF degradation pilot scale plant.
The central element of the equipment was a vertical cylindrical vessel (V-1) of 8
litres capacity (90 mm of inner diameter and 1.2 m height). The fluid was heated in V-
1 by an internal electrical resistance, able to reach temperatures up to 400 ºC. The
HTF flowed through the piping system by means of a centrifugal pump Sterling
(ZTND) (P-1) located at the bottom of the deposit. The flow was regulated by a valve

Chapter 5
246
system that allowed part of the oil to be recirculated to V-1. A valve for sampling was
placed at the bottom of V-1 prior to the inlet to P-1.
P-1 impulsedthe thermal fluid through a tubular oven (Fisher 3Kw) (O-1), which
was used to heat the fluidagain, before reaching the heat exchanger unit (H-1). H-1
was made of copper. Table 1 lists the main mechanical details of the heat exchanger.
O-1 was capable of heating the fluid up to 500 ºC.
Table 1. Heat exchanger geometric characteristics: inner diameter (Din), outer diameter (Dext), equivalent length (Leq) and roughness (ε) of the internal pipes.
Din (m) (·103) 7.92
Dout (m) (·103) 9.52
L eq(m) 10.48
ε (·104) 0.15
The fluid was later cooled down by air in a heat exchanger. The air was provided
by a centrifugal fan (Sodeca CMA-426-2M) with a maximum air flow of 850 m3/h.
After the cooling, the HTF passed through a Coriolis mass flow measurement unit
(Promass 80) that continuously registered the flow rate and density. There was a by-
pass to the Coriolis unit in order to let the systemreach the steady state, avoiding
device breakdowns due to high temperatures. Finally, the fluid was redirected to V-1,
closing the loop.
A nitrogen generator (model ZEFIRO 3, CINCL®) was used to provide a
continuous flow of nitrogen (99.999 %) to guarantee an inert atmosphere in the pilot
plant. Nitrogen was introduced into the system through a pipe located in the upper side
of V-1.
The piping system and the vessel used were all made of stainless steel and
thermally insulated in order to minimize heat losses.

Chapter 5
247
The installation had several controllers and indicators. K-type thermocouples were
used to measure temperatures: one was put inside V-1, other two were placed at the
inlet and outlet of P-1; finally, another one was situated into O-1. Finally, other four
thermocouples were used to measure both, the fluid and air temperature, at the inlet
and the outlet of H-1.There were several pressure taps located all over the installation
to measure pressure drop in different sections of the circulation loop. A P18L
transducer was used to measure the relative humidity of air.
Three proportional-integral-derivative (PID) controllers were used to control de
plant. The first one was used to set the temperature of V-1, regulated by the internal
electrical resistance placed inside the vessel. The second one was used to control the
temperature of O-1 by adjusting the power supplied to the oven through the 3 kW
resistance. Finally, a controller was used to set the temperature at the exit of H-1 by
varying the air flow.
Furthermore, the experimental setup was connected to a computer allowing a
remote control of it. Using specific software developed by Adepro engineering
company (Spain), temperatures, differential pressures, fluid density and fluid flow
were processed and recorded every five minutes. Finally, data obtained were
processed with a VBA-excel application to evaluate the main physic properties and its
evolution withtime, as commented next.
5.4. Mathematical model of the thermal performance of HTF in the pilot plant
With data collected from the pilot plant, a pseudo-state model was developed able
to calculate the overall heat transfer coefficient in the heat exchanger,the specific heat
capacity and viscosity of the HTF coming from the heat exchanger with time on
stream. For this purpose, a VBA-Excel application was developed. Likewise, a non-
linear procedure for estimating the parameter in the model that accounted for the
decay/growth with time on stream of the process variables under study was considered
and solved according to the corresponding VBA-Excel application.

Chapter 5
248
Details of the partial calculations are shown in the following sections
5.4.1. Overall heat transfer coefficient determination
The heat flow transferred between a cold and a hot fluid into a heat exchanger is
defined as follows:
� = �� ∙ � ∙ ∆�� (2)
whereQ is the heat transferred(W), A is the heat transfer surface area (m2), ∆Tml is the
log mean temperature difference(ºC) and UF is the overall heat transfer coefficient
(W/(m2·ºC)).
The heat exchanger performance may be evaluated using Eq. (2),being the overall
heat transfer coefficient (UF) defined as follows:
�� = � (� ∙ ∆��)� (3)
Eq. (3) is only valid when the operation is carried out under a steady state
condition. In other words, the flow of the hot and cold streams and their inlet
temperatures must be virtually constant.
5.4.2. HTF specific heat capacity calculation
The heat duty gained or lostwas estimated for both fluids as:
���� = ���� ∙ ���� ∙ (� − ) (4)
and
���� = ���� ∙ ���� ∙ (�� − �) (5)
where QHTF and Qair are the heat duty (W)for HTF and air, respectively; CHTF and
Cairare the corresponding specific heat capacity in (J/(kg·ºC)); mHTFand mairare the
corresponding mass flow rates (kg/h);Ti, To, ti and to are the inlet and outlet
temperatures (ºC) for the hot (HTF) and the cold (air) fluids, respectively.

Chapter 5
249
Under steady state conditions,the heat duties for both fluids are balanced according
to the first thermodynamic law(Qair= QHTF= Q).
This way, Eqs. (4) and (5) are equivalent:
���� ∙ ���� ∙ (�� − �) = ���� ∙ ���� ∙ (� − ) (6)
andCHTFcan be calculated from:
���� = ���� (���� ∙ (� − ))� (7)
On the other hand, air heat duty can also be defined as:
���� = ���� ∙ ∆ ��� (8)
wheremairis the dry air mass flow (kg/s) and ∆Hairis the enthalpy of the air
(J/kg)calculated as the difference between the enthalpies of the inlet and outlet air
stream, Ho and Hi, respectively:
∆ ��� = − � (9)
These enthalpies can be calculated as a function of absolute humidity in the air as
follows:
= 4184.1 ∙ (0.24 ∙ + ()*+ ∙ (595 + 0.46))(10)
� = 4184.1 ∙ (0.24 ∙ � + ()*+ ∙ (595 + 0.46�))(11)
whereXABSis the absolute humidity in the air surrounded the pilot plant (%).
5.4.3. Viscosity determination
The viscosity determination was performed according to the Newton’s algorithm.
As a first approximation, the model supposed a valueof viscosity for the HTF. Then,
the fluid mean velocity in the air exchanger tube was calculated (Eq. (12)).

Chapter 5
250
/ = 0.1∙�234∙523467∙89:;
(12)
whereρHTF is the working fluid density (kg/m3) and Din is the heat exchanger inlet
diameter tube (m).
Friction factors were calculated for the laminar flow by the Poiseuille equation (Eq.
(13)) whereas the Chen equation was used for the turbulent regime(Eq. (14)):
< = => 16� (13)
?@A = −4 ∙ log(
?E.F0G1 ∙ (
H89:) −
1.0I1JKL ∙ log[ ?
J.OJ1F ∙ PH89:Q
?.?0RO + 1.O10GKLS,UVUW] (14)
Pressure drop of the fluid in the heat exchanger was calculated by the Fanning
equation (Eq. (15)):
Y = (Z234[\Z2349)5]^ + 2 ∙ < ∙ /J ∙ _
89: (15)
wherePHTFiand PHTFoare the working fluid pressure at the inlet and outlet of the heat
exchanger and L is the equivalent length of the pipe where the fluid passed through
(m).F represented a factor that should be zero or close to zero when the iterative
process represented by the Newton’s algorithm is completed.
5.5. Deactivation/activation model.
The variationof the calculated variables (µ, Cp and U) with time on stream was
evaluated in terms of deactivation/activation depending on their decay/growth over
time, respectively. These expressions are well-known in describing deactivation of
catalysts in chemical reactions [16].
Four different deactivation/activation modelswere considered:
`� = >ab\cd∙� (16)

Chapter 5
251
`� = 1 (1 + ef ∙ �)� (17)
`� = �cd (18)
`� = 1 (1 + �cd)� (19)
whereai is the estimated variable (µ, cp and U), kw is the deactivation/activation
constant and t is time.
5.5.1. Parameter estimation.
A VBA-Excel application was developed to solve this model [17], which was
based on the Marquardt-Levenberg algorithm for nonlinear regression [18-20].
The weighted sum of the squared differences between the observed (Exp) and the
calculated (Pr) variables was minimized according to the following equation described
elsewhere [18]:
gg = ∑ ∑ (i�Z� − i�jk�)lJ�lm?��m? (20)
wherei represents the number of equations to be fitted, j the specific experimental data
and n and m the total number of equations and experiments, respectively.
AF-test is a statistical test in which a F-distribution under the null hypothesis is
established. The procedure was based on the comparison between the tabulated F-
value (F-test) and Fc, which was defined elsewhere[19]:
Yn =∑ ∑ (o9pqr );stuW:9uW
∑ ∑ (o9pqvo9wxy)t;(:∙svy)
stuW:9uW (21)
wherep represents the total number of parameters.
If Fc is larger than F(p, n-p, 1-α), assuming a value of α = 0.05, 95% confidence
level, the regression was considered to be meaningful, although there is no guarantee
that the model is statistically suitable since the meaningfulness of each parameter in
the model must be also evaluated.
Hence, a complementary test, named t-test, was used. The t-test considers that the
statistical hypothesis test follows a Student’s t distribution and allows to verify if the

