
Cinética Química (Química 2º de bachillerato)
56
Ciné%ca química Química 2º de bachillerato Francisco José Navarro Rodríguez
-
Upload
frodriguez30 -
Category
Documents
-
view
320 -
download
15
description
Libro digital de la unidad. Presenta contenidos y ejemplos numéricos y conceptuales resueltos.
Transcript of Cinética Química (Química 2º de bachillerato)
Ciné%ca química
Química 2º de bachillerato
Francisco José Navarro
Rodríguez

Ejemplos de reacciones químicas con diferente velocidad
• Formación de estalac%tas o estalagmitas en cuevas. • Oxidación de hierro • Destrucción de la capa de ozono • Envejecimiento celular • Descomposición de los alimentos
Muy rápida (explosiva) a temperatura ambiente: Na (s) + H
2O(l) →NaOH(aq) + 1⁄2H
2(g)
Muy lenta a temperatura ambiente H2(g)+I
2(g)→2HI(g)
Muy lenta a tª ambiente y muy rápido a 500 °C H2(g) +1⁄2 O
2(g) → H
2O (l) ∆G°=-‐236kJ

Hipótesis
1. ¿Existen diferentes velocidades? 2. ¿Afectarán las magnitudes termoquímicas a la
velocidad de la reacción química? 3. ¿Conocemos sus mecanismos de reacción? 4. ¿Podemos controlar estos mecanismos de reacción? 5. ¿Podemos acelerar o frenarlas? 6. ¿Qué ocurre a escala molecular para que los reac%vos
se transformen en productos? 7. …

• Cuando conocemos: – Ajuste estequiométrico que nos permite realizar un balance de materia (o balance de masa)
– Balance energé%co, que nos permite saber: • ∆H energía intercambiada (calor a P cte) • ∆S variación en el grado de desorden • ∆G revela porqué el proceso ocurre espontáneamente en un determinado sen%do y no en otro
– Ciné%ca de la reacción, es decir su velocidad y su mecanismo. Podremos así mejorar las condiciones de trabajo que permitan (o no) que ese proceso tenga lugar. (Ej. Conservación de alimentos)

La espontaneidad es independiente de su velocidad
• C(s)+O2(g)CO2(g) – Espontánea – Afortunadamente muy lenta (inapreciable), y gracias a este hecho, los diamantes no se convierten en CO2
• C(diamante)C(grafito) – Espontánea: todos los diamantes acabarán como grafito.
– Extremadamente lento, hasta ser inapreciable

2. Estudio dinámico de las r.q. La velocidad de una reacción química se define como el cambio en la concentración de los reac%vos (o productos) con respecto al %empo en dicho proceso.
aA+bBcC+dD
2H2(g)+02(g)2H2O(g) Por cada mol de O2 que reacciona (desaparece), desaparecen 2 moles de H2 y se formarán 2 moles de H2O.
Por cada “a” moles de A que reaccionan (desaparecen), también desaparecen “b” moles de B y se formarán “c” moles de C “d” moles de D
€
nAo − nAa
=nBo − nB
b=nCc
=nDd
nAo= moles iniciales de A nA= moles de A en el instante t nAo-‐nA= moles que han reaccionado.
€
vH2= 2v02 = vH2 0
→12vH2
= v02 =12vH2 0
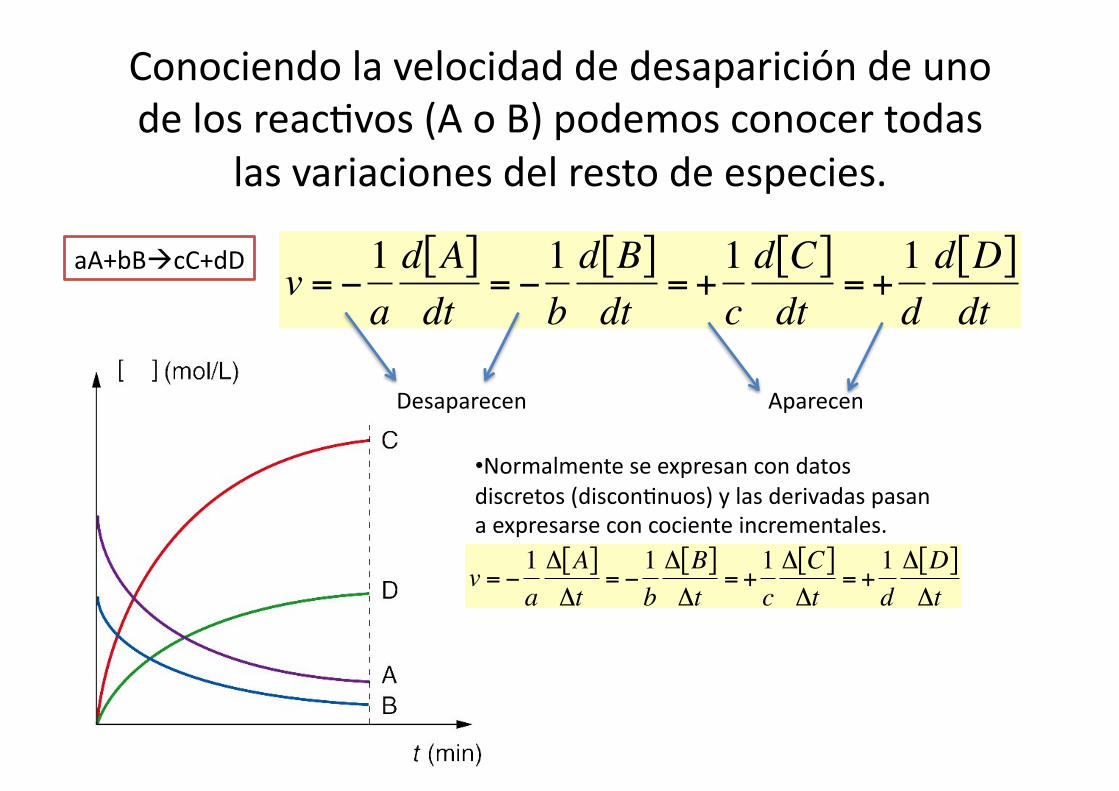
Conociendo la velocidad de desaparición de uno de los reac%vos (A o B) podemos conocer todas
las variaciones del resto de especies.
aA+bBcC+dD
€
v = −1ad A[ ]dt
= −1bd B[ ]dt
= +1cd C[ ]dt
= +1dd D[ ]dt
Desaparecen Aparecen
• Normalmente se expresan con datos discretos (discon%nuos) y las derivadas pasan a expresarse con cociente incrementales.
€
v = −1aΔ A[ ]Δt
= −1bΔ B[ ]Δt
= +1cΔ C[ ]Δt
= +1dΔ D[ ]Δt
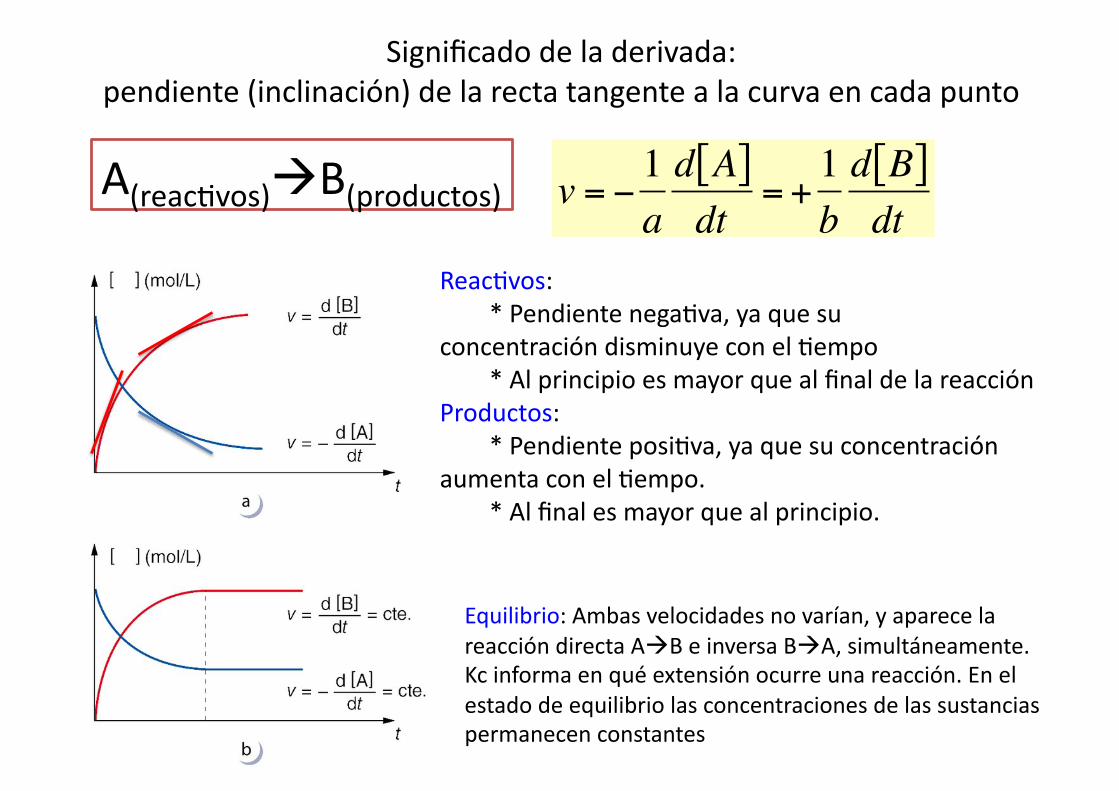
€
v = −1ad A[ ]dt
= +1bd B[ ]dt
Significado de la derivada: pendiente (inclinación) de la recta tangente a la curva en cada punto
Reac%vos: * Pendiente nega%va, ya que su concentración disminuye con el %empo * Al principio es mayor que al final de la reacción Productos: * Pendiente posi%va, ya que su concentración aumenta con el %empo. * Al final es mayor que al principio.
A(reac%vos)B(productos)
Equilibrio: Ambas velocidades no varían, y aparece la reacción directa AB e inversa BA, simultáneamente. Kc informa en qué extensión ocurre una reacción. En el estado de equilibrio las concentraciones de las sustancias permanecen constantes

