
TP aminoacidurias 2017 - qbpatologica.files.wordpress.com · El pool de aminoácidos está formado...
26
Química Biológica Patológica 2017 Licenciatura en Bioquímica Área de Química Biológica 78 TP Nº 6: DIAGNÓSTICO BIOQUÍMICO DE AMINOACIDURIAS Dra. Ethel Viviana Larregle Objetivos: - Presentar conocimientos básicos del metabolismo de los aminoácidos. - Establecer las bases bioquímicas de la patogenia y orientar en el diagnóstico y tratamiento de las aminoacidurias. - Conocer y realizar los test diagnósticos para detectar aminoacidurias. Introducción Las proteínas de la dieta ingresan al tracto gastrointestinal, donde por acción de enzimas proteolíticas y peptidasas, son transformadas en péptidos pequeños, dipéptidos y aminoácidos libres. Estos se absorben por sistemas de transporte específicos en las células intestinales, donde continúa la hidrólisis para dar aminoácidos libres, los cuales pasan a la sangre portal. Los aminoácidos son posteriormente utilizados para biosíntesis de otros aminoácidos y otros compuestos nitrogenados celulares, además de servir como fuente de energía, pero principalmente su destino es la síntesis de proteínas corporales. Por ejemplo, los aminoácidos se requieren para la síntesis de porfirinas, colina y etanolamina de los fosfolípidos, y glicosaminas del tejido conectivo, además de la síntesis de aminas biógenas como (serotonina y adrenalina) y ácidos nucleicos. Figura 1. Resúmen del metabolismo de aminoácidos. El metabolismo de los aminoácidos (figura 1) está regulado por hormonas (insulina, somatotrofina, glucagón, tiroxina, corticosterona y testosterona), coenzimas y metabolitos secundarios que modulan actividades enzimáticas. Los procesos de absorción están controlados genéticamente, siendo el mecanismo de captación de aminoácidos independiente del correspondiente a péptidos. Por otro lado, las concentraciones plasmáticas de un aminoácido pueden determinar la no absorción de otros relacionados estructuralmente, por ejemplo, la hiperfenilalaninemia inhibe la
Transcript of TP aminoacidurias 2017 - qbpatologica.files.wordpress.com · El pool de aminoácidos está formado...

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 78
TP Nº 6: DIAGNÓSTICO BIOQUÍMICO DE AMINOACIDURIAS
Dra. Ethel Viviana Larregle Objetivos:
- Presentar conocimientos básicos del metabolismo de los aminoácidos.
- Establecer las bases bioquímicas de la patogenia y orientar en el diagnóstico y tratamiento de las aminoacidurias.
- Conocer y realizar los test diagnósticos para detectar aminoacidurias.
Introducción Las proteínas de la dieta ingresan al tracto gastrointestinal, donde por acción de
enzimas proteolíticas y peptidasas, son transformadas en péptidos pequeños, dipéptidos y aminoácidos libres. Estos se absorben por sistemas de transporte específicos en las células intestinales, donde continúa la hidrólisis para dar aminoácidos libres, los cuales pasan a la sangre portal. Los aminoácidos son posteriormente utilizados para biosíntesis de otros aminoácidos y otros compuestos nitrogenados celulares, además de servir como fuente de energía, pero principalmente su destino es la síntesis de proteínas corporales.
Por ejemplo, los aminoácidos se requieren para la síntesis de porfirinas, colina y etanolamina de los fosfolípidos, y glicosaminas del tejido conectivo, además de la síntesis de aminas biógenas como (serotonina y adrenalina) y ácidos nucleicos.
Figura 1. Resúmen del metabolismo de aminoácidos.
El metabolismo de los aminoácidos (figura 1) está regulado por hormonas (insulina,
somatotrofina, glucagón, tiroxina, corticosterona y testosterona), coenzimas y metabolitos secundarios que modulan actividades enzimáticas. Los procesos de absorción están controlados genéticamente, siendo el mecanismo de captación de aminoácidos independiente del correspondiente a péptidos. Por otro lado, las concentraciones plasmáticas de un aminoácido pueden determinar la no absorción de otros relacionados estructuralmente, por ejemplo, la hiperfenilalaninemia inhibe la

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 79
captación de triptofano. El pool de aminoácidos está formado por las proteínas dietarias, el recambio de las
proteínas corporales, los aminoácidos circulantes en sangre y las pequeñas cantidades de intermediarios intracelulares.
Los aminoácidos del plasma se filtran en el glomérulo y son reabsorbidos por el túbulo renal; normalmente constituyen solamente el 1 a 3% del nitrógeno urinario total. El patrón de excreción en sujetos normales presenta variaciones individuales, pero la cantidad excretada es constante y muy pequeña.
DEFINICIÓN: Las AMINOACIDURIAS son un grupo de enfermedades que se caracterizan por la
eliminación en orina de grandes cantidades de aminoácidos, como consecuencia de dos factores:
a) Alteración de los mecanismos de transporte, fundamentalmente a nivel renal, sin reabsorción tubular de los aminoácidos filtrados.
b) Alteración en los sistemas enzimáticos involucrados en su metabolización. En las AMINOACIDURIAS la concentración de aminoácidos en orina está
aumentada de 10 a 100 veces el valor normal; el aumento puede deberse a un aminoácido específico o ser generalizado.
AMINOACIDURIAS CAUSADAS POR ALTERACIONES ENZIMATICA S
HIPERFENILALANINEMIAS
Las hiperfenilalaninemias se caracterizan por una elevación persistente de la
concentración plasmática de fenilalanina, lo que produce una alteración de la homeostasis metabólica con diferentes consecuencias clínicas, siendo la principal el desarrollo alterado del sistema nervioso central (cuadro 1).
METABOLISMO NORMAL DE FENILALANINA Fenilalanina es un aminoácido esencial para los mamíferos. Es requerido en mayor
proporción para la síntesis proteica durante el desarrollo temprano. Fisiológicamente se degrada a tirosina por acción de la FENILALANINA
HIDROXILASA (figura 2), reacción irreversible limitada al hígado, riñón y páncreas. La misma requiere de oxígeno molecular, NADH y del cofactor Dihidrobiopterina, que actúa en forma reducida: Tetrahidrobiopterina..
