“ANÁLISIS PROTEÓMICO DE LA APOPTOSIS EN Entamoeba …
140
INSTITUTO POLITÉCNICO NACIONAL ESCUELA NACIONAL DE MEDICINA Y HOMEOPATÍA SECCIÓN DE ESTUDIOS DE POSGRADO E INVESTIGACIÓN MAESTRÍA EN CIENCIAS EN BIOMEDICINA MOLECULAR TESIS QUE PARA OBTENER EL GRADO DE: MAESTRÍA EN CIENCIAS EN BIOMEDICINA MOLECULAR PRESENTA: Q.B.P. TANIA DOMÍNGUEZ FERNÁNDEZ MÉXICO, D.F; 2012 “ANÁLISIS PROTEÓMICO DE LA APOPTOSIS EN Entamoeba histolytica”
Transcript of “ANÁLISIS PROTEÓMICO DE LA APOPTOSIS EN Entamoeba …

INSTITUTO POLITÉCNICO NACIONAL
ESCUELA NACIONAL DE MEDICINA Y HOMEOPATÍA SECCIÓN DE ESTUDIOS DE POSGRADO E
INVESTIGACIÓN
MAESTRÍA EN CIENCIAS EN BIOMEDICINA MOLECULAR
TESIS
QUE PARA OBTENER EL GRADO DE:
MAESTRÍA EN CIENCIAS EN BIOMEDICINA MOLECULAR
PRESENTA:
Q.B.P. TANIA DOMÍNGUEZ FERNÁNDEZ
MÉXICO, D.F; 2012
“ANÁLISIS PROTEÓMICO DE LA
APOPTOSIS EN
Entamoeba histolytica”

DIRECTOR DE TESIS:
D. en C. DAVID GUILLERMO PÉREZ ISHIWARA
ASESORES:
D. en C. CONSUELO GÓMEZ GARCÍA
D. en C. ELIZABETH PÉREZ HERNÁNDEZ
D. en C. DORIS ATENEA CERECEDO MERCADO
D. en C. VIRGINIA SÁNCHEZ MONROY



Este trabajo se desarrolló en el Laboratorio de Biomedicina Molecular I de la
Escuela Nacional de Medicina y Homeopatía del Instituto Politécnico Nacional,
bajo la dirección del Dr. David Guillermo Pérez Ishiwara.

Los recursos empleados para desarrollar esta investigación fueron proporcionados
por el Consejo Nacional de Ciencia y Tecnología (CONACYT) con el proyecto
número 83197 y por la Secretaría de Investigación y Posgrado (SIP) con el
proyecto 29113499 y 20120527. Adicionalmente la autora del trabajo fue apoyada
con la Beca del CONACYT y la beca del Programa Institucional de Formación de
Investigadores (PIFI).

La ciencia es el alma de la prosperidad de las naciones y la fuente de todo
progreso.
Louis Pasteur
(1822-1895)

AGRADECIMIENTOS
El presente trabajo de tesis de maestría se debe al esfuerzo y a la orientación,
de la autor y del director de tesis, respectivamente.
Quiero agradecer al Dr. David Guillermo Pérez Ishiwara por su gran apoyo y
sugerencias para este trabajo y finalmente por conducirme en el área de la
investigación.
A mis asesores de tesis: Dra. Consuelo Gómez García, por su compañerismo en
el laboratorio y otras circunstancias, por sus comentarios y sugerencias para
este proyecto.
Dra. Elizabeth Pérez Hernández, por sus valiosas sugerencias en el proyecto,
sin duda sus conocimientos aportaron gran información al trabajo.
Dra. Virginia Sánchez Monroy, al ser parte y autora de los antecedentes del
proyecto, agradezco sus comentarios para el mismo.
Y finalmente a la Dra. Doris Atenea Cerecedo Mercado, una gran mujer a
quién agradezco haya aceptado estar en mi comité evaluador.
Mi reconocimiento a todos ustedes quienes juzgaron este proyecto con sus
valiosas sugerencias y comentarios durante el desarrollo del trabajo.
También quiero agradecer a todos mis amigas (os) y compañeras (os) que me
acompañaron durante el tiempo que estuve en el posgrado, sin duda fueron
parte de esto.
Finalmente agradezco al Profr. Gregorio Domínguez Herrera y a la Profra.
María Guadalupe Fernández Villalobos, mis padres, quienes son un pilar
fundamental para mi, gracias por su apoyo desinteresado.

CONTENIDO
Lista de abreviaturas ............................................................................................. I
Lista de tablas ...................................................................................................... III
Lista de figuras ..................................................................................................... IV
Resumen .............................................................................................................. VI
Abstract ................................................................................................................ VII
1. INTRODUCCIÓN ......................................................................................................................... 1
1.1. Historia ............................................................................................................................... 1
1.2. Ciclo Biológico ................................................................................................................... 3
1.3. Morfología .......................................................................................................................... 4
1.3.1. Trofozoíto ................................................................................................................... 4
1.3.2. Prequíste ......................................................................................................................... 6
1.3.3. Quíste .............................................................................................................................. 6
1.3.4. Metaquíste ...................................................................................................................... 6
1.4. Epidemiología ........................................................................................................................ 7
1.5. Mecanismos de patogenicidad y virulencia ...................................................................... 8
1.6. Procesos involucrados en la patogénesis ........................................................................ 9
1.6.1. Adhesión ....................................................................................................................... 10
1.6.2. Fagocitosis.................................................................................................................... 10
1.6.3. Lisis ................................................................................................................................ 11
1.6.4. Invasión ......................................................................................................................... 12
1.7. Mecanismos de evasión de la respuesta inmune .......................................................... 13
1.8. Muerte celular programada ............................................................................................... 15

1.8.1. Moléculas implicadas en el proceso de apoptosis:efectores intracelulares.......... 18
1.8.1.1. La familia de las caspasas ...................................................................................... 18
1.8.1.2. Proteína p53.............................................................................................................. 20
1.8.1.3. Familia de proteínas Bcl-2 ...................................................................................... 20
1. 9. Antecedentes ...................................................................................................................... 27
1.9.1. MCP en Leishmania .................................................................................................... 27
1.9.2 MCP en Trypanosomas ............................................................................................... 29
1.10 MCP en E. histolytica .................................................................................................... 30
1. 10. Proteómica ........................................................................................................................ 36
1. 11. Herramientas de la proteómica ...................................................................................... 36
1. 12. Análisis proteómico de E. histolytica ............................................................................. 38
2. JUSTIFICACIÓN ....................................................................................................................... 40
3. HIPÓTESIS ................................................................................................................................. 41
4. OBJETIVOS ............................................................................................................................... 42
5. ESTRATÉGIA EXPERIMENTAL ............................................................................................ 43
6. MATERIALES Y MÉTODOS ................................................................................................... 44
6.1. Cultivo de E. histolytica ...................................................................................................... 44
6. 2. Inducción de la apoptosis con G418 .............................................................................. 44
6.3. Obtención de proteínas ..................................................................................................... 44
6.4. Lisis de los trofozoítos ....................................................................................................... 45
6. 5. Precipitación proteíca ....................................................................................................... 45
6.6. Cuantificación proteíca ...................................................................................................... 45
6.7. Primera dimensión - isoelectroenfoque ........................................................................... 46
6.8. Segunda dimensión SDS-PAGE ...................................................................................... 49
6. 9. Visualización proteíca y análisis de imágenes .............................................................. 49

6.10. Digestión con tripsina de proteínas en el gel .............................................................. 51
6. 11. MALDI-TOF (MS); MALDI TOF/TOF (MS/MS) ............................................................ 52
6. 13. Papel de las proteínas identificadas ............................................................................. 54
7. RESULTADOS .......................................................................................................................... 55
8. DISCUSIÓN ................................................................................................................................ 86
10. PERSPECTIVAS ................................................................................................................... 103
11. BIBLIOGRAFÍA ..................................................................................................................... 104
12. ANEXOS ................................................................................................................................. 116

I
Lista de abreviaturas
WHO Organización Mundial de la Salud
ATP Adenosin trifosfato
Ca2+ Calcio
AIF Factor Inductor de Apoptosis
CHAPS [3-[(3-Colaamidopropil)dimetilamonio]-1
propanosulfonato]
DNA Ácido desoxirribunucleíco
DTT Ditiotreitol
Gal Galactosa
H Hora
K+ Ión potasio
M Molar
kDa Kilodaltones
MALDI Desorción/ionización mediante láser inducida por matriz
MCP Muerte Celular Programada
µg Microgramo
µl Microlitro
Ml Mililitro

II
NO Óxido nítrico
PBS Amortiguador de Fosfatos
PI Punto Isoeléctrico
PM Peso Molecular
PS Fosfatidil serina
TOF Tiempo de vuelo
NADP Nicotinamida-Adenina-Dinucleótido-Fosfato
IPG Gradientes de pH inmovilizados
CPA Cisteín proteasa
GDP Guanosin difostato
G418 Geniticina 418
AFLPs Polimorfismos en la longitud de fragmentos amplificados
MS Espectrometría de masas
TCA Acido triclo- roacético
IEF Isoelectroenfoque
E-64 Epóxido-64
PS Fosfatidilserina
Pb Pares de bases

III
Lista de tablas
Tabla 1. MCP en diferentes parásitos protozoarios…………………………………….. 25
Tabla 2. Protocolo de isoelectroenfoque Ettan IPGphor III…………………………….
56
Tabla 3. Protocolo de isoelectroenfoque…………………………………………………
57
Tabla 4. Volumen densitométrico de cada spots………………………………………..
75
Tabla 5. Peso molecular, punto isoeléctrico y expresión cualitativa de cada spot
monitoreado en los diferentes tiempos de inducción……………………………….......
76
Tabla 6. Resultados de huella peptídica (MALDI TOF) y MALDI TOF-TOF……........
78
Tabla 7. Proteínas diferenciales obtenidas de los perfiles proteómicos de
trofozoítos de E. histolytica inducidos a MCP con G418, identificadas por MALDI
TOF/ MADI TOFTOF………………………………………………………………………..
85
Tabla 8. Lista de proteínas de E. histolytica identificadas en geles bidimensionales
teñidos con azul de Coomassie, por Leitsch y colaboradores en el 2005…………….
90
Tabla 9. Lista de algunas proteína de E. histolytica reportadas por J. Tolstrup y
colaboradores en el 2006…………………………………………………………………..
91

IV
Lista de figuras
Figura 1. Proceso patogénico en E. histolytica. ……………………………………........ 9
Figura 2. Esquema comparativo de las características morfológicas de la muerte
celular por apoptosis y por necrosis……………………………………...........................
17
Figura 3. Vías apoptóticas………………………………………………………...............
24
Figura 4. Primera dimensión isoelectroenfoque………………………………...............
48
Figura 5. Perfil proteómico de trofozoítos de E. histolytica sin inducir a MCP con
G418…………………………………………………………………….................................
59
Figura 6. Imagen del perfil proteómico de trofozoíto sin inducir a MCP………………..
61
Figura 7. Perfil proteómico de 1 ½ h (90 min) de inducción a MCP…………………….
62
Figura 8. Perfil proteómico de 3 h de inducción a MCP………………………………….
63
Figura 9. Perfil proteómico de 6 h de inducción a MCP………………………………….
64
Figura 10. Perfil proteómico de 9 h de inducción a MCP………………………………..
65
Figura 11. Comparación del perfiles proteómicos. (A) trofozoítos sin inducir a MCP
(B) trofozoítos expuestos 1 ½ h a G418……………………………………………………
67
Figura 12. Comparación del perfiles proteómicos. (A) trofozoítos expuestos 1 ½ h a
G418 (B) trofozoítos expuestos 3 h a G418……………………………………………….
68

V
Figura 13. Comparación del perfiles proteómicos. (A) trofozoítos expuestos 3 h a
G418 (B) trofozoítos expuestos 6 h a G418……………………………………………….
69
Figura 14. Comparación del perfiles proteómicos. (A) trofozoítos expuestos 6 h a
G418 (B) trofozoítos expuestos 9 h a G418……………………………………………….
70
Figura 15. Etiquetas de los spots diferenciales, utilizando Ludesi REDFIN3………….
71
Figura 16. Ampliación de los spots expresados diferencialmente de los perfiles
proteómicos de trofozoítos: sin inducir a MCP (control); y a distintos tiempos de
inducción de MCP (1 ½, 3, 6 y 9 h)…………………………………………………………
73

VI
RESUMEN
Durante el proceso de infección, Entamoeba histolytica utiliza diversos
mecanismos para evadir la respuesta inmune; uno de los mecanismos descritos
es la inducción de la apoptosis a leucocitos, macrófagos y células hepáticas,
empleando moléculas como lectinas, lipasas y toxinas. Se ha demostrado que la
muerte celular programada (MCP) en este parásito también se induce in vitro: i)
mediante el aminoglicósido G418; ii) por especies reactivas de oxído nítrico y iii)
por especies reactivas de oxígeno. In vivo, nuestro grupo de investigación
demostró en un modelo de hámster chino, que el producto de la interacción entre
el hospedero y el parásito induce la apoptosis tanto en los neutrófilos como en los
trofozoítos, lo que sugiere que durante el proceso fisiopatogénico de la
enfermedad se desarrolla una respuesta del huésped contra el parásito, con la
consecuente activación de mecanismos de defensa del parásito. Recientemente,
demostramos mediante cDNA-AFLPs que la MCP inducida por fármacos
representa un mecanismo activo que involucra la expresión de genes específicos,
como la Glutaminil-tRNAsintetasa, las Subunidades ribosomales 40S y 18S, el Sir-
2, las Graininas 1-2 y la Saponina-like que están implicados en la regulación de
rutas pro- y anti-apoptóticas. En este trabajo presentamos el análisis proteómico
comparativo de la MCP inducida con G418. Los resultados mostraron la expresión
de proteínas diferenciales en los diversos estadios de inducción de la MCP,
identificando hasta el momento por huella péptidica y Maldi TOF TOF, proteínas
del citoesqueleto como actina, proteínas del metabolismo energético como la
alcohol deshidrogenasa y la gliceraldehído-3-fosfato deshidrogenasa e
interesantemente una proteína de unión a Ca2+ como la grainina 2. Estas
proteínas en otros sistemas se ha demostrado que actúan como proteínas pro- y
anti-apoptóticas.

VII
ABSTRACT
During the infection process, Entamoeba histolytica uses different mechanisms to
evade the immune response; one of them is the induction of apoptosis in
leukocytes, macrophages and liver cells, using molecules such as lectins, lipases,
and toxins. It has been shown that programmed cell death (PCD) in this parasite is
also induced in vitro: i) using the aminoglycoside G418, ii) by reactive species of
nitric oxide and iii) by reactive oxygen species. In vivo, our research group
demonstrated in a Chinese hamster model, that after interaction between host and
parasite, apoptosis in both neutrophils and in trophozoites, were induced
suggesting that during the pathophysiological interaction a host response against
the parasite is developed, with the subsequent activation of parasite defense
mechanisms. Recently, we demonstrated by cDNA-AFLPs that PCD induced by
drug represents an active mechanism that involves the expression of specific
genes, such as glutaminyl-tRNAsintetasa, the 40S and 18S ribosomal subunits, the
Sir-2, and Graininas 1-2 and saponin-like that are involved in the regulation pro-
and anti-apoptotic routes. In this assay we present a comparative proteomic
analysis of PCD induced by G418. The results showed differential protein
expression in various stages of induction of PCD, by Peptide fingerprinting and
Maldi TOF TOF, we identified actin, alcohol dehydrogenase, glyceraldehyde-3-
phosphate dehydrogenase and a grainina 2. Some of them acting as pro- and anti-
apoptotic proteins.

1
1. INTRODUCCIÓN Las enfermedades parasitarias son una de las causas de morbilidad más
importantes a nivel mundial, las cuales reflejan deficiencias sanitarias,
características de áreas pobres donde priva el hacinamiento y el mal manejo de
las aguas y de las excretas. La amebiasis representa la tercera enfermedad
parasitaria a nivel mundial y en México, al ser endémica, constituye un problema
de salud pública muy importante (Conde-Bonfil 1992; WHO 1997).
Entamoeba histolytica, es el agente causal de la amebiasis, reside y se multiplica
en el intestino grueso causando en la mayoría de los casos una infección
asintomática en el lumen del intestino; otras manifestaciones clínicas incluyen la
diarrea y la disentería, sin embargo, ocasionalmente E. histolytica penetra la
mucosa intestinal, y genera una colitis ulcerativa o bien se disemina hacia otros
órganos como el pulmón y el cerebro. Sin embargo, el órgano principal es el
hígado, donde induce la formación de abscesos (Tillack 2007; Baxt 2008) .
1.1. Historia
E. histolytica fue descrito por Rösel van Rosenhof desde el siglo XVII y XVIII,
siendo Lösch quien describió la disentería amebiana en San Petersburgo, Rusia
(Bernal-Redondo 2001). En 1875 descubrió en un campesino de 24 años que
sufría de disentería, unos microorganismos móviles que poseían ecto y
endoplasma y contenían glóbulos rojos. El investigador inoculó 4 perros por vía
rectal y oral con las heces del paciente y logró reproducir en uno de ellos, la
disentería, con ulceraciones en la mucosa intestinal y amibas en el exudado. El
paciente enfermo murió a los 7 meses y la necropsia demostró numerosas y
extensas ulceraciones de la mucosa del colon, donde de nuevo observó los
microorganismos. No obstante estos hallazgos, el autor no consideró a la amiba

2
como el agente etiológico, sino como un coadyuvante mecánico que impedía la
curación de las lesiones originadas por otro agente causal (Jackson, Gathiram, et
al.,1985).
Koch en 1883, revisando necropsias en una epidemia de cólera, demostró las
amibas en la submucosa de la pared intestinal, en los capilares cercanos a la
pared de abscesos hepáticos y en el exudado de lesiones del hígado. Los
hallazgos de Koch fueron confirmados por Kartulis (1885 – 1887), al demostrar la
presencia de amibas en 150 necropsias de casos de disentería. A este autor se le
considera el primero en afirmar que la amiba era el agente etiológico de la
disentería tropical y que el absceso del hígado era una secuela de la disentería
experimental. Councilman y Lafleur (1891), consideraron la disentería como una
entidad clínica caracterizada por lesiones asociadas a la amiba. Usaron por
primera vez las expresiones disentería amibiana, absceso amibiano del hígado y
propusieron para el agente causal el nombre de Amoeba dysenteriae. Heuber en
1903, hizo la descripción de los quistes de esta amiba y Schaudinn de los
trofozoítos, este asignó el nombre de E. histolytica por su capacidad de lisar tejido
y producir úlceras intestinales (Bernal-Redondo 2001); este autor diferenció dos
especies: Entamoeba histolytica o amiba patógena y Entamoeba coli o no
patógena. Para mostrar esta diferencia de patogenicidad ingirió quistes y sufrió
como consecuencia dos crisis disentéricas, lo que para mucho fue la causa de su
temprana muerte. Posteriormente, se adoptó el nombre genérico Entamoeba, que
había sido propuesto desde el siglo pasado.
Los trabajos definitivos sobre la patogenicidad de E. histolytica, fueron realizados
en 1913 por Musgrave y Clegg y por Walker y Sellards, quienes suministraron
quistes de E. histolytica y quistes de E. coli a voluntarios sanos y obtuvieron la
disentería solo aquellos que ingirieron E. histolytica.
En 1914 se realizaron los trabajos inmunológicos por Izar quien preparó antígenos
acuosos de E. histolytica a partir de materias fecales y obtuvo reacciones positivas

3
de fijación del complemento. En 1924 Boeck y Drbohlav lograron cultivar con éxito
E. histolytica en un medio artificial con base de huevo, que contenía la flora
microbiana de las materias fecales. Con este tipo de cultivo Craig, en 1927,
preparó un antígeno con fines diagnósticos para fijación del complemento.
En 1925 Brumpt, mediante experimentos en gatos y en humanos, diferenció una
amiba patógena, E. dysenteriae, equivalente a E. histolytica (Pimenta, Diamond, et
al., 2002) y una no patógena, E. dispar. Diamond en 1961 obtuvo por primera vez
un cultivo axénico, es decir, libre de bacterias, el cual ha sido utilizado para
preparar antígenos con alto grado de pureza para diversas reacciones serológicas.
En 1993, Diamond y Clark, re-describieron la existencia de dos especies
diferentes: E. histolytica, patógena y E. dispar, no patógena, morfológicamente
iguales, pero con diferencias inmunológicas, bioquímicas y genéticas (Botero,
1994).
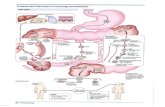
1.2. Ciclo Biológico
El ciclo de vida de E. histolytica se desarrolla a través de una alternancia de
crecimiento trofozoítico y de la formación periódica de quistes (Calzado, Verde et
al. 2007). Esto comienza después de la ingestión de un quiste no móvil, por el
huésped humano. La forma móvil del parásito o trofozoíto surge después de la
digestión de la pared del quiste durante el paso a través del intestino delgado y
coloniza el intestino grueso, donde prolifera, mientras se alimentan de
la flora bacteriana residente. Para completar el ciclo de vida, los trofozoítos se
desarrollan en quistes cuadri-nucleados y pasan hacia el ambiente exterior
con las heces del huésped. E. histolytica puede existir como un
comensal en el intestino del ser humano, o causar disentería o abscesos
extraintestinales. La proliferación de los trofozoítos en la luz del intestino se
produce en un ambiente polixénico donde coexiste con diferentes especies de
bacterias anaerobias y microaerófilas. En contraste, durante la invasión del

4
epitelio intestinal o en las lesiones extraintestinales como abscesos hepáticos, las
amibas proliferan en un ambiente libre de otros microorganismos (Mukherjee,
Clark, et al., 2008).
1.3. Morfología
La división metaquística en el ciclo de vida de E. histolytica forma un quíste
maduro siendo una amiba metaquística, con cuatro núcleos quísticos. Cada amiba
metaquística produce finalmente ocho amibas por una serie de divisiones
nucleares y citoplasmáticas. Cada núcleo quístico se divide, resultando que ocho
núcleos son producidos, que se separan y forman ocho amibas (Dutta 1960).
1.3.1. Trofozoíto
El trofozoíto o forma móvil, es extraordinariamente pleomórfico. Se multiplica por
fisión binaria y es muy sensible al jugo gástrico y a los agentes externos. Su
hábitat comprende la luz y la pared del colon y específicamente el ciego y el recto
(Pumarola 1991). Su tamaño es muy variable y oscila entre 10 y 60 µm. Las
formas más pequeñas corresponden a las no invasivas y se encuentran
habitualmente en los casos asintomáticos. Los de mayor tamaño son las formas
invasivas, que a diferencia de los anteriores no aparecen en la luz intestinal y
poseen en el endoplasma restos celulares o hematíes.
El trofozoíto presenta una membrana citoplásmica dividida en dos porciones: una
externa llamada ectoplasma y una porción interna denominada endoplasma (Petri,
1993)
La pared celular periférica del trofozoíto, recibe el nombre de ectoplasma, es
hialina, transparente, retráctil y casi sin granulaciones (Pumarola 1991). Los

5
pseudópodos son prolongaciones del ectoplasma y proporcionan una movilidad al
parásito de aproximadamente 50 µN/s (Romero, 1993).
El endoplasma tiene una estructura granular fina y, en las formas mayores,
abundantes vesículas y vacuolas, que contienen restos celulares y hematíes
intactos o en vías de degradación, pero no bacterias (Pumarola, 1991).
El núcleo es esférico con un acumulo de cromatina pequeña y puntiforme en el
centro, encerrados en una cápsula llamados cariosoma o endosoma (Petri 1993).
También presenta cromatina adherida a la cara interna de la membrana nuclear,
distribuida en forma más o menos homogénea (Romero, 1993).
El trofozoíto se nutre por fagocitosis a expensas de los tejidos disueltos y
hematíes, y se ayuda de los pseudópodos. Al microscopio electrónico no se
detecta aparato de Golgi, microtúbulos, mitocondrias, ni retículo endoplásmico
rugoso (Pumarola, 1991).
Si las condiciones del medio ambiente no son propicias, el trofozoíto empieza a
cambiar de forma, deja de emitir pseudópodos, el ectoplasma y el endoplasma ya
no se diferencian, de manera que casi desaparece el primero, se pierde la forma
irregular y se hace esférico, al tiempo que aparece una pared gruesa llamada
pared quísitica (Petri, 1993).
También se expulsa al exterior todo el contenido de las vacuolas y empieza a
formar material de reserva como vacuolas de glucógeno y barras cromatoidales. El
cambio de trofozoíto concluye con la formación de un quíste (Romero, 1993).

