
Ensayo clínico-expo-3
49
Ensayo Clínico Integrantes: Enciso Avilés Zaira Selina Gonzáles Benítez Mariana López Díaz Alba Miramontes González Claudia Patricia Monroy Parra Frida Margarita Tovar Gómez María Carolina Unidad Académica de Medicina Academia de Farmacología 1
-
Upload
andrea-ortega -
Category
Health & Medicine
-
view
45 -
download
0
Transcript of Ensayo clínico-expo-3

1
Ensayo ClínicoIntegrantes:
- Enciso Avilés Zaira Selina
- Gonzáles Benítez Mariana
- López Díaz Alba
- Miramontes González Claudia Patricia
- Monroy Parra Frida Margarita
- Tovar Gómez María Carolina
Unidad Académica de Medicina
Academia de Farmacología

2 Según la OMS
- Es cualquier estudio de investigación que asigna de manera prospectiva participantes humanos o grupos de humanos a una o más intervenciones sanitarias a fin de evaluar los efectos en los resultados sanitarios.
(*incluye ensayos de fase I a fase IV) Objetivo
Evaluar la eficacia de
intervenciones sanitarias, médicas ó
quirúrgicas.
Who.int. OMS | Bienvenido a la ICTRP de la OMS [Internet]. [cited 27 May 2015]. Available from: http://www.who.int/ictrp/es/
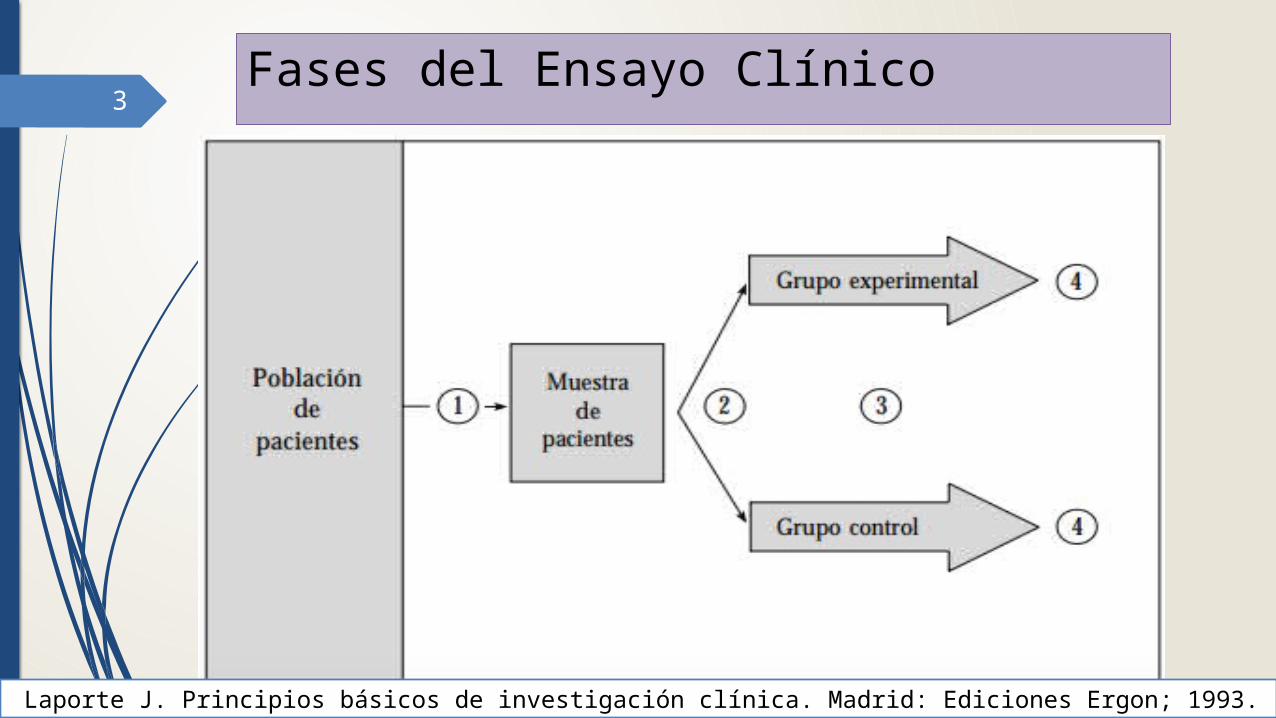
3Fases del Ensayo Clínico
Laporte J. Principios básicos de investigación clínica. Madrid: Ediciones Ergon; 1993.
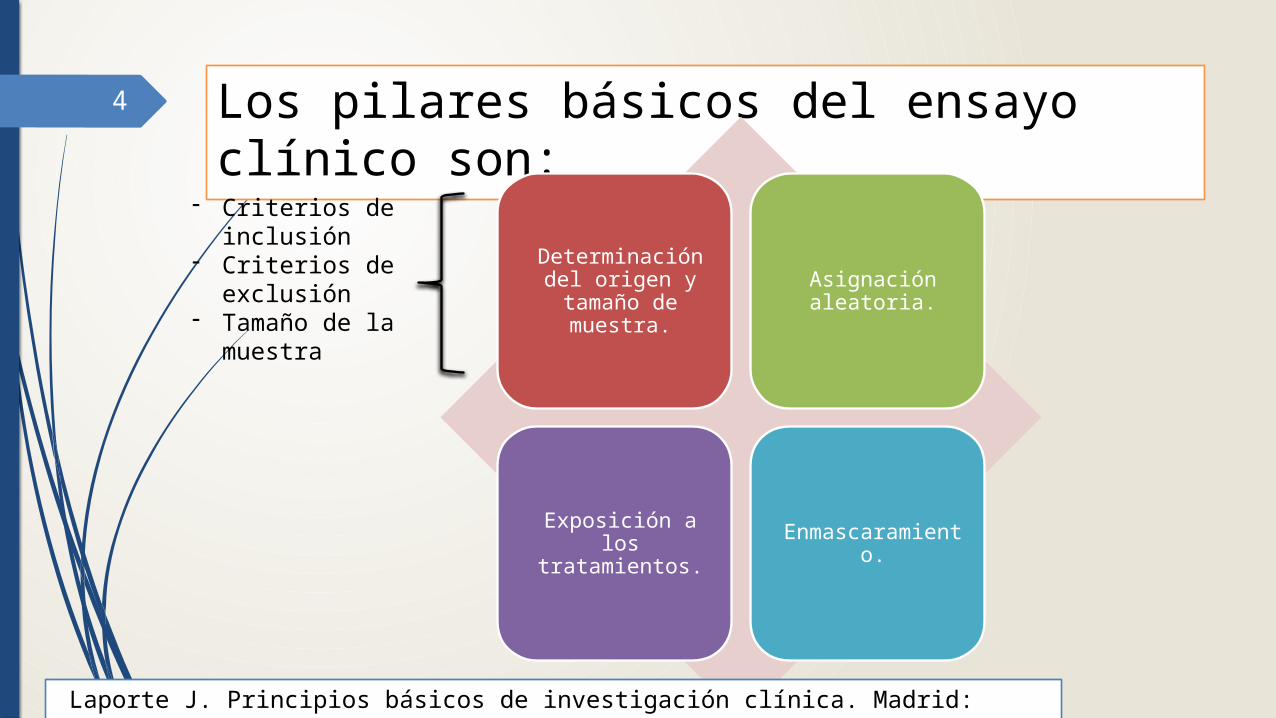
4 Los pilares básicos del ensayo clínico son:
Determinación del origen y tamaño
de muestra.
Asignación aleatoria.
Exposición a los tratamientos.
Enmascaramiento.
- Criterios de inclusión
- Criterios de exclusión
- Tamaño de la muestra
Laporte J. Principios básicos de investigación clínica. Madrid: Ediciones Ergon; 1993.

