
EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y …
70
EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y CLOROFILÍN EN UN MODELO IN VITRO DE CÉLULAS HUMANAS DE CÁNCER DE COLON. CAMILA RUBIO PATIÑO TRABAJO DE GRADO Presentado como requisito parcial Para optar al titulo de BIÓLOGA GERMÁN DARÍO DÍAZ, M.D., MSSc, Ph.D. Director PONTIFICIA UNIVESIDAD JAVERIANA FACULTAD DE CIENCIAS CARRERA DE BIOLOGÍA Bogotá, D.C. Octubre 27, 2006
Transcript of EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y …

EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y CLOROFILÍN
EN UN MODELO IN VITRO DE CÉLULAS HUMANAS DE CÁNCER DE COLON.
CAMILA RUBIO PATIÑO
TRABAJO DE GRADO Presentado como requisito parcial
Para optar al titulo de
BIÓLOGA
GERMÁN DARÍO DÍAZ, M.D., MSSc, Ph.D. Director
PONTIFICIA UNIVESIDAD JAVERIANA FACULTAD DE CIENCIAS CARRERA DE BIOLOGÍA
Bogotá, D.C. Octubre 27, 2006

NOTA DE ADVERTENCIA Artículo 23 de la Resolución No 13 de Julio de 1946 “La Universidad no se hace responsable por los conceptos emitidos por sus alumnos en sus trabajos de tesis. Solo velará por que no se publique nada contrario al dogma y a la moral católica y por que las tesis no contengan ataques personales contra persona alguna, antes bien se vea en ellas el anhelo de buscar la verdad y la justicia”.

EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y CLOROFILÍN
EN UN MODELO IN VITRO DE CÉLULAS HUMANAS DE CÁNCER DE COLON.
CAMILA RUBIO PATIÑO
APROBADO
---------------------------------------- Germán Darío Díaz, M.D., MSSc, Ph.D.
Director
---------------------------------------- ---------------------------------------- Dr. Pablo Moreno, Ph.D. Dra. Albis Hani M.D. Jurado Jurado

EFECTOS DE LA COMBINACIÓN DE 5-FLUOROURACILO Y CLOROFILÍN
EN UN MODELO IN VITRO DE CÉLULAS HUMANAS DE CÁNCER DE COLON.
CAMILA RUBIO PATIÑO
APROBADO
---------------------------------------- ---------------------------------------- Ángela Umaña, Ph.D. Andrea Forero, Bióloga Decana Académica Directora de Carrera Facultad de Ciencias Básicas Biología

v
AGRADECIMIENTOS
Mis más sinceros agradecimientos:
Al Doctor Germán Darío Díaz, por permitirme aprender de su experiencia, por su apoyo al
acompañarme y ayudarme a dar mis primeros pasos como científica.
A la Doctora Sandra Gutiérrez por brindarme un lugar de trabajo en el Centro de
Investigaciones Odontológicas de la Pontificia Universidad Javeriana.
A Adriana García y Sandra Perdomo por su paciencia y valiosa colaboración en todo
momento.
A Juan Pablo Botero por su ayuda en el análisis de datos.
A mi familia y amigos por el ánimo y la confianza que tuvieron en mi durante la realización
de este proyecto de investigación.
A Michael Alberico por ayudarme a encontrar el camino.

vi
Tabla de Contenido
1. Introducción 1
2. Marco teórico y revisión de literatura 2
2.1. Carcinogénesis 2
2.2. Epidemiología del Cáncer de Colorectal 4
3. Etiología 5
3.1. Factores Genéticos 5
3.2. Características Moleculares del Cáncer Colorectal 7
3.2.1. Mutaciones en APC y β-Catenina 7
3.2.2. Metilaciones 9
3.2.3. Dieta 11
3.2.4 Microbiota 13
3.3. Modelo Secuencia Adenoma-Carcinoma 14
3.3.1. Bases moleculares de la Secuencia Adenoma-Carcinoma 16
3.3.2. Características Histopatológicas 18
3.3.3. Bases Celulares y Moleculares del Cáncer Colorectal 18
3.4. Prevención y Tratamiento 21
3.4.1. 5-Fluoracilo 21
3.4.2. Quimioprevención 24
3.4.3. Clorofilín 26
4. Formulación del problema 29
5. Justificación 29

vii
6. Objetivos 30
7. Hipótesis 30
8. Materiales y Métodos 31
9. Resultados 35
10. Discusión 42
11. Conclusiones 50
12. Recomendaciones 51
13. Bibliografía Citada 52

Resumen
El Cáncer Colorectal (CCR) es una patología gastrointestinal con una alta
morbimortalidad en los países occidentales y que se esta presentando y diagnosticando
de forma mayor en países conocidos como de baja incidencia, como Colombia. A pesar
de que el 5-Fluorouracilo (5 FU) es la quimioterapia de elección para el CCR, no existe
una estrategia óptima de tratamiento establecida. Por esta razón, se evaluó la actividad
antiproliferativa de la combinación de 5FU y Clorofilín (CHL), un agente antitumoral
de origen natural, sobre células tumorales humanas de colon Caco-2. Se evaluó la
viabilidad celular y morfología de células tratadas con diferentes dosis de los dos
compuestos antitumorales: 0.25 mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13 µg/ml de 5
FU, y sus posibles combinaciones. El 5 FU de manera individual, tuvo un efecto pobre
sobre la viabilidad celular, a diferencia de cuando fue utilizado en combinación con el
CHL, donde se observó una actividad sinérgica sobre la capacidad anti-proliferativa de
los dos agentes. La combinación de las concentraciones más bajas no presentó
disminución de la viabilidad celular. Se confirmó que las células Caco-2 tratadas
mueren por apoptosis.
Palabras Clave: apoptosis, cáncer de colon, Clorofilín, 5 Fluorouracilo

Abstract
Colorectal cancer (CCR) is a gastrointestinal pathology with a high incidence in
occidental countries and that is being diagnosed in a bigger manner in countries known
with low incidence, as Colombia. Eventhough 5-Fluorouracil (5FU) is the election
chemotherapy for CCR, there is no optimum strategy established. For this reason, the
antiproliferative activity of the combination of 5FU and Chlorophyllin (CHL), an
antiproliferative agent of natural origin, was evaluated over human colon tumoral cells,
Caco-2. We evaluated cell viability and morphology of cells treated with different doses
of both antitumoral compounds: 0.25 mM and 0.5 mM of CHL, 0.039 µg/ml and 0.13
µg/ml of 5 FU and their possible combinations. 5 FU, by itself, had no effect over cell
viability, except when used in combination with CHL, where there was synergy over the
anti-proliferative effect of both agents. The combination of the lower doses did not
show a decrease of cell viability nor a decrease of the adherent cell yield. It was
confirmed that Caco-2 cells died through the induction of apoptosis.
Key Words: apoptosis, colon cancer, Chlorophyllin, 5 Fluorouracil

1
1. Introducción
El cáncer es una enfermedad multifactorial, donde intervienen factores genéticos y
epigenéticos en su desarrollo. El cáncer de colon específicamente, es una patología
muy propia de países industrializados que se esta presentando en áreas conocidas de
baja incidencia, como América latina. Por ejemplo, en Colombia, el cáncer colorectal
se está diagnosticando en forma más frecuente, posicionándose en forma más
próxima al cáncer gástrico. Debido a esto, se hace necesario investigar sobre esta
patología, con miras a entender su funcionamiento a nivel molecular para generar
estrategias de prevención y tratamiento.
El cáncer colorectal se presenta tanto de forma hereditaria y no hereditaria. Entre las
formas hereditarias más conocidas se encuentran la poliposis adenomatosa familiar
(FAP) y el cáncer colorectal hereditario sin poliposis (HNPCC). El cáncer colorectal
surge como resultado de una serie de cambios histológicos que se acompañan de
alteraciones genéticas. La identificación de estas anormalidades genéticas ha llevado
a conocer más profundamente el modelo de la secuencia adenoma-carcinoma. Este
modelo describe el proceso de carcinogénesis partiendo de lesiones adenomatosas,
pasando por el estado de carcinoma y finalmente la metástasis.
Para que haya carcinogénesis, la célula colónica requiere adquirir una ventaja
selectiva que le permita la expansión clonal e inestabilidad genética que hace que
algunos genes se vean afectados, sobretodo aquellos responsables de la progresión
tumoral y la transformación maligna de las células. En el caso de los oncogenes como
K-RAS, generalmente es suficiente una mutación somática en un alelo para conferir
una ventaja selectiva en el crecimiento de la célula. En cuanto a algunos genes
supresores de tumores como APC y p53, algunos recientemente descritos, también se
presenta esta característica de haploinsuficiencia. Sin embargo, en la mayoría de los
genes supresores de tumores se requiere que haya una mutación tanto en el alelo
materno como en el paterno para que la célula adquiera una ventaja selectiva. Este

2
proceso se conoce como pérdida de la heterocigocidad (LOH). Esta situación
generalmente se da por la deleción de un alelo a causa de rearreglos cromosómicos,
acompañada de una mutación intragénica de otro alelo. Un oncogen (K-RAS) y por lo
menos dos genes supresores de tumores (APC y p53) son los blancos más
importantes de las mutaciones génicas que permiten desatar la carcinogénesis
colorectal. Debido a la alta tasa de mutación que presenta el cáncer colorectal, la
quimioprevención, que consiste en la utilización de moléculas que pueden prevenir
y/o controlar el cáncer mediante la interferencia de los factores que intervienen en la
iniciación, es una línea muy prometedora.
El 5-Fluorouracilo (5FU) es la quimioterapia de elección para el CCR. Este es un
metabolito sintetizado a partir del Uracilo, cuyo mecanismo de acción es la inhibición
de la enzima Timidilato Sintasa, inhibiendo la síntesis de DNA e induciendo la
apoptosis dependiente de p53 por medio de la vía intrínseca. Sin embargo, no existe
una estrategia óptima de tratamiento establecida para el 5FU. Por esta razón, se
evaluó la actividad antiproliferativa de la combinación de 5FU y Clorofilín (CHL), un
agente antitumoral de origen natural, sobre células tumorales humanas de colon
Caco-2, al ser administrados a diferentes concentraciones sobre la línea tumoral de
colon Caco-2. Se buscará evidenciar si su combinación genera algún tipo de
sinergismo, si alguno, para proponer nuevas formas terapéuticas para la prevención y
tratamiento del cáncer de colon.
2. Marco Teórico y Revisión de Literatura
2.1. Carcinogénesis
En el cáncer de colon hereditario los genes que más se encuentran mutados son el
APC, el gen que codifica para la β-Catenina y en el caso del HNPCC, los genes
encargados de la reparación del DNA. En cuanto al no hereditario son muchos más
los genes que se encuentran mutados, ya que su aparición depende de carcinógenos
externos presentes en el medio ambiente, la dieta, fármacos, entre otros, generando

3
diversos cambios en el DNA. Estos cambios en el DNA se dan por procesos tales
como la metilación y silenciación de genes permitiendo la pérdida de la función de
genes supresores de tumores y la activación de oncogenes, tipo k-ras y c-myc (Fodde
et al, 2001; Vogelstein & Kinzler, 2004).
La progresión neoplásica es un proceso multifactorial caracterizado por la presencia
de inestabilidad genética, que conduce a una perdida del control de la proliferación y
diferenciación celular. Esta es un proceso en el que las células proliferan sin control,
generando clones que pueden invadir otros tejidos. Esta pérdida del control en la
proliferación de los sistemas regulatorios transcripcionales responsables de la
homeostasis celular, que se refleja en el comportamiento de las células en forma
órgano específica (Cooper, 2000; Volgestein & Kinzler, 2004).
Para que se de el correcto mantenimiento de los procesos de desarrollo del
organismo, es necesaria una coordinación y un balance entre el ciclo celular y la
apoptosis. El cáncer resulta cuando los clones de células mutadas sobreviven y
proliferan de manera inapropiada, rompiendo este balance. Los diversos estudios
realizados en este ámbito sugieren que hay una señalización entre la maquinaria de
muerte y proliferación, ya que se ha observado que las mutaciones que promueven la
entrada inapropiada de las células al ciclo celular usualmente también promueven la
apoptosis. Estas observaciones sugieren una importante función de la comunicación
entre estas maquinarias para prevenir la supervivencia de los clones de células
aberrantes (Guo & Hay, 1999).
Adicionalmente, los oncogenes y los genes supresores de tumores están relacionados
de alguna forma con algunos aspectos de la transducción de las señales mitogénicas y
los puntos de chequeo del ciclo celular. La transducción de señales mitogénicas
incluye numerosas oncoproteínas, tales como los factores de crecimiento secretados
extracelularmente, los receptores de los factores de crecimiento, las cascadas de
transducción de señales citoplasmáticas y los factores de transcripción (Ver tabla 1).

4
Un estímulo oncogénico que activa la proliferación tiene el potencial de romper el
balance entre ciclo celular y apoptosis. Sin embargo, este potencial normalmente se
mantiene bajo control, ya que una proliferación excesiva también lleva a la
producción de señales de muerte (Schafer, 1998).
Uno de los problemas más importantes del cáncer, es el comportamiento biológico
de tumores benignos y los malignos. Los tumores benignos se mantienen confinados
a su lugar original de aparición y no invaden el tejido normal ni se dispersan a las
otras partes del cuerpo. Los tumores malignos son capaces de invadir y rodear el
tejido normal y finalmente pueden dispersarse al resto del cuerpo por medio del
proceso de metástasis. En el caso del cáncer colorectal, los tumores malignos son
conocidos como adenocarcinomas, ya que se trata de células cancerosas epiteliales
(Cooper, 2000).
2.2. Epidemiología del Cáncer Colorectal
En el intestino se presentan entre el 3 y 6% del total de los tumores que se desarrollan
en el tracto gastrointestinal. El 90% de los casos de cáncer de colon son esporádicos o
no hereditarios y en su mayoría son del tipo adenocarcinoma, que se desarrollan a
partir de células glandulares de la pared del intestino. En el caso del cáncer colorectal,
este ocupa el segundo lugar como causa de mortalidad por cáncer y para países como
Canadá, Finlandia y otros europeos es la primera causa (revisado en Ángel et al,
2004). Por ejemplo, en el Reino Unido se presentan 30.942 casos nuevos y 17.000
muertes al año por esta causa. En Estados Unidos se ha reportado un incremento en la
incidencia anual de cáncer de colon (98.757 a 131.200) en 24 años (1972-1997). En
promedio se presentan 133.000 casos nuevos de cáncer colorectal responsable por
55.300 muertes por año (revisado en Ángel et al, 2004).
En contraste, el centro y sur de América, Asia y África, se caracterizan por ser áreas
de bajo riesgo. En Colombia, según estadísticas del año 1995, se reportó una tasa de

