
REPARTO PREFERENCIAL DE ÁCIDOS … · Barridos de formulación con ácidos carboxílicos y sus...
205
REPÚBLICA BOLIVARIANA DE VENEZUELA UNIVERSIDAD DEL ZULIA FACULTAD EXPERIEMNTAL DE CIENCIAS DIVISIÓN DE ESTUDIOS PARA GRADUADOS POSTGRADO EN QUÍMICA: NIVEL DOCTORADO REPARTO PREFERENCIAL DE ÁCIDOS CARBOXÍLICOS EN SISTEMAS SURFACTANTE/AGUA/ACEITE POR HPLC Tesis Doctoral presentada para optar al título de Doctora en Química Autor: Bélgica Balbina Bravo Tovar Maracaibo, Diciembre 2004
Transcript of REPARTO PREFERENCIAL DE ÁCIDOS … · Barridos de formulación con ácidos carboxílicos y sus...

REPÚBLICA BOLIVARIANA DE VENEZUELA UNIVERSIDAD DEL ZULIA
FACULTAD EXPERIEMNTAL DE CIENCIAS DIVISIÓN DE ESTUDIOS PARA GRADUADOS
POSTGRADO EN QUÍMICA: NIVEL DOCTORADO
REPARTO PREFERENCIAL DE ÁCIDOS CARBOXÍLICOS EN SISTEMAS
SURFACTANTE/AGUA/ACEITE POR HPLC
Tesis Doctoral presentada para optar al título de Doctora en Química
Autor: Bélgica Balbina Bravo Tovar
Maracaibo, Diciembre 2004

A G R A D E C I M I E N T O S … … … . .
Especialmente a los Profesores Nelson Márquez y Jean Louis
Salager, quienes dirigieron este trabajo.
Al honorable jurado evaluador del trabajo, Profesores: Dr.
Nelson Márquez (LPS-LUZ-Venezuela), Dr. Jean Louis Salager (FIRP-
ULA-Venezuela), Dr. Jean Lachaise (LFC-GSD-UPPA-Francia), Dr.
Fredy Ysambertt (LPS-LUZ-Venezuela), Dr. Alexander Moronta
(CESUC-LUZ-Venezuela), Dra. Dinorah Ávila (LPN-LUZ-Venezuela),
Dra. Marinela Colina (LA-LUZ-Venezuela).
Esta tesis se realizó dentro del cuadro de los siguientes
programas y proyectos:
1. Programa de Cooperación Postgraduados (PCP) Francia-
Venezuela
2. Proyecto Agenda Petróleo FONACIT Nº 97003719
3. Proyecto Apoyo directo a grupos ADG-FONACIT G-97000828
4. Proyecto CONDES-LUZ Nº CC-0340-04
5. Proyecto CONDES-LUZ Nº CC-0504-02
6. Proyecto CONDES-LUZ Nº CC-0505-02
A todas las personas de los laboratorios LPS, FIRP, LFC, que de
una u otra forma contribuyeron a este trabajo.
A LUZ, FONACIT, FUNDADESARROLLO-LUZ y CONDES por el
apoyo financiero brindado para el desarrollo de esta tesis.

ii
C O N T E N I D O

iii
C o n t e n i d o ...................................................................................................... ii L i s t a d e a b r e v i a t u r a s .................................................. vi L i s t a d e f i g u r a s ........................................................................viii R e s u m e n ............................................................................................................ xi A b s t r a c t ......................................................................................................... xiv
I n t r o d u c c i ó n .......................................................................................... 1
Referencias bibliográficas ..........................................................................................................................5
C a p i t u l o I .................................................................................................... 8
Generalidades de los ácidos carboxílicos...............................................................................................9 Referencias bibliográficas ........................................................................................................................18
C a p i t u l o I I .............................................................................................. 19
Generalidades de la cromatografía líquida de alta resolución (HPLC).......................................20 Aspectos fundamentales ............................................................................................. 24 Termodinámica del proceso cromatográfico.............................................................. 27
Referencias bibliográficas ........................................................................................................................32
C a p i t u l o I I I ......................................................................................... 33
Sistemas automatizados de analisis. ......................................................................................................34 Instrumentación .......................................................................................................... 36
Derivatización de ácidos carboxílicos...................................................................................................41 Referencias bibliográficas ........................................................................................................................44
C a p i t u l o I V ............................................................................................ 46
Comportamiento de fase y fraccionamiento........................................................................................47 Surfactantes y sus propiedades................................................................................................................47 Sistemas surfactante-agua-aceite (SOW) .............................................................................................53 B a l a n c e H i d r o f í l i c o - L i p o f í l i c o ( H L B ) . ...................................................................56 C o m p o r t a m i e n t o d e f a s e d e s i s t e m a s s u r f a c t a n t e - a g u a - a c e i t e .........58 R e l a c i ó n R d e W i n s o r ................................................................................................................60 F o r m u l a c i ó n y v a r i a b l e s d e f o r m u l a c i ó n ................................................................66 T e o r í a d e c o r r e l a c i o n e s n u m é r i c a s p a r a l a f o r m u l a c i ó n ó p t i m a ....70 T e o r í a d e r e p a r t o d e s u r f a c t a n t e s e n s i s t e m a s a g u a / a c e i t e .................76 Referencias bibliográficas ........................................................................................................................82
C a p i t u l o V ................................................................................................ 84
Antecedentes. ...............................................................................................................................................85 Referencias bibliográficas ........................................................................................................................93
C a p i t u l o V I ............................................................................................ 96 Desarrollo de métodos de análisis de ácidos carboxílicos grasos (FAs) por HPLC................97 Sección experimental .................................................................................................................................97 Materiales ......................................................................................................................................................97 Reactivos. ......................................................................................................................................................98

iv
Procedimiento Experimental....................................................................................................................98 Análisis de ácidos carboxílicos por HPLC-fase reversa. .................................................................98 Efecto de la temperatura en la retención de los ácidos carboxílicos y surfactantes no-iónicos en la separación por HPLC-fase reversa. ..............................................................................98 Efecto de la polaridad de la fase móvil en la retención de los ácidos carboxílicos por HPLC-fase reversa......................................................................................................................................99 Resultados y discusión.............................................................................................................................100
Desarrollo de métodos de análisis de ácidos carboxílicos grasos por HPLC........... 100 Efecto de la temperatura en la retención de compuestos por HPLC. ....................... 104 Efecto de un solvente polar en la fase móvil en la retención de los ácidos grasos. . 112
Conclusiones...............................................................................................................................................116 Referencias bibliográficas ......................................................................................................................117
C a p i t u l o V I I ...................................................................................... 119
Desarrollo de un sistema en linea para la derivatización y análisis de ácidos carboxílicos grasos (FAs) por HPLC...........................................................................................................................120 Sección experimental ...............................................................................................................................120 Materiales ....................................................................................................................................................121 Reactivos. ....................................................................................................................................................121 Procedimiento Experimental..................................................................................................................122 Desarrollo de un sistema de análisis continuo de ácidos carboxílicos. .....................................122 Resultados y discusión.............................................................................................................................123
Condiciones de reacción ....................................................................................... 125 Desarrollo del sistema en línea ............................................................................. 126 Condiciones de análisis por HPLC ....................................................................... 127 Reactivo derivatizante........................................................................................... 127
Conclusiones...............................................................................................................................................131 Referencias bibliográficas ......................................................................................................................132
C a p i t u l o V I I I .................................................................................. 133
Comportamiento de fase en la formulación con ácidos carboxílicos grasos (FAS) ..............134 Sección experimental ...............................................................................................................................135 Materiales ....................................................................................................................................................135 Reactivos. ....................................................................................................................................................135 Procedimiento Experimental..................................................................................................................135 Barridos de formulación con ácidos carboxílicos y sus mezclas. ...............................................135 Evaluación del tipo de aceite en el sistema surfactante-agua-aceite. .........................................137 Evaluación del tipo y concentración del co-surfactante en el sistema óptimo. .......................137 Resultados y discusión.............................................................................................................................138
Efecto del tipo y concentración de ácido carboxílico en el comportamiento de fase.................................................................................................................................. 141 Efecto del tipo y concentración del co-surfactante (alcohol) en el comportamiento de fase ........................................................................................................................... 154 Longitud de cadena del alcano de la fase aceite (ACN)........................................... 160
Conclusiones...............................................................................................................................................163 Referencias bibliográficas ......................................................................................................................164 C a p i t u l o I X ...............................................................................................................165

v
Influencia de la formulación en el reparto de los ácidos carboxílicos en sistemas ácido/agua/aceite .......................................................................................................................................166 Sección experimental ...............................................................................................................................166 Equipos.........................................................................................................................................................166 Materiales ....................................................................................................................................................167 Reactivos. ....................................................................................................................................................167 Procedimiento Experimental..................................................................................................................167 Evaluación del reparto de los surfactantes ácidos carboxílicos en sistemas de Winsor a formulación óptima. .................................................................................................................................167 Resultados y discusión.............................................................................................................................169
Proceso termodinámico en el reparto de FAs en sistemas microemulsión/agua/aceite.................................................................................................................................. 179
Conclusiones...............................................................................................................................................181 Referencias bibliográficas ......................................................................................................................182 Conclusiones generales ...........................................................................................................................183 Recomendaciones generales ..................................................................................................................184 D i s e m i n a c i ó n d e l c o n o c i m i e n t o ................185

vi
L I S T A D E A B R E V I A T U R A S

vii
FAS: Ácidos carboxilicos grasos
HPLC: Cromatografía líquida de alta resolución
FR: Fase reversa
UV-Vis: Ultravioleta visible
FIA: Anáisis de inyección en flujo
RP: Reverse Phase
min.: minutos
s.: Segundos
SCFA: Análisis en flujo continuo segmentado

viii
L I S T A D E F I G U R A S

ix
Figura I.1. Disociación de alcoholes y ácidos carboxílicos y su comportamiento energético. .................................................................................................... 10
Figura I.2. Reacción de saponificación......................................................................... 11 Figura I.3. Fraccionamiento de ácidos grasos por el proceso de destilación al
vacío. ......................................................................................................... 15 Figura I.4. Fabricación de jabón por el proceso continuo (Procter & Gamble) ...... 16 Figura II.1. Representación esquemática del proceso de adsorción en
cromatografía ........................................................................................... 21 Figura II.2. Representación esquemática del proceso de partición en
cromatografía ........................................................................................... 22 Figura II.3. Representación esquemática del proceso de intercambio iónico en
cromatografía ........................................................................................... 22 Figura II.4. Representación esquemática del proceso de exclusión por tamaño en cromatografía .......................................................................................... 23 Figura II.5. Proceso de separación en cromatografía líquida. ................................. 26 Figura III.1. Principio del análisis de inyección en flujo............................................ 38 Figura IV.1. Representación clásica de una molécula de surfactante ...................... 48 Figura IV.2. Las propiedades fundamentales de los surfactantes Adsorción y
Asociación (A), dan origen a las micelas (B). ........................................ 50 Figura IV.3. Variación de la tensión superficial versus la concentración de
surfactante para determinar la Concentración Micelar Crítica. ........ 51 Figura IV.4. Representación de un diagrama ternario .............................................. 59 Figura IV.5. Energías de interacción que intervienen en el concepto R de Winsor 61 Figura IV.6. Representación de un Diagrama Ternario Winsor I............................ 62 Figura IV.7. Representación de un Diagrama Ternario Winsor II. ......................... 63 Figura IV.8. Representación de un Diagrama Ternario Winsor III......................... 65 Figura IV.9. Procedimientos experimentales para la determinación de la
formulación óptima ................................................................................. 72 Figura IV.10. Diagrama hipotético del efecto de la interacción ion-dipolo de los
grupos carboxílicos en la distancia intermolecular en una interfase aire/agua. ................................................................................................. 75
Figura IV.11. Modelo equilibrio / disociación. ............................................................ 78 Figura VI.1. Espectros de absorción UV-Vis de los ácidos carboxílicos grasos
estudiados. .............................................................................................. 100 Figura VI.2. Separación por HPLC de la mezcla de ácidos C12, C14, C16 y C18.102 Figura VI.3. Separación de la mezcla de ácidos C8, C10, C12, C14, C16 y C18. .. 103 Figura VI.4. Diagrama de van’t Hoff para los ácidos carboxílicos grasos estudiados.
................................................................................................................. 106 Figura VI.5. Dependencia del factor de retención con la temperatura. ................. 107 Figura VI.6: Cromatograma HPLC del surfactante NP20 a diferentes temperaturas
................................................................................................................. 108 Figura VI.7. Diagrama de van't Hoff para los primeros doce etoxímeros del
surfactante nonilfenol etoxilado (NP20). ............................................. 109 Figura VI.8. Parámetros termodinámicos, en el proceso cromatográfico de los FAs
y el surfactante NP20............................................................................. 111 Figura VI.9. Dependencia del Ln k' con el N° de átomos de carbono de los ácidos
carboxílicos............................................................................................. 113

x
Figura VI.10. Variación del ln k' en función del contenido de acetonitrilo en la fase móvil para los diferentes ácidos estudiados. ...................................... 115
Figura VII.1. Diagrama esquemático del sistema HPLC-derivatización en línea de ácidos grasos.......................................................................................... 124
Figura VII.2. Reacciones químicas generales de los ácidos grasos de cadena larga con el reactivo derivatizante.. .............................................................. 125
Figura VII.3. Variación de la intensidad del pico con la longitud de la cadena del ácido graso. ............................................................................................ 128
Figura VII.4. Cromatogramas de los derivados de ácidos grasos. ........................... 130 Figura VIII.1. Barrido de hidróxido de sodio para el ácido láurico........................ 142 Figura VIII.2. Mapa del sistema Winsor obtenido para el ácido láurico. .............. 145 Figura VIII.3. Mapa del sistema Winsor obtenido para el ácido mirístico ........... 146 Figura VIII.4. Mapa del sistema Winsor obtenido para el ácido palmitito ........... 147 Figura VIII.5. Variación del volumen de fase media con la concentración de ácido
graso en el sistema ácido/agua/aceite................................................. 149 Figura VIII.6. Variación del volumen de fase de una mezcla mirístico/palmítico a
diferentes proporciones ...................................................................... 150 Figura VIII.7. Variación del volumen de fase de una mezcla láurico/mirístico a
diferentes proporciones ...................................................................... 151 Figura VIII.8. Variación del volumen de fase media con la concentración de ácido
en una mezcla mirístico/láurico en el sistema ácido/agua/aceite .... 152 Figura VIII.9. Variación del pH de la fase acuosa con la longitud de la cadena
alquílica del ácido graso...................................................................... 153 Figura VIII.10. Concentración de n-butanol necesaria para obtener sistemas
trifásicos en sistemas ácido/agua/aceite........................................... 155 Figura VIII.11. Efecto del 2-butanol en la formulación del sistema
mirístico/palmítico (25/75)................................................................ 155 Figura VIII.12. Efecto del 2-butanol en la formulación del sistema
mirístico/palmítico (50/50)................................................................ 156 Figura VIII.13. Efecto del n-butanol sobre la concentración del ácido en la fase
oleica en los sistemas a formulación óptima. .................................. 159 Figura VIII.14. Variación del volumen de fase media con la concentración de ácido
en el sistema ácido/agua/aceite ......................................................... 161 Figura VIII.15 Variación del volumen de fase del sistema con la concentración del
ácido mirístico, empleando tolueno como fase oleica....................... 162 Figure IX.1. Curva de calibración para los ácidos obtenida por HPLC. ............... 170 Figura IX.2 Cromatograma del ácido láurico a diferente concentración. ............ 171 Figura IX.3 Coeficiente de reparto del ácido láurico .............................................. 172 Figura IX.4. Coeficiente de reparto del ácido mirístico. .......................................... 173 Figura IX.5. Concentración de los diferentes ácidos en la fase oleica (n-heptano) 174 Figura IX.6. Coeficiente de reparto del C14 en diferentes fases oleicas. ................ 176 Figura IX.7. Coeficiente de reparto de los diferentes ácidos estudiados ................ 176 Figura IX.8. Efecto de alcoholes en el coeficiente de reparto. ................................. 178 Figura IX.9 Variación de la energía libre de transferencia del ácido.................... 180

xi
R E S U M E N

xii
Bélgica Balbina Bravo Tovar. Estudio del reparto preferencial de ácidos carboxílicos en sistemas surfactante/agua/aceite. Laboratorio de Petroquímica y Surfactantes, Departamento de Química, Facultad de Ciencias, La Universidad del Zulia, Maracaibo, Zulia-Venezuela, E-mail: [email protected]
En los últimos años se ha generado una intensa actividad de investigación, para desarrollar
procesos de recuperación de petróleo. El empleo de soluciones alcalinas produce bajas
tensiones interfaciales en ciertos sistemas crudo-agua, debido a las interacciones de los
agentes alcalinos con los componentes ácidos del crudo (ácidos carboxílicos,
carboxifenoles, porfirinas entre otras), las cuales generan sustancias con alta actividad
interfacial. El equilibrio de disociación de los ácidos carboxílicos grasos (FAs) producido
por una variación del pH del medio produce dos especies que se comportan como
surfactantes noiónico (ácido) y aniónico (carboxilato), respectivamente. Estas especies en
un sistema agua/aceite sufren un reparto preferencial entre las fases del sistema originando
una disminución de la tensión interfacial y la formación de una tercera fase denominada
trifásico o microemulsion donde se encuentra la mayor concentración del anfifilo. El
desarrollo de un método analítico para la identificación y cuantificación simultánea de una
variedad de FAs es deseable tanto en sistemas agua-microemulsión-aceite, como en otros
campos (cosmético, farmacéutico, productos alimenticios). La determinación de estos FAs
se ha desarrollado principalmente por cromatografía gaseosa (GC) de sus derivados ésteres
de metilo. Sin embargo, como una alternativa a la GC, la cromatografía líquida de alta
precisión (HPLC) proporciona una buena sensibilidad y selectividad. En este trabajo se
reportan varios métodos de análisis por HPLC para el estudio de los FAs sin ningún
tratamiento previo y haciendo uso de su débil absorción a 214 nm. Además, se evalúa el
efecto de la temperatura en la retención de los FAs así como de surfactantes noiónicos
polietoxilados en el proceso cromatográfico a partir del cual se pudo determinar el
comportamiento termodinámico en dicho proceso. En ambos casos se observó un
comportamiento lineal entre los parámetros termodinámicos de retención y el grado de
etoxilación (surfactante noiónico) o cadena alquílica (FAs) de estos compuestos. No
obstante, debido a que la mayoría de las muestras de FAs no presentan ninguna absorción
útil en las regiones del visible o ultravioleta (UV) para su detección en HPLC, con
frecuencia se emplean técnicas de derivatización pre-columna para aumentar la sensibilidad

xiii
y selectividad de la detección. Por consiguiente, es necesario establecer un método en línea
simple, rápido y con una completa separación de estos compuestos para su análisis
sistemático. Se emplearon varios reactivos (fenilhidrazina, PH; 2,4-dinitrofenilhidrazina,
2,4-DNPH; Cloruro de bencilo, CB) para la derivatización de FAs por irradiación con
microondas (MW), y su separación y cuantificación por HPLC. Se optimizó el flujo del
solvente, la cantidad de reactivos, el tiempo de irradiación, y las condiciones
cromatográficas. El análisis continuo por el sistema MW-HPLC-UV proporcionó alta
sensibilidad, mínimo gasto de reactivos y tiempos cortos de análisis. En los sistemas
aceite/agua, los FAs se comportan como compuestos anfifílicos, es decir, que al encontrarse
en contacto con una fase acuosa y otra orgánica sufren un reparto preferencial a través de
las transiciones de Winsor por modificación del pH del medio, obteniendo un sistema de
mucha importancia en el ámbito industrial como lo es el sistema trifásico o WIII, el cual
contiene la mayor cantidad de las especies no-iónicas y aniónicas del ácido de partida y es
el estado de mayor solubilización y de mínima tensión interfacial. Para el estudio del
reparto preferencial de los ácidos carboxílicos por el método analítico desarrollado, se
prepararon diferentes sistemas ácido/agua/aceite a través de los barridos de formulación,
para evaluar el efecto de algunas variables de formulación en el reparto de estos
compuestos. Se obtuvieron amplios rangos de sistemas trifásicos a diferentes
concentraciones de FAs. El empleo de alcoholes lipofílicos como co-surfactantes, facilita la
solubilización de las sales formadas en los sistemas y contribuye a la formulación optima.
Los resultados mostraron una tendencia lineal del volumen de microemulsión con la
concentración del ácido. La cantidad de FAs en la microemulsión es mayor mientras que su
concentración en las fases acuosa y oleica en exceso disminuye. El coeficiente de reparto
permite obtener información termodinámica del proceso de transferencia del ácido en las
fases. Se obtuvo un valor positivo de energía, que representa la resistencia de transferencia
de un grupo metileno de la fase aceite al agua.
Palabras claves: Ácidos carboxílicos grasos (FAs), HPLC, análisis en línea, formulación.

xiv
A B S T R A C T

xv
Bélgica Balbina Bravo Tovar. Estudio del reparto preferencial de ácidos carboxílicos en sistemas surfactante/agua/aceite. Laboratorio de Petroquímica y Surfactantes, Departamento de Química, Facultad de Ciencias, La Universidad del Zulia, Maracaibo, Zulia-Venezuela, E-mail: [email protected] In the last years an intense investigation activity has been generated, to develop petroleum
recovery processes. The employment of alkaline solutions produces tension interfacial
decreases in certain crude-water systems, due to the interactions of the alkaline agents with
the acids components of crude oil (carboxylic acids, carboxyphenols, porphyrins etc),
which generate substances with high interfacial activity. Dissociation equilibrium of the
fatty carboxylic acids (FAs) by pH variation produces two species that behave as nonionic
(acid) and anionic (carboxylate) surfactant, respectively. These species in a water/oil
system suffer a preferential partition in the phases to produce a decrease of the interfacial
tension and the formation of a third phase so-called three-phase or microemulsión, where
they have the highest concentration of anphiphile compound. The development of an
analytical method for the simultaneous identification and quantification of a variety of FAs
is desirable in formulations of water/microemulsión/oil systems and in other fields
(pharmaceutical cosmetic, food products). The determination of FAs has been developed
mainly by gas chromatography (GC) of methyl esters. As an alternative to GC, high
performance liquid chromatography (HPLC) has better sensitivity and selectivity. In this
work, several analytical methods are reported by HPLC to study FAs without any previous
treatment and using their weak absorption band at 214 nm. Furthermore, the temperature
effect in the retention of the FAs and nonionic ethoxylated surfactantes in the
chromatographic process was evaluated. Thermodynamic behavior in this process was
determined. In both cases a lineal behavior between the thermodynamic parameters of
retention and the ethoxylation grade (surfactante nonionic) or alkyl group length (FAs) of
these compounds was observed. Nevertheless, because most FAs show no useful absorption
in the visible and ultraviolet (UV) regions for detection in HPLC, frequently pre-column
derivatization techniques are used to increase the sensitivity and selectivity of detection.
Therefore, the establishment of a simpler and more rapid on-line method with complete
separation capability is needed for the screening of large numbers of samples. It intends the
utility of various reagents (2-nitrophenyhydrazine hydrochloride, 2-NPH.HCl; Benzyl

xvi
chloride, BC) for the derivatization of different FAs by microwaves radiation (MW), and
their separation and quantification by HPLC. The flow of the solvent, the quantity of
reagents, the time of irradiation, and the chromatographic conditions were optimized. The
continuous analysis using the MW-HPLC-UV system provided high sensitivity, minimum
expense of reagents and short analysis times. In the water/oil systems the acids behave as
anphiphylic compounds. That means, when it is in contact with a water phase and other
organic phase for pH modification, they suffer a preferential partition through the Winsor
transitions, obtaining a very important system at industrial level well-known as WIII
system, the one which contains the biggest quantity of the nonionic and anionic species of
the departure acid. For the preferential partition study of the carboxylic acids for the
developed analytical method, various acid/water/oil systems with different FAs by the
formulation scan technique was performed, to evaluate the effect of some formulation
variables in the partition of these compounds. Wide ranges of three-phase systems to
different FAs concentrations were obtained. The employment of lipophilic alcohols like co-
surfactants, it facilitates the solubilization of the salts formed in the systems and it
contributes to the optimal formulation. The results showed a lineal tendency of the
microemulsion volume with the acid concentration. The quantity of FAs in the
microemulsión is bigger while its concentration in the excess, of water and oil phases
diminishes. The partition coefficient allows obtaining thermodynamic information of the
process of transfer of the acid in the phases. A positive energy value that represents the
transfer resistance of a methylene group of the oil to water phases was obtained.
Key word: fatty acid (FAs), Reverse Phase-HPLC analysis, on-line-HPLC, scan formulation.

xvii

I N T R O D U C C I Ó N

2
En los últimos años se ha generado una intensa actividad de investigación, para
desarrollar procesos de recuperación de petróleo, comúnmente denominados procesos de
recuperación mejorada o terciaria. Uno de estos procesos es la inyección de soluciones micelares
de surfactantes a los pozos de producción, con la finalidad de reducir la tensión interfacial entre
el crudo y el fluido de drenaje.1 Al lograr disminuir la tensión interfacial crudo-agua hasta
aproximadamente 10-3 mN/m se consiguen las condiciones que permiten la movilización del
crudo mediante un derrame difásico.
Se ha demostrado que es posible obtener bajas tensiones interfaciales, en ciertos sistemas
crudo-agua usando únicamente soluciones alcalinas.2-6 En estos casos, el valor de la tensión
interfacial depende en forma determinante del pH de la solución acuosa. La literatura reporta que
éste fenómeno es debido a las interacciones de los agentes alcalinos con los componentes ácidos
del crudo las cuales generan sustancias con alta actividad interfacial.2,7 Estos componentes han
sido identificados principalmente como ácidos carboxílicos y en menor extensión como
carboxifenoles, porfirinas o fracciones de asfaltenos. 8-9
La conducta de fase de los sistemas surfactante/aceite/agua es usualmente referida con un
modelo matemático, denominado “modelo de Winsor”.10 Estos sistemas se pueden clasificar en
cuatro tipos, conocidos como Winsor tipo I, tipo II, tipo III y las microemulsiones tipo IV. En los
sistemas Winsor tipo I, o aceite en agua, la microemulsión contiene un exceso de fase oleica y
micelas de surfactante en la fase microemulsión. En la fase microemulsión, la solubilidad del
aceite está dada por la partición del surfactante en micelas dispersadas en la fase continua acuosa.
Para el sistema Winsor II o agua en la microemulsión aceite, un exceso de la fase acuosa está en
equilibrio con la fase microemulsión. En este caso, el agua está solubilizada en las micelas
inversas, dispersadas en la fase continua oleica. Ajustando las variables de formulación
(salinidad, temperatura, entre otras), se obtiene un sistema Winsor III o fase media. Este sistema
contiene una nueva fase media, termodinámicamente estable que contiene todo el surfactante y
una mezcla de agua y aceite; esta fase media coexiste con las fases en exceso de agua y aceite.
Finalmente, al adicionar una cantidad suficiente de surfactante, el sistema Winsor tipo III cambia
a un sistema Winsor tipo IV, donde todo el surfactante, aceite y agua coexisten en una sola fase.11

3
Estudios realizados12-16 han demostrado que las propiedades de los sistemas aceite-
salmuera-ácido dependen del pH. Otras variables que pueden influir, en el equilibrio, o en la
actividad interfacial de estas especies son el tipo de ácido, presencia de alcoholes, electrolito,
entre otros. Cuando se emplean surfactantes de tipo jabón, preparados a partir de la neutralización
de un ácido carboxílico con un álcali (como el hidróxido de sodio), el equilibrio que se establece
indica que en el producto final se obtendrá tanto el carboxilato correspondiente como una parte
del ácido carboxílico utilizado originalmente, el cual no reacciona.17-21 Este caso resulta
interesante desde el punto de vista fisicoquímico; debido a que el carboxilato se comporta como
un surfactante aniónico hidrofílico, mientras que el ácido carboxílico no disociado se comporta
como un surfactante no iónico lipofílico. Evidentemente, el comportamiento de fases que se
obtenga dependerá de las cantidades relativas de las especies presentes en el sistema.
En un sistema agua-aceite puede ocurrir un reparto del ácido entre ambas fases y para
fines prácticos es importante la determinación de la composición de la mezcla de ácido
carboxílico presente en cada fase ya que, estas especies son las responsables de la disminución de
la tensión interfacial y, en consecuencia de la recuperación mejorada de los crudos pesados.
Hasta ahora se han empleado las titulaciones ácido-base para la determinación de la
concentración de las especies en juego en las tres fases del sistema Winsor III.22 Sin embargo,
técnicas analíticas como la cromatografía de gas (CG)23-28 y la cromatografía líquida de alta
resolución (HPLC)29-34 proporcionan la ventaja de emplear poca cantidad de muestra, así como
de una determinación más precisa de la concentración de dichas especies.
Algunos análisis por HPLC están limitados por la técnica de detección. El límite de
resolución por absorbancia óptica en la región ultravioleta puede aproximarse a 10-9g.34 Por lo
tanto, la elección de la longitud de onda representa un compromiso entre las propiedades ópticas
de la muestra y la absorción de fondo del solvente. En el caso de los ácidos carboxílicos, el
cromóforo carboxilo presenta una banda débil de absorción cerca de 200 nm debido a la
transición n-π* por los electrones de valencia.35 Por otro lado, el aumento en la longitud de la
cadena alquílica ocasiona un pequeño desplazamiento batocrómico en la posición de la banda.
Este hecho resulta adecuado para la detección de estos ácidos a 214 nm. Sin embargo, se han
reportado una amplia variedad de métodos de esterificación para ácidos carboxílicos, entre los

4
más comunes36-38 la condensación con alcohol en condiciones ácidas o la reacción con haluros de
alquilo en condiciones básicas.
Se puede emplear para la detección de estos productos una variedad de detectores, pero
para las separaciones de los ácidos grasos derivados se emplea detección espectrofotometría UV
o fluorimétrica.36-43 En la actualidad, la síntesis orgánica asistida con microondas ha evidenciado
un alto repunte, en parte debido a su velocidad de reacción, selectividad y fácil manipulación.44-52
No obstante, el diseño de nuevas técnicas y tratamientos de muestras para el análisis rápido de
compuestos de este tipo resulta imperioso desde el punto de vista experimental. Las técnicas de
inyección en flujo o análisis en línea han permitido mejorar la sensibilidad y selectividad de los
procesos analíticos.
Por lo antes expuesto, este trabajo persigue separar mezclas de ácidos carboxílicos de
cadena larga por HPLC para evaluar el efecto de algunas variables de formulación en el reparto
de estos ácidos en sistemas ácido/agua/aceite. Para ello, se discuten y explican los métodos de
separación adecuados para el análisis de una mezcla de ácidos carboxílicos grasos por HPLC en
fase reversa, empleando dos fases estacionarias y varias mezclas de solventes polares como fase
móvil. Además, se evalúa el efecto de la temperatura en la retención de estos compuestos en el
proceso cromatográfico, para lo cual se estudian también los surfactantes nonilfenol
polietoxilados a objeto de establecer las comparaciones respectivas (capítulo IV). Por otro lado,
debido a la baja absorción que poseen estos compuestos en la región UV, se propone un método
para la preparación de sus derivados con radiación microonda diseñando un sistema en línea
acoplado al cromatógrafo líquido para su análisis secuencial (capítulo V). En los capítulos VII y
VIII, se estudia el comportamiento de fase de estos compuestos en sistemas aceite-agua
aprovechando su carácter anfifílico, con la finalidad de encontrar el sistema a formulación óptima
(máxima solubilización), cuyas fases son analizadas empleando el método desarrollado por
HPLC para determinar su reparto en los sistemas microemulsión-aceite-agua.

5
REFERENCIAS BIBLIOGRÁFICAS [1] Salager, J., Rev. Inst. Mex. Pet. 1979, 11, 59-61.
[2] Bourrel, M.; Graciaa, A.; Schechter, R.; Wade, W.; J. Colloid Interf. Sci., 1979, 72, 161-
169.
[3] Salager, J.; Morgan, J.; Schechter, R.; Wade, W.; Vasquez, E.; J. Soc. Petrol. Eng. 1979,
19, 107-114.
[4] Bourrel, M.; Salager, J.; Schechter, R.; Wade, W.; J. colloid Interf. Sci., 1980, 75, 451-459.
[5] Trujillo, E. J. Soc. Petrol. Eng. 1983, 645, 08-15.
[6] Mc Cafferty, F. J. Canadian Pet. Technol., 1976, 15, 71-78.
[7] Jan, L.; Sharma, M.; Chang, Y.; Chang, M.; Yen, T. AICHE Symposium series, 1982, 78,
97-103.
[8] Layrise, I.; Rivas, H.; Acevedo, S. J. Dispers. sci. Technol. 1984, 5, 1-9.
[9] Ovalles, C.; García, M.; Lujano, E.; Aular, W.; Bermúdez, R.; Cotte, E.; Fuel 1998, 77, 3-
11.
[10] Winsor, P. Solvent Properties of Amphiphilic Compounds. Butterworth London, 1954.
[11] Lohateeraparp, P.; Wilairuengsuwan, P.; Saiwan, C.; Sabatini, D.; Harwell, J. J.
Surfactants and detergents. 2003, 6, 15-24.
[12] Seifert, W.; Howells, W. Anal. Chem., 1969, 41, 554-562.
[13] Seifert, W. Anal. Chem., 1969, 41, 562-568.
[14] Strassner, J. J. Petrol. Technol. 1968, 03, 45-52.
[15] Chiwetelu, C.; Hornof, V.; Neale, G.; Chem. Eng. Sci. 1990, 45, 627-634.
[16] Ramakrishnan, T.; Wasan, D.; Soc. Petrol. Eng. J. 1983, 08, 602-609.
[17] Schick M. Nonionic surfactants. Marcel Dekker, INC., New York. 1967.
[18] Hummel D., Handbook of surfactants analysis. John Willey & Sons, LTD, England, 2000.
[19] Kemp D., Vellaccio F., Organic Chemistry. Worth Publishers, INC. 1980.
[20] Rappoport Z., Handbook of table for organic compounds identification, 3ed., CRC Press.
Florida 1967.
[21] Weast R., Astle M., Handbook of chemistry and physicals, CRC Press. Florida 1983.
[22] Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. L. Proceedings 4th World Surfactants
Congress, R. de Llúria, Ed., A.E.P.S.A.T. Barcelona, España. 1996, 2, 244-249.
[23] Bevilacqua, A.; Califano, A. Food Chemistry, 1992, 43, 354-361.

6
[24] Chocrane, G. J. Chromatogr. 1975, 3, 440-447.
[25] Hsiau-hsu, W.; Santoro, N.; Miller, R.; Tuovinen, O. J. gen. Microbiol. 1984, 130, 1051-
1058.
[26] Seppänen-Laakso, T.; Laakso, I.; Hiltunen R. Anal. Chim. Acta 2002, 465, 39-62.
[27] Meier-Augenstein, W. Anal. Chim. Acta 2002, 465, 63-79.
[28] Ackman, R. Anal. Chim. Acta 2002, 465, 175-192.
[29] Kaliszan, R.; Haber, P.; Baczek, T.; Siluk, D.; Valko, K. J. Chromatogr. A. 2002, 965,
117-127.
[30] Helaleh, M.; Tanaka, K.; Taoda, H.; Hu, W.; Hasebe, K.; Haddad, P. J. Chromatogr. A.
2002, 956, 201-208.
[31] Tanaka, K.; Ding, M.; Helaleh, M.; Taoda, H.; Takahashi, H.; Hu, W.; Hasebe, K.;
Haddad, P.; Fritz, J.; Sarzanini, C. J. Chromatogr. A. 2002, 956, 209-214.
[32] Nimura, N.; Fujiwara, T.; Watanabe, A.; Sekine, M.; Furuchi, T.; Yohda, M.; Yamagishi,
A.; Oshima, T.; Homma, H. Anal. Biochem. 2003, 315, 262-269.
[33] Hanko, V; Rohrer, J. Anal. Biochem. 2004, 324, 29-38.
[34] Snyder, L.; Kirkland, J. Introduction to Modern Liquid Chromatography. 2ed. John Wiley
& Sons, INC. 1979.
[35] Silverstein, R.; Bassler, G.; Morril, T. Spectrometric identification of Organic Compounds,
3rd (ed.) New York:Wiley. 1974.
[36] Mototeru, Y. Anal. Chim. Acta 2002, 465, 227-236.
[37] Amet, Y.; Adas, F.; Berthou, F. Anal. Chim. Acta 2002, 465, 193-198.
[38] Brondz, I. Anal. Chim. Acta 2002, 465, 1-37.
[39] Ohba, Y.; Kuroda, N.; Nakashima, K. Anal. Chim. Acta 2002, 465, 101-109.
[40] Toyo’oka, T. Anal. Chim. Acta 2002, 465, 111-130.
[41] Rozhkov, V.; Vorob’ov, S.; Lobatch, A.; Kuvshinov, A.; Shevelev, S. Synthetic Comm.,
2002, 32, 467-474.
[42] Greence, T.; Wuts, P. Protective Groups in Organic Synthesis, John Wiley & Sons. New
York 1999, Chapter 5.
[43] Mulzer, J. In Comprehensive Organic Synthesis; Trost, B.; Fleming, I.; Heathcock, C.
Eds., Pergamon Press. New York, 1991, 6, 324-337.
[44] Ganzler, K.; Salgo, A.; Valko, K.; J. Chromatogr. 1986, 371, 299-304.

7
[45] Yoshida, H.; Hiroka, N.; Kajimoto, G. J. Food Sci. 1990, 55, 1412-1418.
[46] Taketomi, T.; Hara, A.; Uemura, K.; Kurahashi, H.; Sugiyama, E. Biochem. Biophys. Res.
Común. 1996, 224, 462-469.
[47] Dayal, B.; ENTEL, N. Lipids. 1998, 33, 333-341.
[48] Dasgupta, A.; Banerjee, P.; Malik, S. Chem. Phys. Lipids. 1992, 62, 281-287.
[49] Khan, M.; Williams, J. Lipids. 1993, 28, 953-961.
[50] Carrapiso, A.; Garcia, C. Lipids. 2000, 35, 1167-1173.
[51] Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J-L. A. Petit. Tetrahedron. 1999, 55, 10851-
10858.
[52] Kabza, K.; Chapados, B.; Gestwicki, J.; McGrath, J. J. Org. Chem. 2000, 65, 1210-1213.

8
C A P I T U L O I

9
GENERALIDADES DE LOS ÁCIDOS CARBOXÍLICOS
A los compuestos que contienen el grupo carboxilo (abreviado -COOH o CO2H) se les
denomina ácidos carboxílicos. El grupo carboxilo es el origen de una serie de compuestos
orgánicos entre los que se encuentran los haluros de ácido (RCOCl), los anhídridos de ácido
(RCOOCOR), los ésteres (RCOOR´), y las amidas (RCONH2). El grupo carboxilo, -COOH, es
formalmente una combinación de un grupo carbonilo y de un hidroxilo. Algunos ácidos alifáticos
se conocen desde hace cientos de años y sus nombres comunes reflejan sus orígenes históricos. El
ácido carboxílico más simple, el ácido fórmico, es el causante de la irritación causada por la
picadura de las hormigas (del latín formica, hormiga). El ácido acético se aisló del vinagre, cuyo
nombre en latín es acetum (agrio). El ácido propiónico se consideró como el primer ácido graso,
y su nombre deriva del griego protos pion (primera grasa). El ácido butírico se obtiene por
oxidación del butiraldehído, que se encuentra en la mantequilla (en latín butyrum). Los ácidos
caproico, caprílico y cáprico se encuentran en las secreciones cutáneas de las cabras (capri en
latín)1-2. Los ácidos de cadena lineal se pueden obtener por hidrólisis de las grasas o aceites
triglicéridos. Los ácidos lineales hasta C5, son líquidos incoloros de olor picante y sabor ácido,
solubles en agua, etanol y éter, y destilan a presión ordinaria sin descomponerse. Los ácidos
lineales siguientes, hasta C9 siguen siendo líquidos a 20 °C, pero perdiendo mucho su fluidez;
esto es, van siendo cada vez más viscosos, de olor repugnante a mantequilla rancia, poco solubles
en agua, pero solubles en éter y etanol. 1-4
Los ácidos grasos con más de diez átomos de carbonos se llaman ácidos grasos superiores
y son sólidos sin color y sin sabor, prácticamente insolubles en agua pero solubles en alcohol y
éter; además, el calor los descompone. La solubilidad en los alcoholes, es debido a la formación
de enlaces de hidrógeno con ellos. Los ácidos carboxílicos son relativamente débiles y su
constante de disociación varía entre 10-3 y 10-5. A partir de C8 la constante de disociación se
mantiene esencialmente constante a 10-5. 1-4 En el petróleo crudo y en el aceite de madera se
encuentran ácidos carboxílicos, policíclicos con alto grado de insaturación (nafténicos, abiéticos,
entre otros).

10
La disociación de un ácido o un alcohol implica, en ambos casos, la ruptura heterolítica de
un enlace O-H, pero cuando la disociación se produce sobre el ácido carboxílico se genera un ión
carboxilato con la carga negativa repartida por igual sobre dos átomos de oxígeno, mientras que
la ionización de un alcohol genera un ión alcóxido, en el que la carga negativa se encuentra casi
en su totalidad sobre un solo átomo de oxígeno. La deslocalización de la carga en el ión
carboxilato hace que éste sea mucho más estable que un ión alcóxido y por tanto, la disociación
de un ácido carboxílico es menos endotérmica que la de un alcohol (Figura I.1).1-3,5
Figura I.1. Disociación de alcoholes y ácidos carboxílicos y su comportamiento
energético.

11
Cuando se hidroliza la grasa con NaOH, se obtiene glicerina (propanotriol) y las
correspondientes sales sódicas de los ácidos carboxílicos de cadena larga (Figura I.2). Estas sales
son lo que se conoce como jabón (saponificación, del latín saponis que significa jabón).
Figura I.2. Reacción de saponificación.
En la reacción de saponificación, el enlace con el grupo saliente se rompe en un segundo
paso del mecanismo (pérdida de ion metóxido). Este segundo paso es muy exotérmico y por tanto
el estado de transición de este segundo paso se asemejará al reactivo y no al producto de la
reacción. En este estado de transición, el enlace con el grupo saliente apenas se ha comenzado a
romper. En general una base fuerte puede funcionar como grupo saliente si se elimina en un paso
muy exotérmico, convirtiendo un intermedio inestable y con carga negativa, en una molécula
estable.5
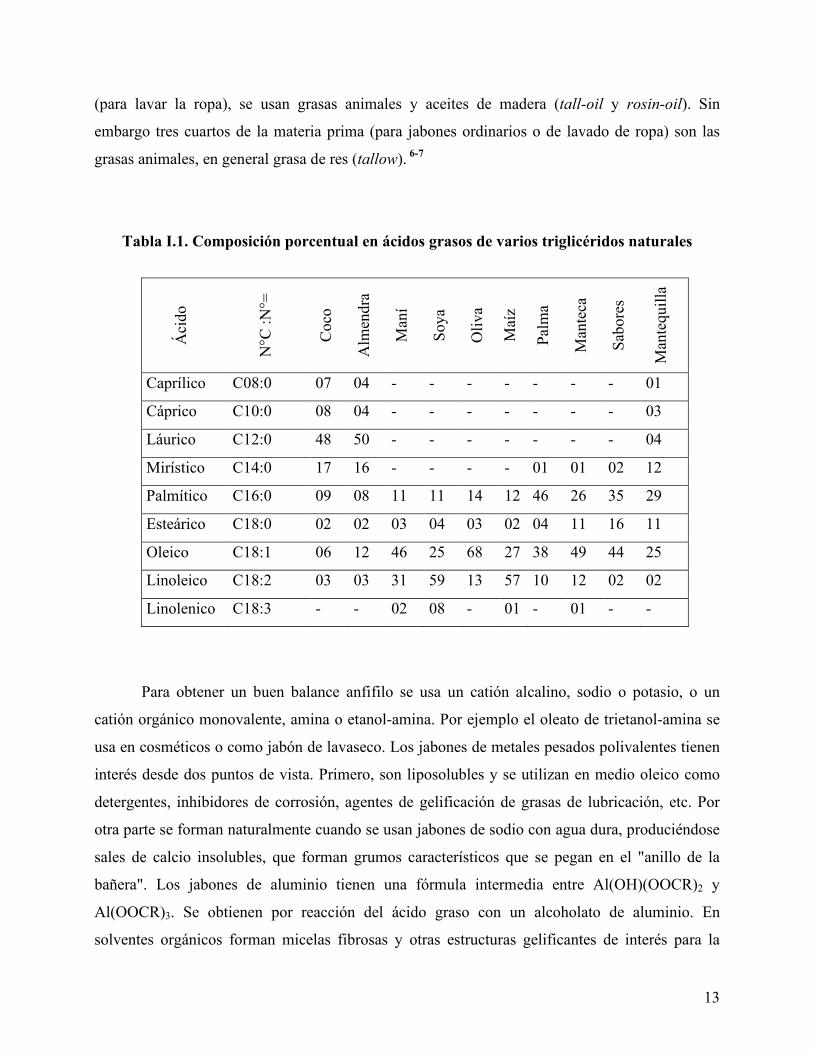
La Tabla I.1 indica la composición porcentual en ácidos grasos de varios aceites y grasas
vegetales (coco, palma, olivo, entre otros) y animales (tocino, manteca). Estos ácidos tienen una
cadena lineal con un número par de átomos de carbono generalmente de 12 a 18, los cuales

12
pueden ser saturados o contener uno o varios doble enlaces. En la primera columna, se indica el
nombre común del ácido y en la segunda el número de átomos de carbono y después de los dos
puntos el número de dobles enlaces. Los datos de la tabla son indicativos, ya que pueden variar
según las condiciones ambientales y climáticas, o la alimentación del ganado. Sin embargo
algunas diferencias aparecen claramente, en particular acerca del contenido relativo de ácidos
saturados o insaturados.6
De la insaturación depende la estabilidad, ya que los insaturados pueden enranciarse o
polimerizarse, y también la posibilidad de reacción sobre el o los dobles enlaces. Por ejemplo
puede notarse que el aceite de coco y el aceite de almendra de palma contienen un alto porcentaje
de ácido láurico (C12), el cual es la base de sustancias espumantes. Sin embargo, también
contienen ácidos cortos (C6-C8) que pueden producir irritaciones de la piel. Los ácidos grasos
naturales, así como los sintéticos, particularmente en el rango C12-C18, son una fuente
importante de materias surfactantes de todo tipo. Los ácidos grasos permiten introducir en los
surfactantes un grupo lipofílico (con extremidad levemente hidrofílica) que no es tóxico y que
por lo tanto puede usarse en productos farmacéuticos o alimenticios
En la Tabla I.1 puede verse también la correspondencia entre los triglicéridos naturales
(aceites y grasas) y el contenido en ácidos grasos. De manera general se puede decir que los
carboxilatos de cadena corta (C10-C12)6-7 son rápidamente solubles en agua, producen espuma y
toleran el agua dura. Sin embargo son también irritantes para las pieles sensibles. Por otra parte
los carboxilatos en C18 saturados o insaturados no son irritantes y pueden usarse en jabones y
cremas fáciles, pero se solubilizan en agua muy lentamente.
Un buen balance anfifilo lo tiene el palmitato de sodio. Los jabones de aceite de palma
son la base de los jabones de tocador. Se mezclan con jabones de aceite de oliva ("Palmolive",
"Jabón de Marseille") para mayor suavidad y menor irritabilidad, y con un poco de jabón de
aceite de coco o de almendra de palma para aumentar el poder espumante y disolver eventuales
jabones de calcio que pueden formase con agua dura. El aceite de ricino (80% de ácido
ricinoléico) se usa para jabón en barra transparente. El aceite de coco (50% de ácido láurico) se
usa para jabones de marina que deben espumar en el agua salada. Para los jabones en escamas
13
(para lavar la ropa), se usan grasas animales y aceites de madera (tall-oil y rosin-oil). Sin
embargo tres cuartos de la materia prima (para jabones ordinarios o de lavado de ropa) son las
grasas animales, en general grasa de res (tallow). 6-7
Tabla I.1. Composición porcentual en ácidos grasos de varios triglicéridos naturales
Áci
do
N°C
:N°=
Coc
o
Alm
endr
a
Man
í
Soya
Oliv
a
Maí
z
Palm
a
Man
teca
Sabo
res
Man
tequ
illa
Caprílico C08:0 07 04 - - - - - - - 01
Cáprico C10:0 08 04 - - - - - - - 03
Láurico C12:0 48 50 - - - - - - - 04
Mirístico C14:0 17 16 - - - - 01 01 02 12
Palmítico C16:0 09 08 11 11 14 12 46 26 35 29
Esteárico C18:0 02 02 03 04 03 02 04 11 16 11
Oleico C18:1 06 12 46 25 68 27 38 49 44 25
Linoleico C18:2 03 03 31 59 13 57 10 12 02 02
Linolenico C18:3 - - 02 08 - 01 - 01 - -
Para obtener un buen balance anfifilo se usa un catión alcalino, sodio o potasio, o un
catión orgánico monovalente, amina o etanol-amina. Por ejemplo el oleato de trietanol-amina se
usa en cosméticos o como jabón de lavaseco. Los jabones de metales pesados polivalentes tienen
interés desde dos puntos de vista. Primero, son liposolubles y se utilizan en medio oleico como
detergentes, inhibidores de corrosión, agentes de gelificación de grasas de lubricación, etc. Por
otra parte se forman naturalmente cuando se usan jabones de sodio con agua dura, produciéndose
sales de calcio insolubles, que forman grumos característicos que se pegan en el "anillo de la
bañera". Los jabones de aluminio tienen una fórmula intermedia entre Al(OH)(OOCR)2 y
Al(OOCR)3. Se obtienen por reacción del ácido graso con un alcoholato de aluminio. En
solventes orgánicos forman micelas fibrosas y otras estructuras gelificantes de interés para la

