







Idiomas
Páginas
Jurídico


Charla 6. Hoy trataremos sobre: Algunos rasgos mendelianos simples en algunas genealogías La Herencia multifactorial: Modelo umbral. Estudios con gemelos. Impronta genética: Síndrome de Prader Willi y Síndrome de Angelman:
mecanismos genéticos y técnicas de análisis Síndrome del X-Frágil
Genética y Envejecimiento celular y orgánico. Algunos tipos de enfermedades genéticas de envejecimiento prematuro.
GENÉTICA DEL DIAGNÓSTICO PRENATAL

Prognatismo (dominante)
Felipe IV (Velázquez) Carlos II (Coello) Alfonso XIII (Casas)
RASGOS MENDELIANOS (MONOGÉNICOS) EN EL HOMBRE

La endogamia acabó con los Austrias * Los matrimonios emparentados durante generaciones provocaron alteraciones genéticas * Carlos II sufrió esa situación en forma de diversas enfermedades como la hidropesía Carlos II, el Hechizado, fue el último rey de la dinastía de los Habsburgo que gobernó en España y su muerte en 1700 dejó paso a los Borbones.

Hoyuelo en el mentón
con hoyuelo sin hoyuelo
M > m
RASGOS MENDELIANOS
(MONOGÉNICOS) EN EL HOMBRE
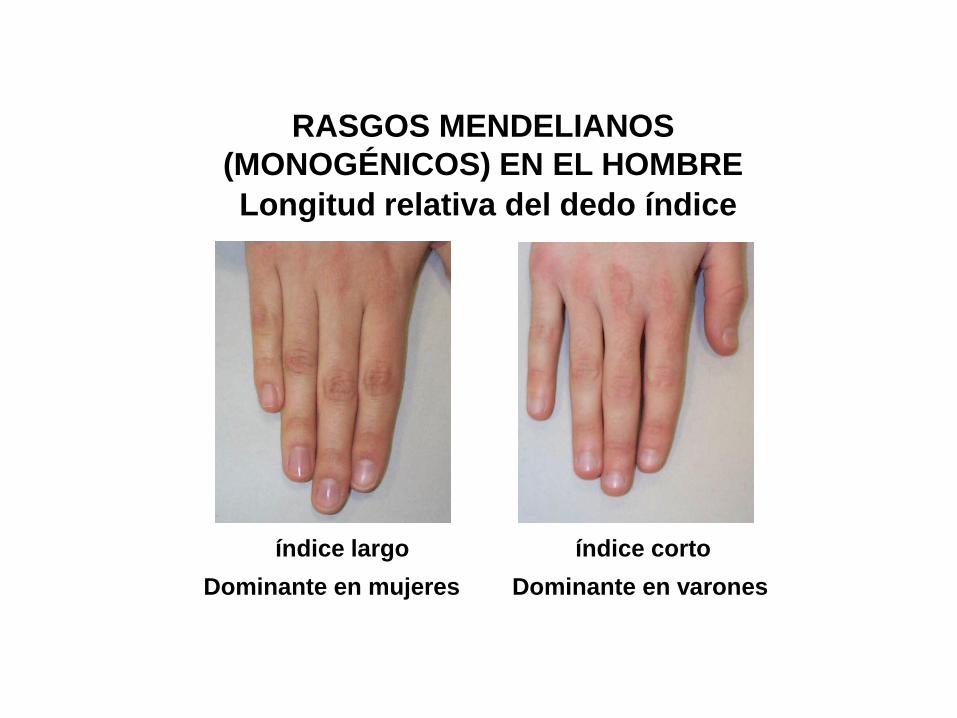
Longitud relativa del dedo índice
índice largo índice corto
RASGOS MENDELIANOS
(MONOGÉNICOS) EN EL HOMBRE
Dominante en mujeres Dominante en varones

Genética multifactorial
100%
genético
100%
ambiental
diabetes
Tipo 1
Fibrosis
quística
Pierna
rota