5
mortalidad de 2.52 x 100.000 humanos solo por cáncer de colon. Unas 1.000
personas murieron ese año por esta causa, por lo cual el cáncer de colon se ubicó en
un séptimo lugar de causa de muertes por cáncer. En el año 1997, la tasa de
mortalidad por cáncer colorectal fue de 5.5 x 100.000 para las mujeres (ocupando el
cuarto lugar en causas de muerte por cáncer para este género) y de 5.9 x 100.000 para
los hombres (ocupando el quinto lugar). Se calcula que ese año solo en Bogotá
murieron 350 personas por cáncer colorectal, ocupando este un cuarto lugar de
muertes por cáncer después del cáncer de estómago, pulmón, y leucemia (revisado en
Ángel et al, 2004).
3. Etiología
3.1. Factores Genéticos
Entre la formas hereditarias de cáncer colorectal la más conocida es la poliposis
adenomatosa familiar (FAP). Sin embargo, esta solo es responsable del 1% de los
cánceres colorectales. La otra forma hereditaria es el síndrome de lynch o cáncer
colorectal hereditario sin poliposis (HNPCC) que representa alrededor del 4 al 6%,
siendo esta más difícil de diagnosticar por la falta de un marcador fenotípico como
son los pólipos. Los síndromes familiares de poliposis son desórdenes autosómicos
dominantes poco comunes. Tienen gran potencial de transformación maligna en
cáncer de colón con una frecuencia del 100%. En este caso el defecto genético se
encuentra en el gen APC en el cromosoma 5q21. Generalmente los pacientes generan
muchos adenomas colónicos formando una alfombra de estos en la superficie mucosa
(Robbins et al, 1999).
El HNPCC es una enfermedad causada por mutaciones en cualquiera de los cuatro
genes responsables de la reparación del DNA. Estos genes son: hMSH2 (cromosoma
2p22), hMLH1 (3p21), hPMS1 (2q31-33) y hPMS2 (7p22). Son conocidos como
“caretaker genes” ya que se encargan de cuidar que el DNA se encuentre en buen
estado durante los distintos puntos de restricción del ciclo celular. Su función radica

6
en detectar la presencia de inestabilidad de microsatélites a través del genoma. En
algunos casos, hay personas que nacen con un alelo mutado en alguno de estos genes.
A pesar de esto, tienen el otro alelo en forma silvestre que compensa la actividad. Sin
embargo, en el colon hay una alta tasa de mutación somática, que puede convertir el
alelo silvestre en uno mutado, causando así la perdida de función del gen. Este tipo de
mutaciones esporádicas forman parte de una vía alternativa para la formación de
cáncer de colon (revisado en Robbins et al, 1999).
Uno de los ejemplos de mutaciones causadas por la inestabilidad de microsatélites
son las mutaciones en PIK3CA observadas en un gran espectro de tumores, que
causan una ganancia de función en la actividad fosfatidilinositol-3 kinasa (PI3K). Las
PI3Ks son una gran familia de kinasas de lípidos que juegan un papel muy
importante en funciones celulares como la proliferación, diferenciación, quimiotaxis,
entre otras. Las PI3Ks pueden ser activadas por medio de la interacción con residuos
de fosfotirosinas de receptores de actividad tirosin kinasas o por unión de ras a su
subunidad catalítica p110. La PI3K activa por fosforilación al grupo 3´OH de los
fosfolípidos de inositol para generar el fosfatidilinositol 3,4,5 trifosfato (PIP3) que
activa proteínas de supervivencia como el complejo AKT/PKB (Velho et al, 2005).
Vale la pena resaltar que las mutaciones en PIK3CA ocurren en su mayoría en los
exones 9 y 20 de este gen, afectando principalmente los dominios kinasa de la
proteína y provocando cambios de amino ácidos como E542K, E545K y H1047R.
Estos cambios a nivel in vitro inducen un aumento en la actividad kinasa de la PI3K
que lleva a la activación de AKT e induce transformación oncogénica. Esto ha sido
visto hasta ahora en cultivos de fibroblastos embrionarios de pollo (Velho et al,
2005).

7
3.2. Características Moleculares del Cáncer Colorectal
3.2.1. Mutaciones en APC y β-Catenina
El gen del APC ha sido ubicado en la región 21 del brazo largo del cromosoma 5. La
proteína para la que codifica promueve la migración celular y la adhesión. Su función
más importante es inhibir la transcripción mediada por β-Catenina/TCF por medio de
la unión de APC a la β-Catenina, llevando a la degradación de la última por medio de
la vía ubiquitin-proteosoma. Al reducirse la afinidad del gen APC por la β-Catenina
por medio de la secreción de la proteína Wnt y el rompimiento del complejo, la β-
Catenina se acumula en el citoplasma y luego se transloca al núcleo donde arma un
complejo con el factor de células T (TCF) (Ver Figura 2). Cuando esto sucede se
pierde el contacto intercelular y aumenta la transcripción de genes promotores de
crecimiento, lo cual lleva al desarrollo carcinogénico. El complejo β-Catenina/TCF
juega un papel crítico en la regulación del desarrollo y proliferación del epitelio
intestinal (Guo et al, 2004; Robbins et al, 1999).
La mayoría de mutaciones somáticas del APC ocurren en el exón 15, muy conocido
como MCR (mutation cluster region) y son el 45% de las mutaciones de los tumores
esporádicos de colon. Por eso se le atribuye a esta región un estado de
hipermutabilidad, lo cual le confiere una ventaja selectiva a la formación de tumores.
Sin embargo, también se sabe que las mutaciones relacionadas con FAP están
distribuidas entre los codones 168 del exón 4 y el codón 1680 del exón 15
(Diergaarde et al, 2003; Frayling et al, 1998; Kapitanovic et al, 2004).

8
Figura 1. Esquema del papel de APC en la vía Wnt/ β-Catenina (Seminario de cátedra de la Dra.
Montserrat Corominas, Universidad de Barcelona, Barcelona, España, 2005).
Por otro lado, existen otras frecuencias de variantes subpolimórficas en APC en la
población con un efecto de penetrancia significante que pueden predisponer a la
incidencia de adenomas y carcinomas colorectales. Un ejemplo de esto pueden ser las
mutaciones en líneas germinales conocidas como I1307K y E1317Q que confieren un
riesgo incrementado de generación de tumores colorectales en la población general.
Las mutaciones en el gen supresor de tumores APC que resultan en perdida de
función, se piensa que son eventos de iniciación claves en cáncer colorectal familiar y
esporádico (Diergaarde et al, 2003; Frayling et al, 1998; Kapitanovic et al 2004).
En algunos pacientes con múltiples adenomas no tienen mutaciones en APC, por lo
cual su fenotipo seguramente resulta de variaciones en otros sitios del genoma. Una
de estas variaciones puede ser las mutaciones que se han observado en la β-Catenina
que la hacen resistente al efecto del APC por medio de una ganancia de función. En
cualquiera de los dos casos, pérdida de función de APC o ganancia de función de β-
Catenina, el resultado será una acumulación de β-Catenina que aumentará la
transcripción de genes como c-Myc y ciclina D1, llevando a la proliferación de
células epiteliales mutadas y la formación de adenomas (Michor et al. 2005). Es
importante anotar, que la activación de la vía de la β-Catenina juega un papel muy

9
importante durante la iniciación y la promoción de las diferentes etapas de la
carcinogénesis de colon (Arango et al, 2001; Frayling et al, 1998; Yamada et al
2003).
El desarrollo y diferenciación celular del epitelio intestinal también está relacionado
con la actividad de proteínas de homeodominios relacionados con la zona caudal
llamados Cdx1 y Cdx2. Estos son factores de transcripción requeridos para la
expresión de ciertos genes específicos del intestino, promueven el desarrollo del
epitelio intestinal durante la embriogénesis y promueven la expresión de la
morfología celular columnar madura. Por medio de la realización de estudios se ha
observado que Cdx1 inhibe la proliferación celular y se ha determinado que esto es
debido a la inhibición de la actividad kinasa de la ciclina D1 durante la fase G1 (Guo
et al, 2004). La expresión de Cdx1 y Cdx2 puede inhibir la actividad transcripcional
de la vía Wnt/ β-Catenina. Sin embargo, se ha observado que para la expresión de
Cdx1 en las criptas intestinales se necesita de la vía de señalización Wnt/ β-Catenina,
por lo cual se cree que Cdx1 actúa como un mecanismo de retroalimentación
negativa. Este es un mecanismo muy común de regulación en las vías de señalización
(Guo et al, 2004).
3.2.2. Metilaciones
La metilación de citosinas en regiones ricas en dinucleotidos de CpG son
modificaciones epigenéticas del DNA. Usualmente estas islas de CpG marcan los
promotores de muchos genes. El genoma cancerígeno está caracterizado en muchos
casos por la silenciación de ciertos genes supresores de tumores, resultado de estas
metilaciones aberrantes en las islas CpG. La DNA metiltransferasa mantiene estable
los patrones de metilación del DNA después de la replicación por medio del
reconocimiento y metilación del DNA hemimetilado. La metilación puede estar
relacionada con la alta frecuencia de mutaciones en las cuales se cambia C por T en
genes de tumores humanos (Li et al, 2003).

10
La inactivación de los genes relacionados con la reparación del DNA esta asociada
con la inestabilidad cromosómica y un fenotipo mutador. Por otro lado, también la
hipermetilacion de las islas CpG en el extremo 5’ de el gen hMLH1 se encuentra en
la mayoría de canceres colorectales esporádicos primarios y esta ampliamente
relacionada con la inestabilidad microsatélite. A esto se ha relacionado con un
fenotipo mutador o a exposición de cancerígenos. Este fenómeno ha sido observado
en condiciones fisiológicas tales como la inactivación del cromosoma X y en el
imprinting genómico. La metilación aberrante de islas CpG se ha observado en
desórdenes genéticos como en el síndrome X frágil, neoplasia y células en
envejecimiento (Herman et al, 1998; Toyota et al, 1999).
La edad es conocida como un factor de predisposición al cáncer y la metilación
relacionada con la misma podría ser un evento temprano en el proceso de
tumorogenesis. Según Toyota et al, 1999, por medio de la amplificación de islas CpG
se examinaron 30 secuencias metiladas de DNA y se observó que 19 de ellas habían
sido metiladas progresivamente de una forma dependiente de la edad en colon normal
llamada hipermetilación de tipo A. Por otro lado 7 de las secuencias fueron metiladas
en una manera propia del cáncer (hipermetilación de tipo C), pero en su mayoría
ocurrió en células colónicas normales durante el proceso de envejecimiento (Toyota
et al, 1999).
La hipermetilación de tipo A usualmente resulta por procesos fisiológico más que
por alteraciones genéticas, ya que es muy frecuente y afecta un gran número de
células, está presente en todos los individuos, no solo pacientes con cáncer; y este
proceso es específico de ciertos genes y tejidos. Esta metilación, como muchos
procesos celulares, depende de un equilibrio entre factores positivos (de metilación) y
factores negativos (protectores) y este balance puede favorecer metilaciones de novo
que se ven reflejadas en hipermetilación progresiva a través de divisiones celulares
repetidas (Toyota et al, 1999).

11
A diferencia de la hipermetilación de tipo A, la de tipo C es relativamente infrecuente
en cáncer colorectal y nunca está presente en la mucosa normal del colon. Este tipo
muestra un patrón revelador, que sugiere la presencia de un fenotipo hipermetilador.
La hipermetilación de tipo C no se observa en tumores que no tenga un fenotipo
mutador de islas CpG. Como muchos genes son candidatos potenciales de metilación
de promotores, el fenotipo mutador de islas CpG puede tener consecuencias
patofisiológicas en la neoplasia por medio de la inactivación de genes supresores de
tumores, genes supresores de metástasis, inhibidores de angiogénesis, y una
deficiencia de reparación de DNA por medio de la inactivación del promotor del gen
hMLH1 (Toyota et al, 1999).
3.2.3. Dieta
Se ha sugerido que ciertos hábitos alimentarios pueden afectar la incidencia de cáncer
colorectal. Más aún, el hecho de que algunos y no todos los pólipos se conviertan en
adenocarcinomas, sugiere la presencia de carcinógenos en el contenido del lumen
intestinal (Bruce et al, 2000). Se han estipulado cuatro hipótesis fundamentales en
cuanto a los factores de la dieta que influyen en el riesgo de contraer cáncer de colon.
En primer lugar se ha estipulado que el riesgo de contraer cáncer de colon está
relacionado epidemiológicamente con el consumo de carne frita, probablemente por
la presencia de aminas aromáticas heterocíclicas (Bruce et al, 2000; Sesink et al,
1999).
La segunda hipótesis dice que en el tracto gastrointestinal, la acción de agentes
nitrogenados como NO y N2O3 junto con aminas pueden formar compuestos N-
nitrosos. Estos aunque se ha probado que son mutagénicos in vitro, no han sido
probados in vivo (Bruce et al, 2000; Sesink et al, 1999).
La tercera hipótesis, propuesta por Burkitt en 1971, ha sido muy bien documentada y

12
se basa en la relación que existe entre el bajo consumo de fibras vegetales, el alto
consumo de grasas animales, colesterol y proteínas y la frecuencia elevada de cáncer
intestinal. Se ha observado que la ingestión alta de grasa en la dieta produce aumento
en la excreción de ácidos biliares y de colesterol, que son transformados por las
bacterias del colon en ácidos biliares secundarios y esteroles fecales, que pueden ser
metabolizados en esteroides carcinógenos, como los fenantrenos (Bruce et al, 2000;
Sesink et al, 1999).
Por otro lado los productos de la pirolisis en comidas cocinadas pueden iniciar el
cáncer de colon y la ingestión de grasas puede promoverlo. Es por esto la importancia
que se le atribuye al consumo de frutas y vegetales, ya que estos contienen fibras
vegetales y fitoquímicos que actúan como antioxidantes e inhiben la carcinogénesis.
Por otro lado, las fibras vegetales aumentan el tránsito intestinal y disminuyen la
acumulación de los alimentos en el mismo (disminuyendo la exposición del colon a
carcinógenos), absorben carcinógenos, modifican la microflora intestinal, disminuyen
el pH del colon e incrementan las concentraciones de ácidos grasos de cadenas
cortas, entre otras cosas (Bruce et al, 2000; Sesink et al, 1999; Williams et al, 1996).
La cuarta hipótesis sugiere que el consumo excesivo de energía en la dieta resulta en
el exceso de triglicéridos, ácidos grasos y desarrollo de resistencia a insulina, que
resulta en la necesidad de mayores niveles de insulina para disponer de glucosa en
plasma. La hiperinsulinemia actúa como un factor de crecimiento y promotor de
tumores. El exceso de energía dietaria eleva los niveles intravasculares de insulina y
substratos de energía tales como carbohidratos y lípidos en sangre. La exposición
excesiva a hormonas y el exceso de energía disponible para las células epiteliales
estimulan las vías de señalización celular que no afectaría en nada a las células
normales, pero si incrementa la proliferación de las células con problemas de control
de ciclo celular. Este fenómeno también incrementa la formación de ROI
(intermediarios reactivos del oxigeno) que pueden llegar a ser dañinos para el
hospedero (Bruce et al, 2000; Hope et al, 2005; Sesink et al, 1999).