Chapter 5
252
estimate of the parameter bfi differs from a reference value (generally zero). Thus, a
parameter is meaningful (at α = 0.05) each time that the following inequality occurs:
�n� = z{|9z@}({|)99 > �(� − b, 1 −
�J) (22)
where[V(bf)] ii represents the diagonal i th term of the covariance matrix used in the last
step of the n-linear regression procedure.
5.3. RESULTS AND DISCUSSION
5.3.1. Heat transfer fluids characterization for their use as thermal fluids in
parabolic trough plants
The heat transfer fluids chosen for their study were: two ionic liquids (1-Butyl-
3methylimidazolium tetrafluoroborate ([BMIM][BF4]) and 1-ethyl-3-
methylimidazolium tetrafluoroborate ([EMIM][BF4])), two molten salts (Hitec XL
(60% NaNO3, 40% KNO3, Ca(NO3)2-tetrahydrate) and solar salt (60% NaNO3, 40%
KNO3, Ca(NO3)2-tetrahydrate), a commercial HTF (Mobiltherm 605) and a new oil
extracted from the microalgae NannocholorpsisGaditana (NG oil). These different
kinds of fluids were selected due to their excellent properties and their different
nature. Ionic liquids and molten salts have exceptional thermal properties since it is
possible that they can be used as thermal storage media [12, 15]. On the other hand,
algae are one of the most attractive biomass feedstock to produce high-valuable
products. In this context, the oil extracted from them,which has been widely studied as
a transport fuel [21], could be used as a valuable HTF.At the best of our knowledge,
the use of the oil extracted directly from algae as a heat transfer fluid has not been
reported yet.
Table 2 shows the main properties that the new generation of HTFs must meet to
be used in the parabolic trough technology field [22].

Chapter 5
253
Table 2.Heat transfer fluids requirements according to National Renewable Energy Laboratory [22].
Heat transfer fluid requirements
Storage density > 1.9 MJ/m3
Freezing point ≤ 0 ºC
High temperature stability ≥ 430 ºC
Vapor presure < 1 atm
Material compatibility Carbon and stainlesssteel
Viscosity Similar toTherminol VP-1
The commercial HTF Therminol®VP-1 was taken as the reference material.
Density, degradation temperature, melting point, heat capacity, heat storage density
and viscosity of the HTFsconsidered in this study together withTherminol® VP-1 are
summarized in Table 3.

Chapter 5
254
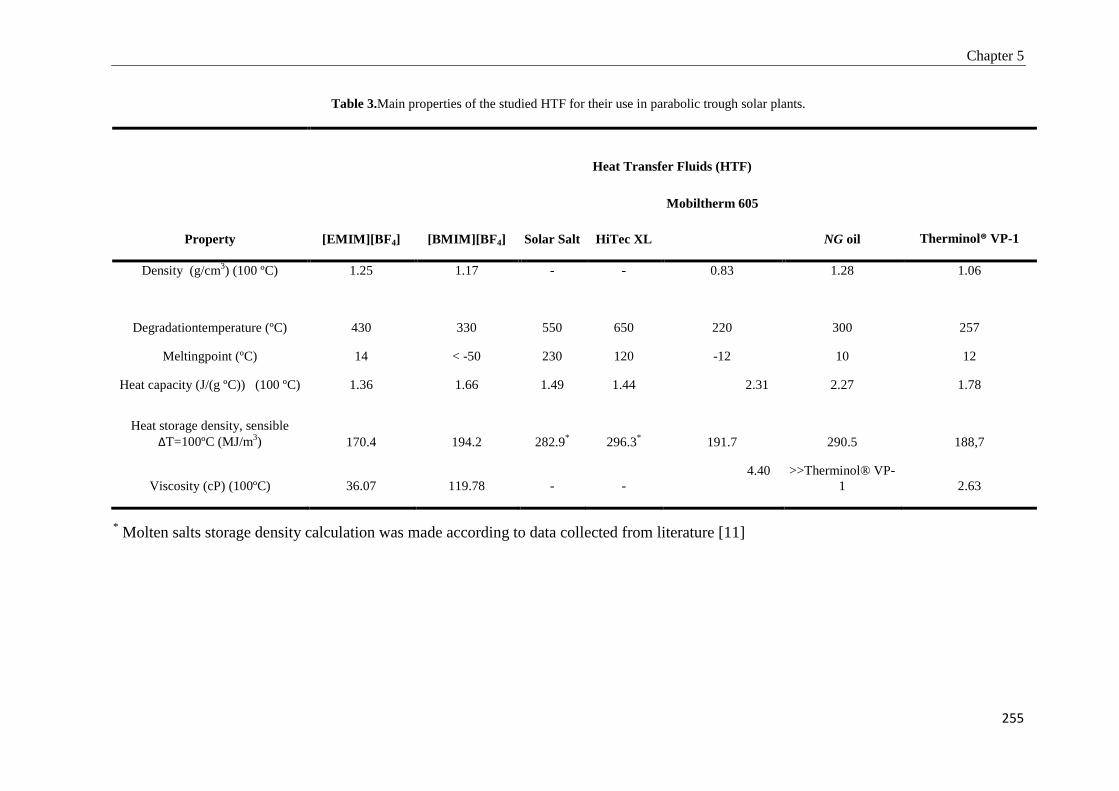
Chapter 5
255
Table 3.Main properties of the studied HTF for their use in parabolic trough solar plants.
Property
Heat Transfer Fluids (HTF)
[EMIM][BF 4] [BMIM][BF 4] Solar Salt HiTec XL
Mobiltherm 605
NG oil Therminol® VP-1
Density (g/cm3) (100 ºC) 1.25 1.17 - - 0.83 1.28 1.06
Degradationtemperature (ºC) 430 330 550 650
220 300 257
Meltingpoint (ºC) 14 < -50 230 120 -12 10 12
Heat capacity (J/(g ºC)) (100 ºC) 1.36 1.66 1.49 1.44 2.31 2.27 1.78
Heat storage density, sensible ΔT=100ºC (MJ/m3) 170.4 194.2 282.9* 296.3*
191.7 290.5 188,7
Viscosity (cP) (100ºC) 36.07 119.78 - - 4.40 >>Therminol® VP-
1 2.63
* Molten salts storage density calculation was made according to data collected from literature [11]

