
Capitulo 3 Crystal
60
3 Crecimiento de cristales Únicamente cuando una fuerza motora que provoca que un sistema se aparte de su condición de equilibrio es que se forma un núcleo de un cristal y comienza el crecimiento. Fases sólida, fundida, solución y vapor se puede distinguir como fases del ambiente en el que los cristales pueden crecer. Estos ambientes se pueden clasificar de forma sistemática dependiendo de si son fases condensadas o fases diluidas, y si está involucrada o no la interacción. En la fuerza impulsora, la transferencia de calor y masa están unidas, pero el grado de su respectiva contribución depende del tipo de ambiente de fase envuelto. La interfaz entre el cristal y el ambiente de la fase es el único lugar donde el crecimiento o la disolución se llevan a cabo. Se puede considerar el problema de la interfaz mediante la clasificación de su estructura en interfaces atómicamente rugosas y lisas. Se tiene un mecanismo de crecimiento de tipo 1
-
Upload
anonymous-djq7mdvq -
Category
Documents
-
view
18 -
download
1
description
cristalografia
Transcript of Capitulo 3 Crystal

3Crecimiento de cristales
Únicamente cuando una fuerza motora que provoca que un sistema se aparte de su condición de equilibrio es que se forma un núcleo de un cristal y comienza el crecimiento. Fases sólida, fundida, solución y vapor se puede distinguir como fases del ambiente en el que los cristales pueden crecer. Estos ambientes se pueden clasificar de forma sistemática dependiendo de si son fases condensadas o fases diluidas, y si está involucrada o no la interacción. En la fuerza impulsora, la transferencia de calor y masa están unidas, pero el grado de su respectiva contribución depende del tipo de ambiente de fase envuelto. La interfaz entre el cristal y el ambiente de la fase es el único lugar donde el crecimiento o la disolución se llevan a cabo.
Se puede considerar el problema de la interfaz mediante la clasificación de su estructura en interfaces atómicamente rugosas y lisas. Se tiene un mecanismo de crecimiento de tipo adhesivo para una interfaz atómicamente rugosa, mientras que en las interfaces atómicamente lisas los cristales crecen ya sea por la nucleación bi-dimensional y crecimiento de capa o por un mecanismo de crecimiento de espiral. Basándose en los análisis anteriores se puede sistematizar las relaciones mutuas entre morfologías poliédricas, en tolva, y dendríticas.
3.1 Equilibrio termodinámico frente a cinética termodinámicaEl crecimiento de cristal es el proceso del nacimiento y el desarrollo de una fase sólida
con una estructura regular de un estado desordenado e irregular, y por lo tanto pueden considerarse como una fase de transición de primer orden.
1

La ciencia que trata con las transiciones de fase es la termodinámica. Usando la termodinámica, se puede discutir cual fase con el tiempo eventualmente se formará cuando un material (de composición c y la fase p) se mantiene bajo las mismas condiciones durante un tiempo infinito, y la fase alcanza el estado de mínima energía (estado de equilibrio) bajo condiciones termodinámicas dadas (temperatura y presión). Experimentalmente, se prepara un diagrama de fase (diagrama de fase de equilibrio), y se pude utilizar los datos de este diagrama para extraer información sobre el sistema. Si el sistema permanece durante un largo período en las mismas condiciones, tales como en los procesos geológicos, se asume que ha alcanzado el equilibrio, y puede ser un tema de discusión termodinámico.
Al tratar con los sistemas de metales, aleaciones, cerámicas, y silicatos, así como el crecimiento de cristales individuales, los diagramas de fase son el requisito previo en la comprensión de lo básico de las relaciones de fases. Hay muchas formas de diagrama de fases. Cuando se trata con crecimiento de cristales cuyos diagramas de fase no se conocen, primero se tiene que preparar un diagrama de fase preliminar.
A diferencia del equilibrio termodinámico, las principales preocupaciones de este libro son el mostrar cómo las transiciones de fase están implicadas en el crecimiento y disolución que se efectúan a un nivel atómico, y explicar cómo la morfología, la perfección y la homogeneidad de los cristales individuales y las texturas de los agregados policristalinos se determinan a través de estos procesos. La transición de fase y la formación de un cristal, el producto final, no se producen instantáneamente. Junto con nuestra curiosidad de cómo se produce el crecimiento de un cristal en un nivel atómico, la ciencia y la tecnología de este tema también han hecho grandes progresos durante el siglo XX, debido a las fuertes demandas de las industrias de semiconductores y opto-electrónicos, en los que cristales individuales y películas delgadas se utilizan como materiales básicos. Los cristales reales contienen varios defectos de la red, y no pueden ser utilizados para fines industriales a menos que estos defectos, que generalmente ocurren durante el proceso de crecimiento, estén controlados. Un buen ejemplo es el láser de rubí. Si se utiliza un cristal de rubí que contiene dislocaciones no controlados (véase la Sección 3.7), el rayo láser se emitirá sólo una vez. Sólo después que se logró la síntesis exitosa de monocristales de rubí con una densidad de dislocaciones controladas, el láser de rubí se volvió útil en la práctica. La razón por la que la sílice ha llegado a desempeñar el papel principal en la industria de los semiconductores se debe a la síntesis de monocristales de silicio libre de dislocaciones. Aunque se logró el control de defectos de red en monocristales en el siglo XX para los sistemas de un solo componente como el silicio, todavía se está muy lejos de una
2

situación similar en la síntesis de monocristales de compuestos semiconductores o de cuarzo. Si se pudiera entender los fenómenos relacionados con el crecimiento y la disolución que se produce en sistemas complicados y complejos, tales como cristales minerales que constituyen materiales sólidos terrestres y extra-terrestre, o los formados como resultado de actividades biológicas, tales como dientes, los huesos y conchas, seríamos capaces de ofrecer nuevas perspectivas a estas disciplinas. Estos fenómenos serán un objetivo de investigación para la ciencia del crecimiento de los cristales en el siglo XXI.
3.2 Fuerza Motora
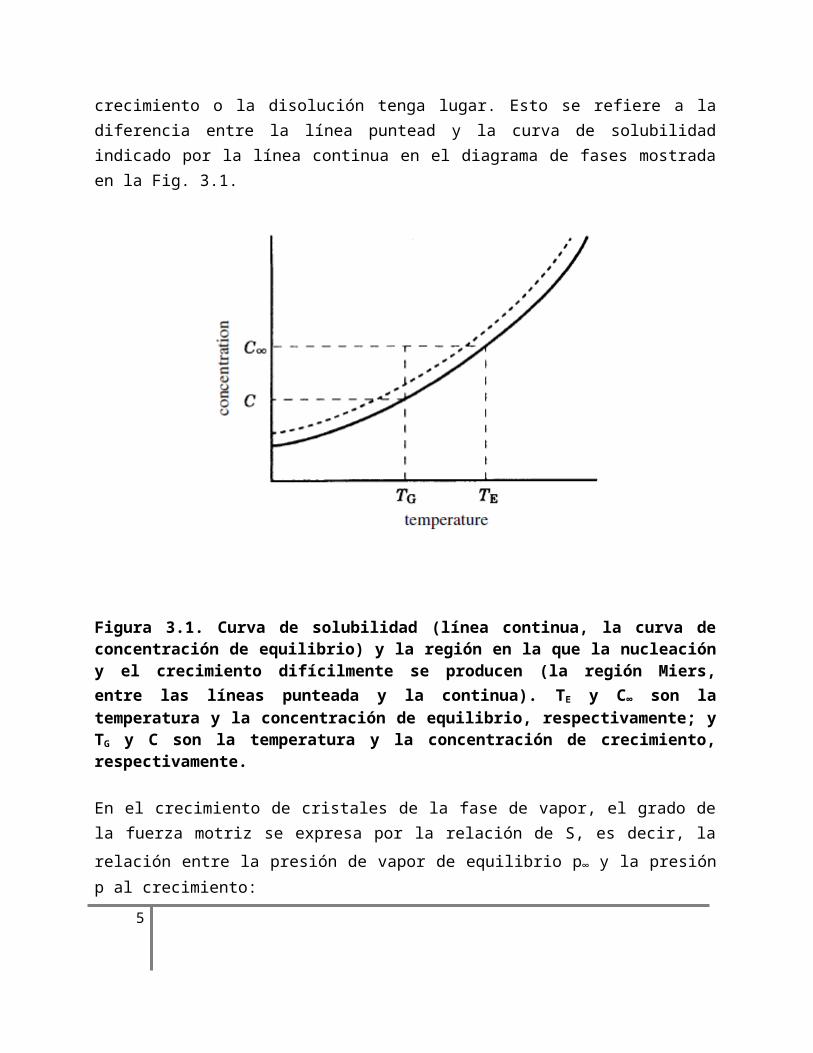
El estado de equilibrio corresponde al estado cuando el intercambio de calor y masa entre el material inicial y el producto se convierte en cero en la interface de dos fases. Esto corresponde al estado donde ni la temperatura ni el grado de concentración (a través del enfriamiento o evaporación) cambian en un punto dado en una curva de solubilidad en el diagrama de fase de un sistema en solución. En esta situación, ni el crecimiento ni la disolución de un cristal se produce. Para lograr el crecimiento o disolución de un cristal, es necesario alcanzar una situación lejos del estado de equilibrio, ya sea mediante la reducción de la temperatura o por concentración del componente por evaporación. El grado de la salida desde el estado de equilibrio corresponde a la fuerza motora para que el crecimiento o la disolución tenga lugar. Esto se refiere a la diferencia entre la línea puntead y la curva de solubilidad indicado por la línea continua en el diagrama de fases mostrada en la Fig. 3.1.
3

Figura 3.1. Curva de solubilidad (línea continua, la curva de concentración de equilibrio) y la región en la que la nucleación y el crecimiento difícilmente se producen (la región Miers, entre las líneas punteada y la continua). TE y C∞ son la temperatura y la concentración de equilibrio, respectivamente; y TG y C son la temperatura y la concentración de crecimiento, respectivamente.
En el crecimiento de cristales de la fase de vapor, el grado de la fuerza motriz se expresa por la
relación de S, es decir, la relación entre la presión de vapor de equilibrio p∞ y la presión p al crecimiento:
S = p / p∞ =J / J∞
Donde J y J∞ son flujos entrantes de la fase de vapor al cristal en el crecimiento y el equilibrio, respectivamente.
En el caso de la solución de crecimiento, la fuerza impulsora corresponde a la diferencia entre
la concentración de C∞ a la temperatura de equilibrio TE de la solución saturada y la concentración de C a la temperatura de crecimiento TG y se expresa como sigue:
∆C = C∞ - C,
S = C∞ / C (relación de supera-saturación del soluto)
σ = (C∞ / C)/ C (sobresaturación relativa) σ = S -1
En el crecimiento del fundido, la fuerza impulsora puede ser evaluada como la diferencia entre las temperaturas de equilibrio y crecimiento:
4
∆T = T∞ - T, (sobre-enfriamiento).
El grado de fuerza motriz se puede generalizar por la diferencia de los potenciales químicos entre las dos fases:
∆μ = μ (g) - μ∞ (g) = μ (g) - μ∞
(C)
Aquí, μ (g), μ∞
(g), y μ∞ (C) son, respectivamente, los potenciales químicos del vapor
sobresaturado (o el ambiente fase tal como la fase solución), la fase saturada, y la fase sólida. De esto, una fuerza motriz generalizada se puede expresarse como
∆μ = kTB ln (p/p∞) = kTB ln S
Donde k es la constante de Boltzmann, y TB es la temperatura absoluta. Se va a expresar la
fuerza impulsora en términos de la fuerza impulsora generalizada, ∆μ = kT, en este libro.
3.3 Transferencia de masa y calor
Como puede verse de la expresión para la fuerza impulsora en términos de las diferencias de potenciales químicos, que están relacionados a las diferencias en temperatura y concentración, los dos procesos de transporte, transferencia de calor y transferencia de masa van unidos en el crecimiento de cristales. El grado de contribución del proceso de transporte respectivo se determina por el grado de condensación de la fase del medio ambiente (ambiente). Para el crecimiento de cristales en un ambiente de fase diluido, se requiere un proceso de condensación, y así la transferencia de masa juega un rol esencial. La contribución de calor generado por la cristalización en este caso es pequeña comparado con el de transferencia de masa. Sin embargo, para la cristalización en una fase condensada, tal como una fase fundida, la transferencia de calor juega el papel esencial, y la contribución de la transferencia de masa será muy pequeño, debido a la diferencia en la concentración (densidad) entre las fases sólida y líquida es muy pequeño, más es decir 1 ó 2%. Por ello es necesario clasificar los tipos de ambiente de fases y estar familiarizado con sus respectivas características de este punto de vista.
5

