
Inmunodeficiencias primarias - Dra. med. Carmen Zarate Hernández Asesora: Dra. Atenea Cáceres...
76
Dra. med. Carmen Zarate Hernández Asesora Dra. Atenea Cáceres Papadakis R3 Pediatría
-
Upload
juan-c-ivancevich -
Category
Health & Medicine
-
view
220 -
download
1
Transcript of Inmunodeficiencias primarias - Dra. med. Carmen Zarate Hernández Asesora: Dra. Atenea Cáceres...

Dra. med. Carmen Zarate Hernández Asesora Dra. Atenea Cáceres Papadakis R3 Pediatría

Se han identificado más de 250 genes, los cuales son los causantes de las diferentes formas de inmunodeficiencias primarias. Un problema importante que afecta los pacientes es el intervalo de tiempo entre el inicio de los síntomas y el diagnóstico. El diagnóstico temprano es vital para prevenir de manera significativa la morbi-mortalidad, especialmente desde que el reemplazo de inmunoglobulinas es eficaz en la inmunodeficiencia humoral.
Heather Lehman, Vivian Hernandez-Trujillo, Mark Ballow, Diagnosing primary immunodeficiency: a practical approach for the non-immunology Vol. 31, No. 4, 2015, 697–706
Dra. Cáceres CRAIC Mty

HISTORIA
Ogden Bruton (1952): asociaciones entre la ausencia de inmunoglobulinas y la susceptibilidad a las infecciones.
Charles Janeway (1953) y Robert Good (1956): agammaglobulinemia
Ortega Mc. Generalidades sobre inmunodeficiencias primarias. Universitaria Medica 2005; 46: 48.
Dra. Cáceres CRAIC Mty

HISTORIA
Cooper y colaboradores (1969): desarrollo independiente de los sistemas del timo y la bolsa de Fabricio responsables de la inmunidad celular y humoral.
70’s identificación de las células B y T como poblaciones separadas.
Diferenciación de los linfocitos B a células plasmáticas secretorias de inmunoglobulinas e hizo posible el delineamiento de los defectos funcionales de las células T y B.
Ortega Mc. Generalidades sobre inmunodeficiencias primarias. Universitaria Medica 2005; 46: 48.
Dra. Cáceres CRAIC Mty

1. Inmunodeficiencias combinadas 2. Otros síndromes de inmunodeficiencias
bien definidos 3. Deficiencias predominantemente de
anticuerpos 4. Enfermedades con desregulación
inmunológica 5. Defectos congénitos de número, función o
ambos del sistema fagocitario 6. Defectos en la inmunidad innata 7. Trastornos autoinflamatorios 8. Deficiencias del sistema de complemento. 9. Fenocopias de inmunodeficiencias
Ortega Mc. Generalidades sobre inmunodeficiencias primarias. Universitaria Medica 2005; 46: 48.
Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Señales de alerta para detección temprana:
1. Dos o más neumonías en el ultimo año. 2. Cuatro o más otitis en el ultimo año 3. Historia familiar de inmunodeficiencias 4. Estomatitis de repetición o candidiasis
por más de dos meses 5. Abscesos de repetición o ectima 6. Un episodio de infección grave
(meningitis, sepsis, osteoartritis). 7. Infecciones o parasitosis de repetición 8. Alergia respiratoria 9. Colagenosis o enfermedad autoinmune 10. Fenotipo clínico de inmunodeficiencia
Dra. Cáceres CRAIC Mty