6
1.3.2. Prequíste En el estado prequístico, el trofozoíto tiene aproximadamente el mismo tamaño
que el quíste. El citoplasma contiene inclusiones de restos celulares pero
usualmente contiene depósitos difusos de glucógeno y ocasionalmente cuerpos
cromatoides. Los cuerpos cromatoides están compuestos de ribosomas; sin
embargo, su papel funcional es incierto. La forma prequística es uninucleada y el
núcleo es agrandado, el cual contiene un cariosoma.
1.3.3. Quíste El quiste o elemento infectante es redondo u oval y de 10-25 µm de tamaño.
Posee una pared lisa de 0.6 µm y es resistente al jugo gástrico, factores
ambientales externos y cifras habituales de cloro del agua (entre 0,5 y 0,2 mg/L)
(Pumarola, 1991). Posee de 1 a 4 núcleos, según la fase de maduración. Los
quistes jóvenes tienen 1 o 2 núcleos, algunos cuerpos cromáticos y vacuolas de
glucógeno. Cuando el quiste madura, posee 4 núcleos y desaparecen los cuerpos
cromáticos. Solo los quistes maduros son infecciosos. Los cuerpos cromáticos
contienen principalmente ácidos nucleicos y fosfatos (Pumarola, 1991).
1.3.4. Metaquíste Durante el proceso de enquistamiento, la amiba contiene 4 núcleos que son muy
activos. La amiba cuadri-nucleada escapa de la pared del quiste a través de un
poro pequeño. Afuera del quíste, los núcleos de la amiba cuadrinucleada
comienzan a separarse de alrededor del citoplasma y someterse a la división para
formar ocho trofozoítos metaquísticos uninucleados. Los trofozoitos resultantes
son siempre más pequeños que los trofozoitos presentes en el intestino de una

7
persona infectada. Los trofozoítos metaquísticos siguen alimentándose y
creciendo para colonizar el intestino (Bruckner, 1992).
1.4. Epidemiología La Organización Mundial de la Salud reportó que E. histolytica causa
aproximadamente 50 millones de casos y 100, 000 muertes (Gaucher, 2002) al
año, principalmente en el centro y sur de América, África y la India, así como una
considerable morbilidad manifestada como características clínicas intestinales o
extraintestinales invasoras (Walsh, 1986).
La gran mayoría de las infecciones son adquiridas en países en desarrollo; por
ejemplo, en un estudio realizado por Rashidul Haque et al, se observó que el 39%
de los niños de un barrio pobre de la zona urbana de Dhaka, Bangladesh, había
una nueva infección por E. histolytica durante un estudio de un año (Haque,
2006).
A nivel mundial, la amebiasis es la tercera causa más común de muerte debido a
una infección parasitaria, después de la malaria y la esquistosomiasis. (Tanyuksel,
2003). Las infecciones por amebiasis son endémicas en la mayoría de las zonas
con climas templados y tropicales en países en desarrollo. La prevalencia de la
amebiasis varía en los distintos países dependiendo de las condiciones
socioeconómicas. (Tanyuksel, 2003). En los países industrializados, la amebiasis
se produce en relaciones sexuales en hombres homosexuales, inmigrantes,
turistas que viajan a zonas endémicas de infección y en individuos
inmunodeficientes, VIH positivos.
La prevalencia global de la infección por E. histolytica en los países
industrializados, como los Estados Unidos, se ha estimado entre un 4% por año a
pesar de la presencia de algunos grupos de alto riesgo. Los estudios

8
epidemiológicos han demostrado que el bajo nivel socioeconómico y las
condiciones insalubres son factores significativos de riesgo. Además, las personas
de edad temprana que viven en países en desarrollo tienen un mayor riesgo que
aquellas que están en regiones desarrolladas. Por ejemplo, en México, 11% de la
población estudiada de 5 a 9 años fueron positivos para la infección, siendo el
porcentaje superior en las niñas (9,34%). Investigaciones seroepidemiológicas de
la amebiasis en algunas zonas tropicales de México indican que, mientras que la
prevalencia de anticuerpos anti-amebianos es relativamente baja en zonas donde
la transmisión de la epidemia no se ha informado, durante las epidemias una tasa
de incidencia de 50% es común, llegando a 80% en caso de epidemia. En los
países desarrollados como Italia, Japón, y Estados Unidos, la prevalencia de la
infección por Entamoeba es entre el 4 y 21% en los hombres que practican sexo
oral-anal con otros hombres, pero la mayoría de las infecciones se deben a la
especie no invasiva, E. dispar, que no requiere tratamiento (Tanyuksel, 2003).
1.5. Mecanismos de patogenicidad y virulencia Mucho se ha avanzado en el conocimiento de la biología de E. histolytica, a partir
de la aparición de los primeros medios sintéticos para su cultivo y su posterior
perfeccionamiento, sin embargo, el papel de la respuesta inmune en el control de
la enfermedad no está bien establecido, menos aún el singular comportamiento
del parásito observado por décadas, que con frecuencia actúa, como comensal y
más raramente como invasor. En general, la capacidad patogénica depende entre
otras cosas, de las sustancias tóxicas que libera, ya sea enzimas o citotoxinas, de
su asociación con otras bacterias, y del estado inmunológico y nutricional del
huésped.
Aunque el término "patogénesis", definido como los mecanismos implicados en el
inicio, evolución y resultado final de un proceso patológico, se refiere tanto a

9
factores del huésped y del parásito, nos centraremos principalmente en los
mecanismos parasitarios que pueden estar relacionados con la amebiasis
invasiva.
1.6. Procesos involucrados en la patogénesis En la actualidad se han identificado diferentes moléculas asociadas a la virulencia
del parásito como son las lectinas, las proteasas, las toxinas y las lipasas.
Figura 1.- Proceso patogénico en E. histolytica. Tomado de TRENDS in Parasitology.

10
1.6.1. Adhesión Para que ocurra la invasión intestinal por los trofozoítos de E. histolytica es
necesario el contacto célula-célula y la adherencia a las células del huésped,
mediante moléculas de superficie del parásito llamadas adhesinas (Canales-
Trevino, 1986). Estas se adhieren a las células epiteliales de la mucosa intestinal y
a las células inflamatorias (Leroy, 2000). Durante este proceso se da una
interacción entre las lectinas del parásito Gal / GalNAc con glicoproteínas (mucina)
que se encuentran en el moco en la luz intestinal del hospedero, que son ligandos
de alta afinidad por la lectina amebiana (Tanyuksel, 2003). Las lectinas son
glicoproteínas, que reaccionan con los azúcares de las paredes celulares. La
lectina Gal/GalNAc facilita la adherencia a la célula diana (como se observa en la
figura 1), la resistencia al complemento y la citotoxicidad (Haque, 2001).
Las cepas virulentas de E. histolytica contienen varias lectinas, de las cuales dos
lectinas de superficie están fuertemente relacionadas con la virulencia del parásito.
La lectina de 140 kDa está unida a la membrana y tiene una especificidad para los
oligosacáridos de N-acetil glucosamina (GlcNAc), mientras que la lectina de 220
kDa tiene afinidad por la galactosa y por la N-acetilgalactosamina (GalNAc).
(Meza, Cazares, et al., 1987).
1.6.2. Fagocitosis La fagocitosis se caracteriza por la ingestión de restos celulares, células vivas,
fragmentos inertes y eritrofagocitosis. Existen mecanismos moleculares muy
específicos donde intervienen proteínas de superficie (Arroyo, 1987), cisteín
proteasas (CPs) (Ankri, 1998) y componentes del citoesqueleto. La fagocitosis es
acompañada por la formación del fagolisosoma (Stuart, 2005) dirigido por
GTPasas y proteínas Rab, que actúan como moduladores en la regulación de la
fusión vesicular con las membranas de la célula blanco.

11
El citoesqueleto de E. histolytica le permite realizar dos funciones importantes
relacionadas con la patogénesis: la locomoción y la fagocitosis. Esto se logra
mediante la polimerización de la actina, la cadena pesada de la miosina II y un
conjunto de proteínas ligadoras de calcio, además de otros mecanismos de
señalización intracelular (Meza, 2006). La lectina Gal-GalNac, a través de una
serie de cinasas transmembranales relacionadas, induce cambios estructurales en
el trofozoíto y en la célula blanco a través de mecanismos no identificados, para
formar una placa de anclaje. Después, la miosina pesada tipo II y la actina
interactúan “contrayendo” el trofozoíto, que de esta manera avanza. Durante el
proceso de fagocitosis, el reconocimiento de la célula blanco depende
nuevamente de la lectina. Así, el trofozoíto penetra rápidamente por la mucosa y la
submucosa, extendiendo la invasión a capas más profundas. Las cisteín proteasas
juegan un papel importante en esta etapa, dado que permiten degradar casi todos
los componentes de la matriz extracelular y su actividad depende de la virulencia
del trofozoíto. Los principales componentes destruidos por las proteasas son
laminina, colágeno tipo I y IV, y fibronectina. A medida que avanza la invasión, la
úlcera se extiende profundamente en un área mayor de la submucosa, dado que
el tejido subyacente al epitelio interglandular ofrece menor resistencia, lo que da
lugar a las típicas úlceras en botón. Para obtener el hierro necesario para su
supervivencia, el trofozoíto procesa los eritrocitos fagocitados por medio de
hemoglobinasas. En este momento se aprecia una notable infiltración
caracterizada por neutrófilos, macrófagos y algunos linfocitos (Romero-Diaz,
2007).
1.6.3. Lisis La lisis de las células epiteliales ocurre a través de la acción del amoebaporo, y de
la degradación de la matriz por acción de cisteín proteasas y otras enzimas líticas
(Carrero, 2007). Varias proteínas que forman el poro amebiano (30, 14 y 5 kDa) se
han descrito. Una proteína amibiana de 30 kD se purificó y se demostró que

12
participa en la lisis de los eritrocitos insertándose en la bicapa lipídica y creando
poros. Otra proteína de 14 kDa que forma poros fue descrita como la proteína que
forma el canal iónico. De éstos, la proteína de 5 kDa, es la más importante y
principal formadora del amoebaporo (Leippe, et al., 1991). Esta proteína está
compuesta por 77 aminoácidos, incluyendo 6 residuos de cisteína. Al igual que
otros polipéptidos que penetran la membrana, también tiene una conformación
helicoidal, todos α- hélices (Leippe, et al., 1992).
Las cisteína proteasas se sabe que son importantes para la patogénesis de E.
histolytica al degradar proteínas extracelulares. Aproximadamente 20 genes, que
codifican para las cisteín proteasas se han identificado. Recientemente, se ha
demostrado que la expresión diferencial de estos 20 genes varía entre E.
histolytica y E. dispar, explicando al menos un mecanismo que permite a E.
histolytica invadir al huésped (Stauffer, et al., 2003).
1.6.4. Invasión Después de la adherencia a la célula huésped a través de Gal-lectina prosigue un
proceso de invasión (figura 1). Se entiende que la invasividad es la capacidad de
destruir localmente los tejidos colonizados, en este caso depende de la liberación
de CPs, del amoebaporo, de las colagenasas, de las fosfatasas acidas y de otras
enzimas hidrolíticas, así como de las hidrolasas (Espinosa-Cantellano, 2000);
(Ackers, 2006).
La invasión es seguida por un proceso de inflamación agudo que recluta
neutrófilos, células plasmáticas, eosinófilos, macrófagos y linfocitos, los cuales
contribuyen al daño tisular por liberación descontrolada de sus productos tóxicos
(Olivos-Garcia, 2004).

13
La mucosa es destruida y el parásito se propaga en el tejido, formando una típica
úlcera, asociada con diarrea conteniendo moco y sangre (Hughes 2000).
Posteriormente, puede producirse la diseminación de los trofozoítos a diferentes
órganos y tejidos (Espinosa-Cantellano, 2000).
1.7. Mecanismos de evasión de la respuesta inmune Durante el curso de la invasión en el colon o la dispersión hacia el hígado, los
trofozoítos de E. histolytica están en continua exposición con el sistema del
complemento. Los trofozoítos activan la vía clásica y alterna del complemento en
la ausencia de anticuerpos antiamebianos a través de una CP de 56 kDa (Reed,
1990). Esta CP degrada a las fracciones C3, C4, C5 y C9, evitando así efectos
fisiológicos de anafilatoxinas C3a, C5a, así como el incremento de la
permeabilidad vascular y la contracción del musculo liso; de igual manera generan
la supresión de la proliferación de las células T, además de limitar la quimiotaxis
de los neutrófilos (Reed, 1989).
Por otra parte, se ha descrito que la lectina Gal/GalNac inhibe también el
ensamblaje de C8 y C9 del complemento de ataque a la membrana C5b-9, lo que
impide la lisis del parásito mediada por el complemento (Braga, 1992).
Otro mecanismo de evasión de la respuesta inmune usado por Entamoeba, es la
digestión proteolítica de anticuerpos IgA e IgG por proteasas (Kelsall, 1993). Los
anticuerpos IgA se desarrollan durante el episodio de infección intestinal, previo
y/o concomitante con el episodio hepático, es decir invasivo, donde se sintetizan
anticuerpos IgG, sin embargo el problema básico que el paciente tiene es que
aunque la cantidad de anticuerpos específicos IgA e IgG contra la amiba
aumentan, estos son por un lado degradados por las proteasas amibianas, y por
otro removidos de su superficie, a través del recambio membranal del trofozoíto,

14
mediante la formación del casquete y muda de los anticuerpos unidos a su
membrana, ya que se deshace de ellos, evitando así su acción (Calderon, 1980).
In vitro e in vivo, algunos parásitos internalizan los complejos inmunes de la
membrana y recuperan su forma y movilidad al cabo de dos horas y ya no se
inmovilizan al ser re-expuestos al suero inmune (Biagi, 1966) (Cole, 1953); (Neal,
1971).
Por otro lado, varios estudios han documentado que la amiba interfiere en la
actividad anti-amebiana de los neutrófilos, interrumpiendo las actividades de la
NADPH oxidasa, y resistiendo a través de la peroxirredoxina de 29 kDa (Guerrant,
1981) (Arbo, 1990). También se ha demostrado in vitro que E. histolytica puede
inducir la apoptosis de los neutrófilos, siendo este mecanismo un importante
evento en la resolución de la inflamación y por lo tanto de la sobrevivencia del
parásito. Sim y colaboradores en el 2005, investigaron los aspectos
ultraestructurales de la muerte de neutrófilos provocada por E. histolytica, al
incubar neutrófilos humanos de sangre periférica, en ausencia o presencia de
trofozoítos de E. histolytica. Las evidencias de microscopía electrónica de
transmisión (TEM) muestran que los neutrófilos incubados con E. histolytica se
observaron con características de apoptosis, tales como la compactación de la
cromatina y condensación de la envoltura nuclear. Por el contrario, los neutrófilos
incubados en ausencia de la amiba tenían protuberancias en la superficie celular y
heterocromatina nuclear irregular. Estos datos sugieren que la apoptosis de
neutrófilos inducida por Entamoeba contribuye a prevenir la inflamación de los
tejidos, limitando los daños en las lesiones donde la amiba invade (Sim, 2005).

15
1.8. Muerte celular programada La apoptosis o muerte celular programada (MCP), está referida a una forma
específica de muerte celular, proceso que ocurre en los tejidos en condiciones
fisiológicas normales. El nombre de apoptosis fue propuesto por Kerr y Searle
(1972), utilizando un concepto griego de Apo (απο), desde, y Ptosis (πτοσισ),
caída o prolapso de un órgano o parte de él. Glücksmann (1951), fue el primero en
observar la muerte de células durante el desarrollo.
Desde 1972, se han publicado diferentes observaciones referidas a la muerte
celular programada, Kerr y Searle (1972) propuso que la muerte celular
programada era un evento morfogénico normal en el desarrollo de los organismos
multicelulares, describiendo las características morfológicas de células
desintegradas en variados tipos de tejidos (Van Furth, 1998).
La apoptosis constituye por lo tanto un fenómeno fundamental en el desarrollo
embrionario (Bellairs 1961), en la metamorfosis (Goldsmith, 1966), en la atrofia
tisular y la regresión tumoral (Wyllie, 1973; Ucker, 1991) tanto en animales
vertebrados como invertebrados.
Trabajos de Jacobson y colaboradores en 1997 y Lam en el 2004 han descrito que
la MCP ha sido reconocida como un proceso fundamental en los organismos
multicelulares, tanto en animales como plantas. Recientemente, procesos de
muerte celular programada han sido reportados en eucariotes unicelulares, lo que
implica que algunos componentes de la maquinaria de MCP existían a principios
de la evolución de organismos eucariotes.
La apoptosis se caracteriza por una serie de alteraciones morfológicas y
bioquímicas. Ocurre en dos fases: primero un marcaje de la muerte celular,
seguido por una fase de ejecución, caracterizado por dramáticos cambios

16
morfológicos muy característicos en la estructura celular, sugiriendo la presencia
de una maquinaria común en los diferentes organismos (Hsu, 1997).
Los cambios morfológicos que ocurren en las células son: agregación de la
cromatina formando núcleos picnóticos, observables al microscopio óptico;
condensación nuclear y citoplasmática y partición del citoplasma en pequeñas
vesículas llamadas cuerpos apoptóticos que contienen ribosomas, mitocondrias
morfológicamente intactas y material nuclear. Las alteraciones en la disminución
en el volumen celular y la exposición de proteínas específicas en la membrana
plasmática resultan en el reconocimiento y la fagocitosis de células apoptóticas,
evitando así una respuesta inflamatoria (Hsu, 1997) (figura 2).
Ultraestructuralmente, se pierden las uniones estrechas así como de otras
proteínas de la membrana celular. In vivo, estos cuerpos apoptóticos son
rápidamente reconocidos y fagocítados por macrófagos o células epiteliales
adyacentes (Savill, 1993). In vitro, los cuerpos apoptóticos quedan como restos de
células fragmentadas que luego se hinchan y finalmente se rompen, esta fase
terminal en las células in vitro es llamada necrosis secundaria (Wyllie, 1981) .
La muerte celular se puede producir por necrosis, cuando el daño es letal o se
produce una muerte accidental. La necrosis (del griego nekrós “muerte”) es la
muerte patológica de las células o tejidos del organismo. Se origina por una lesión
aguda, irreversible, derivada de una situación no fisiológica o condición patológica
y que no puede ser reparada por mecanismos de adaptación y de resistencia. Ésta
se produce debido a agentes nocivos, condiciones o circunstancias determinadas,
como un aporte insuficiente de sangre al tejido (isquemia), defiencia de oxígeno
(hipoxia), un traumatismo, la exposición a la radiación ionizante, la acción de
sustancias químicas o tóxicos o, por ejemplo, por una infección o por el desarrollo
de una enfermedad autoinmune. Esta forma de muerte celular se califica como un
proceso violento ya que las células se hinchan, las estructuras celulares se
deterioran, y las funciones críticas para la vida se paralizan. La pérdida de

17
viabilidad se asocia a la ruptura de la membrana plasmática con la consecuente
lisis celular y liberación al exterior del contenido citoplasmático y orgánulos,
dañando al tejido en el que se encuentra. La liberación del contenido celular puede
provocar a su vez reacciones inflamatorias importantes (Degterev A, 2008). Los
cambios morfológicos que se observan en una célula necrótica se observan en la
Figura 2.
Figura 2. Esquema comparativo de las características morfológicas de la muerte celular por
apoptosis y por necrosis. Los diagramas muestran los cambios morfológicos que se observan en
las células apoptóticas y en las necróticas. En las micrografías obtenidas mediante microscopía de
transmisión electrónica se compara el núcleo de una célula normal (panel superior) con el de una
célula en la que se ha iniciado el proceso de apoptosis (panel inferior). Tomado de Lizarbe, 2007.

18
Es evidente que la muerte por apoptosis es más limpia que la necrosis; se
detectan cambios morfológicos particulares y la membrana celular, que no se
destruye, engloba a los cuerpos apoptóticos o material celular (Figura 2). Cabe
enfatizar nuevamente que no se produce inflamación ya que las células
fagocitarias reconocen, captan y eliminan los cuerpos apoptóticos. La apoptosis se
puede definir entonces como “el conjunto de reacciones bioquímicas que tienen
lugar en la célula y que determinan su muerte de una forma regulada en respuesta
a una serie de acontecimientos fisiológicos o patológicos”. En este caso, una serie
de estímulos o señales hacen que la célula programe su propia muerte (Vaux D.L,
et al., 1992; Hengartner M.O, et al., 1994)
1.8.1. Moléculas implicadas en el proceso de apoptosis: efectores intracelulares
La apoptosis es un mecanismo evolutivamente conservado. Se ha demostrado
que los mecanismos moleculares de la apoptosis dependen primordialmente de
una familia de proteasas. Estas proteasas tienen la característica en su secuencia
de tener una cisteína en el sitio activo. Esta cisteína va a interactuar, rompiendo
los enlaces peptídicos de los residuos de ácido aspártico en las moléculas diana.
Por eso, las proteínas que inician la apoptosis se denominan caspasas, que van a
mediar los principales cambios morfológicos.
1.8.1.1. La familia de las caspasas Las caspasas son una familia muy regulada y bien conservada de cisteín
proteasas intracelulares. Una vez que se activa la caspasa iniciadora, los procesos
de caspasas efectoras, río abajo son responsables de la apoptosis. Dado que la
señalización de apoptosis mediada por caspasas es irreversible, la activación de

19
procaspasas y caspasas deben ser reguladas en forma precisa para prevenir la
muerte no deseada de la célula huésped (Lu, 2008).
Las caspasas implicadas en la apoptosis se dividen generalmente en dos
categorías: las caspasas iniciadoras, que incluyen a la caspasa 2, 8, 9 y 10, y las
caspasas efectoras, que incluyen a la caspasa 3, 6 y 7. La tercera subfamilia de
caspasas es la de las caspasas procesadoras de citocinas; caspasa 1, 4, 5, 12,
13, 14 (Earnshaw W.C, et al., 1999).
Se ha descrito que las caspasa 1 se activa por la ausencia de factores de
crecimiento, que hidroliza a la caspasa 3 in vitro y que promueve el procesamiento
y activación de la interleucina-1b (IL-1b), sustancia implicada en la muerte
neuronal. En cuanto a la caspasa 2, se sabe que se activa tras su unión a una
molécula adaptadora que a su vez se une a una parte del complejo de
señalización del receptor de factor de necrosis tumoral (TNF). La caspasa activa
puede procesar su propio precursor.
La caspasa 9, que en situación fisiológica se encuentra en su forma inactiva en el
citosol, se activa tras la salida de citocromo c desde la mitocondria. Ante un
determinado daño se produce una alteración en la membrana mitocondrial que
desencadena la salida de citocromo C al citosol. El citocromo C forma entonces un
complejo con el factor activador de proteasa (Apaf-1), dATP y procaspasa 9, lo
que conduce a la activación de la caspasa. Una vez activa, la caspasa 9 puede
activar otras caspasas.
Las caspasas 3, 6 y 7 son las principales responsables de los cambios
morfológicos y bioquímicos que ocurren en las células apoptóticas (Earnshaw
W.C, et al., 1999). Entre sus sustratos se encuentran un factor responsable de la
fragmentación del ADN. Además las caspasas eliminan la poli ADP-ribosa
polimerasa (PARP), molécula implicada en la maquinaria celular que repara el
daño en el ADN. Por otro lado, activan la vía que conduce a la condensación de la