La cristalización de estado sólido, o recristalización, * es el proceso mediante el cual tanto los materiales de inicio y el producto final son agregaciones de partículas con la misma estructura cristalina y el engrosamiento de tamaños de grano es el único proceso que se produce por cristalización. En este caso, los límites de grano se vuelven móviles, y el engrosamiento de los granos se produce debido a la liberación de energía contenida que se almacena en los límites de grano por la energía térmica resultante del calentamiento. La recristalización primaria es el nombre que se le da al proceso que describe la situación anterior al estado en el que se alcanza un estado con textura que consiste en partículas de tamaño casi igual, mientras que la recristalización secundaria describe la situación en la que los granos particulares crecen rápidamente mucho mas debido a la difusión de impurezas en los límites de grano por un calentamiento adicional de la muestra. Sin embargo, la situación será diferente de esto cuando la precipitación de la fase B se produce en el estado sólido en una solución sólida de un componente (A, B).
En contraste a la cristalización en estado sólido, la cristalización a partir de fases de vapor, solución y fundido corresponde a las fases ambiente que tienen estructuras aleatorias, pueden ser clasificados además en fases condensadas y diluidas. Fases de vapor y de soluciones son fases diluidas, en el que el proceso de condensación de transferencia de masa juega un papel esencial en el crecimiento de cristales. En la fase de fusión condensada, sin embargo, la transferencia de calor juega el papel esencial. Además de calor y transferencia de masa, un factor adicional, la interacción soluto-solvente, se debe tenerse en cuenta.
Dentro de los tres tipos de fases ambiente, el crecimiento de cristales a partir de la masa fundida pura de un material de fusión congruente (es decir, las fases sólidas y líquidas tienen la misma composición) se llama "crecimiento en estado fundido." En esta situación, las interacciones con otros componentes no están involucradas. Un concepto similar se aplica a la deposición física de vapor (de transporte) (PVD, PVT) en el que la cristalización se produce a través de la simple evaporación por calentamiento y condensación por enfriamiento. En la fase de solución, sin embargo, no son componentes de soluto y disolvente, que interactúan mutuamente para formar una solución. Esto puede ser comparado con la deposición química de vapor (transporte) (CVD, CVT), que implica reacciones químicas. La fuerza de la energía de interacción soluto-disolvente está estrechamente relacionada con la solubilidad del componente soluto, la nucleación, y las tasas de crecimiento de cristales.
6

Los criterios básicos a considerar en el crecimiento de cristales de vapor, solución, y se funden fases son, por tanto, si la fase se condensa o diluir, y si la fase implica una interacción soluto-disolvente o no.
En la fase condensada, la transferencia de masa no está implicada de manera significativa, y el crecimiento de cristales se produce por la reorganización de los átomos en la interface sólido-líquido a través de la eliminación del calor. La transferencia de calor es la fuerza motriz necesaria para la reordenación atómica que se produzca; la temperatura de crecimiento es alta, y, como resultado, la interfaz sólido-líquido tiende a ser más áspero que el formado en una fase diluida ambiente (véase la Sección 3.6). * En química, el término recristalización tiene un significado diferente; que implica el proceso de purificación por solución y recristalización.
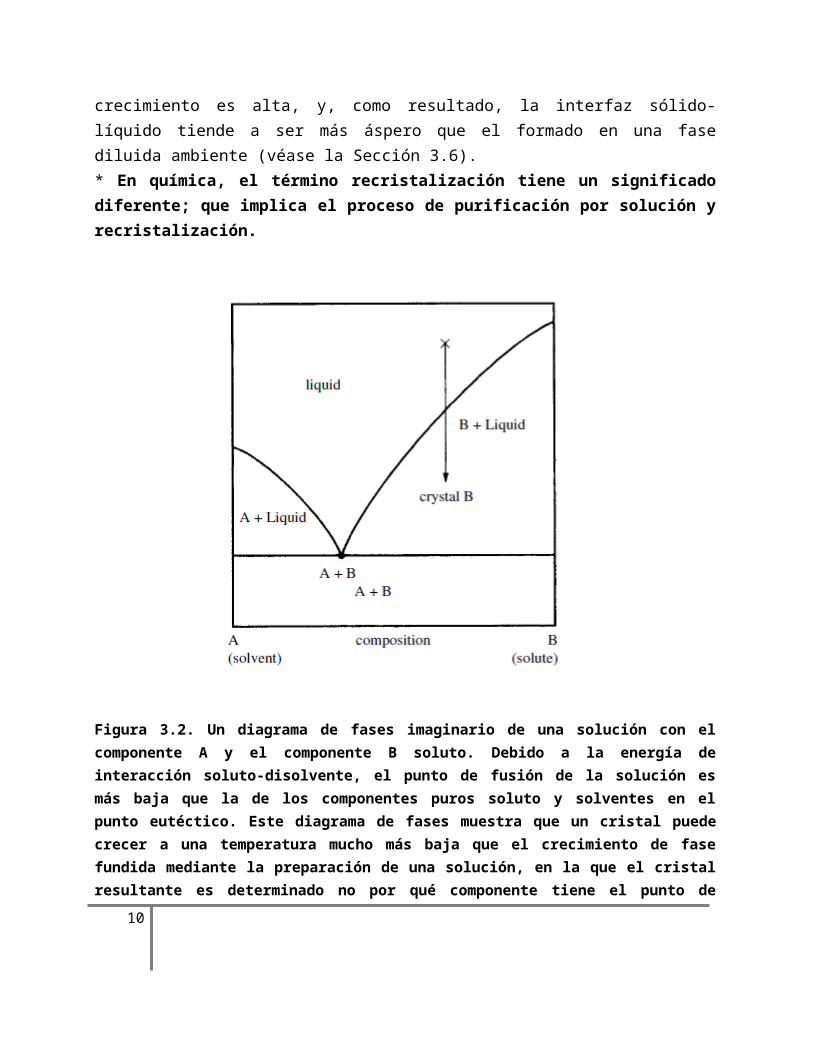
Figura 3.2. Un diagrama de fases imaginario de una solución con el componente A y el componente B soluto. Debido a la energía de interacción soluto-disolvente, el punto de fusión de la solución es más baja que la de los componentes puros soluto y solventes en el punto eutéctico. Este diagrama de fases muestra que un cristal puede crecer a una temperatura mucho más baja que el crecimiento de fase fundida mediante la preparación de una solución, en la que el cristal resultante es determinado no
7

por qué componente tiene el punto de fusión más alto, sino por la composición de la solución. Estas
son las características esenciales del crecimiento en una solución.
En una fase diluida, átomos o iones son transportados desde lejos, y, a la llegada a la superficie del cristal, que se incorporan en el cristal. Hay un proceso de solvatación involucrado en la cristalización en la fase de solución o en CVT. (Para una explicación de solvatación, consulte la sección 3.4.) El papel esencial de la fuerza motriz es la transferencia de masa; temperatura de crecimiento es mucho menor que en el crecimiento de la masa fundida, y la interfaz sólido-líquido tiende a ser más lisa que en la fase condensada.
La Tabla 3.1 resume las diferencias esenciales en el crecimiento de cristales para las diversas fases del ambiente. Crecimiento de cristales que tienen lugar en los procesos geológicos y en los mecanismos biológicos también está indicado para referencia. Dado que el tema principal de este libro es entender los fenómenos que ocurren en sistemas complicados y complejos, nos centramos nuestra discusión sobre la solución y CVT crecimiento en lugar de crecimiento de los cristales de fusión mucho más simple o crecimiento PVT.
8

