Malformaciones del sistema digestivo
41
MALFORMACIONES DEL SISTEMA DIGESTIVO Cervera Varía Cristhian La Torre Gálvez Cristian Gómez Echeandia Sofía Panta Quezada Kristell Velásquez Montenegro Ángela
-
Upload
kristell-panta-quezada -
Category
Documents
-
view
7 -
download
2
description
formacion embrionaria del sistema digestivo malformaciones
Transcript of Malformaciones del sistema digestivo

MALFORMACIONES DEL SISTEMA DIGESTIVO
Cervera Varía CristhianLa Torre Gálvez CristianGómez Echeandia SofíaPanta Quezada Kristell
Velásquez Montenegro Ángela

Malformaciones del aparato digestivo
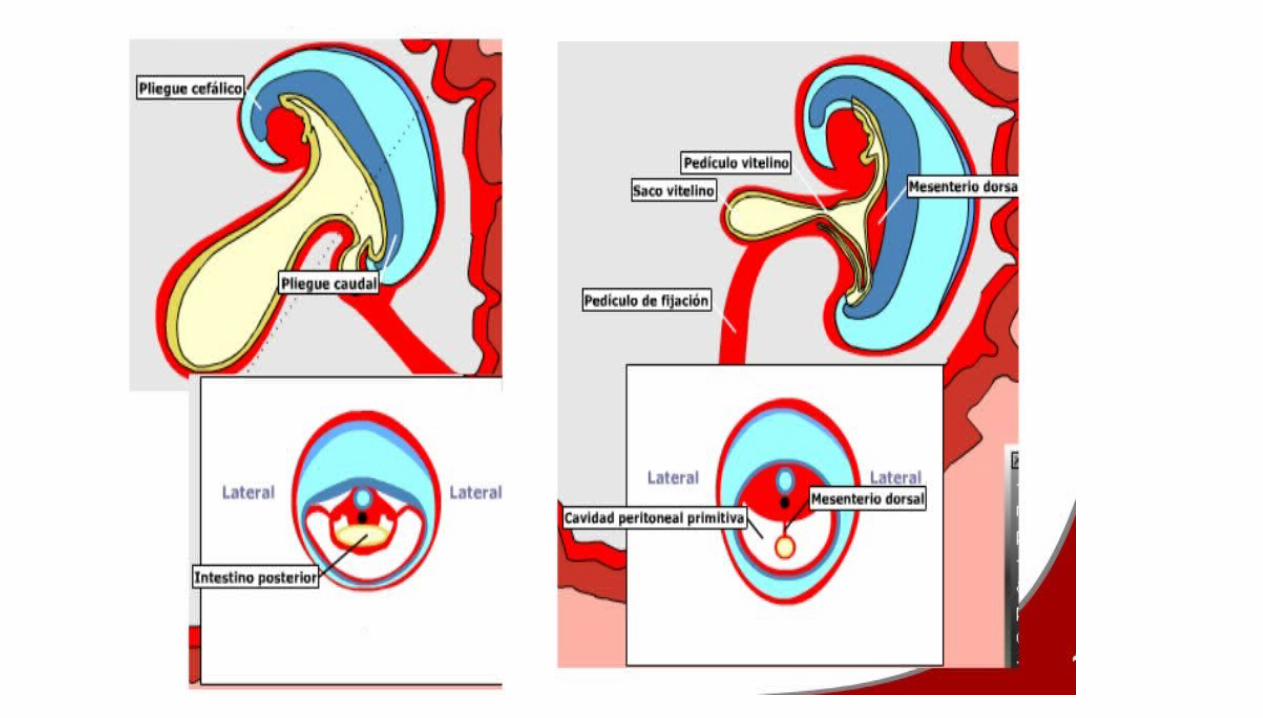
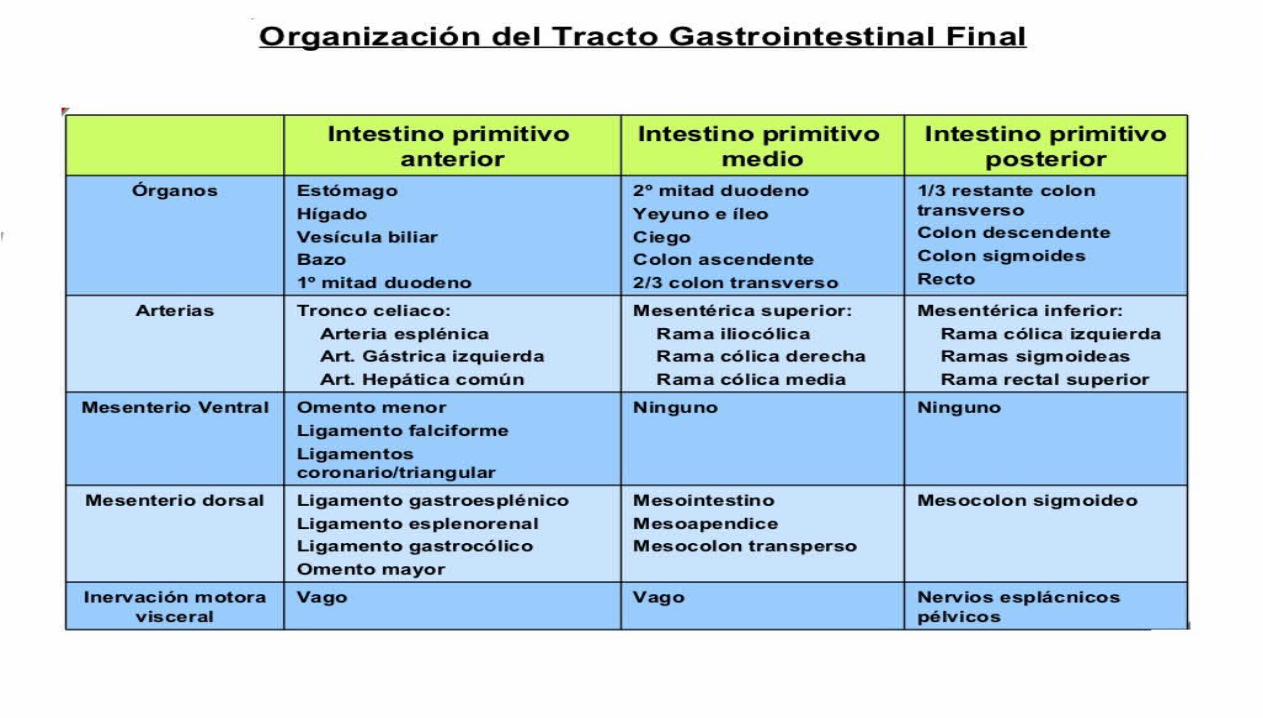
Desarrollo del tubo intestinal: El plegamiento cefalocaudal y lateral del embrión hace una porción de la cavidad del saco vitelino revestida de endodermo se incorpore al embrión para formar el intestino primitivo. Otras dos porciones de la cavidad revestida de endodermo, el saco vitelino y el alantoides permanecen fuera del embrión.En las partes cefálicas y caudal del embrión, el intestino primitivo forma un tubo con el extremo ciego, el intestino anterior y el intestino posterior, respectivamente. La parte central, el intestino medio, queda temporalmente conectada al saco vitelino a través del conducto vitelino o pedículo del saco vitelino.

