
Tratado de Trombosis
508
-
Upload
javier-andres-perez -
Category
Documents
-
view
68 -
download
1
description
Tratado de Trombosis
Transcript of Tratado de Trombosis


TRATADO DE TROMBOSIS


Tratado de trombosis
Luis Fernando García–Frade Ruiz
Médico Internista del Hospital Ángeles del Pedregal.Licenciatura como Médico Cirujano en la
Facultad Mexicana de Medicina de la Universidad “La Salle”.Diplomado en Diabetes, Hipertensión y Obesidad,
Centro Médico Nacional “Siglo XXI”.Especialidad en Medicina Interna en la Universidad “La Salle”,
Hospital Ángeles del Pedregal.
EditorialAlfil

Tratado de trombosis
Todos los derechos reservados por:� 2015 Editorial Alfil, S. A. de C. V.Insurgentes Centro 51–A, Col. San Rafael06470 México, D. F.Tels. 55 66 96 76 / 57 05 48 45 / 55 46 93 57e–mail: [email protected]
ISBN 978–607–741–124–6
Dirección editorial:José Paiz Tejada
Revisión editorial:Irene Paiz, Berenice Flores
Ilustración:Alejandro Rentería
Diseño de portada:Arturo Delgado
Impreso por:Solar, Servicios Editoriales, S. A. de C. V.Calle 2 No. 21, Col. San Pedro de los Pinos03800 México, D. F.Octubre de 2014
Esta obra no puede ser reproducida total o parcialmente sin autorización por escrito de los editores.
Los autores y la Editorial de esta obra han tenido el cuidado de comprobar que las dosis y esquemasterapéuticos sean correctos y compatibles con los estándares de aceptación general de la fecha dela publicación. Sin embargo, es difícil estar por completo seguros de que toda la información pro-porcionada es totalmente adecuada en todas las circunstancias. Se aconseja al lector consultar cui-dadosamente el material de instrucciones e información incluido en el inserto del empaque de cadaagente o fármaco terapéutico antes de administrarlo. Es importante, en especial, cuando se utilizanmedicamentos nuevos o de uso poco frecuente. La Editorial no se responsabiliza por cualquier alte-ración, pérdida o daño que pudiera ocurrir como consecuencia, directa o indirecta, por el uso y apli-cación de cualquier parte del contenido de la presente obra.

Colaboradores
Dra. Yolanda AburtoDepartamento de Terapia Endovascular del Instituto Nacional de Neurología yNeurocirugía. Terapia Endovascular Neurológica, Centro Integral de Enferme-dad Vascular Cerebral, Hospital Ángeles del Pedregal.Capítulo 8
Dr. Marlon Patricio Aguirre EspinosaCardiólogo Ecocardiografista del Instituto Nacional de Cardiología “IgnacioChávez”.Capítulo 24
Dr. Antonio Arauz GóngoraClínica de Enfermedad Vascular Cerebral. Instituto Nacional de Neurología yNeurocirugía “Manuel Velasco Suárez”.Capítulo 9
Dr. Raúl Ariza AndracaMédico Internista. División de Estudios de Posgrado, Facultad de Medicina,UNAM.Capítulo 19
Dra. Leonor A. Barile FabrisJefa del Departamento de Reumatología del Hospital de Especialidades del Cen-tro Médico Nacional “Siglo XXI”, IMSS. Investigadora Nacional. Profesora deReumatología.Capítulo 19
V

VI (Colaboradores)Tratado de trombosis
Dr. Fernando Barinagarrementeria AldatzHospital Ángeles de Querétaro. Querétaro, México.Capítulo 9
Dr. René Bourlon CuéllarMédico Internista y Profesor Adjunto del Curso de Medicina Interna del HospitalÁngeles del Pedregal. Consejero Titular del Consejo Mexicano de Medicina In-terna.Capítulo 23
Dra. Christianne Bourlon de los RíosMédico Residente en la subespecialidad de Hematología, Instituto Nacional deCiencias Médicas y Nutrición “Salvador Zubirán”.Capítulo 23
Dra. María Teresa Bourlon de los RíosMédico Residente en la subespecialidad de Oncología Médica, Instituto Nacio-nal de Ciencias Médicas y Nutrición “Salvador Zubirán”.Capítulo 23
Dra. Rocío Catana HernándezDepartamento de Reumatología del Hospital de Especialidades del Centro Médi-co Nacional “Siglo XXI”, IMSS.Capítulo 19
Dr. José Mauricio Cedillo FernándezResidente de Medicina Interna, Hospital Ángeles del Pedregal.Capítulo 2
Dr. Carlos F. Cuevas GarcíaDirector del Hospital de Especialidades “Dr. Bernardo Sepúlveda G.”, CentroMédico Nacional “Siglo XXI”, IMSS. Profesor Titular de Posgrado de Neurolo-gía, UNAM.Capítulo 10
Dr. Víctor A. de la Garza EstradaMédico Internista. Jefe de la División de Medicina del Hospital Ángeles del Pe-dregal.Capítulo 18
Dr. Marcos César Gallegos SolórzanoMedicina Interna y Neumología, avalado por la Universidad Nacional Autónomade México. Médico Adscrito al Centro de Investigación de Enfermedades Infec-

VIIColaboradores
ciosas del Instituto Nacional de Enfermedades Respiratorias, SSA. Subjefe deUrgencias del Hospital Ángeles del Pedregal. Profesor Adjunto del Curso de Pos-grado de Neumología en la Universidad Nacional Autónoma de México.Capítulo 13
Dr. Luis Fernando García–Frade RuizEgresado de la Facultad Mexicana de Medicina de la Universidad La Salle. Médi-co Internista en el Hospital Ángeles del Pedregal.Capítulos 1, 3, 4, 5, 14, 15, 18, 20, 21, 28
Dr. Rafael Gutiérrez CarreñoMédico Cirujano, UNAM. Fellow del Arizona Heart Institute, Arizona, EUA.Exsecretario Académico, Facultad de Medicina, ULSA. Exjefe de EducaciónMédica, Hospital Ángeles del Pedregal. Expresidente de la Sociedad Mexicanade Angiología y Cirugía Vascular.Capítulo 11
Dr. Rafael Hurtado MonroyJefe del Departamento de Hematología del Hospital Ángeles del Pedregal.Miembro de la Sociedad Americana de Hematología y de la Sociedad Americanade Oncología.Capítulo 2
Dr. Ángel LeeTerapia Endovascular Neurológica, Centro Integral de Enfermedad Vascular Ce-rebral, Hospital Ángeles del Pedregal. Consultante de Neurocirugía, InstitutoNacional de Ciencias Médicas y Nutrición “Salvador Zubirán”.Capítulo 8
Dr. René Iván Lizola MargolisSociedad Mexicana de Angiología y Cirugía Vascular. Metepec, Estado de Mé-xico.Capítulo 11
Dra. Julieta Lomelín GascónMédico Interno de Pregrado. Facultad Mexicana de Medicina, Universidad “LaSalle”.Capítulo 18
Dra. Ana Olga López AisaMédico Cirujano egresada de la Facultad Mexicana de Medicina de la Universi-dad “La Salle”. Diplomado en Excelencia para Asistentes Administrativas, Psi-cólogos Industriales Asociados, S. C. Maestría en Administración de Organiza-

VIII (Colaboradores)Tratado de trombosis
ciones de la Salud, Universidad “La Salle”. Coordinador Médico en ClínicaBariátrica.Capítulo 22
Dra. Mónica Mendieta HernándezSociedad Mexicana de Angiología y Cirugía Vascular. México, D. F.Capítulo 11
Dra. Tania Teresa Mora AriasReumatología y Medicina Interna, Hospital Ángeles del Pedregal.Capítulo 2
Dr. Luis Fernando Mundo GallardoMédico Gastroenterólogo y Endoscopista del Hospital Ángeles del Pedregal.Fellow del American College of Gastroenterology.Capítulo 16
Dr. Víctor Hugo Navarro CejaMédico Adscrito al Servicio de Angiología y Cirugía Vascular del Centro MédicoNacional “Siglo XXI”, IMSS. Coordinador del Comité de Flebología de la Socie-dad Mexicana de Angiología y Cirugía Vascular, A. C. Médico Adscrito del Hos-pital Ángeles del Pedregal.Capítulo 12
Dr. Federico Javier Ortiz IbarraDirector General de Laboratorios Diagnomol.Capítulo 27
Dr. Juan Carlos Peláez PiedrahitaMédico Cardiólogo del Hospital Ángeles del Pedregal.Capítulo 6
Dra. Irene Pérez PáezMédico Internista.Capítulo 10
Dr. Eduardo Perusquia OrtegaJefe del Departamento de Neurofisiología del Hospital Ángeles del Pedregal.Capítulo 8
Dr. Óscar Quiroz CastroJefe del Departamento de Imagen del Hospital Ángeles del Pedregal. Expresi-dente de la Sociedad Mexicana de Radiología e Imagen.Capítulo 25

IXColaboradores
Enf. Ivonne Ramírez DíazEnfermera General con práctica en la medicina privada.Capítulo 28
Dr. Ignacio Rodríguez BrionesCardiólogo y Electrofisiólogo egresado del Instituto Nacional de Cardiología“Ignacio Chávez”. Profesor de Pregrado de la Facultad de Medicina, UASLP. Di-rector del Centro de Investigación “Cardioarritmias e Investigación” en San LuisPotosí, S. L. P.Capítulo 7
Dr. Francisco Javier Roldán GómezCardiólogo Ecocardiografista del Instituto Nacional de Cardiología “IgnacioChávez”. Investigador Nacional.Capítulos 14, 24
Dr. Bernardo Ronzón FernándezPatólogo Clínico. Jefe del Laboratorio y Banco de Sangre del Hospital Ángelesdel Pedregal. Profesor de Patología Clínica, UNAM.Capítulo 26
Dr. Jorge SantosNeurocirugía y Terapia Endovascular Neurológica, Centro Médico “La Raza”,Instituto Mexicano del Seguro Social.Capítulo 8
Dra. Judith Sandra SarminaCirujana Oftalmóloga con alta especialidad en Glaucoma, egresada de la UNAMy del Instituto de Oftalmología “Conde de Valenciana”. Jefe del Servicio de Of-talmología del Hospital Regional “Lic. Adolfo López Mateos”, ISSSTE. Profe-sor Titular del Curso de Posgrado de Oftalmología.Capítulo 17
Dr. Emmanuel Solís AyalaResidente de Medicina Interna, Hospital Ángeles del Pedregal.Capítulo 14
QFB Ema Valenzuela MéndezGerente de Biología Molecular, Laboratorios Diagnomol.Capítulo 27
Dr. Salvador Vargas CruzEspecialista en Cirugía General egresado del Hospital de Especialidades del Cen-tro Médico Nacional “Siglo XXI”, IMSS. Miembro de la Asociación Mexicana

X (Colaboradores)Tratado de trombosis
de Cirugía General y de la Asociación Latinoamericana de Cirugía Endoscópica.Miembro de la Sociedad Médica del Hospital Ángeles del Pedregal. Fellow delAmerican College of Gastroenterology.Capítulo 16
Dr. Pablo Vargas ViverosMédico Internista del Hospital Ángeles del Pedregal.Capítulo 2
Dra. Berenice Vicente HernándezResidente de Medicina Interna, Hospital Ángeles del Pedregal.Capítulo 28
Dra. Miriam Villada MenaEspecialidad en Medicina Critica en la Universidad “La Salle”. Medico Intensi-vista de la unidad de Terapia Intensiva del Hospital Ángeles del Pedregal. Jefede Áreas Críticas del Hospital Ángeles Clínica Londres. Profesor Titular de laEspecialidad en Medicina Crítica, UNAM. Profesor Titular de Posgrado de laFacultad Mexicana de Medicina de la Universidad “La Salle”.Capítulo 22
Dr. Marco Antonio ZentenoJefe del Departamento de Terapia Endovascular del Instituto Nacional de Neuro-logía y Neurocirugía. Profesor Titular del Curso en la Universidad Nacional Au-tónoma de México. Miembro de la Academia Nacional de Medicina.Capítulo 8

Contenido
Prefacio XV. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Rafael Hurtado MonroyIntroducción XVII. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
1. Sistema normal de la coagulación 1. . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
2. Trombofilia 11. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Pablo Vargas Viveros, Tania Teresa Mora Arias,José Mauricio Cedillo Fernández, Rafael Hurtado Monroy
3. Fármacos antitrombóticos 31. . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
4. Tromboprofilaxis 65. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
5. Enfermedad vascular 81. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
6. Angina inestable e infarto agudo del miocardio 91. . . . . . . . . . . Juan Carlos Peláez Piedrahita
7. Fibrilación auricular y trombosis 105. . . . . . . . . . . . . . . . . . . . . . Ignacio Rodríguez Briones
8. Enfermedad carotídea 119. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Eduardo Perusquia Ortega, Ángel Lee, Yolanda Aburto,Jorge Santos, Marco Antonio Zenteno
XI

XII (Contenido)Tratado de trombosis
9. Ataque isquémico transitorio, infarto cerebralaterotrombótico e infarto lacunar 149. . . . . . . . . . . . . . . . . . . . . . Fernando Barinagarrementeria Aldatz, Antonio Arauz Góngora
10. Trombosis venosa cerebral 163. . . . . . . . . . . . . . . . . . . . . . . . . . . . Carlos F. Cuevas García, Irene Pérez Páez
11. Insuficiencia arterial periférica 191. . . . . . . . . . . . . . . . . . . . . . . . Rafael Gutiérrez Carreño, Mónica Mendieta Hernández,René Iván Lizola Margolis
12. Trombosis venosa profunda 205. . . . . . . . . . . . . . . . . . . . . . . . . . . Víctor Hugo Navarro Ceja
13. Tromboembolia pulmonar 215. . . . . . . . . . . . . . . . . . . . . . . . . . . . Marcos César Gallegos Solórzano
14. Trombosis venosa yugular 241. . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz, Emmanuel Solís Ayala,Francisco Javier Roldán Gómez
15. Enfermedad vascular renal 251. . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
16. Trombosis mesentérica 269. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando Mundo Gallardo, Salvador Vargas Cruz
17. Oftalmología y trombosis 283. . . . . . . . . . . . . . . . . . . . . . . . . . . . . Judith Sandra Sarmina
18. Trombosis en el paciente oncológico 295. . . . . . . . . . . . . . . . . . . . Víctor A. de la Garza Estrada,Luis Fernando García–Frade Ruiz, Julieta Lomelín Gascón
19. Enfermedad inmunitaria y trombosis. Síndrome deanticuerpos antifosfolípidos 303. . . . . . . . . . . . . . . . . . . . . . . . . . . Raúl Ariza Andraca, Leonor A. Barile Fabris,Rocío Catana Hernández
20. Embarazo y trombosis 315. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
21. Anticonceptivos orales, terapia hormonal sustitutiva ytrombosis 335. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz
22. Trombosis en el paciente crítico 347. . . . . . . . . . . . . . . . . . . . . . . . Miriam Villada Mena, Ana Olga López Aisa
23. Trombocitopenia inducida por heparina. Síndrome de HIT 363René Bourlon Cuéllar, María Teresa Bourlon de los Ríos,Christianne Bourlon de los Ríos

XIIIContenido
24. Ecocardiografía en el paciente con trombosis 377. . . . . . . . . . . . Marlon Patricio Aguirre Espinosa,Francisco Javier Roldán Gómez
25. Imagenología en el paciente con trombosis 387. . . . . . . . . . . . . . . Óscar Quiroz Castro
26. Laboratorio clínico en trombosis 405. . . . . . . . . . . . . . . . . . . . . . . Bernardo Ronzón Fernández
27. Biología molecular en la trombosis 431. . . . . . . . . . . . . . . . . . . . . Ema Valenzuela Méndez, Federico Javier Ortiz Ibarra
28. Fármacos, alimentos y hierbas en el paciente contrombosis 445. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Luis Fernando García–Frade Ruiz, Ivonne Ramírez Díaz,Berenice Vicente Hernández
Índice alfabético 459. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .

XIV (Contenido)Tratado de trombosis

PrefacioRafael Hurtado Monroy
Agradezco la invitación del Dr. Luis Fernando García–Frade Ruiz para escribirel prefacio de la segunda edición del Manual de trombosis y terapia antitrombó-tica.
Persisto en el abordaje de la implementación de las medidas de prevencióncomo la mejor forma de ejercer la medicina, ya que con el paso de los años con-cluyo con tristeza y preocupación que las recomendaciones internacionales paraevitar las enfermedades trombóticas y sus consecuencias no se llevan a cabo, apesar de la difusión de que son las primeras causas de muerte por estas enferme-dades, en paralelo con los accidentes.
El desarrollo y la aplicación de los factores de riesgo para el surgimiento dela trombosis son la piedra angular para establecer las medidas profilácticas perti-nentes para cada paciente, ya sea en el hospital o de manera externa, de todas lasespecialidades médicas y quirúrgicas.
Los avances tecnológicos y moleculares en el conocimiento de la trombofilia(primaria y secundaria) definen en forma precisa la conducta terapéutica que pue-de decidir la vida o la muerte de un paciente, por lo que es aquí donde resulta clavela importancia de las investigaciones recientes, sobre todo del desarrollo de nue-vos medicamentos con excelente especificidad y sensibilidad, con el progresivodesplazamiento de las anteriores formas de tratamiento, todo con el objeto de am-pliar la seguridad farmacológica en mayor beneficio de los pacientes.
Aunque hay una creciente y robusta aportación de información en las diferen-tes fuentes de difusión en la medicina, creo que sólo algunas proporcionan cono-cimiento y en realidad pueden hacer cambios válidos en la conducta médica, por
XV

XVI (Prefacio)Tratado de trombosis
lo que debemos tener mucho cuidado al seleccionar los avances más relevantes,sin los sesgos comerciales, que son una muy mala moda de nuestros tiempos.
La elaboración de esta obra es el resultado no sólo del éxito de la obra que laantecedió, Manual de trombosis y terapia antitrombótica, sino más bien de lanecesidad de la constante actualización de los conceptos etiológicos, fisiopatoló-gicos y terapéuticos en la enfermedad trombótica, que no rara vez comprometela vida del paciente y nos obliga a incrementar nuestro nivel de conocimientos,sobre todo cuando estamos en la atención primaria e incluso en la atención tercia-ria de la medicina, es decir, desde el diagnóstico temprano y preciso hasta el trata-miento sofisticado y sobre todo oportuno.

IntroducciónLuis Fernando García–Frade Ruiz
Con entusiasmo elaboramos esta nueva edición, dada la gran aceptación que tan-to en México como fuera de él obtuvo el Manual de trombosis y terapia antitrom-bótica. De este modo, tras la actualización de los procesos fisiopatológicos, la in-corporación de nuevos agentes antitrombóticos, el reordenamiento de los temasy la suma de ideas innovadoras se da ahora lugar a este nuevo texto, que cuentacon la siempre valiosa y entusiasta colaboración de los más selectos expertos enel área, para lograr satisfacer las necesidades académicas respecto al tema en unsolo texto.
En los últimos años nos ha tocado vivir de manera muy afortunada un gran mo-vimiento en pro de ampliar nuestra atención y conocimiento en relación con elfenómeno trombótico, en el que la participación de la industria farmacéutica através de la proyección de nuevos agentes antitrombóticos ha generado un sinnú-mero de actividades de educación médica continua, tras lo cual la trombosis esya hoy un fenómeno mucho más presente entre los médicos de todas las especiali-dades.
El objetivo de este Tratado de trombosis es continuar con la gran tarea que suantecesor realizó, proporcionar de una manera práctica el conocimiento, en reali-dad complejo, que hoy en día encierran la trombosis y su adecuado tratamiento,bajo la invaluable escritura de médicos líderes en cada uno de los capítulos.
La epidemia que hoy se vive en relación con la trombosis, la cual la colocacomo la principal causa de mortalidad en México y en los países desarrollados,sólo se podrá combatir a través de la constante educación médica, misma que,como indica el texto, involucra a todas las especialidades médicas. Será la colec-
XVII

XVIII (Introducción)Tratado de trombosis
tividad profesional quien, con base en el reconocimiento de un claro problemade salud, pueda exigir los recursos materiales y humanos para hacer frente a talenfermedad. El trabajo multidisciplinario es fundamental en la adecuada y actua-lizada atención del paciente con trombosis, en donde el abordaje diagnóstico pro-fundo determina por completo tanto el tratamiento como la duración del mismo.
La elaboración de diagnósticos genéticos en reconocimiento de trombofiliasprimarias se ha ido haciendo cada vez más accesible, lo que, junto a una mayordifusión de las trombofilias secundarias, debe llevarnos a los profesionales de lasalud a una mayor tromboprofilaxis tanto primaria como secundaria.
El uso adecuado de los recientes anticoagulantes orales conlleva un profundoconocimiento de la enfermedad y de cada uno de ellos, con el fin de obtener susgrandes beneficios a través de su practicidad y no sus devastadoras complicacio-nes, generadas por un uso inadecuado de ellos.
En el presente Tratado de trombosis intentamos que el lector realice un viajedesde la fisiología normal de la coagulación, pasando por todos y cada uno de losmúltiples escenarios clínicos que la trombosis puede presentar, su típica enferme-dad venosa, sus presentaciones oftalmológicas, sus múltiples herramientas diag-nósticas y la acción precisa de sus tratamientos, hasta la fina y moderna aplica-ción de la biología molecular para la realización de diagnósticos específicosdentro de una medicina civilizada.
De la misma forma, incorporamos de manera práctica, pero a la vez novedosa,en un mismo capítulo la inflamación endotelial con su respectiva aterotrombosis,así como la trombosis “pura”, con el fin de facilitar el reconocimiento y el manejode sus múltiples factores de riesgo, su diagnóstico y su tratamiento a manera deuna sola enfermedad vascular. Es así que sólo queda decir que:
Quien logre ver lo no visible, curará de verdad.

Agradezco a todos los médicos que hicieron posible este libroy a Editorial Alfil por su apoyo incondicional.
Este libro está dedicado a ti, que piensas en tu paciente.

XX Manual de trombosis y terapia antitrombótica

Dedicado a todos los médicos y a sus familias,que en su labor diaria reflejan “el haber nacido para dar”.


Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
1Sistema normal de la coagulación
Luis Fernando García–Frade Ruiz
Desde el punto de vista didáctico el sistema de la coagulación se divide en tresgrandes fases: fase vascular, fase plaquetaria y fase plasmática. Dichos procesossuceden en realidad de manera simultánea y constante en el organismo con el finde evitar la pérdida extravascular de sangre y llevar a cabo la reparación del vasosanguíneo lesionado.
CLASIFICACIÓN
El sistema de la coagulación se divide en hemostasia primaria y hemostasia se-cundaria. La hemostasia primaria se refiere a la respuesta celular, mientras quela secundaria comprende los factores solubles circulantes de la coagulación queconvergen en una cascada de reacciones enzimáticas para generar fibrina comoproducto final.
Los componentes celulares y proteínicos de la coagulación se encuentran efi-cazmente coordinados y son interdependientes.
FASE VASCULAR
Ante la lesión de la pared vascular de un vaso sanguíneo se activa de manera in-mediata la primera fase de la coagulación mediante un mecanismo de vasocons-tricción, llamado fase vascular, el cual de forma mecánica disminuye la pérdidasanguínea a través de la disrupción vascular.
1

2 (Capítulo 1)Tratado de trombosis
La contracción vascular se debe a reflejos nerviosos, espasmo miógeno localy factores humorales procedentes del tejido traumatizado y de las plaquetas san-guíneas.
En los vasos pequeños las plaquetas son responsables de la mayor parte de lavasoconstricción por liberación de la sustancia vasoconstrictora tromboxano A2.Cuanto mayor sea el traumatismo que sufra el vaso, mayor será la intensidad delespasmo. Este espasmo vascular se puede prolongar durante varios minutos o in-cluso horas, y en el transcurso de este tiempo ocurren los procesos de tapona-miento plaquetario y fase plasmática.
FASE PLAQUETARIA
Las plaquetas son minúsculos discos redondos u ovalados de 2 a 4 �m de diáme-tro. Se forman en la médula ósea a partir de los megacariocitos, que son célulasextremadamente grandes de la serie hematopoyética de la médula ósea, que sesegmentan en la propia médula o poco después de abandonarla. La concentraciónnormal de plaquetas en la sangre es de 150 000 a 450 000/�L. Las plaquetas care-cen de núcleo y contienen en su citoplasma factores activos, como:
1. Moléculas de actina y miosina, semejantes a las que se encuentran en lascélulas musculares.
2. Residuos de retículo endoplásmico y de aparato de Golgi, que sintetizan di-versas enzimas y almacenan grandes cantidades de iones de calcio.
3. Sistemas enzimáticos capaces de formar ATP y ADP.4. Sistemas enzimáticos que sintetizan prostaglandinas.5. Una proteína llamada factor estabilizador de la fibrina.6. Un factor de crecimiento que hace que las células endoteliales, las del mús-
culo liso vascular y los fibroblastos se multipliquen y crezcan, lo cual pro-duce la reparación de la pared vascular lesionada.
Las plaquetas tienen una vida media de 8 a 12 días, al final de los cuales parecenhaber agotado su proceso vital.
En condiciones normales 1 x 1012 plaquetas fluyen de manera continua a travésde 1 000 m2 de superficie vascular en el organismo sin adherirse ni agregarse; noobstante, una disrupción de la integridad de la pared vascular da lugar a interac-ciones complejas entre las plaquetas circulantes, las células endoteliales y las es-tructuras subendoteliales.
De manera simultánea a la fase vascular se activa la segunda fase de la coagu-lación, llamada fase plaquetaria o hemostasia primaria, la cual tiene la finalidadde formar un tapón plaquetario sobre la lesión endotelial. Durante los primerosminutos posteriores a la lesión vascular estas dos primeras fases de la coagulación

3Sistema normal de la coagulaciónE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
intentan inhibir la pérdida extravascular de sangre a través de la fase plaquetaria,la cual se divide a su vez en tres grandes pasos:
� Adhesión plaquetaria.� Liberación granular.� Agregación plaquetaria.
Adhesión plaquetaria
Tras la lesión de la pared vascular el vaso sanguíneo expone hacia el interior dela luz vascular el colágeno subendotelial, sitio en donde las plaquetas se adhierena través de su receptor, la glucoproteína Ia/IIa.
Dicha unión entre las plaquetas y el colágeno subendotelial se refuerza a travésde la glucoproteína adhesiva conocida como factor de von Willebrand, la cual se“ancla” en las plaquetas a través del receptor de glucoproteína Ib/IX en un extre-mo y del colágeno subendotelial por el otro.
Enseguida se unen a las plaquetas los llamados agonistas plaquetarios, comola adrenalina, la trombina y la colágena, tras lo cual se activan dos enzimas de lamembrana plaquetaria: la fosfolipasa C y la fosfolipasa A2, que catalizan la libe-ración de ácido araquidónico a partir de dos fosfolípidos de membrana: el fosfati-dil inositol y la fosfatidil colina.
El ácido araquidónico forma endoperóxidos a través de la ciclooxigenasa (lacual puede ser inhibida por los AINE). Estos endoperóxidos pueden formar trom-boxano A2 en presencia de la tromboxano sintetasa o formar prostaciclina (PGI2)en presencia de la prostaciclín sintetasa.
El tromboxano A2 aumenta a su vez la actividad de la fosfolipasa C, que esti-mula la activación plaquetaria, mientras que la prostaciclina la inhibe, como semuestra en la figura 1–1.
Liberación granular
Por otro lado, la hidrólisis del fosfolípido de membrana, el fosfatidil inositol 4–5bifosfato (PIP2), produce diacilglicerol (DAG) e inositol trifosfato (IP3). Este úl-timo interviene en el movimiento de calcio en el citosol plaquetario y estimulala fosforilación de las cadenas ligeras de miosina, la que interactúa con la actinapara facilitar el movimiento de los gránulos plaquetarios.
El DAG activa la proteincinasa C, que a su vez fosforila otras proteínas quesirven para la secreción de las granulaciones plaquetarias.
Tras la activación plaquetaria las plaquetas secretan al plasma el contenido desus gránulos, los cuales contienen los siguientes elementos:

4 (Capítulo 1)Tratado de trombosis
Figura 1–1. A partir del ácido araquidónico se forman endoperóxidos en presencia dela ciclooxigenasa, misma que puede ser inhibida por los antiinflamatorios no esteroi-deos (AINE). En presencia de la tromboxano sintetasa se forma tromboxano A2, potenteactivador plaquetario. La formación de prostaciclina (PGI2) en presencia de la prostaci-clín sintetasa inhibe la activación plaquetaria.
Ácido araquidónico
Ciclooxigenasa (–) AINE
Endoperóxidos
Tromboxanosintetasa
Prostaciclínsintetasa
Aumenta laactividad defosfolipasa C
(plaquetas)(–): inhibido por
Inhibe laactivaciónplaquetaria
6 ceto PGF(células endoteliales)
1�Tromboxano B2
Tromboxano A2 Prostaciclina (PGI2)
1. Gránulos densos: calcio, serotonina y adenosín difosfato (ADP).2. Gránulos alfa: factor de crecimiento de las plaquetas (PDGF) y factor pla-
quetario–4.
Agregación plaquetaria
El ADP liberado a partir de los gránulos densos se une a un receptor que cuandose activa modifica la configuración del complejo del receptor plaquetario, la glu-coproteína IIb/IIIa, a la cual se une el fibrinógeno y las plaquetas entre sí, forman-do de manera final un tapón plaquetario en el sitio de la lesión endotelial.
El PDGF estimula el crecimiento y la migración de los fibroblastos y de lascélulas musculares lisas de la pared vascular, lo cual genera el proceso de repara-ción del vaso sanguíneo lesionado.
Mientras se llevan a cabo los mecanismos mencionados las proteínas plasmá-ticas de la coagulación se activan de manera simultánea para iniciar la fase plas-mática de la coagulación, también llamada hemostasia secundaria. La superficiefosfolipídica de las plaquetas desempeña un papel fundamental, ya que sobre ellase activan las proteasas de la cascada de la coagulación para formar el coágulode fibrina.

5Sistema normal de la coagulaciónE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Agregación plaquetaria bajo condiciones patológicas
Las plaquetas desempeñan una función básica en la patogénesis de la aterotrom-bosis aguda, la cual es el sustrato patológico de los eventos vasculares agudos,como el infarto agudo del miocardio y el evento cerebrovascular. En el caso dela formación del trombo patológico existe una disfunción local de las células en-doteliales asociadas con mecanismos inflamatorios. Las plaquetas participan enla formación del trombo sobre la placa aterosclerótica rota y en la formación dela placa por sí misma. Las plaquetas liberan ligandos adhesivos, como la P–selec-tina, y proveen una superficie fosfolipídica para el reclutamiento de células mo-nonucleares y linfocitos, los cuales contribuyen al proceso inflamatorio localasociado con la aterosclerosis. Además, los gránulos plaquetarios contienen fac-tores de crecimiento, como el factor de crecimiento derivado de las plaquetas, elcual es importante para la proliferación celular en la expansión de la lesión ateros-clerótica.
FASE PLASMÁTICA
En la década de 1960 se propuso un modelo de la cascada de la coagulación com-puesto por una serie de pasos secuenciales, donde la activación de un factor dela coagulación guía a la activación de otro para concluir en la generación de trom-bina.
La fase plasmática de la coagulación, o hemostasia secundaria, tiene la finali-dad de formar trombina suficiente para convertir el fibrinógeno en fibrina y refor-zar así el tapón plaquetario creado en la hemostasia primaria a través de un segun-do tapón formado por fibrina. En la fase plasmática intervienen los llamadosfactores de la coagulación, que en su mayor parte son formas inactivas de enzimasproteolíticas. Cuando son convertidas a las formas activas sus acciones enzimáti-cas causan reacciones en cascada del proceso de coagulación. Como se indica enel cuadro 1–1, la mayor parte de estos factores se designan con números romanos.Desde el punto de vista didáctico, la fase plasmática de la coagulación se divideen cuatro reacciones:
Reacción I
Fase intrínseca o de contacto de la coagulación
El factor XII (Hageman), el cininógeno de alto peso molecular (HMWK) y la pre-calicreína (Fletcher) forman un complejo con el colágeno vascular. Tras la uniónde estos elementos el factor XII se convierte en su forma activa en el factor XIIa,que en seguida convierte a la PK en calicreína y en el factor XI en su forma acti-

6 (Capítulo 1)Tratado de trombosis
Cuadro 1–1. Factores de la coagulación sanguínea y sus sinónimos
Factor Sinónimos
Factor I Fibrinógeno
Factor II ProtrombinaFactor III Tromboplastina (tisular)Factor IV Calcio
Factor V Proacelerina, globulina aceleradora, factor lábil, cofactorde tromboplastina
Factor VI Ya no se emplea
Factor VII Proconvertina, factor estable, acelerador plasmático paraconversión de protrombina
Factor VIII Factor antihemofílico, globulina antihemofílica, factor anti-hemofílico A
Factor IX Factor Christmas, factor antihemofílico BFactor X Factor Stuart–Prower, factor Stuart
Factor XI Antecedente plasmático de tromboplastinaFactor XII Factor Hageman, factor de contactoFactor XIII Factor estabilizante de fibrina, factor Laki–Lorand
Precalicreína Factor FletcherCininógeno de alto peso molecular Factor Fitzgerald, HMWK
vada XIa. A su vez, la calicreína acelera la conversión del factor XII en XIIa yel factor XIa participa en las reacciones subsecuentes.
Reacción II
Vía extrínseca o dependiente de un factor tisular
En esta reacción se forma un complejo entre el factor VII, el calcio y el factortisular. Este último es una lipoproteína que se encuentra en todas las membranascelulares y que queda expuesto tras una lesión tisular.
Por otro lado, los factores II, VII, IX y X, llamados “dependientes de vitaminaK”, requieren calcio y vitamina K para adquirir su actividad biológica. Estas pro-teínas plasmáticas se sintetizan en el hígado, donde una carboxilasa que dependede vitamina K cataliza una modificación traslacional específica que añade un se-gundo grupo carboxilo a ciertos residuos de ácido glutámico.
Una pareja de estos residuos de ácido dicarboxiglutámico se une al calcio, elcual fija estas proteínas a las superficies de los fosfolípidos cargados negativa-mente y les confiere su actividad biológica.
La inhibición de la reacción bioquímica señalada es el mecanismo a través delcual actúan los anticoagulantes orales (warfarina y acenocumarina).

7Sistema normal de la coagulaciónE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Reacción III
Vía común
En esta reacción el factor X es activado por las proteasas formadas en las dos reac-ciones previas. En una reacción se forma un complejo que depende de calcio en-tre los factores VIII, IX y X, donde primero se convierte el factor IX en IXa gra-cias al factor XIa sintetizado en la vía intrínseca. Después el factor X es activadopor el factor IXa en concierto con el factor VIII. La activación de los factores IXy X proporciona una unión importante entre las vías intrínseca y extrínseca de lacoagulación.
Reacción IV
En esta reacción la protrombina se convierte en trombina en presencia del factorV, el calcio y los fosfolípidos. La trombina desempeña varias funciones, comola conversión de fibrinógeno en fibrina y la activación de los factores V, VIII yXIII (factor estabilizador de la fibrina), y estimula la adhesión y la agregaciónplaquetarias.
Tras la liberación de fibrinopéptidos A y B de las cadenas � y � del fibrinóge-no, la molécula modificada, ahora llamada monómero de fibrina, se polimerizaen un gel insoluble. El polímero de fibrina es estabilizado por el enlace cruzadode cadenas mediante el factor XIIIa.
De esta manera la hemostasia secundaria concluye con la formación de un ta-pón de fibrina sobre el tapón plaquetario, producto de la hemostasia primaria.
FIBRINÓLISIS
La lisis del coágulo y la reparación del vaso comienzan inmediatamente despuésde la formación del tapón hemostático definitivo. La fibrinólisis es esencial pararemover los coágulos intravasculares durante el proceso de reparación y evitarla trombosis. El depósito intravascular de fibrina también se asocia con el desa-rrollo de aterosclerosis. Un sistema fibrinolítico efectivo tiende a proteger contrael proceso crónico de enfermedad vascular aterosclerótica y el proceso de trom-bosis aguda.
Existen tres activadores principales del sistema fibrinolítico: los fragmentosdel factor de Hageman, la urocinasa (UK) y el activador tisular del plasminógeno(tPA). Este último se difunde desde las células endoteliales y convierte al plasmi-nógeno, absorbido en el tapón de fibrina, en plasmina.

8 (Capítulo 1)Tratado de trombosis
Las células endoteliales liberan un inhibidor del activador del plasminógeno(PAI–1), el cual bloquea la actividad del tPA.
Es necesario que exista un equilibrio entre los mecanismos cuyo objetivo esla formación de los coágulos y los anticoagulantes naturales (inhibidores de lacoagulación), con el fin de que la sangre mantenga de manera constante una ade-cuada fluidez. Los anticoagulantes naturales son la proteína C, la proteína S, laantitrombina III y el inhibidor de la vía del factor tisular (TFPI), el cual inhibe alfactor tisular en la vía externa o reacción II de la fase plasmática de la coagulación.
Proteína C
La proteína C es convertida en una proteasa activa después de unirse a la trombo-modulina, una proteína de la célula endotelial. La proteína C activada inactivalos factores V y VIII, lo cual es favorecido por la proteína S.
Antitrombina III
La antitrombina forma complejos con todos los factores de la coagulación, ex-cepto con el factor VII. La heparina no fraccionada potencia la acción de la anti-trombina III.
Función del endotelio en la hemostasis
El hígado produce la mayoría de las proteasas de serina, así como las proteínasC, S y antitrombina III, pero el endotelio participa de manera crítica en la modula-ción de la hemostasia. Las células endoteliales son pequeñas productoras de losfactores hemostáticos. Desde el punto de vista anticoagulante, expresan TFPI,heparán, trombomodulina, receptores endoteliales para la proteína C, óxido nítri-co, activador tisular del plasminógeno (tPA) y ciclooxigenasa. En su función pro-coagulante el endotelio produce el inhibidor del activador del plasminógeno(PAI–1), tromboxano, factor de von Willebrand, receptores de proteasa activadosy, quizá, factor tisular.
La expresión de estos factores hemostáticos varía de acuerdo con el sitio delárbol vascular. Por ejemplo, el TFPI se expresa de manera predominante en elendotelio de la microvasculatura, la trombomodulina en los vasos sanguíneos decualquier calibre y en todos los tejidos, excepto en el cerebro, mientras que el fac-tor de von Willebrand se expresa en toda la circulación venosa y el óxido nítricoen la circulación arterial.

9Sistema normal de la coagulaciónE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Antitrombina III (�)
Figura 1–2. Sistema normal de la coagulación y su valoración clínica. (–): inhibido por;( � ): inhibe a.
Fase vascular
Fase plaquetaria
Fase plasmática
Lesión endotelial
Vasoconstricción
Adhesión
Heparina
Vía intrínsecaReacción 1
Vía extrínsecaReacción 2
Vía comúnReacción 3
FibrinaReacción 4
Reparación vascular Fibrinólisis
Liberacióngranular
Agregación(–)
AINEAbciximabClopidogrel
Valoraciónclínica
Tiempo desangradoAgregometría
TPT
TP
TT
Dímero D
ESTUDIO CLÍNICO DE LA COAGULACIÓN
La valoración clínica de los procesos fisiológicos comentados comprenden lossiguientes estudios (figura 1–2):
Fase plaquetaria
La hemostasia primaria o fase plaquetaria se valora a través de la cuenta plaqueta-ria y del tiempo de sangrado.
Las trombocitopatías se valoran mediante una agregometría plaquetaria, lacual consiste en obtener plasma rico en plaquetas mediante centrifugación para

10 (Capítulo 1)Tratado de trombosis
someterlo a método de absorbencia, a través del cual se agregan agonistas plaque-tarios, como la ristocetina, la epinefrina, el ADP, la serotonina, el factor activadorde plaquetas, la colágena, el fibrinógeno, el ácido araquidónico y el tromboxano,con el fin de medir el porcentaje de agregación con cada uno de los reactivos. Laagregometría plaquetaria con estimulación con ADP se puede utilizar para medirlos efectos inhibitorios del clopidogrel y la estimulación con ácido araquidónicopara medir los efectos de la Aspirina�.
Fase plasmática
� La vía intrínseca o de contacto se valora mediante el tiempo parcial de trom-boplastina (TPT).Valor normal de TPT: de 25 a 40 seg.
� La vía extrínseca o dependiente de un factor tisular se valora mediante eltiempo de protrombina (TP).Valor normal de TP: de 9.50 a 14.00 seg.Porcentaje de actividad: de 75 a 100.
� La vía común se valora mediante el tiempo de trombina (TT).Valor normal de TT: de 15 a 25 seg.
REFERENCIAS
1. Handin IR: Hemorragia y trombosis. En: Principios de medicina interna. 15ª ed. México,McGraw–Hill, 2002.
2. Guyton A: Hemostasia y coagulación normal. En: Tratado de fisiología médica. 8ª ed. Mé-xico, McGraw–Hill, 1992.
3. Leavell B, Thorup O: Hemostasia normal. En: Hematología clínica. 4ª ed. México,McGraw–Hill, 1978.
4. Aird W: Coagulation. Crit Care Med 2005;33:485–487.5. Hoffman M, Monroe D: Coagulation 2006: a modern view of homeostasis. Hematol Oncol
Clin N Am 2007;21:1–11.6. Levi M: Platelets. Crit Care Med 2005;33:523–525.7. Mann K: Thrombin formation. Chest 2003:124.8. Shore LL: Platelet inhibitors and monitoring platelet function: implications for bleeding.
Hematol Oncol Clin N Am 2007;21:51–63.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
2Trombofilia
Pablo Vargas Viveros, Tania Teresa Mora Arias,José Mauricio Cedillo Fernández, Rafael Hurtado Monroy
INTRODUCCIÓN
La trombofilia es una afección clínica que define a los pacientes con mayor riesgode trombosis arterial o venosa, o ambas, y sustituye al término hipercoagulabili-dad. Los mecanismos de trombosis fueron descritos por Rudolph Virchow en elsiglo XIX y continúan vigentes en la fisiopatología general: daño endotelial, esta-sis vascular y cambios en la composición de la sangre.
Es fundamental el conocimiento de la trombofilia, ya que constituye la segun-da causa de muerte en la población general. Se calcula que más de dos millonesde personas mueren al año por un evento trombótico, sea arterial o venoso, y supresentación, prevención y tratamiento mantienen una estrecha relación con lamayoría de las especialidades médico–quirúrgicas. Gran parte de estos sucesostrombóticos se pueden prevenir con una terapia antitrombótica adecuada, mien-tras que los cuadros de recurrencia se previenen al elegir un tratamiento antitrom-bótico específico. Entre 80 y 90% de los episodios inexplicables de trombosis ve-nosa (no traumática y no quirúrgica) y cerca de 65% de las trombosis arterialesse asocian a un defecto en las proteínas de la coagulación o en las plaquetas.
La trombofilia puede ser hereditaria (primaria) o adquirida (secundaria) (cua-dro 2–1). La trombofilia hereditaria se debe sospechar en los pacientes con trom-bosis recurrente o en caso de trombosis en lugares poco frecuentes (mesentérica,portal, cerebral). De igual forma, en los casos en que se cuente con menos de 45años de edad y no se presenten factores de riesgo aparentes, además de que se trate
11

12 (Capítulo 2)Tratado de trombosis
Cuadro 2–1. Clasificación de la trombofilia
Primaria Secundaria
1. Proteína deficiente 1. Síndrome de trombosis y anticuerpos antifosfolípidos
� Antitrombina III 2. Síndrome de trombosis y anticoagulante de lupus
� Proteínas C y S 3. Obesidad
� Plasminógeno 4. Aterosclerosis y tabaquismo
� Factor XII 5. Inmovilización prolongada
� Cofactor de la heparina 6. Cirugía mayor2. Mutaciones 7. Trauma
� Factor V Leiden (A506G) 8. Embarazo y puerperio
� Gen de protrombina (G20210A) 9. Ingestión de estrógenos3. Otros 10. Cáncer
� Síndrome de la plaqueta pega-josa
11. Insuficiencia cardiaca congestiva12. Hiperlipidemia
� Hiperhomocisteinemia 13. Viajes prolongados (aéreos o terrestres)
� Aumento de glucoproteína ricaen histidina (GRH)
14. Viajes cortos con trombosis previa15. Síndrome nefrótico
� Variantes de integrina �2–�1 16. Síndrome de trombocitopenia inducida por heparina
� Factor VIII aumentado
de una mujer con antecedentes de abortos múltiples o mortinatos. La prevalenciade trombofilia puede ser mayor de 60% en los casos hereditarios.
EPIDEMIOLOGÍA Y FACTORES DE RIESGOPARA LA TROMBOSIS VENOSA
La trombosis venosa profunda (TVP) y el embolismo pulmonar (TEP) tienen unaincidencia de cerca de uno por cada 1 000 adultos por año, siendo mayor entrelas mujeres. La trombosis venosa es una enfermedad cuya incidencia aumentacon la edad; ocurre en uno de cada 10 000 pacientes al año antes de los 40 añosde edad y aumenta después de los 45 años hasta llegar a ser de cinco a seis casosal año por cada 1 000 pacientes a los 80 años de edad. Los factores de riesgo parapadecer trombosis venosa se exponen en el cuadro 2–2, igual que las característi-cas endógenas del paciente, como la obesidad, los factores genéticos y los facto-res desencadenantes, como la cirugía, la inmovilidad o el embarazo.
La trombosis venosa tiende a ocurrir por la interacción de factores intrínsecosy por la combinación de factores de riesgo genéticos y adquiridos, que se alteranpor la presencia de factores de riesgo desencadenantes. Se sabe que cerca de lamitad de los sucesos trombóticos son secundarios a factores desencadenantes y

13TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 2–2. Factores de riesgo para trombosis venosa
Modificables No modificables Temporales
Obesidad Alteraciones genéticas de la coagulación Inmovilidad
Anticonceptivos orales Síndrome antifosfolípidos Embarazo/puerperioTratamiento hormonal de
reemplazoAntecedentes personales de trombosisAntecedentes familiares de trombosis
CirugíaHospitalización
Hiperhomocisteína Más de 45 años de edad CáncerTraumaViajes largos
que, de ellos, en 65% de los casos existía más de una causa precipitante: hospitali-zación, cáncer y cirugía, con 52, 48 y 42%, respectivamente. Las trombofilias he-reditarias se pueden dividir en las que condicionan la pérdida de la función inhibi-toria de la coagulación y en las que causan la activación de la coagulación. Lassegundas son más frecuentes y son originadas por el factor V Leiden y la variantede la protrombina G20210A. El riesgo relativo y la incidencia de trombosis enlas principales trombofilias hereditarias (primarias) se mencionan en el cuadro2–3.
CLASIFICACIÓN
La enfermedad trombótica se clasifica en dos tipos:
1. La que ocurre en el terreno venoso (de flujo y presión bajos).2. La que se presenta en el árbol arterial (de presión y flujo altos).
Cuadro 2–3. Riesgo relativo de trombosisy prevalencia de trombofilia hereditaria
Factor de riesgo Incidenciade TEV (%)
TEV recu-rrente (%)
Poblaciónsana
Riesgo relativode trombosis
Resistencia a la proteínaC activada (factor VLeiden)
20 40 a 50 3 a 7 3 a 7 heterocigotos50 a 100 homo-cigotos
Mutación del gen de pro-trombina G20210A
3 a 8 15 a 20 1 a 3 2 a 8 heterocigotos
Deficiencia de proteína C 2 a 5 5 a 10 0.2 a 0.5 6 a 10Deficiencia de AT 1 a 2 2 a 5 0.02 a 0.04 5Deficiencia de proteína S 1 a 3 5 a 10 0.1 a 1 2
TEV: tromboembolismo venoso.

14 (Capítulo 2)Tratado de trombosis
Cuadro 2–4. Trombofilia hereditaria
Localización vascular frecuente
Defecto Venosa Arterial
Mutación Leiden ���� AMutación gen de protrombina ���� ADeficiencia de AT ���� ADeficiencia de proteína C ���� �
Deficiencia de proteína S ���� �
Hiperhomocisteinemia �� ����
Síndrome de la plaqueta pegajosa �� ���
Deficiencia de plasminógeno ��� A
����: muy frecuente; ���: frecuente; ��: poco frecuente; �: rara, A = ausente.
La composición del trombo (rico en plaquetas en la trombosis arterial y en fibrinaen la trombosis venosa) y la presencia de daño en el endotelio vascular (ateroma)en trombosis arterial constituyen una diferencia entre dichas ubicaciones. En estesentido, las diversas entidades de la trombofilia hereditaria muestran tendenciascon características particulares hacia la presencia de trombosis arterial o venosa,y se indican en el cuadro 2–4.
Trombofilia primaria
La trombosis ocurre a causa de un amplio espectro de condiciones que se caracteri-zan por trombosis in situ; es variable la presencia de manifestaciones embólicas,y las características clínicas dependen del sitio afectado, la extensión y el tamañodel trombo y la causa subyacente.
A diferencia de la trombofilia secundaria, en la hereditaria o primaria se rela-cionan mutaciones en los genes que codifican para los anticoagulantes endóge-nos (inhibidores: proteína C, S y antitrombina), manifestaciones clínicas antes delos 45 años de edad, ausencia o mínima influencia de factores ambientales ytrombosis en sitios anatómicos poco frecuentes.
En los últimos años aumentó el conocimiento de la coagulación y su relaciónentre los factores genéticos y ambientales y la trombosis. La mayoría de estosavances conciernen a variaciones en los genes de los factores de la coagulación,los inhibidores, la fibrinólisis y los receptores plaquetarios de membrana. Desdeel punto de vista funcional, la interacción de estos factores comprende el balanceentre los mecanismos procoagulantes y los anticoagulantes, lo cual sirve para evi-tar la generación masiva de trombina cuando se activa la cascada de la coagula-ción. Este balance se determina de acuerdo con el nivel funcional de los factoresde coagulación, la fibrinólisis y los mecanismos inhibitorios.

15TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Deficiencia de antitrombina
La antitrombina (AT) es el principal inhibidor fisiológico de la trombina y otrosfactores de la coagulación activados; su deficiencia cuantitativa o cualitativa con-duce a una mayor actividad enzimática procoagulante y a la producción de fibri-na, con gran propensión a la trombosis. También inactiva otros factores, comoXa, IXa, XIa y XIIa, y la calicreína. Se conocen tres fenotipos en la deficienciade AT. Los pacientes con el tipo I (clásica) tienen una reducción proporcional delnivel plasmático de AT funcional y antigénica, resultante de la deficiencia cuanti-tativa de la proteína normal. Alrededor de dos terceras partes de los casos se pre-sentan entre los 10 y los 35 años de edad. La disminución en la síntesis, la secre-ción defectuosa o la inestabilidad en la antitrombina en el tipo I ocurren pordeleciones genómicas, cambios de un nucleótido e inserciones o deleciones enel gen de la AT, lo cual resulta en una proteína truncada e inestable. A su vez, eltipo I se subdivide en heterocigoto (disminución de AT) y homocigoto (ausenciade AT). Los pacientes con el tipo II tienen un nivel plasmático antigénico normalo casi normal, con bajo nivel de actividad (defecto funcional de la molécula), aun-que también ocurre por mutaciones puntuales específicas que conducen a sustitu-ciones de un solo aminoácido, produciendo una proteína disfuncional. Se recono-cen más de 80 diferentes mutaciones causantes de los tipos I y II. El patrón deherencia es autosómico dominante y la mayoría de los afectados son heterocigo-tos, con niveles plasmáticos de AT entre 40 y 60% del nivel normal, trombosisarterial y venosa, y una mayor frecuencia en el embarazo y el posparto. La preva-lencia de deficiencia hereditaria de AT en la población general con eventos trom-bóticos va de 3 a 8%. El tipo III se caracteriza por niveles normales de AT funcio-nal y antigénica, pero está alterada la interacción entre la AT y la heparina. Se hanidentificado múltiples moléculas anormales de AT con defectos a nivel del sitiode unión a la heparina, que resultan en reducción aislada de la actividad de estecofactor. Estas variantes por lo general tienen mutaciones en el extremo amino-terminal de la molécula de AT. Los individuos afectados presentan niveles plas-máticos del cofactor AT–heparina de aproximadamente 50%.
El riesgo de trombosis ante la deficiencia de AT se presenta en aproximada-mente 0.5 a 1% de la población.
Deficiencia de proteína C
La proteína C es una proteína dependiente de la vitamina K que inhibe el sistemade coagulación primario mediante la inactivación de los factores V y VIII:C, co-factores requeridos para la activación de la trombina y del factor Xa, por lo quela deficiencia de este inhibidor de la coagulación genera una formación descon-

16 (Capítulo 2)Tratado de trombosis
trolada de fibrina y ocurre en 4% de los casos de trombosis. Se conocen dos for-mas de deficiencia de esta proteína.
1. El tipo I, con disminución proporcional de la actividad funcional y antigé-nica.
2. El tipo II, con defecto cualitativo (funcional).
Se conocen más de 160 mutaciones que condicionan una terminación prematuraen la síntesis de la proteína, una inestabilidad de la molécula (tipo I) y alteracionesen la activación o la función (tipo II). Se hereda con carácter autosómico domi-nante y la mayoría de los casos son heterocigotos, con manifestaciones de trom-boflebitis superficial, trombosis arterial cerebral, necrosis cutánea por warfarina,púrpura neonatal fulminante (homocigotos) y trombosis portal idiopática recu-rrentes, con inicio en la adolescencia. Este grupo de pacientes corren el riesgo depresentar estados procoagulantes al iniciar la terapia con warfarina, sobre todonecrosis cutánea, que se debe a que la warfarina produce una rápida disminuciónde la proteína C, más rápida que la del resto de los factores dependientes de vita-mina K, lo cual resulta en estados de hipercoagulabilidad transitorios.
Deficiencia de proteína S
Esta proteína es el cofactor principal de la proteína C activada (PCA), por lo quesu deficiencia produce un efecto similar al de la deficiencia de la proteína C, aun-que, a diferencia de esta última, la proteína S circula parcialmente en el plasmaunida a la proteína ligadora de C4b y sólo la porción libre (de 35 a 40%) funcionacomo cofactor de PCA.
La deficiencia congénita de proteína S se hereda de forma autonómica domi-nante y se presenta en 10% de los pacientes menores de 45 años de edad con trom-bosis venosas. Se han descrito las deficiencias cuantitativa (tipo I), cualitativa(tipo II) y de tipo III (tipo IIa), que se caracteriza por un nivel plasmático normaly bajo nivel de proteína libre. Su frecuencia y manifestaciones clínicas se compa-ran con la deficiencia de proteína C.
Resistencia a la proteína C activada (RPCA)
La resistencia hereditaria a la PCA se identifica como el factor de riesgo más im-portante para trombosis venosa, con presencia hasta en 60% de los pacientes; asi-mismo, constituye la causa más importante de pérdida fetal tardía y preeclam-psia. La principal causa se presenta por mutaciones conocidas del sustrato para

17TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
la PC (factor Va); sin embargo, en años recientes se identificaron la resistenciaa la proteína C activada (RPCA) en ausencia de factor V, las mutaciones del factorV no Leiden y las alteraciones inespecíficas (RPCA adquirida) o relacionadascon otras alteraciones, como en el caso de la deficiencia de proteína S, en la cualno existe el efecto cofactor sobre la proteína C, pero sí una actividad deficientede la proteína C.
Otra causa es el efecto bloqueador o inhibidor de los anticuerpos antifosfolípi-dos, en particular los anticuerpos anti–�2 glucoproteína, que impiden la interac-ción entre PCA y factor V (FV), conduciendo a un estado de RPCA.
Mutación del factor V (mutación de Leiden, factor V Leiden)
Tiene una prevalencia 10 veces mayor que la suma de las frecuencias de todaslas trombofilias hereditarias conocidas, presentándose entre 20 y 60% de los pa-cientes con eventos trombóticos. Consiste en la mutación del gen G1691A delfactor V (A506G: inclusión de arginina por glicina en la posición 506). La muta-ción se presenta principalmente entre las poblaciones con ascendencia caucásica;su defecto no permite la acción proteolítica de la proteína C para inhibir el factorVa, por lo que en esta condición existe una mayor cantidad de factor Va dentrodel complejo de protrombinasa, aumentando la generación de trombina.
El riesgo relativo de trombosis en heterocigotos es de 5 a 10 veces, mientrasque es de 50 a 100 veces en los homocigotos, en comparación con los individuossanos.
Los episodios tromboembólicos que se asocian con la mutación de Leiden sonen su mayoría venosos.
De las mujeres que desarrollan un primer suceso trombótico, 30% tienen fac-tor V Leiden. De las mujeres con trombosis idiopática del embarazo y el primerevento trombótico, 35% tienen factor V Leiden. En 68% de los pacientes con lamutación existen antecedentes de un evento trombótico, en 10% de trombosis ar-terial cerebrovascular y en 8% de infarto del miocardio. La edad promedio de pre-sentación es a los 45 años.
La combinación de otros factores de riesgo trombóticos, sean genéticos o ad-quiridos, es común debido a la alta frecuencia de esta mutación, en la que el riesgoparece ser sinérgico cuando se relaciona con uno o más factores de riesgo. Pormucho, la combinación más frecuente de factores de riesgo es la mezcla entre elfactor V Leiden y el uso de anticonceptivos orales, ya que se observa un riesgorelativo de 34.7% de sufrir trombosis venosa vs. 6.9% de las pacientes con lamutación sin el uso de anticoncepción hormonal.
Se han descrito varias mutaciones del residuo Arg306 del factor V. Éstas inclu-yen el reemplazo de Arg 306 por Tre (factor V Cambridge) o por glicina (factorV Hong Kong), entre muchas otras menos frecuentes.

18 (Capítulo 2)Tratado de trombosis
Resistencia a la proteína C activadaen ausencia de factor V Leiden
Se sabe que 5% de los pacientes con RPCA presentan una ausencia de la mutaciónArg–Gln 506. Dichos pacientes pueden tener anormal el gen del factor V o el pro-ducto del factor V, o no conocerse una relación directa con el gen del factor V.
Como se mencionó, se reportan dos variantes de mutaciones en el FV; la pri-mera se denomina FV Cambridge, que consiste en el reemplazo de Arg por Thren la posición 306. La segunda mutación, en un locus similar, resulta en un reem-plazo de Arg por Gly 306, y se describe como FV Hong Kong; es más frecuenteentre las poblaciones orientales, aunque para ambas formas de mutaciones el fe-notipo de resistencia para la PCA es intermedio.
Mutación del gen de la protrombina
Es una mutación que ocurre por la sustitución de glicina por arginina en la posi-ción 20210 del nucleótido (G20210A). La protrombina (factor II) es el precursorde la trombina, la cual rompe las moléculas de fibrinógeno y permite la formaciónde fibrina, el producto final de la coagulación. La trombina tiene retroalimenta-ción positiva en la activación de la hemostasia secundaria, pero también poseeactividades anticoagulantes y fibrinolíticas, por lo que cualquier trastorno de estamolécula puede resultar en múltiples alteraciones de la hemostasia. Esta mutaciónse asocia con aumento del riesgo de trombosis, principalmente enfermedad trom-boembólica venosa (ETV), aunque en menor grado que la mutación de Leiden.
Otra variante común en el gen de la protrombina es el A19911G, el cual tam-bién se asocia a un incremento de los niveles de protrombina. El defecto conduceal incremento en la producción de protrombina y predisposición al desarrollo detrombosis, aunque el mecanismo exacto aún se desconoce.
Variante de integrina �2–�1
La integrina �2–�1 (glicoproteína Ia–IIa CD49b/CD29) es la proteína de la mem-brana plaquetaria más estudiada entre los receptores de la colágena, ya que fun-ciona como un receptor para las plaquetas y para otro tipo de células que requie-ren adhesión a la colágena. En condiciones normales se expresa en un nivelmínimo (1 000 a 3 000 copias por plaqueta), en comparación con otros receptoresque también tienen la función de adhesión plaquetaria y que se expresan en mayorcantidad. Aunque el nivel mínimo de la integrina varía y se correlaciona conadhesividad a las colágenas, los estudios recientes indican que las secuencias

19TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
polimórficas de DNA dentro del gen de la integrina que está unido a la superficieplaquetaria para la expresión de ella y la variación o mutación genética producendos variantes polimórficas: los de integrina de baja expresión (variante 807C)con implicación clínica en hemorragia mayor que los que tienen la variante 807Ten el polimorfismo (segunda variante, apreciada en las personas que expresanmás cantidad de integrina); precisamente en este grupo se tiene sobreexpresiónde integrina 807T–873G polimórfico de la integrina �2–�1, que es la variable quese presenta en personas de menos de 50 años de edad con historia previa de infartodel miocardio. Esto constituye un factor de riesgo (genético) para el desarrollode cardiopatía isquémica en personas jóvenes.
Síndrome de la plaqueta pegajosa
En 1983 se describió como un padecimiento hereditario con patrón autosómicodominante en pacientes con trombosis cerebrovascular, y en 1986 se describióen pacientes con enfermedad coronaria. Se caracteriza por una hiperreactividado hiperagregabilidad plaquetaria. A pesar de que es fácil de diagnosticar, tratary prevenir consecuencias, la mayoría de los clínicos no conocen este síndrome.Existen tres tipos diferentes; el diagnóstico se basa en la sospecha clínica (histo-rial de trombosis arterial en individuos jóvenes) y la reactividad plaquetaria enla agregometría con diferentes reactivos.
� Tipo I: hiperagregabilidad plaquetaria a dos concentraciones de un reacti-vo (epinefrina).
� Tipo II: hiperagregabilidad plaquetaria a una concentración de dos reacti-vos (epinefrina, ADP).
� Tipo III: hiperagregabilidad plaquetaria a una concentración de un reacti-vo (ADP).
La prueba repetida un tiempo después demuestra resultados similares.
Hiperhomocisteinemia
Esta enfermedad puede ser una alteración congénita o adquirida, manifestada porniveles plasmáticos elevados de homocisteína y tendencia a la trombosis arterialy venosa. La homocisteína es un aminoácido no esencial que resulta del metabo-lismo de la metionina. La hiperhomocisteinemia es una consecuencia de la defi-ciencia congénita de la enzima � sintetasa, que es fundamental en el metabolismode la homocisteína, así como en individuos con polimorfismos para el gen

20 (Capítulo 2)Tratado de trombosis
MTHFR (metiltetrahidrofolato reductasa) en la posición 677. El estado homoci-goto condiciona hiperhomocisteinemia grave, trombosis arterial y venosa, retra-so mental y defectos neurológicos, visuales y esqueléticos. Sin embargo, losadultos con el trastorno heterocigoto sólo manifiestan hiperhomocisteinemia deleve a moderada y trombosis arterial o venosa. Sólo 25% de los casos se presentanantes de los 16 años de edad y la mitad antes de los 28 años. La frecuencia delestado heterocigoto en la población general es de 0.4 a 1.4%. El estado adquiridosecundario a deficiencias nutricionales de cofactores que se requieren en el meta-bolismo de la homocisteína, incluso de la piridoxina, la cobalamina y el ácido fó-lico, es más frecuente, aunque también se encuentra un aumento en los pacientescon cáncer, sobre todo de mama, gliomas, leucemia mieloide aguda, tabaquismoe hipertensión, entre otras causas.
El mecanismo mediante el cual este fenómeno produce trombosis es multifac-torial; destacan un aumento de la trombomodulina y de los receptores endotelia-les de la proteína C, una disminución de la capacidad de la proteína C activadapara inactivar al factor V y un incremento de la activación y la agregación plaque-tarias. Se ha visto que la administración de vitamina B12, ácido fólico y vitaminaB6 disminuye las cantidades de homocisteína, pero no se sabe que este suceso dis-minuya el riesgo de recurrencia de trombosis.
Fibrinógeno
Es el precursor de la fibrina; está compuesto por dímeros de tres subunidades(A�, B� y �) que se codifican en el cromosoma 4. La disminución del fibrinógeno�, y sobre todo la disminución de la razón fibrinógeno �/fibrinógeno total, se rela-cionó con un aumento del riesgo de trombosis. La producción anormal de fibrinó-geno puede resultar en disfibrinogenemia. El fibrinógeno anormal por lo regularpresenta una producción anormal de fibrina, pero mientras la mayoría de los pa-cientes con disfibrinogenemia son clínicamente asintomáticos, algunos indivi-duos se presentan con hemorragia y otros con trombofilia —ocasionalmente conambas, hemorragia y trombosis. Sin embargo, el mecanismo exacto por el cualla fibrina anormal resulta en trombosis aún se desconoce.
Se estima que la prevalencia de disfibrinogenemia congénita en pacientes conhistoria de ETV es de 0.8%. La verdadera prevalencia de trombosis entre pacien-tes con disfibrinogenemia se desconoce, pero se estima entre 10 y 20%.
Factor VII
En la cascada de la coagulación la asociación del factor VIIa con el factor tisularaumenta la actividad proteolítica mediante la atracción de sitos de unión para am-

21TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
bos sustratos (factor X y IX) y la enzima (factor VIIa) en una proximidad estrechamediante cambios conformacionales, aumentando la actividad enzimática delfactor VIIa. Existen numerosos estudios que demuestran que la deficiencia defactor VII puede causar un aumento significativo del riesgo de hemorragia. Tam-bién hay evidencia de que el aumento de la concentración plasmática de factorVII se relaciona con un incremento del riesgo de trombosis. En este caso la activi-dad fibrinolítica medida mediante el tiempo de lisis del coágulo diluido se asociacon aumento del factor VII y riesgo de eventos fatales en los individuos afecta-dos.
Factor VIII
Es el cofactor del factor IX activado para la activación recurrente del factor X.Los niveles plasmáticos de FVIII se determinan por las concentraciones del fac-tor de von Willebrand (vWF), el cual a su vez depende del tipo ABO sanguíneo.Los niveles del vWF y del factor VIII son mayores en los individuos con tipo san-guíneo A o B.
Los niveles de FVIII superiores a 150 UI/dL aumentan cinco veces el riesgode trombosis, en comparación con las personas con valores menores a 100 IU/dL.Los niveles altos de FVIII constituyen un factor de predicción de trombosis y nosólo un factor de riesgo.
Factor IX
Se ha descrito que los niveles aumentados de factor IX pueden incrementar elriesgo de trombosis. El factor IX es una serina–proteasa circulante que funcionacomo componente esencial de la coagulación; se sabe que sus niveles circulantesaumentan con la edad, y los valores superiores a 129 U/dL son un factor de riesgofrecuente en la población holandesa.
Factor XI
Con base en los hallazgos del estudio de trombofilia de Leiden se determinó queel grado de elevación del nivel plasmático de factor XI (> percentil 90) se correla-ciona con el riesgo de eventos trombóticos venosos.
Deficiencia de factor XII
El factor XII, o factor de Hageman, es un cimógeno de una serina–proteasa queinicia la activación de la fase de contacto (intrínseca) de la coagulación. La defi-

22 (Capítulo 2)Tratado de trombosis
ciencia severa de este factor (actividad del factor XII menor de 1% de lo normal)se hereda con carácter autosómico recesivo. Los individuos afectados presentanun marcado aumento del tiempo de tromboplastina parcial activado (TTPa) y dela predisposición a trombosis, debido a la reducción de la actividad fibrinolíticadel plasma, aunque la importancia de esta alteración es menor en cuanto a la ten-dencia trombofílica.
Aumento de lipoproteína a
La lipoproteína a es un factor de riesgo hereditario para tromboembolismo, queinhibe la unión del plasminógeno a la superficie celular, reduciendo así la produc-ción de plasmina y la subsecuente lisis del coágulo.
Se demostró que además de su actividad antifibrinolítica también se une einactiva el inhibidor de la vía del factor tisular, que es un regulador endógenomayor de la coagulación mediada por el factor tisular.
Los valores mayores de 300 mg/L se asocian con un mayor riesgo de enferme-dad tromboembólica venosa.
Deficiencia de plasminógeno
Los dos principales componentes del sistema fibrinolítico son el plasminógenoy el activador tisular del plasminógeno (tPA). El plasminógeno se convierte enla enzima activa plasmina por acción de tPA en presencia de fibrina. La plasmina,por su parte, digiere el coágulo de fibrina, formando los productos solubles de ladegradación de la fibrina. Una deficiencia de tPA o de plasminógeno puede redu-cir la capacidad para remover los coágulos y contribuir a la enfermedad trombo-embólica.
En la deficiencia de plasminógeno existen defectos cuantitativos (tipo 1, hipo-plasminogenemia en el heterocigoto y aplasminogenemia en el homocigoto) ydefectos funcionales (tipo 2, displasminogenemia). Hay reportes de trombosis enindividuos jóvenes, en quienes la concentración de plasminógeno es menor de40% de los valores control.
Activador del plasminógeno tisular
Se ha reportado que los niveles plasmáticos elevados de tPA antigénico en pa-cientes con tromboembolismo venoso se correlacionan con el desarrollo de trom-bosis recurrente en los siguientes dos a seis años.

23TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Inhibidor del activador del plasminógeno
El inhibidor del activador del plasminógeno tipo 1 (PAI–1) inhibe al activadordel plasminógeno tisular (tPA). Los niveles altos de PAI–1 se pueden asociar conaumento del riesgo de trombosis arterial debido a la inhibición de la fibrinólisis,aunque los datos no son claros.
Defecto del gen de la trombomodulina
La trombomodulina es uno de los componentes principales de la vía de anticoa-gulación de la proteína C. El complejo anticoagulante de la proteína C consisteen la trombina (factor II) como enzima, la trombomodulina como cofactor y laproteína C como sustrato. Conforme progresa la formación de un coágulo latrombina se une a la trombomodulina como proteína integral de la membrana dela superficie endotelial. La unión de la trombina con la trombomodulina induceun cambio conformacional en la trombina, que cambia su especificidad de sustra-to y adquiere la habilidad para activar la proteína C y no promover más la activa-ción plaquetaria y la unión a fibrinógeno.
El defecto genético en la producción de trombomodulina afecta también la víaanticoagulante de la proteína C y predispone al desarrollo de trombosis en los pa-cientes portadores. Sin embargo, la detección bioquímica de los defectos de latrombomodulina es obstaculizada por su localización subendotelial.
Glucoproteína rica en histidina
La glucoproteína rica en histidina (GRH) es una glucoproteína de una cadena.Las plaquetas almacenan GRH en los gránulos alfa y la secretan con la activaciónde la trombina. La GRH se ha mostrado que interactúa con las proteínas plasmáti-cas que participan en la coagulación sanguínea y la fibrinólisis; sin embargo, elsignificado fisiológico de estas interacciones moleculares permanece incierto. Seha encontrado que su deficiencia conduce a trombofilia, aunque hace falta infor-mación que sustente este hecho, ya que sólo se ha demostrado en pocos pacientescon otros factores de riesgo para trombosis.
Trombofilia secundaria (adquirida)
Las causas adquiridas de trombofilia son casi siempre las secundarias a enferme-dades autoinmunitarias (síndrome de anticuerpos antifosfolípidos y anticoagu-

24 (Capítulo 2)Tratado de trombosis
lante de lupus) y a las enfermedades malignas (adenocarcinomas secretores demucina y leucemia promielocítica aguda), en las que incluso la trombosis puedeser la primera manifestación clínica. En los pacientes con enfermedad trombóticaarterial se deben considerar la aterosclerosis preexistente y las causas heredita-rias, que se asocian con hipercolesterolemia, dislipidemias, diabetes mellitus,hipertensión arterial sistémica e hiperhomocisteinemia. Entre las causas adquiri-das se encuentran la enfermedad de Buerger, la arteritis y el síndrome antifosfolí-pidos (hay que descartar la presencia de lupus eritematoso generalizado). En losúltimos años se informó que el síndrome de trombocitopenia inducida por hepa-rina también presenta trombosis como parte del cuadro clínico. Existen múltiplesreportes epidemiológicos para designar los factores de riesgo en el desarrollo dela enfermedad trombótica y la prevalencia de cada uno de ellos, cuya interpreta-ción no es fácil, ya que no siempre se señalan los grupos con mayor riesgo recono-cido (como ocurre en el grupo de los pacientes quirúrgicos) o se incluye a la po-blación general sin estratificación.
Síndrome de anticuerpos antifosfolípidos
Los anticuerpos antifosfolípidos (Aa–F) constituyen un grupo heterogéneo deautoanticuerpos dirigidos contra una amplia variedad de objetivos específicosafines, que reconocen varias combinaciones de fosfolípidos o proteínas unidasa fosfolípidos, o ambos. El término síndrome antifosfolípidos se usa para descri-bir la asociación clínica entre anticuerpos antifosfolípidos, trombosis vascular,trombocitopenia y alteraciones obstétricas. Algunos de los objetivos antigénicosincluyen cardiolipina, �2 glucoproteína I, protrombina, cininógeno de alto y bajopeso molecular, anexina V, proteína C activada y proteína S. Los subgrupos másfrecuentes de Aa–F son el anticoagulante de lupus y los anticuerpos anticardioli-pina y anti �2 glucoproteína I, cuya división se basa en el método de detecciónen el laboratorio clínico. La �2 glucoproteína es una proteína de 54 kDa que estácompuesta por cinco dominios, que van del I al V. El dominio V es causante dela unión aniónica a los fosfolípidos. La hipótesis de mayor aceptación respectoa los mecanismos celulares y moleculares en la génesis de la trombosis implicala activación de células endoteliales mediante la unión del Aa–F con los fosfolípi-dos de la membrana celular, lo cual activa la célula endotelial e induce un estadoprocoagulante y proinflamatorio. Se ha indicado que la familia de la anexina A2interactúa con el complejo anti–�2 glucoproteína Ab/�2 glucoproteína en la su-perficie de la célula endotelial, para mediar su activación. Otra teoría proponeque los Aa–F interfieren o modulan la función de proteínas unidas a fosfolípidosen la fase plasmática de la coagulación. En otra hipótesis se implica la activacióndel complemento en la patogénesis de la trombosis basada en el aumento de los

25TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
productos activados del complemento en el plasma de los pacientes con eventosisquémicos cerebrovasculares que tenían síndrome antifosfolípidos, en compara-ción con los pacientes con eventos cerebrales no relacionados con el síndrome.Se ha encontrado un posible papel en la desregulación de la activación plaqueta-ria que contribuye a las manifestaciones trombóticas; este hecho se ha demostra-do por los niveles elevados de metabolitos del tromboxano en la orina de los pa-cientes con síndrome antifosfolípidos, en comparación con los controles. Elsíndrome antifosfolípidos es la causa más frecuente de trombofilia adquirida;consiste en dos síndromes clínicos diferentes, síndrome de trombosis y anticoa-gulante de lupus (STAL), y síndrome de trombosis y anticuerpos anticardiolipina(STACL), también conocido como antifosfolípidos. El STACL es más comúnque el STAL (5:1), aunque ambos se relacionan con trombosis arterial, trombosisvenosa, pérdida fetal y trombocitopenia, en orden descendente de frecuencia. ElSTACL se clasifica en varios tipos de acuerdo con su frecuencia y su agresividaden los diversos órganos y sistemas, pero se consideran otras manifestaciones me-nos frecuentes:
a. Síndrome de Snedon: livedo reticularis en asociación con trombosis arte-rial o venosa recurrente, valvulopatías y trombosis cerebrovascular con hi-pertensión arterial esencial.
b. Púrpura necrosante con trombosis venosa profunda recurrente, úlceras enlos tobillos y enfermedad de Degos (vasculopatía multisistémica).
c. Trombosis venosa en sitios poco frecuentes (síndrome de Budd–Chiari).d. Trombosis microvascular diseminada (síndrome antifosfolípidos catastró-
fico).
A pesar de que el STAL y el STACL fueron descritos en su inicio en pacientescon lupus, en la actualidad se sabe que son más frecuentes en otros padecimientosautoinmunitarios, como ocurre en la artritis reumatoide, la enfermedad de Behçety la púrpura trombocitopénica autoinmunitaria.
Existen dos clases de síndrome de trombosis y anticoagulante de lupus: el pri-mario, que es más frecuente y se presenta con trombosis sin asociación con pade-cimientos preexistentes, y el secundario, que se asocia con el diagnóstico de lupuseritematoso generalizado; casi siempre se caracterizan por pérdida fetal, mani-festaciones neuropsiquiátricas, trombosis renal, trombosis cutánea, trombocito-penia y anemia hemolítica autoinmunitaria. También se asocia con otras enfer-medades autoinmunitarias, malignidad, infección, inflamación y consumo dealgunos medicamentos. La trombosis venosa es más frecuente que la arterial;puede involucrar las extremidades inferiores, los vasos mesentéricos, renales, he-páticos y portales, y la vena cava. Es raro que ocurran los eventos arteriales, perocuando suceden lo hacen de diferente manera que en el STACL, en el que la ocu-rrencia de eventos venosos y arteriales tiene la misma frecuencia.

26 (Capítulo 2)Tratado de trombosis
Trombocitopenia inducida por heparina
Resulta de una reacción alérgica idiosincrásica en la que se desarrollan anticuer-pos IgG a la heparina, que activan receptores Fc y la formación de complejo confactor plaquetario 4, el cual se libera del interior de los gránulos � de las plaque-tas. Después de su activación induce la liberación de procoagulantes y, por ende,trombogénesis. La trombocitopenia aparece entre cinco y ocho días después delinicio de la heparina. El diagnóstico de este tipo de trombocitopenia se debe sos-pechar cuando la cuenta plaquetaria desciende 50% en relación con el valor pre-vio de 24 h. Tiene una frecuencia de 1 a 3% en los pacientes en tratamiento profi-láctico con heparina no fraccionada, en dosis que van de 250 000 a 50 000 U/día;se incluyen pequeñas dosis para permeabilización de catéteres. En 51% de los ca-sos la trombocitopenia se acompaña de un evento trombótico de predominio ve-noso, mientras que en el 49% restante sólo se presenta trombocitopenia aisladasin evento trombótico. La trombosis arterial tiene un principio abrupto de isque-mia distal que conduce a obstrucción y necrosis.
Neoplasias mieloproliferativas JAK–2 positivas y trombosis
Las complicaciones trombóticas son bien conocidas en los pacientes con neopla-sias mieloproliferativas (NMP): mielofibrosis primaria, policitemia vera y trom-bocitemia esencial. En los últimos años se describió una mutación que aumentala función del gen que codifica para la tirosincinasa JAK–2 y que resulta en lasustitución de valina por fenilalanina en la posición 617 (V617F); desde su des-cripción se han reportado múltiples casos de trombosis esplácnica, incluso en in-dividuos sin neoplasia mieloproliferativa manifiesta. El mecanismo por el cualse incrementa el riesgo de trombosis no está claro y no es uniforme para todoslos portadores de la mutación. Existen evidencias de laboratorio que sugieren quela mutación JAK–2 V617F confiere un aumento de la activación de leucocitosy plaquetas en las NMP. Sin embargo, la búsqueda de la mutación en pacientescon trombosis sin NMP manifiesta permanece sin indicación precisa, pero parecejustificarse en los casos de trombosis venosas esplácnica y cerebral idiopáticas.
Diagnóstico de trombofilia hereditaria
El estudio de cualquier trombofilia se debe basar en decisiones individuales paracada caso, según la posibilidad de encontrar un padecimiento hereditario. Los es-tudios de laboratorio que incluye el diagnóstico son los que determinan las condi-ciones más frecuentes (estudios de alta prioridad): resistencia a PCA, factor V

27TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Leiden, mutación del gen de la protrombina, nivel sérico de homocisteína, factorVIII, anticuerpos antifosfolípidos y anticoagulante de lupus. El tiempo óptimopara la determinación de estas alteraciones es de seis meses después del eventotrombótico, ya que los resultados de los exámenes que se practican de maneramás temprana pueden resultar falsos positivos y negativos, debido a que la trom-bosis por sí misma causa una disminución de protrombina y un aumento de factorVIII, así como un incremento de los niveles de homocisteína. En estos pacientesse debe cambiar la anticoagulación oral por heparina de bajo peso molecular almenos dos semanas antes de realizar los estudios. También es de gran importan-cia descartar cualquier condición adquirida que pueda simular o agravar un esta-do de trombofilia hereditaria.
Por último, hay que estudiar a los familiares en primer grado de los pacientesque resulten positivos para el diagnóstico, ya que quizá requieran seguimiento alargo plazo y profilaxis antitrombótica en caso de presentar alguna anormalidad.
Recurrencia de la trombosis venosa
La trombosis venosa es una condición crónica con una recurrencia de 5 a 7% alaño después del primer episodio. Existe un mayor riesgo entre los casos que serelacionan con cáncer y uno menor en los casos de riesgo temporal, como los queocurren durante una cirugía o después de ella. Los estudios recientes indican queexiste 60% mayor riesgo de recurrencia entre los hombres. El riesgo es más gran-de entre los primeros 6 y 12 meses después de suspender el tratamiento anticoa-gulante. La deficiencia de proteína C, proteína S y antitrombina confiere un riesgodos veces mayor de recurrencia, así como valores elevados de dímero D despuésdel tratamiento anticoagulante inicial.
REFERENCIAS
1. Ardissino D, Mannucci PM et al.: Prothrombotic genetic risk factors in young survivorsof myocardial infarction. Blood 1999;94:46–51.
2. Austin SK, Lambert JR: The JAK2 V617F mutation and thrombosis. Br J Haematol 2008;143(3):451.
3. Bezemer I et al.: Predictive genetic variants for venous thrombosis: what‘s new? Sem He-matol 2007;44(2):85–90.
4. Bick RL, Kaplan H: Syndromes of thrombosis and hypercoagulability: congenital and ac-quired thrombophilias (1997 ICATH Meeting). Clin Appl Thrombosis Hemostasis 1998;4(1):25–50.
5. Bick RL: The antiphospholipid thrombosis syndrome: a common multidisciplinary medi-cal problem. Clin Appl Thrombosis Hemostasis 1997;3(4):270–283.
6. Bonna KH: Homocysteine lowering and cardiovascular events after acute myocardial in-farction. N Engl J Med 2006;354(15):1578–1588.

28 (Capítulo 2)Tratado de trombosis
7. Caprini JA, Arcelus JI, Hasty JH: Clinical assessment of venous thromboembolic riskin surgical patients. Semin Thromb Hemost 1991;17:304–312.
8. Carlsson LE et al.: The alpha–2 gene coding sequence t807/A873 of the platelet collagenreceptor integrin alpha2–beta1 might be a genetic factor for the development of stroke inyounger patients. Blood 1999;93:55–83
9. Cushman M: Epidemiology and risk factors for venous thrombosis. Sem Hematol 2007;44(2):62–67.
10. Dipaola J et al.: Low platelets alpha2–beta1 levels in type 1 von Willebrand disease correla-tion with impaired platelet function in a high shear stress system. Blood 1999;93:3578–3582.
11. Feero WG: Genetic thrombophilia. Primary Care 2004;31(3):685–709.12. Ferro D et al.: Coexistence of antiphospholipid antibodies and endotelial perturbation in
SLE patients with ongoing prothrombotic state. Circulation 1997;95:1425.13. Galli M et al.: Antiphospholipid antibodies and associated clinical manifestations. Hema-
tology 1996;1:125–132.14. Galli M, Barbui T: Review: antiprothrombin antibodies: detection and clinical signifi-
cance in the antiphospholipid syndrome. Blood 1999;93:2149–215715. Gallus AS: Management options for thrombophilias. Semin Thromb Haemost 2005;31(1):
118–126.16. Gatt A et al.: Hyperhomocysteinemia and venous thrombosis. Semin Hematol 2007;44(2):
70–78.17. Gennari L, Blanco A et al.: The concomitant presence of lupus anticoagulant, anticardioli-
pin antibodies and anti �2–glycoprotein I antibodies could be associated with acquired acti-vated protein C resistance in non–systemic lupus erythematosus patients. Br J Haematology2003;121:527–529.
18. Gennari L, Blanco A et al.: Activated protein C (APC) resistance considerations about theimportance of using the original and modified methods in thrombophilia disease. Thrombo-sis Hemostasis 2000;84:138–139
19. Giannakopoulos B et al.: Current concepts on the pathogenesis of the antiphospholipidsyndrome. Blood 2007;109:2.
20. Greer IA: Inherited thrombophilia and venous thromboembolism. Best Pract Res ClinObstet Gynecol 2003;17(3):413–425.
21. Hillarp A, Dahlback B: APC resistance and factor V: a single point mutation in the factorV gene responsible for venous thrombosis. Clin Hemostasis 1996;10(9):11.
22. Horkko S et al.: APL are directed against epitopes of oxidized phospholipids. Recognitionof cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. JClin Inv 1996;98:815.
23. Khan S, Dickerman J: Hereditary thrombophilia. Thrombosis J 2006;4:15.24. Legnani C, Cosmi B, Cini M, Frascaro M, Guazzaloca G et al.: High plasma levels of
factor VIII and risk of recurrence of venous thromboembolism. Br J Haematol 2004;124(5):504.
25. Loscalzo J: Homocysteine trials–clear outcomes for complex reasons. N Engl J Med 2006;354(15):1629–1632.
26. Majluf CA, Hurtado Monroy R et al.: The incidence of protein C deficiency in thrombo-sis–related portal hypertension. Am J Gastroenterology 1996;91:976–980.
27. Mammen EF: Ten years experience with the “sticky platelet syndrome”. Clin Appl ThrombHemost 1995;1:66–70.
28. Nojima J, Kuratsune H et al.: Acquired activated protein C resistance is associated with

29TrombofiliaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
the coexistence of antiprothrombin antibodies and lupus anticoagulant activity in patientswith systemic lupus erythematosus. Br J Haematol 2002;118:577–583.
29. Nossent Y, Jeroen CJ et al.: Plasma coagulation factor levels in venous thrombosis. SeminHematol 2007;44:85–90.
30. Orozco H, Guraleb E, Hurtado Monroy R et al.: Deficiency of protein C in patients withportal vein thrombosis. Hepatology 1988;8:1110–1112.
31. Picard V, Dautzenberg MD, Villoutreix BO, Orliaguet G, Alhenc Gelas M et al.: Anti-thrombin Phe229Leu: a new homozygous variant leading to spontaneous antithrombin po-lymerization in vivo associated with severe childhood thrombosis. Blood 2003;102(3):919–925.
32. Ridker PM et al.: Mutation in the gene coding for coagulation factor V and the risk of myo-cardial infarction, stroke and venous thrombosis in apparently healthy men. N Engl J Med1995;332(14):91.
33. Rodger B et al.: Thrombosis and thrombophilia: diagnosis and management Hem OncolClin N Am 2003;17:1.
34. Santoro SA: Platelet surface collagen receptor polymorphisms: variable receptor expres-sion and thrombotic/hemorrhagic risk. Blood 1999;35:3575–3577.
35. Xavier SG et al.: Screening of JAK2 V617F mutation thrombosis. Int Jnl Lab Hem 2011;33:117–124.
36. Vaarala O et al.: Anticardiolipin antibodies and risk of myocardial infarction in a prospec-tive cohort of middle age men. Circulation 1995;91:23.
37. Warkentn TE, Chong BH, Greinacher A: Heparin–induced thrombocytopenia: towardsconsensus. Thromb Haemost 1998;94:895–901.
38. Watson H: Sex hormones and thrombosis. Semin Hematol 2007;44(2):98–101.39. Zivelin A, Gitel S et al.: Extensive venous and arterial thrombosis associated with an inhibi-
tor to activated protein C. Blood 1999;94:895–901.

30 (Capítulo 2)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
3Fármacos antitrombóticos
Luis Fernando García–Frade Ruiz
Como parte de las herramientas terapéuticas para la profilaxis y el tratamiento dela enfermedad trombótica se encuentran las medidas mecánicas, los agentes far-macológicos y los procedimientos invasivos. Las medidas mecánicas compren-den la ambulación temprana, los ejercicios antitrombóticos, el uso de mediascompresivas y la compresión neumática intermitente (ver los detalles en el capí-tulo 4).
Como parte de las medidas farmacológicas se incluyen los agentes que actúande manera primordial sobre la función plaquetaria, los que ejercen su efecto sobrealgún o algunos elementos de la fase plasmática del sistema de la coagulación(como los inhibidores indirectos y directos de la trombina) y los que actúan sobreel sistema fibrinolítico, como son los trombolíticos.
Los distintos procedimientos invasivos incluyen las trombectomías, la coloca-ción de filtro en la vena cava inferior, la colocación de stents, etc. En el presentecapítulo se detallan los distintos agentes farmacológicos antitrombóticos, inclu-yendo los recientes anticoagulantes orales, que constituyen, sin duda, un avancea la par de los constantes descubrimientos de causas específicas de los estadostrombofílicos primarios y secundarios.
A pesar de la variedad de recursos terapéuticos antitrombóticos, las heparinascontinúan siendo un medicamento de uso importante en el tratamiento de los es-tados trombóticos establecidos y su profilaxis, además de que su administraciónen cualquier forma ha disminuido la morbimortalidad de la enfermedad trombo-embólica venosa.
31

32 (Capítulo 3)Tratado de trombosis
AGENTES CON ACCIÓN EN LA FASEPLAQUETARIA DE LA COAGULACIÓN
En los últimos 40 años se ha desarrollado el estudio clínico de los agentes antipla-quetarios, que comenzó con el descubrimiento de la adhesión y la agregación pla-quetarias bajo la influencia del adenosín difosfato (ADP). Las investigaciones delas distintas vías bioquímicas involucradas en los procesos de la activación pla-quetaria fueron estimuladas por el descubrimiento de que el ácido acetilsalicílicoprolongaba el tiempo de sangrado.
Acido acetilsalicílico
El ácido acetilsalicílico (ASA) es un derivado del árbol Saliz alba, un sauce blan-co. El ASA se ha utilizado en el campo farmacológico como antipirético desdemediados del siglo XIX, pero fue en la década de 1940 cuando se descubrieronsus efectos hemorrágicos. Veinte años después se reconoció que sus efectos eranmediados a través de las plaquetas, y 20 años más tarde se demostró que dichosefectos antiplaquetarios estaban mediados por las prostaglandinas.
Como se detalla en el capítulo 1, las plaquetas se activan a través de un grannúmero de agonistas, entre los cuales está el tromboxano A2 (TXA2), que es delos más potentes. El TXA2 se forma cuando el ácido araquidónico es activado porla ciclooxigenasa 1 (COX–1). El ASA y otros fármacos antiinflamatorios no es-teroideos (AINE) inhiben la COX–1, con lo que previenen la generación deTXA2 y otras prostaglandinas. El ASA muestra varias propiedades como agenteantiplaquetario, ya que se puede administrar por vía oral, sus efectos son irrever-sibles, no se requiere monitoreo de sus niveles séricos y posee un rápido iniciode acción, de cerca de 30 min. El ASA se absorbe rápidamente a partir del estóma-go y el intestino proximal, alcanzando un pico máximo a las dos horas de su in-gestión y una vida media de 2 a 15 h de acuerdo con la dosis.
Es importante mencionar que el ASA no inhibe por completo la función pla-quetaria, debido a que las altas concentraciones de otros agonistas plaquetarios,como el ADP, el colágeno y la trombina, pueden activar a las plaquetas por víasalternas.
No obstante, el efecto del ASA sobre la COX–1 es irreversible (acetilación enla posición 529 de la enzima). El efecto de algunos AINE, como el ibuprofeno,es reversible y puede provocar un débil efecto antiplaquetario e incluso antagoni-zar la acción de bajas dosis de ASA. El efecto del ASA sobre la actividad de laCOX–1 es de 50 a 100 veces mayor que sobre la COX–2.
Como el efecto del ASA sobre la COX–1 es irreversible, una dosis única diariade ASA es suficiente para inhibir la función plaquetaria a lo largo de su vida. La

33Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
dosis de ASA que se requiere para inhibir la función plaquetaria es relativamentebaja si se compara con la necesaria para alcanzar su efecto antiinflamatorio. Losefectos antitrombóticos del ASA se saturan en dosis de aproximadamente 100mg; sin embargo, los estudios de farmacodinamia sugieren que con sólo 30 a 50mg de ASA al día es suficiente para inhibir por completo la producción de trom-boxano plaquetario.
La incidencia de efectos adversos del ASA, en especial los relacionados consangrado del tracto digestivo, se correlaciona con la frecuencia y las dosis admi-nistradas.
El principal efecto adverso del ASA es la hemorragia. El sangrado del tubodigestivo alto se puede atribuir tanto a los efectos antiplaquetarios como a la inhi-bición de la citoprotección de la mucosa gástrica.
La incidencia de sangrado gastrointestinal mayor es de uno a dos por cada1 000 pacientes al año.
En la actualidad se recomienda el uso de un inhibidor de bomba de protonespara la protección gástrica en pacientes que reciben bajas dosis de ASA para laprevención secundaria de eventos cardiovasculares y que cuentan con historia desangrado gastrointestinal o úlcera péptica, así como en los pacientes de alto ries-go (como infección por Helicobacter pylori, edad mayor de 65 años, síntomas dedispepsia o reflujo gastroesofágico, y aquellos en terapia concomitante con corti-costeroides). De este modo es como de manera reciente se cuenta con una presen-tación dual que incluye ASA 81 mg con esomeprazol de 20 mg en una sola tomaal día.
Existe resistencia a los efectos antiplaquetarios del ASA en algunas subpobla-ciones, quizá por mutaciones en la COX–1, por aumento en la liberación de TXA2
a partir de la COX–2, hiperactividad plaquetaria (diabetes) y aumento de la adhe-sión plaquetaria (cambios en el receptor del colágeno), entre otros. Los estudiosrecientes muestran que las mujeres fumadoras y de edad avanzada presentan unamayor resistencia a los efectos antiplaquetarios del ASA.
Las contraindicaciones para el uso del ASA son las siguientes:
� Hipersensibilidad a salicilatos y a otras sustancias similares.� Enfermedad acidopéptica activa.� Antecedentes de asma bronquial inducida por salicilatos o sustancias de ac-
ción similar, sobre todo antiinflamatorios no esteroideos.� Uso en combinación con metotrexato en dosis de 15 mg por semana o más.� Diátesis hemorrágica.� Último trimestre del embarazo.� Insuficiencia renal o hepática.
El ASA continúa siendo el antiagregante plaquetario por excelencia en la mayorparte de las indicaciones. La combinación de ASA con otros antiagregantes pla-

34 (Capítulo 3)Tratado de trombosis
quetarios incrementa el riesgo de sangrado, tal como se ha demostrado en variosestudios (MATCH, PROFESS, ACTIVE A).
Tienopiridinas
El ADP, un componente de los gránulos densos de las plaquetas (capítulo 1), acti-va a las mismas a través de distintos receptores en su superficie. La unión delADP con los receptores P2Y1 resulta en una movilización de calcio intracelular,mientras que la activación de los receptores P2Y12 conduce a la inhibición de laformación de adenosín monofosfato cíclico (AMPc) y a la liberación granular.Ambos receptores requieren ser activados para que se lleve a cabo la agregaciónplaquetaria. Los metabolitos de las tienopiridinas, la ticlopidina y el clopidogrelinhiben de manera selectiva el ADP que induce la agregación plaquetaria. La ti-clopidina ha sido virtualmente abandonada debido a sus dos principales efectosadversos: neutropenia (1 a 2.4%) y púrpura trombocitopénica trombótica (uno decada 3 000 pacientes).
Clopidogrel
El metabolismo hepático del clopidogrel por la vía del citocromo P450 (CYP3A4y CYP3A5) conduce a la generación de un metabolito activo que induce la altera-ción irreversible del receptor P2Y12. Hasta 50% de éste se elimina a través de laorina y 46% mediante las heces. No se requiere ajustar la dosis en pacientes confalla renal, pero sí se debe tener precaución en los que tienen falla hepática. Aligual que el ASA, el clopidogrel se administra en una sola dosis al día. El iniciode su efecto antiplaquetario es lento, aunque se puede alcanzar con dosis crecien-tes del fármaco. La inhibición plaquetaria se puede detectar a las dos horas de laadministración de altas dosis por vía oral. Después de cuatro a siete días de terapiadiaria (75 mg/día) se alcanzan niveles estables de inhibición plaquetaria. Para lo-grar un máximo efecto el paciente puede recibir una dosis inicial de 300 a 600mg y después la dosis estándar de 75 mg al día. La función plaquetaria regresaa la normalidad después de siete días de la última dosis.
La contraindicación absoluta para el uso de clopidogrel es la presencia de he-morragia, sea gastrointestinal, retiniana, retroperitoneal o intracraneal. Se debeutilizar con precaución en pacientes con riesgo de sangrado, como trauma, ciru-gía o presencia de enfermedad acidopéptica. El clopidogrel se asocia con una in-cidencia de sangrado gastrointestinal de 2%.
Sin embargo, el uso de terapia antiplaquetaria con ASA o clopidogrel en algu-nos pacientes ocasiona una alta incidencia de retrombosis después de una revas-cularización coronaria o una colocación de stent, por lo que se estudia la posibili-

35Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
dad del fenómeno de resistencia al clopidogrel, el cual podría ser resultado dedistintos polimorfismos en el receptor P2Y12, de diferencias en el metabolismohepático a través del citocromo P450 o de interacciones farmacológicas, por loque quizá a corto plazo surgirá la indicación de medir los efectos de estos agentesen los pacientes a través de la agregometría plaquetaria, que en su caso deberácontener mediciones específicas para estos dos antiagregantes, tal como trombo-xano y ADP, con el fin de evitar trombosis, y sobre todo de las prótesis endovas-culares.
Prasugrel
El prasugrel es un inhibidor irreversible del receptor P2Y12, con rápida y completaabsorción por vía oral. Después de su absorción intestinal es rápidamente hidroli-zado a través de esterasas a un metabolito intermedio que requiere oxidación de-pendiente de CYP para generar su compuesto activo. En un estudio de 13 608 pa-cientes con angina inestable e infarto del miocardio, quienes fueron sometidosa una intervención coronaria percutánea programada, el prasugrel disminuyó demanera significativa el riesgo de recurrencia de infarto y trombosis del stent, encomparación con el clopidogrel, por lo que fue aceptado por la Food and DrugAdministration (FDA) para su indicación en pacientes con angina inestable e in-farto del miocardio con intervención coronaria percutánea. La dosis de manteni-miento es de 10 mg al día, pero se sugiere reducir a 5 mg al día en pacientes conun peso corporal menor de 60 kg, con el fin de disminuir el riesgo de sangrado.Se debe evitar su uso en pacientes de edad avanzada, excepto en situaciones demuy alto riesgo. La activación de clopidogrel y de prasugrel requiere isoenzimasCYP, por lo que su actividad puede resultar afectada por polimorfismos genéti-cos.
Ticagrelor
Es un agente oral que actúa como inhibidor directo y reversible del receptorP2Y12, con un inicio de acción más rápido y mayor efecto inhibitorio sobre la fun-ción plaquetaria que el clopidogrel. El ticagrelor se absorbe rápidamente en el in-testino. Su vida media es de siete a ocho horas, y tiene la característica de no re-querir biotransformación hepática para adquirir su forma activa. El estudiomulticéntrico PLATO comparó el ticagrelor y el clopidogrel para la prevenciónde eventos cardiovasculares en 18 624 pacientes. En dicho estudio se concluyóque en los pacientes con síndrome coronario agudo con o sin elevación del seg-mento ST el tratamiento con ticagrelor, en comparación con clopidogrel, redujode manera significativa el rango de muerte por causas vasculares, infarto del mio-cardio e infarto cerebral sin un aumento del riesgo de sangrado mayor, pero con

36 (Capítulo 3)Tratado de trombosis
incremento del riesgo de sangrado no relacionado con el procedimiento corona-rio.
Inhibidores de la glucoproteína IIb/IIIa
La glucoproteína IIb/IIIa (GpIIb/IIIa) es el receptor más abundante de la superfi-cie plaquetaria (capítulo 1); sirve como un receptor primario en la mediación dela agregación plaquetaria. Cuando las plaquetas son estimuladas por cualquiervariedad de agonistas plaquetarios ocurre un cambio conformacional dependien-te de calcio en la GpIIb/IIIa, a la cual se une el fibrinógeno con una alta afinidad.Como el fibrinógeno es bivalente, su unión con la GpIIb/IIIa conduce a una unióncruzada de plaquetas.
Se han desarrollado varios antagonistas de la GpIIb/IIIa, como el abciximab,el tirofibán y la eptifibatida. El abciximab es un fragmento Fab de anticuerpo mo-noclonal humano 7E3, que inhibe al receptor de vitronectina. Su efecto inhibito-rio sobre la función plaquetaria es evidente a los 10 min de su administración, conun incremento del tiempo de sangrado. Tiene una vida media corta de 10 a 30 min,pero puede permanecer en la circulación unido a las plaquetas durante más de 10días. El tiempo de sangrado se normaliza de una a cuatro horas después de la inte-rrupción de su administración. Los fragmentos Fab y el abciximab libre son depu-rados en los riñones; sin embargo, la dosis del fármaco no requiere, en teoría, unajuste de acuerdo con la función renal.
Contrario a lo que ocurre con el ASA y el clopidogrel, el efecto antiplaquetariode los inhibidores de la GpIIb/IIIa es reversible, por lo que su efecto sólo está pre-sente cuando el fármaco se encuentra unido al receptor. Sus efectos, además, seencuentran directamente relacionados con el porcentaje de receptores a los cualesse unen, siendo necesaria la inhibición de al menos 80% de ellos para poder alcan-zar una completa inhibición de la agregación plaquetaria. Después de suspenderla administración del antiplaquetario (bolo IV y posterior infusión IV), la funciónplaquetaria regresa a la normalidad dentro de las siguientes 48 h.
La eptifibatida es un heptapéptido cíclico sintético que inhibe la agregaciónplaquetaria de acuerdo con la dosis, en la que dicha inhibición es reversible trassuspender la infusión del fármaco. Posee una vida media de cerca de 2.5 h y 50%de ella se depura por vía renal.
El tirofibán es un peptidomimético sintético de bajo peso molecular que mi-metiza al fibrinógeno y se une a la GpIIb/IIIa con alta afinidad. Al igual que laeptifibatida, su inhibición plaquetaria depende de la dosis y es reversible al sus-pender su infusión. Posee una vida media de alrededor de dos horas, y 65% sedepura por vía renal. Debido a la corta vida media de estos fármacos y al potencialde expresión de GpIIb/IIIa en la superficie plaquetaria posterior a su activación,su administración se realiza a través de infusión intravenosa continua.

37Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 3–1. Dosis recomendadas para los inhibidores de la GpIIb/IIIa
Agente Dosis en bolo Dosis demantenimiento
Comentario
Tirofibán 0.4 �g/kg/mindurante 30min
0.1 �g/kg/min durante 48a 72 h y continuar de12 a 24 h después deICP
Pacientes con insuficiencia renalgrave (depuración de creatinina< 30 mL/min) deben recibir lamitad de la dosis usual
Eptifibatida 180 �g/kg du-rante 1 a 2min
2 �g/kg/min durante 72 hy continuar otras 24 hdespués de ICP
Si la creatinina es de 2 a 4 mg/dL,reducir bolo a 135 �g/kg/min, se-guido de infusión a 0.5 �g/kg/min
Abciximab 0.25 mg/kg/min durante10 a 60 minantes deICP
0.125 �g/kg/min (máximo10 �g/min) durante 12horas
No se requiere ajustar a dosis renaly la cuenta plaquetaria requierevigilarse a las 4 h después delbolo y luego a las 24 h; si éstadisminuye a menos de 100 000o más de 25% de la cuenta pre-tratamiento, se debe suspenderla infusión del fármaco
Abreviaturas: ICP = intervención coronaria percutánea.
Se ha reportado trombocitopenia grave durante las primeras horas de adminis-tración de estos fármacos en algunos pacientes, lo cual es más frecuente con eluso de abciximab (aproximadamente en 5% de los casos), pudiendo resultar enun sangrado profundo, en cuyo caso el manejo consiste en transfusión de plaque-tas para restablecer la cuenta plaquetaria y la función de las mismas. La adminis-tración de desmopresina también ha mostrado ser de utilidad para normalizar eltiempo de sangrado.
Ante el riesgo potencial de sangrado tras el uso de estos agentes se debe tenerprecaución con la dosis, así como con el uso concomitante de otros antiplaqueta-rios o anticoagulantes. Las dosis recomendadas para los inhibidores de la GpIIb/IIIa se muestran en el cuadro 3–1.
Inhibidores de la fosfodiesterasa
Dipiridamol
Un agente de esta clase de antiplaquetarios es el dipiridamol, que de manera típicase ha utilizado en forma concomitante con la warfarina sódica como profilaxisantitrombótica en los pacientes con prótesis valvular cardiaca. En la actualidadse comercializa en combinación con ASA y se encuentra indicado como profila-xis secundaria en pacientes con evento cerebrovascular isquémico de tipo atero-trombótico. El dipiridamol afecta la función plaquetaria a través de un complejo

38 (Capítulo 3)Tratado de trombosis
mecanismo de acción que conduce a un incremento de nucleótidos cíclicos intra-plaquetarios. El incremento en el AMPc/GMPc conduce a la inhibición del trans-portador de adenosina y de fosfodiesterasas. El incremento plaquetario de GMPcpotencia los efectos del óxido nítrico y estimula la producción de prostaciclina.El dipiridamol inhibe la agregación plaquetaria de manera reversible.
Cilostazol
El cilostazol es un potente inhibidor selectivo de la isoenzima fosfodiesterasa–3,que conduce a la inhibición de agonistas plaquetarios y de TXA2. A diferenciadel ASA, el cilostazol inhibe la agregación plaquetaria inducida por el ADP y laepinefrina, pero no disminuye la producción endotelial de prostaciclina. El cilos-tazol se indica de manera principal en la enfermedad arterial periférica, en dosisde 100 mg dos veces al día; está contraindicado en pacientes con insuficienciacardiaca congestiva, ya que su uso se asocia con un efecto proarritmogénico.
FÁRMACOS CON ACCIÓN ANTIPLAQUETARIA INDIRECTA
Varios agentes con acción primordial sobre algún elemento de la fase plasmáticade la coagulación también pueden, de manera indirecta, actuar sobre la funciónplaquetaria. Al suceder ambas fases de manera simultánea en el organismo, exis-ten elementos que las unen, como es el caso de la trombina, para la que existeninhibidores directos de la misma, sean de forma intravenosa, subcutánea o víaoral, como el dabigatrán.
Otro ejemplo lo constituye la heparina no fraccionada, que en altas dosis pro-longa el tiempo de sangrado por su efecto antitrombina y porque provoca fallaen la acción del factor de von Willebrand sobre las plaquetas, con lo que se afectala adhesión de las mismas.
AGENTES CON ACCIÓN EN LA FASEPLASMÁTICA DE LA COAGULACIÓN
Inhibidores indirectos de la trombina
Los inhibidores indirectos de la trombina comprenden la heparina no fracciona-da, la heparina de bajo peso molecular, el fondaparinux, el idraparinux y los anta-gonistas de la vitamina K.

39Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Heparina no fraccionada
Desde hace más de 50 años la heparina ha sido el fármaco con acción anticoagu-lante de mayor uso en la práctica clínica. En 1937 Crafford demostró una reduc-ción de eventos trombóticos en los pacientes que recibieron heparina intrave-nosa.
La heparina utilizada hasta la década de 1970 fue de origen porcino o bovino;en la actualidad se conoce como heparina no fraccionada. Existe una amplia hete-rogeneidad en los efectos farmacológicos de las heparinas no fraccionadas(HNF), que dependen de la especie de la cual se obtienen.
La HNF posee una actividad anticoagulante que depende de un pentasacáridocon alta afinidad por la antitrombina III (AT–III). La unión de la heparina con laAT–III propicia un cambio conformacional en esta última, con lo que se activasu efecto inhibitorio sobre la trombina, el factor Xa y otras proteasas de serina(capítulo 1). Las dosis altas de HNF pueden inhibir la trombina a través del cofac-tor II (un cofactor plasmático independiente de la AT–III). Además, la HNF seune a numerosas proteínas plaquetarias y plasmáticas, células endoteliales y leu-cocitos, con lo que suprime la función plaquetaria e incrementa la permeabilidadvascular.
Desde el punto de vista clínico y terapéutico, existen dificultades para estanda-rizar la acción anticoagulante de la HNF, lo cual condiciona dosis y efectos diver-sos en cada paciente. Dichos efectos se deben a la escasa absorción de la heparinaa través de su administración subcutánea, así como a su afinidad y unión a diver-sas proteínas plasmáticas, plaquetarias y endoteliales. Estas características con-dicionan la incapacidad para sugerir una dosis estándar y la necesidad de diferen-tes requerimientos según la vía de administración y las características deresistencia de cada paciente. Las diferencias en la respuesta a la HNF que se ob-servan en cada paciente van desde la ineficacia terapéutica hasta una respuestaexagerada a una dosis convencional, lo cual genera la necesidad de vigilar el efec-to anticoagulante por medio de pruebas de laboratorio, en especial del tiempoparcial de tromboplastina activada.
La biodisponibilidad y la bioactividad de la HNF varían incluso con el horario,por lo que presentan variaciones diurnas, lo cual condiciona la posibilidad de queexistan periodos prolongados de anticoagulación subclínica incluso en los casoscon infusión continua de HNF.
Entre sus complicaciones destaca el riesgo de sangrado, que es mayor en lasmujeres y en los pacientes de 65 años de edad en adelante.
Otra complicación importante asociada con el uso de HNF es la trombocitope-nia, que presenta graves implicaciones de pronóstico de morbimortalidad cuandose desarrolla en el tratamiento de pacientes con trombosis venosa profunda. Latrombocitopenia inducida por HNF requiere un diagnóstico oportuno y un trata-

40 (Capítulo 3)Tratado de trombosis
Cuadro 3–2. Limitaciones en la eficacia de la heparina no fraccionada
Limitación Mecanismo
Respuesta anticoagulante variable La heparina se une a varias proteínas de la fase aguda yproteínas liberadas a partir de plaquetas activadas ycélulas endoteliales
Depuración dependiente de dosis La heparina se une a sitios sobre el endotelio y macrófa-gos que deben ser saturados antes de aparecer en lacirculación
Actividad disminuida en presenciade plaquetas
El factor plaquetario 4, liberado a partir de plaquetas acti-vadas, neutraliza la heparina
Incapacidad para inactivar el fac-tor Xa dentro del complejo pro-trombinasa
El factor Xa se une a la superficie plaquetaria y es relati-vamente resistente a la inactivación por el complejoheparina–antitrombina
miento adecuado, que puede consistir en la suspensión de la heparina y el uso detransfusiones de concentrados plaquetarios o plasmaféresis. Además, puede ocurrirel síndrome de trombocitopenia o trombosis inducida por heparina (HIT), o am-bos.
Otras complicaciones que se pueden presentar con el uso de la HNF son osteo-porosis, incremento de las enzimas hepáticas, hipoaldosteronismo secundario,reacciones de hipersensibilidad y necrosis cutánea.
Las limitaciones en la eficacia de la HNF se incluyen en el cuadro 3–2.
Heparinas de bajo peso molecular
En la segunda mitad de la década de 1970 Anderson y Thumberg demostraronla actividad y la relación de los diferentes pesos moleculares de las heparinas conel efecto sobre diferentes proteasas de serina plasmáticas, demostrando que unapequeña cadena de polímeros tenía afinidad por la AT–III y se unía a ella paradesarrollar su actividad anticoagulante. A partir de estos descubrimientos se estu-diaron de manera sistemática el efecto y la acción de las heparinas fraccionadas;así, en 1978 apareció la primera heparina de bajo peso molecular (HBPM) conactividad antitrombótica.
Las HBPM tienen su origen en la HNF mediante una despolimerización con-trolada y el uso de sustancias químicas —como el óxido nitroso—, la hidrólisisalcalina, la división con peróxidos o la acción enzimática con heparinasa. LasHBPM tienen un peso molecular que varía de 4 000 a 6 000 Da. Las diferentesHBPM son distintas en su peso molecular, su contenido de glucosaminoglicanosy su actividad anticoagulante antifactor Xa y antitrombina. Algunas ventajas queofrecen las HBPM en relación con la HNF son una mayor biodisponibilidad (más

41Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
de 90% de la dosis administrada por vía subcutánea), una mayor vida media plas-mática y una respuesta antitrombótica predecible con base en el peso del pa-ciente.
Su capacidad para inhibir el factor Xa unido a las plaquetas y la resistencia ala inhibición del factor plaquetario 4 explica su mayor efecto antitrombótico, asícomo su menor efecto sobre la función plaquetaria y poca disposición para afec-tar la permeabilidad vascular, lo cual contribuye a un menor efecto anticoagulan-te, con la consecuente disminución del riesgo de sangrado.
Mientras que en la HNF la proporción de efecto antifactor Xa:antitrombina esde 1:1, en las HBPM va de 1.9:1 a 3.6:1. La posibilidad de hemorragia, osteoporo-sis y trombocitopenia inducida por heparina es menos frecuente con las HBPM(menos de 7%). Las HBPM proveen una alternativa en los casos de trombocito-penia inducida por HNF, ya que no presentan reacción cruzada.
Las HBPM inducen con mayor frecuencia una elevación de las enzimas hepá-ticas (10% de los casos), en comparación con la HNF (3% de los casos). La res-puesta terapéutica de la HBPM depende también de variables propias de cada in-dividuo, como en el caso de las pacientes no fumadoras que presentan una mayoractividad anti–Xa.
Fondaparinux
El fondaparinux es una heparina sintética (pentasacárido) que inhibe al factor Xade forma selectiva por unión con la antitrombina. Posee una excelente biodispo-nibilidad por vía subcutánea, con un rápido inicio en su acción y una vida mediaprolongada (de 14 a 20 h). Su acción depende de los niveles de antitrombina en-dógena.
El fondaparinux es sintético, se encuentra libre de contaminantes virales y ani-males, posee una vida media prolongada, constituye una estructura molecular ho-mogénea y pura, tiene un efecto predecible de acuerdo con la dosis, no interactúacon el factor plaquetario 4 y no ejerce reacción cruzada con anticuerpos contrala heparina, en comparación con otros agentes antitrombóticos; sin embargo, pro-voca una mayor incidencia de trombocitopenia inducida por heparina, lo cual noocasionan las HBPM.
Su uso ha sido aprobado por la FDA para la prevención de eventos tromboem-bólicos venosos en pacientes que son sometidos a cirugía ortopédica. El pentasa-cárido atraviesa tanto la barrera placentaria como la hematoencefálica, por lo quese cuestiona su uso en pacientes embarazadas y en las personas de edad avanzadacon compromiso cerebral.
Debido a su vida media larga se debe administrar una vez al día; igual que lasHBPM, no requiere monitoreo mediante pruebas de laboratorio, ya que no afecta

42 (Capítulo 3)Tratado de trombosis
el tiempo de protrombina ni el de tromboplastina. No se ha establecido un antído-to para este agente, y no se conocen hasta el momento interacciones farmacológi-cas relevantes con su uso.
Idraparinux
El idraparinux es un derivado sintético de la molécula de fondaparinux que resul-ta un fármaco atractivo, ya que sus ventajas en la dosificación y su farmacociné-tica perfectamente predecible hacen que se pueda administrar por vía subcutáneauna vez por semana, sin necesidad de controles de laboratorio. Tiene una vida me-dia de 130 h, y el resto de sus características farmacocinéticas y su mecanismode acción son similares a los descritos para el fondaparinux.
Se han realizado los estudios van Gogh DVT y PE con idraparinux en pacien-tes con trombosis venosa profunda y embolismo pulmonar, respectivamente. Endichos estudios se comparó el idraparinux en dosis de 2.5 mg por vía subcutáneauna vez a la semana vs. el tratamiento estándar; las conclusiones indican que, aun-que el idraparinux no resultó inferior al tratamiento estándar en el grupo de TVP,sí lo fue en el estudio de embolismo pulmonar. El estudio AMADEUS, que estabacomparando el idraparinux y la warfarina para la prevención de embolismo enpacientes con fibrilación auricular, se cerró debido a un exceso de hemorragiasclínicamente relevantes en el grupo que recibió idraparinux.
Antagonistas de la vitamina K
La vitamina K induce una conversión postraslacional de residuos de glutamatoa �–carboxiglutamato en un número limitado de proteínas, de las cuales las mejorconocidas son los factores de la coagulación II, VII, IX y X y las proteínas C, Sy Z.
El �–carboxiglutamato une el calcio a dichas proteínas, con lo que los factoresde la coagulación sufren un cambio conformacional, requerido para unirse a va-rios cofactores activos sobre las superficies celulares. La forma reducida de la vi-tamina K (KH2) actúa como coenzima. La oxidación de la vitamina KH2 por partedel oxígeno a epóxido de vitamina K (KO) provee energía para fijar dióxido decarbono a la posición � de un residuo de glutamato. La vitamina KO después esreciclada, primero por la vitamina KO reductasa a vitamina K (quinona) y des-pués por la vitamina K reductasa a vitamina KH2 (hidroquinona). Es necesarioque cada molécula de vitamina K sea reciclada cientos de veces antes de ser meta-bolizada.
Los antagonistas de la vitamina K inhiben la vitamina KO reductasa; es posi-ble que hagan lo mismo con la vitamina K reductasa.

43Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Las dos formas de vitamina K son la filoquinona (vitamina K1) y la menaqui-nona (vitamina K2). Las filoquinonas se encuentran en las verduras, como la espi-naca, la coliflor y el brócoli. La escasez de estos alimentos en la dieta puede cau-sar una deficiencia de vitamina K, mientras que el consumo de cantidadesexcesivas puede disminuir los efectos de los anticoagulantes orales (capítulo 28).Las menaquinonas se encuentran en varios alimentos, como en el yogurt y losproductos orgánicos; además, son producidas por la flora bacteriana del colon ytal vez por el intestino delgado.
Farmacocinética y farmacodinamia de la warfarina
Existen dos grupos distintos de antagonistas de la vitamina K:
1. Los derivados de la 4–hidroxicumarina (warfarina sódica y acenocumari-na).
2. Los derivados del indano–1,3–diona (fenindiona).
Los derivados cumarínicos son los antagonistas de la vitamina K de elección, de-bido a que causan menos efectos adversos no hemorrágicos.
La warfarina es una mezcla racémica de estereoisómeros (formas R y S). Laforma S es la más potente, con un pico máximo de absorción a los 90 min y unavida media de 36 a 42 h. La warfarina se une a las proteínas (principalmente albú-mina), por lo que cualquier fármaco o químico que también se une a la albúminapuede desplazar a la warfarina de sus sitios de unión e incrementar así la formabiológicamente activa.
La forma S de la warfarina se metaboliza en el hígado a través del sistema enzi-mático P450 (CYP2C9). Su interferencia con las enzimas CYP2C9 mediante va-rios fármacos y la presencia de mutación en el gen que codifica para una de lasenzimas comunes CYP2C9 pueden interferir en gran medida con el metabolismode la warfarina.
El efecto anticoagulante de la warfarina es mediado por la inhibición de la vita-mina K dependiente de la �–carboxilación de los factores de la coagulación II,VII, IX y X. Esta inhibición resulta en la síntesis de dichos factores, pero biológi-camente inactivos. La warfarina también inhibe la vitamina K dependiente de �–carboxilación de las proteínas C y S. La proteína C circula como proenzima, quees activada sobre las células endoteliales por el complejo trombina–trombomo-dulina. La proteína C activada en presencia de la proteína S inhibe los factoresV y VIII activados; por lo tanto, los antagonistas de la vitamina K provocan unaparadoja bioquímica al inducir un efecto anticoagulante a través de la inhibiciónde procoagulantes (factores II, VII, IX y X) y un efecto trombogénico potente tras

44 (Capítulo 3)Tratado de trombosis
inhibir la síntesis de inhibidores naturales de la coagulación (proteínas C y S). Poresta razón se deben utilizar HFN o HBPM los primeros cuatro o cinco días de iniciode la terapia con warfarina en los pacientes que padecen enfermedad trombótica.
El efecto anticoagulante de la warfarina se alcanza hasta que los factores dela coagulación son depurados de la circulación, con un efecto pico entre 36 y 72h posteriores a su administración. La relación entre dosis y respuesta tras la tera-pia con warfarina varía de acuerdo con cada individuo, por lo que la dosis se debemonitorear con precaución para prever la sobredosificación.
Las interacciones farmacológicas, alimentarias y herbales de los agentes anti-trombóticos se revisan a detalle en el capítulo 28.
Tecarfarina
La tecarfarina es un reciente antagonista de la vitamina K no metabolizado porel citocromo P450, por lo que sus interacciones farmacológicas son menores. Enun estudio fase II se investigaron la seguridad, la tolerabilidad y la eficacia de latecarfarina, que parece alcanzar porcentajes de tiempo de permanencia en el in-tervalo terapéutico superiores a los obtenidos con la warfarina; sin embargo, ha-cen falta más estudios para establecer su utilidad.
Monitoreo mediante pruebas delaboratorio y rango terapéutico
El examen de laboratorio utilizado para medir los efectos de la warfarina sobreel sistema de la coagulación es el tiempo de protrombina. El tiempo de protrombi-na es sensible a la disminución de la actividad de los factores II, VII y X, peroinsensible a la reducción de la actividad del factor IX. Con el fin de promover laestandarización del tiempo de protrombina para un adecuado monitoreo de la te-rapia con warfarina, la Organización Mundial de la Salud desarrolló una referen-cia internacional y recomendó que el tiempo de protrombina se exprese medianteel índice normalizado internacional (INR: International Normalized Ratio).
La dosis usual de la warfarina al inicio de la terapia es de 5 mg al día durantelos primeros dos días, y posteriormente se ajusta de acuerdo con el INR. La HNFo la HBPM se continúan hasta el quinto día de inicio de la terapia con warfarina,una vez que la prolongación del INR se encuentra dentro de los rangos terapéuti-cos (INR 2.0 a 3.0) durante al menos dos días consecutivos. Cuando el efecto anti-coagulante de la warfarina se encuentre estable se deberá realizar el monitoreodel INR cada una a tres semanas durante la terapia; sólo en los pacientes en quie-nes se mantenga un INR estable durante los siguientes meses se podrá espaciarel seguimiento cada tres meses.

45Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Efectos adversos de los antagonistas de la vitamina K
Efectos hemorrágicos
El principal efecto adverso de los antagonistas de la vitamina K es el sangrado.El sangrado que ocurre durante una terapia oral bien controlada casi siempre sedebe a cirugía, trauma o lesiones locales, como la presencia de úlcera péptica ocarcinoma. El sangrado espontáneo puede ocurrir cuando la warfarina se admi-nistra en dosis excesivas, que resultan en una marcada prolongación del INR.
Los principales factores de riesgo para el desarrollo de complicaciones hemo-rrágicas graves con el uso de warfarina son la edad mayor de 65 años, el sexo fe-menino, el sangrado gastrointestinal previo, el alcoholismo, la falla renal crónicay la presencia de cáncer.
Manejo del paciente con un INR prolongado
El manejo depende tanto del grado de prolongación del INR como de las circuns-tancias clínicas. Las opciones terapéuticas, dependiendo del caso, incluyen:
1. Interrupción temporal de la terapia anticoagulante.2. Administración de vitamina K.3. Transfusión de plasma fresco congelado.
En el cuadro 3–3 se muestran las recomendaciones a seguir en cada caso.
Efectos no hemorrágicos
El uso de cumarina puede provocar necrosis cutánea, una rara pero grave compli-cación que requiere la interrupción inmediata de la terapia; se presenta casi siem-pre entre 3 y 10 días después del inicio del tratamiento. Es más común entre lasmujeres y con mayor frecuencia involucra áreas con abundante tejido subcutá-neo, como el abdomen, las nalgas, los muslos y las mamas. Dicha complicaciónse asocia con el desarrollo de trombosis microvascular por una disminución enlos niveles de proteína C. En el cuadro 3–4 se muestran las contraindicacionesdel uso de anticoagulantes.
Inhibidores directos del factor Xa
Rivaroxabán
El factor Xa cataliza la conversión de protrombina a trombina (capítulo 1). El ri-varoxabán constituye el primer inhibidor oral directo del factor Xa, al cual inhibe

46 (Capítulo 3)Tratado de trombosis
Cuadro 3–3. Manejo del paciente con INR prolongado
Situación clínica Guía
INR mayor al rango terapéutico ymenor de 5.0, sin presencia dehemorragia significativa
Disminuir la dosis u omitir la siguiente dosis. Monitoreofrecuente del INR
INR entre 5.0 y 9.0 sin presencia dehemorragia significativa
Omitir la siguiente dosis o dos dosis, monitorear el INRcon más frecuencia y disminuir la dosis cuando elINR esté en rangos terapéuticos
Alternativamente, omitir la siguiente dosis y dar vitaminaK1 (� 5 mg vía oral), sobre todo si el paciente tieneriesgo de hemorragia
Los pacientes que requieren una conversión más rápidapor cirugía urgente: vitamina K1 (2 a 4 mg vía oral); siel INR se mantiene alto a las 24 h hay que dar unadosis adicional de vitamina K1 (1 a 2 mg vía oral)
INR mayor de 9.0 sin presencia dehemorragia significativa
Omitir la warfarina; dar vitamina K1 (5 a 10 mg vía oral)con la expectativa de que el INR se reduzca entre 24y 48 h. Monitorear de cerca el INR; si el INR no dismi-nuye de manera sustancial en 24 h, monitorearlo conmás frecuencia y dar dosis adicional de vitamina K sies necesario
Hemorragia grave con cualquiernivel de INR
Omitir la warfarina, dar vitamina K1 (10 mg lentamenteen infusión IV), suplementar con PFC o concentradode protrombina. El factor VII recombinante puede seruna alternativa a este último, dependiendo de la ur-gencia. La vitamina K1 puede repetirse cada 12 h
Hemorragia que pone en riesgo lavida
Omitir la warfarina, dar vitamina K1 (10 mg IV en infusiónlenta) y concentrado de protrombina. El factor VIIrecombinante puede ser una alternativa a este último;repetir si es necesario, dependiendo del INR
INR: International Normalized Ratio; PFC = plasma fresco congelado.
de manera específica y altamente selectiva. El rivaroxabán no ejerce un efectodirecto sobre la trombina, sino que regula la generación de ésta a través de la inhi-bición de la actividad del factor Xa sin necesidad de unirse a la antitrombina, a
Cuadro 3–4. Contraindicaciones para anticoagulación
� Presencia de sangrado� Trombocitopenia inducida por heparina
� Sangrado reciente� Trauma mayor reciente o cirugía� Enfermedad cerebrovascular hemorrágica� Trombocitopenia (< 50 000/mm3)
� Prueba de guayaco positiva en materia fecal� Neoplasias del sistema nervioso central, aneurismas o malformación vascular

47Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
diferencia del fondaparinux. El fármaco oral se absorbe rápidamente después desu administración y la concentración máxima se alcanza de 2 a 4 h con una vidamedia de 7 a 11 h. Con dosis de rivaroxabán de 10 mg la absorción oral es casicompleta y la biodisponibilidad oral es alta (80 a 100%), independientemente delas condiciones de ayuno/alimento. Para el comprimido de 20 mg en condicionesde ayuno y por el grado reducido de absorción se determinó una biodisponibili-dad oral de 66%, por lo que los comprimidos de 15 y 20 mg deben tomarse conalimentos.
Aproximadamente dos tercios del fármaco activo se convierten a su formainactiva en el hígado (CYP3A4 y P–gp) y se eliminan por vías renal y fecal. Eltercio restante se elimina a través de la orina sin cambios.
Con la administración simultánea de inhibidores del CYP3A4 y P–gp (comoazoles–antimicóticos o inhibidores de la proteasa VIH) puede aumentar la con-centración plasmática de rivaroxabán, con riesgo de hemorragia. Tanto el tiempode protrombina como el tiempo parcial de tromboplastina activada se ven afecta-dos con la administración del fármaco; sin embargo, no es necesario monitorearlos parámetros de coagulación durante el tratamiento.
En la actualidad se recomienda su uso como profilaxis en la enfermedad trom-boembólica venosa, tanto en el reemplazo total de cadera (estudios RECORD1y RECORD2) como de rodilla (estudios RECORD3 y RECORD4). La dosis pro-filáctica en cirugía ortopédica es de 10 mg al día, con su inicio entre 6 y 10 horasdespués de la cirugía, siempre que se haya alcanzado la hemostasia. El estudioROCKET AF realizado para evaluar la profilaxis antitrombótica en pacientescon fibrilación auricular no valvular involucró a 14 264 pacientes que recibieronrivaroxabán en dosis de 20 mg una vez al día (Ud) (15 mg Ud en pacientes condepuración de creatinina entre 30 y 49 mL/min) o dosis ajustada de warfarina(INR 2.0 a 3.0). El promedio en la clasificación de CHADS2 fue de 3.5. La mayo-ría de los pacientes (54.8%) tuvieron evento vascular cerebral (EVC) previo, is-quemia cerebral transitoria (ICT) o embolismo sistémico. El punto primario deeficacia (EVC isquémico o embolismo sistémico) ocurrió en 188 pacientes conrivaroxabán (1.7%/año) y en 241 pacientes con warfarina (2.2%/año). La hemo-rragia mayor y no mayor clínicamente relevante ocurrió en 1 475 pacientes conrivaroxabán y en 1 449 pacientes con warfarina.
En dicho estudio se concluyó que el rivaroxabán fue no inferior a la warfarinaen la prevención de EVC o embolismo sistémico en pacientes con fibrilación au-ricular no valvular, sin diferencia en el riesgo de sangrado mayor y no mayor clí-nicamente relevante.
El programa EINSTEIN incluye los estudios del uso del rivaroxabán en EP,TVP y prevención secundaria (EXTENSION). Los dos últimos estudios ya fina-lizaron; en ellos se incluyeron 3 449 pacientes en quienes se comparó el uso derivaroxabán (dosis de 15 mg dos veces al día durante las tres primeras semanas,

48 (Capítulo 3)Tratado de trombosis
seguidos de 20 mg una vez al día) vs. enoxaparina, seguida de antagonista de lavitamina K durante 3, 6 o 12 meses en pacientes con TVP aguda y sintomática.
De forma paralela se realizó el estudio EXTENSION, que comparó el uso derivaroxabán en dosis de 20 mg una vez al día vs. placebo durante un periodo adi-cional de 6 a 12 meses en los pacientes que habían completado de 6 a 12 mesesde tratamiento para la ETV. Los resultados publicados indicaron que el uso derivaroxabán es tan efectivo como la terapia estándar, con un perfil de seguridadsimilar para el tratamiento de la TVP aguda, y que cuando se continúa el trata-miento el uso del anticoagulante es efectivo en la prevención de recurrenciascomparado con el placebo, con un riesgo aceptable de sangrado.
Actualmente el rivaroxabán se encuentra aprobado por la FDA para la profila-xis de cirugía ortopédica mayor, fibrilación auricular no valvular y trombosis ve-nosa profunda establecida.
Su administración está contraindicada en los siguientes casos:
� Hipersensibilidad al rivaroxabán o a alguno de sus excipientes.� Hemorragia activa.� Enfermedad hepática significativa asociada con coagulopatía.� Mujeres embarazadas y lactantes.� Depuración de creatinina inferior a 15 mL/min.
Apixabán
El apixabán es un reciente inhibidor, potente y altamente selectivo del factor Xa,con una vida media de entre 9 y 14 h, que es metabolizado en el hígado a travésdel CYP3A4. En el estudio ADVANCE–2 se administró apixabán como trombo-profilaxis en 3 057 pacientes con reemplazo electivo total de rodilla, en dosis de2.5 mg dos veces al día, con inicio a la mañana siguiente después de la cirugíadurante 10 a 14 días, y se comprobó que es una alternativa, en comparación conla enoxaparina, que no presenta incremento del riesgo de sangrado. El estudioAVERROES comparó el apixabán en dosis de 5 mg dos veces al día vs. ácido ace-tilsalicílico en dosis de 81 a 324 mg una vez al día en 5 600 pacientes con fibrila-ción auricular y uno o varios factores de riesgo de EVC, que no toleran los antago-nistas de la vitamina K o no son aptos para su uso. El estudio se interrumpió antesde lo previsto a causa de una importante reducción de los eventos embólicos conapixabán, con un perfil de seguridad aceptable.
El estudio ARISTOTLE incluyó a 18 201 pacientes para la tromboprofilaxisen fibrilación auricular no valvular, quienes recibieron apixabán en dosis de 5 mgdos veces al día (2.5 mg dos veces al día con � 2 de los siguientes criterios: edad� 80 años, peso corporal � 60 kg o creatinina sérica � 1.5 mg/dL) o dosis ajus-tadas de warfarina (INR 2.0 a 3.0). El promedio en la clasificación de CHADS2

49Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
fue de 2.1. Se anotó que 19% de los pacientes tenían antecedentes de EVC isqué-mico o ICT. El punto primario de eficacia (EVC isquémico o embolismo sistémi-co) ocurrió en 212 pacientes con apixabán (1.27%/año) y en 265 pacientes conwarfarina (1.6%/año). La hemorragia mayor ocurrió en 327 pacientes con apixa-bán (2.13%/año) y en 462 pacientes con warfarina (3.09%/año). Dicho estudioconcluyó que el apixabán fue superior a la warfarina en la prevención de EVCisquémico o embolismo sistémico en pacientes con fibrilación auricular no val-vular, con menos sangrado y menos mortalidad.
Edoxabán
El edoxabán es un inhibidor oral altamente selectivo del factor Xa. De acuerdocon los estudios fase III (STARS E–3 y STARS J–V) realizados en Japón, en losque se comparó el edoxabán en dosis de 30 mg una vez al día vs. enoxaparina endosis de 20 mg por vía subcutánea dos veces al día en pacientes sometidos a reem-plazo total de rodilla y de cadera, respectivamente, el uso de edoxabán es una al-ternativa al uso de enoxaparina, con un perfil de seguridad aceptable. Ya finalizóun estudio fase III (ENGAGE AF–TIMI 48) que compara el edoxabán (30 y 60mg una vez al día) y la warfarina en la prevención de embolismo en aproximada-mente 16 500 pacientes con fibrilación auricular. De igual forma, se encuentraen desarrollo el estudio HOKUSAI VTE para la prevención secundaria de enfer-medad tromboembólica venosa. En Japón ya está aceptada la comercializacióndel edoxabán para su uso en profilaxis de cirugía ortopédica mayor.
Betrixabán
El betrixabán es otro inhibidor directo del factor Xa, el cual se elimina de maneraprincipal por la bilis, por lo que ha sido estudiado en pacientes con distintos gra-dos de disfunción renal (excepto en diálisis). El betrixabán ha completado el estu-dio de fase II (EXPLORE–Xa) en pacientes con fibrilación auricular o aleteo novalvulares y al menos un factor de riesgo para EVC, comparado con dosis ajusta-das de warfarina.
INHIBIDORES DIRECTOS DE LA TROMBINA
Entre los inhibidores directos de la trombina se encuentran varios fármacos, loscuales difieren en sus sitios específicos de unión con la trombina y poseen distin-tas formas de liberación, metabolismo y excreción. Los inhibidores directos dela trombina son la hirudina (aprobada para el tratamiento del síndrome de HIT),

50 (Capítulo 3)Tratado de trombosis
la bivalirudina (aprobada para su uso en angioplastia), el argatrobán (aprobadopara el tratamiento del síndrome de HIT), el ximelagatrán (retirado del mercado),el AZD0837 (en estudio) y el dabigatrán.
Hirudina recombinante: lepirudina
La lepirudina (hirudina recombinante) muestra una notable eficacia terapéuticapara el síndrome de HIT; su uso fue aprobado en 1997 en la Comunidad Europea,en 1998 por la FDA y en 1999 en Canadá.
La lepirudina es un inhibidor bivalente de la trombina que se une de manerasimultánea al sitio activo (sitio catalítico de la trombina) y accesorio (fibrinó-geno), lo que genera un complejo irreversible con la trombina e inhibe todas susfunciones biológicas. Su administración intravenosa tiene un tiempo de inicio de8 a 12 min, con una vida media de aproximadamente una hora. A través de su ad-ministración subcutánea alcanza una concentración máxima entre tres y cuatrohoras; no se transporta al sistema nervioso central ni a la leche materna, y cercade 50% de los pacientes que reciben este fármaco durante más de cinco días desa-rrollan anticuerpos contra él. Se depura principalmente por vía renal y no se haestablecido el método más adecuado para el monitoreo de sus niveles terapéuti-cos, ya que el tiempo de trombina no es lineal, el tiempo de protrombina es insen-sible y el tiempo parcial de tromboplastina no puede ser determinado con altasdosis del medicamento.
Su principal efecto adverso es el sangrado, que se produce al formarse un com-plejo irreversible con la trombina para el cual aún no existe un antídoto.
El esquema de administración de la lepirudina es el siguiente:
1. Obtención del tiempo parcial de tromboplastina (TPT) basal: no iniciar encaso de un índice TPT de 2.5 o mayor.
2. Inicial: bolo intravenoso de 0.4 mg/kg/h (en 15 a 20 seg).3. Mantenimiento: 0.15 mg/kg/h (infusión continua).4. Ajustar dosis en caso de falla renal:
� Disminuir 50% para creatinina entre 1.6 a 2.0� Disminuir de 70 a 75% para creatinina entre 2.1 a 3.0� Disminuir de 80 a 85% para creatinina entre 3.1 a 6.0 (nivel sanguíneo
terapéutico de 0.5 a 1.5 mg/mL).
Bivalirudina
Sobre la estructura de la hirudina se han desarrollado una variedad de péptidossintéticos, como la bivalirudina, que es un péptido bivalente que se une a la trom-

51Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
bina tanto en el sitio del fibrinógeno como en el sitio catalítico, con el fin de dis-minuir la trombosis dependiente de las plaquetas.
A diferencia de la hirudina, la bivalirudina provoca sólo una inhibición tran-sitoria del sitio activo de la trombina, lo cual la convierte en un inhibidor de bajaafinidad. Su unión con la trombina es reversible y posee una vida media corta,que le confiere una mayor seguridad de uso que la lepirudina. Se metaboliza demanera principal en el hígado. Se encuentra aprobada como terapia alternativaa la heparina en pacientes que se someten a angioplastia, así como en el tratamien-to de pacientes con angina inestable.
Argatrobán
El argatrobán fue descubierto y desarrollado por el profesor S. Okamoto, en To-kio, en la década de 1970. Consiste en una mezcla compleja y sintética de isóme-ros R y S derivada de la L–arginina. El argatrobán fue utilizado en Japón en eldecenio de 1980 para la enfermedad arterial oclusiva periférica, pero actualmentese utiliza para la isquemia cerebral aguda y como anticoagulante en los pacientescon deficiencia de antitrombina o con síndrome de HIT. En la actualidad su usoen EUA incluye el manejo del síndrome de HIT y la intervención coronaria per-cutánea. El argatrobán realiza una inhibición competitiva y reversible del sitiocatalítico activo de la trombina. En esencia, inhibe las reacciones inducidas porla trombina, que incluyen la activación de los factores V, VIII y XIII, y de la pro-teína C; asimismo, interfiere con la agregación plaquetaria. Es altamente selecti-vo para la trombina y tiene una excelente constante de inhibición.
Posee un rápido inicio de acción (cerca de 30 min) y se metaboliza en el hígadoa través de hidroxilación y aromatización mediante el sistema del citocromoP450 microsomal, por lo que la dosis se debe ajustar en los pacientes con fallahepática. El monitoreo de la terapia con argatrobán se puede hacer a través delTPT. Este fármaco prolonga el INR, por lo que se debe monitorear en pacientesque reciben antagonistas de la vitamina K de manera concomitante.
El esquema de administración del argatrobán incluye:
1. Obtención del TPT basal.2. Dosis inicial: 2 �g/kg/min mediante infusión intravenosa continua.3. Obtención del TPT a las dos horas, después ajustar la dosis para mantener
el TPT de 1.5 a 3 veces el valor basal (no exceder una dosis de 10 �g/kg/mino un TPT de 100 seg).
Melagatrán y ximelagatrán
El melagatrán y su profármaco oral —el ximelagatrán— dieron inicio a una nue-va era en la profilaxis y en la terapia de la enfermedad tromboembólica.