14
fabricación de grasas lubricantes. Los jabones de plomo, manganeso, cobalto y cinc se usan como
agentes secantes en pinturas, mientras que los jabones de cobre tienen propiedades fungicidas.
Los estearatos de cinc y de magnesio se usan en polvos faciales. Se preparan por precipitación a
partir de una solución de jabón de sodio, mediante introducción de una sal soluble del metal
pesado.7
La fabricación del jabón ordinario o para lavar se realiza mediante una operación continua
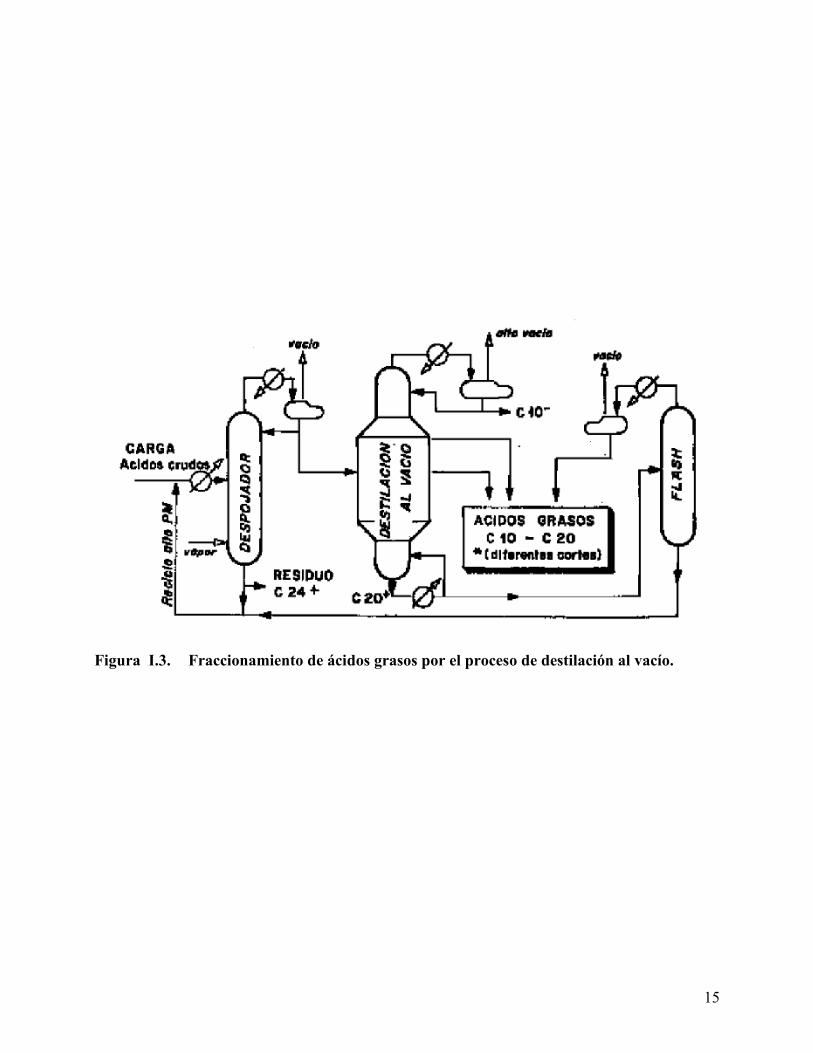
con un tiempo de residencia del orden de 2 h. El proceso no está basado sobre la saponificación
directa sino sobre una hidrólisis del triglicérido, seguido de la neutralización de los ácidos grasos.
Esto permite separar el glicerol, y fraccionar los ácidos grasos por destilación al vacío (Figura
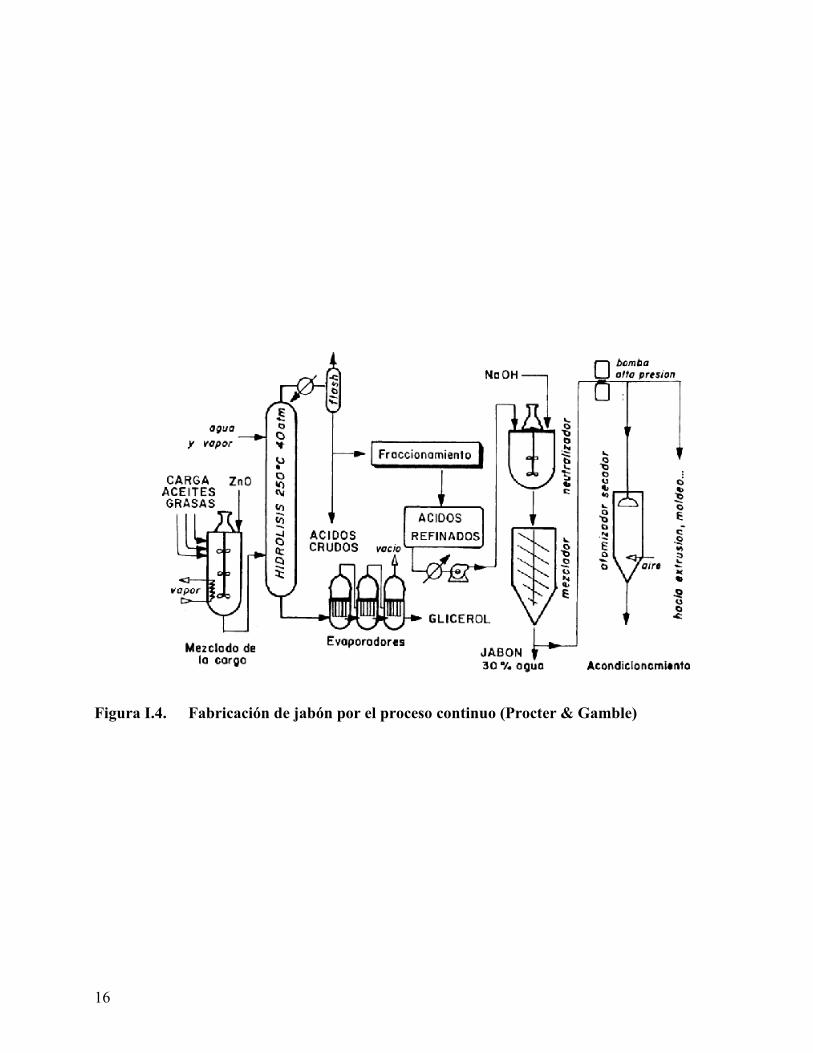
I.3). En el mezclador se introduce también el catalizador alcalino (insoluble en agua) que es
óxido de cinc o de magnesio en el proceso Procter y Gamble (Figura I.4). Otros procesos usan
ácidos sulfónicos en medio ácido. La carga se introduce a través de un distribuidor para dispersar
las gotas de aceite. En ciertos procesos (Monsanto) se fabrica una fina emulsión de aceite en agua
al pasar los dos fluidos a través de un molino coloidal). 6-7
En la Tabla I.2 se muestran los ácidos más corrientes con su estructura y su nombre
común. La casi totalidad (en peso) de los ácidos grasos naturales tienen un número par de átomos
de carbono. Se encuentran ácidos con número impar de átomos de carbono en muchos lípidos,
pero siempre en cantidad mínima. Los ácidos insaturados naturales ocurren en general en
configuración cis, a pesar de que la conformación trans sea termodinámicamente más estable.2
En general, los ácidos carboxílicos están ampliamente distribuidos en la naturaleza y se
encuentran con frecuencia en bacterias. Los ácidos mirístico y palmítico, por ejemplo, se
encuentran en microorganismos.8-11 Los FAs son también fuentes de energía y aquellos que
contienen 20 átomos de carbono así como tres a cinco dobles enlaces son precursores
biosintéticos.12 Por otro lado, el exceso de ácidos grasos libres en el plasma es el responsable de
la resistencia a la insulina encontrada en los músculos de individuos obesos.13 Así como estas
existen muchas otras aplicaciones de estos compuestos citadas en la literatura,14-21 dejando claro
su importante función biológica.
15
Figura I.3. Fraccionamiento de ácidos grasos por el proceso de destilación al vacío.
16
Figura I.4. Fabricación de jabón por el proceso continuo (Procter & Gamble)
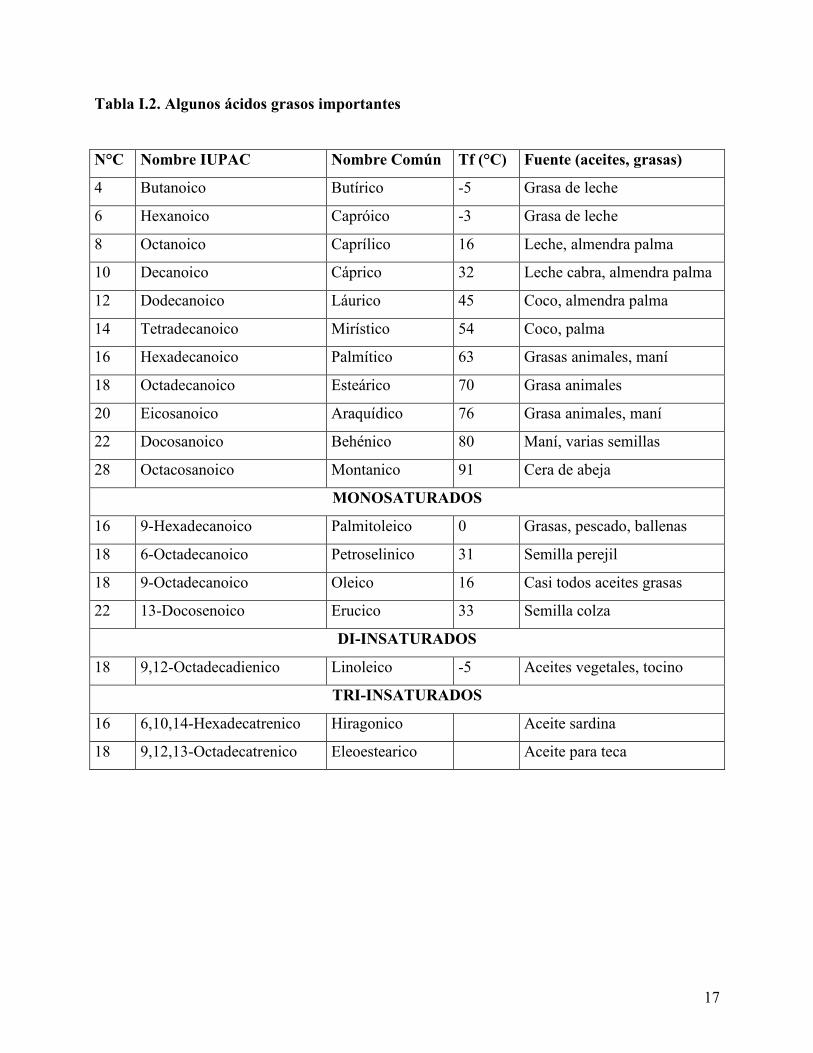
17
Tabla I.2. Algunos ácidos grasos importantes
N°C Nombre IUPAC Nombre Común Tf (°C) Fuente (aceites, grasas)
4 Butanoico Butírico -5 Grasa de leche
6 Hexanoico Capróico -3 Grasa de leche
8 Octanoico Caprílico 16 Leche, almendra palma
10 Decanoico Cáprico 32 Leche cabra, almendra palma
12 Dodecanoico Láurico 45 Coco, almendra palma
14 Tetradecanoico Mirístico 54 Coco, palma
16 Hexadecanoico Palmítico 63 Grasas animales, maní
18 Octadecanoico Esteárico 70 Grasa animales
20 Eicosanoico Araquídico 76 Grasa animales, maní
22 Docosanoico Behénico 80 Maní, varias semillas
28 Octacosanoico Montanico 91 Cera de abeja
MONOSATURADOS
16 9-Hexadecanoico Palmitoleico 0 Grasas, pescado, ballenas
18 6-Octadecanoico Petroselinico 31 Semilla perejil
18 9-Octadecanoico Oleico 16 Casi todos aceites grasas
22 13-Docosenoico Erucico 33 Semilla colza
DI-INSATURADOS
18 9,12-Octadecadienico Linoleico -5 Aceites vegetales, tocino
TRI-INSATURADOS
16 6,10,14-Hexadecatrenico Hiragonico Aceite sardina
18 9,12,13-Octadecatrenico Eleoestearico Aceite para teca

18
REFERENCIAS BIBLIOGRÁFICAS
[1] Kemp D., Vellaccio F., Organic Chemistry. Worth Publishers, INC. 1980.
[2] Rappoport Z., Handbook of table for organic compounds identification, 3ed., CRC Press.
Florida 1967.
[3] Weast R., Astle M., Handbook of chemistry and physicals, CRC Press. Florida 1983.
[4] Hummel, D. Handbook of surfactants analysis. John Willey & Sons, LTD, England, 2000.
[5] Bruwster, R. Química Orgánica. Compañía editorial continental, S.A. México. D.F.1970.
[6] Salager, J. L., Surfactantes: I. Generalidades y materia prima (cuaderno FIRP), 2004,
S301-PP, 10-12.
[7] Salager, J. L.; Fernández, A. Surfactantes: III. Surfactantes aniónicos (cuaderno FIRP),
2004, S302-PP, 2-6.
[8] Brondz, I. J. Chromatogr. 1985, 342, 13-18.
[9] Paic, M.; Lee, K.; Shin, H. J. Chromatogr. B, 1999, 721, 3-10.
[10] Nimz, E.; Morgan, S. J. Chromatogr. Sci. 1993, 31, 145-153.
[11] Martin, M.; Alonso, L.; Juarez, M.; Fontecha, J. Chromatographia. 1988, 25, 87-93.
[12] Ferrannimi, E.; Barreto, E.; Bevilacqua, S.; DeFronzo, R. J. Clin. Invest. 1983, 73, 1737-
1743.
[13] Thompson, A.; Lim-Fraser, M.; Kraegen, E.; Cooney, G. Am. J. Physiol. Endocrinol.
Metab. 2000, 279, E577-E587.
[14] Schmitz, C.; Craig, D.; Biden, T. J. Biol. Chem. 1999, 274, 24202-24210.
[15] Adlof, R.; Copes, L.; Walter, E. Lipids. 2001, 36, 315-322.
[16] Sassaki, G.; Cruz, L.; Gorin, P.; Lacomini, M. Lipids. 2001, 36, 167-173.
[17] Lin, J.; McKeon, T.; Stafford, A. J. Chromatogr. A. 1995, 699, 85-93.
[18] Marcato, B.; Cechin, G. J. Chromatogr. A. 1996, 730, 83-89.
[19] Mehta, A.; Oeser, A.; Carlson, M. J. Chromatogr. B. 1998, 719, 9-17.
[20] Chen, S.; Chen, K.; Lien, H. J. Chromatogr. A. 1999, 849, 357-364.
[21] Momchilova, S.; Nikolova, B. J. Liquid Chromatogr. Rel. Technol. 2000, 23, 1319-1326.

19
C A P I T U L O I I

20
GENERALIDADES DE LA CROMATOGRAFÍA LÍQUIDA DE ALTA RESOLUCIÓN (HPLC).
Resulta difícil definir rigurosamente el término “Cromatografía” debido a la variedad de
sistemas y técnicas a los que se ha aplicado. De hecho, esta denominación se ha mantenido por
razones históricas ya que fue a principios de siglo, cuando el botánico Mikhail Tswett,1 hizo
pasar soluciones que contenían pigmentos vegetales (clorofilas y xantofilas) a través de columnas
de vidrio empacadas con carbonato de calcio, finamente dividido, logrando separar especies que
se observaban como bandas coloreadas sobre la columna, por lo que llamo al método
“Cromatografía” (del griego chroma que significa color y graphein que significa describir).
Sin embargo, la evidencia escrita más antigua para el uso de un proceso cromatográfico se
encuentra en el viejo testamento.2 En Éxodo 15:25 está escrito: “Y llegaron a Mara, y no
pudieron beber las aguas de Mara, porque eran amargas.......... Entonces Moisés clamó a Jehová y
Jehová le mostró un árbol; y lo echo en las aguas, y las aguas se endulzaron”. Se sabe bien que
las aguas de primavera de la Península de Sinai y del Desierto de Negev en Israel contienen
CaCl2 disuelto que hace el agua amarga y picante. En la región también hay muchas plantas cuya
madera y corteza son ricas en oxalato de potasio, por lo que el hecho descrito en Éxodo puede
atribuirse a un posible proceso de intercambio iónico, en la que la solución acuosa es deionizada
con una pieza de madera impregnada con oxalato.
No obstante, los avances desde entonces permiten describir ahora esta técnica como un
proceso de separación en la cual los componentes de una mezcla se reparten selectivamente entre
una fase móvil (gas, líquido o fluido supercrítico) y otra fija o estacionaria (soporte sólido,
soporte recubierto de una película líquida delgada o soporte con una superficie enlazada
químicamente). Cuando estas fases se escogen de forma adecuada, los componentes de una
mezcla se separan gradualmente en bandas en la fase móvil.
Tradicionalmente, se ha clasificado las diferentes modalidades de la cromatografía de
acuerdo a la naturaleza de la fase móvil. Así, se tiene la Cromatografía Gaseosa (GC) si la fase
móvil es un gas y si es un líquido, Cromatografía Líquida (LC); en este último grupo se anexan la

21
Cromatografía en Capa Fina (TLC), la Cromatografía Líquida en Columna abierta (CC) y la
Cromatografía Líquida de Alta Precisión (HPLC). En todos estos casos, los principios que
gobiernan la separación son fundamentalmente los mismos.3-4
Debido a las limitaciones de la GC para analizar la mayoría de los compuestos orgánicos
conocidos (compuestos de bajo peso molecular), la popularidad de la LC, en su modo HPLC, ha
ido aumentando gradualmente en la medida en que las nuevas tecnologías han permitido el uso de
una mejor instrumentación (detectores, bombas, etc.) y empaques de columnas de mayor
eficiencia. Las razones de esta popularidad son su sensibilidad, fácil adaptación a las
determinaciones cuantitativas exactas, posibilidad de separación de especies no volátiles o
termolábiles y sobretodo, su gran potencial de aplicación para el análisis de compuestos de
interés industrial: aminoácidos, proteínas, hidrocarburos, drogas, plaguicidas, agentes
tensoactivos, especies organometálicas y una cierta variedad de sustancias inorgánicas. La
cromatografía en fase líquida sobre columna puede ser subdividida en cuatro tipos de
cromatografía de acuerdo al material usado como fase estacionaria y al proceso de separación.4-7
a) Cromatografía de adsorción. En este tipo de cromatografía líquida la fase
estacionaria es un adsorbente y la separación se basa en etapas de adsorción y
desorción sucesivas. Como ejemplo de este tipo de cromatografía tenemos las
columnas empacadas con sílica gel y alúmina neutra (Figura II.1).
Figura II.1. Representación esquemática del proceso de adsorción en cromatografía

22
b) Cromatografía de partición o cromatografía líquida-líquida. La separación se basa
en la partición de los componentes de la muestra entre las dos fases líquidas, la una la
fase móvil y la otra la fase estacionaria. Ejemplo: columnas de octilsilano, columnas
de amino, ciano, etc. (Figura II.2).
Figura II.2. Representación esquemática del proceso de partición en cromatografía
c) Cromatografía de intercambio iónico. La fase estacionaria presenta una superficie
compuesta de iones de carga opuesta a los constituyentes de la muestra. Su utilización
se limita a la separación de muestras que contienen compuestos iónicos o ionizables.
La fase móvil esta constituida por una solución tampón acuosa, de manera que el pH
y la polaridad puedan ser reguladas para modificar los tiempos de elusión de los
componentes que atraviesan la columna (Figura II.3).
Figura II.3. Representación esquemática del proceso de intercambio iónico en
cromatografía

23
d) Cromatografía de exclusión o filtración de geles. La fase estacionaria esta
constituida por un material poroso de dimensiones bien definidas los cuales actúan
como filtros de acuerdo a las dimensiones de las moléculas de la muestra, de modo
que las moléculas muy pequeñas viajen a través de los poros del empaque lo cual
retarda mas su permanencia en la columna, mientras que las de mayor tamaño viajan
más rápido por las cavidades entre las partículas. Este tipo de cromatografía se emplea
para la determinación de la masa molecular promedio de los componentes de una
muestra (Figura II.4).
Figura II.4. Representación esquemática del proceso de exclusión por tamaño en cromatografía

24
Aspectos fundamentales
La separación cromatográfica clásica se produce por la diferencia en el reparto de un
compuesto soluble entre la fase móvil y la fase estacionaria, es decir, este proceso es el resultado
de diferencias localizadas en la distribución de los compuestos de la mezcla en la fase móvil. Así,
cuando la muestra y la fase móvil son forzadas a atravesar la fase estacionaria se producen
distintos tipos de interacción entre cada uno de los componentes: interacciones hidrofóbicas,
puentes de hidrógeno, interacciones dipolares, electrostáticas, etc.
Estas interacciones son responsables de la mayor o menor afinidad de cada uno de los
componentes de la muestra por la fase móvil o la fase estacionaria, de manera que el componente
más afín por la fase estacionaria se retiene más y tarda más en eluir, y el más afín por la fase
móvil se retiene menos y eluye antes; de esta forma, en la columna se establecerá un equilibrio
que involucra la fracción de cada especie “disuelta” por cada fase en equilibrio.4
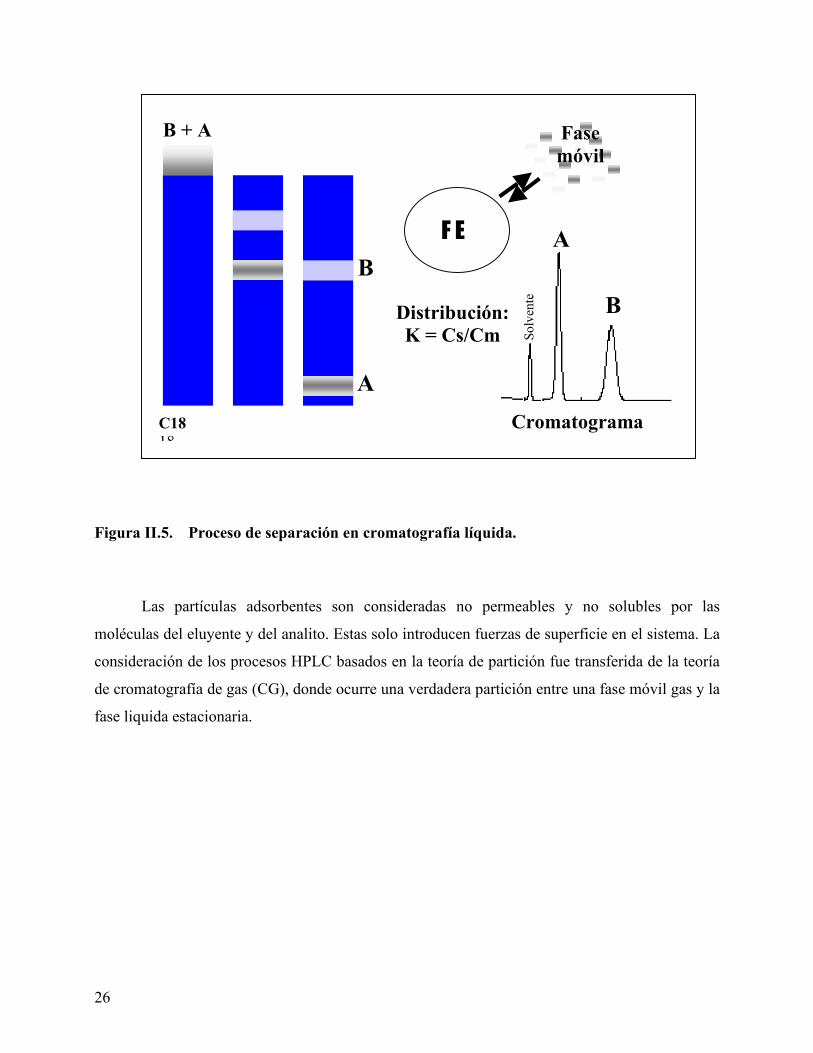
En la Figura II.5, se observa el proceso de separación para un caso hipotético de dos
componentes A y B, que se reparten de manera diferente entre un soporte (FE) y una fase móvil
(s). En cada caso, en la columna se establecerá un equilibrio (gobernado por su respectiva
constante de equilibrio K: KA, KB) que involucra la fracción de cada especie “disuelta” por cada
fase en equilibrio. Sin embargo, dependiendo del empaque de la columna, puede no solo haber
fuerzas de partición entre solutos y la fase estacionaria. Por ejemplo, si esta fase esta compuesta
de partículas adsorbentes como sílice, las fuerzas serán de adsorción (Cromatografía de
Adsorción); si es un relleno con grupos terminales contrarios al signo de la carga eventual de los
solutos, las fuerzas serán iónicas (Cromatografía de Intercambio Iónico). 4-7
El notable aumento de calidad del material de relleno y el avance en la instrumentación,
llevaron al nacimiento y rápido crecimiento de la Cromatografía Líquida de Alta Precisión con la
cual es posible obtener reproducibilidad y rapidez en el análisis. Actualmente, el método
dominante es la cromatografía de fase enlazada, en la cual la separación obedece a la diferencia
en números y naturaleza de los grupos funcionales que parten de la muestra problema.4 Esta

25
puede clasificarse en Fase Normal y Fase Reversa de acuerdo a la polaridad relativa de la fase
móvil y de los grupos funcionales químicamente ligados a la matriz.
Específicamente, si la fase móvil la integran líquidos orgánicos (n-heptano, n-hexano,
entre otras), de baja polaridad, la fase estacionaria debe ser relativamente polar (Sílice, amino por
ejemplo) y a esta técnica se denomina fase normal. Si la fase móvil la constituyen líquidos
acuosos o polares en general (agua, metanol, etc.), y la fase estacionaria es apolar (hidrocarburos
por ejemplo), se denominará fase reversa, técnica que será empleada en este trabajo para el
análisis de ácidos grasos.
La HPLC como primer intento de separación y análisis de los componentes en mezclas
complejas presenta ventajas primordiales debido a su amplio rango de aplicabilidad, y a la
facilidad con la que puede modificarse el factor capacidad (k'), variando las fases móviles.4,8-9
Este k', es un parámetro que expresa la relación entre el tiempo de retención por la fijación del
componente en la fase estacionaria (Tr) y el tiempo de transito de ese mismo componente en la
fase móvil (Tm), el cual es característico para cada componente de la muestra.
Por otro lado, existen dos aproximaciones básicas para la descripción del fenómeno de
retención en HPLC desde el punto de vista termodinámico: uno se basa en la teoría de partición y
el otro se basa en la adsorción. La partición es un cambio en concentración en el sistema debido a
la distribución de los componentes entre dos (o mas) fases, y la absorción es el cambio en
concentración en el sistema en presencia de una interfase con otra fase y es debida a las fuerzas
de superficie; dicha fase es una forma de materia que es por todo uniforme en composición
química y estado físico.
26
Figura II.5. Proceso de separación en cromatografía líquida.
Las partículas adsorbentes son consideradas no permeables y no solubles por las
moléculas del eluyente y del analito. Estas solo introducen fuerzas de superficie en el sistema. La
consideración de los procesos HPLC basados en la teoría de partición fue transferida de la teoría
de cromatografía de gas (CG), donde ocurre una verdadera partición entre una fase móvil gas y la
fase liquida estacionaria.
C18 18 C12
B + A
B
A
B
A
Cromatograma
Distribución: K = Cs/Cm
F E
Fase móvil
Solv
ente

27
Termodinámica del proceso cromatográfico
La descripción usual de la cromatografía liquida en base a la partición; considera la
suposición de la existencia de la fase liquida separada la cual se encuentra cerca de la superficie
del adsorbente. Las fases químicamente enlazadas son las consideradas usualmente, siendo una
de las más populares la octadecilsilica (RP-C18), donde cadenas alquílicas relativamente largas
(21 Ǻ) están químicamente enlazadas a la superficie de la sílice. El principio del concepto de
partición es que las moléculas del analito pueden penetrar entre estas cadenas alquílicas. Este
proceso es termodinámicamente considerado como la disolución de las moléculas del analito en
la fase alquílica superficial.
Sin embargo, existen dos restricciones para la aplicación de esta aproximación en la
descripción termodinámica:
1. Las cadenas enlazadas tienen un cierto espesor (ca. 3.8 Ǻ de diámetro) y
dependiendo de la densidad del enlace la capa puede no comportarse como un
liquido.
2. Las capas mono-moleculares no deberían considerarse como una fase en
termodinámica clásica.9
En la actualidad, existe cierta controversia en la aplicación de la teoría de partición para
describir los datos obtenidos por HPLC. Por ello es necesario también, considerar la teoría
termodinámica de adsorción de la retención en HPLC.8-9 Una descripción clásica de los procesos
de adsorción se basa en la teoría de exceso de adsorción de Gibbs, la cual considera básicamente
dos sistemas de adsorción hipotéticos con el mismo volumen, temperatura, presión y área
superficial del adsorbente. La única diferencia radica en que el primer sistema no muestra
adsorción alguna en la superficie (no hay fuerzas superficiales).
Después de alcanzar un equilibrio en ambos sistemas, se mide la concentración de los
componentes en la mayor parte de la solución sobre el adsorbente. El primer sistema obviamente,

28
tendrá una concentración original (Co) del componente A (considerando una solución binaria,
Vo), y en el segundo sistema se observara una concentración diferente, (Ce) del mismo
componente. El exceso de adsorción (Γ) se define como una cantidad en exceso del componente
concentrado en el adsorbente por unidad del área superficial (mS), es decir:
( )mS
VoCoCe !=" (1)
La dependencia del exceso de adsorción de Gibbs en la concentración del analito en el
equilibrio es lo que se conoce usualmente como isoterma de adsorción. Para las mezclas binarias
simples una isoterma de adsorción puede ser considerada por la siguiente ecuación:
( ) ( )( )xK
xxK
S
Vo
11
11
!+
!!=" (2)
donde x es la concentración en equilibrio del analito en fracción molar, K es la constante de
equilibrio termodinámica. K es una medida de la diferencia de energía de interacción de las
moléculas de eluente y analito con la superficie adsorbente, y puede ser expresada de la siguiente
forma:
RT
GK
!= exp (3)
donde ∆G es la diferencia de la energía libre de Gibbs del analito y el eluyente, R es la constante
de los gases, y T es la temperatura absoluta.

29
Si se asume que la columna está en equilibrio, esto significa que en cualquier momento y
en cualquier parte de la columna, las condiciones son infinitamente cercanas al equilibrio
termodinámico. Ahora bien, considerando la masa dinámica del analito en las pequeñas secciones
de la columna es posible obtener la ecuación de balance de masa para la parte dx de la columna a
una distancia x del inyector. Esta ecuación de balance de masa tiene una forma diferencial, y
tiene una solución exacta solo para un sistema binario (ecuación 4).
dc
dSVV
R
!+=
0 (4)
Esta solución establece una conexión entre el volumen de retención del analito (VR) y su
exceso de isoterma de adsorción. Esta expresión es la ecuación básica de retención en
cromatografía de adsorción y es la que generalmente se emplea para la explicación
termodinámica de la mayoría de los efectos cromatográficos.9
En general, se dice que el volumen de retención del componente es la suma del volumen
muerto (volumen total de la fase liquida en la columna cromatográfica), el producto del área
superficial del adsorbente y la derivada de su exceso de isoterma de adsorción. Por lo general, la
teoría de retención de HPLC basada en la adsorción de soluciones puede ayudar a establecer las
relaciones de los valores de retención medibles (Vr, tr, y k’) con los parámetros termodinámicos,
tales como la constante de equilibrio de adsorción (K), o la energía libre de Gibbs. El factor
capacidad (definido anteriormente) viene expresado como:
0
0'
V
VVk
r!
= (5)
Tomando como referencia la ecuación básica de retención, tenemos:

30
dC
d
V
Sk
!=
0
' (6)
Para concentraciones bajas de analito (dC), en ausencia de interacciones analito-analito en
solución y en la superficie, la ecuación anterior podría ser aplicada para la descripción de la
isoterma de adsorción. A muy bajas concentraciones el límite de la derivada del exceso de
adsorción por concentración es igual a:
( )1lim0
0
!="
#k
S
V
dc
d
c
(7)
De estas dos últimas ecuaciones se puede concluir que el factor capacidad puede
expresarse de la siguiente forma:
1' != Kk (8)
Por otro lado, la constante de equilibrio termodinámica es un parámetro energético. Su
logaritmo es igual a la diferencia de la energía libre de Gibbs del analito y el solvente en el
sistema de adsorción:
( ) ( )1'lnln +=!
"!
=!
= kR
S
RT
H
RT
GK (9)
donde ∆H es la diferencia de la entalpía de adsorción del analito y el solvente y ∆S es la
diferencia correspondiente a su entropía. Si el solvente interacciona con la superficie del

31
adsorbente más fuerte que el analito, este será de preferencia adsorbido. El valor de la energía
libre de Gibbs será negativo y la constante de equilibrio será menor a 1. La ecuación (8) deduce
que el factor capacidad de ese analito será negativo. Esto significa que el analito se moverá a
través de la columna más rápido que el eluente. Por tanto, el analito el cual tiene una adsorción
negativa no penetrará en la parte del volumen adyacente a la superficie adsorbente y en
consecuencia, este analito ocupara menor volumen mientras se mueve a través de la columna y se
moverá más rápido que el eluente.8-10

32
REFERENCIAS BIBLIOGRÁFICAS
[1] Tswett, M. Ver. Dtsch. Bot. Ges. 1906, 24, 316-384.
[2] La Biblia, Viejo Testamento. Éxodos 15:25.
[3] Yost, R.; Ettre, L.; Conlon, R. Practique de la Chromatographie Liquide, Technique et
Documentation. Paris, 1981.
[4] Kyrkland & Snyder. "Introduction to HPLC", M. Dekker, Inc., New York, N.Y., 1983.
[5] Rouessac, F.; Rouessac, A. Chemical Analysis: Modern Instrumental Methods and
Techniques. 4ª Edición. Editorial John Wiley & Sons, 2001.
[6] Skoog, D.; Holler, F.; Nieman, T. Principios de Análisis Instrumental. 5ª Edición.
Editorial McGraw-Hill, 2001.
[7] Rubinson, K.; Rubinson, J. Análisis Instrumental. 1ª Edición. Editorial Prentice-Hall,
2001.
[8] Levin, S. http://www.forumsci.co.il/HPLC/program.html; 1997.
Kazakevich, Y.; McNair, H. http://hplc.chem.shu.edu/NEW/HPLC_Book/index.html.
2000.
[9] Snyder L. R. Principle of Adsorption Chromatography. M. Dekker, Inc., New York, 1968.
[10] Jandera, P.; Colin, H.; Gulochon, G. Anal. Chem. 1982, 54, 435-440.

33
C A P I T U L O I I I

34
SISTEMAS AUTOMATIZADOS DE ANALISIS.
En la actualidad la química analítica está enfocada hacia tres grandes objetivos: la
automatización, sistematización y miniaturización de los análisis, para ello es fundamental
emplear técnicas o métodos de bajo costo, que permitan el análisis de un gran número de
muestras en tiempos cortos y en los cuales la intervención del operador sea mínima. Por esta
razón, hasta los momentos se han estudiado diversos sistemas automáticos y automatizados, estas
terminologías han sido establecidas por la IUPAC, los primeros son dispositivos que no
modifican su funcionamiento como resultado de una señal de retroalimentación procedente de un
transductor analítico, y los segundos son aquellos que controlan el curso del análisis debido a que
se incorporan uno o varios sistemas de retroalimentación. Entre los sistemas automáticos se
encuentran: los discontinuos, en los cuales las muestras se mantienen como entidades separadas,
y los de flujo continuo, en estos sistemas las muestras llegan a formar parte de un efluente, donde
se llevan a cabo diversas operaciones unitarias (preparación de muestra, disolución, separación,
medición, entre otras). El método de flujo continuo más empleado en la actualidad es el de
inyección en flujo ó método de flujo continuo no segmentado, FIA del anglosajón flow injection
analysis.
El trabajo manual de las soluciones sigue siendo el talón de Aquiles de la instrumentación
analítica moderna. Sin embargo, está siendo sustituido actualmente por el FIA, que permite el
manejo automatizado de soluciones de muestra y reactivo con un control determinante de las
condiciones de reacción. FIA fue en principio descrito en Dinamarca por Ruzicka y Hansen en
1975.1 La técnica ha crecido desde entonces en una disciplina cubierta por seis monografías y
más de 11.000 artículos de investigación.
El alcance del método creció desde el análisis en serie de muestras a una herramienta para
el realce del funcionamiento de instrumentos espectroscópicos y electroquímicos. La técnica de
inyección en flujo llegó a ser lo más recientemente aplicado en la biología para el estudio de
células vivas por microscopia de fluorescencia. Otros campos incluyen la supervisión de procesos
químicos en tiempo real, biotecnología, inmuno-ensayos incluyendo reacciones de anticuerpos,

35
transformación de los analitos en compuestos que generen una respuesta adecuada para su
cuantificación, entre otros.2-13
FIA es una técnica de flujo continuo empleada para el análisis rápido automatizado de
muestras líquidas. Sin embargo, debido a la gran flexibilidad y diversidad de FIA, es difícil de
capturar su amplio alcance y muchas facetas en una sola definición. Hay, en general, dos tipos de
definiciones, las cuales se clasifican como "académico" y las de "valor industrial".
1. Desde el punto de vista académico, es una forma de reunir información de la
disminución de la concentración en una zona inyectada y bien definida de un
líquido, dispersado en una corriente continua de un portador no segmentado.
2. Desde el punto de vista industrial es una tecnología analítica sencilla y versátil
para automatizar el análisis químico, basado en la manipulación física y química
de una zona dispersada de la muestra formada de la inyección de la muestra en un
portador de corriente para su detección. No obstante, FIA ha avanzado más allá del
análisis químico húmedo automatizado de una sustancia ya analizada. Las
innovaciones iniciales utilizando FIA incluyó:
• Eliminar las burbujas del flujo analítico
• Disminuir el diámetro interno de la tubería del reactor
• La inyección precisa de las muestras en el flujo analítico
El resultado fue picos analíticos con tiempos muy rápidos de subida y recuperación y
completo desastre entre muestras. Cuando se desarrollo la tecnología, muchos otros aspectos de
FIA se descubrieron y esto ha resultado ser extraordinariamente beneficioso para los laboratorios
analíticos rutinarios. En FIA, los tiempos de inicio y de cierre son tan cortos que es muy práctico
cambiar entre métodos analíticos mejor que con los canales requeridos con SFA.
Los cortos tiempos de análisis, debidos a la alta reproducibilidad de los volúmenes de muestra
y tiempos de residencia, permite que las muestras sean analizadas cerca del tiempo real. Además,

36
debido al análisis de las muestras cerca del tiempo real, la calidad de los datos se puede controlar
y tomar así las acciones correctivas durante el análisis, y no al final del ensayo cuando es
demasiado tarde. FIA es realmente más análogo a la HPLC moderna que a SFA. De hecho, FIA
ha sido llamada "HPLC sin la columna y altas presiones". En la Tabla III.1 se muestra esta
analogía y en la Tabla III.2 sus características de productividad.
Por otro lado, debido a la ausencia de burbujas de aire en el sistema, la contribución del
ruido por el flujo analítico es mínimo; de esta forma la relación señal / ruido en FIA es mejor que
en SFA. La discusión anterior muestra como la tecnología de análisis de inyección en flujo brinda
una poderosa y flexible capacidad de análisis automatizados de iones. Como una analogía a la
HPLC, FIA usa modernos principios de dinámica de fluidos para efectuar la introducción de la
muestra, mezclado, reacciones, calentamiento, diálisis, digestión y extracción, entre otros.
Instrumentación
En un analizador de inyección de flujo, un volumen pequeño y fijo de una muestra líquida
se inyecta como una zona distinta que utiliza un dispositivo de inyección en un líquido portador
que fluye por un estrecho tubo o conducto. La zona de la muestra se dispersa progresivamente en
el portador, inicialmente por convección, y posterior por la difusión axial y radial, cuando se
transporta por el conducto en condiciones laminares de flujo. En la Figura III.1 se muestra el
principio del análisis de inyección en flujo, cuyo punto de confluencia se representa con la letra
D.
Se puede agregar reactivo en varios puntos de la confluencia y éstos se mezclan con la
zona de la muestra bajo la influencia de dispersión radial, para producir la especie reactiva o
perceptible que puede ser percibida por cualquier variedad de dispositivos de detección. La altura
o el área de la señal formada del pico así obtenido se pueden utilizar para cuantificar el analito
después de la comparación con los picos obtenidos para soluciones de concentraciones conocidas
que contienen el analito.
37
Tabla III.1. Comparación de las Características de FIA, HPLC y SFA
Parámetro FIA µHPLC SFA
introducción de Muestra Inyección Inyección Aspiración
volumen de Muestra µL µL mL
Flujo analítico No segmentado No segmentado Segmentado
Conductos múltiples <1 mm d.i. <1 mm d.i. 1-2 mm d.i.
Retraso de la fase Ninguno Ninguno Significativo
Data de reducción normal Integración Integración Altura
Condiciones de mezclado Laminar Laminar Turbulento
Tabla III.2. Características de productividad de FIA
Inicio rápido ~5 minutos
Análisis rápido Es típico de 20 a 60 s
Rendimiento por muestras Es típico 60 a 120 muestras por hora
Rangos amplios de trabajo Partes por trillón a porcentajes
Completa resolución de línea base No aditivo entre muestras
Amplio rango dinámico 2 a 3 décadas es típico
Rápido cierre ~5 min.
Método de cambio rápido ~10 min.
Auto dilución inteligente Las muestras fuera de escalas son diluidas automáticamente
utilizando la relación correcta sin la intervención del operador
Control de calidad de los datos Tiempo real cercano al control de la calidad de los datos

38
Figura III.1. Principio del análisis de inyección en flujo.
El proceso entero de la muestra / inyección uniforme, el transporte, la adición de reactivo,
la reacción y detección se pueden alcanzar muy rápidamente (segundos), utilizando cantidades
mínimas de muestra y reactivos, y con excelente reproducibilidad (Ej. el coeficiente de variación,
CV, generalmente <2%). Aunque no se puede lograr durante este proceso el equilibrio completo,
la cuantificación es posible porque tanto los estándares como las muestras son dispersados a la
misma extensión y procesados en una manera idéntica.
FIA difiere notablemente del otro método continuo muy común de análisis de flujo, el
análisis en flujo continuo segmentado (SCFA), una técnica que implica la separación de la
muestra y una solución del lavado por burbujas aéreas para evitar la contaminación entre
muestras. En SCFA, aplican las condiciones turbulentas de flujo, ocurre una completa dispersión
de la muestra, y se alcanza una condición de estado constante antes de la detección del analito.
Por tanto, el análisis de muestras en SCFA es generalmente más lento que en FIA.

39
El proceso de FIA se puede agrupar en tres etapas:
1. La muestra se mide fuera y se inyecta en el portador de corriente (FIA). Este paso se
realiza generalmente con una válvula de inyección de muestra.
2. Procesamiento de la muestra: el propósito de este paso es transformar el analito en una
especie que pueda ser medida por el detector y manipular su concentración en una
gama que sea compatible con el detector.
3. Descubrimiento: es donde el analito, o un derivado de el, produce un pico de la señal
que se utiliza para la cuantificación.
En resumen, FIA puede:
• Diluir por factores hasta decenas de miles, y puede concentrar por varios cien.
• Realizar la química en un analito para producir una especie perceptible.
• Transferir un analito de un medio a otro, por ejemplo de una muestra de gas a un portador
de FIA, y viceversa.
• Hacer extracción de solvente, y modificación de matriz o eliminación de matriz.
• Además, ser utilizado también para enriquecer (concentrar) un analito. En este caso, el
mismo dispositivo (MSD) utilizado para la dilución se puede utilizar también para el
enriquecimiento del analito configurándolo a una válvula de inyección y haciendo uso de
una membrana.
Los requisitos mayores para controlar un proceso con sistemas FIA son la sencillez,
robustez, y la certeza instrumental y química. En los sistemas FIA, la muestra que implica
acondicionamiento o modificación, o la determinación de multi-parámetros puede implicar
muchas líneas de bombeo y una configuración múltiple moderadamente compleja. La certeza de

40
los sistemas FIA desplegados para aplicaciones a largo plazo, puede ser también sumamente
dependiente de la estabilidad de los reactivos utilizados.
El desempeño a largo plazo de estos sistemas quizás sea aumentado por el uso de
reactivos de fase sólida o de producción de reactantes in situ, ambos son posibles en un régimen
de análisis de inyección en flujo. Muchas investigaciones en esta técnica están dirigidas a la
medida de una sola especie, pero existe la necesidad de desarrollar multi-sistemas. Además, la
especificidad y la sensibilidad de muchos procedimientos enzimáticos y de inmuno-ensayo
utilizados en análisis clínicos y bioquímicos se podrían adaptar muy ventajosamente a las
aplicaciones continuas de control o selección en el sistema FIA que se utilice.9,11,14
En conclusión, FIA ha invadido el mundo científico para hacer más ventajosa la
investigación. Es decir, se ha incursionado en el mercado con tales ventajas que permite
disminuir en muchos casos casi al máximo la manipulación o tratamiento de una muestra antes de
ser analizada, hasta su total detección y cuantificación. Por tanto FIA, acoplado a las técnicas
analíticas existentes, proporciona la mejor solución hasta el momento a la mayor parte de los
problemas instrumentales y metodológicos que enfrenta día a día el investigador.

41
Derivatización de ácidos carboxílicos
Las técnicas de separación cromatográficas en combinación con sistemas de detecciones
selectivas y sensibles han ganado una enorme popularidad en todos los campos de la química
analítica. Sin embargo, incluso con esta variedad de sistemas de detección-separación disponibles
no siempre es posible alcanzar el límite de detección deseado con la precisión necesaria sin
alguna manipulación analítica o derivatización.
Los dos objetivos generales de una técnica de derivatización son:
1. Para incrementar la sensibilidad de detección, normalmente introduciendo cromóforos o
fluoróforos o al obtener un compuesto diferente con alta respuesta; y
2. Para incrementar la selectividad, al aplicar una reacción de derivatización específica y
selectiva para derivar solo el compuesto o compuestos de interés y poder detectarlos
entonces selectivamente en una matriz compleja.
Cuando la adición de reactivo es luz, el procedimiento es llamado reacción fotoquímica o
fotoderivatización. Así como alguna reacción de derivatización, esta puede realizarse fuera de
línea o en línea, siendo este último modo el más preferido. Cualquier detector común en
cromatografía puede ser usado para mostrar los productos de derivatización. Tanto los
fotorreactores de bombilla como los comerciales son usados con frecuencia, permitiendo una
gran variedad de esquemas de derivatización, cada uno apropiado para cada tipo de muestra en
particular.
Existen varios tipos de reacciones que pueden ser iniciadas por activación fotoquímica,
aunque la aplicabilidad de alguno de estos es limitada a aquellos compuestos que tienen la
estructura molecular apropiada para absorber suficiente energía de luz. Estas reacciones pueden
ser: oxidación y reducción, fotólisis y foto-hidrólisis, fotoionización, rearreglos moleculares,
adición y eliminación, ciclización, dimerización y polimerización, entre otros.15-19

42
La elección del agente derivatizante está influenciada por parámetros tales como:
absorción y/ o actividad de fluorescencia, conducta cromatográfica del compuesto resultante,
condiciones de reacción requeridas para la conversión cuantitativa del compuesto, así como,
estabilidad química, disponibilidad comercial y manejo seguro del agente derivatizante.
Las técnicas de derivatización para análisis por HPLC han recibido especial atención
debido a que ellas proporcionan alta sensibilidad de detección de los compuestos al enlazar un
grupo cromóforo que resulta en un producto con fuerte absorción en el UV. Existen dos
alternativas para la derivatización de ácidos carboxílicos grasos, por reacción pre o post
columna,20-22 usando un sistema FIA en línea o por pasos. La derivatización pre-columna es la
alternativa que se emplea con más frecuencia, con el objeto de incrementar la sensibilidad de
detección.
No obstante, la derivatización de FAs requiere algunas condiciones mínimas de reacción
para obtener el producto deseado. La mayoría de las reacciones de derivatización reportan el uso
de solvente y calentamiento en un baño de agua.23-24 Por otro lado, la irradiación con microondas
puede reemplazar el calentamiento convencional, debido a que permite la irradiación de la
mezcla de reacción en sistemas continuos o no continuos, al mismo tiempo que permite el control
eficiente del poder energético aplicado. Además, la irradiación con microondas puede ser
empleada para acelerar la reacción química hasta en unas mil veces sobre los métodos
convencionales. Las condiciones experimentales, tales como cantidad de reactivo, temperatura de
reacción y tiempo de reacción, son algunas veces críticos para aumentar la eficiencia de la
reacción y la formación del producto, por lo que, los procedimientos de purificación y
derivatización son algunas veces complicados y de gran consumo de tiempo.
El calentamiento por microondas involucra la absorción directa de energía por la muestra
digiriéndose. Las microondas son energía electromagnética, la cual es una radiación no ionizante
que causa movimiento molecular por la migración de los iones y la rotación de dipolos, pero no
produce cambios en la estructura molecular. La energía microondas tiene un rango de frecuencia

43
entre 300 a 300.000 MHz. El sistema microondas puede ser capaz de dar salida a los vapores sin
avisar o dañar el sistema electronico.6
En años recientes, se han reportado un gran numero de métodos para la preparación de
esteres de ácidos carboxílicos utilizando irradiación con microondas.25-35 Ganzler y col.,36 fueron
los primeros en reportar un método de extracción con microondas. Este método resulto efectivo y
económico en solvente y tiempo. Sin embargo, la irradiación con microondas puede provocar la
oxidación y modificación de los FAs.
Por otro lado, la oxidación de los FAs insaturados disminuye durante la derivatización con
irradiación microonda, comparado a las técnicas de calentamiento usuales.30 Tomando como
referencia los trabajos que reportan la irradiación microondas como una ventaja en las reacciones
de obtención de derivados, se desarrollo un sistema en línea para la obtención de derivados de
FAs con irradiación microondas y su análisis por HPLC en fase reversa. Para esto se
establecieron las condiciones necesarias de reacción empleando los siguientes agentes
derivatizantes: Fenilhidrazina, 2,4-dinitrofenilhidrazina, y cloruro de bencilo.

