Trabajo Práctico: PROTEÍNAS Extracción, cuantificación y ... · La proteína es soluble cuando...
18
Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud. Cátedra: Química Biológica I Año 2017 33 Trabajo Práctico: PROTEÍNAS Extracción, cuantificación y separación electroforética OBJETIVO -Determinar la concentración de proteínas solubles en una muestra biológica mediante el método de Biuret. - Separar las proteínas presentes en el suero humano por un método electroforético. FUNDAMENTO TEÓRICO Las proteínas son las más variadas de todas las macromoléculas, y cada célula contiene varios miles de proteínas diferentes. Las funciones de las proteínas incluyen servir como componentes estructurales de células y tejidos (p.ej., colágeno) , actuar en el transporte y almacenamiento de pequeñas moléculas (p.ej., en el transporte de oxigeno por la hemoglobina), transmitir información entre células ( p.ej., hormonas proteicas), proporcionar una defensa frente a la infección (p.ej., anticuerpos), actuar como elementos esenciales en sistemas motiles y contráctiles (p.ej., actina), almacenar aminoácidos como elemento nutritivo (p.ej., caseína). Sin embargo su principal función es la de actuar como catalizadores biológicos, denominados enzimas, las cuales participan en la mayoría de las reacciones químicas en los sistemas biológicos. Las proteínas están constituidas por la concatenación de unas sustancias químicas denominadas aminoácidos (unidades monoméricas). Por hidrolisis de proteínas se han identificado 20 aminoácidos distintos. Cada aminoácido consiste en un átomo de carbono (llamado carbono α) ligado a un grupo carboxilo (COO - ), un grupo amino (NH + 3) , un átomo de hidrogeno y una cadena lateral característica cuya propiedades físicas y químicas determinan los papeles de cada aminoácido en la estructura y función proteica. Los aminoácidos se clasifican basándose en la polaridad de su cadena lateral. Dentro del conjunto de todos los aminoácidos naturales existen unos que pueden ser sintetizados por las células del organismo humano a partir de materiales sencillos que contengan C, O, H y N, pero otros tienen adquirirse necesariamente con la dieta. Estos últimos se denominan aminoácidos esenciales para la especie humana y son: valina, leucina, isoleucina, treonina, metionina, fenilalanina, triptófano, lisina e histidina (solo para lactantes). Todos los aminoácidos a excepción de la glicina, poseen átomos de carbono asimétricos y en consecuencia presentan actividad óptica, es decir, presentan estero isomería (enantiomeros D y L). En las proteínas solo se encuentran aminoácidos de la seria L. El valor de pH al cual el aminoácido no tiene carga neta se llama punto isoeléctrico (pI), a valores de pH < pI la molécula tendrá carga positiva mientras que la carga será negativa si pH > pI. La forma neutra se debe a la α α
Transcript of Trabajo Práctico: PROTEÍNAS Extracción, cuantificación y ... · La proteína es soluble cuando...

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
33
Trabajo Práctico: PROTEÍNAS Extracción, cuantificación y separación electroforética
OBJETIVO -Determinar la concentración de proteínas solubles en una muestra biológica mediante el método de Biuret. - Separar las proteínas presentes en el suero humano por un método electroforético. FUNDAMENTO TEÓRICO Las proteínas son las más variadas de todas las macromoléculas, y cada célula contiene varios miles de proteínas diferentes. Las funciones de las proteínas incluyen servir como componentes estructurales de células y tejidos (p.ej., colágeno) , actuar en el transporte y almacenamiento de pequeñas moléculas (p.ej., en el transporte de oxigeno por la hemoglobina), transmitir información entre células ( p.ej., hormonas proteicas), proporcionar una defensa frente a la infección (p.ej., anticuerpos), actuar como elementos esenciales en sistemas motiles y contráctiles (p.ej., actina), almacenar aminoácidos como elemento nutritivo (p.ej., caseína). Sin embargo su principal función es la de actuar como catalizadores biológicos, denominados enzimas, las cuales participan en la mayoría de las reacciones químicas en los sistemas biológicos. Las proteínas están constituidas por la concatenación de unas sustancias químicas denominadas aminoácidos (unidades monoméricas). Por hidrolisis de proteínas se han identificado 20 aminoácidos distintos. Cada aminoácido consiste en un átomo de carbono (llamado carbono α) ligado a un grupo carboxilo (COO-), un grupo amino (NH+
3), un átomo de hidrogeno y una cadena lateral característica cuya propiedades físicas y químicas determinan los papeles de cada aminoácido en la estructura y función proteica. Los aminoácidos se clasifican basándose en la polaridad de su cadena lateral. Dentro del conjunto de todos los aminoácidos naturales existen unos que pueden ser sintetizados por las células del organismo humano a partir de materiales sencillos que contengan C, O, H y N, pero otros tienen adquirirse necesariamente con la dieta. Estos últimos se denominan aminoácidos esenciales para la especie humana y son: valina, leucina, isoleucina, treonina, metionina, fenilalanina, triptófano, lisina e histidina (solo para lactantes). Todos los aminoácidos a excepción de la glicina, poseen átomos de carbono asimétricos y en consecuencia presentan actividad óptica, es decir, presentan estero isomería (enantiomeros D y L). En las proteínas solo se encuentran aminoácidos de la seria L. El valor de pH al cual el aminoácido no tiene carga neta se llama punto isoeléctrico (pI), a valores de pH < pI la molécula tendrá carga positiva mientras que la carga será negativa si pH > pI. La forma neutra se debe a la
