
Coagulopatìas hereditarias hematologia brenda valdes
73
Dr. Luis Hernández Ayala E.M. Brenda Valdés Arias Coagulopatìas hereditarias 8ª A 31/03/14 Hematología
-
Upload
brendisbre -
Category
Documents
-
view
394 -
download
1
Transcript of Coagulopatìas hereditarias hematologia brenda valdes

Dr. Luis Hernández Ayala E.M. Brenda Valdés Arias
Coagulopatìas hereditarias
8ª A 31/03/14
Hematología

Grupo heterogéneo de enfermedades hemorrágicas que afectan a diferentes mecanismos de la coagulación, se agrupan dentro de las enfermedades de tendencia hemorrágica.
ORIGEN:
DEFINICIÓN
Primario Secundario
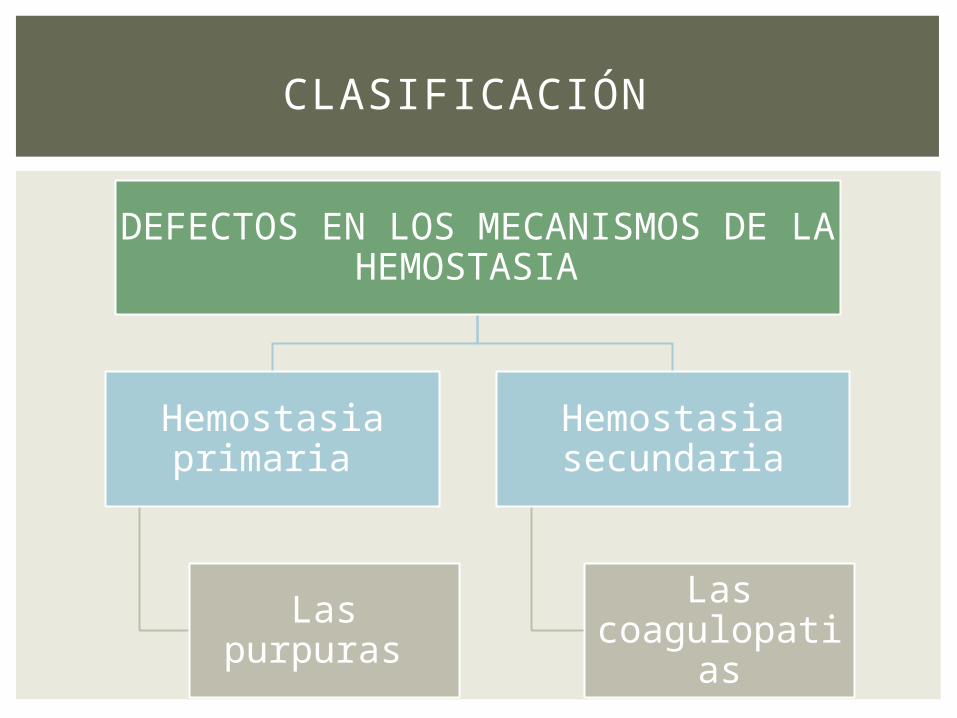
DEFECTOS EN LOS MECANISMOS DE LA HEMOSTASIA
Hemostasia primaria
Las purpuras
Hemostasia secundaria
Las coagulopatias
CLASIFICACIÓN
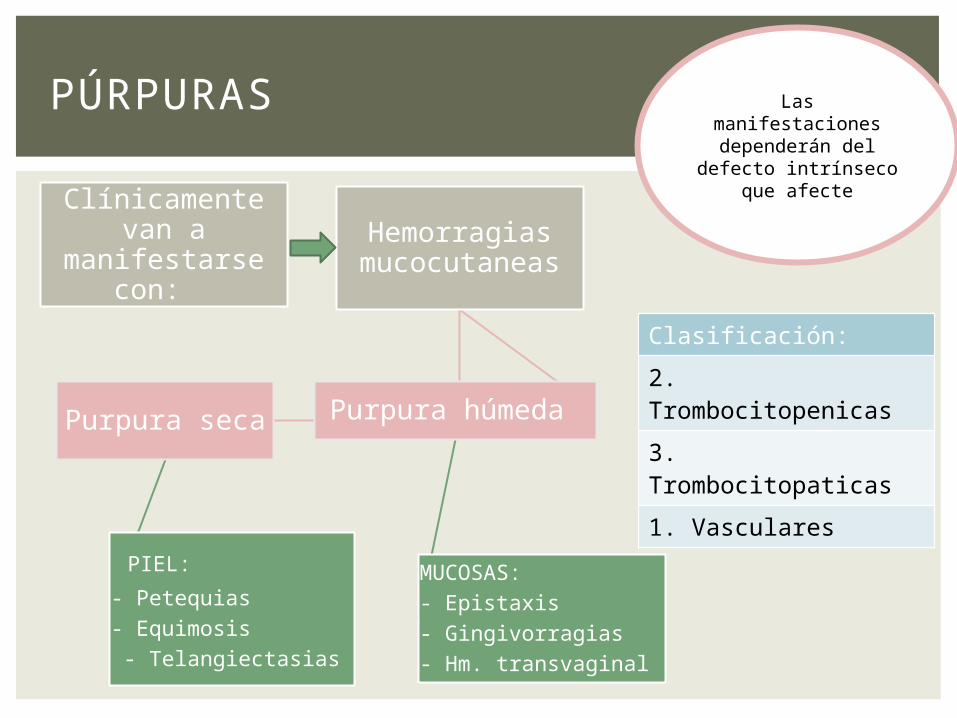
Clínicamente van a manifestarse
con:
Hemorragias mucocutaneas
Purpura seca
PIEL:
- Petequias - Equimosis - Telangiectasias
Purpura húmeda
MUCOSAS:- Epistaxis- Gingivorragias- Hm. transvaginal
PÚRPURAS Las manifestaciones dependerán del
defecto intrínseco que afecte
Clasificación:
2. Trombocitopenicas
3. Trombocitopaticas
1. Vasculares