44
REFERENCIAS BIBLIOGRÁFICAS
[1] Ruzicka, J.; Hansen, E. Anal. Chim. Acta 1975, 78, 145-147.
[2] Frenzel, W.; Fresenius, J. Anal. Chem. 1992, 342, 817-21.
[3] Luque, M.; Valcárcel, M. Analyst 1984, 109, 413-419.
[4] Schmid, R.; Künnecke, W. J. Biotechnol 1990, 14, 3-31.
[5] Song, Z.; Hou, S. Anal. Chim. Acta 2003, 488, 71–79.
[6] Asan, A.; Isildak, I.; Andac, M.; Yilmaz, F. Talanta 2003, 60, 861-866.
[7] Mizutani, F.; Hirata, Y.; Yabuki, S.; Iijima, S. Sensors and Actuators B 2003, 91, 195–
198.
[8] Pobo’zya, E.; Halkob, R.; Krasowskia, M.; Wierzbickia, T.; Trojanowicza, M. Water
Research 2003, 37, 2019–2026.
[9] Ruzicka, J.; Hansen, E. Anal Chem. 2000, 72, 212A-217A.
[10] Nielsen, S.; Hansen, E. Anal. Chim Acta., 2000, 422, 47-62.
[11] Hansen, H. Talanta, 2000, 51, 607-608.
[12] Wang, J.; Hansen, E. Anal. Lett. 2000, 33, 2747-2766.
[13] Wang, J.; Hansen, E. Anal. Chim. Acta, 2000, 424, 223-232.
[14] Ruzicka, J.; Hansen, E. Flow Injection Analysis, J. Wiley and Sons, 1981.
[15] Lores, M.; Cabaleiro, R. Trends in analytical chemistry, 1999, 18, 6-14.
[16] Li, L.; Liu, M.; Da, S.; Feng, Y. Talanta 2004, 62, 643–648.
[17] Porschmann, J.; Plugge, J.; Toth, R. J. Chromatogr. A, 2001, 909, 95–109.
[18] Fan, L.; Chen, H.; Zhang, J.; Chen, X.; Hu, Z. Anal. Chim. Acta 2004, 501, 129–135.
[19] Sobolevsky, T.; Revelsky, A.; Revelsky, I.; Miller, B.; Oviedo, V. J. Chromatogr. B,
2004, 800, 101–107.
[20] Toyo’oka, T. Anal. Chim. Acta 2002, 465, 111-.118.
[21] Miwa, H. Anal. Chim. Acta 2002, 465, 237-255.
[22] Rosenfeld, J. Anal. Chim. Acta 2002, 465, 93-101.
[23] Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J.
Tetrahedron 1986, 27, 279-282.
[24] Raner, K.; Strauss, Ch. J. Org. Chem. 1992, 57, 6231-6234.

45
[25] Montaser, A.; Malean, A.; Huiying, L.; Mermet, J. Inductively coupled plasma mass
spectrometry. (Ed); Wiley-VCH; Chapter 2. 1998, 33-79.
[26] Yoshida, H.; Hiroka, N.; Kajimoto, G. J. Food Sci. 1990, 55, 1412-1418.
[27] Taketomi, T.; Hara, A.; Uemura, K.; Kurahashi, H.; Sugiyama, E. Biochem. Biophys. Res.
Común. 1996, 224, 462-469.
[28] Dayal, B.; Entel, N. Lipids. 1998, 33, 333-341.
[29] Dasgupta, A.; Banerjee, P.; Malik, S. Chem. Phys. Lipids. 1992, 62, 281-287.
[30] Khan, M.; Williams, J. Lipids. 1993, 28, 953-961.
[31] Carrapiso, A.; García, C. Lipids. 2000, 35, 1167-1173.
[32] Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J-L. A. Petit. Tetrahedron 1999, 55, 10851-
10859.
[33] Kabza, K.; Chapados, B.; Gestwicki, J.; McGrath, J. J. Org. Chem. 2000, 65, 1210-1213.
[34] Sterbová, D.; Matejícek, D.; Vlcek, J.; Kubán, V. Anal. Chim. Acta 2004, 513, 435–444.
[35] Priego, F.; Ruiz, J.; García, J.; Luque, M. Anal. Chim. Acta 2004, 517, 13–20.
[36] Ganzler, K.; Salgo, A.; Valko, K. J. Chromatogr. 1986, 371, 299-304.

46
C A P I T U L O I V

47
COMPORTAMIENTO DE FASE Y FRACCIONAMIENTO
En los últimos años se ha generado una intensa actividad de investigación, para
desarrollar procesos de recuperación de petróleo, comúnmente denominados procesos de
recuperación mejorada o terciaria. Uno de estos procesos es la inyección de soluciones micelares
de surfactantes a los pozos de producción, con la finalidad de reducir la tensión interfacial entre
el crudo y el fluido de drenaje. Al lograr disminuir la tensión interfacial crudo-agua hasta
aproximadamente 10-3 mN/m se consiguen las condiciones que permiten la movilización del
crudo mediante un derrame difásico.
En este capitulo, se explican las teorías necesarias para comprender el comportamiento de
fase de los ácidos carboxílicos grasos en sistemas aceite-agua producido por su carácter
anfifílico. Luego, se presentan las condiciones óptimas para la preparación de sistemas
ácido/agua/aceite con comportamiento trifásico, cambiando las variables de formulación hasta
encontrar el sistema a formulación óptima (máxima solubilización), cuyas fases son analizadas
empleando el método desarrollado por HPLC que se discutió en el primer capitulo. El análisis de
las fases se realizó con el fin de determinar el reparto de estos compuestos en los sistemas
microemulsión-aceite-agua. Finalmente, se discute la utilidad del coeficiente de reparto para
obtener información termodinámica del proceso de transferencia de la molécula del ácido del
aceite al agua.
Surfactantes y sus propiedades
Un surfactante es una sustancia anfifílica (doble afinidad), es decir, que posee una
dualidad polar-apolar, por lo cual tiene una actividad superficial o interfacial cuando está
presente una cierta concentración en un sistema agua-aceite, teniendo la propiedad de adsorberse
en la superficie o interfase del sistema en el cual se solubiliza, alterando la energía libre de estas
superficies o interfases.

48
La molécula típica de un anfifilo tiene dos partes (Figura IV.1): 1) Un grupo polar que
contiene heteroátomos como O, S, P o N, que se encuentran en grupos de tipo alcohol, ácido,
sulfato, sulfonato, fosfato, amina, amida, etc. 2) Un grupo apolar o poco polar que es
generalmente un hidrocarburo de tipo alquil o alquil-arilo (puede contener eventualmente átomos
de halógeno u oxígeno).1
Figura IV.1. Representación clásica de una molécula de surfactante
Generalmente los surfactantes se clasifican de acuerdo a su ionización en medio acuoso
en:1-3
a) Surfactantes aniónicos: Se disocian en un anión anfifilo y un catión, en general un metal
alcalino o un amonio cuaternario. Son buenos espumantes, humectantes, emulsionantes y
dispersantes. Su producción representa alrededor del 55 % de los surfactantes producidos
en el mundo. Los jabones (sales de sodio de ácidos grasos) pertenecen a este rubro.
Sales de ácidos carboxílicos: R–COO–Na+
Alquilbenceno sulfonatos: R–C6H4SO3–Na+
Alquil sulfatos: R–OSO3–Na+
b) Surfactantes catiónicos: Son aquellos que se disocian en un catión anfifilo y un anión,
generalmente del tipo halogenuro. Se caracterizan por ser poco eficientes en las

49
aplicaciones más populares, sin embargo poseen otras propiedades interesantes como su
poder bactericida.
Sales de aminas grasa: R–NH3+Cl–
Sales de amonios cuaternarios: R–N(CH3) 3+ Cl–
c) Surfactantes noiónicos: En solución acuosa no se disocian, puesto que poseen grupos
hidrofílicos del tipo alcohol, fenol, éter o amida. Representan aproximadamente el 40% de
la producción anual mundial.2 Los ácidos grasos pertenecen a este rubro.
Monoglicéridos: R–COOCH2CHOHCH2OH
Alcoholes etoxilados: R–(OC2H4)nOH
Alquilfenoles polietoxilados: R– (C6H4)–(OC2H4)nOH
d) Surfactantes anfotéricos: La porción que posee actividad interfacial tiene carga positiva,
negativa o ambas dependiendo del pH del medio. Su empleo es muy reducido.
Aminoácidos de cadena larga: RNH2+CH2COO–
Sulfobetainas: R–N+(CH3)2CH2CH2SO3–
En los surfactantes se distinguen dos propiedades fundamentales:1 la adsorción, donde los
surfactantes al encontrarse en solución tienden a migrar hacia las interfases de carácter
polar/apolar, para satisfacer su doble afinidad, alcanzando así un estado de mínima energía y, la
asociación de los surfactantes, en forma de micelas a partir de cierta concentración conocida
como la Concentración Micelar Crítica (CMC), en este punto el surfactante alcanza una posición
favorable desde el punto de vista termodinámico (Figura IV.2).
Por ejemplo, en la Figura IV.3 se muestra la variación de la tensión superficial en función
de la concentración del surfactante.2,4 A partir del valor que corresponde al agua pura (72 mN/m

50
o dina/cm), se observa una disminución de la tensión superficial con el aumento de concentración
de surfactante; en esta primera zona (I), la mayoría de las moléculas de surfactante se adsorben en
la superficie agua-aire, y la concentración superficial crece rápidamente.
A partir de un cierto valor, la superficie está ocupada por una capa monomolecular de
surfactante, y la tensión interfacial decrece linealmente con el logaritmo de la concentración;
según la isoterma de Gibbs, esto indica que la concentración superficial permanece constante. En
esta segunda zona (II) la superficie es por lo tanto saturada y las moléculas de surfactante que se
añaden deben solubilizarse en la fase acuosa, lo que es poco favorable desde el punto de vista
energético, por la presencia del grupo no-polar L.
Figura IV.2. Las propiedades fundamentales de los surfactantes Adsorción y Asociación (A), dan origen a las micelas (B).
La CMC de un surfactante depende a la vez de su grupo hidrofílico (tipo, tamaño,
contraión) y de su grupo lipofílico (longitud, ramificación). En medio acuoso, la CMC disminuye
cuando el número de átomos de carbono del lipófilo del surfactante aumenta. La tendencia
general para grupos lipofílicos lineales puede representarse mediante una expresión del tipo:
Log CMC = A – BN (1)
Monómeros
Adsorci ón
(A)
(B)
Micelas
Asociaci ón
ACEITE O AIRE
AGUA
Monómeros
Adsorci ón
(A)
(B)
Micelas
Asociaci ón
ACEITE O AIRE
AGUA
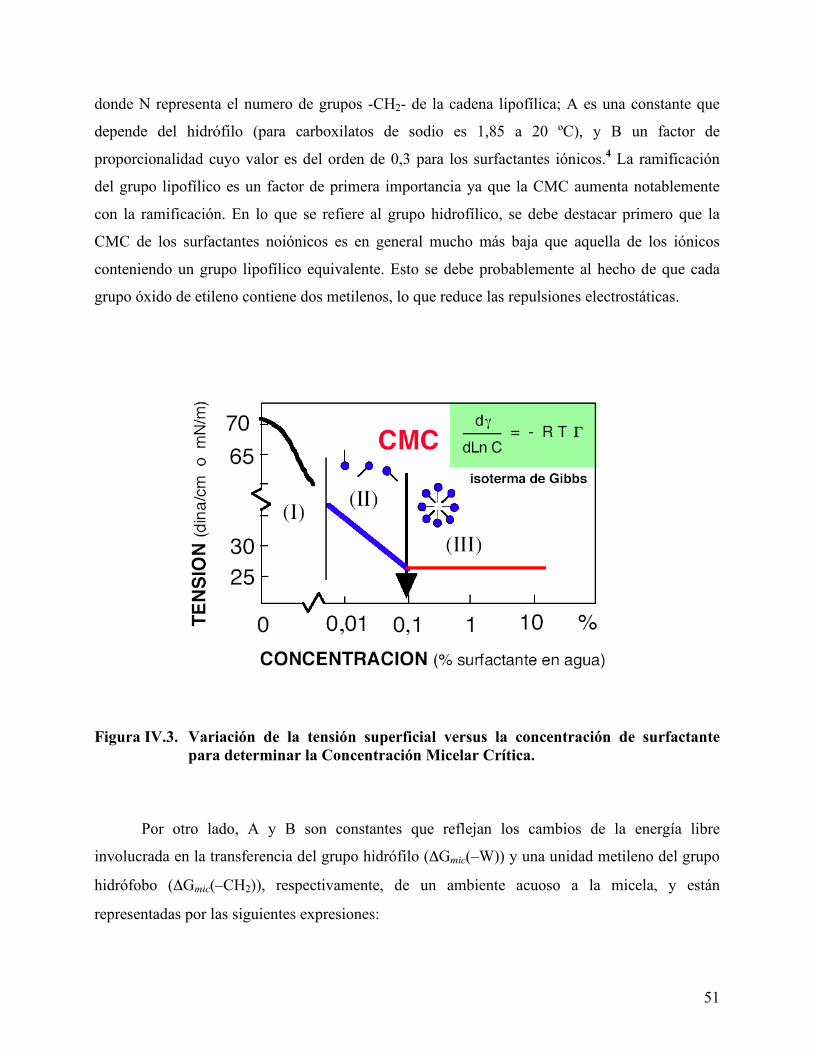
51
donde N representa el numero de grupos -CH2- de la cadena lipofílica; A es una constante que
depende del hidrófilo (para carboxilatos de sodio es 1,85 a 20 ºC), y B un factor de
proporcionalidad cuyo valor es del orden de 0,3 para los surfactantes iónicos.4 La ramificación
del grupo lipofílico es un factor de primera importancia ya que la CMC aumenta notablemente
con la ramificación. En lo que se refiere al grupo hidrofílico, se debe destacar primero que la
CMC de los surfactantes noiónicos es en general mucho más baja que aquella de los iónicos
conteniendo un grupo lipofílico equivalente. Esto se debe probablemente al hecho de que cada
grupo óxido de etileno contiene dos metilenos, lo que reduce las repulsiones electrostáticas.
Figura IV.3. Variación de la tensión superficial versus la concentración de surfactante para determinar la Concentración Micelar Crítica.
Por otro lado, A y B son constantes que reflejan los cambios de la energía libre
involucrada en la transferencia del grupo hidrófilo (ΔGmic(–W)) y una unidad metileno del grupo
hidrófobo (ΔGmic(–CH2)), respectivamente, de un ambiente acuoso a la micela, y están
representadas por las siguientes expresiones:
, ,, ,

52
( )5.55log
3.2+
+!"!=
RT
kWGA
mic (2)
( )RT
CHGB
mic
3.2
2!!"!
= (3)
Con los valores reportados de B y de estas ecuaciones se puede inferir que el cambio en la
energía libre ΔG(-CH2-) involucrado en la transferencia de una unidad metileno del grupo
hidrófobo de un ambiente acuoso al interior de la micela es negativo, favoreciendo así la
micelización, lo cual se considera por el hecho que la CMC disminuye con el incremento en la
longitud del grupo hidrófobo.
El campo de aplicación de los surfactantes es quizás el más interdisciplinario de la ciencia
y la tecnología moderna; pues aunque haya sólo un número reducido de fenómenos
fundamentales (adsorción y asociación por ejemplo), el número de aplicaciones es muy elevado y
de considerable variedad. Es debido a esto que se pueden encontrar sustancias surfactantes en
aplicaciones industriales y domésticas como: detergentes, productos farmacéuticos, cosméticos y
de limpieza, productos de la industria papelera, pinturas, polímeros, procesos químicos, entre
otros.

53
Sistemas surfactante-agua-aceite (SOW)
Los sistemas surfactante-agua-aceite pueden presentarse bajo diferentes formas; pueden
ser monofásicos como las soluciones micelares o las microemulsiones o polifásicas con la
presencia de varias fases liquidas (macroemulsiones difásicas o polifásicas), y de mesofases
como los cristales líquidos. En las microemulsiones es frecuente que una de las fases sea una
solución micelar o una microemulsión. La naturaleza del sistema depende de factores físicos,
especialmente aquellos que han actuado durante su formación, así como de factores
fisicoquímicos; la influencia de estos últimos corresponde al efecto de las variables de
formulación, es decir de la naturaleza de las sustancias que conforman el sistema, y de sus
respectivas concentraciones o proporciones; adicionalmente se considera la influencia de la
temperatura, la que puede afectar notablemente las interacciones fisicoquímicas.
El problema de la formulación es extremadamente complejo por diversas razones:
1. La formulación de un sistema surfactante-agua-aceite no garantiza, de manera única, la
obtención de un cierto tipo de emulsión. Es decir, que las operaciones sucesivas para
preparar la emulsión pueden igualmente influir en el tipo y las propiedades de la emulsión
obtenida.
2. Se puede obtener resultados idénticos con sistemas diferentes y se sabe que ciertos tipos
de efectos pueden compensarse con otros.
3. Las formulaciones que han sido un éxito desde el punto de vista comercial son en general
extremadamente complejas y han sido obtenidas por tanteo.
4. La comprensión teórica se limita a algunos modelos que son muy simplificados para
reflejar en general la realidad. Sin embargo, ellos pueden servir como guía cualitativa o
aproximativa.

54
5. Las industrias que utilizan surfactantes lograron sus formulaciones a expensas de un
trabajo laborioso de tanteos y, por consiguiente, la mayor parte del “saber hacer” no se ha
publicado; un ejemplo son las patentes sobre los agentes emulsificantes.
No obstante, aunque se crea que la formulación no es importante, se sabe que ella puede
ser determinante en ciertas condiciones:
1. La formulación determina de manera única el comportamiento de fase del
sistema, al igual que todas las propiedades al equilibrio, particularmente la
adsorción a la interfase, la composición de las fases presentes, la tensión
interfacial y/o superficial, entre otras.
2. Si bien es cierto que los procesos utilizados en la producción de sistemas fuera
de equilibrio, como las macroemulsiones, pueden influir en el resultado final,
para una formulación dada, el problema consiste en determinar cuando y como.
El propósito de los procesos de recuperación, es obtener tensiones interfaciales muy bajas,
que permitan una recuperación eficiente, según el criterio del número capilar. Varios autores5-7
han establecido que a altos valores del número capilar es posible la disminución de la tensión
interfacial. Cuando se realiza un barrido de formulación, la tensión interfacial puede pasar por un
mínimo, para un cierto valor del parámetro de barrido.8 Este mínimo se puede encontrar dentro
del rango de ocurrencia de la zona trifásica.
La formulación está relacionada con la naturaleza de los componentes en sistemas
surfactante-agua-aceite (SOW), mientras que la proporción o cantidades respectivas de estos son
consideradas como variables de composición. Su problema es muy complejo por diversas
razones:9-10
1. Una formulación para un sistema SOW no garantiza de manera única la obtención de
un cierto tipo de emulsión.

55
2. Con sistemas diferentes se pueden obtener resultados idénticos ya que algunos efectos
pueden compensarse con otros.
3. Las formulaciones exitosas son el producto, generalmente, de infinidad de pruebas de
ensayo y error y las industrias que lo logran generalmente no lo publican.
4. Los modelos que han tratado de explicar algún comportamiento son muy simples para
reflejar lo real y su uso es sólo de forma aproximada o cualitativa.
En el caso más simple, es decir un sistema ternario SOW, las variables de formulación son
al menos 5 en un diagrama real: 3 variables químicas estándar, temperatura y presión. Pero la
realidad es que en tal sistema hay muchas más variables de formulación puesto que con
frecuencia sus componentes son mezclas muy complejas, con el fin de producir un sinergismo o
para ajustar alguna propiedad. Así por ejemplo, la fase acuosa normalmente puede contener
electrolitos diferentes en naturaleza y concentración y la fase aceite puede ser desde un simple
alcano puro hasta una compleja mezcla de un crudo.
Además de esto, se debe considerar la presencia de aditivos: co-surfactantes, co-solventes,
hidrótopos o coloides protectores que se introducen en el sistema para obtener algún efecto
deseado. Estas variables contribuyen a un completo balance de afinidad en la interfase, un hecho
reconocido y tratado de explicar por diferentes teorías que a continuación se discuten.
A continuación se discutirán los diferentes modelos utilizados para dar cuenta de la
influencia del surfactante a la interfase de un sistema disperso.

56
B a l a n c e H i d r o f í l i c o - L i p o f í l i c o ( H L B ) .
La primera de estas teorías fue una aproximación que implícitamente lo que trata es de
reducir el número de variables que se toman en cuenta hasta solo una o dos; esto funciona solo si
las variables seleccionadas son las más importantes y/o si las otras variables de formulación son
constantes o tienen un efecto despreciable sobre el problema particular.
El concepto HLB11 se basa en un método experimental que consiste en atribuir un cierto
número HLB a los agentes emulsionantes a partir de datos relativos a la estabilidad de una
emulsión. Depende esencialmente del surfactante, aunque en la determinación experimental
original se tomó en cuenta también la naturaleza de la fase aceite, así que este número HLB
representa implícitamente varios parámetros y da cuenta del balance hidrofílico-lipofílico del
sistema.
La determinación experimental del número HLB es muy tediosa e inexacta, y no es de
gran uso. Sin embargo, hoy día el HLB es un parámetro característico del surfactante que se
calcula directamente del peso relativo de la parte hidrofílica y lipofílica de este. Así por ejemplo,
para surfactantes no-iónicos polietoxilados, el HLB se estima usando la ecuación empírica.
totalmolecular peso x 5
OE de cadena la de molecular peso x 100HLB = (4)
Por lo tanto, el valor del HLB no puede tomar en cuenta el efecto de variables que afectan
la físico-química en sistemas SOW como de hecho se conoce, lo cual indica la inexactitud de su
valor. Otra desventaja del número HLB es que surfactantes con el mismo HLB pueden exhibir un
comportamiento bastante diferente, particularmente si estos contienen (o son) productos
mezclados que muestran un fenómeno de fraccionamiento. Las numerosas relaciones que se han
propuesto para relacionar el HLB de un surfactante con sus propiedades12 indican que este
número parece tener una significación fundamental escondida. Estas relaciones funcionan

57
perfectamente para una familia de surfactantes pero ellas presentan incoherencias cuando se
realizan comparaciones entre familias.
A pesar de estas y otras limitaciones, y de su carácter empírico, la escala HLB es aun
ampliamente usada, probablemente debido a su extrema simplicidad y porque su valor es un
"cálculo estimado" que para muchos es suficientemente bueno. Por ejemplo el HLB para los
ácidos carboxílicos y sus respectivos carboxilatos de sodio son las siguientes, donde n
corresponde al número de átomos de carbono en la molécula.
HLB ácido carboxílico = 9,1 – 0,475 (n = 1) (5)
HLB carboxilato de sodio = 26,1 – 0,475 (n = 1), (6)
Por otro lado, el HLB de la mezcla viene dado por la siguiente relación, donde α, es un
valor que depende del tipo de ácido
HLBmezcla = (1-α)HLBHA + αHLBA- (7)
Los ácidos carboxílicos tienen muy bajo HLB, por lo que su mezcla puede ser hidrofílica
o lipofílica (o diferente a la formulación óptima) dependiendo de las cantidades relativas de cada
uno. Sin embargo en la actualidad, es más correcto hablar de balance de afinidad del surfactante
hacia el aceite y el agua, debido a que la naturaleza de estas fases así como la temperatura juegan
un papel muy importante.

58
C o m p o r t a m i e n t o d e f a s e d e s i s t e m a s s u r f a c t a n t e - a g u a - a c e i t e
Se define como la descripción cualitativa y/o cuantitativa del número de fases que
presenta un sistema al equilibrio cuando se mezclan varias sustancias. Para una composición
dada, la naturaleza de los compuestos presentes determina el comportamiento de fase del sistema
al equilibrio y las propiedades asociadas, como la composición de las fases, la tensión interfacial,
las cantidades de agua y de aceite solubilizados en una posible microemulsión, entre otros. 5,13-14
Los casos más importantes de comportamiento son: (1) una situación monofásica donde
se obtiene la solubilidad total de todos los componentes, a menudo en forma de microemulsión;
(2) una situación o comportamiento difásico donde existe un equilibrio entre una fase acuosa y
una fase orgánica (aceite) y que puede producir una emulsión cuando se agita el sistema; (3)
finalmente, un caso de extrema importancia ya que corresponde a la formulación óptima, es el
comportamiento trifásico.15
El comportamiento de fase se representa en general sobre un diagrama donde se considera
la composición del sistema, el cual posee diferentes zonas separadas por las fronteras que limitan
los diferentes casos; se incluye frecuentemente una indicación suplementaria que precisa la
composición de las fases en equilibrio llamada líneas o triángulos de reparto.
Como los sistemas reales suelen ser extremadamente complicados, sólo se estudia el caso
de sistemas ternarios SOW, donde existe únicamente dos variables de composición
independientes y se puede representar el comportamiento de fase sobre un gráfico bidimensional.
Para evitar resaltar alguno de los tres componentes en particular, se utiliza en general un
diagrama triangular equilátero que se representa a temperatura y presión constante, similar al que
se utiliza para describir los procesos de destilación y de extracción líquido-líquido14 (Figura
IV.4). En este diagrama un punto interior "s" representa la composición del sistema, y la altura
desde este punto al lado opuesto es proporcional a la cantidad del compuesto cuya representación
está sobre la punta opuesta a este lado.

59
Específicamente en el caso de la Figura IV.4 se tiene un sistema ternario ideal que
representa el comportamiento de fase cuando dos de los componentes, agua (W) y aceite (O) son
inmiscibles, y un tercero (S) es miscible en ambos por su carácter anfifílico. Para tal sistema se
tienen tres tipos de diagramas, conocidos como diagramas de Winsor, los cuales se describirán
mas adelante.
Figura IV.4. Representación de un diagrama ternario
A
C
B
Concentración
en C
S
Graduación de la
concentración en C
100%
(S)
(W)(O)
A
C
B
Concentración
en C
S
Graduación de la
concentración en C
100%
(S)
(W)(O)

60
R e l a c i ó n R d e W i n s o r
Winsor12 estableció, en sus trabajos pioneros a finales de los años 50, que el
comportamiento de fase de sistemas surfactante-agua-aceite dependía no sólo de valores
específicos de las variables de formulación, sino también de la situación físico-química en la
interfase, introduciendo la relación de la energía de interacción entre el surfactante y la fase
aceite por un lado, y la energía de interacción entre el surfactante y la fase acuosa por el otro,
como un modelo para medir el efecto de la formulación. Esta relación entre los diferentes
componentes no fue medida a través de algún procedimiento experimental, sino estimada por un
balance de energías de interacción entre las moléculas de surfactante localizadas en la interfase y
las localizadas en el seno de las fases oleica y acuosa.
La relación original de energías de interacción de Winsor se establece como:
CW
CO
A
AR = (8)
donde los símbolos utilizados por Winsor representan:
ACO: energía de interacción por unidad de área interfacial entre el anfifilo y la fase oleica.
ACW: energía de interacción por unidad de área superficial entre el anfifilo y la fase acuosa.
Posteriormente, se definió una expresión más completa:
HHWWCW
LLOOCO
AAA
AAAR
!!
!!= (9)
donde:
AOO: energía de interacción entre las moléculas del aceite.

61
ALL: energía de interacción entre las cadenas lipofílicas del surfactante.
AWW: energía de interacción entre las moléculas de agua.
AHH: energía de interacción entre los grupos hidrofílicos del surfactante.
Una relación modificada aún más amplia puede escribirse como el cociente de todas las
interacciones netas entre el surfactante y el aceite de un lado, y del otro las interacciones netas
entre el surfactante y el agua,8 como se aprecia en la Figura IV.5.
AL
O
AL
L AH
O
AO
O
AH
L
AH
H
AW
W
AW
H
AH
L
Figura IV.5. Energías de interacción que intervienen en el concepto R de Winsor
Un cambio en la relación R se asocia esencialmente a toda transición del comportamiento
de fase y a la aparición de los fenómenos correspondientes,16 así:
1. Si R < 1, las interacciones hidrofílicas son las más fuertes, lo que implica más
penetración del solvente del lado de la "cabeza polar" del surfactante a la interfase, lo que
produce una curvatura y una micela de tipo S1 (Figura IV.6). En la zona difásica del diagrama de
Winsor I, la pendiente de las líneas de reparto indica que el anfifilo se encuentra principalmente

62
en la fase acuosa. El punto crítico de la curva binodal se encuentra a la derecha, cerca de la fase
oleica. Es decir, un sistema en el cual la composición global se separa en 2 fases cuyas
composiciones corresponden a los puntos de intersección entre la línea de reparto y la curva
binodal: de un lado la fase acuosa (W) que contiene la mayor parte del anfifilo y una cierta
cantidad de aceite solubilizado, y del otro la fase oleica (O) que contiene básicamente el aceite.
Para indicar que se trata de un sistema bifásico donde el surfactante se encuentra en la fase
inferior acuosa, se utiliza el símbolo nemotécnico 2.
Figura IV.6. Representación de un diagrama ternario Winsor I.
2. Si R > 1, las interacciones lipofílicas son las más fuertes y la interfase se curva en
sentido contrario al anterior por lo que se forma una micela inversa de tipo S2. Como este es un
caso contrario al anterior, si hay una gran cantidad de agua se forma un sistema bifásico
compuesto por una solución micelar o una microemulsión inversa de tipo S2, y una fase en
exceso de agua cuya situación corresponde a un comportamiento bifásico 2 en un diagrama
ternario de Winsor de tipo II (Figura IV.7).

63
Micela S2
Tipo II
W
S
O
1!
2!
Figura IV.7. Representación de un diagrama ternario Winsor II.
3. Si R = 1, indica que las tendencias hidrofílicas y lipofílicas se equilibran y se obtiene el
caso Winsor III con dos tipos de estructuras posibles: (a) una laminar plana, o de micelas
cilíndricas, que incorpora alternativamente agua y aceite; (b) otra fluctuante que contiene con
igual probabilidad estructuras locales que se asemejan a las micelas S1 y S2. A esta situación le
corresponde un diagrama ternario de Winsor tipo III (Figura IV.8), que es el más complejo y
comprende 3 zonas distintivas: una zona trifásica (3Φ) rodeada de 3 zonas difásicas (2Φ) y de
una zona monofásica (1Φ).
Los sistemas cuya composición global está representada por un punto en la zona trifásica
se separan en 3 fases: una fase "W" que es esencialmente agua, una fase "O" que es
esencialmente aceite y una microemulsión media "M", de densidad intermedia que contiene
prácticamente todo el anfifilo, situación muy importante para medir un reparto porque existe la
seguridad de que en las fases en exceso no hay micelas, ya que estas se concentran en la fase
media (microemulsión).
La formulación óptima (3 ó III) corresponde a un equilibrio de afinidad del surfactante
por las fases agua y aceite.15 Desafortunadamente, en el tiempo de Winsor la situación de

64
modelar interacciones moleculares no era plenamente accesible como para realizar un cálculo
exacto de las energías de interacción. Como consecuencia, la relación R no permite cálculos
numéricos y sólo se usa para estimar tendencias. Sin embargo, esta relación introdujo un alcance
muy importante, el concepto de un único, global y completo parámetro de formulación que
proporciona el efecto de todas las distintas variables de formulación actualmente manipuladas por
el formulador.
Esto fue extremadamente importante para el avance de posteriores investigaciones puesto
que se evidenció que el fenómeno observado no está relacionado con los valores numéricos
específicos de las variables de formulación, sino con alguna condición físico-química completa a
ser satisfecha por y dependiendo de estas variables. Los sistemas ternarios de Winsor aunque
representan una primera aproximación de la descripción de los sistemas reales, toleran con
frecuencia un gran número de compuestos, específicamente cuando se tiene una mezcla de
aceites, una mezcla de electrolitos y una mezcla de surfactantes.
Estos compuestos se pueden agrupar en general en seudo-compuestos, o especies
químicas mezcladas que pueden comportarse como substancias puras en lo que respecta a los
fenómenos estudiados, aproximación que normalmente no es válida pero permite reducir el
número de variables que se manejan.
La existencia de un diagrama de tipo WI, WII, o WIII depende de la naturaleza físico-
química de los compuestos, definida esta por las variables de formulación.17 Generalmente, se
consideran 3 grupos de variables para describir el estado de un sistema SOW, las cuales se
definirán en la próxima sección.
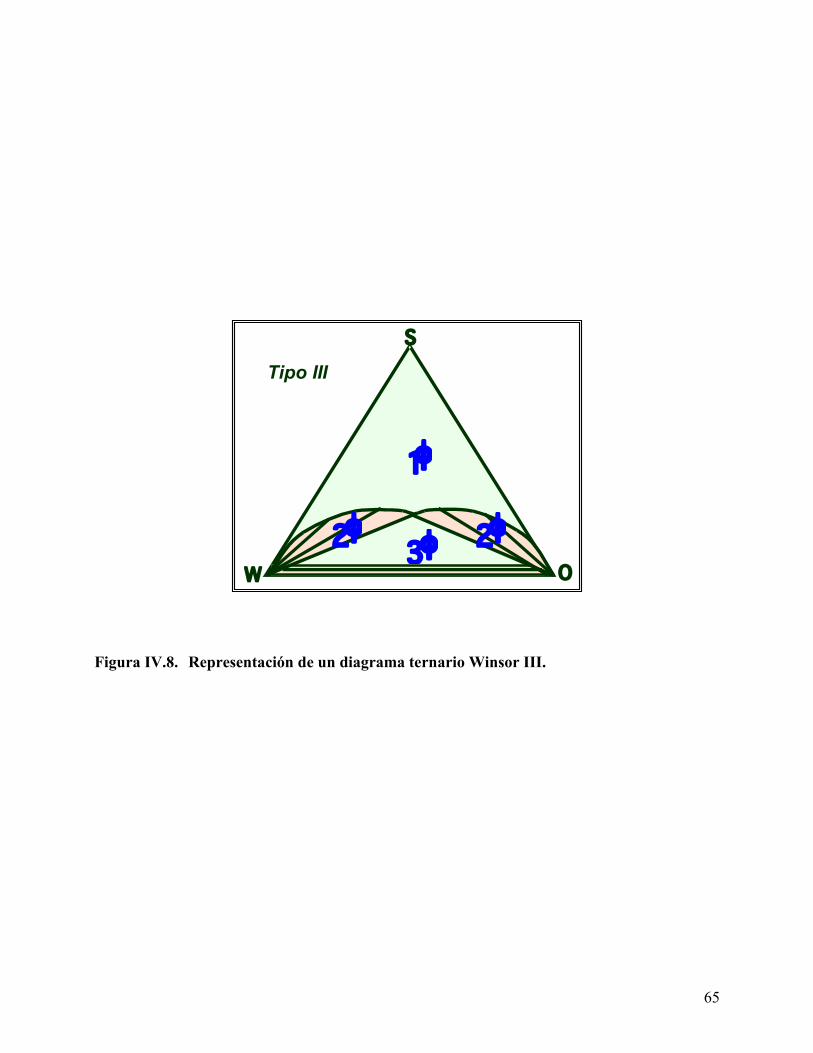
65
Tipo III
W
S
O
1!
3! 2! 2!
Figura IV.8. Representación de un diagrama ternario Winsor III.

66
F o r m u l a c i ó n y v a r i a b l e s d e f o r m u l a c i ó n
Los sistemas reales surfactante-salmuera-crudo son muy complejos, y en ellos se pueden
considerar las siguientes variables: tipo de crudo, tipo y concentración de electrolito presente en
la salmuera, tipo y concentración de surfactante y co-surfactantes, los cuales son en general
mezclas complejas que contienen un gran numero de sustancias químicas. A fin de establecer una
relación cuantitativa entre los diferentes factores capaces de influir en el comportamiento de fase
de éstos sistemas, se han simplificado al caso sistemas surfactante-salmuera-alcano. Por otro
lado, La existencia de un diagrama de tipo WI, WII, o WIII depende de la naturaleza físico-
química de los compuestos, definida esta por las variables de formulación.14,15 Así pues, se
consideran 3 grupos de variables para describir el estado de un sistema SOW:
Variables físicas.- El estudio de la influencia de las variables físico-químicas, sobre el
comportamiento de fase de los sistemas anfifilo-agua-aceite, es de gran importancia en cuanto a
la recuperación mejorada de crudo y otras aplicaciones de esos sistemas. Puesto que los sistemas
líquidos son relativamente incompresibles, la presión no produce un efecto notable; por el
contrario, la temperatura puede producir un efecto importante ya que puede afectar prácticamente
todas las interacciones moleculares involucradas. Así por ejemplo, la temperatura modifica la
miscibilidad relativa entre el agua y el aceite, y puede además tener un efecto considerable sobre
la afinidad del surfactante para estas fases, sobre todo en el caso de surfactantes no iónicos.
Variables de naturaleza.- Se refieren a la naturaleza química de los componentes básicos
de los sistemas estudiados, entre los cuales se distinguen tres: agua, aceite y surfactante. La fase
acuosa se caracteriza por la salinidad (electrolitos), que se define como los gramos de NaCl por
100 mL de fase acuosa (esencialmente el porcentaje en peso de NaCl) y eventualmente otras
substancias hidrosolubles, mientras que para describir la naturaleza de la fase oleica se utilizan
los términos ACN (“Alkane Carbon Number”) si es un hidrocarburo del tipo alcano lineal, o el
EACN (“Equivalent Alkane Carbon Number”) para otros tipos de aceites o mezclas de estos, las
cuales parecen comportarse más o menos como los seudo-compuestos; en ocasiones, sin
embargo, para aceites polares parece necesario ajustar un parámetro adicional que corresponde a
su aromaticidad. En cuanto al surfactante, por lo general se caracteriza por su tipo, su estructura

67
molecular y su peso molecular. Este consiste de una mezcla de especies químicas complejas con
un comportamiento más o menos ideal, cuya caracterización se realiza a través de parámetros
empíricos como el HLB, los cuales se relacionan con sus estructuras químicas.
Variables de composición.- Sirven para ubicar, sobre el diagrama ternario, la posición de
un punto representativo del sistema. Se puede considerar como tales: la relación agua-aceite
(WOR) y la concentración del surfactante.
Mediante un barrido de naturaleza se puede estudiar la influencia de estas variables de
formulación sobre la fisicoquímica de los sistemas. Este tipo de estudio se realiza por el método
del barrido unidimensional. Este consiste en la realización de una serie de sistemas, en los cuales
se va cambiando solo una variable capaz de modificar la afinidad del surfactante por las distintas
fases, manteniendo constante todas las demás variables involucradas. Este método permite
analizar el comportamiento de fase (transición del sistema) y la variación en las propiedades del
sistema.
Una formulación óptima ha sido definida como un estado en el cual, el conjunto de valores de
variables externas y de naturaleza es tal que se produce un mínimo en el valor de la tensión
interfacial, así como una serie de otros fenómenos. Entre los fenómenos característicos de la
formulación óptima se destacan:
- Comportamiento trifásico
- Máxima solubilización: A medida que la formulación tiende a la correspondiente del
sistema trifásico de un barrido, las microemulsiones de los sistemas 2 y 2 solubilizan
cada vez mayor cantidad de aceite y de agua respectivamente.
- El coeficiente de reparto del surfactante entre las fases acuosa y aceite sufre un cambio
considerable en la transición: 2 ↔ 3 ↔ 2 , este coeficiente es aproximadamente unitario
en los sistemas trifásicos.
Para el estudio del fraccionamiento se requiere, como ya se ha explicado, la presencia de un
sistema óptimo (W III) y este se obtiene mediante un procedimiento experimental conocido como

68
barrido de formulación, que no es mas que una exploración para determinar el comportamiento
de fase de una sola composición (fija) para así conocer el tipo de diagrama con una precisión
aceptable. Para ello, se va modificando la variable en estudio hasta conseguir el sistema deseado
(generalmente el de mayor importancia, W III o lo que es lo mismo, la formulación óptima).
Las modificaciones más importantes (barridos) que pueden realizarse son:
1. Barrido de composición, el cual permite determinar de manera sistemática el
comportamiento de fase de un sistema SOW de constituyentes dados y de composición variable.
El método normalmente es demasiado largo ya que involucra preparar una serie de sistemas
ternarios en donde las composiciones comprenden los nudos de una malla que recubre el
diagrama. Para efectos prácticos, se prefiere utilizar el método de dilución del sistema
monofásico para la alícuota agua-aceite apropiada.
2. Barrido de formulación, en este caso se busca producir un cambio de naturaleza, de forma
continua, de uno de los constituyentes. La transición de diagramas WI—WIII—WII (o viceversa)
se puede producir modificando, de manera sistemática en una serie de tubos, una de las siguientes
variables:
La salinidad de la fase acuosa (tipo y concentración del electrolito).
La naturaleza de la fase oleica, ACN o EACN.
La naturaleza del surfactante: su afinidad relativa por el agua y por el aceite (HLB,
número de óxidos de etileno, etc.).
La naturaleza de un co-surfactante, alcohol por ejemplo, sus tipos y concentración.
Si la variación es sólo en uno de estos componentes, se le llama Barrido Unidimensional.
3. Barrido de temperatura, aunque se puede considerar como una variable de formulación
y agrupar dentro del grupo precedente, se prefiere considerar como un barrido aparte por su
complejidad para expresarlo, ya que no es ni una variable de composición ni una variable que
describe la naturaleza de algún compuesto del sistema ternario. Esta variable puede a la vez

69
modificar el diagrama de fase (por co-solubilización debido a la agitación térmica), o bien
producir un efecto físico-químico que afecte las propiedades de uno de los compuestos.
Particularmente, la temperatura posee un efecto importante sobre los surfactante no iónicos ya
que puede afectar la solvatación y por ende la interacción con la fase oleica.
Para realizar un barrido normalmente se toma como composición de prueba un punto a las
condiciones que dé una alta probabilidad para ubicarse en las zonas 2Φ y 3Φ (Figura IV.8). Esto
se consigue normalmente entre 1-3% de surfactante y un WOR (relación agua-aceite) entre 0,5-
5,0 siendo el caso más general un WOR =1. A las condiciones de concentración de surfactante y
WOR escogida, se preparan una serie de tubos en los que se cambia sistemáticamente la variable
objeto de estudio (ACN, salinidad de la fase acuosa, alcohol, temperatura, etc.). A cada tubo le
corresponderá un tipo de diagrama determinando simplemente la fase rica en surfactante, lo cual
se deduce rápidamente por simple observación visual ya que esta fase contiene micelas (micelas
hinchadas) y presenta una turbidez azulada (efecto Tyndal).
Cuando esto no ocurre, la difusión del haz de luz de un láser de baja intensidad detecta la
presencia de micelas y ayuda a definir la fase rica en surfactante. Como tubo óptimo de estos
barridos se toma aquel donde la microemulsión de la fase media solubiliza igual cantidad de agua
y aceite, un criterio que es equivalente a la presencia de un mínimo de tensión interfacial. Este
procedimiento se seguirá para el estudio del comportamiento de fase de los ácidos carboxílicos
grasos en sistemas surfactante/agua/aceite, como se detallara más adelante.

70
T e o r í a d e c o r r e l a c i o n e s n u m é r i c a s p a r a l a f o r m u l a c i ó n ó p t i m a
La formulación óptima tiene mucho interés práctico no solo para la baja tensión y la alta
solubilización (en el caso de las investigaciones para la recuperación mejorada de crudos con
soluciones de surfactantes), sino también para emulsiones y espumas. En teoría, una formulación
es óptima cuando R =1 (caso Winsor III); esto es una situación físico-química precisa en la que la
afinidad del surfactante por la fase aceite equilibra exactamente su afinidad por la fase acuosa.
Puesto que las afinidades podrían cambiar con alguna de las variables de formulación, fue
necesario conocer como esto podría pasar desde el punto de vista práctico, es decir, relacionar los
datos numéricos al conjunto de variables tales como la salinidad del agua, temperatura o
naturaleza de la fase aceite.
En la práctica este estado óptimo particular se puede localizar por diferentes técnicas:10
1. Midiendo la tensión interfacial entre la microemulsión y las fases acuosa o aceite en
exceso (Figura IV.9A); el estado óptimo se define como aquel donde las dos tensiones son
lo más pequeñas e iguales:
*ee
ãleicao fase
ã
acuosa fase
ã==
µµ (10)
El comportamiento de fase ya mencionada muestra como las tensiones interfaciales de la zona
trifásica pasan por un mínimo el cual es un valor extremadamente bajo (del orden de 10–3 a 10–4
mN/m). Como esta condición es favorable para producir la movilización del petróleo residual en
un proceso de recuperación mejorada, a esta formulación se le llama óptima18-19 y se indica como
γ*.
2. Midiendo los volúmenes de fase en exceso y microemulsión, el estado óptimo
corresponde a volúmenes iguales de agua y de aceite solubilizado en la solución micelar;
estos volúmenes solubilizados dependen de la cantidad y tipo de surfactante presente en el

71
sistema. Para poder comparar estos volúmenes en diversos sistemas se define un
parámetro de solubilización, SP, con respecto al agua y al aceite:
S
O
O
V
VSP = y
S
W
W
V
VSP = (11)
donde Vo y Vw son los volúmenes de aceite y agua solubilizados, y Vs es el volumen o masa de
surfactante en la fase micelar sin tomar en cuenta la posible presencia de alcohol u otros co-
surfactantes.
De acuerdo al gráfico de la Figura IV.9B la formulación óptima corresponde esencialmente al
punto donde las dos curvas que indican la variación de los parámetros de solubilización en
función de la variable de barrido se cruzan5 esto es:
*
WOSPSPSP == (12)
3. Cuando las afinidades del surfactante con las fases acuosa y aceite se igualan, la relación
R se hace igual a 1; este equilibrio impone un HLB cercano de 10 según la definición de
Griffin.11
4. Cuando se observa un cambio brusco del coeficiente de reparto del surfactante entre las
fases en exceso acuosa y aceite se está en presencia de un estado óptimo.
Como se observa de los diferentes procedimientos para medir el estado óptimo, la situación
de equilibrio de afinidades puede cambiar con alguna de las variables de formulación; así, fue
necesario conocer este cambio desde el punto de vista práctico, es decir relacionando la data
numérica al conjunto de variables (salinidad del agua, temperatura o naturaleza de la fase aceite).
Extensos estudios experimentales realizados en este sentido para ambos grupos de
surfactantes, aniónicos y no-iónicos, mostraron que la formulación óptima se obtiene cuando se
satisface una cierta condición entre las variables de formulación; a esta condición se le denominó
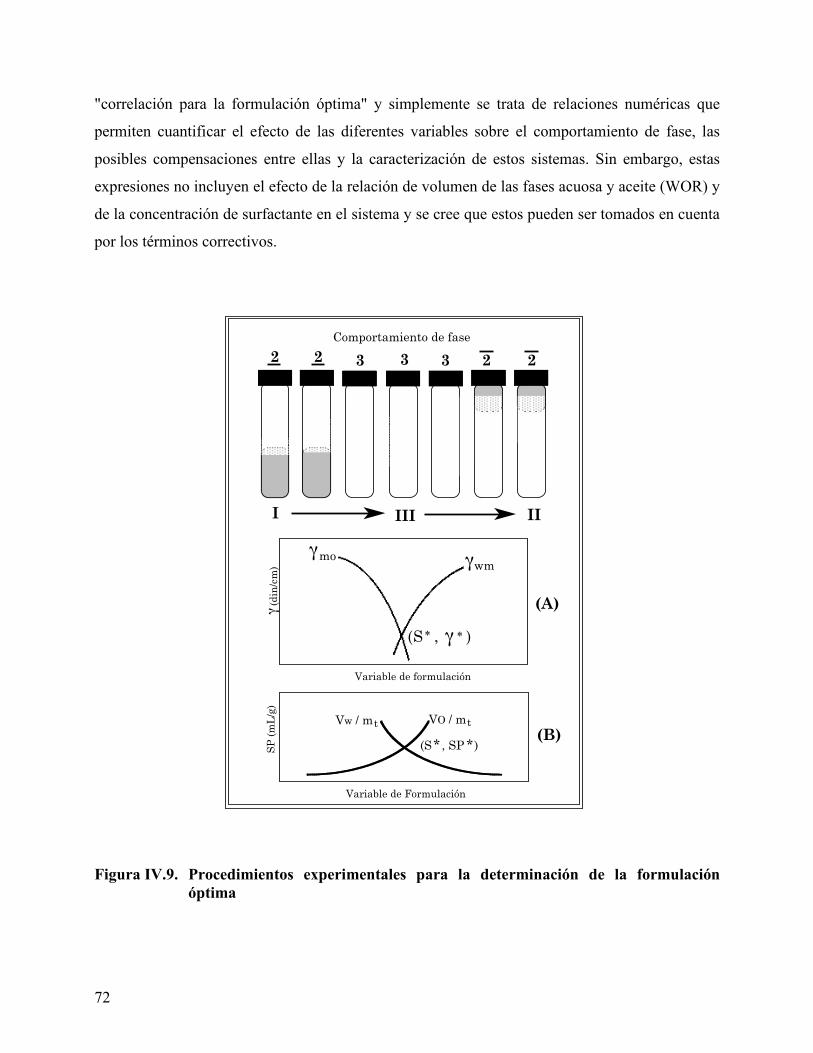
72
"correlación para la formulación óptima" y simplemente se trata de relaciones numéricas que
permiten cuantificar el efecto de las diferentes variables sobre el comportamiento de fase, las
posibles compensaciones entre ellas y la caracterización de estos sistemas. Sin embargo, estas
expresiones no incluyen el efecto de la relación de volumen de las fases acuosa y aceite (WOR) y
de la concentración de surfactante en el sistema y se cree que estos pueden ser tomados en cuenta
por los términos correctivos.
Figura IV.9. Procedimientos experimentales para la determinación de la formulación óptima
I IIIII
2 22 2 3 33
SP
(m
L/g
)
Variable de Formulación
Vw / mt VO / mt
(S , SP )* *(B)
!mo !wm
(S , ! )* *
(A)
Variable de formulación
! (d
in/c
m)
Comportamiento de fase

73
Estudios realizados20-24 han demostrado que las propiedades de los sistemas aceite-salmuera-
ácido dependen del pH. Otras variables que pueden influir, bien sobre el equilibrio, o bien sobre
la actividad interfacial de estas especies son el tipo de ácido, presencia de alcoholes, electrolito,
entre otros. Los surfactantes generalmente se emplean como mezclas. Esto se debe bien sea a la
presencia de impurezas en el producto final (por ejemplo, parte de los reactivos empleados para
preparar el surfactante) o a que el surfactante se obtiene como una mezcla de productos, como en
el caso de los surfactantes polietoxilados. En cualquiera de estos casos, es importante conocer a
priori cuál será el comportamiento del sistema; es decir, si la mezcla de productos se comportará
en forma colectiva o si es necesario considerar el efecto de cada uno de los compuestos por
separado.
Cuando se emplean surfactantes de tipo jabón, preparados a partir de la neutralización de un
ácido carboxílico con un álcali (como el hidróxido de sodio), el equilibrio que se establece indica
que en el producto final se obtendrá tanto el carboxilato correspondiente como una parte del
ácido carboxílico utilizado originalmente, el cual no reacciona. Este caso resulta interesante
desde el punto de vista fisicoquímico; el carboxilato es un surfactante aniónico hidrofílico,
mientras que el ácido carboxílico no disociado se comporta como un surfactante no iónico
lipofílico.
Evidentemente, el comportamiento de fases que se obtenga dependerá de las cantidades
relativas de las especies presentes en el sistema. La formulación está directamente relacionada
con la naturaleza de los componentes en los sistemas surfactante-agua-aceite (SOW), mientras
que la proporción o cantidades respectivas de estos son consideradas como variables de
composición.9
Todos los estudios que involucran una fase aceite y una acuosa coinciden en son
esencialmente inmiscibles, y que la presencia de un tercer componente con propiedades
anfifilicas puede reducir esta inmiscibilidad. Eventualmente los tres componentes pueden ser co-
subilizados en una microemulsión la cual es un sistema de una sola fase. En este trabajo, la data
experimental y conceptos asociados son producidos con sistemas modelos que pueden ser
analizados fácilmente. La fase oleica es una fase hidrocarbonada limpia (heptano), el surfactante

74
es un ácido graso bien definido en el rango de C8-C16, y la fase acuosa contiene NaOH y
eventualmente NaCl para hacer la fuerza iónica coincidir en una salinidad dada, que es expresada
en % p/v de NaCl o NaOH equivalente en la fase acuosa.
Por otro lado, las mezclas de surfactantes aniónicos y noiónicos permiten producir sistemas
surfactante-agua-aceite, en el cual el comportamiento de fase es insensible a la temperatura, lo
cual es de gran interés desde el punto de vista de ciertas aplicaciones. Además, es posible
disponer con estas mezclas de un grado de libertad para formular un sistema con una doble
insensibilidad, a la vez con respecto a la temperatura y a la composición.
La forma en la cual las moléculas de surfactantes se alinean a una interfase es un factor
importante en sistemas que involucran películas interfaciales solubles e insolubles. Por otro lado,
la presencia de una carga neta en un grupo polar de un surfactante afecta también su rearreglo en
una interfase. En el caso de los ácidos carboxílicos grasos, estos se ionizan por un incremento en
el pH del sistema. Si todas las moléculas del ácido graso en estudio se ionizan, entonces, la
repulsión entre las moléculas de carga similar en la monocapa puede resultar en una expansión de
la monocapa a altos valores de pH, lo cual en algunos casos puede producir una película débil e
inestable y producir así un sistema WI (Figura IV.10). 25-27
El comportamiento de fases en un barrido de este tipo exhibe una transición del tipo
!
!
"" 232 . Esto se debe a que inicialmente (es decir, cuando el volumen de hidróxido de sodio
agregado es cero) el surfactante presente es esencialmente lipofílico (ácido carboxílico). Por lo
tanto todo el surfactante se encuentra en la fase aceite y el sistema es del tipo !
2 . Al ir agregando
hidróxido de sodio, comienza a formarse la sal (carboxilato de sodio), la cual se comporta como
un surfactante hidrofílico. En cierto punto, la combinación de los dos surfactantes presentes en la
interfase produce la solubilización de las fases agua y aceite y se observa la aparición de una fase
media (microemulsión). Al continuar la adición de hidróxido de sodio, predomina la especie
hidrofílica y el sistema se comporta como un sistema !2 .