α α

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
34
formación de una sal interna llamada Zwitterion, esto ocurre porque el protón del grupo carboxilo es abstraído por el grupo amino NH2 que está en posición alfa y quedando este como grupo amonio NH3+. El punto isoeléctrico de las proteínas está influenciado por las cadenas laterales, ya que los grupos aminos y ácidos principales participan en el enlace peptídico. Los aminoácidos están unidos por enlaces peptídicos entre el grupo α amino de un aminoácido y el grupo α carboxilo del siguiente. La propiedad fundamental del
Enlace peptídico es que todos los átomos que lo forman han de estar en un mismo plano, por lo que la cadena proteica solo puede girar por sus Cα pero nunca por el enlace peptídico. La unión de miles de aminoácidos forman lo que se conoce como cadena poli peptídica, la cual contiene dos extremos distintivos, uno llamado N terminal y otro C terminal. Los poli péptidos se sintetizan desde el extremo amino al carboxilo terminal, y la secuencia de aminoácidos en un poli péptido se escribe (por convención) en el mismo orden. Cada proteína consiste en una secuencia específica de aminoácidos, determinada por el orden de los nucleótidos en un gen. Esta secuencia permite interacciones entre aminoácidos constituyentes que darán origen a configuraciones tridimensionales características, cruciales para la función proteica. Cada tipo de proteína puede tener diferentes estructuras de organización:
Estructura primaria: Composición cuantitativa y secuencia lineal de aminoácidos.
Estructura secundaria: estructura local de la cadena polipetidica. Se determina por las interacciones mediante puentes de hidrogeno entre el oxígeno del grupo carbonilo de una cadena peptídica y el hidrogeno de amida de otro puente peptídico cercano. Existen dos tipos de estructuras secundarias: Hélice α posee una cadena peptídica fuertemente enrollada y con las cadenas laterales de los residuos aminoácidos extendiéndose hacia fuera del eje de la espiral; Hoja plegada β es una estructura extendida que se forma cuando dos partes de una cadena poli peptídica se encuentran una junto a otra con enlaces puente hidrogeno entre ellas, estas a su vez, pueden estar orientadas bien paralela o anti paralelamente entre sí.
Estructura terciaria: conformación tridimensional, plegada y biológicamente activa. La estructura está estabilizada por interacciones entre grupos funcionales de las cadenas laterales: puentes di sulfuros covalentes, puentes de hidrógeno, puentes salinos e interacciones hidrofobicas.
Estructura cuaternaria: complejo ensamblaje de dos o más cadenas peptídicas que se mantienen unidas por interacciones no covalentes, en algunos casos, covalentes. La mayoría de las proteínas mayores de 50 da constan de más de una cadena y se conocen como proteínas diméricas, trimétricas o multimérica.
Según su conformación nativa las proteínas pueden clasificarse en Fibrosas o Globulares:

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
35
Fibrosas: típicas estructuras secundarias, formadas por fibras ordenadas a lo largo de un eje. Insolubles en agua y en soluciones acuosas, gran resistencia física: acción mecánica (esqueletos, transmisión de esfuerzos) o de protección. Ej.: colágeno, elastina, queratina.
Globulares: cadenas plegadas de tal modo que toman formas esféricas o globulares compactas (estructura terciaria). Solubles en agua o soluciones acuosas. Papeles muy dinámicos en el organismo. Pertenecen a esta categoría, las enzimas, los anticuerpos, algunas hormonas, las proteínas con función de transporte, etc.
Algunas proteínas no se encuentran claramente en ninguno de estos grupos de clasificación: por ejemplo, pueden presentar estructura de tipo fibroso, pero ser solubles en soluciones salinas, como las globulares, que es el caso de la miosina del músculo y el fibrinógeno del plasma sanguíneo. Según su composición química, las proteínas pueden ser:
Simples: su hidrólisis produce sólo aminoácidos, como la insulina o el colágeno.
Conjugadas: su hidrólisis produce, además de aminoácidos, otros compuestos inorgánicos u orgánicos, clasificándose entonces en:
Nucleoproteínas. Fosfoproteínas. Glicoproteínas. Metal proteínas. Lipoproteínas.
La solubilidad de las proteínas es variable y depende de la distribución y de la proporción de grupos polares y no polares de la molécula. La proteína es soluble cuando ocurre interacción proteína-agua y tiende a ser insoluble cuando ocurre interacción proteina-proteina. Cualquier condición que altere esas interacciones alterará la solubilidad. Por lo tanto la solubilidad de una proteína es función de la composición iónica del medio, de la fuera iónica y del pH.
La adición de un disolvente orgánico, como acetona o alcohol, a una solución de proteína en agua, disminuye la constante dieléctrica del disolvente, desplaza también algunas de las moléculas de agua asociadas con la proteína, y reduce la concentración del agua presente en la solución. Estos efectos tienden a disminuir la solubilidad de la proteína y causar su precipitación en solución.
En presencia de pequeñas concentraciones de sal, la proteína en solución de agua pura disminuye su coeficiente de actividad y aumenta su solubilidad (disolución salina o “salting in”) debido a las fuerzas de atracción entre los iones de la proteína y los iones de la sal.