5 Tipos de Ensayos Clínicos
- Ensayo Clínico Abierto - Ensayo Clínico a Ciego Simple- Ensayo Clínico a Doble Ciego- Ensayo Clínico a Triple Ciego- Ensayo Clínico Cruzado- Ensayo Clínco Paralelo- Ensayo Clínico Secuencial- Fase I
- Fase II (Tx para cáncer de mama avanzado en combinación con otro fármaco)
- Fase III- Fase IV
Laporte J. Principios básicos de investigación clínica. Madrid: Ediciones Ergon; 1993.

6 ¿Qué son los registros de ensayos?
- La OMS considera el registro de ensayos como: “la publicación de un conjunto de datos consensuados internacionalmente sobre el diseño, la conducción y la administración de ensayos clínicos.”
Who.int. OMS | Bienvenido a la ICTRP de la OMS [Internet]. [cited 27 May 2015]. Available from: http://www.who.int/ictrp/es/
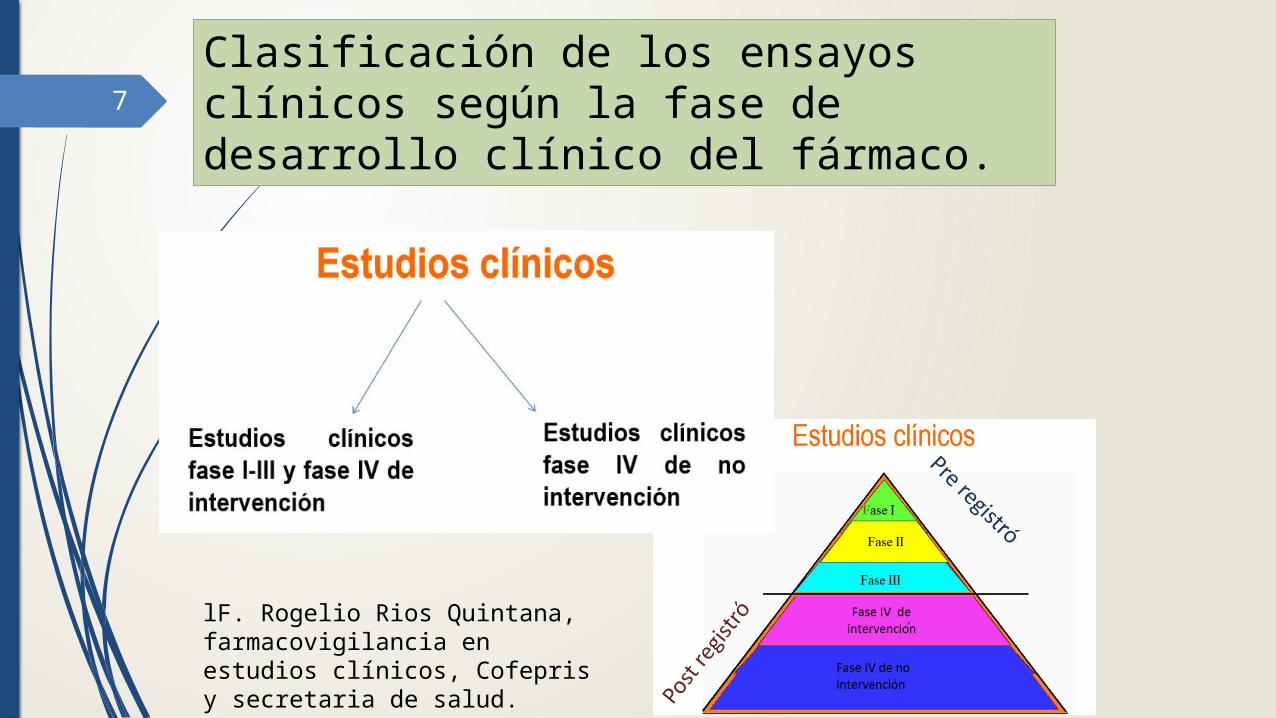
7
Clasificación de los ensayos clínicos según la fase de desarrollo clínico del fármaco.
lF. Rogelio Rios Quintana, farmacovigilancia en estudios clínicos, Cofepris y secretaria de salud.

8 - FASE I:- Suponen la primera administración de un
nuevo fármaco a seres humanos.
ESTUDIOS FARMACOCINETICOS
(DOSIS : UNICAS, REPETIDA, MULTIPLES) ESTUDIOS DE
TOLERABILIDAD Y SEGURIDAD
ESTUDIOS FARMACODINAMICOS
ESTUDIOS DE BIODISPONIBILIDAD Y BIOEQUIVALENCIA.
Josep Eladi Baños Díez, Magí Farré Albaladejo,Principios de farmacologia clinica, Elsevier España, 2002

9- FASE II :- Se realizan en pacientes que padecen la
enfermedad o la entidad clínica para la que va utilizarse el nuevo fármaco.
- SUS OBJETIVOS: Obtención de la información preliminar sobre la eficacia
Establecimiento de una relación entre dosis y respuesta
Obtener mas información sobre la seguridad del nuevo fármaco.
Josep Eladi Baños Díez, Magí Farré Albaladejo,Principios de farmacologia clinica, Elsevier España, 2002

10- FASE III ( ESTUDIOS TERAPEUTICOS CONFIRMATORIOS) :
• Tienen por objetivo principal evaluar la eficacia y la seguridad del tratamiento experimental en una muestra de pacientes mas representativa de la población.
Josep Eladi Baños Díez, Magí Farré Albaladejo,Principios de farmacologia clinica, Elsevier España, 2002