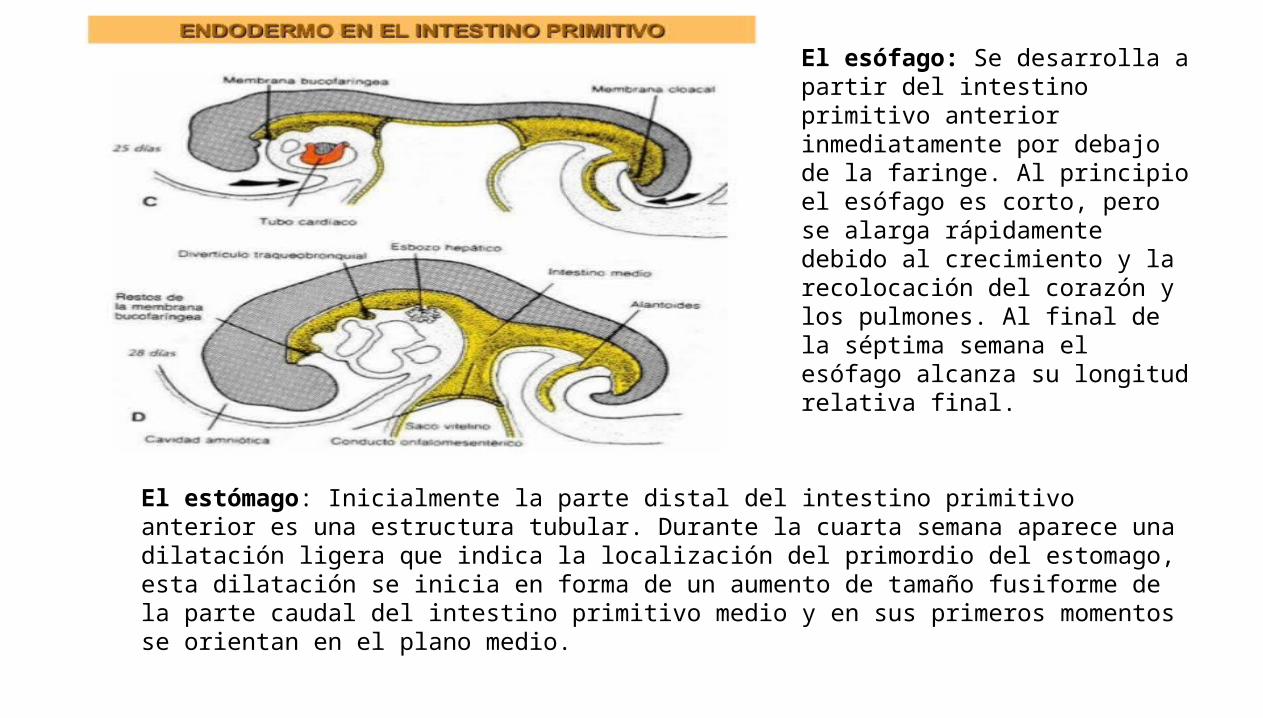
El esófago: Se desarrolla a partir del intestino primitivo anterior inmediatamente por debajo de la faringe. Al principio el esófago es corto, pero se alarga rápidamente debido al crecimiento y la recolocación del corazón y los pulmones. Al final de la séptima semana el esófago alcanza su longitud relativa final.
El estómago: Inicialmente la parte distal del intestino primitivo anterior es una estructura tubular. Durante la cuarta semana aparece una dilatación ligera que indica la localización del primordio del estomago, esta dilatación se inicia en forma de un aumento de tamaño fusiforme de la parte caudal del intestino primitivo medio y en sus primeros momentos se orientan en el plano medio.
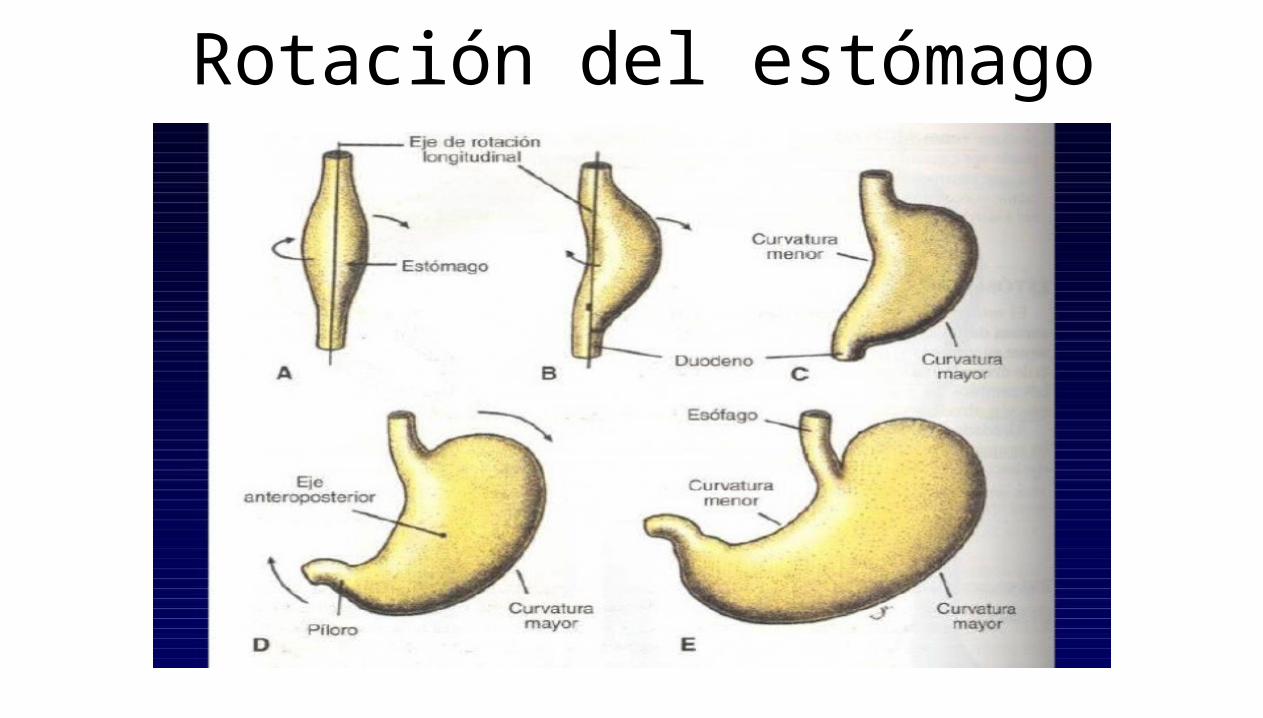
Rotación del estómago



El duodeno: Al comienzo de la cuarta semana se empieza a desarrollar el duodeno a partir de la parte caudal del intestino primitivo anterior. La unión de las dos partes del duodeno es distal al origen del colédoco. El duodeno en fase de desarrollo crece con rapidez y forma un asa con forma de C que se proyecta ventralmente. En el transcurso de la quinta y sexta semana, la luz del duodeno se va estrechando cada vez más y se oblitera temporalmente debido a la proliferación de sus células epiteliales, para cuando se ha alcanzado esta época de gestación, ha desaparecido la mayor parte del mesenterio ventral del duodeno.

Hígado y aparato biliar: Se originan de una evaginación ventral, el divertículo hepático, en la parte distal del intestino primitivo anterior al comienzo de la cuarta semana. El divertículo hepático aumenta rápidamente de tamaño y se divide en dos partes a medida que crece entre las capas del mesogastrio ventral, la parte craneal más grande del divertículo hepático es el primordio del hígado y la parte más caudal y pequeña se convierte en la vesícula biliar.