Ahora bien en concentración elevadas de sales muy solubles como sulfato de amonio, se observa precipitación salina o “salting out” de las proteínas, la cual depende de la disminución de la actividad del agua, lo que a su vez disminuye las interacciones solubilizantes entre el agua y los grupos de la proteína. Es decir, que cuando aumenta mucho la cantidad de iones extraños, la interacción proteína-proteína se hace mayor que la interacción proteína-agua, baja la movilidad de las cargas proteicas y las proteínas precipitan.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
36
Desnaturalización Es la modificación que ocurre en la estructura nativa de una proteína, alterando así sus propiedades. Envuelve alteraciones de la estructura 2°,3° y 4° de las proteínas. La estructura 1° se mantiene. Las alteraciones que se observan son disminución de la solubilidad y/o pérdida de la actividad biológica. La disminución de la solubilidad puede ser explicada por la exposición de radicales hidrofóbicos que perjudican la interacción proteína-agua y favorecen la interacción proteína-proteína. La floculación y la coagulación son manifestaciones visibles de la alteración estructural causada por agentes desnaturalizantes. Agentes desnaturalizante:
Calor: la agitación térmica afecta las interacciones que estabilizan la estructura tridimensional de las proteínas (puentes H, interacción hidrofóbica)
Ácidos: se combinan con proteínas con carga + formando complejos insolubles. Sales de metales pesados: se combinan con proteínas de carga formando proteinatos
insolubles. Solventes orgánicos: presentan corriente dieléctrica inferior al agua entonces la
atracción entre cargas opuestas es alta.(precipitación)
Caracterización de aminoácidos Debido a la presencia de diferentes aminoácidos en las uniones peptídicas las proteínas reaccionan con una variedad de compuestos formando productos coloreados. Existen reacciones de coloración que son específicas para aminoácidos y son importantes, tanto para la detección como para el dosaje de aminoácidos y proteínas. Existen también reacciones generales porque sirven para caracterizar grupos comunes a todas las proteínas como grupos amino o uniones peptídicas (ninhidrina y biuret)
Reacción Xantoproteica: Es debida a la formación de un compuesto aromático nitrado de color amarillo, cuando las proteínas son tratadas con ácido nítrico concentrado. La prueba da resultado positivo en aquellas proteínas con aminoácidos portadores de grupos bencénicos, tirosina, fenilalanina, triptófano. Una vez realizada la prueba se neutraliza con un álcali y vira a un color anaranjado.
Reacción de aminoácidos azufrados: Se pone de manifiesto por la formación de un precipitado negruzco de sulfuro de plomo. Se basa esta reacción en la separación mediante un álcali, del azufre de los aminoácidos, el cual al reaccionar con una solución de acetato de plomo, forma el sulfuro de plomo.
Determinación cuantitativa de proteínas mediante el método de Biuret: Se basa en la formación de un complejo coloreado entre el Cu2+ y los grupos NH de los enlaces peptídicos en medio básico. 1 Cu2+ se acompleja con 4 NH. La intensidad de coloración es directamente proporcional a la cantidad de proteínas (enlaces peptídicos) y la reacción es bastante específica, de manera que pocas sustancias interfieren. La sensibilidad del método es muy baja y sólo se recomienda para la cuantificación de proteínas en preparados muy concentrados. Este método es sencillo y su sensibilidad está dentro de un rango de concentración del orden de miligramos.
Determinar la concentración de proteínas en una muestra biológica es una técnica de rutina básica cuando se aborda un esquema de purificación de una proteína concreta, cuando se quiere conocer la actividad específica de una preparación enzimática, para el diagnóstico de enfermedades, así como para otros muchos propósitos. Existen diferentes métodos para la cuantificación de proteínas. Muchos de estos métodos se basan en: a) la propiedad intrínseca

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
37
de las proteínas para absorber luz en el UV, b) para la formación de derivados químicos, o c) la capacidad que tienen las proteínas de unir ciertos colorantes. En la Tabla I se recogen los métodos más usados así como sus respectivas sensibilidades. Cada uno de estos métodos tiene sus ventajas e inconvenientes, las principales se recogen en la Tabla II. Tabla I. Principales métodos para la cuantificación de proteínas y sus rangos de sensibilidad
Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
38
Tabla II. Principales métodos para la cuantificación de proteínas, principales ventaja e inconveniente
PARTE EXPERIMENTAL Extracción de material biológico
Pesar 1 g de material biológico: hígado, pollo y carne. Homogeneizarlo en un Potter con 10 ml de agua destilada. Centrifugar 5 minutos a 3000 rpm. Separar el sobrenadante. (muestra).