11 - FASE IV:
- Incluye todos aquellos realizados tras la comercialización del medicamento.
- OBJETIVO:
Establecer su eficacia y seguridad a largo plazo
Estudios de uso terapéutico, o de farmacovigilancia
Josep Eladi Baños Díez, Magí Farré Albaladejo,Principios de farmacologia clinica, Elsevier España, 2002
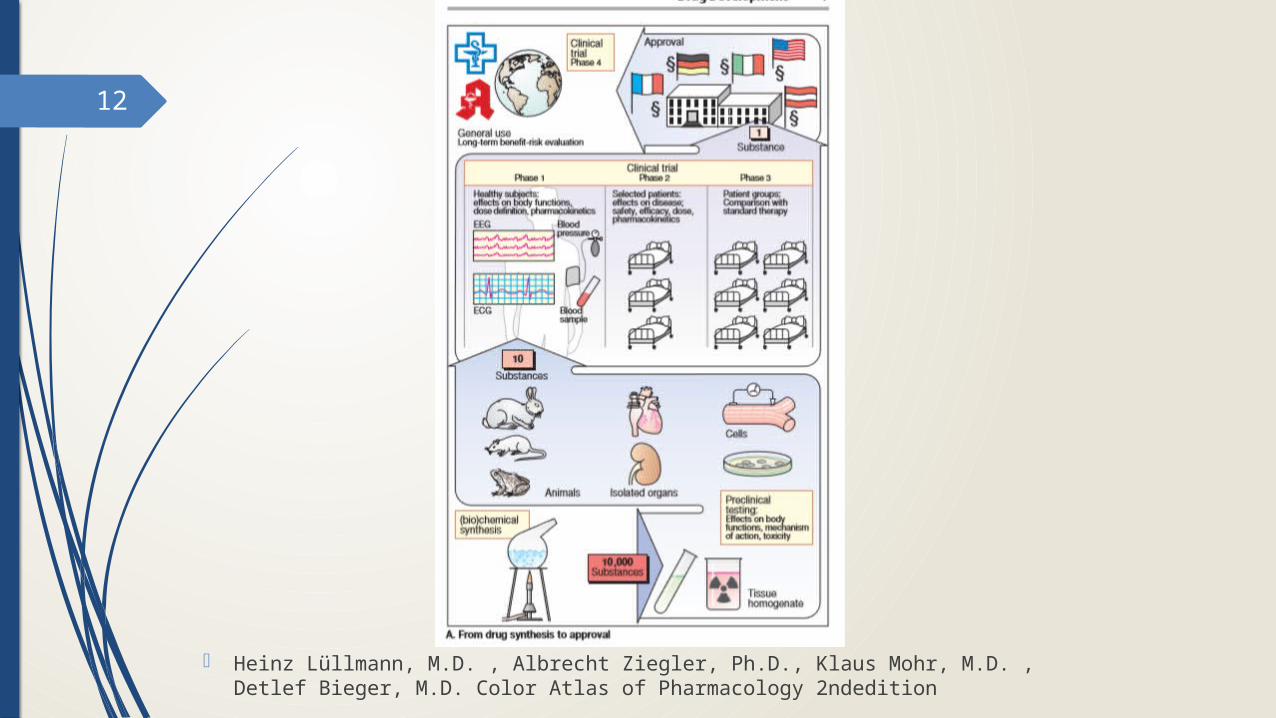
12
- Heinz Lüllmann, M.D. , Albrecht Ziegler, Ph.D., Klaus Mohr, M.D. , Detlef Bieger, M.D. Color Atlas of Pharmacology 2ndedition

13
NORMAS DE BUENA PRACTICA CLINICA( bpc) conjunto de documentos que aseguran que los
ensayos clínicos se realizan siguiendo procedimientos estándar que aseguran su validez y fiabilidad científica.
Asegurar que los estudios de farmacología y toxicología en animales se realizan con absoluto rigor con el fin que los resultados sean creíbles.
Buena arma para evitar el fraude científico.
Josep Eladi Baños Díez, Magí Farré Albaladejo,Principios de farmacologia clinica, Elsevier España, 2002

NOM-220-SSA1-2012 Instalación y
Operación de la farmacovigilancia 14
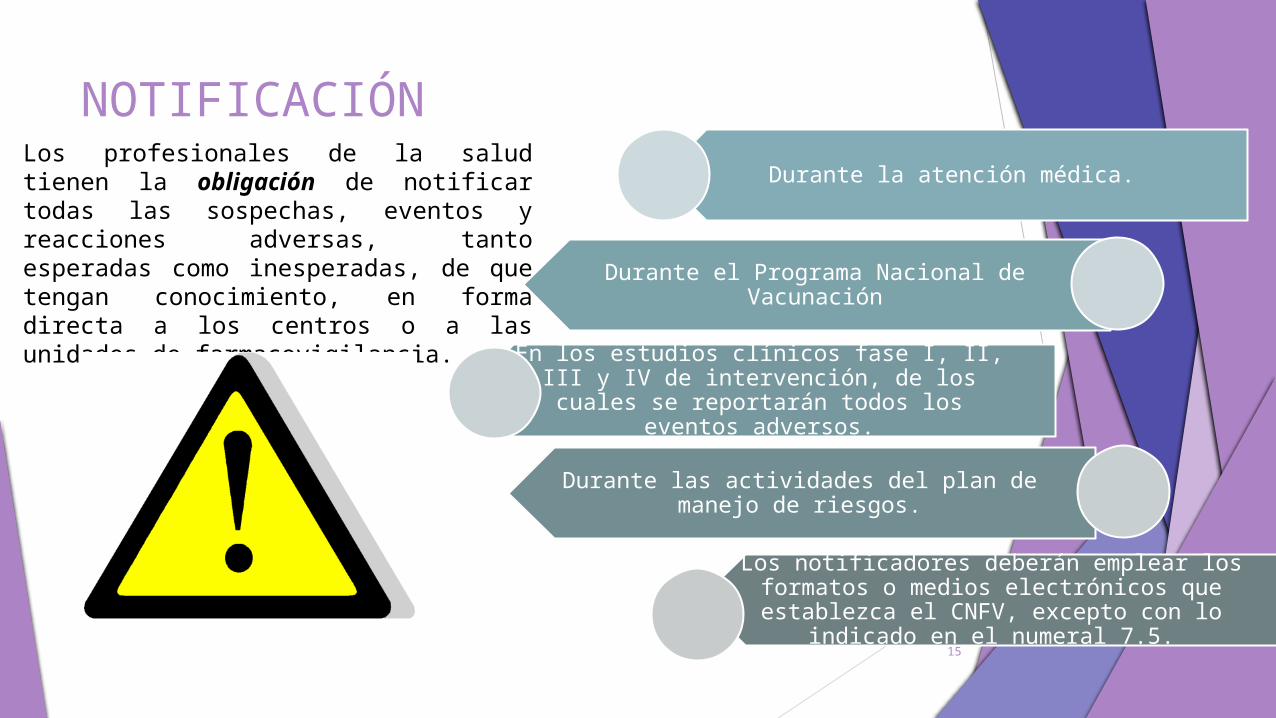
NOTIFICACIÓNLos profesionales de la salud tienen la obligación de notificar todas las sospechas, eventos y reacciones adversas, tanto esperadas como inesperadas, de que tengan conocimiento, en forma directa a los centros o a las unidades de farmacovigilancia.
Durante la atención médica.
Durante el Programa Nacional de Vacunación
En los estudios clínicos fase I, II, III y IV de intervención, de los cuales se reportarán
todos los eventos adversos.
Durante las actividades del plan de manejo de riesgos.
Los notificadores deberán emplear los formatos o medios electrónicos que
establezca el CNFV, excepto con lo indicado en el numeral 7.5.
15