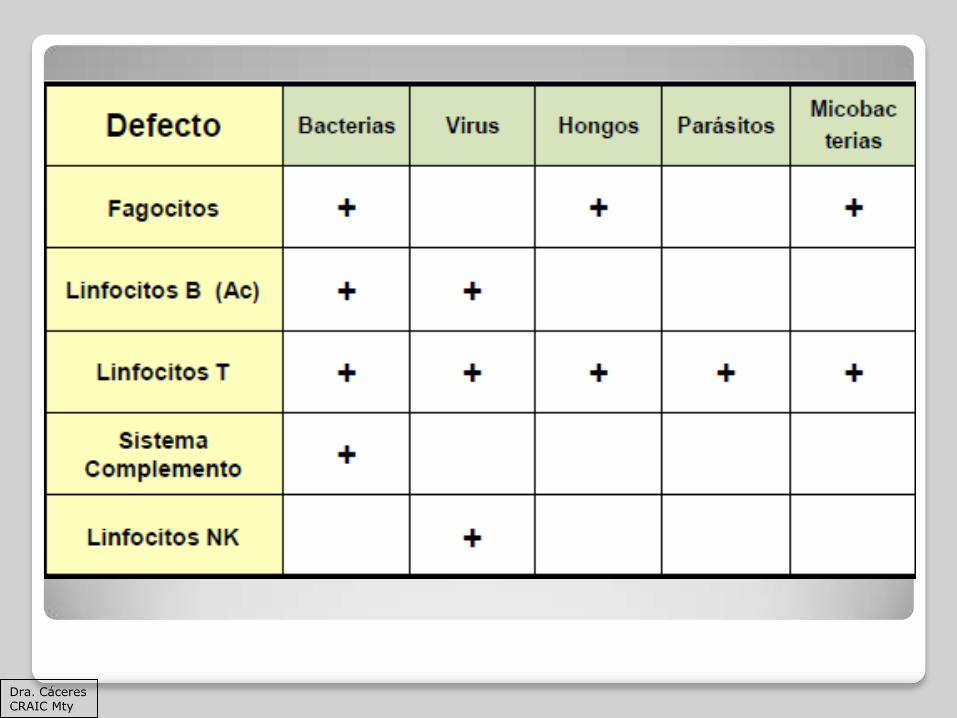
La infección por ciertos organismos puede orientar a considerar una inmunodeficiencia primaria
Aspergillus spp.
Candida spp
Mycobacterium avium
P jirovecci
Enterovirus (recurrente o grave)
Celular o combinada, defecto de fagocitos
Celular o combinada, defecto fagocitos, HIV
Deficiencia humoral o combinada
Dra. Cáceres CRAIC Mty
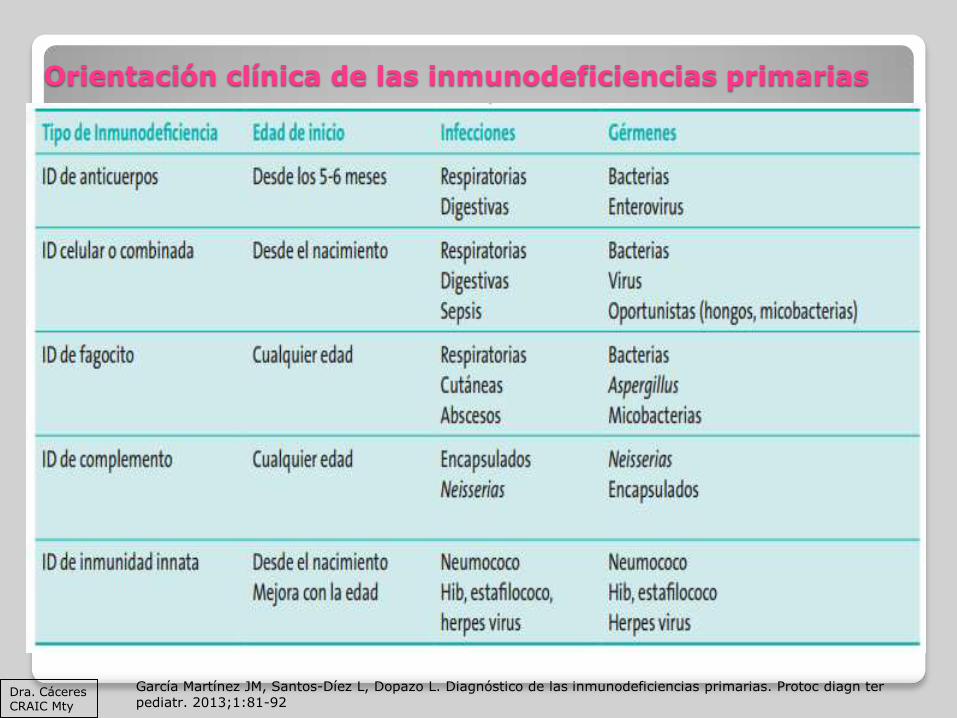
Orientación clínica de las inmunodeficiencias primarias
García Martínez JM, Santos-Díez L, Dopazo L. Diagnóstico de las inmunodeficiencias primarias. Protoc diagn ter pediatr. 2013;1:81-92
Dra. Cáceres CRAIC Mty
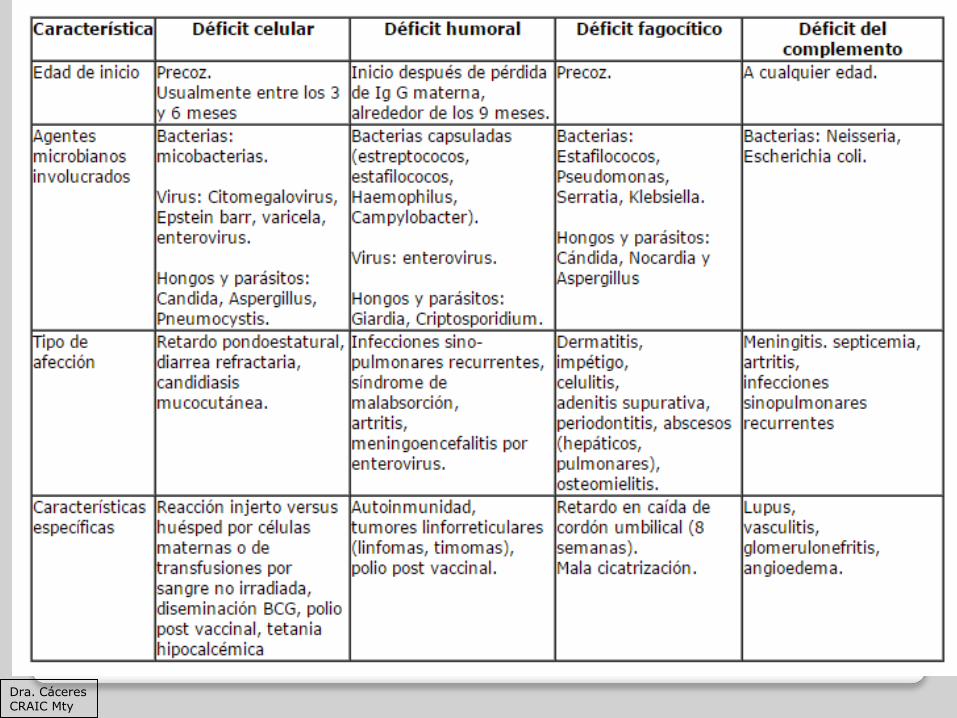
Dra. Cáceres CRAIC Mty

Casanova JL, Fieschi C, Zhang SY, Abel L. Revisiting human primary immunodeficiencies. J Intern Med 2008;264:115-27.
Dra. Cáceres CRAIC Mty

Valoración de Inmunoglobulinas
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Son las más frecuentes
Se manifiestan con infecciones bacterianas recurrentes desde los 4 a 6 meses de vida en paralelo a la declinación fisiológica de la IgG de la madre.
La historia es el aspecto más importante de la evaluación clínica
Ortega Mc. Generalidades sobre inmunodeficiencias primarias. Universitaria Medica 2005; 46: 48.
Dra. Cáceres CRAIC Mty