Ejemplo 1 : En la reacción 2A+B 3C+D, par%mos de 1 mol de A y 1 mol de B en un recipiente de 1 litro. Al cabo de 10 minutos, tenemos 0,9 moles de B. Calcular la velocidad de esta reacción respecto a A, B, C y D
€
v = −12Δ A[ ]Δt
= −Δ B[ ]Δt
= +13Δ C[ ]Δt
= +Δ D[ ]Δt
€
nBo − nB1
=nC3
= 0,1→ nC = 0,3
A los 10 minutos tendremos 0,9 moles de B, y sólo 0,8 de A (desaparece el doble de rápido). También tendremos 0,3 moles de C y 0,1 mol de D
€
nBo − nB1
=1− 0,9 = 0,1
€
nAo − nA2
= 0,1→ nAo − nA = 0,2→ nA = 0,8€
nAo − nA2
=nBo − nB1
=nC3
=nD1
€
nBo − nB1
=nD1
= 0,1→ nD = 0,1
Especies (moles)
Tiempo (min) Variación moles Δn 0 10
nA 1 1-‐0,1x2=0,8 -‐0,2
nB 1 0,9 -‐0,1
nC 0 0+0,1x3=0,3 +0,3
nD 0 0+0,1=0,1 +0,1
En 10 minutos, desaparecen 0,2 mol de A, desaparecen 0,1 mol de B, y aparecen 0,3 mol de C y aparecen 0,1 mol de D
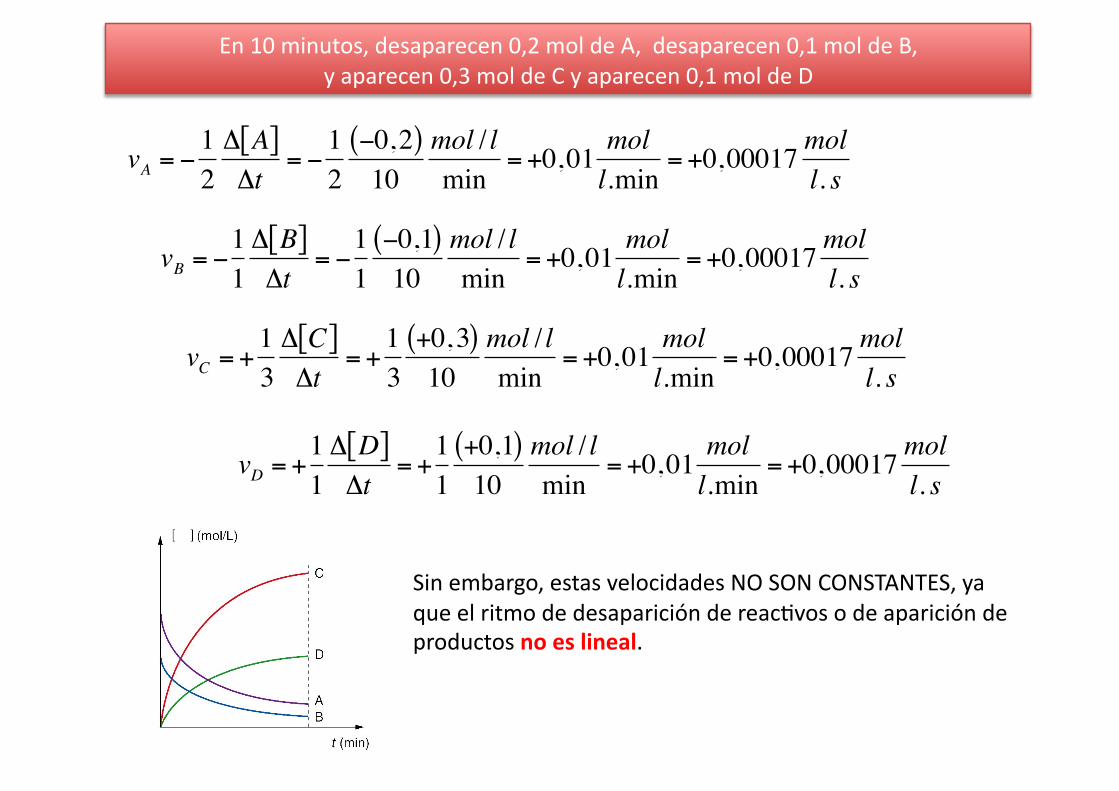
En 10 minutos, desaparecen 0,2 mol de A, desaparecen 0,1 mol de B, y aparecen 0,3 mol de C y aparecen 0,1 mol de D
€
vA = −12Δ A[ ]Δt
= −12−0,2( )10
mol / lmin
= +0,01 moll.min
= +0,00017 moll. s
€
vB = −11Δ B[ ]Δt
= −11−0,1( )10
mol / lmin
= +0,01 moll.min
= +0,00017 moll. s
€
vC = +13Δ C[ ]Δt
= +13
+0,3( )10
mol / lmin
= +0,01 moll.min
= +0,00017 moll. s
€
vD = +11Δ D[ ]Δt
= +11
+0,1( )10
mol / lmin
= +0,01 moll.min
= +0,00017 moll. s
Sin embargo, estas velocidades NO SON CONSTANTES, ya que el ritmo de desaparición de reac%vos o de aparición de productos no es lineal.

A + B 2C + D Ejemplo 2

3. Ecuación ciné%ca (ecuación de velocidad o ley de velocidad o ley ciné%ca)
€
v = f (concentración, temperatura, catalizador)Reacciones HOMOGÉNEAS (UNA SOLA FASE)
EXPERIMENTALMENTE SE COMPRUEBA QUE: 1. La velocidad disminuye con el %empo, debido a la disminución de la concentración de los
reac%vos. Debemos por tanto relacionar v con la [ ] de los reac%vos. 2. Está afectada por la temperatura de la reacción (suelen aumentar con la temperatura) 3. Aumenta o disminuye con la presencia / ausencia de catalizadores.
Reacción general: aA+bBproductos Ecuación ciné%ca:
v=k [A]α[B]β
k= constante de velocidad (unidades variables) v= velocidad de reacción (mol/L.s) α= orden de reacción respecto de A β= orden de reacción respecto de B
α+β= Orden global de reacción

¿Cómo se determina el orden de una reacción química?
Método analí%co • α y β, no %enen NADA que ver con los coeficientes
estequiométricos. • α se determina manteniendo constante la [B] y
variamos [A] v=k´[A]α, siendo k´=k[B]β • Tomando logaritmos: log v=log k´+α.log[A]. • Representando log v frente a log[A], se ob%ene la
pendiente α • β se calcula igual, y de este modo el orden (α+β) Método por tanteo • Mantenemos la [B] y duplicamos [A]...
– Si la v se hace doble, significa que α=1 – Si v se cuadruplica, entonces α=2
v=k [A]α[B]β