Las provenientes de no profesionales de la salud podrán notificar directamente las sospechas RAM a cualquier Unidad de Farmacovigilancia, ya sea a través de un profesional de la salud, vía telefónica o por cualquier otro medio de que dispongan.
www.cofepris.Gob.mx
(55) 50805200 extensión 1432
Oklahoma No. 14 Col. Nápoles, C.P 03810 D.F
16
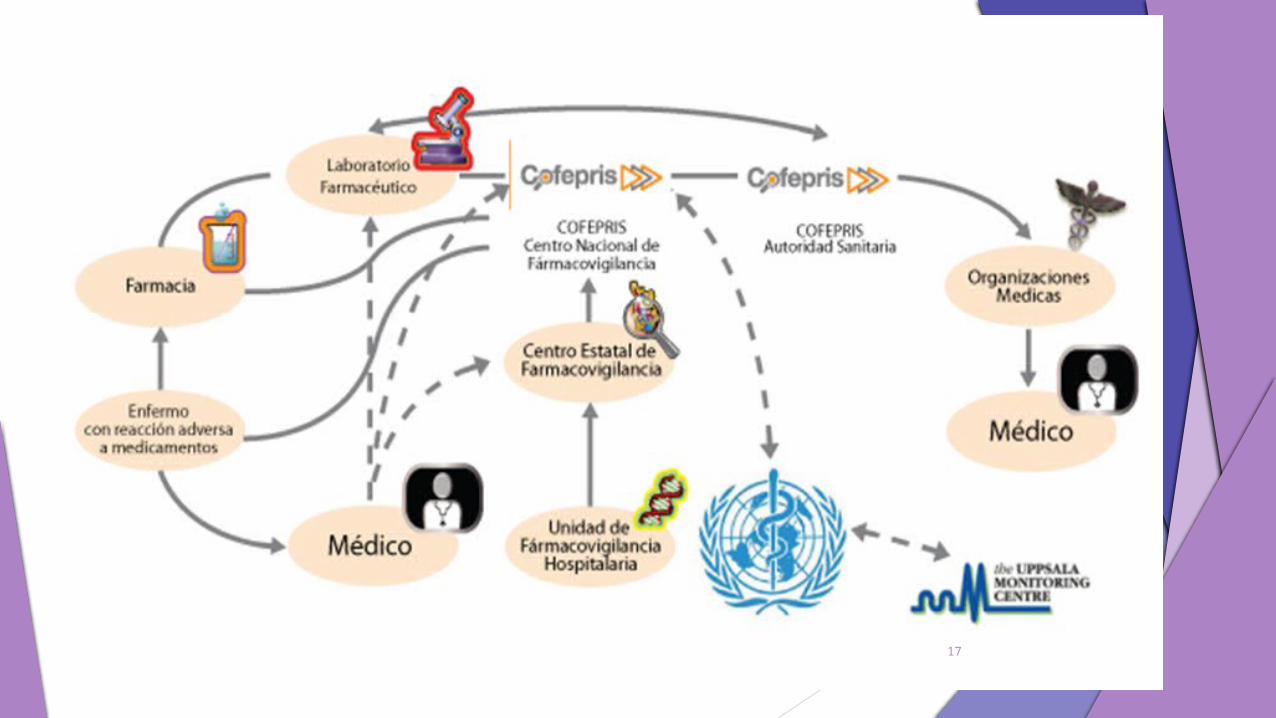
17
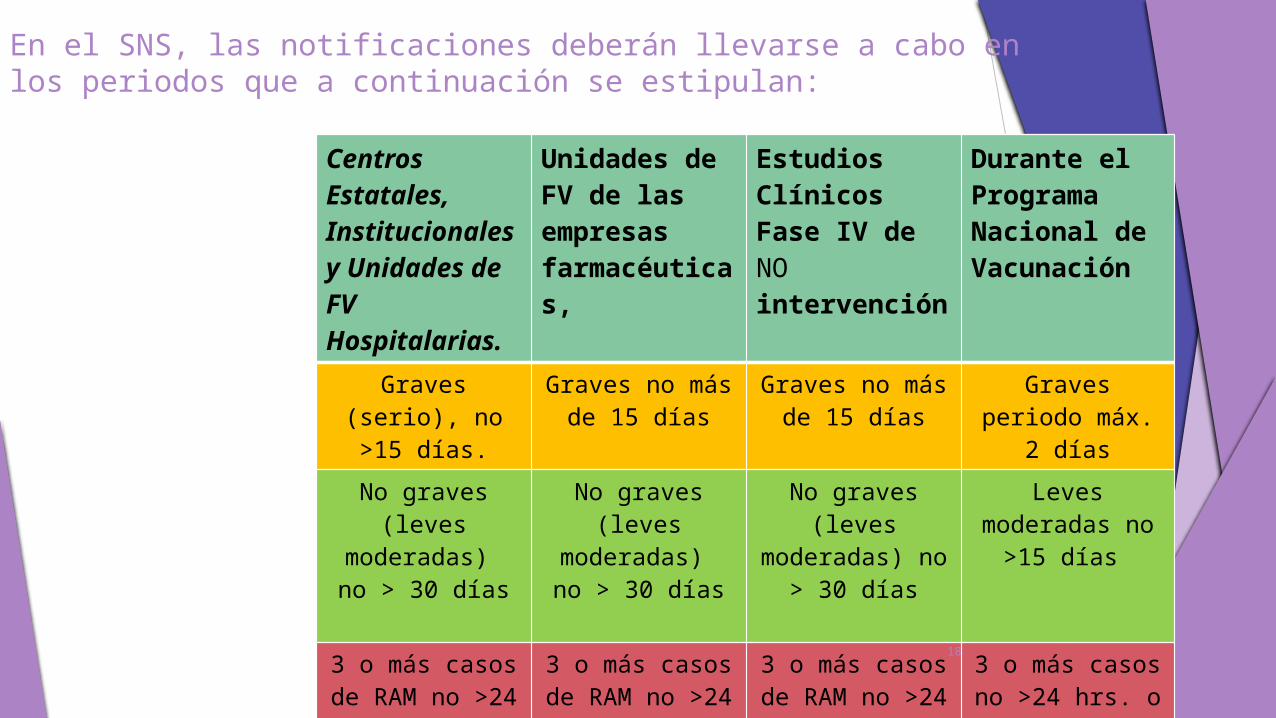
En el SNS, las notificaciones deberán llevarse a cabo en los periodos que a continuación se estipulan:
Centros Estatales, Institucionales y Unidades de FV Hospitalarias.
Unidades de FV de las empresas farmacéuticas,
Estudios Clínicos Fase IV de NO intervención
Durante el Programa Nacional de Vacunación
Graves (serio), no >15 días.
Graves no más de 15 días
Graves no más de 15 días
Graves periodo máx. 2 días
No graves (leves
moderadas) no > 30 días
No graves (leves
moderadas) no > 30 días
No graves (leves
moderadas) no > 30 días
Leves moderadas no
>15 días
3 o más casos de RAM no >24
horas.
3 o más casos de RAM no >24
horas. O sig. Día hábil
3 o más casos de RAM no >24
horas.
3 o más casos no >24 hrs. o al
sig. día hábil
18

Estudios clínicos fases I, II, III y IV de intervención.Graves que sucedan en el extranjero
incluirse en el reporte de seguridad final del estudio.
Graves (serio), no > 15 días RAM que sucedan en sujetos dentro del territorio nacional.
No graves (leves, moderados) , en sujetos dentro del territorio nacional, deberán incluirse en el reporte de seguridad final del estudio.
19

REGLAMENTO DE LA LEY GENERAL DE SALUD EN MATERIA DE INVESTIGACION
PARA LA SALUD
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

Disposiciones generales
• Articulos 1-12.
ARTICULO 3o.- La investigación para la salud comprende el desarrollo de acciones que contribuyan:
• Conocimiento de procesos biológicos y psicológicos en los seres humanos así como de los vínculos entre las causas de enfermedad, la práctica médica y la estructura social
• Prevención y control de los problemas de salud;
• Conocimiento y evaluación de los efectos nocivos del ambiente en la salud
• Estudio de técnicas y métodos que se recomienden o empleen para la prestación de servicios de salud, y A la producción de insumos para la salud.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

Organizaciones relacionadas
• Secretaria de salud• Comisión interinstitucional de investigación en salud• SEP• Consejo nacional de ciencia y tecnología• Instituciones de educación superior• Sistema nacional de registro de la investigación y desarrollo tecnológico• Consejo de salubridad general
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