Purpura ejemplo1. Vasculares P. De Henoch, p. senil, p. simple,
2. Trombocitopenicas P. Trombocitopenica autoinmune, p. TBP por secuestro esplénico, p. TBP secundaria coagulación intravascular diseminada.
3. Trombocitopaticas Enf. De Bernard Soulier, trombastenia de Glanzmann, p. TBP sec ingesta de aspirina.
DIVISIÓN DE LAS PURPURAS
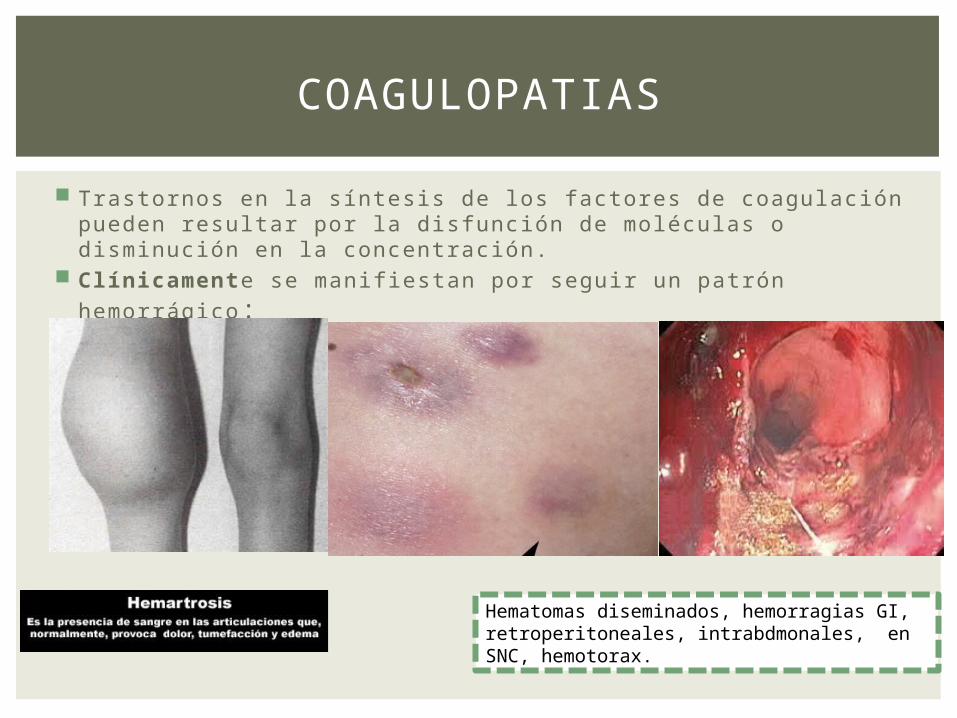
Trastornos en la síntesis de los factores de coagulación pueden resultar por la disfunción de moléculas o disminución en la concentración.
Clínicamente se manifiestan por seguir un patrón hemorrágico :
COAGULOPATIAS
Hematomas diseminados, hemorragias GI, retroperitoneales, intrabdmonales, en SNC, hemotorax.

Tipo Nombre Enfermedades
Purpuras Vasculares Telangiectasia hemorrágica hereditariaP. De Henoch, p. senil, p. simple,
Trombocitopenicas PT. Amegacariocitica, PT autoinmune, PT postransfusional, PT hiperesplenismo, PT post mielosupresion.
Trombopaticas Enf. De Bernard Soulier, trombastenia de Glanzman, p. TBP sec ingesta de aspirina, Sx de plaqueta gris, deficiencia de cuerpos densos.
CLASIFICACIÓN DE LAS ENFERMEDADES HEMORRÁGICAS
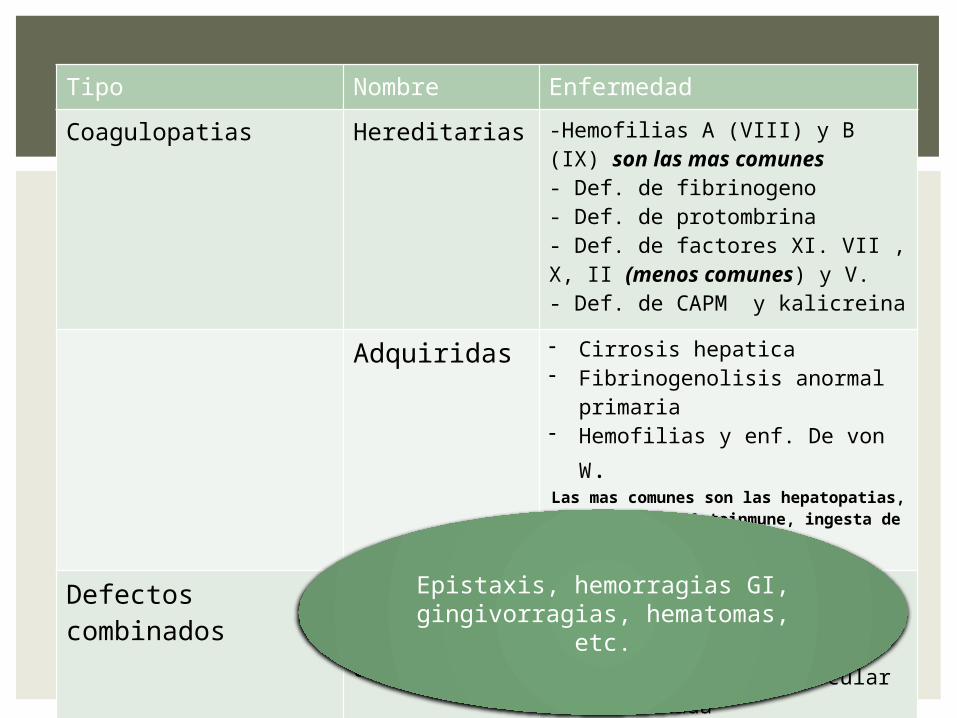
Tipo Nombre Enfermedad
Coagulopatias Hereditarias -Hemofilias A (VIII) y B (IX) son las mas comunes - Def. de fibrinogeno- Def. de protombrina- Def. de factores XI. VII , X, II (menos comunes) y V.- Def. de CAPM y kalicreina
Adquiridas - Cirrosis hepatica- Fibrinogenolisis anormal
primaria- Hemofilias y enf. De von W.Las mas comunes son las hepatopatias, uremias, enf. Autoinmune, ingesta de
medicamentos.
Defectos combinados
Hereditarios y adquiridos
- Afibrinogenemias- Disifibrinogenemias- Cirrosis hepática- Coaguacion intravascular
diseminadaEpistaxis, hemorragias GI,
gingivorragias, hematomas, etc.
HEMOFILIAS A Y B
Son coagulpatias hereditarias Defecto en los genes del qX ligada al X Hemofilia A deficiencia del factor VIIIHemofilia B deficiencia del factor IX
Hemofilia A y B
La intensidad es dependiente del factor deficiente:

Mundial:
hemofilia A (clasica) 1:10 000 Hombres 80%
hemofilia B (enf. Chritsmas) 1:40 000 Hombres 20-25%
En México: 2 000 casosde los cuales 45% tienen Hemofilia grave
INCIDENCIA
400 000 hemofílicos
Factor VIII: codificación del Cr. X (q28)Se han ID > 600 mutaciones > 40 genes anormales Concentración plasmática 1nmol/L Los Px con deleciones presentan HEMOFILIA
GRAVE
La S celular del factor VIII hígado, bazo, GL y riñón trasplante de hígado.
ALTERACIÓN GENÉTICA FVIII
Gen del FIX (región distal qX)
En plasma circula como una glicoproteína en forma de zymogeno.
Vida media: 1 día
Se han detectado 10 polimorfismos: 7 intragenicos y 3 extragenicos en el Gen del FIX
4% representa a mutaciones por delecion dando origen a proteínas inactivas
ALTERACIÓN GENÉTICA FIX

Mujeres portadoras y los hombres la manifiestan. 70-75% tienen antecedentes de la enfermedad. 25-30% presentan mutaciones de novo. Esta asociada al síndrome de Turner. Relacionad c/ concentraciones plasmáticas del FIX y FVIII < 50
UI/dL
HERENCIA

GRAVEDAD DE LAS HEMOFILIAS
Tipo Actividad del factor deficiente
Grave <1 UI/dL
Moderada 1-4UI/dL
Leve 5-25 UI/dL
Subhemofilicos/portadores
5-50 UI/dL
Valores normales
50-150 UI/dL
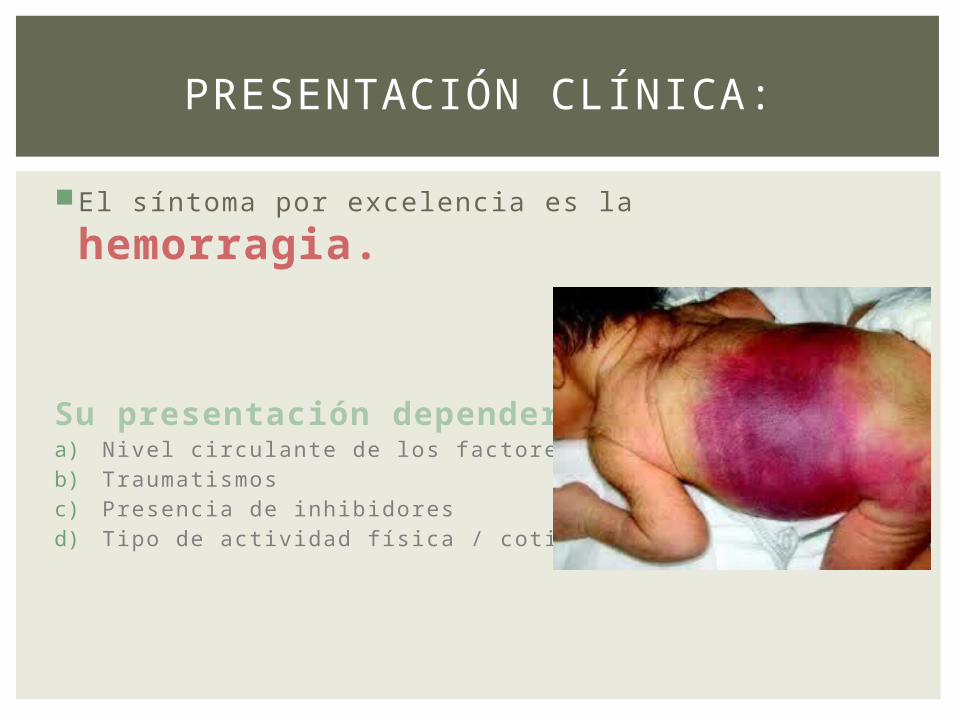
El síntoma por excelencia es la hemorragia.
Su presentación dependerá:a) Nivel circulante de los factores.b) Traumatismosc) Presencia de inhibidores d) Tipo de actividad física / cotidiana.
PRESENTACIÓN CLÍNICA:
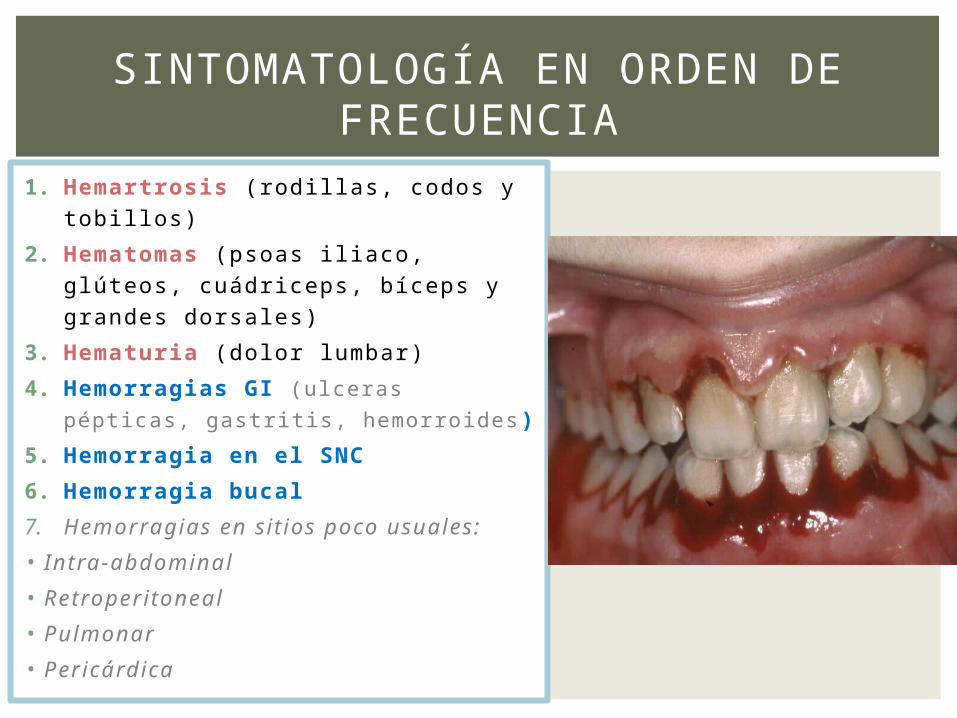
1. Hemartrosis (rodillas, codos y tobillos)
2. Hematomas (psoas iliaco, glúteos, cuádriceps, bíceps y grandes dorsales)
3. Hematuria (dolor lumbar)
4. Hemorragias GI (ulceras pépticas,
gastritis, hemorroides)
5. Hemorragia en el SNC
6. Hemorragia bucal
7. Hemorragias en sitios poco usuales:
• Intra-abdominal
• Retroperitoneal
• Pulmonar
• Pericárdica
SINTOMATOLOGÍA EN ORDEN DE FRECUENCIA