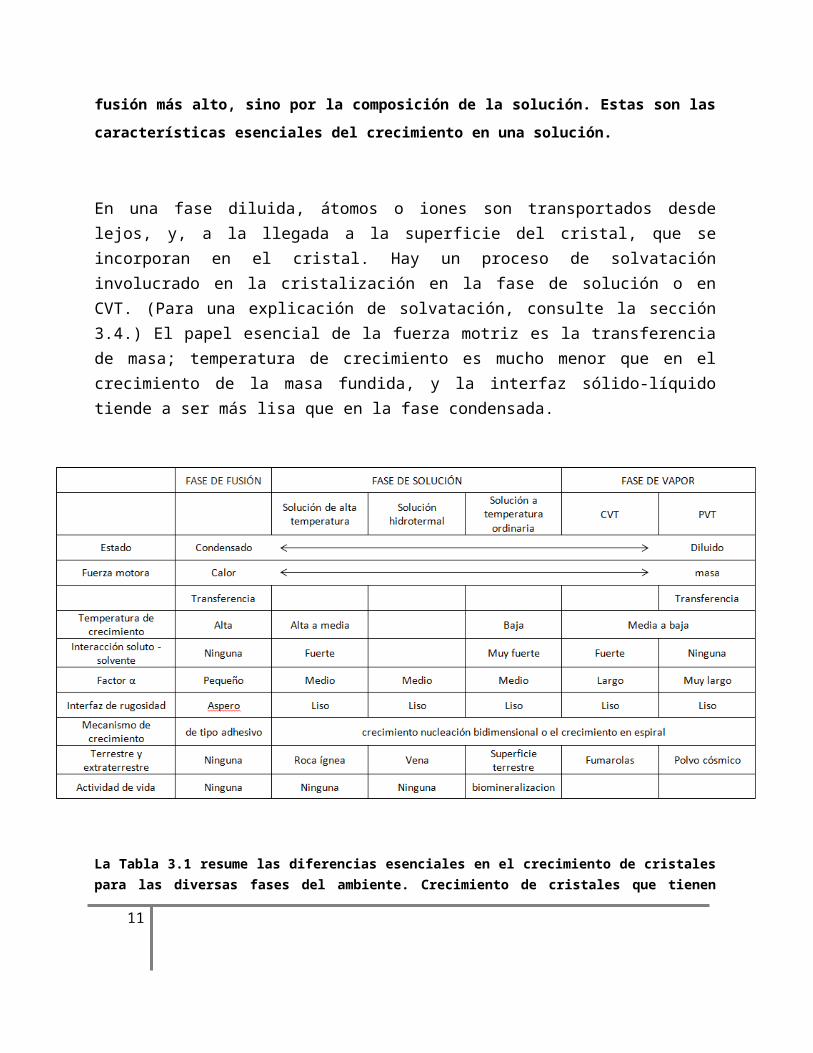
Figura 3.3. Varias características de difusión y convección asociados con el crecimiento de cristales en solución (a) en un vaso de precipitados y (b) alrededor de un cristal. El cristal se representa por el área sombreada. Que se muestran son: la capa límite de difusión (db); la difusión mayor (D); la convección debido a la diferencia térmica o la gravedad (T); Marangoni convección (M); convección flotabilidad impulsada (B); flujo laminar, flujo turbulento (F); Efecto Berg (ser); interfaz suave (S); interfaz rugosa (R); unidad de crecimiento (g). El apego y el desapego del soluto (línea continua) y el disolvente (línea abierta) se ilustran en (b).
3.4 Ejemplos de transferencia de masa
Vamos a considerar en esta sección cómo procede la transferencia de masa, utilizando el crecimiento de cristales de una fase de solución como un ejemplo representativo de crecimiento de los cristales en la que el calor y transferencia de masa se acoplan. Vamos a utilizar el crecimiento de cristales iónicos en solución acuosa en un vaso de precipitados como ejemplo (Fig. 3.3).
Para la solución de NaCl acuosa, que es una fase diluida ambiente, todos los componentes de soluto de Na, Cl-, o NaCl, (NaCl) n están unidos con el componente disolvente H2O. Si el componente soluto está presente en un estado iónico aislado, o si las moléculas de NaCl o grupos de (NaCl) n están presentes, dependerá de la fuerza impulsora. Aunque la situación no
9
se entiende completamente, se espera que, como la sobresaturación aumenta, la proporción de racimos se incrementará. Aunque puede haber varios estados de unión entre estos grupos y H2O, en general se considera que hay dos regiones que rodean el componente disolvente: una estructura formando una región con una fuerte unión, y la otra estructura que forma una región rompible con mucho más débil unión entre el moléculas de disolvente.
Figura 3.4. Ilustración esquemática que explica el gradiente de concentración alrededor de un cristal en crecimiento y la presencia de la capa límite de difusión. Difusión bulk indicada por capa límite de difusión (para A o B). El gradiente de concentración es (σB-σS), donde σB está la sobresaturación mayor y σS es la sobresaturación superficie.
Cuando la temperatura de la solución acuosa se reduce, o la evaporación ocurre, una sobresaturadas resultados estatales. Si la fuerza impulsora del sistema excede la barrera de energía necesaria para la nucleación se lleve a cabo (para una discusión, véase la sección 3.6), la nucleación del componente soluto se produce en el sistema. Acompañando a la nucleación, una región más diluida aparecerá que rodea el núcleo, y la difusión de la fase ambiente mayor se producirá debido a la diferencia de concentración. Esto se llama volumen o mayor difusión. El gradiente de concentración no es lineal desde la fase de mayor ambiente, lejos de los núcleos y la superficie de los núcleos o cristal. En la región cercana a la superficie de los núcleos o el cristal, el gradiente de concentración es agudo, mientras que entre la solución a granel ambiente lejos de la superficie casi no muestra gradiente. Esto ya estaba claro desde las investigaciones efectuadas antes de la década de 1930 (ver Fig. 3.4). La región con un gradiente fuerte se llama la capa límite de difusión, y el gradiente de concentración en esta capa
10
desempeña un papel esencial en el crecimiento de cristales. Para el crecimiento de los cristales iónicos en una solución acuosa, el espesor de esta capa es del orden de 100 μm.
Figura 3.5. La barrera de energía en diversas etapas de crecimiento solución. Torcedura K; Difusión superficial SD; De solvatación DEADS; DBL = capa límite de difusión.
La concentración en la superficie de un cristal (un núcleo) no es la misma que la concentración de equilibrio. En los días antes de que pudiéramos medir la concentración superficial directa, el motor de una fase a granel fue evaluada por la sencillez asumiendo la concentración de equilibrio, pero ahora se supone que la concentración de la superficie es ligeramente superior a este valor. Por lo tanto, la fuerza motriz para el crecimiento de cristales se considera ahora a ser la diferencia y su gradiente entre la concentración en la región más externa de la capa límite de difusión y la concentración en la superficie (superficie sobresaturación).
Partículas cristalizan que llegan a la superficie se difundirá en la (difusión superficial) de superficie. Mientras esto ocurre, algunos pueden regresar a la fase ambiente, mientras que algunos serán arrebatados a torceduras o pasos (véase la Sección 3.6) en la superficie y se incorporarán en el cristal. Cuando estas partículas se incorporan en el cristal, se disocia el componente disolvente. Este proceso se llama desolvatación. En crecimiento solución, este proceso va a determinar la tasa de crecimiento. En ciertos puntos en estos procesos, es necesario superar las barreras de energía necesarias para subir los respectivos pasos (Fig. 3.5).
11

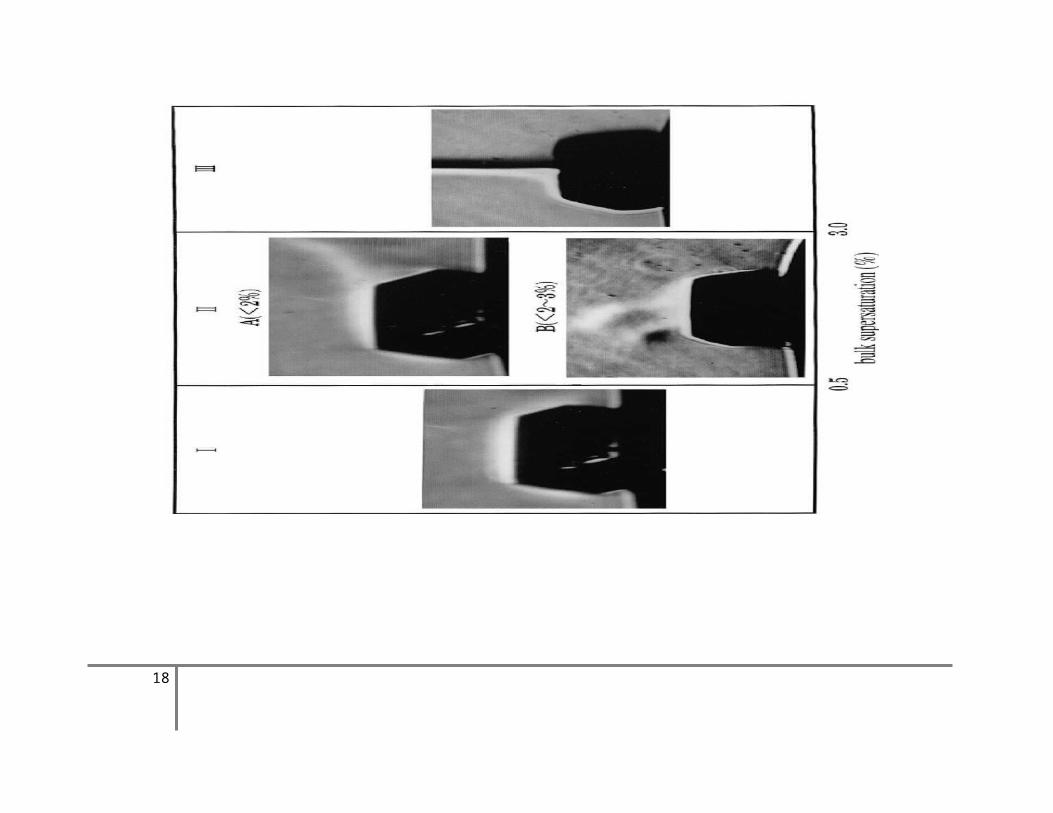
El componente disolvente individual por el proceso de desolvatación vuelve a la fase ambiente. Como resultado, la concentración en la capa límite de difusión se reducirá. A través de la competencia entre el suministro de componente soluto de la fase en masa y la del componente de disolvente por el proceso de desolvatación, el espesor de la capa límite de difusión variará de acuerdo con el cambio en la concentración, o convección flotabilidad impulsada ocurrirá cuando los cristales están creciendo en el campo de gravedad de la Tierra. Este cambio está relacionado con la sobresaturación a granel. En la Fig. 3,6, cambios en el comportamiento de la capa límite de difusión en torno a un creciente Ba (NO3) 2 de cristal en solución acuosa se ilustran en relación con la sobresaturación a granel. Al 0,5%, el espesor de la capa límite de difusión simplemente aumenta, mientras que a 0,5% 3,0%, aparece inestable convección elevar, y en el 3,0% aparece una constante de convección penacho flotabilidad impulsado por el aumento perpendicularmente desde el centro de la cara superior. En σ >8,0%, aparecen aumento penachos de convección, la creación de un agotamiento considerable en la raíz, lo que afecta en gran medida la perfección del cristal.
Variación de la concentración en la fase a granel es isotrópica alrededor de un cristal. Considere la posibilidad de un cristal que crece en un campo de concentración equi-distribución concéntrica. En un cristal poliédrico delimitada por las esquinas, bordes y caras, esperamos que la diferencia de concentración en una esquina a ser mayor que en un borde, que es a su vez mayor que en una cara, y por lo tanto las esquinas y los bordes tienen un mayor estado de concentración que es el centro de una cara. Esto crea la difusión superficial. Las diferencias de concentración en las esquinas, bordes y centros de la cara de un cristal en crecimiento serán diferentes dependiendo de la sobresaturación mayor o caudal a lo largo de la superficie. La diferencia en la concentración en diferentes sitios en una cara se demostró por Berg [3] por medio de una técnica interferométrica, que ahora se conoce como el efecto Berg o fenómeno Berg. Este es un fenómeno importante en el análisis de la morfología de los cristales.
12
Figura 3.6. Fotografías Schlieren mostrando los cambios de espesor de la capa límite de difusión y el comportamiento de la flotabilidad - impulso de la convección que muestra en relación al bulk sobresaturado [1], [2]. La figura muestra la cara (111) de un cristal de Ba(NO3)2 de una solución acuosa. En la región I, solo el espesor de la capa límite de difusión se incrementa; en la región II,
13

se observa la convección lateral inestable (IIA) y el levantamiento de la plima intermitentemente (IIB); y en la región III se observa la convección constante de la flotabilidad - impulso.
14

3.5 El flujo laminar y convección
Al igual que la difusión, convección es también un factor importante para la masa o la transferencia de calor. La convección es un modo de flujo en un fluido que crea la rotación o la circulación del fluido, y se distingue de flujo laminar y el flujo turbulento. Si hay una diferencia de temperatura entre el fluido en la parte superior y la parte inferior del vaso de precipitados se muestra en la Fig. 3.3, la convección se produce debido a la diferencia de concentración resultante de la diferencia de temperatura. La convección también se producirá cuando existe un gradiente de concentración, o puede surgir debido a la flotabilidad. Convección debido a la tensión superficial de una solución se llama Marangoni convección. La diferencia en la tensión superficial puede ser debida a una diferencia de temperatura o a la concentración de especies químicas en la superficie. La convección puede ser tomada para significar la convección térmica, o una combinación de los dos. En contraste con estos procesos naturales de convección, diversos métodos, tales como la rotación de los cristales de siembra o el propio crisol, se adoptan cuando el crecimiento de cristales individuales con el fin de controlar la convección y para efectuar la homogeneización de la fase líquida. Puesto que es deseable alcanzar una fase líquida homogénea con el fin de hacer crecer cristales individuales homogéneos, la convección se produce en la fase líquida ha sido ampliamente investigado por medio de simulación por ordenador o varias otras técnicas de visualización.
† El valor adimensional más representativa de la dinámica de fluidos es Re, la relación de la magnitud de la fuerza de inercia y la viscosidad, Re l / l / v, donde es la densidad, es la viscosidad, v es la viscosidad dinámica, l es la longitud representativa de la materia en el flujo, y es la tasa de flujo representativo.
El flujo laminar, que es un flujo direccional, los cambios en el flujo turbulento cuando el número de Reynolds crítico (Re) † excede 200. Cuando hay un flujo de la solución en torno a un cristal en crecimiento, el espesor de la capa límite de difusión, el efecto Berg, y Cómo se suministra el componente soluto se verá afectado dependiendo de la tasa de flujo del flujo laminar o de la naturaleza del flujo turbulento, todos los cuales influyen en la tasa de crecimiento, la perfección, y la morfología del cristal.
3.6 Nucleación
Los procesos de crecimiento de los cristales se dividen en las siguientes tres etapas.
(1) Se aplica una fuerza de accionamiento, que hace que el proceso de proceder por la formación de un estado sobresaturado o sobre enfriado.
(2) Las partículas más pequeñas que un tamaño crítico, que puede ser referido como racimos, se forman en el sistema. Algunos de estos grupos, de los muchos que vienen en varias ocasiones juntos y parte de nuevo, puede por casualidad crecer más grande que el tamaño crítico. El proceso que describe la situación hasta el punto en que las partículas alcanzan el tamaño crítico puede ser llamado la etapa de nucleación