75
Figura IV.10. Diagrama hipotético del efecto de la interacción ion-dipolo de los grupos carboxílicos en la distancia intermolecular en una interfase aire/agua.
Bajo pH
Pelicula no ionizada
Aire
Agua
pH = pKa
Interaccion ion-dipolo(maxima al pKa)
Aire
Agua
Alto pH
Repulsion ionica entregrupos polares
Aire
Agua

76
T e o r í a d e r e p a r t o d e s u r f a c t a n t e s e n s i s t e m a s a g u a / a c e i t e
Existe una herramienta muy útil en el estudio de sistemas surfactante-agua-aceite llamada el
modelo de seudo-fases. Este modelo supone que la microemulsión esta constituida por una
seudo-fase acuosa, más una seudo-fase aceite, mas la interfase. Dado que la microemulsión es un
sistema bicontinuo, las seudo-fases acuosa y aceite se suponen con las mismas características que
las fases acuosa y aceite del sistema global y por consiguiente se incorporarán a éstas.19, 28-29, 30-32
Así, el sistema se representa como una fase aceite (constituida por la fase aceite original más
la seudo-fase aceite), una fase acuosa (constituida por la fase acuosa original más la seudo-fase
acuosa) y la interfase. En trabajos anteriores27,33-34 se ha encontrado que esta suposición
simplifica la aplicación del modelo de equilibrio-disociación sin introducir errores conceptuales
en el estudio.
Existen diversos sistemas que presentan sensibilidad al pH del medio; en particular, aquellos
que contienen sustancias que pueden ser transformados en otras especies a través de una
variación del pH, tales como las combinaciones ácido carboxílico/carboxilato o aminas grasas y
sus sales. En estos sistemas, el surfactante estará constituido por la mezcla de ambas especies, los
cuales en general presentan afinidades distintas.
En el caso de sistemas ácido (HA)-carboxilato (A-), el equilibrio en fase acuosa está
controlado por la constante de disociación, Ka, y así se puede escribir:
[ ] [ ][ ]
W
WW
a
HA
A HK
!+
= (13)
El índice “w” indica que las especies consideradas están en la fase acuosa. Rearreglando la
expresión anterior, se obtiene que:

77
[ ][ ][ ]
[ ] [ ][ ]HA
A log H log
HA
A HloglogKa
!+
!+
+== (14)
[ ] [ ][ ]W
Wa
HA
A logKlogH log
!
+ +!=! (15)
Empleando un valor típico de Ka para los ácidos carboxílicos tal como ≈10-6, se obtiene:
[ ][ ]W
W
HA
A log6pH
!
=! (16)
De aquí se observa que al aumentar el pH, la concentración de la especie hidrofílica aumenta,
lo cual eventualmente podría llevar a una transición del tipo WII-WIII-WI; esto se debe a que al
aumentar la concentración de la especie más hidrofílica, aumenta la interacción de la mezcla de
surfactantes con la fase acuosa.
Un sistema trifásico óptimo (Figura IV.11) en el caso de los compuestos ionizables, posee
cuatro componentes: salmuera, aceite y dos surfactantes (HA) y (A-).27 Por otro lado, el
equilibrio: HA → A- + H+, se produce exclusivamente en la fase acuosa y en la fase media. En la
microemulsión, se encuentra la mayor concentración de la especie disociada, mientras que la
especie no disociada se encuentra en equilibrio en las fases oleica y acuosa de acuerdo a su
reparto preferencial (Kr). No obstante, las cantidades de HA y A- en la fase acuosa, son
despreciables desde el punto de vista del balance de masa, y corresponden a la CMC de la mezcla
de surfactantes.
Para un sistema como el de la figura IV.11, se tiene que: por ser la neutralización de un
ácido relativamente débil con una base fuerte, se debe considerar la disociación del agua:
pH pKa

78
KW = [H+ ]W * [OH- ]W (en fase acuosa) (17)
KW = [H+ ]M * [OH- ]M (en fase media) (18)
Figura IV.11. Modelo equilibrio / disociación.
A un mismo pH:
[H+ ]M = [H+ ]W (19)
Parte o todo el ácido contenido en el sistema va a ser neutralizado por el álcali contenido
en la fase acuosa. Como el álcali añadido es NaOH, la neutralización se puede representar:
HA + NaOH A-Na+ + H2O (20)
Esta ecuación se puede escribir como ecuaciones parciales para tener en cuenta el reparto
del ácido entre las fases y su disociación:
[HA]O
[HA]M [H+]M [A-]M
Ka+
[HA]W [H+]WKa
+
Fase oleica
Fase media
Fase acuosa [A-]W
HAO
HAM
A-M
HAW
A-W
[HA]O
[HA]M [H+]M [A-]M
Ka+
[HA]W [H+]WKa
+
Fase oleica
Fase media
Fase acuosa [A-]W
HAO
HAM
A-M
HAW
A-W

79
HAW HW+ + AW
- (21)
HAO HAW (22)
La constante de disociación del ácido (Ka), en el agua se define:
[ ] [ ][ ]
W
WW
a
HA
HAK
+!
= (23)
y para la emulsión en forma análoga, donde KMa es la constante de disociación del ácido en la
interfase:
[ ] [ ][ ]
M
MM
Ma
HA
H AK
+!
= (24)
Combinando las ecuaciones 19, 23 y 24, se tiene:
[ ][ ]
[ ][ ]
aW
W
-
M
M
-
MaK
1
HA
A
HA
A
K
1= (25)
La composición de la interfase puede suponerse, en primera aproximación, semejante a la
de las primeras micelas. Se sabe que las primeras micelas de una mezcla de surfactante no
contienen la misma composición que la solución acuosa con la cual están en equilibrio. Por lo
tanto KMa es diferente de Ka.
En realidad se sabe que las primeras micelas colectan más de la especie lipofílica que lo
que existe en solución acuosa. Por lo tanto, para un mismo pH se espera que la interfase contenga
una fracción no disociada mayor que la solución. En consecuencia, la constante de disociación
aparente, KMa, es probablemente menor que Ka. Esto se justifica con el modelo de seudo-fase
utilizado para interpretar los fenómenos de comportamiento de fase. En este modelo se supone

80
que se extraen de la microemulsión todas las fases en exceso presentes, quedando solo el
surfactante a la interfase.
Así, de las ecuaciones (17 a la 23), y por la identidad de las fases agua en exceso y dentro
de la microemulsión, entonces resulta la relación 25 de identidad de disociación. El uso del
modelo seudo-fase, asume que no hay agua ni aceite en la fase media lo que significa un volumen
de fase media cercano a cero (Vφm = 0).
En cuanto a la fase aceite en exceso, ésta contiene una cierta cantidad de ácido en
equilibrio de reparto con el agua:
[ ][ ]
W
O
r
HA
HAK = (26)
Constante de fraccionamiento en el sistema:
[ ][ ]
W
O
f
A
HAK
!= (27)
y además en la fase media:
[ ][ ]
M
M
M
A
HAK
!= (28)
Nótese que si se quiere regresar a la microemulsión basta incorporar a la seudo-fase los
valores apropiados de fases en exceso con su surfactante (lo que importa sólo para el balance).35
El balance molar de carga en el agua y la microemulsión está dado por:
[Na+]NaOHW + [Na+]NaCl + [H+]W = [A-]W + [A-]m + [OH-]W + [Cl-]NaCl (29)

81
El balance de masa (en moles), para el ácido presente inicialmente en el sistema se puede
escribir como:
[HA]TOTAL = [HA]O + [HA]m + [HA]W + [A-]m + [A-]W (30)
En estos sistemas, siempre ocurre un reparto de las especies en las distintas fases como
resultado de las distintas afinidades que muestran las especies presentes en el sistema (el ácido es
lipofílico, mientras que el carboxilato es hidrofílico). Este reparto preferencial se expresa a través
del coeficiente de reparto del ácido (Kr) entre el aceite y el agua mediante la siguiente ecuación:
[ ][ ]HAw
HAoKr= (31)
donde [Haw], es la concentración del ácido en la fase acuosa y [HAo], es la concentración del
ácido en la fase oleica.27-29
El Kr, es un parámetro de primaria importancia que depende de las variables de
formulación. Sin embargo, no se han reportado aplicaciones del análisis de FAs en sistemas de
Winsor por HPLC, ni su uso en el cálculo del coeficiente de reparto (Kr). Esta información es de
gran importancia en el área de fenómenos interfaciales, ya que se ha demostrado en estudios
previos28-32 que el Kr esta relacionado con la influencia de las variables de formulación (tipo y
concentración de anfifilo, tipo de aceite, etc.) en los sistemas surfactante/aceite/agua.

82
REFERENCIAS BIBLIOGRÁFICAS
[1] Schick, M. Nonionic surfactants. Marcel Dekker, INC., New York, 1967,
[2] Salager, J. L., Surfactantes en solución acuosa (cuaderno FIRP), 1993, S201-A, 1-3
[3] Hummel D., Handbook of surfactants analysis. John Willey & Sons, LTD, England, 2000.
[4] Rosen, M. Surfactants and interfacial phenomena. New York, John Wiley & Sons, 1978,
5-76
[5] Wade, W.; Morgan, J.; Schechter, R.; Jacobson, J.; Salager, J-L. SPE 9824. J. Soc. Pet.
Eng., 1978, 18, 242-
[6] Taba, J. J. Soc. Pet. Eng., 1969, 9, 3-8
[7] Mekose, J.; Brandner, C. J. Can Petrol. Technol. 1974, 4, 54-59
[8] Chan, K.; Shan, D. Symposium on oil fuel and geothermal chemistry, Houston, Texas,
Paper SPE 7969, 1979
[9] Davies, J. Proceeding 2nd International Congress Surface Activity. Butterworths, London,
1957, 1, 426-437.
[10] Ysambertt, F. Thèse Docteur. Université de Pau et des Pays de l'Adour. Octubre, 1997.
[11] Griffin, W. J. Soc. Cosm. Chem. 1949, 1, 311-319.
[12] Winsor, P. Solvent Properties of Amphiphilic Compounds. Butterworth London, 1954
[13] Salager, J. Rev. Inst. Mex. Pet. 1979, 11, 59-65.
[14] Salager, J. Formulación, composición y fabricación de emulsiones para obtener las
propiedades deseadas. Estado del arte (cuaderno FIRP). 1999, S747-A, 2-6.
[15] Salager, J. Encyclopedia of Emulsions Technology. Marcel Dekker, Inc., New Cork. 1988,
3, 247-280.
[16] Shinoda, K.; Kunieda, H. in: Becher, P. (ed.), Encyclopedia of Emulsions Technology.
Marcel Dekker, Inc., New York. 1985, Vol. 1.
[17] Salager, J. L. Micro y Macroemulsiones. Informe Técnico FIRP 8401. 1984
[18] Reed, R.; Healy, R. Some Physicochemical Aspects of Microemulsion Floo ding.
Academies Press. New York. 1977.
[19] Bourrel, M.; Schechter, R. Microemulsions and Related Systems; Marcel Dekker, New
York. 1988.
[20] Seifert, W.; Howells, W. Anal. Chem. 1969, 41, 554-561.

83
[21] Seifert, W. Anal. Chem. 1969, 41, 562-569.
[22] Strassner, J. J. Petrol. Technol. 1968, 03, 45-52.
[23] Chiwetelu, C.; Hornof, V.; Neale, G. Chem. Eng. Sci. 1990, 45, 627-634.
[24] Ramakrishnan, T.; Wasan, D. J. Soc. Petrol. Eng. 1983, 08, 602-609.
[25] Kanicky, J.; Shah, D. J. Colloid and Interf. Sci. 2002, 256, 201-208.
[26] Whiddon, Ch.; Bunton, C.; Söderman, O. J. phys. Chem. B. 2003, 107, 1001-1009.
[27] Antón, R.; Salager, J. J. Colloid Int. Sci. 1990, 140, 75-81.
[28] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1995, 100,
225-231.
[29] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1998, 131, 45-
49.
[30] Graciaa, A.; Lachaise, J.; Sayous, J.; Grenier, P.; Yiv, S.; Schechter, R.; Wade, W. J.
Colloid Interf. Sci., 1983, 93, 474-481.
[31] Márquez, N.; Antón, R.; Usubillaga, A.; Salager, J-L. Sep. Sci. Technol, 1993, 28, 1769-
1775.
[32] Márquez, N.; Antón, R.; Usubillaga, A.; Salager, J-L. Sep. Sci. Technol, 1993, 28, 2387-
2394.
[33] Chan, M.; Yen, T. J. Chem. Eng. 1982, 60, 305-312.
[34] Trujillo, E. J. Soc. Petrol. Eng. 1983, 645, 08-14.
[35] Antón, R. Thèse de Doctorat: Contribution a l’étude du comportement de phase des
systèmes: mélanges de surfactifs-eau-huile. 1992.

84
C A P I T U L O V

85
ANTECEDENTES.
En la separación por HPLC-FR de analitos ionizables como es el caso de los ácidos
carboxílicos grasos de cadena larga, es necesario controlar adecuadamente el pH de las fases
móviles convencionales usadas. Para un ácido débil, HA, su disociación en la fase móvil es
gobernada por el equilibrio de disociación. Si el equilibrio de disociación es mas rápido que el
equilibrio de adsorción-desorción de los analitos entre la fase móvil y la fase estacionaria, el
factor de retención (K) del ácido débil es el peso promedio de las especies neutras (KHA) y las
especies ionizadas (KA-). Por otro lado, es posible determinar el tiempo de retención de un ácido
débil al relacionar su constante de disociación con el factor de retención de la siguiente forma:1
pH
a
RR
pH
a
R10K1
HAtAt10Kt
+
+=
!
(1)
La bibliografía muestra diversos métodos de análisis por cromatografía de ácidos
carboxílicos con diferentes grados de complejidad2-4 entre los que se puede mencionar la
determinación de ácidos benzoicos sustituidos adicionando CO2 a la fase móvil MeOH/H2O, con
lo cual se obtuvo una relación inversa del pH de la fase móvil con la retención de estos
compuestos; resultando más afectados aquellos ácidos con menor pKa (Wen y col.5).
Otra forma de analizar ácidos grasos es mediante la formación de sus derivados
fluorogénicos para su determinación por cromatografía líquida (Chi-Yu y col.6); y de sus
epóxidos en presencia de sus correspondientes dioles para su determinación por cromatografía de
gas-masa (Newman y col.7). Por otro lado, también se ha reportado el uso combinado de la
cromatografía liquida y de gas para el análisis de esteres de ácidos grasos con el propósito de
evitar la presencia de picos interferentes (Meulenaer y col.8).
Un procedimiento de análisis muy particular fue el desarrollado por Van Beek y col.,9
quienes combinaron las modalidades de la cromatografía liquida: fase reversa, fase normal y

86
argentométrica, para el aislamiento preparativo de ácidos tipo 6-alquilsalicilicos. Para lo cual
emplearon dos columnas en serie con una misma fase móvil, con la que se obtuvo una separación
casi lineal y simultanea de seis ácidos de este tipo. Sin embargo, este procedimiento representa un
incrementado costo de análisis.
Por otro lado, Tosió y col.,10 utilizaron los factores de retención en HPLC-FR para el
análisis de la reacción de formación de complejos de esteres con iones potasio en medio metanol-
agua. Estos ésteres se inyectaron como analitos y los iones potasio se adicionaron al eluyente. Por
lo general, para controlar los factores de retención con frecuencia se usan en el eluyente reactivos
de interacción; además, los cambios en los factores de retención se atribuyen a un equilibrio de
reacción entre los analitos de interés y el reactivo de interacción en el eluyente.
Este estudio demostró que el factor retención de estos ésteres disminuyó con el
incremento de la concentración de los iones potasio en el eluyente, efecto que se atribuye al
cambio en las especies de los ésteres de un ligando neutro a un complejo cargado positivamente.
Además, sus resultados indican que las reacciones de equilibrio en la fase móvil no son afectadas
por el tipo de columna empleada (cadena hidrocarbonada del empaque). Para este estudio, los
autores utilizaron una serie de ecuaciones y un método matemático para obtener los valores de las
constantes de los ésteres en su forma libre y acomplejada como se muestra a continuación.
Cuando se supone una reacción 1:1, la reacción con su equilibrio se describen en las
ecuaciones (11) y (12):
K+ + L KL+ (2)
[ ][ ] [ ]L K
KLK
ML +
+
= (3)

87
donde L denota el éster y KML es una constante de formación de complejos. El factor de
retención para un cierto éster, K, es afectado por la KL del éster libre, y la del éster acomplejado
KML, como se describe en la ecuación (13):
[ ][ ] [ ]
[ ][ ] [ ] MLL
KKLL
KLK
KLL
LK
+
+
+ ++
+= (4)
La siguiente ecuación (5) se deriva de la combinación de la constante de equilibrio (3) y el
balance de masa para L con la ecuación (4):
[ ][ ][ ] ML
ML
ML
L
ML
KKK1
KKK
KK1
1K
+
+
+ ++
+= (5)
En otro estudio, Melchior y Gäb,11 evaluaron la alta selectividad y eficiencia de la
cromatografía electrocinética micelar (MEKC) con una solución de bórax-dodecilsulfato de sodio
(SDS) en la separación de ácidos grasos hidroxi y hidroxiperóxido y sus ácidos grasos
insaturados no oxidados de los cuales se derivan. La utilización de eluyentes altamente acuosos
en HPLC-FR hace posible una derivatización enzimática post-columna, la cual en combinación
con detección de fluorescencia o quimioluminicencia mejora tanto la selectividad como la
sensibilidad.
La MEKC, con sistemas reguladores complejos es efectiva en la separación de isómeros
con estructura similar. Este estudio muestra que con el uso de bórax y un flujo normal es posible
la reducción del tiempo de análisis, además mejora simultáneamente la eficiencia y selectividad
para los hidroperóxidos lípidos de una variedad de ácidos grasos.

88
Koning y col.,12 idearon un equipo automático para la determinación de la composición de
ácidos grasos (como metilesteres, FAMEs) en grasas y aceites y su distribución cis/trans (CTME)
frecuentemente encontrados en la industria alimenticia. Con este equipo el único procedimiento
manual es la pesada de la muestra y adición de heptano, el resto del trabajo, que incluye adición
de reactivo, agitación, instalación de la muestra y la inyección final en el cromatógrafo de gas la
realiza dicho robot en un tiempo no mayor de 10 min. Este sistema automatizado, el cual es
rustico y fácil de emplear, evita en gran parte el contacto manual con la muestra y es actualmente
usado para el análisis de un gran número de muestras.
Bruzzoniti y col.,13 evaluaron la conducta cromatográfica de ácidos carboxílicos en tres
columnas diferentes de intercambio aniónico. Las columnas analíticas producidas se
caracterizaron por contener alquilaminas con cero, uno o dos grupos hidroxilo en el sitio
funcional de intercambio aniónico. Se usaron los ácidos carboxílicos fumárico, maléico, oxálico,
malónico, succínico, glutárico, adípico, málico, tartárico, entre otros. Se estudió la función de las
tres fases estacionarias empleando NaOH como eluyente así como acetonitrilo, metanol y n-
propanol.
El estudio demostró que la conducta cromatográfica fue similar para las tres columnas
empleadas, pero los diferentes solventes orgánicos produjeron variaciones en la selectividad. Para
caracterizar estas diferencias los autores realizaron medidas de tamaño de partícula tanto en agua
pura como en presencia de cada solvente orgánico. Los estudios demostraron que en presencia de
metanol, el látex es más pequeño que en un sistema de agua pura, es decir, el metanol reduce el
número de moléculas de agua requeridas para solvatar el látex. Contrariamente, en presencia de
acetonitrilo las dimensiones del látex fueron tan altas que produjeron hinchazón de las partículas
del látex.
En este caso, la densidad de carga es más baja que en agua pura y en consecuencia los
tiempos de retención son más cortos. Por otro lado, en presencia de n-propanol el tamaño del
látex permaneció constante, indicando que no hubo cambios en las propiedades electrostáticas de
la resina. En consecuencia, los efectos de n-propanol en la retención de los analitos podrían

89
relacionarse principalmente a las interacciones hidrofóbicas. Finalmente, existe otra variedad de
alternativas para el análisis por cromatografía de estos compuestos citados en la literatura.14-34
Los ácidos grasos enlazados (BFAs) y los FAs, quedan por lo general, contaminados con
sustancias no lípidos después de la extracción de la fuente natural. Wuthier,35 recomienda el uso
de la cromatografía de partición entre un líquido inmovilizado en la fase estacionaria (Sephadex
G-25) y una fase móvil inmiscible, para remover estos contaminantes de la mezcla. El uso de la
cromatografía en fase reversa es descrito por Hirsch,36 Ellingboe,37 y Nystrøm.38 En
cromatografía de adsorción,39-43 las mezclas de lípidos se separan de acuerdo a las polaridades de
los compuestos, cuyo orden de separación es: hidrocarburos saturados, hidrocarburos insaturados,
ceras, aldehídos de cadena larga, triglicéridos, alcoholes de cadena larga, ácidos grasos libres,
diglicéridos, monoglicéridos, y otros.
En conclusión, el fraccionamiento puede ser realizado por los diferentes modos de HPLC
(adsorción, partición, intercambio iónico) o por cromatografía de fluidos supercríticos (SFC),44-47
la cual ha sido utilizada solo para una magnitud limitada de análisis de FAs y sus derivados. Por
otro lado, la extracción en fase sólida,48-53 es una técnica que permite disminuir los tiempos,
tratamientos y solventes, especialmente si está conectada a la derivatización simultánea en el
análisis de FAs.
Sin embargo, hasta ahora no se han estudiado los efectos de retención de los ácidos grasos
de cadena larga del tipo cáprico, caprílico, láurico, mirístico, esteárico, palmítico, por HPLC-FR.
No obstante, de la revisión bibliográfica se deduce que es conveniente utilizar soluciones de
acetato o de fosfato para controlar el pH de las fases móviles a emplear, que influye directamente
en los equilibrios de disociación y de adsorción-desorción de estos ácidos, afectando así los
tiempos de retención en el análisis. Es importante resaltar que el desarrollo de un método de
análisis para estos compuestos que no implique procedimientos tediosos es necesario para
análisis de rutina. Además, el desarrollo de este método será aplicado a la determinación de la
concentración de las especies de ácidos repartidas en las fases acuosa, oleica y microemulsión de
un sistema ácido-agua-aceite a formulación óptima.

90
Como se mencionó anteriormente, los sistemas ternarios consisten de un compuesto
anfifílico, agua y aceite, el cual puede formar una microemulsión en equilibrio con un exceso de
aceite, exceso de agua o ambos. Red y col.54 definen la microemulsión como una solución
micelar estable y traslúcida de aceite y agua que puede contener electrolitos, y uno o más
compuestos anfifílicos.
Salager55 estudió la influencia de la variación de la concentración de sal (NaCl) y surfactante
en sistemas surfactante–agua–aceite. Concluyó que estos sistemas presentan la característica de
exhibir una tensión interfacial ultra–baja para ciertas formulaciones óptimas. Este mínimo de
tensión interfacial se acompaña en general de la aparición de un sistema trifásico en el cual la
mayoría del surfactante está en la fase media (microemulsión); las fases agua y aceite contienen
concentraciones de surfactante del orden de la CMC.
Otros investigadores56,57, han estudiado la influencia de los parámetros de formulación para
promover la transición I–III–II con surfactantes nonilfenol etoxilados. El comportamiento de fase
de sistemas surfactantes noiónico/salmuera/aceite no es cualitativamente diferente a los
surfactantes aniónicos. Los factores tienden a incrementar la afinidad del surfactante mas por la
fase oleica que por la acuosa, originando transiciones I–III–II.
Así mismo, se han realizado estudios (Graciaa y col.58) de la influencia del alcano en la fase
oleica para sistemas con surfactantes no-iónicos, encontrándose para alcanos debajo del límite
superior de fase, el surfactante se reparte en el aceite mientras que para alcanos encima del límite
inferior de fase, el surfactante se reparte en la fase acuosa. De esta manera la ramificación del
alcano incrementa la CMC en la fase acuosa, y decrece también el HLB.
Andérez J. M. y col.59 estudiaron el comportamiento de fase de sistemas
surfactante/aceite/salmuera/alcohol conteniendo surfactantes aniónicos o noiónicos comerciales,
acorde a las técnicas de barridos de formulación clásicos. La concentración del surfactante fue
cambiada desde menos del 3% hasta la desaparición de las tres fases. La formulación óptima con
surfactantes aniónicos no cambió significativamente con la concentración del surfactante; por el
contrario, el incremento en la concentración del surfactante noiónico (nonilfenol etoxilado)

91
decrece la hidrofilicidad aparente del surfactante; y también tiende a depender de la naturaleza de
la fase oleica.
Saito Y. y col.60 estudiaron los efectos de la distribución de cadenas de óxido de etileno y
cadena alquílica en la estabilidad de emulsiones O/W. Concluyeron que la mezcla de
emulsificadores teniendo anchas distribuciones de cadenas de OE y cadenas alquílicas son más
efectivas para mejorar la estabilidad de emulsiones O/W, y que las temperaturas por encima de la
PIT forman emulsiones más estables que aquellas por debajo de la PIT.
Márquez N. y col.61 estudiaron la partición de surfactantes alquilfenol etoxilados en
sistemas microemulsión-aceite-agua conteniendo especies octil, nonil, decil, dodecil y dinonil
fenol. Encontró que el coeficiente de partición entre las fases acuosa y aceite obedece una ley que
puede ser expresada numéricamente por la variación en el logaritmo del coeficiente de partición
como una función lineal del EON y el ACN en la cadena alquílica. Los resultados permitieron la
estimación de la energía de transferencia de un grupo EO y de un grupo CH2 desde el aceite al
agua.
En otro trabajo62 estudió la influencia de la cadena hidrófoba del surfactante en la
partición de surfactantes alquilfenol etoxilado en sistemas microemulsión-aceite-agua. Se
encontró que el reparto del surfactante es ligeramente afectado por la estructura hidrófoba del
surfactante; mientras más ramificada sea la cadena alquílica de éste, tiende a repartirse menos en
la fase oleica. Los resultados corroboran las tendencias de la variación de la CMC y parámetros
del surfactante. Esto corresponde un argumento adicional a considerar en el coeficiente de
partición y al momento de la formulación físico-química.
Salager J. L. y col.63 estudiaron la influencia de n-pentanol en el comportamiento de fase
en sistemas surfactantes polietoxilados-agua-aceite. Encontraron que el aumento del contenido de
alcohol origina una transición retrógrada W I - W III - W I debido a que se incrementa el reparto
del surfactante en la fase oleica por efecto de la lipofilicidad interfacial del alcohol, el cual resulta
en una disminución de estas sustancias en la interfase el cual vuelve hidrofílico debido al balance
de masas.

92
Ysambertt F. y col.64 estudiaron la influencia del ACN en el comportamiento de fase en
sistemas surfactante polietoxilado-agua-aceite. Encontraron que el aumento del ACN contribuye
en el aumento en la interacción interfacial entre el surfactante y el aceite, el cual resulta en una
transición W I - W III - W II. Sin embargo, el aumento del contenido de benceno en la fase oleica
origina una transición retrógrada W I - W III - W I debido a la partición de oligómeros lipofílicos
en la fase oleica, el cual afecta el comportamiento interfacial hidrofílico de los surfactantes
restantes.
Por su parte, Chan K.65 reportó que el mecanismo molecular para que ocurra un mínimo en
la curva de tensión interfacial, es una función de la concentración del surfactante, salinidad y el
ACN de la fase oleica para soluciones diluidas de sulfonatos de petróleo. Con los tres efectos
estudiados, la tensión interfacial mínima ocurre a la concentración que es la CMC del surfactante
en la fase acuosa después de equilibrarse con la fase oleica. La tensión interfacial mínima
también coincide con el coeficiente de partición unitario. Esto implica que la interacción de
moléculas de surfactante es igual con la sal y aceite; en la interfase experimentan igual afinidad
para el aceite y sal y consecuentemente producen una alta concentración en la interfase.

93
REFERENCIAS BIBLIOGRÁFICAS
[1] Mussini, P.; Mussini, T.; Rondinini, S. Pure Appl. Chem. 1997, 69, 1007-1012.
[2] Balcan, M.; Anghel, D.; Voicu, A.; Balcan, D. Colloids Surf. A: Physicochem. Engng
Aspects, 2002, 204, 141-151.
[3] Donghyun, C.; Jeongmin, H.; Soojin, P.; Taihhyun, C. J. Chromatogr. A, 2003, 986, 199-
206.
[4] Jandera, P.; Colin, H.; Gulochon, G. Anal. Chem. 1982, 54, 435-440.
[5] Wen, D.; Olesik, S. J. Chromatogr. A. 2001, 931, 41-48.
[6] Chi-Yu, L.; Hsin-Lung, W.; Su-Hwei, C.; Hwang-Shang, K. Chromatographia. 2000, 51,
315-322.
[7] Newman, J.; Hammock, B.; J. Chromatogr., A. 2000, 925, 223-229.
[8] Meulenaer, B.; Van, G.; Vanhoutte, B.; Huyghebaert, A. J. Chromatogr. A, 2000, 896,
239-246.
[9] Van, B.; Wintermans, M. J. Chromatogr. A, 2001, 930, 109-116.
[10] Toshio, T.; Ikuhiro, I.; Shoji, M. J. Chromatogr., A, 2001, 932, 165-171.
[11] Melchior, D.; Gäb, S. J. Chromatogr. A, 2000, 894, 145-149.
[12] Koning, S; Van der Meer, B.; Alkema, G.; Jansen, H.; Brinkman, U. J. Chromatogr., A,
2001, 922, 391-399.
[13] Bruzzoniti, M.; Mentasti, E.; Pohi, C.; Riviello, J.; Sarzanini, C. J. Chromatogr., A, 2001,
925, 99-106.
[14] Nimura, N.; Fujiwara, T.; Watanabe, A.; Sekine, M.; Furuchi, T.; Yohda, M.; Yamagishi,
A.; Oshima, T.; Homma, H. Anal. Biochem. 2003, 315, 262-269.
[15] Hanko, V; Rohrer, J. Anal. Biochem. 2004, 324, 29-38.
[16] Waddington, E.; Sienuarine, K.; Puddey, I.; Croft, K. Anal. Biochem. 2001, 292, 234-244.
[17] Grechishkina, O. J. Chromatogr. A. 2002, 948, 65-67.
[18] Sanli, N.; Fonrodona, G.; Barrón, D.; Özkan, G.; Barbosa, J. J. Chromatogr. A. 2002, 975,
299-309.
[19] Shinoda, K.; Saito, H.; J. Colloid Interface Sci. 1968, 26, 70-75.
[20] Merdas, A.; Gindre, M.; Ober, R.; Nicot, C.; Urbach, W.; waks, M. J. Phys. Chem. 1996,
100, 15180-15186.

94
[21] Penfold, J.; Staples, E.; Tucker, I.; Cummins, P. J. Phys. Chem. 1996, 100, 18133-18137.
[22] Menge, U.; Lang, P.; Findenegg, G. Colloids Surf. A: Physicochem. Engng Aspects. 2000,
163, 81-90.
[23] Zhu, P.; Snyder, L.; Dolan, J.; Djordjevic, N.; Hill, D.; Sander, L.; Waeghe, T. J.
Chromatogr. A. 1996, 756, 21-39.
[24] Zhu, P.; Dolan, J.; Snyder, L. J. Chromatogr. A. 1996, 756, 41-50.
[25] Lee, W.; Cho, D.; Chun, B.; Chang, T.; Ree, M. J. Chromatogr. A. 2001, 910, 51-60.
[26] Cassia R.; Zanon V.; Trombetta, F.; Martinelli, M.; Bastos, E. Anal. Chim. Acta 2004,
505, 223–226.
[27] Mitakos, A.; Panderi, I. Anal. Chim. Acta 2004, 505, 107–114.
[28] Sánchez, A.; Rodríguez A.; López, J.; Paseiro, P. J. Chromatogr. A, 2004, 1032, 7–15.
[29] Chen, S.; Chuang, Y. Anal. Chim. Acta 2002, 465, 145-152.
[30] Tran, T.; Christophersen, B. Biochim. Biophy. Acta 2002, 1583, 195-203.
[31] Mototeru, Y. Anal. Chim. Acta 2002, 465, 227-.234.
[32] Amet, Y.; Adas, F.; Berthou, F. Anal. Chim. Acta 2002, 465, 193-199.
[33] Brondz, I. Anal. Chim. Acta 2002, 465, 1-7.
[34] Ohba, Y.; Kuroda, N.; Nakashima, K. Anal. Chim. Acta 2002, 465, 101-108.
[35] Wuthier, R. J. Lipids Res. 1966, 7, 558-563.
[36] Hirsch, J. J. Lipids Res. 1963, 4, 1-9.
[37] Ellingboe J.; Nystrøm, E.; Sjøvall, J. J. Lipids Res. 1970, 11, 266-274.
[38] Nystrøm, E.; Sjøvall, J. Meth. Enzymol. 1975, 35B, 378-384.
[39] Rouser, G.; Kritchevsky, G.; Yamamoto, A. Lipid Chromatographic Analyses, 2nd Edition,
Vol. 3, Marcel Dekker, New York, 1976, 713-776.
[40] Carroll, K. Lipid Chromatographic Analysis, 2nd Edition, Vol. 1, Marcel Dekker, New
York, 1976, 173-214.
[41] Singleton, W.; Gray, M.; Brown, M.; White, J. J. Am. Oil. Chem. Soc. 1965, 42, 53-60.
[42] Comfurius, P.; Zwaal, R. Biochim. Biophys. Acta. 1977, 488, 36-41.
[43] Kaluzny, M.; Duncan, L.; Merritt, M.; Epps, D. J. Lipid Res. 1985, 26, 135-142.
[44] Alkio, M.; González, C.; Jantti, M.; Aaltonen, O. J. Am. Oil Chem. Soc. 2000, 77, 315-
321.
[45] Hawthorne, S.; Millar, D.; Nivens, D.; White, D. Anal. Chem. 1992, 64, 405-411.

95
[46] Cummins, M.; Wells, R. J. Chromatogr. B. 1997, 694, 11-18.
[47] Laakso, P. en: Christie, W. (Ed.), Advances in Lipid Methodology, Vol. 1, Oily Press, Ayr.
1992, 81-92.
[48] Battistutta, F.; Buiatti, S.; Zenarola, C.; Zironi, R. J. High Resolut. Chromatogr. 1994, 17,
662-667.
[49] Pan, L.; Pawliszyn, J. Anal. Chem. 1997, 69, 196-201.
[50] Hirschlag, H.; Koster, R. Anal. Chem. 1998, 362, 274-279.
[51] Abalos, M; Bayona, J.; Pawliszyn, J. J. Chromatogr. 2000, 873, 107-112.
[52] Wilson, R.; Smith, P.; Wilson, P.; Shepherd, M.; Riemersma, R. Anal. Biochem. 1997,
248, 76-81.
[53] Pan, L.; Adams, M.; Pawliszyn, J. Anal. Chem. 1995, 67, 4396-4404.
[54] Reed, R.; Healy, R. Some Physicochemical Aspects of Microemulsion Floo ding.
Academies Press. New York. 1977.
[55] Salager, J. Encyclopedia of Emulsions Technology. Marcel Dekker, Inc., New Cork. 1988,
3, 247-280.
[56] Bourrel, M.; Salager, J.; Schechter, R.; Wade W. J. colloid Interf. Sci., 1980, 75, 451-457.
[57] Bourrel, M.; Koukounis, Ch.; Schechter, R.; Wade, W. J. Dispers. Sci. Technol. 1980, 1,
13-19.
[58] Graciaa, A.; Barakat, Y.; Emary, M.; Fortney, L.; Schechter, R.; Yiv, S.; Wade, W. J.
colloid Interf. Sci. 1982, 89, 209-216.
[59] Andérez, M.; Salager, J.; Graciaa, A.; Lachaise, J. 8th International Symposium on
Surfactants in Solution. 1990, 6, 10–15.
[60] Saito, Y.; Sato, T.; Anazawa, I. J. Amer. Oil Chem. Soc. 1990, 67, 145–148.
[61] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1995, 100,
225-231.
[62] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1998, 131, 45-
49.
[63] Salager, J.; Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J. Langmuir. 1995, 11, 37-41.
[64] Ysambertt, F.; Antón, R.; Salager, J. Coll. Surf. A. 1997, 125, 131-136.
[65] Chan, K.; Shah, D. J. Disp. Sci. Technol. 1980, 1, 55–95.

96
C A P I T U L O V I

97
DESARROLLO DE MÉTODOS DE ANÁLISIS DE ÁCIDOS CARBOXÍLICOS GRASOS (FAs) POR HPLC.
El análisis de FAs se realizó empleando la modalidad fase reversa, en la cual se emplea
una columna no polar debido al carácter lipofilico de estos compuestos. Por otro lado, el uso de
fases móviles acuosas para el análisis hace necesario controlar adecuadamente el pH, debido a la
disociación que tiene lugar durante el proceso de separación. En el siguiente capitulo se proponen
varias metodologías de análisis y separación de una mezcla de FAs; así como el estudio de la
influencia de la temperatura y polaridad de la fase móvil en la retención de dichos compuestos en
el proceso cromatográfico.
SECCIÓN EXPERIMENTAL
Equipos
El análisis de los ácidos carboxílicos de cadena larga se realizó en un cromatógrafo
líquido modular de la Waters, constituido por los siguientes módulos (marca Waters):
Inyector universal U6K
Computador PC 5500 con el software Millenium; para el procesamiento de los
resultados.
Detector de arreglo de fotodiodos (PDA), 996
Controlador de solventes 600
Bomba cuaternaria distribuidora de solventes, M6000A
Columnas cromatográficas analíticas de fase reversa: (250 x 4.6 mm; de Merk) y
Lichropher C8 de 5-µm ( 250 x 4.6 mm; de Merk)
Materiales
Sistemas de purificación de solventes y muestras de la Waters Millipore, con filtros de
0,22 y 0,45 µm acuosos y orgánicos.

98
Sistema de desgasificación de solventes con Helio.
Baño ultrasónico, marca Bransonic modelo 1200.
Jeringas de desgasificación de 10 mL y de inyección de 25 µL, marca Hamilton
Balanza analítica, marca ACCULAB, sensibilidad ± 0,0001 g
Material de vidrio en general.
Reactivos.
Todos los solventes utilizados para la preparación de las fases móvil y de las muestras fueron
de grado HPLC, metanol (MeOH), acetonitrilo (ACN), isopropanol, n-Heptano (Baker
Chemicals). Todos desgasificados con helio y sonicados.
Se estudiaron las siguientes muestras: ácido caprílico (C8), ácido cáprico (C10), ácido láurico
(C12), ácido mirístico (C14), ácido palmítico (C16) y ácido esteárico (C18), todos marca Merck.
Etoxyl (Venezuela) suministró los surfactantes nonilfenol etoxilados, empleados en el estudio
de retención, para comparación con los ácidos carboxílicos
Procedimiento Experimental
Análisis de ácidos carboxílicos por HPLC-fase reversa.
El estudio se inició evaluando diferentes proporciones de mezclas de solventes
constituidas por metanol, agua y acetonitrilo con polaridades diferentes como fases móvil en
forma isocrática. Se evaluaron dos fases estacionarias: octilsilano (RP-8) y octadecilsilano (RP-
18), hasta establecer el método óptimo para el estudio de mezclas de los ácidos carboxílicos.
Todos los ácidos se prepararon en metanol a una concentración de 0,1 M y se inyectaron
alícuotas de 10 µL en el cromatógrafo líquido. Se empleó un flujo 1 mL/min.
Efecto de la temperatura en la retención de los ácidos carboxílicos y surfactantes no-iónicos
en la separación por HPLC-fase reversa.

