
UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS...
53
UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS. DEPARTAMENTO DE CIENCIA DE LOS ALIMENTOS Y TECNOLOGÍA QUÍMICA. Centro de Investigación y Desarrollo en Grasas y Aceites. CIDGRA. PROFESOR PATROCINANTE DIRECTOR Prof. Q.F. Lilia Masson S. Prof. Q.F. Lilia Masson S. “IMPLEMENTACIÓN DE UNA METODOLOGÍA ANALÍTICA PARA LA CUANTIFICACIÓN DE ACRILAMIDA EN PAPAS CHIPS POR HPLC MS/MS.” MEMORIA PARA OPTAR AL TITULO DE QUIMICO FARMACEUTICO LUIS GUSTAVO EDUARDO HERNÁNDEZ MORENO Santiago-Chile 2007
Transcript of UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS...

UNIVERSIDAD DE CHILE
FACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS. DEPARTAMENTO DE CIENCIA DE LOS ALIMENTOS Y
TECNOLOGÍA QUÍMICA. Centro de Investigación y Desarrollo en Grasas y Aceites. CIDGRA.
PROFESOR PATROCINANTE DIRECTOR Prof. Q.F. Lilia Masson S. Prof. Q.F. Lilia Masson S.
“IMPLEMENTACIÓN DE UNA METODOLOGÍA ANALÍTICA PARA LA CUANTIFICACIÓN DE ACRILAMIDA EN PAPAS
CHIPS POR HPLC MS/MS.”
MEMORIA PARA OPTAR AL TITULO DE QUIMICO FARMACEUTICO
LUIS GUSTAVO EDUARDO HERNÁNDEZ MORENO Santiago-Chile
2007

ii
AGRADECIEMENTOS
Quiero agradecer profundamente a la Profesora Lilia Masson por todo su apoyo y entrega de
conocimientos que contribuyeron a la realización de esta memoria. A Erik Petersson por
compartir sus conocimientos y toda su experiencia en el tema de esta memoria. Finalmente a
todo el grupo de trabajo del Centro de Investigaciones y Desarrollo en Grasas y Aceites
(CIDGRA) del Departamento de Ciencia de los Alimentos y Tecnología Química de la Facultad
de Ciencias Químicas y Farmacéuticas Conrado Camilo, Cristián Encina y José Reinaldo Muñoz
por el apoyo y camaradería demostrados durante toda la realización de la memoria.

iii
TABLA DE CONTENIDOS
Página SUMMARY iv RESUMEN v 1. INTRODUCCIÓN 1 2. ACRILAMIDA 3 2.1 Estructura química, propiedades y aplicaciones 3 2.2 Mecanismo de formación 4 2.3 Efectos sobre la salud 6 2.4 Determinación 8 3. OBJETIVOS 10 3.1 Objetivo General 10 3.2 Objetivos Específicos 10 4. MATERIALES Y METODOS 11 4.1 Fundamentos Técnicos 11 4.2 Reactivos 13 4.3 Materiales 13 4.4 Equipos 14 4.5 Determinación por HPLC MS-MS 16 4.6 Cálculo de resultados 21 5. RESULTADOS 23 5.1 Determinación de exactitud 24 5.2 Determinación de precisión 27 5.3 Especificidad 32 5.4 Límite de detección y límite de cuantificación 35 5.5 Linealidad y rango 36 5.6 Muestras reales 37 6. DISCUSIÓN 40 7. CONCLUSION 42 8. BIBLIOGRAFIA 43 9. ANEXOS 46

iv
SUMMARY ANALYTICAL METHOD FOR ACRYLAMIDE DETERMINATION IN POTATOES CHIPS BY HPLC MS/MS In April of the 2002 researchers of the University of Stockholm and from the Swedish National
Food Administration announced to the world that in different foods mainly rich in starch,
normally consumed by the population and processed at high temperatures as frying, baking ,
extruding, acrylamide was formed. The foods with the higher content were potatoes chips,
“french fries”, cookies , crackers, breads, breakfast cereals, the amounts were in ppb.
Acrylamide has been classified as “probable carcinogenic for humans” (Grupo 2A) by the
International Agency of Research on Cancer - IARC , and its exposition cause damage to the
nervous system in human and animals.
Due this communication, it was necessary to develop methods enough sensible, robust at a
reasonable cost , which could be applied to determine acrylamide in different matrixes of food
with a low detection level (ug/kg).
During this research the methodology for determination acrylamide in foods by HPLC MS/MS ,
was implemented at the CIDGRA, Centre of Fats and Oils R&D Department of Food Science
and Chemistry Technology, Faculty of Chemistry and Pharmaceutical Sciences, University of
Chile. The precision and accuracy of the method was determined. The method was apply to
quantify acrylamide in potatoes chips from the commerce and the potatoes chips elaborated at
the laboratory according the research programmed during the development of the UE HEATOX
Project Nº 506820.
As a conclusion, it is possible to say that all the objectives were reached, and the Method is
working at CIDGRA Centre , which is the first step to start with the research for determining
the acrylamide content of the foods more consumed in Chile which potentially could be a source
of acrylamide.

v
RESUMEN
En el año 2002 la Administración Nacional de Alimentos de Suecia detectó altas
concentraciones de acrilamida que se formaba durante el calentamiento a altas temperaturas de
alimentos ricos en almidón, como las papas fritas, pan, galletas, alimentos extruídos, etc.
La acrilamida es genotóxico y ha sido clasificada como “probable carcinogénico para humanos”
(Grupo 2A) por la Agencia Internacional de Investigación sobre Cáncer IARC, y esta exposición
causa daño al sistema nervioso en seres humanos y animales
Con motivo de este hallazgo, surgió el requerimiento urgente de desarrollar un método analítico
sensible, robusto y de un costo razonable, que pueda cuantificar acrilamida en diferentes
matrices de alimentos con bajos niveles de detección (µg/Kg).
Durante el desarrollo de esta memoria se implementó en el Centro de Investigación y Desarrollo
en Grasas y Aceites (CIDGRA), Departamento de Ciencia de los Alimentos y Tecnología
Química de la Facultad de Ciencias Químicas y Farmaceúticas de la Universidad de Chile, la
metodología analítica para cuantificar acrilamida por HPLC MS/MS. Se determinó la precisión y
exactitud en el análisis de acrilamida por HPLC MS/MS. Finalmente se aplicó la metodología
para el análisis de papas fritas chips comerciales y elaboradas en el laboratorio dentro del
Proyecto Heatox 506820.
En conclusión se puede decir que se logró una respuesta total a los objetivos planteados siendo
implementado en el Centro de Investigación y Desarrollo en Grasas y Aceites (CIDGRA), la
metodología analítica para la extracción de acrilamida en papas chips y su cuantificación por
HPLC MS/MS. Lo cual abre las puertas, para el comienzo de la investigación, de los alimentos
de consumo habitual en nuestro país y que pudieran contener acrilamida, y así tomar las medidas
necesarias en cuanto a políticas de salud, para el bienestar de nuestra población.

1
1. INTRODUCCIÓN
La acrilamida (2- propenamida) es un compuesto hidrofílico de bajo peso molecular conocida
principalmente por su uso como monómero en la producción de poliacrilamida se emplea en
plásticos y como medio de electroforesis.
En el año 2002 la Administración Nacional de Alimentos de Suecia detectó altas
concentraciones de acrilamida que se formaba durante el calentamiento a altas temperaturas de
alimentos ricos en almidón, como las papas fritas, pan, galletas, alimentos extruídos, etc.
Este descubrimiento es de interés público, porque la acrilamida es genotóxico y ha sido
clasificada como “probable carcinogénico para humanos” (Grupo 2A) por la Agencia
Internacional de Investigación sobre Cáncer (IARC) (IARC, 1994, De Wilde et al. 2005)
El descubrimiento se origina a partir de resultados de la formación de un aducto específico en
humanos entre la hemoglobina y la acrilamida, más adelante también encontrada por los
científicos de la Universidad de Estocolmo en ratas alimentadas con papas fritas (Tareke et al.,
2000).
Las conclusiones a la fecha muestran que la acrilamida se forma durante la preparación culinaria
e industrial de alimentos, ricos en carbohidratos que se fríen, hornean o extruyen a temperaturas
que exceden los 120º C, como producto de la reacción de la asparagina y glucosa que
corresponde a la conocida reacción de Maillard.
Con motivo de este hallazgo, surgió el requerimiento urgente de desarrollar un método analítico
sensible, robusto y de un costo razonable, que pueda cuantificar acrilamida en diferentes
matrices de alimentos con bajos niveles de detección (µg/Kg). Diferentes métodos se habían
desarrollado en el pasado para determinar monómero de acrilamida, especialmente en agua,
fluidos biológicos y alimentos. Se basaban en técnicas de cromatografía líquida o de gas. Sin
embargo, estos métodos como tales no eran apropiados, en alimentos cocinados, para analizar
acrilamida en bajos niveles. En particular, carecen de selectividad y el grado adicional de certeza
analítica requerida para confirmar la presencia de esta pequeña molécula como es la acrilamida
en la compleja matriz de un alimento (Paleologos et al. 2005). Es así como a raíz de este
hallazgo diversos grupos de investigación en el mundo inician trabajos para desarrollar
metodologías analíticas que cumplan con estos requisitos de determinación de alta sensibilidad.
El hallazgo de altos niveles de acrilamida en alimentos de consumo habitual despertó
preocupación e interés mundial. La Universidad de Chile a través de la Facultad de Ciencias

2
Químicas y Farmacéuticas y el grupo de investigación y desarrollo CIDGRA. Participa en el
proyecto internacional HEATOX Nº 506820, integrado por 24 instituciones de 14 países y
financiado por la Unión Europea. El enfoque del proyecto HEATOX permite la participación de
grupos multidisciplinarios ya que considera investigaciones en metodologías analíticas,
mecanismos de formación, modificación de procesos tecnológicos y aspectos relacionados con el
riesgo para la salud.
El grupo de trabajo del Centro de Investigaciones y Desarrollo en Grasas y Aceites (CIDGRA)
Departamento de Ciencia de los Alimentos y Tecnología Química de la Facultad de Ciencias
Químicas y Farmacéuticas participa en la línea de investigación que tiene por objetivo introducir
modificaciones al proceso de fritura de papas chips para reducir al mínimo las cantidades de
acrilamida y obtener alimentos más seguros. Adicionalmente, este grupo de trabajo implementó
la metodología analítica para determinar acrilamida en papas chips, aplicando el método de Erik
V. Petersson (2006), que emplea cromatografía líquida y espectrometría de masa/masa.
La presente memoria, se financiará con recursos del proyecto HEATOX Nº 506820.

3
2. ACRILAMIDA
2.1 ESTRUCTURA QUÍMICA, PROPIEDADES Y APLICACIONES
Sinónimos: 2-Propenamida, etilén carboxamida, amida acrílica, amida vinílica.
Fórmula : CH2CHCONH2
Peso molecular : 71.09
Punto de ebullición : 125°C
Punto de fusión : 87.5°C
Es un monómero intermediario usado en la síntesis de poliacrilamidas. El monómero es un polvo
blanco cristalino soluble en agua, etanol, metanol, dimetileter y acetona. No es soluble en
heptano y benceno. Es estable a temperatura ambiente, pero puede polimerizar violentamente
cuando se funde o se expone a agentes oxidantes.
Los usos principales de la poliacrilamida son para la producción de plásticos, como agente
floculante en el tratamiento de agua de bebida y en el procesamiento de pulpa y papel. También
se emplea para retirar sólidos suspendidos de aguas industriales de desecho antes de la descarga,
reuso o descarte (Masson L. et al 2005).
Otras aplicaciones son como aditivo en cosmética, agente para el acondicionamiento de suelos y
en la formulación de mezclas para encementado de túneles. El humo del tabaco es una fuente
conocida de exposición a acrilamida.

