
Práctica 1.-Determinación y observación de principios ... · PDF...
20
Práctica 1.-Determinación y observación de principios inmediatos en alimentos: 1.1 Azúcares reductores (tinción con reactivo de Fehling), 1.1 Almidón (tinción con Lugol) 1.2 Lípidos (tinción con Sudán III) 1.3 Proteínas (reacción de Biuret). Práctica 2.-Estudio de la mitosis en células de raíz de cebolla: Tinción con fucsina ácida y/o orceína Práctica 3.- Observación de bacterias: Tinción de Gram (serrapio dental, yogur) Práctica 4.-.- Extracción de ADN. Práctica 5.- Observación de vídeos animados sobre el proceso de la síntesis proteica Practica 6.- Búsqueda de información en libros o revistas científicas o de divulgación de los siguientes temas • Proyecto genoma humano • Clonación en animales • Plantas transgénicas. Análisis de sus repercusiones biológicas, económicas y sociales.
Transcript of Práctica 1.-Determinación y observación de principios ... · PDF...

Práctica 1.-Determinación y observación de principios inmediatos en alimentos:1.1 Azúcares reductores (tinción con reactivo de Fehling),1.1 Almidón (tinción con Lugol)1.2 Lípidos (tinción con Sudán III)1.3 Proteínas (reacción de Biuret).
Práctica 2.-Estudio de la mitosis en células de raíz de cebolla: Tinción con fucsina ácida y/o orceína
Práctica 3.- Observación de bacterias: Tinción de Gram (serrapio dental, yogur)
Práctica 4.-.- Extracción de ADN.
Práctica 5.- Observación de vídeos animados sobre el proceso de la síntesis proteica
Practica 6.- Búsqueda de información en libros o revistas científicas o de divulgación de los siguientes temas
• Proyecto genoma humano
• Clonación en animales
• Plantas transgénicas. Análisis de sus repercusiones biológicas, económicas y sociales.

IntroducciónMicroscopios
M. simple Lupas monocularesLupas binoculares
• Estereomicroscopios• De fluorescencia
M. Compuesto: • De campo oscuro• Microscopia por luz reflejada• De luz ultravioleta• De contraste de fases• De polarización
*De Barrido (MEB)Microscopia electrónica *Microscopio confocal de barrido láser
*De Transmisión (TLM)
MICROSCOPIO ÓPTICO (o de campo luminoso).En estos tipos de microscopios el área observada está ampliamente iluminada y los objetos que se estudianaparecen más oscuros que el fondo.Normalmente alcanzan hasta unos 1000 aumentos, aunque con oculares poderosos esta cifra puede llegar aincrementarse en dos veces. El limite útil de este aumento es un máximo 2000 y la razón de estelímite de amplificación, se debe al poder de resolución, entendido como la capacidad de distinguirdos puntos adyacentes como distintos y separados. Este poder de resolución se da enfunción de la longitud de onda de la luz utilizada y de la apertura numérica que posea el sistemade lentes empleado. Así, puede afirmarse que no siempre las amplificaciones mayores son las de másutilidad, ya que pueden no ser tan claras como otras menores.Dentro de la microscopia óptica podemos distinguir, según el número y posición de las lentes el microscopiosimple y el compuesto.
MICROSCOPIO SIMPLE (Lupa)Está provisto de una lente o sistema de lentes convergentes dispuestas de manera que proporcionan unaimagen virtual, derecha y mayor que el objeto, que a su vez está situado entre la lente y el foco. La ampliacióndel microscopio simple es bastante limitada y suele utilizarse para la disección de pequeños animales o para ladisociación de piezas histológicas.
Este tipo de microscopio se denomina también lupa. Las lupaspueden ser monoculares o binoculares.La lupa binocular permite aumentar la imagen de objetos y constade los siguientes elementos:-Oculares: son dos lentes (una para cada ojo) a través de lascuales se realiza la observación. Existen lupas con un solo ocular.-Objetivo: lente más cercana al objeto, normalmente en el interiorde una estructura de plástico, redondeada y de color negro. Aveces existen dos posiciones, con diferentes aumentos.-Platina: estructura de vidrio circular, donde se coloca lapreparación y objeto que se va a visualizar. Encima de ella existenunas pinzas para sujetar a la muestra.-Mando de enfoque: rueda o tornillo de enfoque, que se regularapara obtener una mayor nitidez de la muestra. Al moverle sedesliza todo el bloque de la lupa hacia abajo o hacia arriba.-Interruptor: para conectarse a la red.-Fuente de luz (bombilla). Suelen existir dos bombillas que sesitúan una cerca del objetivo y la otra debajo de la platina. Enfunción de la muestra a observar se seleccionará una u otra através de una llave de tres posiciones. A veces existe una “rueda”para controlar la intensidad de la luz.-Base y brazo: donde se acoplan el resto de las partes de la lupabinocular. En el brazo existe un tornillo que controla la posición detodo el conjunto de objetivos, oculares y fuente de luz. Todo el

conjunto se tendrá que subir o bajar en función del tamaño de la muestra u objeto a observar
MICROSCOPIO COMPUESTOA diferencia del simple, en este tipo de microscopios se combinan dos lentes o sistemas de lentes
convergentes de amplificación de imagen, colocados en los extremos del tubo el denominadoobjetivo, situado más cerca del objeto a observar; y el ocular más cercano al ojo del observador.Antes de pasar a hablar de los diferentes tipos de microscopios compuestos consideramos
importante hacer referencia a los cabezales monoculares, binoculares y triloculares, en orden demenor a mayor especialización en este tipo de técnica. El cabezal monocular consta de un soloocular, llevando consigo el inconveniente de producir la fatiga visual en observaciones prolongadas.Este problema se solventó con la aparición del cabezal binocular, el cual permite la visión con los dosojos, siendo importante alcanzar una adecuada fusión de la imagen. El cabezal trilocular además deposeer las ventajas del anterior posibilita fotografiar el objeto de estudio.
Pié: Pieza maciza y pesada para asegurar la estabilidad del aparato y para soportar las demás piezas quecomponen al microscopio; en ocasiones está provisto por una charnela, que permite la inclinación dela parte superior del microscopio.
Platina: Pieza metálica redonda o cuadrada, donde se colocan las preparaciones; tiene en el centro unaabertura circular por la que pasarán los rayos luminosos procedentes del sistema de iluminación.Puede estar adosada a un carro con dos tornillos de cremallera que permitan movimientos de