13
3.2.4. Microbiota
Se conoce que el colon humano es un ecosistema microbiano complejo, alojando
cientos de especies bacterianas. Este micro ecosistema se forma básicamente por la
interacción entre la fisiología del individuo y las bacterias introducidas en el mismo
después del nacimiento, manteniéndose constante después de alcanzar la edad adulta.
Sin embargo la flora intestinal puede ser modificada como resultado del ambiente en
el cual vive el organismo, que depende de factores tales como la dieta, el uso de
antibióticos y la edad. En el colon ocurren procesos tan importantes como la
degradación de carbohidratos y otros nutrientes. Los polisacáridos que no son
digeridos al entrar al colon, son reducidos a ácidos grasos de cadena corta (Short
Chain Fatty Acids- SCFA) y luego son absorbidos por difusión pasiva. Ejemplo de
estos ácidos grasos son el butirato, propionato y acetato. Sin embargo esta conversión
es dependiente de la disponibilidad del sustrato y es más abundante en el lado
derecho del colon (Hope et al, 2005).
Los SCFA juegan un papel importante en el mantenimiento de la integridad de la
capa epitelial del colon y la energía necesaria para esto es tomada en un 70% del
butirato. Esta podría ser la razón por la cual la mayoría de los canceres colorectales se
dan en la parte distal o izquierda del colon, donde la concentración de SCFAs es
menor y el transito por este segmento es más lento, permitiendo que las bacterias y el
material tengan mayor contacto con el lumen del colon. También se ha demostrado
que el butirato induce diferentes niveles de apoptosis, detención del ciclo celular y
diferenciación celular en diferentes modelos animales y líneas celulares de cáncer de
colon (Hope et al, 2005; Williams et al, 1996).
Se cree que organismos tales como las bacterias productoras de ácido láctico (LAB)
confieren beneficios como la estimulación del sistema inmune por mecanismos no
patológicos, evitan la colonización del intestino por especies patógenas y aun más
importante protegen contra tumores en el colon por medio de influencia metabólica,

14
inmunológica y de funciones protectoras del colon. Todo esto se ha visto demostrado
por la prevención de la formación de tumores por carcinógenos en animales modelos
que aumentan el volumen de consumo de LAB. Se sabe que la actividad metabólica
de la microbiota, es responsable de la formación de productos como los (ROI), que
ya fueron nombrados anteriormente (revisado en Hope et al, 2005).
Las especies bacterianas más predominantes en estudios de cáncer de colon son la E.
coli y especies relacionadas. Sin embargo, no se sabe si la presencia de estas bacterias
es debida a la patología o si son la causa de la misma. En los estudios realizados en
mucosa de pacientes enfermos de cáncer y pacientes no enfermos muestran que en los
primeros hay presencia de bacterias asociadas a la mucosa y bacterias asociadas a
intramucosa, mientras que en los últimos no se encontró una cantidad significativa de
bacterias aeróbicas (revisado en Hope et al, 2005).
Diferentes estudios demuestran que la microbiota intestinal afecta el contenido de
mucina, grueso, composición y estructura de la capa mucosa pre-epitelial en el colon.
También se ha demostrado que las bacterias comensales pueden inducir patrones
complejos de glicosilación en el colon, cambiando la distribución celular y subcelular
de glicanos. Las alteraciones en la superficie de glicosilación del hospedero es una
característica universal del proceso neoplásico, en cambios que ocurren en pólipos
adenomatosos y metastáticos, cáncer colorectal y otros desordenes asociados
(revisado en Hope et al, 2005).
3.3. Modelo Secuencia Adenoma-Carcinoma
La forma más ilustrativa de entender el proceso neoplásico en el colon es por medio
de la secuencia adenoma-carcinoma descrita por Volgestein (Figura 1). Esta decribe
el desarrollo de carcinomas partiendo de lesiones adenomatosas que son masas
tumorales benignas que protruyen al lumen del colon y que son muy comunes en la
población adulta. Los adenomas, a veces llamados pólipos, son derivados del epitelio

15
colónico normal y se pueden formar como resultado de la maduración anormal de la
mucosa e inflamación. (Robbins et al, 1999; Williams et al, 1996).
Todos los adenomas aparecen como el resultado de displasia proliferativa epitelial,
que puede variar de leve a severo. Hay evidencia de que los adenomas son una lesión
precursora a la formación de adenocarcinomas colorectales invasivos. Se requiere un
periodo aproximado de 10 años para que el adenoma pase a estado de
adenocarcinoma y de 3 a 5 años para que se pueda producir el proceso de metástasis
(Figura 1). Se reporta, que en algunos casos se puede observar cambios displásicos en
la mucosa del colon sin pasar previamente por el estado de pólipo (Hope et al, 2005;
Robbins et al, 1999; Li et al, 2003).
Los pólipos hiperplásicos han sido menos caracterizados que los adenomas, pero
están epidemiológicamente relacionados con el cáncer colorectal. En la mucosa
normal del colon las células epiteliales van madurando a medida que migran hacia la
luz intestinal a través de la columna críptica hasta que alcanzan su estado de
diferenciación y mueren por apoptosis. En la formación de pólipos hiperplásicos la
migración celular es más lenta y las células maduras se mantienen en la superficie.
Por tanto, los individuos que desarrollan pólipos hiperplásicos, tienen un mayor
riesgo de desarrollar cáncer colorectal, ya que estos pólipos presentan inestabilidad
genética representada por múltiples mutaciones en oncogenes, genes supresores de
tumores y genes de estabilidad que pueden ser heredadas (mutaciones germinales) o
adquiridas (mutaciones somáticas). Las mutaciones somáticas pueden darse de dos
formas: por inestabilidad de microsatélites o inestabilidad cromosómica. En el primer
caso, se da un aumento en la tasa de mutaciones puntuales y en el segundo se da un
aumento en la tasa de cambios aberrantes en la organización cromosómica (Robbins
et al, 1999; Vogelstein & Kinzler, 2004; Young et al, 2001).

16
Figura 2. Diagrama de la secuencia adenoma-carcinoma descrita por Volgestein. Se describen cada
uno de los pasos que intervienen en la transformación de una célula colónica normal a displásica
(Fearon, Vogelstein, 1990).
3.3.1. Bases Moleculares de la Secuencia Adenoma-Carcinoma
Los genes que son protagonistas en el desarrollo de la secuencia Adenoma-
Carcinoma son:
APC (5q21): El evento genético más temprano y más prevalente conocido en el
desarrollo del cáncer colorectal, es la alteración de la vía de señalización del
Adenomatosus Polyposis Coli (APC). Se sabe que esta vía se encuentra afectada hasta
en un 95% de los casos de cáncer colorectal (Ver Mutaciones en APC y β-Catenina)
(Michor et al. 2005).
K-RAS (12p12): Las proteínas para la cual codifica este gen está anclada a la cara
citoplasmática de la membrana celular y se une a los nucleótidos de guanina GTP y
GDP y se cree que funciona como un “interruptor” en los eventos de señalización
transmembranales de crecimiento celular y diferenciación (Singh et al, 1997). Las
mutaciones en los genes ras están fuertemente implicados en el proceso neoplásico.
Los proto-oncogenes ras constituyen una familia de genes altamente conservados

17
(HRAS1, KRAS2 y NRAS). Evidencia reciente indica que la activación de los proto-
oncogenes, junto con la inactivación de genes supresores de tumores, induce al
fenotipo maligno en células colónicas. Las mutaciones de los genes ras, permite que
su identificación temprana en tejido no carcinómico provea de un marcador clínico
que permita identificar pacientes con mayor riesgo de contraer cáncer de colon en la
población (Burmer & Loeb, 1989; Fodde et al, 2001).
DCC (18q21): Deleted in colon cancer es un gen que codifica para una proteína de
adhesión que se expresa normalmente en la mucosa, per su expresión se reduce
considerablemente en lesiones displásicas. Hay evidencia que sugiere que DCC es un
gen supresor de tumores. Esta incluye mutaciones observadas en algunos cánceres
que presentan pérdida alélica en 18q y la disminución de la expresión de la proteína
DCC en líneas celulares de cáncer de colon (Bapat et al, 1997; Robbins et al, 1999).
p53 (17p13): El gen p53 codifica para una proteína de localización nuclear que
funciona como regulador de la transcripción de un número muy elevado de genes. Por
ejemplo, regula la actividad de los genes p21 y Bax. Sin embargo, si el gen de la p53
se encuentra mutado, se producirán alteraciones en la proliferación y muerte celular
(Robbins et al, 1999; Tanaka et al, 2000). Aproximadamente el 50% de todos los
tumores humanos 8 presentan mutaciones en el gen p53. La pérdida de función de la
proteína codificada por este gen se observa en su mayoría en los carcinomas de colon,
más no en los adenomas. Esto sugiere que esta mutación ocurre en las etapas tardías
de la carcinogénesis (Hayward et al, 2004; Russo et al, 2002).
C-MYC: Este oncogen se encuentra entre los genes activados por la β-Catenina y
codifica para la proteína que lleva el mismo nombre. Esta proteína es estimulante de
crecimiento celular y proliferación (Robbins et al, 1999).

18
3.3.2. Características Histopatológicas
El cáncer de colon se clasifica de acuerdo a la extensión de las células cancerosas por
la mucosa del colon, por áreas vecinas y/o a distancia. Tiene la siguiente
clasificación:
·Etapa 0 o carcinoma in situ. Células cancerosas solo en tejidos superficiales del
colon.
·Etapa I. (cáncer del colon Dukes A) Células cancerosas fuera de la capa mas interna
del colon a la segunda y tercera capas e implica la pared interior del colon, pero no a
la pared exterior del colon.
·Etapa II. (Cáncer del colon Dukes B). Células cancerosas diseminadas fuera del
colon a los tejidos vecinos, pero no a los ganglios linfáticos.
·Etapa III. (Dukes C). Células cancerosas diseminadas fuera del colon y a los
ganglios linfáticos vecinos, pero no a otros órganos .
·Etapa IV. (Dukes D). Células cancerosas diseminadas fuera del colon y por otros
órganos del cuerpo.
·Recurrente. Cuando vuelven a aparecer células cancerosas una vez recibido
tratamiento, pueden aparecer en el colon o en otra parte del cuerpo (hígado,
pulmones) (López, 2002).
3.3.3. Bases Celulares y Moleculares del Cáncer Colorectal
El ciclo celular es un proceso complejo relacionado con el crecimiento y
proliferación de las células, el desarrollo de los organismos y la regulación de los

19
mecanismos de reparación de los daños en el DNA (Vermeulen et al, 2003).
El correcto funcionamiento del ciclo celular requiere del trabajo de diferentes
complejos enzimáticos que controlan el proceso. El trabajo de estos complejos
ciclinas/CDKs es dirigir a la célula de una fase a otra del ciclo celular. Los complejos
Ciclina D/CDK6 y Ciclina D/CDK4 son importantes para la entrada a la fase G1, el
complejo Ciclina E/CDK2 se requiere para la entrada de la célula a la fase S y el
complejo Ciclina A/CDK2 es importante durante la fase S. La Ciclina A se acopla a
la CDK1 en la etapa tardía de G2 para promover la entrada de la célula a la Mitosis,
que es regulada por el complejo Ciclina B/CDK1. Estos complejos son regulados por
moléculas conocidas como Inhibidores de Kinasas dependientes de Ciclinas (CKI),
tales como los miembros de la familia INK4 (p15, p16, p18, p19) y de la familia
Cip/Kip (p21, p27, p57) (revisado en Vermeulen et al, 2003).
Debido a la importancia del ciclo celular, existen puntos de chequeo a través del
mismo en donde se asegura que la célula está lista para entrar al ciclo (R), que el
DNA no presente daños para poder entrar a la fase de síntesis (G1/S) o que ha sido
replicado de forma correcta (G2/M). Cuando hay errores en el DNA, la célula no
puede pasar de los puntos de restricción del ciclo y la célula entra en un estado de
parada. Por medio de la inducción de la p53 (guardián del genoma) se activa todo un
mecanismo de corrección que de no tener solución la célula estará destinada a sufrir
apoptosis. Entre estos mecanismos de corrección se encuentra la inducción de la p21,
que por medio de la unión al antígeno nuclear de proliferación celular (PCNA),
inhibe la síntesis de DNA, permitiéndole a la célula corregir sus daños (Schafer
1998; Tanaka et al, 2000; Williams et al, 1996).
La apoptosis es una forma de muerte celular. Se diferencia de la muerte por necrosis
por las características morfológicas específicas como el encogimiento celular, perdida
del contacto entre células, condensación de la cromatina y degradación del DNA
entre nucleosomas. La apoptosis es un fenómeno necesario para el mantenimiento de

20
la homeostasis celular. En el adulto la apoptosis es importante para el mantenimiento
del número de células en aquellos tejidos que sufren renovación celular constante.
Adicionalmente, la muerte celular programada permite eliminar células
potencialmente peligrosas y células dañadas, incluyendo aquellas que contienen
mutaciones que pueden llevar al desarrollo del cáncer (Tanaka et al, 2000; Cooper,
2000). El proceso de muerte celular programada presenta una fase iniciadora, una
amplificadora y una ejecutora. La fase iniciadora, conocida también como la ruta de
los receptores de muerte, consiste en la activación de receptores de membrana (por
ejemplo FAS/CD95) por medio de la unión de un ligando (FASL/CD95L). Tal unión
induce el reclutamiento de las proteínas de dominio de muerte de unión a FAS
(FADD), múltiples moléculas de procaspasa 8, resultando en la activación de la
caspasa 8 que puede ser inhibida por c-FLIP (Hengartner, 2000; Evan & Vousden,
2001). La fase amplificadora funciona por medio de la respuesta de la mitocondria a
señales extracelulares e insultos como daños en el DNA, por medio de la activación
de miembros proapoptóticos de la familia Bcl-2 (Bax, Bad, Bim y Bad). Estas
moléculas forman dímeros que permiten por medio de la formación de canales, la
salida de otras tales como el citocromo c. Este se une al Apaf-1 y a la procaspasa 9,
formando el apoptosoma (Hengartner, 2000; Evan & Vousden, 2001).
Por último está la fase ejecutora de la apoptosis, que es en donde convergen la vía
iniciadora y la amplificadora a nivel de la activación de la caspasa 3. Esta molécula
puede ser activada por la caspasa 9 y la caspasa 8 e inhibida por las proteínas
inhibidoras de la apoptosis (IAPs), que a su vez son inhibidas por Smac/DIABLO. A
partir del proceso de activación de la caspasa 3, se desata una cascada de eventos que
terminan por producir una muerte ordenada de la célula (Hengartner, 2000; Evan &
Vousden, 2001).
Las células epiteliales intestinales (IEC) se derivan de células madre que yacen en la
base de las criptas. Las IEC migran a través de las criptas y llegan al lumen y mueren
entre 3 y 4 días. Esta muerte se da por un tipo de apoptosis conocida como anoikis.

21
Este proceso consiste en que las células pierden el anclaje, se despegan y finalmente
sufren exfoliación. Varios factores intrínsecos, incluyendo la expresión de proteínas
de la familia Bcl-2 y varios factores ambientales tales como pérdida del contacto
célula-célula y célula-matriz extracelular, composición de la matriz extracelular,
ácidos grasos de cadena corta, expresión de integrinas y citoquinas contribuyen a la
inducción de apoptosis en la superficie del lumen. Adicionalmente, se conoce la
muerte celular inducida por desprendimiento (DICD) que es una forma reconocida de
apoptosis en IEC humanas recientemente aisladas (Grossmann et al, 1998; Lamprecht
& Lipkin, 2002).
En el caso de las IEC, las caspasas traducen y aumentan la señal apoptótica por medio
de la activación de otras caspasas. Estas proteasas inducen apoptosis por medio de la
proteólisis de importantes proteínas estructurales y funcionales, tales como la poli
(ADP-ribosa) polimerasa (PARP) y las láminas A. En las IEC se expresan las
Caspasas 1 y 10, pero permanecen inactivas durante la DICD, mientras que las
Caspasas efectoras 6 y 3 si se encuentran activas, asociándolas al clivaje de PARP y
de las láminas A Adicionalmente, la disminución en la expresión de Bcl-2 y el
aumento en la expresión de Bax y Bak en IEC a medida que van llegando a la
superficie del lumen, sugiere que estas células se vuelven más susceptibles al
ambiente pro apoptótico de la superficie (revisado en Grossmann et al, 1998).
3.4. Prevención y tratamiento
La quimioterapia hace referencia a la administración de medicamentos antitumorales
en forma sistémica por vía venosa u oral. Estos medicamentos entran al torrente
sanguíneo y se distribuyen a todos los tejidos del cuerpo.
3.4.1. 5-Fluorouracilo
El 5-fluorouracilo (5- FU) fue desarrollado por Heidelberger et al, 1957 y desde