99
Para el estudio de la influencia de la temperatura en la retención de los ácidos carboxílicos
se empleó un horno para columna modelo Eppendorf-CH-30. Se cambió la temperatura de la
columna de 288 a 323 K y se empleó acetonitrilo como fase móvil. Se inyectaron al sistema
cromatográfico alícuotas de 10 µL de la mezcla de ácidos grasos (preparada en metanol al 0,2 %
p/v). En el caso de los surfactantes noiónicos polietoxilados se empleó una columna Lichrospher
NH2 (5µm, 250 x 4 mm), y se preparó a la misma concentración.
Efecto de la polaridad de la fase móvil en la retención de los ácidos carboxílicos por HPLC-
fase reversa.
Debido al carácter lipofílico de los ácidos carboxílicos, su afinidad por la fase estacionaria
no polar (RP-18) incrementa con la longitud de la cadena alquílica del ácido. El aumento en la
polaridad de la fase móvil influye en dicho comportamiento. Por tanto, se evaluó el efecto de
adicionar progresivamente acetonitrilo a la fase móvil en la retención de los ácidos carboxílicos
en la fase estacionaria no polar. El tiempo de retención (tr) de los ácidos se obtuvo de cinco
determinaciones individuales (n = 5).
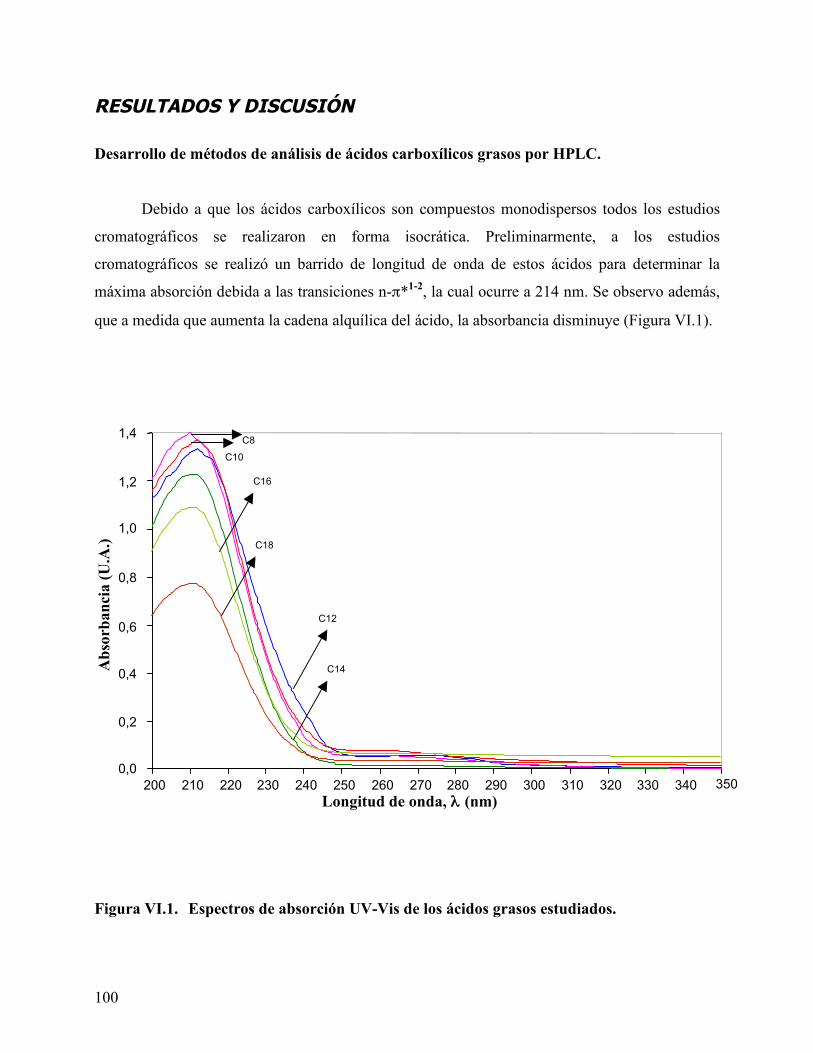
100
RESULTADOS Y DISCUSIÓN
Desarrollo de métodos de análisis de ácidos carboxílicos grasos por HPLC.
Debido a que los ácidos carboxílicos son compuestos monodispersos todos los estudios
cromatográficos se realizaron en forma isocrática. Preliminarmente, a los estudios
cromatográficos se realizó un barrido de longitud de onda de estos ácidos para determinar la
máxima absorción debida a las transiciones n-π*1-2, la cual ocurre a 214 nm. Se observo además,
que a medida que aumenta la cadena alquílica del ácido, la absorbancia disminuye (Figura VI.1).
Figura VI.1. Espectros de absorción UV-Vis de los ácidos grasos estudiados.
200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Longitud de onda, λ (nm)
Abs
orba
ncia
(U.A
.)
C8 C10
C12
C14
C18
C16
350

101
Es importante destacar que debido a las propiedades surfactante de estos ácidos y por la
variedad en el número de átomos de carbono de la cadena lipofílica, es difícil encontrar una fase
móvil donde toda la mezcla sea soluble. Por otro lado, los ácidos de mayor cadena alquílica C16
y C18, son poco solubles en acetonitrilo. Por tanto, se evaluó con metanol y se observó que todos
los ácidos son solubles (debido a la formación de puentes de hidrogeno). Se comenzó el estudio
fraccionando mezclas de ácidos carboxílicos desde C8 hasta C18, en una columna octilsilano. Se
evaluaron varias fases móviles y se logro la separación de los ácidos C14, C16, C18 con una fase
móvil ACN/H2O (80/20). Este análisis se realizó acidificando la fase móvil (pH = 4) con una
solución de ácido acético para mantener los ácidos en su forma ionizada (Supresión iónica). Sin
embargo, no se obtuvo buena resolución para los ácidos C8 y C10, en consecuencia, esta fase
estacionaria no sería adecuada para la separación efectiva de estos ácidos cuando se analicen en
un sistema surfactante-agua-aceite que contenga una mezcla de estos.
En vista de lo observado se continuó el estudio pero empleando la fase estacionaria
octadecilsilano (C18), con la finalidad de aumentar el factor de retención y la eficiencia en la
separación de los ácidos C8 y C10, sin afectar la resolución de los otros ácidos (C12, C14, C16 y
C18). Las condiciones óptimas de separación de la mezcla de ácidos C8, C10, C12, y C14 se
alcanzó con una fase móvil 70/30 ACN/H2O (pH 4)., por lo cual se eligió el metanol en la fase
móvil. Empleando una mezcla acetonitrilo/metanol 95:5 como fase móvil, se observó la
separación de C8, C10, C12 y C14 pero con muy poca resolución. Debido a la solubilidad de toda
la mezcla de ácidos en metanol, se incorporó a la fase móvil. Sin embargo, no se obtuvo buena
resolución. Por tanto, se incorporó nuevamente el agua (modificador polar) para reducir la
solubilidad de los ácidos en la fase móvil y aumentar su interacción con la fase estacionaria.
Después de varias mezclas de acetonitrilo/metanol/agua acidulada con ácido acético, se
encontró que la mezcla ACN/MeOH/H2O, 80:10:10 resulto óptima para fraccionar la mezcla
C12, C14, C16 y C18 (todas las muestras se inyectaron disueltas en metanol), como se observa en
la Figura VI.2. Sin embargo, para optimizar un método de análisis se debe evitar el uso de pasos
engorrosos que encarezcan el análisis. Tomando en cuenta esta idea, se ensayaron diferentes
mezclas de metanol/acetonitrilo sin el uso de la supresión iónica, es decir empleando la fase
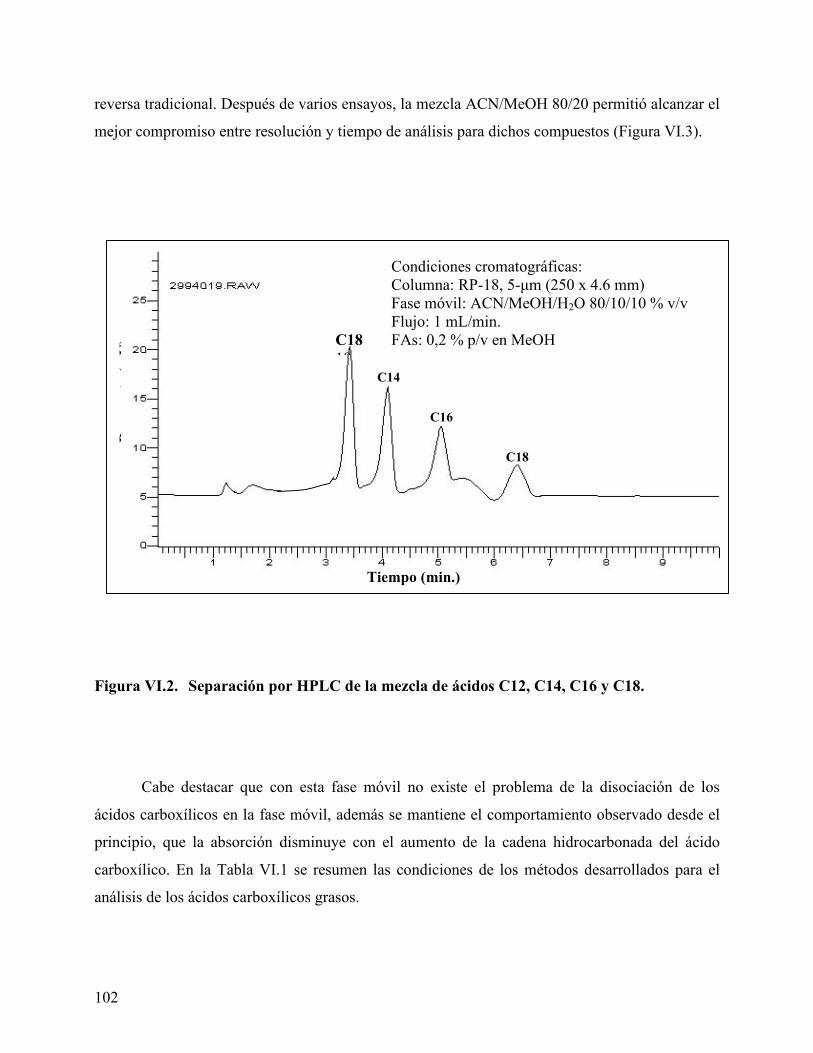
102
reversa tradicional. Después de varios ensayos, la mezcla ACN/MeOH 80/20 permitió alcanzar el
mejor compromiso entre resolución y tiempo de análisis para dichos compuestos (Figura VI.3).
Figura VI.2. Separación por HPLC de la mezcla de ácidos C12, C14, C16 y C18.
Cabe destacar que con esta fase móvil no existe el problema de la disociación de los
ácidos carboxílicos en la fase móvil, además se mantiene el comportamiento observado desde el
principio, que la absorción disminuye con el aumento de la cadena hidrocarbonada del ácido
carboxílico. En la Tabla VI.1 se resumen las condiciones de los métodos desarrollados para el
análisis de los ácidos carboxílicos grasos.
Tiempo (min.)
Res
pues
ta (m
V)
Condiciones cromatográficas: Columna: RP-18, 5-µm (250 x 4.6 mm) Fase móvil: ACN/MeOH/H2O 80/10/10 % v/v Flujo: 1 mL/min. FAs: 0,2 % p/v en MeOH C18
18 C12 C14
C16
C18
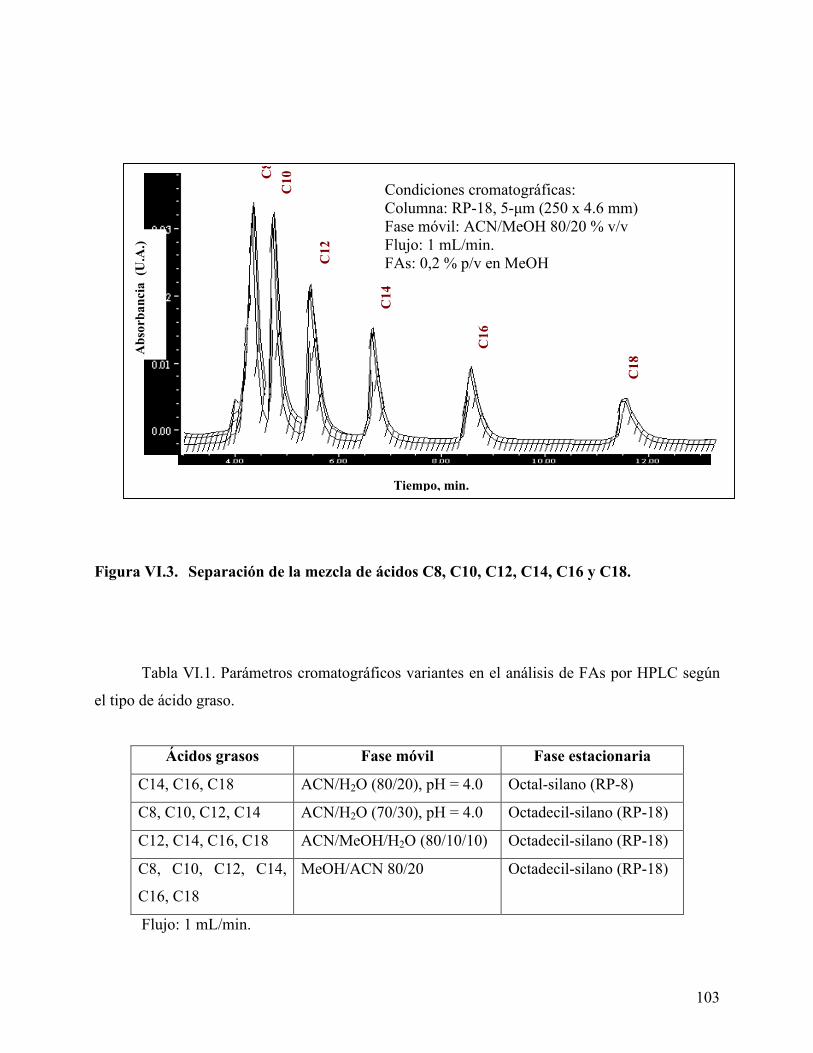
103
Figura VI.3. Separación de la mezcla de ácidos C8, C10, C12, C14, C16 y C18.
Tabla VI.1. Parámetros cromatográficos variantes en el análisis de FAs por HPLC según
el tipo de ácido graso.
Ácidos grasos Fase móvil Fase estacionaria
C14, C16, C18 ACN/H2O (80/20), pH = 4.0 Octal-silano (RP-8)
C8, C10, C12, C14 ACN/H2O (70/30), pH = 4.0 Octadecil-silano (RP-18)
C12, C14, C16, C18 ACN/MeOH/H2O (80/10/10) Octadecil-silano (RP-18)
C8, C10, C12, C14,
C16, C18
MeOH/ACN 80/20 Octadecil-silano (RP-18)
Flujo: 1 mL/min.
C8
C10
C12
C14
C16
C18
T im e ( m in .)
Abs
orba
nce
Condiciones cromatográficas: Columna: RP-18, 5-µm (250 x 4.6 mm) Fase móvil: ACN/MeOH 80/20 % v/v Flujo: 1 mL/min. FAs: 0,2 % p/v en MeOH
Tiempo, min.
Abs
orba
ncia
(U
.A.)

104
Efecto de la temperatura en la retención de compuestos por HPLC.
Tomando como referencia la teoría de la termodinámica en el proceso cromatográfico,3-
4,se realizaron estudios del comportamiento de retención de diferentes compuestos en
cromatografía líquida, evaluando el efecto de la temperatura y del solvente sobre dicho
comportamiento. Como se mencionó anteriormente, el proceso cromatográfico es un proceso de
distribución consecutivo de solutos entre las fases móvil y estacionaria cuando se mueven a
través de la columna.
La constante de equilibrio (K) de moléculas de soluto entre la fase estacionaria y la fase
móvil se puede relacionar a los cambios de energía libre (ΔGº), entalpía (ΔHº) y entropía (ΔSº)
asociado al traslado de un mol de analito de la fase móvil a la fase estacionaria. Este cambio de
energía viene expresado por la siguiente expresión (la cual se asocia a los procesos
cromatográficos mediante el uso del factor de retención):
KlnRTºSTºHºG !="!"=" (1)
De esta forma, la conducta de retención de un soluto en un sistema cromatográfico puede
expresarse a través del factor capacidad como sigue:
!+"
+"
=!+"
#= lnR
ºSln
RT
ºG'ln
RT
Hº- k (2)
donde, R es la constante de los gases, T es la temperatura absoluta y φ es la relación de fase de la
columna (es decir, la relación del volumen de fase estacionaria a la fase móvil). Las variables
termodinámicas pueden entonces obtenerse de los datos de la curva de retención vs. el inverso de

105
la temperatura (1/T), conocida como curva de Van Hoff. Si el diagrama de Van Hoff es lineal,
entonces se puede asumir que la entalpía y la entropía son constantes y, que el mecanismo del
proceso de retención no cambia por encima del rango de temperatura en estudio. Así, ΔHº puede
obtenerse de la pendiente y ΔSº del intercepto de la línea de regresión, siempre que se conozca φ.
El factor capacidad es adimensional e independiente de algunos parámetros geométricos
de la columna o el sistema HPLC. Esto puede considerarse como una característica
termodinámica del sistema eluente-compuesto-adsorbente. Por otro lado, la ecuación (2), indica
que un incremento en la temperatura disminuirá el valor de k', de tal forma que el tiempo de
retención actual también disminuirá. Para la mayoría de los sistemas esta disminución no excede
el 50% de la reducción del tiempo de retención del componente a temperatura ambiente.
En este estudio se empleo la siguiente definición de relación de fase:5,6-14
φ = (VG – VM) / VM (3)
donde VG y VM son el volumen geométrico (columna vacía) y muerto de la columna (esto se
determinó empleando la velocidad de flujo y el tiempo de elusión de compuestos no retenidos,
metanol o acetonitrilo), respectivamente. La relación de fase definida en la ecuación (3) es una
cantidad adimensional y se puede determinar rápidamente, debido a que VG se calcula
simplemente de la longitud y el diámetro interno de la columna.5,6-14 Empleando este
procedimiento, se obtuvo para la columna en estudio un valor de φ= 0,54.
Para este estudio se inyectaron al sistema cromatografito alícuotas de 10 µL de la mezcla
de ácidos grasos (preparada en metanol al 0,2 % p/v) y se empleo acetonitrilo como fase móvil.
Todas las medidas se hicieron bajo condiciones isocráticas. El tiempo de retención (tr) de los
ácidos se obtuvo de cinco determinaciones individuales (n = 5). Se cambió la temperatura de la
columna de 288 a 323 K. En la Figura VI.4 se muestra, para todos los ácidos, la variación del ln
k’ en función del inverso de la temperatura absoluta (1/T). La pendiente positiva indica una
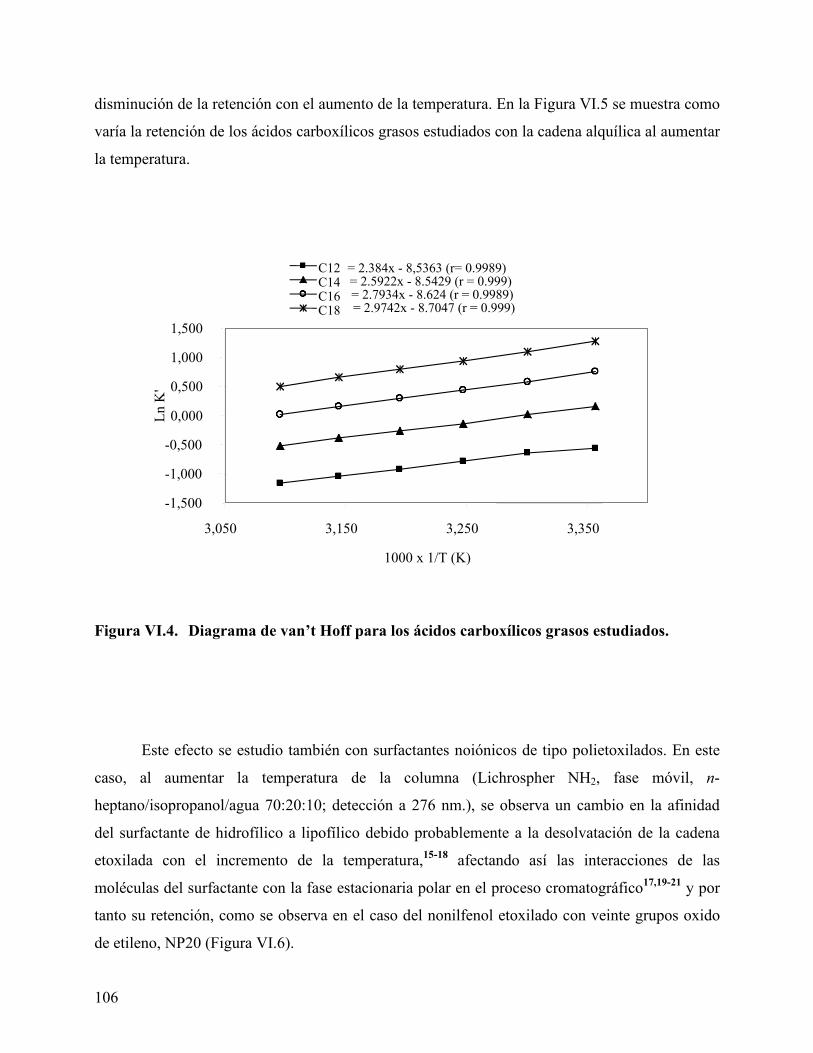
106
disminución de la retención con el aumento de la temperatura. En la Figura VI.5 se muestra como
varía la retención de los ácidos carboxílicos grasos estudiados con la cadena alquílica al aumentar
la temperatura.
Figura VI.4. Diagrama de van’t Hoff para los ácidos carboxílicos grasos estudiados.
Este efecto se estudio también con surfactantes noiónicos de tipo polietoxilados. En este
caso, al aumentar la temperatura de la columna (Lichrospher NH2, fase móvil, n-
heptano/isopropanol/agua 70:20:10; detección a 276 nm.), se observa un cambio en la afinidad
del surfactante de hidrofílico a lipofílico debido probablemente a la desolvatación de la cadena
etoxilada con el incremento de la temperatura,15-18 afectando así las interacciones de las
moléculas del surfactante con la fase estacionaria polar en el proceso cromatográfico17,19-21 y por
tanto su retención, como se observa en el caso del nonilfenol etoxilado con veinte grupos oxido
de etileno, NP20 (Figura VI.6).
C12C14C16C18 = 2.9742x - 8.7047 (r = 0.999)
= 2.7934x - 8.624 (r = 0.9989)= 2.5922x - 8.5429 (r = 0.999)= 2.384x - 8,5363 (r= 0.9989)
-1,500
-1,000
-0,500
0,000
0,500
1,000
1,500
3,050 3,150 3,250 3,350
1000 x 1/T (K)
Ln
K'
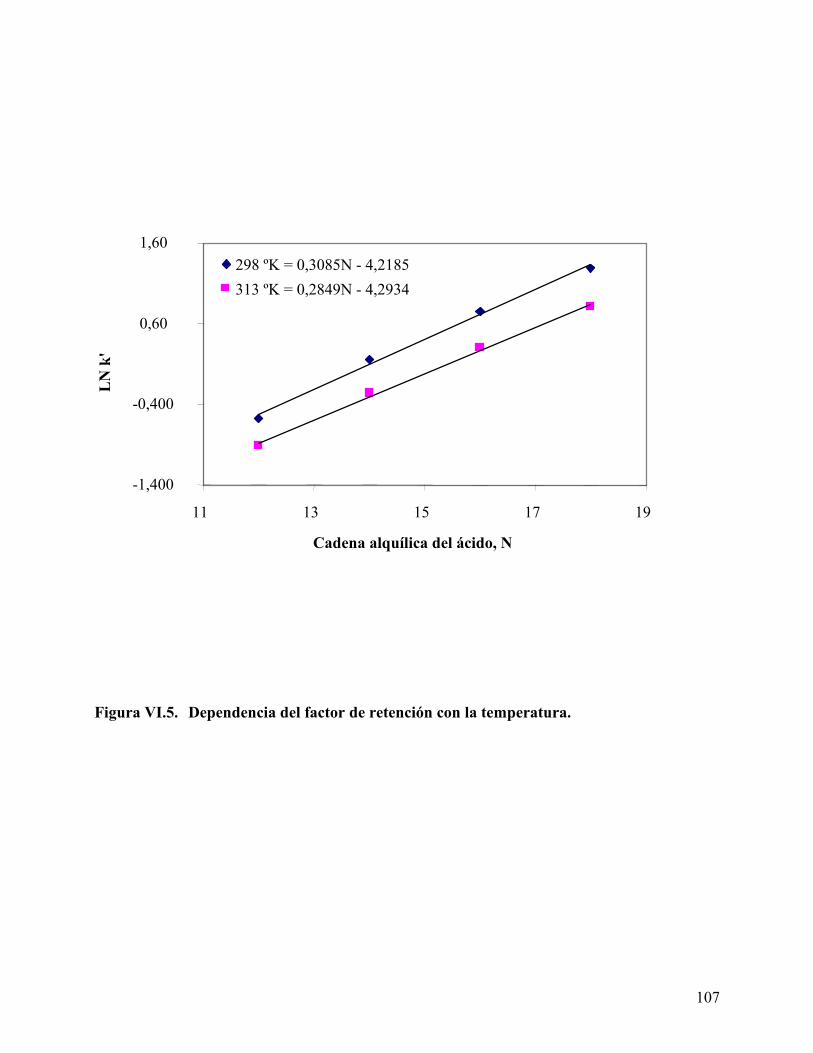
107
Figura VI.5. Dependencia del factor de retención con la temperatura.
-1,400
-0,400
0,600
1,600
11 13 15 17 19
Cadena alquílica del ácido, N
LN
k'
298 ºK = 0,3085N - 4,2185 (0.9949) 313 ºK = 0,2849N - 4,2934 (0.9963)
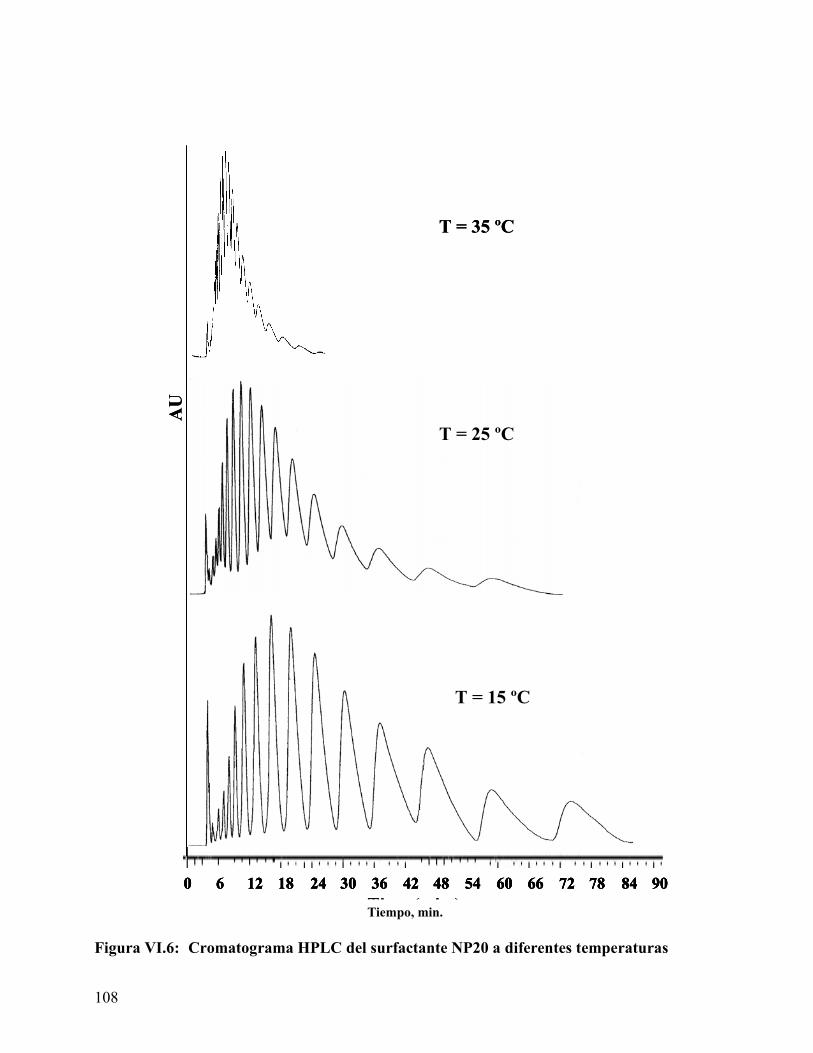
108
Figura VI.6: Cromatograma HPLC del surfactante NP20 a diferentes temperaturas
Time (min.)0 6 12 18 24 30 36 42 48 54 60 66 72 78 84 90
T = 15 ºC
AU
T = 25 ºC
T = 35 ºC
Time (min.)0 6 12 18 24 30 36 42 48 54 60 66 72 78 84 900 6 12 18 24 30 36 42 48 54 60 66 72 78 84 900 6 12 18 24 30 36 42 48 54 60 66 72 78 84 90
T = 15 ºC
AU
T = 25 ºC
T = 35 ºC
Tiempo, min.
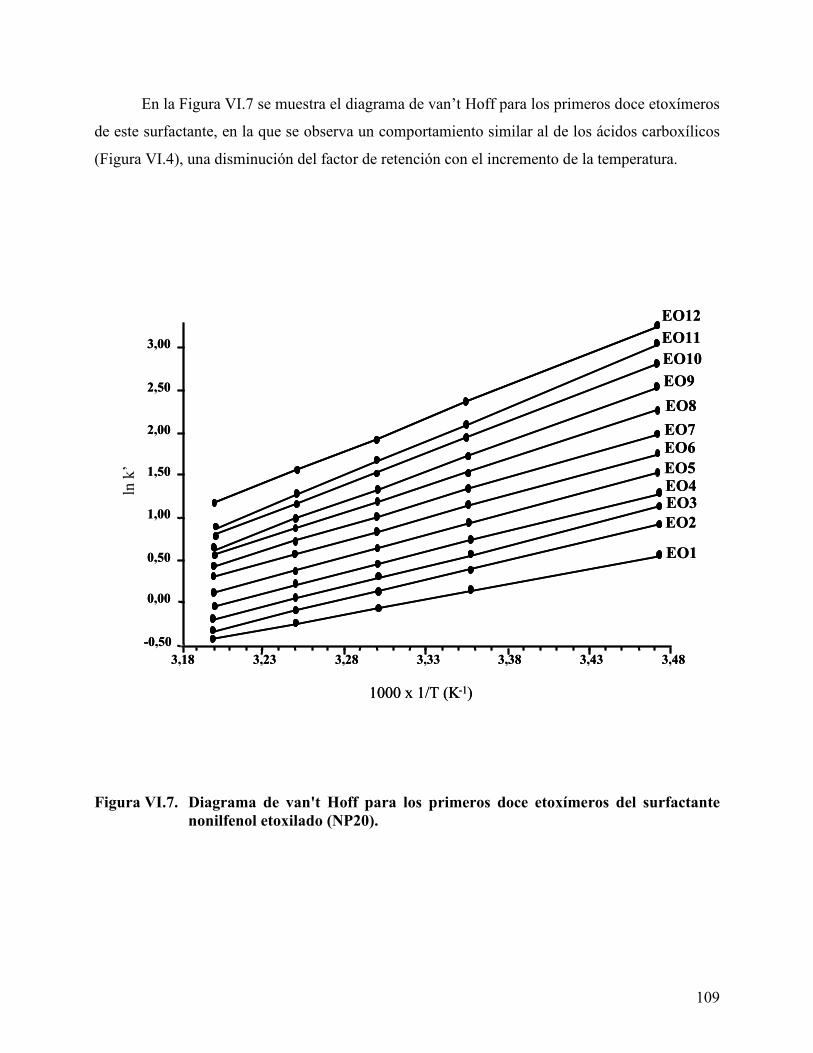
109
En la Figura VI.7 se muestra el diagrama de van’t Hoff para los primeros doce etoxímeros
de este surfactante, en la que se observa un comportamiento similar al de los ácidos carboxílicos
(Figura VI.4), una disminución del factor de retención con el incremento de la temperatura.
Figura VI.7. Diagrama de van't Hoff para los primeros doce etoxímeros del surfactante
nonilfenol etoxilado (NP20).
1000 x 1/T (K - 1 )
ln k
’
- 0.50
0.00
0.50
1.00
1.50
2.00
2.50
3.00
3.18 3.23 3.28 3.33 3.38 3.43 3.48
EO1 EO2
EO11 EO10 EO9
EO12
EO5 EO6 EO7 EO8
EO4 EO3
1000 x 1/T (K - 1 )
- 0,50
0,00
0,50
1,00
1,50
2,00
2,50
3,00
3,18 3,23 3,28 3,33 3,38 3,43 3,48
EO1 EO2
EO11 EO10 EO9
EO12
EO5 EO6 EO7 EO8
EO4 EO3

110
Por otro lado, se calculó la entalpía y entropía de transferencia del soluto de la fase móvil
a la fase estacionaria para ambos tipos de compuestos anfifilicos: ácidos carboxílicos (Figura
VI.8A) y nonilfenol etoxilado (Figura VI.8B). Como se observa en la Figura, en ambos casos el
ΔHº es negativo, lo que corrobora que la adsorción del soluto (cadena alquilita en la fase no polar
y etoxímeros en la fase polar) es favorable desde el punto de vista energético.
No obstante, para los ácidos carboxílicos el ΔSº se mostró esencialmente independiente
del ácido, mientras que para el nonilfenol, los valores negativos variantes del ΔSº, indican un
incremento en el orden molecular resultante de la asociación del analito con los ligandos
hidrofílicos de la fase estacionaria. En todos los casos, se observó un comportamiento lineal entre
los parámetros termodinámicos de retención y el grado de etoxilación o cadena alquílica para los
nonilfenol etoxilados y ácidos carboxílicos, respectivamente.
La disminución promedio en ΔHº y ΔGº por grupo metileno adicional en los ácidos
carboxílicos es 0,60 y 0,74 kJ/mol (estos valores se obtuvieron a partir de los datos de la Figura
IV.8A), respectivamente. La conducta observada es consistente con la selectividad de los grupos
alquílicos de la fase estacionaria (fase reversa) para separar mezclas de ácidos de acuerdo a su
cadena hidrófoba. Es importante resaltar que esta información termodinámica se obtuvo a partir
de la ecuación (2), ampliamente reportada en la literatura22-27.
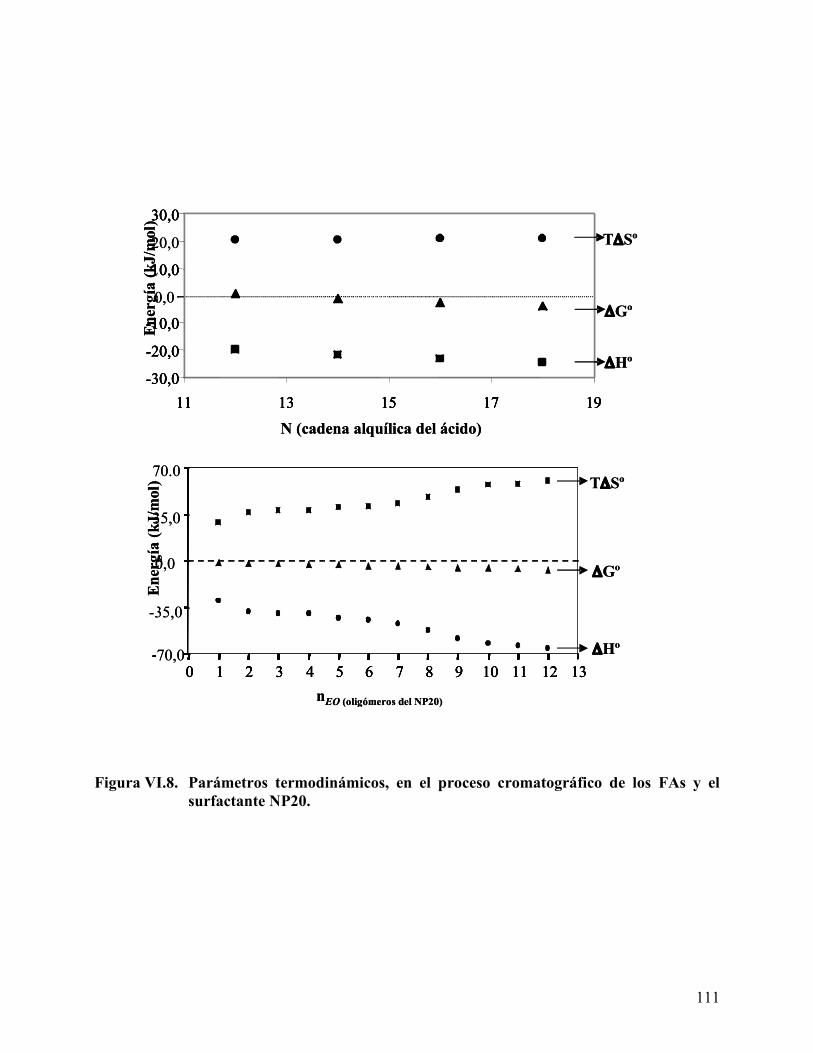
111
Figura VI.8. Parámetros termodinámicos, en el proceso cromatográfico de los FAs y el
surfactante NP20.
N (cadena lipofilica )
-
-
-
-
-
-
-30,0
-20,0
-10,0
10,0
20,0
30,0
11 13 15 17 19
N (cadena alquílica del ácido)
-70,0
-35,0
0,0
35,0
70.0
0 1 2 3 4 5 6 7 8 9 10 11 12 130 1 2 3 4 5 6 7 8 9 10 11 12 13
nEO (oligómeros del NP20)
En
erg
ía (
kJ
/mol)
-30,0
-20,0
-10,0
0,0
10,0
30,0
11 13 15 17 19
!Hº
!Gº
T!Sº
!Hº
!Gº
T!Sº
En
erg
ía (
kJ
/mol)
N (cadena lipofilica )
-
-
N (cadena lipofilica )
-
-
-
-
-
-
-
-
-
-
-30,0
-20,0
-10,0
10,0
20,0
30,0
11 13 15 17 19
N (cadena alquílica del ácido)
-70,0
-35,0
0,0
35,0
70.0
0 1 2 3 4 5 6 7 8 9 10 11 12 130 1 2 3 4 5 6 7 8 9 10 11 12 13
nEO (oligómeros del NP20)
En
erg
ía (
kJ
/mol)
-30,0
-20,0
-10,0
0,0
10,0
30,0
11 13 15 17 19
!Hº
!Gº
T!Sº
!Hº
!Gº
T!Sº
En
erg
ía (
kJ
/mol)

112
Efecto de un solvente polar en la fase móvil en la retención de los ácidos grasos.
Como se mencionó anteriormente, a mayor longitud de la cadena alquílica del ácido más
hidrófobo es este, y por tanto menos soluble en la fase móvil polar, la cual tiene que escogerse en
función del tipo de ácido. En el siguiente estudio se evaluó la influencia en la retención de estos
compuestos con la variación de la polaridad de la fase móvil. Con metanol puro como fase móvil,
los ácidos grasos de cadena menor a 10 átomos de carbono emergen en un solo pico. No obstante,
con la adición de acetonitrilo, la polaridad de la fase móvil se puede ajustar para producir la
separación adecuada como se mostró en la Figura VI.3.
Todos los estudios cromatográficos se realizaron en forma isocrática para garantizar
reproducibilidad. Para el estudio de la selectividad se inicio el análisis con 100% de metanol
como fase móvil, observándose que los ácidos de cadena mas corta (C8, C10) eluyen junto al
frente del solvente. Esto es debido a que la polaridad del metanol no es lo suficientemente alta
como para permitir una mayor interacción de los ácidos estudiados con la cadena alquílica de la
fase estacionaria.
En la Figura VI.9, se muestra la variación del logaritmo del factor de retención con el
número de átomos de carbono de la cadena alquílica (N) de los ácidos estudiados. De los datos de
la Figura VI.9 se obtiene la siguiente expresión:
ln k’ = a N + b (4)
En este caso a = 0.18 y b = -2.5. Esto indica que la retención o factor capacidad (como ln
k’) incrementa de forma lineal con la longitud de la cadena del ácido la cual interactúa con la fase
estacionaria. Además, el valor del intercepto b depende de la proporción acetonitrilo/metanol en
la fase móvil e indica la retención característica del grupo carboxílico.
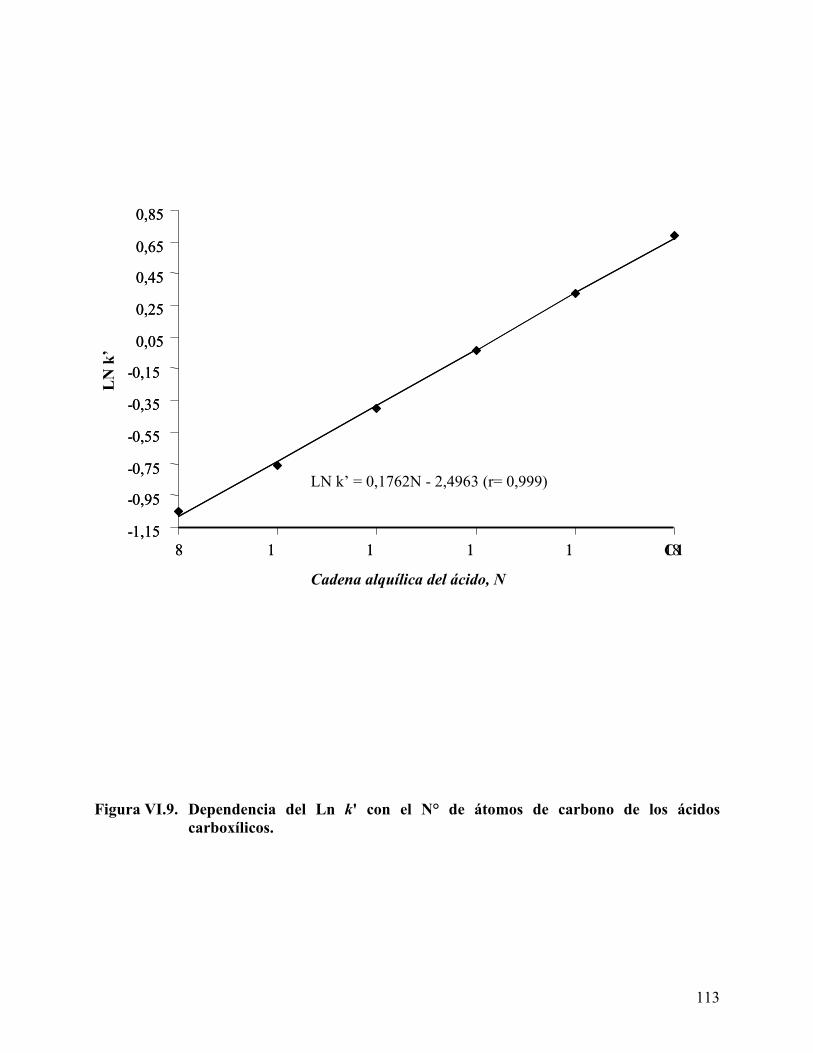
113
Figura VI.9. Dependencia del Ln k' con el N° de átomos de carbono de los ácidos carboxílicos.
- 1,15
- 0,95
- 0,75
- 0,55
- 0,35
- 0,15
0,05
0,25
0,45
0,65
0,85
8 10
12
14
16
18
LN
k’
Cadena alquílica del ácido, N
LN k’ = 0,1762N - 2,4963 (r= 0,999)
- 1,15
- 0,95
- 0,75
- 0,55
- 0,35
- 0,15
0,05
0,25
0,45
0,65
0,85
8 10
12
14
16
C18 18 C12

114
En la Figura VI.10 se muestra la variación del ln k’ en función del contenido de
acetonitrilo en la fase móvil para los diferentes ácidos. Como se observa en esta figura, la
retención de todas las especies incrementa con el contenido de acetonitrilo en la fase móvil, es
decir cuando esta se hace más polar. Estos datos pueden ser representados por una función lineal
de la siguiente forma:
ln k’ = c X + d (5)
donde X es la fracción volumen de acetonitrilo en la fase móvil y, c y d son parámetros
determinados por la regresión lineal y dependen de las otras condiciones de análisis. Se encontró
que estos parámetros varían linealmente con N. Por tanto las ecuaciones previas (4) y (5) se
pueden relacionar en una expresión bilineal del ln k’ como una función de X y N, con α = -2.45,
β= -0.115, γ= 0.174 y δ= 0.0555, a 298 K:
ln k = α + βX + γN + δXN (6)
En el caso de los surfactantes no-iónicos polietoxilados, el acetonitrilo disminuye su
tiempo de retención en fase reversa.6 Con los ácidos estudiados se produce el efecto contrario,
debido probablemente a su comportamiento hidrófobo. Para una determinación individual de
estos compuestos se puede escoger entre usar solo metanol o mezclas de metanol/acetonitrilo
como fase móvil. Sin embargo, es importante tomar en cuenta que el acetonitrilo produce un
ensanchamiento en los picos así como el efecto tailling (“cola de novia”), lo cual no es favorable.
No obstante, es un método alternativo a las mezclas acuosas que normalmente se emplean en fase
reversa pero que implicaría el empleo de una solución buffer para regular el pH de la fase móvil y
evitar así la disociación de estos compuestos durante el análisis.
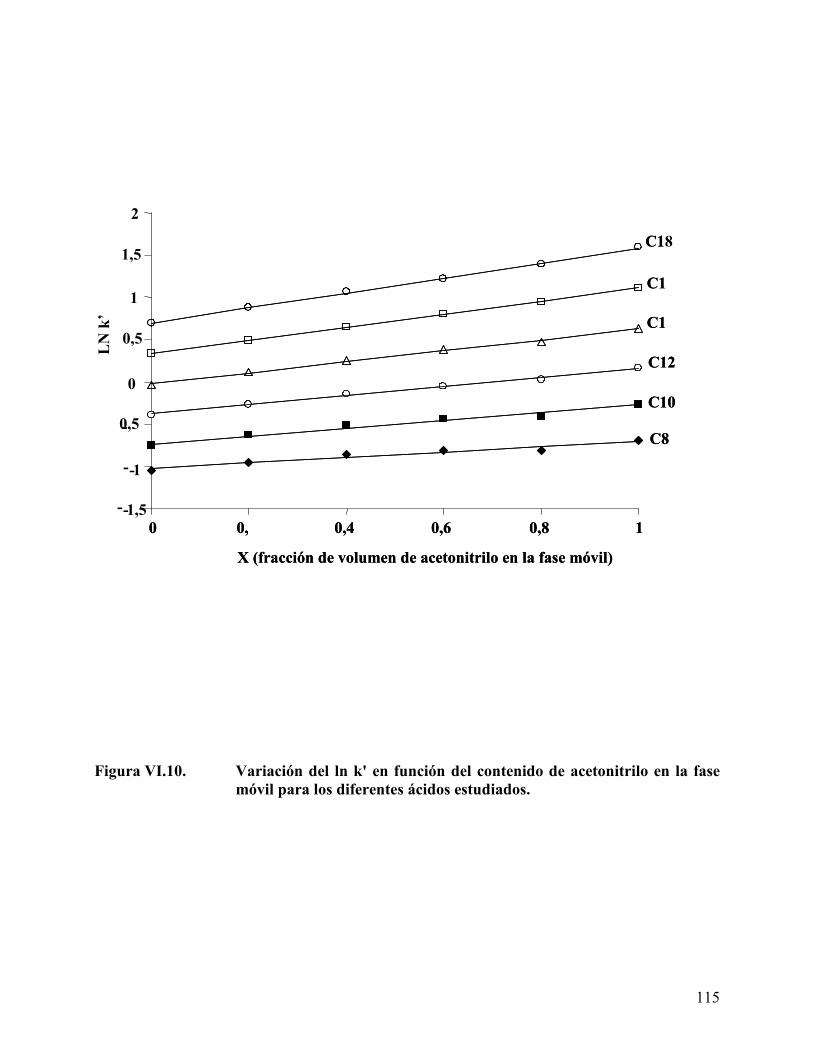
115
Figura VI.10. Variación del ln k' en función del contenido de acetonitrilo en la fase móvil para los diferentes ácidos estudiados.
- 1,5
- 1
-
0 0,2
0,4 0,6 0,8 1
X (fracción de volumen de acetonitrilo en la fase móvil)
C8
C10
C12
C14
C16
C18
0,5
0
0,5
1
1,5
2
-
-
-
0 0,2
0,4 0,6 0,8 1
X (fracción de volumen de acetonitrilo en la fase móvil)
LN
k’
C8
C10
C12
C14
C16
C18 18 C12

116
CONCLUSIONES
La afinidad de los ácidos carboxílicos grasos por la fase estacionaria octadecilsilano
aumenta linealmente con la longitud de la cadena alquílica del ácido y con la polaridad de la fase
móvil, aumentando sus tiempos de retención Los métodos de análisis desarrollados pueden ser
aplicados en diferentes campos de interés.
Al aumentar la temperatura de la columna, la retención de estos compuestos tiende a
disminuir. No obstante, ΔSº es esencialmente independiente del ácido, mientras que el ΔHº de
transferencia de una molécula de ácido de la fase móvil a la fase estacionaria es negativa y
disminuye cuando la longitud de la cadena alquílica o la cadena etoxilada (en el caso de los
surfactantes no-iónicos en una fase estacionaria polar) es mas grande.

117
REFERENCIAS BIBLIOGRÁFICAS
[1] Kemp D., Vellaccio F., Organic Chemistry. Worth Publishers, INC. 1980.
[2] Rappoport Z., Handbook of table for organic compounds identification, 3ed., CRC Press.
Florida 1967.
[3] Levin, S. http://www.forumsci.co.il/HPLC/program.html; 1997.
Kazakevich, Y.; McNair, H. http://hplc.chem.shu.edu/NEW/HPLC_Book/index.html. 2000
[4] Snyder L. R. Principle of Adsorption Chromatography. M. Dekker, Inc., New York, 1968
[5] Jandera, P.; Colin, H.; Gulochon, G. Anal. Chem. 1982, 54, 435-440.
[6] Jandera, P.; Holcapek, M.; Theodondis, G. J. Chromatog. A. 1998, 813, 299-309.
[7] Krastulovic, A.; Colin, H.; Guiochon, G. Anal. Chem. 1982, 54, 2438-2447.
[8] Kazakevich, Y.; Mcnair, H. J. Chromatogr. Sci. 1993, 31, 317-328.
[9] Mockel, H. J. Chromatogr. A. 1994, 675, 13-19.
[10] Oumada, F.; Roses, M.; Bosch, E. Talanta. 2000, 53, 667-673.
[11] Kazakevich, Y.; Mcnair, H. J. Chromatogr. A. 2000, 872, 49-53.
[12] Rustamov, I.; Farcas, T.; Ahmed, F.; Chan, F.; LoBrutto, R.; McNair, H.; Kazakevich, Y.
J. chromatogr. A. 2001, 913, 49-54.
[13] Dominguez, J.; Diez-Masa, J.; Davankov, V. Pure Appl. Chem. 2001, 73, 969-976.
[14] Rimmer, C.; Simmons, C.; Dorsey, J. J. Chromatogr. A. 2002, 965, 219-226.
[15] Shinoda, K.; Saito, H. J. Colloid Interface Sci. 1968, 26, 70-75.
[16] Merdas, A.; Gindre, M.; Ober, R.; Nicot, C.; Urbach, W.; Waks, M. J. Phys. Chem. 1996,
100, 15180-15186.
[17] Penfold, J.; Staples, E.; Tucker, I.; Cummins, P. J. Phys. Chem. 1996, 100, 18133-18137.
[18] Menge, U.; Lang, P.; Findenegg, G. Colloids Surf. A: Physicochem. Engng Aspects. 2000,
163, 81-90.
[19] Zhu, P.; Snyder, L.; Dolan, J.; Djordjevic, N.; Hill, D.; Sander, L.; Waeghe, T. J.
Chromatogr. A. 1996, 756, 21-39.
[20] Zhu, P.; Dolan, J.; Snyder, L. J. Chromatogr. A. 1996, 756, 41-50.

118
[21] Lee, W.; Cho, D.; Chun, B.; Chang, T.; Ree, M. J. Chromatogr. A. 2001, 910, 1-60.
[22] Vigh, G.; Varga, Z. J. Chromatogr. 1980, 196, 1-10.
[23] Petrovic, S.; Lomic, S. Chromatographia. 1989, 27, 378-384.
[24] Silveston, R.; Kronberg, B. J. Chromatogr. A. 1994, 659, 43-49.
[25] Ludolph, B.; jeng, C.; Chu, A.; Langer, S. J. Chromatogr. A. 1994, 660, 3-12.
[26] Philipsen, H.; Claessens, H.; Lind, H.; Klumperman, B.; German, A. J. Chromatogr. A.
1997, 790, 101-108.
[27] Ranatunga, R.; Carr, P. Anal. Chem. 2000, 72, 5679-5686.