Historia familiar de hemofilia ( muestra de sangre del c.
umbilical)
Sin historia familiar de hemofiliaa. En pacientes graves: al año de vida con
evidenciahemorragias en sitios de presiónb. Moderados o leves: en la adultes presentan
hemorragias posterior a una cirugia, traumatismo, etc.
DIAGNOSTICO
BH no muestra hallazgos característicos amenos que presente anemia por la hemorragias constantes y sn mas comunes en niños.
LABORATORIO
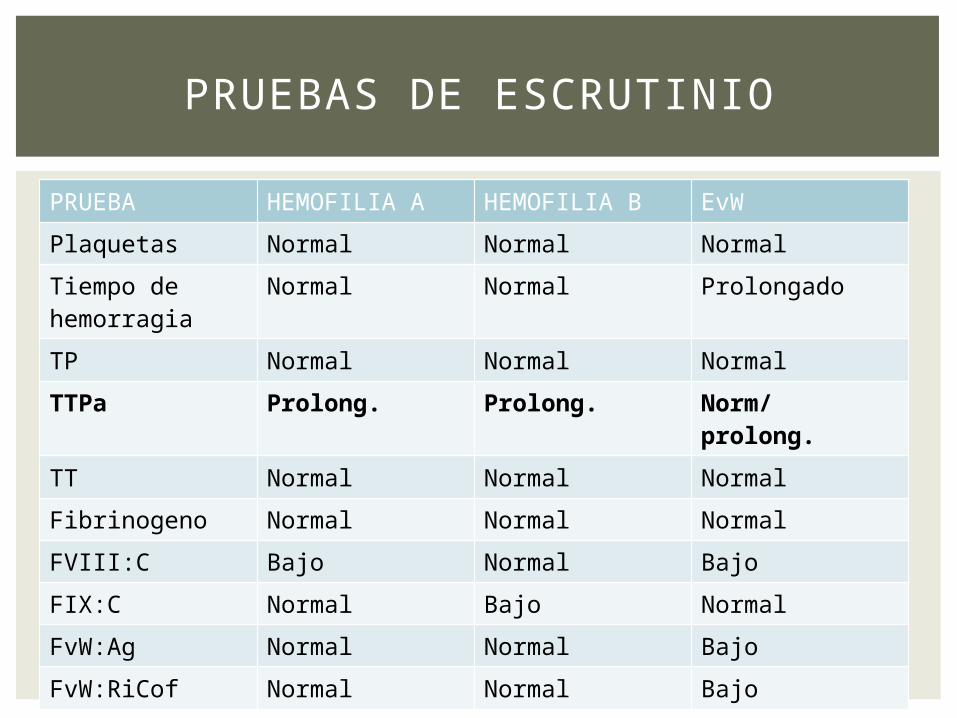
PRUEBA HEMOFILIA A HEMOFILIA B EvW
Plaquetas Normal Normal Normal
Tiempo de hemorragia
Normal Normal Prolongado
TP Normal Normal Normal
TTPa Prolong. Prolong. Norm/ prolong.
TT Normal Normal Normal
Fibrinogeno Normal Normal Normal
FVIII:C Bajo Normal Bajo
FIX:C Normal Bajo Normal
FvW:Ag Normal Normal Bajo
FvW:RiCof Normal Normal Bajo
PRUEBAS DE ESCRUTINIO

MATERIAL DE REACCION CRUZADA:
A. MRC positivo: Nivel plasmático existe una sustancia con similitud antigénica al factor de coagulación anormal
B. MRC negativa: indican la ausencia de la proteína antigénica y siguiere que la síntesis del factor es baja
CUANTIFICACIÓN DE FACTORES

CoagulométricosMiden el tiempo que
tarda en formarse un coagulo a través de un sensor que capta cambios de luz del plasma.
Cromogenicos Miden la reacción
producida por la liberación de un compuesto mediante la fotometría.
MÉTODOS DE CUANTIFICACIÓN DE FACTORES:
Las proteinas de F:VII:C y F:IX:C se determinan por ensayo inmunologgico del antigeno del F:VIII y FIX en el ELISA
Detectando las moléculas normales y anormales.