(3) Una vez que se supera el tamaño crítico, la partícula puede crecer más grande. Este proceso corresponde a la etapa de crecimiento en su sentido más estricto.
Entre los grupos que se forman, los que superan el tamaño crítico no lo hacen se disocian y puede crecer más grande. Como se muestra esquemáticamente en la Fig. 3.7, esto es porque la densidad de los enlaces libres por unidad de área en la superficie disminuye a medida que aumenta el tamaño, y finalmente alcanza un valor crítico. Figura 3.7 (b) muestra esta relación en términos de cambios de energía. La energía de nucleación es la suma de la energía gastada en la formación de una partícula por átomos de coagulantes, -G v, que es proporcional a r3, y la energía obtenida mediante la creación de la superficie, G s, que es proporcional a r2, a saber, G-G v G s. Como puede verse en la Fig. 3.7 (b), la energía necesaria para que se produzca la nucleación aumenta a medida que r aumenta, alcanza un máximo a rc y disminuye a partir de entonces. El núcleo que alcanza rc se llama el núcleo crítico, y rc se conoce como el radio del núcleo crítico.
Dado que existe un valor crítico para que se produzca la nucleación, la nucleación o crecimiento apenas se producen, excepto en una región estrecha lejos de la curva de solubilidad, que corresponde a la situación de equilibrio. Esta región estrecha a lo largo de la curva de solubilidad se llama la región Miers (ver Fig. 3.1).
La nucleación se puede clasificar como (i) "nucleación espontánea" o "nucleación homogénea", en el que es necesaria ninguna intervención por otros factores para que tenga lugar la nucleación, y (ii) "nucleación heterogénea", que se produce debido a la presencia de una pared de la superficie de un vaso, o partículas extrañas, o un componente de la impureza. Se requiere un motor mucho mayor para el primero que lo segundo. Hay una diferencia de unas pocas decenas de grados centígrados en términos de sobre enfriamiento entre los dos en el crecimiento de masa fundida. La mayoría de los fenómenos de nucleación en los sistemas reales pueden ser considerados como heterogéneos. En contraste con esto, se acepta que la nucleación homogénea juega el papel esencial en la nucleación en condiciones de micro gravedad.

Figura 3.7. (A) Los cambios en la densidad de enlace colgando por unidad de superficie como el tamaño de una partícula aumenta. Cuando el número de unidades que constituyen el núcleo aumenta desde cuatro hasta dieciséis, la densidad de enlace colgando por unidad de superficie disminuye de 8 a 16/9. (B) Los cambios en la nucleación de energía G como el radio de un núcleo r cambios. G alcanza un máximo en r c. La energía necesaria para alcanzar el volumen G v es proporcional a r3, mientras que la energía obtenida en la creación de la superficie, G s, es proporcional a r2.
La energía crítica para la nucleación está determinada por la energía libre de la interfaz (la superficie de un núcleo) y la fuerza de conducción. Cuanto menor sea la energía crítica, más fácil es para la nucleación a tener lugar, por lo que las impurezas con similitud química cristal, pequeñas partículas no resueltos, los pasos, y las irregularidades o arañazos en la superficie de la pared del vaso puede todos actúan como sitios de nucleación heterogénea. El método de la decoración en la microscopía electrónica (ver sección 5.2), lo que nos permite observar las medidas en una escala nanométrica, y el método epitaxial para el cultivo de películas delgadas cristalinas individuales (ver sección 7.4), son técnicas que utilizan los fenómenos de nucleación heterogénea teniendo en cuenta la energía en los pasos y las interfaces. El uso de cristales de siembra permite el crecimiento para iniciar de inmediato, sin pasar por la etapa de nucleación. Nucleación secundaria, un método ampliamente utilizado en el campo de la cristalización industrial, es esencialmente el mismo que el crecimiento de cristales usando semillas, aunque tradicionalmente un término de la nucleación se ha utilizado.
Es el propósito principal de la cristalización industrial para obtener numerosos pequeños cristalitos de igual tamaño y forma. Para lograr esto, un método práctico ha sido adoptado en la que grandes cristales de siembra se introducen en un recipiente de reacción y agitado. Las superficies de los cristales de siembra se separan por agitación, por lo que proporciona numerosos cristalitos diminutos en el sistema que actúan como cristales de siembra. Por lo tanto, se consigue el objetivo de obtener numerosos cristalitos de igual tamaño. Fenómenos similares, cabe esperar que se produzca en el movimiento ascendente de un magma que contiene fenocristales [4] (ver sección 8.5).
En la termodinámica de equilibrio, las fases finales estables son determinados por un rango de temperatura-presión dada. Sin embargo, no siempre es posible lograr la nucleación y el crecimiento de la fase estable desde el principio cuando el sistema se mantiene bajo esta condición. En muchos casos, la fase más temprana que aparece por la nucleación es una fase metaestable, que es diferente de la fase estable. Este fenómeno se conoce como la regla de Ostwald paso [5]. Esto puede ser entendido como resultado de la competencia entre la fuerza motriz y los términos de energía libre de la interfaz. Cuando el término fuerza impulsora es pequeño, la contribución del término de energía de superficie se vuelve
más importante. Como resultado de esta competición, la nucleación de una fase metaestable se producirá antes de la de la fase estable. Antes de la aparición de la fase estable, la fase metaestable formado anteriormente se puede desarrollar de una manera estable, y se comporta como si se trata de la fase estable. Sin embargo, una vez que la fase estable aparece en el sistema, la fase metaestable Formado anterior comienza a disolverse o transformar, y la fase estable crece. Si se mantienen las condiciones que suprimen la aparición de una fase estable, la fase metaestable puede llegar a ser estable [6]. La formación de aragonita (una fase de alta presión de CaCO3) existente en los cuerpos vivos, o de diamantes CVD (una fase de alta presión de carbono) bajo 1 atm de presión son tales ejemplos.
En esta sección hemos explicado la relación entre una fase ambiente a granel y un cristal mayor (donde se supone que el cristal para tener un ideal, la estructura regular). Desde la década de 1930, las investigaciones teóricas y experimentales han indicado que un verdadero cristal contiene defectos de orden de celosía, y que estos defectos tienen un efecto significativo sobre las propiedades físicas del cristal. Esta comprensión ha llevado a la actual industria de semiconductores días.
3.7 Defectos en la red
Las propiedades físicas de los materiales sólidos que son en gran medida influenciadas por la presencia de defectos de orden celosía en monocristales reales se llaman "propiedades structuralsensitive", y se distinguen de las "propiedades intrínsecas", que están determinados por los elementos que constituyen el cristal, por ejemplo, el enlaces químicos, la estructura, etc. Color, plasticidad, deslizamiento y propiedades semiconductoras son propiedades estructurales sensibles, mientras que la densidad, dureza, elasticidad y propiedades ópticas, térmicas y magnéticas son las propiedades intrínsecas. Propiedades estructurales sensibles serán diferentes para cada muestra individual. Hay casos en que incluso las propiedades intrínsecas están influenciados por defectos de orden celosía

Figura 3.8. Explicación de las dislocaciones en relación a deslizarse. La flecha sólida, b, corresponde a la vector de Burgers de la dislocación. SV es la dislocación de tornillo, WE es la dislocación de borde, y VW es una dislocación mixta. El área sombreada representa un plano de deslizamiento.
Los defectos de la red se clasifican como (i) "defectos puntuales", como las vacantes, átomos intersticiales, átomos de impurezas de sustitución, y los átomos de impurezas intersticiales, (ii) "defectos de línea", como borde, tornillo, y dislocaciones mixtas, y ( iii) "defectos planas", tales como fallas de apilamiento, aviones individuales, y los límites de grano.
Dentro de estas tres categorías, se prestó atención a los primeros defectos puntuales; esto surgió de la curiosidad intelectual sobre el origen del color azul profundo visto en grandes monocristales incoloros de NaCl se producen en los depósitos de sal. Desde este punto de partida, se desarrolló la idea de centros de color. Desde los centros de color son defectos puntuales, que modifican los estados electrónicos alrededor de los defectos y afectan el calor y la conductividad electrónica; en consecuencia, se han convertido directamente conectado con el desarrollo de la industria de los semiconductores.
El concepto de defectos de línea fue introducida en teoría para dar una respuesta a la discrepancia entre los valores teóricos y experimentales de la tensión de corte para crear deslizamiento (deslizamiento). Le tomó casi veinte años para demostrar la presencia de dislocaciones en cristales. Glide es un fenómeno en el que parte de un cristal se desplaza (sin perder fuerza adhesiva sobre un plano de deslizamiento) por una distancia (ya sea un espaciado reticular o un múltiplo de la misma) en la dirección de deslizamiento. El valor teórico de la módulo de cizallamiento, suponiendo que deslizamiento se produce instantáneamente en un cristal perfecto ideal, se calculó a ser hasta 1.000 veces el valor medido experimentalmente. De esta surgido una teoría que considera el fenómeno de deslizamiento a estar vinculado a un movimiento de dislocaciones. La teoría asume que deslizamiento se produce porque las dislocaciones se mueven en el plano de deslizamiento (ver Fig. 3.8).
Si se asume que el deslizamiento se produce en el plano de deslizamiento en la dirección de y por la cantidad indicada por la flecha sólida, vemos que la red está distorsionada a lo largo de la parte delantera del deslizamiento, SVWE. El modo de distorsión del plano de celosía es diferente entre la parte perpendicular a la flecha, WE, y que paralelamente a la flecha, SV, y de nuevo a la parte intermedia VW. A WE, el plano reticular está distorsionada, como si de un plano medio de celosía se inserta como un filo de la navaja, y al SV continúa el próximo avión de celosía en la forma de una escalera de caracol. El primero se llama una dislocación de borde, y el segundo se refiere como una dislocación de tornillo. VW se llama una dislocación mixta, ya que posee ambos componentes.
La dirección y el tamaño de la flecha sólida en la Fig. 3.8 representan el vector que caracteriza a la dislocación; esto se llama el vector de Burgers. Esto puede ser de tamaño de celosía, o una fracción o número entero múltiplo entero del tamaño del enrejado. Desde el plano reticular está distorsionada por la zona estrecha alrededor de una línea de dislocación, un campo de deformación se concentra allí. Esta región se llama el núcleo