traslación y si los tornillos están graduados, para medir desplazamientos La preparación se sujeta enlas platinas fijas, con dos palanquitas móviles. En las patinas con carro, se fija por un reborde
Tubo óptico: En él está instalado el sistema óptico. Está constituido por dos cuerpos. Uno el externo en elque se encuentra la cremallera y otro interno adosado al anterior donde se encuentra el objetivo. Eltubo se mueve verticalmente para poder enfocar el objetivo, con movimiento rápido mediante elmacrométrico y movimiento lento mediante el micrométrico.
Objetivos: Están formados por varias lentes para corregir aberraciones, deben tratarse con mucho cuidado,un golpe puede variar la posición de las lentes y averiarlas, se atornillan al revólver del portaobjetos Objetivos en seco: Entre la lente y el objetivo sólo hay aire. Poseen gran profundidad de foco, lo
que permite observar diferentes planos paralelos del objeto Objetivos de inmersión: Debe interponerse entre la lente frontal y la preparación un líquido
cuyo índice de refracción sea superior al aire (aceite de cedro) lo que permite una mayorluminosidad, son objetivos de gran aumento y gran poder de definición (Bacteriología)
Oculares: Los forman dos lentes separadas por un diafragma y van montados en la extremidad superior deltubo
El sistema de iluminación se compone de una lámpara o espejo, un condensador y un diafragma; tiene lamisión de iluminar los objetos por medio de luz transmitida.
• El espejo es redondo, con una cara plana y otra cóncava, los expertos sugieren usar la parteplana para objetivos de poco aumento y fuentes de luz directa, y la parte cóncava paraobjetivos con gran aumento y fuentes de luz indirecta; el espejo se puede mover y adaptar adiferentes posiciones según la fuente luminosa.
• El condensador consta de un sistema de lentes de gran abertura sujetos a una montura,colocados entre la platina y el espejo, puede subirse y bajarse a voluntad y tiene la finalidadde concentrar los rayos de luz para dirigirlos hacia la preparación.
• El diafragma se encuentra unido al condensador y regula la cantidad de rayos luminosos queinciden sobre la preparación
---------------------------------------------------------------------------------------------------------------------------------------------Poder de definición: El objetivo debe presentar con corrección los contornos de la imagenPoder penetrante: El objetivo debe permitir sin variar el enfoque ver varios planos del espesor deuna preparación.El poder de resolución: El poder de resolución es la distancia mínima a que deben estar situadosdos puntos para verlos con claridadLa resolución máxima del microscopio óptico es de 0,2 μ = 200nm, la de 1 ojo humano 0,1 mm
_______________________________________________________________________________________• ¿Cómo se calcula o número de aumentos?Como el microscopio compuesto tiene dos sistemas de aumentos el ocular y el objetivo. Paracalcular el aumento total tendremos que multiplicar el aumento propio del objetivo por elaumento del ocular
______________________________________________________________________________________

• Ópticas:_Condensador (8): Concentra los rayos de luz sobre lapreparación._Objetivos (3): Aumentan el tamaño de la muestra y proyectan laimagen al ocular. Se encuentran montados en el revólver._Ocular (1): Aumenta la imagen y la proyecta sobre la retina delobservador._Diafragma (6): Regula la cantidad de luz que llega alcondensador._Fuente de luz (7): Puede ser un foco o un espejo que refleje laluz hacia el condensador.• Mecánicas:_Base: Pieza rígida encargada de otorgar estabilidad a todo elconjunto._Brazo: Pieza que une al sistema óptico con la base._Platina (9): Plataforma horizontal con un agujero central quepermite el paso de la luz. Los portaobjetos con las preparacionesse colocan sobre ella y se fijan con un par de pinzas. Un sistemade tornillos permite desplazar la platina y la muestra haciaadelante, atrás, izquierda y derecha._Revólver (2): Sistema sobre el que se encuentran los objetivos, algirar permite seleccionar cualquiera de los objetivos disponibles._Tornillos macrométrico y micrométrico (4 y 5): Tornillos quepermiten enfocar la muestra, desplazando la platina hacia arriba oabajo._Tubo: Cámara oscura unida al brazo
MICROSCOPIO CONTRASTE DE FASESEl microscopio de contraste de fases proporciona una imagen clara y detallada de células vivas sin teñir. En unmicroscopio óptico ordinario, el contraste se debe a que los materiales distintos absorben diferentescantidades de luz. Sin embargo, en el microscopio de contraste de fases se debe a los distintos índices derefracción entre las partes de los microorganismos y el fondo. Las únicas diferencias estructurales entre unmicroscopio óptico ordinario y uno de contraste de fases son los sistemas de las lentes del objetivo y delcondensador, que en el segundo están dotados de unos anillos opacos especiales.El microscopio de contraste de fases se fundamenta en el hecho conocido de que las ondas de luz viajan adistintas velocidades en los materiales que poseen índices de refracción diferentes. Por tanto, los rayos de luzque pasan a través del espécimen no se encuentran en la misma fase que los que pasan a su alrededor.Imaginemos las olas del océano viniendo hacía la playa desde diferentes direcciones. Las olas se suman oanulan unas a otras, dependiendo de que sus crestas lleguen al mismo tiempo o no. De forma similar, elmicroscopio de contraste de fases combina los rayos de luz procedentes del espécimen y los que llegan de susalrededores. Los rayos se suman o se anulan unos a otros produciendo diferentes intensidades de luz yaumentando, en consecuencia, el contraste. Se denomina interferencia al resultado de la combinación derayos de luz de distinta fase.
MICROSCOPIO DE CAMPO OSCUROUtiliza un haz enfocado de luz muy intensa en forma de un cono hueco concentrado sobre el espécimen. Elobjeto iluminado dispersa la luz y se hace así visible contra el fondo oscuro que tiene detrás, como laspartículas de polvo iluminadas por un rayo de sol que se cuela en una habitación cerrada. Por ello lasporciones transparentes del espécimen quedan oscuras, mientras que las superficies y partículas se venbrillantes, por la luz que reciben y dispersan en todas las direcciones, incluida la del eje óptico que conecta elespécimen con la pupila del observador. Esta forma de iluminación se utiliza para analizar elementosbiológicos transparentes y sin pigmentar, invisibles con iluminación normal, sin fijar la muestra, es decir, sinmatarla