Método de unidades Bethesda
Se sospecha cuando hay prolongación del TTPa + ciertas condiciones clínicas:
1) Px con dx de hemofilia que no responde a tratamiento sustitutivo con dosis adecuada.
2) Px sin HC previa de hemorragia y que presentan de manera súbita hemorragias graves.
3) Px con hemofilia B que presentan anafilaxia al concentrados de factor transfundido.
INHIBIDORES EN HEMOFILIA
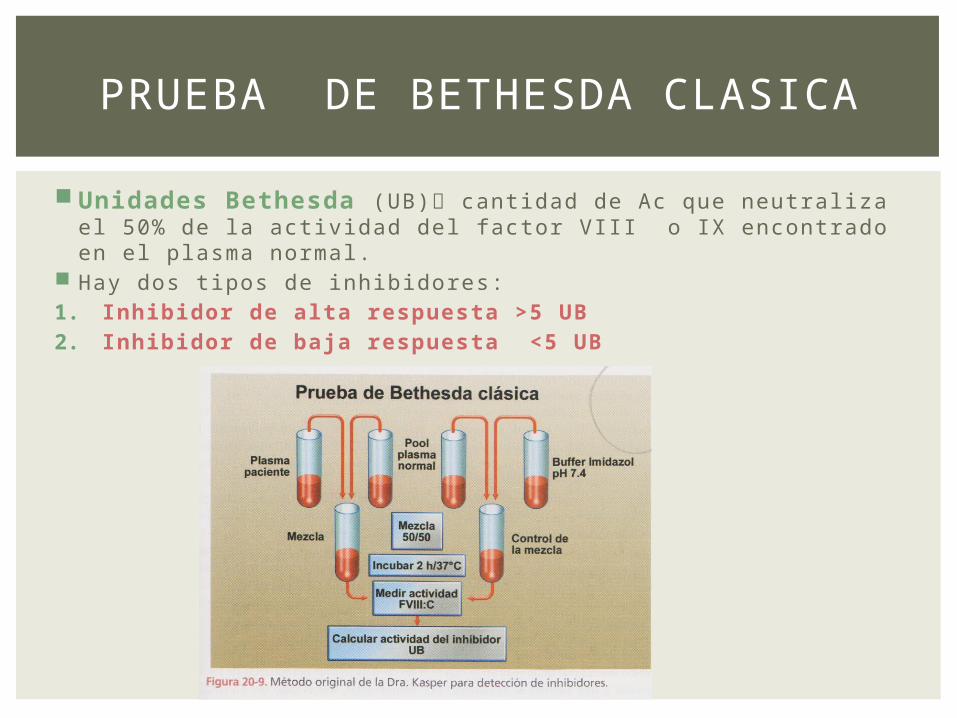
Unidades Bethesda (UB) cantidad de Ac que neutraliza el 50% de la actividad del factor VIII o IX encontrado en el plasma normal.
Hay dos tipos de inhibidores:1. Inhibidor de alta respuesta >5 UB2. Inhibidor de baja respuesta <5 UB
PRUEBA DE BETHESDA CLASICA
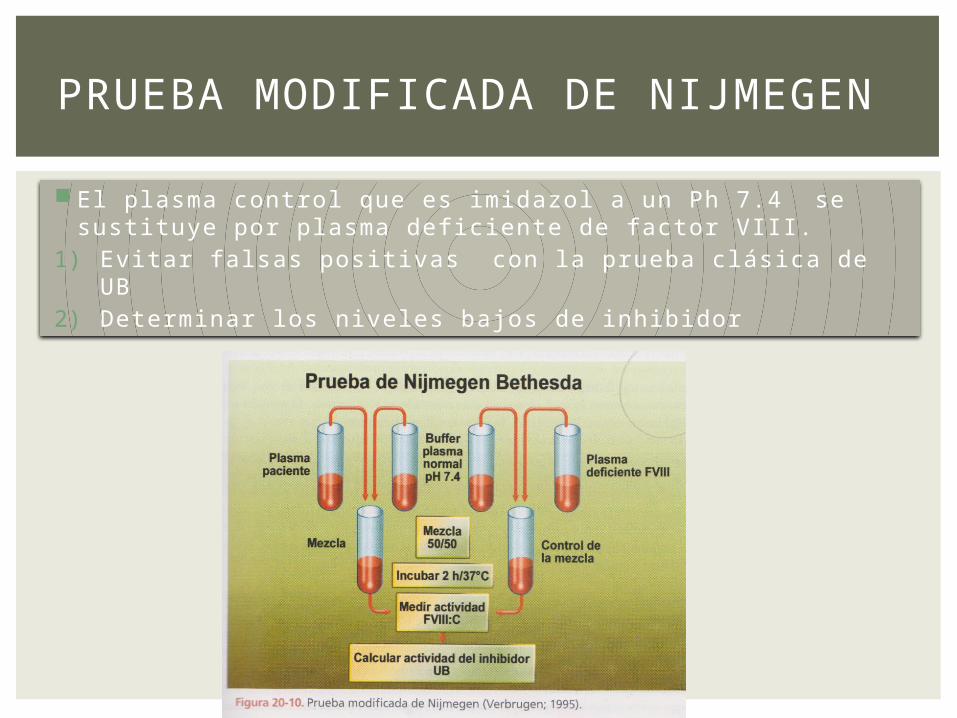
El plasma control que es imidazol a un Ph 7.4 se sustituye por plasma deficiente de factor VIII.
1) Evitar falsas positivas con la prueba clásica de UB 2) Determinar los niveles bajos de inhibidor
PRUEBA MODIFICADA DE NIJMEGEN

Otras deficiencias hereditarias de la coagulaciónDeficiencias combinadas de FV:C/FVIII.CENFERMEDAD DE VON WILLEBRANDHemofilia adquirida
DIAGNÓSTICOS DIFERENCIALES