Ejemplo 3 Determina los órdenes de reacción total y parciales de las reacciones anteriores: H2 (g) + I2 (g) → 2 HI (g) v = k ·∙ [H2] ·∙ [I2] H2 (g) + Br2 (g) → 2 HBr (g) v = k ·∙ [H2] ·∙ [Br2]1/2
H2 (g) + I2 (g) → 2 HI (g) v = k ·∙ [H2] ·∙ [I2] – Reacción de segundo orden (1 + 1) – De primer orden respecto al H2 y de primer orden respecto al I2.
H2 (g) + Br2 (g) → 2 HBr (g) v = k ·∙ [H2] ·∙ [Br2] ½
– Reacción de orden 3/2 (1 + ½) – De primer orden respecto al H2 y de orden ½ respecto al Br2.
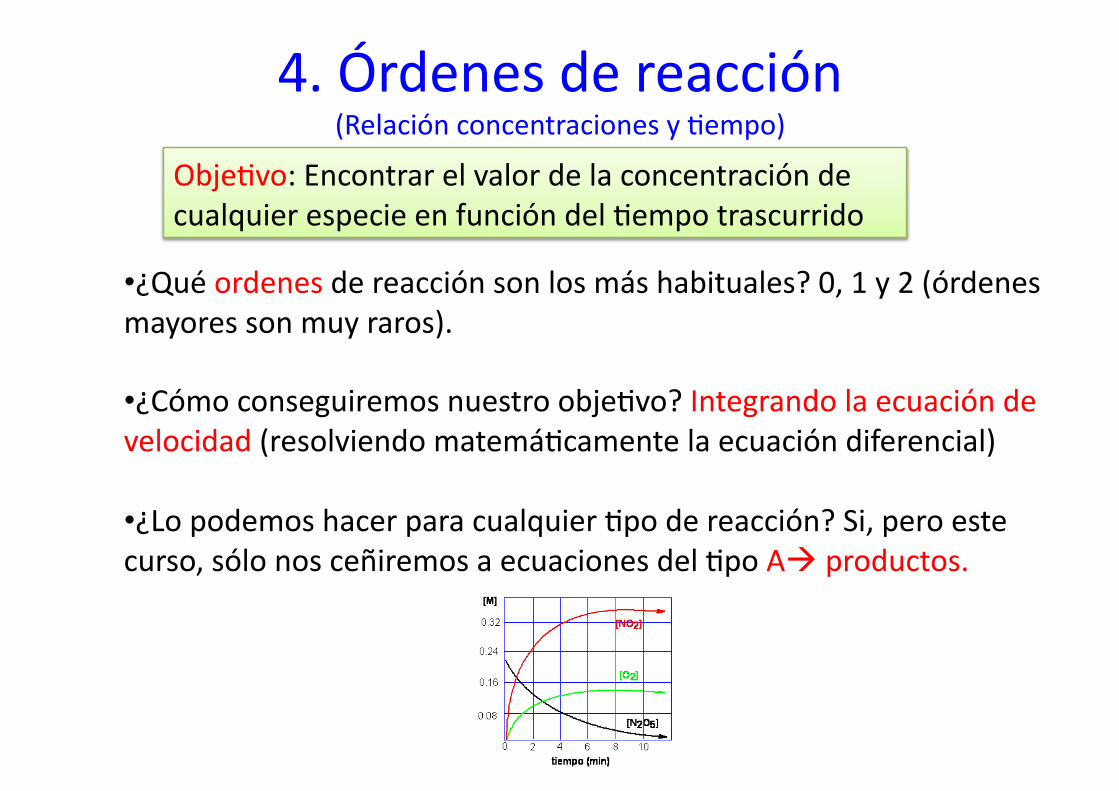
4. Órdenes de reacción (Relación concentraciones y %empo)
Obje%vo: Encontrar el valor de la concentración de cualquier especie en función del %empo trascurrido
• ¿Qué ordenes de reacción son los más habituales? 0, 1 y 2 (órdenes mayores son muy raros).
• ¿Cómo conseguiremos nuestro obje%vo? Integrando la ecuación de velocidad (resolviendo matemá%camente la ecuación diferencial)
• ¿Lo podemos hacer para cualquier %po de reacción? Si, pero este curso, sólo nos ceñiremos a ecuaciones del %po A productos.
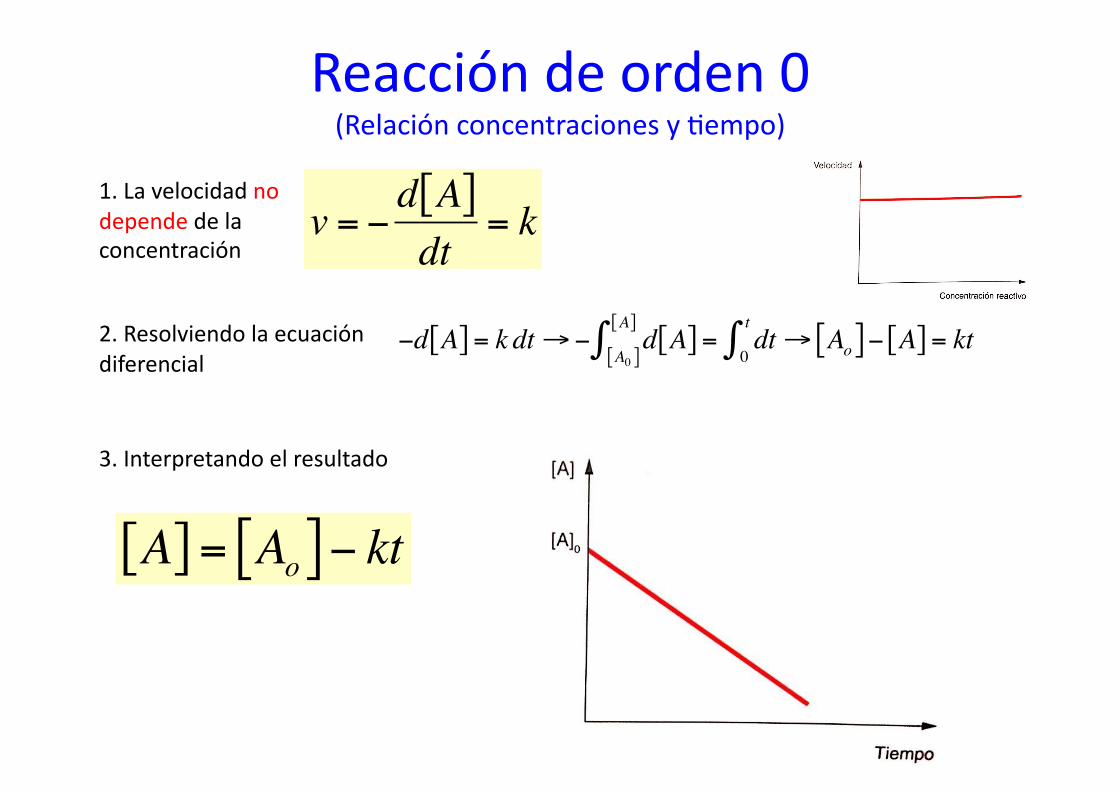
Reacción de orden 0 (Relación concentraciones y %empo)
1. La velocidad no depende de la concentración
€
v = −d A[ ]dt
= k
2. Resolviendo la ecuación diferencial
3. Interpretando el resultado
€
A[ ] = Ao[ ]− kt€
−d A[ ] = k dt→− d A[ ] = dt→ Ao[ ]− A[ ] = kt0
t∫A0[ ]
A[ ]∫
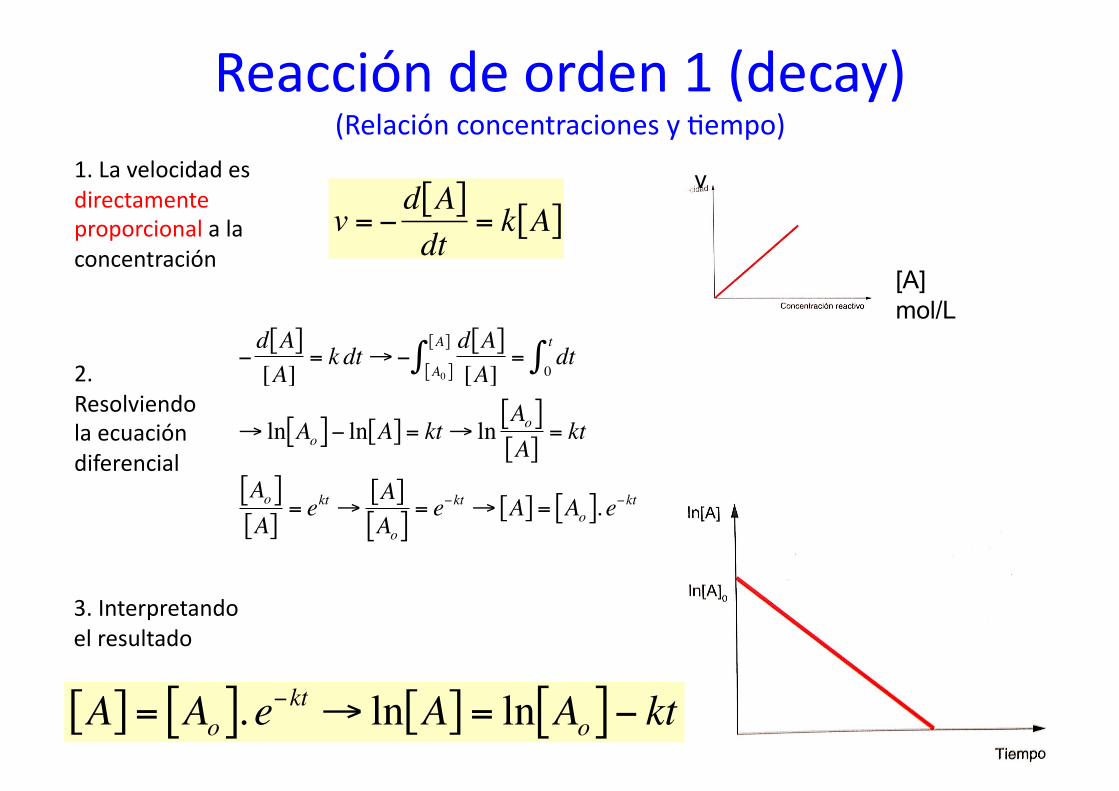
Reacción de orden 1 (decay) (Relación concentraciones y %empo)
1. La velocidad es directamente proporcional a la concentración
€
v = −d A[ ]dt
= k A[ ]
2. Resolviendo la ecuación diferencial
€
3. Interpretando el resultado
€
A[ ] = Ao[ ]. e−kt → ln A[ ] = ln Ao[ ]− kt
€
−d A[ ][A]
= k dt→−d A[ ][A]
= dt0
t∫A0[ ]
A[ ]∫
→ ln Ao[ ]− ln A[ ] = kt→ lnAo[ ]A[ ]
= kt
Ao[ ]A[ ]
= ekt → A[ ]Ao[ ]
= e−kt → A[ ] = Ao[ ]. e−kt
v
[A] mol/L
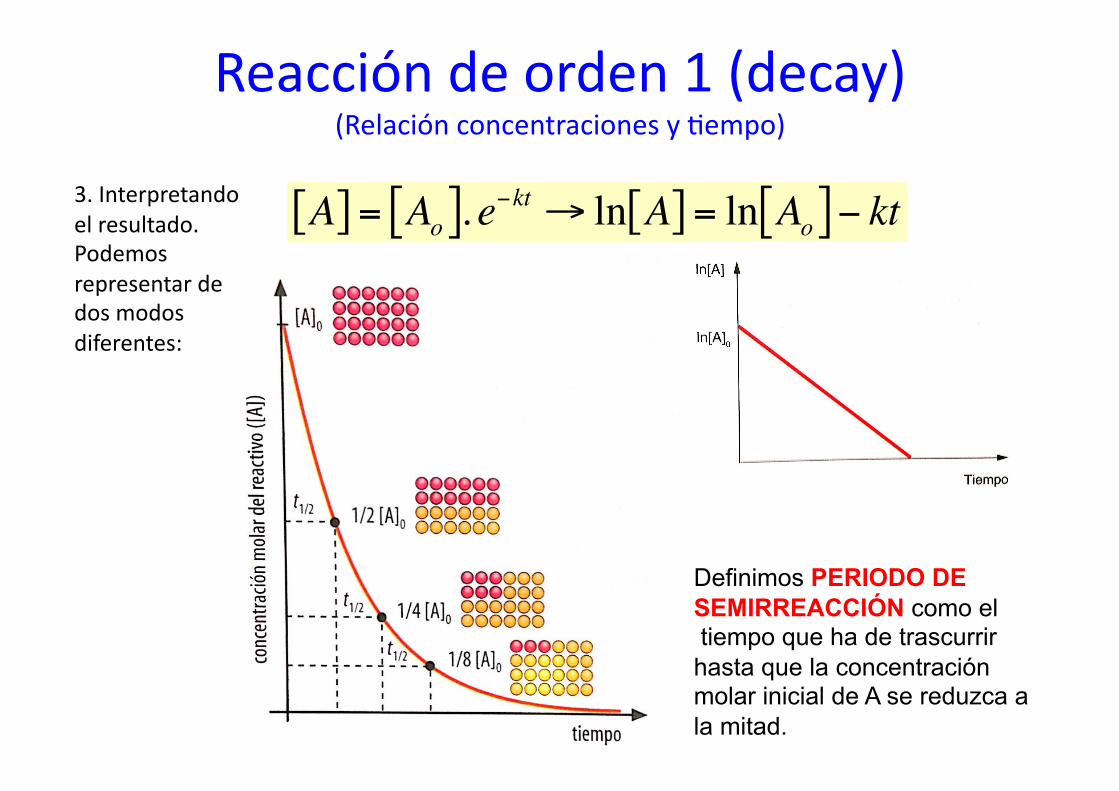
Reacción de orden 1 (decay) (Relación concentraciones y %empo)
3. Interpretando el resultado. Podemos representar de dos modos diferentes:
€
A[ ] = Ao[ ]. e−kt → ln A[ ] = ln Ao[ ]− kt
Definimos PERIODO DE SEMIRREACCIÓN como el tiempo que ha de trascurrir hasta que la concentración molar inicial de A se reduzca a la mitad.
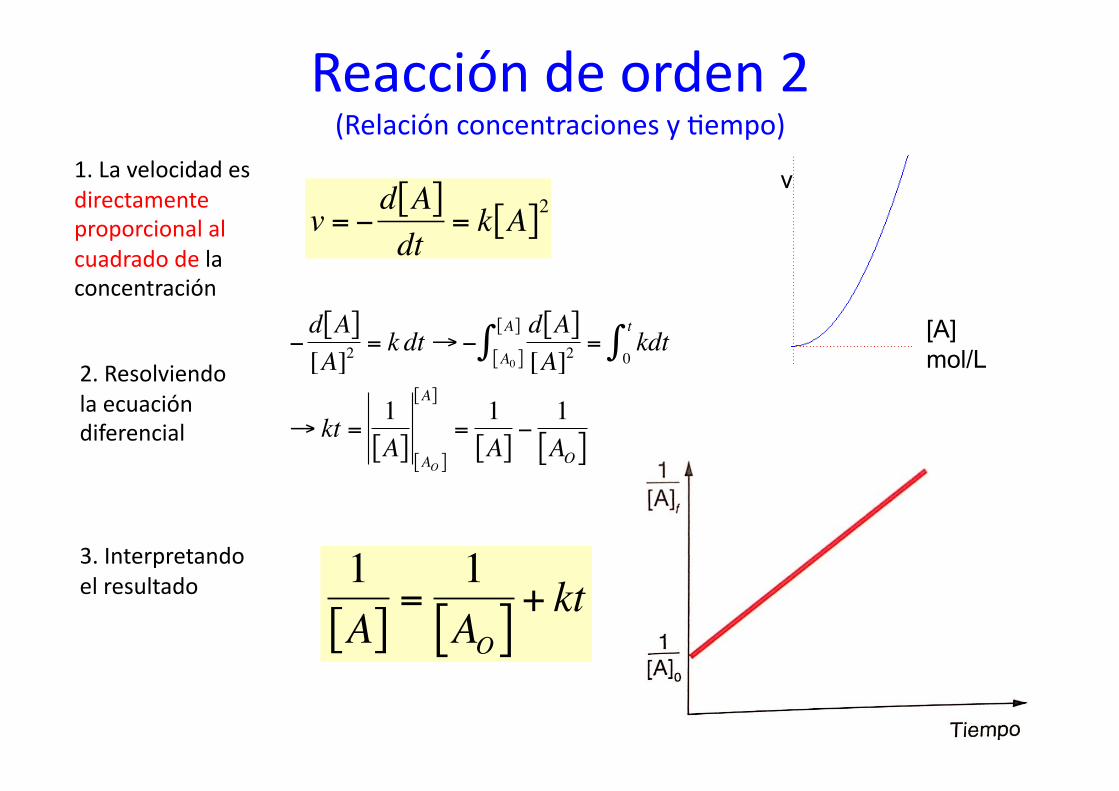
Reacción de orden 2 (Relación concentraciones y %empo)
1. La velocidad es directamente proporcional al cuadrado de la concentración
€
v = −d A[ ]dt
= k A[ ]2
2. Resolviendo la ecuación diferencial
€ 3. Interpretando el resultado
€
1A[ ]
=1AO[ ]
+ kt€
−d A[ ][A]2
= k dt→−d A[ ][A]2
= kdt0
t∫A0[ ]
A[ ]∫
→ kt =1A[ ] AO[ ]
A[ ]
=1A[ ]−1AO[ ]
v
[A] mol/L

Ejemplo 4 : Determinar el orden de reacción : CH3-‐Cl (g) + H2O (g) → CH3-‐OH (g) + HCl (g) usando los datos de la tabla.
Tanteo
v = k ·∙ [CH3-‐Cl ]n ·∙ [H2O]m
En las experiencias 1 y 2 vemos que no cambia [H2O] luego el cambio de “v” se debe al cambio de [CH3-‐Cl ]. Como al doblar [CH3-‐Cl] se dobla la velocidad podemos deducir que el orden de reacción respecto del CH3-‐Cl es “n=1”.
En las experiencias 1 y 3 vemos que no cambia [CH3-‐Cl] luego el cambio de “v” se debe al cambio de [H2O]. Como al doblar [H2O] se cuadruplica la velocidad podemos deducir que el orden de reacción respecto del H2O es “m=2”.
v = k ·∙ [CH3-‐Cl ] ·∙ [H2O]2
Y el orden total de la reacción es “n+m=3”. El valor de “k” se calcula, despejando k, a par%r de cualquier experiencia y resulta 181’4 mol–2l2s –1.