• I. Se ajustará a los principios científicos y éticos que la justifiquen
• II.- Se fundamentará en la experimentación previamente realizada en animales , laboratorios o en otros hechos científicos.
• III.- Se deberá realizar sólo cuando el conocimiento no pueda obtenerse por otro medio idóneo
• IV.- Deberán prevalecer siempre las probabilidades de los beneficiados esperados sobre los riesgos predecibles;
• V.- Contará con consentimiento informado y por escrito del sujeto de investigación o su representante legal.
Aspectos Éticos de la Investigación en Seres Humanos13-27
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

• VII. Contará con el dictamen favorable de las Comisiones de Investigación, Ética y la de Bioseguridad
• VIII. Se llevará a cabo cuando se tenga la autorización del titular de la institución de atención a la salud y, en su caso, de la Secretaría.
VI.- Deberá ser realizada por profesionales de la salud bajo la responsabilidad de una institución de atención a la salud que actúe bajo la supervisión de las autoridades sanitarias competentes y cuente con recursos humanos y materiales necesarios, que garanticen el bienestar del sujeto de investigación
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

I.- Investigación sin riesgo: no se realiza intervención o modificación intencionada en las variables fisiológicas, psicológicas y sociales de los individuos (cuestionarios, entrevistas, revisión de expedientes clínicos y otros)
II. Investigación con riesgo mínimo: Estudios prospectivos emplean riesgo de datos a través de procedimientos comunes en exámenes físicos o psicológicos de diagnósticos o tratamiento rutinarios, (pesar al sujeto, colección de excretas y secreciones externas, obtención de placenta durante el parto, obtención de saliva, extracción de sangre por punción venosa en adultos en buen estado de salud, con frecuencia máxima de dos veces a la semana y volumen máximo de 450 Ml.)
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

• III.- Investigación con riesgo mayor que el mínimo: las probabilidades de afectar al sujeto son significativas, se consideran: estudios radiológicos y con microondas, ensayos con los medicamentos , ensayos con nuevos dispositivos, procedimientos quirúrgicos, extracción de sangre 2% del volumen circulante en neonatos, y otras técnicas invasoras o procedimientos mayores.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

De la Investigación en Menores de Edad o Incapaces. 34-39
• Asegurar estudios semejantes en personas de mayor de edad y en animales inmaduros, excepto cuando se trate de estudiar condiciones propias de la etapa neonatal o padecimientos específicos de ciertas edades.
ARTICULO 38.- Las investigaciones de riesgo y con probabilidad de beneficio directo para el menor o el incapaz, serán admisibles cuando;I.- El riesgo se justifique por importancia del beneficio II.- El beneficio sea igual o mayor a otras alternativas ya establecidas para su diagnóstico y tratamiento.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

ARTICULO 39.- las investigaciones clasificadas como riesgo y sin beneficio directo al menor o al incapaz, serán admisibles:• Si riesgo mínimo:-altas probabilidades de obtener conocimientos sobre la condición del sujeto, que sean de gran importancia para comprender el trastorno o para lograr su mejoría en otros sujetos.• Si riesgo mayor al mínimo:-grandes probabilidades de entender, prevenir o aliviar un problema grave que afecte la salud y el bienestar, y haya supervisión estricta para determinar si aumenta la magnitud de los riesgos
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

De la investigación en Mujeres en Edad Fértil, Embarazadas, durante el Trabajo de Parto, Puerperio, Lactancia y Recién Nacidos; de la utilización de Embriones, Obitos y Fetos y de la Fertilización Asistida. 40-56.
riesgo mayor que el mínimo que se realicen en mujeres en edad fértil, deberán tomarse medidas para:
Certificar que las mujeres no están embarazadas, previamente a su aceptación como sujetos de investigación, y Disminuir en lo posible las posibilidades de embarazo durante el desarrollo de la investigación.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

• ARTICULO 47.- Las investigaciones en mujeres embarazadas, con beneficio terapéutico relacionado con el embarazo, se permitirán cuando:
• I. Tengan por objeto mejorar la salud de la embarazada con un riesgo mínimo para el embrión o feto, o
• II.- Estén encaminadas a incrementar la viabilidad del feto, con un riesgo mínimo para la embarazada.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

Investigación Farmacológica65-71
• actividades científicas encaminadas al estudio de medicamentos y productos biológicos para uso en humanos, respecto de los cuales no se tenga experiencia previa en el país, que no hayan sido registrados por la Secretaría y, por lo tanto, no sean distribuidos en forma comercial, así como los medicamentos registrados y aprobados para su venta, cuando se investigue su caso con modalidades, indicaciones, dosis o vías de administración diferentes de las establecidas, incluyendo en empleo en combinaciones.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

COMISIONES98-112
• Comisión de Ética • Comisión de Bioseguridad • Comisión de investigación• Las finalidades principales serán las siguientes:• I. Proporcionar asesoría a los titulares o responsables de la institución• II. Auxiliar a los investigadores para la realización óptima de sus estudios, y• III. Vigilar la aplicación de este Reglamento y demás disposiciones aplicables.
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos

De la Investigación que incluya a la utilización de animales de experimentación. 121-126
• ARTICULO 122.- Las investigaciones se diseñarán a modo de evitar al máximo el sufrimiento de los animales.
• ARTICULO 123.- Cuando sea necesario sacrificar a un animal se empleará un procedimiento que asegure su muerte sin sufrimiento.
• ARTICULO 125.- Los bioterios de producción o mantenimiento crónico serán supervisados por profesionales calificado y competente en la materia y deberán permitir el crecimiento, maduración, reproducción y comportamiento normal de los animales, de conformidad con las normas que la propia institución emita.REGLAMENTO de la Ley General de Salud en Materia de
Investigación para la Salud. Estados Unidos Mexicanos

Las ratas. Cobayas o
conejillos de Indias
Conejos Perros
REGLAMENTO de la Ley General de Salud en Materia de Investigación para la Salud. Estados Unidos Mexicanos
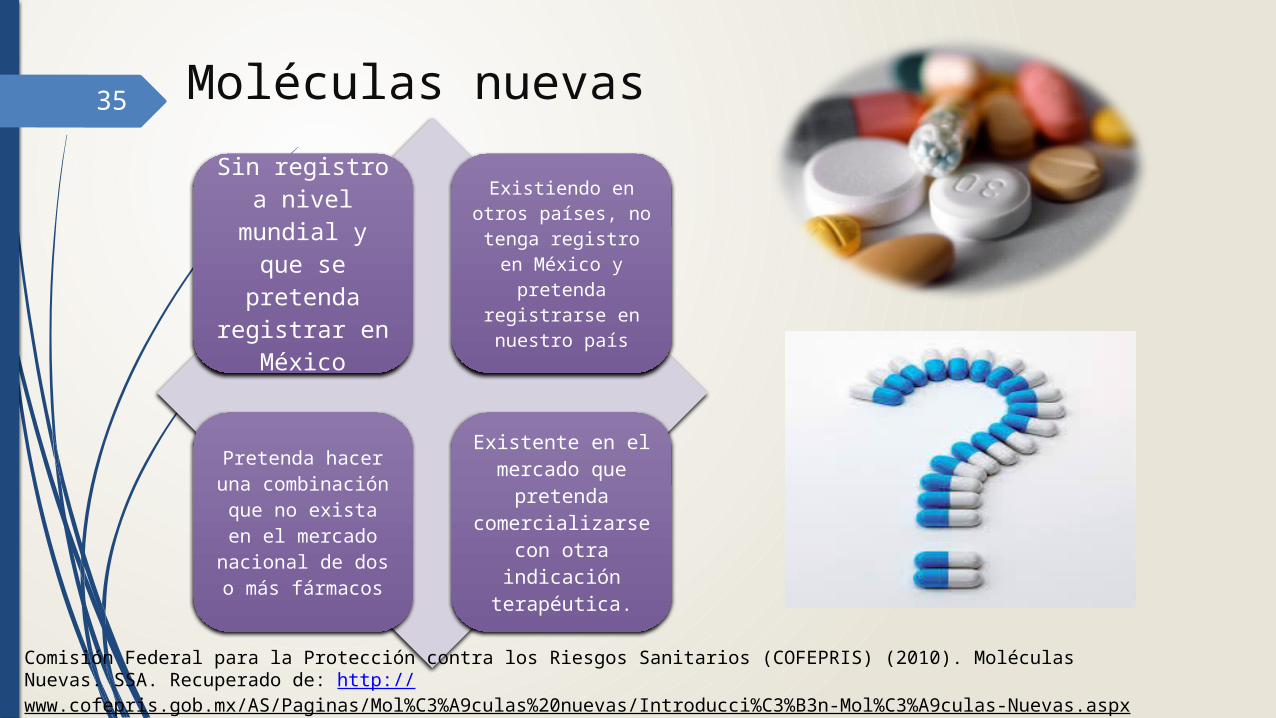
35 Moléculas nuevas
Sin registro a nivel mundial
y que se pretenda
registrar en México
Existiendo en otros países, no tenga registro en México y
pretenda registrarse en nuestro país
Pretenda hacer una
combinación que no exista en el mercado
nacional de dos o más fármacos
Existente en el mercado que
pretenda comercializars
e con otra indicación
terapéutica.
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