Algunas inmunodeficiencias humorales específicas presentan susceptibilidad a gérmenes específicos: ◦ Agamamaglulinemia de Bruton se asocia a
meningoencefalitis por enterovirus ◦ Híper Ig M asociada a X se asocia a infección por
Pseudomonas jeroveci y Cryptosporidium ◦ Síndrome linfoproliferativos asociado a X a
infecciones por virus Epstein Bar.
Ortega Mc. Generalidades sobre inmunodeficiencias primarias. Universitaria Medica 2005; 46: 48. Dra. Cáceres CRAIC Mty

Enfermedad de Bruton agammaglobulinemia ligada al cromosoma X
Mutación en el gen que codifica a la tirosina cinasa de Bruton.
Afecta la diferenciación temprana de linfocitos B.
Descrita en 1952 por el Dr. Ogden C Bruton, fue la primer inmunodeficiencia primaria descrita en la que se conoció la inmunopatogenia.
hun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J 2008;49(1):28-36
Dra. Cáceres CRAIC Mty

Antes del primer año de vida, el cuadro clínico se distingue por infecciones bacterias recurrentes en las vías aéreas altas y bajas, disminución marcada o ausencia de inmunoglobulinas séricas y linfocitos B ausentes en sangre periférica.
El diagnóstico temprano repercute en la calidad de vida a largo plazo de estos pacientes.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated
data from pediatric patients at Severance Hospital. Yonsei Med J 2008;49(1):28-36
Dra. Cáceres CRAIC Mty

FISIOPATOGENIA
La proteína que resulta afectada es la tirosina cinasa de Bruton que funciona como un transductor de señal del medio intracitoplasmático al medio intranuclear, la cual esta íntimamente relacionada con las vías de transmisión de señales que regulan la proliferación y diferenciación del linfocitos V.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric
patients at Severance Hospital. Yonsei Med J 2008;49(1):28-36
Dra. Cáceres CRAIC Mty

FISIOPATOGENIA
Las mutaciones presentes condicionan a un
defecto en la diferenciación de linfocito pro B a linfocito pre-B, lo que afecta las formas celulares maduras, debido a que la producción de linfocitos B periféricos e inmunoglobulinas se ausenta o disminuye de manera importante.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J 2008;49(1):28-36
Dra. Cáceres CRAIC Mty

Lo que resulta…
Bloqueo de la diferenciación de la célula pro B a pre B en médula ósea
˂1% linfocitos B circulantes
Notarangelo, L. Primary immunodeficiencies. Allergy Clin Immunol 2010;125:S182-94.
Dra. Cáceres CRAIC Mty

MANIFESTACIONES CLÍNICAS
Infecciones bacterianas recurrentes a los seis meses de edad ya que a dicha edad ocurre una disminución fisiológica de las inmunoglobulinas maternas.
La mayoría cursa con sinutis o neumonías de repetición.
Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations
of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J 2008;49(1):28-36
Dra. Cáceres CRAIC Mty

TX:
GAMMAGLOBUINA INTRAVENOSA
DX:
INMUNOGLOBULINAS POR DEBAJO DE DOS
DESVIACIONES ESTÁNDAR
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

INMUNODEFICIENCIA CELULAR Son menos comunes que los defectos humorales. Infecciones oportunistas en edad temprana, diarrea crónica, candidiasis y falla de medro. Si hay una fuerte sospecha, evitar vacunas de virus vivos. La evaluación inicial incluirá citometría de flujo para deficiencia cuantitativa de células T, B y natural killer.
Heather Lehman, Vivian Hernandez-Trujillo, Mark Ballow, Diagnosing primary immunodeficiency: a practical approach for the non-immunology Vol. 31, No. 4, 2015, 697–706
Dra. Cáceres CRAIC Mty

La edad de inicio es precoz antes de los 6 meses de vida siendo los patógenos predominantes micobacterias, virus, hongos y gérmenes oportunistas.
Los niños afectados tienen infecciones comunes inusuales y graves, típicamente es un lactante o niño que no sobrevive sin un tratamiento médico temprano.
Dra. Cáceres CRAIC Mty