MICROSCOPIO DE LUZ REFLEJADAEstos microscopios se usan principalmente para observar preparados transparentes y líquidos. El ámbito deuso es por ejemplo el análisis de sangre, células, pruebas en plantas. Los microscopios clásicos de luz reflejadatienen una distancia de trabajo muy ínfima, por debajo de 4 mm. Por ello, esta clase de microscopios sonaptos para preparados muy finos
MICROSCOPIO ELECTRÓNICO.El microscopio electrónico ha revolucionado el conocimiento de la biología o la medicina. Tiene la ventaja dealcanzar una extraordinaria amplificación. Puede dar un poder de resolución hasta mil veces mayor que elóptico debido a que emplea un haz de electrones en lugar de un haz de fotones.Existen dos tipos principales de microscopios electrónicos: el microscopio electrónico de barrido y el detransmisión
DE BARRIDOEn el microscopio electrónico de barrido (MLB) o microscopia de exploración electrónica (SEM), loselectrones inciden desde arriba sobre la preparación por ello la muestra puede ser de cualquier grosor otamaño.Emplea dos técnicas preparatorias:
Secado por congelación. Secado por punto crítico. Después se cubre la muestra con una capa de metal (oro o
platino).
DE TRANSMISIÓN
En este tipo de microscopia electrónica, el haz de electrones atraviesa al material que se desea observar.El modo de operar de este tipo de microscopio es similar al del microscopio óptico, ya que está basado enel hecho de que la manera de actuar de un campo electromagnético sobre un haz de electrones es análogaa la acción de la lente de cristal sobre el haz de fotones. La imagen, sin embargo, se forma sobre unapantalla fluorescente como lo haría en una pantalla de televisor.El haz de electrones pasa a través de la muestra estudiada y posteriormente, a través de unas lenteselectromagnéticas que dan lugar a una imagen amplificada. Esta imagen pasa a su vez por una lenteproyectora hasta una pantalla de material fluorescente, que brilla al recibir el impacto de los electrones.Debajo de la pantalla se sitúa la cámara para fotografiar la imagenNo obstante, al igual que en el microscopio óptico, el microscopio electrónico de transmisión tiene lalimitación de ser útil sólo para un grosor determinado del objeto. Las películas de muestra, además de serdelgadas, no deben poseer materiales que puedan dispersar o absorber electrones, deben ser losuficientemente fuertes como para poderlas manipular, y lo bastante estables, como para no volatilizarse,a causa del bombardeo en el vacío.El interior del microscopio debe hallarse en vacío, ya que el aire impide la movilidad de tos electrones. Estose efectúa mediante las bombas de vacío. Estas condiciones imposibilitan la observación demicroorganismos vivos, y de sus procesos fisiológicos. Las técnicas que más se utilizan para este tipo demicroscopio electrónico son: tinción negativa, microtomía y congelación.

PREPARACIÓN DEL OBJETO DE ESTUDIO PARA MICROSCOPIA ÓPTICALa preparación del objeto de estudio puede resultar un proceso simple o. por el contrario, ser bastante
complejo, dependiendo de la naturaleza y características de aquello que queremos observar.La preparación de una muestra para estudiarla al microscopio óptico es diferente según la naturaleza delmaterial que ha de ser observado: orgánico o inorgánico. Si es orgánico, según queramos observarpropiedades que sólo se manifiestan en estado vivo, o si queremos observar morfología y estructuras,que no se modifican cuando sobreviene la muerte celular.
- Preparaciones húmedasPara observar organismos acuáticos microscópicos (algas, larvas, etc.). Se pone una gota del líquido quelos contiene sobre el portaobjetos y se pone el cubreobjetos con cuidado para que no aparezcan burbujas deaire
- Gota pendiente:Se utilizan portaobjetos excavados sobre los que se pone el cubreobjetos, que lleva adherido la gota del
líquido que ha de ser observado. Así podemos observar el movimiento de los microorganismos en el mediolíquido, sin que estén sometidos a la presión entre portaobjetos y cubreobjetos.Esta técnica presenta varios problemas que hay que tener en cuenta:
1. La gota constituye una lente trémula que desvía los rayos de luz e interfiere con la iluminación, estoresulta muy molesto cuando se emplea microscopio de contraste de fases.
2. Los portaobjetos excavados actúan como una lente divergente que puede modificar la realidad de loque se está observando.
- Examen en fresco con nigrosina:Esta técnica consiste en añadir nigrosina, se prefiere a la tinta china, al objeto de estudio.Con este método podemos distinguir bacterias incoloras sobre fondo negro. Se utiliza sobre todo para el
estudio de detalles estructurales como cápsulas y flagelos. Se utiliza el objetivo de inmersión.En el examen en fresco se pueden utilizar dos tipos de medios: los líquidos fisiológicos o los líquidos de
adición. Los líquidos fisiológicos son los que conservan las condiciones más parecidas a aquellas en las quese desenvuelve el microorganismo vivo. Los líquidos de adición se utilizan para aclarar los objetos deestudio poco transparentes (insectos, pelos, fragmentos vegetales) y así hacerlos visibles. Como líquidosde adición se utilizan el lactofenol o el clorofenol.
b) Coloración vital.Permite poner de relieve detalles estructurales sin matar al organismo. En general no tiñen, sino que
se acumulan en determinadas zonas de la célula. Como todo colorante es una sustancia tóxica; por lo quehay que emplear bajas concentraciones. Como colorantes vitales se utilizan el azul de metileno, elrojo congo, el verde Janus.La coloración en vivo se puede hacer de dos maneras:
1. Por difusión: en el espacio entre portaobjetos y cubreobjetos de una preparación en fresco, sepone una gota de colorante que penetra en la muestra por capilaridad.
2. Mezclando una gota de colorante con el material que ha de ser examinado en el portaobjetos ycolocando luego el cubreobjetos.
ESTUDIO "IN VITRO".Consiste en la observación de células y tejidos muertos. Para ello se realizan una serie de pasos, cornoson la fijación, la inclusión, el corte (microtomía), la tinción y el montaje.
I. Fijación:Mecanismo que consiste en matar a la célula lo más rápidamente posible, para permitir que se
mantengan las propiedades fisiológicas y morfológicas del organismo vivo. Los fijadores solidifican elcoloide protoplasmático mediante coagulación o precipitación, convirtiéndolo en un gel insoluble. Lafijación evita que el tejido se pudra y se desintegre, produciendo puentes entre proteínas y diferentesmateriales de los tejidos. 24 horas después se procederá a la inclusión.Hay diferentes tipos de fijadores:
- Físicos: calor (húmedo o seco), frío (congelación rápida).- Químicos: según la base fijadora: alcohol metílico, dicromato de potasio, formol 10% tamponado,
etc. La muestra que va a ser fijada no debe tener más de tres milímetros de espesor, porque, delo contrario, el fijador, que penetra por difusión no actuaría por igual en todas las células de la muestra.Además, el líquido fijador debe exceder en 50:1 al de la muestra. Hay que tener en cuenta que los líquidosfijadores son volátiles y que el recipiente donde se vaya a dar la fijación debe ser cerrado. Existe untiempo adecuado de fijación, que no debe excederse; una vez concluido, se lava la muestra para quitar elexceso de fijador.Además de mantener las propiedades del objeto de estudio, los fijadores, dependiendo de los casos,pueden servir para endurecerlo o ablandarlo, o aumentar su afinidad tintorial.