36Comité de Moléculas Nuevas (CMN)
- Apoyo al análisis y evaluación de:
- Productos farmacéuticos nuevos
-Con nuevas indicaciones
- Evaluados por grupos de especialistas
Para: establecer los elementos adicionales a la evaluación y autorización de un producto
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

37 ¿Cómo funciona?
Solicitud de reunión a CMN
Presentación de la molécula nueva ante el Comité
Solicitud de más información
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

38¿Cómo solicitar una reunión ante el CMN?
Rubro Información
1. Datos del solicitante
• Razón social• Representante legal• Dirección• Teléfono• E-mail
2. Datos de la molécula
• Nombre genérico• Denominación distintiva (si
aplica)• Forma farmacéutica• Concentraciones• Vía de administración• Consideración de uso
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

39Rubro Información
3. Clasificación de la molécula
• Tipo de solicitud • Tipo de molécula• Origen de la molécula
4. Información de la molécula
• Indicación solicitada• Resumen de evidencia pre-
clínica y clínica (estudios fase I, II y III)
• Resumen de plan de manejo de riesgos y de eventos adversos
• Resumen de metodología analítica
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

40
Rubro Información
5. Estatus regulatorio • Nuevo registro a nivel mundial• Registro en otros países
indicando agencia regulatoria y fecha de autorización
6. Propuesta de fecha probable de reunión
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

41 Autorización de protocolo de investigación
La investigación clínica en México forma parte de los requisitos para el registro de insumos para la salud ya que provee información sobre el riesgo-beneficio del producto sobre nuestra población.
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

42 ¿Qué tipo de autorizaciones se otorgan por el área?
- Protocolos de investigación de medicamentos para empleo en seres humanos
- Protocolos de investigación de nuevos recursos (dispositivos médicos)
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx
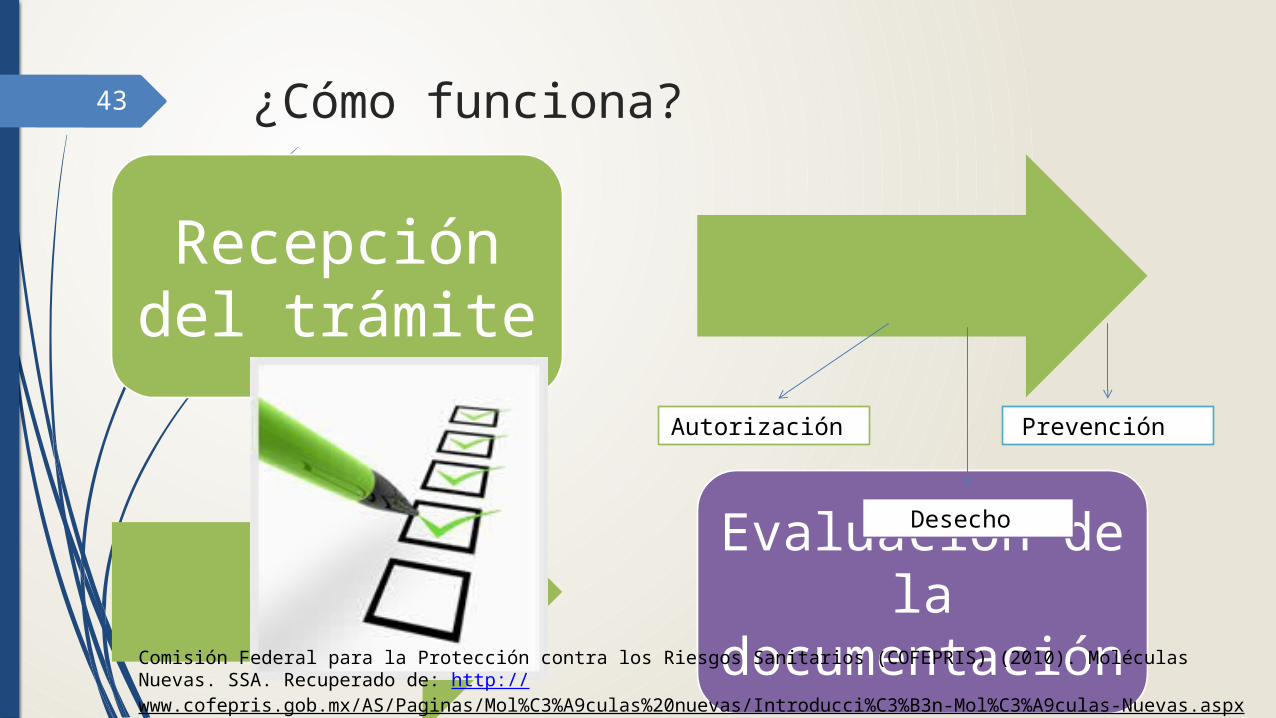
43 ¿Cómo funciona?
Recepción del trámite
Evaluación de la
documentación
Autorización Prevención
Desecho
Comisión Federal para la Protección contra los Riesgos Sanitarios (COFEPRIS) (2010). Moléculas Nuevas. SSA. Recuperado de: http://www.cofepris.gob.mx/AS/Paginas/Mol%C3%A9culas%20nuevas/Introducci%C3%B3n-Mol%C3%A9culas-Nuevas.aspx

