
análisis de proteínas
24
Análisis de Proteínas Electroforesis, Transferencia e Inmunodetección advansta
-
Upload
violeta-hp -
Category
Documents
-
view
237 -
download
1
Transcript of análisis de proteínas

Análisis de Proteínas Electroforesis, Transferencia e Inmunodetección
advansta

Western Blot es una técnica analítica, ampliamente utilizada, para el estudio de proteínas.
Este método, descrito por primera vez por Towbin, et. al1, permite la detección de una sola
proteína dentro de una muestra biológica. La especificidad de Western Blot se logra me-
diante la utilización de un anticuerpo que reconoce y se une a un epítopo único de la pro-
teína de interés.
Con la técnica de Western Blot se puede estimar el tamaño de una proteína, confirmar la
presencia de modificaciones post-traduccionales como la fosforilación,
y ser utilizado para comparar cuantitativamente los niveles
de proteína entre muestras.
Esta guía de laboratorio describe los pasos
involucrados en la realización de un Western
blot, incluyendo la preparación de muestras,
la separación de proteínas, la transferencia y
la detección.
Análisis de Proteínas
Análisis de Proteínas: Electroforesis, Transferencia e Inmunoprecipitación.
Página 1
Un manual de laboratorio de la empresa Advansta
1. Towbin, H. et. al. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some appli-
cations. Proc Natl Acad Sci U S A. 76(9):4350-4.
Índice de Contenidos Página
1. Western Blot: Visión General 2
2. Preparación de las muestras 4
3. Electroforesis en gel de Poliacrilamida 5
4. Transferencia Electroforética 11
5. Hibridación Anticuerpos 13
6. Detección 15
7. Solución de Problemas 17
8. Herramientas y Referencias Técnicas 21

Consulte los capítulos siguientes para obtener más detalles acerca de cada paso.
1. Western Blot: Visión General
Página 2
Western Blot: Visión General
a. Preparación de la muestra
Extracción de las proteínas de
las muestras biológicas me-
diante disrupción mecánica o
química.
Los materiales de partida incluyen
planta, tejidos animales, células
cultivadas, levadura, o bacterias.
La disrupción mecánica
con un homogenizador
disgrega los tejidos.
Procesos adicionales
logran el fraccionamien-
to subcelular
Se usan tampones con-
teniendo detergentes
para la lisis celular
Figura 1. Preparación de muestras
b. Electroforesis en gel de
Acrilamida
Las proteínas en el extracto se
separan de acuerdo a su ta-
maño mediante electroforesis.
Se prepara un gel de acrilami-
da, bisacrilamida.
El dodecilsulfato de sodio
(SDS) añadido al gel se une a
las proteínas y confiere, de
forma proporcional a la masa
de cada proteína, una carga
negativa a cada una de ellas.
Figura 2. Electroforesis en Acrilamida
Se añade un coloran-
te de carga. Así, la
migración de la mues-
tra se puede monitori-
zar.
Se utiliza una pipeta para
cargar las muestras en los
pocillos del gel Una fuente de ali-
mentación propor-
ciona el voltaje
El gel se coloca dentro de un tanque
de electroforesis lleno de tampón,
que conducirá la corriente. La proteí-
nas con carga negativa migran des-
de el ánodo.
c. Transferencia electroforéti-
ca a una membrana
Las proteínas son transferidas
electroforéticamente a un
soporte rígido o membrana,
dónde quedan inmovilizadas.
El gel y la membrana se coloca
entre papel de filtro y almohadillas
de esponja..
Se aplica voltaje en el
tanque de transferencia y
las proteínas migran desde
el gel a la membrana.
Figura 3. Transferencia de Proteínas a la Membrana
Un cartucho aplica presión y
mantiene un estrecho contacto
entre el gel y la membrana.

a. Hibridación del Anticuerpo
La membrana con las proteí-
nas inmovilizadas debe tratar-
se para bloquear las zonas
con ausencia de proteínas y
así evitar la unión no específi-
ca de los anticuerpos.
A continuación se incuba con
un anticuerpo primario dirigi-
do contra el epítopo específi-
co de la proteína diana.
Tras varios lavados de la mem-
brana, se adiciona un anti-
cuerpo secundario marcado
que se unirá de forma especí-
fica al anticuerpo primario,
proporcionando una forma
de detección.
Figura 4. Hibridación de Anticuerpo
e. Detección de las Bandas
Después de un lavado de la
membrana para eliminar el
anticuerpo no unido, se pro-
cede a detectar la localiza-
ción de la banda en la mem-
brana. Para la detección de
quimioluminiscencia, se utiliza
un anticuerpo secundario
conjugado con el enzima pe-
roxidasa (HRP); la adición de
un sustrato de HRP provoca
una reacción enzimática que
da como resultado final la
emisión de luz. Dicha luz pue-
de ser detectada en una pelí-
cula de rayos X, o de forma
digital, mediante un sistema
de captación de imágenes.
La detección de fluorescen-
cia se basa en la captación
de luz emitid por sustancias
fluoróforas conjugadas con el
anticuerpo secundario.
Análisis de Proteínas
Página 3
Figura 5. Detección quimioluminiscente de las bandas de proteínas
Los materiales de partida incluyen
tejidos vegetales, tejidos animales,
células en cultivo, levadura, o bac-
terias.

Una adecuada preparación de la muestra es
esencial para tener éxito en un ensayo de Wes-
tern Blot. En la mayoría de ensayos, las proteínas
se extraen mediante procesos químicos y/o
mecánicos. El método elegido dependerá del
tipo de muestra de partida, de la localización
subcelular de la proteína y de las condiciones
óptimas que requiera el anticuerpo para recono-
cer el epítopo proteico. En la tabla 1 se resumen
los métodos mecánicos que habitualmente se
utilizan en la extracción de proteínas para inmu-
notransferencia.
A menudo, en muestras tisulares de plantas y ani-
males, se requiere el uso de un proceso mecáni-
co para la disrupción de la compleja matriz celu-
lar. Disrupción mecánica que suele preceder a un
proceso químico de lisis celular, mediante el uso
de solución tampón que contiene detergentes,
que va dirigida a enriquecer la proteína de interés
dentro del extracto final. Las células en cultivo,
frecuentemente, se suelen lisar utilizando una so-
lución tampón con detergente y sin la aplicación
de métodos mecánicos.
La estructura química de los detergentes permite
romper las membranas celulares y solubilizar las
proteínas. Estas substancias tienen una parte po-
lar y otra no polar y se pueden clasificar según las
características del grupo polar: iónicos si el grupo
polar tiene carga positiva o negativa, no iónicos si
no poseen carga o zwiteriónicos si poseen carga
negativa y positiva y la carga neta es 0.
La elección de la substancia detergente, depen-
de en parte de la localización, dentro de la célu-
la, de la proteína de interés, citoplasmática, uni-
da a la membrana, dentro de orgánulos celulares
como el núcleo y las mitocondrias, etc...Las pro-
teínas citoplasmáticas se pueden unir a las proteí-
nas del citoesqueleto, afectando así a los proce-
sos de fraccionamiento subcelular.
En la tabla 2 se clasifican las soluciones tampón y
detergentes, más utilizados habitualmente, según
el tipo de proteína y localización subcelular.
2. Preparación de la muestra
Página 4
Preparación de la muestra
Tabla 1. Métodos físicos para la dirupción de muestras
Método Descripción Tipo de muestra
Licuadora La muestra se tritura
mediante cuchillas
rotatorias
Gran cantidad de
tejido
Homogenizador
Dounce
Tubo de vidrio con
pistón ajustado maja
las células por ac-
ción de corte.
Células tisulares; útil
para protocolos de
enriquecimiento de
proteínas mitocon-
driales o nucleares
Homogenizador de
ultrasonidos
Ondas de sonido
emitidas por una
sonda para romper
las membranas celu-
lares
Células, tejidos, bac-
terias.
Pulverización en
Nitrógeno líquido
La muestra se pulve-
riza utilizando un
mortero y una almi-
rez refrigerados
Tejido animal o o
vegetal
Perlas de vidrio La ruptura celular se
produce por agita-
ción de las perlas de
vidrio
Levaduras
Tipo/Localización
Proteína de interés
Detergente o Solu-
ción Tampón
TComentarios
Nativa (no desnatu-
ralizada)
Detergente suave
no iónico
Evitar detergentes
desnaturalizantes
(SDS, Deoxicolatos)
Citoplasmáticos
(soluble)
Tris-HCl Puede combinarse
con métodos mecá-
nicos, tales como el
homogenizador
Dounce
Citoplasmático
(unida a citoesque-
leto)
Tris-Triton Los detergentes
Triton son suaves y
no iónicos
Membrana mitocon-
drial o nuclear
NP-40, RIPA
(multiples detergen-
tes), Triton X-100
Para antígenos con
niveles bajos de
expresión pueden
ser necesarios pro-
cesos de enriqueci-
miento
Lisado total de célu-
las
NP-40, RIPA,
Triton X-100
Tabla 2. Soluciones Tampón y detergentes, según el tipo de pro-
teína de interés y la localización subcelular