Analí+camente (método general)
También puede calcularse usando logaritmos:
log v = log k + n ·∙ log [CH3-‐Cl ] + m ·∙ log [H2O]
Aplicamos dicha expresión a cada experimento: (1) log 2,83 = log k + n ·∙ log 0,25 M + m ·∙ log 0,25 M
(2) log 5,67 = log k + n ·∙ log 0,50 M + m ·∙ log 0,25 M (3) log 11,35 = log k + n ·∙ log 0,25 M + m ·∙ log 0,50 M
Si restamos dos ecuaciones en las que se mantenga constante uno de los reac%vos, podremos obtener el orden de reacción parcial del otro. Así, al restar (1) – (2) eliminamos “k” y [H2O] :
log (2,83/5,67) = n ·∙ log (0,25/0,50)
Análogamente restando (1) – (3) eliminamos “k” y [CH3-‐Cl]
log (2,83/11,35) = m ·∙ log (0,25/0,50) log (2,83/5,67) log (2,83/11,35)
n = —————— = 1 ; m = ——————— = 2 log (0,25/0,50) log (0,25/0,50)
v = k ·∙ [CH3-‐Cl ]n ·∙ [H2O]m


Ejemplo 6 : El oxido nítrico, NO, reacciona con hidrógeno formando óxido nitroso, N2O: 2NO(g) + H2(g) → N2O (g) + H2O (g). En una serie de experimentos se han obtenidos los siguientes resultados: Determinar la ecuación de la velocidad y calcular el valor de la constante de velocidad.
Por la simple inspección de los datos se puede ver que, cuando se duplica [H2], manteniendo constante [NO] (exper. 1ª y 2ª), la velocidad se hace también doble, es decir, que “v” es proporcional a [H2]1.
En cambio, cuando se man%ene constante [H2] y se duplica [NO] (exper. 1ª y 3ª), la velocidad se mul%plica por 4 (=22), es decir, que la “v” es proporcional a [NO]2. Por tanto, la ecuación de velocidad será:

2NO(g) + H2(g) → N2O (g) + H2O (g).

5. Mecanismos de reacción y molecularidad
Mecanismo de reacción: Secuencia de etapas elementales que explican el modo en que los reac%vos se transforman en los productos
• ¿Qué explican los mecanismos de reacción? • El hecho de que en reacciones con los mismos coeficientes estequiométricos (a, b, c, d…) no coincidan con los órdenes de reacción (α, β, γ…). • Las reacciones globales, en muchos casos, están formadas realmente por varias etapas sencillas
Reacciones elementales: Los órdenes de reacción coinciden con los coeficientes estequiométricos. Ejemplo: 1A+1Bproductos v=k[A]1[B]1

Molecularidad (sólo para reacciones elementales): Número de moléculas que intervienen en el proceso
1A+1B1AB v=k[A]1[B]1 Reacción elemental y molecularidad=2 (1 de A y 1 de B) Así hablamos de reacciones unimoleculares, bimoleculares, trimoleculares (raras), etc…
1A2+1B22 AB v=k[A2]1
NO es una reacción elemental ( está formada por etapas), y no se define la molecularidad
€
A2 → 2A*
A* + B2 → AB+ B*
A* + B* → AB
⎫
⎬ ⎪
⎭ ⎪
suma⎯ → ⎯ ⎯ A2 + B2 → 2AB
Para explicar la cinética de las reacciones NO elementales, suponemos una secuencia de etapas que sí lo son en la que se forman productos intermedios (*) no observados. Sólo podemos observar los reactivos y los productos.
v=k[A2]1 Etapa limitante, pues la ecuación cinética responde a ella
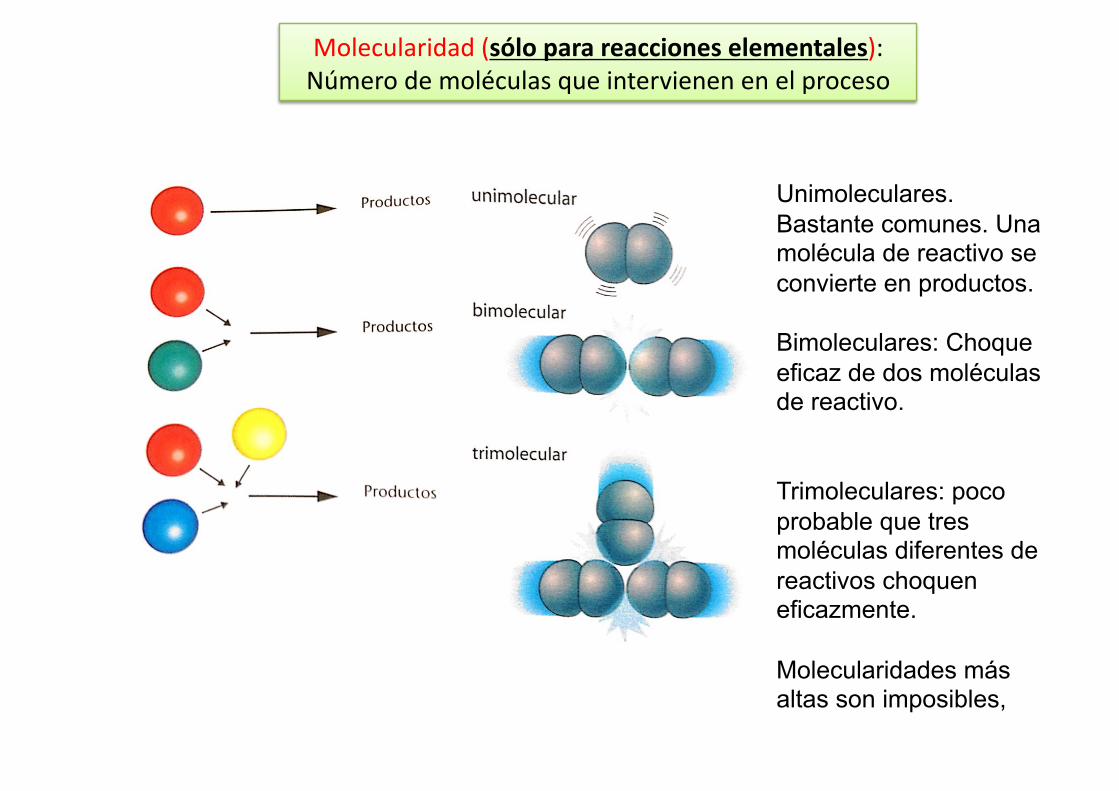
Unimoleculares. Bastante comunes. Una molécula de reactivo se convierte en productos.
Bimoleculares: Choque eficaz de dos moléculas de reactivo.
Trimoleculares: poco probable que tres moléculas diferentes de reactivos choquen eficazmente.
Molecularidades más altas son imposibles,
Molecularidad (sólo para reacciones elementales): Número de moléculas que intervienen en el proceso

Ejemplo de mecanismo de reacción La reacción
NO2 (g) + CO (g) → NO (g) + CO2 (g) sucede en dos etapas:
1ª etapa (lenta): 2 NO2 → NO + NO3
2ª etapa (rápida): NO3 + CO → NO2 + CO2
• La reacción global es la suma de las dos. • NO3 es un intermedio de reacción.
• La velocidad está condicionada por la etapa lenta. En nuestra ejemplo, en la etapa lenta intervienen dos moléculas de NO2,, luego v = k ·∙ [NO2]2
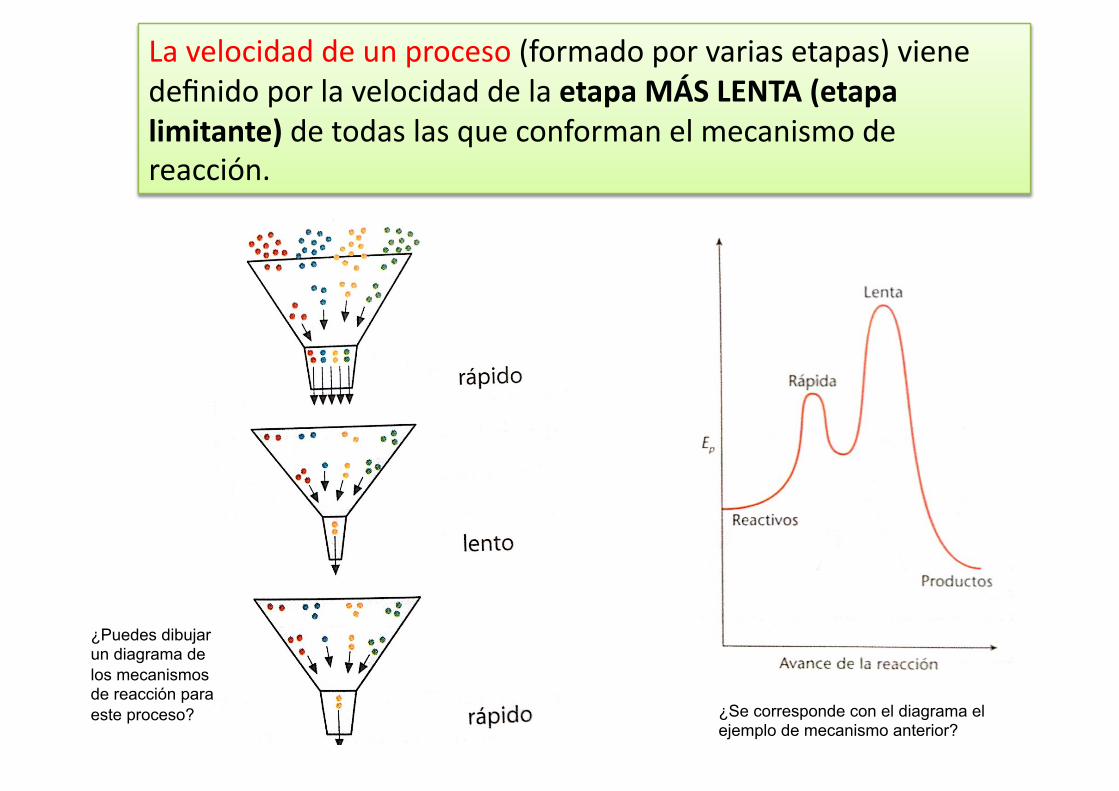
La velocidad de un proceso (formado por varias etapas) viene definido por la velocidad de la etapa MÁS LENTA (etapa limitante) de todas las que conforman el mecanismo de reacción.
¿Se corresponde con el diagrama el ejemplo de mecanismo anterior?
¿Puedes dibujar un diagrama de los mecanismos de reacción para este proceso?

6. Teorías de las reacciones químicas ¿Cómo conocemos los mecanismos de reacción? ¿Qué factores afectan a la velocidad de las r.q.?
Hechos: 1. En las rq “suele” producirse la rotura de enlaces en los reac%vos y formación de enlaces nuevos en los productos de reacción. La naturaleza de los enlaces químicos es un factor determinante. 2. El estado �sico (estado de agregación, grados de división..) afecta. 3. Otros factores: temperatura, catalizadores…
Reacción de formación de agua:
€
H +(aq) +OH −
(aq) → H2O(l )1. Instantánea en esta reacción de neutralización ácido-base
€
2H2(g) +O2(g) → H2O(l) 2. No se produce sin salto de una chispa
En la primera no hay que romper enlaces, y la velocidad es muy grande, mientras que en la segunda, que requiere romper enlaces y reagrupar para formar otros, la velocidad es mucho menor.