44
Etapa postcomercializacion:
Estudios fase IV
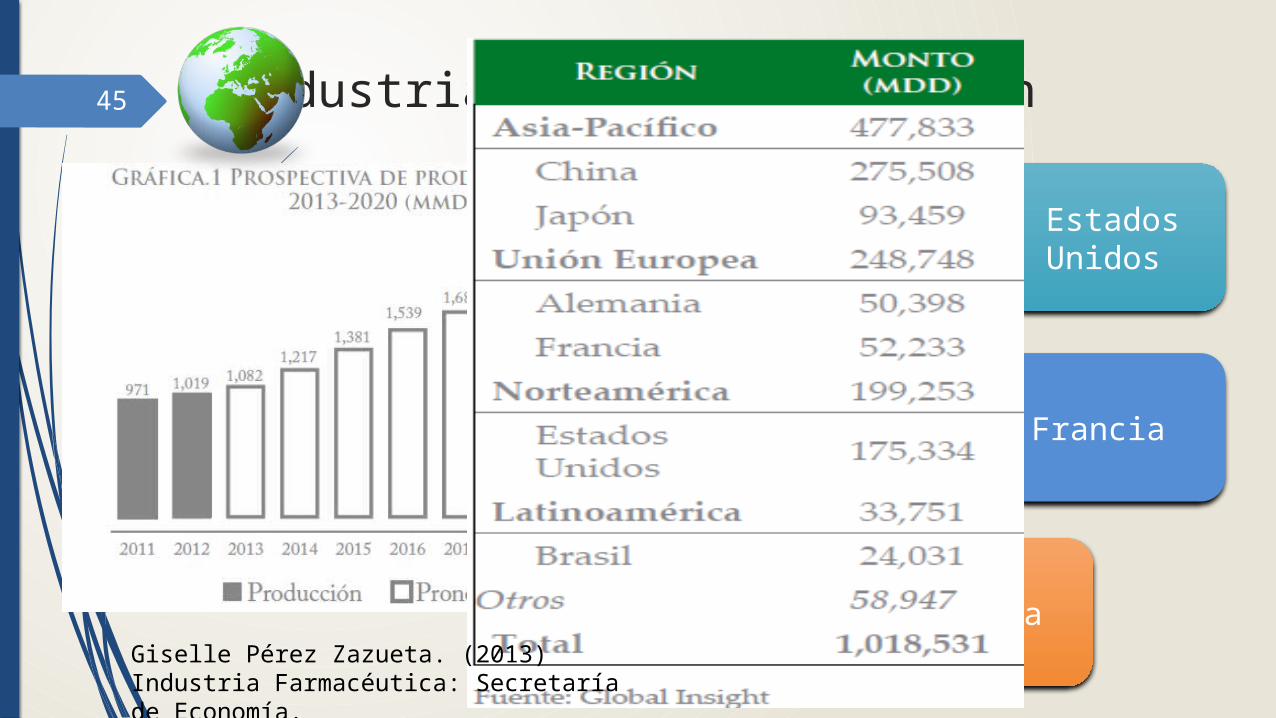
45 Industria Global: Producción
China
Alemania
Francia Japón
Estados Unidos
Giselle Pérez Zazueta. (2013) Industria Farmacéutica: Secretaría de Economía.
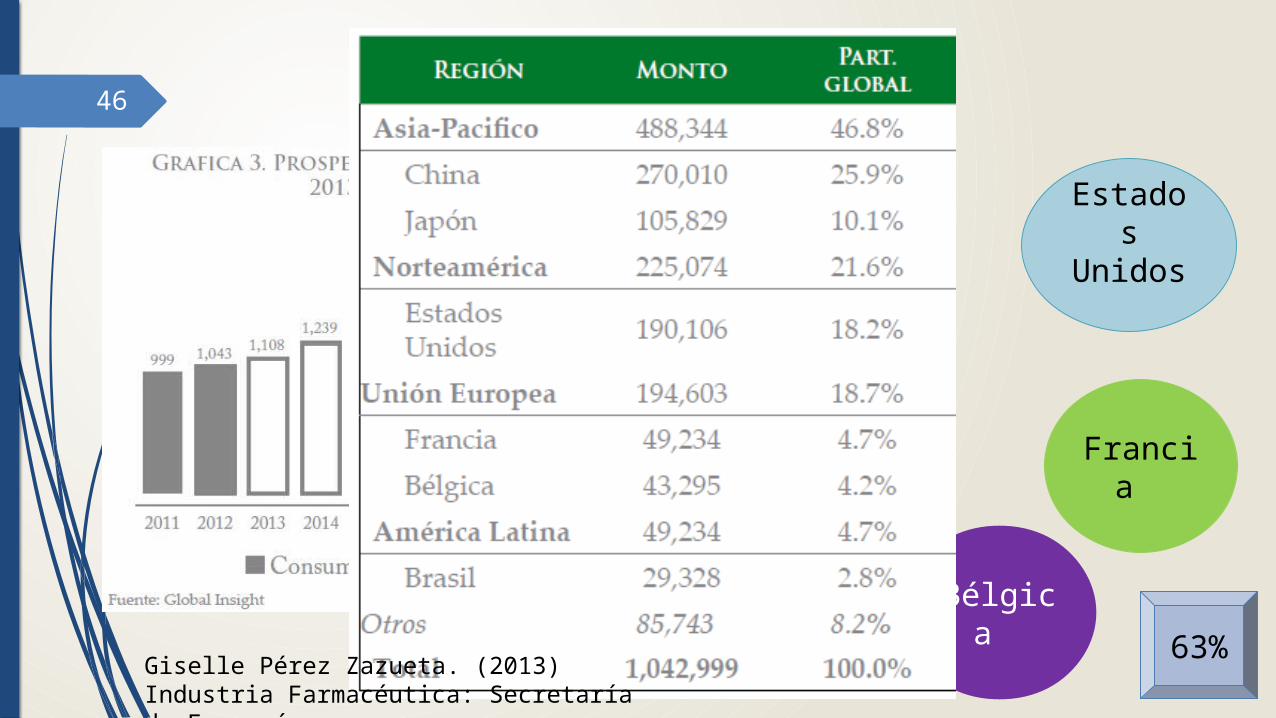
46 Consumo global
China
Bélgica
Japón Francia
Estados Unidos
63%Giselle Pérez Zazueta. (2013) Industria
Farmacéutica: Secretaría de Economía.