52 (Capítulo 3)Tratado de trombosis
El melagatrán inhibe de manera reversible el sitio catalítico de la trombina. Esun dipéptido univalente de bajo peso molecular, diseñado para mimetizar el fibri-nopéptido A. Asimismo, inhibe la agregación plaquetaria dependiente de trombi-na en muy bajas concentraciones. A diferencia de otros inhibidores directos dela trombina, se une de manera escasa a las proteínas plasmáticas (< 15%). Des-pués de su administración intravenosa o subcutánea alcanza un estado estable alos 30 min y tiene una vida media de dos a cuatro horas. Su excreción es por víarenal. El TPT es una medida adecuada de monitoreo, con un incremento de 1.5a 2.5 veces el control. El INR no se debe utilizar como monitor de su actividad.
El ximelagatrán fue el primer inhibidor oral directo de la trombina comerciali-zado en Europa para la prevención de eventos cerebrovasculares en la fibrilaciónauricular. Fue tan eficaz como la warfarina en dosis ajustadas, se asoció a meno-res índices de hemorragia que la warfarina y mostró un índice terapéutico másamplio (estudios SPORTIF), con una dosificación fija y sin necesidad de contro-lar la anticoagulación; sin embargo, fue retirado del mercado por su hepatotoxici-dad.
AZD0837
El AZD0837 es un anticoagulante oral que aún se encuentra en investigación; esconvertido a su forma activa, AR–H067637, un inhibidor directo y selectivo dela trombina, con una semivida de 9 a 14 h. Ya finalizó la fase II de estudio en pa-cientes con fibrilación auricular no valvular, en quienes la dosis de 300 mg unavez al día mostró una disminución similar de los eventos trombóticos, con menorriesgo de sangrado vs. dosis ajustadas de AVK.
Dabigatrán
La trombina constituye un agonista de la activación plaquetaria, además de quees esencial en el paso de fibrinógeno a fibrina en la fase plasmática de la coagula-ción. El etexilato de dabigatrán inhibe la trombina de forma directa y reversible,y es hidrolizado por esterasas plasmáticas a su forma activa —dabigatrán— des-pués de su absorción intestinal. El etexilato de dabigatrán requiere un medio áci-do para su adecuada absorción, motivo por el que la cápsula contiene microesfe-ras cubiertas por el fármaco y un centro de ácido tartárico. Dicha característicaexplica la mayor incidencia de dispepsia, en comparación con la warfarina; sibien el uso concomitante de inhibidores de la bomba de protones reduce la biodis-ponibilidad cerca de 20%, no es clínicamente significativo, por lo que no es nece-sario realizar un ajuste de la dosis. Tiene una semivida de 12 a 17 h y se elimina

53Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
principalmente por vía renal (80%), sin participación del CYP450, por lo que pre-senta una baja interacción con los fármacos y los alimentos. Su administraciónes oral en dosis fija y no requiere monitoreo por estudios de laboratorio. El dabi-gatrán es un sustrato de la glucoproteína–p (p–gp), lo que ocasiona que algunosfármacos, como el verapamilo, la claritromicina y la amiodarona, interactúen conél, requiriéndose para esta última un ajuste de la dosis en los pacientes que seránintervenidos quirúrgicamente.
El etexilato de dabigatrán se recomienda para la prevención primaria de la en-fermedad tromboembólica venosa en reemplazo total de cadera y de rodilla (estu-dios RE–NOVATE y RE–MODEL, respectivamente).
La dosis recomendada en cirugía ortopédica mayor es de 220 mg una vez aldía (dos cápsulas de 110 mg). Su administración se inicia con la mitad de la dosis(110 mg) entre una y cuatro horas posteriores a la cirugía; a partir de esto se conti-núa con la dosis de mantenimiento.
La población especial en cirugía ortopédica incluye los pacientes mayores de75 años de edad, los pacientes con depuración de creatinina de 30 a 50 mL/miny los pacientes bajo tratamiento con amiodarona, quienes deben recibir 75 mg en-tre una y cuatro horas posteriores a la cirugía y continuar con 150 mg una vez aldía.
De acuerdo con la última recomendación de las guías del American Collegeof Chest Physicians de 2012, los pacientes deben recibir tratamiento profilácticodurante 35 días posteriores a la cirugía.
Después de los estudios de fase II de intervalo de dosis (PETRO) y de exten-sión a largo plazo (PETRO–EX), en 2009, se publicaron los resultados del estu-dio RE–LY para la tromboprofilaxis en pacientes con fibrilación auricular no val-vular, que incluyó a 18 113 pacientes, quienes recibieron una de tres opciones:dabigatrán en dosis de 110 mg dos veces al día, dabigatrán en dosis de 150 mgdos veces al día o dosis ajustadas de warfarina (INR 2.0 a 3.0). El promedio enla clasificación de CHADS2 fue de 2.1. Se anotó que 20.1% de los pacientes te-nían antecedentes de EVC isquémico o ICT. El punto primario de eficacia (EVCo embolismo sistémico) ocurrió en 183 pacientes con dabigatrán en dosis de 110mg dos veces al día (1.54%/año), en 134 pacientes con dabigatrán en dosis de 150mg dos veces al día (1.11%/año) y en 202 pacientes con warfarina (1.71%/año).El dabigatrán en ambas dosis no fue inferior a la warfarina; incluso la dosis de150 mg dos veces al día fue superior. La hemorragia mayor se presentó en 342pacientes con dabigatrán en dosis de 110 mg dos veces al día (2.87%/año), en 399pacientes con dabigatrán en dosis de 150 mg dos veces al día (3.32%/año) y en421 pacientes con warfarina (3.57%/año). En dicho estudio se concluyó que eldabigatrán en dosis de 110 mg dos veces al día no fue inferior a la warfarina enla prevención de EVC isquémico y embolismo sistémico en pacientes con fibrila-ción auricular no valvular, mientras que el dabigatrán en dosis de 150 mg dos ve-

54 (Capítulo 3)Tratado de trombosis
Cuadro 3–5. Normas para suspender el uso de dabigatrán en pacientesque serán sometidos a procedimientos invasivos o cirugía
Depuración decreatinina (mL/min)
Cirugía mayor oalto riesgo de sangrado
Cirugía no mayor
> 80 48 h antes 24 h antes80 a 50 48 a 72 h antes 24 a 48 h antes30 a 50 96 h 48 a 72 h antes
Tomado de Alikhan R et al.: Emerg Med J 2013;0:1–6.
ces al día fue superior a la warfarina, sin diferencia en el riesgo de hemorragiamayor, por lo que en 2010 fue aprobado por la FDA.
El dabigatrán está contraindicado en los pacientes con una depuración de crea-tinina menor de 30 mL/min.
La dosis de dabigatrán recomendada en México para la fibrilación auricularno valvular es de 150 mg cada 12 h.
La población especial en fibrilación auricular no valvular incluye los pacientesmayores de 80 años de edad, los pacientes con riesgo elevado de sangrado o fallarenal moderada (depuración de creatinina de 30 a 50 mL/mm) y los pacientes bajotratamiento con verapamilo, quienes deben recibir 110 mg cada 12 h.
El estudio RE–COVER comparó el dabigatrán en dosis de 150 mg dos vecesal día con la warfarina (INR 2.0 a 3.0) durante seis meses en 2 564 pacientes contrombosis venosa profunda proximal aguda y sintomática o tromboembolia pul-monar. Las conclusiones de este estudio de no inferioridad indicaron que en lospacientes con enfermedad tromboembólica venosa el dabigatrán es tan efectivocomo la warfarina, con un perfil de seguridad similar.
En el cuadro 3–5 se muestran las recomendaciones de suspensión del dabiga-trán en caso de cirugía, y en el cuadro 3–6 se incluyen las características farmaco-cinéticas de los nuevos anticoagulantes orales.
Nuevos anticoagulantes orales y tiempos de coagulación
El tiempo de protrombina (TP/INR) mide la vía extrínseca o dependiente de unfactor tisular, por lo que es sensible a la deficiencia de los llamados factores de-pendientes de vitamina K (II, VII, IX y X), cuya activación se ve inhibida por losAVK y en los cuales el INR refleja el grado de anticoagulación de dichos fárma-cos. Sin embargo, el TP no resulta de utilidad para estimar el grado de anticoagu-lación con los nuevos anticoagulantes. El TPT mide las vías intrínseca y comúnde la fase plasmática de la coagulación, y así la integridad de todos los factores,excepto el VII y el XIII. El TPT no se muestra lineal a ciertas concentraciones

55Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 3–6. Características farmacocinéticasde los nuevos anticoagulantes orales
Dabigatrán Rivaroxabán Apixabán
Profármaco Etexilato de dabigatrán No NoPico máximo (h) 0.5 a 2 2 a 4 3 a 4Vida media plasmática (h) 12 a 14 9 a 13 8 a 15Eliminación renal 80% 33% 25%Hígado CYP3A4 No Sí SíSustrato gp–p Sí Sí SíUnión a proteínas 35% 95% 87%
Tomado de Palladino M, Thomson L, Swift B et al.: Implementing the new oral anticoagulants intothe hospital formulary. Am J Hematol 2012.
de dabigatrán, por lo que puede ser una herramienta cualitativa mas no cuantita-tiva del inhibidor directo de la trombina. Si bien el rivaroxabán también modificael TPT, éste tampoco es sensible para su adecuado monitoreo. El tiempo de trom-bina (TT) mide el paso de fibrinógeno a fibrina, por lo que se altera en presenciadel inhibidor directo de la trombina. Un TT en rango normal puede traducir pocao nula presencia de dabigatrán.
Los inhibidores directos de la trombina, incluido el dabigatrán, muestran unarelación tanto lineal como dependiente de la dosis con el tiempo de ecarina(ECT). El ECT proporciona información basada en los niveles plasmáticos de da-bigatrán; sin embargo, no se encuentra aún disponible en forma comercial. ElECT se realiza utilizando como reactivo el veneno de la serpiente Echis carina-tus, el cual promueve la formación de protrombina a meizotrombina, un precur-sor de la trombina que resulta inhibido por el dabigatrán.
El apixabán, que es un inhibidor directo del factor Xa similar al rivaroxabán,también modifica los tiempos de coagulación; sin embargo, es la medición de laactividad anti–factor Xa la manera de realizar el adecuado monitoreo de ambosanticoagulantes. En el cuadro 3–7 se muestran las utilidades de los tiempos decoagulación en cada uno de los nuevos anticoagulantes orales.
Cuadro 3–7. Pruebas de laboratorio: nuevos anticoagulantes orales
Utilidad Dabigatrán Rivaroxabán Apixabán
Fuerte TEC Antifactor Xa Antifactor XaTT
TPT TPT,TPDébil TP/INR
TEC: tiempo de ecarina; TT: tiempo de trombina; TPT: tiempo parcial de tromboplastina; TP: tiempode protrombina; INR: índice normalizado internacional. Tomado de Palladino M, Thomson L, SwiftB et al.: Implementing the new oral anticoagulants into the hospital formulary. Am J Hematol 2012.

56 (Capítulo 3)Tratado de trombosis
Presencia de hemorragia con losnuevos anticoagulantes orales
Hasta ahora no se cuenta con un antídoto específico para ninguno de los nuevosanticoagulantes orales; no obstante, se trabaja un antídoto para el dabigatrán, abase de anticuerpos monoclonales.
En el caso del dabigatrán se puede utilizar carbón activado, siempre que hayasido ingerido dentro de las dos horas previas. Como el dabigatrán presenta unabaja unión con las proteínas plasmáticas, se puede dializar en caso de hemorragiaclínicamente significativa, ya que 62% del producto es removido a las dos horasy 68% a las cuatro horas del procedimiento. Existen reportes sobre la recomenda-ción de utilizar concentrados de complejo de protrombina (PCC) y factor VII re-combinante (VIIa).
Para el rivaroxabán se puede utilizar también carbón activado, que disminuyesu absorción gastrointestinal; sin embargo, a diferencia del dabigatrán, el rivaro-xabán y el apixabán no se pueden dializar, debido a su alta unión a las proteínasplasmáticas. Es probable que los PCC sean de utilidad para ambos anticoagulan-tes.
EFECTO ANTITROMBÓTICO DE LAS ESTATINAS
Las estatinas (inhibidores de la hidroximetilglutaril coenzima A reductasa) pre-vienen la trombosis venosa y arterial. Las estatinas disminuyen la actividad dela trombina a través de la inhibición de la expresión del factor tisular sobre la su-perficie celular de los monocitos y los macrófagos. Todas las estatinas, exceptola pravastatina, incrementan la expresión del activador tisular del plasminógeno(t–PA) y disminuyen la expresión del inhibidor del activador del plasminógeno(PAI–1) por parte de las células endoteliales vasculares y del músculo liso.
Las estatinas también inhiben la activación y la agregación plaquetarias a tra-vés de la alteración de la composición de la membrana lipídica de las plaquetas,la disminución del tromboxano y el aumento en la síntesis de óxido nítrico.
Las estatinas reducen también la producción de moléculas inflamatorias, cuyoefecto antiinflamatorio sobre la pared endotelial es independiente de su efecto hi-polipemiante (cuadro 3–8).
TROMBOLÍTICOS
Desde la primera publicación acerca de los trombolíticos, en 1959, su uso tera-péutico se ha extendido para el manejo de la enfermedad trombótica de cualquierlocalización.

57Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 3–8. Efectos antitrombóticos de las estatinas
Mecanismo de acciónInhibición de la generación de trombinaEstimulación de la fibrinólisis (incremento del t–PA o disminución del PAI–1, o ambos)Inhibición de la actividad plaquetaria
Inhibición de la proliferación del músculo liso vascularDisminución de la viscosidad sanguíneaActividad antiinflamatoria
El objetivo de la terapia trombolítica es lograr una pronta recanalización delvaso sanguíneo (arteria o vena) trombosado.
Todos los agentes trombolíticos activan el plasminógeno a plasmina, la cualactúa para degradar los coágulos de fibrina, el fibrinógeno y otras proteínas plas-máticas, incluidos los factores V, VIII, IX, XI y XII, y los componentes del com-plemento. La plasmina se inactiva a través de varios inhibidores, como la a2–anti-plasmina y la �2–macroglobulina.
Los agentes trombolíticos utilizados con mayor frecuencia son la urocinasa(UK), la estreptocinasa (SK), la alteplasa (activador tisular del plasminógeno re-combinante [rt–PA]) y la anistreplasa (complejo activador de estreptocinasa ani-soilado o APSAC). Los agentes más recientes incluyen la reteplasa (r–PA), latenecteplasa (TNKt–PA), la saruplasa (scu–PA), la lanoteplasa (n–PA) y la estafi-locinasa (STAR).
La estreptocinasa es una enzima antigénica bacteriana, mientras que la uroci-nasa es un derivado de células vasculares, células renales y orina.
Los agentes trombolíticos no sólo activan el sistema fibrinolítico, sino queademás inducen un estado procoagulante. Durante la terapia trombolítica en elinfarto agudo del miocardio se observa una elevación de los marcadores de hiper-coagulabilidad, como el PAI–1, el t–PA, el fibrinógeno, el factor XII, el complejotrombina–antitrombina, la calicreína, la plasmina y el dímero–D.
La estreptocinasa provoca un efecto procoagulante mayor que el rt–PA en pa-cientes con infarto agudo del miocardio, con incremento en la activación de trom-bina, con un prolongado aumento en la actividad de calicreína y con elevados ni-veles de dímero–D.
En la trombosis coronaria aguda se activan las fases plaquetaria y plasmáticade la coagulación, así como el sistema fibrinolítico. El trombo coronario es ricoen plaquetas, por lo que la activación y la agregación plaquetarias se acentúandespués de la terapia trombolítica, de ahí la utilidad del manejo concomitante deagentes fibrinolíticos con antiplaquetarios.
En el cuadro 3–9 se muestran las distintas aplicaciones clínicas de los agentestrombolíticos y sus dosis.

58 (Capítulo 3)Tratado de trombosis
Cuadro 3–9. Aplicaciones clínicas de los agentes trombolíticos
Trombolítico Aplicación clínica Dosis
Estreptocinasa IAM 1.5 mU IV en 1 h o 20 kU intracoronario seguido de 2kU/min durante 60 min
TEP 250 kU IV en 0.5 h seguido de 100 kU/h durante 24 h(72 h si hay TVP concomitante)
TVP 250 kU IV en 0.5 h seguido de 100 kU/h durante 72 hAnistreplasa IAM 30 U IV en 3 a 5 minAlteplasa IAM 15 mg en bolo IV, seguido de 50 mg en infusión en 30
min; luego 35 mg en los siguientes 60 min (> 67 kg)Para � 67 kg ajustar a peso (dosis máxima de 100mg)
TEP 100 mg IV en 2 hEVC 0.9 mg/kg IV en 60 min con 10% de la dosis como bolo
inicial (90 mg máximo como dosis total)Reteplasa IAM 10 U IV en 2 min, esperar 28 min y luego repetirTenecteplasa IAM 30 a 50 mg IV en bolo (basado en el peso)
IAM = infarto agudo del miocardio; IV = intravenoso; TEP = tromboembolia pulmonar; TVP = trom-bosis venosa profunda; kU = 1 000 unidades; EVC = evento cerebrovascular isquémico.
CONTRAINDICACIONES PARAEL USO DE TROMBOLÍTICOS
Las contraindicaciones para el uso de agentes trombolíticos incluyen:
� Cirugía mayor reciente (10 días).� Punción reciente de un órgano o vaso sanguíneo no compresible (10 días).� Sangrado gastrointestinal o genitourinario reciente (10 días).� Trauma reciente (10 días).� Cirugía intracraneal o intraespinal, traumatismo craneoencefálico grave o
EVC recientes (tres meses).� Hipertensión arterial sistémica descontrolada, con una presión sistólica ma-
yor de 180 mmHg o una presión diastólica mayor de 110 mmHg.� Neoplasia intracraneal, malformación arteriovenosa o aneurisma.� Alta probabilidad de trombo intracardiaco.� Endocarditis bacteriana subaguda.� Pericarditis aguda.� Diátesis hemorrágica.� Disfunción hepática grave (la reteplasa se puede utilizar con seguridad, de-
bido a su metabolismo renal).� Embarazo.

59Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 3–10. Complicaciones de la terapia trombolítica
Complicación Etiología Manejo
Hemorragia Fibrinogenólisis Ácido �–aminocaproico, PFC, crioprecipita-dos
Disfunción plaquetaria Transfusión plaquetaria (requiere ser deter-minada de acuerdo con el tiempo desangrado)
Embolización Trombólisis parcial Continuar infusión de trombolítico, asociarotros agentes adyuvantes
Hipotensión, rash, fiebre Anafilaxia Líquidos intravenosos, vasopresores, glu-cocorticoides, antihistamínicos
PFC = plasma fresco congelado.
� Oftalmopatía hemorrágica (como retinopatía diabética).� Uso de anticoagulantes (sobre todo warfarina).� Tromboflebitis séptica o fístula arteriovenosa séptica y trombosada.� Alergia al agente trombolítico (en especial estreptocinasa o anistreplasa).� Administración de estreptocinasa o anistreplasa en los dos años previos o
más (los anticuerpos pueden persistir hasta 54 meses).� Edad mayor de 75 años (contraindicación relativa).
El cuadro 3–10 incluye el manejo de las posibles complicaciones surgidas conel uso de los agentes trombolíticos.
CONCLUSIÓN
Nos está tocando ser testigos de los grandes avances no sólo respecto a un conoci-miento más amplio del fenómeno trombótico, sino de la evolución que han tenidoa la par los recientes agentes antitrombóticos. Los estudios que hasta el momentose han realizado con ellos, dentro del tan amplio abanico que involucra a la enfer-medad trombótica, conducirán con probabilidad a corto plazo a conocer su utili-dad en todas las demás áreas, incluyendo la enfermedad aterotrombótica y quizálas trombofilias primarias. Las finas diferencias entre los recientes agentes quizágeneren indicaciones específicas para su uso, lo cual nos obliga a los médicos detodas las especialidades a mantenernos actualizados en tan importante entidadnosológica en nuestros días, la enfermedad trombótica.
REFERENCIAS
1. Messmore H, Jeske W, Wehrmacher W et al.: Antiplatelet agents: current drugs and fu-ture trends. Hematol Oncol Clin N Am 2005;19:87–117.

60 (Capítulo 3)Tratado de trombosis
2. Billett H: Antiplatelet agents and arterial thrombosis. Clin Geriatr Med 2006;22:57–74.3. Fernández J, Sadaniantz B, Sadaniantz A: Review of antithrombotic agents used for
acute coronary syndromes in renal patients. Am J Kidney Dis 2003;42:446–455.4. Pineo G, Hull R: Vitamin K antagonists and direct thrombin inhibitors: present and future.
Hematol Oncol Clin N Am 2005;19:69–85.5. Owens C, Belkin M: Thrombosis and coagulation: operative management of the anticoa-
gulated patient. Surg Clin N Am 2005;85:1179–1189.6. Frenkel E, Shen Y, Haley B: The direct thrombin inhibitors: their role and use for rational
anticoagulation. Hematol Oncol Clin N Am 2005;19:119–145.7. Hyers TM, Agnelli G, Hull RD et al.: Antithrombotic therapy for venous thromboembolic
disease. Chest 1998;114:561S–578S.8. Mullen M, McGarvey M, Kasner S: Safety and efficacy of thrombolytic therapy in post-
operative cerebral infarctions. Neurol Clin 2006;24:783–793.9. Smalling R: Role of fibrinolytic therapy in the current era of ST–segment elevation myo-
cardial infarction management. Am Heart J 2006;151:S17–S23.10. Cohen M, Arjomand H, Pollack C: The evolution of thrombolytic therapy and adjunctive
antithrombotic regimens in acute ST–segment elevation myocardial infarction. Am J EmergMed 2004;22:14–23.
11. Sixth ACCP Consensus Conference on Antithrombotic Therapy. Chest 2001;119(1).12. Ansell J, Hirsh J, Poller L: The Seventh ACCP Conference on Antithrombotic and Throm-
bolitic Therapy: evidence–based guidelines. Chest 2004;126(3).13. Scazziota A, Altman R: El mecanismo de la hemostasia normal. Rev Iberoam Tromb He-
most 1994;7:95–109.14. Lobato ME, Ruiz AGJ: Proteína C, proteína S y trombomodulina: uno de los mecanismos
antitrombóticos naturales. Rev Invest Clin 1990;42:54–62.15. Shafer A: The hipercoagulable states. Ann Intern Med 1985;102:814–828.16. Gensini GF, Prisco D, Falciani M, Comeglio M, Colella A: Identification of candidates
for prevention of venous thromboembolism. Sem Thromb Hemostas 1997;23:55–67.17. Subar M: Thromboembolic disease and anticoagulation in the elderly. Clin Geriatric Med
2001;17(1).18. Whiteman T, Hassouna H: Hypercoagulable states. Hematol Oncol Clin N Am 2000:14(2).19. Hurtado R: Hipercoagulabilidad (trombofilia). Rev Med La Salle 1998;29(3):211–217.20. De Estefano V, Finazzi G, Manucci PM: Inherited thrombophilia: pathogenesis, clinical
syndromes and management. Blood 1996;87:3531–3144.21. Ruiz AGJ: Contribuciones mexicanas al conocimiento de las alteraciones trombogénicas en
el síndrome de anticuerpos antifosfolípidos. Rev Iberoam Tromb Hemost 1997;10:69–71.22. Ruiz AGJ: Resistencia a la proteína C activada como causa de trombofilia. Rev Invest Clin
1996;48;223–229.23. Goldhaber S, Savage D, Garrison R et al.: Risk factors for pulmonary embolism: the Fra-
mingham Study. Ann Intern Med 1983;74:1023–1028.24. Hirsh J, Fuster V: Guide to anticoagulant therapy: heparin. Circulation 1994;89:1449–1468.25. Clagett G, Anderson F, Het J, Levine M et al.: Prevention of venous thromboembolism.
Chest 1995;108:3125–3134.26. Hyers T, Hull R, Weg J: Antithrombotic therapy for venous thromboembolic disease.
Chest 1995;108:3355–3515.27. Bick RL, Fareed J: Current status of thrombosis: a multidisciplinary medical issue and ma-
jor American health problem–beyond the year 2000. Clin Appl Thrombosis Hemostasis1997;3(Suppl 1):S1–S5.

61Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
28. Gutiérrez R, Sánchez C: Embolia pulmonar. Rev Med La Salle 1998;29(3):219–224.29. Cesarman G, Villazón S, Zúñiga J: Absence of factor V Leiden mutation in Mazateco In-
dians. Int J Haematol 1996;64(Suppl 1):S111.30. Gross JS, Neufeld RR, Lobow LS, Gerber I, Rodstein M: Autopsy studies of the elderly
institutionalized patients: review of 234 autopsies. Arch Intern Med 1988;148:173–176.31. Lindblad B, Sternby NH, Bergqvist D: Incidence of venous thromboembolism verified
by necropsy over 30 years. Br Med J 1991;302:709–711.32. Ibrahim E, Iregui M, Prentice D, Sherman G, Kollef M et al.: Deep vein thrombosis dur-
ing prolonged mechanical ventilation despite prophylaxis. Crit Care Med 2002:30(4).33. Selby R, Geerts W: Venous thromboembolism prophylaxis after trauma: dollars and sense.
Crit Care Med 2001:29(9).34. Anderson FA, Wheeler B, Goldberg RJ, Hosmer DW: The prevalence of risk factors for
venous thromboembolism among hospital patients. Arch Intern Med 1992;152:1660–1664.35. Anderson DR, Wells PS, Stiel I, MacLeod B, Simms M et al.: Thrombosis in the emer-
gency department. Use of clinical diagnosis model to safely avoid the need for urgent radio-logical investigation. Arch Intern Med 1999;159:477–482.
36. Gensini GF, Prisco D, Falciani M, Comeglio M, Colella: Identification of candidates forprevention of venous thromboembolism. Sem Thromb Hemostas 1997;23:55–67.
37. Joynt G, Kew J, Gomersall C, Leung V, Liu E: Deep venous thrombosis caused by femo-ral venous catheters in critically ill adult patients. Chest 2000;117(1):178–183.
38. Kyrle P, Minar E, Hirschl M, Bialonczyk C, Stain M et al.: High plasma levels of factorVIII and the risk of recurrent venous thromboembolism. N Engl J Med 2000;343(7):457–462.
39. Lobato ME, Majluf CA: Trombofilia, tromboembolia y el uso de las heparinas de bajopeso molecular. Rev Invest Clin 2000;52(5):529–545.
40. Clayton JK, Anderson JA, McNicol GP: Preoperative prediction of postoperative deepvein thrombosis. Br Med J 1976;2:910–912.
41. Rocha E, Alfaro MJ, Páramo JA, Canadell JM: Preoperative identification of patientsat high risk of deep venous thrombosis despite prophylaxis in total hip replacement. ThrombHaemostas 1988;59:93–95.
42. Andres R, Miles A: Venous thromboembolism and pregnancy. Obstet Gynecol 2001:28(3).43. Rosendaal FR: Venous thrombosis: a multicausal disease. Lancet 1999;353:1167–1173.44. Davis J: Prevention, diagnosis, and treatment of venous thromboembolic complications of
gynecologic surgery. Am J Obstet Gynecol 2001;184(4).45. Amiral J: Molecular markers in thrombosis and hemostasis. Clin Appl Thrombosis Hemos-
tasis 1997;3:71–81.46. Geerts W, Heit J, Clagett P, Pineo G, Colwell C et al.: Prevention of venous thromboem-
bolism. Chest 2001;119:132S–175S.47. Bergqvist D, Benoni G, Björgell O, Fredin H, Hedlundh U et al.: Low–molecular–
weight heparin (enoxaparin) as prophylaxis against venous thromboembolism after totalhip replacement. N Engl J Med 1996;335(10):696–700.
48. Comp P, Spiro T, Friedman R, Whitsett T, Johnson G et al.: Prolonged enoxaparin thera-py to prevent venous thromboembolism after primary hip or knee replacement. J Bone JointSurg Am 2001;83(3):336–349.
49. Bergqvist D, Agnelli G, Cohen A, Eldor A, Nilsson P et al.: Duration of prophylaxisagainst venous thromboembolism with enoxaparin after surgery for cancer. N Engl J Med2002;346(13):975–980.
50. Hirsh J: The optimal duration of anticoagulant therapy for venous thrombosis. N Engl JMed 1995;332(25):1710–1712.

62 (Capítulo 3)Tratado de trombosis
51. Schulman S, Granqvist S, Holmström M, Carlsson A, Lindmarker P et al.: The durationof oral anticoagulant therapy after a second episode of venous thromboembolism. N EnglJ Med 1997;336(6):393–398.
52. Kearon C, Gent M, Hirsh J, Weitz J, Kovacs M et al.: A comparison of three months ofanticoagulation with extended anticoagulation for a first episode of idiopathic venousthromboembolism. N Engl J Med 1999;340(12):901–907.
53. Ressel G: Practice bulletin on preventing deep venous thrombosis and pulmonary embo-lism. Am Acad Fam Phys 2001;63(11).
54. Brandjes DP, Buller HR, Heijboer H et al.: Randomized trial of effect of compression stock-ings in patients with symptomatic proximal–vein thrombosis. Lancet 1997;349:759–762.
55. Hull RD, Raskob GE, Rosenbloom D et al.: Optimal therapeutic level of heparin therapyin patients with venous thrombosis. Arch Intern Med 1992;152:1589–1595.
56. Raschke RA, Reilly BM, Guidry JR et al.: The weight–based heparin dosing nomogramcompared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med1993;119:874–881.
57. Levine M, Gent M, Hirsh J et al.: A comparison of low–molecular–weight heparin admin-istered primary at home with unfractionated heparin administered in the hospital for proxi-mal deep–vein thrombosis. N Engl J Med 1996;334:677–681.
58. Koopman MM, Prandoni P, Piovella F et al.: Treatment of venous thrombosis with intra-venous unfractionated heparin administered in the hospital as compared with subcutaneouslow–molecular–weight heparin administered at home. The Tasman Study Group. N Engl JMed 1996;334:682–687.
59. Schulman S, Rhedin AS, Lindmarker P et al.: A comparison of six weeks with six monthsof oral anticoagulant therapy after a first episode of venous thromboembolism. N Engl JMed 1995;332:1661–1665.
60. Hull RD, Pineo GF, Valentine KA: Treatment and prevention of venous thromboembol-ism. Semin Thromb Hemost 1998;24(Suppl 1):21–31.
61. Goldhaber SZ: Thrombolytic therapy in venous thromboembolism. Clinical trials and cur-rent indications. Clin Chest Med 1995;16:307–320.
62. Levine MN, Raskob G, Landefeld S, Kearon C: Hemorrhagic complications of anticoag-ulant treatment. Chest 2001;119:1088–1218.
63. Laster J, Elfrink R, Silver D: Reexposure to heparine of patients with heparin–associatedantibodies. J Vasc Surg 1989;9:677–681.
64. Warkentin TE: Heparin–induced thrombocytopenia: a ten–year retrospective. Ann RevMed 1999;50:129–147.
65. Greinacher A, Lubenow N: Recombinant hirudin in clinical practice: focus on lepirudin.Circulation 2001;103:1479–1484.
66. Booth SL, Centurelli MA: Vitamin K: a practical guide to the dietary management of pa-tients on warfarin. Nutr Rev 1999;57:288–296.
67. Triplett DA: Current recommendations for warfarin therapy. Use and monitoring. Med ClinN Am 1998;82:601–611.
68. Baker W: Thrombolitic therapy. Clinical applications. Hematol Oncol Clin N Am 2003;17:283–311.
69. Sanson BJ, Lensing AW, Prins MH et al.: Safety of low–molecular–weight heparin inpregnancy: a systematic review. Thromb Haemost 1998;81:668–672.
70. Weitz J, Hirsh J, Samara M: New anticoagulant drugs. The Seventh ACCP Conferenceon Antithrombotic and Thrombolytic Therapy: evidence–based guidelines. Chest 2004:126.
71. Steinhubl S, Bhatt D, Brennan D et al.: Ácido acetilsalicílico para prevenir enfermedad

63Fármacos antitrombóticosE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
cardiovascular: asociación de la dosis de ácido acetilsalicílico y clopidogrel con trombosisy hemorragia. Ann Intern Med 2009;150:379–386.
72. García–Frade RLF: Pasado, presente y futuro en la terapia antitrombótica. Med Int Mex2010;26(4):383–389.
73. Bhatt D: Prasugrel in clinical practice. N Engl J Med 2009;361(10):940–942.74. Schömig A: Ticagrelor–is there need for a new player in the antiplatelet–therapy field? N
Engl J Med 2009;361(11):1108–1111.75. Wallentin L, Becker R, Budaj A et al.: Ticagrelor versus clopidogrel in patients with acute
coronary syndromes. N Engl J Med 2009;361(11):1045–1057.76. The Van Gogh Investigators: Idraparinux versus standard therapy for venous thromboem-
bolic disease. N Engl J Med 2007;357:1094–1110.77. Ellis D, Usman MH, Milner P: The first evaluation of a novel vitamin K antagonist, tecar-
farin (ATI–5923), in patients with atrial fibrillation. Circulation 2009;120:1029–1035.78. Geerts W, Bergqvist D, Pineo G et al.: Prevention of venous thromboembolism: American
College of Chest Physicians evidence–based clinical guidelines (8ª edition). Chest 2008;133:381–453.
79. Lassen M, Raskob G, Gallus A et al.: Apixaban versus enoxaparina for thromboprophy-laxis after knee replacement (ADVANCE–2): a randomized double–blind trial. Lancet2010;375(9717):807–815.
80. Eriksson B, Borris L, Friedman R et al.: Rivaroxaban versus enoxaparin for thrombopro-phylaxis after hip arthroplasty. N Engl J Med 2008;358:2765–2775.
81. Kakkar A, Brenner B, Dahl O et al.: Extended duration rivaroxaban versus short–termenoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: adouble blind, randomized controlled trial. Lancet 2008;372:31–39.
82. Lassen M, Ageno W, Borris L et al.: Rivaroxaban versus enoxaparin for thromboprophy-laxis after total knee arthroplasty. N Engl J Med 2008;358:2776–2786.
83. Turpie A, Bauer K, Davidson B et al.: Comparison of rivaroxaban–an oral direct factorXa inhibitor–and subcutaneous enoxaparin for thromboprophylaxis after total knee re-placement (RECORD4: a phase III study). European Federation of National Associationsof Orthopedics and Traumatology 2008 Meeting. Niza, 29 de mayo a 1 de junio de 2008.Abstract F85.
84. Eriksson B, Dahl O, Rosencher N et al.: Dabigatran etexilate versus enoxaparin for pre-vention of venous thromboembolism after total hip replacement: a randomized, double–blind, noninferiority trial. Lancet 2007;370:949–956.
85. Eriksson B, Dahl O, Rosencher N et al.: Oral dabigatran etexilate vs. subcutaneous enoxa-parin for the prevention of venous thromboembolism after total knee replacement: the RE–MODEL randomized trial. J Thromb Haemost 2007;5:2178–2185.
86. Connolly S, Ezekowitz M, Phil et al.: Dabigatran versus warfarin in patients with atrialfibrillation. N Engl J Med 2009;361(12):1131–1159.
87. Schulman S, Kearon C, Kakkar A et al.: Dabigatran versus warfarin in the treatment ofacute venous thromboembolism. N Engl J Med 2009;361:2342–2362.
88. Burness C, Scott L: Acetylsalicylic acid/esomeprazole fixed–dose combination. DrugsAging 2012;29(3):233–242.
89. Guyatt G, Akl E, Crowther M et al.: Antithrombotic therapy and prevention of thrombo-sis. 9ª ed. American College of Chest Physicians evidence–based clinical practice guide-lines. Chest 2012;141;7S–47S.
90. Tendera M, Syzdó M, Parma Z: ARISTOTLE re–lys on the rocket. What’s new in strokeprevention in patients with atrial fibrillation? Cardiol J 2012;19(1):4–10.

64 (Capítulo 3)Tratado de trombosis
91. Tan Ru San, Mark Yan Yee Chan, Teo Wee Siong et al.: Stroke prevention in atrial fibri-llation: understanding the new oral anticoagulants dabigatran, rivaroxaban, and apixaban.Thrombosis 2012:1–10.
92. Banerjee A, Lane D, Torp Pedersen C et al.: Net clinical benefit of new oral anticoagu-lants (dabigatran, rivaroxaban, apixaban) versus no treatment in a “real world” atrial fibri-llation population: a modelling analysis based on a nationwide cohort study. Thromb Hae-most 2012;107:1–6.
93. Hvilsted L, Bjerregaard T, Graungaard T et al.: Primary and secondary prevention withnew oral anticoagulant drugs for stroke prevention in atrial fibrillation: indirect comparisonanalysis. Br Med J 2012;345:e7097.
94. The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembol-ism. N Engl J Med 2010.
95. Schulman S, Kearon C, Kakkar A et al.: Dabigatran versus warfarin in the treatment ofacute venous thromboembolism. N Engl J Med 2009;361:2342–2362.
96. Eikelboom J, Quinlan D, Connolly S et al.: Dabigatran efficacy–safety assessment forstroke prevention in patients with atrial fibrillation. J Thromb Haemost 2012;10:966–968.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
4Tromboprofilaxis
Luis Fernando García–Frade Ruiz
INTRODUCCIÓN
La mayoría de los eventos trombóticos primarios y recurrentes se pueden preve-nir a través de una apropiada terapia antitrombótica primaria y secundaria, res-pectivamente.
La mayoría de las patologías que afectan al ser humano pueden ser prevenidas;la diferencia con la mayor parte de las trombosis venosas es que éstas son precedi-das por un claro escenario de factores de riesgo, lo que las hace claramente preve-nibles, como el caso de los pacientes quirúrgicos, hospitalizados, ortopédicos,cardiacos, etc. Entre las medidas profilácticas para el fenómeno trombótico se en-cuentran las terapias mecánica y farmacológica. Cabe mencionar que la sustitu-ción de algunas de ellas por métodos no mencionados en este capítulo puede ori-ginar resultados inciertos; es decir, la adecuada selección de los instrumentos autilizar para una adecuada profilaxis se basa en la estratificación de riesgo, lasindicaciones, las contraindicaciones y la disponibilidad de los mismos. Más ade-lante se mencionan las Guías de terapia antitrombótica y prevención de trombo-sis del American College of Chest Physicians, en su novena edición del año 2012,mismas que fueron elaboradas por un grupo de expertos, con base en resultadosde estudios multicéntricos.
Hay varias condiciones clínicas que se encuentran asociadas a un incrementodel riesgo de trombosis arterial o venosa; las más comunes se muestran en el cua-dro 4–1.
65

66 (Capítulo 4)Tratado de trombosis
Cuadro 4–1. Causas clínicas de la trombosis
Condiciones clínicas: arterial Condiciones clínicas: venosa
Aterosclerosis Cirugía general
Tabaquismo Cirugía ortopédicaHipertensión ArtroscopiaDiabetes mellitus Trauma
Colesterol LDL MalignidadHipertrigliceridemia InmovilizaciónHistorial familiar positivo Sepsis
Falla ventricular izquierda Falla cardiaca congestivaAnticonceptivos orales Síndrome nefróticoEstrógenos Obesidad
Lipoproteína (a) Venas varicosasPolicitemia Síndrome posflebíticoSíndromes de hiperviscosidad Anticonceptivos orales
Trombocitemia EstrógenosTrombocitemia
Los factores de riesgo para el desarrollo de enfermedad tromboembólica veno-sa en los pacientes hospitalizados no quirúrgicos se muestran en el cuadro 4–2.De manera particular, la presencia de enfermedades malignas y respiratorias cró-nicas se asocia a un alto riesgo de enfermedad trombótica. La mayoría de los pa-cientes hospitalizados son mayores de 40 años de edad, lo que representa un fac-tor de riesgo independiente para trombosis venosa. Los pacientes mayores de 60años de edad son los que tienen más alto riesgo.
Las enfermedades cardiovasculares confieren un mayor riesgo para el desarro-llo de eventos trombóticos, como infarto agudo del miocardio, cardiomiopatía is-quémica y no isquémica, cardiomiopatía crónica dilatada idiopática y falla car-diaca congestiva (de manera particular en los estadios III y IV de la New YorkHeart Association).
La falla respiratoria aguda también incrementa el riesgo de enfermedad trom-boembólica venosa, como las exacerbaciones agudas de una enfermedad pulmo-nar obstructiva crónica, la neumonía nosocomial o adquirida en la comunidad, elcáncer pulmonar, la enfermedad pulmonar intersticial y la hipertensión pulmonar.
La restricción en la movilización representa uno de los factores más importan-tes para el desarrollo de eventos trombóticos en el paciente hospitalizado no qui-rúrgico. Los pacientes confinados a guardar reposo en cama por menos de cincodías presentan 21% de ocurrencia de eventos trombóticos venosos, en compara-ción con 36% en aquellos con reposo por más de 10 días.
El riesgo de trombosis venosa en pacientes hospitalizados con enfermedadesmédicas sin terapia profiláctica incluyen la enfermedad médica (no quirúrgica)

67TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 4–2. Factores de riesgo relacionados con la enfermedadtromboembólica venosa en pacientes médicos hospitalizados
Riesgo alto
� Historial de trombosis venosa profunda o tromboembolia pulmonar
� Historial familiar de trombosis
� Infección aguda
� Malignidad
� Edad > 75 años
� Insuficiencia cardiaca congestiva
� Enfermedad cerebrovascular
� Inmovilización prolongada (> 4 días)
� Embarazo o posparto
� Enfermedad pulmonar aguda o crónica
� Enfermedad inflamatoria aguda
� Enfermedad intestinal inflamatoria
� Estado de choque
Riesgo posible
� Paraproteinemia
� Enfermedad de Behçet
� Desórdenes del plasminógeno
� Síndrome nefrótico
� Policitemia
� Hemoglobinuria paroxística nocturna
� Homocisteinemia
� Disfibrinogenemia
� Desórdenes mieloproliferativos
� Edad > 41 años
� Sepsis
Riesgo probable
� Altas dosis estrogénicas
� Obesidad (índice de masa corporal > 25)
� Venas varicosas
� Síndrome de trombocitopenia inducida por heparina
� Trombofilia congénita o adquirida
� Deficiencia de antitrombina
� Anticoagulante lúpico positivo
� Anticuerpos antifosfolípidos
� Deficiencia de proteína S
� Deficiencia de proteína C
� Factor V de Leiden positivo
� Anticuerpos anticardiolipina elevados
� Mutación del gen de protrombina G20210A positivo

68 (Capítulo 4)Tratado de trombosis
en general (10 a 26%), el infarto agudo del miocardio (17 a 34%), la enfermedadcerebrovascular (11 a 75%), la insuficiencia cardiaca congestiva (20 a 40%) y elcuidado médico intensivo (25 a 42%).
ESTRATIFICACIÓN DE RIESGO
La estratificación de riesgo para el desarrollo de tromboembolismo venoso en elpaciente quirúrgico, propuesta por el American College of Chest Physicians, semuestra en los cuadros 4–3 y 4–4. Los pacientes son clasificados con base en laedad, el tipo de cirugía y la presencia o ausencia de factores de riesgo adicionalespara tromboembolismo.
Profilaxis no farmacológica
Ambulación temprana
La ambulación temprana debe ser parte de la rutina del cuidado posquirúrgico en
Cuadro 4–3. Estratificación de riesgo para la enfermedadtromboembólica venosa en el paciente quirúrgico
Nivelde riesgo
Edad(años)
Tipo decirugía
Factoresadicionales
Incidenciade TVP
proximal(%)
Incidenciade TEP
(%)
Bajo < 40 Menor Ninguno 0.4 < 0.5Moderado 2 a 4 1 a 2
A Cualquiera Menor PresentesB < 40 Mayor NingunoC De 40 a 60 No mayor Ninguno
Alto 4 a 8 2 a 4A > 40 No mayor � otrosB > 60 Mayor NingunoC > 40 Mayor Presente
Muy alto 10 a 20 4 a 10A Artroplastia de cadera o
rodilla, cirugía por fracturade cadera, trauma mayoro lesión del cordón espinal
B > 40 Mayor ETV previaCáncerEstado hiper-
coagulable
TVP = trombosis venosa profunda; TEP = tromboembolia pulmonar; ETV = enfermedad tromboem-bólica venosa.

69TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 4–4. Profilaxis antitrombótica en procedimientos de cirugía general
Riesgo Profilaxis recomendada
Riesgo bajoProcedimiento menor en menores de 40 años de edadsin factor de riesgo adicional
Movilización temprana
Riesgo moderadoProcedimiento menor con factores de riesgo adicionalespara trombosis; cirugía no mayor en pacientes de entre40 y 60 años de edad sin factor de riesgo adicional;cirugía mayor en pacientes menores de 40 años sinfactores de riesgo adicionales
Bajas dosis de heparina no frac-cionada, HBPM, ME o CNI
Riesgo altoCirugía menor en mayores de 60 años de edad confactor de riesgo adicional; cirugía mayor en pacientesmayores de 40 años o con factor de riesgo adicional
Heparina no fraccionada, HBPMo CNI
Riesgo altoCon riesgo mayor que el usual para sufrir sangrado
Profilaxis mecánica con ME oCNI
Riesgo muy altoMúltiples factores de riesgo
Método farmacológico efectivo(heparina no fraccionada oHBPM) combinada con méto-dos mecánicos (ME o CNI)
HBPM = heparina de bajo peso molecular; ME = medias elásticas; CNI = compresión neumáticaintermitente.
todos los pacientes, a menos que exista una contraindicación absoluta. Los ries-gos y beneficios de la ambulación temprana se encuentran bien establecidos, enespecial en los pacientes con cirugía ortopédica de los miembros inferiores. Laambulación temprana y la terapia física posterior a fractura de cadera se asociana una corta estancia hospitalaria, escasa complicaciones y una baja mortalidad alos seis meses. La ambulación temprana posquirúrgica es una adecuada medidaprofiláctica para la enfermedad tromboembólica venosa en los pacientes que sonsometidos a procedimientos quirúrgicos de bajo riesgo, sea por cirugía general,ginecológica o urológica, en quienes también se indica el uso de medias elásticascomo medida de rutina.
Medias elásticas
Las medias elásticas fueron las primeras medidas en reducir los eventos trombo-embólicos venosos en 1952. Su beneficio se atribuye a la mejoría del flujo venosoy a la reducción de daño en las paredes de los vasos sanguíneos causado por ladilatación venosa pasiva que ocurre durante la cirugía.
Se estima que la reducción del riesgo relativo con el uso de las medias elásticases de al menos 60% en cirugías general, neurológica y ginecológica.
Se requiere que las medias estén correctamente ajustadas y que sean colocadasa partir del periodo preoperatorio, con su uso continuo durante la estancia en el

70 (Capítulo 4)Tratado de trombosis
hospital y el periodo de rehabilitación. Se recomienda continuar la profilaxis enlos pacientes que aun egresados del hospital continuarán con relativa inmoviliza-ción.
La única contraindicación mayor para el uso de medias elásticas es la presen-cia de enfermedad vascular periférica.
Ejercicios antitrombóticos de los miembros inferiores
Se debe instruir al paciente para la realización de ejercicios antitrombóticos delos miembros inferiores, los cuales consisten en flexión dorsoplantar del pie, conla pierna soportando el pie, estirada y elevada aproximadamente entre 7 y 10� dela cadera. Los ejercicios se deben realizar de manera continua durante tres a cincominutos o hasta que se fatiguen los músculos de la pierna, con el inicio de dolormuscular. Posteriormente se debe ejercitar la pierna contralateral de la mismamanera. Es necesario indicarle al paciente que no debe mantenerse en posiciónsedente sin estirar las piernas o deambular más de 20 min.
Ejercicios antitrombóticos de los miembros superiores
Se debe instruir al paciente para la realización de ejercicios de los miembros su-periores, los cuales consisten en cerrar la mano, con el brazo extendido y elevadoa la altura de la cabeza. Se puede sostener una bola de tenis en la mano mientrasse aprieta. Se debe realizar el ejercicio de manera continua durante tres a cincominutos o hasta que se fatiguen los músculos del brazo, con el inicio de dolormuscular. Posteriormente se debe ejercitar el brazo contralateral de la misma ma-nera. El paciente debe realizar estos ejercicios entre cuatro y seis veces al día.
Compresión neumática intermitente
Existen dos tipos principales de compresión neumática intermitente (CNI) parala prevención de la enfermedad tromboembólica venosa. Uno de ellos provee unacompresión neumática secuencial a nivel de la pierna, en las pantorrillas o en losmuslos. El segundo es una “bomba” a nivel de los pies, ya que comprime el plexovenoso plantar. Ambas formas de compresión son equivalentes en cuanto a efica-cia. El principal mecanismo a través del cual disminuye la incidencia de eventostrombóticos es el efecto directo de bombeo de sangre venosa, con lo que se reducela estasis. No generan complicaciones, excepto molestia para el paciente y riesgode trastornos en la piel. Se desconoce el número de horas que se requiere tenerla compresión en cuestión de efectividad; sin embargo, se recomienda su uso demanera continua mientras el paciente se encuentre inmóvil.
La CNI es el método de elección para la profilaxis de enfermedad tromboem-bólica en los pacientes con contraindicación para el uso de anticoagulantes.

71TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Filtro en la vena cava inferior
Las indicaciones para la colocación de filtro en la vena cava inferior (VCI) inclu-yen:
1. Presencia de contraindicación absoluta para la terapia anticoagulante.2. Presencia de hemorragia bajo terapia anticoagulante que pone en riesgo la
vida.3. Falla a una adecuada anticoagulación.
El uso de filtro en VCI es seguro y efectivo para reducir la incidencia de trombo-embolia pulmonar (TEP) hasta entre 0.3 y 3.8% en pacientes con presencia decontraindicación para la terapia anticoagulante. Los riesgos de la colocación delfiltro en VCI incluyen migración del filtro, trombosis venosa profunda recurren-te, trombosis de la VCI y síndrome posflebítico.
En el periodo perioperatorio la situación más frecuente para la colocación defiltro en VCI como medida profiláctica de TEP fatal es cuando el paciente requie-re una cirugía de urgencia posterior a un reciente diagnóstico de enfermedadtromboembólica venosa (<4 semanas), en el cual el riesgo de recurrencia es ma-yor durante el primer mes de tratamiento y se debe interrumpir la terapia anticoa-gulante.
La colocación de un filtro temporal extraíble puede ser una adecuada opciónterapéutica, ya que puede ser removido una vez que el paciente no presenta con-traindicación para la anticoagulación.
Profilaxis farmacológica
Se indican los métodos de prevención de enfermedad tromboembólica venosa deacuerdo con las Guías de terapia antitrombótica y prevención de trombosis delAmerican College of Chest Physicians (ACCP) en su novena edición.
Profilaxis en el paciente hospitalizadocon enfermedad médica (no quirúrgica)
Cuando los pacientes ingresan en el hospital deben ser clasificados de acuerdocon su riesgo tromboembólico a través de la identificación de factores de riesgo.Las guías recomiendan que los pacientes hospitalizados con enfermedad aguday riesgo incrementado de trombosis deben recibir heparina de bajo peso molecu-lar (HBPM), bajas dosis de heparina no fraccionada dos o tres veces al día, o fon-daparinux. En los pacientes con enfermedad médica y bajo riesgo de trombosis

72 (Capítulo 4)Tratado de trombosis
no se recomienda la profilaxis, mientras que en los pacientes con riesgo incre-mentado de trombosis y presencia de sangrado o riesgo del mismo se recomiendael uso de profilaxis mecánica a través de medias o CNI, y una vez que disminuyael riesgo de sangrado, si persiste el riesgo de trombosis, el empleo de profilaxisfarmacológica.
Paciente en estado crítico
Las guías no recomiendan el uso del ultrasonido como medida de detección derutina para trombosis venosa profunda (TVP) en el paciente en estado crítico; su-gieren el uso de HBPM o de heparina no fraccionada más que la ausencia de profi-laxis.
En los pacientes bajo tratamiento con HBPM y depuración de creatinina me-nor de 30 mL/min se recomienda disminuir la dosis.
Paciente con cáncer
En los pacientes ambulatorios que padecen cáncer, sin la presencia de algún otrofactor de riesgo para enfermedad tromboembólica venosa (ETV), las guías no re-comiendan el uso de terapia farmacológica como profilaxis; se mencionan comofactores de riesgo adicional la trombosis venosa previa, la inmovilización, la tera-pia hormonal, los inhibidores de la angiogénesis, la talidomida y la lenalidomida.En los pacientes ambulatorios con presencia de un tumor sólido, alguno de losfactores mencionados y bajo riesgo de sangrado se recomienda la profilaxis conHBPM o heparina no fraccionada.
En personas con inmovilidad crónica que se encuentran en su hogar no se reco-mienda el uso rutinario de tromboprofilaxis.
Viajes prolongados
Para los viajeros de largas distancias y riesgo incrementado de ETV (incluyendoETV previa, cirugía reciente o trauma, malignidad activa, embarazo, uso de es-trógenos en edad avanzada, movilidad limitada, obesidad o desorden trombofíli-co conocido) las guías sugieren la ambulación frecuente, la ejercitación del mús-culo de las piernas y el uso apropiado de medias compresivas, que proveen de 15a 30 mmHg de presión en el tobillo durante el viaje. No se sugiere el uso de ácidoacetilsalicílico (ASA) o de anticoagulantes.
Personas con trombofilia asintomática
En personas con trombofilia asintomática (sin historia previa de ETV) las guíasno recomiendan el uso de profilaxis mecánica o farmacológica de uso diario.

73TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Profilaxis de enfermedad tromboembólica venosaen el paciente quirúrgico no ortopédico
Para la prevención de la TVP no se recomienda el uso exclusivo de ASA comomedida profiláctica. La profilaxis anticoagulante se debe realizar con precauciónen todos los casos que involucren punción espinal o catéteres epidurales paraanestesia regional o analgesia.
Cirugía general, gastrointestinal, urológica, ginecológica,bariátrica, vascular, plástica o reconstructiva
Para pacientes de cirugía general y abdominopélvica, y muy bajo riesgo de ETV(escala de Caprini: 0), se recomienda sólo la ambulación temprana, mientras queen aquellos con riesgo bajo (Caprini: 1 a 2) se sugiere la profilaxis mecánica, depreferencia con CNI. En los pacientes con riesgo moderado para ETV (Caprini:3 a 4) que no se encuentran en alto riesgo de sangrado mayor se recomienda eluso de HBPM, heparina no fraccionada o profilaxis mecánica, de preferencia conCNI. En los pacientes con riesgo moderado para ETV y alto riesgo de complica-ciones hemorrágicas se sugiere el uso de profilaxis mecánica, de preferenciaCNI, mientras que para los pacientes con alto riesgo de ETV que no tienen altoriesgo de complicaciones hemorrágicas se recomienda el uso de HBPM o hepari-na no fraccionada más el uso de profilaxis mecánica, como medias o compresivaso CNI. Por el contrario, en aquellos con alto riesgo además de complicacioneshemorrágicas se recomienda la profilaxis mecánica, de preferencia con CNI; unavez que disminuya el riesgo de sangrado se debe iniciar con la terapia farmacoló-gica.
Para los pacientes de alto riesgo con cáncer que van a ser sometidos a cirugíaabdominal o pélvica se recomienda extender la tromboprofilaxis con HBPM du-rante cuatro semanas.
Cirugía cardiaca
Para el paciente que se somete a cirugía cardiaca se recomienda el uso de profila-xis mecánica, de preferencia con el uso óptimo de la CNI, más que la ausenciade profilaxis o la profilaxis farmacológica. Si la estancia en el hospital se prolon-ga por una o más complicaciones no hemorrágicas se sugiere la tromboprofilaxiscon HBPM o heparina no fraccionada.
Cirugía torácica
Los pacientes con riesgo moderado para ETV que no se encuentran en alto riesgode hemorragia se sugiere el uso de HBPM, heparina no fraccionada o profilaxismecánica con uso óptimo de la CNI sobre la ausencia de profilaxis.

74 (Capítulo 4)Tratado de trombosis
Para los pacientes con alto riesgo para ETV y sin alto riesgo de hemorragia sesugiere el uso de profilaxis tanto farmacológica como mecánica.
Para aquellos con alto riesgo de complicaciones hemorrágicas se recomiendael uso de profilaxis mecánica, de preferencia CNI, y una vez que disminuya elriesgo de sangrado iniciar la terapia farmacológica.
Craneotomía
Para los pacientes con craneotomía se recomienda la profilaxis mecánica, de pre-ferencia CNI, más que la ausencia de profilaxis o la profilaxis mecánica. En lospacientes con alto riesgo para ETV (craneotomía por enfermedad maligna) se su-giere agregar profilaxis farmacológica a la profilaxis mecánica, una vez estable-cida la adecuada homeostasis y disminuido el riesgo de sangrado.
Cirugía espinal
En los pacientes con alto riesgo de ETV (incluidos aquellos con enfermedad ma-ligna o cirugía con abordaje anteroposterior) se sugiere agregar la profilaxis far-macológica a la mecánica, una vez establecida la adecuada homeostasis y dismi-nuido el riesgo de sangrado.
Pacientes con trauma mayor: lesión cerebraltraumática, lesión espinal aguda o traumática
Para los pacientes con trauma mayor se sugiere el uso de HBPM, heparina nofraccionada o profilaxis mecánica, de preferencia CNI, sobre la ausencia de pro-filaxis.
En los pacientes con alto riesgo de ETV (incluidos aquellos con lesión agudadel cordón espinal, lesión cerebral traumática y cirugía espinal por trauma) se su-giere agregar profilaxis mecánica a la farmacológica cuando no se encuentre con-traindicada por lesión del miembro inferior. En los que se encuentre contraindi-cado el uso de heparina se sugiere utilizar de preferencia CNI y sustituirla por laterapia farmacológica una vez que se resuelva la contraindicación. En los pacien-tes con trauma mayor no se recomienda la colocación de filtro en VCI como trom-boprofilaxis primaria.
Prevención de enfermedad tromboembólicavenosa en cirugía ortopédica
En los pacientes que van a ser sometidos a artroplastia total de cadera (ATC) oa artroplastia total de rodilla (ATR) se recomienda el uso de uno de los siguientes

75TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
agentes durante un lapso mínimo de 10 a 14 días: HBPM, fondaparinux, apixa-bán, dabigatrán, rivaroxabán, bajas dosis de heparina no fraccionada (BDHNF),dosis ajustada de antagonista de vitamina K (AVK), ASA o CNI.
En los pacientes que son sometidos a cirugía por fractura de cadera se reco-mienda el uso de uno de los siguientes agentes durante al menos 10 a 14 días:HBPM, fondaparinux, BDHNF, dosis ajustada de AVK, ASA o CNI. (Un miem-bro del panel consideró que el uso de ASA solo no debía de ser una opción en estasdos últimas recomendaciones.)
Para los pacientes que son sometidos a cirugía ortopédica mayor (reemplazototal de cadera, reemplazo total de rodilla o cirugía por fractura de cadera) y reci-ben HBPM como tromboprofilaxis se recomienda iniciar a las 12 h o más del pre-operatorio o a las 12 h o más del posoperatorio.
Para los pacientes que van a ser sometidos a reemplazo total de cadera o reem-plazo total de rodilla se sugiere utilizar de manera concomitante a la terapia mecá-nica la HBPM, en lugar de otros agentes.
En pacientes que son sometidos a cirugía por fractura de cadera se recomiendael uso de HBPM en preferencia a otros agentes y de manera concomitante a laprofilaxis mecánica.
Para los pacientes a quienes se les realiza cirugía ortopédica mayor se sugiereextender el periodo de tromboprofilaxis en el paciente ambulatorio por 35 díasa partir del día de la cirugía, más que por sólo 10 a 14 días. A la vez, se recomiendala terapia tanto farmacológica como mecánica (CNI) mientras el paciente se en-cuentre en el hospital. En los pacientes con riesgo incrementado de sangrado sesugiere el uso de CNI.
En los pacientes que se someten a cirugía ortopédica mayor y declinan o nocooperan con las inyecciones, las guías recomiendan el uso de apixabán o dabiga-trán (alternativamente rivaroxabán o dosis ajustadas de AVK si no se dispone deapixabán o dabigatrán), más que otras formas de profilaxis.
Para los pacientes que se someten a artroscopia de rodilla sin una historia pre-via de ETV se sugiere no brindar tromboprofilaxis.
Manejo perioperatorio de la terapia antitrombótica
En los pacientes que requieren una interrupción temporal de los AVK antes dela cirugía se recomienda suspender el uso de los AVK aproximadamente cincodías antes de la cirugía y reanudarlos de 12 a 24 h después de la intervención,siempre que se tenga una adecuada hemostasia.
En los pacientes con válvulas cardiacas mecánicas, fibrilación auricular oETV con alto riesgo de tromboembolismo se sugiere la anticoagulación “puente”durante la interrupción de los AVK, no así para los de bajo riesgo de tromboembo-lismo.

76 (Capítulo 4)Tratado de trombosis
Para los pacientes que requieren un procedimiento dental menor se sugierecontinuar los AVK con la coadministración de un agente hemostático oral o sus-pender los AVK entre dos y tres días antes del procedimiento. En los pacientesque requieren un procedimiento dermatológico menor se sugiere continuar losAVK y optimizar la hemostasia local. En los pacientes que requieren cirugía porcatarata se sugiere continuar el uso de los AVK.
Los pacientes que se encuentran recibiendo ASA para prevención secundariade enfermedad cardiovascular y van a ser sometidos a un procedimiento dentalmenor, dermatológico o a cirugía de catarata se sugiere continuar el antiagre-gante.
En pacientes con riesgo de moderado a alto de sufrir eventos cardiovascularesque reciben ASA y requieren cirugía no cardiaca se sugiere continuar el antiagre-gante, mientras que en aquellos con bajo riesgo de eventos cardiovasculares sesugiere suspender el uso del ASA entre 7 y 10 días antes de la cirugía.
Cirugía de bypass coronario
En pacientes que se encuentran bajo terapia con ASA se sugiere continuar su usopara la cirugía, mientras que en los que se encuentran con terapia antiagregantedual se recomienda continuar el uso de ASA y suspender el clopidogrel/prasugrelcinco días antes de la cirugía.
Cirugía en pacientes con prótesis endovasculares coronarias
En los pacientes con stent coronario que reciben terapia dual y requieren cirugíase recomienda diferir la cirugía durante al menos seis semanas después de la colo-cación del stent de metal y al menos seis meses después de colocar un stent libera-dor de fármaco. En los pacientes que requieran la cirugía antes de estos periodosse sugiere continuar la terapia dual alrededor del tiempo de la cirugía.
Terapia antitrombótica en enfermedad valvular
La tromboprofilaxis en la fibrilación auricular no valvular se detalla en el capí-tulo 7.
Pacientes con enfermedad valvular mitral reumática
Los pacientes con enfermedad reumática de la válvula mitral, ritmo sinusal y diá-metro de la aurícula izquierda menor de 55 mm se sugiere no utilizar terapia anti-trombótica, mientras que en aquellos con aurícula mayor de 55 mm se sugiere el

77TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
uso de AVK —índice normalizado internacional (INR) de 2.0 a 3.0—, igual queen los pacientes con presencia de trombo en la aurícula, fibrilación auricular oembolismo sistémico previo.
Pacientes con foramen oval permeable y aneurisma septal atrial
En pacientes con foramen oval permeable (FOP) o aneurisma atrial asintomáti-cos no se sugiere terapia antitrombótica. En los pacientes con EVC criptogénicoy FOP o aneurisma atrial se sugiere el uso de ASA (50 a 100 mg/día); si inclusocon el manejo con antiagregante plaquetario presentan eventos recurrentes, se su-giere el uso de AVK y considerar el cierre. En aquellos con EVC criptogénico yFOP con evidencia de ETV se recomienda el uso de AVK durante tres meses yconsiderar el cierre.
En los pacientes con bioprótesis de la válvula aórtica que se encuentran en rit-mo sinusal y no presentan otra indicación para el uso de AVK se sugiere el usode ASA (50 a 100 mg/día) los primeros tres meses, mientras que en aquellos conbioprótesis en posición mitral se recomienda el uso de AVK durante los primerostres meses.
Después de los tres meses de la colocación de válvulas bioprotésicas se sugiereel uso de ASA.
En pacientes con válvulas mecánicas se sugiere el uso de AVK. En aquelloscon prótesis aórtica se sugiere un INR de 2.5 (2.0 a 3.0), mientras que en los pa-cientes con prótesis en posición mitral se sugiere un INR de 3.0 (2.5 a 3.5), igualque en aquellos con doble prótesis valvular.
En los pacientes con prótesis valvular mecánica aórtica o mitral con bajo ries-go de sangrado se recomienda agregar bajas dosis de ASA (50 a 100 mg/día) aluso de AVK.
Prevención primaria y secundariade la enfermedad cardiovascular
Para las personas de 50 años de edad o mayores sin enfermedad cardiovascularsintomática se sugieren bajas dosis de ASA (75 a 100 mg/día).
Para los pacientes con enfermedad coronaria establecida —definida como pa-cientes al año de un síndrome coronario agudo—, previa revascularización, este-nosis coronaria mayor de 50% por angiografía coronaria o diagnóstico de isque-mia cardiaca se sugiere el uso indefinido de un antiagregante plaquetario, ASAo clopidogrel.
Para los pacientes que cursan el primer año con un síndrome coronario agudoen quienes no se considera una intervención percutánea se recomienda la terapia

78 (Capítulo 4)Tratado de trombosis
antiplaquetaria dual (ticagrelor en dosis de 90 mg dos veces al día más ASA oclopidogrel más ASA). Se prefiere el uso de ticagrelor más ASA, en lugar deASA más clopidogrel.
Para los pacientes en quienes se planea la colocación de un stent se recomiendala terapia antiagregante dual (ticagrelor en dosis de 90 mg dos veces al día másASA, clopidogrel más ASA o prasugrel en dosis de 10 mg al día más ASA).
Para los pacientes con infarto del miocardio anterior y presencia de trombo enel ventrículo izquierdo o alto riesgo de sufrir trombo (FEVI < 40%, anormalidaden la movilidad de la pared anteroapical) y en quienes no se planea colocar unstent se indica el uso de warfarina más dosis bajas de ASA durante los primerostres meses. Posteriormente se recomienda suspender la warfarina y continuar conterapia antiagregante dual hasta los 12 meses y posteriormente continuar con unsolo antiagregante plaquetario.
En los pacientes con las mismas características en quienes se coloca un stentde metal se recomienda la terapia triple (warfarina, ASA y clopidogrel) duranteun mes y continuar con warfarina y un solo antiagregante durante los dos siguien-tes meses.
Posteriormente se debe suspender la warfarina y continuar con terapia antia-gregante dual hasta los 12 meses. En caso de colocar un stent liberador de fárma-co se recomienda la terapia triple durante tres a seis meses y posteriormente laterapia antiagregante plaquetaria dual hasta los 12 meses.
Terapia antitrombótica posterior auna colocación de stent coronario
Para los pacientes que reciben un stent de metal se recomienda la terapia antiagre-gante dual con ASA (75 a 325 mg/día) y clopidogrel (75 mg/día) durante el pri-mer mes. Durante los siguientes 11 meses se recomienda continuar con la terapiadual con ASA (75 a 100 mg/día) y clopidogrel (75 mg/día), y después de los 12meses continuar con un solo antiagregante plaquetario. En caso de colocar unstent liberador de fármaco se sugiere utilizar la terapia dual recomendada para elprimer mes del stent de metal pero durante los primeros tres a seis meses; poste-riormente se debe continuar hasta los 12 meses para luego utilizar un solo antia-gregante plaquetario de manera indefinida.
Para los casos anteriores se sugiere el uso de cilostazol en dosis de 100 mg dosveces al día sólo si se usa como terapia dual y ante la presencia de intoleranciao alergia al ASA o al clopidogrel.
Para los pacientes con enfermedad coronaria que son sometidos a una inter-vención coronaria percutánea, pero no se les coloca stent, se sugiere utilizar tera-pia antiagregante plaquetaria dual con ASA y clopidogrel durante el primer mesy posteriormente continuar con un solo antiagregante.

79TromboprofilaxisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
REFERENCIAS
1. Bick R, Haas S: Thromboprophylaxis and thrombosis in medical, surgical trauma and obs-tetric/gynecologic patients. Hematol Oncol Clin N Am 2003;17:217–258.
2. Spyropoulos A: Emerging strategies in the prevention of venous thromboembolism in hos-pitalized medical patients. Chest 2005;128.
3. The Sixth ACCP Consensus Conference on Antithrombotic Therapy. Chest 2001;119(Suppl 1).
4. Ansell J, Hirsh J, Poller L: The Seventh ACCP Conference on Antithrombotic and Throm-bolitic Therapy: evidence–based guidelines. Chest 2004;126(Suppl 3):S204–S233.
5. Henderson M, White R: DVT prophylaxis and anticoagulation in the surgical patient. MedClin N Am 2003;87:77–110.
6. Merli G: Prevention of thrombosis with warfarin, Aspirin, and mechanical methods. Clini-cal Cornerstone 2005;7(4):49–56.
7. Michota F: Venous thromboembolism prophylaxis in the medically ill patient. Clin ChestMed 2003;24:93–101.
8. Guyatt G, Akl E, Crowther M et al.: Antithrombotic therapy and prevention of thrombo-sis. 9ª ed. American College of Chest Physicians Evidence–Based Clinical Practice Guideli-nes. Chest 2012;141:7S–47S.

80 (Capítulo 4)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
5Enfermedad vascular
Luis Fernando García–Frade Ruiz
Es importante resaltar que el fenómeno trombótico constituye el factor desenca-denante de la isquemia final en el proceso aterosclerótico; por otra parte, la trom-bosis sin placa aterosclerótica de base (a la que llamaremos “trombosis pura”)puede resultar de trombofilias primarias o secundarias, que constituyen en con-junto las principales causas de morbimortalidad en todo el mundo e involucrana todas las especialidades médicas.
Por este motivo, el fenómeno trombótico precedido o no por una inflamaciónendotelial focal, sus múltiples causas, su gran gama de manifestaciones clínicasde acuerdo con el territorio afectado —arterial o venoso— y su amplio espectroactual de tratamiento farmacológico, debe constituir para los médicos la principalpatología en nuestros días desde un concepto integral de diagnósticos específicosy múltiples manejos.
En el presente capítulo se intenta, de manera ambiciosa, integrar varias patolo-gías hasta ahora estudiadas por separado de manera didáctica en una sola, asícomo simplificar las múltiples variables a las que los médicos se enfrentan en re-lación con el fenómeno aterotrombótico y trombótico puro desde un enfoque ce-lular, para facilitar su posterior entendimiento de intervención farmacológica.Por ejemplo, la deficiencia estrogénica se asocia con disfunción endotelial y ate-rotrombosis, mientras que la terapia hormonal sustitutiva incrementa el riesgo detrombosis pura, lo que resalta la importancia de la medicina multidisciplinaria enla actualidad.
81

82 (Capítulo 5)Tratado de trombosis
BASES CIENTÍFICAS DE LA INFLAMACIÓNEN LA ATEROGÉNESIS
El endotelio vascular es un sistema metabólicamente activo que ayuda a mante-ner la hemostasia vascular. Además de ser una barrera anatómica para la circula-ción, el endotelio modula el tono vascular, regula el crecimiento celular local yel depósito de material extracelular, protege a los vasos de sustancias y célulastóxicas que se encuentran en la circulación y regula las respuestas inflamatoriasy de reparación del daño local, este último a través de la liberación de agonistasdel sistema fibrinolítico (t–PA) y de la liberación del factor de crecimiento de lasplaquetas a partir de los gránulos alfa.
Algunas condiciones, como la diabetes mellitus, la hipertensión arterial sisté-mica, la dislipidemia, la obesidad y el tabaquismo, entre otras, se asocian con dis-función endotelial, la cual resulta en inflamación, oxidación de lipoproteínas,proliferación de células del músculo liso, depósito de material extracelular, acu-mulación de material rico en lípidos, activación plaquetaria y formación de untrombo. De esta forma, la disfunción endotelial contribuye al proceso de ateros-clerosis–trombosis (aterotrombosis) y a la presencia de enfermedad del sistemavascular, incluyendo las circulaciones coronaria, cerebral, mesentérica y perifé-rica.
La aterosclerosis es una enfermedad multifactorial que involucra a la inflama-ción crónica desde su inicio hasta su progresión y eventual ruptura de la placa,por lo que las funciones homeostáticas del endotelio se ven alteradas, promovien-do así una respuesta inflamatoria. Las moléculas de adhesión expresadas en elendotelio inflamado reclutan leucocitos, incluyendo los monocitos, los cualespenetran en el interior de la íntima y predisponen a la pared vascular a una vascu-litis o al incremento de lípidos. Los mediadores inflamatorios incrementan lacaptura de partículas de lipoproteínas y la formación de macrófagos llenos de lí-pidos. Las células T también penetran en la íntima y secretan citocinas, que poste-riormente amplifican la respuesta inflamatoria y promueven la proliferación y lamigración de células del músculo liso. Más adelante los mediadores inflamato-rios pueden debilitar la capa fibrosa protectora del ateroma, guiando a la trombo-sis.
El sistema inmunitario contribuye a la formación de la aterosclerosis; las célu-las T y los macrófagos colaboran en la generación de la placa, su extensión y suruptura, así como a la oxidación de las lipoproteínas de baja densidad (oxLDL).Las oxLDL son las moléculas pivote en el desarrollo de la aterosclerosis, por loque deben ser consideradas como cruciales en el proceso proinflamatorio. Unavez que las oxLDL son retenidas en la capa íntima de las arterias activan las célu-las endoteliales, las que incrementan la expresión de moléculas de adhesión y la

83Enfermedad vascularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
secreción de citocinas, mismas que contribuyen al reclutamiento de leucocitoscirculantes. Una vez que los monocitos/macrófagos infiltran la placa ateroscleró-tica toman las oxLDL y forman las “células espumosas”, las cuales participan enla secreción de mediadores inflamatorios.
Tanto la trombosis como la inflamación se encuentran íntimamente relaciona-das. Tras la lesión vascular las plaquetas cubren el subendotelio expuesto y pro-veen la superficie requerida para la unión de micropartículas derivadas de los leu-cocitos que contienen factor tisular. Además, las plaquetas liberan partículas quemedian las interacciones entre los leucocitos y entre éstos y las células endotelia-les. Las plaquetas activadas incrementan la adhesión de leucocitos al endotelioy promueven la activación leucocitaria a través del depósito de quimiocinas enel endotelio. Así, las plaquetas participan de manera central tanto en la trombosiscomo en la inflamación.
La trombosis ocurre, entonces, como consecuencia de dos procesos: rupturade la placa y erosión endotelial. Tras la ruptura de la placa aterosclerótica se expo-nen desde su región central al torrente sanguíneo elementos protrombóticos,como fosfolípidos y factor tisular. La exposición de este último pone en claro queno sólo existe activación plaquetaria, sino la fase plasmática de la coagulacióna través de la vía del factor tisular (vía extrínseca), sobre la cual los antiagregantesplaquetarios no tienen mayor efecto. Las células inmunitarias activadas producenmoléculas inflamatorias y enzimas proteolíticas, que a su vez activan células enla región central de la placa, transformándola de un estado estable a una estructu-ra inestable con riesgo de fractura y formación de trombosis; desde el punto devista clínico, esto se conoce como síndrome coronario agudo.
DISLIPIDEMIA E INFLAMACIÓN
Otras lipoproteínas, como las lipoproteínas de muy baja densidad (VLDL) y laslipoproteínas de densidad intermedia, también poseen un considerable potencialaterogénico. Estas lipoproteínas pueden conducir a modificaciones oxidativas,como las lipoproteínas de baja densidad (LDL). En adición, algunas evidenciassugieren que las partículas beta VLDL pueden por sí mismas activar funcionesinflamatorias en las células endoteliales. Las lipoproteínas de alta densidad(HDL) protegen contra la aterosclerosis. Las HDL también pueden transportarenzimas antioxidantes, las cuales pueden disminuir las oxLDL y neutralizar susefectos proinflamatorios.
Los altos niveles de LDL y de colesterol total se encuentran inversamente re-lacionados con la vasodilatación dependiente del endotelio. Los cambios pospran-diales en las concentraciones de los remanentes de quilomicrón, ricos en coleste-

84 (Capítulo 5)Tratado de trombosis
rol, promueven la aterogénesis en varios grados de desarrollo. La hipertrigliceri-demia posprandial se asocia con un estado inflamatorio e incremento en los nive-les de factor de necrosis tumoral alfa (TNF–�), interleucina (IL) 6, ICAM–1 yVCAM–1.
El sistema renina–angiotensina desempeña un papel importante en la disfun-ción endotelial, en la que el receptor de angiotensina tipo II (AT1R) conduce a lavasoconstricción y activación neurohumoral, y se asocia con un incremento dela liberación de especies de oxígeno reactivo, apoptosis de células vasculares yaumento de la expresión del receptor de oxLDL, moléculas de adhesión y citoci-nas proinflamatorias. La hipercolesterolemia eleva la respuesta vasoconstrictorade la AT1R. La existencia de la enzima convertidora de angiotensina en las lesio-nes ateroscleróticas sugiere quizá la capacidad de generación local de angiotensi-na II y de sustancias proinflamatorias. La hipercolesterolemia incrementa el an-giotensinógeno plasmático, mientras que el antagonismo del AT1R mejora lahipercolesterolemia asociada a la disfunción endotelial.
HIPERTENSIÓN ARTERIAL SISTÉMICA E INFLAMACIÓN
La hipertensión arterial constituye, junto con los lípidos, un factor de riesgo clá-sico para la presencia de aterosclerosis. La inflamación que tiene lugar en la ate-rosclerosis puede participar en la fisiopatología de la hipertensión, lo que repre-senta una unión entre las dos enfermedades. La angiotensina (AII), además de suspropiedades vasoconstrictoras, puede incrementar la inflamación de la íntima yla expresión de citocinas proinflamatorias, como IL–6 y la molécula de adhesiónde leucocitos VCAM–1 sobre las células endoteliales. Algunos de los beneficiosclínicos de la terapia inhibidora de la enzima convertidora de angiotensina pue-den derivarse de la interrupción de algunas vías inflamatorias.
DIABETES E INFLAMACIÓN
La hiperglucemia asociada a diabetes puede conducir a la modificación de ma-cromoléculas, por ejemplo, la formación de productos de glucosilación avanzada(AGE). Por unión a la superficie de receptores, como los RAGE (receptor paraAGE), las proteínas AGE–modificadas pueden aumentar la producción de citoci-nas proinflamatorias y otras vías inflamatorias en las células endoteliales vascu-lares. A la par de la hiperglucemia, el estado diabético promueve el estrés oxida-tivo.

85Enfermedad vascularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
OBESIDAD E INFLAMACIÓN
La obesidad no sólo predispone a la resistencia a la insulina y la diabetes, sinoque contribuye a la dislipidemia aterogénica. Los altos niveles de ácidos grasoslibres originados de la grasa visceral alcanzan el hígado a través de la circulaciónportal y estimulan la síntesis de lipoproteínas VLDL ricas en triglicéridos porparte de los hepatocitos. El resultado en la elevación de las VLDL puede dismi-nuir las HDL por un aumento en el cambio de HDL a VLDL. El tejido adiposotambién puede sintetizar citocinas, como TNF–� e IL–6. En este sentido, la obe-sidad por sí misma promueve la inflamación y potencia la aterogénesis, indepen-dientemente de sus efectos sobre la resistencia a la insulina o a las lipoproteínas.
BIOMARCADORES DE INFLAMACIÓNY RIESGO DE EVENTO CARDIOVASCULAR
Varios estudios prospectivos han encontrado una asociación entre el incrementodel riesgo vascular y el aumento de los niveles basales de citocinas —la IL–6 yTNF–�—, así como en los niveles de las moléculas de adhesión celular ICAM–1,P selectina y E selectina, y reactantes de fase aguda —proteína C reactiva (PCR),fibrinógeno y amiloide A sérico. Los niveles elevados de IL–6 y de PCR se aso-cian no únicamente con el desarrollo posterior de aterosclerosis, sino de diabetesmellitus tipo 2, aun en los pacientes sin presencia de resistencia a la insulina.
La IL–6 es una citocina circulante cuyos niveles se incrementan en respuestaa la presencia de infarto agudo del miocardio, angina inestable, intervención co-ronaria percutánea y reestenosis tardía. La IL–6 estimula la agregación plaqueta-ria y la expresión de factor tisular, los receptores LDL en los macrófagos, la PCRy el fibrinógeno.
La PCR es un marcador clásico de fase aguda que pertenece a la familia pentra-xina de las proteínas de la respuesta inmunitaria. La PCR es producida en el híga-do en respuesta a la IL–6, la IL–1� y el TNF–�; posee una vida media larga sinvariaciones con el ciclo circadiano. Tiene la habilidad de unirse y activar al com-plemento, inducir la expresión de varias moléculas de adhesión y del factor tisu-lar, mediar la captura de LDL por parte de los macrófagos endoteliales e inducirel reclutamiento de monocitos al interior de la pared arterial. La hiperglucemiapotencia los efectos de la PCR sobre la activación de las células endoteliales; sinembargo, la intervención farmacológica a través de estatinas, glitazonas, bosen-tán, antagonistas del receptor de endotelina, etc., atenúa dicho proceso. La evi-dencia acumulada sugiere que los niveles de PCR constituyen uno de los mayores

86 (Capítulo 5)Tratado de trombosis
predictores de aterosclerosis y muerte vascular, por lo que la PCR ofrece un valorpronóstico que supera el del colesterol LDL.
La IL–18 fue originalmente identificada en los macrófagos y en las células deKupffer como un factor capaz de inducir la producción de interferón gamma(IFN–�) por parte de las células T. La IL–18 se expresa de manera importante enla placa aterosclerótica, participa en todos los estadios del desarrollo de la placay promueve la producción de IFN–� y varias funciones proaterogénicas. Las in-tervenciones que reducen los niveles de IL–18 pueden beneficiar la estabilidadde la placa y la consiguiente reducción de eventos trombóticos agudos. La reduc-ción de peso genera una disminución de los niveles circulantes de IL–18.
FACTORES DE RIESGO PARA ENFERMEDAD VASCULAR
Parece ser, entonces, que es crucial identificar en cada uno de los pacientes duran-te la consulta la presencia de factores de riesgo para el desarrollo de enfermedadvascular, en virtud de que constituye una de las principales causas de morbimor-talidad en todo el mundo. Si se consideran dentro de la enfermedad vascular tantola aterotrombosis como la trombosis pura, se obtendrá de manera práctica quetoda trombosis en el territorio arterial podrá ser secundaria a la presencia de unaplaca aterosclerótica previa (aterotrombosis), siempre que se presenten factoresde riesgo para la misma; de lo contrario, al igual que en la trombosis del territoriovenoso, habrá que considerar y estudiar los factores de riesgo para la trombosispura.
Los factores de riesgo para la aterotrombosis incluyen diabetes mellitus, hi-pertensión arterial sistémica, dislipidemia, obesidad, tabaquismo, sedentarismo,síndrome metabólico, antecedentes familiares, sexo masculino y edad, entreotros.
Los factores de riesgo para la trombosis pura incluyen todas las trombofiliasprimarias y secundarias.
MANEJO FARMACOLÓGICODE LA ENFERMEDAD VASCULAR
En general, se puede decir que el tratamiento de la enfermedad vascular se resu-me en un tratamiento antiinflamatorio endotelial con control de la coagulación—que se divide en antiagregantes plaquetarios y anticoagulantes. El manejo dela aterotrombosis se lleva a cabo con tratamiento antiinflamatorio endotelial y an-

87Enfermedad vascularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
tiagregantes plaquetarios, mientras que la trombosis pura requiere manejo conanticoagulantes.
TRATAMIENTO ANTIINFLAMATORIO ENDOTELIAL
Las estatinas disminuyen los niveles de PCR independientemente de su efecto so-bre los lípidos, por lo que algunas han mostrado regresión de la placa y otras sóloestabilización de la misma. Las estatinas pueden afectar la actividad de los me-diadores inflamatorios de los monocitos circulantes antes de que penetren en lapared vascular, se diferencien a macrófagos y contribuyan al proceso de la ateros-clerosis; además, disminuyen los marcadores de inflamación —PCR, IL–6,TNF–� e IL–8— y la activación de monocitos, e inhiben la adhesión de fibrinó-geno a la GP IIb/IIIa, con lo que se reduce la agregación plaquetaria.
La disfunción endotelial es una de las manifestaciones iniciales de la ateroscle-rosis, que ocurre aun sin evidencia angiográfica de la enfermedad. Una caracte-rística importante de esta disfunción es la falla en la síntesis, liberación y activi-dad del óxido nítrico endotelial, mismo que inhibe varios procesos aterogénicos.Las estatinas incrementan la producción de óxido nítrico por estimulación de laóxido nítrico sintetasa endotelial, además de que elevan la expresión de t–PA yreducen la de endotelina–1, un potente vasoconstrictor y mitógeno.
Otro mecanismo importante mediante el cual las estatinas parecen mejorar lafunción endotelial es a través de sus propiedades antioxidantes, ya que inhibenla producción de superóxido y radicales hidroxi a través de la atenuación de an-giotensina II.
Las estatinas también aumentan las células progenitoras endoteliales, favore-ciendo la angiogénesis y, en consecuencia, la neovascularización. Disminuyenel TXA2, la composición lipídica de la membrana plaquetaria y la expresión defactor tisular de los macrófagos, todo ello de manera independiente a su efectopositivo sobre el perfil de lípidos. La mayoría de los efectos de las estatinas pare-cen ocurrir a través de la inhibición de isoprenoides, en especial del Rho GTPasaen las células de la pared vascular, lo que conduce a una mayor expresión de genesprotectores contra la aterosclerosis.
A través de la interrupción en la expresión de moléculas de adhesión y de cito-cinas los inhibidores de la enzima convertidora de angiotensina ejercen efectosantiinflamatorios tanto en el desarrollo de la aterosclerosis como en la ruptura dela placa.
Por otro lado, el papel que desarrollan los agonistas de PPAR y los fibratos enla atenuación de la respuesta inflamatoria endotelial aún continúa en controver-sia.

88 (Capítulo 5)Tratado de trombosis
El efecto cardioprotector de los ácidos grasos omega 3 puede resultar de su dis-minución en la activación endotelial o en la expresión de moléculas de adhesión,además de que disminuye tanto los triglicéridos como la agregación plaquetaria.
En cuanto al manejo de la coagulación en relación con los antiagregantes pla-quetarios, para el caso de la aterotrombosis, y de los anticoagulantes, para latrombosis pura, se sugiere revisar el capítulo correspondiente para cada caso.
CONCLUSIÓN
La inflamación endotelial conduce al desarrollo de un cuerpo extraño en la paredvascular arterial, con la participación silenciosa de varias patologías que hasta elmomento eran estudiadas por separado y que parecen estar más relacionadas delo que se consideraba. Tal cuerpo extraño genera, de manera consecuente, infla-mación y su posterior trombosis. Por lo anterior, resulta importante resaltar la ne-cesidad de integrar, desde el punto didáctico, a la inflamación endotelial con susrespectivos factores de riesgo y la manera en que éstos interactúan entre sí, conla trombosis pura, con el fin de considerar a la enfermedad vascular como unasola y facilitar la correcta y rápida identificación de factores de riesgo en la con-sulta diaria, además de intentar ofrecer mucho más a los pacientes con el menornúmero de fármacos posible, de modo que la correcta elección de un antihiper-glucemiante o antihipertensivo incluya un beneficio extra que la sola reducciónde la glucosa o de la presión arterial. Es importante mencionar que los nuevosanticoagulantes orales se prueban en la enfermedad coronaria, lo que con proba-bilidad conducirá a que aumente la población con esta indicación.
Los avances en la medicina obligan a los médicos a extender su campo de co-nocimientos, en los que afecciones como la enfermedad vascular involucran hoypor hoy a todas las especialidades y de manera particular a la medicina internacomo integradora diagnóstica y terapéutica, lo que exige una actualización conti-nua.
REFERENCIAS
1. García–Frade LF, Arredondo J: Algo más que inflamación vascular. Med Int Mex 2010;26(6):597–607.
2. Paoletti R, Gotto A, Hajjar D: Inflammation in atherosclerosis and implications for thera-py. Circulation 2004;109:20–26.
3. Libby P, Ridker P, Maseri A: Inflammation and atherosclerosis. Circulation 2002;105:1135–1143.
4. Szmito P, Wang C, Weisel R et al.: New markers of inflammation and endothelial cell ac-tivation. Circulation 2003;108:1917–1923.

89Enfermedad vascularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
5. Wagner D, Burger P: Platelets in inflammation and thrombosis. Arterioscler Thromb VascBiol 2003;23:2131–2137.
6. Liu Z, Tan Y, Beecham G et al.: Notch activation induces endothelial cell senescence andpro–inflammatory response: implication of notch signaling in atherosclerosis. Atheroscle-rosis 2012;225:296–303.
7. Kaplan Z, Jackson S: The role of platelets in atherothrombosis. Hematology 2011;61:52–61.
8. Sitia S, Tomasoni L, Atzeni F et al.: From endothelial dysfunction to atherosclerosis. Auto-immunity Rev 2010:830–834.
9. Liao J, Laufs U: Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005;45:89–118.
10. Babelova A: Anti–atherosclerotic mechanisms of statin therapy. Curr Opin Pharmacol2013;13:1–5.
11. Wang C, Liu P, Liao J: Pleiotropic effects of statins: molecular mechanisms and clinicalresults. Trends Mol Med 2008;14(1):37–44.

90 (Capítulo 5)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
6Angina inestable e infarto
agudo del miocardioJuan Carlos Peláez Piedrahita
INTRODUCCIÓN
El síndrome coronario agudo es una entidad patológica que incluye desde la angi-na inestable, con o sin cambios electrocardiográficos, hasta la presencia de un in-farto agudo del miocardio. En la actualidad se calcula que a nivel internacionales una de las principales causas de morbimortalidad, la cual no respeta sexo ninivel socioeconómico, por lo que se trata de un problema de salud con un altocosto económico. Es importante notar que la presencia de este tipo de evento enEUA rebasa el medio millón de casos por año y que aproximadamente 60 millo-nes de adultos son portadores de enfermedad aterosclerótica. En Europa se calcu-la que por cada 1 000 internamientos tres se relacionan con un evento coronario.1
En México el Registro Nacional de Síndromes Coronarios Agudos (RENASI-CA) ha reportado que la presencia de isquemia miocárdica es la principal causade muerte en las personas de la tercera edad y la segunda causa entre la poblacióngeneral.2 Si a esto se le agrega que actualmente se considera que la enfermedadcardiovascular secundaria a aterosclerosis es la principal causa de muerte entrelas mujeres en México, entonces nos encontramos ante un enorme problema desalud pública.
El RENASICA II identificó que el síndrome coronario agudo resulta del efec-to sumatorio de múltiples factores de riesgo bien conocidos y estudiados (cuadro6–1). La realización de cambios en los que son modificables es un tema que aquíno se trata. Lo ideal sería disminuir al máximo la presencia de los factores modifi-
91

92 (Capítulo 6)Tratado de trombosis
Cuadro 6–1. Factores de riesgo de mayor prevalenciaen la población mexicana (RENASICA II)
Factores de riesgo %
Tabaquismo 65Hipertensión arterial 50Diabetes mellitus 43
Hipercolesterolemia 26Antecedente de infarto 23
cables, pero pretender eliminarlos en su totalidad es muy difícil. Por lo tanto, esindispensable identificar de manera oportuna la enfermedad y manejarla de for-ma eficiente. Más adelante se mencionan las medidas disponibles para tratar deevitar la culminación en necrosis o por lo menos mitigar el daño.
PLACA INESTABLE: ANGINA INESTABLE–INFARTO
El síndrome coronario agudo implica la presencia de enfermedad ateroscleróticacoronaria, que ante la formación de un trombo in situ presenta un cuadro de angi-na inestable o culmina en un infarto agudo del miocardio. Existen múltiples fac-tores intrínsecos de la vasculatura que pueden facilitar la formación de este trom-bo, como son la turbulencia y lentitud del flujo sanguíneo en el sitio de la placaateromatosa, la fractura de la placa, el tabaquismo, la inflamación, la disfunciónendotelial, el aumento en los niveles de fibrinógeno y la disminución en la pro-ducción de óxido nítrico. En la mayor parte de los casos la presencia de enferme-dad aterosclerótica y de placa inestable favorece la activación y agregación pla-quetarias, lo cual conlleva al desarrollo de trombosis, isquemia y posiblementenecrosis.3
La aterosclerosis se ha reportado en los adolescentes mediante la presencia deestrías de grasa, que constituyen los primeros indicios. Se podría considerar quesu existencia es un estado normal en el ser humano, pero que su progresión acele-rada y exagerada en presencia de otras situaciones ambientales conlleva a la en-fermedad aterosclerótica. El siguiente paso en la evolución de la enfermedad, unavez presente la placa, es un proceso que la vuelve inestable, lo cual puede ocurrirde una de dos formas: por erosión del endotelio que cubre la capa fibrosa de laplaca, por ruptura o fractura de la capa fibrosa, o ambas. La primera causa se pre-senta con mayor frecuencia en la mujer, mientras que la segunda es la causa máscomún entre la población general.
Una vez fracturada la placa se inician una serie de eventos que generan un ciclode inflamación, activación y agregación plaquetaria. No sólo es la actividad de

93Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
la plaqueta la que propaga este ciclo, sino también los cambios agudos que se pre-sentan en el endotelio del vaso involucrado. Es importante notar que esta acciónde trombosis es el resultado del trabajo en equipo de los participantes; la plaquetase activa y se agrega, y a su vez ella misma aumenta el proceso con la expresiónde CD40 en su superficie, que a nivel del endotelio atrae a un mayor número decélulas inflamatorias, con lo que ocasiona una mayor ruptura de la placa, atraemás plaquetas activadas e incrementa el tamaño del trombo.
No toda placa inestable resulta en isquemia de manera necesaria.3 Una placacrítica en relación con el grado de estenosis puede no producir un evento isquémi-co importante que genere isquemia miocárdica, tal como se ha demostrado en unnúmero importante de placas ateromatosas que de manera intermitente aumentande tamaño (mayor grado de estenosis), no necesariamente por descontrol en losniveles de lípidos o factores externos a la placa, y presentan un mecanismo porel cual ellas mismas alternan entre los dos estados “estable” e “inestable”. Unaplaca incipiente puede presentar ruptura de su capa fibrosa, lo cual, como se sabe,favorece la formación de un trombo en ese sitio, pero al ser pequeña o generarun grado insignificante de estenosis su inestabilidad pasa inadvertida, sin la pre-sencia de sintomatología isquémica. Esta misma placa se “repara” volviendo aun estado de estabilidad, pero lo importante es que aumentó su tamaño y, por lotanto, su grado de estenosis. La repetición de este ciclo puede terminar en el desa-rrollo de una placa significativa, en la cual se repite el ciclo de ruptura–reparacióny se le agrega la presencia de turbulencia y la disminución en la velocidad del flu-jo sanguíneo en el sitio de la placa, favoreciendo la formación de un trombo queocasiona una oclusión completa del vaso, produciendo angina inestable e infarto.Dichos eventos han sido demostrados con evidencias histopatológicas.3 Lo ante-rior demuestra que el paciente con múltiples factores de riesgo, aun tratado y con-trolado de manera adecuada, puede desarrollar obstrucciones críticas o bien en-fermedad aterosclerótica subclínica grave.
ANGINA INESTABLE
En relación con la angina inestable, se consideran tres tipos de pacientes:
1. El que presenta la enfermedad o angina inestable por primera vez.2. El que tiene el antecedente de angina o un infarto agudo del miocardio, o
de ambos.3. El paciente con enfermedades o situaciones que complican el manejo de la
terapia antitrombótica y se sabe que es isquémico.
Dicha clasificación parece ser de importancia, ya que los riesgos de morbimorta-lidad son totalmente distintos entre ellos.

94 (Capítulo 6)Tratado de trombosis
El primer caso se trata de un paciente probablemente “virgen” a cualquier tipode tratamiento antitrombótico, lo que de forma inicial otorga la libertad para se-leccionar un tratamiento de manera rápida, con una mayor posibilidad de éxito.El segundo caso implica a un paciente con manejo posiblemente inadecuado (porfalta de apego), ineficiente (por factores particulares del paciente, como resisten-cia al tratamiento) o incompleto. El tercer caso es el de un paciente con otros fac-tores de riesgo que pueden complicar el manejo antitrombótico.
El cuadro clínico que se comenta a continuación es el de un paciente que pre-senta un síndrome coronario agudo diagnosticado, como angina inestable sin cul-minar en un infarto agudo. Como se mencionó, se consideran tres tipos de pacien-tes que se pueden presentar con angina inestable. La identificación del grupo alque pertenece el paciente determina el tratamiento a seguir. El primer tipo de pa-ciente es aquel sin antecedente de un evento previo de isquemia, por lo que proba-blemente no cuenta con un tratamiento antitrombótico. El manejo de este pacien-te implica el uso de dos tipos de medicamentos: la antiagregación plaquetaria conmedicamentos por vía oral y el uso de algún tipo de anticoagulación por vía pa-renteral, sea heparina no fraccionada o heparina de bajo peso molecular (HBPM),siempre y cuando no exista contraindicación para su uso.
Se inicia con una dosis de carga de ácido acetilsalicílico de 500 mg por vía oral.De manera simultánea se agrega un segundo antiagregante plaquetario, como elclopidogrel, el prasugrel o el ticagrelor. Cada uno de estos medicamentos tienebeneficios, limitaciones, efectos segundarios y complicaciones de uso (ver el ca-pítulo 3).
La heparina no fraccionada se ha utilizado desde hace más de 20 años por víaintravenosa en presencia de angina inestable.4,5 Su uso cotidiano ha hecho de sumanejo algo sencillo pero laborioso, ya que se requiere una vigilancia estrechade los tiempos de coagulación (TPT) para disminuir el riesgo de sangrado. Se po-dría pensar que la aparición de la HBPM ha venido a cambiar el tratamiento médi-co, pero no es así, ya que su uso conlleva el inconveniente de su tiempo de acción.Al administrarse por vía subcutánea no hay forma de revertir su efecto sin la pre-sencia de riesgo, además de que es necesario esperar hasta 12 h para que disminu-ya su efecto. Por el contrario, una vez que es suspendida la administración de laheparina no fraccionada por vía endovenosa desaparece su efecto en un lapso deuna a tres horas, permitiendo en caso necesario llevar a cabo algún tipo de inter-vención sin un riesgo de sangrado importante. Una tercera modalidad terapéuticaes el uso de HBPM por vía endovenosa. Si bien esta vía de administración no esuna práctica común ni ampliamente conocida, se ha reportado en la literatura queposee una mayor eficacia y mejores resultados en el paciente con evento isquémi-co. El estudio ATOLL, que incluyó 910 pacientes, mostró que la presencia demuerte por infarto del miocardio, hemorragias importantes, complicaciones delinfarto, infarto recurrente y revascularización urgente disminuyó en el grupo tra-

95Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
tado con enoxaparina.4 Además, el uso de enoxaparina no alteró la tasa de éxitode los procedimientos invasivos realizados.
Entre los fármacos específicamente diseñados para la antiagregación plaque-taria el clopidogrel es el medicamento que más se ha estudiado y usado a nivelinternacional. La dosis de carga es de 300 a 600 mg por vía oral en dosis única;esta variación en la dosis de impregnación depende de los estudios en los cualesse base el médico. Una de las limitaciones de este medicamento es el riesgo desangrado, que es directamente proporcional a la dosis utilizada. La dosis de cargapermite que el medicamento tenga un efecto antiagregante a las tres o cuatro ho-ras. El clopidogrel en dosis de 600 mg de dosis de impregnación y 75 mg/día pue-de demorar hasta cinco días en mostrar un efecto aproximado de 50% en la inhibi-ción de ADP para la agregación plaquetaria. Otro obstáculo es la existencia deuna población en la cual no existe el efecto deseado, que oscila entre 15 y 30%de aquellos que son tratados con este medicamento.6,7 Sus beneficios indican quees fácil de obtener, está disponible en presentación con dosis de carga en una odos tabletas y su costo es accesible, debido a la presencia de fármacos genéricosen el mercado.
El prasugrel, igual que el clopidogrel, es una tienopiridina inhibidora de losreceptores P2Y12.8 Es un profármaco que se metaboliza a su forma activa. Se creócomo una segunda generación de este tipo de medicamentos con la intención defacilitar su uso y asegurar un efecto antiagregante más completo y seguro que suantecesor. El prasugrel llega a tener concentraciones plasmáticas pico a los 30min de su administración y alcanza un estado estable de acción a los tres días dehaberse iniciado, lo cual le confiere ventajas claras sobre el clopidogrel. La dosisde carga de este medicamento es de 60 mg con una dosis diaria de 10 mg (éstaes su presentación habitual, dado que no existe hasta el momento una tableta conla dosis de impregnación). Se ha demostrado en algunos estudios que la tasa desangrado es ligeramente mayor (incluyendo hemorragias importantes que ponenen peligro la vida) y que el riesgo de reestenosis aguda por presencia de tromboses menor. En el estudio TRITON se apreció que la presencia de eventos recurren-tes de isquemia miocárdica se redujo 30% con el uso de prasugrel, en compara-ción con el clopidogrel; sin embargo, cuando se compararon los dos medicamen-tos no hubo diferencias significativas en la mortalidad global.9–12
El ticagrelor es una nueva generación de antiagregante plaquetario que, a dife-rencia de los dos anteriores, no requiere ser metabolizado para ejercer su efecto.El estudio PLATO demostró ser más potente que el clopidogrel en la prevenciónde nuevos eventos isquémicos, sin ninguna diferencia en la incidencia de hemo-rragias.14 Este medicamento alcanza un nivel plasmático máximo entre 1.5 y 3h después de una dosis de carga de 180 mg; la dosis de mantenimiento es de 90mg dos veces al día. En el estudio RESPOND se demostró que el uso de ticagreloren el paciente que no respondía a clopidogrel tuvo un efecto antiagregante por

96 (Capítulo 6)Tratado de trombosis
debajo de los niveles asociados con riesgo isquémico; este efecto se apreció tam-bién en los pacientes con un efecto adecuado del clopidogrel.6 Una de las desven-tajas de este medicamento es la necesidad de prescribir dos tomas al día, lo cualpuede dificultar el adecuado apego al tratamiento.
Se cuenta actualmente con varias herramientas terapéuticas para alterar de ma-nera efectiva y rápida la formación de trombos que pueden desencadenar un dañomiocárdico importante y poner en riesgo la vida del paciente.
Pero volvamos a los tres tipos de pacientes: el que debuta con la enfermedad,el isquémico conocido y el isquémico conocido complicado.15 El primer caso co-rresponde a un paciente que permite utilizar cualquiera de las herramientas men-cionadas, siempre bajo la vigilancia que este tipo de antitrombóticos requieren.
¿Cómo saber si el fármaco es el indicado para el paciente?
Desde 1994 se demostró que el éxito en el manejo del efecto antiagregante pla-quetario consiste en la vigilancia del mismo mediante estudios de laboratorio. Elproblema es, ¿quién realmente lo hace de manera rutinaria en sus pacientes? Lamayor parte de los estudios identifican a los pacientes que no tiene una respuestaadecuada al antiagregante, como el clopidogrel, que ha sido designado como el“villano”, luego de que se presenta un nuevo evento isquémico agudo. Quizá ha-bría que realizar estudios en los pacientes para determinar el nivel de antiagrega-ción y modificar el tratamiento, ya que esperar un nuevo evento isquémico sólopone en peligro la vida. Tanto la agregometría plaquetaria como el tiempo de san-grado permiten valorar la función plaquetaria. La agregometría plaquetaria per-mite identificar a los pacientes que se encuentran en mayor riesgo de presentarun nuevo evento isquémico por falta de efecto o de apego al antiagregante plaque-tario.
Otra forma de identificar a los pacientes con probabilidad de no responder alclopidogrel consiste en la realización de un estudio genético para determinar lapresencia o ausencia de riesgo elevado para nuevos eventos isquémicos por va-riantes del CYP2C19.11,12 Aún no se ha definido cuáles alteraciones disminuyenla eficacia del clopidogrel y si éstas afectan de alguna manera a los otros antiagre-gantes que funcionan de manera similar. Entonces, el paciente “virgen” a trata-miento se convierte en el paciente isquémico conocido con un nuevo evento is-quémico.
El paciente con diagnóstico de isquemia miocárdica o enfermedad coronariapor aterosclerosis, que presenta eventos recurrentes de isquemia sintomática oasintomática, nos coloca ante una situación difícil. Este paciente tratado con va-rios medicamentos para controlar los factores de riesgo y las alteraciones quepuede presentar la placa limita los cambios que se pueden realizar en el manejo

97Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
médico, sin aumentar los riesgos concomitantes del tratamiento, como el san-grado.
Lo primero es identificar si se trata de un nuevo evento o no, y si este nuevoevento es progresión de una placa preexistente. En caso de ser un nuevo evento,en otras palabras, la generación de una placa de novo significa que la aterosclero-sis ha avanzado y el manejo no consiste de manera necesaria en un cambio en eltratamiento con antiagregante, sino en un cambio en el tratamiento de otros facto-res de riesgo. Si es continuación de la misma enfermedad, entonces se trata de unproblema en el manejo médico establecido. Finalmente, en estos dos casos se tra-ta probablemente de una placa inestable. Al inicio se debe determinar si existeo no un efecto antiagregante adecuado, y de manera simultánea identificar loscambios en el estilo de vida y otros factores de riesgo que requieran ajustes ensu tratamiento. En este tipo de paciente se han realizados estudios que demues-tran que el cambio de un antiagregante a otro no aumenta el riesgo de sangradoni la posibilidad de un nuevo trombo; por el contrario, puede ayudar a protegeral paciente y a evitar la propagación del evento isquémico.6
Como parte del manejo del paciente durante el evento agudo uno no debe apo-yarse sólo en los medicamentos por vía oral, sino que se requiere una terapia quepermita alcanzar un efecto anticoagulante rápido, eficaz, confiable y fácil de me-dir. Como se mencionó, el uso de heparina no fraccionada lleva más de 20 añosy continúa siendo útil. La aparición de la HBPM ofrece una alternativa más enel manejo de este tipo de paciente. No obstante, en algunos estudios no se hademostrado una clara ventaja en su empleo, ya que no ha habido diferencias im-portantes en la mortalidad, el sangrado o la necesidad de transfusiones. Este me-dicamento en cualquiera de sus dos presentaciones (fraccionada o no) tiene indi-caciones precisas. El paciente que presenta un riesgo intermedio o alto deberárecibir de manera inmediata una dosis de heparina siempre y cuando no exista unacontraindicación. Para la heparina no fraccionada se considera una dosis en bolode 60 UI/kg de peso, seguida de una infusión de 10 a 12 UI/kg. La dosis de laHBPM es de 1 mg/kg dos veces al día por vía subcutánea. En ambos casos se con-tinúa durante al menos dos a cuatro días o hasta que el paciente sea revasculariza-do. Una de las indicaciones para el uso exclusivo de heparina no fraccionada esel paciente considerado con un riesgo bajo a intermedio o aquel con riesgo eleva-do, pero en quien el manejo será conservador por la presencia de comorbilidades.Es importante notar que los pacientes no deberán ser alternados entre uno y otrotratamiento, debido al elevado riesgo de sangrado.
El manejo de este tipo de paciente puede ser conservador, siempre y cuandono exista una evolución tórpida. El paciente se puede manejar con modificacio-nes en el tratamiento médico con o sin el aval de un estudio de laboratorio quedemuestre el efecto del antiagregante. Además, se debe establecer un manejo in-tegral con otros medicamentos, como la heparina en cualquiera de sus presenta-

98 (Capítulo 6)Tratado de trombosis
ciones, para brindarle al paciente una mayor cobertura y tratar de evitar que laarteria se ocluya de forma completa y se genere un infarto. También se han utili-zado los antagonistas IIb/IIIa en pacientes con angina inestable.
Se han desarrollado nuevos anticoagulantes orales (rivaroxabán, apixabán ydabigatrán, entre otros) con la finalidad de eliminar los riesgos de utilizar warfa-rina o acenocumarina, simplificando tanto su administración como su control.Para algunos de ellos ya existe la indicación y aceptación de uso en presencia defibrilación auricular no valvular, principal causa de embolia cerebrovascular enla población mayor de 70 años de edad. Hace poco se autorizó el uso de rivaroxa-bán en la etapa aguda del síndrome coronario en dosis de 2.5 mg dos veces al díaen combinación con ácido acetilsalicílico y clopidogrel en dosis habituales. Eluso del rivaroxabán no sólo se indica en presencia de angina inestable tratada demanera conservadora, sino que la indicación se extiende a los pacientes a los quedurante la etapa aguda se les realiza una intervención coronaria con angioplastiacon o sin implante de stent. No se ha determinado si una vez superado el eventocoronario agudo se deberá o podrá seguir con este medicamento. Aun no se hademostrado si existe o no algún beneficio a largo plazo. La disposición de estetipo de herramienta constituye un paso importante, ya que en el paciente con en-fermedad aterosclerótica avanzada o en quienes no se obtiene el efecto antiagre-gante plaquetario adecuado parece ser que la base del manejo en un futuro próxi-mo será la anticoagulación oral.
INFARTO AGUDO DEL MIOCARDIO
Trombólisis
La culminación de un síndrome coronario agudo es la presencia de un infarto,evento que aún confiere una tasa de morbimortalidad muy elevada.16 Su manejoes complejo y su éxito depende del tiempo de acción. Para esto se han desarrolla-do varios protocolos de manejo, todos con la intención de restablecer el flujo co-ronario a la brevedad, con un menor riesgo de complicaciones para el paciente.
Una de las primeras herramientas farmacológicas que se presentaron para elmanejo del síndrome coronario agudo fueron los fármacos trombolíticos, los cua-les se usan en situaciones de urgencia y en cierto tipo de pacientes. Es cierto quese empleaban única y exclusivamente en los eventos de infarto agudo, en los quese documentaba de manera fehaciente la presencia de lesión miocárdica, exclu-yendo al paciente con un infarto de la cara inferior, debido al riesgo de rupturamiocárdica. La estreptocinasa, en uso desde 1982, fue el primero de estos medi-camentos y, por lo tanto, el de mayor riesgo de complicaciones. Este tipo de me-

99Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 6–2. Contraindicaciones para el uso de trombolíticos
Absolutas:� Evento cerebrovascular o malformación de cualquier tipo un año atrás� Neoplasia intracraneal conocida� Hemorragia activa
� Sospecha de disección de aortaRelativas:
� Hipertensión severa no controlada o historia crónica de hipertensión arterial severa
� Historia de evento cerebrovascular previo o patología intracraneal conocida no cubierta enlas contraindicaciones
� Uso actual de anticoagulantes en dosis terapéuticas
� Trauma reciente (en un lapso de dos a cuatro semanas)� Reanimación cardiopulmonar traumática o prolongada (> 10 min)� Cirugía mayor (< 3 semanas)
� Punción vascular no compresible� Para estreptocinasa o APSAC: exposición previa a las mismas (especialmente entre cinco
días y dos años)
� Embarazo� Úlcera péptica activa
dicamentos cuentan con múltiples consideraciones para su uso, por lo que se en-cuentra limitada su aplicación (cuadro 6–2).
Dichos medicamentos actualmente se pueden utilizar en dos circunstancias:como monoterapia inicial en situaciones en las que no existe la posibilidad de en-viar en tiempo y forma al paciente a un procedimiento intervencionista (angio-plastia con o sin colocación de stent), y como coadyuvante en el procedimientointervencionista.
La fibrinólisis como monoterapia ha caído en desuso, pues los estudios handemostrado que la reperfusión mecánica por medio de un cateterismo con angio-plastia con o sin la instalación de un stent conlleva una mayor sobrevida y un me-nor número de complicaciones. Uno de los factores primordiales del uso de estetipo de tratamiento es el tiempo de evolución del episodio isquémico. Existen es-tudios que sustentan su utilidad, aunado a un procedimiento invasivo, ya que me-jora la posibilidad de mantener un flujo coronario adecuado y disminuye de ma-nera importante la reestenosis aguda por presencia de un trombo, siendo suprincipal riesgo el sangrado. El paciente a quien se le administra un trombolíticosegún los protocolos establecidos al ser invadido, aunque sea por punción, pre-senta un riesgo muy elevado de sangrado incontrolable, lo cual obviamente au-menta la morbimortalidad.
El uso de trombolíticos en pacientes con angina inestable se encuentra contra-indicado, ya que se ha demostrado que aumenta el riesgo de presentar un infartoagudo.

100 (Capítulo 6)Tratado de trombosis
Abciximab, eptifibatida y tirofibán
Forman parte de una familia de medicamentos dirigidos al receptor glucoprotei-co (GP) IIb/IIIa, que se localiza en la superficie de las plaquetas y es un importan-te factor en el desarrollo de trombos. Su uso ha cambiado el panorama del pacien-te con un síndrome coronario agudo que concluye en un infarto, y ha demostradouna disminución de la tasa de morbimortalidad. Algunos estudios importantes ymetaanálisis, como GUSTO y TIMI, entre otros, han demostrado que este tipode medicamentos más la intervención coronaria dentro de la ventana establecidade manejo (que puede ser, de forma ideal, dentro de las primeras tres a cuatro ho-ras de haberse iniciado el evento isquémico) mejoran de manera sustancial la po-sibilidad de éxito y la recuperación del paciente.2 Los pacientes más beneficiadoscon el uso de este tipo de medicamentos son aquellos con elevación de la troponi-na I, desnivel del segmento ST, calificación TIMI mayor de 3 y diabetes. Estosmedicamentos no son inocuos, ya que confieren el riesgo principal de presentarhemorragias de difícil control, que pueden poner en riesgo la vida del paciente.
Abciximab
Es un anticuerpo monoclonal que se adhiere al receptor GP IIb/IIIa de manerapermanente. La dosis inicial es en bolo, basada en el peso corporal del paciente.Se calcula una dosis de 0.15 a 0.30 mg/kg de su peso corporal, que habitualmentese administra al iniciar el procedimiento intervencionista. Es importante notarque existen múltiples protocolos en los que al paciente se le administra 50% dela dosis en el área de urgencias antes de realizar el cateterismo y el 50% restanteen el procedimiento o continuando con la dosis de mantenimiento. La dosis demantenimiento también se basa en el peso corporal, y se calcula en 0.125 �g/kg/min, hasta un máximo de 10 �g/min. La infusión habitualmente se administra du-rante 12 a 96 h, lo cual dependerá de las características de la oclusión, la presenciao ausencia de trombo y su extensión, y la aparición de hemorragias.17,18
Eptifibatida
Este péptido tiene una vida media corta, por lo que deja de surtir efecto despuésde dos a cuatro horas de su suspensión. La impregnación es de dos dosis de 180�g/kg seguidas de 2 �g/kg/min durante 24 h.21
Una observación importante en cualquiera de los medicamentos antagonistasde la GP IIa/IIIb en la mayor parte de los estudios publicados es que requierenel uso concomitante de heparina, ácido acetilsalicílico y algún antiagregante pla-quetario, como el clopidogrel, que siempre se menciona en primera estancia porser el más antiguo. Si a esto se le suma que al implantar un stent en una placa ines-

101Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
table, que implica la presencia de un cuerpo extraño en la circulación, se favoreceaún más la agregación plaquetaria y la posibilidad de desarrollar un trombo demanera aguda, por esto es fundamental utilizar todas las herramientas farmacoló-gicas que se tengan a la mano para evitarlo.22,23
Tirofibán
Es un medicamento sintético con una afinidad muy alta por el receptor de las pla-quetas, produciendo un efecto a los cinco minutos de haberse iniciado su admi-nistración y siendo reversible una vez suspendido de cuatro a seis horas posterio-res. Una de sus indicaciones principales es su administración antes (upstream) derealizar cualquier procedimiento intervencionista, sea cateterismo o cirugía derevascularización. La dosis de impregnación es de 0.4 �g/kg/min durante 30 min,seguido de 0.10 �g/kg/min en infusión durante 12 a 24 h. Varios estudios, comoel On–Time, y metaanálisis, demostraron que el uso de tirofibán en pacientes coneventos coronarios agudos mejora de manera significativa el resultado de la inter-vención coronaria, pero también aumenta el riesgo de sangrado, aunque no demanera significativa.19,20
Consideraciones finales
Igual que en cualquier otra enfermedad, no existe un manejo mágico, ya que éstedependerá de la circunstancia y el paciente que está bajo el cuidado del médico.El uso de antagonistas de la GP IIb/IIIa, antiagregantes plaquetarios y anticoagu-lantes se ha discutido mucho, y se ha demostrado que en ciertas poblaciones existeun beneficio.23 El paciente que presenta una elevación importante de la troponinaI se beneficia del uso en conjunto de antagonistas GP IIb/IIIa y antiagregantesplaquetarios. El paciente con un riesgo elevado por presencia de trombos tambiénrecibe beneficios, además de que se debe continuar con anticoagulación a basede heparina, después de completar el esquema de hasta 96 h con la infusión deantagonistas de la GP IIb/IIIa. La intervención coronaria es un procedimiento dediagnóstico, terapéutico y de pronóstico, ya que permite visualizar la lesión coro-naria. Se ha demostrado que en el paciente sometido a una angioplastia con balóncon o sin instalación de un stent se rescata mayor masa muscular. Si a este proce-dimiento se le agrega el uso de un bloqueador de la GP IIb/IIIa el éxito es aúnmayor. La combinación de tratamiento médico con diferentes fármacos y la reali-zación de un procedimiento invasivo probablemente en conjunto constituyen elmejor camino, siempre teniendo flexibilidad para cambiar la actuación según lasnecesidades del paciente. Todo se concentra en cómo resolver el problema en ple-no cuando la vida del paciente se encuentra en riesgo.24–26

102 (Capítulo 6)Tratado de trombosis
REFERENCIAS
1. Bui QT, Reddy VS, Jacobs JR, Begelman SM, Frederick PD et al.: Previous myocardialinfarction as a risk factor for in–hospital cardiovascular outcomes (from the National Regis-try of Myocardial Infarction 4 and 5). Am J Cardiol 2013;111(12):1694–1700.
2. Juárez HU, Jerjes SC, The RENASICA II Investigators: Risk factors, therapeutic approa-ches, and in–hospital outcomes in Mexicans with ST–elevation acute myocardial infarc-tion: The RENASICA II Multicenter Registry. Clin Cardiol 2013;36(5):241–248.
3. Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT et al.: Healed plaque rupturesand sudden coronary death: evidence that subclinical rupture has a role in plaque progres-sion. Circulation 2001;103(7):934–940.
4. Schumacher WA, Lee EC, Lucchesi BR: Augmentation of streptokinase–induced throm-bolysis by heparin and prostacyclin. J Cardiovasc Pharmacol 1985;7(4):739–746.
5. Nicolini FA, Nichols WW, Saldeen TG, Khan S, Mehta JL: Adjunctive therapy with lowmolecular weight heparin with recombinant tissue–type plasminogen activator causes sus-tained reflow in canine coronary thrombosis. Am Heart J 1992;124(2):280–288.
6. Gurbel PA, Bliden BS, Butler K, Antonino J et al.: Response to ticagrelor in clopidogrelnonresponders and responders and effect of switching therapies. The RESPOND Study.Circulation 2010;121:1188–1199.
7. Fox KA, Mehta SR, Peters R et al.: Clopidogrel in unstable angina to prevent recurrentischemic events trial. Benefits and risks of the combination of clopidogrel and Aspirin inpatients undergoing surgical revascularization for non–ST–elevation acute coronary syn-drome: the Clopidogrel in Unstable Angina to prevent Recurrent Ischemic Events (CURE)Trial. Circulation 2004;110:1202–1208.
8. Dobesh PP: Pharmacokinetics and pharmacodynamics of prasugrel, a thienopyridine P2Y12
inhibitor. Pharmacotherapy 2009;29(9):1089–1102.9. Smith P, Goodnough L, Levy JH, Poston RS, Short MA et al.: Mortality benefit with pra-
sugrel in the TRITON–TIMI 38 Coronary Artery Bypass Grafting (CABG) Cohort: risk–adjusted retrospective data analysis. J Am Coll Cardiol 2012;60(5):388–396.
10. TRITON–TIMI 38 Investigators: Prasugrel versus clopidogrel in patients with acute coro-nary syndromes. N Engl J Med 2007;357:2001–2015.
11. Goswami S, Cheng–Lai A, Nawarskas J: Clopidogrel and genetic testing: is it necessaryfor everyone? Cardiol Rev 2012;20(2):96–100.
12. Salas I, Olalde RM, Santiago GD, Corona de la Peña N, Valencia SJ: The impact ofCYP3A5*1/*3, PIA1/A2 and T744C polymorphisms on clopidogrel and acetylsalicylicacid response variability in Mexican population. Thromb Res 2012;130(3):e67–e72.
13. Loh JP, Pendyala LK, Kitabata H, Torguson R, Chen F et al.: Safety of reloading prasu-grel in addition to clopidogrel loading in patients with acute coronary syndrome undergoingpercutaneous coronary intervention. Am J Cardiol 2013;111(6):841–845.
14. Steg PG, Harrington RA, Emanuelsson H, Katus HA et al., for the PLATO Study Group:Stent thrombosis with ticagrelor versus clopidogrel in patients with acute coronary syndro-mes: an analysis from the prospective randomized PLATO trial. Circulation 2013.
15. Angiolillo DJ: The evolution of antiplatelet therapy in the treatment of acute coronary syn-dromes: from Aspirin to the present day. Drugs 2012;72(16):2087–2116.
16. Herrett E, George J, Denaxas S, Bhaskaran K, Timmis A et al.: Type and timing of her-alding in ST–elevation and non–ST–elevation myocardial infarction: an analysis of syndro-mes prospectively collected electronic healthcare records linked to the National Registryof Acute Coronary. Eur Heart J Acute Cardiovasc Care. 9 de mayo de 2013.

103Angina inestable e infarto agudo del miocardioE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
17. Brener SJ, Dambrink JH, Maehara A, Chowdhary S, Gershlick AH et al.: Benefits ofoptimizing coronary flow before stenting in primary percutaneous coronary intervention forST–elevation myocardial infarction: insights from INFUSE–AMI. EuroIntervention 2013.Pii:20130401–06.
18. Berglund U, Nilsson L, Janzon M: Abciximab bolus with optional infusion in interventionfor ST–elevation myocardial infarction. Scand Cardiovasc J 2013;47(4):230–235.
19. Zhang Y, Gao C, Liu H, Wang X, Yang H et al.: Routine early versus deferred provisionaltirofiban treatment in patients with acute coronary syndrome undergoing percutaneous co-ronary intervention. Clin Exp Pharmacol Physiol 2013;40(4):289–294.
20. Jia Z, Guo M, Zhang YQ, Liang HQ, Song Y: Short–term effect of upstream administra-tion in comparison to deferred injection of tirofiban on patients with acute ST–segment ele-vation myocardial infarction undergoing primary percutaneous coronary intervention. J In-terv Cardiol 2013.
21. Puri A, Bansal A, Narain VS, Sethi R, Dwivedi SK et al.: Comparative assessment of pla-telet GpIIb/IIIa receptor occupancy ratio with eptifibatide/tirofiban in patients presentingwith ACS and undergoing PCI. Indian Heart J 2013;65(2):152–157.
22. Kristensen SD, Würtz M, Grove EL, de Caterina R, Huber K et al.: Contemporary useof glycoprotein IIb/IIIa inhibitors. Thromb Haemost 2012;107(2):215–224.
23. Bagai A, Cantor WJ, Tan M, Tong W, Lamy A et al.: Clinical outcomes and cost implica-tions of routine early PCI after fibrinolysis: one–year follow–up of the Trial of Routine An-gioplasty and Stenting after Fibrinolysis to Enhance Reperfusion in Acute Myocardial In-farction (TRANSFER–AMI) study. Am Heart J 2013;165(4):630–637. Epub 2013.
24. Díaz JF, Cardenal R, Gómez MA, Sánchez GC: Safety and efficacy of tirofibán as ad-junctive therapy for patients with ST–elevation myocardial infarction: a comparison versusplacebo and abciximab. Cardiovasc Hematol Agents Med Chem 2011;9(3):147–153.
25. Schneider DJ: Current issues with glycoprotein IIb–IIIa antagonists. Curr Drug Targets2011;12(12):1813–1820.
26. Tricoci P, Newby LK, Hasselblad V, Kong DF, Giugliano RP et al.: Upstream use ofsmall–molecule glycoprotein IIb/IIIa inhibitors in patients with non–ST–segment elevationacute coronary syndromes: a systematic overview of randomized clinical trials. Circ Car-diovasc Qual Outcomes 2011;4(4):448–458.

104 (Capítulo 6)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
7Fibrilación auricular y trombosis
Ignacio Rodríguez Briones
INTRODUCCIÓN
La fibrilación auricular (FA) es la arritmia cardiaca clínicamente significativamás común, cuya presentación se incrementa día a día en la población de todoel mundo. La FA afecta a las poblaciones de todas las edades, pero en especiala los ancianos, a quienes les confiere un mayor riesgo de evento vascular cerebral(EVC). Mediante estrategias preventivas adecuadas se pueden reducir de maneraimportante las consecuencias negativas de esta arritmia, mismas que se revisanen el presente capítulo.
EPIDEMIOLOGÍA
La FA es una de las principales patologías cardiológicas en la actualidad tanto porla frecuencia de su presentación en la población de mayor edad como por ser unacausa frecuente de atención en los servicios de urgencias y de hospitalización.
De manera reciente se le aplicó el término de “epidemia”, debido al gran incre-mento de su prevalencia. Tanto la prevalencia como la incidencia de la FA se hanelevado por razones aún no bien establecidas. El aumento en la incidencia de losfactores etiológicos de la arritmia, como la población añosa y la alta prevalenciade enfermedades cardiovasculares, explica sólo parcialmente este fenómeno.
Muchos estudios poblacionales y diversos análisis han afirmado que la FA esuna enfermedad que predomina en la población mayor, afectando aproximada-mente a 20% de la población mayor de 80 años de edad.1
105

106 (Capítulo 7)Tratado de trombosis
Riesgo de trombosis en la fibrilación auricular
La fisiopatología de la FA involucra sobre todo los cambios estructurales de lasaurículas, mismos que ocasionan un aumento del estrés, como en la inflamaciónde sus paredes, y en los focos ectópicos que se localizan en especial en la desem-bocadura de las venas pulmonares. También existe la asociación genética en al-gunas familias, en las que se ha identificado que una región del cromosoma 10es la responsable de la arritmia.2
La contracción no coordinada de la aurícula izquierda conduce a la estasis san-guínea y las anormalidades en la coagulación, en las plaquetas y en la fibrinólisis.Las dos terceras partes de los EVC son causados por tromboembolismo desde elinterior de la orejuela de la aurícula izquierda en pacientes con FA.3 El tromboem-bolismo de la aurícula izquierda, la aterosclerosis coexistente de las grandes arte-rias y las anormalidades valvulares comprenden el resto.
El incremento de hasta 17 veces el riesgo de complicaciones tromboembólicasy EVC en pacientes con FA y enfermedad cardiaca reumática ha sido descritodesde hace muchos años. Aun en FA de origen no reumático el riesgo tromboem-bólico se incrementa de manera significativa hasta cinco o seis veces más, encomparación con controles sanos, y es responsable de hasta 25% de todos losEVC.3–5 La FA paroxística recurrente se asocia a un riesgo de EVC comparablecon el de la FA permanente.6
Los pacientes con FA que presentan un EVC tienen mayores mortalidad, dis-capacidad y estancia hospitalaria, y menos posibilidad de egreso hospitalario quelos pacientes que presentan un EVC en ausencia de arritmia.7–11
El tiempo necesario para la formación de un trombo intracavitario es incierto,pero se considera que casi siempre surge en un lapso no mayor de 72 h despuésde haberse iniciado la FA; sin embargo, esto no es concluyente, ya que se ha re-portado que los episodios de FA paroxística más cortos pueden predisponer a laformación de trombos auriculares, lo que reafirma la importancia de la anticoa-gulación independientemente del tipo de FA, sea paroxística, persistente o per-manente.12
Después de varios días la FA provoca un proceso de remodelación y fibrosis,con la posterior dilatación auricular, lo que predispone a una mayor estasis san-guínea y un mayor riesgo de trombosis. Otro factor fisiopatológico para la forma-ción de trombos lo constituye el trauma o daño a nivel de la pared vascular, el cualse relaciona con los cambios estructurales de la aurícula. De manera independien-te a la reducción y las alteraciones en el flujo sanguíneo, la sola presencia de dis-función endotelial o endocárdica es un factor de riesgo para la formación de trom-bos. Dicho daño genera un aumento en la liberación del factor de von Willebrand,mismo que desempeña un papel primordial en la agregación plaquetaria. Los pa-cientes con FA presentan un mayor nivel del factor en relación con aquellos que

107Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 7–1. Factores de riesgo para eventostromboembólicos en fibrilación auricular
Clínicos� Evento vascular o embólico previo , o isquemia cerebral transitoria� Hipertensión arterial
� Edad mayor de 65 años� Diabetes mellitus� Insuficiencia cardiaca
� Enfermedad arterial coronaria� Válvula protésica
Ecocardiográficos� Enfermedad valvular reumática (estenosis mitral)� Falla ventricular izquierda� Incremento de tamaño de la aurícula izquierda
� Trombo auricular izquierdo� Contraste espontáneo severo� Disfunción mecánica auricular izquierda (velocidades de vaciado menores de 20 cm/seg)� Ateroma aórtico complejo
se encuentran en ritmo sinusal, y suele ser aún mayor en los que presentan creci-miento auricular izquierdo.
Los pacientes que padecen la arritmia comprenden un grupo heterogéneo, porlo que el riesgo de EVC varía de acuerdo con la población. Los factores de riesgoclínicos y ecocardiográficos para el EVC en pacientes con FA están bien estable-cidos. La consideración tanto de las comorbilidades como de sus contraindica-ciones permite identificar a los pacientes candidatos al uso de anticoagulantes(cuadro 7–1).
El riesgo de EVC en la FA “aislada” (es decir, sin causa aparente) es bajo, deaproximadamente 0.5% por año.12 La historia previa de un evento embólico y laedad son los factores de predicción más importantes que conllevan a un incre-mento del riesgo de EVC en los pacientes con FA no valvular.13 El riesgo anualde fenómenos embólicos asociados a FA en las personas mayores de 60 años deedad es de 1.5%, pero se incrementa a 23.5% en los octogenarios.
Estratificación del riesgo tromboembólico
Los beneficios de la terapia antitrombótica en relación con la reducción del riesgode EVC requieren ser balanceados, considerando el riesgo de sangrado, la difi-cultad del monitoreo y los costos. Además de decidir entre utilizar antiagregantesplaquetarios, anticoagulantes o no utilizar terapia alguna, varios esquemas de es-tratificación han sido desarrollados para evaluar el riesgo de EVC sin el uso deanticoagulantes.

108 (Capítulo 7)Tratado de trombosis
Cuadro 7–2. Estratificación del riesgo tromboembólicoen el paciente con fibrilación auricular CHADS2
Criterios CHADS2 Puntuación
Insuficiencia cardiaca (congestive heart failure) 1Hipertensión arterial (hypertension) 1Edad (age) > 75 años 1
Diabetes mellitus 1Evento vascular cerebral o isquemia cerebral transitoria (stroke) 2
Uno de los diversos esquemas que existen para la estratificación de riesgo esel CHADS2,15 el cual es el más utilizado y validado, si bien todos los esquemasde riesgo poseen una utilidad predictiva modesta.
La escala CHADS2 equivale al acrónimo de cinco factores clínicos: insufi-ciencia cardiaca congestiva, hipertensión arterial sistémica, edad mayor de 75años, diabetes mellitus y EVC previo o antecedente de isquemia cerebral transito-ria (ICT). La presencia de cada factor otorga un punto, excepto por el antecedentede EVC o ICT, en los que se otorgan dos puntos. Así, los pacientes con una pun-tuación CHADS2 de 0 son considerados de bajo riesgo, aquellos con una califica-ción de 1 son considerados de riesgo intermedio y los que tienen una puntuaciónde 2 o más se consideran de riesgo alto (cuadros 7–2 y 7–3).
Las puntuaciones pueden ser traspoladas a eventos por 100 personas por añoy al número necesario a tratar (NNT) para prevenir un evento tromboembólicoen 100 personas/año. El NNT para prevenir un EVC declina rápidamente confor-me el CHADS2 se incrementa; así, para prevenir un EVC 417 pacientes con unapuntuación de 0 deben de ser tratados con warfarina, en comparación con 125 pa-cientes con una puntuación de 1 y menos de 50 pacientes con una calificaciónCHADS2 de 3 o más.
Cuadro 7–3. Escala de riesgo de tromboembolismo y eventocerebrovascular en pacientes con fibrilación auricular (CHA2DS2–VASc)
Criterios CHA2DS2–VASc Puntuación
Insuficiencia cardiaca (congestive heart failure) 1Hipertensión arterial (hypertension) 1
Edad (age) > 75 años 2Diabetes mellitus 1Evento vascular cerebral o isquemia cerebral transitoria (stroke) 2Infarto del miocardio, arteriopatía periférica, placa aórtica (vascular disease) 1
Edad (age) de 65 a 74 años 1Sexo (sex) femenino, más de 65 años de edad 1

109Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Los pacientes con una puntuación CHADS2 de 0 tienen un beneficio absolutomuy pequeño al usar anticoagulante, mientras que los pacientes con calificacio-nes de 2 o más tienen un riesgo basal de EVC que garantiza que la anticoagulaciónserá bien tolerada.
Otro esquema de riesgo tromboembólico desarrollado de manera más reciente—el CHA2–DS2–VASc— incorporó los factores de riesgo adicionales (sexo fe-menino, edad de 65 a 74 años, mayor puntaje a los pacientes mayores de 75 añosde edad y presencia de enfermedad vascular). Dicho esquema representa un in-tento de mejorar al CHADS2 y ampliar el número de pacientes que se pueden be-neficiar a través de la terapia preventiva. El CHA2–DS2–VASc fue adoptado porla Sociedad Europea de Cardiología16 en su más reciente publicación y consenso,mientras que en la novena edición las guías del American College of Chest Physi-cians se basan en la clasificación de CHADS2; sin embargo, se considera que siel paciente tiene otros factores de riesgo que no pertenecen a la escala CHADS2se puede indicar el uso de anticoagulantes. En general el CHA2–DS2–VASc pa-rece ser más útil para proveer una estratificación de riesgo adicional en pacientescon una puntuación CHADS2 baja (p. ej., de 0 o 1).
Estratificación del riesgo de sangrado
Otro de los objetivos que se busca al estratificar a los pacientes con FA, ademásde establecer su riesgo de trombosis, es el de su seguridad, la cual está relacionadacon el riesgo de sangrado al utilizar un anticoagulante o un antiagregante plaque-tario, por lo que se han propuesto diversos esquemas de estratificación, como esel riesgo de sangrado HAS–BLED, el cual otorga puntos por cada factor de riesgopresente. Un paciente con un puntaje HAS–BLED alto (mayor de 3) no por fuerzacuenta con contraindicación para el uso de terapia antitrombótica, simplementeindica que se requiere un monitoreo más estrecho.17
Dos aspectos importantes que pueden limitar o indicar que hay que ser másprecavidos son la edad del paciente y la función renal; así, una depuración de 30a 50 mL/min incrementa el riesgo de sangrado, y una depuración menor de 30mL/min puede llegar a contraindicar el uso de anticoagulantes, en especial losnuevos anticoagulantes orales.
TROMBOPROFILAXIS EN LA FIBRILACIÓN AURICULAR
La prevención del EVC en la FA puede ser abordado mediante la restauración yel mantenimiento del ritmo sinusal, terapia anticoagulante y aislamiento mecáni-co de la orejuela de la aurícula izquierda. Cinco estudios aleatorizados recientesfallaron en mostrar una disminución del riesgo embólico con una estrategia de

110 (Capítulo 7)Tratado de trombosis
control del ritmo comparado con el control de la frecuencia cardiaca.18–22 Los es-tudios AFFIRM21 y RACE22 han resaltado la utilidad de ampliar la anticoagula-ción en los pacientes con FA; sin embargo, antes de estos estudios se presumíaque la estrategia de control del ritmo prevenía el desarrollo de EVC embólicos.Ambos estudios demostraron que el riesgo tromboembólico no se reduce a pesarde la aparente restauración y mantenimiento del ritmo sinusal. La mayoría de losEVC en el grupo control ocurrieron cuando el nivel de anticoagulación era sub-óptimo o bien los pacientes no estaban anticoagulados. Además, los estudios re-saltaron que con frecuencia la FA puede ser asintomática, así como la escasa efi-cacia de los fármacos antiarrítmicos actuales para mantener el ritmo sinusal yprevenir recurrencias de la arritmia.
Varios estudios han mostrado tanto la seguridad a largo plazo como la eficaciaen la reducción del EVC al aislar u ocluir el apéndice auricular izquierdo, sea porvía quirúrgica o percutánea. En la actualidad la terapia con anticoagulantes conti-núa siendo la piedra angular en la prevención del EVC en los pacientes con FA.
TRATAMIENTO FARMACOLÓGICO
Muchos anticoagulantes y antiplaquetarios están actualmente disponibles para suuso como tromboprofilaxis en el tromboembolismo relacionado con la fibrila-ción auricular. Durante décadas la warfarina y la acenocumarina fueron los úni-cos anticoagulantes disponibles. El advenimiento de nuevos anticoagulantes ora-les —inhibidores directos de la trombina o inhibidores del factor Xa— hapermitido que se expandan las opciones terapéuticas en los pacientes con FA.23
La selección de la terapia depende de las ventajas relativas y desventajas de losmedicamentos, la experiencia del médico y la evidencia detrás de cada uno deestos fármacos para un paciente en particular (cuadro 7–4).
Tratamiento con warfarina o acenocumarina
Los antagonistas de la vitamina K, como la warfarina, han constituido el métodoprincipal de la anticoagulación en los pacientes con FA, la cual reduce 68% elriesgo de EVC y la mortalidad total 33%, en comparación con el placebo. Ade-más, los EVC que ocurren en pacientes tratados con niveles terapéuticos son me-nos severos que aquellos que se presentan en pacientes no anticoagulados.
El índice normalizado internacional (INR, por sus siglas en inglés) recomen-dado de entre 2.0 y 3.0 maximiza la prevención del EVC y minimiza el riesgo desangrado. Este rango de INR también es el más apropiado para los pacientes ma-yores y aquellos con historia previa de EVC.

111Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 7–4. Opciones terapéuticas antitrombóticaspara pacientes con fibrilación auricular
Agente Dosis Ventajas Desventajas
ASA 81 a 325 mg una vez aldía
Fácil de usar; menossangrado en compa-ración con la warfari-na
Mínima eficacia en preven-ción de EVC
ASA másclopido-grel
81 mg de ASA más 75mg de clopidogrel aldía
Más efectiva que ASAsola al disminuir elEVC
Más sangrado que ASA,menor protección quewarfarina
Warfarina Una vez al día; mante-ner un INR de 2 a 3
Alta eficacia en preven-ción de EVC, reversi-ble, económica, sepueden monitorearsus niveles
Requiere monitoreo fre-cuente y ajuste de dosis;presenta muchas interac-ciones con otros fárma-cos y los alimentos
Dabigatrán 150 mg/12 h en meno-res de 80 años deedad con depuración> 50 mL/min, 110 mgen > 80 años de edadcon depuración de 30a 50 mL/min
No hay monitoreo de ru-tina, dosis oral fija
No se puede monitorear suefecto no reversible; nohay seguridad a largoplazo; se debe tener pre-caución en pacientes conIRC
Rivaroxa-bán
20 mg/24 h con depura-ción > 50 mL/min, 15mg/24 h con depura-ción de 30 a 50 mL/min
No monitoreo de rutina,una dosis fija al día
No se puede monitorear suefecto no reversible; nohay seguridad a largoplazo: se debe tener pre-caución en pacientes conIRC y daño hepático
Apixabán 5 mg/12 h, 2.5 mg/12 hcon depuración < 30mL/min y edad mayorde 80 años o pesomenor de 60 kg
No monitoreo de rutina,dosis oral fija, puededisminuir la mortali-dad comparado conwarfarina
No se puede monitorear suefecto no reversible; nohay seguridad a largoplazo; se debe tener pre-caución en pacientes conIRC avanzada
ASA: ácido acetilsalicílico; EVC: evento vascular cerebral; INR: índice normalizado internacional;IRC: insuficiencia renal crónica.
La principal desventaja de la warfarina es la gran interacción que tiene conotros fármacos, incluidos los antibióticos más comunes, la amiodarona y las esta-tinas. La ingestión de alimentos ricos en vitamina K también puede alterar elefecto de la warfarina, por lo que los pacientes deben ser aconsejados en cuantoa mantener un constante cuidado en su dieta. Existen variaciones genéticas en elmetabolismo y en la actividad de la warfarina que contribuyen a las diferenciasindividuales en las dosis.24
Por todos estos factores, la terapia crónica con warfarina se asocia con una ne-cesidad de monitoreo y ajuste de las dosis. Para ayudar con las necesidades deun monitoreo frecuente se han desarrollado clínicas especializadas en anticoagu-

112 (Capítulo 7)Tratado de trombosis
lación, con el fin de contar con la menor variación posible en los manejos de estospacientes y así tener el menor número de eventos adversos relacionados con eluso de la warfarina. Otra de las principales complicaciones —quizá más devasta-dora— es el riesgo de hemorragia cerebral e intracraneal, la cual ocasiona unamortalidad de hasta 50%.
Inhibidores directos de la trombina
El dabigatrán es el único inhibidor directo de la trombina disponible desde queel ximelagatrán fue retirado del mercado debido a su hepatotoxicidad. Este fár-maco representa el primer anticoagulante oral en América y Europa desde la war-farina. Las ventajas que tiene sobre la warfarina son las dosis orales fijas, dos ve-ces al día, sin la necesidad de monitoreo mediante pruebas de laboratorio.
La eficacia del dabigatrán para la prevención del EVC fue establecida graciasa los resultados del estudio RE–LY, en el que los pacientes fueron aleatorizadosa recibir dabigatrán en dos dosis posibles —de 110 o de 150 mg más cada 12 h—y un tercer brazo más recibió warfarina, con el objetivo de mantener un INR de2.0 a 3.0. El dabigatrán en la dosis más baja no fue inferior a la warfarina en laprevención del EVC, además de una tasa significativamente menor de sangradomayor, mientras que la dosis de 150 mg cada 12 h mostró una tasa similar de san-grado a la de la warfarina, con menor hemorragia intracraneal y una menor inci-dencia de EVC isquémico.
En la actualidad se encuentra aprobado su uso en EUA, México y Europa. Ennuestro país las dosis recomendadas son 150 mg cada 12 h en pacientes menoresde 80 años de edad, con una depuración de creatinina mayor de 50 mL/min, mien-tras que en los pacientes mayores de 80 años de edad o con depuraciones menoresde 50 mL/min se recomienda la dosis de 110 mg cada 12 h. En los pacientes confalla renal avanzada con depuración de creatinina menor de 30 mL/min su usose encuentra contraindicado, debido al elevado riesgo de sangrado.
La dispepsia constituye un evento secundario que resulta por el tipo de encap-sulado especial a base de ácido tartárico, lo cual se requiere para una adecuadaabsorción del profármaco. Se ha demostrado que la administración conjunta conbloqueadores de la bomba de protones, como el pantoprazol, no interfiere de ma-nera importante con su biodisponibilidad.25
Otras ventajas de este fármaco es que alcanza su eficacia entre dos y tres horasposteriores a su ingesta oral y no requiere monitoreo de su efecto. Debido a sueliminación renal, se debe evitar en individuos que padezcan enfermedad renalsevera, y se debe utilizar con precaución en los pacientes con alteración renal demoderada a grave. No existe un método directo para monitorear el efecto anticoa-gulante ni para revertir efectivamente el efecto del mismo en los pacientes consangrado. El dabigatrán afecta el tiempo parcial de tromboplastina; sin embargo,

113Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
los niveles de éste no se correlacionan de manera directa con la actividad anticoa-gulante.
El estudio RELY–ABLE,26 mismo que incluyó a los pacientes manejados condabigatrán durante el estudio RE–LY, continuó con la misma dosis durante al me-nos dos años después de finalizado el estudio; hasta el momento ha mostrado losmismos beneficios y seguridad encontrados durante el estudio RE–LY. Un meta-análisis publicó que el dabigatrán provoca una mayor incidencia de infartos quela warfarina; sin embargo, el análisis del estudio RE–LY al respecto, y despuésde una nueva revisión de los casos de infarto silente no reportados, observó quedicho incremento de infartos en el grupo de dabigatrán no fue significativo.
Una ventaja del dabigatrán sobre otros anticoagulantes es que al unirse en unalto porcentaje a las proteínas plasmáticas las hace un producto dializable.27
Las guías del American College of Cardiology,28 las de la American HeartAssociation, las canadienses,29 las del CHEST30 y las europeas31 sugieren su usocomo primera opción en los pacientes con fibrilación auricular cuya recomenda-ción sea la anticoagulación oral.
El análisis de costo–efectividad del dabigatrán en comparación con la warfari-na resalta las potenciales ventajas de disminuir el riesgo de isquemia cerebral yhemorragia intracraneal. Otros estudios demostraron que el dabigatrán tiene unmejor costo–beneficio, especialmente en los pacientes con alto riesgo de EVC yun mayor riesgo de sangrado. El costo–beneficio se reduce en los pacientes conmenor riesgo de EVC y un bajo riesgo de sangrado.32
Inhibidores del factor Xa
Rivaroxabán
Los inhibidores del factor Xa, como el rivaroxabán y el apixabán, inhiben direc-tamente el factor Xa, convirtiendo a la protrombina en trombina activada, efectocompartido con las heparinas de bajo peso molecular, como la enoxaparina. Estosagentes tampoco requieren monitoreo de la coagulación o ajuste de dosis.
El rivaroxabán se encuentra aprobado para su uso en la prevención del EVCen pacientes con FA, con base en los resultados del estudio ROCKET–AF,33 elcual comparó una dosis diaria de 20 mg vs. warfarina en pacientes con fibrilaciónauricular y con una puntuación promedio de CHADS2 de 3.2. Este nuevo anti-coagulante oral no mostró inferioridad ante la warfarina en la prevención delEVC, con una tasa similar de sangrado y un menor riesgo de hemorragia intracra-neal y sangrado fatal.
EL rivaroxabán se recomienda en dosis de 20 mg al día en los pacientes confunción renal normal y de 15 mg por día en aquellos con función renal entre 15

114 (Capítulo 7)Tratado de trombosis
y 50 mL/min. Se debe utilizar con precaución en los pacientes con depuracionesmenores de 30 mL/min; se encuentra contraindicado en aquellos con depuracio-nes menores de 15 mL/min.
Tampoco se recomienda su uso en los pacientes con enfermedad hepática sig-nificativa. Cabe mencionar que el estudio ROCKET–AF no incluyó pacientescon función renal menor de 30 mL/min, por lo que en este subgrupo no se encuen-tra probada su seguridad.
Existen pocos datos de seguimiento a largo plazo debido al poco tiempo quelleva en el mercado mundial.
El rivaroxabán puede afectar el tiempo de protrombina y el INR, pero los nive-les de éstos no se correlacionan con el grado de anticoagulación. No se ha mostra-do una manera efectiva de revertir su efecto.
Debido a su vida media corta y a que sólo se administra una vez al día, tienela desventaja de que se debe cumplir muy bien con el horario, así como adminis-trarlo después del alimento, ya que si se consume en ayuno su biodisponibilidadpuede disminuir hasta 20%.34
Apixabán
Es otro de los inhibidores del factor Xa. Este fármaco se comparó en dosis de 5mg vs. la warfarina en el estudio ARISTOTLE, mostrando ser superior a la warfa-rina en la prevención del EVC y el embolismo sistémico, así como una menorincidencia de sangrado y EVC hemorrágico. Dicho estudio también mostró unbeneficio al disminuir la mortalidad después de un seguimiento de 1.8 años vs.la warfarina, con una P = 0.047.
Se recomiendan las dosis bajas del fármaco —2.5 mg cada 12 h— en los pa-cientes que cuenten con dos de los siguientes tres factores: edad mayor de 80 años,peso igual o menor de 60 kg y creatinina sérica de 1.5 a 2.5 mg/dL.35 En los pacien-tes con creatinina sérica mayor de 2.5 mg/dL no se recomienda su uso, ya que estetipo de pacientes no fueron incluidos en los protocolos de investigación.
El otro estudio importante del apixabán en pacientes con fibrilación auriculares el AVERROES, en el cual se comparó el apixabán con el ácido acetilsalicílico(ASA) en pacientes con contraindicación para recibir warfarina. El apixabán dis-minuyó de manera significativa el riesgo de EVC al compararse con el ASA, sinaumentar de manera significativa el riesgo de sangrado o de hemorragia intracra-neal.
Agentes antiplaquetarios
Aunque no son tan efectivos como la warfarina, los agentes antiplaquetarios tie-nen alguna eficacia en la reducción del EVC. El ASA confiere una reducción de

115Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
20% del riesgo de EVC, con un incremento concomitante del riesgo de sangrado,por lo que no queda claro el beneficio neto cuando se ponen en balance ambosaspectos. En las guías del ACCP–9 dicho balance lleva a la recomendación de quees preferible que los pacientes con un CHADS2 de 0 no tomen ASA.36
La dosis óptima de ASA aún no está clara, aunque la dosis de 81 a 325 mg pue-de ser efectiva, y tampoco existe evidencia de que exista una mayor eficacia enmayores dosis, si bien se prefiere una dosis menor para minimizar los efectos se-cundarios.
El ASA combinado con una dosis completa de warfarina es a menudo utilizadoen los pacientes que además cuentan con indicaciones para anticoagulación,como la enfermedad arterial coronaria. Los pacientes seleccionados con ciertascondiciones, como síndrome coronario agudo o colocación de stent coronario,pueden requerir una combinación de antiagregantes plaquetarios con warfari-na.37
La terapia antiplaquetaria dual con ASA y clopidogrel en pacientes con FA fueevaluada en los estudios ACTIVE. El estudio ACTIVE W comparó la warfarinacon el ASA–clopidogrel, apreciándose que el anticoagulante fue superior a la te-rapia dual para la prevención del EVC. Los índices de infarto fueron menores enel grupo de warfarina y el índice de sangrado fue similar en ambos grupos, porlo que la combinación de ASA–clopidogrel no se debe emplear en lugar de la an-ticoagulación en los pacientes que puedan tomar warfarina.38
El estudio ACTIVE A fue llevado a cabo con pacientes que no eran elegiblespara warfarina, por lo que se comparó el uso de ASA vs. ASA más clopidogrel;asimismo, se demostró una mayor eficacia en la prevención del EVC en el grupode la terapia dual que en el grupo con ASA solo; sin embargo, en la terapia dualse duplicaron los eventos de sangrado.
REFERENCIAS
1. Kannel WB, Abbott RD, Savage DD, McNamara PM: Epidemiological features of chro-nic atrial fibrillation: The Framingham Study. N Engl J Med 1982:306(17):1018–1022.
2. Lyp GY, Rumley A, Dunn FG: Plasma fibrinogen and fibrin D–dimer in patients with atrialfibrillation: effects of cardioversion to sinus rhythm. Am J Cardiol 1995;51(3):245–251.
3. Kamath S, Chin BS, Blan AD: A study of platelets activation in paroxysmal, persistent,and permanent atrial fibrillation. Blood Coagul Fibrinol 2002;13(7):627–636.
4. Hart RG, Haerin JL: Atrial fibrillation and stroke: concepts and controversies. Stroke2001;32(3):803–808.
5. Wolf PA, Abbott RD, Kannel WB: Atrial fibrillation as an independent risk factor forstroke: the Framingham Study. Stroke 1991;22(8):983–988.
6. Flegel KM, Shipley MJ, Rose G: Risk of stroke in non–rheumatic atrial fibrillation. Lancet1987;1(8532):526–529.
7. Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis ofpooled data from live randomized controlled trials. Arch Intern Med 1994;154:1449–1457.

116 (Capítulo 7)Tratado de trombosis
8. Hart RG, Pearce LA, Rothbart RM, McAnulty JH: Stroke with intermittent atrial fibri-llation: incidence and predictors during Aspirin therapy. Stroke Prevention in Atrial Fibri-llation Investigators. J Am Coll Cardiol 2000;35(1):183–187.
9. Saxena R, Lewis S, Berge E, Sandercock PA: Risk of early death and recurrent stroke andeffect of heparin in 3 199 patients with acute ischemic stroke and atrial fibrillation in theInternational Stroke Trial. Stroke 2001;32(10):2333–2337.
10. Lin HJ, Wolf PA, Kelly Hayes M et al.: Stroke severity in atrial fibrillation. the Framing-ham Study. Stroke 1996;27(10):1760–1764.
11. Marini C, De Santis F, Sacco S et al.: Contribution of atrial fibrillation to incidence andoutcome of ischemic stroke: results from a population–based study. Stroke 2005;36(6):1115–1119.
12. Kimura K, Minematsu K, Yamaguchi T: Atrial fibrillation as a predictive factor for se-vere stroke and early death in 15 831 patients with acute ischaemic stroke. J Neurol Neuro-surg Psychiatry 2005;76(5):679–683.
13. Steger C, Pratter A, Martinek Breguel M et al.: Stroke patients with atrial fibrillationhave a worse prognosis than patients without data from the Austrian Stroke Registry. EurHeart J 2004:25(19):1734–1740.
14. Kopecky SL, Gersh BJ, McGoon MD: The natural history of line atrial fibrillation. A pop-ulation–based study over three decades. N Engl J Med 1987;317(11):669–674.
15. Gage BF, Waterman AD, Shanon W: Validation of clinical classification schemes for pre-dicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001;285(22):2864–2870.
16. Lip GY Nieuwlaat R, Pisters R: Refining clinical risk stratification for predicting strokeand thromboembolism in atrial fibrillation using a novel risk factor–based approach: theEuro Heart Survey on Atrial Fibrillation. JAMA 2001;285(22):2864–2870.
17. Lip GY, Frison L, Halperin JL: Comparative validation of a novel risk score for predictingbleeding risk in anticoagulated patients with atrial fibrillation: The HAS–BLED. J Am CollCardiol 2011;57:173–180.
18. RJ, Pearce LA, McBride R, Rothbart MD: Factors associated with ischemic stroke dur-ing Aspirin therapy in atrial fibrillation: analysis of 2 012 participants in the SPAF I–III clin-ical trials. Stroke 1999;30(6)1223–1229.
19. Stroke Prevention in Atrial Fibrillation Study. Final results. Circulation 1991;84(2)527–539.
20. Van Walraven C, Hart RJ, Wells GA: A clinical prediction rule to identify patients withatrial fibrillation and a low risk for stroke while taking Aspirin. Arch Intern Med 2003;163(8):936–943.
21. Van Gelder I, Hagens V, Bosker H: A comparison of rate control and rhythm control inpatients with recurrent persistent atrial fibrillation. N Engl J Med 2002;347(23):1825–1833.
22. Wyse DG, Waldo AL, Di Marco JP: A comparison of rate control and rhythm control inpatients with atrial fibrillation. N Engl J Med 2002;347(23):1825–1833.
23. Lip GYH: Recommendations for thromboprophylaxis in the 2012 focused update of theESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013;11:615–626.
24. Ezekowitz MD, Brideges SL, James KE: Warfarin in the prevention of stroke associatedwith nonrheumatic atrial fibrillation. N Engl J Med 1992;327(20):1406–1412.
25. Connolly SJ, Ezekowitz MD, Yusuf S: Dabigatran versus warfarin in patients with atrialfibrillation. N Engl J Med 2009;361(12):1139–1151.
26. Legrand M, Mateo J, Aribaud A: The use of dabigatran in elderly patients. Arch InternMed 2012;172(5):397–402.

117Fibrilación auricular y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
27. Fuster V, Ryden LE, Cannom DS: 2011 ACCF/AHA/HRA focused updates incorporatedinto the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrilla-tion. J Am Coll Cardiol 2011;57:e101.
28. Skanes AC, Healey JS, Cairns JA: Focused 2012 update of the Canadian CardiovascularSociety Atrial fibrillation guidelines: recommendations for stroke prevention and raterhythm control. Can J Cardiol 2012;28:125–136.
29. You JJ, Nieuwlaat R, Pisters R: Antithrombotic therapy and prevention of thrombosis. 9ªed. American College of Chest Physicians evidence–based clinical practice guidelines.Chest 2012;141:e531S–e575S.
30. Camm AJ, Lip GY, de Caterina R: 2012 focused update of the ESC Guidelines for themanagement of atrial fibrillation: an update of the 2010 ESC guidelines for the managementof atrial fibrillation. Europace 2012;14:1385–1413.
31. Lip GYH: Recommendations for thromboprophylaxis in the 2012 focused update of theESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013;11:615–626.
32. Patel MR, Mahaffey KW, Garg J: Rivaroxaban versus warfarin in nonvalvular atrialfibrillation. N Engl J Med 2011;365:883–891.
33. Granger CB, Alexander JH, McMurray JJ: Apixaban versus warfarin in patients withatrial fibrillation. N Engl J Med 2012;365:981–982.
34. Connoly SJ, Eikelboom J, Joyner C: Apixaban in patients with atrial fibrillation. N EnglJ Med 2011;364:806–817.
35. Lip GYH: The role of Aspirin for stroke prevention in atrial fibrillation. Nat Rev Cardiol2011;8:602–606.
36. Sweeney KG, Gray DP, Steele R: Use of warfarin in non–rheumatic atrial fibrillation: acommentary from general practice. Br J Gen Pract 1995;45:153–158.
37. Haeley JS, Hart RG, Pogue J: Risks and benefits of oral anticoagulation compared withclopidogrel plus Aspirin in patients with atrial fibrillation: Clopidogrel Trial with Irbesartanfor Prevention of Vascular Events (ACTIVE–W). Stroke 2008;39:1482–1486.

118 (Capítulo 7)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
8Enfermedad carotídea
Eduardo Perusquia Ortega, Ángel Lee, Yolanda Aburto,Jorge Santos, Marco Antonio Zenteno
En el siglo IV a.C. Hipócrates describió la asociación entre enfermedad de la arte-ria carótida y hemiplejía contralateral,1 y en 1855 Gull reconoció la enfermedadvascular extracraneal como causa de infarto cerebral.2 Sin embargo, fue hasta 1953cuando de Bakey realizó la primera endarterectomía carotídea (EC) exitosa3 y has-ta 1977 cuando Mathias reportó la primera angioplastia carotídea (AC).4
INTRODUCCIÓN
La frecuencia de la aterosclerosis carotídea depende de la población estudiada,pero se considera una prevalencia de estenosis carotídea � 50%. Los datos delestudio de Framingham indican que a los 75 años de edad es de 9% entre los varo-nes y de 7% entre las mujeres.5 Por otro lado, su prevalencia es de 35% en indivi-duos con soplo carotídeo,6 de 22% en pacientes con enfermedad coronaria7 y de14% en pacientes con enfermedad arterial periférica.8
Alrededor de un tercio de las lesiones ateroscleróticas extracraneales se locali-zan en la bifurcación de la carótida, y se estima que esta ubicación es responsablede entre 20 y 25% de todos los eventos isquémicos cerebrales.9 Una lesión carotí-dea puede ser tratada de forma farmacológica o quirúrgica, lo cual se denominaprevención primaria cuando el paciente no ha presentado síntomas isquémicosy prevención secundaria cuando el paciente ya es referido por un infarto cerebral.Por esta razón, el diagnóstico y el manejo de las lesiones carotídeas son puntosparticularmente importantes dentro del campo de la patología vascular.
119

120 (Capítulo 8)Tratado de trombosis
Es capital tener en mente que los objetivos de la prevención en estos pacientesconsisten en limitar la progresión de la aterosclerosis y reducir el riesgo de infartocerebral y de eventos vasculares en otros territorios. Por consiguiente, la revascu-larización de la carótida (endovascular o quirúrgica) es únicamente un aspectodel esquema de tratamiento de la arteriopatía carotídea, ya que también es necesa-rio lograr un control estricto de los factores de riesgo y administrar un tratamientoantitrombótico.
El tema de la aterosclerosis carotídea y su tratamiento es extremadamente am-plio, por lo que este capítulo se limita a tratar el marco general de la enfermedady los elementos básicos para poder decidir en qué casos recurrir a un tratamientoarmado (quirúrgico o endovascular). Para aspectos más fundamentales acerca dela anatomía vascular, la aterogénesis y el manejo médico de la aterosclerosis, sesugiere recurrir a las obras de referencia.
IMPORTANCIA DEL PROBLEMAY SU ABORDAJE TERAPÉUTICO
El evento vascular cerebral (EVC) es la tercera causa de mortalidad en el mundo,antecedido por los padecimientos cardiacos y el cáncer. Se calcula que para el año2050 se incrementará 25% el número de individuos afectados, debido a los cam-bios en la edad, los hábitos y la distribución étnica.10
Se considera que cerca de 25% de los EVC se relacionan con enfermedad oclu-siva de la arteria carótida común e interna cervical.9 El doble efecto trombóticoy embólico debe ser considerado en la etiopatogenia y la fisiopatología de las le-siones de asiento extracraneal.
El desarrollo de la terapia endovascular ha sido particularmente espectacularen el campo de la enfermedad vascular cerebral. En general, ha surgido como unaalternativa al tratamiento quirúrgico “clásico”, es decir, a la cirugía con bisturí.Por otro lado, la neurocirugía vascular se ha beneficiado con técnicas como losabordajes key–hole, la especialización del manejo anestésico y de terapia inten-siva, etc. La terapia endovascular ha establecido en algunos casos un rango equi-parable y en otros casos superior, en cuanto al manejo de mínima invasión de laenfermedad vascular cerebral.
El recurso indiscriminado a la EC en décadas anteriores llevó a los diferentesgrupos a un análisis profundo en 1985 y a numerosos ensayos aleatorizados paradefinir el verdadero papel de esta técnica. El NASCET11 y el ACAS12 demostra-ron que la cirugía reduce sustancialmente el riesgo de infarto e incrementa la su-pervivencia en pacientes sintomáticos con estenosis carotídea > 50% y estenosismayores de 60% en asintomáticos.11–13 La EC constituye el estándar de oro en elmanejo de la enfermedad aterosclerótica carotídea extracraneal.

121Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
En los últimos años la AC surgió como alternativa en los pacientes con altoriesgo de complicaciones quirúrgicas, lo que ahora permite definir en muchos ca-sos la complementariedad de ambas técnicas, más que una actitud excluyente. LaAC es un procedimiento que permite restablecer el calibre de la arteria carótida,y existen dos variantes: la angioplastia con balón y la angioplastia con colocaciónde stent.
Si bien la indicación primordial de dicho procedimiento es la estenosis de ori-gen aterosclerótico, se han reportado casos de angioplastia con otras indicacio-nes, como displasia fibromuscular,14 arteritis de Takayasu,15,16 neurofibromato-sis,17 estenosis posirradiación18–20 y aneurismas de la carótida cervical.21
La AC comenzó como una simple recalibración del vaso con un balón (angio-plastia percutánea transluminal con balón);22 sin embargo, la angioplastia simplecon balón tiene varios inconvenientes, incluidos la disección de la íntima y el em-bolismo distal por fractura de la placa. Apoyados en la experiencia y el éxito dela angioplastia coronaria23 y periférica24 se comenzaron a realizar angioplastiascon colocación de stent en la década de 1980, y desde entonces ha habido un au-mento exponencial del número de AC realizadas en todo el mundo.25
Esto explica el interés de tantas especialidades por la AC. El número de pa-cientes potenciales es elevado y los intereses económicos prevalecen sobre lapreocupación científica; además, acceder a la bifurcación carotídea es relativa-mente fácil, lo cual ha originado que cualquier cirujano se sienta capaz de operaruna carótida y cualquier cardiólogo o radiólogo de colocar un stent. De ahí el grandebate acerca de la justificación o no justificación de recurrir a un procedimientonuevo como alternativa a un procedimiento establecido que en teoría es eficaz,eficiente y con un riesgo aceptable.
Pero, ¿a quién compete el territorio carotídeo? Se ha venido pregonando desdehace tiempo, y ahora coinciden en esto varios autores y asociaciones.26,27 La ate-rosclerosis carotídea es una enfermedad cerebrovascular, no cardiovascular ovascular periférica (por cierto, afecta al sistema nervioso central). El tratamientoendovascular de la estenosis carotídea no es una cuestión meramente técnica; setrata de un padecimiento cuyo manejo integral (diagnóstico, manejo médico ymanejo endovascular o quirúrgico) es asunto de un grupo multidisciplinario enneurociencias. En varios lugares algunos cardiólogos intervencionistas se dedi-can a colocar stents carotídeos de manera indebida. No es de imaginar la reacciónde estos mismos cardiólogos si los neurorradiólogos intervencionistas hicieranangioplastias coronarias o los neurocirujanos realizaran esternotomías. Si el neu-rocirujano vascular domina la técnica de la anastomosis cuando realiza un bypassextracraneal e intracraneal, ¿por qué no se dedica a operar puentes coronarios?
Conceptualmente, el fenómeno de invasión por parte de especialidades noadecuadas muestra que todos olvidan que el aspecto técnico de cualquier inter-vención representa únicamente una de las responsabilidades del médico, quien

122 (Capítulo 8)Tratado de trombosis
también debe determinar qué tipo de paciente debe someterse a dicha interven-ción, proveer el tratamiento antes y después del procedimiento, y manejar laseventuales complicaciones. Determinar si una lesión carotídea deber ser tratadao no requiere conocimiento del riesgo del tratamiento, de los síntomas de un in-farto cerebral, de la interpretación de imágenes neurorradiológicas y de la histo-ria natural del padecimiento cuando se trata médicamente. Incluso para el espe-cialista en neurociencias no es fácil seleccionar de manera apropiada a loscandidatos a tratamiento. Las complicaciones de las intervenciones neurovascu-lares son complejas y la mayoría no se presentan durante la realización de angio-grafías coronarias. Los EVC por émbolos distales requieren conocimiento yexperiencia en neurología vascular. Los episodios de hipoperfusión e hiperperfu-sión que aparecen después de una AC implican el conocimiento de los cuidadoscríticos del paciente neurológico.28
Si bien en el campo de los aneurismas cerebrales las publicaciones internacio-nales han definido poco a poco el papel actual de la terapia endovascular, aún nose han establecido las bases para dicha técnica en el campo de la enfermedad vas-cular isquémica extracraneal.
Antes de definir qué pacientes se deben tratar y cuáles no se debe aclarar unconcepto muy trillado, pero que amerita una mayor precisión.
ESTENOSIS CAROTÍDEA“SINTOMÁTICA” Y “ASINTOMÁTICA”
Según la Real Academia Española de la Lengua, en el campo de la medicina unsíntoma es un “fenómeno revelador de una enfermedad”, y en el lenguaje comúnes la “señal, indicio de algo que está sucediendo o va a suceder”. Además, aunqueparezca una perogrullada, se puede decir que toda carótida que ahora es sintomá-tica ¡algún día fue asintomática!, por lo que, a pesar de la tan citada división decarótidas sintomáticas y carótidas asintomáticas, parece más lógica una divisiónentre placas estables y placas inestables, porque finalmente lo que interesa es loque le va a pasar al paciente y si es un paciente con riesgo de presentar un eventoo sin riesgo de padecerlo.
Aun así, si se quisiera conservar la vieja división entre carótida sintomática ycarótida asintomática, la pregunta sería: ¿para quién es asintomática?
Se considera que una carótida es sintomática cuando se presenta un evento ce-rebrovascular isquémico en el territorio que depende de ese vaso, y se consideraasintomática cuando no ha dado síntomas isquémicos, es decir, que se descubrede manera incidental, por ejemplo durante un ultrasonido (US) de rastreo por laexistencia de un soplo cervical o durante la evaluación vascular por un evento enotro territorio (que puede ser coronario).

123Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
CARÓTIDA Y COGNICIÓN
Un punto que se ha ignorado hasta ahora en casi todos los estudios es la valora-ción neuropsicológica de los pacientes con estenosis carotídea. Sin embargo, elhecho ha atraído la reciente atención de algunos grupos. Existen algunas pruebasde que la estenosis carotídea se asocia con el deterioro cognitivo.29
La Universidad de San Francisco30 estudió una cohorte de 4 006 pacientesdiestros, mayores de 65 años de edad y sin antecedentes de infarto cerebral isqué-mico, ataque isquémico transitorio o EC. A todos se les realizó una prueba Mini–Mental modificada, un estudio neuropsicológico simple. Se definió la presenciade alteraciones cognitivas con un puntaje menor de 80 en esta prueba y la existen-cia de deterioro con una disminución promedio de más de un punto por año. Seles dio seguimiento a los pacientes durante cinco años. El estudio demostró quelas personas con estenosis carotídea del lado izquierdo mayor de 75% tienen ma-yor posibilidad de presentar alteraciones del estado cognitivo y un deterioro dela función a lo largo del tiempo respecto de las personas sin enfermedad carotí-dea. Esta asociación permanece aun cuando los investigadores ajustaron los da-tos tomando en cuenta otros factores de riesgo y la enfermedad carotídea derecha;esto indica que lo que realmente se asocia con los problemas cognitivos es la este-nosis carotídea izquierda y no la aterosclerosis generalizada.30
Otra investigación realizada por la universidad de Tromsø, en Noruega,31 de-mostró que la estenosis carotídea se asocia con un rendimiento neuropsicológicomenor. El estudio incluyó a 189 pacientes con estenosis carotídea demostrada porultrasonido y a 201 individuos sin enfermedad carotídea. También se registraronlas lesiones isquémicas por resonancia magnética (hiperintensidades de la sus-tancia blanca e infartos lacunares y corticales) y se evaluaron la atención, la velo-cidad psicomotora, la memoria, el lenguaje, la velocidad de procesamiento de lainformación, las funciones motoras y la depresión. En los pacientes con estenosiscarotídea los resultados en cuanto a atención, velocidad psicomotora y memoriafueron desfavorables.
En las funciones motoras, el lenguaje, la velocidad de procesamiento de la in-formación y la depresión no hubo diferencias significativas. Esta diferencia nopudo explicarse por la presencia de una mayor proporción de lesiones “silencio-sas” mediante la resonancia magnética, lo cual dificulta que el deterioro cogniti-vo se explique por émbolos “silenciosos”.
Es necesario tratar de identificar a los pacientes que se verían beneficiados conun tratamiento quirúrgico, con respecto de los que no obtendrían mejoría. Entrelos posibles enfoques para identificar a los pacientes con alto riesgo de ACV yque presentan una mayor probabilidad de beneficiarse con la práctica de una ciru-gía cabe destacar el Doppler transcraneal (identificación de pacientes con signosde microembolias),32 la evaluación hemodinámica (identificación de pacientes

124 (Capítulo 8)Tratado de trombosis
con flujo colateral inadecuado)33 y las técnicas de imagen (identificación de pla-cas ateroscleróticas inestables).34 Por consiguiente, en los pacientes con eventosisquémicos transitorios o con ACV isquémicos no discapacitantes el procedi-miento debe realizarse en el transcurso de dos semanas desde que aparecen lossíntomas.35
Una vez más se hace hincapié en que se trata de una patología neurovascularque requiere un equipo multidisciplinario, en el cual el cirujano vascular o el car-diólogo intervencionista quedarían reducidos a “ejecutores de procedimientos”,ya que no es una patología cardiológica o “vascular pura”, como las varices.
Asimismo, aunque se mantenga la división de carótidas “sintomáticas” y“asintomáticas”, es indispensable abordar la noción de placa “estable” y placa“inestable”.
LA PLACA DE ATEROSCLEROSIS CAROTÍDEA
Imagen de la placa
Ningún estudio de imagen es completamente fidedigno para visualizar al mismotiempo:
1. La placa.2. El diámetro residual del vaso (porcentaje de estenosis).3. Las consecuencias hemodinámicas de dichas anomalías, tanto a nivel del
vaso cervical como a nivel intracraneal.
Los estudios de imagen se deben complementar, y no excluir, y menos debe repo-sar el diagnóstico y el manejo de un paciente sobre un solo estudio. ¡Es como sise pretendiera tratar una leucemia exclusivamente con los resultados de la biome-tría hemática!
Por ejemplo, en un caso hipotético, la estenosis de la carótida cervical puedeser mostrada por las velocidades posestenosis en el Doppler carotídeo a color. Anivel intracraneal el Doppler transcraneal permite observar la apertura de colate-rales basales del polígono de Willis, la angiografía por sustracción digital facilitael descubrimiento de la apertura de colateralidad leptomeníngea a nivel de la con-vexidad y los estudios de perfusión pueden evaluar la hipoperfusión cortical (TCperfusión, técnicas de perfusión de RM, SPECT y PET) y predecir el éxito o fra-caso de una cirugía de revascularización.
La imagen está al servicio del paciente, no del bisturí, y los estudios tienen unvalor diagnóstico, pero también pronóstico y auxiliar en la toma de decisiones.

125Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Ultrasonido carotídeo
Indicaciones
� Pacientes sintomáticos. Evento neurológico de naturaleza vascular (infartocerebral, TIA, amaurosis fugax).
� Pacientes asintomáticos. Masa cervical pulsátil, soplo cervical, valoraciónprequirúrgica (bypass coronario).
Modalidades de realización:
� Imagen modo B en escala de grises.� Doppler de flujo en color (axial y transversal).� Velocidades espectrales (plano longitudinal).
Hallazgos36
Engrosamiento íntima–mediaSe trata de la distancia que existe entre la interfase luz vascular y la íntima y la inter-fase media y la adventicia. Es un engrosamiento de estas dos estructuras que cons-tituye un marcador de riesgo vascular y que traduce la existencia de enfermedadaterosclerótica en el lecho coronario, periférico y cerebrovascular. La Asociaciónde Cardiólogos de EUA la recomienda como marcador de aterosclerosis.37
Existencia de placa ateromatosaEl ultrasonido proporciona información acerca de la extensión, la localización yel grado de estenosis en las lesiones de más de 40% de estrechamiento de la luzarterial.38 El tamaño de la placa, es decir, el grosor de la misma, que determinael porcentaje de amputación de la luz arterial, corresponde a las alteraciones he-modinámicas de las velocidades ya conocidas. También hay que determinarcómo es la superficie de la placa y su ecogenicidad.
Parámetros que se deben tomar en cuenta
Durante el ultrasonido se deben determinar:
� La presencia de placa en las imágenes de escala de grises o de imágenes deDoppler a color.
� La velocidad sistólica pico (PSV o peak systolic value) de la arteria carótidainterna (ACI).

126 (Capítulo 8)Tratado de trombosis
Cuadro 8–1. Clasificación de la placa39
Hemodinámica(% estenosis)
Morfológica Superficie
H1, leve (< 50%) P1, homogénea S1, lisaH2, moderada (de 50 a 69%) P2, heterogénea S2, irregular (defecto < 2 mm)H3, severa (de 70 a 95%) S3, ulcerada (defecto > 2 mm)
H4, crítica (de 95 a 99%)H5, oclusiva (100%)
Estos datos permiten distribuir las placas de acuerdo con las diversas clasificacio-nes, como la de Thiele (cuadro 8–1).39
También permiten estratificar la estenosis en una de las seis clases recomenda-das por la Sociedad de Radiólogos en Ultrasonido, de EUA (cuadro 8–2).
Ultrasonido y otras modalidades de diagnóstico
En estenosis mayores de 70% los estudios hechos en laboratorios vasculares con-fiables reportan entre 87 y 98% de sensibilidad, con 59 a 75% de especificidad,41
en comparación con la angiografía por sustracción digital, que continúa siendola modalidad de referencia. Las cifras de la angiorresonancia magnética (MRA)con medio de contraste van de 92 a 96% de sensibilidad y de 58 a 76% de especifi-cidad.41
Cuadro 8–2. Clasificación de las estenosis40
Arteria normal PSVACI < 125 cm/segNo hay placaNo hay engrosamiento intimal
Estenosis < 50% PSVACI < 125 cm/segPlacaEngrosamiento intimal
Estenosis de 50 a 69% PSVACI < 125 � 230 cm/segPlaca visible
Estenosis � 70% � preoclusión PSVACI > 230 cm/seg
Placa visibleEstrechamiento de la luz
Preoclusión Estrechamiento marcado de la luz en el ultrasonido Dopplera color
Oclusión completa No hay luz detectable en el ultrasonido Doppler de escalade grises
No hay flujo en el ultrasonido Doppler espectral y el ultraso-nido Doppler a color

127Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
A pesar de estas cifras, que denotan que los métodos de imagen no son exclu-yentes entre sí sino complementarios, hay grupos con argumentos de costo queindican que los pacientes pueden ser sometidos a un procedimiento de correcciónde la estenosis carotídea.42,43 Pero esto no parece adecuado, ya que el ultrasonidono permite evaluar las lesiones altas (por arriba de C3) ni valorar la circulaciónintracraneal y, por lo tanto, una estenosis intracraneal asociada, la existencia y elvalor de la circulación colateral, entre otros. Incluso el grupo del estudio NAS-CET recomienda la valoración de la colateralidad por angiografía para evaluarel riesgo de infarto cerebral y de ataque isquémico transitorio, ya que el riesgode evento isquémico casi se quintuplica en ausencia de colateralidad por angio-grafía.44
Los autores tratan un aneurisma incidental gigante de la circulación posterioren una paciente con angiodisplasia bilateral de la carótida cervical que un ciruja-no vascular había programado para EC, pensando que se trataba de una estenosisateromatosa.
Los estudios recientes demuestran que basar la indicación quirúrgica sólo enlos resultados del ultrasonido puede resultar insuficiente y costoso para el pacien-te. En una investigación de una universidad escocesa se observó que hasta en 14%de los pacientes que hubieran sido programados para cirugía con base en el ultra-sonido la cirugía hubiera sido innecesaria.45
Angiotomografía (CTA)
Las ventajas de la angiotomografía para la valoración de la estenosis carotídeaincluyen su carácter “no invasor” y “ambulatorio”, y una ventaja del ultrasonidorelacionada con su amplia disponibilidad en muchos sitios, así como una ventajade la MRA, que equivale a su carácter anatómico.46
Tomando como referencia la angiografía por sustracción digital, en cuanto ala cualidad para predecir de la angiotomografía, en el estudio de la estenosis caro-tídea mayor de 70% la sensibilidad es perfecta, aunque con una especificidad de63%; el valor predictivo negativo en los grados de estenosis menores de 70% escasi de 100%,47 es decir, cuando hay una estenosis grave, ésta se ve; cuando nose ve una estenosis severa en el estudio, es que no la hay; pero, si se ve algo queparece una estenosis severa, puede que en realidad no la haya.
A pesar de las bondades aparentes de la angiotomografía, no hay que olvidarque, aunque se trata de un método excelente para la detección de la enfermedadcarotídea grave, sobre todo la oclusiva, los estudios publicados acerca de la an-giotomografía en enfermedad carotídea indican que una cuarta parte de los estu-dios publicados reúnen criterios metodológicos satisfactorios,48 por lo que “notodo lo que brilla es oro” y no es posible basarse exclusivamente en esta modali-dad para valorar a un paciente.

128 (Capítulo 8)Tratado de trombosis
Este método de rastreo aún tiene algunos puntos no satisfactorios, es insufi-ciente en estenosis no graves49 y dificulta la evaluación de las ulceraciones y lacalidad de la placa en general; no obstante, se cree que puede mejorar estos aspec-tos en el futuro.50
Angiorresonancia (MRA)
Las ventajas de la MRA en la evaluación de la estenosis carotídea son similaresa las de la angiotomografía, en especial su carácter “no invasor”.
Al principio la MRA recurrió a la técnica de tiempo de vuelo (time–of–flight),pero hace poco tiempo se introdujo la técnica de angiorresonancia magnética conmedio de contraste (contrast–enhanced MR angiography, o CEMRA). Sin em-bargo, la CEMRA tiende a sobreestimar la estenosis, principalmente por la exis-tencia de variabilidad interobservador. Para la detección de estenosis grave, enun estudio comparativo contra la angiografía por sustracción digital la CEMRAtuvo una sensibilidad de 93.0% y una especificidad de 80%, con una clasificacióninadecuada de los pacientes en 15.0% de los casos. Aun combinando con un ultra-sonido, la proporción de pacientes mal clasificados asciende a 10%, por lo queson discordantes ambos estudios, lo cual hace necesaria la realización de una an-giografía por sustracción digital en 25% de los casos.51 En lesiones moderadasla MRA sobreestima el grado de estenosis de manera significativa y es un predic-tor inadecuado de la misma; sin embargo, la CEMRA ofrece la opción de obser-var de manera simultánea el arco aórtico y los vasos del cuello, lo cual proporcio-na variaciones anatómicas y revela lesiones en tandem inadvertidas, lo cualpuede de ser de utilidad para la planeación preoperatoria del tratamiento.52
Angiografía por sustracción digital (DSA)
Función de la angiografía
Se ha dicho mucho que la angiografía es un estudio “invasor”; sin embargo, hoypor hoy sigue siendo el método de referencia para la toma de decisiones en patolo-gía carotídea y el método de detección y rastreo. En realidad, para un paciente esmás invasor tomar una decisión inadecuada que realizar un estudio cuya morbili-dad neurológica en la práctica clínica es extremadamente baja (menor de 1%).53
En general el ultrasonido, la angiotomografía y la angiorresonancia muestranuna exactitud similar en el diagnóstico de la estenosis carotídea sintomática. Nin-guna técnica por sí sola es lo suficientemente exacta para reemplazar a la angio-grafía por sustracción digital. Dos técnicas no invasoras usadas en combinación,con la ayuda de una tercera en caso de discordancia parecen ofrecer resultadosmás exactos, pero pueden aún conducir a errores diagnósticos.54

129Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 8–3. Comparación entre los diferentesmétodos de imagen de la placa carotídea
US CTA MRA DSA
% de estenosis (sí estenosis leve) + + – +% de estenosis (sí estenosis grave) ++ ++ + ++Inestabilidad de la placa ++ – ¿? –Ulceración de la placa ++ – ¿? +Observador dependiente – + + ++Ambulatorio ++ ++ ++ +
%: grado, porcentaje; ++: exactitud adecuada, ventaja considerable; +: exactitud pasable, ventajamediocre; –: no exacto, desventaja; ¿?: se desconoce, en curso de investigación.
Calidad y características de la placa
Además del grado de estenosis, la irregularidad superficial de la placa es un factorde riesgo independiente de EVC.55–57 Este hecho es bien conocido, pero general-mente ignorado por muchos grupos al momento de tomar una decisión.
Las placas de los pacientes asintomáticos son radicalmente diferentes de lasde los pacientes sintomáticos.58 La estabilidad de la placa está influida por facto-res mecánicos locales y hemodinámicos, como la movilidad de la placa (plaquemotion)38 y la tensión de fricción (shear stress).59
Además, existen métodos computarizados para determinación, y algunos fac-tores ultrasonográficos, como la ecogenicidad de la placa, que permiten detectarlas placas carotídeas de alto y de bajo riesgo, que por demás se presentan en pa-cientes sintomáticos y en pacientes asintomáticos.
La morfología de la placa ha sido objeto de estudio desde hace más de 10años,60 así como otros factores, como la hemorragia intraplaca y su ulceración,el mayor número de células musculares apoptósicas y la capa fibrosa más delga-da.61
También hay factores como la no calcificación de la placa, que se asocia conun menor número de eventos isquémicos que la placa calcificada.62
¿Después de una década y media sonsólo válidos los criterios de NASCET?
Aunque los criterios de NASCET se han difundido y respetado cual palabra divi-na, son cuestionables.63,64 Los estudios multicéntricos que se mencionan másadelante tienen un valor intrínseco importante, aunque se debe considerar que enesa época sólo se tomó como criterio de tratamiento el diámetro residual (porcen-

130 (Capítulo 8)Tratado de trombosis
taje de estenosis) calculado en la angiografía por sustracción digital, que propor-ciona una visión exclusivamente en un plano (dos dimensiones). La placa es im-portante y no sólo la “cantidad” de estenosis, lo cual constituye un tema abordadoanteriormente por otros autores. Dicho de una manera más coloquial, un autoraustraliano afirmó que hay que “fijarse más en la dona y no tanto en el hoyo”.64
Ante todo, hay que recordar que el porcentaje de estenosis que sirvió como cri-terio de selección en el estudio NASCET se determinó mediante una única moda-lidad de imagen (angiografía).
En la actualidad la detección de la estenosis carotídea es cada día más frecuen-te y está relacionada con el incremento en el número de estudios de imagen reali-zados para diversos padecimientos (diagnóstico de la estenosis carotídea sólocomo hallazgo) o mediante el rastreo durante las evaluaciones preventivas del es-tado de salud (“chequeo”). Es más o menos frecuente que una estenosis grave ouna oclusión completa diagnosticadas por el ultrasonido sean en realidad vasosnormales por angiografía, con casos de estrechamiento angiográfico importantecon hallazgos normales en el Doppler.
Además, gracias a la experiencia se sabe que una estenosis que se cuantificaen determinado porcentaje (50%, más de 70%, etc.) podrá desaparecer o incre-mentarse de acuerdo con la proyección en la que se visualice el vaso.
Por lo tanto, la determinación de la estenosis debe establecerse después de unminucioso estudio de las imágenes y confrontación entre los hallazgos de las dife-rentes modalidades.
Un criterio hasta ahora ignorado por los diversos grupos de trabajo a nivel in-ternacional, y sobre el que nuestro grupo ha insistido en foros nacionales e inter-nacionales, es la inestabilidad de la placa, por lo que los estudios recientes hanhecho énfasis en la valoración multimodal de la placa.65
En realidad, lo que importa es la probabilidad de aparición de un evento isqué-mico por trombosis local o por embolismo distal hacia el parénquima cerebral.
Existen placas con una oclusión de la luz vascular mínima que llegan a produ-cir un evento isquémico. Dichas placas escaparían a la indicación de tratamientoquirúrgico o endovascular si se tomaran exclusivamente en cuenta los criteriosdel estudio NASCET. Estas placas tienen una calidad distinta de la de las placasque no embolizan. La luz vascular está preservada, pero existen alteraciones dela pared vascular que hacen particularmente frágil e inestable a esta placa. Loscriterios de inestabilidad de la placa son conocidos desde hace muchos años porlos ultrasonografistas y, sin ser exhaustivos, se refieren a parámetros, como la he-morragia intraplaca y su ecogenicidad, el movimiento de la placa (plaque mo-tion), el trombo fresco adherido a la pared y la ulceración de su superficie.
Se ve entonces que no entran exclusivamente en juego los criterios cuantitati-vos (porcentaje de estenosis), sino que los criterios cualitativos que se han venidopregonando desde hace algunos años (inestabilidad de la placa) deben ser tam-

131Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
bién tomados en cuenta, a pesar de haber sido excluidos en la mayoría de los estu-dios realizados sobre la estenosis carotídea. Esto ha llevado a acuñar el conceptode síndrome luminal (lumen = luz, en latín) y de síndrome parietal (parietalis =relativo a la pared, en latín).
Existen pacientes en los que la indicación se plantea con respecto a criterioscuantitativos y de porcentaje de estenosis, interesando exclusivamente el diáme-tro de la luz arterial, en cuyo caso se trata de un síndrome luminal.
Hay otros pacientes en cuyo caso se sugiere la indicación con respecto a crite-rios cualitativos y de naturaleza de la placa, interesando exclusivamente la cali-dad de la pared arterial, en cuyo caso se trata de un síndrome parietal.
Endarterectomía carotídea
Desde la publicación en 1991 de los resultados del NASCET11 se ha consideradoa la EC como el estándar de oro de la estenosis carotídea extracraneal; sin embar-go, esto debe considerarse con cautela, ya que NASCET excluye a un numerosogrupo de pacientes que por factores de riesgo específicos no son candidatos idó-neos para la cirugía, lo cual no permite una evaluación objetiva de la enfermedadfuera de ensayos clínicos.11,66
El NASCET estratificó dos grandes grupos de acuerdo con el grado de esteno-sis:
� Grupo 1: de 30 a 69%.� Grupo 2: de 70 a 99%.
Los criterios de exclusión utilizados fueron una EC previa, lesión intracranealquirúrgicamente inaccesible y falla pulmonar, hepática u orgánica; los criteriosde exclusión temporales fueron hipertensión, diabetes, angina inestable, infartodel miocardio de menos de seis meses de evolución, EC contralateral de cuatromeses, síntomas de déficit neurológico progresivo y procedimiento quirúrgicomayor en los últimos 30 días.
En el ACAS12 los pacientes fueron distribuidos de manera aleatoria para reci-bir tratamiento médico o quirúrgico; en él las complicaciones de ictus ipsilateraly muerte a los cinco años fueron de 5.1% para EC y de 11% para el grupo contratamiento médico.
¿Por qué sería necesario otro tipo de procedimiento?
Primero, por que las cifras de morbimortalidad manejadas por NASCET en losensayos clínicos (mortalidad de 1%) son mucho menores que las que se observanen el “mundo real”, es decir, en hospitales diferentes a los de los ensayos, dondeesta mortalidad es hasta dos veces mayor.67,68

132 (Capítulo 8)Tratado de trombosis
Un punto capital a considerar es que los pacientes del estudio NASCET fueronpacientes “poco enfermos”. ¿Qué significa esto? Que se excluyó a los que real-mente estaban “muy enfermos”, que por sus patologías concomitantes eran “ma-los candidatos” para la cirugía. Por ello, las indicaciones y los resultados de lacirugía en este subgrupo de pacientes no se han establecido.
A continuación se mencionan los factores que hacen que sea poco aconsejableque un paciente se someta a EC.
Edad
Los pacientes mayores de 85 años de edad tienen 300% mayor probabilidad demorir que los mayores de 70 años. Goldstein69,70 reportó 7.5% de pacientes asin-tomáticos mayores de 75 años de edad vs. 1.8% de pacientes menores de 75 añosde edad. Por otro lado, el riesgo de infarto del miocardio fue de 6.6% en las perso-nas sintomáticas mayores de 75 años de edad y de 2.3% en las que tenían menosde 75 años. Sin embargo, un análisis estratificado de NASCET71 demuestra quela reducción del riesgo absoluto en el manejo quirúrgico fue de 28.9% en los pa-cientes mayores de 75 años, de 15.1% entre los 65 y los 74 años de edad y de 9.7%en los pacientes menores de 65 años. Por lo tanto, aunque parece que la EC bene-ficia a los individuos mayores, parece razonable pensar que la AC puede ofrecerbeneficios similares a estos pacientes mayores con una menor tasa de complica-ciones.69
Insuficiencia cardiaca congestiva
Los pacientes con insuficiencia cardiaca tienen un mayor riesgo de padecer uninfarto perioperatorio y muerte con EC. Una revisión multicéntrica de pacientesmostró una morbimortalidad de 8.6% en pacientes con IC, en comparación con2.3% de pacientes sin esta enfermedad.
Enfermedad coronaria grave y bypass coronario simultáneo
Constituye el factor más importante a considerar en la evaluación de riesgo perio-peratorio de la cirugía carotídea. La coexistencia de ambas enfermedades (coro-naria y carotídea) determina dilemas de manejo,72,73 ya que la reparación carotí-dea no está exenta de un alto riesgo de complicaciones en la otra. En el análisisde NASCET se determina que quizá la puesta en condiciones generales y cardia-cas ideales mejoró los resultados de la cirugía. La presencia de padecimiento arte-rial carotídeo significativo desempeña un papel importante en el incremento delriesgo para EVC o embolismo en pacientes sometidos a cirugía de bypass.74 Sinembargo, se ha demostrado que la estenosis carotídea mayor de 75% de estenosis

133Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
debe considerarse como un factor independiente de EVC durante la cirugía. Enun estudio multicéntrico el riesgo de manejo conjunto de ambas modalidades fuede 18.7%, comparado con la cirugía carotídea aislada (2.1%).70 Por otro lado, larealización de EC antes de la corrección del problema cardiaco también presentaaltos niveles de riesgo de complicaciones durante la operación.73,75 Por ejemplo,en estos subgrupos de alto riesgo las complejas maniobras de tipo quirúrgico pue-den ser perfectamente sustituidas por una técnica de mínima invasión, como laAC. En resumen, la AHA reporta que el AMI y la muerte se presentan en 16.4%con la técnica combinada de cirugía carotídea y coronaria, en 26.2% cuando laendarterectomía precede a la corrección cardiaca y en 16.4% cuando la cirugíacardiaca precede a la EC.76 Por el contrario, lo reportado por Lopes77 muestra queel rango de morbilidad, mortalidad y EVC en procedimientos combinados conAC fue de 8%, con 2% de EVC; estos resultados contrastan y validan lo antes se-ñalado, en razón de la superioridad de la técnica endovascular carotídea sobre lasdiferentes alternativas de revascularización carotídea.
Lesiones en tandem
La presencia de lesiones distales de mayor gravedad que las proximales constitu-yó un criterio de exclusión en NASCET. En pacientes sintomáticos el incrementode la morbilidad con lesión en sifón distal ipsilateral al sitio de la EC fue de13.9%, contra 7.9% de los pacientes sin estenosis distal. Por el contrario, en laserie reportada en pacientes con lesiones en tandem mediante AC,78 la lesión pro-ximal se consideró suboclusiva.
Trombo intraluminal ipsilateral
En el análisis estratificado de NASCET con la coexistencia de un coágulo y unaplaca de ateroma (identificados mediante DSA) se produjo un EVC en 10.7% delos pacientes bajo tratamiento médico y en 12% de los pacientes operados. Porel contrario, la alternativa endovascular permite el uso de trombólisis intraarte-rial, así como su asociación con agentes antiplaquetarios, como los inhibidoresde la GP IIb/IIIa.79
Oclusión carotídea contralateral
La asociación de carótida sintomática ipsilateral y oclusión carotídea contralate-ral se ha documentado con un alto grado de morbilidad, ya que la estratificaciónde NASCET al respecto mostró que el manejo médico y el seguimiento a dosaños80 con manejo médico ocasionaron infarto en 69.4% de los pacientes, lo cual

134 (Capítulo 8)Tratado de trombosis
fue reducido mediante EC a 14.3%. Sin embargo, el riesgo agregado de la cirugíase debe sobre todo al uso de shunting. En este subgrupo la AC representa una al-ternativa a la EC, ya que con los sistemas modernos de filtro la oclusión sólo seefectúa durante segundos en estenosis suboclusivas, pues en oclusiones de 70%se recurre a la colocación de stent primario sin angioplastia.81–83
Reestenosis posterior a la endarterectomía carotídea
La existencia de reestenosis posterior a una cirugía ha sido cada vez más recono-cida,84 y su tratamiento mediante técnica quirúrgica tradicional conlleva un ries-go mayor. Por ejemplo, la Clínica Mayo reportó que la morbilidad y la mortalidaden cirugía de reestenosis era de hasta 10.8%, casi cinco veces más que la EC pri-maria,84,85 así como la presencia de TIA a 30 días se eleva a 4.8% en lugar de 0.8y 1% de la EC primaria. Asimismo, se encontró un incremento significativo(17%) de parálisis de nervios craneales durante la reoperación. No así en lo repor-tado por Lanzino,86 que indica que de 18 pacientes después de la EC y estenosiscarotídea recurrente sólo uno desarrolló TIA sin infarto perioperatorio. El estudiode ARCHeR87 muestra que la AC en manejo de reestenosis EVC + AMI + muertefue de 0.7%.
Estenosis posterior a la radiación
En la estenosis inducida por radiación el proceso inflamatorio de la pared asocia-do con cicatrización y una escasa definición de planos quirúrgicos hace que lacirugía sea una mala opción;88,89 por el contrario, la AC ha demostrado ser un me-jor método,18,20 incluso después de la EC.19
Angioplastia carotídea
Antecedentes
Como ya se indicó, el éxito de la angioplastia en otros lechos vasculares incitóa varios grupos médicos a realizar procedimientos de AC en pacientes no candi-datos a cirugía.90,91 A continuación se resumen algunos de los ensayos clínicosrealizados en todo el mundo acerca de la AC.92,93
CAVATASEs el primer estudio aleatorizado que comparó la AC con la EC; es el estudio deangioplastia transluminal carotídea vertebral (Carotid and Vertebral ArteryTransluminal Angioplasty Study, o CAVATAS).9 En él se reclutaron 504 pacien-

135Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
tes, de los cuales sólo a una quinta parte se les colocó un stent y en el que la tasade EVC a los 30 días fue de 10% para la AC y de 9.9% para la EC. En cuanto alas complicaciones “menores”, hubo 1.2% de hematomas en la serie endovascu-lar contra 6.7% en la serie quirúrgica, y hasta 8.7% de los pacientes presentaronparálisis de los nervios craneales.
World RegistryEste registro mundial reclutó al principio a 6 734 pacientes en 42 centros en todoel mundo95 y se reporta cada dos años.96,97 En su último reporte, con un total de11 243 pacientes, informó una morbilidad (EVC) y una mortalidad a los 30 díasde 4.75%. Hubo 1.2% de EVC mayores, 2.14% de EVC menores y 0.77% demortalidad no relacionada directamente con el procedimiento. Aunque no se tratade un estudio aleatorizado, 4.75% se comparan favorablemente con 5.6% del es-tudio NASCET, tomando en cuenta que la mayoría de los pacientes incluidos enel estudio World Registry hubieran sido excluidos tanto del ACAS como delNASCET, por su alto perfil de riesgo.
CRESTEs un estudio clínico aleatorizado diseñado para comparar la eficacia de la ECcon la de la AC con colocación de stent y la ayuda de un dispositivo de proteccióncontra los émbolos, en cuanto a la prevención de infarto cerebral, infarto del mio-cardio y muerte, con una población sintomática (con un porcentaje de estenosismayor de 50% por angiografía o más de 60% por ultrasonido).98 Este estudio fueformulado por el National Institute of Neurological Disorders, y el primer pa-ciente se reclutó en diciembre del2000.99 Está previsto que el estudio reclute a2 500 pacientes en EUA, e incluirá también pacientes asintomáticos con esteno-sis superior de 60% por angiografía o de 70% por ultrasonido. Se utiliza un stentautoexpandible de nitinol (Acculink Carotid Stent System) y un sistema de protec-ción (RX Accunet Embolic Protection System) de la compañía Guidant (SantaClara, California, EUA). Se reclutará a 4 000 pacientes en 60 centros de EUAdespués de una fase de “aprobación” del centro por parte del Comité.100 Hastaahora no se han publicado resultados de este estudio.
CARESSEl estudio CARESS (Carotid Artery Endarterectomy Stenting Study) es un regis-tro observacional paralelo tanto de EC como de AC. Su objetivo es representarla enfermedad carotídea del mundo real, y es el único ensayo que recluta a pacien-tes de bajo, mediano y alto riesgo.101 En los resultados publicados recientemen-te102,103 se había tratado a 397 pacientes, a 254 con cirugía y a 143 con AC. Másde 90% de los pacientes tenían una estenosis mayor de 75%, de los cuales 68%eran sintomáticos y 32% asintomáticos. No hubo una diferencia significativa en

136 (Capítulo 8)Tratado de trombosis
la tasa combinada de infarto cerebral y mortalidad a los 30 días entre la cirugía(2%) y la terapia endovascular (2%). Tampoco hubo una diferencia significativaen el punto final secundario, es decir, mortalidad por cualquier causa, infarto ce-rebral o del miocardio a los 30 días entre la cirugía (3%) y la AC (2%).92
SAPPHIREEl estudio Stenting and Angioplasty with Protection in Patients at High Risk forEndarterectomy (SAPPHIRE)104 es el primer ensayo clínico aleatorizado quecompara la EC con la AC con protección distal. El estudio incluyó un grupo depacientes distribuidos de manera aleatoria entre cirugía y terapia endovascular(334 pacientes, de los cuales se trató a 310). Dentro del mismo estudio se creóun registro de pacientes a los cuales se les realizó angioplastia, porque el grupoquirúrgico contraindicó la EC, así como un registro de pacientes operados, quefueron contraindicados por el grupo endovascular.
Se reclutó a pacientes con estenosis asintomática mayor de 80% o estenosissintomática superior a 50%, ambas con “alto riesgo quirúrgico” (enfermedad car-diaca o pulmonar grave, oclusión carotídea contralateral, parálisis del nervio la-ríngeo contralateral, radiación o cirugía cervical previa, estenosis recurrente pos-terior a la EC y una edad mayor de 80 años).
Se emplearon materiales de la compañía Cordis (Precise Nitinol Self–Expand-ing Stent y AngioGuard XP Emboli Capture Guidewire System). Un punto inicialindicaba que un neurólogo examinara a los pacientes después del procedimiento.El punto final primario incluía muerte, cualquier infarto cerebral e infarto delmiocardio 30 días después del procedimiento.
El hallazgo principal de este estudio es que con el uso de un dispositivo de pro-tección la AC no resulta inferior a la EC en cuanto a la prevención de infarto cere-bral, muerte o infarto del miocardio en pacientes en los que la cirugía constituyeun riesgo mayor.
En un análisis secundario el índice acumulado de infarto cerebral, muerte e in-farto del miocardio, así como el índice acumulado de parálisis de nervios cranea-les y de duración de la estancia hospitalaria, fueron inferiores en los pacientes tra-tados con AC, no así en los que fueron tratados con cirugía.
Hay varios puntos criticables de este estudio que han sido analizados por va-rios autores.93,105 Por ejemplo, las AC del registro no aleatorizado (406) fueronmuchas más que las del registro aleatorizado (334), lo cual no habla a favor delentorno quirúrgico en el que se realizó el estudio. El estudio se terminó antes detiempo por una dificultad para reclutar pacientes, que los autores atribuyen a unamayor disponibilidad de otros registros de stents carotídeos no aleatorizados.Más de 20% de los pacientes de cada grupo de tratamiento tenían estenosis recu-rrente y, tomando en cuenta el mecanismo de reestenosis después de la EC (hiper-plasia de la íntima), esta alta proporción crea un sesgo favorable para la AC.

137Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
A pesar de esto, el estudio SAPPHIRE aporta pruebas en favor de la funciónde la AC en el manejo de la enfermedad carotídea en pacientes sintomáticos ypacientes asintomáticos con un alto riesgo de morbilidad y mortalidad por cirugía.
ARCHeREl ensayo clínico ARCHeR (Acculink for Revascularization of Carotids in HighRisk Patients)87 estudió a pacientes de alto riesgo quirúrgico tratados con AC.Los puntos finales primarios y criterios de inclusión (porcentajes de estenosis ycriterios de alto riesgo quirúrgico) fueron similares a los del estudio SAPPHIRE.Se utilizó el stent Acculink� y el sistema de protección Accunet� (Guidant)para la AC de los 437 pacientes reclutados en más de 40 centros de EUA.
La tasa de infarto cerebral y muerte a los 30 días fue de 6.6% y la de infartocerebral, muerte e infarto del miocardio fue de 7.8%. Estos resultados se compa-ran de manera muy favorable con los del grupo quirúrgico del estudio NASCET.
SPACEEl estudio SPACE106 (Stent–Protected Percutaneous Angioplasty of the Carotidvs. Endarterectomy) es un estudio aleatorizado que se realizó en Alemania, Suizay Austria. Se asignó de manera aleatoria a 1 200 pacientes con estenosis carotídeasintomática a los 180 días de un ataque isquémico transitorio o de un EVC nograve (escala de Rankin modificada = 3) con AC (n = 605) o EC (n = 595) comotratamiento. El punto final primario fue el EVC ipsilateral o la muerte hasta 30días después del procedimiento. Se debía mostrar que la AC no es inferior encomparación con la EC. El margen de no inferioridad fue menor de 2.5% y se es-peró una tasa de eventos de 5%. Finalmente se incluyó a 1 183 pacientes en elanálisis. La tasa de muerte o EVC ipsilateral a los 30 días fue de 6.84% con la ACy de 6.34% con la EC (diferencia absoluta de 0.51%, 90% IC –1.89 a 2.91%). Elvalor de “p” para la no inferioridad fue de 0.09.
En resumen, el estudio SPACE no pudo demostrar la no inferioridad de la ACen comparación con la EC, en cuanto a la tasa de complicaciones durante el pro-cedimiento. Se esperan los resultados a 6 y a 24 meses, pero por el momento nojustifican la amplia indicación de la AC.
EVA–3SEl estudio EVA–3S107 (EC vs. AC en estenosis sintomática severa) es un estudiofrancés que compara ambas técnicas en pacientes con una estenosis de al menos60%. El estudio se suspendió de manera prematura después de haber incluido a527 pacientes, porque la incidencia de cualquier EVC o muerte fue de 3.9% des-pués de la EC y de 9.6% después de AC.
Algunos podrían argumentar que el EVA–3S ha marcado el punto final en lacontroversia. Aunque los resultados son sorprendentes hay varios puntos débiles,

138 (Capítulo 8)Tratado de trombosis
y aunque los resultados son impactantes hay varios puntos que es necesario men-cionar. Primero, mientras los centros del SPACE tenían que demostrar haber he-cho al menos 25 AC para poder participar, en el estudio francés solamente se exi-gía haber cumplido con cinco procedimientos. Es obvio que al reclutar centrossecundarios con experiencia marginal no se puede esperar gran cosa. Segundo,el estudio no encontró diferencias entre los centros reclutados y además intentaconvencer de que esto demuestra que no hay problema en cuanto a la calidad delos centros. Sin embargo, como la mayoría de los centros tienen bajo reclutamien-to y poca experiencia, lo peor no puede ser empeorado. En cambio, el estudioSPACE mostró una clara relación según los centros, pues demostró que los sitioscon reclutamiento amplio y un número elevado de pacientes tuvieron una bajatasa de complicaciones. Tercero, en el estudio SPACE no hay diferencias entrelos centros que utilizan sistema de protección y los que no. El uso de dispositivosadicionales en manos de intervencionistas inexpertos agrega complicaciones yen el estudio EVA–3S dichos dispositivos eran obligatorios. Por suerte, estas de-bilidades en el estudio EVA–3S no le proporcionan validez. Además de este pun-to, el uso de antiagregantes fue heterogéneo, ya que se usó un solo antiagreganteen algunos casos.108
Incluso el autor principal del estudio EVA–3S no cree que los resultados delmismo deban marcar “la extinción de la AC”.109 También puntualiza que es nece-sario mejorar la seguridad del tratamiento endovascular antes de que pueda ser pro-puesto como alternativa a la EC en pacientes con estenosis carotídea sintomática.
Aun así, ni el SPACE ni el EVA–3S demostraron qué método protege mejoral paciente contra un EVC. El seguimiento previsto en los dos años siguientes enSPACE y un análisis de estos datos proporcionarán más pruebas.110
Pro–CASEl estudio Pro–CAS83 (Prospective Registry of Carotid Angioplasty and Stent-ing) es un registro prospectivo de AC realizado en Alemania y Austria para reco-lectar datos técnicos y de resultados en AC tratados en 38 centros participantesfuera de cualquier ensayo terapéutico. Este registro ofrece una oportunidad únicade documentar los resultados tal como se obtienen en el “mundo real”, ya que esuna población de pacientes no seleccionados con los métodos y materiales quese emplean de manera rutinaria en condiciones “normales”, sin las restriccionesde un protocolo científico previamente elaborado.
Se registraron al egreso los detalles técnicos, los medicamentos durante el pro-cedimiento y la evolución clínica de los pacientes. Durante los cuatro años queduró el estudio se realizaron 3 267 AC, de las 3 853 planeadas. Hubo 56% de ca-rótidas sintomáticas (1 827 pacientes) y 44% de asintomáticas (1 433). Se colocóun stent en 98% de los casos (3 127), siendo el 89% de estos (2 784). El éxito téc-nico fue de 98% (3 207 casos), con cifras de mortalidad de 0.6% (n = 18), EVC

139Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
mayor de 1.2% (n = 38) y EVC menor de 1.3% (n = 41). La tasa combinada demortalidad y EVC fue de 2.8% (n = 90).
Este estudio prospectivo multicéntrico ofrece el único cuadro realista acercade las posibilidades y limitaciones de la AC en la comunidad, y no basado en losreportes de los centros especializados que participan en los estudios clínicos.
Además de los puntos señalados, es necesario mencionar que casi siempre elcosto y el uso de recursos con la colocación de stent son sustancialmente menoresque los que se utilizan para una EC.111
Complicaciones de la angioplastia carotídea
Las complicaciones de la AC pueden dividirse en complicaciones durante el pro-cedimiento y después de él.112
Angioplastia carotídea e hipotensión
En las primeras series, cuando se usaba el stent de Palmaz� en AC, la bradicardiay la hipotensión eran frecuentes hasta en 5.3% de los casos. Los stents expandi-bles con balón, por su mayor fuerza radial, aumentan la respuesta de los barorre-ceptores del bulbo carotídeo.92 Con el uso de los stents autoexpandibles dicho fe-nómeno disminuyó, en comparación con los stents expandibles con balón.113 Seha observado que las complicaciones isquémicas son hasta cinco veces más fre-cuentes cuando se presenta hipotensión durante la AC que cuando no la hay.114
El uso de stents autoexpandibles, el no inflado de balón si no es necesario, asícomo el monitoreo estrecho del paciente y el tratamiento oportuno de la hipoten-sión, han disminuido esta complicación.
Hiperperfusión
La definición de “síndrome de hiperperfusión” varía entre los diversos autores;115
sin embargo, en general se define como un cuadro de cefalea, vómito, hiperten-sión arterial, confusión, convulsiones y déficit neurológico focal116 causado porun aumento del flujo sanguíneo cerebral después de un procedimiento de revas-cularización,117 que se puede acompañar de sangrado en el parénquima o en elespacio subaracnoideo (sangrado por reperfusión). La incidencia global de estascomplicaciones es baja: de 1.1% del síndrome de hiperperfusión y de 0.67% delsangrado por reperfusión.
Es importante considerar que el síndrome de hiperperfusión puede conducira un sangrado intracraneal por reperfusión. Los pacientes con mayor riesgo depresentar hiperperfusión presentan las siguientes características: estenosis grave

140 (Capítulo 8)Tratado de trombosis
de la carótida ipsilateral, oclusión o estenosis grave contralateral, escasa colatera-lidad intracraneal, elevación de las velocidades de las arterias cerebrales mediasdespués del procedimiento e hipertensión transoperatoria.118
Es necesaria una estrecha vigilancia después del procedimiento, considerandoque el sangrado posterior a la reperfusión se presenta en el periodo temprano des-pués de la colocación del stent.119,120
Reestenosis
Aunque se trata de un problema muy debatido en otros lechos vasculares,121,122
este problema no se presenta con frecuencia en las lesiones de la bifurcación caro-tídea. En uno de los registros más importantes de AC (World Registry) la inciden-cia de este fenómeno es de 2.7, 2.6 y 2.4% al año, a los dos años y a los tres años,respectivamente. Willfort–Ehringer y col.123 reportaron 2.0% de reestenosis in-trastent en una serie de 303 stents carotídeos vigilados durante dos años.
Consideraciones prácticas antesde un procedimiento de angioplastia
Evaluación previa del paciente
Antes de someter a un paciente a la colocación de un stent carotídeo conviene rea-lizarle una amplia valoración clínica y paraclínica.
Se recomienda que en la toma de decisiones participe un grupo multidiscipli-nario, el cual debe incluir un neurólogo, un intensivista, un neuroanestesiólogo,un neurorradiólogo y un neurocirujano, que es quién realizará el procedimiento.
Valoración clínica
Historial clínico completo que incluya:
� Antecedentes patológicos y no patológicos relevantes (hipertensión arte-rial, diabetes mellitus, tabaquismo, obesidad, insuficiencia orgánica —car-diaca, respiratoria, renal— y alergia al medio de contraste).
� Toma de signos vitales, en especial la frecuencia cardiaca y la presión arte-rial basal.
� Historia del padecimiento actual, en la que se correlacione la sintomatolo-gía presentada con la localización de la lesión, para establecer un vínculocausal.
� Valoración neuropsicológica completa.

141Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Valoración paraclínica:
� Estudios de laboratorio; en especial biometría hemática, tiempo de coagu-lación, función renal y glucemia.
� Estudios de gabinete, como:� Electrocardiograma —prueba de esfuerzo u otros estudios similares en
función del contexto.� Radiografía de tórax —pruebas funcionales respiratorias o valoración
por parte de un especialista en caso de ser necesario.� Estudios de imagen:
� Resonancia magnética de cráneo (secuencias T1, T2, difusión, perfu-sión, FLAIR).
� Estudio ultrasonográfico de vasos del cuello realizado por un expertoen ultrasonido neurovascular.
� Ultrasonido Doppler transcraneal (valoración de la permeabilidad yla colateralidad a nivel del polígono de Willis).
Una vez que se cuente con esta valoración se podrá programar la fecha para llevara cabo el procedimiento.
CONCLUSIÓN
En algunos centros la AC es un procedimiento rutinario para la revascularizaciónde la carótida.
La Brain Attack Coalition lo recomienda en diversas circunstancias para pa-cientes con alto riesgo quirúrgico;124 asimismo, indica que la colocación del stentla lleven a cabo médicos con entrenamiento y dominio de la teoría y de la técnicatanto en angiografía cerebral como en patofisiología cerebrovascular, hemodiná-mica cerebral e intervenciones neurovasculares.124 Es evidente que ningún car-diólogo o radiólogo intervencionista reúne estas condiciones.
La American Heart Association y el American Stroke Association Council re-conocen que la AC no es inferior a la EC en casos de reestenosis después de laEC o de estenosis posterior a la radiación.125
Un panel conjunto de la American Society of Interventional and TherapeuticNeuroradiology, la American Society of Neuroradiology y la Society of Interven-tional Radiology recomienda la AC en pacientes sintomáticos con estenosis de70% o en pacientes asintomáticos con estenosis de 90% con un alto riesgo quirúr-gico o que rechacen la cirugía.27 Incluso la administración sanitaria de EUA hareconocido a través de los Centers for Medicare and Medicaid Services que la AC

142 (Capítulo 8)Tratado de trombosis
con protección distal es razonable y necesaria en pacientes sintomáticos con este-nosis � 70%.126
Los resultados del estudio EVA–3S contradicen a todos los estudios hasta aho-ra realizados, ya que la tasa de complicaciones es muy alta y no coinciden conninguno de los numerosos estudios publicados. No obstante, los resultados deeste estudio no afectarán las recomendaciones y las guías ya emitidas ni las apro-baciones reguladoras de los dispositivos.127
Debido al reclutamiento poco riguroso de sus intervencionistas, el estudioEVA–3S hace insistir en la necesidad de ser más estrictos y estandarizados encuanto a los criterios de entrenamiento requeridos por los intervencionistas quese consagren a la colocación de stents en la carótida,108 lo cual constituye una ne-cesidad reconocida y estipulada por varias asociaciones.128,129
El trabajo colectivo que aún queda pendiente es trabajar en los datos disponi-bles en la actualidad para clasificar a los pacientes que se verán beneficiados conun método o con el otro. Finalmente, esta reflexión llevará a identificar los facto-res que se asocian con una mayor tasa de complicaciones durante la realizaciónde una AC. Dichos factores serán relativos al paciente (p. ej., la presencia de pla-cas de ateroma aórtico importantes), a la placa (p. ej., placa inestable y su morfo-logía en general), a la experiencia del operador (p. ej., formación teórica y prác-tica, pericia y prudencia), al procedimiento mismo y a los materiales anexos (p.ej., dispositivos adecuados y seguimiento del protocolo establecido), así comoal monitoreo, la vigilancia y el tratamiento médico antes, durante y después delprocedimiento (p. ej., un correcto esquema antiagregante y el manejo de la hipo-tensión). Sólo de esta manera se concentran los esfuerzos para mejorar la canti-dad y la calidad de lo aprendido, tanto en la teoría como en la práctica, con la fina-lidad de ofrecer siempre la mejor opción y sin olvidar el lema que se aprende enlas primeras correrías como médicos, antes de decidir si se aplica o se declina untratamiento: primum non nocere.
REFERENCIAS
1. Robicsek F, Roush TS, Cook JW, Reames MK: From Hippocrates to Palmaz–Schatz. Thehistory of carotid surgery. Eur J Vasc Endovasc Surg 2004;27:389.
2. Thompson JE: The development of carotid artery surgery. Arch Surg 1973;107:643–648.3. DeBakey MCE, Cooley D, Morris GJ: Surgical considerations of occlusive disease of in-
nominate, carotid, subclavian and vertebral arteries. Ann Surg 1959;149:690–710.4. Mathias K: A new catheter system for percutaneous transluminal angioplasty (PTA) of ca-
rotid artery stenoses. Fortschr Med 1977;95:1007–1011.5. Pasternak RC, Criqui MH, Benjamin EJ et al.: Atherosclerotic Vascular Disease Confer-
ence. Writing Group I: Epidemiology. Circulation 2004;109:2605–2612.6. Louridas G, Junaid A: Management of carotid artery stenosis. Update for family physi-
cians. Can Fam Phys 2005;51:984–989.

143Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
7. Ernst CB SJ (eds.): Current therapy in vascular surgery. 4ª ed. St. Louis, Mosby, 2001:102.8. Simons PCG, Ale A, Bert CE, Diederick EG et al.: Carotid artery stenosis in patients with
peripheral arterial disease: The SMART study. J Vasc Surg 1999;30:519.9. Dyken M: Stroke risk factors in prevention of stroke. En: Norris JW (ed.): Prevention of
stroke. Nueva York, Springer Verlag, 1991:83–102.10. Taylor TNDP, Torner JC: Projected number of strokes by subtype in the year 2050 in the
United States (abstract). Stroke 1998;29:322.11. Beneficial effect of carotid endarterectomy in symptomatic patients with high–grade caro-
tid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. NEngl J Med 1991;325:445–453.
12. Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for theAsymptomatic Carotid Atherosclerosis Study. JAMA 1995;273:1421–1428.
13. Warlow C: MRC European Carotid Surgery Trial: interim results for symptomatic patientswith severe (70–99%) or with mild (0–29%) carotid stenosis. Lancet 1991;337:1235.
14. Finsterer J, Strassegger J, Haymerle A, Hagmuller G: Bilateral stenting of symptomaticand asymptomatic internal carotid artery stenosis due to fibromuscular dysplasia. J NeurolNeurosurg Psychiatry 2000;69:683–686.
15. Bali HKBM, Bhatta YK, Sandhu MS: Single stage bilateral common carotid artery stent-ing in a patient of Takayasu arteritis. Neurol India 2001;49:87–90.
16. Takahashi JC, Sakai N, Manaka H et al.: Multiple supra–aortic stenting for Takayasu ar-teritis: extensive revascularization and two–year follow–up. Am J Neuroradiol 2002;23:790–793.
17. Smith TP, Halbach VV, Fraser KW, Teitelbaum GP, Dowd CF et al.: Percutaneous trans-luminal angioplasty of subclavian stenosis from neurofibromatosis. Am J Neuroradiol1995;16:872–874.
18. Houdart E, Mounayer C, Chapot R, Saint MJP, Merland JJ: Carotid stenting for radi-ation–induced stenoses: a report of 7 cases. Stroke 2001;32:118–121.
19. Ting A: Carotid stenting for irradiation associated carotid stenosis 3 years after previouscarotid endarterectomy. Hong Kong Med J 2003;9:51–53.
20. Harrod KP, Kadkhodayan Y, Derdeyn CP, Cross DTII: Outcomes of carotid angioplastyand stenting for radiation–associated stenosis. Am J Neuroradiol 2005;26:1781–1788.
21. Binaghi S, Chapot R, Rogopoulos A, Houdart E: Carotid stenting of chronic cervical dis-secting aneurysm: a report of two cases. Neurology 2002;59:935–937.
22. Kerber CW, Loehden OL: Catheter dilatation of proximal carotid stenosis during distalbifurcation endarterectomy. Am J Neuroradiol 1980;1:348–349.
23. Serruys PW, de Jaegere P, Kiemeneij F et al.: A comparison of balloon–expandable–stentimplantation with balloon angioplasty in patients with coronary artery disease. N Engl JMed 1994;331:489–495.
24. Tsetis D, Belli AM: Guidelines for stenting in infrainguinal arterial disease. Cardiovasc In-terventional Radiol 2004;27:198.
25. Pelz DM, Lownie SP: Carotid angioplasty and stenting: current status. CMAJ 2000;162:1451–1454.
26. Caplan L: Stroke is best managed by neurologists. Stroke 2003;34:2763.27. Barr JD, Connors JJ, III, Sacks D et al.: Quality improvement guidelines for the perfor-
mance of cervical carotid angioplasty and stent placement: developed by a collaborative pa-nel of the American Society of Interventional and Therapeutic Neuroradiology, the Ameri-can Society of Neuroradiology, and the Society of Interventional Radiology. Am JNeuroradiol 2003;24:2020–2034.

144 (Capítulo 8)Tratado de trombosis
28. Johnston SC: Who belongs inside the carotid arteries? Neurology 2005;64:188–189.29. Rao R: The role of carotid stenosis in vascular cognitive impairment. Eur Neurol 2001;46:
63–69.30. Johnston SC, O’Meara ES, Manolio TA et al.: Cognitive impairment and decline are
associated with carotid artery disease in patients without clinically evident cerebrovasculardisease. Ann Intern Med 2004;140:237–247.
31. Mathiesen EB, Waterloo K, Joakimsen O, Bakke SJ, Jacobsen EA et al.: Reduced neu-ropsychological test performance in asymptomatic carotid stenosis: the Tromso Study. Neu-rology 2004;62:695–701.
32. Spence JD, Tamayo A, Lownie SP, Ng WP, Ferguson GG: Absence of microemboli ontranscranial Doppler identifies low–risk patients with asymptomatic carotid stenosis. Stroke2005;36:2373–2378.
33. Silvestrini M, Vernieri F, Pasqualetti P et al.: Impaired cerebral vasoreactivity and risk ofstroke in patients with asymptomatic carotid artery stenosis. JAMA 2000;283:2122–2127.
34. Touze E, Toussaint JF, Coste J et al.: Reproducibility of high–resolution MRI for the iden-tification and the quantification of carotid atherosclerotic plaque components: consequen-ces for prognosis studies and therapeutic trials. Stroke 2007;38:1812–1819.
35. Rothwell PM: Endarterectomy for symptomatic carotid stenosis in relation to clinical sub-groups and timing of surgery. Lancet 2004;363:915–924.
36. Gaitini D, Soudack M: Diagnosing carotid stenosis by Doppler sonography: state of theart. J Ultrasound Med 2005;24:1127–1136.
37. Barth JD: An update on carotid ultrasound measurement of intima–media thickness. AmJ Cardiol 2002;89:32.
38. Meairs SSW: Ultrasound imaging and Doppler sonography. En: Stroke: physiopathology,diagnosis and management. Nueva York, Churchill Livingstone, 1998:298.
39. Thiele BL, Hobson RW, Bandyk DF, Baker WH, Sumner DS et al.: Standards in nonin-vasive cerebrovascular testing. Report from the Committee on Standards for NoninvasiveVascular Testing of the Joint Council of the Society for Vascular Surgery and the North Am-erican Chapter of the International Society for Cardiovascular Surgery. J Vasc Surg 1992;15:495–503.
40. Grant EG, Benson CB, Moneta GL et al.: Carotid artery stenosis: gray–scale and DopplerUS Diagnosis–Society of Radiologists in Ultrasound Consensus Conference. Radiology2003;229:340–346.
41. Filis KA, Arko FR, Johnson BL et al.: Duplex ultrasound criteria for defining the severityof carotid stenosis. Ann Vascular Surg 2002;16:413.
42. Buskens E, Nederkoorn PJ, Buijs van der Woude T et al.: Imaging of carotid arteries insymptomatic patients: cost–effectiveness of diagnostic strategies. Radiology 2004;233:101–112.
43. Moore WS: For severe carotid stenosis found on ultrasound, further arterial evaluation isunnecessary. Stroke 2003;34:1816–1817.
44. Henderson RD, Eliasziw M, Fox AJ, Rothwell PM, Barnett HJM: Angiographicallydefined collateral circulation and risk of stroke in patients with severe carotid artery steno-sis. Stroke 2000;31:128–132.
45. Collins P, McKay I, Rajagoplan S, Bachoo P, Robb O et al.: Is carotid duplex scanningsufficient as the sole investigation prior to carotid endarterectomy? Br J Radiol 2005;78:1034–1037.
46. Feasby TE, Findlay JM: CT angiography for the assessment of carotid stenosis. Neurology2004;63:412–413.

145Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
47. Josephson SA, Bryant SO, Mak HK, Johnston SC, Dillon WP et al.: Evaluation of ca-rotid stenosis using CT angiography in the initial evaluation of stroke and TIA. Neurology2004;63:457–460.
48. Koelemay MJW, Nederkoorn PJ, Reitsma JB: Systematic review of computed tomo-graphic angiography for assessment of carotid artery disease. Stroke 2004;35:2306–2312.
49. Conceicao C PT, Evangelista P: Angio–TC da doenca carotídea ateromatosa. Acta MedPort 2003;16:149–153.
50. Barry OT, Alistair A, Sandi C, Donald: Atherosclerotic plaque at the carotid bifurcation:CT angiographic appearance with histopathologic. Am J Neuroradiol 1999;20:897–901.
51. U–King–Im JM, Trivedi RA, Graves MJV: Contrast–enhanced MR angiography for ca-rotid disease: diagnostic and potential clinical impact. Neurology 2004;62:1282–1290.
52. Muhs B, Gagne P, Wagener J et al.: Gadolinium–enhanced versus time–of–flight mag-netic resonance angiography: what is the benefit of contrast enhancement in evaluating ca-rotid stenosis? Ann Vascular Surg 2005;19:823.
53. Johnston D: Low rate of complications of cerebral angiography in routine clinical practice.Neurology 2001;57:2012–2014.
54. Patel SG, Collie DA, Wardlaw JM et al.: Outcome, observer reliability, and patient prefer-ences if CTA, MRA, or Doppler ultrasound were used, individually or together, instead ofdigital subtraction angiography before carotid endarterectomy. J Neurol Neurosurg Psy-chiatry 2002;73:21–28.
55. Rothwell PM, Gibson R, Warlow CP: Interrelation between plaque surface morphologyand degree of stenosis on carotid angiograms and the risk of ischemic stroke in patients withsymptomatic carotid stenosis. Stroke 2000;31:615–621.
56. Eliasziw M, Streifler JY, Fox AJ, Hachinski VC, Ferguson GG et al.: Significance ofplaque ulceration in symptomatic patients with high–grade carotid stenosis. North Ameri-can Symptomatic Carotid Endarterectomy Trial. Stroke 1994;25:304–308.
57. Mas J: Internal carotid artery revascularization. Rev Esp Cardiol 2007;60:861–871.58. Carr S, Farb A, Pearce WH, Virmani R, Yao JST: Atherosclerotic plaque rupture in
symptomatic carotid artery stenosis. J Vascular Surg 1996;23:755.59. Schulz URP: Association between arterial bifurcation anatomy and angiographic plaque
ulceration among 4 627 carotid stenoses. Cerebrovasc Dis 2003;15:244–251.60. Hayward JK, Davies AH, Lamont PM: Carotid plaque morphology: a review. Eur J Vasc
Endovasc Surg 1995;9:368.61. Dhume AS, Soundararajan K, Hunter WJ, Agrawal DK: Comparison of vascular
smooth muscle cell apoptosis and fibrous cap morphology in symptomatic and asymptoma-tic carotid artery disease. Ann Vasc Surg 2003;17:1.
62. Nandalur KR, Baskurt E, Hagspiel KD, Phillips CD, Kramer CM: Calcified carotid ath-erosclerotic plaque is associated less with ischemic symptoms than is noncalcified plaqueon MDCT. Am J Roentgenol 2005;184:295–298.
63. Biasi G: Is it time to reconsider the selection criteria for conventional or endovascular repairof carotid artery stenosis in the prevention of cerebral ischemia? J Endovasc Ther 2001;8:339–340.
64. Hankey GJ: Stroke prediction and prevention by carotid endarterectomy: keep an eye onthe doughnut and not just the hole. Cerebrovasc Dis 1999;9:345–350.
65. Nighoghossian N, Derex L, Douek P: The vulnerable carotid artery plaque: current imag-ing methods and new perspectives. Stroke 2005;36:2764–2772.
66. Ouriel K, Hertzer NR, Beven EG et al.: Preprocedural risk stratification: identifying anappropriate population for carotid stenting. J Vascular Surg 2001;33:728.

146 (Capítulo 8)Tratado de trombosis
67. Hsia DC, Krushat WM, Moscoe LM: Epidemiology of carotid endarterectomies amongMedicare beneficiaries. J Vasc Surg 1992;16:201.
68. Wennberg DE, Lucas FL, Birkmeyer JD, Bredenberg CE, Fisher ES et al.: Variationin carotid endarterectomy mortality in the Medicare population: trial hospitals, volume, andpatient characteristics. JAMA 1998;279:1278–1281.
69. Goldstein LB, McCrory DC et al.: Multicenter review of preoperative risk factors forcarotid endarterectomy in patients with ipsilateral symptoms. Stroke 1994;25:1116–1121.
70. Goldstein LB, Samsa GP, Matchar DB, Oddone EZ: Multicenter review of preoperative riskfactors for endarterectomy for asymptomatic carotid artery stenosis. Stroke 1998;29:750–753.
71. Alamowitch S, Eliasziw M, Algra A, Meldrum H, Barnett HJM: Risk, causes, and pre-vention of ischaemic stroke in elderly patients with symptomatic internal–carotid–arterystenosis. Lancet 2001;357:1154.
72. Paciaroni M, Eliasziw M, Kappelle LJ, Finan JW, Ferguson GG et al.: Medical com-plications associated with carotid endarterectomy. Stroke 1999;30:1759–1763.
73. Harbaugh RE, Moayeri N, Hsu L: Carotid–coronary artery bypass graft conundrum. Neu-rosurgery 1998;43:926–931.
74. Faggioli GLCG, Ricotta JJ: The role of carotid screening before coronary artery bypass.J Vascular Surg 1990;12:724–731.
75. Del Sette M, Eliasziw M, Streifler JY, Hachinski VC, Fox AJ et al.: Internal borderzoneinfarction: a marker for severe stenosis in patients with symptomatic internal carotid arterydisease. Stroke 2000;31:631–636.
76. Moore WS, Barnett HJM, Beebe HG et al.: Guidelines for carotid endarterectomy: a mul-tidisciplinary consensus statement from the ad hoc Committee, American Heart Associa-tion. Stroke 1995;26:188–201.
77. Lopes DK, Lanzino G, Wakhloo AK, Guterman LR, Hopkins LN: Stent placement forthe treatment of occlusive atherosclerotic carotid artery disease in patients with concomitantcoronary artery disease. J Neurosurg 2002;96:490–496.
78. Kim SHM, Robert A, Lanzino G, Wakhloo AK, Guterman LR et al.: Carotid angioplas-ty and stent placement in patients with tandem stenoses (abstract of oral presentation). Neu-rosurgery 1998;43:708.
79. Ho DSW, Chui M, Wang Y, Ho SL, Cheung RTF: Intracarotid abciximab injection to abortimpending ischemic stroke during carotid angioplasty. Cerebrovasc Dis 2001;11:300–304.
80. Gasecki AP, Ferguson GG, Hachinski V, Barnett HJ: Long–term prognosis and effectof endarterectomy in patients with symptomatic severe carotid stenosis and contralateralcarotid stenosis or occlusion: results from NASCET. J Neurosurg 1995;83:778–782.
81. Willfort EA, Ahmadi R, Gruber D et al.: Arterial remodeling and hemodynamics in carotidstents: a prospective duplex ultrasound study over 2 years. J Vascular Surg 2004;39:728.
82. Men SPL, Pelz DM: Carotid stenting without angioplasty. Can J Neurological Sci 2002;29:175–179.
83. Theiss W, Hermanek P, Mathias K et al.: Pro–CAS: a prospective registry of carotid an-gioplasty and stenting. Stroke 2004;35:2134–2139.
84. Meyer FBPD, Fode NC: Surgical treatment of recurrent carotid artery stenosis. J Neuro-surg 1995;80:781–787.
85. Abu Rahma AF, Jennings TG, Wulu JT, Tarakji L, Robinson PA: Redo carotid endarter-ectomy versus primary carotid endarterectomy. Stroke 2001;32:2787–2792.
86. Lanzino GMR, Lopes DK, Wakhloo AK, Guterman LR, Hopkins LN: Percutaneoustransluminal angioplasty and stent placement for recurrent carotid artery stenosis. J Neuro-surg 1999;90:688–694.

147Enfermedad carotídeaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
87. Wholey M: ARCHeR (Acculink for Revascularization of Carotids in High–Risk Patients).Clin Cardiol 2003;26:296–299.
88. Loftus CM, Biller J, Hart MN, Cornell SH, Hiratzka LF: Management of radiation–in-duced accelerated carotid atherosclerosis. Arch Neurol 1987;44:711–714.
89. Melliere DBJ, Berrahal D, Desgranges P, Cavillon A: Management of radiation–inducedocclusive arterial disease: a reassessment. J Cardiovasc Surg 1997;38:261–269.
90. Diethrich EB, Ndiaye M, Reid DB: Stenting in the carotid artery: initial experience in 110patients. J Endovascular Surg 1996;3:42–62.
91. Roubin GS, Yadav S, Iyer SS, Vitek J: Carotid stent–supported angioplasty: a neurovas-cular intervention to prevent stroke. Am J Cardiol 1996;78:8–12.
92. Wholey MH, Wholey M: History and current status of endovascular management for theextracranial carotid and supra–aortic vessels. J Endovasc Ther 2004;11:II43–II61.
93. Das SBB, Getch CC, Awad IA, Batjer HH: Update on current registries and trials of caro-tid artery angioplasty and stent placement. Neurosurg Focus 2005;18:e2.
94. Cavatas I: Endovascular versus surgical treatment in patients with carotid stenosis in theCarotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS): a randomizedtrial. Lancet 2001;357:1729.
95. Wholey MH, Wholey M, Bergeron P, Edward B, Diethrich MH et al.: Current globalstatus of carotid artery stent placement. Catheter Cardiovasc Diagn 1998;44:1–6.
96. Wholey MH, Wholey M, Gary SK, Edward B, Diethrich MH et al.: Global experiencein cervical carotid artery stent placement. Catheter Cardiovasc Interv 2000;50:160–167.
97. Wholey MH, Al–Mubarek N, Wholey M: Updated review of the Global Carotid ArteryStent Registry. Catheter Cardiovasc Interv 2003;60:259–266.
98. Hobson RWII, Brott T, Ferguson R et al.: CREST: Carotid Revascularization Endarterec-tomy Versus Stent Trial. Cardiovascular Surg 1997;5:457.
99. Hobson RW II, Brott TG, Howard G, Roubin GS, Ferguson RD, for the CREST Execu-tive Committee: Organizing the Carotid Revascularization Endarterectomy Versus StentingTrial (CREST): National Institutes of Health, Health Care Financing Administration, andIndustry Funding. Curr Control Trials Cardiovasc Med 2001;2:160–164.
100. Hobson RW II: Update on the Carotid Revascularization Endarterectomy Versus StentTrial (CREST) protocol. J Am Coll Surg 2002;194:S9.
101. Steering C: Carotid Revascularization with Endarterectomy or Stenting Systems (CA-RESS): investigator selection. J Endovascular Ther 2001;8:547–549.
102. Steering C: Carotid Revascularization Using Endarterectomy or Stenting Systems (CA-RESS): phase I clinical trial. J Endovascular Ther 2003;10:1021–1030.
103. Steering C: Carotid Revascularization Using Endarterectomy or Stenting Systems (CA-RESS): phase I clinical trial: 1–year results. J Vasc Surg 2005;42:213.
104. Yadav JS, Wholey MH, Kuntz RE et al.: Protected carotid–artery stenting versus endar-terectomy in high–risk patients. N Engl J Med 2004;351:1493–1501.
105. Thomas DJ: Protected carotid artery stenting versus endarterectomy in high–risk patients:reflections from SAPPHIRE. Stroke 2005;36:912–913.
106. SPACE CG: 30 days results from the SPACE trial of stent–protected angioplasty versuscarotid endarterectomy in symptomatic patients: a randomized non–inferiority trial. Lancet2006;368:1239–1247.
107. Mas JL, Beyssen B et al.: Endarterectomy versus stenting in patients with symptomatic se-vere carotid stenosis. N Engl J Med 2006;355:1660–1671.
108. Furlan A: Carotid–artery stenting: case open or closed? N Engl J Med 2006;355:1726–1729.109. Mas JL: Recent carotid stenting trials. Lancet Neurol 2007;6:295–296.

148 (Capítulo 8)Tratado de trombosis
110. Forsting M: Shortcomings and promises of recent carotid–stenting trials. Lancet Neurol2007;6:101–102.
111. Gray WA, White HJ Jr, Barrett DM, Chandran G, Turner R et al.: Carotid stenting andendarterectomy: a clinical and cost comparison of revascularization strategies. Stroke 2002;33:1063–1070.
112. Raithel D: Complications of carotid artery stenting. J Cardiovasc Surg 2005;46:261–265.113. Wholey MH, Wholey M, Tan WA, Eles G, Jarmolowski C et al.: A comparison of bal-
loon–mounted and self–expanding stents in the carotid arteries: immediate and long–termresults of more than 500 patients. J Endovasc Ther 2003;10:171–181.
114. Lowell F, Satler JRL: Limiting the complications of carotid stenting. Catheter CardiovascInterv 2001;54:524–525.
115. Van Mook WN, Schurink GW, van Oostenbrugge RJ, Mess WH, Hofman PA et al.:Cerebral hyperperfusion syndrome. Lancet Neurol 2005;4:877–888.
116. Meyers PM, Phatouros CC, Malek AM, Lempert TE, Dowd CF et al.: Cerebral hyper-perfusion syndrome after percutaneous transluminal stenting of the craniocervical arteries.Neurosurgery 2000;47:335–343.
117. Ko NU, Achrol AS, Chopra M et al.: Cerebral blood flow changes after endovascular treat-ment of cerebrovascular stenoses. Am J Neuroradiol 2005;26:538–542.
118. Abou CA, Yadav JS, Reginelli JP, Bajzer C, Bhatt D et al.: Intracranial hemorrhage andhyperperfusion syndrome following carotid artery stenting: risk factors, prevention, andtreatment. J Am Coll Cardiol 2004;43:1596.
119. Al–Mubarak N: Subarachnoidal hemorrhage following carotid stenting with the distal–balloon protection. Catheter Cardiovasc Interv 2001;54:521–523.
120. McCabe DJH, Brown MM, Clifton A: Fatal cerebral reperfusion hemorrhage after carotidstenting. Stroke 1999;30:2483–2486.
121. Ellozy SH: Drug–eluting stents in peripheral vascular disease: eliminating restenosis. MtSinai J Med 2003;70:417–419.
122. Dietz U, Lambertz H: Combining short stent implantation and drug eluting stenting forroutine use yields a low restenosis rate. Curr Control Trials Cardiovasc Med 2005;6:18.
123. Willfort EA, Ahmadi R, Gschwandtner ME, Haumer M, Lang W et al.: Single–centerexperience with carotid stent restenosis. J Endovascular Therapy 2002;9:299–307.
124. Alberts MJ, Selman WR, Shephard T, Hadley MN, Brass LM, Brain Attack Coalition:Recommendations for comprehensive stroke centers: a consensus statement from the BrainAttack Coalition. Stroke 2005;36:1597–1616.
125. Sacco RL, Albers G, Alberts MJ, Benavente O, Furie K: Guidelines for prevention of strokein patients with ischemic stroke or transient ischemic attack: a statement for healthcare profes-sionals from the American Heart Association/American Stroke Association Council on Stroke:co–sponsored by the Council on Cardiovascular Radiology and Intervention: the AmericanAcademy of Neurology affirms the value of this guideline. Circulation 2006;113:e409–e449.
126. www.cms.hhs.gov/apps/media/press/release.asp?127. Qureshi A: Carotid angioplasty and stent placement after EVA–3S Trial. Stroke 2007;38:
1993–1996.128. Wallace R: Quality improvement guidelines for adult diagnostic neuroangiography. Am J
Neuroradiol 2000;21:146–150.129. Connors J: Carotid artery angioplasty and stent placement: quality improvement guide-
lines to ensure stroke risk reduction. J Vasc Interv Radiol 2003;14:1095–1097.130. Smith CM: Origin and uses of primum non nocere–above all, do no harm! J Clin Pharma-
col 2005;45:371–377.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
9Ataque isquémico transitorio,
infarto cerebral aterotrombóticoe infarto lacunar
Fernando Barinagarrementeria Aldatz, Antonio Arauz Góngora
GENERALIDADES
La enfermedad vascular cerebral (EVC) es un grupo heterogéneo de enfermeda-des que tienen en común el desarrollo final de isquemia cerebral (transitoria odefinitiva) o de una hemorragia cerebral. Sin embargo, las anormalidades pri-mordiales se encuentran a nivel de los vasos sanguíneos, ya que son éstos las es-tructuras primariamente enfermas que condicionan las manifestaciones neuroló-gicas.1 Desde el punto de vista clínico, es muy importante conocer losmecanismos fisiopatológicos que afectan el sistema circulatorio, incluido el co-razón, y las causas de estas anormalidades. De esta forma, la EVC debe ser clasi-ficada de acuerdo con el mecanismo fisiopatogénico, como EVC asociada a en-fermedad de los grandes vasos, a enfermedad de los pequeños vasos o bien amecanismos cardioembólicos. Existen, sin embargo, otros mecanismos poten-cialmente productores de isquemia cerebral, los cuales están fuera de los objeti-vos del presente capítulo.1,2
Las arterias de gran calibre (grandes vasos) incluyen los vasos supraaórticos(arterias carótidas y vertebrales en su porción cervical) y las arterias cerebralesintracraneales en su porción proximal (carótidas y vertebrales intracraneales y ar-terias cerebrales media, anterior y posterior). La principal causa de afectación delos grandes vasos es la aterosclerosis. Los pacientes habitualmente son portado-res de los factores de riesgo tradicionales: hipertensión arterial, diabetes, taba-quismo, obesidad e hiperlipidemia.1–3
149

150 (Capítulo 9)Tratado de trombosis
La EVC de pequeños vasos implica la afectación de las pequeñas arterias pe-netrantes que nacen de las grandes arterias intracraneales, particularmente las ar-terias lenticuloestriadas dependientes de la arteria cerebral media, las arterias ta-lamoperforantes dependientes de la arteria cerebral posterior y las arteriaspontinas paramedianas dependientes de la arteria basilar. Un punto importantees que estas arterias son terminales e irrigan las estructuras profundas del cerebro(ganglios basales, cápsula interna y tallo cerebral). La principal anormalidad fi-siopatológica en estos casos es la formación de lipohialinosis, habitualmente aso-ciada a microateroma relacionado a hipertensión arterial o diabetes.4
Respecto a la enfermedad cardioembólica, son múltiples las fuentes embolíge-nas que se pueden asociar al desarrollo de un ataque isquémico transitorio o deun infarto cerebral. Una de las más frecuentes, y que en los últimos años ha cobra-do creciente importancia, es la fibrilación auricular.1,2
ATAQUE ISQUÉMICO TRANSITORIO
Conceptos clínicos
Es un concepto clínico descrito desde la década de 1950 a raíz de los trabajos deMillikan y posteriormente de Fisher. Durante muchos años fue definida por la Or-ganización Mundial de la Salud como un trastorno neurológico con una duraciónde hasta 24 h. Este lapso quedó muy arraigado en la comunidad médica e inclusohoy erróneamente se considera igual. En 2002 una comisión especial se dio a latarea de revisar los criterios de diagnóstico y se consideraron los resultados obte-nidos mediante estudios de imagen.5 Es importante tomar en cuenta que el térmi-no correcto es el de ataque isquémico transitorio y no el de isquemia cerebral tran-sitoria. Si bien en la mayoría de los casos la isquemia que sufre el paciente escerebral, ésta puede ser retiniana o incluso, aunque poco frecuente, espinal. Porello el término considera exclusivamente el mecanismo isquémico y su caráctertransitorio y no la estructura afectada.2
Respecto al lapso de tiempo, en la actualidad no se define un periodo estableci-do, sino la exclusión de infarto cerebral en los estudios de imagen. El uso crecien-te de la imagen por resonancia magnética (IRM) con técnicas de difusión (DWI,por sus siglas en inglés) ha demostrado que un porcentaje de pacientes con mani-festaciones transitorias muestran anormalidades en la IRM con DWI, que son de-bidas a daño isquémico permanente. La mayoría de los eventos de ataque isqué-mico transitorio (AIT) duran menos de una hora, de los cuales dos tercios duranmenos de 10 min.5,6
Desde el punto de vista clínico, las manifestaciones clínicas del ataque isqué-mico transitorio se deben dividir por sus manifestaciones clínicas en aquellas con

151Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
isquemia en el territorio carotídeo o en el territorio vertebrobasilar. Entre lasmanifestaciones del territorio carotídeo es muy importante recordar que la enfer-medad carotídea se puede manifestar con trastornos visuales monoculares, espe-cialmente en forma de amaurosis fugax o ceguera monocular transitoria, lo cuales debido al compromiso de la arteria oftálmica, primera rama de la arteria caróti-da interna.
Un punto de controversia frecuente en la práctica clínica, especialmente conlos colegas otorrinolaringólogos, es el llamado vértigo monosintomático. El tallocerebral y el cerebelo son estructuras que contienen numerosas funciones y habi-tualmente las anormalidades vasculares de esta región condicionan cuadros clíni-cos, con frecuencia polisintomáticos, en los que en caso de vértigo éste suele estaracompañado por otras manifestaciones clínicas, como son diplopía, disartria, dis-fagia, ataxia y trastornos de la coordinación. De acuerdo con la experiencia, esexcepcional que el vértigo monosintomático sea debido a enfermedad vascularcerebral.
Hallazgos de imagen cerebral enel ataque isquémico transitorio
Aproximadamente un tercio de los pacientes con AIT clínicamente definido tie-nen evidencia de infarto cerebral en los estudios de imagen cerebral. El estudioideal en pacientes con AIT es la IRM con DWI, que permite diferenciar las lesio-nes isquémicas agudas de las crónicas. Desde el punto de vista imagenológico,la principal característica de los “infartos asociados a AIT” es su tamaño peque-ño, pues 96% de los infartos asociados a AIT son menores de 1 mL. La probabili-dad de encontrar una imagen de infarto en los estudios aumenta con la mayor du-ración del AIT.7 La definición más reciente de AIT se basa en la presencia oausencia de isquemia tisular. La nueva definición se refiere al AIT como el episo-dio de déficit neurológico causado por una lesión focal en el cerebro, la retina yla médula espinal, sin evidencia de infarto cerebral. Esta definición, basada enel estado del tejido, cambia la subjetividad del tiempo a la objetividad de la evi-dencia de lesión cerebral.6,7
Predicción del riesgo de infarto cerebraldespués de ataque isquémico transitorio
Tradicionalmente se ha dicho que el riesgo de un infarto cerebral después de unAIT es elevado (aproximadamente 10% en los siete días siguientes). La mayoríadel restante 90% de los pacientes no tienen un riesgo elevado de infarto. La dife-renciación del grupo de alto riesgo y de bajo riesgo es elemental.8

152 (Capítulo 9)Tratado de trombosis
Cuadro 9–1. Escalas ABCD2 y ABCD2–I
Variable Puntaje
A Edad (age) mayor de 60 años 1 puntoB Presión arterial (blood pressure) > 140/90 mmHg en el momento del
ingreso1 punto
C Manifestaciones clínicas (clinical)Debilidad unilateralTrastorno del lenguaje sin debilidad
2 puntos1 punto
D DuraciónDe 1 a 59 minMás de 60 min
1 punto2 puntos
D Diabetes 1 puntoABCD2–II Imagen (evidencia de infarto en técnicas de difusión o tomografía com-
putarizada)3 puntos
Los puntajes de 1 a 3 son de bajo riesgo, 4 y 5 son de mediano riesgo y 6 o 7 son de alto riesgo.En realidad, los puntajes superiores a 4 conllevan un riesgo de 5.6 a 23% a los siete días del evento.
Los pacientes de alto riesgo serán beneficiados con tratamientos específicos,que incluyen la administración de antiplaquetarios o anticoagulantes, o endarte-rectomía carotídea. El gasto en todos esos recursos de diagnóstico y terapéuticosen pacientes de bajo riesgo será innecesario.
Se han desarrollado varias escalas de pronóstico para la predicción del riesgode infarto temprano después del AIT. Hay dos tipos de escalas: las clínicas y las“clínicas–plus”; las primeras se basan en factores de predicción clínicos; entrelas segundas la más popular es la denominada ABCD2 (cuadro 9–1).9 Las escalasclínicas–plus calculan el riesgo con base en factores de predicción clínicos y ha-llazgos de imagen, como la ABCD2–I y la ABCD3–I.10,11
Algunos puntos prácticos respecto a la estratificación son, por ejemplo, quelos pacientes con escala ABCD2 > 3 puntos deben ser hospitalizados y estudiadosde manera urgente. Otros factores considerados de alto riesgo, además de la esca-la ABCD, son la evidencia de isquemia en los estudios de imagen y de enferme-dad de los grandes vasos. La incorporación de los hallazgos radiográficos en laestratificación de riesgo puede aumentar el valor pronóstico. La presencia de in-farto cerebral en la tomografía computarizada (TC) o en las DWI en pacientes conAIT aumenta drásticamente el riesgo subsecuente de infarto cerebral, teniendolas DWI un mayor valor predictivo que la TC. De manera similar, la imagen neu-rovascular puede mejorar la precisión en la estratificación de riesgo.
Fisiopatogenia del ataque isquémico transitorio
Hay algunas reglas clínicas que son especialmente útiles al estudiar a un pacientecon AIT. Siempre se debe considerar como primera posibilidad que sea debido

153Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
a enfermedad aterosclerosa extracraneal, especialmente si el paciente tiene facto-res de riesgo de aterosclerosis. Son dos los mecanismos por los que la enfermedadaterosclerosa extracraneal puede producir un AIT. El denominado embolismoarteria–arteria es el más común. En él una placa aterosclerosa localizada en laporción cervical de las arterias carótidas o vertebrales desprende material fibri-noplaquetario que es alojado en algunos de los vasos intracraneales o retinianos,ocluyendo de forma transitoria la circulación de la región en cuestión. Habitual-mente estos episodios son aislados y suelen ser seguidos por el desarrollo de in-farto cerebral.8,9
El segundo mecanismo consiste en falla hemodinámica. Una lesión ateroscle-rosa produce una lesión hemodinámicamente significativa, que en circunstanciasasociadas a hipotensión (farmacológica, postural, hipovolemia) produce las ma-nifestaciones deficitarias propias del AIT. Con frecuencia éstas suelen ser recu-rrentes y estereotipadas; es decir, se presentan de la misma manera en cada episo-dio. Este mecanismo es menos frecuente que el embolismo arteria–arteria.
En caso de descartar enfermedad de los vasos extracraneales hay que conside-rar la posibilidad de embolismo cardiogénico. Entre las potenciales fuentes car-dioembólicas hay que estar especialmente atentos a la presencia de fibrilaciónauricular paroxística. No son pocos los casos de pacientes que son evaluados porAIT en quienes son normales todos los estudios; sin embargo, en los próximosmeses puede ser detectada por algún método la presencia de fibrilación auricular.
Protocolo de estudio
La mayoría de los EVC isquémicos son debidos a lesión en uno o más de los vasoscerebrales. Hace muchos años se pensaba que el estudio Doppler carotídeo eraadecuado para visualizar la vasculatura cerebral. Sin embargo, la amplia disponi-bilidad actual de angiografía por resonancia magnética (ARM) y angiotomogra-fía (ATC) combinadas aumentan la sensibilidad y la especificidad, lo cual hacambiado el paradigma de la visualización de la vasculatura cerebral.
La resonancia magnética (RM) y la ATC pueden mostrar la mayoría de lasgrandes y medianas arterias y algunos de los pequeños vasos del arco aórtico, elcuello y las áreas intracraneales de forma no invasiva y detallada. Estas técnicasson muy sensibles para determinar áreas de estenosis, oclusión, irregularidadvascular, disecciones, aneurismas y malformaciones arteriovenosas. Estas técni-cas permiten también visualizar las estructuras venosas y a menudo detectar fís-tulas arteriovenosas durales. Para la visualización óptima en ambas se requiereel uso de material de contraste intravenoso, lo cual puede limitar el uso en pacien-tes con enfermedad renal.
La RM tiene la ventaja de que no necesita radiación ionizante y se puede reali-zar como parte de una IRM cerebral. Sin embargo, ni la IRM ni la RM se pueden

154 (Capítulo 9)Tratado de trombosis
llevar a cabo en pacientes con marcapasos, y los pacientes con claustrofobia re-querirán sedación. La ATC se puede realizar en pacientes con marcapasos, aun-que ello expone al paciente a una significativa cantidad de radiación. Una técnicaespecial de RM, llamada tiempo de vuelo (TOF: time of flight), se puede realizarsin el uso de contraste; ofrece una vista gruesa de los vasos intracraneales y extra-craneales, y permite determinar si están permeables o con enfermedad severa.
Tanto las técnicas de IRM como las de TC pueden ser usadas para determinarla perfusión cerebral, lo cual es útil en los casos en los que se desea evaluar cuan-do una lesión estenótica proximal produce isquemia distal. Estas lesiones puedenproducir isquemia limítrofe (AIT), particularmente en circunstancias en las quela presión arterial está reducida. Éste es un mecanismo común de AIT (e infartos)en pacientes perioperatorios o aquellos con graves padecimientos médicos.13
Imagen cardiaca
Actualmente hay dos técnicas de imagen cardiaca: el ecocardiograma transtorá-cico (ETT) y el ecocardiograma transesofágico (ETE). La IRM cardiaca (IRMc)aún está en etapa de desarrollo. El ETT y el ETE están ampliamente disponibles,por lo que son pruebas rutinarias en la mayor parte del mundo. El ETT es seguro,no invasivo y visualiza la mayoría de las estructuras cardiacas y válvulas de unamanera detallada y completa. Sin embargo, el ETT visualiza escasamente la ore-juela de la aurícula izquierda y el arco aórtico. En estas dos áreas las lesiones pue-den conducir al desarrollo de AIT o infarto cerebral.12
El ETE es un procedimiento de mínima invasión que a menudo requiere unaligera sedación. Es muy sensible para detectar pequeños coágulos en la orejuelaauricular izquierda, así como lesiones aterotrombóticas en el arco aórtico. Tantoel ETT como el ETE permiten visualizar lesiones valvulares, como las vegetacio-nes que pueden producir AIT.
INFARTO CEREBRAL ATEROSCLEROSO
La aterosclerosis es la principal enfermedad de la arterias grandes y de medianotamaño que irrigan al encéfalo, y tiende a desarrollarse en los puntos de bifurca-ción arterial (como en la bifurcación carotídea), de curvatura (en el arco aórtico)y de confluencia arterial (arteria basilar). A nivel extracraneal tiende a afectar labifurcación carotídea, la porción proximal de la carótida interna y el origen delas arterias vertebrales. En las arterias intracraneales la aterosclerosis a menudoafecta la porción terminal de las arterias carótida interna y basilar, y en menor gra-do las arterias cerebrales medias, cerebrales anteriores y cerebrales posteriores.

155Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
La isquemia cerebral relacionada con aterosclerosis es resultado de una oclusióntrombótica (aterotrombosis) o tromboembólica (embolismo arteria–arteria) delos vasos cerebrales. En los últimos años se ha reconocido que las placas ateroma-tosas del arco aórtico también son fuente importante de isquemia cerebral a travésde embolismo arteria–arteria, mecanismo que también se denomina aortoembo-lismo.13,14
La etiología aterosclerosa en un infarto cerebral (IC) se sospecha en pacientescon factores de riesgo vascular; se puede confirmar a través de estudios no invasi-vos que permiten evaluar el estado de las principales arterias. Las arterias cervi-cales, carótidas y vertebrales se pueden evaluar de manera adecuada medianteultrasonido de alta resolución, con ARM, ATC o angiografía cerebral, como yase comentó. La mayor parte de las lesiones ateromatosas son asintomáticas, yaque son de pequeño tamaño. Sin embargo, estas placas se hacen sintomáticas, ge-nerando isquemia en el parénquima cerebral por diferentes mecanismos, que acontinuación se detallan.15
1. Alteración hemodinámica: una importante reducción del calibre vascularcarotídeo conlleva una disminución de la presión de perfusión y del flujosanguíneo a nivel distal; por el tipo de circulación terminal del cerebro, losterritorios más comprometidos son los más alejados, los correspondientesa las arteriolas corticales, denominadas zonas limítrofes, que resultan afec-tados dependiendo del grado de estenosis y de la capacidad de la circulacióncolateral.
2. Oclusión vascular: las complicaciones de la placa, como la hemorragia in-traplaca y las ulceraciones de la superficie que favorecen la generación deun trombo mural, conducen al rápido aumento del tamaño de la placa, hastallegar a ocluir por completo la arteria.
3. Embolia arteria–arteria: se produce la oclusión de un vaso intracranealdebido a un fenómeno embólico formado por material que se ha generadoen la placa ateromatosa:a. Agregados fibrinoplaquetarios formados en la superficie. La mayoría de
las veces dichos agregados terminan autodisolviéndose en poco tiempo,constituyendo el sustrato fisiopatológico del ataque isquémico transitorio.
b. Trombo rojo estructurado: sobre un agregado fibrinoplaquetario se aña-den otros elementos formes de la sangre. La mayor parte de las veces sonlos responsables de los infartos cerebrales de origen aterotrombótico.
c. Material procedente de la ruptura de la placa, con alto componente de fi-brina y cristales de colesterol.
La embolia arteria–arteria puede también asociarse a otras patologías arteriales,como la disección arterial cervicocerebral, que es causa frecuente de infarto cere-bral en las personas menores de 45 años de edad sin factores de riesgo vascular.

156 (Capítulo 9)Tratado de trombosis
En todos los casos el IC ocurre con mayor frecuencia si la placa aterosclerosaestá ulcerada, por la facilitación a la agregación plaquetaria que produce la expo-sición de material subendotelial a la luz del vaso. La embolia arteria–arteria esquizá el mecanismo mas frecuente de IC en el territorio carotídeo.
La sintomatología clínica no permite diferenciar si la fuente de émbolos es car-diaca o carotídea. Desde luego, las manifestaciones neurológicas dependerán delterritorio y el lado afectado. Los IC aterotrombóticos suelen ser de tamaño de me-diano a grande y pueden afectar territorios corticales y subcorticales, siendo obje-tivables mediante pruebas de neuroimagen que corroboran la presunción clínicainicial. Una vez confirmado el diagnóstico y pasada la fase aguda del infarto cere-bral, lo siguiente es decidir sobre la mejor estrategia de prevención secundaria,que puede incluir endarterectomía, hipoglucemiantes y antihipertensivos en al-gunos casos, y antiagregantes y estatinas en todos.16 La endarterectomía carotí-dea reduce el riesgo de recurrencia de infarto cerebral de origen embólico en pa-cientes con lesiones ateromatosas de la bifurcación carotídea, mediante laresección de la placa de ateroma y la reconstrucción de la luz arterial. Hasta ahoralos estudios epidemiológicos y los ensayos clínicos han dejado en claro que lospacientes asintomáticos con lesión aterosclerosa tienen un riesgo menor de infar-to cerebral, en comparación con aquellos que ya han sufrido un infarto cerebralatribuido a lesión aterosclerosa. Por lo tanto, en los pacientes sintomáticos conestenosis severa (entre 70 y 99%) está indicada la realización de endarterectomía,ya que confiere un beneficio a largo plazo, al reducir el riesgo absoluto de un in-farto cerebral (13%) y la muerte (16%) en los siguientes dos años. El número nece-sario a tratar para evitar un nuevo evento recurrente es de ocho casos. Los pacientescon estenosis leves (< 50%) de la luz de la arteria se benefician más del tratamientomédico, por lo que no deben ser sometidos a este procedimiento.16,17
En los pacientes con estenosis moderadas (50 a 69%) se ha mostrado una re-ducción significativa del riesgo absoluto de infartos y de muerte de 5.3%; sin em-bargo, en este grupo no se encontraron beneficios en las mujeres ni en los pacien-tes con síntomas retinianos. La recomendación general es determinar el grado deestenosis, así como las características de la placa, antes de tomar la decisión dedar un tratamiento médico o hacer una endarterectomía. La colocación de stentcon protección distal es otra opción de tratamiento en casos seleccionados. En laactualidad existe un gran debate acerca de los pacientes que deben ser tratadoscon endarterectomía o con stent.
Los antiagregantes plaquetarios son la piedra angular en la prevención secun-daria del IC ateroscleroso. La decisión de su uso en pacientes asintomáticos y sineventos vasculares previos debe balancear entre el potencial beneficio de reduc-ción del riesgo de IC y el riesgo de complicaciones hemorrágicas. Se sabe queel ácido acetilsalicílico (ASA) es relativamente seguro y barato, y se encuentraampliamente disponible, por lo que es el agente con más indicaciones en la pre-

157Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
vención primaria en individuos con alto riesgo vascular sin fibrilación auricular,a pesar de que se cuenta con poca información de su efecto en prevención prima-ria. El riesgo de hemorragia con su uso es relativamente bajo; por cada 1 000 pa-cientes tratados en cinco años ocurre una hemorragia intracerebral y tres gastro-intestinales.17,18 Contrarios a la evidencia que apoya el uso de antiagregantes enprevención secundaria, los datos de prevención primaria muestran un efecto muydiscreto o pequeño, por lo que no está indicado en todos los casos. En contraparte,un metaanálisis de antiagregantes (comparados contra control) demostró una re-ducción de 22% del riesgo combinado de IC recurrente, de 21.4 de infarto delmiocardio y de 17.8% de muerte vascular, así como un beneficio absoluto en 36pacientes de 1 000 que fueron tratados. De forma similar, se previenen 17 IC re-currentes en cada 1 000 pacientes tratados. Las opciones aprobadas en preven-ción secundaria son el ASA, el clopidogrel y la combinación de ASA/dipiridamolde liberación prolongada. Las recomendaciones actuales de la guías de preven-ción sugieren que:18
1. La monoterapia con ASA en dosis de 50 a 325 mg (clase I, nivel de eviden-cia A), la combinación de ASA en dosis de 25 mg más dipiridamol de libera-ción prolongada en dosis de 200 mg dos veces al día (clase I, nivel de evi-dencia B) o el clopidogrel en dosis de 75 mg al día (clase IIa, nivel deevidencia B) son opciones aceptables como terapia inicial, pero la decisiónsobre alguno en especifico debe ser individualizada.
2. En los pacientes alérgicos al ASA el clopidogrel es una opción razonable.3. La combinación de ASA y clopidogrel incrementa el riesgo de hemorragia,
por lo que no se recomienda su uso rutinario en pacientes con IC o AIT, amenos que tengan una indicación específica, como colocación de stent co-ronario o síndrome coronario agudo.
4. En pacientes que desarrollan un nuevo evento vascular mientras se encuen-tran en tratamiento con ASA no existen evidencias de que el incremento dela dosis brinde protección adicional. Aunque con frecuencia se consideranotras opciones de antiagregantes, no se ha estudiado el efecto de un antia-gregante solo o combinado en este grupo de pacientes (clase IIB, nivel deevidencia C).
En relación con el uso de estatinas, los hallazgos del estudio SPARCL19 mostra-ron que los pacientes con IC previa tratados con atorvastatina en dosis de 80 mgtuvieron una reducción de la recurrencia de IC o AIT de 23% (P < 0.001) y unadisminución de 26% de otros eventos cardiovasculares (coronarios y periféri-cos). Los metaanálisis posteriores, que incluyen los resultados del estudioSPARCL, confirman una reducción del riesgo de recurrencia de al menos 18%.Asimismo, varios estudios en los últimos años han puesto en evidencia que la sus-pensión de las estatinas se asocia a la recurrencia de eventos cerebrovasculares

158 (Capítulo 9)Tratado de trombosis
a corto plazo y que cuando se suspende el tratamiento con estatinas en la fase agu-da de IC se puede asociar a un incremento del riesgo de muerte o de dependenciafuncional. Debido a los datos anteriores, se recomienda iniciar con estatinas, depreferencia atorvastatina, en todos los pacientes con IC ateroscleroso, y mantenersu administración mientras sea posible.
Lesiones aterosclerosas del arco aórtico
Está bien establecido que la enfermedad aterosclerosa de la aorta torácica es unfactor de riesgo independiente para infarto cerebral.20 El grosor de la placa de ate-roma y su morfología (ulceración, calcificación, sus características de movilidado su estado de protrusión) se relacionan estrechamente con un mayor riesgo deisquemia cerebral. Las placas > 4 mm representan, al parecer, un riesgo de IC mu-cho mayor. Los estudios reportados utilizan principalmente ETE para su identifi-cación y caracterización.21
La presencia de placas aterosclerosas en el arco aórtico no sólo representan unmayor riesgo de infarto cerebral, sino también un peor pronóstico por la mayorfrecuencia de recurrencia, sobre todo en los pacientes con placas de ateroma �4 mm de diámetro. Recientemente, en el estudio SPARCL se buscó la localiza-ción de la aterosclerosis en diferentes regiones del arco aórtico y se demostró unamayor prevalencia en el arco (28%) y la aorta descendente (38%), asociada espe-cialmente a una mayor edad. En la búsqueda de diferencias raciales en la enfer-medad aterosclerosa, Di Tullio y col. han sugerido que el riesgo de infarto cere-bral en aterosclerosis aórtica puede ser similar en diferentes grupos étnicos.22 Sinembargo, los estudios posteriores han mostrado que los pacientes negros tienenuna significativa menor prevalencia de enfermedad aterosclerosa extracraneal,aun cuando la frecuencia de los principales factores de riesgo, principalmente hi-pertensión y diabetes, son mayores en este grupo étnico.
Aunque se reconoce como una potencial fuente de infarto cerebral, el aortoem-bolismo es poco estudiado en la práctica clínica diaria, quizá porque el mejormétodo para su reconocimiento es el ecocardiograma transesofágico, que no seencuentra disponible en todos los centros hospitalarios. Se debe sospechar en pa-cientes con factores de riesgo vascular, infarto cerebral y ausencia de lesiones ca-rotídeas que justifiquen la sintomatología.
El tratamiento de los pacientes con infarto cerebral secundario a aortoembolis-mo aún no está definido. Se han propuesto los antiagregantes solos o combinadoso incluso la anticoagulación oral. Hasta que existan evidencias claras acerca delmejor tratamiento los antiagregantes plaquetarios, las estatinas y en algunos ca-sos los hipoglucemiantes y los antihipertensivos podrán ser sugeridos en estos ca-sos.20,22

159Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Enfermedad de los pequeños vasos cerebrales
La microangiopatía cerebral representa una parte importante de los eventos vas-culares isquémicos, además de que puede ocasionar también deterioro cognitivoy demencia. Se calcula que de 20 a 30% de los IC se deben a enfermedad cerebralde los pequeños vasos.23 El infarto lacunar (IL) es un infarto de un tamaño nosuperior a 20 mm, localizado en el territorio de distribución de una arteriola per-forante cerebral que da lugar a uno de los cinco síndromes clínicos lacunares: he-miparesia motora pura, síndrome sensitivo puro, síndrome sensitivo motor, dis-artria–mano torpe y hemiparesia atáxica. La hipertensión arterial (HTA) es unimportante factor de riesgo independiente para los EVC en general, así como elprincipal factor de riesgo para los IL, puesto que la prevalencia de la HTA es ma-yor en los IL (> 70%) que en los otros subtipos de EVC. Asimismo, la presenciade IL múltiples se asocia significativamente con la presencia de HTA. La diabetesmellitus es también un factor de riesgo y probablemente causa de algunos IL. Suprevalencia es también mayor en los IL que en los otros subtipos de IC, confir-mándose como un factor de riesgo independiente para IL, principalmente cuandose presenta en forma de IL múltiples.
La RM constituye en la actualidad el estudio de elección para la confirmaciónde los IL. Su eficacia diagnóstica es significativamente mayor que la de una to-mografía, principalmente en los IL de localización en el puente y en la cápsulainterna.
La evolución de los IL muestra un curso clínico paradójico con pronósticofavorable en el corto plazo, caracterizado por una baja mortalidad temprana ypoca discapacidad en el momento del egreso hospitalario, pero con incrementodel riesgo de muerte, recurrencia y demencia en el seguimiento a mediano y largoplazos. En la fase aguda la mortalidad en los IL varía de 0 a 2% a los 30 días, mien-tras que el riesgo de recurrencia precoz muestra una media de 7.7%, con un rangode 2 a 12%. Sin embargo, a los cinco años la recurrencia es de 22.4% y se debeprincipalmente a nuevos infartos lacunares (50 a 72%) y con menos frecuenciaa hemorragias intracerebrales (10%). Si bien los infartos lacunares iniciales sue-len ocasionar una ligera limitación funcional, los IL recurrentes o múltiples pue-den ser responsables de un estado lacunar o de una demencia vascular.24
La progresión asintomática de la enfermedad de los pequeños vasos cerebraleses un hallazgo típico en la evolución de los IL, por lo que la enfermedad de lospequeños vasos cerebrales debe ser considerada como una condición potencial-mente severa, más que un trastorno benigno, ya que los pacientes con IL requie-ren un adecuado y riguroso manejo y seguimiento.
Los antiagregantes plaquetarios, el cuidadoso control de la presión arterial, elcontrol de las cifras de glucosa, el uso de estatinas y la modificación del estilo devida son los elementos clave en la prevención secundaria del IL. Se recomienda

160 (Capítulo 9)Tratado de trombosis
la combinación de ASA, dipiridamol más ASA o clopidogrel solo, igual que enel resto de los infartos cerebrales. Recientemente se dieron a conocer los resulta-dos del estudio Secondary Prevention of Small Subcortical Strokes (SPS3),25 unensayo clínico de fase III cuyo objetivo fue analizar la utilidad de la terapia antia-gregante plaquetaria en pacientes con IL, utilizando antiagregación sola o doble(ASA 325 mg/día o ASA más clopidogrel 75 mg/día), así como dos tipos de con-trol de la tensión arterial sistólica: intensiva (< 130 mmHg) o usual (130 a 149mmHg), para prevenir la recurrencia y el deterioro cognitivo de la población ana-lizada. Los resultados del estudio no demostraron que la combinación de anti-agregantes plaquetarios fuera superior que el ASA solo, y la terapia dual se asocióa una mayor frecuencia de hemorragias y más alta mortalidad. Por otro lado, losdiferentes controles de la presión arterial estudiados tampoco mostraron diferen-cias significativas. Queda claro que los pacientes con IL deben recibir un antia-gregante plaquetario; hasta ahora las evidencias sugieren que se debe utilizar mo-noterapia.
A lo largo de este capítulo se ha mostrado la importancia de reconocer el AIT,los diferentes subtipos de IC ateroscleroso y de pequeños vasos, sus manifesta-ciones clínicas y la importancia de la prevención secundaria. Los pacientes conAIT, IC ateroscleroso o IL requieren de un tratamiento combinado con antiagre-gantes plaquetarios, estatinas y control estricto de factores de riesgo vascular,especialmente de hipertensión arterial y diabetes mellitus.
REFERENCIAS
1. Barinagarrementeria F, Arauz A (eds.): Temas selectos de enfermedad vascular cerebral.Elsevier, 2012
2. Petty GW, Brown R, Whisnant JP, Sicks J, O’Fallon M et al.: Ischemic stroke subtypes.A population–based study of functional outcome, survival, and recurrence. Stroke 2000;31:1062–1068.
3. Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB et al.: Classification of subtypeof acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial ofOrg 10172 in Acute Stroke Treatment. Stroke 1993;24:35–41.
4. Wardlaw JM, Smith C, Dichgans M: Mechanisms of sporadic cerebral small vessel dis-ease: insights from neuroimaging. Lancet Neurol 2013;12:483–487.
5. Siket MS, Edlow J: Transient ischemic attack: an evidence–based update. Emerg MedPract 2013;15:1–26.
6. Thomassen L, Waje Andreassen U, Moen G, Logallo N, Naess H: Clinical implicationsof increased use of MRI in TIA. Acta Neurol Scand 2012.
7. Purroy F, Jiménez CPE, Mauri CG et al., from the PROMAPA study: Stroke Project,Cerebrovascular Diseases Study Group, Spanish Neurological Society: Predictive value ofbrain and vascular imaging including intracranial vessels in transient ischaemic attack pa-tients: external validation of the ABCD3–I score. Eur J Neurol 2013;10. 1111/ene.12141.
8. Easton JD, Albers GW, Caplan LR, Saver JL, Sherman DG: Discussion: reconsidera-tion of TIA terminology and definitions. Neurology 2004;62:S29–S34.