4
2.2 MECANISMO DE LA FORMACIÓN DE LA ACRILAMIDA. Durante el calentamiento de los alimentos, los azúcares reductores reaccionan con aminoácidos
iniciando una cascada de eventos químicos que conducen al pardeamiento de alimentos,
conocido como la reacción de Maillard.
Se sabe que este proceso genera compuestos reactivos como monocarbonilos y dicarbonilos los
cuales son los responsables de la reacción de pardeamiento.
Para entender mejor el mecanismo de formación de la acrilamida desde la asparragina, se
investigó la capacidad de otros compuestos carbonilos, para generar acrilamida en un sistema
modelo de papas snack (Zyazak et. al. 2003)
Se encontró que una gran variedad de fuentes de carbonilos, podían generar acrilamida de la
asparagina bajo calor. Mientras la cadena del azúcar es más corta, la molécula se tensiona, para
formar una estructura cíclica hemiacetal y posteriormente el carbonilo llega a estar más
fácilmente disponible para el ataque nucleofílico alfa-amino de la asparagina. Así, las cadenas
más cortas de azúcares llegan a ser más reactivas.
En base de los resultados de la sustitución por isótopo y los estudios de carbonilos descritos
arriba, se propone el mecanismo exhibido en la figura 2. El grupo libre alfa-amino de la
asparagina reacciona con un azúcar fuente de grupo carbonilo, formando una base de Schiff.
Bajo calor, la base Schiff se descarboxila (facilitado por la descolocación de la carga negativa,
que permite la formación de la base de Schiff), formando un producto que puede reaccionar por
dos vías. Se puede hidrolizar para formar 3-aminopropionamida que se puede degradar más aún,
vía la eliminación del amoníaco para formar la acrilamida cuando se somete a calor.
Alternativamente, la base descarboxilada de Schiff puede descomponerse directamente para
formar acrilamida vía la eliminación de una imina (Zyazak et. al. 2003).
Los factores más importantes que determinan la cinética de formación de acrilamida y su
posterior degradación son la composición de las papas y las variables de proceso. La papa aporta
los precursores en una concentración dependiente de su variedad, de las condiciones del suelo,
del periodo de cosecha y de las condiciones de almacenamiento postcosecha (Low et al., 2006).
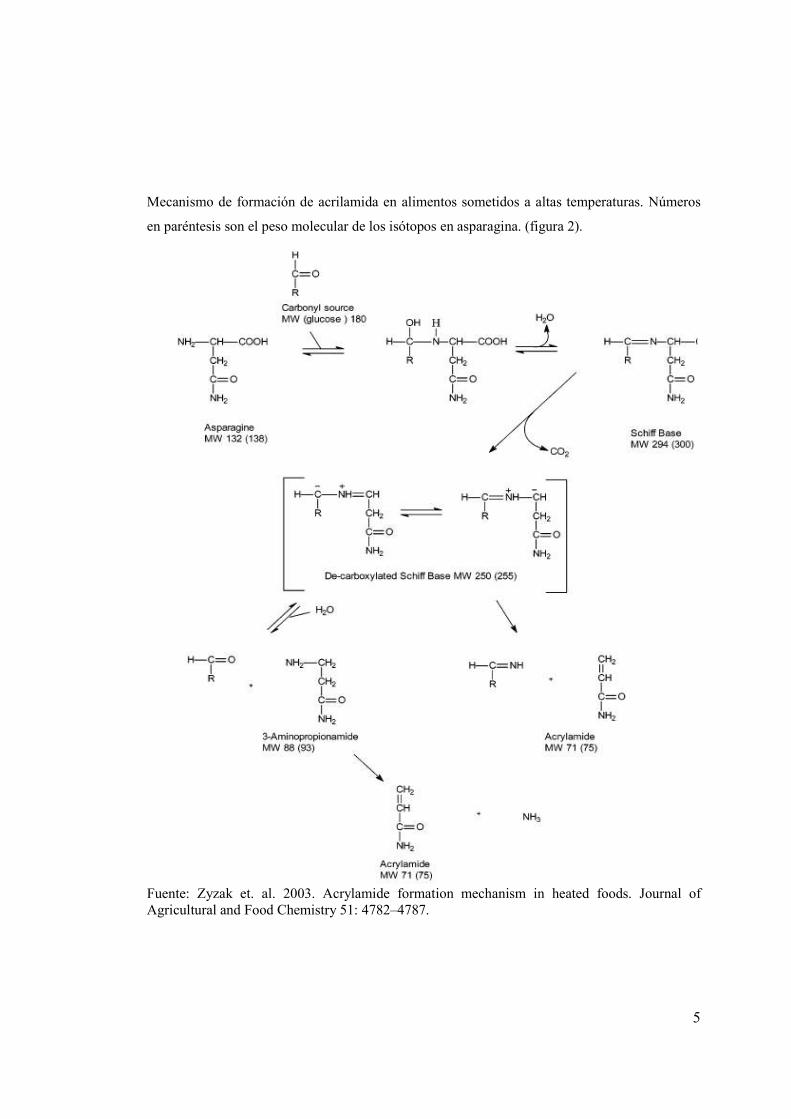
5
Mecanismo de formación de acrilamida en alimentos sometidos a altas temperaturas. Números
en paréntesis son el peso molecular de los isótopos en asparagina. (figura 2).
Fuente: Zyzak et. al. 2003. Acrylamide formation mechanism in heated foods. Journal of Agricultural and Food Chemistry 51: 4782–4787.

6
2.3 EFECTOS SOBRE LA SALUD
La acrilamida es genotóxico y ha sido clasificada como “probable carcinogénico para humanos”
(Grupo 2A) por la Agencia Internacional de Investigación sobre Cáncer (IARC) (IARC, 1994).
(De Wilde et al. 2005)
La exposición a la acrilamida causa daño al sistema nervioso en seres humanos y animales
(Lopachin y Lehning, 1994; Tilson, 1981), también se considera una toxina antireproductiva
(Costa et al., 1992; Dearfield et al., 1988) con características mutágenas y carcinógenas en
sistemas experimentales in-vitro e in-vivo de algunos mamíferos estudiados (Dearfield et al.,
1995).
Se han identificado y cuantificado aductos de hemoglobina (Hb), con el fin de la supervisión in
vivo de la dosis de productos químicos o metabolitos reactivos. La cuantificación de aductos
formados con aminas terminales, como por ejemplo el aminoácido valina de las cadenas de
hemoglobina se han convertido en una herramienta poderosa, para la dosimetría de agentes
químicos genotóxicos carcinógenos. Este método se ha aplicado, para el monitoreo de las dosis
en las personas ocupacionalmente expuestas a la acrilamida (AA).La AA reacciona por la
adición de Michael con los grupos NH2 terminal de las cadenas de hemoglobina durante la
formación de N-(2-carbamoiletill) valina (CEV) (figura1). LA AA se metaboliza a glycidamida,
un epoxido (figura 3) que es reactivo para el ADN y se asume que puede ser el agente
mutagénico e iniciativo de cáncer, mientras que la AA es probablemente la causa principal de los
efectos neurotóxicos producidos por su exposición a AA (Tareke, et al. 2000).
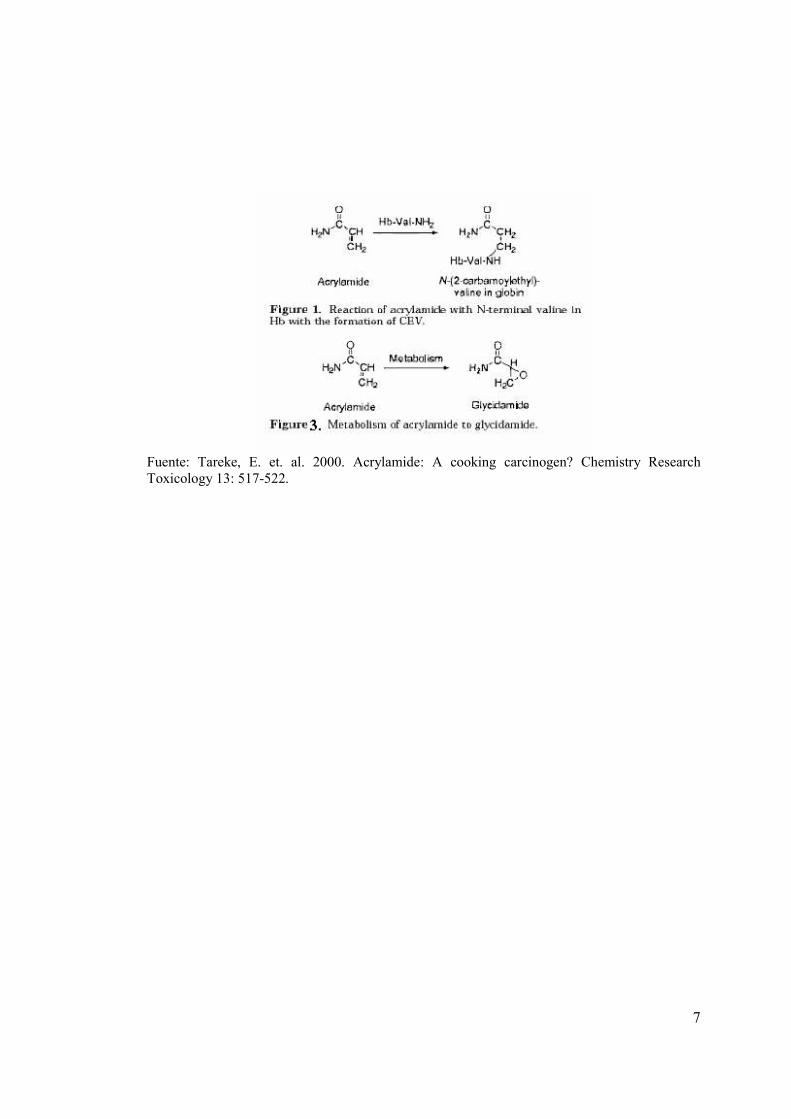
7
Fuente: Tareke, E. et. al. 2000. Acrylamide: A cooking carcinogen? Chemistry Research Toxicology 13: 517-522.