22
entonces se ha revelado su actividad antitumoral convirtiéndolo en un tratamiento
quimioterapéutico importante para muchos tipos de cáncer, principalmente el
colorectal. El 5-fluorouracilo es un antimetabolito sintetizado a partir del uracilo, por
medio de la sustitución de un Hidrógeno por una Fluorina en su quinta posición
(Figura 3). La citotoxicidad del 5FU se debe a la incorporación de fluoronucleotidos
en el RNA y el DNA y a la inhibición de la enzima Timidilato Sintasa (TS) (Longley
et al, 2003; Tanaka et al, 2000; Papamichael, 2000).
Figura 3. Estructura química del Uracilo y el 5 Fluorouracilo (tomada de Tanaka et al, 2000).
Tanto el Uracilo como el 5FU se incorporan entre las bases de los ácidos nucleicos
de las células tumorales, provocando daños a nivel del DNA, bloqueando su
replicación y su reparación. Este proceso ocurre cuando el 5FU es fosforilado y
entonces convertido a otros metabolitos activos. Las posibles vías que puede tomar la
fosforilación del 5FU incluyen por un lado la conversión en FUMP
(5─fluororidina─5’monofosfato) y posteriormente en FUDP (5─fluorouridina─5’
difosfato) y por último en FUTP (5─fluororidin─5’trifosfato). El FUTP se incorpora
en el RNA causando su disfunción (Tanaka et al, 2000). Es importante recordar que
el FUDP se puede convertir en FdUMP
(5─fluorouridina─2’─deoxiuridina─5’─monofosfato) por medio de FdUDP
(5─fluoro─2’─deoxiuridina─5’difosfato). En este caso, el FdUMP forma un
complejo con la timidilato sintasa (TS) inhibiendo su actividad. La TS cataliza la
metilación reductora de la deoxiuridina monofosfato (dUMP) a deoxitimidina
monofosfato (dTMP), con el 5,10 metilenotetrahidrofolato (CH2THF) como donador
del grupo metilo (Tanaka et al, 2000). De esta forma el FdUMP se une al sitio de

23
unión de nucleótidos (sitio de unión de la dUMP) de la TS, generándose un complejo
ternario entre la TS, FdUMP y CH2THF (Ver Figura 4). El cambio de dUMP a dTMP
genera timidilato, precursor esencial de la síntesis de DNA, la inhibición de la
timidilato sintasa lleva a daños en el DNA, bloqueando su síntesis e induciendo la
apoptosis de las células tumorales de forma dependiente de P53 por medio la vía
intrínseca. Es de gran importancia el hecho de que el 5FU es agente terapéutico no
dependiente de la dosis sino más que todo del tiempo. (Bunz et al, 1999; Hayward et
al, 2004; Longley et al, 2003; Papamichael, 2000; Russo et al, 2002; Tanaka et al,
2000).
Figura 4. Mecanismo de inhibición de la Timidilato Sintasa por el 5FU (Tomada de Longley et al,
2003).
Uno de los principales problemas que presenta la terapia basada en el 5FU, es que el
80% del 5FU administrado es catalizado en el hígado por la Dihidropirimidina
dehidrogenasa (DPD), convirtiendo al 5FU en dihidrofluorouracilo (DHFU). Por otro
lado, los pacientes deficientes en DPD experimentan una alta toxicidad en respuesta
al tratamiento con 5FU, debido a su catabolismo disminuido y consecuente
exposición prolongada al quimioterapéutico ( revisado en Longley et al, 2003).
Existen otras enzimas relacionadas con el metabolismo del 5FU, incluyendo la
fosforibosil-transferasa, uracil dehidrogenasa, uracil fosforilasa, uridin sintasa,

24
fosfatadin difosfoesterasa, ribonucleotidil reductasa, y timidin kinasa. Muchas de
estas enzimas proveen blancos racionales para el desarrollo de drogas e
investigaciones clínicas (Wang et al, 2004).
3.4.2. Quimioprevención
Durante las últimas décadas la investigación se ha basado en la identificación de
nuevas terapias para el tratamiento del cáncer. Sin embargo, la tendencia ha ido
modificándose con la aparición de la quimioprevención que consiste en la utilización
de sustancias químicas, naturales o sintéticas que buscan evitar o revertir el desarrollo
de la carcinogénesis. Por medio de la utilización de anticarcinógenos el cáncer puede
prevenirse o controlarse mediante la interferencia de los factores que intervienen en la
iniciación (cambios genéticos iniciales), promoción (progresión de la célula iniciada
en el proceso carcinogénico con cambios fenotípicos) o progresión. La identificación
de carcinógenos y anticarcinógenos naturales, le permite conocer más el proceso de la
carcinogénesis y abre una ventana para la oncología, ya que esta vía podría reducir
notablemente la incidencia de esta enfermedad a nivel mundial. Ya que el cáncer es
un proceso tan heterogéneo en cuanto al número de eventos celulares que lo provocan
en las diferentes líneas celulares, un anticancerígeno funcionará para un tipo de
células pero no es seguro que funcionará para otro. En estudios realizados en
personas que mantienen una alimentación a base de dieta mediterránea, se observó
que aquellos que consumían altos niveles de aceite de oliva, presentaban una menor
incidencia de cáncer de colon en comparación con pacientes que consumían aceites a
base de maíz y girasol u otro tipo de aceites no marinos (revisado en Rao et al, 1998).
Inicialmente se había observado que el aceite de oliva tenía efectos anticancerígenos,
pero no se sabía exactamente cual era el componente que producía tal efecto. Ahora
se sabe que el escualeno, que es una pieza clave en la biosíntesis del colesterol, ácidos
biliares y esteroles, es el componente del aceite de oliva que tiene actividad

25
antitumorigénica. Este compuesto tiene características muy positivas en comparación
con anticancerígenos sintéticos como que se produce endógenamente y está presente
en la alimentación humana. Se ha observado que en animales como los tiburones,
que tienen niveles basales de escualeno en sus tejidos, son inmunes al cáncer. Es
posible que su efecto inhibitorio se deba a la modulación de la vía biosintética del
colesterol, por medio de la reducción de intermediarios tales como el mevalonato,
geranil pirofosfato y el farnesil pirofosfato. Este último es un recurso para la
farnesilación de oncogenes tales como ras, permitiendo su activación completa. Por
lo tanto la inhibición de esta vía o de sus intermediarios, evita la activación de
proteínas oncogénicas como agentes de transducción de señales en la regulación de la
actividad transformante de las células (revisado en Rao et al, 1998).
Otro agente quimiopreventivo muy importante para el cáncer de colon es la Vitamina
D3 y sus análogos. Es de gran importancia pues es un quimiopreventivo que actúa
por medio de vías alternas a p53, que se encuentra mutada en una gran variedad de
cánceres de colon. Su metabolito activo es la 1 α, 25- 1,25(OH)2 D3. Su acción
preventiva ha sido demostrada en modelos animales, donde la ausencia de 1 α, 25-
1,25(OH)2 D3 permite hiperproliferación celular y baja migración de células maduras
por la cripta del colon. El papel básico de la 1 α, 25- 1,25(OH)2 D3 es la inhibición
de crecimiento de las células colónicas tumorales y promover la diferenciación
celular. Este metabolito activo se comporta como una hormona esteroidea y regula la
transcripción génica uniéndose a un receptor nuclear de Vitamina D conocido como
VDR. Este es un receptor dependiente de ligando que se une a sitios específicos del
DNA conocidos como VDRE que activan o reprimen la transcripción de genes
reguladores (revisado en Díaz et al, 2000).
En las células tratadas con 1 α, 25- 1,25(OH)2 D3 la apoptosis ocurre por un proceso
de diferenciación que conlleva a la apoptosis. Las células tumorales de adenomas
colónicos son más sensibles a la 1 α, 25- 1,25(OH)2 D3 en etapas tempranas del
proceso neoplásico, mientras que las células de carcinoma responden a ella en etapas

26
tardías del proceso. Uno de los compuestos análogos de la 1 α, 25- 1,25(OH)2 D3 es
el EB1089, que mantiene el poder antiproliferativo de este metabolito activo, pero
reduce la hipercalcemia observada en los pacientes. Se sabe que actúa bloqueando la
progresión celular manteniendo a las células en fase G1 por medio de la regulación de
la p21 (Díaz et al, 2000). Cabe anotar que durante el tiempo de exposición se ve
aumentada la concentración de Bak, que es una proteína proapoptótica de la familia
de Bcl2. Bak aumenta en respuesta a EB1089 más que con 1 α, 25- 1,25(OH)2 D3 en
la superficie de la cripta, con una mayor expresión en la células maduras de la
superficie. Los estudios realizados demuestran que EB1089 puede ser un
quimiopreventivo eficiente para los individuos con alto riesgo de desarrollar cáncer
colorectal (Díaz et al, 2000).
3.4.3. Clorofilín
Por otro lado se ha encontrado el efecto quimiopreventivo de la molécula clorofilín,
que es antimutagénica y anticarcinógena. Es una molécula derivada de la clorofila
que arma complejos con carcinógenos aromáticos como las aminas heterocíclicas,
aflatoxinas y carbonos aromáticos policíclicos, reduciendo la toma de carcinógenos
por el colon. Esta molécula altera el espectro mutacional en la vía de la β- Catenina
en tumores de modelos animales e induce a apoptosis acompañada de diferenciación
celular por medio de una vía independiente de citocromo-c. (Díaz et al, 2003).
Como se discutió anteriormente, la dieta es un determinante del riesgo de contraer
diversos tipos de cáncer como el de colon. Muchos constituyentes químicos de los
vegetales han sido purificados y se ha demostrado que protegen contra la
carcinogénesis en diversos modelos animales (Neault & Tajmir-Riahi, 1999).
Las clorofilas y sus derivados hidrosolubles (clorofilinas) son constituyentes de la
dieta humana y se ha encontrado que son anticarcinógenos efectivos. Las clorofilinas
son una mezcla de sales de cobre de la clorofila (Ver Figura 5) que también son

27
potentes antimutagénicos y anticarcinógenos al ser administradas durante la
exposición a carcinógenos. Estudios mecanísticos sugieren que el clorofilín podría
actuar como un interceptor de moléculas por medio de la formación de complejos
moleculares con carcinógenos impidiendo su absorción, reduciendo la formación de
aductos en el DNA. Se conoce que es un potente inhibidor del citocromo p450, que se
relaciona con la activación de varios carcinógenos ambientales. En el caso de la
Aflatoxina B1, esta sufre catálisis oxidativa por el citocromo p450 convirtiéndose en
aflatoxina 8,9 epoxide, que puede formar aductos con el DNA por medio de unión
covalente con el átomo de N7 de la guanina. También actúa como antioxidante
inhibiendo la peroxidación de lípidos. Una de las ventajas del uso de CHL es que al
ser derivado de la clorofila, el clorofilín está al alcance de la población con una dieta
rica en vegetales (Carter et al, 2004; Díaz et al, 2003; Egner et al, 2001; Neault &
Tajmir-Riahi, 1999).
Figura 5. Estructura química dela Clorofila y el Clorofilín. El Clorofilín se diferencia de la Clorofila
por la ausencia del grupo Fitol, la presencia de Cu en lugar de Mg y 3 Na+ (Tomado de Carter et al,
2004).
Se ha observado como el clorofilín arma complejos con carcinógenos aromáticos
como las aminas heterocíclicas, aflatoxinas y carbonos aromáticos policíclicos,
reduciendo la toma de carcinógenos por el colon. Esta molécula altera el espectro
mutacional en la vía de la β- Catenina en tumores de modelos animales. En estudios

28
realizados en la línea celular de cáncer de colon HCT116, se demostró que el CHL
induce a apoptosis acompañada de diferenciación celular por medio de la caspasa 8 y
6 que lleva a la formación de tBid, que con la ayuda de Bak y otros miembros de
Bcl2, facilitan la liberación de AIF. La apoptosis mediada por clorofilín en este
estudio, demostró no ser mediada por la liberación de citocromo c. En resumen, el
CHL activa la vía apoptótica de los receptores de muerte, llevando al clivaje de la
caspasa 8, liberación de AIF por la mitocondria y la actuación de eventos que llevan a
la destrucción de las láminas nucleares. (Ver Figura 6) (Carter et al, 2004; Díaz et al,
2003).
Figura 6. Modelo para el mecanismo de apoptosis inducido por CHL en células HCT116 (Tomado de
Díaz et al, 2003).
Por otro lado, se ha demostrado en células HCT116 que el CHL induce fuertemente,
entre otros, a la E-Cadherina y la fosfatasa alcalina, dos indicadores de diferenciación
celular. La expresión de E-Cadherina se localiza principalmente en la membrana
plasmática. Se da un decremento de la ß-Catenina a nivel nuclear en más de un 50%.
Esto podría explicarse por una redistribución de la ß-Catenina fuera del núcleo hacia
el citoplasma y una subsecuentemente traslocación a la membrana plasmática donde
ayuda al mantenimiento de la integridad del tejido. Esta nueva vía de redistribución
celular es un potencial mecanismo de quimioprevención y quimioterapia del cáncer

29
colorectal (Carter et al, 2004).
4. Formulación del Problema
El Cáncer Colorectal es una patología gastrointestinal con una alta morbimortalidad
en los países occidentales y que se esta presentando y diagnosticando de forma mayor
en países conocidos como de baja incidencia, como Colombia (Ángel et al, 2004)..
Se planteó la siguiente pregunta en este trabajo:
¿Existe sinergia antiproliferativa al combinar el 5-Fluorouracilo y el Clorofilín, en un
modelo in vitro de células humanas de cáncer colorectal?
5. Justificación
El cáncer es una patología que se puede presentar en forma muy heterogénea con
diferente incidencia en todo el mundo, presentándose como una causa importante de
mortalidad junto con las enfermedades cardiovasculares (Ángel et al, 2004).
Teniendo en cuenta la alta incidencia del cáncer colorectal a nivel mundial, es
importante poder realizar investigación que pueda ofrecer mejores alternativas de
tratamiento. Se argumenta que los agentes quimiopreventivos pudieran ofrecer una
mayor protección potencial a largo plazo que los quimioterapéuticos. De igual forma,
las terapias, donde se combinen medicamentos convencionales para el tratamiento del
cáncer con medicamentos de origen natural, muestran resultados prometedores en el
tratamiento contra el cáncer. Sin embargo, se ha observado que la quimioterapia
basada en el 5FU ha sido la quimioterapia de elección para el tratamiento del cáncer
colorectal, a pesar de que su tasa de respuesta como monoterapia es menor al 20%.
Esto sugiere que no se ha establecido una estrategia óptima de tratamiento.
Adicionalmente, este medicamento tiene una vida media in vivo reportada de tan solo
10 a 20 minutos. Esta característica farmacológica, produce que este agente

30
antitumoral deba utilizarse a dosis altas para mantener niveles séricos terapéuticos
(Tanaka et al, 2000; Torrance et al, 2000).
La posibilidad de poder combinar 5FU con productos de origen natural, permitirá
ampliar el conocimiento sobre el mecanismo de acción del 5FU. Por medio de la
utilización de anticarcinógenos de origen natural, la quimioprevención busca reducir
notablemente la incidencia del cáncer a nivel mundial. Bajo este contexto es
importante buscar otros medicamentos como el clorofilín que puedan presentar una
sinergia al ser combinado con el 5FU, para mejorar la calidad de vida de los pacientes
y la efectividad de los tratamientos. El análisis de los efectos que el 5FU y el CHL
puedan tener sobre la línea celular Caco-2, representa una aproximación fundamental
para conocer su posible sinergia y potenciación como compuestos antitumorales.
6. Objetivos
Objetivo General
Evaluar la actividad antiproliferativa de la combinación de 5FU y CHL sobre células
tumorales humanas de colon, Caco-2.
Objetivos específicos
• Determinar la viabilidad celular de células Caco-2 tratadas con diferentes
concentraciones de 5FU (0.039, 0.13 µg/ml) y CHL (0.25, 0.5 mM).
• Determinar la morfología de células Caco-2 tratadas con 5FU y CHL.
7. Hipótesis
Ho: No existe una sinergia sobre la actividad antitumoral al combinar 5-fluorouracilo
y Clorofilín sobre la línea celular Caco-2.