Chapter 5
256
3.1.1. Thermal stability.
As was mentioned above, fast-scans do not provide reliable information about a
material long-term thermal stability [23]. However, it is often reported as an
appropriate value for establishing comparison among materials.
According to values listed in Table 3, molten salts were the most thermal stable
compounds.NG oil, HTF Therminol® VP-1, Mobiltherm 605 and [BMIM][BF4]
showed the lowest degradation temperature (Td) whereas the value of Td of
[EMIM][BF 4] kept in the limit of the required value.
Molten salts are eutectic mixtures, which imply that they are really stable in the
fluid state. Furthermore, the studied molten salts are composed by metallic nitrates
conferring a great thermal resistance[12]. On the other hand, ionic liquids thermal
decomposition depends on the nature of the anion rather than that of the cation, and
specially decreases withincreasinghydrophilicity of the anion[24]. Thus,
[EMIM][BF4] showed higher Td value due to the higher hydrophilicity of the
corresponding anion. Besides, ionic liquids can be easily contaminated (water, sodium
ion, silver ion and chloride),which may have an important effect on its thermal
behaviour [13].Finally, Mobiltherm 605 is a paraffinic mineral oil which Tdis
typicallyrangingfrom 150 to 315 ºC [25].
These results were corroborated by long-term experiments. Figure 2 shows the
long-term stability experiments for the ionic liquid [BMIM][BF 4]. The corresponding
Tdmeasured by fast scan turned out to be 330 ºC. However, it can be seen that after 5
hours at 300 ºC the liquid had lost almost the 20 wt. % of its initial weight.
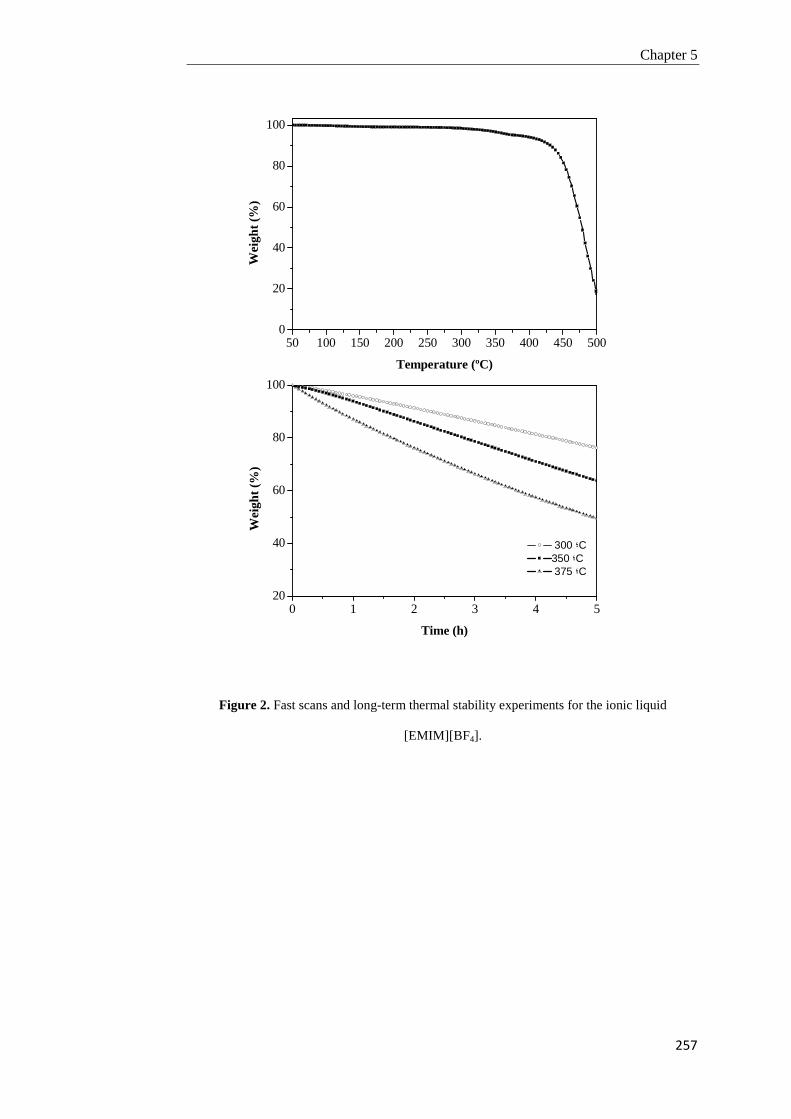
Chapter 5
257
50 100 150 200 250 300 350 400 450 5000
20
40
60
80
100
0 1 2 3 4 520
40
60
80
100
Wei
ght (
%)
Temperature (ºC)
Wei
ght (
%)
Time (h)
C؛ 300 C؛ 350C؛ 375
Figure 2. Fast scans and long-term thermal stability experiments for the ionic liquid
[EMIM][BF 4].
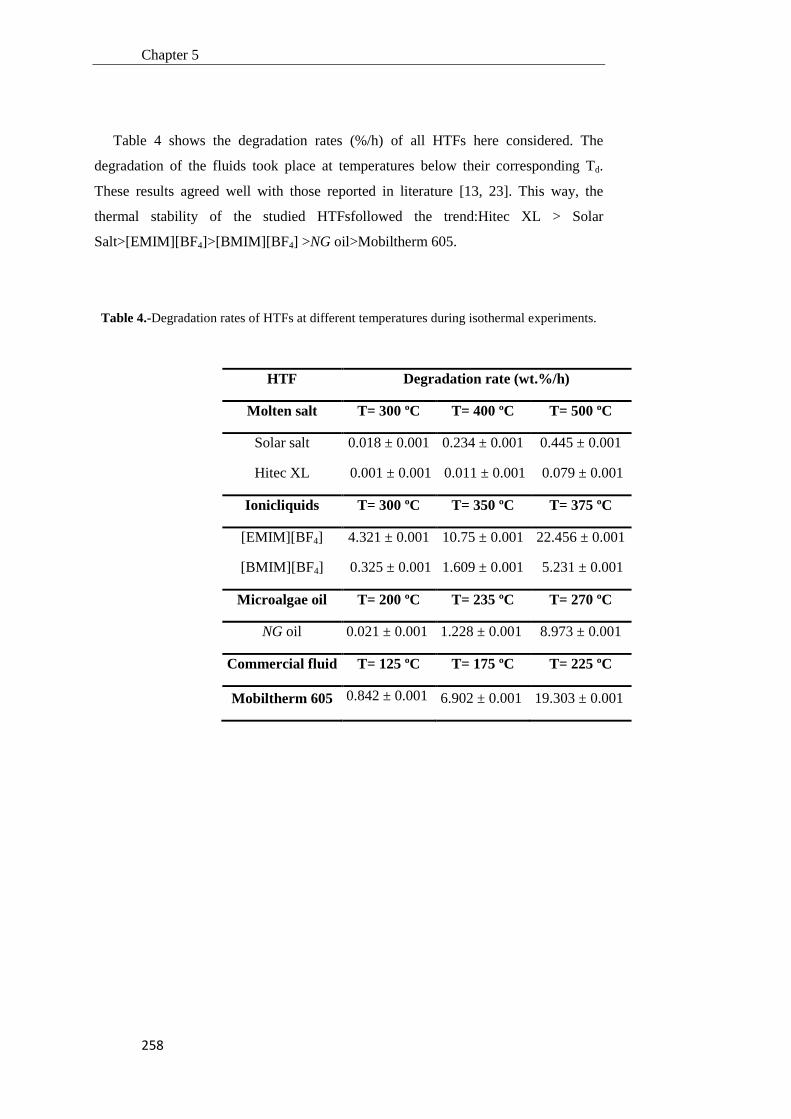
Chapter 5
258
Table 4 shows the degradation rates (%/h) of all HTFs here considered. The
degradation of the fluids took place at temperatures below their corresponding Td.
These results agreed well with those reported in literature [13, 23]. This way, the
thermal stability of the studied HTFsfollowed the trend:Hitec XL > Solar
Salt>[EMIM][BF4]>[BMIM][BF 4] >NG oil>Mobiltherm 605.
Table 4.-Degradation rates of HTFs at different temperatures during isothermal experiments.
HTF Degradation rate (wt.%/h)
Molten salt T= 300 ºC T= 400 ºC T= 500 ºC
Solar salt 0.018 ± 0.001 0.234 ± 0.001 0.445 ± 0.001
Hitec XL 0.001 ± 0.001 0.011 ± 0.001 0.079 ± 0.001
Ionicliquids T= 300 ºC T= 350 ºC T= 375 ºC
[EMIM][BF 4] 4.321 ± 0.001 10.75 ± 0.001 22.456 ± 0.001
[BMIM][BF 4] 0.325 ± 0.001 1.609 ± 0.001 5.231 ± 0.001
Microalgae oil T= 200 ºC T= 235 ºC T= 270 ºC
NG oil 0.021 ± 0.001 1.228 ± 0.001 8.973 ± 0.001
Commercial fluid T= 125 ºC T= 175 ºC T= 225 ºC
Mobiltherm 605 0.842 ± 0.001 6.902 ± 0.001 19.303 ± 0.001

Chapter 5
259
5.1.2. Melting point/freezing point.
Melting temperatures (Tm) were evaluated instead of freezing temperatures due to
the fact that some of the fluids tend to super-cool. The targeted value of Tm would be
below 0ºC, which would allowthese fluids to be used in cold weather regions. Among
the selected HTFs,only [BMIM][BF4] and Mobiltherm 605 maintained their liquid
state at these operating conditions. Molten salts had the highest Tm, being their use in
parabolic trough plants difficult unless freeze protection could be used to keep them in
the liquid state during the process [26]. On the other hand, the ionic liquid
[EMIM][BF 4] and the NG oil showed an acceptable value as long asa minimum
heating is provided to the plant.
5.1.3. Heat capacity
Heat capacity measurements were made by MDSC. Heat capacity affects directly
to the storage capacity of the thermal fluids. All fluids were within an acceptable
range close to Therminol® VP-1 heat capacity. Molten salts and NG oil showed the
lowest values being Mobiltherm 605 the fluid with the highest one.
5.1.4. Density
Density is a necessary value for the calculation of sensible heat storage. The higher
the density, the higher the capacity of the compound to storage heat.The molten salts
density at the targeted temperature (100 ºC) was not evaluated since they are still
solids at this temperature. The ionic liquids density has been reported in
literature[13].NG oil density is higher than that ofMobiltherm 605 andTherminol® VP-
1but it is lower than those of the ionic liquids.
3.1.5. Storage capacity
Storage capacity is calculated as a function of the HTF density, heat capacity and
usable liquid temperature range. These properties determine the use of HTF as thermal
storage media and heat transfer fluids for solar power plants [15]. As it can be
observed, all the studied HTFshad a value of the storage capacity above that of