II. Inclusión
Para poder cortar la pieza ésta debe tener una cierta consistencia. Para endurecerla, la incluimos en unmaterial que llegue a todas las estructuras celulares. Esta debe ser una sustancia con plasticidad como laparafina., el colodión o la gelatina. La inclusión nos permite conservar la muestra durante largo tiempo. Lomás corriente es utilizar la parafina:a) Deshidratación: proceso necesario para que, a pesar de la insolubilidad de la parafina, ésta puedaimpregnar la pieza. Consiste en hacer pasar la muestra por una gradación creciente de alcoholes.b) Aclaración: se baña la pieza tres veces durante unos 30-40 minutos en un disolvente de la parafina,
como el xilol, el benzol o el toluol.c) Impregnación en parafina: Para evaporar el disolvente de la parafina y que ésta pueda penetrar enla pieza, se la incluye en parafina fundida dos veces durante 5 horas cada vez. Para que la parafina estéfundida debemos tenerla 24 horas a 56-60°Cd) Inclusión definitiva: La pieza se mete en parafina fundida depositada en moldes, normalmenteen 'casquetes" de parafina, para que al enfriarse podamos obtener bloques de parafina que incluyen lapieza.
II Corte
La obtención de cortes para estudios microscópicos se realiza mediante unos aparatos llamados. Hay dediferentes tipos que se eligen dependiendo de la textura del material que se ha de estudiar. Paracortes de células vegetales de un órgano duro se utiliza el micrótomo de mano o de Ranvier. Para el restode órganos vegetales y para órganos animales se utiliza el micrótomo de rotación o de Monot, o el dedeslizamientoSe pueden obtener diferentes grosores de corte. Finos de 5-10 μm de grosor. Los cortes de 0.5-5 μm de
grosor se realizan a partir de material incluido en plástico y no en parafinaLos cortes obtenidos se planchan en la superficie de un baño María a 45°C con un 55 de gelatina, y sepresentan con el portaobjetosTambién se pueden realizar cortes a partir de material no incluido en parafina mediante congelación. Es
muy rápida, sólo permite cortes de 60-80 μm de grosorLos cortes una vez obtenidos se tiñen para evitar que se sequen
IV Tinción
Para teñir los cortes no deben tener parafina porque no penetraría el colorante. La parafina se eliminasumergiendo los cortes 15 minutos en xilol. Se elimina el xilol haciéndolos pasar por alcohol absoluto,alcohol de 90°, alcohol de 70° y agua destilada. Una vez eliminada la parafina, se procede a la tinción.Hay muchos tipos de tinciones:
a) según su origen-Naturales: proceden de animales o vegetales: eosina, carmín de cochinilla, etc.-Artificiales: proceden del carbón mineral. Son los que más se utilizan.
b) Según su naturaleza:-Ácidos: tiñen lo básico. Ejemplo: eosina-Básicos: tiñen lo ácido. Ejemplo: azul de metileno.-Neutros: tanto la parte aniónica como la catiónica son colorantes. Existen dos tipos de técnicas decoloración:
-Vitales o en vivo: el colorante no provoca la muerte celular. Ejemplos: azul de metileno, rojo neutro.-Supravitales: los colorantes se aplican cuando el material ya ha muerto a causa de la fijación.
Hay tinciones que son bastante rutinarias y frecuentes, como la hematoxilina eosina en animal. Elgreen-safranina en vegetal o el Gram para bacterias.Una vez teñidos los cortes, se deshidratan para quitarles el agua y que ésta no produzca cambios en larefringencia. Para ello se pasan los cortes por alcohol de gradación creciente, hasta acabar en alcoholpuro. Después se aclaran con dos baños de xilol.
V. Montaje:
Hay varios tipos de montaje:
o -Gota pendiente: ya explicado anteriormente. Se untan los bordes del cubreobjetos convaselina para conseguir adherencia y que no se seque la muestra, si se va a observar durantemás de una hora.
o Extensión o frotis: Se extiende la gota sobre el portaobjetos con ayuda de otro portaobjetos,muy rápidamente para que no se dé coagulación. El líquido extendido se fija, colorea y monta demanera normal. Se suele utilizar en el estudio de sangre y de bacterias.
o Aplastamiento: es la técnica más común. Los cortes se depositan en el portaobjetos, si no se

quiere conservar la preparación, no añadiremos medio de montaje, sólo pondremos elcubreobjetos, pero la preparación no durara más de una hora ya que su contenido líquido seevaporará por el calor emitido por el foco luminoso. Si la preparación se quiere conservar,utilizaremos un buen medio de montaje que debe cumplir una serie de propiedades comotener un buen índice de refracción, un pH neutro, y un secado rápido.
Tradicionalmente se ha venido utilizando el bálsamo de Canadá, que actualmente ha sido sustituido porresinas sintéticas, como el Eulcitt.Gracias al medio de montaje, la preparación se conservará durante mucho tiempo. Una vez añadida la
gota de medio de montaje. Se cubre con el cubreobjetos y se espera a su secado.
VI Etiquetaje:Las preparaciones deben etiquetarse, si es que se quieren conservar o se van a utilizar posteriormente. En
la etiqueta se pone el nombre del material, la técnica utilizada y la fecha de realización.