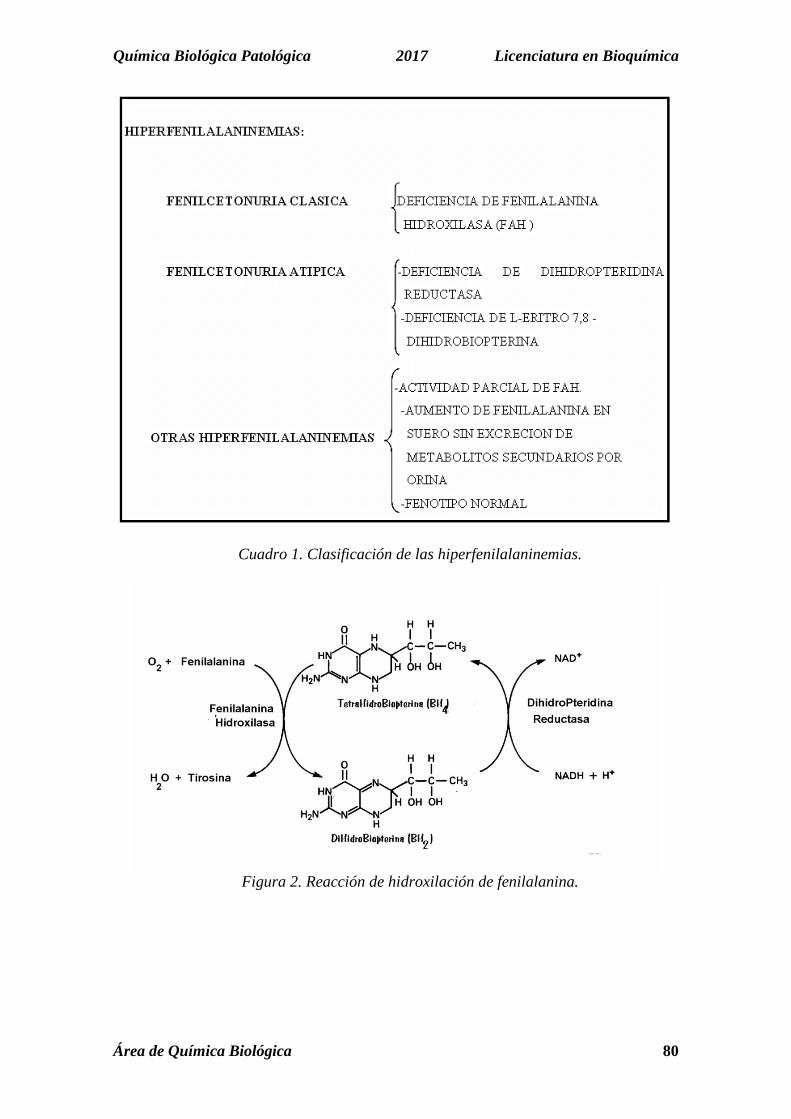
Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 80
Cuadro 1. Clasificación de las hiperfenilalaninemias.
Figura 2. Reacción de hidroxilación de fenilalanina.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 81
VIAS METABÓLICAS DE FENILALANINA
Figura 3: Las Vías Metabolicas de Fenilalanina (excluyendo su incorporación a
proteínas) son: (1) Su hidroxilación, mediada por FAH (mayor), (2) Transaminación (menor) y (3) su decarboxilación (menor). Tomado de Scriver y col., 1989
En fenilcetonuria adquiere importancia la transaminación, para dar Acidos
Fenilacético, Fenilpirúvico y Fenil-láctico, el primero es el responsable del olor característico de la orina
FENILCETONURIA CLÁSICA Enfermedad debida a la deficiencia de FENILALANINA HIDROXILASA. Se
hereda en forma Autosómica Recesiva y tiene una frecuencia estimada de 1:25000. Cuadro Clínico: El 60% de los pacientes son agresivos e hiperactivos, presentan hipertonicidad
muscular y temblores, a veces hay alteraciones electroencefalográficas y convulsiones. La piel, cabellos y ojos tienen menor pigmentación debido a la menor síntesis de
Melanina y puede presentarse eczema. La orina y la piel tiene olor característico a ratón. Sin tratamiento se produce el retraso mental.
Un caso especial es el de hijos de mujeres fenilcetonúricas, los cuales presentan microcefalia, retraso del desarrollo, cardiopatías congénitas y anormalidades esqueléticas a pesar de no ser ellos fenilcetonúricos. Esto puede evitarse instaurando una dieta restringida en fenilalanina en la mujer desde el primer trimestre de embarazo o antes. La hiperfenilalaninemia de la madre es la responsable del cuadro, ya que fenilalanina atraviesa placenta y se acumula en el feto al superar su capacidad de metabolizarla.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 82
Causas sugeridas de la disfunción cerebral: -Defectuosa mielinización, con modificación de la relación sulfátidos -cerebrósidos
de mielina. -Síntesis alterada de neurotransmisores. Al ser fenilalanina precursor de tirosina, Esta
se hace esencial para el fenilcetonúrico. Teniendo en cuenta que tirosina es precursor de aminas biógenas como dopamina y adrenalina, además de tiroxina y melanina, se puede explicar la depleción de catecolaminas. Fenilalanina además inhibe el transporte de tirosina en membranas presinápticas. Serotonina también se encuentra disminuida, en parte porque fenilalanina inhibe su absorción intestinal y, por otro lado, los metabolitos de fenilalanina inhiben a las enzimas 5-OH Triptofano Descarboxilasa, Acido Glutámico Descarboxilasa y la DOPA Descarboxilasa, disminuyendo así las concentraciones de GABA, Adrenalina, Noradrenalina y Dopamina.
Diagnóstico: 1) Hiperfenilalaninemia: Valores de phe en sangre mayores a1 mM, en pacientes
con dieta normal. VALORES NORMALES: hasta 0.15 mM en recién nacidos. hasta 0.12 mM en niños
y adultos. La fenilalaninemia puede determinarse por: -Test de Guthrie y Sun. -cromatografía. -fluorometría 2) Pesquisa neonatal: Permite la identificación de pacientes fenilcetonúricos y la
rápida instauración del tratamiento. Se basa en la medición de los niveles de phe en manchas de sangre recogidas sobre papel de filtro.
3) Prueba de sobrecarga de fenilalanina: permite clasificar las hiperfenilalaninamias y detectar heterocigotos.
4) Determinación de Ácido fenilpirúvico y derivados en orina: un valor positivo tiene valor presuntivo para el diagnóstico de la fenilcetonuria, pueden producirse falsos negativos ya que se requieren valores de fenilalanina plasmática mayores a 15 mg/dl para dar positivo.
5) Exclusión de deficiencia de tetrahidrobiopterina por análisis de biopterina y neopterina en orina.
6) Actividad enzimática en biopsia hepática: ausencia total o valores de 9- 20 % del normal.
7) Análisis de ADN: se puede hacer con sondas de oligonucleótidos para las mutaciones conocidas para fenilcetonuria, por RFLP, por PCR.
8)Diagnóstico prenatal: se realiza a través del análisis de ADN de células líquido amniótico.