El intestino primitivo medio
Los derivados del intestino primitivo medio son los siguientes:
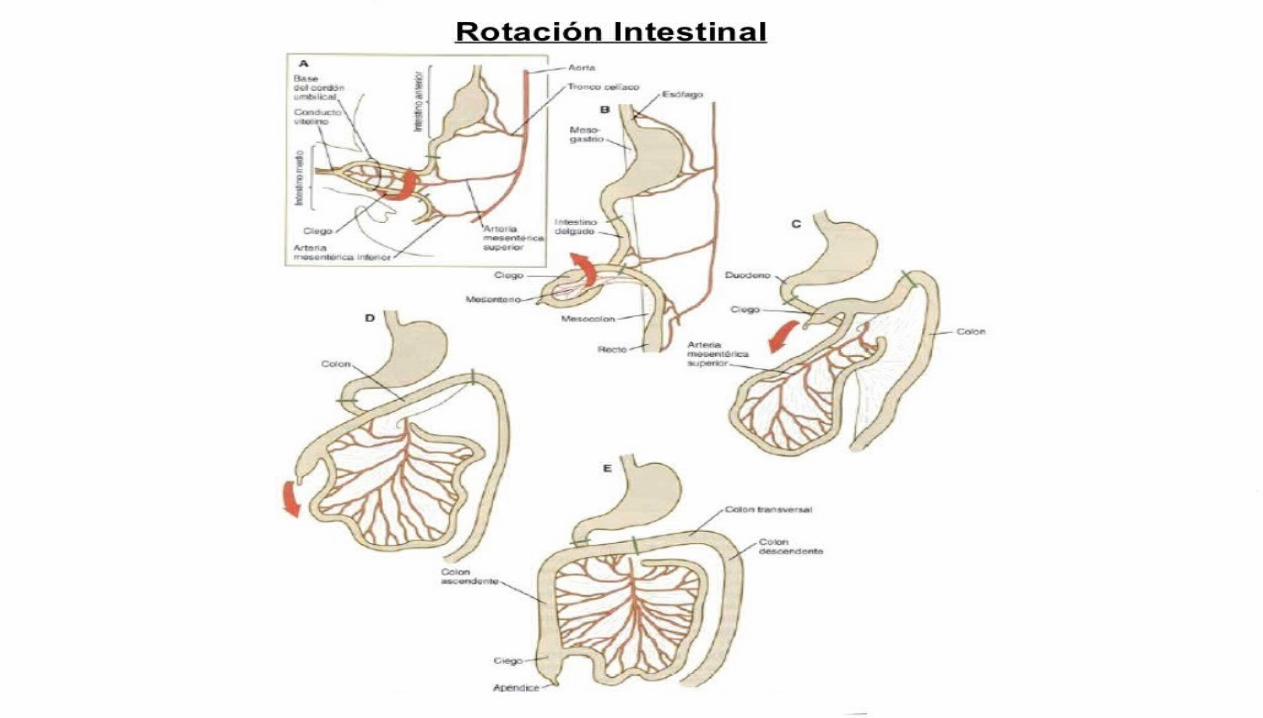
El intestino delgado que incluye el duodeno distal hasta la abertura del colédoco.El ciego y el apéndice: El primordio del ciego y el apéndice, el ensanchamiento cecal, aparece durante la sexta semana como una elevación del borde antimesenterico de la rama caudal del asa del intestino primitivo medio. El vértice del ensanchamiento cecal no crece con tanta rapidez como el resto de esta estructura por lo tanto, el apéndice es inicialmente un pequeño divertículo del ciego.Estos derivados del intestino primitivo medio están vascularizados por medio de la arteria mesentérica superior.





Desarrollo del páncreas: El páncreas se desarrolla entre las capas del mesenterio desde las yemas pancreáticas dorsal y ventral del endodermo, que se origina a partir del extremo caudal del intestino primitivo anterior. La mayor parte del páncreas procede de la gran yema pancreática dorsal que aparece al inicio y se desarrolla a una corta distancia por encima de la yema ventral.
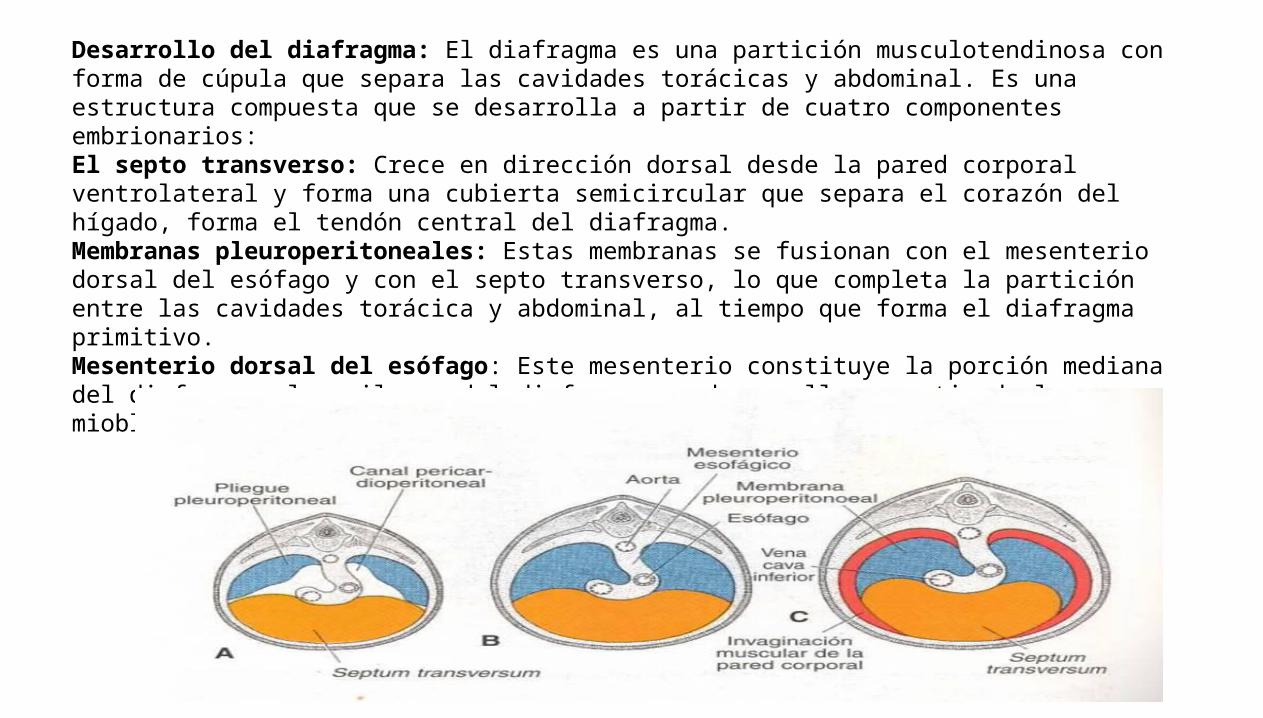
Desarrollo del diafragma: El diafragma es una partición musculotendinosa con forma de cúpula que separa las cavidades torácicas y abdominal. Es una estructura compuesta que se desarrolla a partir de cuatro componentes embrionarios:El septo transverso: Crece en dirección dorsal desde la pared corporal ventrolateral y forma una cubierta semicircular que separa el corazón del hígado, forma el tendón central del diafragma.Membranas pleuroperitoneales: Estas membranas se fusionan con el mesenterio dorsal del esófago y con el septo transverso, lo que completa la partición entre las cavidades torácica y abdominal, al tiempo que forma el diafragma primitivo.Mesenterio dorsal del esófago: Este mesenterio constituye la porción mediana del diafragma, los pilares del diafragma se desarrolla a partir de los mioblastos que crecen en el mesenterio dorsal del esófago.

Métodos de diagnóstico prenatal Ecografía
Tiene unos momentos clave de aplicación:
En el primer trimestre. Nos permite detectar algunas imágenes consideradas “marcadores de cromosomopatías”. La más válida y aceptada en los últimos años es la conocida como la translucencia nucal. Este método se aplica para poder reconocer si el feto tuviera trisomías (síndrome de Down, trisomía 13 y trisomía 18), en cuyo caso suele presentar una nuca más gruesa que un embrión normal.
En el segundo trimestre. Especialmente alrededor de la “semana 20”, es la época más adecuada para el diagnóstico de la mayoría de malformaciones de tipo anatómico.