161Ataque isquémico transitorio, infarto cerebral aterotrombótico...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
9. Easton JD, Saver JL, Albers GW et al.: Definition and evaluation of transient ischemicattack: a scientific statement for healthcare professionals from the American Heart Associa-tion/American Stroke Association Stroke Council; Council on Cardiovascular Surgery andAnesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovas-cular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. Stroke2009;40:2276–2293.
10. Van Rooij FG, de Leeuw FE, van Dijk EJ: Early stroke risk and ABCD2 score perfor-mance in tissue– vs. time–defined TIA: a multicenter study. Neurology 2012;78:224.
11. Song B, Fang H, Zhao L, Gao Y, Tan S et al.: Validation of the ABCD3–I score to predictstroke risk after transient ischemic attack. Stroke 2013;44:1244–1248.
12. Arias S, Rodríguez M, López A, Santamaría M, Fernández PG et al.: Is necessary toperform a transthoracic echocardiogram in all the patients with cryptogenic stroke duringhospitalization? Rev Neurol 2013;56:510–514.
13. Kim JM, Jung KH, Sohn CH, Moon J, Han MH et al.: Middle cerebral artery plaque andprediction of the infarction pattern. Arch Neurol 2012;69:1470–1475.
14. Van Gils MJ, Vukadinovic D, van Dijk AC, Dippel DW, Niessen WJ et al.: Carotid athe-rosclerotic plaque progression and change in plaque composition over time: a 5–year fol-low–up study using serial CT angiography. AJNR Am J Neuroradiol 2012;33:1267–1273.
15. Cicha I, Wörner A, Urschel K, Beronov K, Goppelt Struebe M et al.: Carotid plaquevulnerability: a positive feedback between hemodynamic and biochemical mechanisms.Stroke 2011;42:3502–3510.
16. Villarreal CJ, Murillo BL, Góngora RF, Leyva RA, Barinagarrementeria F et al.: Guíaclínica para el tratamiento quirúrgico (endarterectomía) y endovascular (angioplastia conprotección distal y colocación de stent) para la prevención secundaria de la isquemia cere-bral asociada a enfermedad aterosclerosa carotídea. Rev Invest Clín 2010;62:170–180.
17. Burgazli KM, Bilgin M, Kavukcu E, Mericliler M, Bohl N et al.: Which is a better treat-ment for carotid artery stenosis: stenting or endarterectomy? Eur Rev Med Pharmacol Sci2013;17:1025–1032.
18. Barinagarrementeria F, Arauz A, Ruiz SJL, Cantú C, Leyva A et al.: Antiplaquetariosen la prevención del infarto cerebral o isquemia cerebral transitoria aterotrombótica. RevInvest Clín 2010;62:135–140.
19. Goldstein LB, Amarenco P, Szarek M et al.: Effects of intense low–density lipoproteincholesterol reduction in patients with stroke or transient ischemic attack: the Stroke Preven-tion by Aggressive Reduction in Cholesterol Levels (SPARCL) Trial. Stroke 2008;38:3198–3204.
20. Fujimoto S, Yasaka M, Otsubo R, Oe H, Nagatsuka K et al.: Aortic arch atheroscleroticlesions and the recurrence of ischemic stroke. Stroke 2004;35:1426–1429.
21. Grupta V, Nanda N, Yesilbursa D, Huang W, Grupta V et al.: Racial differences in thora-cic aorta atherosclerosis among ischemic stroke patients. Stroke 2003;34:408–412.
22. Di Tulio M, Sacco R, Savoia MT et al.: Aortic atheroma morphology and the risk of ische-mic stroke in a multiethnic population. Am Heart J 2000;139:329–336.
23. Arboix A, Marti Vilalata JL: Enfermedad vascular cerebral de pequeño vaso. En: Barina-garrementeria F, Arauz A (eds.): Temas selectos en enfermedad vascular cerebral. Elsevier,103–127.
24. Arboix A, Altés E, García EL, Massons J: Clinical study of lacunar infarcts in non–hyper-tensive patients. J Stroke Cerebrovasc Dis 2003;12:232–236.
25. Benavente OR, Hart RG, McClure LA, Szychowski JM et al.: Effects of clopidogreladded to Aspirin in patients with recent lacunar stroke. N Engl J Med 2012;367:817–825.

162 (Capítulo 9)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
10Trombosis venosa cerebral
Carlos F. Cuevas García, Irene Pérez Páez
INTRODUCCIÓN
La trombosis venosa cerebral (TVC) es una condición clínica neurológica pocofrecuente que representa cerca de 0.5% de todos los casos de enfermedad vascu-lar cerebral (EVC) y casi siempre afecta a adultos jóvenes y niños. Tiene una inci-dencia anual estimada de tres a cuatro casos por cada millón de habitantes y au-menta a siete casos por cada millón de habitantes infantiles.3 Entre la poblaciónadulta más de 75% de los casos corresponden al sexo femenino. Las manifesta-ciones clínicas de la TVC y su modo de inicio son variables, por lo que representaun reto diagnóstico para el clínico.
En 1825 se describió el primer caso de TVC, lo cual dio pauta a la posteriorpublicación de reportes de casos aislados post mortem, los cuales tenían en co-mún una presentación clínica manifestada con cefalea, papiledema, crisis con-vulsivas, déficit focal, coma y muerte. En esa época los principales factores deriesgo identificados fueron los procesos infecciosos;7 posteriormente se descu-brieron e implementaron métodos como la angiografía y la tomografía axial com-putarizada de cráneo, que mejoraron el estudio y el tratamiento de la enfermedad.En la actualidad la resonancia magnética (IRM) y la angiografía por resonanciamagnética (angio–IRM) constituyen los mejores métodos diagnósticos de la en-fermedad.
A pesar de estos avances, la proporción de casos en los que la etiología no logradetectarse aún es muy alta y el pronóstico, aunque mejor que lo que era antes, nologra ser todavía el óptimo. Más de 80% de todos los pacientes actuales tienen
163

164 (Capítulo 10)Tratado de trombosis
un buen pronóstico neurológico. El tratamiento, que debe iniciarse tan prontocomo se establezca el diagnóstico, consiste en revertir la causa subyacente siem-pre que se conozca y controlar las crisis convulsivas y la hipertensión endocra-neana, así como en el uso de agentes antitrombóticos. La heparina constituye elagente de elección y los recientes estudios confirman su seguridad, incluso en pa-cientes con lesiones parenquimatosas hemorrágicas. Hoy en día se discute el usode heparina de bajo peso molecular. La indicación de trombólisis local, la defe-nestración del nervio óptico para mejorar el papiledema y otras pautas terapéuti-cas se discuten brevemente en este capítulo.
CIRCULACIÓN VENOSA CEREBRAL
Las venas cerebrales se dividen en un grupo superficial y un grupo profundo. Lasvenas superficiales drenan las superficies corticales a través de cuatro grupos devenas “puente”:
1. El grupo sagital superior, que drena el seno longitudinal superior.2. El grupo esfenoidal, que drena el seno esfenoparietal y el seno cavernoso.3. El grupo tentorial, que converge en los senos del tentorio.4. El grupo falcine, que drena al seno longitudinal inferior y el seno recto.4
El sistema venoso profundo colecta dentro de canales que cursan a través de lasparedes de los ventrículos y las cisternas basales y converge a nivel de la venacerebral interna, la vena basal de Rosenthal y la gran vena de Galeno. El territoriocubierto por estas venas incluye no sólo los plexos coroideos y la sustancia grisdel tálamo y del cuerpo estriado, sino también la sustancia blanca periventricular,el cuerpo calloso, el hipocampo y las áreas corticales del sistema límbico (el cín-gulo y el giro hipocampal), la corteza visual, el diencéfalo y parte del cerebelo.La vena basal se conecta con la gran vena de Galeno y con el seno petroso supe-rior. En una persona adulta se puede conectar también con el seno cavernoso yel plexo pterigoideo, y en algunas ocasiones con el seno lateral. Debido a todaslas interconexiones del sistema venoso, sólo la obstrucción simultánea de la granvena de Galeno y las venas basales da por resultado la obstrucción completa dela circulación venosa profunda.5
EPIDEMIOLOGÍA DE LA TROMBOSIS VENOSA CEREBRAL
La TVC representa < 1% de la enfermedad vascular cerebral. La incidencia anualestimada va de tres a cuatro casos por cada millón de personas en general y másde siete casos por millón en niños. La presentación de casos más frecuente es enla tercera década de la vida, y cerca de 75% de los pacientes adultos afectados son

165Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
mujeres.3 En el Estudio Internacional de Trombosis de las Venas Cerebrales y Se-nos Durales (ISCVT) se reportó una mortalidad de 8%.9
La trombosis del seno transverso es la más frecuente, seguida de la del senosagital y la del seno sigmoides. Cerca de la mitad de los casos involucran a másde un seno.8
FACTORES DE RIESGO
La TVC se describió por primera vez en el siglo XIX, y la mayoría de las prácticasse llevaron a cabo post mortem. En aquellos tiempos los procesos infecciososeran la principal causa de este padecimiento; sin embargo, las publicaciones pos-teriores postularon causas no infecciosas para el desarrollo de TVC, aunque enla actualidad se han descrito más de 100 causas.1
En 1888 William Gowers describió la relación de la TVC con condicionescomo marasmo y caquexia; después Colmes y Sargent describieron un caso deTVC asociado con trauma, y en 1941 Martin y Sheehan hicieron la asociación deTVC con el puerperio, que ya había sido descrita antes por Gowers. En 1953 Bar-nett y Hylland publicaron una de las primeras series de casos, en la que reportaronel hallazgo de 39 casos (todos post mortem), en la que se señalaba que la etiologíade la trombosis venosa intracraneal era similar a la etiología de la trombosis venosade cualquier otro sitio y que ocurría bajo circunstancias similares. En este trabajose señalaron factores predisponentes a las enfermedades cardiacas, como caque-xia y marasmo (los que están asociados con estados de deshidratación), posciru-gía, postrauma y algunas discrasias sanguíneas.10
En la actualidad se han estudiado los diversos factores que pueden causar opredisponer a TVC. En el ISCVT cerca de 44% de los pacientes tuvieron más deuna causa o factor predisponente, y en cerca de 22% de los casos se logró identifi-car la presencia de alguna trombofilia. Es importante puntualizar que una vez quese logra identificar un factor de riesgo no se puede descartar la presencia de unomás, en especial las trombofilias congénitas.9 En el cuadro 10–1 se muestran losprincipales factores asociados con TVC.
Dentro de los denominados estados protrombóticos, la deficiencia de anti-trombina y la disfibrinogenemia fueron las primeras trombofilias descritas en fa-milias cuyos miembros eran afectados por trombosis venosa. Más tarde se identi-ficaron deficiencias heterocigotas en las proteínas C y S como causas detrombofilias hereditarias. Subsecuentemente se descubrió la presencia del factorV de Leiden, considerado hasta el momento como la trombofilia más frecuente.Otras de las trombofilias hasta ahora descritas son la homocistinuria y la muta-ción puntual específica del gen de la protrombina.12

166 (Capítulo 10)Tratado de trombosis
Cuadro 10–1. Factores de riesgo relacionados con trombosis venosa central
Estados protrombóticos:Factores genéticosDeficiencia de antitrombinaDéficit de proteínas C y S
Mutación del factor V de LeidenMutación del gen de la protrombina (G20210A)Mutación del gen de la metilentetrahidrofolatorreductasa
Factores adquiridosAnticuerpos antifosfolípidosSíndrome nefrótico
HiperhomocisteinemiaEmbarazoPuerperio
Malignidad:Del sistema nervioso centralTumores sólidos fuera del sistema nervioso centralHematológicos
Trastornos del sistema nervioso central:Fístula duralMalformación AV
Hematológicos:PolicitemiaAnemia
Vasculitis:Lupus eritematoso sistémicoEnfermedad de Behçet
Artritis reumatoideOtros trastornos inflamatorios sistémicos:
Enfermedad inflamatoria intestinal
SarcoidosisInfección:
Sistema nervioso central
OídoSinusitisBocaCara
CuelloMecánicos:
Punción lumbar
Traumatismo cranealNeurocirugía
Fármacos:Anticonceptivos oralesTerapia de reemplazo hormonal

167Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 10–1. Factores de riesgo relacionadoscon trombosis venosa central (continuación)
EsteroidesFármacos citotóxicosCirugíaDeshidratación
Fuente: Ferro, Canhão: Stroke 2004;35:664.
La deficiencia de proteínas C y S y de antitrombina secundaria a mutacionesen los genes que codifican para esas proteínas se ha reconocido como causa deTVC y representa menos de 10% de todas las causas. Los reportes recientes indi-can una incidencia cercana a 20% de TVC asociado con la presencia del factorV de Leiden. Asimismo, la mutación en la región 3’ no traducida del gen de laprotrombina (conversión de G en A en la posición 20210:G20210A) se encuentraasociada con un incremento en las concentraciones plasmáticas de protrombinay con un riesgo importante de desarrollar trombosis venosa cerebral.11
La incidencia de resistencia de la proteína C activada en la presencia de trom-bosis venosa en la población europea va de 21 a 64%. El uso concomitante de an-ticonceptivos incrementa el riesgo de trombosis entre 30 y 50 veces más. Dhal-back y Svensson describieron TVC en pacientes con presencia de resistencia ala proteína C activada (R–PCA) que además tenían otro factor de riesgo, como eluso de anticonceptivos. Esto indica que se requieren factores de riesgo adicionalesen la mayoría de los casos para que los pacientes desarrollen TVC en presenciade R–PCA. A partir de ese estudio se han publicado otros más, que han reportadouna incidencia de factor V de Leiden entre 10 y 21% de los casos de TVC.13,14
Los diversos estudios muestran una asociación entre hiperhomocisteinemia(HHC), enfermedad vascular arterial y tromboembolismo venoso, la cual se haidentificado hasta en 27 a 47% de los pacientes con TVC. Un trabajo publicado porMartinelli, que incluyó a 121 pacientes con un primer episodio de TVC, reportóque la HHC incrementa hasta cuatro veces más el riesgo de desarrollar TVC.15
Los factores genéticos y nutricionales son determinantes para el metabolismode la homocisteína. Se reconoce como factor de riesgo la presencia de la muta-ción C67737 en el gen de la metilentetrahidrofolatorreductasa (MTHFR), que dacomo resultado una variante termolábil, lo cual reduce la actividad de la enzimahasta 50%. Entre 10 y 13% de la población general son homocigotos para estamutación.11
Los niveles de folato, vitamina B12 y con menor frecuencia vitamina B6 se en-cuentran relacionados en forma inversa con la homocisteína, de tal forma quecualquiera que presente una deficiencia nutricional de estas vitaminas presentaun riesgo elevado de desarrollar HHC. La interacción entre el medio ambientey los factores genéticos es importante para el incremento de los niveles de homo-

168 (Capítulo 10)Tratado de trombosis
cisteína, ya que los trastornos metabólicos hereditarios se manifiestan más en losindividuos con un deficiente estado nutricional. Los casos de TVC durante el em-barazo y el puerperio descritos en México corresponden a pacientes de bajo nivelsocioeconómico y con deficiencias nutricionales, en las cuales se han correlacio-nado los bajos niveles de folato con la presencia de anemia, de tal forma que laescasa ingestión de folatos es “causa” del incremento en la frecuencia de TVCen México.16,17
La presencia de anticuerpos anticardiolipina (aCL) o de anticoagulante lúpico(LA) constituye un factor importante para el desarrollo de TVC. Se han identifi-cado aCL en cerca de 5% de la población con TVC.9 La TVC relacionada con lapresencia de aCL suele presentarse en pacientes muy jóvenes con un cuadro clíni-co más extenso y con mayor afección del sistema venoso profundo.18
La TVC puede ser uno de los mecanismos de afección del sistema nerviosocentral (SNC) en pacientes con lupus eritematoso sistémico (LES). Los pacientescon anticoagulante lúpico (AL), trombocitopenia y eventos vasculares están pre-dispuestos a desarrollar TVC. Debido a que la TVC puede llegar a ser una de lasformas de presentación del LES (1% de los casos), en todo paciente con TVC sedebe descartar la presencia del AL y de LES.19
La enfermedad de Behçet es una enfermedad vascular sistémica inflamatoriacon una variedad de manifestaciones clínicas que puede afectar venas de diferen-te tamaño con tendencia a la trombosis. La TVC es una complicación frecuentede la enfermedad (cerca de 30% de los pacientes con afección neurológica desa-rrollarán TVC), que a veces se presenta incluso como la manifestación inicial,sobre todo en los pacientes jóvenes.25
Durante el último trimestre del embarazo y el periodo posterior al parto existeun incremento en el riesgo de trombosis asociado con el incremento de R–PCAdurante el embarazo o con una disminución de los niveles de proteína C activadaposterior a la cesárea. La frecuencia de trombosis durante el parto y el pospartoes de casi 12 casos por cada 100 000 nacimientos. Entre los factores relacionadoscon el parto y el puerperio, los diversos estudios muestran que la operación cesá-rea y la hipertensión vinculada con el embarazo son factores importantes para eldesarrollo de TVC.20
Los anticonceptivos orales (AO) son ampliamente utilizados por millones demujeres de todo el mundo, y los grandes estudios epidemiológicos han confirma-do que su uso, en particular los de tercera generación, confiere un riesgo elevadopara el desarrollo de tromboembolismo venoso. En un trabajo de Martinelli y col.se mostró que las mujeres que los consumen presentan un riesgo de hasta cuatroveces más de desarrollar TVC. Además, se ha observado que este riesgo se incre-menta en presencia de alguna alteración protrombótica.21,22
La frecuencia de TVC secundaria a un proceso infeccioso ha disminuido hasta6% en las grandes series.3 La otitis y la mastoiditis pueden tener como complica-

169Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ción trombosis del seno sigmoideo o transverso adyacente. Los principales agen-tes causales son Staphylococcus aureus, Streptococcus spp. y Escherichia coli,con un cuadro clínico característico que incluye picos febriles, cefalea, otalgia,edema y sensibilidad aumentada en el mastoideo. Sin embargo, cuando la trom-bosis afecta en forma significativa la reabsorción del LCR o el flujo venoso cere-bral se puede desarrollar lo que se conoce como hidrocefalia ótica, que incluyecefalea intensa, náusea, vómito, parálisis del nervio craneal VI y papiledema.23
La tuberculosis se asocia con TVC, sin presentar necesariamente infección di-recta del SNC, y con meningitis tuberculosa. Entre los mecanismos que se postu-lan están el daño directo del endotelio, las alteraciones del flujo sanguíneo y lasalteraciones del estado de coagulabilidad.24
Existen pocas series de pacientes con TVC relacionado con cáncer, y se men-cionan como etiologías oncológicas la compresión por metástasis, los estados dehipercoagulabilidad relacionados con el mismo cáncer y la toxicidad vinculadacon la quimioterapia. Existe un aumento en la incidencia de estados de hipercoa-gulabilidad en pacientes con meningiomas y gliomas de alto grado de diferencia-ción. En los pacientes con tumores primarios del SNC es muy importante el diag-nóstico temprano de TVC a través de estudios de imagen no invasivos. En elestudio publicado por Raizer y col. los procesos hematológicos fueron más fre-cuentes en la población joven, en comparación con los tumores sólidos, y el diag-nóstico de TVC se realizó en forma más temprana en los procesos hematológicos,lo cual se debe a que la TVC de los tumores sólidos por lo general es secundariaa lesiones metastásicas y, por lo tanto, la presentación suele ser tardía.26
La trombosis de los senos venosos asociada con leucemia fue descrita por pri-mera vez por Gowers en el siglo XIX. En algunos reportes se menciona que másde 50% de las trombosis relacionadas con LLA ocurren en el SNC. La TVC esel resultado de la combinación de factores protrombóticos y de las condicionesclínicas subyacentes. Cerca de 60% de los niños con LLA presentan dos factoresde riesgo de manera concomitante, mientras que 40% presentan tres. Los eventostrombóticos se asocian con frecuencia con el uso de L–asparaginasa (L–ASP) du-rante la inducción de la remisión, ya que la L–ASP puede reducir las concentra-ciones plasmáticas de las proteínas aglutinantes, en especial de la antitrombina.27
Entre las causas mecánicas de la trombosis de los senos venosos se encuentranel traumatismo craneoencefálico y el daño directo sobre los senos venosos, asícomo la lesión de las venas yugulares, que en ocasiones se origina durante la colo-cación de catéteres. Los procedimientos neuroquirúrgicos constituyen otra causade TVC, como la punción lumbar, cuando al haber una baja presión de LCR pos-terior a la punción se provoca un cambio hacia abajo sobre el cerebro con la consi-guiente tracción de las venas corticales y de los senos. La deformidad de las pare-des de las venas también puede inducir trombosis.3,28 Existen muchas otrascausas de TVC, como la hipotensión intracraneal, el uso de talidomida y tamoxi-

170 (Capítulo 10)Tratado de trombosis
feno1 y la administración de eritropoyetina, que provoca un aumento en las cifrasdel hematócrito.29,30 No obstante, en 15% de los pacientes no se logra identificarla causa de la TVC, de tal forma que todo paciente que presenta la enfermedadrequiere un amplio estudio para descartar todas las posibles causas.
FISIOPATOLOGÍA
Para comprender la sintomatología descrita es necesario ver este padecimientocomo dos mecanismos diferentes. Por una parte existe trombosis de las venas ce-rebrales por una obstrucción parcial, ya sea por un trombo o por compresión ex-trínseca que al final progresa a una oclusión completa, lo cual genera edema locale infartos venosos, que pueden generar grandes hematomas. Por otro lado se tieneel desarrollo de hipertensión intracraneal, ya que al haber trombosis de los senosvenosos se produce un aumento de la presión venosa, lo cual altera la absorcióndel líquido cefalorraquídeo y con ello un aumento en la presión intracraneal. Noexiste dilatación de los ventrículos ni datos de hidrocefalia, ya que no se desarro-lla un gradiente de presión entre los espacios subaracnoideos en la superficie delcerebro y los ventrículos.3,6
CUADRO CLÍNICO
La forma en que se presenta el cuadro clínico de la TVC depende de la edad delpaciente, el tiempo de presentación, la localización de la TVC y la presencia delesiones a nivel del parénquima. El surgimiento de la TVC suele ser agudo (hastados días) en 30% de los casos, y casi siempre se asocia con signos de focalización,como la TVC secundaria a procesos infecciosos o de origen obstétrico. Las cau-sas subagudas (que van desde días hasta un mes) se presentan en 50% de los ca-sos, por lo general asociadas con procesos inflamatorios sistémicos y con trastor-nos de la coagulación, los cuales se vinculan también con las formas crónicas(duración de más de un mes) en 20% de los casos.31 Los pacientes con un curso máscrónico o una presentación más retrasada pueden presentar datos de hipertensiónintracraneal y papiledema en el fondo de ojo, lo cual constituye un hallazgo fre-cuente.30
Existe una gran variedad de manifestaciones clínicas en los pacientes conTVC. En el ISCVT los síntomas más frecuentes fueron cefalea en 89%, paresiasen 37%, crisis convulsivas generalizadas en 30%, crisis convulsivas parciales en20%, papiledema en 28% y alteraciones del estado mental en 22% de los casos(cuadro 10–2).9

171Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 10–2. Manifestaciones clínicas más frecuentes
CefaleaParesiasCrisis convulsivasPapiledemaPérdida visualDiplopíaEstupor y comaAlteraciones del estado mentalAfasiaSignos motores bilateralesSíntomas sensorialesOtros síntomas de focalización
Fuente: Ferro, Canhão: Stroke 2004;35:664.
En el estudio presentado por Gosk–Bierska y col. el síntoma más frecuente fuela cefalea (87%) y el signo más común fue el papiledema (55%). En este mismoestudio 25% de los pacientes tuvieron un examen neurológico normal al momen-to del diagnóstico.8 Los signos neurológicos suelen desarrollarse en la mitad delos pacientes. Las crisis convulsivas se presentan en 40% de los casos y suelenser limitadas y focalizadas en 50% de ellos, pudiendo llegar a generalizarse o in-cluso presentar un estado epiléptico.3 La cefalea es el síntoma principal, por loque cuando un paciente presenta sólo este dato no se descarta la presencia deTVC.
Existen cuatro formas de presentación de la TVC:32
1. Déficit focal o crisis parciales. Cuando se presenta la asociación de cefaleay crisis parciales, además de alteración del estado de alerta, debilidad de al-guna extremidad o pérdida sensorial se debe sospechar TVC.
2. Hipertensión intracraneal aislada. Se manifiesta con cefalea, náusea, vó-mito, papiledema, pérdida visual momentánea y parálisis del nervio cranealVI.
3. Encefalopatía subaguda difusa. Se caracteriza por la disminución del ni-vel de conciencia y en algunas ocasiones crisis convulsivas, sin una locali-zación clara de signos reconocibles de HIC. Algunos casos pueden simularencefalitis o trastornos metabólicos.
4. Oftalmoplejía dolorosa. Es causada por lesión de los nervios craneales III,IV y VI.
La trombosis aislada de los diferentes senos y venas cerebrales resulta tambiénen diferentes manifestaciones clínicas. En la trombosis del seno cavernoso pre-dominan los signos oculares con datos de dolor ocular, quemosis, proptosis y pa-rálisis del oculomotor. La oclusión de las venas corticales produce déficit motor

172 (Capítulo 10)Tratado de trombosis
o sensorial y crisis convulsivas. La oclusión del seno sagital muestra deficienciamotora en forma bilateral y las crisis convulsivas son típicas. Los pacientes conobstrucción del seno lateral casi siempre muestran cuadros de hipertensión intra-craneal; sin embargo, se menciona que cuando existe afección del seno transver-so izquierdo el cuadro es seguido de afasia.1
La trombosis de las venas cerebrales internas, la vena basal (vena de Rosenthal)o la vena de Galeno afectan de 3 a 8% de los pacientes, lo que es más frecuenteentre las mujeres. Las características típicas de la trombosis del sistema profundoson disfunción del diencéfalo —reflejada con cuadros más graves con la presen-cia de coma—, déficit motor —por lo general bilateral— y alteración de los refle-jos oculares, que confiere un escaso pronóstico. No obstante, puede haber síndro-mes parciales en donde no hay alteración del estado de alerta o signos defocalización, lo cual puede conducir a una omisión del diagnóstico en sus formastempranas. El espectro de los síntomas refleja el grado de congestión venosa, quedepende no sólo de la extensión del trombo en el sistema venoso profundo sinotambién del territorio involucrado y del establecimiento de venas colaterales. Sedebe sospechar afección del sistema venoso cerebral profundo en pacientes (so-bre todo mujeres) con lesiones dentro de los ganglios basales o el tálamo, en espe-cial si la lesión es bilateral.33
DIAGNÓSTICO
Como se observa en el cuadro 10–1, las causas y los factores que predisponen apadecer TVC son múltiples. Cuando la TVC ocurre en ausencia de una causa ob-via y conocida se requiere realizar un protocolo amplio de estudio, debido a quela causa subyacente puede requerir tratamiento específico, además de la anticoa-gulación (flujograma de estudio de los pacientes con TVC). Una adecuada histo-ria clínica y un examen físico exhaustivo son primordiales para establecer eldiagnóstico. Se debe prestar especial interés a la historia reciente o previa a trau-ma craneal o cervical, cáncer, deshidratación, úlceras orales o genitales recurren-tes, abortos repetitivos y problemas de sangrado y de coagulación.
A pesar de un exhaustivo protocolo de estudio, se estima que no se logra identi-ficar una causa específica de TVC entre 20 y 35% de los pacientes. En dichos pa-cientes que padecen “TVC idiopática” se recomienda el seguimiento a largo pla-zo y una nueva realización del protocolo de estudio, debido a que la causasubyacente puede hacerse evidente meses o años después de la presentación ini-cial. Además de los estudios radiológicos, que son fundamentales para el diag-nóstico, deben practicarse otros estudios especiales y de rutina para cada pacientecon sospecha de TVC.

173Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Estudios de rutina
Deben incluir biometría hemática completa con diferencial, velocidad de sedi-mentación globular, electrólitos séricos, pruebas de función renal, tiempos decoagulación, electroforesis de proteínas séricas y anticuerpos antinucleares.También pueden incluir perfil de trombofilia con búsqueda de factor V de Leiden,anticuerpos antifosfolípidos, proteínas C y S, anticuerpos anticardiolipina, anti-cuerpos anti–ANCA, etc. Sin embargo, cada profesional debe decidir con todocuidado la extensión de la investigación de laboratorio, el tiempo y la selecciónde los estudios de acuerdo con cada paciente, sin olvidar el riesgo–beneficio y loscostos. Respecto de esto se han publicado guías de recomendación del BritishCommittee for Standards in Hematology, que aportan un abordaje razonable paraevaluar a los pacientes con sospecha de un estado hipercoagulable. De acuerdocon estas guías, el protocolo de estudio de la coagulación se puede dividir en dosetapas. La primera etapa incluye la solicitud de proteína C activada, factor V deLeiden, antitrombina III, proteínas C y S, fibrinógeno, anticuerpos antifosfolípi-dos y homocisteína. Sería ideal solicitar estos estudios antes de iniciar la anticoa-gulación formal, dado que se pueden alterar los resultados. Cuando este primerrastreo resulta negativo la sospecha de que el paciente presenta una trombofiliacongénita es alta, sobre todo si existen antecedentes familiares positivos de trom-bosis, si el paciente es joven y si existen eventos tromboembólicos recurrentes.En estos casos se deben realizar los estudios de la segunda etapa, que incluyenbúsqueda de la mutación G20210A de la protrombina, disfibrinogenemia, plas-minógeno, activador del plasminógeno y niveles de inhibición del factor activa-dor del plasminógeno. Todas estas pruebas se deben realizar en un centro hemato-lógico experimentado. Debido a que las anomalías de la coagulación presentanniveles fluctuantes, se recomienda repetir todo el protocolo de estudio unos me-ses después del evento agudo, en especial en los pacientes en los que el estudioinicial resultó negativo para la presencia de trombofilia. Finalmente, hay queconsiderar causas raras de estudio, como la deficiencia hereditaria de la peroxida-sa del glutatión plasmático (GPx–3), una enzima que se encuentra involucradaen la reducción de los lípidos y los peróxidos de hidrógenos en el plasma, y quese ha vinculado causalmente con TVC en pacientes jóvenes.
Estudio del líquido cefalorraquídeo
El estudio del líquido cefalorraquídeo (LCR) es un elemento útil para el diagnós-tico de TVC. Se debe realizar siempre que existan síntomas de hipertensión endo-craneana y que se haya descartado por imagen la presencia de abscesos cerebra-les, hemorragia cerebral masiva o infarto cerebral extenso. El examen del LCR

174 (Capítulo 10)Tratado de trombosis
a menudo reporta resultados normales. La presión de apertura es elevada en almenos 40% de los pacientes, y otras anormalidades detectadas pueden ser la ele-vación de proteínas, la presencia de células rojas o leucocitosis. El LCR reflejainflamación parameníngea, por ejemplo, la elevación de leucocitos con predomi-nio de polimorfonucleares o células mononucleares, glucosa normal y cultivosnegativos en la tercera parte de los pacientes con TVC.
Electroencefalograma
El electroencefalograma (EEG) presenta anormalidades entre 68 y 80% de lospacientes con TVC, las cuales son inespecíficas. La anomalía más frecuente esla presencia de ondas lentas asimétricas o generalizadas. El mayor valor del EEGradica en que puede demostrar descargas epileptiformes superimpuestas subclí-nicas que quizá requieran terapia anticonvulsiva específica.
Doppler transcraneal
La trombosis del seno longitudinal superior o de las venas basales profundas, deGaleno, Labbe o Rosenthal se asocia con un aumento en las velocidades de flujoen el sistema venoso profundo. Por esto se indica que la evaluación seriada conDoppler transcraneal (DTC) puede ser de utilidad en el monitoreo de los cambiosen el flujo venoso y la respuesta terapéutica. Sin embargo, los datos actuales deque se dispone en relación con la utilidad real del DTC son limitados y los resulta-dos en la confiabilidad de dicho estudio aún deben ser confirmados.
Diagnóstico radiológico
Los estudios de imagen son cruciales para establecer el diagnóstico de TVC. Enla mayoría de los hospitales la TAC de cráneo es el primer examen de gabineteque se realiza ante la sospecha de TVC; no obstante, dicho estudio no suele serconfiable para diagnosticar la presencia de la enfermedad, sobre todo si no seefectúa con medio de contraste. Hoy en día la IRM y la venografía con IRM co-mienzan a utilizarse con mayor frecuencia, en especial en los centros médicos.En algunos casos la angiografía convencional constituye el estudio final paraconfirmar el diagnóstico. De hecho, antes del advenimiento de la IRM la angio-grafía convencional representaba el estándar de oro.
Los hallazgos en imagen para el diagnóstico de TVC se dividen en signos di-rectos e indirectos. Los signos indirectos incluyen anormalidades en el parénqui-ma cerebral (principalmente infartos venosos), presencia de sistema venoso cola-teral y anormalidades en el seno mastoideo. Dichos signos son similares en la

175Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
TAC y la IRM, y con frecuencia son sutiles y poco específicos. Los signos direc-tos son los que se observan secundarios a la interrupción del flujo venoso y a suoclusión, o a la visualización del trombo.
IMAGEN DEL PARÉNQUIMA CEREBRAL
Tomografía axial computarizada de cráneo
La tomografía axial computarizada de cráneo (TAC) simple revela anormalida-des inespecíficas en la mayoría de los pacientes con TVC. Además, puede ser“normal” hasta en 25 a 40% de los casos en los que posteriormente se demuestraTVC. Lo anterior es notable en los pacientes con aumento aislado de la presiónintracraneana. Sin embargo, la TAC simple inicial permite descartar otras patolo-gías, como tumores, cisticercosis, etc.
La administración de medio de contraste aumenta la confiabilidad de la TACen el diagnóstico de TVC. No obstante, sin importar si la TAC es contrastada ono, puede ayudar a confirmar el diagnóstico de TVC.
Los principales hallazgos en la TAC que pueden aumentar la sospecha de TVCson:
1. Signos indirectos:� Evidencia de erosión de las estructuras del oído medio y cambios en la
región mastoidea, sobre todo en los pacientes con trombosis séptica delseno lateral.
� Cambios parenquimatosos que incluyen infartos venosos (hemorrágicoso no hemorrágicos), edema cerebral difuso y reforzamiento del falx y deltentorio. Hasta 30% de los pacientes con TVC presentan infartos, de loscuales un gran número son hemorrágicos. No hay características patog-nomónicas al visualizar los infartos venosos hemorrágicos, ya que puedenvariar de grandes hematomas parenquimatosos a pequeñas hemorragiaspetequiales. Casi siempre se describen como hemorragias intraparenqui-matosas. Sin embargo, los datos que pueden aumentar la sospecha de quedichas lesiones sean infartos venosos incluyen su multiplicidad, su loca-lización subcortical, que no sigan un trayecto arterial y que tengan unaapariencia bien definida. Asimismo, el compromiso bilateral de los tála-mos y los ganglios basales es un indicador de TVC. También se observacon frecuencia un reforzamiento intenso del tentorio o del falx, o de am-bos, después de la administración del medio de contraste, que indica es-tasis venosa o colaterales de las venas durales. En ocasiones se puede ob-

176 (Capítulo 10)Tratado de trombosis
servar un reforzamiento giral aislado o una hiperdensidad linear que enocasiones son malinterpretados como hemorragia subaracnoidea. Lapresencia de ventrículos pequeños “como hendiduras” es un hallazgo co-mún y se atribuye a un aumento de la presión intracraneana y edema rela-cionado, aunque es un signo inespecífico y difícil de interpretar, en espe-cial en los pacientes jóvenes con TVC en quienes así se observa elsistema ventricular sin ser patológico.
� Puede observarse hidrocefalia y compresión del cuarto ventrículo en pa-cientes con infartos venosos cerebelosos.
2. Signos directos:� Signo de la cuerda. En 2 a 25% de los pacientes se puede observar el
trombo fresco como un foco de hiperdensidad dentro de la vena o el senoocluido en la TAC simple. La mayoría de las veces se trata de un hallazgosutil y se observa mejor cuando se afecta el seno longitudinal superioro el seno recto. Este signo tiene poca especificidad para la TVC.
� Signo delta (vacío). Se observa en la TAC después de la administracióndel medio de contraste como un triángulo brillante que rodea a un núcleocentral hipodenso. Representa el reforzamiento al contraste de las venascolaterales y las paredes dilatadas de los senos que rodean al coágulo, elcual no refuerza. Lo anterior se observa en 25 a 52% de los pacientes contrombosis del seno longitudinal superior. Se pueden hacer algunos ha-llazgos similares en la trombosis del seno recto o lateral. Para su observa-ción es necesario hacer cortes menores de 5 mm de grosor en la TAC paraincrementar el rango de detección.
En la trombosis del seno cavernoso la TAC revela múltiples defectos de llenadoy bulging de los senos después de la inyección del material de contraste.
Imagen de resonancia magnética
La imagen de resonancia magnética (IRM) constituye la modalidad diagnósticade elección cuando se sospecha TVC. La capacidad multiplanar y la ausencia deartefactos óseos hacen que la IRM sea más sensible que la TAC para detectaranormalidades en el parénquima, hemorragias petequiales, formación de trom-bos y flujo sanguíneo. Debido a esto aporta pruebas definitivas para el diagnósti-co de TVC, aunque los hallazgos dependen de la secuencia de IRM utilizada, asícomo del estadio y del tiempo de la trombosis.
Imagen de resonancia magnética estándar en T1 y T2
El signo directo más importante en la IRM estándar es la ausencia de flujo (signode la ausencia de flujo) en las secuencias T1 y T2. Las alteraciones en el flujo san-

177Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
guíneo y la degradación de los productos de la hemoglobina en las venas contrombosis producen los cambios observados en las imágenes en T1 y T2 que indi-can la TVC. La apariencia y la intensidad de signos del trombo intraluminal en eltiempo dependen de los efectos paramagnéticos de la degradación de los productosde la sangre, de una manera similar a la observada en la hemorragia intraparenqui-matosa.
De este modo se pueden observar algunos de los siguientes signos:
� En el estadio agudo muy temprano (de uno a cinco días) existe ausencia deflujo y el trombo aparece isointenso en T1 e hipointenso en T2, por la pre-sencia de oxihemoglobina en los glóbulos rojos intactos. Este patrón se ob-serva rara vez debido al retraso habitual en la realización de la IRM.
� Unos cuantos días más tarde (estadio subagudo) el trombo comienza a serhiperintenso, inicialmente en T1 (días 6 a 9) y luego en T2 (de 10 a 15 días),debido a la conversión de la oxihemoglobina a metahemoglobina. La au-sencia de flujo persiste. En los senos grandes la hiperintensidad del tromboprocede de la periferia al centro. Esto puede observarse como un trombo hi-pointenso rodeado de un anillo circular hiperintenso, denominado signoblanco o diana. Este signo está presente en 15% de los casos. El incrementoen la intensidad de señal en T1 y en T2 es el hallazgo más frecuente en laIRM de los pacientes con TVC. Este patrón puede tardar entre 21 y 35 díasdespués del inicio de la trombosis.
� En el estadio crónico (más de 21 a 35 días) es más variable el patrón de laIRM. El seno con trombosis puede seguir parcial o totalmente trombosado,o recanalizarse, produciendo cambios subsecuentes en la señal del flujo.
En la mayoría de los pacientes el trombo crónico aparece heterogéneo y comien-za a ser progresivamente isointenso en T1 e isointenso a hiperintenso en T2. Estoshallazgos pueden tardar años y ser malinterpretados como una TVC recurrenteo aguda. Es importante subrayar que el uso de las secuencias T1 y T2 para el diag-nóstico de la TVC tienen sus debilidades. Dichas secuencias estándar son relati-vamente poco sensibles, dado que los cambios en los signos de la trombosis sonvariables y a menudo sutiles. Los falsos negativos o positivos no son raros, sobretodo en casos hiperagudos o crónicos, en oclusiones de venas o senos pequeñoso cuando existe disminución en la velocidad del flujo sanguíneo sin una verdade-ra oclusión. Además, los senos laterales y la parte anterior del seno longitudinalsuperior son poco visualizados debido a la orientación axial transversa de la ob-tención de las imágenes.
La administración de gadolinio aumenta la sensibilidad para demostrar el sig-no delta, análogo al observado en la TAC contrastada, así como también el engro-samiento meníngeo y el reforzamiento de las venas corticales. Asimismo, el usode cortes coronales y variaciones en el tiempo de repetición puede permitir una

178 (Capítulo 10)Tratado de trombosis
menor visualización de los senos laterales y diferenciar el seudorreforzamientodel flujo lento de las oclusiones verdaderas.
El T2 en la IRM es sensible a cambios en la mucosa del seno aéreo mastoideo.Las anormalidades mastoideas, que van desde incremento en la intensidad de T2y el engrosamiento de la mucosa hasta la acumulación de líquido dentro de lasceldas aéreas, se han descrito hasta en 39% de los pacientes con trombosis delseno lateral. Las anormalidades mastoideas son ipsilaterales al seno trombosadoy ninguno de los pacientes presenta signos de enfermedad ótica o mastoiditis. Porlo anterior, se indica que los cambios mastoideos en la IRM en trombosis no sépti-ca del seno lateral pueden ser secundarios a un aumento en la presión venosa delas venas que drenan las celdas mastoideas con la subsiguiente congestión vascu-lar, edema y trasudación de líquido. Esta observación en la IRM puede constituirla presencia no sospechada o reconocida de una trombosis venosa.
Imagen de resonancia magnética de edemacerebral venoso e infarto venoso
Muchas veces la TVC produce edema cerebral localizado, edema e infarto veno-so debido al aumento de la presión venosa. Lo anterior se origina por extensiónretrógrada del trombo desde los senos venosos hasta las venas corticales, puentesy medulares. Existe una escasa relación entre la extensión y la situación de laslesiones parenquimatosas cerebrales y la localización y el grado de trombosis ve-nosa, lo cual quizá refleje variaciones en la circulación venosa colateral. Ademásde los cambios parenquimatosos, en la IRM pueden observarse otras consecuen-cias de la TVC, como edema cerebral generalizado y dilatación de las venas medu-lares o colaterales. La IRM es más sensible que la TAC para detectar y caracterizarcambios parenquimatosos presentes entre 40 y 70% de los pacientes con TVC.
El edema venoso o el infarto se observan en imágenes de T2 como un área dehiperintensidad (figura 10–1), que casi siempre representa edema citotóxico ovasogénico reversible, el cual puede persistir hasta dos años y rara vez precedea un infarto verdadero. Lo extenso de la anormalidad parenquimatosa es un buenindicador pronóstico de la TVC. El cambio en la intensidad parenquimatosa estípicamente subcortical, pero puede comprometer la corteza subyacente. La he-morragia relacionada que se observa en T2 como una hipointensidad en los esta-dios tempranos ocurre con mayor frecuencia que en la enfermedad isquémicaoclusiva y puede comportarse como un gran hematoma subcortical, el cual, a di-ferencia de lo que ocurre en el infarto arterial, tiende a diseminarse del centro ala periferia o en forma de pequeñas hemorragias petequiales. Cuando ocurre uninfarto venoso puede haber un reforzamiento con el gadolinio, como se observaen las lesiones neoplásicas, y dar la apariencia de tumor–like. La localización delos cambios parenquimatosos no está confinadas a un territorio vascular y es me-

179Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 10–1. El edema venoso o el infarto se observa en imágenes de T2 como un áreade hiperintensidad. Paciente de 33 años de edad con una lesión hiperintensa de T2 enla región parietal derecha, que no sigue un trayecto venoso y que incluso presenta áreashipointensas sugerentes de hemorragia dentro de la lesión.
nos consistente que en un infarto arterial. Otras características que ayudan a dis-tinguir el edema o el infarto venoso de la enfermedad arterial oclusiva son la pre-sencia de múltiples sitios y un mayor grado de edema cerebral desde el estadiotemprano, así como su proximidad con un seno venoso ocluido (figura 10–2).
Una trombosis del seno longitudinal superior se sospecha en presencia de cam-bios parenquimatosos cercanos a la línea media en las regiones frontales, parieta-les u occipitales. Dichos cambios pueden ser unilaterales o bilaterales, y casisiempre asimétricos. Una anormalidad en la parte posterior del lóbulo temporalaumenta la probabilidad de un infarto secundario a un trombo dentro del senotransverso no dominante y en la vena de Labbé.
El edema o infarto venoso unilateral o bilateral que involucra la sustancia blan-ca periventricular, el cuerpo estriado, los tálamos o el cerebelo superior puede sersecundario a trombosis de las venas cerebrales internas o del seno recto (figura10–3). Dichas lesiones bilaterales profundas pueden confundirse con tumores,daño hipóxico isquémico, enfermedades mitocondriales y una variedad de altera-ciones metabólicas o infartos arteriales.
Imagen de resonancia magnéticacon secuencias en T2 ecoplanares
La disminución del flujo venoso cerebral en la TVC promueve un cambio localen la curva de oxigenación de la hemoglobina hacia la formación de deoxihemo-

180 (Capítulo 10)Tratado de trombosis
Figura 10–2. Un mayor grado de edema cerebral desde el estadio temprano, así comosu proximidad a un seno venoso ocluido, aumentan la sospecha de una trombosis venosacerebral. Esta imagen representa una lesión parenquimatosa localizada en forma parasa-gital, en la región parietal derecha que inicialmente fue interpretada como un tumor. Si sepone un poco más de atención y se observa el seno longitudinal superior (flechas) seobserva que se encuentra trombosado e hiperintenso, lo que sugiere que se trata de unedema o un infarto venoso secundario a oclusión del seno longitudinal superior.
globina, la cual produce un “efecto de susceptibilidad magnética” que da comoresultado una pérdida de la intensidad de señal (darkening), observable en lasimágenes de T2. Se ha descubierto que las imágenes en T2 pueden detectar la pre-sencia de un coágulo intravenoso durante la fase aguda y subaguda, que aparecencomo un área de hipointensidad dentro del seno o la vena afectada, y que los senostrombosados se observan más fácilmente en secuencias T2 que en cualquier otrasecuencia de IRM. Las secuencias T2 permiten la visualización directa de los se-nos involucrados que están distendidos y con coágulos ricos en deoxihemoglobi-na, y de los infartos venosos asociados, que muchas veces son hemorrágicos.
Difusión con imagen de resonancia magnética
Las anormalidades en las secuencias de difusión en los pacientes con TVC sonvariables e inespecíficas. Los hallazgos incluyen áreas mixtas heterogéneas deintensidades altas y señales bajas, lesiones hiperintensas multifocales (figura10–3) similares a las observadas en las lesiones vasculares isquémicas arterialeso hiperintensidades en los coágulos. La medición cuantitativa del coeficienteaparente de difusión (CAD) de las lesiones en propagación ayuda a diferenciar

181Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 10–3. Mujer de 52 años que después de tener diarrea por ocho días inicia conmanifestaciones neurológicas. A. Aquí se ejemplifica la presencia de lesiones bilatera-les talámicas cercanas a la línea media (flechas negras), que son secundarias a trombo-sis profunda de las venas cerebrales internas y seno recto. B. Incluso con la presenciadel signo delta en la presa de Herófilo (flechas blancas). C. En la secuencia de difusiónse observan cambios en el metabolismo del agua (flechas blancas).
A B
C
un edema citotóxico de un vasogénico. La mayor ventaja del CAD yace en su ca-pacidad para detectar anormalidades sutiles o congestión venosa y edema cere-bral antes de que se hagan visibles las lesiones parenquimatosas en T1 o T2, o ensecuencias FLAIR de la IRM. Se ha sugerido que la presencia de lesiones hiperin-tensas en secuencias de difusión indica un movimiento restringido de las molécu-las de agua dentro del coágulo y en venas ocluidas al momento del diagnóstico,que pueden servir para predecir una baja tasa de recanalización del coágulo a tresmeses de evolución después del evento.

182 (Capítulo 10)Tratado de trombosis
Venografía con resonancia magnética
La venografía con resonancia magnética (VRM) ha sobrepasado a la angiografíaconvencional para establecer el diagnóstico de TVC, debido principalmente aque es un método no invasor. La ausencia de flujo e intensidad dentro de una venao un seno y la ausencia de opacificación de un seno o vena indican trombosis in-traluminal. El trombo ocluido aparece como hiperintenso la mayoría de las veces.Sin embargo, vale la pena considerar que la VRM en ocasiones no muestra unseno transverso ausente de manera congénita o hipoplásico, lo cual lleva a undiagnóstico erróneo de trombosis venosa. Además, la saturación del flujo sanguí-neo cuando las imágenes son paralelas al seno, en especial al seno longitudinalsuperior, puede originar una falsa imagen de pérdida de la intensidad de señal yun diagnóstico erróneo de oclusión de un seno.
Angiografía convencional
La angiografía de los cuatro vasos cerebrales, que fue el estándar de oro para eldiagnóstico de TVC, permite la visualización de toda la fase venosa. Sin embar-go, su utilidad actual ha disminuido por tratarse de un procedimiento invasor ypor la existencia de las nuevas modalidades con IRM. Su uso está confinado a lospacientes en los que la IRM y la venografía con IRM son inconclusas, en especialen los pacientes con trombosis aislada de las venas corticales y en los casos enlos que se contemple la administración intraseno de terapia trombolítica o recana-lización intervencionista.
La angiografía fue durante muchos años la llave para llegar al diagnóstico deTVC. El signo angiográfico más característico de la TVC es la ausencia parcialo completa de llenado de las venas o de los senos. Algunos signos indirectos sonla dilatación de las venas colaterales con apariencia tortuosa y el retardo en el lle-nado venoso. La interpretación de la angiografía puede ser difícil en casos de va-riaciones anatómicas, como en la hipoplasia unilateral o en la ausencia de un senotransverso.1,32
TRATAMIENTO
En el ISCVT la mayoría de los pacientes fueron anticoagulados (83.3%) con he-parina intravenosa (64%) y con heparina de bajo peso molecular (34.9%), y pos-teriormente con anticoagulación oral (ACO). Pocos pacientes recibieron HBPMa dosis profilácticas (1.4%) o antiagregantes plaquetarios (5.9%). Trece pacien-tes fueron tratados con trombólisis local, de los cuales cinco murieron (38.5%)o quedaron con alguna discapacidad a los seis meses posteriores al evento.9

183Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Existen varias guías para el manejo de la TVC que tratan de abordar al pacienteen forma integral, es decir, brindándole manejo a la etiología de la TVC (cuandose logra identificar la causa) en conjunto con el tratamiento antitrombótico y sin-tomático. El principio del tratamiento antitrombótico en la TVC consiste en larecanalización del seno o la vena ocluida, en la prevención de la propagación deltrombo y en el tratamiento del estado protrombótico subyacente, con el fin de pre-ver nuevos eventos trombóticos en otra región del cuerpo, como es el caso delembolismo pulmonar. El manejo sintomático está dirigido a los síntomas de laHIC o al manejo de las crisis convulsivas.1,32
Tratamiento trombótico
Desde hace más de 50 años la heparina se ha utilizado cada vez más debido a laspruebas que se tienen de su seguridad y eficacia, incluso en las lesiones hemorrá-gicas. Esto se confirmó en el primer estudio aleatorizado que comparó dosis ajus-tadas de heparina intravenosa contra placebo, en el que se observó una drásticadiferencia entre los dos grupos: ocho pacientes en el grupo de heparina y uno enel grupo de placebo presentaron una recuperación total. Se registró una muerteen el grupo placebo y ninguna en el grupo de heparina.40
Un segundo estudio comparó dosis ajustadas a peso de heparina de bajo pesomolecular con placebo en 60 pacientes con TVC. Se observó una escasa respuestadespués de tres semanas en 6 de los 30 pacientes (20%) tratados con HBPM, encomparación con 7 de los 29 pacientes controles (24%). Después de tres mesestres pacientes (10%) del grupo de HBPM y seis pacientes (21%) del grupo place-bo presentaron una escasa respuesta, que correspondía a una reducción de riesgorelativo no significativo de 11% a favor del tratamiento activo. No se observaronnuevos datos o empeoramiento de HIC en los 15 pacientes con datos de hemorra-gia anteriores al tratamiento. Se reportó un caso de sangrado de tubo digestivoen el grupo de HBPM y un caso de embolismo pulmonar en el grupo placebo.41
Un metaanálisis comparó la eficacia de dosis subcutáneas de HBPM con la efi-cacia de dosis ajustadas de heparina no fraccionada para embolismo venoso ex-tracerebral y se probó la superioridad de la HBPM al presentar menos complica-ciones de sangrado. Otras de las ventajas incluyeron la ruta de administración yla ausencia de monitoreo de resultados de laboratorio para hacer el ajuste de lasdosis. Una posible ventaja de la heparina no fraccionada permite que en los pa-cientes críticos el tiempo de tromboplastina tienda a normalizarse en una a doshoras después de descontinuar la infusión en caso de complicación o de reque-rirse alguna intervención quirúrgica.42
Las pruebas actuales muestran que los pacientes con TVC sin contraindicacio-nes para la anticoagulación deben ser tratados con dosis ajustadas a peso de

184 (Capítulo 10)Tratado de trombosis
HBPM (180 antifactor Xa U/kg/24 h administradas en dos inyecciones al día) ocon dosis ajustadas de heparina intravenosa hasta alcanzar al menos dos veces eltiempo de tromboplastina. La HIC relacionada con TVC no es una contraindica-ción para iniciar el manejo con heparina, y se prefiere el uso de HBPM en casosno complicados de TVC.43,44
Aún no se dispone de estudios controlados acerca del beneficio y la duraciónóptima de la anticoagulación oral (ACO) en pacientes con TVC, pero la mayoríade los autores recomiendan continuar con ACO después de la fase aguda. Existenestudios que han mostrado que la recanalización ocurre dentro de los primeroscuatro meses posteriores a la TVC, de tal forma que estos datos pueden proveeruna guía de la duración de la ACO.44 Al contrario de lo que ocurre con los pacien-tes con trombosis venosa extracerebral, se deberá alcanzar un INR de entre 2.0y 3.0 a lo largo de tres meses si existe un factor de riesgo reversible y a lo largode 6 a 12 meses si el evento es idiopático, pero será indefinido si se trata del se-gundo evento de TVC o en casos de trombofilias hereditarias.46
Algunos reportes señalan que la terapia trombolítica tiene el potencial de pro-veer una rápida restitución del flujo venoso y efectos positivos con el uso de trom-bólisis local para el manejo de TVC. En 1971 Vines y Davis reportaron el uso deurocinasa para el tratamiento de TVC. Diez años más tarde Di Rocco y col. trata-ron en forma exitosa a cinco pacientes con urocinasa intravenosa y heparina. En1988 Scott y col. reportaron el primer caso de terapia fibrinolítica local en pacien-tes jóvenes con trombosis extensa del seno sagital. En épocas recientes el activa-dor del plasminógeno tisular recombinante (rTPA) se ha utilizado ampliamentey con buenos resultados (en combinación con heparina) para el manejo de estospacientes.45
Sin embargo, en la actualidad no existen pruebas que recomienden la trombó-lisis local como tratamiento de primera línea para la TVC, pues se tienen buenosresultados con la heparina no fraccionada. Además, este tipo de tratamiento re-sultaría muy costoso en los países de bajos recursos, donde la TVC parece ser másfrecuente. Así, la única indicación hasta el momento para el uso de trombólisislocal es en los casos en los que el estado del paciente merme a pesar del uso deanticoagulantes, además de que requiere personal calificado para su ejecución.El fármaco utilizado (urocinasa o rTPA), la dosis, la ruta y el método de adminis-tración no se han establecido por completo.44,45
Tratamiento sintomático
El tratamiento sintomático incluye el uso de antiepilépticos, el manejo de la hi-pertensión intracraneal, el uso de antibióticos para casos de origen infeccioso, larehidratación, el control de la agitación psicomotriz y las medidas analgésicas.1

185Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
No existen datos que sustenten el uso de antiepilépticos en forma profilácticaen los pacientes con TVC, y hay autores que consideran que sólo deben indicarseen presencia de crisis convulsivas. No obstante, las guías más recientes mencio-nan que pueden ser una buena opción para los pacientes con déficit neurológicofocal o lesiones parenquimatosas en las imágenes de RM o de TC, aunque la dura-ción del tratamiento aún no se ha establecido.44,47
En los pacientes con HIC aislada en los que el papiledema pone en riesgo lavisión se puede requerir una punción lumbar para remover el LCR antes de iniciarel manejo con heparina y mejorar así la cefalea y la visión. Si la presión intracra-neal aumenta mucho se debe dar manejo antiedema, sedación, elevación de la ca-beza y tratamiento con glicerol y manitol en la unidad de cuidados intensivos. Noexisten reportes acerca del beneficio de los esteroides en estos casos, aun cuandohay lesión parenquimatosa. En los pacientes con datos de herniación por una le-sión hemisférica unilateral se indica la hemicraneotomía descompresiva.1
PRONÓSTICO
La TVC es un tipo de enfermedad vascular cerebral poco frecuente, que en mu-chas ocasiones se describió con un pronóstico impredecible. En el pasado la TVCse diagnosticaba casi exclusivamente a través de autopsias, por lo que se creía queera un padecimiento que conducía a la muerte la mayoría de las veces. En las pri-meras series de angiografía cerebral la mortalidad fue de 30 a 50%. En las seriesmás recientes se reportó una mortalidad de 4 a 33%. En el estudio ISCVT 4.3%de los pacientes murieron en la fase aguda de la TVC y 3.4% murieron dentro delos primeros 30 días posteriores a la aparición de los síntomas. Las causas másfrecuentes de muerte fueron la herniación transtentorial debida a lesión hemorrá-gica, la presencia de edema difuso y las lesiones bilaterales. Otras causas demuerte fueron las complicaciones médicas y el embolismo pulmonar.48
En el estudio ISCVT el estado de coma, la hemorragia cerebral y la presenciade malignidad fueron importantes factores pronósticos de muerte o de alguna dis-capacidad. Otros factores de mal pronóstico fueron el sexo masculino, la edadmayor de 37 años, las alteraciones del estado mental, la trombosis del sistema ve-noso profundo y la presencia de infección en el SNC.9
Los pacientes con TVC presentan un bajo riesgo de mortalidad. Hoy es posibledetectar a los pacientes con un mayor riesgo de muerte, por lo que en ellos se debemantener un monitoreo más estrecho y proporcionar una terapia más agresiva encaso de deterioro clínico.48
Las crisis convulsivas y la presencia de nuevos eventos trombóticos son lascomplicaciones más frecuentes durante el seguimiento. Más de dos tercios de los

186 (Capítulo 10)Tratado de trombosis
pacientes con TVC presentan una recanalización completa o parcial dentro de losprimeros meses después del inicio del cuadro. No hay pruebas de que la recanali-zación ocurra entre el tercero y el sexto meses de evolución; sin embargo, el bene-ficio de que la recanalización se presente en los primeros meses o más tarde aúnse desconoce.49,50 Las recurrencias posteriores al primer evento son poco fre-cuentes y ocurren en 2.8% de los pacientes. Las mujeres que presentan un eventodurante el embarazo poseen un bajo riesgo de recurrencia en los embarazos si-guientes. A excepción de los abortos espontáneos, no suelen presentarse otrascomplicaciones durante los nuevos embarazos o después de ellos. Estos hallaz-gos apoyan la idea de que el antecedente de TVC no es un impedimento para em-barazarse.50
A partir de la primera descripción realizada por Robins han participado variosfactores en la modificación de la historia de la TVC, entre los que se incluyen losestudios con alta sensibilidad, que facilitan el diagnóstico temprano de la enfer-medad.
Tales estudios mejoran en gran medida el pronóstico de los pacientes, puescontribuyen a iniciar de manera más temprana el manejo con anticoagulantes, locual, sin duda, ha revolucionado y cambiado por completo la historia natural dela enfermedad (figura 10–4).
Figura 10–4. Flujograma de pacientes con trombosis venosa cerebral.
Factor de riesgo evidente para trombosis+
cuadro clínico sugestivo de TVC(cefalea, HIC y/o déficit focal)
Realizar estudio de imagenRM + VRM
Trombosis venosa cerebral
Sí No
Descartar la presencia de factor agregadoRealizar perfil trombofílico completo Buscar otra alternativa diagnóstica
Brindar medidas generalesIniciar tratamiento trombótico

187Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
REFERENCIAS
1. Bousser MG, Ferro JM: Cerebral venous thrombosis: an update. Lancet Neurol 2007;6:162–170.
2. Kimber J: Cerebral venous sinus thrombosis. QJM 2002;95:137–142.3. Stam J: Thrombosis of the cerebral veins and sinuses. N Engl J Med 2005;352:1791–1798.4. Rhoton AL: The cerebral veins. Neurosurgery 2002;51:159–205.5. Van der Schaaf I, van Gijn J: The spectrum of presentations of venous infarction caused
by deep cerebral vein thrombosis. Neurology 2005;65:192–196.6. Grover M: Cerebral venous thrombosis as a cause of acute headache. J Am Board Fam
Pract 2004;17:295–298.7. Bousser MG, Chiras J, Bories J, Castaigne P: Cerebral venous thrombosis: a review of
38 cases. Stroke 1985;16:199–213.8. Gosk Bierska I, Wysokinski W, Brown RD, Karnicki K, Grill D et al.: Cerebral venous
sinus thrombosis: incidence of venous thrombosis recurrence and survival. Neurology2006;67:814–819.
9. Ferro JM, Canhão P, Stam J, Bousser MG, Barinagarrementeria F: Prognosis of cere-bral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein andDural Sinus Thrombosis (ISCVT). Stroke 2004;35:664–670.
10. Barnett HJ, Hyland HH: Non–infective intracranial venous thrombosis. Brain 1953;76:36–49.
11. Hillier CE, Collins PW, Bowen DJ, Bowley S, Wiles CM: Inherited prothrombotic riskfactors and cerebral venous thrombosis. QJM 1998;91:677–680.
12. Seligsohn U, Lubetsky A: Genetic susceptibility to venous thrombosis. N Engl J Med 2001;344:1222–1231.
13. Dulli DA, Luzzio CC, Williams EC, Schutta HS: Cerebral venous thrombosis and activa-ted protein C resistance. Stroke 1996;27:1731–1733.
14. Zuber M, Toulon P, Marnet L, Mas JL: Factor V Leiden mutation in cerebral venousthrombosis. Stroke 1996;27:1721–1723.
15. Martinelli I, Battaglioli T, Pedotti P, Cattaneo M, Mannucci PM: Hyperhomocysteine-mia in cerebral vein thrombosis. Blood 2003;102:1363–1366.
16. Cantú C, Alonso E, Jara A et al.: Hyperhomocysteinemia, low folate and vitamin B12 con-centrations, and methylene tetrahydrofolate reductase mutation in cerebral venous throm-bosis. Stroke 2004;35:1790–1794.
17. Cantú C, Barinagarrementeria F: Cerebral venous thrombosis associated with pregnan-cy and puerperium. Review of 67 cases. Stroke 1993;24:1880–1884.
18. Carhuapoma JR, Mitsias P, Levine SR: Cerebral venous thrombosis and anticardiolipinantibodies. Stroke 1997;28:2363–2369.
19. Vidailhet M, Piette JC, Wechsler B, Bousser MG, Brunet P: Cerebral venous thrombosisin systemic lupus erythematosus. Stroke 1990;21:1226–1231.
20. Lanska DJ, Kryscio RJ: Risk factors for peripartum and postpartum stroke and intracra-nial venous thrombosis. Stroke 2000;31:1274–1282.
21. Dentali F, Crowther M, Ageno W: Thrombophilic abnormalities, oral contraceptives, andrisk of cerebral vein thrombosis: a meta–analysis. Blood 2006;107:2766–2773.
22. Martinelli I, Rosendaal FR, Vandenbrouck JP: Oral contraceptives are a risk factor forcerebral vein thrombosis. Thromb Haemost 1996;76:477–478.
23. Iseri M, Aydin O, Ustundag E, Keskin G, Almac A: Management of lateral sinus throm-bosis in chronic otitis media. Otol Neurotol 2006;27:1098–1103.

188 (Capítulo 10)Tratado de trombosis
24. Fiorot JA Jr, Felício AC, Fukujima MM, Rodrigues CA, Morelli VM et al.: Tuberculosis:an uncommon cause of cerebral venous thrombosis? Arq Neuropsiquiatr 2005;63:852–854.
25. Wechsler B, Vidailhet M, Piette JC, Bousser MG, Dell Isola B et al.: Cerebral venousthrombosis in Behçet’s disease: clinical study and long–term follow–up of 25 cases. Neurol-ogy 1992;42:614–618.
26. Raizer JJ, DeAngelis LM: Cerebral sinus thrombosis diagnosed by MRI and MR venogra-phy in cancer patients. Neurology 2000;54:1222–1226.
27. Mazzoleni S, Putti MC, Simioni P, Sainati L, Tormene D et al.: Early cerebral sinovenousthrombosis in a child with acute lymphoblastic leukemia carrying the prothrombinG20210A variant: a case report and review of the literature. Blood Coagul Fibrinolysis2005;16:43–49.
28. Canhão P, Batista P, Falcão F: Lumbar puncture and dural sinus thrombosis–a causal orcasual association? Cerebrovasc Dis 2005;19:53–56.
29. Lage JM, Panizo C, Masdeu J, Rocha E: Cyclist’s doping associated with cerebral sinusthrombosis. Neurology 2002;26;58:665.
30. Finelli PF, Carley MD: Cerebral venous thrombosis associated with epoetin alfa therapy.Arch Neurol 2000;57(2):260–262.
31. Ferro JM, Lopes MG, Rosas MJ, Fontes J, VENOPORT Investigators: Delay in hospitaladmission of patients with cerebral vein and dural sinus thrombosis. Cerebrovasc Dis 2005;19:152–156.
32. Crassard I, Bousser MG: Cerebral venous thrombosis. J Neuroophthalmol 2004;24:156–163.
33. Van den Bergh WM, van der Schaaf I, van Gijn J: The spectrum of presentations of venousinfarction caused by deep cerebral vein thrombosis. Neurology 2005;26(65):192–196.
34. Idbaih A, Boukobza M, Crassard I, Porcher R, Bousser MG et al.: MRI of clot in cere-bral venous thrombosis: high diagnostic value of susceptibility–weighted images. Stroke2006;37:991–995.
35. Majoie CB, van Straten M, Venema HW, den Heeten GJ: Multisection CT venographyof the dural sinuses and cerebral veins by using matched mask bone elimination. Am J Neu-roradiol 2004;25:787–791.
36. Idbaih A, Boukobza M, Crassard I, Porcher R, Bousser MG et al.: MRI of clot in cere-bral venous thrombosis: high diagnostic value of susceptibility–weighted images. Stroke2006;37:991–995.
37. Favrole P, Guichard JP, Crassard I, Bousser MG, Chabriat H: Diffusion–weightedimaging of intravascular clots in cerebral venous thrombosis. Stroke 2004;35:99–103.
38. Lalive PH, De Moerloose P, Lovblad K, Sarasin FP et al.: Is measurement of D–dimeruseful in the diagnosis of cerebral venous thrombosis? Neurology 2003;28(61):1057–1060.
39. Kosinski CM, Mull M, Schwarz M, Koch B, Biniek R et al.: Do normal D–dimer levelsreliably exclude cerebral sinus thrombosis? Stroke 2004;35:2820–2825.
40. Einhäupl KM, Villringer A, Meister W, Mehraein S, Garner C et al.: Heparin treatmentin sinus venous thrombosis. Lancet 1991;338:597–590.
41. De Bruijn SF, Stam J: Randomized, placebo–controlled trial of anticoagulant treatment withlow–molecular–weight heparin for cerebral sinus thrombosis. Stroke 1999;30:484–488.
42. Van Dongen CJJ, van den Belt AGM, Prins MH, Lensing AWA: Fixed–dose subcuta-neous low molecular weight heparins versus adjusted–dose unfractionated heparin for ve-nous thromboembolism. Cochrane Database Syst Rev 2004;4:CD001100.
43. Stam J, De Bruijn SF, DeVeber G: Anticoagulation for cerebral sinus thrombosis. Coch-rane Database Syst Rev 2002;(4):CD002005.

189Trombosis venosa cerebralE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
44. Einhäupl K, Bousser MG, De Bruijn SF, Ferro JM, Martinelli I et al.: EFNS guidelineon the treatment of cerebral venous and sinus thrombosis. Eur J Neurol 2006;13:553–559.
45. Bousser MG: Cerebral venous thrombosis: nothing, heparin or local thrombolysis. Stroke1999;30:481–483.
46. Büller HR, Agnelli G, Hull RD, Hyers TM, Prins MH et al.: Antithrombotic therapy forvenous thromboembolic disease. The Seventh ACCP Conference on Antithrombotic andThrombolytic Therapy. Chest 2004;126:401–428.
47. Ferro JM, Correia M, Rosas MJ, Pinto AN, Neves G, Cerebral Venous Thrombosis Por-tuguese Collaborative Study Group (VENOPORT): Seizures in cerebral vein and dural si-nus thrombosis. Cerebrovasc Dis 2003;15:78–83.
48. Canhão P, Ferro JM, Lindgren AG, Bousser MG, Stam J et al.: Causes and predictorsof death in cerebral venous thrombosis. Stroke 2005;36:1720–1725.
49. Baumgartner RW, Studer A, Arnold M, Georgiadis D: Recanalization of cerebral ve-nous thrombosis. J Neurol Neurosurg Psychiatry 2003;74:459–461.
50. Dentali F, Gianni M, Crowther MA, Ageno W: Natural history of cerebral vein thrombo-sis: a systematic review. Blood 2006;108:1129–1134.

190 (Capítulo 10)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
11Insuficiencia arterial periférica
Rafael Gutiérrez Carreño, Mónica Mendieta Hernández,René Iván Lizola Margolis
La insuficiencia arterial periférica (IAP) de difícil apreciación clínica en etapastempranas del padecimiento es una de las afecciones más frecuentes en la patolo-gía angiológica. Generalmente se asocia con enfermedad vascular y en ocasioneses parte del síndrome metabólico, afectando las arterias coronarias, la circulacióncarotídea y vertebral, las arterias de las extremidades superiores e inferiores, asícomo la circulación mesentérica y renal. Es un problema que debe ser tratado demanera global, pues, aunque en ocasiones se divida para su estudio en diferentesespecialidades por razones académicas, debe ser visto como un todo. La ateros-clerosis y la arteriosclerosis son las principales causas, junto con la microangio-patía y la macroangiopatía diabéticas, que conducen a las llamadas “patías” deldiabético en el cerebro, los ojos, los oídos, los nervios, el corazón, el riñón, el tubodigestivo y las extremidades.
¿Por qué se ha incrementado el índice de amputaciones no traumáticas? Lomás importante es la presencia de la diabetes mellitus en México, en donde, segúnla Encuesta Nacional de Salud y Nutrición 2006, se aprecia en 9.5% en la pobla-ción general; asimismo, al ocupar el primer lugar en el mundo en obesidad infan-til se han incrementado las enfermedades crónico–degenerativas, que en conjun-to con las enfermedades vasculares del cerebro, el corazón, los ojos, los riñonesy las extremidades ocupan el primer lugar en mortalidad, de acuerdo con lo repor-tado por la Secretaría de Salud.
¿Por qué a pesar de estas “patías” los pacientes llegan a la atención de su saluden etapas avanzadas y con complicaciones, si sus extremidades tienen pulsos y
191

192 (Capítulo 11)Tratado de trombosis
Cuadro 11–1. Factores de riesgo vascular
No modificable (hasta 2013) Modificables
Genes Tipo de alimentación
Historia familiar Tabaquismo y adiccionesEdad Hipertensión arterialSexo Ejercicio físico
Menopausia ObesidadAndropausia Hiperlipidemias
Homocisteína elevada
Fibrinógeno elevadoAcido úrico elevado
sus arterias laten? Muchos de estos padecimientos comienzan en la microcircula-ción, en la cual existe hipercoagulabilidad, inflamación por lesión arterial, fibri-nólisis disminuida y disminución en la tromborresistencia endotelial y en la fun-ción plaquetaria.
Entonces, ¿se está ejerciendo la medicina tardía?, ¿la medicina de complica-ciones? Las evidencias así lo indican. Hay un entorno en el que los factores deriesgo desencadenantes (cuadro 11–1) de estas enfermedades degenerativas sehan incrementado con una preponderancia en los hábitos alimenticios, en los quela obesidad y la diabetes ocupan un alarmante primer lugar.
Las arterias se van ocluyendo de forma paulatina (figura 11–1), disminuyendoel aporte sanguíneo necesario para que los órganos se mantengan viables. Las
Figura 11–1. Esquema en corte transversal de una arteria afectada por aterosclerosis.

193Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 11–2. Insuficiencia arterial aguda. Causas
1. Cardiacas:Fibrilación auricularInfarto miocárdicoAneurisma ventricular
EndocarditisPrótesis valvularesMixoma en la aurícula izquierda
Miocardiopatía hipertrófica o congestivaAnillo mitral calcificadoProlapso de la válvula mitral
2. Arteriales:Placa de ateroma ulcerada en aorta torácica o abdominal, trombo mural en aneurisma
3. Paradójicas:Trombos del sistema venoso
Tromboflebitis en extremidades con comunicación interauricular o interventricular
principales causas en etapas agudas se enumeran en el cuadro 11–2 y en las etapascrónicas en el cuadro 11–3.
Cuadro 11–3. Insuficiencia arterial en extremidades. Causas trombóticas
1. Degenerativas:Diabetes mellitus. Aterosclerosis
Necrosis quística de la capa media arterialDisplasia fibromuscular
2. Traumáticas:Trauma, fractura ósea, contusiones
Poscateterismo diagnóstico o terapéuticosSíndrome de compresión neurovascular
3. Hematológicas:Enfermedades mieloproliferativasPúrpura trombocitopénica trombóticaDislipoproteinemia
Coagulación intravascular diseminada4. Vasculitis:
Enfermedad de Buerger
Tromboangeítis obliteranteSíndrome antifosfolípidosLupus eritematoso sistémico
PoliarteritisArteritis de TakayasuToxicomanías y otras arteritis

194 (Capítulo 11)Tratado de trombosis
Cuadro 11–4. Oclusiones topográficas arteriales
Femoral 46%
Iliaca 18%Aórtica 14%Poplítea 11%
Mesentérica 6%Humeral 3%Renal 2%
Existen diversas interrogantes, además de la frecuencia en la localización delos diversos territorios arteriales (cuadro 11–4).
CLÍNICA
Los síntomas del paciente están relacionados con el déficit de todos los nutrientesy oxígeno que llevan las arterias. Sin embargo, al principio se puede confundircon neuropatía radicular, compresión lumbar, hernia discal, enfermedades reu-máticas u otros tipos de neuropatías y miopatías que condicionan dolor.
Se ha tratado de definir los estadios clínicos que se muestran en el cuadro 11–5.Aun cuando la claudicación intermitente es el síntoma más común, varía de
acuerdo con la distancia que logra caminar el paciente. En los pacientes con clau-dicación intermitente el dolor se puede localizar en la región de las nalgas y lascaderas, los muslos, las pantorrillas y los pies. La localización del dolor se asociacon mucha precisión al nivel de la estenosis arterial:
1. Nalgas y caderas: enfermedad aortoiliaca.2. Muslos: arteria femoral común o aortoiliaca.3. Dos tercios superiores de las pantorrillas: arteria femoral superficial.4. Tercio inferior de las pantorrillas: arteria poplítea.5. Claudicación del pie: arteria tibial o peronea.
Cuadro 11–5. Isquemia en extremidades. Datos clínicos
Precoz Tardía
Dolor Hipoestesia o anestesiaPalidez CianosisFrialdad Flictenas
Impotencia funcional Rigidez muscularAusencia de pulsos Gangrena

195Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Otros síntomas de la enfermedad vascular periférica pueden incluir cambios enla piel y en la coloración, atrofia dérmica o subdérmica, pérdida del vello cutáneo,disfunción eréctil en los varones, heridas en puntos de presión que no cicatrizan,adormecimiento, debilidad o pesadez en los músculos, palidez al elevar las pier-nas y uñas de los pies más gruesas y opacas.
La severidad de los síntomas se relaciona con la gravedad de la enfermedadvascular y el pronóstico. Las clasificaciones más conocidas son la de Fontaine,la de Rutherford y la TASC (Inter–Society Consensus for the Management of Pe-ripheral Arterial Disease), publicada en 2007. Para el pie del diabético se cuentacon la del Hospital de Tampico y la de San Elian.
El dolor en reposo implica un padecimiento crónicamente avanzado e isque-mia crítica, que con frecuencia marca el inicio de úlceras isquémicas y de gangre-na (figura 11–2).
DIAGNÓSTICO
La historia clínica completa es la base, seguida de la palpación de las pulsacionesarteriales en los sitios anatómicos. Se debe tomar la tensión arterial en las cuatro
Figura 11–2. Fotos clínicas de casos con IAP
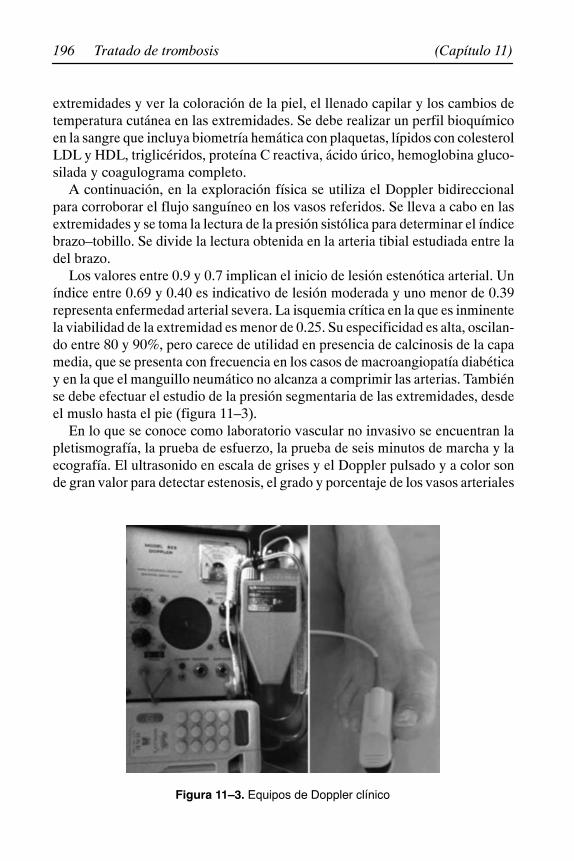
196 (Capítulo 11)Tratado de trombosis
extremidades y ver la coloración de la piel, el llenado capilar y los cambios detemperatura cutánea en las extremidades. Se debe realizar un perfil bioquímicoen la sangre que incluya biometría hemática con plaquetas, lípidos con colesterolLDL y HDL, triglicéridos, proteína C reactiva, ácido úrico, hemoglobina gluco-silada y coagulograma completo.
A continuación, en la exploración física se utiliza el Doppler bidireccionalpara corroborar el flujo sanguíneo en los vasos referidos. Se lleva a cabo en lasextremidades y se toma la lectura de la presión sistólica para determinar el índicebrazo–tobillo. Se divide la lectura obtenida en la arteria tibial estudiada entre ladel brazo.
Los valores entre 0.9 y 0.7 implican el inicio de lesión estenótica arterial. Uníndice entre 0.69 y 0.40 es indicativo de lesión moderada y uno menor de 0.39representa enfermedad arterial severa. La isquemia crítica en la que es inminentela viabilidad de la extremidad es menor de 0.25. Su especificidad es alta, oscilan-do entre 80 y 90%, pero carece de utilidad en presencia de calcinosis de la capamedia, que se presenta con frecuencia en los casos de macroangiopatía diabéticay en la que el manguillo neumático no alcanza a comprimir las arterias. Tambiénse debe efectuar el estudio de la presión segmentaria de las extremidades, desdeel muslo hasta el pie (figura 11–3).
En lo que se conoce como laboratorio vascular no invasivo se encuentran lapletismografía, la prueba de esfuerzo, la prueba de seis minutos de marcha y laecografía. El ultrasonido en escala de grises y el Doppler pulsado y a color sonde gran valor para detectar estenosis, el grado y porcentaje de los vasos arteriales
Figura 11–3. Equipos de Doppler clínico

197Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 11–4. Duplex Doppler color. Estenosis arterial en IAP
afectados y la velocidad del flujo, con una sensibilidad de 92 a 95% y una especi-ficidad de 97 a 98% (figura 11–4).
Otros métodos son la angiografía mediante tomografía computarizada o reso-nancia magnética y la arteriografía convencional (figura 11–5).
Figura 11–5. AngioTAC en arterias de miembros inferiores

198 (Capítulo 11)Tratado de trombosis
MANEJO MÉDICO
En los pacientes con enfermedad arterial periférica el manejo médico tiene un do-ble objetivo: mejorar la situación funcional de la extremidad y prevenir los even-tos secundarios a la distribución multifocal de la enfermedad. Como primera in-dicación terapéutica está la eliminación total de los factores de riesgo de laenfermedad arterial, tales como control de la diabetes, de la hipertensión arterial,uso de antineuríticos, suspensión de inhalantes tóxicos, oxigenoterapia habitualo hiperbárica, uso de bota hiperbárica, nutrición adecuada y una explicación am-plia y explicita al enfermo y a la familia de lo que representan la IAP y el pie dia-bético. Los fármacos utilizados en la IAP están dirigidos al tratamiento especí-fico de la claudicación y a la prevención secundaria de los eventos vasculares(cuadro 11–6).
En casos seleccionados se utilizan los fibrinolíticos, como el activador tisulardel plasminógeno (t–PA), el complejo activador SK–plasminógeno acilado (AP-SAC) y la reteplasa (r–PA).
En caso de que se asocie patología trombótica venosa se pueden utilizar losantagonistas de la vitamina K o los nuevos anticoagulantes orales (figura 11–6).
El manejo con prostaglandinas y prostaciclinas PGE1, PGE2 y PGI3 es por víaparenteral. Su mecanismo de acción se basa en la inhibición de la agregación pla-quetaria y de la activación leucocitaria, con un efecto vasodilatador importantey alivio de la hipertensión pulmonar. La infusión intravenosa intermitente a largoplazo (varias semanas) de prostanoides y análogos de la prostaciclina, como eliloprost y el cisaprost, ha demostrado una reducción del dolor de reposo y mejoríade las ulceraciones isquémicas. Las prostaglandinas y los tromboxanos desempe-ñan un papel esencial en la fisiopatología cardiovascular. Los prostanoides mo-dulan la patogénesis de enfermedades vasculares, como la trombosis y la ateros-clerosis, mediante una serie de procesos, como la agregación plaquetaria, lavasodilatación, la vasoconstricción y la respuesta inflamatoria local.
Cuadro 11–6. Manejo terapéutico de eventos vasculares
Antiplaquetarios y hemorreológicos
Acido acetilsalicílicoDipiridamolTriflusal
TiclopidinaClopidogrelPrasugrelTicagrelor y cangrelor
Sulodexida Pentoxifilina y cilostazol

199Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 11–6. Esquema de la cascada de la coagulación y sitios de acción de nuevosanticoagulantes.
XII XIIa
XI XIa
IX IXa
VII
DabigatránXimelagatránBivalirudinaArgatrobán
Activa o transforma
Inhibe
TrombinaDrotrecogin alfa
Proteína C
F. tisular
TifacoginVIIa
VIIIa calcio
Xa Rivaroxabán
Va calciofosfolípido
II
RazaxabánX
fosfolipido
F. tisularCalcio
La bioingeniería genética con el factor de crecimiento PDGF–BB humano re-combinante se ha utilizado en el manejo de las lesiones del pie diabético.
En la angiogénesis terapéutica diversos estudios han demostrado la posibili-dad de mejorar la perfusión tisular mediante el uso de factores de crecimiento(proteínas recombinantes), genoterapia o tratamiento celular.
Aunque los resultados de los trabajos afirman la mejoría de la perfusión tisular,serán necesarios estudios aleatorios y comparativos que demuestren cuál de lostratamientos ofrece un mejor intervalo de seguridad terapéutica en los casos norevascularizables.
En el futuro el manejo de la enfermedad arterial periférica podrá encaminarsea modalidades terapéuticas que incrementen la formación y el desarrollo de nue-vos vasos, con lo que se mejorará la viabilidad de los tejidos isquémicos. En estesentido, se han desarrollado diversos estudios con el común denominador de laangiogénesis terapéutica.
El empleo de factores de crecimiento, de genoterapia y de células progenitorasendoteliales ha arrojado resultados esperanzadores.
Con el descubrimiento de las células progenitoras endoteliales en el adulto ysu funcionalidad dentro del sistema vascular se ha incrementado el interés por eltratamiento celular.

200 (Capítulo 11)Tratado de trombosis
MANEJO QUIRÚRGICO
La simpatectomía lumbar tuvo un lugar como paliativo y para provocar vasodila-tación en las arterias sin rigidez; sin embargo, su utilidad deja de ser la deseadacuando el sistema nervioso autónomo, en este caso la cadena lumbar, se encuen-tra lesionado por la neuropatía. Se ha sugerido también el uso de neuroestimula-dores a nivel de la médula espinal.
Los puentes o derivaciones (figura 11–7) con injertos sintéticos o venas a va-sos, de acuerdo con el angiosoma correspondiente, y a vasos distales, contribuyenal salvamento de las extremidades; sin embargo, es importante mencionar que au-mentan el flujo arterial al área isquémica, pero no curan la enfermedad de fondo.
La alternativa a la cirugía convencional es la denominada cirugía de mínimainvasión. Dicha técnica emplea la cirugía endovascular con el uso de arteriografíadiagnóstica, guías, balones de angioplastia, en algunas ocasiones el láser y la ate-rectomía con la colocación de stent o férulas, así como la endoprótesis cubiertaen casos seleccionados (figura 11–8).
Los procedimientos de revascularización de los tejidos isquémicos conllevanel riesgo de síndrome de reperfusión, que implica una hipoxia prolongada, conmiopatía y rabdomiólisis posteriores con salida del potasio intracelular, hiperca-lemia y mioglobinuria con un alto índice de falla renal, así como también lentifi-cación en el flujo venoso, presencia de trombosis venosa y embolia pulmonar, loque en conjunto requiere un manejo integral en las áreas de cuidados especiales(figura 11–9).
Para el manejo del pie diabético se puede emplear la clasificación de Wagner,de la Universidad de Texas, o bien la de San Elian, las cuales permiten emitir unpronóstico. Se requiere una nutrición adecuada y en caso necesario oxigenotera-pia con la posibilidad del uso de oxígeno hiperbárico. El control estricto del ma-nejo metabólico es fundamental.
Figura 11–7. Arteriografía de control en cirugía de revascularización por angiosomasy a arterias distales en la pierna y el pie.

201Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 11–8. Cirugía endovascular con angioplastia y stents en arteria femoral.
Se debe tomar en cuenta el manejo local de las lesiones a través de los protoco-los del manejo de heridas, mediante hidrogel, alginato, esponja, colágeno, hidro-coloides, apósitos impregnados con yodo, plata o antimicrobianos, y los sistemasVAC (vacuum assisted closure), entre otros.
AMPUTACIÓN
La amputación se indica cuando la enfermedad vascular se encuentra muy avan-zada y hay presencia de dolor en reposo, isquemia crítica o gangrena, así comocuando no se ha alcanzado la mejoría o el control con el manejo farmacológico,
Figura 11–9. Esquema de complicaciones en el síndrome de reperfusión a nivel de célu-las, músculo, riñón, corazón y pulmón.
MioglobinuriaEmbolia
Gangrena
K+

202 (Capítulo 11)Tratado de trombosis
la cirugía convencional o la endovascular no fueron efectivas, existe inviabilidadde la extremidad o se encuentre en riesgo la vida del paciente. La amputación sedeberá realizar en un punto en donde exista una “adecuada circulación” para quela cicatriz se lleve a cabo de primera intención. Hasta 85% de las amputacionesde los miembros inferiores relacionadas con la diabetes son precedidas por unaúlcera en el pie. Luego de una amputación mayor la sobrevida a tres años es de50% y a cinco años es de 40%. En 42% de los pacientes se produce una amputa-ción contralateral entre uno y tres años posteriores a la primera amputación, y en56% entre tres y cinco años posteriores.
Existen amputaciones menores de ortejos, transmetatarsales, de Syme, infra-condíleas y supracondíleas, así como desarticulaciones de la cadera.
La rehabilitación se debe realizar a la brevedad, aun cuando genera un alto cos-to económico, social y emocional. El alcance de esta patología lo expresa la Orga-nización Mundial de la Salud con la siguiente frase: “Cada 30 seg alguien pierdeuna extremidad debido a la diabetes”.
Para concluir, hay quien dice que la “diabetes es el fracaso del éxito”, pero¿habrá en esta epidemia, además de lo ya conocido, algunos priones, virus o fac-tores no detectados? Entonces, ¿estamos realizando medicina tardía?, ¿medicinade complicaciones? “No hay peor error que hacer lo mismo siempre y esperar re-sultados diferentes”, decía Einstein.
REFERENCIAS
1. Gutiérrez CAR: Perspectiva para el estudio y control de la aterosclerosis humana. Medici-na (Mex) 1976;1214:331–336.
2. Gutiérrez CAR et al.: Síndrome miopático metabólico renal. Rev Mex Angio 1977;55:5–8.3. Gutiérrez CAR et al.: Ateroesclerosis. Algunos aspectos patogénicos. Rev Med IMSS
1979;18:411–414.4. Gutiérrez CAR et al.: Arteriopatía del diabético. Correlación clínico–patológica. Rev Mex
Angiol 1984;12:10–13.5. Gutiérrez CAR et al.: Apósito hidrocoloide, tratamiento en pie diabético, úlceras isquémi-
cas y úlceras venosas. Rev Mex Angiol 1991;19:29–35.6. Gutiérrez CAR et al.: Tratamiento de la enfermedad arterial periférica en pacientes diabé-
ticos. En: Rull J, Zorrilla E, Jadzinsky M, Santiago J: Diabetes mellitus, complicaciones cró-nicas. McGraw–Hill, 1992:156–165.
7. Gutiérrez CAR et al.: Ateroesclerosis. ¿Es posible su regresión? Rev Mex Angiol 1995;23:84–86.
8. Gutiérrez CAR et al.: Insuficiencia arterial aguda de extremidades. Rev Mex Angiol 2001;29(2):54–59.
9. Gutiérrez CAR et al.: Insuficiencia arterial periférica. En: Cárdenas CA (ed.): Temas selec-tos de medicina interna. México, El Manual Moderno, 2003:25–32.
10. Gutiérrez CAR: Afección del sistema vascular en el diabético. 3ª Reunión Internacionalsobre Diabetes. Una visión económica, sociocultural y familiar del problema. UNAM,2009:383–385.

203Insuficiencia arterial periféricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
11. Navarro CVH: Insuficiencia arterial periférica. En: García–Frade RLF: Manual de trom-bosis y terapia antitrombótica. México, Alfil, 2008:193–208.
12. Aragón SJ, Ortiz R: El pie diabético. España, Elsevier, 2001.13. Martínez de Jesús F: Pie diabético. México, McGraw–Hill, 2010.14. Ferrufino MAL, Rodríguez TM, Escotto SI, Rodríguez RN: Angioplastia infrapoplítea:
correlación entre el vaso tratado y el angiosoma lesionado. Rev Mex Angiol 2012;40(4):123–134.
15. Salazar PJR, Serrano LR: Ultrasonido Doppler de miembros pélvicos en la insuficienciaarterial crónica: lo que el radiólogo debe reportar. An Radiol Mex 2011;11(3):174–177.
16. Serrano RE, Serrano LJA, Sánchez NNE, Muñoz PJA, Hernández SJG et al.: Diagnós-tico etiológico de la insuficiencia arterial aguda secundaria a embolismos. Rev Mex Angiol2004;32(2):35–43.
17. Rojas RGA, Cervantes CJ, Reyes E, Huerta M, Ramírez SOA et al.: Insuficiencia arte-rial embólica aguda. Rev Mex Angiol 2002;30(1):7–10.
18. Enríquez VME, Bobadilla FNO, Rodríguez JOA, Guerra MA, Carrasco NL et al.:Plasma rico en plaquetas para el tratamiento de úlceras isquémicas del paciente diabético.Rev Mex Angiol 2012;40(2):51–56.
19. Padilla L, Rodríguez TJM, Escotto SI, De Diego J, Rodríguez N et al.: Trasplante autólo-go de células mononucleares progenitoras derivadas de la médula ósea para inducir angio-génesis en pacientes con isquemia severa en extremidades inferiores. Acta Méd Grupo Án-geles 2011;9(2):57–62.
20. http://www.hapmd.com/home/hapmdcom/public_html/wp–content/uploads/2009/03/cirugiaacademicas–cx/20110614_pie_diab__tico_marcos_velasco.ppt.
21. Águila MR, Marquina RM: Estado actual de la enfermedad arterial periférica oclusiva(EAPO). Acta Med Grupo Ángeles 2007;5(4):187–196.
22. Águila MR, Marquina RM: Tratamiento endovascular de la enfermedad arterial oclusivaperiférica (EAPO) sector infrainguinal. Acta Med Grupo Ángeles 2007;5(4):197–208.
23. GE Enfermedad arterial periférica–IMSS. www.imss.gob.mx/profesionales/guiasclinicas/Documents/007GE.pdf.

204 (Capítulo 11)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
12Trombosis venosa profunda
Víctor Hugo Navarro Ceja
INTRODUCCIÓN
Por definición, la trombosis venosa es una condición clínica en la que los compo-nentes sanguíneos inician el proceso de la coagulación en el interior de una venaen cualquier parte del organismo.
Cuando la trombosis se localiza en el sistema venoso superficial se denominatromboflebitis, y cuando se localiza en el sistema venoso profundo se le llamatrombosis venosa profunda (TVP).
Es importante establecer la diferencia entre ambas condiciones, ya que lascaracterísticas clínicas, el manejo y las posibles complicaciones son muy diferen-tes. En la tromboflebitis la afección ocurre en un segmento de una vena superfi-cial, ocasionando inflamación, dolor y eritema en la zona afectada. La posibili-dad de tromboembolia pulmonar (TEP) en el curso de una tromboflebitis esprácticamente nula.
Sin embargo, cuando se presenta una TVP la complicación más temida es laTEP, condición que puede poner en peligro la vida del paciente. Por tal motivo,es importante detectar a los pacientes de manera inmediata para iniciar su trata-miento con base en anticoagulantes y reposo absoluto, con lo que se favorece laadherencia del trombo a las paredes de la vena y se evita así la posibilidad de em-bolismo a distancia.
Es errónea la creencia de que una trombosis venosa profunda puede causar em-bolismo cerebral o infarto agudo del miocardio, ya que en condiciones normalesla circulación arterial no se encuentra conectada con la venosa.
205

206 (Capítulo 12)Tratado de trombosis
EPIDEMIOLOGÍA
Aunque no se conoce la prevalencia real de la TVP por la gran cantidad de casosque no se diagnostican o que no generan una sintomatología relevante como paraacudir a consulta, en EUA se estima que aproximadamente 600 000 personas sonhospitalizadas con este diagnóstico cada año y que mueren alrededor de 200 000por complicaciones tromboembólicas de la misma enfermedad, sea que se esta-blezca o no el diagnóstico. Los ancianos son los más susceptibles a sufrir la enfer-medad, con tasas de mortalidad de hasta 20%.
Tanto la prevalencia como la recurrencia de la TVP son mayores en los hom-bres que en las mujeres.
En relación con la prevalencia de la enfermedad durante el embarazo, se es-tima que afecta de 0.5 a 2% de las embarazadas, siendo entre tres y cinco vecesmás común durante el puerperio que en el embarazo y hasta 16 veces más fre-cuente en los embarazos que terminan en cesárea. La TEP se considera una delas principales causas de mortalidad materna (ver a detalle en el capítulo corres-pondiente).
La TVP se relaciona también con el reposo prolongado y los procedimientosquirúrgicos, siendo más frecuente en los pacientes mayores de 40 años de edad,cuando el procedimiento quirúrgico dura más de 30 min, cuando el paciente pre-senta factores de riesgo adicionales como obesidad, varices o trombofilias cono-cidas, y ante cirugías ortopédicas, principalmente de cadera y rodilla, en las queel paciente debe recibir tromboprofilaxis prequirúrgica, ya que la enfermedad sepresenta hasta en 40% de los pacientes que no la reciben (ver a detalle en el capí-tulo correspondiente).
Recientemente se describió la famosa “trombosis de la clase turista” a raíz deuna serie de casos que presentaron TVP después de un viaje prolongado en avión;sin embargo, la trombosis se puede presentar al viajar en cualquier medio detransporte cuya duración sea mayor de cuatro horas, dado que ello favorece la es-tasis sanguínea, componente esencial de la tríada de Virchow.
FACTORES DE RIESGO
Hay que recordar que la tríada de Virchow (estasis sanguínea, lesión de la pareddel vaso e hipercoagulabilidad) es de utilidad para describir los factores de riesgopara el desarrollo de TVP (cuadro 12–1). Se debe tener en cuenta que no es posi-ble separar dichos factores, ya que en ocasiones se entrelazan, e incluso un solofactor de riesgo puede condicionar la presencia de los tres componentes de latríada.

207Trombosis venosa profundaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 12–1. Factores de riesgo para trombosis venosaprofunda relacionados con la tríada de Virchow
Estasis sanguínea Lesión de la pared Defectos de la coagulación
Reposo prolongado (más detres días)
TraumatismosEdad
Enfermedades propias de lasangre
Viajes largos (mayores de 4h)
Cirugía mayor (en especial derodillas y de cadera)
Anticonceptivos oralesEmbarazo y puerperio
Sedentarismo Catéteres intravenosos Historial familiar de trombosis
Inmovilización de una extre-midad
Obesidad
Uso de sustancias intraveno-sas
Tabaquismo
CáncerEnfermedades inflamatorias
(lupus, esclerodermia, etc.)
Varices Síndrome nefróticoParálisis
El reposo prolongado (refiriéndonos al reposo en cama durante varios días),como sucede en el paciente posquirúrgico o en el paciente que cursa con algunaenfermedad que lo obliga a permanecer en cama durante varios días, constituyeun factor de riesgo importante para el desarrollo de TVP, ya que la bomba muscu-lar de la pantorrilla (que es uno de los mecanismos que impulsan la sangre de re-greso hacia el corazón) no funciona de forma adecuada, lentificando el retornovenoso y la subsecuente formación de trombos que luego se propagan. Se calculaque el riesgo de TVP en un paciente encamado es de 13% a los ocho días de repo-so. El mismo mecanismo describe el alto riesgo de TVP en los pacientes que pre-sentan parálisis de la musculatura de las extremidades inferiores y en los pacien-tes obesos, en quienes el gran sobrepeso dificulta el retorno venoso.
En los pacientes con presencia de varices en los miembros inferiores la sangrese estanca en un sistema venoso superficial que ha perdido sus funciones tantode contractilidad como de función valvular, lo que genera los respectivos meca-nismos que inician el sistema de la coagulación.
La pared de un vaso sanguíneo se puede lesionar por traumatismo directo o porla generación de sustancias que dañan el endotelio, tras lo cual se activa la faseplaquetaria de la coagulación con la consecuente formación de trombos, en unintento por detener la hemorragia (capítulo 1). Se considera que una intervenciónquirúrgica mayor de 30 min de duración constituye un riesgo incrementado detrombosis venosa, debido a la liberación de factores procoagulantes durante lacirugía o a mecanismos aún no bien entendidos que favorecen la formación detrombos.
La edad constituye otro factor de riesgo para el desarrollo de TVP, ya que suincidencia aumenta conforme se incrementa la edad a partir de los 40 años.
Existen también alteraciones propias de la sangre que afectan la hemostasia(trombofilias), sea por insuficiencia, por defecto o por exceso en la producción

208 (Capítulo 12)Tratado de trombosis
de uno o varios de los factores de la coagulación, los cuales pueden tener una fun-ción anticoagulante o procoagulante. Algunos ejemplos de estas alteraciones sonla deficiencia de las proteínas C o S, la deficiencia de antitrombina III o la muta-ción del factor V (llamado factor V Leiden), por mencionar algunos (ver a detalleen el capítulo correspondiente). El antecedente familiar de TVP debe hacer pen-sar en la posibilidad de una causa hereditaria, y así iniciar la búsqueda y tamizajeadecuados para establecer patrones de herencia.
Desde hace tiempo se sabe que el uso de hormonales orales o parenterales, confines anticonceptivos en minidosis o como terapia hormonal sustitutiva, favoreceel desarrollo de trombosis venosa (ver a detalle el capítulo correspondiente).
No es rara la trombosis durante el embarazo, lo cual se puede deber a la produc-ción de sustancias placentarias generadoras de trombosis. La enfermedad trom-bótica venosa es más frecuente durante el puerperio, lo cual también puede estarasociado con la formación placentaria de elementos procoagulantes, mismos quepermanecen en la circulación varios días después del parto (ver capítulo corres-pondiente).
El cáncer puede conducir a la presencia de trombosis venosa por la producciónde elementos procoagulantes, así como también el uso de quimioterapia, la cualsuele ser demasiado irritante para las paredes de las venas, por lo que siempre sedebe administrar en una vena de alto flujo (ver el capítulo correspondiente).
En ciertas enfermedades sistémicas, como el lupus eritematoso sistémico (LES)o la esclerodermia, se generan factores que pueden activar el sistema de la coagu-lación (como el anticoagulante lúpico o los anticuerpos anticardiolipina), por loque ante estas condiciones es importante mantener al paciente bajo terapia anti-coagulante antes de que se presente una trombosis venosa (ver el capítulo corres-pondiente).
SIGNOS Y SÍNTOMAS
Las manifestaciones clínicas de la TVP son de escasa certeza diagnóstica, por loque no se deben utilizar como única prueba para establecer o descartar su presen-cia. De manera habitual, el cuadro clínico inicia de manera súbita con la presenciade dolor de moderada intensidad en la extremidad afectada.
El signo de Homans consiste en la presencia de dolor al hiperextender la panto-rrilla, mientras que en el signo de Bankroft se desencadena el dolor al comprimirlos músculos de la pantorrilla.
Estos signos se pueden acompañar de edema, el cual suele ser progresivo y porlo regular se extiende hasta el nivel donde se encuentra la trombosis. Por ejemplo,cuando se trata de una trombosis venosa a nivel de la vena poplítea el edema seencuentra hasta la pierna, mientras que si se trata de una trombosis a nivel iliofe-

209Trombosis venosa profundaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 12–1. Trombosis venosa profunda a nivel iliofemoral izquierdo.
moral el edema se manifiesta en toda la extremidad inferior. Aunque no es fre-cuente, también se puede encontrar coloración cianótica, hipotermia y retardo enel llenado capilar de la extremidad afectada (figura 12–1). La obstrucción severaal retorno venoso de la extremidad que condiciona necrosis de los tejidos se cono-ce como flegmasia cerúlea dolens (figura 12–2).
Figura 12–2. Flegmasia cerúlea dolens en el miembro inferior izquierdo.

210 (Capítulo 12)Tratado de trombosis
ABORDAJE DIAGNÓSTICO
Ante la sospecha de TVP se deben realizar algunos estudios de laboratorio y gabi-nete para confirmar el diagnóstico e iniciar el tratamiento a la brevedad.
El dímero D es un producto de la degradación de la fibrina. Su presencia enaltas concentraciones no establece el diagnóstico de TVP, pero si se encuentra enbajas concentraciones y la clínica es de baja probabilidad se descarta la presenciade trombosis venosa (su valor predictivo negativo es de 98%).
El ultrasonido Doppler constituye el estudio no invasivo de elección en estospacientes. Es de gran utilidad para establecer la presencia de trombosis a niveliliofemoral; sin embargo, su utilidad es muy limitada por debajo de la rodilla, enla que la valoración de los vasos tibiales es muy subjetiva y depende de la expe-riencia del explorador.
La angiotomografía y la angiorresonancia son pruebas que se utilizan en la ac-tualidad; sin embargo, su sensibilidad y especificidad aún son bajas, y resultandemasiado costosas para utilizarlas de manera rutinaria.
La flebografía continúa siendo el patrón de oro contra el cual se comparan elresto de los estudios; tiene la ventaja de delinear todo el árbol venoso y las víascolaterales, que de otra manera no se identifican, siendo indispensable su valora-ción cuando se planea un abordaje quirúrgico de la trombosis venosa. Las des-ventajas que presenta incluyen la posibilidad de reacciones alérgicas al medio decontraste y necrosis cutánea en el sitio de la aplicación. Con los medios de con-traste de que se dispone actualmente (hipoosmolares) han disminuido estas com-plicaciones y son más seguros de utilizar. Por estas razones, la flebografía no seencuentra indicada como primera línea de diagnóstico, y se debe reservar sólopara los pacientes en quienes no se ha podido establecer el diagnóstico a travésde otros estudios y cuando se planea una intervención quirúrgica o la colocaciónde un dispositivo en la vena cava inferior.
TRATAMIENTO
El tratamiento de la TVP se debe iniciar de manera inmediata en cuanto se esta-blece el diagnóstico, con el objetivo de limitar la extensión de la trombosis, evitarla TEP y disminuir las secuelas postrombóticas a largo plazo.
Medidas generales
Las medidas generales tradicionales consisten en indicar reposo al paciente, ele-var la parte de lo pies de la cama a 30� y colocar un soporte elástico compresivo

211Trombosis venosa profundaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
en la extremidad afectada para disminuir el edema y favorecer el retorno venoso.No se deben realizar maniobras para exprimir la extremidad ni de bombeo, ya quese puede ocasionar una TEP.
Heparina
De manera inicial se debe administrar heparina, sea no fraccionada o de bajo pesomolecular (HBPM). Existen diversos esquemas para la administración de hepa-rina no fraccionada, pero el más fácil de manejar es el que se calcula con base enel peso corporal; se administra una dosis de impregnación en bolo intravenosode 80 a 100 U/kg de peso corporal y posteriormente en infusión intravenosa conti-nua en dosis de 18 a 20 U/kg/h. Se debe medir el tiempo parcial de tromboplastina(TPT) a las seis horas de iniciada la infusión y ajustar la dosis hasta alcanzar unTPT de 70 a 90 seg.
La enoxaparina (HBPM) se debe administrar a razón de 1 mg/kg/día de mane-ra subcutánea, con la ventaja de que no necesita controlarse a través de pruebasde laboratorio, ya que los riesgos de sangrado son mínimos y no prolonga lostiempos de coagulación (véase capítulo XVIII: Indicaciones para medición de losniveles de anti–Xa con HBPM).
Trombólisis
Este procedimiento está encaminado a lisar el trombo, y se puede utilizar tantopor vía sistémica como de manera local en el sitio del trombo. Su manejo es deli-cado y se debe administrar siempre en una unidad de terapia intensiva para moni-torear de manera estrecha al paciente y detectar las posibles complicaciones demanera oportuna. Las tasas de permeabilización son mejores que las obtenidasa través de las medidas generales y el uso de heparina solamente; no obstante,existen contraindicaciones para su uso. No se debe utilizar en los casos con trom-bosis de más de ocho días de evolución, en pacientes con cirugía abdominal ma-yor o craneal en los seis meses previos ni en pacientes con riesgo de sangrado porcualquier causa o que hayan recibido un traumatismo contuso reciente (ver el ca-pítulo correspondiente).
Los tres medicamentos disponibles son la estreptocinasa, el factor activadorrecombinante del plasminógeno tisular (rTPA) y la urocinasa. En México sólo sedispone de los dos primeros, y sus dosis se revisan en el capítulo correspondiente.
Se debe administrar heparina de manera simultánea con el uso de estreptocina-sa, ya que se puede generar un proceso de coagulación difusa como efecto para-dójico. Con el uso de rTPA y urocinasa se inicia la administración de heparina

212 (Capítulo 12)Tratado de trombosis
al terminar su infusión, misma que se debe continuar durante al menos tres díasen lo que se logra establecer la anticoagulación oral con derivados de la warfarina(antagonistas de la vitamina K).
Nuevos anticoagulantes orales
Algunos estudios demuestran la no inferioridad del rivaroxabán y del dabigatránfrente a los “estándares de oro” en cuanto al manejo agudo de la TVP y la TEP,y en la prevención secundaria en estudios extendidos, por lo que hoy en día seaprueban para esta indicación.
TRATAMIENTO QUIRÚRGICO
La realización de trombectomía venosa en el manejo de la TVP ha sido muy con-trovertido. De hecho, el enfoque tradicional indica que no es de utilidad; sin em-bargo, se ha realizado en casos seleccionados con buenos resultados. El procedi-miento consiste en hacer una incisión en la región inguinal y abordar la venafemoral común; se refieren ésta y sus ramas, y se realiza un corte longitudinal enla pared del vaso, a través del cual se extraen los trombos (figuras 12–3 y 12–4).A través de este procedimiento quirúrgico se han obtenido buenos resultados en
Figura 12–3. Trombo en la vena femoral común izquierda.

213Trombosis venosa profundaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 12–4. Trombos obtenidos en el paciente de la figura 12–3.
cuanto a la prevención de las secuelas tardías, como la formación de varices se-cundarias y la aparición de lesiones ulcerativas en las extremidades inferiores. Elprocedimiento cuenta con indicaciones específicas y no se debe realizar despuésde 10 días de la trombosis en pacientes con compromiso hemodinámico o en loscasos en que los riesgos superan los beneficios.
REFERENCIAS
1. Panacek EA, Douglas KJ: Deep venous thrombosis and thrombophlebitis. En: Harwood–Nuss’ clinical practice of emergency medicine. EUA.
2. Merli G: Diagnostic assessment of deep vein thrombosis and pulmonary embolism. Am JMed 2005;118:3S–12S.
3. Nelson S, Greer I: Thrombophilia and the risk for venous thromboembolism during preg-nancy, delivery and puerperium. Obstet Gynecol Clin N Am 2006;33:413–427.
4. Doyle N, Monga M: Thromboembolic disease in pregnancy. Obstet Gynecol Clin N Am2004;31:319–344.
5. Rosovsky R, Kuter D: Catheter–related thrombosis in cancer patients: pathophisiology,diagnosis and treatment. Hematol Oncol Clin N Am 2005;19:183–202.
6. Segal JB, Streiff MB, Hofmann LV, Thornton K, Bass EB: Management of venousthromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007;146(3):211–222.
7. Weitz J, Hirsh J, Samara M: New anticoagulant drugs. The Seventh ACCP Conferenceon Antithrombotic and Thrombolytic Therapy: evidence–based guidelines. Chest 2004:126.
8. Hyers TM, Agnelli G, Hull RD et al.: Antithrombotic therapy for venous thromboembolicdisease. Chest 1998;114:561S–578S.
9. Sixth ACCP Consensus Conference on Antithrombotic Therapy. Chest 2001;119(1).

214 (Capítulo 12)Tratado de trombosis
10. Ansell J, Hirsh J, Poller L: The Seventh ACCP Conference on Antithrombotic and Throm-bolytic Therapy: evidence–based guidelines. Chest 2004;126(3).
11. Subar M: Thromboembolic disease and anticoagulation in the elderly. Clin Geriatric Med2001;17(1).
12. Hyers T, Hull R, Weg J: Antithrombotic therapy for venous thromboembolic disease.Chest 1995;108:3355–3515.
13. Rocha E, Alfaro MJ, Páramo JA, Canadell JM: Preoperative identification of patientsat high risk of deep venous thrombosis despite prophylaxis in total hip replacement. ThrombHaemostasis 1988;59:93–95.
14. Andres R, Miles A: Venous thromboembolism and pregnancy. Obstet Gynecol 2001;28(3).15. Rosendaal FR: Venous thrombosis: a multicausal disease. Lancet 1999;353:1167–1173.16. Hirsh J: The optimal duration of anticoagulant therapy for venous thrombosis. N Engl J
Med 1995;332(25):1710–1712.17. Baker W: Thrombolytic therapy clinical applications. Hematol Oncol Clin N Am 2003;17:
283–311.18. Koopman MM, Prandoni P, Piovella F et al.: Treatment of venous thrombosis with intra-
venous unfractionated heparin administered in the hospital as compared with subcutaneouslow–molecular–weight heparin administered at home. The Tasman Study Group. N Engl JMed 1996;334:682–687.
19. Schulman S, Rhedin AS, Lindmarker P et al.: A comparison of six weeks with six monthsof oral anticoagulant therapy after a first episode of venous thromboembolism. N Engl JMed 1995;332:1661–1665.
20. Hull RD, Pineo GF, Valentine KA: Treatment and prevention of venous thromboembol-ism. Semin Thromb Hemost 1998;24(Suppl 1):21–31.
21. Goldhaber SZ: Thrombolytic therapy in venous thromboembolism. Clinical trials and cur-rent indications. Clin Chest Med 1995;16:307–320.
22. The EINSTEIN Investigators: Oral rivaroxaban for symptomatic venous thromboembol-ism. N Engl J Med 2010.
23. Schulman S, Kearon C, Kakkar A: Extended use of dabigatran, warfarin, or placebo invenous thromboembolism. N Engl J Med 2013;368:709–718.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
13Tromboembolia pulmonar
Marcos César Gallegos Solórzano
INTRODUCCIÓN
En pleno siglo XXI la tromboembolia pulmonar (TEP) constituye uno de losdiagnósticos más difíciles y fáciles a la vez que existen en el campo de la medici-na interna y sus ramas. No en vano es conocida como la “gran enmascaradora”.La TEP es una causa común de atención de urgencia neumológica y cardiológica.Se presenta secundaria a la obstrucción de un trombo del lecho arterial pulmonar,que puede poner en riesgo la vida por una disfunción ventricular derecha (DVD),la cual es potencialmente reversible. El diagnóstico temprano cobra mucha im-portancia debido a que el tratamiento oportuno es altamente efectivo; pues porun lado puede mejorar la sobrevida de los pacientes debido a la restauración delflujo de los vasos pulmonares ocluidos y por otro puede prevenir la recurrenciade la misma enfermedad.
La asociación inevitable con la trombosis venosa profunda (TVP) merece es-pecial atención, ya que en conjunto comparten el síndrome de tromboembolismovenoso (STV). El presente capítulo se enfoca en la epidemiología, los factoresde riesgo, la historia natural y la fisiopatología, los criterios de gravedad y el pro-nóstico, el abordaje diagnóstico, el tratamiento y la prevención de la TEP.
EPIDEMIOLOGÍA
En la mayor parte de los casos, mas no en todos, la TEP es consecuencia de laTVP. Hasta 50% de los pacientes con TVP tienen TEP,1 mientras que 70% de los
215

216 (Capítulo 13)Tratado de trombosis
pacientes con TEP presentan TVP.2 La mortalidad de la TEP va de 7 a 11%; asi-mismo, la TEP recurrente es más común tras un episodio de TEP que de TVP (60vs. 20%).3 En México no existe literatura fidedigna que respalde los datos de mor-talidad y de prevalencia; sin embargo, en EUA la prevalencia de TEP en pacienteshospitalizados fue de 0.4% durante el periodo de 1978 a 1999.4 Se ha estimadouna prevalencia de 40 a 53 casos por cada 100 000 personas al año, con una inci-dencia anual de 600 000 casos.5 Sin embargo, en un estudio regional de la ciudadde Malmo, en Suecia, con una población de 230 000 habitantes, se analizaron2 356 autopsias realizadas en 1987 y se reveló que 25% (n = 595) cursaban conSTV y 18.3% (n = 231) presentaban TEP; cabe resaltar que sólo se logró estable-cer el diagnóstico pre mortem en 2% (n = 48).6 En México se han encontrado da-tos muy semejantes.7 De esta manera se aprecia que la verdadera incidencia dela TEP es difícil de evaluar, en vista de lo poco específica que es su presentaciónclínica.8 En nuestro país constituye una de las complicaciones más habituales enla práctica médica nacional.9
El estudio multicéntrico internacional más grande que describe la mortalidadde la TEP a tres meses es el International Cooperative Pulmonary Embolism Re-gistry (ICOPER), que documentó una mortalidad de 17% en 2 454 pacientes conTEP, la cual fue la causa de muerte en 45% de los casos. Los factores de pronósti-co asociados a muerte fueron una edad mayor de 70 años (HR 1.6, IC 95% 1.1a 2.3), cáncer (HR 2.3, IC 95% 1.5 a 3.5), insuficiencia cardiaca congestiva (HR2.4, IC 95% 1.5 a 3.7), enfermedad pulmonar obstructiva crónica (HR 1.8, IC95% 1.2 a 2.7), hipotensión arterial sistólica (HR 2.9, IC 95% 1.7 a 5.0), taquip-nea (HR 2.0, IC 95% 1.2 a 3.2) y DVD por ecocardiograma (HR 2.0, IC 95% 1.3a 2.9).10 En Boston, EUA, los pacientes con TEP que acuden de forma ambulato-ria al hospital tienen una mortalidad de 11.1% a 90 días.11 Asimismo, otro estudiode EUA mostró que en los pacientes que acudían a urgencias con TEP la mortali-dad general fue de 5.4% a 30 días, y la atribuida a la TEP sólo de 1%,12 lo cualse explica por las comorbilidades asociadas y las secuelas tardías de la TEP, lahipertensión pulmonar tromboembólica crónica13 y el síndrome postrombóti-co,14 los cuales conllevan un pronóstico adverso.
La incidencia del STV aumenta conforme a la edad, con una edad media depresentación de la TEP de 62 años; 60% de los casos pertenecen a las personasmayores de 60 años de edad.18
FACTORES DE RIESGO
El STV comparte la fisiopatología de la enfermedad aterotrombótica coronaria,descrita por Rudolph Virchow, la cual incluye inflamación, hipercoagulabilidad

217Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–1. Factores de riesgo para tromboembolia pulmonar
Idiopática:Edad (> 65 años)Viaje largoTrombofiliaObesidadTabaquismoHipertensiónSíndrome metabólicoContaminación ambiental
Secundaria:InmovilizaciónPosquirúrgicaTraumaAnticonceptivos oralesEmbarazoCáncerEnfermedad médica aguda (neumonía adquirida en la comunidad, insuficiencia cardiaca)
Adaptado de la referencia 51.
y daño endotelial.15 Los factores de riesgo asociados a STV son el tabaquismo,la hipertensión arterial, la diabetes y la obesidad.16
El estado inflamatorio crónico (aumento de proteína C reactiva) se asocia tam-bién a STV.17
Existe en la actualidad la categorización arbitraria de los factores de riesgoasociados a STV en dos grupos:
1. Idiopático (primario, permanente o irreversible).2. Secundario (provocado, transitorio o reversible) (cuadro 13–1).
Esta aproximación es más didáctica que lógica, pero permite identificar los facto-res modificables en el manejo médico (vide infra).
En el estudio ICOPER la proporción de pacientes con causa idiopática fue de20%.10
La realidad es que los fenómenos trombóticos resultan de la interdependenciaentre los factores dependientes del paciente y los factores asociados a una situa-ción clínica determinada (cuadro 13–2).
Resulta de gran importancia conocer todos estos factores de predisposiciónpara el momento de tomar decisiones en la prevención primaria de la enferme-dad; sin embargo, este anhelo parece un ideal, ya que en la realidad un estudiointernacional multicéntrico (más de 300 hospitales) demostró que sólo 58.5% delos pacientes con riesgo de STV debido a causas médicas y 39.5% debido a causasquirúrgicas recibieron profilaxis.19

218 (Capítulo 13)Tratado de trombosis
Cuadro 13–2. Factores de riesgo para tromboembolia pulmonar
Factor Dependiente Asociado
Factores con gran asociación (OR > 10):Fractura o reemplazo de cadera o tibia XCirugía general mayor XDaño de columna lumbar o médula espinal X
Factores con moderada asociación (OR 2 a 9):Artroscopia XCatéteres venosos centrales XQuimioterapia XEnfermedad cardiaca o pulmonar crónica XTerapia de reemplazo hormonal XCáncer XEnfermedad vascular cerebral paralítica XEmbarazo y posparto X
Factores con baja asociación (OR < 2):Reposo en cama > 3 días XInmovilidad (viaje aéreo o en auto) XEdad mayor de 65 años XCirugía laparoscópica XObesidad XVenas varicosas X
Adaptado de la referencia 33.
HISTORIA NATURAL
En cuanto a la mencionada relación entre TEP y TVP, la historia natural se abordaa raíz de los estudios iniciales de esta asociación en la década de 1960 tras cirugíasortopédicas; desde entonces se documentó que el factor inicial es una TVP de lapantorrilla en 30% de los casos, de los cuales en un tercio la TVP se resolvió es-pontáneamente tras pocos días, no se produjeron complicaciones en 40% de loscasos y 25% presentaron TVP proximal y TEP.20 El riesgo de TVP después decualquier cirugía es alto durante las primeras dos semanas, pero existe el riesgoaun después de dos a tres meses. La terapia anticoagulante profiláctica (AP) re-duce este riesgo; cuanto mayor sea ésta, menor será la incidencia de TVP. Muchosde los pacientes con TVP sintomática cursan con trombos proximales, mientrasque de 40 a 50% de los casos se complicarán con TEP, la mayor parte sin un cua-dro clínico aparente.5
La TEP ocurre entre tres y siete días después de la TVP; es fatal en 10% de loscasos una hora después del inicio de los síntomas. La presentación usual incluyeestado de choque o hipotensión (5 a 10% de los pacientes); en 50% de los casossin estado de choque se presenta con datos de DVD, según las pruebas de labora-

219Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
torio y los estudios de gabinete.21 La mayoría de las muertes (90%) ocurren enlos pacientes no manejados médicamente, debido a una TEP no reconocida,22
mientras que en los pacientes con tratamiento se espera una mortalidad menor de10%.5 En 50% de los pacientes con TVP/TEP que no reciben anticoagulaciónpuede recurrir la trombosis en un periodo de tres meses,2 mientras que en aquelloscon TVP/TEP que finalizan el tratamiento anticoagulante (3 a 12 meses) el riesgofuturo de TEP fatal es de 0.19 a 0.49 eventos por cada 100 personas al año.23
FISIOPATOLOGÍA
Las consecuencias de la TEP son principalmente hemodinámicas; aparecencuando de 30 a 50% de la circulación de las arterias pulmonares está ocluida porun trombo.24 Los trombos grandes solitarios o pequeños múltiples incrementansúbitamente las resistencias vasculares pulmonares a tal nivel que la poscarga nopuede ser manejada por el ventrículo derecho (VD) y se puede presentar muertesúbita por disociación electromecánica.25
En los pacientes que sobreviven a esta etapa, a pesar de la DVD, se inicia laactivación del sistema simpático, con respuestas inotrópica y cronotrópica y elmecanismo de Frank–Starling, produciendo un aumento de la presión de la arte-ria pulmonar (PAP); esto contribuye a mejorar el flujo sanguíneo pulmonar, per-mitiendo un llenado ventricular izquierdo y una mejoría del gasto cardiaco, quesumados a la vasoconstricción sistémica estabilizan la presión arterial sisté-mica.26
La hipotensión asociada a la TEP afecta la perfusión coronaria del ventrículoderecho (VD) y, por ende, su función. Hay pacientes que presentan pérdida de laestabilidad hemodinámica dentro de las primeras 24 a 48 h por trombos recurren-tes o por DVD.27 En ocasiones la respuesta simpática no es suficiente para evitarla DVD; esto es secundario a un incremento de la demanda de oxígeno del mio-cardio y a una disminución de la perfusión coronaria, lo cual explica la mortalevolución de la enfermedad.28
Asimismo, la TEP produce cambios respiratorios que suelen ser secundariosa las alteraciones hemodinámicas en las que el bajo gasto cardiaco resulta en de-saturación de la sangre venosa mezclada que ingresa a la circulación pulmonar;existen también alteraciones en la ventilación perfusión, debido a áreas ventila-das poco perfundidas, aumentando el espacio muerto; en los casos en los que seafecta un mayor número de vasos arteriales pulmonares se producen cortocircui-tos intrapulmonares por mezcla de sangre no oxigenada, produciéndose hipoxe-mia, que con obstrucciones mayores de 50% del lecho pulmonar conllevan a hi-poxemia refractaria.29 Un tercio de los pacientes cursan con cortocircuitos

220 (Capítulo 13)Tratado de trombosis
extrapulmonares por foramen oval permeable debido a un gradiente de presióninvertido de la aurícula de derecha a izquierda, produciendo también hipoxemiarefractaria, con el riesgo adicional de embolización paradójica, es decir, a la cir-culación general.30
Hay trombos que se fragmentan tras un evento y dan lugar a trombos pequeñosde 2 mm que se alojan en la circulación distal, es decir, en las ramas segmentariaso subsegmentarias, las cuales tal vez no produzcan cambios hemodinámicos; sinembargo, por ser múltiples pueden causar hemorragia alveolar con hipoxemia,disnea y hemoptisis.31 En algunos casos entre 24 y 48 h después se puede presen-tar infarto pulmonar, caracterizado por hipoxemia con dolor pleural, hemoptisisy derrame pleural mínimo, de tipo exudativo. El dolor es consecuencia de la irri-tación pleural secundaria a la oclusión vascular distal a los émbolos; este cuadroes conocido como síndrome de infarto pulmonar, en el que de 10 a 15% de lospacientes presentan infartos lobares y 10% infartos menores de 3 cm; lo usual(70%) es que se encuentren en las arterias segmentarias. El infarto pulmonar seencuentra en 30 a 50% de los casos y el derrame en 20 a 30%.32 Todo pacientecon enfermedad cardiopulmonar previa presentará mayor gravedad de la enfer-medad por los cambios mencionados.33
GRAVEDAD Y EVALUACIÓN DEL PRONÓSTICO
Desde 2008 las guías y consensos internacionales abogan por evitar en la TEPel empleo de los términos “masiva”, “submasiva” y “no masiva”, debido a quees recomendable evaluar el impacto de la gravedad por el riesgo de muerte. Lagravedad se basa, entonces, en la presencia de marcadores de riesgo; el inconve-niente es la necesidad de estudios de gabinete y laboratorio, que por sí mismosevitan estadificar el riesgo de muerte con escalas meramente clínicas. Esta estadi-ficación de riesgo propuesta por la Sociedad Americana de Cardiología (AHA,por sus siglas en inglés) es práctica y sencilla, ya que emplea los marcadores deriesgo (cuadros 13–3 y 13–4).33
Su gran utilidad radica en que distingue la TEP de riesgo alto de la muerte (ma-siva), la cual implica 5% de los pacientes sintomáticos, que además requieren unaestrategia de diagnóstico y terapéutica inmediata, debido al riesgo de mortalidadde 15%.34 El tratamiento con trombólisis o embolectomía no se debe retrasar. Porotro lado, la TEP que no es de riesgo alto se clasifica en dos de acuerdo con lapresencia o ausencia de marcadores de DVD o de daño miocárdico:
1. TEP de riesgo intermedio de muerte (submasiva): se diagnostica con undato de DVD o un marcador de daño miocárdico; corresponde a 30% de los

221Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–3. Principales marcadores útiles para evaluarel riesgo de mortalidad en tromboembolia pulmonar
Marcadores clínicos 1. Choque2. Hipotensión*
Marcadores de DVD 1. Dilatación del VD, hipocinesia o sobrecarga de presiónpor ecocardiografía
2. Dilatación del VD en tomografía computarizada espiral3. BNP elevado4. Elevación de la presión del VD en CCVD
Marcadores de daño miocárdico 1. Troponina cardiaca T o I positivas
Presión arterial sistólica < 90 mmHg o caída de la presión arterial sistólica > 40 mmHg por más de15 minutos que no es causada por una arritmia, hipovolemia o sepsis. TEP: tromboembolia pulmo-nar; DVD: disfunción del ventrículo derecho; VD: ventrículo derecho; BNP: péptido natriuréticocerebral; CCVD: cateterismo cardiovascular derecho. Adaptado de la referencia 33.
pacientes sintomáticos. Se debe manejar en el ambiente hospitalario y sepuede beneficiar de la terapia trombolítica según los estudios clínicos másrecientes.
2. TEP de bajo riesgo de muerte (no masiva): representa la mayoría de los ca-sos de TEP; en ella no hay marcadores de DVD o de daño al miocardio(mortalidad menor de 1%).35 Se caracteriza por estancias hospitalarias cor-tas o incluso manejo ambulatorio.
Tromboembolia pulmonar de alto riesgo de mortalidad
Anteriormente la clasificación se basaba en los hallazgos angiográficos por el nú-mero de lóbulos afectados según las ramas obstruidas (índice de Miller),38 lo cualha quedado en desuso.35 En el estudio ICOPER la mortalidad de los pacientes con
Cuadro 13–4. Escala de riesgo de acuerdo con la mortalidadesperada por tromboembolia pulmonar
Riesgo de mortalidadi d TEP
Marcadores de riesgo Implicacionesdasociado a TEP Clínicos
(choque/hipotensión)
DVD* Daño almiocardio
detratamiento
No alta Alta (> 15%) +++ ++ + Trombólisis o em-bolectomía
Intermedia (3 a 15%) – + � Ingreso a hospitalBaja (< 1%) – – – Egreso a domici-
lio o tratamien-to en casa
TEP: tromboembolia pulmonar; DVD: disfunción ventrículo derecho. Adaptado de la referencia 33.

222 (Capítulo 13)Tratado de trombosis
presión arterial sistólica (PAS) por debajo de 90 mmHg fue de 52.4% (IC 95%43.3 a 62.1%) vs. 14.7% (IC 95% 13.3 a 16.2%) en el grupo con PAS mayor.10
En el Management Strategy and Prognosis of Pulmonary Embolism Registry(MAPPET), que incluyó 1 001 pacientes, se mostró una mortalidad de 8.1% enpacientes con PAS > 90 mmHg vs. 25% en aquellos con PAS < 90 mmHg, y vs.65% aquellos que requirieron apoyo cardiovascular avanzado.34 Las escalas degravedad, como la de Ginebra o el índice de gravedad del embolismo pulmonar(PESI, por sus siglas en inglés), consideran que la hipotensión es un factor de pre-dicción de pronóstico adverso.39,40 La definición de la AHA parece atractiva,como hipotensión (PAS < 90 mmHg) durante al menos 15 min, o requerimientode apoyo inotrópico, no asociada a arritmias, hipovolemia o sepsis.35
Tromboembolia pulmonar deriesgo intermedio de mortalidad
Se requieren pruebas de gabinete que demuestren la presencia de DVD, como lassiguientes:
1. El ecocardiograma identifica a los pacientes con riesgo de pronóstico ad-verso, mediante la búsqueda de DVD. En un metaanálisis realizado porSánchez y col. se apreció una razón de momios (OR) de 2.53 (IC 1.17 a 5.5)para mortalidad en pacientes con TEP y DVD.41
2. La tomografía computarizada tiene la finalidad de evaluar la dilatación delVD (existe discrepancia en la literatura, debido a que tiene el inconvenientede que no valora la funcionalidad).33
Asimismo, se requiere la elevación de biomarcadores, como las troponinas, lascuales se asocian a pronósticos adversos; Becattini y col. mostraron en un metaa-nálisis que la OR de mortalidad en pacientes con TEP y elevación de troponinasfue de 5.9 (IC 95% 2.68 a 12.95);42 asimismo, las elevaciones del péptido natriu-rético cerebral (BNP, por sus siglas en inglés) y de la prohormona (pro–BNP)constituyen factores adversos; en el metaanálisis mencionado41 se encontró quelas OR de muerte a corto plazo en pacientes con TEP y elevación de BNP y pro–BNP fueron de 9.51 (IC 95% 3.16 a 28.64) y de 5.74 (IC 95% 2.18 a 15.1), respec-tivamente.
Mediante electrocardiografía (ECG) se han descrito una gran cantidad de sig-nos, que incluyen taquicardia sinusal, arritmias auriculares, ondas Q de bajo vol-taje en DIII y AvF, patrón S1Q3T3, patrón Qr en V1, P pulmonar, desviación deleje a la derecha, elevación y depresión del ST, QT prolongado y bloqueo de larama derecha incompleto. A pesar de ello, en la actualidad no se toman en cuentacomo marcadores de sobrevida.35

223Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Por lo tanto, la AHA35 propone como definición una TEP sin hipotensión perocon DVD o daño miocárdico. La DVD requiere al menos uno de los siguienteshallazgos:
1. Dilatación del VD (ventana apical de cuatro cámaras por ecocardiogramaque mida la relación del diámetro VD/VI > 0.9, o la presencia de DVD).
2. Dilatación del VD (relación del diámetro del VD/VI > 0.9 en tomografía).3. Elevación del BNP (> 90 pg/mL).4. Elevación del pro–BNP (> 500 pg/mL).5. Cambios electrocardiográficos (bloqueo de la rama derecha completo o in-
completo de novo, elevación o depresión del ST, o inversión de la onda T).
El daño miocárdico se define por cualquiera de los siguientes elementos:
1. Elevación de troponina I (> 0.4 ng/mL).2. Elevación de troponina T (> 0.1 ng/mL).
Tromboembolia pulmonar de bajo riesgo de mortalidad
Abarca a los pacientes con TEP y presión arterial normal, sin elevación de bio-marcadores y sin DVD por imagen.
Esta clasificación no considera la coexistencia de comorbilidades, de tal man-era que un evento no masivo se puede asociar a un alto riesgo de complicacionesy de mortalidad en un paciente con enfermedad cardiopulmonar previa.
La gravedad también se puede evaluar por métodos clínicos, los cuales sonfactores de predicción de mortalidad independientes de los biomarcadores o a losestudios de imagen.
Se recomienda emplear la escala de Ginebra y el PESI o el PESI simplificado(cuadros 13–5 y 13–6).
ABORDAJE PARA EL DIAGNÓSTICO
Debido a que lo más importante es diagnosticar la TEP, el diagnóstico por clínicaes muy variable (cuadro 13–7), por lo que se recomienda emplear las probabilida-des o sospechas preprueba de la enfermedad; ya que una alta probabilidad pre-prueba de TEP indica la necesidad de un tratamiento específico y una baja proba-bilidad preprueba justifica evitarlo y que además conlleve un bajo riesgo demortalidad, aun cuando fuera ambulatorio.

224 (Capítulo 13)Tratado de trombosis
Cuadro 13–5. Factores de predicción clínicos de mortalidada 30 días en pacientes con tromboembolia pulmonar (PESI)
Variable Puntos
Edad 1 punto por añoSexo masculino 10Cáncer 30Insuficiencia cardiaca 10Enfermedad pulmonar crónica 10Frecuencia cardiaca > 110/min 20Presión arterial sistólica < 100 mmHg 30Frecuencia respiratoria > 30/min 20Temperatura < 36 �C 20Alteraciones del sensorio 60SaO2 < 90% 20Categorías de riesgo (% de mortalidad a 30 días):
Clase I, < 65 puntos (0%)Clase II, 66 a 85 puntos (1%)Clase III, 96 a 105 puntos (3.1%)Clase IV, 106 a 125 puntos (10.4%)Clase V, > 126 puntos (24.4%)
TEP: tromboembolia pulmonar; SaO2: saturación por pulsioximetría. Adaptado de las referencias44 y 45.
La sospecha o probabilidad surge en 90% de los casos debido a la presenciade síntomas, como son disnea, dolor torácico y síncope, solos o en combina-ción.43 El síncope es poco común; indica una reserva hemodinámica disminuida,invariablemente acompañado de hipotensión o choque. El dolor pleurítico cono sin disnea es la presentación más frecuente. La disnea aislada súbita se presentapor TEP de grandes ramas de la arteria pulmonar, la cual conlleva mayores mani-
Cuadro 13–6. Factores de predicción de mortalidad a 30 días en pacientescon tromboembolia pulmonar. PESI, forma simplificada
Variable:Edad > 80 añosCáncerInsuficiencia cardiaca o enfermedad pulmonar crónicaFrecuencia cardiaca > 110/minPresión arterial sistólica < 100 mmHgSaO2 > 90%
Interpretación: se da un punto por cada variable.0 puntos: bajo riesgo de mortalidad a 30 días (1.2%)> 1 punto: alto riesgo de mortalidad a 30 días (11.3%)
TEP: tromboembolia pulmonar, SaO2: saturación por pulsioximetría. Adaptado de la referencia 46.
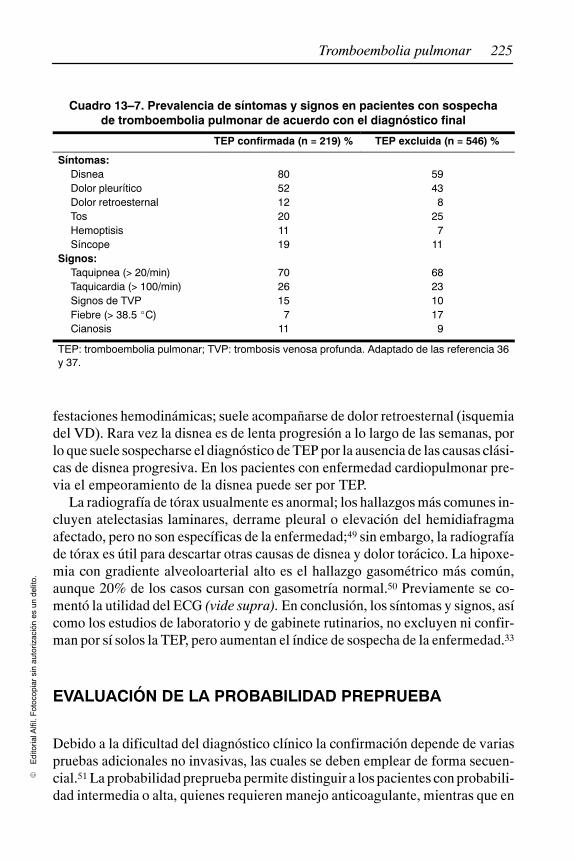
225Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–7. Prevalencia de síntomas y signos en pacientes con sospechade tromboembolia pulmonar de acuerdo con el diagnóstico final
TEP confirmada (n = 219) % TEP excluida (n = 546) %
Síntomas:Disnea 80 59Dolor pleurítico 52 43Dolor retroesternal 12 8Tos 20 25Hemoptisis 11 7Síncope 19 11
Signos:Taquipnea (> 20/min) 70 68Taquicardia (> 100/min) 26 23Signos de TVP 15 10Fiebre (> 38.5 �C) 7 17Cianosis 11 9
TEP: tromboembolia pulmonar; TVP: trombosis venosa profunda. Adaptado de las referencia 36y 37.
festaciones hemodinámicas; suele acompañarse de dolor retroesternal (isquemiadel VD). Rara vez la disnea es de lenta progresión a lo largo de las semanas, porlo que suele sospecharse el diagnóstico de TEP por la ausencia de las causas clási-cas de disnea progresiva. En los pacientes con enfermedad cardiopulmonar pre-via el empeoramiento de la disnea puede ser por TEP.
La radiografía de tórax usualmente es anormal; los hallazgos más comunes in-cluyen atelectasias laminares, derrame pleural o elevación del hemidiafragmaafectado, pero no son específicas de la enfermedad;49 sin embargo, la radiografíade tórax es útil para descartar otras causas de disnea y dolor torácico. La hipoxe-mia con gradiente alveoloarterial alto es el hallazgo gasométrico más común,aunque 20% de los casos cursan con gasometría normal.50 Previamente se co-mentó la utilidad del ECG (vide supra). En conclusión, los síntomas y signos, asícomo los estudios de laboratorio y de gabinete rutinarios, no excluyen ni confir-man por sí solos la TEP, pero aumentan el índice de sospecha de la enfermedad.33
EVALUACIÓN DE LA PROBABILIDAD PREPRUEBA
Debido a la dificultad del diagnóstico clínico la confirmación depende de variaspruebas adicionales no invasivas, las cuales se deben emplear de forma secuen-cial.51 La probabilidad preprueba permite distinguir a los pacientes con probabili-dad intermedia o alta, quienes requieren manejo anticoagulante, mientras que en

226 (Capítulo 13)Tratado de trombosis
Figura 13–1. Algoritmo de diagnóstico ante sospecha preprueba de tromboemboliapulmonar. DDD: dímero D; USC: ultrasonido por compresión; TEP: tromboembolia pul-monar; ATCM: angiografía por tomografía computarizada multicorte. Adaptado de lareferencia 51.
Probabilidadpreprueba
USC o ATCM
Baja ointermedia oimprobable
Alta oprobable
DDD
Positiva
Negativa
Diagnósticode TEP
DescartaTEP
> 500 �g/L
< 500 �g/L
los pacientes con probabilidad preprueba baja el diagnóstico de TEP se puededescartar únicamente con la determinación de dímero D (DDD) en rangos norma-les (figura 13–1).
Los modelos actuales validados y evaluados de forma prospectiva47,48 incor-poran datos clínicos de la historia clínica, signos y síntomas, pero no incluyen ga-sometría, radiografía de tórax ni ECG debido a sus bajas sensibilidad y especifici-dad para el diagnóstico de TEP. Ambas formas predicen la probabilidadpreprueba de TEP y tienen la misma aceptabilidad, pero no son iguales, debidoal grupo poblacional en el que fueron validadas. La escala de Ginebra (cuadro13–8)47 se emplea en áreas con una prevalencia de TEP de 20%, mientras que laescala de Wells (cuadro 13–9) se debe emplear en los pacientes hospitalizados.51
Una vez evaluada la probabilidad preprueba se sigue el algoritmo que se sugiereen la figura 13–1, con los debidos estudios de gabinete y laboratorio para evaluarel rendimiento diagnóstico de cada una de las pruebas (cuadro 13–10).
Dímero D
El DDD es un producto de la degradación de la fibrina cuya concentración se in-crementa en pacientes con fenómenos trombóticos venosos; cuando es empleadoa través de la técnica de ELISA cuantitativa es altamente sensible (95%) para ex-cluir TEP, usualmente a valores menores de 500 �g/L; es por ello que en pacien-

227Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–8. Reglas de predicción clínica para tromboemboliapulmonar. Escala de Ginebra revisada
Variable Puntos
Factores predisponentes:Edad > 65 años 1TVP o TEP previa* 3Cirugía o fractura en el último mes 2Cáncer 2
Síntomas:Dolor de extremidad inferior unilateral 3Hemoptisis 2
Signos:Frecuencia cardiaca
75 a 94/min 395 /min 5
Dolor en extremidad inferior a la palpación o edema unilateral 4Probabilidad clínica:
Baja (0 a 3 puntos)Intermedia (4 a 10 puntos)Alta (> 11 puntos)
TVP: trombosis venosa profunda; TEP: tromboembolia pulmonar. Adaptado de la referencia 47.
Cuadro 13–9. Reglas de predicción clínica para tromboemboliapulmonar. Escala de Wells en sus dos modalidades
Variable Puntos
Factores de predisposición:TVP o TEP previa* 1.5Cirugía reciente o inmovilización 3.5Cáncer 1
Síntomas:Hemoptisis 1
Signos:Frecuencia cardiaca
> 100/min 1.5Datos clínicos de TVP 3Juicio clínicoDiagnóstico alternativo menos probable que TEP 3
Probabilidad clínica tres niveles:Baja (0 a 1 punto)Intermedia (2 a 6 puntos)Alta (> 7 puntos)
Probabilidad clínica a dos niveles:TEP poco probable (0 a 4 puntos)TEP probable (> 4 puntos)
TVP: trombosis venosa profunda; TEP: tromboembolia pulmonar. Adaptado de la referencia 48.