20
cromatina y participan en la destrucción de la lámina nuclear y de las proteínas del
citoesqueleto (Ravichandran K.S, 2007).
1.8.1.2. Proteína p53 La proteína p53 es la que ordena la ejecución de la apoptosis en respuesta a los
agentes que dañan el DNA. Se ha demostrado que la p53 requiere del concurso
de las proteínas p63 y p73 para realizar su acción. Estas proteínas actuarían en
conjunto o en forma paralela a la p53 para inducir la apoptosis. Estudios genéticos
en animales deficientes en estas proteínas así lo han confirmado.
Se ha sugerido que drogas que en realidad son pequeñas moléculas pueden ser
diseñadas para activar a p53 impidiendo la unión a un regulador negativo llamada
Mdm 2. La proteía Mdm 2 se liga a un amino terminal de p53 en una relación
proteína-proteína que bloquea la actividad de p53. Normalmente la proteína p53
actúa como un factor de transcripción, determinando la expresión de una batería
de genes que detienen el ciclo celular o inician el proceso apoptótico (Flores E.R,
et al., 2002).
1.8.1.3. Familia de proteínas Bcl-2 La familia de proteínas Bcl-2 incluye tanto miembros inductores como inhibidores
de apoptosis (Antonsson B, 2001). Los miembros de esta familia se caracterizan
por poseer dominios homólogos a Bcl-2 conocidos como dominios BH, los cuales
se enumeran del 1 al 4. Según la presencia de estos dominios, estas proteínas se
dividen en tres subfamilias: una con miembros de función antiapoptótica
denominada subfamilia Bcl-2 que incluye a los miembros Bcl-2, Bcl-xl, Bcl-w, Mcl-1
y A1, todos con los cuatros dominios BH, y dos subfamilias de miembros
proapoptóticos: la subfamilia Bax que incluyen a Bax, Bok y Bak con tres dominios

21
BH y la subfamilia BH3 que incluye a Bim, Bid, Bmf, Bad, Hrk, PUMA y NOXA que
como su nombre lo indica sólo poseen un dominio BH3 (Becerra, 2009).
En general, hay dos vías por las cuales las proteasas de la familia caspasa
pueden ser activadas: una es la muerte inducida por la señal, mediada por
receptor de muerte, y la otra es la inducida por el estrés, mediada por mitocondria
(es decir, vía dependiente de la caspasa 9) como se muestra en la figura 3 (Ting-
Jun FAN, 2005).
La vía mitocondrial es activada por estrés intracelular, como por ejemplo el
iniciado por daño al ADN o el desemsamblado de microtúbulos. Por diversas vías,
estos inductores llevan a la activación de un grupo de proteínas denominadas
“solo BH3”, miembros de la familia del oncogén Bcl-2, que se traslocan a la
mitocondria e inhiben a miembros antiapoptóticos de la familia y/o activan
específicamente a dos miembros proapoptóticos: Bak y Bax. Esta activación lleva,
por mecanismos aún no esclarecidos totalmente, a la liberación de diversas
proteínas apoptóticas al citosol, entre las que destacan citocromo c y Diablo. El
primero forma un complejo con la proteína APAF1 y dATP que recluta a la
caspasa 9. Este complejo, denominado apoptosoma, induce la activación de esta
caspasa iniciadora que a su vez corta a las caspasas efectoras 3, 6 y 7, cuya
función final es el corte especifico de una multitud de sustratos celulares que
culminan con la muerte celular apoptótica. Ejemplos de estos sustratos son la
lamina B y el inhibidor de la nucleasa CAD, la cual lleva al corte especifico del
ADN en regiones internucleosomales y al desensamblado del núcleo (Shi, Y,
2002).
Con el fin de prevenir la activación no regulada de las caspasas, las células
eucariotas poseen una familia de inhibidores, conocidos como IAPs (por Inhibitor
of Apoptosis Proteins) que bloquea la actividad de estas enzimas. Existen diversos
miembros de esta familia, entre los que destacan la XIAP, que posee una alta
afinidad por diversas caspasas y la survivina, proteína muy pequeña e interesante,

22
dado a que también tiene una participación en el ciclo celular para que la
apoptosis se lleve a cabo, es necesario que estas proteínas se inhiban y es aquí
en donde participa Diablo (o SMAC). Al ser liberado de la mitocondria, Diablo se
une a las IAPs, permitiendo la activación de las caspasas y, por ende, la apoptosis
(Shi, Y, 2002). A pesar de que la función de Diablo dependiente de las IAPs ha
sido la más estudiada, la descripción de una isoforma de esta proteína, generada
por splicing alternativo, carente del dominio de unión a IAPs, de localización
citosólica y con capacidad de potenciar la apoptosis sugiere claramente que
existen otros mecanismos para la función de Diablo que hasta la fecha no han sido
elucidados (Roberts, 2001) (Figura 3).
Por otro lado, una ruta de señalización del proceso de apoptosis tiene su origen en
la membrana celular a través de lo que se conoce como vía extrínseca o de
receptores de muerte. Estos receptores pertenecen a la superfamilia del receptor
de TNF (TNFR) cuyos miembros tienen en común un dominio extracelular rico en
cisteína, un dominio transmembrana y una secuencia en su dominio citoplasmático
para acoplar el receptor con el resto de la maquinaría apoptótica. Estos receptores
también participan en el control de otros procesos como la respuesta inmune e
inflamatoria, la homeostasis ósea y el desarrollo y diferenciación de estructuras
epiteliales.
La señal se inicia tras la unión de los correspondientes ligandos (citoquinas pro-
apoptóticas y proinflamatorias como FasL, TNF, etc.) a sus respectivos receptores
de muerte (Fas, TNFR1, TRAILR1, etc.) (Ashkenazi, A, 1998; Danial. N.N, 2004).
La unión del ligando provoca la homotrimerización del receptor y de este modo, el
receptor de muerte es capaz de reclutar proteínas adaptadoras hacia la membrana
celular. Este proceso implica la interacción homofílica entre los dominios de
muerte DD (Death Domain) de los receptores con los de las moléculas
adaptadoras (proteínas puente entre el receptor y la caspasa) como la proteína
FADD (Fas Associated Death Domain). Las moléculas adaptadoras poseen los
dominios efectores de muerte DEDs capaces de interaccionar homofílicamente

23
con algunos miembros de la familia de las caspasas provocando su activación.
Así, se forma un complejo de señalización de muerte (DISC; Death-Inducing
Signaling Complex) que contiene a la proteína FADD y las caspasas 8 o 10. La
activación de la procaspasa-8 requiere su asociación con la molécula adaptadora
FADD a través de los dominios DED, situándose la caspasa-8 en la vía apoptótica
mediada por receptores de muerte (Muzio, M, et al., 1996; Medema, J. P, et al.,
1997). La caspasa-8 se activa, dirigiendo de esta forma la ejecución de la
apoptosis ya que, a su vez, activa a las caspasas ejecutoras. Además, la caspasa
3 puede eliminar un dominio de la proteína Bid amplificando la señal de muerte
celular ya que causa un daño a la mitocondria.
Además, a través de la vía de señalización de los receptores de muerte, la
activación de las caspasas puede ser regulada por la proteína FLIP (Flice-like
Inhibitory Protein). Ésta contiene el dominio DED, lo que le permite unirse al
predominio de la procaspasa-8 bloqueando la interacción de dicha caspasa con
los complejos receptor-proteína adaptadora e interfiriendo en el proceso
apoptótico (Lizarbe, 2007).

24
Figura 3. Vías apoptóticas. La apoptosis puede llevarse a cabo a través de la vía extrínseca,
mediada por receptores de muerte o, por la vía intrínseca, en donde participa activamente la
mitocondria al liberar diversas proteínas pro-apoptóticas. Estas vías no son excluyentes y
convergen en la activación de las caspasas, que son las principales ejecutoras de la apoptosis.
Tomado de Lizarbe, 2007.

25
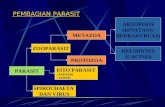
1.9. ANTECEDENTES 1.9.1. MCP en protozoarios La muerte celular programada (MCP) ha sido considerada un mecanismo de
desarrollo, diferenciación y control de proliferación de metazoarios. Sin embargo,
evidencias encontradas indican que MCP está también presente en organismos
unicelulares en determinadas condiciones, como los que se resumen en la Tabla 1
(Christensen S. T, 1998; Bruchhaus, et al., 2007). Las formas de MCP así como
procesos de apoptosis y necrosis han sido identificados en varias bacterias,
levaduras, dinoflagelados, ciliados y parásitos protozoarios como Trypanosoma
(Nguewa et al., 2004), Leishmania (Nguewa et al., 2004), Plasmodium (Al-Olayan
et al., 2002) y Entamoeba histolytica (Ramos, et al., 2007).
Tabla 1. MCP en diferentes parásitos protozoarios.
Parásitos protozoarios Señal de la MCP
Plasmodium spp.
Fragmentación de DNA
Exposición de fosfatidilserina
Desintegración parasitaria
Leishmania spp.
Condensación de la cromatina
Fragmentación del DNA
Liberación de citocromo C
Exposición de fosfatidilserina
Activación de proteasas
Trypanosoma cruzi
Fragmentación del DNA
Condensación de la cromatina
Pérdida del potencial transmembranal
mitocondrial
Activación de proteasas
Trichomonas vaginalis
Condensación de la cromatina
Fragmentación del DNA

26
Cuerpos apoptóticos
Giardia lambia
Condensación de la cromatina
Fragmentación del DNA
Cuerpos apoptóticos
Vacuolización citoplasmática
Blastocystis hominis
Vacuolización citoplásmica
Fragmentación del DNA
Contracción celular
Dyctyostelium discoideum
Vacuolización
Condensación de la cromatina
Condensación citoplásmica
Degradación del DNA
Exposición de fosfatidilserina
E. histolytica
Condensación de la cromatina
Fragmentación del DNA
Incremento de Ca 2 + citosólico
Disminución de K + intracelular
En protozoarios, el patrón de eventos de la MCP es similar al observado en las
células de metazoarios; se comparten eventos como la destrucción de la
organización estructural normal del núcleo debido al colapso de la cromatina en
masas electrodensas y, en muchos casos, la ruptura del DNA nuclear en
fragmentos oligonucleosomales. Se considera que la falta de regulación en la
homeostasis del calcio conlleva a importantes eventos de muerte celular en los
parásitos, ya que la mayoría muere en presencia de concentraciones altas de
calcio más que en ausencia de este ión (Fernández-Presas, 2000).
Es evidente que la MCP no está limitada a los eucariontes superiores, si no que
es un evento generalizado, presente en la mayoría de los seres vivos. A
continuación se describen evidencias de MCP en algunos parásitos más
estudiados.

27
1.9.1. MCP en Leishmania Leishmania es un parásito protozoario miembro del orden Kinetoplastida. Es el
agente causante de una de las parasitosis humanas más importantes que puede
conducir, de acuerdo a las especies, desde lesiones mucocutáneas y/o hasta
infección visceral generalizada. La forma promastigote del parásito, flagelado y
extracelular, reside en el intestino medio del vector insecto, la hembra de
flebótomo (Phlebotomus). Se diferencia de una proliferación procíclica no
infecciosa a promastigotes metacíclicos infecciosos que no se dividen. Después
de la picadura por el mosquito, el parásito infecta a los macrófagos del huésped
mamífero y se diferencia en amastigotes que pueden sobrevivir y dividirse en los
fagolisosomas de los macrófagos (Zangger, et al., 2002).
La carencia de nutrientes es uno de los factores que activan la MCP; se analizaron
los cambios de MCP en cultivos de promastigotes (en el intestino del insecto) y
amastigotes (en los vertebrados) de diferentes especies de Leishmania en la fase
estacionaria, donde mostraron un patrón de fragmentación del DNA en múltiples
tamaños oligonucleosomales, siendo esta consistentemente más fuerte en los
promastigotes de L. donovani que los amastigotes axénicos, sugiriendo que los
promastigotes son mas susceptibles a la fragmentación del DNA que los
amastigotes, resultado de la falta de nutrientes (Lee, et al., 2002).
Además, también hay estudios que evidencian la MCP en respuesta a las drogas.
Lee y colaboradores (2002), demostraron que durante la MCP en L. donovani, en
parasitos que fueron tratados con pentostam o anfotericina B, se presentaron
algunas características como la fragmentación del DNA en el núcleo, la
externalización de fosfatidilserina, la disminución del potencial de membrana, así
como la probable activación de caspasas (Lee, et al., 2002).
La fosfatidilserina (PS) es una proteína que se localiza en la superficie de las
células apoptóticas, éstas, son reconocidas por fagocitos vecinos participando en

28
eliminación de la inflamación. Una característica ventajosa del proceso y que
resulta un concepto interesante es que la apoptosis puede funcionar sin la muerte.
Leishmania spp. son capaces de evadir la actividad de los fagocitos y ellos
mismos establecerse como parásitos intracelulares obligados mediante la
exposición de PS y la inhibición de la actividad de los macrófagos, es decir, la
exposición de PS participa en la internalización de amastigotes. El reconocimiento
del resto por lo macrófagos induce la transformación de la secreción del Factor de
crecimiento (TGF-β) y la síntesis de interleucina 10 (IL-10), inhibe la producción de
NO, y aumenta la susceptibilidad del crecimiento intracelular de Leishmania
(Freitas, et al., 2001).
Estudios realizados por Moreira (1995) han demostrado que la muerte de
promastigotes de Leishmania amazonesis se encuentra modulada por iones de
calcio, ya que en los promastigotes sometidos a choque térmico se induce un
incremento de tres a cuatro veces los niveles de calcio. Se ha encontrado, en
cerca de 20% de estos parásitos, un patrón de rompimiento de DNA
internucleosomal típico y características morfológicas sugerentes de MCP. Las
lesiones tipo apoptóticas observadas en leishmanias son claramente de origen
nuclear, y escasamente se observan en los organelos citoplásmicos, incluyendo el
mitocondrión (Moreira, et al., 1995). Se ha observado también una separación
extensa entre el cuerpo y la membrana flagelar, patrón distinto al observado en
promastigotes que mueren por necrosis; también encontrado durante el choque
térmico. Finalmente, se ha señalado que las endonucleasas dependientes de Ca 2+ responsables de la apoptosis actúan exclusivamente en el núcleo, lo cual
concuerda con el hallazgo de que el mitocondrión se encuentra intacto cuando las
alteraciones nucleares ya son detectadas (Moreira, et al., 1995).
Una de las ventajas evolutivas conferidas por la MCP podría incluir la selección
constante de los parásitos más aptos, y la estricta regulación del ciclo celular y la
diferenciación celular en respuesta a los cambios ambientales (Chandrima Shaha,
2006).

29
1.9.2 MCP en Trypanosomas Se ha observado que Trypanosoma cruzi tiene la capacidad de autorregular su
crecimiento. Los epimastigotes proliferantes de T. Cruzi en cultivo, sufren MCP
masiva durante la diferenciación de G0/G1, deteniendo a los parásitos en la fase
de tripomastigotes (fase estacionaria de crecimiento). La muerte puede ser
prevenida o acelerada mediante la modificación de las condiciones de cultivo,
sugiriendo que los tripanosomas reciben señales extracelulares para la regulación
de su sobrevida y/o diferenciación (Ameisen J. C., 1995). La falta de renovación
del medio de cultivo en parásitos crecidos en medio monofásico puede propiciar
que una porción de epimastigotes se diferencie en tripomastigotes metacíclicos
infectivos, mientras que un número significativo de epimastigotes muere. Los
parásitos muertos aparecen como células esféricas con un largo flagelo y
muestran patrones citoplásmicos y nucleares que sugieren apoptosis, como la
fragmentación del DNA en escalera (Billaut-Mulot, 1994).
La inducción de MCP por drogas ha sido estudiado últimamente, Poliana Deolindo
y colaboradores (2005) determinaron el tipo de muerte inducida a epimastigotes
de T. cruzi por el veneno de Bothrops jararacá, que es una serpiente de la familia
Viperidae (Bochner & Struchiner 2003). El crecimiento del parásito fue inhibido
después del tratamiento con el veneno, observándose que el 50% del crecimiento
fue inhibido con 10 mg / ml. Ultraestructuralmente se observó la desorganización
del cinetoplasto y del mitocondrión, además de la condensación citoplásmica y la
pérdida del potencial de membrana mitocondrial. Al aumentar el tiempo de
incubación, la fosfatidilserina se expuso en la parte externa de la membrana
plasmática, seguido de la activación de caspasas, y la fragmentación del DNA.
Estos resultados indican que el estrés inducido en los epimastigotes por el
veneno, activa un proceso de muerte celular programada, similar a la apoptosis de
los metazoos, lo que conduce a los parásitos a la muerte (Poliana Deolindo,et al.,
2005).

30
Por otro lado, el tratamiento de los parásitos T. cruzi con el antibiótico geneticina
(G418) indujo la muerte celular de epimastigotes mostrando una selectiva
localización del factor de elongación 1-α en el núcleo de los parásitos muertos
(Billaut-Mulot, et al., 1996), sugiriendo la participación de este factor en la
regulación genética de la MCP.
1.10 MCP en E. histolytica Un fenómeno importante en el proceso de fisiopatogénesis de Entamoeba
histolytica es la inducción de la MCP (Stauffer, 2003) a una variedad de células del
huésped, incluyendo los leucocitos (Berninghausen, 1997), los macrófagos, las
células hepáticas (Seydel, 1998), por mecanismos dependientes de contacto
mediados a través de la interacción de galactosa-N-acetil-D-galactosamina del
parásito y la lectina de superficie del huésped (Bruchhaus, et al., 2007),
permitiendo el establecimiento del patógeno y la infección, así como la evasión a
la respuesta inmune.
Por otro lado se ha demostrado que la MCP también se presenta en E. histolytica,
inducida por especies reactivas de oxido nítrico (Ramos, et al., 2007). Los
parásitos que se incubaron durante 1 h con NOs desarrollaron 6 h más tarde la
MCP, resultando la fragmentación del DNA y niveles bajos de ATP. El inhibidor de
la caspasa Z-VAD-FMK inhibió totalmente la actividad de cisteín proteasas (CPA),
pero no tuvo ningún efecto sobre la apoptosis. Por otro lado, Ghosh, utilizando
peróxido de hidrógeno (Ghosh, 2010); demostró varias características
morfológicas en el parásito cuando se expone a H2O2 que son idénticas al fenotipo
de MCP de otros metazoos, observando una contracción de la célula, la
fragmentación del DNA, el aumento de las especies reactivas de oxígeno cuando
se expone a 2,0 mM de H2O2. Aunque en el genoma del parásito no se han
encontrado homólogos de caspasas de mamíferos, si codifica para una gran
cantidad de cisteín proteasas que pueden asumir la función de las caspasas. Sin

31
embargo, el estudio indica la existencia de cisteín proteasas independientes de
muerte celular programada en el parásito ya que el E-64 que es un inhibidor
específico de la cisteín proteasa no pudo rescatar a las células de una MCP como
apoptosis inducida por H2O2.
Nuestro grupo de trabajo determinó que el tratamiento con antibióticos como
aminoglicósido G418 (Villalba, 2007) y la emetina (Villalba, 2007a) induce MCP
en este parásito. Las micrografías electrónicas de los trofozoítos tratados con
G418 revelaron características de MCP, los trofozoítos sin tratamiento presentaron
un núcleo de 4-7 µm de diámetro, con un endosoma central y presencia de
cromatina periférica en la membrana nuclear; después de la incubación con G418,
la amiba mostró una forma redondeada mostrando condensación nuclear,
formación de cuerpos de inclusión y vacuolización citoplasmática, con la pérdida
aparente de la membrana nuclear; algo muy importante es que durante el tiempo
de exposición al fármaco, la membrana plasmática se mostró integra, siendo esto
una característica de la apoptosis.
Las primeras evidencias durante la apoptosis, son lesiones ultraestructurales en
la organización de la cromatina conduciendo a la aparición de fragmentos de DNA
(300 y/o 50 kb) (Walker et al., 1991; Tomei et al., 1993), en este estudio se
observó un patrón de DNA en escalera en gel de electroforesis, resultando
fragmentos de 180-200 pb y múltiples de estos. Es obvio que la incubación de
trofozoítos de E. histolytica con G418 da lugar a un fragmentación del DNA pero
no se detectó por análisis de gel electroforético, ya que se observó un DNA
degradado y las bandas en escalera eran débiles, sin embargo, el DNA
condensado y el corte sin desintegración de la membrana fue observado por
microscopía electrónica de transmisión. Mediante la prueba de TUNEL que es
altamente sensible para confirmar un proceso de suicidio intracelular, se observó
una reacción positiva en los trofozoitos induccidos a MCP con G418. E-64 que fue
un inhibidor específico de cisteín protesas, bloqueo la degradación del DNA, esto
se demostró por geles electroforéticos, TUNEL y la ultraestructura de los

32
trofozoítos por microscopia electrónica, lo cual sugiere fuertemente que al menos
una de las cisteín proteasas reportadas participan en la inducción de MCP por
G418. Estos resultados contrastan con los publicados por Ramos et al. (2007),
donde el autor especula que especies de oxído nítrico induce apoptosis
independiente de cisteín proteasa. Esta afirmación se basa en el hecho de que el
tratamiendo con E-64 falló en abolir la muerte de trofozoítos, sin embargo, no se
llevaron a cabo experimentos para determinar los efectos a niveles
ultraestructurales y moleculares de la MCP.
Durante las etapas tempranas de la apoptosis en eucariotes, se produce la
translocación de PS, desde el interior a la capa externa de la membrana
plasmática (Gatti et al., 1998). Para determinar este fenómeno se utilizó citometría
de flujo y anexina V-FITC que se une con afinidad a PS, siendo esto negativo para
trofozoítos tratados y no tratados con G418, ya que una de las razones es que la
PS forma menos del 10% de los lípidos de membrana de E. histolytica, reportado
por Aley et al. (1980), mientras que Martin et al. (1993) no detectó PS como
constituyente de la membrana de E. histolytica, mediante espectroscopia 31P-
NMR, sin embargo reporta que los principales fosfolípidos es fosfatidilcolina y dos
especies de fosfatidiletanolamina. Debido a los resultados se sugiere que la
abundancia de PS es insuficiente para la detección por el método usado, o que la
membrana plásmatica de E. histolytica no contiene PS o que PS interactúa con
otros componentes que bloquean su interacción con anexina V.
Una de las características bioquímicas que se producen por la MCP en trofozoítos
tratados con G418 es la disminución de Ki+ del 90% en comparación con los
trofozoítos no tratados, esto observado mediante citometría de flujo. En este
estudio también se mostró que en E. histolytica inducida a MCP por G418 hubo un
incremento dos veces los niveles de ROS intracelular. A su vez, el estrés oxidativo
causan incremento de los niveles de Ca2+ citosólico (Jiang et al., 1994),
observándose en este estudio un significante incremente de [Ca2+]i en
trofozoítos inducidos a MCP. Como consecuencia de la sobreproducción de ROS

33
y la pérdida de Ki+, la disminución del pHi fue observado en trofozoítos inducidos a
MCP.
Recientemente, Sánchez y colaboradores (2010), investigaron la expresión
diferencial de genes durante de la inducción de MCP. Mediante cDNA-AFLPs se
comprobó que el proceso de muerte en los trofozoítos inducido por G418 está
asociado con un mecanismo activo que involucra la síntesis de proteínas
específicas, incluyendo: Glutaminil-tRNA sintetasa, Subunidades ribosomales 40S
y 18S, Sir-2 y Graininas que pudieran estar implicadas en la regulación de rutas
apoptóticas como reguladores negativos de MCP, por ejemplo, las graininas,
donde el fenómeno se caracteriza por un incremento temprano de calcio citosólico,
el hecho de que la expresión de las graininas disminuya al paso del tiempo de
interacción con las células inflamatorias , sugieren fuertemente que los trofozoítos
activan mecanismos moleculares que disparan la recaptura de calcio citosólico, es
decir , una respuesta compensatoria para disminuir el calcio intracelular libre,
probablemente teniendo una actividad anti-apoptótica. y a tiempos largos de
interacción aumentan genes con probable actividad apoptótica suprimiendo la
expresión de gene anti-apoptóticos, Por otro lado, se observó la expresión del gen
Saponina-like, que pudiera estar actuando como un regulador positivo de
apoptosis, activando algunas rutas de muerte celular programada. Estos
resultados dan evidencia de los primeros genes identificados durante estadios
tempranos de MCP en E. histolytica, que pudieran estar regulando negativa o
positivamente a la apoptosis; concluyendo que este evento molecular es regulado
genéticamente.
De esta manera se sugirió que los trofozoítos de E. histolytica puede inducir la
apoptosis de la célula huésped, lo que correlaciona con la virulencia del parásito.
Y por otro lado, son capaces de evadir la respuesta inflamatoria contribuyendo a
la supervivencia de otros parásitos. Los estudios mencionados anteriormente han
demostrado que trofozoítos de E. histolytica son sometidos a MCP in vitro, sin
embargo, no estaba claro si éste proceso sucede in vivo de la célula huésped. En

34
este caso, Villalba y colaboradores (2011) estudiaron la MCP de trofozoítos de E.
histolytica como parte de un evento in vivo relacionado con la reacción inflamatoria
y la interacción huésped-parásito, con la finalidad de entender mejor el mecanismo
de MCP después de la interacción entre células inflamatorias y amebas. Se diseñó
un nuevo modelo in vivo utilizando una bolsa de diálisis que contenía trofozoítos
de E. histolytica, colocada quirúrgicamente dentro de la cavidad peritoneal de
hámsters y se dejó interactuar con los componentes de exudado peritoneal del
huésped. Las amebas recogida de las bolsas se examinaron entonces por
ensayos de TUNEL y microscopía electrónica de transmisión, siendo muy evidente
la condensación nuclear y la fragmentación del DNA tanto de los trofozoítos como
de las células inflamatorias, esto se observó después de la exposición al exudado
peritoneal, que se compone principalmente de los neutrófilos y los macrófagos.
Los resultados mostraron que el 90 %de las células inflamatorias y el 70 % de los
trofozoítos fueron positivos para los ensayos de TUNEL a las 6 h; sugiriendo que
la producción de óxido nítrico por las células inflamatorias podrían estar
involucrados en la MCP de trofozoítos y que la población restante de los
trofozoítos que sobreviven son resistentes a la MCP (Villalba, 2011). El hecho de
que haya un porcentaje de trofozoítos sobreviviendo después de la interacción
podría representar un mecanismo de virulencia y patogenicidad, ya que los
parásitos más aptos podrían sobrevivir evadiendo el sistema inmune, y así
propagar su genoma, garantizando la supervivencia y el establecimiento de la
infección. Estos resultados son útiles para una mejor comprensión del mecanismo
de MCP mediante un sistema in vivo, ya que la MCP parece jugar un papel
destacado en la interacción huésped-parásito.
Un estudio molecular de los genes encontrados en estados tempranos de MCP en
E. histolytica anteriormente mencionados in vitro e in vivo, permitirá elucidar de
que manera podrían estar implicados en la apoptosis y a su vez en la participación
en la interacción huésped-parásito para identificar blancos terapéuticos y ser de
gran ayuda para desarrollar estrategias terapéuticas efectivas, activando la MCP
en E. histolytica para combatir la amebiasis.