Tratamiento: Se administra una dieta restringida en fenilalanina, desde el primer mes de vida para
evitar el retraso mental. Esta debe continuarse hasta los 5-6 años donde se completa la mielinización, manteniendo posteriormente una dieta de bajo contenido de fenilalanina.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 83
FENILCETONURIAS ATIPICAS Los enfermos presentan hiperfenilalaninemia, con deterioro neurológico progresivo,
que no responde al tratamiento dietario y mueren a los pocos años de vida. La actividad de Fenilalanina Hidroxilasa es normal, pero hay deficiencia de
tetrahidrobiopterina, ya sea debida a síntesis alterada o a deficiencia de Dihidropteridina reductasa.
Cuadro Clinico: Se presentan convulsiones, hipotonía, pérdida de peso, trastornos en la marcha. Ya que tetrahidrobiopterina es cofactor también de la Triptofano-5 - Hidroxilasa
existe alteración en la síntesis de neurotransmisores, lo que ocasiona los síntomas neurológicos.
Diagnóstico: a) Deficiencia de dihidropteridina reductasa 1) Sobrecarga de fenilalanina: los pacientes no forman tirosina luego de la carga de
fenilalanina (prueba orientativa). 2) Administración de tetrahidrobiopterina: debería producir descenso de los valores
plasmáticos de fenilalanina, lo que no ocurre en la forma clásica. 3) Administración de Dihidrobiopterina: no produce modificación de los valores
plasmáticos de fenilalanina, ya que la deficiencia está en la reductasa. 4) Dosaje de neopterina y biopterina urinarias, antes y después de administración de
tetrahidrobiopterina y dihidrobiopterina, en enfermos está aumentada la excreción de ambos. Esto puede realizarse por cromatografía bidimensional en papel, por electroforesis, o por HPLC.
5) Actividad enzimática en biopsia de hígado o en cultivo de fibroblastos. La actividad de la enzima en amniocitos se utiliza para diagnóstico prenatal a partir del 2° trimestre.
6) Medición de serotonina y Acido 5-OH- indolacético en orina y líquido cefalorraquídeo.
7) Análisis de ADN: se realiza por RFLP, sirve además para diagnóstico prenatal b) Síntesis defectuosa de tetrahidrobiopterina 1) Los enfermos presentan una relación anormalmente elevada en orina de neopterina
/ biopterina, comparada con controles normales (300-600 veces). 2) Los niveles séricos de fenilalanina disminuyen cuando se administra sepiapterina,
dihidrobiopterina o tetrahidrobiopterina; se incrementan los valores de tirosina y disminuyen los de neopterina en un 80 %.
3) Por biopsia hepática se demuestra actividades normales de Fenilalanina hidroxilasa y Dihidropteridina reductasa.
4) La alteración puede estar a nivel de la sepiapterina sintetasa- Mg dependiente, de la D- 7,8- dihidroneopterina trifosfato sintetasa y de la Guanosina trifosfato ciclohidrolasa.
5) Análisis de ADN: los genes que codifican para las enzimas involucradas no han sido clonados, mapeados o caracterizados aún.
6) Diagnóstico prenatal: se realiza la medición de actividad de sepiapterina sintetasa en eritrocitos fetales obtenidos por fetoscopía. Los niveles de pterinas en líquido amniótico pueden indicar síntesis alterada pero no diferenciar la deficiencia enzimática,

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 84
ni heterocigotas de homocigotas. Tratamiento: Es fundamentalmente dietario, se administra tetrahidrobiopterina, DOPA, Carbidopa
(inhibidor de la descarboxilasa), y 5-OH-triptofano. TIROSINEMIAS La tirosina es un aminoácido no esencial, derivado de la conversión de alimentos o
de proteínas endógenas. Se encuentra en plasma, orina, líquido cefalorraquídeo y tejidos, se filtra en el glomérulo renal y es reabsorbido por los túbulos proximales, perdiéndose un 2% de lo filtrado.
Metabolismo normal de tirosina: Tirosina es utilizada para la síntesis proteica, de hormonas tiroideas, de
neurotransmisores, y pigmentos. Es oxidada por los tejidos a fumarato y acetoacetato, en una cadena de reacciones donde el paso fundamental está catalizado por la Tirosina Amino Transferasa, enzima inducible por glucocorticoides, glucagón y catecolaminas (figura 4)
Las alteraciones hereditarias del metabolismo de tirosina se caracterizan por
tirosinemia, tirosinuria y excreción urinaria de ácidos fenólicos. TIROSINEMIA NEONATAL La tirosinemia neonatal es la alteración más común del metabolismo de tirosina, se
asocia con inmadurez gestacional, presumiblemente la enzima p-OH fenilpirúvico oxidasa hepática no está totalmente desarrollada al nacer y no alcanza a metabolizar la tirosina dietaria. Suele corregirse con vitamina C y con ingesta reducida de proteínas (2 - 3 g/ kg/ día).
Cuadro Clínico: Suele presentarse letargia, movimientos motores erráticos y muy raramente con
ligero retardo mental; estos síntomas desaparecen con el tratamiento Cuadro Bioquimico: En plasma se encuentran elevados los niveles de tirosina, fenilalanina. En orina se
encuentran aumentados ácidos p- OH- fenilacético, p- OH- feniláctico, p- OH- fenilpirúvico, N- acetiltirosina, p- tiramina y tirosina.