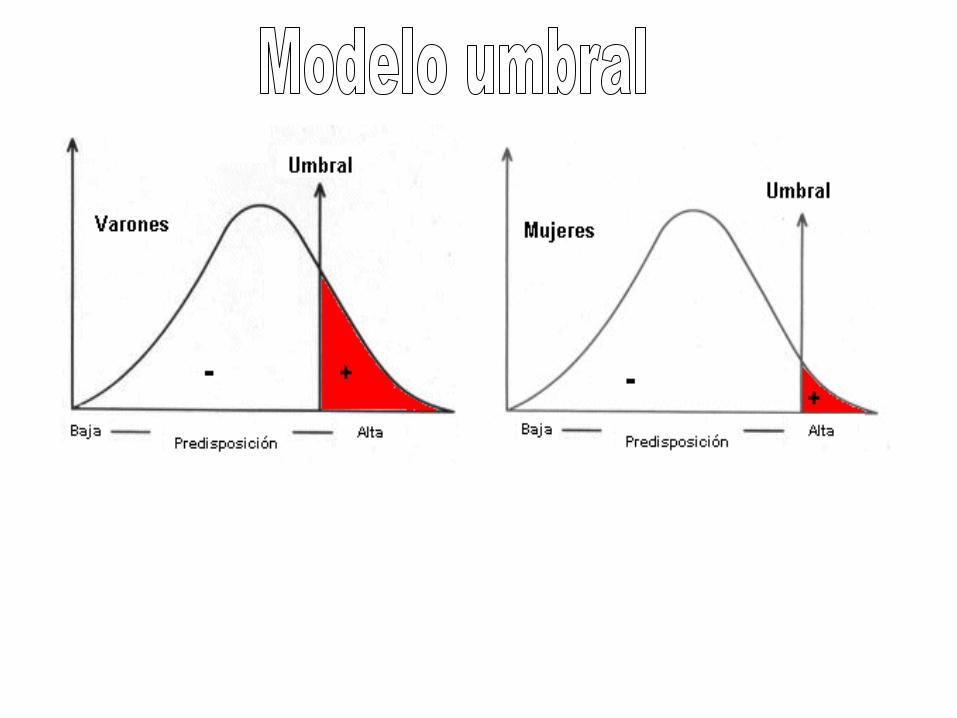
Modelo del Umbral

la comparación del grado de concordancia para el rasgo en estudio en uno y otro tipo de gemelos, permite descubrir el peso de los factores genéticos en la ocurrencia del mismo (ver tabla). Alta concordancia en los gemelos idénticos, indica mayor influencia de los factores genéticos en la determinación del rasgo.
Rasgo
% de
concordancia
en gemelos
idénticos
% de
concordancia
en gemelos
fraternos
Labio y
paladar
hendido
40 5
Estenosis
pilórica 22 4
Pié bot 32 3
Luxación
congénita de
cadera
33 4
ESTUDIOS CON GEMELOS
El Pie Bot o pie Zambo o pie equino varo congénito es una deformidad de los ejes del pie por el cual el mismo no apoya en los tres puntos de apoyo normal
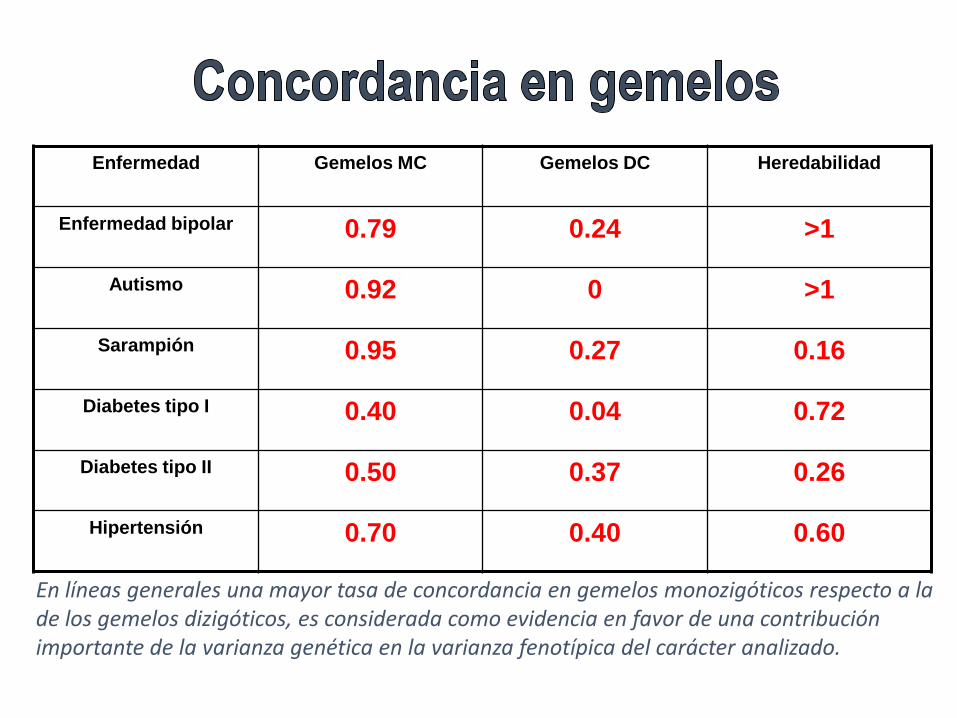
Enfermedad Gemelos MC Gemelos DC Heredabilidad
Enfermedad bipolar 0.79 0.24 >1
Autismo 0.92 0 >1
Sarampión 0.95 0.27 0.16
Diabetes tipo I 0.40 0.04 0.72
Diabetes tipo II 0.50 0.37 0.26
Hipertensión 0.70 0.40 0.60
En líneas generales una mayor tasa de concordancia en gemelos monozigóticos respecto a la de los gemelos dizigóticos, es considerada como evidencia en favor de una contribución importante de la varianza genética en la varianza fenotípica del carácter analizado.