35
Recientemente, se está analizando en el mismo modelo experimental in vivo, la
expresión diferencial global de genes implicados en la MCP de E. histolytica,
mediante microarreglos de expresión, resultando que bajo estas condiciones la
regulación de la expresión de 39 genes de trofozoítos de E. histolityca disminuyó a
partir de los 30 min y aun más a los 90 min de interacción con las células
inflamatorias, algunos de estos genes se relacionan con mecanismos
antiapoptóticos, por ejemplo, las graininas, comprobando algunos resultados del
trabajo de Sánchez en el 2010.
Considerando que la MCP podría ser un importante evento en la patogénesis de la
amibiasis y que se aislaron y caracterizaron genes involucrados en la
regularización de las fases tempranas de este fenómeno. Se ha postulado la
inducción de ciertas proteínas efectoras específicas (Sánchez, et al., 2010)
responsables de los cambios morfológicos y ultraestructurales además de los
cambios bioquímicos que representa la fase de ejecución ocurrida entre el tiempo
de 9 y 12 h después de la incubación con G418 (Villalba 2007, 2011).
En el estudio publicado por Sánchez y colaboradores en el 2010 se evaluó el
grado de la síntesis de proteínas de novo durante la MCP in vitro (Sánchez, et al.,
2010); los resultados sugieren que los trofozoítos inducidos a MCP producen una
fuerte síntesis proteíca desde los primeros minutos, es decir que fue evidente el
incremento del rango de incorporación de metionina [35S] teniendo un pico máximo
de 5348 cpm/ml a los 90 minutos de incubación con G418, para posteriormente
regresar a niveles basales de incorporación después de las 3 horas. Derivado de
estos estudios, es clara la importancia no solo de conocer la expresión genética de
los trofozoítos durante la MCP si no también conocer que proteínas efectoras
están participando en la ejecución de este mecanismo, siendo el uso de la
proteómica una alternativa viable para la obtención de dicho conocimiento.

36
1. 10. Proteómica
El proteoma está formado con todas las proteínas que se expresan a partir del
genoma de un organismo (Wilkins MR, et al., 1996). La proteómica nos permite la
identificación de las proteínas, el conocimiento de su estructura primaria en su
secuencia de aminoácidos, la identificación de sus modificaciones
postraduccionales, su localización y la cuantificación de la expresión proteica
(proteómica cuantitativa) (Aebersold R, et al., 2003, Hanash S, et al., 2003, Steen
H, et al., 2004).
El término para esta nueva disciplina fue acuñado por el Dr. Fred Sanger a
mediados de los 80 como una rama de la genética y biología molecular. Este
término fue utilizado a mediados de los 90 como una rama o disciplina adicional a
la naciente genómica. Estos términos junto con otras “ómicas” se refieren al
estudio de sets completos de genes, proteínas, metabolitos, enzimas, rasgos
fenotípicos, etc… Todas estas disciplinas han evolucionado rápidamente gracias a
los avances tecnológicos en la última década. Este conocimiento nos ha ayudado
a entender mejor cómo el genoma y su expresión tiene impacto en la génesis de la
enfermedad, así como en su comportamiento biológico y por lo tanto en el
pronóstico clínico. La utilización de este conocimiento ha mejorado el diagnóstico y
clasificación de muchas enfermedades humanas, teniendo un impacto real y
práctico en la medicina clínica (Velásquez-Fernández. 2011).
1. 11. Herramientas de la proteómica
Una de las principales herramientas de la proteómica es la electroforesis
bidimensional en geles de poliacrilamida (2D-PAGE) (Anderson NL, et al., 2000).
Esta técnica permite separar miles de proteínas en un solo experimento, y
constituye actualmente el método más eficiente para la separación de mezclas
muy complejas de proteínas. Esta tecnología ha existido por tres décadas y está

37
basada en una separación de las proteínas en función de la carga, seguida de una
separación de las proteínas en función de su masa molecular. La separación de la
primera dimensión se realiza mediante isoelectroenfoque, durante el cual las
proteínas se separan en un gradiente de pH hasta alcanzar una posición en la que
su carga neta es cero, es decir, su punto isoeléctrico (pI). En una segunda
dimensión, las proteínas se separan por su peso molecular (PM) mediante
electroforesis. Posteriormente para la visualización de las proteínas se utiliza la
tinción con azul de Coomassie, con plata o tinciones más sensibles como SYPRO
Ruby y Deep Purple (Gil C, et al., 2003).
La otra herramienta clave para la proteómica es la espectrometría de masas
(Mass Spectrometry, MS), que lleva a cabo la identificación de las proteínas
(Wittke S, Mischak H, et al., 2005). Para analizar las proteínas mediante MS deben
ser convertidas a péptidos mediante proteólisis, generalmente con tripsina. Los
péptidos generados se convierten en iones en fase gaseosa mediante técnicas de
ionización suave, como láser asistida por matriz de desorción / ionización (Matrix
Assited Laser Desorption Ionisation, MALDI). Posteriormente los iones son
separados de acuerdo a su relación masa-carga (M/Z) en un analizador de masas
tipo ToF (Time of Flight). Los datos obtenidos son analizados mediante software
disponibles en la red como MASCOT en www.matrixscience.com, este software
hace búsquedas en base de datos para determinar la secuencia de las proteínas
(Aebersold R, et al., 2003).
La proteómica incluye diversos campos de investigación:
• La identificación de las proteínas expresadas por un organismo en una
condición dada (proteómica descriptiva o estructural, todas las proteínas
expresadas en un momento y en un contexto).
• La identificación de los cambios en el nivel de expresión de proteínas
asociadas a cambios en las condiciones del organismo (proteína

38
comparativa, cuáles proteínas cambian cuando un organismo se somete a
condiciones diferentes).
• La identificación de conjuntos funcionales de proteínas, es decir, grupos de
proteínas que se localizan en un mismo sitio celular y que operan en mutua
interacción (interacciones proteína-proteína, proteómica funcional)
• La identificación de las proteínas que forman un organelo (este enfoque
conduce a la elaboración de un mapa molecular de la célula).
1. 12. Análisis proteómico de E. histolytica
El proteoma de E. histolytica en cultivos axénicos se ha analizado por
electroforesis de gel en dos dimensiones (2-DE), empleando un procedimiento
práctico y eficaz para la solubilización de las proteínas de E. histolytica.
Aproximadamente 900 especies de proteínas en el intervalo de pH entre 4 y 7
fueron detectados por tinción con Coomassie Blue. 95 spots fueron degradados
enzimáticamente con tripsina y se sometieron a espectrometría de masas. Los
datos resultantes de las huellas peptídicas, es decir las masas de péptidos fueron
comparados con las bases de datos disponibles en el genoma de E. histolytica y
Centro Nacional de Información Biotecnológica (NCBI) para la identificación y
categorización de las proteínas. 63 de las proteínas identificadas se postula que
se relacionan con el citoesqueleto, la superficie, la glucólisis, el metabolismo del
DNA / RNA, el sistema ubiquitina-proteasoma, el tráfico vesicular y transducción
de señales. Este estudio constituye uno de los primeros que demuestran que los
genes correspondientes son en efecto expresados en E. histolytica y proporciona
una base para posteriores estudios proteómicos de este parásito (Tolstrup 2006).
Por otro lado, para identificar los factores de virulencia de E. histolytica, se definió
por primera vez los fenotipos de dos cepas: HM-1: IMSS (cepa virulenta) y E.
histolytica Rahman (cepa que se informa menos virulento que el HM-1: IMSS). Se
utilizó electroforesis- 2D (DIGE) diferencial para comparar el proteoma de Rahman

39
y HM-1: IMSS, identificando proteínas que fueron diferencialemente expresadas
cerca de un nivel de cinco veces más, entre los dos microorganismos. Esto incluía
dos proteínas con propiedades antioxidantes (Peroxirredoxina y superóxido
dismutasa), y tres proteínas de función desconocida; grainina 1, 2 y unas proteína
que contiene un LIM-dominio (Paul H. Davis, et al., 2006).
Otro resultado de análisis proteómico en ameba, señala que la glucosa tiene un
papel en la modulación de la virulencia de este parásito, se examinó si la
virulencia del parásito podría ser influenciada por la privación de glucosa (GS). El
comportamiento migratorio del parásito y su capacidad para matar las células de
eucariotes y la lisis de los eritrocitos está fuertemente reforzado por la GS. Con el
fin de obtener información sobre el mecanismo que subyace la GS en los efectos
sobre potenciar la virulencia, se analizaron las diferencias en los niveles de
expresión de proteínas en el control de la glucosa y privación en los trofozoitos,
por análisis proteómico cuantitativo, observado que río arriba hay una proteína de
unión al elemento regulador 3 (URE3- BP), un factor de transcripción que modula
la virulencia E.histolytica, y la proteína rica en lisina-1 (KRiP1), que se induce
durante la el desarrollo de absceso hepático, se reguló por la GS. Se analizaron
también las fracciones de membrana de E. histolytica, determinando que la
subunidad ligera LgL1 de lectina Gal / GalNAc es regulada por la GS.
Sorprendentemente, el amoebapore A (AP-A) y la cisteína proteinasa A5 (CP-A5),
dos importantes factores de virulencia de E. histolytica, fueron fuertemente
reguladas por la GS. Mientras que el efecto potenciador de la virulencia por GS en
E. histolytica se conserva en cepas silenciadas para AP-A y PP-A5, y la perdida
en LgL1 y KRiP1 en cepas bajamente reguladas (Ayala Tovy, et al., 2011).
Sin embargo, hasta la fecha de la realización de esta tesis no se han reportado
otros estudios comparativos que muestren la expresión diferencial de proteínas en
algún mecanismo celular relacionado con el proceso de MCP en patogenicidad del
microorganismo.

40
2. JUSTIFICACIÓN
La MCP es un factor importante en este parásito, ya que no solo es capaz de
inducir apoptosis a las células efectoras del sistema inmunológico si no también es
capaz de inducir y regular su propia muerte celular, siendo esto un mecanismo de
evasión de la respuesta inmune. Por lo tanto, este mecanismo podría ser un
elemento importante dentro de la fisiopatogénesis de la amebiasis. El mecanismo
de MCP podría conferir ventajas adaptativas al parasito de limitar el número de
parásitos en el huésped, habilitando la propagación de tan sólo aquellos parásitos
más aptos, regulando así el tamaño de la población, favoreciendo la
supervivencia, proliferación y replicación del resto de la población de parásitos,
permitiendo por lo tanto, el establecimiento de este patógeno y a su vez la
infección.
El proceso de MCP in vivo reduciría de esta manera el proceso inflamatorio
evitando que células inflamatorias lleguen al sitio de infección, es decir evadiendo
la vigilancia del sistema inmunológico. Recientemente, nuestro grupo demostró
que la MCP es un proceso que involucra la expresión de genes anti-apoptóticos
en fases tempranas de la inducción del fenómeno, seguido por la expresión de
genes pro-apoptóticos en fases intermediarias que dan lugar a la codificación de
moléculas efectoras y/o proteínas específicas de la fase de ejecución que
permiten los cambios bioquímicos, morfológicos y ultraestructurales que marcan y
promueven la MCP.
Por esto, el estudio o el análisis proteómico de la apoptosis en E. histolytica
permitirá identificar las proteínas específicamente expresadas durante el proceso,
lo que a su vez permitirá conocer las moléculas efectoras, así como las proteínas
reguladoras de este fenómeno. Este conocimiento podría ser empleado en el
desarrollo de estrategias que contribuyan a generar drogas o terapias efectivas en
contra de la amebiasis, basadas en la activación MCP y por otro lado permitiría
entender los mecanismos moleculares que el parásito pone en juego durante la
interacción huésped-parásito.

41
3. HIPÓTESIS
En virtud de que se ha demostrado que la MCP es un evento genéticamente
controlado en el que en fases tempranas se inducen eventos anti-apoptóticos y
pro-apoptóticos, mediante un análisis proteómico durante distintos estadios de la
MCP, se podrán identificar diversas proteínas que participan en dichos
mecanismos.

42
4. OBJETIVOS
Objetivo General
Determinar el perfil proteómico de la MCP en Entamoeba histolytica
inducida por G418.
Objetivos Específicos
Caracterizar el perfil proteómico de trofozoítos inducidos y sin inducir a
MCP.
Identificar proteínas específicas expresadas durante las distintas fases de la
MCP.
Identificar proteínas específicas que pudieran estar involucradas en
mecanismos pro-apoptóticos y anti-apoptóticos.

43
5. ESTRATÉGIA EXPERIMENTAL
CA - (HM1:IMSS) E. histolytica
Trofozoítos sin inducir a MCP Trofozoítos inducidos a MCP con G418 a diferentes tiempos.
1-D: Migración de proteínas por su PI.
2-D: Migración de proteínas por PM.
Extracción y Purificación de proteínas
Separación de proteínas:
1-D 2-D
Visualización proteíca y análisis de la imagen (gel).
Identificación
(MALDI-TOF)
Huella peptídica
Homología proteíca (Base de datos)

44
6. MATERIALES Y MÉTODOS
6.1. Cultivo de E. histolytica
Se utilizó la clona A de la cepa HM1:IMSS, y se cultivó axenicalmente en medio
TYI-S-33 suplementado con penicilina 100 U y estreptomicina 100 µg/ml a 37 °C.
Se cultivaron 8 millones de trofozoítos para cada tiempo de interacción con el
antibiótico.
6. 2. Inducción de la apoptosis con G418
Una vez observados los cultivos confluentes, se indujo la MCP adicionando G418
(10 µg/ml) a diferentes tiempos de exposición al antibiótico (0, 1 ½, 3, 6, 9 h).
6.3. Obtención de proteínas
Terminados los distintos tiempos de incubación, los trofozoítos se cosecharon
colocando las cajas de cultivo durante 10 min en hielo para despegarlos, se
centrifugaron a 1500 rpm/10 min y se lavaron con 10 ml de PBS pH 6.8 tres veces,
y un lavado final de 10 ml deTris 10 mM pH 6.8 para eliminar el exceso de las
sales contenidas en el PBS, ya que estas interfieren en la conductividad de la
muestra y en el enfoque de las proteínas. Una vez lavadas las células, se hicieron
alícuotas de cada condición, a estas se le adicionó 40 µl de inhibidores de
proteasas (complete de Roche) y se almacenaron a -20 °C hasta su uso.

45
6.4. Lisis de los trofozoítos
Se tomaron alícuotas de cada condición para lisar los trofozoítos por
congelamiento/descongelamiento, se colocaron 100 µl de buffer de lisis o de
muestra (urea 7M, tiourea 2M, 4% p/v CHAPS, 40 mM p/v ditiotreitol (DTT) y 2%
v/v IPG buffer de 3-10 NL(GE Healthcare) (anexo 1), se homogenizó y se congeló
en nitrógeno líquido, se trituró con un pistilo tipo simple Grinding, una vez
descongelada la muestra se volvió a congelar y se a triturar dos veces más. Este
procedimiento se realizó de manera independiente para cada muestra.
6. 5. Precipitación proteíca
Una vez lisadas las células de cada condición se procedió a concentrar y/o
precipitar las proteínas con ácido tricloroacético(TCA) -acetona fría.
Cada muestra se aforó a 900 µl con agua milliQ y se adicionaron 100 µl de una
solución de 77% de TCA y 23% de acetona fría, se realizó homogenización por
inversión y se dejó 30 minutos a 20°C. Se centrifugó a 15000 g por 10 minutos, se
decantó el sobrenadante y la pastilla se lavó tres veces con 400 µl de acetona fría
centrifugando a las mismas condiciones 5 minutos, se adicionaron 40 µl de
inhibidores de proteasas (complete de Roche) para evitar que las proteínas se
degradaran y posteriormente 250 µl de buffer de hidratación (urea 7 M, tiourea 2
M, 2 % p/v CHAPS, 10 mM DTT y 0.2% v/v IPG buffer de 3-10 NL(GE Healthcare)
(anexo 2). Las alícuotas se guardaron a -20°C hasta su uso.
6.6. Cuantificación proteíca
Para la cuantificación se usó el kit 2-D Quant (GE Healthcare), de acuerdo a las
especificaciones del proveedor (anexo 3) el cual está diseñado para la

46
determinación precisa de la concentración de proteína en las muestras a ser
analizadas por técnicas de alta resolución de electroforesis, tales como
electroforesis 2-D, SDS-PAGE o IEF. Los reactivos utilizado en la preparación de
muestras, para IEF incluyen detergentes, agentes reductores, agentes
caotrópicos y anfólitos, que son incompatibles con otros ensayos de proteínas. El
procedimiento consta en precipitar las proteínas, dejando las sustancias que
interfieren en la solución. El ensayo se basa en la unión específica de los iones de
cobre a la proteína. El precipitado proteico se resuspendió en una solución que
contiene cobre y el cobre no unido se mide con un agente colorimétrico. La
densidad del color es inversamente proporcional a la concentración de proteínas.
El ensayo tiene una relación lineal a la concentración proteínas en un intervalo de
0-50 mg.
El procedimiento es compatible con muestras en las que se hayan empleado
reactivos para su preparación tales como SDS al 2%, DTT al 1%, urea 8 M, 2 M
tiourea, CHAPS al 4% y 2% y 2% IPG buffer. Para la determinación de la
concentración proteica se utilizaron 10 µl de cada muestra. Una vez determinada
la concentración se tomó el volumen equivalente a 400 µg de proteína y se
completó con 250 µl buffer de hidratación, que es el volumen adecuado para las
tiras de poliacrilamida de 13 cms.
6.7. Primera dimensión - isoelectroenfoque
La primera dimensión consistente en la separación por punto isoeléctrico, se llevó
a cabo en el equipo Ettan IPGphor III (AmershamBiosciences), se usaron tiras de
13 cm en un rango de pH de 3-10 NL (ImmobilineDryStrip, GE Healthcare). Para la
rehidratación pasiva de la tira, se colocó la muestra ya cuantificada (figura 4) en la
tira durante 14 h para absorber todas las proteínas que hubiera en cada muestra.
La rehidratación de la tira IPG se llevó a cabo de la siguiente manera: se pipeteó
el volumen apropiado de solución de rehidratación y muestra, en cada soporte,

47
como se indica en la figura 4 A, y se colocó lentamente en un punto del soporte de
las tiras, se evitó hacer burbujas y/o eliminarlas para garantizar una homogénea
absorción de la muestra; posteriormente se procedió a colocar la tira, para esto se
quitó el plástico protector de la IPG (figura 4 B), al exponerse el lado donde está
el gel, esta debe quedar hacia abajo, haciendo contacto con la muestra. La
muestra debe expandirse a lo largo de toda la tira para asegurar nuevamente una
absorción homogénea. Por último, se levantó suavemente y se bajó la banda
deslizándola hacia atrás y hacia adelante a lo largo de la superficie de la solución,
inclinando ligeramente el soporte de tiras para asegurar la completa hidratación
del gel, como se ilustra en la figura 4 C. En seguida, se aplicó aceite (DryStrip
Cover Fluid GE Healthcare) sobre la tira para evitar que la muestra se evapore y
se cristalice la urea, se pipeteó el aceite y se colocó gota a gota sobre un extremo
de la tira hasta llegar a la mitad de esta, y continuar con el otro extremo, hasta
que la tira IPG quedó cubierta, se cerró la caja IPG, y se llevó a cabo una
hidratación pasiva por 14 h.
Pasado el tiempo de hidratación se colocó el colector o charola de porcelana en el
equipo Ettan IPGphor III. La pequeña protusión en forma de T encaja en la
apertura del Ettan IPGphor III haciendo una fácil colocación del colector (figura 4
D). Posteriormente, se colocó la tira o las tiras IPG en el colector como se ilustra
en la figura 5 E, la tira debe ir con la parte del gel hacia arriba con el extremo
anódico (+) de la tira sobre la marca correspondiente grabada en la pista del Ettan
IPGphor III. Finalmente se humedecieron las almohadillas con agua milliQ y se
colocaron en los extemos de la tira o tiras IPG (ver figura 5 F), y se cubrió con el
uso del aceite (Immobiline DryStrip) posteriormente se colocaron los electrodos.
Se cerró la tapa del equipo: Ettan IPGphor III.

48
Figura 4. Primera dimensión isoelectroenfoque.
La separación por isoelectroenfoque se llevó a cabo de acuerdo a las
especificaciones del manual de Amersham Biosciences, y se procedió al
corrimiento en 1-D, donde las proteínas se localizaron en base a su PI.
Se estableció el programa y/o los parámetros deseados para la migración
electroforética en primera dimensión, que consiste en un inicio la aplicación de 500
V durante 1 h, posteriormente dos gradientes de 1000 V, uno por 1 h, y otro de
2:30 h, finalmente un mantenimiento de 8000 V con duración entre 25 y 55 min.
Este programa de isoelectroenfoque tiene una duración de 4:55-5:25 hs.
Cabe mencionar que al terminar el procedimiento, las tiras pueden ser
congeladas por 2 meses a – 20°C y de 6 meses a un año a – 80°C, mientras que
en el presente proyecto, una vez terminado el corrimiento en 1-D, las tiras se
almacenaron no mas de una semana para continuar con el siguiente paso.