119
C A P I T U L O V I I

120
DESARROLLO DE UN SISTEMA EN LINEA PARA LA DERIVATIZACIÓN Y ANÁLISIS DE ÁCIDOS CARBOXÍLICOS GRASOS (FAs) POR HPLC.
Los requisitos mayores para controlar un proceso con sistemas FIA son la sencillez,
robustez, y la certeza instrumental y química. En los sistemas FIA, la muestra que implica
acondicionamiento o modificación, o la determinación de multi-parámetros puede implicar
muchas líneas de bombeo y una configuración múltiple moderadamente compleja. La certeza de
los sistemas FIA desplegados para aplicaciones a largo plazo, puede ser también sumamente
dependiente de la estabilidad de los reactivos utilizados.
Las técnicas de separación cromatográficas en combinación con sistemas de detecciones
selectivas y sensibles han ganado una enorme popularidad en todos los campos de la química
analítica. Sin embargo, incluso con esta variedad de sistemas de detección-separación disponibles
no siempre es posible alcanzar el límite de detección deseado con la precisión necesaria sin
alguna manipulación analítica o derivatización.
SECCIÓN EXPERIMENTAL
Equipos
La reacción de derivatización y análisis por HPLC de los ácidos carboxílicos de cadena
larga se realizó empleando los siguientes equipos:
Inyector universal U6K
Computador PC 5500 con el software Millenium; para el procesamiento de los
resultados.
Detector de arreglo de fotodiodos (PDA), 996
Controlador de solventes 600
Bomba cuaternaria distribuidora de solventes, M6000A
Bomba de alta presión isocrática 510

121
válvula de inyección (Rheodyne modelo 5020)
válvula de paso (Supelco modelo 5011)
Horno microondas doméstico (Kenmore) equipado con un magnetrón de 2450 MHz con
un poder máximo nominal de 400 watts
Columna cromatográfica analítica de fase reversa:C18 de 5-µm (250 x 4.6 mm; de Merk)
Materiales
Sistemas de purificación de solventes y muestras de la Waters Millipore, con filtros de
0,22 y 0,45 µm acuosos y orgánicos.
Sistema de desgasificación de solventes con Helio.
Baño ultrasónico, marca Bransonic modelo 1200.
Jeringas de desgasificación de 10 mL y de inyección de 25 µL, marca Hamilton
Balanza analítica, marca ACCULAB, sensibilidad ± 0,0001 g
Reactor (coil) hecho de mangueras de teflón.
Material de vidrio en general.
Reactivos.
• Todos los solventes utilizados para la reacción y el análisis por HPLC son de grado
HPLC, metanol (MeOH), acetonitrilo (ACN). Todos desgasificados con gas helio y
sonicados.
• Se estudiaron las siguientes muestras: ácido caprílico (C8), ácido cáprico (C10), ácido
láurico (C12), ácido mirístico (C14), ácido palmítico (C16) y ácido esteárico (C18), todos
marca Merck

122
• Se emplearon los siguientes agentes derivatizantes: fenilhidrazina (PH), 2,4-
dinitrofenilhidrazina (2,4-DNPH) y cloruro de bencilo (BC), (pureza sobre el 99%); de la
Merck Company.
Procedimiento Experimental
Desarrollo de un sistema de análisis continuo de ácidos carboxílicos.
Actualmente es importante el desarrollo de técnicas que minimicen la manipulación
directa de las muestras en cualquier análisis cotidiano. Los ácidos carboxílicos presentan una
pobre absorción en la región del ultravioleta, lo cual conlleva a la preparación de derivados para
mejorar la sensibilidad de los métodos de análisis. No obstante, los métodos clásicos de
preparación de derivados implican pre-tratamientos de las muestras que alargan el procedimiento
de análisis. Para solventar este inconveniente, se desarrollo un sistema en línea que permite
obtener compuestos derivados de los ácidos carboxílicos, empleando irradiación microondas y su
análisis por HPLC.
Para la reacción de derivatización de los ácidos carboxílicos se empleó un horno
microondas doméstico (Kenmore) equipado con un magnetrón de 2450 MHz con un poder
máximo nominal de 400 watts. El reactor (coil) se introdujo al microondas través de los orificios
de las ventanas del horno para no romper las paredes. En la Figura V.1 se muestra el esquema del
sistema empleado. En la Tabla VII.1 se muestran las condiciones de las reacciones de
derivatización.
Debido a que los ácidos carboxílicos son compuestos monodispersos, todas las medidas
cromatográficas se hicieron en condiciones isocráticas. El tiempo de retención (tr) de los ácidos
se obtuvo de cinco determinaciones individuales (n = 5).

123
RESULTADOS Y DISCUSIÓN
La mayoría de los FAs tienen una pobre absorción en la región del ultravioleta (UV)
ampliamente empleada en los sistemas de detección en cromatografía líquida. No obstante, se han
reportado una amplia variedad de métodos con el objeto de incrementar su absorción en la región
UV-Vis. Entre los mas empleados se encuentran la esterificación, la condensación con alcoholes
en condiciones ácidas o la reacción con haluros de alquilo en condiciones básicas1-6.
Entre los reactivos empleados para la derivatización de ácidos carboxílicos se destacan la
fenilhidrazina, 2,4-dinitrofenilhidrazina, y el cloruro de bencilo7-8. En esta sección se muestra el
desarrollo de un sistema en línea para la obtención de derivados de ácidos carboxílicos grasos
empleando irradiación microondas y acoplado en línea al sistema HPLC-PDA para la separación
de los ácidos derivados.
En la Figura VII.1 se muestra el sistema continuo desarrollado para la derivatización en
línea de los ácidos grasos. Inicialmente se llena el loop de la válvula de inyección con una
solución que contiene 0.0591 mmoles de cada ácido en acetonitrilo y el reactivo derivatizante
(BC, PH y 2,4-DNPH respectivamente) (3A). Al cambiar de posición la válvula de inyección, el
contenido del loop es insertado en el medio de arrastre (acetonitrilo y llevado al reactor (coil))
localizado dentro del horno microondas. En este sistema, el tubo de teflón (coil) fue enrollado
alrededor de un tubo de ensayo.
Alrededor de 30 segundos después de la inyección de la muestra, justo cuando la muestra
entra en el reactor (coil), el microondas fue encendido a 400W por 1 min. Es importante destacar
que debido al bajo poder del microondas usado, no hubo un calentamiento excesivo, y por tanto
no hubo formación de burbujas. Luego del proceso de derivatización, el loop de la válvula de
(3B) es automáticamente llenado y al cambiar su posición, los productos de la reacción se
analizaron y separaron por HPLC.

124
Figura VII.1. Diagrama esquemático del sistema HPLC-derivatización en línea de ácidos grasos.
7
2B
1
2A3A
45
3B98
11
10
6
7
2B
1
2A3A
45
3B98
11
10
6
ACN (1) Bomba (2A) y (2B) Válvula de inyección (3A) 100 µL y (3B) 10 µL (4) FAs + reactivo; Reactor, MW (450 watts) (5) Válvula de paso (6) 92/8 MeOH/H2O (7) Pre-columna (8) Columna RP-18 (9) Detector PDA (10) Computador (11)

125
Condiciones de reacción
El método propuesto se basa principalmente en la reacción del grupo carbonilo del FAs
con los tres diferentes reactivos derivatizantes, como puede observarse en la Figura VII.2, en las
cuales se empleo el acetonitrilo como solvente de reacción. Los ácidos grasos derivatizados
presentan una fuerte absorción a las longitudes de onda respectivas (Tabla VII.1), cuyos
resultados están en buen acuerdo con los reportados por Miwa7. Algunos autores5 han reportado
que se obtienen mejores rendimientos cuando se adiciona el reactivo derivatizante en exceso. El
empleo de las hidrazinas sustituidas fue con la finalidad de asegurar la monocondensación, ya
que por lo general, la producción de una azina implica la condensación de 2 moles del compuesto
carbonílico.
Figura VII.2. Reacciones químicas generales de los ácidos grasos de cadena larga con el reactivo derivatizante. (I) fenilhidrazona del ácido graso (II) 2,4-dinitrofenilhidrazona del ácido graso y (III) benzoil-ester del ácido graso.
H2NNHC6H5+ ACN C=NNHC6H5
Rn
HO
C=O
Rn
HO
H2NNHC6H5N2O4C=O
Rn
HO
+ACN
C=NNHC6H5N2O4
Rn
HO
Rn-C-O-C-C6H5
= =O O
C=O
Rn
HO
C6H5C-Cl+ ACN
=O
(I)
(II)
(III)
H2NNHC6H5+ ACN C=NNHC6H5
Rn
HO
C=O
Rn
HOH2NNHC6H5+ ACN C=NNHC6H5
Rn
HO
C=NNHC6H5
Rn
HO
C=O
Rn
HO
C=O
Rn
HO
H2NNHC6H5N2O4C=O
Rn
HO
+ACN
C=NNHC6H5N2O4
Rn
HOH2NNHC6H5N2O4C=O
Rn
HO
C=O
Rn
HO
+ACN
C=NNHC6H5N2O4
Rn
HO
C=NNHC6H5N2O4
Rn
HO
Rn-C-O-C-C6H5
= =O O
C=O
Rn
HO
C6H5C-Cl+ ACN
=O
Rn-C-O-C-C6H5
= =O O
Rn-C-O-C-C6H5
= =O O
C=O
Rn
HO
C=O
Rn
HO
C6H5C-Cl+ ACNACN
=O
(I)
(II)
(III)
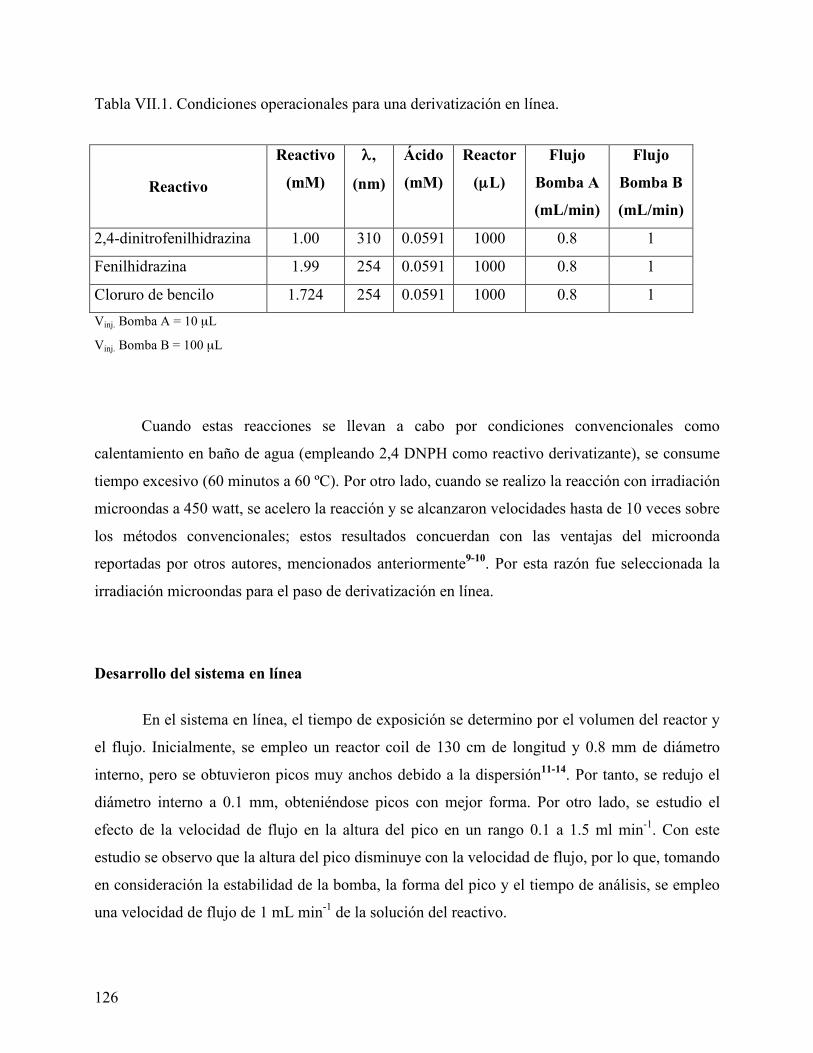
126
Tabla VII.1. Condiciones operacionales para una derivatización en línea.
Reactivo
Reactivo
(mM)
λ ,
(nm)
Ácido
(mM)
Reactor
(µL)
Flujo
Bomba A
(mL/min)
Flujo
Bomba B
(mL/min)
2,4-dinitrofenilhidrazina 1.00 310 0.0591 1000 0.8 1
Fenilhidrazina 1.99 254 0.0591 1000 0.8 1
Cloruro de bencilo 1.724 254 0.0591 1000 0.8 1 Vinj. Bomba A = 10 µL
Vinj. Bomba B = 100 µL
Cuando estas reacciones se llevan a cabo por condiciones convencionales como
calentamiento en baño de agua (empleando 2,4 DNPH como reactivo derivatizante), se consume
tiempo excesivo (60 minutos a 60 ºC). Por otro lado, cuando se realizo la reacción con irradiación
microondas a 450 watt, se acelero la reacción y se alcanzaron velocidades hasta de 10 veces sobre
los métodos convencionales; estos resultados concuerdan con las ventajas del microonda
reportadas por otros autores, mencionados anteriormente9-10. Por esta razón fue seleccionada la
irradiación microondas para el paso de derivatización en línea.
Desarrollo del sistema en línea
En el sistema en línea, el tiempo de exposición se determino por el volumen del reactor y
el flujo. Inicialmente, se empleo un reactor coil de 130 cm de longitud y 0.8 mm de diámetro
interno, pero se obtuvieron picos muy anchos debido a la dispersión11-14. Por tanto, se redujo el
diámetro interno a 0.1 mm, obteniéndose picos con mejor forma. Por otro lado, se estudio el
efecto de la velocidad de flujo en la altura del pico en un rango 0.1 a 1.5 ml min-1. Con este
estudio se observo que la altura del pico disminuye con la velocidad de flujo, por lo que, tomando
en consideración la estabilidad de la bomba, la forma del pico y el tiempo de análisis, se empleo
una velocidad de flujo de 1 mL min-1 de la solución del reactivo.

127
El reactor se colocó en el horno microondas domestico cerca al sistema de separación (ver
Figura VII.1); se emplearon tiempos de irradiación de 1 a 60 segundos observándose que después
de 40 segundos la intensidad de la señal fue casi constante. Por otro lado, se ensayaron altos flujo
para disminuir el tiempo de análisis pero se encontró que con un flujo mayor a 0.8 mL min-1 se
obtenían picos más pequeños.
Condiciones de análisis por HPLC
La separación de los seis ácidos grasos estudiados fue obtenida en la columna RP-18 de 5
µm, la cual dio los mejores resultados en el capitulo anterior, y además es la mas recomendada
para este tipo de análisis5-6 En principio se ensayó con metanol puro como fase móvil, pero se
obtuvo muy pobre resolución. Por lo que se adiciono el modificador polar agua a la fase móvil y
se logro una mejor resolución; la proporción optima fue 92/8 (MeOH/H2O). Cabe destacar que
esta fase móvil resultó apropiada para el análisis de la mezcla de FAs con los tres reactivos
derivatizantes, por tanto se ahorro tiempo de optimización de fase móvil que generalmente se
consume en los estudios por HPLC.
Reactivo derivatizante
Como es bien conocido, la mayoría de los solventes usados como fase móvil en HPLC
presentan una fuerte absorción lo cual puede interferir con el pico del ácido monitoreado a 214
nm; sin embargo, cuando estos compuestos son derivatizados se aumenta la sensibilidad y la
selectividad del método empleado. La intensidad de la señal de los FAs sin derivar después de
su separación se comparó con la intensidad de la señal después de obtener sus derivados con
los tres reactivos derivatizantes respectivamente.
En la Figura VII.3 se muestra que al adicionar el paso de la obtención de derivados en el
análisis de FAs por HPLC-PDA se mejora la sensibilidad (hasta 7 veces mayor al emplear
PH). En general, se observo una relación inversa entre la absorción del producto y el numero
de átomos de carbono de la cadena alquílica del ácido, es decir que la absorción disminuye
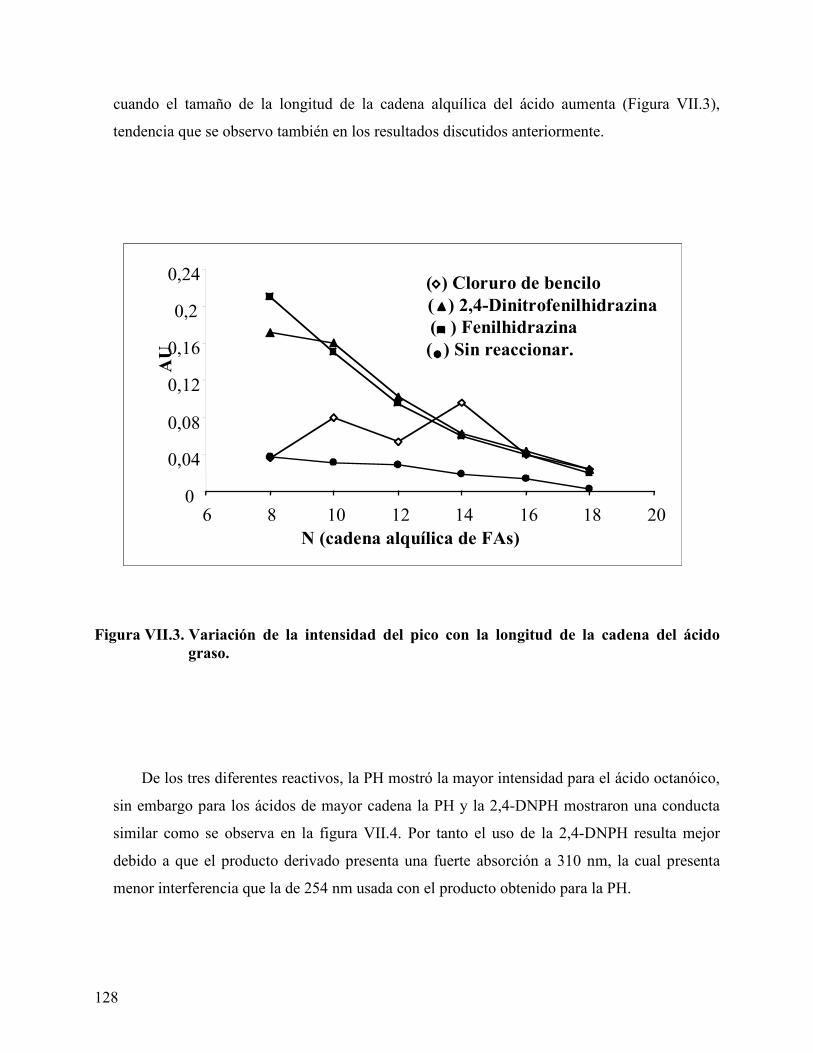
128
cuando el tamaño de la longitud de la cadena alquílica del ácido aumenta (Figura VII.3),
tendencia que se observo también en los resultados discutidos anteriormente.
Figura VII.3. Variación de la intensidad del pico con la longitud de la cadena del ácido graso.
De los tres diferentes reactivos, la PH mostró la mayor intensidad para el ácido octanóico,
sin embargo para los ácidos de mayor cadena la PH y la 2,4-DNPH mostraron una conducta
similar como se observa en la figura VII.4. Por tanto el uso de la 2,4-DNPH resulta mejor
debido a que el producto derivado presenta una fuerte absorción a 310 nm, la cual presenta
menor interferencia que la de 254 nm usada con el producto obtenido para la PH.
0
0,04
0,08
0,12
0,16
0,2
0,24
6 8 10 12 14 16 18 20
N (cadena alquílica de FAs)
AU
(?) Cloruro de bencilo
(| ) 2,4-Dinitrofenilhidrazina
(? ) Fenilhidrazina
(?) Sin reaccionar.
0
0,04
0,08
0,12
0,16
0,2
0,24
6 8 10 12 14 16 18 20
N (cadena alquílica de FAs)
AU
( ) Cloruro de bencilo
( ) 2,4-Dinitrofenilhidrazina
( ) Fenilhidrazina
( ) Sin reaccionar.
0
0,04
0,08
0,12
0,16
0,2
0,24
6 8 10 12 14 16 18 20
N (cadena alquílica de FAs)
AU
(?) Cloruro de bencilo
(| ) 2,4-Dinitrofenilhidrazina
(? ) Fenilhidrazina
(?) Sin reaccionar.
0
0,04
0,08
0,12
0,16
0,2
0,24
6 8 10 12 14 16 18 20
N (cadena alquílica de FAs)
AU
( ) Cloruro de bencilo
( ) 2,4-Dinitrofenilhidrazina
( ) Fenilhidrazina
( ) Sin reaccionar.

129
La principal ventaja de la reacción con 2,4-DNPH fue que la cantidad de reactivo que no
reacciona fue muy baja como se observa en la Figura VII.4. En esta figura la línea base
presenta algunos picos pequeños que se atribuyen a co-productos. Se encontró que el uso del
BC comparado con los otros reactivos empleados no presenta ventaja alguna debido a que los
productos no mostraron una tendencia lineal como se observa en la Figura VII.4.
Además, la absorción de los productos derivados es baja, resultando ser tres veces mayor
que sin el procedimiento de derivatización. Las precisiones (desviación estándar relativa,
RSD) y la exactitud (error relativo) del método se determinaron basándose en la relación de
área del pico para el análisis de cada FAs a 0.059, 0.079 y 0.119 mM. En todos los casos RSD
y RE estuvieron por debajo de 4.
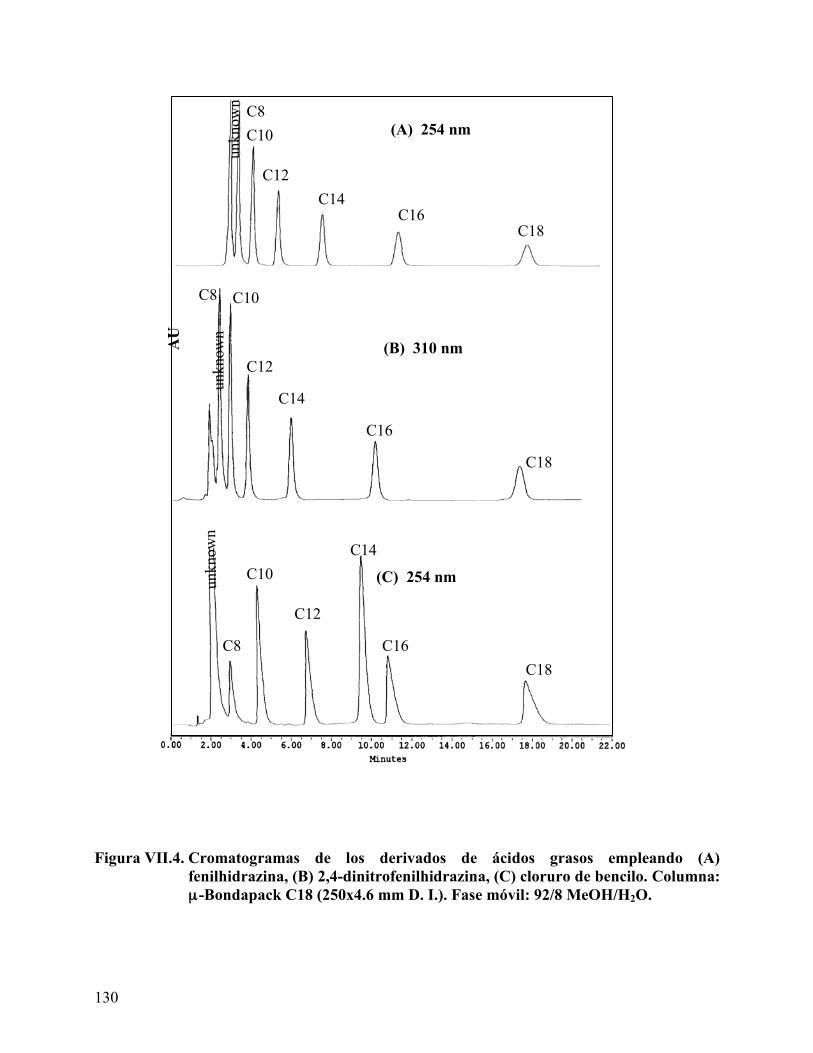
130
Figura VII.4. Cromatogramas de los derivados de ácidos grasos empleando (A) fenilhidrazina, (B) 2,4-dinitrofenilhidrazina, (C) cloruro de bencilo. Columna: µ-Bondapack C18 (250x4.6 mm D. I.). Fase móvil: 92/8 MeOH/H2O.
(A) 254 nm
(B) 310 nm
(C) 254 nm
AU
(A) 254 nm
(B) 310 nm
(C) 254 nm
AU
C8
C18
C18
C18
C16
C16
C16
C14
C14
C14
C12
C12
C12
C10
C10
C10
C8
C8
unknown
unknown
unknown

131
CONCLUSIONES
La adición del paso de derivatización al análisis de ácidos grasos por HPLC-PDA
incrementa la sensibilidad en casi 7 veces y permite detectarlos a 254 y 310 nm. De los tres
reactivos usados el mejor fue la 2,4-DNPH.
La irradiación con microonda a 450 W, resulto ser más eficiente para la reacción de
derivatización que las condiciones convencionales, con una disminución del tiempo de reacción
de 10 veces. El método desarrollado por análisis continuo empleando el sistema MW-HPLC-UV
proporciona alta sensibilidad, reduce la cantidad de reactivo a emplear y permite obtener tiempos
cortos de análisis.

132
REFERENCIAS BIBLIOGRÁFICAS
[1] Rozhkov, V.; Vorob’ov, S.; Lobatch, A.; Kuvshinov, A.; Shevelev, S. Synthetic Comm.
2002, 32, 467-471.
[2] Greence, T.; Wuts, P. Protective Groups in Organic Synthesis, John Wiley & Sons. New
York. 1999, Chapter 5.
[3] Mulzer, J. In Comprehensive Organic Synthesis; Trost, B.; Fleming, I.; Heathcock, C.
Eds., Pergamon Press. New York. 1991, 6, 324-337.
[4] Vandenabeele T.; Mion, L.; Garrelly, L.; Commeyras, A. Adv Environ Res. 2001, 6, 45-
55.
[5] Latorre, A.; Rigol, A.; Lacorte, S.; Barceló, D. J Chromatogr. A. 2003, 991, 205–215.
[6] Lima, E.; Abadía, D. Anal. Chim. Acta. 2002, 465, 81–91.
[7] Miwa, H. Anal. Chim. Acta. 2002, 465, 237-255.
[8] Rosenfeld, J. Anal. Chim. Acta. 2002, 465, 93-101.
[9] Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J.
Tetrahedron. 1986, 27, 279-282.
[10] Raner, K.; Strauss, Ch. J. Org. Chem. 1992, 57, 6231-6234.
[11] Ruzicka, J.; Hansen, E. Anal Chem. 2000, 72, 212A-217A.
[12] Nielsen, S.; Hansen, E. Anal. Chim Acta., 2000, 422, 47-62.
[13] Hansen, H. Talanta. 2000, 51, 607-608.
[14] Ruzicka, J.; Hansen, E. Flow Injection Analysis. J. Wiley and Sons. 1981.

133
C A P I T U L O V I I I

134
COMPORTAMIENTO DE FASE EN LA FORMULACIÓN CON ÁCIDOS CARBOXÍLICOS GRASOS (FAS)
En este estudio se tratara el caso de una mezcla de dos surfactantes que presentan un
antagonismo de carga, sobre un amplio rango de pH, que cubre esencialmente todos los pH
utilizados en las aplicaciones prácticas. Existe sin embargo en la naturaleza, los surfactantes
aniónicos y catiónicos cuya actividad depende del pH, en particular los primeros surfactantes
utilizados: los jabones. Por otra parte, el petróleo contiene resinas cuya actividad es sensible al
pH, particularmente de tipo ácidos1-4. De forma general, estos surfactantes que dependen del pH
se clasifican en dos categorías:
- Primero aquellos de los cuales la disociación, y como consecuencia el carácter iónico,
aumentan con el pH; el caso mas representativo es el de los ácidos grasos, es decir, los
ácidos carboxílicos de cadena hidrocarbonada larga lineales. A pH relativamente
alcalino, ellos se ionizan para formar la sal frecuentemente muy hidrofílica, llamada
carboxilato o jabón.
- Segundo, aquellos en los que la ionización aumenta cuando el pH disminuye; ellos son
susceptibles de captar un protón, como las aminas grasas.
El caso de los ácidos grasos es el objetivo central de la presente investigación, debido a que
ellos se presentan prácticamente todo el tiempo bajo la forma de una mezcla ácido-carboxilato,
cuya composición depende del pH. Como el ácido es un surfactante lipofílico, y el carboxilato un
surfactante hidrofílico, es evidente que una variación del pH produce un cambio de la
formulación fisicoquímica. El problema es mucho más complejo en presencia de una fase acuosa
y una oleica, donde se produce adicionalmente una partición de las diferentes especies.

135
SECCIÓN EXPERIMENTAL
Materiales
Balanza analítica, marca ACCULAB, sensibilidad ± 0,0001 g
Estufa, marca memmert, temperatura máxima de 250 ºC.
Baño de agua circulante refrigerante/ calentador, marca Vev R de la Merck.
Plancha de calentamiento/ agitador, marca CORNING.
pH-metro, marca Orión.
Material de vidrio en general.
Reactivos.
Ácidos carboxílicos (CAX), con 8, 10, 12, 14, 16 y 18 átomos de carbono
Como fase oleica se emplearon los siguientes solventes: n-heptano, tolueno, benceno (grado
HPLC, J.T. Beaker) y kerosén
Como co–surfactantes se usaron los alcoholes: n–propanol (grado P.A., Mallinckrodt), n–
bupanol (grado P.A., Merck), n–pentanol (grado P.A., Merck), n–hexanol (grado P.A., Riedel–De
Haën). Agua destilada y desionizada Se empleó NaCl, (99%, Merck) y NaOH (99%, Merck) para los barridos de formulación.
Los barridos se realizaron a 25 oC en un baño termostatado marca LAUDA.
Procedimiento Experimental
Barridos de formulación con ácidos carboxílicos y sus mezclas.
Para el estudio del reparto de los ácidos se requiere, la presencia de un sistema óptimo (W
III) y este se alcanza mediante un procedimiento experimental conocido como barrido de
formulación. Para ello se va modificando la variable en estudio hasta conseguir el sistema

136
deseado (generalmente el de mayor importancia, W III o lo que es lo mismo, la formulación
óptima).
Se realizaron barridos de formulación empleando ácidos carboxílicos solos y sus mezclas
hasta obtener el sistema de Winsor III (WIII) a formulación óptima. El sistema WIII está
constituido por tres fases llamadas: acuosa, oleica y microemulsión. En dichos sistemas el ácido
tiende a repartirse (fraccionarse) en las tres fases, donde la mayor concentración de este se
encuentra en la microemulsión.
Los sistemas aceite/anfifilo/agua se estudiaron de acuerdo a la técnica de barrido
unidimensional5-6. Para cada serie, una serie de tubos de ensayos graduados fueron preparados,
conteniendo cada uno aceite y agua en cantidades idénticas (WOR = 1) y el anfifilo, pero
variando la naturaleza de alguno de los componentes progresivamente de un tubo de ensayo al
próximo. En este trabajo, la concentración del ácido graso, el tipo de ácido graso, la
concentración de alcoholes lipofílicos como fase externa y la fase oleica son las variables de
estudio. En todos los casos se emplearon soluciones de NaOH a la concentración adecuada para
la formación de la especie carboxilato que actúa como surfactante aniónico, así como soluciones
de NaCl, en proporción tal de mantener la misma concentración de iones sodio en el sistema.
Los tubos de ensayos se prepararon mezclando 5 o 10 mL de la fase oleica (un
hidrocarburo: n-heptano, tolueno, benceno, kerosén) con 5 o 10 mL de fase acuosa (una mezcla
de solución de hidróxido de sodio y cloruro de sodio) y Xg del ácido graso. El anfifilo se preparó
en el aceite. Luego, los tubos bien cerrados con tapa de baquelita se mantienen a 25 ºC en un
baño termostatado. Una vez preparados los tubos, estos se agitaron suavemente hasta observar
una solución homogénea, y se dejaron en reposo por una semana para garantizar el alcance del
equilibrio en el sistema.
El ácido producto de la mezcla (SC) se determina según la expresión:
SCCX
1
i=!
=
i
i
(1)

137
donde C es el número de átomos de carbonos del ácido y Xi representa la fracción en peso del
ácido en la mezcla. Después de alcanzado el equilibrio, se observó el comportamiento de fase y
se registraron los volúmenes de cada fase para calcular los parámetros de solubilidad y
determinar el sistema óptimo.
Evaluación del tipo de aceite en el sistema surfactante-agua-aceite.
Se evaluó el efecto que ejercen diferentes aceites empleados como fase oleica en los
barridos de formulación. Para ello se empleó el procedimiento de barrido unidimensional. Una
vez equilibrados los sistemas, se observó el comportamiento de fase registrándose los volúmenes
de cada fase para determinar el sistema óptimo.
Evaluación del tipo y concentración del co-surfactante en el sistema óptimo.
Una vez formado el sistema ácido/agua/aceite, se adicionó alcohol como fase externa. Los
barridos se realizaron cambiando la longitud de la cadena alquílica del alcohol. En principio se
realiza un barrido de alcohol hasta obtener el sistema trifásico para cada ácido estudiado,
determinándose la concentración de alcohol necesaria para alcanzar este punto. Por otro lado, una
vez obtenido los sistemas trifásicos a formulación optima, se evaluó el efecto de adición de los
diferentes alcoholes en dichos sistemas.

138
RESULTADOS Y DISCUSIÓN
La presencia de ácidos carboxílicos en algunos crudos venezolanos, crea la posibilidad de
usar la inyección de soluciones alcalinas. Este proceso consiste en aprovechar los ácidos
orgánicos naturales presentes en ciertos crudos, para generar el surfactante in situ mediante su
neutralización parcial o total por una solución alcalina.
Estos ácidos al reaccionar con el de la fase acuosa forman la sal del ácido carboxílico, cuya
mezcla ácido carboxílico-carboxilato tiene una alta actividad interfacial. Este proceso consta
básicamente de dos etapas. Primero las sales surfactantes se forman en la interfase por la reacción
de las especies ácidas contenidas en el crudo con el álcali presente en la solución acuosa. En la
segunda etapa, al surfactante formado y adsorbido a la interfase agua/crudo, disminuye la tensión
interfacial, lo que permite la emulsionación y/o se adsorbe sobre las rocas del reservorio y altera
la mojabilidad. En estos sistemas la tensión interfacial depende esencialmente de la relación
ácido/sal en el sistema lo cual fija el pH del mismo.
Es importante, sin embargo, tomar ciertas precauciones en los estudios que implican un
barrido de pH con sistemas sensibles a los electrolitos. En efecto, si a una concentración de sal
(NaCl) constante, se varia el pH con un ácido fuerte (HCl) o una base fuerte (NaOH), es decir
una sustancia completamente ionizada, se estaría cambiando la formulación del sistema, debido a
que se estaría cambiando la fuerza iónica del medio, es decir, su salinidad7. Por otro lado, se ha
reportado que un aumento de la concentración de NaOH produce un aumento de la hidrofilicidad,
debido a la formación de una mayor proporción de carboxilato (transición WII-WIII-WI), que
luego disminuye con el aumento de la fuerza iónica dominante (transición WI-WIII-WII).
Este fenómeno fue reportado por Qutubuddin y col.8, quien, para evitar este hecho, mantuvo
la salinidad constante al variar el pH. Este hecho se reporto para sistemas con surfactantes
insensibles al pH (sal de sodio de un sulfonato de petróleo), en los cuales se demostró que la
formulación óptima no depende de la concentración molar de iones Na+, y tampoco de la
naturaleza del anión7.

139
Cuando se realizan estudios de comportamiento de fase, se debe fijar la concentración de
surfactante a utilizar. En principio esta concentración debe ser lo suficientemente grande para
obtener un sistema trifásico y lo suficientemente bajo para no encontrarse en el caso de un
sistema monofásico, ya que no se podría medir la solubilización. En la práctica una concentración
del orden de 0,5 a 5% es en general suficiente. No obstante, siempre se encuentran excepciones a
la regla.
Dado que la CMC de un carboxilato aumenta cuando la concentración de electrolito
disminuye, se podría pensar que a concentración de surfactante por debajo de la CMC, no se
obtienen sistemas trifásicos. Esto se puede explicar, mediante los barridos de ácidos carboxílicos
variando su concentración en un sistema ácido/agua/aceite; con lo cual se obtiene el grafico de
volumen de microemulsión en función de la cantidad total de ácido en todo el sistema, (HAt).
En el caso de un surfactante convencional como un sulfonato, esta variación es una recta que
pasa por el punto cero de la ordenada y abcisa CMC, cuando la composición de la fase
microemulsión es constante en todo el triangulo de partición trifásico, y cuando la línea de
partición diafásico al limite inferior del trifásico esta al nivel de la CMC7,9. En este trabajo, se
ensayaron una serie de sistemas ácido carboxílico/salmuera/aceite, cuyas diferencias radican
principalmente en:
1. Longitud de la cadena lipofílica del ácido carboxílico
2. Concentración inicial de ácido carboxílico e el sistema
3. Mezcla de ácidos carboxílicos
Todos estos sistemas se encuentran influenciados por la variación del pH en la fase acuosa.
Esto se puede explicar mejor si se considera que existe una variación de las cantidades relativas
de dos surfactantes (ácido carboxílico-carboxilato), en función de la cantidad de NaOH agregado
al sistema. La cantidad de NaOH necesaria para alcanzar un sistema óptimo, depende de la
lipofilicidad del surfactante (ácido carboxílico) y de la concentración inicial del mismo en el
sistema. A continuación se muestran los resultados obtenidos del efecto de las variables de

140
formulación en el comportamiento de fase de los ácidos carboxílicos en los sistemas ácido-agua-
aceite.

141
Efecto del tipo y concentración de ácido carboxílico en el comportamiento de fase
Primeramente se realizo un barrido de pH para el ácido láurico variando la concentración
de hidróxido de sodio en el sistema, encontrándose un sistema óptimo con hidróxido de sodio
0,6M, y una mezcla de sec-butanol y n-pentanol al 4% en el sistema. En la figura VIII.1 se
muestran estos resultados. A partir de estos resultados, se empleo una solución de hidróxido de
sodio a esta concentración para obtener los sistemas trifásicos.
En trabajos reportados7, se encontró que la salinidad del sistema depende de la
concentración de iones Na+, independientemente de si los mismos provienen de la disociación del
cloruro de sodio o del hidróxido de sodio, y si se emplea solo la solución del álcali se podría
producir una transición retrograda, por lo que es necesario trabajar a salinidad constante. Para
esto, se empleo una solución de cloruro de sodio de la misma concentración que la solución de
hidróxido de sodio para la formulación de los barridos.
Una vez optimizado las concentraciones de NaOH y NaCl necesarias para los sistemas en
estudio, se realizaron barridos de formulación variando la concentración de ácido en el sistema.
Para todos los sistemas ácido/agua/aceite de esta primera parte, se empleo una mezcla de n-
pentanol y 2-butanol como fase externa con el objeto de ayudar a la mejor solubilización, una
relación agua/aceite igual a 1 (WOR = 1) y n-heptano como fase oleica.
En las Tablas VIII.1 a la VIII.3 se muestran como ejemplo los parámetros empleados para
el barrido de formulación de los ácidos láurico, mirístico y palmítico, respectivamente. Para la
preparación de los sistemas se emplearon soluciones de hidróxido de sodio y de cloruro de sodio
de la misma concentración molar. Fue necesaria la preparación de más de una treintena de tubos
de cada ácido para la obtención de los sistemas trifásicos. En las tablas se muestra el
comportamiento de fase de los sistemas representados, el cual presenta una transición WII-WIII-
WI a medida que la cantidad de NaOH agregada aumenta.
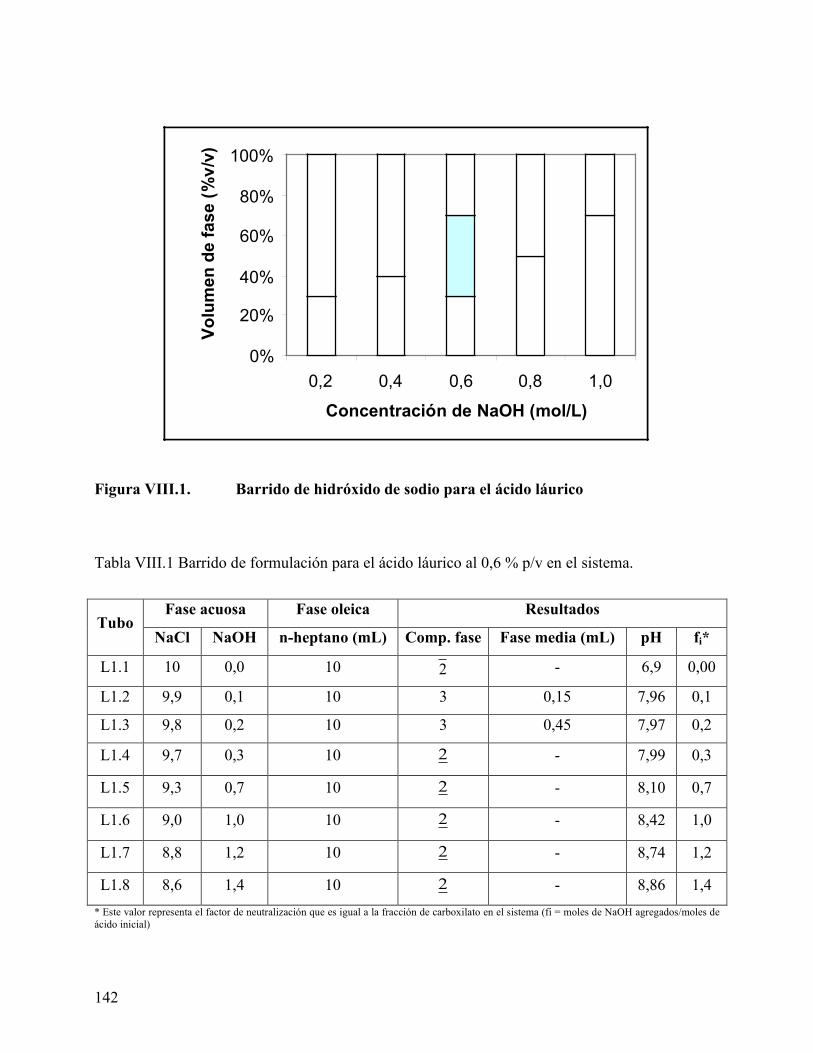
142
Figura VIII.1. Barrido de hidróxido de sodio para el ácido láurico
Tabla VIII.1 Barrido de formulación para el ácido láurico al 0,6 % p/v en el sistema.
Fase acuosa Fase oleica Resultados
Tubo NaCl NaOH n-heptano (mL) Comp. fase Fase media (mL) pH fi*
L1.1 10 0,0 10 2 - 6,9 0,00
L1.2 9,9 0,1 10 3 0,15 7,96 0,1
L1.3 9,8 0,2 10 3 0,45 7,97 0,2
L1.4 9,7 0,3 10 2 - 7,99 0,3
L1.5 9,3 0,7 10 2 - 8,10 0,7
L1.6 9,0 1,0 10 2 - 8,42 1,0
L1.7 8,8 1,2 10 2 - 8,74 1,2
L1.8 8,6 1,4 10 2 - 8,86 1,4 * Este valor representa el factor de neutralización que es igual a la fracción de carboxilato en el sistema (fi = moles de NaOH agregados/moles de ácido inicial)
0%
20%
40%
60%
80%
100%
0,2 0,4 0,6 0,8 1,0
Concentración de NaOH (mol/L)
Vo
lum
en
de f
ase (
%v
/v)
0%
20%
40%
60%
80%
100%
0,2 0,4 0,6 0,8 1,0
Concentración de NaOH (mol/L)
Vo
lum
en
de f
ase (
%v
/v)
143
Tabla VIII.2 Barrido de formulación para el ácido mirístico al 0,8 % p/v en el sistema.
Tubo Fase acuosa Fase oleica Resultados
NaCl NaOH n-heptano (mL) Comp. fase Fase media (mL) pH fi*
M3.1 9,8 0,2 10 2 - 7,81 0,17
M 3.2 9,6 0,4 10 2 - 7,96 0,34
M 3.3 9,3 0,7 10 2 - 8,07 0,60
M 3.4 9,2 0,8 10 3 1,1 8,25 0,68
M 3.5 9,1 0,9 10 3 1,5 8,49 0,77
M 3.6 9,0 1,0 10 2 - 8,63 0,86
M 3.7 8,8 1,2 10 2 - 10,63 1,03
M 3.8 8,5 1,5 10 2 - 11,69 1,28 * Este valor representa el factor de neutralización que es igual a la fracción de carboxilato en el sistema (fi = moles de NaOH agregados/moles de ácido inicial)
Tabla VIII.3 Barrido de formulación para el ácido palmítico al 1,6 % p/v en el sistema.
Tubo Fase acuosa Fase oleica Resultados
NaCl NaOH n-heptano (mL) Comp. fase Fase media (mL) pH fi*
P3.1 8,8 1,2 10 2 - 8,4 0,58
P 3.2 8,6 1,4 10 2 - 8,6 0,67
P 3.3 8,4 1,6 10 3 11.3 8,84 0,77
P 3.4 8,2 1,8 10 2 - 8,91 0,86
P 3.5 8,0 2,0 10 2 - 9,00 0,96 * Este valor representa el factor de neutralización que es igual a la fracción de carboxilato en el sistema (fi = moles de NaOH agregados/moles de ácido inicial)