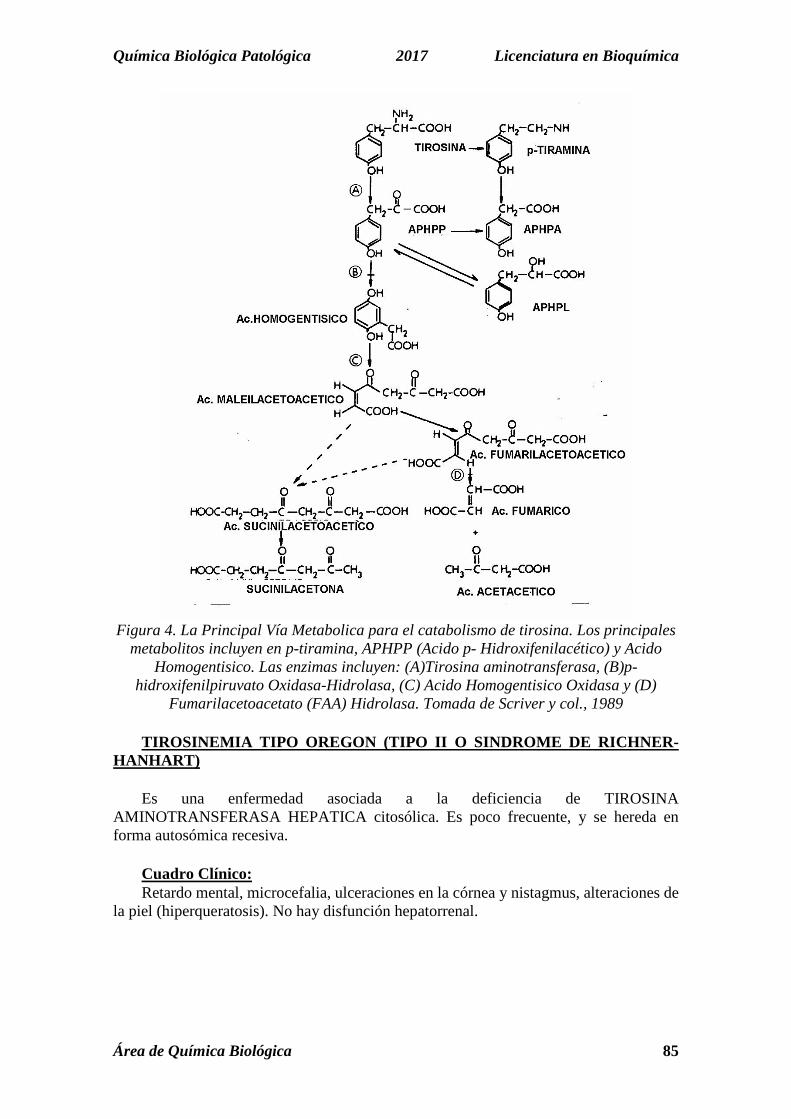
Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 85
Figura 4. La Principal Vía Metabolica para el catabolismo de tirosina. Los principales
metabolitos incluyen en p-tiramina, APHPP (Acido p- Hidroxifenilacético) y Acido Homogentisico. Las enzimas incluyen: (A)Tirosina aminotransferasa, (B)p-
hidroxifenilpiruvato Oxidasa-Hidrolasa, (C) Acido Homogentisico Oxidasa y (D) Fumarilacetoacetato (FAA) Hidrolasa. Tomada de Scriver y col., 1989
TIROSINEMIA TIPO OREGON (TIPO II O SINDROME DE RICH NER-
HANHART) Es una enfermedad asociada a la deficiencia de TIROSINA
AMINOTRANSFERASA HEPATICA citosólica. Es poco frecuente, y se hereda en forma autosómica recesiva.
Cuadro Clínico: Retardo mental, microcefalia, ulceraciones en la córnea y nistagmus, alteraciones de
la piel (hiperqueratosis). No hay disfunción hepatorrenal.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 86
Diagnóstico: Presentan hipertirosinemia e hipertirosinuria. Aumenta además la excreción urinaria
de p- OH- fenilpirúvico por acción de la transaminasa mitocondrial sobre tirosina, lo que determina aumento de p-OH-feniláctico urinario.
TIROSINEMIA HEREDITARIA HEPATORRENAL Se debe a la deficiencia de ACIDO p-OH- FENILPIRUVICO OXIDASA. Se hereda
en forma autosómica recesiva. Existen dos formas: AGUDA y CRONICA. Ambas se deben al mismo alelo mutado o a variantes alélicas.
Cuadro clínico: FORMA AGUDA: Se manifiesta en los primeros meses de vida, con vómitos,
diarreas, disnea, hemorragias, hepatomegalia, raquitismo, hipoglucemia. Se produce la muerte en el 90 % de los enfermos no tratados entre el primero y octavo mes de vida.
FORMA CRONICA: Muestra cirrosis hepática severa y nefropatía; puede desarrollar carcinoma hepático. Puede haber un retraso mental leve a moderado. Hay hiperplasia de islotes de Langerhans e hipoglucemia.
Cuadro Bioquímico: La característica principal es la tirosinuria acompañada de ácidos p-OH-fenilpirúvico
y p-OH- feniláctico. También aparecen signos de alterada función renal: glucosuria, proteinuria, hiperfosfaturia con raquitismo. Hay además aminoaciduria generalizada. En la forma aguda se agrega hipermetioninemia debida al daño hepático. En la forma crónica aumenta la síntesis y excreción de δ-aminolevulínico, con síntomas semejantes a la porfiria aguda.
Otros hallazgos: leucopenia, trombocitopenia, anemia moderada, disminución de los factores de coagulación producidos por el hígado.
En plasma está elevada la concentración de Acido p- OH- fenilpirúvico. Diagnóstico: 1) Detección de metabolitos de tirosina en orina: test (+) con los reactivos de
Benedict, Cloruro Férrico y α-nitrosonaftol. 2) Determinación de actividad enzimática en biopsia hepática. 3) Diferenciar de fructosemia, galactosemia y hepatitis neonatal. 4) Diagnóstico de Heterocigotas: no hay método bioquímico para la detección de los
mismos. Tratamiento: Dieta restringida en fenilalanina y tirosina, previene los síntomas clínicos y revierte
el cuadro bioquímico, pero no se logra superar el daño hepático. Suele administrarse VitD y remover fructosa y galactosa de la dieta.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 87
TIROSINEMIA TIPO l En esta enfermedad se encuentra deficiente la FUMARILACETOACETATO
HIDROLASA. Se hereda en forma autosómica recesiva y afecta a ambos sexos. Cuadro clínico: Se presenta en forma aguda o crónica , con cuadros semejantes al descripto en
tirosinemia hepatorrenal. Cuadro Bioquímico: Esta aminoaciduria cursa con anemia normocítica, leucocitosis y aumento del
número de plaquetas.Además se observa bilirrubinemia y enzimas hepáticas aumentadas, colesterol disminuido, tiempo de protrombina aumentado, α-fetoproteína aumentada.
En plasma aumentan tirosina y metionina, mientras que en orina aumentan tirosina y metabolitos, Acido δ- aminolevulínico y catecolaminas, hay glucosuria, fosfaturia, aminoaciduria generalizada.Por otro lado, orina y plasma contienen succinilacetona.
Diagnóstico: Se hace determinación de succinilacetona urinaria, y actividad de
fumarilacetoacetato hidrolasa en tejidos. Los heterocigotas presentan la mitad de la actividad normal en fibroblastos y linfocitos. DIAGNOSTICO PRENATAL
Se realiza sobre muestras de líquido amniótico, células amnióticas y de vellosidades coriónicas, con los siguientes métodos:
- Determinación directa de la actividad enzimática. - Medición de succinilacetona por GC/MS (cromatografía gaseosa/ espectrometría
de masa). - Capacidad de succinilacetona de inhibir a la enzima Acido δ- aminolevulínico
Dehidratasa. Tratamiento: Dieta de bajo contenido de fenilalanina, tirosina y metionina.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 88
ALBINISMO Grupo de enfermedades que afectan al sistema pigmentario de melanina (ver figura
5).
