
UNIVERSIDAD NACIONAL AUTÓNOMA DE NICARAGUA FACULTAD DE...
125
UNIVERSIDAD NACIONAL AUTÓNOMA DE NICARAGUA FACULTAD DE CIENCIAS DEPARTAMENTO DE QUÍMICA MONOGRAFIA “DESARROLLO DE UN MÉTODO PARA EL ANÁLISIS DE DIURÉTICOS POR CROMATOGRAFÍA MICELAR” PREVIA OPCIÓN AL TITULO DE LICENCIADO EN QUÍMICA AUTOR: BR. DANIEL ANTONIO RIVERA CASTILLO TUTOR: Msc. FABIO PALAVICCINI LEÓN, NICARAGUA, DICIEMBRE DEL AÑO 2002
Transcript of UNIVERSIDAD NACIONAL AUTÓNOMA DE NICARAGUA FACULTAD DE...

UNIVERSIDAD NACIONAL AUTÓNOMA DE NICARAGUA FACULTAD DE CIENCIAS
DEPARTAMENTO DE QUÍMICA
MONOGRAFIA
“DESARROLLO DE UN MÉTODO PARA EL ANÁLISIS DE DIURÉTICOS POR CROMATOGRAFÍA MICELAR”
PREVIA OPCIÓN AL TITULO DE LICENCIADO EN QUÍMICA
AUTOR:
BR. DANIEL ANTONIO RIVERA CASTILLO
TUTOR:
Msc. FABIO PALAVICCINI
LEÓN, NICARAGUA, DICIEMBRE DEL AÑO 2002

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
1
INDICE MARCO TEORICO
Dedicatoria Agradecimiento……………………………………………………..…………………..1 Objetivos Generales…………………………………………………………………….2 Objetivos Específicos…………………………..………………………………………2
1.1. Diazotacion y Acoplamiento ……………………………….…………….…………3 1.1.1. Enlace de la muestra con un reactivo dianzonio…………..……………..…4 1.1.2. Diazotacion del analito y acoplamiento ……..………………..…………....7
1.2. Uso de tensiactivos en Química Analítica ……………..…………………….……11
1.2.1. Modificación de las propiedades físicas y químicas en medios micelares…………………………………………………………..……….11
1.2.2. Efecto de los tensiactivos en las reacciones cromogénicas de diazotiacion y enlace ………………………………………………………13
1.3. La cromatografía liquida micelar ………………………………………………….19 1.3.1. Características de la técnica …………………………….…………………19 1.3.2. Predicción de la retención …………………………………………………20
1.4. Clasificación de los diuréticos ………..…………………………………………...22
1.4.1. Diuréticos de alta eficacia ………………………………….……………..23 1.4.2. Diuréticos de eficacia intermedia ………………………..………………..24 1.4.3. Diuréticos de baja eficacia …………………………..…………………….25
II. OBJETO Y PLAN DE TRABAJO …………………………..……..…………….….27 III. PARTE PRÁCTICA ………………………………………….…..…………………27 3.1. Reactivos…………………..………………………………………………………….27 3.2. Instrumentación………….…………..……………………………………………….30 3.3. Procedimientos.………………………………………………………………………30
3.3.1. Hidrólisi……………………………………………………………………….30 3.3.2.Diazotacion y Enlace…………………..……………………………………..31
IV. OPTIMIZACION DE LA ETAPA DE HIDRÓLISIS……………..……………....32 4.1. Mecanismo de Hidrólisis…………………………………………………………….32 4.2. Presencia de Tensiactivos en la Hidrólisis………………..…………………………34 4.3. Estudio de las condiciones de Hidrólisis………………………………………………34 4.4. Reproducibilidad………………………………………………………..……………..43 V. REACCION DE DIAZOTACION Y ACOPLAMIENTO………………….…….44

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
2
5.1. Estabilidad de los Azocolorantes……………………………..………………………44 5.2.Naturaleza de los Azocolorantes formados………..…………………………………47
5.2.1. Comportamiento absorciometrico…………………….……………………..47 5.2.2. Comportamiento Acido – Base de los azocolorante…..……………………61 5.2.3. Comportamiento cromatografico de los azocolorantes……………………..75
VI. DETERMINACION CROMATOGRAFICA DE DIURETICOS………………77 6.1. Elección del pH y del modificador…………………………………………………….77 6.2. Influencia de la concentración del tensiactivos y del modificador……………..…..82 6.3. Optimización de la fase móvil………………………..……………………………….84
6.3.1. Introducción……………..…………………………………………………….84 6.3.2. Procedimiento de optimización……………….………………………..…….84 6.3.3. Resultados…….…………………………………………..……………….......90 6.4. Análisis de Diuréticos en orina…………..…………………………………...108
VII. RESUMEN Y CONCLUSIONES………….………………………..…………….116 VIII. BIBLIOGRAFIA…………………………………..…..………………………….118

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
3
Dedicatoria Dedico esta tesis a todas las personas que con gran interés contribuyeron con ánimo, con conocimientos, ayuda en todos los años de estudio. A mi madre: Juana Castillo de Rivera. A mi Esposa: Marianela Solórzano Balitán. A mi hermano: Julio Rivera Castillo. A mis hijos e hijas. A mi amigo: Dr. Sergio López Grillo. Por haber sembrado los sabios consejos del estudio, educación y moral, que hoy se concretan en esta tesis.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
4
Agradecimiento Doy gracias en primer lugar a Dios, nuestro señor creador de todas las cosas. Por darme la vida, la sabiduría, por concluir esta tesis y por estar conmigo siempre en todo momento. Gracias también a mis profesores que desde el primer año de clase hasta el momento me brindaron conocimiento y animo para concluir mis estudios con éxitos. Gracias a todos mis amigos y personal de la facultad que de una u otra forma son parte de mi éxito.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
5
I. OBJETIVOS I.1. OBJETIVOS GENERALES:
Determinar diuréticos mediante la técnica de Cromatografía Liquida Micelar utilizando reacciones de Diazotación.
1.2. ESPECIFICOS:
1) Realizar la hidrólisis de diuréticos.
2) Desarrollar una reacción de diazotación en medio Micelar.
3) Determinar Diuréticos mediante Cromatografía Líquida Micelar.
4) Utilizar las coloraciones de diazotación en diuréticos para su determinación.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
6
I MARCO TEÓRICO I.1.- DIAZOTACIÓN Y ENLACE Las arilaminas reaccionan con nitrito en exceso en medio ácido para formar el ion diazonio correspondiente, en una reacción llamada de diazotación. El nitrito que no ha reaccionado ha de ser eliminado antes de añadir la sustancia, con la que ha de reaccionar. Una vez eliminado el nitrito, se realiza el acoplamiento entre la sustancia y el ion diazonio para originar el azocolorante. Las reacciones que tienen lugar son las siguientes: Ar-NH2 + HNO2 + H+ Ar-N+ = N + 2 H2O Sustancia ion diazonio diazotable Ar-N+ = N + Ar´- H Ar – N = N Ar´ + H+ Sustancia azocolorante
Los compuestos azo producidos en la reacción de enlace absorben en el visible, con una longitud de onda de máxima absorción y absortividad molar, que dependen de la estructura del producto formado, del pH y del disolvente. La absortividad molar generalmente es muy elevada y permite la determinación de una gran variedad de sustancias (muestras), en concentraciones del orden del �g/ml. Para que la reacción sea rápida y cuantitativa, la sustancia, Ar´-H, debe poseer un anillo aromático muy activado. Los sustratos más utilizados son fenoles o anilinas, pero también se pueden utilizar feniléteres, cuando el ion diazonio es muy reactivo. Para que el acoplamiento se realice, es necesario que las posiciones orto o para, activadas del anillo no se encuentren directamente bloqueadas por sustitución, ni presenten un apantallamiento estérico, producido por un sustituyente voluminoso en una posición adyacente. Las determinaciones basadas en una reacción de acoplamiento con una sal de diazonio pueden dividirse en tres grupos:
a) Enlace de la muestra con un reactivo diazonio. b) Conversión de la muestra en una sal de diazonio y enlace con una sustancia
adecuado. c) Conversión de la muestra orgánica en ácido nitroso, que es determinado
posteriormente mediante una reaccion de enlace. .
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
7
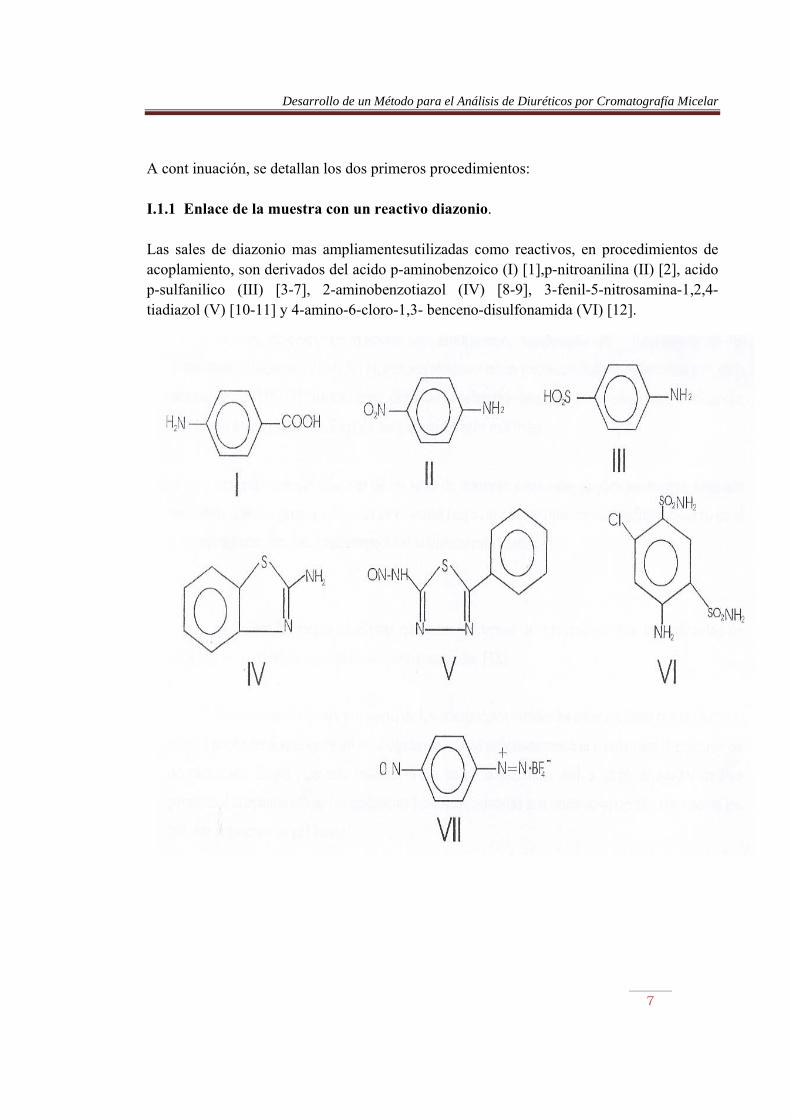
A cont inuación, se detallan los dos primeros procedimientos: I.1.1 Enlace de la muestra con un reactivo diazonio. Las sales de diazonio mas ampliamentesutilizadas como reactivos, en procedimientos de acoplamiento, son derivados del acido p-aminobenzoico (I) [1],p-nitroanilina (II) [2], acido p-sulfanilico (III) [3-7], 2-aminobenzotiazol (IV) [8-9], 3-fenil-5-nitrosamina-1,2,4-tiadiazol (V) [10-11] y 4-amino-6-cloro-1,3- benceno-disulfonamida (VI) [12].

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
8
Estos reactivos son transformados en sales de diazonio por reacción con exceso de nitrito sódico, en un medio HCI 1-2 M. La reacción se produce, usualmente, en un baño de hielo y el exceso de nitrito se elimina por reacción con ácido sulfámico o sulfamato amónico. El reactivo así formado debe utilizarse inmediatamente, debido a que la mayoría de las sales de diazonio no son estables. La diazotación de dichos reactivos es un proceso rápido, que se completa generalmente en 5 a 10 min. La diazotación del 2-aminobenzotiazol es más lenta, requiriendo de 30 a 40 min, pero el compuesto formado es una sal de diazonio muy reactiva, que permite la derivatización de un mayor número de compuestos que con otros diazobencenos derivados de compuesto anilínicos. El 2-aminobenzotiazol también puede obtenerse a partir de (V) por tratamiento con ácido perclórico del 30-35% en etanol [10]. Existen diversos flúorboratos de arildiazonio, incluyendo el fluorborato de p-nitrobencenodiazonio (VII) [13-15], que son sólidos estables y no necesitan ser preparados para cada determinación [16-17]. Sin embargo, éstos se acoplan más lentamente que otras sales de diazonio, y sólo son útiles para el análisis de las sustancias más reactivas. Las reactividades relativas de las sales de diazonio sustituidas pueden predecirse, teniendo en cuenta que los grupos que activan el anillo respecto a la sustitución electrofílica desactivan al correspondiente ion diazonio, respecto al acoplamiento (Tabla 1) La Tabla 2 muestra el efecto que tienen diversos sustituyentes sobre la velocidad de acoplamiento de iones fenildiazonio para-sustituidos [18]. El ion diazonio formado a partir de los compuestos citados ha de reaccionar con la muestra, el cual puede ser fenol, un indol o cualquier sustancia suficientemente activada para la reacción de acoplamiento. El pH para esta reacción varía según la sustancia. Así, a un pH alrededor de 8 se produce el acoplamiento de las sustancias fenólicas, mientras que otros compuestos, tales como los índoles requieren un pH ácido.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
9
Tabla 1.- Efecto de los sustituyente del anillo bencénico sobre la sustitución electrofílica.
Activación en dirección orto, para
Desactivación en dirección meta
Desactivación débil en dirección orto, para
fuerte: O-, -OH -NR2, -NH2, -NHR moderada: -OR, NO, -SR -NHCOR, -OCOR débil: -C6H5, -R, -COO-
fuerte: -NR3
+, -NO2, -CN -SO3H moderada: -CHO, -COR, -COOH, -COONH- -CCI, -CF3
-CI, -Br, -I, -F
Tabla 2.- Velocidades relativas de acoplamiento para los iones fenildiazonio para-sustituidos. Sustituyente Velocidad relativa
-OCH3 -CH3 -H -Br -SO3
-
-NO2
0.1 0.4 1 13 13
1300 La fuente más usual de interferencia en este tipo de determinaciones son las impurezas presentes en la muestra, que también se puedan enlazar con la sal de diazonio y producir colorantes que absorban a la longitud de onda de medida. Las interferencias de este tipo se evitan utilizando procedimientos de separación, previamente a la obtención del azocolorante. A pesar de que las sales de diazonio pueden dar lugar a una gran variedad de reacciones laterales, la mayoría de ellas no originan productos coloreados y, por lo tanto, no afectan a los resultados, siempre que se use suficiente exceso de reactivo para asegurar la conversión cuantitativa de la muestra en el producto enlazado.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
10
I.1.2. DIAZOTACIÓN DEL ANALITO Y ACOPLAMIENTO Este tipo de procedimiento es el más frecuente. Las sustancias más usuales son el (1-naftil) etilendiamina (NED), VIII) [19] y el 2-naftol (IX). El primero (VIII) es el más utilizado en análisis cuantitativo, ya que generalmente forma compuestos solubles con altos valores de absortividad molar. El 2-naftol, frecuentemente, da lugar a productos insolubles y es más utilizado en ensayos cualitativos. Cuando el compuesto que se determina se diazota, es muy importante optimizar las condiciones experimentales (acidez, concentración de los reactivos y tiempo de reacción), debido a que las sales de diazonio son usualmente inestables, y cualquier pérdida por descomposición de la sal de diazonio o por la producción de reacciones laterales, disminuye la sensibilidad y la presión del análisis.
La reacción de las aminas aromáticas con el ácido nitroso, para formar sales de diazonio, puede llevarse a cabo independientemente de la presencia de otros sustituyentes sobre el anillo. El mecanismo de la reacción es el siguiente [20 - 21]:

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
11
Los reactivos nitrosantes más utilizados son el ion ácido nitroso (H2O-NO+), sales de nitrosilo (Br-NO y CI-NO) y el trióxido de dinitrógeno (NO2-NO). La reacción debe verificarse en medio ácido, para que estos iones se encuentren en concentraciones apreciables. La formación de una sal de diazonio es rápida para valores de pH en el intervalo de 0-3. cuando esta reacción es inusualmente lenta, o cuando la sal de diazonio es muy inestable, es necesario optimizar el pH para aumentar la velocidad de reacción y minimizar los efectos de descomposición. La velocidad de reacción también se ve incrementada si el pH se ajusta con HCl, en lugar de H2SO4, debido a que la especie CI-NO es un agente nitrosante más poderoso que el HSO4-NO además, la velocidad puede incrementarse por adición de NaBr

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
12
o KBr, probablemente debido a la formación de Br-NO, que es un agente nitrosante aún mejor que el CI-NO [21]. Por su parte, la reacción de enlace requiere un disolvente polar para disolver los productos iónicos, siendo el agua y el etanol los más utilizados. Un riguroso control de pH es muy importante para que esta reacción sea rápida y cuantitativa. Ya que sólo la arilamina libre o el anión fenolato son activos en el acoplamiento, un pH demasiado bajo inhibiría la reacción al convertir las arilaminas en iones muestra, y los iones fenolato neutros. Así, las muestra sólo reaccionarán rápidamente por encima de pH 5 y los fenoles requerirán un pH tan alto como sea posible. En cambio, a pH 9 o mayor, el ion diazonio se convierte en una especie no reactiva, de acuerdo con el siguiente equilibrio:
Por tanto, el acoplamiento con sustancia amina se debe llevar a cabo en la zona de pH 5-9, y el de los fenoles, en la zona de pH 9-10. el efecto del pH sobre la velocidad de la reacción pueden utilizarse para conseguir la diazotación selectiva de una arilamina, en presencia de una amina alifática, sin que esta última reaccione. Por debajo de pH = 3, la amina alifática, más básica, se encontrará protonada en mayor grado y, por lo tanto, no reaccionará con el agente nitrosante, mientras que existirá suficiente concentración de arilamina libre, menos básicas, para que tenga lugar su diazotación. El pH más adecuado debe encontrarse experimentalmente para cada caso. Debido a que la sal de diazonio se forma a pH bajo, será necesario realizar un ajuste del pH antes de que se produzca el enlace. Así, por ejemplo, en el análisis de sulfonamidas diazotadas con el reactivo de Bratton-Marshall (VIII) [22-23], la diazotación debe realizarse en medio muy ácido y el acoplamiento a pH debidamente ácido. Sin embargo, debido a la mayor absortividad molar de los azocolorantes protonados, antes de medir al absorbancia debe acidularse nuevamente hasta un valor de pH = 1-2. Los procedimientos pueden hacerse específicos, en la mayoría de aplicaciones, por medio de la realización de extracciones o separaciones cromatográficas, previas a la reacción de acoplamiento. La separación puede realizarse, en muchos casos, después del acoplamiento.
Ar – N = N Ar – N = N – OH Ar – N = N – O ión diazonio ácido diazótico ión diazotado Especies no reactivas
H – H –
H + H+

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
13
Existe también la posibilidad de convertir algunos compuestos en especies diazotables, para continuar después con el procedimiento normal así, por ejemplo, el cloranfenicol (X) contiene un grupo nitro, que puede ser reducido a amina con cinco metálico, cloruro de estaño o ditionito sódico. La reducción a amina, seguida de la reacción de diazotación y acoplamiento con el reactivo de Bratton-Marshall [24-26], ha sido utilizada para la determinación de este producto farmacéutico. Por otra parte, el cloracepato potasio (XI) [27] y el clordiazepóxido (XII) pueden ser convertidos mediante hidrólisis en medio muy ácido a 2-amino-4-clorobenzofenona (XIII), que a continuación puede determinarse utilizando la reacción de Bratton-Marshall.
X

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
14
I.2.- USO DE TENSIOACTIVOS EN QUÍMICA ANALÍTICA En presencia de tensioactivos, el equilibrio químico, la cinética y las propiedades espectrales pueden modificarse, lo que pueden ser aprovechado para mejorar las características de los procedimientos analíticos. Los cambios se observan sobre un amplio. Intervalo de concentraciones de tensioactivos, por debajo y por encima de la concentración micelar crítica (cmc). Por encima de la cmc, los monómeros de tensioactivos se agregan espontáneamente en estructuras globulares denominadas micelas. Los tensioactivos inducen alteraciones favorables en las constantes de equilibrio y en las propiedades espectrales, inhiben reacciones no deseadas, tales como las de hidrólisis y fotólisis, estabilizan intermedios de reacción, co-solubilizan muestras polares y no polares, reactivos derivatizantes y productos, y aumentan la velocidad de las reacciones mediante la catálisis micelar. A continuación, se discuten estos efectos en relación a la mejora de los procedimientos espectrofotométricos para la determinación de especies orgánicas. I.2.1 Modificación de las propiedades físicas y químicas en medios micelares Los iones y moléculas orgánicos se pueden asociar a los agregados de tensioactivos, tanto por interacciones electrostáticas como hidrofóbicas. En una disolución acuosa micelar las partes no polares de la molécula de un soluto interaccionan hidrofóbicamente con la cadenas hidrocarbonadas del tensioactivo. Si elsoluto y el tensioactivo son ambos iones, la interacción electrostática también se produce. La especies conjugadas de un par ácido-base se asocian a las micelas son diferentes fuerzas. Simultáneamente, se producen cambios en la constante dieléctrica y en el potencial superficial que experimentan las especies químicas. Todo ello puede producir el desplazamiento de la constante de equilibrio del par ácido-base. De la misma forma, otros equilibrios, como los de formación de complejos, redox, extracción y solubilidad, pueden modificarse. Como consecuencia de la modificación de las constantes de protonación de los compuestos, el valor de pH óptimo al cual una reacción de derivatización tiene lugar, podría ser diferente para las disoluciones micelares y acuosas. Es frecuente que una de las especies del para ácido-base conjugado sea activa y la otra no reaccione. Por ello, el desplazamiento de la constante de protonación modificará el intervalo de pH al cual se produce la reacción. Puesto que los reactivos e intermedios que intervienen en muchas reacciones de derivatización cromogénica son iones, se requiere un medio polar para llevarlas a cabo. Sin embargo, muchas muestras de origen natural e industrial, y muchos colorantes orgánicos

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
15
que se usan para mejorar las características espectrales de los analitos, tienen un pronunciado carácter no polar, y su solubilidad en medios polares, como agua o mezclas alcohol-agua, es limitada. En las disoluciones micelares, la solubilidad de las especies no polares se ve incrementada, sin disminuir la solubilidad de las especies polares e iónicas, lo que permite manejar un mayor número de analitos, reactivos y muestras. El uso de medios organizados puede también mejorar los métodos espectrofotométricos, produciendo desplazamientos batocrómicos e incrementos de la sensibilidad en los espectros de moléculas orgánicas. Los cambios espectrales se producen cuando los cromóforos se asocian a las micelas mediante fuerzas hidrofóbicas o electrostáticas, y son consecuencia de cambios en el microambiente que rodea al grupo cromóforo. Los cambios espectrales pueden producirse también por un desplazamiento en la constante de protonación, y cuando se realiza un derivatización cromogénica, los incrementos de sensibilidad pueden ser el resultado deun mayor rendimiento de la reacción. Frecuentemente, se producirán diversos efectos simultáneamente, y se tendrán que realizar experiencias encaminadas a evaluar las contribuciones de las diversas causas a los cambios espectrales observados. Por otra parte, en los procedimientos cinéticos o en los de inyección en lujo continuo, los incrementos de sensibilidad puede ser causados por un aumento en la velocidad de las reacciones. Los dos principales factores responsables de la catálisis en las disoluciones micelares son los cambios en la reactividad, al introducir micelas en las disoluciones acuosas, y el efecto de la concentración. La asociación de los reactivos e intermedios con las micelas, sus lugares de solubilización y la orientación juegan un importante papel en la catálisis [28]. En las catálisis micelar se origina con frecuencia una estabilización electrónica favorable de un estado de transición cargado, al asociarse este a una micela iónica de signos opuestos. El efecto de orientación a nivel molecular puede también influir sobre la estereoselectividad de los procesos químicos, alternando así los caminos de reacción. Los compuestos hidrofóbicos que son eléctricamente neutros son preferentemente solubilizados por las micelas, independientemente de su carga. En este caso, los reactivos son reunidos y concentrados localmente en un volumen pequeño de la disolución y habitualmente se observa una catálisis positiva. Sin un reactivo tiene carga opuesta a la de la micela, será electrostáticamente atraído hacia ella y así, se acercará a cualquier otro reactivo hidrofóbricos, contribuyendo ello a la