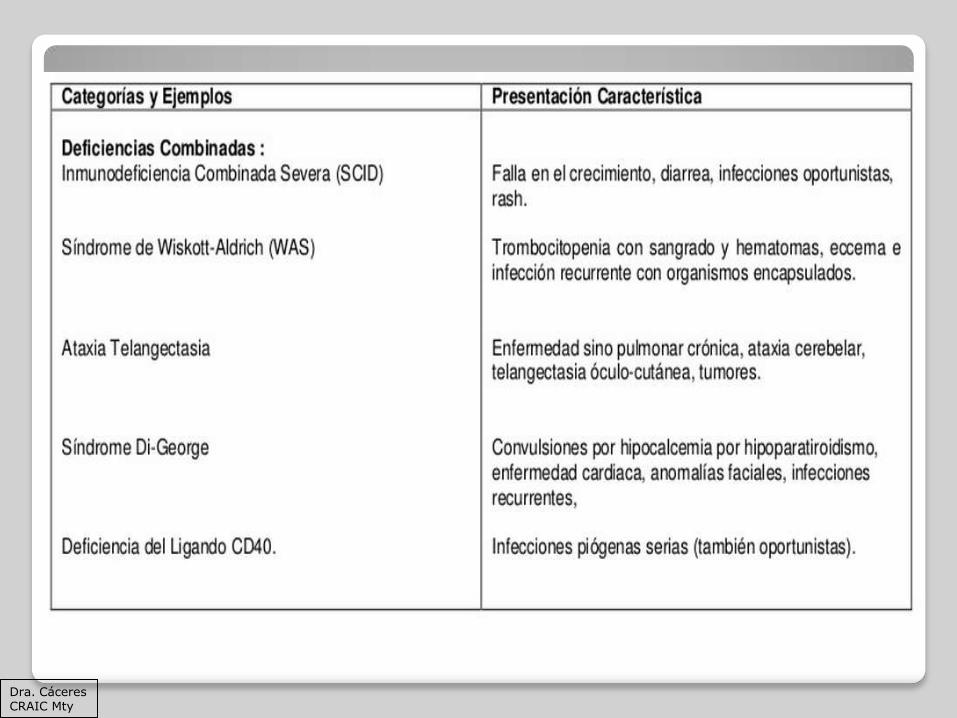
La mayoría puede presentarse como:
Dismorfia facial (Sx de Di George) o displasia ectodérmica
Apariencia grave
Falla en el crecimiento
Cardiopatía congénita
Dermatosis: eritema del pañal, eccema, rash como en el síndrome de Omenn, telangectasias, ausencia de uñas, cabello o sudor (displasia ectodérmica)
Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty
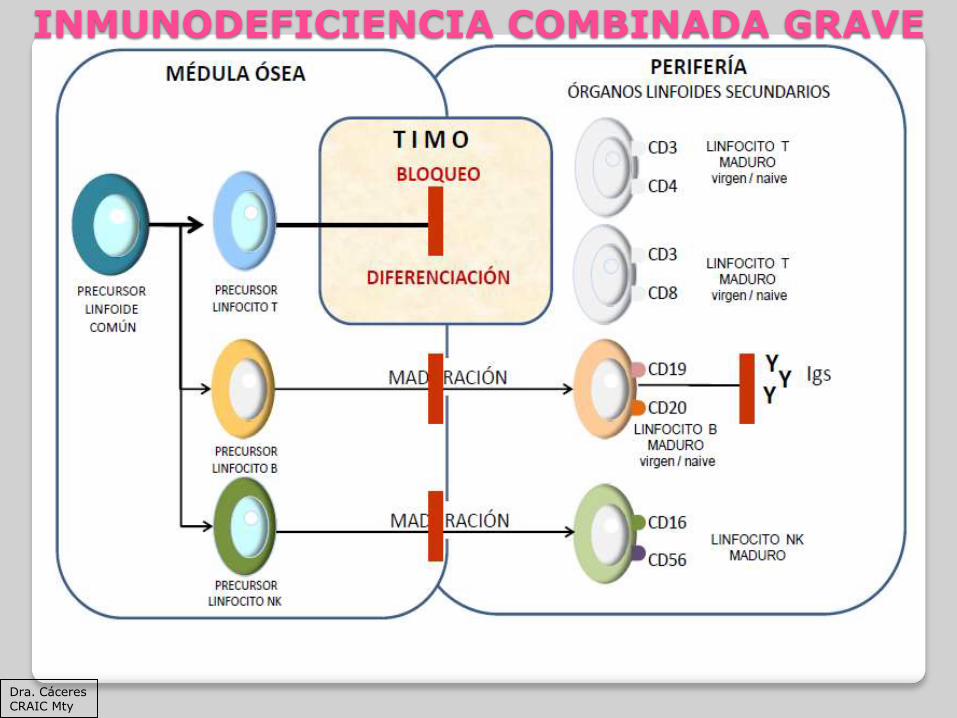
INMUNODEFICIENCIA COMBINADA GRAVE
Dra. Cáceres CRAIC Mty

Síndrome de WisKott Aldrich
Ligada al cromosoma X
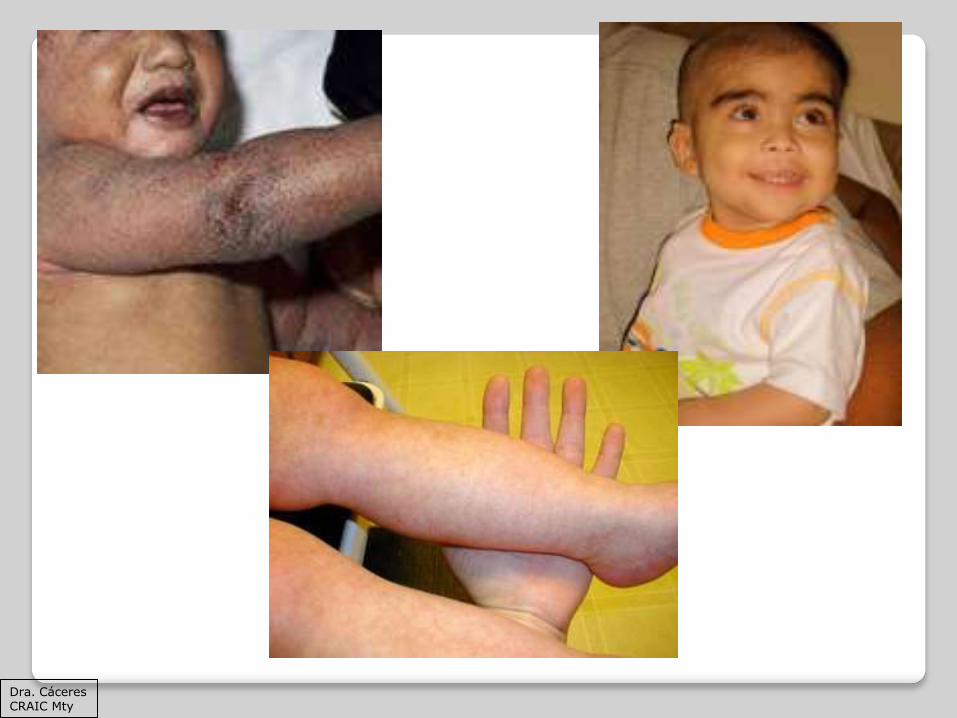
Se distingue por microtrombocitopenia congénita, eccema e infecciones recurrentes, así como por un mayor riesgo de linfomas y autoinmunidad.
Incidencia mundial: es de 1 a 10 en un millón de recién nacidos vivos
Expectativa de vida es de 15 años sin trasplante de medula ósea.
Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich syndrome: immunodeficiency resulting from
defective cell migration and impaired immunostimulatory activation. Immunobiology 2009;214(9-10):778-790.
Pai SY, DeMartiis D, Forino C, Cavagnini S, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant 2006;38(10):671-679.
Dra. Cáceres CRAIC Mty