31
Ha: Existe una sinergia sobre la actividad antitumoral al combinar 5-fluorouracilo y
Clorofilín sobre la línea celular Caco-2.
8. Materiales y métodos
A. Tipo de estudio
Este estudio es de tipo experimental descriptivo
B. Muestra
La línea celular Caco-2, obtenida de la American Type Culture Collection (ATCC),
se ha utilizado en estudios que buscan profundizar en la reprogramación genética que
acompaña la maduración celular colónica epitelial. Esta línea celular fue establecida a
partir de un adenocarcinoma de colon bien diferenciado obtenido de un paciente de
72 años de edad. Las células Caco-2 son p53 mutadas. Esta línea celular alcanza su
confluencia entre los 3 y 6 días de cultivo y su estado de diferenciación a los 20 días
de cultivo. Las células Caco-2 sufren de manera espontánea arresto en el ciclo celular
dependiente de la inhibición por contacto, sirviendo como modelo de los cambios
fenotípicos que sufren las células colónicas a medida que migran a través del axis de
las criptas. (Fogh et al, 1977; Mariadason et al, 2003; Morin et al, 1996).
C. Variables de Estudio
En este estudio las variables independientes fueron las diferentes concentraciones de
5FU, CHL y sus combinaciones y el tiempo de exposición a ellas. La variable
dependiente fue el efecto de las diferentes concentraciones sobre la viabilidad y
proliferación de las células Caco-2.

32
E. Reactivos
Los reactivos utilizados para este estudio fueron: 5FU de Ebewe Pharma y CHL de
Sigma (C6003), medio de cultivo DMEM (Gibco) suplementado con 10% de suero
fetal bovino (Gibco), penicilina (100U/ml) (Gibco) y estreptomicina (100µg/ml)
(Gibco), CellTiter 96 AQueous One Soultion Cell Proliferation Assay Kit (Promega),
solución de Naranja de Acridina (AO) (Sigma) y solución de Bromuro de Etidio 500
µg/ml (EB) (Sigma).
F. Equipos
Los equipos utilizados en este estudio fueron: Incubadora de CO2 Napco-serie 5400,
cabina de flujo laminar Labconco clase II, centrifuga refrigerada Eppendorf-serie
5403, microscopio de luz invertido Anxiovert 25, espectofotómetro Humareader-
Human, microscopio de fluorescencia Leitz.
G. Métodos
Cultivos Celulares:
La línea celular de adenocarcinoma de colon, Caco-2, fue cultivada en cajas de
cultivo T25 cm2, alimentadas con medio DMEM (Gibco), y suplementado con 10%
de suero fetal bovino (SFB) (Gibco), penicilina (100U/ml) (Gibco) y estreptomicina
(100µg/ml) (Gibco). Se realizó cambio de medio cada tercer día. Se realizaron los
respectivos pasajes celulares por disociación con tripsina al 0.3% (Gibco) cuando se
obtenía una confluencia de aproximadamente el 80%.

33
Tratamiento de las células Caco-2 con CHL y 5FU:
Para conocer el efecto del 5FU y el CHL sobre las células Caco-2, se utilizo una
solución stock de 5FU a una concentración de 50 µg/ml. La solución stock de CHL se
preparó a una concentración de 2mM en medio de cultivo DMEM completo.
Posteriormente se procedió a tratar las células Caco-2 con diferentes concentraciones
tanto de CHL como de 5FU. Para esto, se realizaron las respectivas diluciones de las
soluciones stock en medio de cultivo, previo a su aplicación para obtener las
soluciones de trabajo, a saber: 0.25 mM y 0.5 mM de CHL y 0.039 µg/ml y 0.13
µg/ml de 5FU. Se realizó una combinación de las concentraciones de trabajo de la
siguiente manera: 0.13 µg/ml de 5FU+ 0.5 mM de CHL, 0.13 µg/ml de 5FU + 0.25
mM de CHL, 0.039 µg/ml de 5FU + 0.5 mM de CHL y 0.039 µg/ml de 5FU+ 0.25
mM de CHL. Posteriormente, se procedió a realizar el tratamiento de las células para
realizar las pruebas de viabilidad y morfología apoptótica.
Ensayo de la Viabilidad Celular:
La viabilidad celular fue evaluada mediante la utilización de un ensayo colorimétrico
como es el MTS (Cell Titer 96, Promega®) cuyo principio se basa en la reducción de
sales de formazan por parte de la mitocondria en células vivas. Se utilizaron cajas de
96 pozos (Cellstar®) sembrándose 19750 células por pozo en triplicado. Las células
se trataron por 12, 24 y 36 horas respectivamente con las diferentes concentraciones
de CHL y 5FU antes descritas: 0.25 mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13
µg/ml de 5FU, 0.13 µg/ml de 5FU+ 0.5 mM de CHL, 0.13 µg/ml de 5FU + 0.25 mM
de CHL, 0.039 µg/ml de 5FU + 0.5 mM de CHL y 0.039 µg/ml de 5FU+ 0.25 mM de
CHL. Posterior al tratamiento, se retiró el medio con el 5FU y CHL y se agregaron
500 µl de medio completo a cada pozo. Seguidamente se agregó 10 µl por pozo de
MTS (Cell Titer 96, Promega®) y se procedió a incubar las células por tres horas a
37ºC. Luego se procedió a realizar la lectura de la absorbancia del medio en cada
pozo en un espectrofotómetro a 492nm (Humareader-Human) por triplicado.

34
Recuento de células adherentes y flotantes:
Las células Caco-2, fueron sembradas (180000 células) por triplicado en cajas de
cultivo 12 pozos y cultivadas hasta lograr el 60% de confluencia. Las células se
trataron por 12 y 36 horas con las diferentes concentraciones de CHL y 5FU: 0.25
mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13 µg/ml de 5FU, 0.13 µg/ml de 5FU+ 0.5
mM de CHL, 0.13 µg/ml de 5FU + 0.25 mM de CHL, 0.039 µg/ml de 5FU + 0.5 mM
de CHL y 0.039 µg/ml de 5FU+ 0.25 mM de CHL. Posteriormente, se realizó un
recuento de las células flotantes y adherentes. Las flotantes, se obtuvieron a las 12 y
36 horas respectivamente, se tomó el medio de cada pozo en tubos eppendorf y se
centrifugaron. Posteriormente, el precipitado celular se lavo con PBS estéril y se
resuspendieron en 200 µl de DMEM completo. En cuanto a las células adheridas, se
lavaron con PBS estéril, se agregó tripsina al 0.3% (Gibco) y se incubo a 37º C hasta
obtener su desprendimiento. Una vez desprendidas se inhibió el efecto de la tripsina
agregando DMEM completo, se transfirieron a tubos eppendorf centrifugándose por 3
minutos a 2500 rpm, se descartó el sobrenadante teniendo cuidado de no perder el
precipitado el cual fue resuspendido en DMEM completo, procediéndose a realizar el
conteo celular en un hemocitómetro, como ha sido descrito por (Davis & Mcateer,
1994).
Morfología Apoptótica con Naranja de Acridina y Bromuro de Etidio
Para conocer el efecto de las diferentes concentraciones del 5FU y del CHL sobre
posibles cambios en la morfología de estas células a las 12 y 36 horas de tratamiento,
se utilizó una doble tinción con Naranja de Acridina (AO) y Bromuro de Etidio (EB).
Fluorocromos que tienen la capacidad de atravesar la membrana celular de acuerdo a
cambios en su permeabilidad. En este caso, la AO puede ingresar al citoplasma
celular tiñendo las células sanas y apoptóticas tempranas, las cuales presentan
cambios citoplasmáticos y condensación del DNA. En cambio, el EB se caracteriza

35
por teñir células en fases avanzadas del proceso apoptótico, como es la alteración de
la permeabilidad lo que facilita la entrada del fluorocromo. Para comprobar este
efecto, se sembraron 180000 células en cajas de 12 pozos por triplicado. Se trataron
las células con las diferentes concentraciones y combinaciones de CHL y 5FU antes
descritas. Se tomó el medio de cada pozo en un tubo eppendorf y se centrifugaron
para obtener las células flotantes por pozo y se lavaron con PBS. Las células
adheridas, se lavaron con PBS y se desprendieron con tripsina al 0.3% (Gibco),
procediéndose a incubar por 5 minutos a 37ºC. Posteriormente, se inactivo la tripsina
con medio completo y se centrifugó por 5 minutos a 2500 rpm. El precipitado celular
fue resuspendido con 5µg/ml de AO y 5µg/ml de EB en PBS para hacer una
observación directa en un microscopio (40X) de fluorescencia marca Leitz.
Análisis de Datos
Para los datos que no presentaron una distribución normal y debido al tamaño
pequeño de la muestra, se realizó una Kruskal-Wallis, que es una prueba no
paramétrica que define si existen o no diferencias significativas entre variables (en
este caso tratamientos). Para aquellos datos que presentaron una distribución normal
se realizó una anova sencilla para determinar si existían diferencias significativas
entre tratamientos.
9. Resultados
1) Efecto del CHL y 5FU sobre la viabilidad de las células Caco-2:
Para determinar el efecto del CHL y 5FU sobre las células Caco-2, a las
concentraciones descritas: 0.25 mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13 µg/ml de
5FU, 0.13 µg/ml de 5FU+ 0.5 mM de CHL, 0.13 µg/ml de 5FU + 0.25 mM de CHL,
0.039 µg/ml de 5FU + 0.5 mM de CHL y 0.039 µg/ml de 5FU+ 0.25 mM de CHL, se
realizó una prueba de viabilidad celular con MTS, midiéndose la absorbancia para
determinar el nivel de formación de formazan a diferentes tiempos de tratamiento: 12,

36
24 y 36 horas. Después de 12 horas, se observó con respecto al control, una mayor
disminución en la viabilidad de las células tratadas con 0.5 mM de CHL (62.33%),
seguida por las dosis con 0.25 mM de CHL (81.36%) 0.13 µg/ml de 5FU + 0.25 mM
de CHL (76.64%) (Ver Figura 7). Sin embargo, no se observaron diferencias
significativas entre el control y las diferentes dosis de CHL y 5FU utilizadas (p=
0.0605).
Ensayo de MTS en células Caco-2 luego de 12 Horas de tratamiento con CHL y 5FU
0
20
40
60
80
100
120
140
control
0,13 µg/ml 5FU
0,039 µg/ml 5FU
0,5 mM CHL
0,25 mM CHL
0,5 mM CHL + 0,13 µg/ml 5FU
0,25 mM CHL + 0,13 µg/ml 5FU
0,5 mM CHL + 0,039 µg/ml 5FU
0,25 mM CHL + 0,039 µg/ml 5FU
Concentraciones de CHL y 5 FU
Via
bilid
ad (%
)
Caco-2
Figura 7. Viabilidad de las Células Caco-2 luego de12 horas de tratamiento con CHL y 5FU. Se observó una disminución en la viabilidad de las células tratadas, pero no se observaron diferencias significativas con las diferentes dosis de CHL y 5FU utilizadas (p= 0.0605).
Después de 24 horas de tratamiento con las diferentes concentraciones de CHL y
5FU, se observó una disminución en la viabilidad celular. Por ejemplo, la dosis mas
alta de CHL (0.5 mM) indujo una mayor disminución de la viabilidad (50%) que la
dosis de 5FU (0.13 µg/ml), que indujo un 20%. Al observar el efecto de las diferentes
combinaciones, se encontró que las más efectivas fueron: 0.039 µg/ml de 5FU + 0.5
mM de CHL y 0.13 µg/ml de 5FU+ 0.5 mM de CHL, ambas combinaciones
disminuyeron la viabilidad celular a menos del 60% con respecto al control. La
combinación de 0.13 mM de 5FU + 0.25 µg/ml de CHL logró una disminución del

37
50%, en contraste a la concentración de 0.039 µg/ml de 5FU+ 0.25 mM de CHL
donde no se observó una disminución mayor al 10% (figura 8). Aplicando la prueba
de KW a los resultados obtenidos luego de 24 horas de tratamiento, se encontraron
diferencias significativas entre los tratamientos de 5FU y CHL utilizados (p =
0.0371).
Ensayo de MTS en células Caco-2 luego de 24 Horas de tratamiento con CHL y 5FU
0
20
40
60
80
100
120
140
control
0,13 µg/ml 5FU
0,039 µg/ml 5FU
0,5 mM CHL
0,25 mM CHL
0,5 mM CHL + 0,13 µg/ml 5FU
0,25 mM CHL + 0,13 µg/ml 5FU
0,5 mM CHL + 0,039 µg/ml 5FU
0,25 mM CHL + 0,039 µg/ml 5FU
Concentraciones de CHL y 5 FU
Viab
ilida
d (%
)
Caco-2
Figura 8. Viabilidad de las Células Caco-2 luego de 24 horas de tratamiento con CHL y 5FU. Se presentó una disminución en la viabilidad celular frente a las dosis de CHL, tanto por si solo como en combinación con el 5FU. Se encontraron diferencias significativas entre los tratamientos con CHL y 5FU utilizadas (p=0.0371).
A las 36 horas de tratamiento, se observó una disminución en la viabilidad celular
siendo mayor en las células tratadas con la combinación de 5FU y CHL. Se observó
una disminución mayor al 50% en el porcentaje de viabilidad de las células sometidas
a los tratamientos con 0.5 mM de CHL (35.85%), 0.13 µg/ml de 5FU + 0.5 mM de
CHL (38.09%), 0.13 µg/ml de 5FU + 0.25 mM de CHL (32.79%) y 0.039 µg/ml de
5FU + 0.5 mM de CHL (44.70%). El tratamiento con las concentraciones más bajas
de 5FU (0.039 µg/ml) y CHL (0.25 mM), no presentó una disminución mayor al 15%

38
de la viabilidad celular en comparación con el control (Figura 9). También se observa
que al comparar los resultados a las 12, 24 y 36 horas, el uso de 5FU per se a
concentraciones de 0.13 µg/ml y 0.039 µg/ml, tiene un efecto del 20% y 2% con
respecto al control, respectivamente. Sin embargo, cuando el 5FU se utiliza en
combinación con el CHL se observa una disminución en la viabilidad celular, evento
que sugiere algún tipo de efecto sinérgico por parte del clorofilín (Figuras 7, 8 y 9).
Aplicando la prueba de KW, para conocer la significancia de los resultados obtenidos
luego de 36 horas de tratamiento, este mostró diferencias significativas de los
tratamientos de 5FU y CHL utilizados (p = 0.0075).
Ensayo de MTS en células Caco-2 luego de 36 Horas de tratamiento con CHL y 5FU
0
20
40
60
80
100
120
140
control
0,13 µg/ml 5FU
0,039 µg/ml 5FU
0,5 mM CHL
0,25 mM CHL
0,5 mM CHL + 0,13 µg/ml 5FU
0,25 mM CHL + 0,13 µg/ml 5FU
0,5 mM CHL + 0,039 µg/ml 5FU
0,25 mM CHL + 0,039 µg/ml 5FU
Concentraciones de CHL y 5 FU
Viab
ilida
d (%
)
Caco-2
Figura 9. Viabilidad de las Células Caco-2 luego de 36 horas de tratamiento con CHL y 5FU. Se presentó una disminución en el porcentaje de viabilidad de las células tratadas con las combinaciones de CHL y 5FU. Se presentaron diferencias significativas entre tratamientos con un (p =0.0075).
2) Efecto del CHL y 5FU sobre el número de células Caco-2 flotantes y
adherentes:
Para complementar los resultados obtenidos en el ensayo de viabilidad celular, se
realizó un recuento de células Caco-2 adherentes y flotantes después de transcurridas