6a. Teorías de colisiones • Para que se produzca rq es preciso que los átomos o moléculas de
los reac%vos COLISIONES O CHOQUEN de modo eficaz, y para ello es preciso que se cumple que: – CONDICIÓN 1. Las moléculas de los reac%vos choquen con suficiente energía
como para romper sus enlaces. Estas moléculas se llaman moléculas ac%vadas y la energía mínima necesaria para el choque se llama energía de ac%vación Ea (Cuanta menor Ea, mayor velocidad de reacción).
En el caso a) la E>Ea y se produce la reacción. En el caso b), solo hay choque pero no aparecen sustancias nuevas y por tanto no se produce la reacción
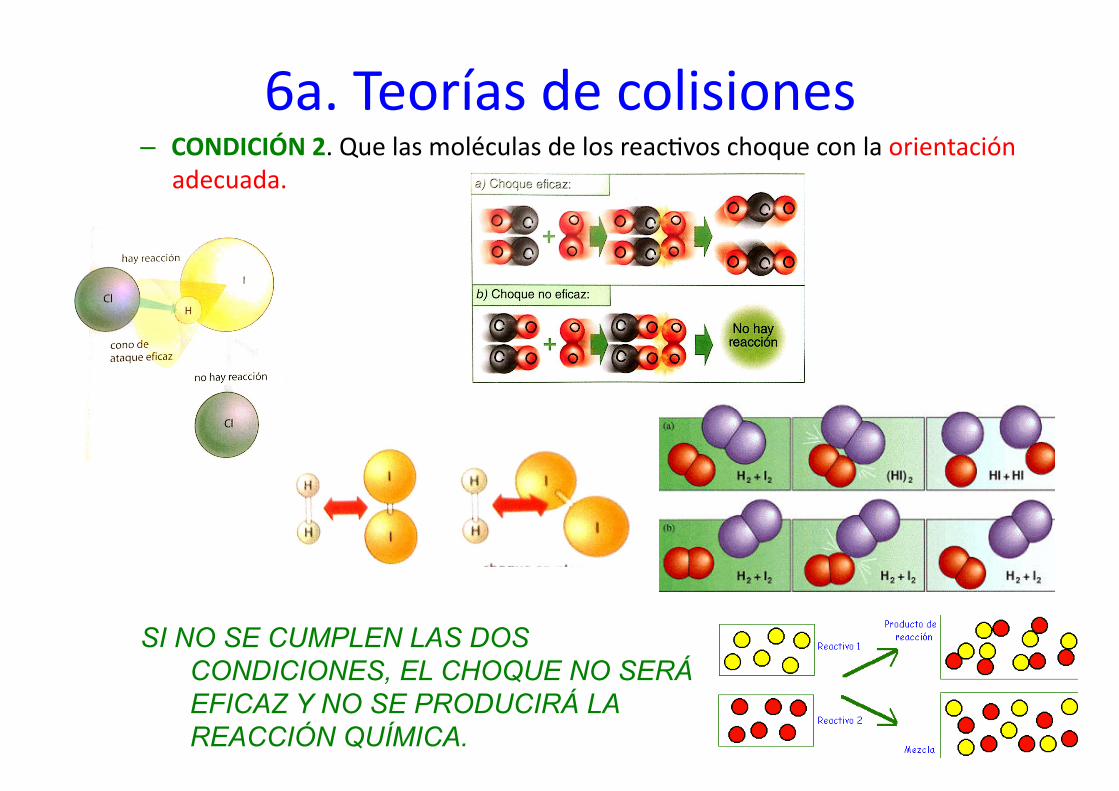
6a. Teorías de colisiones – CONDICIÓN 2. Que las moléculas de los reac%vos choque con la orientación
adecuada.
SI NO SE CUMPLEN LAS DOS CONDICIONES, EL CHOQUE NO SERÁ EFICAZ Y NO SE PRODUCIRÁ LA REACCIÓN QUÍMICA.
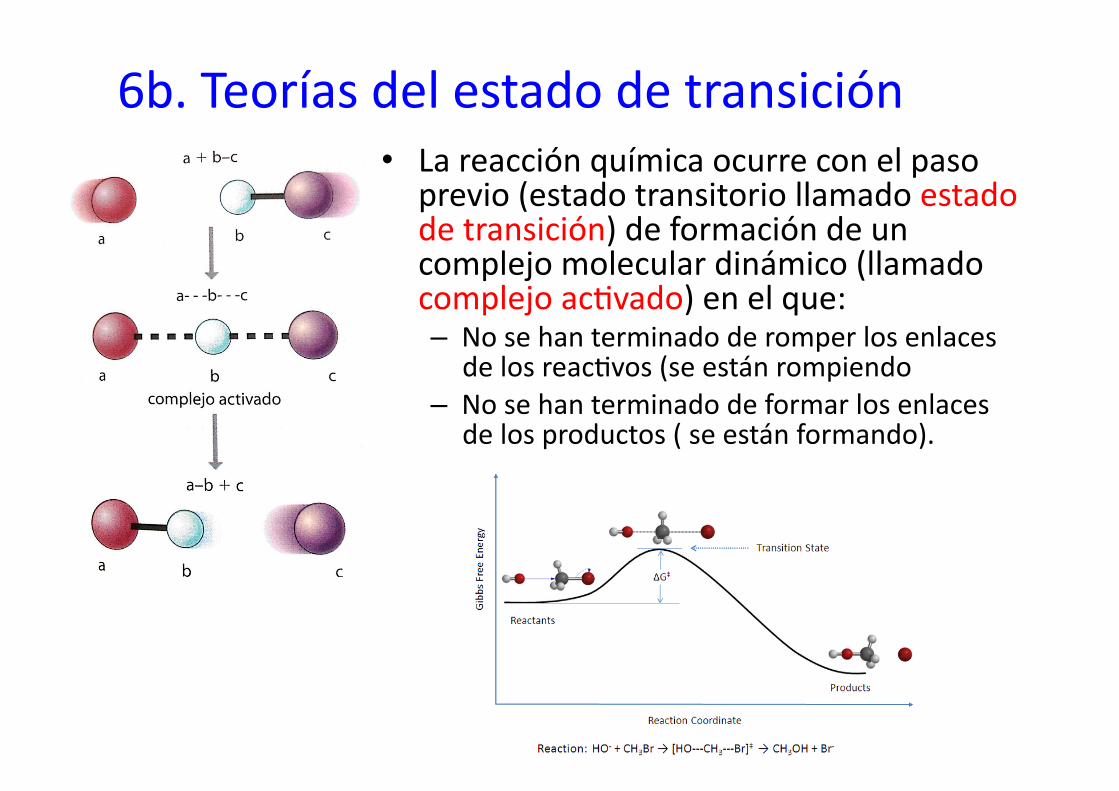
6b. Teorías del estado de transición • La reacción química ocurre con el paso
previo (estado transitorio llamado estado de transición) de formación de un complejo molecular dinámico (llamado complejo ac%vado) en el que: – No se han terminado de romper los enlaces
de los reac%vos (se están rompiendo – No se han terminado de formar los enlaces
de los productos ( se están formando).
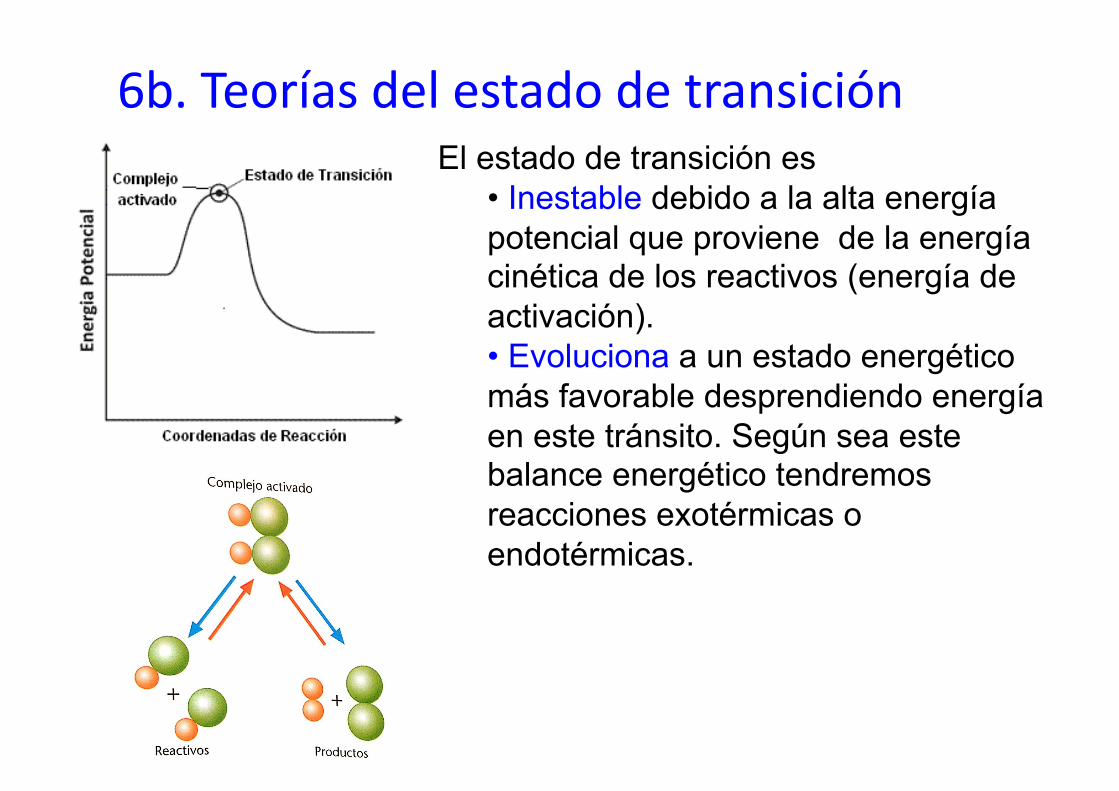
6b. Teorías del estado de transición El estado de transición es
• Inestable debido a la alta energía potencial que proviene de la energía cinética de los reactivos (energía de activación). • Evoluciona a un estado energético más favorable desprendiendo energía en este tránsito. Según sea este balance energético tendremos reacciones exotérmicas o endotérmicas.
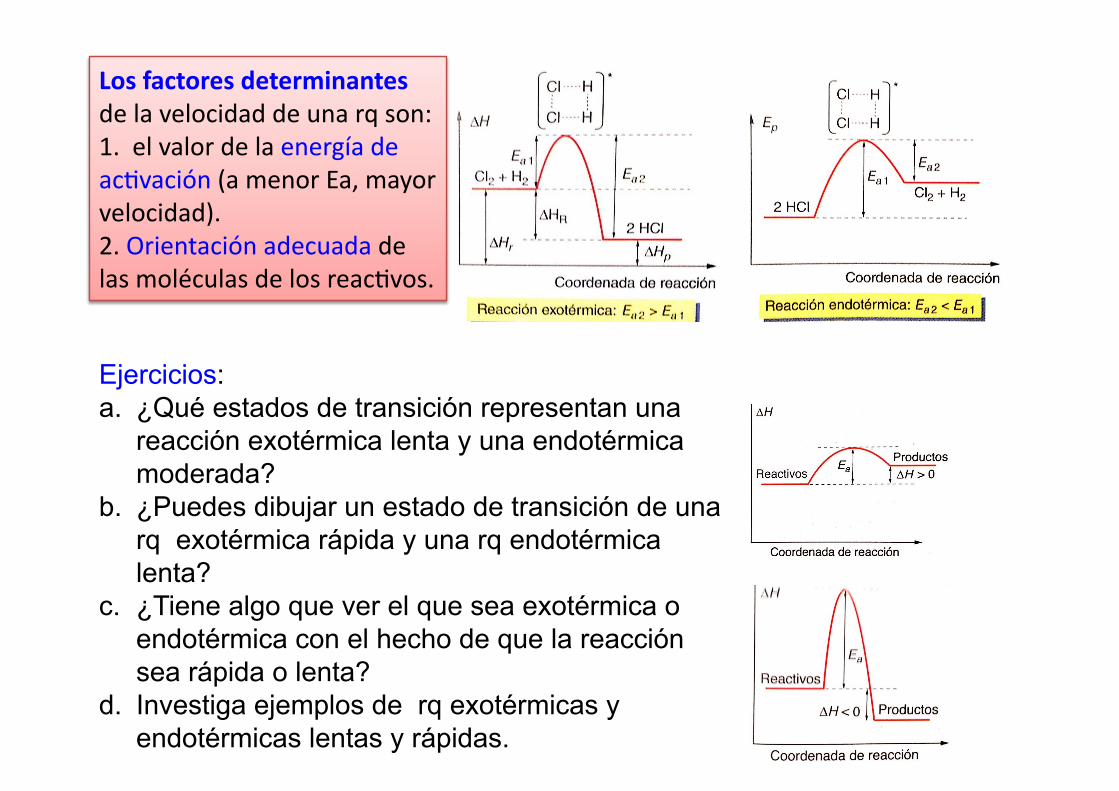
Los factores determinantes de la velocidad de una rq son: 1. el valor de la energía de ac%vación (a menor Ea, mayor velocidad). 2. Orientación adecuada de las moléculas de los reac%vos.
Ejercicios: a. ¿Qué estados de transición representan una
reacción exotérmica lenta y una endotérmica moderada?
b. ¿Puedes dibujar un estado de transición de una rq exotérmica rápida y una rq endotérmica lenta?
c. ¿Tiene algo que ver el que sea exotérmica o endotérmica con el hecho de que la reacción sea rápida o lenta?
d. Investiga ejemplos de rq exotérmicas y endotérmicas lentas y rápidas.