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
16
aceleración de la reacción. Finalmente, puede esperarse una inhibición o catálisis negativa por reacción entre una especies que está enlazada a las micelas y otra que es electrostáticamente repelida por los agregados. En realidad, este efecto de repulsión puede usarse para evitar interferencias. La catálisis micelar no es útil tal sólo para acelerar reacciones lentas, adaptando la escala de tiempo del experimento científico a las necesidades de los instrumentos, sino también para suavizar las condiciones experimentales requeridas para las reacciones (reduciendo la temperatura o la concentración de un ácido mineral). Esto tiene interés cuando se trabaja con analitos o reactivos lábiles y para simplificar procedimientos analíticos manuales y automatizados. Además, un aumento en la velocidad de una reacción cronogénica y una reducción de la velocidad de una reacción lateral competitiva, incluida la inhibición de la hidrólisis del producto, puede aumentar la absortividad molar aparente y, por lo tanto, la sensibilidad y fiabilidad del procedimiento. Los efectos de los tensioactivos en diversas reacciones cromogénicas de derivatización se examinan a continuación. En algunos casos, se modifica sólo un único parámetro, para mejorar el procedimiento analítico, pero la situación más frecuente es la modificación simultánea de diversas propiedades físicos-químicas de la disolución y de los reactivos, intermedios y productos. El correspondiente procedimiento analítico se beneficiará de algunos de los efectos producidos, pero otros serán perjudiciales. I.2.2- EFECTO DE LOS TENSIOACTIVOS EN LAS REACCIONES CROMOGÉNICAS DE DIAZOTACIÓN Y ENLACE. Modificación de las propiedades ácido-base de los Azocolorantes de N-(1-naftil) Etilendiamina Los iones diazonio, formados por reacción del ácido nitroso con las arilaminas, tienen carácter electrofílico y se acoplan con sustratos activados, tales como N- (1-naftil) etilendiamina (NED), para formar azocolorantes intensamente coloreados (esquema 1).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
17
Esquema 1.- Enlace de arilaminas diazotadas con reactivos
El grupo alquilamino primario de los azocolorantes de NED se protona en medio débilmente alcalino, y el grupo alquil-arilamin segundario se protona en medio débilmente ácido, dando lugar a un desplazamiento batocrómico y a un incremento de la sensibilidad. Por eso, en medio neutro y en medio ácido, los azocolorantes de NED son siempre cationes, con una o dos cargas positivas. Los azocolorantes de NED se modifica en presencia de tensioactivos, lo que se ha estudiado espectrofortométricamente [29-31]. Para22 ello, se consideran las siguientes constantes de asociación:
donde A y HA (las cargas no se indican) son el azocolorante monoprótico y diprótico, respectivamente; el subíndice en es el número de agregación (número de moléculas de tensioactivo que se unen entre sí), y [Sn] es la concentración de agregados del tensioactivo. La constante de protonación del compuesto de asociación azocolorante-agregado de tensioactivo puede definirse como:

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
18
Se ha comprobado que, cuando una disolución ácida de un azocolorante de NED se valora con hidróxido sódico, en presencia de una concentración constante de tensioactivo, el punto de inflexión de la curva de valoración viene dado por la ecuación:
donde K es la constante de protonación del par ácido-base, en ausencia de tensioactivo, y Kap es la constante de protonación aparente, que es función de la concentración de tensioactivo. Para valores elevados de [Sn], se alcanza el límite PHI = log KC, lo que se ha utilizado para determinar el valor de esta constante. La constante log KC se puede determinar por valoración de una disolución tamponada del colorante con una disolución de tensioactivo. Finalmente, log KC puede calcularse teniendo en cuenta la siguiente relación:
que indica que la diferencia entre log KC y log K es una medida de la capacidad selectiva del tensioactivo para unirse a la forma diprótica del colorante, respecto de la forma monoprótica. Así, por ejemplo, cuando se valoró con NaOH con azocolorante formado por la anilina diazotada y NED, en ausencia de tensioactivo, se obtuvo un valor de log K= 3.30, mientras que al incrementar la concentración de dodecilsulfato sódico (SDS, XIV), log Kap se modificó desde este valor hasta log KC = 4.15 [30]. La mayor variación en log Kap tuvo lugar casi completamente antes de la micelación. Por el contrario, con el Triton X-100 (XV), el mayor cambio en log Kap se produjo para concentraciones mayores que la cmc. El cloruro de N-cetilpiridinio (CMPC, XVI) originó una situación intermedia, con una importante parte del cambio antes, y la otra después de la micelación. Se obtuvieron los valores log KC = 1.25 y 0.8, para Triton X-100 y CMPC, respectivamente.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
19
Los valores de Kap indicaron que el azocolorante diprótico, que tiene una carga positiva sobre el grupo amino en posición para, se asocia más fuertemente a las micelas aniónicas de SDS que las especies monopróticas, mientras que para el Triton X-100 y para el CMPC, las especies monopróticas mostraron una asociación más fuerte. Otros estudios, referentes a otros azocolorantes de NED, también mostraron que log KC sigue el orden SDS > agua > Triton X-100 > CMPC [31]. CATÁLISIS DE LAS REACCIONES DE ACOPLAMIENTO ENTRE EL ION DIAZONIO Y NED Las reacciones de acoplamiento de arilaminas diazotadas con NED son catalizadas positivamente en un medio micelar de SDS. El efecto se puede explicar por asociación de los cationes diazonio y NED con las micelas aniónicas. El medio ácido de la reacción, tanto la forma monoprótica como diprótica del NED deben hallarse fuertemente asociadas a las micelas por interacciones electrostáticas e hidrofóbicas; sin embargo, la forma diprótica estará más fuertemente asociada a causa de la carga positiva adicional. Ello produce que el grupo amino secundario del NED se protone a un pH más elevado que en agua; consecuentemente, en el medio ácido en el que se produce la reacción y, en presencia del tensioactivo aniónico, la concentración de la forma monoprótica del NED es más baja que en agua. Como sólo esta forma del NED reacciona con el agente electrofílico, a un pH dado la presencia de SDS debería reducir la velocidad de la reacción. En las experiencias realizadas, se observó lo contrario, lo cual indicó que, en comparación con la protonación adelantada del grupo amino secundario del NED,

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
20
predomina el efecto de la concentración micelar. En una disolución no micelar, las reacciones de diazotación y acoplamiento, y la etapa de medida de la absorbancia, se realizan cada una a un valor de pH distinto (ver apartado I.1.b). Los efectos de desplazamiento de las constantes de protonación de los azocolorantes a valores de pH más altos y de catálisis micelar de las reacciones de acoplamiento con NED, en presencia de SDS, se utilizaron para desarrollar un procedimiento espectrofotométrico simplificado para la determinación de arilaminas, donde no es necesario realizar un cambio de pH [29,31,32]. Se realizó un estudio para establecerse un intervalo óptimo de pH, en el cual el acoplamiento se produzca con una velocidad suficientemente grande y al mismo tiempo, el azocolorante se protone [31]. Se estableció el límite más bajo, pHmin, como el pH al cual el acoplamiento se completa en un 99% en un minuto, y el límite superior como pHmáx = (log Kap – 2), el cual corresponde a un 99% de protonación del colorante. Al examinar un grupo numeroso de sustancias diazotables, no se encontró en agua un intervalo de pH óptimo, o en todo caso éste fue muy pequeño. En cambio, en el medio micelar de SDS, el pHmáx fue más alto que el pHmin, para todas las arilaminas estudiadas, con una diferencia de 1 a 4 unidades de pH. Se observó que estas arilaminas podían ser diazotadas y acopladas para valores de pH entre 1 y 1.6, donde todos los intervalos óptimos de pH se solapaban. Por eso, finalmente, se decidió efectuar las etapas de acoplamiento y medida en una disolución de HC1 0.06M, que se consigue al adicionar los reactivos necesarios para completar la derivatización, a la disolución 0.15 M de HCI empleada para diazotar la arilamina. Este procedimiento simplificado se adaptó también a un procedimiento de inyección en flujo [33]. Acoplamiento De La 2,4,6-trimetilanilina Diazotada con Fenoles Se ha propuesto también el uso de la 2,4,6-trimetilanilina (TMA) diazotada, para determinar derivados de fenoles, por acoplamiento en un medio micelar [34]. Cuando los fenoles se acoplan con una arilamina diazotada (Esquema 2), se observan, generalmente, valores altos e inestables de absorbancia para la disolución del blanco (preparada en ausencia de fenol).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
21
Esquema 2.- Acoplamiento de la 2,4,6-trimetilanilina diazotada con fenoles.
La señal elevada es causada por la hidrólisis del ion diazonio, que produce el correspondiente fenol, en un proceso conocido como hidroxi-dediazoniación, el cual es seguido del acoplamiento del fenol producido, con el exceso de reactivo en el medio básico. Las posiciones orto- y para- del 2,4,6-trimetilfenol resultante de la hidroxi-dediazoniación de la TMA diazotada, están bloqueadas por los grupos metilo, los cuales impiden su acoplamiento con el exceso de reactivo. Eso permite que la disolución del blanco origine valores muy bajos de absorbancia, incluso a pH elevado. Sin embargo, a causa de los sustituyentes metilo hidrofóbicos de la TMA, este reactivo es sólo ligeramente soluble en agua, pero la reacción puede llevarse a cabo en un medio micelar. Se observó también una catálisis micelar fuertemente positiva en la derivatización de fenoles con la TMA diazotada [34]. Las velocidades de reacción siguieron el orden:
NCPC > SDS > Triton X-100 > agua La catálisis positiva en el medio micelar se puede explicar sobre la base de la asociación hidrofóbica de los iones diazonio y fenoles con las micelas. Sin embargo, a causa de los efectos opuestos de las fuerzas electrostáticas, los iones diazonio están más fuertemente asociados a las micelas aniónicas de SDS, mientras que los iones fenolato se asocian con más fuerza a las micelas catiónicas del CMPC. Puede ser, que por esta razón, y por el predominio de las fuerzas hidrofóbicas, las micelas de carga opuesta no produzcan diferencias importantes en la catálisis micelar. Además, en el medio micelar de CMPC, la protonación de los iones fenolato se produce a un valor de pH aún más bajo que en agua, lo que debería incrementar más la velocidad de la reacción en presencia de CMPC, mientras que en el medio mecilar de SDS ocurre lo contrario.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
22
I.3.- LA CROMATOGRAFÍA LÍQUIDA MICELAR I.3.1) CARACTERÍSTICAS DE LA TÉCNICA La Cromatografía Líquida Micelar (CLM) constituye una importante respuesta a la cromatografía líquida convencional en fase inversa. Se basa en el uso de fases móviles que contienen una disolución de tensioactivo en agua, en una concentración superior a la cmc. El uso de estas fases móviles aumenta el número de posibles de interacciones de un soluto en el interior de la columna cromatográfica, y ofrece así nuevas formas de separar mezclas complejas. Los monómeros de tensioactivo que constituyen las micelas están en equilibrio con la fase estacionaria, donde pueden ser adsorbidos, y con la fase móvil, donde pueden existir como entidades individuales o agruparse en micelas. La adsorción de los monómeros de tensioactivo sobre la superficie de una fase estacionaria alquílica puede suceder mediante los mecanismos [35]: Adsorción hidrofóbica: por la afinidad que existe entre la zona hidrocarbonada de las moléculas de tensioactivo y la cola alquílica del silano enlazado a la matriz de sílice. De este modo, la cabeza iónica del tensioactivo queda en contacto con la disolución polar y columna adquiere una cierta capacidad de intercambio iónico, que no se observa en cromatografía líquida convencional en fase inversa. Este tipo de interacción es la más importante.
Adsorción silanofílica: por afinidad entre la cabeza polar de las moléculas de tensioactivo y los grupos silanol residuales de la superficie de la fase estacionaria. Así, las cadenas hidrocarbonadas quedan orientadas hacia la fase móvil, debido a lo cual se incrementa la hidrofobicidad de la columna. Para la mayoría de los tensioactivos y de las fases estacionarias, superado un determinado nivel de concentración, la cantidad de tensioactivo adsorbido sobre la fase estacionaria permanece constante. Los tensioactivos no sólo modifican las propiedades de retención de la fase estacionaria: también producen nuevas interacciones de los solutos con las micelas presentes en la fase móvil, a nivel tanto electrostático, como hidrofóbico. El aprovechamiento de estas interacciones mediante su conveniente exaltación o inhibición, proporcionan una gran versatilidad a la técnica y la hacen virtualmente adaptable a todo tipo de solutos. Posiblemente, esta flexibilidad es la característica más notable de la CLM. En general, se puede decir que las interacciones del soluto con las micelas son las fuerzas dominantes que gobiernan la retención.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
23
Otra ventaja importante que aporta el uso de medios organizados micelares, como fases móviles, es la solubilización en medio acuoso de sustancias apolares, reduciéndose en muchos casos el tiempo necesario para la preparación de las muestras o incluso, evitando etapas previas de eliminación de interferentes o de componentes de la matriz, que pudieran precipitar en el interior de la columna cromatográfica, como ocurre con las muestras fisiológicas. Es posible incluso separar mezclas de sustancias hidrofílicas e hidrofóbicas con una misma fase móvil. También es interesante el ahorro en disolventes orgánicos que supone el uso de las disoluciones acuosas micelares. Además, éstas son inocuas y no inflamables. Desde el punto de vista de la detección, el uso de micelas también resulta beneficioso. Muchos compuestos experimentan en medios micelares un incremento en su absortividad molar, acompañado de un desplazamiento batocrómico. En algunos casos se ha observado también un aumento en la luminiscencia debido a la mayor rigidez del medio micelar, que inhibe las relajaciones moleculares e incrementa la eficiencia de los procesos luminiscentes. Además, la mayor parte de los tensioactivos no absorben luz de forma apreciable en el ultravioleta cercano y tampoco fluorescen. Sólo en casos excepcionales existe una fuerte absorción en el ultravioleta (sulfonatos de alquilbenceno), o una fluorescencia apreciable de la fase móvil (Brij-35, aunque se debe a las impurezas que suele contener más que a su propia naturaleza). Las micelas, que suelen medir menos de 10 nm, resultan demasiado pequeñas como para que se produzca dispersión de luz a las longitudes de onda de detección habituales. El uso de las fases móviles micelares también plantea algunos inconvenientes. Por un lado, la eficacia en CLM, es con frecuencia, mucho más reducid que en cromatografía líquida convencional en fase inversa, pudiendo los picos cromatográficos llegar a ser muy anchos y asimétricos. Por otro lado, la producción de espuma propia de las disoluciones micelares dificulta la desgasificación de las fases móviles, la preparación de dichas fases y en general, su manejo. I.3.2) PREDICCIÓN DE LA RETENCIÓN En cromatografía líquida, la predicción de la retención permite la optimización rápida de la separación de mezclas complejas de solutos. En la bibliografía, han aparecido diferentes artículos sobre la modelización de la retención en fases micelares puras. En todos ellos se llega a una ecuación similar, partiendo de puntos de vista diferentes [36-39].

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
24
donde K´ es el factor de capacidad, [M] es la concentración de monómeros de tensioactivo formando micelas, KAM representa la constante de asociación del soluto con la micela, y KSW, la intensidad de la asociación del soluto con la fases estacionaria. La relación entre estos coeficientes y los equilibrios de reparto del soluto puede verse en la figura1:
miscela
Fase estacionaria
Figura 1.- Equilibrios de reparto en Cromatografía Líquida Micelar. Torres Lapasió et al. Extendieron el modelo dado por la Ecuación (6) a fases móviles conteniendo concentraciones variables de tensioactivo y modificador [40]:
siendo ϕ la concentración de modificador., las constante KAD y KMD mide la variación relativa debida a la presencia del modificador, que se produce en la concentración de soluto

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
25
presente en la pseudofase acuosa y en la micela, respectivamente, tomando como referencia la disolución micelar pura. Estas variaciones son debidas a los cambios que origina el modificador en cada fase o pseudofase, tales como la disminución de la polaridad de la fase acusa y la modificación de las interacciones del soluto con la micela. I.4.- CLASIFICACIÓN DE LOS DIURÉTICOS Los diuréticos son fármacos que actúan sobre el riñón, estimulando la excreción renal de agua y de electrolitos. Se clasifican en función de su eficacia diurética, entendida como la cantidad máxima de sal que se pierde en orina por efecto del diurético [41-43]. La Figura 2 indica el lugar de acción de los principales diuréticos: los más eficaces son los diuréticos del asa, que inducen pérdidas de sodio superiores al 15% del total salino filtrado en el glomérulo. Los diuréticos de eficacia intermedia, entre los que se encuentra las benzotiadiazinas y sus variantes heterocíclicos, actúan preferentemente en la porción inicial del túbulo contorneado distal. Estos diuréticos inducen fracciones de eliminación de sodio del 5 al 10%. Finalmente, con los diuréticos de baja eficacia, la eliminación de sodio es inferior al 5%. En este grupo existen diuréticos que actúan por mecanismos muy diversos, como los ahorradores de potasio, entre los que se encuentran el triantereno y la amilorida, y los diuréticos que actúan antagonizando a la aldosterona, como la espironolactona. Los inhibidores de la anhidrasa carbónica se encuentran también en este grupo. A continuación, se hace referencia al lugar de acción, efecto farmacológico y características farmacocinéticas de los distintos tipos de diuréticos.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
26
Figura 2.- Lugar de acción de los diuréticos en la nefrona. I.4.1) DIURÉTICOS DE LA ALTA EFICACIA Los diuréticos de alta eficacia o del asa producen diuresis, con pérdida abundante de agua, Na+ y Cl-. Las diuresis son rápidas, copiosas y de corta duración. Estos diuréticos aumentan también la eliminación de K+, ya que al aumentar la carga de Na+ que llega al túbulo distal, aumenta el intercambio con K+ a este nivel. La pérdida de K+ es, sin embargo, inferior a la que producen las tiazidas, para una determinada acción natriurética. Los más utilizados son los derivados del sulfamoilbenzoato, tales como la furosemida y la bumetanida, y los derivados del ácido fenoxiacético, como el ácido etacrínico. El lugar crítico donde los diuréticos de alta eficacia ejercen su acción diurética es la rama ascendente gruesa del asa de Henle.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
27
Los diuréticos del asa se absorben rápidamente por vía oral, aunque en grado variable. La biodisponibilidad de la furosemida es del 50-60%, mientras que la de la bumetanida se acerca al 100%. La furosemida y bumetanida se eliminan por metabolismo en un 50%, mientras que el ácido etacrínico lo hace en una proporción de 2/3. en caso de insuficiencia renal, la semivida biológica se prolonga, pero también disminuye la capacidad del fármaco para llegar al lugar de acción. La vida media de eliminación es, en general, corta. Así, la de la furosemida es de unos 90 min. La aparición del efecto es muy rápida: por vía oral, el efecto se inicia a los 10-20 min y la acción máxima aparece a los 20-40 min, con una duración de 4 a 6 h. Por vía intravenosa, el efecto aparecen a los 2 min, lo cual puede ser útil en caso de urgencia, tal como en el edema agudo de pulmón. I.4.2) DIURÉTICOS DE EFICACIA INTERMEDIA Las benzotiadiazinas, entre las que se encuentran la benzotiazida, la bendroflumetiazida y la hidroclorotiazida (tiazidas e hidrotiazidas) son los fármacos más característicos de este grupo, pero existen en él también otros diuréticos, las ftalimidinas, como la clortalidona, y las clorobenzamidas, tal como la xipamida, que difieren de las tiazidas en la naturaleza de anillo heterocíclico. Sin embargo, su acción farmacológica es equivalente. El desarrollo de estos fármacos partió del intento de obtener inhibidores más potentes de la anhidrasa carbónica. Ello permitió descubrir que es ciertas bencenodisulfonamidas, el cierre del anillo entre el grupo acilamino y el correspondiente orto-sulfamoilo alteraba las características de la diuresis, provocando un aumento en la concentración de CI-. El prototipo de estos fármacos, descubierto en 1957, fue la clorotiazida, la cual a pesar de producir una menor inhibición de la anhidrasa carbónica, presentaba una mayor actividad diurética. A partir de la clorotiazida, se desarrollaron las hidrotiazidas, en las cuales ya no existe un doble enlace entre C3 y N4 (ver esquema 3), y diuréticos en los que el anillo benzotiadiazínico se ha sustituido por otros heterociclos.
Esquema 3.- Estructura general de las tiazidas

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
28
El lugar crítico donde los diuréticos de eficacia intermedia ejercen su acción diurética es en la porción inicial del túbulo contorneado distal. Las benzotiadiazinas pueden actuar sobre diversos lugares de la nefrona. También pueden inhibir la anhidrasa carbónica, por lo que es posible que actúen adicionalmente en el túbulo proximo. El lugar exacto de acción de estos diuréticos tiene poca repercusión en la acción diurética, aunque contribuyen a explicar la mayor pérdida de K+ en orina. En efecto, si hay una menor disponibilidad de protones es el túbulo distal, el intercambio de Na+ con el H+ se ve disminuido y al compensarse con un mayor intercambio de Na+ con el K+, se produce una mayor pérdida de éste en la orina. Las tiazidas se absorben bien por vía oral. La biodisponibilidad suele oscilar entre el 60% y el 95%. La clorotiazida se absorbe mal y tiene una biodisponibilidad de tan sólo un 10%. El comportamiento farmacocinético condiciona la duración y magnitud del efecto diurético y es la principal causa de diferencias entre benzotiadiazinas. Las vidas medias son variables y dependen de la tasa de secreción y aclaración tubular. La semivida biológica de eliminación oscila entre las 3 h, para la bendroflumetiazida y las 8-10 h de la hidroflumetiazida. En la xipamida es de 7 h, y 40-64 h para la clortalidona. Lógicamente, al alargarse la semivida, se prolonga a la duración de la acción y la frecuencia de administración puede espaciarse más, pudiendo efectuarse incluso cada 2 ó 3 días. I.4.3) DIURÉTICOS DE BAJA EFICACIA En los diuréticos de baja eficacia, la eliminación de sodio es inferior al 5%. El lugar de acción de los mismos es variable, así, los ahorradores de potasio actúan sobre el último segmento del túbulo distal y los inhibidores de la anhidrasa carbónica actúan en diversos segmentos tubulares. AHORRADORES DE POTASIO El lugar crítico de la acción de estos diuréticos se ubica a nivel del túbulo contorneado distal y porción inicial del tubo colector. A este nivel inhiben la reabsorción de sodio, con lo que reducen el intercambio de Na+ con el K+, y disminuyen la eliminación de éste. Su valor reside, sobre todo, en su capacidad de interferir los procesos de pérdida de potasio. Existen dos clases de ahorradores de potasio: los inhibidores de la aldosterona y los inhibidores directos del transporte de Na+. Así, estos fármacos actúan mediante dos mecanismos: mientras que los del tipo de la espironolactona (inhibidores de la aldosterona) actúan desde el medio intersticial, los del tipo de la amilorida y triantereno lo hacen desde la luz tubular. La espironolactona posee una estructura esteroide similar a la de la