Rasgos multifactoriales en el hombre
Normales
• Estatura
• Peso
• Color de
piel
• CI`?
Patologías
• Enfermedades comunes de la edad adulta:
HTA, Diabetes, Dislipemias,
Arteriosclerosis, Obesidad.
• Enfermedades Autoinmunes
• Asma
• Patologías Psiquiátricas
• Cáncer
• Susceptibilidad a infecciones
• Malformaciones congénitas simples

IMPRONTA GENÉTICA
Síndrome de Prader Willi y S. Angelman

IMPRONTA GENÉTICA
Expresión diferencial de ciertos genes
dependiendo del sexo del progenitor
• Modificación epigenética: alteración reversible de la
cromatina que afecta a la expresión de un gen pero
no a su secuencia de ADN
• Centro de impronta: región del ADN que controla la
impronta de una región cromosómica

Ciertos genes están modificados de distinta manera
dependiendo de su origen materno o paterno
IMPRONTA GENÉTICA
Gen ON
Gen OFF
IMPRONTA GENÉTICA

A*
B XY
A
B*
Hombre A
B* Y
A*
B X
Gametos
Eliminar la impronta anterior y establecer
un nuevo marcado específico de sexo
Gametogénesis
A
B XY
A
B
Hombre A
B*
Gametos
A
B*
X
Y
A
B* X
A*
B X
A*
B XX
A
B*
Mujer
A*
B X
A*
B X
XX
Mujer
A
B
A
B
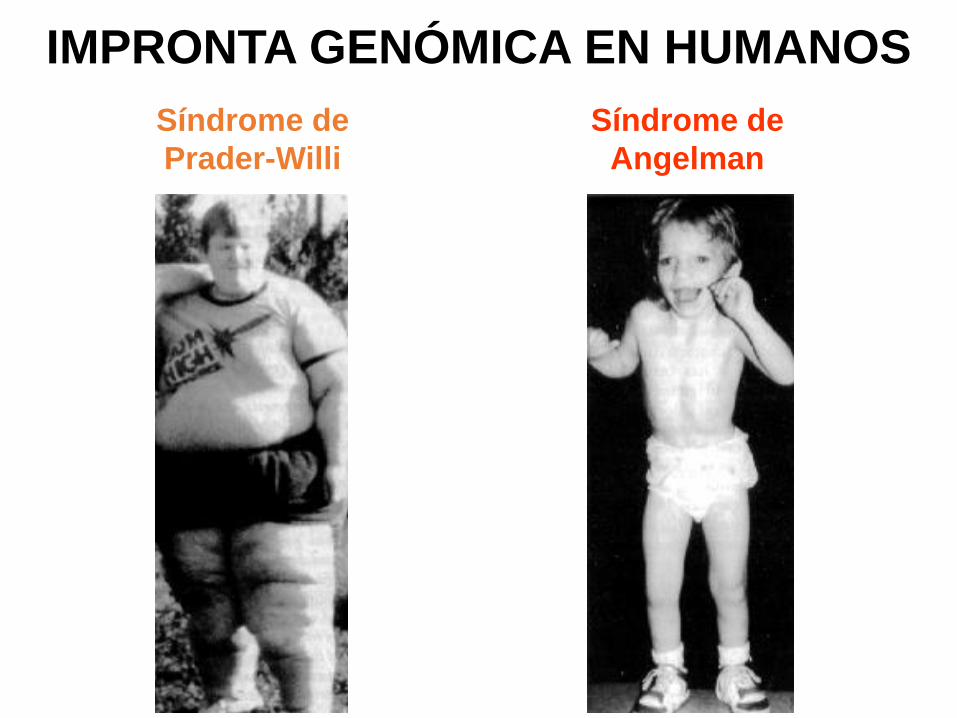
IMPRONTA GENÓMICA EN HUMANOS
Síndrome de
Prader-Willi
Síndrome de
Angelman

SINDROMES DE PRADER WILLI Y ANGELMAN
El síndrome de Prader-Willi y el de Angelman son enfermedades diferentes asociadas con microdeleciones en la misma región del cromosoma 15. Si la microdeleción ocurre en el 15 paterno el fenotipo es el del síndrome de Prader-Willi, mientras que si la microdeleción tiene lugar en el 15 materno, el resultado es un fenotipo de síndrome de Angelman (ver figura)