8
2.4 DETERMINACIÓN DE ACRILAMIDA.
Debido a la alta solubilidad y alta reactividad (Mottran y Wedzicha, 2002) y también, por la
carencia de un grupo cromóforo, la acrilamida no es fácilmente detectable (Jezussek y
Schieberle, 2003).
Diferentes métodos se han desarrollado en los últimos años para determinar el monómero de la
acrilamida, especialmente en agua, líquidos biológicos, y alimentos crudos (azúcar, cultivos de
campo, setas). La mayoría son métodos clásicos basados en la Cromatografía Líquida de Alta
Resolución (HPLC) o las técnicas cromatográficas con gas (Cromatografía Gaseosa) (Bologna et
al., 1999; Castle, 1993; EPA, 1996; Tekel et al., 1989).
Sin embargo, debido a la complejidad de las matrices de los alimentos, estos métodos como tal
no son suficientes para el análisis de la acrilamida en alimentos procesados. Particularmente,
carecen de selectividad y del grado adicional de la certeza del analito requerido, para confirmar
la presencia de una molécula tan pequeña como la acrilamida, en la matriz compleja del
alimento.
El primer método de análisis de la acrilamida en diversos alimentos cocidos y procesados se
publicó en mayo de 2002. Se basa en el uso de la dilución isotópica en cromatografía líquida con
espectrometría de masas (LC-MS/MS) (Rosén y Hellenäs, 2002). Desde entonces, se han
publicado en revistas científicas o se han presentado en las reuniones internacionales, varios
métodos analíticos que describen el análisis de acrilamida en alimentos procesados (Wenzl et al.,
2003). Estos métodos se basan principalmente en el empleo de la Espectrometría de Masas (MS)
como la técnica determinativa, antecedido de una separación cromatográfica por LC (Ahn et al.,
2002; Becalski et al., 2003; Hartig et al., 2002; Höfler et al., 2002; Gutsche et al., 2002; Nemoto
et al., 2002; Tareke et al., 2002), o Cromatografía Gaseosa, donde se derivatiza el analito (Gertz
y Klostermann, 2002; Höfler et al., 2002; Ono et al., 2003; Tareke et al., 2002), o, el análisis del
compuesto directamente (Biedermann et al., 2002; Tateo y Bononi, 2003).
El grupo de trabajo creado para los métodos analíticos que se convocó durante la reunión sobre
la acrilamida (JIFSAN/NCFST, 2002) y Clarke et al., (2002) concluyeron que la mayoría de los
laboratorios aplican el método de LC-MS/MS para el análisis de la acrilamida. La ventaja de los

9
métodos basados en LC-MS/MS, es que la acrilamida se puede analizar sin la derivatización
anterior (ejemplo, bromación), que simplifica y apresura considerablemente el análisis.
Se planteo un estudio optimizado del método de determinación de acrilamida mediante LC-
MS/MS. Este estudio ha demostrado que la acrilamida se puede extraer eficientemente de varias
matrices alimenticias usando agua pura. El uso de una mezcla de etanol-agua como solvente de
extracción, o la eliminación de grasa con un solvente orgánico, tiene un efecto no significativo
sobre la extracción de acrilamida (Petersson et al. 2006).
El método propuesto por Petersson et al. (2006) consiste en el uso de muestras desintegradas,
agua como el solvente de la extracción y agitación de la muestra horizontalmente (100 RPM) a
25 °C durante 45 minutos. Este es un método simple, sin solventes orgánicos, que permite un
alto rendimiento de procesamiento de muestras puesto que se pueden extraer en paralelo.

10
3. OBJETIVOS
3.1 Objetivo General
Cuantificar el contenido de acrilamida en papas fritas chips.
3.2 Objetivos específicos
Implementar en el Centro de Investigación y Desarrollo en Grasas y Aceites CIDGRA,
Departamento de Ciencia de los Alimentos y Tecnología Química de la Facultad de Ciencias
Químicas y Farmaceúticas de la Universidad de Chile, la metodología analítica, para la
extracción de acrilamida en papas chips y su cuantificación por HPLC MS/MS.
Aplicar la metodología para el análisis de papas fritas chips comerciales y elaboradas en el
laboratorio, para las investigaciones del Proyecto Heatox 506820.
Determinar la precisión y exactitud en el análisis de acrilamida por HPLC MS/MS.

11
4. MATERIALES Y MÉTODOS
4.1 FUNDAMENTOS TÉCNICOS
Una porción de prueba (papas fritas 1 gramo) se extrae con agua y se le agrega acrilamida
marcada (Deuterada). El extracto se centrifuga y al sobrenadante se le realiza una purificación y
limpieza con dos columnas de extracción en fase sólida (SPE).
El primer SPE, (Isolute Multimode), contiene una base de sílica con grupos C-18, que actúan
como intercambiadores de aniones y cationes, la acrilamida no es retenida por la columna de
extracción, y pasa a través de ella para ser colectada. La razón para el uso de esta columna es
para retener muchos posibles componentes de la matriz (compuestos no polares como también
aniones y cationes) sin la retención de acrilamida, esta columna “Multimode” se usa como un
filtro químico.
El segundo SPE, (ENV+), contiene un polímero con relativamente alta capacidad para captar
acrilamida. El extracto se pasa a través de la columna, se lava con agua y finalmente se eluye la
acrilamida con metanol al 60%. El propósito de este paso, además de aumentar la limpieza del
extracto, es concentrar el extracto y obtener bajos niveles de límite de cuantificación (Petersson
et al. 2006).
Estructura de Isolute ENV+
Descripción: Copolimero Hidroxilado poliestireno-divinilbenceno.
Partícula promedio: tamaño 90µm
Porosidad Nominal: 800 Å
Comentarios: Extracción de drogas muy polares y metabolitos que no son retenidos por C8 o
C18.

12
Luego de la evaporación del metanol el extracto se analiza por cromatografía líquida (HPLC-
MS-MS). Para este propósito se instala una columna con carbono gráfito como fase estacionaria
con un factor de retención (k) relativamente alto (k=4 cuando solventes no orgánicos se agregan
en la fase móvil), comparada con otras columnas comercialmente disponibles.

13
4.2 REACTIVOS
Acetonitrilo Grado HPLC
Metanol grado HPLC
Acido Acético glacial (cas 64-19-7)
Agua grado HPLC
Estándares
Aldrich
Acrilamida 99% (148571) (CAS 79-06-1)
Acrilamida-2,3,3-D3 98% átomo D (636568)
4.3 MATERIALES
Papas fritas chips elaboradas en el laboratorio y papas chips comerciales.
Tubos Falcon de polipropileno 50 ml con tapa.
Tubos Falcon de polipropileno 15 ml con tapa.
Pipetas pasteur
Probeta de 40 ml
Material de vidrio (Clase A)
Matraz volumétrico de 50 ml
Matraz volumétrico de 100 ml
Viales de vidrio autosampler
Micropipetas
Precision calibrada
100- 1000 microlitros
1000-5000 microlitros

14
4.4 EQUIPOS
HPLC MS/MS
Cromatografo líquido consiste de:
Inyector capaz de inyectar 10µ L de la muestra. Bomba del HPLC capaz de mantener un flujo de
la fase móvil de 0.4 ml/minuto
Columna del HPLC.
Hypercarb, 5µm, 50x2.1 mm, con una pre-columna 5 µm, 10x2.1 mm (Thermo Hypersil-
Keystone). La fase estacionaria de esta columna es carbón grafito. Las columnas de otros
proveedores que demuestren las mismas características que la columna especificada puede ser
también aplicada, la igualdad de las características de la columna tiene que ser demostrada.
Espectrómetro de masa.
Espectrómetro de masa triple quadrupole que funciona en electrospray positivo con modo de
monitoreo de reacción múltiple, sistema para obtener resolución de la unidad.
Adquisición de datos y sistema del análisis
Colección de datos y software "masslynx" de evaluación especifico.
Sistemas de extracción de fase sólida
Dispositivo con sistema de vacío para extracción en fase sólida (Vacuum manifold).
SPE column “Multimode”:
Isolute Multimode, 1000 mg / 6 ml, from IST (International Sorbent Technology Ltd), Hengoed,
Mid Glamorgan, UK.
SPE column “ENV+”:
Isolute ENV+, 500 mg / 6 ml, from IST (International Sorbent Technology Ltd), Hengoed, Mid
Glamorgan, UK.

15
Balanza analítica
Sensibilidad 0.01 mg
Mezclador Horizontal
(Capacidad tubos Falcon 50 ml)
Centrifuga
Para tubos Falcón (velocidad 8000 rpm)
Equipo para evaporación del solvente (La temperatura de evaporación no debe exceder los 40
ºC). Se necesita corriente de nitrógeno o vacío.

16
4.5 DETERMINACIÓN POR HPLC-MS-MS Las muestras extraídas, fueron leídas en HPLC MS/MS (LC perkinelmer), facilitado por la
Profesora Betty San Martín del Laboratorio de Farmacología, Facultad de Ciencias Veterinarias
y Pecuarias, de la Universidad de Chile.
Condiciones de HPLC-MS-MS
Para un análisis adecuado es de vital importancia que el instrumento esté en buenas condiciones
y que todos los parámetros instrumentales estén optimizados.
Se utiliza la columna de Hypercarb y la fase móvil con un flujo de 400 µL/min. La columna se
sostiene en un espacio temperado. Se inyecta un volumen de 10 µL.
Se optimizan todos los parámetros como, diversas temperaturas, flujos de gas, voltajes, y
posición de la señal para la detección de la acrilamida en el flujo de la fase móvil. Optimice la
energía de colisión para cada uno de las transiciones siguientes: m/z 72>55, 72>54, 72>44 y
75>58. Se utilizan los ajustes óptimos del tiempo de detección para obtener la mejor
sensibilidad, evitando cualquier interferencia y para obtener cromatógramas con por lo menos 15
puntos de referencias por canal sobre el pico. La acrilamida y el estándar interno se detectan con
las transiciones m/z 72>55 y 75>58 para los propósitos cuantitativos, y m/z 72>55, 72>54 y
72>44 para la confirmación de la identidad de la acrilamida. Usando el instrumento Quattro
Ultima de Micromass el siguiente ajuste de parámetro fue aplicado con éxito:
Parámetros HPLC
Columna HPLC Hypercarb column 50x2.1mm equipped with a Hypercarb pre-column (10x2.1 mm)
Temperatura de Columna 25 º C Volumen de Inyección 10 µL Fase Móvil 0.1% ácido acético en agua Flujo de Fase Móvil 400 µL/min Tiempo de Corrida total 15 min

17
Parámetro MS
Desolvation gas N2, 600 dm3/h
Desolvation temperature 400°C Nebulising gas N2, fully open
Cone gas N2, 200 dm3/h
Collision gas Argon, 2,3 mbar Ion source temp 125°C
Capillary voltage 2 kV Cone voltage 20 V Hexapole voltage 10 V Collision energies: 72>55 and 75>58 9 eV 72>44 20 eV 72>54 16 eV Dwell time 0,15 sec Inter channel delay 0,03 sec
Se inyecta el extracto de cada muestra por lo menos dos veces, preferiblemente tres veces.
Inyecte las soluciones estándar por lo menos tres, preferiblemente cuatro veces: antes, entre y
después del conjunto de extractos de muestra. Utilice un tiempo de pasada de 15 minutos para
las muestras para permitir que los componentes de la matriz se enjuaguen de la columna. En
caso de necesidad lave la columna con acetonitrilo al 80% en agua, según lo descrito más
adelante "lavado de la columna del HPLC", entre el conjunto de muestras, o después del final de
la jornada.
Sistema adecuado
La respuesta del LC/MS puede variar día a día o en períodos más largos. También la columna
del HPLC puede deteriorarse después de haber sido utilizada varias veces, o apenas una vez,
dependiendo del número de inyecciones y del tipo de muestras analizaba. Por lo tanto, el sistema
se debe comprobar antes de cada serie de análisis:
La columna se debe equilibrar con la fase móvil y el espectrómetro de masa por ejemplo 30
minutos. Se inyecta por lo menos tres veces una de las soluciones estándar para comprobar la
respuesta del equipo de LCMS así como el tiempo de retención, forma máxima y anchura