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
29
aldosterona, e impide que la aldosterona promueva la síntesis de proteínas necesarias para facilitar la reabsorción de Na+. En forma micronizada, la espironolactona por vía oral presenta una biodisponibilidad del orden del 90% y una vida máxima de 3 h. La del triantereno y amilorida por vía oral es de 50%. La espironolactona se fija a las proteínas plasmáticas en un 90%, mientras que el triantereno lo hace en un 50-55%, y la amilorida no se une a las proteínas. El triantereno es extensamente metabolizado en el hígado. La amilorida se elimina inalterada en la orina, y la espironolactona se metaboliza en canrenona, a la que se debe de 1/3 a 3/4 de la actividad biológica antialdosterónica. El triantereno tiene una semivida biológica de 2-4 h y la amilorida de 6-9 h. La espironolactona tarda de uno a dos días en actuar, debido al tiempo necesario para que se agote la reserva de proteínas inducidas por la aldosterona. INHIBIDORES DE LA ANHIDRASA CARBÓNICA Son derivados sulfamídicos, que inhiben la anhidrasa carbónica presente en las células de los túbulos renales, sobre todo, en el túbulo contorneado proximal. La acetazolamida ha sido la más estudiada. Tras la administración de acetazolamida, el volumen de orina aumenta rápidamente y el pH de ésta, usualmente ácido, se hace alcalino. La inhibición proximal de la anhidrasa carbónica intracelular impide la secreción de protones, lo cual inhibe la reabsorción del CI- y del HCO3
- (vía formación del ácido carbónico y CO2). En secciones ulteriores de la nefrona, el Cl- es reabsorbido, pero el HCO3
- sólo se absorbe en parte. Ello aumenta la eliminación de agua, HCO3- y Na+,
consiguientemente, la orina se vuelve alcalina y se produce acidolisis, que reduce la eficacia de la siguiente dosis del diurético. Dado que en el líquido tubular queda más Na+, la pérdida de K+ en el túbulo distal resulta favorecida por aumentar el intercambio de Na+ y K+. Los efectos de los diuréticos inhibidores de la anhidrasa carbónica sobre el transporte de H+ y HCO3-, también tiene lugar en otros lugares del organismo.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
30
II.- MATERIALES, EQUIPOS Y REACTIVOS II.1.- REACTIVOS La Tabla 3 muestra las estructuras de los diuréticos estudiados que formaron azocolorantes, junto con las abreviaturas utilizadas a lo largo de esta practica y los Laboratorios farmacéuticos que nos suministró. La Tabla 4 corresponde a diuréticos que tras someterlos a hidrólisis y derivatizarlos no formaron azocolorantes. Se prepararon disoluciones madre de cada diurético en concentración de 100 �g/ml, disolviendo 0.010 g del compuesto en 10 ml de etanol, con ayuda de una baño de vibratorio. A continuación, se añadió aproximadamente 70 ml de agua y se introdujo la disolución nuevamente en el ultrasonidos. Finalmente, se llevó a 100 ml y se hizo uso una vez más del vibriatorio, para asegurar la completa disolución del diurético. El porcentaje de etanol en estas disoluciones fue aproximadamente del 10%. Las sucesivas diluciones se efectuaron con agua. De esta forma, el contenido final en etanol fue muy bajo, garantizando la formación de micelas, al añadir el SDS. Otros reactivos utilizados fueron: nitrito sódico (MERK), ácido sulfámico (Beyker), clorhidrato de N- (1-naftil) etilendiamina, dodecilsulfto sódico (Merck, Darmstadt, Alemania), HCI, etanol, hidrógenofosfato disódico (MERK), ácido acético, ácido fosfórico, tetraborato sódico (Beyker), 1-propanol y 1-petanol (MERK).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
31
Tabla 3.- Diuréticos que forman azocolorantes
Grupo Compuesto
I
4 – amino – 6 –
clorobenceno – 1 , 3 – disulfonamida
Hidroclorotiazida
(HCTA)
Clorotiazida
(CTA)
Triclorometiazida
(TCM)
Altiazida
(ALT)
II
Hidroflumetiazida
(HFM)
Bendroflumetiazida
(BENDRO)
III
Furosemida
(FURO)
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
32
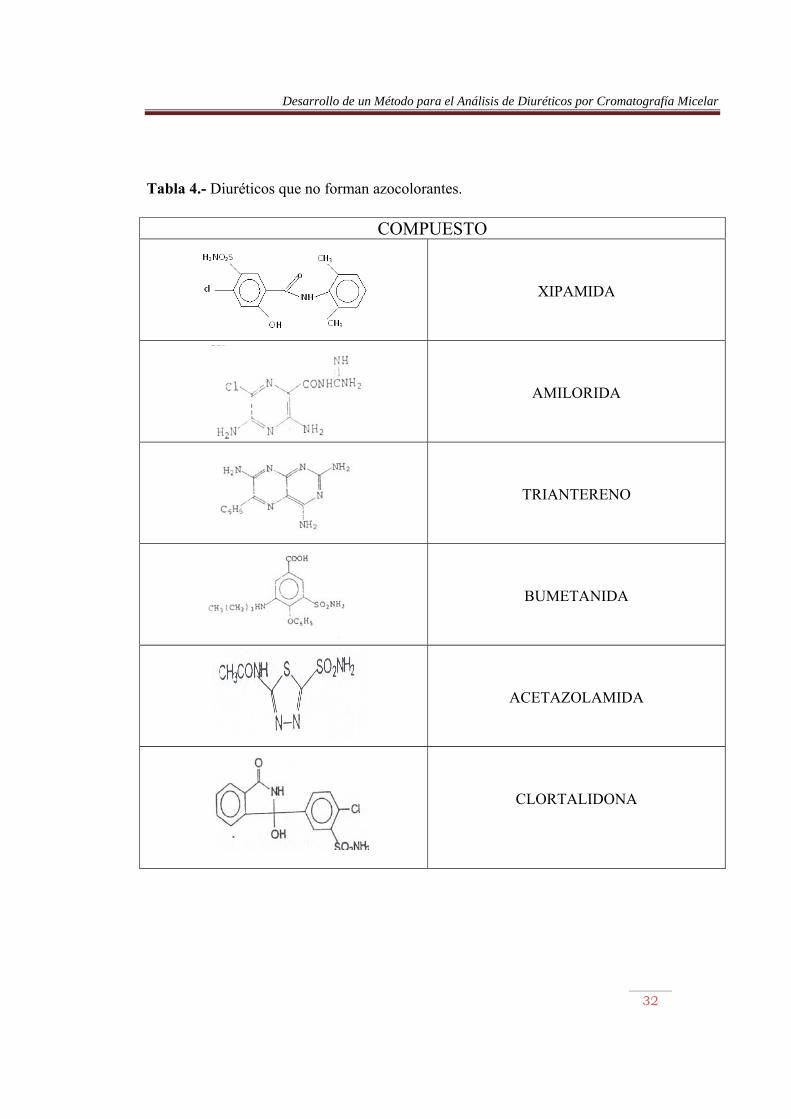
Tabla 4.- Diuréticos que no forman azocolorantes.
COMPUESTO
XIPAMIDA
AMILORIDA
TRIANTERENO
BUMETANIDA
ACETAZOLAMIDA
CLORTALIDONA

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
33
II.2.- INSTRUMENTACIÓN Para realizar la hidrólisis de los diuréticos se utilizó un baño termostático Selecta, modelo Precis-Term. Los espectros se obtuvieron con un espectrofotómetro UV-vis Perkin Elmer, modelo Lambda 16. las medidas de pH se efectuaron con un pH-metro Crison modelo 2001, con electrodo combinado de vidrio-calomelanos. El estudio cromatográfico se realizó con un equipo de Perkin Elmer, de la serie 295, provisto de una bomba cuaternaria y de una válvula Rheodyne con un bucle de 20 �l, como fase estacionaria se empleó una columna ODS-2 (5 �m, 12.0 cm x 4.6 mm) de Merck (Alemania), precedida por una precolumna de las mismas características y de 3.0 cm de longitud. La fase móvil y las disoluciones de los diuréticos se inyectaron a través de filtros de 0.45 :m y 0.22 :m de membrana de Nylon, respectivamente (Micron Separations, Westboro, MA, USA). El caudal de la fase móvil fue de 1 mI/min. La detección se realizó mediante un detector UV-Visible Perkin Elmer LC 295, utilizando longitudes de onda de 525 a 550 nm. Los cromatogramas fueron adquiridos por una estacion de datos PE Nelson, NCI 900 y procesados en una computadora mediante el progrma TurboChrom 6.1.2, de Perkin Elmer. II.3.- PROCEDIMIENTOS II.3.1 HIDRÓLISIS Para realizar la hidrólisis se utilizaron tubos de ensayo de aproximadamente 16 ml de capacidad, con tapón roscado, a los que se les practicó una perforación. Para evitar proyecciones de la disolución sometida a hidrólisis, se introdujo un trozo de varilla de vidrio en cada tubo. La hidrólisis se efectuó mezclando 1 ml de HCI 1.65 M y 10 mI del diurético, en concentración de 10 �g/mI. Así la concentración de HCI en la mezcla de reacción fue 0.15 M. Los tubos se introdujeron en un baño termostático a 100ºC durante 60 min. Una vez trascurrido este tiempo, los tubos se enfriaron en un baño de agua, a temperatura ambiente durante 2 min, con el fin de detener el proceso hidrolítico. Se comprobó que el volumen en el interior de los tubos, una vez terminado el tratamiento, no varió significativamente. A continuación, se abrieron los tubos y se pipeteó 10 mI de la mezcla hidrolizadas, introduciéndose en aforados de 25 mI, para proceder a la derivatización del diurético hidrolizado. En el Esquema 4 se muestran los tubos empleados en la etapa de hidrólisis.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
34
Esquema 4.- Erlenmeyer utilizado en la etapa de hidrólisis. II.3.2. DIAZOTACIÓN Y ENLACE Para realizar la diazotación de las arilaminas obtenidas en la hidrólisis, se adicionó a la disolución anterior 2.5 mI de una disolución que contenía SDS 0.5 M y HCI 0.15 M, y 1 mI de nitrito sódico 0.15 M. Al cabo de 5 min, se añadió 1 mI de ácido sulfámico 0.3 M, para eliminar el exceso de nitrito. Finalmente, transcurridos 10 min, se adicionó 0.5 mI de NED 0.03 M, con lo que se formó el azocolorante y se aforó con agua, hasta completar el volumen a 25 mI.
TAPON CON ORIFICIO DE 0.4 CM
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
35
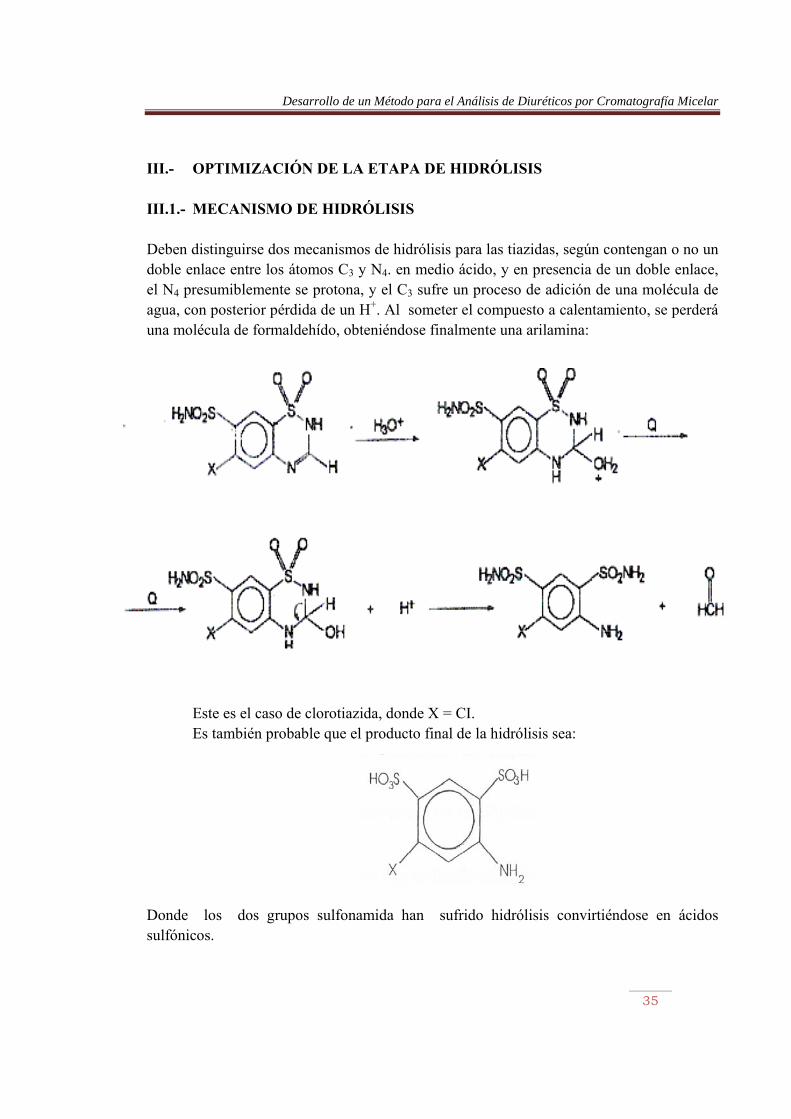
III.- OPTIMIZACIÓN DE LA ETAPA DE HIDRÓLISIS III.1.- MECANISMO DE HIDRÓLISIS Deben distinguirse dos mecanismos de hidrólisis para las tiazidas, según contengan o no un doble enlace entre los átomos C3 y N4. en medio ácido, y en presencia de un doble enlace, el N4 presumiblemente se protona, y el C3 sufre un proceso de adición de una molécula de agua, con posterior pérdida de un H+. Al someter el compuesto a calentamiento, se perderá una molécula de formaldehído, obteniéndose finalmente una arilamina:
Este es el caso de clorotiazida, donde X = CI. Es también probable que el producto final de la hidrólisis sea:
Donde los dos grupos sulfonamida han sufrido hidrólisis convirtiéndose en ácidos sulfónicos.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
36
Cuando existe un sustituyente R en el C3, el mecanismo de hidrólisis puede ser similar al de los acetales: donde X = Cl para los diuréticos del Grupo I y X = CF3 para los diuréticos del Grupo II (ver Tabla 3). Por otra parte, el mecanismo de hidrólisis de la furosemida puede ser el siguiente:

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
37
En cualquier caso, la arilamina formada puede ser diazotada y enlazada con N- (1-naftil) etilendiamina, para formar un azocolorantes (ver Esquema 1 en apartado I.2). III.2.- PRESENCIA DE TENSIOACTIVO EN LA HIDRÓLISIS En el estudio de optimización realizado con diversas arilaminas [29], se encontró que las concentraciones de SDS y HCI más adecuadas para llevar a cabo las reacciones de diazotación y acoplamiento, en un medio micelar, eran, 0.05 M y 0.15 M, respectivamente (apartado I.2.b). en el procedimiento desarrollado y aplicado al análisis de sulfonamidas, se preparaban las disoluciones de los analitos en ese medio. Inicialmente, se prepararon las disoluciones de diuréticos de la misma forma. EL SDS facilita la disolución de los diuréticos estudiados, razon por la cual se estudio la posibilidad de realizar la reaccion de hidrolisis en tensiactivo, sin embargo, se encontró un inconveniente, al someter las disoluciones de SDS a intenso calentamiento, éste probablemente sufre una descomposición, como parece indicarlo el aumento de viscosidad de las disoluciones al ser enfriadas. En ocasiones, incluso, se observó la formación de una sustancia gelatinosa, que sólo mediante un nuevo calentamiento se licuaba. Por ello, se optó por disolver e hidrolizar los diuréticos en ausencia del tensioactivo. III.3.- ESTUDIO DE LAS CONDICIONES DE HIDRÓLISIS Los diuréticos sufren hidrólisis en medio HCI, pero a temperatura ambiente esta hidrólisis es insignificante tal y como demuestra la baja absorbancia de sus disoluciones sometidas a derivatización a esta temperatura (ver Tabla 5). Debido a la lenta hidrólisis experimentada a baja temperatura se procedio calentar las disoluciones para acelerar este proceso. Para esto se optó por realizar un calentamiento a 100 ºC, con el fin de obtener los compuestos hidrolizados con la mayor rapidez posible. Por lo tanto, el único parámetro que se consideró que debía optimizarse fue la concentración de HCI.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
38
Tabla 5.- Absorbancia de los azocolorantes formados a partir de los diuréticos mantenidos a temperatura ambiente en medio HCI. Diurético � (nm)a Ab ACDS HCTA CTA TCM ALT HFM BENDRO FURO
532.4 532.4 534.8 533.5 531.6 528.4 525.2 537.2
1.604±0.002 0.012±0.001 0.0140±0.0008 0.084±0.0013 0.2634±0.0015 0.0029±0.0009 0.0529±0.0016 0.0532±0.0004
a Longitud de onda del máximo. b Valores medios de experiencias realizadas por triplicado.
El ACDS es el único diurético que origina una absorbancia apreciable, sin necesidad de ser sometido a hidrólisis, debido a que el compuesto no hidrolizado presenta ya la estructura de una arilamina. Se comprobó que las disoluciones de este diurético no resultan afectadas por un intenso calentamiento a 100ºC. La Figura 3 muestra la evolución de la absorbancia para el ACDS sometido a calentamiento a 100ºC, en medio HCI. La absorbancia del azocolorante formado, no sometiendo a calentamiento previo al diurético y calentando éste durante 60 min, fue de 0.54±0.003 y 0.562±0.004, respectivamente (Tabla 6). Es decir, se produjo un incremento en la absorbancia de tan sólo un 3%. Por lo tanto, el ACDS no es afectado por el fuerte calentamiento.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
39
Figura 3.- Evolución de la absorbancia del azocolorante de 4-amino-6-
clorobenceno-1,3-disulfonamida (ACDS), formado tras someter a calentamiento (100ºC) la disolución del diurético, en presencia de HCI
1) 0 . 15 M, 2) 0 . 3 M, 3) 0 . 5 M.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
40
Tabla 6.- Sensibilidad del ACDS frente al calentamiento.
Número de réplica Sin calentamiento Calentamientoa 1 2 3 4 5 6 7
0.5483 0.5419 0.5482 0.5406 0.5437 0.5456 0.5437
0.5549 0.5635 0.5620 0.5627 0.5670 0.5606 0.5627
Media 0.545±0.003 0.562±0.004
a Temperatura: 100ºC. El resto de diuréticos se sometieron también a calentamiento a 100ºC. Se utilizaron concentraciones de HCI 0.15 M, 0.3 M y 0.5 M. Se calentó durante tiempos variables, interrumpiendo la hidrólisis mediante enfriamiento, y se derivatizó el compuesto hidrolizado para formar los azocolorantes. Las Figuras 4 y 5 muestran el progreso de los procesos de hidrólisis de clorotiazida, triclorometiazida, hidroflumetiazida y bendroflumetiazida. Las absorbancias se midieron a la longitud de onda del máximo del espectro del azocolorante, obtenido tras hidrolizar el diurético durante 10 min. Se puede observar que al incrementarse la concentración de HCI se aceleró la hidrólisis, pero en ningún caso parece haberse completado este proceso al cabo de 90 min.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
41
Figura 4.- Evolución de la absorbancia del azocolorante de: a) Clorotiazida y b) triclorometiazida, formado tras someter a calentamiento (100 ºC) la disolución del diurético, en presencia de HC1: 1) 0 .15 M, 2) 0 . 3 M, 3) 0 . 5 M.
a

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
42
Figura 5.- Evolución de la absorbancia del azocolorante de: a) Hidroflumetiazida y b) bendroflumetiazida, formado tras someter a calentamiento (100 ºC) la disolución del
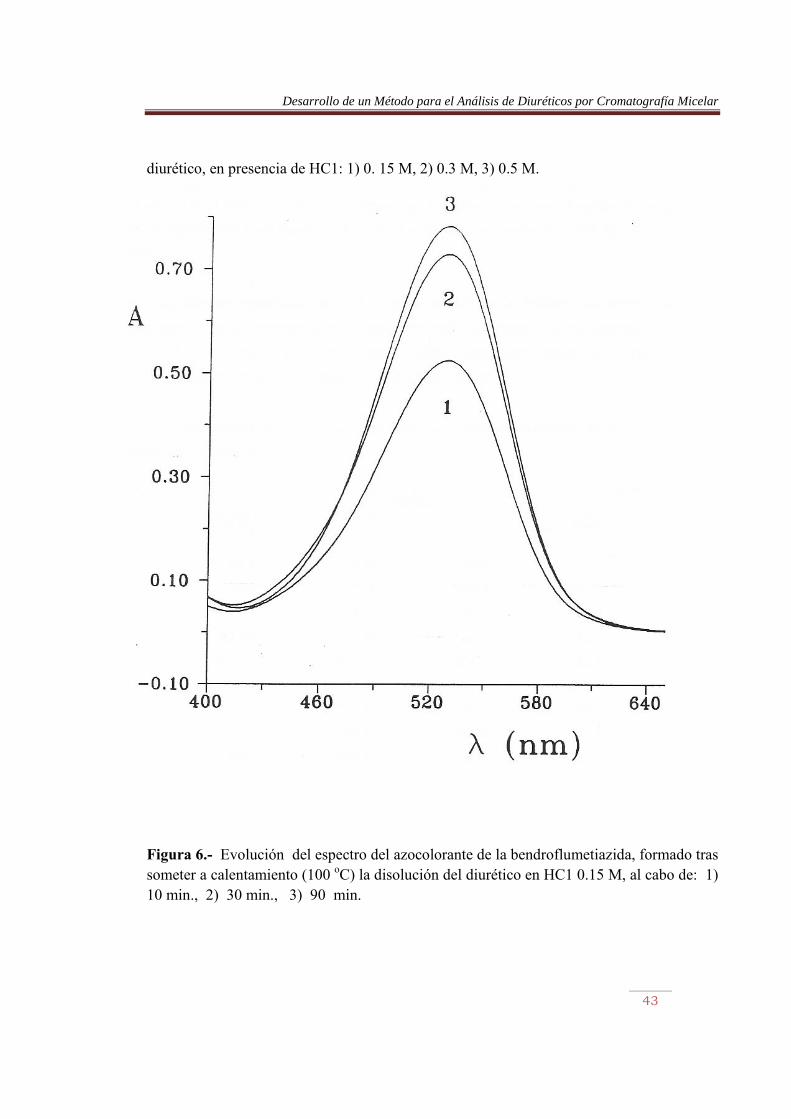
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
43
diurético, en presencia de HC1: 1) 0. 15 M, 2) 0.3 M, 3) 0.5 M.
Figura 6.- Evolución del espectro del azocolorante de la bendroflumetiazida, formado tras someter a calentamiento (100 oC) la disolución del diurético en HC1 0.15 M, al cabo de: 1) 10 min., 2) 30 min., 3) 90 min.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
44
La Figura 6 muestra la evolución del espectro del azocolorante de la bendroflumetiazida formado tras someter el diurético a diversos tiempos de calentamiento en HCI 0.15 M. El máximo de absorción formado a 528.4 nm se ve incrementado, sin sufrir prácticamente desplazamiento. Lo mismo se observó con el resto de diuréticos examinados. La Tabla 7 muestra la longitud de onda del máximo del espectro del azocolorantes formado, al someter a los diuréticos a distintos tiempos de calentamiento. Para no alargar excesivamente la etapa inicial de hidrólisis, se optó por someter los diuréticos a calentamiento durante 60 min. Se eligió una concentración de HCI 0.15 M, debido a que ésta es la adecuada para llevar a cabo la derivatización, y no es necesario realizar un cambio de pH. Tabla 7.- Desplazamiento de la longitud de onda (nm) de los azocolorantes de los diuréticos sometidos a hidrólisis. t (min)a Diurético ACDS HCTA CTA TCM 0 10 20 30 40 50 60 70 80 90 100
533.2 532.4 532.4 532.4 531.6 532.4 532.4 531.6
532.8 532.8 532.8 532.0 532.3 532.0 532.0 532.8 532.8 531.2
538.4 530.4 531.2 533.6 534.4 532.0 530.4 532.8 531.2 532.8
531.2 533.6 532.0 531.2 532.8 531.2 533.6 532.8 531.2
a Tiempo de calentamiento a 100ºC.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
45
Tabla 7.- (continuación). t (min)a Diurético ALT HFM BENDRO FURO 10 15 20 30 40 45 50 60 70 75 80 90 100
532.4 532.4 532.4 532.4 532.4 532.4
528.8 528.8 528.8 528.8 528.8 528.8 528.8 528.8 528.8 528.8
528.4 529.2 529.2 530.0 529.2 529.2 528.4 529.2 529.2
537.2 536.4 536.4
a Tiempo de calentamiento a 100ºC Los tubos de hidrólisis, una vez terminado el calentamiento, se sumergieron en baño de agua a temperatura ambiente. Se efectuó una serie de experiencias para estudiar la influencias del tiempo de enfriamiento sobre la estabilidad de los diuréticos hidrolizados y por lo tanto, sobre la absorbancia de los azocolorantes. En la Tabla 8 se indican los valores de absorbancia de los azocolorantes formados a partir de los diuréticos hidrolizados. Los tiempos indicados corresponden al período que transcurre entre el inicio del enfriamiento y el inicio de la derivatización. Tabla 8.- Influencia del tiempo de enfriamiento.
Diurético t (min)a HCTA BENDRO FURO 2 5 15 30 45 60
0.356 0.357 0.361 0.356 0.359 0.357
0.321 0.330 0.320 0.339 0.327 0.322
0.483 0.486 0.481 0.491 0.479
Los valores de absorbancia indican que los diuréticos hidrolizados son suficientemente estables y no precisan ser derivatizados inmediatamente después de ser hidrolizados.
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
46
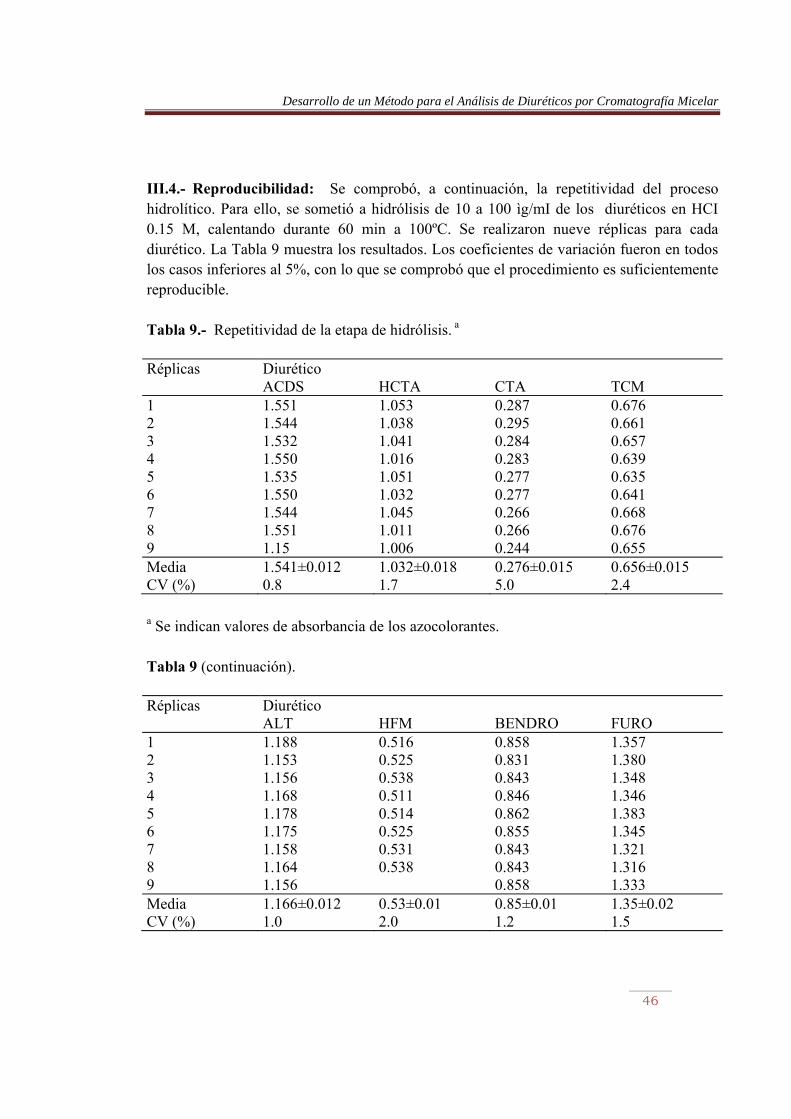
III.4.- Reproducibilidad: Se comprobó, a continuación, la repetitividad del proceso hidrolítico. Para ello, se sometió a hidrólisis de 10 a 100 ìg/mI de los diuréticos en HCI 0.15 M, calentando durante 60 min a 100ºC. Se realizaron nueve réplicas para cada diurético. La Tabla 9 muestra los resultados. Los coeficientes de variación fueron en todos los casos inferiores al 5%, con lo que se comprobó que el procedimiento es suficientemente reproducible. Tabla 9.- Repetitividad de la etapa de hidrólisis. a
Réplicas Diurético ACDS HCTA CTA TCM 1 2 3 4 5 6 7 8 9
1.551 1.544 1.532 1.550 1.535 1.550 1.544 1.551 1.15
1.053 1.038 1.041 1.016 1.051 1.032 1.045 1.011 1.006
0.287 0.295 0.284 0.283 0.277 0.277 0.266 0.266 0.244
0.676 0.661 0.657 0.639 0.635 0.641 0.668 0.676 0.655
Media CV (%)
1.541±0.012 0.8
1.032±0.018 1.7
0.276±0.015 5.0
0.656±0.015 2.4
a Se indican valores de absorbancia de los azocolorantes. Tabla 9 (continuación). Réplicas Diurético ALT HFM BENDRO FURO 1 2 3 4 5 6 7 8 9
1.188 1.153 1.156 1.168 1.178 1.175 1.158 1.164 1.156
0.516 0.525 0.538 0.511 0.514 0.525 0.531 0.538
0.858 0.831 0.843 0.846 0.862 0.855 0.843 0.843 0.858
1.357 1.380 1.348 1.346 1.383 1.345 1.321 1.316 1.333
Media CV (%)
1.166±0.012 1.0
0.53±0.01 2.0
0.85±0.01 1.2
1.35±0.02 1.5