49
6.8. Segunda dimensión SDS-PAGE
Posteriormente, a la separación por isoelectroenfoque, se llevó a cabo la segunda
dimensión, que es la separación por peso molecular (PM) en geles
desnaturalizantes de poliacrilamida al 12% y 10% (anexo 4) de 16 x 18 cm. La
segunda dimensión consistió de los siguientes pasos: preparación del gel de
segunda dimensión llevando la polimerización de la acrilamida toda la noche, se
equilibró la tira con un buffer de equilibrio (anexo 5), el equilibrio se lleva acabo
tratando las tiras IPG con buffers reductores (Tris-HCl 1,5 M pH 8,8, urea 6 M,
glicerol y trazas de azul de bromofenol) que contengan SDS e iodoacetamida/DTT,
con el fin de permitir la solubilización de las proteínas previamente enfocadas y de
una completa interacción de las proteínas con el dodecil sulfato sódico (sodium
dodecyl sulfate, SDS) de la segunda dimensión; el primer paso es colocar la tira
con 10 ml del buffer de equilibrio adicionando DTT (100 mg) para mantener el
estado reducido de las proteínas desnaturalizadas, este paso se realizó durante
15 minutos en agitación y a temperatura ambiente, posteriormente se quitó la
solución y a continuación se añadió iodoacetamida (250 mg) a 10 ml del buffer de
equilibrio para evitar una posible reoxidación de las proteínas. Se repite la
incubación de 15 min con agitación a temperatura ambiente. La o las tiras quedan
listas para usar en la segunda dimensión.
Se colocaron las tiras de geles y se selló con agarosa al 0.1 %. Las condiciones
de corrida para la electroforesis fueron a 100 Volts. Una vez que las proteínas
entraron al gel separador se disminuyó la corriente a 80 Volts durante 8 horas.
6. 9. Visualización proteíca y análisis de imágenes
Los geles de cada muestra se tiñeron con azul de Coomassie coloidal para el
analisis de la imagen, para esto, cada gel se colocó en solución fijadora (30%
etanol, 5 % ácido acétido glacial) durante 1 h, posteriormente se retiró la solución

50
fijadora y se cubrió el gel con azul coomassie coloidal de Bio-Rad, durante 1 h.
después fue retirado y se hicieron 3 lavados con agua milliQ con la finalidad de
desteñir, para obtener una mejor tinción se repitieron los pasos a partir de la
adición del colorante, una vez teñidos, los geles se mantuvieron en agua milliQ.
Los perfiles proteícos de cada muestra se escanearon con el Scanner LAS4000
(GE Healthcare) para obtener las imágenes digitalizadas y exportadas en formato
TIFF para importarlas a continuación al software Ludesi REDFIN3 con el que se
realizó el procesamiento de las imágenes y el análisis bioinformático para ver la
cantidad y porcentaje de spots, la intensidad de cada spot por densitometria,
además de la normalización: esto es el proceso de hacer que el volúmen de estos
spots sean disponibles y/o comparables entre la imagen del gel en la parte de
diferencias técnicas en la tinción, el escaneo, el volumen de la muestra, y así
sucesivamente.
Las imágenes importadas a este programa se depuraron y optimizaron mediante
un filtrado previo al análisis. A continuación, se creó un archivo “experimento”, en
el que el programa incluye todos los geles que se vayan a comparar para detectar
a continuación las manchas proteicas existentes en cada gel y emparejarlas con
las correspondientes manchas en el resto de los geles. Este proceso requiere una
revisión manual pormenorizada y, en ocasiones, la edición y corrección de alguna
de las manchas o de los emparejamientos detectados automáticamente. A
continuación el programa densitometría las manchas proteicas, lo que permite
obtener medidas del nivel de expresión de cada una de ellas. Para compensar las
posibles variaciones entre geles debidas a diferencias en la eficacia de la tinción
en los diversos niveles de concentración en cada gel, se aplica el método de
normalización como ya se ha mencionado anteriormente. Este método calcula una
curva en el diagrama de dispersión que minimiza la distancia a todos sus puntos y
que sirve para calcular el factor de normalización para cada mancha.

51
El software realiza la prueba de ANOVA, que nos permite comparar las medias de
una misma variable en más de dos grupos, cuando existe normalidad en la
distribución de los datos y homogeneidad en las varianzas muestrales.
Se compararon los perfiles proteícos de cada tiempo de inducción a MCP con el
perfil proteómico de trofozoítos sin MCP, con el fin de identificar las proteínas
diferenciales, para esto se tomó en cuenta sólo a aquellos spots que
desaparezcan y/o aparezcan en cada condición, cabe mencionar que otro
parámetro que se tomó en cuenta fue la concentración; solo nos enfocamos en
aquellas proteínas que estuvieran en alta concentración , es decir, al ser
visualizadas con el azul de Coomassie coloidal y que la detección fuera intensa y
no ténue. Una vez determinadas las proteínas diferenciales de cada perfil proteíco
contemplando los parámetros anteriormente mencionaddos.
6.10. Digestión con tripsina de proteínas en el gel
Una vez que se observó las manchas en el gel de 2-D teñido con azul de
Coomassie coloidal, se procedió al corte de esa o esas manchas de interés, con
una punta de pipeta recortada (aproximadamente 1.5mm) se tomó cada mancha y
deposito en un tubo eppendorf (previamente rotulado con el número de la mancha
seleccionada). adicionando 100 µl de agua MilliQ y se analizaron las proteínas
mediante espectrometría de masas MALDI TOF/MALDI TOF-TOF en colaboración
con la Unidad de Proteómica de la Universidad Complutense de Madrid, para
identificar las proteínas diferenciales presentes en cada uno de los spots
seleccionados que pudieran estar participando en el proceso de apoptosis de E.
histolytica.
Las proteínas seleccionadas en el gel fueron reducidas, alquiladas y digeridas con
tripsina empleando el protocolo de digestión de Schevchenko (Schevchenko, et
al., 1996), con ligeras variaciones. Brevemente, los spots fueron lavados dos
veces con agua.

52
Se añadió acetonitrilo hasta que cubra la mancha (20 µl) y se incubó 5 minutos
para que el gel se deshidrate y libere el colorante (se repitió 2 veces). Se secó a
vacío en “speed-vac” por 30 min.
Con la finalidad de que las manchas pasen por un proceso de reducción y
alquilación, las bandas se cubrieron con una solución de DTT 10 mM en NH4
HCO3 (25 mM) y se incubó por un tiempo de 30 minutos a 56 °C. Posteriormente,
se dejó a temperatura ambiente, donde se eliminó el DTT y se añadió Acetonitrilo
que se retiró rápidamente para incubar con Iodoacetamida 55 mM en NH4 HCO3
(25 mM) durante 15 minutos en oscuridad.
Finalmente, se realizó la digestión con tripsina añadiendo 20 µl del tampón de
digestión (12.5 ng/µl de tripsina (Roche Molecular Biochemicals) en 25 mM de
NH4 HCO3, pH 8.5) y se incubó el tubo eppendorf a 37 °C toda la noche, para
favorecer la acción de la enzima.
6. 11. MALDI-TOF (MS); MALDI TOF/TOF (MS/MS)
La mayor parte de las manchas proteicas de interés se identificaron mediante
análisis con MALDI-TOF por huella peptídica (PMF), o bien MALDI-TOF/TOF, por
secuenciación peptídica.
Una vez finalizada la digestión, se mezcló una alícuota de cada digestión con una
alícuota de matriz (ácido α-ciano-4-hidroxicinámico (Sigma Aldrich) 3 mg/mL en
acetonitrilo al 50% y el de ácido trifluoroacético (TFA) al 0.1%). La mezcla (1μl) se
deposita en un portamuestras MALDI y se dejó cristalizar a temperatura ambiente.
El análisis MALDI-TOF MS se realizó en el espectrómetro de masas 4800 Plus
MALDI TOF/TOF Analyzer, con fuente de ionización tipo MALDI y dos
analizadores tipo tiempo de vuelo en tandem y cámara de colisión (CID) para
fragmentación (Applied Biosystems, MDS Sciex Toronto, Canadá) en la Unidad de
Proteómica del Parque Científico de Madrid, Universidad Complutense de Madrid.

53
Los espectros de masas se miden en modo reflector positivo (analizándose iones
cargados positivamente), con un voltaje de aceleración iónica de 20.000 V.
Para obtener una mayor exactitud, los espectros MALDI-TOF se calibran
internamente empleando como referencia señales de masa de dos iones
provenientes de la autolisis de la tripsina, lo que garantiza un error menor a 30
ppm en la masas peptídicas detectadas.
El análisis de espectrometría de masas MALDI-TOF/TOF produce la huella de las
masa peptídicas y los péptidos observados con una señal ruido superior a 10,
pueden ser recogidos y representado como una lista de pesos moleculares
monoisotópicos.
De los espectros MS se seleccionan los precursores más apropiados para el
análisis MS/MS con la CID conectada (empleando gas atmosférico) a un voltaje de
1 kV en modo ion reflector y con una ventana de masa para el precursor de ± 5
Da. El modelo de la placa y la calibración por defecto fueron optimizados para el
procesamiento de espectros MS-MS.
El posterior análisis para la identificación de proteínas, se eliminaron los valores
de masas contaminantes (tripsinas, queratinas, aductos de sodio o señales
resultantes de la oxidación de metioninas), para buscar en una base de datos no
redundante (NR; ~4 x 106 entradas; Nacional Center for Biotechnology
Information) (secuencias 2012/05/08, 17919084; residuos 6150218869) y sin
restricción taxonómica además de la base de Entamoeba histolytica (secuencias
070.612; 1366; residuos 456887) de NCBI donde se realizaron búsquedas
empleando también MASCOT 2.3 (Matrix Science/www.matrixscience.com) a
través del servidor Global Protein v3.5 de Applied Biosystems. Los parámetros de
búsqueda fueron los siguientes:
• Carbamidometil cisteín como modificación fija y metionina oxidada como
variable de modificación.

54
• Tolerancia de la masa péptido fue de 50 para las búsquedas de PMF y 80-
100 ppm de MS/MS y búsquedas combinadas (PMF + MS/MS).
• Un sitio pérdido de corte de tripsina
• La tolerancia de fragmentos MS-MS de 0.3 Da
• En total la identificación de proteínas, la probabilidad de puntuación fue
mayores que la puntuación fijada por la MASCOT como significativa con un
valor de p > 0.05.
6. 13. Papel de las proteínas identificadas
Mediante la literatura se obtuvo información sobre la función de dichas proteínas
identificadas por MALDI-TOF/MALDI TOF-TOF y así determinar si las proteínas
diferenciales pudieran estar relacionadas con eventos pro-apoptóticos o anti-
apoptóticos.

55
7. RESULTADOS
Mucho se ha avanzado en el conocimiento de la biología de E. histolytica, sin
embargo, el papel de la respuesta inmune en el control de la enfermedad no está
bien establecido, menos aún el singular comportamiento del parásito, que con
frecuencia actúa, como comensal y más raramente como invasor.
Durante el proceso de infección, E. histolytica está expuesta a la respuesta
inmunológica del hospedero, no obstante se ha demostrado que utiliza diversos
mecanismos para evadir la respuesta inmune; por ejemplo induce la apoptosis de
leucocitos (Berninghausen, 1997), macrófagos y células hepáticas (Seydel, 1998),
empleando moléculas como lectinas, lipasas y toxinas. Recientemente, se ha
demostrado que la muerte celular programada (MCP) también se induce en este
parásito in vitro: i) mediante el aminoglicósido G418 y la emetina (Villalba, 2007 a,
b); ii) por especies reactivas de oxído nítrico (Ramos, et al., 2007) y iii) por
especies reactivas de oxígeno (Ghosh, 2010). In vivo, nuestro grupo de
investigación demostró en un modelo de hámster chino que el producto de la
interacción entre el hospedero y el parásito se induce la apoptosis tanto en los
neutrófilos como en los trofozoítos, lo que sugiere que durante el proceso
fisiopatogénico se desarrolla una respuesta del huésped contra el parásito, con la
consecuente activación de mecanismos de defensa del mismo (Villalba, 2011).
Recientemente, también demostramos mediante cDNA-AFLPs y RT-PCR en
tiempo real que la MCP inducida por fármacos representa un mecanismo activo
que involucra la expresión de genes específicos, incluyendo: la Glutaminil-tRNA
sintetasa, las Subunidades ribosomales 40S y 18S, el Sir-2, las Graininas 1-2 y la
Saponina-like que en otros sistemas, se ha visto que están implicados en la
regulación de rutas pro- y anti-apoptóticas (Sánchez, et al., 2010). En este trabajo
presentamos el análisis proteómico comparativo de la MCP activada con G418,
con la finalidad de poder identificar proteínas que participen en el proceso de
apoptosis in vitro.

56
El estudio se inició con la estandarización del protocolo del isoelectroenfoque de
las proteínas de trofozoítos sin inducir a MCP, es decir sin exposición a G418. Se
emplearon cultivos confluentes para obtener los extractos, lisar las células y
extraer las proteínas mediante precipitación con ácido tricloroacético-acetona fría.
Se realizó la cuantificación proteica y se tomaron 400 µg de proteína para la
hidratación de la muestra, durante 12-14 h. Se evaluaron dos programas de
isoelectroenfoque para la separación de las proteínas (ver tabla 2 y 3).
Tabla 2. Protocolo de isoelectroenfoque Ettan IPGphor III. (protocolo corto)
Intervalos de Ph Modo de voltaje Voltaje (V) Tiemp (h:min)
3-10 NL 1. voltaje inicial 500 1: 00
2. Gradiente 1000 1:00
3. Gradiente 1000 2:30
4. Voltaje final 8000
0:25-0:55
Total 4:55-5:25

57
Tabla 3. Protocolo de isoelectroenfoque. (protocolo largo)
Intervalos de pH Modo de voltaje Voltaje (V) Tiempo (h:min)
3-10 NL 1. Voltaje inicial 1 120 1:00
2. Voltaje inicial 2 500 1:00
3. Gradiente 1000 2:00
4. Gradiente 4500 6:00
5. Voltaje final 4500 10:00
Total 20:00
Una vez finalizado el isoelectroenfoque, las proteínas ya enfocadas en su PI se
separaron en base a su PM, para lo cual, las tiras se colocaron en geles de
poliacrilamida al 12%, la migración de las proteínas se inició al aplicar un voltaje
de 100 durante 45 min, y se separaron por 8 h a un voltaje de 80 volts. Con la
finalidad de observar el perfil proteómico de trofozoítos sin inducir a MCP, los
geles se tiñeron con Azul de Coomassie coloidal (Bio-Rad), al comparar la
separación de las proteínas, empleando los dos protocolos de isoelectroenfoque,
se observó una separación y enfoque similar en ambos, por lo que se optó por
seleccionar el primer programa (Tabla 2), en virtud de que se requiere menos
tiempo para su ejecución.
Se notó que las proteínas resueltas no se separaban correctamente en el gel, es
decir que se observaban aglomeradas en la parte de arriba del gel, como se
puede ver en la figura 5 A. Por lo tanto, se procedió a utilizar geles al 10 %,
observándose que estas proteínas de alto peso molecular se resolvían de mejor
manera, obteniendo spots mejor definidos, como se puede ver en la figura 5 B.

58
Una vez que el patrón de trofozoítos sin inducir a MCP fueron reproducibles, se
inició la inducción de los trofozoítos a MCP con G418 (10 µg/ml), el antibiótico se
adicionó a cada caja de cultivo confluente con aproximadamente 8 millones de
trofozoítos, a diferentes tiempos de exposición (0. 1 ½, 3, 6 y 9 h). Posteriormente
se obtuvieron los extractos de cada tiempo para lisar las células y obtener así las
proteínas mediante precipitación con ácido tricloroacético-acetona fría. Se
cuantificaron los extractos de proteínas de cada tiempo de exposición con G418,
por el método 2-D Quant (GE Healthcare) y se tomaron 400 µg de proteína para la
hidratación de la muestra y realizar el corrimiento electroforético en 1-D,
posteriormente se realizó el corrimiento electroforético en 2-D. Una vez terminado
los geles bidimensionales, estos fueron teñidos con Azul de Coomassie coloidal
(Bio-Rad) para la visualización proteica y el posterior análisis de las imágenes.
Cabe mencionar un punto importante, Entamoeba histolytica contiene grandes
cantidades de cisteín proteasas, que tienen que ser inhibida rápidamente para
obtener resultados reproducibles y fiables. Por lo tanto, la presencia del inhibidor
específico de cisteín proteasas antes y después directamente en los ciclos de
congelación-descongelación del extracto de E. histolytica fue altamente
beneficioso tanto para la resolución y reproductibilidad en el análisis 2-DE, desde
luego la extracción de proteínas en general como primer paso. Para evaluar la
resolución y la reproducibilidad de los mapas proteómicos, se comparó el total de
spots en cada condición haciendo mínimo tres experimentos independientes.
Las imágenes de los geles bidimensionales de cada tiempo se obtuvieron con el
Scanner LAS4000 y se analizaron con el software Ludesi REDFIN3 (figura 6) para
obtener la cantidad de spots, el porcentaje de spots de cada condición y la
intensidad de los mismos mediante densitometría.

59
Figura 5. Perfil proteómico de trofozoítos de E. histolytica sin inducir a MCP con G418. (A) gel al
12 %. (B) gel al 10 %.

60
La imagen analizada con el software del perfil proteómico de los trofozoítos de E.
histolytica sin inducir a MCP presentó 142 spots predominantes, se identificaron
un grupo de 18 spots de 50-37 kDa con aproximadamente un pH entre 5 a 9; 51
spots en el rango de 37-20 kDa, 41 spots con un pH de 5 a 8 y 10 spots con pH
aproximado de 9; también se identificaron spots con bajo peso molecular entre 20-
10 kDa, 49 spots con un pH de 4 a 8 y 9 spots con pH de 9 a 10 (figura 6). De
acuerdo con algunos parámetros que nos brindó el software Ludesi REDFIN3, el
volumen de cada spot a través de unidades de pixelaje para determinar la
densitometría y cabe señalar que se observaron varios spots de menor intensidad
que no fueron considerados. A partir de esta imagen control (gel maestro) ¿se
analizaron las imágenes de los perfiles de trofozoítos inducidos a MCP con G418,
utilizando el software Ludesi REDFIN3 las imágenes de los mapas proteómicos
con MCP se colocalizaron para ir monitoreando los spots que se localizaron en el
gel maestro (mapa proteómico de trofozoítos sin MCP) con las imágenes de los
mapas proteómicos con las inducciones a MCP. Donde se observó que después
de los 90 min de inducir el proceso de apoptosis con G418, se detectaron 140
spots, manteniéndose esencialmente el mismo perfil proteómico (figura 7). A las 3
h de exposición con el antibiótico hubo un total de 148 spots, en general el patrón
de proteínas se mantuvo pero se logró detectar aproximadamente un grupo de 7
proteínas ácidas (pH de 5): dos spots de 100 kDa, tres spots entre el rango de 75
a 50 kDa, dos spots con un peso molecular entre 50 a 37 kDa, y una proteína
básica (pH de 9) de aproximadamente 37 kDa (figura 8). A las 6 h hay un total de
152 spots, aparentemente se mantuvo el mismo patrón del perfil proteómico de 3 h
de inducción, además de un spot de 250 kDa y pH 5 (figura 9). El perfil de 9 h de
inducción a MCP tuvo un total de spots de 156, el grupo de proteínas ácidas del
perfil proteico de 3 h y 6 h de inducción fue más evidente en esta condición (figura
10).

61
Figura 6. Imagen del perfil proteómico de trofozoíto sin inducir a MCP. Análisis de imagen con el
software Ludesi REDFIN3.
Flechas naranja, spots en el rango de 50-37 kDa; flechas moradas, spots ubicados en el rango de
37-20 kDa; flechas verdes spots en el rango de 20-10 kDa.

62
Figura 7. Perfil proteómico de 1 ½ h (90 min) de inducción a MCP.
Flechas naranja, spots en el rango de 50-37 kDa; flechas moradas, spots ubicados en el rango de
37-20 kDa; flechas verdes, spots en el rango de 20-10 kDa.

63
Figura 8. Perfil proteómico de 3 h de inducción a MCP.
Flechas rojas, spots ubicados en el rango de 250-75 kDa; flechas naranja, spots en el rango de 50-
37 kDa; flechas moradas, spots ubicados en el rango de 37-20 kDa; flechas verdes, spots en el
rango de 20-10 kDa.

64
Figura 9. Perfil proteómico de 6 h de inducción a MCP.
Flechas rojas, spots ubicados en el rango de 250-75 kDa; flechas naranja, spots en el rango de 50-
37 kDa; flechas moradas, spots ubicados en el rango de 37-20 kDa; flechas verdes, spots en el
rango de 20-10 kDa.

65
Figura 10. Perfil proteómico de 9 h de inducción a MCP.
Flechas rojas, spots ubicados en el rango de 250-75 kDa; flechas naranja, spots en el rango de 50-
37 kDa; flechas moradas, spots ubicados en el rango de 37-20 kDa; flechas verdes, spots en el
rango de 20-10 kDa.

66
Posteriormente, para observar la expresión diferencial de los spots, se utilizó el
programa Ludesi REDFIN3. Con herramientas que nos brinda el software los spots
detectados fueron etiquetados (datos no mostrados), como se muestra en la figura
figura 11 (donde solo se ilustra aquellos spots a los cuales se fue monitoreando
individualmente su comportamiento), una vez etiquetados los spots se realizó la
comparación de los perfiles proteómicos para ver aquellos spots diferenciales que
pudieran aparecer o desaparecer en las diferentes condiciones. Comparando el
perfil de trofozoítos sin G418 con el perfil de 1 ½ h de inducción a MCP, se
observó la desaparición del spot 114 de aproximadamente 10 kDa con pH de 7
(figura 11). A las 3 h de exposición con el antibiótico, comenzaron a ocurrir
cambios significativos, aparecieron tres spots con un pI de 5 dentro del rango de
75 y 37 kDa, nos referimos a los spot con los ID siguientes: 65, 77 y 2. Por otro
lado, se detecto también al spot 11 con aproximadamente un pI de 8 y un PM
experimental de 35 kDa (figura 12). A las 6 hde inducción de la apoptosis
aparecen tenuemente el spot 16 con aproximadamente 250 kDa y pH 5; y el spot
94 con un pH de 7 y un PM de 15 kDa aproximadamente. Además en esta
condición desparecieron los spots 2 y 11, por otro lado también desapareció un
spot que se observaba desde el perfil proteómico control, es decir en el perfil de
trofozoítos sin inducir a MCP, y durante el tiempo de 1 ½ h y 3 h de inducción, no
fue hasta las 6 h que el spot 86 con PM experimental de 18 kDa y un pI de 8
aproximadamente, se ausentó (figura 13). En el perfil de 9 h de inducción
comparado con el de 6 h, fue más evidente el spot 16, mientras que los spots 65 y
77 que aparecieron tenuemente a las 3 h de MCP, fueron más evidentes a las 9 h
de inducción a MCP; en esta condición aparecieron notablemente los spots 117
(PM experimental de 72 kDa y pH de 8), 45 (PM de 38 kDa y pH de 6
experimental), 115 (PM de 37 kDa y pH de 6.5 experimental), 78 (PM de 35 kDa y
pH de 6 experimental ) y 83 (PM de 23 kDa y pH de 7 experimental). En contraste,
desaparecieron los spots 15 (PM de 23 kDa y pH de 5 experimental) y 13 (PM de
16 kDa y pH de 9), ambos spots fueron aparentemente notables en todas las
condiciones y se ausentaron hasta las 9 h de incubación con el G418 (figura 14),

67
mientras que el spot 94, el cual estuvo presente a las 6h de inducción a MCP
aparentemente desaparece en esta condición.
Figura 11. Etiquetas de los spots diferenciales, utilizando el software Ludesi REDFIN3.

68
Figura 12. Comparación del perfiles porteómicos. (A) trofozoítos sin inducir a MCP (B) trofozoítos
expuestos 1 ½ h a G418. El círculo rojo indica la desaparición del spot.

69
Figura 13. Comparación del perfiles porteómicos. (A) trofozoítos expuestos 1 ½ h a G418 (B)
trofozoítos expuestos 3 h a G418. Los círculos negros indican la presencia de nuevos spots.

70
Figura 14. Comparación del perfiles porteómicos. (A) trofozoítos expuestos 3 h a G418 (B)
trofozoítos expuestos 6 h a G418. Los círculos rojos indican la desaparición de spots mientras que
los círculos negros la presencia de nuevos spots.

71
Figura 15. Comparación del perfiles proteómicos. (A) trofozoítos expuestos 6 h a G418 (B)
trofozoítos expuestos 9 h a G418. Los círculos rojos indican la desaparición de spots y los círculos
negros la presencia de nuevos spots.