Síndrome de WisKott Aldrich
El gen WASP participa en la proliferación de la actina.
Ausencia de microfilamentos en las plaquetas y son rápidamente destruidas por el bazo.
Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich syndrome: immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology 2009;214(9-10):778-790.
Pai SY, DeMartiis D, Forino C, Cavagnini S, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant 2006;38(10):671-679. Dra. Cáceres
CRAIC Mty

Las diferentes mutaciones en el gen WASP producen espectros
clínicos diferentes
Se han descrito el síndrome de WisKott Aldrich clásico, la trombocitopenia intermitente o ligada al cromosoma X y la neutropenia ligada al cromosoma X
La triada clásica del síndrome de WisKott Aldrich es microtrombocitoenia, eccema e infecciones recurrentes.
Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia RM.
Recent advances in understanding the pathophysiology of
Wiskott-Aldrich syndrome. Blood 2009;113(25):6288-6295.
Dra. Cáceres CRAIC Mty

La cantidad de plaquetas puede variar pero suele estar por debajo del nivel normal este dato es la piedra angular para diagnosticar síndrome de WisKott Aldrich clásico y trombocitopenia ligada al cromosoma X.
Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia RM. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood 2009;113(25):6288-6295.
CLÍNICA
Petequias Equimosis STDB Epistaxis Trombocitopenia
Dra. Cáceres CRAIC Mty

El eccema es una manifestación característica durante la lactancia y la infancia
En la mayoría de los casos se manifiesta en forma grave y resistente al tratamiento
Las enfermedades autoinmunes: anemia hemolítica, púrpura de Henoch Schönlein y neutropenia.
Los tumores malignos hematológicos.
Fallecen por hemorragia infecciones graves o enfermedad maligna.
Román Jiménez MG, Yamazaki Kakashimada Ma, Blanca Galicia L. Síndrome de WisKott Aldrich. Rev Alerg Mexico. 2010.57:171-174.
Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty
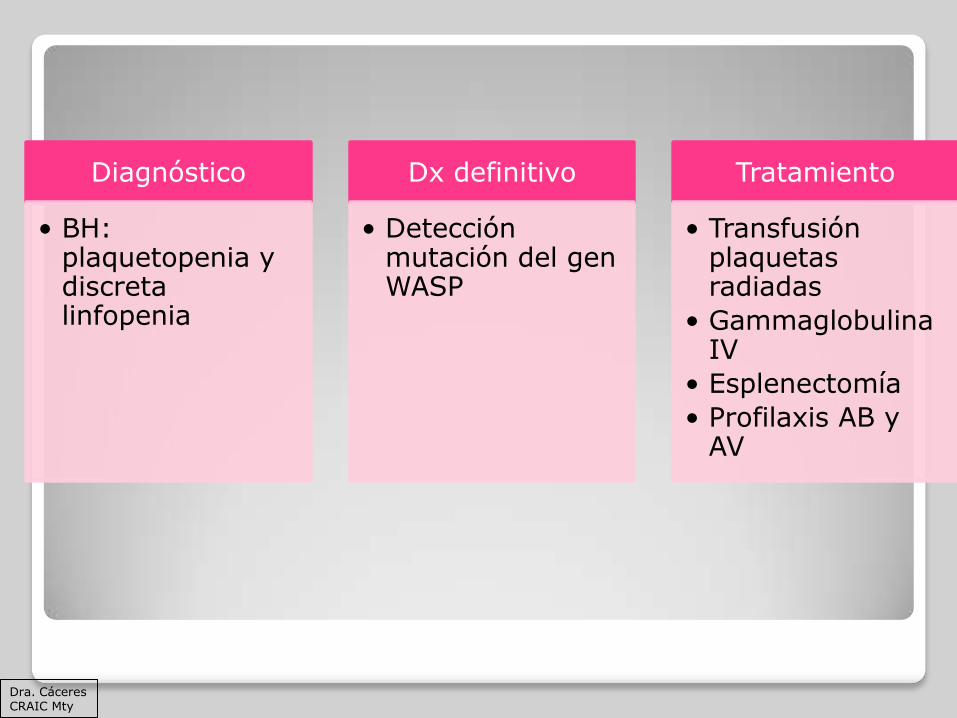
Diagnóstico
• BH: plaquetopenia y discreta linfopenia
Dx definitivo
• Detección mutación del gen WASP
Tratamiento
• Transfusión plaquetas radiadas
• Gammaglobulina IV
• Esplenectomía
• Profilaxis AB y AV
Dra. Cáceres CRAIC Mty

Síndrome de Di George
Síndrome congénito caracterizado por un espectro de malformaciones que incluye la ausencia del timo y las glándulas paratiroides, lo que da lugar a inmunodeficiencia de células T e hipocalcemia.
García E, Camacho J, Gómez MJ, Del Castillo E, Martínez MJ, López JP. Transient congenital hypoparathyroism and 22q11 deletion. J Pediatr
Endocrinol Metab 2000; 13: 659-61.
Dra. Cáceres CRAIC Mty