Tabla 3. Inhibidores de fosfatasas y proteasas utilizans en la ex-
tracción de proteínas.
Análisis de Proteínas
Página 5
Con la rotura celular, se liberan enzimas proteasas
que pueden degradar la proteína de interés. Por
tanto, es importante evitar, durate la preparación
de la muestra, cualquier actividad proteásica.
La siguiente estrategia ayuda a evitar la degra-
dación de proteínas:
Evitar excesivos ciclos de congelación/
descongelación.
Trabajar rápido y mantener las muestras en
frío durante todo el proceso.
Añadir inhibidores de la proteasa en el
tampón de lisis.
Además de inhibir la actividad de la proteasa,
puede ser interesante reducir la actividad de la
fosfatasa alcalina, en particular en estudios de
fosforilación.
En la tabla 3 se muestran los inhibidores deprotea-
sa y fosfatasa de uso más habitual.
3. Electroforesis en Acrilamida
Inhibidores Protea-
sas
Concentración en
el Tampón de lisis
Enzimas diana
Aprotinina 1-2 µg/ml Proteasas de Serina
EDTA 1-10 mM Mg++/Mn++ Metalopro-
teasas
EGTA 1-10 mM Ca++ Metaloproteasas
Leupeptin 1-2 µg/ml Proteasas de Serinas,
Cisteinas
Pepstatina A 1µg/ml Proteasas de Aspartico
PMSF (17-170 µg/ml)
O,1—1 mM
Proteasas de Serina
Inhibidores
Fosfatasas
Concentración en
el Tampón de lisis
Enzimas diana
- Glicerofosfato 1 mM Fosfatasas Serina/
Treonina
EDTA 1-10 mM Mg++/Mn++ Metalopro-
teasas
EGTA 1-10 mM Ca++ Metaloproteasas
Leupeptin 1-2 µg/ml Proteasas de Serinas,
Cisteinas
Pepstatina A 1µg/ml Proteasas de Asparti-
co
PMSF (17-170 µg/ml)
O,1—1 mM
Proteasas de Serina
Las proteínas del extracto se separan mediante
electroforesis en gel de poliacrilmaida (PAGE). En
primer lugar, las proteínas, se revisten con carga
negativa, mediante la acción del detergente Do-
decilsulfato Sódico (SDS), así Las proteínas, se
pueden separar en el gel según su tamaño (SDS-
PAGE).
Medición de proteína total
Previamente a la electroforesis, es importante de-
terminar la concentración de proteínas por las
siguientes razones:
1. Asegurar la carga de la cantidad correcta de
proteínas en el gel.
La cantidad de proteína óptima a cargar de-
penderá de la prevalencia de la proteína de
interés y de la sensibilidad del anticuerpo pri-
mario.
Cargar mucha cantidad puede dar lugar a un
mayor ruido de fondo y unión no específica de
los anticuerpos. A menudo, para determinar la
carga de proteína apropiada, se requiere
hacer experimentos preliminares con cantida-
des seriadas de proteínas.
Nota: con el kit Afyon ™ SDS-PAGE de preparación de
muestra, se puede controlar la cantidad de carga de la pro-
teína mediante la cantidad de resina utilizada, evitando la
necesidad de determinar la concentración de proteína. Por
ejemplo, para 5 µg de proteína se utilizan 20 µl de resina Af-
yon.
Información para pedidos
K-02101-025 Afyon ™ SDS-PAGE ssmple preparation kit

2. Realización de Western Blots cuantitativos o
semi-cuantitativos.
Si la cantidad de proteína cargada por carril
es la misma, se pueden comparar los niveles
relativos de la proteína de interés. Un experi-
mento de Western Blot cuantitativo es útil para
estudio de cambios de expresión de proteínas
o de modificaciones proteicas como la fosfori-
lación.
Descripción del método
Existen diversos métodos para medir la proteína
total de una muestra. Los más habituales son los
métodos Lowry, Bradford y Ácido Bincocinico
(BCA) (Tabla 4). Estos ensayos colorimétricos se
basan en las reacciones que se producen entre
las proteínas de la muestra y los reactivos de de-
tección.
Con el fin de determinar la concentración de pro-
teínas totales en una muestra, se debe realizar
una curva estándar en cada ensayo. Esto se logra
con la realización de un ensayo de concentra-
ción crecientes, de proteína pura. El estándar utili-
zado con mayor frecuencia es la albúmina de
suero bovino (BSA), aunque se puede utilizar cual-
quier proteína producida en el laboratorio o dis-
ponible comercialmente.
En la figura 6 se muestra un ejemplo de una curva
estándar. Los valores de absorvancia se trazan en
función del contenido conocido de la proteína
estándar utilizada. El contenido de la proteína de
interés en la muestra (línea verde) se puede de-
terminar mediante la extrapolación de la absor-
vancia obtenida a la cantidad o concentración
de proteína correspondiente.
Elección de un ensayo de proteínas
Hay muchos kits de determinación de proteínas
disponibles comercialmente. La elección depen-
de de muchos factores:
El equipo de lectura disponible en el labora-
torio (espectrofotómetro, lector de placas,
etc…).
Los reactivos del tampón de lisis - revisar el
protocolo del fabricante para identificar
sustancias interferentes.
La cantidad de muestra disponible.
La facilidad/rapidez del ensayo.
Página 6
Tabla 4. Métodos físicos para la dirupción de muestras
Ensayo Descripción Ventajas Inconvenientes
Bradford El colorante
azul de Coo-
masie se une a
las proteínas y
sufre un cam-
bio de absor-
vancia
La realización
de los ensayos
son general-
mente rápidos
y sencillos
Interferencia
de SDS o de
otros deter-
gentes a
altas con-
centraciones
.
Rango lineal
pequeño.
BCA Reducción de
iones de Cu2+
mediante inter-
ación con las
proteínas, el
BCA se une a
los iones de
Cu1+ y forman
un producto
coloreado que
se puede me-
dir espectrofo-
metricamente
(reacción de
Biuret)
Compatible
con la mayo
-
ría de deter-
gentes Comercial-
mente dispo-
nible en for-
mato com-
patible con
agentes
reductores.
Menos varia-
ción proteí-
na-proteína
que en el
método
Bradford.
Interferencia
con agentes
quelantes o
reductores del
Cobre
Lowry Similar al BCA
(reacción de
Biuret)
Muy bien cita-
da en literatura
El tiempo de
ensayo pue-
de ser mayor
que en otros
métodos No es prácti-
co para
grupos gran-
des de
muestras
Se pueden
formar preci-
pitados
Figura 6. Ejemplo de la curva estándar de un ensayo de pro-
teínas. Se representa la absorvancia de las soluciones estándar
que contienen la proteína conocida (línea roja). La concentra-
ción de la proteína de interés se puede determinar extrapolan-
do la absorvancia obtenida en la curva patrón o estándar
(línea verde de puntos).
Proteína (mg)
Ab
sorv
an
cia
Electroforesis en Acrilamida