39
12 y 36 horas de tratamiento con CHL y 5FU a las concentraciones descritas: 0.25
mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13 µg/ml de 5FU, 0.13 µg/ml de 5FU+ 0.5
mM de CHL, 0.13 µg/ml de 5FU + 0.25 mM de CHL, 0.039 µg/ml de 5FU + 0.5 mM
de CHL y 0.039 µg/ml de 5FU+ 0.25 mM de CHL. Después de transcurridas 12 horas
de tratamiento, se presentó un aumento en el porcentaje de células flotantes y una
disminución de las células adherentes a las diferentes concentraciones utilizadas de
5FU y CHL. Los tratamientos que mostraron el mayor aumento en el porcentaje de
células flotantes fueron: 0.13 µg/ml de 5FU + 0.5 mM de CHL (233.33%), seguido
por 0.039 µg/ml de 5FU + 0.5 mM de CHL (172.73%). Aplicando una prueba de
Anova, se demostraron diferencias significativas de las diferentes concentraciones de
5FU y CHL utilizados para la inducción de células flotantes (p=0.00001) y para las
adherentes (p=0.00007) (Figura 10).
Recuento de células Caco-2 flotantes y adherentes luego de 12 horas de tratamiento con CHL y 5FU
0
100
200
300
Control
0,13 µg/ml 5 FU
0,039 µg/ml 5 FU
0,5 mM CHL
0,25 mM CHL
0,13 µg/ml 5 FU + 0,5 mM CHL
0,13 µg/ml 5 FU+0,25 mM CHL
0,039µg/ml 5 FU+0,5mM CHL
0,039 µg/ml 5 FU+0,25mM CHL
Concentraciones de CHL y 5 FU
% d
e cé
lula
s
células flotantescélulas adherentes
Figura 10. Recuento de células Caco-2 flotantes y adherentes luego de 12 horas de tratamiento con CHL y 5FU. Las células tratadas con las dosis que contenían 0.5 mM de CHL, presentaron un mayor aumento de células flotantes. Se presentaron diferencias significativas entre tratamientos entre células flotantes (p=0.00001) y adherentes (p=0.00007).
Después de 36 horas de tratamiento con las diferentes concentraciones de CHL y
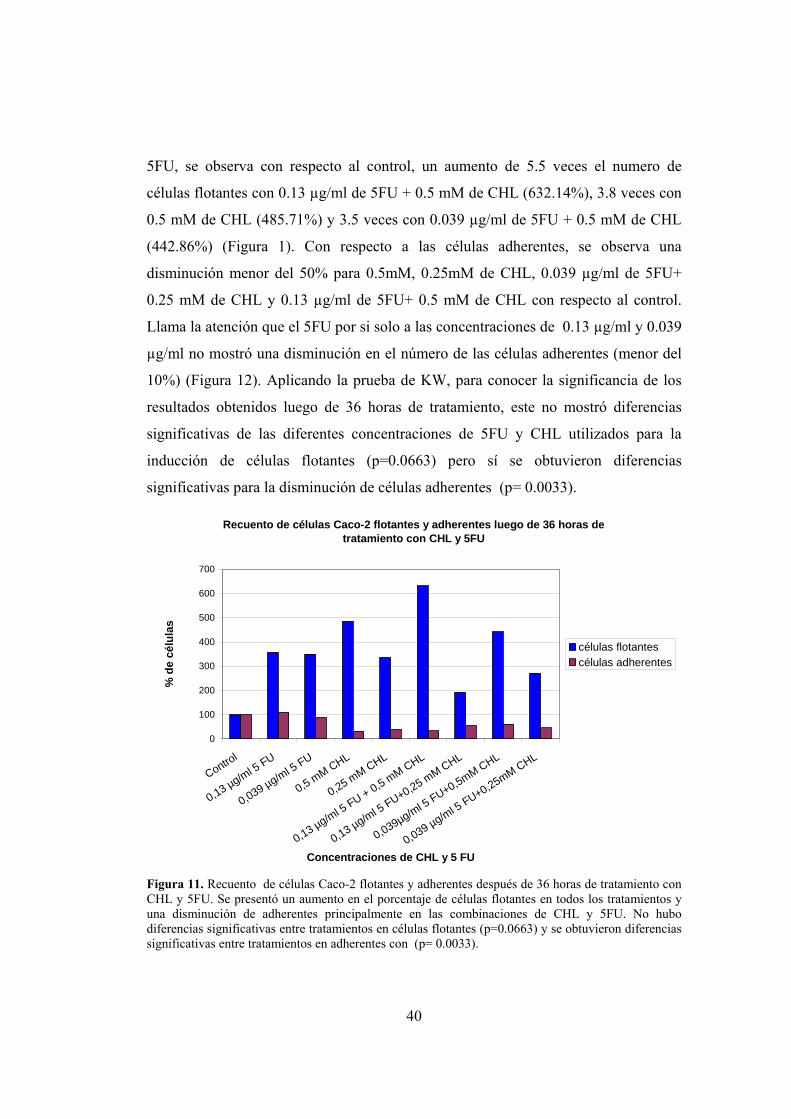
40
5FU, se observa con respecto al control, un aumento de 5.5 veces el numero de
células flotantes con 0.13 µg/ml de 5FU + 0.5 mM de CHL (632.14%), 3.8 veces con
0.5 mM de CHL (485.71%) y 3.5 veces con 0.039 µg/ml de 5FU + 0.5 mM de CHL
(442.86%) (Figura 1). Con respecto a las células adherentes, se observa una
disminución menor del 50% para 0.5mM, 0.25mM de CHL, 0.039 µg/ml de 5FU+
0.25 mM de CHL y 0.13 µg/ml de 5FU+ 0.5 mM de CHL con respecto al control.
Llama la atención que el 5FU por si solo a las concentraciones de 0.13 µg/ml y 0.039
µg/ml no mostró una disminución en el número de las células adherentes (menor del
10%) (Figura 12). Aplicando la prueba de KW, para conocer la significancia de los
resultados obtenidos luego de 36 horas de tratamiento, este no mostró diferencias
significativas de las diferentes concentraciones de 5FU y CHL utilizados para la
inducción de células flotantes (p=0.0663) pero sí se obtuvieron diferencias
significativas para la disminución de células adherentes (p= 0.0033).
Recuento de células Caco-2 flotantes y adherentes luego de 36 horas de tratamiento con CHL y 5FU
0
100
200
300
400
500
600
700
Control
0,13 µg/ml 5 FU
0,039 µg/ml 5 FU
0,5 mM CHL
0,25 mM CHL
0,13 µg/ml 5 FU + 0,5 mM CHL
0,13 µg/ml 5 FU+0,25 mM CHL
0,039µg/ml 5 FU+0,5mM CHL
0,039 µg/ml 5 FU+0,25mM CHL
Concentraciones de CHL y 5 FU
% d
e cé
lula
s
células flotantescélulas adherentes
Figura 11. Recuento de células Caco-2 flotantes y adherentes después de 36 horas de tratamiento con CHL y 5FU. Se presentó un aumento en el porcentaje de células flotantes en todos los tratamientos y una disminución de adherentes principalmente en las combinaciones de CHL y 5FU. No hubo diferencias significativas entre tratamientos en células flotantes (p=0.0663) y se obtuvieron diferencias significativas entre tratamientos en adherentes con (p= 0.0033).

41
3) Tratamientos con CHL y 5FU inducen apoptosis de células Caco-2
En oncología, uno de los aspectos muy importantes a tener en cuenta durante la
evaluación de un fármaco y/o su posible combinación con otros, con la finalidad de
observar una posible disminución en la proliferación celular, es la forma como esta se
logra. En lo posible, lo ideal es semejar una muerte celular lo mas fisiológica posible,
donde la presencia de daño por necrosis no sea el común denominador para explicar
la efectividad de los compuestos a estudio, Teniendo en cuenta que muchos de los
compuestos no son completamente ubicuos para las células sanas. Por tal motivo, se
hace necesario, la evaluación directa de la morfología que presentan las células antes
y después de ser tratadas. En este estudio, se comprobó que las células Caco-2
tratadas murieron por apoptosis. Esta forma de muerte celular se observó a las 12 y
36 horas posterior al tratamiento con CHL y 5FU a las concentraciones descritas:
0.25 mM y 0.5 mM de CHL, 0.039 µg/ml y 0.13 µg/ml de 5FU, 0.13 µg/ml de 5FU+
0.5 mM de CHL, 0.13 µg/ml de 5FU + 0.25 mM de CHL, 0.039 µg/ml de 5FU + 0.5
mM de CHL y 0.039 µg/ml de 5FU+ 0.25 mM de CHL. Con la doble tinción de
naranja de acridina (AO) y bromuro de etidio (EB), se observó que las células
exhibían características morfológicas de la apoptosis, que incluían: condensación
citoplasmática y nuclear, disrupción de la membrana nuclear y formación de cuerpos
apoptóticos (Figura 12).
Figura 12. Tinción de células Caco-2 con naranja de AO y EB. Las células exhibieron características morfológicas de la apoptosis, que incluían: condensación citoplasmática y nuclear, disrupción de la membrana nuclear y formación de cuerpos apoptóticos.

42
10. Discusión
El cáncer colorectal es una enfermedad multifactorial de alta incidencia mundial, de
los cuales 5 al 10% de los casos son de origen familiar, por ejemplo, el cáncer
colorectal hereditario sin poliposis (HNPCC) y la poliposis adenomatosa familiar
(FAP) ) (Aranda-Aguilar, 2004). Sin embargo, la mayoría de cánceres colorectales
son esporádicos, y están altamente influidos por factores medioambientales, como
son los hábitos alimentarios. Además, es importante considerar la susceptibilidad
genética de cada individuo a desarrollar este tipo de cáncer (Bapat et al, 1997; Weitz
et al, 2005). Desde su descubrimiento en 1957, el 5 FU ha demostrado tener actividad
antitumoral en células de adenocarcinoma de colon. Se sabe que su mecanismo de
acción se efectúa por medio de la inhibición de la TS, afectando la síntesis de DNA e
induciendo apoptosis principalmente por la activación del gen p53 (Rustum, 2003).
Sin embargo, este compuesto a pesar de tener una vida media in vivo de tan solo 10 a
20 minutos, puede producir sobre los pacientes sometidos a este fármaco, efectos
secundarios tales como: inmunosupresión, anemia, y trastornos gastrointestinales
como diarrea crónica, entre otros. A pesar de los efectos secundarios que puede
generar la administración del 5FU, a la fecha, no hay un medicamento con la
actividad antitumoral para el tratamiento del cáncer colorectal como lo es el 5FU.
Razón por la cual se ha explorado la posibilidad de utilizar el 5FU en combinación
otro tipo de medicamento antitumoral, con miras de disminuir la presencia de efectos
secundarios y aumentar la sobrevida a cinco años, pero sin reducir la capacidad
antitumoral (Longley et al, 2003; Tanaka et al, 2000).
La capacidad antitumoral del 5FU, también se observa en combinación con otros
agentes antitumorales con el propósito de potenciar su actividad anti-proliferativa
para modular el crecimiento tumoral (Longley et al, 2003; Tanaka et al, 2000). Dos
de los agentes antitumorales usados en combinación con el 5FU son el Leucovirín
(LV) e Interferon-α (Houghton et al, 1993; Matherly et al, 1990). Con la combinación
de LV+5FU, se observó una mejor respuesta antitumoral en comparación con la

43
utilización del 5FU por si solo (23% frente a 11% respectivamente). Por su parte, la
combinación de 5FU con Interferón-α no mostró mejor actividad antitumoral en
comparación con la combinación de 5FU+LV (Houghton et al, 1993). En adición, los
efectos secundarios observados, y la supervivencia a cinco años no mejoró (Longley
et al, 2003). Son la generación de efectos secundarios y la baja sobrevida a 5 años, lo
que ha llevado a utilizar otro tipo de medicamentos de origen natural de comprobada
actividad anti-carcinógenica. Se reporta que la utilización de anticarcinógenos
naturales, podría reducir notablemente los efectos secundarios del 5FU y aumentar el
porcentaje de supervivencia de los pacientes (Jordan & Stein, 2003). Entre estos
agentes naturales, se encuentran el ácido graso Omega-3. Ácido graso que al
combinarse con el 5FU y ser aplicado sobre células Caco-2 se obtiene una
disminución en la proliferación celular asociado a un aumento en la muerte celular
por apoptosis (Jordan & Stein, 2003). De forma similar, el Geraniol, un compuesto de
origen natural, también ha sido estudiado en células Caco-2. En este caso, su
combinación (5FU+Geraniol) mejora la sensibilidad de estas células tumorales a la
actividad anti-proliferativa del 5FU (Carnesecchi et al, 2002). El clorofilín, un
derivado hidrosoluble de la clorofila, ha demostrado tener actividades anti-
mutagénicas y anti-tumorales (Egner et al, 2001). Por ejemplo, el CHL ha
demostrado actividad anti-proliferativa en células tumorales de colon, HCT116, que
expresan la variedad silvestre del gen p53 (Díaz et al, 2003). A la fecha, no esta
reportado en la literatura los efectos anti-proliferativos de la combinación del 5FU
con CHL. Habiéndose demostrado las bondades del clorofilín sobre las células
HCT116, como es la inducción de apoptosis por regulación del nivel post-
transcripcional de las proteínas BAK, AIF, Caspasa-8, y E-Cadherina, surge la
pregunta sobre cual es el efecto de la combinación de 5FU+CHL, sobre la regulación
de la proliferación celular (Díaz et al, 2003; Torrance et al, 2000).
Con el presente estudio experimental, se logró conocer los efectos anti-proliferativos
que tiene la combinación de 5FU y CHL. Efecto que fue observado por medio de la
prueba de viabilidad celular, MTS (7, 8 y 9) y apoptosis por la doble tinción con los

44
fluorocromos AO/EB (figuras 10,11 y 12) en células de adenocarcinoma de colon
Caco-2. Las concentraciones de 5FU utilizadas (0.039 µg/ml y 0.13 µg/ml) han sido
reportadas por la literatura como efectivas para producir una disminución en la
proliferación celular e inducción de apoptosis (Arango et al, 2001; Gugliemi et al,
1995; Jordan & Stein, 2003; Tanaka et al, 2000). Por otra parte, las concentraciones
de CHL utilizadas (0.25 mM y 0.5 mM) son las reportadas con un efecto
antiproliferativo sobre células tumorales de colon, HCT116 (Díaz et al., 2003).
Con la utilización individual del 5FU y CHL se obtuvo una disminución en la
proliferación celular (figuras 7, 8 y 9). El tratamiento con CHL (0.5 mM) mostró a
las 12 horas una disminución de la viabilidad celular (40%) (figura 8). Sin embargo,
esta viabilidad disminuyó en más del 50% a las 24 y 36 horas de tratamiento (8 y 9).
La menor concentración (0.25mM) de CHL, produjo a las 12 horas una disminución
en la viabilidad celular del 20%, del 42% a las 24 horas y del 17% a las 36 horas de
tratamiento (figuras 7, 8 y 9). Llama la atención, que a las 36 horas el efecto del CHL
(0.25mM), disminuye. Este efecto podría explicarse, en primera instancia por pérdida
de su actividad farmacológica probablemente por termolabilidad. Es decir, que
dependiendo del tiempo de incubación, el CHL (0.25mM), pudiera disminuir o
aumentar la viabilidad de las células Caco-2. Por otra parte, es importante tener en
cuenta la presencia del gen p53 mutado en estas células Caco-2, hecho que podría
explicar en parte el efecto observado (Mariadason et al, 2003). Estos resultados están
acorde con lo publicado por Díaz et al., 2003; quienes reportan que 0.5mM de CHL
es una concentración que disminuye la proliferación de células tumorales de colon.
Por otra parte, al utilizar en forma individual el 5FU, se observó que el 5FU no
disminuye la viabilidad celular como lo hace el CHL (7, 8 y 9). Las concentraciones
de 5FU utilizadas (0.13 µg/ml y 0.039 µg/ml) demostraron poca actividad anti-
proliferativa (12 h: 0.2% - 0%; 24 h: 20% - 5%; 36 h: 20% - 2%) sobre las células
Caco-2, respectivamente (figuras 7, 8 y 9). Estos resultados, están de acuerdo con lo
reportado por Ashktorab et al., 2005; quienes utilizando la línea tumoral de colon,
HT29 (mutada para p53), demuestran que el 5FU per se no tiene un efecto