Chapter 5
260
Therminol® VP-1. Therefore, all the candidates could be considered as useful thermal
storage media.
3.1.6. Viscosity
The parabolic trough-based technology is a flowing system.Thus,the viscosity of
the HTF used in this process is of special interest in order to reduce pumping,
operation and maintenance costs. The viscosity of the reference HTF (Therminol® VP-
1) is 2.48 cSt measured at 40 ºC.NG oil had the highest viscosity of all HTFs
considered. Molten salts were not liquids at ambient temperature, which make them
not suitable for this technology unless freeze protection methods are used. Ionic
liquids under study were closer to the targeted viscosity although still seems too high
to be considered viable candidates without causing problems in the HTF loop. Finally,
Mobiltherm 605 showed the closest viscosity value to that of Therminol® VP-1.
On the other hand, the best HTF should be an inexpensive and nontoxic liquid with
excellent thermo-physical properties and a long service life [25]. The obtained
resultsindicated that the thermal fluids with better propertieswere the ionic liquids, and
more specifically the [EMIM][BF4]. However, there are still several drawbacks to deal
with for its implementation in large scale parabolic trough plants. Availability and
costs are two main issues that a fluid must meet. Ionic liquids and algae oil are still in
the development phase, which imply high production costs and low availability[15].
Furthermore, its preparationprocedures are difficult and tedious. On the other hand,
molten salts are inexpensive and are easily produced although theirhigh melting points
add complexity and larger operating costs [12]. Therefore, the commercial HTF
Mobiltherm 605 was selected in the following study due to its great availability and
similar properties to the commercial fluid used in parabolic trough solar plants. The
main characteristics of Mobiltherm 605 are shown in Table 5.

Chapter 5
261
Table 5.-MOBILTHERM 605 properties.
Properties MOBILTHERM 605
Viscosity (µ) ASTM D 445
40 ºC (cSt)
100 ºC (cSt)
30.4
5.4
Freezing point, ºC, ASTM D97 -12
Flash point (ºC), ASTM D92 230
Maximumoperationtemperature (ºC) 316
Density (kg/l), ASTM D4052 0.86
5.3.2. Pilot plant assembly and tuning.
5.3.2.1. Selection of the operating conditions.
The degradation of a HTF mainly depends on two variables: operation time and
working temperature. The selected procedure to establish the most suitable operating
conditions to evaluate the HTF degradation in the pilot scale plant was based in fast
scans and long-term stability experiments performed by the TGA technique. In
addition, an operation time of 15 days was chosen to evaluate the thermal degradation
of the HTF in the pilot plant.
Figure 3 shows the fast scans and the long-term stability experiments carried out
with Mobiltherm 605. It can be observed that at temperatures above 175 ºC,
Mobiltherm 605 started to decompose. Thus, the temperature set in the vessel (V-1)
was kept in the temperature range between 125-180 ºC where slight degradation was
appreciated. Secondly, the thermal fluid was heated up in the tubular oven (O-1)at 180
ºC in order to keep the temperature of the thermal fluid and accelerate its degradation
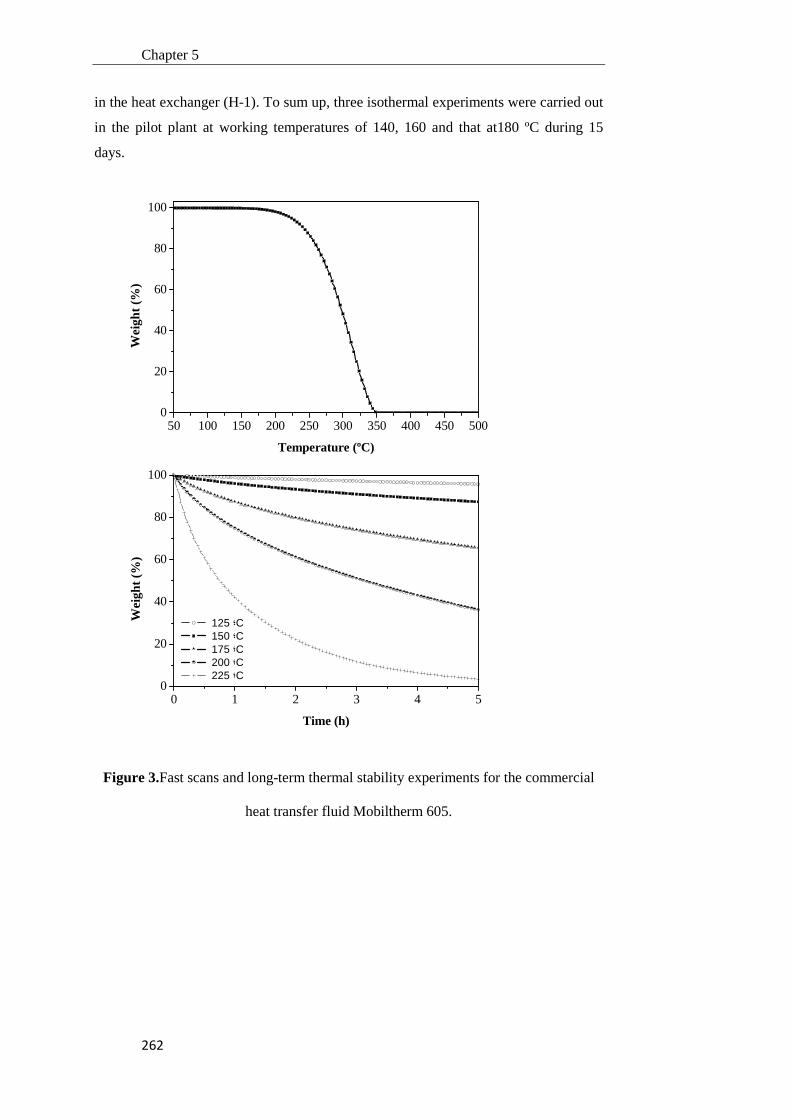
Chapter 5
262
50 100 150 200 250 300 350 400 450 5000
20
40
60
80
100
0 1 2 3 4 50
20
40
60
80
100
Wei
ght (
%)
Temperature (ºC)
Wei
ght (
%)
Time (h)
C؛ 125 C؛ 150 C؛ 175 C؛ 200 C؛ 225
in the heat exchanger (H-1). To sum up, three isothermal experiments were carried out
in the pilot plant at working temperatures of 140, 160 and that at180 ºC during 15
days.
Figure 3.Fast scans and long-term thermal stability experiments for the commercial
heat transfer fluid Mobiltherm 605.
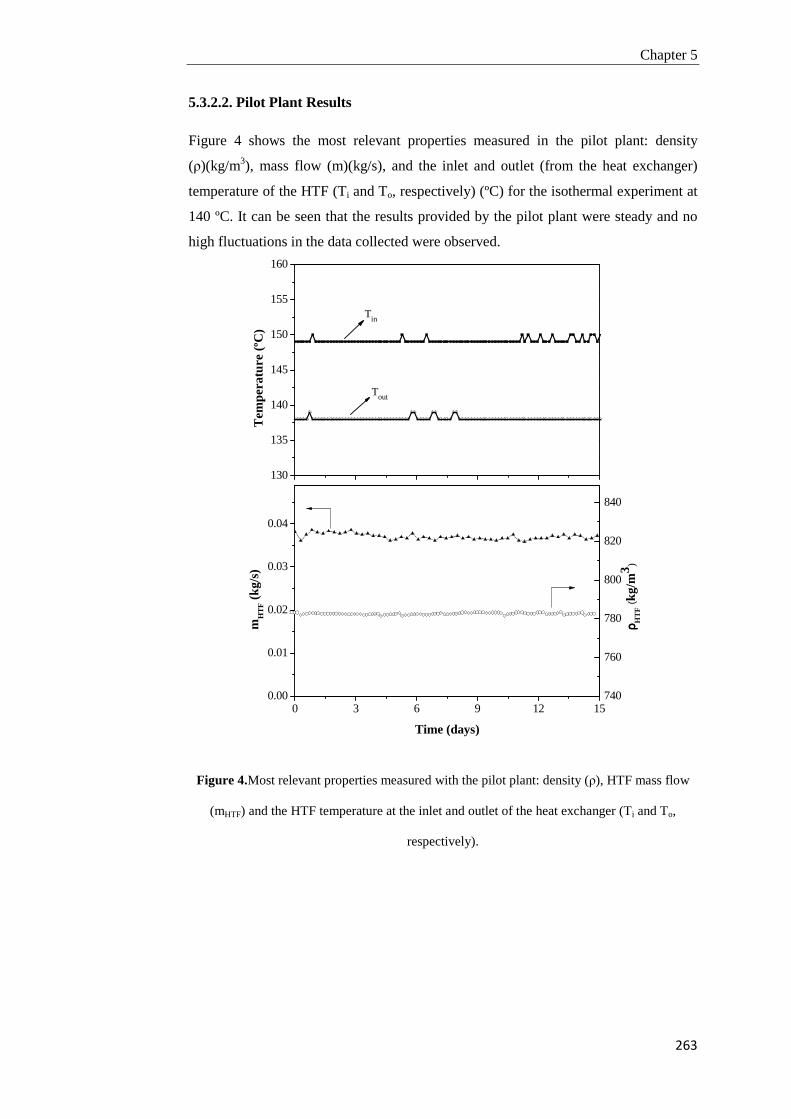
Chapter 5
263
130
135
140
145
150
155
160
0 3 6 9 12 150.00
0.01
0.02
0.03
0.04
Tout
T
empe
ratu
re (
ºC)
Tin
ρρ ρρH
TF (k
g/m
3 )
mH
TF (
kg/s
)
Time (days)
740
760
780
800
820
840
5.3.2.2. Pilot Plant Results
Figure 4 shows the most relevant properties measured in the pilot plant: density
(ρ)(kg/m3), mass flow (m)(kg/s), and the inlet and outlet (from the heat exchanger)
temperature of the HTF (Ti and To, respectively) (ºC) for the isothermal experiment at
140 ºC. It can be seen that the results provided by the pilot plant were steady and no
high fluctuations in the data collected were observed.
Figure 4.Most relevant properties measured with the pilot plant: density (ρ), HTF mass flow
(mHTF) and the HTF temperature at the inlet and outlet of the heat exchanger (Ti and To,
respectively).