dislocación, y puede ser un sitio preferencial para la disolución o adsorción a tener lugar. De hecho, la verificación experimental más temprana de la presencia de dislocaciones en cristales individuales se logró mediante la producción de pozos de grabado de punto de fondo, correspondientes a los afloramientos de dislocaciones, y posteriormente grabado el espécimen después de aplicar el estrés. Un cambio en la forma del pozo de ataque desde el punto de fondo a los hoyos de grabado de fondo plano se observó en el afloramiento original de la dislocación, y el punto de fondo fueron observadas en la posición en que la dislocación había viajado. Las dislocaciones pueden ahora ser observados directamente por diversas técnicas, tales como la topografía de rayos X o la microscopía electrónica de transmisión.
El concepto de dislocaciones se introdujo en teoría, en la década de 1930 por E. Orowan y GI Taylor, y de inmediato tuvo un papel esencial en la comprensión de las propiedades plásticas de materiales cristalinos, pero tomó otros veinte años para entender plenamente la importancia de dislocaciones en el crecimiento de cristales. Como se describe en la Sección 3.9, no fue sino hasta 1949 que la teoría del crecimiento en espiral, en el que se asume que el crecimiento de una interfaz suave para proceder de una manera paso de caracol, con el paso de servir como una fuente de paso perpetúa a sí misma, era propuesto [7]. Dislocaciones mismos juegan un papel en la promoción del crecimiento, pero también pueden ser inducidos en crecimiento, cristales existentes en las distintas etapas. Desajuste del enrejado puede resultar donde las ramas laterales de un cristal dendrítico se unen, cuando una inclusión atrapado durante el crecimiento está encerrada, o a lo largo de la frontera de dos cristales con un poco diferentes orientaciones. Desajustes parámetro de red entre los cristales de acogida y de huéspedes en el crecimiento epitaxial también son orígenes de dislocaciones inducida crecimiento. Cuando un verdadero cristal crece en un sistema no controlado, que por lo general tienen una densidad de dislocación del orden de 10.810 líneas / cm2. En la utilización de cristales individuales para los dispositivos, es necesario crecer cristales individuales que contienen una densidad de dislocaciones reducida; sin embargo, la distribución de las dislocaciones en cristales individuales puede servir como un registro fiable del proceso de crecimiento.
Dislocaciones inducida crecimiento pueden multiplicarse durante el proceso de post-crecimiento. Si el estrés se aplica a las dislocaciones fijas por las impurezas, las dislocaciones inducidos multiplican para formar anillos de dislocación de bucle. Este es un mecanismo importante en la multiplicación de dislocaciones, y se refiere como una fuente de Frank-Read. De esta manera, dislocaciones inducida crecimiento se mueven durante el período post-crecimiento y forman acumulaciones energéticamente más estables de dislocaciones. Estructuras secundarias, tales como ovillos de dislocación, redes o estructuras de panal de dislocaciones, límites de grano de ángulo pequeño, límites de torsión, y dislocaciones sacabocados, son ejemplos típicos. Por otro lado, dislocaciones inducida crecimiento en el crecimiento de cristales de la fase de solución generalmente se distribuyen en paquetes procedentes del centro del cristal o de inclusiones atrapadas durante el crecimiento. Ellos también se originan en la superficie de un cristal de siembra o de bandas de crecimiento. Dislocaciones inducida crecimiento generalmente se ejecuta casi perpendicularmente a la superficie de cultivo.
Además de los defectos retícula de orden, defectos mucho más grandes a menudo pueden ser inducidos en un cristal en crecimiento durante o después del crecimiento; por ejemplo,

la inclusión de fases sólido, líquido o de vapor. Las inclusiones atrapadas simultáneamente durante el proceso de crecimiento se llaman inclusiones syngenetic, mientras que los inducidos después del crecimiento es completa se llaman inclusiones epigenéticos. Inclusiones syngenetic estado sólido proporcionan evidencia de las composiciones y las condiciones termodinámicas de las fases ambiente en el momento del crecimiento de cristales. Si un líquido inclusión fueron atrapados como una sola fase, el componente disuelto en el líquido se disocia en forma de vapor o fases sólidas debido a la disminución de la temperatura y la presión, y daría lugar a inclusiones de dos fases de vapor-líquido o de múltiples -Fase inclusiones en el que las fases de vapor-líquido-sólido coexisten. Sobre la base de la temperatura a la que dos fases cambio a una sola fase en la calefacción, la temperatura en el momento de la inclusión fue atrapado se puede evaluar, después de primero calibrar el efecto de la presión.
Una fase se encuentra a menudo entre los minerales de silicato o sulfuro en manchado, estrella, laminar o texturas de celosía en la fase de acogida. La fase se mezcló originalmente como una solución sólida homogénea solo con la fase de acogida en el momento de crecimiento, y más tarde precipita o exsolved la fase de una componente de la solución sólida dentro de la fase cristalina de acogida como la temperatura disminuye. Este fenómeno se llama la precipitación en los campos físicos o metalúrgicos y exsolution en
disciplinas geológicas. La fase exsolved tiene una relación definida cristalográfica con la fase de acogida para mantener la energía mínimo interfaz entre las dos fases.
3.8 Interfaces
Después de racimos alcanzan un tamaño crítico en el sistema y la etapa de nucleación esta completa, comienza la etapa de crecimiento en un sentido más estrecho. Puesto que la estructura ya está formado, el componente de soluto será incorporado en el cristal a expensas de una energía mucho menor que la necesaria para la nucleación. Aquí, la estructura de la interfaz entre las fases sólida y líquida que apareció como resultado de la nucleación se vuelve importante. Esto es porque el mecanismo de crecimiento será diferente dependiendo de si la interfaz es atómicamente áspera o lisa. La interfaz áspera consiste enteramente de torceduras, y la interfaz lisa comprende una superficie plana (terraza), un pequeño número de pasos, y un sitio donde un paso está doblado, es decir, un punto de torsión.

Figura 3.9. Cristal Kossel
Kossel [8] y Stranski [9] fueron los primeros en llamar la atención sobre la estructura de la interfaz, después de considerar los resultados experimentales obtenidos por Volmer [10], que demostró la existencia de difusión superficial. De este modo fue posible discutir el mecanismo de crecimiento de los cristales a un nivel atómico, a partir de estos análisis.
Supongamos que las entidades componentes de ambos un cristal y la de crecimiento son cubos simples. Este tipo de cristal modelo se llama un cristal Kossel, y se muestra en la Fig. 3.9. El {100} cara está completamente pavimentado por la unidad, y la superficie es atómicamente plana. Esta cara se denomina el "plano completo". El {111} cara, sin embargo, consta de torceduras, como puede verse en la Fig. 3.9, y tiene una superficie irregular, por lo que se llama un "plano incompleto." En contraste con {111}, {110} corresponde a un rostro compuesto enteramente de pasos. Kossel no dio un nombre en particular a este tipo de cara cristalina.
Hay tres sitios diferentes que tienen diferente energía de fijación en un cristal Kossel. Una unidad constituyente de forma cúbica simple en la superficie de un {100} está conectado a la cara del cristal en cinco de las seis caras; en el borde de una cara {100} o en un en la cara {110}, que está conectado a cuatro caras de seis posibles; mientras que en la esquina de una cara {100} o en la superficie de una cara incompleta {111} , que está conectado a tres de las seis caras. Estos sitios pueden ser llamados una cara lisa (cara terraza), un paso y una torcedura, respectivamente. Podemos entender inmediatamente que la energía de unión para la unidad de crecimiento de al cristal disminuye en este orden. Dado que en un sitio de torcedura la energía de fijación es igual a la media de la energía de unión entre las unidades constituyentes vecinas, un sitio de torcedura se llama sitio medio de cristal.

Figura 3.10. Esquema que muestra el modelo de crecimiento capa por capa debido a la nucleación bidimensional. Esta figura asume el modo de nucleación para ser el modelo mono-nuclear. Otros modelos, como los modelos de poli-nuclear o de nacimiento y propagación, como se explica en el texto, también pueden ser considerados.
Teniendo en cuenta la energía de unión, vemos que en una cara incompleta una unidad de crecimiento que incide será incorporada inmediatamente en el cristal. Esto indica (i) que el mecanismo de crecimiento de una cara incompleta será de tipo crecimiento adhesivo, (ii) que la interfaz avanzará de manera homogénea, y (iii) que la velocidad de avance de la interfaz (la tasa de crecimiento normal) será linealmente relacionada a la fuerza impulsora.
En contraste, una unidad de crecimiento de llegar a una superficie plana sobre una terraza interfaz completa puede no ser incorporado inmediatamente en el cristal. Se puede difundirse en la superficie o dejar la interfaz, volviendo a la fase inicial. Si hay pasos o torceduras, pasos que se pliegan, sobre la superficie de terraza, una unidad de crecimiento de llegar será incorporada en el cristal. Como resultado, la cara crecerá en dos dimensiones de difusión del paso. Si el paso alcanza el borde de la cara y la superficie se vuelve completamente plana, se requiere una nueva fuente de paso para continuar el crecimiento. Kossel [8] y Stranski [9] asumieron que tendrá lugar la nucleación bidimensional. Este mecanismo se denomina crecimiento de la capa por capa. La figura 3.10 muestra este modelo de forma esquemática.
En esta teoría, ya que la tasa de crecimiento de una cara se controla por la tasa de nucleación bidimensional, debemos esperar que exista la presencia de una fuerza de conducción crítica y que el crecimiento puede tener lugar solo si se supera esta. Por debajo de este valor, no habrá crecimiento. Como posibles modos de nucleación de dos dimensiones, se pueden considerar tres modelos diferentes.
(1) Un modelo de mono-nuclear, que permite sólo una nucleación, la próxima nucleación toma lugar sólo después de que toda la superficie está cubierta por capas de crecimiento procedentes del núcleo anterior.
(2) Un modelo polinuclear, que permite llevar a cabo muchas nucleaciones en una superficie.
(3) Un modelo de nacimiento y propagación, lo que permite la nucleación y el avance de una capa de crecimiento a la vez en una superficie.
En todos estos modelos, asumimos nucleación bidimensional.
Tabla 3.2 Términos usados para expresar los estados de las interfaces
Kossel, Stranski cara completa, cara incompletaBurton, Cabrera, Frank rostro singular, la cara no singular Hartman, Perdok cara plana (cara F), salió cara (cara S ), la cara retorcida (cara
K)

Terminología actual cara lisa, cara rugosa
Muchas teorías sobre estructuras de interfaz se han expuesto, y en consecuencia muchos términos diferentes se han introducido; resumimos éstos en la Tabla 3.2. En lugar de referirse a completos y rostros incompletos, Burton, Cabrera y Frank [11] utilizaron los términos rostros singulares y no singulares, en el sentido de una cara que muestra un mínimo aguda (cúspide) y uno que no muestra ni mínima, respectivamente, en un diagrama polar de la energía superficial. Esta clasificación se basa en el cálculo de la mecánica estadística, y está estrechamente relacionado con el debate sobre la forma de equilibrio (véase la Sección 4.3). Hartman y Perdok [12] cristal clasificado en F (para la cara plana), S (escalonada), y K (cara retorcido), en función de los números de las cadenas de bonos periódicas (CNS) que se encontraron en una estructura cristalina mediante la conexión fuerte bonos. Está claro que estas caras corresponden a las caras de un cristal Kossel {100}, {110}, y {111}, respectivamente (Fig. 3.9). Hoy en día, los términos interfaz suave y áspero se usan comúnmente junto con rostros perfectos e imperfectos, y rostros singulares y no singulares.
Como puede verse desde el cristal tipo Kossel se muestra en la Fig. 3.9, la rugosidad de una interfaz depende de que considerando a que se enfrenten, es decir, en las direcciones cristalográficas. También varía dependiendo de la especie de cristal, la temperatura, y la fuerza impulsora. El aumento de la temperatura o la fuerza impulsora causará una interfaz fácil de cambiar en una interfaz áspera. El punto de transición que existe en pasar de una interfaz suave a una áspera cuando aumenta la temperatura se denomina una transición de rugosidad. La existencia de transiciones de rugosidad ha sido demostrada por simulación por ordenador. Una transición de rugosidad resultante de aumentar la fuerza impulsora se llama una transición de rugosidad cinética. Dependiendo del grado de rugosidad de una interfaz, el tipo de mecanismo de crecimiento o el crecimiento frente a los cambios de relación de conducción de fuerza, y por lo tanto la rugosidad de la interfaz es un concepto esencialmente importante en el análisis de la morfología de los cristales y elemento de segmentación (véase la Sección 3.14).
Las predicciones teóricas más tempranas sobre el estado de las interfaces fueron hechas por Burton, Cabrera, y Frank [11], que demostró que la interfaz sería, en la mayoría de los casos áspera a la temperatura de crecimiento para los cristales de metal. Jackson [13] sugirió un sistema en el que las fases sólida y líquida se separan mediante una interfaz con un espesor de una sola capa, y calcula los cambios de energía como una función de la relación de ocupación del sitio de la unidad constituyente en la interfaz. Cuando la relación de ocupación del sitio fue del 50%, la interfaz era áspera, mientras que 0 o 100% el sitio de ocupación correspondió a una interfaz suave. En t su cálculo, que utiliza los llamados Jacks en α factor de definido por:

Figura 3.11. Predicción de la rugosidad de la interfaz obtenido por (a) Jackson [13] y (b) Temkin [14], [15]. (A) El eje horizontal muestra la relación entre la ocupación del sitio de la superficie, y la vertical de x representa el cambio en la energía libre. Los números son factores. (B) El eje vertical indica la fuerza impulsora, y el eje horizontal muestra el factor. A es el área estable para una interfaz lisa; B es el área estable para una interfaz áspera.
Donde ɛ es el factor de orientación, L es el calor de fusión, k es la constante de Boltzmann, y Tm es el punto de fusión. Figura 3.11 (a) muestra los resultados de cálculo de Jackson, e indica que hay dos tipos de curvas en función del factor α. Para materiales con α<2, sólo hay un mínimo de 50% de ocupación del sitio de la superficie, lo que indica que la interfaz energéticamente prefiere ser áspera. Por otro lado, en un material con α>3, hay dos mínimos de energía en el sitio de ocupación 0 y 100%, lo que indica que la interfaz será suave.
Los factores α son diferentes para diferentes materiales. Los cristales metálicos tienen generalmente α<2; los cristales semiconductores tienen 2<α<3; y en óxidos, silicatos, y cristales de polímero, que tienen estructuras cristalinas complejas, α>3. Debido a la presencia de ɛ (correspondiente a la relación entre dos y números de coordinación tridimensionales zs/ z), los factores α serán diferentes en diferentes direcciones cristalográficas en el mismo cristal. Para los cristales de hielo, el (0001) cara tiene α>3, mientras que para la cara (10 1-1 0) es 2<α<3, lo que indica que (10 0) es una cara más áspera que (0001).
En contraste con el tratamiento termodinámico de equilibrio realizado por Jackson, Temkin [14], [15] considerado el problema de la transición de rugosidad de una interfaz en relación con la fuerza de accionamiento y el factor α. Su resultado se muestra en la Fig. 3.11 (b), en el que la zona A corresponde a un área donde se espera una interfaz suave, mientras que la zona B corresponde al esperado para una interfaz áspera. Al aumentar la fuerza motriz, una interfaz suave se transforma en una interfaz áspera: una transición de rugosidad cinética.
Para Jackson el factor α es el valor medido en el punto de fusión; la interacción soluto-disolvente no se tiene en cuenta. Esto corresponde a la situación de crecimiento de los cristales en la fase de fusión. Bennema y Gilmer [16] el factor α de Jackson se da de la

siguiente de modo que se puede aplicar a la fase de solución, que implica una interacción soluto-disolvente:
αG = ɛ {φsf - ½ (φss + φff)}/KTG
αG es un generalizado del factor α, φss y φff son la unión de las energías en las fases sólida y de fluidos, respectivamente, φsf es la energía de interacción soluto-disolvente, k es la constante de Boltzmann, y TG es la temperatura de crecimiento, que es menor que el punto de fusión. En el crecimiento de cristales de la fase de fusión, αG se convierte en la misma que en la expresión de Jackson. En esta generalización, sin embargo, αG se da en términos de la temperatura de crecimiento y la energía de interacción soluto-disolvente, por lo que es diferente de Jackson del factor α. Experimentos informáticos extensos han sido realizados desde la década de 1980 para investigar cómo los cambios en la estructura de interfaz como Δu/kT cambios nudos, para determinar si la transición de rugosidad se lleva a cabo, y establecer la forma en la tasa de crecimiento en comparación con la relación fuerza motriz (y por tanto el mecanismo de crecimiento) cambian dependiendo en los cambios en el factor α, el factor α G o Δu/kT. Los resultados fueron los esperados; Fig. 3.12 muestra dos ejemplos de los resultados de experimentos de ordenador.
Los análisis anteriores suponen que la estructura de interfaz no cambia en todas las condiciones de crecimiento. Se ha demostrado experimentalmente usando cristales de Si que la estructura de interfaz puede ser reconstruido en algunas fases ambiente. Los átomos en el interior del cristal están unidos de forma tridimensional y simétricamente a los átomos vecinos, mientras que los de la superficie no. Los átomos de la superficie están unidos con átomos en la estructura, pero no con los de la fase ambiente. En consecuencia, están en un estado de energía más alto que los átomos en el cristal. Bajo ciertas condiciones, se requiere la "reconstrucción de la superficie" para relajar el estado de alta energía; esto se logra mediante la formación de la unión parcial, por ejemplo la formación de una estructura en lugar del original estructura 1x1 el 3x3. Se ha demostrado que las estructuras reconstruidas aparecen en Si 2x1 o 7x7, y en Ir (100) una estructura 1x1 cambia a una estructura de 1x5. En consecuencia la estructura del cristal se enfrenta a dicha modificación a consecuencia de tal reconstrucción, y que exhiben una morfología de la superficie diferente de caras no reconstruidas ordinarios (véase la Sección 4.4).

Figura 3.12. Los cambios en la rugosidad de la interfaz como resultado de cambiar y, tomados de los resultados de un experimento ordenador [17]. (A) - (d) indica cambios en el estado de la interfaz para variar α , manteniendo Δu/kT constante; (E) muestra un paso creado por una dislocación de tornillo en equilibrio, Δu/kT=0, y (f) - (h) muestra la forma en que avanza bajo Δu/kT=1.5, con α constante.

Figura 3.13. Mecanismo de crecimiento espiral.
3.9 crecimiento Espiral
En el modelo de crecimiento de la capa por capa formulado por Kossel [8] y Stranski [9], la tasa de crecimiento está limitado por la nucleación bidimensional. El crecimiento no se producirá a menos que se supere la barrera de energía necesaria para la nucleación de dos dimensiones, lo que indica que debe haber un valor crítico de la fuerza motriz para que tenga lugar el crecimiento. En el caso de crecimiento de los cristales de la fase de vapor, este valor se estima en alrededor de 25-50% en términos de sobresaturación. En los casos reales, observamos que cristales crecen bajo sobresaturación a porcentajes tan bajos como 1%. Esta gran discrepancia entre los valores teóricos y experimentales se origina en el hecho de que Kossel y Stranski asumieron cristal perfecto. Los cristales reales, sin embargo, son imperfectos, que contiene impurezas y dislocaciones. El Modelo de crecimiento espiral de Frank ha sido propuesta para explicar el mecanismo de crecimiento de cristales reales. Poco después de la propuesta de este modelo, la primera evidencia para apoyar la prueba del modelo se obtuvo en caras {10 1 -1 0} de los cristales naturales berilo [18], seguido por observaciones de los patrones de paso espiral en muchas caras de una amplia variedad de cristales . Así, la teoría del crecimiento en espiral estaba firmemente establecido [19].
Cuando una dislocación de tornillo aflora en una interfaz lisa, se crea un paso en la superficie que tiene la altura cero en el núcleo de la dislocación y una altura correspondiente a un vector de Burgers en el borde de la cara. La etapa de crecimiento a partir de un paso tal avanza como una escalera espiral alrededor de la dislocación, que actúa como un apoyo, como se muestra en la Fig. 3.13. Esto es debido a la diferencia en la velocidad angular de avance del paso de la espiral en el centro y que en el borde. Desde la dislocación de tornillo es una fuente paso auto perpetúa, no es necesario superar la barrera de energía para la nucleación de dos dimensiones. Los cristales pueden crecer por este mecanismo por debajo de la fuerza impulsora fundamental para el crecimiento de capa por capa. La tasa de crecimiento y la fuerza motriz se relacionan de la siguiente manera:
R=A (Δu/ kT)2
donde A es una constante, y Δu/ kT es la fuerza impulsora.

Tipo adhesivo Crecimiento de nucleación Crecimiento en espiral bidimensional
) R = A ( )2
Rugoso En transición liso transición termodinámica rugosa transición cinética rugosa
Figura 3.14. Las diferencias entre los mecanismos de crecimiento en las interfaces rugosas y lisas, muestran la relación entre la tasa de crecimiento y la fuerza motriz en el mecanismo de crecimiento de tipo adhesivo, el mecanismo de crecimiento de nucleación en dos dimensiones, y el mecanismo de crecimiento en espiral. También se muestra la transición de rugosidad de una
superficie lisa a una interfaz rugosa. R es la tasa de crecimiento, /kT es la fuerza motriz, y A y
B son constantes.
El crecimiento en espiral es un mecanismo que se espera solamente en las interfaces lisas. La ayuda proporcionada por las dislocaciones de tornillo no son necesarias en el crecimiento de las interfaces rugosas, donde opera un crecimiento de tipo adhesivo.
Como se explicó anteriormente, se establecieron tres modelos fundamentales de mecanismo de crecimiento de los cristales en relación con la rugosidad de las interfaces; éstos se ilustran en la Fig. 3.14. En la actualidad, no hay ningún otro mecanismo de crecimiento conocido que es esencialmente diferente de estos tres. Por lo tanto, vamos a analizar la morfología de los cristales, el tema principal de este libro, basado en estos tres mecanismos de crecimiento.
3.10 Mecanismo de Crecimiento y morfología de los cristales
La figura 3.15 muestra la tasa de crecimiento de R frente a la fuerza motriz /kT para los
tres modelos de mecanismos de crecimiento. Esta figura ilustra los dos puntos siguientes.
(1) Cuando como el motor aumenta, una interfaz se vuelve más rugosa.
(2) Dos puntos de flexión aparecen en /kT * y /kT **, ya que las curvas de R frente a
las relaciones /kT esperados para los tres modelos de mecanismos de crecimiento son
diferentes. Una interfaz se vuelve rugosa y el mecanismo de crecimiento anterior será de
tipo adhesivo /kT **, mientras que la interfaz será lisa y el crecimiento será controlado

principalmente por el mecanismo de crecimiento por debajo de la espiral /kT *. Entre
/kT* y /kT **, la interfaz será lisa, pero el mecanismo de crecimiento será principalmente
por la nucleación bidimensional. Figura 3.15 es un diagrama esquemático de R frente a la
relación /kT esperada en una dirección cristalográfica en una fase ambiente imaginario
Figura 3.15. Áreas donde se espera que las interfaces rugosas y lisas. La tasa de crecimiento en comparación con los que impulsan las relaciones de fuerza que se espera para los tres modelos de crecimiento se indican en la tasa de crecimiento, R (vertical), eje frente a la fuerza motriz (/ kT) diagrama. La curva A muestra el mecanismo de crecimiento de espiral; B representa el mecanismo de crecimiento de dos dimensiones de nucleación; C denota el mecanismo de tipo adhesivo.
Las posiciones de /kT * y /kT ** son diferentes, incluso en la misma dirección
cristalográfica de la misma especie de cristal, en función de la diferencia de fase ambiente.
En la fase condensada en estado fundido, la posición de /kT ** de un material con
< 2 se espera estar muy cerca del origen (a saber, la interfaz será rugosa), mientras que la
posición será a un valor mucho más largo en el caso de crecimiento de la fase de vapor,
que es la fase más diluida ambiente. En crecimiento solución, la posición de /kT **
estará en entre las dos posiciones (Fig. 3.16). Dependiendo de la diferencia de energía de
interacción soluto-disolvente, las posiciones de /kT ** serán diferentes de material a
material que crece de la fase de solución con el mismo componente de disolvente. Esto implica que cuando el cristal crece a partir de la fase de fusión de la interfaz, será rugosa, asimismo un cristal de la misma especie se espera que tenga una interfaz lisa y el

crecimiento en espiral cuando el cristal crece a partir de la fase de vapor. En otras palabras, esto implica que un cristal poliédrico limitada por caras planas se espera que más generalmente cuando un cristal crece a partir de la fase de vapor que cuando crece a partir de la fase de fusión. También, dependiendo de las especies de cristal, la implicación es que la morfología será diferente, incluso si los cristales crecen de la misma solución acuosa,
porque los valores de la /kT ** son diferentes para las diferentes especies cristalinas
debido a soluto-disolvente interaccion