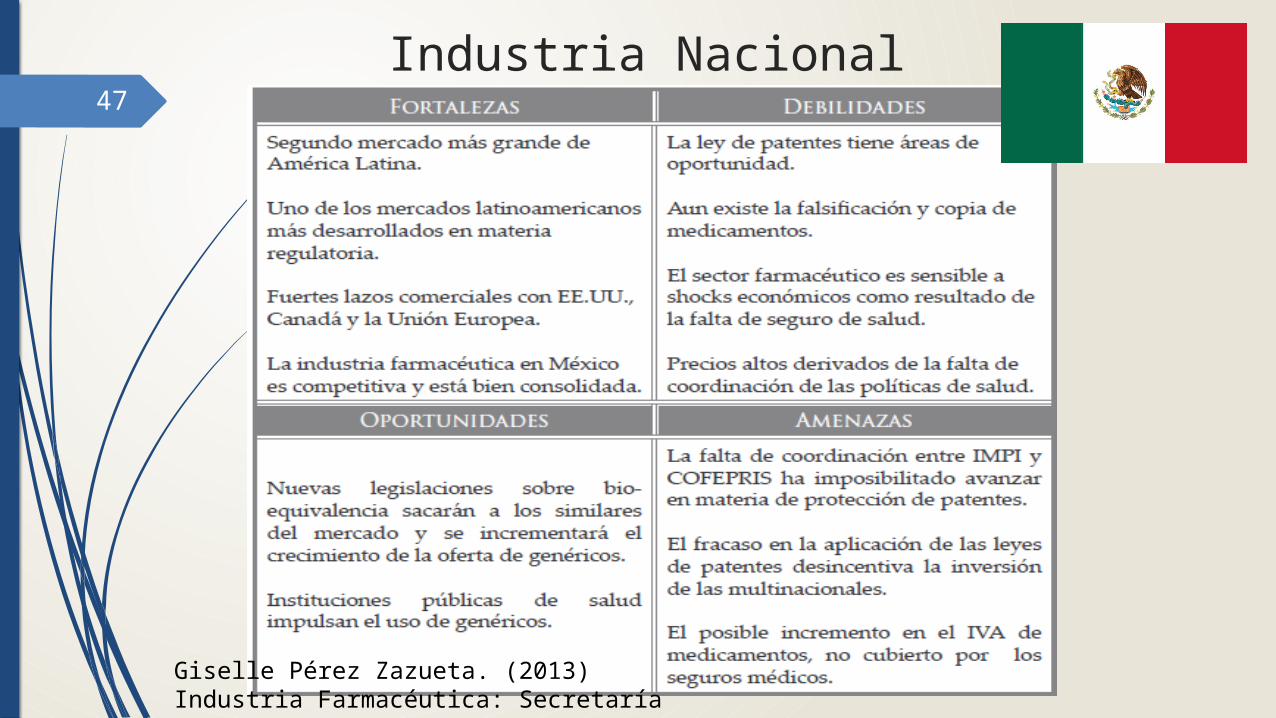
47
Industria Nacional
Giselle Pérez Zazueta. (2013) Industria Farmacéutica: Secretaría de Economía.
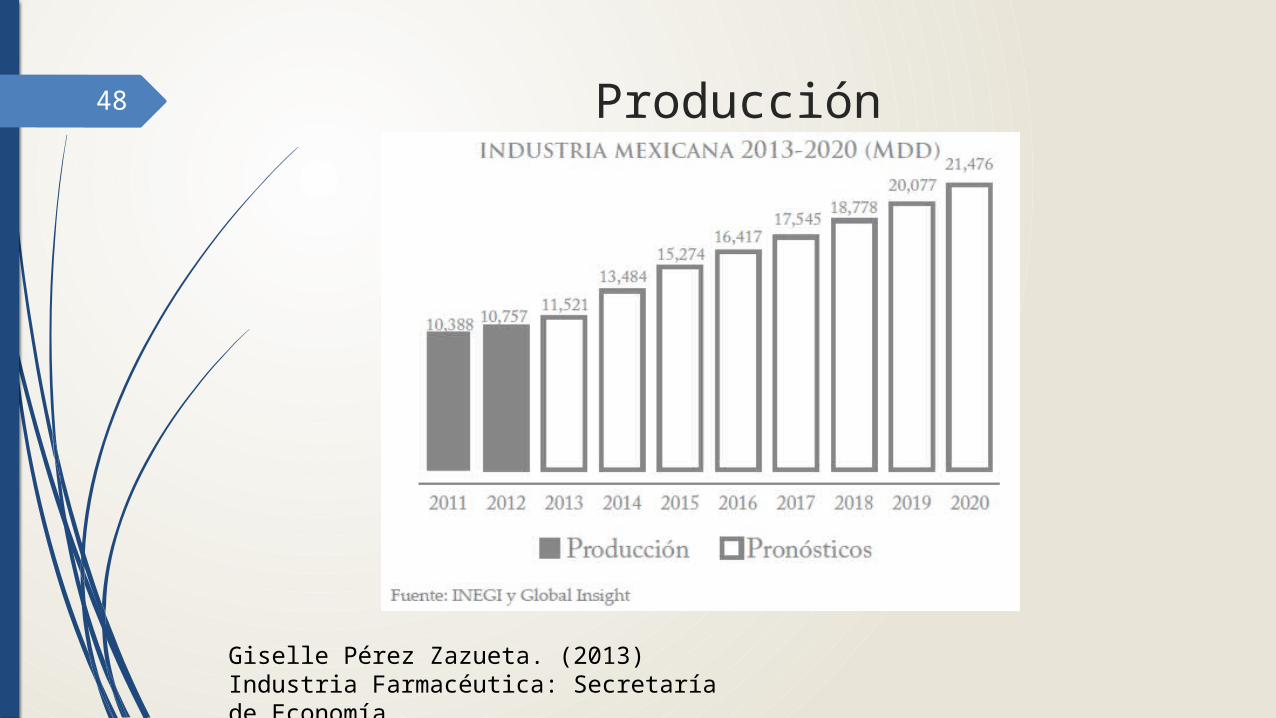
48 Producción
Giselle Pérez Zazueta. (2013) Industria Farmacéutica: Secretaría de Economía.
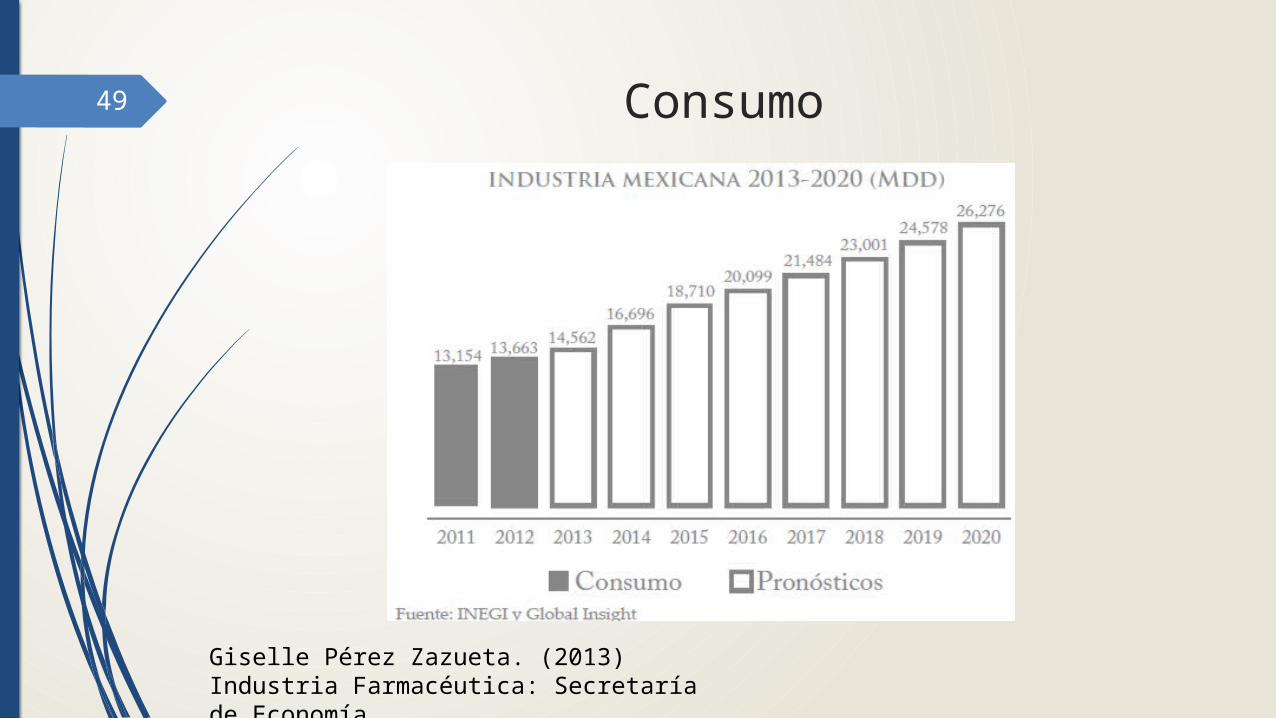
49 Consumo
Giselle Pérez Zazueta. (2013) Industria Farmacéutica: Secretaría de Economía.


