Figura 7. Mecanismo de desnaturalización por SDS de las proteí-
nas.
Análisis de Proteínas
Página 7
Reactivo Propósito
Glicero. Aportan Viscosidad/densidad para arrastrar
la muestra la fondo del pocillo y evitar que se
extienda a los pocillos adyacentes.
Azul de Bromofe-
nol
Molécula colorante pequeña: Da color a la muestra para ayudar a
la carga.
Migra por delante de las proteínas, de
modo que se puede hacer el segui-
miento del movimiento de las muestras
en el gel.
SDS Detergente desnaturalizante Cola hidrofóbica que rodea la esque-
leto peptídico.
Unión a la proteína de forma regular,
aportando carga negativa de forma
proporcional al tamaño de la proteí-
na.
Evita las interacciones hidrofóbicas y
rompe los enlaces de hidrógeno. Destruye la estructura secundaria/
terciaria y lineariza la proteína.
Beta Mercaptoe-
tanol o DTT
Agentes reductores Evita la oxidación de cisteínas.
Completan la linearización de la pro-
teína al romper los puentes disulfuro.
Tampón Tris Peps-
tatina A
Mantiene el pH adecuado
Tampón de carga de muestras
Una vez se tienen las muestras de proteína, se
pueden conservar congeladas, teniendo cuida-
do en evitar ciclos repetitivos de congelación/
descongelación. Alternativamente, se pueden
utilizar inmediatamente, combinándolas con un
tampón de carga y cargar la muestra en un gel
de electroforesis. Los componentes del tampón
de carga varían, dependiendo de las condicio-
nes deseadas. Los ingredientes típicos de un
tampón de carga para detectar proteínas bajo
condiciones desnaturalizantes/reductoras se des-
criben en la Tabla 5. No se utilizarán ni SDS, beta
mercaptoetanol o ditiotreitol, si se necesitan con-
diciones no desnaturalizantes /no reductoras.
Previamente a la carga del gel, las muestras se
someten a una incubación a 95ºC durante 5-10
minutos, para asegurar que la desnaturalización/
reducción es total. En condiciones no desnaturali-
zantes/no reductoras, esta incubación no es ne-
cesaria. En la Figura 7 se ilustra cual es el mecanis-
mo de desnaturalización del SDS.
Nota: Los tampones de carga permiten ahorrar tiempo y
garantizan que las muestras se separan de forma reproduci-
ble, carril a carril y gel a gel. Advansta dispone de tampones
de carga reductores y no reductores.
Información para pedidos
R– 03018-B10 Non-reducing protein sample loading buffer (2x)
R-03019-B10 Reducing protein sample loading buffer (2x)
La proteína se encuentra en su
estado conformacional, mante-
niendo su carga intrínseca
SDS
El SDS se une a la proteína y da
lugar a una linearización y a una
uniformidad de la carga negativa
de ésta.
Tabla 5. Componentes del tampón de carga.

Gel de Electroforesis
Si se requieren condiciones naturales para que el
anticuerpo pueda detectar el epítopo, en la
electroforesis no se añadirá SDS ni en el gel ni en
el tampón de electroforesis. En esta situación,
también conocida como PAGE nativo, las proteí-
nas mantienen su estructura tridimensional y la
migración en el gel depende de su masa: rela-
ción de carga de la proteína por encima de su
tamaño. Sin embargo, la mayoría de ensayos
Western Blot, se realizan en geles de poliacrilami-
da conteniendo SDS (SDS-PAGE) en condiciones
desnaturalizantes. Bajo estas condiciones desna-
turalizantes y de reducción, las proteínas carga-
das negativamente, cuando se someten a un
campo eléctrico, migran al electrodo positivo.
Debido a que el SDS iguala la carga en todas las
proteínas, la tasa de migración viene determina-
da por su peso molecular. Las moléculas más pe-
queñas migran más rápidamente que aquellas
con pesos moleculares mayores.
Los geles de SDS-PAGE están disponibles comer-
cialmente o se pueden elaborar en el propio la-
boratorio. Los geles pre-fabricados son muy
prácticos pero debido al coste, no son tan renta-
bles como los elaborados por el usuario. En la ta-
bla 6, se describen los componentes químicos de
un gel de SDS-PAGE.
Elección del grosor del gel y del tamaño de poro
El tipo de muestra y el peso molecular de la pro-
teína de interés dictan el espesor y tamaño de
poro del gel.
Los espaciadores de gel (ver Figura 8) con-
trolan el espesor del gel. Están disponibles en
diversos tamaños.
Los geles con mayor espesor, pueden acep-
tar mayor volumen de carga. También se
procesarán más lentamente y requieren
tiempos de transferencia mas largos que los
geles más delgados
El porcentaje del gel, según la cantidad de
acrilamida y bisacrilamida, determina el ta-
maño del poro. A más porcentaje menos
tamaño de poro.
El tamaño de poro determina la ratio de se-
paración de las proteínas. (tabla 7).
Página 8
Electroforesis en Acrilamida
Tabla 6. Componentes del gel de acrilamida en las electroforesis
de proteínas
Reactivos Propósito
Acrilamida Moléculas que polimerizan para formar
cadenas.
Bisacrilamida Forman uniones con las cadenas de acri-
lamida para formar un entramado.
SDS Mantiene la linearidad y la uniformidad
de carga de las proteínas según la elec-
troforesis.
Tampón Tris Mantiene el pH adecuado.
Persulfato de Amonio Generación de radicales libres que cata-
lizan la reacción de polimerización.
TEMED Aumenta la generación de radicales
libres por el persulfato de amonio.
Porcentaje Rango Peso Molecular (kDa)
7,5 25-500
10 15-300
12 10-200
15 10-45
20 5-40
Tabla 7. Rango de separación de proteínas según el porcentaje
de acrilamida.

Análisis de Proteínas
Página 9
Elaboración del gel
Si el gel de acrilamida esta elaborado en el labo-
ratorio, los distintos reactivos se mezclan y se vier-
te en el molde, formado por el conjunto de dos
cristales, espaciadores lateral, espaciador inferior,
peines y un sistema de sujeción (Figura 8), dónde
la acrilamida se polimeriza en aproximadamente
30 minutos. Para mejorar la nitidez de la banda el
gel resolutivo se corona con una acrilamida de
menor porcentaje (stacking gel).
Diseño Experimental; Controles y Estándares
Antes de la carga del gel, es importante diseñar
el experimento incorporando los controles y
estándares necesarios para la validación de los
resultados. Los tipos de estándares y controles utili-
zados incluyen:
1. Marcadores de Peso Molecular.
Son una mezcla de proteínas purificadas de
peso molecular conocido.
Se utilizan para verificar si la proteína de in-
terés está dentro del rango de tamaño apro-
piado.
Disponible en diversos intervalos de pesos
moleculares, así como teñidos o incoloros.
Las versiones preteñidas se utilizan para
controlar el progreso de la electroforesis y
comprobar la eficacia de la posterior trans-
ferencia.
Pueden estar marcadas por la detección
por fluorescencia o quimioluminiscencia.
2. Controles Positivos.
Para verificar si el anticuerpo primario se une
a la proteína correcta.
Generalmente, si está disponible, son una
muestra de la proteína de interés purificada.
Puede ser una línea celular que sobreexpre-
se la proteína de interés.
Puede ser una muestra de tejido del que se
conozca que expresa altos niveles de la pro-
teína de interés. Se pueden utilizar otras es-
pecies como tejido de control si el anticuer-
po tiene reactividad cruzada apropiada.
1. Colocar los espaciadores de la parte inferior y laterales entre
los cristales.
2. Tras fijación de los lados, verter el gel resolutivo y esperar que
el gel se polimerice.
3. Colocar el peine en la parte superior del gel.
4. Verter el ―stacking gel‖ y quitar el peine una vez el gel haya
polimerizado. Lavar los pocillos con ddH20. El gel está listo
para cargar las muestras y la electroforesis.
Controles de carga
Como norma general se utilizan proteínas con
niveles de expresión constante para todas las
muestras. Similar a los ―housekeeping genes‖
en Biología Molecular.
Algunos tratamientos pueden alterar la expre-
sión de estas proteínas ―housekeeping‖.
Se utiliza para verificar que la carga de proteí-
na por carril es más o menos igual, así los nive-
les de proteínas se pueden comparar de forma
cuantitativa.
La expresión cuantitativa de la proteína de in-
terés puede normalizarse con la del control de
carga para cada muestra.
Péptidos de bloqueo
Algunos proveedores ofrecen péptidos de blo-
queo para sus anticuerpos.
El péptido de bloqueo se mezcla con el anti-
cuerpo primario antes de la incubación con la
membrana y evita la uníón del anticuerpo a la
proteína diana. Por tanto no se detectará nin-
guna banda.
Estos estudios demuestran que el anticuerpo
utilizado se une de forma específica a la proteí-
na de interés.
Figura 8. Preparación del gel de electroforesis para la separa-
ción de proteínas.