Figura 3.16. Las diferentes posiciones de /kT * y /kT ** en función de las fases ambientales. (A)
la fase de vapor; (B) en fase de solución; (C) fundir fase.
La figura 3.15 muestra R frente /kT para una dirección cristalográfica. Las posiciones de
/kT * y /kT ** son diferentes en diferentes direcciones cristalográficas del mismo
cristal. La cara cristalina clasificada como la cara más importante en orden de importancia morfológica (es decir, la que muestra la cúspide más aguda en el diagrama polar de la energía de interfaz) parcela (, y pertenece a una categoría de la cara más importante F en el análisis PBC, y tiene la densidad reticular más alto en la ley BFDH (Bravais-Friedel-
Donnay-Harker); véase el capítulo 4) se supone que tiene los valores más grandes de /kT
* y /kT **. Como el orden de importancia morfológica disminuye, estos valores se
redujeron entre los rostros categorizados como caras F. Se espera que la posición del /kT
** de K o S se enfrenta en el análisis de PBC para estar más cerca del origen.
3.11 inestabilidad morfológica
La interfaz es el único lugar donde el crecimiento de cristales (y disolución) lleva a cabo. Hemos clasificado la interfaz en rugoso y liso en la sección anterior, y hemos presentado los mecanismos de crecimiento esperados. En el crecimiento de cristales de la fase de fusión, una interfaz rugosa tomará una forma curvada que sigue un plano curvo de igualdad de temperatura, mientras que en el crecimiento de las fases de vapor o solución, que va a seguir una superficie curva siguiente líneas de igual concentración. En contraste, una interfaz lisa toma una forma recta truncar líneas equipotenciales térmica o equiconcentration. Vamos a discutir en esta sección si la forma de interfaz se puede mantener durante todo el crecimiento o no, qué tipo de inestabilidad se llevará a cabo cuando la forma de interfaz no se mantiene, y cómo estas inestabilidades afectar a la morfología de los cristales. No, qué tipo de inestabilidad se llevará a cabo cuando la forma de interfaz no se mantiene, y cómo estas inestabilidades afectar a la morfología de los cristales.

Figura 3.17. Relación entre la interfaz y líneas de igual concentración en la fase ambiente cuando se produce la inestabilidad morfológica.
Supongamos que una pequeña protuberancia aparece en una interfaz curvada rugosa, y que por alguna razón se altera la morfología interfaz. Los intervalos entre las líneas de igual temperatura o concentración vuelto más estrecho en el bulto; Por lo tanto, el gradiente de temperatura o la concentración se vuelve más nítida, lo que resulta en una tasa de crecimiento cada vez mayor (Fig. 3.17). Las situaciones son las mismas para el crecimiento de la masa fundida, en el que la transferencia de calor juega el papel esencial, y para el vapor diluido o solución de crecimiento, en el que la transferencia de masa juega el papel esencial. Cuando se producen cambios en el gradiente de la temperatura y la concentración, la morfología interfaz pierde su estabilidad, y el bulto se verá reforzada. Inestabilidad morfológica es más fuerte en las interfaces rugosas, curvas que en las interfaces lisas y planas. A menos que la cooperación de las fuerzas resulta en la supresión de la inestabilidad de la interfaz, la inestabilidad se verá reforzada a medida que avanza de crecimiento. Como resultado, la estructura celular periódica, aparecerá matrices de cúspides, varilla o estructura laminar y, además, la morfología dendrítica. Varios patrones derivados de la inestabilidad morfológica se indican en la figura. 3.18. Ahora se entiende que la morfología dendrítica de cristales de nieve, lo que atrajo la atención de los primeros observadores de cristales, los resultados de los cambios morfológicos derivados de la inestabilidad morfológica. Las seis ramas simétricas son debido a la estructura. El problema de la inestabilidad morfológica se resolvió teóricamente por Mullins y Sekerka [20], que propuso una teoría lineal que demuestra que la morfología de un cristal esférica creciente en la masa fundida sobre enfriada se desestabiliza debido a la difusión térmica; la teoría tratada cuantitativamente con y dio análisis lineal de la inestabilidad en la interfaz de solidificación unidireccional. La morfología interfaz es inestable para una variedad de razones; debemos considerar primero el sobre enfriamiento constitucional, es decir, el cambio de punto debido a un cambio en el contenido de impurezas de fusión. En el proceso de solidificación de una aleación, incluso si la cristalización se inicia desde una interfaz sólido-líquido plana con contenido de impurezas homogénea, concentraciones de impurezas en el sólido y el líquido va a cambiar a medida que avanza de crecimiento (como los avances de interfaz) debido a la compartimentación de los componentes de impurezas entre las fases sólida y liquida. Los componentes de impurezas con una partición (distribución) coeficiente menor que la unidad se concentrará en la interfaz

Figura 3.18. El proceso de cambios en la morfología debido a la interfaz de inestabilidad. (A) - (e) a partir de una interfaz lisa, se ve que la morfología cambia de una matriz cúspide, a una estructura de varilla, y a una estructura dendrítica.
La tasa de concentración será determinada por las constantes de difusión del componente de la impureza, la tasa de crecimiento del cristal, y el coeficiente de distribución del componente de impureza. Esto se traduce en un estado de sobre enfriamiento aparente debido a la relación entre el gradiente de temperatura entre el cristal y la masa fundida y el gradiente de concentración, como se ilustra esquemáticamente en la Fig. 3.19. Esto se conoce como sobre enfriamiento constitucional, y proporciona una explicación para el crecimiento de cristales a partir de una fase de fusión. Como resultado, se explican las causas de la varilla, celular, o estructura laminar observado como la estructura de solidificación en las aleaciones. Sin embargo, el mismo concepto puede ser aplicable a crecimiento vapor o solución en la presencia de componentes de impureza. Por qué las formas poliédricas delimitadas por las interfaces lisas puede crecer, manteniendo sus formas poliédricas, no fue tenido en cuenta adecuadamente hasta que la teoría del crecimiento capa PORCAPA (que considera el proceso atómico de crecimiento de los cristales) formulado por Kossel y Stranski apareció.
Si el crecimiento

está totalmente controlado por difusión, el cristal debe tener una forma esférica, sin caras planas. La aparición de caras cristalinas planas se explica por la introducción del concepto del mecanismo de crecimiento de la capa por capa en una interfaz lisa. Como un ejemplo de la violación de la inestabilidad morfológica, podemos mencionar el efecto Berg. Como Berg demostró (véase la Sección 3.4), la fuerza motriz a través de una cara del cristal es mayor en las esquinas o en los bordes que en la cara, y es más bajo en el centro de la cara. Hay un valor crítico para la nucleación de dos dimensiones que se produzca. Si la fuerza impulsora en las esquinas y los bordes es menor que este valor crítico, el crecimiento de esta cara será controlado por el mecanismo de crecimiento de espiral Fig. 3.15), y la capa de crecimiento procedente de dislocaciones de tornillo en la zona central de la cara avanzará hacia el exterior, y así la estabilidad poliédrica de la morfología podrá ser mantenido. Si la mayor parte de fuerza aumenta por encima del valor crítico para la nucleación bidimensional que tendrá lugar en
las esquinas o bordes (el área entre /kT * y /kT ** en la Fig. 3.15), dislocaciones
afloramiento cerca de los bordes o esquinas, o nucleación bidimensional en estos lugares, actuará como fuentes para las capas de crecimiento, que luego avanzar hacia el interior a la zona central de la cara. Esto corresponde a la formación de una cara de la tolva. Si la fuerza motriz aumenta aún más, la nucleación tridimensional se llevarán a cabo en los lugares donde la fuerza impulsora es máxima, es decir, en las esquinas del cristal. Se espera que este saliente aparecerá en la dirección de las esquinas, y que será posiblemente convertirse en un cristal dendrítico por la misma razón que la inestabilidad morfológica se produce en una interfaz rugosa.
Tomando el efecto Berg en cuenta, Kuroda et al. [21] demostraron que el límite entre la zona donde la morfología poliédrica permanece estable y la región donde se viola la estabilidad, y la tolva o formas dendríticas aparece, cambiará dependiendo del tamaño del cristal. La figura 3.20 muestra los resultados.
3.12 Fuerza impulsora y morfología de los cristales
Si podemos predecir cambios morfológicos en base a los análisis de estados de interfaz, en la R frente relaciones para los tres modelos de mecanismo de crecimiento, y en la estabilidad morfológica de interfaces, como se discutió en las secciones 3.2 a 3.9, y luego
graficar los resultados en un frente ∆µ/kT diagrama R como se da en la Fig. 3.15, creamos la figura. 3. 21.

Figura 3.20. Tamaño dependencia de la frontera entre las regiones morfológicamente estables e inestables cuando se toma en consideración [21] el efecto Berg.
La Figura 3.21 es un diagrama esquemático que muestra una sola dirección cristalográfica.
Por debajo de la condición de fuerza motriz ∆µ/kT*, el mecanismo de crecimiento en espiral es el mecanismo de control, y se espera un cristal poliédrico limitada por caras
planas. Por encima ∆µ/kT**, la interfaz será áspero, el mecanismo de crecimiento será de
tipo adhesivo, y se espera una morfología dendrítica. En la región entre ∆µ/kT* y
∆µ/kT**, una morfología tolva se espera debido al mecanismo de nucleación bidimensional. De esta manera, la lógica coherente se ha establecido entre morfologías dendríticas y poliédricas de cristales, que se consideran como problemas completamente independientes en las primeras etapas de estudio de la morfología de los cristales. Esta gama de morfología, de poliédrico a dendrítica, es exhibida por cristales individuales. Si
∆µ/kT aumenta aún más, el ritmo de aumento de nucleación, lo que resulta en la agregación de muchos cristales. Dependiendo de las condiciones, aparecerá agregados policristalinos esféricas debido a que irradia el crecimiento de un centro. En el proceso de formación de un esferulıtica, divergente, pajarita, o la morfología semi-esferulıtica, o bien se pueden formar agregados policristalinos o aparecerá una agregación policristalino azar.
Mecanismo de crecimiento en
Dendrita
Región estable
Mecanismo de
crecimiento
Región inestable
Tam
año
de c
rista
l (cm
)

Figura 3.21. Los cambios en la morfología de los cristales, mostrados en una tasa de crecimiento frente a la conducción diagrama de fuerza (ver Fig. 3.15), asumiendo un cristal delimitada por el {111} única cara.
Las posiciones de ∆µ/kT* y ∆µ/kT** en el diagrama será diferente para diferentes direcciones cristalográficas. Cuanto mayor sea la importancia morfológica, el más grande de estos valores serán. En otras palabras, un cristal en crecimiento está limitado por las caras que tienen diferentes fuerzas impulsoras, y así cada cara presenta una rugosidad interfaz diferente.
En el caso de un cristal delimitada por ambas interfaces lisas y rugosas, la interfaz áspera desaparecerá a medida que avanza de crecimiento, y el cristal eventualmente tomar una forma poliédrica limitada por una interfaz suave solo, a menos que se suprime la tasa de crecimiento de la interfaz áspera debido a las condiciones ambientales. Puesto que el crecimiento de un cristal poliédrico es controlado por el mecanismo de crecimiento de espiral, la tasa de crecimiento normal de la cara está determinada por la altura, la separación, y la tasa de avance de los pasos de la espiral de crecimiento. Los factores que determinan estas tres características, por lo tanto el control de la Tracht y el habitus de cristales poliédricos. Esto se analizará en el capítulo 4.
Tasa
de
crec
imie
nto R
Nucleación bidimensional
Áspero
Fuerza motriz
Espiral
Liso