Tiene dos etapas clínicas típicas y muy diferentes.
• En su primera etapa se caracteriza por: bajo peso al nacer, hipotonía y retraso del crecimiento.
• En la segunda etapa: los pacientes se desarrollan muy bien hasta que aparece la compulsión por la comida con un apetito voraz. Obesidad.
• También presentan retraso mental, que no suele ser severo. Talla baja, manos y pies pequeños e hipogenitales
• Ocurre en 1 de cada 15000 personas, en ambos sexos y en cualquier raza
• nariz fina, boca con los extremos hacia abajo, ojos almendrados, frente prominente y diámetro bifrontal disminuido, escroto hipoplásico, criptorquidia bilateral, pene pequeño o hipoplasia de los labios menores. Durante el primer año de vida se observa retraso del desarrollo sicomotor y la hipotonía va mejorando. Suelen sentarse alrededor de los 12 meses y caminar a los 24 meses. El lenguaje es tardío y se desarrolla entre los 18 meses y los 5 años.


Síndrome de Angelman El síndrome de Angelman se caracteriza:
• Ataxia (carencia de la coordinación de movimientos musculares)
• Afasia (trastorno, defecto o pérdida de la facultad de expresión hablada, escrita o mímica)
• Sonrisa mantenida sin causa y ataques paroxísticos de risa, movimientos anormales de las extremidades
• Hiperactividad
• Crisis convulsivas de cualquier tipo, que aparecen normalmente antes de los 3 años de edad, rasgos faciales inusuales del tipo de: microcefalia (cabeza anormalmente pequeña)
• Macrostomía (orificio bucal grande), mandíbula grande y protusión de la lengua y anomalías musculares.
• Estrabismo (desviación de uno de los ojos de su dirección normal, por lo que los ejes visuales no pueden dirigirse en un mismo tiempo al mismo punto) en el 30-60% de los casos e hipopigmentación de la piel y los ojos
• Retraso mental grave aunque no progresivo
• Retraso psicomotor (retraso en la adquisición de las habilidades que requieren la coordinación de la actividad muscular y mental) severo.

• La frecuencia de la enfermedad se estima en 1 por cada 12.000-20.000 nacidos vivos
• El diagnóstico clínico se realiza entre los 3 y los 7 años de edad, cuando se hacen evidentes los rasgos clínicos característicos de la enfermedad
• Las convulsiones parecen disminuir con la edad y la esperanza de vida no parece estar significativamente acortada, estando descritos numerosos casos de supervivencia en la cuarta década.
• El tratamiento es sintomático usándose anticonvulsivos, mientras duren las convulsiones. La fisioterapia es útil en general para mejorar la estabilidad de la marcha
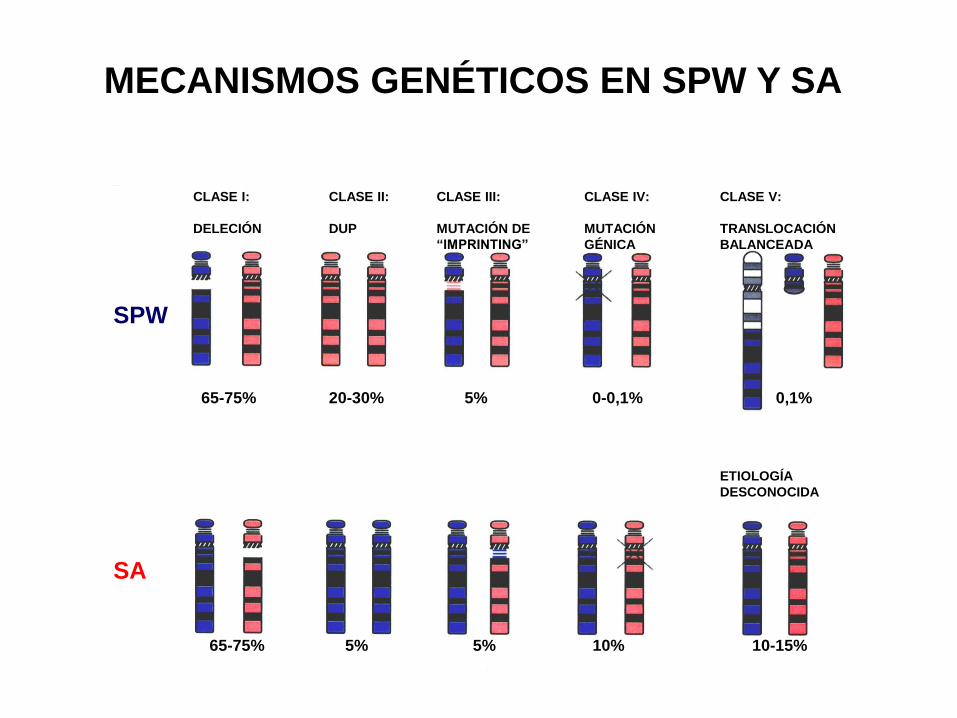
65-75% 20-30% 5% 0-0,1% 0,1%
65-75% 5% 5% 10% 10-15%
MECANISMOS GENÉTICOS EN SPW Y SA
CLASE I:
DELECIÓN
CLASE II:
DUP
CLASE III:
MUTACIÓN DE
“IMPRINTING”
CLASE IV:
MUTACIÓN
GÉNICA
CLASE V:
TRANSLOCACIÓN
BALANCEADA
ETIOLOGÍA
DESCONOCIDA
SPW
SA
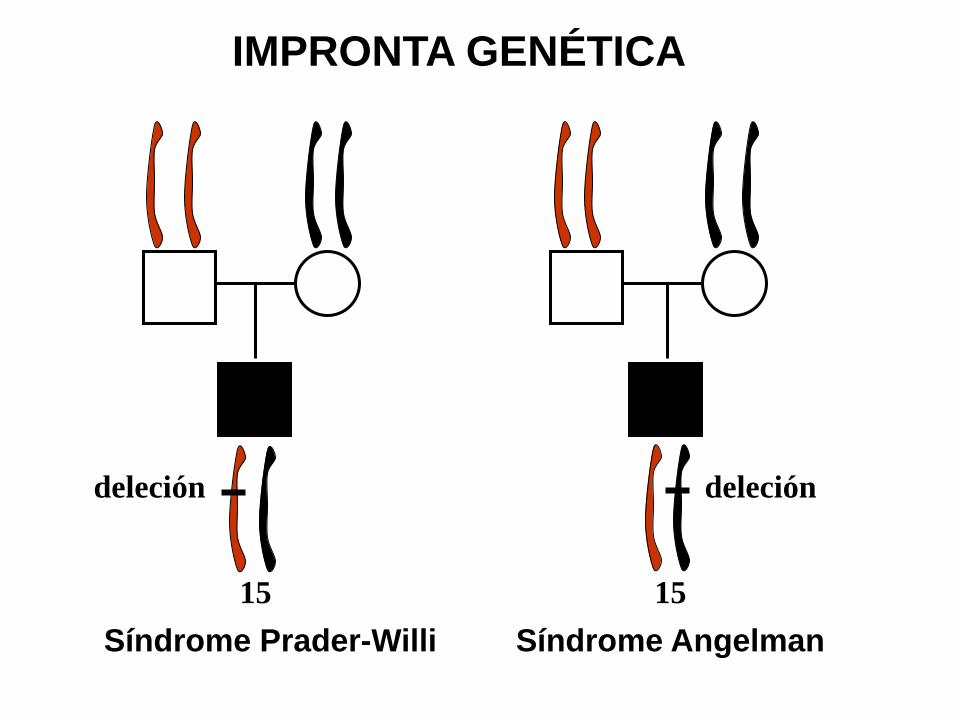
deleción deleción
15 15
Síndrome Prader-Willi Síndrome Angelman
IMPRONTA GENÉTICA