Chapter 5
264
As described in section 2.4, three main parameters were estimated by means of the
corresponding mathematical model: viscosity (µ) in kg/(m·s), heat capacity (Cp)
J/(kg·ºC) and overall heat transfer coefficient (UF) in J/(m2·ºC·s).These parameters
were normalized respecting their initial value.
The viscosity is an important parameter when evaluating the operative life of a
HTF as it has a direct impact on its heat transfer capacity. Furthermore, viscosity is
important to design heat transfer applications because the pressure drop and the
resulting pumping power depend on its value[27]. The HTF heat capacity (Cp) and the
HTF overall heat transfer coefficient (UF) were also estimated to assess the heat
transfer efficiency and performance of the fluid[28], which are directly related to the
storage capacity of it[15].
There are many researchers focusing on the development of high performance heat
transfer fluids [29]. However, there are not published any standard procedure to
predict the life cycle of a HTF. The limits values have been mainly set byboth users
and manufacturers experience. Nevertheless, there are more and more standards
appearing concerning to oil in-service predictive maintenance. For instance, ASTM D
4378-03[30]and ASTM D 6224[31] are related to In-Service monitoring of mineral
Turbine Oils for Steams and Gas turbines and lubricating oil for auxiliary power
plants equipment, respectively.Thus, it is generallyaccepted a variation in viscosity of
± 15 %respect to its initial value, before an oil in-service could be considered out of its
usable range.
Figure 5 shows the estimated parameters normalized respecting their initial value
(solid line) together with the deactivation model prediction (dotted line), described in
section 2.5,for thethree experiments performedat140 ºC, 160 ºC and 180 ºC. It can be
appreciated the good fitting reached by the deactivation/activation model. The results
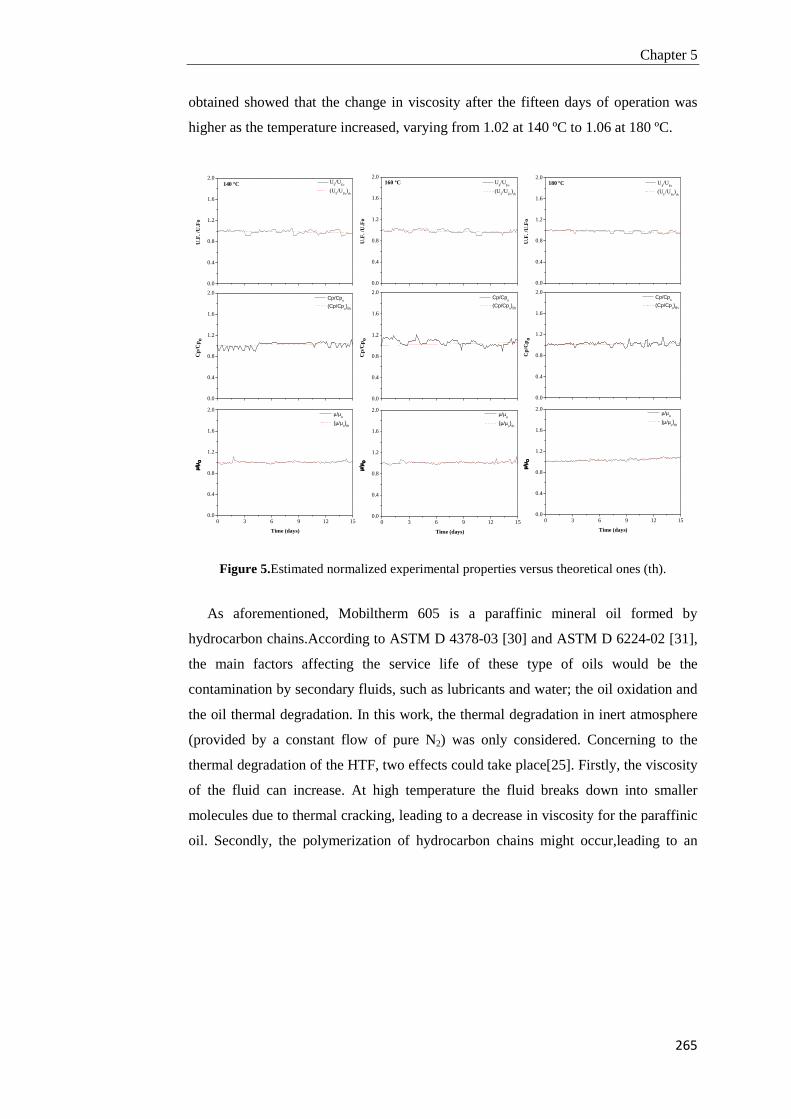
Chapter 5
265
0.0
0.4
0.8
1.2
1.6
2.0
0.0
0.4
0.8
1.2
1.6
2.0
0 3 6 9 12 150.0
0.4
0.8
1.2
1.6
2.0
UF/U
Fo
(UF/U
Fo)
th
U.F
. /U
.Fo
160 ºC
Cp/Cpo
(Cp/Cpo)th
Cp/
Cp o
µ/µ
µ/µ
µ/µ
µ/µ οο οο
Time (days)
µ/µo
(µ/µο)
th
0.0
0.4
0.8
1.2
1.6
2.0
0.0
0.4
0.8
1.2
1.6
2.0
0 3 6 9 12 150.0
0.4
0.8
1.2
1.6
2.0
UF/U
Fo
(UF/U
Fo)
th
U.F
. /U
.Fo
180 ºC
Cp/Cpo
(Cp/Cpo)th
Cp/
Cp o
µ/µ
µ/µ
µ/µ
µ/µ οο οο
Time (days)
µ/µo
(µ/µο)
th
0.0
0.4
0.8
1.2
1.6
2.0
0.0
0.4
0.8
1.2
1.6
2.0
0 3 6 9 12 150.0
0.4
0.8
1.2
1.6
2.0
UF/U
Fo
(UF/U
Fo)
th
U.F
. /U
.Fo
140 ºC
Cp/Cpo
(Cp/Cpo)th
Cp/
Cp o
µ/µ
µ/µ
µ/µ
µ/µ οο οο
Time (days)
µ/µo
(µ/µο)th
obtained showed that the change in viscosity after the fifteen days of operation was
higher as the temperature increased, varying from 1.02 at 140 ºC to 1.06 at 180 ºC.
Figure 5.Estimated normalized experimental properties versus theoretical ones (th).
As aforementioned, Mobiltherm 605 is a paraffinic mineral oil formed by
hydrocarbon chains.According to ASTM D 4378-03 [30] and ASTM D 6224-02 [31],
the main factors affecting the service life of these type of oils would be the
contamination by secondary fluids, such as lubricants and water; the oil oxidation and
the oil thermal degradation. In this work, the thermal degradation in inert atmosphere
(provided by a constant flow of pure N2) was only considered. Concerning to the
thermal degradation of the HTF, two effects could take place[25]. Firstly, the viscosity
of the fluid can increase. At high temperature the fluid breaks down into smaller
molecules due to thermal cracking, leading to a decrease in viscosity for the paraffinic
oil. Secondly, the polymerization of hydrocarbon chains might occur,leading to an

Chapter 5
266
increase in viscosity. Therefore, opposite effects could be taken place, producing no
net changes in viscosity even though the HTF degradationwas occurring.
The viscosity of Mobiltherm 605 increased during operationat all the temperatures
essayed. This fact points out that the polymerization path was the predominant effect.
In addition, at the highest temperature tested the maximum variation of the viscosity
turned out to be ~ 6 %,indicating the high thermal stability of Mobiltherm 605.
The heat capacity (Cp) and overall heat transfer coefficient (UF) followed opposite
trends. Cp increased with time, being the final value of 1.02, 1.03 and 1.12 for 140,
160 and 180 ºC, respectively. On the other hand, UF decreased with time. However, no
influence of temperature was found and thefinal value of this parameter was 0.95.
The activation/deactivation constants and the resulting parameters obtained by the
non-linear regression procedure described in section2.5.1, are summarized in Table 6.
As aforementioned, the discrimination of kinetic parameters was done applying the F-
test and the t-test at the 95% confidence level. In terms of statistical results, F-test
considered the regression to be suitable in all cases since the corresponding values to
the Fc/Ftest ratio was larger than 1. The t-test was also used for evaluating each
parameter in the model. As shown in Table 6, the values of tc/t-test ratio were also
larger than 1, showing the statistical significance of the proposed models and their
corresponding parameters.