Cuando un cristal crece bajo condiciones no controladas, como en el crecimiento de cristales de minerales naturales, la nucleación se produce primero bajo condiciones de alta fuerza motriz, que disminuirán a medida que avanza de crecimiento. Esta implihat un cristal que es originalmente dendríticas eventualmente tomar una forma poliédrica limitada por una cara plana. Si un cristal poliédrico limitado por caras planas es atravesada, se observará un esqueleto que muestra la forma dendrítica en el interior. Los cambios abruptos en las condiciones de conducción de fuerza se producen con frecuencia en la cristalización natural, como en la elevación de magma, y por lo tanto esperan encontrar cristales poliédricos recubiertos de una textura fibrosa debido al crecimiento dendrítico, o cristales que muestran varios cambios en la sección transversal. La figura 3.22 presenta un resumen de estas texturas internas.
Figura 3.22. Texturas registradas dentro de un solo cristal debido a cambios en las condiciones de crecimiento.
3.13 Morphodroms
Hemos discutido en la Sección 3.12 de que existe una relación mutua entre esferulıtica, dendrítica, tolva, y los cristales poliédricos, con respecto a la fuerza impulsora. Vamos a ver cómo aparecen estas relaciones mutuas en sistemas reales, utilizando como ejemplos

representativos, cristales de baja temperatura de nieve (de crecimiento en fase de vapor) y cristales de silicato de alta temperatura que crecen en las fases de solución de silicato.
Figura 3.23. Morphodrom de cristales de nieve: Diagrama de Nakaya [22].
El diagrama de fase morfológica en el que las variaciones morfológicas de un cristal se ilustran en relación con las condiciones de crecimiento se denomina "morphodrom". Esta palabra fue usada originalmente por Kern et al. para ilustrar las variaciones de Tracht y Habitus de cristales iónicos poliédricas que crecen en solución acuosa (ver ref. [29], en el capítulo 4), pero, de hecho, existen ejemplos mucho más antiguas que muestran la variación morfológica en la forma de un diagrama de fase morfológica. El ejemplo más antiguo es famoso diagrama de Nakaya de cristales de nieve [22]; Nakaya representa la morfología de los cristales de nieve artificial en relación con sobreenfriamiento y sobresaturación de H2O. Diagrama de Nakaya se muestra en la Fig. 3.23. Se ve claramente en este diagrama que los cristales de nieve adquieren una morfología poliédrica en la región de baja sobresaturación de H2O, mientras que asumen una morfología dendrítica en la región de la fuerza de conducción elevada. Un análisis de la razón por la cual los cristales de nieve poliédricos cambian su habitus alternativamente de platy a prismática dependiendo del grado de sobreenfriamiento se dará en el Capítulo 4. El diagrama Nakaya muestra claramente que la
Dendrítica
Sector y la placa
Placa gruesa
Placa espacial
Aguja
Aguja irregular
Columna
Desplazamiento
Sobr
esat
uram
ient
o (%
)
Temperatura (0C)
Temperatura (0C)

morfología de los cristales de nieve cambios de poliédrico a dendríticas como la sobresaturación ( motor) aumenta.
Figura 3.24. Morphodroms de minerales de silicato [23]. (A) resultados resumidos en muestras enfriadas: anortita serie (An) -albita (Ab). (B) la observación Resumido obtenida por método in situ: diópsido sistema (Di) -anortita (An).
Las figuras 3.24 (a) y (b) muestran morphodroms de cristales de silicato que crecen de soluciones de silicato: Fig. 3.24 (a) muestra los resultados de las observaciones de los productos enfriados, y la fig. 3.24 (b) resume los resultados obtenidos por una alta temperatura en el método de la observación in situ de crecimiento [23]. En estos casos también, ya que aparte de la licuefacción de componente solución sólida, se ve que la morfología de los cristales de cambios de poliédrico, a través de la tolva, a dendrítica, a continuación, esferulıtica. Las predicciones que se describen en la Sección 3.12 de este modo se confirmaron mediante experimentos.
PoliédricoTolvaDendríticaEsferulitica
(0C)
% molar de An(b)
% en peso de An(a)
(0C)

3.13 Elemento de partición
Cuando los átomos de impurezas o iones están presentes en una fase ambiente, se comportan de manera diferente a partir del componente de cristalización al llegar a las interfaces.
¿Hasta qué punto estos componentes de impureza se incorporan en el cristal en crecimiento serán diferentes dependiendo de las condiciones termodinámicas, es decir, la temperatura y la presión, así como en las propiedades químicas de cristal como el tamaño iónico y propiedades y el tamaño espacial de posiciones de sustitución en el cristal, y también será diferente en función de la tasa de crecimiento y direcciones cristalográficas. Los átomos y iones que son difíciles de incorporar en un cristal en crecimiento serán eliminadas de cristal y se acumulan en la interfaz completamente cuando la tasa de crecimiento es lento, pero a tasas de crecimiento más altas algunos serán encerradas por el cristal en crecimiento. Dado que la tasa de crecimiento es dependiente de la estructura de la interfaz, y por lo tanto sobre el mecanismo de crecimiento, la concentración de componentes de impureza que se incorporan en el cristal será diferente dependiendo de la dirección cristalográfica, es decir, sus contenidos serán diferentes dependiendo de los sectores de crecimiento (véase la Sección 6.1).
Para qué lado (fase ambiente o cristal) y en qué medida los componentes de impureza concentrado es llamado el "distribución (partición) del elemento", y la relación está representada por la distribución (partición) de coeficientes. Si las concentraciones del componente i impureza en la fase ambiente y en el cristal se expresan como cL y cS, respectivamente, la relación cL/cS = k es el coeficiente de distribución. También se llama el "coeficiente de segregación", k se determina por parámetros termodinámicos tales como la temperatura y la presión, pero no es el mismo que el k calculado en condiciones de equilibrio, donde la tasa de crecimiento es cero (esto k se expresa como k0). Podemos definir el coeficiente de distribución durante el crecimiento como el coeficiente de distribución eficaz, keff. Elementos de impurezas con keff < 1 son aquellos elementos que son difíciles de incorporar en el cristal en esta relación, y así se barren hacia fuera para y se concentran en la interfase; elementos con keff > 1 se comportan de la manera opuesta.
Elementos con keff < 1 que son barridas a la interfaz forman una capa concentrada con un gradiente que depende de la tasa de crecimiento, R, si suponemos una capa de difusión con un espesor δ. Bajo la condición de equilibrio donde R = 0, no hay gradiente, pero al enfriamiento rápido, con R = ∞, el gradiente es la más empinada, y en un finito R el gradiente es intermedio. En R = ∞, concentraciones de impurezas son iguales en la fase ambiente y en el cristal, y keff = 1, mientras que en condiciones de equilibrio, donde R = 0, keff = k0. Suponiendo la constante de difusión de una impureza sea D,
Burton, Prim, y Slichter [24] expresaron keff por la siguiente fórmula:

Esta es la famosa ecuación de BPS; muestra que keff depende de R, pero no lo hace muestran que depende de la orientación.
El primer ejemplo que demostró que elemento de partición tiene la dependencia de la orientación fue la observación de la distribución de átomos de impurezas Bi en un cristal único de Si crecido a partir de la masa fundida. Los átomos de Bi se distribuyen principalmente en el sector de crecimiento interfaz suave. Esto se explicó cómo debido a una tasa de avance mucho más grande de pasos de crecimiento en una interfaz suave que el R tasa de crecimiento normal de una interfaz áspera. Si la tasa de avance es rápido, las impurezas que llegan a la interfaz serán capturados por las etapas de crecimiento de avance antes de que sean arrastrados a la fase ambiente. Si extendemos este concepto, se predice que el particionado impureza dependerá de la orientación cristalográfica, y que los sectores de crecimiento de una cara con la más alta importancia morfológica contendrán más impurezas con keff < 1. Distribución impureza en un cristal único no puede ser homogénea y dependerá de los sectores de crecimiento.
Elementos de impurezas con keff < 1 se concentrarán en la interfaz. Esto afecta a la estabilidad morfológica de la interfaz a través de la tasa de crecimiento y los fenómenos de sobreenfriamiento constitucionales, y puede modificar la suavidad de la interfaz. Como resultado, las fluctuaciones en la concentración de impurezas van a aparecer en forma de bandas de crecimiento.
3.15 Inclusiones
Cristalitos muy pequeñas de los mismos o diferentes especies pueden precipitar sobre la superficie de crecimiento de un cristal. Si estos son expulsados de ocluidos o en el cristal en crecimiento se determina por la relación entre las fuerzas de atracción y repulsión con el cristal en crecimiento, de manera similar a la partición de elementos de impurezas. A tasas de crecimiento más altas, es más probable que los precipitados se ocluidos en el cristal. Se convierten en inclusiones sólidas, que sirven como buenos indicadores de la composición química de la ambiente y condiciones de fase de crecimiento, la temperatura y la presión. Inclusiones sólidas en cristales de diamante naturales han sido ampliamente investigado, de la cual dos tipos de fases ambiente, ultramaficas y eclogiticos, han sido consideradas como las fases probables ambiente en el que se formaron los diamantes.
Cuando la fase de ambiente es una fase de solución de baja viscosidad, tal como una solución hidrotérmica, la oclusión del líquido madre (el líquido en el que el cristal crece) en un cristal en crecimiento se produce en los lugares donde las ramas dendríticas conjugado, o a través de voladizo de macro- etapas de crecimiento, o en lugares donde avanzan las macro-etapas cumplan. Cuando el crecimiento procede aún más en la pared de una inclusión líquido madre de acogida, que se disociará en inclusiones más pequeñas
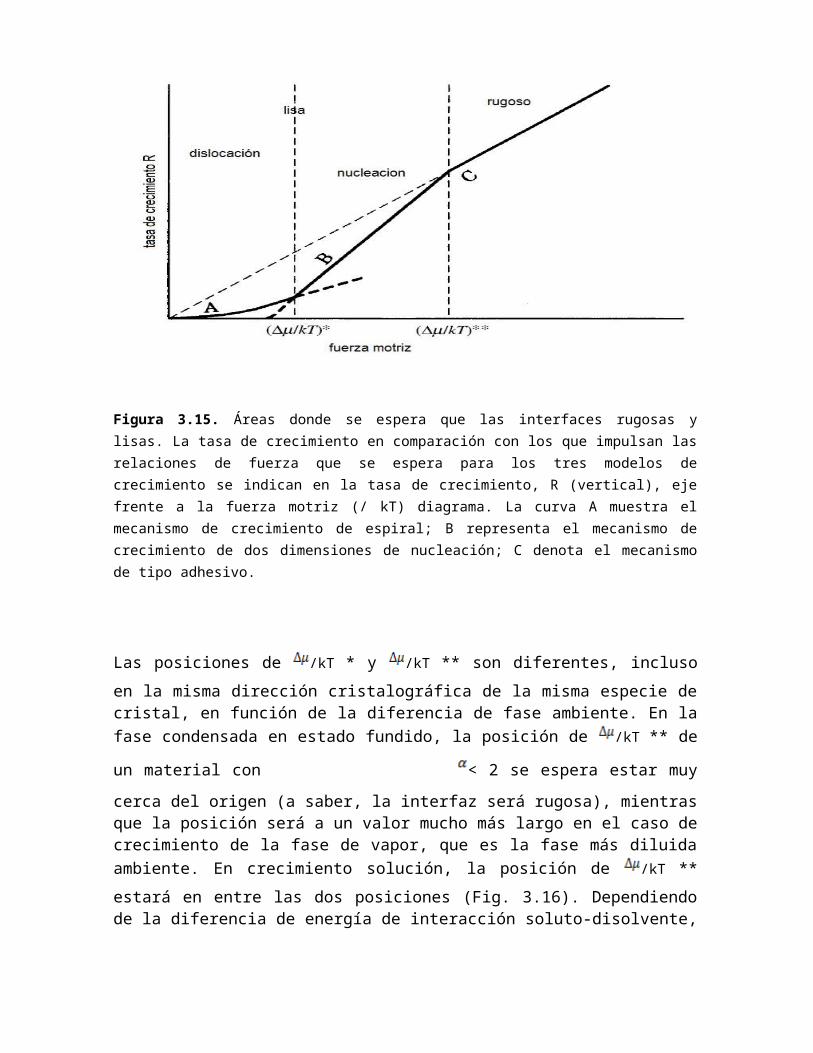
dispuestas por estricción, o se convertirá en un cristal negativo delimitada por caras cristalográficas.


