TRATAMIENTO

Incrementar el nivel plasmático del factor deficiente
mediante la administración de concentrados crudos o
purificados el factor requerido.
“ Toda hemorragia en un px hemofílico es
considerada una urgencia” tratamiento
inmediato
I. Medidas locales (taponamiento, compresas frias)
II. Medidas generales (analgesicos, antifibrinliticos)
III. Medidas famacologicas (desmopresina)
OBJETIVO DEL TRATAMIENTO
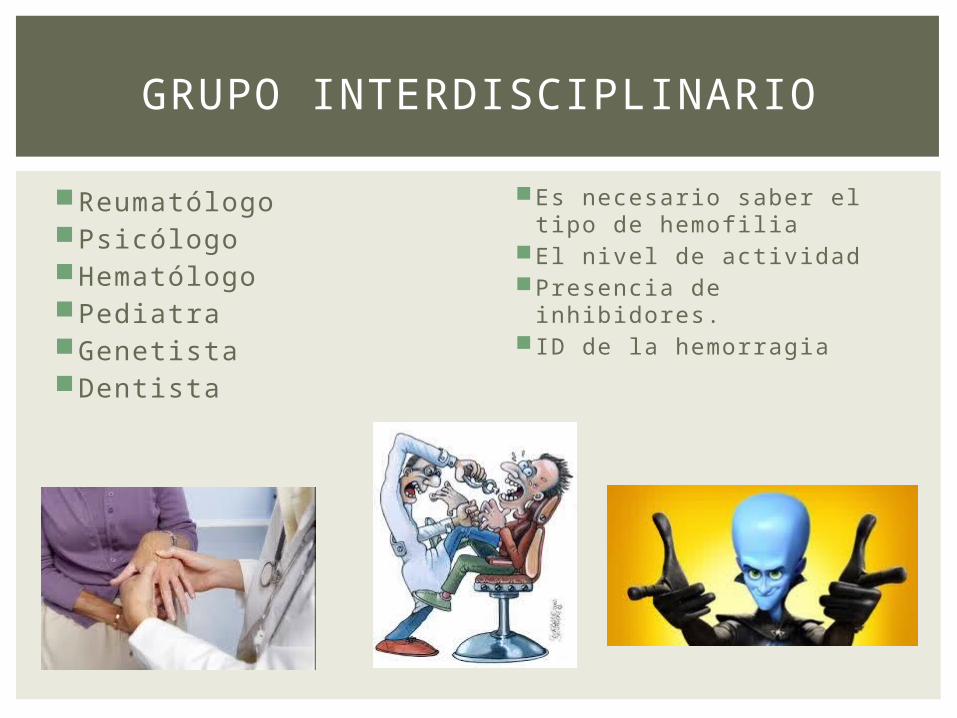
ReumatólogoPsicólogoHematólogoPediatraGenetista Dentista
GRUPO INTERDISCIPLINARIO
Es necesario saber el tipo de hemofilia
El nivel de actividadPresencia de
inhibidores. ID de la hemorragia
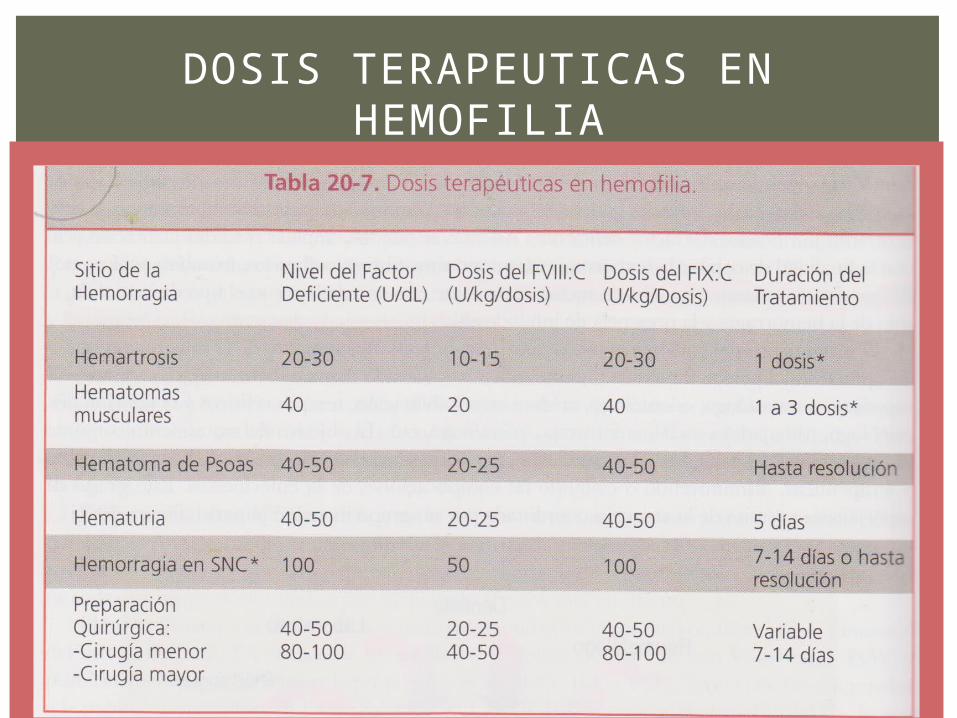
DOSIS TERAPEUTICAS EN HEMOFILIA

Hemofilia A Concentrados liofilizados
de FVIII, de acuerdo con la cantidad de proteínas, en baja pureza, pureza intermedia y alta pureza.
Hemofilia B Plasma fresco congelado y
posteriormente concentrados liofilizados derivados del plasma (concentrados de complejo protrombinico.
TRATAMIENTO

Concentrados de factor IX
Proplex TProfilnine SDBerinineImmunineOctanine FAimafix
CONCENTRADO DE FACTOR VIII
Factor 8YHeamate PKoate InmunateEmoclot Doctanate

En mexico el aporte de los concentrados de FVIII o
FIX no se encuentran disponibles en grandes
cantidades y además tienen una alto costo, la mayor
disponibilidad de productos con los que se cuentan
son los CRIOPRECIPITADOS.
TERAPIA TRANSFUSIONAL

Desmopresina: análogo sintético de la hormona antidiurética, y su acción consiste en l iberar FVI I I y FvW de los sit ios donde se almacenan, permit iendo su l iberación a la circulación.
Utilizado en: Hemofilia leve y EvW Dosis: 0.3 mg/kg/dosis IV diluido en 50cc de sol.
FisiológicaVida media de 8-12hrs /efecto 30 min
TRATAMIENTO FARMACOLÓGICO
Intranasal 300mg efecto a los 60 min.

Antifrbrinoliticos: inhiben la activación del plasminogeno y la actividad de la plasmia previene la lisis del coagulo.
Hemorragias de mucosas, orales y dentales.
Ac. Aminocaproico 75mg/kg cada 6 hrs Ac. Tranexamico 25mg/kg/ohrs
Aprotinina
TRATAMIENTO FARMACOLÓGICOS

Hemostáticos locales: Fibrinas adhesivasBismutoCementosColágenaCelulosa oxidadaTrombina tópica.
TRATAMIENTO FARMACOLÓGICO