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
47
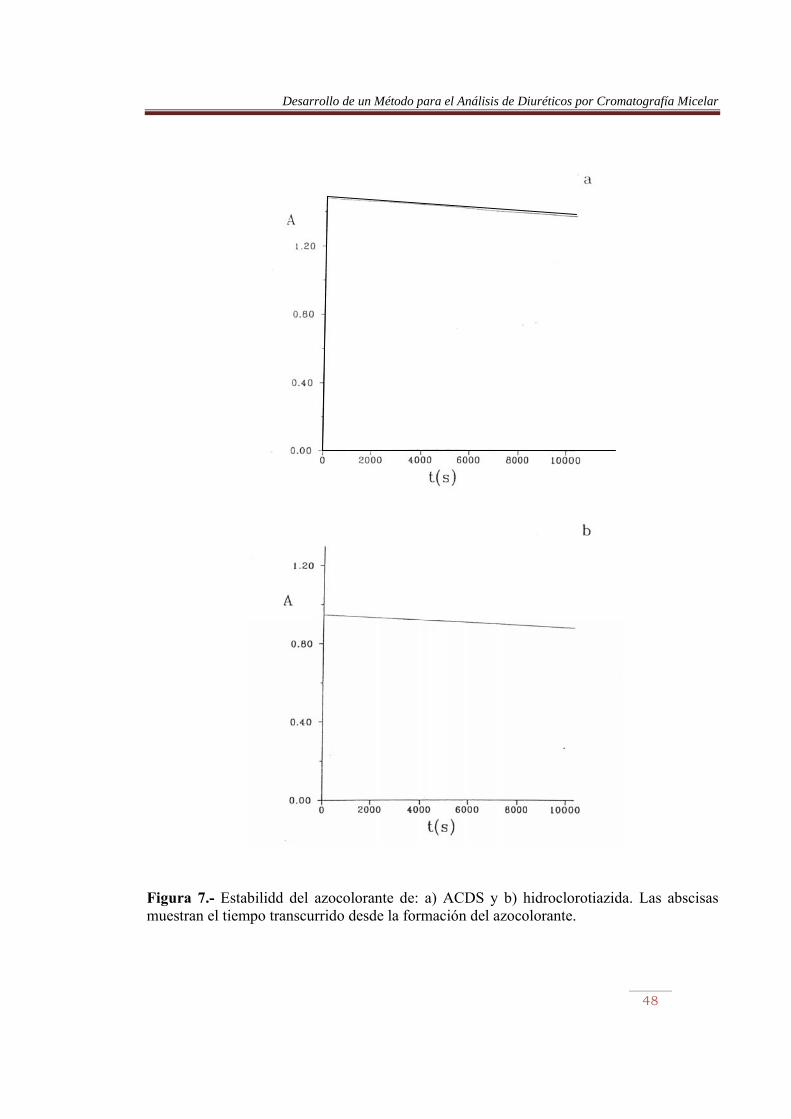
IV.- REACCIÓN DE DIAZOTACIÓN Y ACOPLAMIENTO IV.1.- ESTABILIDAD DE LOS AZOCOLORANTES Las condiciones experimentales óptimas (pH, concentración de reactivos y tiempo de reacción) para derivatizar una arilamina mediante diazotación y acoplamiento, en un medio micelar de SDS, han sido ampliamente estudiadas [29-31], por lo que no se abordó este estudio, y se utilizaron las condiciones recomendadas en la bibliografía. Sin embargo, se consideró interesante examinar la estabilidad de los azocolorantes formados con los productos de hidrólisis de los diuréticos. Las experiencias se llevaron a cabo con ACDS, hidroclorotiazida, bendroflumetiazida y furosemida. Se siguió la evolución de la absorbancia de los azocolorantes con el tiempo, durante un período de aproximadamente 3 h (Figuras y 8). El porcentaje de descomposición al cabo de las 3 h fue el 7.4%, 7.4%, 9.6% y 11.6% para ACDS, hidroclorotiazida, bendroflumetiazida y furosemida, respectivamente. Estos resultados indican que se produce una ligera descomposición de los derivados, por lo que aunque no es necesario inyectarlos en el cromatógrafo inmediatamente después de su formación, no deben conservarse durante largos períodos de tiempo.
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
48
Figura 7.- Estabilidd del azocolorante de: a) ACDS y b) hidroclorotiazida. Las abscisas muestran el tiempo transcurrido desde la formación del azocolorante.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
49
Figura 8.- Estabilidad del azocolorante de: a) Bendroflumetiazida y b) furosemida. Las abscisas muestran el tiempo transcurrido desde la formación del azocolorante.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
50
IV.2.- NATURALEZA DE LOS AZOCOLORANTES FORMADOS El proceso hidrolítico para los diuréticos que poseen una estructura básica común, probablemente origina un mismo azocolorantes, tal como se mostró en el apartado IV.1. por ello, finalmente, la hidrólisis de los diuréticos estudiados tan sólo da lugar a tres compuestos distintos: los diuréticos del grupo I originan el azocolorante XVII, los del grupo II dan lugar a la estructura XVIII y la furosemida a la XIX (ver también Tabla 3).
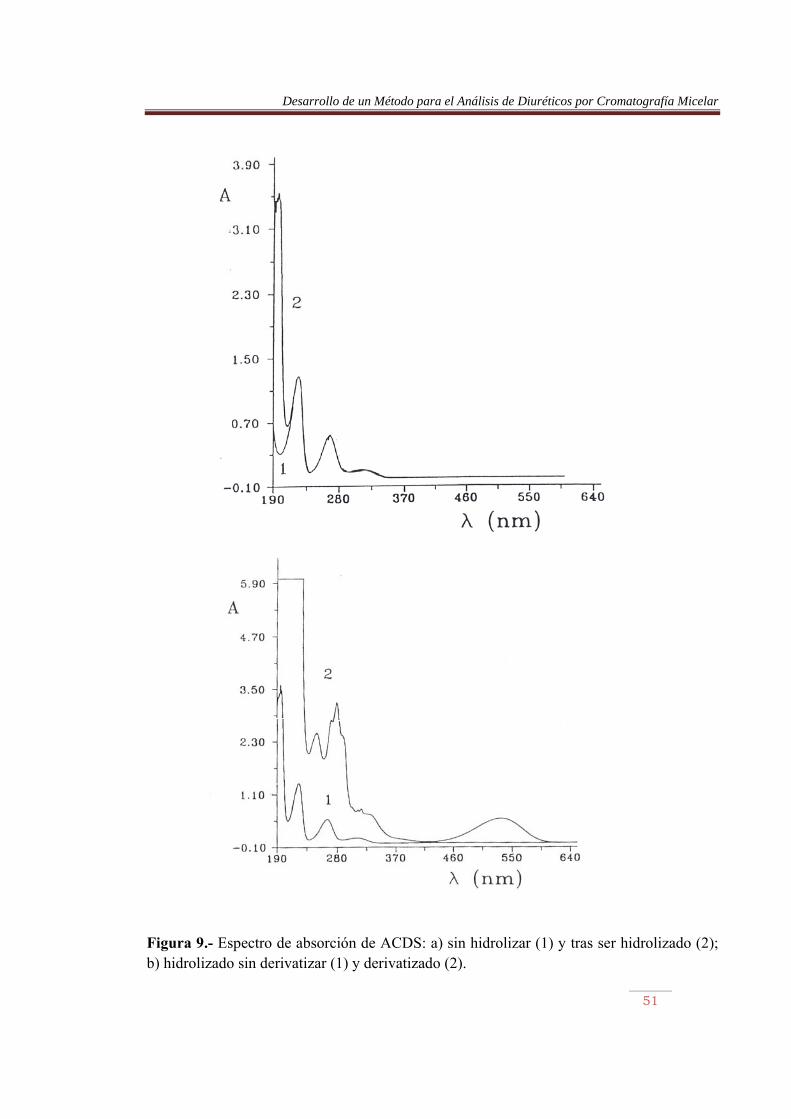
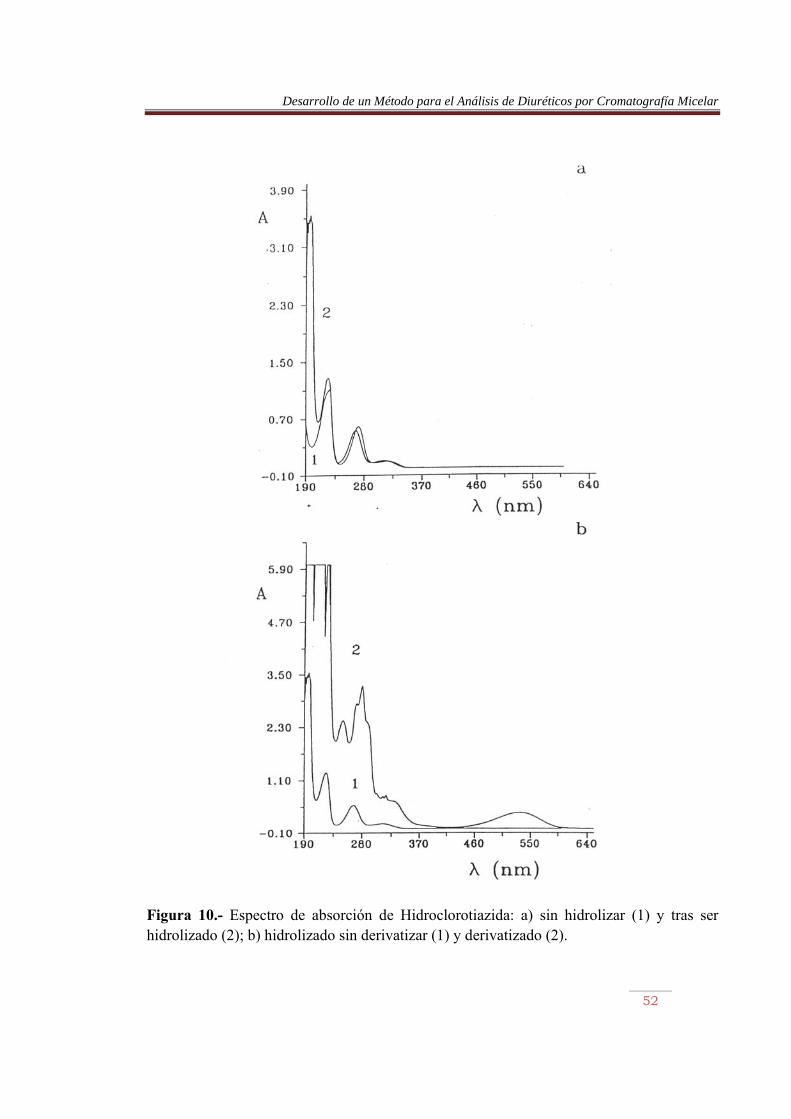
Se realizaron diversas experiencias, en las que se intentó comprobar la existencia de estos tres compuestos. En primer lugar, se obtuvieron los espectros de absorción de los azocolorantes y sus absortividades molares. En segundo lugar, se valoran los azocolorantes frente a NaOH, siguiendo el curso la volumetría ácido-base mediante medidas de absorbancia y pH, y por último, se obtuvo su comportamiento de retención con diversas fases móviles micelares. V.2.1) PRUEBAS DE ABSORCIÓN DE DIURETICOS Se obtuvieron los espectros de absorción de los diuréticos sin derivatizar y tras someterlos a hidrólisis, diazotación y acoplamiento. Los distintos espectros se muestran en las Figuras 9 a 16.
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
51
Figura 9.- Espectro de absorción de ACDS: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
52
Figura 10.- Espectro de absorción de Hidroclorotiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
53
Figura 11.- Espectro de absorción de clorotiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
54
Figura 12.- Espectro de absorción de triclorometiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
55
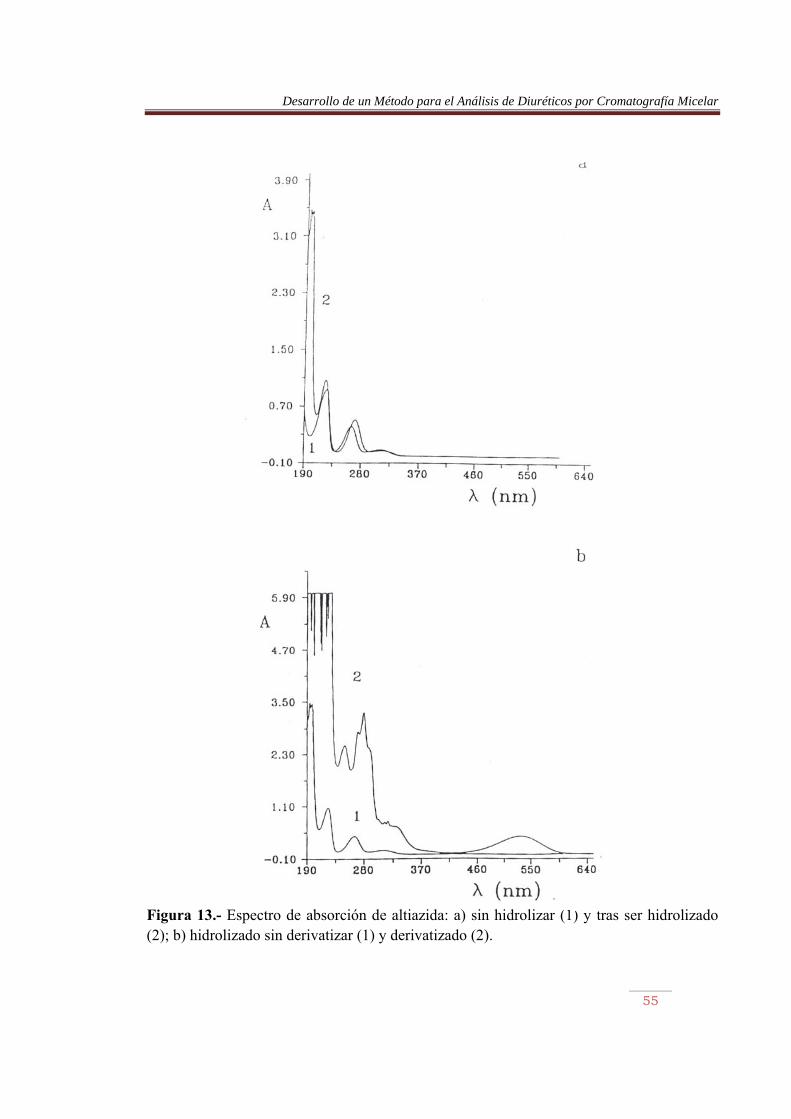
Figura 13.- Espectro de absorción de altiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
56
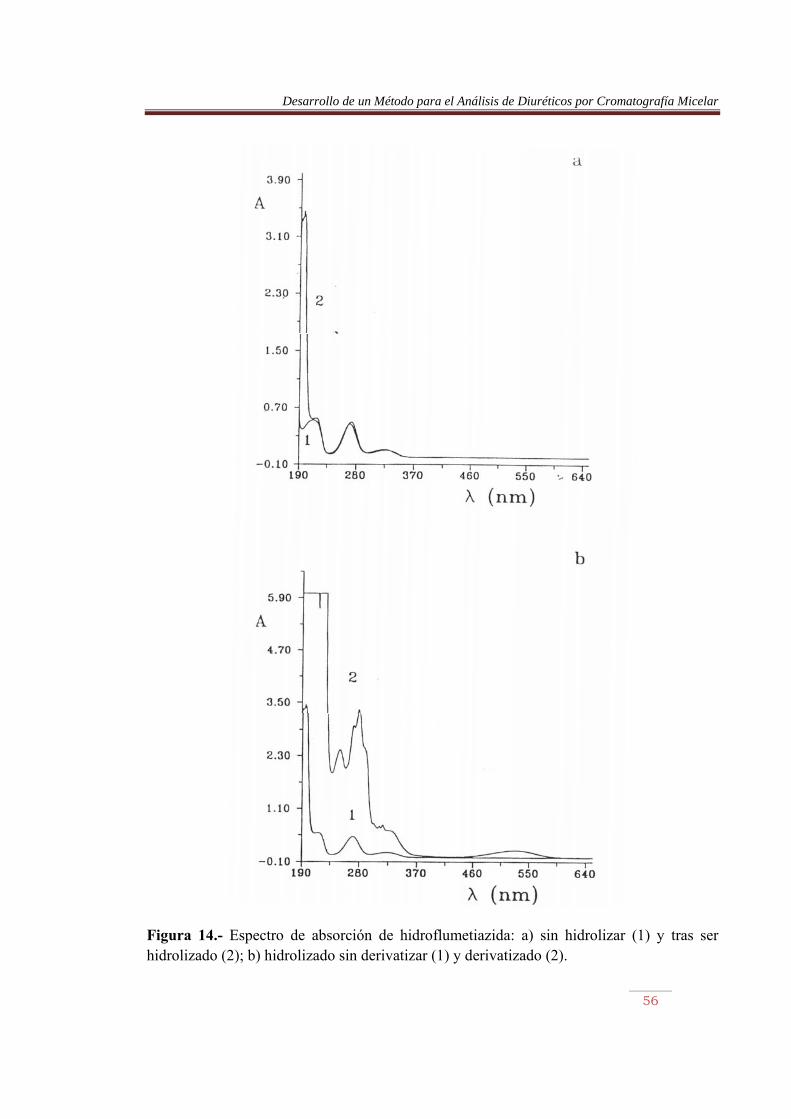
Figura 14.- Espectro de absorción de hidroflumetiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
57
Figura 15.- Espectro de absorción de bendroflumetiazida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
58
Figura 16.- Espectro de absorción de furosemida: a) sin hidrolizar (1) y tras ser hidrolizado (2); b) hidrolizado sin derivatizar (1) y derivatizado (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
59
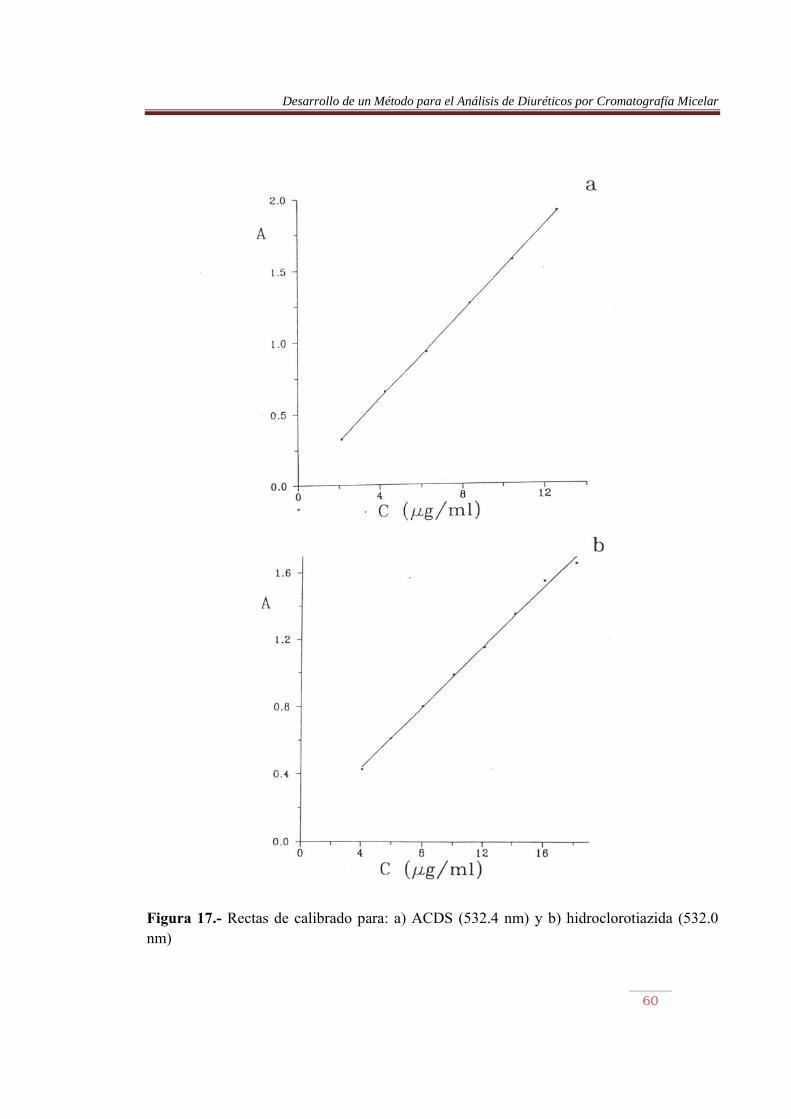
En las Figuras 9 a 16, parte a, se comparan los espectros de los diuréticos sin hidrolizar y tras ser sometidos a calentamiento. Se observa en este último caso la aparición de una nueva banda sobre 195 nm, permaneciendo el resto sin prácticamente desplazamiento. La furosemida mostró el mayor desplazamiento. Por otro lado, en las misma figuras, parte b, se comparan los espectros de los diuréticos hidrolizados, previamente y tras ser sometidos al proceso de derivatización. Se observan cambios importantes en los espectros, pero lo más llamativo es la aparición de una banda en la región del visible, entre 450 y 600 nm, aproximadamente. La detención de los azocolorantes se basa en la medida de la absorbancia de esta banda. En la Tabla 10 se muestran las longitudes de onda de máxima absorción de los azocolorantes en el visible, junto con su absortividad molar. Las Figuras 17 a 20 muestran las recetas de calibrado abtenidas. Tabla 10.- Longitud de onda, absortividad molar y coeficiente de regresión de los azocolorantes. Compuesto �máx (nm) (1 mol-1 cm-1) x10-4 r ACDS HCTA CTA TCM ALT HFM BENDRO FURO
532.4 532.0 532.0 531.2 533.2 528.8 529.2 536.4
4.26 2.67 0.57 3.46 4.08 1.49 3.22 3.88
0.99990 0.9987 0.9992 0.99990 0.9997 0.9990 0.9993 0.9996
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
60
Figura 17.- Rectas de calibrado para: a) ACDS (532.4 nm) y b) hidroclorotiazida (532.0 nm)