Métodos de diagnóstico prenatal Radiografía
Hay tres aspectos que se deben considerar cuando se realizan estudios de rayos X
durante el embarazo.
La edad gestacional (cuando es posible determinarla).
La región del cuerpo donde se tomará la radiografía.
La dosis
De la primera a la octava semana después de la concepción, el embrión es muy resistente a las malformaciones causadas por los rayos X. Sin embargo es sensible a los efectos letales de los rayos X, y si recibe dosis mayores a los 5 rads, podrían ser causa de un aborto o de retardo en el crecimiento. De la semana 8ª a la 15ª de embarazo, el embrión o feto es sensible a los efectos de la radiación en su sistema nervioso central. Se ha estimado que la dosis debe ser mayor a los 30 rads. Los estudios de RX para diagnóstico en general NO alcanzan estos niveles.
En la semana 20ª de embarazo se considera que el bebé está completamente desarrollado y es más resistente a los efectos de las radiaciones. De hecho en el último trimestre del embarazo el bebé no es más vulnerable a las radiaciones que la mamá.

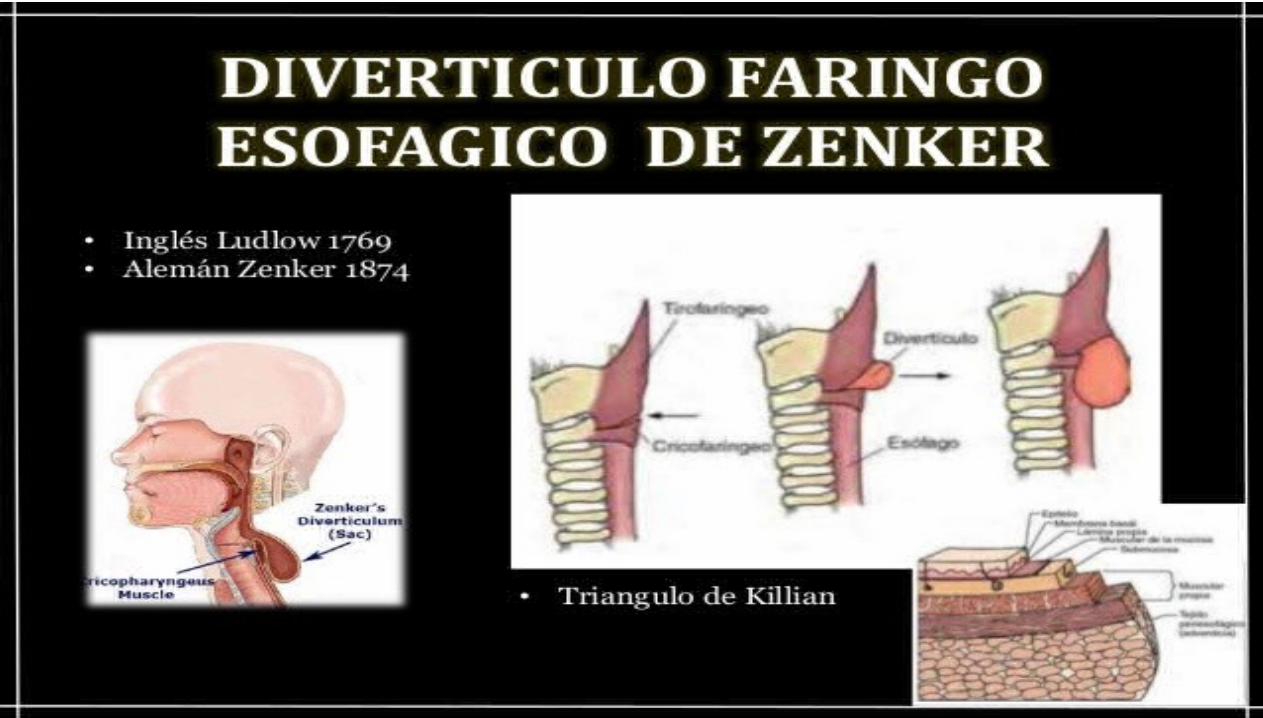
ATRESIA ESOFÁGICA Y FÍSTULA TRAQUEOESOFÁGICA
La Atresia Esofágica y las Fístulas Traqueoesofágicas ocurren en cada 3000 a 5000 nacidos vivos y afectan por igual a los dos sexos.
Por razones desconocidas, en ocasiones la separación del tubo esofágico se lleva de manera incompleta, de arriba abajo, lo
que da lugar a atresia. En la mayor parte de los casos, el esófago posterior no se separa totalmente de la tráquea, lo
que da lugar a distintas variedades de Fístula Traqueoesofágicas o a hendiduras. Las anomalías de la
tráquea (atresias, estenosis y hendiduras) pueden coexistir con defectos del esófago.
Los signos clínicos más típicos para el diagnóstico son:- Aumento de secreciones y saliva en boca y faringe: sialorrea.- Crisis de sofocación, tos y cianosis.

Las anomalías asociadas graves son en orden de frecuencia:
- Anomalías cardiovasculares: ductus permeable, CIV, Coartación aórtica, Tetralogía
de Fallot.
- Anomalías digestivas: atresia duodenal, atresia anal, onfalocele. Merece especial
mención la asociación VATERL (V=vertebrales, A=anales, TE=fístula TE, R=radiales
y/o renales)
- Anomalías urológicas: hidronefrosis, agenesia renal, riñón poliquístico, etc.
- Otras. Cromosómicas: trisomía 21, trisomía 13, 15, 18; neurológicas: meningocele,
hidrocefalia, craneoestenosis, etc.
Entre anomalías asociadas leves, las más frecuentes son las óseas: costovertebrales,
presencia de 11 ó 13 costillas, hemivértebras, agenesia sacra etc. Y otras como la
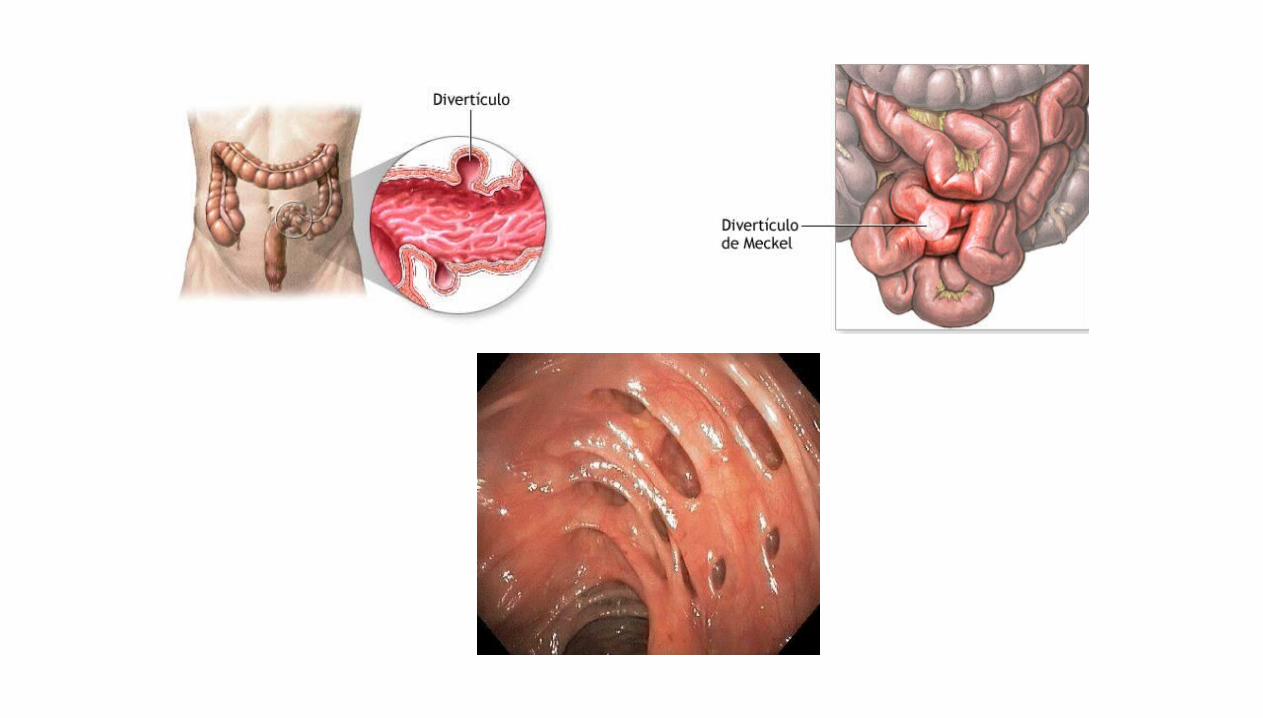
sindactilia y el divertículo de Meckel.