COMPLICACIONES
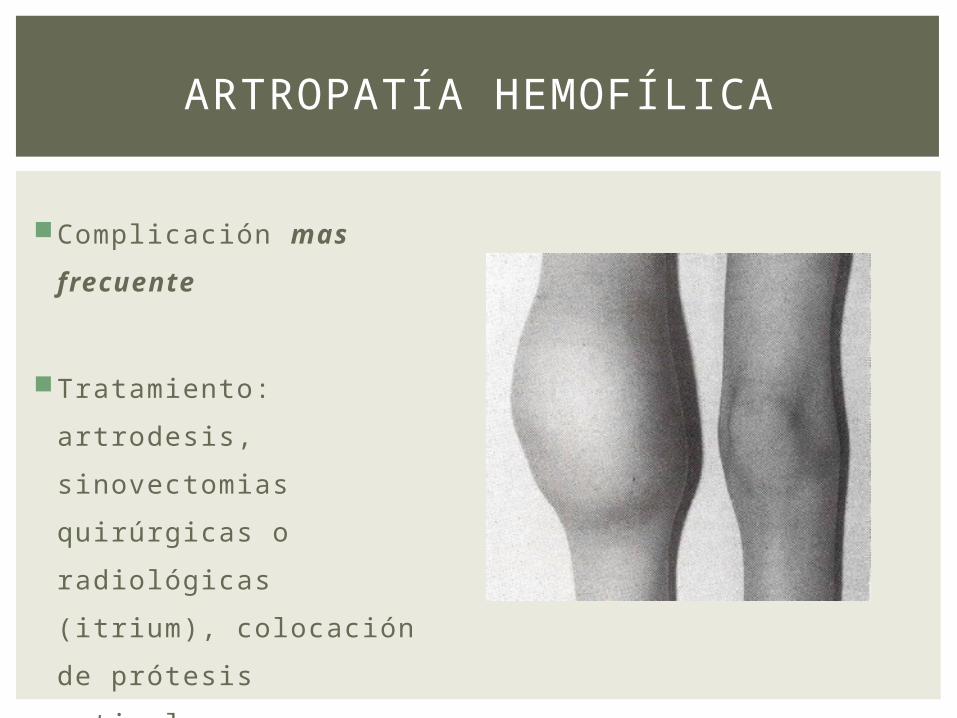
Complicación mas
frecuente
Tratamiento: artrodesis,
sinovectomias
quirúrgicas o
radiológicas (itrium),
colocación de prótesis
articular.
ARTROPATÍA HEMOFÍLICA
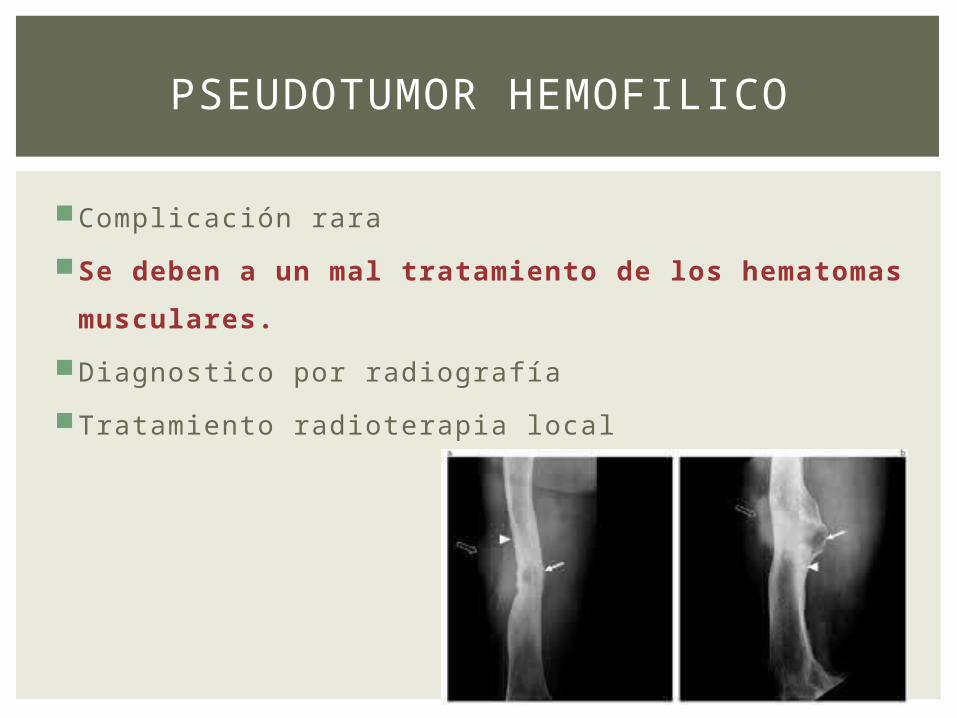
Complicación rara
Se deben a un mal tratamiento de los
hematomas musculares.
Diagnostico por radiografía
Tratamiento radioterapia local
PSEUDOTUMOR HEMOFILICO
VHIVHB y VHC
CONTAMINACIÓN VIRAL

OTROS DEFECTOS RAROS EN LA CAGULACION

Afibrinogenemias, hipofibrinogenemias o
desfibrinogenemias
Clínica: hematomas, hemartrosis y hemorragias
sistémicas desde la infancia
Diagnostico: prolongación del TP, TTPa, TT, TH
(defecto en la agregación plaquetaria)
Tratamiento: crioprecipitados
Defectos del fibrinogeno

Aparecen en: hipoprombinemia o en
disprotrombinemia ( son las mas comunes)
Diagnostico: prolongacion del TP y TTPa con
disminucion FII
Tratamiento: plasma fresco congelado o CCP
Deficiencia de protrombina

Se presenta mas en judíos Ashkenazi
Autosómico recesivo
Población general: incidencia 1 :100 000
Clínica: hemorragias leves seguidos de largos
periodos sin hemorragias, y son comunes las
hemorragias postquirurgicas (cavidad oral)
Tratamiento: plasma fresco congelado o
concentrados purificados
Deficiencia del FXI

Incidencia de 1 : 500 000
Clinica: hemorragias a cualquier edad
Diagnóstico: TP, TTPa prologados, y
disminución del FX
Tratamiento: plasma fresco congelado o CCP
DEFICIENCIA CONGENITA DEL FX

TROMBOFILIA

Diversos trastornos adquiridos o hereditarios que
presentan condiciones en las que hay riesgo de
trombosis venosa o arterial.
Estados de hipercogulabilidad o estados
pretromboticos.
GENERALIDADES

TROMBOSIS ARTERIAL

Factores de riesgo: a) Hombreb) Historia familiarc) Hiperlipidemiad) Tabacoe) HTAf) DMg) Hiperuricemia h) Eritrocitosisi) Incremento del F VII y fibrinogeno.
TROMBOSIS ARTERIAL

Los trombos venosos son menos firmes, es mas fácil que s fragmenten y originen émbolos que puedan causar daño a distancia. tromboembolia venosa.
“El factor de riesgo mas importante es la hipercoagulabilidad.”
TROMBOSIS VENOSA