144
Cuando se observa la formulación de los barridos, por ejemplo para el sistema con ácido
mirístico, se tiene que la formulación con más bajo pH corresponde a un sistema al cual se le ha
agregado poco hidróxido de sodio. A este pH el ácido debe estar presente en su mayor parte en la
forma no disociada (HA). Por tanto, estamos en presencia de un surfactante muy lipofílico y la
cantidad de ácido disociado presente en la interfase es probablemente baja. De ahí que el
comportamiento de fase sea ( 2 ). Al aumentar la cantidad de hidróxido de sodio, el pH aumenta y
por consiguiente la proporción de la sal del ácido formada también aumenta.
Siendo el carboxilato una sal hidrofílica, una parte de este puede permanecer en la fase
acuosa, en forma monomérica y en equilibrio con cierta cantidad de sal en la interfase. Un
aumento en el pH (mayor cantidad de NaOH agregado) conlleva a un aumento de la sal presente
en la fase acuosa y por ende, un aumento en la sal presente en la interfase; la cual solubiliza cierta
cantidad de fase acuosa y fase aceite produciendo a un determinado pH (dependiente de las
condiciones) un sistema trifásico o (3). Al seguir agregando NaOH (aumentando el pH),
predomina la especie hidrofílica (carboxilato) y por tanto el sistema presenta un comportamiento
(2 ). La cantidad de NaOH necesaria para alcanzar un sistema óptimo, depende de la lipofilicidad
del surfactante (ácido carboxílico) y de la concentración inicial del mismo en el sistema (en el
caso de los sistemas estudiados).
En las Figuras VIII.2, VIII.3 y VIII.4, se muestran el mapa de Winsor obtenido para los
ácidos láurico, mirístico y palmítico, respectivamente. Como se puede observar de las figuras, es
posible obtener un amplio rango de sistemas trifásicos. Por ejemplo, para el ácido mirístico se
obtienen sistemas trifásicos en el rango de concentración de 0,8 a 2,0 %p/v del ácido. En todos
los estudios se observo una transición de !
!
"" 232 .
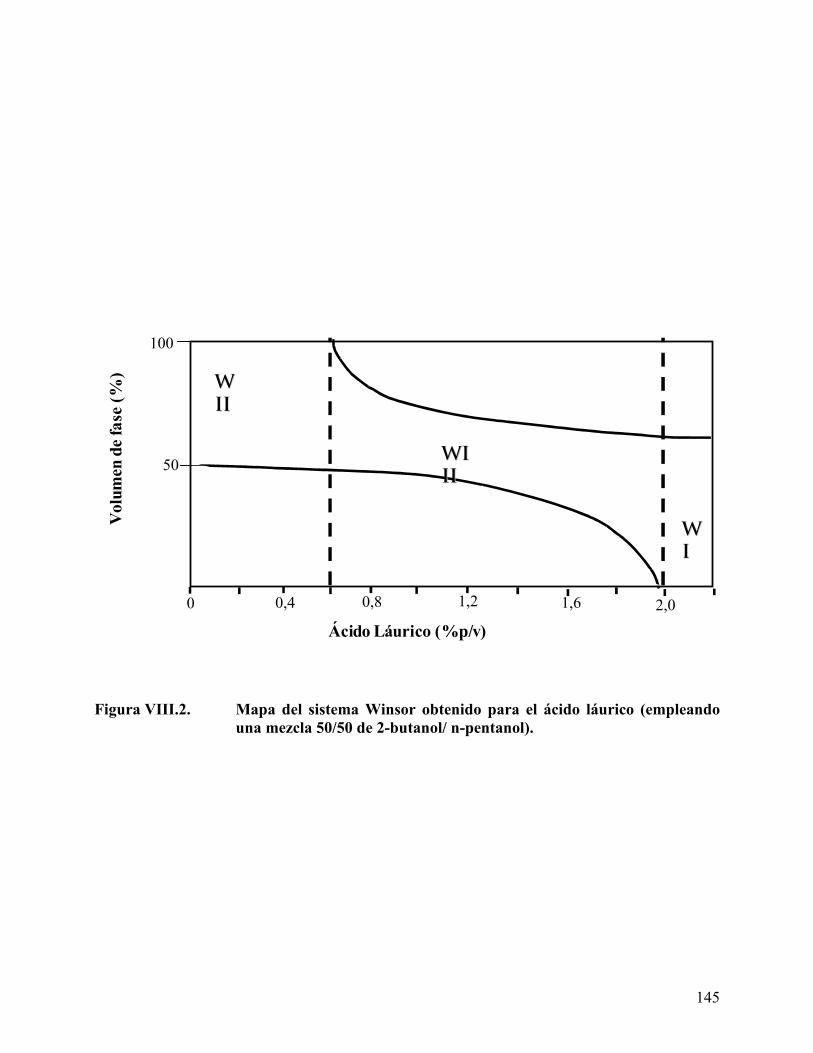
145
Figura VIII.2. Mapa del sistema Winsor obtenido para el ácido láurico (empleando una mezcla 50/50 de 2-butanol/ n-pentanol).
Ácido Láurico (%p/v)
WI
WI
WIII
WIII
WII
WII
50
100
0 0,4 0,8 1,2 1,6 2,0
Vol
umen
de f
ase
(%)
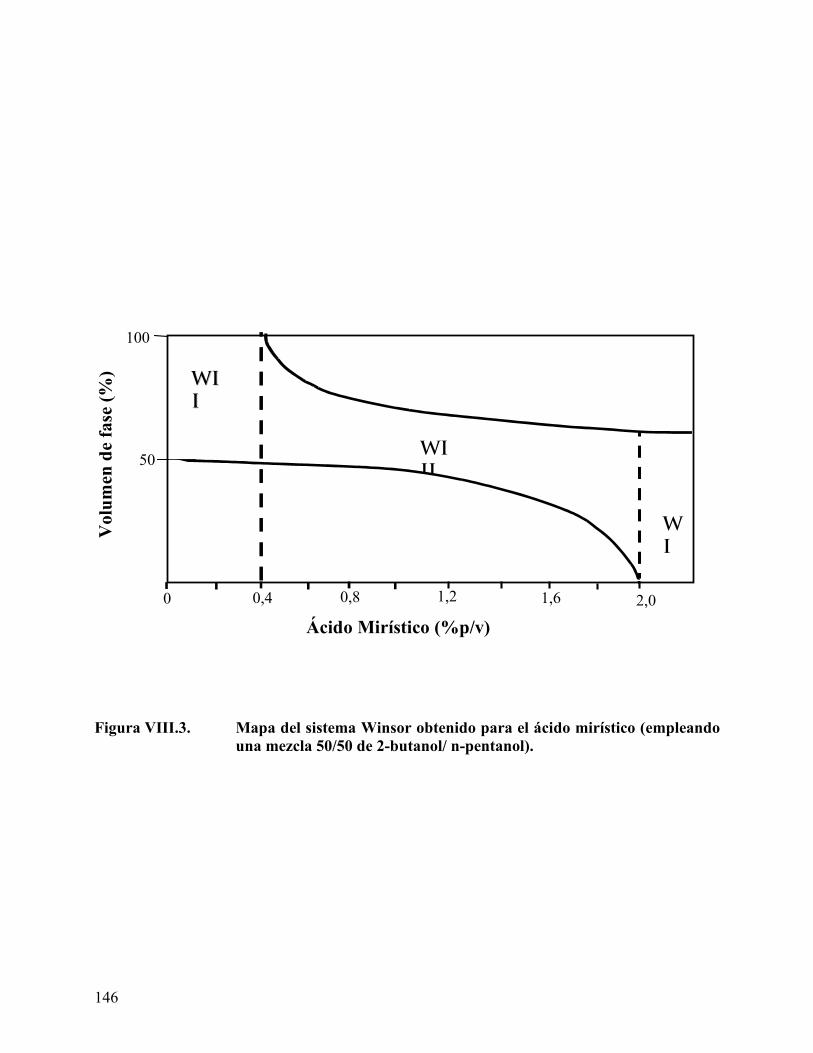
146
Figura VIII.3. Mapa del sistema Winsor obtenido para el ácido mirístico (empleando
una mezcla 50/50 de 2-butanol/ n-pentanol).
WIII
WI
1,6 2,0 1,2
WII
50
100
0 0,4 0,8
WII
Ácido Mirístico (%p/v)
Vol
umen
de
fase
(%)
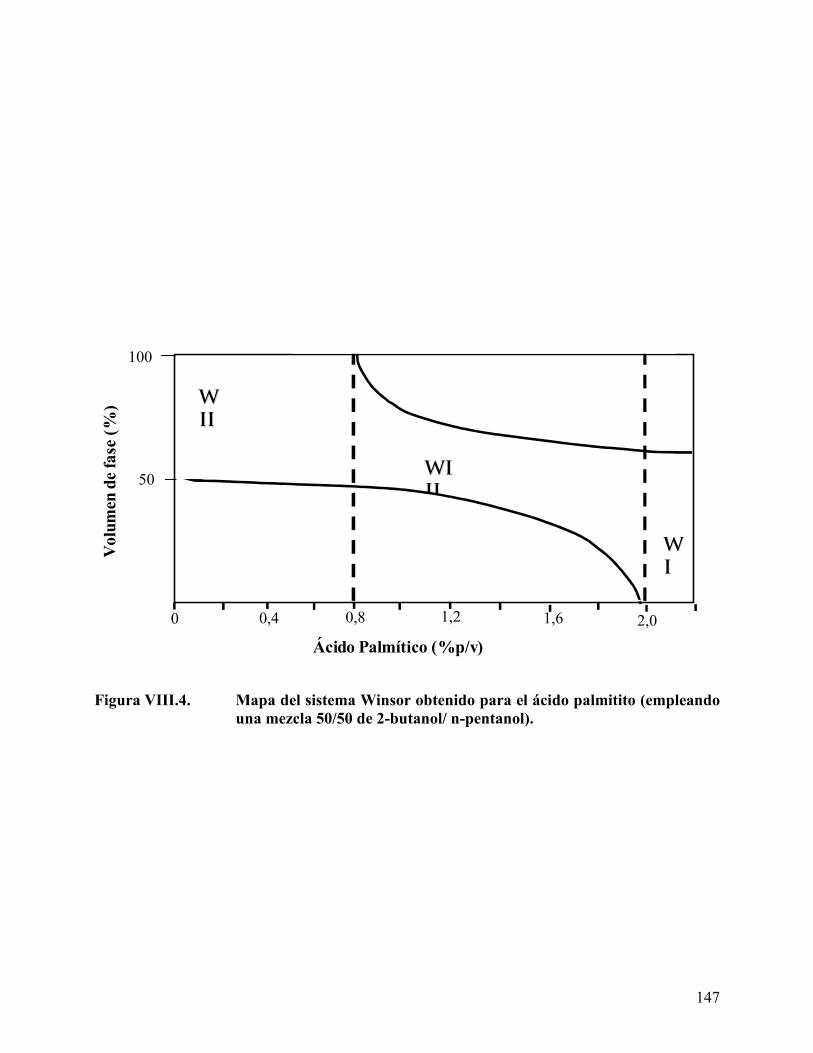
147
Figura VIII.4. Mapa del sistema Winsor obtenido para el ácido palmitito (empleando
una mezcla 50/50 de 2-butanol/ n-pentanol).
WI
WIII
WII
50
100
0 0,4 0,8 1,2 1,6 2,0
WI
WIII
WII
Vol
umen
de f
ase
(%)
Ácido Palmítico (%p/v)

148
En la figura VIII.5, se muestra la relación del volumen de fase media con la concentración
del ácido en el sistema para láurico, mirístico y palmítico, respectivamente. Se observa en esta
figura un comportamiento lineal, que muestra como aumenta el volumen de fase media con la
concentración del ácido. Se observa que al punto donde la linea se intercepta con el eje de las
abcisas (lo que corresponde a Vfm = 0), el valor de la concentración del ácido no coincide con la
CMC de éste (la cual es un valor mucho mas bajo). El valor de la absisa corresponde a la
cantidad de ácido que no está en la fase acuosa, ni en la fase media (cuyo volumen es cero), sino
que este valor representa la cantidad de ácido que queda en la fase aceite.
Por otro lado, en las Figuras VIII.6 y VIII.7, se muestra la transición !
!
"" 232 del
comportamiento de fases para un barrido de hidróxido de sodio en una mezcla mirístico/palmítico
y mirístico/láurico, respectivamente. Con estas mezclas es posible obtener un amplio rango de
sistemas trifásicos.
La formulación optima (sistema de tres fases donde la microemulsión esta en equilibrio
con las fases acuosa y oleica en exceso) fue alcanzada al 2% para la mezcla 50/50 de
mirístico/palmítico y al 1% para la mezcla 25/75, respectivamente; la tabla 5 muestra estos
resultados. Como se puede observar fue necesario el empleo de una mezcla de alcoholes
lipofílicos como co-surfactantes para ayudar a la solubilización de la sal del ácido formada. En la
Figura VIII.8 se muestra la comparación de los volúmenes de fase media de la mezcla
mirístico/láurico 50/50 y 25/75, respectivamente. Como puede apreciarse no hay una variación
significativa en el volumen de fase media para ambos sistemas, observándose el aumento lineal
del volumen de fase con la concentración del ácido.
En todos los casos se observo que a medida que se aumenta la concentración inicial de
ácido carboxílico en el sistema, el volumen de fase media también aumenta. Esto es debido a que
un incremento de la concentración del surfactante aumenta la solubilización de las fases en el
sistema. En la mayoría de los casos a mayor concentración de ácido en el sistema se observa una
mayor cantidad de sólido precipitado, esto es debido a la formación del carboxilato cuya

149
solubilidad es ayudada por el co-surfactante. En todos los casos se observo una transición de
!
!
"" 232 .
Por otro lado, los resultados de la tabla VIII.3, muestran que a medida que aumenta la
cadena hidrocarbonada del surfactante (ácido carboxílico), aumenta el volumen de fase media
óptimo. Esto se debe a que un aumento de la cadena lipofílica del surfactante, conlleva a un
incremento en la actividad de éste con la fase oleica, lo que se traduce en un aumento en el
parámetro de solubilización para un sistema óptimo.
C16 = 4,2233x - 0,3353
R2 = 0,9998
C14 = 3,7147x - 1,4676
R2 = 0,9995
C12= 3,0077x - 1,3664
R2 = 0,999
0,0
0,8
1,6
2,4
3,2
4,0
4,8
5,6
6,4
7,2
8,0
8,8
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2
% p/v ácido graso
Vfm
(m
L)
Figura VIII.5. Variación del volumen de fase media con la concentración de ácido graso en el sistema ácido/agua/aceite

150
Figura VIII.6. Variación del volumen de fase de una mezcla mirístico/palmítico a diferentes proporciones
0 20 40 60 80
100
Vf (
%)
75/25 50/50 25/75
Concentración de C14/C16 (%p/v)

151
Figura VIII.7. Variación del volumen de fase de una mezcla láurico/mirístico a diferentes proporciones
0
20
40
60
80
100
Vf
(%)
Concentración de C14/C12 (%p/v)
75/25 50/50 25/75
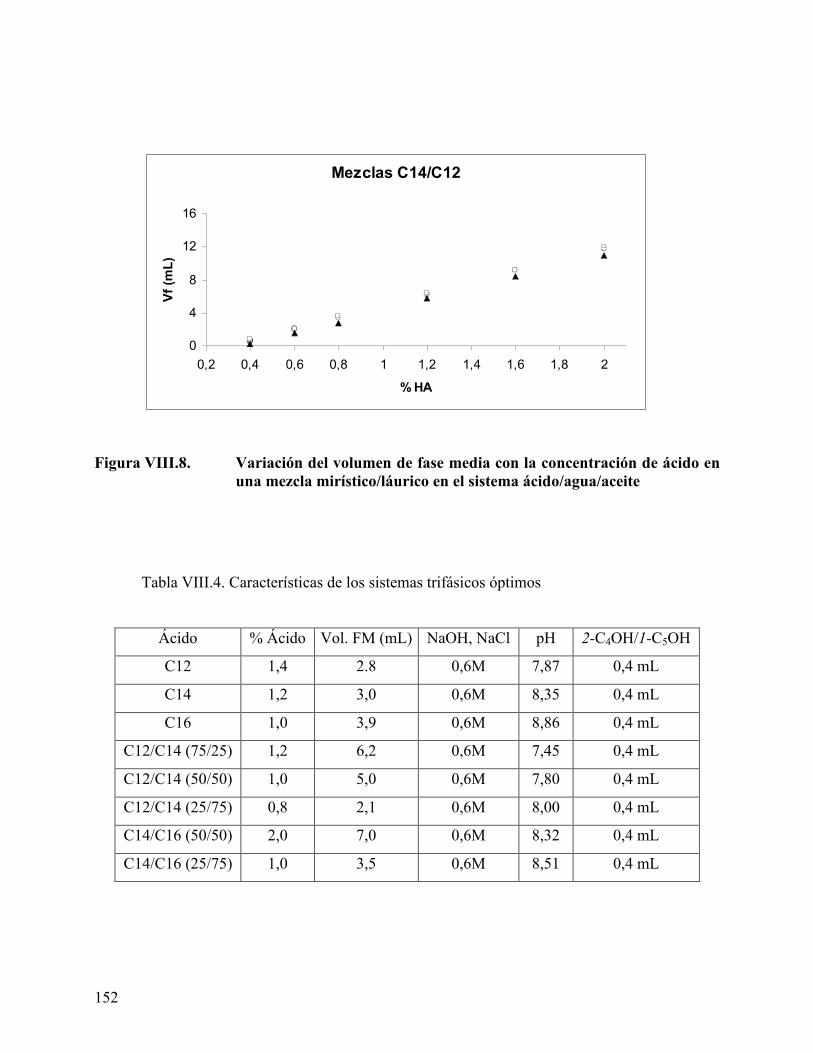
152
Mezclas C14/C12
0
4
8
12
16
0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2
% HA
Vf
(mL
)
Figura VIII.8. Variación del volumen de fase media con la concentración de ácido en una mezcla mirístico/láurico en el sistema ácido/agua/aceite
Tabla VIII.4. Características de los sistemas trifásicos óptimos
Ácido % Ácido Vol. FM (mL) NaOH, NaCl pH 2-C4OH/1-C5OH
C12 1,4 2.8 0,6M 7,87 0,4 mL
C14 1,2 3,0 0,6M 8,35 0,4 mL
C16 1,0 3,9 0,6M 8,86 0,4 mL
C12/C14 (75/25) 1,2 6,2 0,6M 7,45 0,4 mL
C12/C14 (50/50) 1,0 5,0 0,6M 7,80 0,4 mL
C12/C14 (25/75) 0,8 2,1 0,6M 8,00 0,4 mL
C14/C16 (50/50) 2,0 7,0 0,6M 8,32 0,4 mL
C14/C16 (25/75) 1,0 3,5 0,6M 8,51 0,4 mL
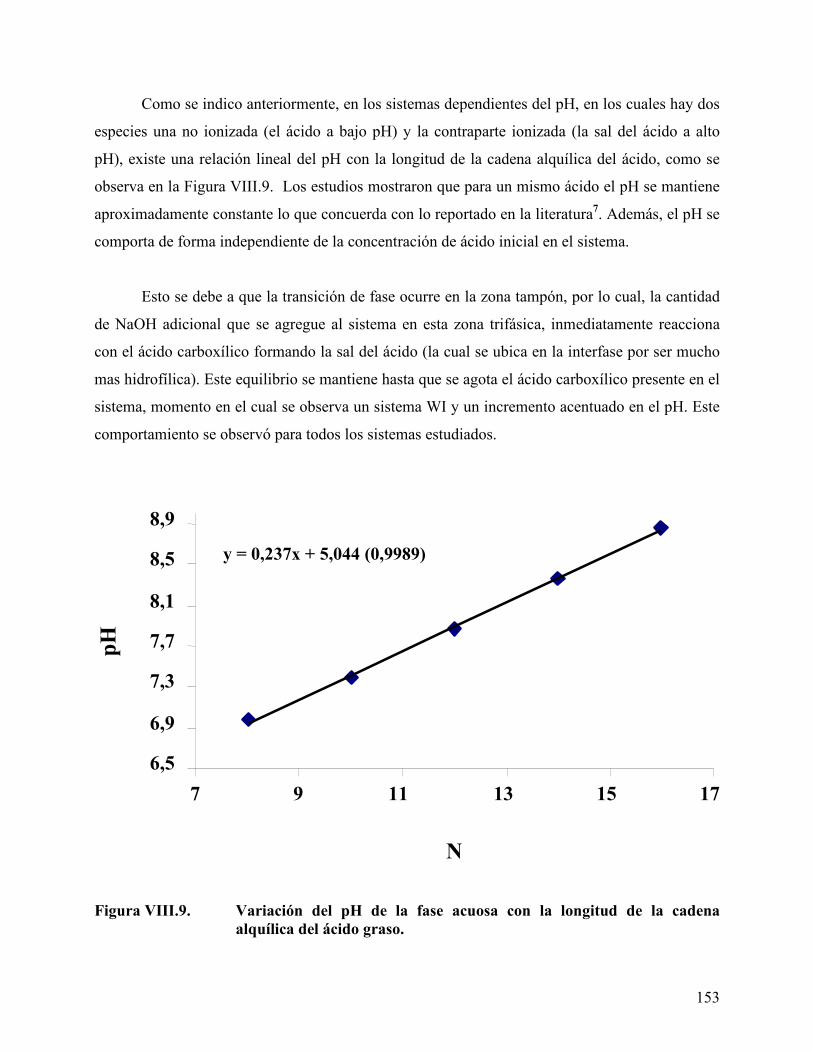
153
Como se indico anteriormente, en los sistemas dependientes del pH, en los cuales hay dos
especies una no ionizada (el ácido a bajo pH) y la contraparte ionizada (la sal del ácido a alto
pH), existe una relación lineal del pH con la longitud de la cadena alquílica del ácido, como se
observa en la Figura VIII.9. Los estudios mostraron que para un mismo ácido el pH se mantiene
aproximadamente constante lo que concuerda con lo reportado en la literatura7. Además, el pH se
comporta de forma independiente de la concentración de ácido inicial en el sistema.
Esto se debe a que la transición de fase ocurre en la zona tampón, por lo cual, la cantidad
de NaOH adicional que se agregue al sistema en esta zona trifásica, inmediatamente reacciona
con el ácido carboxílico formando la sal del ácido (la cual se ubica en la interfase por ser mucho
mas hidrofílica). Este equilibrio se mantiene hasta que se agota el ácido carboxílico presente en el
sistema, momento en el cual se observa un sistema WI y un incremento acentuado en el pH. Este
comportamiento se observó para todos los sistemas estudiados.
Figura VIII.9. Variación del pH de la fase acuosa con la longitud de la cadena alquílica del ácido graso.
y = 0,237x + 5,044 (0,9989)
6,5
6,9
7,3
7,7
8,1
8,5
8,9
7 9 11 13 15 17
N
pH

154
Efecto del tipo y concentración del co-surfactante (alcohol) en el comportamiento de fase
Por razones prácticas, los surfactantes se usan a menudo junto con un alcohol, sea por el
papel físico de éste o bien por su influencia físico-química. Por ejemplo los alcoholes tienden a
reducir la CMC de un surfactante, es decir que favorece la formación de micelas. Sin embargo,
esto depende del tipo (peso molecular y ramificación) y concentración del alcohol en el sistema.
Shinoda (1954)10 encontró mediante una expresión para determinar la CMC, que cuanto más
lipofílico es el alcohol, mas importante es el descenso de la misma.
Este fenómeno se explica por la formación de micelas mixtas surfactante-alcohol, en las
cuales la inserción de las moléculas de alcohol permite reducir las fuerzas repulsivas entre los
grupos hidrofílicos de las moléculas vecinas de surfactante, lo que resulta en un descenso de la
energía de formación de las micelas, y por lo tanto una reducción de la CMC. Inicialmente, se
evaluó el efecto del n-butanol en los sistemas ácido/agua/aceite a diferentes concentraciones de
hidróxido de sodio (0,2 a 0,8 M) y a 0,5% de ácido en el sistema, con la finalidad de obtener
sistemas trifásicos sin adición de sal al sistema. Los resultados mostraron una tendencia lineal de
la concentración de n-butanol con la cadena lipofílica del ácido en todo el rango de NaOH
estudiado (Figura VIII.10).
Por otro lado, en la Figura VIII.10, se observa que el porcentaje de n-butanol necesario
para obtener sistemas trifásicos, disminuye con la concentración de hidróxido de sodio en el
sistema, así como con la cadena alquílica del ácido. Lo que se traduce, en que los ácidos de
mayor cadena son mas rápidamente desplazados de la fase oleica por acción del co-surfactante,
aumentando por consiguiente la concentración de la mezcla de surfactantes (ácido-carboxilato) en
la interfase.
Así mismo, se evaluó el efecto de este alcohol en los sistemas a formulación óptima para
las mezclas mirístico/palmítico, observándose que el volumen de fase media disminuye con la
concentración de alcohol en el sistema y a expensas del volumen de la fase oleica (Figuras
VIII.11 y VIII.12).
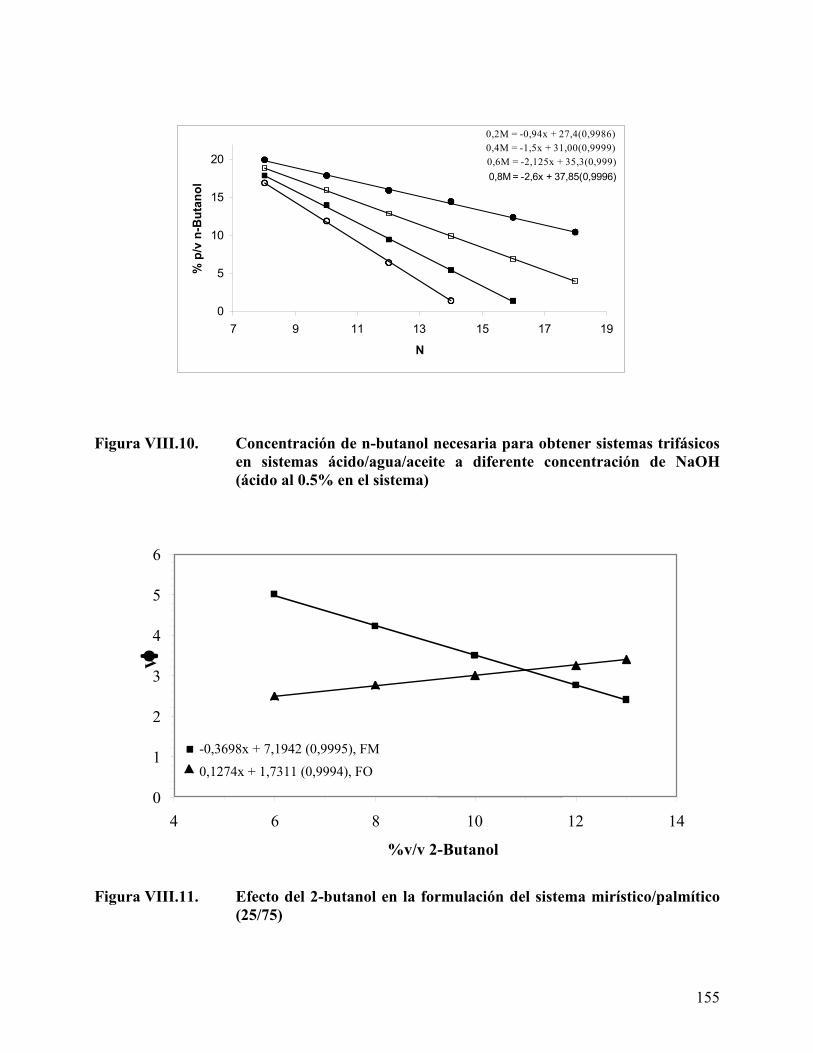
155
Figura VIII.10. Concentración de n-butanol necesaria para obtener sistemas trifásicos en sistemas ácido/agua/aceite a diferente concentración de NaOH (ácido al 0.5% en el sistema)
Figura VIII.11. Efecto del 2-butanol en la formulación del sistema mirístico/palmítico (25/75)
0,8M = -2,6x + 37,85(0,9996)
0,6M = -2,125x + 35,3(0,999)
0,4M = -1,5x + 31,00(0,9999)
0,2M = -0,94x + 27,4(0,9986)
0
5
10
15
20
7 9 11 13 15 17 19
N
% p
/v n
-Bu
tan
ol
0
1
2
3
4
5
6
4 6 8 10 12 14
%v/v 2-Butanol
V!
-0,3698x + 7,1942 (0,9995), FM
0,1274x + 1,7311 (0,9994), FO
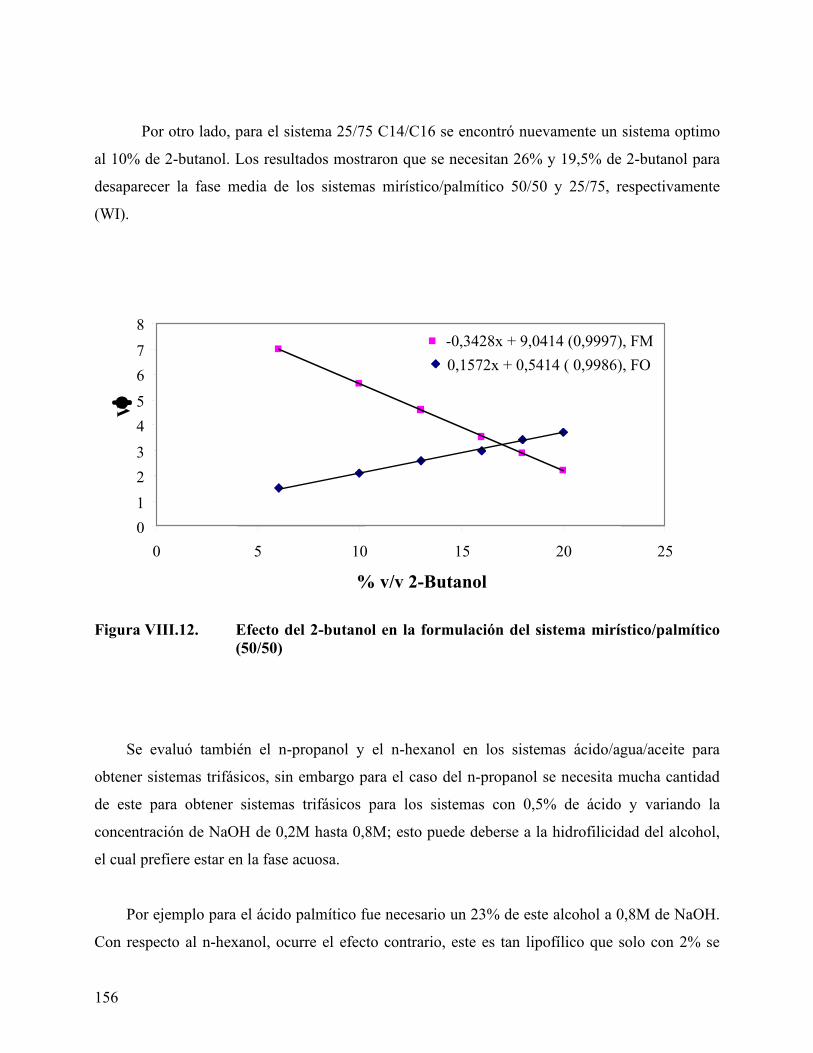
156
Por otro lado, para el sistema 25/75 C14/C16 se encontró nuevamente un sistema optimo
al 10% de 2-butanol. Los resultados mostraron que se necesitan 26% y 19,5% de 2-butanol para
desaparecer la fase media de los sistemas mirístico/palmítico 50/50 y 25/75, respectivamente
(WI).
Figura VIII.12. Efecto del 2-butanol en la formulación del sistema mirístico/palmítico (50/50)
Se evaluó también el n-propanol y el n-hexanol en los sistemas ácido/agua/aceite para
obtener sistemas trifásicos, sin embargo para el caso del n-propanol se necesita mucha cantidad
de este para obtener sistemas trifásicos para los sistemas con 0,5% de ácido y variando la
concentración de NaOH de 0,2M hasta 0,8M; esto puede deberse a la hidrofilicidad del alcohol,
el cual prefiere estar en la fase acuosa.
Por ejemplo para el ácido palmítico fue necesario un 23% de este alcohol a 0,8M de NaOH.
Con respecto al n-hexanol, ocurre el efecto contrario, este es tan lipofílico que solo con 2% se
0,1572x + 0,5414 ( 0,9986), FO
-0,3428x + 9,0414 (0,9997), FM
0
1
2
3
4
5
6
7
8
0 5 10 15 20 25
% v/v 2-Butanol
V!

157
obtuvo un sistema trifásico para el ácido láurico a 0,6M de NaOH, este alcohol debe desplazar
casi de inmediato al ácido que se encuentra en la fase oleica ya que no es posible observar la zona
trifásica.
Cabe destacar, que estos sistemas se prepararon en ausencia de sal (NaCl), observándose
la formación de una turbidez en la fase acuosa de dichos sistemas antes de la formación de la
zona trifásica. Este fenómeno lleva a pensar que se puede estar en presencia de una transición:
23232 !!!! . Sin embargo, la primera zona trifásica no se observa. Además, los
resultados muestran que es necesario la presencia de sal en dichos sistemas para la obtención de
los trifásicos con baja concentración de los alcoholes estudiados. Esto puede explicarse por el
fenómeno del salting out, el cual indica que la sal presente en la fase acuosa siempre será mas
soluble que el carboxilato formado con la variación del pH en el sistema, produciendo la
formación de una zona trifásica como consecuencia del desplazamiento del carboxilato por
adición de sal (NaCl) y del ácido por la adición del alcohol.
Para corroborar esta hipótesis, se repitieron los sistemas con ácido láurico y mirístico
(para los cuales se obtuvieron sistemas trifásicos con una mezcla de 2-butanol y n-pentanol), en
presencia de NaCl y adicionando n-butanol gradualmente, encontrándose que solo con un 3% y
4% de éste aproximadamente se alcanzaron los sistemas trifásicos para los ácidos láurico y
mirístico, respectivamente. Esto representa una ventaja, ya que no es necesaria una mezcla de
alcoholes ni alta concentración de estos para obtener sistemas trifásicos, lo cual disminuye el uso
de reactivos y por tanto de costos a la hora de formular con estos ácidos.
Por otro lado, las propiedades de las soluciones de surfactantes, en particular las que
tienen relación con la solubilización, varían con la temperatura9. Para los surfactantes iónicos, la
concentración de surfactante solubilizado en el agua en equilibrio con un precipitado sólido
aumenta lentamente con la temperatura hasta un cierto valor, llamado temperatura de Kraft, a
partir del cual la solubilización se produce en forma de micelas, y aumenta considerablemente.
Además, un aumento de la temperatura tiende a aumentar el carácter hidrofílico del surfactante.
Aprovechando esta característica, se empleo la temperatura para facilitar la homogenización de
las fases al preparar los sistemas, ya que al formarse el carboxilato con la adición de NaOH se

158
observo la precipitación de la sal del ácido. Sin embargo, no se observo en todos los casos la
formación del sistema trifásico. Para el caso del sistema al 1% de ácido y barrido de n-butanol,
los sistemas con solo 5% del alcohol al calentarlos formaron sistemas trifásicos, de los cuales
solo el sistema con NaOH 0,8M permaneció estable luego de alcanzar la temperatura ambiente y
el equilibrio.
Se evaluó también el efecto de adición de alcoholes a los sistemas a formulación óptima
para los diferentes ácidos estudiados. Se emplearon los alcoholes n-propanol, n-butanol, n-
pentanol y n-hexanol para el estudio. Los resultados mostraron que para los ácidos mirístico y
palmitito se necesita menor e igual cantidad de n-pentanol y n-hexanol para desaparecer la
microemulsión (Tabla VIII.5). Estos alcoholes lipofílicos deben aumentar la afinidad del
surfactante por la fase oleica, favoreciendo el sistema bifásico.
Tabla VIII.5. Concentración (%v/v) de alcohol necesario en los sistemas a formulación
óptima para la formación de sistemas bifásicos.
Alcohol Ácido Láurico Ácido Mirístico Ácido Palmítico
n-Propanol 1,5 2,0 1,5
n-Butanol 3,5 2,0 2,0
n-Pentanol 1,5 1,0 1,0
n-Hexanol 2,0 1,0 1,0
En la Figura VIII.13, se muestra el efecto del n-butanol en los sistemas a formulación
óptima para los ácidos láurico, mirístico y palmítico, respectivamente. Se observa de la figura que
a medida que se incrementa la cantidad de alcohol en el sistema, aumenta la concentración del
ácido en la fase oleica, lo que hace pensar que se estaría produciendo una transición retrograda.
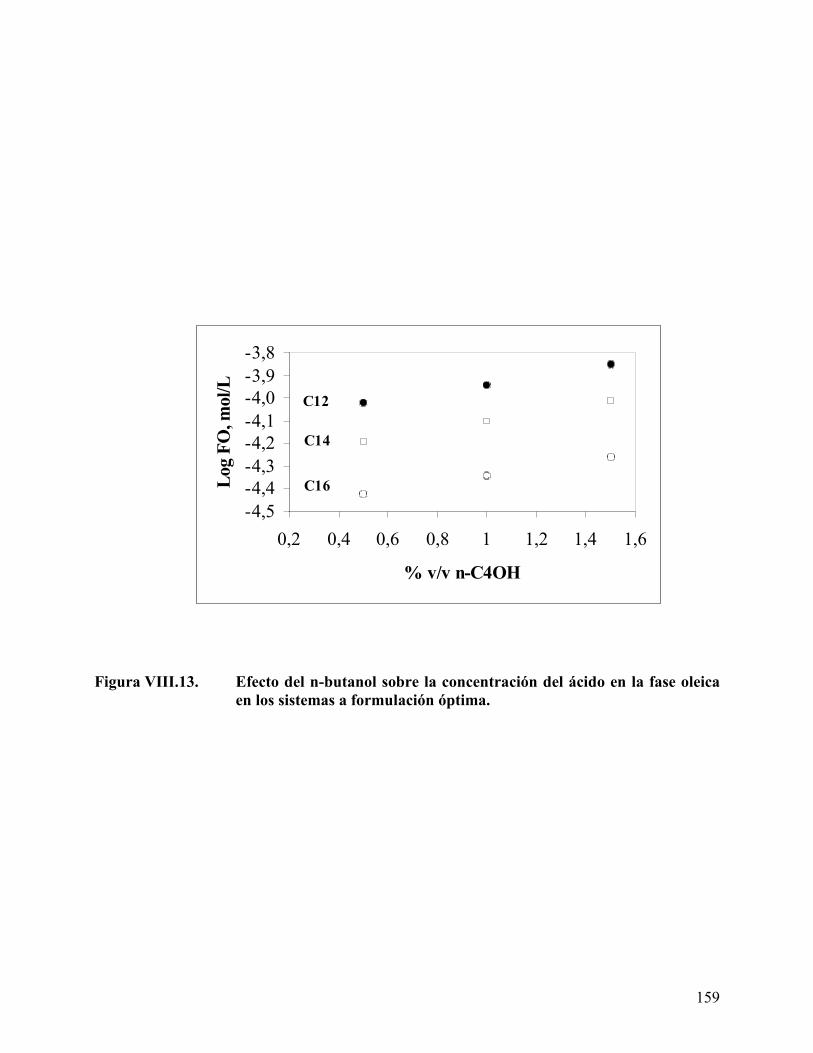
159
Figura VIII.13. Efecto del n-butanol sobre la concentración del ácido en la fase oleica en los sistemas a formulación óptima.
-4,5
-4,4
-4,3
-4,2
-4,1
-4,0
-3,9
-3,8
0,2 0,4 0,6 0,8 1 1,2 1,4 1,6
% v/v n-C4OH
Log F
O, m
ol/
L
C12
C14
C16
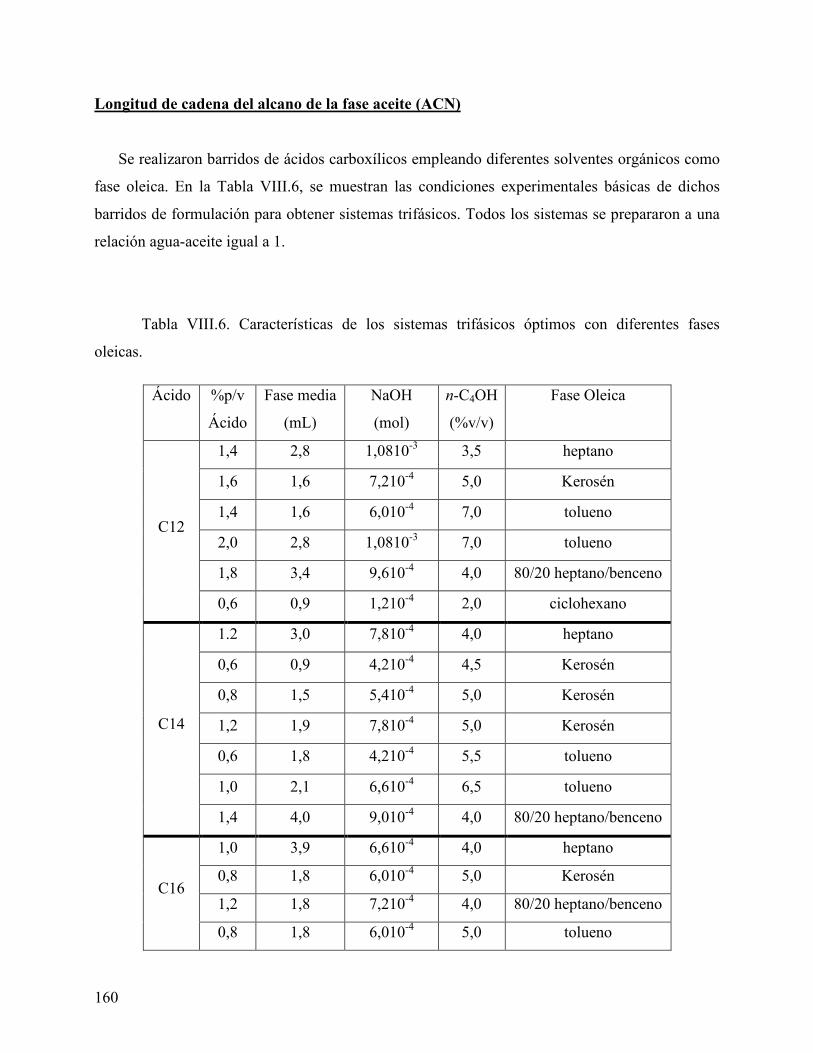
160
Longitud de cadena del alcano de la fase aceite (ACN)
Se realizaron barridos de ácidos carboxílicos empleando diferentes solventes orgánicos como
fase oleica. En la Tabla VIII.6, se muestran las condiciones experimentales básicas de dichos
barridos de formulación para obtener sistemas trifásicos. Todos los sistemas se prepararon a una
relación agua-aceite igual a 1.
Tabla VIII.6. Características de los sistemas trifásicos óptimos con diferentes fases
oleicas.
Ácido %p/v
Ácido
Fase media
(mL)
NaOH
(mol)
n-C4OH
(%v/v)
Fase Oleica
1,4 2,8 1,0810-3 3,5 heptano
1,6 1,6 7,210-4 5,0 Kerosén
1,4 1,6 6,010-4 7,0 tolueno
2,0 2,8 1,0810-3 7,0 tolueno
1,8 3,4 9,610-4 4,0 80/20 heptano/benceno
C12
0,6 0,9 1,210-4 2,0 ciclohexano
1.2 3,0 7,810-4 4,0 heptano
0,6 0,9 4,210-4 4,5 Kerosén
0,8 1,5 5,410-4 5,0 Kerosén
1,2 1,9 7,810-4 5,0 Kerosén
0,6 1,8 4,210-4 5,5 tolueno
1,0 2,1 6,610-4 6,5 tolueno
C14
1,4 4,0 9,010-4 4,0 80/20 heptano/benceno
1,0 3,9 6,610-4 4,0 heptano
0,8 1,8 6,010-4 5,0 Kerosén
1,2 1,8 7,210-4 4,0 80/20 heptano/benceno C16
0,8 1,8 6,010-4 5,0 tolueno
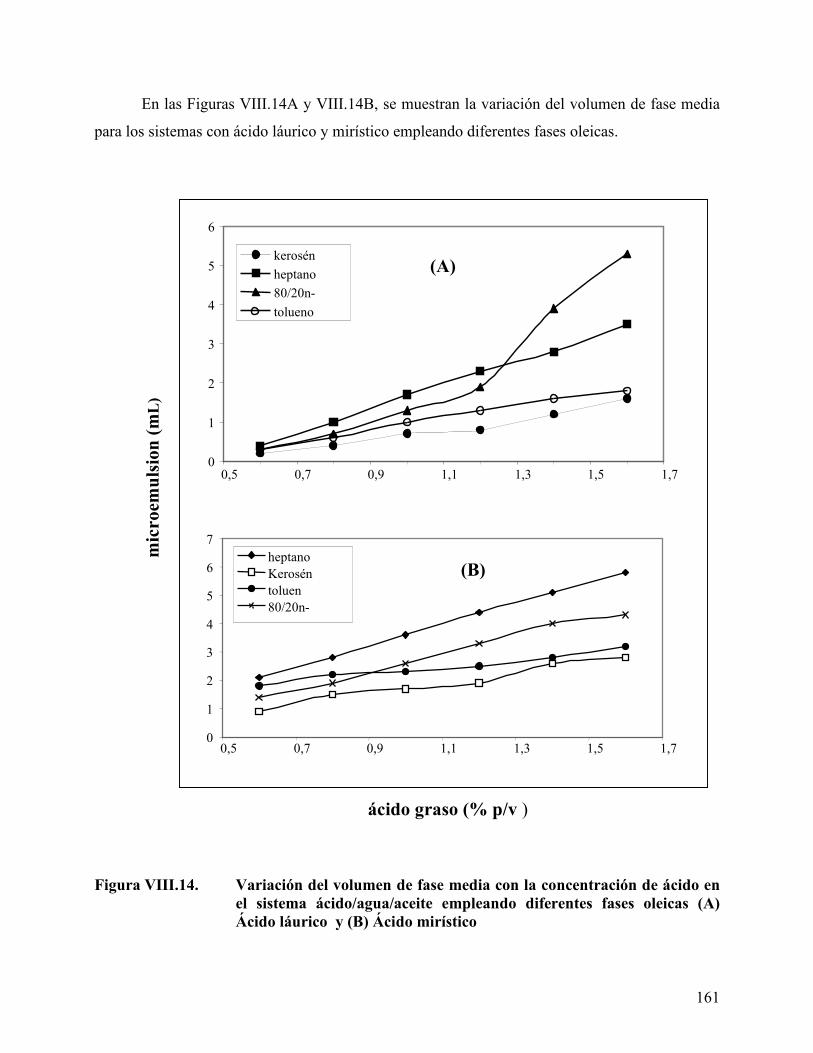
161
En las Figuras VIII.14A y VIII.14B, se muestran la variación del volumen de fase media
para los sistemas con ácido láurico y mirístico empleando diferentes fases oleicas.
Figura VIII.14. Variación del volumen de fase media con la concentración de ácido en el sistema ácido/agua/aceite empleando diferentes fases oleicas (A) Ácido láurico y (B) Ácido mirístico
mic
roem
ulsi
on (m
L)
ácido graso (% p/v )
0
1
2
3
4
5
6
0,5 0,7 0,9 1,1 1,3 1,5 1,7
kerosén heptano 80/20n-C7/Bz tolueno
(A)
0
1
2
3
4
5
6
7
0,5 0,7 0,9 1,1 1,3 1,5 1,7
heptano Kerosén tolueno 80/20n-C7/Bz
(B)

162
Se puede remarcar las siguientes observaciones para las fases oleicas diferentes del
heptano:
1. No existe una tendencia completamente lineal del volumen de fase media con la
concentración del ácido en el sistema. Sin embargo, se observa en todos los casos un
aumento del volumen de fase.
2. Todas las curvas se encuentran por debajo de la del sistema con heptano, con un orden de
menor a mayor aproximado de: kerosén, tolueno, 80/20 heptano/benceno y heptano. Este
orden puede atribuirse a la polaridad de la fase.
En la Figura VIII.15 se muestra la curva de solubilidad del ácido mirístico obtenida
graficando el volumen de fase (acuosa y oleica) con la concentración del ácido en el sistema. En
esta figura, se observa que el sistema óptimo se obtiene a 1,0 % p/v de ácido, lo que representa el
sistema de mínima tensión interfacial.
FO = -1,2759x + 5,1972 (0,997)
FA = -0,5747x + 4,4676 (0,989)
2,0
2,5
3,0
3,5
4,0
4,5
5,0
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2
% p/v ácido mirístico
Fa
se (
mL
)
FO = -1,2759x + 5,1972 (0,997)
FA = -0,5747x + 4,4676 (0,989)
2,0
2,5
3,0
3,5
4,0
4,5
5,0
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 2,2
% p/v ácido mirístico
Fa
se (
mL
)
Figura VIII.15 Variación del volumen de fase del sistema con la concentración del ácido mirístico, empleando tolueno como fase oleica.

163
CONCLUSIONES
Para la obtención de sistemas trifásicos es necesario el uso de n-butanol para ayudar la
solubilización, así como de NaCl para ayudar a la especie carboxilato a ubicarse en la interfase
con la especie ácido no disociada y formar así la microemulsión.
En estos sistemas el aumento en la concentración del ácido produce un aumento lineal del
volumen de la microemulsion. Además, El pH óptimo aumenta con la cadena lipofílica del ácido
y es casi esencialmente independiente de la cantidad de ácido inicial añadida al sistema.
En los sistemas ácido/agua/aceite, a un pH dado el ácido carboxílico se disocia
parcialmente y se reparte entre las fases del sistema, en el cual la especie no disociada se
encuentra en la fase oleica mientras que la especie disociada (carboxilato) se encuentra en la fase
acuosa. Por tanto, el comportamiento de fase de estos sistemas en función del pH esta
determinado por la mezcla de las especies anfifílicas presentes.

164
REFERENCIAS BIBLIOGRÁFICAS
[1] Bourrel, M.; Graciaa, A.; Schechter, R.; Wade, W. J. Colloid Interf. Sci. 1979, 72, 161-
169.
[2] Jan, L.; Sharma, M.; Chang, Y.; Chang, M.; Yen, T. AICHE Symposium series. 1982, Nº
212, 78, 97-101.
[3] Layrise, I.; Rivas, H.; Acevedo, S. J. Dispers. Ssci. Technol. 1984, 5, 1-7.
[4] Ovalles, C.; García, M.; Lujano, E.; Aular, W.; Bermúdez, R.; Cotte, E. Fuel 1998, 77, 3-
11.
[5] Bourrel, M.; Schechter, R. Microemulsions and Related Systems; Marcel Dekker, New
York, 1988.
[6] Salager, J-L.; Morgan, J.; Schechter, R.; Wade, W.; Vasquez, E. J. Soc. Petrol. Eng. 1979,
19, 107-112.
[7] Antón, R.; Salager, J. J. Colloid Int. Sci. 1990, 140, 75-81.
[8] Qutubuddin, S.; Miller, C.; Fort, T. J. Colloid Interf. Sci. 1984, 101, 46-53.
[9] Salager, J-L.; Antón, R. Ionic microemulsions. Surfactant science series, Marcel Dekker,
New York, 1989.
[10] Shinoda, K.; Kunieda, H. in: Becher, P. (ed.). Encyclopedia of Emulsions Technology.
Marcel Dekker, Inc., New Cork. 1985, Vol. 1.

165
C A P I T U L O I X

166
INFLUENCIA DE LA FORMULACIÓN EN EL REPARTO DE LOS ÁCIDOS CARBOXÍLICOS EN SISTEMAS ÁCIDO/AGUA/ACEITE
Un sistema trifásico óptimo en el caso de los compuestos ionizables, posee cuatro
componentes: salmuera, aceite y dos surfactantes (HA) y (A-)1. Por otro lado, el equilibrio: HA
→ A- + H+, se produce exclusivamente en la fase acuosa y en la fase media. En la microemulsión,
se encuentra la mayor concentración de la especie disociada, mientras que la especie no disociada
se encuentra en equilibrio en las fases oleica y acuosa de acuerdo a su reparto preferencial (Kr).
No obstante, las cantidades de HA y A- en la fase acuosa, son despreciables desde el punto de
vista del balance de masa, y corresponden a la CMC de la mezcla de surfactantes.
En estos sistemas, siempre ocurre un reparto de las especies en las distintas fases como
resultado de las distintas afinidades que muestran las especies presentes en el sistema (el ácido es
lipofílico, mientras que el carboxilato es hidrofílico). Este reparto preferencial se expresa a través
del coeficiente de reparto del ácido (Kr) entre el aceite y el agua.
Esta información es de gran importancia en el área de fenómenos interfaciales, ya que se
ha demostrado en estudios previos2-3,4-7 que el Kr esta relacionado con la influencia de las
variables de formulación (tipo y concentración de anfifilo, tipo de aceite, entre otros.) en los
sistemas surfactante/aceite/agua.
SECCIÓN EXPERIMENTAL
Equipos
El análisis de las fases de los sistemas ácido carboxílico/agua/aceite se realizó en el
cromatógrafo líquido modular (Waters), descrito en el capitulo IV.

167
Materiales
Balanza analítica, marca ACCULAB, sensibilidad ± 0,0001 g
Estufa, marca memmert, temperatura máxima de 250 ºC.
Material de vidrio en general.
Reactivos.
Ácidos carboxílicos (CAX), con 8, 10, 12, 14, 16 y 18 átomos de carbono
Se empleó metanol y acetonitrilo como fase móvil (grado HPLC (J.T. Beaker) Agua destilada y desionizada
Procedimiento Experimental
Evaluación del reparto de los surfactantes ácidos carboxílicos en sistemas de Winsor a
formulación óptima.
Para el estudio del reparto de los ácidos se requiere, la presencia de un sistema óptimo (W
III) y este se alcanza mediante un procedimiento experimental conocido como barrido de
formulación. Para ello se va modificando la variable en estudio hasta conseguir el sistema
deseado (generalmente el de mayor importancia, W III o lo que es lo mismo, la formulación
óptima).
Los sistemas a formulación óptima obtenidos, en los cuales se evalúan tanto las variables
de composición como de formulación, se analizaron siguiendo el siguiente procedimiento: se
separaron cada una de las fases componentes del sistema (media, acuosa y oleica), se midió el
pH de las fases acuosas, y luego se secaron las tres fases utilizando un rotavapor o en la
estufa. Los ácidos carboxílicos de cada fase se disolvieron en metanol y se inyectaron en el
cromatógrafo liquido alícuotas de 10 µL. Para su estudio se utilizó el método de
fraccionamiento mas simple por HPLC optimizado en el capitulo anterior, con el cual se
obtiene el cromatograma de las tres fases en juego, y se calcula el coeficiente de reparto (Kr)

168
del ácido carboxílico en el sistema empleado. Con estos datos se determina la concentración
del ácido carboxílico en las tres fases.
La constante de reparto viene dada por la ecuación (1):
Kr = Co/Cw. (1)
Donde Co es la concentración del ácido no disociado en la fase oleica (HAo) y Cw es la
concentración del ácido no disociado en la fase acuosa (HAw).