Estenosis Esofágica• Ocurren en uno de cada 25000 a 50000 nacidos vivos.
• Después de que se forma el tabique traqueoesofágico y se alargan
tanto el esófago como el árbol traqueobronquial, las células
mesenquimatosas del divertículo respiratorio quedan incluidas en la
pared del esófago en forma de epitelio respiratorio glándulas y
cartílago. Este foco ectópico puede causar estenosis que no da de sí,
la estenosis también suele ser debida a una falta de recanalización
completa del esófago durante la octava semana de desarrollo, pero
también puede ser falta de desarrollo de los vasos sanguíneos
esofágicos de la zona afectada.

Síntomas
• Si la estenosis es severa se desarrollará disfagia para líquidos en primeros días/semanas de vida. Infecciones respiratorias recurrentes, saliveo incesante o fallo de medro pueden darse además. Pueden asociarse anomalías congénitas como la atresia esofágica, síndrome de Down, anomalías cardiacas, ano-rectales, atresia duodenal, etc. (si bien su frecuencia es muy inferior a las asociadas a atresia de esófago).

DUPLICACIONES
• Embriológicamente se deben a trastornos en la recanalización, cuando el estado sólido del tubo digestivo se convierte en hueco, por la aglutinación de vacuolas del sector dorsal del intestino anterior, en lugar de originar un sólo tubo origina dos.
• La duplicación esofágica representa 10 a 20% de malformaciones esofágicas y 15 a 20% de duplicaciones del tubo digestivo.
• En los sintomáticos se manifiestan por datos de compresión extrínseca, como disfágia; así como con nauseas, vómito, o bien por sangrado de tubo digestivo.
Malformaciones del estómago

Hipertrofia congénita del píloro
Definición Etiología
Obstrucción del músculo pilórico
Tratamiento quirúrgico
Causa desconocida
Hiperacidez gástrica Inervación pilórica anormal
Motilidad anormal (disminución de las células ganglionares)
Péptido C Oxido Nítrico
Factores genéticos
Masculino Sangre: AO
Factores ambientales
Alimentación Eritromicina
Tabaquismo materno
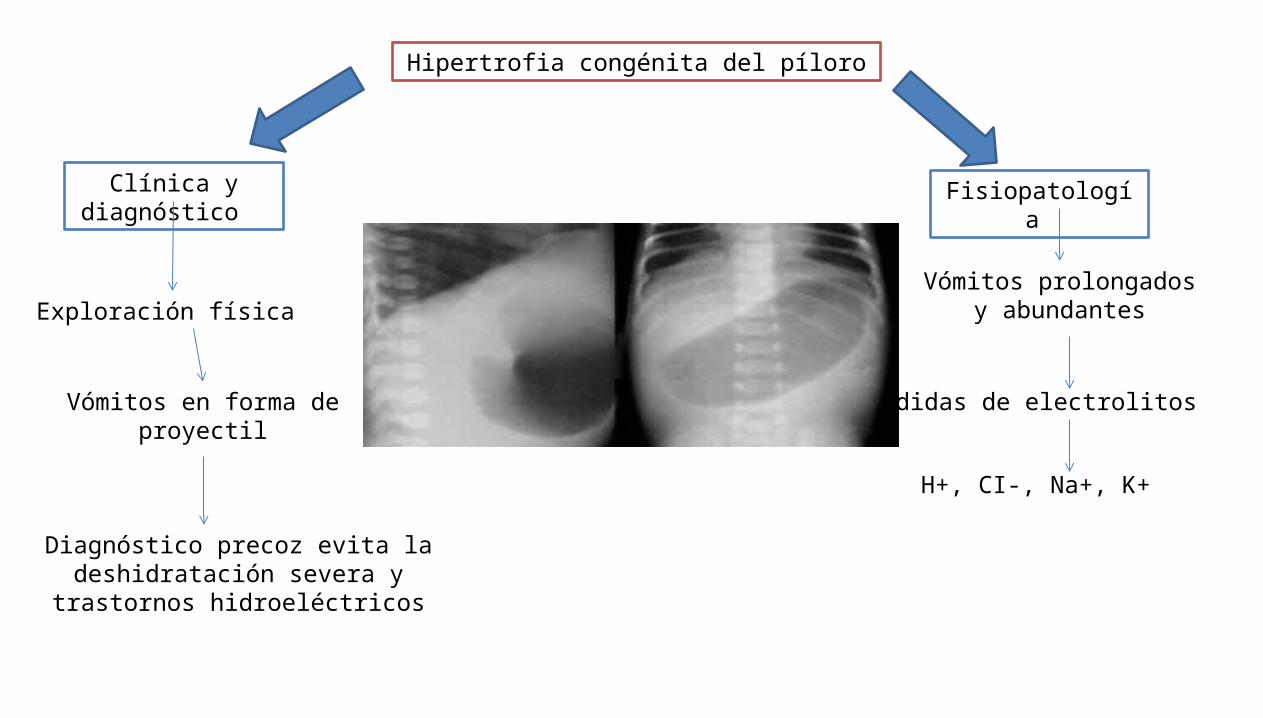
Hipertrofia congénita del píloro
Clínica y diagnóstico
Fisiopatología
Vómitos prolongados y abundantes
Pérdidas de electrolitos
H+, CI-, Na+, K+
Exploración física
Vómitos en forma de proyectil
Diagnóstico precoz evita la deshidratación severa y trastornos
hidroeléctricos