72
Se ampliaron los spots monitoreados para observar los cambios de la inducción
del proceso de apoptosis, donde se puede observar la desaparición o aparición de
cada spot diferencial, el software nos brida herramientas para demostrar la
expresión de los spots mediante densitometría el volumen de cada spot además
de graficar estos valores densitométricos (figura 16).
Los spots seleccionados fueron analizados cuantitativamente mediante
densitometría en unidades de pixelaje. El valor de densitometría de cada spot fue
graficado, la primera barra del lado izquierdo corresponde al control, y la segunda,
tercera, cuarta y quinta barra de izquierda a derecha corresponde al tiempo de
inducción de apoptosis de 1 ½ h, 3 h, 6 h y 9 h, respectivamente (figura 16). El
volumen de cada spots diferencial seleccionado se puede observar en la tabla 4.
Mientras que en la tabla 5 se muestra un resumen del peso molecular, punto
isoeléctrico experimental y del monitoreo de la intensidad de detección observada
en el transcurso de la inducción a MCP de los spots seleccionados.
Cabe mencionar que en nuestro caso realizamos el Análisis de Expresión
Diferencial para poder trabajar con un conjunto de datos reducido que incluyese
sólo aquellas manchas con un valor p <0.05 en la prueba de ANOVA, además
este software cuenta también con la prueba de Mann-Whitney.
,

73

74
Figura 16. Ampliación de los spots expresados diferencialmente de los perfiles proteómicos de
trofozoítos: sin inducir a MCP (control); y a distintos tiempos de inducción de MCP (1 ½, 3, 6 y 9 h).
Del lado derecho se gráfica el volumen de su comportamiento de cada spots, la barra roja indica el
control y las barras verdes de izquierda a derecha indican los tiempos de inducción a MCP 1 ½, 3,
6 y 9 respectivamente. La línea horizontal representa el valor de corte que discrimina un valor
densitométrico positivo para cada spot.

75
Tabla 4. Volumen densitométrico de cada spot
ID CONTROL 0 h
1 ½ h 3 h 6 h 9 h
114 2092.60 1187.36 929.11 693.06 738.06
65 6607.68 3245.01 2924.40 3744.10 26031.11
77 12874.88 5182.33 3421.59 4549.23 53364.00
2 9982.08 30257.02 27343.46 20460.24 17463.80
11 10941.22 7945.30 43740.12 16047.54 2685.13
16 47715.91 150907.03 35933.51 155366.58 154480.73
94 1674.41 1869.31 1600.79 2170.23 2000.43
86 3207.65 5288.49 4746.75 3774.60 954.88
117 1391.64 1400.84 938.58 1581.92 3242.88
115 1020.22 1058.54 1426.20 857.20 1746.00
45 1247.18 1485.57 1411.41 1463.54 4062.40
78 1884.64 2014.03 1848.47 3055.92 2466.39
83 5734.81 3761.82 3651.03 4890.08 5499.21
15 49164.54 79444.74 65215.60 67976.33 22031.41
13 287151.00 159028.66 317176.41 124286.34 21007.25
Se recortaron las secciones del gel correspondientes a cada proteína
seleccionada, se colocaron en tubos eppendorf adicionando 100 µl de agua MilliQ
para analizar las proteínas mediante MALDI-TOF y MALDI-TOF/TOF, en
colaboración con la Unidad de Proteómica de la Universidad Complutense de
Madrid.

76
Tabla 5. Peso molecular, punto isoeléctrico experimental y expresión cualitativa de
cada spot monitoreado en los diferentes tiempos de inducción
ID de spot
PM (kDa)
PI 0 h (S/A)
1 ½ h 3 h 6 h 9h
114 12 7 +++ + + + -
65 51 5 - + ++ ++ +++
77 42 5 - + ++ ++ +++
2 37 5 - + +++ - -
11 35 8 - - +++ - -
16 250 5 - - - ++ +++
94 15 7 - - - +++ +
86 18 8 +++ +++ ++ - -
117 72 8 - - - - +++
115 37 6.5 - - - - +++
45 38 6 - - - - +++
78 35 6 - - - ++ +++
83 23 7 +++ - - ++ +++
15 23 5 + +++ ++ ++ -
13 16 9 +++ ++ +++ ++ -
Los símbolos representan: +, spot presente;++, detección moderada; +++, detección alta; -, spot
ausente).
De los 15 spots seleccionados, 7 que ya fueron identificados y caracterizados por
sus perfiles peptídicos (tabla 6) y comparados con una base de datos no
redundante (NR; ~4 x 106 entradas; Nacional Center for Biotechnology
Information), donde se realizaron búsquedas empleando también MASCOT 2.3
(Matrix Science/www.matrixscience.com) a través del servidor Global Protein v3.5
de Applied Biosystems. Mediante el análisis de la fragmentación peptídica MALDI

77
TOF-TOF, se secuenciaron las proteínas obteniendo: El spot con ID: 83, que se
identificó como una alcohol deshidrogenasa con un PM de 180.76 kDa y PI de
6.34; el spot 11 que correspondió como gliceraldehído-3-fosfatasa deshidrogenasa
con PM de 36.333 kDa y PI de 7.04; los spots 16, 65, 77 y 2 identificados como
actina con pesos moleculares de 42.042 KDa, 42.042 kDa, 42.345 kDa, 42.345
kDa y puntos isoeléctricos de 5.26, 5.26, 5.19 y 5.19, respectivamente y el spot 13
que fue identificado como Grainina 2 con PM de 24. 220 kDa con un PI de 8.35.
Cabe mencionar que los PM y pI de las proteínas identificadas son datos teóricos.
En la tabla 7, se resume los datos de PM y pI experimentales y teóricos de cada
spot con su ID y el nombre de los spots ya identificados por espectrometría de
masas MALDI TOF, MALDI TOF-TOF.

78
Tabla 6. Resultados de huella peptídica (MALDI TOF) y MALDI TOF-TOF.

79

80

81

82

83

84

85
Tabla 7. Proteínas diferenciales obtenidas de los perfiles proteómicos de
trofozoítos de E. histolytica inducidos a MCP con G418, identificadas por MALDI
TOF/ MADI TOF-TOF.
ID PROTEÍNA IDENTIFICADA PM (T/E) PI (T/E)
83 Alcohol deshidrogenasa 18.076/23 6.34/7
11 Gliceraldehído-3-fosfato deshidrogenasa 36. 333/35 7.04/8
16 Actina 42. 042/250 5.26/5
65 Actina 42. 042/51 5.26/5
77 Actina 42. 345/42 5.19/5
2 Actina 42. 345/37 5.19/5
13 Grainina 2 24.220/16 8.35/9

86
8. DISCUSIÓN
Anteriormente se consideraba que la muerte celular programada (MCP) existía
exclusivamente en eucariontes superiores, sin embargo, estudios recientes
demuestran que diversos eucariotas unicelulares pueden ejecutar mecanismos de
muerte celular programada, por lo que se cree constituye un fenómeno biológico
generalizado en todos los seres vivos. Es por eso que este mecanismo se ha
convertido en un campo interesante en los parásitos protozoarios, no sólo por sus
implicaciones evolutivas, sino también debido a la participación que este proceso
pudiera tener en la interacción huésped-parásito.
Con base en los estudios realizados en Trypanosoma (Nguewa et al., 2004),
Leishmania (Nguewa et al., 2004) y Plasmodium (Al-Olayan et al., 2002) entre
otros se ha sugerido que la MCP puede contribuir a la regulación de la densidad
celular de los parásitos, tanto en los huéspedes vectores como en los mamíferos;
a la diferenciación de los distintos estadios de algunos parásitos; a la respuesta al
estrés y como un mecanismo que module la respuesta inmune del huésped.
Diferentes eventos celulares característicos de la apoptosis de eucariotes
superiores han sido observados ante ciertos estímulos tales como: la exposición
de fosfatidilserina en la superficie de la membrana celular; la fragmentación del
DNA genómico y diversos cambios morfológicos, así como la despolarización de la
membrana mitocondrial. Sin embargo, debido a que los microorganismos no
presentan todas las características propias de la apoptosis típica, se sigue
discutiendo la existencia de mecanismos suicidas de muerte en estos organismos.
En E. histolytica, durante el proceso patogénico, es decir durante el proceso de
invasión y durante la colonización del tejido, los trofozoítos se ven continuamente
expuestos a la respuesta inmune que monta el huésped en contra del parásito. Sin
embargo, diversos estudios han mostrado que E. histolytica posee varios
mecanismos de evasión para dicha respuesta: Los trofozoítos activan la vía
clásica y alterna del complemento en la ausencia de anticuerpos anti-amebianos a

87
través de una CP de 56 kDa (Reed, 1990), degradando a las fracciones C3, C4,
C5 y C9, evitando así efectos fisiológicos de anafilatoxinas C3a, C5a (Reed,
1989); inducen la digestión proteolítica de anticuerpos IgA e IgG por proteasas
(Kelsall, 1993); presentan un recambio membranal importante, mediante la
formación del casquete y muda de los anticuerpos unidos a su membrana, ya que
se deshace de ellos, evitando así su acción (Calderon, 1980); interfieren la
actividad anti-amebiana de los neutrófilos, interrumpiendo las actividades de la
NADPH oxidasa, y resistiendo a través de la peroxirredoxina de 29 kDa (Guerrant,
1981)(Arbo, 1990); y también se ha demostrado in vitro que E. histolytica puede
inducir la apoptosis de los neutrófilos; Sim y colaboradores en el 2005,
evidenciaron mediante microscopía electrónica de transmisión (TEM) en que los
neutrófilos incubados con trofozoitos se observaron características de apoptosis,
tales como la compactación de la cromatina, la condensación de la envoltura
nuclear, manteniendo la integridad de la membrana celular, siendo este
mecanismo un importante evento en la resolución de la inflamación y por lo tanto
de la sobrevivencia del parásito (Sim, 2005). Se ha descrito que induce apoptosis
en leucocitos (Stauffer, 2003) (Berninghausen, 1997), en los macrófagos y células
blanco como las hepáticas (Seydel, 1998). En contraparte, también se ha
observado la MCP en este parásito, in vitro: mediante antibióticos como G418 y
emetina (Villalba, 2007 a y b), evidenciado por nuestro grupo de trabajo en el
2007; por especies reactivas de oxído nítrico descrito por Ramos y colaboradores
en el 2007 y por especies reactivas de oxígeno según Ghosh y colaboradores en
el 2010. De estos trabajos se puede resumir que el fenómeno presenta diversos
cambios morfológicos y bioquímicos como: la fragmentación del DNA, la
condensación de la cromatina nuclear, la acidificación del pH intracelular, el
incremento de los niveles de Ca2+citosólico, y el aumento en las especie reactivas
de oxígeno.
En nuestro grupo de investigación también demostramos que la MCP inducida por
G418 representa un mecanismo activo que involucra por un lado la síntesis de
novo de proteínas, y por el otro lado el encendido de genes específicos a tiempos

88
tempranos, tales como la Glutaminil-tRNAsintetasa, las Subunidades ribosomales
40S y 18S, el Sir-2, las Graininas 1-2 y la Saponina-like, los cuales pudieran estar
implicados en la regulación de rutas pro y anti-apoptóticas (Sánchez, 2010). Esta
hipótesis se sustenta por el hecho de que en otras especies, genes similares
participan en el fenómeno de MCP, sugiriendo que es un evento controlado y
modulado genéticamente.
Mucho se ha avanzado en el conocimiento de la biología de E. histolytica, sin
embargo, la singular relación huésped-parásito aún no está bien establecida.
In vivo, nuestro grupo de investigación demostró mediante un modelo
experimental de interacción huésped parásito, que consistió en la colocación de
una bolsita de diálisis con trofozoitos de E. histolytica en la cavidad peritoneal de
hámster chinos, que existen factores difusibles del huésped y del parásito que
inducen la apoptosis tanto en los neutrófilos como en los trofozoítos, lo que
sugiere que durante el proceso fisiopatogénico se desarrolla una respuesta del
huésped contra el parásito, con la consecuente activación de mecanismos de
defensa del parásito que agreden al huésped (Villalba 2011). Así, como
mecanismos de protección contra la apoptosis ante dicho evento.
Con lo anterior, está claro que E. histolytica, además de sufrir eventos de MCP, es
capaz de inducir o prevenir la MCP en las células efectoras del sistema
inmunológico, tanto in vivo como in vitro.
Con la finalidad de profundizar en el conocimiento de los posibles mecanismos
que participan en el mecanismo de MCP en la relación huésped-parásito, nuestro
grupo se encuentra analizando la expresión diferencial global de genes implicados
en la MCP de E. histolytica, mediante microarreglos de expresión (datos no
mostrados), utilizando el mismo modelo experimental in vivo. Los resultados
sugieren que bajo estas condiciones se modifica la expresión de 124 genes, de
estos, 39 genes se observaron con una menor expresión basal, en comparación
con 85 genes que fueron altamente regulados, como por ejemplo las proteínas de
unión a calcio que aumentó a los 30 min y fue disminuyendo a los 90 min y aún

89
mas a los 180 min de interacción con las células inflamatorias, (Pérez Ishiwara,
D.G., comunicación personal).
Los datos obtenidos con el análisis global de expresión que están en proceso de
publicación, sugieren que el evento inducido in vivo e in vitro pudiera seguir
procesos moleculares similares y ser un importante evento en la patogénesis de la
amebiasis, del cual se han aislado y caracterizado algunos genes involucrados en
la regulación de las fases tempranas de MCP inducida por G418, tanto in vitro
(Sánchez, et al., 2010) como in vivo (Pérez Ishiwara, D.G., 2012 comunicación
personal). La caracterización de la expresión de estos genes permite sentar las
bases para caracterizar las moléculas efectoras, es decir aquellas responsables
del inicio y la ejecución de la MCP que darán lugar a los cambios bioquímicos,
ultraestructurales y morfológicos que ocurren después de 6 h de la incubación con
G418, es decir la fase resolutiva de la apoptosis (Villalba en el 2007). Al respecto
en un análisis proteico inicial realizado por Sánchez en el 2010, se observó la
inducción de la síntesis de novo de proteínas durante la MCP (Sánchez, et al.,
2010), resultando que a los 90 min hay un pico máximo de síntesis de proteínas, lo
que sugiere la inducción a esos tiempos de proteínas inductoras y reguladoras del
proceso tanto pro- como anti-apoptoticos.
Es por eso que nos dimos a la tarea de investigar que proteínas específicas están
participando en el fenómeno de apoptosis. En este trabajo presentamos el análisis
proteómico comparativo de los diversos estadíos de la MCP inducida con G418 in
vitro. Inicialmente, se estandarizó la técnica de isoelectroenfoque de las proteínas
de trofozoítos sin exponerlos al G418, es decir, sin inducir MCP. Las proteínas de
los geles bidimensionales fueron visualizadas con azul de Coomassie coloidal
(Bio-Rad) obteniendo un patrón reproducible con aproximadamente 110 spots,
cabe mencionar que el total de spots encontrados es el promedio de tres
experimentos biológicos independientes y reproducibles (datos no mostrados). El
hecho de que se hayan detectado alrededor de 110 spots no indica que este fuera
el número total de spots encontrados, debido a que existen tinciones con mayor
sensibilidad que el azul de Coommasie coloidal, como la tinción de plata o

90
métodos de fluorescencia y radiactividad, que tienen una sensibilidad de 5-10 ng
(Shevchenko, et al., 1996), 1 ng (Rabilloud, et al., 2001) y 1 pg, respectivamente.
Uno de los primeros estudios proteómicos de E. histolytica, como evidencia de lo
anteriormente mencionado, son los trabajos de Leitsch en el 2005, donde
obtuvieron alrededor de 1500 spots que fueron detectados por tinción de plata, de
los cuales 10 spots fueron identificados por espectrometría de masas matriz
asistida por láser de desorción / ionización-tiempo de vuelo (MALDI-TOF) o
secuenciación de proteínas (Tabla 7).
Tabla 8. Lista de proteínas de E. histolytica identificadas en geles bidimensionales
teñidos con azul de Coomassie, por Leitsch y colaboradores en el 2005.
Número Proteína identificada PM PI (T/E)
1 Actina 42.000 5.20/5.6
2 Calreticulim 47.700 4.7/5.1
3 Bip/Grp 78 71.600 5.05/5.5
4 Hsp70 71.526 5.30/5.75
5 Fragmento de actina
˜ 35.000
/5.75
6a-6b Piruvato fosfatasa dicinasa 98.060 6.00/6.4-6.6
7 Fructusa 1,6 bifosfato
aldosa
36. 286 6.10/6.8
8 Deshidrogenasa-1 NADP-
dependiente de alcohol
38.567 6.10/6.85
9 Fosfogliceratocinasa 46.700 8.20/7.1
10 Superoxidodismutasa 22.030 6.30/7
(PM), peso molecular y (PI) punto isoeléctrico. T, teórico; E, experimental.

91
J. Tolstrup y colaboradores en el 2006, caracterizaron 100 de los spots obtenidos
en los geles bidimensionales y fueron identificados como proteínas relacionadas
con el citoesqueleto, la glucólisis, el metabolismo del DNA/RNA, el sistema
proteasomal, el tráfico vesicular y la traducción de señales (tabla 8).
Tabla 9. Lista de algunas proteína de E. histolytica reportadas por J. Tolstrup y
colaboradores en el 2006.
Proteína identificada # de acceso PM (T/E) PI (T/E)
Citoesqueleto:
Actina
Actina
Actina
Actina
Actina
Actina
Actina
Factor despolimerizador de actina
Actoforina
Coactosina
Cinesinalike
Cadenas ligeras de miosina unida a calcio
XP_656738
XP_656738
XP_656738
XP_656738
XP_656610
XP_656610
XP_656610
XP_654122
XP_651689
XP_650926
XP_651294
XP_657577
41.7/47
41.7/47
41.7/37
42.0/39
33.3/40
33.3/33
33.3/50
15.2/13
15.7/13
16.1/17
64.0/15
16.9/14
5.0/5.7
5.0/5.7
5.0/5.6
5.2/5.8
5.3/5.5
5.3/5.8
5.3/6.5
5.4/5.5
5.3/5.5
5.0/5.3
6.5/4.7
4.7/4.8

92
Proteínas asociadas a superficie:
Proteína con repetidos ricos en leucina como
BspA
Cysteín proteasa (EhCP-C3)
XP_652259
XP_655128
19.7/24
65.5/11
5.0/5.9
5.5/5.2
Glicólisis:
Aldehído- alcohol 2-
deshidrogenasa
Enolasa
Enolasa
Enolasa
Enolasa
Fructuosa- 1,6 bifosfato-aldosa
Fosforilasa glicógeno
Alcohol deshidrogenasa dependiente de NADP
Piruvatocinasa
XP_657577
XP_649161
XP_649161
XP_649161
XP_649161
XP_650373
XP_655667
XP_653507
XP_649089
95.6/38
47.4/40
47.4/39
47.4/39
47.4/40
36.2/30
105/120
39.2/37
36.4/22
6.9/6.6
5.9/6.2
5.9/6.3
5.9/6.4
5.9/6.5
6.1/6.7
6.2/6.0
6.1/6.7
6.5/6.5
Metabolismo del
DNA/RNA:
Box helicasa DEAD/DEAH
Sububidad catalítica alfa
de DNA polimerasa
XP_649638
XP_657373
47.7/24
130/120
5.7/5.0
8.7/6.1

93
Vía de ubiquitinación:
Proteína de choque
térmico 70 kDa
Proteína de choque térmico 70
Subunidad alfa de
proteosoma
Subunidad alfa de proteosoma
Ubiquitina
Proteína relacionada con
ubiquitina
XP_653218
XP_650458
XP_65639
XP_653086
XP_650527
XP_651112
74.0/63
72.0/63
26.9/25
28.2/42
8.7/10
8.6/10
5.1/5.8
5.3/5.9
5.9/6.0
5.6/4.1
6.6/6.1
5.0/5.3
Traducción de señales:
Proteíncinasa
Fostatasaserina/threonina
Tráfico vesicular:
Inhibidor de disociación Rab- GDP
Familia Rab
GTPasa-RabK1
Familia Rab GTPasa-RabK1
Familia Rab GTPasa-
RabF4
Familia Rab GTPasa-RabX28
Familia Rab GTPasa-
RabX28
XP_655027
XP_656538
XP_655898
XP_652298
XP_652298
XP_654217
XP_650164
XP_650164
138/130
46.8/23
55.0/38
24.4/22
24.4/15
23.7/24
21.7/21
21.7/22
5.7/5.2
8.3/6.3
5.4/6.0
5.0/6.6
5.0/4.5
6.6/5.6
5.6/5.4
5.6/6.4

94
Familia de Rho GTPasa
Inhibidor de intercambio
Rho-GDP
XP_650536
XP_654522
22.5/22
20.0/17
5.8/5.5
5.5/5.9
En este trabajo también se realizaron tinciones con plata (datos no mostrados) en
geles bidimensionales de trofozoítos sin inducir a MCP, obteniendo un total de 286
spots, cabe mencionar que el numero total de spots obtenidos es menor que el
esperado, ya que algunos antecedentes reportan la detección de un mayor
número de spots detectados tanto con tinciones de azul de Coomassie como con
plata. Consideramos que la diferencia puede deberse a dos hechos
fundamentales: i) los autores citados anteriormente, sugieren que la utilización de
rangos de pH más estrechos, permite una mejor separación de las proteínas y ii) a
diferencia de los otros trabajos, la concentración de proteína utilizada para la
técnica de isoelectroenfoque, en este trabajo fue menor, 400 µg a diferencia de los
autores mencionados que usaron 500 µg. Por lo tanto, debido a que en el presente
trabajo en primera instancia nos interesaba detectar y seleccionar proteínas en los
trofozoítos de E. histolytica cuya expresión se apagara o encendiera al inducir
MCP con G418, decidimos utilizar tiras de poliacrilamida de un rango más amplio
de pH (3-11 NL), lo anterior con la finalidad de ampliar el rango de posibilidades
para la detección de las proteínas. Una vez que se estandarizó la técnica de
isoelectroenfoque y se obtuvieron resultados reproducibles, se procedió a realizar
la inducción de MCP de los trofozoitos a tiempos tempranos (1 ½ h y 3 h), medios
(6 h) y tardíos (9h) con G418 con la finalidad de detectar aquellos spots
diferenciales de cada tiempo.
El perfil proteómico de los trofozoitos sin tratamiento, presento 142 spots
predominantes, a la 1 ½ h de exposición con G418 se detectaron un total de 140
spots, a las 3 h 148 spots, al tiempo de 6 h se detectaron 152 spots y finalmente a

95
las 9 h 156 spots (cabe señalar que se observaron un número importante de spots
de menor intensidad que no fueron considerados), indicando que el fenómeno de
MCP por G418 en trofozoítos de E. histolytica conlleva la síntesis de moléculas
que pudieran participar en la MCP.
Villalba en el 2007, caracterizó los cambios morfológicos y bioquímicos de
trofozoítos de E. histolytica inducidos a MCP con G418 in vitro durante 3, 6, y 9 h,
reportando que a partir de las 6 h es donde se observaron cambios morfológicos
evidentes de MCP, los cuales evolucionaron paulatinamente hacia las 9 h.
Como comentamos con anterioridad, este evento se demostró que es controlado
genéticamente (Sánchez y colaboradores, 2010) ya que hay una expresión de
genes anti-apoptóticos a los 30 min de exposición con G418, incluyendo:
Glutaminil-tRNAsintetasa, Subunidades ribosomales 40S y 18S, Sir-2 y las
Graininas; así como también la detección de gen de la saponina-like que es un
gen apoptótico. De manera paralela, se observó la síntesis de proteínas de novo a
los 90 min posteriores a la inducción de los trofozoítos incubados con G418. Lo
que sugiere que dicha expresión puede ser el reflejo de la inducción de la
expresión de genes específicos.
Por tanto, encontrar aquellas moléculas responsables de los cambios
mencionados anteriormente, fue el objetivo de este trabajo. Se compararon los
geles bidimensionales de los distintos tiempos de inducción a MCP probados (0, 1
½, 3, 6 y 9 h) para detectar los spots diferenciales (ID: 114, 65, 77, 2, 11, 16, 94,
86, 117, 115, 45, 78, 83, 15 y 13), de estos solo los spots: 83, 11, 16, 65, 77, 2 y
13 ya fueron identificados mediante huella peptídica (MALDI TOF) y fragmentación
peptídica (MALDI TOF-TOF):