7. Factores que influyen en la velocidad de una reacción química (estudio cualita%vo)
1. Naturaleza de los reac%vos y energía de ac%vación. Ejemplo 1: Na con agua, reacción explosiva. Ca con agua, reacción mucho más lenta. Otros metales con el agua, no reaccionan.
Ejemplo 3: El CuSO4 y el Fe, en estado sólido no reaccionan, mientras que en disolución el proceso es relativamente rápido y provoca la formación de Cu y FeSO4 (desplazamiento de metal)
Ejemplo 2: HCl(aq) y NaOH(aq) para dar agua y NaCl: reacción instantánea. Fe(s) + O2(g) para dar FeO y Fe2O3: lenta a tª ambiente.
• Si las sustancias covalentes producen reacciones relaQvamente lentas a tª ambiente ya que hemos de romper enlaces covalentes. Ejemplo 4: 2CH3-‐CH3 (g) + 7 O2 (g) 4 CO2 (g) + 6 H2O (g) Ea alta, v pequeña
• Si las sustancias son iónicas en disolución (enlaces iónicos ya rotos) reaccionan muy rápidamente: Ejemplo 5: FeCl3 (aq) + CrCl2 (aq) FeCl2 (aq) + CrCl3 (aq) Ea pequeña, v alta. Sólo transferencia de 1 electrón.

2. Estado �sico y grado de división de los reac%vos.
Ejemplo 2: Los metales no arden, pero finamente divididos (pulverizados), arden con facilidad.
Ejemplo 1: El CuSO4 y el Fe, en estado sólido no reaccionan, mientras que en disolución el proceso es relativamente rápido y provoca la formación de Cu y FeSO4 (desplazamiento de metal)
Ejemplo 3: Si deseamos quemar leña lentamente ¿usamos un leño grande o virutas de madera? Ejemplo 4: ¿quién se disuelve antes? ¿sal gorda o sal fina? ¿un terrón de azúcar o azúcar granulado? Ejemplo 5: ¿quién se oxida antes, un tornillo de hierro o limaduras de hierro?
Favoreciendo el el contacto entre reac%vos, favorecemos la reacción. • En reacciones homogéneas para favorecer la superficie de contacto, aumentaremos la concentración. • En las reacciones heterogéneas, la reacción sólo se desenvuelve en la superficie de contacto sólido-‐gas o sólido-‐líquido. Por eso las sustancias sólidas finamente divididas (carbón en polvo de minas) o líquidos pulverizados (gasolina en motores), reaccionan mucho más rápidamente.

Las condiciones más favorables para que se produzcan choques eficaces son que las moléculas de los reac%vos estén: * En estado gaseoso. En esta caso la velocidad dependerá del nº de enlaces a romper y del nº de moléculas a colisionar. * En disolución, formando iones. * En estado sólido finamente divididos (favorecemos la superficie de contacto)

3. Concentración de las sustancias reaccionantes. Por la misma razón que son más frecuentes los accidentes de tráfico en las, «horas punta», cuanto mayor sea el número de moléculas de los reactivos presentes en un mismo volumen más fácilmente podrán colisionar.
€
↑ Reactivos[ ]⇒↑%choques⇒⇒↑%choques eficaces⇒↑v
Reacción general: aA+bBproductos Ecuación ciné%ca: v=k [A]α[B]β
Valores de α y β: 0, 1, 2…o valores fraccionarios…pero siempre al aumentar la concentración de los reacQvos, aumentará la velocidad de reacción.
Un ejemplo: Cl2(g) + H2(g) 2 HCl(g) v=k[H2][Cl2]
Por lo tanto, al duplicar la concentración molar de cualquiera de los dos reactivos, la velocidad de la reacción también se duplica. (k sólo depende de la temperatura…)
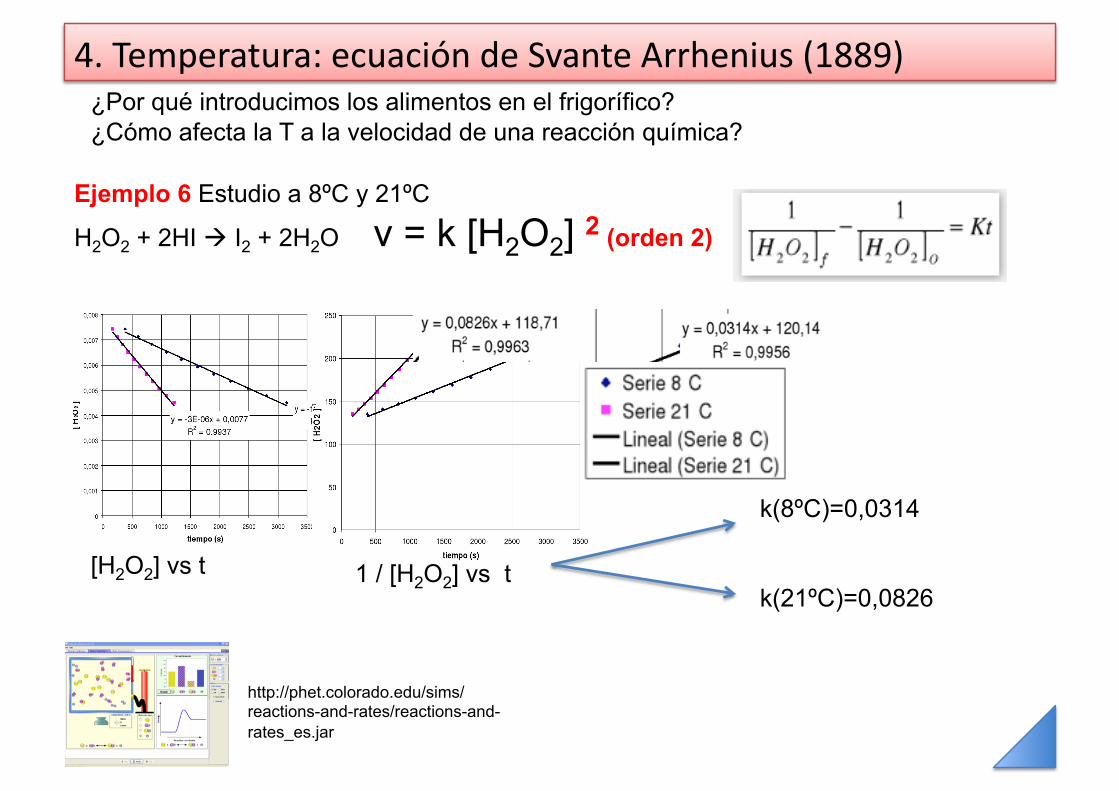
4. Temperatura: ecuación de Svante Arrhenius (1889) ¿Por qué introducimos los alimentos en el frigorífico? ¿Cómo afecta la T a la velocidad de una reacción química?
Ejemplo 6 Estudio a 8ºC y 21ºC
H2O2 + 2HI I2 + 2H2O v = k [H2O2] 2 (orden 2)
[H2O2] vs t 1 / [H2O2] vs t
k(8ºC)=0,0314
k(21ºC)=0,0826
http://phet.colorado.edu/sims/reactions-and-rates/reactions-and-rates_es.jar

4. Temperatura: ecuación de Svante Arrhenius (1889)
v=k(T) [A]α[B]β ¿A qué se debe que aumente la velocidad de modo tan notable con la temperatura?
€
Ec =32
KT =12
mv 2 K = cte.de Botzman
Al aumentar T, aumenta la Ec de las moléculas, y por tanto tendrán más velocidad, y aumentará el % de moléculas con E>Eac%vación y aumentará la velocidad de la reacción.
Ecuación de Svante Arrhenius deducida a partir de datos experimentales:
€
k = A.e−EaRT =
A
eEaRT
k= constante de velocidad; A= factor de frecuencia. Recoge la proporción de choque eficaces respecto del total de choques. Ea= energía de activación (kJ/mol) R= constante de los gases (kJ/molK) T= temperatura (K)
Aproximadamente: Por cada 10ºC de aumento de la temperatura, se duplica la v de reacción.

4. Temperatura: ecuación de Svante Arrhenius (1889)
€
k = A.e−EaRT =
A
eEaRT
Para k ( y v) es preciso: • Ea (depende de cada rq) • T • A (depende de cada rq)
La fracción de moléculas que sobrepasan la energía de activación es mayor. Así, a T2>T1 hay un mayor porcentaje de moléculas con energía suficiente para producir la reacción (área sombreada) que a T1 T2 zona rosa mas zona azul T1 sólo zona azul
T2>T1
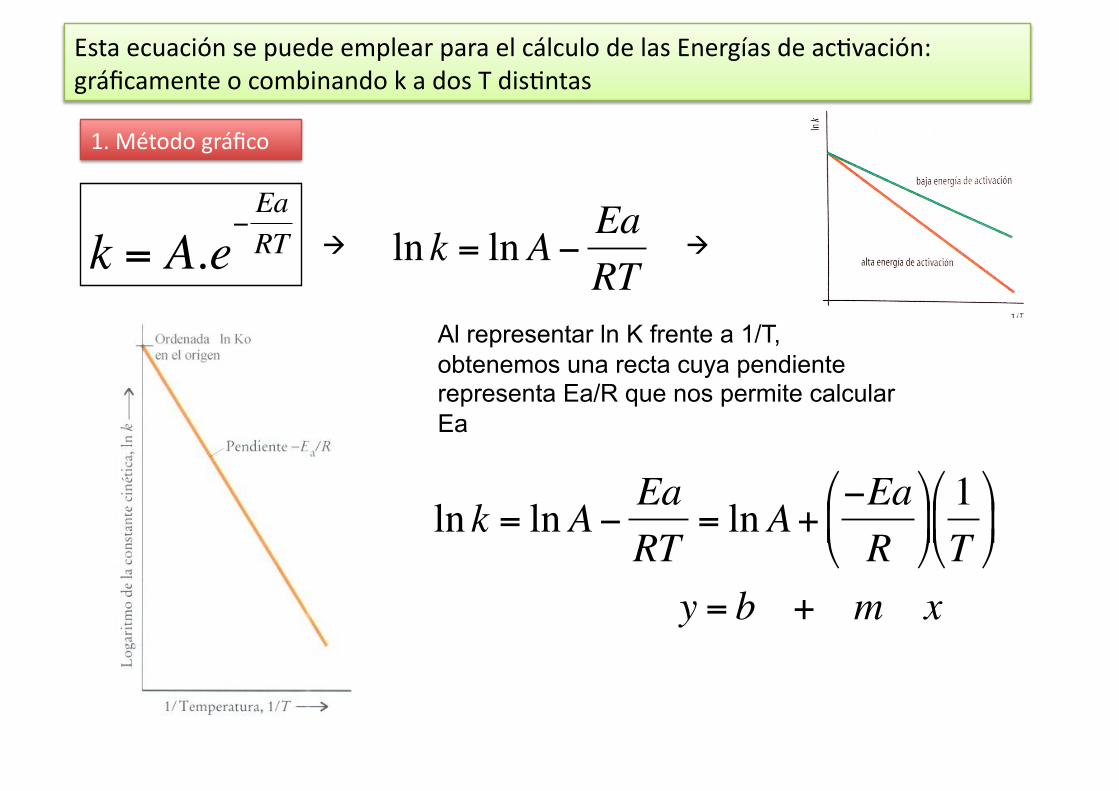
€
ln k = lnA− EaRT
€
k = A.e−EaRT
Esta ecuación se puede emplear para el cálculo de las Energías de ac%vación: gráficamente o combinando k a dos T dis%ntas
1. Método gráfico
Al representar ln K frente a 1/T, obtenemos una recta cuya pendiente representa Ea/R que nos permite calcular Ea
€
ln k = lnA− EaRT
= lnA+−EaR
⎛
⎝ ⎜
⎞
⎠ ⎟ 1T⎛
⎝ ⎜
⎞
⎠ ⎟
y = b + m x

