







Idiomas
Páginas
Jurídico


TEORIA DE FUNCIONALES
DE LA DENSIDAD
Simone Barbonetti
Guadalupe Yazmin Rosas Pinon
Juan Manuel Vargas Alcaraz
Blanca Angelica Vega Alanis
1

¿QUE ES LA TEORIA DEL FUNCIONAL DE LA DENSIDAD?
La Teoría del Funcional de la Densidad (DFT,
por sus siglas en inglés), aplicada a sistemas
electrónicos, es un procedimiento variacional
alternativo a la solución de la ecuación de
Schrödinger, donde el funcional de la energía
electrónica es minimizado con respecto a la densidad
electrónica.
2

FECHAS IMPORTANTES EN EL DESARROLLO DE DFT
1920s: Modelo de Thomas-Fermi
1951: Método X-α de Slater
1964: Teoremas I y II de
Hohenberg-Kohn
1965: Método de Kohn-Sham
∼1980: Dificultades numéricas
resultas: LDA en la química
1988: GGAs, DFT se vuelve útil
1993: DFT en el paquete de
programas Gaussian
1998: Walter Kohn premio Nobel
de química 3

Los métodos tradicionales dentro de las teorías
de la estructura electrónica de la materia, en
particular la teoría de Hartree-Fock y los
derivados de este formalismo, se basan en una
función de onda multielectrónica.
Si bien esta resolución de la
ecuación de Schrödinger
permite describir de forma
exacta el comportamiento de
los sistemas muy pequeños, su
capacidad de predicción se ve
limitada por el hecho de que sus ecuaciones son
demasiado complejas de resolver numéricamente
o menos aun analíticamente. 4

PRIMEROS INTENTOS
Enrico Fermi y Llewellyn Thomas en los años 20
pensaron en la energía cinética como función de
la densidad usando las expresiones clásicas de
interacciones electrón-electrón y electrón-núcleo.
Paul Dirac en el 1928 añade un funcional de
energía de intercambio mejorando la teoría pero
dejándola todavía demasiado imprecisa.
5

LA TEORIA DE FUNCIONALES DE DENSIDAD
La base teórica para la DFT fue dada en 1964 por
Hohenberg y Kohn, quienes mostraron que la energía
es un funcional de la densidad y que además la
densidad del sistema minimiza este funcional.
6

Otro desarrollo más importante fue dado el 1965,
cuando Kohn y Sham presentaron una forma de
aproximar al funcional universal. Para lograr
este propósito, Kohn y Sham recurrieron a un
sistema ficticio el cual está constituido por un
sistema de electrones no interactuantes.
El sistema se representa con un determinante
(Slater) cuyos elementos son funciones que
representan a cada uno de los electrones del
sistema (orbitales).
Así la energía cinética corresponde a una suma
de energías cinéticas individuales y la densidad
electrónica a la suma de densidades orbitales.
7

La DFT se volvió muy popular para cálculos de
física del estado sólido ya desde los años ’70, pero
no lo bastante precisa para química cuántica
hasta los ‘90, cuando se refinaron en gran medida
las aproximaciones usadas en la teoría. Ahora la
DFT es un método fundamental para los cálculos
de estructura electrónica en ambos campos.
8

FUNDAMENTO TEÓRICO
ψ (n electrones, 3n coordenadas espaciales y
n coordenadas de espín) deriva a una
función de la densidad electrónica libre de
espín, ρ(1)
Hohenberg y Kohn encontraron que existe
una relación entre E0 y ρ0 de modo que:
9