Cuantificación por el método del Biuret
Blanco Testigo Problema
Agua destilada 0,1 ml -- --
Sobrenadante -- -- 0,1 ml
Testigo 0,3 g % -- 0,1 ml --
Biuret 3 ml 3 ml 3 ml
Incubar 30 minutos a t ambiente y leer a 540 nm, llevando a 0 con el tubo blanco. Calcular la concentración del problema en g % de proteínas solubles. Calcular el % p/p en cada material biológico.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
39
SEPARACIÓN ELECTROFORÉTICA Se denomina electroforesis al transporte de partículas en un campo eléctrico. Cualquier ión o molécula cargada eléctricamente migrará cuando se someta a la acción de un campo eléctrico. A un pH determinado, muchas moléculas biológicas poseen carga eléctrica, cuya magnitud depende del pH y composición del medio en que se encuentren. Con técnicas electroforéticas, es posible separar los diferentes componentes de una mezcla de aminoácidos, proteínas, ácidos nucleicos y otras biomoléculas cargadas Se basa: en las diferentes velocidades de migración que experimentan en un campo eléctrico las distintas partículas cargadas eléctricamente o bien en el tamaño de la partícula que se desplaza. La velocidad de migración depende principalmente de la densidad de carga y de la intensidad del campo eléctrico aplicado. Tipos de electroforesis Electroforesis de frente móvil o libre Las sustancias a separar se introducen en un tubo en forma de U, disueltas en un tampón de pH y fuerza iónica adecuados. Se colocan dos electrodos en sendos brazos del dispositivo, entre los que se crea un campo eléctrico, provocando que las moléculas de proteína cargadas emigren hacia los electrodos de polaridad opuesta. Las diferentes proteínas se desplazan a velocidades diferentes de acuerdo con sus cargas y coeficientes de fricción respectivos, formándose nubes (o frentes) que se desplazan en la disolución tampón. Este desplazamiento puede seguirse con diversos sistemas ópticos que ponen de manifiesto las sustancias según se van separando. Para impedir la mezcla por convección de las proteínas que emigran se necesita un sofisticado aparato y a su vez se precisa el empleo de muestras muy grandes. Esta es la razón por la que la electroforesis de frente móvil ha sido sustituida por la electroforesis de zona. Electroforesis de zona En esta técnica, la muestra está obligada a desplazarse sobre un soporte sólido de papel, celulosa o gel. La pequeña cantidad necesaria de muestra permite que las moléculas migren en discretas zonas o bandas. La electroforesis zonal de biomoléculas cargadas, generalmente se lleva a cabo en una disolución estabilizada con un medio que sirve de soporte.
1. Electroforesis en papel: La muestra se aplica sobre una tira de papel de filtro humedecido con una disolución tampón, bien en el centro o en cualquiera de los extremos del papel, dependiendo de las sustancias a separar y del pH del tampón. Los extremos de la tira se sumergen en dos recipientes separados que contienen la disolución tampón y en los que se hallan colocados los electrodos. Se conectan los electrodos a una fuente de tensión continua y se aplica una diferencia de potencial durante el tiempo adecuado. Al aplicar una corriente directa, las moléculas cargadas de la mezcla emigran hacia los electrodos de polaridad opuesta formando bandas en el papel. La velocidad de emigración de una molécula, depende preferentemente de su carga. Completada la electroforesis, se desconecta la fuente de tensión y se secan las tiras sobre una superficie, limpia, seca y lisa (cristal, azulejo, etc...). El revelado se

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
40
realiza mediante la acción de los colorantes adecuados. La electroforesis en papel se emplea principalmente en la separación de sustancias de bajo peso molecular como aminoácidos y péptidos. La difusión que se produce puede disminuirse aumentando la diferencia de potencial entre los electrodos con lo que se reduce el tiempo de desarrollo. Este tipo de electroforesis se denomina de “alto voltaje”. El revelador para localizar las sustancias sería la ninhidrina en el caso de péptidos y aminoácidos. Las electroforesis en papel también pueden hacerse en tiras de acetato de celulosa, que son químicamente más homogéneas y tienen la ventaja de ser transparentes, con lo que las sustancias a separar, una vez teñidas o reveladas, pueden cuantificarse por espectrofotometría. Es muy útil en los laboratorios clínicos por su fácil manipulación y su rápido desarrollo. La principal aplicación es la separación de proteínas del suero, conociéndose el resultado como proteinograma. Las fracciones proteicas del suero que se separan mediante este tipo de electroforesis son la albúmina y las globulinas α1, α2, β y γ.
2. Electroforesis en gel: Un gel es un estado intermedio entre el sólido y el líquido. Este tipo de electroforesis se halla entre los métodos más resolutivos y convenientes empleados en la separación de macromoléculas. Los geles de uso más generalizado son: poliacrilamida y agarosa. Éstos poseen poros de diferentes dimensiones moleculares que delimitan la velocidad de traslado y moléculas trasladadas durante el proceso electroforético. De esta forma, la separación no se produce sólo por las diferentes cargas de las moléculas, sino también por las diferencias de tamaño. Los geles están formados por un reticulado de polímeros (constituyendo una red enmarañada) y el líquido intersticial en el que se encuentra inmerso esta red.
Existe una gran variedad de tipos de electroforesis en gel : Electroforesis en gel en una dimensión (continuo o discontinuo) o PAGE-nativa o SDS-PAGE o Isoelectroenfoque. Según las aplicaciones u objetivos que se pretendan conseguir en una práctica o experimento, se utilizará un tipo u otro de electroforesis en gel. Los geles discontinuos mejoran la resolución de la electroforesis pues se consigue agudizar las bandas en gran medida por el uso de esta técnica conocida como electroforesis a pH discontinuo que precisa de dos geles y dos tampones diferentes. La PAGE-nativa se utiliza para separar proteínas en condiciones no desnaturalizantes, por tanto, en función de la carga intrínseca de las mismas. La SDS-PAGE es muy útil para calcular el peso molecular de una proteína concreta ya que separa las proteínas según su tamaño. El isoelectroenfoque es un tipo de PAGE en una dimensión que se basa en la separación de moléculas de acuerdo a sus diferentes puntos isoeléctricos. Electroforesis en gel en dos dimensiones, combina el isoelectroenfoque (primera dimensión) con la SDS-PAGE (segunda dimensión). Es una técnica muy resolutiva, y el mejor método analítico para separar proteínas que existe hoy en día.