169
RESULTADOS Y DISCUSIÓN
Los ácidos grasos son ácidos débiles que presentan en solución acuosa un equilibrio de
disociación. Es evidente que el pH puede afectar considerablemente este equilibrio, ya que la
concentración de ácido no disociado en el agua es muy débil y se puede considerar
frecuentemente que es constante e igual a la solubilidad del ácido en el agua. A un pH neutro o
ácido, la concentración de ión hidrogeno es relativamente grande, y de hecho la fracción ionizada
es esencialmente nula, por lo que es necesario alcanzar un pH alcalino para tener una fracción
notable de carboxilato.
Si se ajusta una solución alcalina a un sistema ácido-agua-aceite equilibrada, el álcali va a
neutralizar el ácido no disociado presente en la fase acuosa y va por tanto a reducir su
concentración, que ya es débil. Una cierta cantidad de ácido va entonces a pasar de la fase oleica
a la fase acuosa, justa para restablecer el equilibrio de partición. El ácido presente en la fase
oleica actuará por tanto como una reserva. Este reparto se calcula empleando la ecuación descrita
anteriormente (1).
El reparto del ácido no disociado se midió entre las fases en exceso acuosa y aceite del
sistema trifásico a formulación óptima o, en los sistemas mas cercanos a dicha formulación pero
a una misma concentración de ácido para la comparación respectiva. Como es de esperarse, la
fase aceite en exceso contiene una mayor concentración de ácido comparada a la encontrada en la
fase acuosa (Tabla IX.1). De hecho, la concentración del ácido (en su forma asociada) en la fase
acuosa esta limitada por su solubilidad en agua o la CMC.
Las concentraciones del ácido en cada fase se obtienen a partir de una curva de
calibración que relaciona el área o altura del pico con la concentración del ácido. En la Figura
IX.1, se muestra las curvas de calibración respectivas para los ácidos láurico, mirístico y
palmítico. Estas curvas presentan un coeficiente de correlación muy cercano a la unidad, lo que
las caracteriza como buenas curvas para la cuantificación de la especie en estudio.
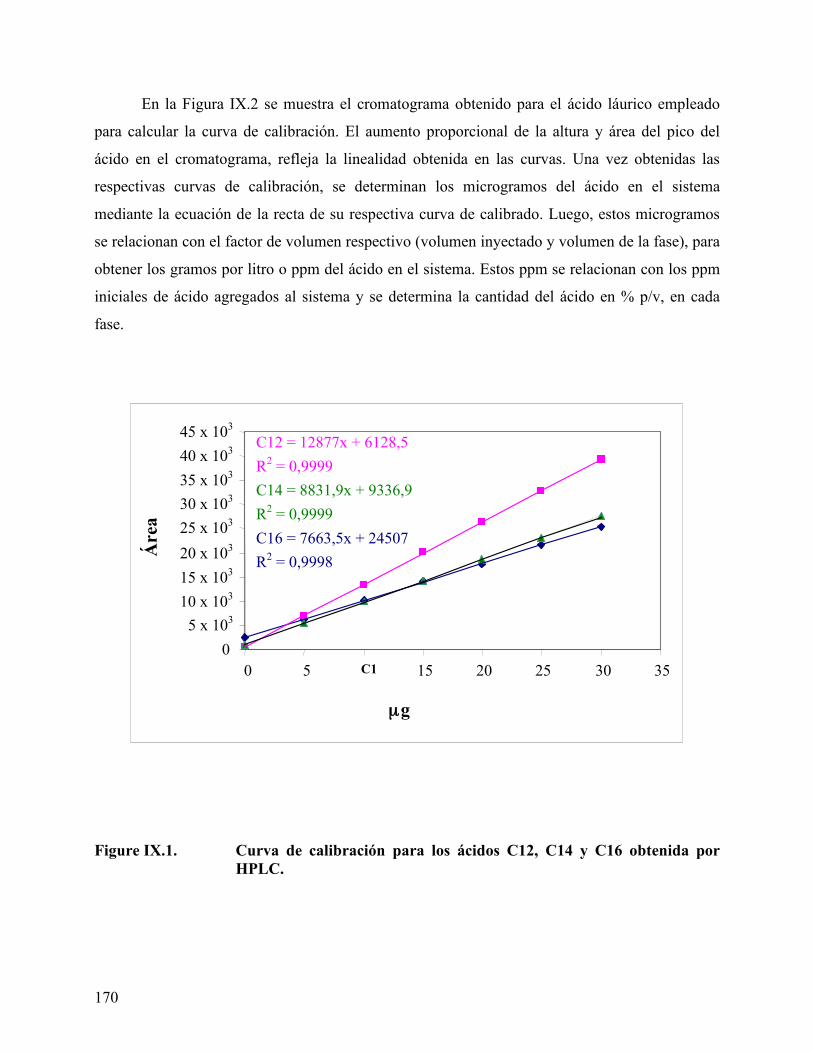
170
En la Figura IX.2 se muestra el cromatograma obtenido para el ácido láurico empleado
para calcular la curva de calibración. El aumento proporcional de la altura y área del pico del
ácido en el cromatograma, refleja la linealidad obtenida en las curvas. Una vez obtenidas las
respectivas curvas de calibración, se determinan los microgramos del ácido en el sistema
mediante la ecuación de la recta de su respectiva curva de calibrado. Luego, estos microgramos
se relacionan con el factor de volumen respectivo (volumen inyectado y volumen de la fase), para
obtener los gramos por litro o ppm del ácido en el sistema. Estos ppm se relacionan con los ppm
iniciales de ácido agregados al sistema y se determina la cantidad del ácido en % p/v, en cada
fase.
Figure IX.1. Curva de calibración para los ácidos C12, C14 y C16 obtenida por HPLC.
C16 = 7663,5x + 24507 R2 = 0,9998
C12 = 12877x + 6128,5 R2 = 0,9999 C14 = 8831,9x + 9336,9 R2 = 0,9999
0 5 x 103
10 x 103 15 x 103 20 x 103
25 x 103 30 x 103 35 x 103 40 x 103 45 x 103
0 5 C14 15 20 25 30 35
µg FAs
Áre
a
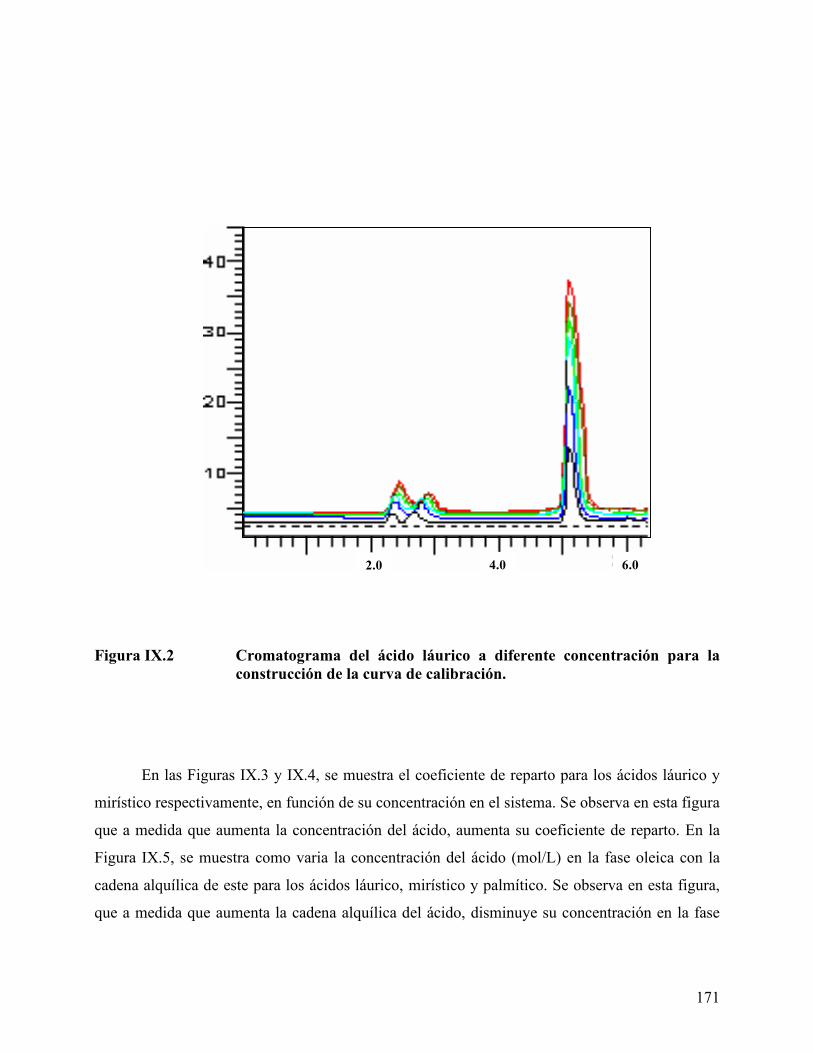
171
Figura IX.2 Cromatograma del ácido láurico a diferente concentración para la construcción de la curva de calibración.
En las Figuras IX.3 y IX.4, se muestra el coeficiente de reparto para los ácidos láurico y
mirístico respectivamente, en función de su concentración en el sistema. Se observa en esta figura
que a medida que aumenta la concentración del ácido, aumenta su coeficiente de reparto. En la
Figura IX.5, se muestra como varia la concentración del ácido (mol/L) en la fase oleica con la
cadena alquílica de este para los ácidos láurico, mirístico y palmítico. Se observa en esta figura,
que a medida que aumenta la cadena alquílica del ácido, disminuye su concentración en la fase
4.0 2.0 6.0
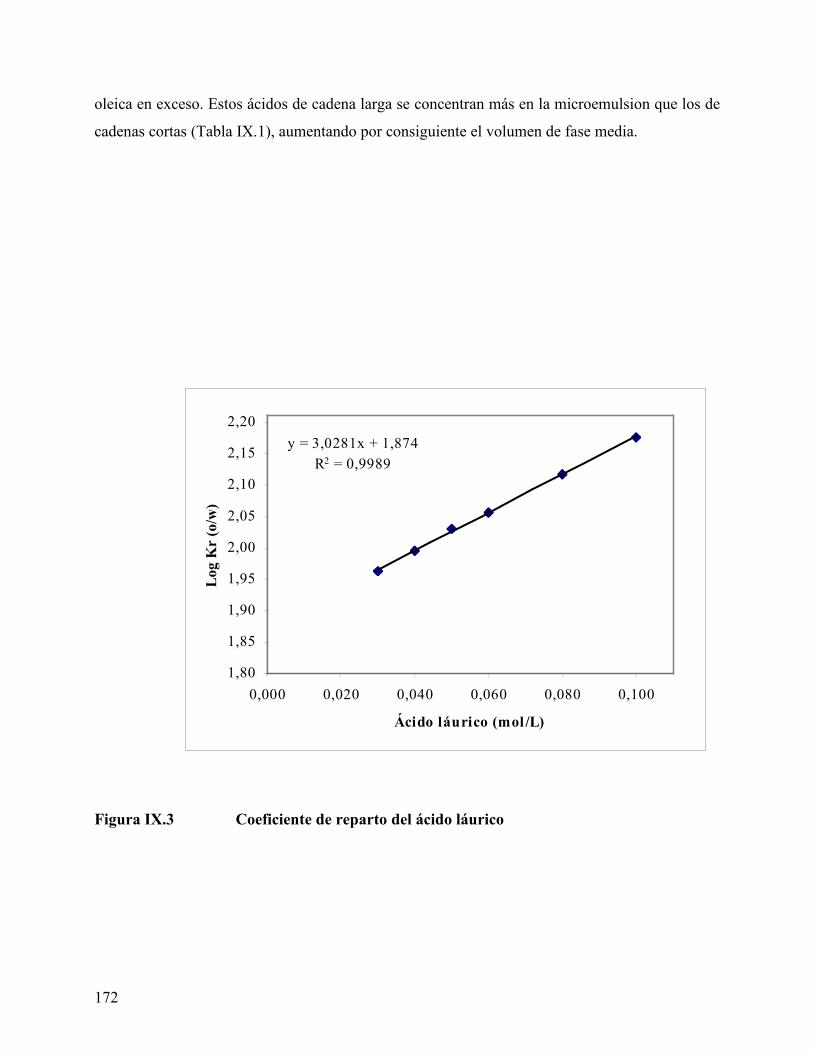
172
oleica en exceso. Estos ácidos de cadena larga se concentran más en la microemulsion que los de
cadenas cortas (Tabla IX.1), aumentando por consiguiente el volumen de fase media.
Figura IX.3 Coeficiente de reparto del ácido láurico
y = 3,0281x + 1,874
R2 = 0,9989
1,80
1,85
1,90
1,95
2,00
2,05
2,10
2,15
2,20
0,000 0,020 0,040 0,060 0,080 0,100
Ácido láurico (mol/L)
Log
Kr
(o/w
)
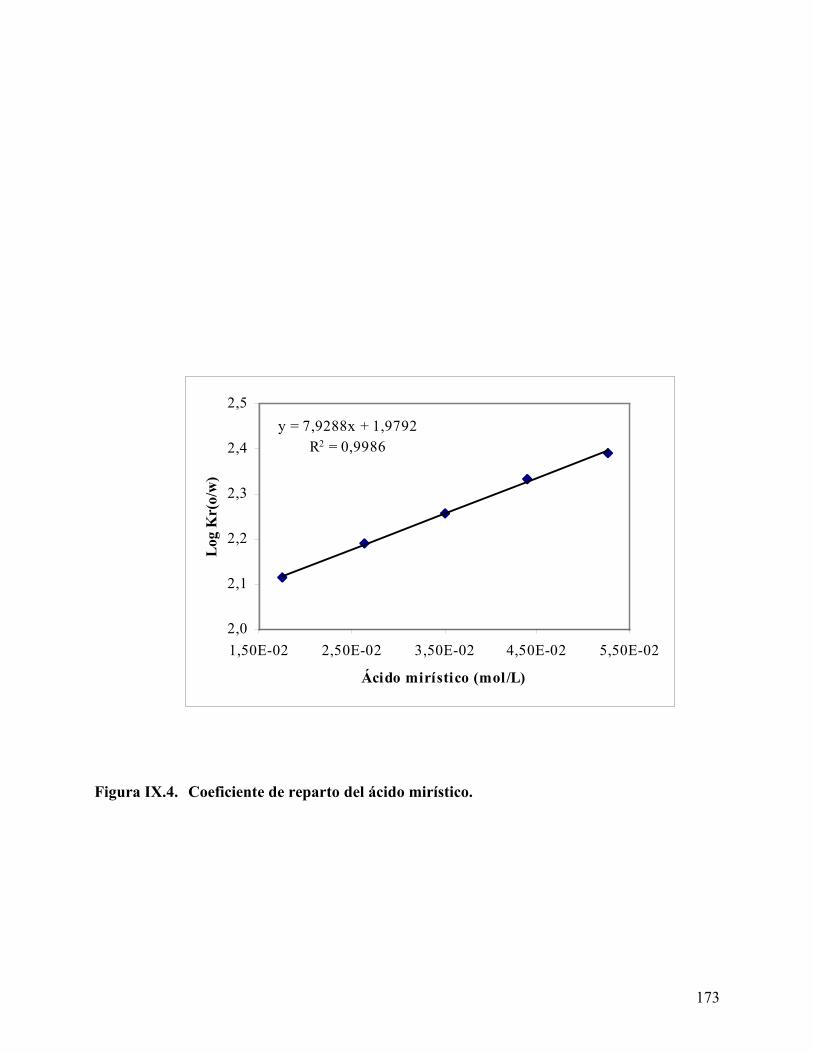
173
Figura IX.4. Coeficiente de reparto del ácido mirístico.
y = 7,9288x + 1,9792
R2 = 0,9986
2,0
2,1
2,2
2,3
2,4
2,5
1,50E-02 2,50E-02 3,50E-02 4,50E-02 5,50E-02
Ácido mirístico (mol/L)
Log
Kr(
o/w
)
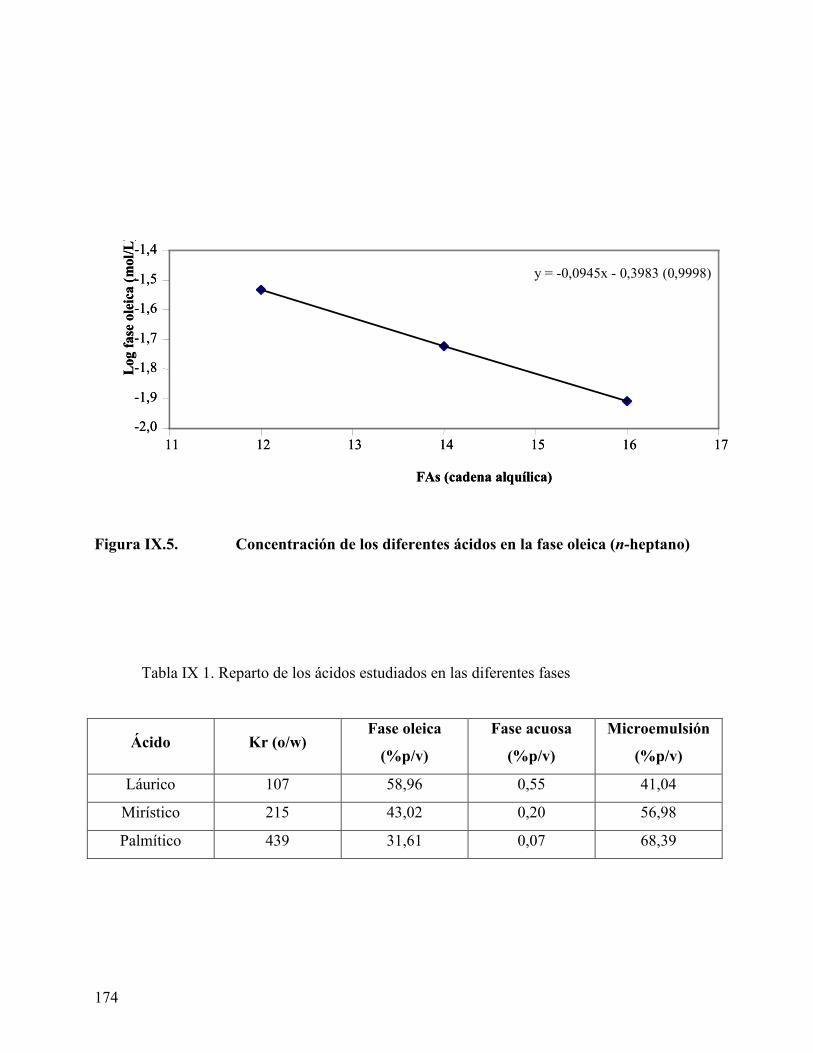
174
Figura IX.5. Concentración de los diferentes ácidos en la fase oleica (n-heptano)
Tabla IX 1. Reparto de los ácidos estudiados en las diferentes fases
Ácido Kr (o/w) Fase oleica
(%p/v)
Fase acuosa
(%p/v)
Microemulsión
(%p/v)
Láurico 107 58,96 0,55 41,04
Mirístico 215 43,02 0,20 56,98
Palmítico 439 31,61 0,07 68,39
y = -0,0945x - 0,3983 (0,9998)
-2,0
-1,9
-1,8
-1,7
-1,6
-1,5
-1,4
11 12 13 14 15 16 17
FAs (cadena alquílica)
Log f
ase
ole
ica (
mol /
L)
y = -0,0945x - 0,3983 (0,9998)
-2,0
-1,9
-1,8
-1,7
-1,6
-1,5
-1,4
11 12 13 14 15 16 17
FAs (cadena alquílica)
Log f
ase
ole
ica (
mol /
L)
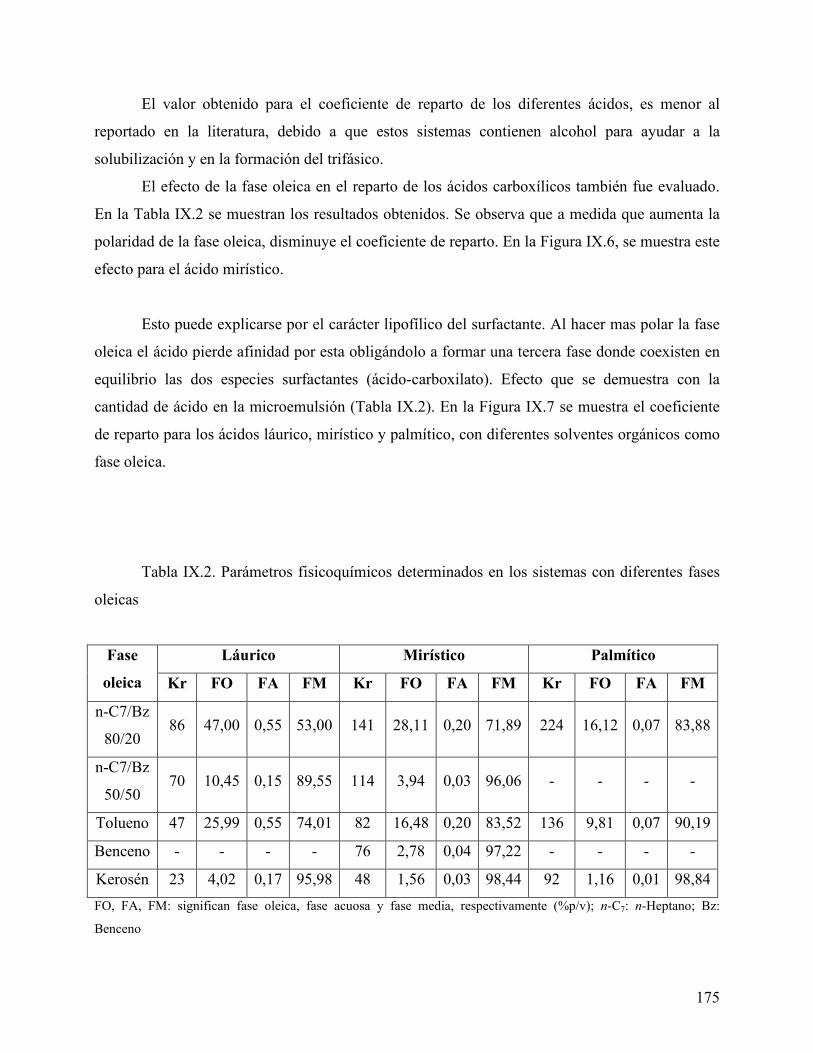
175
El valor obtenido para el coeficiente de reparto de los diferentes ácidos, es menor al
reportado en la literatura, debido a que estos sistemas contienen alcohol para ayudar a la
solubilización y en la formación del trifásico.
El efecto de la fase oleica en el reparto de los ácidos carboxílicos también fue evaluado.
En la Tabla IX.2 se muestran los resultados obtenidos. Se observa que a medida que aumenta la
polaridad de la fase oleica, disminuye el coeficiente de reparto. En la Figura IX.6, se muestra este
efecto para el ácido mirístico.
Esto puede explicarse por el carácter lipofílico del surfactante. Al hacer mas polar la fase
oleica el ácido pierde afinidad por esta obligándolo a formar una tercera fase donde coexisten en
equilibrio las dos especies surfactantes (ácido-carboxilato). Efecto que se demuestra con la
cantidad de ácido en la microemulsión (Tabla IX.2). En la Figura IX.7 se muestra el coeficiente
de reparto para los ácidos láurico, mirístico y palmítico, con diferentes solventes orgánicos como
fase oleica.
Tabla IX.2. Parámetros fisicoquímicos determinados en los sistemas con diferentes fases
oleicas
Láurico Mirístico Palmítico Fase
oleica Kr FO FA FM Kr FO FA FM Kr FO FA FM
n-C7/Bz
80/20 86 47,00 0,55 53,00 141 28,11 0,20 71,89 224 16,12 0,07 83,88
n-C7/Bz
50/50 70 10,45 0,15 89,55 114 3,94 0,03 96,06 - - - -
Tolueno 47 25,99 0,55 74,01 82 16,48 0,20 83,52 136 9,81 0,07 90,19
Benceno - - - - 76 2,78 0,04 97,22 - - - -
Kerosén 23 4,02 0,17 95,98 48 1,56 0,03 98,44 92 1,16 0,01 98,84 FO, FA, FM: significan fase oleica, fase acuosa y fase media, respectivamente (%p/v); n-C7: n-Heptano; Bz:
Benceno
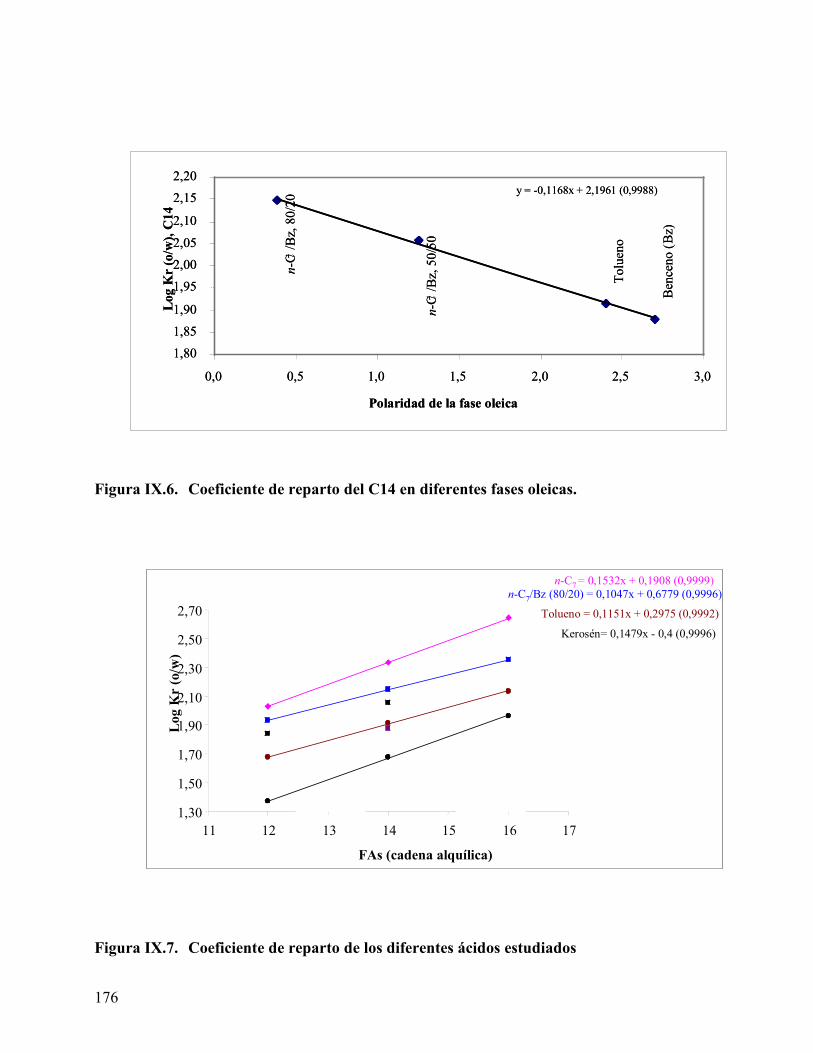
176
Figura IX.6. Coeficiente de reparto del C14 en diferentes fases oleicas.
Figura IX.7. Coeficiente de reparto de los diferentes ácidos estudiados
n- C
7/B
z, 8
0/2
0
n-C
7/B
z, 5
0/5
0
Ben
ceno (
Bz )
Tolu
eno
y = -0,1168x + 2,1961 (0,9988)
1,80
1,85
1,90
1,95
2,00
2,05
2,10
2,15
2,20
0,0 0,5 1,0 1,5 2,0 2,5 3,0
Polaridad de la fase oleica
Lo
g K
r(o
/w),
C1
4
n- C
7/B
z, 8
0/2
0
n-C
7/B
z, 5
0/5
0
Ben
ceno (
Bz )
Tolu
eno
y = -0,1168x + 2,1961 (0,9988)
1,80
1,85
1,90
1,95
2,00
2,05
2,10
2,15
2,20
0,0 0,5 1,0 1,5 2,0 2,5 3,0
Polaridad de la fase oleica
Lo
g K
r(o
/w),
C1
4
Kerosén= 0,1479x - 0,4 (0,9996)
Tolueno = 0,1151x + 0,2975 (0,9992)
n-C7/Bz (80/20) = 0,1047x + 0,6779 (0,9996)
n-C7 = 0,1532x + 0,1908 (0,9999)
1,30
1,50
1,70
1,90
2,10
2,30
2,50
2,70
11 12 13 14 15 16 17
FAs (cadena alquílica)
Log K
r(o
/w)
Kerosén= 0,1479x - 0,4 (0,9996)
Tolueno = 0,1151x + 0,2975 (0,9992)
n-C7/Bz (80/20) = 0,1047x + 0,6779 (0,9996)
n-C7 = 0,1532x + 0,1908 (0,9999)
1,30
1,50
1,70
1,90
2,10
2,30
2,50
2,70
11 12 13 14 15 16 17
FAs (cadena alquílica)
Log K
r(o
/w)
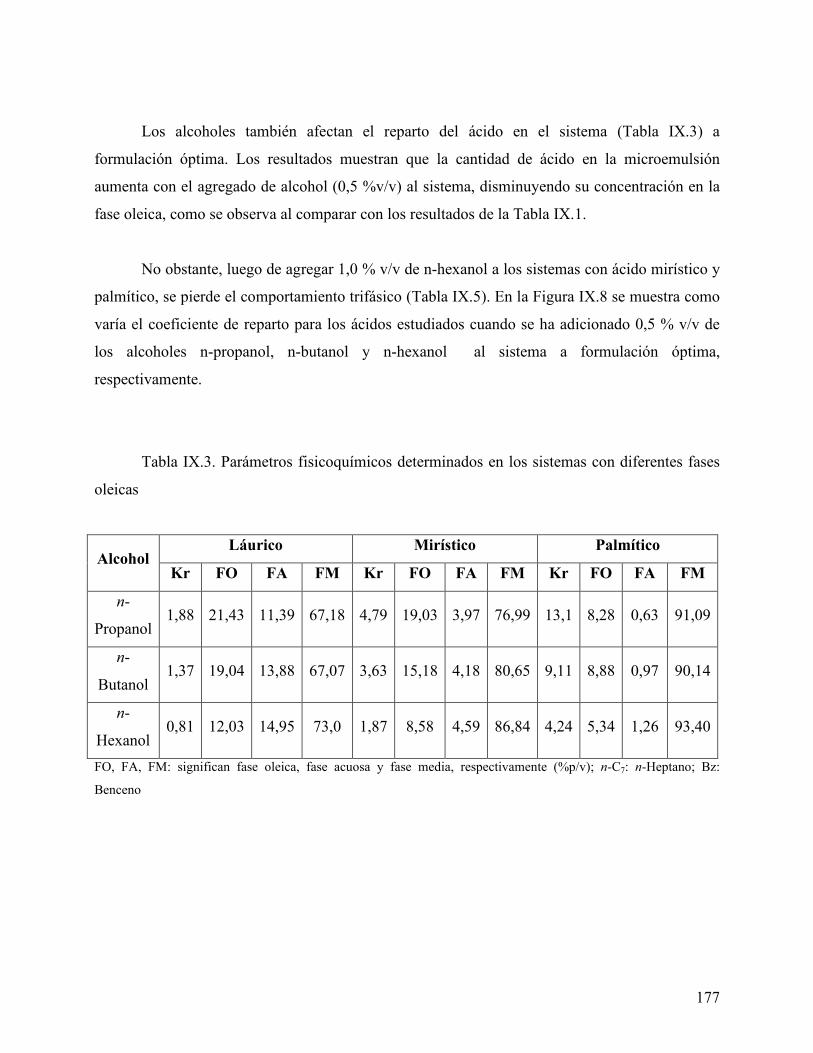
177
Los alcoholes también afectan el reparto del ácido en el sistema (Tabla IX.3) a
formulación óptima. Los resultados muestran que la cantidad de ácido en la microemulsión
aumenta con el agregado de alcohol (0,5 %v/v) al sistema, disminuyendo su concentración en la
fase oleica, como se observa al comparar con los resultados de la Tabla IX.1.
No obstante, luego de agregar 1,0 % v/v de n-hexanol a los sistemas con ácido mirístico y
palmítico, se pierde el comportamiento trifásico (Tabla IX.5). En la Figura IX.8 se muestra como
varía el coeficiente de reparto para los ácidos estudiados cuando se ha adicionado 0,5 % v/v de
los alcoholes n-propanol, n-butanol y n-hexanol al sistema a formulación óptima,
respectivamente.
Tabla IX.3. Parámetros fisicoquímicos determinados en los sistemas con diferentes fases
oleicas
Láurico Mirístico Palmítico Alcohol
Kr FO FA FM Kr FO FA FM Kr FO FA FM
n-
Propanol 1,88 21,43 11,39 67,18 4,79 19,03 3,97 76,99 13,1 8,28 0,63 91,09
n-
Butanol 1,37 19,04 13,88 67,07 3,63 15,18 4,18 80,65 9,11 8,88 0,97 90,14
n-
Hexanol 0,81 12,03 14,95 73,0 1,87 8,58 4,59 86,84 4,24 5,34 1,26 93,40
FO, FA, FM: significan fase oleica, fase acuosa y fase media, respectivamente (%p/v); n-C7: n-Heptano; Bz:
Benceno
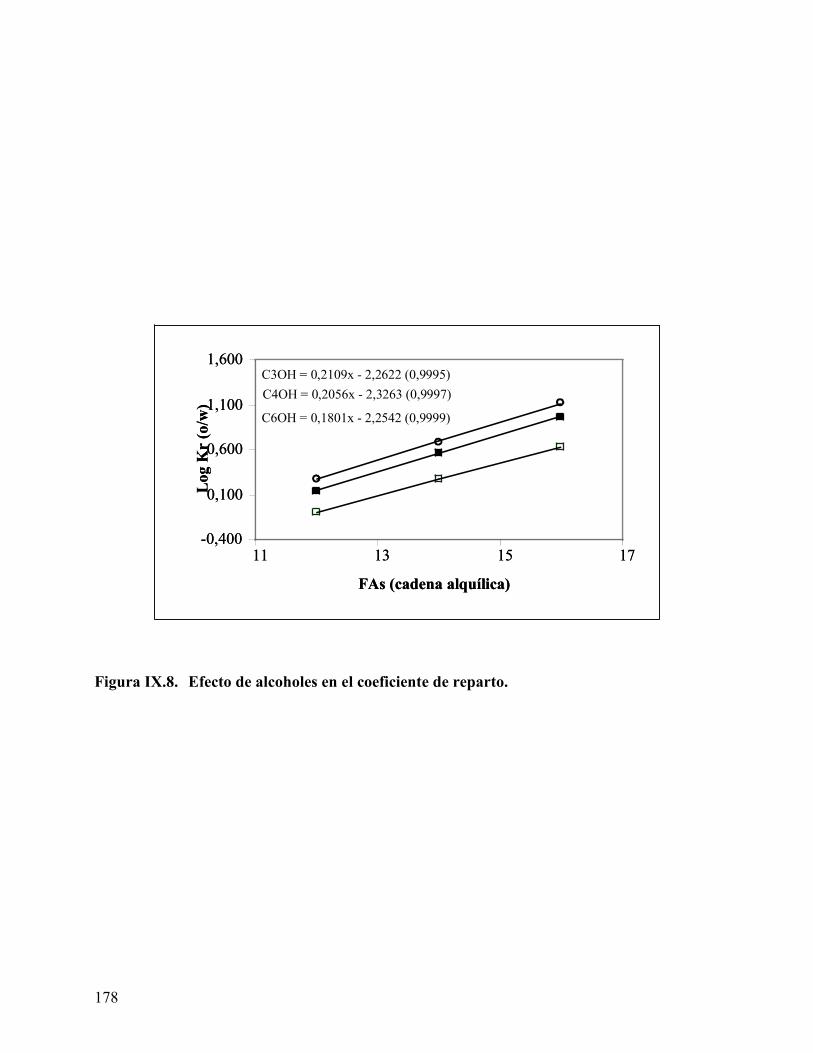
178
Figura IX.8. Efecto de alcoholes en el coeficiente de reparto.
C6OH = 0,1801x - 2,2542 (0,9999)
C4OH = 0,2056x - 2,3263 (0,9997)
C3OH = 0,2109x - 2,2622 (0,9995)
-0,400
0,100
0,600
1,100
1,600
11 13 15 17
FAs (cadena alquílica)
Lo
g K
r(o
/w)
C6OH = 0,1801x - 2,2542 (0,9999)
C4OH = 0,2056x - 2,3263 (0,9997)
C3OH = 0,2109x - 2,2622 (0,9995)
-0,400
0,100
0,600
1,100
1,600
11 13 15 17
FAs (cadena alquílica)
Lo
g K
r(o
/w)

179
Proceso termodinámico en el reparto de FAs en sistemas microemulsión/agua/aceite
Como fue sugerido por Cratin8, la variación en el coeficiente de reparto puede producir
interesante información termodinámica. En el caso de un sistema trifásico, el potencial químico
del surfactante puede escribirse de la siguiente forma: wwww
CRT !µµ ln* += (1)
oooo
CRT !µµ ln* += (2)
donde *µ es el potencial químico estándar en los estados de referencia dados, C es la
concentración y φ es el coeficiente de actividad. Los subíndices “w” y “o” se refieren a las fases
acuosa y oleica en exceso, respectivamente. Subsecuentemente, las fases aceite y acuosa se
encuentran en equilibrio en un sistema trifásico. Por otro lado, la concentración del surfactante en
las fases en exceso es baja (típicamente del orden de la CMC), por lo que se puede asumir que los
coeficientes de actividad son igual a la unidad. En consecuencia, el coeficiente de partición del
surfactante entre las fases acuosa y oleica se puede representar como:
wo
o
w
C
CRTKRT
**lnln µµ !== (3)
La diferencia del potencial químico estándar contiene solo términos de energía, y de
acuerdo a Cratin8, esto puede escribirse como una sumatoria de las contribuciones parciales, en
consecuencia, los términos energía pueden resumirse de acuerdo a la primera ley de la
termodinámica.
Cada energía potencial estándar es así dividida en tres términos, que corresponde a la
contribución del grupo lipofílico (N), del grupo hidrofílico (que es el mismo en todos los casos y
se desprecia, a) y de otros factores tales como naturaleza del aceite o la temperatura. En la Figura
IX.9 se muestra la variación lineal del logaritmo del coeficiente de reparto con la longitud de la
cadena alquílica del ácido (N). Por lo tanto se puede obtener la contribución de varios grupos
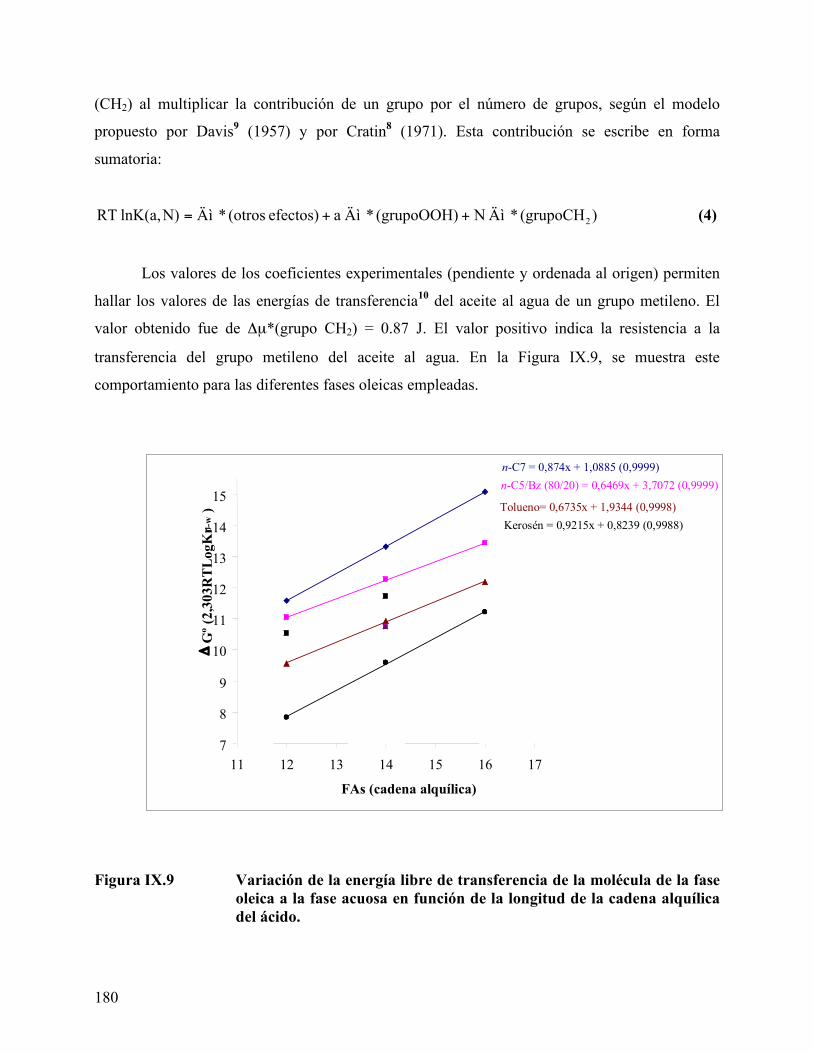
180
(CH2) al multiplicar la contribución de un grupo por el número de grupos, según el modelo
propuesto por Davis9 (1957) y por Cratin8 (1971). Esta contribución se escribe en forma
sumatoria:
)(grupoCH*Äì N(grupoOOH)*Äì a efectos) (otros*ÄìN)lnK(a, RT 2++= (4)
Los valores de los coeficientes experimentales (pendiente y ordenada al origen) permiten
hallar los valores de las energías de transferencia10 del aceite al agua de un grupo metileno. El
valor obtenido fue de Δµ*(grupo CH2) = 0.87 J. El valor positivo indica la resistencia a la
transferencia del grupo metileno del aceite al agua. En la Figura IX.9, se muestra este
comportamiento para las diferentes fases oleicas empleadas.
Figura IX.9 Variación de la energía libre de transferencia de la molécula de la fase oleica a la fase acuosa en función de la longitud de la cadena alquílica del ácido.
n-C5/Bz (80/20) = 0,6469x + 3,7072 (0,9999)
Tolueno= 0,6735x + 1,9344 (0,9998)
n-C7 = 0,874x + 1,0885 (0,9999)
Kerosén = 0,9215x + 0,8239 (0,9988)
7
8
9
10
11
12
13
14
15
11 12 13 14 15 16 17
FAs (cadena alquílica)
!G
º(2
,303R
TL
ogK
ro-w
)
n-C5/Bz (80/20) = 0,6469x + 3,7072 (0,9999)
Tolueno= 0,6735x + 1,9344 (0,9998)
n-C7 = 0,874x + 1,0885 (0,9999)
Kerosén = 0,9215x + 0,8239 (0,9988)
7
8
9
10
11
12
13
14
15
11 12 13 14 15 16 17
FAs (cadena alquílica)
!G
º(2
,303R
TL
ogK
ro-w
)

181
CONCLUSIONES
Los resultados mostraron que a medida que aumenta la cadena alquílica del ácido,
disminuye su concentración tanto en la fase oleica en exceso como en la fase acuosa. Los ácidos
de cadena larga se concentran más en la microemulsion que los de cadenas cortas.
Finalmente, la medida del coeficiente de reparto de las especies surfactantes permite
determinar experimentalmente las contribuciones de cada uno de los términos al balance
hidrofílico-lipofílico total, lo que hace muy fácil extender la aplicación de estas correlaciones en
otros sistemas.
Los valores positivos de energía obtenidos indican la resistencia a la transferencia de un
grupo metileno del aceite al agua. Esta información termodinámica se produce a partir del
coeficiente de reparto.

182
REFERENCIAS BIBLIOGRÁFICAS [1] Salager, J-L.; Antón, R. Ionic microemulsion. Surfactant science series, Marcel Dekker,
New York, 1989.
[2] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1995, 100,
225-231.
[3] Márquez, N.; Antón, R.; Graciaa, A.; Lachaise, J.; Salager, J. Coll. Surf. A, 1998, 131, 45-
49.
[4] Antón, R.; Salager, J. J. Colloid Int. Sci. 1990, 140, 75-81.
[5] Graciaa, A.; Lachaise, J.; Sayous, J.; Grenier, P.; Yiv, S.; Schechter, R.; Wade, W. J.
Colloid Interf. Sci., 1983, 93, 474-481.
[6] Márquez, N.; Antón, R.; Usubillaga, A.; Salager, J-L. Sep. Sci. Technol, 1993, 28, 1769-
1775.
[7] Márquez, N.; Antón, R.; Usubillaga, A.; Salager, J-L. Sep. Sci. Technol, 1993, 28, 2387-
2394.
[8] Cratin, P. in S. Ross (Ed.), Chemistry and Physics at interfaces-II, American Chemical
Society, Washington, DC, 1971, vol. 97.
[9] Davies, J. Proceeding 2nd International Congress Surface Activity. Butterworths, London,
1957, 1, 426-432.
[10] Salager, J.; Marquez, N.; Graciaa, A.; Lachaise, J. Langmuir. 2000,16, 5534-5539.

183
CONCLUSIONES GENERALES
Se demostró que la cromatografía líquida de alta resolución (HPLC), es una técnica analítica
eficaz para el análisis y separación de mezclas de ácidos carboxílicos grasos de 8 a 18
átomos de carbono, empleando una mezcla metanol/acetonitrilo como fase móvil en forma
isocrática y en una columna octadecilsilano.
La adición del paso de derivatización en el análisis de estos compuestos aumenta la
sensibilidad del método de detección.
El desarrollo de un sistema de derivatización y análisis por HPLC en línea de estos
compuestos, proporcionó: menor tiempo de análisis, poco tratamiento de la muestra, menor
tiempo de reacción, menor gasto de reactivos y mayor sensibilidad.
Debido al carácter lipofílico de estos compuestos cuando se encuentran en contacto con una
fase acuosa y otra oleica, se produce un aumento del coeficiente de reparto (Kr) que varia
linealmente con la cadena alquílica y la concentración del ácido en el sistema. Por otro lado,
el aumento de la polaridad de la fase oleica, así como la longitud de la cadena alquílica del
alcohol que actúa como co-surfactante, produce una disminución del Kr.
Los resultados obtenidos permitieron calcular la energía libre de transferencia de una
molécula de surfactante de una fase a la otra.

184
RECOMENDACIONES GENERALES
En la actualidad, los ácidos presentes en los crudos pesados son objeto de muchas
investigaciones. Los métodos desarrollados en este capitulo pueden servir de base para la
evaluación de estos compuestos en diferentes matrices.
Es importante evaluar los parámetros necesarios para el diseño del sistema en línea a nivel
industrial, para lo cual se recomienda evaluar el efecto de la potencia del microonda en las
reacciones de derivatización de los ácidos grasos, para determinar la potencia que produzca
mayores rendimientos de los productos derivados.
Para la determinación exacta de los sistemas a formulación óptima se recomienda el método
de la tensión interfacial, para evaluar el porcentaje de error cometido en la selección de estos
sistemas por el método de los volúmenes de fase.
Finalmente, se recomienda aplicar los parámetros de formulación óptima en sistemas reales
que contengan crudos pesados, a objeto de trasladar estos resultados a escala industrial.

185
D I S E M I N A C I Ó N D E L C O N O C I M I E N T O

186
El trabajo desarrollado en este capitulo arrojo las siguientes publicaciones y presentaciones en congresos:
1. Bélgica Bravo, Gerson Chávez, Fredy Ysambertt, Nelson Márquez, Jean Lachaise,
Alain Graciaa, Raquel Antón y Jean Louis Salager. Thermodynamic parameters of the
chromatographic equilibrium distribution process of amphiphilic compound by HPLC.
Part I: Fatty acids. Ciencia, 2004, Vol. 12, Nº 4, (Oct-Dec)
2. Bélgica Bravo, Gerson Chávez, Ana Cáceres, Fredy Ysambertt y Nelson Márquez.
Thermodynamic parameters of the chromatographic equilibrium distribution process
of amphiphilic compound by HPLC. Part II: Nonylphenol polyethoxylated. Ciencia,
2004, Vol. 12, Nº 4, (Oct-Dec)
3. Bélgica Bravo, Gerson Chávez, Nolberto Piña, Fredy Ysambertt, Nelson Márquez,
Ana Cáceres. Developing an on-line derivatization of FAs by microwave irradiation
coupled to HPLC separation with UV detection. Talanta. 2004, 64, 1329-1334
4. Nelson Márquez, Bélgica Bravo, Gerson Chávez, Fredy Ysambertt. Desarrollo de un
método de cromatografía en fase reversa para el estudio de mezclas de ácidos
carboxílicos lipofílicos”,. VI Congreso venezolano de Química (CVQ), Universidad
de Margarita, UNIMAR, del 2 - 6 de Noviembre de 2003.
5. Bélgica Bravo, Gerson Chávez, Fredy Ysambertt, Nelson Márquez, Ana Cáceres. On-
line analysis of fatty carboxylic acid by RP-HPLC-UV, 12th International Conference
on Flow Injection Analysis Mérida-Venezuela del 07-13 de Diciembre de 2003.
6. Nelson Márquez, Bélgica Bravo, Gerson Chávez, Ana Caceres, Roberto Bauza.
Formulation scan of fatty acids in surfactant/oil/water systems. Philadelphia, PA.
228th ACS National Meeting. Philadelphia, Agosto de 2004.
7. Nelson Marquez, Belgica Bravo, Gerson Chavez, Fredy Ysambertt, Ana Cáceres,
Roberto Bauza. Thermodynamic parameters of the chromatographic equilibrium
distribution of fatty acid compounds by HPLC. 230th ACS National Meeting.
Washington-USA, Agosto 2005.

187
A MI DIOS REDENTOR
A MIS PADRES
A MI ESPOSO


