Figura 5. Biosíntesis de melanina.
Cuadro clínico: Fotofobia, nistagmus, agudeza visual disminuida, ausencia de melanina en la piel,
cabellos y ojos. Trastornos de la coagulación y del sistema de macrófagos, como consecuencia se producen procesos infecciosos.
Los melanocitos están normalmente distribuidos, pero no tienen capacidad para sintetizar melanina a partir de tirosina y TIROSINASA.
� Existen dos clases de albinismo: el albinismo ocular y el oculocutáneo. ALBINISMO OCULOCUTANEO a) Tirosinasa negativo: es la forma clásica. El bulbo capilar del paciente incubado
con tirosina no tiene pigmento visible a la luz o al microscopio. Los melanocitos contienen melanosomas an los estados l y ll que no son
pigmentados. La mutación provoca ausencia total de TIROSINASA. Los niveles séricos de
tirosina, cobre y MSH son normales. La piel y los ojos son rosados, el cabello blanco. b) Tirosinasa positivo: afecta piel y ojos. El bulbo piloso incubado con tirosina
deposita melanina. Se sintetiza melanina pero no se libera. El defecto se desconoce, se sugiere una alteración post- tirosinasa.
El pelo presenta color blanco o amarillo.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 89
c) Sindrome de Hermansky- Pudlak o Albinismo con diátesis hemorrágica d) Sindrome de Chediak- Higashi, albinismo con alteración de enzimas
lisosomales, a nivel de microtúbulos. ALBINISMO OCULAR La forma clásica está ligada al cromosoma X. El epitelio del iris es escaso, se observa
fotofobia y nistagmus. En el epitelio retinal y en la piel se observan macromelanosomas ALCAPTONURIA La alcaptonuria es una enfermedad hereditaria poco frecuente, en la cual no puede
metabolizarse el ACIDO HOMOGENTISICO, producto intermediario del metabolismo de fenilalanina y tirosina, lo que ocasiona la tríada de características de la enfermedad: Aciduria, Ocronosis y Artritis.
La enzima deficiente es la ACIDO HOMOGENTISICO OXIDASA, que se encuentra en hígado y riñón.
La enzima está involucrada en la reacción de oxidación, descarboxilación y migración de la cadena lateral del Ácido Homogentísico, requiere O2, iones ferrosos y ácido Ascórbico para mantener el hierro reducido.
La enfermedad se hereda en forma Autosómica Recesiva. Cuadro Clinico Durante la infancia los pacientes son asintomáticos. Hay depósitos de pigmento
ocronótico intra y extracelular. Es característico la aparición de Acido homogentísico en orina (normalmente sólo
se encuentran trazas). No aumenta en sangre, ya que riñón lo excreta activamente. Esto explicaría la aparición de OCRONOSIS luego de varios años.
OCRONOSIS es la pigmentación de cartílagos y tejidos, con un pigmento color ocre. Se observa en esclerótica, cartílago de orejas, conjuntiva y córneas.
En tejido conectivo produce alteraciones químicas que conducen a la ARTRITIS OCRONOTICA. Están afectados los cartílagos traqueales, laríngeos, costales, tendones y ligamentos. El pigmento aparece en la zona genital y axilar, es amarronado y la ropa se mancha.
La orina es color normal a pH ácido, pero oscurece rápidamente en orinas alcalinas o si hay menor cantidad de Vit C y otros agentes reductores normales en orina.
No se eliminan aminoácidos por orina. En la figura 6 se muestra el mecanismo propuesto de formación del pigmento
ocronótico.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 90
Figura 6. Mecanismo propuesto para la formación del pigmento ocronótico.
Diagnóstico: a) Aparición de color oscuro en la orina alcalinizada. b) Con el reactivo de Benedict en medio ácido , de color naranja vira a marrón. c) Reducción de Molibdato. d) Reacción con Cloruro Férrico, da color púrpura. e) Con Nitrato de Plata, da coloración negra. -CONFIRMACION: a) Cromatografía en capa fina. b) Espectrofotometría de Ácido Maleilacetoacético. Pronóstico: Los pacientes que desarrollan ocronosis, generalmente sufren de invalidez entre los
50 y 70 años, por cambios degenerativos de las articulaciones. Tratamiento No existe. Producida la ocronosis la terapia es correctora de la artritis. Se restringe fenilalanina y tirosina de la dieta y se administra vitamina C. La
administración de ascorbato bloquea la acción inhibitoria de ácido homogentísico sobre la enzima lisil hidroxilasa del metabolismo del colágeno.
ALTERACIONES DEL METABOLISMO DE AMINOACIDOS DE CADE NA
RAMIFICADA Los aminoácidos neutros, leucina, isoleucina y valina (esenciales alifáticos), son
catabolizados por mecanismos semejantes: 1) Transaminación. 2) Descarboxilación oxidativa de α-cetoácidos. 3) Deshidrogenación del acil-CoA saturado.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 91
La deficiencia de deshidrogenasa, que integra un complejo enzimático, conduce a la acumulación de α-Cetoácidos, que se acumulan en sangre y pasan a orina, la que adquiere un olor característico: ENFERMEDAD CON OLOR A JARABE DE ARCE.
En sangre y orina aumenta la concentración de isoleucina, leucina y valina, ya que la transaminación es reversible, sobre todo en tejidos extrahepáticos.
Es un desorden autosómico recesivo, con una incidencia de 1:120000. Cuadro Clínico: Existen cinco fenotipos reconocidos, probablemente producidos por alelos
mutados diferentes que ocupan el mismo locus, lo que ocasiona la heterogeneidad clínica.
La forma clásica presenta: Cetoacidosis severa, en los primeros días de vida, con signos neurológicos como convulsiones, rigidez, vómitos y respiración irregular.
Sin tratamiento se produce el coma y muerte. Tratamiento: Dieta con pequeñas cantidades de los aminoácidos involucrados (los tres esenciales),
desde los primeros días, incluyendo pocas cantidades de proteínas naturales para promover el desarrollo. Administrar tiamina, que estabiliza el complejo enzimático
AMINOACIDURIAS POR ALTERACION DEL TRANSPORTE TRAVES DE
MEMBRANAS CISTINURIA Enfermedad autosómica recesiva, donde se encuentra afectado el transporte de
aminoácidos a nivel del epitelio intestinal y del túbulo renal. Se excretan por orina los aminoácidos CISTINA, ORNITINA, LISINA Y
ARGININA, (COLA) ,que comparten el sistema de transporte. Sus niveles plasmáticos son normales o bajos y no hay depósitos de cistina en los tejidos.