Ejemplo 7 :¿Cual es el valor de la energía de activación para una reacción si la constante de velocidad se duplica cuando la temperatura aumenta de 15 a 25 ºC? ·
Sabemos que k2 (298 K) = 2 x k1 (288 K)
(1) ln k1 = ln A – Ea/RT1 (2) ln 2 k1 = ln A – Ea/RT2
Sustituyendo R = 8,31 Jmol–1 K, T1 = 288 K y T2 = 298 K y restando (2) – (1):
Despejando EA se obtiene:
€
ln k1 = lnA− EaRT1
ln k2 = lnA− EaRT2
⎫
⎬ ⎪ ⎪
⎭ ⎪ ⎪
→ ln k1 − ln k2 =EaR
1T2−1T1
⎛
⎝ ⎜
⎞
⎠ ⎟ → ln k1
k2=EaR
1T2−1T1
⎛
⎝ ⎜
⎞
⎠ ⎟
2. Método analí%co
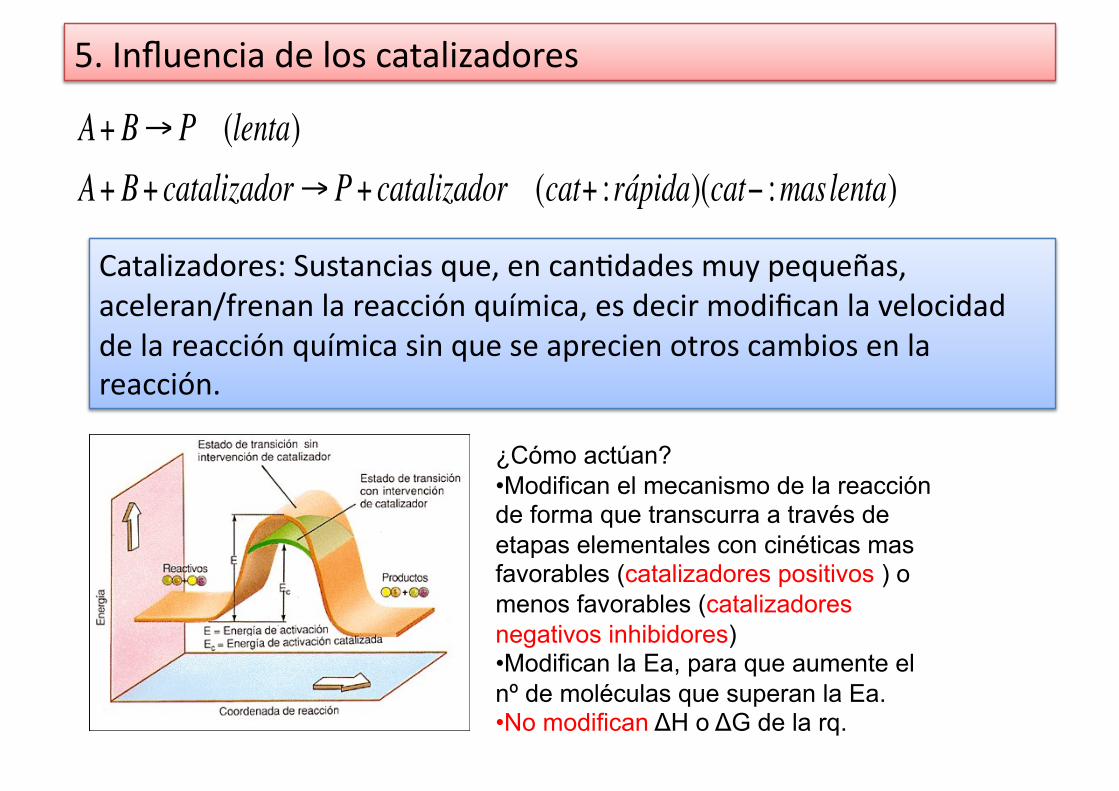
5. Influencia de los catalizadores
€
A+ B→ P (lenta)A+ B+ catalizador→ P+ catalizador (cat+ : rápida)(cat− :maslenta)
Catalizadores: Sustancias que, en can%dades muy pequeñas, aceleran/frenan la reacción química, es decir modifican la velocidad de la reacción química sin que se aprecien otros cambios en la reacción.
¿Cómo actúan? • Modifican el mecanismo de la reacción de forma que transcurra a través de etapas elementales con cinéticas mas favorables (catalizadores positivos ) o menos favorables (catalizadores negativos inhibidores) • Modifican la Ea, para que aumente el nº de moléculas que superan la Ea. • No modifican ΔH o ΔG de la rq.

• No interviene en los ajustes estequiométricos aunque si interviene en la reacción, debido a que las cantidades consumidas del catalizador, se regeneran al final (no se gasta).
• Si se “envenena el catalizador” deja de ser eficaz.
€
A+ B→ P
€
v = k[A][B]vcat = kcat[A][B]
⎫ ⎬ ⎭
€
Catalizador kcat > kInhibidor kinh < k
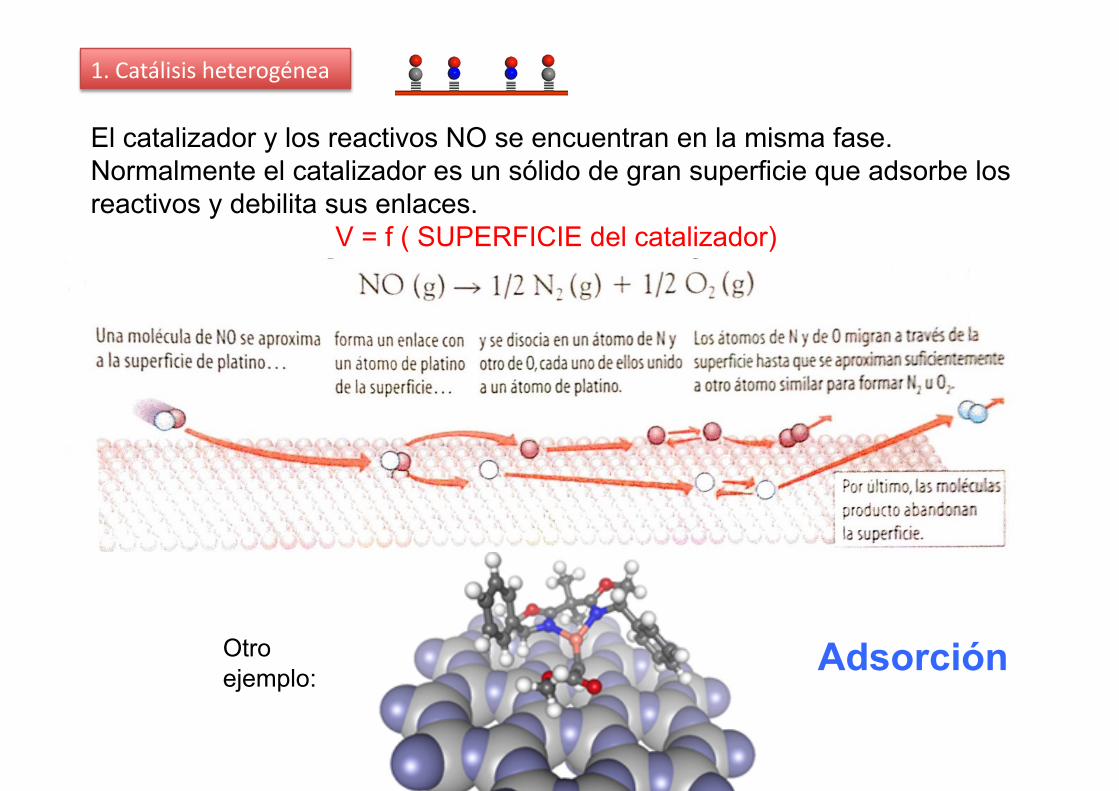
1. Catálisis heterogénea
El catalizador y los reactivos NO se encuentran en la misma fase. Normalmente el catalizador es un sólido de gran superficie que adsorbe los reactivos y debilita sus enlaces.
V = f ( SUPERFICIE del catalizador)
Otro ejemplo:
Adsorción
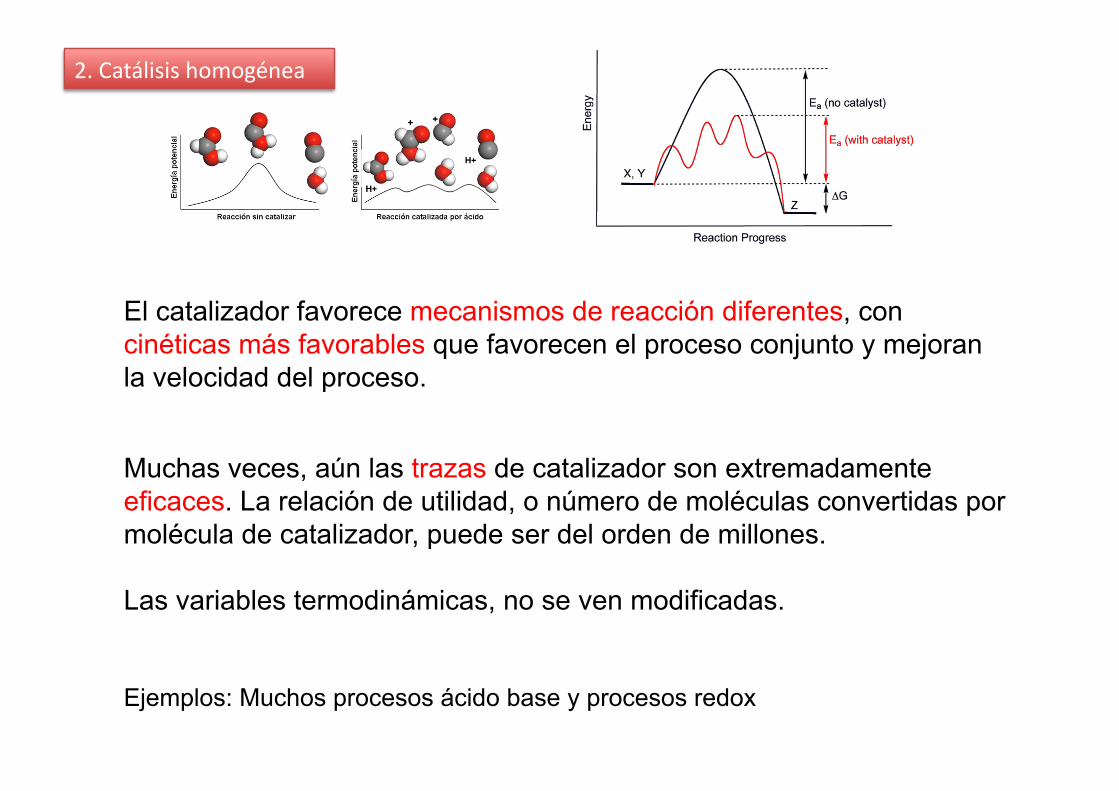
2. Catálisis homogénea
Muchas veces, aún las trazas de catalizador son extremadamente eficaces. La relación de utilidad, o número de moléculas convertidas por molécula de catalizador, puede ser del orden de millones.
Las variables termodinámicas, no se ven modificadas.
El catalizador favorece mecanismos de reacción diferentes, con cinéticas más favorables que favorecen el proceso conjunto y mejoran la velocidad del proceso.
Ejemplos: Muchos procesos ácido base y procesos redox