228 (Capítulo 13)Tratado de trombosis
Cuadro 13–10. Rendimiento de algunas pruebas en el algoritmode diagnóstico para confirmar o descartar la presencia
de tromboembolia pulmonar
Para confirmar TEP Razón de verosimilitud (IC 95%)
Gammagrama pulmonar ventilatorio perfusorio no normal 18.3 (10.3 a 32.5)ATCM 24.1 (12.4 a 46.7)USC de venas proximales positivo 16.2 (5.6 a 46.7)
Para descartar TEP
Gammagrama pulmonar ventilatorio perfusorio normal 0.05 (0.03 a 0.10)ATCM negativa 0.11 (0.06 a 0.19)ACTM negativa y USC de venas proximales negativa 0.04 (0.03 a 0.06)USC de venas proximales negativo 0.67 (0.50 a 0.89)DDD por ELISA < 500 mg/L 0.08 (0.04 a 0.18)
La razón de verosimilitud (likelihood ratio) para confirmar TEP es positiva, para descartarla esnegativa. Es la razón de que un resultado de una determinada prueba corresponda en un pacientecon la enfermedad, en comparación con la razón de que el resultado de la prueba corresponda aun paciente sin enfermedad. Por ejemplo, una razón de 20 significa que el paciente tiene 20 vecesmás probabilidad de tener TEP que de no tenerla; por el contrario, una razón negativa (0.10) signi-fica que el paciente tiene 10 veces menos probabilidad de tenerla. TEP: tromboembolia pulmonar;ACTM: angiografía con tomografía computarizada multidetector; USC: ultrasonido con compre-sión; DDD: determinación de dímero D. Adaptado de referencia 51.
tes con probabilidad preprueba baja o intermedia un valor < 500 �g/L descartala presencia de TEP.
Existen varios tipos de pruebas para la determinación del DDD, pero es impor-tante mencionar que éste se debe obtener mediante técnica de ELISA de formacuantitativa.
El DDD es poco útil en algunos grupos de pacientes, como en aquellos conprobabilidad preprueba alta, pacientes hospitalizados por otra causa diferente aTEP, edad mayor de 65 años, embarazo, insuficiencia renal y periodo posteriora la cirugía.52
Ultrasonido por compresión para diagnósticode trombosis venosa profunda
La imagen ultrasonográfica ha reemplazado a la venografía para el diagnósticode TVP; es el estudio de elección en pacientes en quienes se contraindica la reali-zación de angiografía con tomografía computarizada multidetector (ACTM). Elultrasonido por compresión (USC) se puede emplear tanto en el abordaje inicialcomo en el árbol de manejo (figura 13–1).

229Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Angiografía con tomografía computarizada multidetector
La ATCM ha sustituido al gammagrama ventilatorio perfusorio (V/Q) como elestudio por imagen más útil para el diagnóstico de TEP, siendo importante quesea multidetector, ya que posee una mayor sensibilidad para tal fin.53
Su uso es seguro, ya que en un metaanálisis que incluyó más de 20 estudioscon más de 4 000 pacientes54 se demostró que en pacientes en quienes se descartóTEP mediante ATCM negativa y que además no recibieron anticoagulación latasa de TVP/TEP a los tres meses fue de 1.4% (IC 95% 1.1 a 1.8), con una tasade TEP de alto riesgo de 0.51% (IC 95% 0.33 a 0.76), datos muy semejantes alos que se obtenían antes con el empleo de angiografías con resultados norma-les.55
TRATAMIENTO
Una vez establecido el diagnóstico (cuadros 13–8 y 13–9, figura 13–1) y evalu-ada la gravedad de la TEP (cuadros 13–3 a 13–6) se deben tomar en cuenta lassituaciones clínicas (figura 13–2).
Figura 13–2. Tres formas de manejo de la tromboembolia pulmonar. A y B son nuevaspropuestas de manejo basadas en ensayos clínicos. A. Con etexilato de dabigatrán enel estudio RECOVER.56 B. Con rivaroxabán en el estudio EINSTEIN.57 HNF: heparinano fraccionada, HBPM: heparina de bajo peso molecular; AVK: antagonistas de vitami-na K; INR: radio normalizado internacional. Adaptado de referencia 51.
Agudo Intermedio Crónico
Entrelazar
Tratamiento estándar
InicialHNF, HBPM,Fondaparinux> 5 días
MantenimientotempranoAVK INR 2.0 a 3.0> 3 meses
Prevención secundariaa largo plazoAVK INR 2–0 a 3–0*> 3 meses, años o indefinido
Nuevos esquemas potenciales con medicamentos nuevos anticoagulantesA
BEntrelazar
Empleo de un único medicamento

230 (Capítulo 13)Tratado de trombosis
Tratamiento de tromboembolia pulmonarcon riesgo de mortalidad intermedio o bajo
Toda TEP no masiva debe incluir tratamiento temprano y de prevención a largoplazo. El tratamiento de primera línea se debe administrar ante la sospecha inter-media o alta de TEP, sin necesidad de esperar los resultados confirmatorios.63 Elmanejo debe incluir las heparinas en cualquiera de sus dos formas —no fraccio-nada (HNF) y de bajo peso molecular (HBPM)— o fondaparinux.63 Las hepari-nas actúan uniéndose a la antitrombina, un anticoagulante natural, provocandola inactivación acelerada de la trombina y de otros factores de la coagulación acti-vados (factor X, FXa).
Las HNF se administran de forma intravenosa, inicialmente en bolo, con dosisde mantenimiento; desafortunadamente, tienen un efecto farmacológico pocoprevisible y de gran variabilidad interindividual (determinado por la unión delfármaco a las proteínas plasmáticas), por lo que se requieren ajustes en la dosisde acuerdo con los resultados del tiempo de tromboplastina tisular parcial activa-do (TTP) (cuadro 13–11).
La gran ventaja de las HBPM (cuadro 13–12) es que su administración es sub-cutánea, en dosis ajustadas al peso del paciente, sin necesidad de monitorear elefecto.64 Su mecanismo de acción es muy semejante al de las HNF, pero es máspronunciado en el FXa, en comparación con la trombina; ambas poseen la mismaequivalencia clínica.65
El fondaparinux es idéntico al componente más pequeño de la heparina, conel mismo sitio de acción, pero es de origen sintético.
Este medicamento no es inferior a las heparinas.66
Ambos grupos de medicamentos se depuran por vía renal, por lo que se sugiereespecial precaución en pacientes con depuración de creatinina menor de 30 mL/min. En dichos casos se recomienda reducir la dosis o aumentar el intervalo de
Cuadro 13–11. Ajuste de la dosis de heparina no fraccionadacon base en el tiempo de tromboplastina parcial activado
TTP Cambio en la dosis
< 35 seg (< 1.2 veces el control) 80 UI/kg en bolo, aumentar la infusión de manteni-miento a 4 UI/kg/h
38 a 45 seg (1.2 a 1.5 veces el control) 40 UI/kg en bolo, aumentar la infusión de manteni-miento a 2 UI/kg/h
46 a 70 seg (1.5 a 2.3 veces el control) Sin cambios, mantener dosis de mantenimiento71 a 90 seg (2.3 a 3.0 veces el control) Reducir la infusión de mantenimiento a 2 UI/kg/h> 90 seg (> 3 veces el control) Detener la infusión por 1 h y después reducir la
dosis de mantenimiento a 3 UI/kg/h
Adaptado de la referencia 62.

231Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–12. Posología subcutánea de las heparinas de bajopeso molecular y fondaparinux en tromboembolia pulmonar
Enoxaparina 1 mg/kg cada 12 h o 1.5 mg/kg/díaDalteparina 200 UI/kg/díaTinzaparina 175 UI/kg/díaFondaparinux 5 mg/día (peso < 50 kg)
7.5 mg/día (peso de 50 a 100 kg)10 mg/día (peso > 100 kg)
TEP: tromboembolia pulmonar. Adaptado de la referencia 33.
administración, con monitoreo de la actividad del FXa o en su caso empleo deHNF.64
El empleo de heparinas o fondaparinux se debe entrelazar en el momento deldiagnóstico con los antagonistas de vitamina K (AVK) durante al menos cincodías.63 Los primeros se suspenden una vez que la concentración anticoagulanteinducida por los AVK alcanza un índice normalizado internacional (INR, por sussiglas en inglés) de 2.0 durante al menos dos días consecutivos. Se sugiere queen pacientes con cáncer se utilice HBPM durante tres meses, más que con losAVK.63
Los AVK bloquean el último paso de la biosíntesis de cuatro factores de la coa-gulación (II, VII, IX y X) en el hígado. Debido a las diferentes vidas medias enplasma de estos factores, el estado de anticoagulación se consigue entre cuatroy siete días después de iniciado el medicamento. Los AVK se clasifican de acuer-do con su vida media en corta (acenocumarina), intermedia (warfarina) y larga(fenprocumona, no disponible en México). Se debe tomar en cuenta que existeuna variabilidad genética en el metabolismo de estos fármacos.67 La dieta que serequiere es restringida en vitamina K, además de diversas interacciones farmaco-lógicas, por lo que se requiere un monitoreo estrecho cuando se emplean para al-canzar un INR de 2.0 a 3.0. La dosis inicial recomendada de warfarina es de 5 a7.5 mg, y la de acenocumarina es de 6 a 8 mg,33 evitando excederse y ajustándolade acuerdo con el INR a las 72 h de iniciado el manejo.
Todos los fármacos anticoagulantes producen hemorragia, la cual es más acen-tuada al inicio del manejo (trauma o hemorragias ocultas graves, como intracra-neales, abdominales o torácicas). La hemorragia asociada a los AVK se incre-menta con la edad, por lo que existen escalas de predicción de riesgo de sangrado(cuadro 13–13): la de RIETE (Registro Informatizado de la Enfermedad Trom-boembólica Venosa) y la de HEMORR2HAGES; ésta última no se emplea enTVP ni TEP.
La seguridad de estos medicamentos depende de la adherencia de los pacientesal tratamiento y de evitar las interacciones farmacológicas asociadas, así comoel consumo de alcohol. Asimismo, es importante evitar las dosis altas de inicio

232 (Capítulo 13)Tratado de trombosis
Cuadro 13–13. Escalas clínicas de predicción de hemorragiacuando se emplea tratamiento anticoagulante
Escala/variable HEMORR2HAGES*58 Puntos
Enfermedad hepática o renal 1Abuso de alcohol 1Cáncer 1Edad > 75 años 1Hipertensión descontrolada 1Anemia 1Riesgo aumentado de caídas 1Enfermedad vascular cerebral 1Trombocitopenia o disfunción plaquetaria 1Hemorragia previa 2
RIETE**59
Hemorragia reciente 2Creatinina > 1.2 mg/dL 1.5Hemoglobina < 13 g/dL (hombres) o < 12 g/dL (mujeres) 1.5Cáncer 1Resolución de la TEP clínica 1Edad > 75 años 1
Interpretación (riesgo por 1 000/pacientes/año): * 0: 1.9; 1 punto: 2.5; 2 puntos: 5.3; 3 puntos: 8.4;4 puntos: 10.4 y > 5 puntos: 12.3. ** 0: 0.3; 1 a 3 puntos: 2.6; > 4 puntos: 7.3. TEP: tromboemboliapulmonar; RIETE: Registry of Patients with Thromboembolism. Adaptado de la referencia 51.
para evitar la reacción paradójica de un estado protrombótico debido a la deple-ción de proteína C, la cual es un inhibidor de la coagulación dependiente de vita-mina K que posee una vida media muy corta. La trombocitopenia inducida porheparina es otra complicación importante que desencadena fenómenos trombo-embólicos venoarteriales (los cuales son más comunes con HNF, poco comunescon HBPM y raros con fondaparinux). Aún queda en duda la realización de escru-tinio de medición de plaquetas y de la función plaquetaria.51
La duración del tratamiento se debe evaluar según el riesgo de recurrencia delos eventos y el riesgo de hemorragia. En el estudio RIETE59 el riesgo de recu-rrencia con tratamiento anticoagulante fue de 7%. Se sugiere seguir las recomen-daciones de las guías internacionales del Noveno Reporte del Colegio Americanode Tórax (cuadro 13–14),63 que indican que una duración de tres meses es tanefectiva como el tratamiento de 6 a 12 meses, y que en los eventos asociados aun factor secundario es aún menor la recurrencia. La medición del DDD es unaalternativa al mes de tratamiento, ya que su disminución justifica la suspensióndel AVK64 o bien la realización de un USC para evaluar los trombos residuales.65
Desafortunadamente, se ha prestado poca atención a tales propuestas.Actualmente existen nuevos anticoagulantes orales con mecanismos de acción
diferentes a los mencionados, como son los inhibidores de FXa (rivaroxabán) o

233Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–14. Duración del tratamiento anticoagulantepara tromboembolia pulmonar
Escenario clínico Duración de tratamientorecomendado*
Nivel derecomendación
Primera TEP asociada a un factorsecundario
3 meses 1A
Primera TEP asociada a un factorprimario
Al menos tres meses 1A
Al final de los tres meses de manejo Valorar el tratamiento a largo plazo 1CEn ausencia de contraindicación Tratamiento a largo plazo 1ADurante un seguimiento a largo plazo Evaluar el riesgo–beneficio periódi-
camente1C
TEP recurrente o trombofilia Tratamiento a largo plazo 1ATEP secundaria a cáncer Tratamiento a largo plazo 1A
Uso de HBPM durante tres a seismeses, después AVK conformeel cáncer se considere activo
Se pueden emplear dabigatrán/ri-varoxabán
2C
Recomendaciones de acuerdo con las guías de práctica clínica del American College of Chest Phy-sicians. Recomendaciones G1, significa que el beneficio es mayor que el perjuicio de emplear eltratamiento. A se refiere a que la calidad metodológica se sustenta en varios ensayos clínicos con-trolados y C se sustenta en estudios observacionales, series de casos o ensayos clínicos controla-dos con sesgos.* El tratamiento a largo plazo debe tomar en cuenta los riesgos de sangrado (cuadro 13–13).TEP: tromboembolia pulmonar. Adaptado de la referencia 63.
de trombina (dabigatrán), en los que se evitan los cuidados de las heparinas y esprobable que puedan reemplazar a los AVK. Asimismo, se administran en dosisfija, no requieren seguimiento mediante pruebas de laboratorio y tienen pocas in-teracciones farmacológicas y alimentarias.
El ensayo clínico asignado al azar doble ciego RECOVER56 involucró pacien-tes con TEP/TVP inicialmente manejados con anticoagulación parenteral duran-te nueve días (8 a 11). El etexilato de dabigatrán administrado en dosis de 150 mgcada 12 h, sin monitoreo, no fue inferior a la warfarina (INR meta de 2 a 3) enlaza-do a la HBPM. Se encontró la misma seguridad en su uso.
En el estudio EINSTEIN57 el empleo de rivaroxabán en dosis de 15 mg cada12 h durante tres semanas, continuando con 20 mg/día y sin monitoreo, no fueinferior a los AVK enlazado con HBPM, sino que mostró la misma seguridad. In-cluso se evaluó la profilaxis secundaria con rivaroxabán (20 mg/día), la cual fuemejor que con placebo, con una reducción de 82% de TVP/TEP (HR 0.18, IC95% 0.09 a 0.39, p < 0.001), sin aumento del riesgo de hemorragia mayor; no ocu-rrió lo mismo con la hemorragia menor, que fue de 5.4%, en comparación con1.2% con placebo.

234 (Capítulo 13)Tratado de trombosis
Tratamiento de la tromboemboliapulmonar de alto riesgo de mortalidad
La DVD es la principal causa de muerte. El tratamiento con altos volúmenes desoluciones intravenosas ha mostrado ser perjudicial más que benéfico, por lo quesólo se recomienda en pacientes normotensos. El uso de norepinefrina mejora laeficiencia del VD, además de que aumenta la PAS, por lo que puede ser de utili-dad en pacientes con hipotensión. El empleo de dobutamina o dopamina puedeocasionar una mayor caída de la presión arterial, por lo que se deben emplear concautela.33 Aún no se recomienda en esta enfermedad el empleo de antagonistasde endotelina, inhibidores de prostaglandinas o inhibidores de fosfodiesterasa,ya que se encuentran en fases I a II de estudio.
La hipoxemia suele mejorar únicamente si se incrementa la fracción inspiradade oxígeno de 30 a 40% (cánula nasal o mascarilla Venturi). Rara vez se requiereintubación endotraqueal; sin embargo, cuando se requiera se debe evitar la hiper-insuflación dinámica, porque complica el retorno venoso y disminuye el gastocardiaco del VD, por lo que se debe mantener una presión de apertura alveolar< 30 cmH2O, siguiendo las maniobras de protección pulmonar ya conocidas.66
Todo paciente debe iniciar además con el tratamiento mencionado (vide su-pra). La trombólisis67–69 ha mostrado a través de varios ensayos clínicos que re-suelve rápidamente la obstrucción por el trombo, mejorando las condiciones he-modinámicas y respiratorias; produce una reducción de 30% de la PAP y mejora15% el índice cardiaco del VD, lo cual se observa mediante ecocardiografía des-pués de tres horas.67 En México existen varios tipos de agentes (cuadro 13–15),cada uno con sus características propias, pero igualmente seguros y con la mismaeficacia.70–72 Todos deben ser administrados a través de un vaso venoso periféri-co, evitando las punciones arteriales.33 La heparina no se debe administrar conestreptocinasa ni con urocinasa; sin embargo, no existe contraindicación de su ad-ministración junto con el activador de plasminógeno tisular recombinante (rtPA).
Cuadro 13–15. Tratamientos y posología de los agentestrombolíticos para tromboembolia pulmonar
Estreptocinasa Dosis de carga: 250 000 UI por 30 minDosis de mantenimiento: 100 000 UI/hora por 12 a 24 hRégimen rápido: 1.5 millones UI para 2 h
Urocinasa Dosis de carga: 4 400 UI/kg por 10 minDosis de mantenimiento: 4 400 UI/kg/h por 12 a 24 hRégimen rápido: tres millones UI para 2 h
Activador de plasminógeno ti-sular recombinante (rtPA)
100 mg en 2 h o 0.6 mg/kg en 15 min (dosis máxima: 50 mg)
Adaptado de la referencia 33.

235Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 13–16. Metaanálisis de estudios con trombólisisen pacientes con tromboembolia pulmonar
Puntoprimario
Estudios que incluyeron pacientescon TEP masiva
Estudios que excluyeronpacientes con TEP masiva
Trombóli-sis (n/N/%)
Heparina(n/N/%)
OR(IC 95%)
Trombóli-sis (n/N/%)
Heparina(n/N/%)
OR(IC 95%)
TEP recurrenteo muerte
12/128/9.4 24/126/19 0.45 (0.22 a0.92)
13/246/5.3 12/248/4.8
1.07 (0.50a 2.30)
TEP recurrente 5/128/3.9 9/126/7.1 0.61 (0.23 a1.62)
5/246/2 7/248/2.8 0.76 (0.28a 2.08)
Muerte 8/128/6.2 16/126/12.7
0.47 (0.20 a110)
8/246/3.3 6/248/2.4 1.16 (0.44a 3.05)
Hemorragiamayor
28/128/21.9
15/126/11.9
1.98 (1.00 a3.92)
6/246/2.4 8/248/3.2 0.67 (0.24a 1.86)
TEP: tromboembolia pulmonar; n: número de pacientes con el punto primario en estudio; N: totalde pacientes; OR: razón de momios. Adaptado de la referencia 60.
La efectividad de la trombólisis se puede evaluar mediante datos clínicos y eco-cardiográficos en las primeras 36 h,73 obteniéndose la mayor efectividad cuandose administra en las primeras 48 h de inicio de los síntomas, pero puede ser útilaun entre 6 y 14 días después de iniciado el cuadro.33
A pesar de estos datos y de la rapidez con que actúan dichos medicamentos,una semana después la efectividad en la función del VD y la gravedad de la obs-trucción vascular son similares a las conseguidas con la heparina70 (cuadro13–16).
El principal riesgo de la trombólisis es la hemorragia, por lo que es importantetener en consideración las contraindicaciones de su uso (cuadro 13–17), ya queconlleva un riesgo de hemorragia de 13% y de hemorragia mortal de 1.8%.33 Latrombólisis no está indicada en los pacientes con bajo riesgo de mortalidad; enlos pacientes con riesgo de mortalidad intermedia existe discrepancia acerca desu uso.63
La embolectomía quirúrgica y la embolectomía percutánea con catéter son al-ternativas con pocas indicaciones en el contexto de la TEP aguda; están indicadasen los pacientes con contraindicación de trombólisis, o que ésta fuese fallida, asícomo también en pacientes con trombo intracardiaco.33,63 Los filtros venosossuelen colocarse de manera infrarrenal en la vena cava inferior; protegen de unarecurrencia de TEP, pero no están exentos de complicaciones tempranas (trombo-sis en el sitio de inserción en 10% de los casos) y tardías (TVP recurrente en 20%de los casos y síndrome posflebítico en 40%), aun con anticoagulantes. Hasta22% de los pacientes presentarán oclusión del filtro en un lapso de cinco años y33% en nueve años.33 No existen estudios ni ensayos que comparen su empleocon el tratamiento convencional ni que soporten su uso rutinario; sólo existe un

236 (Capítulo 13)Tratado de trombosis
Cuadro 13–17. Contraindicaciones de trombólisis
Absolutas:Enfermedad vascular hemorrágica o enfermedad vascular cerebral de origen desconocidoEnfermedad vascular cerebral en los últimos seis mesesTraumatismo craneoencefálico o tumores cerebralesTrauma reciente en las últimas tres semanas (ortopedia/cirugía)Hemorragia gastrointestinal en el último mesHemorragia conocida o evidente
Relativas:Ataque isquémico transitorio en los últimos seis mesesTerapia anticoagulante oralEmbarazo o puerperio en la primer semanaPunciones vasculares no compresiblesReanimación traumáticaHipertensión refractaria (presión arterial sistólica > 180 mmHg)Enfermedad hepática avanzadaEndocarditis infecciosaÚlcera péptica activa
Adaptado de la referencia 61.
ensayo en la literatura que no ha mostrado cambios en la sobrevida a ocho añosde seguimiento;74 sin embargo, son una opción cuando existe contraindicaciónde anticoagulación.63
PREVENCIÓN
Es indudable que la HBPM es efectiva y segura en dosis fijas y pequeñas, siemprey cuando se tomen en cuenta los factores de riesgo tradicionales ya mencionados(cuadro 13–1) y particularmente los factores secundarios. Los beneficios se hanmostrado en la cirugía ortopédica (reemplazo de cadera o de rodilla) con un usoextendido hasta por cinco semanas.51
REFERENCIAS
1. Moser KM, Fedullo PF, Littejohn JK et al.: Frequent asymptomatic pulmonary embolismin patients with deep venous thrombosis. JAMA 1994;271:223–225.
2. Dalen JE: Pulmonary embolism: what have we learned since Virchow? Natural history, pa-thophysiology, and diagnosis. Chest 2002;122:1440–1456.
3. Stein PD, Kayali F, Olson RE: Estimated case fatality rate of pulmonary embolism, 1979to 1998. Am J Cardiol 2004;93:1197–1199.
4. Stein PD, Beemath A, Olson RE: Trends in the incidence of pulmonary embolism anddeep venous thrombosis in hospitalized patients. Am J Cardiol 2005;95:1525–1552.

237Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
5. Dalen JE, Alpert JS: Natural history of pulmonary embolism. Prog Cardiovasc Dis 1975;17:259–270.
6. Nordstrom M, Lindblad B: Autopsy–verified venous thromboembolism within a definedurban population, the city of Malmo, Sweden. APMIS 1998;106:378–384.
7. Sigler L, Romero T, Meillón LA et al.: Tromboembolia pulmonar en autopsias en un pe-riodo de 10 años. Rev Med IMSS 1996;34:7–11.
8. Karwinski B, Svendsen E: Comparison of clinical and postmortem diagnosis of pulmona-ry embolism. J Clin Pathol 1989;42:135–139.
9. Jerjes SC, Ramírez RA, Gutiérrez FP et al.: Un enfoque diferente del estado actual dela trombólisis en tromboembolia pulmonar. Arch Inst Cardiol Mex 1998;68:166–180.
10. Goldhaber SZ, Visani L, de Rosa M: Acute pulmonary embolism: clinical outcomes inthe International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet 1999;353:1386–89.
11. Spencer FA, Goldberg RJ, Lessard D et al.: Factors associated with adverse outcomes inoutpatients presenting with pulmonary embolism: the Worcester Venous Thromboembol-ism Study. Circ Cardiovasc Qual Outcomes 2010;3:390–394.
12. Ng AC, Chung T, Yong AS et al.: Long–term cardiovascular and noncardiovascular mor-tality of 1 023 patients with confirmed acute pulmonary embolism. Circ Cardiovasc QualOutcomes 2011;4:122–128.
13. Piazza G, Goldhaber SZ: Chronic thromboembolic pulmonary hypertension. N Engl JMed 2011;364:351–360.
14. Kahn SR: The post–thrombotic syndrome. Hematology Am Soc Hematol Educ Prog 2010;2010:216–220.
15. Piazza G, Goldhaber SZ: Venous thromboembolism and atherothrombosis: an integratedapproach. Circulation 2010;121:2146–2150.
16. Sim DS, Jeong MH, Kang JC: Current management of acute myocardial infarction: expe-rience from the Korea Acute Myocardial Infarction Registry. J Cardiol 2010;56:1–7.
17. Folsom AR, Lutsey PL, Astor BC et al.: C–reactive protein and venous thromboembol-ism. A prospective investigation in the ARIC cohort. Thromb Haemost 2009;102:615–619.
18. Hansson PO, Welin L, Tibblin G et al.: Deep vein thrombosis and pulmonary embolismin the general population. “The Study of Men Born in 1913”. Arch Intern Med 1997;157:1665–1670.
19. Cohen AT, Tapson VF, Bergmann JF et al.: Venous thromboembolism risk and prophy-laxis in the acute hospital care setting (ENDORSE study): a multinational cross–sectionalstudy. Lancet 2008;371:387–394.
20. Kakkar VV, Howe CT, Flanc C et al.: Natural history of postoperative deep–vein throm-bosis. Lancet 1969;2:230–232.
21. Wood KE: Major pulmonary embolism: review of a pathophysiologic approach to the gol-den hour of hemodynamically significant pulmonary embolism. Chest 2002;121:877–905.
22. Laporte S, Mismetti P, Decousus H et al.: Clinical predictors for fatal pulmonary embo-lism in 15 520 patients with venous thromboembolism: findings from the Registro Informa-tizado de la Enfermedad Tromboembólica Venosa (RIETE) Registry. Circulation 2008;117:1711–1716.
23. Douketis JD, Gu CS, Schulman S, Ghirarduzzi A et al.: The risk for fatal pulmonary em-bolism after discontinuing anticoagulant therapy for venous thromboembolism. Ann InternMed 2007;147:766–774.
24. McIntyre KM, Sasahara AA: The hemodynamic response to pulmonary embolism in pa-tients without prior cardiopulmonary disease. Am J Cardiol 1971;28:288–294.

238 (Capítulo 13)Tratado de trombosis
25. Morpurgo M, Marzagalli M: Death in pulmonary embolism. En: Morpurgo M (ed.): Pul-monary embolism. Nueva York, Marcel Dekker, 1994:107–114.
26. Molloy WD, Lee KY, Girling L et al.: Treatment of shock in a canine model of pulmonaryembolism. Am Rev Respir Dis 1984;130:870–874.
27. Hull RD, Raskob GE, Hirsh J et al.: Continuous intravenous heparin compared with inter-mittent subcutaneous heparin in the initial treatment of proximal–vein thrombosis. N EnglJ Med 1986;315:1109–1114.
28. Wiedemann HP, Matthay RA: Acute right heart failure. Crit Care Clin 1985;1:631–661.29. Goldhaber SZ, Elliott CG: Acute pulmonary embolism. Part I. Epidemiology, pathophy-
siology, and diagnosis. Circulation 2003;108(22):2726–2729.30. Kasper W, Geibel A, Tiede N, Just H: Patent foramen ovale in patients with haemodynam-
ically significant pulmonary embolism. Lancet 1992;340:561–564.31. Chabra A, Batra K et al.: Pulmonary embolism in segmental and subsegmental arteries:
optimal technique, imaging appearances, and potential pitfalls. App Radiol 2007;2:34–40.32. Talbot S, Worthington BS, Roebuck EJ: Radiographic signs of pulmonary embolism and
pulmonary infarction. Thorax 1973;28:128–132.33. Torbicki A, Perrier A, Konstantinides S et al.: Guidelines on the diagnosis and manage-
ment of acute pulmonary embolism. Eur Heart J 2008;28:2276–2315.34. Kasper W, Konstantinides S, Geibel A et al.: Management strategies and determinants of
outcome in acute major pulmonary embolism: results of a multicenter registry. J Am CollCardiol 1997;30:1165–1171.
35. Jaff MR, McMurtry S, Archer SL et al.: Management of massive and submassive pulmo-nary embolism, iliofemoral deep vein thrombosis, and chronic thromboembolic pulmonaryhypertension. Circulation 2011;123:1788–1830.
36. Miniati M, Prediletto R, Formichi B et al.: Accuracy of clinical assessment in the diagno-sis of pulmonary embolism. Am J Respir Crit Care Med 1999;159:864–871.
37. Stein PD, Saltzman HA, Weg JG: Clinical characteristics of patients with acute pulmo-nary embolism. Am J Cardiol 1991;68:1723–1724.
38. Miller GA, Sutton GC, Kerr IH et al.: Comparison of streptokinase and heparin in treat-ment of isolated acute massive pulmonary embolism. Br Med J 1971;2:681–684.
39. Aujesky D, Obrosky DS, Stone RA et al.: Derivation and validation of a prognostic modelfor pulmonary embolism. Am J Respir Crit Care Med 2005;172:1041–1046.
40. Wicki J, Perrier A, Perneger TV et al.: Predicting adverse outcome in patients with acutepulmonary embolism: a risk score. Thromb Haemost 2000;84:548–552.
41. Sánchez O, Trinquart L, Colombet I et al.: Prognostic value of right ventricular dysfunc-tion in patients with haemodynamically stable pulmonary embolism: a systematic review.Eur Heart J 2008;29:1569–1577.
42. Becattini C, Vedovati MC, Agnelli G: Prognostic value of troponins in acute pulmonaryembolism: a meta–analysis. Circulation 2007;116:427–433.
43. Wells PS, Ginsberg JS, Anderson DR et al.: Use of a clinical model for safe managementof patients with suspected pulmonary embolism. Ann Intern Med 1998;129:997–1005.
44. Aujesky D, Roy PM, Le Manach CP et al.: Validation of a model to predict adverse out-comes in patients with pulmonary embolism. Eur Heart J 2006;27:476–481.
45. Aujesky D, Obrosky DS, Stone RA et al.: Derivation and validation of a prognostic modelfor pulmonary embolism. Am J Resp Crit Care Med 2005;172:1041–1046.
46. Jiménez D, Aujesky D, Moores L et al., for the RIETE investigators: Simplification of thepulmonary embolism severity index for prognosticating patients with acute symptomaticpulmonary embolism. Arch Intern Med 2010;170:1383–1389.

239Tromboembolia pulmonarE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
47. Le Gal G, Righini M, Roy PM et al.: Prediction of pulmonary embolism in the emergencydepartment: the revised Geneva score. Ann Intern Med 2006;144:165–171.
48. Wells PS, Anderson DR, Rodger M et al.: Derivation of a simple clinical model to catego-rize patients probability of pulmonary embolism: increasing the models utility with the Sim-pliRED D–dimer. Thromb Haemost 2000;83:416–420.
49. Elliott CG, Goldhaber SZ, Visani L et al.: Chest radiographs in acute pulmonary embo-lism. Results from the International Cooperative Pulmonary Embolism Registry. Chest2000;118:33–38.
50. Stein PD, Goldhaber SZ, Henry JW et al.: Arterial blood gas analysis in the assessmentof suspected acute pulmonary embolism. Chest 1996;109:78–81.
51. Goldhaber SZ, Bounameaux H: Pulmonary embolism and deep vein thrombosis. Lancet2012;379:1835–1846.
52. Righini M, Perrier A, De Moerloose P et al.: D–dimer for venous thromboembolism diag-nosis: 20 years later. J Thromb Haemost 2008;6:1059–1071.
53. Perrier A, Roy PM, Sánchez O et al.: Multidetector–row computed tomography in sus-pected pulmonary embolism. N Engl J Med 2005;352:1760–1768.
54. Moores LK, Jackson WL Jr, Shorr AF et al.: Meta–analysis: outcomes in patients withsuspected pulmonary embolism managed with computed tomographic pulmonary angio-graphy. Ann Intern Med 2004;141:866–874.
55. Van Beek EJ, Brouwerst EM, Song B et al.: Clinical validity of a normal pulmonary an-giogram in patients with suspected pulmonary embolism–a critical review. Clin Radiol2001;56:838–842.
56. Schulman S, Kearon C, Kakkar AK et al.: Dabigatran versus warfarin in the treatmentof acute venous thromboembolism. N Engl J Med 2009;361:2342–2352.
57. Bauersachs R, Berkowitz SD, Brenner B et al.: Oral rivaroxaban for symptomatic venousthromboembolism. N Engl J Med 2010;363:2499–2510.
58. Gage BF, Yan Y, Milligan PE et al.: Clinical classification schemes for predicting hemor-rhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006;151:713–719.
59. Ruiz GN, Suárez C, González R et al.: Predictive variables for major bleeding events inpatients presenting with documented acute venous thromboembolism. Findings from theRIETE Registry. Thromb Haemost 2008;100:26–31.
60. Wan S, Quinlan DJ, Agnelli G et al.: Thrombolysis compared with heparin for the initialtreatment of pulmonary embolism: a meta–analysis of the randomized controlled trials. Cir-culation 2004;110:744–749.
61. Van de WF, Ardissino D, Betriu A et al.: Management of acute myocardial infarction inpatients presenting with ST–segment elevation. The Task Force on the Management ofAcute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2003;24:28–66.
62. Raschke RA, Gollihare B, Peirce JC: The effectiveness of implementing the weight–ba-sed heparin nomogram as a practice guideline. Arch Intern Med 1996;156:1645–1649.
63. Guyatt G, Akl EA, Crowther M et al.: Antithrombotic therapy and prevention of thrombo-sis. 9ª ed: American College of Chest Physicians evidence–based clinical practice guide-lines. Chest 2012;141(2 Suppl):7S–47S.
64. Le Gal G, Kovacs MJ, Carrier M et al.: Validation of a diagnostic approach to excluderecurrent venous thromboembolism. J Thromb Haemost 2009;7:752–759.
65. Prandoni P, Lensing AW, Prins MH et al.: Residual venous thrombosis as a predictive fac-tor of recurrent venous thromboembolism. Ann Intern Med 2002;137:955–960.

240 (Capítulo 13)Tratado de trombosis
66. Brower RG, Matthay MA, Morris A et al.: Ventilation with lower tidal volumes ascompared with traditional tidal volumes for acute lung injury and the acute respiratory dis-tress syndrome. N Engl J Med 2000;342:1301–1308.
67. Goldhaber SZ, Haire WD, Feldstein ML et al.: Alteplase versus heparin in acute pulmo-nary embolism: randomized trial assessing right–ventricular function and pulmonary perfu-sion. Lancet 1993;341:507–511.
68. Tissue plasminogen activator for the treatment of acute pulmonary embolism. A collabora-tive study by the PIOPED Investigators. Chest 1990;97:528–533.
69. Levine M, Hirsh J, Weitz J et al.: A randomized trial of a single bolus dosage regimen ofrecombinant tissue plasminogen activator in patients with acute pulmonary embolism.Chest 1990;98:1473–1479.
70. Urokinase–Streptokinase Embolism trial. Phase 2 results. A cooperative study. JAMA 1974;229:1606–1613.
71. Meyer G, Sors H, Charbonnier B et al.: Effects of intravenous urokinase versus alteplaseon total pulmonary resistance in acute massive pulmonary embolism: a European multicen-ter double–blind trial. The European Cooperative Study Group for Pulmonary Embolism.J Am Coll Cardiol 1992;19:239–245.
72. Goldhaber SZ, Kessler CM, Heit J et al.: Randomized controlled trial of recombinant tis-sue plasminogen activator versus urokinase in the treatment of acute pulmonary embolism.Lancet 1988;2:293–298.
73. Meneveau N, Seronde MF, Blonde MC et al.: Management of unsuccessful thrombolysisin acute massive pulmonary embolism. Chest 2006;129:1043–1050.
74. Decousus H, Leizorovicz A, Parent F et al.: A clinical trial of vena caval filters in the pre-vention of pulmonary embolism in patients with proximal deep–vein thrombosis. Preven-tion du Risque d’Embolie Pulmonaire par Interruption Cave Study Group. N Engl J Med1998;338:409–415.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
14Trombosis venosa yugular
Luis Fernando García–Frade Ruiz, Emmanuel Solís Ayala,Francisco Javier Roldán Gómez
El propósito de este capítulo es comentar dos casos clínicos de trombosis venosayugular y posteriormente revisar la información existente en la literatura.
CASO CLÍNICO 1
Paciente femenina de 56 años de edad dedicada al hogar, con los siguientes ante-cedentes de importancia:
Historia
Antecedentes heredofamiliares
Padre y madre finados portadores de diabetes mellitus e hipertensión arterial sis-témica. Padre finado por evento cerebrovascular isquémico. Se ignora la causade muerte de la madre. Cuenta con nueve hermanos, dos de ellos portadores dehipertensión arterial sistémica y uno con diabetes mellitus tipo 2.
Antecedentes personales no patológicos
Tabaquismo y consumo de alcohol negados. Como ejercicio, realiza bicicleta fijacinco horas a la semana.
241

242 (Capítulo 14)Tratado de trombosis
Antecedentes personales patológicos
Quirúrgicos: amigdalectomía en la infancia, histerectomía hace 12 años por mio-matosis uterina. Transfusionales: negados. Hospitalizaciones: internamientohace 12 años por probable evento de tromboembolia pulmonar; no recuerda larealización de estudios en aquella ocasión; refiere haber recibido sólo manejo conácido acetilsalicílico durante aproximadamente un año posterior al evento. En-fermedades: hipertensión arterial sistémica de cinco años de diagnóstico, mane-jada con enalapril en dosis de 10 mg al día, con un adecuado control de las cifrasde tensión arterial.
Antecedentes ginecoobstétricos
Menarca: a los 12 años. Ciclos regulares: 28 por cinco días. Fecha de la últimamenstruación: hace 12 años. Gesta: cuatro, para: cuatro; abortos: 0, cesáreas: 0.Tratamiento hormonal sustitutivo: comenzó tres semanas previas a su padeci-miento; actual manejo con estrógenos conjugados con vía de administracióntransdérmica.
Padecimiento actual
Inició su padecimiento tres días previos a su valoración médica con cefalea occi-pital de tipo punzante, intermitente, acompañada de mareo a los cambios de posi-ción. Presentaba también otalgia derecha con irradiación a hombro ipsilateral,con mejoría parcial de los síntomas con la administración de analgésicos conven-cionales.
Clínica
Exploración física
Paciente con signos vitales en rangos normales, con índice de masa corporal de31 kg/m2; con presencia de masa en la cara lateral derecha del cuello, de consis-tencia semisólida, no desplazable, no dolorosa a la palpación, con aumento de latemperatura local; el resto de la exploración física, incluyendo exploración neu-rológica y fondo de ojo, sin alteraciones.
Estudios
A su ingreso al hospital se le realizaron biometría hemática completa, pruebas defunción hepática, electrólitos séricos, tiempos de coagulación y química sanguí-

243Trombosis venosa yugularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
nea, todo en parámetros normales, con cifras de colesterol total de 220 mg/dL.Se le realizó tomografía axial computarizada (TAC) de cuello con medio de con-traste intravenoso, que mostró oclusión de la vena yugular interna derecha.
Se complementó el estudio con ultrasonido Doppler de los vasos del cuello,el cual corroboró la presencia de trombosis en la vena yugular interna derecha,con oclusión de 80% de la luz del vaso sanguíneo; el resto de los vasos del cuellosin alteraciones del flujo.
Se realizaron electrocardiograma en reposo de 12 derivaciones y telerradio-grafía de tórax, sin encontrar alteraciones. Se realizó perfil de trombofilia, queincluyó proteína C, proteína S, antitrombina III, factor V Leiden y perfil inmuni-tario, todo en rangos de normalidad.
Diagnóstico
Trombosis venosa yugular interna derecha espontánea.
Tratamiento
La paciente recibió tratamiento anticoagulante con heparina no fraccionada y seinicia anticoagulación oral con acenocumarina, manteniendo el índice normali-zado internacional (INR) entre 2.0 y 3.0. Se agregó al tratamiento estatina y secontinuó el control antihipertensivo con inhibidor de la enzima convertidora deangiotensina.
Evolución
A los tres meses se realizó un ultrasonido Doppler de los vasos del cuello, quemostró disminución del trombo, con una oclusión aproximada de 50% de la luzdel vaso. A los mismos tres meses se repitió el perfil de trombofilia, con la sus-pensión de la acenocumarina dos semanas previas, resultando nuevamente den-tro de los parámetros normales.
Al año de tratamiento con anticoagulación se realizó nuevamente un ultraso-nido Doppler de control, que mostró ausencia de trombosis en la vena yugularinterna derecha, sin alteraciones del flujo, por lo que se decidió suspender el trata-miento anticoagulante y se inició el manejo con antiagregante plaquetario. Desdeentonces no se sabe nada de la paciente. Desde el momento de su internamientola paciente rechazó la posibilidad de realizar estudios para descartar enfermedadmaligna y al año del evento no mostró datos clínicos en relación con la misma.

244 (Capítulo 14)Tratado de trombosis
CASO CLÍNICO 2
Historia
Paciente masculino de 66 años de edad, sin antecedentes heredofamiliares de im-portancia para el padecimiento actual. Cuenta con tabaquismo positivo a razónde 20 cigarrillos al día durante 20 años, con un índice tabáquico de 20.
Contaba con diagnóstico de hipertiroidismo en tratamiento con tiamazol, en-fermedad pulmonar obstructiva crónica en tratamiento con salmeterol/fluticaso-na y tiotropio, así como hipertensión arterial sistémica controlada con nebivolol.Indicó el antecedente de neumonía adquirida en la comunidad que requirió mane-jo intrahospitalario con colocación de catéter en la región subclavia derecha parala administración de líquidos y fármacos durante siete días, con egreso del hospi-tal en adecuadas condiciones cuatro semanas previas al inicio de su padecimientoactual.
Padecimiento actual
Inició su padecimiento tres días previos a su ingreso, presentando astenia, adina-mia, cefalea frontal bilateral de tipo opresivo y constante, con irradiación a la re-gión occipital. Presenta de manera concomitante aumento de volumen en laregión cervical derecha y eritema, con dolor a la palpación y sensación de indura-ción sobre el trayecto del vaso sanguíneo.
Clínica
En el momento de su ingreso presentaba signos vitales estables. Durante la explo-ración física se destacó un aumento de volumen del lado derecho de cuello y delhueco supraclavicular ipsilateral, con eritema localizado, induración, signo de lacuerda positivo, pulso carotídeo derecho con disminución de la amplitud y ausen-cia de soplo.
Estudios
Se le realizó una TAC de cuello con contraste, la cual demostró la presencia detrombosis de la vena yugular externa derecha y de la vena subclavia ipsilateral.
Se continuó con el protocolo del diagnóstico con TAC toracoabdominal, pan-endoscopia y colonoscopia, sin la presencia de factores asociados.

245Trombosis venosa yugularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Tratamiento
Se inició anticoagulación formal, al principio con enoxaparina en dosis terapéuti-cas de 60 mg por vía subcutánea cada 12 h y posteriormente con acenocumarinaen dosis inicial de 4 mg hasta alcanzar el INR deseado. Se dio de alta al pacientepor mejoría clínica; se continuaron el seguimiento y la anticoagulación formalmediante consulta externa.
REVISIÓN DE LA LITERATURA
Discusión
Alrededor del decenio de 1920 la trombosis de la vena yugular fue descrita comouna complicación de la infección orofaríngea aguda. En 1936 Lemierre describióla primera serie de casos de tromboflebitis séptica de la vena yugular.
Aunque es menos común que la trombosis venosa profunda de los miembrospélvicos, la trombosis venosa de miembros superiores se encuentra en aumentocon 4% de los casos. Hasta ahora el principal factor de predisposición lo constitu-ye la presencia de un catéter venoso central, el que se asocia hasta en 75% de loscasos.
Definición
La trombosis de la vena yugular se refiere a la presencia de un trombo intralumi-nal que ocurre en cualquier sitio desde el origen de la vena yugular por debajodel cráneo hasta el punto en donde se une la vena subclavia.
Causas
El tratamiento hormonal sustitutivo en las mujeres menopáusicas y su relacióncon la enfermedad tromboembólica se revisan a detalle en el capítulo correspon-diente. Las causas de trombosis de la vena yugular interna se muestran en el cua-dro 14–1.
Si bien existe poca información acerca de casos reportados, se han llevado acabo algunos estudios que indican que las etiologías más frecuentes son maligni-dad (32.7%), catéteres venosos (22.4%) y estados protrombóticos, en 8.2% de loscasos. De manera contrastante, se encuentra el estudio realizado por Marinella y

246 (Capítulo 14)Tratado de trombosis
Cuadro 14–1. Causas de trombosis venosa yugular
Catéter venoso o de Swan–Ganz en la vena subclavia
Administración de fármacos intravenosos usando la vena yugular como accesoSíndrome de LemierreInfecciones profundas del cuello
Infecciones necrosantes de tejidos blandosComplicación quirúrgica de la disección del cuelloMalignidad de la cabeza y el cuello
Malignidad a distancia que produce hipercoagulabilidadEstado hipercoagulable secundario a trombofiliaCualquier cirugía de cuello que involucre la retracción prolongada de la vena yugular
TraumaAsociación a inducción de la ovulación con gonadotropinasCausas espontáneas, secundarias a malignidad o estados hipercoagulables no diagnosticados
col., en el que reportan como etiología los siguientes factores: catéteres venosos(72%), infección (28%), malignidad extratorácica (22%), malignidad torácica(21%), falla renal (21%) y falla cardiaca (8%).
No sólo se ha estudiado la asociación que existe con los factores hereditarios,sino también de los factores adquiridos; en este caso la edad desempeña un papela considerar, presentando dos grupos etarios: niños pequeños y personas mayo-res. Se menciona que, cuanto mayor sea la edad, mayor será el riesgo de presentartrombosis asociada a los catéteres.
También se pueden mencionar como factores de predisposición una cuentaelevada de plaquetas en el momento de la colocación del catéter y trombosis pre-vias.
Lo último a considerar son los factores externos asociados a la presencia detrombosis, como el tipo de catéter utilizado, ya que se reporta que los que estánfabricados de silicón o poliuretano se asocian a un menor riesgo de trombosis quelos de polietileno.
De igual manera, se presenta un riesgo incrementado cuanto mayor es el númerode lúmenes; el sitio de inserción del catéter continua siendo un punto controver-sial, puesto que las series y los estudios clínicos controlados sólo se han realizadocomparando catéteres subclavios vs. femorales, en los que la trombosis femoralse encuentra de manera más frecuente.
Manifestaciones clínicas
Las manifestaciones clínicas de la trombosis de la vena yugular se muestran enel cuadro 14–2.

247Trombosis venosa yugularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 14–2. Manifestaciones clínicas de la trombosisvenosa yugular en orden de frecuencia
FiebreLeucocitosisDolor cervical
Masa en el cuelloSigno del cordónSepsis
Complicaciones pleuropulmonaresSíndrome de la vena cava superiorQuilotórax
En cuanto a las manifestaciones clínicas, se debe realizar la división entre losdos tipos de trombosis venosa que se pueden presentar: las clínicamente mani-fiestas y las que cursan de manera subclínica.
El cuadro clínico, asociado en orden de frecuencia, consiste en fiebre, leucoci-tosis, dolor cervical en la región trombosada, masa palpable en el cuello, signodel cordón, sepsis, complicaciones pleuropulmonares, síndrome de vena cava su-perior y quilotórax.
Pruebas de laboratorio
Con frecuencia la causa de la trombosis es obvia (p. ej., la presencia de un catétercentral); sin embargo, algunos casos requieren una investigación más profundadel sistema de la coagulación para la detección de estados protrombóticos.
Estudios de gabinete
En el pasado se utilizó la venografía como la regla de oro para el diagnóstico dela trombosis de la vena yugular. Sus desventajas son la utilización de medio decontraste y el riesgo potencial de desprendimiento del coágulo, con la subsecuen-te embolia pulmonar.
El ultrasonido es un estudio seguro y no invasivo. Los hallazgos ultrasonográ-ficos incluyen dilatación venosa, coágulo intraluminal y falta de respuesta a lamaniobra de Valsalva (en la que se esperaría un cambio en el volumen intralumi-nal secundario a un aumento en el retorno venoso). El ultrasonido Doppler puedeser útil para detectar cambios en el flujo secundarios a la presencia del trombodurante la fase aguda de formación del coágulo.
La TAC con medio de contraste intravenoso se considera el estudio de elecciónen caso de sospecha de trombosis de la vena yugular. Sus hallazgos incluyen baja

248 (Capítulo 14)Tratado de trombosis
densidad del trombo intraluminal y distensión venosa yugular proximal altrombo.
Para el diagnóstico se requiere de manera principal una alta sospecha clínica;como se mencionó, existen cuadros que pueden cursar de manera asintomática,por lo que la pericia y la experiencia clínica del médico son determinantes. Paradefinir una trombosis venosa clínicamente manifiesta se requiere la presencia deun diagnóstico por imagen (ultrasonido o venografía) y síntomas, como dolor, ca-lor, edema, cambios de coloración violáceos o circulación colateral visible.
En cuanto a los estudios de imagen, en los rayos X se puede encontrar una ima-gen que rodea al catéter venoso central o una imagen de trombo mural. El estudiode imagen más utilizado para el diagnóstico de trombosis yugular es el Doppler,puesto que no es invasivo y cuenta con una sensibilidad que varía de 56 a 100%y una especificidad de 77 a 100%. Se puede realizar la TAC de cuello con contras-te como estudio inicial.
Tratamiento
Una vez realizado el diagnóstico se debe considerar el uso de terapia anticoagu-lante. No existen estudios disponibles para guiar al médico en esta área. La mayo-ría de los reportes en los que se ha utilizado terapia trombolítica (vía catéter colo-cado directamente junto al trombo) han involucrado a pacientes con trombosisque se extiende dentro del seno sigmoides; sin embargo, la seguridad de la terapiatrombolítica no ha sido definida.
Los casos asociados con infección profunda del cuello requieren drenaje de lascolecciones y desbridamiento de todo el tejido infectado. La fascitis cervical ne-crosante requiere desbridamiento extenso.
Un análisis de Cochrane mostró que no existe beneficio en el uso profilácticode heparina o antagonistas de la vitamina K en la prevención de mortalidad enlos pacientes con trombosis sintomática; sin embargo, el uso de antagonistas dela vitamina K sí resulta en una disminución significativa de la sintomatología.
Los pacientes son manejados durante un curso corto con heparina o análogos,seguidos de antagonistas de la vitamina K durante tres a seis meses.
El uso de heparina o análogos debe ser durante al menos cinco días. Si bienla duración óptima del tratamiento con warfarina se desconoce, el periodo de tresa seis meses se ha asociado con una baja recurrencia.
En cuanto al inicio con anticoagulantes orales, se recomienda alcanzar comoobjetivo un INR de 2.5, con grado de evidencia 1A, así como iniciarlos junto conla anticoagulación parenteral, la que se deberá mantener durante cinco días, hastaque el INR se encuentre por encima de 2.0 durante al menos dos días consecutivos(grado de evidencia 1C).

249Trombosis venosa yugularE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Complicaciones
Las posibles complicaciones que se pueden presentar incluyen embolismo pul-monar, trombosis de la vena subclavia, trombosis del seno sagital superior, sín-drome de la vena cava superior, edema de vías respiratorias y tromboflebitis sép-tica. Esta última puede conducir a síndrome de respuesta inflamatoria sistémica,embolismo séptico, empiema, artritis séptica, falla renal, disfunción hepática yedema cerebral.
Resumen
La trombosis venosa yugular constituye una causa poco común de trombosis ve-nosa. Su causa más frecuente es el uso de catéter venoso central; sin embargo, enlos casos de trombosis espontánea habrá que descartar estados de hipercoagulabi-lidad, como trombofilia y enfermedad maligna. Sus principales manifestacionesclínicas son fiebre, leucocitosis, presencia de masa en el cuello y dolor cervical.La tomografía axial computarizada con medio de contraste intravenoso y la ultra-sonografía Doppler constituyen los estudios de gabinete de elección para su diag-nóstico. Sin embargo, se requieren más estudios para establecer las guías de trata-miento en estos casos de enfermedad trombótica.
REFERENCIAS
1. García–Frade RLF, Hurtado R: Alteraciones hematológicas en el embarazo. En: Manualde diagnóstico para el consultorio. 1ª ed. México, Litoral, 2004.
2. Dacey M: Internal jugular thrombosis. Kent County Hospital. www.google.com.3. Vázquez F: Bilateral internal jugular thrombosis associated with thrombophilia after ova-
rian induction for infertility. Medicina (Buenos Aires) 2002;62(4):328–330.4. Cushman M: Estrogen plus progestin and risk of venous thrombosis. JAMA 2004;292(13):
1573–1580.5. Mammen E: Oral contraceptive pills and hormonal replacement therapy and thromboem-
bolic disease. Hematol/Oncol Clin N Am 2000;14(5):1045–1059.6. Moores L, Bilello K, Murin S: Sex and gender issues and venous thromboembolism. Clin
Chest Med 2004;25:281–297.7. Humphries K, Gill S: Risks and benefits of hormone replacement therapy: the evidence
speaks. CMAJ 2003;168(8):1001–1010.8. Whiteman T, Hassouna H: Hypercoagulable states. Hematol/Oncol Clin N Am 2000;14
(2):355–377.9. Erkoc R, Uzun K, Yuca K, Etlik O, Dogan E et al.: Internal jugular vein thrombosis two
different etiologies. Eur J Gen Med 2005;2(3):123–128.10. Marie I, Levesque H, Cailleux N, Primard E, Peillon C et al.: Upper extremity deep ve-
nous thrombosis. A report of 49 cases. Rev Med Interne 1998;19:399–408.

250 (Capítulo 14)Tratado de trombosis
11. Marinella MA, Kathula SK, Markert RJ, Ohio D: Spectrum of upper extremity deep ve-nous thrombosis in a community teaching hospital. Heart Lung 2000;29:113–117.
12. Van Rooden CJ, Tesselaar ET, Osanto S, Rosendaal FR, Huisman MV: Deep veinthrombosis associated with central venous catheters a review. J Thromb Haemost 3:2409–2419.
13. García–Frade RLF, Mas AP: Trombosis venosa yugular interna espontánea. Comunica-ción de un caso y revisión de la bibliografía. Med Int Mex 2005;21:477–480.
14. Shaham D, Sklair Levy M: Lemierre’s syndrome presenting as multiple lung abscesses.Clin Imaging 2000;24:197–119.
15. Rocha L, Núñez C, Suazol C, González M: Tromboflebitis séptica de la vena yugular in-terna o síndrome de Lemierre. Rev Chil Cir 2010;62(5):439–440.
16. Tait C, Baglin T, Watson H, Laffan M, Makris M et al.: Guidelines on the investigationand management of venous thrombosis at unusual sites. Br J Hematol 2012;159:28–38.
17. Keeling D, Baglin T, Tait C, Watson H, Perry D et al.: Guidelines on oral anticoagulationwith warfarin. Fourth edition. Br J Hematol 2011;154:311–324.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
15Enfermedad vascular renal
Luis Fernando García–Frade Ruiz
OCLUSIÓN AGUDA DE LA ARTERIA RENAL
Trombosis traumática de la arteria renal
En 1861 von Recklinghausen describió la trombosis traumática de la arteria renalen un joven que tras una caída sufrió un infarto renal segmentario, y hasta 1971Rohl reportó la primera revascularización exitosa de la arteria renal en un pacien-te con trombosis traumática.
El trauma abdominal es la principal causa de trombosis aguda de la arteria re-nal, la cual es unilateral la mayoría de las veces y ocurre con mayor frecuenciadel lado izquierdo, pero puede ser bilateral. La trombosis de la arteria renal se pre-senta entre 1 y 3% de los casos de trauma abdominal grave, ocasionados con másfrecuencia por accidentes automovilísticos, riñas callejeras y actividades atléti-cas. Hay dos mecanismos a través de los cuales se produce la trombosis arterialposterior al trauma. El primero se refiere a una desaceleración del riñón en direc-ción anterior, lo cual provoca un estrechamiento de la arteria renal en relación conla posición que guarda con la aorta, que genera una disrupción de la íntima conla consecuente exposición del endotelio e inicio de la cascada de la coagulación.Dicha lesión suele ocurrir con mayor frecuencia en el riñón izquierdo debido aque en el derecho el duodeno limita su desplazamiento. El segundo mecanismopostula que después del trauma los vasos renales se comprimen contra los cuer-pos vertebrales y generan daño al endotelio con la consecuente activación de losprocesos de la coagulación y formación de trombo.
251

252 (Capítulo 15)Tratado de trombosis
La principal complicación de la trombosis aguda es la isquemia renal grave ysu evolución a infarto. El tiempo máximo isquémico tolerado por el riñón antesde que se inicie un daño irreversible es de entre 60 y 90 min, pero el tiempo preci-so es incierto.
Los signos y síntomas clínicos más comunes asociados con la trombosis de laarteria renal incluyen dolor abdominal (en especial en el flanco), náusea, vómitoy fiebre. También puede presentarse hipertensión arterial sistémica de recienteinicio y de gravedad. En caso de anuria se debe sospechar la presencia de trombo-sis arterial renal bilateral o unilateral del único riñón que está funcionando. Lahematuria es macroscópica o microscópica, y puede estar ausente en 25% de loscasos, mientras que la proteinuria leve es frecuente. Los estudios de laboratoriopueden mostrar la elevación de la deshidrogenasa láctica, creatinfosfocinasa,transaminasas séricas y fosfatasa alcalina.
La tomografía computarizada (TAC) contrastada es el estudio de gabinete deelección para los pacientes con sospecha de trauma renal. En caso de trombosisde la arteria renal se observa ausencia de realce del parénquima renal, así comode la excreción del medio de contraste. En caso de que el diagnóstico permanezcaincierto después de los estudios no invasivos se tendrá que recurrir a la arteriogra-fía renal, que proporciona el diagnóstico definitivo y la localización y extensiónde la oclusión. Una vez que se tiene el diagnóstico se debe considerar la prontarevascularización, para evitar el infarto renal.
El tiempo de isquemia es el principal factor que determina el éxito en la tera-péutica a utilizar. Se reporta un rango de éxito de 80% cuando la reparación vas-cular se realiza dentro de las siguientes 12 h al inicio del evento; sin embargo,dicho rango disminuye a 57% cuando la reparación se realiza entre las siguientes12 a 18 h y a 0% para tiempos mayores de isquemia.
Debido a que en la mayoría de los pacientes el diagnóstico se integra despuésdel tiempo óptimo de reparación, muchos de ellos requieren una nefrectomía tar-día. Otros factores que influyen en el pronóstico del paciente son la extensión deldaño renal, la presencia de circulación colateral, la dificultad técnica del procedi-miento quirúrgico y la presencia de daño a otros órganos.
Existen reportes de estrategias endovasculares, pero es frecuente que coexis-tan lesiones renales y extrarrenales en el paciente traumatizado, lo cual obliga arealizar una exploración quirúrgica.
En general, se debe intentar la revascularización en todos los pacientes contrombosis bilateral de la arteria renal o en los que padecen trombosis del únicoriñón funcional. En los pacientes con trombosis unilateral y riñón funcional con-tralateral el procedimiento quirúrgico con trombectomía o la resección del seg-mento arterial afectado con reemplazo vascular no conducen por fuerza a mejoresresultados que el tratamiento médico, por lo que la indicación quirúrgica en estoscasos continúa siendo incierta.

253Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 15–1. Causas de trombosis no traumática de la arteria renal
Causas asociadas con daño o disrupción endotelial� Aterosclerosis� Aneurisma de la arteria renal� Aneurisma disecante� Displasia fibromuscular� Consumo de cocaína
Inflamación vascular� Poliarteritis nodosa� Arteritis de Takayasu� Enfermedad de Behçet
Estados hipercoagulables adquiridos� Síndrome antifosfolípidos� Trombocitopenia inducida por heparina� Hiperhomocisteinemia� Síndrome nefrótico
Estados hipercoagulables hereditarios� Mutación del factor V de Leiden
Otras causas� Anemia de células falciformes� Ejercicio aeróbico� Presión del cinturón de seguridad del automóvil
Fuente: Brenner: Brenner & Rector’s the kidney. 7ª ed. Saunders, 2004
Trombosis no traumática de la arteria renal
La trombosis no traumática de la arteria renal es un padecimiento poco frecuentey la mayoría de las veces no se sospecha su presencia de manera temprana. Enel cuadro 15–1 se muestran las causas de trombosis no traumática de la arteriarenal obtenidas a partir de reportes de casos.
El trombo de la arteria renal se encuentra formado principalmente por plaque-tas y se desarrolla en los sitios de lesión de la pared vascular en presencia de flujossanguíneos de alta velocidad. Los trombos más frecuentes son los que se formanen sitios de placas ateromatosas preexistentes, lo cual subraya el enfoque trombó-tico en la enfermedad aterosclerótica.
Los estados hipercoagulables hereditarios (como la deficiencia de proteínasC y S), excepto la mutación del factor V de Leiden, constituyen un factor de ries-go para el desarrollo de trombos en la circulación venosa, aunque no en el territo-rio arterial.
Como se menciona en el capítulo 1, el daño o la disrupción de la superficie en-dotelial de la pared vascular (en este caso de la arteria renal) promueve la adhe-sión y agregación plaquetaria, lo cual conduce al desarrollo de trombosis. En adi-ción al fenómeno aterosclerótico, la formación de trombos se puede llevar a caboen sitios de displasia fibromuscular, aneurismas de la arteria renal y estados infec-

254 (Capítulo 15)Tratado de trombosis
ciosos e inflamatorios, mientras que los pacientes con síndrome nefrótico o conhiperhomocisteinemia rara vez cursan con trombosis de la arteria renal.
Los padecimientos relacionados con el abuso de la cocaína han aumentado enla última década, pero el infarto renal constituye una rara complicación con pocoscasos reportados en la literatura médica hasta el momento. Los efectos tóxicoscomunes de la cocaína sobre el sistema vascular se manifiestan con isquemiamiocárdica y eventos cerebrovasculares, aunque se han reportado casos raros denecrosis de extremidades superiores, piel y músculo, disección aórtica, trombosismesentérica e infarto renal.
Los efectos vasculares de la cocaína se atribuyen sobre todo al vasoespasmosecundario a estimulación adrenérgica masiva; no obstante, hay pruebas de quela cocaína contribuye a la formación del trombo después de provocar un aumentoen el tromboxano, con la subsecuente activación plaquetaria. En todo caso, sedebe investigar la presencia de un estado hipercoagulable agregado (enfermedadautoinmunitaria, enfermedad valvular cardiaca, trombofilia, etc.).
La oclusión aguda de la arteria renal en los pacientes con trasplante de riñónes causante de hasta la mitad de las pérdidas tempranas del injerto. Dicha oclusiónocurre casi siempre varias semanas después de la cirugía y suele ser secundariaa factores técnicos de la anastomosis quirúrgica, riñones de donadores pediátri-cos y presencia de anticuerpos antifosfolípido, entre otros.
Tromboembolismo de la arteria renal
La tromboembolia de las arterias renales tiene su origen la mayoría de las vecesen el corazón. La presencia de enfermedad cardiaca y arritmias (en especial lafibrilación auricular) se asocia con tromboembolismo, aunque con un riesgo rela-tivamente bajo hacia la vasculatura renal.
De todos los tromboembolismos secundarios a la fibrilación auricular sólo 2%involucran a las arterias renales, mientras que 61% ponen en riesgo la circulaciónde las extremidades superiores y 29% comprometen las arterias mesentéricas.Entre las enfermedades que se relacionan con el desarrollo de tromboembolismode las arterias renales se incluyen las arritmias, el infarto del miocardio, la cardio-patía congestiva, la enfermedad valvular reumática, la prótesis valvular, la endo-carditis bacteriana, el tromboembolismo paradójico, el aneurisma aórtico y renal,las complicaciones de la cateterización intraarterial, la embolia tumoral y la em-bolia grasa. El trombo se puede formar en el corazón después de un infarto delmiocardio, como un trombo mural sobre el sitio del infarto, en un ventrículo contrastorno en su movilidad o en la aurícula. Los trombos también pueden desarro-llarse en las paredes de la aorta o de la arteria renal, sobre lesiones ateroscleróticasy en aneurismas, y viajar hacia el riñón.

255Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
La trombosis también puede ocurrir luego de reparaciones endovasculares deaneurismas aórticos o de estenosis de la arteria renal. Las complicaciones renalessignificativas después de la colocación de prótesis endovasculares en casos de es-tenosis de la arteria renal son de 9% e incluyen trombosis de la arteria renal, disec-ción y hemorragia.
Características clínicas del tromboembolismo renal
Las características clínicas del embolismo de la arteria renal son variables y de-penden del tamaño y de la extensión del émbolo. El embolismo arterial bilateralo de un riñón solitario funcional ocasiona insuficiencia renal aguda y anuria.
Los pacientes experimentan algún grado de dolor abdominal o en el flanco,náusea y vómito. Asimismo, se puede presentar hematuria macroscópica. Al mo-mento de la exploración física es posible encontrar, además del dolor abdominalo en el flanco, datos de irritación peritoneal. La fiebre y los escalofríos son fre-cuentes y el paciente puede mostrar hipertensión arterial sistémica grave, lo cualconstituye el problema clínico en varios casos.
Se debe explorar al paciente con todo cuidado, con el fin de buscar datos clíni-cos de embolización en otros sitios (en especial el cerebro y las extremidades su-periores), así como la presencia de arritmias cardiacas, prótesis valvulares o car-diopatía isquémica.
Los estudios de laboratorio suelen mostrar una elevación de la deshidrogenasaláctica, hematuria microscópica, proteinuria leve y leucocitosis. Una concentra-ción disminuida en el sodio urinario indica la existencia de hipoperfusión renal.
Los estudios de imagen útiles para establecer el diagnóstico de tromboembo-lismo renal incluyen la tomografía computarizada (TAC) helicoidal contrastada,considerada el estudio de gabinete de elección por la rapidez con la que se puederealizar. La angiografía por resonancia magnética muestra las anormalidades enla perfusión renal y constituye una mejor alternativa a la TAC, siempre y cuandose tenga disponible, ya que es preferible evitar la exposición al medio de contrasteyodado. El estudio de medicina nuclear con tecnecio 99m muestra ausencia o unaimportante disminución en la perfusión del riñón afectado, mientras que el ultra-sonido de los vasos renales con técnica Doppler a color tiene un valor limitado,pues la calidad del estudio depende del operador.
La arteriografía renal es el método definitivo para el diagnóstico de la oclusiónde la arteria renal, además de que permite administrar terapia trombolítica en casode enfermedad tromboembólica.
Tratamiento para la oclusión arterial renal aguda
Tanto la trombólisis intraarterial como los procedimientos vasculares quirúrgi-cos se utilizan para disolver o remover el trombo de la arteria renal. La preserva-

256 (Capítulo 15)Tratado de trombosis
ción de la función renal en los procedimientos quirúrgicos es inversamente pro-porcional al tiempo de la isquemia, y la mayoría de las intervenciones resultanútiles cuando se realizan dentro de las siguientes 12 h después del inicio de la is-quemia. En general se prefiere el tratamiento trombolítico intraarterial (con es-treptocinasa, urocinasa o activador tisular del plasminógeno); no obstante, se de-ben considerar sus probables riesgos, como la hemorragia y la embolizacióndistal. El manejo con anticoagulación representa la mejor opción en los casos enlos que el defecto de perfusión sea pequeño y la función renal se encuentre con-servada.
ATEROSCLEROSIS DE LA ARTERIA RENAL
La estenosis aterosclerótica de la arteria renal es la enfermedad más frecuente delas arterias renales y se asocia sobre todo con dos síndromes clínicos: enfermedadrenal isquémica e hipertensión arterial. La identificación y el manejo de la nefro-patía isquémica continúa siendo un tema controversial entre el personal clínico,ya que se desconoce su prevalencia en la población general, aunque se reporta unacifra de 16 a 28% en los pacientes con enfermedad aórtica abdominal y de 22 a45% en los que padecen enfermedad vascular periférica. De 19 a 24% de los pa-cientes que se someten a angiografía coronaria presentan una prevalencia de le-siones de alto grado de la arteria renal, de los cuales 7% son bilaterales. La preva-lencia de las lesiones vasculares está relacionada con la edad y la presencia defactores de riesgo para aterosclerosis, incluidos el tabaquismo, la hipertensión ar-terial sistémica, la dislipidemia y la diabetes mellitus. Es común que la arteria re-nal esté involucrada en los pacientes con enfermedad coronaria o insuficienciacardiaca, lo cual influye en su supervivencia.
Aún no se ha establecido el grado de oclusión arterial que se requiere para eldesarrollo de hipertensión, por lo que su presencia no es universal en los pacientescon la enfermedad; por otro lado, la función renal se puede mantener en pacientescon compromiso bilateral de las arterias renales, por lo que se requiere un altogrado de sospecha para establecer el diagnóstico.
La detección de enfermedad renovascular se indica en los pacientes con fallacardiaca y edema pulmonar, así como en los pacientes jóvenes con historial dehipertensión arterial sistémica.
La arteriografía renal es el estudio ideal para establecer el diagnóstico; sin em-bargo, su práctica se relaciona con serios efectos adversos, sobre todo ante la pre-sencia de enfermedad vascular avanzada e insuficiencia renal, por lo que los estu-dios iniciales en el paciente con sospecha de la enfermedad deberán ser depreferencia no invasivos. El gammagrama renal ofrece una importante informa-

257Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ción funcional y hemodinámica de la estenosis de la arteria renal con una sensibi-lidad y especificidad aproximadas de 90 y 95%, respectivamente. La ultrasono-grafía con Doppler tiene una sensibilidad de 92 a 98%, pero presenta limitacionesque dependen del grado de obesidad abdominal, de la presencia de gas intestinaly de la experiencia del operador. La sensibilidad y la especificidad de la angiogra-fía por resonancia magnética son de 83 a 100%. La angiografía por tomografíacomputarizada tiene una sensibilidad de 92% y una especificidad de 84%; sin em-bargo, presenta la desventaja de que requiere infusión de material de contraste.
El daño del parénquima renal (fibrosis tubulointersticial) secundario a la ne-fropatía isquémica es el principal determinante de la función renal. No obstante,varios estudios realizados en los últimos 20 años reportaron que dicha funciónrara vez mostraba una mejoría significativa después de la revascularización, porlo que la terapia intervencionista a través de cirugía o angioplastia tenía el fin dereducir el rango de disminución de la función renal, más que para mejorarla. Loanterior se explica ante la presencia de otras condiciones concomitantes que con-ducen al daño renal, como el desarrollo paralelo de nefrosclerosis, el daño a lapared de las arterias renales de mediano y pequeño calibres, y la enfermedad renalateroembólica de cristales de colesterol. Aun así, los estudios recientes indicanque de 25 a 30% de los pacientes con insuficiencia renal pueden recuperar la fil-tración glomerular después de la revascularización, mientras que de 19 a 25% delos pacientes continuarán con disminución de la función renal.
La enfermedad renal aterosclerótica se asocia con otras complicaciones car-diovasculares, por lo que su presencia se debe considerar como un marcador deriesgo para la mortalidad.
La revascularización quirúrgica o percutánea se indica en los pacientes condisminución de más de 75% del diámetro de la arteria renal, deterioro progresivode la función renal con un adecuado control de la presión arterial, insuficienciarenal irreversible en presencia de agentes antihipertensivos (excepto IECA)como consecuencia de la estenosis crítica y falla renal causada por trombosis aór-tica o de la arteria renal unilateral o bilateral, en la que los riñones permanecenviables por la presencia de circulación colateral.
La revascularización quirúrgica continúa siendo el tratamiento de elecciónpara el paciente con nefropatía de origen aterosclerótico, ya que logra la estabili-zación de la función renal entre 71 y 92% de los pacientes. Dicho procedimientotiene un rango de mortalidad de 3 a 13%, donde la aterosclerosis difusa y la insufi-ciencia cardiaca son los principales factores de riesgo. Las lesiones mayores delas arterias coronarias y de las carótidas se deben reparar antes de la manipulaciónde la vasculatura renal.
La angioplastia percutánea es una opción atractiva por su baja morbimortalidad,aunque parece ser técnicamente útil en cerca de 50% de los casos y tiene una inci-dencia de reestenosis de 5 a 38%. La colocación de prótesis endovascular tiene una

258 (Capítulo 15)Tratado de trombosis
incidencia de reestenosis de 10 a 16% durante el primer año, pero disminuye en losaños siguientes. Las posibles complicaciones del procedimiento endovascular in-cluyen formación de hematoma, disección arterial, trombosis arterial y enferme-dad ateroembólica. Sólo de 6 a 16% de los pacientes con nefropatía isquémica quese someten a revascularización logran normalizar sus niveles de presión arterial.
TROMBOSIS DE LA VENA RENAL
Hunter describió la trombosis de la vena renal (TVR) y en 1840 Rayer estableciósu asociación con el síndrome nefrótico. Aunque la TVR puede ser causada portrauma o por la presencia de una enfermedad maligna, es común en el pacientenefrótico.
Etiología
La incidencia de TVR en la población general es difícil de establecer, pero se es-tima que es significativa en los pacientes con nefropatía membranosa y en los quepadecen síndrome nefrótico. Otras causas de TVR son traumatismo, deshidrata-ción (sobre todo en los niños), terapia con esteroides, neoplasia retroperitonealcon compresión venosa, trasplante renal, consumo de anticonceptivos orales yabuso de cocaína.
Fisiopatología de la trombosis de la vena renal
Un factor importante que predispone la aparición de TVR en los pacientes consíndrome nefrótico es la presencia de un estado hipercoagulable. Varios investi-gadores han reportado alteraciones importantes en los factores de la coagulación,como disminución en los niveles de los factores IX, X y XII a través de sus pérdi-das renales secundarias a su pequeño tamaño molecular. También se ha observa-do un aumento en los niveles de los factores II, VII y X, lo cual parece ser secun-dario a un incremento en su síntesis hepática como respuesta a la disminución dela presión oncótica provocada por la hipoalbuminemia. En general, la mayoríade estas alteraciones tienden a normalizarse con la remisión clínica del síndromenefrótico. Los niveles elevados de fibrinógeno aumentan la viscosidad sanguíneaen los pacientes con el síndrome, lo cual conduce a un estado protrombótico. Lospacientes con síndrome nefrótico muestran una disminución en las concentracio-nes plasmáticas de plasminógeno, lo cual se correlaciona con bajos niveles de al-búmina sérica y con la magnitud de la proteinuria.

259Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Un grupo de investigación observó que los pacientes con síndrome nefróticotienen elevados los niveles del inhibidor del plasminógeno �2–antiplasmina, e in-dicó que dicha alteración puede contribuir al desarrollo y la persistencia de TVR.
Asimismo, se han demostrado alteraciones en los inhibidores de la coagula-ción. La antitrombina–III (AT–III) es una �2–globulina que inhibe la trombinay los factores activados IX, X, XI y XII. Existe una importante correlación entrelos niveles séricos disminuidos de AT–III y la excreción de proteínas urinarias,por lo que se concluye que el desarrollo de trombosis en los pacientes nefróticosquizá está relacionado con la deficiencia de AT–III, causada por un aumento enlas pérdidas urinarias, donde sus bajas concentraciones séricas son insuficientespara inactivar los factores procoagulantes.
Además, existe una disminución en los niveles de proteína S, constituyendo unfactor de riesgo agregado para el desarrollo de complicaciones tromboembólicas.
De igual forma, se presentan anormalidades en la fase plaquetaria de los pa-cientes nefróticos, en quienes son frecuentes la trombocitosis y un aumento enla agregación plaquetaria ante ADP y colágena, no así ante la epinefrina. La dis-función plaquetaria se correlaciona con el grado de disminución en la albúminasérica y con la gravedad de la proteinuria.
En conclusión, el síndrome nefrótico representa un estado patológico en el quela disminución sérica en la concentración de albúmina secundaria a su pérdidapor vía renal desencadena a través de un aumento en su síntesis hepática un incre-mento en ciertos factores de la coagulación, fibrinógeno e inhibidores del plasmi-nógeno, que agregado a las pérdidas renales de anticoagulantes naturales, a unaumento en la agregación plaquetaria y en ocasiones al estado de deshidrataciónsecundario al uso de diuréticos predispone al paciente a un estado procoagulante,con el consiguiente desarrollo de eventos trombóticos, en especial TVR.
Manifestaciones clínicas de la trombosis de la vena renal
El espectro clínico de la TVR varía de acuerdo con el paciente. La rapidez de laoclusión venosa y el desarrollo de circulación venosa colateral determinan la pre-sentación clínica y la subsiguiente función renal; no obstante, los pacientes quedesarrollan TVR casi siempre tienen dos maneras de presentación clínica: la agu-da y la crónica. La presentación crónica es la más frecuente y suele cursar sin sín-tomas, mientras que la forma aguda ataca casi siempre a los pacientes jóvenes coninicio súbito de dolor persistente y en ocasiones dolor tipo cólico en el flanco, conhipersensibilidad en el ángulo costovertebral y hematuria macroscópica.
La severidad de la sintomatología, la presencia de anuria y la magnitud del de-terioro de la función renal dependen de varios factores. El compromiso previo dela función renal, la ausencia de circulación colateral y la presencia de un trombo

260 (Capítulo 15)Tratado de trombosis
grande que involucre ambas venas renales puede resultar en una presentación clí-nica florida, así como en un rápido deterioro de la función renal.
La TVR aguda ocurre hasta en 6% de los pacientes con trasplante de riñón ycontribuye con la tercera parte de los eventos de falla del injerto durante los pri-meros 90 días. Hace poco tiempo se reconoció que la presencia de la mutacióndel factor V de Leiden representa un factor de riesgo significativo para el desarro-llo de TVR y de eventos tromboembólicos en los pacientes con trasplante renal.
La presencia del factor V de Leiden heterocigoto es una causa frecuente deTVR cuando ocurre in utero y en pacientes neonatos, y puede ser un factor de pre-disposición en los adultos, en especial en el contexto del síndrome nefrótico. Lamutación confiere un riesgo particular para la trombosis venosa del trasplante re-nal, por lo que se debe buscar su detección en los pacientes que están en lista deespera para el procedimiento.
Existen reportes de casos de desarrollo de TVR vinculados con el uso de anti-conceptivos orales, en cuyo caso se debe investigar la presencia de un desordenbioquímico protrombótico, como la hiperhomocisteinemia.
Los tumores abdominales constituyen una causa común de TVR, la cual sueletener una presentación de tipo crónico. Recientemente se reportó la asociaciónentre TVR y el abuso de cocaína. Las complicaciones relacionadas con el uso decocaína son múltiples, ya que afecta a varios órganos y sistemas, como el cardio-vascular y el cerebrovascular, que son los más dañados. Dichas complicacionesincluyen infarto del miocardio, arritmias, disección aórtica, miocarditis, cardio-miopatía, convulsiones, migraña, infarto cerebral y hemorragia intracraneal.
Los riñones se afectan con menos frecuencia y existen pocos reportes de dañorenal agudo secundario al abuso de cocaína; las lesiones existentes consisten eninsuficiencia renal aguda secundaria a rabdomiólisis o hipertensión maligna, in-farto renal, desequilibrio electrolítico y TVR. Los probables mecanismos involu-crados en el desarrollo de TVR tras el abuso de cocaína incluyen daño vascularpor vasoconstricción, daño endotelial directo y alteración en la expresión de cito-cinas endoteliales.
Manifestaciones radiológicasde la trombosis de la vena renal
Las manifestaciones radiológicas de la TVR aguda están bien establecidas, a di-ferencia de la presentación crónica, donde suelen ser mínimas. La oclusión com-pleta de la vena renal provoca un rápido crecimiento del riñón afectado durantelas primeras 24 h, alcanzando sus máximas dimensiones una semana después dela trombosis. Durante los próximos dos meses disminuye el tamaño renal, dandocomo resultado un riñón pequeño y atrofiado.

261Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
El ultrasonido inicial del paciente con TVR aguda revela un aumento en el ta-maño renal. El pielograma intravenoso muestra en la mayoría de los pacientes lacirculación colateral y una opacidad variable con alguna visualización del riñón.Con frecuencia se puede distinguir la pelvis renal distorsionada, estrecha y borro-sa. Las manifestaciones clínicas agudas más estas características radiológicasconstituyen el diagnóstico de TVR.
La venocavografía con cateterización selectiva de la vena renal sirve paradiagnosticar la TVR. Una venografía renal normal muestra un sistema venoso in-tralobular completo a nivel de la vena arcuata. En presencia de un flujo venosorenal normal todo el medio de contraste se “lava” en tres segundos o menos.
Por el contrario, una venografía renal anormal demuestra la presencia de untrombo en el lumen como un defecto de llenado rodeado de medio de contraste.En el caso de una obstrucción venosa parcial se puede hallar circulación colateral,la cual refleja la cronicidad del padecimiento.
La ultrasonografía renal es un procedimiento útil en el diagnóstico de la TVR,establecido mediante la demostración directa del trombo en el interior de la venarenal y en la vena cava inferior, la dilatación de la vena renal proximal al sitio dela oclusión y la pérdida de la estructura renal con aumento de su tamaño.
La ultrasonografía con Doppler es una técnica útil y no invasora que mideesencialmente la velocidad del flujo venoso renal. La apariencia de la trombosisde la vena renal principal a través de este estudio incluye la presencia de ecogeni-cidad sólida y un defecto en el llenado completo o parcial en el interior de la venarenal, así como la ausencia o escasez de signos de flujo venoso intrarrenal en 70%de los riñones involucrados. Sin embargo, este estudio presenta una alta inciden-cia de falsos negativos en el diagnóstico de la TVR.
La TAC es un procedimiento útil e inocuo en la evaluación del paciente conTVR aguda. La infusión de medio de contraste intravenoso puede ser de ayudapara identificar el trombo. Los hallazgos radiográficos incluyen aumento de ta-maño y distensión de la vena renal afectada con la identificación de los trombosen su interior, los cuales en ocasiones se extienden al interior de la vena cava infe-rior. La resonancia magnética es el procedimiento diagnóstico de elección parael diagnóstico de la TVR, ya que es inofensivo y permite una adecuada identifica-ción del flujo sanguíneo, de las paredes vasculares y de los tejidos circundantes,además de que no necesita medio de contraste.
Tratamiento de la trombosis de la vena renal
La función renal puede mejorar en gran medida en los pacientes con TVR agudatratados con terapia anticoagulante. Las pruebas indican que la terapia anticoagu-lante reduce la incidencia de nuevos eventos tromboembólicos y muchas veces

262 (Capítulo 15)Tratado de trombosis
revierte el deterioro de la función renal. Los pacientes llegan a mostrar recanali-zación de la vena renal y en ocasiones la disolución completa del trombo.
La heparina constituye la terapia inicial de elección. Los pacientes con untrombo renal de gran tamaño y embolismo pulmonar concomitante tienen un au-mento en la depuración de heparina, por lo que muchas veces requieren altas do-sis. Después de cinco a siete días de terapia con heparina (de preferencia en infu-sión continua) se debe iniciar el manejo con anticoagulantes orales a largo plazo,sin olvidar sus interacciones farmacológicas.
Existen reportes de buenos resultados de pacientes con TVR secundaria a sín-drome nefrótico tratados de manera ambulatoria con heparina de bajo peso mole-cular en administración subcutánea cada 12 h a largo plazo.
Un estudio con siete pacientes reporta que la trombectomía directa percutáneacon o sin uso de trombólisis en la TVR aguda se asocia con una rápida mejoríaen la función renal y una baja incidencia en la morbilidad, por lo que dicho proce-dimiento se debe considerar en los pacientes con evento agudo y deterioro de lafunción renal, o en los que padecen embolismo pulmonar recurrente incluso conterapia anticoagulante.
Trombosis traumática de la vena renal
La TVR secundaria a traumatismo se acompaña casi siempre de trombosis de laarteria renal, en cuyo caso la historia de traumatismo previo más dolor agudo eintenso en el flanco y una masa palpable a la exploración física sugieren el diag-nóstico. Existen escasos reportes de trombosis traumática de la vena renal sin pre-sencia de daño renal o parenquimatoso concomitante. La TVR traumática es unacondición poco común y las guías de tratamiento aún no están bien establecidas.El tratamiento general tiene el objetivo el preservar la función renal y evitar lapropagación del trombo. Desde siempre los pacientes han sido tratados con ne-frectomía o trombectomía, pero en la actualidad el tratamiento ha cambiado a unaconducta más conservadora a través de terapia anticoagulante, la cual se debe uti-lizar con precaución en los pacientes traumatizados. Las recomendaciones actua-les consisten en mantener la terapia con anticoagulación al menos durante seismeses. En el caso de un riñón único o de insuficiencia renal aguda se debe consi-derar el uso de terapia trombolítica directa.
ENFERMEDAD VASCULAR RENALEN EL SÍNDROME ANTIFOSFOLÍPIDOS
Las manifestaciones renales del síndrome antifosfolípidos (SAF) ocurren encualquier parte de la vasculatura renal, incluidos los grandes vasos, ambas arte-rias o venas, arterias o arteriolas intraparenquimatosas y los capilares glomerula-

263Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
res. Las consecuencias clínicas del riesgo renal incluyen hipertensión renovascu-lar, infarto renal, atrofia cortical, insuficiencia renal aguda, proteinuria yenfermedad renal en fase terminal.
Los pacientes con el síndrome padecen estenosis y oclusión de la arteria renal,ambos en el contexto de enfermedades reumáticas, en especial LES, o en el SAFprimario. No obstante, la naturaleza de la estenosis arterial en el SAF es poco cla-ra y la respuesta a la terapia anticoagulante indica un proceso trombótico. La ate-rosclerosis acelerada y el incremento en los niveles de endotelina con la consi-guiente vasoconstricción se han considerado factores probables.
La lesión vascular intrarrenal más frecuente en los pacientes con anticuerposantifosfolípidos es la microangiopatía trombótica.
En el síndrome antifosfolípidos catastrófico se presenta una disfunción multi-orgánica grave secundaria a trombosis de los pequeños vasos y fenómenos isqué-micos que dominan el cuadro clínico. La hipertensión arterial, la trombosis de laarteria o de la vena renal y la microangiopatía trombótica son comunes en la pre-sentación catastrófica del SAF.
El manejo de la enfermedad renal en el SAF hasta el momento ha sido empíricoy algunos autores han utilizado combinaciones de terapia anticoagulante con an-tiagregantes plaquetarios y nifedipino, inmunosupresores, inhibidores de la enzi-ma convertidora de angiotensina, esteroides, etc.
Se ha reportado TVR en los pacientes con anticuerpos antifosfolípidos positi-vos con LES y en los que padecen SAF primario, incluso como causa de TVRbilateral en el periodo posparto. En estos casos el manejo consiste en terapia anti-coagulante.
ENFERMEDAD RENAL ATEROEMBÓLICA
La enfermedad renal ateroembólica, también llamada embolismo de colesterol,constituye una parte del síndrome sistémico de la embolización de cristales decolesterol, que consiste en una enfermedad clínica poco diagnosticada, con lacual se deben diferenciar varias condiciones tromboembólicas, además de su aso-ciación con procedimientos endovasculares y con el uso de terapia anticoagulan-te y trombolítica.
El daño renal resulta de la embolización de los cristales de colesterol a partirde las placas ateroscleróticas presentes en las grandes arterias, como la aorta. Lasarterias renales tienen su origen en la aorta abdominal y reciben una gran cantidadde flujo sanguíneo, lo cual las convierte en el blanco principal de los émbolos decristales de colesterol.
La enfermedad renal ateroembólica es una causa común de insuficiencia renalen los pacientes de edad avanzada y se debe diferenciar de otros desórdenes em-

264 (Capítulo 15)Tratado de trombosis
bólicos, como fibrilación auricular, mixoma auricular izquierdo y endocarditisbacteriana.
Los pacientes con síndrome de embolización de colesterol muchas veces tie-nen historial de enfermedad cardiovascular isquémica, aneurisma aórtico, enfer-medad cerebrovascular, falla cardiaca congestiva o insuficiencia renal. Entre losprincipales factores predisponentes de la enfermedad se incluyen la presencia deaterosclerosis, la hipertensión arterial sistémica, el tabaquismo, la hipercoleste-rolemia, la diabetes mellitus, la edad mayor de 55 años, la raza blanca, el sexomasculino, el trauma en la placa ateromatosa, el procedimiento vascular invasivoque involucre a la aorta abdominal proximal al origen de las arterias renales, laarteriografía, la angioplastia, la anticoagulación con heparina y la terapia trom-bolítica.
Manifestaciones clínicas dela enfermedad renal ateroembólica
Las manifestaciones aparecen entre 1 y 14 días posteriores al evento inicial. Lasmanifestaciones sistémicas suelen ocurrir en menos de la mitad de los casos e in-cluyen la presencia de fiebre, mialgias, cefalea y pérdida de peso. Las manifesta-ciones cutáneas, como livedo reticularis y gangrena del pie, surgen entre 40 y50% de los pacientes, y constituyen las manifestaciones extrarrenales más fre-cuentes de la enfermedad. Otros sitios afectados suelen ser el tejido subcutáneo,el músculo esquelético, la próstata, el páncreas, el hígado, el cordón espinal, elsistema nervioso central, la retina y el tracto gastrointestinal. El embolismo entejidos distintos del renal indica el diagnóstico.
La afección renal aparece en 50% de los pacientes, donde la hipertensión arte-rial constituye la manifestación más frecuente. La insuficiencia renal suele teneruna presentación subaguda a lo largo de varias semanas, aunque puede llegar aser aguda y cursar con oliguria.
La falla renal en estos casos muchas veces se atribuye a otras condiciones,como nefropatía por contraste, necrosis tubular aguda, nefritis intersticial induci-da por fármacos y depleción de volumen intravascular.
El diagnóstico específico de la enfermedad se realiza a través de la demostra-ción histológica por biopsia o autopsia de los cristales de colesterol en las arteriasy arteriolas del órgano blanco.
En los estudios de laboratorio se suele encontrar una elevación de los nivelesséricos de BUN y creatinina. Al momento de la presentación inicial 80% de lospacientes presentan niveles séricos de creatinina � 2.0 mg/dL. El sedimento uri-nario no suele ser diagnóstico. La presencia de cilindros granulosos y hialinosocurre en cerca de 40% de los casos, mientras que de 33 a 40% de los pacientes

265Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
tienen hematuria y de 50 a 60% proteinuria en rangos no nefróticos. La fracciónde excreción de sodio suele ser mayor de 1%.
Tratamiento de la enfermedad renal ateroembólica
No se cuenta aún con una terapia efectiva para la enfermedad ateroembólica re-nal. El uso de terapia anticoagulante es controversial, ya que puede precipitar unmayor número de eventos embólicos. No obstante la alta mortalidad, varios estu-dios reportan un curso clínico favorable en los pacientes con la enfermedad. Envarios casos la función renal mejora en gran medida aun después de un prolonga-do periodo de insuficiencia renal.
Dos estudios clínicos reportan una sobrevivencia de 87 y 69% después del usode terapia agresiva a través del inicio inmediato de terapia anticoagulante y luegode posponer los procedimientos aórticos, control de la presión arterial y la fallacardiaca, manejo con diálisis y un adecuado soporte nutricional.
RESUMEN
La trombosis traumática de la arteria renal se presenta entre 1 y 3% de los casosde trauma abdominal grave, donde el dolor en el flanco, las náuseas y la fiebreconstituyen las manifestaciones clínicas más habituales. La TAC es el estudio deelección en caso de trauma renal. La indicación quirúrgica en estos casos conti-núa en controversia, salvo en los pacientes con riesgo renal bilateral o de un soloriñón funcional.
La trombosis no traumática de la arteria renal suele tener su origen en lesionesvasculares previas, como placa aterosclerótica, displasia fibromuscular o aneu-risma. El tromboembolismo renal suele tener su origen en enfermedades cardia-cas, como arritmia, valvulopatía, prótesis valvular y cardiopatía congestiva o is-quémica, donde la presencia de dolor en el flanco, náusea, fiebre e hipertensiónarterial es indicativa del diagnóstico. La angiorresonancia magnética o la TAC,el estudio de medicina nuclear y el ultrasonido Doppler son los estudios de gabi-nete inocuos a elegir; sin embargo, la arteriografía renal es la que establece eldiagnóstico. El tratamiento de la oclusión arterial renal aguda se basa en el usode terapia trombolítica o intervención quirúrgica, las cuales se deben realizaridealmente en las primeras 12 h después de establecido el evento.
La trombosis de la vena renal (TVR) puede ser una consecuencia de traumatis-mo, síndrome nefrótico, neoplasia retroperitoneal, uso de esteroides, anticoncep-tivos orales y trasplante renal, además del reciente reporte del abuso de cocaína.

266 (Capítulo 15)Tratado de trombosis
La TVR puede tener dos tipos de presentación clínica: aguda y crónica. Esta últi-ma suele ser la más frecuente y cursar sin síntomas, mientras que la aguda puedegenerar dolor en el flanco y hematuria macroscópica. El ultrasonido renal resultaútil en el diagnóstico de la enfermedad, mientras que la resonancia magnéticaconstituye el estudio de gabinete no invasivo de elección. El tratamiento consisteen terapia con anticoagulación a largo plazo.
La enfermedad renal ateroembólica forma parte del síndrome sistémico de laembolización de cristales de colesterol, una enfermedad clínica poco diagnosti-cada, de la cual se deben diferenciar varias condiciones tromboembólicas, ade-más de su asociación con procedimientos endovasculares y con el uso de terapiaanticoagulante y trombolítica. Sus manifestaciones clínicas varían de acuerdocon los tejidos afectados, y su diagnóstico se establece mediante el estudio histo-lógico que demuestra los cristales de colesterol en las arterias y arteriolas del ór-gano blanco. Su tratamiento permanece en controversia, pero la terapia de sostény el uso de anticoagulación parecen ofrecer buenos resultados.
REFERENCIAS
1. Tillou A, Romero J, Asensio J et al.: Renal vascular injuries. Surg Clin N Am 2001;81(6).2. Yudd M, Llach F: Disorders of the renal arteries and veins. En: Brenner & Rector’s the kid-
ney. 7ª ed. EUA, Saunders, Elsevier, 2004:1571–1614.3. Zoccali C, Mallamaci F, Finocchiaro P: Atherosclerotic renal artery stenosis: epidemiolo-
gy, cardiovascular outcomes, and clinical prediction rules. J Am Soc Nephrol 2002:13.4. Saleem T, Singh M, Murtaza M et al.: Renal infarction: a rare complication of cocaine
abuse. Am J Em Med 2001:19(6).5. Alcázar J, Rodicio J: Ischemic nephropathy: clinical characteristics and treatment. Am J
Kidney Dis 2000;36(5).6. Textor S: Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004;15:1974–1982.7. Lerman L, Textor S: Pathophysiology of ischemic nephropathy. Urol Clin N Am 2001;28(4).8. Mesnard L, Delahousse M, Raynaud A et al.: Delayed angioplasty after renal thrombosis.
Am J Kid Dis 2003;41(6).9. Ghantous V, Eisen T, Sherman A et al.: Evaluating patients with renal failure for renal
artery stenosis with gadolinium–enhanced magnetic resonance angiography. Am J KidneyDis 1999;33(1).
10. Conlon P, O’Riordan E, Kalra P: New insights into the epidemiologic and clinical man-ifestations of atherosclerotic renovascular disease. Am J Kidney Dis 2000;35(4).
11. Kau E, Patel R, Shah O: Isolated renal vein thrombosis after blunt trauma. Urology 2004;64(4):e14–e15.
12. Kim V, Spandorfer J: Epidemiology of venous thromboembolic disease. Emerg Med ClinN Am 2001;19.
13. Asghar M: Renal vein thrombosis. Eur J Vasc Endovasc Surg 2007;34(2):217–223.14. Thomas D, Roberts H: Hypercoagulability in venous and arterial thrombosis. Ann Intern
Med 1997;126(8):638–644.15. Wüthrich R: Factor V Leiden mutation: potential thrombogenic role in renal vein, dialysis
graft and transplant vascular thrombosis. Curr Opin Nephrol Hypertens 2001;10:409–414.

267Enfermedad vascular renalE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
16. Chan H, Douketis J, Nowaczyk M: Acute renal vein thrombosis, oral contraceptive use,and hyperhomocysteinemia. Mayo Clin Proc 2001;76:212–214.
17. Zoghby Z, Sekhon I, Millar D et al.: Cocaine, loin pain, and renal vein thrombosis. AmJ Kidney Dis 2007;49:859–861.
18. Cai S: Color Doppler ultrasonography appearances of renal vein thrombosis and its diag-nostic value. Chin Med Sci J 2007;22(1):17–21.
19. Wu CH: Successful outpatient treatment of renal vein thrombosis by low–molecular weightheparins in 3 patients with nephrotic syndrome. Clin Nephrol 2006;65(6):433–440.
20. Kim H: Catheter–directed thrombectomy and thrombolysis for acute renal vein thrombo-sis. J Vasc Interv Radiol 2006;17(5):815–822.
21. Amigo MC: Kidney disease in antiphospholipid syndrome. Rheum Dis Clin N Am 2006;32:509–522.
22. Modi K, Rao V: Atheroembolic renal disease. J Am Soc Nephrol 2001;12(8).23. Scolari F, Tardanico R, Zani R et al.: Cholesterol crystal embolism: a recognizable cause
of renal disease. Am J Kidney Dis 2000;36(6):1089–1109.

268 (Capítulo 15)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
16Trombosis mesentérica
Luis Fernando Mundo Gallardo, Salvador Vargas Cruz
INTRODUCCIÓN
La isquemia mesentérica constituye un evento poco frecuente como causa de ab-domen agudo. La literatura médica reporta un caso por cada 1 000 ingresos hos-pitalarios, con una mortalidad que va de 40 a 100%, dependiendo del sitio y lamagnitud de la obstrucción vascular. El pronóstico depende de la rapidez con laque se establece el diagnóstico y de su manejo oportuno.
En general, se estima que un tercio de los casos de isquemia mesentérica sonsecundarios a embolismo, otro tercio se deben a trombosis arterial y el resto a is-quemia no oclusiva, con un pequeño porcentaje debido a trombosis venosa.
Se le llama isquemia intestinal a la incapacidad de la circulación mesentéricapara mantener el metabolismo del intestino.
La circulación mesentérica recibe cerca de 25% del total del gasto cardiaco ysu oculta posición anatómica dificulta su evaluación. Se debe diferenciar la circu-lación esplácnica de la circulación mesentérica. La primera se refiere a la circula-ción en la que los nervios esplácnicos corren paralelos a sus respectivas arteriase irrigan a todos los órganos de la cavidad abdominal, mientras que la circulaciónmesentérica se refiere sólo a la circulación intestinal, la cual se encuentra confor-mada por el tronco celiaco, la arteria mesentérica superior (AMS) y la arteria me-sentérica inferior (AMI). Es importante señalar que la circulación mesentéricacolateral es muy amplia y que protege de alguna manera al intestino contra la is-quemia. Dicha circulación colateral se encuentra formada por las arcadas pan-creaticoduodenales superior e inferior, la arteria serpiginosa media (antes arco de
269

270 (Capítulo 16)Tratado de trombosis
Riolano) y la arteria marginal de Drummond. En condiciones normales la circula-ción intestinal es controlada por la presión arterial sistémica y por mecanismoslocales autónomos, como las catecolaminas, las cuales inducen vasoconstricciónde las vénulas mesentéricas poscapilares regulando así el volumen esplácnico;por otro lado, los estímulos alfaadrenérgicos y betaadrenérgicos producen vaso-constricción y vasodilatación, respectivamente.
Cabe mencionar que la mucosa intestinal es tan lábil a la isquemia que una dis-minución grave en su flujo sanguíneo durante un periodo de unas tres horas sueleser suficiente para producir cambios histopatológicos muy evidentes, incluso esposible advertir cambios ultraestructurales de la mucosa a los 10 min de la isque-mia. El daño tisular ocurre cuando el aporte de oxígeno y otros nutrientes vitalespara el metabolismo y la arquitectura celular es interrumpido, ya sea en formaabrupta o progresiva. No importa cuál sea la etiología de la isquemia, ya que lasconsecuencias suelen ser las mismas e ir desde trastornos funcionales reversibleshasta la muerte del tejido. El efecto inmediato posterior a la oclusión es la hipoxiatisular, seguida de una lesión secundaria a reperfusión cuando se restablece elflujo. Es por esto que se produce un aumento significativo en la permeabilidadde la mucosa intestinal con la consecuente translocación bacteriana, causante dela septicemia que se suele presentar en estos pacientes. Este daño celular a causade la reperfusión consiste sobre todo en una acidosis intracelular, con disfunciónmitocondrial y formación de elementos oxidantes citotóxicos o radicales libres(superóxido, radicales hidroxilo y peróxido de hidrógeno) producto del oxígenomolecular, y de citocinas inflamatorias, como el factor de necrosis tumoral, inter-leucinas, factor activador plaquetario y leucotrienos. En cuestión de horas el in-testino se torna edematoso y hemorrágico, con alteración en su circulación capi-lar, lo cual incrementa la presión hidrostática intramural, misma que agrava eledema. En una etapa posterior se presenta infarto o necrosis, que son los términossinónimos para indicar la muerte del tejido, que puede evolucionar a gangrena,la cual es una lesión en la que existe invasión bacteriana e infiltración de leucoci-tos polimorfonucleares, que conduce a un síndrome de respuesta inflamatoria sis-témica o incluso a la coagulación intravascular diseminada.
Durante la exploración quirúrgica el intestino infartado presenta una colora-ción oscura violácea muy característica, donde la serosa es opaca con membranasde fibrina de color cobrizo y en la luz intestinal existe líquido hemático. Cuandohay gangrena el color del intestino se torna negro purpúreo o gris verdoso conadelgazamiento y friabilidad de su pared, la cual exuda material fibrinopurulen-to, e incluso puede haber sitios de perforación.
Aún existe confusión en cuanto a la terminología relacionada con la isquemiaintestinal. De acuerdo con el mecanismo de la obstrucción, algunos autores la cla-sifican en aguda o crónica, oclusiva o no oclusiva, y arterial o venosa. Sin embar-go, en fechas recientes la American Gastroenterological Association publicó un

271Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 16–1. Clasificación de la isquemia intestinal
1. Isquemia mesentérica aguda
a. Oclusión arterial mayorb. Oclusión arterial menorc. Émbolo mayor
d. Trombosis venosa mesentéricae. Vasoconstricción esplácnica (isquemia mesentérica no oclusiva)
2. Isquemia mesentérica crónica o angina intestinal
3. Colitis isquémica
artículo en el que clasifica la isquemia intestinal en tres grandes categorías (cua-dro 16–1). A continuación se mencionan los trastornos relacionados con eventosde isquemia intestinal:
� Lupus eritematoso sistémico.� Síndrome antifosfolípidos.� Trombofilias.� Poliarteritis nodosa.� Artritis reumatoide.� Dermatomiositis.� Síndrome de Sjögren.� Esclerosis sistémica.� Tuberculosis.� Sífilis.� Ascaridiasis.� Lepra.� Fiebre tifoidea.� Síndrome poscoartación.� Hipertensión maligna.� Feocromocitoma.� Diabetes mellitus.� Aterosclerosis.� Telangiectasia hemorrágica hereditaria.� Seudoxantoma elástico.� Síndrome de Ehlers–Danlos.� Aneurismas aórticos.� Papilosis atrófica maligna.� Angioqueratoma difuso.� Púrpura trombocitopénica trombótica.� Tromboangeítis obliterante.� Púrpura anafilactoide.

272 (Capítulo 16)Tratado de trombosis
� Amiloidosis.� Policitemia vera.� Granuloma de Wegener.� Radiaciones.� Traumatismos.� Hernias estranguladas.� Carcinoma.� Trombosis venosa mesentérica focal.� Anticonceptivos orales.� Digitálicos.� Anticoagulantes bishidroxicumarínicos.� Alcaloides del cornezuelo de centeno.� Potasio con capa entérica.
La forma más frecuente de isquemia intestinal es la oclusión aguda de la AMSsecundaria a trombosis o a embolismo. La trombosis venosa mesentérica es me-nos frecuente y casi siempre se relaciona con una coagulopatía subyacente, mien-tras que la isquemia mesentérica no oclusiva se asocia con un síndrome de bajogasto cardiaco.
En el manejo de la isquemia intestinal se debe considerar la naturaleza de laoclusión, y la extensión y gravedad del daño. Hasta hace algunos años la cirugíaconstituía la piedra angular del tratamiento, pero los avances tecnológicos en re-lación con la terapia endovascular han modificado la conducta en el manejo ini-cial de estos pacientes.
El tratamiento endoluminal ha ganado terreno en los últimos 10 años con lacolocación de catéteres en el tronco celiaco y la infusión intraluminal de fárma-cos vasodilatadores, como la papaverina, o de agentes trombolíticos, con resulta-dos promisorios. No obstante, cuando se comparan los resultados que se obtienencon la cirugía con los de la terapia endovascular se encuentra un mayor índice derecurrencia en los grupos manejados con cateterismo; a pesar de ello, 93% de lospacientes mejoran de sus síntomas y la morbimortalidad perioperatoria es signifi-cativamente menor, además de que se puede volver a intentar el procedimiento.
También se ha intentado el tratamiento a través del uso de bloqueadores delsistema renina angiotensina y de inhibidores de los radicales libres, y se han teni-do resultados alentadores, aunque poco claros.
Un aspecto muy importante, y que pocos suelen considerar, es el apoyo nutri-cional que debe formar parte del manejo integral de estos enfermos. La mayoríade las veces se trata de pacientes sometidos a un ayuno prolongado y que requie-ren alimentación parenteral. La controversia se suscita al reiniciar la toleranciaa la vía oral, ya que, como se sabe, la atrofia de las vellosidades intestinales im-pide el inicio de dicha tolerancia por medio de una dieta normal, por lo que debe

273Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
realizarse a través de fórmulas específicas, sean peptídicas, elementales, semiele-mentales o poliméricas diluidas. Algunos estudios han demostrado que las dietasenterales ricas en lípidos pueden causar isquemia intestinal, porque aumentan lasdemandas metabólicas y de oxígeno de la mucosa.
El pronóstico de la enfermedad depende de la rapidez con que se establece eldiagnóstico. En 90% de los pacientes con síntomas de menos de 12 h de evoluciónel intestino aún es viable.
ISQUEMIA ARTERIAL MESENTÉRICA AGUDA
La obstrucción aguda de la AMS constituye la causa más común de isquemia in-testinal, debido al grosor de su calibre y a sus características anatómicas (emergeen ángulo agudo de la aorta). En 55% de los casos dicha obstrucción se debe aémbolos provenientes de las cavidades izquierdas del corazón (por cardiopatíareumática o fibrilación auricular, o después de un infarto agudo del miocardio)y 45% a trombosis.
En la trombosis de la AMS preexiste una estenosis aterosclerótica, por lo quela obstrucción del flujo sanguíneo ocurre dentro de los primeros centímetros de laarteria, lo cual causa una isquemia intestinal desde el ligamento de Treitz hastala parte media del colon transverso. Por el contrario, la oclusión embólica se ani-da en los sitios naturales de estrechamiento vascular; el primero de ellos es la sali-da de la primera gran rama de la AMS, que es la arteria cólica media, por lo quesu oclusión ocasiona isquemia en el íleon y en el colon proximal, no así en el ye-yuno proximal.
La presentación clínica de la isquemia intestinal embólica y trombótica es lamisma, ya que en ambas el efecto final es la interrupción del flujo sanguíneo. Laaparición súbita e intensa de dolor, aunada al antecedente de enfermedad cardia-ca, como estenosis mitral o fibrilación auricular, debe sugerir una causa embóli-ca. En el caso de la trombosis el dolor suele ser más insidioso y menos drástico,y casi siempre con evidencia de aterosclerosis generalizada manifestada por elantecedente de dolor abdominal posprandial. La manifestación más frecuente esla presencia de dolor abdominal tipo cólico, muy intenso, fuera de proporción conlos hallazgos durante la exploración física y de predominio periumbilical. El do-lor secundario a necrosis intestinal suele ser constante y sostenido, 25% de lospacientes presentan vómito y quizá diarrea, y la diaforesis suele ser muy marca-da. Durante la exploración física los datos no son relevantes, pues el abdomen seencuentra blando y sin datos de alarma, e incluso hay peristalsis. Conforme avan-za la isquemia (de 6 a 12 h) aparecen la resistencia muscular, la distensión abdo-minal y la pérdida de los movimientos intestinales con irritación peritoneal.

274 (Capítulo 16)Tratado de trombosis
El examen coprológico muestra al principio sangre oculta en heces, pero des-pués las evacuaciones pueden llegar a ser vinosas. La leucocitosis impera desdeel principio, igual que la acidosis metabólica.
En las radiografías no se aprecian datos específicos, pero sí permiten observarla dilatación de asas de intestino y el engrosamiento de su pared debida al edemade la mucosa; sin embargo, las placas simples de abdomen y de tórax proporcio-nan información para excluir la presencia de otras patologías o complicaciones,como la perforación de una víscera hueca.
Sólo un tercio de los pacientes presentan la tríada de dolor abdominal, fiebrey sangre oculta en heces. El ultrasonido Doppler constituye una herramienta fá-cilmente disponible, pero los pacientes con la enfermedad presentan una impor-tante dilatación de asas intestinales, secundaria a la producción de gas por el sufri-miento tisular, lo cual le confiere a este estudio poco valor diagnóstico.
Se han realizado esfuerzos para identificar la presencia de algún marcador sé-rico específico que indique con oportunidad la presencia de isquemia mesentéri-ca; entre los que se han estudiado están la cuenta leucocitaria, el pH, los electróli-tos séricos, el lactato sérico y diversas enzimas (como la creatinincinasa, lafosfatasa alcalina y la deshidrogenasa láctica). Hace poco tiempo un grupo suizodocumentó la alteración del dímero D en etapas tempranas de la isquemia mesen-térica aguda con resultados alentadores. También se ha empleado el análisis dellíquido peritoneal obtenido mediante paracentesis; sin embargo, no ha tenidoaceptación. Se ha empleado además la determinación del pH intestinal intramurala través de un catéter (tonómetro), pero tampoco ha sido aprobada del todo.
Los estudios experimentales en animales han demostrado la presencia de ta-quiarritmias intestinales detectadas a través de sensores electrofisiológicos colo-cados en la pared intestinal, que indican la presencia de hipoperfusión visceral.
La laparoscopia constituye otra herramienta que se ha probado para el diag-nóstico de la enfermedad isquémica intestinal; no obstante, se sabe que el aumen-to de la presión intraabdominal reduce el flujo de la circulación esplácnica, porlo que es importante mantener presiones muy bajas y realizar el procedimientoen el menor tiempo posible.
Para realizar el diagnóstico definitivo se emplea la arteriografía. Si bien estatécnica no resulta útil en la evaluación de la isquemia de la mucosa intestinal, síconfirma la oclusión vascular aguda e incluso ayuda a establecer el origen y lasdiferencias entre una trombosis, una embolia y una isquemia no oclusiva. La em-bolia ocurre casi siempre en la arteria cólica media, lo cual da lugar al signo an-giográfico del menisco invertido, mientras que la trombosis se produce la mayo-ría de las veces en el origen de la AMS.
En el año 2000 la American Gastrointestinal Association (AGA) concluyó quela TC tiene muy poco valor en los casos de isquemia mesentérica aguda, no asíen los de trombosis venosa. De hecho, su empleo preoperatorio en los pacientes

275Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
con signos de peritonismo sólo retrasa el tratamiento, además de que hoy en díaexisten muchos centros hospitalarios que no cuentan con la infraestructura paradicho estudio y entonces se tiene que trasladar al paciente, con el consecuenteprogreso de la enfermedad; por eso la intervención quirúrgica inmediata es el pro-cedimiento recomendado en estos casos.
Para el tratamiento casi siempre es necesaria la cirugía, para lo cual se debepreparar al paciente con rehidratación intravenosa, administración de antibióti-cos, anticoagulación, colocación de sondas nasogástrica y vesical, así como mo-nitoreo hemodinámico.
En cuanto al tratamiento quirúrgico, se recomienda intentar la reparación arte-rial con embolectomía, tromboendarterectomía o puentear con vena safena autó-loga, ya que limitarse sólo a la resección intestinal muy amplia puede ser mortal.
Un yeyuno íntegro durante la exploración abdominal indica que el origen dela oclusión es un émbolo anidado a nivel de la arteria cólica media. Si, por el con-trario, todo el intestino delgado y el lado derecho del colon se encuentran isqué-micos, se infiere la presencia de una oclusión secundaria a trombosis en el origende la AMS.
Una vez que se lleva a cabo la reconstrucción vascular se debe valorar la viabi-lidad del intestino a través de métodos clínicos o con el apoyo de equipos. Desdeel punto de vista clínico, el color del intestino, la presencia del pulso arterial me-sentérico y los movimientos peristálticos son de ayuda para valorar el intestino,pero estos criterios son subjetivos, ya que dependen de la experiencia del cirujanoy, por lo tanto, no son del todo fiables. En un estudio de Sheridan se reportó quela evaluación de la viabilidad intestinal realizada a través de medios clínicos sóloes acertada en 58% de los casos. Existen dos procedimientos objetivos para deter-minar la viabilidad intestinal durante la cirugía: el ultrasonido Doppler de los va-sos mesentéricos y la fluorescencia intestinal al administrar fluoresceína sódicaa través de una vía vascular periférica. Con esta última técnica se puede apreciarla luminosidad que produce dicha sustancia en el tejido viable con el empleo deuna lámpara de Wood y un fluorómetro.
Una vez que se determina la viabilidad intestinal se realiza la resección del seg-mento isquémico. Sin embargo, en ocasiones no es posible determinarla y ellopone en riesgo una anastomosis. Ante esta situación lo prudente es programar unasegunda intervención entre 24 y 36 h posteriores para definir la viabilidad. Otraopción interesante consiste en realizar un estoma, ya sea a manera de yeyunosto-mía, ileostomía o colostomía, para apreciar en forma directa el color de la mucosadel intestino, evitar una anastomosis riesgosa y otorgarle al paciente la oportuni-dad de mejorar para restablecer posteriormente el tránsito intestinal. Para lo ante-rior, es necesario considerar que, cuanto más proximal sea el estoma, más difícilserá mantener neutro el equilibrio hídrico, lo cual puede ser también riesgosopara el paciente.

276 (Capítulo 16)Tratado de trombosis
La obstrucción aguda de la AMS tiene una mortalidad de 47 a 100% y sus com-plicaciones pueden ser varias, como síndrome de intestino corto, reoclusión, san-grado posoperatorio y desarrollo de fístulas enterocutáneas.
ISQUEMIA MESENTÉRICA NO OCLUSIVA
Ende describió la isquemia mesentérica no oclusiva en 1958 en una paciente coninsuficiencia cardiaca. Esta enfermedad tiene una incidencia de 10 a 30% en per-sonas de ambos sexos, de 72 años de edad promedio, y constituye la forma másletal de isquemia mesentérica aguda.
El punto cardinal en este trastorno es la vasoconstricción o la disminución delflujo sanguíneo mesentérico, que puede resultar de un bajo gasto cardiaco (pre-sión sanguínea menor de 40 mmHg o flujo sanguíneo menor de 30 mL/min porcada 100 g de tejido), hipovolemia, hipotensión, hemoconcentración o uso de fár-macos vasopresores; todos estos factores son consecuencia de un infarto del mio-cardio, arritmias cardiacas, insuficiencia cardiaca congestiva, insuficiencia aórti-ca, cirugía cardiovascular, enfermedad renal, enfermedad hepática, estado dechoque, deshidratación grave o abuso de diuréticos. Los fármacos que poseen unefecto vasoconstrictor directo sobre el músculo liso de la AMS, como la digital,se asocian con el desarrollo de isquemia intestinal no oclusiva.
El cuadro clínico es indistinguible de la isquemia oclusiva y se acompaña dedolor abdominal difuso y gradual, al que posteriormente se le agregan datos deirritación peritoneal, leucocitosis y acidosis metabólica. El diagnóstico debe sos-pecharse en un paciente de edad avanzada, cardiópata y con el cuadro clínico su-gestivo. La confirmación diagnóstica se realiza a través de la arteriografía, en laque se observa un adelgazamiento tenue y progresivo de la arteria distal y de susramas.
El tratamiento tiene el principal objetivo de resolver el origen del bajo gastocardiaco, es decir, tratar la insuficiencia cardiaca y las arritmias, rehidratar al pa-ciente y eliminar el uso de fármacos vasopresores. El manejo debe incluir la des-compresión gástrica por medio de una sonda nasogástrica y la administración deantibióticos. En estos casos se emplea la arteriografía, además de método diag-nóstico como herramienta terapéutica, ya que a través del catéter es factible admi-nistrar medicamentos vasodilatadores, como la papaverina. Algunos grupos mé-dicos han agregado al manejo médico la anestesia peridural continua, ya que alproducir vasodilatación esplácnica se mejora el flujo circulatorio. La cirugía seindica en estos casos cuando después del manejo endovascular existen datos per-sistentes de abdomen agudo. Durante la cirugía se debe extirpar el segmento in-testinal no viable sin la remoción del catéter de la angiografía, ya que a través de

277Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
él se pueden aplicar medicamentos vasodilatadores para mejorar el flujo arterialmesentérico y la irrigación. La mortalidad es más elevada (90%) que en la isque-mia aguda debido a las patologías subyacentes del paciente.
TROMBOSIS VENOSA MESENTÉRICA
La trombosis venosa mesentérica fue descrita por Warren y Eberhard en 1935;afecta entre 3 y 7% de los casos de isquemia intestinal aguda. Casi siempre atacaa los pacientes jóvenes del sexo femenino con una edad promedio de 47 años(rango de 30 a 60 años). Aunque puede ser de origen desconocido (primaria en20% de los casos), lo más común es que exista algún trastorno patológico previo(secundaria en 80% de los casos), como:
� Cirugía abdominal.� Pancreatitis.� Enfermedad inflamatoria intestinal.� Diverticulitis.� Apendicitis.� Sepsis intraabdominal.� Trauma cerrado de abdomen.� Trombosis venosa profunda periférica.� Neoplasias.� Deficiencia de proteína C.� Deficiencia de proteína S.� Deficiencia de antitrombina III.� Deficiencia de factor activador de plasminógeno.� Anemia de células falciformes.� Anticuerpos anticardiolipina.� Hiperfibrinogenemia.� Enfermedades mieloproliferativas.� Embarazo.� Hipertensión portal.� Esplenectomía y esplenomegalia.� Escleroterapia endoscópica de varices.� Anticonceptivos orales.� Oclusión intestinal
Existe un reporte en la literatura médica de un caso de trombosis venosa mesenté-rica asociada al empleo de anticonceptivos vaginales.

278 (Capítulo 16)Tratado de trombosis
El desarrollo y el daño histopatológico de esta enfermedad son menos abrup-tos que los que se observan en la trombosis arterial; sin embargo, en etapas másavanzadas puede conducir a una oclusión arterial al interrumpir el flujo capilar,lo cual complica la diferenciación entre los dos padecimientos.
La enfermedad se presenta de manera insidiosa. El paciente refiere malestarabdominal difuso, más que dolor, unos días antes de la aparición de franco dolorabdominal inespecífico, que no se localiza en un solo sitio del abdomen y sueleacompañarse de náusea y vómito. Cerca de la mitad de los pacientes presentanevacuaciones diarreicas sanguinolentas o vinosas. Es común la presencia de asci-tis y una gran distensión abdominal, producto de la dilatación de asas de intestinocomo respuesta a la isquemia. La irritación peritoneal aparece una vez que existenecrosis intestinal. La leucocitosis con hemoconcentración es parte de los hallaz-gos en los estudios de laboratorio. En las radiografías simples de abdomen se ob-serva una imagen de vidrio despulido, lo cual indica la presencia de líquido librey engrosamiento de la pared intestinal, que son resultado del edema de la mucosa.
El diagnóstico se establece con la sospecha clínica ante un paciente con coagu-lopatía de base o factor predisponente. En el pasado se realizaba una paracentesisen la que si se obtenía líquido vinoso fétido se establecía el diagnóstico de trom-bosis venosa mesentérica.
La TC es de gran ayuda y es capaz de mostrar la localización del coágulo en lavena mesentérica superior. A través de la arteriografía es más complicado realizarel diagnóstico definitivo, ya que sólo ofrece datos indirectos, como la presencia deespasmo arterial y el estrechamiento de las ramas periféricas del mesenterio.
La angiotomografía helicoidal (multicorte) y la angiorresonancia magnéticacon reconstrucción en tercera dimensión constituyen, según los reportes, los estu-dios de imagenología no invasores de elección en los pacientes con alta sospechaclínica de trombosis mesentérica sin datos de peritonitis.
El tratamiento debe incluir la mejoría de las condiciones generales del pacien-te antes de la cirugía, igual que en los casos de oclusión arterial aguda.
En los casos de obstrucción venosa mesentérica el intestino presenta un tonorojizo púrpura oscuro a consecuencia de la ingurgitación vascular que se fusionade manera gradual con el intestino normal sin evidencia de un límite exacto entreel tejido sano y el isquémico, contrario a lo que sucede en los casos de oclusiónarterial, en los que se precisa con cierta facilidad el tejido isquémico del viable.
La resección intestinal debe ser generosa y amplia, y prever una trombosis re-sidual que disemine algún coágulo. Si se recurre a una anastomosis primaria debeconsiderarse que los cabos del intestino a reconectar deben estar libres de datosde inflamación o edema, ya que esto puede conducir a una dehiscencia, por lo queen su lugar se puede recurrir a un estoma.
La anticoagulación durante la operación y después de ella es necesaria, puesal menos un tercio de los pacientes presentan una recurrencia dentro del primer

279Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
mes del evento inicial. Dicha anticoagulación puede prolongarse durante más deseis meses.
Debido a que el segmento intestinal afectado es más corto que en la enferme-dad arterial, el pronóstico es más favorable en estos pacientes.
La administración de medicamentos con efecto vasodilatador a través de tera-pia endovascular constituye una herramienta útil en estos casos. Dicho tratamien-to ha disminuido la mortalidad a 70% en la década de 1980 y a 50% en la últimadécada. La papaverina es la más utilizada en la actualidad; es un fármaco opiáceono adictivo obtenido de la planta de amapola, el cual funciona como un inhibidorde la fosfodiesterasa y se administra en infusión intraarterial a razón de 30 a 60mg/h, para relajar el músculo liso endotelial. En épocas recientes se documentóque el iloprost, un derivado sintético del epoprostenol, es un potente agente fibri-nolítico con efecto antiagregante plaquetario que favorece el flujo de la arteriamesentérica superior.
ISQUEMIA MESENTÉRICA CRÓNICA
La isquemia mesentérica crónica, también conocida como angina intestinal, fuedescrita en la década de 1920 por Mikkelsen. Esta afección patológica es pocofrecuente y se relaciona con la aterosclerosis mesentérica. La gran circulación co-lateral que desarrollan los pacientes con la enfermedad, además de las redes vas-culares de intercomunicación que existen entre el tronco celiaco y la AMS y lasque hay entre esta última y la AMI, dificulta la presencia de angina intestinal. Porello se requiere la afección aterosclerótica de al menos dos de las tres arterias ma-yores intestinales para la aparición de los síntomas.
Existen otras alteraciones que pueden dar lugar a una angina intestinal, entrelas que destacan el traumatismo abdominal y los aneurismas aórticos, del troncoceliaco o de las arterias mesentéricas.
El síntoma cardinal es el dolor abdominal opresivo y posprandial con predo-minio en el epigastrio, que casi siempre aparece entre 15 y 30 min posteriores alconsumo de alimento. A las dos o tres horas el dolor alcanza su máximo nivel, paradespués disminuir paulatinamente. El dolor suele ser tan intenso que el pacientecomienza a comer en menor cantidad, por el temor de que se presente un eventodoloroso. Por esta razón ocurre la pérdida de peso, la cual suele ser de 10 kg enpromedio, lo cual sugiere la presencia de una enfermedad oncológica.
Si bien el dolor se acompaña de otros síntomas, como distensión abdominal,náusea, vómito, evacuaciones esteatorreicas y en ocasiones constipación, éstosson opacados por la intensidad del dolor.
La naturaleza del dolor obedece a que durante el estado posprandial se requiereun mayor aporte de oxígeno al estómago y el yeyuno, por lo que en condiciones

280 (Capítulo 16)Tratado de trombosis
normales se incrementa el flujo mesentérico, lo cual no ocurre en la isquemia cró-nica, y da como resultado la liberación de metabolitos causantes del dolor.
Durante la exploración física se encuentran signos de aterosclerosis generali-zada, como son la disminución de los pulsos periféricos y los soplos en la regióncarotídea, femoral o epigástrica.
En los exámenes de laboratorio es frecuente encontrar sangre oculta en heces.El diagnóstico clínico se debe sospechar en un paciente con datos de ateroscle-
rosis generalizada, con presencia de dolor abdominal posprandial muy intenso,temor al consumo de alimento (sitofobia) y pérdida de peso.
La confirmación diagnóstica se logra a través de la arteriografía. Si bien la en-doscopia, la TC y los estudios contrastados no son de utilidad para establecer eldiagnóstico, sí lo son para descartar la presencia de otra patología. En estos casoses útil el ultrasonido Doppler, ya que permite determinar el flujo arterial, dondeuna velocidad diastólica mayor de 45 cm/seg o un pico sistólico mayor de 275cm/seg poseen una alta especificidad para establecer la presencia de estenosis dela AMS; sin embargo, estos datos se deben comparar con los hallazgos clínicosy angiográficos. Es importante considerar, como en cualquier estudio ultrasono-gráfico, que los resultados suelen depender de la persona que realiza el estudioy de la cantidad de gas intestinal, que puede impedir una adecuada ventana sono-gráfica.
El tratamiento es siempre quirúrgico y la indicación es mitigar el dolor incapa-citante. Existe controversia respecto a cómo tratar a los pacientes asintomáticoscon evidencia angiográfica de compromiso de las tres grandes arterias abdomina-les, ya que hay grupos que indican la revascularización profiláctica, pero otroscuestionan el riesgo. Los objetivos de la revascularización mesentérica son lograrmitigar los síntomas, mejorar el estado nutricional del enfermo y evitar el infartointestinal. Lo anterior se puede lograr a través de la terapia endovascular descrita.
Si se opta por la cirugía convencional, el procedimiento más recomendado enestos casos consiste en realizar un bypass de la AMS o del tronco celiaco con teji-do autólogo o con una prótesis.
La reconstrucción de una sola arteria (casi siempre la AMS) a través de un by-pass de la aorta infrarrenal es el procedimiento más aceptable, debido a lo relati-vamente sencillo de su técnica, a su vida media larga y a que suele ser suficientepara el alivio de los síntomas. Cunningham reportó que 86% de los pacientes so-metidos a revascularización permanecen sin dolor durante un lapso de cinco añosdespués del procedimiento.
REFERENCIAS
1. Boley SJ, Brandt LJ, Veith FJ: Ischemic disease of the intestine. Curr Probl Surg 1978;15:1–85.

281Trombosis mesentéricaE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
2. Schneider TA, Longo WE, Ure T, Vernava AM III: Mesenteric ischemia: acute arterialsyndromes. Dis Colon Rectum 1994;37:1163–1174.
3. Yasuhara H: Acute mesenteric ischemia: the challenge of gastroenterology. Surg Today2005;35:185–195.
4. Bayraktar Y, Harmanci O: Etiology and consequences of thrombosis in abdominal ves-sels. World J Gastroenterol 2006;12(8):1165–1174.
5. Joh JH, Kim DI: Mesenteric and portal vein thrombosis: treated with early initiation of an-ticoagulation. Eur J Vasc Endovasc Surg 2005;29:204–208.
6. Voora D, Vijayan A: Mesenteric vein thrombosis associated with intravaginal contracep-tives. J Thrombosis Thrombolysis 2003;15:105–108.
7. Bradbury MS, Kavanagh PV, Bechtold RE et al.: Mesenteric venous thrombosis: diagno-sis and noninvasive imaging. RadioGraphics 2002;22:527–541.
8. Sheridan WG, Lowdes RH, Williams GT, Young HL: Determination of a critical levelof tissue oxygenation in acute intestinal ischemia. Gut 1991;33:762–766.
9. AGA technical review on intestinal ischemia. Gastroenterology 2000;118:954–968.10. Crissinger KD, Tso P: The role of lipids in ischemia/reperfusion induced changes in muco-
sa permeability in developing piglets. Gastroenterology 1992;102:1693–1699.11. Vellar ID, Doyle JC: Acute mesenteric ischemia. Aust NZ J Surg 1977;47:54–61.12. Acosta S, Nilsson TK, Bjorck M: Preliminary study of D–dimer as a possible marker of
acute bowel ischaemia. Br J Surg 2001;88:385–388.13. Rha SE, Ha HK, Lee SH et al.: CT and MR imaging findings of bowel ischemia from vari-
ous primary causes. RadioGraphics 2000;20:29–42.14. Wison C, Gupta R, Gilmour DG, Imrie CW: Acute superior mesenteric ischaemia. Br J
Surg 1987;74:279–281.15. Taourel PG, Deneuville M, Pradel JA, Regent D, Cruel JB: Acute mesenteric ischemia:
diagnosis with contrast–enhanced CT. Radiology 1996;199:632–636.16. Levy P, Krausz MM, Manny J: The of second–look procedure in improving survival time
for patients with mesenteric venous thrombosis. Surg Gynecol Obstet 1990;170:287–291.17. Grendell JH, Ockner RK: Mesenteric venous thrombosis. Gastroenterology 1982;82:
358–392.18. Reinus JF, Brandt LJ, Boley SJ: Ischemic diseases of the bowel. Gastroenterol Clin North
Am 1990;19:319–343.19. Moawad J, Gewertz BL: Chronic mesenteric ischemia: clinical presentation and diagno-
sis. Surg Clin N Am 1997;77:357–369.

282 (Capítulo 16)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
17Oftalmología y trombosis
Judith Sandra Sarmina
La alteración trombótica por excelencia en el ojo es la oclusión de la vena de laretina (OVC). Aunque es uno de los padecimientos más drásticos del ojo, su fisio-patología no es del todo clara. Suele afectar a ambos sexos por igual y general-mente sucede en personas mayores de 65 años de edad.1–3 Cuando el cuadro sepresenta en personas jóvenes se debe sospechar una causa inflamatoria.1,4 Dichopadecimiento presenta una prevalencia de 0.1 a 0.4% a nivel mundial, pero algu-nos estudios reportan frecuencias de 25 a 42%.5–13 Es más frecuente la forma uni-lateral, pero hasta cinco años después del primer evento se puede desarrollar unaOVC en el ojo contralateral en 7% de las personas.5,13,14
PATOLOGÍA
La oclusión suele ser resultado tanto de causas locales como sistémicas; sin em-bargo, no es fácil realizar un estudio histopatológico recién sucedido el eventoobstructivo, ya que el ojo sólo se enuclea de manera secundaria a complicacionesposteriores a la oclusión, como el glaucoma neovascular. La evaluación histopa-tológica de ojos enucleados por oclusión de la vena central de la retina (OVCR)muestra oclusión en la lámina cribosa o por detrás ella;2,3,15–18 ocurre en esta zonaporque tanto la luz de la vena como de la arteria central de la retina es más peque-ña que la de la órbita y los vasos se encuentran rodeados por una adventicia co-mún.19 La lámina cribosa es una estructura de tejido conectivo que además de dar
283

284 (Capítulo 17)Tratado de trombosis
soporte al nervio óptico también limita su expansión y movimiento; lo mismo su-cede con sus vasos.
ETIOLOGÍA
Se desconoce la etiología exacta del padecimiento, pero se sabe que se asocia avarias afecciones sistémicas, como hipertensión arterial (HAS), aterosclerosis,padecimientos cardiovasculares, diabetes mellitus (DM), hiperviscosidad san-guínea, policitemia, anemia e hiperlipidemia, entre las más comunes.20–24
Por mucho, la más importante de todas estas causas la constituye la ateroscle-rosis23 relacionada con la HAS. La prevalencia de HAS en pacientes con OVCRes de aproximadamente 60%.20,22,24 La DM también se asocia a la OVCR, perovaría su prevalencia según el estudio.23,25–27
La asociación más frecuente es con el glaucoma primario de ángulo abierto(otro padecimiento ocular) entre 40 y 70%.28,29 En realidad no se sabe con exacti-tud si el glaucoma predispone a la OVCR o si al disminuir el flujo de sangre seorigina el glaucoma. La oclusión de la vena central de la retina se clasifica de lasiguiente manera:26
Clasificación
No isquémica o perfundida
Isquémica o no perfundida
CUADRO CLÍNICO DE LA OCLUSIÓN DE LA VENACENTRAL DE LA RETINA NO ISQUÉMICA
La oclusión de la vena central de la retina no isquémica se caracteriza por ser me-nos agresiva que la isquémica, si bien ésta se presenta en general en personas queson cinco años más jóvenes.11,23 Con frecuencia se presenta disminución de laagudeza visual, de hasta 20/50, pero puede existir agudeza visual normal, en cuyocaso suele ser un hallazgo de la exploración hasta cuenta dedos. Los pacientes sequejan de visión borrosa de inicio súbito. Durante la exploración se encuentrandatos muy similares a los de la forma isquémica pero menos extensos, como sonvasos tortuosos y dilatados, y hemorragias retinianas que pueden abarcar el poloposterior o ser periféricas, o ambas. La variación de la agudeza visual general-

285Oftalmología y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
mente se debe a edema macular o a hemorragias sobre el área macular. Tambiénse puede encontrar edema de papila, pero es menos frecuente. Los reflejos pupila-res y los campos visuales suelen ser normales.
En la fluorangiografía (FAG) se observa la tortuosidad de los vasos y el edemade disco o mácula, o ambos, pero no existe disminución de la perfusión capilar,lo cual la diferencia de la forma isquémica.
El curso natural es benigno, con mejoría de la agudeza visual y de la sintomato-logía. Las hemorragias y el edema mejoran con los meses; sin embargo, algunospacientes permanecen con edema macular quístico, lo cual no permite una mejo-ría completa de la agudeza visual,25,26 aunque suelen conservar una visión útil.En estos casos no suele haber neovascularización, que es la complicación másfrecuente de la forma isquémica. Hasta 10% de los pacientes evolucionan a unaforma “no perfundida” con formación de neovasos, por lo que requieren vigilancia.
Tratamiento
En el seguimiento del paciente es necesaria la observación del fondo de ojo cadacuatro semanas, con especial énfasis en la agudeza visual, los reflejos pupilares,el estudio de los campos visuales y la FAG cuando se requiera. Dependiendo delcurso de la enfermedad y de la presencia de factores de riesgo es como surge unaevolución al tipo isquémico (9 a 12% de los casos). En caso de que se presenteesta última se aplican láser y antiangiogénicos en presencia de edema macular.
OCLUSIÓN DE LA VENA CENTRALDE LA RETINA ISQUÉMICA
En estos casos se presenta una disminución brusca, significativa e indolora de laagudeza visual de 20/400 con movimiento de manos. Desde el punto de vista clí-nico, se presentan grandes hemorragias que confluyen y abarcan todo el polo pos-terior. Algunas de ellas pueden ser en flama, porque siguen la capa de fibras ner-viosas. También se pueden presentar hemorragias puntiformes que se encuentranen capas más profundas de la retina. Algunas veces esas hemorragias pueden al-canzar el vítreo, ocasionando mayores síntomas y complicaciones del cuadro.Suele existir tortuosidad venosa con dilatación y exudados blandos (o algodono-sos, como se les conocía anteriormente), los cuales se sabe que constituyen infar-tos de la capa de las fibras nerviosas. También en este caso puede haber edemade disco, pero es difícil de valorar, ya que puede estar cubierto por las hemorra-gias; en este caso el edema macular es más frecuente que en la perfundida. Puede

286 (Capítulo 17)Tratado de trombosis
Figura 17–1. Oclusión de la vena central de la retina no perfundida.
existir defecto pupilar aferente hasta en 88% de los casos, mientras que los cam-pos visuales se encuentran anormales hasta en 90%. La fluorangiografía muestravasos muy tortuosos, con fuga de fluoresceína, sobre todo en el área macular yalrededor de las venas tortuosas y las zonas no perfundidas, lo que le confiere altoriesgo a este tipo de oclusión, ya que puede conducir a complicaciones muy gra-
Figura 17–2. Oclusión de la vena central de la retina no perfundida; fluorangiografía concierres capilares.