DISOMÍA UNIPARENTAL: Las dos copias de un par
cromosómico proceden del mismo progenitor y el
otro no aporta ninguno
Prader-Willi
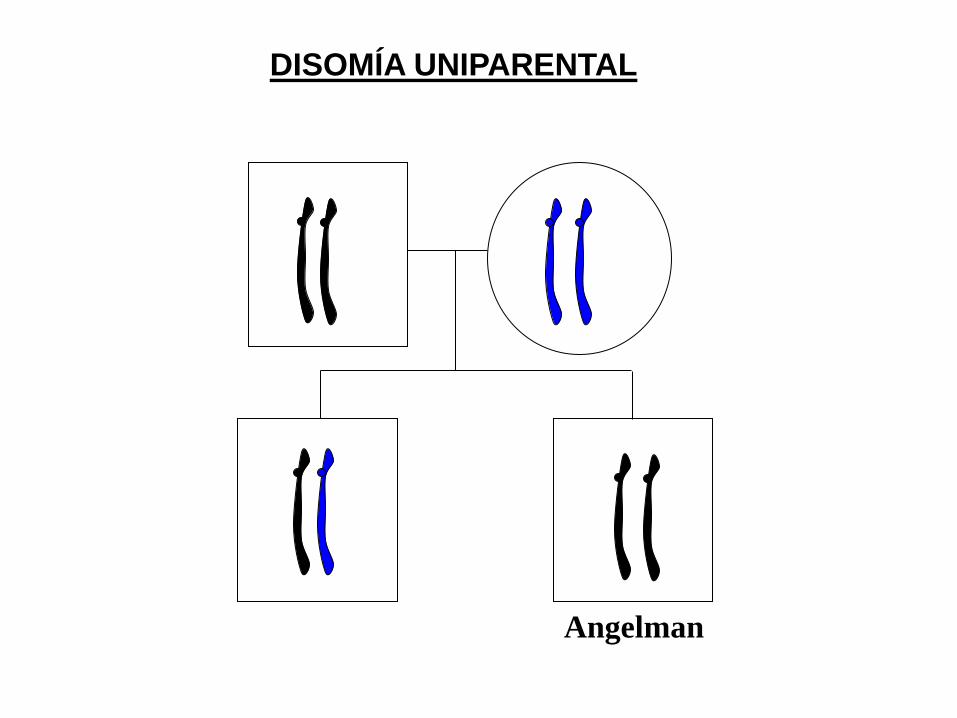
DISOMÍA UNIPARENTAL
Angelman