2
1. Vaso de precipitado.2. Vidrio de reloj.3. Espátula.4. Abrazadera.5. Gradilla para tubos de ensayo.6. Probeta.7. Bureta.8. Pie o soporte de hierro.9. Mechero Bunsen.10. Tubo de ensayo.11. Trípode.12. Mortero.13. Matraz de fondo curvo.14. Embudo de filtro.15. Matraz cónico o Erlenmeyer.16. Varilla de agitación.17. Rejilla.18. Matraz de fondo piano.19. Termómetro.20. Embudo de separación.21. Pipeta.22. Capsula de porcelana.23. Escobilla.24. Cuentagotas.

Jose Seijo Ramil PRÁCTICAS 3
Determinación y observación de principios inmediatos en alimentosAzúcares reductores (tinción con reactivo de Fehling),Almidón (tinción con Lugol)Lípidos (tinción con Sudán IIProteínas (reacción de Biuret)
Objetivos:∙ Identificación de principios inmediatos mediante ensayos simples de laboratorio
∙ Caracterización dos distintos tipos de moléculas en función de sus propiedades químicas
∙ Valorar a importancia das técnicas de identificación para detectar fraudes alimenticios
1.1 a) Identificación de azúcares reductores:
Reacción de FehlingGeneralmente las pruebas de un análisis cualitativo se basan en la obtención de compuestos coloreados queponen de manifiesto la presencia de determinadas sustancias cuando reaccionan con ciertos reactivos.El reactivo utilizado con el fin de poner de manifiesto la capacidad reductora de un azúcar sedenomina licor de Fehling, y consiste en una mezcla de dos reactivos: el Fehling A (sulfato cúprico) decolor azul y el Fehling B (tartrato sódico‐potásico) incoloro. Se procede de la siguiente manera
Preparación del licor de FehlingLICOR DE FHELING.
Solución A Solución B
CuS0 4 -------- 3,5 g Tartrato sódico potásico---- 1,8 gAgua hasta 50 m1 KOH-------------------------- 7,7 g
Agua hasta 50 ml
Se mezclan volúmenes iguales de A y B._________________________________________________________________
Preparar una disolución de NaOH al 10 %.Disponemos de lentejas de NaOHPreparar una disolución de HCl al 10 %.Disponemos de ácido clorhídrico al 37% y d = 1,18 g/cc
___________________________________________________________________________________
Material:Tubos de ensayoVaso de precipitados de 250ccPinzasTrípodeRejilla de amiantoDos pipetasBalanzaEspátula
Productos químicos:Glucosa, sacarosa, almidón.CuS04
Tartrato sódico potásicoKOHLugol (6 g de yoduro de potasio en 100 cm3 de agua más2 g de yodo).NaOH al 20%.HCl al 10%.Papel indicador

Jose Seijo Ramil PRÁCTICAS 4
Protocolo① Preparamos, para utilizar todos, una disolución de glucosa, otra de sacarosa y una tercera de almidón. Paraello disolvemos (3‐5 g de cada sustancia en 100 cc de agua)
② Tomamos 2 cc de cada una de las sustancias a investigar (glucosa, sacarosa y almidón) y las depositamos entres tubos de ensayo, que marcamos y colocamos en la parte inferior de la gradilla. Las utilizaremos paracomprobar su carácter reductor frente al licor de Fehling
③ Tomamos 2 cc de cada una de las sustancias a investigar (glucosa, sacarosa y almidón) y las depositamos entres tubos de ensayo, que marcamos y colocamos en la parte superior de la gradilla
④ A continuación se agrega 2 cc de líquido Fehling a cada una de las disoluciones de glucosa, sacarosa y almidón,de la parte inferior de la gradilla. Las disoluciones adoptarán entonces un color azul intenso.
⑤ Se calientan suavemente los tubos de ensayo en la llama del mechero (procura que no entre en ebulliciónpues el líquido saldría proyectado y podría quemar al que lo maneja y acompañantes).
a. Si el azúcar es reductor al cabo de unos instantes aparecerá un precipitado de óxido de cobre (I) de colorrojo ladrillo.
b. Los azúcares no reductores como la sacarosa y los polisacáridos (como el almidón) no dan positiva lareacción de Fehling.
Fundamento• En ausencia de sustancias reductoras, el hidróxido de cobre (I) de color azul se transforma en óxido de
cobre (II) de color negro• En presencia de un agente reductor como es el caso de un monosacárido o disacárido que tiene un
grupo carbonilo. El hidróxido de cobre (II) pasa a hidróxido de cobre (I).El hidróxido de cobre (I), pierdeuna molécula de agua y origina el óxido de cobre (I) de color rojo ladrillo
⑥: A los dos tubos de ensayo que contienen sustancias que no han reducido el reactivo de Fehling, añadir 1 cc deHCl al 10% para efectuar la hidrólisis de los enlaces glucosídicos
⑦: Calentar durante 15 minutos al baño maría
⑧ Añadir hidróxido de sodio al 20% hasta que el medio se vuelva básico (se comprueba mediante el papelindicador)
⑨ Repetir la prueba Fehling
1.1 b) Almidón (tinción con Lugol)
⑩ Con los tres tubos de ensayo que nos quedan en la parte superior de la gradilla con glucosa, sacarosa yalmidón, vamos a comprobar si forman un complejo azul oscuro con el yodo
Para ello añadimos unas gotas de Lugol a los tres tubos. El que contiene almidón se tiñe con el yodo porque lasmoléculas de yodo quedan atrapadas en la hélice de glucosas
Entregar las respuestas a las siguientes cuestiones:1) ¿En qué se basa el empleo del licor de Fehling para caracterizar monosacáridos?2) ¿Cómo nos permite o reactivo de Fehling distinguir entre los diferentes hidratos de carbono?3) ¿Podrías mediante la reacción de Fehling distinguir entre almidón y celulosa?4) Si dispusieras de dos soluciones en dos tubos de ensayo, uno con glucosa y otro con sacarosa, pero sin
etiquetar, ¿cómo determinarías en el laboratorio la identidad de cada tubo?5) ¿Cómo demostrarías no laboratorio el poder reductor dos glúcidos? Indica el método y el material.6) ¿Qué tipo de proceso tiene lugar en la tinción del almidón con Lugol?, ¿es una reacción química? ¿Podrías
explicarlo?7) ¿Cómo podrías investigar si una sustancia desconocida es una proteína?8) ¿Por qué la reacción de Biuret es positiva con las proteínas y no con los aminoácidos?