La cistina y los aminoácidos dibásicos parecen compartir el sistema de bajo Km y alta afinidad, que estaría defectuoso en pacientes cistinúricos, esto ocasiona pérdidas de la capacidad reabsortiva del túbulo. En intestino los aminoácidos no absorbidos son transformados por las bacterias de la flora intestinal; lisina, ornitina y arginina son descarboxilados produciendo diaminas como cadaverina y putrescina.
Según el sistema de transporte alterado existen distintas anormalidades. Se pueden encontrar tres tipos distintos de acuerdo a la absorción intestinal.
TIPO 1: No hay transporte de ornitina, lisina y arginina. TIPO 2: No se transporta ninguno de los cuatro aminoácidos. TIPO 3: No se transporta cistina. L- Cisteína se absorbe en forma normal, por un mecanismo distinto a cistina.
Pequeños oligopéptidos y dipéptidos también se absorben normalmente, éstos suplen la carencia nutricional ocasionada por la alteración y proveen los aminoácidos esenciales.
Cuadro Clínico: Si bien los cuatro aminoácidos se excretan en elevada proporción en homocigotas,
cistina es la responsable del cuadro clínico. Debido a su baja solubilidad (figura 7)

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 92
precipita formando cálculos uretrales y renales, que conducen a obstrucciones, infecciones e insuficiencia renal, lo que se asocia con hipertensión.
La enfermedad puede manifestarse en el primer año de vida, pero hace pico a los 20 - 30 años. Los cálculos son radio-opacos, debido a la densidad de grupos sulfuros.
Figura 7. Solubilidad de cistina según el pH del medio.
Diagnóstico: 1) Observación del sedimento urinario: cálculos, cristales hexagonales de color
amarillo-marrón. Los cálculos se forman con una excreción de cistina mayor de 300 mg / g de creatinina en orina. La excreción normal es de 75 a 125 mg / g de creatinina.
2) Test del Nitroprusiato de Sodio: es sensible, pero también da positivo en pacientes con homocistinuria.
3) Confirmar la presencia de cistina por cromatografía en capa fina o en columna. Tratamiento: a) Restricción dietaria para disminuir la producción y excreción de estos
aminoácidos. b) Alcalinización de la orina con bicarbonato o citrato para aumentar la solubilidad c) Aumentar el volumen de orina, bebiendo hasta 4 litros de agua por día. d) Disminuir la excreción de cistina con la administración de D- penicilamina (β-
β′- dimetilcisteina), que forma un compuesto más soluble en orina. Este tratamiento tiene el riesgo de provocar proteinuria y síndrome nefrótico.
ENFERMEDAD DE HARTNUP Enfermedad autosómica recesiva, donde se encuentra alterado el sistema de
transporte para aminoácidos neutros, monoaminos, mono -carboxílicos en intestino y túbulo renal, ocasionando una aminoaciduria generalizada.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 93
Cuadro Clínico: Está aumentada la excreción 5 a 20 veces de alanina, serina, treonina, fenilalanina,
isoleucina, valina, triptofano. Prolina, metionina y arginina son normales. A nivel intestinal, no son absorbidos, y permanecen por más tiempo siendo
susceptibles de ataque microbiano; éstos producen compuestos que sí se absorben. Por ejemplo, luego de una dieta rica en triptofano, se excretan grandes cantidades de ácido indolacético e indoxilsulfato (indicán) por orina y heces, las que toman color amarillento.
Triptofano es precursor de Niacina, como consecuencia de su absorción alterada disminuyen los niveles de niacina y se producen los síntomas dermatológicos y neurológicos del tipo Pelagra.
La absorción deficiente a nivel intestinal y renal conducirían a: - Formación de productos de descomposición que serían tóxicos para el sistema
nervioso central. - Disminuida la concentración de Nicotinamida, causando síntomas de pelagra. - Menor disponibilidad de aminoácidos esenciales, produciendo desnutrición - Menor disponibilidad de triptofano, y por lo tanto de serotonina. SINDROME DE FANCONI Se presenta con aminoaciduria generalizada, por inhibición del transporte tubular. Cuadro clínico - Disfunción compleja de los túbulos proximales, caracterizado por clearence renal
aumentado de fosfato, α-cetoácidos, bicarbonato, glucosa, ácido úrico, y alterada reabsorción de sodio cloruros, agua, bicarbonato, lo que altera el equilibrio electrolítico, pudiendo ser extremadamente severo.
- Metabolismo óseo alterado, presentado como raquitismo en niños y osteomalacia en adultos.
- Hipocalcemia e hiperparatiroidismo asociado. Tratamiento Administrar vitamina D. El cuadro renal mejora al corregir la hipocalcemia. Se
sugiere una anormalidad en el metabolismo renal de 25 (OH) D3 o 1, 25 diOH D3, o incapacidad del túbulo renal para responder al segundo compuesto.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 94
BIBLIOGRAFÍA:
1. Brand E, Harris MM and Biloon S .1930. Cystinuria. The excretion of a cystine complex which decomposes in the urine with the liberation of free cystine. J.Biol.Chem. 86: 315- 331
2. Peter KF Chiu, Eddie SY Chan, Simon SM Hou & CF Ng. 2008. Case Report: Cystinuria: a rare diagnosis that should not be missed. Hong Kong Med J Vol 14 No 5: 399-401. www.hkmj.org
3. J. A. Orts Costa, A. Zúñiga Cabrera, J. Martínez de la Cámara y Salmerón. 2003.Actualización de la cistinuria: aspectos clínicos, bioquímicos y genéticos. An. Med. Interna (Madrid) Vol. 20, N.º 6, pp. 317-326.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 95
TP DE LABORATORIO: DIAGNOSTICO BIOQUIMICO DE LAS AMINOACIDURIAS
Objetivos:
- Desarrollar técnicas cualitativas y cuantitativas, que sirven para el diagnóstico diferencial de las distintas aminoacidurias.
Muestra a utilizar Se utilizará una muestra de orina de 24 horas refrigerada. En caso de no ser analizada
de inmediato, conservar a -20°C. La muestra debe estar límpida, deben descartarse aquellas que muestren contaminación fecal o bacteriana.
MARCHA DE LABORATORIO PARA EL DIAGNOSTICO DE
AMINOACIDURIAS PRUEBA DE LA NINHIDRINA Sirve para detectar un exceso de aminoácidos en orina; es la indicada para
aminoaciduria generalizada, ya que el aumento de un aminoácido puede dar negativa la prueba. En caso de ser positiva debe confirmarse con un estudio cromatográfico de la orina.