Inicio de Electroforesis
Las muestras se cargan en los pocillos del gel me-
diante una pipeta. Normalmente se cargan de 20
a 40 µg de proteína total por carril. Es importante
conocer de antemano, el volumen que se puede
cargar por pocillo, para así evitar sobrecargas
que pueden dar lugar a la pérdida de muestra o
derrames en pocillos adyacentes. Una vez finali-
zada la carga, el gel se somete a un campo eléc-
trico generado por una fuente de alimentación. El
tampón utilizado durante la electroforesis, contie-
ne Tris (mantiene el pH), Glicina (conductor elec-
tricidad) y SDS (mantiene las proteínas cargadas
negativamente). El progreso en el gel se monitori-
za observando el frente coloreado por el coloran-
te presente en el tampón de carga y la posición
de los marcadores de tamaño siempre que estos
estén teñidos.
La técnica de electroforesis más habitualmente
utilizada es la de Laemmli. En este método, los
valores de pH de la capa de gel de apilamiento
o concentración (Stacking Gel) y el gel de resolu-
ción (Resolving Gel) son diferentes. La figura 9 ilus-
tra el flujo de iones durante la electroforesis.
Página 10
Electroforesis en Acrilamida
Muestra (pH 6,8)
Gly
Proteinas
CL-
Stacking Gel
pH 6,8
Resolving Gel
pH 8,8 Gly
Proteinas
Tampón de Electroforesis (pH 8,3)
La glicina tiene carga negativa en el tampón de electrofo-
resis a pH 8,3.
Con la aplicación de un campo eléctrico, la glicina, en el
Stacking Gel, se aleja del electrodo negativo.
La glicina empieza a perder su carga a pH 6,8 y ralentiza
su movimiento. Los iones de cloruro avanzan por delante
de la Gly. El aumento de la resistencia, obliga a las proteí-
nas a alejarse, de forma apilada, del electrodo negativo.
La glicina, debido a un mayor pH del ―Resolving Gel‖, au-
menta su carga negativa y se mueve por delante de las
proteínas.
Las proteínas se separan según su peso molecular por el
Figura 9. Concentración de bandas de proteínas por el flujo
iónico en el sistema discontinuo de Laemmli.

Análisis de Proteínas
Página 11
Una vez la electroforesis del gel ha terminado, las
proteínas se transfieren a una membrana. La
membrana es un soporte sólido que une e inmovi-
liza las proteínas, permitiendo así que la hibrida-
ción de un anticuerpo las pueda detectar. La
mayoría de los sistemas de transferencia utilizan la
generación de un campo eléctrico para condu-
cir a las proteínas desde el electrodo negativo al
positivo. Los sistemas de transferencia están dispo-
nibles en formato semiseco o húmedo. Los siste-
mas semisecos son más rápidos y requeiren menor
cantidad de volumen de Tampón que los siste-
mas húmedos, sin embargo no son apropiados
para transferencia de proteínas de gran tamaño
(>70 kDa) y pueden causar, según el sistema de
detección, un mayor ruido de fondo. Ambos
métodos requieren, para una transferencia efecti-
va de las proteínas, un estrecho contacto entre el
gel y la membrana.
Aunque ambas funcionan bien en experimentos
de inmunotransferencia, las diferencias en sus ca-
racterísticas puden influir a la hora de la elección.
La membrana de PVDF, ofrece una mayor resis-
tencai mecánica, resistencia la SDS y una mayor
unión que la nitrocelulosa. La membrana de nitro-
celulosa tiene la ventajas de proporcionar un me-
nor ruido de fondo y que no requieren un pre-
tratamiento con metanol. Recientemente se han
desarrollado, con el objetivo de reducir el ruido
de fondo al utilizar métodos de detección de fluo-
rescencia, membranas PVDF de baja autofluores-
cencia.
Previamente a la transferencia, tanto el gel como
la membrana se equilibran en una solución
tampón pre-enfriada. Típicamente, el tampón de
transferencia contiene Tris, Glicina, SDS (opcional)
y metanos (para membranas de nitrocelulosa). La
figura 10 ilustra como se ensamblan el gel y la
membrana en un transferencia semiseca o húme-
da.
4. Transferencia a una Membrana
Nota: Advansta suministra membranas de transferencia pre
-cortadas de nitrocelulosa o PVDF aptas de tamaño de mini-
gel. El uso de membranas pre-cortadas permite ahorrar tiem-
po y elimina la contaminación accidental de las membranas
con guantes o tijeras contamindas.
Información para pedidos
L-08001-010 Membrana PVDF baja fluorescencia 7x9 cm
L-08002-010 Membrana Nitrocelulosa 0,45 µm 8x10 cm
L-08003-010 Membrana Nitrocelulosa 0,22 µm 8x10 cm
Figura 8. Transferencia de proteínas a una membrana.

A continuación se describen algunas sugerencias
para la consecución exitosa de una transferen-
cia:
Evitar la manipulación de las membranas con
las manos desnudas. Las proteínas y aceites de
la piel pueden adherirse a la membrana y pue-
den dar lugar a manchas no deseadas en la
membrana.
Las burbujas de aire entre el gel y la membra-
na pueden causar transferencia defectuosa y
desigual. Se pueden eliminar haciendo rodar
suavemente una pipeta Pasteur por encima
del ―sándwich‖ formado por el gel/
membrana/papel de filtro.
No permitir un sobrecalentamiento durante la
transferencia. Es aconsejable utilizar tampón
frio, un sistema de refrigeración o hacer la
transferencia en una cámara fría. Disminuir el
voltaje o el tiempo si fuera necesario.
Ajustar las condiciones de transferencia al ta-
maño de la proteína. Las proteínas más peque-
ñas se transferirán más rápidamente y pueden
atravesar la membrana. Reducir o eliminar el
SDS o utilizar una membrana con un tamaño
de poro menor puede ayudar a una mayor
retención de la proteína. Incrementar la canti-
dad de SDS y disminuir la concentración de
Metanol pueden facilitar la transferencia de
proteínas de gran tamaño.
La aparición de marcadores de tamaño prete-
ñidos en la membrana es indicación que se ha
completado la transferencia.
El colorante Ponceau S puede utilizarse para
teñir la membrana y asegurarse de la eficacia
de la transferencia. La membrana debe ser
desteñida antes del proceso de transferencia.
La tinción con Coomassie puede usarse para
la tinción del gel una vez realziada la transfe-
rencia y comprobar los residuos de proteína en
el gel.
Página 12
Nota: Advansta ofrece soluciones optimizadas de bloqueo
y de lavado para su línea de reactivos de detección, Wes-
ternBright, así como soluciones tampón en polvo de PCS, TBS,
...que facilitan una rápida y fácil preparación
Información para pedidos
R-03024-D50 AdvanWashTM, 500 ml
R-03023-D20 AdvanBlockTM-PF, 200 ml
R-01038-020 AvantTM Buffer Pouches - PBS
R-01039-020 AvantTM Buffer Pouches - TBS
Bloqueo
El bloqueo de la membrana se lleva a cabo incu-
bando con una solución diseñada para reducir los
lugares de unión no específicos al anticuerpo. A me-
nudo, son soluciones a base de proteínas, como la
leche descremada en polvo o la albúmina de suero
bovino (BSA). La primera elección, cuando se inicia
un nuevo protocolo, es la utilización de la leche des-
cremada en polvo, ya que es más rentable que el
BSA, sin embargo y debido a la presencia de biotina
en las fosfoproteínas, puede que no sea la mejor
elección cuando se van a utilizar anticuerpos fosfo-
específicos o en métodos de detección de biotina.
Es preferible, para estas y otras situaciones, el uso
de agentes bloqueantes no proteicos.
El agente bloqueante se diluye en TBS (Tampón
Tris salino) o en PBS (Tampón Fosfato salino); se
puede añadir también detergente Tween 20.
Una incubación durante una hora
(temperatura ambiente o a 4ºC) suele ser sufi-
ciente para un bloqueo de los lugares de unión
no específicos del anticuerpo. Si los niveles del
ruido de fondo son demasiados altos, se pue-
den seleccionar otras soluciones de bloqueo
alternativas.
Electroforesis en Acrilamida