Jose Seijo Ramil PRÁCTICAS 5
1.2: Reconocimiento de grasasTubos de ensayoAguaAceiteSudán III
El Sudán III es un colorante específico de grasas.①Para prepararlo disolver un poco de Sudán II en polvo en alcohol de 70º, hasta que la solución quedesaturada, luego se filtra
② Coloca 2 ml de aceite en un tubo de ensayo y añade 2 ml de agua. Apreciarás que ambos líquidos no semezclan, se producen dos fases: una superior de aceite y otra inferior de agua
③Añade unas gotas de Sudan III, agita y espera unos minutos. Observarás que la fase superior, el aceite,aparece teñida de rojo mientras que la inferior permanece incolora
____________________________________________________________________________________________1.3: Reconocimiento de Proteínas Reacción de Biuret
Tubos de ensayoNaOH al 10%Cu SO4
①Disolución de clara de huevo (proporción 1 a 6)
② Coloca 2 ml de la disolución proteica en un tubo de ensayo
③ Añade 2 ml de una disolución de NaOH al 10% y agita.
④ Añade 4 0 5 gotas de una disolución de Cu SO4
Obtendrás una coloración añil indicativa de la presencia de proteínas
El resultado se debe a una reacción típica de los enlaces peptídicos, en la cual los átomos de Cu se unen alos grupos amino, lo que provoca la coloración rosa‐violácea
____________________________________________________________________________________________

Jose Seijo Ramil PRÁCTICAS 6
Estudio de la mitosis en la raíz de cebolla: Tinción con fucsina ácida y/o orceínaObjetivos:∙ Observación de cromosomas y estudio de las etapas de la mitosis
∙ Adquirir experiencia en la utilización de técnicas de procesamiento de tejidos para su estudio con microscopía óptica
∙ Conocer o manejo do microscopio óptico
∙ Conocer y manejar as unidades de medida das células
∙ Valorar a contribución decisiva da microscopía al estudio y conocimiento de la célula
ProcedimientoPara observar la mitosis en células vegetales se utilizan tejidos meristemáticos, en los que las células semultiplican constantemente, por lo que será fácil "sorprender" algunas células en distintas fasesmitóticas.Observación de la mitosis en el ápice de la raíz cebolla o de otros vegetales con bulbo.Como material biológico, en la práctica se describe el procedimiento a partir de raicillas de cebolla, pero
también se podrían utilizar las raíces obtenidas a partir de otros bulbos (ajos, puerros, jacintos, tulipanes,etcétera).Para conseguir raíces en crecimiento, se coloca el bulbo de cebolla (al que previamente se habrán eliminadolas raíces secas) sobre un vaso de precipitados (si es necesario se sujeta mediante unos palillos). Se llenael vaso con agua hasta que toque superficialmente la zona donde van a crecer las raicillas. Al cabo de 2 o 3días empezarán a crecer las raíces.
① Elige una raíz joven (de unos 2 cm.), lávala con agua y corta la punta a 5 mm del extremo.
② Coloca la punta de la raíz en un vidrio de reloj. Para ablandar los tejidos e iniciar la tinción, se cubre la raízcon orceína acética y clorhídrico 1 N en la proporción de 9:1, es decir, 9 gotas de orceína por cada gota de HCl.(Orceína A)
③ La hidrólisis debe hacerse en caliente, a unos 60 °C. Para ello, calienta el vidrio de reloj a la llama delmechero, justo hasta que empiecen a desprenderse tenues vapores, teniendo mucho cuidado de que nohierva (al retirar el vidrio no debe quemar la mano).Retíralo para que se enfríe durante 8 minutos y repite esta operación 3 veces, añadiendo más orceína si hicierafalta, de modo que el líquido cubra en todo momento la punta de la raíz.¾5. Saca la raicilla con la agujaenmangada y colócala sobre el portaobjetos.
④Con la cuchilla corta el ápice a unos 2 mm. El resto se descarta ya que las células meristemáticas seencuentran en el extremo, bajo la cofia.
⑤ Añade una gota de ácido acético al 45% (orceína B) para terminar de ablandar los tejidos.
⑥ Limpia con papel de filtro los posibles residuos del colorante.
●Microscopio ●Cubreobjetos ●Aguja enmangada●Vidrio de reloj ●Cubreobjetos ●Cebolla●Mechero ●Pinzas●Portaobjetos ●Vaso de precipitados●Portaobjetos ●Cuchilla o escalpelo ●Ácido clorhídrico 1 N
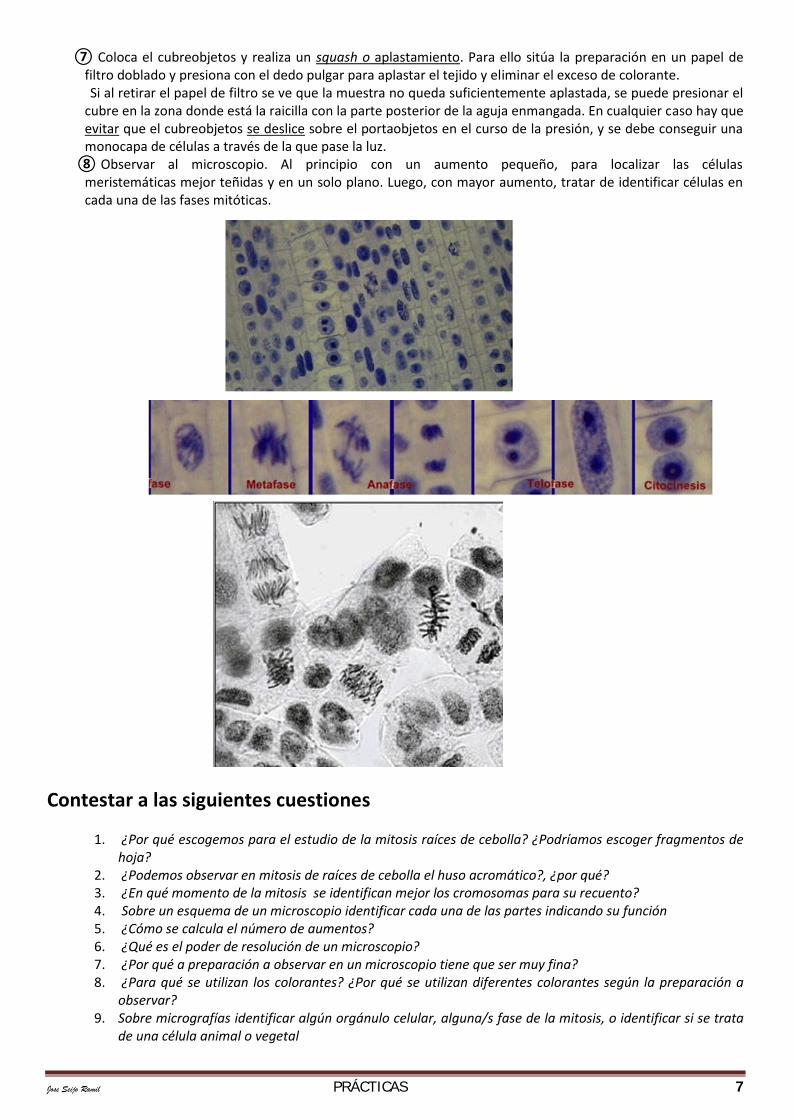
Jose Seijo Ramil PRÁCTICAS 7
⑦ Coloca el cubreobjetos y realiza un squash o aplastamiento. Para ello sitúa la preparación en un papel defiltro doblado y presiona con el dedo pulgar para aplastar el tejido y eliminar el exceso de colorante.Si al retirar el papel de filtro se ve que la muestra no queda suficientemente aplastada, se puede presionar el
cubre en la zona donde está la raicilla con la parte posterior de la aguja enmangada. En cualquier caso hay queevitar que el cubreobjetos se deslice sobre el portaobjetos en el curso de la presión, y se debe conseguir unamonocapa de células a través de la que pase la luz.
⑧ Observar al microscopio. Al principio con un aumento pequeño, para localizar las célulasmeristemáticas mejor teñidas y en un solo plano. Luego, con mayor aumento, tratar de identificar células encada una de las fases mitóticas.
Contestar a las siguientes cuestiones
1. ¿Por qué escogemos para el estudio de la mitosis raíces de cebolla? ¿Podríamos escoger fragmentos dehoja?
2. ¿Podemos observar en mitosis de raíces de cebolla el huso acromático?, ¿por qué?3. ¿En qué momento de la mitosis se identifican mejor los cromosomas para su recuento?4. Sobre un esquema de un microscopio identificar cada una de las partes indicando su función5. ¿Cómo se calcula el número de aumentos?6. ¿Qué es el poder de resolución de un microscopio?7. ¿Por qué a preparación a observar en un microscopio tiene que ser muy fina?8. ¿Para qué se utilizan los colorantes? ¿Por qué se utilizan diferentes colorantes según la preparación a
observar?9. Sobre micrografías identificar algún orgánulo celular, alguna/s fase de la mitosis, o identificar si se trata
de una célula animal o vegetal