96
Dos de las proteínas identificadas, Alcohol deshidrogenasa con ID: 83 y
Gliceraldehído-3-fosfato deshidrogena con ID: 11, son enzimas importantes en la
vía metabólica de la amiba; la proteína 83, Alcohol deshidrogenasa, se expresó
basalmente en el control (trofozoítos sin G418) y su expresión incrementó en los
tiempos de 6 y 9 h de inducción a MCP. Las alcohol deshidrogenasas (ADHs) son
enzimas que catalizan la oxidación reversible de alcoholes a los correspondientes
aldehídos o cetonas, con la consiguiente reducción de NAD o NADP.
En un estudio de Espinosa en el 2010 en E. histolytica se demostró el efecto
inhibitorio de NO en la actividad de las CPs y la alcohol deshidrogenasa 2, los
resultados indican que la ADH2, es una enzima fundamental para la sobrevivencia
del parásito.
Por otro lado, comparaciones proteómicas entre E. histolytica y E. dispar
evidenciaron la presencia y actividad de alcohol deshidrogenasa 3 (EhADH3), la
cual fue localizada en la superficie de los trofozoítos. Aunque la sobreexpresión de
dicha proteína no fue capaz de asociarse a un incremento en la virulencia, si se
determinó que EhADH3 es más abundante en E. histolytica HM-1: IMSS que
cualquier cepas de E. dispar (Paul H., Davis, 2009).
En otros sistemas de eucariotes superiores, se ha encontrado una asociación de
la ADH con la apoptosis en la disfunción miocárdica (Zhang X, et al ., 2004). Se
observó que durante la miocardiopatía alcohólica, uno de los metabolitos
principales del alcohol es el acetaldehído, este induce la disfunción miocárdica
contráctil, el alargamiento de los cardiomiocitos, liberación de Ca2+, daño
mitocondrial y finalmente la apoptosis. Sugiriendo que la sobreexpresión de la
ADH en la disfunción cardiaca esta íntimamente relacionada con el daño celular
vía apoptosis (Rui Guo, Jun Ren., 2010).
Debido a lo anteriormente mencionado, y a los patrones de detección del alcohol
deshidrogenasa en los geles bidimensionales, podemos sugerir que de alguna

97
manera a tiempos tardíos de la MCP la ADH participa como una proteína
apoptótica, marcando a los trofozoítos a la MCP.
La glicerol-3-fosfato deshidrogenasa (GAPDH ) con ID: 11,por su parte, es una
enzima de la glucólisis, GAPDH ejerce entre otras funciones, por ejemplo,
participar en la endocitosis, en la reparación del DNA (debido a que es una uracil
DNA glucosilada y que remueve uracilos) , en la transcripción, en la exportación
de RNA al núcleo y al parecer puede participar también en la apoptosis (Kim JE,
2003).
Diversos estudios han indicado un papel para la GAPDH en la apoptosis o estrés
oxidativo. El papel en la apoptosis fue inicialmente demostrado en células
granulares de cerebelo (CGCs) (Ishitani, et al., 1998, 1996; Sunaga, et al., 1995).
Las CGCs maduras que sufren una muerte por apoptosis inducida por la edad, fue
asociado con la expresión incrementada de GAPDH. Este efecto fue demostrado
por el hecho que la inhibición de la GADPH mediante secuencias en antisentido
suprimen la acumulación de GADPH antes de la apoptosis (Ishitani, et al., 1998;.
Saunders, et al., 1999; Sawa, et al., 1997). Otros resultados indican que la
translocación al núcleo de GAPDH puede jugar un papel en la apoptosis y estrés
oxidativo, probablemente como enzima reparadora del DNA o como acarreadora
de moléculas pro-apoptóticas (Dastoor, et al., 2001).
En nuestro análisis esta GAPDH fue detectada en el perfil proteómico a las 3 h de
inducción de MCP con G418, sugiriendo, que la GAPDH pudiera compensar el
proceso apoptosis ya que podría participar en la reparación del DNA y actuar
como una proteína anti-apoptótica.
Una de las consecuencias de la activación del proceso de muerte celular consiste
en la fragmentación y desacoplamiento de las principales estructuras celulares,
como componentes del citoesqueleto. Las caspasas degradan a la mayoría de los
constituyentes del citoesqueleto de la célula; los sustratos incluyen componentes
de los microfilamentos de actina, tales como la actina misma y sus proteínas

98
asociadas, incluyendo la miosina, espectrinas, α-actinina, gelsolina y filamina.
Asimismo, diferentes proteínas microtubulares son también sustrato de las
caspasas, incluyendo la tubulina y las proteínas asociadas a los microtúbulos,
tales como Tau, la cadena intermediaria de dineina citoplasmática y pl50Glued
(Rojas Masyelly, et al., 2009). Adicionalmente, las proteínas de filamentos
intermediarios tales como vimentina, queratinas y laminina nuclear son también
moléculas diana de las caspasas.
La proteólisis de estos constituyentes del citoesqueleto probablemente contribuye
al redondeamiento y retracción de la célula que es observado en los estados
tempranos de la apoptosis (Taylor, et al., 2008). Morfológicamente los trofozoítos
de E. histolytica inducidos a apoptosis a partir de las 3 h de incubación con G418,
se observan redondeados, siendo este evento muy evidente a las 12 h, según
datos de Villalba en el 2007. Otra consecuencia de la desestabilización del
citoesqueleto es el “blebbing” o burbujeo de la membrana (otra característica
distintiva de la apoptosis) en el cual el citoplasma celular se particulariza en
pequeñas membranas que son expulsadas al medio. Un sustrato de caspasas que
está fuertemente implicado es esta fase de la apoptosis es el efector Rho ROCKI,
un regulador de la dinámica del citoesqueleto de actina. La eliminación de la
región C-terminal de ROCKl a través del corte por caspasa resulta en la activación
constitutiva de esta cinasa, conduciendo a la fosforilación de la cadena ligera de
miosina y la contracción de la actina (Taylor, et al., 2008).
Por otro lado, aunque la fragmentación nuclear es la principal característica de la
apoptosis, no es claro porque el núcleo se fragmenta y se dispersa a través de los
cuerpos apoptóticos durante este modo de muerte celular. La fragmentación
nuclear resulta de la desintegración de la lámina nuclear y del colapso de la
envoltura nuclear. El primero de estos eventos involucra la proteólisis de las
lamininas A, B, y C por las caspasas. El citoesqueleto de actina tiene también un
rol importante en la fragmentación nuclear. La lámina nuclear está rodeada por
una malla de actina, la cual está asociada con la envoltura nuclear. La inhibición

99
tanto de ROCKI, la cinasa de cadena ligera de miosina, o la interrupción de los
filamentos de actina evita la fragmentación nuclear asociada con la apoptosis. La
degradación de la lámina no es suficiente para causar fragmentación nuclear en
ausencia de la fuerza contráctil del citoesqueleto de actina, pero podría debilitar la
lamina nuclear, permitiendo que la envoltura nuclear se desgarre, luego de la
fragmentación del núcleo (Rojas Masyelly, et al., 2009).
La actina fue una las proteínas prominentemente detectadas a tiempos tardíos en
los geles bidimensionales, lo que sugiere que su detección en fases tardías del
proceso pudiera deberse a la sobreexpresión de actina como un intento de la
célula para re-establecer el citoesqueleto.
Las actinas con ID: 77 y 65, fueron detectadas a tiempos tardíos (9h) en los geles
bidimensionales con PM de 42 kDa y 51 kDa, respectivamente. Mientras que la
actina con ID: 16 con PM de 250 kDa fue evidente a las 6 h y 9 h de inducción a
apoptosis..
La actina con ID: 65, que tuvo un PM experimental de 51 kDa y PM teórico de 42.
042 kDa, es interesante; el hecho de haber identificado diferentes spots como
actinas, con diferentes PM pudiera deberse a que la actina es una proteína muy
versátil que tiene la capacidad de asociarse con otras proteínas, las cuales afectan
la velocidad de polimerización y destrucción de filamentos; algunas proteínas,
como la profilina, se unen a las proteínas de actina libres y favorecen su unión a
filamentos prexistentes, también hay proteínas que median en la interacción de los
filamentos de actina con otras proteínas relacionadas, como es el caso de la
tropomiosina, que media la interacción entre actina y miosina. También las
proteínas de anclaje permiten la unión de los filamentos de actina a estructuras
celulares como las membranas o a otros componentes del citoesqueleto (Molist, et
al., 2011). Este hecho concuerda también con la actina con ID: 16, que teniendo
un PM teórico de 42. 042 kDa, en los geles bidimensionales se observó un PM
experimental aproximado de 250 kDa, indicando posiblemente la formación de
complejos de actina con otras proteínas del citoesqueleto.

100
La aparición a tiempos tardíos de la MCP de las actinas con ID: 77 (9 h), 65 (9 h) y
16 (6 h y 9 h) indican que podrían tener un papel importante, como proteína anti-
apoptótica tratando de re-establecer la conformación del citoesqueleto y/o el
rescate del acoplamiento del citoesqueleto,
Por otro lado, la actina con ID: 2 fue apareciendo en las primeras horas de
inducción (1 ½ h y 3 h) a MCP con G418, este spot con PM teórico de 42.345 kDa
pero con PM experimental aproximado de 37 kDa observado en el gel
bidimensional, indican quizás una proteína no polimerizada (G-actina),
comprometiendo a los trofozoítos a la MCP.
De manera muy interesante en este trabajo se identificó el spot ID: 13 que
correspondió a la proteína grainina 2, hecho que correlaciona con el fenotipo
bioquímico de la MCP producto de la liberación del calcio intracelular (Villalba,
2007), y con los resultados obtenidos a nivel genómico, tanto mediante AFLPs-
RTPCR (Sánchez, 2010) como mediante análisis de expresión global donde se
detectó la sobreexpresión de las graininas 1 y 2. Se observó que la grainina 2
aparece notablemente desde las primeras fases hasta las 6 h de inducción a MCP,
y se dejó de detectar a las 9 h de incubación con G418. Los hallazgos obtenidos
sugieren fuertemente que la proteína grainina 2, participa como factor anti-
apoptotico en la MCP tratando de recapturar el calcio liberado. Hasta antes de
estos estudios se tenía evidencia de la sobreexpresión del gen, sin embargo ahora
podemos mencionar que dicha inducción claramente conduce a la síntesis de la
proteína grainina 2 que por su patrón de expresión participa en el fenómeno de
MCP inducida con G418 en trofozoítos de E. histolytica, tratando de compensar la
liberación del Ca2+ intracelular, recapturándolo para limitar la activación de
diversos procesos celulares, entre ellos los relacionados con las fases activadoras
del proceso de MCP y por lo tanto la adopción sin retorno del compromiso hacia
la muerte.
Analisis tipo Blast y alineamientos con otras proteínas demuestran que la grainina
2 tiene homología con calmodulinas (CaM) de otros protozoarios como T. brucei y

101
T. gondii (Moreno y Docampo, 2003) jugando un papel importante en la MCP
activando mecanismos moleculares que disparan la recapturación de calcio
citosólico, es decir, una respuesta compensatoria para disminuir el calcio
intracelular libre (Sánchez, 2010).
De los 15 spots diferenciales y seleccionados, 8 spots con los siguientes ID:.114,
94, 86, 117, 115, 45, 78 y 15 en estos momentos están secuenciandose por Maldi
TOF-TOF.

102
9. CONCLUSIÓN
Nuestros resultados del análisis proteómico de la MCP en E. histolytica in vitro,
nos permitió elucidar y conocer la expresión de algunas proteínas en las distintas
fases de la MCP inducida por G418.
Se observaron 15 spots diferenciales, los cuales fueron analizados por huella
peptídica y MALDI TOF-TOF con la finalidad de identificar que proteínas son las
que están participando en proceso de apoptosis. Hasta el momento solo se han
identificado 7 spots:
El spot 83 como alcohol deshidrogenasa, se expresó aparentemente tanto en el
control (trofozoítos sin G418) como en los tiempos de 6 y 9 h de inducción a MCP;
sugiriendo un papel apoptótico. Mientras que el spot 11 (gliceraldehído-3-fosfato
deshidrogenasa) solo se expresó a las 3 h del fenómeno, tratando de compensar
la MCP como proteína anti-apoptótica, en la reparación del DNA.
Además, se identificaron por fragmentación peptídica a los spots 77, 2, 65 y 16
como proteínas del citoesqueleto, tales spots fueron determinados como actina
que probablemente en otros sistemas se ha demostrado que tienen un papel
apoptótico, ya que la actina 65, 77 y 16 fueron más evidentes hasta las 9 h de
exposición con el antibiótico, mientras que el spot 2 con PM de 42.345 kDa y PI
de 5.19 se expresó durante las fases iniciales (1 ½ y 3 h) de MCP.
En el perfil proteómico de la ameba se identificó por fragmentación peptídica al
spot 13 como grainina 2, esta proteína una vez inducida la apoptosis desapareció
a las 9 h de exposición con el antibiótico, sugiriendo que la amiba tiene
mecanismos anti-apoptóticos a fases iniciales de la MCP.

103
10. PRESPECTIVAS
• Continuar la investigación sobre proteínas específicas de la MCP de E.
histolytica para conocer el papel fundamental y como están participando en
dicho mecanismo.
• Realizar cepas mutantes con la inhibición de la expresión de proteína (s)
que pudieran tener un papel anti-apoptótico, con la finalidad de detectar
posibles blancos terapéuticos en contra de la amibiasis basada en la MCP
en la ameba.
• Realizar cepas mutantes con la sobreexpresión de proteínas que pudieran
tener un papel apoptótico en E. histolytica, con la finalidad de detectar
posibles blancos terapéuticos en contra de la amibiasis basada en la MCP
en la ameba.

104
11. BIBLIOGRAFÍA Ackers, J. P. M., D. (2006). "Progress in research on Entamoeba histolytica
pathogenesis." Curr Opin Microbiol 9(4): 367-373.
Aebersold R, et al. (2003). "Constellations in a cellular universe." Nature 422:115-
115.
Aebersold R, Mann M. (2003). "Mass spectrometry-based proteomics." Nature.
422:198-207.
Aley, S. B., Scott, W. A. & Cohn, Z. A. (1980). "Plasma membrane of Entamoeba
histolytica." J Exp Med 152, 391–404.
Al-Olayan, E. M., Williams, G. T. & Hurd, H. (2002). "Apoptosis in the malaria
protozoan, Plasmodium berghei: a possible mechanism for limiting intensity
of infection in the mosquito." Int J Parasitol 32, 1133–1143.
Ameisen JC, ldziorek T, Billaut Mulot O, Loyens U Tissier JP, Potemtier A, Ouissi.
(1995). "Apoptosis in a unicellular eukaryote Trypanosoma cruzi:
Implications for the evolutionary origin and role of programmed cell death in
the control of cell proliferation, differentiation, and survival." Cell Death and
Differentiation 2: 285-300.
Ana M, Fernández-Presas. (2000). "Apoptosis en protozoarios y en la célula
huésped inducida por protozoarios." Revista Mexicana de Patología Clínica,
Vol 47, Núm. 2.
Anderson NL, Matheson AD, Steiners. (2000). "Proteomics: applications in basic
and applied biology." Curr Opin Biotech 11:408-412.
Antonsson B. (2001). "Bax and other pro-apoptotic Bcl-2 family «killerproteins» and
their victim the mitochondrion." Cell Tissue Res 306: 347-61.
Ankri, S. S., T. Mirelman, D. (1998). "Antisense inhibition of expression of cysteine
proteinases does not affect Entamoeba histolytica cytopathic or haemolytic
activity but inhibits phagocytosis." Mol Microbiol 28(4): 777-785.
Arbo, A. H., M. Ramirez, A. Ignacio Santos, J. (1990). "Entamoeba histolytica
inhibits the respiratory burst of polymorphonuclear leukocytes." Arch Invest
Med (Mex) 21 Suppl 1: 57-61.

105
Arroyo, R. O., E. (1987). "Localization and identification of an Entamoeba
histolytica adhesin." Mol Biochem Parasitol 23(2): 151-158.
Ashkenazi A. y Dixit V.M. (1998). "Death receptors signaling and modulation."
Science 281, 1305-1308.
Aust-Kettis, A. S., K. G. (1978). "Dynamics of the interaction between Entamoeba
histolytica and components of the immune response. I. Capping and
endocytosis; influence of inhibiting and accelerating factors; variation of the
expression of surface antigens." Scand J Immunol 7(1): 35-44.
Aust-Kettis, A. T., R. Sundqvist, K. G. (1981). "Dynamics of the interaction between
Entamoeba histolytica and components of the immune response. III. Fate of
antibodies after binding to the cell surface." Scand J Immunol 13(5): 473-
481.
Ayala Tovy, Rivka Hertz, Rama Siman-Tov, Sylvie Syan, Daniela Faust, Nancy
Guillen, Serge Ankri. (2011). “Glucose Starvation Boosts Entamoeba
histolytica Virulence." PLoS Negl Trop Dis 5(8): e1247.
doi:10.1371/journal.pntd.0001247.
Bakker BM, Bro C, Kotter P, Luttik MA, van Dijken JP, Pronk JT. (2000). J.
Bacteriol. 182, 4730-4737.
Bansal, D. A., P. Kerneis, S. Frileux, P Boche, O. Baglin, A. C. Dubost, G.
Leguern, A. S. Prevost, M. C. Bracha, R. Mirelman, D. Guillen, N.
Labruyere, E. (2009). "An ex-vivo human intestinal model to study
Entamoeba histolytica pathogenesis." PLoS Negl Trop Dis 3(11): e551.
Baxt, L. A. S., U. (2008). "New insights into Entamoeba histolytica pathogenesis."
Curr Opin Infect Dis 21(5): 489-494.
Becerra Vanessa, Hernán J., P. (2009). "Apoptosis neuronal: la diversidad de
señales y de tipos celulares." Colomb Med. 2009; 40: 124-33.
Bellairs, R. (1961). "Cell death in chick embryos as studied by electron
microscopy." J Anat 95(Pt 1): 54-60 53.
Bernal-Redondo, R. (2001). "Entamoebosis-amibiasis intestinal Entamoeba
histolytica/Entamoeba dispar." Médico del Hospital Infantil de México 58:
217 - 219.

106
Berninghausen, O. L., M. (1997). "Necrosis versus apoptosis as the mechanism of
target cell death induced by Entamoeba histolytica." Infect Immun 65(9):
3615-3621.
Biagi, F. B., F. Ortega, P. S. (1966). "Remobilization of Entamoeba histolytica after
exposure to immobilizing antibodies." Exp Parasitol 18(1): 87-91.
Billaut-Mulot, O., Fernández-Gómez, R., Loyens, M., Ouaissi, A. (1996).
"Trypanosoma cruzi elongation factor 1-α: nuclear localization in parasites
undergoin apoptosis." Gene. 174:19-26.
Billaut-Mulot O, Schoneck R, Fernández-Gómez R, Taibi A, Capron A, Pommier V,
Plumas-Marty B, Loyens M, Ouissi A. (1994). "Molecular an immunological
characterization of a Trypanosoma cruzi protein homologous to mammalian
elongation 1 gamma." Biol Cell 82:39-44.
Botero (1994). Parasitología humana Medellín, Col, CIB.
Braga, L. L. N., H. McCoy, J. J. Adal, K. Wiedmer, T. Pham, C. Sims, P. J. Petri,
W. A., Jr. (1992). "Inhibition of the complement membrane attack complex
by the galactose-specific adhesin of Entamoeba histolytica." Arch Med Res
23(2): 133.
Bruckner, D. A. (1992). "Amebiasis." Clin Microbiol Rev 5(4): 356-369.
Calderon, J. T.-G., R. (1980). "Loss of susceptibility to antibody and complement
mediated lysis in E. histolytica." Arch Invest Med (Mex) 11(1 Suppl): 241-
244.
Calzado, C., J. Verde, et al. (2007). "Inhibición del proceso de enquistamiento de
Entamoeba invadens por Castela texana." Ciencia UNAL 10.
Canales-Trevino, M. L. (1986). "[Experimental amebiasis in the guinea pig: a model
for resistance]." Arch Invest Med (Mex) 17 Suppl 1: 223-228.
Carrero, J. C. C.-R., C. Aguilar-Diaz, H. Diaz-Gallardo, M. Y. Laclette, J. P.
Morales-Montor, J. (2007). "The role of the secretory immune response in
the infection by Entamoeba histolytica." Parasite Immunol 29(7): 331-338.
Chandrima Shaha. (2006). "Apoptosis in Leishmania species & its relevance to
disease pathogenesis. Indian J Med Res 123, pp 233-244.Christenser ST,

107
et al. (1998). Signaling in unicelular eukaryotes." Int. Rev. Cytol. 177:181-
253.
Cole, B. A. K., J. F. (1953). "Immobilization of Endamoeba histolytica in vitro by
antiserum produced in the rabbit." Proc Soc Exp Biol Med 83(4): 811-814.
Conde-Bonfil, M. C. d. l. M.-Z., C. (1992). "[Entamoeba histolytica: a standing
threat]." Salud Publica Mex 34(3): 335-341.
Danial N.N. y Korsmeyer S.J. (2004). "Cell death: critical control points." Cell 116,
205-219.
Dastoor Z, Dreyer JL.(2001). "Potential role of nuclear translocation of
glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative
stress." J Cell Sci. 114(Pt 9):1643-53.
Degterev A. y Yuan J. (2008) "Expansion and evolution of cell death programmes."
Nat Rev Mol Cell Biol. 9, 378-390.
Denault, J. B. S., G. S. (2002). "Caspases: keys in the ignition of cell death." Chem
Rev 102(12): 4489-4500.
Dutta, G. P. (1960). "Metacystic divisions in the life-cycle of Entamoleba histolytica
Shaudinn, 1903, in man." National Instirute of Sciences of India 27.
Earnshaw W.C., Martins L.M. y Kaukmann S.H. (1999). "Mammalian caspases:
structure, activation, substrates, and functions during apoptosis". Annu Rev
Biochem. 68, 383-424.
Espinosa-Cantellano, M. M.-P., A. (2000). "Pathogenesis of intestinal amebiasis:
from molecules to disease". Clin Microbiol Rev 13(2): 318-331.
Falkowski, A. H., R. Han, V. Richardson, B. (2002). "Apoptosis in the preterm and
near term ovine fetal brain and the effect of intermittent umbilical cord
occlusion". Brain Res Dev Brain Res 136(2): 165-173.
Freitas Balanco JM, et al., (2001). "Apoptotic mimicry by an oblígate intracelular
parasite downregulates macrophage macrophage microbicidal activity." Curr
Biol 11:1870-3.
Flores, E.R. et al., (2002). "p63 and p73 are requiered for p53 dependenty
apoptosis in response to DNA damage." Nature 416:560-564.

108
Gaucher, D. (2002). "Construction and immunogenicity of a codon-optimized
Entamoeba histolytica Gal-lectin-based DNA vaccine." Vaccine 20(27-28):
3244-3253.
Gatti, R., Belletti, S., Orlandini, G., Bussolati, O., Dall’Asta, V. & Gazzola, G. C.
(1998). "Comparison of Annexin V and Calcein-AM as early vital markers of
apoptosis in adherent cells by confocal laser microscopy." J Histochem
Cytochem 46, 895–900.
Ghosh, A. S. D., S. Raha, S. (2010). "Hydrogen peroxide-induced apoptosis-like
cell death in Entamoeba histolytica." Parasitol Int 59(2): 166-172.
Gil C. (2003) "La metodología proteómica: una herramienta para la búsqueda de
función." Actualidad. 35:12-20.
Goldsmith, M. (1966). "The anatomy of cell death (Abstr.)." J. Cell Bio 31: 41.
Guerrant, R. L. B., J. Ravdin, J. I. Sullivan, J. A. Mandell, G. L. (1981). "Interaction
between Entamoeba histolytica and human polymorphonuclear neutrophils."
J Infect Dis 143(1): 83-93.
Hanash S. (2003). "Disease proteomics." Nature. 422:226-232.
Haque, R. (2001). "Amebiasis and mucosal IgA antibody against the Entamoeba
histolytica adherence lectin in Bangladeshi children." J Infect Dis 183(12):
1787-1793.
Haque, R. (2006). "Diagnosis of amebiasis in Bangladesh." Arch Med Res 37(2):
273-276.
Hengartner M.O. y Horvitz H.R. (1994) "C. elegans cell survival gene ced-9
encodes a functional homolog of the mammalian proto-oncogene bcl-2."
Cell 76, 665-676.
Hsu, C. A. R., A. K. Su-Li, X. Gerald, T. M. Dawson, M. I. Schiffer, C Reichert, U.
Shroot, B. Poirer, G. C. Fontana, J. A. (1997). "Retinoid induced apoptosis
in leukemia cells through a retinoic acid nuclear receptor-independent
pathway." Blood 89(12): 4470-4479.
Hughes, M. A. P., W. A., Jr. (2000). "Amebic liver abscess." Infect Dis Clin North
Am 14(3): 565-582, viii.