287Oftalmología y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 17–3. Tomografía de coherencia óptica macular con edema (flecha).
500
400
300
200
100
0 m�
Overlay: ILM–RPE Transparency: 50%
Espesor mácula: cubo macular 512 x 128 OD OS
ID:
DOB:
Sexo:
Doctor:
CZMI590313502
8/29/1946
Masculino
Fecha examen:
Hora examen:
Técnico:
Fuerza de señal:
10/22/2012
1:00 PM
Operador Cirrus
5/10
CZM
Fovea: 276, 59
ILM–RPE
ILM
RPE
Distribución
99%95%5%1%
Central subfieldthicknees ( m)
Cube volume(mm )3
Cube averagethickness ( m)��
de normales
Espesor ILM–RPE( m)�
ILM–RPE 83 2318.3
60
65
83
529
310
195 133 305 385

288 (Capítulo 17)Tratado de trombosis
ves, como glaucoma neovascular, con pérdida total de la visión. La FAG no suelerealizarse en fases muy tempranas, ya que la sangre oculta la fluorescencia y nopermite observar las alteraciones vasculares ni retinianas (hipofluorescencia porbloqueo). Por lo anterior, se debe revisar clínicamente el fondo de ojo y tan prontose observe la presencia de reabsorción de las hemorragias se debe indicar la FAG,para así poder decidir el tratamiento a seguir.
El pronóstico de la OVCR isquémica generalmente es malo para la función,ya que existe disminución de la agudeza visual que no mejora; si ésta no se diag-nostica y trata a tiempo, se producirán neovascularización, dolor y glaucoma neo-vascular con pérdida de la visión a no percepción de la luz. Se pueden formar agu-jeros maculares, membranas epirretinianas, exudados circinados y membranasfibrovasculares que pueden ocasionar tracción y desprendimiento de la retina.
Las hemorragias desaparecen en el transcurso de los meses, aunque algunaspuntiformes pueden tardar años en hacerlo. Los exudados blandos y los microa-neurismas tienden a desaparecer en meses y el árbol venoso se ve menos tortuosoy dilatado.
La velocidad con que se recupera la retina depende del tratamiento, de las con-diciones generales del paciente y de la retina misma, relacionado con la revascu-larización y la recuperación del flujo normal.
Tratamiento
Se deben vigilar y tratar los factores sistémicos de riesgo, como la HAS, la DM,la aterosclerosis, etc. En el Central Vein Occlusion Study (CVOS), un estudio clí-nico aleatorizado de 728 ojos con OVCR, se evaluaron la historia natural y losresultados de la fotocoagulación con laser argón. En dicho estudio se reportó neo-vascularización retiniana en 12% de los pacientes tratados y en 18% de los no tra-tados;13 además, se reportaron cuatro factores de riesgo asociados a la apariciónde neovasos:
1. Área de no perfusión mayor de 30 diámetros papilares.2. Cantidad de hemorragias en la retina.3. Menos de un mes de evolución.4. Sexo masculino.
Por ello se revisa al paciente con la variedad no perfundida cada tres a cuatrosemanas, con especial cuidado en la valoración del ángulo y el iris. Si en dichaexploración se encuentra la presencia de neovasos, se deben fotocoagular en for-ma agresiva los cuatro cuadrantes hasta la periferia.30 En el tratamiento del edemamacular ya no se utiliza la fotocoagulación en parrilla, ya que con frecuencia pro-

289Oftalmología y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
voca lesiones que pueden afectar la agudeza visual, que fotocoagula en el áreamacular. Hoy en día se inyectan antiangiogénicos intravítreos (ranibizumab o be-vacizumab), cuyo efecto consiste en disminuir la vascularización secundaria a lahipoxia de la retina y con ello reducir el edema, lo cual mejora de forma impor-tante la agudeza visual.31
OCLUSIÓN DE LA RAMA SUPERIORO INFERIOR DE LA VENA RETINIANA
Esta entidad afecta una rama venosa superior o inferior y es menos agresiva quela OVCR. La oclusión puede afectar una zona periférica o todo un cuadrante. Suincidencia es similar a la de la OVCR. La mayoría de las oclusiones de rama sontemporales; aún no se determina si es porque en dicha zona se producen síntomasy se diagnostican con mayor facilidad o porque en realidad son más raras las nasa-les. Suelen afectar por igual a hombres y mujeres, y son más frecuentes entre los60 y los 70 años de edad.
Patología
Se sabe que dichos padecimientos son muy frecuentes en la intersección arterio-venosa y superior, ya que donde se cruzan la arteria y la vena comparten la adven-ticia y, de este modo, la luz de la vena puede disminuir hasta la tercera parte,32
conllevando a la formación de trombosis, isquemia y neovascularización.6 Losfactores de riesgo más frecuentes son la HAS, la DM, la hiperlipidemia, el glau-coma, el tabaquismo, la aterosclerosis7 y la longitud axial corta.7,11–13
Manifestaciones clínicas
Generalmente son de inicio súbito. El paciente refiere visión borrosa o un defectode campo, además de que se puede observar una hemorragia intrarretiniana dis-tribuida de forma segmentaria. La localización del bloqueo determina la distribu-ción de la hemorragia. Si es a nivel del disco se afectan dos cuadrantes, pero sies más periférico al disco se puede afectar un cuadrante o menos. Si el bloqueovenoso es más periférico que las venas que drenan la mácula, ésta no se afectani existe disminución de la agudeza visual. Puede pasar hasta un año para que lahemorragia se reabsorba.
Se debe realizar el diagnóstico oftalmoscópico, pero tan pronto se reabsorbala hemorragia se debe confirmar mediante la observación de las alteraciones vas-culares retinianas en la fluorangiografía.
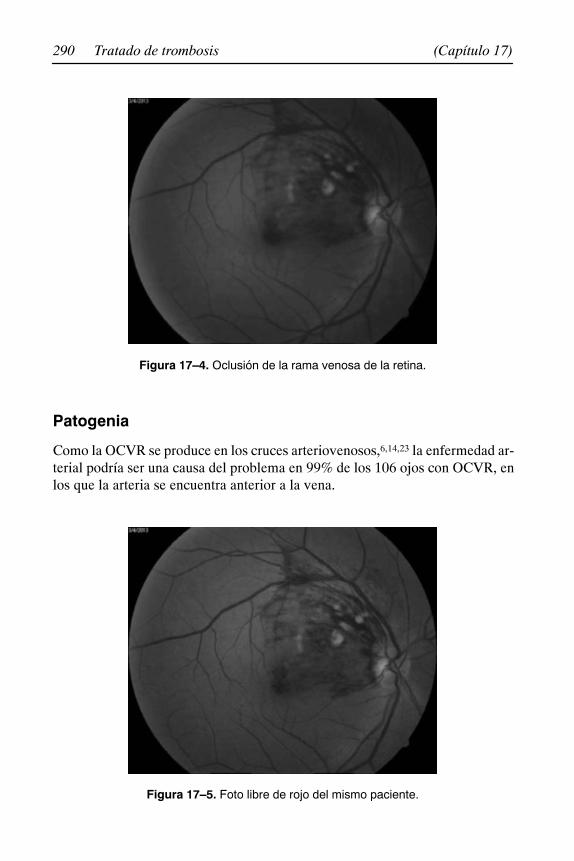
290 (Capítulo 17)Tratado de trombosis
Figura 17–4. Oclusión de la rama venosa de la retina.
Patogenia
Como la OCVR se produce en los cruces arteriovenosos,6,14,23 la enfermedad ar-terial podría ser una causa del problema en 99% de los 106 ojos con OCVR, enlos que la arteria se encuentra anterior a la vena.
Figura 17–5. Foto libre de rojo del mismo paciente.
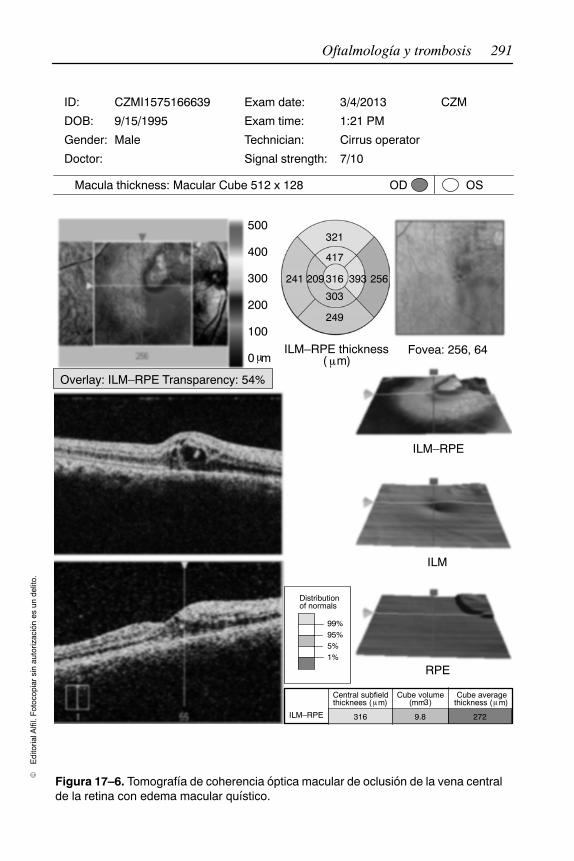
291Oftalmología y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
� Figura 17–6. Tomografía de coherencia óptica macular de oclusión de la vena centralde la retina con edema macular quístico.
Macula thickness: Macular Cube 512 x 128 OD OS
ID:
DOB:
Gender:
Doctor:
CZMI1575166639
9/15/1995
Male
Exam date:
Exam time:
Technician:
Signal strength:
3/4/2013
1:21 PM
Cirrus operator
7/10
CZM
500
400
300
200
100
0 m�
Overlay: ILM–RPE Transparency: 54%
Fovea: 256, 64
ILM–RPE
ILM
RPE
Distribution
99%95%5%1%
Central subfieldthicknees ( m)
Cube volume(mm )3
Cube averagethickness ( m)��
of normals
ILM–RPE thickness( m)�
ILM–RPE 316 2729.8
321
417
316
303
249
241 209 393 256

292 (Capítulo 17)Tratado de trombosis
En el lugar de la obstrucción la luz de la vena se puede encontrar disminuidahasta 33% en el cruce.2,32–34
Tratamiento
El tratamiento médico se intentó con el uso de anticoagulantes pero no funcionó,de tal manera que el tratamiento actual se basa en tratar las complicaciones queafectan la visión, como son el edema macular, la falta de perfusión macular y elhemovítreo.9,24,26,30
El Branch Vein Occlusion Study30,34 demostró que la panfotocoagulación pro-filáctica puede disminuir la neovascularización y que en caso de que ya existanneovasos puede reducir la posibilidad de hemovítreo. Se pueden desarrollar neo-vasos o hemovítreo hasta tres años después del evento inicial, pero lo más comúnes que esto suceda en los primeros 6 a 12 meses. En caso de tracciones o membra-nas epirretinianas se puede realizar una vitrectomía para retirar las tracciones. Sesabe que los corticosteroides intravítreos funcionan de manera adecuada para eledema macular, pero tienen el inconveniente de producir glaucoma secundario.Actualmente se realizan protocolos para valorar el uso de los antiangiogénicosintravítreos en el edema macular, y se han observado resultados óptimos.31
En conclusión, se tiene muy clara la asociación de dicho padecimiento oftal-mológico con afecciones sistémicas, de tal manera que se debe trabajar en equipopara mejorar el pronóstico visual de los pacientes, ya que se requiere un controlmuy estricto de los padecimientos de base, así como la comunicación con el oftal-mólogo tan pronto se reporten alteraciones visuales que interfieran en el adecua-do control y tratamiento del paciente, todo con el fin de mejorar su pronósticovisual.
REFERENCIAS
1. Coats G: Further cases of thrombosis of the central vein. Royal London Ophthalm HospReports 1906;16:516.
2. Green WR, Chance CC, Hutchins GM et al.: Central vein occlusion: a prospective histo-pathologic study of 29 eyes in 28 cases. Retina 1981;1:27.
3. Hogan MJ, Alvarado JA, Weddell JJ: Histology of the human eye: an atlas and textbook.Filadelfia, W. B. Saunders, 1971:565–592.
4. Dodson PM, Kritzinger EE: Underlying medical conditions on young patients and ethnicdifferences in retinal vein occlusions. Trans Ophthalmol Soc UK 1985;104:114.
5. Hayreth SS, Zimmermann MB, Podjasky P: Characteristic incidence of various types ofretinal vein occlusion and their recurrence and demographic. Am J Ophthalmol 1994;117:429–441.
6. Mitchell T, Smith W et al.: Prevalence and associations of retinal vein occlusion in Austra-lia. The Blue Mountains Eye Study. Arch Ophthalmol 1996;114:1243–1247.

293Oftalmología y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
7. Group TCVOS: Baseline and early natural history report: The Central Vein Occlusion Stu-dy. Arch Ophthalmol 1993;111:1087–1995.
8. Fekrat S, Finkestein D: Venous occlusive disease. En: Regilo CD, Brown GC, Flynn HW(eds.): Vitreoretineal disease: the essentials. 1ª ed. Nueva York, 1999.
9. Gutman FA: Evaluation of the patient with central retinal vein occlusion. Ophthalmology1983;90:481–483.
10. Fong AC, Schatz H: Central retina vein occlusion in young adults. Surv Ophthalmol 1993;38:88.
11. Zeramo VA, Cox TA et al.: Vision–related quality of life in people with central retineal veinocclusion using 25–item National Eye Institute visual function questionarie. Arch Ophthal-mol 2003;121:1297–1302.
12. Klein R et al.: The epidemiology of retineal vein occlusion: The Beaver Dam Eye Study.Trans Am Ophthalmol Soc 2000:133–143.
13. Hayreh SS: Central retineal vein occlusion. Ophthalmol Clin N Am 1998;11:559–590.14. Pliskeewicz K, Pournaras C, Roth A: Thrombose veineuse oculaire et pathologie vascul-
aire general. Klin Monatsbl Augenheilkd 1984;184:367.15. Zegarra H, Gutmann FA, Zakar N et al.: Partial occlusion of the central retinal vein. Am
J Ophthalmol 1983;96:330.16. Zegarra H, Gutmann FA, Conforto J: The natural course of central vein occlusion. Trans
Am Acad Ophthalmol Otolaryngol 1979;86:1931.17. McGrath M, Wechsler F, Humgar AB et al.: Systemic factors contributing to retinal vein
occlusion. Arch Intern Med 1978;138:216.18. Priluck IA, Robertson DM, Hollenhost RW: Long–term follow–up of occlusion of the
central vein in young adults. Am J Ophthalmol 1980;990:190.19. Abu el–Azrar AM et al.: Hypercoagulable states in patients with retineal vein occlusion.
Doc Ophthalmol 1998;95:133–143.20. Ariturk N, Oge Y, Erkan D et al.: Relations between retinal vein occlusions and axial
length. Br J Ophthalmol 1996;80:633–636.21. Bhatt AK, Quinland PM et al.: The risk of systemic vascular diseases and mortality in
patients with central retinal vein occlusion. Ophthalmology 1990;97:1543–1548.22. Hayret SS, Zimmerman B et al.: Systemic diseases associated with various types of retinal
vein occlusion. Am J Ophthalmol 2001;10:61–77.23. Martin SC, Butcher A et al.: Cardiovascular risk assessment in patients with the retineal
vein occlusion. Br J Ophthalmol 2002;86:776.24. Browning DJ, Scott AQ et al.: The risk of missing angle neovascularization by omitting
screening gonioscopy in acute central retinal vein occlusion. Ophthalmology 1998;105:776–784.
25. Grey JM, Tunc M et al.: Laboratory evaluations of hipercoagulable factors in patients withcentral retinal vein occlusion who are less than 56 years of age. Ophthalmology 2002;109:126–131.
26. Gottlieb JL, Blize JP et al.: Activated protein C resistant factor V Leiden, and central reti-nal vein occlusion in young adults. Arch Ophthal 1998;116:577–579.
27. Driden RM: Central retinal vein occlusion and chronic simple glaucoma. Arch Ophthalmol1965;73:659.
28. Hayreth SS, Rojas P, Podhajsky P et al.: Ocular neovascularization with retinal ocularocclusion. Ophthalmology 1983;90:488.
29. Hayreth SS, Rojas P, Podhajsky P et al.: Ocular neovascularization with retinal ocular oc-clusion. Ophthalmology 1983;90:488.

294 (Capítulo 17)Tratado de trombosis
30. Hayreth SS, Rojas P, Branch Vein Occlusion Study Group: Argon laser scatter photocoa-gulation for prevention of neovascularization and vitreous hemorrhage in branch vein oc-clusion. Arch Ophthalmol 1986;104:34–41.
31. Zhu D, Jin ZY,Tao Y, Jonas JB: Meta–analysis of the effect of intravitreal bevacizumabin branch retinal vein occlusion. J Ocul Pharmacol Ther 2013 (Epub).
32. Duker JS, Brown GC: Anterior location of crossing artery in branch retinal vein occlusion.Arch Ophthalmol 1989;107:998–1000.
33. Weinberg D, Dodwell DG, Fern SA: Anatomy of arteriovenous crossings in retinal branchretinal vein occlusion. Am J Ophthalmol 1990;109:298–302.
34. Zhao J, Sastry SM, Sperduto RD et al.: Arteriovenous crossing patterns in branch retinalvein occlusion: the Eye Disease Case Control Study Group. Ophthalmology 1993;100:423–428.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
18Trombosis en el paciente oncológicoVíctor A. de la Garza Estrada, Luis Fernando García–Frade Ruiz,
Julieta Lomelín Gascón
INTRODUCCIÓN
Pocas entidades patológicas explican con tanta claridad la conocida tríada de Vir-chow para la formación de trombos intravasculares como el cáncer. A principiosdel siglo XX el médico Alemán Rudolf Virchow propuso los tres factores que sonindispensables para la trombosis, aunque no como se conocen ahora. Esos tresfactores son:
1. Fenómeno de flujo sanguíneo interrumpido.2. Fenómeno relacionado con irritación de los vasos y su vecindad.3. Fenómeno de coagulación de la sangre.
A partir de un consenso internacional esa tríada cambió a la nomenclatura moder-na, que es como se conoce hoy en día y que consta de estasis sanguínea, lesiónendotelial o de la pared de los vasos, y estado de hipercoagulabilidad.1 Así, estastres condiciones son indispensables para que se formen trombos (o coágulos) enel interior de los vasos, y es en el cáncer donde se conjuntan las tres.
Los fenómenos trombóticos en el cáncer pueden ser de varios tipos:
1. Trombosis venosa profunda.2. Embolia pulmonar.3. Tromboflebitis superficial.4. Trombosis venosa central.
295

296 (Capítulo 18)Tratado de trombosis
5. Trombosis arterial.6. Endocarditis trombótica no bacteriana.2,3
Para el tema que ocupa el capítulo —trombosis en el paciente oncológico—, laatención se centrará en las dos primeras: trombosis venosa profunda y emboliapulmonar, las que en conjunto son conocidas como enfermedad tromboembólicavenosa (ETEV), con alguna mención de las otras formas.
La ETEV es una complicación frecuente del cáncer, aunque también se puedemanifestar antes de que lo haga una neoplasia maligna, como un fenómeno para-neoplásico. Como curiosidad histórica, la tromboflebitis superficial migratoriafue descrita por el Dr. Trousseau hace casi 150 años, y ahora se le conoce con elsíndrome que lleva su nombre. Él describió (y también lo padeció en carne pro-pia) un fenómeno que acompañaba a la caquexia y que se debía a una situaciónde coagulación espontánea.2–4
Como complicación del cáncer, la ETEV es causa de gran morbilidad y morta-lidad en los pacientes con tumores malignos, y tiene mucha relación con los pro-pios tumores, los sitios de localización, los tratamientos a los que son sometidos,los catéteres intravenosos que a menudo se colocan para la administración de losagentes quimioterápicos o antibióticos, y las características de los propios pa-cientes.2–5
FISIOPATOLOGÍA
En línea con la tríada de Virchow, la enfermedad tromboembólica venosa en elcáncer se presenta de la siguiente manera:2–6
� Estasis sanguínea: sea por características propias del tumor en cuanto a ta-maño, localización o tipo, o por situaciones propias de los pacientes, comoinmovilización prolongada por el cáncer o después de una cirugía, el hechoes que estos pacientes tienen gran propensión a que se les “estanque” la san-gre en cualquier sitio, hecho que favorece la formación de un trombo in situque puede producir alteraciones locales o emigrar a sitios distantes.
� Lesión del endotelio o de la pared vascular: por las características del tu-mor, como tamaño, tipo, grado de invasión que produce, y por los procedi-mientos a los que se les puede someter, no es nada infrecuente que se lesio-nen los vasos, situación que también dará lugar a que se formen trombos.
� Estado de hipercoagulabilidad: los tumores malignos elaboran y secretanuna serie de sustancias que tienen actividad procoagulante reconocida. Deellas la principal es el factor tisular, que, al igual que en otros estados no

297Trombosis en el paciente oncológicoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
neoplásicos, es la iniciadora de la coagulación. Varía la magnitud con la quese expresa la secreción de este factor tisular según el tipo de tumor de quese trate; por ejemplo, son muy altas las concentraciones en los tumores delcerebro y del páncreas, y menores en el cáncer de mama.4 La expresión delfactor tisular está bajo control transcripcional de varios oncogenes y de ge-nes supresores de tumores, como los pertenecientes a las familias EGFR,RAS, TP53 y PTEN.4
El factor tisular es una glucoproteína de membrana que funciona como receptorpara el factor VII y como sitio de activación del mismo en su forma activada,VIIa, para formar luego un complejo con el factor tisular y los fosfolípidos demembrana, y activar así al factor X. La actividad del complejo factor tisular–fac-tor VIIa puede ser inhibida por el inhibidor del factor tisular (TFPI) en conjuntocon el factor Xa.4,7–9
Éste es el principal punto de inicio de la activación de la coagulación y es unimportante activador celular del sistema de la coagulación en el paciente con cán-cer. En las células vasculares sanas la expresión del factor tisular está controladade manera precisa. El factor de necrosis tumoral alfa, la interleucina 1� y otrascitocinas aumentan la expresión del factor tisular como respuesta a estímulos in-flamatorios. En las células tumorales el factor tisular se expresa de manera cons-tante. El factor tisular se encuentra en la mayor parte de los tejidos con cáncer ysu expresión pudiera ser una característica de la malignidad de una célula.4,7–9
El procoagulante del cáncer es una proteasa de cisteína que activa al factor Xen ausencia de factor VII. El procoagulante se expresa en tejidos embrionariosy en varias células malignas; en vista de que no se encuentra en los tejidos sanos,sirve para diferenciar las células malignas. El procoagulante del cáncer se usa in-cluso como marcador tumoral detectado por pruebas de inmunoabsorción (técni-ca de ELISA).4,7–9
El TFPI es una proteinasa inhibidora compuesta por 276 aminoácidos que ac-túa sobre todo en dos dominios: en el primero se une e inhibe al factor VIIa y enel segundo se une e inhibe el factor X. El TFPI puede inhibir la actividad del fac-tor VIIa–factor tisular a través de la formación de un complejo cuaternario com-puesto por el factor Xa, el TFPI, el factor VIIa y el factor tisular. Los pacientescon varios tipos de cáncer presentan concentraciones plasmáticas aumentadasdel TFPI, como se ve en la enfermedad maligna de colon y páncreas.4–7,9 El TFPIno se expresa en las células tumorales, sino que se produce en las células endote-liales vasculares del tumor. Las citocinas inflamatorias producidas por las célulastumorales, como el factor de necrosis tumoral alfa y la interleucina 1�, aumentanla expresión del TFPI.4,7–9
Las células tumorales expresan todas las proteínas que regulan el sistema fibri-nolítico, como la urocinasa, los activadores del plasminógeno, los inhibidores del

298 (Capítulo 18)Tratado de trombosis
activador del plasminógeno 1 y 2 y el receptor del activador del plasminógeno.El aumento en las concentraciones plasmáticas de los inhibidores del activadordel plasminógeno y la alteración en la actividad fibrinolítica en los pacientes concáncer indican que hay otro mecanismo protrombótico relacionado con la enfer-medad maligna.
La disminución del sistema fibrinolítico en el paciente con cáncer no se sumapor fuerza al estado trombofílico en esta situación, y su función participa en elcrecimiento tumoral y el desarrollo de metástasis.4,7–9
La trombina se genera como resultado de su liberación a partir de células tu-morales y de la cascada de la coagulación, además de que favorece el desarrollode trombosis y ejerce otros efectos sobre el cáncer. También el estado procoagu-lante en el paciente con cáncer se puede generar por anormalidades en las plaque-tas. Las células tumorales inducen la activación y agregación plaquetaria por con-tacto celular directo o a través de la liberación de agonistas plaquetarios, comoADP, trombina y otras proteasas.
El aumento en la adhesión plaquetaria, la acelerada formación de tromboplas-tina y el consumo reducido de protrombina son anormalidades uniformes en unaserie de tumores sólidos.4,7–9
Prevalencia de la enfermedadtromboembólica venosa en cáncer
La ETEV se llega a presentar hasta en 20% de los pacientes con cáncer en algúnmomento de su evolución, según las distintas series analizadas.3 Sin embargo, enestudios de autopsia se ha encontrado que hasta la mitad de los pacientes que fa-llecen por algún cáncer tienen datos de trombosis en alguna parte de su organis-mo, incluso sin haber mostrado manifestaciones clínicas.4 Se debe tener muy pre-sente a la ETEV, ya que es la segunda causa de muerte en los pacientes con cáncer,por debajo de la propia enfermedad oncológica como motivo de la defunción;este fenómeno fue tomado en cuenta con toda seriedad apenas en los últimos cin-co años, debido a la importancia que tiene.3,4
Por su parte, la trombosis venosa profunda, sobre todo la pélvica y de las extre-midades inferiores, manifestada como fenómeno paraneoplásico, es un problemamenos sencillo de aclarar en cuanto a su prevalencia. Se dice que hasta 20% delos pacientes que tienen trombosis venosa profunda sin causa aparente en el trans-curso de un año empezarán a mostrar manifestaciones de algún tumor maligno.3,4
Sin embargo, este dato es difícil de corroborar. Hacen falta marcadores clíni-cos o biológicos para determinar en qué pacientes con trombosis venosa profundasin causa aparente está justificado hacer todo un escrutinio diagnóstico despuésde haber presentado este problema.

299Trombosis en el paciente oncológicoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Factores de riesgo para enfermedadtromboembólica venosa en cáncer
Son varios los factores que participan en la presentación de la ETEV en los pa-cientes con cáncer. Los más importantes son los siguientes:
� Relacionados con el tumor: aquí se aprecia la etapa de la enfermedad;cuanto más avanzada esté, mayor riesgo habrá de presentar ETEV. El tipoy la localización del tumor influyen muchísimo en la ocurrencia de ETEV.Los que con mayor frecuencia la producen son los tumores malignos del ce-rebro, las neoplasias hematológicas y los adenocarcinomas de páncreas,útero, ovario, próstata, estómago y riñón.3,4
� Relacionados con el paciente: los factores de riesgo para ETEV en pacien-tes con cáncer atribuidos al propio paciente son la edad (> 65 años), el sexo,la raza (menos en razas asiáticas), el estado de desempeño, el grado de mo-vilidad, las comorbilidades, las trombosis previas y las mutaciones de genesprotrombóticos.3,4,6 Un ejemplo de este último caso es el de mujeres concáncer de mama, que no suele ser un problema que origine ETEV, salvo enaquellas que tienen la mutación V de Leiden, que corren más riesgo de sufrirla complicación.10
� Relacionados con el tratamiento: la cirugía sigue siendo la piedra angulardel tratamiento en muchos de los tipos de cáncer. Por sí misma confiere unriesgo aumentado de ETEV debido a varios factores, como sitio del proce-dimiento, tamaño del tumor, duración de la intervención (> 2 h de anestesia)e inmovilidad posterior al acto quirúrgico (> 4 días de reposo en cama).4
La quimioterapia es otro factor de riesgo para ETEV en pacientes con cáncer. Seha encontrado que este riesgo aumenta hasta siete veces la posibilidad cuando serecibe quimioterapia farmacológica. Los fármacos más implicados con este ries-go aumentado son el tamoxifeno, la doxorrubicina, la talidomida y su derivadolenalidomida (usadas en cierto tipo de leucemias) y el bevacizumab, un anticuer-po monoclonal que está siendo muy utilizado en ciertas neoplasias que antes te-nían muy mal pronóstico, como algunas de pulmón y colon.4 Se dice que el meca-nismo de acción por el que ocurre la complicación de ETEV es por aumento enla concentración de moléculas procoagulantes y disminución de los anticoagu-lantes endógenos, por inducción de apoptosis del tumor y las células endoteliales,y liberación de citocinas, lo que aumenta la expresión y la actividad del factortisular; por activación de las plaquetas y por inducción en la expresión del factortisular de macrófagos monocitos.3,4,6
No hay que olvidar que muchos pacientes con cáncer tienen mayor propensióna presentar infecciones más o menos graves. Por tal motivo, suelen colocarse ca-

300 (Capítulo 18)Tratado de trombosis
téteres intravenosos centrales y otros dispositivos centrales, como los “puertos”,que también son muy utilizados para la administración de los fármacos quimiote-rápicos. Cuando no se manejan debidamente estos catéteres se convierten en elorigen de la formación de trombos y pueden dar lugar a trombosis locales y a em-bolia pulmonar. Ante la sospecha de una ETEV originada en un catéter centralsiempre se debe retirar y reinstalar uno nuevo si todavía se requiere.4,11
Manifestaciones clínicas de laenfermedad tromboembólica venosa
No es infrecuente que la enfermedad tromboembólica venosa se presente sin nin-guna manifestación clínica. Cuando sucede las manifestaciones más comunesson edema (80%), dolor (75%) y eritema de extremidades (25%) cuando se tratanada más de trombosis venosa profunda. En caso de embolia pulmonar los datosclínicos son disnea (85%), dolor torácico (40%), taquipnea (30%) y taquicar-dia.23 No se debe olvidar que la primera manifestación de una ETEV es la muertesúbita, que llega a ocurrir hasta en 20% de los pacientes.3,4,6
Tratamiento de la enfermedad tromboembólica venosa
Una vez presentada el manejo de la enfermedad tromboembólica venosa es elmismo que si se tratara de cualquier otro paciente: anticoagulación (intravenosao subcutánea, seguida de la oral para mediano y largo plazos) y medidas generalesde sostén. En casos especiales, sobre todo cuando la enfermedad trombótica estámuy generalizada o en casos de contraindicación de anticoagulación, se debe co-locar un filtro en la vena cava.12
En cuanto a la anticoagulación, se suele recurrir a la heparina por vía intrave-nosa, aunque en la actualidad se están prefiriendo ya a las heparinas de bajo pesomolecular. El surgimiento de los nuevos compuestos anticoagulantes por vía orales una opción que, aunque todavía no está autorizada en la indicación del trata-miento de la ETEV en cáncer, es muy posible que se vaya a lograr en un futurono muy lejano. Sin embargo, se deben tomar en cuenta las complicaciones hemo-rrágicas que pueden presentarse por el uso de estos compuestos y que en la actua-lidad están en la mira de las autoridades regulatorias internacionales, como laFood and Drug Administration y la European Medicines Evaluation Agency.14
En fechas recientes se publicaron dos guías para el manejo de la tromboembo-lia venosa. Una de ellas se hizo a partir de una revisión sistemática hecha por Se-gal y col. en 2007, en la que se concluyó que hay pruebas que apoyan el uso deheparinas de bajo peso molecular y no de anticoagulantes orales en pacientes se-

301Trombosis en el paciente oncológicoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
leccionados, aunque el costo de las primeras sea muy superior al de los anticoagu-lantes.15 La otra guía fue elaborada por un grupo conformado especialmente paraanalizar la enfermedad tromboembólica venosa del Colegio Americano de Médi-cos y la Academia Americana de Médicos Familiares (2007); en ella se concluyeque hay pruebas para apoyar la recomendación del uso de heparinas de bajo pesomolecular sobre los anticoagulantes orales, con el fin de evitar la enfermedadtromboembólica venosa, profunda y pulmonar en pacientes con cáncer seleccio-nados.16
Prevención de la enfermedad tromboembólicavenosa en pacientes con cáncer
Tal vez la prevención sea más importante que el propio tratamiento de esta com-plicación en los pacientes con cáncer. Para ello lo primero que se debe tener enconsideración es que el problema existe, que es muy frecuente, que se debe guar-dar un alto grado de sospecha de que ocurra y que se puede prevenir con relativafacilidad.4,13 Dicho en otras palabras, si se piensa que es muy alta la probabilidadde que ocurra enfermedad tromboembólica venosa en un paciente con cáncer, porla edad, las comorbilidades, el tipo de tumor, el tipo de cirugía, las quimioterapiasa las que se va a someter o por la presencia de un catéter, lo mejor es iniciar cuantoantes con anticoagulación, de preferencia con heparinas de bajo peso molecular,que se administran por vía subcutánea.4,13,15,16
CONCLUSIÓN
Ahora es bien sabido que el paciente oncológico tiene un riesgo muy elevado depadecer enfermedad tromboembólica venosa, sea como complicación a lo largode su evolución, con o sin tratamiento, o como una manifestación en forma desíndrome paraneoplásico. Esto se debe a una serie de alteraciones en los compo-nentes de las distintas fases de la coagulación normal y a la presencia de algunassustancias procoagulantes. Entre los tumores que más se relacionan con enferme-dad tromboembólica venosa están el cáncer de cerebro, próstata, estómago, pul-món, páncreas y ovario, y algunas neoplasias hematológicas. También hay otrosfactores propios del paciente y de los tratamientos que reciba, que lo hacen pro-penso a presentar esta ominosa complicación, que es la segunda causa de muerteen los pacientes con cáncer. En estos momentos se prefiere el uso de heparinasde bajo peso molecular, sobre todo enoxaparina y dalteparina, para el tratamientoy la prevención de la enfermedad tromboembólica venosa en la enfermedad neo-plásica maligna.

302 (Capítulo 18)Tratado de trombosis
REFERENCIAS
1. http://en.wikipedia.org/wiki/Virchow’s_triad.2. Bauer K: Pathogenesis of the hypercoagulable state associated with malignancy. Literature
Rev Curr 2013;124.3. Karimi M, Cohan N: Cancer–associated thrombosis. Open Cardiovasc Med J 2010;4:78–
82.4. Young A, Chapman O, Connor C, Poole C, Rose P et al.: Thrombosis and cancer. Nat
Rev Clin Oncol 2012;9:437–449.5. Khorana AA: Venous thromboembolism and prognosis in cancer. Thrombosis Res 125(6):
490–493.6. Shelke AR, Khorana AA: Cancer–associated thrombosis: an update. Haematology 2011;8
(1–2):e39–e45.7. Kohli M, Kaushal V, Metha P: Role of coagulation and fibrinolytic system in prostate can-
cer. Semin Thromb Hemost 2003;29(3):301–308.8. Marek Z, Ewa S, Leo R: Tissue factor–dependent coagulation activation and impaired fi-
brinolysis in situ in gastric cancer. Semin Thromb Hemost 2003;29(3):291–299.9. Zangari M, Saghafifar F, Metha P: The blood coagulation mechanism in multiple mye-
loma. Semin Thromb Hemost 2003;29(3):275–281.10. Gaber JE, Halabi S, Tolaney SM, Kaplan E, Archer L et al.: Factor V Leiden mutation
and thromboembolism risk in women receiving adjuvant tamoxifen for breast cancer. JNCI2012;13:942–949.
11. Saber W, Moua T, Williams EC, Verso M, Agnelli G et al.: Risk factors for catheter–re-lated thrombosis (CRT) in cancer patients: a patient–level data (IPD) meta–analysis of clini-cal trials and prospective studies.
12. Agnelli G, Verso M: Management of venous thromboembolism in patients with cancer. JThromb Haemost 2011;9(Suppl 1):316–324.
13. Lee AYY, Levine MN, Baker RI, Bowden C, Kakkar AK et al.: Low–molecular–weightheparin versus a coumarin for the prevention of recurrent venous thromboembolism in pa-tients with cancer. N Engl J Med 2003;349(2):146–153.
14. Southworth MY, Reichman ME, Unger EF: Dabigatran and postmarketing reports ofbleeding. N Engl J Med 2013;368:1272–1274.
15. Segal JB, Streiff MB, Hoffmann LV, Thornton K, Bass EB: Management of venousthromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007;146(3):211–222.
16. Snow V, Qaseem A, Barry P, Hornbake ER, Rodnick JE et al.: Management of venousthromboembolism: a clinical practice guideline from the American College of Physiciansand the American Academy of Family Physicians. Ann Intern Med 2007;146(3):204–210.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
19Enfermedad inmunitaria y trombosis.
Síndrome de anticuerposantifosfolípidos
Raúl Ariza Andraca, Leonor A. Barile Fabris,Rocío Catana Hernández
El estado trombofílico es el resultado de un desequilibrio entre los mecanismosprocoagulantes y anticoagulantes naturales, en el que van a predominar los meca-nismos procoagulantes. Sus causas son múltiples; pueden ser hereditarias oadquiridas, las cuales pueden ser producidas por mecanismos o enfermedades au-toinmunitarias. No existe una estadística que ubique en un lugar a estas enferme-dades en la epidemiología de la trombosis, pero en general se acepta que su preva-lencia es baja, sobre todo cuando se compara con el resto de las enfermedadesgeneradoras de trombosis. La lista de padecimientos con patogenia inmunitariao autoinmunitaria que pueden inducir trombosis se modifica con frecuencia, yaque periódicamente se identifican nuevas moléculas que interfieren en la hemos-tasia y que se encuentran asociadas con varias enfermedades autoinmunitarias;sin embargo, no son entidades específicas, ya que la enfermedad eje es el padeci-miento asociado. En el cuadro 19–1 se anotan las más frecuentes e importantes.
La entidad más sobresaliente y estudiada es el síndrome de antifosfolípidos(SAF), cuyos aspectos más relevantes se comentan a continuación.
El SAF es un trastorno autoinmunitario que puede ser primario o secundario(asociado a enfermedades predominantemente autoinmunitarias) y se caracterizaclínicamente por eventos trombóticos venosos y arteriales, y morbilidad obstétri-ca, aunado a la presencia persistente de un grupo de autoanticuerpos que se diri-gen contra proteínas fijadoras de fosfolípidos.1,2 La importancia del SAF es queafecta sobre todo a población joven, es una causa de abortos y pérdida fetal, y sudetección y un tratamiento adecuado pueden prevenir la morbimortalidad asocia-da con el síndrome.
303

304 (Capítulo 19)Tratado de trombosis
Cuadro 19–1. Afecciones con patogeniaautoinmunitaria que generan trombofilia
1. Lupus eritematoso sistémico2. Síndrome antifosfolípidos primario y secundario3. Resistencia adquirida de proteína C activada
4. Resistencia adquirida a la proteína S5. Resistencia adquirida de antitrombina III6. Hiperhomocisteinemia secundaria a enfermedades autoinmunitarias
7. Inhibidores de factores de coagulación
Los anticuerpos antifosfolípidos aluden a un grupo de autoanticuerpos que sedirigen contra una amplia variedad de fosfolípidos. Los sucesos que precedierona la integración del síndrome fueron:
1. En el siglo pasado la prueba de Wasserman fue ampliamente utilizada parael diagnóstico de sífilis. Se observó que en pacientes con lupus eritematososistémico (LES) en ocasiones resultaba la prueba positiva en ausencia desífilis; a dicho hallazgo se le otorgó una gran importancia, al grado que du-rante un tiempo se utilizó como un criterio de diagnóstico para LES.
2. En 1952 Conley y Hartmann reportaron dos pacientes con LES que teníanelevación del tiempo de protrombina y describieron un inhibidor de la coa-gulación que interfiere con la conversión de protrombina en trombina.
3. En 1963 Bowie y col. describieron a ocho pacientes con LES y evidenciade un “anticoagulante circulante”, de los cuales cuatro presentaron eventostromboembólicos, lo que permitió documentar por vez primera la asocia-ción paradójica de tiempos de protrombina prolongados con eventos trom-bóticos.
4. En 1972 Feinstein y Rapaport acuñaron el término de anticoagulante lúpico(AL), al reportar a dos pacientes con LES y pruebas de laboratorio que suge-rían un efecto anticoagulante.
5. Posteriormente Graham Hughes realizó la vinculación del anticoagulantelúpico y pruebas seroluéticas falsas positivas en pacientes con abortos es-pontáneos y pérdidas fetales.
6. Posteriormente, mediante el desarrollo de anticuerpos monoclonales, se de-mostró que los anticuerpos antifosfolípidos se dirigen contra los fosfolípi-dos que tienen una carga aniónica (y a veces switeriónica).
7. Finalmente, Nigel Harris y col. desarrollaron por medio de radioinmunoa-nálisis de fase sólida el método para la detección de anticuerpos anticardio-lipina en pacientes con LES y complicaciones trombóticas, que posterior-mente demostró una fuerte correlación entre los niveles de anticuerposanticardiolipina con trombosis venosa y arterial.1,3–5

305Enfermedad inmunitaria y trombosis. Síndrome de anticuerpos...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
EPIDEMIOLOGÍA
La prevalencia de anticuerpos antifosfolípidos en la población sana es de 1 a 5%,con riesgo de trombosis menor de 1%; sin embargo, en mujeres con historia depérdida fetal es superior a 10%,6 en mujeres jóvenes con infarto cerebral se hainformado una frecuencia de 17%7 y en pacientes con LES ha sido de 30 a 40%.Cuando se asocia a otras condiciones trombogénicas el riesgo de trombosis se in-crementa en forma significativa; se han encontrado riesgos de OR tan elevadoscomo de 201 en casos de consumo de anticonceptivos y de 87 cuando se asociaa tabaquismo.8
MECANISMOS DE LA TROMBOFILIA
Los estudios in vivo han demostrado que los anticuerpos antifosfolípidos (aFL)aumentan la formación de trombos en la circulación venosa y arterial; a pesar desu nombre, los antifosfolípidos no interaccionan directamente con los fosfolípi-dos; su objetivo es la �2–glucoproteína I(�2GPI), una proteína plasmática congran afinidad por los fosfolípidos aniónicos. Los complejos aFL–�2GPI son ca-paces de interferir en un gran número de procesos fisiológicos; sin embargo, nose conocen con exactitud los mecanismos trombogénicos que operan en el SAF,pero diversos estudios han demostrado una serie de alteraciones; entre las másimportantes se encuentran:3–5
1. Activación de células endoteliales. La unión de los anticuerpos antifosfo-lípidos con las células endoteliales activa a estas últimas, las cuales secretanmoléculas proinflamatorias, como selectina E, moléculas de adhesión celu-lar (ICAM–1), moléculas de adhesión vascular (VCAM–1) y moléculas deadhesión plaquetaria (PECAM–1), además de que aumenta el factor activa-dor de plaquetas.
2. Incremento del estrés oxidativo y oxidación del endotelio.3. Modificación de la producción endotelial de prostaglandinas. Existe
una disminución de la producción de prostaciclina (PGI–2), cuyo efecto esvasodilatador y antiagregante plaquetario, así como un aumento de trombo-xano A2 (TXA–2), que tiene efectos contrarios: vasoconstrictor y agregan-te plaquetario.
4. Aumento en la activación y la agregación plaquetarias. Los anticuerposantifosfolípidos inhiben la antitrombina, se unen y activan las plaquetas, yaumentan la expresión de glucoproteínas de la membrana plaquetaria,como las glucoproteínas GPIIb y GPIIIa.

306 (Capítulo 19)Tratado de trombosis
5. Disfunción de la �2GPI. Esta glucoproteína se une fisiológicamente a losfosfolípidos con carga negativa y es un regulador de la coagulación; los an-ticuerpos en su contra impiden su función hemostática normal. Además, la�2GPI se expresa en el tejido placentario, la unión con su anticuerpo resultaen inhibición del crecimiento y diferenciación del trofoblasto, al produciruna placenta defectuosa, incremento de citocinas inflamatorias y activacióndel complemento, causas de disfunción placentaria y pérdidas fetales.
6. Inhibición de la activación de proteínas C y S generada por interferen-cia con la trombomodulina.
7. Activación del complemento. La activación de las células endoteliales tam-bién resulta en el incremento de la síntesis de factor tisular (FT), el iniciadormás potente de la cascada de la coagulación in vivo. Los estudios en modelosmurinos otorgan un papel muy relevante a la activación del sistema delcomplemento en la producción de trombosis y de pérdidas embriofetales.
8. Disminución de la producción de anexina V. Proteína intracelular, tam-bién conocida como proteína anticoagulante placentaria 1, que se une a losfosfolípidos aniónicos expuestos en la superficie celular que la torna notrombogénica.
9. Reacción cruzada con glucosaminoglucanos. La unión de anticuerposantifosfolípidos con los glucosaminoglucanos origina una pérdida de laspropiedades antitrombóticas del endotelio.2,9
De esta forma, los tres elementos implicados principalmente en la génesis delSAF son las disfunciones endotelial, plaquetaria y del complemento, cuya conju-gación produce un estado procoagulante a nivel de la hemostasia primaria (pla-quetas) y secundaria (cascada de la coagulación). La activación del complementocierra el circuito con el resultado final de trombosis y pérdida del embarazo.5,10
PRUEBAS PARA EL DIAGNÓSTICO
Los anticuerpos antifosfolípidos están dirigidos contra fosfolípidos con carga ne-gativa, así como contra las proteínas fijadoras de los fosfolípidos, los cofactoreso una combinación de condiciones. Antes se denominaba anticuerpos antifosfolí-pidos verdaderos a los anticuerpos anticardiolipina, los anticuerpos antifosfati-dilserina y los anticuerpos antifosfatidiletanolamina, mientras que a los anticuer-pos que para su detección requieren una proteína que actúa como cofactor (�2glucoproteína I, protrombina, factor X) se les llamaba seudoantifosfolípidos.
Los anticuerpos que se detectan con más frecuencia en la práctica clínica sonel anticoagulante lúpico, los anticuerpos anticardiolipina y los anticuerpos contrala �2 glucoproteína I (�2GPI).3,11,12

307Enfermedad inmunitaria y trombosis. Síndrome de anticuerpos...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Los anticuerpos antifosfolípidos se pueden identificar mediante las siguientespruebas:
a. VDRL falsa positiva.b. Determinación de anticoagulante lúpico.c. Anticuerpos anticardiolipina.
Las tres pruebas tienen especificidad y sensibilidad diferentes; los anticuerposque detectan son distintos y se dirigen contra diferentes epítopes.
a. La prueba menos específica es la de VDRL, ya que es indispensable que elsujeto reaccione en forma negativa a las pruebas de Treponema pallidum.
b. El anticoagulante lúpico constituye anticuerpos con diferentes especifici-dades a fosfolípidos y puede tratarse de anticuerpos IgG o IgM que interfie-ren con las reacciones de coagulación que dependen de fosfolípidos.8 Laspruebas para determinar anticoagulante lúpico evalúan la capacidad de losanticuerpos antifosfolípidos para prolongar el tiempo parcial de trombo-plastina activado (TTPa) mediante la inhibición de la conversión de pro-trombina o por activación del factor X. La Sociedad Internacional de Trom-bosis y Hemostasia (The International Society on Thrombosis andHemostasis, ISTH) y el Subcomité de Estandarización Científica de Anti-coagulante Lúpico/Anticuerpos Antifosfolípidos (Lupus Anticoagulant/An-tiphospholipid Antibody Scientific and Standardization Committee, SSC)destacaron la importancia de contar con una determinación de anticoagu-lante lúpico estandarizada, por lo que en 1991 publicaron cuatro recomenda-ciones para su identificación, las cuales se han actualizado en 1995 y 2009.1. Prolongación de un ensayo de coagulación dependiente de fosfolípidos.
Se sospecha la presencia de anticoagulante lúpico cuando existe un alar-gamiento del TTPa, del tiempo de Russell o del tiempo de coagulacióncon caolín, por lo que se deben realizar por lo menos dos de estas pruebas.
2. Pruebas de actividad inhibidora con mezcla de plasma normal. La pruebase debe repetir añadiendo plasma normal en una relación de 1:1; si laprueba se normaliza se debe interpretar como deficiencia de factores decoagulación; por el contrario, si los tiempos permanecen prolongados seinterpretará como una prueba positiva para anticoagulante lúpico o paraalgún otro inhibidor endógeno de la coagulación.
3. Evidencias de que la actividad inhibidora es dependiente de fosfolípidos.La presencia de anticoagulante lúpico se confirma con la normalizaciónde la prueba de coagulación anormal al añadir plaquetas o plasma conexceso de fosfolípidos, los cuales se unirán a los anticuerpos.
4. Distinción de otras coagulopatías que pueden dar resultados similares.5. Los anticuerpos anticardiolipina detectan tres isotipos de inmunoglobu-

308 (Capítulo 19)Tratado de trombosis
linas (IgG, IgM e IgA) que se pueden detectar por radioinmunoanálisisde fase sólida o mediante la prueba de inmunoabsorbencia ligada a enzi-mas (técnica de ELISA). Cuando se utiliza radioinmunoanálisis de fasesólida los títulos diagnósticos son � 20 UI. La prueba con ELISA varíade acuerdo con el laboratorio, por lo que se requiere estandarizarla encada uno de ellos y considerarla positiva si los títulos son mayores de dosdesviaciones estándar, siendo el isotipo IgG el que se presenta con mayorfrecuencia.13,14
MANIFESTACIONES CLÍNICAS
El SAF primario y el secundario son más comunes en el sexo femenino. Más de60% de los pacientes presentan fenómenos trombóticos y el porcentaje más altolo ocupan las trombosis venosas profundas de las extremidades inferiores(38.9%).12 Las trombosis venosas pueden surgir en cualquier vena, pero es fre-cuente la tromboembolia pulmonar, y hay informes de hipertensión arterial pul-monar por tromboembolias de repetición no diagnosticadas. Las trombosis arte-riales más comunes ocurren en el cerebro (20 a 30%), pero se pueden producir,igual que las trombosis venosas, en cualquier territorio arterial. Los infartos cere-brales pueden carecer de síntomas, pero ser repetitivos y conducir a demencia porinfartos múltiples.16
A pesar del gran interés que despierta este síndrome, ha sido controvertido de-finir los criterios de inclusión para pacientes en estudios clínicos. En 1999 enSapporo, en el marco de un consenso internacional, se establecieron criterios parala inclusión de pacientes, lo que contribuyó de manera significativa a establecerel peso de los datos experimentales obtenidos de los distintos estudios clínicosy proporcionó un enfoque diagnóstico más completo para el SAF.1,14,15 En 2004,durante el Congreso Internacional de Anticuerpos Antifosfolípidos, celebrado enSidney, se acordó una modificación de los criterios de Sapporo, que fue publicadaen 2006. Cabe recordar que dichos criterios fueron diseñados originalmente paraestudios clínicos y no para uso diagnóstico, por lo que en un futuro podrían cam-biar para su uso clínico rutinario.2,5
Los criterios actualizados y revisados para diagnosticar el SAF se incluyen enel cuadro 19–2.
Además de las manifestaciones clínicas incluidas en los criterios de clasifica-ción para el síndrome, también pueden surgir otras alteraciones que pueden aler-tar al clínico a sospechar el diagnóstico; entre ellas se encuentran1 la valvulopatíacardiaca, la trombosis glomerular, la corea, la epilepsia, la migraña, la livedo reti-cularis, la mielitis transversa, la polirradiculoneuritis y la neuritis óptica. Recien–

309Enfermedad inmunitaria y trombosis. Síndrome de anticuerpos...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 19–2. Criterios de clasificación para síndrome antifosfolípidos
Criterios clínicos1. Trombosis vascular. Uno o más episodios de trombosis arterial o venosa, o ambas, en
cualquier órgano o tejido, confirmados por estudios de imagen o histopatología (exceptotrombosis venosa superficial)Clasificar a los pacientes con síndrome antifosfolípidos con presencia o ausencia de fac-
tores de riesgo adicionales, como pacientes con un primer episodio de infarto agudodel miocardio o evento vascular cerebral después de los 55 (hombres) o 65 años deedad (mujeres) en presencia de cualquiera de los factores de riesgo establecidos paraenfermedad cardiovascular; pacientes con neoplasias y pacientes con otros factoresimportantes de riesgo de trombosis (inmovilización prolongada, etc.). Las trombofiliascongénitas también deben ser consideradas
2. Morbilidad obstétrica:a. Una o más muertes fetales (> 10 semanas de gestación) de fetos morfológicamente
normales, documentadas por ultrasonido o por examen directo del fetob. Uno o más nacimientos prematuros (< 34 semanas de gestación) de neonatos morfoló-
gicamente normales debido a eclampsia o preeclampsia severa o insuficiencia placen-taria severa (peso de la placenta < del percentil 10 e infartos placentarios que afectan >20% de la placenta)
c. Tres o más abortos tempranos (< 10 semanas de gestación) excluyendo causas mater-nas anatómicas u hormonales y causas paternas y maternas cromosómicas
Se recomienda clasificar en los estudios clínicos a los pacientes en los tipos 2a, 2b o 2cCriterios de laboratorio
1. Anticuerpos anticardiolipina aCL isotipo IgG o IgM, o todos, título moderado o alto (> 40GPL o MPL, o > del percentil 99), en al menos dos ocasiones separadas más de 12 se-manas y realizadas mediante técnica de ELISA estandarizada
2. AL en al menos dos oportunidades separadas más de 12 semanas (criterios de ISTH)3. Anticuerpos anti–�2GPI isotipo IgG o IgM, o ambos (títulos > del percentil 99) en al menos
dos oportunidades separadas más de 12 semanas, mediante técnica de ELISA estandari-zada
Categorizar a los pacientes de acuerdo con el tipo de aFL presente:I: cualquier combinación de aFLIIa: sólo ALIIb: sólo aCLIIc: sólo anti–�2GPI
AL: anticoagulante lúpico; ISTH: The International Society on Thrombosis and Hemostasis.
temente se describieron lesiones cutáneas con pérdida del tejido elástico, deno-minadas anetoderma (piel “aguada”).4 Las mujeres presentan con más frecuenciaartritis, livedo reticularis y migraña, mientras que los hombres padecen infartodel miocardio, epilepsia y trombosis arterial de las extremidades.15
Existe una variante que se presenta en sólo 1% de los casos; se trata de una for-ma muy grave de curso acelerado, con falla multiorgánica y elevada mortalidad,conocida como síndrome de anticuerpos antifosfolípidos catastrófico (tambiénconocido como CAPS, por sus siglas en inglés). Se caracteriza por trombosismúltiples que se producen en un tiempo reducido y afectan de forma predominan-

310 (Capítulo 19)Tratado de trombosis
te los vasos de pequeño calibre. Debido a sus particulares características clínicas,se han propuesto criterios de clasificación que incluyen evidencia de afección detres o más órganos o tejidos, evolución clínica en una semana o menos, confirma-ción histopatológica de trombosis de pequeños vasos y presencia de aFL. Si se cum-plen las cuatro características se determina que el paciente presenta CAPS.5,17,18
TRATAMIENTO
El tratamiento del SAF tiene la condición de que, de acuerdo con la manifestaciónu objetivo que se presente o persiga, puede incluir antiagregantes plaquetarios,anticoagulantes o ambos; es decir, puede estar dirigido a trombo venoso o a trom-bo arterial. Por otro lado, la variabilidad clínica complica su manejo, ya que suhistoria natural es variable, las recurrencias sin tratamiento son frecuentes y tien-den a repetirse en los mismos territorios.5
El tratamiento se divide en dos etapas: el agudo y el que se lleva a largo plazo.El manejo agudo se lleva a cabo con anticoagulación con heparinas (no fraccio-nada y de bajo peso molecular), con continuación con anticoagulantes por víaoral. No obstante, de acuerdo con el sitio de la trombosis existen variantes tera-péuticas, por ejemplo, el empleo de fármacos antiagregantes plaquetarios, blo-queadores de los receptores de glucoproteína plaquetaria IIb/IIIa y endocánulasvasculares en casos de infarto agudo del miocardio. El tratamiento a largo plazoaún es controvertido, debido a que los estudios han mostrado resultados inconsis-tentes. Una meta del tratamiento es evitar nuevos eventos trombóticos. A conti-nuación se comentan los aspectos más importantes.
1. Duración de la anticoagulación. En varios estudios se ha demostrado quelos pacientes con títulos altos de anticardiolipinas y condiciones de riesgopara trombosis tienen un riesgo elevado de sufrir nuevos episodios trombó-ticos, por lo que se recomienda la anticoagulación durante un tiempo pro-longado, a pesar de que aún se carece de estudios que permitan recomendarsi el tratamiento debe ser indefinido o debe tener un curso perentorio. Lapostura más razonable es mantener la anticoagulación si las condiciones delpaciente lo permiten, es decir, buen apego al tratamiento, ausencia de con-traindicaciones y bajo riesgo de hemorragia.
2. Intensidad de la anticoagulación. Para prevenir nuevos episodios detrombosis se recomendó durante algunos años mantener altos los niveles deanticoagulación con cociente normalizado internacional mayor de 3 (INR3 a 4); posteriormente una corriente de opinión entre autores de gran pesodefendió que los pacientes con SAF y trombosis de mayor riesgo (arteriales

311Enfermedad inmunitaria y trombosis. Síndrome de anticuerpos...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
y recidivantes) debían recibir un tratamiento más intenso que aquellos queiniciaban con una trombosis venosa profunda; no obstante, esta práctica seestá abandonando debido al riesgo de hemorragia, a que la recomendaciónse sustentaba en ensayos clínicos con ciertas limitaciones metodológicas ya que en al menos dos estudios con mejor diseño no se logró demostrar laprevención de los eventos trombóticos. En la última guía de tratamientopara el SAF, publicada en 2012, se recomienda mantener como objetivo te-rapéutico un INR en rangos de 2.0 a 3.0; sin embargo, el estado clínico indi-vidual del paciente ayudará a tomar la decisión terapéutica.19,20
3. Prevención primaria. En este rubro tampoco se cuenta con estudios quepermitan hacer recomendaciones absolutas. En general, se recomienda elácido acetilsalicílico en dosis bajas (75 a 100 mg/día), durante un tiempoindefinido, en los pacientes que cursan de manera persistente con títulos al-tos de anticardiolipina y anticoagulante lúpico. En un ensayo clínico recien-te no se demostró que dicho tratamiento fuera superior al placebo, pero elestudio presentó limitaciones metodológicas, y por el momento no es posi-ble efectuar una recomendación definitiva.13
SÍNDROME ANTIFOSFOLÍPIDOS EN EL EMBARAZO
El vínculo del SAF con el embarazo es muy estrecho, al grado que las pérdidasfetales forman parte de los criterios de diagnóstico. La morbilidad del SAF duran-te el embarazo incluye los periodos preembriónico, fetal y neonatal. El periodopreembriónico abarca desde la concepción hasta la cuarta semana de gestación,el periodo embriónico desde la quinta hasta la novena semanas y el periodo fetaldesde la décima semana hasta la resolución del embarazo. La asociación de anti-cuerpos antifosfolípidos con la pérdida del embarazo se reconoció desde hacetiempo, pero hasta el momento no se ha establecido con precisión qué complica-ción es la más común; la mayoría de las series indican que la muerte fetal es lacomplicación más frecuente en el segundo y el tercer trimestres.
Además, se pueden presentar preeclampsia, insuficiencia placentaria (por mi-croangiopatía y trombosis), parto pretérmino y tromboflebitis materna en el puer-perio.10
El tratamiento del SAF durante el embarazo pretende mejorar la morbimortali-dad maternofetal; las propuestas terapéuticas más importantes son el empleo deácido acetilsalicílico (AAS) y heparina; los glucocorticoides y la inmunoglobu-lina no han mostrado ser superiores al tratamiento antiagregante/antitrombóticoen SAF obstétrico.
Se ha encontrado que las dosis bajas de AAS son capaces de prevenir la muertefetal mediante la inhibición de la ciclooxigenasa plaquetaria sin afectar la síntesis

312 (Capítulo 19)Tratado de trombosis
de prostaglandinas. Por otra parte, en un estudio clínico controlado se encontróque la frecuencia de nacidos vivos aumentó significativamente cuando se agregóheparina al tratamiento con AAS. Sin embargo, el esquema óptimo de combina-ción de ambos fármacos tampoco está definitivamente establecido. Aunque la re-comendación más aceptada es la de asociar Aspirina� y heparina en todas las pa-cientes con morbilidad obstétrica previa, los resultados de los dos únicos ensayosclínicos realizados son discordantes.
En general, para las mujeres con SAF con pérdida recurrente del embarazo (>3) se recomienda la administración prenatal de heparina en combinación con do-sis bajas de Aspirina�, iniciando el tratamiento tan pronto como se confirme elembarazo. Para las mujeres con SAF y antecedentes de preeclampsia o insufi-ciencia placentaria se recomiendan dosis bajas de Aspirina�, mientras que parael posparto se debe considerar la tromboprofilaxis.13,18
El SAF es una de las entidades que más atención han recibido en los últimosaños en el campo de las enfermedades inmunitarias y la trombosis, aunque conti-núa envuelto en una constante polémica en relación con su patogenia, manifesta-ciones clínicas, diagnóstico y tratamiento, por lo que las investigaciones futurasseguramente proporcionarán datos más específicos que permitirán enfrentar me-jor a la enfermedad.
REFERENCIAS
1. Ortel TL: Antiphospholipid syndrome: laboratory testing and diagnostic strategies. Am JHematol 2012;87(Suppl 1):S75–S81.
2. Gardiner C, Hills J, Machin SJ: Diagnosis of antiphospholipid syndrome in routine clini-cal practice. Lupus 2013;22:18–25.
3. Huerta E, Ariza ACR: Síndrome antifosfolípidos. En: Ariza ACR (ed.): Medicina internaen la mujer. México, McGraw–Hill, 2004:125–144.
4. Shoenfeld Y, Khamastha M (eds.): Antiphospholipid syndrome. Lupus 2003;12:495–573.5. Ruiz IG, Martínez BA, Egurbide MA: The antiphospholipid syndrome in the 21st century.
Med Clin (Barc) 2009;133(10):390–396.6. Shekelle PG, Woolf SH, Eccles M, Grimshaw J: Clinical guidelines: developing guide-
lines. Br Med J 1999;318:593–596.7. Urbanus RT, Siegerink B, Roest M, Rosendal FR, de Groot PG et al.: Antiphospholipid
antibodies and risk of myocardial infarction and ischaemic stroke in young women in theRATIO study: a case–control study. Lancet Neurol 2009;8:998–1005.
8. Giannakopoulos B, Krilis S: The pathogenesis of antiphospholipid syndrome. N Engl JMed 2013;368:1033–1044.
9. Li Z, Krilis SA: Anti–beta(2)–glycoprotein I antibodies and the antiphospholipid syndro-me. Autoimmun Rev 2003;2(5):229–234.
10. Galarza MC, Kourilovitch MR, Pérez FOM et al.: Obstetric antiphospholipid syndrome.Autoimmun Rev 2012;11(4):288–295.
11. Teruya J, West AG, Suell MN: Lupus anticoagulant assays: questions answered and to beanswered. Arch Pathol Lab Med 2007;131(6):885–889.

313Enfermedad inmunitaria y trombosis. Síndrome de anticuerpos...E
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
12. Cabral AR, Amigo MC, Cabiedes J, Alarcón SD: The antiphospholipid/cofactor syndro-mes: a primary variant with antibodies to beta 2–glycoprotein–I but no antibodies detectablein standard antiphospholipid assays. Am J Med 1996;101(5):472–481.
13. Keeling D, Mackie I, Moore GW et al.: Guidelines on the investigation and managementof antiphospholipid syndrome. Br J Haematol 2012;157(1):47–58.
14. Baker WF, Bick RL, Fareed J et al.: Controversies and unresolved issues in antiphospholi-pid syndrome pathogenesis and management. Hematol Oncol Clin N Am 2008;22:155–174.
15. Cervera R, Piette JC, Font J et al.: Antiphospholipid syndrome: clinical and immunologicmanifestations and patterns of disease expression in a cohort of 1 000 patients. ArthritisRheum 2002;46(4):1019–1027.
16. Biggioggero M, Meroni PL: The geoepidemiology of the antiphospholipid antibody syn-drome. Autoimmun Rev 2010;9(5):A299–A304.
17. Asherson RA, Cervera R, de Groot PG et al.: Catastrophic antiphospholipid syndrome:international consensus statement on classification criteria and treatment guidelines. Lupus2003;12(7):530–534.
18. Gómez PJA, Cervera R, Espinosa G et al.: Pregnancy and puerperium are high suscepti-bility periods for the development of catastrophic antiphospholipid syndrome. AutoimmunRev 2006;6(2):85–88.
19. Miyakis S, Lockshin MD, Atsumi T et al.: International consensus statement on an updateof the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Hae-most 2006;4(2):295–306.
20. Khamashta MA, Cuadrado MJ, Mujic F et al.: The management of thrombosis in the an-tiphospholipid–antibody syndrome. N Engl J Med 1995;332(15):993–997.

314 (Capítulo 19)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
20Embarazo y trombosis
Luis Fernando García–Frade Ruiz
El embarazo constituye un factor de riesgo para la enfermedad tromboembólica,el cual confiere un aumento de tres a cinco veces el riesgo de trombosis venosaprofunda (TVP) en relación con el estado de no gravidez. La tromboembolia pul-monar (TEP) constituye una causa importante de muerte materna en los paísesdesarrollados. La enfermedad tromboembólica representa 11% de las causas demuerte materna en EUA y es la causa más común en Reino Unido.
Los cambios fisiológicos que ocurren durante el embarazo colocan a la mujeren un riesgo particularmente alto de enfermedad trombótica (cuadro 20–1). Elestado de hipercoagulabilidad que se presenta en las pacientes es resultado delincremento en los niveles de fibrinógeno y de varios factores de la coagulación(II, VII, VIII, IX, X y XII), así como disminución de los niveles de los anticoagu-lantes naturales, como la proteína S. En el embarazo normal se presenta tambiénuna resistencia adquirida a la proteína C y una disminución en la fibrinólisis. Estaalteración en el sistema de la coagulación persiste a lo largo del embarazo y hastaseis semanas después del parto.
La tríada clásica de Virchow —hipercoagulabilidad, estasis venosa y dañovascular— se presenta durante el embarazo no complicado y el parto, en el cualexiste una importante disminución en la velocidad del flujo venoso en los miem-bros inferiores, con una reducción aproximada de 50% hacia las semanas 25 a 29de la gestación, alcanzando su menor velocidad en la semana 36 y regresando asus rangos normales hasta las seis semanas después del parto. Además, algún gra-do de daño endotelial vascular o trauma a los vasos pélvicos son inevitables du-rante el parto vaginal o abdominal.
315

316 (Capítulo 20)Tratado de trombosis
Cuadro 20–1. Cambios hemostáticos durante el embarazo
Condiciones que promueven la trombosis Condiciones que disminuyen la trombosis
Activación de los factores II, VII, VIII, IX, X y XII Expansión del volumen plasmático
Disminución de la actividad fibrinolítica Disminución de los factores XI y XIIIResistencia adquirida a la proteína CTrombofilia hereditaria
Neutralización de la trombina por la antitrom-bina
Anticuerpos antifosfolípidosDaño endotelial asociado con el partoEstasis venosa de las extremidades inferiores
TROMBOFILIA Y TROMBOEMBOLISMO VENOSORELACIONADO CON EL EMBARAZO
La presencia de trombofilia constituye un importante riesgo adicional para el de-sarrollo de trombosis durante el embarazo.
Algunas de las formas de trombofilias hereditarias o adquiridas contribuyena más de la mitad de los eventos tromboembólicos maternos. En adición al trom-boembolismo materno, la trombofilia se encuentra asociada a un aumento en laprevalencia de múltiples efectos adversos, como abortos, restricción del creci-miento intrauterino, preeclampsia y muerte intrauterina que resultan de la trom-bosis de la circulación uteroplacentaria.
Múltiples desórdenes trombofílicos se han asociado con desórdenes trombóti-cos durante el embarazo, como la mutación del factor V de Leiden, la mutacióndel gen de protrombina G20210A, la deficiencia de proteínas C y S, la deficienciade antitrombina, la hiperhomocisteinemia y el síndrome de anticuerpos antifos-folípidos.
El factor V de Leiden se identifica en 20 a 40% de las pacientes embarazadasque presentan trombosis venosa. La resistencia a la proteína C activada tambiénpuede ser causada por otras alteraciones distintas al factor V de Leiden, incluyen-do el síndrome de anticuerpos antifosfolípidos y otros defectos genéticos en lamolécula del factor V, como el factor V de Cambridge o el haplotipo HR2. Noobstante, el factor V de Cambridge es poco común, el haplotipo HR2 es frecuentey proporciona un alto riesgo de trombosis venosa en las pacientes que lo presen-tan.
Al menos 30% de los episodios trombóticos que involucran la mutación delfactor V de Leiden ocurren durante el puerperio. Se requieren más estudios paraestablecer la relación entre la mutación del factor V con el desarrollo de compli-caciones obstétricas severas; sin embargo, se estima que ante la presencia de lamutación y del embarazo se incrementa ocho veces el riesgo de complicaciones

317Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
tromboembólicas. El riesgo absoluto se calcula en aproximadamente uno de 500casos en presencia de la mutación del factor V de Leiden.
Hasta 40% de las mujeres presentan una resistencia adquirida a la proteína Cactivada durante el embarazo, lo cual se relaciona con un incremento de los facto-res V y VIII.
La variante en el gen de protrombina G20210A se encuentra presente en 2%de la población europea. Dicho gen suele estar presente en aproximadamente 6%de las pacientes que cursan con enfermedad tromboembólica venosa durante elembarazo. El riesgo absoluto se calcula en uno de 200 casos en presencia de lamutación del gen y en 4.6 de 100 casos que además presentan la mutación del fac-tor V de Leiden. La mutación del gen de protrombina se ha reportado en al menos20% de los casos que cuentan con historia familiar de enfermedad trombótica.
Existe un alto riesgo de tromboembolismo durante el embarazo y el puerperioen la mujer que padece deficiencia de proteína C, proteína S o antitrombina.
La deficiencia de la proteína S participa en la génesis de la trombosis de mane-ra especial durante el puerperio. El riesgo calculado de trombosis en una mujerembarazada con deficiencia de antitrombina tipo I es de 1 por cada 2.8 casos, detipo II es de 1 por cada 42 y de deficiencia de proteína C es de 1 por cada 113casos.
La mutación de metiltetrahidrofolato reductasa (MTHFR C677T) se asociacon la presencia de hiperhomocisteinemia. Este genotipo por sí mismo no se en-cuentra directamente relacionado con trombosis venosa, pero predispone al desa-rrollo de trombosis arterial y venosa en coexistencia con deficiencia de vitaminaB. El genotipo interactúa con la deficiencia de vitamina B, lo que provoca hiper-homocisteinemia. Aproximadamente 10% de la población europea es homocigo-ta para esta variante genética. La MTHFR no parece incrementar el riesgo de en-fermedad tromboembólica venosa durante el embarazo, quizá por la disminuciónfisiológica de los niveles de homocisteína o bien por los efectos de la administra-ción de ácido fólico. La asociación específica entre la mutación de la trombomo-dulina y trombosis durante el embarazo aún no se ha establecido.
SÍNDROME DE ANTICUERPOS ANTIFOSFOLÍPIDOS
La presencia de anticoagulantes circulantes adquiridos fue descrita por primeravez por Conley en 1948. Quince años después Bowie describió el fenómeno detrombosis en pacientes con lupus eritematoso sistémico (LES) y la presencia deanticoagulantes circulantes. Los anticuerpos antifosfolípidos (APLA) se encuen-tran en un rango de 1 a 5% en la población sana y son mucho más comunes enlos pacientes que padecen desórdenes autoinmunitarios, como el LES. La inci-

318 (Capítulo 20)Tratado de trombosis
dencia de APLA en la población obstétrica de bajo riesgo es de aproximadamente2%, en comparación con una incidencia en embarazos complicados, que es de 11a 61%. El síndrome incluye no sólo el anticoagulante lúpico y los anticuerpos an-ticardiolipina (ACLA), sino también los más recientes subgrupos de anticuerposreconocidos como los anticuerpos contra beta–2–glucoproteína–I, anticuerposcontra fosfatidilserina, fosfatidiletanolamina, fosfatidilglicerol, fosfatidilinosi-tol, fosfatidilcolina y anti–annexin V (proteína anticoagulante placentaria). Laprevalencia de anticuerpos antifosfolípidos aumenta con la edad, de manera par-ticular en aquellos con enfermedades crónicas. Los eventos trombóticos asocia-dos con la presencia de APLA incluyen trombosis tanto del sistema venoso comodel arterial, trombosis coronaria, trombosis cerebrovascular, isquemia cerebraltransitoria, trombosis de la retina y trombosis placentaria. Los criterios de diag-nóstico del síndrome de anticuerpos antifosfolípidos (APS) se revisan en el capí-tulo correspondiente.
Las complicaciones durante el embarazo asociadas con APLA incluyen pérdi-da recurrente del embarazo, preeclampsia, restricción en el crecimiento intraute-rino y trombosis. Las características clínicas del síndrome en orden de frecuenciason:
1. Abortos frecuentes en el primer trimestre del embarazo secundarios a trom-bosis placentaria o vasculitis.
2. Pérdida fetal recurrente en el segundo o el tercer trimestres.3. Trombocitopenia materna.
Aproximadamente 30% de las mujeres con APS presentan trombosis venosa du-rante el embarazo.
La presencia de ACLA también se encuentra asociada con un síndrome pos-parto peculiar que consiste en la presencia de picos febriles, dolor torácico de tipopleurítico, disnea y derrame pleural, infiltrados pulmonares en parche, cardio-miopatía y arritmias ventriculares, el cual de manera característica se presentadentro de los primeros 10 días posparto. La mayoría de las pacientes afectadasse recuperan de manera espontánea, por lo que no se requiere más que terapia sin-tomática.
La paciente que presenta ACLA tiene entre 50 y 75% de probabilidades de pér-dida fetal, mientras que a través del manejo anticoagulante óptimo se puede in-crementar la probabilidad de alcanzar un parto a término normal de aproximada-mente 97%.
Las recomendaciones para el manejo del APS durante el embarazo se encuen-tran asociadas con un riesgo significativo tanto para la madre como para el feto.Como resultado, la paciente sólo debe ser tratada si existe evidencia de que lascomplicaciones mediadas por APLA exceden los riesgos propuestos por el trata-miento. El uso de corticosteroides se debe reservar para la mujer que presenta

319Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
APS complicado con trombocitopenia clínicamente importante o LES. En las pa-cientes que se encuentran bajo tratamiento con corticosteroides a largo plazo sedebe vigilar el probable desarrollo de complicaciones, como diabetes gestacionalo hipertensión arterial sistémica.
Al inhibir la Aspirina� la formación de tromboxano se reduce el riesgo detrombosis vascular mediada por las plaquetas. La Aspirina� se puede utilizar du-rante el embarazo con mínimas complicaciones tanto para el producto como parala madre. La Aspirina� se puede continuar hasta el parto, y no existe evidenciade que su uso en bajas dosis incremente el riesgo de hemorragia epidural durantelos procedimientos de anestesia local.
El uso de heparina mejora los resultados en las pacientes con la enfermedad.La combinación de bajas dosis de Aspirina� (81 mg/día) con heparina no frac-cionada (HNF) (5 000 U subcutáneas cada 12 h) o heparina de bajo peso molecu-lar (HBPM) mejora los resultados, en comparación con el uso exclusivo de Aspi-rina�, en las mujeres que presentan abortos recurrentes.
La paciente con APS que se encuentra bajo terapia con anticoagulación oraly Aspirina� y que cuenta con el antecedente de trombosis arterial o venosa con-firmada en embarazos previos debe recibir desde el momento de la confirmacióndel embarazo manejo a través de HBPM en lugar de los antagonistas de la vitami-na K, además de que se puede continuar con la administración de Aspirina�. Parareducir el riesgo de sangrado en el momento del parto se recomienda inducir elparto vaginal a las 37 semanas, seguido por la sustitución de HBPM por antago-nistas de la vitamina K tan pronto se alcance la hemostasia. Si la paciente cuentacon el antecedente de trombosis y no es tratada con dosis terapéuticas de antago-nistas de la vitamina K en el momento de la prueba positiva de embarazo se reco-mienda el uso de HBPM en dosis profiláctica a lo largo del embarazo, debido alalto riesgo de trombosis recurrente durante el mismo y el puerperio.
El uso de warfarina se debe evitar durante el embarazo, de manera particulardurante el primer trimestre, por sus efectos teratogénicos. Su uso se puede consi-derar durante el segundo trimestre y de manera temprana en el tercer trimestredel embarazo sólo en las pacientes con contraindicación para recibir manejo conHNF o HBPM; no obstante, el uso de warfarina en estas etapas del embarazo pue-de estar asociado con anormalidades del sistema nervioso central, como atrofiaóptica, retraso mental, retraso en el desarrollo, convulsiones y microcefalia. Eluso de warfarina se debe evitar después del inicio del tercer trimestre debido aque atraviesa la placenta y causa anticoagulación fetal, con el riesgo potencial desangrado fatal durante el parto, incluida la hemorragia del sistema nervioso cen-tral. No se recomienda la administración de inmunoglobulina intravenosa en lapaciente embarazada que padece APS.
En los últimos años se ha sugerido que la trombofilia manifiesta es con fre-cuencia un desorden multigenético. La coexistencia de mutación del factor V de

320 (Capítulo 20)Tratado de trombosis
Leiden con deficiencia de proteína C o S, con deficiencia de antitrombina y muta-ción de protrombina, con mutación de metiltetrahidrofolato reductasa o con anti-cuerpos antifosfolípidos incrementa el riesgo de trombosis.
La presencia de trombofilia por sí sola, aun en el contexto de un estado pro-trombótico como es el embarazo, no necesariamente resulta en un evento trom-bótico. La trombosis clínica es una enfermedad multifactorial que resulta de lainteracción entre factores congénitos y adquiridos, como son los cambios en lahemostasia y en el flujo venoso que se presentan durante el embarazo. El riesgode desarrollar trombosis clínica depende del tipo de trombofilia que se padece,el número de trombofilias presentes, los eventos tromboembólicos previos y losfactores de riesgo adicionales, como son la obesidad y la inmovilización.
La detección general de trombofilia en las mujeres embarazadas no es apoyadapor la literatura y no puede ser recomendada. No obstante, la detección debe serconsiderada en la mujer con antecedentes personales o familiares de tromboem-bolismo venoso, así como en la mujer con complicaciones obstétricas, como sonabortos recurrentes, restricción del crecimiento intrauterino, preeclampsia seve-ra, desprendimiento placentario y muerte fetal intrauterina, que sugieren la pre-sencia de una trombofilia. La detección de trombofilia tiene un valor limitado enla mujer embarazada porque el resultado no modifica el manejo clínico inmedia-to. Si se decide realizar la detección, ésta se debe hacer antes de iniciar la terapiaanticoagulante. De manera alternativa, se puede realizar un mes después de habersuspendido la anticoagulación. La búsqueda de trombofilia durante el embarazonecesita ser interpretada con precaución, ya que los niveles de proteína S dismi-nuyen de manera fisiológica, existe resistencia a la proteína C activada en 40%de los embarazos sin mutación del factor V de Leiden y los niveles de antitrombi-na pueden estar disminuidos en presencia de una enfermedad trombótica aguda.En adición, la coexistencia de condiciones médicas, como el síndrome nefrótico,está asociada a niveles disminuidos de antitrombina y la enfermedad hepáticapuede causar disminución en los niveles de proteínas C y S. La detección de losgenotipos del factor V de Leiden y de la protrombina G20210A pueden ser inter-pretados con seguridad tanto en el transcurso del embarazo como en presenciade enfermedad trombótica aguda.
FACTORES DE RIESGO CLÍNICOS ADICIONALES
Cesárea
La incidencia de trombosis venosa en el periodo posparto es afectada por la víade resolución del parto. La cesárea se complica con trombosis venosa en un por-

321Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
centaje de 0.4 por cada 1 000, en comparación con un porcentaje de 0.173 porcada 1 000 partos vaginales. La mujer mayor de 35 años de edad que se sometea una cesárea de urgencia tiene mayores probabilidades de presentar una trombo-sis venosa en el periodo posparto, aunque no un mayor riesgo de TEP que la mujerque se somete a una cesárea electiva.
Historia previa de trombosis venosaprofunda o tromboembolia pulmonar
Entre 5 y 15% de las mujeres con historia de trombosis durante el embarazo seencuentran en riesgo de eventos recurrentes en embarazos subsecuentes.
Reposo en cama y obesidad
El Royal College of Obstetricians and Gynecologists considera como factores deriesgo adicionales la edad mayor de 35 años, el peso corporal aproximado de 80kg, más de cuatro embarazos, venas varicosas, preeclampsia, cirugía e inmovili-zación por más de cuatro días antes del parto.
Edad materna en el momento del parto
La incidencia de TVP aumenta tanto antes como después del parto en las mujeresmayores de 35 años de edad. Una edad mayor de 35 años se asocia además conun aumento del riesgo de TEP, con una incidencia de 0.4 por cada 1 000 vs. 0.11por cada 1 000 embarazos en mujeres menores de 35 años, por lo que una edadmayor de 35 años duplica tanto el riesgo de TVP como de TEP.
Otros factores de riesgo
Otros factores de riesgo adicionales para el desarrollo de enfermedad trombóticaincluyen estancia prolongada en la sala de labor, inmovilidad después del parto,paraplejía, deshidratación, condiciones inflamatorias e infecciosas, hemorragiaobstétrica mayor, uso de fármacos intravenosos, hiperemesis gravídica y la pre-sencia de condiciones médicas como síndrome nefrótico, anemia de células falci-formes, enfermedades mieloproliferativas o hiperestimulación ovárica.
ESTRATIFICACIÓN DE RIESGOEN EL PUERPERIO QUIRÚRGICO
La estratificación de riesgo para el desarrollo de enfermedad tromboembólica ve-nosa en el periodo posparto en pacientes sometidas a cesárea es la siguiente:

322 (Capítulo 20)Tratado de trombosis
� Bajo riesgo: paciente menor de 35 años de edad, embarazo no complicadoy cirugía cesárea electiva.
� Riesgo moderado: paciente mayor de 35 años de edad, obesidad, presenciade venas varicosas, infecciones recurrentes, inmovilización por más de cua-tro días antes del parto, preeclampsia y cesárea de emergencia.
� Alto riesgo: presencia de tres o más factores de riesgo moderados, enfer-medad trombofílica hereditaria o síndrome de anticuerpos antifosfolípidos,historia familiar o personal de enfermedad tromboembólica venosa, histo-ria de TEP y cesárea–histerectomía.
TROMBOEMBOLISMO VENOSO EN EL EMBARAZO
La enfermedad tromboembólica venosa (ETV) incluye la TVP y la TEP.El embarazo se asocia a un aumento del riesgo de 5 a 10 veces para ETV. La
trombosis venosa ocurre con una frecuencia de 0.5 a 2 por cada 1 000 embarazos.El riesgo relativo de ETV durante las primeras seis semanas posparto es de 10 a20 veces mayor que durante el embarazo. De 15 a 24% de las pacientes embaraza-das que presentan TVP no tratadas desarrollan TEP, con una mortalidad subse-cuente de 15%; las dos terceras partes de estas muertes ocurren en los 30 min si-guientes al evento embólico. La recurrencia de TEP en embarazos subsecuenteses de 4 a 15%.
Los síntomas clásicos de ETV suelen ser menos específicos durante el embara-zo. El edema y el dolor de los miembros pélvicos, disnea, taquipnea, taquicardiay palpitaciones son comunes durante el embarazo normal, por lo que se debemantener una alta sospecha clínica y así realizar el diagnóstico de manera apro-piada.
La TVP suele afectar en la mayoría de los casos el miembro pélvico izquierdo(en 90% de los casos vs. 55% en las mujeres no embarazadas) posiblemente porcompresión de la vena iliaca izquierda.
La TEP se diagnostica con mayor frecuencia en el periodo posparto, de maneraparticular en el puerperio quirúrgico.
Los estudios de gabinete a realizar en una mujer embarazada con sospecha deTEP pueden resultar problemáticos en lo que concierne la exposición fetal a laradiación. La radiación ionizante confiere riesgos tanto teratogénicos como on-cogénicos al producto, por lo que se han examinado los riesgos fetales de los pro-cedimientos radiológicos utilizados en la evaluación de la trombosis venosa.
La combinación de radiografía simple de tórax, gammagrama de ventilación/perfusión (V/Q) pulmonar y angiografía pulmonar provocan una exposición fetalmenor de 0.5 rad, con un riesgo de cáncer en los primeros 10 años de vida de apro-ximadamente 0.2%.

323Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
El estudio de medicina nuclear por sí solo no parece incrementar los efectosadversos durante el embarazo. La resonancia magnética parece no provocar dañofetal a corto plazo; sin embargo, se desconocen los efectos sobre el producto alargo plazo.
La medición del dímero D es una importante herramienta diagnóstica de ETVen las mujeres no embarazadas, aunque en el embarazo normal o complicado au-mentan los niveles de dicho marcador, por lo que carece de valor diagnóstico enestas pacientes. No obstante, una prueba negativa descarta la presencia de ETV.
En caso de sospecha de TVP durante el embarazo se sugiere realizar una ultra-sonografía de miembros pélvicos (sensibilidad 97%, especificidad 94%). Encaso de resultar negativa con baja sospecha clínica quizá no sea necesaria mayorevaluación; sin embargo, algunos expertos recomiendan en estos casos realizarultrasonidos seriados cada siete días para descartar una probable propagación deltrombo. En caso de alta sospecha clínica de TVP las opciones para realizar undiagnóstico definitivo son la venografía o la venografía por resonancia magnéti-ca (alto grado de sensibilidad y especificidad para la detección de trombosis enla vena iliaca vs. ultrasonografía).
Ante la presencia de lumbalgia más edema de miembro pélvico se debe sospe-char la presencia de trombosis de la vena iliaca, por lo que se debe realizar veno-grafía o venografía por resonancia magnética para el diagnóstico definitivo, yaque la ultrasonografía no es sensible para la detección de trombos a ese nivel.
La gammagrafía V/Q es el estudio inicial recomendado después de la radio-grafía simple de tórax en las pacientes con sospecha de TEP. No obstante, al serla TVP y la TEP dos desórdenes muy relacionados y que comparten un mismomanejo inicial, algunos expertos recomiendan realizar ultrasonografía (para evi-tar la exposición fetal a la radiación) como una prueba inicial en la evaluaciónde la paciente embarazada en la que se sospecha TEP. Las pacientes que resultancon estudio ultrasonográfico positivo para TVP deben recibir tratamiento, mien-tras que en aquellas con estudio negativo para trombosis deberán realizarse otrosestudios.
La gammagrafía V/Q es la piedra angular para el diagnóstico de TEP, y se pue-de realizar con seguridad durante el embarazo. El tecnecio 99m se excreta por laleche materna, por lo que si a la paciente se le realiza el estudio posterior al partodeberá utilizar fórmula durante dos días para la alimentación del neonato. En laspacientes que aun con estudio de medicina nuclear persisten sin un diagnósticodefinitivo y la sospecha clínica de TEP es baja se recomienda realizar ultrasono-grafía de miembros pélvicos; por el contrario, si la sospecha es moderada o altase deberá realizar angiografía pulmonar, considerada la regla de oro para el diag-nóstico de TEP.
La tomografía helicoidal ha adquirido popularidad para el diagnóstico de TEPy ha reemplazado inclusive a la medicina nuclear y a la angiografía pulmonar en

324 (Capítulo 20)Tratado de trombosis
algunos casos; sin embargo, algunos expertos consideran que la sensibilidad dela tomografía para la detección de TEP aún es muy baja, sobre todo para la detec-ción de embolismo de pequeños vasos. Falta aún realizarse más estudios para re-comendar la tomografía helicoidal como una prueba inicial útil en las pacientesembarazadas con sospecha de TEP.
La angiografía con gadolinio por resonancia magnética muestra una sensibili-dad de 100% y una especificidad de 95% para TEP; no obstante, el embolismode pequeños vasos puede pasar inadvertido. El uso de gadolinio no ha sido apro-bado durante el embarazo, ya que atraviesa la barrera placentaria y es considera-do un medicamento clase C.
TRATAMIENTO DEL TROMBOEMBOLISMOVENOSO EN EL EMBARAZO
Durante el embarazo se debe considerar la seguridad de la terapia antitrombóticatanto para el producto como para la madre.
La HNF y la HBPM no atraviesan la barrera placentaria, por lo que no causanhemorragia fetal o defectos en el momento del nacimiento; no obstante, se puedepresentar hemorragia uteroplacentaria.
La seguridad en el uso tanto de HNF como de HBPM durante el embarazo haquedado bien establecida. En contraste, los derivados de cumarina atraviesan labarrera placentaria y pueden causar malformaciones y hemorragia fetal, por loque se deben evitar durante el embarazo.
La HBPM es la terapia antitrombótica de elección durante el embarazo, aun-que su alto costo puede ser una limitación en algunos casos; sin embargo, ofrecelas siguientes ventajas: la dosis se establece en relación con el peso corporal dela paciente sin necesidad de monitoreo de los tiempos de coagulación y se relacio-na con una baja incidencia tanto de trombocitopenia como de osteoporosis indu-cidas por heparina.
En el caso de utilizar HNF se recomienda realizar una cuenta plaquetaria demanera periódica durante las tres primeras semanas de terapia, así como adminis-trar suplementos con adecuadas dosis de vitamina D y calcio para disminuir elriesgo de osteopenia.
El tratamiento de la trombosis venosa se debe realizar al menos durante tresa seis meses. Si el evento ocurre en etapas tempranas del embarazo, después deconcluir el tratamiento para la trombosis venosa, se debe continuar con terapiaprofiláctica con heparina no fraccionada o HBPM hasta al menos seis semanasdespués del parto.
La HBPM se debe suspender 24 h antes de la inducción electiva del parto paradisminuir el riesgo de sangrado durante el mismo. Para disminuir el riesgo de he-

325Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
matoma epidural la anestesia se debe realizar 12 h después de la última adminis-tración de HBPM en dosis profiláctica y 24 h después de una dosis de anticoagu-lación total.
Después del parto la heparina puede ser sustituida por anticoagulantes orales.Ni la warfarina ni las heparinas son secretadas por la leche materna.
En el caso de TEP inestable durante el embarazo se requieren más estudiospara establecer la seguridad y la eficacia del uso de agentes trombolíticos, embo-lectomía y colocación de filtro en la vena cava inferior. Algunos reportes indicanque la terapia trombolítica (con tPA) se asocia con una menor incidencia de pérdi-da fetal que la embolectomía (pérdida fetal de 20 a 40%), por lo que esta últimase debe reservar para los casos en que se amenaza la vida de la paciente.
Las indicaciones para la colocación de filtro en vena cava inferior durante elembarazo son las mismas que para la paciente no embarazada: paciente con ETVaguda y que presenta contraindicación para terapia anticoagulante, paciente conepisodio de ETV no obstante la administración de terapia anticoagulante óptimay paciente críticamente enferma en riesgo de embolismo recurrente que pudieraser fatal.
Las recomendaciones establecidas durante la Octava Conferencia de TerapiaAntitrombótica del American College of Chest Physicians (ACCP) para el mane-jo de la enfermedad tromboembólica venosa durante el embarazo son:
� Dosis ajustadas de HBPM a lo largo del embarazo o HNF intravenosa (boloseguido de infusión continua para mantener el TPTa en rango terapéutico)durante al menos cinco días, seguidas de dosis ajustadas de HNF o HBPMpor el resto del embarazo.
� Se recomienda el uso de HBPM sobre la HNF y los antagonistas de la vita-mina K.
� Los anticoagulantes se deben administrar al menos hasta las seis semanasposparto.
� Se recomienda suspender la anticoagulación 24 h antes de la inducción.
En el cuadro 20–2 se muestran las recomendaciones del ACCP para la prevenciónde enfermedad tromboembólica durante el embarazo ante distintas condicionesclínicas y de acuerdo con los siguientes términos:
� Heparina no fraccionada: minidosis de 5 000 IU subcutáneas cada 12 h.Para dosis moderada se requiere dosis ajustada (subcutánea cada 12 h) hastaun nivel de anti–Xa de 0.1 a 0.3 IU/mL. Para dosis ajustada se requiere dosissubcutánea cada 12 h, hasta un rango terapéutico de TPTa.
� Heparina de bajo peso molecular: para dosis profiláctica se recomiendaenoxaparina (40 mg subcutánea cada 24 h), dalteparina (5 000 IU subcutá-nea cada 24 h) o cualquier HBPM ajustada a un nivel anti–Xa de 0.2 a 0.6

326 (Capítulo 20)Tratado de trombosis
Cuadro 20–2. Pacientes con aumento del riesgode enfermedad tromboembólica venosa
Características Recomendación
ETV previa con factor de riesgo transitorio, nofactores de riesgo en ese momento
Vigilancia y anticoagulantes posparto
Si el evento previo fue durante un embarazo,relacionado con estrógenos o se encuen-tran presentes en ese momento factores deriesgo (como obesidad)
Profilaxis anticoagulante prenatal
Pacientes con un episodio idiopático de ETVque no se encuentran recibiendo anticoagu-lación a largo plazo
HBPM profiláctica o minidosis de HNF o dosismoderada de HNF o vigilancia más anticoa-gulantes posparto
Pacientes con episodio previo de ETV y trom-bofilia (confirmada por laboratorio) o fuertehistoria familiar de trombosis y que no reci-ben terapia anticoagulante a largo plazo
Dosis intermedia o profiláctica de HBPM o mi-nidosis o dosis moderada de HNF más anti-coagulantes posparto
En deficiencia de antitrombina, heterocigotopara protrombina G20210A o FVL u homo-cigoto para estas condiciones con historiade ETV
Dosis intermedia profiláctica de HBPM o dosismoderada de HNF
Paciente con dos o más episodios de ETV ymujer que recibe anticoagulantes a largoplazo
Dosis ajustada de HBPM o dosis ajustada deHNF seguida de anticoagulantes a largoplazo posterior al parto
Todas las mujeres con evento previo de ETV Usar medias elásticas antes y después delparto
ETV: enfermedad tromboembólica venosa; HNF: heparina no fraccionada; HBPM: heparina debajo peso molecular; FVL: factor V de Leiden.
IU/mL. Para dosis ajustada se requiere dosis ajustada al peso, por ejemplo,dalteparina en dosis de 200 IU/kg una vez al día o enoxaparina en dosis de1 mg/kg cada 12 h por vía subcutánea.
� Anticoagulantes posparto: warfarina durante cuatro a seis semanas (INR2.0 a 3.0), inicialmente junto con HNF o HBPM.
En el cuadro 20–3 se muestran las recomendaciones para la profilaxis en la pa-ciente embarazada que presenta aumento del riesgo de pérdida del embarazo.
La novena edición del ACCP realiza las siguientes recomendaciones:
� Se recomienda evitar el uso de los nuevos anticoagulantes orales duranteel embarazo.
� No se recomienda el uso rutinario de tromboprofilaxis en las mujeres quese encuentran bajo reproducción asistida.
� En las pacientes que desarrollan síndrome de hiperestimulación ovárica se-vero se recomienda el uso de HBPM profiláctica durante tres meses.

327Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 20–3. Profilaxis en la paciente embarazadacon riesgo aumentado de pérdida del embarazo
Características Recomendación
Mujer con tres o más abortos y mujer conpreeclampsia severa o recurrente o muerteintrauterina inexplicable
Detección de trombofilia congénita y APLA
Pacientes embarazadas con APLA e historiade dos o más pérdidas de embarazo tem-prano o una o más pérdidas de embarazotardío, preeclampsia
Anteparto: Aspirina� más minidosis o dosismoderada de HNF o HBPM profiláctica
Paciente homocigoto para la variante (C677T)de MTHFR
Suplementos de ácido fólico antes de la con-cepción y tan pronto como sea posible du-rante el embarazo
Paciente con trombofilia congénita y abortosrecurrentes, pérdida tardía o en el segundotrimestre, preeclampsia severa o recurrente
Terapia con bajas dosis de Aspirina� más mi-nidosis de heparina o HBPM profiláctica.Anticoagulantes posparto
Paciente con APLA e historia de trombosisvenosa, que de manera común está bajoterapia anticoagulante a largo plazo
Dosis ajustada de HPM o HNF más bajas do-sis de Aspirina�; reiniciar anticoagulación alargo plazo posterior al parto
La paciente con APLA sin historia previa detrombosis o pérdida del embarazo se debeconsiderar de alto riesgo para trombosis ypérdida del embarazo
Vigilancia, minidosis de HNF, HBPM profilácti-ca o bajas dosis de Aspirina� (75 a 162mg/día), o todas
APLA: anticuerpos antifosfolípidos; HNF: heparina no fraccionada; HBPM: heparina de bajo pesomolecular; MTHFR: metiltetrahidrofolato.
Cesárea
Para las mujeres que se someten a cesárea sin factores de riesgo adicionales serecomienda sólo la ambulación temprana, mientras que en aquellas con riesgo in-crementado de ETV se recomienda el uso de HBPM profiláctica o tromboprofila-xis mecánica cuando exista contraindicación para la anticoagulación.
Antecedente de enfermedad tromboembólica venosa
Para todas las mujeres con antecedente de ETV se sugiere profilaxis durante lasprimeras seis semanas posparto con dosis profilácticas o intermedias de HBPMo antagonistas de la vitamina K (AVK) con índice internacional normalizado(INR) de 2.0 a 3.0.
En las pacientes con bajo riesgo de recurrencia de ETV (episodio único porfactor transitorio no relacionado con el embarazo o estrógenos) se recomiendasólo la vigilancia durante la etapa preparto, mientras que en aquellas con riesgomoderado o alto de recurrencia se recomienda el uso de HBPM en dosis profilác-tica o intermedia.

328 (Capítulo 20)Tratado de trombosis
Paciente que ya recibe antagonistas de vitamina K
Para las mujeres embarazadas que reciben AVK de forma crónica se recomiendael uso de HBPM en dosis ajustada o a 75% de la dosis terapéutica durante el em-barazo.
Paciente con trombofilia primaria conocida
Las mujeres embarazadas sin historia previa de ETV que se sabe que son homoci-gotas para la mutación del factor V de Leiden o del gen de protrombina 20210Ae historia familiar positiva para ETV, se sugiere la profilaxis preparto con dosisprofiláctica o intermedia de HBPM y profilaxis posparto durante seis semanascon HBPM o AVK, mientras que en aquellas sin antecedente familiar de ETV serecomienda la vigilancia en la etapa preparto y la anticoagulación durante seissemanas posparto.
En las pacientes embarazadas con cualquiera de las otras trombofilias, sin his-toria de ETV, se sugieren la vigilancia preparto y la profilaxis posparto conHBPM profiláctica.
En las pacientes que no tienen deficiencia de proteínas C o S se deben utilizarAVK.
PACIENTE CON PRÓTESIS VALVULAR CARDIACA
En las embarazadas con prótesis valvular se recomienda uno de los siguientes es-quemas:
a. Dosis ajustada de HBPM dos veces al día, ob. Dosis subcutánea ajustada de HNF cada 12 h, oc. HBPM o HNF hasta la semana 13 con sustitución por AVK hasta cerca del
parto cuando se reanudan la HBPM o la HNF.
EVENTO CEREBROVASCULARISQUÉMICO EN EL EMBARAZO
El evento vascular cerebral (EVC) es una condición poco común durante el em-barazo y el puerperio, con una importante morbimortalidad materna y fetal, y una

329Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
incidencia de 5 a 15 por cada 100 000 partos. Hasta 5% de las muertes maternascorresponden a EVC. La mujer que padece un evento durante el embarazo pre-senta un rango de mortalidad de 0 a 38%. En adición a la mortalidad materna, lascomplicaciones por enfermedad cerebrovascular en esta población incluyen par-to prematuro y mortalidad fetal; entre 42 y 63% de las sobrevivientes presentanalguna deficiencia neurológica residual.
Los eventos suelen presentarse alrededor del parto y más de 60% de ellos den-tro de las seis semanas siguientes.
Embolismo cardiogénico
El embolismo cardiogénico constituye la etiología más frecuente de EVC isqué-mico durante el embarazo. Los mecanismos cardiogénicos se dividen en anorma-lidades estructurales (congénitas o adquiridas), alteraciones en la conducción yfalla inotrópica.
Las anormalidades estructurales incluyen los defectos congénitos, como de-fecto septal auricular, foramen oval permeable y aneurisma septal auricular. Losdefectos valvulares congénitos o adquiridos y las prótesis valvulares constituyenotras causas estructurales de EVC isquémico.
La endocarditis infecciosa se encuentra asociada a embolismo cerebral en20% de los casos, en los que Streptococcus viridans y Staphylococcus aureus sonlos agentes infecciosos más frecuentes durante el embarazo.
Las endocarditis no infecciosas (Libman–Sacks o marántica) pueden tambiénresultar en embolización al sistema nervioso central. La endocarditis de Libman–Sacks se encuentra en la tercera parte de las pacientes con síndrome de anticuer-pos antifosfolípidos.
La presencia de arritmias cardiacas, como la fibrilación auricular, representaun riesgo de embolismo sistémico de 10 a 23% y de embolismo cerebral de 2 a10%. La fibrilación auricular que se presenta durante el embarazo es habitual-mente secundaria a enfermedad valvular o alteraciones metabólicas.
La miocardiopatía periparto es una causa extremadamente rara de isquemiacerebral en las pacientes embarazadas. Su etiología se desconoce. El mecanismode la isquemia cerebral es secundario a hipoperfusión relacionada con falla en lafunción cardiaca o como resultado de embolización de trombos murales a los va-sos cerebrales.
El estudio de la paciente embarazada con EVC isquémico en la que se sospe-cha un origen cardiogénico debe incluir la realización de electrocardiograma,monitor Holter cardiaco de 24 h o telemetría para la detección de arritmias o tras-tornos de la conducción cardiaca, así como ecocardiograma, de preferencia trans-esofágico, el cual se considera seguro durante el embarazo.

330 (Capítulo 20)Tratado de trombosis
Anormalidades hematológicas
La presencia de anormalidades hematológicas como las trombofilias anterior-mente descritas, incluyendo el síndrome de anticuerpos antifosfolípidos, puedenresultar en EVC isquémico.
La crisis de células falciformes es común en las pacientes embarazadas y pue-den ocasionar isquemia cerebral por aumento en la viscosidad sanguínea y esta-sis. La púrpura trombocitopénica trombótica es tres veces más frecuente en laspacientes embarazadas; suele cursar con isquemia cerebral y encefalopatía demanera secundaria a la oclusión de arteriolas cerebrales por microtrombos.
Otra causa poco común de EVC isquémico durante el embarazo es el embolis-mo de líquido amniótico, que de manera típica se presenta con disnea, cianosis,estado de choque y convulsiones, en cuyo caso las manifestaciones neurológicassuelen presentarse por un daño hipóxico difuso, aunque puede ocurrir embolismoparadójico con manifestaciones neurológicas focales.
Enfermedad arterial
Las cuatro alteraciones arteriales más comunes asociadas con embolismo son laaterosclerosis, la enfermedad de Takayasu, la enfermedad de moya moya y la di-sección arterial.
La aterosclerosis es responsable de 15 a 25% de los infartos cerebrales queocurren durante el embarazo. Las mujeres afectadas con aterosclerosis se en-cuentran de manera habitual alrededor de los 30 años, son fumadoras y padecenhipertensión arterial sistémica, diabetes mellitus o ambas. La enfermedad de Ta-kayasu es una arteriopatía progresiva que involucra tanto la aorta como sus ramasy que se presenta de manera habitual en las mujeres menores de 45 años de edad.La enfermedad de moya moya es una vasculopatía progresiva que afecta las arte-rias carótidas intracraneales y sus ramas terminales, la cual puede progresar du-rante el embarazo y conducir a un EVC.
TRATAMIENTO DEL EVENTO VASCULAR CEREBRALISQUÉMICO AGUDO EN LA PACIENTE EMBARAZADA
El tratamiento del EVC isquémico agudo incluye el manejo médico de emergen-cia básico. En adición, el uso de terapia trombolítica en las pacientes embaraza-das se encuentra limitado hasta el momento como reporte de casos. La adminis-tración de tPA durante el embarazo no constituye una contraindicación absoluta;sin embargo, es un medicamento clase C.

331Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
La anticoagulación puede prevenir la propagación del trombo de manera efec-tiva tanto en la trombosis arterial como en la venosa. Las pacientes candidatas aanticoagulación son aquellas con fibrilación auricular, hipercoagulabilidad,trombosis venosa, disección arterial aguda y prótesis valvular cardiaca.
El manejo anticoagulante en la paciente embarazada se debe realizar como semenciona previamente a través de HNF o HBPM, y se debe posponer el uso deantagonistas de la vitamina K hasta el periodo posparto. En caso de un periodode gestación menor de 34 semanas se deben administrar esteroides para madura-ción pulmonar fetal si se considera el parto temprano e iniciar la administraciónde heparina seis horas después del parto vaginal o 12 h posteriores a la cesárea.
TROMBOSIS VENOSA CEREBRAL EN EL EMBARAZO
La paciente embarazada se encuentra en un riesgo aumentado de presentar trom-bosis venosa intracraneal por los mecanismos descritos previamente, con una in-cidencia de 40 a 50 por cada 10 000 nacimientos. Los senos sagital y transversoson los afectados con mayor frecuencia, y es más común que esto ocurra en elpuerperio que durante el embarazo. Al ocurrir una trombosis venosa intracranealse altera la reabsorción de líquido cefalorraquídeo, lo que conlleva un aumentode la presión intracraneal. Las pacientes pueden presentar cefalea, convulsiones,manifestaciones neurológicas focales y disminución del estado de conciencia.Además, la presencia de náusea y vómito, papiledema y deterioro del estado neu-rológico debe sugerir la presencia del trastorno. El diagnóstico de trombosis ve-nosa intracraneal requiere tanto de la evaluación clínica como de la demostracióndel trombo a través de un estudio de imagen apropiado. La venografía por tomo-grafía computarizada o por resonancia magnética es el estudio más sensible; noobstante, el seno trombosado puede ser detectado a través de una tomografía oresonancia magnética simples. La heparina es la piedra angular del tratamientoen las pacientes embarazadas (ver a detalle en el capítulo 10).
SEGURIDAD DE LA HEPARINA DE BAJO PESOMOLECULAR DURANTE EL EMBARAZO
Las ventajas que ofrece el uso de heparina de bajo peso molecular (HBPM) du-rante el embarazo son:
1. La HBPM no atraviesa la barrera placentaria.

332 (Capítulo 20)Tratado de trombosis
2. La aplicación puede ser una sola vez al día, con una respuesta anticoagu-lante más predecible que la de la HNF.
3. El riesgo de osteoporosis es menor con la HBPM que con la HNF.4. La incidencia de síndrome de trombocitopenia inducida por heparina es
menor con la HBPM que con la HNF.5. En la mayoría de los casos el monitoreo de la actividad de anti–Xa no es
necesario.
INDICACIONES PARA MEDICIÓN DE LOS NIVELES DEANTI–XA CON HEPARINA DE BAJO PESO MOLECULAR
� Falla renal.� Hipotensión o escasa perfusión tisular.� Obesidad (> 80 kg o índice de masa corporal > 30 kg/m2).� Bajo peso (< 50 kg o índice de masa corporal < 22 kg/m2).� Cambios en los eventos clínicos (sangrado, trombosis y otros).� Embarazo.
REFERENCIAS
1. Bilello KL, Murin S: Sex and gender issues and venous thromboembolism. Clin Chest Med2004;25:281–297.
2. Heilmann L, Schneider D, Tempelhoff G: Antithrombotic therapy in high–risk pregnan-cy. Hematol/Oncol Clin N Am 2000;14(5):1133–1150.
3. American College of Chest Physicians: Sixth ACCP Consensus Conference on Antithrom-botic Therapy. Chest 2001;119(1).
4. Nelson S, Greer I: Thrombophilia and the risk for venous thromboembolism during preg-nancy, delivery and puerperium. Obstet Gynecol Clin N Am 2006;33:413–427.
5. Dentali F, Crowther M: Acquired thrombophilia during pregnancy. Obstet Gynecol ClinN Am 2006;33:375–388.
6. Bick R: Antiphospholipid thrombosis syndromes. Hematol Oncol Clin N Am 2003;17:115–147.
7. Gei A, Vadhera R, Hankins G: Embolism during pregnancy: thrombus, air, and amnioticfluid. Anesthesiol Clin N Am 2003;21:165–182.
8. Stone S, Morris T: Pulmonary embolism and pregnancy. Crit Care Clin 2004;20:661–677.9. Bates S, Greer I, Hirsh J: Use of antithrombotic agents during pregnancy: the Seventh
ACCP Conference on Antithrombotic and Thrombolitic Therapy. Chest 2004;126.10. Turan T, Stern B: Stroke in pregnancy. Neurol Clin 2004;22:821–840.11. Sibai B, Coppage K: Diagnosis and management of women with stroke during pregnancy/
postpartum. Clin Perinatol 2004;31:853–868.12. Danik S, Fuster V: The obstetrical patient with a prosthetic heart valve. Obstet Gynecol
Clin N Am 2006;33:481–491.

333Embarazo y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
13. Seshadri N, Goldhaber S, Elkayam U et al.: The clinical challenge of bridging anticoa-gulation with molecular–weight–heparin in patients with mechanical prosthetic heartvalves: an evidence–based comparative review focusing on anticoagulation options in preg-nant and non–pregnant patients. Am Heart J 2005;150:27–34.
14. Elkayam U, Singh H, Irani A, Akhter MW, from Westminster Publications: Anticoagula-tion in pregnant women with prosthetic heart valve. J Cardiovasc Pharmacol Ther 2004;9:107–115.
15. Naylor D, Olson M: Critical care obstetrics and gynecology. Crit Care Clin 2003;19:127–149.
16. Chasen S: Peripartum and perioperative management of the anticoagulated patient. ObstetGynecol Clin N Am 2006;33:493–497.
17. Bick R, Haas S: Thromboprophylaxis and thrombosis in medical, surgical, trauma and obs-tetric/gynecologic patients. Hematol Oncol Clin N Am 2003;17:217–258.
18. Ginsberg J, Greer I, Hirsh J: Use of antithrombotic agents during pregnancy. Chest 2001;119.
19. Brenner B: Thrombophilia and adverse pregnancy outcome. Obstet Gynecol Clin N Am2006;33:443–456.
20. Doyle N, Monga M: Thromboembolic disease in pregnancy. Obstet Gynecol Clin N Am2004;31:319–344.
21. Stone S, Morris T: Pulmonary embolism during and after pregnancy. Crit Care Med 2005;33(Suppl):S294–S300.
22. Johnston J, Brill Edwards P, Ginsberg J: Cost–effectiveness of prophylactic low molecu-lar weight heparin in pregnant women with a prior history of venous thromboembolism. AmJ Med 2005;118:503–514.
23. Soubra S, Guntupalli K: Critical illness in pregnancy: an overview. Crit Care Med 2005;33(Suppl):S248–S255.
24. Guyatt G, Akl E, Crowther M et al.: Antithrombotic therapy and prevention of thrombo-sis. 9ª ed. American College of Chest Physicians evidence–based clinical practice guide-lines. Chest 2012;141:7S–47S.

334 (Capítulo 20)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
21Anticonceptivos orales, terapia
hormonal sustitutiva y trombosisLuis Fernando García–Frade Ruiz
A principios de la década de 1960, poco tiempo después de la introducción de losanticonceptivos orales (ACOS), se reportó el primer caso de trombosis venosaprofunda (TVP) y tromboembolia pulmonar (TEP) en una mujer que los consu-mía. Más tarde su uso se vinculó con infarto agudo del miocardio (IAM) y eventocerebrovascular (ECV). Dicha asociación se confirmó en 1985 tras una críticaevaluación y estudios epidemiológicos, donde los ahora llamados ACOS de pri-mera generación (50 �g de estrógeno y progestágenos) mostraron un riesgo in-crementado de seis a ocho veces de padecer TEP, de dos a cuatro veces de sufrirIAM y de dos a seis veces de sufrir ECV. El aumento en el riesgo de TVP y TEPse relacionó con el componente estrogénico, mientras que los progestágenos serelacionaron con cambios en el metabolismo de los lípidos y los carbohidratos(aterosclerosis).
Por lo anterior, en el decenio de 1970 se realizaron dos cambios en la fórmula:se disminuyó la dosis de estrógeno de 50 �g a entre 30 y 35 �g y se agregó levo-norgestrel como progestágeno, dando lugar a los ahora llamados ACOS de se-gunda generación. Este cambio en la formulación disminuyó el riesgo de IAM,ECV, TVP y, sobre todo, de TEP.
Durante la década de 1980 se realizó otro cambio en la fórmula para mejorar laseguridad de los ACOS, donde el contenido estrogénico disminuyó de 30 a 20 �gy se introdujeron tres nuevos progestágenos: desogestrel, gestodene y norgesti-mato. De manera sorprendente, varios estudios epidemiológicos indican que es-tos ACOS de tercera generación elevan el riesgo de padecer TVP y TEP, en com-paración con los de segunda generación.
335

336 (Capítulo 21)Tratado de trombosis
Los progestágenos de los ACOS contribuyen también a un incremento en elriesgo de sufrir enfermedad trombótica. Las progestinas de tercera generaciónaumentan el riesgo de TVP más que las de segunda generación; no obstante, lasprimeras aumentan las lipoproteínas de alta densidad, lo cual indica que puedenconferir un bajo riesgo para el desarrollo de trombosis arterial. Los ACOS másrecientes contienen acetato de ciproterona o drospirenona.
MECANISMOS TROMBÓTICOSDE LOS ANTICONCEPTIVOS ORALES
Los efectos protrombóticos potenciales de los ACOS incluyen un incremento enlos niveles de los factores de la coagulación y una disminución en los niveles deantitrombina y proteína S. Entre los efectos procoagulantes de los ACOS se en-cuentran el incremento en los niveles de protrombina, factor VII, factor VIII, fac-tor X, fibrinógeno y plasminógeno, y una disminución en los niveles del factorV. Dichos cambios son más pronunciados después del uso de los ACOS de tercerageneración que los de segunda generación.
Los estrógenos causan una activación del sistema fibrinolítico dosis–depen-diente con cambios en los niveles del plasminógeno, del activador tisular delplasminógeno y del complejo plasminógeno–plasmina.
La base molecular de la resistencia adquirida a la proteína C durante el uso delos ACOS se desconoce. La disminución de los niveles de proteína S y del cofac-tor de la proteína C activada sólo explica de manera parcial la resistencia a la pro-teína C activada que se encuentra en las mujeres que los consumen.
EFECTO DE LOS ANTICONCEPTIVOSORALES SOBRE EL PERFIL DE LÍPIDOS
Los estrógenos tienen un efecto favorable sobre los lípidos séricos y las lipopro-teínas, ya que producen un aumento de las lipoproteínas de alta densidad (HDL)y una disminución de las lipoproteínas de baja densidad (LDL) y del colesteroltotal. Por otro lado, los progestágenos disminuyen las HDL y el colesterol total,y provocan un aumento de las LDL.
FACTORES DE RIESGO
Entre los factores de riesgo para el desarrollo de trombosis asociada con el usode anticonceptivos orales se encuentran los que se incluyen en el cuadro 21–1.

337Anticonceptivos orales, terapia hormonal sustitutiva y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 21–1. Factores de riesgo para trombosis asociadacon el consumo de anticonceptivos orales
Uso inicialMayor edadDosis de estrógeno
Tipo de progestágenoTabaquismoHipertensión arterial sistémica
ObesidadEstado de saludMutación del factor V de Leiden
Otras trombofilias hereditarias
Uso inicial
El mayor riesgo de trombosis surge durante el primer año de uso de los ACOSindependientemente del tipo de formulación.
Mayor edad
El riesgo de trombosis venosa asociada con el uso de ACOS se incrementa en laspacientes mayores de 35 años de edad.
Dosis de estrógeno y tipo de progestágeno
Los estrógenos poseen un efecto dosis–dependiente sobre el riesgo de trombosis.El riesgo de trombosis venosa es mayor luego del uso de ACOS de tercera genera-ción que de los de segunda generación.
Tabaquismo
El tabaquismo es el principal factor de riesgo independiente para el desarrollo deIAM en las pacientes que consumen ACOS. Aun en pacientes fumadoras (� 15cigarrillos al día) que no consumen hormonales el riesgo de IAM se incrementade 3 a 11 veces.
Sin embargo, el uso combinado de ACOS con tabaquismo genera un sinergis-mo con un aumento de 30 veces del riesgo relativo de IAM.

338 (Capítulo 21)Tratado de trombosis
Hipertensión arterial sistémica
La hipertensión arterial sistémica representa otro factor de riesgo independientepara el desarrollo de IAM. El uso de ACOS en pacientes con historial de hiperten-sión arterial sistémica incrementa 17 veces el riesgo relativo de sufrir IAM.
Otros
Otros factores incrementan el riesgo de trombosis venosa al vincularse con el usode ACOS, como el aumento en el índice de masa corporal, la inmovilización, etc.
La recurrencia de trombosis venosa en las mujeres que continúan usandoACOS después de un evento trombótico es de 28 por cada 1 000 casos al año,mientras que el riesgo de recurrencia en las mujeres que los suspenden es de 12.9por cada 1 000 casos al año.
Susceptibilidad al estado protrombótico
En 1993 Dahlbäck describió la resistencia a la proteína C activada como una con-dición protrombótica hereditaria que disminuye la respuesta anticoagulante a laproteína C activada. Un año más tarde las bases moleculares identificaron unamutación en el factor V de la coagulación (la sustitución de adenina por guaninaen el nucleótido 1691), en uno de los sitios donde la proteína C activada se acoplacon el factor V y lo inactiva. Dicho factor se conoce como el factor V de Leiden;su prevalencia es de 5% en las personas de raza blanca y de 20 a 40% en los pa-cientes con TVP o TEP.
La presencia de la mutación incrementa en gran medida el riesgo de trombosisen las mujeres que consumen ACOS. El riesgo de enfermedad trombótica estáaumentado por un factor de 4 en las mujeres que consumen ACOS, pero que nopresentan la mutación; por un factor de 7 en las que padecen la mutación, perono consumen ACOS; y por un factor incrementado de 35 en las que presentan lamutación del factor V de Leiden y consumen ACOS. De las pacientes que presen-tan TVP con el consumo de ACOS entre 14 y 40% padecen la mutación.
No obstante, otros defectos protrombóticos hereditarios, como la mutación delgen de protrombina G20210A —que se encuentra entre 3 y 4% de la poblacióngeneral de raza blanca y entre 12 y 16% de los pacientes que presentan TVP—,la metiltetrahidrofolato reductasa (MTHFR 677TT), la deficiencia de proteína C,proteína S o antitrombina, presentan una menor prevalencia que la mutación delfactor V de Leiden, y las pacientes que los padecen y consumen ACOS adquierenun mayor riesgo de desarrollo de enfermedad trombótica.

339Anticonceptivos orales, terapia hormonal sustitutiva y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
En los últimos años se ha descrito una resistencia adquirida a la proteína C porparte de las mujeres embarazadas y de las que usan ACOS. Dicha resistencia esindependiente de las mutaciones descritas. Esta forma de resistencia adquiridase presenta en 12% de la población sana y es independiente del tiempo de uso deACOS, del tipo de formulación y de la dosis de estrógeno o progestágeno; sin em-bargo, los ACOS de tercera generación generan mayor resistencia que los de se-gunda.
TROMBOSIS ARTERIAL RELACIONADACON LOS ANTICONCEPTIVOS ORALES
No existe un incremento significativo en el riesgo de ECV isquémico en las pa-cientes que consumen ACOS con bajas dosis de estrógenos (< 50 �g); sin embar-go, el riesgo relativo de ECV hemorrágico se incrementa tres veces en las pacien-tes de 35 años de edad o mayores con historial de tabaquismo que consumenACOS. La presencia de hipertensión arterial sistémica tiene mayor efecto sobreel EVC hemorrágico que sobre el isquémico entre las pacientes que consumenhormonales.
Los principales factores de riesgo para ECV isquémico vinculados con el usode ACOS son la presencia de trombofilias hereditarias, el tabaquismo, la hiper-tensión arterial sistémica, la hipercolesterolemia, la diabetes mellitus, la migrañay la obesidad. El riesgo de ECV en las pacientes que consumen ACOS y padecenla mutación del factor V de Leiden parece incrementarse 11 veces, mientras queel riesgo asociado con MTHFR 677 TT es cinco veces mayor que en las mujeressin el polimorfismo.
Las pacientes que consumen ACOS y tienen factores de riesgo (como taba-quismo e hipertensión arterial sistémica) para cardiopatía isquémica tienen un es-pecial riesgo de sufrir IAM. Los ACOS también incrementan el riesgo de enfer-medad arterial periférica.
¿SE DEBE DETECTAR LA TROMBOFILIA ANTES DEINICIAR LA TERAPIA CON ANTICONCEPTIVOS ORALES?
Aunque la presencia de trombofilias hereditarias incrementa el riesgo de trombo-sis venosa en pacientes que consumen ACOS, la detección de trastornos, comola deficiencia de antitrombina, proteína C y proteína S, no es una medida prácticadada su baja prevalencia en la población general y lo compleja que resulta suidentificación.

340 (Capítulo 21)Tratado de trombosis
La mutación del factor V de Leiden tiene una mayor prevalencia en la pobla-ción general, como se mencionó; sin embargo, su detección resulta costosa y seargumenta que su identificación sólo podría prever pocos casos de embolia pul-monar masiva, mientras que los beneficios potenciales contraceptivos de losACOS favorecen a una gran cantidad de mujeres.
Una adecuada historia clínica que haga hincapié en los antecedentes familiaresy una detallada exploración física pueden ayudar a seleccionar a las pacientes enlas que la detección temprana de trombofilias hereditarias antes de la administra-ción de ACOS puede ser de utilidad. No obstante, una historia familiar de trom-boembolismo venoso tiene un bajo valor predictivo positivo para defectos gené-ticos trombofílicos.
¿SE CONOCEN TODAS LAS TROMBOFILIAS?
A través de los años se han identificado cada vez más mutaciones que pueden in-crementar el riesgo de trombosis. Aunque existe una lista de mutaciones del fac-tor V —cada una lleva el nombre de la ciudad donde se estableció la mutación:Leiden, Hong Kong, Cambridge, Liverpool, etc.—, se espera que con el adveni-miento de sofisticados laboratorios de análisis se identifiquen más mutaciones ynuevos factores trombofílicos.
SÍNDROME DE HIPERESTIMULACIÓN OVÁRICA
Los procedimientos de fertilización in vitro comprenden la estimulación ováricamediante la administración de análogos de hormona de liberación de gonadotro-pinas y gonadotropinas exógenas. Una hiperestimulación ovárica se caracterizapor la presencia de derrame pleural, ascitis y disminución del volumen intravas-cular, lo cual complica entre 1 y 2% de los ciclos de fertilización in vitro. La hiper-estimulación ovárica se asocia con un estado de hipercoagulabilidad y con un in-cremento en el riesgo de sufrir trombosis venosa y trombosis arterial.
TERAPIA HORMONAL SUSTITUTIVA Y TROMBOSIS
Las mujeres menores de 50 años de edad tienen una menor incidencia de enfer-medades cardiovasculares (en especial aterosclerosis) que los hombres de su mis-ma edad. Antes de los 50 años la relación de enfermedad cardiovascular es de 1:2entre mujeres y hombres, pero es de 1:1 a los 80 años de edad. Una disminución

341Anticonceptivos orales, terapia hormonal sustitutiva y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
en esta relación se asocia con el inicio de la menopausia. Estas observaciones in-dican que las hormonas sexuales femeninas, sobre todo el estradiol y la progeste-rona, protegen contra la enfermedad cardiovascular.
La enfermedad arterial coronaria por aterosclerosis es menos frecuente en lasmujeres premenopáusicas. La aterosclerosis se relaciona con el metabolismo delos lípidos, en especial con las lipoproteínas de alta densidad (HDL) y las lipopro-teínas de baja densidad (LDL). La mujer premenopáusica suele presentar un per-fil de lípidos en mejores condiciones que un hombre de su misma edad o que lamujer posmenopáusica. Además, existen cambios en el sistema hemostático rela-cionados con la edad. Los niveles plasmáticos del fibrinógeno y del factor VIIaumentan con la edad, y ambos han sido descritos como factores de riesgo inde-pendientes para el desarrollo de trombosis arterial.
La terapia hormonal sustitutiva (THS) se puede administrar en forma oral,transdérmica o transvaginal en las mujeres que ya pasaron la menopausia o estánen ella. En América del Norte se administran con mayor frecuencia en forma deestrógenos de equino conjugados, mientras que en Europa se recurre al estradiolsintético. Usualmente se agrega un progestágeno al tratamiento (combinado o se-cuencial) para evitar la hiperplasia quística endometrial (o foco endometrial enla mujer con histerectomía previa).
El uso de THS incrementa el riesgo de trombosis venosa, tal como lo informóel reporte inicial del Women’s Health Initiative Trial publicado en julio de 2002.Este estudio prospectivo, doble ciego, evaluó los riesgos y beneficios de la THSen más de 16 000 mujeres posmenopáusicas. El estudio concluyó su desarrollo demanera temprana, debido a que los efectos adversos excedieron los beneficios, yaque las mujeres que recibieron la terapia hormonal presentaron un riesgo duplicadode eventos trombóticos venosos, incluidas TVP y TEP; no obstante, dicho riesgodisminuye con el tiempo de uso. Las mujeres con antecedentes de trombosis veno-sa también tienen un mayor riesgo de recurrencia con el uso de THS.
No existe una diferencia significativa del riesgo de trombosis venosa entre lasdistintas fórmulas y concentraciones de estrógeno, así como tampoco en su víade administración. Los efectos de la THS sobre el sistema de la coagulación sonsimilares a los que ejercen los ACOS (cuadro 21–2).
La THS causa una significativa disminución de los inhibidores de la coagula-ción, excepto de la proteína S libre, y un incremento en los fragmentos de pro-trombina y dímero D. Estos cambios son más pronunciados en las mujeres quedesarrollan TVP recurrente, lo cual indica que algunas mujeres son “hiperreacti-vas” a los efectos estrogénicos sobre el sistema de la coagulación.
La actividad trombogénica de los estrógenos orales quizá esté relacionada consu metabolismo hepático en su primer paso luego de su absorción, el cual puedeser evitado por la administración parenteral o transdérmica. Los efectos de losestrógenos transdérmicos sobre el sistema de la coagulación no son los mismos

342 (Capítulo 21)Tratado de trombosis
Cuadro 21–2. Efectos de la terapia hormonalsustitutiva en el sistema de la coagulación
Incremento Disminución Efecto no demostrado
Plasminógeno Antitrombina Resistencia a la proteína C activadaActivador tisular del plasminógeno Proteína S FibrinógenoDímero D Proteína C Factor X
Proteína C reactiva PAI–1 Activación plaquetaria
que se generan con los estrógenos orales. El efecto de los estrógenos transdérmi-cos sobre el riesgo de TVP aún no es claro, pero el riesgo de recurrencia en muje-res con trombosis venosa previa parece ser menor con el uso de éstos que con losorales.
Las dosis orales habituales de THS provocan una disminución de las LDL de10 a 15% y un aumento de las HDL de 10 a 15%, y del nivel de triglicéridos de10 a 20%.
OTROS FACTORES DE RIESGO
El riesgo de eventos trombóticos con el uso de THS y de anticonceptivos oraleses mayor durante el primer año de su uso, lo cual indica que puede existir un sub-grupo de mujeres con desórdenes genéticos o adquiridos que las colocan en unriesgo particularmente alto.
La mujer que padece trombofilia tiene más riesgo de padecer trombosis veno-sa si utiliza THS. La mujer que padece la mutación del factor V de Leiden y usaTHS presenta 15 veces mayor riesgo de sufrir trombosis.
El aumento de edad incrementa el riesgo de trombosis asociado con el uso deTHS. Otros factores de riesgo adicional que aumentan el riesgo de trombosis in-cluyen fracturas de miembros pélvicos, obesidad, cirugía reciente y enfermeda-des concomitantes.
TERAPIA HORMONAL SUSTITUTIVAY TROMBOSIS ARTERIAL
La THS incrementa el riesgo de ECV isquémico. Las mujeres que padecen la mu-tación del factor V de Leiden o la mutación del gen de protrombina 20210A yutilizan THS tienen dos veces más riesgo de sufrir ECV isquémico que las queno la utilizan.

343Anticonceptivos orales, terapia hormonal sustitutiva y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
La THS no se indica para prevención secundaria en la mujer con enfermedadcerebrovascular; por el contrario, la terapia con estrógenos puede empeorar unalesión ocasionada por isquemia.
El mecanismo a través del cual la THS incrementa el riesgo de ECV aún noes claro; no obstante, los estrógenos tienen un efecto sobre las neuronas que incre-menta su sensibilidad a la isquemia a través de la modulación de neurotransmiso-res, lo cual genera efectos proinflamatorios.
El mayor riesgo de ECV isquémico tras el uso de THS se presenta durante losprimeros seis meses de tratamiento. La THS no ofrece prevención primaria en lacardiopatía isquémica, sino que aumenta el riesgo de infarto agudo del miocardiodurante el primer año de su uso, y se ha demostrado que no ofrece protección car-diovascular en las mujeres con cardiopatía isquémica previa.
Las pacientes que padecen la mutación del gen de protrombina 20210A y usanTHS presentan un riesgo 11 veces mayor de sufrir infarto agudo del miocardiono fatal, en comparación con las que no la utilizan. Se sugiere que la inflamaciónpuede contribuir al desarrollo de eventos cardiovasculares tempranos en la mujercon THS. Los niveles de proteína C reactiva (PCR) se incrementan de maneraprogresiva a lo largo de la terapia con hormonales.
La PCR secretada en el hígado en respuesta a las citocinas inflamatorias incre-menta la expresión del factor tisular de monocitos, el cual es un potente estimula-dor de la actividad plaquetaria y de trombosis local. La elevación de la PCR du-rante el curso temprano de la THS también puede ser resultado de una placaateromatosa vulnerable.
El uso de THS en la mujer posmenopáusica puede potenciar el riesgo aterogé-nico por aumento en los factores de la coagulación, incluido el factor VII, y pordisminución en los niveles de antitrombina III.
No obstante, la THS también ejerce mecanismos cardioprotectores, como dis-minución en los niveles de fibrinógeno e incremento en la actividad fibrinolíticaa través de la disminución del PAI–1.
El balance de los estrógenos sobre el efecto coagulante y la actividad fibrinolí-tica depende de la existencia de factores de riesgo cardiovasculares o de polimor-fismos genéticos de factores hemostáticos individuales, o de ambos.
EFECTO DE LOS ESTRÓGENOS SOBRE EL FIBRINÓGENO
La disminución en los niveles de fibrinógeno fue uno de los mecanismos por loscuales se había propuesto que la THS disminuía el riesgo trombótico en ciertaspacientes. Sin embargo, después de varios estudios se indica que los estrógenosno actúan a través del fibrinógeno para mejorar el riesgo trombótico.

344 (Capítulo 21)Tratado de trombosis
EFECTO DE LOS ESTRÓGENOS SOBRE EL FACTOR VII
Las elevaciones plasmáticas del factor VII constituyen un factor de riesgo cardio-vascular.
La presencia de polimorfismos en el factor VII puede afectar su secreción efi-ciente, la influencia de los triglicéridos sobre el mismo, sus propiedades de unióna las proteínas nucleares y su actividad transcripcional.
La edad, el incremento en el índice de masa corporal, los anticonceptivos ora-les, la THS y la menopausia elevan los niveles o alteran la actividad del factorVII. Los niveles de factor VII se correlacionan también con los niveles de coleste-rol sérico. Aún queda por establecerse el impacto que tiene la THS sobre el riesgocardiovascular en presencia de varios polimorfismos genéticos del factor VII; sinembargo, la influencia de los estrógenos sobre los triglicéridos podría influir enla regulación del gen de transcripción del factor.
EFECTO DE LOS ESTRÓGENOS SOBRE EL INHIBIDORDEL ACTIVADOR DEL PLASMINÓGENO TIPO 1
El plasminógeno tipo 1 (PAI–1) está presente en las células endoteliales y en lasdel músculo liso de las arterias sanas, y en altas cantidades en las arterias ateroma-tosas.
El PAI–1 inhibe el tPA y la urocinasa, e impide así la activación de plasminóge-no a plasmina (capítulo 1). Los niveles altos de PAI–1 inhiben la actividad fibrino-lítica y pueden constituir un factor de riesgo para eventos coronarios agudos.
Se han identificado tres polimorfismos del PAI–1, de los cuales sólo la variante4G/5G ha sido investigada por su posible función en la enfermedad arterial. Di-cha variante se correlaciona con la presencia de altos niveles de PAI–1 en presen-cia de hipertrigliceridemia. Los pacientes con enfermedad cardiovascular pre-sentan con frecuencia un incremento en el genotipo 4G/5G y altos niveles dePAI–1.
Los estrógenos disminuyen en general los niveles del PAI–1; sin embargo, és-tos tienen distintos efectos sobre la fibrinólisis en ciertas subpoblaciones de mu-jeres, como en las que tienen síndrome de resistencia a la insulina y en las quemuestran altos niveles de PAI–1 determinado genéticamente por la presencia delalelo 4G. La transcripción del alelo 4G es regulada por los triglicéridos, los cualesse elevan con la THS, por lo que un incremento en los niveles del PAI–1 podríaresultar en una alteración del sistema fibrinolítico endógeno y en un aumento deriesgo aterotrombótico en la población con riesgo cardiovascular.

345Anticonceptivos orales, terapia hormonal sustitutiva y trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ESTRÓGENOS Y PLAQUETAS
Los megacariocitos y las plaquetas expresan el receptor de estrógenos �, lo cualaumenta la posibilidad de un efecto hormonal directo sobre esta línea celular. Es-tas pruebas indican que los compuestos hormonales activan las plaquetas.
MODULADORES SELECTIVOSDEL RECEPTOR ESTROGÉNICO
En la actualidad se utilizan dos moduladores selectivos del receptor estrogénico(SERM). El tamoxifeno se ha empleado para la prevención y terapia adyuvantedel cáncer de mama, y el raloxifeno ha sido aprobado para la prevención de laosteoporosis. Estos agentes poseen efectos antiestrogénicos, pero también ejer-cen efectos agonistas parciales sobre ciertos receptores, por lo que pueden provo-car algunos efectos adversos y riesgos asociados con otros estrógenos.
El uso de SERM incrementa el riesgo relativo de trombosis venosa, ya que,al igual que otros estrógenos, induce cambios en las concentraciones de los inhi-bidores de la coagulación y su efecto es más pronunciado en las mujeres que pa-decen trombofilia.
RESUMEN
Todas las formas de terapia hormonal que se utilizan habitualmente están relacio-nadas con un incremento en el riesgo de trombosis. Dicho riesgo es mayor duran-te los primeros 6 a 12 meses de terapia. Cierto subgrupo de mujeres tienen un ries-go particularmente alto, como las que padecen alguna trombofilia y tienenhistorial de trombosis venosa, mayor edad y obesidad, o son fumadoras. Estosriesgos se deben tener presentes y considerar los beneficios potenciales cuandose prescriban hormonales.
REFERENCIAS
1. Walker I: Exogenous sex hormones and thrombophilia. Obstet Gynecol Clin N Am 2006;33:467–479.
2. Mitchell J, Walsh J, Wang Cheng R et al.: Postmenopausal hormone therapy: a conciseguide to therapeutic uses, formulations, risks, and alternatives. Prim Care Clin Office Pract2003;30:671–696.