45
antitumoral mayor al mostrado por el ácido acetil salicílico (ASA). Sin embargo, la
combinación 5FU +ASA, demostró tener un efecto antiproliferativo aún mayor que el
ASA en forma individual.
La combinación de 5FU con CHL (0.13 µg/ml de 5FU+ 0.5 mM de CHL, 0.13 mM
de 5FU + 0.25 µg/ml de CHL, 0.039 µg/ml de 5FU + 0.5 mM de CHL y 0.039 µg/ml
de 5FU+ 0.25 mM de CHL), mostró a las 12 horas un pobre efecto anti-proliferativo,
menor al 50% (figura 8). Contrario al efecto observado a las 24 y 36 horas, donde se
logró y mantuvo una actividad anti-proliferativa mayor al 50%, excepto con la
combinación de 0.039 µg/ml de 5FU+0.25 mM de CHL (figuras 8 y 9). Estos
resultados, indican que la actividad anti-proliferativa mostrada por la combinación de
5FU y CHL son tiempo y concentración dependientes. Se evidencia, que a las 24 y 36
horas de tratamiento, la combinación de las concentraciones de CHL con 0.13 mM
de 5FU, tiene una actividad anti-proliferativa mayor al 50%. Llama la atención, que
luego de 12 horas de tratamiento la combinación de la concentración menor tanto de
5FU (0.039 µg/ml) como de CHL (0.25 mM) inducen un aumento en la reducción de
formazan, hecho que podría sugerirse como un indicador de que la combinación de
5FU y CHL a estas concentraciones inducen proliferación celular. Sin embargo, a las
24 y 36 horas, hay actividad anti-proliferativa, pero de tan solo un 15%
aproximadamente. Observación que debe ser tenida en cuenta para poder plantear
hipótesis que lleven a entender el mecanismo de acción e interacción de de estos
medicamentos.
Como una forma de poder corroborar los resultados obtenidos por la prueba de
viabilidad con el MTS, se realizó un estudio de la cinética celular, haciendo un
recuento de las células tanto adherentes como flotantes, evaluándose a las 12 y 36
horas de tratamiento con 5FU y CHL y su combinación, pues estas células son reflejo
directo del estrés que les causa el estar expuestas al 5FU y CHL. Es conocido, que las
células flotantes, en un sistema in vitro, son el reflejo de la inducción de muerte
celular programada por apoptosis (Díaz et al., 2000). Llama la atención que a las 12

46
horas de tratamiento la combinación de 5FU y CHL (0.13 µg/ml de 5FU + 0.5 mM de
CHL), indujo un 150% más de células flotantes con respecto al control e incluso con
respecto a la mayor concentración de CHL (0.5 mM). Concentración que mostró
poder disminuir la viabilidad en un 40% a este mismo punto control. A las 36 horas
de tratamiento, se observa un mayor número de células flotantes con concentraciones
de 0.13 µg/ml de 5FU + 0.5 mM de CHL, seguido por 0.5 mM de CHL y el 0.039
µg/ml de 5FU + 0.5 mM de CHL. La concentración de 0,25mM de CHL + 0.039
µg/ml de 5FU no disminuyó en el número de células adherentes, al igual que no
produjo una disminución de la viabilidad celular en ninguno de los tres tiempos
analizados. Esto nos sugiere, que aunque la prueba de MTS nos muestra la viabilidad
celular, esta no refleja el posible mecanismo responsable de la disminución de la
viabilidad celular, sea por apoptosis, necrosis y/o control directo sobre el ciclo
celular. En los tres casos, nos muestra una disminución en la reducción de formazan
reflejando una disminución en el número de células. Este hecho, nos llevó a
determinar la morfología celular de las células en cultivo y así poder tener un
acercamiento más directo sobre el probable efecto del uso del 5FU y CHL. Para
determinar, si la morfología de las células flotantes y adheridas era compatible con la
inducción de muerte celular programada por apoptosis sobre las células Caco-2, se
realizó una doble tinción con AO y EB, observándose que tanto las células flotantes y
adherentes tenían características que semejaban la morfología de células apoptóticas,
tal cual como ha sido descrita por Díaz et al., 2000; 2003.
Los resultados obtenidos en este estudio, sugieren una baja sensibilidad de las células
Caco-2 al tratamiento individual con 5FU pero no con CHL. Por el contrario, la
combinación de 5FU+CHL se observa una sinergia entre los compuestos para inducir
inhibición del crecimiento de las células Caco-2, que se ve reflejado por la inducción
de muerte celular por apoptosis. Aunque en este trabajo de investigación no se realizó
un estudio del contenido del DNA en estas células, Caco-2, podría sugerirse, con base
en la literatura, que la acción sinérgica en su actividad anti-proliferativa podría ser
debida a efectos de estos medicamentos sobre el ciclo celular induciendo alteraciones

47
en cualquiera de sus puntos control para inhibir el crecimiento celular observado en
las células Caco-2. Esta sinergia observada entre 5FU y CHL se hace atractiva, para
un posible uso en quimioterapia debido a la resistencia al efecto del 5FU en cierto
tipo de tumores colorectales (Longley et al, 2003; Tanaka et al, 2000). Se conocen
varios tipos de resistencia a la quimioterapia con 5FU que han sido demostrados en
estudios experimentales (Longley et al, 2003; Tanaka et al, 2000). El principal tipo
de resistencia es el que resulta de una regulación traduccional y post-traduccional de
la TS (Chu et al., 1991). En este estudio, Chu et al., 1991, establece un modelo de
resistencia, donde se demuestra que los niveles proteicos de TS, no siempre obedecen
a un incremento en el nivel de expresión de su mRNA, indicando una posible
regulación traduccional y/o post-traduccional de la enzima TS. Este mecanismo
postulado por Chu et al, 1991, es conocido como el Modelo de Autorregulación
Traduccional de la Timidilato Sintasa, en el cual la TS es capaz de unirse a su propio
mRNA inhibiendo su traducción. La resistencia al efecto anti-tumoral resulta cuando
el 5FU induce una acumulación de FdUMP y dUMP al interior de la célula. Esta
resistencia, se refleja mediante la interacción de estos sustratos con el mRNA de la
TS produciendo una pérdida en la capacidad autorreguladora de la enzima (Kitchens
et al, 1999).
Se reportan otros modelos, que se postulan para explicar la resistencia a este agente
antitumoral (Kitchens et al, 1999). La gran mayoría de estos estudios, muestran un
aumento en la actividad transcripcional de genes que controlan la transición G1/S,
entre ellos la DNA polimerasa α, el antígeno nuclear de proliferación celular
(PCNA), la Ribonucleótido Reductasa, la Dehidrofolato Reductasa (DHFR) y la TS
(Kasahara et al, 2000). Estos resultados podrían aportar y sugerir posibles respuestas
al efecto observado con la combinación de las menores concentraciones de 5FU y
CHL (0.039 µg/ml de 5FU+ 0.25 mM de CHL) luego de 12 horas de tratamiento. Por
ejemplo, el 20% de aumento en la viabilidad observado (figura 8), podría ser
sugestivo de un aumento en el número de células en cultivo, reflejando actividad
proliferativa que pudiera explicarse por una estimulación sobre los genes que regulan

48
la transición G1/S, tal cual lo reporta Kasahara et al, 2000. Obviamente, hay que
realizar los estudios de contenido de DNA, para mirar como es el comportamiento en
G1/S y poder estudiar la actividad de algunos de sus genes que regulan este punto del
ciclo celular.
Un aspecto importante a considerar, en la actividad del 5FU, es su estatus de las
células Caco-2 para p53. Se ha reportado, que las líneas celulares con un p53 mutado
son resistentes a la apoptosis inducida por 5FU, causando un aumento en el umbral de
sensibilidad frente a este agente (Longley et al, 2003; Tanaka et al, 2000). Las células
de adenocarcinoma de colon Caco-2 son mutantes para p53 y por lo tanto, resistentes
a la muerte inducida por Fas (principal mecanismo de inducción de apoptosis del
5FU). Esta propiedad, en estas células Caco-2, podría ser causante de lo que se
observa al utilizar en forma individual el 5FU (0.13 µg/ml y 0.039 µg/ml) donde no
se observa una actividad antiproliferativa (figuras 7, 8 y 9). Sin embargo, es
interesante observar que al combinar 5FU con las diferentes concentraciones de CHL
se observa una mejor actividad anti-proliferativa (figuras 7, 8 y 9). El punto a discutir,
es si el efecto anti-proliferativo es producido por sinergia del CHL sobre el
mecanismo de acción del 5FU o si realmente el CHL es el único responsable por esta
actividad anti-proliferativa. Si el CHL, fuera responsable de la actividad anti-
proliferativa, lo estaría haciendo en forma independiente a la vía de p53, mecanismo
que esta reportado en otras líneas celulares tumorales de colon (Bunz et al, 1999;
Longley et al, 2003; Tillman et al, 1999). En este estudio se demuestra, que la
combinación de 5FU y CHL es sinérgica. Queda por elucidar como se lleva a cabo
esta “cooperación”, si alguna, para aumentar el efecto anti-proliferativo sobre las
células Caco-2. Un posible mecanismo podría estar relacionado con el factor de
transcripción E2F1 (Comunicación personal Dr. G. Darío Díaz). En este caso, el CHL
podría estar regulando de manera positiva a E2F1, causando una sobre-expresión del
mismo y generando apoptosis por regulación de proteínas pro-apoptóticas como BAK
y AIF al igual que activación de caspasas o por la activación de p73, proteína a la que
se le atribuyen funciones similares a p53, por acción de E2F-1 (Stiewe y Pützer,

49
2000; Díaz et al., 2003; Díaz et al., manuscrito en preparación). Sin embargo, es
conocido, que la sobre-expresión de E2F1, también puede incrementar la expresión
de la TS, activando genes que intervienen en la transición de la fase G1/S como la
DHFR y el PCNA, como fue demostrado por Kasahara et al., 2000.
Cualquiera de los anteriores mecanismos moleculares, podría al menos en parte,
servir para sugerir el mecanismo del sinergismo observado al combinar 5FU y CHL.
Combinación que abre puertas a la necesidad que se tiene en la farmacología
oncológica, de encontrar un medicamento “mágico”, que pudiera tener una actividad
anti-proliferativa actuando sobre el control del ciclo celular y/o la inducción de la
muerte celular, con una mínima inducción de efectos secundarios y mejorando la
sobrevida a 5 años y porque no curar el cáncer en forma total. Sin embargo, no hay
que olvidar, que la literatura es tácita al afirmar, que el cáncer es un producto
multifactorial donde aspectos genéticos y hábitos influenciados por el medio
ambiente son las variables a controlar para prevenir en lo posible cambios en la
homeostasis celular en los órganos que componen el cuerpo humano.

50
11. Conclusiones
A partir de los resultados obtenidos en este estudio, se puede concluir:
1. La combinación de 5FU y CHL produce un efecto sinérgico sobre la capacidad
antiproliferativa en las células Caco-2.
2. El efecto antiproliferativo de 5FU y CHL se evidenció por una disminución en la
viabilidad celular reflejada, por lo menos en parte, por la inducción de muerte celular
por apoptosis.
3. La actividad antiproliferativa individual del 5FU y del CHL fue mayor para el
CHL.

51
12. Recomendaciones
Con base en los resultados obtenidos con 5FU y CHL sobre células Caco-2, se
sugiere realizar estudios de contenido de DNA y estudiar los genes que se conocen
son importantes en la regulación de ciclo y muerte celular por apoptosis. De igual
forma, es importante aplicar métodos matemáticos que nos permitan concluir la
presencia de sinergia entre ambos compuestos.
.

52
Bibliografía
ALBERTS B.; JOHNSON A.; LEWIS J. 2002. Molecular biology of the cell. Fourth
Edition. New York; Garland Science.
ÁNGEL L.A.; GIRALDO A.; PARDO C.E. 2004. Mortalidad por cánceres del
aparato digestivo en Colombia entre 1980 y 1998, análisis de tendencias y
comparación regional. Revista Facultad Medicina Universidad Nacional Colombia.
Vol.52 No. 1 19-36.
ARANDA-AGUILAR E. 2004. Tratamiento del cáncer de colon estadios II, III y IV.
Oncología. 27 (4) 258-261.
ARANGO D; CORNER G.A; WADLER S.; CATALANO P.J.; AUGENLICHT L.H.
2001. c-myc/p53 Interaction Determines Sensitivity of Human Colon Carcinoma
Cells to 5-Fluorouracil in Vitro and in Vivo. Cancer Research. Vol.61 4910–4915.
ASHKTORAB H.; DAWKINS F.W.; MOHAMED R.; LARBI D.; SMOOT D.T.
2005. Apoptosis induced Aspirin and 5-Fluorouracil in human colonic
adenocarcinoma cells. Digestive Diseases and Sciences. Vol. 50 No. 6 1025-1032.
BAPAT B.; COUTURE J.; GALLINGER S.; GRYFE R.; REDSTON M.;
SWALLOW C. 1997. Molecular Biology of Colorectal Cancer. Current Problems in
Cancer. Vol 21 No.5 235-299.
BRUCE R.W.; GIACCA A.; MEDLINE A. 2000. Possible mechanisms relating diet
and risk of colorectal cancer. Cancer Epidemiology. Vol.9 1271-1279
BUNZ F.; HWANG P.M.; TORRANCE C.; WALDMAN T.; ZHANG Y.;
DILLEHAY L.; WILLIAMS J.; LENGAUER C.; KINZLER K.W.; VOGELSTEIN
B. 1999. Disruption of p53 in human cancer cells alters responsens to therapeutic

53
agents. Journal of Clinical Investigation. Vol.104 263-269.
BURMER G.C.; LOEB L.A. 1989. Mutations in KRAS2 oncogene during
progressive stages of human colon carcinoma. Proc. Natl. Acad. Sci. vol 86 2403-
2407.
CARNESECCHI S.; LANGLEY K.; EXINGER F.; GOSSE F.; RAUL F. 2002.
Geraniol, a component of plant essential oils, sensitizes human colonic cancer cells to
5-Fluorouracil treatment. The Journal of Pharmacology and Experimental
Therapeutics. Vol.301 No.2 625-630.
CARTER O; BAILEY G.S.; DASHWOOD R.H. 2004. The Dietary Phytochemical
Chlorophyllin alters E─Cadherin and ß─Catenin Expression in Human colon Cancer
Cells. American society for Nutritional Sciences. 3441S─3444S.
CHU E.; KOELLER D.M.; CASEY J.L.; DRAKE J.C.; CHABNER B.A.; ELWOOD
P.C.; ZINN S.; ALLEGRA C.J. 1991. Autoregulation of human thymidylate synthase
messenger RNA translation by thymidylate synthase. Proc. Natl. Acad. Sci. vol. 88
8977-8981.
COOPER G.M. 2000. The Cell a Molecular approach. Second Edition. Sinauer
Associates, Inc., Washington DC.
DAVIS J.; MCATEER J.A. 1994. Basic cell culture technique and the maintenance
of cell lines. 126-131. Basic cell culture; a practical approach. Oxford University
Press. New York, USA.
DIAZ G. D.; LI Q.; DASHWOOD R. 2003. Caspase 8 and apoptosis-inducing Factor
Mediate a Citochrome c-independent Pathway of Apoptosis in Human Colon Cancer
Cells Induced by Dietary Phytochemical Chlorophyllin. Cancer Research. Vol 63.