18
máxima del pico. La respuesta debe ser similar antes como después de la optimización. Si no, la
interfase necesita limpiarse y / o el espectrómetro de masa necesita re-optimizarse. El tiempo de
retención no debe ser menor a 1.7 min. (En un flujo de 0.4 ml/minuto), y la anchura máxima
media debe ser bajo de 0.2 min. El alargamiento del pico ocurre incluso para las columnas
nuevas pero la distancia máxima al borde del pico (medido en la altura del 10%) no debe ser más
de dos veces la distancia del máximo al borde del pico principal. Para una ilustración mejor, un
cromatógrama típico para los productos de la patata se demuestra a continuación.
Las columnas que no satisfagan estos requisitos pudieran ser reacondicionadas lavándose. Si el
re-acondicionamiento no es el acertado, la columna tiene que ser cambiada. Las tres inyecciones
de la solución de estándar deben dar resultados casi idénticos.
Debe inyectarse agua pura para comprobar si hay posible contaminación del sistema. No debe
detectarse ningún rastro de acrilamida.
prov 70 Havrefras (omanalys)
0.00 0.50 1.00 1.50 2.00 2.50 3.00 3.50 4.00 4.50 5.00Time0
100
%
0
100
%
0
100
%
0
100
%
0
100
%
analys 020326 037 Sm (Mn, 2x3) MRM of 5 Channels ES+ 74.8 > 57.8
3.11e5Area
2.1365316
analys 020326 037 Sm (Mn, 2x3) MRM of 5 Channels ES+ 71.8 > 71.75
1.59e6Area
2.15318210
analys 020326 037 Sm (Mn, 2x3) MRM of 5 Channels ES+ 71.8 > 54.85
8.39e5Area
2.15174304
analys 020326 037 Sm (Mn, 2x3) MRM of 5 Channels ES+ 71.8 > 53.85
1.99e4Area
2.153869
analys 020326 037 Sm (Mn, 2x3) MRM of 5 Channels ES+ 71.8 > 43.95
1.14e4Area
2.152294

19
Lavado de la columna del HPLC
Si el funcionamiento de la columna del HPLC es perceptiblemente peor de lo esperado o
requerido (véase que la "conveniencia del sistema") puede ser restaurada lavándose. La columna
se puede lavar con acetonitrilo al 80% en agua con un flujo de 0.4 ml/minuto por 30 minutos en
línea con el LCMS. Esto se puede hacer rutinariamente después de cada día de trabajo o aún
entre cada lote de inyecciones. Cerciórese de dar tiempo para el equilibrio con la fase móvil.
Para casos más severos el siguiente procedimiento de lavado, preferiblemente realizado sin
LCMS, puede ser usado:
Disponga de la pre-columna. Cambie el sentido del lavado y limpie la columna a la temperatura
ambiente 0.2 ml/minuto en un orden consecutivo:
(a) dos horas con una mezcla de 50% tetrahidrofurano (THF), 10% amoníaco y 40% agua. Por
favor, considere que algunos polímeros usados en HPLC no son resistentes al THF.
(b) 30 minutos con metanol puro.
(c) 30 minutos con la fase móvil para el equilibrio.
Control de Calidad
Para cada lote de muestras se utilizan los controles siguientes:
Estándares de la calibración
Los estándares de la calibración que empiezan con la concentración más baja se analizan al
principio de cada lote y la curva de calibración que resulta, se utiliza para cuantificar ese lote de
muestras. Al final de cada jornada un estándar del punto medio se debe analizar para supervisar
la desviación del instrumento. Los valores aceptables son el ± el 5% de la concentración
original.
Muestra en blanco:
El Puré de papas, en caso de los productos de la papa, tiene que ser analizadas con cada lote de
muestras.

20
Muestras con acrilamida agregada.
Puré de papas u otro material apropiado con acrilamida a un nivel adecuado para el alimento
bajo investigación, deben ser analizadas con cada lote de muestras.
Materiales de referencia
Materiales certificados de referencia.
Materiales de referencia del laboratorio
Las matrices o las muestras apropiadas de control con acrilamida agregada (spiked), se
recomiendan para el uso como materiales de referencia internos del laboratorio.
Carta de control
Los resultados para las muestras spiked, así como para los materiales de referencia del
laboratorio (CRM) respectivamente serán supervisados en cartas de control. Los resultados
aceptables deben ser dentro de los límites de 3 veces la desviación de estándar intermedia de la
reproducibilidad del método.
Duplicados
Cada muestra se debe medir por lo menos en duplicado. La diferencia absoluta entre valores de
replicas para una muestra dada, hechos por un operador, con un instrumento dentro del intervalo
de tiempo más corto, debe estar en 95 % de todos los casos dentro de la desviación de estándar
de la capacidad de repetición.

21
4.6CALCULO DE RESULTADOS
La identificación de los picos es confirmada por comparación de la razón de picos de áreas para
m/z 54/55, y 44/55 de las soluciones estándar.
La calibración se realiza con un estándar interno y este se aplica para la determinación de
acrilamida. Esta calibración nesecita de la determinación de un factor de respuesta Rf el cual se
define en la siguiente ecuación:
Rf = ASAA × C[d3]AA Ecuación 1 A[d3]AA × CSAA
Donde: ASAA Área del pico de acrilamida como trazas de masa MRM m/z 72/55 en los estándares de calibración. A[d3]AA Área del pico de acrilamida-D3 marcada como trazas de masa m/z 75/58. C[d3]AA Concentración de acrilamida-D3 de la solución estandar interno. CSAA Concentración de acrilamida de la solución de estandar de calibración.
Calcular para cada muestra el promedio de la cantidad de acrilamida que fue extraída (XAA), para
N número de replicas de muestra que fueron inyectadas usando la siguiente ecuación.
XAA = 1 1ΣN AAA × X[d3]AA Ecuación 2
N A[d3]AA × Rf
Donde: XAA Valor absoluto de la cantidad de acrilamida (ng) que fue extraída de la muestra. AAA Area del pico de acrilamida para transición 72/55 de la muestra. A[d3]AA Area del pico de acrilamida-D3 para la transición 75/58 de la muestra. X[d3]AA Valor absoluto de la cantidad de acrilamida-D3 (ng) agregado a la muestra.

22
Rf El factor de respuesta determinado por el análisis de los estandares de acrilamida y acrilamida-D3.
La concentración de acrilmida en la muestra Cm (µg/Kg) es obtenida de la ecuación 3.
Cm (µg/Kg) = XAA Ecuación 3 WS
Donde XAA Cantidad absoluta (ng) de acrilamida que fue extraida de la muestra. WS Peso de la muestra en gramos

23
5. RESULTADOS
Primeramente se comenzó con la revisión bibliográfica, para familiarizarse lo más posible con
este tema de gran importancia, como lo es la acrilamida y su estrecha relación con la
alimentación a nivel mundial, ya que afecta directamente la salud todos los consumidores, por su
condición de probable cancerigeno y causante de daños al sistema nervioso. Por otro lado
también se comienza con la búsqueda de referencias para la metodología analítica mas adecuada
para la cuantificación de acrilamida. Hecho esto se empieza a buscar los materiales y los equipos
necesarios para comenzar con los análisis de cuantificación por HPLC MS/MS. Se empieza a
trabajar en las preparaciones de soluciones patrones de acrilamida, tanto en al solución Standard
interno (acrilamida deuterada), como en el Standard de acrilamida de alta pureza (≤ 99 %).
Se comienza a trabajar con el equipo HPLC MS/MS de la Facultad de Ciencias Veterinarias y
Pecuarias, de la Universidad de Chile, en el Laboratorio de Farmacología con la Profesora Betty
San Martín. En este laboratorio se comienza con los primeros análisis de muestra. El equipo se
pone en marcha ajustando los parámetros para máxima respuesta en la señal de acrilamida con la
ayuda del Doctor Erik Petersson de la Universidad de Uppsala, Institute of Chemistry,
Department of Analytical Chemistry. Suecia y de la Agencia Nacional de Administración de
Alimentos Uppsala , Suecia.
24
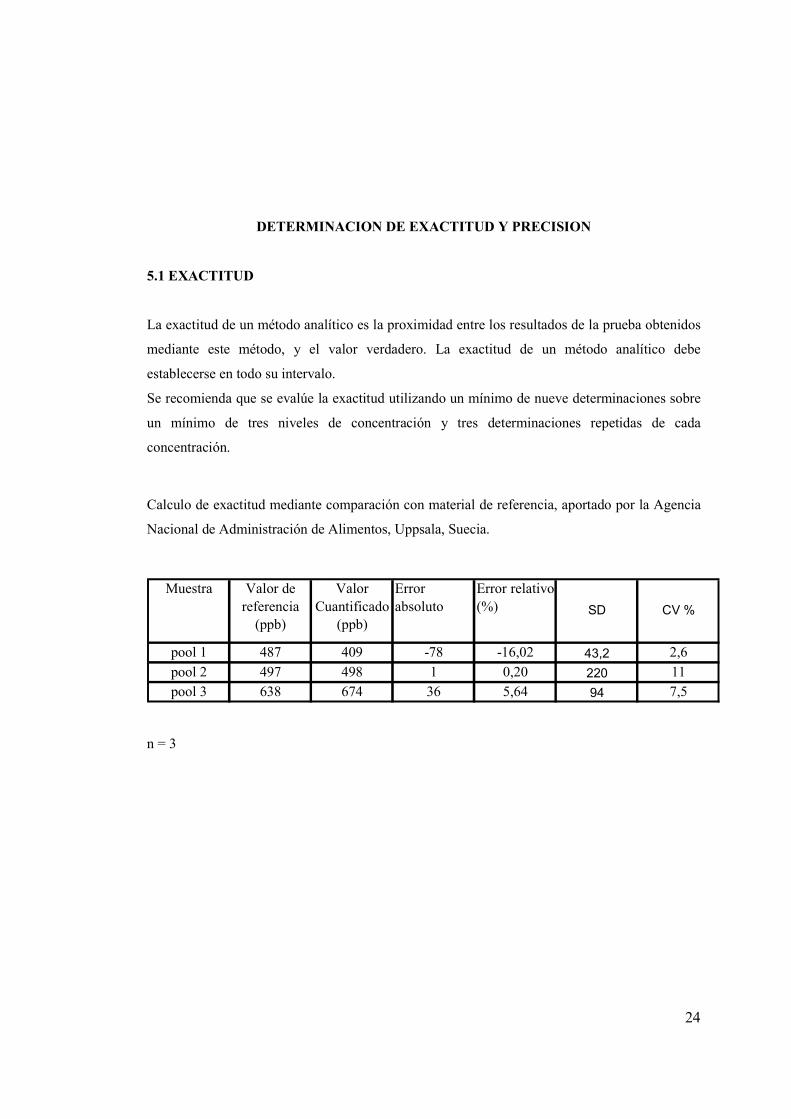
DETERMINACION DE EXACTITUD Y PRECISION
5.1 EXACTITUD
La exactitud de un método analítico es la proximidad entre los resultados de la prueba obtenidos
mediante este método, y el valor verdadero. La exactitud de un método analítico debe
establecerse en todo su intervalo.
Se recomienda que se evalúe la exactitud utilizando un mínimo de nueve determinaciones sobre
un mínimo de tres niveles de concentración y tres determinaciones repetidas de cada
concentración.
Calculo de exactitud mediante comparación con material de referencia, aportado por la Agencia
Nacional de Administración de Alimentos, Uppsala, Suecia.
pool 1 487 409 -78 -16,02 43,2 2,6
pool 2 497 498 1 0,20 220 11
pool 3 638 674 36 5,64 94 7,5
SD CV %
Muestra Error absoluto
Error relativo(%)
Valor de referencia
(ppb)
Valor Cuantificado
(ppb)
n = 3
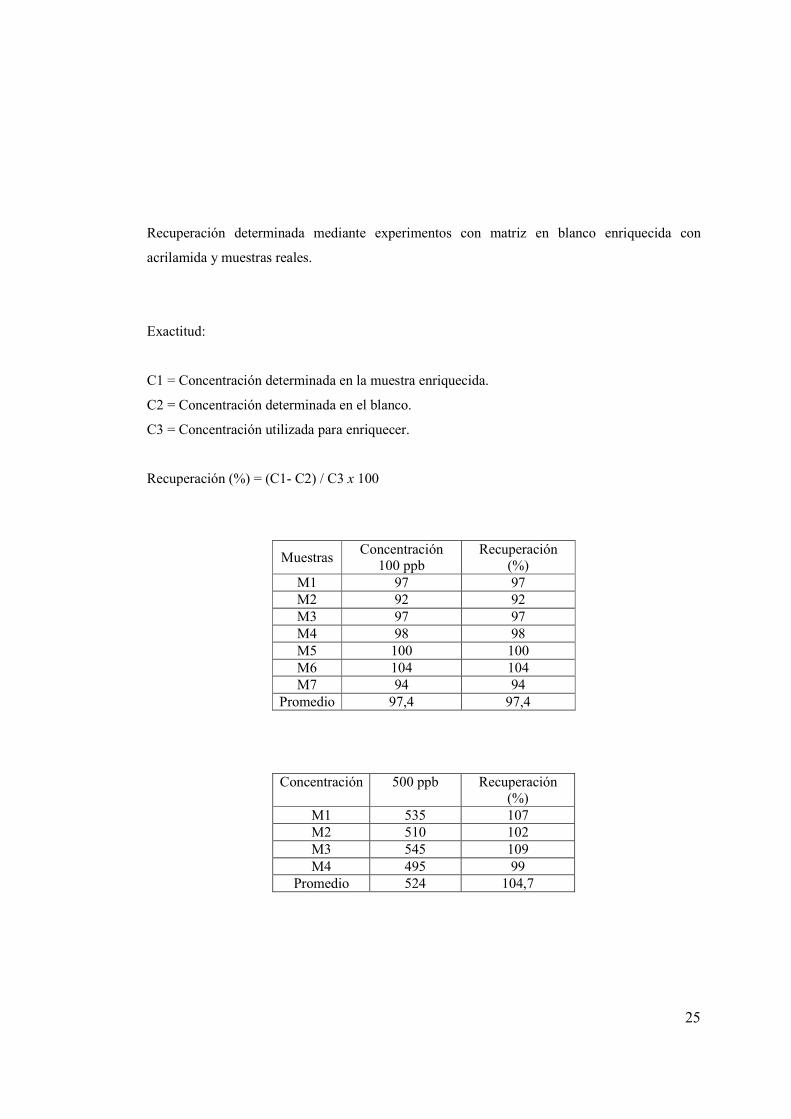
25
Recuperación determinada mediante experimentos con matriz en blanco enriquecida con
acrilamida y muestras reales.
Exactitud:
C1 = Concentración determinada en la muestra enriquecida.
C2 = Concentración determinada en el blanco.
C3 = Concentración utilizada para enriquecer.
Recuperación (%) = (C1- C2) / C3 x 100
Muestras Concentración
100 ppb Recuperación
(%) M1 97 97 M2 92 92 M3 97 97 M4 98 98 M5 100 100 M6 104 104 M7 94 94
Promedio 97,4 97,4
Concentración 500 ppb Recuperación (%)
M1 535 107 M2 510 102 M3 545 109 M4 495 99
Promedio 524 104,7