≡
1) ≡
2) problema cíclico,
volver a ab initio
Problemática
10
Dos potenciales externos no podrían dar
un mismo valor de ρ0
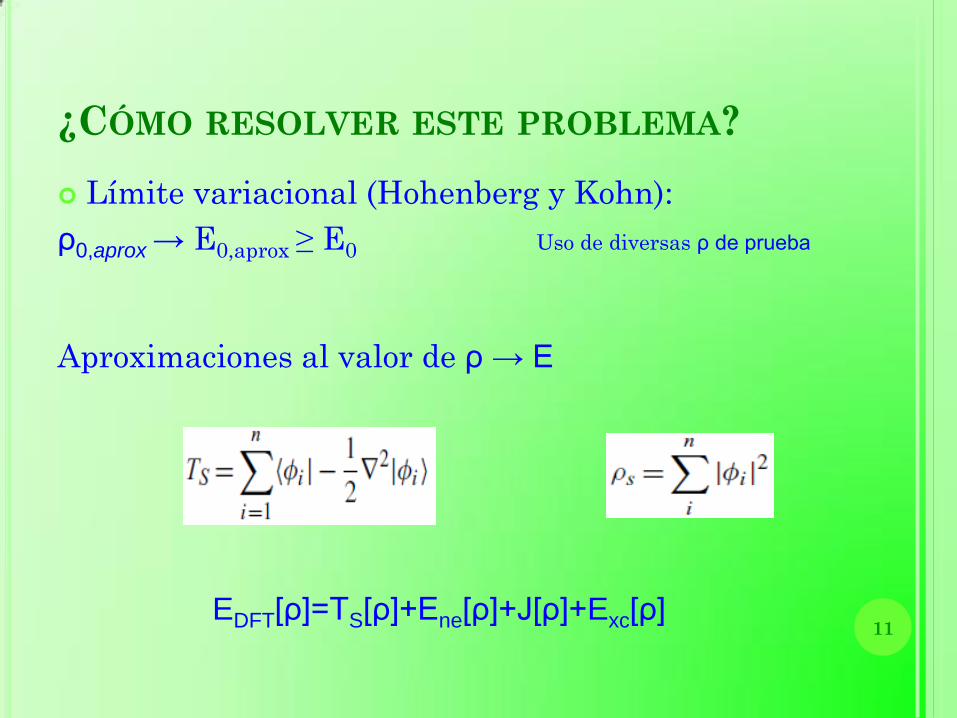
¿CÓMO RESOLVER ESTE PROBLEMA?
Límite variacional (Hohenberg y Kohn):
ρ0,aprox → E0,aprox ≥ E0 Uso de diversas ρ de prueba
Aproximaciones al valor de ρ → E
EDFT[ρ]=TS[ρ]+Ene[ρ]+J[ρ]+Exc[ρ] 11

La calidad de los resultados DFT depende de cómo se defina el término Vxc
Generalized Gradient Approximation (GGA)
Toma en cuenta la densidad electrónica r y su gradiente (variación de
la densidad con la posición.
Funciona bien para sólidos y para moléculas
drxgrEGGA
X
3/4r
3/4 rrx
Local Density Approximation (LDA)
La densidad se modela como la de un gas electrónico homogéneo con
densidad electrónica r Funciona bien para sólidos pero no para moléculas
drrcE X
LDA
X
3/4r
(subestima 10%) 3/1
4
3
2
3
Xc
Si: 3/1
4
3
2
3
xg
LDA
X
GGA
X EE 12

B3LYP: Funcional Híbrido
VWN
C
LYP
C
Becke
X
HF
X
Slater
X ECCEBEEAAE 11 88
A, B y C son parámetros obtenidos empíricamente de modo que los
resultados ajusten 56 energias de atomización, 42 potenciales de
ionización, 8 afinidades protónicas, y 10 energías atómicas
correspondientes a elementos de la primera fila
A=0.80, B=0.72 y C=0.81
13

APLICACIONES Y COMPARACIÓN CON OTROS
MÉTODOS
VENTAJAS APLICACIONES DESVENTAJAS
Las ecuaciones son
más simples de resolver
que las ecuaciones de
muchos cuerpos de
mecánica cuántica u
otras aproximaciones.
Permiten tratar
sistemas más grandes y
calcular más
propiedades.
En principio es una
teoría exacta; solo se
puede aplicar de forma
aproximada, lo que
hace que sus
resultados sean menos
precisos que otros
métodos.
Diferentes
aproximaciones a la
energía de intercambio
y correlación pueden
dar resultados
diferentes. 14

VENTAJAS APLICACIONES DESVENTAJAS
Para muchos métodos más
sofisticados se utiliza como
punto de partida los resultados
de DFT
Los resultados son muy
satisfactorios, y que, por su
bajo costo computacional, es la
única forma de abordar
sistemas más allá de cierta
complejidad.
Sus detractores apuntan a
que es un método
semiempírico más, y que no es
tan fiable como los métodos ab
initio «clásicos».
En física se le considera
método ab initio .
No se requiere ningún tipo
de parámetro adicional ni
ajuste obtenido de resultados
experimentales.
En química por el contrario,
suele guardarse el término ab
initio para métodos derivados
de la teoría cuántica de
muchos cuerpos que por lo
general son más precisos, más
costosos computacionalmente
y cuyo nivel de aproximación
puede ser ajustado.
15

Ventajas con respecto a
cálculos ab-initio
Desventajas con respecto a
cálculos ab-initio
La función de onda de un sistema de N
electrones tiene 3N variables.
La densidad electrónica tiene 3 variables
(x,y,z) para cualquier sistema sin importar
su tamaño DFT reduce
considerablemente el costo computacional
(N3 máx, MP2: N4, CI: N6 ó N7).
Se obtienen propiedades de significado
químico como electronegatividad
(potencial químico) y dureza (blandura).
No existe una forma universal conocida
para expresar la E como funcional de la
densidad. La estrategia a seguir es emplear
aproximaciones, pero no hay un modo
sistemático de mejorar los resultados
obtenidos (a diferencia de ab-initio).
DFT está formulada para sistemas en su
estado base, por lo que su extensión a
estados excitados no es obvia.
16