54
1254-1261.
DIAZ G.D.; PARASKEVA C.; THOMAS M.G.; BINDERUP L.; HAGUE A. 2000.
Apoptosis is Induced by the Active Metabolite of Vitamine D3 and its Analogue
EB1089 in colorectal Adenoma and Carcinoma Cells: Possible implications for
Prevention and Therapy. Cancer Research. vol. 60 2304-2312.
DIERGAARDE B.; VAN GELLOF W.L.; VAN MUIJEN G.N.P; KOK F.J;
KAMPMAN E. 2003. Dietary factors and the ocurrence of truncating APC mutations
in sporadic colon carcinomas: a Dutch population-based study. Carcinogenesis.
Vol.24 no.2 283-290
DIERGAARDE B; VRIELING A; VAN KRAATS A; VAN MUIJEN G.N.P.; KOK
F.J.; KAMPMAN E. 2003. Cigarrette smoking and genetic alterations in sporadic
colon carcinomas. Carcinogenesis. Vol.24 no.3 565-571.
EGNER P.A.; WANG J.B.; ZHU Y.R.; ZHANG B.C.; WU Y.; ZHANG Q.N.; QIAN
G.S.; KUANG S.Y.; GANGE S.J.; JACOBSON L.P.; HELZLSOUER K.J.; BAILEY
G.S.; GROOPMAN J.D.; KENSLER T.W. 2001. Chlorophyllin intervention reduces
aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc. Natl. Acad.
Sci. vol. 98 no. 25. 14601-14606.
EVAN G.I; VOUSDEN K.H. 2001. Proliferation, cell cycle and apoptosis in cancer.
Nature. Vol 411. 342-348.
FEARON ER, VOGELSTEIN B.1990. A genetic model for colorectal tumorigenesis.
Cell. 1;61(5):759-67.
FODDE R.; SMITS R.; CLEVERS H. 2001. APC Signal Transduction and Genetic
Instability in Colorectal Cancer. Nature Reviews/Cancer. vol.1 55-67.

55
FOGH J., FOGH J.M., ORFEO T. 1977. One hundred and twenty seven cultured
human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 59: 221-
226.
FRAYLING I.M.; BECK N.E.; ILYAS M.; DOVE-EDWIN I.; GOODMAN P.;
PACK K.; BELL J.A.; WILLIAMS C.B.; HODGSON S.V.; THOMAS H.J.W.;
TALBOT I.C.; BODMER W.F.; TOMLINSON I.PM.T. 1998. The APC variants
I1307K and E1317Q are associates with colorectal tumors, but not always with a
family history. Proc. Natl. Acad. Sci. vol.95 10722-10727.
GROSSMANN J.; MOHR S.; LAPETINA E.G.; FIOCCHI C.; LEVINE A.D. 1998.
Sequential and rapid activation of select caspasas during apoptosis of normal
intestinal epithelial cells. The American Physiological Society. G1117-G1124.
GUGLIEMI A.; ASCHELE C.; MORI A.; BALDO C.; RUSSO P.; DEBERNARDIS
D.; VALENTI P.; BRUNO S.; TAVERNA M.; ROSSO R.; SOBRERO A. 1995. In
Vitro Synergism between 5-Fluorouracil and natural β Interferon in human colon
carcinoma cells. Clinical Cancer Research. Vol. 1 1337-1344.
GUO M. & HAY B.A. 1999. Cell proliferation and apoptosis. Current Opinion in
Cell Biology. Vol. 11 745-752.
GUO R.J.; HUANG E.; EZAKI T.; PATEL N.; SINCLAIR K.; WU J.; KLEIN P.;
SUH E.R.; LYNCH J.P. 2004. Cdx1 Inhibits Human Colon Cancer Cell Proliferation
by Reducing β-Catenin/T-cell Factor Transcription Activity. The Journal of
Biological Chemistry. Vol:279 No. 35 36865-36875.
HAYWARD R.L.; MACPHERSON J.S.; CUMMINGS J.; MONIA B.P.; SMYTH
J.F.; JODRELL D.I. 2004. Enhanced oxaliplatin-induced apoptosis following

56
antisense Bcl-xl down-regulation is p53 and Bax dependent: Genetic evidence for
specificity of the antisense effect. Molecular Cancer Therapeutics. vol. 3 no. 2 169–
178.
HENGARTNER M.O. 2000. The biochemistry of apoptosis. Nature. vol 407 . 770-
776.
HERMAN J.G.; UMAR A.; POLYAK K.; GRAFFJ.R.; AHUJA N.; ISSA P.P.J.;
MARKOWITZ S.; WILLSON J.K.V.; HAMILTON S.R.; KINZLER K.W.; KANE
M.F.; KOLODNER R.D.; VOGELSTEIN B.; KUNKEL T.A.; BAYLIN S.B. 1998.
Incidence and functional consequences of hMLH1 promoter hypermethylation in
colorectal carcinoma. Proc. Natl. Acad. Sci. vol. 95 6870-6875.
HOPE M.E.; HOLD G.L.; KAIN R.; EL-OMAR E.M. 2005. Sporadic colorectal
cancer- role of the comensal microbiota. FEMS microbiology Letters 244 1-7.
HOUGHTON J.A.; MORTON C.L.; ADKINS D.A.; RAHMAN A. 1993. Locus of
the interaction among 5-Fluorouracil, leucovirin, and interpheron-alpha 2a in colon
carcinoma cells. Cancer Research. Vol. 4243-4250.
JORDAN A.; STEIN J. 2003. Effect of an omega-3 fatty acid containing lipid
emulsion alone and in combination with 5- fluorouracil (5FU) on growth of the colon
cancer cell line Caco-2. European Journal of Nutrition. Vol 42 324-331.
KAPITANOVIK S.; CACEV T.; RADOSEVIC S.; SPAVENTI S.; SPAVENTI R.;
PAVELIC K. 2004. APC gene loss of heterozygocity, mutations, E1317Q, and
I1307K germ-line variants in sporadic colon cancer in Croatia. Experimental and
Molecular Pathology vol. 77 193-200.
KASAHARA M.; TAKAHASHI Y.; NAGATA T.; ASAI S.; EGUCHI T.; ISHII Y.;

57
FUJII M.; ISHIKAWA K. 2000. Thymidylate synthase expression correlates closely
with E2F1 expression in colon cancer. Clinical Cancer Research. Vol. 6 2707-2711.
KITCHENS M.E.; FORSTHOEFEL A.M.; BARBOUR K.W. 1999. Mechanisms of
acquired resistance to thymidylate synthase inhibitors: the role of enzyme stability.
LAMPRECHT S.A. & LIPKIN M. 2002. Migrating colonic crypt epithelial cells:
primary targets for transformation. Carcinogenesis. vol.23 no. 11 1777-1780
LI H; MYEROFF L.; SMIRAGLIA D.; ROMERO M.F.; PRETLOW T.P.;
KASTURI L.; LUTTERBAUGH J.; RERKO R.M.;CASEY G.; ISSA J.P.; WILLIS
J.; WILLSON J.K.V; PLASS C.; MARKOVITZ S. 2003. SLC5A8, a sodium
transporter, is a tumor suppressor gene silenced by methylation in human colon
aberrant crypt foci and cancers. Proc. Natl. Acad. Sci. vol.100 no.14 8412-8417.
LONGLEY D.B.; HARKIN D.P.; JOHNSTON P.G. 2003. 5-Fluorouracil:
Mechanisms of Action and Clinical Strategies. Nature Reviews/ Cancer. vol 3. 330-
338.
LOPEZ G. 2002. Manual De Patología colo-recto-anal. Editorial Gente Nueva.
Bogotá.
MARIADASON J.M.; ARANGO D.; CORNER G.A.; ARAÑES M.J.; HOTCHKISS
K.A.; YANG W.; AUGENLICHT L.H. 2003. A Gene Expression Profile That
Defines Colon Cell Maturation in Vitro. Cancer Research 62, 4791–4804.
MATHERLY L.H.; CZAJKOWSKI C.A.; MUENCH S.P.; PSIAKIS J.T. 1990. Role
for cytosolic folic-binding proteins in the compartmentation of endogenous
tetrahydrofolates and the 5-formyltetrahydrofolate-mediated enhancement of 5-
fluoro-2´-deoxyuridine antitumor activity in vitro. Cancer Research. Vol.50. 3262-

58
3269.
MICHOR F.; IWASA Y.; LENGAUER C.; NOWAK M.A. 2005. Dynamics of
colorectal cancer. Seminars in Cancer Biology. 10pp
MORIN P.J.; VOGELSTEIN B.; KINZLER K.W. 1996. Apoptosis and APC in
colorectal tumorigenesis. Proc. Natl. Acad. Sci. vol93. 7950-7954.
NEAULT J.F. & TAJMIR-RIAHI H.A.1999. Structural Analysis of DNA-
Chlorophyll Complexes by Fourier Transform Infrared Difference Spectroscopy.
Biophysical Journal. Vol. 76 2177-2182.
NICOLETTI I; MANNUCCI R; MIGLIORATI G; RICCARDI C & GRIGNANI F.
1997. Common methods for measuring apoptotic cell death by flow cytometry. The
Purdue Cytometry CD-ROM Volume 3, J. Parker, C. Stewart, Guest Eds., J.Paul
Robinson, Publisher. Purdue University, West Lafayette, ISBN 1-890473-02-2
PAPAMICHAEL D. 2000. The Use of Thymidylate Synthase Inhibitors in the
treatment of Advanced Colorectal Cancer: Current Status. Stem Cells. vol. 18 166-
175.
RAO V.C.;NEWMARK H.L.; REDDY B.S. 1998. Chemopreventive effect of
squalene on colon cancer. Carcinogenesis. vol. 19 no. 2. 287-290.
ROBBINS S.L.; KUMAR V.; COTRAN R.; COLLINS T. 1999. Pathologic Basis of
Disease. Sixth Edition. Philadelphia W.B.Saunders. 1425pp
RUSSO P.; OTTOBONI C.; MALACARNE D.; CRIPPA A.; RIOU J.F.;
O´CONNOR P.M. 2002. Nonpeptidomimetic Farnesyltransferase Inhibitor RPR-
115135 Increases Cytotoxixity of 5-Fluorouracil: Role of p-53. The Journal of

59
Pharmacology and Experimental Therapeutics. Vol.300 no.1 220-226.
RUSTUM Y.M. 2003. Fluoropyrimidines in Cancer Therapy. Humana Press.
Totowa, New Jersey 07512, USA. 318 p.
SESINK A.L.A; TERMONT D.S.M.L.; KLEIBEUKER J.H.; VAN DER MEER.
1999. Red meat and Colon Cancer: The cytotoxic and Hyperproliferative Effects of
Dietary Heme. Cancer Research Vol. 59 5704-5709.
SCHAFER K.A. 1998. The Cell Cycle: a Review. Veterinary Pathology. Vol 35 461-
478.
SINGH J.; RIVENSON A.; TOMITA M.; SHIMAMURA S.; ISHIBASHI N. &
REDDY B.S. 1997. Bifidobacterium longum, a lactic acid─producing intestinal
bacterium inhibits colon cancer and modulates the intermediate biomarkers of colon
carcinogenesis. Carcinogenesis. vol. 18 no. 4 833─841.
STIEWE T.; PÜTZER B.M. 2000. Role of the p53-homologue p73 in E2F1-induced
apoptosis. Nature Genetics. Vol. 26 464-469.
TANAKA F.; FUKUSE T; HIROMI W.; FUKUSHIMA M. 2000. The History,
Mechanism and clinical use of Oral 5-Fluorouracil Derivative Chemotherapeutic
Agents. Current Pharmacological Biotechnology. vol.1 137-164.
TILLMAN D.M.; PETAK I.; HOUGHTON J.A. 1999. AFas-dependent component
in 5-Fluorouracil/Leucovirin-induced cytotoxicity in colon carcinoma cells. Clinical
Cancer Research. Vol.5 425-430.
TORRANCE C.J.; JACKSON P.E.; MONTGOMERY E.; KINZLER K.W.;
VOGELSTEIN B.; WISSNER A.; NUNES M.; FROST P.; DISCAFANI C.M. 2000.

60
Combinatorial Chemoprevention of Intestinal Neoplasia. Nature Medicine. vol.6.
no.8 1024-1028.
TOYOTA M; AHUJA N; OHE-TOYOTA M; HERMAN J.G; BAYLIN S.B; ISSA
J.P. 1999. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad.
Sci. vol. 96 8681-8686.
VELHO S.; OLIVIERA C.; FERREIRA A.; FERREIRA A.C.; SURIANO G.;
SCHWARTZ S.J.; DUVAL a.; CARNEIRO F.; MACHADO J.C.; HAMELIN R.;
SERUCA R. 2005. The prevalence of PIK3CA mutations in gastric and colon
cancer. European Journal of Cancer vol. 41 1649-1654.
VERMEULRN K.; VAN BOCKSTAELE D.R.; BERNEMAN Z.N. 2003. The cell
cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell
Proliferation 36, 131-149.
VOLGESTEIN B. & KINZLER K. 2004. Cancer genes and the pathways they
control. Nature Medicine. Vol.10 no.8 789-799.
WANG T.L.; DIAZ L.A.; ROMANS K.; BARDELLI A.; SAHA S.; GALIZIA G.;
CHOTI M.; DONEHOWER R.; PARMIGIANI G.; SHIH I.M.; IACOBUZIO-
DONAHUE C.; KINZLER K.W.;VOGELSTEIN B.; LENGAUER C.;
VELCULESCU V.E. 2004. Digital karyotyping identifies thymidilate synthase
amplification as a mechanism of resistance to 5-fluoracil in metastasic colorectal
cancer patients. Proc. Natl. Acad. Sci. vol.101 no. 9. 3089-3094.
VELHO S.; OLIVEIRA C.; FERREIRA A; FERREIRA A. C.; SURIANO G.;
SCHWARTZ S. JR; DUVAL A.; CARNEIRO F; MACHADO J.C; HAMELIN R.;
SERUCA R. 2005. The prevalence of PIK3CA mutations in gastric and colon cancer.
European Journal of Cancer 41. 1649–1654.

61
WEITZ J.; KOCH M.; DEBUS J.; HOHLER T.; GALLE P.R.; BUCHLER M.W.
2005. Colorectal cancer. The Lancet. 153-165.
WILLIAMS A.C.; HAGUE A.; ELDER J.E.E.; PARASKEVA C. 1996. In vitro
models for studying colorectal carcinogenesis: cellular and molecular events
including APC and Rb cleavage in the control of proliferation, differentiation and
apoptosis. Bioquímica et Biophysica Acta 1288 F9-F19.
WILLSON J.K. V.; GREEN S.B.; ISSA J.P. ; MARKOWITZ S.D. 2002. HLTF
gene silencing in human colon cancer. Proc. Natl. Acad. Sci. Vol: 99 No: 7 4562-
4567.
YAMADA Y.; OYAMA T.; HIROSE Y.; HARA A.; SUGIE S.; YOSHIDA K.;
YOSHIMI N.; MORI H. 2003. β- Catenin mutation is selected during malignant
transformation in colon carcinogénesis. Carcinogenesis. vol.24 no.1. 91-97.
YOUNG J.; BIDEN K.G.; SIMMS L.A.; HUGGARD P.; KARAMATIC R.; EYRE
H.J; SUTHERLAND G.R.; HERATH N.; BARKER M.; ANDERSON G.J;
FITZPATRICK D.R.; RAMM G.A.; JASS J.R.; LEGGETT B.A. 2001. HPP1: A
transmembrane protein-encoding gene commonly methylated in colorectal polyps and
cancers. Proc. Natl. Acad. Sci . vol.98 no.1 265-270.


