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
61
Figura 18.- Rectas de calibrado para: a) Clorotiazida (532.0 nm) y b) triclorometiazida (531.2 nm)
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
62
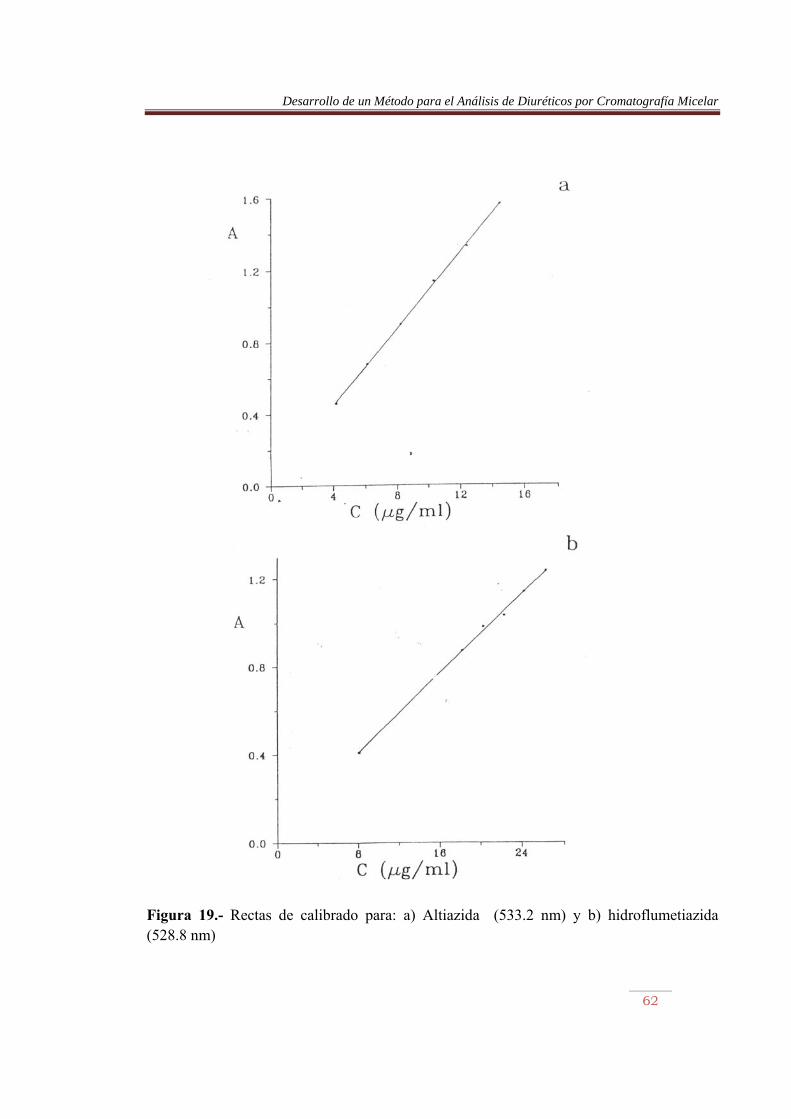
Figura 19.- Rectas de calibrado para: a) Altiazida (533.2 nm) y b) hidroflumetiazida (528.8 nm)

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
63
Figura 20.- Rectas de calibrado para: a) Bendroflumetiazida (529.2 nm) y b) furosemida (536.4 nm)

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
64
Se observa que la longitud de onda de los azocolorantes de hidroclorotiazida, clorotiazida, triclorometiazida y altiazida es prácticamente la misma, coincidiendo con la del ACDS (532.2±0.7nm), mientras con hidroflumetiazida (528.8 nm), y bendroflumetiazida (529.2nm) se obtienen valores similares entre sí. Por su parte, la furosemida presentó un valor de 536.4 nm. Sin embargo, las absortividades molares de los azocolorantes fueron muy distintas entre sí, incluso comparando las de los compuestos que por hidrólisis originaron, presumiblemente, un mismo producto, lo que puede indicar que el grado de avance de dicha reacción, o porcentaje de producto formado que puede originar el azocolorantes, difiere de un diurético a otro. Así, comparando la absortividad molar de hidroclorotiazida, clorotiazida, triclorometiazida con la del ACDS, se obtiene una disminución en la absortividad molar del 37%, 87%, 19% y 4%, respectivamente. Por su parte, la absortividad molar de la hidroflumetiazida es aproximadamente la mitad de la bendroflumetiazida. IV.2.2) Comportamiento ácido-base de los azocolorantes Para obtener las constantes de protonación de los azocolorantes , se realizaron valoraciones ácido-base con NaOH, siguiendo la absorbancia a la longitud de onda del máximo, y el pH de las disoluciones. La disolución de azocolorantes se preparó mezclando 25 mI de una disolución del diurético de 100: g/ml y 75 mI de SDS 0.4 M, conteniendo HCI 0.15 M. Se diazotó con 10 mI de nitrito sódico 0.15 M y transcurridos 5 min, se añadió 10 mI de ácido sulfámico 0.3 M; 10 min después de adicionó 5ml de NED 0.03 M y 10 mI de HCI 2 M, y se aforó con agua a 250 ml. Se tomaron alícuotas de 50 ml de esta disolución, a las que se añadió 10 ml de un tampón universal, preparado con ácido acético 0.1 M, ácido fosfórico 0.1 M y tetraborato sódico decahidrato 0.1 M, y se valoraron con una disolución de NaOH 0.25 M en SDS 0.12 M. El tampón universal impidió que se produjeran variaciones bruscas de pH en algún punto de la curva de valoración. Las experiencias se realizaron por duplicado. A partir de los datos A-pH se obtuvieron las constantes de protonación de los azocolorantes. Para ello, la absorbancia se corrigió respecto a la dilución. La observación de las curvas de valoración indicó la presencia de dos equilibrios ácido-base consecutivo, con unas constantes de protonación bien diferenciadas. Ello permitió la determinación de cada constante por separado. De este modo, se ha podido utilizar un método matemático que supone la presencia de una única protonación, y que se detalla a continuación. La absorbancia de una disolución que contenga un sistema ácido monoprótico vendrá dada

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
65
por la siguiente ecuación:
Siendo 0 y 1 las absortividades molares de las formas básica y ácida del par, respectivamente, C la concentración total del sistema ácido-base y K la constante de protonación que se pretende determinar. En el caso en que 1 > 0 (Figura 21a), se cumple:
de la que se obtiene:
Por otro lado, si 0 > 1 (Figura 21b):
Y:
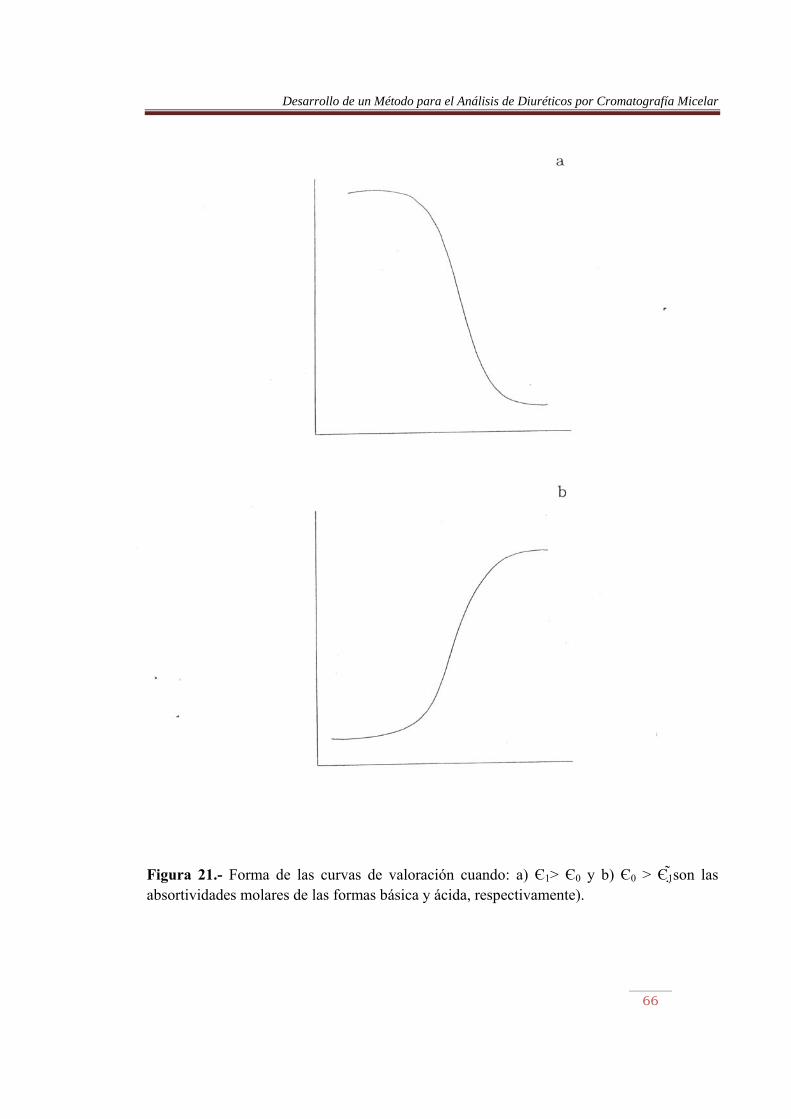
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
66
Figura 21.- Forma de las curvas de valoración cuando: a) Є1> Є0 y b) Є0 > Є1 son las absortividades molares de las formas básica y ácida, respectivamente).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
67
Las Ecuaciones (10) y (12) son ecuaciones lineales del tipo: Y = a + b x (13) Por lo que ajustando por mínimo cuadrados y frente a x, se obtendrá, en ambos casos, la constante de protonación a partir de la ordenada en el origen. Las Figuras 22 a 26 muestran curvas de valoración obtenidas con cada uno de los diuréticos. Las Figuras 27 a 30 muestran algunos de los espectros obtenidos a diversos valores de pH. Las Tablas 11 y 12 recogen los valores de las constantes de protonación obtenidos, con indicación de la calidad de los ajustes. La observación de los espectros de las Figuras 27 a 30 muestran su evolución al aumentar el pH. A pH débilmente ácido el compuesto presenta dos bandas, una sobre 450-460 nm y otra sobre 540-560 nm (ver parte a de las figuras). Al aumentar el pH dentro de la zona ácida de la escala de acidez, la primera banda de intensidad menor, crece, mientras que la banda a mayor longitud de onda disminuye. Ello produce la formación de un punto isobéstico sobre 480-490 nm. Un incremento posterior del pH invierte el proceso anterior, la banda a 540-560 nm crece y se desplaza ligermanete a menores longitudes de onda, mientras que la banda a 450-460 nm disminuye (ver parte b de las figuras). Las valoraciones se realizaron, por tanto, a una longitud de onda próxima a la de máxima absorción de la especie diprotonada del azocolorante y de la especie neutra.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
68
Figura 22.- Valoración ácido-base del azocolorante de ACDS (532.4 NM).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
69
Figura 23.- Valoración ácido-base del azocolorante de: a) Hidroclorotiazida (532 nm) y b) clorotiazida (532 nm).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
70
Figura 24.- Valoración ácido-base del azocolorante de: a) Triclorometiazida (531.2 nm) y b) altiazida (533.2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
71
Figura 25.- Valoración ácido - base del azocolorante de: a) Hidroflumetiazida (528.8 nm) y b) bendroflumetiazida (529.2 nm).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
72
Figura 26.- Valoración ácido-base del azocolorante de furosemida (536.4 nm).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
73
Figura 27.- Espectros de absorción del azocolorante de ACDS a diveresos valores de pH: a) 4.03 (1), 5.02 (2), 6.03 (3); b) 10.26 (1), 12.01 (2).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
74
Figura 28.- Espectros de absorción del azocolorante de triclorometiazida a diversos valores de pH: a) 3.40 (1), 4.00 (2), 5.27 (3), 7.01 (4); b) 10.04 (1), 11.06 (2), 12.00 (3)
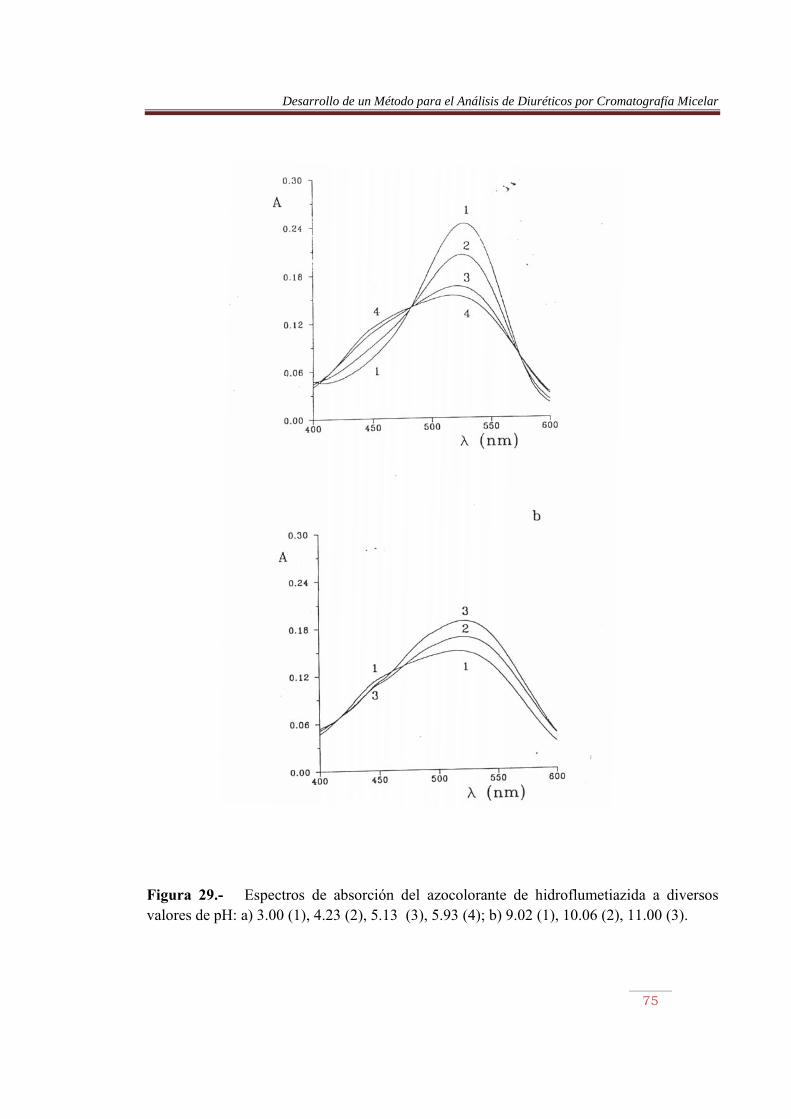
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
75
Figura 29.- Espectros de absorción del azocolorante de hidroflumetiazida a diversos valores de pH: a) 3.00 (1), 4.23 (2), 5.13 (3), 5.93 (4); b) 9.02 (1), 10.06 (2), 11.00 (3).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
76
Figura 30.- Espectros de absorción del azocolorante de furosemida a diversos valores de pH: a) 5.00 (1), 7.06 (2), 8.03 (3); b) 11.02 (1), 12.00 (2).
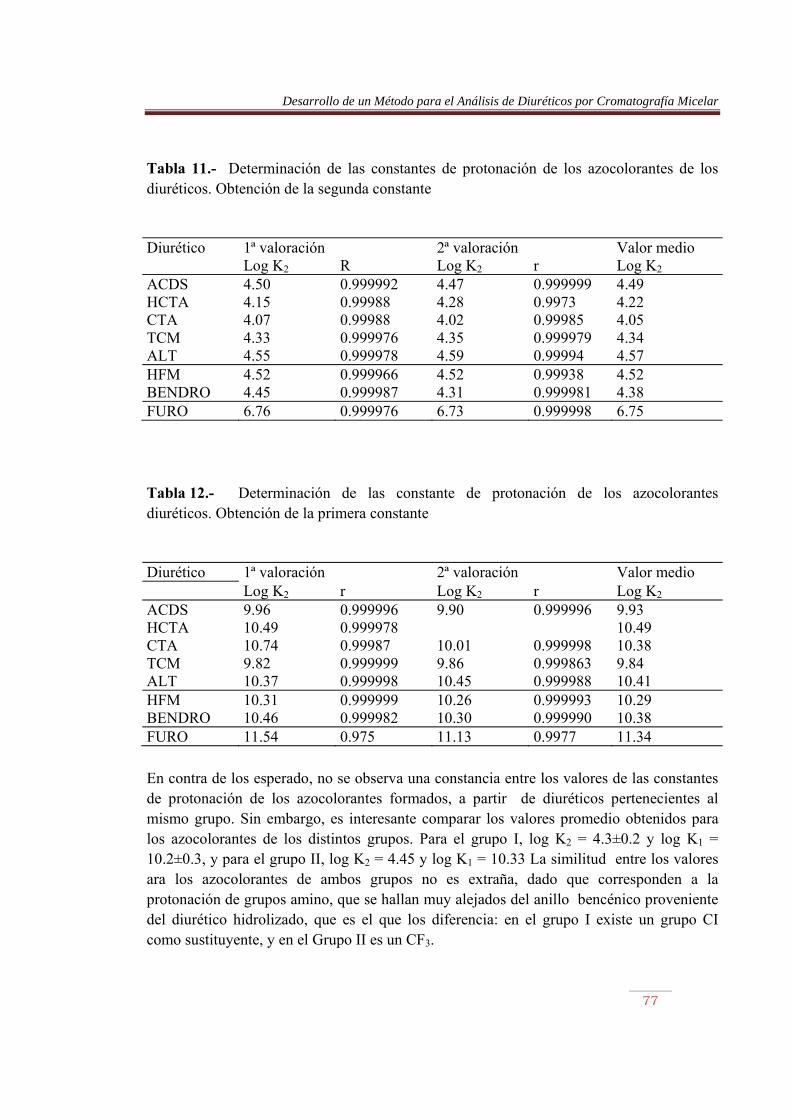
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
77
Tabla 11.- Determinación de las constantes de protonación de los azocolorantes de los diuréticos. Obtención de la segunda constante Diurético 1ª valoración 2ª valoración Valor medio Log K2 R Log K2 r Log K2 ACDS HCTA CTA TCM ALT
4.50 4.15 4.07 4.33 4.55
0.999992 0.99988 0.99988 0.999976 0.999978
4.47 4.28 4.02 4.35 4.59
0.999999 0.9973 0.99985 0.999979 0.99994
4.49 4.22 4.05 4.34 4.57
HFM BENDRO
4.52 4.45
0.999966 0.999987
4.52 4.31
0.99938 0.999981
4.52 4.38
FURO 6.76 0.999976 6.73 0.999998 6.75 Tabla 12.- Determinación de las constante de protonación de los azocolorantes diuréticos. Obtención de la primera constante Diurético 1ª valoración 2ª valoración Valor medio Log K2 r Log K2 r Log K2 ACDS HCTA CTA TCM ALT
9.96 10.49 10.74 9.82 10.37
0.999996 0.999978 0.99987 0.999999 0.999998
9.90 10.01 9.86 10.45
0.999996 0.999998 0.999863 0.999988
9.93 10.49 10.38 9.84 10.41
HFM BENDRO
10.31 10.46
0.999999 0.999982
10.26 10.30
0.999993 0.999990
10.29 10.38
FURO 11.54 0.975 11.13 0.9977 11.34 En contra de los esperado, no se observa una constancia entre los valores de las constantes de protonación de los azocolorantes formados, a partir de diuréticos pertenecientes al mismo grupo. Sin embargo, es interesante comparar los valores promedio obtenidos para los azocolorantes de los distintos grupos. Para el grupo I, log K2 = 4.3±0.2 y log K1 = 10.2±0.3, y para el grupo II, log K2 = 4.45 y log K1 = 10.33 La similitud entre los valores ara los azocolorantes de ambos grupos no es extraña, dado que corresponden a la protonación de grupos amino, que se hallan muy alejados del anillo bencénico proveniente del diurético hidrolizado, que es el que los diferencia: en el grupo I existe un grupo CI como sustituyente, y en el Grupo II es un CF3.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
78
Las constantes de protonación para la furosemida mostraron valores muy distintos: log K2 = 6.75 y log K1 = 11.34. Esta diferenciación puede deberse a la sustitución de un grupo sulfonamida por un grupo carboxilo en el anillo bencénico del diurético hidrolizado, respecto a las moléculas de los diuréticos del Grupos I y II. Es interesante también, comparar los valores obtenidos con valores bibliográficos: la constante de protonación para la anilina es log K = 4.6 y para el etilamonio es log K = 10.6. Por otro lado, para el ácido sulfámico, log K = 1.0 y para el grupo sulfonamida, sustituyente de un anillo bencénico, es log K = 2. Para el ácido benzoico, log K = 4.2, valor similar al del anilio. IV.2.3) Comportamiento cromatográfico de los azocolorantes En el Capítulo VI se expone detalladamente el estudio sobre el comportamiento Cromatográfico de los azocolorantes. Aquí tan sólo se muestra en la Figura 31, un cromatograma que se ha obtenido con una fase móvil de SDS 0.05 M y 12.5% de propanol. El primer pico se obtiene con ACDS, hidroclorotiazida, clorotiazida, triclorometiazida y altiazida; el segundo pico corresponde a hidroflumetiazida y bendroflumetiazida; y el tercero a la furosemida. Con todas las fases móviles utilizadas se obtuvieron tan sólo estos tres picos.
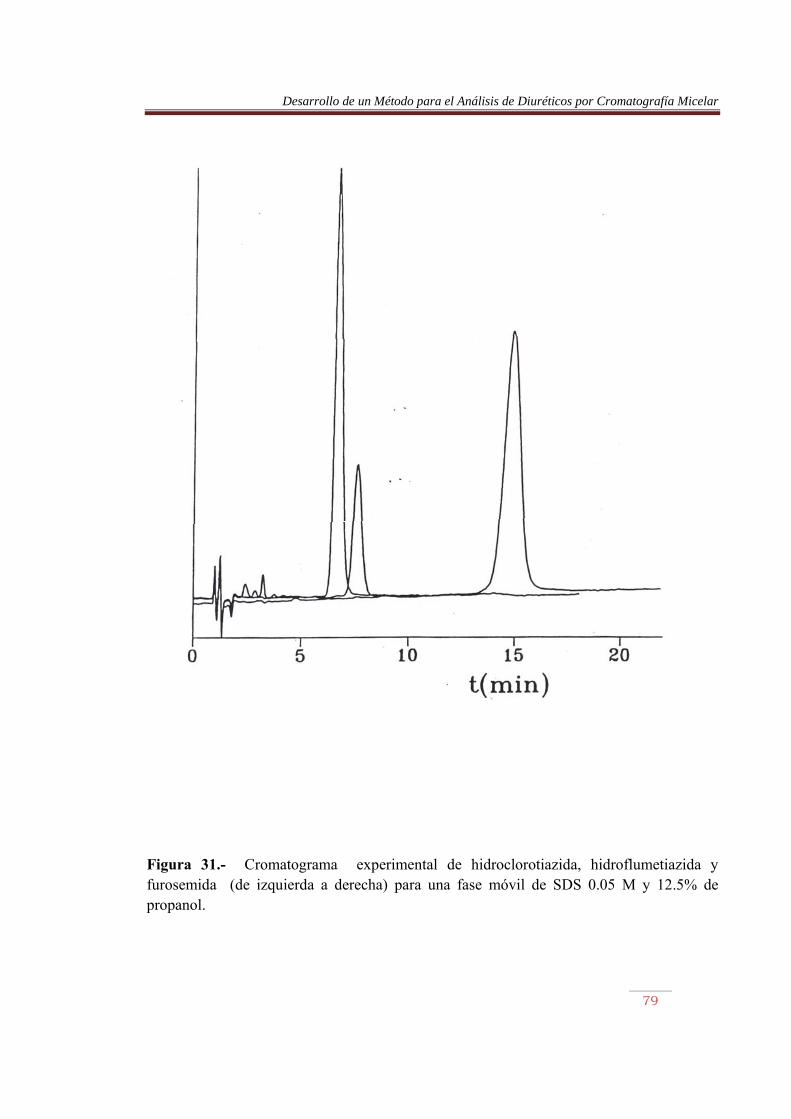
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
79
Figura 31.- Cromatograma experimental de hidroclorotiazida, hidroflumetiazida y furosemida (de izquierda a derecha) para una fase móvil de SDS 0.05 M y 12.5% de propanol.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
80
V.- DETERMINACIÓN CROMATOGRÁFICA DE DIURÉTICOS El objetivo principal de esta Tesina es la determinación de diuréticos en orina, mediante la formación de azocolorantes, de cloro-rojo-violeta. La derivatización de estos fármacos origina un aumento en la selectividad, cuando son analizados en muestras fisiológicas. A continuación, se muestran una serie de estudios realizados para encontrar la composición óptima de la fase móvil, que debe emplearse en estos análisis. V.1.- SELECCIÓN DEL PH Y DEL MODIFICADOR En el apartado V.2.b se obtuvieron las constantes de protonación de los azocolorantes. Las tiazidas mostraron valores de log K2 = 4-4.6 y log K1 = 9.8-10.4. Por su parte, las constantes de protonación de la furosemida fueron: log K2 = 6.8 y log K1 = 11.3. La Figura 32 muestra los diagramas de predominio lineal respecto del pH, para los sistemas ácido-base de azocolorante de tiazida y de furosemida. Figura 32.- Diagramas de predominio lineal de los azocolorantes.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
81
El intervalo de pH de trabajo, para una columna C18, es pH = 3-7. En este intervalo tiene lugar un equilibrio de protonación entre la especie diprotonada y la monoprotonada de las tiazidas y furosemida. A pH = 7 predominará la especie monoprotonada de las tiazidas, mientras que para la furosemida, la concentración de las especies diprotonada y monoprotonada es similar. Por otro lado, a pH = 3 predominará la especie dicargada tanto para las tiazidas como ara la furosemida. Se realizó una serie de experiencias en las que se inyectaron en el cromatógrafo los azocolorantes de los diuréticos hidrolizados y del ACDS, eluyendo con fases móviles de SDS y propanol a dos valores de pH: 3 y 7. En la Tabla 13 se comparan los valores de factor de capacidad, K´, eficacia, N, y factor de asimetría (B/A, medio al 10% de la altura del pico) obtenidos a ambos valores de pH). Se observa que la retención es mayor a pH = 3, lo que se debe, probablemente, a que la especie positiva dicargada del azocolorantes es atraída más fuertemente hacia la fase estacionaria modificada con el tensioactivo aniónico, que hacia la micela. Por otro lado, con frecuencia, la eficacia es también superior a pH = 3. Por todo ello, se eligió este valor de pH para la separación de los azocolorantes. Se constató en otra serie de experiencias que al utilizar fases móviles puras (conteniendo sólo el tensioactivo), la retención de los diuréticos derivatizados era excesiva, con tiempos de retención del orden de los 30 min (Tabla 14). Por ello, se consideró necesaria la adición de un modificador para aumentar la fuerza eluyente de la fase móvil. Para seleccionar el modificador más adecuado, se realizaron series de experiencias en las que se utilizó los alcoholes: propanol, butanol y pentanol. Las Tablas 14 y 15 recogen, los parámetros cromatográficos para propanol y butanol, respectivamente. Debe señalarse que la columna utilizada en esta serie y siguiente no es la misma que la empleada en la serie anterior del estudio del pH.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
82
Tabla 13.- Parámetros cromatográficos a pH = 3 y pH = 7 Compuesto Composición fase móvil pH 3 7 Componente Composición SDS (M) 0.05 0.05 Propanol (%) 5 5 K´ N B/A K´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
15.0 14.5 14.7 13.9 14.7 17.3 18.1 58.3
1150 1100 1100 100 1120 770 855
1.0 1.1 1.0 1.1 1.0 0.8 1.1
7.6 7.5 7.8 7.5 7.5 8.6 8.6 7.4
700 900 1100 1000 900 900 900 100
0.9 0.9 1.0 1.0 1.0 1.0 1.0 1.1
SDS (M) 0.05 0.05 Propanol (%) 10 10 K´ N B/A K´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
9.4 9.6 9.4 9.9 9.5 11.8 11.4 28.9
1700 1700 980 1500 837 1808 1600 1500
1.0 1.1 1.4 1.1 1.4 1.1 1.1 1.1
5.05 4.97 5.1 5.0 5.1 5.8 5.6 8.3
1500 1400 1500 1500 1500 1600 3000 1600
1.0 1.0 1.1 1.0 1.0 1.0 0.9 1.0
SDS (M) 0.1 0.1 Propanol (%) 7.5 7.5 K´ N B/A K´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
5.7 5.75 5.6 5.5 5.4 6.4 6.6 17.6
744 990 921 914 480 800 818 528
0.9 1.1 1.1 1.1 1.6 1.0 1.1 1.0
3.3 3.4 3.15 3.5 3.4 4.28 3.4 5.2
1000 1100 1000 1300 1000 1000 800 900
1.0 1.2 1.2 1.0 1.0 1.0 0.9 1.2