Electroforesis capilar Aunque la electroforesis en gel en sus diversas formas resulta un método común y efectivo para la separación de moléculas, el proceso de separación puede durar varias horas, siendo difícil de cuantificar y automatizar. La técnica aquí presentada se lleva a cabo en el interior de tubos capilares (1– 10 µm de diámetro). Estos capilares disipan el calor rápidamente permitiendo la aplicación de campos eléctricos elevados, reduciendo así los tiempos de separación.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
41
PARTE EXPERIMENTAL Electroforesis en tiras de acetato de celulosa La electroforesis en acetato de celulosa se usa para la separación y caracterización de proteínas y otras moléculas. Durante la corrida en acetato las moléculas se moverán según su densidad de carga. El soporte consiste en tiras delgadas de acetato de celulosa, con propiedades de adsorción mínimas, por lo que se evita la formación de colas (cometas) y se logra una separación en bandas bien definidas. Además de la buena resolución este método tiene otras ventajas: la separación es muy rápida, se pueden emplear pequeñas cantidades de muestras, pueden recuperarse y aislar los componentes de la muestra. El estimulo eléctrico se realiza a través de un solución buffer cuyo pH es escogido según la mezcla que se quiere separar. Debido a la naturaleza anfolítica de las proteínas, la carga eléctrica se modifica con el pH del medio, definiéndose entonces como punto isoeléctrico (pI), al pH en el cual la molécula no tiene carga eléctrica neta y entonces no migrará en un campo eléctrico. Si el pH se encuentra por debajo del pI, predominan las cargas positivas y a la inversa a un pH superior al pI.
Objetivo: Separar mediante electroforesis las proteínas contenidas en el suero sanguíneo. La muestra biológica que se utiliza es suero sanguíneo humano, los pI de las proteínas del suero van desde 4,9 (albúmina) hasta 7,4 (gama globulinas) por lo que se utiliza una solución tampón Veronal sódico (dietilbarbiturato de sodio 40 mM pH 8,5)

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
42
En suero humano la composición normal es: Albúmina........... 54-62% α globulinas....... 9-15% β globulinas....... 8-13% γ globulinas....... 14-19% Soporte: tiras de acetato de celulosa 1. Acondicionamiento de la tira: Las tiras de acetato de celulosa se conservan en metanol al 40%, por esto es necesario lavarlas primero con abundante agua y luego sumergirlas en la cuba con el buffer de electroforesis Veronal sódico por 10 minutos. Escurrir el exceso de buffer y colocar la tira sobre el soporte de la cuba, con la superficie opaca hacia arriba, debiendo quedar bien fija y plana. 2. Siembra: Mezclar la muestra biológica con el colorante (azul de bromo fenol) sobre una placa de vidrio. Sembrar la muestra en la zona cátodo y aplicar 2 mA por siembra. Observar el frente de la corrida. 3. Revelado: Una vez finalizada la corrida, que dura aproximadamente 40 minutos, se sumerge la tira 5 minutos en el colorante, que puede ser Amidoschwartz (0,5% en metanol, agua, ácido acético 4,5: 4,5: 1) o Ponceau S (rojo Ponceau 0,5% en ácido tricloroacético, agua 5:95). Las tiras luego se decoloran en solución decolorante (metanol, agua, ácido acético 47,5: 47,5: 5), de tal manera que finalmente sólo retienen el color las bandas de proteínas. Observar la tira, graficar y reconocer las bandas proteicas. 4- Cuantificación de las fracciones proteicas. Cortar las bandas coloreadas, tras la corrida electroforética. Colocar cada una en tubos correctamente rotulados ( α ,β, albumina, ), disolver el acetato de celulosa en ácido Acético al 80%. A Continuación proceder a la lectura espectrofotométrica a una longitud de onda de 495 nm. Electroforesis en gel de poliacrilamida PAGE La electroforesis en geles es una técnica de separación de proteínas y puede aplicarse también en ácidos nucleicos. En ambos casos la separación ocurre a través de un gel cuyo tamaño de poro es regulado según ciertas características propias de cada corrida. Si lo que se va a separar son proteínas y fragmentos pequeños de ácidos nucleicos se utiliza un gel preparado con acrilamida y bis acrilamida, mientras que para fragmentos grandes de ADN y ARN se emplean geles de agarosa. En la electroforesis en geles de poliacrilamida (PAGE), la mezcla de proteínas es mezclada con un detergente aniónico (El dodecil sulfato de sodio –SDS-) conocido también como lauril sulfato, es un detergente aniónico capaz de romper las interacciones no covalentes, el cual se agrega en la preparación de la muestra con un agente reductor, Mercaptoetanol para reducir los puentes disulfuro de tal manera de desnaturalizar la proteína cargándose negativamente,

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
43
siendo en general, la cantidad de SDS unido proporcional, al peso molecular del polipéptido e independiente de su secuencia. La unión es aproximadamente de una molécula de SDS cada dos aminoácidos, cubriendo en exceso toda carga neta (al pH en que se está trabajando), siendo entonces la relación carga/masa similar para todas las proteínas, pudiéndose separar estas por su masa molecular (y no por su densidad de carga) por acción de los poros del gel. También se agrega un colorante que permite observar el frente de la corrida. La relación carga / masa es similar en todas las proteínas de la mezcla. Aquí como la densidad de carga es uniforme las proteínas se separaran por su tamaño. La electroforesis SDS-PAGE es un sistema de corrida discontinuo formado por dos geles con tamaño de poro distinto, en un primer gel (de siembra o compactador) de poro mayor y menor pH, y un segundo gel donde ocurre la separación. Luego de transcurrida la corrida se deben revelar los geles empleando distintas sustancias, radiación o luz uv. Así, los complejos SDS- proteína formados se someten a electroforesis sobre gel de poliacrilamida. La dirección de la corrida es vertical descendente. Las bandas se visualizan cuando se tiñen con colorante. Las proteínas pequeñas se desplazan rápidamente y las proteínas grandes permanecen arriba cerca del punto de aplicación. El desplazamiento es proporcional al logaritmo de su masa pudiendo realizarse una curva de calibración de pesos moleculares.