26
Criterios de funcionamiento y otros requisitos que deben cumplir los métodos cuantitativos de
análisis.
Veracidad de los métodos cuantitativos
En el caso de análisis repetido de un material de referencia certificado, los intervalos de
referencia de la desviación entre la fracción de la masa media determinada experimentalmente,
con corrector de recuperación, y el valor auténtico estarán dentro de los siguientes límites:
Veracidad mínima de los métodos cuantitativos
Fracción de masa Intervalo
≤ 1 µg/Kg > 1 µg/Kg a 10 µg/Kg
≥ 10 µg/Kg
-50% a + 20% -30% a +10% -20% a + 10%
Cuando no se dispone de tales CRM, se acepta una valoración de la veracidad de las mediciones
mediante recuperación de adiciones de cantidades conocidas de uno o varios analitos a una
matriz en blanco. Los datos corregidos mediante la recuperación media sólo son aceptables si
entran dentro de los intervalos expuestos en el cuadro anterior.
Los datos obtenidos de exactitud son aceptables según los criterios de aceptación, ya que se esta
dentro de los límites permitidos en el caso de materiales de referencia certificado, para masa
mayor igual a 10 µg/Kg se esta entre el -20% a +10%, siendo los valores para las muestras
entregadas pool 1, pool 2 y pool 3, son -16,02 %; 0,20 %; y 5,64% respectivamente. En el caso
de ensayos de recuperación también son aceptados, debido a que para una concentración de 100
ppb y 500 ppb se obtuvieron en promedio un 97,4% y 104,7% de recuperación respectivamente
lo que cae dentro del rango establecido en el recuadro anterior.

27
5.2 PRECISIÓN: REPETIBILIDAD Y REPRODUCIBILIDAD
Es el grado de concordancia entre los resultados de las pruebas individuales cuando se aplica el
método repetidamente a múltiples muestreos de una muestra homogénea. La precisión de un
método analítico habitualmente se expresa como la desviación estándar o la desviación estándar
relativa (coeficiente de variación) de una serie de mediciones. La precisión puede ser una
medida del grado de reproducibilidad o repetibilidad del método analítico en condiciones
normales de operación. En este contexto, la reproducibilidad se refiere al uso del procedimiento
analítico en diferentes laboratorios, diferentes analistas, diferentes días, diferentes temperaturas,
etc. La repetibilidad se refiere a la utilización del procedimiento analítico en un laboratorio
durante un periodo corto realizado por el mismo analista con el mismo equipo.
Para el análisis realizado bajo condiciones de repetibilidad, el coeficiente de variación intra-
laboratorio debe estar entre la mitad y dos tercios del valor, derivado de la ecuación modificada
de Horwitz.
Para los análisis realizados bajo condiciones intermedias de reproducibilidad (intra-laboratorio),
la desviación de estándar no será mayor que el valor derivado de la ecuación modificada de
Horwitz.
Por debajo de un contenido de la acrilamida de 120 µg/kilogramo, la desviación de estándar se
fija a 22 % del contenido de la acrilamida. Sobre ese valor, se calcula según la ecuación 4, que
incluye el promedio de la acrilamida contenido en la muestra respectiva, expresada como
cociente total sin dimensiones (1µg/kg ~1 ppb = 1.10-9).
σ: desviación estándar; : contenido promedio del analito (µg/kg).

28
Los ejemplos para la desviación de estándar modificada de Horwitz son:
El coeficiente de variación (CV), para el análisis repetido de un material de referencia o
enriquecido, en condiciones de reproducibilidad, no superará el nivel de cálculo mediante la
ecuación de Horwitz, a saber:
CV = 2 (1 – 0,5 log C)
Donde C es la fracción de masa expresada como potencia (exponente) de 10 (por ejemplo, 1
mg/g = 10-3 ). El cuadro siguiente recoge varios ejemplos.
CV de reproducibilidad de los métodos cuantitativos en un intervalo de fracciones de masa de
analito.
Fracción de masa CV de reproducibilidad (%)
1 µg/kg (*)
10 µg/kg (*)
100 µg/kg 23
1000 µg/kg 16
(*) Con fracciones de masa inferiores a 100 µg/kg, la aplicación de la ecuación de Horwitz
conduce a valores inaceptablemente elevados. Por ello, los coeficientes de variación de las
concentraciones inferiores a 100 µg/kg serán lo más bajo posible (Diario Oficial de las
Comunidades Europeas 17.8.2002).
Contenido promedio de acrilamida. Desviación Standard modificada por Horwitz
µg/kg µg/kg
100 22.0
200 40.8
500 88.8
1000 160.0
2000 288.2
29
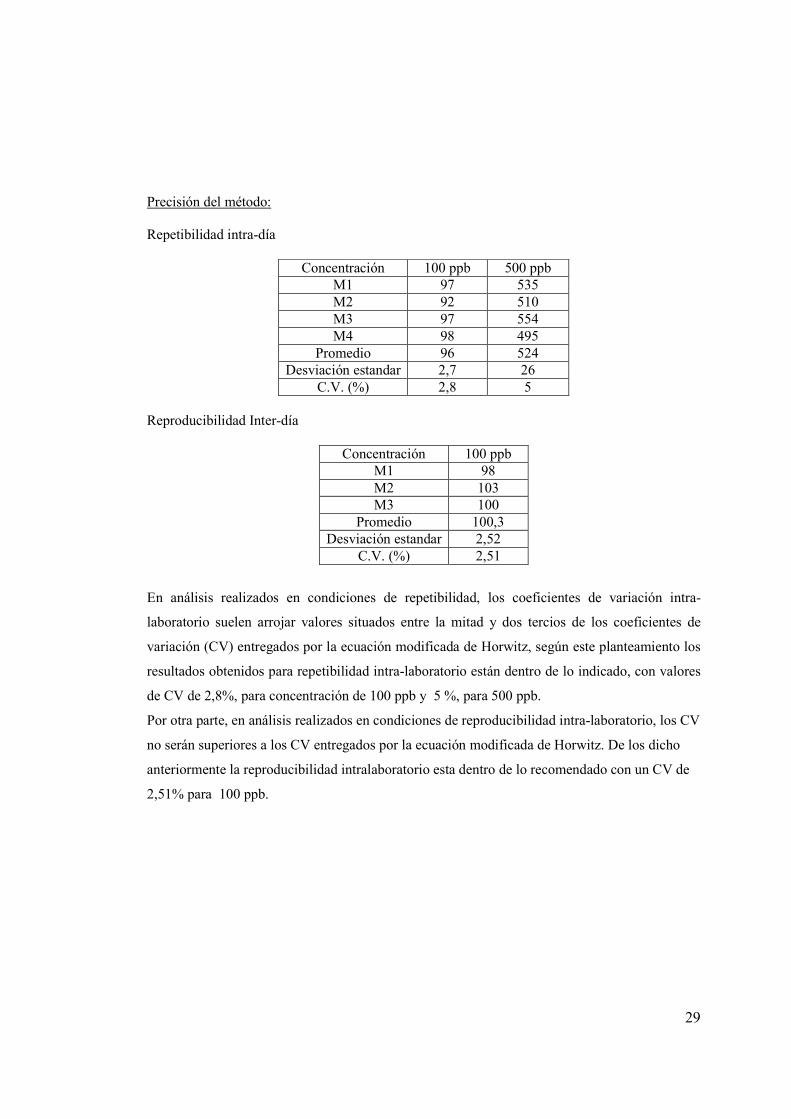
Precisión del método: Repetibilidad intra-día
Concentración 100 ppb 500 ppb M1 97 535 M2 92 510 M3 97 554 M4 98 495
Promedio 96 524 Desviación estandar 2,7 26
C.V. (%) 2,8 5 Reproducibilidad Inter-día
Concentración 100 ppb M1 98 M2 103 M3 100
Promedio 100,3 Desviación estandar 2,52
C.V. (%) 2,51
En análisis realizados en condiciones de repetibilidad, los coeficientes de variación intra-
laboratorio suelen arrojar valores situados entre la mitad y dos tercios de los coeficientes de
variación (CV) entregados por la ecuación modificada de Horwitz, según este planteamiento los
resultados obtenidos para repetibilidad intra-laboratorio están dentro de lo indicado, con valores
de CV de 2,8%, para concentración de 100 ppb y 5 %, para 500 ppb.
Por otra parte, en análisis realizados en condiciones de reproducibilidad intra-laboratorio, los CV
no serán superiores a los CV entregados por la ecuación modificada de Horwitz. De los dicho
anteriormente la reproducibilidad intralaboratorio esta dentro de lo recomendado con un CV de
2,51% para 100 ppb.