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Tabla 14.- Influencia de la concentración de propanol sobre la retención forma del pico . Compuesto Composición fase móvil
Componente Concentración SDS (M)
Propanol (%) 0.05 0
0.05 2.5
0.05 5
k´ N B/A k´ N B/A k´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
28.1 27.7 28.4 27.8 36.6 35.5
226 179 111 202 168 90
1.1 1.3 1.4 1.1 1.02 0.96
16.8 17.0 17.1 16.9 16.8 21.1 60.4
634 569 599 596 665 406 77
1.1 1.2 0.91 1.1 1.1 1.1 1.0
13.3 13.3 13.2 13.2 13.3 15.9 15.9 38.8
970 1014 994 1049 970 782 843 296
1.0 1.1 1.1 1.1 1.1 1.1 1.0 1.2
SDS (M) Propanol (%)
0.05 7.5
0.05 10
0.05 12.5
k’ N B/A k´ N B/A k´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
10.8 10.7 10.7 10.8 10.8 12.5 12.5 27.6
1242 1283 1314 1221 839 1335 1224 691
1.0 1.1 1.1 1.1 1.3 1.2 1.1 1.2
8.1 8.0 8.1 8.1 8.1 9.2 9.2 18.8
1502 1539 1504 1510 1499 1435 1356 1440
1.1 1.1 1.1 1.0 1.1 1.1 1.2 1.0
6.7 6.6 6.6 6.6 6.6 7.5 7.5 15.4
1476 1530 1462 1440 1520 1452 1386 1814
1.0 1.1 1.1 1.1 1.1 1.1 1.1 0.6
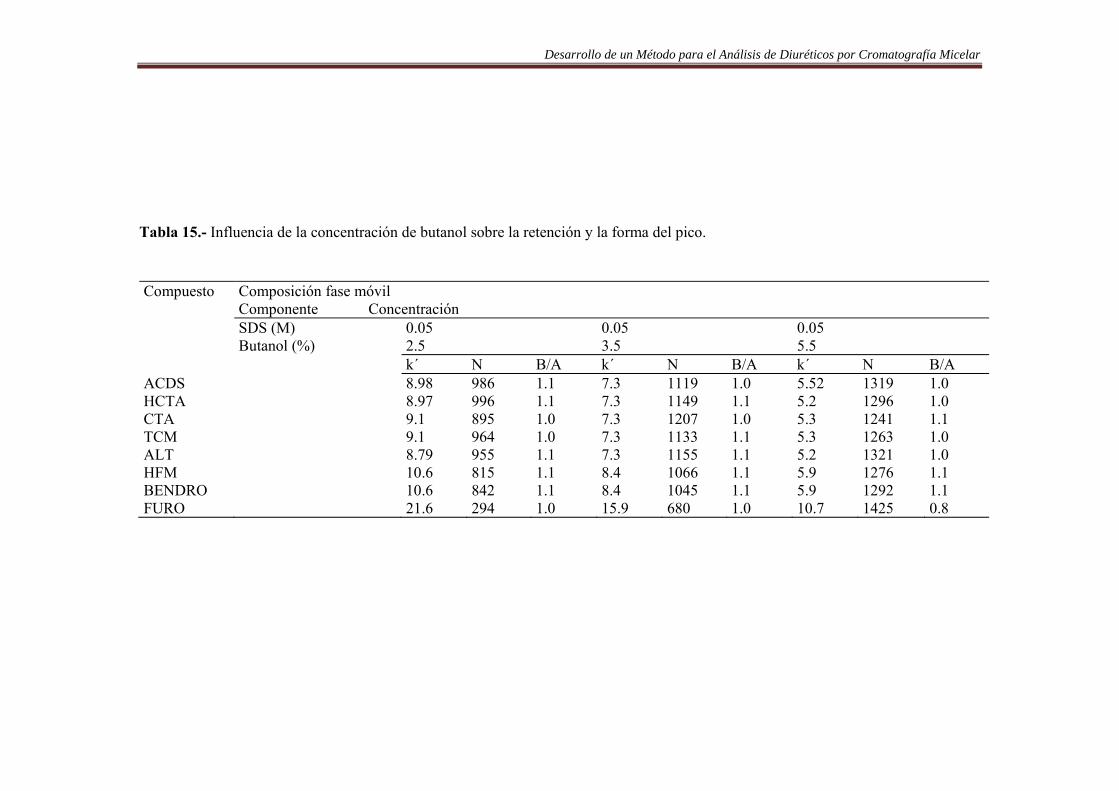
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Tabla 15.- Influencia de la concentración de butanol sobre la retención y la forma del pico. Compuesto Composición fase móvil
Componente Concentración SDS (M)
Butanol (%) 0.05 2.5
0.05 3.5
0.05 5.5
k´ N B/A k´ N B/A k´ N B/A ACDS HCTA CTA TCM ALT HFM BENDRO FURO
8.98 8.97 9.1 9.1 8.79 10.6 10.6 21.6
986 996 895 964 955 815 842 294
1.1 1.1 1.0 1.0 1.1 1.1 1.1 1.0
7.3 7.3 7.3 7.3 7.3 8.4 8.4 15.9
1119 1149 1207 1133 1155 1066 1045 680
1.0 1.1 1.0 1.1 1.1 1.1 1.1 1.0
5.52 5.2 5.3 5.3 5.2 5.9 5.9 10.7
1319 1296 1241 1263 1321 1276 1292 1425
1.0 1.0 1.1 1.0 1.0 1.1 1.1 0.8

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
85
De la observación de las Tablas 14 y15, se hace evidente el mayor poder eluyente del butanol respecto al propanol. Así, por ejemplo, para hidroclorotiazida, k´= 17.0, para una fase móvil de SDS 0.05 M y 2.5% de propanol, y k´= 9.0, para una fase móvil de SDS 0.05 M y 2.5$ de butanol. Además, se obtuvieron los datos de retención con una fase móvil de SDS 0.05 M y 2% de pentanol. Los valores de factor de capacidad fueron para hidroclorotiazida, altiazida, bendroflumetiazida, y furosemida: k´ = 0.07, 6.03, 7.15 y 13.2, respectivamente. El pentanol modifica excesivamente la retención, incluso a concentraciones bajas del mismo, mientras que propanol y butanol permiten un mayor control de la fuerza eluyente. Se decidió continuar las experiencias con propanol, que es el alcohol más usualmente utilizado en Cromatografía Líquida Micelar. V.2.- Influencia de la concentración del tensioactivo y del modificador El comportamiento eluyente de los azocolorantes es el usual: un incremento en la concentración de SDS produce una disminución en la retención (Tabla 16), junto con un empeoramiento de la eficacia. Nos se observa un deterioro especial de la asimetría de los picos cromatográficos. Por otro lado, un aumento en la concentración del alcohol origina una disminución de la retención, junto con una mejora de la eficacia (Tabla 14). Sin embargo, debe resaltarse el gran poder eluyente de ambos, tensioactivo y modificador, para estos compuestos. Así, para la hidroclorotiazida, k´ = 44.0 en una fase móvil de SDS 0.02 M y 2.5% de propanol, que disminuyó hasta un valor de k´ = 7.7, cuando la concentración del tensioactivo se incrementó a 0.1 M. Para el mismo compuesto, k´= 27.7, para una fase móvil de SDS 0.05 M, sin modificador, que disminuyó hasta un valor de k´ = 6.6, cuando la concentración del modificador se incrementó hasta el 12.5%.
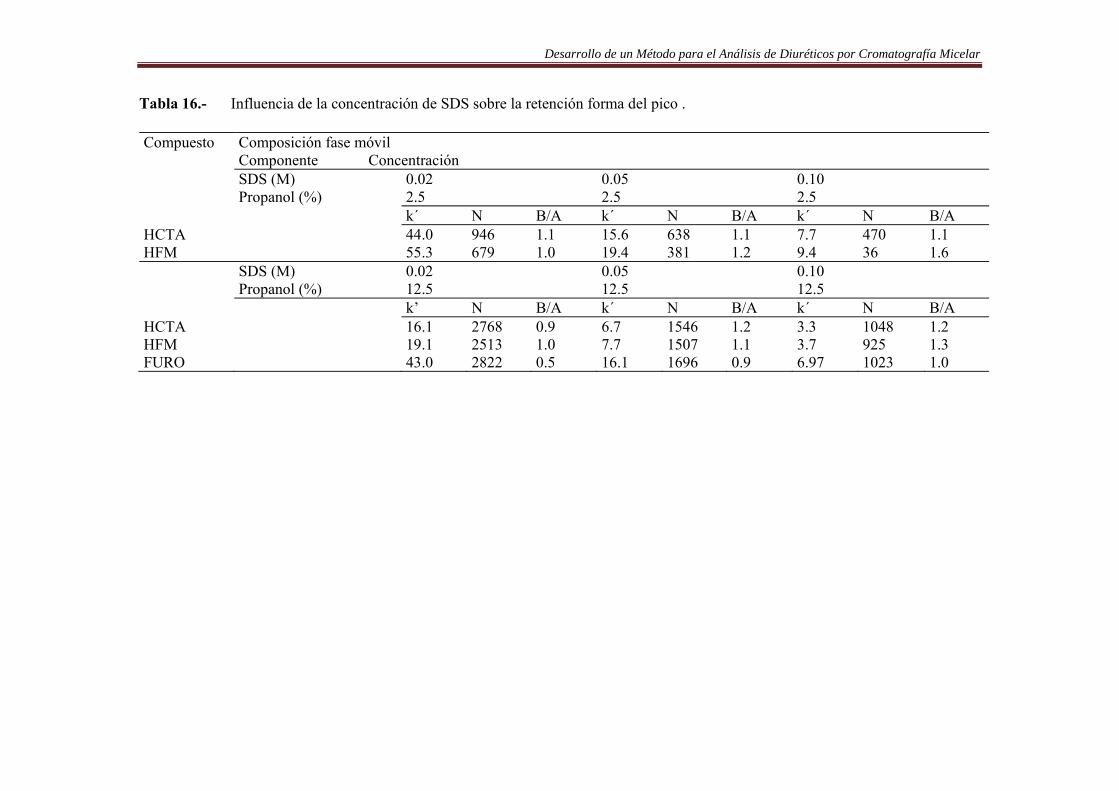
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Tabla 16.- Influencia de la concentración de SDS sobre la retención forma del pico . Compuesto Composición fase móvil
Componente Concentración SDS (M)
Propanol (%) 0.02 2.5
0.05 2.5
0.10 2.5
k´ N B/A k´ N B/A k´ N B/A HCTA HFM
44.0 55.3
946 679
1.1 1.0
15.6 19.4
638 381
1.1 1.2
7.7 9.4
470 36
1.1 1.6
SDS (M) Propanol (%)
0.02 12.5
0.05 12.5
0.10 12.5
k’ N B/A k´ N B/A k´ N B/A HCTA HFM FURO
16.1 19.1 43.0
2768 2513 2822
0.9 1.0 0.5
6.7 7.7 16.1
1546 1507 1696
1.2 1.1 0.9
3.3 3.7 6.97
1048 925 1023
1.2 1.3 1.0

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
87
En las Tablas 14 y 15 se puede constatar que los ocho diuréticos, tras el tratamiento de hidrólisis, originan tan sólo tres compuestos (apartado IV.1 y Tabla 3 en apartado III.1), ya que los valores de factor de capacidad, para fases móviles de muy distinta composición, son iguales dentro de cada grupo de diuréticos. V.3.- OPTIMIZACIÓN DE LA FASE MÓVIL V.3.1) INTRODUCCIÓN Uno de los problemas que debe resolver rutinariamente el cromatografista es la obtención de la fase móvil óptima, que le permita la separación de los compuestos que componen una mezcla, en un tiempo de análisis mínimo. Esta tarea puede ser compleja cuando en el problema intervienen dos o más variables. El proceso de optimización puede enfocarse de dos modos: secuencial o interpretativamente. En una estrategia secuencial, no se conocen a priori los datos de retención de los solutos analizados, y cada serie de fases móviles se diseña, teniendo en cuenta la retención observada al hacer uso de los eluyentes precedentes. Por el contrario, en una estrategia interpretativa, las experiencias se diseñan antes del proceso de optimización. En cromatografía líquida, una optimización interpretativa puede ser mucho más eficiente y fiable, pero para ser aplicada, se debe conocer previamente el comportamiento de retención de cada componente de la mezcla de solutos, a ser posible mediante una expresión matemática. Torres Lapasió et al., en diversas publicaciones han propuesto y demostrado que la Ecuación (7) (apartado I.3.a) describe adecuadamente la retención de los solutos cromatografiados con fases móviles de tensioactivo y modificador [40,44-47]. En base al estudio previo mostrado en el apartado V.2, se aplicó un proceso de optimización interpretativo, para obtener la fase móvil más adecuada para el análisis de los diuréticos en muestras de orina. V.3.2) PROCEDIMIENTOS DE OPTIMIZACIÓN El proceso de optimización consistió en las siguientes etapas
a) obtención de las ecuaciones de retención para cada soluto existente en la mezcla. b) Búsqueda de la fase móvil óptima, con la ayuda de los diagramas de curvas de nivel de
las funciones globales de resulución. c) c ) Simulación del cromatograma para la fase móvil óptima.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
88
ECUACIONES DE RETENCIÓN La Ecuación (7) puede se reescrita como:
Donde [M] y ϕ son la concentración de tensioactivos formando micelas y modificador, respectivamente. En lugar de la concentración micelar, es posible utilizar la concentración total de tensioactivo, sin incrementar el error de la predicción. CRITERIOS DE OPTIMIZACIÓN Se han empleado dos criterios de optimización: resolución posicional y grado de solapamiento (ver Figura 33 para explicación de los símbolos): El criterio de resolución posicional se base en la ecuación:
Siendo ti y ti+1 los tiempos de retención de los solutos i e i+1, y k´i y k´i+1 los correspondiente factores de capacidad. Por su parte, el criterio de grado de solapamiento viene dad por
donde Wi es el área total de un pico dado y W´i el área del mismo, solapada por la suma de todos los demás picos.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
89
Las dos funciones anteriores varían entre 0 y 1, indicando la proximidad a la unidad la bondad de la separación. Los productos normalizados de estas funciones individuales se utilizaron para describir la separación global de todos los picos del cromatograma:
donde Xi,i+1 es Si,i+1 para Oi, en la Ecuación (17), n-1 se sustituye por n. La función de resolución, r, se maximiniza para obtener la fase móvil óptima. Los diagramas de contorno (mapas de curvas de nivel o topogramas) de esta función se emplearon para encontrar la posición de los máximos y la forma de la superficie de respuesta en sus proximidades.
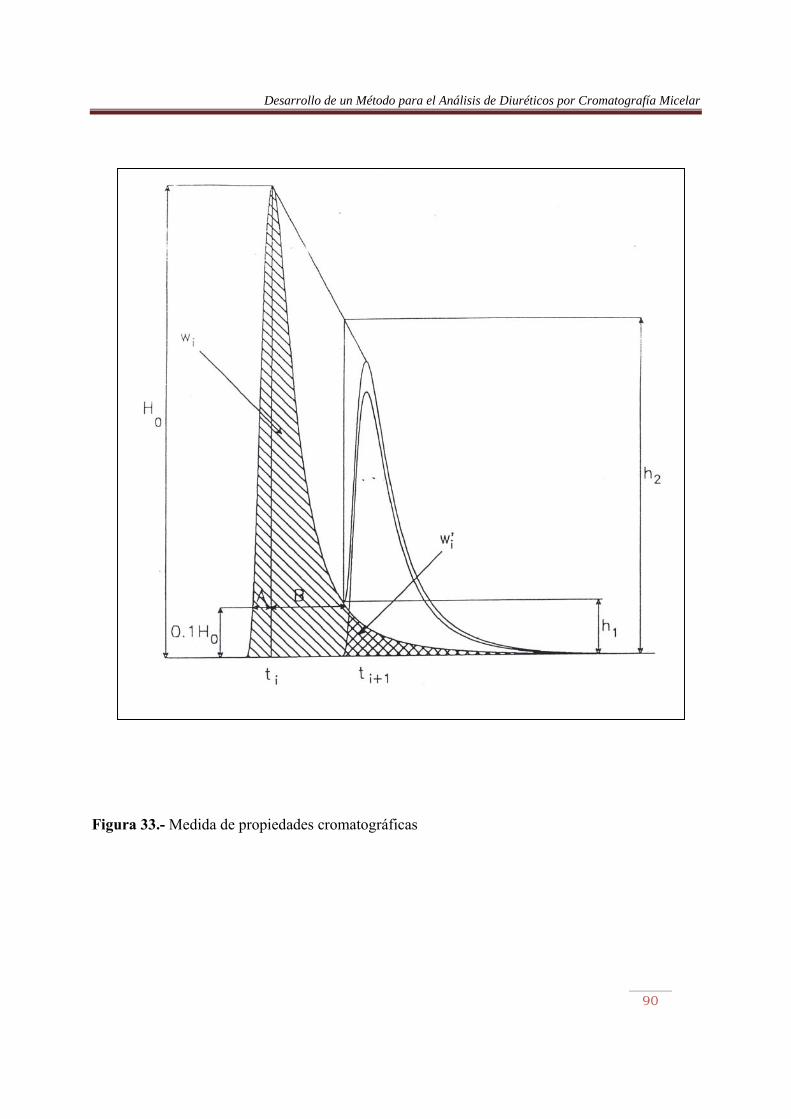
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
90
Figura 33.- Medida de propiedades cromatográficas