Utilizando proteínas de peso molecular conocido, y midiendo las distancias de corrida, podemos graficar log pM /distancia recorrida, lo que da una recta. Si medimos la distancia recorrida por una muestra incógnita podremos determinar el pM (llamado aparente) de la proteína. Para esto las proteínas deberán poder verse, para lo cual primero serán coloreadas, siendo el colorante más usado el Coomassie Brilliant Blue R250, con posterior decoloración en un sistema de solventes.
Objetivo: Calcular gráficamente la masa molecular aparente de proteínas por comparación con proteínas patrones, en un gráfico de distancia vs. Log PM aparente.
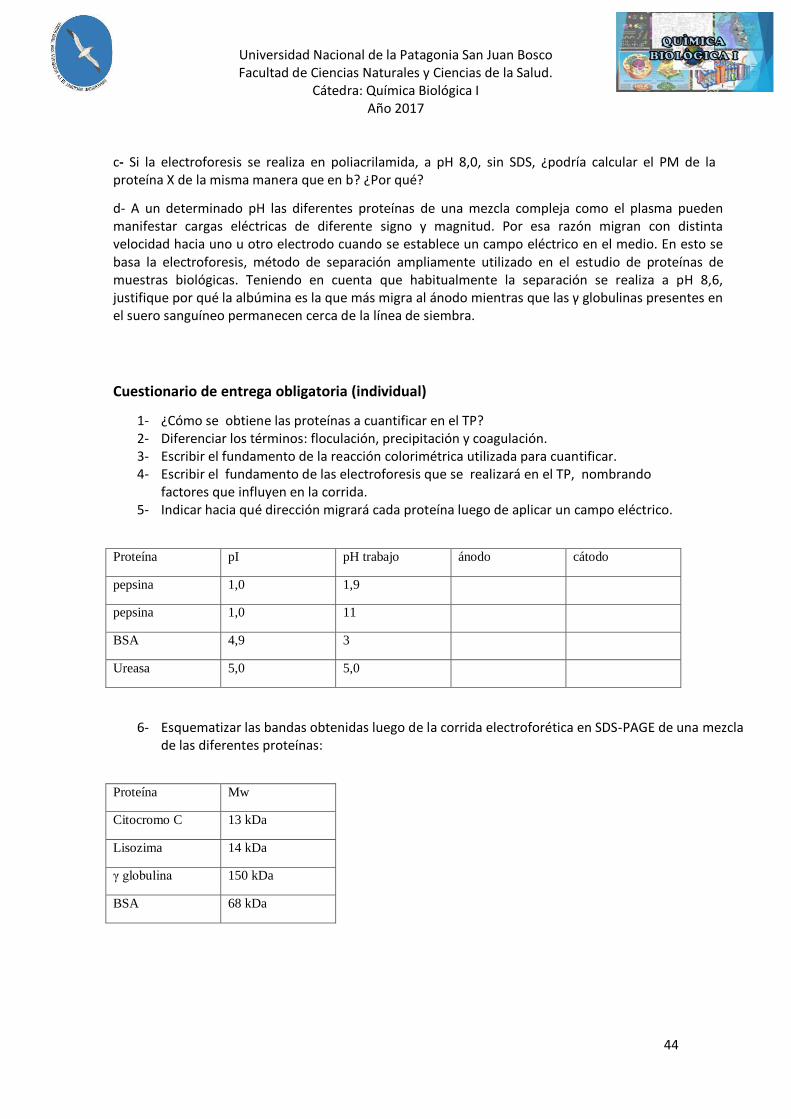
Se determinó el PM de una proteína X analizándola simultáneamente con proteínas patrones, mediante electroforesis en gel de poliacrilamida. La corrida electroforética se llevo a cabo a pH 8,0 y en presencia de SDS. Los resultados fueron los siguientes:
PROTEINA DISTANCIA RECORRIDA (cm) PM (Da)
Seroalbúmina 3,0 68.000
Quimiotripsina 4,7 23.000
Citocromo C 5,7 12.000
Proteína X 4,1 ¿?
a- Haga un diagrama del gel indicando la posición de las distintas proteínas. Indique en el diagrama el punto de siembra de la muestra y la posición del ánodo y cátodo. b- Calcule el PM de la proteína X.
Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
44
c- Si la electroforesis se realiza en poliacrilamida, a pH 8,0, sin SDS, ¿podría calcular el PM de la proteína X de la misma manera que en b? ¿Por qué?
d- A un determinado pH las diferentes proteínas de una mezcla compleja como el plasma pueden manifestar cargas eléctricas de diferente signo y magnitud. Por esa razón migran con distinta velocidad hacia uno u otro electrodo cuando se establece un campo eléctrico en el medio. En esto se basa la electroforesis, método de separación ampliamente utilizado en el estudio de proteínas de muestras biológicas. Teniendo en cuenta que habitualmente la separación se realiza a pH 8,6, justifique por qué la albúmina es la que más migra al ánodo mientras que las γ globulinas presentes en el suero sanguíneo permanecen cerca de la línea de siembra.
Cuestionario de entrega obligatoria (individual)
1- ¿Cómo se obtiene las proteínas a cuantificar en el TP? 2- Diferenciar los términos: floculación, precipitación y coagulación. 3- Escribir el fundamento de la reacción colorimétrica utilizada para cuantificar. 4- Escribir el fundamento de las electroforesis que se realizará en el TP, nombrando
factores que influyen en la corrida. 5- Indicar hacia qué dirección migrará cada proteína luego de aplicar un campo eléctrico.
Proteína pI pH trabajo ánodo cátodo
pepsina 1,0 1,9
pepsina 1,0 11
BSA 4,9 3
Ureasa 5,0 5,0
6- Esquematizar las bandas obtenidas luego de la corrida electroforética en SDS-PAGE de una mezcla de las diferentes proteínas:
Proteína Mw
Citocromo C 13 kDa
Lisozima 14 kDa
γ globulina 150 kDa
BSA 68 kDa

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
45
Bibliografía
Rivera J. M., Pertierra A. Fundamentos de Bioquímica estructural. Alfaomega S. A. México. 2005. Pág.: 51-80.
Baynes J., Dominiczak . Bioquímica Médica. 4° Ed. Elseiver España. 2015 . Pág.: 6-20.
Bradford MM (1976): A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72: 248-254.
Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985): Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
46
Trabajo Práctico: Ácidos nucleicos ADN y ARN
Extracción, identificación y reconocimiento.
OBJETIVO
-Extraer ácidos nucleicos de tejido vegetal.
-Reconocer los componentes estructurales del ARN y ADN.
FUNDAMENTO TEÓRICO
Los ácidos nucleicos son compuestos nitrogenados de elevado peso molecular que se pueden encontrar en las células asociados a proteínas formando complejos llamados núcleo-proteínas. Estas proteínas tienen carácter básico mientras que los ácidos nucleicos son ácidos. Se conocen dos grupos de ácidos nucleicos: ARN (ácido ribonucleico) y el ADN (ácido desoxirribonucleico). La hidrólisis controlada de ARN y ADN produce nucleótidos. Si la hidrólisis continúa se producen nucleósidos y finalmente fosfatos, azúcares y bases púricas y pirimidínicas.
La función biológica de los ácidos nucleicos es fundamental:
El ADN está asociado con el material genético de las células, pudiendo encontrarse como una sola cadena en algunos microorganismos o formando parte de núcleo-proteínas de los cromosomas en organismos superiores. Almacena información genética y sirve de molde para la síntesis de proteínas. Se encuentra mayoritariamente en el núcleo y en muy poca cantidad en mitocondria y cloroplastos. Contiene dos purinas (adenina y guanina) y dos pirimidinas ( citosina y timina). El azúcar presente en sus estructuras es una desoxirribosa
El ARN participa en la síntesis de proteínas y se distribuye en toda la célula, mayormente en citoplasma en forma soluble o formando parte de los ribosomas. Contiene dos purina (adenina y guanina) y dos pirimidinas (citosina y uracilo). El azúcar presente en su estructura es una ribosa.
La polimerización de nucleótidos para formar ácidos nucleicos implica la formación de enlaces fosfodiester entre el 5´fosfato de un nucleótido y el 3´hidroxio de otro. Los oligonucleótidos son pequeños polímeros que contienen solo unos pocos nucleótidos; los poli nucleótidos mayores que componen el ARN y ADN celular pueden contener miles o millones de nucleótidos. Una cadena polinucleotidica tiene un sentido, con un extremo de la cadena terminando en un grupo 5´fosfato y el otro en un grupo 3´OH. Los poli nucleótidos siempre se sintetizan en la dirección 5´a 3´, añadiéndose un nucleótido libre al grupo de 3´OH de la cadena en formación.
Los nucleótidos no solo son importantes como ladrillos de construcción de los ácidos nucleicos; también desempeñan papeles vitales en otros procesos celulares por ejemplo: el adenosin 5´trifosfato (ATP) que representa la principal forma de energía química dentro de la célula.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
47
El protocolo básico de extracción de ADN consiste en el tratamiento del homogeneizado con disolventes orgánicos (fenol, cloroformo o ambos)
Para evidenciar la presencia de ácidos nucleicos en extractos celulares vegetales se requiere células en cantidad suficiente y en medio libre de interferencias que puedan causar lisis celular o hidrólisis de los ácidos nucleicos por nucleasas. Una completa extracción de ácidos nucleicos es fundamental para la determinación indirecta.