Reactivos Solucion de Ninhidrina: 1 g de ninhidrina, disolver en 500 ml de etanol 95%.
Envasar en frasco color caramelo y guardar en heladera. Técnica: En tubo de hemólisis colocar:
- 1ml de reactivo de ninhidrina - 3 gotas de orina Mezclar en vórtex. Examinar el color de la reacción durante 2 a 5 min. La presencia de color azul a
púrpura luego de los 2 min señala un posible exceso de uno o más aminoácidos.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 96
DETERMINACION DE ALFACETOACIDOS ( PRUEBA DE DNFH) Se basa en la propiedad del grupo alfa-ceto para reaccionar con 2,4- dinitro-
fenilhidrazina (DNFH), dando un compuesto de color amarillo, cuya intensidad depende de la concentración del mismo. Para cuantificar se agrega álcali que intensifica el color y aclara la solución.
Reactivos: -Solucion Testigo de Ácido Fenilpirúvico 50 mg% : 5mg de ácido fenilpirúvico ,
llevar a 10 ml con agua destilada. -Dinitrofenilhidrazina (DNFH) 0.2 % en HCl 2N: 200 mg de DNFH disolver en
HCl 2N enrasar a 100 ml . Envasar en frasco oscuro y guardar en heladera. Antes de usar dejar que tome temperatura ambiente.
-NaOH 0.4 N: 4.8 g de NaOH llevar a 300 ml con agua destilada. Técnica: En tubos de ensayo colocar: Reactivos Muestra Testigo Blanco
Orina 1 ml ----------- -----------
H2O destilada ----------- ----------- 1 ml
Testigo ----------- 1 ml -----------
DNFH 1 ml 1 ml 1 ml
Agitar y dejar a temperatura ambiente 30 min.
Interpretación:
Turbidez ligera NEGATIVO Turbidez (+) 50 mg % Turbidez (++) 100 mg % Ppdo. amarillo (+++) 200 mg % Ppdo rojo (++++) 500 mg %
En caso de ser positiva la reacción puede cuantificarse; para ello agregar a todos los
tubos 10 ml de NaOH 0,4 N. Mezclar por inversión. Leer a 505 nm. El cálculo se realiza teniendo en cuenta el valor del testigo:
Esta prueba sirve para el screening de enfermedades congénitas asociadas con alta
excreción de cetoácidos como fenilcetonuria, enfermedad de la orina de jarabe de arce y tirosinosis.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 97
Falsos Positivos: La presencia de cuerpos cetónicos en orina da positiva la reacción. Suele ser positiva además en los primeros días de vida , ya que están aumentada
normalmente la excreción de ácido pirúvico, acetoacético y beta-glutárico. DETERMINACION DE CISTINA : MODIFICACION DE BRAND Cistina en medio alcalino se reduce por el cianuro a cisteína y los grupos sulfhidrilos
reaccionan con el nitroprusiato de sodio, dando color rojo oscuro. Cistina está presente normalmente en cantidades muy pequeñas en orina. La intensidad de color es proporcional a la concentración de cistina en la muestra.
Reacción:
Reactivos:
- Cianuro de sodio 5%: 5 g en 100 ml de agua destilada. Estable a temperatura ambiente 1 mes.
- Na2 [Fe(CN)5 NO].2H2O (Nitroprusiato de sodio) al 15 %: 1.5 g en 10 ml de agua. Preparar en el momento de usar.
Técnica: En tubo de hemólisis colocar: 1.5 ml de orina 3 gotas de NH4OH 0.9 ml de nitroprusiato de sodio al 15 % 50 µl de NaCN ( OJO!) Mezclar y esperar 5 min.; no dejar más de 20 min., ya que la sensibilidad de la
reacción disminuye con el transcurso del tiempo. En presencia de cistina aparecerá color ROJO. Otros compuestos pueden dar lugar a interpretaciones erróneas; por ejemplo
homocistina, y drogas como N- Acetilcisteína, 2- mercaptoetanosulfonato, penicilamina, captopril, y otras drogas que contengan grupos sulfhídricos.
Falsas reacciones negativas pueden deberse a precipitación de la cistina en orinas congeladas, o a contaminación bacteriana.
PRUEBA DEL NITROSONAFTOL Los ácidos fenólicos polihidroxilados y 5- hidroxindoles reaccionan con
nitrosonaftol en presencia de los ácidos nítrico y nitroso dando color rojo o naranja. Reactivos: -Acido Nítrico 2.63 N: 1 volumen de Acido Nítrico concentrado + 5 volúmenes de
agua destilada.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 98
-Nitrito de sodio 2.5%: 2.5 g en 100 ml de agua destilada. Guardar en heladera. -1-Nitrosonaftol 0.1%: 0.1 g en 100 ml de etanol 95%. Guardar en heladera. Técnica: En tubo de hemólisis colocar: -1 ml de Ácido nítrico 2.63N -1 gota de solución de nitrito de sodio -10 gotas de reactivo Nitrosonaftol. Agitar con vórtex. -3 gotas de orina y mezclar nuevamente. Tomar nota del color formado en 2 - 5 min.. Interpretación: La aparición de color rojo o naranja indica la presencia de cantidades excesivas de
tirosina y/o tirosilil derivados, por ejemplo Acido p- hidroxipirúvico, p- hidroxifenilactico y p- hidroxifenilacético. Estos indican metabolismo alterado de tirosina, como tirosinemia hereditaria o neonatal transitoria. Otras enfermedades hepáticas pueden dar alteración en el metabolismo de tirosina, entre ellas galactosemia o fructosuria.
N-Acetiltirosina da resultado positivo, por lo que hay que tener en cuenta si el paciente está recibiendo alimentación parenteral conteniendo este aminoácido.
TEST DE CLORURO FERRICO El test del cloruro férrico es el más usado para la detección de fenilcetonuria. El
cloruro férrico forma con el fenilpiruvato contenido en la orina de fenilcetonúricos, un complejo color verde.
Reactivos:
- Cloruro Férrico 10%: solución acuosa. Guardar en frasco oscuro en heladera. - Ácido sulfúrico 25%( p/v) Técnica: En tubo de hemólisis colocar: 0,5ml de orina reciente 2 gotas de Ácido sulfúrico 25% 10 gotas de FeCl3 10%. Observar el desarrollo de color durante 2 min.. VERDE OSCURO o AZUL
VERDOSO indican la presencia de Ácido fenilpirúvico o compuestos relacionados. El color se aclara lentamente y vira al amarillo.
Para hacer el diagnóstico presuntivo de fenilcetonuria esta prueba debe ser soportada por un test de dinitrofenilhidrazina positivo. No es aplicable a recién nacidos ya que estos excretan muy bajas cantidades de fenilpirúvico por orina.