109
H Zangger, et al. (2002). "Cell death in Leishmania induced by stress and
differentiation: programmed cell death or necrosis?" Cell Death Differ.9:
1126-1139.
Ishitani, R., Sunaga, K., Hirano, A., Saunders, P., Katsube, N. and Chuang, D. M.
(1996). "Evidence that glyceraldehyde-3-phosphate dehydrogenase is
involved in age-induced apoptosis in mature cerebellar neurones in culture."
J. Neurochem. 66, 928-935.
Ishitani, R., Tanaka, M., Sunaga, K., Katsube, N. and Chuang, D. M.
(1998)."Nuclear localization of overexpressed glyceraldehyde-3-phosphate
dehydrogenase in cultured cerebellar neurones undergoing apoptosis." Mol.
Pharmacol. 53, 701-707.
Iris Bruchhaus, Thomas Roeder, Annika Rennenberg y Volker T. Heussler. (2007).
"Protozoan parasites: programmed cell death as a mechanism of
parasitism." TRENDS in Parasitology Vol. 23 No. 8.
Jackson, T. F., V. Gathiram, et al. (1985). "Seroepidemiological study of antibody
responses to the zymodemes of Entamoeba histolytica." Lancet 1(8431):
716-719.
Jiang, S., Chow, S. C., Nicotera, P. & Orrenius, S. (1994). "Intracellular Ca2+
signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase
inhibitor thapsigargin." Exp Cell Res 212, 84–92.
Kelsall, B. L. R., J. I. (1993). "Degradation of human IgA by Entamoeba histolytica."
J Infect Dis 168(5): 1319-1322.
Kim JW, Danng CV. (2005). "Multipaceted roles of gycolytic enzymes. TRENDS
Biochem. Sci. 30: 142-150.
Lee, N, et al., (2002). "Programmed cell death in the unicelular protozoan parasite
Leishmania." Cell Death Differ. 9: 53-64.
Leroy, A. (2000). "Entamoeba histolytica disturbs the tight junction complex in
human enteric T84 cell layers." FASEB J 14(9): 1139-1146.
Leippe, M., Ebel, S., Schoenberger, O.L., Horstmann, R.D., Muller-Eberhard, H.J.
(1991). "Pore-forming protein of pathogenic Entamoeba histolytica." Proc.
Natl. Acad. Sci. USA. 88: 7659-7663.

110
Leippe, M., E. Tannich, R. Nickel, G. van der Goot, F. Pattus, R. D. Horstmann, y
H. J. Mu¨ller-Eberhard. (1992). "Primary and secondary structure of the
pore-forming peptide of pathogenic Entamoeba histolytica." EMBO J.
11:3501–3506.
Lizarbe Iracheta Mª Antonia. (2007). "El suicidio y muerte celular."
Rev.R.Acad.Cienc.Exact.Fís.Nat. (Esp); 101
Lu, H. F. C., Y. S. Yang, J. S. Chen, J. C. Lu, K. W. Chiu, T. H. Liu, K. C. Yeh, C.
C. Chen, G. W. Lin, H. J. Chung, J. G. (2008). "Gypenosides induced G0/G1
arrest via inhibition of cyclin E and induction of apoptosis via activation of
caspases-3 and -9 in human lung cancer A-549 cells." In Vivo 22(2): 215-
221.
Martin, J. B., Bakker-Grunwald, T. & Klein, G. (1993). "31P-NMR analysis of
Entamoeba histolytica. Occurrence of high amounts of two inositol
phosphates." Eur J Biochem 214, 711–718.
Medema J. P., Scaffidi C., Kischkel F.C., Shevchenko A., Mann M., Krammer P.H.
y Peter M.E. (1997). "FLICE is activated by association with the CD95
death-inducing signaling complex (DISC)." EMBO J 16, 2794-2804.
Meza, I., F. Cazares, et al. (1987). "Use of antibodies to characterize a 220-
kilodalton surface protein from Entamoeba histolytica." J Infect Dis 156(5):
798-805.
Meza, I. T.-R., P. Vargas, M. A. (2006). "The cytoskeleton of Entamoeba
histolytica: structure, function, and regulation by signaling pathways." Arch
Med Res 37(2): 234-243.
Moreira CM, Del Portillo AH, Milder RV, Balanco JM, Barcinski M. (1995). "Heat
shock of the unicellular organism Leishmania amazonensis." J Cell Physiol
167:305-313.
Moreno, S.N.J., Docampo, R. (2003). "Calcium regulation in protozoan parasites."
Current Opinion in Microbiology 6, 359-364.
Mukherjee, C., C. G. Clark, et al. (2008). "Entamoeba shows reversible variation in
ploidy under different growth conditions and between life cycle phases."
PLoS Negl Trop Dis 2(8): e281.

111
Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A. (1996).
"FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to
the CD95 (Fas/APO-1) death-inducing signaling complex." Cell 85, 817-827.
Neal, R. A. (1971). "Pathogenesis of amoebiasis." Gut 12(6): 483-486.
Nguewa, P. A., Fuentes, M. A., Valladares, B., Alonso, C. & Pe´ rez, J. M. (2004).
"Programmed cell death in trypanosomatids: a way to maximize their
biological fitness?" Trends Parasitol 20, 375–380.
Olivos-Garcia, A. N.-A., M. Tello, E. Martinez, R. D. Gonzalez-Canto, A. Lopez-
Vancell, R. Garcia de Leon, M. C. Montfort, I. Perez-Tamayo, R. (2004).
"Inflammation, complement, ischemia and amoebic survival in acute
experimental amoebic liver abscesses in hamsters." Exp Mol Pathol 77(1):
66-71.
Paul H. Davis, Xiaochun Zhang, Jianhua Guo, R. Reid Townsend1 and Samuel L.
Stanley, Jr., (2006) “Comparative proteomic analysis of two Entamoeba
histolytica strains with different virulence phenotypes identifies peroxiredoxin as an
important component of amoebic virulence." Molecular Microbiology, 61,
1523–1532.
Paul H. Davis, et al., (2009) " Proteomic Comparison of Entamoeba histolytica and
Entamoeba dispar and the Role of E. histolytica Alcohol
Dehydrogenase 3 in Virulence." PLoS Negl Trop Dis 3(4): e415.
doi:10.1371/journal.pntd.0000415.
Petri, W. A., Jr. (1993). "Molecular mechanisms of invasion by Entamoeba
histolytica." Semin Cell Biol 4(5): 305-313.
Pimenta, P. F., L. S. Diamond, et al. (2002). "Entamoeba histolytica Schaudinn,
1903 and Entamoeba dispar Brumpt, 1925: differences in their cell surfaces
and in the bacteria-containing vacuoles." J Eukaryot Microbiol 49(3): 209-
219.
Pumarola (1991). Microbiología y Parasitología Médica
Rabilloud, et al.( 2001).Proteómica 1:699-704).
Ravichandran K.S. y Lorenz U. (2007). "Engulfment of apoptotic cells: signals for a
good meal." Nat Rev Immunol. 7, 964-974.

112
Reed, S. L. G., I. (1990). "Lysis of complement-sensitive Entamoeba histolytica by
activated terminal complement components. Initiation of complement
activation by an extracellular neutral cysteine proteinase." J Clin Invest
86(6): 1815-1822.
Reed, S. L. K., W. E. McKerrow, J. H. Gigli, I. (1989). "Cleavage of C3 by a neutral
cysteine proteinase of Entamoeba histolytica." J Immunol 143(1): 189-195.
Roberts, Darren L. (2001). "The Inhibitor of Apoptosis Protein-Binding Domain of
Smac Is Not Essential for Its Proapoptotic Activity." J Cell Biol 153:221-228.
Romero-Diaz, M. G., C. Lopez-Reyes, I. Martinez, M. B. Orozco, E. Rodriguez, M.
A. (2007). "Structural and functional analysis of the Entamoeba histolytica
EhrabB gene promoter." BMC Mol Biol 8: 82.
Romero, R. (1993). "Microbiología y Parasitología Humana, Bases Etiológicas de
las Enfermedades Infecciosas." México.
Rui Guo, Jun Ren. (2010) "Alcohol Dehydrogenase Accentuates Ethanol-Induced
Myocardial Dysfunction and Mitochondrial Damage in Mice: Role of
Mitochondrial Death Pathway." PLoS ONE 5(1): e8757.
Sánchez, V. (2010). "Entamoeba histolytica: Differencial gene expression during
programmed cell death and identification of early pro-and anti-apoptotic
signals." Experimental Parasitology.
Saunders, P. A., Chen, R. W. and Chuang, D. M. (1999). "Nuclear translocation of
glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal
apoptosis." J. Neurochem. 72, 925-932.
Sawa, A., Khan, A. A., Hester, L. D. and Snyder, S. H. (1997). "Glyceraldehyde-3-
phosphate dehydrogenase: nuclear translocation participates in neuronal
and nonneuronal cell death." Proc. Natl. Acad. Sci. USA 94, 11669-11674.
Savill, J. F., V. Henson, P. Haslett, C. (1993). "Phagocyte recognition of cells
undergoing apoptosis." Immunol Today 14(3): 131-136.
Seydel, K. B. S., S. L., Jr. (1998). "Entamoeba histolytica induces host cell death in
amebic liver abscess by a non-Fas-dependent, non-tumor necrosis factor
alpha-dependent pathway of apoptosis." Infect Immun 66(6): 2980-2983.

113
Shevchenko, A., Wilm, M., Vorm, O. & Mann, M. (1996). "Mass spectrometric
sequencing of proteins from silver-stained polyacrylamide gels." Anal.
Chem. 68, 850–858.
Shi Y. (2002). "A conserved tetrapeptide motif: Potentiating apoptosis through IAP-
binding.” Cell Death Differ 9, 93-95.
Shi Y. (2002). "Mechanisms of caspase activation and inhibition during apoptosis."
Mol Cell 9:459-470.
Sim, S. Y., T. S. Park, S. J. Im, K. I. Kong, Y. Ryu, J. S. Min, D. Y. Shin, M. H.
(2005). "NADPH oxidase-derived reactive oxygen species-mediated
activation of ERK1/2 is required for apoptosis of human neutrophils induced
by Entamoeba histolytica." J Immunol 174(7): 4279-4288.
Simpson David C, Smith Richard D. (2005). "Combining capillary electrophoresis
with mass spectrometry for applications in proteomics." Electrophoresis.
Apr;26(7-8):1291–1305.
Stauffer, W. R., J. I. (2003). "Entamoeba histolytica: an update." Curr Opin Infect
Dis 16(5): 479-485.
Steen H, Mann M. (2004). "The ABC’s (and XYZ’s) of peptide sequencing." Nat
Rev Mol Cell Biol. 5(9):699-711.
Stuart, L. M. E., R. A. (2005). "Phagocytosis: elegant complexity." Immunity 22(5):
539-550.
Sunaga, K., Takahashi, H., Chuang, D. M. and Ishitani, R. (1995).
"Glyceraldehyde-3-phosphate dehydrogenase is over-expressed during
apoptotic death of neuronal cultures and is recognized by a monoclonal
antibody against amyloid plaques from Alzheimer’s brain."Neurosci Lett.200,
133-136.
Tanyuksel, M. (2003). "Laboratory diagnosis of amebiasis." Clin Microbiol Rev
16(4): 713-729.
Tillack, M. B., L. Irmer, H. Freitas, M. Gomes, M. A. Tannich, E. Bruchhaus, I.
(2007). "The Entamoeba histolytica genome: primary structure and
expression of proteolytic enzymes." BMC Genomics 8: 170.

114
Ting-Jun FAN, L.-H. H., Ri-Shan CONG, Jin LIANG (2005). "Caspase Family
Proteases and Apoptosis." Acta Biochim Biophys Sin 37: 719-727.
Tomei, L. D., Shapiro, J. P. & Cope, F. O. (1993). "Apoptosis in C3H/ 10T1/2
mouse embryonic cells: evidence for internucleosomal DNA modification in
the absence of double-strand cleavage." Proc Natl Acad Sci U S A 90, 853–
857.
Ucker, D. S. (1991). "Death by suicide: one way to go in mammalian cellular
development?" New Biol 3(2): 103-109.
Van Furth, R. (1998). "Human monocytes and cytokines." Res Immunol 149(7-8):
719-720.
Vaux D.L., Weissman I.L. y Kim S.K. (1992) "Prevention of programmed cell death
in Caenorhabditis elegans by human bcl-2."Science 258, 1955-1957.
Velásquez-Fernández. (2011). "Genómica y proteómica: Impacto clínico en
cáncer." Vol. 33 Supl. 1 – 2011.
Vera Rosenkranz y Michael Wink. (2008). "Alkaloids Induce Programmed Cell
Death in Bloodstream Forms of Trypanosomes (Trypanosoma b. brucei)."
Molecules 13, 2462-2473.
Villalba, J. D. G., Gómez, C., Medel, O. Sanchez, V. Carrero, J. C. Shibayama, M.
Ishiwara, D. G. (2007). "Programmed cell death in Entamoeba histolytica
induced by the aminoglycoside G418." Microbiology 153(Pt 11): 3852-3863.
Villalba, J. D, G., R. Rojas, C. Gómez, V. Carrero, J. C. Shibayama, (2007).
"Emetine produce Entamoeba histolytica Death by Inducing a Programmed
Cell Death." American Journal of Infectious Diseases 3 (2): 110-114.
Villalba, J. D, Pérez-Ishiwara, G., Serrano-Luna, J., Tsutsumi, et al. (2011). "In vivo
programmed cell death of Entamoeba histolytica trophozoites in a hamster
model of amoebic liver abscess." Microbiology 157, 1489–1499.
Von Jagow G, Klingenberg M. (1970). Eur. J. Biochem. 12, 583-592.
Walker, P. R., Smith, C., Youdale, T., Leblanc, J., Whitfield, J. F. & Sikorska, M.
(1991). "Topoisomerase II reactive chemotherapeutic drugs induce
apoptosis in thymocytes." Cancer Res 51, 1078–1085.

115
Walsh, J. A. (1986). "Amebiasis in the world." Arch Invest Med (Mex) 17 Suppl 1:
385-389.
WHO (1997).
Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I, Hochstrasser
DF, et al. (1996). "Progress with proteome projects: why all proteins
expressed by a genome should be identified and how to do it." Biotechnol
Genet Eng Rev 13:19-50.
Wittke S, Mischak H, et al. (2005). "Discovery of biomarkers in human urine and
cerebroespinal fluid by capillary electrophoresis coupled to mass
spectrometry: Towars new disgnostic and therapeutics approaches."
Electrophoresis 26: 1476-1487.
Wyllie, A. H. B., G. J. Hargreaves, A. D. (1981). "Chromatin changes in apoptosis."
Histochem J 13(4): 681-692.
Wyllie, A. H. K., J. F. Currie, A. R. (1973). "Cell death in the normal neonatal rat
adrenal cortex." J Pathol 111(4): 255-261.
Zhang X, Li SY, Brown RA, Ren J. (2004). "Ethanol and acetaldehyde in alcoholic
cardiomyopathy: from bad to ugly en route to oxidative stress." Alcohol 32:
175–186.

116
12. ANEXOS
Anexo 1 Buffer de lisis o de muestra
Reactivos Concentración final Cantidad
Urea (PM 60.06) 7M 10.5 g
Tiourea 2 M 3.8 g
CHAPS 4 % 1.0 g
IPG Buffer 2 % 500 µl
DTT (PM 154.2) 40 Mm 154 mg
Agua MilliQ -- Para 25 ml (16 requeridos)
Notas:
- Pesar la urea y disolverla en aproximadamente 10 ml de agua y
desionizarla por los menos 2 h, en agitación con la resina AG 501-X8 (Bio-
Rad) en una proporción de 5g de resina por 100 ml de solución.
- No adicionar las IPG buffer.
- Hacer alícuotas de 2 ml y almacenar a -20°C.
- Para utilizar una alícuota adicionarle 40 µl de las IPG buffer.
Anexo 2 Buffer de hidratación
Reactivos Concentración final Cantidad
Urea (PM 60.06) 7M 10.5 g
Tiourea 2 M 3.8 g
CHAPS 2 % 1.0 g

117
IPG Buffer 0.5 % o 2 % 500 µl
1 % azul de bromofenol 0.002 % 154 mg
Agua MilliQ -- Para 25 ml (13.5 requeridos)
Notas:
- Pesar la ures y disolverla en aproximadamente 10 ml de agua y deionizarla
por lo menos 2 h, en agitación con la resina AG 501-X8 (Bio-Rad) en una
proporción de 5g de resina por 100 ml de solución.
- No adicionar las IPG buffer
- No adicionar azul de bromofenol
- Hacer alícuotas de 2 ml y almacenar a -20 °C
- Para utilizar una alícuota adicionarles 15 µl de las IPG buffer y 7 mg de
DTT.
Anexo 3
Protocolo Introducción
El volumen de ensayo es de 1,5 ml, que es compatible con la mayoría de las
cubetas del espectrofotómetro. Sin embargo, el volumen de ensayos mayores o
menores pueden ser acomodados por la ampliación proporcionalmente de los
volúmenes de la muestra, la curva estándar y los reactivos del ensayo.
La precisión de este ensayo se determina principalmente por la técnica del
usuario. Pipeteo cuidadoso de las soluciones de la curva estándar, la muestra y
reactivos es esencial. Si la muestra es viscosa, puede ser difícil la precisión
al pipetear volúmenes por debajo de 10 μl. Resultados más precisos pueden
obtenerse diluyendo una muestra concentrada en agua antes del ensayo.

118
Se recomienda la realización del ensayo por duplicado.
A diferencia de la mayoría de los ensayos de proteínas, la absorbancia de la
solución de ensayo disminuye con la concentración de proteína en aumento. No
reste la lectura del blanco a partir de la lectura de muestra. El rango de volumen
del ensayo es de 1-50 μl y el rango lineal para la cuantificación es 0-50 µg. El
ensayo tiene un umbral de sensibilidad de 0.5 μg.
Sugerencia: Colocar siempre los tubos de microcentrífuga en el rotor de la
centrífuga con la tapa de bisagra hacia afuera. De esta manera el pellet siempre
quedará en el mismo lado del tubo para que pueda ser dejado sin tocar, y
minimizar la pérdida.
El ensayo se debe realizar a temperatura ambiente.
Requerido, pero no proporcionado:
- Tubos de microcentrifuga de 2 ml.
- Mezclador vortex
- Microcentrifuga
- Espectrofotómetro de luz visible (ejem. Novaspec II o Ultrospec 1000pro)
Preparaciones preliminares:
Previo a realizar el ensayo, preparar un volumen apropiado de reactive color de
trabajo, mezclando 100 partes de reactivo de color A con 1 parte del reactivo de
color B. Cada ensayo individual requiere de 1 ml de reactivo de color de trabajo.
El reactivo de color de trabajo puede ser guardado a 4-8 °C durante un máximo de
una semana o siempre que la absorbancia óptica (A480) de la solución
permanezca por debajo de 0. 025 a 480 nm.

119
Procedimiento estándar
1. Preparación de una curva estándar de acuerdo a al tabla de abajo, usando 2
mg/ml de la solución estándar de la albumina de suero bovino proporcionada en el
kit. Coloque seis tubos y añadir una solución de acuerdo a la siguiente tabla.
Tabla 10. Tubo 1 es el blanco de ensayo, que no contiene proteínas.
Preparación de la curva estándar: Numero de tubo 1 2 3 4 5 6
Volumen de 2 mg/ml
de la solución estándar de BSA
0 μl 5 μl 10 μl 15 μl 20 μl 25 μl
Cantidad de proteína 0 μg 10 μg 20 μg 30 μg 40 μg 50 μg
Nota: La precisión del ensayo no se ve afectado por el volumen de la muestra así como el volumen
de muestra es de 50 μl o menos. Por consiguiente, no es necesario una dilución estándar o
soluciones de muestra para un volumen constante.
2. Preparar los tubos que contienen 1-50 µl de la muestra a ser analizada. Se
recomienda hace duplicado. El intervalo rango útil del ensayo es 0.5-50 µg.
3. Añadir 500 µl de precipitante a cada tubo (incluyendo los tubos de la curva
estándar). Se usó vortex brevemente e incubar los tubos 2-3 min a temperatura
ambiente.
4. Añadir 500 µl de co-precipitación a cada tubo y mezclar brevemente por
vortex o inversión.
5. Centrifugar los tubos a 10 000 x g durante 5 min. Esto sedimentos la proteína.

120
6. Sacar los tubos de la centrífuga, tan pronto como la centrifugación se completa.
Un pequeño precipitado debe ser visible. Decantar el sobrenadantes. Proceder
rápidamente con el paso siguiente para evitar la resuspensión o dispersión de los
gránulos.
7. Vuelva a colocar los tubos en la microcentrífuga como antes, con la tapa de
bisagra y el precipitado hacia afuera. Centrifugar los tubos de nuevo para llevar
todo el líquido restante a la parte inferior del tubo. Un breve pulso es suficiente.
Utilizar una micropipeta para eliminar el sobrenadante restante. No debe haber
ningún líquido visible que quede en los tubos.
8. Añadir 100 µl de solución de cobre y 400 µl de agua destilada o desionizada a
cada tubo. Dar voxtex brevemente para disolver la proteína precipitada.
9. Añadir 1 ml de reactivo de color de trabajo a cada tubo (Ver "preliminar
preparados" para la preparación del reactivo de color de trabajo). Asegurar
instantánea mezcla al introducir el reactivo tan rápidamente como posible. Mezclar
por inversión.
10. Incubar a temperatura ambiente durante 15-20 minutos.
11. Leer la absorbancia de cada muestra y estándar a 480 nm utilizando agua
como referencia. La absorbancia debe leerse dentro de 40 minutos de la adición
de reactivo de color de trabajo (paso 9).
Nota: A diferencia de la mayoría de los ensayos de proteínas, la absorbancia de la
solución del ensayo disminuye con la concentración de proteína en aumento. No
restar la lectura del blanco a partir de la lectura de la muestra o utilizar el ensayo
blanco como referencia.

121
12. Generar una curva estándar trazando la absorbancia del estándar contra la
cantidad de proteína. Utilice esta curva estándar para determinar la concentración
de proteína de las muestras.
Anexo 4 Gel separador
Reactivos 10% 12%
Agua milliQ 16.4 ml 13.6 ml
1.5 M Tris-HCl, pH 8.8 10 ml 10 ml
20% (w/v) SDS 200 µl 200 µl
Acrilamida/Bis-acrilamida (30%/0.8% w/v) 13.2 ml 16 ml
10% (w/v) persulfato de amonio 200 µl 200 µl
TEMED 20 µl 20 µl
Cantidad total 40.02 ml 40.02 ml
Gel concentrador
Reactivos 4%
Agua milliQ 6.15 ml
0.5 M Tris-HCl, pH 8.8 2.5 ml
20% (w/v) SDS 50 µl
Acrilamida/Bis-acrilamida (30%/0.8% w/v) 1.34 ml
10% (w/v) persulfato de amonio 50 µl
TEMED 10 µl
Cantidad total 10.01 ml

122
Anexo 5 Buffer de equilibrio (200 ml)
Reactivos Concentración final Cantidad
Urea (PM 60.06) 6 M 72.1 g
1.5 M Tris-HCl, pH 8.8 75 Mm 10 ml
Glicerol (87%) 29.3 % 69 ml
SDS 2 % 4 g
1 % azul de bromofenol 0.002 % 400 µl
Agua milliQ -- 40 ml
Cantidad total Aforar a 200 ml