Un ejemplo de catálisis homogénea
H2O2 (aq) + I-1 (aq) H2O (aq) + IO-1 (aq) IO-1 (aq) + H2O2 (aq) H2O (aq) + I-1 + O2 (g)
Proceso global:
2 H2O2 (aq) 2 H2O (l) + O2 (g)
Descomposición del agua oxigenada en presencia de iones yoduro (catalizador)
Ea sin catalizador: 75 kJ/mol.
Ea con catalizador:
Yoduro: 56,25 kJ/mol
Platino cololidal: 48,75 kJ/mol
Enzima catalasa: 22,91 kJ/mol (multiplica la velocidad por 1022)
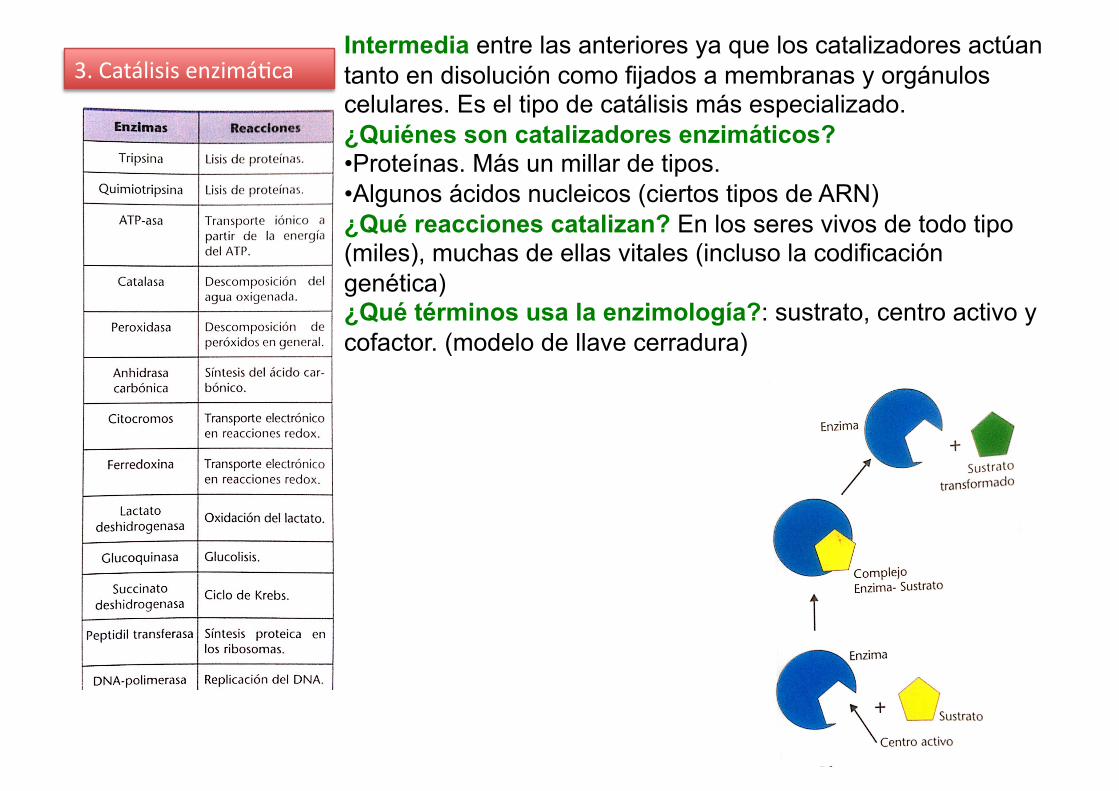
3. Catálisis enzimá%ca Intermedia entre las anteriores ya que los catalizadores actúan tanto en disolución como fijados a membranas y orgánulos celulares. Es el tipo de catálisis más especializado. ¿Quiénes son catalizadores enzimáticos? • Proteínas. Más un millar de tipos. • Algunos ácidos nucleicos (ciertos tipos de ARN) ¿Qué reacciones catalizan? En los seres vivos de todo tipo (miles), muchas de ellas vitales (incluso la codificación genética) ¿Qué términos usa la enzimología?: sustrato, centro activo y cofactor. (modelo de llave cerradura)

• La mayoría de los procesos industriales u%lizan catalizadores sólidos.
• Estos sólidos, de composición compleja poseen tres componentes elementales: – la fase ac%va – el soporte – el promotor.
• La fase acQva es la responsable de la ac%vidad catalí%ca. – Puede ser una sola fase química o un conjunto de ellas. – Puede ella sola puede llevar a cabo la reacción en las condiciones establecidas.
– Puede tener un costo muy elevado – Puede ser muy sensible a la temperatura.
8. U%lización de catalizadores en procesos industriales

• El soporte es la matriz sobre la cual se deposita la fase ac%va y el que permite op%mizar sus propiedades catalí%cas. – Dispersa, estabilizarla y le proporcionar buenas propiedades mecánicas a la “fase ac%va”.
– Puede ser poroso y por lo tanto presentar un área superficial por gramo elevada.
• El promotor es aquella sustancia que incorporada a la fase ac%va o al soporte en pequeñas proporciones permite mejorar las caracterís%cas de un catalizador en cualquiera de sus funciones de ac%vidad, selec%vidad o estabilidad.
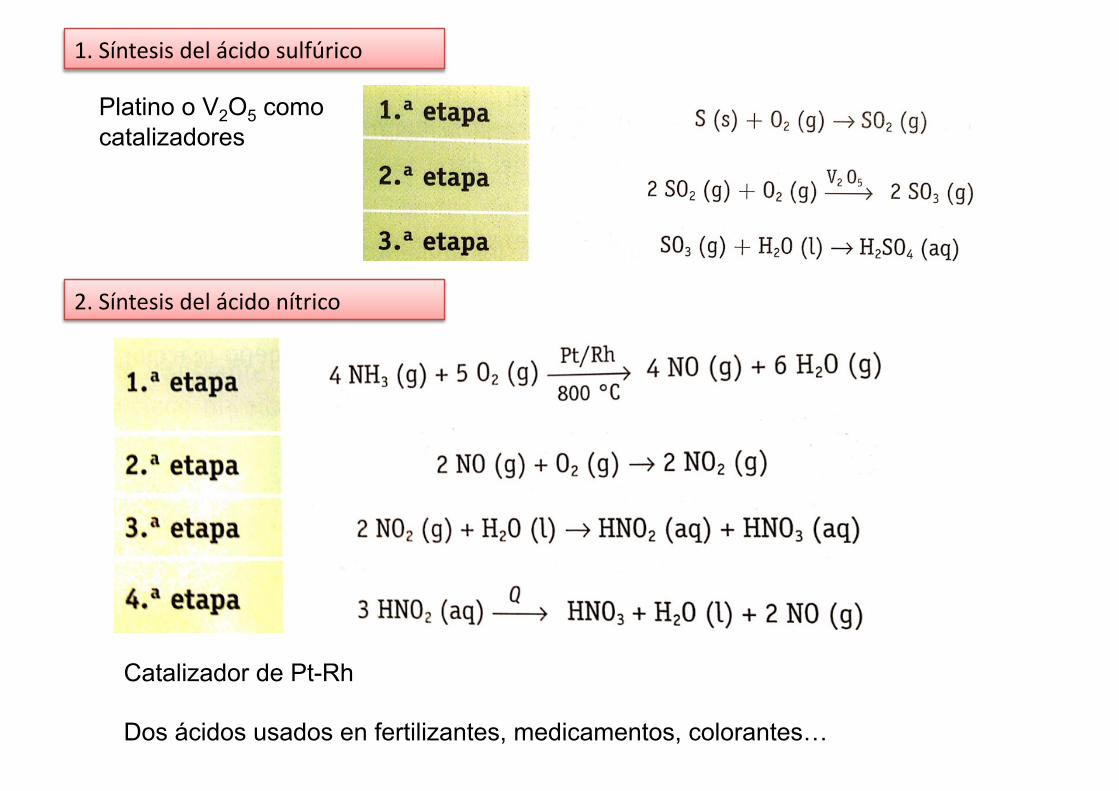
1. Síntesis del ácido sulfúrico
2. Síntesis del ácido nítrico
Platino o V2O5 como catalizadores
Catalizador de Pt-Rh
Dos ácidos usados en fertilizantes, medicamentos, colorantes…

3. Síntesis del amoniaco (IMPORTANTE)
Exotérmica y espontánea (termoquímica favorable), pero lentísima a temperatura ordinaria (cinética desfavorable) y por tanto…¡nulo rendimiento! ¿qué hacer?: • Temperatura, baja el rendimiento (favorece inversa) aunque aumente la velocidad. • Temperatura, se hace mucho más lenta • Solución: catálisis heterogénea con hierro y trazas de aluminio y potasio a 500ºC. El proceso catalítico supone la ADSORCIÓN de los reactivos en la superficie del catalizador.
N2(g) + 3H2(g) → 2NH3(g)
ΔHº = -46,2 kJ/mol ΔGº< 0 v=k[N2]2[H2] (orden 3)
Otros catalizadores: Mo, Al2O3
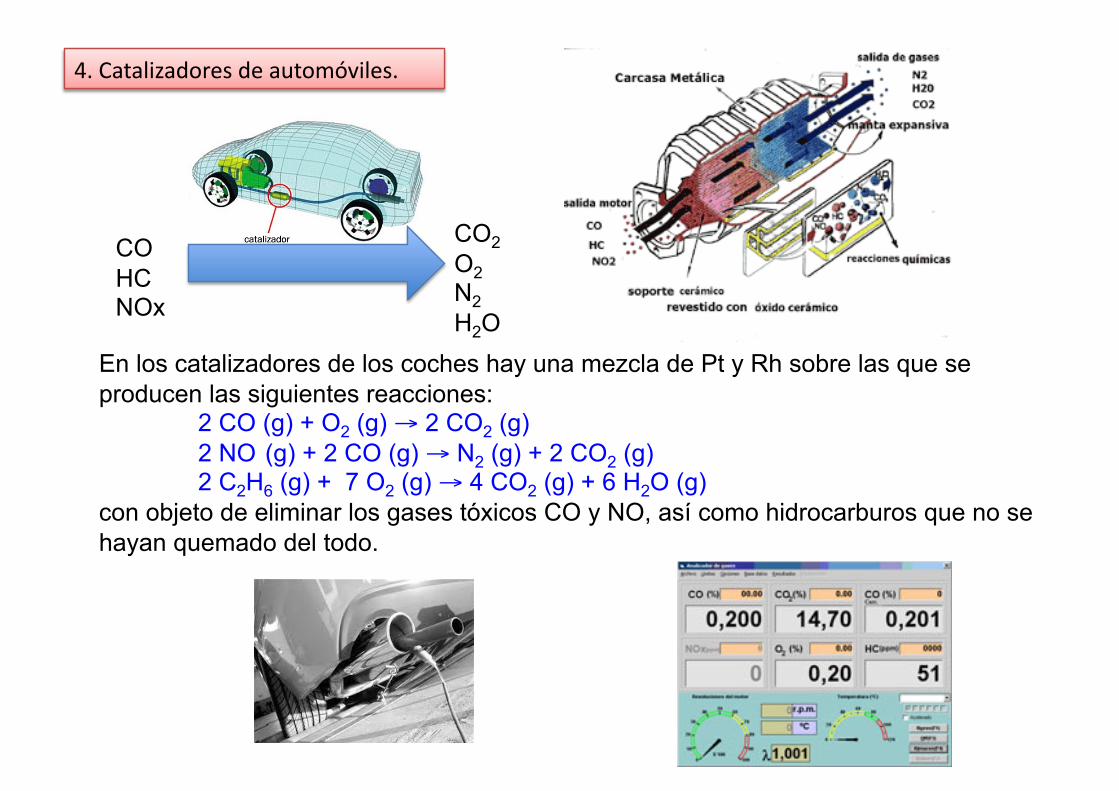
4. Catalizadores de automóviles.
CO HC NOx
CO2 O2 N2 H2O
En los catalizadores de los coches hay una mezcla de Pt y Rh sobre las que se producen las siguientes reacciones:
2 CO (g) + O2 (g) → 2 CO2 (g) 2 NO (g) + 2 CO (g) → N2 (g) + 2 CO2 (g) 2 C2H6 (g) + 7 O2 (g) → 4 CO2 (g) + 6 H2O (g)
con objeto de eliminar los gases tóxicos CO y NO, así como hidrocarburos que no se hayan quemado del todo.
![[Química 2º Bachillerato] Ejercicios + Soluciones: Equilibrio químico y cinética química](https://static.fdocuments.es/doc/165x107/55721019497959fc0b8ca660/quimica-2o-bachillerato-ejercicios-soluciones-equilibrio-quimico-y-cinetica-quimica.jpg)

![[PPT]Cinética química - Universidad Autónoma de Madrid · Web view4. Cinética química Química (1S, Grado Biología) UAM 4. Cinética química Química (1S, Grado Biología)](https://static.fdocuments.es/doc/165x107/5b1b99ce7f8b9a1e258eee6f/pptcinetica-quimica-universidad-autonoma-de-madrid-web-view4-cinetica.jpg)