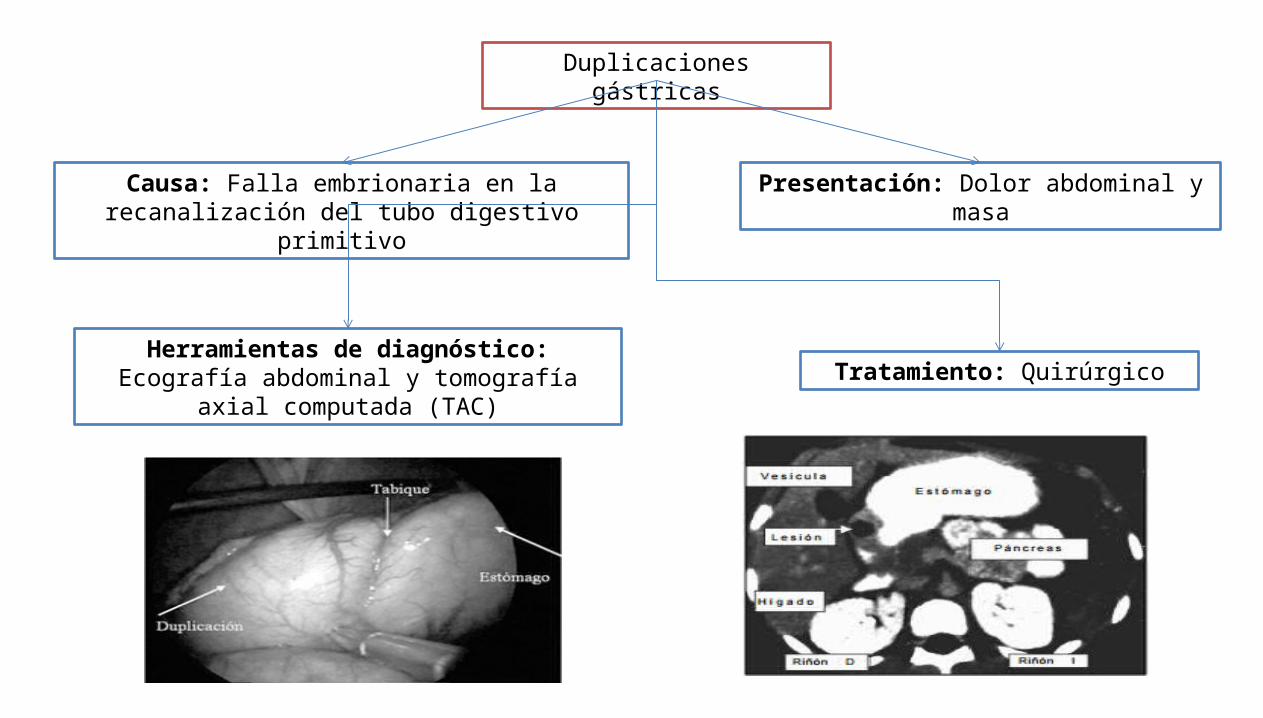
Duplicaciones gástricas
Causa: Falla embrionaria en la recanalización del tubo digestivo primitivo
Presentación: Dolor abdominal y masa
Herramientas de diagnóstico: Ecografía abdominal y tomografía axial computada (TAC) Tratamiento: Quirúrgico
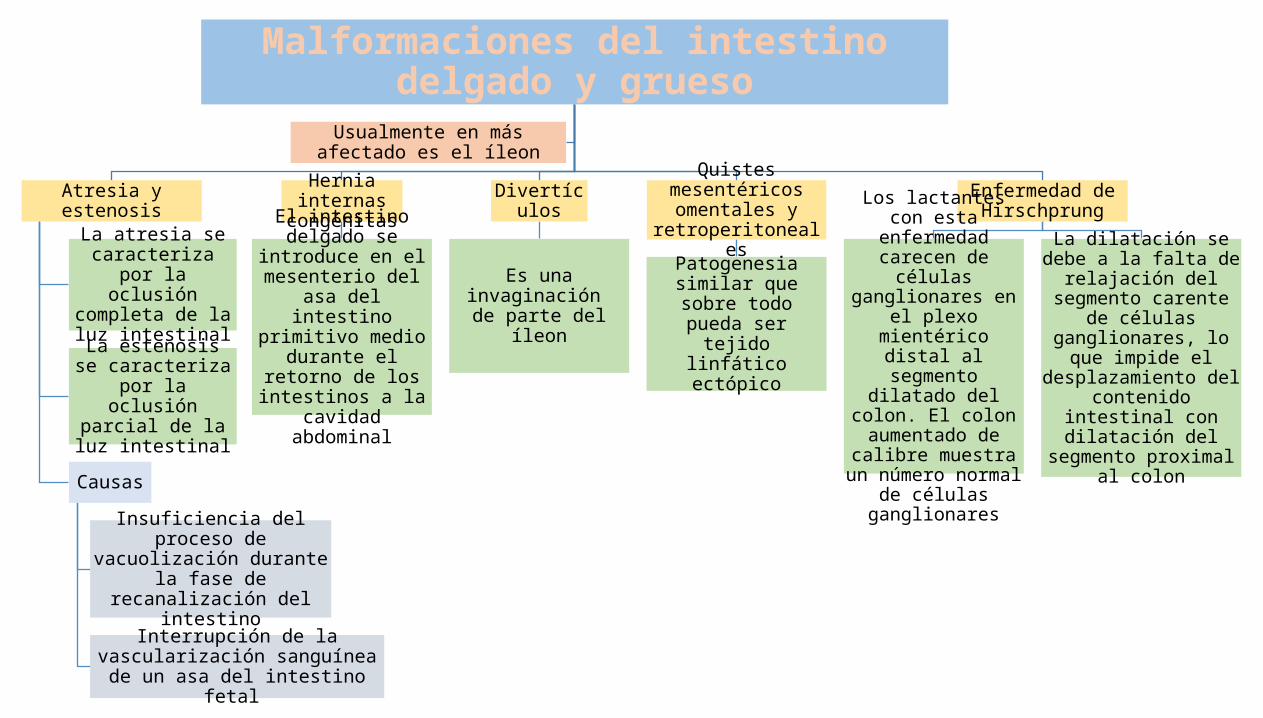
Malformaciones del intestino delgado y grueso
Atresia y estenosis
La atresia se caracteriza por la
oclusión completa de la luz intestinal
La estenosis se caracteriza por la
oclusión parcial de la luz intestinal
Causas
Insuficiencia del proceso de vacuolización durante la fase
de recanalización del intestino
Interrupción de la vascularización sanguínea de un asa del intestino fetal
Hernia internas congénitas
El intestino delgado se introduce en el
mesenterio del asa del intestino primitivo medio durante el
retorno de los intestinos a la cavidad
abdominal
Divertículos
Es una invaginación de parte del íleon
Quistes mesentéricos omentales y
retroperitoneales
Patogenesia similar que sobre todo pueda
ser tejido linfático ectópico
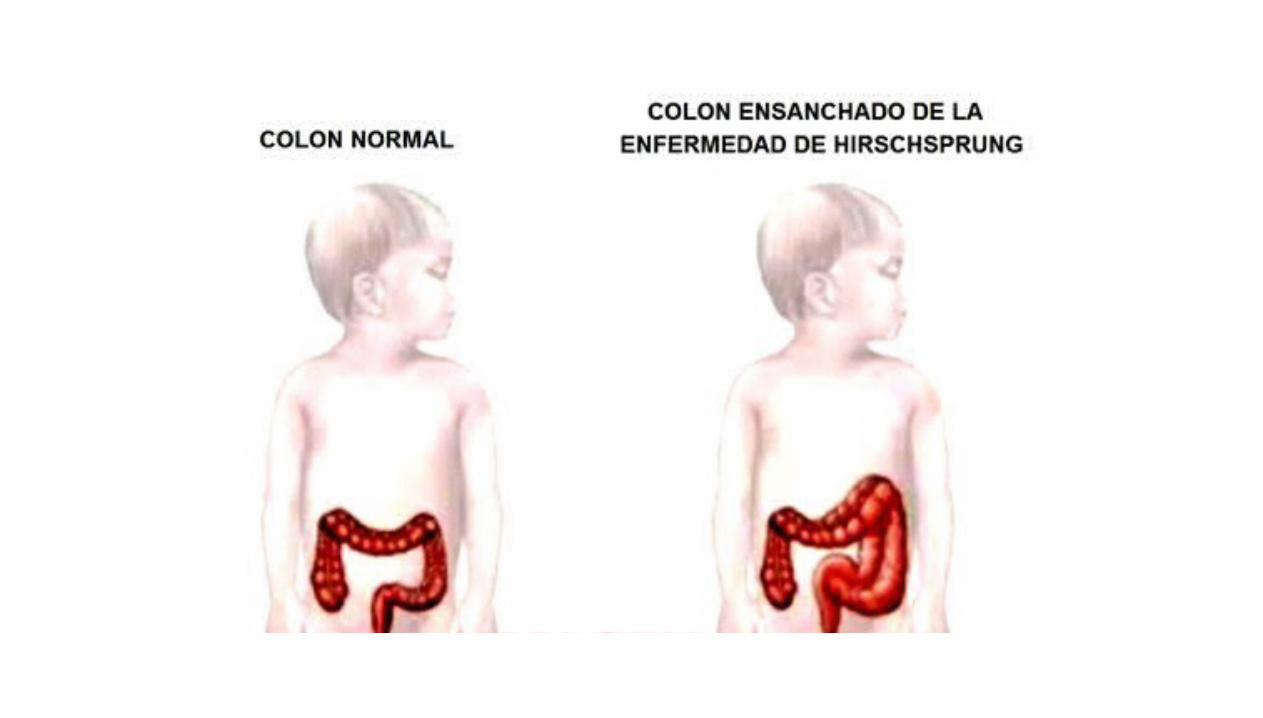
Enfermedad de Hirschprung
Los lactantes con esta enfermedad carecen de células ganglionares en
el plexo mientérico distal al segmento
dilatado del colon. El colon aumentado de calibre muestra un número normal de
células ganglionares
La dilatación se debe a la falta de relajación del segmento carente de
células ganglionares, lo que impide el