Síndrome de Di George
60s el Dr. Angelo Di George describió a pacientes con hipoparatiroidismo, timo hiperplásico o ausencia de timo y defectos cardiacos conotroncales junto a labio y/o paladar hendido.
70s el Dr. Robert Shprintzen, síndrome velocardiofacial.
García E, Camacho J, Gómez MJ, Del Castillo E, Martínez MJ, López JP. Transient congenital hypoparathyroism and 22q11 deletion. J
Pediatr Endocrinol Metab 2000; 13: 659-61.
Dra. Cáceres CRAIC Mty

Síndrome de Di George
En la década de los 80´s, se determinó que más del 90 por ciento de todos los pacientes que presentaban las características de los síndromes de Di George y Shprintzen (síndrome velocardiofacial) tenían una deleción cromosómica en la región 22q11.
García E, Camacho J, Gómez MJ, Del Castillo E, Martínez MJ, López JP. Transient congenital hypoparathyroism and 22q11 deletion. J Pediatr Endocrinol
Metab 2000; 13: 659-61.
Dra. Cáceres CRAIC Mty

El 90 % de los pacientes carecen de una pequeña parte del cromosoma 22 en la región q11.
Que abarca cerca de 30 genes individuales y es responsable de defectos del desarrollo en estructuras específicas de todo el cuerpo.
Weinzimer SA. Endocrine aspects of the 22q11.2 deletion syndrome. Genet-Med 2001; 3: 19-22.
Dra. Cáceres CRAIC Mty

Se estima que la ausencia de 22q11 se produce en uno de cada 3000 a 4000 recién nacidos vivos.
La mayoría son episodios esporádicos
10% hereditario, por un gen autosómico dominante.
Weinzimer SA. Endocrine aspects of the 22q11.2 deletion syndrome. Genet-Med
2001; 3: 19-22.
Dra. Cáceres CRAIC Mty

Clínica
IMPLANTACIÓN BAJA OREJAS
FILTRUM
CORTO
TELECANTO
CON FISURAS
PALPEBRALES CORTAS
HIPOCALCEMIA DEFECTOS
TRACTO SALIDA CORAZÓN
DÉFICIT INMUNOLÓGICO
(HIPOPLASIA TIMO)
Dra. Cáceres CRAIC Mty

Clínica
ANOMALÍAS CARDIACAS
PALADAR HENDIDO TALLA BAJA
DEDOS LARGOS E
HIPEREXTENDIBLES
NARIZ PROMINENTE
ALAS NASALES HIPOPLÁSICAS
MICROGNATIA MICROCEFALIA ANOMALÍAS OCULARES
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Clínica
El retardo mental, CI <70 presente en un 50% de los casos
La importancia de una correcta fenotipificación de los afectados con la ausencia de 22q11 radica en el pronóstico resultante.
Markert ML, Devlin BH, Alexieff MJ, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus
transplantation: outcome of 44 consecutive transplants. Blood . May 15 2007;109(10):4539-47
Dra. Cáceres CRAIC Mty
La sobrevida puede estar gravemente comprometida por la hipocalcemia de difícil corrección y un compromiso inmunológico importante que se complica con infecciones graves; mientras que en el síndrome velocardiofacial el cuadro clínico puede ser menos grave. Markert ML, Devlin BH, Alexieff MJ, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood . May 15 2007;109(10):4539-47
Dra. Cáceres CRAIC Mty

El diagnóstico confirmatorio se realiza mediante técnica FISH siendo el tratamiento multidisciplinario.
Markert ML, Devlin BH, Alexieff MJ, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood . May 15 2007;109(10):4539-47
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Las manifestaciones se inician a edad temprana.
Los signos de defecto de los fagocitos se manifiestan en diversas regiones:
PIEL
• ABSCESOS
• EGC
NÓDULOS LINFÁTICOS TUMEFACTOS
EN LA ENFERMEDAD GRANULOMA-
TOSA
DEFECTOS
DE ADHERENCIA
•RETRASO EN EL DESPRENDI-MIENTO DEL CORDÓN UMBILICAL
INFECCIONES PULMONARES
•ASPERGILO
•ABSCESOS
FIEBRE SIN CAUSA
EXPLICABLE
GINGIVITIS
ÚLCERAS ALBINISMO
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Enfermedad granulomatosa crónica
1954 por Janeway y se denominó "enfermedad granulomatosa fatal de la infancia".
Incidencia mundial de 1 por cada 250 000 recién nacidos vivos.
◦ Álvarez-Cardona A, Yamazaki-Nakashimada MA, Espinosa-Padilla SE. Enfermedad
granulomatosa crónica. Alergia Rev Mex. 2009;56(5):165-74.
Dra. Cáceres CRAIC Mty

•Más del 60 % de los enfermos presenta una herencia recesiva ligada al cromosoma X; entre el 30 y el 40 % la heredan de forma autosómica recesiva; y en el 10 % de novo.
Dra. Cáceres CRAIC Mty

Enfermedad granulomatosa crónica
Mutaciones en cualquiera de los genes que codifican para las subunidades que conforman la NADPH oxidasa, la glicoproteína gp91phox y las proteínas p22phox, p47phox, p67phox, p40phox y p21rac.
La deficiencia de la NADPH oxidasa impide la formación de los radicales libres de oxígeno en el fagocito activado, la muerte de los microorganismos fagocitados y la fragmentación del material ingerido.
◦ Álvarez-Cardona A, Yamazaki-Nakashimada MA, Espinosa-Padilla SE. Enfermedad granulomatosa crónica.
Alergia Rev Mex. 2009;56(5):165-74. Dra. Cáceres CRAIC Mty