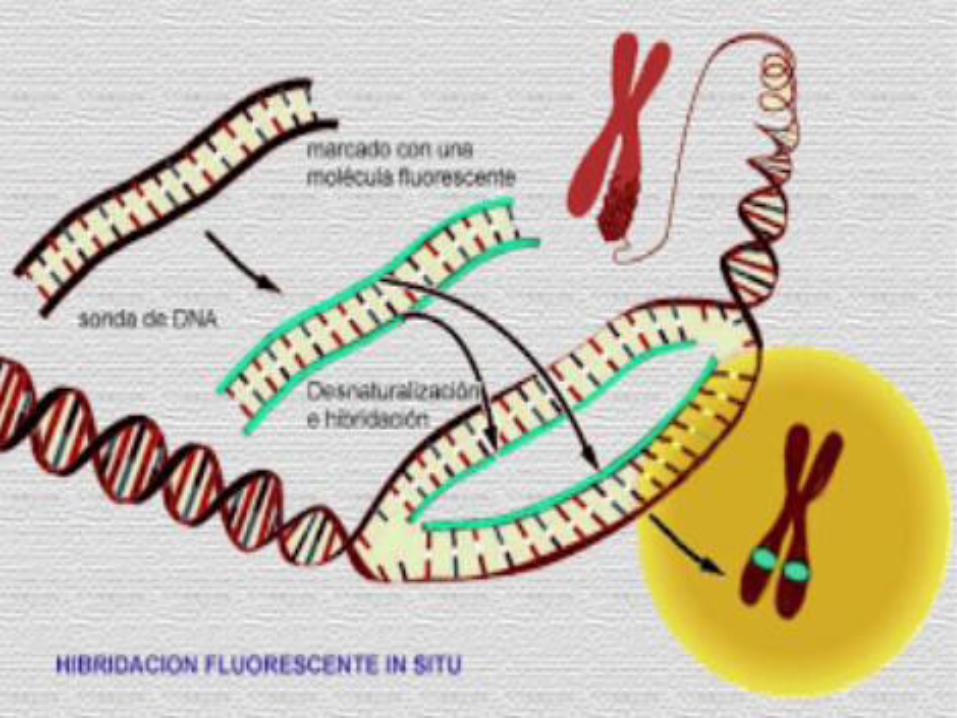

1 2 3 4 5 6 7 8 9 10 11
Banda materna
Banda paterna
1 2 3 4 5 6
Banda materna
Banda paterna
SPW
SA
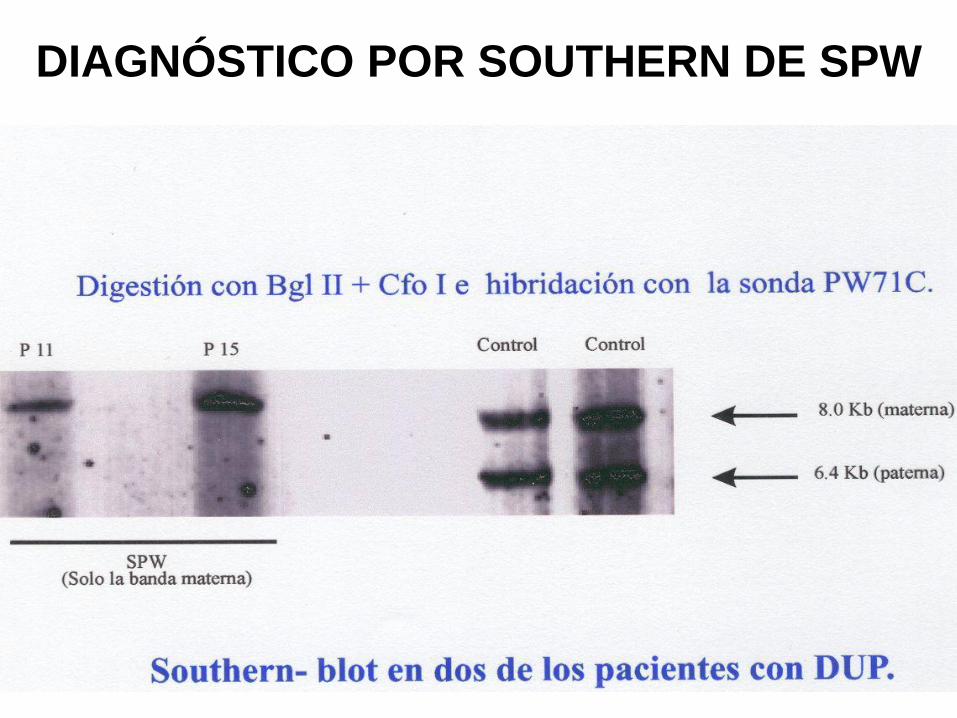
DIAGNÓSTICO POR SOUTHERN DE SPW