DETERMINACIÓN CUANTITATIVA DE ÁCIDO FENILPIRÚVICO Se basa en la reacción de cloruro férrico con el Ácido fenilpirúvico, para dar un
complejo azul verdoso. Este tiene un máximo de intensidad a los 2- 3 min. y luego se pierde. Para evitar este fenómeno la reacción se lleva a cabo a bajas temperaturas, en la oscuridad y en presencia de iones férricos.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 99
El buffer glicina- HCl 0.2 M previene la hidrólisis de la sal doble sulfato de hierro y amonio; el nitrato de uranilo se agrega para remover sustancias que causan turbidez.
Reacción:
Reactivos: -Buffer glicina pH 2.2: (a) Glicina 0.2 M : 1.5 g, llevar a 100 ml con agua destilada.
(b) HCl 0.2 M : 1.66 ml de HCl, llevar a 100 ml con agua destilada. (c) Mezclar 100 ml de (a) + 88 ml de (b), llevar a 300 ml. Agregar 3.4 g de NaCl antes de enrasar.
-Nitrato de uranilo al 4%: 4 g. en 100 ml de buffer glicina. -Sulfato de hierro y amonio al 9.8 % : 9.8 g en 100 ml de buffer glicina. -Cloruro férrico al 10 % en agua destilada. -Soluciones standard de fenilpiruvato de sodio: 100 mg %, 75 mg %, 50 mg % y 25
mg%. Técnica:
Expresión de Resultados Con los valores de los testigos se obtiene una curva de calibración. A partir de ella
se calculan los mg % de fenilpiruvato de sodio. Este valor debe multiplicarse por 0.88, que es el factor que lo transforma en mg % de Ácido fenilpirúvico, que surge de la relación de sus pesos moleculares.
El valor obtenido se refiere a la cantidad de creatinina presente en la muestra de orina expresada en g%. Se expresa de este modo para referirlo a un parámetro constante, ya que creatinina se filtra a nivel glomerular y se elimina por orina sin reabsorberse, en forma

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 100
constante y en las circunstancias más diversas. VALORES NORMALES: 0 - 4mmoles/ mol creatinina 0 - 5.8 mg Acido fenilpirúvico / g creatinina VALORES DE FENILCETONURIA: 300 - 1000 mmoles / mol creatinina 400 - 1500 mg / g creatinina. DETERMINACIÓN DE CREATININA En solución alcalina, la creatinina reacciona con el ácido pícrico y forma una
sustancia de color rojo, que permite su cuantificación. La naturaleza del pigmento formado sería la siguiente (*):
(*) Vasillades J: Reaction of Alkaline Sodium Picrate with Creatinine: I. Kinetics and Mechanism of Formation of the Mono-Creatinine Picric Acid Complex. 1976. Clin.
Chem. 22(10), 1664-1671. Reactivos: - Solución standard de creatinina 0.5 mg / ml en H2O destilada. - Reactivo alcalino: a) Acido Pícrico al 1% : 0.25 g en 25 ml de Agua destilada.
b) NaOH 10 %: 0.5 g en 5 ml de agua destilada. Mezclar antes de usar: 22.5 ml de a) + 4.5 ml de b) y diluir a 150 ml con agua destilada.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 101
Técnica:
Cálculos:
En donde la concentración del testigo debe estar expresado en las siguientes
unidades: g/dl. Valores Normales de Creatinina en Orina: 15-25 mg/Kg/24 hs 0,9 - 1,5 g/24 hs. CROMATOGRAFÍA DE AMINOÁCIDOS EN ORINA La ninhidrina produce la reacción que caracteriza al grupo alfa -amino. Por el calor
y en presencia de ninhidrina el aminoácido es oxidado a aldehído, conteniendo un átomo de carbono menos, resultando así la liberación de NH3 y CO2.
A pH bajo, el amoniaco, la ninhidrina y el producto de la reacción (hidrantina), reaccionan para formar un compuesto coloreado que permite detectar y cuantificar los aminoácidos en los procesos cromatográficos.
Reactivos: NINHIDRINA 300 mg % EN METANOL SOLVENTE DE CORRIDA: Butanol : Acético : Agua ( 80: 20: 20 ) TESTIGOS: fenilalanina, glicina, lisina, valina, ornitina, etc. 1 mg / ml en agua
destilada o solución fisiológica.

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 102
Técnica: a) Procesamiento de la Muestra La muestra se debe purificar por columna de intercambio iónico. Se utilizan resinas
DOWEX 50 W - X 8 9 grano 20-50 forma H+), activadas con Acido antes de usar. • Sembrar 10 ml de orina de 24 horas. • Lavar con agua destilada. • Eluir con 10 ml de Hidróxido de amonio 1 N. • Extraer la décima parte del eluido y llevarlo a sequedad en estufa a 37°C b) Cromatografía en Capa Fina. • Retomar el extracto seco con 1 ml de isopropanol al 10 %. • Sembrar en banda la placa de Sílica gel G- 60, previamente activada en estufa
120 °C 1 hora, la muestra y testigos. • Colocar en la cuba con el solvente de corrida (colocar el solvente unos 15 min.
antes para saturar el ambiente). Dejar correr 180 min.. • Sacar la placa y dejar secar a temperatura ambiente. Revelar con Ninhidrina,
llevar a estufa 5 min..

Química Biológica Patológica 2017 Licenciatura en Bioquímica
Área de Química Biológica 103
GLOSARIO: Eccema: Dermatitis superficial de causa desconocida. En el primer estadio pede ser
pruriginoso, eritematoso, pápulo-vesicular, edematoso y húmedo. Posteriormente se convierte en costroso, escamoso, engrosado y liquenificado.
Microcefalia: anomalía congénita caracterizada por tamaño anormalmente pequeño de la cabeza en relación con el resto del cuerpo y por subdesarrollo del cerebro que conduce a cierto grado de retraso mental. La cabeza se encuentra más de dos desviaciones estándar por debajo del tamaño medio para la edad, sexo, la raza, y el periodo de gestación.
Nistagmo (en ingles:Nystagmus): movimiento involuntario y rítmico de los ojos. Las oscilaciones pueden ser horizontales, verticales, rotatorias o mixtas.
Pelagra: enfermedad producida por la deficiencia de la conversión del precursor triptófano en niacina. Se caracteriza por una dermatitis descamativa que afecta especialmente a la piel expuesta al sol, junto con glositis, inflamación de las membranas mucosas, diarrea y trastornos mentales tales como depresión, confusión, desorientación, alucinaciones y delirio.


