30
Repetibilidad Intra-día Curva Calibración
Concentración agregada ng/g
Concentración promedio cuantificada ng/g
Desviación estándar
n CV %
0 <10
5 5,0 0,8 3 15,6
10 10,4 0,7 3 6,6
50 52,1 1,2 3 2,3
100 103,9 2,5 3 2,5
500 512,3 13,7 3 2,7
1000 1015,7 37,2 3 3,7
2000 2024,0 29,0 3 1,4
5000 4941,7 102,2 3 2,1 Reproducibilidad Inter-día Curva Calibración
Concentración agregada ng/g
Concentración promedio cuantificada ng/g
Desviación estándar
n CV %
0 <10
2 1,8 0,4 4 22
5 5,5 0,9 4 17
10 9 2 4 16
50 48 3 4 6
100 100 3 4 3
500 497 12 4 2
1000 1029 48 4 5
2000 1977 58 4 3
5000 5047 56 4 1 Las curvas de calibración con rango de 0 a 5000 ng/ml entregan coeficientes de correlación ≥
0.999 lo cual nos indica un buen resultado. Cuando se agregaron concentraciones conocidas a
muestras en blanco para la curva de calibración el coeficiente de variación fluctúa de 1,4 a 15,6
% en condiciones de repetibilidad Intra-dia y de 1 a 22% incondiciones Inter-día.

31
Reproducibilidad Inter-analista.
Analista 1 Analista 2 Analista 3 Concentración (ppb) 100 ppb 100 ppb 100 ppb
M1 97 92 97 M2 94 100 98 M3 104 97 98
Promedio 98 96 97 Desviación estandar 5,1 4 0.58
C.V. (%) 5,2 4,2 0,59 Resultado Analistas 98 96 97
Promedio 97 Desviación estandar 1
C.V. (%) 1 La reproducibilidad Inter-analista nos entrega un coeficiente de variación de 1% lo cual esta
dentro de lo recomendado por la ecuación de Horwitz.

32
5.3 ESPECIFICIDAD
Los documentos de Validación de Procedimientos Analíticos y el texto suplementario
Metodología, ambos de la Conferencia Internacional Tripartita Sobre Armonización (ICH) , que
tratan sobre procedimientos analíticos incluidos como parte de las solicitudes de registro
presentada en la UE, Japón y EE.UU. definen especificidad como la capacidad de evaluar de
manera inequívoca el analito en presencia de aquellos componentes cuya presencia resulta
previsible, como impurezas, productos de degradación y componentes de la matriz. También
afirman que cuando se utilizan los procedimientos cromatográficos, deberán presentarse
cromatogramas representativos para demostrar el grado de selectividad y los picos deberán
identificarse adecuadamente. Las pruebas de pureza de picos (por ejemplo, utilizando redes de
iodos o espectrometría de masa) pueden resultar útiles para demostrar que el pico
cromatográfico del analito no puede atribuirse a más que un solo componente (USP 29).
33
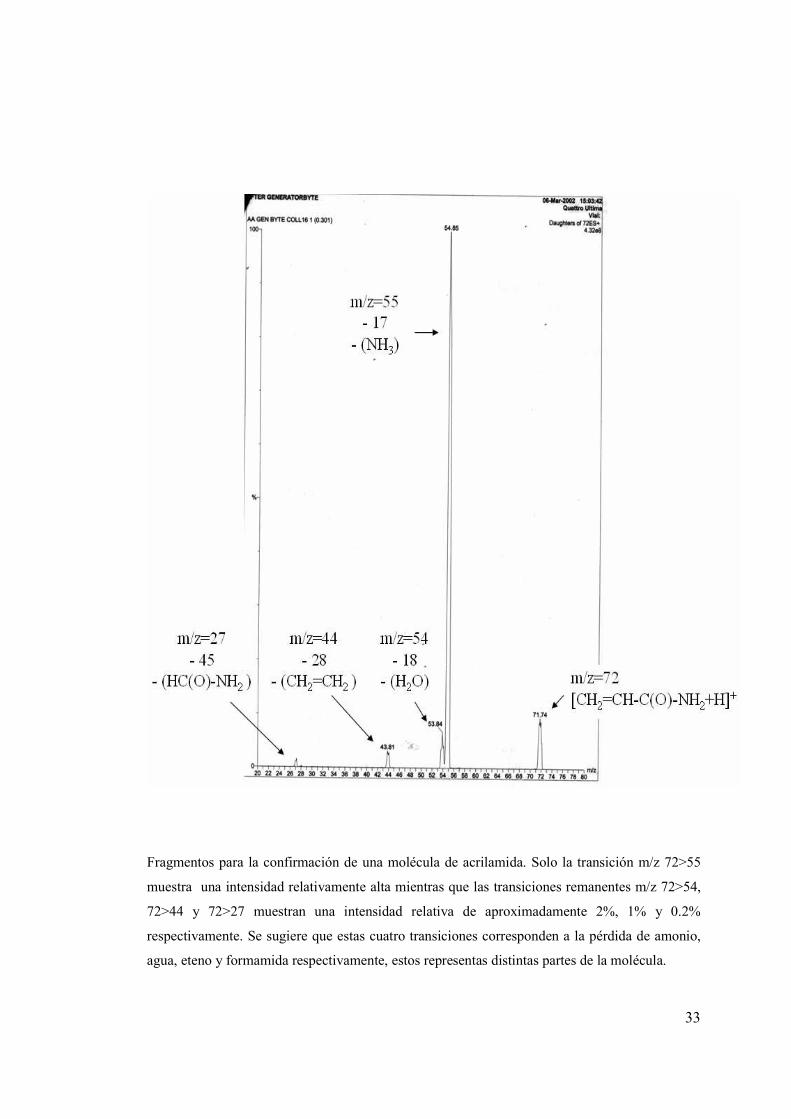
Fragmentos para la confirmación de una molécula de acrilamida. Solo la transición m/z 72>55
muestra una intensidad relativamente alta mientras que las transiciones remanentes m/z 72>54,
72>44 y 72>27 muestran una intensidad relativa de aproximadamente 2%, 1% y 0.2%
respectivamente. Se sugiere que estas cuatro transiciones corresponden a la pérdida de amonio,
agua, eteno y formamida respectivamente, estos representas distintas partes de la molécula.

34

35
5.4 LÍMITE DE DETECCIÓN Y LÍMITE DE CUANTIFICACIÓN.
Límite de detección:
Es una característica de las pruebas de límite. Es la cantidad mínima de analito en una muestra
que puede detectarse, aunque no necesariamente cuantificarse, en las condiciones experimentales
indicadas. Las pruebas de límite simplemente comprueban que la cantidad de analito se
encuentra por encima o por debajo de un nivel determinado. Se establece la concentración
mínima a la que puede cuantificarse confiablemente un analito. Una relación señal-ruido
habitualmente aceptable es de 2:1 o 3:1. El límite de detección generalmente se expresa en forma
de concentración de analito en la muestra.
Límite de cuantificación:
Es un a característica de las valoraciones cuantitativas de compuestos que se encuentran en baja
concentración en la matriz de una muestra. Es la mínima cantidad de analito en una muestra que
se puede determinar con precisión y exactitud aceptables en las condiciones experimentales
indicadas. Se establece la concentración mínima a la que puede cuantificarse confiablemente un
analito. Una relación señal-ruido habitualmente aceptable es de 10:1. El límite de cuantificación
generalmente se expresa en forma de concentración de analito en la muestra.
El límite de la detección (LOD) y el límite de la cuantificación (LOQ) para la acrilamida en
alimentos se pueden extrapolar del señal/ruido (S/N) obtenidos para las respuestas de las trazas
en el MRM m/z 72>55 y m/z 72>54 respectivamente 72> 44. El límite de la detección (S/N=3)
se espera que este entre 15 y 30 el µg/kg. Se espera que el límite de la cuantificación (S/N = 10)
este entre 40 y 70 el µg/kg .
Si LOQ se define como el nivel validado mas bajo con la recuperación y la precisión aceptables,
LOQ estara entre 2 y 5 el µg/kg para la transición m/z 72>55 según la tabla presentada en la
sección repetibilidad y reproducibilidad.

36
5.5 LINEALIDAD Y RANGO
La linealidad de un método analítico es su capacidad para obtener resultados de prueba que sean
proporcionales ya sea directamente, o por medio de una trasformación matemática definida, a la
concentración de analito en muestras en un intervalo dado. El rango es el intervalo entre los
niveles inferior y superior del analito, que han demostrado pueden ser determinados con un
adecuado nivel de precisión, exactitud y linealidad.
La curva de calibración debe ser lineal sobre el rango de concentraciones medidas. Compruebe
la homogeneidad de variaciones en el nivel de concentración bajo y alto del analito. Un
aceptable coeficiente de correlación (valor r2) es ≥ 0.99.
Concentarción agregada ng/g
0 5 10 50 100 500 1000 2000 5000
Curva Calibración Acrilamida
y = 0,0034x
R2 = 0,999
0,000
5,000
10,000
15,000
20,000
0 1000 2000 3000 4000 5000 6000
Concentración AA
Razón A
A/D
-AA