Jose Seijo Ramil PRÁCTICAS 8
OBSERVACIÓN DE BACTERIAS (TINCIÓN GRAM)Objetivos:
• Utilización de técnicas sencillas para la observación de microorganismos
• Conocer el manejo del microscopio óptico para la observación de bacterias (importancia del objetivo deinmersión)
• Conocer y manejar las unidades de medida de las células y establecer comparaciones entre tamaños deprocariotas y de eucariotas
• Reconocer los distintos modelos de la pared bacteriana
MATERIALES• Microscopio• Aceite de inmersión• Cubeta de tinciones• Frasco lavador• Safranina al 0,5%• Cristal violeta al 1%• Solución diluida de yodo (lugol)• Alcohol‐acetona 1:1 o alcohol 95
TÉCNICA①Preparar los frotis bacterianos.
②Teñir con cristal violeta 1 min.
③Lavar con abundante agua el exceso de colorante.
④ Cubrir con Lugol 1 min.
⑤ Lavar con agua el exceso de Lugol.
⑥Decolorar con alcohol‐acetona o simplemente con alcohol hasta que la preparación deje de perdercolor (30 seg.)
⑦Lavar con abundante agua para eliminar el resto de disolvente.
⑧ Teñir con safranina 1 min.
⑨Lavar con agua para eliminar el colorante de contraste.
⑩ Secar la preparación.
⑪ Examinar al microscopio fijándose sobre todo en el color de cada preparación.
FUNDAMENTOEsta tinción fue desarrollada empíricamente por Christian Gram en 1884.A pesar del tiempo transcurrido, la tinción apenas se ha modificado y es uno de los primeros pasos que se
realiza para cualquier identificación bacteriana.

Jose Seijo Ramil PRÁCTICAS 9
La técnica es capaz de diferenciar dos grandes grupos de eubacterias: Gram positivas (G+) y Gramnegativas (G‐).La tinción de Gram requiere cuatro soluciones:
1: Primer colorante: E s un colorante básico que en contacto con las células cargadasnegativamente, reacciona con ellas coloreándolas. El más utilizado es el cristal violeta.
2: Solución mordiente: Fija las tinciones y aumenta la afinidad entre el colorante y las células. Los mordientesempleados suelen ser sales metálicas, ácidos o bases, como por ejemplo el Lugol.
3: Agente decolorante: es un disolvente orgánico, por ejemplo alcohol‐acetona (1:1).
4: Colorante de contraste: Es un colorante básico de distinto color que el primer colorante, como porejemplo la safranina o la fucsina.
_______________________________________________________________________________________________Los dos grupos bacterianos que distingue esta técnica difieren en el color con el que finalmenteadquieren. Las bacterias Gram + se teñirán de azul por el cristal violeta y no perderán esta coloracióndurante los pasos sucesivos. Las bacterias Gram‐ perderán la coloración inicial del cristal violeta en lossiguientes pasos y se teñirán de rosa debido a la safranina.
______________________________________________________________________________________________
La diferencia está determinada por la composición de su envoltura celular. Las Gram + poseen una malla depeptidoglicano en su parte más externa, mientras que las Gram‐, recubriendo una fina capa depeptidoglicano, presentan una membrana externa que envuelve toda la célula.
______________________________________________________________________________________________Una de las precauciones al realizar esta tinción es la de trabajar con cultivos en fase exponencial. De locontrario se pueden obtener resultados falsos. P.ej. las bacterias Gram + en fase estacionaria puedenaparecer como Gram‐.