Anormalidades que condicionan estados de trombofilia

*ANGIOGRAFIA* ( flebogragia o venografia o
arteriografia) con medios de contraste
Otros: pletismogragia de impedancia, gammagrafia
c/radioisopos, TAC o RM.
Tratamiento: anticoagulantes, antiplaquetarios y
agentes fibrinoliticos.
DIAGNOSTICO DE LA TROMBOSIS

Los indicadores clínicos sugieren un estado de trombofilia primaria cuando:
1) Trombosis en sujetos <40años
2) Hiistoria familiar de trombosis
3) Trombosis recurrente sin factores
precipitantes
4) Trombosis en sitios raros
5) Resistencia al tratamiento anti-trombotico
convencional
ESTADOS DE TROMBOFILIA PRIMARIA

ESTADOS DE TF PRIMARIAS

En EUA es una de las causas de trombosis arterial en
20% o venosa en 13%.
Estado de hiperagregabilidad de las
plaqueta.
En México este síndrome es mas frecuente y ocurre
50 -60 % de los mestizos mexicanos que tiene un
marcador clinico de trombofilia.
Tratamiento: antiplaquetarios revierten la
hiperagregabilidad y disminuyen el riesgo de
trombosis.
SINDROME DE LAS PLAQUETAS PEGAJOSAS

Alteración en la estructura del gen que codifica
la S de la molécula del FV de coagulación, el
cual conservando su función procoagulante, es
resistente a su inhibición por la proteína C
activada.
La mas frecuente es: Mutación puntual en el
residuo 506 del gen del FV (13% de mexicanos)
“mutación de tipo Leiden”
Resistencia a la proteína C reactiva

Incrementó de las concentraciones de FVIII, Ac anti-
fosfolipidos, anticoagulantes lupicos, hepatopatías,
embarazo, empleo de anovulatorios, etc.
En México es mas frecuente la forma ADQUIRIDA que
primaria.
Resistencia a la proteína C reactiva ADQUIRIDA

67 y 11% de los pacientes mestizos mexicanos con
marcador clínico causa estados de
hiperhomocistinemia subministrar
suplementos de ac. Fólico para evitarla
Esta mutacion aparece tambien en el 80% de los
mestizos mexicanos sin trombofilia.
Mutación C677T del gen dela MTHFR

Autosómico codominante
2-5% de los pacientes con marcadores clínicos.
En México es muy rara su aparición
La proteína S es dependiente de vitamina K
Deficiencia heredada de proteína S

La AT-III es responsable de 75% de la actividad inhibitoria de
la trombina.
Autosómica dominante
En mexico es causa frecuente de trombofilia.
Heterocigotos 1:2 000 y tienen concentraciones <
50%
50% de estos heterocigotos sufren un episodio
vasooclusivo en la adolescencia.
El estado homocigoto para la deficiencia de AT-
III es incompatible con la vida.
Deficiencia heredada de antitrombina III

TROMBOFILIAS SECUNDARIAS

TROMBOFILIAS SECUNDARIAS

Las mas frecuentes sonPostoperatorio Traumatismo Postparto Neoplasias malignasTratamiento con estrógenosEnfermedades autoinmunes (sx anifosfolipido y LES)
GENERALIDADES

Es un periodo “trombogenico” ya que hay:
1) Disminución en la actividad fribrinolitica
2) Estasis de venas profundas
3) Dilatación de venas intrabdominales
4) Hiperfibrinogenemia
Neoplasias malignas:
La tromboflebitis migratoria y el síndrome de Trousseau
trombofilia venosa
PERIODO POSTPARTO

Tiene características clínicas asociadas:
Trombosis arterial o venosa
Perdida fetal repetida
Trombocitopenia
El SAF primario se acompaña de:
Migraña
Livedo reticularis
Síndrome de Evans (anemia hemolít ica y purpura trombocitopenica
autoinmunes s imultaneas)
Corea
Hipertensión pulmonar
SÍNDROME DE AC ANTIFOSFOLIPIDOS (SAF)

Anormalidades funcionales en:
Complejo del FVIII
Anticoagulantes lupicos
Ac anti fosfolipidos
Deficiencia de prostaccilcinas
La mas frecuente es el fenotipo es la resistencia a la
proteína C
LUPUS ERITEMATOSO GENERALIZADO

Pacientes con factores de riesgo (<45años)
Se deben de hacer las pruebas para encontrar si es
primaria o secundaria, buscando la mutación de
Leiden del gen del FV por medio de biología
molecular (origen primario)
Evaluación de pacientes con riesgo trombotico alto

HeparinasAnticoagulantes orales Inhibidores directos de la trombinaFibrinoliticosAntiplaquetarios (aspirina)Otros antiplaquetarios (clopidogrel)
Profilaxis y tratamiento

Inhiben la coagulación a su interacción con la
antitrombina III potencian la formación de
complejos entre la antitrombina III y las proteasa de
serina IXa, Xa, XIa
Administracion IV o subcutanea
Vida media 1 hora
HBPM: actividad antitrombotica y anticoagulante
Heparinas

Derivados de la cumarina o de la indandiona son
antagonistas de la vitamina k
Factores dependientes de vit K: II,VII, IX, X
Otros: Inhibidores directos del FXa (rivaroxaban)
Anticoagulantes orales
Inhibidores directos de la trombina
Hirudinas y fragmentos de hirudinas
Aun no se utilizan en la practica diaria

Primera generación: estreptoquinasa (SK) y urocinasa
Segunda generación: activador tisular del
plasminogeno
Tercera generación: activador de plasminogeno tipo
urocinasa de una sola cadena y el complejo activados
acilado de plasminogeno / estreptoquinasa
Deben usarse entre las primeras 6 horas del proceso
vasooclusivo
Dosis inicial de 250 000 U SK
Dosis de mantenimiento 100 000 U/h durante 12-48 hrs
Fibrinoliticos

Aspirina por exelencia: inbhibe la cliclooxigenasa plaquetaria reduciendo la producción del tromboxano A2.
Tratamiento de elección en el Síndrome de plaqueta pegajosas.
Dosis: 100 mg cada 24 -48 hrs
Antiplaquetarios
Clopidogrel antiagregante plaquetarioPropanolol, nifedipina y diltiacem efecto
atiagregante
Otros antiplaquetarios

GRACIAS!


