37
5.6 MUESTRAS REALES.
Con los parámetros de precisión y exactitud ya definidos se precedió a realizar análisis a
muestras reales.
Los niveles de acrilamida se cuantificaran en papas (variedad Panda) sometidas a pretratamiento
y papas control sin pretratamiento, antes del proceso de fritura con el objetivo de reducir el
contenido de acrilamida, en las papas que son pre-tratadas.
Estos pretratamiento se realizarán por Cristián Encina Ingeniero en alimentos que trabajara con
fritura al vacío en papas pre-tratadas y en papas control (sin pre-tratamiento). Y por José
Reinaldo Muñoz Ingeniero Agroindustrial que trabajara con fritura a temperatura ambiente en
papas pre-tratadas y en papas control (sin pre-tratamiento). También se trabajará con algunas
muestras obtenidas del mercado nacional preferentemente las de mayor consumo como por
ejemplo papas fritas Lays, papas fritas en tarro de marcas conocidas tales como Lays, Pringles y
Kryspo y otras del mercado internacional papas fritas Picadilli. Algunas de estas muestras serán
comparadas con laboratorios certificados como la Agencia Nacional de Alimentos, Uppsala,
Suecia y el Centro Nacional de Tecnología y Seguridad Alimentaria CNTA, Laboratorio del
Ebro.
38
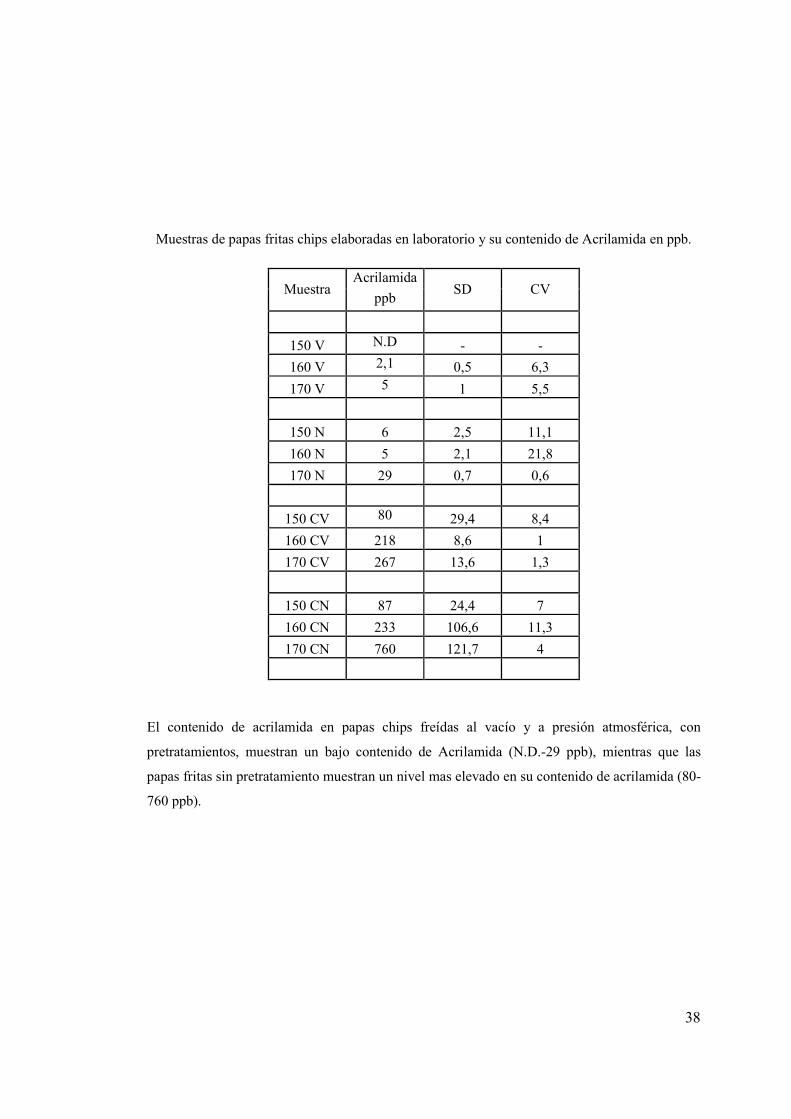
Muestras de papas fritas chips elaboradas en laboratorio y su contenido de Acrilamida en ppb.
Acrilamida
Muestra ppb
SD CV
150 V N.D - -
160 V 2,1 0,5 6,3
170 V 5 1 5,5
150 N 6 2,5 11,1
160 N 5 2,1 21,8
170 N 29 0,7 0,6
150 CV 80 29,4 8,4
160 CV 218 8,6 1
170 CV 267 13,6 1,3
150 CN 87 24,4 7
160 CN 233 106,6 11,3
170 CN 760 121,7 4
El contenido de acrilamida en papas chips freídas al vacío y a presión atmosférica, con
pretratamientos, muestran un bajo contenido de Acrilamida (N.D.-29 ppb), mientras que las
papas fritas sin pretratamiento muestran un nivel mas elevado en su contenido de acrilamida (80-
760 ppb).

39
Contenido de acrilamida en papas fritas chips comerciales y algunos alimentos expresados en
ppb (µg/kg).
Muestras AA ppb SD CV % Galleta (Cookies) 76 22,6 7,7 Pan marraqueta ( white bread, no loaf) 21 9,7 11,9 Sopaipilla (fried product with mashed pupkin ) 36 7,1 5,1 Berlín (Fried Wheat dough) 22 4,6 5,6 Papas fritas Lays tarro lote 1 (pot.Crisps) 589 160,9 6,8 Papas fritas Lays tarro lote 2 545 124,4 5,7 Papas fritas Kryspo tarro lote1 (like Pringles) 164 48,1 7,3 Papas fritas Kryspo tarro lote 2 382 78,2 5,1 Papas fritas Pringles tarro lote 1 793 165,1 5,2 Papas fritas Pringles tarro lote 2 570 134,0 5,9 Papas fritas Lays artesanales (Pot.crisps) 828 140,8 4,3 Papas fritas Chips Limón (P.crisps lemon) 879 63,5 1,2 Papas fritas Picadilli 2159 765,8 8,9
El contenido de acrilamida en papas chips comerciales, muestran un contenido de acrilamida con
un rango de entre 164 ppb para papas fritas Kryspo tarro del lote 1, y 2159 ppb, para las papas
fritas Picadilli. Por otra parte, para otro tipo de alimentos como por ejemplo: Galletas, Pan
(marraqueta), Sopaipillas y Berlín el contenido de acrilamida fue muy bajo 76 ppb, 21 ppb , 36
ppb y 22 ppb respectivamente, en comparación con las papas fritas.

40
6. DISCUSIÓN
El objetivo principal de este trabajo fue el desarrollo de una metodología analítica para la
cuantificación de acrilamida en papas chips por HPLC MS/MS, que verificaría inequívocamente
la presencia de acrilamida en dichas papas. La elección de cromatografía liquida de alta
resolución (HPLC) se debe a las propiedades hidrofílicas de la acrilamida, y el detector de masa
(MS-MS), para un alto grado de verificación de las transiciones que podrían encontrarse.
Electrospray positivo probó ser el modo más sensible que presentaba el instrumento. Todos los
parámetros fueron optimizados para obtener como una posible gran señal al ión [M+1]+ en
solución acuosa. Las transiciones de los iones hijos con la más alta respuesta fueron
identificadas por escaner. Para cada transición la energía de colisión fue optimizada.
Solo la m/z transición 72>55 muestra una intensidad relativamente alta mientras que las
transiciones m/z remanentes 72>54, 72>44 y 72>27 muestran una intensidad relativa de
aproximadamente 2%, 1% y 0.2% respectivamente. Se sugiere que estas cuatro transiciones
corresponden a la pérdida de amonio, agua, eteno y formamida respectivamente, estos
representas distintas partes de la molécula. Esto fue comprobado por el hecho que la acrilamida
deuterada (CD2CDCONH2) entrega los correspondientes fragmentos m/z 75>58, 75>57,75>44 y
75>30, los cuales son optimizados en iguales colisiones de energía correspondientes a las
transiciones para la acrilamida.
Para propósitos de análisis de acrilamida en papas chips el instrumento fue utilizado en modo
MRM montado para registro de m/z 72>55, 72>54, 72>44, 72>72 y 72>58. Para la
cuantificación de acrilamida fue usada la razón m/z 55/58, y las concentraciones fueron
calculadas con una curva de estandards de calibración. Las curvas de calibración con rango de 0
a 5000 ng/ml entregan coeficientes de correlación ≥ 0.999 lo cual indica un buen resultado.
Cuando se agregaron concentraciones conocidas a muestras en blanco para la curva de
calibración el coeficiente de variación fluctuó de 1.4 a 15.6 % en condiciones de repetibilidad
Intra-dia y de 1 a 22% incondiciones Inter.-día. El límite de detección LOQ está entre 2 y 5 el
µg/kg para la transición m/z 72>55.
En análisis realizados en condiciones de repetibilidad, los coeficientes de variación intra-
laboratorio suelen arrojar valores situados entre la mitad y dos tercios de los coeficientes de
variación (CV) entregados por la ecuación modificada de Horwitz, según este planteamiento los

41
resultados obtenidos para repetibilidad intra-laboratorio están dentro de lo indicado, con valores
de CV de 2,8%, para concentración de 100 ppb y 5 %, para 500 ppb.
Por otra parte, en análisis realizados en condiciones de reproducibilidad intra-laboratorio, los CV
no serán superiores a los CV entregados por la ecuación modificada de Horwitz. De los dicho
anteriormente la reproducibilidad intra-laboratorio esta dentro de lo recomendado con un CV de
2,51% para 100 ppb.
La reproducibilidad Inter-analista entrega un coeficiente de variación de 1% lo cual esta dentro
de lo recomendado por la ecuación de Horwitz.
Finalmente las muestras reales a las que se les cuantificaron el contenido de acrilamida, dieron
resultados acorde a los datos que se encuentran en la literatura.

42
7. CONCLUSION
En conclusión se puede decir que se logró una respuesta total a los objetivos planteados siendo
Implementado en el Centro de Investigación y Desarrollo en Grasas y Aceites (CIDGRA),
Departamento de Ciencia de los Alimentos y Tecnología Química de la Facultad de Ciencias
Químicas y Farmaceúticas de la Universidad de Chile, la metodología analítica para la
extracción de acrilamida en papas chips y su cuantificación por HPLC MS/MS. Se determino
con éxito la precisión y exactitud y se aplicó la metodología para el análisis de papas fritas chips
comerciales y elaborados en el laboratorio dentro del Proyecto Heatox 506820.

43
8. BIBLIOGRAFIA
Ahn JS, Castle L, Clarke DB, Lloyd AS, Philo MR, Speck DR. 2002. Verification of the findings of acrylamide in heated foods. Food Addit. Contam. 19, 1116–1124. Becalski, A.; Lau, B.P.Y.; Lewis, D. y Seaman, S., 2003. Acrylamide in foods: Occurrence, sources and modelling. J. Agric. Food Chem 51: 802–808. Becalski, A.; Lau, B.P.Y.; Lewis, D. y Seaman, S., Hayward S., Sahagian M., Ramesh M., y Leclerc Y., 2004. Acrylamide in french fries: Influence of Free Amino Acids and Sugars J. Agric. Food Chem 2004 52, 3801–3806. Biedermann M, Biedermann-Brem S, Noti A, Grob K, Egli P, Mändli H. 2002. Two GC- MS methods for the Analysis of Acrylamide in Foods. Mitt. Lebensm. Hyg 93, 638–652. Bologna LS, Andrawes FF, Barvenik FW, Lentz RD, Sojka REJ. 1999. Analysis of residual acrylamide in field crops. Chrom. Sci 37: 240–244. Castle, L., 1993. Determination of acrylamide monomer in mushrooms grown on polyacrylamide gel. J. Agric. Food Chem 41: 1261–1263. Costa, L.G.; Deng, H., Greggotti, C.; Manzo, L.; Faustman, E.M.; Bergmark, E. y Calleman, C.J., 1992. Comparative studies on the neuro and reproductive toxicity of acrylamide and its epoxide metabolite glycidamide in the rat. Neurotoxicology 13: 219–224. Clarke DB, Kelly J, Wilson LA. 2002. Assessment of performance of laboratories in determining acrylamide in crisp bread. J. AOAC Int. 85, 1370–1373. Dearfield KL, Abernathy CO, Ottley MS, Brantner JH, Hayes PF. 1988. Acrylamide: Its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutation Research 195: 45–77. Dearfield, K.L.; Douglas, G.R.; Ehling, U.H.; Moore, M.M.; Sega, G.A. y Brusick, D.J., 1995. Acrylamide: A review of its genotoxicity and an assessment of heritable genetic risk. Mutation Research 330: 71–99. De Wilde T, De Meulenaer B., Mestdagh F., Govaert Y, Vandeburie S, Ooghe W, Fraselle S, Demeulemeester K, Peteghem CA, Calus A, Degroodt JM, y Verhe R., 2005. Influence of Storage Practices on Acrylamide Formation during Potato Frying. J. Agric. Food Chem. 53: 6550-6557.
Diario Oficial de las Comunidades Europeas 17.8.2002. Consejo en cuanto al funcionamiento de los métodos analíticos y la interpretación de resultados. C(2002)3044. L221/8-L221/36
EPA SW 846, Method 8032A, U.S. Environmental Protection Agency.Washington DC, 1996. FAO/WHO Consultation on the Health Implications of Acrylamide in Food. Summary Report of a Meeting held in Rome, 8–17 Frebuary, WHO, Italy, 2005.