OTRAS APLICACIONES DEL MÉTODO DE
DFT
Deducción de estructuras moleculares y cálculo de frecuencias
vibracionales
Cálculo de diversas energías, como puede ser energía de ionización,
afinidad electrónica, energía electrónica de excitación
Cálculo de frecuencias poblacionales, momentos dipolares,
Intensidades de IR y espectroscopía RAMAN
Cálculo de desplazamientos químicos en RMN y constantes de
acoplamiento nucleares spin-spin
Modelado de cúmulos de agua y otros sistemas enlazados por
puentes de hidrógeno.
17
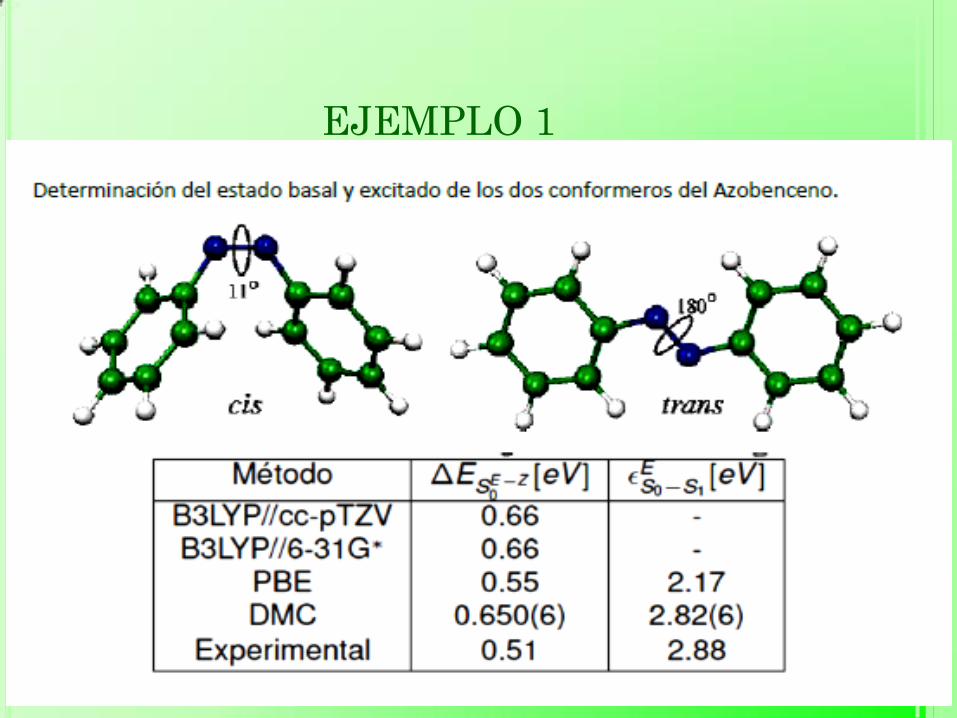
EJEMPLO 1
18

Dureza y blandura:
La primera derivada del potencial químico se conoce como dureza absoluta
de una especie química (η) y su inverso como blandura (S):
2
2
1 1 1
2 2 2
ES
N N
2
IP AE
Desde un punto de vista práctico puede obtenerse como:
Y para aislante o semiconductores la expresión anterior aproxima a:
2
LUMO HOMOE E
Desde un punto de vista químico las derivadas primera y segunda de
la energía son las que revisten mayor importancia
La Dureza Química es una propiedad global del sistema y mide la
resistencia impuesta por este al cambio en su distribución electrónica.
En este contexto la dureza es un descriptor de la reactividad.
19

Dureza y Blandura:
Para valores enteros de N es necesario usar la aproximación de diferencias
finitas ya que la derivada de la energía con respecto a N es discontinua:
donde I = {E(N-1) – E(N)} es el primer potencial de ionización;
A = {E(N+1)-E(N)} es la afinidad electrónica.
1
2I A
1
2S
20

21
Referencias bibliográficas
Fiolhais C., Noguera F., Marques M. (2003) A Primer in Density Functional
Theory. Editorial Springer, Alemania.
Scholl D., Steckel J. (2009) Density Functional Theory: A practical
introduction. Editorial Wiley, E.U.A.
Dubeck´y M., Derian R., Mitas L., Stich I. (2010) Ground and excited electronic
states of azobenzene: a quantum Montecarlo study. Journal of Chemical
Physics, 133(24): 244-301.
Hongo K., Watson M., Sánchez-Carrera R., Iitaka T., Azpuru-Guzik A. (2010)
Failure of the conventional density functionals for the prediction of
molecular cristal polymorphism: A quantum Montecarlo study. Journal of
Physical Chemical Letters, 1(12): 1789-1794
Koch W., Holthausen M. (2001) A chemist´s guide to density functional theory.
Editorial Wiley, 2ª edición, Alemania.
Levine I. (2001) Química cuántica. Editorial Pearson Educación, 5ª Edición,
España.
Top Related