346 (Capítulo 21)Tratado de trombosis
3. Warren M: A comparative review on the risks and benefits of hormone replacement thera-py regimens. Am J Obst Gynecol 2004;190:1141–1167.
4. Moores L, Bilello K, Murin S: Sex and gender issues and venous thromboembolism. ClinChest Med 2004;25:281–297.
5. Staren E, Omer S: Hormone replacement therapy in postmenopausal women. Am J Surg2004;188:136–149.
6. Mammen E: Oral contraceptive pills and hormonal replacement therapy and thromboem-bolic disease. Hematol Oncol Clin N Am 2000;14(5):1045–1059.
7. Petitti D: Combination estrogen–progestin oral contraceptives. N Engl J Med 2003;349:1443–1450.
8. Vandenbroucke J, Rosing J, Bloemenkamp K et al.: Oral contraceptives and the risk ofvenous thrombosis. N Engl J Med 2001;344(20):1527–1534.
9. Bushnell C: Oestrogen and stroke in women: assessment of risk. Lancet Neurol 2005;4:743–751.
10. Savelli S, Kerlin B, Springer M et al.: Recommendations for screening for thrombophilictendencies in teenage females prior to contraceptive initiation. J Pediatr Adolesc Gynecol2006;19:313–316.
11. Blickstein I: Thrombophilia and women’s health: an overview. Obstet Gynecol Clin N Am2006;33:347–356.
12. Manson J, Hsia J, Johnson K et al.: Estrogen plus progestin and the risk of coronary heartdisease. N Engl J Med 2003;349(6):523–534.
13. Burkman R, Schlesselman J, Zieman M: Safety concerns and health benefits associatedwith oral contraception. Am J Obstet Gynecol 2004;190:5–22.
14. Reed S, Newton K, LaCroix A: Indications for hormone therapy: the post–women’s healthinitiative era. 2004;33:691–715.
15. Braunstein J, Kershner D, Bray P: Interaction of hemostatic genetics with hormone ther-apy. Chest 2002;121.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
22Trombosis en el paciente crítico
Miriam Villada Mena, Ana Olga López Aisa
El tromboembolismo venoso, que incluye tanto la trombosis venosa profunda(TVP) como la embolia pulmonar (EP), es una complicación común en los pa-cientes críticos. Los pacientes críticamente enfermos poseen varios factores deriesgo para la TVP, como son la necesidad de cirugía, la presencia de catéteresarteriales y venosos, la inmovilidad y el uso de sedantes y agentes paralizantes.1,2
Los pacientes pueden ingresar a la unidad hospitalaria con formación de trombosde forma crónica y encontrarse asintomáticos; con el tiempo, de 20 a 30% de lostrombos sin tratar en las venas de la pantorrilla se pueden extender en sentido pro-ximal al muslo, lo que representa un riesgo de 40 a 50% de desarrollar EP. Losestudios tempranos sobre la historia natural de la EP sugieren que si no es tratadasu tasa de mortalidad puede ser de al menos 25%.
Dichos pacientes poseen varios factores de riesgo inherentes, como historia deTVP, insuficiencia renal, insuficiencia cardiaca, trauma, sepsis, cáncer, edadavanzada y obesidad.3,4 El riesgo de TVP es de aproximadamente 1% por día en-tre algunos subgrupos de pacientes críticos.5 Cuando se compara con los pacien-tes que no tienen TVP, aquellos con TVP presentan una mayor duración en la ven-tilación mecánica, mayor estancia en la unidad de cuidados intensivos (UCI) uhospitalaria y una mayor mortalidad hospitalaria.6
Las consecuencias clínicas de la TVP incluyen el potencial de ser particular-mente graves, pero pueden no ser reconocidas en la UCI, por lo que la importan-cia de reconocer los factores de riesgo e iniciar la profilaxis antitrombótica es devital importancia. Es posible que los pacientes que reciben tratamiento profilácti-co sean más propensos que otros a padecer complicaciones graves de la terapia
347

348 (Capítulo 22)Tratado de trombosis
anticoagulante, por lo que un primer paso hacia la profilaxis antitrombótica esconcientizar a los médicos intensivistas de los factores de riesgo para TVP, identi-ficar los signos y síntomas tempranos de la trombosis y utilizar las herramientasdiagnósticas de forma temprana.
Devora Cook y col. realizaron un estudio,7 en el que encuestaron a médicosintensivistas canadienses acerca de la importancia de reconocer de forma tempra-na la TVP en pacientes críticos, y demostraron que los intensivistas definen clíni-camente importante la TVP utilizando criterios que son relevantes para los pa-cientes de cuidados críticos. Los criterios que encontraron pueden ser utilizadosen estudios futuros para la prevención y el tratamiento de la TVP en los pacientesen estado crítico (cuadro 22–1).
El desarrollo de TVP en el paciente críticamente enfermo puede tener conse-cuencias únicas, sobre todo en los pacientes que requieren ventilación mecánica,vasopresores o inotrópicos, ya que la reserva cardiopulmonar es crucial en la in-terpretación de las características de una TVP que hace que sea clínicamente im-portante. Tomar la decisión del riesgo que involucra darle o no tratamiento profi-láctico a estos pacientes puede ser mayor para los intensivistas que toman encuenta otras complicaciones de la enfermedad crítica, por ejemplo, sangrado, al-teraciones de la coagulación y riesgo de trombosis. Debe existir consenso sobreel riesgo–beneficio al tratar una TVP y con qué tratarla. Tales factores influyenen la morbilidad y la mortalidad de los pacientes con TVP en la UCI, por lo quetienen que ser evaluados de forma más rigurosa.
Cuadro 22–1. Factores del paciente que afectan la importancia clínicade la trombosis venosa profunda en pacientes de cuidados intensivos
Factores de riesgo del paciente Clasificación (DE)
Sospecha clínica de embolia pulmonar 4.6 (0.7)Comorbilidad cardiopulmonar crónica/aguda 4.5 (0.7)Síntomas en las piernas 4.2 (0.8)Ocurre al recibir anticoagulación terapéutica 4.0 (1.2)Detectado por el examen físico 3.8 (0.7)Ocurre al recibir tromboprofilaxis 3.6 (0.8)Al menos un factor de riesgo de tromboembolismo venoso 3.6 (0.6)Se asocia con el cateterismo venoso central 3.3 (0.9)No reciben profilaxis 3.2 (0.7)No se asocia con cateterismo venoso central 3.1 (0.5)No se recibe anticoagulación terapéutica 3.1 (0.3)No hay factores de riesgo 3.0 (0.4)Asintomática 2.8 (0.5)No se detecta mediante un examen físico 2.8 (0.5)
De acuerdo con las puntuaciones medias consideradas por los intensivistas se clasificaron los fac-tores de los pacientes y las probabilidades de desarrollar trombosis venosa profunda clínicamenteimportante. DE: desviación estándar.7

349Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Cuadro 22–2.Factores de riesgo de tromboembolismovenoso en pacientes hospitalizados
CondiciónEnfermedad infecciosa agudaInsuficiencia cardiaca congestivaa
Infarto agudo del miocardioEnfermedad respiratoria agudaEnfermedad vascular cerebralEnfermedad reumática (p. ej., artritis aguda)Enfermedad inflamatoria intestinal
Características clínicasTromboembolismo venoso previoEdad avanzada (> 75 años)Cirugía o trauma recienteInmovilidad o paresiaObesidad (IMC > 30)b
Cateterización venosa centralEstados trombofílicos hereditarios o adquiridosVaricesTerapia con estrógenos
a La insuficiencia cardiaca congestiva se define como lo hace la New York Heart Association clasesIII o IV de la enfermedad.b El índice de masa corporal (IMC) es el peso en kilogramos dividido entre el cuadrado de la alturaen metros.
EVALUACIÓN DEL RIESGO
El riesgo de tromboembolismo venoso está relacionado con la presencia o ausen-cia de factores de riesgo (cuadro 22–2), el cual aumenta si varios de ellos se en-cuentran presentes, como suele ser en la mayoría de los pacientes hospitalizados.Debido a que las decisiones relativas a la profilaxis dependen del riesgo inicial,todos los pacientes deben ser sometidos a una evaluación de riesgo en el momen-to de ingresar al hospital y una nueva evaluación cuando sucedan cambios en suestado, como posterior a la transferencia a la UCI, después de una cirugía o antegravedad por disfunción de un órgano vital. Se han propuesto diversos sistemaspara estimar el riesgo sobre la base del número y el tipo de los factores de riesgopresentes en el paciente hospitalizado,8,9 pero ninguno de ellos ha sido validadode manera prospectiva. Si bien no se cuenta con datos disponibles para funda-mentar las recomendaciones específicas para la profilaxis, éstas se basan en laedad del paciente y la duración prevista de la hospitalización. La profilaxis seconsidera necesaria en los pacientes hospitalizados que son mayores de 40 añosde edad, tienen movilidad limitada de tres días o más y presentan al menos un fac-tor de riesgo.10

350 (Capítulo 22)Tratado de trombosis
VENTILACIÓN MECÁNICA Y TROMBOSIS
La trombosis arterial y venosa es una causa importante de aumento de la morbili-dad y la mortalidad. La trombosis arterial es una causa común de infarto del mio-cardio, evento vascular cerebral isquémico y gangrena de miembros, mientrasque la trombosis venosa incluye trombosis venosa profunda, que puede ser com-plicada por el síndrome postrombótico y la EP, y puede ser fatal o conducir a hi-pertensión pulmonar crónica.
Muchos pacientes con ventilación mecánica y episodios repentinos de hipo-tensión, taquicardia o hipoxia pueden estar cursando con un EP y no ser detecta-dos.11 La falta de sospecha de un EP puede contribuir a tener dificultades en laprogresión de la ventilación mecánica, aumentando la estancia hospitalaria y elriesgo de desarrollar neumonía asociada a la ventilación mecánica.12
Los pacientes con mayor probabilidad de desarrollar un EP con repercusiónhemodinámica importante son los que tienen una comorbilidad cardiopulmonaraguda o crónica que limita su tolerancia. La poca reserva cardiopulmonar puedetener consecuencias graves o mortales si se presenta un evento de EP, indepen-dientemente del diagnóstico en el momento del ingreso.
Profilaxis
La patogénesis de la trombosis venosa se relaciona principalmente con la hiper-coagulabilidad y la estasis venosa. Los tratamientos profilácticos disponibles seutilizan para mejorar el retorno venoso y reducir la hipercoagulabilidad de la san-gre.
TERAPIAS NO FARMACOLÓGICAS
La deambulación y los ejercicios, como la extensión de los pies, mejoran el flujovenoso, por lo que deben fomentarse. Las medias de compresión graduada y losdispositivos de compresión neumática también reducen la estasis venosa y soneficaces en la reducción del riesgo de tromboembolismo venoso posoperato-rio.13,14 En una revisión se demostró que el uso de la compresión neumática redu-ce en alrededor de 50% el riesgo de tromboembolismo venoso en los pacienteshospitalizados después de una cirugía.14 La profilaxis no farmacológica es unaelección para los pacientes con alto riesgo de hemorragia tras el uso de anticoagu-lantes, incluidos los pacientes con hemorragia gastrointestinal activa reciente, los

351Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
pacientes con evento cerebrovascular hemorrágico y aquellos con defectos he-mostáticos, como trombocitopenia grave. La suspensión del uso de medias decompresión por malestar, transporte dentro del hospital o visitas al baño puedelimitar su eficacia.
Profilaxis farmacológica
La profilaxis eficaz se puede realizar con heparina no fraccionada, heparinas debajo peso molecular (HBPM) o fondaparinux (cuadro 22–3). La información ini-cial sobre el valor de la profilaxis en los pacientes hospitalizados provino de ensa-yos clínicos con pacientes sometidos a cirugía.15 El único ensayo grande mostróque el uso de dosis bajas de HBPM se asoció con una disminución en la incidenciade EP fatal después de una cirugía general.16 Un metaanálisis posterior de 74 en-sayos mostró que las dosis bajas de heparina reducen la tasa de tromboembolismovenoso posoperatorio, EP y EP fatal en 67, 47 y 64%, respectivamente.17 El trata-miento con heparina dio lugar a un aumento de 2% de la incidencia de complica-ciones hemorrágicas, principalmente en el sitio quirúrgico.17 La mayoría de estosestudios identificaron la trombosis en pacientes asintomáticos mediante pruebasvasculares no invasivas. A pesar de la importancia clínica de la trombosis, los
Cuadro 22–3. Medicamentos y dosis para la profilaxis del tromboembolismovenoso en pacientes médicos hospitalizadosa
Fármacosb Dosis Comentario
Heparina no fraccionada 5 000 U por vía subcutánea ca-da 8 hc
Heparinas de bajo pesomolecular� Enoxaparina 40 mg por vía subcutánea una
vez al díaEs más costosa que la heparina;
20 mg al día no son efectivos� Dalteparina 5 000 U por vía subcutánea
una vez al díaEs más costosa que la heparina
� Fondaparinuxd 2.5 mg por vía subcutánea unavez al día
Es más costoso que la heparina
a La profilaxis anticoagulante no se debe usar si hay un riesgo de sangrado excesivo, tal como enpacientes con hemorragia activa, hemorragia gastrointestinal reciente, evento cerebrovascular he-morrágico o defectos hemostáticos, como trombocitopenia severa.b La heparina no fraccionada y las heparinas de bajo peso molecular no se deben utilizar en pacien-tes con trombocitopenia inducida por heparina actual o anterior.c También se ha utilizado una dosis de 5 000 U por vía subcutánea cada 12 h. La opinión de losexpertos favorece las ocho horas de dosificación, aunque los regímenes de 12 h no se han compa-rado directamente.d El fondaparinux está aprobado por la Administración de Alimentos y Medicamentos para la profila-xis en los pacientes quirúrgicos, pero el mismo régimen ha sido utilizado en pacientes médicos.

352 (Capítulo 22)Tratado de trombosis
estudios de la historia natural de la enfermedad tromboembólica venosa indicanque existe una relación directa entre los trombos en venas pequeñas, los grandestrombos proximales y la EP.
Los pacientes críticos suelen tener múltiples factores de riesgo con una altatasa de tromboembolismo venoso. Las pruebas de pequeños ensayos aleatoriza-dos indican que la profilaxis con heparina no fraccionada o HBPM es eficaz.15
Una revisión sistemática de 26 estudios con 73 000 pacientes que habían presen-tado infarto agudo del miocardio mostró una reducción significativa de alrededorde 50% de la tasa de EP en pacientes que recibieron terapia con heparina en cual-quier dosis y casi la misma reducción en otros siete ensayos con dosis bajas deheparina.18
El riesgo de tromboembolismo venoso es alto en los pacientes hospitalizados,pero se puede reducir significativamente cuando los pacientes reciben profilaxisapropiada. La HBPM, el fondaparinux, la warfarina y la compresión mecánicaintermitente han demostrado su eficacia en varios estudios clínicos. Por desgra-cia, las medidas profilácticas parecen estar subestimadas, tanto en EUA como enestudios internacionales.19,20 La profilaxis anticoagulante es más eficaz que laprofilaxis mecánica de los miembros inferiores, pero los riesgos tanto de trombo-sis como de sangrado deben ser considerados en cada paciente.21
Después de una cirugía de cadera o de rodilla el riesgo de trombosis venosaes de 50% o superior sin profilaxis. El paciente con trauma múltiple o con lesiónde la médula espinal también representa un alto riesgo.
La superioridad de la profilaxis con HBPM ante la heparina no fraccionada hasido demostrada en varios estudios. La profilaxis con fondaparinux es tambiéneficaz para el reemplazo total de cadera. En otros casos, como la cirugía abdomi-nal, las dosis bajas de heparina no fraccionada parecen ser adecuadas, aunque sedeben considerar las ventajas de la HBPM en la mayoría de las indicaciones.
Un estudio a gran escala que utilizó resultados de venografías indicó que, sinmedidas preventivas, el riesgo de tromboembolismo venoso entre pacientes críti-camente enfermos y pacientes hospitalizados pueden ser tan alto como 15%. Losensayos prospectivos, aleatorizados, bien diseñados y realizados en esta pobla-ción han demostrado que la enoxaparina, la dalteparina y el fondaparinux son,cada uno, superiores al placebo en la prevención aguda de tromboembolismo ve-noso.22,23
En los pacientes médicos no quirúrgicos la enoxaparina una vez al día es al me-nos tan eficaz como la heparina no fraccionada cada ocho horas; en los pacientescon evento cerebrovascular la profilaxis con enoxaparina se asocia con tasas sig-nificativamente más bajas de TVP que la profilaxis con 5 000 U de heparina nofraccionada cada 12 h.24 Los pacientes con evento cerebrovascular o insuficien-cia cardiaca congestiva se encuentran en un riesgo particularmente alto; en el últi-mo grupo el grado de disfunción del ventrículo izquierdo se puede correlacionar

353Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
con el riesgo de trombosis.25 La profilaxis extendida con enoxaparina (duranteaproximadamente 38 días) en comparación con 7 a 10 días de tratamiento parecereducir la tasa de TVP entre personas con enfermedades médicas crónicas, ade-más de que existe una disminución en la morbilidad. Prolongar la profilaxis hademostrado su eficacia en determinadas poblaciones de alto riesgo, como los pa-cientes sometidos a reemplazo total de cadera o cirugía oncológica.
Al igual que con la terapia para la enfermedad tromboembólica venosa, sedebe considerar una disminución de la dosis para la profilaxis con HBPM en lospacientes con insuficiencia renal avanzada, de lo contrario se debe utilizar la he-parina estándar. Combinar los métodos anticoagulantes y mecánicos es razona-ble, sobre todo en los pacientes quirúrgicos y en estado crítico, ya que tienen unriesgo mayor. Se recomienda el uso de filtros en la vena cava inferior como profi-laxis primaria en los pacientes con trauma y en los que son sometidos a neurociru-gía.26
La efectividad de la enoxaparina se mide por la actividad del antifactor Xa(aFXa). La concentración máxima de la actividad aFXa ocurre entre tres y cuatrohoras después de la inyección de enoxaparina subcutánea (SC).27,28 Los nivelesde actividad aFXa de 0.1 a 0.4 U/mL se consideran eficaces en cuanto a actividadantitrombótica.29,30
La dosis estándar europea de 40 mg de enoxaparina SC una vez al día a menu-do se utiliza como profilaxis de ETV en pacientes en estado crítico.27 Priglingery Mayr y col.29,31 demostraron la escasa eficacia de esta dosis para el logro de losniveles anticoagulantes recomendados de aFXa. Este estudio coincide con el ha-llazgo de que 40 mg produjeron niveles subterapéuticos de aFXa en pacientes enestado crítico. Las razones para esta aparente resistencia a la heparina son intri-gantes y no del todo claras. Algunos autores sugieren que los mecanismos fisioló-gicos o farmacológicos de algunos agentes, por ejemplo, los vasopresores, pue-den alterar la absorción SC de heparina a través de la vasoconstricción de losvasos sanguíneos periféricos mediada por adrenérgicos.28,29,32 Otros tienen lateoría de que la presencia de edema puede disminuir la absorción SC de heparina.
Sin embargo, no hay diferencias en la actividad aFXa tras la inyección subcu-tánea de 2 500 UI de dalteparina como profilaxis de TVP entre los pacientesingresados en la UCI con y sin edema. Otros sostienen que la presencia de disfun-ción múltiple de órganos puede alterar el metabolismo de fármacos, la distribu-ción y la unión a albúmina y proteínas de fase aguda.29
El monitoreo periódico de los niveles de aFXa se recomienda en poblacionesespeciales (p. ej., en pacientes embarazadas, niños, pacientes con lesión renalaguda o en quienes están en los extremos de peso corporal).33 La actividad aFXatres horas después de enoxaparina tuvo una correlación negativa con el índice demasa corporal,31 debido a la reducción del flujo sanguíneo en los tejidos grasos,resultando en una disminución de la absorción de fármacos.29

354 (Capítulo 22)Tratado de trombosis
La dosis estándar de 40 mg de enoxaparina produjo niveles subterapéuticos deaFXa en pacientes críticamente enfermos. Una dosis de 60 mg o 1.5 veces la dosisestándar brinda un mejor resultado en los niveles de aFXa. Una dosis inadecuadapuede ser uno de los mecanismos posibles de elevación de la tasa de falla de eno-xaparina en pacientes que se encuentran en la UCI.34
Una dosis basada en el peso generó mejores niveles anti–Xa, sin riesgo aparen-te de bioacumulación, por lo que puede ser más apropiada, conveniente y eficaz.Un nuevo estudio con 1 mg/kg una vez al día de enoxaparina ha sido aprobadode manera reciente por el Ministerio de Sanidad danés. En la actualidad se realizaun estudio en pacientes con lesión renal aguda con hemofiltración venovenosacontinua, el cual tendrá puntos finales clínicos, como la ocurrencia de ETV, san-grado, tiempo de estancia hospitalaria, número de días sin ventilador y mortali-dad.35 A pesar de que la tromboprofilaxis reduce la incidencia de tromboembolismovenoso en pacientes con enfermedad médica aguda, la correspondiente reducciónen la tasa de mortalidad por cualquier causa no se ha demostrado.
TROMBOPROFILAXIS EN LA UNIDAD DE CUIDADOSINTENSIVOS COMO INDICADOR DE CALIDAD
Desde hace algo más de 30 años la medicina intensiva es una especialidad queha permitido mejorar la atención del paciente crítico. Durante estos años se hanproducido cambios importantes en el manejo de estos pacientes, introduciéndoseavances científicos y tecnológicos especialmente en el monitoreo y el soporte dela disfunción orgánica. Ello ha llevado, sin lugar a dudas, a mejorar la efectividadde la medicina actual, aunque se cobra como precio el hecho de que es menos se-gura y más peligrosa. En palabras de Chantler: “La medicina ha pasado de sersimple, poco efectiva y relativamente segura a convertirse en compleja, efectivapero potencialmente peligrosa”.36 La medicina intensiva es el máximo exponentede ello.
El reto en los próximos años debe ser moldear la efectividad de la medicinaintensiva con el resto de las dimensiones de la calidad y, en el caso de que la segu-ridad entre en conflicto con alguna de ellas, priorizarla para cumplir el aforismohipocrático: “lo primero, no dañar”.
Hasta hace poco tiempo los sistemas sanitarios no se han centrado en la medidade la calidad, lo cual hace que sea difícil monitorear de forma efectiva la calidady la seguridad, responder a la pregunta ¿con qué frecuencia los pacientes recibenlos cuidados adecuados? o comprobar si ciertas iniciativas en la mejora de la cali-dad resultan efectivas. Los sistemas de monitoreo permiten medir y evaluar deforma periódica y planificada los aspectos relevantes de la asistencia mediante

355Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
el uso de indicadores de calidad, que conforman la unidad básica de un sistemade control. Los indicadores de calidad son instrumentos de medida que identifi-can la presencia de un fenómeno o suceso y su intensidad, por lo que deben serfiables, objetivos, aceptables, relevantes y basados en la evidencia. La finalidaddel monitoreo es identificar problemas o situaciones de mejora potencial o biendesviaciones de la práctica estandarizada. Los indicadores actúan como señalesde alarma que advierten acerca de esta posibilidad. Los indicadores de calidaddel paciente crítico se han traducido al inglés y han sido publicados en la páginaweb de la Sociedad Europea de Medicina de Cuidados Intensivos (ESICM, porsus siglas en inglés); estos indicadores han sido referenciados por otras socieda-des científicas internacionales.37,38 En la actualidad la SEMICYUC está colabo-rando en el proyecto Safety Task Force, con el objetivo de definir indicadoresconsensuados que permitan, junto con otras herramientas, evaluar la calidad y laseguridad en las UCI.
El objetivo principal es proporcionar a los profesionales de la salud instrumen-tos para analizar la adecuación de la asistencia al paciente crítico, así como selec-cionar los indicadores básicos o relevantes que puedan ser aplicados en la mayo-ría de los servicios de medicina intensiva, independientemente del nivel decomplejidad del hospital y de la patología específica que atiendan; en este casoexiste un sinnúmero de indicadores del paciente crítico, entre los cuales se en-cuentra el indicador que mide la profilaxis antitrombótica. Cuando la evolucióndel indicador es negativa o los resultados están por debajo del estándar el respon-sable del indicador debe proponer las actuaciones más adecuadas, sea iniciar di-rectamente acciones de mejora o realizar un estudio de las causas que han llevadoa este mal resultado.39
En EUA se han llevado a cabo varios proyectos en un esfuerzo para optimizarla prevención y el tratamiento del tromboembolismo venoso agudo. El proyectode mejoramiento de la atención quirúrgica patrocinado por los centros para servi-cios de Medicare y Medicaid, la Asociación Médica de EUA y otras organizacio-nes relacionadas con la salud, ha incluido la profilaxis de la enfermedad trombo-embólica venosa como un área objetivo para la mejora;40 asimismo, el ForoNacional de la Calidad, junto con la Comisión Conjunta de Acreditación de Orga-nizaciones de Salud, ahora llamada Comisión Conjunta (Joint Commission), estáen proceso de finalizar las medidas de rendimiento para garantizar el riesgo detromboembolismo y la profilaxis en pacientes hospitalizados.41 La tasa de EP fa-tal no ha servido para asegurar el uso óptimo de la profilaxis en estos pacientes.42
Las intervenciones hospitalarias pueden aumentar el uso de anticoagulaciónprofiláctica. Los programas educativos basados en evidencias proporcionan da-tos hospitalarios específicos que demuestran el problema del tromboembolismovenoso y el éxito que puede tener el aumento del uso de la profilaxis por partede los clínicos, como se muestra en un estudio que evaluó el uso de la profilaxis

356 (Capítulo 22)Tratado de trombosis
Cuadro 22–4.
Nombre delindicador
Prevención de la enfermedad tromboembólica
Dimensión SeguridadJustificación El uso de medidas profilácticas de la TVP durante la estancia en la UCI se
asocia a un descenso de la morbimortalidad debida a enfermedad trom-boembólica
Fórmula Número de pacientes con profilaxis de TVP
Número de pacientes ingresadosX 100
Explicación detérminos
Profilaxis de TVP; uso durante toda la estancia hospitalaria de:� Heparina fraccionada� Heparina no fraccionada� Fondaparinux� Anticoagulación completa� Sistemas de compresión (neumática o no) de las extremidades inferiores
Población � Todos los pacientes hospitalizados durante el periodo de estudio.Criterios de exclusión:
� Absolutos: pacientes ingresados para procedimientos con hospitaliza-ción de un día o menos
� Uso de profilaxis farmacológica: existencia de contraindicaciones parael uso de anticoagulación
� Uso de medidas mecánicas: presencia de lesiones en las extremidadesinferiores
Tipo ProcesoFuente de datos Documentación clínicaEstándar 90%
En el estudio de la SEMICYUC (2007) la media alcanzada fue de 77.4%Comentarios El grupo redactor aconseja la medición de este indicador por periodos
TVP: trombosis venosa profunda; UCI: unidad de cuidados intensivos; SEMICYUC: Sociedad Es-pañola de Medicina Intensiva, Crítica y Unidades Coronarias. Fuentes: Crowther MA, Cook DJ:Preventing venous thromboembolism in critically ill patients. Semin Thromb Hemost 2008;34(5):469–474. Chan CM, Shorr AF: Venous thromboembolic disease in the intensive care unit. SeminRespir Crit Care Med 2010;31(1):39–46. Cook DJ, Crowther MA: Thromboprophylaxis in the inten-sive care unit: focus on medical–surgical patients. Crit Care Med 2010;38(Suppl 2):S76–S82.
en 15 hospitales de la comunidad antes y después de un programa de educaciónmédica continua (cuadro 22–4).43
COSTO–BENEFICIO DE LA PROFILAXISTROMBOEMBÓLICA EN PACIENTESCRÍTICAMENTE ENFERMOS
En los pacientes en estado crítico el tromboembolismo venoso en desarrollo con-lleva un mayor tiempo de estancia en la UCI, mayor duración de la ventilación

357Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
mecánica y mayor mortalidad hospitalaria.44 En consecuencia, el tromboembo-lismo venoso no sólo es asociado con una morbilidad y una mortalidad graves,sino que también tiene importantes implicaciones para la utilización de recursossanitarios.
El uso apropiado de la profilaxis para prevenir el tromboembolismo venosoen pacientes con riesgo se ha identificado como una de las intervenciones másimportantes de seguridad del paciente por parte de los hospitales.45 Sin embargo,la variabilidad es considerable con el uso de dicha profilaxis en la práctica. Laprevención se logra habitualmente con medicamentos anticoagulantes. Dado quelas decisiones importantes sobre intervenciones farmacológicas se hacen con co-nocimiento de su consecuencia económica, se necesitan herramientas útiles enel análisis económico para guiar a los médicos y los responsables administrativossobre el valor de los medicamentos y sus consecuencias.46 Sin embargo, muchasevaluaciones de los nuevos fármacos no se producen de forma prospectiva conlos estudios de eficacia y varios de ellos son patrocinados por los fabricantes, au-mentando la posibilidad de sesgo.
Se llevó a cabo una revisión sistemática de los análisis económicos de las estra-tegias de prevención del tromboembolismo venoso en pacientes hospitalizados47
y se encontró que las HBPM parecen ser la estrategia económicamente más atrac-tiva para la prevención del tromboembolismo venoso en la mayoría de los pacien-tes médicos y quirúrgicos, mientras que el fondaparinux es económicamente másatractivo para los pacientes ortopédicos. Sin embargo, los estudios pueden tenerel riesgo de sobreestimar la eficacia y subestimar los efectos secundarios, comoel sangrado. Aproximadamente dos tercios de todas las evaluaciones fueron fi-nanciadas directamente por el fabricante de los nuevos medicamentos, por lo queestos fármacos eran más atractivos desde el punto vista económico, en compara-ción con otras estrategias. En el futuro se recomienda planificar un análisis pros-pectivo de costo–efectividad de la intervención de alta calidad junto con ensayos;asimismo, que éstos sean diseñados, realizados, analizados y reportados indepen-dientemente de patrocinadores de la industria farmacéutica.
Para concluir, la novena edición del American College of Chest Physicians yla última Campaña para Sobrevivir a la Sepsis recomiendan el uso de la heparinano fraccionada o HBPM para combatir la TVP en pacientes en estado crítico. Latromboprofilaxis mecánica con dispositivos de compresión intermitente o gra-duada se recomienda en pacientes con un riesgo elevado de hemorragia o contra-indicaciones para la heparina.48,49
En las UCI de Europa una dosis estándar de 40 mg al día de enoxaparina subcu-tánea (SC) se emplea a menudo para la profilaxis de la trombosis.50 Dicha dosisprodujo niveles significativamente más altos de anti–Xa y proporcionalmentemenor incidencia de TVP.51 Los pacientes que recibieron 1 mg/kg/día de enoxa-parina consiguieron niveles constantes de anti–Xa a partir del primer día de admi-

358 (Capítulo 22)Tratado de trombosis
nistración, lo cual podría ser ventajoso en una población en la que la ocurrenciade TVP puede tener graves consecuencias. Los niveles pico de anti–Xa no su-peran los niveles aceptables máximos anti–Xa de 0.4 UI/mL para las dosis de 40mg dos veces al día y 1 mg/kg/día de enoxaparina. Los niveles de anti–Xa másaltos son útiles en pacientes en estado crítico, ya que pueden conferir mejor trom-boprofilaxis. En un estudio anterior se estableció que los pacientes eran más pro-pensos a mantener niveles de anti–Xa dentro del intervalo terapéutico durante pe-riodos más largos cuando se administran dosis más altas de enoxaparina.52 Unadosis basada en el peso produjo mejores niveles de anti–Xa sin riesgo aparente debioacumulación y puede, por lo tanto, ser más apropiada, conveniente y eficaz.35
En un estudio reciente realizado por Cook y col.4 la hemorragia mayor ocurrióen 5.5% de los pacientes ingresados en la UCI asignados a recibir tromboprofila-xis con HBPM. Muchos pacientes críticamente enfermos experimentan episo-dios de sangrado, la mayoría de los cuales son menores. La hemorragia grave omortal en la UCI se cree que está asociada con las pruebas de coagulación globa-les prolongadas y un bajo recuento de plaquetas, y no al uso de anticoagulantesprofilácticos o agentes antiplaquetarios.53
El reto para el intensivista que atiende pacientes graves con sepsis, trauma, fa-lla cardiovascular, pulmonar u orgánica múltiple, y que desarrollan trombosis esintegrar cuadros clínicos e identificar los factores de riesgo específicos que tienentrascendencia en la mejoría terapéutica y en la disminución de la morbimortali-dad secundaria. Se deben realizar pruebas de diagnóstico tempranas ante la sos-pecha de trombosis o en pacientes con muchos factores de riesgo en el momentode su ingreso en la UCI; de igual forma, los cuidados se deben enfocar en la pre-vención y el diagnóstico temprano, así como en evitar procedimientos invasivosinnecesarios.
REFERENCIAS
1. Committee on Quality of Health Care in America: Crossing the quality chasm: a new healthsystem for the 21st Century. Washington, DC, National Academy Press, 2001.
2. Patel R, Cook DJ, Meade MO, Griffith LE, Mehta G et al., for the Burden of Illness inVenous Thrombo Embolism in Critical Care (BITEC) Study Investigators, Canadian Criti-cal Care Trials Group: Burden of illness in venous thromboembolism in critical care: a mul-ticenter observational study. J Crit Care 2005;20:341–347.
3. Geerts WH, Bergqvist D, Pineo GF, Heit JA, Samama CM et al., American College ofChest Physicians: Prevention of venous thromboembolism: American College of ChestPhysicians evidence–based clinical practice guidelines (8ª Edition). Chest 2008;133:381S–453S.
4. Cook DJ, Crowther MA: Thromboprophylaxis in the intensive care unit: focus on medi-cal–surgical patients. Crit Care Med 2010;38:S76–S82.
5. Shorr AF, Williams MD: Venous thromboembolism in critically ill patients. Observationsfrom a randomized trial in sepsis. Thromb Haemost 2009;101:139–144.

359Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
6. Cook D, Meade M, Guyatt G, Walter S, Heels Ansdell D et al., PROTECT Investigatorsfor the Canadian Critical Care Trials Group and the Australian and New Zealand IntensiveCare Society Clinical Trials Group: Dalteparin versus unfractionated heparin in criticallyill patients. N Engl J Med 2011;364:1305–1314.
7. Cook et al.: Clinically important deep vein thrombosis in the intensive care unit: a surveyof intensivists. The Canadian Critical Care Trials Group. Res Crit Care 2004;8(3).
8. Cohen AT, Alikhan R, Arcelus JI et al.: Assessment of venous thromboembolism risk andthe benefits of thromboprophylaxis in medical patients. Thromb Haemost 2005;94:750–759.
9. Haas SK: Venous thromboembolic risk and its prevention in hospitalized medical patients.Semin Thromb Hemost 2002;28:577–584.
10. Charles W, Francis MD: Prophylaxis for thromboembolism in hospitalized medical pa-tients. N Engl J Med 2007;356:1438–1444.
11. McKelvie PA: Autopsy evidence of pulmonary thromboembolism. Med J Aust 1994;160:127–128.
12. Hirsch DR, Ingenito EP, Goldhaber SZ: Prevalence of deep venous thrombosis amongpatients in medical intensive care. JAMA 1995:274:335–337.
13. Agu O, Hamilton G, Baker D: Graduated compression stockings in the prevention of ve-nous thromboembolism. Br J Surg 1999;86:992–1004.
14. Amaragiri SV, Lees TA: Elastic compression stockings for prevention of deep vein throm-bosis. Cochrane Database Syst Rev 2000;3:CD001484.
15. Geerts WH, Pineo GF, Heit JA et al.: Prevention of venous thromboembolism: the Se-venth ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126(Suppl):338S–400S.
16. Prevention of fatal postoperative pulmonary embolism by low doses of heparin: an interna-tional multicentre trial. Lancet 1975;2:45–51.
17. Collins R, Scrimgeour A, Yusuf S, Peto R: Reduction in fatal pulmonary embolism andvenous thrombosis by perioperative administration of subcutaneous heparin: overview ofresults of randomized trials in general, orthopedic, and urologic surgery. N Engl J Med 1988;318:1162–1173.
18. Collins R, MacMahon S, Flather M et al.: Clinical effects of anticoagulant therapy in sus-pected acute myocardial infarction: systematic overview of randomized trials. Br Med J1996;313:652–659.
19. Bratzler DW, Raskob GE, Murray CK, Bumpus LJ, Piatt DS: Underuse of venousthromboembolism prophylaxis for general surgery patients: physician practices in the com-munity hospital setting. Arch Intern Med 1998;158:1909–1912.
20. Cohen AT, Tapson VF, Bergmann JF et al.: Venous thromboembolism risk and prophyla-xis in the acute hospital care setting (ENDORSE study): a multinational cross–sectional stu-dy. Lancet 2008;371:387–394.
21. Nicolaides AN, Fareed J, Kakkar AK et al.: Prevention and treatment of venous thrombo-embolism: international consensus statement. Int Angiol 2006;25:101–161.
22. Samama MM, Cohen AT, Darmon JY et al.: A comparison of enoxaparin with placebofor the prevention of venous thromboembolism in acutely ill medical patients. N Engl J Med1999;341:793–800.
23. Leizorovicz A, Cohen AT, Turpie AGG, Olsson CG, Vaitkus PT et al.: Randomized pla-cebo–controlled trial of dalteparin for the prevention of venous thromboembolism inacutely ill medical patients. Circulation 2004;110:874–879.
24. Sherman DG, Albers GW, Bladin C et al.: The efficacy and safety of enoxaparin versusunfractionated heparin for the prevention of venous thromboembolism after acute ischae-

360 (Capítulo 22)Tratado de trombosis
mic stroke (PREVAIL Study): an open–label randomized comparison. Lancet 2007;369:1347–1355.
25. Hull RD, Schellong S, Tapson V et al.: Extended–duration venous thromboembolism pro-phylaxis in acutely ill medical patients with recent reduced mobility: the EXCLAIM study.En: Program and abstracts of the XXI Congress of the International Society on Thrombosisand Haemostasis. Ginebra, 6 a 12 de julio de 2007:O–S–001. (Abstract.)
26. Rogers FB, Shackford SR, Ricci MA, Wilson JT, Parsons S: Routine prophylactic venacava filter insertion in severely injured trauma patients decreases the incidence of pulmona-ry embolism. J Am Coll Surg 1995;180:641–647.
27. Horlocker TT, Heit JA: Low molecular weight heparin: biochemistry, pharmacology, per-ioperative prophylaxis regimens, and guidelines for regional anesthetic management.Anesth Analg 1997;85:874–885.
28. Dörffler Melly J, de Jonge E, Pont AC, Meijers J, Vroom MB et al.: Bioavailability ofsubcutaneous low–molecular–weight heparin to patients on vasopressors. Lancet 2002;359:849–850.
29. Mayr AJ, Dünser M, Jochberger S, Fries D, Klingler A et al.: Antifactor Xa activity inintensive care patients receiving thromboembolic prophylaxis with standard doses of enox-aparin. Thromb Res 2002;105:201–204.
30. Levine MN, Planes A, Hirsh J, Goodyear M, Vochelle N et al.: The relationship betweenanti–factor Xa level and clinical outcome in patients receiving enoxaparin low molecularweight heparin to prevent deep vein thrombosis after hip replacement. Thromb Haemost1989;62:940–944.
31. Priglinger U, Delle Karth G, Geppert A, Joukhadar C, Graf S et al.: Prophylactic anti-coagulation with enoxaparin: is the subcutaneous route appropriate in the critically ill? CritCare Med 2003;31:1405–1409.
32. Cook D, Crowther M, Meade M, Rabbat C, Griffith L et al.: Deep venous thrombosisin medical–surgical critically ill patients: prevalence, incidence, and risk factors. Crit CareMed 2005;33:1565–1571.
33. Duplaga BA, Rivers CW, Nutescu E: Dosing and monitoring of low–molecular– weightheparins in special populations. Pharmacotherapy 2001;21:218–234.
34. Robinson S, Zincuk A, Strøm T, Bjerregaard T, Rasmussen B et al.: Enoxaparin, effec-tive dosage for intensive care patients: double–blinded, randomized clinical trial. Crit Care2010;14:R41. http://ccforum.com/content/14/2/R41.
35. Sian Robinson et al.: A comparative study of varying doses of enoxaparin for thrombopro-phylaxis in critically ill patients: double–blinded, randomized controlled trial. Crit Care2013;17:R75.
36. Chantler C: The role and education of doctors in the delivery of health care. Lancet 1999;3(353):1178–1181.
37. Braun JP, Bause H, Bloos F, Geldner G, Kastrup M et al.: NeQuI (Quality Network inIntensive Care Medicine). Ger Med Sci 2010;8:Doc23.
38. Ray B, Samaddar DP, Todi SK, Ramakrishnan N, John G et al.: Quality indicators forICU: ISCCM guidelines for ICUs in India. Indian J Crit Care Med 2009;13:173–206.
39. Martín MC, Saura RM, Cabré L, Ruiz J, Blanch L et al., Grupos de Trabajo de la Socie-dad de Medicina Intensiva Crítica y Unidades Coronarias (SEMICYUC), Sociedad Españo-la de Enfermería Intensiva y Unidades Coronarias (SEEIUC) y Fundación Avedis Donabe-dian (FAD): Indicadores de calidad en el enfermo crítico. Med Intens 2008;32:23–32.
40. Surgical Care Improvement Project: SCIP process and outcome measures. http://www.medqic.org/scip 2005.

361Trombosis en el paciente críticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
41. National Quality Forum: NQF project brief: national voluntary consensus standards for theprevention and care of venous thromboembolism (including deep–vein thrombosis and pul-monary embolism). Enero 2006. http://www.qualityforum.org/projects/ongoing/vte/index.asp.
42. Tapson VF: Acute pulmonary embolism. N Engl J Med 2008;358. www.nejm.org.43. Anderson FA Jr, Wheeler HB, Goldberg RJ, Hosmer DW, Forcier A et al.: Changing
clinical practice: prospective study of the impact of continuing medical education and qual-ity assurance programs on use of prophylaxis for venous thromboembolism. Arch InternMed 1994;154:669–677.
44. Patel R, Cook DJ, Meade MO, Griffith LE, Mehta G et al., for the Burden of Illness inVenous Thrombo Embolism in Critical Care (BITEC) Study Investigators; Canadian Criti-cal Care Trials Group: Burden of illness in venous thromboembolism in critical care: a mul-ticenter observational study. J Crit Care 2005;20:341–347.
45. Shojania KG, Duncan BW, McDonald KM, Wachter RM, Markowitz AJ: Evidencereport/Technology Assessment Number 43: Making health care safer: a critical analysisof patient safety practices: summary. http://www. ahrq.gov/clinic/ptsafety/summary.htm.
46. Olin G, Machlin R, Agency for Healthcare Research and Quality, Center for Cost and Fi-nancing Studies: Health care expenses in the US civilian population. http://www.meps.ahrq.gov/papers/rf_01–r035/statisticaltables.htm.
47. Thirugnanam S, Pinto R, Cook DJ, Geerts WH, Fowler RA: Economic analyses of ve-nous thromboembolism prevention strategies in hospitalized patients: a systematic review.Crit Care 2012;16:R43. http://ccforum.com/content/16/2/R43.
48. Kahn SR, Lim W, Dunn AS, Cushman M, Dentali F et al., American College of ChestPhysicians: Prevention of VTE in nonsurgical patients: antithrombotic therapy and preven-tion of thrombosis. 9ª ed. American College of Chest Physicians Evidence–Based ClinicalPractice Guidelines. Chest 2012;141:e195S–e226S.
49. Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H et al., Surviving Sepsis Cam-paign Guidelines Committee including The Pediatric Subgroup: Surviving Sepsis Cam-paign: international guidelines for management of severe sepsis and septic shock, 2012. In-tens Care Med 2013;39:165–228.
50. Horlocker TT, Heit JA: Low molecular weight heparin: biochemistry, pharmacology, per-ioperative prophylaxis regimens, and guidelines for regional anesthetic management.Anesth Analg 1997;85:874–885.
51. Riha GM, Van PY, Differding JA, Schreiber MA: Incidence of deep vein thrombosis isincreased with 30 mg twice daily dosing of enoxaparina compared with 40 mg daily. Am JSurg 2012;203:598–602.
52. Robinson S, Zincuk A, Strom T, Larsen TB, Rasmussen B et al.: Enoxaparin, effectivedosage for intensive care patients: double–blinded, randomized clinical trial. Crit Care2010;14:R41.
53. Arnold DM, Donahoe L, Clarke FJ, Tkaczyk AJ, Heels Ansdell D et al.: Bleeding duringcritical illness: a prospective cohort study using a new measurement tool. Clin Invest Med2007;30:E93–E102.

362 (Capítulo 22)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
23Trombocitopenia inducida por
heparina. Síndrome de HITRené Bourlon Cuéllar, María Teresa Bourlon de los Ríos,
Christianne Bourlon de los Ríos
INTRODUCCIÓN
La trombocitopenia es una complicación bien conocida de la terapia con hepari-na, que se presenta entre 4 y 10 días después de iniciada la administración del fár-maco. Existen dos variantes del síndrome de HIT (heparin–induced thrombocy-topenia): tipo I y tipo II (cuadro 23–1).
� Tipo I. Esta forma carece de consecuencias clínicas; se caracteriza por unacaída en la cuenta plaquetaria moderada (100 000/�L) que ocurre en los pri-meros dos días de administración de la heparina y generalmente vuelve a
Cuadro 23–1. Diferencias entre los tiposde trombocitopenia inducida por heparina
Variables HIT tipo I HIT tipo II
Frecuencia de presentación en pacien-tes tratados con heparina
10 a 20% 1 a 3%
Inicio (días de terapia con heparina) 1 a 4 días 5 a 10 díasNadir de la cuenta plaquetaria 100 000/�L 30 000 a 50 000/�LMediada por anticuerpos No SíEventos tromboembólicos Ninguno 30 a 80%Secuelas hemorrágicas Nunca Raras
Manejo Observación Suspender la heparina e iniciar la anti-coagulación alternativa y terapiasadicionales
363

364 (Capítulo 23)Tratado de trombosis
la normalidad aun continuando la terapia con este fármaco. El mecanismomediante el cual se produce un descenso plaquetario no es de origen inmu-nitario, y parece estar mediado por un efecto de la heparina sobre la agrega-ción plaquetaria. Se recomienda la observación, aunque no es necesariosuspender la heparina.1
� Tipo II. Esta variante de HIT se denomina trombocitopenia asociada conheparina, trombocitopenia y trombosis asociadas con heparina, y síndromedel coágulo blanco (haciendo referencia al trombo arterial rico en plaquetasque caracteriza a la enfermedad). Constituye la forma más grave de HIT yes una patología inmunitaria mediada por anticuerpos (contra el complejofactor plaquetario 4 y heparina). Las cuentas plaquetarias son más bajas (de30 000 a 50 000/�L) que en el tipo I, y se presentan entre 4 y 10 días despuésde iniciada la heparina, con una frecuencia baja (de 1 a 3% de los pacientescon heparina). Se caracteriza por trombocitopenia y eventos trombóticosque pueden poner en peligro la vida y las extremidades. Se presenta con ma-yor frecuencia (5%) en pacientes con heparina no fraccionada que en quie-nes reciben heparina de bajo peso molecular (< 1%).
DEFINICIÓN
El diagnóstico de HIT se define con base en tres criterios:2
1. El paciente recibe heparina o tuvo una exposición reciente a ella.2. La presencia de al menos una característica clínica del síndrome (tromboci-
topenia con o sin trombosis).3. Pruebas de laboratorio de anticuerpos anti–HIT (contra el complejo PF4–
heparina).
HISTORIA
Weismann y Tobin describieron este síndrome por primera vez en 19583 con elcaso de un embolismo arterial asociado con heparina con coloración asalmonadaque en el examen histopatológico careció de glóbulos rojos en su interior; sin em-bargo, en ese momento no se relacionó este suceso con la trombocitopenia. Losreportes de casos posteriores documentaron la coexistencia de trombocitopenia.En 1973 Rodees y col. sugirieron la existencia de un mecanismo inmunitario im-plicado en la fisiopatología de la HIT, dado que el suero de los pacientes con elsíndrome causaba agregación plaquetaria.4

365Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
EPIDEMIOLOGÍA
La incidencia de HIT y el contexto clínico están influidos por la fórmula de hepa-rina (convencional o de bajo peso molecular).
Se estima que entre 10 y 20% de los pacientes que reciben heparina no fraccio-nada presentan una caída de la cuenta plaquetaria inferior al rango normal o unareducción de 50% de la cuenta normal basal. No obstante, la mayoría de los casosrepresentan un cuadro de HIT tipo I. La incidencia del síndrome de HIT tipo IIes de 0.2 a 3% de los pacientes expuestos a heparina durante más de cuatro días.
Según algunos estudios recientes la incidencia del síndrome de HIT tipo II esmayor con el uso de heparina no fraccionada que con el uso de heparina de bajopeso molecular (HBPM); asimismo, el porcentaje de pacientes con anticuerposIgG dependientes de heparina es mayor en los pacientes tratados con heparinaconvencional.5,6 Un hallazgo interesante es que los pacientes sometidos a cirugíacardiaca presentan con mayor frecuencia Ig anti–PF4/heparina que los pacientescon intervenciones ortopédicas. No obstante, el porcentaje de pacientes con anti-cuerpos positivos que desarrollan enfermedad es mayor en los pacientes someti-dos a cirugía ortopédica (4.9%) que los sometidos a cirugía cardiaca (1%).7 Estesíndrome es raro entre los pacientes pediátricos y obstétricas y los que recibenhemodiálisis crónica. Después de las complicaciones hemorrágicas el síndromede HIT constituye la complicación más importante del uso de heparina no frac-cionada (afecta a 3% de los pacientes).
FISIOPATOLOGÍA
El síndrome de HIT tipo II es causado por la presencia de anticuerpos (Ig–HIT).Los anticuerpos de esta afección pueden ser IgG, IgM o IgA, no están dirigidoscontra la heparina por sí sola y reconocen el complejo antigénico del factor 4 pla-quetario (PF4) y heparina. Se especula que la unión del PF4 con la heparina gene-ra un cambio conformacional de la molécula dando origen a un epítope antigéni-co. Dichos anticuerpos están presentes en casi todos los pacientes con estediagnóstico y son patógenos en los animales. No obstante, las inmunoglobulinasse pueden encontrar en pacientes expuestos a heparina en diversas ocasiones,pero no necesariamente en enfermedad clínicamente significativa.
Los complejos antígeno y anticuerpo resultantes activan las plaquetas, las cua-les se agregan y son removidas de la circulación antes de tiempo, dando lugar ala trombocitopenia inducida por heparina. Las plaquetas activadas también libe-ran micropartículas procoagulantes que activan las células endoteliales y los mo-nocitos. La activación de las células endoteliales resulta en la liberación de inter-
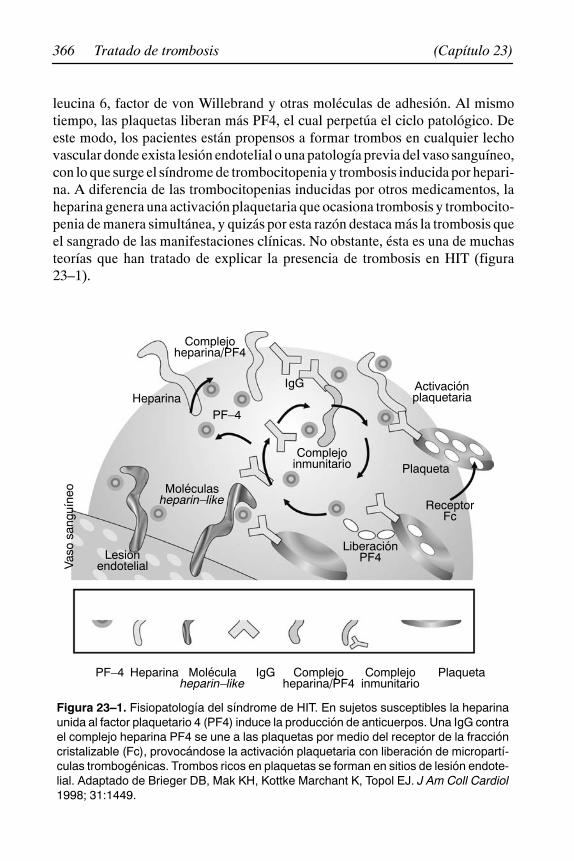
366 (Capítulo 23)Tratado de trombosis
leucina 6, factor de von Willebrand y otras moléculas de adhesión. Al mismotiempo, las plaquetas liberan más PF4, el cual perpetúa el ciclo patológico. Deeste modo, los pacientes están propensos a formar trombos en cualquier lechovascular donde exista lesión endotelial o una patología previa del vaso sanguíneo,con lo que surge el síndrome de trombocitopenia y trombosis inducida por hepari-na. A diferencia de las trombocitopenias inducidas por otros medicamentos, laheparina genera una activación plaquetaria que ocasiona trombosis y trombocito-penia de manera simultánea, y quizás por esta razón destaca más la trombosis queel sangrado de las manifestaciones clínicas. No obstante, ésta es una de muchasteorías que han tratado de explicar la presencia de trombosis en HIT (figura23–1).
Figura 23–1. Fisiopatología del síndrome de HIT. En sujetos susceptibles la heparinaunida al factor plaquetario 4 (PF4) induce la producción de anticuerpos. Una IgG contrael complejo heparina PF4 se une a las plaquetas por medio del receptor de la fraccióncristalizable (Fc), provocándose la activación plaquetaria con liberación de micropartí-culas trombogénicas. Trombos ricos en plaquetas se forman en sitios de lesión endote-lial. Adaptado de Brieger DB, Mak KH, Kottke Marchant K, Topol EJ. J Am Coll Cardiol1998; 31:1449.
PF–4 Heparina Moléculaheparin–like
IgG Complejoheparina/PF4
Complejoinmunitario
Plaqueta
Lesiónendotelial
Moléculasheparin–like
Complejoheparina/PF4
Activaciónplaquetaria
ReceptorFc
LiberaciónPF4
IgG
Complejoinmunitario Plaqueta
Heparina
Vas
o sa
nguí
neo
PF–4

367Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Los trombos observados en esta afección son ricos en plaquetas y se denomi-nan trombos blancos, de donde se deriva el nombre de síndrome de trombo blanco.
ETIOLOGÍA
Si bien la mayoría de los pacientes que desarrollan HIT han estado expuestos aheparina IV o SC, la dosis requerida para causar este síndrome parece pequeña,pues se han reportado casos con sólo 250 U (catéteres permeabilizados o catéterescubiertos con heparina).
CUADRO CLÍNICO
La mayoría de las veces se presenta un cuadro de trombocitopenia con o sin trom-bosis.
Trombocitopenia
Una caída en la cuenta plaquetaria basal mayor de 50% o una trombocitopeniaabsoluta entre 4 y 10 días después de iniciada la exposición en ausencia de otrascausas de trombocitopenia debe considerarse como HIT hasta demostrar lo con-trario. Los pacientes que hayan recibido heparina en los 100 días previos puedentener caídas de la cuenta plaquetaria mucho más rápidas. La mayoría de los pa-cientes mantienen una cuenta plaquetaria mayor de 30 000/�L y sólo 20% llegana tener cuentas menores (el nadir promedio es de 60 000/�L), por lo que el san-grado espontáneo es inusual. A pesar de que en ocasiones la trombocitopeniapuede ser grave, las complicaciones por sangrado no son frecuentes. Las presen-taciones mayores de más de dos semanas de iniciado el tratamiento son inusuales(puesto que el desarrollo de anticuerpos se da entre los cinco y los ocho días detratamiento y rara vez después de este periodo) y las presentaciones más tempra-nas indican que el paciente estaba previamente sensibilizado. A veces las plaque-tas son < 10 000/�L, y es muy raro que los pacientes presenten sangrado aun conestas cifras plaquetarias, ya que la mayoría de las veces se recuperan entre 4 y 14días después de haber suspendido la heparina.
Trombosis
Los pacientes con HIT tienen un mayor riesgo de trombosis que de sangrado, aunen asociación con plaquetopenias graves, así como de desarrollar nuevas trombo-

368 (Capítulo 23)Tratado de trombosis
sis y extensión de las ya existentes. Puede ser la manifestación inicial y presentar-se en el periodo de trombocitopenia o varios días después de que se haya resuelto,incluso con la descontinuación de la terapia con heparina. La mayoría de loseventos trombóticos suelen ser venosos, aunque se presentan arteriales.
Las situaciones clínicas incluyen trombosis venosa profunda (TVP), trombo-embolia pulmonar (TEP), trombosis arterial de las extremidades, gangrena veno-sa de los miembros, EVC trombótico, trombosis venosa cerebral, infarto agudodel miocardio, infarto mesentérico e infarto renal. Las dos manifestaciones másfrecuentes son TVP y TEP. En general, las complicaciones trombóticas se presen-tan entre 20 y 50% de los pacientes,8 pero los pacientes con HIT tienen un riesgo30 veces mayor que la población general,9 el cual se mantiene días o semanas des-pués de suspendida la heparina e incluso cuando la cuenta plaquetaria se norma-liza.
Otras manifestaciones
Son menos frecuentes que las anteriores e incluyen lesiones cutáneas inducidaspor heparina, infarto hemorrágico adrenal (asociado con una mortalidad de 8 a20%, independientemente del tratamiento), reacciones sistémicas agudas (esca-lofríos, disnea y paro cardiorrespiratorio secundarios a la administración de bolosde heparina IV), isquemia de miembros con riesgo de amputación (con una inci-dencia de 5 a 10%) y en raras ocasiones amnesia transitoria global.10
Según los reportes de casos, la mayoría de los pacientes (40%) debutan contrombocitopenia, en otro grupo (cerca de 30%) se presentan trombocitopenia ytrombosis concomitantemente, y en otro más (30%) se manifiesta la trombosisantes de la aparición de la trombocitopenia.
Las trombosis se pueden encontrar en cualquier lecho vascular y predominanen los pacientes ortopédicos y médicos. Las trombosis arteriales son más frecuen-tes que las venosas en pacientes con antecedentes de cirugía cardiovascular pre-via. Hasta el momento se desconoce por qué las trombosis se presentan en unospacientes y en otros no. En estudios transversales se ha encontrado una relaciónde las manifestaciones trombóticas con la elevación de marcadores de activaciónplaquetaria, el incremento en la producción de trombina, los títulos mayores deanticuerpos contra el complejo PF4–heparina y el descenso de la cuenta plaqueta-ria > 70% de la basal.
DIAGNÓSTICO
El primer paso es la detección del síndrome, que aparece como trombocitopeniao trombosis, o de lesiones cutáneas en los sitios de inyección asociadas con el uso

369Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
de heparina en los últimos 4 a 10 días o con la administración de HBPM duranteun tiempo prolongado. Al principio el diagnóstico es sobre todo clínico, puestoque quizá no se dispone de los ensayos de laboratorio.
Laboratorio
Se puede realizar la detección en suero de anticuerpos HIT–Ig mediante un ensa-yo funcional específico y sensible (ensayo de la liberación plaquetaria de seroto-nina), la inducción de la agregación plaquetaria con heparina o un ELISA paraanticuerpos PF4/heparina. Asimismo, se pueden detectar anticuerpos circulantesIgA, IgG e IgM.
� El ensayo funcional constituye la regla de oro para medir la activación pla-quetaria. Se utilizan plaquetas de donadores sanos radiomarcadas con 14C–serotonina y se agrega el suero del paciente con heparina; se considera unresultado positivo cuando existe liberación de serotonina. Este estudio tieneuna sensibilidad de 90% y una especificidad que oscila entre 77 y 100%.
� En el ensayo de inducción de agregación plaquetaria se utilizan plaquetasde donadores sanos a las que se les agrega suero del paciente, para medirla agregación antes y después de agregar la heparina. Un ensayo positivoes el que no muestra agregación plaquetaria en ausencia de heparina, agre-gación con dosis bajas de heparina y ausencia de agregación a altas dosis.La prueba es muy específica (> 90%), pero carece de sensibilidad.
� La sensibilidad del inmunoensayo es buena (> 97%), aunque su especifici-dad es limitada (de 74 a 86%), porque hace detecciones en pacientes queno tienen HIT. El valor predictivo positivo de la prueba es bajo (oscila entre10 y 93%), mientras que el valor predictivo negativo es bueno (> 95%). Adiferencia de los anteriores, éste no es un estudio funcional. En esta pruebase le agrega suero del paciente a los complejos de heparina/PF4; se determi-na si el paciente tiene anticuerpos contra el complejo cuando se agrega unanticuerpo y se observa aglutinación.
Índice de sospecha clínica
Se establece entre 4 y 10 días después de iniciada la heparina, con base en la pre-sencia de trombocitopenia, una nueva trombosis (con la exclusión de otras cau-sas, como púrpura postransfusional, fármacos distintos a la heparina, etc.) y alzaplaquetaria al descontinuar la heparina. La reunión de los tres criterios equivalea una alta sospecha, la presencia de uno o dos constituye una sospecha intermediay la ausencia de ellos equivale a una sospecha baja.

370 (Capítulo 23)Tratado de trombosis
La detección de anticuerpos es útil cuando la sospecha de la enfermedad es altao intermedia, debido a que un resultado negativo tiene un alto valor predictivonegativo y sugiere otro diagnóstico.11 En general, no se recomienda cuando lasospecha clínica es baja. El ensayo funcional puede ser útil cuando la sospechaclínica es intermedia y la serología es positiva, puesto que un resultado positivoincrementa la posibilidad de estar ante un cuadro de HIT (figura 23–2).
Figura 23–2. Algoritmo diagnóstico para síndrome de HIT. La trombocitopenia absoluta(plt < 150 000) o relativa (reducción de > 50% de basal) en un paciente con heparinano fraccionada o heparina de bajo peso molecular. Se debe estudiar en base a la sospe-cha clínica alta, intermedia o baja. La decisión de continuar con heparina en pacientescon una sospecha clínica baja es dependiente de las circunstancias individuales del pa-ciente. El ensayo funcional es de gran utilidad en pacientes con sospecha intermediay serología positiva, puesto que incrementa notablemente la sensibilidad de la prueba.El inicio de anticoagulación alternativa con no heparínicos es dependiente del riesgo desangrado y trombosis. Adaptado de Arepally GM, Ortel TL: Heparin–induced thrombo-cytopenia. N Engl J Med 2006;355:809–817.
Trombocitopenia en un paciente con heparina o HBPM
Sospecha clínica intermedia
Descontinuar la terapia con
Resultados inmunoensayo
Ac’s positivoscon sospecha
Dx confirmado
Ac’s positivoscon sospecha
Ac’s negativoscon sospecha
Ac’s negativoscon sospecha
Ensayo funcional
Sospecha clínica baja
Descartar otros dignósticos
Se puede continuar laterapia con heparina
Considerar Dxalternativo, HIT
intermedio
Considerar Dxalternativo, se puede
reiniciar heparina
Positivo: HITprobable
intermediaaltaintermediaalta
Negativo: HITindeterminado
heparina
o alta

371Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
DIAGNÓSTICOS DIFERENCIALES
Es importante hacer hincapié en que en otro tipo de trombocitopenias de etiologíainmunitaria, como la púrpura trombocitopénica idiopática y la púrpura postrans-fusional, los pacientes presentan cuentas plaquetarias más bajas (< 10 000/�L)y sangrados clínicamente aparentes.
Se deben excluir otras causas de trombocitopenia, como infección bacteriana,infección medicamentosa (diferente de la heparina) y enfermedad primaria de lamédula ósea, entre otras.
SÍNDROME DE HIT DE INICIO TARDÍO
Hay una variante del síndrome de HIT que se presenta después de la suspensióndel tratamiento con heparina. Su incidencia se desconoce, y casi siempre se pre-senta a los 14 días de la exposición (puede variar entre 9 y 40 días).12
Los siguientes criterios sirven para diagnosticar HIT:
� Exposición a la heparina y alta hospitalaria con curso clínico indolente, enel que el síndrome de HIT quizá no fue reconocido.
� Representación clínica con trombosis venosa y arterial, y desarrollo de latrombocitopenia posterior a otra exposición a la heparina.
� Serología positiva para anticuerpos inducidos por heparina.
SÍNDROME DE HIT DE INICIO TEMPRANO
Los pacientes presentan el cuadro clínico cerca de 10.5 h después de iniciada laheparina.
También se observa en 30% de los pacientes con serología persistentementepositiva, debido a terapia con heparina durante los tres meses previos.13
PREVENCIÓN
La HBPM y otros heparínicos (danaparoide) se relacionan con una incidenciamenor de HIT que la heparina convencional, por lo que se prefiere la heparinaconvencional en caso de que estén indicados. Otras estrategias planteadas consis-

372 (Capítulo 23)Tratado de trombosis
ten en limitar el tiempo de tratamiento con heparina a menos de cinco días y cam-biar a warfarina de manera temprana, para minimizar el tiempo de exposición.
TRATAMIENTO
Se recomienda observar y vigilar a los pacientes que tienen HIT tipo I. No se reco-mienda el tratamiento en los pacientes que tienen anticuerpos sin otras manifesta-ciones de la enfermedad. Existen pocos estudios prospectivos aleatorizados so-bre los cuales basar la decisión terapéutica para el síndrome de HIT tipo II. Elprincipal objetivo es reducir el riesgo de trombosis mediante la disminución dela actividad plaquetaria y la generación de trombina.
1. Suspender la administración de heparina no fraccionada o heparina de bajopeso molecular por cualquier vía de administración y no realizar una hepari-nización de catéteres y retiro de catéteres intravasculares con recubrimientode heparina. La sola suspensión de heparina no es un tratamiento suficiente,pues el paciente tiene un elevado riesgo de desarrollar trombosis.
2. En los pacientes con HIT con trombosis o sin ella se recomienda la anticoa-gulación con un medicamento alterno heparínico (danaparoide) o no hepa-rínico (lepirudina, argatrobán o bivalirudina). Se desconoce el tiempo deanticoagulación al que deben ser sometidos estos pacientes.
3. La warfarina se debe evitar en el episodio agudo, a menos que sea usada encombinación con dosis terapéuticas de danaparoide, lepirudina o argatro-bán, y sobre todo cuando se haya resuelto la trombocitopenia, debido a quese ha vinculado con el empeoramiento de la trombosis venosa, de la gangre-na venosa de las extremidades y de la necrosis cutánea cuando se usa solao combinada con ancrod en el síndrome de HIT agudo.
4. No se indica la transfusión de plaquetas de manera profiláctica, sobre todoporque en el síndrome de HIT es rara la aparición de petequias o de sangradopor trombocitopenia. Además, se han reportado eventos trombóticos poste-riores a la administración de plaquetas.
5. La trombolectomía quirúrgica y la trombólisis local o sistémica pueden serapropiadas para los pacientes seleccionados con tromboembolismo arterialde grandes vasos o tromboembolia pulmonar.
6. La heparina de bajo peso molecular no se debe administrar en pacientes conHIT, ya que existen reportes de falla al tratamiento y otras complicaciones.
7. Prevención. La administración de heparina no fraccionada y heparina debajo peso molecular debe evitarse en los pacientes con historial de HIT tipoII; sin embargo, en circunstancias especiales se pueden considerar excep-

373Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ciones (uso de heparina no fraccionada en un paciente que será sometido acirugía cardiaca si los anticuerpos anti–HIT ya no son detectables).
8. El uso de Aspirina� y la colocación de filtro en la vena cava no constituyenmedidas de elección.
9. Otros. Existen reportes del uso de fondaparinux y bivalirudina para el trata-miento de HIT, aunque se desconoce su seguridad y eficacia.
ANTICOAGULANTES EN HIT
En esta sección se describen las características sobresalientes de los anticoagu-lantes indicados para el tratamiento de HIT.
a. Danaparoide (combinación de heparán sulfato y dermatán sulfato). Esun heparínico que inhibe el factor Xa y en menor medida la trombina. Seexcreta por vía renal y no existe un antídoto específico. La vida media delefecto anti–Xa es de cerca de 25 h y se prolonga en los pacientes con insufi-ciencia renal, por lo que siempre se debe ajustar la dosis. Se ha descrito unaactividad cruzada con los anticuerpos–HIT, por lo que se debe monitoreara los pacientes con caídas inexplicables en la cuenta plaquetaria, así comocon tromboembolismo recurrente o progresivo y sangrado. Si bien hace al-gunos años este medicamento era un pilar en el tratamiento, en la actualidadha sido sustituido por los inhibidores de trombina que se mencionan a conti-nuación.
b. Lepirudina (hirudina recombinante). La hirudina es un inhibidor directode la trombina que se elimina por vía renal (requiere ajuste según la fun-ción), tiene una vida media de 1.3 h y carece de antídoto específico, y suefecto terapéutico se monitorea con tiempo parcial de tromboplastina acti-vada (TPTa). Existen reportes de casos de anafilaxis secundarios a la infu-sión en bolo, después de la sensibilización, por lo que los pacientes no sedeben tratar más de una vez con este medicamento.14 Parece efectivo en lareducción de eventos tromboembólicos; sin embargo, su eficacia para redu-cir la necesidad de amputación o muerte aún no está probada.
c. Argatrobán. Es un inhibidor de trombina sintético con una vida media muycorta (de 40 a 50 min), que tiene una excreción normal aun en pacientes coninsuficiencia renal; sin embargo, se debe ajustar a la función hepática. Parasu monitoreo se usa TPTa. Parece tener un efecto sinergista en la prolonga-ción del INR cuando se usa en conjunto con warfarínicos. Al igual que laleupiridina, parece prevenir nuevos eventos tromboembólicos, aunque noreduce las tasas de amputación o muerte.

374 (Capítulo 23)Tratado de trombosis
d. Bivalirudina. Es otro inhibidor sintético de la trombina que se considerael medicamento de elección para la anticoagulación en los pacientes alérgi-cos a la heparina que serán sometidos a cateterismo coronario y cirugía derevascularización cardiaca.
El tiempo recomendado de anticoagulación es de al menos cuatro semanas, pues-to que el riesgo de trombosis persiste durante dos a cuatro semanas después deiniciado el tratamiento; no obstante, se requieren más estudios para determinarel tiempo óptimo. El tratamiento con inhibidores de trombina puede cambiarsepor warfarínicos sólo cuando las cuentas plaquetarias sean > 150 000/�L. El war-farínico se inicia a dosis bajas y se traslapa con heparínicos durante cinco díashasta conseguir un INR terapéutico durante al menos 48 h.
USO DE HEPARINA DESPUÉSDE UN EPISODIO DE HIT TIPO II
Los pacientes con antecedentes de HIT tipo II que posteriormente se presentanen circunstancias clínicas en las que es necesaria la anticoagulación (revasculari-zación coronaria) pueden ser tratados en el momento agudo con heparina no frac-cionada sin riesgo de complicaciones. Esta premisa se basa en la teoría de que nose presentará una respuesta inmunitaria secundaria antes de tres días de exposi-ción. La intención es usarla sólo en el momento agudo, ya que, como será elimi-nada rápidamente del organismo, los anticuerpos no serán trombogénicos en au-sencia de heparina.
En estas circunstancias no se debe usar HBPM, porque tiene una vida mediamás larga y sus efectos no son completamente reversibles con protamina.
Ante esta situación siempre se debe considerar la administración de otros fár-macos para la anticoagulación y el uso de ensayos de laboratorio para ver si exis-ten anticuerpos detectables en ese momento. Siempre se debe restringir la hepari-na si se va a realizar una cirugía o un procedimiento intravascular, y usar otrasalternativas en el periodo anterior y posterior a la cirugía.15
REFERENCIAS
1. Warkentin TE, Greinacher A: Heparin–induced thrombocytopenia: recognition, treat-ment, and prevention: the Seventh ACCP Conference on Antithrombotic and ThrombolyticTherapy. Chest 2004;126:311S.
2. Lee D, Carter C, Dolan C: Clinical guide–heparin induced thrombocytopenia. TheThrombosis Interest Group of Canada, 2005.

375Trombocitopenia inducida por heparina. Síndrome de HITE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
3. Weismann RE, Tobin RW: Arterial embolism occurring during systemic heparin therapy.AMA Arch Surg 1958;76:219–225.
4. Rhodes GR, Dixon RH, Silver D: Heparin–induced thrombocytopenia with thromboticand hemorrhagic manifestations. Surg Gynecol Obstet 1973;136:409–416.
5. Warkentin TE, Levine MN, Hirsh J et al.: Heparin–induced thrombocytopenia in patientstreated with low molecular weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330.
6. Pohl C, Kredteck A, Bastians B et al.: Heparin–induced thrombocytopenia in neurologicpatients treated with low–molecular–weight heparin. Neurology 2005;64:1285.
7. Napolitano LM, Warkentin TE, Almahameed A, Narsaway SA: Heparin–inducedthrombocytopenia in the critical care center: diagnosis and management. Crit Care Med2006;32: 2898.
8. Greinacher A, Farner B, Kroll H, Kohlman T, Warkentin TE et al.: Clinical featuresof heparin–induced thrombocytopenia including risk factors for thrombosis: a retrospectiveanalysis of 408 patients. Thromb Haemost 2005;94:132–135.
9. Wallis DE, Workman DL, Lewis BE, Steen L, Pifarre R et al.: Failure of early heparincessation as treatment for heparin–induced thrombocytopenia. Am J Med 1999;106:629–635.
10. Arepally GM, Ortel TL: Heparin–induced thrombocytopenia. N Engl J Med 2006;355:809–817.
11. Lo GK, Juhl D, Warkentin TE, Sigoun CS, Bicheler P et al.: Evaluation of pretest clinicalscore (4T’s) for the diagnosis of heparin–induced thrombocytopenia in two clinical settings.J Thromb Haemost 2006;4:759–765.
12. Rice L, Attisha WK, Drexler A, Francis JL: Delayed onset heparin–induced thrombocy-topenia. Ann Intern Med 2002;136:210.
13. Warkentin TE, Kelton JG: Temporal aspects of heparin–induced thrombocytopenia. NEngl J Med 2001;344:1286.
14. Greinacher A, Lubenow N, Eichler P: Anaphylactic and anaphylactoid reactions associa-ted with lepirudin in patients with heparin–induced thrombocytopenia. Circulation 2003;108:2062–2065.
15. Follis F, Schmidt CA: Cardiopulmonary bypass in patients with heparin–induced thrombo-cytopenia and thrombosis. Ann Thorac Surg 2000;70:2173.

376 (Capítulo 23)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
24Ecocardiografía en el
paciente con trombosisMarlon Patricio Aguirre Espinosa, Francisco Javier Roldán Gómez
INTRODUCCIÓN
Cuando se tiene un paciente con sospecha clínica de oclusión arterial aguda sedebe pensar en la posibilidad de un evento embólico de origen cardiogénico. Lostrombos intracavitarios, las vegetaciones valvulares y los tumores intracardiacos,como el mixoma, son las principales causas de embolismo cardiaco. Tambiénpuede existir embolismo arterial por la presencia de placas de ateroma localiza-das en la aorta o en las arterias carótidas. Los émbolos que migran de las venasde los miembros inferiores o de las venas pélvicas hacia las cavidades derechas,y de ellas a la arteria pulmonar y sus ramas, son los causantes de los episodiosde tromboembolia pulmonar (TEP).
Después de una exploración clínica cuidadosa se debe indicar una evaluaciónecocardiográfica, para descartar la presencia de trombos intracavitarios. Este es-tudio debe ser realizado por cardiólogos entrenados y capaces de diferenciar en-tre lo normal y lo anormal, los artefactos de imágenes reales y las estructuras nor-males de las patológicas. Las dos aproximaciones utilizadas en la investigaciónde un evento sospechoso de cardioembolismo son la transtorácica y la transesofá-gica, cada una con sus respectivas indicaciones.
Las posibles causas de un ecocardiograma transtorácico (ETT) negativo enpresencia de cardioembolismo son la migración del trombo hacia un sitio extra-cardiaco, que el tamaño del trombo sea demasiado pequeño o que el trombo seencuentre localizado en zonas de difícil identificación (como en la orejuela iz-quierda). Con la complementación del ecocardiograma transesofágico (ETE) la
377

378 (Capítulo 24)Tratado de trombosis
sensibilidad para la identificación de trombos intracardiacos aumenta en granmedida y puede documentarse con más exactitud su localización, tamaño y gradode movilidad. Este estudio puede llevarse a cabo con seguridad en condicionesde inestabilidad hemodinámica y con frecuencia condiciona cambios en el trata-miento médico. Cuando un paciente presenta un cuadro clínico de embolismo sis-témico el clínico debe preguntarse:
1. ¿El origen del émbolo es extracardiaco? Para descartar una embolia arterio-arterial se debe investigar la presencia de placas de ateroma en las arteriascarótidas o en la aorta ascendente. En los pacientes jóvenes se deben identi-ficar las causas primarias de trombosis, como la presencia de enfermedadesinmunitarias (lupus eritematoso sistémico, síndrome antifosfolípidos, etc.)o la resistencia a las proteínas C y S.
2. ¿El origen del émbolo es cardiaco? Si es el caso, se deben analizar las condi-ciones predisponentes para la formación de trombos, como son los trastor-nos del ritmo (principalmente fibrilación auricular), las patologías (comola miocardiopatía dilatada) y las valvulopatías (como la estenosis mitral).También se debe descartar la presencia de tumores, vegetaciones, anormali-dades estructurales, áreas de infarto, aneurismas del ventrículo izquierdo oestasias del flujo intracardiaco (visualizado como “contraste espontáneo”en el estudio ecocardiográfico).
3. ¿Existe la posibilidad de un embolismo paradójico? Como se mencionó, lapresencia de trombos en los miembros inferiores se relaciona con embolis-mo pulmonar; sin embargo, en caso de un foramen oval permeable (FOP)los trombos pueden migrar hacia las cavidades izquierdas y provocar unembolismo sistémico (figura 24–1).
CONDICIONES PREDISPONENTES
La fibrilación auricular es uno de los factores de riesgo más importantes de car-dioembolismo, el cual se reduce con la administración de antiagregantes o de an-ticoagulantes en forma crónica (según el caso), así como con la restauración a rit-mo sinusal.
La ecocardiografía permite establecer el riesgo de embolismo al determinarel tamaño de la aurícula izquierda, las características del flujo (contraste espontá-neo) y la función de la válvula mitral (el riesgo es mayor en presencia de esteno-sis). También predice la posibilidad de éxito de una cardioversión (eléctrica o far-macológica) y del mantenimiento a ritmo sinusal.
Antes de una cardioversión es importante descartar la presencia de trombosque pudieran migrar al revertir la arritmia. Para este fin se aconseja la anticoagu-

379Ecocardiografía en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 24–1. Imagen de un ecocardiograma transesofágico que muestra el eje largo dela aurícula derecha (AD) a nivel de la desembocadura de las venas cava superior (VCS)e inferior (VCI). La flecha continua muestra la cabeza de un trombo longilíneo atrapadoen un foramen oval permeable. La flecha discontinua muestra el cuerpo del trombo entránsito ya en la cavidad de la aurícula izquierda (AI).
lación formal durante al menos tres a cuatro semanas o la realización de un ETE.Aun cuando el ETE es un estudio semiinvasivo, ofrece ciertas ventajas, como unamayor resolución y una mayor cercanía del transductor con la orejuela auricularizquierda, la cual representa el sitio más frecuente de formación de trombos intra-cardiacos ante la presencia de fibrilación auricular y no puede ser valorada de for-ma adecuada desde la aproximación transtorácica.
Los hallazgos que se asocian con un mayor riesgo de apoplejía relacionada conla cardioversión eléctrica son:
� Presencia de trombos intracavitarios.� Disminución de la velocidad de vaciado de la orejuela izquierda (menos de
20 cm/seg).� Contraste espontáneo auricular izquierdo denso.� Presencia de placa compleja de ateroma en la aorta.
Si el ETE no demuestra la presencia de factores de riesgo para embolismo la car-dioversión eléctrica se puede realizar con un alto porcentaje de éxito.
Después de una cardioversión efectiva se recomienda la anticoagulación for-mal de seis semanas a seis meses, debido a que el aturdimiento transitorio de laorejuela y del atrio constituye un factor de riesgo para la formación de trombos.
La presencia de miocardiopatía dilatada representa un factor de riesgo para laformación de trombos intracardiacos, debido a la discontinuidad e irregularidad

380 (Capítulo 24)Tratado de trombosis
del endocardio, el flujo lento, las áreas de discinesia de las paredes miocárdicasy la posibilidad de aneurismas en el ventrículo izquierdo.
La estenosis mitral incrementa la presión intraauricular izquierda con el consi-guiente aumento del tamaño y de fibrosis de su pared, lo cual constituye el sustra-to para el desarrollo de fibrilación auricular.
La combinación de estenosis mitral con dilatación de la aurícula y la orejuelaizquierdas, estasis sanguínea, contraste espontáneo y fibrilación auricular es fac-tor para la formación de trombos intraauriculares, que se pueden distinguir fácil-mente mediante el ecocardiograma.
Hasta antes de la era de la anticoagulación al menos 20% de los pacientes conestenosis mitral presentaban un evento de embolia sistémica en algún momentode su evolución.
MASAS INTRACARDIACAS
Los trombos pueden localizarse en cualquiera de las cuatro cámaras cardiacas.En el lado izquierdo, como se sabe, existen condiciones y enfermedades que pre-disponen a la formación de los mismos. En cambio, en las cavidades derechas lostrombos pueden provenir de las venas de los miembros inferiores o de las venaspélvicas, los cuales se consideran émbolos en tránsito que al llegar a la arteria pul-monar o a sus ramas producen una embolia pulmonar.
El tumor cardiaco que se ha relacionado con un alto porcentaje de embolismosistémico es el mixoma, el cual representa 30% de todos los tumores cardiacos.El mixoma puede identificarse fácilmente mediante un ETT como una masa debordes irregulares o en “racimo de uvas” cuya localización habitual es la aurículaizquierda a nivel de la fosa oval (75% de los casos). Asimismo, puede localizarseen el ventrículo izquierdo (20% de los casos) o puede ser de múltiples localizacio-nes (5% de los casos).
En ocasiones es difícil diferenciar un trombo de un tumor. El carcinoma de cé-lulas renales tiende a visualizarse en la desembocadura de la vena cava inferiora la aurícula derecha, con una apariencia lobulada y móvil. Para diferenciarlo serequieren otros estudios de imagen, como la tomografía computarizada de abdo-men.
La vegetación es la lesión patognomónica de la endocarditis. El porcentaje deriesgo de un evento embólico secundario a endocarditis es de 20%. Existen carac-terísticas ecocardiográficas que predicen un riesgo elevado de eventos embóli-cos, como la presencia de una vegetación mayor de 10 mm, una gran movilidady la localización en la válvula mitral. En ocasiones la diferenciación ecocardio-gráfica entre un trombo y una vegetación puede ser difícil, por lo que hay que te-

381Ecocardiografía en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ner en cuenta el contexto clínico del paciente (presencia o ausencia de un cuadroinfeccioso).
ANOMALÍAS ESTRUCTURALES
Los aneurismas y las anormalidades en la movilidad (discinesia) de la pared delventrículo izquierdo secundarias a cardiopatía isquémica se relacionan con la for-mación de trombos. El FOP se relaciona con embolismo paradójico sistémico, so-bre todo si la apertura del foramen es mayor de 5 mm y se asocia con la presenciade aneurisma del septum interauricular. El aneurisma del septum interatrial se defi-ne como un desplazamiento mayor de 15 mm de la membrana de la fosa oval desdeel plano septal. Al formarse un trombo en el sistema venoso dicho aneurisma sedesprende, migra al atrio derecho y pasa a través del FOP hacia las cavidades iz-quierdas (figura 24–1) hasta terminar en la oclusión arterial. En los pacientes consíndrome de Eisenmenger esta situación se observa con más frecuencia.
Las válvulas protésicas también constituyen un factor de riesgo para la forma-ción de trombos, sobre todo las prótesis mecánicas. La demostración de trombosintracavitarios en la prótesis valvular es difícil; sin embargo, el ETE ha facilitadosu diagnóstico. El objetivo principal del ecocardiograma debe ser la valoraciónde la función valvular (estenosis o insuficiencia) y la exclusión de otras situacio-nes que predispongan a la formación de émbolos (como la disfunción ventricularizquierda). La frecuencia de trombosis valvular es de 0.2 a 1.8% anual y es másfrecuente en la posición tricuspídea, seguida de la mitral y la aórtica. La cinefluo-roscopia ayuda a visualizar la limitación de los discos protésicos; no obstante, elETE es el principal método diagnóstico para evaluar la función protésica, la pre-sencia de trombo o depósito de panus a nivel valvular.
INDICACIONES DEL ECOCARDIOGRAMA
La etiología cardioembólica abarca de 15 a 20% de los eventos cerebrovascularesisquémicos y es más frecuente en los pacientes menores de 45 años de edad (de23 a 50%). En los pacientes ancianos con diagnóstico de oclusión arterial agudalas lesiones estenóticas arteriales pueden coexistir con fuentes cardioembólicas,por lo que hay que tener mucho cuidado al momento de evaluar a este grupo depacientes. En 40% de los pacientes con infarto cerebral o evento isquémico tran-sitorio que presentan un ETT normal se puede detectar una fuente cardioembóli-ca a través del ETE.

382 (Capítulo 24)Tratado de trombosis
Cuadro 24–1. Indicación ecocardiográfica en pacientescon evento cerebrovascular u otras embolias arteriales
1. Pacientes de cualquier edad con oclusión brusca de un vaso periférico mayor o una arteriavisceral. Clase I
2. Pacientes jóvenes (edad < 45 años) con evento cerebrovascular. Clase I
3. Pacientes mayores (edad > 45 años) con eventos neurológicos sin evidencia de enfermedadcerebrovascular o cualquier otro mecanismo etiológico evidente. Clase I
4. Enfermos en los que una decisión terapéutica clínica (anticoagulación, etc.) puede dependerdel resultado de un ecocardiograma. Clase IIa
5. Pacientes con sospecha de enfermedad embólica y con enfermedad cerebrovascular dediscutible significado. Clase IIa
6. Pacientes con eventos neurológicos y enfermedad cerebrovascular intrínseca suficiente paracausar el evento clínico. Clase IIb
7. Pacientes en los que el resultado del ecocardiograma no significa ningún cambio en la deci-sión para prescribir terapéutica anticoagulante o en la aproximación diagnóstica o terapéu-tica. Clase III
La ecocardiografía es un adecuado método de estudio para valorar las estruc-turas valvulares, las cavidades cardiacas y la función ventricular. La técnicatransesofágica es superior a la transtorácica para la valoración del tabique inter-auricular (FOP y aneurisma), el contraste espontáneo, las prótesis cardiacas, laspequeñas vegetaciones y los trombos localizados en la orejuela izquierda. LaETE es especialmente útil para el diagnóstico de aterosclerosis aórtica.
En el cuadro 24–1 se muestran las recomendaciones para la realización de unestudio ecocardiográfico de acuerdo con las guías clínicas de la Sociedad Espa-ñola de Cardiología.
ECOCARDIOGRAFÍA Y TROMBOEMBOLIA PULMONAR
La utilidad básica del ETT en los pacientes con sospecha de tromboembolia pul-monar (TEP) incluye la confirmación del diagnóstico al observar de manera di-recta la presencia de trombos en las cavidades derechas o en la arteria pulmonar,la observación de la repercusión hemodinámica sobre las cavidades derechas yla estratificación del riesgo de mortalidad o de tromboembolia recurrente, asícomo guiar las decisiones terapéuticas (anticoagulación, colocación de filtro enla vena cava inferior, trombólisis y procedimientos hemodinámicos o quirúrgi-cos). El nivel de evidencia del ETT en esta patología es IIa.
Tromboembolia pulmonar aguda
El estudio ecocardiográfico ayuda en la estratificación de riesgo del paciente conembolia pulmonar aguda. La observación de acinesia moderada o severa del ven-

383Ecocardiografía en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 24–2. Imagen de un ecocardiograma transesofágico en un paciente con fibrila-ción auricular que muestra la presencia de un trombo ovoideo (flecha) adherido a la re-gión dorsal de la aurícula derecha (AD).
AD
AI
trículo derecho, la dilatación de su cavidad, la determinación del grado de hiper-tensión arterial pulmonar y la identificación de trombos en tránsito son hallazgosque determinan un alto riesgo de mortalidad o de tromboembolismo recurrente.Todos estos datos permiten definir la estrategia terapéutica adecuada. En la figura24–2 se muestra un trombo en la cavidad auricular derecha de un paciente confibrilación auricular detectado a través de ETE.
De acuerdo con la magnitud de la TEP, los hallazgos del ecocardiograma vandesde resultados normales hasta alteraciones importantes, como la acinesia y ladilatación del ventrículo derecho, y los datos de interdependencia ventricular(que predispone a una disminución del gasto cardiaco por reducción del llenadoventricular izquierdo por desplazamiento del septum interventricular). Otro ha-llazgo frecuente es el movimiento paradójico del septum interventricular, defini-do como disincronía de la contractilidad del septum en relación con la pared librey posterior del ventrículo izquierdo durante la sístole. En los pacientes con FOPse presenta un cortocircuito que favorece la desaturación arterial al aumentar lapresión de la aurícula derecha y superar la presión de la aurícula izquierda, lo cualaumenta el riesgo de tromboembolia paradójica sistémica.
Los datos ecocardiográficos de disfunción ventricular derecha incluyen unarelación del diámetro entre el ventrículo derecho y el ventrículo izquierdo > 1 enla vista apical de las cuatro cámaras, diámetro del ventrículo derecho mayor de30 mm y movimiento septal paradójico. Se ha descrito un signo denominado “deMcConnel” en presencia de TEP, caracterizado por acinesia de la pared libre del

384 (Capítulo 24)Tratado de trombosis
ventrículo derecho con movimiento normal del ápex. Este signo tiene una sensi-bilidad de 77% y una especificidad de 94% para diferenciar si la disfunción ven-tricular derecha es secundaria a TEP aguda o por otras causas, como hipertensiónpulmonar primaria. El ETE tiene la capacidad de identificar la presencia de trom-bos a nivel de la arteria pulmonar.
Tromboembolia pulmonar crónica
La TEP crónica produce hipertensión arterial pulmonar e hipertrofia ventricularderecha de manera secundaria. En etapas más avanzadas el ventrículo derechopuede llegar a claudicar con dilatación de su cavidad y de su anillo tricuspídeo,lo cual condiciona insuficiencia valvular secundaria.
Los hallazgos ecocardiográficos en caso de TEP crónica están directamenterelacionados con el grado de hipertensión arterial pulmonar; ellos son la hipertro-fia de la pared libre del ventrículo derecho, la posible dilatación de su cavidad,la insuficiencia tricuspídea de diferentes grados y la apertura del foramen ovalsi éste era permeable con cortocircuito derecha–izquierda.
El ecocardiograma permite calcular de manera no invasora la presión en la ar-teria pulmonar a través del cálculo del gradiente de regurgitación sistólica trans-tricuspídea, lo cual posee una elevada confiabilidad en manos expertas y una altacorrelación con la presión pulmonar medida a través de cateterismo.
REFERENCIAS
1. Otto CM: Source of embolus. En: Echocardiografic evaluation of cardiac masses and po-tential cardiac. 2ª ed. Filadelfia, Elsevier, 2000:251–370.
2. Feigenbaum H, Armstrong W, Ryan TH: Ecocardiografía. Masas, tumores y fuentes deémbolos. En: Feigenbaum H. 6ª ed. Panamericana, 2006:701–733.
3. Kury J, Crespo L, Necoechea J: Valvulopatías. En: Vargas J: Tratado de cardiología. Mé-xico, Intersistemas, 2006:443–502.
4. Armstrong W: Ecocardiografía. En: Braunwald E: Tratado de cardiología. 7ª ed. Madrid,Elsevier, 2006:187–270.
5. Lin S, Wong J: Prótesis valvulares cardiacas. En: Marso S, Griffin B, Topol E: Cardiología.Madrid, Marbán, 2004:232–245.
6. Oliveira FJ, Viana L, Vieira de Melo R, Faic AF, Torreaõ J et al.: Chagas disease is anindependent risk factor for stroke: baseline characteristics of a Chagas disease cohort.Stroke 2005;36:2015–2017.
7. Murphy SM, McIntyre D, Cronin S, Moroney J: Is transthoracic echocardiography use-ful in investigation of patients with suspected cardioembolic stroke? J Neurol NeurosurgPsychiatry 2007;78(2):212.
8. De Bruijn S, Agema W, Lammers G, van der Wall E, Wolterbeek R et al.: Transesopha-geal echocardiography is superior to transthoracic echocardiography in management of pa-tients of any age with transient ischemic attack or stroke. Stroke 2006;37:2531–2534.

385Ecocardiografía en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
9. Muir KW, McNeish L, Grosset, Donald G, Metcalfe: Visualization of cardiac embolifrom mitral valve papillary fibroelastoma. Stroke 1996;27(6):1133–1134.
10. Goldhaber S: Echocardiography in the management of pulmonary embolism. Ann InternMed 2002;136:691–700.
11. Evangelista MA, Alonso GA, Martín DR, Moreno YM, Oliver RJ et al.: Guías de prácti-ca clínica de la Sociedad Española de Cardiología en ecocardiografía. Rev Esp Cardiol2000;53(5):663–683.
12. Mylonakis E, Calderwood S: Infective endocarditis in adults. N Engl J Med 2001;345(18):1318–1330.
13. Habid G: Management of infective endocarditis. Heart 2006;92:124–130.14. Heidenreich PA: Transesophageal echocardiography (TEE) in the critical care patient.
Cardiol Clin 2000;18(4):789–805.15. Selim M: Perioperative stroke. N Engl J Med 2007;356:706–713.16. Grem M, Bergqvist D, Eriksson H, Lindblad B, Sternby N: Prevalence and risk of pul-
monary embolism in patients with intracardiac thrombosis: a population–based study of23 796 consecutive autopsies. Eur Heart J 2005;26:1108–1114.
17. Ercan E, Tengiz I, Sekuri C, Sahin F, Aliyev E et al.: Cardiac thrombi in a patient withprotein–C and S deficiencies: a case report. Thromb J 2(2):1–4.
18. Popescu B, Radulescu B, Ginghina C: Free–floating left atrial thrombus formed in a fewweeks in a patient with a normal mitral prosthesis. Heart 2007;93(2):246.

386 (Capítulo 24)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
25Imagenología en el paciente
con trombosisÓscar Quiroz Castro
INTRODUCCIÓN
La patología trombótica tiene diferentes causas y un sinnúmero de localizacio-nes; puede ser de origen arterial o venoso y, dependiendo de su localización, sudiagnóstico puede tener un mayor grado de dificultad.
El radiólogo desempeña un papel importante en el diagnóstico de esta patolo-gía, ya que de manera habitual es el primero en corroborarlo, lo cual se debe engran parte a que la enfermedad no se sospecha clínicamente.
Como parte de las herramientas de diagnóstico existen múltiples métodos deimagen que pueden ser utilizados, como son las placas simples y la flebografíapor rayos X, el ultrasonido Duplex y el Doppler a color, la resonancia magnética(RM), la tomografía computarizada (TAC) multicorte, la medicina nuclear, la to-mografía por emisión de positrones (PET–CT), la angiografía convencional, laangiotomografía (angio–TAC) y la angiorresonancia.
TROMBOSIS DE LAS VENAS YSENOS VENOSOS CEREBRALES
Esta trombosis constituye una patología rara pero importante en los pacientes conenfermedad vascular cerebral, la cual afecta a pacientes jóvenes. Su incidenciaanual es de tres a cuatro por cada millón de habitantes; 80% son mujeres. Su pre-
387

388 (Capítulo 25)Tratado de trombosis
Figura 25–1. Mujer de 54 años con trombosis del seno longitudinal superior; se observahiperintensa en la resonancia magnética y se muestra como un defecto de llenado enla venografía por tomografía computarizada.
sentación clínica y su evolución son variables, cursa de forma habitual con datosde hipertensión endocraneal, como son cefalea, mareo, náusea y trastornos visua-les. Se puede presentar déficit neurológico focal o parálisis de los nervios cranea-les. A diferencia de los eventos vasculares cerebrales (EVC) arteriales, su evolu-ción es más lenta y subaguda.
En la actualidad la venografía por RM combinada con la imagen por resonan-cia magnética (IRM) normal constituye el método de elección para el diagnósticode esta patología, con una sensibilidad diagnóstica de 100%.1 Los signos especí-ficos son la ausencia de flujo de uno o más senos venosos durales y la presenciade defectos de llenado en su interior. En las secuencias convencionales se puedeobservar hiperintensidad de señal en el interior de los senos venosos dependiendodel grado de metabolismo de la hemoglobina en el interior del coágulo.
Sin embargo, en reportes recientes se menciona que la angiografía venosa contomografía computarizada multicorte alcanza hasta 100% de sensibilidad y espe-cificidad en esta patología (figuras 25–1 a 25–3).2
INFARTO CEREBRAL
La lesión isquémica cerebral es el resultado de una serie de eventos en cascadade la depleción de energía hasta la muerte celular, en la que se presentan una va-riedad de eventos intermedios, como el exceso de aminoácidos excitatorios ex-tracelulares, la formación de radicales libres y la inflamación.

389Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 25–2. Paciente masculino de 23 años de edad con cefalea, trastornos visualesy afasia amnésica. La resonancia magnética muestra hiperintensidad del seno lateralizquierdo. La venografía por resonancia se muestra con ausencia de flujo y la venogra-fía por tomografía demuestra el defecto de llenado en el interior del mismo.
Después de la oclusión del flujo arterial se observa una zona central con muybaja perfusión, rodeada de una zona de disfunción causada por disturbios iónicosy metabólicos, pero la integridad estructural se encuentra conservada (zona de
Figura 25–3. Paciente femenino de 29 años de edad con cefalea y crisis convulsivas.La angiotomografía demuestra un defecto de llenado en el seno lateral izquierdo. La re-sonancia mostró un coágulo hiperintenso en su interior y la angiorresonancia demostróausencia de flujo en este seno.

390 (Capítulo 25)Tratado de trombosis
Figura 25–4. Perfusión cerebral. Obstrucción de la arteria carótida interna izquierda quecondiciona una zona de isquemia medial ipsilateral en el territorio cerebral.
penumbra). Dependiendo del flujo sanguíneo residual y de la duración de la is-quemia el tejido en penumbra puede evolucionar hacia el infarto.
Existe un periodo de ventana aproximado de tres horas en el que el tejido cere-bral isquémico se puede recuperar, de acuerdo con los datos del Ninds R–TPA Stu-dy Group.3
Los métodos para el diagnóstico temprano del infarto cerebral incluyen:
1. IRM con secuencias de perfusión difusión, la cual puede detectar el eventoisquémico a los 30 min de su inicio en cualquier localización del cerebro.
2. La TAC con perfusión puede detectar el evento isquémico a la hora de suinicio en el territorio de la arteria cerebral media, ya que en la fosa posteriory el tallo cerebral su sensibilidad es muy baja (figura 25–4).4
TROMBOSIS VENOSA PROFUNDADE LOS MIEMBROS INFERIORES
La incidencia anual de la trombosis venosa profunda (TVP) es de uno a dos porcada 1 000 habitantes, en la cual, incluso cuando se tratan de manera adecuadalos pacientes, de 1 a 8% desarrollan tromboembolismo pulmonar, 40% desarro-llan síndrome posflebítico y 4% presentan hipertensión arterial pulmonar.
Sólo en 10 a 25% de los pacientes en quienes se sospecha TVP desarrollan laenfermedad.5

391Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 25–5. Mapa de perfusión en tomografía con infarto en territorio de la arteria cere-bral media derecha, el cual afecta el lóbulo temporal.
Ultrasonido Doppler a color y con compresión
Constituye actualmente la prueba de diagnóstico de imagen de elección para laTVP, en la que la ausencia de compresibilidad constituye el signo más fidedigno,aunque, si bien el Doppler a color permite la detección del segmento venosotrombosado, para la detección de trombosis de las venas profundas proximalesdesde la ingle hasta la pantorrilla tiene una sensibilidad de 97%, mientras quepara las venas de la rodilla hacia abajo desciende a 73%.
Teniendo en cuenta que sólo en 10 a 20% de los pacientes el trombo se extiendehacia el territorio proximal y que sólo en 1 a 2% de los pacientes con ultrasonidonegativo inicial se demuestra trombosis en los estudios subsecuentes, no es nece-sario realizar ultrasonidos seriados (figuras 25–5 y 25–6).
TROMBOEMBOLISMO PULMONAR
Definición
El tromboembolismo pulmonar (TEP) es el síndrome que se origina como com-plicación de numerosos y diferentes padecimientos a partir de la formación de untrombo en el sistema venoso, el cual se emboliza a través del corazón derechohasta alojarse en la circulación arterial pulmonar.

392 (Capítulo 25)Tratado de trombosis
Figura 25–6. Ultrasonido Doppler a color.
Epidemiología
A pesar de los importantes avances en la profilaxis, el diagnóstico y el tratamien-to, la TEP se mantiene como un grave problema de salud mundial.
Cuadro 25–1. Manifestaciones de la emboliapulmonar en la radiografía de tórax
Infiltrado 22 (54%)
Derrame pleural 21 (51%)Atelectasias 11 (27%)Elevación diafragmática 7 (17%)Dos o más de los mencionados 18 (44%)
Sólo oligohemia focal 1 ( 2%)Cardiomegalia y falla congestiva 7 (17%)Hipertensión arterial pulmonar 2 (5%)
Normal

393Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
En EUA su incidencia es de 600 000 casos, con una mortalidad aproximadade 50 000 a 200 000 personas por año.
La dimensión de este problema se ha reflejado en las últimas cuatro décadas,debido a la incidencia sostenida en estudios de autopsia (15%) y el porcentaje altode diagnóstico erróneo premórtem (30%).6
Angiotomografía computarizada pulmonar
Es el primer estudio de imagen en los casos de sospecha de TEP aguda. El signocardinal es un defecto de llenado en la arteria pulmonar que ocluye parcial o com-pletamente el vaso y está asociado a un incremento del diámetro del vaso afec-tado.
Los signos específicos de la enfermedad incluyen:
� En caso de TEP aguda: un defecto de llenado que forma ángulos agudos conel vaso.
� En caso de TEP aguda y crónica: el defecto de llenado forma ángulos obtu-sos u oclusión completa del vaso.
Los signos indirectos de TEP incluyen opacificaciones parenquimatosas periféri-cas en forma de cuña, secundarias a infartos pulmonares, así como áreas regiona-les con perfusión asimétrica (perfusión en mosaico).
En la TEP crónica son frecuentes las arterias bronquiales dilatadas, así comolos estrechamientos y las deformaciones vasculares.
Los nuevos aparatos de TAC multicorte se encuentran provistos de un softwarepara reconstrucción de mapas en color de la perfusión arterial pulmonar, que seobtienen simultáneamente al realizar la angio–TAC pulmonar y además puedenreconstruir imágenes en 3D (figuras 25–7 y 25–8).
TROMBOSIS PORTAL Y MESENTÉRICA SUPERIOR
La trombosis venosa portal es una condición rara que de forma habitual se asociacon cirrosis, infecciones, cuadros de pancreatitis o procesos neoplásicos, comoel cáncer de páncreas o el hepatocarcinoma. La TVP se puede presentar en esta-dos posquirúrgicos, de manera principal en los pacientes con esplenectomía porpadecimientos hematológicos o bien en procedimientos quirúrgicos combina-dos, como la gastrectomía total para el manejo del cáncer gástrico.
Es esencial reconocer y establecer la permeabilidad del sistema venoso portalen los pacientes con cirrosis hepática e hipertensión portal, sobre todo con la ca-

394 (Capítulo 25)Tratado de trombosis
Figura 25–7. Corte axial de angiotomografía que muestra tromboembolia pulmonar condefectos de llenado en las ramas inferiores de la arteria pulmonar y la zona de infartopulmonar.
pacidad actual para realizar trasplantes hepáticos, TIPS o el abordaje portosisté-mico quirúrgico. En pacientes con hipertensión portal e historia de sangrado gas-troesofágico es indispensable corroborar la permeabilidad del sistema venosoportal y sus ramas.8
Figura 25–8. Angiotomografía pulmonar que muestra defectos de llenado en las ramasinferiores derechas y zona de infarto secundario con hipoperfusión en el mapa a color.

395Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Los factores de predisposición más comunes de la trombosis de la vena mesen-térica superior en los casos de causas radiológicas no identificadas son la cirugíaabdominal reciente, la infección y los estados de hipercoagulabilidad. Aunque nose ha precisado la relación entre factor de riesgo y sobrevida, la presencia de en-grosamiento de la pared intestinal y la congestión mesentérica en la TAC o en laRM se asocia con el desarrollo de isquemia intestinal. El pronóstico es bueno eneste grupo de pacientes, con una mortalidad de sólo 7%, aunque la isquemia intes-tinal se desarrolla en 21% de los casos.6
Existen diferentes características radiológicas que pueden ayudar a diferen-ciar si una trombosis venosa portal obedece a una causa benigna o maligna. Laidentificación de neovascularidad trombótica, la expansión venosa con dimen-sión del diámetro vascular igual o mayor a 2.3 cm y la invasión directa de la venaporta son indicadores por TAC de extensión tumoral. Para lo anterior se debenconsiderar otros hallazgos, como el efecto de contigüidad del trombo vascularcon la lesión parenquimatosa, además de un importante reforzamiento posteriora la TAC con contraste intravenoso de la trombosis portal. Estos dos últimos ha-llazgos no se aceptan como evidencia absoluta de extensión tumoral maligna enla TVP (figuras 25–9 a 25–12).8,9
Figura 25–9. Reconstrucción portal y mesentérica superior. Reconstrucción de tomo-grafía multiplanar coronal fase venosa que muestra hipodensidad de la vena porta portrombosis de la misma.

396 (Capítulo 25)Tratado de trombosis
Figura 25–10. Corte axial en fase venosa que muestra defectos hipodensos en la venaporta intrahepática.
SÍNDROME DE LERICHE
El síndrome de Leriche es causado por la oclusión aguda o paulatina de la aortaabdominal distal o de su bifurcación, lo que puede ser el resultado de ateromato-sis, arteritis o embolismo (figuras 25–13 y 25–14).
Figura 25–11. Corte axial de tomografía y ultrasonido Doppler a color que muestrantrombosis parcial de vena esplénica en su unión con la vena mesentérica superior.

397Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 25–12. Reconstrucción con tomografía coronal que muestra degeneracióncavernomatosa de la porta y trombosis completa de la vena mesentérica superior y susafluentes. En la imagen a color se observan macroscópicamente las asas de intestinoinfartadas.
ENFERMEDAD ATEROSCLEROSA CORONARIA
El desarrollo tecnológico de las máquinas de TAC multidetectoras ha permitidouna gran resolución anatómica de las arterias coronarias. La sensibilidad diag-
Figura 25–13. Síndrome de Leriche. Angiorresonancia de aorta abdominal que muestrasíndrome de Leriche por trombosis aórtica aguda.
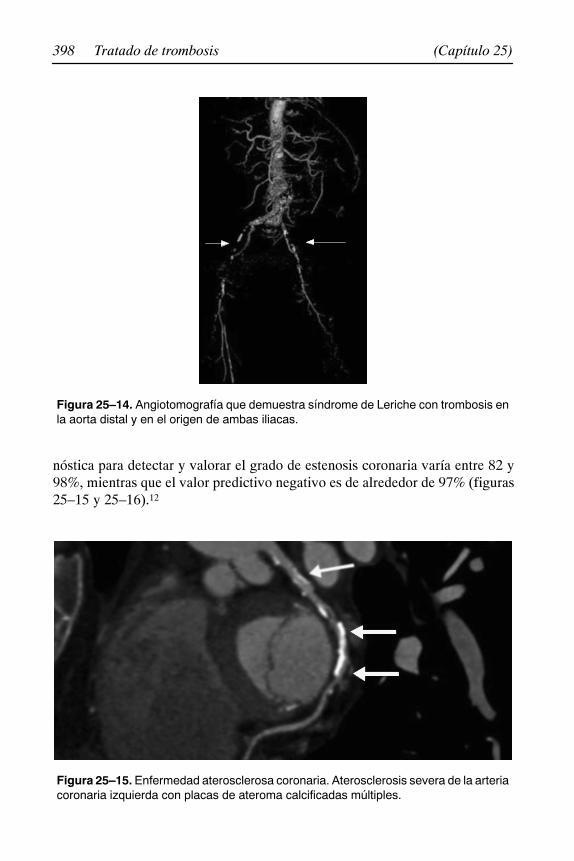
398 (Capítulo 25)Tratado de trombosis
Figura 25–14. Angiotomografía que demuestra síndrome de Leriche con trombosis enla aorta distal y en el origen de ambas iliacas.
nóstica para detectar y valorar el grado de estenosis coronaria varía entre 82 y98%, mientras que el valor predictivo negativo es de alrededor de 97% (figuras25–15 y 25–16).12
Figura 25–15. Enfermedad aterosclerosa coronaria. Aterosclerosis severa de la arteriacoronaria izquierda con placas de ateroma calcificadas múltiples.

399Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 25–16. Arteria coronaria derecha de calibre y flujo normales.
ENFERMEDAD DE TAKAYASU
La enfermedad de Takayasu es una arteriopatía progresiva que involucra tantoa la aorta como a sus ramas y que se presenta de manera habitual en las mujeresmenores de 45 años de edad (figura 25–17).
Figura 25–17. Enfermedad de Takayasu. Angiotomografía que muestra trombosis par-cial de arterias carótida y subclavia izquierdas por enfermedad de Takayasu.

400 (Capítulo 25)Tratado de trombosis
INFARTO RENAL
Definición
Área localizada o global de necrosis isquémica en el riñón. La mayoría son secun-darias a oclusión de la arteria renal (AR).
Información clínica
Los signos y síntomas más frecuentes incluyen:
� Asintomático, dolor en costado, postración (traumático), hematuria.� Hipertensión en el infarto crónico.
Etiología
� Embolia cardiaca: es la más frecuente.� Trombosis:
� Aterosclerosis, panarteritis nodosa.� Aneurisma o disección aórtica.
� Enfermedad de células falciformes.� Traumatismo: puede ser contuso o penetrante.� Cirugía.
Los hallazgos en la TAC incluyen:
� Infarto focal y segmentario:� Área bien delimitada, dorsal o ventral, segmentaria, con una disminu-
ción de la captación.� Limitación por líneas rectas entre el parénquima normal que capta y el
anómalo que no capta.� Muy sugerente de isquemia.
� Infarto global:� Ausencia total de captación renal, sin excreción, sin hematoma perirre-
nal (trombosis de la AR).� Contorno renal conservado.� Ausencia total de contraste, gran hematoma perirrenal (avulsión de la
AR).

401Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 25–18. Infarto renal. Reconstrucción con tomografía que muestra trombosis par-cial de la arteria renal izquierda con zonas de infarto renal secundario.
� +/– estrías medulares: captación “en rueda de carro” (circulación colate-ral) (figura 25–18).10
TROMBOSIS DE LAS VENAS CAVA SUPERIORE INFERIOR, Y YUGULARES
Definición
Oclusión de la vena cava superior (VCS) por compresión extrínseca o trombosisintrínseca.
Información clínica
Los signos y síntomas más frecuentes incluyen edema facial, tos, ortopnea, estri-dor, cefaleas leves y confusión e ingurgitación de las venas cervicales.
Los hallazgos en la TAC incluyen:
� TAC sin contraste:� Calcificaciones mediastínicas en pacientes con tuberculosis, histoplasmo-
sis.� Masa de tejidos blandos mediastínica en las etiologías malignas.

402 (Capítulo 25)Tratado de trombosis
Figura 25–19. Trombosis de las venas cava superior e inferior, y yugulares. Pacientefemenino de 30 años de edad con trombosis de la vena yugular interna, la cava superior,los troncos braquiocefálicos y drenaje por colaterales y sistema ácigos, secundaria asíndrome de anticuerpos antifosfolípidos.
Los hallazgos en la TAC más contraste incluyen:
� Venas colaterales dilatadas.� Adenopatías mediastínicas (linfoma o metástasis).� Trombo en la VCS en pacientes con formación de coágulos.
Figura 25–20. Corte axial de tomografía que muestra trombo parcial en la vena cavainferior.

403Imagenología en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
� Opacificación de partes del parénquima hepático (habitualmente el seg-mento medial izquierdo, lóbulo cuadrado) a través de venas colaterales in-trahepáticas y perihepáticas.
Diagnósticos diferenciales
Mediastinitis fibrosante, aneurisma o disección aórtica (figuras 25–19 y 25–20).
REFERENCIAS
1. Lafitte F, Boukobza M, Guichard JP et al.: MRI and MRA for diagnosis and follow–upof cerebral venous thrombosis (CVT). Clin Radiol 1997;52:672–679.
2. Diagnostic value of multidetector–row CT angiography in the evaluation of thrombosis ofthe cerebral venous sinuses. AJNR Am J Neuroradiol 2007;28:946–952.
3. The National Institute of Neurological Disorders and Stroke Study Group rTPA Stroke Stu-dy Group: Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995;33:1581–1587.
4. Van der Worp HB, van Giin J: Acute ischemic stroke. N Engl J Med 2007;357:572–579.5. Páramo JA, Ruiz E, García R: Trombosis profunda. Rev Med Univ Navarra 2007;51(1):
13–17.6. Moses DC: The complementary roles of chest radiography, lung scanning, and selective
pulmonary angiography in the diagnosis of pulmonary embolism. Circulation 1974;XLXI.7. Kreft B, Strunk H, Flacke S et al.: Detection of thrombosis in the portal venous system:
comparison of contrast–enhanced MRA with intraarterial subtraction angiography. Radiol-ogy 2000;216:86–92.
8. Tublin ME, Dodd GD III, Baron RL: Benign and malignant portal vein thrombosis: dif-ferentiation by CT characteristics. Am J Roentgenol 1997;168:719–723.
9. Mori H, Hayashi K, Uetani M et al.: High attenuation recent thrombus of the portal vein:CT demonstration and clinical significance. Radiology 1987;163:353–356.
10. Federle M: Diagnóstico por imagen de abdomen. Sección 3–IV–3–82. Marbán, 2011.11. Federle M: Diagnóstico por imagen de abdomen. Sección 1–1–46. Marbán, 2011.12. Martuschelli E, Romagnoli A, D’Eliseo A et al.: Accuracy of thin–slice computed tomo-
graphy in the detection of coronary stenoses. Eur Heart J 2004:1043–1048.

404 (Capítulo 25)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
26Laboratorio clínico en trombosis
Bernardo Ronzón Fernández
Existen una gran cantidad de pruebas que se utilizan tanto para el diagnóstico detrombosis como para el seguimiento y la prevención de la enfermedad. Entre laspruebas iniciales que se requieren para el estudio de la trombosis, además de laspruebas básicas como la biometría hemática y los tiempos de coagulación, secuenta con pruebas más específicas, como son resistencia de la proteína C acti-vada, antitrombina III funcional, actividad de la proteína C y la proteína S, factorVIII, detección de la mutación de protrombina 20210GA, mutación del factor Vde Leiden, determinación de homocisteína, anticuerpos anticardiolipina y anti-cuerpos antifosfolípidos. Las pruebas funcionales que resultan positivas se pue-den confirmar con pruebas genéticas en algunos casos o demostrando la anorma-lidad en algún otro miembro de la familia. Por ejemplo, una resistencia a laproteína C activada puede ser confirmada con un análisis de la mutación del fac-tor V de Leiden. Estas pruebas pueden diferenciar el estado homocigoto o hetero-cigoto de la enfermedad, con lo que se puede establecer el pronóstico de la enfer-medad (cuadro 26–1).
Si todas las pruebas anteriores resultan negativas el paciente puede tener algu-na alteración rara que puede ser identificada midiendo los factores IX y XI, la li-poproteína (a), la actividad del plasminógeno, el inhibidor de la actividad delplasminógeno (PAI–1) y el activador tisular del plasminógeno. Estas pruebassólo se recomiendan en individuos con fuertes antecedentes personales o familia-res de trombosis y en quienes las pruebas de inicio resultaron negativas. Debidoa que la etiología de la trombofilia aún no está bien determinada, es posible queincluso estas últimas pruebas resulten negativas.
405

406 (Capítulo 26)Tratado de trombosis
Cuadro 26–1. Causas de alargamiento del tiempo de protrombina
Hereditarias Adquiridas
� Deficiencia de factor VII (el TTP es nor-mal)
� Alteración en la función hepática (TP alargadoen forma más temprana que el TTP)
� Deficiencia de fibrinógeno o de factoresII, V o X (el TTP puede estar tambiénl d )
� Deficiencia de vitamina K (TP alargado en for-ma más temprana que el TTP)
alargado) � Warfarina (TP alargado en forma más tempra-na que el TTP)
� Coagulación intravascular diseminada (TP alar-gado en forma más temprana que el TTP)
� Anticoagulante lúpico (puede o no prolongar elTTP; el TP muy rara vez se alarga)
� Heparina (se alarga el TTP y el TP general-mente es normal)
A continuación se revisan los puntos importantes de estas pruebas.
TIEMPO DE PROTROMBINA
El tiempo de protrombina (TP) mide el tiempo de formación del coágulo desdela activación del factor VII hasta la formación del coágulo de fibrina. Esta pruebamide la vía extrínseca y las vías comunes de la coagulación, mientras que el tiem-po de tromboplastina parcial mide la integridad de la vía intrínseca y la vía comúnde la coagulación, de tal forma que se utiliza para medir los factores VII, II, Vy X. Esta prueba es utilizada también para monitorear el uso de anticoagulantes,como la warfarina.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428.� Limitaciones: cuando hay deficiencia de un factor único, sea de la vía in-
trínseca o de la vía común, ese factor debe estar debajo de 15 a 45% antesde que el TP se vea alargado. Cuando hay deficiencia en varios factores elTP se alarga con disminuciones menos severas de esos factores.
La deficiencia de los factores VIII, IX, XI y XII, precalicreína o cininógenos dealto peso molecular no afecta el TP, aunque sí altera el tiempo de tromboplastinaparcial (TTP). Las alteraciones en el factor XIII no afectan el TP ni el TTP, porlo que se recomienda utilizar su medición directa (cuadro 26–1).

407Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
MONITOREO DE ANTICOAGULANTE
Para monitorear el uso de warfarina como anticoagulante se utiliza el índice nor-malizado internacional (INR: International Normalized Ratio), el cual se obtienemediante la medición del TP. Este índice se estableció para homologar los resul-tados y poder hacer comparaciones válidas utilizando diferentes tipos de reacti-vos, sea con alta o con baja sensibilidad. Esto es debido a que los reactivos tienenuna diferente sensibilidad, por lo que si se usa uno con baja sensibilidad no se ob-tendrá un tiempo alargado, sino hasta que los niveles de los factores de coagula-ción estén muy bajos, mientras que utilizando un reactivo con alta sensibilidadlos tiempos se alargarán con disminuciones ligeras de los factores. Por ello se leasignó a cada reactivo un índice de sensibilidad internacional (ISI), de tal formaque si tienen una baja sensibilidad tendrán valores de ISI mayores por arriba de3, mientras que los reactivos con alta sensibilidad tendrán valores de ISI bajoscercanos a 1. El índice ISI es determinado por los propios fabricantes. Para calcu-lar el INR se utiliza la siguiente fórmula:
INR = (TP del paciente / TP testigo normal)ISI
El valor terapéutico esperado del INR es de 2.0 a 3.0.Existen varios aspectos que pueden alterar la dosis requerida de warfarina para
lograr el nivel de anticoagulación necesario. Hay medicamentos y alimentos queproducen interacción con la warfarina afectando los resultados del TP (cuadro26–2). Existen también situaciones clínicas que disminuyen la dosis requeridapara incrementar el TP, como el hipertiroidismo, la falla hepática, la presencia decáncer, la fiebre o la deficiencia de vitamina K, sea por malabsorción, esteatorrea,malnutrición o el uso de algunos antibióticos. Por otra parte, algunas situaciones,como hipotiroidismo o ciertos polimorfismos genéticos tienden a requerir un in-cremento de la dosis.
TIEMPO DE TROMBOPLASTINA PARCIAL ACTIVADA
El tiempo de tromboplastina parcial activada (TTP) mide el tiempo de coagula-ción desde la activación del factor XII hasta la formación del coágulo de fibrina.Esta prueba mide la integridad de las vías intrínseca y común de la coagulación,mientras que el tiempo de protrombina, como se mencionó anteriormente, midelas vías intrínseca y común de la coagulación. La prolongación de la TTP puedeser causada tanto por deficiencia de factores, especialmente los factores VIII, IX,XI y XII, como por inhibidores, como el anticoagulante lúpico, o anticoagulantesterapéuticos, como la heparina, la hirudina o el argatrobán.

408 (Capítulo 26)Tratado de trombosis
Cuadro 26–2.
Tipo de estudio Potenciación Inhibición Sin efecto
Estudio aleatoriocontrolado
Alcohol (con enferme-dad hepática conco-mitante), amiodarona,esteroides anabóli-cos, cimetidina, clofi-brato, cotrimoxazol,eritromicina, flucona-zol, isoniazida, metro-nidazol, miconazol,omeprazol, fenilbuta-zona, piroxicam, pro-pafenona, proprano-lol, sulfinpirazona
Barbitúricos, carba-mazepina, clordia-zepóxido, colesti-ramina, griseoful-vina, nafcilina, ri-fampicina, sucral-fato, alimentoscon niveles altosde vitamina K/ali-mentación enteral,grandes cantida-des de aguacate
Alcohol, antiácidos,atenolol, bumeta-nida, enoxacina,famotidina, fluoxe-tina, ketorolaco,metoprolol, napro-xeno, nizatidina,psyllium, ranitidina
Estudios aleatorioscontrolados
Acetaminofén, hidratode cloral, ciprofloxaci-na, dextropropoxife-no, disulfiram, itraco-nazol, quinidina, feni-toína, tamoxifeno, te-traciclina, vacunacontra la influenza
Dicloxacilina Ibuprofeno, ketoco-nazol
Estudios observacio-nales
Ácido acetilsalicílico, di-sopiramida, fluoroura-cilo, ifosfamida, keto-profeno, lovastatina,metozalona, moricizi-na, ácido nalidíxico,norfloxacina, ofloxa-cino, propoxifeno, su-lindaco, tolmetin, sali-cilatos tópicos
Azatioprina, ciclospo-rina, etretinato,trazodona
Estudios observacio-nales
Cefamandol, cefazolina,gemfibrozil, heparina,indometacina, sulfiso-xazol
Diltiazem, tabaco,vancomicina
Fuente: Chest 2001;119(Suppl):11S.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428. En el caso de esta
prueba, la toma de muestras a través de un catéter o línea heparinizada pue-de contaminarse fácilmente con la heparina, incluso aunque se haga un pur-gado y se elimine el volumen inicial extraído. Por lo anterior, se recomiendatomar las muestras para las pruebas de coagulación directamente de algunavena periférica, evitando usar el brazo en el que se esté infundiendo hepari-na, hirudina o argatrobán.

409Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
� Utilidad: esta prueba sirve para evaluar la vía intrínseca de la coagulación(factores VIII, IX, XI y XII) y en menor grado la vía común (fibrinógenoy factores II, V y X). Puede detectar el anticoagulante circulante lúpico,pero el TTP no debe ser usado para su escrutinio, ya que puede resultar alte-rado o normal dependiendo del tipo de reactivo que se utilice. También esampliamente utilizado para el monitoreo terapéutico de la anticoagulacióncon heparina no fraccionada, hirudina o argatrobán.
� Limitaciones: para que el TTP se prolongue con la deficiencia de un solofactor, éste tendrá que estar disminuido de 15 a 45%, dependiendo del reac-tivo que se utilice y del factor deficiente. Por otra parte, la elevación del fac-tor VIII acorta el TTP; la elevación es relativamente común al ocurrir du-rante una reacción de fase aguda. La presencia de anticoagulante lúpico yla deficiencia de ciertos factores pueden prolongar el TTP o acentuar sualargamiento cuando hay presencia de heparina, por lo que en estas situa-ciones se recomienda utilizar una prueba alterna, como es la heparina anti–Xa para monitorear el uso de la misma.
� Metodología: el reactivo de TTP, que consiste en fosfolípidos con un acti-vador de la vía intrínseca (sílica, celite, caolín o ácido elágico) y calcio, esañadido a la muestra del paciente midiéndose en segundos la formación delcoágulo. Los fosfolípidos en la prueba de TTP son llamados tromboplastinaparcial, debido a que no está presente el factor tisular. Este factor tisular síse encuentra presente en los reactivos de tromboplastina “completos” quese usan para medir el tiempo de protrombina, ya que este factor tisular esel que activa la vía extrínseca.
� Valores de referencia: varían en forma importante entre los diferentes ti-pos de reactivos que existen en el mercado. El límite normal inferior aproxi-mado es de 25 seg, mientras que el valor normal superior es de 45 seg. Losrecién nacidos tienen un TTP más elevado que los adultos, llegando hasta55 seg; sin embargo, disminuyen paulatinamente hasta alcanzar los valoresde los adultos a los seis meses.
PROTEÍNA C DE COAGULACIÓN
La proteína C, junto con la proteína S como cofactor, es una proteína anticoagu-lante normal. Es dependiente de vitamina K y es sintetizada en el hígado. La vidamedia biológica de la proteína C en el plasma es de 6 a 10 h.
La proteína C se activa por la trombina en presencia de un cofactor celular en-dotelial (trombomodulina) para formar la proteína C activada. Ésta funcionacomo un anticoagulante que inactiva las formas activas de los factores de la coa-

410 (Capítulo 26)Tratado de trombosis
gulación V y VIII (Va y VIIIa). También la proteína C activada mejora la fibrinó-lisis al inactivar al activador–inhibidor del plasminógeno (PAI–1).
Una deficiencia hereditaria de la proteína C conlleva a un estado de hipercoa-gulabilidad, con un aumento del riesgo de trombosis venosa, como es el caso dela deficiencia heterocigota congénita de proteína C, la cual resulta en diátesistrombótica, que se hace evidente desde el periodo neonatal. También existe la de-ficiencia heterocigota congénita, que predispone a eventos trombóticos venososy arteriales. La deficiencia heterocigota hereditaria puede ser de dos tipos: el tipoI es una deficiencia cuantitativa de proteína C y el tipo II resulta de una alteracióncualitativa más que cuantitativa de la proteína.
La deficiencia adquirida de la proteína C puede ocurrir en asociación con:
� Deficiencia de vitamina K.� Anticoagulantes orales.� Enfermedad hepática.� Coagulación intravascular y fibrinólisis/coagulación intravascular disemi-
nada.
Existen dos pruebas para estudiar la proteína C: la de la proteína C funcional yla de la proteína C antigénica.
PROTEÍNA C FUNCIONAL Y PROTEÍNA C ANTIGÉNICA
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras”en la página 428. Se debe determinar
si el paciente se encuentra recibiendo anticoagulantes, como la warfarina,ya que los niveles de la proteína C se reducen por acción de ésta. Para la de-terminación de la proteína C es importante separar el plasma lo más prontoposible y procesar la prueba inmediatamente. En caso de requerir almace-namiento, deberá ser en congelación. Las causas de rechazo de la muestraen el laboratorio incluyen las muestras tomadas más de cuatro horas antes,los tubos no llenados completamente y las muestras coaguladas.
� Utilidad: la proteína C funcional se utiliza como prueba inicial, ya que pue-de detectar deficiencias de la proteína C de tipo I y de tipo II. La proteínaC antigénica se utiliza únicamente si la prueba funcional se encuentra dis-minuida, con el fin de determinar si la deficiencia es de tipo I o de tipo II.Si la prueba antigénica se realiza primero sin la prueba funcional, no se po-drán detectar las deficiencias de tipo II. Dicha prueba también detecta las

411Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
deficiencias de proteína C de origen adquirido, como en los casos de uso deanticoagulantes orales, deficiencia de vitamina K, enfermedad hepática, etc.
� Metodología: existen dos métodos para determinar la actividad de la pro-teína C:� Pruebas cromogénicas: en este ensayo la proteína C presente en el plas-
ma del paciente es activada con una fracción proteica del veneno de ser-piente. La proteína C activada se une a un sustrato sintético que semejael sustrato natural de la proteína C, liberando una sustancia cromogénicaque puede ser medida de manera espectrofotométrica. Con esta pruebaexiste la posibilidad de no detectar ciertas deficiencias de tipo II, las cua-les sí pueden ser detectadas con el método coagulométrico. Estas pruebasno se ven afectadas por la presencia de heparina en rangos de hasta 2U/mL. La ventaja de esta metodología es que no se ve afectada por la pre-sencia de anticoagulante lúpico, niveles de factor VIII, factor V de Lei-den u otras anormalidades de la coagulación que sí afectan la metodolo-gía coagulométrica.
� Pruebas coagulométricas: en esta metodología la proteína C del pa-ciente se activa también con veneno de víbora. La proteína C activada,entonces, degrada los factores Va y VIIIa, prolongando el tiempo detromboplastina parcial. Esta metodología se puede ver afectada por alte-raciones en la coagulación, como la presencia de anticoagulante lúpicoque incrementa el nivel de la proteína C in vitro. También la elevacióndel factor VIII (> 200%) puede dar una falsa disminución en el resultado.Se han reportado valores falsamente disminuidos con esta metodologíaen pacientes con la mutación del factor V de Leiden. La ventaja de estemétodo es que puede detectar todas las variantes de las deficiencias tantode tipo I como de tipo II. Este método no se ve afectado con niveles deheparina de hasta 1 U/mL.
� Valor de referencia: los resultados se reportan como porcentaje de la canti-dad esperada en plasma normal. El valor de referencia es de 70 a 140%. Enel momento del nacimiento los niveles de proteína C son menores y van de17 a 53% de los valores normales en el adulto. Los niveles medios de la pro-teína C aumentan a cerca de 50% de los valores en el adulto a la edad de seismeses, pero se mantienen por debajo del rango del adulto hasta la edad de10 a 16 años.
PROTEÍNA C ANTIGÉNICA
Esta prueba sirve para diferenciar la deficiencia congénita de proteína C de tipoI y de tipo II. También evalúa la importancia de la disminución de la proteína C

412 (Capítulo 26)Tratado de trombosis
funcional, especialmente cuando la disminución pueda ser de carácter congénitomás que adquirido, sea por uso de anticoagulantes orales, deficiencia de vitaminaK, enfermedad hepática, etc.
� Valores de referencia: en los adultos el valor normal es de 70 a 150%. Enlos recién nacidos a término e incluso en los niños prematuros sanos puedehaber un valor disminuido de la proteína C funcional (15 a 50%), que alcan-zará los valores normales al final de la infancia o el inicio de la adolescencia.
� Información adicional: algunas de las causas externas que también pue-den disminuir la proteína C son:� Disminución de la síntesis por enfermedad hepática o por tratamiento
con L–asparaginasa.� Síntesis de una proteína disfuncional debida a deficiencia de vitamina K
o uso de warfarina.� Consumo por trombosis, coagulación intravascular diseminada o ciru-
gía.
Si un paciente tiene cualquiera de las condiciones enlistadas arriba como causasadquiridas y obtiene resultados de proteína C bajos, la prueba debe ser repetidauna vez que las condiciones que la produjeron hayan desaparecido. La confirma-ción de una deficiencia de proteína C hereditaria puede requerir la documenta-ción de deficiencia en un familiar. En el síndrome nefrótico la proteína C puedeestar disminuida, elevada o normal. Hay opiniones encontradas entre estudiosque reportan una elevación de la proteína C con anticonceptivos orales y con elembarazo, y otros estudios en que reportan no haber encontrado diferencias sig-nificativas de los niveles de la proteína C durante el embarazo. Las mujeres tienenvalores ligeramente más bajos de proteína C que los hombres, y las mujeres pre-menopáusicas tienen valores ligeramente más bajos que las posmenopáusicas.
PROTEÍNA S DE COAGULACIÓN
La proteína S es un cofactor necesario para la actividad anticoagulante de la pro-teína C. Es una glucoproteína dependiente de vitamina K que se sintetiza princi-palmente en el hígado. También se sintetiza en las células endoteliales y está pre-sente en las plaquetas. Cerca de 60% del total de antígeno de la proteína S circulaunido a la proteína C4b (C4b–BP), mientras que el restante 40% circula en formalibre. Sólo la proteína S libre tiene actividad anticoagulante.
La deficiencia congénita de la proteína S es una alteración autosómica domi-nante que se encuentra presente en 2 a 6% de los pacientes con trombosis venosa

413Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
y que se puede presentar como cuadros de abortos de repetición, complicacionesdel embarazo (preeclampsia, desprendimiento prematuro de placenta, disminu-ción del crecimiento intrauterino y muerte intrauterina) y posiblemente trombo-sis arterial.
Las deficiencias de proteína S son de tres tipos: deficiencias cuantitativas (tipoI), deficiencias cualitativas (tipo II) y deficiencias con niveles normales de la pro-teína total pero bajos de la fracción libre.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428. Se debe determinar
si el paciente se encuentra recibiendo anticoagulantes, como warfarina o es-trógenos, sea como anticonceptivos orales o reemplazo hormonal sustituti-vo, o que la paciente se encuentre embarazada, ya que los niveles de la pro-teína S disminuyen por acción de los estrógenos, el embarazo o el uso dewarfarina. Si se encuentra tomando warfarina, ésta se deberá suspender porlo menos 10 días antes. Es importante tomar en cuenta el nivel del factorVIII, ya que un nivel elevado de éste (> 200%) es una causa común de unafalsa disminución de la proteína S cuando se usan las pruebas basadas enla determinación del TTP. En estos casos se recomienda hacer la cuantifica-ción del factor VIII en la misma muestra para descartar que su elevación seala causa de la disminución de la proteína S. La elevación del factor VIII ocu-rre en los pacientes como reacción de fase aguda.
� Metodología: ensayos funcionales (proteína S funcional). Es medida porla habilidad para servir como cofactor requerido para la degradación de laactividad de los factores V y VIII por la proteína C.� Proteína S antigénica libre: inmunoensayo con anticuerpos monoclona-
les específicos para la proteína S libre.� Proteína S antigénica total: inmunoensayo que mide tanto la parte unida
a la proteína C4b como la libre mediante la técnica de ELISA.� Valores de referencia: proteína S funcional, de 70 a 122%; proteína S anti-
génica, de 70 a 140%; proteína S antigénica libre, de 70 a 140% en hombresy de 50 a 147% en mujeres.
� Información adicional: la proteína se puede encontrar también disminui-da en condiciones adquiridas, como:� Disminución en la síntesis hepática o por tratamiento con L–asparagi-
nasa.� Síntesis de una proteína disfuncional debida a deficiencia de vitamina K
o por el uso de warfarina.� Consumo por trombosis, coagulación intravascular diseminada o por
procedimientos invasivos.

414 (Capítulo 26)Tratado de trombosis
� Estrógenos, incluyendo anticonceptivos orales, terapia de reemplazo oembarazo. La proteína S se mantendrá disminuida hasta dos meses des-pués del parto o de la suspensión de la terapia.
� Reacción de fase aguda debida a la elevación de la proteína C4b, la cualdisminuye la fracción libre y, por tanto, la función de la proteína S.
� También se puede encontrar disminuida en el síndrome nefrótico y en in-fección por varicela y por VIH.
En casos de enfermedad hepática la proteína S se puede encontrar ocasionalmen-te normal a pesar de la disminución de la proteína C y la antitrombina, las cualestambién son sintetizadas en el hígado. Se ha especulado que esto puede ser debidoa que la proteína S también es sintetizada en las células endoteliales y en los me-gacariocitos, mientras que la proteína C y la antitrombina son sintetizadas exclu-sivamente por el hígado.
En caso de que el primer evento trombótico se presente en un paciente mayorde 50 años de edad se puede posponer la medición de las proteínas C y S, así comode la antitrombina III, ya que la hipercoagulabilidad debida a estas alteracionesse manifiesta mucho antes de la quinta década de la vida.
RESISTENCIA A LA PROTEÍNA C ACTIVADA
La resistencia a la proteína C activada es una condición que lleva a un estado dehipercoagulabilidad, con un riesgo incrementado de trombosis venosa. Utilizan-do el efecto de la proteína C activada en los tiempos de coagulación del paciente,principalmente el tiempo de tromboplastina parcial, se detecta la presencia de re-sistencia a la proteína, como ocurre en los individuos que tienen la mutación enel factor V de Leiden.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “toma de muestras” en la página 428.� Metodología: se utilizan pruebas coagulométricas, principalmente el tiem-
po de tromboplastina parcial, que provee una medición de la habilidad dela proteína C activada de actuar como anticoagulante. La muestra es diluidaa razón de 1:5 con plasma deficiente de factor V. Se realiza un tiempo detromboplastina parcial a la muestra diluida en presencia y ausencia de pro-teína C activada exógena. Esta proteína deberá degradar los factores Va yVIIIa, prolongando así el TTP. Con ambos resultados se calcula una rela-ción, que en pacientes normales es > 2.0, mientras que en los pacientes con

415Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
la mutación del factor V de Leiden, se presenta una relación < 2.0 debidoa que su factor Va mutado se resiste a la degradación por parte de la proteínaC activada. La sensibilidad y la especificidad de la determinación de la re-sistencia utilizando el TTP alcanza el 100% gracias a la dilución que se hacedel plasma con plasma deficiente de factor V.
� Valores de referencia: cociente normalizado de 0.86 a 1.10. La resistenciaa la proteína C prolonga el tiempo de tromboplastina parcial dos veces sobreel valor normal en pacientes sanos, mientras que en los pacientes con la alte-ración el tiempo obtenido es por debajo de dos veces el valor normal. Algu-nos laboratorios lo reportan como una relación entre el resultado de la prue-ba en el paciente, dividido entre el resultado de un pool de plasma normalo control.
� Limitaciones: se pueden obtener resultados falsos con la metodología coa-gulométrica por la presencia de anticoagulante lúpico o por el hecho de queel paciente se encuentre bajo tratamiento con hirudina o argatrobán. En casode que esta prueba resulte positiva se recomienda hacer la determinaciónde la mutación del factor V de Leiden.
MUTACIÓN DEL FACTOR V DE LEIDEN
De 12 a 52% de los pacientes con tromboembolia venosa presentan resistenciaal efecto anticoagulante de la proteína C activada. Aproximadamente 90% de lospacientes con esta resistencia a la proteína C activada tienen una mutación en unnucleótido del gen del factor V de la coagulación, que sustituye una arginina poruna glutamina en la posición 506 de la proteína del factor V, conociéndose comomutación Leiden. Esta mutación se presenta en pacientes con trombosis venosaprofunda, embolia pulmonar, oclusión de la vena central de la retina, trombosiscerebral y trombosis de la vena hepática.
� Espécimen: sangre total.� Tubo colector: tubo con anticoagulante que dependerá de la técnica.� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utilizan pruebas moleculares, como la reacción en cadena
de la polimerasa (PCR). Estas pruebas permiten definir la presencia de lamutación y determinar si es homocigota o heterocigota.
� Valores de referencia: negativo. Homocigoto normal.� Utilidad: esta prueba para detectar la presencia de la mutación Leiden se
debe realizar en los siguientes casos:� Pacientes con sospecha clínica de trombofilia.