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
91
SIMULACIÓN DE LOS CROMATOGRAMAS Se simularon picos cromatográficos para poder utilizar el criterio de optimización de posición –forman (grado de soplamiento) y para comparar los cromatogramas experimentales con los reales. Para predecir y simular el perfil de cada pico., se hizo uso de los valores de eficacia y factor de asimetría, ajustando a un plano los valores correspondiente a las tres fases móviles más próximas a las fase móvil que se deseaba simular, y realizando una interpolación. Las áreas de los picos cromatográficos obtenidos se normalizaron. Se trazaron los picos cromatográficos simulados, haciendo uso de una función gaussiana asimétrica:
donde H0 es la altura del pico Cromatográfico para un soluto determinado y la función F (t-t0) no es constante, sino una función polinómica de la separación a la coordenada del máximo del pico, t0. según se ha demostrado, el uso de una función polinómica lineal aproxima aceptablemente la forma real del pico [48]:
Con esta función, el número de parámetros cromatográficos que definen un pico (eficacia y factor de asimetría) coincide con el número de coeficientes de la función h (t) (C0 y C1). Los pasos a seguir en la deducción de los coeficientes de la ecuación lineal, con fines de simulación son los siguientes:
1º) Obtención de k´0 (factor de capacidad del máximo) a partir de la ecuación de retención (14)).
2º) Interpolación de los valores de N y B/A de cada uno de los picos cromatográficos, a partir de los valores obtenidos con las fases móviles experimentales utilizadas en la modelización.
3º) Cálculo de la anchura de los picos (A+B) al 10% de la altura máxima, en unidades de
k´:

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
92
4º) Obtención de los valores individuales de A y B, a partir de la anchura al 10% de la
altura máxima, a, y del factor de asimetría:(21)
5º) La altura del pico normalizado puede obtenerse de forma rápida asumiendo un perfil triangular: H0 = E/a, donde E es una constante de normalización.
6º) Finalmente, los dos parámetros de la desviación estándar, expresados en unidades de k´, se obtendrían a partir de:
Se comprobó que los volúmenes muertos para las diferentes fases móviles estudiadas eran muy parecidos, por lo que cuando se simularon cromatogramas con las abscisas en unidades de tiempo, se utilizó siempre un valor de tiempo muerto común: el promedio obtenido, tM = 0.87 min.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
93
V.3.3) RESULTADOS Se obtuvieron las ecuaciones de retención de los azocolorantes en un número reducido y seleccionado de fases móviles de SDS y propanol. Para ello, se inyectó hidroclorotiazida, hidroflumetiazida y furosemida en la columna cromatográfica y se eluyó con cinco fases móviles, siguiendo el diseño de la Figura 34a. La Tabla 17 recoge los valores de factor de capacidad obtenidos, mientras que la Tabla 18 muestra el valor de los coeficientes de la Ecuación (14) ( ver los valores obtenidos para el intervalo de variación de SDS 0.05 – 0.1 M). El coeficiente de correlación de los ajustes fue siempre r > 0.99.
Figura 34.- Diseño experimentales. Para comprobar la bondad de la predicción, se obtuvieron los valores de factor de capacidad teórico (calculado con la ecuación de retención obtenida) y se compararon con los valores experimentales. La Tabla 17 muestra los errores de predicción calculados, que indican que el modelo describe adecuadamente la retención de los derivados, a pesar de que el error se incrementa en la zona central del espacio de los parámetros. El error se ha calculado según la siguiente ecuación:

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Taba 17.- Factores de capacidad correspondiente a las fases móviles del diseño de la Figura 34a, y errores obtenidos en
la modelización (Ecuación 14).
Compuesto Composición fase móvil Componente Concentración
SDS (M) Propanol (%)
0.05 2.5
0.05 12.5
0.075 7.5
k´exp k´calc (%) k´exp k´calc (%) k´exp k´calc (%) HCTA HFM FURO
15.6 19.4 32.5
15.9 19.7 34.2
-1.72 -1.53 -5.17
6.66 7.74 16.1
6.56 7.61 15.7
1.45 1.74 2.84
6.67 7.70 18.4
6.18 7.15 14.7
7.29 7.20 19.98
SDS (M)Propanol(%)
0.10 2.5
0.10 12.5
k´exp k´calc (%) k´exp k´calc (%) HCTA HFM FURO
7.70 9.42 28.5
7.8 9.5 29.7
-0.08 -0.69 -4.33
3.33 3.71 6.97
3.30 3.68 6.87
0.80 0.92 1.39

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Tabla 18.- Coeficientes de la Ecuación (14) para fases móviles de SDS y propanol, con indicación del coeficiente de regresión no lineal. Intervalo de concentración de SDS: 0.05 - 0.1 M Compuesto C1 C2 C3 C0 r HCTA HFM FURO
0.989 0.662 -0.298
0.00051 -0.00053 -0.0043
0.169 0.172 0.155
-0.0044 -0.0025 +0.036
0.998 0.998 0.991
Intervalo de concentración de SDS: 0.02-0.1 M Compuesto C1 C2 C3 C0 r HCTA HFM
0.930 0.698
0.00067 0.00021.
0.164 0.161
-0.0057 -0.0045
1.000 1.000
Intervalo de concentración de SDS: 0.02 – 0.05 M Compuesto C0 C1 C2 C3 r HCTA HFM
0.986 0.757
0.00083 0.00055
0.156 0.144
-0.0069 -0.0056
1.000 1.000

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
96
En la Figura 35 se han representado los valores de k´ experimental vs. K´ calculado para los tres azocolorantes. Las ecuaciones de retención, junto con los valores de eficacia y asimetría, se utilizaron para obtener los mapas de contorno de la función de resolución dada por la Ecuación (17), siguiendo el criterio posicional (Figuras 36 y 37) y de grado de solapamiento (Figuras 38 y 39). Debido a las eficacias relativamente bajas que se obtienen en general, en el intervalo estudiado de concentraciones de SDS y propanol, los criterios de posición y de posición-formado mostraron máximos de resolución muy distintos. Para el primero, el máximo correspondió claramente a una fase móvil de SDS 0.05 M y 2.5% de propanol, mientras que para el criterio de grado de solapamiento, se obtuvo una amplia cima de la función de resolución, con un máximo correspondiente a una fase móvil de SDS 0.05 M y 8% de propanol. En la Figura 40 se han superpuesto los cromatogramas experimentales de cada azocolorante, para cada una de las fases móviles utilizadas en la predicción de la retención. Es evidente que la composición de la fase móvil afecta apreciablemente el aspecto del cromatograma y la posibilidad de determinar conjuntamente los tres compuestos. Sin embargo, en los cinco cromatogramas, los tres picos aparecen más o menos resueltos (con la excepción de la fase móvil de SDS 0.1 M y 12.5% de propanol), diferenciándose especialmente en los tiempos de retención. Este resultado se hace también evidente en el examen del topograma obtenido, siguiendo el criterio de grado de solapamiento, en el que la función de resolución decrece con mucha lentitud, especialmente hasta la zona intermedia del mapa. La Figura 40a se corresponde con el máximo del criterio posicional y muestra unos picos cromatográficos con tiempos de elusión más elevados que los obtenidos con concentraciones mayores de SDS y de alcohol. Ello explica, en parte, la obtención de este máximo.
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
97
a
33
K` exp
27
21
15 15 21 27 33
K` exp b
16.2
K` exp
12.8
9.4
6.0 6.0 9.4 12.8 16.2
K` exp

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
98
c
14.3
K` exp
11.5
8.8
6.0 6.0 10.0 14.0 18.0
K` exp d
28.0
K` exp
21.0
14.0
7.0 6.0 14.0 21.0 28.0
K` exp
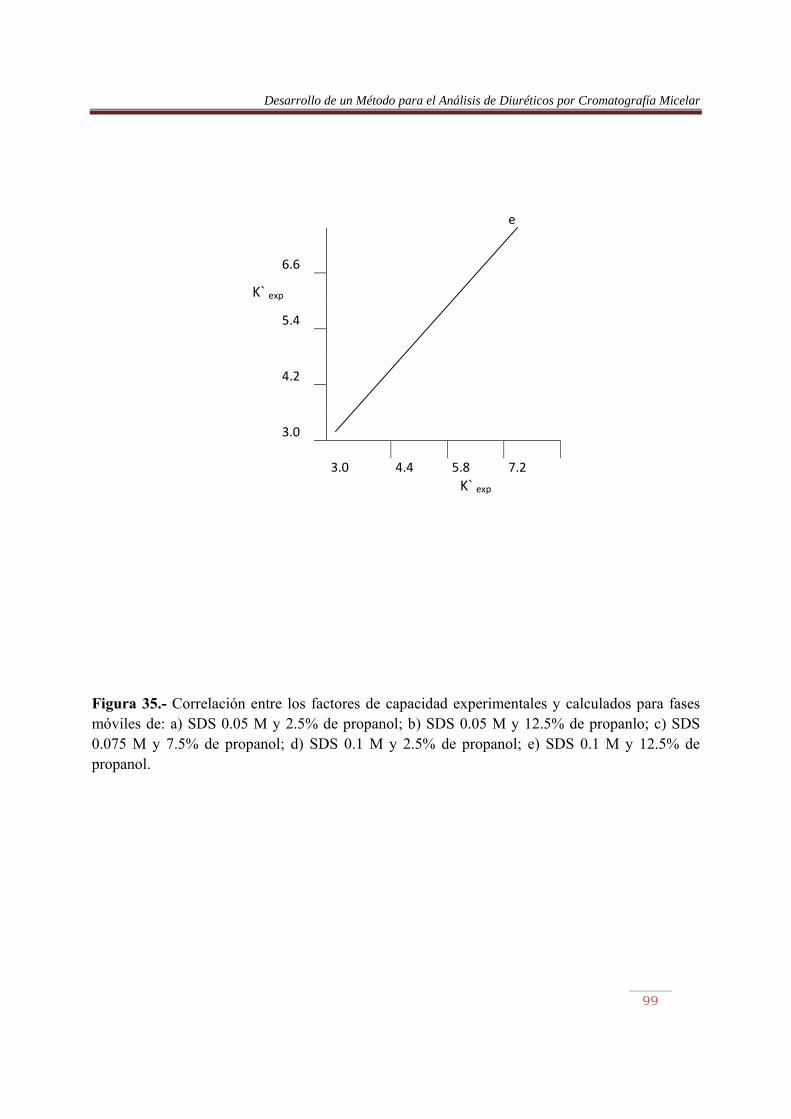
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
99
Figura 35.- Correlación entre los factores de capacidad experimentales y calculados para fases móviles de: a) SDS 0.05 M y 2.5% de propanol; b) SDS 0.05 M y 12.5% de propanlo; c) SDS 0.075 M y 7.5% de propanol; d) SDS 0.1 M y 2.5% de propanol; e) SDS 0.1 M y 12.5% de propanol.
e
6.6
K` exp
5.4
4.2
3.0 3.0 4.4 5.8 7.2
K` exp

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Figura 36.- Mapa de contorno de resolucion posicional.
Propanol
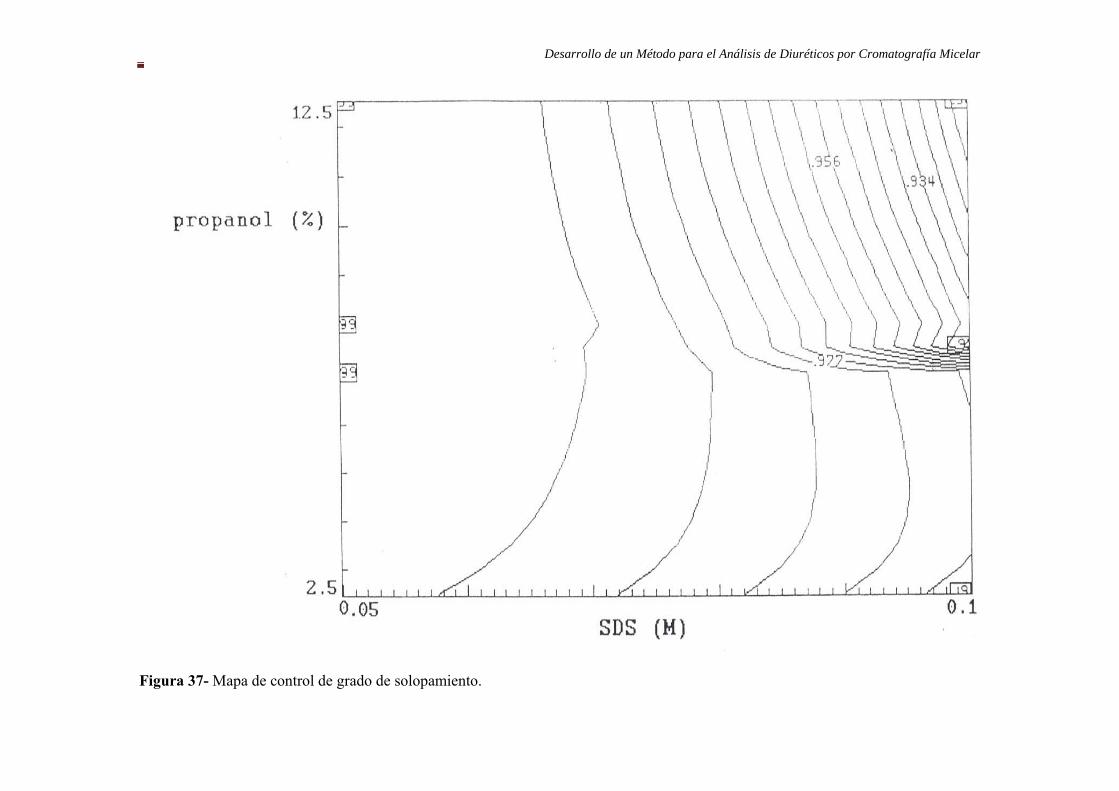
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Figura 37- Mapa de control de grado de solopamiento.
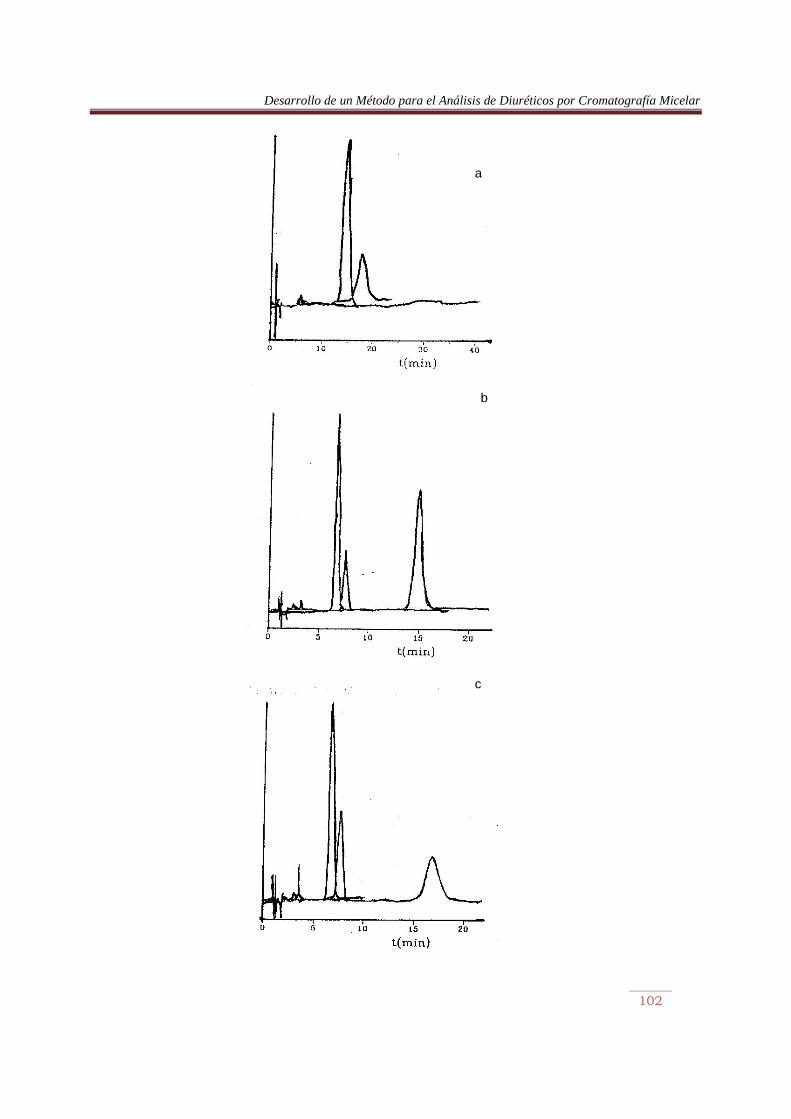
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
102
a
b
c

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
103
Figura 38.- Siuperposición de los cromatogramas experimentales de hidroclotiazida, hidroflumetiazida y furosemida para las fases móviles del diseño de la Figura 34a: a) SDS 0.05 M y 2.5% de propanol; b) SDS 0.05 M y 12.5% de propanol; c) SDS 0.075 M y 7.5% de propanol; d) SDS 0.1 M y 2.5% de propanol; e) SDS 0.1 M y 12.5% de propanol.
d
e

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
104
Los dos criterios de optimización sitúan los máximos para una concentración de SDS baja (la menor ensayada). Para visualizar la resolución alcanzada con distintas concentraciones de propanol, adicioando a una fase móvil que contiene SDS 0.05 M, se simularon una serie de cromatogramas, en los que la concentración de propanol varió entre 2.5% y 12.5%. La Figura 41 muestra estos cromatogramas. Preferiblemente, los picos deben aparecer a menores tiempos de retención, lo que se consigue con un incremento en la concentración de propanol. Sin embargo, debido a que la zona del óptimo de resolución posición-forma corresponde a una meseta (ver Figura 38, no se observan diferencias apreciables en la resolución entre las fases móviles de SDS 0.05 M. Por el contrario, se aprecia un empeoramiento en la resolución de los dos primeros picos al aumentar ligeramente la concentración de SDS (desde 0.05 M a 0.07) (ver Figuras 42 y 43). El deterioro en la resolución es más evidente para las fases móviles que contienen 12.5% de propanol. Esta observación no sugirió que podría obtenerse una resolución aún mayor rebajando la concentración de SDS. Por ello, se preparó otra serie de experiencias de optimización, aprovechando los datos de retención de las fases móviles de SDS 0.1 M conteniendo 2.5% y 12.5% de propanol, a las que se añadió los datos correspondientes a fases móviles de SDS 0.02 M conteniendo 2.5% y 12.5% de propanol. Con ello, se obtuvo finalmente el diseño de experiencias de la Figura 34b. Los datos de retención en las cuatro esquina del rectángulo son suficientes para obtener los coeficientes de la ecuación de retención (ecuación 14). La Tabla 19 recoge los datos de retención para todas las fases móviles utilizadas en el proceso de optimización, mientras que la Tabla 18 indica los coeficientes de la ecuación ajustada para cada azocolorante, en el intervalo de concentración de SDS 0.02 M – 0.1 M.


Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
Tabla 19.- Parámetros de retención para las fases móviles utilizadas en el proceso de modelización. Compuesto Composición fase móvil
Componente Concentración SDS (M)
Propanol(%) 0.02 2.5
0.02 12.5
0.05 2.5
0.05 7.5
K´ N B/A k´ N B/A k´ N B/A k´ N B/A HCTA HFM RURO
44.0 55.3
946 679
1.1 1.0
16.1 19.1 43.0
2768 2513 2822
0.9 1.0 0.52
15.6 19.4 32.5
638 381 161
1.1 1.2 1.6
9.5 11.2 26.6
1351 1205 778
1.1 1.1 1.1
SDS (M) Propanol (%)
0.05 10
0.05 12.5
0.075 7.5
0.075 17.5
k´ N B/A k´ N B/A k´ N B/A k´ N B/A HCTA HFM FURO
SSD 7.7 8.9 19.1
1579 1446 1319
1.0 1.0 1.0
6.7 7.7 16.1
1546 1507 1696
1.2 1.1 0.9
6.7 7.7 18.4
1021 931 624
1.1 1.1 1.0
2.8 3.1 5.8
1255 1062 1409
1.1 1.3 0.8
SDS (M) Propanol (%
0.1 2.5
0.1 12.5
0.125 7.5
0.125 17.5
k´ N B/A k´ N B/A k´ N B/A k´ N B/A HCTA HFM FURO
7.7 9.4 28.5
470 330 36
1.1 1.2 1.6
3.3 3.7 7.0
1048 925 1023
1.2 1.3 1.0
3.8 4.3 8.5
722 684 532
1.2 1.1 1.0
1.6 1.7 3.1
1727 3251 971
1.0 1.0 1.0
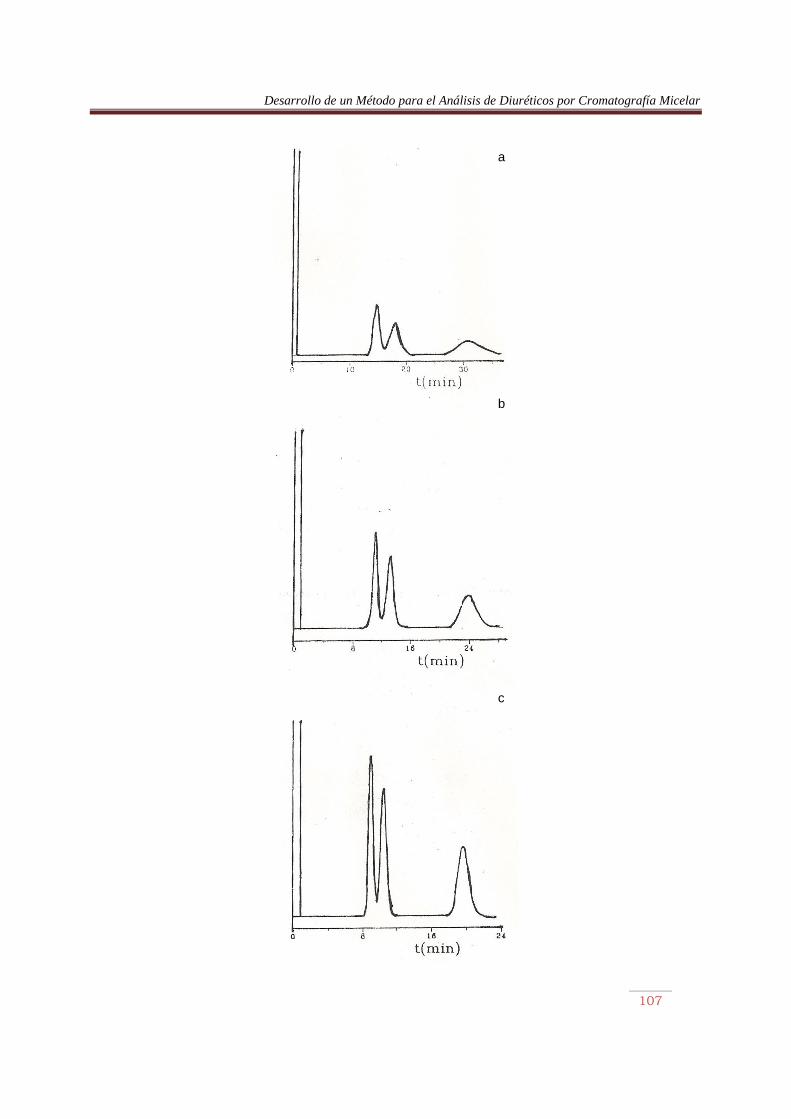
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
107
c
b
a