Una hidrólisis ácida remueve todas las bases púricas (hidrólisis de uniones N-glicosídicas entre pentosa y las bases púricas) sin afectar las uniones fosfodiester del esqueleto nucleotídico, por lo tanto obtenemos como producto ácidos nucleicos sin bases púricas.
Las desoxipentosas debido a la hidrólisis ácida sufren deshidratación dando aldehídos δ-hidroxi-levulínicos. Estos reaccionan con difenilamina dando un producto de color azul. Así, la reacción de difenilamina servirá para caracterizar, indirectamente, la presencia de DNA en células de cebolla.
La pentosa del ADN y ARN puede evidenciarse por la reacción con orcinol. El ácido sulfúrico hidroliza las uniones fosfodiester y N-glicosídicas, liberando ribosa, bases púricas, pirimidínicas y ácido fosfórico.
La pentosa en medio ácido se deshidrata originando furfural que reacciona con el orcinol dando un producto verde que demuestra la presencia de DNA y RNA.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
48
El grupo fosfato se identifica por la reacción para fósforo inorgánico, en medio ácido, con el molibdato (reactivo 1) da fosfomolibdato, color amarillo, que es reducido luego por el ácido ascórbico (reactivo 2) a azul de molibdeno, color azul verdoso. El reactivo 3 (arsenito/citrato) se combina con el exceso de molibdato impidiendo su reacción posterior con otros fosfatos.
PARTE EXPERIMENTAL:
Reactivos
Solución de lisis: 0,1 % de SDS en 25 mM EDTA pH 8,0.
Solución de lisis alternativa: 120 ml H2O, 1,5 g NaCl, 5 g bicarbonato de sodio, 5 ml de detergente.
Alcohol etílico 95%
Reactivo difenilamina: 0,2 g en 20 ml de ácido acético glacial y adicionar 0,5 ml de ácido sulfúrico. (Preparar en el momento).
Reactivo Bial: 1,5 g de orcinol en 500 ml de HCl cc y 20 gotas de cloruro férrico 100 g/l
Extracción de ácido nucleico de material biológico
Pelar y rallar una cebolla mediana con rallador o procesador hasta picarla finamente. Pesar 10 g de cebolla rallada y adicionar 25 ml de solución de lisis y dejar 20 min. a t° amb. Filtrar en gasa y recoger en probeta. Centrifugar 10 minutos, recoger el sobrenadante. Transferir 2 ml de sobrenadante a un tubo de ensayo. Adicionar 2 volúmenes de etanol 95% procurando no mezclar las dos soluciones. En la interfase observar la formación de una fina hebra. Aspirar la interfase con pipeta Pasteur y separarla en un tubo. Precipitar el ADN por calentamiento suave. Observar. Homogeneizar en 2 ml de agua destilada para realizar reacciones.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
49
Reacción para caracterización de ADN (desoxipentosa)
Cuando se trata el ADN con difenilamina en condiciones ácidas, se forma un compuesto azul con una absorción definida máxima a 595 nm. Esta reacción la dan en general las 2-desoxipentosas y no es específica para el ADN. En solución ácida, la estructura lineal de una desoxipentosa se convierte a la forma b- hidroxilevulinoaldehído altamente reactiva y que reacciona con difenilamina para dar un complejo azul.
Tubo A Tubo B
Agua destilada 1 ml -
Extracto - 1 ml
Rvo difenilamina 2 ml 2 ml
Hervir 20 minutos, enfriar y observar.
Reacción para identificación de pentosa (ribosa o desoxirribosa)
La reacción de Bial es una reacción general para las pentosas y se debe a la formación de furfural cuando se calienta la pentosa con ácido clorhídrico concentrado. El orcinol reacciona con furfural en presencia de cloruro férrico como catalizador, dando una coloración verde. Solamente los nucleótidos de purina dan una reacción considerable.
Tubo A Tubo B
Agua destilada 1 ml -
Extracto - 1 ml
Rvo orcinol 2 ml 2 ml
Hervir 10 minutos, enfriar y observar.
Reacción para identificación de grupo fosfato
La presencia del grupo fosfato se determina indirectamente por la presencia de fósforo inorgánico con formación de azul de fosfomolibdeno.
Blanco Problema
Agua Destilada 0,1 ml
Extracto - 0,1 ml
Rvo 1 0,5 ml 0,5 ml
Mezclar y esperar 30 seg.

Universidad Nacional de la Patagonia San Juan Bosco Facultad de Ciencias Naturales y Ciencias de la Salud.
Cátedra: Química Biológica I Año 2017
50
Rvo 2 0,5 ml 0,5 ml
Mezclar y esperar 30 seg.
Rvo 3 0,75 ml 0,75 ml
Mezclar, incubar 15 minutos a 37° y observar el color.
Cuestionario de entrega obligatoria (individual)
1. Dibujar la estructura de las bases púricas. 2. ¿Cómo se llaman las proteínas asociadas al DNA en eucariota? 3. Mencionar nucleótidos de importancia en el metabolismo. 4. Dibujar la estructura del ATP
Bibliografía
Morrison, Boyd. Química Orgánica. Fondo Educativo Interamericano. S.A. México.
1996.
Cooper, G., La célula. Marban libros. España. 2007. Pág.: 48-49.
Rivera J. M., Pertierra A. Fundamentos de Bioquímica estructural.