44
Gertz C, Klostermann S, Kochhar P. 2003. Deep frying: the role of water from food being fried and acrylamide formation. Oléagineux Corps Gras Lipides 10 (4) 297-303.
Gutsche B, Weisshaar R, Buhlert J. 2002. Acrylamid in Lebensmitteln—Ergebnisse aus der amtlichen Lebensmittel¨uberwachung Baden-Württembergs. Deutsche Lebensm. Rund 98, 437–443. Hartig, L.; Hummert, Ch.; Buhlert, J.; Von Czapiewski, K. y Schreiber. 2002. Detection of acrylamide in starch enriched foods with HPLC-MS/MS. 17th Symposium on Liquid Chromatography/Máss Spectrometry. Montreux, Switzerland Höfler, F.; Maurer R. y Cavalli, S., 2002. Schnelle Analyse von Acrylamid in Lebensmitteln mit ASE und LC/MS. GIT Labor-Fachzeitschrift 48: 986–970.
IARC. Acrylamide. In IARC Monographs on the Evaluation of Carcinogen Risk to Humans: Some Industrial Chemicals; International Agency for Research on Cancer: Lyon, France, 1994; Vol. 60: 389-433. JIFSAN/NCFST Workshop “Acrylamide in Food, scientific issues, uncertainties, and research strategies” 28–30th October 2002. Rosemont, USA. Disponible en www.jifsan.umd.edu/acrylamide/acrylamideworkshop. html. Jezussek M, Schieberle P. 2003. A new LC/MS method for the quantitation of acrylamide based on a stable isotope dilution assay and derivatization with 2-mercaptobenzoic acid. Comparison with two GC/MS methods. J. Agric. Food Chem. 51: 7866–7871. Lopachin, R.M. y Lehning, E.J., 1994. Acrylamide induced distal axon degeneration. A proposed mechanism of action. Neurotoxicology 15: 247–260. Masson L, Romero N, Castro J, Robert P. 2005. Informe de avance proyecto HEATOX 506820. Vvageningen, Holanda. Mottram DS, Wedzicha BL, Dodson AT. 2002. Acrylamide is formed in the Maillard reaction. Nature 419: 448–449. Nemoto S, Takatsuki S, Sasaki K, Maitani T. 2002. Determination of acrylamide in food by GC/MS using 13C-labelled acrylamide as internal standard. J. Food Hyg. Soc. Japan 43, 371–376. Ono H, Chuda Y, Ohnishi-Kameyama M, Yada H, Ishizaka M, Kobayashi H, Yoshida, M. 2003. Analysis of acrylamide by LC-MS/MS and GC-MS in processed Japanese foods. Food Add. Contam. 20, 215–220.

45
Paleologos, E.K.;Kontominas M.G. 2005 Determination of Acrylamide ande methacrylamide by normal phase high performance liquid chromatography and UV detection. Journal of Chromatography A, 1077 128-135. Petersson E., Rosén J.,Turne Ch., Danielsson R., Hellenäs K., 2006. Critical factors and pitfalls affecting the extraction of acrylamide from foods An optimisation study. Analytica Chimica Acta 557 (2006) 287–295. Rosén, J.; Hellenäs, K. E., 2002. Analysis of acrylamide in cooked foods by liquid chromatography tandem máss spectrometry. Analyst 127: 880-882. Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S. y Törnqvist, M., 2000. Acrylamide: A cooking carcinogen? Chemistry Research Toxicology 13: 517-522. Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S. y Törnqvist, M., 2002. Analysis of Acrylamide, a Carcinogen Formed in Heated Foodstuffs J. Agric: Food Chem. 2002, 50, 4998-5006. Tateo F, Bononi M. 2003. A GC/MS method for the routine determination of acrylamide in food. Italian J. Food Sci. 15, 149–151.
Tekel J, Farkas P, Kovác M. 1989. Determination of acrylamide in sugar by capillary GLC with alkali flame-ionization detection. Food Addit. Contam. 6: 377–381. Tilson HA. 1981. The neurotoxicity of acrylamide: An overview. Neurobehavioral Toxicol Teratol 3: 445–461. USP FN 29. Farmacopea de los Estados Unidos de América ˝USP NF 2006˝. Validación de Métodos Farmacopeicos. Rockville, MD 20852, Estados Unidos de América. 3328-3331. Wenzl T, De la Calle B, Anklam E. 2003. Analytical methods for the determination of acrylamide in food products. A review. Food Addit. Contam. 20, 885–902. Zyzak, D., Sanders, R. A., Stojanovic, M., Tallmadge, D. H., Ebehart, L., Ewald, D. K., Gruber, D. C., Morsch, T. R., Strothers, M. A., Rizzi, G. P., y Villagran, M. D., 2003. Acrylamide formation mechanism in heated foods. Journal of Agricultural and Food Chemistry 51: 4782–4787.

46
9. ANEXOS
Anexo Nº 1: Abreviaturas empleadas.
Muestra Significado
150 V 150º C con pretratamiento al vacío
160 V 160º C con pretratamiento al vacío
170 V 170º C con pretratamiento al vacío
150 N 150º C con pretratamiento temperatura ambiente
160 N 160º C con pretratamiento temperatura ambiente
170 N 170º C con pretratamiento temperatura ambiente
150 CV 150º C sin pretratamiento al vacío (control vacío)
160 CV 160º C sin pretratamiento al vacío (control vacío)
170 CV 170º C sin pretratamiento al vacío (control vacío)
150 CN 150º C sin pretratamiento temperatura ambiente (control TA)
160 CN 160º C sin pretratamiento temperatura ambiente (control TA)
170 CN 170º C sin pretratamiento temperatura ambiente (control TA)
150 VS 150º C con pretratamiento al vacío mas spiked
160 VS 160º C con pretratamiento al vacío mas spiked
170 VS 170º C con pretratamiento al vacío mas spiked
150 NS 150º C con pretratamiento temperatura ambiente mas spiked
160 NS 160º C con pretratamiento temperatura ambiente mas spiked
170 NS 170º C con pretratamiento temperatura ambiente mas spiked
150 CVS 150º C sin pretratamiento al vacío (control vacío) mas spiked
160 CVS 160º C sin pretratamiento al vacío (control vacío) mas spiked
170 CVS 170º C sin pretratamiento al vacío (control vacío) mas spiked
150 CNS 150º C sin pretratamiento temperatura ambiente (control TA) mas spiked
160 CNS 160º C sin pretratamiento temperatura ambiente (control TA) mas spiked
170 CNS 170º C sin pretratamiento temperatura ambiente (control TA) mas spiked
Puré Puré de papas utilizado como blanco
Puré S Puré de papas utilizado como blanco mas spiked
Lays Control en el mercado
Lays S Control en el mercado mas spiked
Spiked Estándar de acrilamida agregado con concentración conocida

47
Anexo Nº 5: Análisis muestras 22 de Noviembre 2006
Muestra Spiked ug/kg n AA ppb SD CV % Puré 0ml/g 3 Puré S 100ml/g 3 103,6 16,5 4,0 150 V 0ml/g 3 1,3 2,2 43,1 160 V 0ml/g 3 2,1 0,5 6,3 170 V 0ml/g 3 4,5 1,0 5,5 150 N 0ml/g 3 5,7 2,5 11,1 160 N 0ml/g 3 2,4 2,1 21,8 170 N 0ml/g 3 28,8 0,7 0,6 150 CV 0ml/g 3 80,0 29,4 8,4 160 CV 0ml/g 3 218,0 8,6 1,0 170 CV 0ml/g 3 267,2 13,6 1,3 150 CN 0ml/g 3 87 24,4 7 160 CN 0ml/g 3 233 106,6 11.3 170 CN 0ml/g 3 763,9 121,7 4,0 150 TV S 100ml/g 3 96,2 1,4 0,4 160 TV S 100ml/g 3 101,3 170 TV S 100ml/g 3 103,2 1,2 0,3 150 TN S 100ml/g 3 104,9 14,8 3,5 160 TN S 100ml/g 3 96,6 11,0 2,9 170 TN S 100ml/g 3 125,2 2,2 0,4 150 CV S 100ml/g 3 178,5 0,0 0,0 160 CV S 100ml/g 3 321,9 40,7 3,2 170 CV S 100ml/g 3 377,1 27,9 1,9 150 CN S 100ml/g 3 594,8 44,6 1,9 160 CN S 100ml/g 3 663,9 45,3 1,7 170 CN S 100ml/g 3 891,6 150,1 4,2 Crisp 118 0ml/g 3 34,9 24,6 17,6 Crisp 118 S 100ml/g 3 129,9 4,4 0,8 Crisp 125 0ml/g 3 286,1 40,0 3,5 Crisp 125 S 100ml/g 3 372,6 38,8 2,6 Picadilli 0ml/g 3 2018,1 90,4 1,1 Picadilli S 100ml/g 3 2023,7 102,7 1,3

48
Anexo Nº 6: Análisis muestras 07 de Agosto 2007
Muestras Spiked ug/kg n AA ppb SD CV%
Puré (Mashed Potatoe) 4 2,8 3,8 33,8 Puré Spiked 100 4 107,9 36,8 8,5 Galleta (Cookies) 4 73,2 22,6 7,7 Galleta Spiked 100 4 178,9 33,7 4,7 Pan marraqueta ( white bread, no loaf) 4 20,3 9,7 11,9 Pan marraqueta Spiked 100 4 121,3 27,8 5,7 Sopaipilla(fried product with mashedpupkin) 4 35,2 7,1 5,1 Sopaipilla Spiked 100 4 136,3 27,4 5,0 Berlín (Fried Wheat dough) 4 20,5 4,6 5,6 Berlín Spiked 100 4 124,5 23,7 4,8 Papas fritas Lays tarro lote 1 (pot.Crisps) 4 589,3 160,9 6,8 Papas fritas Lays tarro lote 1 Spiked 250 4 810,1 229,3 7,1 Papas fritas Lays tarro lote 2 4 545,7 124,4 5,7 Papas fritas Lays tarro lote 2 Spiked 250 4 788,6 253,0 8,0 Papas fritas Kryspo tarro lote1 (like Pringles) 4 164,5 48,1 7,3 Papas fritas Kryspo tarro lote1 Spiked 250 4 421,2 79,3 4,7 Papas fritas Kryspo tarro lote 2 4 382,2 78,2 5,1 Papas fritas Kryspo tarro lote 2 Spiked 250 4 616,9 145,0 5,9 Papas fritas Pringles tarro lote 1 4 793,2 165,1 5,2 Papas fritas Pringles tarro lote 1 Spiked 250 4 1045,3 212,5 5,1 Papas fritas Pringles tarro lote 2 4 570,1 134,0 5,9 Papas fritas Pringles tarro lote 2 Spiked 250 4 784,1 283,4 9,0 Papas fritas Lays artesanales (Pot.crisps) 4 828,1 140,8 4,3 Papas fritas Lays artesanales Spiked 500 4 1313,4 230,8 4,4 Papas fritas Chips Limón (P.crisps lemon) 4 879,4 205,5 5,8 Papas fritas Chips Limón Spiked 500 4 1298,0 63,5 1,2 Papas fritas Pool 1 (P.crisps pool) 4 408,8 43,2 2,6 Papas fritas Pool 1 Spiked 500 4 888,3 263,8 7,4 Papas fritas Pool 2 4 497,9 220,0 11,0 Papas fritas Pool 2 Spiked 500 4 970,2 237,0 6,1 Papas fritas Pool 3 4 315,0 94,2 7,5 Papas fritas Pool 3 Spiked 500 4 827,2 132,9 4,0 Papas fritas Picadilli 4 2159,3 765,8 8,9 Papas fritas Picadilli Spiked 2000 4 3934,7 997,4 6,3


