Susceptibles a infecciones recurrentes graves causadas por bacterias, hongos y formación de granulomas en cualquier parte del organismo.
Fundamentalmente por: Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia, micobacterias y especies de Aspergillus.
Wintergerst U, Rosenzweig SD, Abinun M, Malech HL. Phagocytes defects. In: Reza N, Notarangelo LD, Aghamohammadi A, ed. Primary immunodeficiency diseases. Berlin: Springer Berlin Heidelberg; 2008. p.143-56.
Álvarez-Cardona A, Yamazaki-Nakashimada MA, Espinosa-Padilla SE. Enfermedad granulomatosa crónica. Alergia Rev Mex. 2009;56(5):165-74.
Ramírez-Vargas NG, Berrón-Ruiz LR, Berrón-Pérez R, Blancas-Galicia L. Diagnóstico de enfermedad granulomatosa crónica; pacientes y portadoras. Rev Alergia Mex. 2011;58(2):120-5
Dra. Cáceres CRAIC Mty
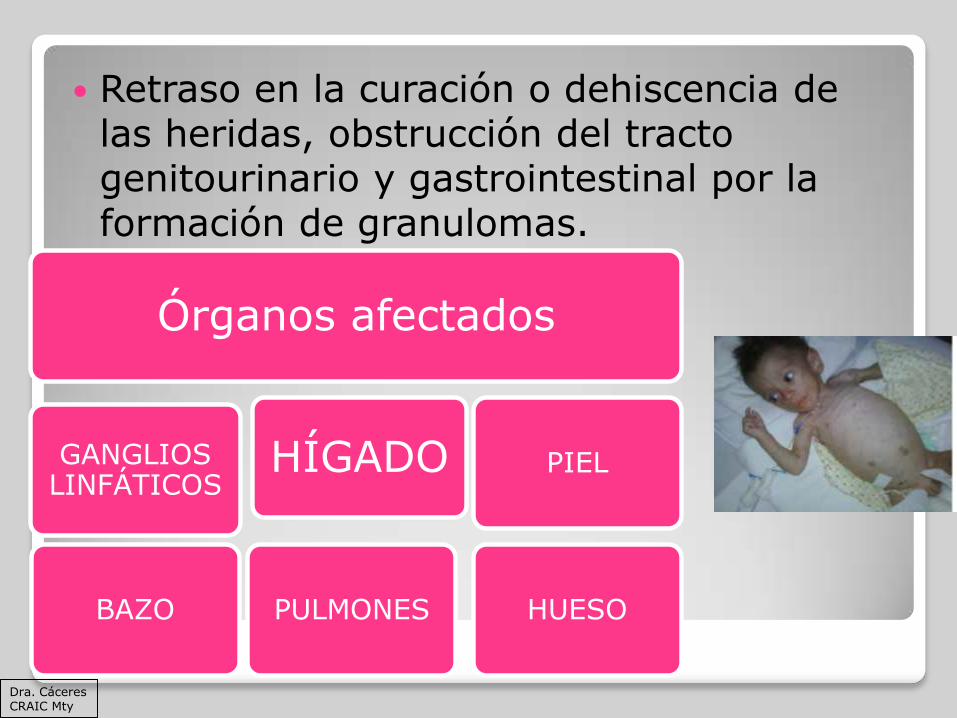
Retraso en la curación o dehiscencia de las heridas, obstrucción del tracto genitourinario y gastrointestinal por la formación de granulomas.
Órganos afectados
GANGLIOS LINFÁTICOS
BAZO PULMONES
PIEL
HUESO
HÍGADO
Dra. Cáceres CRAIC Mty

La vacunación con BCG, puede provocar una reacción inflamatoria localizada o diseminada.
Manifestaciones autoinmunes, lupus eritematoso sistémico o discoide, púrpura trombocitopénica idiopática, sarcoidosis, artritis idiopática juvenil, síndrome antifosfolipídico, pericarditis idiopática recurrente y enfermedad inflamatoria intestinal.
El diagnóstico clínico puede confirmarse con la prueba de nitroazul de tetrazolium (NBT).
Wintergerst U, Rosenzweig SD, Abinun M, Malech HL. Phagocytes defects. In: Reza N, Notarangelo LD, Aghamohammadi A, ed. Primary immunodeficiency diseases. Berlin: Springer Berlin Heidelberg; 2008. p.143-56.
Álvarez-Cardona A, Yamazaki-Nakashimada MA, Espinosa-Padilla SE. Enfermedad granulomatosa crónica. Alergia Rev Mex. 2009;56(5):165-74.
Ramírez-Vargas NG, Berrón-Ruiz LR, Berrón-Pérez R, Blancas-Galicia L. Diagnóstico de enfermedad granulomatosa crónica; pacientes y portadoras. Rev Alergia Mex. 2011;58(2):120-5 Dra. Cáceres
CRAIC Mty

El diagnóstico definitivo o molecular se realiza
con la secuenciación directa de los genes de las subunidades del complejo NADPH oxidasa, en los cuales se detectan eliminaciones, inserciones o mutaciones puntuales.
El defecto molecular identificado resulta en la ausencia de la expresión de las proteínas del citocromo b558.
Wintergerst U, Rosenzweig SD, Abinun M, Malech HL. Phagocytes defects. In: Reza N, Notarangelo LD, Aghamohammadi A,
ed. Primary immunodeficiency diseases. Berlin: Springer Berlin Heidelberg; 2008. p.143-56.
Álvarez-Cardona A, Yamazaki-Nakashimada MA, Espinosa-Padilla SE. Enfermedad granulomatosa crónica. Alergia Rev Mex. 2009;56(5):165-74.
Ramírez-Vargas NG, Berrón-Ruiz LR, Berrón-Pérez R, Blancas-Galicia L. Diagnóstico de enfermedad granulomatosa crónica; pacientes y portadoras. Rev Alergia Mex. 2011;58(2):120-5
Dra. Cáceres CRAIC Mty