Chapter 5
267
Table 6.-Estimated deactivation constant (Kw) at different temperatures
(140, 160 and 180 ºC) for MOBILTHERM 605.
Temperature = 140 ºC
Normalized parameters
K w tc t-test Fc F-test
µ/µo -1.22·10-3 4
Cp/Cpo -3.13·10-3 10 1.96 56702 2.37
UF/UFo 2,12·10-3 6
Temperature = 160 ºC
Normalized parameters
K w tc t-test Fc F-test
µ/µo -1.32·10-3 4
Cp/Cpo -4.33·10-3 13 1.96 58102 2.37
UF/UFo 2.86·10-3 9
Temperature = 180 ºC
Normalized parameters
K w tc t-test Fc F-test
µ/µo -4.85·10-3 25
Cp/Cpo -2.56·10-3 13 1.96 311942 2.37
UF/UFo 2.85·10-3 14
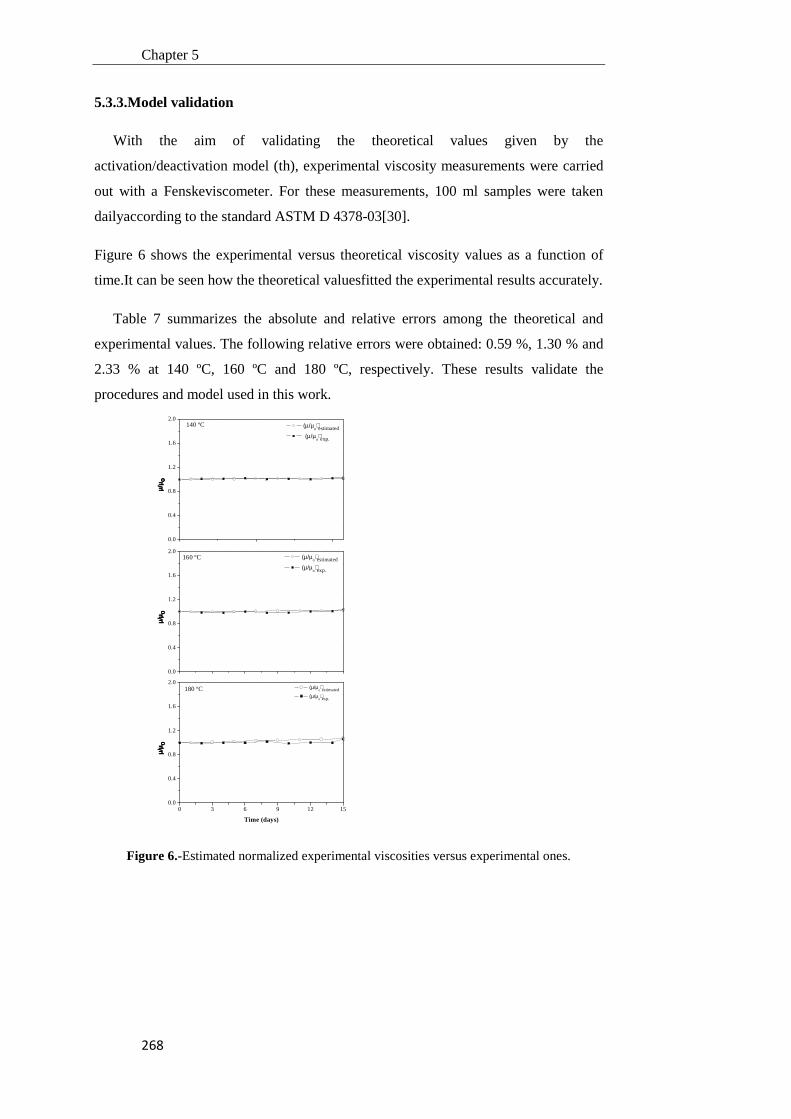
Chapter 5
268
0.0
0.4
0.8
1.2
1.6
2.0
0.0
0.4
0.8
1.2
1.6
2.0
0 3 6 9 12 150.0
0.4
0.8
1.2
1.6
2.0
µ/µ
µ/µ
µ/µ
µ/µ οο οο
(µ/µο)estimated
(µ/µο)exp.
140 ºC
160 ºC
µ/µ
µ/µ
µ/µ
µ/µ οο οο
(µ/µο)estimated
(µ/µο)exp.
180 ºC
µ/µ
µ/µ
µ/µ
µ/µ οο οο
Time (days)
(µ/µο)estimated
(µ/µο)exp.
5.3.3.Model validation
With the aim of validating the theoretical values given by the
activation/deactivation model (th), experimental viscosity measurements were carried
out with a Fenskeviscometer. For these measurements, 100 ml samples were taken
dailyaccording to the standard ASTM D 4378-03[30].
Figure 6 shows the experimental versus theoretical viscosity values as a function of
time.It can be seen how the theoretical valuesfitted the experimental results accurately.
Table 7 summarizes the absolute and relative errors among the theoretical and
experimental values. The following relative errors were obtained: 0.59 %, 1.30 % and
2.33 % at 140 ºC, 160 ºC and 180 ºC, respectively. These results validate the
procedures and model used in this work.
Figure 6.-Estimated normalized experimental viscosities versus experimental ones.
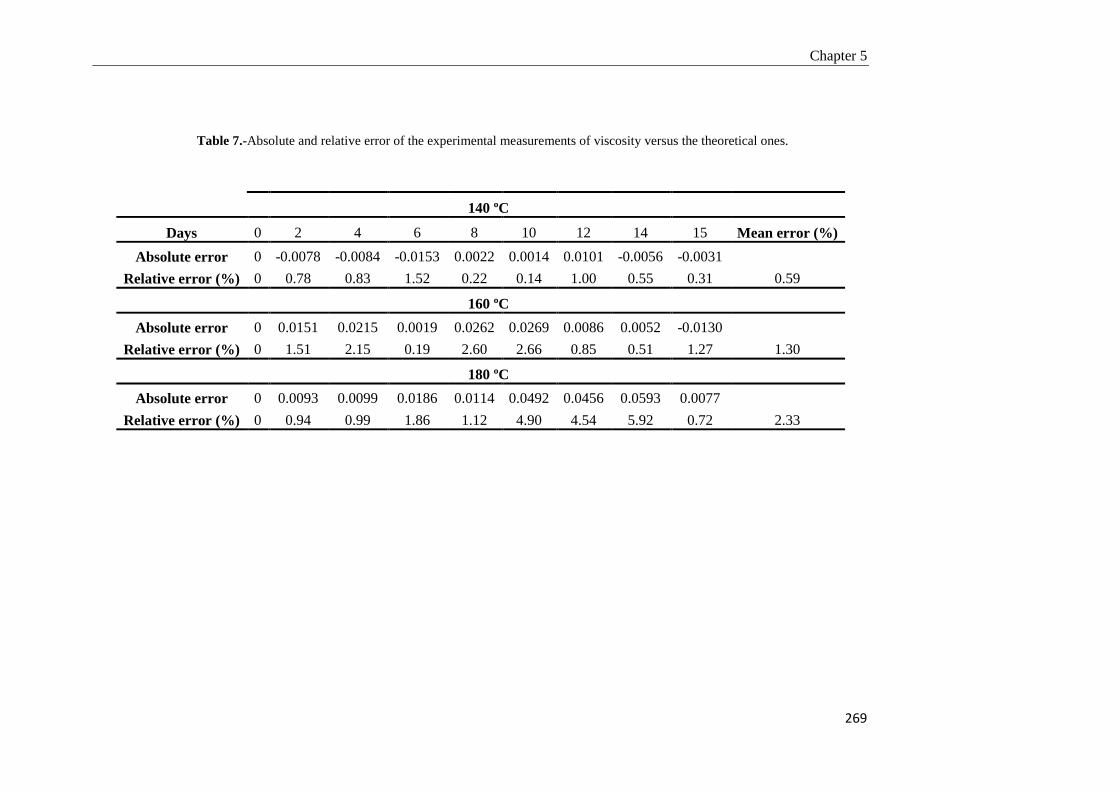
Chapter 5
269
Table 7.-Absolute and relative error of the experimental measurements of viscosity versus the theoretical ones.
140 ºC
Days 0 2 4 6 8 10 12 14 15 Mean error (%)
Absolute error 0 -0.0078 -0.0084 -0.0153 0.0022 0.0014 0.0101 -0.0056 -0.0031
Relative error (%) 0 0.78 0.83 1.52 0.22 0.14 1.00 0.55 0.31 0.59
160 ºC
Absolute error 0 0.0151 0.0215 0.0019 0.0262 0.0269 0.0086 0.0052 -0.0130
Relative error (%) 0 1.51 2.15 0.19 2.60 2.66 0.85 0.51 1.27 1.30
180 ºC
Absolute error 0 0.0093 0.0099 0.0186 0.0114 0.0492 0.0456 0.0593 0.0077
Relative error (%) 0 0.94 0.99 1.86 1.12 4.90 4.54 5.92 0.72 2.33