Análisis de Proteínas
Página 13
Después del proceso de bloqueo, la membrana
se incuba con el anticuerpo primario. En general,
los anticuerpos reconocen una secuencia de
aminoácidos pequeña (epítopo) que queda ex-
puesta al eliminar la estructura tridimensional de
la proteína en condiciones desnaturalizantes y
reductoras. Los geles nativos se pueden utilizar
cuando los anticuerpos requieren de la proteína
plegada para el reconocimiento del antígeno.
Anticuerpos Primarios:
Monoclonal vs Policlonal.
En la transferencia Western se pueden utilizar, tan-
to anticuerpos primarios monoclonales como poli-
clonales. Los dos tipos tienen distintas ventajas y
desventajas y se diferencian según la forma en la
que se sintetizan. El primer paso en la producción
de ambos anticuerpos, es la inyección de un antí-
geno en un animal, para provocar una respuesta
inmune. Los anticuerpos policlonales se obtienen
directamente a partir del suero del animal,
comúnmente conejo, cabra, burro u oveja y son
finalmente purificados y probados. Los anticuer-
pos policlonales reconocen múltiples epítopos del
antígeno y en cambio los monoclonales recono-
cen un único epítopo de la proteína. Para la sínte-
sis de un anticuerpo monoclonal, las células que
sintetizan anticuerpos se aíslan del bazo del ani-
mal y se fusionan con células del mieloma. Lo
hibridomas resultantes secretan anticuerpos al
medio de cultivo, el cual se analiza y determina
la afinidad al antígeno. Los hibridomas con secre-
ción más estable se seleccionan y pueden ser cul-
tivados indefinidamente. Algunas características
de los anticuerpos monoclonales y policlonales se
describen en la Tabla 8.
5. Hibridación del Anticuerpo. Tabla 8. Anticuerpos Policlonales vs Monoclonales.
Anticuerpos
Monoclonales
Anticuerpos
Policlonales
Reconocimiento
epitopo y
sensibilidad
Reconocimiento
de un único epí-
topo
Menos sensibili-
dad debido a
que sólo un anti-
cuerpo se une a
cada proteína
Reconocimiento
múltiples epítopos
en una proteína
Más sensible debi-
do a los multiples
lugares de unión
de anticuerpo en
cada proteína.
Bueno para proteí-
nas poco abun-
dantes.
Reactividad cruza-
da potencial
Reactividad cru-
zada menos
probable.
Potencial para
reconocer otras
proteínas que
contengan el
epítopo.
Reactividad cruza-
da más probable.
Mayor ruido de
fondo potencial
debido al recono-
cimiento de multi-
ples epítopos.
Reactividad cruza-
da con otras espe-
cies más probable.
Tiempo requerido
para su producción
Largo Más corto
Coste de Prepara-
ción
Mayor coste - re-
quiere personal
capacitado y equi-
pamiento especiali-
zado.
Menor coste
Variabilidad Los lotes de un mis-
mo hibridoma son
muy estables.
Propenso a cierta
variabilidad entre
lotes.
Tolerancia a condi-
ciones variables
Poca tolerancia -
puede dejar de
reconocer al antí-
geno en condicio-
nes de desnaturali-
zación/reducción
de la proteína, o
por modificaciones
químicas
(glicosilación fosfori-
lación) o por dife-
rencias en la se-
cuencia peptídica
debido a la presen-
cia de polimorfis-
mos.
Más tolerante a con-
diciones variables.

Página 14
Nota: Advansta ofrece una línea completa de anticuerpos
secundarios marcados para detección mediante quimiolu-
minscencia o fluorecencia.
Información para pedidos
R-05051-050 IgG conjugado APC-cabra-anti-conejo, 50 µl
R-05051-250 IgG conjugado APC-cabra-anti-conejo, 250 µl
R-05052-050 IgG conjugado RPE-cabra-anti-ratón, 50 µl
R-05051-250 IgG conjugado RPE-cabra-anti-ratón, 250 µl
R-05071-500 Anticuerpo Secundario conjugado HRP
cabra-anti-ratón, 500 µl
R-05072-500 Anticuerpo Secundario conjugado APC
cabra-anti-conejo, 500 µl
Incubación de los anticuerpos
El anticuerpo primario se diluye en tampón de
bloqueo, TBST o en un tampón de disolución de
anticuerpo comercial. El fabricante puede reco-
mendar una cantidad inicial, pero a menudo la
concentración óptima de anticuerpo debe deter-
minarse empíricamente. La afinidad del anticuer-
po, la abundancia de la proteína en la muestra y
la sensibilidad del sistema de detección afec-
tarán a la cantidad de anticuerpo a utilizar.
Generalmente, un periodo de incubación
de 1 hora a temperatura ambiente suele ser
suficiente. También es posible una incuba-
ción a 4ºC durante toda la noche.
Durante la incubación, la membrana debe
someterse a agitación suave y sumergida en
la disolución, e incubar en un agitador de
vaivén. Un menor volumen puede utilizarse si
la membrana se sella dentro de una bolsa e
incuba en un agitador orbital.
En algunos casos, la disolución con el anti-
cuerpo, puede reutilizarse. Si se utiliza acida
sódica como conservante, debe ser total-
mente lavada de la membrana, ya que
puede eliminar la actividad de la peroxidasa
e interferir en los métodos de detección.
Después de la incubación con el anticuerpo pri-
mario, se procede a la eliminación del excedente
mediante tampones de lavado (TBS, PBS, TBST o
PBST). En métodos de detección indirectos
(sección 6), es necesario un anticuerpo secunda-
rio apropiado. A continuación se describen las
consideraciones a tener en cuenta a la hora de la
elección de un anticuerpo secundario:
Las especie del primer Ac dicta la elección
del Ac secundario. Si el primer Ac es de co-
nejo, el segundo Ac debe ser anti-conejo, o
sea, deber ser producido en otra especie
distinta a conejo.
Para anticuerpos monoclonales se debe
considerar la clase de Inmunoglobulina (IgG,
IgM, IgA, IgD, IgE) y posiblemente la subclase
(IgG1, IgG2a,…). Un anticuerpo con amplia
especificidad inmunoglobulínica debería ser
suficiente, ya que los anticuerpos específicos
para sub-clases se utilian mayoritariamente,
en experimentos de doble etiquetado u
otras aplicaciones.
Hibridación Anticuerpos