El tratamiento consiste en trasplante de células progenitoras hematopoyéticas
Ramírez-Vargas NG, Berrón-Ruiz LR, Berrón-Pérez R, Blancas-Galicia L. Diagnóstico de enfermedad
granulomatosa crónica; pacientes y portadoras. Rev Alergia Mex. 2011;58(2):120-5
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Se deben evaluar los componentes del complemento en
pacientes con episodios de bacteriemia, meningitis o infecciones sistémicas por Neisseria.
La deficiencia de C1, C4 o C2 pueden presentarse con enfermedad neumocócica recurrente. En algunos casos puede estar asociada a deficiencias de anticuerpos
La deficiencia de C3 es muy rara pero es caracterizada por infecciones bacterianas recurrentes graves como neumonía o bacteremia y desarrollo de glomerulonefritis.
Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford. 1999. pp: 448-458
Dra. Cáceres CRAIC Mty

Las infecciones sistémicas por Neisseria en niños y adolescentes sugieren deficiencias por C5-C7
Autosómica recesiva con excepción de la deficiencia de C1 inhibidor que lo hace de forma autosómica dominante y del defecto de properdina que se transmite de forma recesiva asociada al cromosoma X.
Tienen una mayor susceptibilidad a infecciones,
enfermedades reumáticas, angiodema o pueden permanecer asintomáticos.
Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford. 1999. pp: 448-458 Dra. Cáceres
CRAIC Mty

Las deficiencias de los primeros componentes se asocian frecuentemente con enfermedades autoinmunes y con un incremento en la frecuencia de infecciones bacterianas ya que el aclaramiento de inmunocomplejos y la opsonización se asocian frecuentemente con estos componentes.
Paul ME, Shearer WT. The child who has recurrent infection. Immunol Allergy Clin North Am. 1999; 12 (2):423-436
Dra. Cáceres CRAIC Mty

El diagnóstico de la deficiencia de la vía alterna se realiza mediante ensayos hemolíticos con ensayos funcionales de la vía alterna que utilizan eritrocitos de conejo o cobaya que son fuertes activadores de esta vía en el ser humano.
Dra. Cáceres CRAIC Mty

La evaluación de un defecto de la vía clásica se realiza con el análisis de la capacidad hemolítica del complejo CH-50.
No existe un tratamiento específico, el tratamiento se enfoca a medidas profilácticas y al tratamiento de las infecciones para mejorar la calidad de vida.
Paul ME, Shearer WT. The child who has recurrent infection. Immunol Allergy Clin North Am. 1999; 12 (2):423-436
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

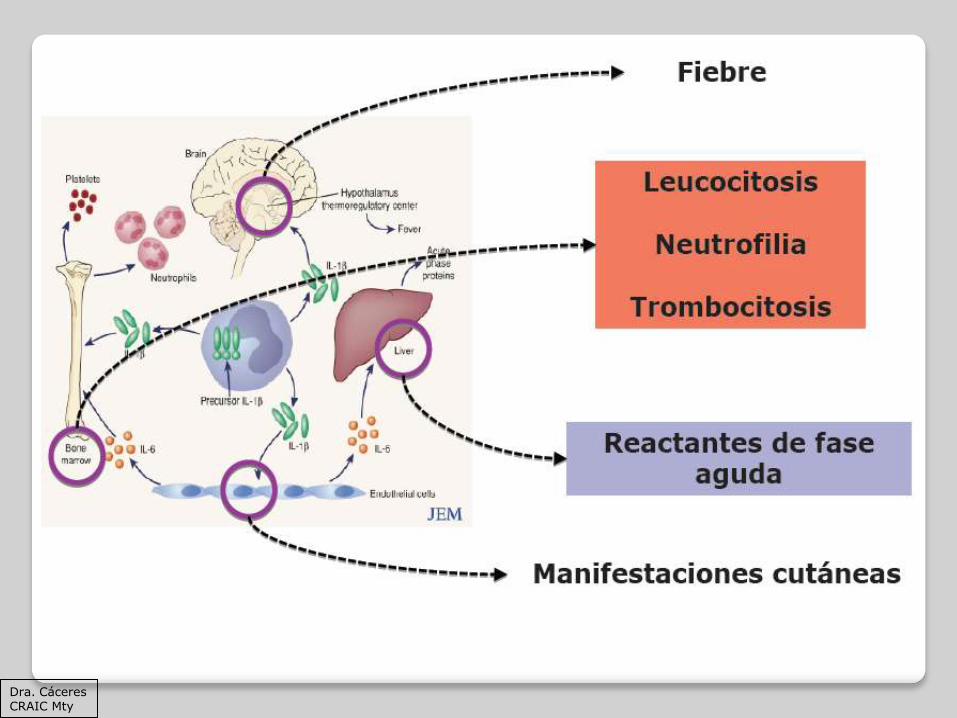
Inflamación
Fiebre
Palidez/ictericia
Petequias
Hepatomegalia
Esplenomegalia
Citopenias
Coagulopatías
Varón/mujer
Infecciones:
BACTERIANAS
Y VIRALES
Manifestaciones no
inmunológicas
Dismorfias
Dentarias
Neurológicas
Cutáneas
Óseas
Cardiacas
Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty

Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty
Dra. Cáceres CRAIC Mty

Tratamiento
GGIV TMO Tx
Génica
Humorales +++ + / - ¿ ?
Combinadas ++ +++ +++
Fagocitarias - ++ +++
Asociadas + / - + / - + / -
Complemento - + / - ¿ ?
Dra. Cáceres CRAIC Mty

• Las inmunodeficiencias están producidas por defectos congénitos o adquiridos de los linfocitos, macrófagos y otros mediadores inmunológicos.
• Las enfermedades se asocian a una predisposición a padecer infecciones.
• Las inmunodeficiencias combinadas graves incluyen defectos de la maduración de los linfocitos T y B.
• El tratamiento de las inmunodeficiencias congénitas supone transfusiones de anticuerpos, trasplantes de medula ósea y células progenitoras.
Dra. Cáceres CRAIC Mty


