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
108
Figura 39- Simulación de la modificación de la retención para hidroclorotiazida, hidroflumetiazida y furosemida (los picos aparecen en este orden de izquierda a derecha), en fases móviles de SDS 0.05 M y concentraciones variables de propanol: a) 2.5%; b) 5%; c) 7.5%; d) 10%; e) 12.5%.
d
e
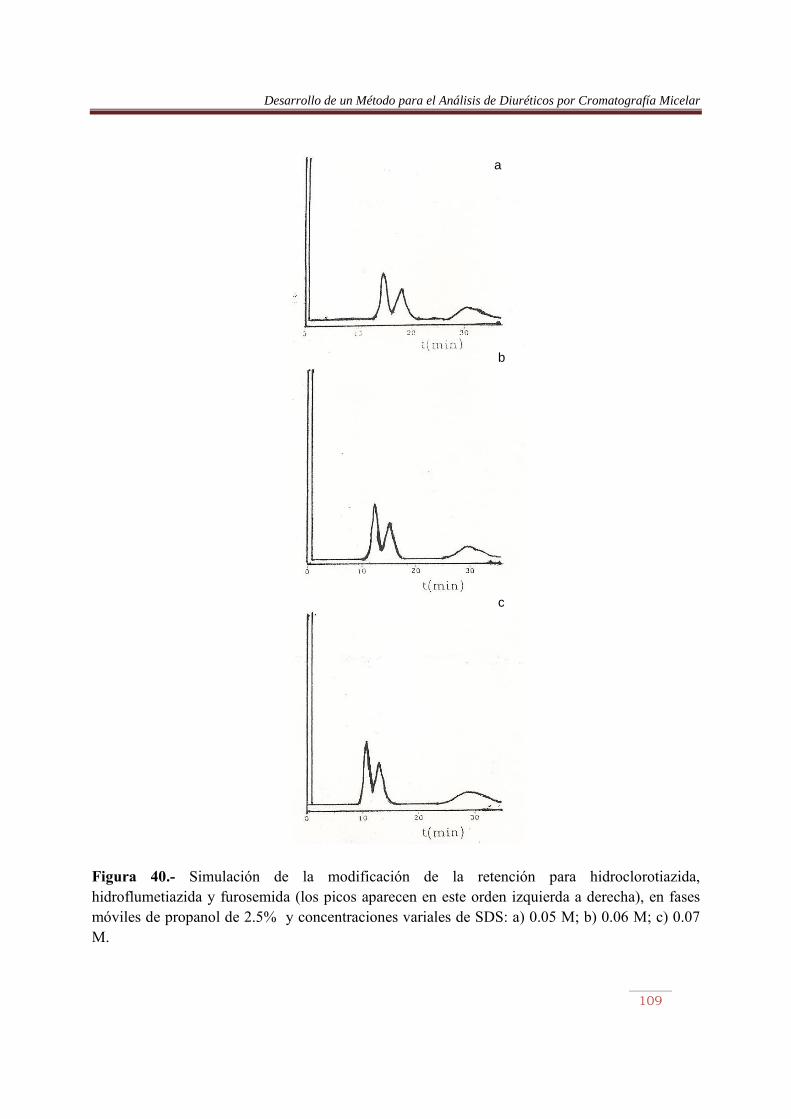
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
109
Figura 40.- Simulación de la modificación de la retención para hidroclorotiazida, hidroflumetiazida y furosemida (los picos aparecen en este orden izquierda a derecha), en fases móviles de propanol de 2.5% y concentraciones variales de SDS: a) 0.05 M; b) 0.06 M; c) 0.07 M.
a
b
c

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
110
Figura 41.- Simulación de la modificación de la retención para hidroclorotiazida, hidroflumetiazida y furosemida (los picos aparecen en este orden de izquierda a derecha), en fases móviles de propanol del 12.5% y concentraciones variables de SDS: a) 0.05 M; b) 0.06 M; c) 0.07 M.
a
c
b

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
111
Según se puede observar en la Tabla 18, se produce una modificación de los coeficientes de la ecuación de retención, al considerar fases móviles con un intervalo de concentraciones de SDS 0.02 – 0.1 M, respecto al intervalo 0.02 –0.05 M. Debe indicarse que no pudo obtenerse el factor de capacidad de la furosemida en una fase móvil de SDS 0.02 M y 2.5% de propanol, debido a su excesiva retención, por lo que no fue posible realizar el ajuste de los datos de este compuesto, siguiendo el diseño de la Figura 34b. El distinto valor de los coeficientes sugirió, que se produce una ligera variación del comportamiento de elusión para fases móviles conteniendo una baja concentración de SDS. De hecho, al utilizar la ecuación de retención obtenida con el diseño de la Figura 34ª, el error de predicción de los factores de retención predichos para la fase móvil de SDS 0.02 M y 2.5% de propanol fue del 15% y 23%, para hidroclorotiazida e hidroflumetiazida, respectivamente; mientras que para una fase móvil de SDS 0.02 M y 12.5% de propanol fue de 0.1%, 11% y 57%, para hidroclorotiazida, hidroflumetiazida y furosemida, respectivamente. El elevado error de predicción obtenido con furosemida se debe también a que la ecuación descriptora no es correcta, como lo indicaban los errores mostrados en la Tabla 17 para este diurético. La excesiva retención de la furosemida a concentraciones de SDS 0.05 M o inferiores, cuando la concentración de alcohol es baja, no permite modelizar adecuadamente su retención. Sin embargo, ello no ha constituido ningún inconveniente para poder predecir y optimizar la resolución de los tres azocolorantes, debido a que el pico de la furosemida aparece siempre a tiempos sensiblemente superiores al de los otros dos azocolorantes. La formación de micelas se ve afectada a partir de una cierta relación concentración de propanol / concentración de SDS, por o que debe evitarse trabajar con concentraciones de SDS demasiado bajas. La retención es excesiva con una fase móvil de SDS 0.02 M y una baja proporción de propanol, lo que obliga a aumentar la cantidad del alcohol. La Figura 44 muestra el cromatograma experimental que se obtendría al cromatografiar una mezcla de hidroclorotiazida, hidroflumetiazida y furosemida con una fase móvil de SDS 0.02 M y 12.5% de propanol. Los picos de los azocolorantes de los dos primeros compuestos aparecen bien resueltos, según se esperaba; sin embargo, la retención de la furosemida es excesiva. Por lo tanto, se prefirió seleccionar una fase móvil de SDS 0.05 M y 10% de propanol como la más adecuada para el análisis de los diuréticos en orina. Esta fase móvil es próxima a la óptima según el criterio de grado de solapamiento y posee una resolución prácticamente igual.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
112
V.4.- ANÁLISIS DE DIURÉTICOS EN ORINA Para comprobar la posibilidad de analizar los diuréticos en orina, se preparó una disolución introduciendo en matraz aforado de 50 ml, 5 mI de una disolución de cada uno de los diuréticos hidroclorotiazida, hidroflumetiazida y furosemida, en concentración de 100 :g/mI, y se aforó con orina. Así, la concentración de los diuréticos en la muestra de orina enriquecida fue de 10 :g/mI. Se tomó 10 mI de la muestra y se mezcló con 1 mI de HCI 1.65 M. La mezcla se sometió a hidrólisis y a diazotación y acoplamiento, según los procedimientos indicados en el apartado III.3. La detección se realizó a 530 nm, longitud de onda próxima a la de máxima absorción para los tres azocolorantes. Además, se sometió a hidrólisis una muestra de orina en ausencia de diuréticos, para comprobar si la matriz originaba alguna señal, tras ser sometida al proceso de hidrólisis y derivatización,y ser cromatografiada.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
113
Figura 42.- Cromatograma experimental de hidroclorotiazida, hidroflumetiazida y furosemida (de izquierda a derecha) para una fase móvil de SDS 0.02 M y 12.5% de propanol

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
114
Por otro lado, se compararon los cromatogramas de la mezcla de los tres diuréticos en presencia y ausencia de orina, y los de los diuréticos en agua inyectados separadamente y en una mezcla conjunta. En la Figura 45 se comparan los cromatogramas de orina en presencia de los diuréticos y en su ausencia, en la Figura 46 se comparan los cromatogramas de los diuréticos en presencia y ausencia de orina. Finalmente, en la Figura 47 se muestran los cromatogramas de los diuréticos en una disolución acuosa, inyectados conjunta o separadamente. Se observan en todos los cromatogramas claramente los picos de los tres diuréticos, que presentan unos tiempos de retención iguales en orina y en agua, independientemente de que los compuestos sean inyectados juntos o por separado. Sin embargo, en los cromatogramas de orina aparecen dos picos a tiempos de retención superior al de la furosemida, e inferior al de la hidroclorotiazida, respectivamente, que corresponden a la presencia de compuestos endógenos que sufren derivatización. Ninguno de estos picos interfiere en los análisis. La Figura 48 muestra los espectros de absorción de orina no derivatizada, y de orina sometida al procedimiento de hidrólisis y derivatización. En este último caso, aparece una banda sobre 550 nm, que indica la formación de un azocolorante. Se desconoce la naturaleza del compuesto endógeno derivatizado. En cromatografía líquida, cuando se inyecta una muestra de orina en la que no se ha eliminado totalmente la matriz, y se detecta a una longitud de onda en el UV, aparece una amplia señal al comienzo de los cromatogramas, debida a las proteínas de la matriz. En el procedimiento propuesto en este trabajo, donde se forman unos derivados que absorben en el visible, desaparece dicha interferencia.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
115
Figura 43.- Cromatograma experimentales de una muestra de orina enriquecida con los diuréticos hidroclorotiazida, hidroflumetiazida y furosemida (a) y de una muestra de orina derivatizasda. Las flechas indican los picos correspondientes a compuestos endógenos que forman azocolorantes.
a
b
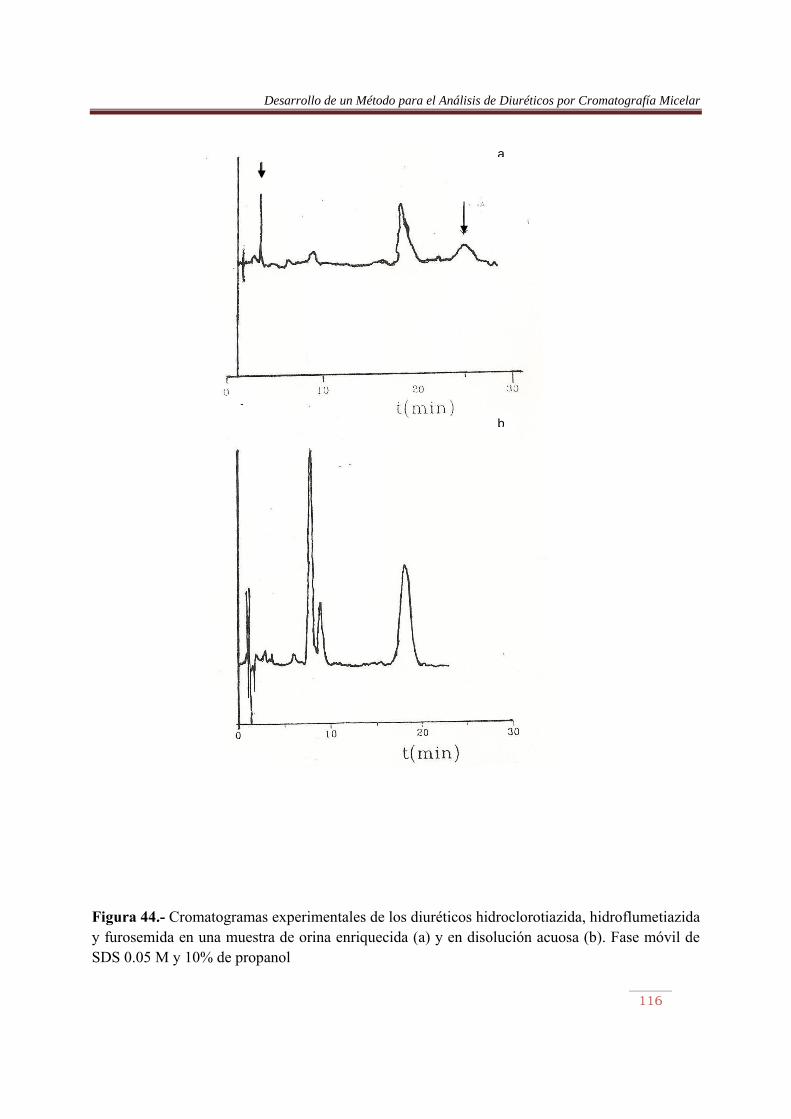
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
116
Figura 44.- Cromatogramas experimentales de los diuréticos hidroclorotiazida, hidroflumetiazida y furosemida en una muestra de orina enriquecida (a) y en disolución acuosa (b). Fase móvil de SDS 0.05 M y 10% de propanol
a
b
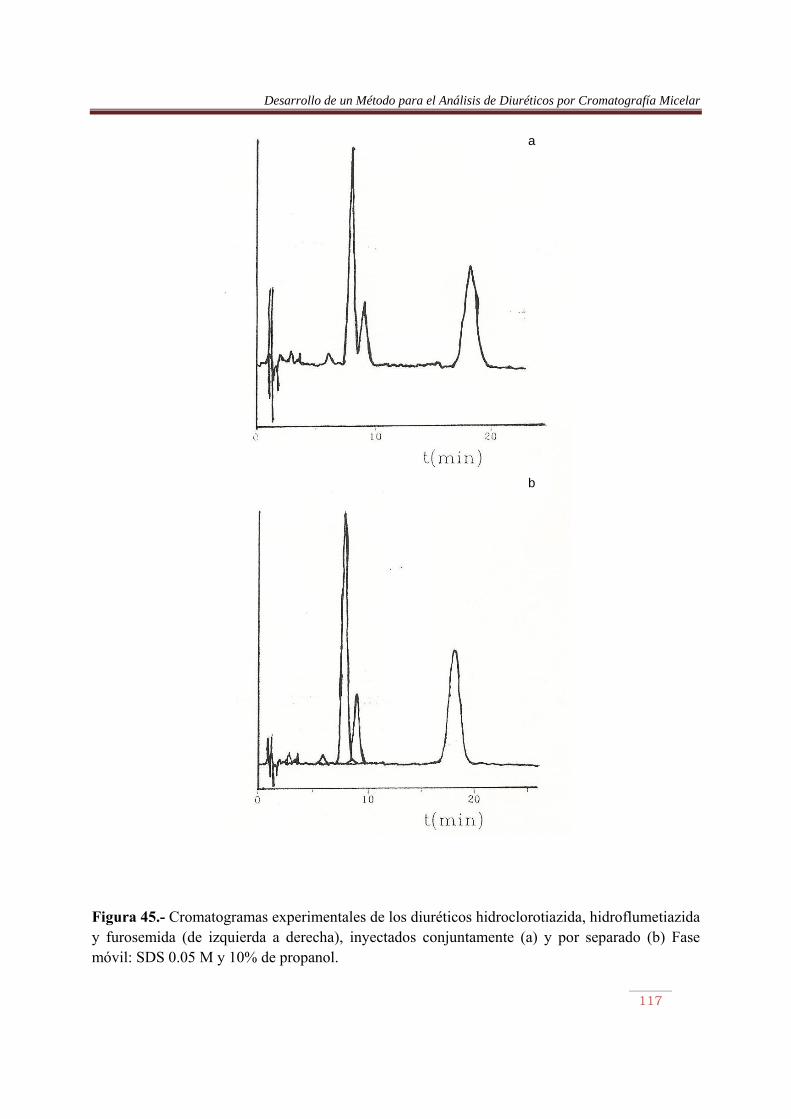
Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
117
Figura 45.- Cromatogramas experimentales de los diuréticos hidroclorotiazida, hidroflumetiazida y furosemida (de izquierda a derecha), inyectados conjuntamente (a) y por separado (b) Fase móvil: SDS 0.05 M y 10% de propanol.
a
b

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
118
Figura 46.- Espectos de absorción de una muestra de orina sin hidrolizar (1) y tras ser sometida a hidrólisis y derivatización (2 ).

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
119
Se estudió también la interferencia de otros diuréticos: acetazolamida, amilorida, bumetanida, clortalidona, triantereno y xipamida. Se obstuvieron los espectros de los compuestos sometidos a hidrólisis y derivatización. Las disoluciones no mostraron absorbancia en el visible, excepto la acetazolamida, que mostró a 550 nm una absorbancia de A = 0.056, en concentración de 10 :g/mI. Este valor de absorbancia es al menos 10 veces inferior al del derivado de hidroflumetiazida, y 20 veces inferior al de la hidroclorotiazida, y 20 veces inferior al de la hidroclorotiazida. Estas experiencias indican que es factible la determinación de los diuréticos del grupo de las tiazidas y de furosemida, en muestras de orina, mediante un procedimiento Cromatográfico que hace uso de fases móviles micelares, tras su hidrólisis y formación de azocolorantes.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
120
VI.- RESUMEN Y CONCLUSIONES: Se consideró la posibilidad de determinar cromatográficamente diuréticos del grupo de las tiazidas (hidroclorotiazida, clorotiazida, triclorometiazida, altiazida, hidroflumetiazida y bendroflumetiazida) y otros diuréticos que contienen un sustituyente sulfonamida en el anillo bencénico, utilizando fases móviles que contienen dodecilsulfato sódico (SDS). Para ello, se formaron azocolorantes de color rojo-violeta, tras hidrolizar los diuréticos en medio ácido y derivatiazarlos por reacción con nitrito sódico y clorhidrato de (1-naftil), etilendiamina (NED). Los estudios realizados indicaron que el proceso hidrolítico origina tan sólo tres arilaminas; La cloraminofenamida es el único diurético que originan una absorbancia apreciable al reaccionar con nitrito y NED, sin necesidad de ser sometido a hidrólisis, debido a que presenta la estructura de una arilamina. Se comprobó que este diurético es altamente estable, incluso tras ser sometido a un calentamiento intenso. Se realizaron diversas experiencias, en las que se constató la existencia de los tres compuestos hidrolizados. Sin embargo, las absortividades molares de los azocolorantes de los diuréticos que por hidrólisis originan un mismo compuestos, fueron muy distintas entre sí, lo que parece indicar que el grado de avance de la reacción de hidrólisis difiere de un diurético a otro. Se realizaron valoraciones ácido-base con NaOH para obtener las constantes de protonación de los azocolorantes, siguiendo la absorbancia a la longitud de onda del máximo y el pH de las disoluciones. La observación de las curvas de valoración indicó la presencia de dos equilibrios ácido-base consecutivos, con unas constantes de protonación bien diferenciadas. Ello permitió la determinación de cada constante por separado. Sin embargo, la prueba más concluyente sobre la formación de tan sólo tres azocolorantes es la obtención de tres picos. Los diuréticos hidrolizados son suficientemente estables y no precisan ser derivatizados inmediatamente después de su hidrólisis. Para elegir la fase móvil óptima, se utilizó un procedimiento de optimización interpretativo, obteniendo los datos de retención de los azocolorantes en un número reducido y seleccionado de fases móviles de SDS y propanol. Se obtuvo una amplia cima de la función de resolución, con un máximo para una fase móvil de SDS 0.05 M y 8% de propanol. Este último criterio es el más adecuado. Se seleccionó una fase móvil de SDS 0.05 M y 10% de propanol como la más adecuada para el análisis de los diuréticos en orina. Para estudiar la posibilidad de analizar los diuréticos en orina, se prepararon muestras de orina enriquecidas con los diuréticos. Se sometió también a hidrólisis una muestra de orina en ausencia de los diuréticos.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
121
Las experiencias mostradas en este trabajo indican, que es factible la determinación en muestras de orina, de los diuréticos del grupo de las tiazidas y de otros diuréticos que por hidrólisis originan una arilamina, mediante un procedimiento Cromatográfico que hace uso de fases móviles micelares, tras su hidrólisis y formación de azocolorantes.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
122
VIII.- BIBLIOGRAFIA 1. W.L. Koltum; J. Am Chem. Soc. 1957, 79, 5681. 2. R.W. Yost, L.S. Ettre y Rd. Colon 1980 Imstrumento a la Cromatografia Liquida Practica
(PERKIN ELMER). Editorial Corporación Perkin Elmer. 3. A. Maciag y R. Schoental; Mikrochemie 1938, 24, 243. 4. F. Feigl y J.R. Amaral; Anal. Chem 1958, 30, 1148. 5. P. Ehrlich; Z. Klin. Med. 18892, 5, 285. 6. H. Pauly; Z. Physiol. Chem. 1904, 42, 508. 7. H. Pauly; Z. Phisiol. Chem. 1905, 44, 159. 8. R.F. Hunter; J. Chem. Soc. 1926, 1385. 9. J. Bartos; Ann. Pharm. Fr. 1971, 29, 147. 10. M. Pesez, J. Bartos y J.F. Burtin; Talanta 1960, 5 213. 11. P. Zirins; Ann. Pharm. Fr. 1961, 19, 604. 12. T. Urbanyi y J.A. Mollica, Jr.; J. Pharm Sci. 1968, 57 13. A. Roe; "Organic Reactions", Vol. 5 (editado por R. Adams), Wiley, NEw York, 1949, pag.
193. 14. K.G. Rutherford, W. Redmond y J. Rigmont; J. Org. Chem. 1961, 26, 5149. 15. G.G. Yakobson, A.I. D'Yachenko y F.A. Bel'chikova; J. Gen. Chem. U.S.S.R. 1962, 32, 842. 16. E. Sawicki, T.R. Hauser y T.W. Stanley; Anal. Chem. 1959, 31, 2063. 17. E. Sawicki, T.W. Stanley y T.R. Hauser; Chemist-Analyst 1959, 48, 30. 18. L.P. Hammett; "Physycal Organic Chemistry", McGraw-Hill, New York, 1940, pag. 314. 19. A.C. Bratton y E.I. Marshall, Jr.; J. Biol. Chem. 1939, 128, 537. 20. J.H. Ridd; Q. Rev. Chem. Soc. 1961, 15, 418. 21. K. A. Connors; "Reaction Mechanisms in Organic Analytical Chemistry", Wiley, New York,
1973, pag. 238. 22. J.P. Dux y C. Rosenblum; Anal. Chem. 1949, 21, 1524. 23. R.A. Kaselis, W. Leibmann, W. Seaman, J.P. Sikels, E.I. Stearns y J.T. Woods; Anal. Chem.
1941, 23, 746. 24. A. Glazko, L. Wolf y W. Dill; Arch Biochem. 1949, 23, 411. 25. S.P. Bessam y S. Stevens; J. Lab. Clin. Med. 1950, 35, 127. 26. J. Levine y H. Fishback; Antibiotic Chemother 1951, 1, 59. 27. P. Gros. y R. Raveux; Chim. Ther. 1969, 4, 312. 28. K. Martinek, A.K. Yatsimirski, A.V. Levashov y I.V. Berezin, "Micellization, Solubilization
and Microemulsions" (editado por K.L. Mittal), Vol. 2, Plenum, New York, 1976, pp. 489-507.
29. G. Ramis Ramos, J.S. Esteve Romero y M.C. García Alvarez-Coque; Anal. Chim. Acta 1989, 223, 327.
30. J.S. Esteve Romero, G. Ramis Ramos y M.C. García Alvarez-Coque; J. Colloid Interface Sci. 1991, 141, 44.
31. J.S. Esteve Romero, M.C. García Alvarez-Coque y G. Ramis Ramos; Talanta 1991, 38, 1285. 32. J.S. Esteve Romero, E.F. Simó Alfonso, M.C. García Alvarez-Coque y G. Ramis Ramos;
Talanta 1993, 40, 1711. 33. J.S. Esteve Romero, G. Ramis Ramos, R. Forteza Coll y V. Cerdá MArtín; Anal Chim Acta
1991, 242, 143. 34. J.S. Esteve Romero, L. Alvarez Rodríguez, M.C. García Alvarez-Coque y G. Ramis Ramos;
Analyst 1994, 199, 1381.

Desarrollo de un Método para el Análisis de Diuréticos por Cromatografía Micelar
123
35. A. Berthod, I. Girard y C. Gonnet, "Ordered Media in Chemical Separations" (editado por W.L. Hinze y D.W. Armstrong), ACS Symposium Series, American Chemical Society, Washington, DC, Vol. 342, 1987, pag. 130.
36. D.W. Armstrong y F. Nome; Anal. Chem. 1981, 53, 1662. 37. M. Arunyanart y L.J. Cline-Love; Anal. Chem. 1984, 56, 1557. 38. J.P. Foley; Anal. Chim. Acta 1990, 231, 237. 39. M.J. Medina Hernández y M.C. GArcía Alvarez-Coque; Analyst 1992, 117, 831. 40. J.R. Torres Lapasió, R.M. Villanueva Camañas, J.M. Sanchis Mallols, M.J. Medina
Hernández y M.C. García Alvarez-Coque; J. Chromatogr. 1993, 639, 87. 41. V. Rimbau; OFFARM, Farmacia y Sociedad 1991, 10, 57. 42. V. Rimbau; OFFARM, Farmacia y Sociedad 1991, 10, 58. 43. J. Florez, J.A. Armijo y A. MEdiavilla; "Farmacología Humana", Ediciones de la Universidad
de NAvarra, Pamplona, 1989. 44. E. Bonet Domingo, J.R. Torres Lapasió, M.J. Medina HErnández y M.C. García Alvarez-
Coque; Anal. Chim. Acta 1994, 287, 201. 45. J.R. Torres Lapasió, R.M. Villanueva Camañas, J.M. Sanchis Mallols, M.J. MEdina
HErnández y M.C. García Alvarez-Coque; J. Chromatogr. 1994, 677, 239. 46. J.R. Torres Lapasió, M.J. Medina HErnández, R.M. Villanueva Camañas y M.C. García
Alvarez-Coque; Chromatographia 1995, 40, 279. 47. M.C. García Alvarez-Coque, J.R. Torres Lapasió y J.J. Baeza Baeza; Anal. Chim. Acta (en
prensa). 48. J.R. Torres Lapasió, J.J. Baeza Baeza y M.C. García Alvarez-Coque; resultados no
publicados.

A LA LIBERTAD A LA UNIVERSIDAD


