Análisis de Proteínas
Página 15
La detección de la proteína se puede hacer me-
diante métodos directos o indirectos, cada uno
con sus ventajas e inconvenientes. Los métodos
directos utilizan un anticuerpo primario conjuga-
do con un marcaje detectable, como un enzima,
biotina o molécula fluorescente. Los anticuerpos
se pueden marcar mediante kits disponibles en le
mercado u obtenerlos ya marcados. Lo métodos
indirectos utilizan dos anticuerpos; un anticuerpo
primario y un anticuerpo secundario contra la es-
pecie del primer anticuerpo. En la detección indi-
recta, es el anticuerpo secundario el que está
conjugado con un marcaje detectable. La figura
11,ilustra los métodos de detección directos e in-
directos y en la Tabla 9 se resumen las ventajas e
inconvenientes de los dos métodos.
Métodos de detección.
Los métodos más comúnmente utilizados para
detectar proteínas son: 1. Quimioluminiscencia, 2.
Fluorescencia y 3. Colorimetría.
1. Quimioluminiscencia.
Los métodos de detección de quimioluminiscen-
cia utilizan comúnmente anticuerpos secundarios
conjugados con peroxidasa (HRP). La reacción
entre el encima y el substrato produce luz, que
puede ser detectada mediante la exposición de
la membrana a una película de rayos X o capta-
da de forma digital utilizando una cámara CCD.
Ventajas: muy sensible, la membrana puede tra-
tarse para re-utilizarse, se pueden hacer multiples
exposiciones en películas de rayos X, fácil de do-
cumentar.
Inconvenientes: requiere una habitación oscura
(película rayos X) o sistemas de imagen caros, se-
micuantitativa - porque se basa en una reacción
enzimática, la película tiene un rango dinámico
limitado, la intensidad de la señal puede variar
con la incubación o el tiempo de exposición, no
son posibles detecciones multiplex (detección de
mas de una proteína, sólo un color de reacción)
6. Detección.
Figura 11 . Detección directa e indirecta de proteínas
Indirecto Directo
Ventajas Amplificación de
señal debido a la
unión de múlti-
ples Ac secunda-
rios a cada Ac
primario.
Versátil - el mis-
mo Ac secunda-
rio puede utilizar-
se para diversos
anticuerpos de
la misma espe-
cie.
Menos pasos.
Menos posibilida-
des de unión no
específica.
Inconvenientes Mayor posibili-
dad de unión no
inespecífica.
Más pasos.
Puede ser más
costoso.
La inmunorreactivi-
dad del Anticuer-
po puede estar
afectada por el
marcaje.
Nota: LucentBlueTM es una película que ha sido optimizada para
la detección de señales de quimioluminiscencia en experimentos
realizados con los substratos WesternBright HRP, incluyedo Western-
BrightTM ECL, Westernbright Quantum y WesternBright Sirius.
Información para pedidos
L07014-100 Película Rayos X LucentBlueTM , 20x25 cm, 100 uds
L07013-100 Película Rayos X LucentBlueTM , 13x18 cm, 100 uds
Tabla 9. Ventajas e Inconvenientes de los métodos de detec-
ción

Página 16
Detección
Nota: WesternBrightTM MCF permite la detección, mediante la
detección fluorescente multicolor, de dos proteínas en una
sola membrana. Perfecto para aquellos experimentos en los
que se necesite comparar la proteína de interés con un control
de carga.
Información para pedidos
K-12021-010 WesternBrightTM fluorescent Western blotting kit
Re-utilización de la membrana
Después de captar la imagen, la membrana pue-
de ser tratada para volver a ser utilizada para la
detección de otra proteína. Este procedimiento
puede conservar las muestras, pero consume mu-
cho tiempo. Con la detección multiplex de fluo-
rescencia, se pueden detectar múltiples proteínas
de forma simultanea, es ideal para comparar la
proteína de interés con un control de carga, o
diferentes isoformas de la proteína. Las membra-
nas PVDF son preferibles a la de nitrocelulosa en
protocolos de lavados para reutilización.
2. Fluorescencia.
El anticuerpo secundario se une a un fluoróforo,
que cuando es ―excitado‖ emite luz. La luz emiti-
da, se detecta mediante un dispositivo capaz de
medir la fluorescencia o mediante una cámara
CCD provista con los filtros de longitud de onda
apropiada. La imagen se digitaliza para su análi-
sis.
Ventajas: sensibilidad (la sensibilidad puede incre-
mentarse utilizando el sistema streptavidina/
biotina), apto para ensayos multiplex con múlti-
ples colorantes, amplio rango dinámico, no se
requiere el uso de una cámara oscura, aporta
datas cuantificables, fácil documentación de re-
sultados, pueden usarse Ac primarios marcados
(para proteínas abundantes) y eliminar pasos.
Inconvenientes: los reactivos y el equipamiento
pueden ser costosos, no es tan sensible como la
quimioluminiscencia (puede no ser apropiado
para proteínas poco abundantes).
3. Colorimetría.
Los métodos colorimétricos utilizan un Ac secun-
dario que ha sido conjugado previamente a un
enzima (peroxidasa o fosfatasa alcalina). El enzi-
ma convierte el colorante en un producto insolu-
ble que es visible en la membrana. La cantidad
de colorante convertido es proporcional al canti-
dad en la muestra. La intensidad de la tinción
puede ser medida por espectrofotometría o por
un densitómetro.
Ventajas: sencilla de realizar, no requiere cámara
oscura, las membranas se pueden documentar
fácilmente mediante fotografía.
Inconvenientes: no es tan sensible como los otros
dos métodos, el color se desvanece con el tiem-
po. Nota: Advansta suministra una línea completa de reactivos
de detección de quimioluminiscencia optimizados para los
diferenentes métodos, concentración de muestra y tipos expe-
rimentales.
Información para pedidos
K-12042-D10 WesternBrightTM Quantum Western Blotting HRP
Substrate (para 1000 cm2 de membrana)
K-12042-D10 WesternBrightTM Sirius Western Blotting HRP Subs-
trate (para 1000 cm2 de membrana)
K-12045-D20 WesternBrightTM ECL Western Blotting HRP Substra-
te (para 2000 cm2 de membrana)
K-12042-D10 WesternBrightTM ECL Spray

Análisis de Proteínas
Página 17
7. Solución de Problemas
Solución de problemas con Western Blot
A. No se puede
ver la proteína
de interés
Preparación
Muestra
(ver 1)
B. Tamaño inco-
rrecto de la
banda
C. Bandas arte-
factuales
D. Problemas en
la electroforesis
E. Ruido de fon-
do excesivo
Transferencia
Inadecuada
(ver 2)
Unión ineficien-
te del Anticuer-
po (ver 3, 4)
Problemas con
los reactivos
(ver 5, 6)
La proteína es
menor de lo
esperado
(ver 7)
La proteína es
mayor a lo es-
perado
(ver 8, 9)
Bandas Múlti-
ples (ver 10)
Las bandas
aparecen muy
altas o muy
bajas en el gel
(ver 11)
Bandas
Incompletas
(ver 12)
Bandas Difusas
(ver 13)
Rayas
Verticales
(ver 14)
Distorsión
Lateral
(ver 15)
Bandas distor-
sionadas
(ver 16)
El gel polimeriza
demasiado
rápido o dema-
siado lento
(ver 17,18,19)
El gel corre de-
masiado rápido
o demasiado
lento
(ver 20,21)
Frente corre
curvado
―smiling‖
(ver 22)
Frente corre
inclinado
(ver 23)
Bloqueo
Insuficiente
(ver 24)
Lavado
inapropiado
(ver 25)
Contaminación
Reactivos
(ver 26)
Elección de
Membrana
(ver 27)
Unión no es-
pecífica del
Anticuerpo
(ver 28)
Revelado de la
película
(ver 29)