Síndrome Deleción Cromosómica
Prader-Willi 15q 11.13 paterno
Angelman 15q 11.13 materno
Langer-Giedion 8q24
DiGeorge 22q11
Smith-Mangenis 17p11.2
Williams 7q1
Aniridia/tumor de Wilms 11p13
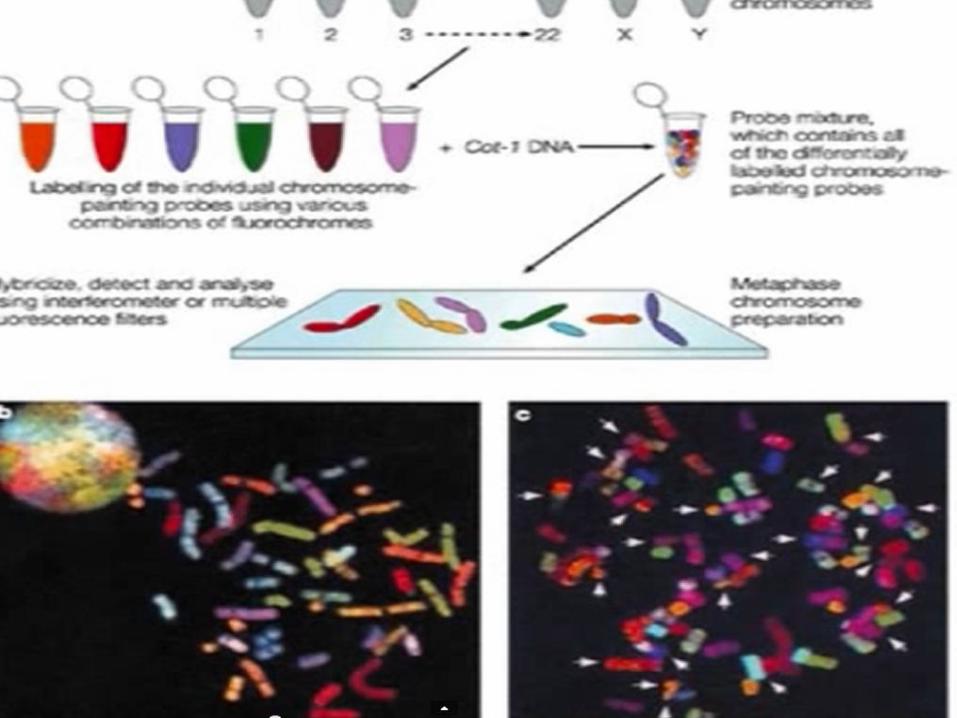
Síndromes por microdeleción

En 1991 se descubrió el gen (llamado FMR1) que causa el X frágil abriendo las puertas al campo de la investigación médica y psicopedagógica. Las aportaciones más importantes han sido la mejora en el diagnóstico prenatal y la identificación de portadores y afectados mediante un análisis de sangre efectuado por un equipo especializado.
Aproximadamente el 15 al 30% de los niños con X frágil tienen autismo y aproximadamente el 6% de los varones autistas tienen el síndrome de X frágil. Las niñas con X frágil usualmente presentan timidez, ansiedad social, dificultades con las matemáticas en la escuela, y problemas de atención.
SÍNDROME X-FRAGIL

En varones: En los recién nacidos las características físicas que más destacan son: Macrocefalia (mayor perímetro craneal) Orejas grandes y/o separadas y, en algunos casos, prolapso de la válvula mitral. Las orejas casi nunca son deformes, sin embargo muestran una hendidura en la parte superior del lóbulo. En el niño los rasgos que destacan, además de los anteriores, son: Cara alargada y estrecha, estrabismo, paladar ojival (alargado y muy arqueado), laxitud articular y pies planos. En el joven, la macrocefalia no suele ser evidente, la cara alargada y estrecha con la mandíbula inferior saliente y paladar ojival, con dientes apelotonados. El macroorquidismo (aumento del tamaño de los testículos) se empieza a hacer evidente con la llegada de la pubertad y se cree que puede ser debido a la estimulación de las gonadotropinas. La laxitud articular es mas frecuente en las articulaciones de los dedos produciéndose una hiperextensibilidad que se detecta al doblar los dedos hacia atrás en dirección a los nudillos produciéndose un ángulo de 90º o superior, aunque también ocasiona debilidad en otras articulaciones como el tobillo o la muñeca. En el varón adulto la macrocefalia ya no se detecta, continúan las orejas grandes y/o prominentes, mandíbula inferior saliente, paladar ojival y dientes apelotonados. La laxitud articular continua en igual proporción que en el varón joven y aumenta en frecuencia el macroorquidismo. El 80% de los varones adultos presentan prolapso de la válvula mitral (en ocasiones se produce una regurgitación de la sangre a través de la válvula durante la sístole).