416 (Capítulo 26)Tratado de trombosis
� Pacientes que tienen determinada la presencia de resistencia a la proteínaC activada o sospecha de esta resistencia por parte de un estudio no con-cluyente.
� Historia familiar de mutación del factor V de Leiden.� Sería apropiada para investigar a las mujeres que contemplan el uso de
anticonceptivos orales o el embarazo, para considerar un método alterna-tivo de anticoncepción o dar un tratamiento profiláctico durante el emba-razo a las mujeres que tienen la mutación.
� El conocimiento de la mutación puede modificar el manejo anticoagu-lante de pacientes con tromboembolismo venoso.
MUTACIÓN G20210A DE LA PROTROMBINA
La mutación G20210A de la protrombina es un polimorfismo de la región 3’–,no traducida del gen de la protrombina, que es de tipo hereditario y que predispo-ne a trombosis venosa. Esta mutación se asocia con un incremento de tres vecesel riesgo de tromboembolia venosa, posiblemente por un aumento en la actividadde la protrombina en el plasma de los portadores. Este riesgo se ve aumentadopor la presencia concomitante de alteraciones en la proteína C, la proteína S, ladeficiencia de antitrombina III o la presencia de la mutación del factor V de Lei-den. Las mujeres portadoras de la mutación G20210A de la protrombina que reci-ben anticonceptivos orales también aumentan el riesgo de presentar una trombo-sis venosa. La recurrencia de la trombosis venosa se duplica en los pacientes queademás de la mutación presentan la mutación del factor V de Leiden.
La prueba consiste en un análisis directo de la mutación G20210A en el DNAgenómico de los leucocitos en sangre periférica del paciente. Los resultados indi-can si la mutación en el individuo afectado es heterocigota u homocigota. La for-ma heterocigota está presente en 2.3% de la población general y en 6.2% de lospacientes con trombosis venosa. También se encuentra presente en 18% de loscasos de trombosis venosa familiar.
� Espécimen: sangre total.� Tubo colector: tubo con anticoagulante de acuerdo con la técnica.� Recolección: ver “Toma de muestras” en la página 428. En caso de requerir
almacenamiento deberá ser en refrigeración a 4 �C o a temperatura ambien-te. No se deben centrifugar ni congelar las muestras.
� Metodología: la investigación de la mutación se realiza utilizando la reac-ción en cadena de la polimerasa (PCR). Esta mutación consiste en el reem-plazo de una guanina en la posición 20210 del nucleótido por una adenina.

417Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Para realizar la prueba se aísla el DNA de la sangre total y el sitio de la muta-ción se amplifica millones de veces por PCR. Con la finalidad de distinguirlos alelos mutados, se utilizan las enzimas de restricción Hind III y se haceuna electroforesis en gel de agarosa para separar las bandas de DNA deacuerdo con su tamaño y así poder detectar la presencia de la mutación porel patrón de las bandas de DNA en el gel. También se puede identificar espe-cíficamente si es heterocigoto u homocigoto.
� Valor de referencia: homocigoto normal. La mutación G20210A de la pro-trombina no debe estar presente.
� Información adicional: el análisis de esta mutación no detectará a indivi-duos con trombofilia causada por otros mecanismos diferentes a los de lamutación de G20210A de la protrombina.
ANTITROMBINA III
La antitrombina es el principal inhibidor fisiológico de la coagulación. La anti-trombina, mediante su acción inhibidora de las proteasas de la coagulación, espe-cialmente trombina, así como de los factores Xa y IXa principalmente, aunquetambién los XIa, XIIa y la calicreína, evita un proceso incontrolado de la coagula-ción. La heparina y otros glucosaminoglicanos aumentan en forma importante laactividad anticoagulante de la antitrombina. La antitrombina es el mediador dela actividad anticoagulante de la heparina.
La disminución de los niveles de antitrombina III se encuentra asociada a unalto riesgo de enfermedades tromboembólicas. Existen dos tipos de deficiencias:las hereditarias y las adquiridas. La hereditaria es una deficiencia genética raracon patrón autosómico dominante que produce alteraciones trombóticas. Los in-dividuos con esta deficiencia generalmente son heterocigotos con actividad dela ATIII menor de 40 y hasta 70%. Existen más de 100 diferentes mutacionesidentificadas que producen dos fenotipos diferentes: el tipo I, en el que están dis-minuidos la actividad y el nivel del antígeno de la antitrombina, y el tipo II, quees más raro y que consiste en una antitrombina disfuncional con baja actividad,pero con un nivel de antígeno normal.
Entre las causas adquiridas de deficiencia de antitrombina III se incluyen:
� Terapia con heparina.� Coagulación intravascular y fibrinólisis, o coagulación intravascular dise-
minada y otras coagulopatías de consumo.� Enfermedad hepática (por síntesis disminuida y consumo aumentado) o con
síndrome nefrótico (pérdida de proteínas por orina).

418 (Capítulo 26)Tratado de trombosis
� Quimioterapia con L–asparaginasa (síntesis disminuida).
En el laboratorio se realizan dos tipos de pruebas: antitrombina III funcional yantigénica.
Antitrombina III funcional
La utilidad de la prueba funcional es para hacer el diagnóstico de la deficiencia,sea adquirida o congénita, así como para monitorear el tratamiento de la deficien-cia. Se recomienda realizar primero la prueba funcional y con base en los resulta-dos hacer posteriormente la prueba antigénica.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: la prueba funcional para medir la actividad de la antitrombi-
na III puede utilizar el método cromogénico, que consiste en una etapa deincubación del plasma del paciente con reactivo factor Xa en presencia deun exceso de heparina. En una segunda etapa se cuantifica la actividad delfactor Xa residual con un sustrato cromogénico, la paranitroanilina, que esmedida cinéticamente, siendo su nivel inversamente proporcional a la acti-vidad de la antitrombina de la muestra. También se puede utilizar el métodoque emplea trombina en vez del factor Xa.
� Valor de referencia: 80 a 120% desde los seis meses de edad hasta la etapaadulta. Los recién nacidos a término pueden presentar niveles bajos (� 35a 40%), alcanzando los niveles del adulto a los 90 días después del naci-miento. Los bebés prematuros (30 a 36 semanas) presentarán niveles bajosque alcanzarán los niveles normales aproximadamente a los 180 días del na-cimiento.
� Limitaciones: la antitrombina III funcional puede verse afectada por:� Heparina (no fraccionada o de bajo peso molecular) > 4 U/mL.� Alfa 1–antitripsina > 4 mg/mL.� Alfa 2–macroglobulina > 10 mg/mL.� Cofactor II de heparina > 4 U/mL.� Hemoglobina > 500 mg/dL (muestras hemolizadas).� Bilirrubinas > 40 mg/dL.� Triglicéridos > 2 300 mg/dL.
La terapia con heparina puede disminuir temporalmente la actividad de la anti-trombina.

419Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
La actividad de la antitrombina en muestras de suero es significativamentemás baja que en el plasma.
Antitrombina III antigénica
Esta prueba se utiliza para confirmar los resultados anormales de la prueba fun-cional; en caso de ser una deficiencia hereditaria establece el tipo de deficienciade antitrombina III, sea tipo I o tipo II. El diagnóstico de la deficiencia hereditariarequiere la determinación tanto de la antitrombina III funcional y antigénica,como de la correlación clínica y los estudios familiares. Actualmente no existenpruebas moleculares que sean de utilidad para su estudio.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utilizan inmunoensayos para la determinación de la anti-
trombina III antigénica. Un ejemplo es el reactivo que utiliza partículas delátex cubiertas con anticuerpos dirigidos contra la antitrombina. En presen-cia de la antitrombina las partículas de látex forman agregados que absor-ben la luz que pasa a través de la muestra. La cantidad de luz absorbida serádirectamente proporcional a la cantidad de antitrombina en la muestra.
� Limitaciones: la antitrombina III antigénica puede resultar afectada en lossiguientes casos:� Heparina (no fraccionada o de bajo peso molecular) > 4 U/mL.� Hemoglobina > 700 mg/dL (muestras hemolizadas).� Bilirrubina > 50 mg/dL.� Lipemia; puede producir una sobreestimación del nivel del antígeno� Factor reumatoide > 800 UI/mL; puede producir una sobreestimación
del nivel del antígeno.� Terapia con heparina que puede disminuir temporalmente los niveles del
antígeno de la antitrombina III.� Valores de referencia: adultos, de 75 a 130%. Los recién nacidos a término
pueden tener niveles bajos (� 35 a 40%), alcanzando los niveles del adultoa los 180 días posnatales. Los prematuros sanos (30 a 36 semanas) presenta-rán niveles bajos que alcanzarán los niveles normales aproximadamente alos 180 días del nacimiento.
DÍMERO D
Los productos de degradación de la fibrina son el resultado de dos fenómenos si-multáneos:

420 (Capítulo 26)Tratado de trombosis
� La transformación del fibrinógeno en fibrina estabilizada después de la ac-ción de la trombina y el factor XIIIa.
� La fibrinólisis, resultado de la disolución del coágulo de fibrina por la plas-mina en fragmentos solubles liberados al torrente sanguíneo. Los productosterminales de la lisis del coágulo son los dímeros D.
Los dímeros D reflejan la presencia de fibrina estabilizada, lo cual se aprovechapara utilizarlo como marcador para el diagnóstico al encontrarse elevados en laenfermedad tromboembólica venosa y la coagulación intravascular diseminada.Los dímeros D pueden estar elevados en otras patologías donde esté presente unaactivación de la coagulación y de la fibrinólisis, como son los casos de cirugía,traumatismo, infección, inflamación, embarazo y neoplasias. Como consecuen-cia, la utilidad para la exclusión de enfermedad tromboembólica venosa es menoren los pacientes hospitalizados debido a una gran cantidad de patologías asocia-das que causan elevación de los dímeros D.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428. En el caso de esta
prueba no se aconseja el uso de jeringa para la toma de muestra, para evitarla formación de coágulos.
� Metodología: para realizar la prueba de dímero D se utilizan inmunoensa-yos, como la turbidimetría, en la que se utilizan partículas de látex revesti-das de anticuerpo antimolécula dímero D. Cuando se mezcla un plasma quecontiene dímero D con las partículas de látex y una solución tampón las par-tículas se aglutinan. El grado de aglutinación es directamente proporcionala la concentración de dímero D contenida en el plasma. Otra técnica es lamedición por ensayo inmunoenzimático, en el que se utilizan anticuerposantidímero D marcados con fosfatasa alcalina, que producen un complejoque es medido por la fluorescencia que emite a 405 nm. El valor de la señalde fluorescencia es proporcional a la concentración del dímero D presenteen la muestra.
� Limitaciones: es importante no usar muestras lipémicas, hemolizadas o ic-téricas. La presencia de factor reumatoide puede producir una falsa eleva-ción de los niveles del dímero D.
� Valores de referencia: < 45 a 500 ng/mL. En el caso de esta prueba no exis-te un estándar internacional, por lo que algunos laboratorios reportan los re-sultados de dímero D en unidades equivalentes de fibrinógeno (UEF). Laequivalencia entre las dos unidades es de aproximadamente 2 UEF = 1 ng/mL. Un resultado normal de dímero D posee un alto valor predictivo negati-vo (95%) para la exclusión de enfermedad tromboembólica venosa.

421Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
HOMOCISTEÍNA
La homocisteína se forma en el metabolismo de la metionina de la dieta. Una vezformada la homocisteína se lleva a cabo una remetilación para formar metionina,proceso que requiere vitamina B12, ácido fólico y riboflavina, además de unatransulfuración que resulta en la síntesis de cisteína y glutatión, proceso que tam-bién requiere vitamina B6 y riboflavina, y por último una oxidación para formarhomocisteína y mezcla de bisulfitos.
Se puede presentar una elevación severa de la homocisteína por errores innatosdel metabolismo en los recién nacidos, generalmente por deficiencia de la cista-tin–� sintetasa. Por otra parte, la homocisteína puede ser considerada un factorde riesgo independiente para el desarrollo de enfermedad cardiovascular. Los pa-cientes con enfermedad cardiovascular, incluyendo enfermedad coronaria, en-fermedad vascular periférica y tromboembolia, tienen elevados los niveles de ho-mocisteína. Sin embargo, existen varias otras condiciones distintas en las quetambién se eleva la homocisteína, como son deficiencia de vitaminas, edad avan-zada, hipotiroidismo, insuficiencia renal, lupus eritematoso sistémico, etc. Tam-bién existen medicamentos que pueden elevar los niveles, como el ácido nicotíni-co, la teofilina, el metotrexato y la L–dopa.
Durante el embarazo la elevación de la homocisteína refleja una deficienciade folatos en la madre, lo que se asocia con una alta incidencia de defectos deltubo neural.
� Espécimen: suero, plasma.� Tubo colector: tubo para suero humano (tapón rojo) o tubo con EDTA (ta-
pón lila), o tubo con heparina de litio (tapón verde).� Recolección: ver “Toma de muestras” en la página 428. En el caso de esta
prueba se recomienda separar inmediatamente el suero o plasma del paque-te globular y colocarlo en hielo antes de procesarlo para evitar el incrementode la homocisteína por síntesis eritrocitaria.
� Metodología: existen varias metodologías para realizar esta prueba, comoinmunoensayo enzimático, inmunoensayo de polarización fluorescente,cromatografía líquida de alta eficacia, espectrometría de masas/cromato-grafía de gas, inmunoanálisis quimioluminiscente de micropartículas, etc.
� Valores de referencia: el valor de referencia variará en relación con el tipode muestra que se tome, sea suero o plasma; en el caso de este último depen-derá del anticoagulante usado. Los valores en los adultos del sexo masculi-no son 25% más altos que en las mujeres premenopáusicas. Los valores ensuero con la técnica de inmunoanálisis quimioluminiscente de micropartí-culas son de 5.4 a 16.2 �mol/L en los hombres y de 4.4 a 13.5 �mol/L enlas mujeres.

422 (Capítulo 26)Tratado de trombosis
� Limitaciones: se recomienda no utilizar muestras que hayan sido inactiva-das con calor o muestras con hemólisis importante.
Existe una elevación tanto en los pacientes con insuficiencia renal como en aque-llos que padecen hipotiroidismo. Los medicamentos que se asocian con eleva-ción de la homocisteína son los corticosteroides, la ciclosporina, la fenitoína, elmetotrexato y el trimetoprim. En las mujeres posmenopáusicas el estradiol redu-ce los niveles de homocisteína 5%, mientras que la terapia con estradiol–proges-terona la reduce 9%. En el caso de los pacientes que cursan con enfermedad coro-naria, infarto del miocardio, enfermedad vascular periférica o tromboembolismovenoso y que presentan niveles elevados de homocisteína se recomienda realizarel análisis de la mutación MTHFR C677.
MUTACIÓN MTHFR C677
El gen de la metiltetrahidrofolato reductasa (MTHFR) modera la producción dela enzima MTHFR. Esta enzima cataliza la reducción de 5,10–metiltetrahidrofo-lato a 5–metiltetrahidrofolato, que es la forma principal de los folatos en el plas-ma. También participa en el proceso que convierte la homocisteína en metionina,necesaria para la formación de muchas proteínas. Los pacientes que tienen unadeficiencia severa de la MTHFR desarrollan homocisteinuria.
� Espécimen: plasma.� Tubo colector: tubo con EDTA (tapón lila).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utilizan pruebas moleculares, como la reacción en cadena
de la polimerasa (PCR). Estas pruebas permiten definir la presencia de lamutación y además determinar si es homocigota o heterocigota.
� Valores de referencia: homocigoto normal. No hay presencia de la muta-ción.
Una mutación MTHFR homocigota puede estar asociada con hiperhomocistei-nemia, un riesgo aumentado para trombosis arterial y venosa, y un riesgo impor-tante para complicaciones obstétricas, como preeclampsia, desprendimiento pre-maturo de placenta, retardo del crecimiento fetal o muerte fetal. La presencia demutación heterocigota con ausencia de deficiencia de vitaminas se asocia gene-ralmente a niveles normales de homocisteína.
ANTICOAGULANTE CIRCULANTE LÚPICO
El anticoagulante circulante lúpico (ACL) constituye un grupo heterogéneo deanticuerpos que se encuentran dirigidos contra fosfolípidos de carga negativa o

423Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
contra complejos formados entre fosfolípidos y proteínas, como la beta 2–gluco-proteína, o factores de la coagulación, como la protrombina. Estos anticoagulan-tes interfieren con las pruebas de coagulación en las que se utilizan fosfolípidos,como el tiempo parcial de tromboplastina, la prueba de veneno de víbora de Rus-sell y otras. La presencia del ACL en un paciente aumenta el riesgo de sufrir cua-dros trombóticos y abortos de repetición. Los pacientes que presentan deficienciadel factor II asociado con el ACL pueden estar en riesgo importante de sangrado.El ACL no se encuentra únicamente en los pacientes con lupus eritematoso sisté-mico, sino también en otros trastornos autoinmunitarios, en respuesta a medica-mentos, en ciertas infecciones y en individuos sin una causa o enfermedad apa-rente.
Esta prueba se utiliza para:
� Evaluar un tiempo de tromboplastina parcial prolongado.� Estudiar un episodio trombótico.� Estudiar los abortos de repetición.� Como parte del estudio del síndrome antifosfolípidos.� Confirmar o excluir la presencia de ACL.� Diferenciar el ACL de los inhibidores de los factores de la coagulación tan-
to específicos como no específicos.� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se realizan dos tipos de prueba con la finalidad de mejorar
la sensibilidad. Una es la prueba de escrutinio de tipo coagulométrico, conuna concentración muy baja de fosfolípidos, por lo que la prueba es muysensible a la presencia de ACL, alargando el tiempo de coagulación en pre-sencia de ésta; y la prueba confirmatoria, que tiene una alta concentraciónde fosfolípidos que neutralizan el ACL, acortando los tiempos de coagula-ción.
� Valores de referencia: negativo para anticoagulante circulante lúpico.También se puede reportar la relación entre la prueba de escrutinio y la con-firmatoria; los valores esperados van de 0.8 a 1.2.
ANTICUERPOS ANTIFOSFOLÍPIDOS
Los fosfolípidos son compuestos polares con una estructura simple con tres com-ponentes básicos: una estructura glicérida (diacilglicerol), un grupo fosfodiéstery una estructura específica para cada fosfolípido, que puede ser colina, serina,

424 (Capítulo 26)Tratado de trombosis
etanolamina e inositol, entre otros. Los fosfolípidos aniónicos (cardiolipina, fos-fatidilserina, fosfatidilinositol y ácido fosfatídico) tienen la capacidad de ligar losanticuerpos antifosfolípidos (aPL), a diferencia de los fosfolípidos neutros. Du-rante los últimos años se demostró que la capacidad de unión de los aPL dependíade una proteína llamada beta 2–glucoproteína. Se ha determinado que la beta2–glucoproteína inhibe la vía intrínseca de la coagulación. Los anticuerpos anti-fosfolípidos consisten en anticuerpos dirigidos contra fosfolípidos cargados ne-gativamente, como son la cardiolipina, la fosfatidilserina, el fosfatidilinositol yel ácido fosfatídico. Generalmente de éstos se utilizaba principalmente la cardio-lipina para estudiar los anticuerpos; sin embargo, actualmente se cuenta con laposibilidad de estudios de otros fosfolípidos, con lo que se mejora la sensibilidaddel diagnóstico en los pacientes con sospecha de síndrome antifosfolípidos.
Dicho perfil incluye los siguientes antígenos:
� Beta 2–glucoproteína.� Cardiolipina con beta 2–glucoproteína.� Fosfatidilserina con beta 2–glucoproteína.� Fosfatidilinositol con beta–2–glucoproteína.� Ácido fosfatídico con beta 2–glucoproteína.� Antígenos mezclados con beta 2–glucoproteína.� Antígenos mezclados sin beta 2–glucoproteína.� Espécimen: suero.� Tubo colector: tubo sin anticoagulante (tapón rojo).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utiliza el método de ELISA, en el que se emplean placas
con micropozos cubiertos con los antígenos altamente purificados de car-diolipina, fosfatidilserina, fosfatidilinositol, ácido fosfatídico y beta 2–glu-coproteína. Si existen anticuerpos presentes en el suero del paciente se uni-rán a sus respectivos antígenos; después de varios lavados se aplica laenzima peroxidasa de rábano picante tanto IgG como IgM y posteriormenteun sustrato para la formación de color, cuya intensidad será directamenteproporcional a los niveles de los anticuerpos.
� Valores de referencia: IgG (U/mL) � 10, IgM (U/mL) � 10.
ANTICUERPOS ANTICARDIOLIPINA
La cardiolipina es un fosfolípido de carga negativa presente en la membrana cito-plasmática de las células humanas y en la membrana interna de las mitocondrias.El ser humano tiene la capacidad de sintetizar anticuerpos contra este lípido, los

425Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
cuales aparecen en el suero durante el curso de algunas enfermedades. La cardio-lipina es uno de los fosfolípidos de mayor importancia en la reacción autoinmuni-taria, ya que por estar localizada casi exclusivamente a nivel de la membrana mi-tocondrial participa en los procesos de oxidación de la cadena respiratoriamediante el transporte de electrones, por lo que los anticuerpos que intervienencon su función podrían determinar la destrucción celular.
La importancia clínica de las cardiolipinas ocurre cuando en ciertas enferme-dades se producen anticuerpos IgG o IgM contra estas moléculas, denominándo-se anticuerpos anticardiolipinas (AAC), las cuales se han correlacionado confenómenos trombóticos por su especificidad al complejo protrombina–fosfolípi-dos. Si se producen anticuerpos contra esos componentes, entonces se producenautoanticuerpos contra las células del organismo humano. Existen diferentes iso-tipos de cardiolipinas; sin embargo, los estudios sugieren que el mejor predictorde estados de hipercoagulabilidad es la IgG, ya que la IgM está relacionada conanemia hemolítica y fármacos. Los individuos que tienen altos niveles de IgG sonpropensos a desarrollar síntomas clínicos; es poco común ver trombosis o pérdi-das fetales con sólo el isotipo IgM.
� Espécimen: suero.� Tubo colector: tubo sin anticoagulante (tapón rojo).� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utiliza la técnica de ELISA, en la que se usan placas con
micropozos cubiertos con los antígenos altamente purificados de cardioli-pina.
� Valores de referencia: negativo (< 10.0 MPL o GPL); zona gris (10.0 a14.9 MPL o GPL); débil positivo (15.0 a 39.9 MPL o GPL); positivo (40.0a 79.9 MPL o GPL); fuertemente positivo (� 80.0 MPL o GPL). Las unida-des MPL y GPL se refieren a unidades fosfolípido IgM e IgG, respectiva-mente. Una unidad GPL o MPL corresponde a 1 ug de anticuerpo IgG oIgM, respectivamente.
PLASMINÓGENO
El plasminógeno es un precursor de la plasmina, la cual lisa los coágulos de fibri-na. La deficiencia hereditaria del plasminógeno es rara y predispone a la forma-ción de trombosis venosa.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).

426 (Capítulo 26)Tratado de trombosis
� Recolección: ver “Toma de muestras” en la página 428.� Metodología: se utilizan ensayos cromogénicos para la determinación. Se
agrega estreptocinasa al plasma del paciente, la cual se une al plasminógenoy forma un complejo que se acopla a un sustrato cromogénico, liberando uncompuesto colorido. La cantidad de color detectada de manera espectrofo-tométrica será proporcional a la cantidad de plasminógeno en la muestra delpaciente.
� Valor de referencia: los resultados funcionales se reportan como un por-centaje de la cantidad esperada en el plasma normal. El valor de referenciava de 75 a 150%. Los niveles del plasminógeno pueden estar elevados enel embarazo. En los recién nacidos los valores son de 60% del valor de losadultos. Estos niveles llegan al valor de los adultos aproximadamente a laedad de seis meses.
� Utilidad: esta prueba se debe utilizar en pacientes con trombosis venosa fa-miliar y sin evidencia de causas más comunes de trombosis. En ocasionesse puede utilizar la medición del plasminógeno para monitorear la terapiatrombolítica, en la que los valores del plasminógeno estarán disminuidos.
INHIBIDOR DEL ACTIVADOR DEL PLASMINÓGENO
Como su nombre lo indica, el inhibidor del activador del plasminógeno (PAI–1)inhibe al activador tisular del plasminógeno. Los niveles elevados del PAI–1 seasocian con un riesgo aumentado de trombosis arterial debido a la inhibición dela fibrinólisis, mientras que los niveles bajos llegan a ser una causa rara de sangra-do debido a una excesiva fibrinólisis.
� Espécimen: plasma.� Tubo colector: tubo con citrato de sodio a 3.2% (tapón azul cielo).� Recolección: ver “Toma de muestras” en la página 428. En el caso de esta
prueba es importante separar el plasma lo más pronto posible para evitar lacontaminación con plaquetas, debido a que éstas contienen PAI–1.
� Valor de referencia: < 25 U/mL.� Metodología: se utilizan ensayos cromogénicos para la determinación, en
los que se añade una cantidad conocida de urocinasa. El PAI–1 presente enel plasma del paciente se une a la urocinasa y la inhibe. La cantidad restantede urocinasa se detecta añadiendo plasminógeno, el cual es convertido aplasmina por la urocinasa residual. Esta plasmina se une a un compuestocromogénico y se mide de manera espectrofotométrica. La cantidad de co-lor producida es inversamente proporcional a la cantidad de PAI–1 en lamuestra. También existen ensayos antigénicos para su determinación.

427Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
� Limitaciones: el PAI–1 es un reactante de fase aguda, por lo que se elevadespués de un evento trombótico; por ello no se debe medir en etapa agudadespués de un cuadro de trombosis. El PAI–1 se eleva también durante elembarazo.
LIPOPROTEÍNA (A) LP(A)
La lipoproteína (a) [Lp(a)] es una proteína plasmática cuya estructura semeja alas lipoproteínas de baja densidad (LDL). La diferencia esencial entre la Lp(a)y las LDL es la presencia de una molécula adicional de apolipoproteína(a)[Apo(a)], que es parecida al plasminógeno y que está unida covalentemente a laApo B–100 por medio de enlaces disulfuro. Debido a su homología con el plas-minógeno, la Lp(a) compite por los sitios de unión en la molécula y las células,por lo que puede interferir con la fibrinólisis y acentuar el riesgo trombótico. LaLp(a) y la Apo(a) han sido identificadas en lesiones ateroscleróticas. La similitudcon el plasminógeno le permite unirse a la fibrina y a las proteínas de las membra-nas celulares.
El efecto aterotrombogénico de la Lp(a) depende de que su concentraciónplasmática sea elevada. Los valores plasmáticos mayores de 30 mg/dL puedenindicar un factor de riesgo independiente para la EAC. Las concentraciones ele-vadas de Lp(a) plasmática se encuentran relacionadas con un riesgo aumentadode infarto del miocardio y cerebral.
� Espécimen: suero.� Tubo colector: tubo sin anticoagulante (tapón rojo).� Recolección: ver “Toma de muestras” en la página 428. En esta prueba es
importante separar el suero lo más pronto posible.� Valor de referencia: < 30 mg/dL.� Metodología: puede incluir métodos espectrofotométricos o nefelométri-
cos.
En la actualidad se recomienda la medición de la lipoproteína (a) en varios sub-grupos de pacientes, en quienes el exceso de lipoproteína(a) puede tener conse-cuencias clínicas importantes. Entre éstos se encuentran:
1. Pacientes con aterosclerosis prematura.2. Pacientes con historia familiar fuerte de enfermedad coronaria prematura.3. Pacientes con colesterol LDL elevado y factor de riesgo mayor o igual a dos
factores de riesgo.

428 (Capítulo 26)Tratado de trombosis
4. Pacientes con angioplastia coronaria, en quienes el exceso de lipoproteí-na(a) puede aumentar el riesgo de reestenosis.
5. Pacientes sometidos a bypass coronario con injerto, en quienes el exceso deLp(a) puede estar asociado a una estenosis del injerto.
ANTICUERPOS HEPARINA–PF4
La trombocitopenia inducida por heparina (HIT) es un efecto adverso del trata-miento con heparina y mediada por anticuerpos anticomplejo factor plaquetario–4(PF4)–heparina (HPIA). La HIT es con frecuencia moderada, pero se pueden desa-rrollar complicaciones trombóticas. El diagnóstico precoz es importante. La de-tección de HPIA mediante técnica de ELISA posee una alta sensibilidad pero unabaja especificidad (títulos bajos sin significación clínica).
� Espécimen: suero.� Tubo colector: tubo sin anticoagulante (tapón rojo).� Recolección: ver “Toma de muestras” más abajo. En esta prueba es impor-
tante separar el suero y congelarlo lo más pronto posible.� Valor de referencia: negativo.� Metodología: inmunoensayo enzimático.� Utilidad: esta prueba detecta la presencia de autoanticuerpos con el com-
plejo heparina–PF4, que causa el HIT. Estos pacientes se encuentran enriesgo de desarrollar trombosis venosa o arterial si se continúa con el ma-nejo con heparina.
TOMA DE MUESTRAS EN GENERAL
El resultado de cualquier prueba va a depender en forma importante de la calidaden la toma de muestras. Existen lineamientos muy bien establecidos para esteproceso; sin embargo, es importante aclarar algunos puntos, sobre todo en el casode las pruebas de coagulación.
Es importante que el torniquete no se deje más de un minuto para evitar la esta-sis, ya que puede dar lugar a errores en los resultados, como concentraciones ele-vadas de parámetros proteicos, elevación del hematócrito, trombocitopenias,hemólisis, etc. Cuando se toman varios tubos para estudios de laboratorio es im-portante seguir cierta secuencia en los tubos para evitar contaminaciones. En pri-mer lugar se deben tomar los tubos con el anticoagulante citrato de sodio (tapón

429Laboratorio clínico en trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
azul), posteriormente los tubos sin anticoagulante (tubos de tapón rojo), luego lostubos con gel separador (tapón dorado), después los tubos con anticoagulante he-parina (tapón verde), siguiendo con los tubos con anticoagulante EDTA (tapónlila) y finalmente los tubos con cualquier otro tipo de anticoagulante, como oxala-to (tapón gris).
En el caso del tubo con citrato de sodio para las pruebas de coagulación es muyimportante tomar la cantidad exacta de sangre para mantener una relación 1:9 an-ticoagulante–sangre, ya que en caso contrario se pueden obtener resultados erró-neos. Generalmente los tubos al vacío están preparados para colectar la cantidadexacta. De igual forma, es importante que en el caso de los tubos con anticoagu-lante se mezcle la sangre con el anticoagulante mediante varias inversiones deltubo, para evitar que se formen coágulos y se afecten los resultados de la prueba.
REFERENCIAS
1. Taube J, Halsall D, Baglin T: Influence of cytochrome P–450 CYP2C9 polymorphismson warfarin sensitivity and risk of over–anticoagulation in patients on long–term treatment.Blood 2000;96(5):1816–1819.
2. Raby A, Moffat K: Anticardiolipin antibody and anti–beta 2 glycoprotein I antibodyassays. Methods Mol Biol 2013;992:387–405.
3. Raby A, Moffat K: Lupus anticoagulant testing. Methods Mol Biol 2013;992:97–108.4. Jacobs D, DeMott W: Laboratory test handbook. Lexi–Comp’s Clinical Reference Libra-
ry. 5ª ed. Cleveland, EUA.5. Ross RH: Haemophilia and haemostasis. 1ª ed. Reino Unido. Blackwell, 2007.6. O’Shaughnessy D: Practical hemostasis and thrombosis. 1ª ed. Reino Unido, Blackwell,
2005.7. Henry JB: El laboratorio en el diagnóstico clínico. 20ª ed. Marbán, 2010.

430 (Capítulo 26)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
27Biología molecular en la trombosis
Ema Valenzuela Méndez, Federico Javier Ortiz Ibarra
INTRODUCCIÓN
La trombosis (del griego thrombos, coágulo) se define como el proceso de forma-ción de un trombo o la oclusión de un vaso sanguíneo. La aparición de trombosisestá relacionada con lesiones de la pared vascular, disminuciones del débito san-guíneo o alteraciones en la coagulación de la sangre.
La patogénesis de la trombosis radica en la obstrucción del flujo de sangre queregresa al corazón desde los órganos, generando dolor, enrojecimiento e inflama-ción, aunque en otros casos el individuo puede no presentar signos o síntomasevidentes.
En el ámbito genético la principal causa de trombofilia hereditaria está asocia-da al desequilibrio de la cascada de la coagulación; el riesgo genético de desarro-llar trombosis se eleva por la influencia de factores ambientales adversos, lo cuala su vez puede generar la aparición de complicaciones, como la embolia pulmo-nar. Los avances en la secuenciación del genoma humano y el desarrollo de méto-dos moleculares han permitido generar un diagnóstico más certero mediante ladetección de genes responsables de enfermedades como ésta, con el fin de ofre-cerle al paciente un tratamiento preventivo aun en un periodo presintomático yademás personalizado. Los pocos genes asociados al aumento de la probabilidadde trombosis son los siguientes:
� Gen F5: codifica para el factor V de la coagulación, que a su vez es cofactoren la formación de trombina a partir de protrombina. Genéticamente uno
431

432 (Capítulo 27)Tratado de trombosis
de los principales focos desencadenantes de trombofilia hereditaria es lamutación Leiden en este gen (mutación 1691G/A). La mutación 1691G/Ase presenta en alrededor de 20% de los casos iniciales y en aproximadamen-te 50% de los pacientes con trombosis recurrente. La identificación de doscopias de esta mutación en un individuo indica que la persona tiene un ries-go 80 veces más alto de presentar trombosis venosa, mientras que si presen-ta sólo una copia de la mutación el riesgo es de ocho veces.
� Gen F2: la protrombina o factor II es uno de los principales componentesdel sistema de la coagulación. La mutación 20210G/A en el gen de la pro-trombina está asociada a un incremento en la expresión de este gen, lo cualaumenta una o dos veces más la concentración de protrombina en la sangre.La mutación 20210G/A también se asocia a trombosis cerebral y emboliapulmonar. En la población general las personas que presentan una copia deesta mutación tienen un incremento de hasta seis veces de riesgo de sufrirtrombosis.
� Gen MTHFR: codifica para la enzima metiltetrahidrofolato reductasa; supolimorfismo más común es la mutación 677C/T. Esta mutación está aso-ciada al incremento del nivel de homocisteína en sangre o hiperhomocistei-nemia, que es uno de los factores de riesgo causante de trombosis venosay endurecimiento de las arterias. La homocisteína es un producto de la des-composición de la metionina. La identificación de esta mutación en perso-nas que presentan episodios de trombosis permite definir la causa y en losfamiliares del paciente se puede prevenir la enfermedad.
La detección de alteraciones en los genes involucrados en los procesos de trom-bosis se realiza en el DNA, pues es la molécula que guarda el código genético detodas las células. Se obtiene de muestras, como sangre total tomada en un tubocon anticoagulante EDTA y exudado de células de la mucosa bucal. A continua-ción se presenta una breve descripción de los métodos moleculares (comercialeso de investigación) utilizados para la detección de los diferentes tipos de anorma-lidades genéticas asociados al fenómeno de trombosis.
Extracción de ácidos nucleicos
En general las técnicas moleculares parten del material genético del individuo(ácido desoxirribonucleico–DNA y ácido ribonucleico–RNA), por lo que es in-dispensable realizar la extracción y el aislamiento de los ácidos nucleicos, garan-tizando una elevada pureza (es decir, que presente una cantidad mínima de proteí-nas o sales que puedan inhibir la reacción) y una concentración adecuada deacuerdo con la metodología que se utilice posteriormente para la detección delgen de interés.

433Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 27–1. Equipo automatizado de extracción de ácidos nucleicos basado en perlasmagnéticas.
Actualmente existen diferentes métodos de extracción y purificación de áci-dos nucleicos, algunos manuales y otros automatizados (figura 27–1); sin embar-go, todos se basan en tres pasos fundamentales:
1. Lisis celular: en esta etapa se rompe la estructura de las células formadapor macromoléculas y se lleva a cabo la liberación de DNA con la ayudade una solución detergente.
2. Degradación de la fracción proteica asociada al DNA: esto se consigueutilizando una proteasa y posteriormente la fracción proteica se elimina.
3. Purificación y recuperación o elución del DNA: los métodos de extrac-ción más utilizados son el de salting out, el de extracción por columna y elde perlas magnéticas, entre otros.
Una vez obtenidos los ácidos nucleicos se procede a realizar la detección de laanormalidad genética asociada al problema trombótico; para esto los métodos co-merciales o de investigación utilizan como base las siguientes técnicas molecula-res.
AMPLIFICACIÓN DE DNA MEDIANTE LA REACCIÓNEN CADENA DE LA POLIMERASA CONVENCIONAL
Esta metodología fue desarrollada por Kary Mullis a mediados de la década de1980; también es conocida como reacción en cadena de la polimerasa (PCR, por

434 (Capítulo 27)Tratado de trombosis
Figura 27–2. Equipos para reacción en cadena de la polimerasa punto final.
sus siglas en inglés) punto final. Consiste en la amplificación de una región dematerial genético, es decir, replica de forma exponencial un fragmento específicode DNA gracias a la actividad de la enzima DNA polimerasa termoestable (TaqDNA polimerasa) en presencia de una mezcla de nucleótidos (dNTP), un par deiniciadores que flanquean la región de interés (primers) y el DNA blanco (DNAextraído de la muestra), todo lo anterior en una solución con concentración deMgCl2 y pH adecuados para que la reacción de PCR se lleve a cabo de forma efi-caz. Asimismo, la reacción se realiza por incubación a diferentes temperaturasdurante varios ciclos, en los cuales las cadenas de DNA se desnaturalizan (95�C), se copian (de 54 a 65 �C) y se vuelven a alinear (72 �C). La cantidad de ci-clos y el tiempo de cada una de las etapas de la PCR dependerán de cada ensayo(figura 27–2).
A su vez, la PCR tiene diferentes variantes en función de su aplicación o formade analizar el producto generado:
� PCR multiplex: es una modificación a la PCR convencional mediante lacual se pueden detectar e identificar simultáneamente múltiples blancos ge-néticos en una sola reacción. Lo anterior se debe a que esta reacción de PCRse compone de varios pares de primers, cada par para un fragmento en parti-cular. Este tipo de reacción permite detectar más de una alteración (muta-ciones, deleciones, polimorfismo de nucleótido simple) en uno a varios ge-nes a partir de una sola muestra.
� PCR anidada (PCR nested): esta reacción se compone de dos reacciones dePCR convencional, las cuales se realizan por separado y cada una con prim-ers distintos. Para esta reacción primero se realiza una PCR con los inicia-

435Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
dores externos, para amplificar una región de DNA de una longitud o tamañomás grande. El producto amplificado de esta primera PCR se utiliza como“molde” para realizar una segunda PCR, en la cual se incluyen primers queamplificarán una región más pequeña dentro de los fragmentos replicadosen la primer PCR. Con este tipo de reacción se incrementa la sensibilidadde detección, pues se reducen las posibles amplificaciones inespecíficas yse incrementa el número de los fragmentos de DNA donde se encuentra laregión de interés.
Además de estas dos variantes de PCR, existen otras que se han diseñado con ob-jetivos particulares, como:
� Transcripción reversa y PCR (RT–PCR): este tipo de reacción utilizaRNA como “molde” inicial, por lo que previa a la reacción de PCR se llevaa cabo una reacción de retrotranscripción (RT) para sintetizar DNA com-plementario (cDNA) a partir de RNA. Para lo anterior, esta reacción incluyeen su mezcla a la enzima transcriptasa reversa.
� PCR de extensión solapada (mutagénesis): este tipo de reacción utilizados pares de primers, un par de un fragmento de DNA clonado y otro parde la molécula normal, de tal forma que el producto amplificado portaráparte de la mutación o del fragmento clonado. Posteriormente se realiza otraPCR para incrementar el producto mutado.
� PCR in situ: se realiza directamente en el tejido o las células, en los que losproductos amplificados se pueden visualizar en el sitio de amplificación.
El resultado de la PCR es un producto amplificado llamado amplicón, el cual pue-de ser analizado por métodos como electroforesis, microarreglos, secuenciación,etc.
La electroforesis es un método analítico que utiliza una corriente eléctrica con-trolada que tiene el objetivo de separar las moléculas de acuerdo con su tamañoy su carga eléctrica. Los métodos electroforéticos más comunes son:
� Electroforesis en gel de agarosa: en este tipo de reacción se utiliza comomatriz una solución de agarosa a una concentración conocida, pues de elladepende el tamaño de las moléculas a separar. Durante la electroforesis elgel de agarosa funge como una red porosa a través de la cual migrarán delpolo negativo al positivo los ácidos nucleicos de menor a mayor peso mole-cular. Los fragmentos de DNA son teñidos con bromuro de etidio, un agenteintercalante de DNA de doble cadena que en presencia de luz ultravioleta(UV) emite fluorescencia. Una vez finalizada la corrida de electroforesis elgel se revela en un transiluminador acoplado a un fotodocumentador, queconsiste en un aparato cerrado con lámparas UV bajo una placa sobre la cual

436 (Capítulo 27)Tratado de trombosis
Figura 27–3. Transiluminador acoplado a un fotodocumentador.
se coloca el gel; en la parte superior tiene una cámara conectada a una com-putadora con la que se tomará una fotografía del gel, que revelará los frag-mentos de diferentes tamaños (figura 27–3).
Para el análisis por electroforesis es necesario que en cada corrida se in-cluya un marcador de peso molecular (MPM) conocido que permita anali-zar los tamaños en peso molecular (PM) de los fragmentos amplificados.Cuando se analizan alteraciones, como polimorfismo de nucleótido simple(SNP, por sus siglas en inglés), en el cual el peso molecular se modifica sóloen una base nitrogenada, no se recomienda este método de análisis, pues suresolución no es lo suficientemente alta para detectarlo (figura 27–4).
� Electroforesis de poliacrilamida en placa: esta reacción es utilizada paraanalizar secuencias de DNA de bajo peso molecular. La técnica consiste enla elaboración de una fina capa de gel de acrilamida adherida a una láminade vidrio por la cual migrará el DNA. Al igual que en un gel de agarosa, lamigración responde a la carga negativa neta de la molécula de DNA y laconcentración del gel. Las secuencias de DNA más pequeñas migran a ma-yor velocidad, mientras que las grandes se quedan atrás. La visualizaciónde las bandas puede hacerse directamente con un transiluminador mediantela tinción con bromuro de etidio (como en geles de agarosa) o nitrato de pla-ta (AgNO3); también se puede realizar a través de autorradiografía, siemprey cuando se hayan utilizado en la PCR primers marcados con radioisótopos(por el riesgo que implica su manejo es un método que no se utiliza de forma
437Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 27–4. Revelado de fragmentos amplificados en un gel de agarosa.
regular) o marcadores fluorescentes. Estos últimos tienen mayor sensibili-dad y se utilizan regularmente cuando se realizan geles para secuenciación.
� PCR en tiempo real (qPCR): a principios de la década de 1990 Higuchiy col. presentaron la primera demostración de PCR en tiempo real. Estareacción permite la amplificación y la detección simultánea de un frag-mento específico de DNA, ya que además del uso de la enzima Taq DNApolimerasa y los iniciadores que flanquean la región de interés se utilizansondas marcadas con un agente fluorescente, el cual permite detectar laseñal de amplificación ciclo a ciclo “en tiempo real” (figura 27–5). El análi-sis se realiza mediante el uso de un software especializado, lo cual evita asu vez el riesgo por contaminación con amplicones en el laboratorio. Lasventajas que presenta este método son mayor sensibilidad y especificidadpor el uso de las sondas complementarias del fragmento de interés. Actual-mente la qPCR se realiza utilizando diferentes reactivos o sondas marcadas,conocidos como químicas, que son tecnologías con marcadores fluorescen-tes, como SybrGreen, Resolight y LC Green (estos dos últimos para curvasde fusión de alta resolución, mejor conocidas como HRM, o high resolutionmelting), y sondas TaqMan, Scorpions, molecular beacon, Fast, Light up,etc., todas ellas marcadas con agentes fluorescentes, como FAM, VIC,HEX, TET, Yellow 55, fluoresceína, Tamra, etc., así como sondas de hibri-dación y simple probes, marcadas con LCRed 640, LCRed 670 y LCRed705. Cada uno de los diferentes formatos de químicas permite realizar ensa-yos para identificación cualitativa, cuantificación absoluta, cuantificación

438 (Capítulo 27)Tratado de trombosis
Figura 27–5. Equipo de reacción en cadenas de la polimerasa en tiempo real.
relativa de expresión genética, detección de mutaciones, discriminaciónalélica, identificación de SNP y análisis de metilación (mediante HRM),entre otros (figuras 27–6 y 27–7). Esta metodología es de las más utilizadas;es aplicada al diagnóstico molecular por su relativo bajo costo y rapidez (nomayor de cuatro horas) en la obtención de resultados.
� Microarreglos de DNA: los microarreglos surgieron a partir de la técnicade Southern blot, que consiste en fijar fragmentos de DNA, previamente
Figura 27–6. Análisis de qPCR cualitativo utilizando sondas Taqman marcadas conFAM.
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.115 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40

439Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 27–7. Análisis de qPCR por curvas melting, utilizando sondas de hibridaciónmarcadas con LCRed 640.
cortados por enzimas de restricción en una membrana de nailon, los cualesson detectados por hibridación con una sonda marcada de DNA de secuen-cia conocida; su desventaja es que utiliza grandes cantidades de DNA. Ac-tualmente la técnica de microarreglos se basa en inmovilizar una colecciónde fragmentos de material genético o sondas (DNA o cDNA) en una basede vidrio, nailon o silicón. Posteriormente se realiza una reacción de hibri-dación reversa entre los fragmentos de DNA proveniente de la muestra deinterés (que por lo regular es un producto de PCR) y las sondas fijadas enla base del microarreglo. Las ventajas de este tipo de técnicas son:� La cantidad de DNA inicial requerida es mucho menor, pues será repli-
cado en la PCR.� El producto amplificado incrementa la sensibilidad y la especificidad de
la prueba.� Se pueden detectar múltiples genes (para análisis de expresión), muta-
ciones, polimorfismos, agentes infecciosos, etc., en una sola reacción.� Durante la amplificación los amplicones quedan marcados con compues-
tos fluorescentes (Cy3, Cy5 o biotina), lo que posteriormente permitiráque la hibridación se detecte por fotoimagen o escáner de fluorescencia.
Todos los microarreglos incluyen controles negativos, positivos y de hibri-dación que permiten validar el resultado de cada ensayo. Las empresas quedesarrollan microarreglos para diagnóstico cuentan también con equipo ysoftware especializado para leer el resultado de la hibridación, evitando asíun resultado de error por interpretación (figura 27–8).

440 (Capítulo 27)Tratado de trombosis
Figura 27–8. Equipo automatizado de hibridación a microarreglos INFINITI�AutoGe-nomics.
� Secuenciación y electroforesis capilar: la técnica de secuenciación con-siste en el análisis detallado de una estructura del DNA, es decir, identificael ordenamiento de cada nucleótido en una cadena de DNA. A lo largo deltiempo se han desarrollado diferentes métodos para obtener la secuencia deDNA:� Método químico de Maxam y Gilbert: esta técnica fue descrita en
1977. Consiste en romper cadenas sencillas de DNA marcadas radiacti-vamente con reacciones químicas específicas para cada una de las cuatrobases (AGCT). Los productos de estas cuatro reacciones se resuelven enfunción de su tamaño por electroforesis en geles de poliacrilamida, en losque la secuencia se puede leer con base en el patrón de bandas radiactivasobtenidas.
� Método enzimático de Sanger y secuenciación automatizada: tam-bién se conoce como método de los terminadores de cadena o didesoxi(dd). Fue diseñado por Sanger y col. en la década de 1970. A partir deldesarrollo de la reacción en cadena de la polimerasa el método se realizócon base en cuatro reacciones de PCR. La mezcla de reacción se compo-ne de un solo primer en dirección 5’ a 3’, la Taq DNA polimerasa, losnucleótidos (dNTP) y los dinucleótidos marcados con una moléculafluorescente (ddNTP) distinta para cada base, además del DNA o cDNAde cadena simple como molde. El producto obtenido son fragmentos de

441Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Figura 27–9. Electroferograma de un análisis de secuenciación.
DNA de cadena sencilla de diferentes tamaños que tienen un terminadordidesoxi. Actualmente el análisis de esta reacción se realiza por secuen-ciación automatizada por electroforesis en gel de poliacrilamida en placao en capilar acoplado a un secuenciador. Durante la electroforesis lasbandas del DNA de cadena sencilla pasan a través de una fuente queexcita las moléculas fluorescentes; la señal de fluorescencia es detectadaa través de un filtro que identifica y asigna la base correspondiente; elresultado obtenido es conocido como electroferograma (figura 27–9).
� Análisis de fragmentos y electroforesis capilar (EC): en la actualidadla electroforesis capilar es una herramienta de separación de biomolécu-
Figura 27–10. Equipo de secuenciación automatizado por electroforesis capilar.

442 (Capítulo 27)Tratado de trombosis
Cuadro 27–1.
Nombre delequipo
Compa-ñía
Metodología Gen/polimorfismodetectado
Utilidadclínica
INFINITI� Fac-tor II Plus
AutoGe-nomics
PCR multiplex e hibridacióna microarreglos
Factor II: C20209T yG20210A
Identifica-ción cuali-
i dINFINITI� Fac-tor V
AutoGe-nomics
PCR multiplex e hibridacióna microarreglos
Factor V de Leiden:G1691A
tativa dela presen-i d lINFINITI� Pa-
nel Factor II–Vde Leiden
AutoGe-nomics
PCR multiplex e hibridacióna microarreglos
Factor II: G20210AFactor V de Leiden:G1691A
cia de lamutación(según elkit utilizaINFINITI�
MTHFR (CE)AutoGe-nomics
PCR multiplex e hibridacióna microarreglos
MTHFR: A1298C yC677T
kit utiliza-do)PermiteINFINITI� Pa-
nel Factor IIPlus–V &MTHFR (CE)
AutoGe-nomics
PCR multiplex e hibridacióna microarreglos
Factor II: C20209T yG20210A. Factor Vde Leiden: G1691AMTHFR: C677T yA1298C
Permitediferenciara pacien-tes conriesgo apadecer
DiagnosticHealthcareThrombosis kit(CE)
AstraBiotechGmbH
PCR multiplex y análisis deSNP por electroforesis engel de poliacrilamida
Factor II: SNP20210G/A. Factor V:SNP 1691G/A.MTHFR: SNP677C/T
padecertrombosisocasiona-do por undesordengenético
Factor II (Proth-rombin)G20210A Kit(CE–IVD)
Roche PCR en tiempo real y análi-sis por curvas melting
Factor II: SNP20210G/A
genético
Factor V deLeiden Kit (CE–IVD)
Roche PCR en tiempo real y análi-sis por curvas melting
Factor V: SNP1691G/A
LightMix�Kit MTHFRC677T (RUO)
Tibmol-biol
PCR en tiempo real y análi-sis por curvas melting
MTHFR: C677T
GeneProofMTHFR C677T(CE–IVD)
Gene-Proof
PCR en tiempo real y análi-sis con sondas marcadas
MTHFR: C677T
ThromboType�plus
HAIN Li-fescience
PCR multiplex y análisis porhibridación a tiras de mem-brana de nailon medianteuna reacción colorimétricacon fosfatasa alcalina
Factor II: SNPG20210A/G. Factor Vde Leiden: SNPG1691A/G. MTHFR:C677T/C y A1298C/A
FluoroType�Factor II
HAIN Li-fescience
PCR en tiempo real y análi-sis por curvas melting
Factor II: G20210A/GSNP
FluoroType�Factor V
HAIN Li-fescience
PCR en tiempo real y análi-sis por curvas melting
Factor V de Leiden:SNP G1691A/G
GenoType�MTHFR
HAIN Li-fescience
PCR multiplex y análisis porhibridación a tiras de mem-brana de nailon medianteuna reacción colorimétricacon fosfatasa alcalina
MTHFR: C677T/C yA1298C/A
PCR: reacción en cadena de la polimerasa; SNP: polimorfismo de nucleótido simple.

443Biología molecular en la trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
las, con capacidad para separar e identificar cientos de componentes deforma simultánea a una mayor velocidad y especificidad. Es una técnicabasada en la migración diferencial de DNA, sujeta a un campo eléctricoa través de un capilar de menos de 50 �m de diámetro; igual que en todaelectroforesis, las moléculas con carga positiva fluyen hacia la terminalnegativa y viceversa. La inducción del alto potencial eléctrico ofrece dosventajas: que la separación es más sensible entre las diferentes moléculas(resolución) y que el tiempo de análisis es más corto (figura 27–10).
La técnica de análisis de fragmentos se basa en la generación de productos dePCR, los cuales se encuentran marcados por diferentes compuestos fluorescen-tes. Durante la EC las moléculas fluorescentes son detectadas por un filtro, per-mitiendo el análisis de múltiples fragmentos mientras se mueven por el campoeléctrico. En el cuadro 27–1 se pueden observar los diferentes equipos comercia-les (con IVD o de investigación–RUO) para la detección de genes asociados atrombosis. Es importante mencionar que el resultado de cualquiera de estas prue-bas le proporciona al médico información que le permite identificar la causa dela trombosis e identificar si los familiares del paciente se encuentran en riesgo depresentar la enfermedad por desórdenes genéticos hereditarios y así estableceruna tromboprofilaxis primaria.
REFERENCIAS
1. Leclerc D, Sibani S, Rozen R: Molecular biology of methylenetetrahydrofolate reductase(MTHFR) and overview of mutations/polymorphisms. En: Ueland PM (ed.): MTHFR poly-morphisms and disease. Austin, Landes Bioscience, 2005. http://www.ncbi.nlm.nih.gov/books/NBK6561/.
2. Simpson EL, Stevenson MD: Thrombophilia testing in people with venous thromboembo-lism: systematic review and cost–effectiveness analysis. HTA 2009;13(2).
3. Varga EA, Sturm AC: Homocysteine and MTHFR mutations: relation to thrombosis andcoronary artery disease. Circulation 2005;111:e289–e293.
4. Kader H, Berman W et al.: Prevalence of factor V G1691A (Leiden), prothrombin G20210Aand methylene tetrahydrofolate reductase C677T thrombophilic mutations in children withinflammatory bowel disease. J Pediatr Gastroenterol Nutr 2002;35(5):629– 635.
5. Liu MS, Amirkhanian VD: DNA fragment analysis by an affordable multiple–channel ca-pillary electrophoresis system. Electrophoresis 2003;24(1–2):93–95.
6. Squizzato A, Micieli E, Galli M: Diagnosis and management of venous thromboembol-ism: results of a survey on current clinical practice. Thromb Res 2010;125(2):134–136.
7. Baglin T, Gray E, Greaves M: Clinical guidelines for testing for heritable thrombophilia.Br J Haematol 2010;149:209–220.
8. Dolnik V: DNA sequencing by capillary electrophoresis (review). J Biochem Biophys Me-thods 1999;41:103–119.
9. Ansorge W, Sproat B et al.: Automated DNA sequencing: ultrasensitive detection of fluo-rescent bands during electrophoresis. Nucleic Acids Res 1987;15(11):4593– 4602.

444 (Capítulo 27)Tratado de trombosis

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
28Fármacos, alimentos y hierbas
en el paciente con trombosisLuis Fernando García–Frade Ruiz, Ivonne Ramírez Díaz,
Berenice Vicente Hernández
No obstante que todos los fármacos tienen el potencial de interactuar con otrosfármacos y alimentos a lo largo de su recorrido por el organismo, abarcando sutránsito en el aparato gastrointestinal, absorción, unión a proteínas transportado-ras, metabolismo, acción y depuración, las interacciones que de manera particu-lar se refieren a los agentes antitrombóticos generan un gran interés por la rele-vancia terapéutica y los riesgos que conllevan un nivel subóptimo y prolongadodel sistema de la coagulación, lo que puede conducir en un extremo a la presenciade trombosis o bien de hemorragia.
Las interacciones farmacológicas con la warfarina pueden resultar en conse-cuencias devastadoras. La toxicidad severa con warfarina se asocia a una alta fre-cuencia de hospitalización, hemorragia gastrointestinal o intracraneal fatales, eincapacidad permanente. La hemorragia intracraneal es una de las mayores com-plicaciones con el uso de la warfarina, la cual ocurre en 2% de los pacientes conterapia a largo plazo. El rango de fatalidad de la hemorragia intracraneal relacio-nada con la warfarina es de 77%.
Con frecuencia al paciente que se encuentra bajo terapia anticoagulante conwarfarina se le prescriben otros medicamentos que aumentan el riesgo de sangra-do; entre los más frecuentemente reportados se encuentran los antibióticos y losantiinflamatorios.
De manera afortunada, una de las ventajas que ofrecen los nuevos anticoagu-lantes orales (NACO) es su baja interacción con los alimentos y fármacos; sinembargo, se considera que quizá más adelante, tras la experiencia con su uso, po-drán manifestarse otras interacciones no conocidas en este momento, sobre todo
445

446 (Capítulo 28)Tratado de trombosis
con algunas hierbas. Por eso es conveniente conocer los fármacos que hasta elmomento se sabe que interactúan con ellos, ya que paradójicamente, siendo lastaquiarritmias una de sus actuales indicaciones, algunos antiarrítmicos de uso co-mún interactúan con ellos.
INTERACCIONES FARMACOLÓGICAS DE LA WARFARINA
El isómero–S más potente de la warfarina se metaboliza por el citocromo P450(CYP) 2C9. Varios fármacos potencian el efecto de la warfarina al inhibir elCYP2C9, incluyendo la amiodarona, el fluconazol, la fluvastatina, la fluvoxami-na, la isoniazida, la lovastatina, la fenilbutazona y la sertralina. El isómero–R dela warfarina se metaboliza por CYP1A2 y CYP3A4. Las quinolonas inhiben aCYP1A2, mientras que los macrólidos inhiben a CYP3A4. Los azoles (metroni-dazol, trimetoprim–sulfametoxazol, voriconazol, fluconazol y miconazol) inhi-ben a CYP1A2 o CYP3A4. La farmacodinamia de la warfarina puede ser influidapor medicamentos que afecten la vitamina K o los factores de la coagulación.
El riesgo de sangrado es mayor cuando de manera concomitante al uso de war-farina se consumen otros anticoagulantes (como la heparina), agentes antiplaque-tarios (ácido acetilsalicílico, clopidogrel y ticlopidina) y antiinflamatorios no es-teroideos (AINE), incluidos los inhibidores selectivos de la ciclooxigenasa 2(COX–2). Se debe evitar el uso de todos estos fármacos junto con warfarina, amenos que se demuestre que su beneficio es mayor que el riesgo de sangrado,como por ejemplo en el caso de prótesis valvulares cardiacas.
Antibióticos
La mayoría de los antibióticos pueden potenciar el efecto de la warfarina por inhi-bición de la flora intestinal que produce vitamina K. La inhibición del metabolis-mo hepático de la warfarina es otro mecanismo mediante el cual varios medica-mentos pueden aumentar el riesgo de sangrado.
Ciprofloxacina
El uso de ciprofloxacina en pacientes bajo terapia con warfarina se relaciona coneventos hemorrágicos fatales o graves que ponen en peligro la vida. La respuestasupraterapéutica se detecta entre cuatro y cinco días posteriores al inicio del anti-microbiano, con valores de tiempo de protrombina e índice normalizado interna-cional (INR) de hasta 38.0 y 10.0, respectivamente. Las fluoroquinolonas inhi-

447Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ben de manera específica las isoenzimas CYP, que metabolizan el isómero–R dela warfarina.
Betalactámicos
La alteración de la flora intestinal que sintetiza vitamina K es uno de los mecanis-mos a través de los cuales el uso de betalactámicos (como penicilinas, cefalospo-rinas, carbapenems y monobactams) interactúan con la warfarina. La warfarinarealiza su efecto anticoagulante al inhibir la síntesis de los factores dependientesde vitamina K (ver capítulo 4). Al reducirse la producción endógena de vitaminaK se puede aumentar el efecto de la warfarina; además, las cefalosporinas semi-sintéticas, como cefamandol, cefoperazona y cefotetán, las cuales contienen unacadena lateral N–metiltiotetrazol, pueden prolongar el tiempo de protrombina.El uso de estos agentes antimicrobianos puede aumentar el riesgo de sangrado enpacientes que se encuentran bajo terapia anticoagulante con warfarina.
Macrólidos
Los fármacos del grupo de los macrólidos (eritromicina y claritromicina) y ketó-lidos (telitromicina), con excepción de los azálidos (azitromicina), se asocian connumerosas interacciones farmacológicas. El mecanismo más común para estasinteracciones involucra la inhibición del sistema CYP450 y PGP. El uso conco-mitante de eritromicina y warfarina se asocia con una prolongación del tiempode protrombina y hematuria. El potencial de esta interacción se reduce de maneraconsiderable con la combinación de warfarina con claritromicina o telitromicina.
Metronidazol
El metronidazol aumenta tanto la vida media como las concentraciones plasmáti-cas del isómero–S de la warfarina.
Trimetoprim–sulfametoxazol
Aproximadamente 60% del trimetoprim se elimina sin cambios en la orina. Elsulfametoxazol se metaboliza de manera principal en el hígado y 30% se eliminaen la orina. El trimetoprim y el sulfametoxazol son inhibidores selectivos deCYP2C8 y CYP2C9, respectivamente, con lo que aumentan el efecto anticoagu-lante de la warfarina.
Isoniazida
La isoniazida provoca inhibición de CYP2C9, CYP2C19 y CYP3A4, por lo queinteractúa de manera importante con la fenitoína, la carbamazepina, el diazepamy la warfarina, aumentando la acción anticoagulante de esta última.

448 (Capítulo 28)Tratado de trombosis
Rifampicina
La rifampicina, igual que otras rifamicinas —como la rifabutina y la rifapenti-na—, aumenta la depuración de la warfarina, con lo que disminuye su concentra-ción plasmática.
Antifúngicos
Los antifúngicos azoles incluyen al ketoconazol y al grupo triazol, químicamenterelacionado con el itraconazol y el fluconazol. Esta clase de antifúngicos causaninhibición del CYP. El ketoconazol y el itraconazol inhiben el CYP3A4 de mane-ra significativa en dosis terapéuticas, mientras que el fluconazol realiza tal efectoen dosis mayores de 200 mg al día. Sin embargo, el fluconazol inhibe el CYP2C9,por lo que interactúa con los inhibidores de la angiotensina II, los AINE, los hipo-glucemiantes orales y la warfarina, aumentando se efecto anticoagulante.
Antiinflamatorios no esteroideos
El uso de AINE en pacientes de edad avanzada se asocia con un riesgo cinco ve-ces mayor de presentar úlcera péptica y sangrado. Dicho riesgo de sangrado au-menta hasta 13 veces, con un rango de mortalidad de 5 a10% cuando se asocianlos AINE con anticoagulantes.
Acetaminofén
Hasta hace poco tiempo se recomendaba el acetaminofén como el analgésico deelección en pacientes bajo terapia anticoagulante con warfarina, pero las recien-tes evidencias demuestran que el consumo de éste aun en bajas dosis aumenta elefecto anticoagulante de la warfarina.
Acido acetilsalicílico
La coadministración de ácido acetilsalicílico y warfarina aumenta el riesgo desangrado. Los mecanismos de tal interacción se deben a los efectos antiplaqueta-rios y al daño a la mucosa gástrica por parte del ácido acetilsalicílico en asocia-ción con el efecto anticoagulante de la warfarina.
El uso de warfarina con bajas dosis de Aspirina� (75 a 100 mg al día) aumentala incidencia de sangrado menor. No obstante, los beneficios de esta combinaciónpueden ser de ayuda para los pacientes con alto riesgo de enfermedad tromboem-bólica (p. ej., los pacientes con prótesis valvular cardiaca).

449Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Inhibidores de la ciclooxigenasa 2
La enzima COX–2 se relaciona de manera principal con la producción de infla-mación y dolor. Los inhibidores de la COX–2 no afectan la función plaquetariay se asocian con menos efectos gastrointestinales que los AINE. El uso concomi-tante de estos agentes con warfarina incrementa el INR y el riesgo de sangrado.El celecoxib es metabolizado por la isoenzima CYP2C9 y puede inhibir el meta-bolismo del isómero–S de la warfarina.
FÁRMACOS CARDIOVASCULARES
Varios medicamentos de uso cardiovascular interactúan con la warfarina aumen-tando su efecto anticoagulante, como la amiodarona, el verapamilo, el diltiazem,el clofibrato, la propafenona, el propranolol y la sulfinpirazona. Esta última ejer-ce un efecto bifásico, seguido por inhibición de la warfarina.
Medicamentos que inhiben el efecto de la warfarina
Son pocos los medicamentos que inhiben el efecto de la warfarina, los cuales in-cluyen a la nafcilina, la rifampicina, la griseofulvina, la colestiramina, los barbi-túricos, la carbamazepina y el sucralfato, así como los alimentos con alto conteni-do de vitamina K y el consumo de grandes cantidades de aguacate o brócoli.
Medicamentos que no ejercen efecto sobre la warfarina
De acuerdo con las evidencias, varios medicamentos no ejercen efecto alguno so-bre la warfarina; ellos incluyen al atenolol, la bumetanida, el felodipino, el meto-prolol, los antiácidos, la famotidina, el psyllium, la ranitidina, el ketorolaco, elnaproxeno, el alcohol, el nitrazepam y la fluoxetina.
Hierbas que interactúan con la warfarina
Las hierbas y sus productos han sido utilizados desde hace varios siglos comoanalgésicos, anticonceptivos, laxantes, sedantes y remedios para la tuberculosis,el resfriado y el cáncer.
En 1888 la Asociación Farmacéutica Americana publicó el Formulario Nacio-nal, que incluye las medicinas basadas en hierbas. En la década de 1920 las hier-

450 (Capítulo 28)Tratado de trombosis
bas fueron reemplazadas por medicamentos farmacéuticos. Debido a que la ma-yoría de los productos herbolarios se encuentran a la venta como suplementosalimenticios, no cumplen con los requisitos que estipula la Farmacopoeia deEUA. En 1995 el Acta Alimenticia de Salud y Educación de EUA permitió quelas preparaciones herbolarias tuvieran dosis sugeridas en las etiquetas y solicitóa los fabricantes que colocaran leyendas médicas específicas, indicando el efectoque el producto provoca en el organismo. Más adelante se estipuló que se debíacolocar en todos los productos, junto a la leyenda anterior, la siguiente frase:“Esta declaración no ha sido evaluada por la FDA”. La Food and Drug Adminis-tration (FDA) espera en un futuro poder contar con mejores y más completos re-portes sobre los efectos adversos tanto de los complementos alimenticios comode las hierbas.
Las razones más frecuentes para el uso de complementos herbolarios y no her-bolarios son la salud en general, el resfriado, la osteoartritis, el aumento de ener-gía, la disminución del colesterol, la prevención de cáncer, la alergia y el manejodel peso corporal.
Las hierbas y otros complementos son utilizados de manera común como tera-pia complementaria o alternativa en todo el mundo. Al menos la mitad de los pa-cientes con condiciones crónicas utilizan una terapia alternativa en conjunto conla prescripción médica.
Los suplementos herbales son utilizados en ocasiones como parte de los ali-mentos comunes por tradición cultural, y sólo la mitad de los pacientes comentancon su médico el uso de tales terapias.
Ginkgo biloba
El ginkgo biloba se obtiene de una de las especies de árbol más antiguas del pla-neta (más de 225 millones de años) y constituye uno de los fitofármacos más utili-zados en Europa. Se utiliza de manera principal para trastornos de la memoria,vértigo, tinnitus, claudicación intermitente y disfunción eréctil. Sus componen-tes flavinoides actúan como antioxidantes y reducen la fragilidad capilar. Sucomponente ginkgolide B antagoniza el factor activador de las plaquetas (FAP),el cual induce la agregación plaquetaria, la degranulación de neutrófilos y la pro-ducción de radicales de oxígeno. El ginkgo biloba se encuentra aprobado en Ale-mania para tratar trastornos de la circulación cerebral y arteriales periféricos conmás de cinco millones de prescripciones al año en ese país, además de que es lahierba más vendida en EUA.
La incidencia de sangrados severos se ha incrementado tras el uso de esta hier-ba, de manera particular en los pacientes que se encuentran bajo tratamiento conAspirina� o warfarina, como resultado de la habilidad del ginkgo para inhibirel FAP.

451Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Hierba de San Juan
La hierba de San Juan se ha utilizado durante más de 2 000 años y es la segundahierba más vendida en EUA. Los extractos de la hierba son antidepresivos atípi-cos. El hipericum inhibe la recaptura de serotonina y antagoniza a la reserpina.Los recientes estudios sugieren que la hiperforina puede ser el ingrediente activode la hierba, el cual inhibe la recaptura de serotonina, dopamina y norepinefrina,e interactúa con los receptores GABA y de glutamato. La efectividad de la hierbase basa en su contenido de hiperforina y no de hipericum.
La hierba de San Juan induce las enzimas del citocromo P450 3A4 y 2C9, ylas glucoproteínas transportadoras, con lo que disminuye los niveles de variosmedicamentos, como la warfarina, la digoxina, la ciclosporina, el indinavir, losestrógenos, la teofilina y los NACO.
Ginseng
El ginseng es la tercera hierba más popular en América. Se ha utilizado en Asiadurante más de 2 000 años y es la hierba mejor estudiada desde el punto de vistacientífico. Su nombre científico, Panax ginseng, alude a su cualidad de “pana-cea”. En la medicina china se utiliza para aumentar la resistencia física, químicay biológica del organismo ante los fenómenos de estrés. Sus principales usos con-sisten en mejorar los síntomas de ansiedad, debilidad, disnea, fatiga y disminu-ción de la libido, y las náuseas. Actualmente las personas lo utilizan para mejorarel desempeño físico, la vitalidad, la función inmunitaria, la disfunción eréctil yla fertilidad; además se utiliza como terapia para la diabetes mellitus, la infecciónpor VIH, el cáncer y la hiperlipidemia.
Los componentes activos más importantes del ginseng constituyen al menos18 ginsenosides, un grupo diverso de saponinas esteroideas que poseen la habili-dad de alcanzar múltiples tejidos blanco produciendo diversos efectos farmaco-lógicos, como bloqueo de los canales de calcio, disminución de radicales libres,aumento en los niveles de insulina, ligera disminución de la glucosa y efecto es-trogénico moderado.
Existen diversos tipos de plantas del ginseng en Asia (Panax ginseng), Améri-ca (Panax quinquefolium) y Japón (Panax japonicus), cada uno con constituyen-tes químicos y propiedades distintas.
El ginseng se comercializa en varias presentaciones a manera de tableta, fruta,té, bebida mineral, extracto, etc. El consumo de esta hierba puede disminuir elINR en pacientes bajo terapia anticoagulante con warfarina.
Ajo
El ajo es una planta medicinal tradicional utilizada en Egipto, Israel, China y Eu-ropa. Los pacientes lo utilizan como agente antiplaquetario, antiparasitario, anti-

452 (Capítulo 28)Tratado de trombosis
fúngico, antiviral, antiinflamatorio y para tratamiento contra el cáncer. Variasmujeres posmenopáusicas lo utilizan para disminuir los niveles de colesterol ycomo prevención de la aterosclerosis.
El ajo posee un débil efecto antihipertensivo e inhibe a la hidroximetilglutarilcoenzima A (MHG Co A) reductasa, activa la fibrinólisis e inhibe la activaciónplaquetaria, por lo que se debe utilizar con precaución junto con otros agentes an-tiplaquetarios, como la Aspirina�, el ginkgo y los AINE. El uso concomitantede ajo y warfarina aumenta el efecto anticoagulante de ésta, con prolongación delINR.
Dong quai
El dong quai es una hierba china que se encuentra a la venta en combinación conotras hierbas, que se utiliza para el manejo de los síntomas climatéricos. El dongquai contiene cumarinas y furocumarinas, por lo que si se consume en combina-ción con la warfarina aumenta el riesgo de sangrado.
En las figuras 28–1 y 28–2 se muestran las interacciones clínicamente signifi-cativas con la warfarina.
NUEVOS ANTICOAGULANTES ORALESY SUS INTERACCIONES CONOCIDAS
En relación con los NACO, no se ha reportado hasta el momento ninguna interac-ción con alimentos. La ruta metabólica de los NACO confiere el potencial de in-teracción con otros fármacos que se involucran con el sistema P450 o con lostransportadores de la glucoproteína P. Así, por ejemplo, se debe tener cuidado enindicar dabigatrán con fuertes inhibidores o inductores de los transportadores dela glucoproteína P y tener precaución con el tratamiento concomitante de rivaro-xabán con inhibidores de la glucoproteína P o del CYP3A4, si bien la coadminis-tración de estos se encuentra contraindicada. Al metabolizarse el apixabán y elrivaroxabán a través del CYP3A4 probablemente compartirán las mismas pre-cauciones.
De acuerdo con algunos estudios, el uso de los siguientes fármacos no provocainteracciones clínicamente significativas: atorvastatina junto con dabigatrán o ri-varoxabán, digoxina y dabigatrán, así como tampoco ranitidina con dabigatránni con rivaroxabán.
El uso de dabigatrán con quinidina, un antiarrítmico y antipalúdico, está con-traindicado. El rivaroxabán se encuentra contraindicado en los pacientes que re-ciben inhibidores de la proteasa del VIH o antimicóticos azólicos, ya que son in-hibidores de la CYP 3A4 y de la glucoproteína P.

453Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
Warfarina
Antibióticos, antivirales y
Probable
Antibióticos, antiviralesy antimicóticos:
cefamandol, cefazolin,
Muy improbable
Antibióticos, antiviralesy antimicóticos:
ciprofloxacina, eritromicina,
Muy probable
sulfisoxazolfluconazol, isoniazida,metronidazol, miconazol
oral y en supositoriosvaginales, voriconazol
Cardiovasculares:bezafibrato, heparina
Antiinflamatorios einmunitarios: levamisol,
metillprednisolona,
Fármacos del sistemanervioso central:
fluoxetina, diazepam,
Otros fármacosetopósido, carbaplatino,
Fármacos cardiovasculares:ácido acetilsalicílico,
nabumetona
quetiapina
levonorgestrel
Antiinflamatorios e inmunitarios:
Fármacos del sistema nervioso
Gastrointestinales y
Suplementos herbales:
Otros fármacos: fluorouracilo,
Figura 28–1. Interacciones clínicamente significativas que aumentan el efecto de lawarfarina.
Fármacos cardiovasculares:amiodarona, clofibrato,
Antiinflamatorios einmunitarios:
fenilbutazona, piroxicam
Fármacos del sistemanervioso central:
alcohol con hepatopatía
Gastrointestinales yalimentos:
cimetidina, omeprazol,
Suplementos herbales:fenugreek, quilinggao,
Otros fármacos: esteroides
diltiazem, fenofibrato,propafenona, propranolol,sulfinpirazona, verapamilo
concomitante, citalopram,entacapone, sertralina
aceite de pescado, mango
tamarindo
fluvastatina, quinidina, ropinirol
alimentos: jugo de toronja,jugo de arándano, orlistat
dancen, dong quai, lycium,barbarum 1, ajo, gingko,papaya, garra del diablo,dancen/metilsalicilatos
antimicóticos: amoxicilina conclavulanato, azitromicina,claritromicina, itraconazol,
levofloxacina, ritonavir,tetraciclina, amoxicilina,
cloranfenicol, gatifloxacina,miconazol tópico, ácidonalidíxico, norfloxacina,ofloxacino, saquinavir y
terbinafina
simvastatina, disopiramida,gemfibrozil, metolazona
acetaminofén, celecoxib,dextropropoxifeno, interferón,
tramadol, indometacina,leflunomida, propoxifeno,
salicilatos tópicos, robecoxib,sulindaco, tolmetin
central: disulfiram, fluvoxamina,fenitoína, felbamato
gemcitabina, levamisol/fluorou-racilo, paclitaxel, tamoxifeno,tolterodina, acarbosa, CFM
(ciclofosfamida, metotrexato,fluorouracilo), danazol,ifosfamida, trastuzumab
anabólicos, zileutón,hormona tiroidea, sildenafil

454 (Capítulo 28)Tratado de trombosis
Warfarina
Antibióticos, antivirales y
Probable
Antibióticos, antiviralesy antimicóticos:
Muy improbable
Antibióticos, antiviralesy antimicóticos:
Muy probable
Fármacos
Fármacos del sistemanervioso central:
Suplementos herbales:
Fármacos cardiovasculares:
Antiinflamatorios e
Fármacos del sistema
Alimentos y fármacos
Suplementos herbales:
Otros fármacos: vacuna de
Fármacos
Antiinflamatorios einmunitarios: mesalamina
Fármacos del sistemanervioso central:
barbitúricos,
Alimentos y fármacos
Suplementos herbales:hierba de San Juan
Otros fármacos:
antimicóticos: dicloxacilina,
Figura 28–2. Interacciones clínicamente significativas que disminuyen el efecto de lawarfarina.
griseofulvina, nafcilina,ribavirina, rifampicina
cardiovasculares:colestiramina
carbamazepina
gastrointestinales:alimentos con alto
contenido de vitamina K,aguacate en grandes
cantidades, coenzima Q10
mercaptopurina, agentesantitiroideos
ritonavir, terbinafina
bosentán, telmisartán
inmunitarios:azatioprina, sulfasalazina
nervioso central:clordiazepóxido
gastrointestinales: leche desoya, sucralfato, grandes
cantidades de brócoli, sushique contenga alga marina
ginseng
la influenza, suplementosmultivitamínicos, raloxifeno,
ciclosporina, etretinato
cloxacilina, teicoplanina
cardiovasculares:furosemida
propofol
té verde
� Inhibidores del CYP3A4 y de la glucoproteína P: claritromicina, azoles yritonavir.
� Inhibidores de la glucoproteína P: quinidina, verapamilo y amiodarona. Ladosis de dabigatrán se reduce en los pacientes con fibrilación auricular quetoman verapamilo, mientras que la dosis en cirugía ortopédica mayor se re-duce en quienes se encuentran tomando amiodarona.

455Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
� Inductores del CYP3A4 y de la glucoproteína P: hierba de San Juan, rifam-picina, pantoprazol, fenitoína y carbamazepina. Si bien el pantoprazol dis-minuye la biodisponibilidad del dabigatrán por disminución del pH, esto notiene un significado clínicamente relevante.
La asociación de un anticoagulante con un antiagregante plaquetario o un AINEincrementa el riesgo de sangrado (figura 28–3).
OTROS AGENTES ANTITROMBÓTICOSY SUS INTERACCIONES
Los antiinflamatorios no esteroideos incrementan el riesgo de complicacionesgastrointestinales cuando se utilizan junto con la Aspirina�. En adición, de ma-nera particular el uso de ibuprofeno puede interferir con los beneficios antitrom-bóticos de la Aspirina� a través de una interacción competitiva con la ciclooxi-genasa 1. Por el contrario, el acetaminofén se considera el analgésico de primeraelección en los pacientes con dolor de leve a moderado que consumen Aspirina�,ya que no contribuye a la gastrotoxicidad ni tampoco interfiere con la agregaciónplaquetaria.
El consumo de tamarindo (Tamarindus indica) aumenta la biodisponibilidaddel ácido acetilsalicílico; la combinación de etanol con este último tiene un efectoagonista sobre el tiempo de sangrado.
El uso concomitante de ácido acetilsalicílico y warfarina aumenta el riesgo desangrado, mientras que el uso de agentes fibrinolíticos y otros agentes antitrom-bóticos junto con el ácido acetilsalicílico no aumenta la incidencia de episodiosde sangrado significativo, a excepción de la combinación con clopidogrel, la cualse asocia con un ligero aumento del riesgo de sangrado, en comparación con lamonoterapia con ácido acetilsalicílico.
Son raras las interacciones farmacológicas clínicamente significativas con eluso de clopidogrel; sin embargo, se describió una importante interacción farma-codinámica entre el clopidogrel y la atorvastatina. La atorvastatina es metaboli-zada por el citocromo P450 3A4 (CYP3A4), la misma enzima que convierte alclopidogrel a su metabolito activo. La atorvastatina compite con el clopidogrelpor el CYP3A4, lo que limita la activación del agente antiplaquetario.
EFECTO DE OTROS FÁRMACOS Y SUPLEMENTOSALIMENTICIOS SOBRE LA FUNCIÓN PLAQUETARIA
La administración de altas dosis parenterales de antibióticos betalactámicos pue-de afectar la función plaquetaria con prolongación del tiempo de sangrado, sien-

456 (Capítulo 28)Tratado de trombosis
Figura 28–3. Interacción de los nuevos anticoagulantes orales.
No asociar:
Nuevosanticoagulantes
orales
Dabigatrán
Rivaroxabán
Apixabán
Disminuye el efecto
Aumenta el efecto
Precaución:�
�
�
�
No asociar:�
�
�
�
No asociar:�
�
�
�
Precaución:�
No se recomienda:�
�
Aumenta
�
�
�
�
Disminuye el efecto
No se recomienda:�
No asociar:�
�
�
�
No asociar:�
�
�
�
Precaución:�
�
�
�
Eritromicina y claritromicinaReducir dosis en FA converapamiloEn cirugía ortopédica mayor:amiodaronaÁcido acetilsalicílico o clopidogrel,aumentan el riesgo de sangradopero sin interacción farmacocinética
Disminuye el efecto
el efecto
Dronedarona
Antimicóticos azolesInhibidores deproteasas del VIHQuinidinaAnticoagulantes
RifampicinaHierba de San JuanCarbamazepinaFenitoína
Aumenta el efecto
RifampicinaHierba de San JuanCarbamazepinaFenitoína
Antimicóticos azolesInhibidores de proteasas del VIHAnticoagulantesÁcido acetilsalicílico o clopidogrel;aumentan el riesgo de sangrado sininteracción farmacocinética
Antimicóticos azolesInhibidores de proteasas del VIHQuinidinaAnticoagulantes
Eritromicina y claritromicina
DronedaronaÁcido acetilsalicílico o clopidogrel;aumenta el riesgo de sangrado sininteracción farmacocinética
RifampicinaHierba de San JuanCarbamazepinaFenitoína
do los más comunes la penicilina y las cefalosporinas. La razón de dicho mecanis-mo es aún desconocida.
Los inhibidores de la 5–fosfodiesterasa se han convertido en un tratamientopopular de la disfunción eréctil. La 5–fosfodiesterasa se expresa en las plaquetasy su inhibición resulta en un aumento en el nivel del GMPc plaquetario; se ob-

457Fármacos, alimentos y hierbas en el paciente con trombosisE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
serva un efecto similar con el óxido nítrico (inhibidor plaquetario). El uso de sil-denafil se asocia con riesgo de sangrado; se han reportado casos de epistaxis, san-grado hemorroidal y hemorragia intracraneal espontánea.
Las infusiones de dextrán pueden alterar la función plaquetaria y prolongar eltiempo de sangrado sin aumentar de manera aparente el riesgo de hemorragia.
La nitroglicerina, el dinitrato de isosorbide y el nitroprusiato disminuyen laagregación plaquetaria in vitro; no obstante, dichos efectos no han sido consis-tentes in vivo.
El propranolol, el metoprolol y el pindolol pueden también inhibir la agrega-ción plaquetaria a través de mecanismos independientes de su efecto betablo-queador.
El clofibrato disminuye la respuesta plaquetaria al adenosín difosfato, el colá-geno y la epinefrina cuando se administra en pacientes con hiperbetalipoprotei-nemia tipo II; asimismo, puede disminuir la respuesta plaquetaria normal al ade-nosín difosfato y a la epinefrina in vitro.
Los inhibidores selectivos de la recaptura de serotonina se han convertido enlos agentes de primera elección para diversos tipos de depresión clínica; su usose asocia con un aumento en el riesgo de sangrado al inhibir la recaptura de sero-tonina a nivel plaquetario. No obstante, dicha complicación es rara; los reportesde sangrado tras su administración han ido en aumento, sobre todo en los pacien-tes con defectos plaquetarios congénitos y en quienes se encuentran en terapiacon agentes antiplaquetarios.
Los ácidos grasos omega 3 inhiben in vitro de manera competitiva a la cicloo-xigenasa (disminuyendo así la síntesis de tromboxano A2 a partir del ácido ara-quidónico en las plaquetas), lo que conduce a una disminución en la agregaciónplaquetaria. Sus efectos sobre el fibrinógeno y los factores de la coagulación aúnson limitados. No obstante que los ácidos grasos omega 3 pueden disminuir elriesgo de evento cerebrovascular trombótico, se sugiere que pueden aumentar elriesgo de hemorragia cerebral.
Las evidencias clínicas sugieren que el riesgo de sangrado con el uso concomi-tante de ácidos grasos omega 3 con agentes antiplaquetarios o anticoagulantes ca-rece de significado clínico; sin embargo, se recomienda suspender su administra-ción entre cuatro y siete días antes de cualquier procedimiento invasivo.
La vitamina E (�–tocoferol), utilizada por sus efectos antioxidantes, inhibe larespuesta plaquetaria al ácido araquidónico por inhibición de la proteincinasa C;sin embargo, su uso no incrementa el riesgo de sangrado.
REFERENCIAS
1. Holbrook A, Pereira J, Labiris S: Systematic overview of warfarin and its drug and foodinteractions. Arch Intern Med 2005;165:1095–1106.

458 (Capítulo 28)Tratado de trombosis
2. Pai M, Momary K, Rodvold K: Antibiotic drug interactions. Med Clin N Am 2006;90:1223–1255.
3. Prybys K: Deadly drugs interactions in emergency medicine. Emerg Med Clin N Am 2004;22:845–863.
4. Ament P, Bertolino J, Liszewski J: Clinically significant drug interactions. Am Fam Physi-cian 2000;61:1745–1754.
5. Wells P, Holbrook A, Crowther R: Interactions of warfarin with drugs and food. Ann In-tern Med 2004;121:676–683.
6. Tariq S: Herbal therapies. Clin Geriatr Med 2004;20:237–257.7. Tesch B: Herbs commonly used by women: an evidence–based review. Am J Obstet Gyne-
col 2003;188.8. Katz H, Gupta A: Oral antifungal drugs interactions: a mechanistic approach to unders-
tanding their cause. Dermatol Clin 2003;21:543–563.9. Petrone K, Katz P: Approaches to appropriate drug prescribing for the older adult. Prim
Care Clin Office Pract 2005;32:755–775.10. Schulman S: Care of patients receiving long–term anticoagulant therapy. N Engl J Med
2003;349:675–683.11. Gaziano M, Gibson M: Potential for drug–drug interactions in patients taking analgesics
for mild–to–moderate pain and low–dose Aspirin for cardioprotection. Am J Cardiol 2006;97:23–29.
12. Hoffman: Hematology: basic principles and practice. 4ª ed. EUA, Elsevier, 2005.13. Mateo J: Nuevos anticoagulantes y su papel en la práctica clínica. Rev Esp Cardiol Supl
2013;13(C):33–41.14. Walenga JM, Adiguzel C: Drug and dietary interactions of the new and emerging oral anti-
coagulants. Int J Clin Pract 2010;64:835–838.15. Aristizábal J, Restrepo A, Uribe W et al.: Practical considerations for the use of new oral
anticoagulants. Rev Colomb Cardiol 2012;19(3):135–141.16. Bays H: Safety considerations with omega–3 fatty acid therapy. Am J Cardiol 2007;99:35–
43.17. Serebruany V: Selective serotonine reuptake inhibitors and increased bleeding risk: are we
missing something? Am J Med 2006;119:113–116.

Edi
toria
l Alfi
l. F
otoc
opia
r si
n au
toriz
ació
n es
un
delit
o.�
Índice alfabético
A
abciximab, 9, 36, 37, 100abdomen agudo, 269, 276aborto, 316
de repetición, 413espontáneo, 186recurrente, 319, 320
absceso cerebral, 173abuso de cocaína, 258, 260, 265acarbosa, 453acenocumarina, 6, 43, 98, 110, 231,
243, 245acetaminofén, 408, 448, 453, 455ácido
acetilsalicílico, 32, 48, 72, 94,98, 100, 111, 114, 156, 198,242, 311, 408, 446, 448, 453,455, 456
araquidónico, 3, 4, 10, 32, 457dicarboxiglutámico, 6elágico, 409fólico, 20, 317, 327, 421
fosfatídico, 424glutámico, 6nalidíxico, 408, 453nicotínico, 421tartárico, 52, 112
acidosisintracelular, 270metabólica, 274, 276
acinesia, 383adenocarcinoma, 24
de estómago, 299de ovario, 299de páncreas, 299de próstata, 299de riñón, 299de útero, 299
adenopatía mediastínica, 402adenosín difosfato, 457adinamia, 244afasia amnésica, 389alcoholismo, 45alergia, 450alteplasa, 57, 58
459

460 (Índice alfabético)Tratado de trombosis
alteracióncognitiva, 123obstétrica, 24protrombótica, 168
amaurosis fugax, 125, 151amiloidosis, 272amiodarona, 53, 111, 408, 446, 449,
453, 454, 456amnesia transitoria global, 368amoxicilina, 453amputación, 201, 373
no traumática, 191anafilaxia, 59ancrod, 372anemia, 166, 168, 232, 284
de células falciformes, 253, 277,321
hemolítica, 425autoinmunitaria, 25
anetoderma, 309aneurisma, 46, 58, 153, 193, 254,
265, 381, 382, 400, 403aórtico, 254, 255, 264, 271
de las arterias mesentéricas,279
del tronco celiaco, 279atrial, 77cerebral, 122de la arteria renal, 253de la carótida cervical, 121del septum
interatrial, 381interauricular, 381
del ventrículo izquierdo, 378disecante, 253incidental gigante, 127renal, 254septal
atrial, 77auricular, 329
ventricular, 193
anginainestable, 85, 91, 92, 93, 94, 98,
131intestinal, 271, 279
angioplastia coronaria, 428angioqueratoma difuso, 271anistreplasa, 57, 58, 59anticoagulación fetal, 319anuria, 252, 255, 259aortoembolismo, 155, 158apendicitis, 277apixabán, 48, 49, 55, 56, 75, 98,
111, 113, 114, 452, 456aplasminogenemia, 22apoplejía, 379argatrobán, 50, 51, 199, 372, 373,
407, 408, 409, 415arritmia, 221, 222, 254, 260, 265,
276auricular, 222cardiaca, 105, 255, 276, 329ventricular, 318
arterioesclerosis, 191arteriopatía
carotídea, 120periférica, 108
arteritis, 24, 396de Takayasu, 121, 193, 253
artritis, 309aguda, 349reumatoide, 25, 166, 271séptica, 249
ascaridiasis, 271ascitis, 278, 340asma bronquial, 33Aspirina�, 10, 312, 319, 327, 373,
448, 450, 452, 455astenia, 244ataque isquémico transitorio, 123,
137, 149, 150, 151, 152, 155,236

461Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
ataxia, 151atelectasia, 392
laminar, 225atenolol, 408, 449ateroma, 14
aórtico, 107ateromatosis, 396aterosclerosis, 5, 7, 12, 24, 66, 82,
83, 84, 85, 86, 87, 91, 92, 97,106, 120, 125, 149, 154, 158,191, 193, 198, 253, 256, 263,264, 271, 284, 288, 289, 330,335, 340, 341, 400, 452aórtica, 158, 382carotídea, 119, 120, 121, 124de la arteria renal, 256difusa, 257generalizada, 123, 273, 280mesentérica, 279
aterotrombosis, 81, 82, 86, 155aguda, 5
atorvastatina, 157, 158, 452, 455atrofia
cortical, 263dérmica, 195óptica, 319
azatioprina, 408, 454AZD0837, 50, 52azitromicina, 447, 453
B
bacteriana, 371betrixabán, 49bevacizumab, 289, 299bezafibrato, 453bivalirudina, 50, 51, 199, 372, 373,
374bosentán, 85, 454bumetanida, 408, 449
C
cáncer, 12, 13, 27, 45, 68, 120, 169,172, 207, 208, 216, 217, 218,224, 227, 232, 233, 295, 296,298, 347, 407, 449, 450, 451,452de cerebro, 301de estómago, 301de mama, 20, 297, 299, 345de ovario, 301de páncreas, 301, 393de próstata, 301de pulmón, 301gástrico, 393pulmonar, 66
cangrelor, 198caquexia, 165, 296carbamazepina, 408, 447, 449, 454,
455, 456carbaplatino, 453carcinoma, 45, 272
de células renales, 380cardioembolismo, 377cardiomegalia, 392cardiomiopatía, 260, 318
crónica dilatada idiopática, 66isquémica, 66
cardiopatíacongestiva, 254, 265isquémica, 19, 255, 265, 339,
343, 381reumática, 273
catarata, 76cefalea, 139, 163, 169, 170, 171,
185, 264, 388, 389frontal bilateral, 244occipital, 242
cefamandol, 408, 447, 453cefazolina, 408, 453cefoperazona, 447

462 (Índice alfabético)Tratado de trombosis
cefotetán, 447ceguera monocular transitoria, 151celecoxib, 449, 453choque, 224cianosis, 225, 330ciclofosfamida, 453ciclosporina, 408, 422, 451, 454cilostazol, 38, 78, 198cimetidina, 408, 453ciprofloxacina, 408, 446, 453ciproterona, 336cirrosis, 393cisaprost, 198cisticercosis, 175citalopram, 453claritromicina, 53, 447, 453, 454,
456claudicación intermitente, 194, 450clavulanato, 453clofibrato, 408, 449, 453, 457clopidogrel, 9, 10, 34, 35, 36, 76,
77, 78, 94, 95, 96, 98, 100, 111,115, 157, 160, 198, 446, 455, 456
cloranfenicol, 453clordiazepóxido, 408, 454cloxacilina, 454coagulación, 1
espontánea, 296intravascular diseminada, 193,
270, 406, 410, 412, 413, 417,420
coágulo, 133, 176, 180, 181, 278,295, 402, 420, 429de fibrina, 4, 57, 406, 407, 420,
425intraluminal, 247intravascular, 7intravenoso, 180
coagulopatía, 48, 272cobalamina, 20cocaína, 253, 254, 258, 260, 265
colestiramina, 408, 449, 454colitis isquémica, 271coma, 163, 171, 172complicación tromboembólica, 106,
206, 259compresión lumbar, 194congestión venosa, 172, 181constipación, 279contracción vascular, 2contusión, 193corea, 308cotrimoxazol, 408crecimiento tumoral, 298crisis convulsiva, 163, 164, 170,
171, 183, 185, 389cumarina, 45, 324
D
dabigatrán, 38, 50, 52, 53, 54, 55,56, 75, 98, 111, 112, 199, 212,229, 233, 452, 454, 456
dalteparina, 231, 301, 325, 326,351, 352, 353
danaparoide, 371, 372, 373danazol, 453daño
celular, 270endotelial, 11, 217, 260, 316hepático, 111hipóxico isquémico, 179isquémico, 150miocárdico, 96, 220, 223parenquimatoso, 262renal, 252, 257, 262, 263tisular, 270vascular, 315
por vasoconstricción, 260defecto
genético trombofílico, 340hemostático, 351

463Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
deficienciade antitrombina, 15, 67de plasminógeno, 14, 22de proteína
C, 13, 14, 15, 16S, 13, 14, 16, 17
de vitamina, 421B, 317K, 43, 406, 407, 410, 411,
412, 413estrogénica, 81neurológica, 329nutricional, 167, 168
déficitfocal, 163neurológico, 139, 151
focal, 388progresivo, 131
deformación vascular, 393demencia
por infartos múltiples, 308vascular, 159
dermatán sulfato, 373dermatomiositis, 271derrame pleural, 220, 225, 318, 340,
392desequilibrio electrolítico, 260deshidratación, 172, 258, 321
grave, 276desmopresina, 37desogestrel, 335desorden
mieloproliferativo, 67trombofílico, 72
desprendimientoplacentario, 320prematuro de placenta, 413, 422
destrucción celular, 425deterioro cognitivo, 123dextrán, 457dextropropoxifeno, 408, 453
diabetes, 33, 84, 85, 100, 131, 149,150, 192, 198, 202, 217gestacional, 319mellitus, 24, 66, 82, 85, 86, 92,
107, 108, 140, 159, 160, 191,193, 241, 256, 264, 271, 284,330, 339, 451
diátesishemorrágica, 33, 58trombótica, 410
diazepam, 447, 453dicloxacilina, 408, 454digital, 276digoxina, 451, 452diltiazem, 408, 449, 453dipiridamol, 37, 38, 157, 160, 198diplopía, 151, 171disartria, 151discinesia, 381discrasia sanguínea, 165disección
aórtica, 260, 400, 403arterial, 258, 330
aguda, 331de aorta, 99
disfagia, 151disfibrinogenemia, 20, 67, 173
congénita, 20disfunción
del complemento, 306endocárdica, 106endotelial, 81, 82, 84, 87, 92,
106, 306eréctil, 195, 450, 451, 456hepática, 58, 249mitocondrial, 270orgánica, 354placentaria, 306plaquetaria, 59, 232, 259, 306renal, 49ventricular

464 (Índice alfabético)Tratado de trombosis
derecha, 215, 383izquierda, 381
dislipidemia, 24, 82, 83, 86, 256aterogénica, 85
dislipoproteinemia, 193disnea, 220, 224, 322, 330disopiramida, 408, 453dispepsia, 33, 52, 112displasia fibromuscular, 121, 193,
253, 265displasminogenemia, 22disrupción vascular, 1disulfiram, 408, 453diverticulitis, 277dobutamina, 234dolor
abdominal, 255, 274, 276, 279,280inespecífico, 278posprandial, 273
cervical, 247, 249incapacitante, 280muscular, 70ocular, 171pleural, 220pleurítico, 224, 225retroesternal, 225torácico, 224, 300, 318
dolor abdominal, 252dopamina, 234doxorrubicina, 299dronedarona, 456drospirenona, 336
E
eclampsia, 309edema, 169, 178, 179, 185, 208,
211, 270, 300, 322, 353cerebral, 178, 179, 181, 249
difuso, 175venoso, 178
citotóxico, 178, 181de disco, 285de papila, 285de vías respiratorias, 249facial, 401macular, 285, 288, 292
quístico, 285pulmonar, 256vasogénico, 178
edoxabán, 49embolia, 201, 274
arterial, 382arterioarterial, 378cardiaca, 400grasa, 254pulmonar, 200, 247, 295, 296,
300, 347, 348, 380, 392, 431,432aguda, 382masiva, 340
sistémica, 380tumoral, 254
embolismo, 130, 132, 269, 272, 396a distancia, 205arterial, 364, 377cardiaco, 377cardiogénico, 153, 329cerebral, 205, 329de colesterol, 263de líquido amniótico, 330de pequeños vasos, 324paradójico, 330, 378
sistémico, 381pulmonar, 12, 183, 185, 222,
249, 262, 378séptico, 249sistémico, 47, 49, 53, 77, 114,
378venoso extracerebral, 183

465Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
embolización paradójica, 220émbolo, 135
de cristales de colesterol, 263empiema, 249enalapril, 242encefalitis, 171encefalopatía, 330
subaguda difusa, 171endocarditis, 193, 380
bacteriana, 58, 254, 264de Libman–Sacks, 329infecciosa, 236, 329trombótica no bacteriana, 296
enfermedadacidopéptica, 33, 34aórtica abdominal, 256aortoiliaca, 194arterial, 196, 198, 279, 290, 330,
344coronaria, 107, 115, 341oclusiva, 179periférica, 38
ateroembólica, 258, 265aterosclerosa, 158
coronaria, 397, 398extracraneal, 153, 158
aterosclerótica, 91, 93, 125, 253carotídea extracraneal, 120coronaria, 92
aterotrombótica, 59coronaria, 216
autoinmunitaria, 23, 25, 254, 304cardiaca, 136, 165, 218, 254,
265, 273reumática, 106
cardioembólica, 150cardiovascular, 66, 76, 77, 91,
105, 309, 340, 341, 421isquémica, 264
carotídea, 119, 123, 127, 135,137, 151
derecha, 123cerebral, 159cerebrovascular, 67, 68, 121,
264, 329, 343, 382hemorrágica, 46
coronaria, 88, 421, 422, 427por aterosclerosis, 96
crónica, 318crónico–degenerativa, 191de Behçet, 25, 67, 166, 168, 253de Buerger, 24, 193de células falciformes, 400de Degos, 25de la arteria carótida, 119de moya moya, 330de Takayasu, 330, 399del sistema vascular, 82embólica, 382hepática, 48, 232, 236, 276, 320,
408, 410, 411, 412, 414, 417infecciosa aguda, 349inflamatoria, 207
aguda, 67intestinal, 166, 277, 349
inmunitaria, 303, 312, 378intestinal inflamatoria, 67isquémica
intestinal, 274oclusiva, 178
maligna, 66, 74, 249, 258, 298mieloproliferativa, 193, 277, 321mitocondrial, 179neoplásica maligna, 301oclusiva de la arteria carótida,
120oncológica, 279ótica, 178pulmonar
aguda, 67crónica, 67, 218, 224grave, 136

466 (Índice alfabético)Tratado de trombosis
intersticial, 66obstructiva crónica, 66, 216,
244renal, 112, 232, 263, 276
ateroembólica, 263, 264, 265,266de cristales de colesterol,
257aterosclerótica, 257isquémica, 256
renovascular, 256respiratoria
aguda, 349crónica, 66
reumática, 194, 263, 349tromboembólica, 22, 51, 245,
255, 315, 325, 356profunda, 301pulmonar, 301venosa, 18, 31, 47, 49, 53, 66,
67, 68, 69, 70, 71, 72, 73,74, 296, 298, 300, 301, 317,321, 322, 325, 327, 352,353, 355, 420
trombofílica hereditaria, 322trombótica, 24, 31, 44, 56, 59,
249, 300, 315, 317, 321, 336,338aguda, 320venosa, 208
valvular, 76, 329cardiaca, 254mitral reumática, 76reumática, 107, 254
vascular, 81, 86, 88, 109, 191,195, 198, 201, 256arterial, 167aterosclerótica, 7cerebral, 120, 149, 151, 163,
164, 185, 232, 236, 349paralítica, 218
de las extremidades, 191de los ojos, 191de los riñones, 191del cerebro, 191del corazón, 191extracraneal, 119hemorrágica, 236isquémica extracraneal, 122periférica, 70, 195, 256, 421,
422renal, 251, 262
enoxacina, 408enoxaparina, 48, 49, 95, 113, 211,
231, 245, 301, 325, 326, 351,352, 353, 354, 357, 358
entacapone, 453epilepsia, 308, 309epinefrina, 38, 259, 457epistaxis, 457eptifibatida, 36, 37, 100eritema, 244
de extremidades, 300eritromicina, 408, 447, 453, 456Escherichia coli, 169esclerodermia, 207, 208esclerosis sistémica, 271escleroterapia endoscópica de vari-
ces, 277esomeprazol, 33espasmo
arterial, 278miógeno, 2vascular, 2
esplenomegalia, 277estado
de choque, 67, 218, 276, 330de coma, 185epiléptico, 171trombofílico, 303
hereditario, 349estafilocinasa, 57

467Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
estasissanguínea, 106, 206, 207, 380vascular, 11venosa, 315, 316, 350
esteatorrea, 407estenosis, 128, 130, 196, 378
ateromatosa, 127aterosclerótica, 273
de la arteria renal, 256carotídea, 119, 120, 121, 122,
123, 127, 128, 130, 131, 132,138extracraneal, 131izquierda, 123sintomática, 128
coronaria, 77de la arteria renal, 255inducida por radiación, 134intracraneal, 127mitral, 107, 273, 378, 380posirradiación, 121recurrente, 136suboclusiva, 134
estreptocinasa, 57, 58, 59, 98, 99,211, 234, 256
estrés oxidativo, 84, 305etopósido, 453etretinato, 408, 454evento
cardiovascular, 33, 76, 157, 343cerebrovascular, 5, 52, 99, 157,
254, 335hemorrágico, 351isquémico, 58, 122, 241, 381
en el embarazo, 328coronario, 91
agudo, 98, 344de tromboembolia pulmonar, 242embólico, 48, 107, 265, 322, 377isquémico, 93, 95, 97, 130, 390
agudo, 96
cerebral, 119transitorio, 124, 381
tromboembólico, 107, 108, 260,261, 304, 320, 373recurrente, 173venoso, 41, 69
trombótico, 11, 15, 26, 27, 39,52, 65, 66, 70, 169, 183, 185,259, 304, 310, 311, 318, 320,338, 364, 372, 427agudo, 86arterial, 303, 410venoso, 303, 341, 410
vascular, 107, 120, 157, 168, 198agudo, 5cerebral, 47, 105, 108, 111,
120, 309, 328arterial, 388isquémico, 350
agudo, 330isquémico, 159
F
fallacardiaca, 246, 256, 265
congestiva, 66, 264cardiovascular, 358congestiva, 392del injerto, 260hemodinámica, 153hepática, 34, 131, 407multiorgánica, 309orgánica, 131
múltiple, 358pulmonar, 131, 358renal, 50, 54, 200, 246, 249, 257,
264, 332crónica, 45
respiratoria aguda, 66ventricular izquierda, 66, 107

468 (Índice alfabético)Tratado de trombosis
famotidina, 408, 449fascitis cervical necrosante, 248felbamato, 453felodipino, 449fenilbutazona, 408, 446, 453fenindiona, 43fenitoína, 408, 422, 447, 453, 455fenofibrato, 453fenprocumona, 231feocromocitoma, 271fibrilación auricular, 52, 75, 77,
105, 106, 107, 109, 110, 150,153, 157, 193, 254, 264, 273,329, 331, 378, 379, 380no valvular, 48, 54, 76, 98paroxística, 153
fibrinólisis disminuida, 192fibrosis tubulointersticial, 257fiebre tifoidea, 271fístula
arteriovenosadural, 153séptica, 59
dural, 166enterocutánea, 276
flegmasia cerúlea dolens, 209fluconazol, 408, 446, 448, 453fluorouracilo, 408, 453fluoxetina, 408, 449, 453fluticasona, 244fluvastatina, 446, 453fluvoxamina, 446, 453fondaparinux, 38, 41, 42, 47, 71,
75, 229, 230, 231, 232, 351, 352,356, 357, 373
foramen oval, 77permeable, 378
fracturade cadera, 68, 69, 75ósea, 193
furosemida, 454
G
gangrena, 195, 201, 270, 350del pie, 264venosa, 368, 372
gatifloxacina, 453gemcitabina, 453gemfibrozil, 408, 453gestodene, 335glaucoma, 284, 289, 292
neovascular, 283, 288primario de ángulo abierto, 284
glicerol, 185glioma, 20granuloma de Wegener, 272griseofulvina, 408, 449, 454
H
Helicobacter pylori, 33hematoma, 135, 170, 258
parenquimatoso, 175perirrenal, 400subcortical, 178
hematuria, 265, 400macroscópica, 259, 266microscópica, 255
hemiparesiaatáxica, 159motora, 159
hemiplejía, 119hemoglobinuria paroxística noc-
turna, 67hemoptisis, 220, 225, 227hemorragia, 20, 33, 41, 48, 94, 95,
99, 100, 207, 232, 235, 255, 256,285, 358, 445abdominal, 231alveolar, 220cerebral, 149, 173, 185de difícil control, 100

469Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
fetal, 324gastrointestinal, 34, 157, 236,
351, 445intracerebral, 157, 159intracraneal, 34, 112, 231, 260,
445espontánea, 457
intraparenquimatosa, 175, 177intraplaca, 155intrarretiniana, 289obstétrica, 321oculta, 231petequial, 175, 176, 178retiniana, 34, 284retroperitoneal, 34subaracnoidea, 176torácica, 231uteroplacentaria, 324
hemorrágico, 175hemovítreo, 292heparán sulfato, 373heparina, 9, 15, 26, 31, 40, 51, 74,
97, 100, 101, 164, 183, 184, 185,231, 233, 234, 235, 248, 262,264, 300, 311, 312, 319, 325,327, 331, 357, 363, 364, 367,368, 369, 371, 372, 374, 406,407, 408, 411, 417, 418, 428,446, 453de bajo peso molecular, 27, 38,
40, 69, 71, 94, 113, 164, 211,229, 230, 231, 262, 300, 301,310, 319, 325, 326, 327, 331,332, 351, 365, 370, 372, 418,419
fraccionada, 356intravenosa, 182no fraccionada, 8, 26, 38, 39, 69,
71, 72, 73, 74, 75, 94, 97, 183,184, 211, 229, 230, 243, 310,319, 324, 325, 326, 327, 351,
352, 357, 364, 365, 370, 372,373, 374, 409, 418, 419
hepatocarcinoma, 393hepatopatía, 453hernia
discal, 194estrangulada, 272
herniación transtentorial, 185hidrato de cloral, 408hidrocefalia, 170, 176
ótica, 169hipercalemia, 200hipercoagulabilidad, 11, 192, 206,
216, 246, 315, 331, 350hipercolesterolemia, 24, 84, 92,
264, 339hiperemesis gravídica, 321hiperestimulación ovárica, 321, 340hiperfibrinogenemia, 277hiperglucemia, 85hiperhomocisteinemia, 12, 13, 14,
19, 24, 166, 167, 253, 254, 260,304, 316, 317, 432
hiperlipidemia, 12, 149, 192, 284,289, 451
hiperplasia quística endometrial,341
hipersensibilidad al rivaroxabán, 48hipertensión, 20, 66, 99, 131, 168
arterial, 92, 99, 107, 108, 139,140, 149, 150, 159, 160, 192,198, 217, 256, 263, 265, 284esencial, 25pulmonar, 383, 384, 390, 392
por tromboembolias derepetición, 308
sistémica, 24, 82, 84, 86, 241,242, 244, 252, 255, 256,264, 319, 330, 337, 339descontrolada, 58
sistólica, 216

470 (Índice alfabético)Tratado de trombosis
descontrolada, 232endocraneana, 164, 173intracraneal, 170, 171, 172, 184maligna, 260, 271portal, 277pulmonar, 66, 198, 384
crónica, 350tromboembólica crónica, 216
refractaria, 236renovascular, 263transoperatoria, 140
hipertiroidismo, 244, 407hipertrigliceridemia, 66, 84, 344hipertrofia ventricular derecha, 384hiperviscosidad sanguínea, 284hipoalbuminemia, 258hipoaldosteronismo, 40hipoperfusión
renal, 255visceral, 274
hipoplasminogenemia, 22hipotensión, 139, 142, 218, 222,
224, 276, 332, 350intracraneal, 169
hipotermia, 209hipotiroidismo, 407, 421, 422hipovolemia, 221, 222, 276hipoxemia, 219, 225, 234
refractaria, 220hipoxia, 350
tisular, 270hirudina, 49, 51, 373, 407, 409, 415histoplasmosis, 401homocisteinemia, 67homocisteinuria, 422homocistinuria, 165
I
ibuprofeno, 32, 408, 455
idraparinux, 38, 42ifosfamida, 408, 453iloprost, 198, 279indinavir, 451indometacina, 408, 453inestabilidad hemodinámica, 378infarto, 92, 93, 98, 113, 252
agudo, 94, 98, 99del miocardio, 5, 57, 58, 66,
68, 85, 91, 92, 93, 98, 205,273, 309, 310, 335, 343,349, 352, 368
arterial, 178, 179cerebral, 35, 119, 122, 125, 135,
136, 137, 150, 151, 152, 153,154, 155, 158, 160, 173, 260,305, 308, 330, 381, 388, 390,427ateroscleroso, 154aterotrombótico, 149de origen embólico, 156isquémico, 123
cortical, 123crónico, 400del miocardio, 17, 19, 35, 94,
108, 131, 135, 136, 137, 157,254, 260, 276, 309, 350, 422,427
hemorrágico adrenal, 368intestinal, 280lacunar, 123, 149, 159lobar, 220mesentérico, 368miocárdico, 193perioperatorio, 132, 134placentario, 309pulmonar, 220, 393recurrente, 94renal, 252, 254, 260, 263, 368,
401segmentario, 251

471Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
venoso, 170, 174, 175, 178, 179,180hemorrágico, 175
infecciónmedicamentosa, 371orofaríngea aguda, 245
inflamación, 83, 84, 85, 88, 92, 205,216endotelial, 88parameníngea, 174por lesión arterial, 192
influenza, 408, 454insuficiencia
aórtica, 276arterial periférica, 191cardiaca, 107, 108, 140, 217,
224, 256, 257, 276, 347congestiva, 12, 67, 68, 132,
216, 276, 349, 352hepática, 33orgánica, 140placentaria, 309, 311, 312renal, 33, 37, 140, 228, 255, 256,
263, 264, 265, 347, 421aguda, 260, 262, 263crónica, 111irreversible, 257
respiratoria, 140tricuspídea, 384valvular secundaria, 384
interferón, 453invasión bacteriana, 270irritación peritoneal, 276, 278isoniazida, 408, 446, 447, 453isosorbide, 457isquemia, 92, 93, 201, 269, 270,
289, 343aguda, 277arterial mesentérica aguda, 273cardiaca, 77cerebral, 149, 155, 329, 330
aguda, 51transitoria, 47, 107, 108, 150,
318crónica, 280de la mucosa intestinal, 274espinal, 150intestinal, 269, 270, 271, 272,
273, 395aguda, 277embólica, 273no oclusiva, 276trombótica, 273
mesentérica, 269, 272, 274aguda, 271, 274, 276crónica, 279no oclusiva, 272, 276
miocárdica, 91, 93, 95, 96, 254no oclusiva, 269, 274oclusiva, 276renal, 252retiniana, 150tisular, 151
itraconazol, 408, 448, 453
K
ketoconazol, 408, 448ketoprofeno, 408ketorolaco, 408, 449
L
L–asparaginasa, 169L–dopa, 421lanoteplasa, 57leflunomida, 453lenalidomida, 72, 299lepirudina, 50, 51, 372, 373lepra, 271lesión

472 (Índice alfabético)Tratado de trombosis
aterosclerosa, 153, 156aterosclerótica, 5, 84, 254, 427
extracraneal, 119aterotrombótica, 154carotídea, 119, 122, 158cerebral, 151
traumática, 74coronaria, 101cutánea, 309
inducida por heparina, 368de la pared vascular, 1, 3del cordón espinal, 68endotelial, 2, 4, 295, 366espinal
aguda, 74traumática, 74
estenótica arterial, 196, 381hemorrágica, 183, 185intracraneal, 131isquémica, 123
aguda, 151cerebral, 388crónica, 151
metastásica, 169miocárdica, 98parenquimatosa, 185, 395
cerebral, 178hemorrágica, 164
renal, 252tisular, 6ulcerativa, 213valvular, 154vascular, 2, 83, 256, 263, 265
isquémica arterial, 180leucemia, 124, 169, 299
mieloide aguda, 20promielocítica aguda, 24
leucocitosis, 174, 247, 249, 255,274, 276, 278
levamisol, 453levofloxacina, 453
levonorgestrel, 335, 453linfoma, 402lipohialinosis, 150livedo reticularis, 264, 308, 309lovastatina, 408, 446lumbalgia, 323lupus, 207
eritematosogeneralizado, 24, 25sistémico, 166, 193, 208, 271,
304, 378, 421
M
macroangiopatía diabética, 191, 196malformación
arteriovenosa, 58, 153vascular, 46
malnutrición, 407manitol, 185marasmo, 165mastoiditis, 168, 178mediastinitis fibrosante, 403melagatrán, 51meningitis tuberculosa, 169mercaptopurina, 454mesalamina, 454metástasis, 169, 402metilprednisolona, 453metolazona, 453metoprolol, 408, 449, 457metotrexato, 33, 421, 422, 453metozalona, 408metronidazol, 408, 446, 447, 453mialgia, 264miconazol, 408, 446, 453microaneurisma, 288microangiopatía, 311
cerebral, 159diabética, 191trombótica, 263

473Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
microateroma, 150microcefalia, 319microembolia, 123microtrombo, 330mielitis transversa, 308mielofibrosis primaria, 26migraña, 260, 308, 309, 339miocardiopatía
congestiva, 193dilatada, 378, 379hipertrófica, 193periparto, 329
miocarditis, 260mioglobinuria, 200, 201miomatosis uterina, 242miopatía, 194, 200mixoma, 193, 377, 380
auricular izquierdo, 264moricizina, 408muerte
celular, 388fetal, 422
intrauterina, 320intrauterina, 327, 413vascular, 86, 157
N
nabumetona, 453nafcilina, 408, 449, 454naproxeno, 408, 449nebivolol, 244necrosis
cutánea, 40, 45, 210, 372por warfarina, 16
intestinal, 273, 278isquémica, 400tubular aguda, 264
nefritis intersticial inducida por fár-macos, 264
nefropatíaisquémica, 256, 257, 258membranosa, 258por contraste, 264
nefrosclerosis, 257neoplasia, 277
hematológica, 299, 301intracraneal, 58, 99maligna, 296mieloproliferativa, 26retroperitoneal, 265
neovascularización, 289neumonía, 350
adquirida en la comunidad, 66,217, 244
nosocomial, 66neuritis óptica, 308neurofibromatosis, 121neuropatía, 194
radicular, 194neutropenia, 34nifedipino, 263nitrazepam, 449nitroglicerina, 457nitroprusiato, 457nizatidina, 408norepinefrina, 234norfloxacina, 408, 453norgestimato, 335
O
obesidad, 12, 13, 66, 67, 72, 82, 85,86, 140, 149, 192, 206, 207, 217,218, 320, 322, 326, 332, 337,339, 342, 345, 347, 349abdominal, 257infantil, 191
obstrucciónvascular, 269venosa, 261

474 (Índice alfabético)Tratado de trombosis
mesentérica, 278oclusión
aguda de la arteria renal, 251arterial, 256, 271, 278, 381
aguda, 278, 377, 381renal aguda, 255, 265
carotídea, 133, 136de la arteria renal, 255embólica, 273intestinal, 277vascular aguda, 274venosa, 259
ofloxacino, 408, 453oftalmopatía hemorrágica, 59oftalmoplejía dolorosa, 171oliguria, 264omeprazol, 408, 453osteoartritis, 450osteoporosis, 40, 41, 345
inducida por heparina, 324otalgia, 169, 242otitis, 168óxido
nítrico, 8, 38, 56, 87, 92, 457nitroso, 40
P
pacientealérgico a la heparina, 374con aborto espontáneo, 304con angina inestable, 35, 51, 98,
99con aterosclerosis, 427con cáncer, 20, 72, 231, 297,
298, 299, 301con cirrosis hepática, 393con claudicación intermitente,
194con claustrofobia, 154
con deficiencia de antitrombina,51
con déficit neurológico, 185con disfibrinogenemia, 20con edema pulmonar, 256con embolia pulmonar, 415
aguda, 382con embolismo pulmonar, 42con enfermedad
aórtica abdominal, 256arterial periférica, 119, 198aterosclerótica, 98cardiopulmonar, 220, 223, 225cardiovascular, 344, 421coronaria, 19, 78, 119, 256hepática, 114renal, 153trombótica arterial, 24vascular cerebral, 387
con estenosisasintomática, 136carotídea, 123, 137, 138mitral, 380
con eventocerebral, 25cerebrovascular, 352, 382
hemorrágico, 351isquémico, 37
coronario agudo, 101isquémico, 94
cerebrovascular, 25trombótico, 17
con fallacardiaca, 256hepática, 51renal, 34, 112
con fibrilación auricular, 42, 48,49, 108, 111, 113, 114, 383,454no valvular, 47, 49, 52, 53
con glioma, 169

475Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
con hemorragia, 351gastrointestinal, 350
con hiperbetalipoproteinemia,457
con hipertensión portal, 393, 394con hipotensión, 234con infarto
agudo del miocardio, 57cerebral, 158, 381del miocardio, 78venoso cerebeloso, 176
con insuficienciacardiaca, 132, 276
congestiva, 38renal, 37, 257, 353, 373, 422
con lesiónateromatosa, 156de la médula espinal, 352parenquimatosa hemorrágica,
164renal aguda, 353, 354
con lupus, 25eritematoso sistémico, 168,
304, 317, 423con meningioma, 169con nefropatía
de origen aterosclerótico, 257isquémica, 258membranosa, 258
con neoplasia, 309mieloproliferativa, 26
con oclusión de la vena centralde la retina, 415
con riesgo de sangrado, 34, 211con síndrome
antifosfolípidos, 25, 309coronario agudo, 35de Eisenmenger, 381de embolización de colesterol,
264nefrótico, 254, 258, 259
con trasplantede riñón, 254, 260renal, 260
con trauma, 353mayor, 74múltiple, 352
con trombo intracardiaco, 235con trombocitopenia inducida
por heparina, 351con tromboembolia
pulmonar, 224, 235venosa, 415
con tromboembolismo venoso,22, 416
con trombofiliacongénita, 327primaria, 328
con tromboflebitis venosa pro-funda, 298
con trombosis, 26, 178, 182, 248,377, 387, 445cerebral, 415cerebrovascular, 19de la vena hepática, 415recurrente, 11, 432séptica, 175sintomática, 248traumática, 251venosa, 412, 416
cerebral, 186extracerebral, 184familiar, 426profunda, 39, 42, 54, 415
con tuberculosis, 401con tumor maligno, 296crítico, 347, 348, 352, 354, 355embarazada, 41, 316, 322, 328,
330, 331, 353en estado crítico, 353, 357, 358isquémico, 96nefrótico, 258, 259

476 (Índice alfabético)Tratado de trombosis
neurológico, 122obeso, 207obstétrica, 365oncológico, 295, 301ortopédico, 357, 368pediátrico, 365quirúrgico, 24traumatizado, 252, 262
paclitaxel, 453panarteritis nodosa, 400pancreatitis, 277, 393pantoprazol, 112, 455papaverina, 272, 276, 279papiledema, 163, 164, 169, 170,
171, 331papilosis atrófica maligna, 271parálisis, 207
de nervios craneales, 134, 388del nervio laríngeo, 136
paraplejía, 321paraproteinemia, 67paro cardiorrespiratorio, 368penicilina, 456pentoxifilina, 198pérdida
embriofetal, 306fetal, 25sanguínea, 1
pericarditis aguda, 58peritonismo, 275peritonitis, 278petequia, 372pie diabético, 198, 199, 200pindolol, 457piridoxina, 20piroxicam, 408, 453poliarteritis, 193
nodosa, 253, 271policitemia, 66, 67, 166, 284
vera, 26, 272polirradiculoneuritis, 308
prasugrel, 35, 76, 78, 94, 95, 198pravastatina, 56preeclampsia, 16, 309, 311, 312,
316, 318, 321, 322, 327, 413,422
propafenona, 408, 449, 453propofol, 454propoxifeno, 408, 453propranolol, 408, 449, 453, 457proptosis, 171prostaciclina, 198protamina, 374proteinuria, 259, 263, 265púrpura
anafilactoide, 271necrosante, 25neonatal fulminante, 16postransfusional, 369, 371trombocitopénica
autoinmunitaria, 25idiopática, 371trombótica, 34, 193, 271, 330
Q
quemosis, 171quetiapina, 453quilotórax, 247quinidina, 408, 452, 453, 454, 456
R
rabdomiólisis, 200, 260raloxifeno, 345, 454ranibizumab, 289ranitidina, 408, 449, 452razaxabán, 199reestenosis aguda, 99reflujo gastroesofágico, 33resfriado, 449, 450

477Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
resistenciaa la heparina, 353a la insulina, 85al clopidogrel, 35
reteplasa, 57, 58, 198retinopatía diabética, 59retraso mental, 20, 319ribavirina, 454riboflavina, 421riesgo
aterotrombótico, 344cardiovascular, 344de amputación, 368de apoplejía, 379de ataque isquémico transitorio,
127de aterosclerosis, 153de cáncer, 322de embolismo, 378
recurrente, 325sistémico, 329
de enfermedadarterial periférica, 339tromboembólica, 417, 448
venosa, 22, 66, 326durante el embarazo, 317
trombótica, 66, 338de evento
cardiovascular, 85cerebrovascular, 108
trombótico, 457embólico, 380isquémico, 127trombótico, 342
venoso, 21de fractura, 83de hematoma epidural, 325de hemorragia, 21, 46, 47, 74,
157, 232, 235, 310, 311, 350,457cerebral, 112, 457
epidural, 319intracraneal, 112, 113, 114mayor, 54, 233
de infarto, 120agudo del miocardio, 343cerebral, 120, 127, 151, 158del miocardio, 132
de isquemia cerebral, 113, 158de osteopenia, 324de osteoporosis, 332de recurrencia de trombosis, 20de reestenosis aguda, 95de ruptura miocárdica, 98de sangrado, 35, 39, 41, 48, 52,
72, 73, 74, 94, 95, 97, 101,107, 109, 110, 112, 113, 114,115, 211, 231, 233, 319, 324,351, 370, 445, 446, 447, 455,457fatal, 113
de tromboemboliaparadójica sistémica, 383venosa, 416
de tromboembolismo, 75, 108,317, 355venoso, 349, 352
posoperatorio, 350de trombosis, 15, 18, 20, 21, 26,
71, 72, 81, 106, 109, 167, 168,305, 309, 320, 337, 340, 342,345, 348, 353, 367, 372, 374arterial, 11, 23, 65recurrente, 319venosa, 11, 65, 66, 316, 337,
338, 339, 341, 352, 410profunda, 315
embólico, 109isquémico, 96renal, 263tromboembólico, 71, 106, 107,
108, 109, 110

478 (Índice alfabético)Tratado de trombosis
trombótico, 343, 427vascular, 85, 125, 158
rifabutina, 448rifampicina, 408, 448, 449, 454,
455, 456rifapentina, 448ritonavir, 453, 454rivaroxabán, 45, 46, 47, 48, 55, 56,
75, 98, 111, 113, 114, 199, 212,229, 232, 233, 452, 456
robecoxib, 453ropinirol, 453
S
salmeterol, 244sangrado, 45, 46, 72, 99, 172, 332,
348, 352, 357, 373, 448de tubo digestivo, 183
alto, 33del tracto digestivo, 33espontáneo, 367gastroesofágico, 394gastrointestinal, 33, 34, 45, 58genitourinario, 58hemorroidal, 457incontrolable, 99intracraneal por reperfusión, 139por reperfusión, 139por trombocitopenia, 372posoperatorio, 276
saquinavir, 453sarcoidosis, 166saruplasa, 57seno
sagital, 165sigmoide, 165
sepsis, 66, 67, 221, 222, 247, 347,358intraabdominal, 277
septicemia, 270sertralina, 446, 453seudoxantoma elástico, 271sífilis, 271, 304sildenafil, 453, 457simvastatina, 453síncope, 224, 225síndrome
antifosfolípidos, 13, 24, 25, 193,253, 262, 271, 304, 309, 311,378, 423, 424catastrófico, 25, 263
coronario, 98agudo, 77, 83, 91, 92, 94, 98,
100, 115, 157de anticuerpos antifosfolípidos,
24, 303, 316, 317, 322, 329,330, 402catastrófico, 309y anticoagulante de lupus, 23
de Budd–Chiari, 25de compresión neurovascular,
193de Ehlers–Danlos, 271de Eisenmenger, 381de embolización de colesterol,
264de hiperestimulación ovárica,
326, 340de hiperperfusión, 139de hiperviscosidad, 66de HIT, 363, 365, 370, 371, 372de infarto pulmonar, 220de intestino corto, 276de la plaqueta pegajosa, 12, 14,
19de la vena cava superior, 247,
249de Lemierre, 246de Leriche, 396, 397, 398de reperfusión, 200, 201

479Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
de resistencia a la insulina, 344de respuesta inflamatoria sisté-
mica, 249, 270de Sjögren, 271de Snedon, 25de trombo blanco, 367de trombocitopenia, 40
inducida por heparina, 12, 24,67, 332
y trombosis inducida porheparina, 366
de tromboembolismo venoso,215
de trombosisy anticoagulante de lupus, 12,
25y anticuerpos
anticardiolipina, 25antifosfolípidos, 12
del coágulo blanco, 364luminal, 131metabólico, 86, 191, 217nefrótico, 12, 66, 67, 166, 207,
253, 258, 259, 260, 262, 265,320, 321, 412, 414, 417
paraneoplásico, 301parietal, 131poscoartación, 271posflebítico, 66, 71, 235, 390postrombótico, 216, 350sensitivo
motor, 159puro, 159
sistémico de la embolización decristales de colesterol, 266
sinusitis, 166sitofobia, 280soplo
carotídeo, 119cervical, 122
Staphylococcus aureus, 169, 329
Streptococcusspp., 169viridans, 329
sucralfato, 408, 449sufrimiento tisular, 274sulfametoxazol, 446, 447sulfasalazina, 454sulfinpirazona, 408, 449, 453sulfisoxazol, 408, 453sulindaco, 408, 453sulodexida, 198
T
tabaco, 408tabaquismo, 12, 20, 66, 82, 86, 92,
140, 149, 192, 207, 217, 241,244, 256, 264, 289, 305, 337,339
talidomida, 72, 169, 299tamoxifeno, 169, 299, 345, 408, 453tapón
de fibrina, 7hemostático, 7plaquetario, 2, 4, 5, 7
taponamiento plaquetario, 2taquiarritmia, 446
intestinal, 274taquicardia, 225, 300, 322, 350
sinusal, 222taquipnea, 216, 225, 300, 322tecarfarina, 44teicoplanina, 454telangiectasia hemorrágica heredita-
ria, 271telitromicina, 447telmisartán, 454tenecteplasa, 57, 58teofilina, 421, 451terbinafina, 453tetraciclina, 408, 453

480 (Índice alfabético)Tratado de trombosis
tiamazol, 244ticagrelor, 35, 78, 94, 95, 198ticlopidina, 34, 198, 446tinzaparina, 231tiotropio, 244tirofibán, 36, 37, 100, 101tolmetin, 408, 453tolterodina, 453toxicomanía, 193tramadol, 453translocación bacteriana, 270trasplante
de riñón, 254, 260hepático, 394renal, 258, 265
trastornoautoinmunitario, 423de la coagulación, 170metabólico, 171
hereditario, 168visual, 388, 389
trastuzumab, 453trauma, 12, 13, 34, 45, 66, 72, 99,
165, 193, 217, 236, 246, 347abdominal, 251, 265cerrado de abdomen, 277cervical, 172craneal, 172renal, 252, 265
traumatismo, 2, 207, 258, 265, 272,400, 420abdominal, 279contuso, 211craneal, 166craneoencefálico, 58, 169, 236
trazodona, 408Treponema pallidum, 307triflusal, 198trimetoprim, 422, 446, 447trombo, 5, 14, 77, 78, 92, 93, 95,
96, 97, 99, 100, 130, 170, 172,
176, 178, 179, 182, 183, 205,207, 211, 215, 218, 219, 220,234, 243, 251, 254, 255, 259,261, 262, 295, 300, 305, 323,347, 352, 366, 378, 380, 381,391, 431arterial, 310, 364auricular, 106
izquierdo, 107blanco, 367coronario, 57crónico, 177de la arteria renal, 253del sistema venoso, 193en tránsito, 383intraauricular, 380intracardiaco, 58, 378, 379intracavitario, 106, 377, 379, 381intraluminal, 133, 177, 245, 248intravascular, 295mural, 155, 193, 248, 254, 329patológico, 5recurrente, 219renal, 262rojo, 155vascular, 395venoso, 310
tromboangeítis obliterante, 193, 271trombocitemia, 66trombocitopatía, 9trombocitopenia, 24, 25, 26, 37, 39,
46, 168, 232, 319, 351, 363, 364,366, 368asociada con heparina, 364esencial, 26inducida por heparina, 26, 41, 46,
232, 253, 324, 363, 428materna, 318
trombocitosis, 259tromboembolia, 421
paradójica sistémica, 383

481Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
pulmonar, 54, 58, 67, 68, 71,205, 215, 217, 218, 221, 222,223, 224, 225, 226, 227, 228,229, 230, 231, 233, 234, 235,308, 315, 321, 335, 368, 372,377, 382aguda, 382crónica, 384
venosa, 300tromboembolismo, 68, 106, 110,
254arterial de grandes vasos, 372de la arteria renal, 254materno, 316paradójico, 254pulmonar, 390, 391recurrente, 373, 383renal, 255, 265venoso, 13, 68, 167, 168, 316,
320, 322, 340, 347, 348, 349,351, 352, 354, 356, 357, 422agudo, 355en el embarazo, 324posoperatorio, 351
trombofilia, 11, 12, 20, 23, 81, 86,165, 173, 206, 207, 217, 233,246, 249, 254, 271, 304, 316,319, 326, 342, 345, 415, 417adquirida, 25, 67, 316asintomática, 72congénita, 67, 309, 327hereditaria, 11, 13, 26, 27, 165,
184, 316, 337, 339, 340, 431,432
primaria, 59tromboflebitis, 205
materna, 311séptica, 59, 249
de la vena yugular, 245superficial, 16, 295
migratoria, 296
venosa profunda, 298, 300trombólisis intraarterial, 255trombomodulina, 409tromborresistencia endotelial, 192trombosis, 7, 13, 14, 15, 16, 18, 22,
23, 24, 27, 35, 51, 67, 81, 82, 83,86, 87, 88, 92, 93, 105, 130, 168,169, 171, 173, 176, 177, 198,205, 206, 207, 208, 219, 243,246, 253, 255, 259, 260, 272,273, 274, 289, 295, 298, 299,300, 303, 306, 310, 311, 312,315, 316, 317, 318, 319, 326,327, 332, 335, 336, 337, 340,342, 347, 348, 351, 352, 357,364, 366, 367, 378, 405, 412,413, 425, 427, 431, 445aguda, 7, 252aórtica, 257
aguda, 397arterial, 11, 14, 15, 19, 20, 25,
26, 56, 251, 258, 269, 278,296, 304, 308, 309, 317, 319,336, 339, 340, 341, 342, 350,368, 371, 413, 422, 426, 428cerebral, 16cerebrovascular, 17de las extremidades, 309, 368renal bilateral, 252
bilateral de la arteria renal, 252cerebral, 11, 432cerebrovascular, 25, 318coronaria, 318
aguda, 57cutánea, 25de la arteria renal, 251, 252, 254,
255, 262de la retina, 318de la vena
renal, 258, 259, 260, 261, 265subclavia, 249

482 (Índice alfabético)Tratado de trombosis
yugular, 244, 245, 246de las venas y senos venosos
cerebrales, 387de pequeños vasos, 310del seno, 169
sagital superior, 249sigmoideo, 169transverso, 165
del sistema venoso profundo, 185en el paciente oncológico, 296esplácnica, 26espontánea, 249femoral, 246glomerular, 308idiopática del embarazo, 17inducida por heparina, 40intraluminal, 182mesentérica, 11, 254, 269, 278microvascular, 45
diseminada, 25no traumática de la arteria renal,
253, 265placentaria, 318portal, 11, 395
idiopática recurrente, 16recurrente, 22renal, 25residual, 278traumática, 251
de la arteria renal, 251, 265de la vena renal, 262
valvular, 381vascular, 24, 309venosa, 12, 14, 15, 16, 17, 19,
20, 25, 27, 56, 65, 66, 72, 165,167, 178, 182, 200, 205, 207,208, 210, 247, 248, 249, 269,274, 304, 309, 316, 317, 319,320, 321, 324, 331, 337, 340,341, 342, 345, 350, 368, 371,414, 416, 422, 425, 428, 432
central, 166, 167, 295cerebral, 26, 163, 164, 167,
186, 368en el embarazo, 331
del trasplante renal, 260esplácnica, 26familiar, 416intracraneal, 165, 331mesentérica, 271, 277, 278
focal, 272portal, 393, 395profunda, 12, 48, 58, 67, 68,
72, 205, 207, 209, 215, 225,227, 228, 245, 295, 296,308, 311, 321, 335, 347,348, 350, 356, 368de los miembros inferiores,
390periférica, 277recurrente, 25, 71
superficial, 309yugular, 241, 243, 246, 247,
249yugular, 248
tromboxano, 10tuberculosis, 169, 271, 449tumor, 175, 179, 296, 299, 378
abdominal, 260cardiaco, 380cerebral, 236del cerebro, 297del páncreas, 297intracardiaco, 377maligno, 296, 298
del cerebro, 299primario del SNC, 169
U
úlceragenital, 172

483Índice alfabéticoE
dito
rial A
lfil.
Fot
ocop
iar
sin
auto
rizac
ión
es u
n de
lito.
�
isquémica, 195oral, 172péptica, 33, 45, 99, 236, 448
ulceración isquémica, 198urocinasa, 57, 184, 211, 234, 256
V
valvulopatía, 25, 265, 378cardiaca, 308
vancomicina, 408varicela, 414varices, 124, 206, 207, 213, 277,
349vasculitis, 82, 166, 193, 318vasculopatía multisistémica, 25vasoconstricción esplácnica, 271vasogénico, 181vegetación valvular, 377vena varicosa, 66, 67, 218, 321, 322verapamilo, 53, 54, 449, 453, 454,
456vértigo monosintomático, 151VIH, 414, 452, 456vitamina
B12, 20, 167, 421B6, 20, 167, 421E, 457
K, 6, 15, 38, 42, 43, 44, 45, 46,48, 51, 54, 75, 110, 111, 198,212, 229, 231, 232, 248, 319,325, 327, 328, 331, 408, 446,447, 449, 454
K1, 46voriconazol, 446, 453
W
warfarina, 6, 16, 42, 43, 44, 46, 47,48, 49, 52, 53, 54, 59, 78, 98,108, 110, 111, 112, 113, 114,212, 231, 233, 248, 319, 325,326, 352, 372, 406, 407, 410,412, 413, 445, 446, 447, 448,449, 450, 452, 453, 455sódica, 37
X
ximelagatrán, 50, 51, 52, 112, 199
Z
zileutón, 453

484 (Índice alfabético)Tratado de trombosis



