A. Problema Causa(s) Solución
1. Preparación Muestra Extracción ineficiente Intentar métodos alternativos; incluir con-
trol positivo en el gel
Baja expresión de la proteína en el tejido
o las células
Cargar más cantidad de proteína total en
el gel; concentrar utilizando Afyon, juntar
múltiples muestras
La proteína se ha degradado durante la
extracción
Usar inhibidores de proteasa en le tampón
de lisis
2. Transferencia indadecua-
da
Tampón de transferencia preparado inco-
rrectamente/demasiado metanol en el
tampón
Revisar el protocolo/disminuir metanol
Proteínas de mayor tamaño necesitan
mas tiempo/voltaje
Repetir con un tiempo mayor/mayor vol-
taje
Contacto insuficiente entre el gel y la
membrana
Revisar la almohadilla de fibra; sustituir si es
muy delgada
3. Unión ineficiente del Anti-
cuerpo primario
Baja afinidad del anticuerpo por la proteí-
na o el anticuerpo es antiguo/débil
Incrementar la concentración del Anti-
cuerpo; comprar un anticuerpo nuevo y
almacenarlo de forma apropiada
El Anticuerpo tiene reactividad cruzada
débil con las especies de interés
Probar con un Anticuerpo primario de di-
ferente origen
El Anrticuerpo se ha eliminado con el lava-
do
Usar menor numero de lavados; disminuir
la concentración de sales el tampón de
lavado
Antígeno enmascarado por el agente
bloqueante (ej.: Leche,…)
Utilizar un agente de bloqueo alternativo
(ej.: BSA,…)
4. Unión ineficiente del Anti-
cuerpo secundario
Elección de especies errónea Usar Anticuerpos dirigidos contra la espe-
cie del Anticuerpo primario
Concentración insuficiente del Anticuerpo
o Anticuerpo antiguo
Aumentar concentración o comprar uno
Anticuerpo nuevo
5. Conjugado/Substrato Inac-
tivo
Reactivos viejos o inestables Mezclar conjugado + substrato en un tu-
bo; o luminiscencia en oscuridad - conse-
guir un nuevo reactivo si no hay señal
Peroxidasa (HRP) inactiva por azida sódi-
ca
Evitar utilizar disoluciones conteniendo
este agente conservante
6. Reactivo de detección
(ECL)
La disolución es antigua o está almacena-
da de forma inapropiada
Comprar reactivo nuevo
B. Problema Causa (s) Solución
7. Proteína más pequeña de
lo esperado
Proteólisis; Congelación/descongelación
de la muestra
Uso de inhibidores de proteasa/muestras
frescas
Proteína variante (Splicing) Consultar literatura/utilizar controles apro-
piados
8. Proteína más larga de lo
esperado y/o bandas múlti-
ples
Proteínas modificadas de forma natural
(glicosilación, fosforilación, acetilación,
etc…)
Consultar literatura para encontrar aditi-
vos que puedan eliminar grupos químicos
Cambio de la expresión de la proteína en
la línea celular
Utilizar cultivos anteriores; incluir un control
positivo
9. Proteína más larga de lo
esperado
Agregado de proteínas - puentes disulfuro
intactos
Uso de DTT en tampón de muestra; pulso
de centrífuga previo a la carga de la
muestra
Página 18
Solución de Problemas
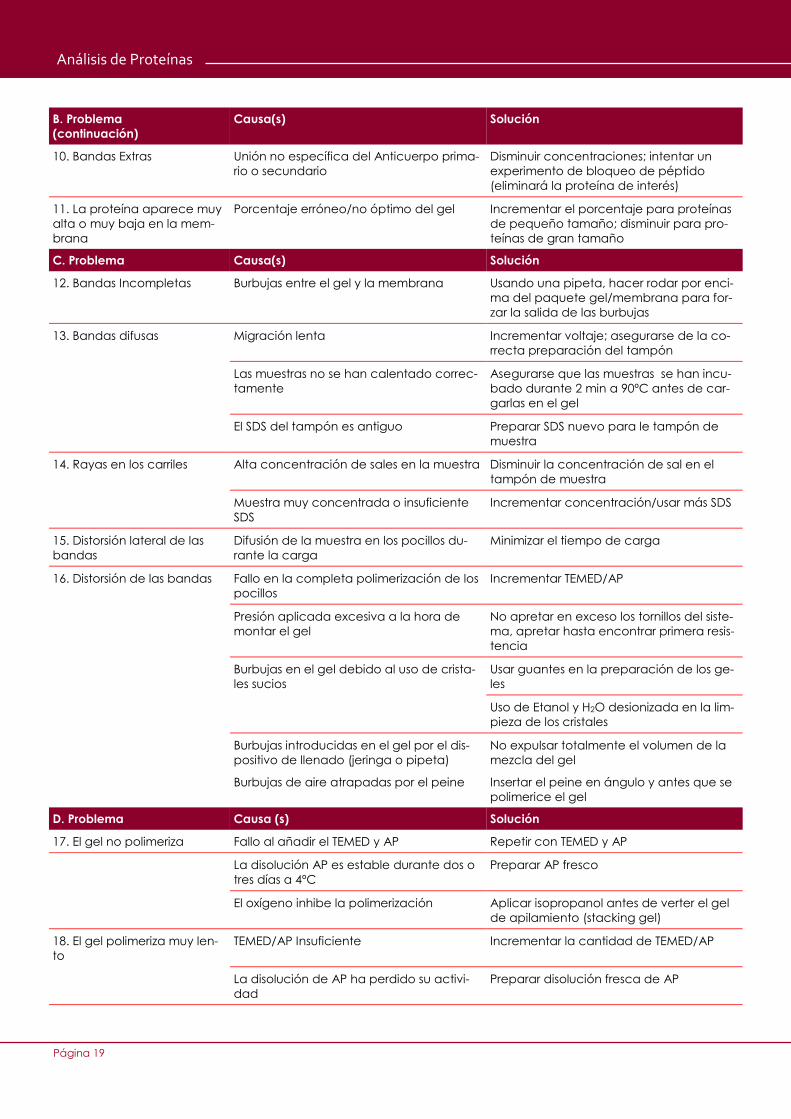
Página 19
B. Problema
(continuación)
Causa(s) Solución
10. Bandas Extras Unión no específica del Anticuerpo prima-
rio o secundario
Disminuir concentraciones; intentar un
experimento de bloqueo de péptido
(eliminará la proteína de interés)
11. La proteína aparece muy
alta o muy baja en la mem-
brana
Porcentaje erróneo/no óptimo del gel Incrementar el porcentaje para proteínas
de pequeño tamaño; disminuir para pro-
teínas de gran tamaño
C. Problema Causa(s) Solución
12. Bandas Incompletas Burbujas entre el gel y la membrana Usando una pipeta, hacer rodar por enci-
ma del paquete gel/membrana para for-
zar la salida de las burbujas
13. Bandas difusas Migración lenta Incrementar voltaje; asegurarse de la co-
rrecta preparación del tampón
Las muestras no se han calentado correc-
tamente
Asegurarse que las muestras se han incu-
bado durante 2 min a 90ºC antes de car-
garlas en el gel
El SDS del tampón es antiguo Preparar SDS nuevo para le tampón de
muestra
14. Rayas en los carriles Alta concentración de sales en la muestra Disminuir la concentración de sal en el
tampón de muestra
Muestra muy concentrada o insuficiente
SDS
Incrementar concentración/usar más SDS
15. Distorsión lateral de las
bandas
Difusión de la muestra en los pocillos du-
rante la carga
Minimizar el tiempo de carga
16. Distorsión de las bandas Fallo en la completa polimerización de los
pocillos
Incrementar TEMED/AP
Presión aplicada excesiva a la hora de
montar el gel
No apretar en exceso los tornillos del siste-
ma, apretar hasta encontrar primera resis-
tencia
Burbujas en el gel debido al uso de crista-
les sucios
Usar guantes en la preparación de los ge-
les
Uso de Etanol y H2O desionizada en la lim-
pieza de los cristales
Burbujas introducidas en el gel por el dis-
positivo de llenado (jeringa o pipeta)
No expulsar totalmente el volumen de la
mezcla del gel
Burbujas de aire atrapadas por el peine Insertar el peine en ángulo y antes que se
polimerice el gel
D. Problema Causa (s) Solución
17. El gel no polimeriza Fallo al añadir el TEMED y AP Repetir con TEMED y AP
La disolución AP es estable durante dos o
tres días a 4ºC
Preparar AP fresco
El oxígeno inhibe la polimerización Aplicar isopropanol antes de verter el gel
de apilamiento (stacking gel)
18. El gel polimeriza muy len-
to
TEMED/AP Insuficiente Incrementar la cantidad de TEMED/AP
La disolución de AP ha perdido su activi-
dad
Preparar disolución fresca de AP
Análisis de Proteínas