En mujeres:
Las mujeres son menos fáciles de identificar por los rasgos físicos típicos, ya que tienen la cara larga y estrecha y las orejas grandes, asociado con el retraso en el aprendizaje o leve retraso mental. También se suelen dar, a veces, hiperextensibilidad en las articulaciones, paladar ojival y prolapso de la válvula mitral. Detección y análisis del X-Frágil En el estudio citogenético (cariotipo) se pueden detectar los sitios «frágiles», folato sensibles, que se expresan en el extremo distal del brazo largo del cromosoma X (Xq27.3) en los individuos afectados (Fig. 1). (Comentar técnica de inducción a la fragilidad cromosómica).
el sitio frágil en Xq27.3

¿¿GENES Y ENVEJECIMIENTO??

Pangeria, Progeria del adulto, síndrome de Werner
• Progeria, síndrome de Hutchinson-Gilford
• Poiquilodermia congénita, síndrome de Rothmund – Thomson
• Síndrome de Cockayne
• Acrogeria, síndrome de Gottron
• Acrometageria
• Metageria
• Dermatosis escleroatrófica y queratósica, síndrome de Huriez
• Xerodermia infantil síndrome de Variot- Cailleau
• Xerodermia osteodisplásica, enanismo tipo Walt Disney
• Síndrome de Store
• Síndrome de Yunis-Varon
• Síndrome de Hoepffner- Dreyer-Rudiger
• Síndrome de Flynn-Aird
• Síndrome de Garit Entre otros pocos más

Aún falta mucho por comprender acerca del proceso de envejecimiento, pero
podemos llegar a la conclusión que existe fuerte evidencia de un control genético del
proceso de envejecimiento, tanto a nivel celular como del organismo en su totalidad.
"El secreto de como prolongar la vida está en el arte de aprender como no
acortarla".
"Más importante que añadir más años a la vida, es dar más vida a los años".
Estamos lejos de encontrar la "fórmula de la eterna juventud".
"El envejecimiento parece ser la única manera posible de vivir mucho tiempo". Daniel-Franois-Esprit Auber (1782-1871), compositor francés
"Cuarenta es la vejez de los jóvenes; cincuenta es la juventud de la vejez". Proverbio francés.

La progeria es un trastorno genético que causa un rápido envejecimiento en los niños, a partir de los dos primeros años de vida.
Síndrome de progeria Hutchinson-Gilford (HGPS), 1 de cada 4 millones de nacidos vivos
La esperanza media de vida para un niño con progeria es de 13 años, pero algunos mueren más jóvenes y algunos viven 20 años o más

Enfermedad genética extremadamente rara (1:4 a 8x106 nacimientos).
Descrita en 1886 Hutchinson y 1897 por Gilford.
Causada por mutación de novo puntual en posición 1824 de gen LMNA
rara vez heredada. LMNA codifica para laminina nuclear que organiza a
la cromatina y dá soporte al núcleo.
Mutación ocurre durante desarrollo embrionario.
Caracterizada por envejicimiento precoz acelerado a etapas muy
tempranas de la infancia.
Presentan tempranamente esclerodermia, retraso del crecimiento,
alopecia, fascies característica.
En etapas tardías presentan piel arrugada, aterosclerosis, falla renal,
presbiacusia, pérdida de la visión y problemas cardiovasculares.
Pocos sobreviven a los 13 años de edad.
Síndrome de Hutchinson-Gilford (progeria)

“la mayoría de pacientes afectados de Progeria presentan una mutación en el exón 11 del gen LMNA, que provoca la producción de una proteína mutada que se denomina progerina. La mutación ocurre en una de las dos copias del gen LMNA que hay en cada célula, por lo que los pacientes producen tanto Lamina A como progerina”.
una sustitución silenciosa de una citosina por una timina en el nucleótido 1824 que activa un sitio de empalme critico cerca al extremo carboxi terminal, lo que origina una proteína truncada con pérdida de 50 aminoácidos
es decir no se codifica la lamina A (72KDa) sino la progerina (67KDa), que se presume incapaz de mediar las funciones de la lamina A y C dentro de la conformación de la membrana nuclear y se ha identificado como causa de la pérdida de la forma y estabilidad en fibroblastos y otras células de los pacientes con progeria Estructura nuclear normal de célula sana
(arriba) frente a desorganizada de célula S. Progeria (abajo)
Top Related