Jose Seijo Ramil PRÁCTICAS 10
Extracción de ADNAprender técnicas sencillas de extracción de moléculas de ADN de un tejido
∙ Identificar a estructura fibrilar do ADN. Existen en: http://www.dnaftb.org/dnaftb/29/concept/index.html
Extracción do DNA de los guisantesMATERIAL
• Guisantes• Detergente lavavajillas• Sal (NaCl)• Agua destilada• Líquido para limpiar lentillas o bien un ablandador de carne• Alcohol de 96• Agua destilada• Probeta de 250 cc• Batidora• Microscopio
MÉTODO① Mezcla en un frasco de batidora 400 g de guisantes con 200 ml de agua destilada y añade 3 cucharadas dedetergente de lavavajillas y una de sal.
② Bate bien y filtra el líquido obtenido (sirve un colador metálico de los que tiene en casa)
③ Pasarlo seguidamente a una probeta.
④ Añade un chorro de líquido para limpiar lentillas y mezcla bien todo.
⑤ Añade 50 ml de alcohol muy cuidadosamente, haciéndolo resbalar por las paredes del vaso para que no semezcle con el filtrado y forme una capa sobre él separada por densidad
⑥Deja reposar durante unos minutos. Se verá que va subiendo y se va mezclando con el alcohol una marañade hilos blancos que corresponde al ADN de los guisantes.
⑦Con un objetivo de máximo aumento observa un fragmento de esos hilos. El DNA debe verse como unoshilos delgados y separados
_______________________________________________________________La solución de lavavajillas y la sal más la acción de la batidora es capaz de romper la pared celular y lasmembranas plasmática y nuclear. El líquido limpialentillas contribuye a eliminar las proteínas que puedancontaminar al ADN. El alcohol separa el ADN que es soluble en agua.
_____________________________________________________________________________________________
Contestar a las siguientes cuestiones:1. ¿Por qué al añadir sal a un homogeneizado se rompen las membranas celulares?2. ¿Se no se eliminan las proteínas, se puede ver también a estructura fibrilar del ADN?, ¿por qué?
______________________________________________________________

Jose Seijo Ramil PRÁCTICAS 11
Observación de vídeos animados sobre el proceso de síntesis proteicaObjetivos:• Caracterizar los distintos tipos de ARN y diferenciarlos del ADN• Resaltar la importancia de la complementariedad de bases y del código genético en la síntesis proteica• Representar e l flujo de información desde una determinada secuencia de bases del ADN a secuencia de
aminoácidos de una proteína• Facilitar el estudio y la comprensión teórica del proceso de traducción• Ver ejemplos de los distintos tipos de mutaciones que afectan a la secuencia de bases del ADN y demostrar
los efectos sobre la proteína traducida
En Internet
http://www.biostudio.com/case_freeman_protein_synthesis.html
http://www.dnaftb.org/dnaftb/22/concept/index.html
Contestar a las siguientes cuestiones:
• Con ayuda de una tabla con el código genético, establecer los pasos que conducirán a la síntesis de undeterminado péptido. ¿Qué efecto tendría la inserción, la sustitución y una deleción de un nucleótido en lacadena del ADN que se transcribe?
• Con ayuda de una tabla con el código genético, escribir todas las secuencias posibles de ADN que codifiquenpara a síntesis de un determinado polipéptido. ¿Por qué pueden existir varias secuencias?
• ¿El número de ARN transferentes es igual, mayor o menor que 20? Justificar la respuesta.
• Enumera los compuestos y estructuras que intervienen en la síntesis proteica, indicando el papel de cadauno en el proceso. ¿Qué se forma al final?

Jose Seijo Ramil PRÁCTICAS 12
Observación de modelos tridimensionales de azúcares, lípidos, proteínas y ácidosnucleicos
Objetivos:• Facilitar la comprensión de la configuración espacial de éstas moléculas
• Agilizar la comprensión teórica del desarrollo de las macromoléculas
• Reconocer y diferenciar representaciones sencillas (tridimensionales y planas) de las diferentes biomoléculas
• Explicar la relación estructura‐función de las biomoléculas. Es de destacar que la funcionalidad de lasproteínas y de los ácidos nucleicos depende de su estructura tridimensional
• Caracterizar la estructura secundaria del ADN
• Utilizar el modelo tridimensional do ADN para explicar su replicación
Direcciones de Internet con imágenes de estas moléculas
http://www.bioxeo.com http://www.ehu.es/biomoleculas/AN/an4.htm
______________________________________________________________
Conteste a las siguientes cuestiones:
• ¿Por qué o almidón, a celulosa y el glucógeno son polisacáridos diferentes se todos están formados pormoléculas de D-glucopiranosa?
• ¿Qué diferencias existen a nivel de la estructura molecular entre un lípido saponificable y otro que no lo es?¿Qué diferencias tridimensionales existen entre los ácidos grasos saturados e insaturados con el mismonúmero de átomos de carbono?
• ¿Qué diferencias hay entre as alfa-hélices y las láminas plegadas de la estructura secundaria de lasproteínas? ¿A qué se deben las distintas estructuras de las proteínas?
• ¿Hacia dónde quedan dirigidos los puentes de hidrógeno entre las bases nitrogenadas en la estructurasecundaria del ADN? ¿Qué moléculas forman el esqueleto del ADN y cuáles las cadenas laterales?
• ¿Qué ventajas supone para la uniformidad y estabilidad de las dos cadenas del ADN que siempre se unanuna base púrica con otra pirimidínica? ¿Causaría algún problema a unión entre dos bases púricas o entre dospirimidínicas?


