D. Problema
(continuación)
Causa(s) Solución
19. El polimeriza demasiado
rápido
Cantidad excesiva de TEMED/AP Reducir la cantidad de TEMED/AP, mante-
niendo la relación entre ambos
20. Tiempo de carrera in-
usualmente largo
Buffer de electroforesis demasiado con-
centrado
Revisar protocolo; diluir el tampón en caso
necesario
Voltaje insuficiente Incrementar voltaje
21. Tiempo de carrera in-
usualmente corto
Tampón demasiado diluido Revisar el protocolo; reemplazar tampón
en caso necesario
22. Frente curvado (smiling) Migración demasiado rápida Disminuir voltaje
Generación de calor Disminuir voltaje; refrigerar
23. Frente inclinado Burbuja atrapada entre los cristales en la
parte inferior del gel
Aguantar el gel en ángulo; colocar una
esquina en el tanque inferior del sistema
de electroforesis; lentamente volver a po-
sición vertical
E. Problema Causa(s) Solución
24. Bloqueo insuficiente La presencia de Biotina en la leche es in-
compatible con el uso de Estreptavidina,
o la leche contiene el antígeno de interés
Usar BSA
Si utiliza AdvanBlock-PF con WesternBright
MCF, algún Anticuerpo primario puede
requerir un bloqueante proteico
Incluir BSA o leche en la disolución Advan-
Block-PF utilizada en la dilución del Anti-
cuerpo primario
La disolución está demasiado diluida Incrementar con un 5% la disolución
Tiempo de bloqueo muy corto Incrementar el tiempo de incubación
Algunos detergentes no son efectivos a
bajas temperaturas
Incubar a temperatura ambiente en vez
de toda la noche a 4ºC
25. Condiciones de lavado
inapropiadas
Número insuficiente de lavados Incrementar el número de lavados o la
duración de cada uno de ellos
Concentración insuficiente de detergente Incrementar la concentración de deter-
gente o usar detergentes más fuertes
(SDS, NP-40)
26. Contaminación de los
reactivos
Crecimiento bacteriano o fúngico en los
tampones
Revisar la turbidez de los tampones; pre-
parar nuevos
27. Elección de tipo de mem-
brana
Las membranas PVDF pueden tener más
ruido de fondo que las de Nitrocelulosa
Elegir membranas de Nitrocelulosa
Algunas membranas tienen alta autofluo-
rescencia
Utilizar únicamente membranas PVDF con
baja autofluorescencia con Western Blots
fluorescentes
Membrana seca Estar seguro que la membrana esta húme-
da durante todo el proceso
28. Unión no específica del
Anticuerpo primario o secun-
dario
Concentración muy alta del Anticuerpo o
no purificado por afinidad
Disminuir la concentración de Anticuerpo;
intentar con Anticuerpo monoclonal o
purificado por afinidad
Mucha cantidad de proteína en el gel Disminuir la cantidad de proteína
29. Sobreexposición de la
imagen
El tiempo excesivo de exposición a la pelí-
cula o al CCD
Reducir los tiempos de exposición; si no es
posible incrementar la dilución de los Anti-
cuerpos o cargar menor cantidad de
muestra
Página 20
Solución de Problemas

Página 21
Análisis de Proteínas
8. Herramientas y Referencias Técnicas
g de Soluto
Molaridad de una disolución =
[Masa molecular de Soluto (g/mol) x (L de disolución)
Tabla 10. Código genético y abreviaciones de los aminoácidos
Peso molecular
(Da)
1 µg 1 nmol
10.000 100 pmol o 6x1013 moléculas 10 µg
20.000 20 pmol o 1,2x1013 moléculas 50 µg
100.000 10 pmol o 6x1012 moléculas 100 µg
150.000 6,7 pmol o 4x1012 moléculas 150 µg
Tabla 11. Conversión entre masa y moles de proteína
Proteina A280 Uds por 1 mg&ml
IgG 1,35
IgM 1,2
IgA 1,3
Proteina A 0,17
Avidina 1,5
Estreptavidina 3.4
Albumina suero bovino 0,7
Tabla 12. Absorvancia a 280 nm de Proteinas habituale
Segunda Posición
U C A G
U
UUU Fenilalanina
UUC (Phe, F)
UUA Leucina
UUG (Leu, L)
UCU
UCC Serina
UCA (Ser, S)
UCG
UAU Tirosina
UAC (Tyr, T)
UAA STOP
UAG
UGU Cisteina
UGC (Cys, C)
UGA STOP
UGG Triptófano
(Trp, W)
C
CUU
CUC Leucina
CUA (Leu, L)
CUG
CCU
CCC Prolina
CCA (Pro, P)
CCG
CAU Histidina
CAC (His, H)
CAA Glutamina
CAG (Gln, Q)
CGU
CGC Arginina
CGA (Arg, R)
CGG
A
AUU Isoleucina
AUC (Ile, I)
AUA
AUG Metionina
(Met, M)
ACU
ACC Treonina
ACA (Thr, T)
ACG
AAU Asparagina
AAC (Asn, N)
AAA Lisina
AAG (Lys, K)
AGU Serina
AGC (Ser, S)
AGA Arginina
AGG (Arg, R)
G
GUU
GUC Valina
GUA (Val, V)
GUG
GCU
GCC Alanina
GCA (Ala, A)
GCG
GAU A. Aspártico
GAC (Asp, D)
GAA A. Glutámico
GAG (Glu, E)
GGU
GGC Glicina
GGA (Gly, G)
GGG
Pri
mera
posic
ión

Página 22
Herramientas y Referencias
M mega 106
K Kilo 103
m mili 10-3
µ micro 10-6
n nano 10-9
K pico 10-12
f femto 10-15
a atto 10-18
Tabla 13. Prefijos métricos
ds doble cadena ( como en dsDNA)
ss cadena sencilla (como en ssDNA)
bp pares de base
kb kilobase; 1.000 bases o pares de base
Da Dalton, unidad de masa molecular
mol mol
M molaridad, moles de soluto por litro de disolución
Tabla 14. Abreviaciones
Masa molecular media Códigos Aminoácidos
A Ala 71,08
C Cys 103,1
D Asp 115,1
E Glu 129,1
F Phe 147,2
G Gly 57,05
H His 137,1
I Ile 113,2
K Lys 128,2
L Leu 113,2
M Met 131,2
N Asn 114,1
P Peo 91,12
Q Gln 128,1
R Arg 156,2
S Ser 87,08
T Thr 101,1
V Val 99,07
W Trp 186,2
Y Tyr 163,2
Radioisótopo Vida media
Carbono-14 (14C) 5.730 años
Iodo-125 (125I) 60 días
Fósforo-32 (32P) 14,3 días
Azufre-35 (35S) 87,4 días
Tritio (3H) 12,4 años
Tabla 16. Masa molecular media de los aminoácidos
Tabla 15.. Vida media de los Radioisótopos más habituales
Copyright © 2011 Advansta. All rights reserved. The Advansta logo is a registered trademark of the Company. AdvanBlock™, AdvanWash™, Afyon™, Avant™, LucentBlue™ and
WesternBright™ are trademarks of the Company. All other trademarks, service marks and tradenames appearing in this brochure are the property of their respective owners.

Advansta Corporation
1455 Adams Drive, Ste. 1160 | Menlo Park, CA 94025
Tel: 650.325.1980 | Fax: 650.325.1904 | Email: [email protected]
www.advansta.com
Ptge. Dos de Maig, 9-11
Tel./Fax: 94 450 26 01
08041 BARCELONA
C/ S. Hermenegildo, 16
Tel.: 91 444 06 62; Fax: 91 594 02
28015 MADRID
www.ecogen.com
Importador
DISTRIBUIDOR LOCAL
Sevilla · Granada · Oviedo · Vigo · Zaragoza · Pamplona · Murcia · Valencia · Islas Canarias


