Chapter 5
270
5.4. CONCLUSIONS
A pilot plant was designed to evaluate the degradation of HTfs to be used in
concentrating solar power plants (CSP). Six different HTFs were characterized:two
ionic liquids (1-Butyl-3methylimidazolium tetrafluoroborate ([BMIM][BF4]) and 1-
ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4])), two molten salts (Hitec
XL (60% NaNO3, 40% KNO3, Ca(NO3)2-tetrahydrate) and solar salt (60% NaNO3,
40% KNO3, Ca(NO3)2-tetrahydrate), a commercial HTF (Mobiltherm 605) and a new
oil extracted from the microalgae NannochloropsisGaditana (NG oil).
Mobiltherm 605 was selected for the assembling and tuning of the pilot plant due
to its great availability and similar properties to the commercial fluid used in parabolic
trough solar plants. The pilot plant behaviour was stable and no high fluctuations of
data collected were detected. Three isothermal experiments were carried out at 140,
160 and 180 ºC for 15 days. The viscosity was selected as the key parameter to follow
the HTF degradation. Mobiltherm 605 viscosity increased with time, indicating that
the polimerization of hydrocarbon chains took place. The variation of viscosity was 6
% at 180 ºC pointing out the high thermal stability of Mobiltherm 605.
Two mathematical models were developed to estimate the most representative
parameters (µ, Cp and UF) with time on stream and predict the behaviour of the
parameters during operation, respectively. The model was validated with experimental
measurements of viscosity obtaining an error lower than 3%. Finally, the statistical
significance of the model was also proved.

Chapter 5
271
5.5. REFERENCES
[1] Sripakagorn A, Srikam C. Design and performance of a moderate temperature
difference Stirling engine. Renew. Energy. 2011;36(6):1728-33.
[2] Du Marchie Van Voorthuysen EH. The promising perspective of Concentrating
Solar Power (CSP). Int. Conf. on Future Power Systems, 2005.
[3] "World Energy Outlook 2004", International Energy Agency, Paris 2004.
[4] El Gharbi N, Derbal H, Bouaichaoui S, Said N. A comparative study between
parabolic trough collector and linear Fresnel reflector technologies. Conference A
comparative study between parabolic trough collector and linear Fresnel reflector
technologies, vol. 6. p. 565-72.
[5] Mills D. Advances in solar thermal electricity technology. Solar Energy.
2004;76(1-3):19-31.
[6] Price H, Lüpfert E, Kearney D, Zarza E, Cohen G, Gee R, et al. Advances in
parabolic trough solar power technology. J. Sol. Energy Eng., Transactions of the
ASME. 2002;124(2):109-25.
[7] Al-Ansary H, Zeitoun O. Numerical study of conduction and convection heat
losses from a half-insulated air-filled annulus of the receiver of a parabolic trough
collector. Sol. Energy. 2011;85(11):3036-45.
[8] Padilla RV, Demirkaya G, Goswami DY, Stefanakos E, Rahman MM. Heat
transfer analysis of parabolic trough solar receiver. Appl. Energy. 2011;88(12):5097-
110.
[9] Sansoni P, Fontani D, Francini F, Giannuzzi A, Sani E, Mercatelli L, et al. Optical
collection efficiency and orientation of a solar trough medium-power plant installed in
Italy. Renew. Energy. 2011;36(9):2341-7.
[10] Laing D, Bahl C, Bauer T, Lehmann D, Steinmann WD. Thermal energy storage
for direct steam generation. Sol. Energy. 2011;85(4):627-33.

Chapter 5
272
[11] Kearney D, Herrmann U, Nava P, Kelly B, Mahoney R, Pacheco J, et al.
Assessment of a molten salt heat transfer fluid in a parabolic trough solar field. J. Sol.
Energy Eng., Transactions of the ASME. 2003;125(2):170-6.
[12] Bradshaw RW, Cordaro JG, Siegel NP. Molten nitrate salt development for
thermal energy storage in parabolic trough solar power systems. Proc. ASME 3rd Int.
Conf. Energy Sustain., vol. 2. p. 615-24.
[13] Valkenburg MEV, Vaughn RL, Williams M, Wilkes JS. Thermochemistry of
ionic liquid heat-transfer fluids. Thermochim. Acta. 2005;425(1–2):181-8.
[14] Blake DM, L.; Hale, M.J.; Price, H.; Kearney, D.; Herrmann, U. New Heat
Transfer and Storage Fluids for Parabolic Trough Solar Thermal Electric Plants. Proc.
11th ColarPACES Int. Symp. Conc. Sol. Power Chem. Energy Technologies. Zurich,
Switzerland 2002.
[15] Wu B, Reddy RG, Rogers RD. Novel ionic liquid thermal storage for solar
thermal electric power systems In: Kleis SJ, Bingham CE editors. Proc. Sol. Forum
2001. Solar Energy: The Power to Choose, Washington, DC. 2001; p. 445-51.
[16] Levenspiel O. Chemical reaction engineering: An introduction to the design of
chemical reactors. In: Wiley, editor. Michigan1962.
[17] Valverde JL, De Lucas A, Carmona M, González M, Rodríguez JF. A generalized
model for the measurement of effective diffusion coefficients of heterovalent ions in
ion exchangers by the zero-length column method. Chem. Eng. Sci. 2004;59(1):71-9.
[18] De Lucas A, Sánchez P, Dorado F, Ramos MJ, Valverde JL. Kinetic model of the
n-octane hydroisomerization on PtBeta agglomerated catalyst: Influence of the
reaction conditions. Ind. Eng. Chem. Res. 2006;45(3):978-85.
[19] Froment GF, Bischof, K.B. Chemical Reactor Analysis and Design. New
York1990.
[20] Marquardt DW. An algorithm for least-squares estimation of nonlinear
parameters. J Soc Ind Appl Math. 1963;11:431-41.
[21] Ross AB, Biller P, Kubacki ML, Li H, Lea-Langton A, Jones JM. Hydrothermal
processing of microalgae using alkali and organic acids. Fuel. 2010;89(9):2234-43.

Chapter 5
273
[22] “Thermal storage for Solar Thermal Parabolic Trough Electric Power Systems”,
National Renewable Energy Laboratory, Request for Proposal Number RCQ-0-30910,
March 27, 2000.
[23] Kosmulski M, Gustafsson J, Rosenholm JB. Thermal stability of low temperature
ionic liquids revisited. Thermochim. Acta. 2004;412(1-2):47-53.
[24] Singh G, Kumar A. Ionic liquids: Physico-chemical, solvent properties and their
applications in chemical processes. Indian J. Chem. – Sect. A Inorg., Phys., Theor.
Anal. Chem.. 2008;47(4):495-503.
[25] Oyekunle LO, Susu AA. Characteristic properties of a locally produced paraffinic
oil and its suitability as a heat-transfer fluid. Pet. Sci. Tech. 2005;23(11-12):1499-509.
[26] Moens L, Blake DM, Rudnicki DL, Hale MJ. Advanced thermal storage fluids
for solar parabolic trough systems. Int. Sol. Energy Conf., 2002; p. 277-83.
[27] Chandrasekar M, Suresh S, Chandra Bose A. Experimental investigations and
theoretical determination of thermal conductivity and viscosity of Al2O3/water
nanofluid. Exp. Therm. and Fluid Sci. 2010;34(2):210-6.
[28] Bergman TL. Effect of reduced specific heats of nanofluids on single phase,
laminar internal forced convection. Int. J. Heat and Mass Transfer. 2009;52(5–
6):1240-4.
[29] Saeedinia M, Akhavan-Behabadi MA, Razi P. Thermal and rheological
characteristics of CuO-Base oil nanofluid flow inside a circular tube. Int. Commun.
Heat and Mass Transfer. 2012;39(1):152-9.
[30] ASTM 4378-03 AD. In-Service Monitoring of Mineral Turbine Oils for Steam
and Gas Turbines. 2003.
[31] ASTM 6224-02 AD. In-Service Monitoring of Lubricating Oil for Auxiliary
Power Plant Equipment. 2003.