desplazamiento del contenido intestinal con dilatación del segmento
proximal al colon
Usualmente en más afectado es el íleon
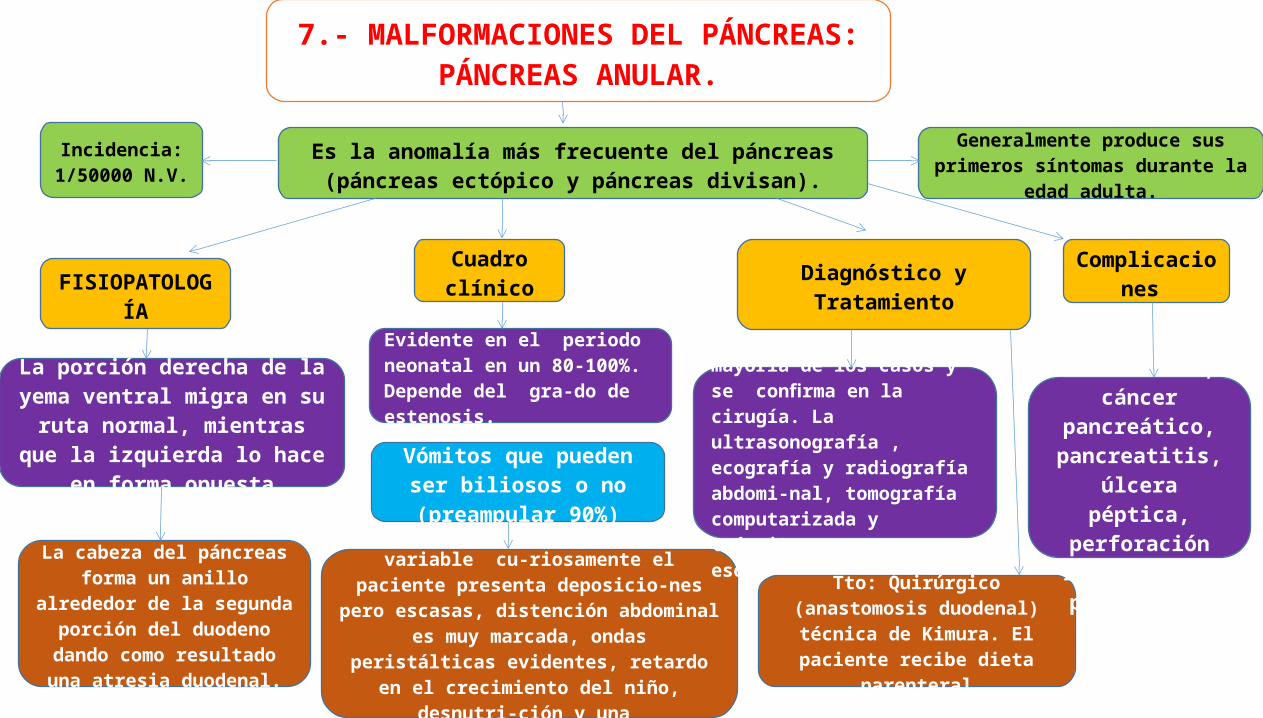
7.- MALFORMACIONES DEL PÁNCREAS: PÁNCREAS ANULAR.
Es la anomalía más frecuente del páncreas (páncreas ectópico y páncreas divisan).
Generalmente produce sus primeros síntomas durante la edad adulta.
FISIOPATOLOGÍA
Incidencia: 1/50000 N.V.
La porción derecha de la yema ventral migra en su ruta normal, mientras que la izquierda lo hace
en forma opuesta
La cabeza del páncreas forma un anillo alrededor de la segunda
porción del duodeno dando como resultado una atresia
duodenal.
Cuadro clínico
Evidente en el periodo neonatal en un 80-100%. Depende del gra-do de estenosis.
Vómitos que pueden ser biliosos o no (preampular
90%)
Diagnóstico y Tratamiento
dolor abdominal de intensidad variable cu-riosamente el paciente presenta deposicio-nes
pero escasas, distención abdominal es muy marcada, ondas peristálticas evidentes, retardo en el crecimiento del niño, desnutri-ción y una
deshidratación asociada al cuadro
Dx. es presuntivo en la mayoría de los casos y se confirma en la cirugía. La ultrasonografía , ecografía y radiografía abdomi-nal, tomografía computarizada y tránsito esofagodudenal.
Tto: Quirúrgico (anastomosis duodenal) técnica de Kimura. El paciente recibe dieta parenteral
Complicaciones
Ictericia obstructiva, cáncer pancreático, pancreatitis, úlcera péptica, perforación
intestinal y peritonitis
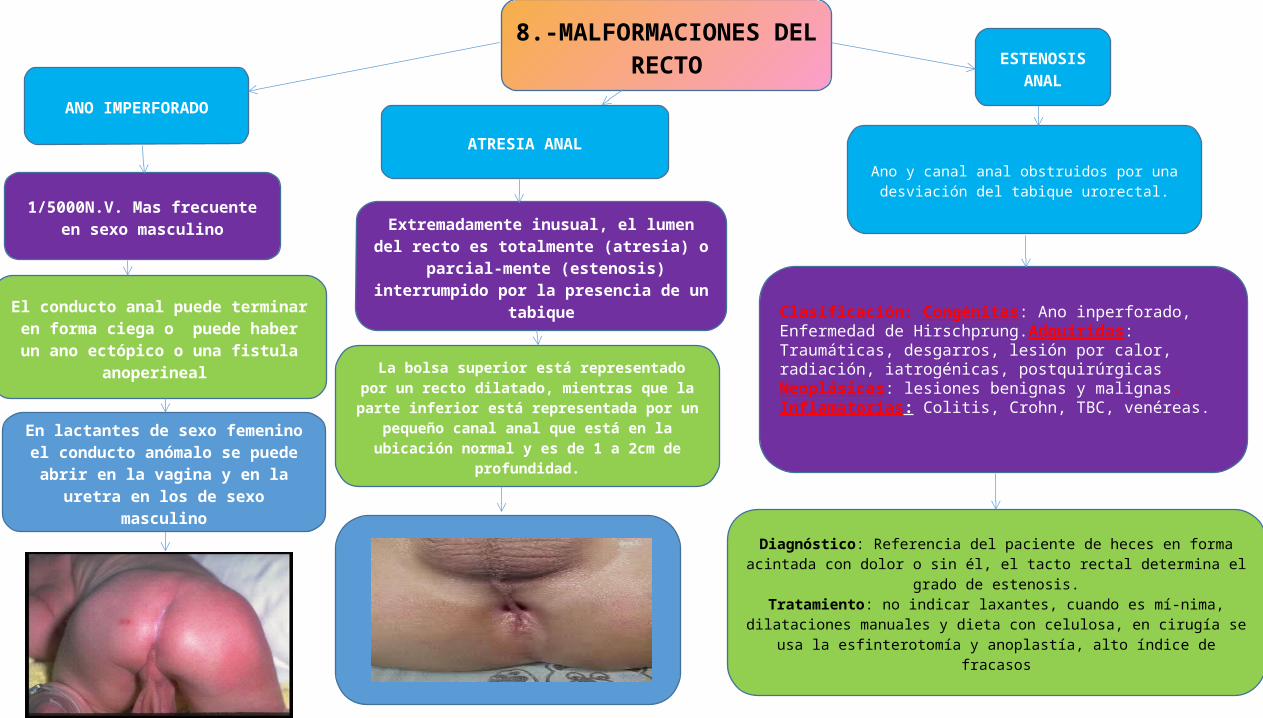
8.-MALFORMACIONES DEL RECTO
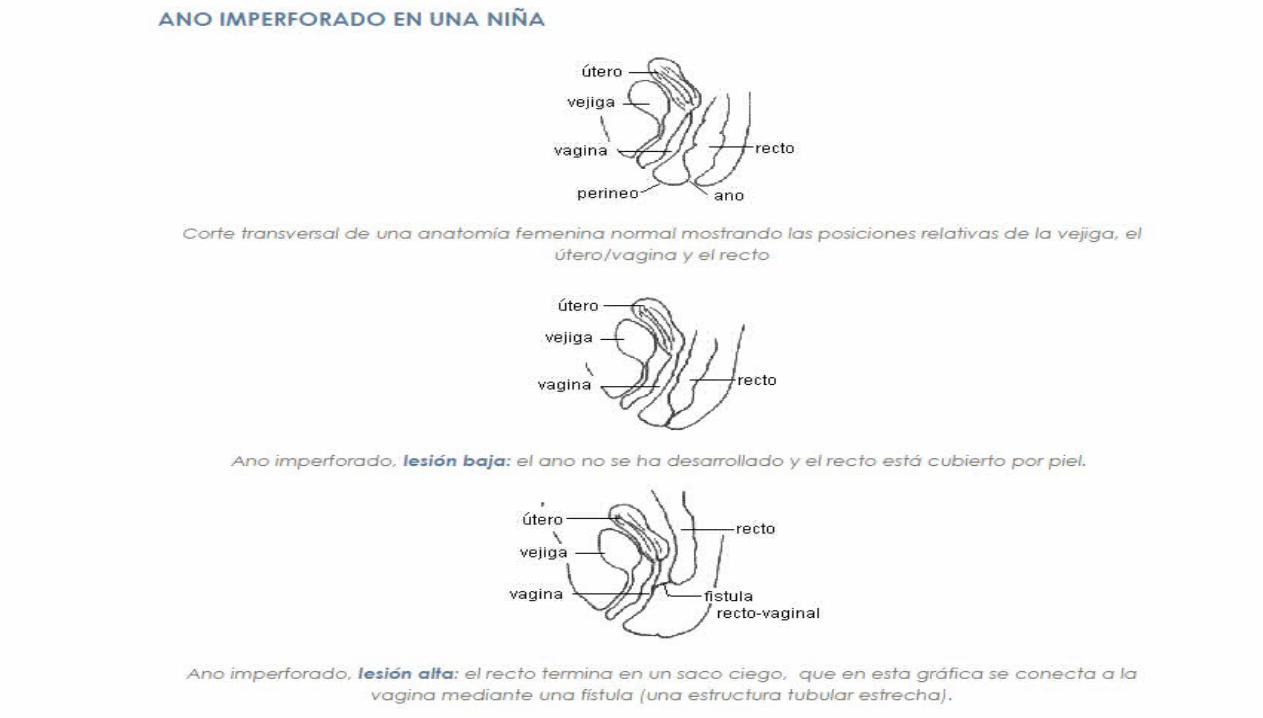
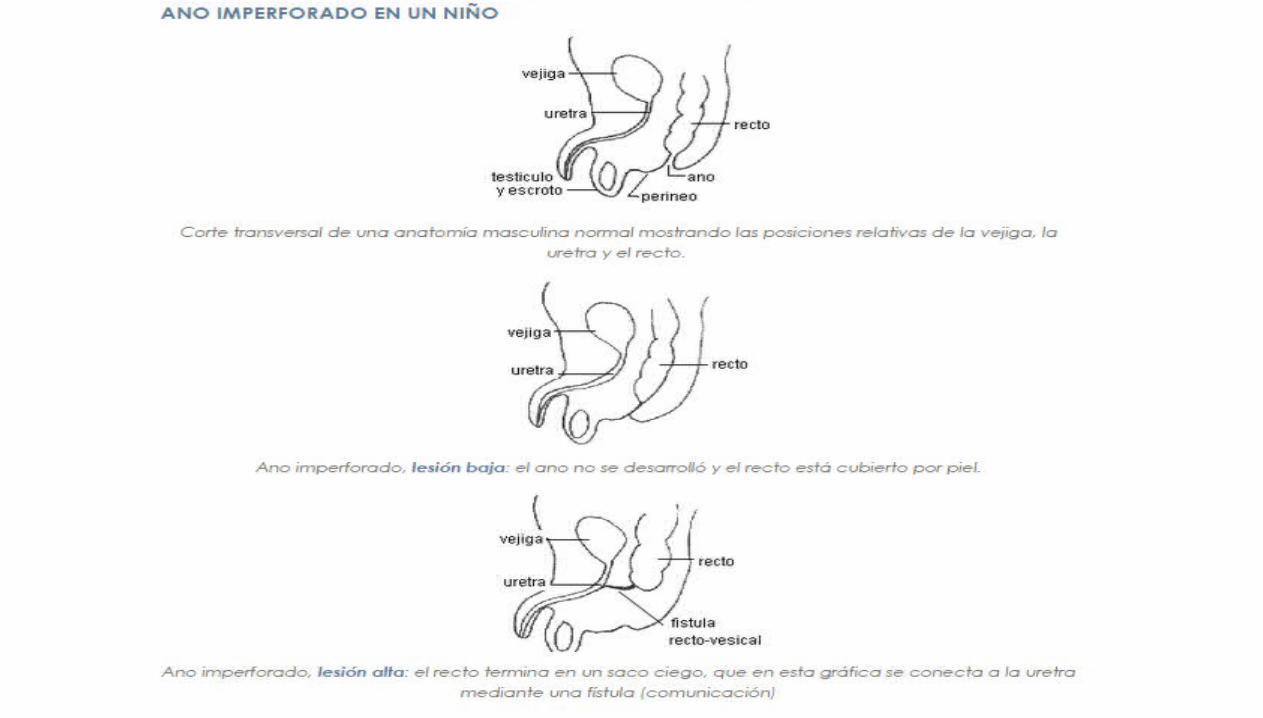
ATRESIA ANAL
ANO IMPERFORADO
ESTENOSIS ANAL
Extremadamente inusual, el lumen del recto es totalmente (atresia) o parcial-mente (estenosis)
interrumpido por la presencia de un tabique
Ano y canal anal obstruidos por una desviación del tabique urorectal.
1/5000N.V. Mas frecuente en sexo masculino
La bolsa superior está representado por un recto dilatado, mientras que la parte inferior está representada
por un pequeño canal anal que está en la ubicación normal y es de 1 a 2cm de profundidad.
El conducto anal puede terminar en forma ciega o puede haber un ano ectópico o una
fistula anoperineal
En lactantes de sexo femenino el conducto anómalo se puede abrir en la vagina y en la
uretra en los de sexo masculino
Clasificación: Congénitas: Ano inperforado, Enfermedad de Hirschprung.Adquiridas: Traumáticas, desgarros, lesión por calor, radiación, iatrogénicas, postquirúrgicas. Neoplásicas: lesiones benignas y malignas. Inflamatorias: Colitis, Crohn, TBC, venéreas.
Diagnóstico: Referencia del paciente de heces en forma acintada con dolor o sin él, el tacto rectal determina el grado de estenosis.
Tratamiento: no indicar laxantes, cuando es mí-nima, dilataciones manuales y dieta con celulosa, en cirugía se usa la esfinterotomía y
anoplastía, alto índice de fracasos