Chapter 6:
GENERAL CONCLUSIONS AND
RECOMMENDATIONS
This chapter lists the main conclusions derived from the research performed in this
Doctoral Thesis. In addition, some recommendations are suggested to be taken into
account in further studies.
6.1. CONCLUSIONS
- Pyrolysis, combustion and gasification characteristics of NG microalgae were
analyzed by TGA-MS. High mass loadings cause heat-transfer problems, whereas
small particle sizes led to less diffusion resistance. Gas flow did not affect pyrolysis
and combustion. Gasification temperature had a direct impact on char conversion and
reactivity. Reactivity increased with decreasing sample weight and increasing
porosity. Low gas flow decreased char conversion. Pyrolysis and combustion main
products were generated in the second degradation step. N-compounds evolution was
associated with the microalgae proteins degradation. SO2 release during combustion
could be related to sulphated polysaccharides decomposition. H2 production was
enhanced by steam concentration.
- Thermal characteristics and gas formation during pyrolysis of Fir Wood,
Eucalyptus Wood, Pine Bark, NG microalgae and three individual components of
lignocellulosic biomass (hemicellulose, lignin and cellulose) were analyzed by TGA-

Chapter 6
274
MS. Pyrolysis of lignocellulosic biomass was divided into four zones: moisture
evolution, hemicellulose decomposition, lignin and cellulose degradation and lignin
decomposition. NG microalgae showed the highest thermal stability. The main
products (CO2, light hydrocarbons and H2O) were generated between 200 and 450 ºC.
H2 was produced at high temperatures (>700 ºC). Kinetic model satisfactorily
predicted the pyrolysis of biomass. Furthermore, the statistical significance of the
model was proved.
- Combustion behavior and gas formation from the oxidation process of fir wood,
eucalyptus wood, pine bark and three individual components of lignocellulosic
biomass (cellulose, hemicellulose and lignin) were analyzed by TGA-MS. Biomass
combustion took place into two main stages: devolatilization stage (Dev. stage) and
oxidation stage (Oxid. stage). Most products detected in the combustion of
lignocellulosic biomass were released during the Dev. stage whereas only NO2,
C2H5O+, CO and CO2 were detected at the Oxid. stage. Nitrogen compounds were
released as CH4N, HCN and NOx. Lignocellulosic biomass combustion was fitted to a
first order reaction model (O1).
- Combustion of microalgae took place into two main stages: devolatilization stage
and oxidation stage. However, up to three sub-steps could be identified during the
microalgae combustion attributed to the decomposition of carbohydrates, proteins and
lipids. The ignition characteristic showed that samples CV and SC required less
amount of energy to develop the combustion process. However, NG sample released a
higher amount of heat during the combustion. The kinetic analysis of microalgae
combustion showed that the most representative mechanism for representing the
process was a first order reaction model (O1). The excellent fitting between the
experimental and theoretical curves (maximum mean error was 3.1%, for NG sample)
confirmed the selection of model O1. CO, CO2 and H2O were the main products
released during combustion. Other compounds detected during the combustion of
microalgae were light hydrocarbons (especially CH4); nitrogen compounds (mainly
released as NO, NO2 and HCN); sulfur compounds (SO, SO2 and COS); hydrogen and

Chapter 6
275
other oxygen containing hydrocarbons (ketones, esters, ethers and carboxylic acids).
Nitrogen compounds were found in higher proportions than sulfur ones.
- Samples W and RP showed the best burning profile by combining a high
combustion characteristic factor (CCF) and a high release of combustion heat (Hcomb).
The kinetic analyses of the oxidation process was performed using pseudo mulit-
component separate-stage models (PMSM). The combustion process was divided into
three stages: Devolatilization stage (correlated with the hemicellulose and cellulose
content in the samples), Oxidation stage (influenced by the initial amount of lignin in
the samples) and Remaining burning (associated to the final char burning and
devolatilization of inorganic matter). The high ash content of CR sample enhanced the
amount of volatiles released during the combustion process lowering its activation
energy. The good fitting of experimental curves with theoretical ones validated the
proposed model (mean error below 3.4 %). H2, CO and CO2 were the main product
obtained from energy crops combustion process. Furthermore, NOx were detected in a
higher proportion than other pollutants such as SOx, chloride compounds (CH3Cl) or
aromatic ones (C6H6).
- Thermal characteristics and gas formation during the pyrolysis and gasfication of
eucalyptus wood, fir wood, pine bark and biomass main components (cellulose, xylan
and lignin) were analyzed by TGA-MS. The presence of indigenous inorganic matter
in the gasification process of biomass samples played an important role compared with
their initial chemical composition. The reactivity of biomass samples was correlated
with their alkali index and was ranked as follows: Xylan > lignin > cellulose and
Eucalyptus wood > fir wood > pine bark. The high relevance of inorganic matter was
proved by the inaccuracy of the results obtained by three standards models (VM, SCM
and RPM) which fail to predict the effect of catalytic active species. A semi-empirical
model was proposed in order to accurately model the gasification process. The
proposed model showed errors below 1 %. Furthermore, the models used in this work
were statistically validated. The high production of H2 and CO showed the
predominance of solid-gas reactions. On the other hand, gas phase reactions as water-

Chapter 6
276
gas shift had a higher relevance in the gasification of fir wood due to its high calcium
content. Methanation reactions also took place especially for eucalyptus wood sample
and was correlated to the catalytic effect of potassium.
- A pilot plant was designed to evaluate the degradation of HTfs to be used in
concentrating solar power plants (CSP). Six different HTFs were characterized: two
ionic liquids (1-Butyl-3methylimidazolium tetrafluoroborate ([BMIM][BF4]) and 1-
ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4])), two molten salts (Hitec
XL (60% NaNO3, 40% KNO3, Ca(NO3)2-tetrahydrate) and solar salt (60% NaNO3,
40% KNO3, Ca(NO3)2-tetrahydrate), a commercial HTF (Mobiltherm 605) and a new
oil extracted from the microalgae Nannochloropsis Gaditana (NG oil).
Mobiltherm 605 was selected for the assembling and tuning of the pilot plant due
to its great availability and similar properties to the commercial fluid used in parabolic
trough solar plants. The pilot plant behaviour was stable and no high fluctuations of
data collected were detected. Three isothermal experiments were carried out at 140,
160 and 180 ºC for 15 days. The viscosity was selected as the key parameter to follow
the HTF degradation. Mobiltherm 605 viscosity increased with time, indicating that
the polimerization of hydrocarbon chains took place. The variation of viscosity was 6
% at 180 ºC pointing out the high thermal stability of Mobiltherm 605.
Two mathematical models were developed to estimate the most representative
parameters (µ, Cp and UF) with time on stream and predict the behaviour of the
parameters during operation, respectively. The model was validated with experimental
measurements of viscosity obtaining an error lower than 3%. Finally, the statistical
significance of the model was also proved.
6.2. RECOMMENDATIONS

LIST OF PUBLICATIONS AND CONFERENCES

List of publications and conferences
279
PUBLICATIONS
López-González, D., Valverde, J.L., Fernandez-Lopez, D., Sanchez-Silva, L. (In
press). Thermogravimetric-mass spectrometric analysis on combustion of
lignocellulosic biomass. Bioresource Technology.
López-González, D., Valverde, J.L., Sánchez, P., Sanchez-Silva, L.
2013.Characterization of different heattransfer fluids and degradation study by using a
pilot plant device operating at real conditions. Energy. 54, PP. 240-250
Sanchez-Silva, L., López-González. D., Garcia-Minguillan, A.M., Valverde, J.L.
2013. Pyrolysis,combustion and gasification characteristics of
Nannochloropsisgaditana microalgae. BioresourceTechnology, 130, pp. 321-331
Sanchez-Silva, L., López-González, D., J. Villaseñor, J., Sánchez, P., Valverde, J.L.
2012.Thermogravimetric-mass spectrometric analysis of lignocellulosic and marine
biomass pyrolysis. Bioresource Technology, 109, pp. 163-172.
CONFERENCES
Keynotes A. de Lucas-Consuegra, J.L. Endrino, J. González-Cobos, D. López, J.A. Díaz, J.L. Valverde.ANQUE’s ICCE. Sevilla (Spain), June 2012. Oral presentations J.A. Díaz, J. González-Cobos, D. López-González, A. Romero, J.L. Valverde. ANQUE’s ICCE. Sevilla (Spain), June 2012. L. Sánchez-Silva, D. López-González, J. González-Cobos, J.A. Díaz, J. Villaseñor, P. Sánchez, J.L. Valverde. ANQUE’s ICCE. Sevilla (Spain), June 2012.

List of publications and conferences
280
Posters: 2 contributions.




![Asociación Española de Valorización Energética de la Biomasa [AVEBIOM] Juan Jesús Ramos Valencia, 30 de mayo de 2.013 PRESENTE Y FUTURO DEL SECTOR DE LA.](https://static.fdocuments.es/doc/165x107/54dfc1954a79595b298b521b/asociacion-espanola-de-valorizacion-energetica-de-la-biomasa-avebiom-juan-jesus-ramos-valencia-30-de-mayo-de-2013-presente-y-futuro-del-sector-de-la.jpg)













